
1 A REPORT ON “STUDY OF SIGNALLING CASCADES IN CANCER AND GROWTH FACTOR SIGNALLING AND...
46
1 A REPORT ON “STUDY OF SIGNALLING CASCADES IN CANCER AND GROWTH FACTOR SIGNALLING AND ONCOGENES” PREPARED IN THE PARTIAL FULFILLMENT OF SPECIAL PROJECT, BIO C491 (APRIL, 2012) NAME OF THE STUDENT IDNO. B SRIMAN 2009B1A8319G PROJECT INSTRUCTOR: Dr. VIJAY SHREE NAYAK
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of 1 A REPORT ON “STUDY OF SIGNALLING CASCADES IN CANCER AND GROWTH FACTOR SIGNALLING AND...

1
A REPORT ON
“STUDY OF SIGNALLING CASCADES IN CANCER AND
GROWTH FACTOR SIGNALLING AND ONCOGENES”
PREPARED IN THE PARTIAL FULFILLMENT OF
SPECIAL PROJECT, BIO C491
(APRIL, 2012)
NAME OF THE STUDENT IDNO.
B SRIMAN 2009B1A8319G
PROJECT INSTRUCTOR: Dr. VIJAY SHREE NAYAK

2
ACKNOWLDGEMENT
First and foremost, I am grateful towards Dr. Vijay shree nayak for providing
such a great opportunity. She has given a constant support, guidance and
motivation throughout the project and providing relevant study materials.
We would like to conclude by thanking our instructor and mentor Dr. Vijay
shree nayak once again for giving us this invaluable learning opportunity of a
lifetime.
We thank Dr.utpal Roy (Instructor in charge) for providing such a
valuable study oriented project.

3
Abstract:
The main aim of this project is to study the different types of cancer,
how cancer is formed and different pathways related to cancer.
During this Project I will review basic research articles and molecular aspects of
cancer. Topics of discussion may include cancer development and progression,
oncogenes and tumour suppressor genes, cell cycle control, apoptosis,
angiogenesis, cell migration/metastasis, and the future of cancer treatment.
To develop a clear appreciation that cancer is not one disease; rather the
mechanism, cause and therapy of cancer are variable depending on the cellular
origin of the disease.
To gain a thorough understanding of the types of molecules and pathways
involved in the process of converting a normal cell into a cancer cell and how
this knowledge has improved over time.
To understand the relationship between knowledge of the detailed mechanism
of cancer, the development of plausible drug therapies and the improvement of
quality of life for individuals with this disease.

4
TABLE OF CONTENTS
ACKNOWLEDGEMENT
ABSTRACT
1. INTRODUCTION
1.1 WHAT IS CANCER??
1.2 DIFFERENT TYPES OF CANCER
1.3 INFLUENTIAL FACTORS AFFECTING CARCINOGENESIS
2. REGULATION OF GENE EXPRESSION
2.1 GENE STRUCTURE
2.2 MOLECULAR TARGETS IN CANCER THERAPHY
3. GROWTH FACTOR SIGNALLING AND ONCOGENES
3.1 EGF PATHWAY
3.2 RAS-MAP KINASE PATHWAY
3.3 JAK-STAT PATHWAY
3.4 HEDGEHOG PATHWAY
3.5 WNT SIGNALLING PATHWYA
4. GROWTH INHIBITION AND TUMOUR SUPRESSOR GENES
4.1 P53 PATHWAY
4.2 RETINOBLASTMO PATHWAY
5. CELL CYCLE
5.1 CDC DEPENDENT KINASE,CDK REGULATION
5.2 PROGRESSION OF CANCER THROUGH CHECK POINTS

5
6. APOPTOPSIS
6.1 MOLECULAR MECHANISM
6.2 APOPTOTOIC DRUG
7. STEM CELL AND CANCER
7.1 DIFFERNTAION AND REGULATION OF CANCER
8. CANCER IN FUTURE
8.1 CANCER VACCINE, CANCER TREATMENT
8.2 TREATING CANCER SYMPOTOMS,
8.3 IMAGING AND CANCER NANOTECHNOLOGY TECHNIQUES
9. CONCLUSIONS AND RECOMMENDATIONS
10. SCOPE FOR FUTURE WORK
11. REFERENCES
12. APPENDIX

6
INTRODUCTION
CANCER:
Cancer known medically as a malignant neoplasm, is a broad group of
various diseases, all involving unregulated cell growth. In cancer, cells divide and
grow uncontrollably, forming malignant tumours, and invade nearby parts of
the body. The cancer may also spread to more distant parts of the body
through the lymphatic system or bloodstream. Not all tumours are
cancerous. Benign tumours do not grow uncontrollably, do not invade
neighbouring tissues, and do not spread throughout the body. There are over
200 different known cancers that afflict humans.
Determining what causes cancer is complex. Many things are known to increase
the risk of cancer, including tobacco use, certain infections, radiation, lack of
physical activity, obesity, and environmental pollutants. These can directly
damage genes or combine with existing genetic faults within cells to cause the
disease.[3] Approximately five to ten per cent of cancers are entirely hereditary.
Cancer can be detected in a number of ways, including the presence of
certain signs and symptoms, screening tests, or medical imaging. Once a
possible cancer is detected it is diagnosed by microscopic examination of a tissue
sample. Cancer is usually treated with chemotherapy, radiation
therapy and surgery. The chances of surviving the disease vary greatly by the
type and location of the cancer and the extent of disease at the start of
treatment. While cancer can affect people of all ages, and a few types of cancer
are more common in children, the risk of developing cancer generally increases
with age. In 2007, cancer caused about 13% of all human deaths worldwide
(7.9 million). Rates are rising as more people live to an old age and as mass
lifestyle changes occur in the developing world
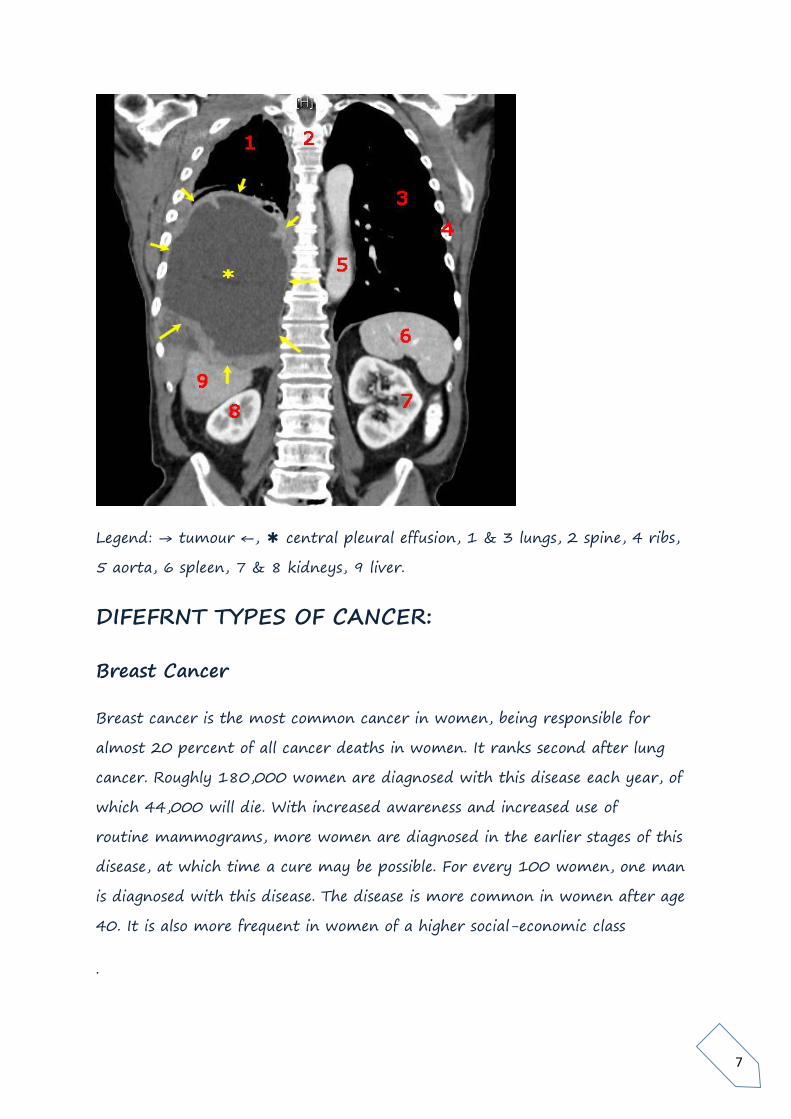
7
Legend: → tumour ←, ✱ central pleural effusion, 1 & 3 lungs, 2 spine, 4 ribs,
5 aorta, 6 spleen, 7 & 8 kidneys, 9 liver.
DIFEFRNT TYPES OF CANCER:
Breast Cancer
Breast cancer is the most common cancer in women, being responsible for
almost 20 percent of all cancer deaths in women. It ranks second after lung
cancer. Roughly 180,000 women are diagnosed with this disease each year, of
which 44,000 will die. With increased awareness and increased use of
routine mammograms, more women are diagnosed in the earlier stages of this
disease, at which time a cure may be possible. For every 100 women, one man
is diagnosed with this disease. The disease is more common in women after age
40. It is also more frequent in women of a higher social-economic class
.

