
Vit Matuska PhD thesis
220
FIVE-MEMBERED SULFUR-NITROGEN RING COMPOUNDS Vit Matuska A Thesis Submitted for the Degree of PhD at the University of St. Andrews 2009 Full metadata for this item is available in Research@StAndrews:FullText at: http://research-repository.st-andrews.ac.uk/ Please use this identifier to cite or link to this item: http://hdl.handle.net/10023/828 This item is protected by original copyright
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of Vit Matuska PhD thesis

FIVE-MEMBERED SULFUR-NITROGEN RING COMPOUNDS
Vit Matuska
A Thesis Submitted for the Degree of PhDat the
University of St. Andrews
2009
Full metadata for this item is available inResearch@StAndrews:FullText
at:http://research-repository.st-andrews.ac.uk/
Please use this identifier to cite or link to this item:http://hdl.handle.net/10023/828
This item is protected by original copyright

Five-Membered Sulfur-NitrogenRing Compounds
A thesis submitted by
Vit Matuska
In partial ful�lment for the award ofDoctor of Philosophy of the University of St Andrews
School of Chemistry, University of St AndrewsNorth Haugh, St Andrews, Fife, KY16 9ST
12 May 2009

DeclarationsI, Vit Matuska, hereby certify that this thesis, which is approximately 50,000 words inlength, has been written by me, that it is the record of work carried out by me and thatit has not been submitted in any previous application for a higher degree.
Date: Signature of candidate:
I was admitted as a research student in September, 2004 and as a candidate for thedegree of Doctor of Philosophy in May, 2009; the higher study for which this is a recordwas carried out in the University of St Andrews between 2004 and 2009.
Date: Signature of candidate:
I hereby certify that the candidate has ful�lled the conditions of the Resolution andRegulations appropriate for the degree of Doctor of Philosophy in the University of StAndrews and that the candidate is quali�ed to submit this thesis in application for thatdegree.
Date: Signature of supervisor:
In submitting this thesis to the University of St Andrews we understand that we aregiving permission for it to be made available for use in accordance with the regulationsof the University Library for the time being in force, subject to any copyright vested inthe work not being a�ected thereby. We also understand that the title and the abstractwill be published, and that a copy of the work may be made and supplied to any bona �delibrary or research worker, that my thesis will be electronically accessible for personalor research use unless exempt by award of an embargo as requested below, and that thelibrary has the right to migrate my thesis into new electronic forms as required to ensurecontinued access to the thesis. We have obtained any third-party copyright permissionsthat may be required in order to allow such access and migration, or have requested theappropriate embargo below.
The following is an agreed request by candidate and supervisor regarding the electronicpublication of this thesis: embargo on both all or part of printed copy and electronic copyfor the same �xed period of 1 year on the following ground: publication would precludefuture publication.
Date: Signature of candidate:
Signature of supervisor:
2

Acknowledgements
I want to thank my Mum, Dad, Brother and my grandparents for a massive supportthroughout the studies. Without them I would have not been able to reach this point. Iwould like to dedicate this thesis to them.
Special thanks to Stu Robertson and Richard Bond, who very patiently introduced me tothe british culture and helped me to feel in Scotland like at home. My academic parentsGil Viana and Blerina Kellezi were always ready to cheer me up and for that I thankthem a lot. I am indebted to Klára Mí£ková who helped me a lot in the very beginning.
I am thankful to my landlords Mr and Mrs Gall, David and Gisele, Tom Lébl andNathaniel for cosy rooms and to my �atmates Mi²o, Alex, Gil, James and Lucia forcreating a relaxed atmosphere at home.
The Woollins, Slawin and Kilian groups - thank you folks for a really enjoyable timeboth in and outside the lab and also for the patience you had with me.
Writing-up of this thesis was easier with LATEX2ε and the KOMA-Script bundle. In ad-dition to the authors I would like to thank Piotr for his help, tips and mainly Konwerter.Paul Waddell checked errors in some parts of this thesis. Grazie Poalo!
I thank Dr. Tomá² Lébl and Melanja Smith for their very kind and instant helpwhenever needed downstairs in the NMR room. Many thanks to Dr. Joe Crayston (CV),Caroline (mass spec.), Sylvia (microanalysis), Marj, Bobby, Brian, Colin the storesman,Artur and Colin the glassblower.
I am grateful to Dr. Petr Kilián who spent a lot of time bringing me up in the lab,showing me the equipment, its maintenance and explaining new techniques.
A big thank is due to Prof. Alex Slawin who was always ready to measure my crystalstructures knowing that they were going to be either S4N4, sulfur, Na2SO4 or they wouldnot di�ract at all.
Finally, I would like to thank my supervisor, Prof. Derek Woollins, for all his help,support, advice, comments and corrections and for the patience he had with me duringthose years.
3

Abstract
A series of 1,3,2,4,5-dithiadiazarsoles with the general formula RAs(S2N2) (R = Me, Et,iPr, tBu, Ph and Mes) have been prepared and characterised by multinuclear NMR, IRand Raman spectroscopies and mass spectrometry. The X-ray structures of PhAs(S2N2)and MesAs(S2N2) were determined.
The low temperature X-ray structures of the half-sandwich 5,1,3,2,4-metalladithia-diazoles Cp*M(S2N2) (M = Co, Ir) were determined and Cp*Rh(S2N2) was prepared.All three metalladithiadiazoles were characterised by multinuclear NMR, IR and Ramanspectroscopies and mass spectrometry. The X-ray structures of complexes [Cp*RhCp*]Cl,[Cp*Rh(µ-S3N2)(µ-S2O3)RhCp*] and Cp*Ir[S2N2(IrCl2Cp*)] obtained during this workwere determined.
The low temperature X-ray structure of Roesky's sulfoxide (S3N2O) is presented to-gether with assignments of its vibrational spectra as suggested by theoretical calculations.
The experimental structures of the metalladithiadiazoles and that of Roesky's sulfoxideare compared with calculated geometries.
A limited amount of simple experiments have been carried out with selected titlecompounds to get an insight into their reactivity.
4

Contents
Declarations 2
Acknowledgements 3
Abstract 4
List of Figures 12
List of Tables 14
Abbreviations 16General abbreviations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16NMR spectroscopy abbreviations . . . . . . . . . . . . . . . . . . . . . . . . . 17Vibrational spectroscopy abbreviations . . . . . . . . . . . . . . . . . . . . . . 17
I. Introduction 19
1. General considerations 201.1. Preparation routes of sulfur-nitrogen heterocycles . . . . . . . . . . . . . 201.2. The multiplicity of S−N bonds . . . . . . . . . . . . . . . . . . . . . . . 221.3. Theoretical calculations . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
1.3.1. Geometry of a molecule . . . . . . . . . . . . . . . . . . . . . . . 241.3.2. Charges and bond orders . . . . . . . . . . . . . . . . . . . . . . . 251.3.3. Aromaticity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
1.3.3.1. Aromaticity of sulfur-nitrogen compounds . . . . . . . . 271.4. General experimental conditions . . . . . . . . . . . . . . . . . . . . . . . 281.5. Experimental details of the key sulfur-nitrogen reagents syntheses . . . . 29
1.5.1. [S3N2Cl]Cl . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 291.5.2. [S4N3]Cl . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 301.5.3. [nBu2Sn(S2N2)]2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
5

Contents
II. Arsenic-Sulfur-Nitrogen Heterocycles 32
2. Introduction 332.1. The toxicity of arsenic . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33
2.1.1. Arsenic in the environment . . . . . . . . . . . . . . . . . . . . . . 332.1.2. Biotransformation of arsenic in nature . . . . . . . . . . . . . . . 342.1.3. Arsenic in the human body . . . . . . . . . . . . . . . . . . . . . 35
2.1.3.1. The principle of toxicity of arsenic . . . . . . . . . . . . 352.1.3.2. Metabolism of arsenic in the human body . . . . . . . . 36
2.2. Commercial availability of starting materials . . . . . . . . . . . . . . . . 382.3. 75As NMR as an analytical tool . . . . . . . . . . . . . . . . . . . . . . . 382.4. Methods of preparation of common starting materials . . . . . . . . . . . 39
2.4.1. Arsenic trihalogenides . . . . . . . . . . . . . . . . . . . . . . . . 392.4.2. Alkylarsonic acids . . . . . . . . . . . . . . . . . . . . . . . . . . . 402.4.3. Arylarsonic acids . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
2.4.3.1. Rosenmund's method . . . . . . . . . . . . . . . . . . . 412.4.3.2. Bart's reaction . . . . . . . . . . . . . . . . . . . . . . . 412.4.3.3. Modi�cations of Bart's method . . . . . . . . . . . . . . 462.4.3.4. Béchamp's reaction . . . . . . . . . . . . . . . . . . . . . 46
2.4.4. Dialkyl- and diarylarsinic acids . . . . . . . . . . . . . . . . . . . 472.4.5. Organohalogenoarsines . . . . . . . . . . . . . . . . . . . . . . . . 48
2.4.5.1. From arsonic and arsinic acids . . . . . . . . . . . . . . . 482.4.5.2. From AsX3 and organometallic reagents . . . . . . . . . 482.4.5.3. Controlled substitution of halogens in AsX3 . . . . . . . 502.4.5.4. From AsX3 and organosilicon and organotin reagents . . 522.4.5.5. From AsX3 by aromatic electrophilic substitution . . . . 542.4.5.6. Special methods of RAsX2 preparation . . . . . . . . . . 552.4.5.7. Halogen exchange reactions of RAsX2 . . . . . . . . . . 58
2.5. Arsenic containing main-group ring compounds . . . . . . . . . . . . . . 592.5.1. Organoarsenic heterocycles . . . . . . . . . . . . . . . . . . . . . . 59
2.5.1.1. Five-membered heterocycles . . . . . . . . . . . . . . . . 592.5.1.2. Six-membered heterocycles . . . . . . . . . . . . . . . . 60
2.5.2. Arsenic homocycles . . . . . . . . . . . . . . . . . . . . . . . . . . 612.5.2.1. Five-membered arsenic homocycles . . . . . . . . . . . . 612.5.2.2. Six-membered arsenic homocycles . . . . . . . . . . . . . 622.5.2.3. Arsafullerenes . . . . . . . . . . . . . . . . . . . . . . . . 63
2.5.3. Arsenic containing analogues of S4N4 . . . . . . . . . . . . . . . . 642.5.3.1. Cyclotetrathiatetrarsocane . . . . . . . . . . . . . . . . . 64
6