8
Many factors are known to increase the risk of development of breast cancer:
Genetic predisposition. A few genetic markers have been linked to development
of breast cancer.
History of breast cancer in the same patient, in the opposite breast
Onset of menstruation in early ages
Late onset of menopause
Radiation exposure
Heavy alcohol consumption
High fat diet
Obesity
First pregnancy after age of 30
Very tall women
Colon Cancer
Colorectal cancer is the third most common cancer in men and women. An
estimated 131,000 Americans are diagnosed with this disease each year and
some 55,000 die as a result of it. Certain genetic factors play a role in the
development of this cancer. The specific cause of colorectal cancer is unknown;
however, environmental, genetic, familial factors and pre-existing Ulcerative
Colitis have been linked to the development of this cancer. It is more common
among African-Americans.
Risk Factors
Age: Average age at the time of diagnosis is between 60-65, and the older we
get the higher our risk of colorectal cancer.
Family History of colorectal cancer increases the risk of developing this illness in
first- degree relatives. Certain familial conditions, like Familial Polyposis, is
associated with a much higher risk.

9
Genetic factors clearly play a role in the development of colorectal cancers.
Several genetic and inherited illnesses carry a very high risk of colorectal cancer:
Familial Polyposis, Turcot syndrome, Gardner syndrome, Peutz-Jeghers
syndrome, Juvenile Polyposis, Cowden's disease, Neurofibromatosis.
Ulcerative colitis , High Dietary Fat and Low Dietary Fiber can each increase the
risk of this cancer.
Signs and Symptoms
This cancer may exhibit no signs in its early stages. Gradually, as the disease
progresses, any of the following may be seen;
Blood in the stool
Diarrhea
Constipation
Bowel obstruction, causing nausea, vomiting and abdominal distention
Abdominal pain
Pelvic pain
Anaemia due to blood Loss
Weight loss
Loss of appetite
Fatigue
Lung cancer:
Lung cancer is the second most common malignancy affecting both sexes.
Roughly 180,000 Americans are diagnosed with this illness ever year. It is
considered the most rapidly increasing cause of death from cancer. Since 1987,
lung cancer has been the leading cause of cancer death in women, surpassing
breast cancer. And while lung cancer incidence has levelled off among men, it
continues to rise steadily among women. The average age of patients with lung
cancer is 60 years. It is more common in African-Americans and Hawaiians.

10
Causes:
Cigarette smoking is the number one cause of this disease. Even passive
inhalation of the smoke increases the chance of developing this illness. Radon
exposure is another cause of lung cancer, killing 14,000 Americans every year.
Asbestos exposure also increases Lung cancer risk. The risk becomes astronomical
in exposed individuals who also smoke.
Signs and Symptoms
Patients do not manifest any signs in the very early stages. Cough, shortness of
breath, chest pain or blood in the sputum is among the early warning signs.
Other signs of this illness could be a change of voice, hoarseness, weakness,
fatigue, and weight loss.
Establishing Diagnosis
When the diagnosis is suspected, patients must be examined carefully by a
qualified physician. A chest x-ray along with studying a sample of sputum are
the very first steps in establishing a diagnosis. If the study sputum does not
confirm the diagnosis, then Bronchoscopy and biopsy are the next steps. In
certain patients, cancer may have already spread to lymph glands in the neck.
In such cases, a Fine needle aspiration of the lymph gland should be performed.
This is a fairly easy procedure. Unfortunately, most doctors avoid the simple
test of sputum study. There is no reason not to perform this test, since it
establishes the diagnosis in about 30% of patients. In a small percentage of
patients, none of the above tests will lead into a diagnosis and there will be a
need to proceed with more invasive procedures, and perhaps surgery.

11
Different Types of Lung cancer
There are two basic kinds of lung cancer:
Small cell or oat cell, that occurs in one-third of all patients with lung cancer
Non-small cell in two-thirds of patients.
This distinction is rather important, because the treatment for the two kinds is
very different. Small cell cancers are primarily treated with Chemotherapy.
Surgery does not play a role in this type of lung cancer. On the other hand,
Non-small cell lung cancer is treated primarily with surgery.
Another type of cancer that can develop in the lungs is Carcinoid Tumour.
RTK (RECEPTOR TYROSINE KINASE) PATHWAY
Four common structural features shared among RTKs:
Extracellular ligand-binding domain
Single trans membrane domain
Cytoplasmic tyrosine kinase domain(s)
Regulatory domains

12
Ligand-induced RTK activation induces Receptor dimerization, leading to
activation of catalytic domains
Receptor auto Tran’s phosphorylation:
Stimulates kinase activity Leads to phosphorylation of additional proteins
involved in receptor signalling pathway .Provides “docking sites” for
downstream signaling proteins (Grb2, PI3-kinase, phospholipase Cg, etc.)
Map kinase pathway:

13
Jak stst pathway:

14
JAK-STAT pathway
Binding of erythropoietin causes dimerization of receptors and recruitment of
soluble JAK kinase to cytosolic domain
JAKs are phosphorylated (activated) and phosphorylate receptors
STATs bind to receptors (SH2) and become phosphorylated
Phosphorylated STATs form dimers activating NLS (nuclear localization)
STAT dimers activate transcription of EPO specific genes
Wnt pathway
Wnt proteins released from or presented on the surface of signaling cells act on
target cells by binding to Frizzled (Fz)/LDL-related protein (LDR) complex at
the cell surface.
Receptors

15
Frizzled receptors, like GPCRs, are transmembrane proteins that span 7 times
the plasma membrane.
Their ligand-binding site is exposed outside the surface of the cell.
Their effector site extends into the cytosol.
Fz receptors transduce signals to intra-cellular proteins including Dsh, GSK-3ß,
Axin, APC and ß-catenin
Nuclear ß-catenin interacts with lymphoid enhancer-binding factor 1/T cell-
specific transcription factor (LEF/TCF) to affect transcription