Contents
2.5.3.2. Cyclotetraarsazenes . . . . . . . . . . . . . . . . . . . . 662.5.3.3. Cyclotetraarsazanes . . . . . . . . . . . . . . . . . . . . 672.5.3.4. Dithiatetrazadiarsocines . . . . . . . . . . . . . . . . . . 692.5.3.5. 1,5,2,4,6,8,3,7-dithiatetrazadiarsocanes . . . . . . . . . . 722.5.3.6. Other AsSN eight-membered heterocycles . . . . . . . . 72
2.5.4. Arsenic containing analogues of S2N2 . . . . . . . . . . . . . . . . 732.5.4.1. As2S2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . 732.5.4.2. As2N2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . 75
2.5.5. Five- and six-membered arsenic-sulfur-nitrogen rings . . . . . . . 772.5.6. AsEN heterocycles (E = Se, Te) . . . . . . . . . . . . . . . . . . . 77
2.5.6.1. Eight-membered As4E4 heterocycles (E = Se, Te) . . . . 772.5.6.2. Four-membered As2E2 heterocycles (E = Se, Te) . . . . 78
2.6. Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
3. Results and Discussion 803.1. Comments on preparative procedures . . . . . . . . . . . . . . . . . . . . 82
3.1.1. Alkyl/aryldihalogenoarsines . . . . . . . . . . . . . . . . . . . . . 823.1.2. 5-alkyl/aryl-1,3λ4δ2,2,4,5-dithiadiazarsoles . . . . . . . . . . . . . 87
3.2. NMR spectroscopy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 893.2.1. 1H and 13C NMR data . . . . . . . . . . . . . . . . . . . . . . . . 893.2.2. 14N NMR data . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92
3.3. Mass spectrometry . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 943.4. IR and Raman spectroscopy . . . . . . . . . . . . . . . . . . . . . . . . . 95
3.4.1. The assignments and their discussion . . . . . . . . . . . . . . . . 963.5. X-ray structures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 105
3.5.1. PhAs(S2N2) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1053.5.2. MesAs(S2N2) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 108
3.6. Cyclic voltammetry . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1093.7. Reactivity of PhAs(S2N2) . . . . . . . . . . . . . . . . . . . . . . . . . . 1103.8. Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 112
4. Experimental 1144.1. Preparation of AsCl3 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1144.2. Preparation of CH3AsI2 . . . . . . . . . . . . . . . . . . . . . . . . . . . 1154.3. Preparation of EtAsI2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1154.4. Preparation of iPrAsCl2 . . . . . . . . . . . . . . . . . . . . . . . . . . . 1174.5. Preparation of tBuAsCl2 . . . . . . . . . . . . . . . . . . . . . . . . . . . 1174.6. Preparation of PhAsCl2 . . . . . . . . . . . . . . . . . . . . . . . . . . . 118
7

Contents
4.7. Preparation of mixture of mesityldihalogenoarsines . . . . . . . . . . . . 1194.8. Preparation of p-phenylenediarsonic acid by Bart's reaction . . . . . . . . 1204.9. Reaction of MeAsI2 with [nBu2Sn(S2N2)]2 . . . . . . . . . . . . . . . . . 1214.10. Reaction of EtAsI2 with [nBu2Sn(S2N2)]2 . . . . . . . . . . . . . . . . . . 1224.11. Reaction of iPrAsCl2 with [nBu2Sn(S2N2)]2 . . . . . . . . . . . . . . . . . 1234.12. Reaction of tBuAsCl2 with [nBu2Sn(S2N2)]2 . . . . . . . . . . . . . . . . 1244.13. Reaction of PhAsCl2 with [nBu2Sn(S2N2)]2 . . . . . . . . . . . . . . . . . 1254.14. Reaction of mesityldihalogenoarsines with [nBu2Sn(S2N2)]2 . . . . . . . . 1264.15. Reaction of PhAs(S2N2) with Pt(COD)Cl2 . . . . . . . . . . . . . . . . . 1274.16. Methylation of PhAs(S2N2) with CH3I . . . . . . . . . . . . . . . . . . . 1274.17. Reaction of PhAs(S2N2) with [Mo(CO)4(piperidine)2] . . . . . . . . . . . 1284.18. Reaction of PhAs(S2N2) with Cp2Ti(CO)2 . . . . . . . . . . . . . . . . . 1284.19. Reaction of PhAs(S2N2) with Cp*Co(CO)2 . . . . . . . . . . . . . . . . . 1284.20. Reaction of PhAs(S2N2) with sulfur . . . . . . . . . . . . . . . . . . . . . 129
III. Transition Metal Sulfur-Nitrogen Ring Compounds 130
5. Introduction 1315.1. Four-membered rings . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1315.2. Five-membered rings . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 134
5.2.1. M(S2N2) complexes containing the (S2N2) 2 � anion . . . . . . . . 1345.2.1.1. Preparation from S4N4 or S2N2 . . . . . . . . . . . . . . 1345.2.1.2. Preparation from other SN rings . . . . . . . . . . . . . 1355.2.1.3. Transmetallation reactions . . . . . . . . . . . . . . . . . 1375.2.1.4. Deprotonation of M(S2N2H) complexes . . . . . . . . . . 1375.2.1.5. Preparation from acyclic SN compounds . . . . . . . . . 138
5.2.2. M(S2N2) complexes containing the (NSSN) moiety . . . . . . . . . 1385.2.3. M(S2N2H) ring complexes . . . . . . . . . . . . . . . . . . . . . . 139
5.2.3.1. Preparation from S4N4 . . . . . . . . . . . . . . . . . . . 1395.2.3.2. Protonation of M(S2N2) complexes . . . . . . . . . . . . 140
5.2.4. M(S3N) ring complexes . . . . . . . . . . . . . . . . . . . . . . . . 1405.2.4.1. Preparation from [S7N] � . . . . . . . . . . . . . . . . . . 1405.2.4.2. Desulfuration of [S4N] � . . . . . . . . . . . . . . . . . . 1415.2.4.3. Preparation from other SN compounds . . . . . . . . . . 141
5.2.5. Structure of the �ve-membered MSN rings . . . . . . . . . . . . . 1415.3. Six-membered rings . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 142
5.3.1. M(S3N2) ring complexes . . . . . . . . . . . . . . . . . . . . . . . 142
8

Contents
5.3.2. M(S2N3) ring complexes bearing the (S2N3) � anion . . . . . . . . 1435.3.3. M(S2N3) ring complexes bearing the (S2N3) 3 � anion . . . . . . . 1435.3.4. M(S2N3) ring complexes bearing the [N2S2N(SO2NH2)] 2 � anion . 144
5.4. Seven-membered rings . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1455.5. Eight-membered rings . . . . . . . . . . . . . . . . . . . . . . . . . . . . 145
5.5.1. Complexes of the (S3N4) 2 � anion . . . . . . . . . . . . . . . . . . 1455.5.2. Complexes of the (S4N3) � anion . . . . . . . . . . . . . . . . . . . 146
5.6. Nine-membered rings . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1465.6.1. Monocyclic nine-membered rings . . . . . . . . . . . . . . . . . . 1465.6.2. Insertion of a metal centre in S4N4 . . . . . . . . . . . . . . . . . 146
5.7. Half-sandwich M(S2N2) complexes . . . . . . . . . . . . . . . . . . . . . . 1485.8. Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 148
6. Results and discussion 1496.1. Syntheses of the complexes . . . . . . . . . . . . . . . . . . . . . . . . . . 149
6.1.1. Cp*Co(S2N2) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1496.1.2. Cp*Rh(S2N2) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1506.1.3. Cp*Ir(S2N2) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 152
6.2. The X-ray structures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1526.2.1. Cp*M(S2N2) (M = Co, Rh, Ir) . . . . . . . . . . . . . . . . . . . 152
6.2.1.1. The in�uence of M in CpM(S2N2) and Cp*M(S2N2) . . . 1566.2.2. Cp*Rh(µ−S3N2)(µ−S2O3)RhCp* and [Cp*RhCp*]Cl . . . . . . . 1586.2.3. Cp*Ir[S2N2(IrCl2Cp*)] . . . . . . . . . . . . . . . . . . . . . . . . 160
6.3. NMR spectroscopy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1636.3.1. 14N NMR spectroscopy . . . . . . . . . . . . . . . . . . . . . . . . 164
6.4. Mass spectrometry . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1656.5. IR and Raman spectroscopy . . . . . . . . . . . . . . . . . . . . . . . . . 1656.6. Protonation of Cp*Co(S2N2) . . . . . . . . . . . . . . . . . . . . . . . . . 1656.7. Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 166
7. Experimental 1687.1. Preparation of [Cp*RhCl2]2 . . . . . . . . . . . . . . . . . . . . . . . . . 1687.2. Preparation of Cp*Co(CO)2 . . . . . . . . . . . . . . . . . . . . . . . . . 1697.3. Preparation of Cp*Co(S2N2) . . . . . . . . . . . . . . . . . . . . . . . . . 1697.4. Cp*Rh(S2N2) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1707.5. [Cp*RhCp*]Cl . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1717.6. [Cp*Rh(µ-S3N2)(µ-S2O3)RhCp*] . . . . . . . . . . . . . . . . . . . . . . 1717.7. Preparation of [Cp*Rh(S2N2H)]PF6 . . . . . . . . . . . . . . . . . . . . . 172
9

Contents
7.8. Cp*Ir(S2N2) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1737.9. Cp*Ir[S2N2(IrCl2)]Cp* . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1737.10. Protonation of Cp*Co(S2N2) with HBF4 . . . . . . . . . . . . . . . . . 1747.11. Protonation of Cp*Co(S2N2) with HCl . . . . . . . . . . . . . . . . . . 1757.12. Computational details . . . . . . . . . . . . . . . . . . . . . . . . . . . 175
IV. Roesky's Sulfoxide, S3N2O 176
8. Introduction 1778.1. Synthesis of Roesky's sulfoxide . . . . . . . . . . . . . . . . . . . . . . . . 1778.2. Structure and reactivity . . . . . . . . . . . . . . . . . . . . . . . . . . . 1798.3. Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 181
9. Results and Discussion 1829.1. The crystal structure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 182
9.1.1. Aromaticity of Roesky's sulfoxide . . . . . . . . . . . . . . . . . . 1849.2. Vibrational spectra . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1849.3. 14N NMR spectroscopy . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1859.4. Cyclic voltammetry . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1859.5. Reactivity of Roesky's sulfoxide . . . . . . . . . . . . . . . . . . . . . . . 1859.6. Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 188
10.Experimental 18910.1. Preparation of S3N2O . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18910.2. Reaction of S3N2O with Cp*Co(CO)2 . . . . . . . . . . . . . . . . . . . . 18910.3. Reaction of S3N2O with Cp2Ti(CO)2 . . . . . . . . . . . . . . . . . . . . 19010.4. Reaction of S3N2O and [(Ph3P)Au(CH3CN)][ClO4] . . . . . . . . . . . . 19010.5. Protonation of S3N2O with HBF4 . . . . . . . . . . . . . . . . . . . . . . 19110.6. Reaction of S3N2O with Woollins' Reagent . . . . . . . . . . . . . . . . . 19110.7. Reaction of (S2N2)CO with Woollins' Reagent . . . . . . . . . . . . . . . 19210.8. Computational details . . . . . . . . . . . . . . . . . . . . . . . . . . . . 192
Conclusion 193
Further work 195
References 196
10

Contents
Appendices 211
A. Crystallographic data 212
11

List of Figures
1.1. Selected S-N compounds with S-N bond lengths . . . . . . . . . . . . . . 24
2.1. Arsenobetaine, arsanilic acid and roxarsone . . . . . . . . . . . . . . . . . 342.2. The structures of syn- and anti - arenediazoates . . . . . . . . . . . . . . 432.3. Ball and stick model of the anion [As@Ni12@As20] 3 � . . . . . . . . . . . 642.4. The structures of realgar (α-As4S4), realgar As4S4(II) and their metal
complexes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 662.5. A schematic demonstration of the structural di�erence between S4N4 and
dithiatetrazadiarsocines . . . . . . . . . . . . . . . . . . . . . . . . . . . . 702.6. Possible conformations of the As2S2 ring . . . . . . . . . . . . . . . . . . 732.7. A part cut out from the polymeric anion [NiAs4S8] 2n �n . . . . . . . . . . . 742.8. Mesomeric structures of bis(1,3,2-dithiarsolylium) dication . . . . . . . . 75
3.1. 1,4-bis(dichloroarsino)benzene, p-phenylenediarsonic acid and Atoxyl . . 863.2. Simulated and recorded 1H NMR spectrum of EtAs(S2N2) . . . . . . . . 913.3. A general presentation of conformers for EtAsI2, iPrAsCl2, EtAs(S2N2)
and iPrAs(S2N2) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 963.4. The X-ray structure of PhAs(S2N2) . . . . . . . . . . . . . . . . . . . . . 1063.5. The stacking of PhAs(S2N2) molecules in the crystal . . . . . . . . . . . 1073.6. The X-ray structure of MesAs(S2N2) . . . . . . . . . . . . . . . . . . . . 1083.7. Cyclic voltammogram of PhAs(S2N2) . . . . . . . . . . . . . . . . . . . . 110
5.1. Possible coordination modes of the N2S and NS2 ligandsonto a metal centre . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 131
5.2. The structure of [Cl2Re(S2N3)(Cl3ReSN3)] 2 �2 . . . . . . . . . . . . . . . . 1445.3. The structures of complexes [(PPh3)(CO)IrCl(fac-S4N4)] and
[PPh4][Cl3Pt(fac-S4N4)] . . . . . . . . . . . . . . . . . . . . . . . . . . . 1475.4. The structures of complexes [(PPhMe2)PtCl2(mer-S4N4)] and
[(PPhMe2)PtCl2(fac-S4N4)] . . . . . . . . . . . . . . . . . . . . . . . . . 1475.5. Numbering of the CpM(S2N2) and Cp*M(S2N2) complexes . . . . . . . . 148
6.1. The X-ray structure of Cp*Co(S2N2) . . . . . . . . . . . . . . . . . . . . 153
12

List of Figures
6.2. The X-ray structure of Cp*Ir(S2N2) . . . . . . . . . . . . . . . . . . . . . 1566.3. The X-ray structure of Cp*Rh(µ−S3N2)(µ−S2O3)RhCp* . . . . . . . . . 1586.4. A Newman projection of Cp*Rh(µ−S3N2)(µ−S2O3)RhCp* . . . . . . . . 1606.5. The X-ray structure of [Cp*RhCp*]Cl ·H2O . . . . . . . . . . . . . . . . 1606.6. The X-ray structure of Cp*Ir[S2N2(IrCl2Cp*)] · nBu2SnCl2 . . . . . . . . 1616.7. Cp*Ir[S2N2(IrCl2Cp*)] and [Cp*Ir[S2N2(IrCl(PPh3)Cp*)]][PF6] . . . . . . 163
9.1. The crystal structure of S3N2O . . . . . . . . . . . . . . . . . . . . . . . 1839.2. The packing of S3N2O molecules in the crystal . . . . . . . . . . . . . . . 1839.3. Cyclic voltammogram of S3N2O . . . . . . . . . . . . . . . . . . . . . . . 1869.4. The structure of the complex Cp2Ti(N3)−O−Ti(N3)Cp2 . . . . . . . . . 187
13