16
17
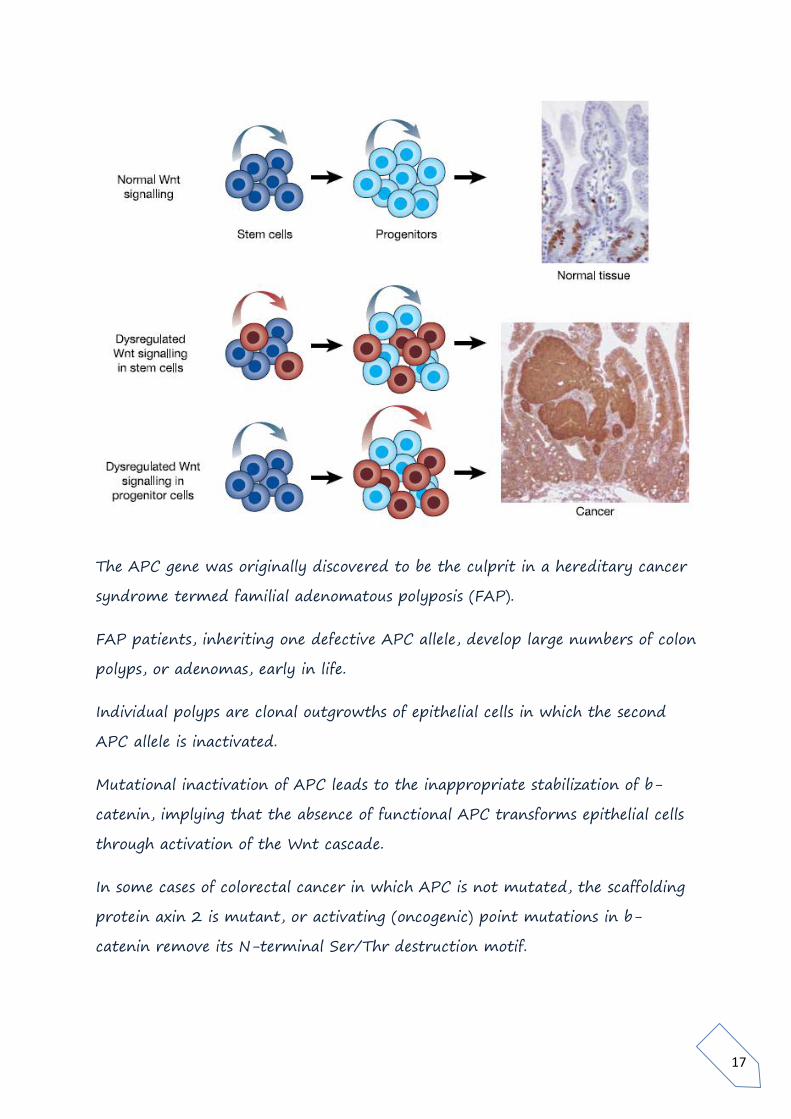
The APC gene was originally discovered to be the culprit in a hereditary cancer
syndrome termed familial adenomatous polyposis (FAP).
FAP patients, inheriting one defective APC allele, develop large numbers of colon
polyps, or adenomas, early in life.
Individual polyps are clonal outgrowths of epithelial cells in which the second
APC allele is inactivated.
Mutational inactivation of APC leads to the inappropriate stabilization of b-
catenin, implying that the absence of functional APC transforms epithelial cells
through activation of the Wnt cascade.
In some cases of colorectal cancer in which APC is not mutated, the scaffolding
protein axin 2 is mutant, or activating (oncogenic) point mutations in b-
catenin remove its N-terminal Ser/Thr destruction motif.

18
Growth inhibition and tumour suppressor genes:
Carcinogenesis or oncogenes is or tumorigenesis is literally the creation
of cancer. It is a process by which normal cells are transformed into cancer cells.
It is characterized by a progression of changes at the cellular, genetic
and epigenetic level that ultimately reprogram a cell to undergo
uncontrolled cell division, thus forming a malignant mass.
Cell division is a physiological process that occurs in almost all tissues and under
many circumstances. Under normal circumstances, the balance between
proliferation and programmed cell death, usually in the form of apoptosis, is
maintained by tightly regulating both processes to ensure the integrity of organs
and tissues. Mutations and epimutations in DNA that lead to cancer (only
certain mutations and epimutations can lead to cancer and the majority of
potential mutations and epimutations will have no bearing) disrupt these
orderly processes by disrupting the programming regulating the processes.
Carcinogenesis is caused by mutation and epimutation of the genetic material of
normal cells, which upsets the normal balance between proliferation and cell
death. This results in uncontrolled cell division and the evolution of those
cells by natural selection in the body. The uncontrolled and often rapid
proliferation of cells can lead to benign tumors; some types of these may turn
into malignant tumors (cancer). Benign tumors do not spread to other parts of
the body or invade other tissues, and they are rarely a threat to life unless they
compress vital structures or are physiologically active, for instance, producing a
hormone. Malignant tumors can invade other organs, spread to distant
locations (metastasis) and become life-threatening.
More than one mutation is necessary for carcinogenesis. In fact, a series of
several mutations to certain classes of genes is usually required before a normal
cell will transform into a cancer cell. On average, for example, 15 "driver
mutations" and 60 "passenger" mutations are found in colon cancers. Mutations
in those certain types of genes that play vital roles in cell division, apoptosis (cell

19
death), and mutations and epimutations (see article Genome instability) in DNA
repair genes will cause a cell to lose control of its cell proliferation.
Oncovirinae, viruses that contain an oncogene, are categorized as oncogenic
because they trigger the growth of tumorous tissues in the host. This process is
also referred to as viral transformation.
Cancer is fundamentally a disease of regulation of tissue growth. In order for a
normal cell to transform into a cancer cell, genes that regulate cell growth and
differentiation must be altered.[3] Genetic and epigenetic changes can occur at
many levels, from gain or loss of entire chromosomes, to a mutation affecting
a single DNA nucleotide, or to silencing or activating microRNA that controls
expression of 100 to 500 genes.[4][5] There are two broad categories of genes
that are affected by these changes. Oncogenes may be normal genes that are
expressed at inappropriately high levels, or altered genes that have novel
properties. In either case, expression of these genes promotes the malignant
phenotype of cancer cells. Tumor suppressor genes are genes that inhibit cell
division, survival, or other properties of cancer cells. Tumor suppressor genes are
often disabled by cancer-promoting genetic changes. Typically, changes in many
genes are required to transform a normal cell into a cancer cell.
There is a diverse classification scheme for the various genomic changes that
may contribute to the generation of cancer cells. Many of these changes
are mutations, or changes in the nucleotide sequence of genomic DNA. There are
also many epigenetic changes that alter whether genes are expressed or not
expressed. Aneuploidy, the presence of an abnormal number of chromosomes, is
one genomic change that is not a mutation, and may involve either gain or loss
of one or more chromosomes through errors in mitosis.
Large-scale mutations involve the deletion or gain of a portion of a
chromosome. Genomic amplification occurs when a cell gains many copies (often
20 or more) of a small chromosomal region, usually containing one or more
oncogenes and adjacent genetic material. Translocation occurs when two

20
separate chromosomal regions become abnormally fused, often at a
characteristic location. A well-known example of this is the Philadelphia
chromosome, or translocation of chromosomes 9 and 22, which occurs
inchronic myelogenous leukemia, and results in production of the BCR-
abl fusion protein, an oncogenic tyrosine kinase.
Small-scale mutations include point mutations, deletions, and insertions, which
may occur in the promoter of a gene and affect its expression, or may occur in
the gene'scoding sequence and alter the function or stability of
its protein product. Disruption of a single gene may also result from integration
of genomic material from a DNA virus or retrovirus, and such an event may
also result in the expression of viral oncogenes in the affected cell and its
descendants.