List of Tables
3.1. Melting and boiling points of Me3SiX and nBu2SnX2 . . . . . . . . . . . 813.2. 1H NMR data of RAsX2 . . . . . . . . . . . . . . . . . . . . . . . . . . . 903.3. 1H NMR data of RAs(S2N2) . . . . . . . . . . . . . . . . . . . . . . . . . 913.4. 13C NMR data of RAsX2 . . . . . . . . . . . . . . . . . . . . . . . . . . . 923.5. 13C NMR data of RAs(S2N2) . . . . . . . . . . . . . . . . . . . . . . . . . 933.6. 14N NMR data of RAs(S2N2) . . . . . . . . . . . . . . . . . . . . . . . . 933.7. Selected IR and Raman wavenumbers of MeAsI2 and MeAs(S2N2) . . . . 983.8. Selected IR and Raman wavenumbers of EtAsI2 and EtAs(S2N2) . . . . . 993.9. Selected IR and Raman wavenumbers of iPrAsCl2 and iPrAs(S2N2) . . . 1003.10. Selected IR and Raman wavenumbers of tBuAsCl2 and tBuAs(S2N2) . . . 1013.11. Selected IR and Raman wavenumbers of PhAsCl2 and PhAs(S2N2) . . . 1033.12. Selected IR and Raman wavenumbers of MesBr and MesAs(S2N2) . . . . 1053.13. Selected bond lengths and angles of PhAs(S2N2) . . . . . . . . . . . . . . 1063.14. Selected bond lengths and angles of MesAs(S2N2) . . . . . . . . . . . . . 1093.15. Experimental and simulated voltammetric data for PhAs(S2N2) . . . . . 110
6.1. Selected bond lengths and angles of CpCo(S2N2), Cp*Co(S2N2)and Cp*Ir(S2N2) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 154
6.2. Selected calculated geometrical data for CpM(S2N2) and Cp*M(S2N2) . . 1546.3. Calculated Wiberg bond orders for the most important bonds
in CpM(S2N2) and Cp*M(S2N2) . . . . . . . . . . . . . . . . . . . . . . . 1576.4. Selected bond lengths and angles for Cp*Rh(µ−S3N2)(µ−S2O3)RhCp* . 1596.5. Selected bond lengths and angles for [Cp*RhCp*]Cl ·H2O . . . . . . . . . 1606.6. Selected bond lengths and angles for Cp*Ir(S2N2), Cp*Ir[S2N2(IrCl2Cp*)]
and [Cp*Ir[S2N2(IrCl(PPh3)Cp*)]][PF6] . . . . . . . . . . . . . . . . . . . 1626.7. 1H NMR data of compounds 47, 49, 51, 52, 53 and 54 . . . . . . . . . . 1636.8. 13C NMR data of compounds 47, 49, 51, 52, 53 and 54 . . . . . . . . . 164
9.1. The bond lengths and angles for S3N2O . . . . . . . . . . . . . . . . . . . 1839.2. Selected IR and Raman wavenumbers of S3N2O . . . . . . . . . . . . . . 1849.3. Voltammetric data for S3N2O . . . . . . . . . . . . . . . . . . . . . . . . 185
14

List of Tables
A.1. Crystal data and structure re�nement for PhAs(S2N2) . . . . . . . . . . . 212A.2. Crystal data and structure re�nement for MesAs(S2N2) . . . . . . . . . . 213A.3. Crystal data and structure re�nement for Cp*Co(S2N2) . . . . . . . . . . 214A.4. Crystal data and structure re�nement for Cp*Ir(S2N2) . . . . . . . . . . 215A.5. Crystal data and structure re�nement for
[Cp*Rh(µ−S3N2)(µ−SSO3)RhCp*]·CH2Cl2 . . . . . . . . . . . . . . . . . 216A.6. Crystal data and structure re�nement for [Cp*RhCp*]Cl ·H2O . . . . . . 217A.7. Crystal data and structure re�nement for
Cp*Ir[S2N2(IrCl2Cp*)] · nBu2SnCl2 . . . . . . . . . . . . . . . . . . . . . 218A.8. Crystal data and structure re�nement for S3N2O . . . . . . . . . . . . . . 219
15

Abbreviations
General abbreviations◦C degrees CelsiusÅ Ångström, 1× 10−10mAc acetylapprox. approximatelyb.p. boiling pointnBu but-1-yltBu tertiary butyl, 2-methylprop-2-ylcalc. calculatedCI chemical ionisationCOD cycloocta-1,5-dieneconc. concentratedCp cyclopentadienyl (C5H5)Cp* 1,2,3,4,5-pentamethylcyclopentadienyl (C5Me5)Cp′ methylcyclopentadienyl (C5H4Me)Cp× 1-ethyl-2,3,4,5-tetramethylcyclopentadienyl (C5Me4Et)Cy cyclohexylDBN 1,5-diazabicyclo[4.3.0]non-5-eneDBU 1,8-diazabicyclo[5.4.0]undec-7-enedecomp. decompositionDME 1,2-dimethoxyethaneDMF N,N -dimethylformamideDMSO dimethylsulfoxideEI electron impactES electrosprayEt ethylFc ferrocenylHOMO Highest Occupied Molecular Orbitalhr, hrs hour, hours
16

Abbreviations
IR infraredLCAO Linear Combination of Atomic OrbitalsLUMO Lowest Unoccupied Molecular Orbitalm.p. melting pointMe methylMes mesityl, 2,4,6-trimethylphenylmin. minute, minutesMS mass spectrometryNBS N -bromosuccinimideNMR Nuclear Magnetic ResonancePh phenylPhth phthalimideiPr iso-propyl, prop-2-ylr.t. room temperaturetoil oil bath temperatureTHF tetrahydrofuranTOF Time Of FlightXRD X-Ray Di�raction
NMR spectroscopy abbreviationsd doubletdd doublet of doubletm multipletqqrt quartet of quartetqrt quartetquart. quarternary (written as a subscript to an atom, e.g. Cquart.)s singlett triplet
Vibrational spectroscopy abbreviationsm mediumms medium-strongmw medium-weak
17

Abbreviations
s strongsh shoulderv veryw weakβ bending (in-plane)γ bending (out-of-plane)δ deformationν stretchνnumber a fundamental frequency; also used for labelling vibrational mode
corresponding to that frequencyρ rockingτ twistω wagQnumber a general label for a vibrational mode if description by commonly
used greek letters cannot be used
18

Part I.
Introduction
19

1. General considerations
This thesis concentrates on the preparation and characterisation of �ve-membered sulfur-nitrogen ring compounds with the emphasis on �ve-membered heterocycles. Since par-ticular projects involved in this thesis deal with elements from di�erent areas of thePeriodic System, it seemed better to compose the thesis along these lines.
The �rst part describes the syntheses of the key sulfur-nitrogen reagents used through-out the research period as well as de�nitions of basic terms in computational chemistry,which are used in the sebsequent parts. The focus of the second part is on 1,3,2,4,5-dithia-diazarsoles with the general formula RAs(S2N2). The third part deals with half-sandwich5,1,3,2,4-metalladithiadiazoles of the Group 9 metals (Co, Rh, Ir) and in the last partthe structure and reactivity of Roesky's sulfoxide (S3N2O) is discussed.
Every part has its own introduction, discussion of results and experimental section.The overall contribution described in the thesis is summarised in a �nal conclusion, afterwhich a short account on future work follows. References are listed at the end of thethesis followed by appendices.
1.1. Preparation routes of sulfur-nitrogenheterocycles
General synthetic routes leading to sulfur-nitrogen heterocycles have been thoroughlyreviewed.1�3 The most useful are the cyclocondensation reactions which start with cheapand commercially available acyclic materials and �nish with ring compounds. These ringcompounds can be further converted into other ring compounds with suitable reagents.1,3
A number of methods will be described in the introductions of particular chapters. Theroutes leading to the most frequent su�ur-nitrogen reagents used during this work aredescribed here.
All syntheses started with S2Cl2, NH4Cl and elemental sulfur, which react together inthe absence of solvents at 150�152 ◦C to give the air-sensitive �ve-membered ring salt
20

1. General considerations
[S3N2Cl]Cl (eq. 1.1).4
2 NH4Cl + 4 S2Cl2 + 8 HCl + 5 S+ S
reflux N
S N
SS+
Cl
Cl-
(1.1)
Although unpleasant and despite relatively low yields, this synthesis can be convenientlyscaled-up to provide good amounts of the material. Interestingly, [S3N2Cl]Cl has notbeen used in many syntheses other than conversions to other purely SN rings. One suchexample is the preparation of [S4N3]Cl (eq. 1.2).4
N
S N
SS+
Cl
Cl- + S2Cl23 2 + 3 SCl2CCl4
reflux
N
SN S
S
NS
Cl-
(1.2)
The bright yellow crystalline compound is also air-sensitive but can be stored for longtime periods with only marginal decomposition. The conversion from [S3N2Cl]Cl to[S4N3]Cl is nearly quantitative.
[S4N3]Cl became extremely popular over the years because it is a safe source of the[S2N2] 2 � anion, which is the building block in a large number of metal-sulfur-nitrogencomplexes (see Part III of this thesis). S4N3Cl reacts with liquid ammonia to form acomplex mixture, which was thoroughly studied by 14N NMR spectroscopy.5 The majorproduct was shown to be the [S3N3] � anion (eq. 1.3), while [S2N2] 2 � was not detected.However, experiments preceding the NMR study con�rmed the formation of metalla-dithiadiazoles (PR3)2M(S2N2) (M = Pt, Pd) from Na[S3N3] (eq. 5.10, page 136)6 in-dicating that [S2N2] 2 � could be formed from [S3N3] � . The role of the metal centre inthe transformation of [S3N3] � was not investigated but it was suggested that [S3N3] �
disproportionates to give S4N4 and [S2N2] 2 � (eq. 1.4).6
7 [S4N3]+ + 6 NH3 9 [S3N3]- + H2S + 16 H+(1.3)
4 [S3N3]- 2 S4N4 + 2 [S2N2]2-(1.4)
Based on the results from previous works7 it is very probable that the disproportionationof [S3N3] � occurs also in the liquid ammonia reaction mixture. The generated [S2N2] 2 �
anion can then be directly utilised in substitution reactions with organometallic dihalo-genides to give various metalladithiadiazoles.
21