21
Tumor suppressor genes
Many tumour suppressor genes effect signal transduction pathways that
regulate apoptosis, also known as "programmed cell death".
Tumor suppressor genes code for anti-proliferation signals and proteins that
suppress mitosis and cell growth. Generally, tumor suppressors are transcription
factors that are activated by cellular stress or DNA damage. Often DNA damage
will cause the presence of free-floating genetic material as well as other signs,
and will trigger enzymes and pathways that lead to the activation of tumor
suppressor genes. The functions of such genes is to arrest the progression of the
cell cycle in order to carry out DNA repair, preventing mutations from being
passed on to daughter cells. The p53 protein, one of the most important
studied tumor suppressor genes, is a transcription factor activated by many
cellular stressors including hypoxia and ultraviolet radiation damage.
Despite nearly half of all cancers possibly involving alterations in p53, its tumor
suppressor function is poorly understood. p53 clearly has two functions: one a
nuclear role as a transcription factor, and the other a cytoplasmic role in
regulating the cell cycle, cell division, and apoptosis.
The Warburg hypothesis is the preferential use of glycolysis for energy to sustain
cancer growth. p53 has been shown to regulate the shift from the respiratory
to the glycolytic pathway.
However, a mutation can damage the tumor suppressor gene itself, or the
signal pathway that activates it, "switching it off". The invariable consequence of
this is that DNA repair is hindered or inhibited: DNA damage accumulates
without repair, inevitably leading to cancer.
Mutations of tumor suppressor genes that occur in germ line cells are passed
along to offspring, and increase the likelihood for cancer diagnoses in subsequent
generations. Members of these families have increased incidence and decreased
latency of multiple tumors. The tumor types are typical for each type of tumor

22
suppressor gene mutation, with some mutations causing particular cancers, and
other mutations causing others. The mode of inheritance of mutant tumor
suppressors is that an affected member inherits a defective copy from one
parent, and a normal copy from the other. For instance, individuals who inherit
one mutant p53 allele (and are therefore heterozygous for mutatedp53) can
develop melanomas and pancreatic cancer, known as Li-Fraumeni syndrome.
Other inherited tumor suppressor gene syndromes include Rb mutations, linked
to retinoblastoma, and APC gene mutations, linked to adenopolyposis colon
cancer. Adenopolyposis colon cancer is associated with thousands of polyps in
colon while young, leading to colon cancer at a relatively early age. Finally,
inherited mutations in BRCA1 and BRCA2 lead to early onset of breast cancer.
Development of cancer was proposed in 1971 to depend on at least two
mutational events. In what became known as the Knudson two-hit hypothesis,
an inherited, germ-line mutation in a tumor suppressor gene would cause
cancer only if another mutation event occurred later in the organism's life,
inactivating the other allele of that tumor suppressor gene.
Usually, oncogenes are dominant, as they contain gain-of-function mutations,
while mutated tumor suppressors are recessive, as they contain loss-of-function
mutations. Each cell has two copies of the same gene, one from each parent,
and under most cases gain of function mutations in just one copy of a
particular proto-oncogene is enough to make that gene a true oncogene. On the
other hand, loss of function mutations need to happen in both copies of a
tumor suppressor gene to render that gene completely non-functional.
However, cases exist in which one mutated copy of a tumor suppressor gene can
render the other, wild-type copy non-functional. This phenomenon is called
the dominant negative effect and is observed in many p53 mutations.
Knudson's two hit model has recently been challenged by several investigators.
Inactivation of one allele of some tumor suppressor genes is sufficient to cause
tumors. This phenomenon is called haplo insufficiency and has been
demonstrated by a number of experimental approaches. Tumors caused

23
by haplo insufficiency usually have a later age of onset when compared with
those by a two hit process.
Cell cycle:
DNA damage and deficient DNA repair in carcinogenesis
The central role of DNA damage and epigenetic defects in DNA repair genes in
carcinogenesis
DNA damage is considered to be the primary cause of cancer. [13] More than
10,000 new naturally occurring DNA damages arise, on average, per human
cell, per day, due to endogenous cellular processes (see article DNA damage
(naturally occurring).

24
Additional DNA damages can arise from exposure to exogenous agents. As one
example of an exogenous carcinogenic agent, tobacco smoke causes increased
DNA damage, and this DNA damages likely cause the increase of lung cancer
due to smoking. In other examples, UV light from solar radiation causes DNA
damage that is important in melanoma, helicobacter pylori infection produces
high levels of reactive oxygen species that damage DNA and contributes to
gastric cancer, and the Aspergillus metabolite, aflatoxin, is a DNA damaging
agent that is causative in liver cancer.
DNA damages can also be caused by endogenous (naturally occurring) agents.
Katsurano et al. indicated that macrophages and neutrophils in an inflamed
colonic epithelium are the source of reactive oxygen species causing the DNA
damages that initiate colonic tumorigenesis, and bile acids, at high levels in the
colons of humans eating a high fat diet, also cause DNA damage and contribute
to colon cancer.
Such exogenous and endogenous sources of DNA damage are indicated in the
boxes at the top of the figure in this section. The central role of DNA damage in
progression to cancer is indicated at the second level of the figure. The central
elements of DNA damage, epigenetic alterations and deficient DNA repair in
progression to cancer are shown in red.
A deficiency in DNA repair would cause more DNA damages to accumulate, and
increase the risk for cancer. For example, individuals with an inherited
impairment in any of 34 DNA repair genes (see article DNA repair-deficiency
disorder) are at increased risk of cancer with some defects causing up to 100%
lifetime chance of cancer (e.g. p53 mutations [74]). Such germ line mutations
are shown in a box at the left of the figure, with an indication of their
contribution to DNA repair deficiency. However, such germ line mutations
(which cause highly penetrant cancer syndromes) are the cause of only about 1
percent of cancers.

25
The majority of cancers are called non-hereditary or "sporadic cancers". About
30% of sporadic cancers do have some hereditary component that is currently
undefined, while the majority, or 70% of sporadic cancers, have no hereditary
component.
In sporadic cancers, a deficiency in DNA repair is occasionally due to a mutation
in a DNA repair gene, but much more frequently reduced or absent expression
of DNA repair genes is due to epigenetic alterations that reduce or silence gene
expression. This is indicated in the figure at the 3rd level from the top. For
example, for 113 colorectal cancers examined in sequence, only four had
a missense mutation in the DNA repair gene MGMT, while the majority had
reduced MGMT expression due to methylation of the MGMT promoter region
(an epigenetic alteration).[23] Five reports present evidence that between 40%
and 90% of colorectal cancers have reduced MGMT expression due to
methylation of the MGMT promoter region.
Similarly, out of 119 cases of mismatch repair-deficient colorectal cancers that
lacked DNA repair gene PMS2 expression, Pms2 was deficient in 6 due to
mutations in the PMS2 gene, while in 103 cases PMS2 expression was deficient
because its pairing partner MLH1 was repressed due to promoter methylation
(PMS2 protein is unstable in the absence of MLH1).[29] In the other 10 cases,
loss of PMS2 expression was likely due to epigenetic overexpression of the
microRNA, miR-155, which down-regulates MLH1.
In further examples [tabulated in the article Epigenetics (see section “DNA
repair epigenetics in cancer”)], epigenetic defects in cancers were found at
frequencies of between 13%-100% for the DNA repair
genes BRCA1, WRN, FANCB, FANCF, MGMT, MLH1, MSH2, MSH4, ERCC1,
XPF, NEIL1 and ATM in cancers including those in breast, ovarian, colorectal,
and the head and neck areas. In particular, two or more epigenetic deficiencies
in expression of ERCC1, XPF and/or PMS2 were shown to occur simultaneously
in the majority of the 49 colon cancers evaluated by Facista et al.