1. General considerations
One class of metalladithiadiazoles, namely the tin analogues dithiadiazastannoles (orstannadithiadiazoles) became versatile reagents in SN synthesis. [nBu2Sn(S2N2)]2 repre-sents a good example of a versatile tin reagent prepared from nBu2SnCl2 and [S4N3]Clin liquid ammonia (eq. 1.5).8
18 nBu2SnCl2 + 28 S4N3Cl + 92 NH3 +NH3 (l)
+ 18 S4N4 + 4 NH4SH + 64 NH4Cl
SnN
SnN
S
NS
S
NS
nBu
nBu
nBu
nBu9
(1.5)
The procedure is relatively time consuming (2�3 days) but this is mainly due to longpuri�cation period (Soxhlet extraction). The time is reasonable with respect to thequantities of the tin reagent obtained.
Both the liquid ammonia method and the use of the tin reagent [nBu2Sn(S2N2)]2 havebeen frequently employed during this work. The experimental details of the syntheses ofthe above SN reagents are described at the end of this introductory part.
1.2. The multiplicity of S−N bondsIt is often important to know whether a chemical bond is single, double or triple. Thiscan be formally decided on the basis of a bond order, which can be calculated accordingto the well known formula
b = n−n∗2
,
where b is the bond order, n is the number of electrons in bonding orbitals and n∗ isthe number of electrons in antibonding orbitals.9 Cystallographic methods showed thattriple bonds are shorter than double, which are shorter than single bonds. Thus, it ispossible to make an assessment of bond multiplicities of a novel compound on the basisof its X-ray structure. Organic molecules are excellent examples, in which the types ofbonds correspond very well with their lengths. Is this also the case for sulfur-nitrogencompounds?
Pauling's original covalent radii of singly bound nitrogen (0.70Å) and other �rst rowelements were corrected by Schomaker and Stevenson (0.74Å), who also provided thecovalent radii for doubly and triply bound main group elements.10 Pauling includedthese corrected values in the 3rd edition of his famous book about chemical bonds but
22

1. General considerations
recommended to use them only for the second period elements.11 Thus, the covalent radiifor singly, doubly and triply bound nitrogen are 0.74, 0.62 and 0.55Å, respectively, andthose of sulfur are 1.04, 0.94 and 0.87Å, respectively.∗
For the calculation of the length of a single bond between atoms A and B, Paulingsuggested to use Schomaker and Stevenson's equation
D(A−B) = rA + rB − c|xA − xB| ,
where rA and rB are Pauling's and - for the second period elements - Schomaker andStevenson's covalent radii of the two elements, xA and xB are Pauling's electronegativitiesof the two elements and c is a correction coe�cient. Pauling suggested the coe�cientto be 0.08Å for any bond involving one or two atoms from the second period of thePeriodic System. This is the case for a S−N single bond, which should be 1.74Å long.The theoretical S−−N bond length calculated by the addition of the covalent radii fordoubly bound nitrogen and sulfur11 is 1.56Å and the S−−−N length is 1.42Å.
The reality is not so straightforward. Classi�cation of S−N bonds on the basis of theirlength is tricky, as the intervals for the lengths of particular bond types are very wide.For example the experimental values of a S−N �single bond� range from 1.74 to 1.61Å(Fig. 1.1).14,15 The experimental values of a S−−N bond lie in the interval 1.56�1.46Å(in INSNI and [S−−N−−S]+, respectively)16,17 and the S−−−N bond lengths were found from1.46 to 1.42Å (in NSPh3 and NSF3, respectively).18�20
The overlap between the double and triple bond length interval is striking. On onehand it was emphasised that the valence description of [S−−N−−S]+ most probably doesnot correspond to reality and that the S−−N bonds in the cation have a signi�cant triplecharacter.17 Nevertheless, the form [S−−N−−S]+ is still commonly used. On the other handit was shown, that the length of a S−−N bond depends on the charge of a S−N molecule,i.e. the total number of electrons within the molecule. For example S4N4 contains nearlyequal S−N bonds with the length of 1.63Å (mean value).21 A withdrawal of one or twoelectrons from the antibonding HOMO orbitals results in a dramatic structural changeand shortening of the bonds to 1.55Å observed in the radical cation [S4N4] ·+ and inthe planar forms of the [S4N4] 2+ cation, respectively.22,23 If similar bond shortening by0.08Å is to be expected upon withdrawing electrons from the hypothetical thio-analogue∗Two recent studies brought a list of new single bond covalent radii for nearly all the elements. Whilethe summary by Cordero et al.12 is based on a vast amount of experimental values published in theCambridge Structural Database, Pyykkö and Atsumi brought a more consistent set of radii based onexperimental or calculated data of several model molecules, which were subsequently optimised byleast-squares �t.13 For nitrogen, both Cordero et al. and Pyykkö and Atsumi published a covalentradius of 0.71Å. The value is very close to the Pauling's original value but since Pauling himselfadopted the corrected value of 0.74Å, 0.74Å will be used throughout this thesis.
The new single bond covalent radius of sulfur was found to be 1.05Å (Cordero) or 1.03Å (Pyykkö).Pauling's original value (1.04Å) o�ers an excellent compromise and will be used throughout thisthesis.
23

1. General considerations
IN
SN
IN
S
NS
N
S
NS
O O1.61 Å
1.56 Å
1.55 Å
1.58 Å
1.60 Å 1.56 Å
(a) (b) (c)
(1.57 Å) (1.53 Å)
1.74 Å
HN
S
S S
S
NH
SS
1.69 Å
Fig. 1.1. Selected S-N compounds with S-N bond lengths: (a) S6(NH)2 (1,4-isomer); (b) S4N4O2; (c) INSNI; the bond lengths in parentheses belongto another INSNI molecule in the unit cell.
of nitrogen dioxide radical (·NS2), then Pauling's S−−N bond length (1.56Å) will changeto 1.48Å, which is very close to the experimental value in the [S−−N−−S]+ cation.17 Asigni�cant triple character then must be put in question. According to Parsons andPassmore, the formal bond order in [S−−N−−S]+ is indeed 2.24
Overall the determination whether a S−N bond is single or double is precarious. Sim-ilarly problematic is the commonly used term �a length between a single and a doubleS−N bond � for molecules where π-electrons delocalisation is possible.
1.3. Theoretical calculationsResearch in chemical synthesis has been for some time strongly supported by the use ofcomputing techniques. Nowadays, there are only a few synthetic chemists who wouldrefuse to bene�t from predictions of structure or electron arrangements within moleculesof their interest. Since some compounds discussed in this thesis were thoroughly analysedby computational methods, a brief section is included about how structure, bonding andaromaticity can be described by the results obtained from theoretical calculations.
1.3.1. Geometry of a moleculeThe structure of a molecule is given by a set of atoms arranged in certain manner in three-dimensional space. A particular set-up of atoms will be stable only if the total energy ofthis set-up will be low. A correlation between energy of a particle and its position in athree-dimensional space is mathematically described by the Schrödinger equation. Oncea mathematical description of a problem exists, calculations can be started to solve theproblem.
The geometry of a molecule is optimised in two steps. The �rst step is usually abinitio quantum mechanical calculations, the target of which is to �nd a solution of the
24

1. General considerations
Schrödinger equation for a multiatomic system, i.e. the energies of atomic orbitals.9 Be-cause the Schrödinger equation cannot be precisely solved for more than a two-particlesystem, the equation used is simpli�ed by several approximations. These approximationslead to inaccurate orbital energy values and therefore it is necessary to apply �post-abinitio� methods (e.g. DFT). The post-ab initio methods elaborate the ab initio resultsby taking into account circumstances neglected or averaged by the previously used ap-proximations and as a result lower orbital energy values are obtained.25
In the second step the ab initio geometry is subjected to molecular mechanics cal-culations (force �eld calculations), which assess how distorted are particular structuralparameters of the ab initio structure - i.e. bond lengths, bond angles, dihedral anglesand non-bonding interactions - from ideal values given by agreed standards.25 The higherthe distorsion, the higher energy contribution and the higher the overall energy of theab initio geometry. The overall energy must correspond to a minimum on a PotentialEnergy Surface (PES) if the ab initio structure is to be considered as stable. During thesemolecular mechanics calculations the vibrational frequencies are also calculated.25,26
1.3.2. Charges and bond ordersThe atomic charges and bond orders are calculated by partitioning the total electrondensity in the regions of the atoms. For both the atomic charge calculation and the bondorder calculation several concepts have been developed. The principles are complex.26
1.3.3. AromaticityAromaticity deserves to be regarded as something special chemistry has been endowedwith. One cannot forget the �rst lectures in organic chemistry where planar ring struc-ture, fully conjugated system of multiple bonds and 4n+2 π-electrons were described asthe basic criteria of aromaticity.27 However, the concept of aromaticity has deeper roots.For complicated systems a more general approach must be opted for and that is wherearomaticity starts to be even more interesting. The three known criteria for aromaticityneed to be generalised and modi�ed to give a set of �ve new ones:28,29
• (4n+2) π-electrons
• Structural criteria
• Energetic criteria
• Electronic criteria
• Magnetic criteria
25

1. General considerations
The origin of the Hückel's rule, (4n+2) π-electrons, is in quantum mechanics and therule is a condition of aromaticity valid for both organic and inorganic compounds.30 Theterm structural criteria refers predominantly to planarity and bond length equalisa-tion in aromatic molecules. Thanks to the delocalisation of π-electrons, the experimen-tally found bond lenghts in aromatic molecules do not have values of localised singleor double bonds but bear an average value between the two.28 The delocalisationenergy represents another possible aromaticity criterion28 since the delocalisation ofthe π-electrons brings lowering of the energy of aromatic systems in comparison withtheir non-delocalised analogues.27 Electronic criteria refer to the description of thedelocalised π-electronic system by electron population analysis.31 The typically highchemical shift of aromatic protons in 1H NMR spectrum27,32,33 points at the magneticproperties of aromatic compounds which are of big signi�cance, especially in the areaof theoretical calculations as is described in the next paragraph.
It was shown that none of the �ve criteria can serve as an indicator of aromaticity onits own.28 The desire to de�ne a single quantity which would be directly connected witharomaticity became stronger and in 1968 a quantity called �Diamagnetic SusceptibilityExaltation� (Λ) was introduced as a unique criterion of aromaticity.34 This quantity isde�ned as the di�erence in magnetic susceptibilities between systems with delocalisedand non-delocalised π-electrons:
Λ = χM − χM′ (1.6)
Since magnetic susceptibility is directly linked to the delocalised π-electrons in aromaticcompounds,35,36 diamagnetic susceptibility exaltation represents the best instrument ofaromaticity description.28,37 Unfortunately, despite being a single quantity describingaromaticity, diamagnetic susceptibility exaltation is not convenient for semi-empiricaltheoretical calculations, as its value �depends strongly on the ring size (area) and requiressuitable calibration standards�.38 Instead, it was suggested to calculate absolute magneticshieldings at the centres of aromatic rings. The obtained values were named Nucleus-Independent Chemical Shifts (NICS).38 The precision of this method was later enhancedwhen functional groups attached to the parent aromatic hydrocarbons were taken intoconsideration. Di�erent functional groups have di�erent magnetic environments whichin�uence the overall environment within the resulting molecule. Therefore it was foundmore exact to calculate NICS at 1 Å above the ring plane.39 For non-planar moleculesleast-squares plane through the ring must be calculated and in addition the use of NICSat 1 Å below the plane was proposed.40 NICS is then denoted as NICS(1), NICS(0) andNICS(-1) for NICS 1 Å above, at the centre and 1 Å below the plane, respectively.39,40 Theconvention says that negative NICS values denote aromaticity and the positive denoteanti-aromaticity.
26

1. General considerations
1.3.3.1. Aromaticity of sulfur-nitrogen compounds
The Hückel's rule was originally postulated for benzene, i.e. a molecule with six equi-valent atoms forming a ring with delocalised π-electrons. Inorganic analogues of benzene(e.g. borazine) are usually formed by di�erent atoms. Di�erent atoms bring inequiva-lent electronic environments to the molecule and therefore the solution of the quantummechanical problem could o�er results signi�cantly di�erent to those obtained for ben-zene.30 This could make Hückel's rule impossible to use for inorganic rings. However,since the semi-emprirical parameters used in the quantum mechanical calculations arevery similar for certain heteroatoms, the Hückel's rule may be applied to some inor-ganic compounds.30 In the case of binary sulfur-nitrogen rings, the Hückel's rule can beapplied. The number of π-electrons can be easily counted according to the method ofBanister:41,42 every sulfur (6 valence electrons) and nitrogen (5 valence electrons) atomuses 2 electrons for the connection with their neighbours by σ bonds and every atomin the ring keeps 2 more electrons as a lone pair. Thus, sulfur has 2 and nitrogen 1electron spare which can be used for π bonding. If the sum of the spare electrons withina SN cycle equals a (4n+2) number, then a high probability arises that the compound isaromatic to some extent.
Additional assurance about aromaticity can be obtained by calculating �TopologicalResonance Energy� (TRE)†, an analogue to delocalisation energy. If TRE > +0.01 β, acompound is aromatic, if +0.01 β > TRE > −0.01 β, a compound is nonaromatic andTRE values lower than −0.01 β indicate antiaromaticity.30,43 It was shown that (4n+2)SN rings give TRE values in the aromatic range. Also, the preference of some (4n+2)SN rings to adopt nonplanar geometry was explained on the basis of the TRE values:nonplanar rings gave signi�cantly lower TRE values than the planar ones.43
Also in case of inorganic rings, the calculation of NICS became very popular amongtheoretical chemists and is frequently used as an instrument of aromaticity determina-tion.31,40,44�46
Except for the extremely useful tool for π-electron counting, Banister also revealed thatthe content of π-electrons in the SN rings is higher than in the aromatic hydrocarbonsof the same ring size.41 The explanation for this comes from the quantum mechanical(Hückel and Extended Hückel) calculations which showed that the energies of sulfur andnitrogen atomic orbitals contain lower coulombic repulsion contributions than those ofcarbon.43 The energy of the molecular orbitals formed by the LCAO is thus lower thanin aromatic hydrocarbons.1,30,47 In addition, the gap between HOMO and LUMO wasfound to be smaller than in aromatic hydrocarbons.1 As a result, the excessive π-electronsoccupy nonbonding or antibonding π orbitals which are more accessible.1,30,47 Since the
†TRE is given in units of resonance integral β, which is also a calculated quantity.
27

1. General considerations
number of electrons in bonding and antibonding orbitals determines the magnitude of abond order,9 the occupation of antibonding orbitals in SN aromatics brings lowering ofthe bond order. This is then re�ected in higher reactivity and lower thermal stability ofSN aromatics in comparison with aromatic hydrocarbons.1,47
1.4. General experimental conditionsThe following general experimental details apply for all the following parts of the thesis.
Unless otherwise stated, all reactions were carried out in an oxygen-free nitrogen at-mosphere using standard Schlenk and syringe techniques. The term �high vacuum� cor-responds to the pressure of 0.3Torr.Dry solvents were used from the Solvent puri�cation system MB-SPS-800 (MBraunGmbH). Less common solvents were dried, puri�ed and stored according to commonprocedures.48
1H, 13C and 31P NMR spectra were recorded on a Jeol GSX spectrometer at the fre-quencies of 270.2, 67.9 and 109.4MHz, respectively. Chemical shifts δ were referencedto external tetramethylsilane and phosphoric acid, respectively. 14N NMR spectra wererecorded on a Bruker Avance II 400 spectrometer at 28.9MHz with δ referenced to exter-nal liquid ammonia. All 13C and 31P NMR spectra are proton-decoupled; for practicalreasons 13C and 31P descriptions will be used instead of 13C{ 1H} and 31P{ 1H}, respec-tively.
Mass spectrometry was performed by the University of St Andrews Mass SpectrometryService and elemental analyses were performed by the St Andrews University School ofChemistry Service.
Cyclic voltammetry was performed using an EcoChemie µAutolab apparatus controlledby GPES 4.2 software. Three-electrode system consisted of a glass-embedded platinumdisc working electrode (area = 0.008 cm2), platinum wire counter electrode and Ag/AgClreference electrode. Typically, 5mm solutions of the analyte in dry CH3CN were placed inan electrochemical cell of 10ml capacity and were provided with 1.0mmol of supportingelectrolyte ([nBu4N][PF6]). A small amount (0.2mg) of solid ferrocene was added asinternal standard. The voltammograms were calibrated for the half-wave potential of theferrocene/ferrocenium couple E1/2 = 0 V. Digital simulation of cyclic voltammogramswas performed using the DigiSim software.
IR spectra were recorded as pressed KBr discs (unless otherwise stated) on a Perkin-Elmer 2000 FT/IR/Raman spectrometer. Raman spectra were recorded in glass capil-laries in the range 3500�100 cm−1 using the same spectrometer.
28