26
When expression of DNA repair genes is reduced, this causes a DNA repair
deficiency. This is shown in the figure at the 4th level from the top. With a DNA
repair deficiency, more DNA damages remain in cells at a higher than usual
level (5th level from the top in figure), and these excess damages cause
increased frequencies of mutation and/or epimutation (6th level from top of
figure). Experimentally, mutation rates increase substantially in cells defective
in DNA mismatch repair[32][33] or in Homologous recombination repair
(HRR).[34] Chromosomal rearrangements and aneuploidy also increase in HRR
defective cells during repair of DNA double strand breaks, or repair of other
DNA damages, incompletely cleared sites of repair can cause epigenetic gene
silencing.[36][37]
Many studies of heavy metal-induced carcinogenesis show that such heavy
metals cause reduction in expression of DNA repair enzymes, some through
epigenetic mechanisms. In some cases, DNA repair inhibition is proposed to be a
predominant mechanism in heavy metal-induced carcinogenicity. For example,
one group of studies shows that arsenic inhibits the DNA repair genes PARP,
XRCC1, Ligase 3, Ligase 4, DNA POLB, XRCC4, DNA PKCS, TOPO2B, OGG1,
ERCC1, XPF, XPB, XPC XPE and P53.[38][39][40][41][42][43] Another
group of studies shows that cadmium inhibits the DNA repair genes MSH2,
ERCC1, XRCC1, OGG1, MSH6, DNA-PK, XPD and XPC[44][45][46][47][48]
The somatic mutations and epigenetic alterations caused by DNA damages and
deficiencies in DNA repair accumulate in field defects. Field defects are normal
appearing tissues with multiple alterations (discussed in the section below), and
are common precursors to development of the disordered and improperly
proliferating clone of tissue in a cancer. Such field defects (second level from
bottom of figure) may have multiple mutations and epigenetic alterations.
It is impossible to determine the initial cause for most specific cancers. In a few
cases, only one cause exists; for example, the virus HHV-8 causes all Kaposi's
sarcomas. However, with the help of cancer epidemiology techniques and
information, it is possible to produce an estimate of a likely cause in many more

27
situations. For example, lung cancer has several causes, including tobacco use
and radon gas. Men who currently smoke tobacco develop lung cancer at a rate
14 times that of men who have never smoked tobacco, so the chance of lung
cancer in a current smoker being caused by smoking is about 93%; there is a 7%
chance that the smoker's lung cancer was caused by radon gas or some other,
non-tobacco cause.[49] These statistical correlations have made it possible for
researchers to infer that certain substances or behaviours are carcinogenic.
Tobacco smoke causes increased exogenous DNA damage, and these DNA
damages are the likely cause of lung cancer due to smoking. Among the more
than 5,000 compounds in tobacco smoke, the genotoxic DNA damaging agents
that occur both at the highest concentrations and which have the strongest
mutagenic effects are acrolein, formaldehyde, acrylonitrile, 1,3-butadiene,
acetaldehyde, ethylene oxide and isoprene.
Using molecular biological techniques, it is possible to characterize the
mutations, epimutations or chromosomal aberrations within a tumor, and rapid
progress is being made in the field of predicting prognosis based on the
spectrum of mutations in some cases. For example, up to half of all tumors have
a defective p53 gene. This mutation is associated with poor prognosis, since
those tumor cells are less likely to go into apoptosis or programmed cell
death when damaged by therapy. Telomerase mutations remove additional
barriers, extending the number of times a cell can divide. Other mutations
enable the tumor to grow new blood vessels to provide more nutrients, or
to metastasize, spreading to other parts of the body. However, once a cancer is
formed it continues to evolve and to produce sub clones. For example, a renal
cancer, sampled in 9 areas, had 40 ubiquitous mutations, 59 mutations shared
by some, but not all regions, and 29 “private” mutations only present in one
region.[

28
Apoptosis:
Multiple mutations
Multiple mutations in cancer cells
In general, mutations in both types of genes are required for cancer to occur.
For example, a mutation limited to one oncogene would be suppressed by
normal mitosis control and tumor suppressor genes, first hypothesised by
the Knudson hypothesis.[93] A mutation to only one tumor suppressor gene
would not cause cancer either, due to the presence of many "backup" genes that
duplicate its functions. It is only when enough proto-oncogenes have mutated
into oncogenes, and enough tumor suppressor genes deactivated or damaged,
that the signals for cell growth overwhelm the signals to regulate it, that cell
growth quickly spirals out of control. Often, because these genes regulate the
processes that prevent most damage to genes themselves, the rate of mutations
increases as one gets older, because DNA damage forms a feedback loop.
Usually, oncogenes are dominant alleles, as they contain gain-of-function
mutations, whereas mutated tumor suppressors are recessive alleles, as they
contain loss-of-function mutations. Each cell has two copies of a same gene, one
from each parent, and, under most cases, gain of function mutation in one
copy of a particular proto-oncogene is enough to make that gene a true
oncogene, while usually loss of function mutation must happen in both copies of
a tumor suppressor gene to render that gene completely non-functional.
However, cases exist in which one loss of function copy of a tumor suppressor

29
gene can render the other copy non-functional, called the dominant negative
effect. This is observed in many p53 mutations.
Mutation of tumor suppressor genes that are passed on to the next generation
of not merely cells, but their offspring, can cause increased likelihoods for
cancers to be inherited. Members within these families have increased incidence
and decreased latency of multiple tumors. The mode of inheritance of mutant
tumor suppressors is that affected member inherits a defective copy from one
parent, and a normal copy from another. Because mutations in tumor
suppressors act in a recessive manner (note, however, there are exceptions), the
loss of the normal copy creates the cancer phenotype. For instance, individuals
that are heterozygous for p53 mutations are often victims of Li-Fraumeni
syndrome, and that are heterozygous for Rb mutations develop retinoblastoma.
In similar fashion, mutations in the adenomatous polyposis coli gene are linked
to adenopolyposis colon cancer, with thousands of polyps in the colon while
young, whereas mutations in BRCA1 and BRCA2 lead to early onset of breast
cancer.
A new idea announced in 2011 is an extreme version of multiple mutations,
called chromothripsis by its proponents. This idea, affecting only 2–3% of cases
of cancer, although up to 25% of bone cancers involve the catastrophic
shattering of a chromosome into tens or hundreds of pieces and then being
patched back together incorrectly. This shattering probably takes place when
the chromosomes are compacted during normal cell division, but the trigger for
the shattering is unknown. Under this model, cancer arises as the result of a
single, isolated event, rather than the slow accumulation of multiple mutations.
Cell types involved in cancer growth
There are several different cell types that are critical to tumour growth. In
particular endothelial progenitor cells are a very important cell population in
tumour blood vessel growth.[84][85] The hypothesis that endothelial progenitor
cells are important in tumour growth, angiogenesis and metastasis has been

30
supported by a recent publication in Cancer Research (August 2010). This
paper argues that endothelial progenitor cells can be marked using the Inhibitor
of DNA Binding 1 (ID1). This novel finding meant that investigators were able
to track endothelial progenitor cells from the bone marrow to the blood to the
tumour-stroma and vasculature. This finding of endothelial progenitor cells
incorporated in tumour vasculature gives evidence for the importance of this
cell type in blood vessel development in a tumour setting and metastasis.
Furthermore, ablation of the endothelial progenitor cells in the bone marrow
lead to a significant decrease in tumour growth and vasculature development.
The continued research into the importance of endothelial progenitor cells may
present novel therapeutic targets.
Oncogenes
Oncogenes promote cell growth through a variety of ways. Many can
produce hormones, a "chemical messenger" between cells that encourage mitosis,
the effect of which depends on the signal transduction of the receiving tissue or
cells. In other words, when a hormone receptor on a recipient cell is stimulated,
the signal is conducted from the surface of the cell to the cell nucleus to affect
some change in gene transcription regulation at the nuclear level. Some
oncogenes are part of the signal transduction system itself, or the
signal receptors in cells and tissues themselves, thus controlling the sensitivity to
such hormones. Oncogenes often produce mitogens, or are involved in
transcription of DNA in protein synthesis, which creates
the proteins and enzymes responsible for producing the products and bio
chemicals cells use and interact with.
Mutations in proto-oncogenes, which are the normally quiescent counterparts
of oncogenes, can modify their expression and function, increasing the amount
or activity of the product protein. When this happens, the proto-oncogenes
become oncogenes, and this transition upsets the normal balance of cell
cycle regulation in the cell, making uncontrolled growth possible. The chance of

31
cancer cannot be reduced by removing proto-oncogenes from the genome, even
if this were possible, as they are critical for growth, repair and homeostasis of
the organism. It is only when they become mutated that the signals for growth
become excessive.
One of the first oncogenes to be defined in cancer research is the Ras oncogene.
Mutations in the Ras family of proto-oncogenes (comprising H-Ras, N-Ras and
K-Ras) are very common, being found in 20% to 30% of all human
tumours.[87] Ras was originally identified in the Harvey sarcoma virus genome,
and researchers were surprised that not only is this gene present in the human
genome but also, when ligated to a stimulating control element, it could induce
cancers in cell line cultures.
Proto-oncogenes
Proto-oncogenes promote cell growth in a variety of ways. Many can
produce hormones, "chemical messengers" between cells that encourage mitosis,
the effect of which depends on the signal transduction of the receiving tissue or
cells. Some are responsible for the signal transduction system and
signal receptors in cells and tissues themselves, thus controlling the sensitivity to
such hormones. They often produce mitogens, or are involved in transcription of
DNA in protein synthesis, which create the proteins and enzymes is responsible
for producing the products and bio chemicals cells use and interact with.
Mutations in proto-oncogenes can modify their expression and function,
increasing the amount or activity of the product protein. When this happens,
they become oncogenes, and, thus, cells have a higher chance to divide
excessively and uncontrollably. The chance of cancer cannot be reduced by
removing proto-oncogenes from the genome, as they are critical for growth,
repair and homeostasis of the body. It is only when they become mutated that
the signals for growth become excessive. It is important to note that a gene
possessing a growth-promoting role may increase carcinogenic potential of a
cell, under the condition that all necessary cellular mechanisms that permit

32
growth are activated.[89] This condition includes also the inactivation of specific
tumor suppressor genes (see below). If the condition is not fulfilled, the cell may
cease to grow and can proceed to die. This makes knowledge of the stage and
type of cancer cell that grows under the control of a given oncogene crucial for
the development of treatment strategies.
Apoptosis:
Apoptosis (is the process of programmed cell death (PCD) that may occur
in multicellular organisms.[4] Biochemical events lead to characteristic cell
changes (morphology) and death. These changes include blebbing, cell
shrinkage, nuclear fragmentation, chromatin condensation, and
chromosomal DNA fragmentation. (See also apoptotic DNA fragmentation.)
In contrast to necrosis, which is a form of traumatic cell death that results
from acute cellular injury, apoptosis generally confers advantages during an
organism's life cycle. For example, the differentiation of fingers and toes in a
developing human embryo occurs because cells between the fingers apoptosis;
the result is that the digits are separate. Unlike necrosis, apoptosis produces cell
fragments called apoptotic bodies that phagocytic cells are able to engulf and
quickly remove before the contents of the cell can spill out onto surrounding
cells and cause damage

33
The process of apoptosis is controlled by a diverse range of cell signals, which
may originate either extra cellular (extrinsic inducers) or intra cellular (intrinsic
inducers). Extracellular signals may include toxins, hormones, growth
factors, nitric oxide or cytokines, that must either cross the plasma membrane
or transduce to effect a response. These signals may positively (i.e., trigger) or
negatively (i.e., repress, inhibit, or dampen) affect apoptosis. (Binding and
subsequent trigger of apoptosis by a molecule is termed positive induction,
whereas the active repression or inhibition of apoptosis by a molecule is
termed negative induction.)

34
A cell initiates intracellular apoptotic signalling in response to a stress, which
may bring about cell suicide. The binding of nuclear receptors
by glucocorticoids, heat, radiation,[15] nutrient deprivation,[15] viral
infection,[15] hypoxia[15] and increased
intracellular calcium concentration,[16] for example, by damage to the
membrane, can all trigger the release of intracellular apoptotic signals by a
damaged cell. A number of cellular components, such as poly ADP ribose
polymerase, may also help regulate apoptosis.[17]
Before the actual process of cell death is precipitated by enzymes, apoptotic
signals must cause regulatory proteins to initiate the apoptosis pathway. This
step allows apoptotic signals to cause cell death, or the process to be stopped,
should the cell no longer need to die. Several proteins are involved, but two
main methods of regulation have been identified: targeting
mitochondria functionality, or directly transducing the signal via adaptor
proteins to the apoptotic mechanisms. Another extrinsic pathway for initiation
identified in several toxin studies is an increase in calcium concentration within
a cell caused by drug activity, which also can cause apoptosis via calcium
binding protease calpain.
Mitochondrial regulation
The mitochondria are essential to multicellular life. Without them, a cell ceases
to respire aerobically and quickly dies. This fact forms the basis for some
apoptotic pathways. Apoptotic proteins that target mitochondria affect them in
different ways. They may cause mitochondrial swelling through the formation of
membrane pores, or they may increase the permeability of the mitochondrial
membrane and cause apoptotic effectors to leak out.[15] These are very closely
related to intrinsic pathway, and tumors arise more frequently through
intrinsic pathway than the extrinsic pathway because of sensitivity.[18] There is
also a growing body of evidence indicating that nitric oxide is able to induce
apoptosis by helping to dissipate the membrane potential of mitochondria and

35
therefore make it more permeable.[14] Nitric oxide has been implicated in
initiating and inhibiting apoptosis through its possible action as a signal molecule
of subsequent pathways that activate apoptosis.[19][citation needed]
Mitochondrial proteins known as SMACs (small mitochondria-derived activator
of caspases) are released into the cytosol following an increase in permeability.
SMAC binds to inhibitor of apoptosis proteins (IAPs) and deactivates them,
preventing the IAPs from arresting the apoptotic process and therefore allowing
apoptosis to proceed. IAP also normally suppresses the activity of a group of
cysteine proteases called caspases,[20] which carry out the degradation of the
cell, therefore the actual degradation enzymes can be seen to be indirectly
regulated by mitochondrial permeability.
Cytochrome c is also released from mitochondria due to formation of a channel,
the mitochondrial apoptosis-induced channel(MAC), in the outer mitochondrial
membrane,[21] and serves a regulatory function as it precedes morphological
change associated with apoptosis.[15] Once cytochrome c is released it binds
with Apoptotic protease activating factor - 1 (Apaf-1) and ATP, which then
bind to pro-caspase-9 to create a protein complex known as an apoptosome.
The apoptosome cleaves the pro-caspase to its active form of caspase-9, which
in turn activates the effector caspase-3.
MAC, also called "Mitochondrial Outer Membrane Permeabilization Pore" is
regulated by various proteins, such as those encoded by the mammalian Bcl-2
family of anti-apoptotic genes, the homologs of the ced-9 gene found in C.
elegans.[22][23] Bcl-2 proteins are able to promote or inhibit apoptosis by
direct action on MAC/MOMPP. Bax and/or Bak form the pore, while Bcl-2,
Bcl-xL or Mcl-1 inhibits its formation.

36
Stem cell cancer:
A new way of looking at carcinogenesis comes from integrating the ideas
of developmental biology into oncology. The cancer stem cell hypothesis proposes
that the different kinds of cells in a heterogeneous tumor arise from a single
cell, termed Cancer Stem Cell. Cancer stem cells may arise from transformation
of adult stem cells or differentiated cells within a body. These cells persist as a
subcomponent of the tumor and retain key stem cell properties. They give rise
to a variety of cells, are capable of self-renewal and
homeostatic control.[98] Furthermore, the relapse of cancer and the emergence
of metastasis are also attributed to these cells. The cancer stem
cell hypothesis does not contradict earlier concepts of carcinogenesis.