1. General considerations
Single crystal X-ray structure data were collected on Rigaku MM007 confocal op-tics/Saturn or Mercury CCD di�ractometers using Mo-Kα radiation (confocal optic,λ = 0.710 73Å), and corrected for absorption. The structures were solved by directmethods and re�ned by full-matrix least-squares methods on F 2 values of all data. Re-�nements were performed using SHELXTL (Version 6.1, Bruker-AXS, MadisonWI, USA,2001).Powder X-ray di�raction patterns were recorded on a Stoe STADI/P di�ractometer usingCuKα1 radiation.
1.5. Experimental details of the key sulfur-nitrogenreagents syntheses
1.5.1. [S3N2Cl]Cl4
All joints were coated with te�on sleeves or te�on tape and slightly greased. Wherete�on coating could not be used, the joints were properly greased.
[NH4]Cl (300 g, 5.61mol) and sulfur �owers (60.0 g, 0.230mol) were placed in a 1 litre�ask. S2Cl2 (300ml, 506 g, 3.75mol) was quickly added, the mixture was mixed a littlebit with a glass rod and the �ask was covered with a lid. The lid was �tted with anair condenser (3.5 cm outer diameter, 75 cm long) and the top of the condenser was�tted with a drying outlet (anhydrous CaSO4). The top of the lid was insulated withglass wool and aluminium foil and the mixture was gently re�uxed. The heating wasadjusted so that the upper level of the condensing S2Cl2 lies just within the bottomjoint of the air condenser (150�152 ◦C). The reaction is �nished usually within 15�20 hours with the lower part of the condenser being covered with a layer of orange/browncrystals of [S3N2Cl]Cl. After cooling down to ambient temperature, the air condenserwas transferred quickly on air onto an empty, predried and weighed 500ml Schlenk �askwith a nitrogen gas �ow. The drying tube was removed, the top of the condenser wasplugged with a well greased stopper and the apparatus was evacuated for 30 minutes todry the product. The nitrogen gas was reintroduced and the dry product was scrapedo� the condenser walls with a spatula attached to a long metal rod. Yield 73 g (40%,based on S2Cl2).IR data: 1406 (mw), 1164 (mw), 1127 (vw), 1013 (ms), 960 (sh), 942 (vs), 705 (vs), 677(sh), 581 (ms), 562 (sh), 472 (m), 458 (m), 432 (mw) cm−1.Raman data: 1017 (m), 934 (w), 730 (m), 582 (m), 460 (w), 405 (s), 370 (ms), 351 (ms),266 (vs), 175 (s) cm−1.
29

1. General considerations
1.5.2. [S4N3]Cl4
All joints were coated with te�on sleeves and slightly greased. In a 250ml �ask equippedwith a stirring bar, dry CCl4 (50ml) was cooled to -10 to -20 ◦C (acetone/dry ice) andunder stirring S2Cl2 (50ml, 84.4 g, 0.630mol) was added in one portion. The solutionwas cooled to -20 ◦C (acetone/dry ice) and [S3N2Cl]Cl (14.0 g, 0.072mol) was added inone portion. The mixture was gently re�uxed (toil = 96 ◦C) for 5 hours. Still warm, themixture was �ltered through a sinter and the canary yellow product was washed twicewith 20ml and 10ml CCl4 respectively. The product was dried under high vacuum for1 hour. Yield 7.0 g (71%).IR data: 1262 (vw), 1165 (vs), 1130 (sh), 1001 (vs), 936 (sh), 794 (vw), 719 (vw), 683(s), 639 (mw), 608 (m), 589 (sh), 565 (s), 527 (sh), 469 (vs), 454 (s) cm−1.Raman data: 1173 (w), 1004 (w), 607 (mw), 568 (m), 447 (vs), 250 (s), 210 (ms) cm−1.
1.5.3. [nBu2Sn(S2N2)]28
Ammonia (200ml) was condensed at −78 ◦C (acetone/dry ice) in a predried 1 litre �askequipped with a stirring bar and [S4N3]Cl (7.0 g, 0.034mol) was added in three portions.The dark red mixture was stirred for 30 minutes at −78 ◦C after which time nBu2SnCl2(20.0 g, 0.066mol) was added in small portions over the period of 5 minutes. After awhile the mixture thickened and changed colour from dark red to brown/orange. Themixture was stirred at −78 ◦C for 2.5 hours, then the cooling bath was removed andthe mixture was allowed to warm up to room temperature overnight with the ammoniabeing evaporated. After the evaporation the mixture was evacuated for an hour toremove residual ammonia. The �ask was then transferred to a glove box together witha Soxhlet apparatus. The crude solid was carefully scraped o� the walls and was placedin a Soxhlet thimble, the thimble was placed into the apparatus and the apparatus wasclosed with stoppers. In this way protected crude [nBu2Sn(S2N2)]2 was moved back toa fume cupboard where the Soxhlet apparatus was attached to a 1 litre �ask containingdry and degassed petroleum ether (650ml). The Soxhlet apparatus was insulated withglass wool and aluminium foil and to the top a water condenser with an oil-bubbler wasattached. The crude solid was extracted for 8.5 hours (toil = 78�80 ◦C), at that point theextracts were nearly colourless. The extract was allowed to cool to room temperature,its volume was reduced to approx. 1
2and the concentrate was placed to freezer overnight,
where bright yellow precipitate separated. This was �ltered o�, washed twice with cold(−40 ◦C) degassed petroleum ether, dried under high vacuum for 1 hour and stored underargon.
The �ltrate was further reduced in volume and the resulting dark orange solution wasplaced in a freezer, where another portion of the material separated. This was isolated
30

1. General considerations
in the same way as the �rst crop. The second crop showed less satisfactory elementalanalyses. Overall yield 14.4 g (67%).Microanalysis (�rst crop): Found C, 29.5; H, 5.6; N, 7.8. Calc. for C8H18SnS2N2: C,29.4; H, 5.6; N, 8.6%.
31

Part II.
Arsenic-Sulfur-Nitrogen Heterocycles
Abstract
The introductory part starts with a brief summary of information on toxicity of arsenicand its compounds. Then, it seems appropriate to provide a small literature review onpreparative methods of basic arsenicals, which are frequently used as starting materialsbut are not commercially available anymore. A short account on the participation ofarsenic in �ve- and six-membered main group heterocycles follows and the introductorypart is ended with an overview of known arsenic-sulfur-nitrogen heterocycles with theemphasis on the analogues of S4N4 and S2N2. Their reactivity is discussed and seleniumand tellurium analogues are also described. Finally, new development within the area ofAsSN heterocycles is presented in the form of a detailed discussion of the results of myresearch.

2. Introduction
The chemistry of arsenic o�ers a lot of adventure. Generations of chemists have beeninvestigating the chemical behaviour of arsenic in the hope that the amount of knowledgewill at least match that of arsenic's lighter neighbours in Group 15, phosphorus andnitrogen. It seems correct to say that these expectations have only been ful�lled partly.Without any deep considerations three main reasons can be named which have hinderedthe chemical research of arsenic: toxicity, lack of commercially available starting materialsand low applicability of arsenic NMR.
2.1. The toxicity of arsenicArsenic and its compounds are very often referred to as strong poisons and carcinogenicsubstances. The use and synthesis of arsenic compounds forms a substantial part of thisthesis. To better understand where is the cause of arsenic's toxicity, a short literaturesurvey is included in the following brief section.
2.1.1. Arsenic in the environmentArsenic is the 20th most abundant element in the Earth's crust and is found in naturemainly in sul�dic ores.49 Its spread into the environment can occur by natural meanssuch as volcanic activity, wind erosion or the biological activity of microorganisms. Thehuman population contributes considerably to the overall arsenic content in the environ-ment through industrial activities such as mining or smelting, but also by burning fossilfuels or by the use of arsenic-based pesticides.49 On the other hand, the possibilities ofnatural arsenic removal from the environment are very limited. The principle of arsenicelimination from water is the oxidation and subsequent coprecipitation with oxidised ironor manganese particles present in oxygen-rich surface water. The precipitate then formssediments which are of high arsenic content. Furthermore, rainfall and dry depositionhelp to remove arsenic from the atmosphere.49
33

2. Introduction
2.1.2. Biotransformation of arsenic in natureArsenic is present everywhere in the environment and all organisms are unwillingly ex-posed to it. Fortunately, every organism has a metabolic pathway in which its body isdetoxi�ed. For example fungi and molds methylate arsenicals and release the correspond-ing alkyl(aryl)arsines as the �nal metabolic products.49 It was also found that fungi inrotting wood, previously treated with arsenic-based preservatives, are able to transformthese arsenicals into trimethylarsine.50,51
The attitude of plants towards arsenicals is diverse. While some plants succumb to thee�ects of on purpose invented weed killers, other plants have an extraordinary toleranceto arsenic and can take up and concentrate arsenic from the environment (e.g. waterhyacinth).52 The high arsenic content in the ash of some plants (e.g. Douglas Fir) waseven interpreted as a biogeochemical indicator of the presence of gold veins buried in thesubsoil.53
Fish and other water fauna metabolise arsenicals to arsenobetaine (Fig. 2.1). This
As+O-
O
H3C
H3C
H3C
NH2
AsO
OHOH
OH
AsO
OHOH
NO2
321
Fig. 2.1. Arsenobetaine (1), arsanilic acid (2) and roxarsone (3)49
highly methylated organoarsenic compound is believed to be the �nal product of thearsenic metabolism in �sh. Analyses have shown that the amount of arsenic in marine �shis greater than in the freshwater �sh.49,54 The possible poisoning of people by the arseniccontained in �sh and seafood is of no concern, since arsenobetaine is not metabolised inthe human body and is excreted unchanged.55
The arsenic tolerance of terrestrial animals is low. Much is known about the arsenicmetabolism of mice, rats and rabbits as they are commonly used for toxicity testing. Ar-senic in their bodies undergoes methylation and is excreted in urine.56,57 Similar metabo-lites were observed in the urine of dogs, pigs and cows.58 Poultry and pigs show increasedweight gain by the addition of arsanilic acid (4-aminobenzenearsonic acid) and roxarsone(4-hydroxy-3-nitrobenzenearsonic acid) (Fig. 2.1) to their food but these are excretedunchanged.59
34