37
Cancer stem cells (CSCs) are cancer cells (found
within tumors or haematological cancers) that possess characteristics associated
with normal stem cells, specifically the ability to give rise to all cell types found
in a particular cancer sample. CSCs are therefore tumorigenic (tumor-forming),
perhaps in contrast to other non-tumorigenic cancer cells. CSCs may generate
tumors through the stem cell processes of self-renewal and differentiation into
multiple cell types. Such cells are proposed to persist in tumors as a distinct
population and cause relapse and metastasis by giving rise to new tumors.
Therefore, development of specific therapies targeted at CSCs holds hope for
improvement of survival and quality of life of cancer patients, especially for
sufferers of metastatic.
Existing cancer treatments have mostly been developed based on animal models,
where therapies able to promote tumor shrinkage were deemed effective.
However, animals could not provide a complete model of human disease. In
particular, in mice, whose life spans do not exceed two years, tumor relapse is
exceptionally difficult to study.
The efficacy of cancer treatments is, in the initial stages of testing, often
measured by the ablation fraction of tumor mass (fractional kill). As CSCs would
form a very small proportion of the tumor, this may not necessarily select for
drugs that act specifically on the stem cells. The theory suggests that
conventional chemotherapies kill differentiated or differentiating cells, which

38
form the bulk of the tumor but are unable to generate new cells. A population
of CSCs, which gave rise to it, could remain untouched and cause a relapse of
the disease.
Both tumor models may play role in the maintenance of tumor. Initially,
tumor growth assures specific CSC (CSC1). With tumor progression, another
CSC (CSC 2) may arise due the clonal selection. Development of new more
aggressive CSC may be result from acquisition additional mutation or epigenetic.
Pathways
The design of new drugs for the treatment of CSCs will likely require an
understanding of the cellular mechanisms that regulate cell proliferation. The
first advances in this area were made with hematopoietic stem cells (HSCs) and
their transformed counterparts in leukemia, the disease for which the origin of
CSCs is best understood. It is now becoming increasingly clear that stem cells of
many organs share the same cellular pathways as leukemia-derived HSCs.
Additionally, a normal stem cell may be transformed into a cancer stem cell
through deregulation of the proliferation and
differentiation pathways controlling it or by inducing on coprotein activity.
Bmi-1
The Poly comb group transcriptional repressor Bmi-1 was discovered as a
common oncogene activated in lymphoma[74] and later shown to specifically

39
regulate HSCs.[75] The role of Bmi-1 has also been illustrated in neural stem
cells.[76] The pathway appears to be active in CSCs of pediatric brain
tumors.[77]
Notch
The Notch pathway has been known to developmental biologists for decades. Its
role in control of stem cell proliferation has now been demonstrated for several
cell types including hematopoietic, neural and mammary [78] stem cells.
Components of the Notch pathway have been proposed to act as oncogenes in
mammary [79] and other tumors.
A particular branch of the Notch signalling pathway that involves the
transcription factor Hes3 has been shown to regulate the number of cultured
cells with cancer stem cell characteristics obtained from glioblastoma
patients.[80]
Sonic hedgehog and Wnt
These developmental pathways are also strongly implicated as stem cell
regulators.[81] Both Sonic hedgehog (SHH) and Wnt pathways are commonly
hyper activated in tumors and are required to sustain tumor growth. However,
the Gli transcription factors that are regulated by SHH take their name
from gliomas, where they are commonly expressed at high levels. A degree
of crosstalk exists between the two pathways and their activation commonly
goes hand-in-hand.[82] This is a trend rather than a rule. For instance, in
colon cancer hedgehog signalling appears to antagonise Wnt.[83]
Sonic hedgehog blockers are available, such as cyclopamine. There is also a new
water soluble cyclopamine that may be more effective in cancer treatment.
There is also DMAPT, a water soluble derivative of parthenolide (induces
oxidative stress, inhibits NF-κB signalling[84]) for AML (leukemia), and possibly

40
myeloma and prostate cancer. A clinical trial of DMAPT is to start in England
in late 2007 or 2008. Finally, the enzyme telomerase may qualify as a study
subject in CSC physiology.[85] GRN163L (Imetelstat) was recently started in
trials to target myeloma stem cells. If it is possible to eliminate the cancer stem
cell, then a potential cure may be achieved if there are no more CSCs to
repopulate a cancer.
Hedgehog pathway:
Embryonic development:
41
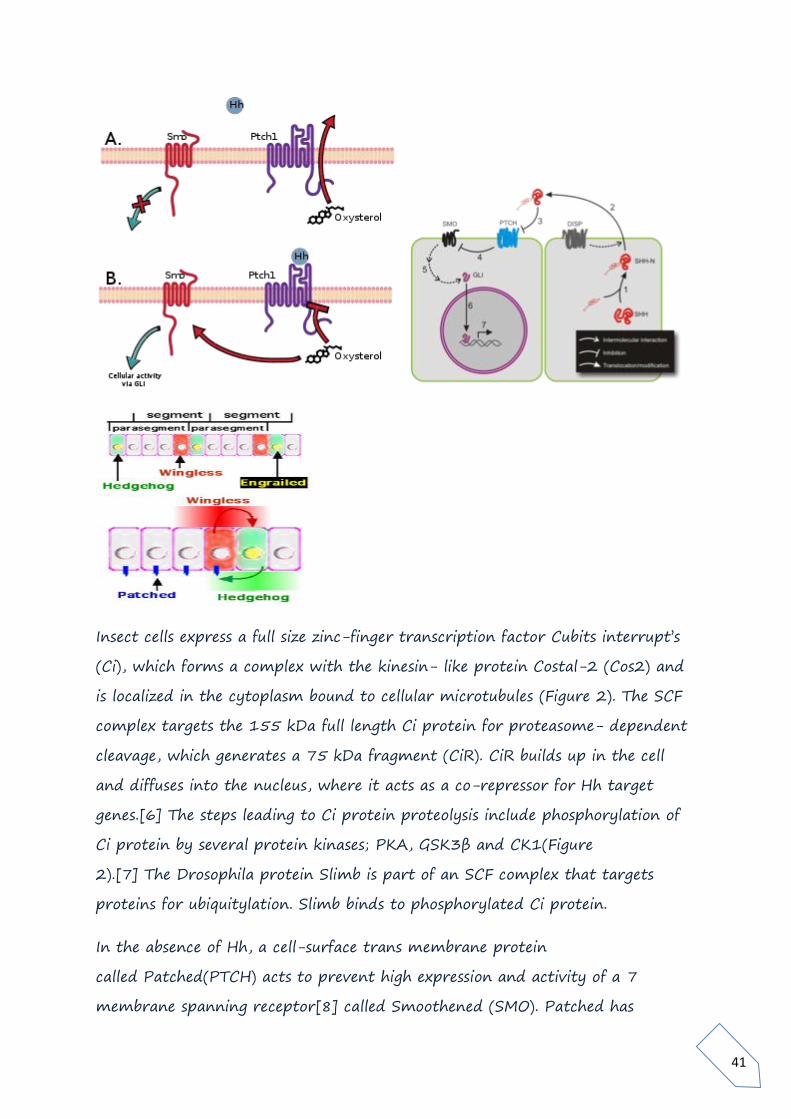
Insect cells express a full size zinc-finger transcription factor Cubits interrupt’s
(Ci), which forms a complex with the kinesin- like protein Costal-2 (Cos2) and
is localized in the cytoplasm bound to cellular microtubules (Figure 2). The SCF
complex targets the 155 kDa full length Ci protein for proteasome- dependent
cleavage, which generates a 75 kDa fragment (CiR). CiR builds up in the cell
and diffuses into the nucleus, where it acts as a co-repressor for Hh target
genes.[6] The steps leading to Ci protein proteolysis include phosphorylation of
Ci protein by several protein kinases; PKA, GSK3β and CK1(Figure
2).[7] The Drosophila protein Slimb is part of an SCF complex that targets
proteins for ubiquitylation. Slimb binds to phosphorylated Ci protein.
In the absence of Hh, a cell-surface trans membrane protein
called Patched(PTCH) acts to prevent high expression and activity of a 7
membrane spanning receptor[8] called Smoothened (SMO). Patched has

42
sequence similarity to known membrane transport proteins. When extracellular
Hh is present (Figure 3), it binds to and inhibits Patched, allowing Smoothened
to accumulate and inhibit the photolytic cleavage of the Ci protein. This process
most likely involves the direct interaction of smoothened and Costal-2 and may
involve sequestration of the Ci protein-containing complex to a micro domain
where the steps leading to Ci protein proteolysis are disrupted.[6] The
mechanism by which Hh binding to Patched leads to increased levels of
Smoothened is not clear (Step 1 in Figure 3). Following binding of Hh to
Patched, Smoothened levels increase greatly over the level maintained in cells
when Patched is not bound to Hh.[9] It has been suggested that phosphorylation
of Smoothened plays a role in Hh-dependent regulation of Smoothened
levels.[10]
In cells with Hh-activated Patched (Figure 3), the intact Ci protein accumulates
in the cell cytoplasm and levels of CiR decrease, allowing transcription of some
genes such as decapentaplegic (dpp, a member of the BMP growth factor
family). For other Hh-regulated genes, expression requires not only the loss of
CiR but also the positive action of uncleaved Ci to act as a transcriptional
activator.[7] Costal-2 is normally important for holding Ci protein in the
cytoplasm, but interaction of Smoothened with Costal-2 allows some intact Ci
protein to go to the nucleus. The Drosophila protein Fused (Fu in Figure 3) is a
protein kinase that binds to Costal-2. Fused can inhibit Suppressor of Fused
(SUFU), which in turn interacts with Ci to regulate gene transcription in some
cell types.[
Hedgehog has roles in larval body segment development and in formation of
adult appendages. During the formation of body segments in the
developing Drosophila embryo, stripes of cells that synthesize the transcription
factor Engrailed can also express the cell-to-cell signalling protein Hedgehog
(green in Figure 4). Hedgehog is not free to move very far from the cells that
make it and so it only activates a thin strips of cells adjacent to the Engrailed-
expressing cells. Only cells to one side of the Engrailed-expressing cells are

43
competent to respond to Hedgehog following interaction of Hh with the
receptor protein Patched .
Cells with Hh-activated Patched receptor synthesize the Wingless protein (red
in Figure 4). If a Drosophila embryo is altered so as to produce Hh in all cells, all
of the competent cells respond and form a broader band of Wingless-expressing
cells in each segment. The wingless gene has an upstream transcription
regulatory region that binds the Ci transcription factor in a Hh-dependent
fashion resulting in an increase in wingless transcription (interaction 2 in Figure
3) in a stripe of cells adjacent to the stripe of Hh-producing cells.[12]
Wingless protein acts as an extracellular signal and patterns the adjacent rows
of cells by activating its cell surface receptor Frizzled. Wingless acts on Engrailed-
expressing cells to stabilize the stripes of engrailed expression. Wingless is a
member of the Wnt family of cell-to-cell signaling proteins. The reciprocal
signaling by Hedgehog and Wingless stabilizes the boundary between Para
segments . The effects of Wingless and Hedgehog on other stripes of cells in each
segment establishes a positional code that accounts for the distinct anatomical
features along the anterior-posterior axis of the segments [13]
The Wingless protein is called "wingless" because of the phenotype of
some wingless fly mutants. Wingless and Hedgehog functioned together
during metamorphosis to coordinate wing formation. Hedgehog is expressed in
the posterior part of developing Drosophila limbs. Hedgehog also participates in
the coordination of eye, brain, gonad, gut and tracheal development. Hedgehog
has been implicated in reduced eye development in the amphipod Gammarus
minus. Specifically, down regulation of hedgehog results in reduced eyes. [14]
Cancer treatment:
The existence of CSCs has several implications in terms of future cancer
treatment and therapies. These include disease identification, selective drug

44
targets, prevention of metastasis, and development of new intervention
strategies.
Normal somatic stem cells are naturally resistant to chemotherapeutic agents-
they have various pumps (such as MDR[citation needed]) that pump out drugs,
DNA repair proteins and they also have a slow rate of cell turnover
(chemotherapeutic agents naturally target rapidly replicating cells)[citation
needed]. CSCs that have mutated from normal stem cells may also express
proteins that would increase their resistance towards chemotherapeutic agents.
These surviving CSCs then repopulate the tumor, causing relapse. By selectively
targeting CSCs, it would be possible to treat patients with aggressive, non-
resectable tumors, as well as preventing the tumor from metastasizing. The
hypothesis suggests that upon CSC elimination, cancer would regress due to
differentiation and/or cell death [citation needed]. What fraction of tumor cells
are CSCs and therefore need to be eliminated is not clear yet.
A number of studies have investigated the possibility of identifying specific
markers that may distinguish CSCs from the bulk of the tumor (as well as from
normal stem cells).[11] Proteomic and genomic signatures of tumors are also
being investigated.[70][citation needed]. In 2009, scientists identified one
compound, Salinomycin, that selectively reduces the proportion of breast CSCs
in mice by more than 100-fold relative to Paclitaxel, a commonly used
chemotherapeutic agent.[71]
The cell surface receptor interleukin-3 receptor-alpha (CD123) was shown to
be overexpressed on CD34+CD38- leukemic stem cells (LSCs) in acute
myelogenous leukemia (AML) but not on normal CD34+CD38- bone marrow
cells.[72] Jin et al., then demonstrated that treating AML-engrafted
NOD/SCID mice with a CD123-specific monoclonal antibody impaired LSCs
homing to the bone marrow and reduced overall AML cell repopulation
including the proportion of LSCs in secondary mouse recipients.[73]

45
He concept of migrating cancer stem cells (MSC).
Stationary cancers stem cells are embedded in begin carcinomas and these cells
are detectable in the differentiated central area of tumor. The important step
toward malignancy is the induction of epithelial mesenchyme transition (EMT)
in the stationary cancer stem cells (SCS), which become mobile or migrating
cancer stem cells. MCS cells divide asymmetrically. One daughter cell starts
proliferation and differentiation. The remaining MCS migrates a short distance
before undergoing new asymmetric division, or starts dissemination through
Blood vessel blood or lymphatic vessels and produces a metastatic.
CONCLUSIONS:
Finally studied different signalling pathways which are related to cancer
and development if cancer.
Different oncogenes and proto oncogenes, which are responsible for the
cancer
And while studying the cell cycle, different check points, and which are
affecting the cell cycle.
Study related to different therapies, drugs, and different several
treatments related to cancer.
Finally how to reduce the cancer using herbs, and many other techniques
are well studied and included in it.
Study of gene expression, and DNA damage, and different tumour
suppressor genes.
Study of different types of molecules and pathways involved in the
process of converting a normal cell into a cancer cell.

46
REFERENCES:
Fearon ER, Vogelstein B (June 1990). "A genetic model for colorectal
tumorigenesis.
Croce CM (January 2008). "Oncogenes and cancer". The New England Journal
of Medicine.
Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel
DP, Linsley PS, Johnson JM (2005).
Microarray analysis shows that some microRNAs downregulate large numbers of
target mRNAs. Nature 433(7027):769-773. PMID 15685193
Epigenetic silencing of miR-137 is an early event in colorectal carcinogenesis.
Cancer Res 70(16):6609-6618. doi: 10.1158/0008-5472.CAN-10-
0622. PMID 20682795
Knudson AG (November 2001). "Two genetic hits (more or less) to cancer
Daniel FI, Cherubini K, Yurgel LS, de Figueiredo MA, Salum FG. (2011). The
role of epigenetic transcription repression and DNA methyl transferases in
cancer. Cancer 117(4):677-687. doi: 10.1002/cncr.25482. Review. PMID
20945317
Kanwal R, Gupta S. (2012). Epigenetic modifications in cancer. Clin Genet
81(4):303-311. doi: 10.1111/j.1399-0004.2011.01809.x. Review. PMID
22082348
Pattani KM, Soudry E, Glazer CA, Ochs MF, Wang H, Schussel J, Sun W,
Hennessey P, Mydlarz W, Loyo M, Demokan S, Smith IM, Califano JA. (2012).
MAGEB2 is activated by promoter DE methylation in head and neck squamous
cell carcinoma.





















{kind=link}
{kind=link}