2. Introduction
2.1.3. Arsenic in the human bodyThe fact that arsenic is toxic to humans is strongly backed up by a large volume ofmedical, toxicological and biochemical studies. It is predominantly the inorganic formsof arsenic to which people are nowadays exposed. The main routes of exposure are in-halation and ingestion. Similarly to other toxic substances, the consequences of exposureto arsenic depend on the scale of the dose and on the period during which the dose wastaken up.
Acute poisoning with huge doses of arsenic leads to a cardiovascular collapse and heavybrain damage with death coming within hours.60
Smaller doses cause acute arsenic poisoning with slower progress. The �rst responsescome from the gastrointestinal tract and take hours to days. They are followed bycardiovascular system shock which takes from minutes to hours and already causes nosmall amount of deaths. The poisoning proceeds further with multiple organ failures(kidney failure, liver enlargement, lung oedem) and also bone marrow damage. Withina week, symptoms on the skin and brain start to develop. Typical for the skin is anincrease of pigmentation, hyperkeratoses and dermatitis. Brain a�ection is distinguishedby perplexity, convulsions or prolonged coma.60
Even if acute arsenic poisoning is successfully warded o�, the �nal consquences in-cluding tumors and cancers may appear. These consequences are tricky because theyalways develop with a latency period and with no respect to the amount of arsenic onewas exposed to.60
Chronic arsenic poisoning develops during a prolonged exposure to arsenic. In the past,arsenic was frequently ingested as a medicine in the form of Fowler's solution (i.e. 1%solution of potassium arsenate prepared by dissolving As2O3 in aqueous KHCO3).61,62
Nowadays it is predominantly people living in areas where drinking water is taken fromwells sunk in arsenic-rich subsoils who su�er from chronic arsenic poisoning. The mostwell known areas are e.g. Bangladesh, the West Bengal state in India, several regions inArgentina and Chile and the southwest coast of Taiwan.61 The consequences of chronicarsenic poisoning are tumors and cancers of various tissues, dermatitis, gangrenes (BlackFoot disease) and other complications.61,63
2.1.3.1. The principle of toxicity of arsenic
There are nowadays two theories explaining the reason why arsenic is toxic to people.The �rst and commonly accepted theory is based on the a�nity of arsenic to sulfur. Inthe human body, arsenic in both oxidation states As III and AsV reacts with the free thiolgroups of proteins, enzymes and cofactors.56,64 This process was successfully simulated.It was shown that the interaction runs e�ciently at neutral pH according to equations
35

2. Introduction
2.1�2.3.64RxAsO(OH)3-x + 2 R'SH RxAs(SR')2(OH)3-x + H2O (2.1)
RxAs(SR')2(OH)3-x RxAs(OH)3-x + R'SSR' (2.2)
RxAs(OH)3-x + (3-x) R'SH RxAs(SR')3-x + (3-x) H2O (2.3)
The stability of the products is variable. While aliphatic As−S compounds are not sostable, As−S heterocycles are characterised by their increased stability. As expected, the�ve- and six membered rings are the most stable.65 The cofactors of the fundamentalenzymes in cell metabolism provide the source of vicinal �SH groups which react witharsenic to form stable heterocycles. The cofactor and thus the activity of the enzymethen become blocked, subsequent biochemical reactions are disrupted, cell metabolismcollapses and the cell dies.66
The second theory is based on the fact that inorganic As III compounds stimulate theproduction of H2O2 in the human body.67 In the presence of Fe 2+, H2O2 may dissociatein free hydroxyl radicals according to the Fenton reaction (eq. 2.4).68
Fe2+ + H2O2 Fe3+ + OH + OH-(2.4)
The radicals oxidise proteins and enzymes, as a consequence of which the cell metabolismcollapses causing death of the cell.66,69 This theory is currently under investigation butrepresents a strong challenge to the previously described �thiol group theory�.
Arsenic in the oxidation state +5 has another toxic e�ect. Due to its similarity tophosphate ion, arsenate can enter the ATP formation process and can replace phosphate.The resulting compounds are unstable, hydrolyse spontaneously and thus cannot serveas energy reservoirs.56,70
2.1.3.2. Metabolism of arsenic in the human body
Arsenic from all sorts of sources gets to the blood by which it is distributed throughthe whole body. Arsenicals are however quickly eliminated from blood and are redis-tributed into the organs. The organs most a�ected with arsenic poisoning are the liver,kidneys, bowel and spleen.60 Also well known is the accumulation of arsenic in hairand nails, a feature frequently utilised in forensics. Hair and nails are skin derivativescomposed of keratins, i.e. structural proteins with high ratio of aminoacids containingthiol groups. Arsenic carried to the hair by blood interacts with these thiol groups andremains stored.71,72
The metabolism of inorganic arsenicals takes place in the kidney.60 Arsenites are oxida-tively methylated to give methylarsonic acid. Methylarsonic acid can be further reduced
36

2. Introduction
to methylarsonous acid and methylated again to dimethylarsinic acid. Inorganic arse-nates must be �rst reduced to arsenites so that they can undergo methylation. Themethylated products are then excreted in urine. Scheme 2.1 summarises the metabolicalprocess.55,56,64
[AsVO4]3- [AsIIIO3]3- [MeAsVO3]2-
[MeAsIIIO2]2- [Me2AsVO2]- [Me2AsIIIO]-
Me3AsVO Me3AsIII
2e-
2e-
2e-
2e-
Me+
Me+
Me+
2e-
Me+
Scheme 2.1 Metabolism of inorganic arsenate in human body64
During the oxidative methylation, a methyl group from a donor is enzymatically trans-ferred onto the As III centre. The following reduction is also an enzymatic process andthe source of electrons are thiols which at the same time are oxidised to disul�des inaccordance with equations 2.1 and 2.2.64,73,74
It was found that soon after the arsenic intake, the unmetabolised inorganic formsare excreted but after 16 hours it is already dimethylarsinic acid as the main metabo-lite. Also, during chronic arsenic poisoning the main metabolite is dimethylarsinic acidwhereas for acute arsenic poisoning the excretion of methylarsonic acid is dominant.This is caused by the inhibition of the methylation by As III.75 Despite the possibility ofreducing dimethylarsinic acid to dimethylarsinous acid and continue the sequence up totrimethylarsine as suggested by Scheme 2.1, no trimethylated species have been detectedin human urine.55,57 An interesting recent discovery is that methylarsonous acid, an in-termediate in human detoxi�cation pathway, is more toxic than inorganic compounds ofAs III which were considered to be the most toxic arsenic substances to people.73
37

2. Introduction
2.2. Commercial availability of starting materials∗
The toxicity of arsenic and its compounds (especially inorganic) represents probably thetoughest obstacle from both practical and organisational points of view. In the pastarsenicals were used as chemical weapons and nowadays the risk of their misuse stillpersists, this time imposed by terrorist organisations. This forces commercial suppliersto introduce strict policies both for the production and distribution. On the industriallevel, the companies producing arsenicals must have special governmental licences whichmust be regularly renewed for no small fees. Preparations of major amounts of arseniccompounds are subject to strict evidence and limits within both the production, stockholdings and dispatching. Waste disposal is strictly monitored and therefore there mustbe investment in technologies which can make the waste arsenic-free. The overall high�nancial demand on the production of arsenicals adds a signi�cant amount to the prices ofthe �nal products which are then di�cult to sell (e.g. 250 g of AsCl3 for over ¿400). Onlymajor companies can a�ord to produce arsenicals and the range of products is usuallyvery narrow. Especially universities �nd this inconvenient. Simple starting materialsneed to be prepared which is not necessarily di�cult but it can be time consuming andit slows down the progress of the research.
The cheapest available material is arsenic trioxide, As2O3, which can be purchased inlarge quantities. It is, however, not useful for synthesis since it is not soluble in commonorganic solvents and it is not particularly reactive at ambient temperatures. Arsonic andarsinic acids are usually poorly available with only phenylarsonic (C6H5As(O)(OH)2),arsanilic (Fig. 2.1 on page 34) and dimethylarsinic (Me2As(O)(OH)) acids being the reg-ular items in the stocks. Arsenic(III) chalcogenides As2E3 (E = Se, Te) can be purchasedbut they �nd application as semiconducting materials rather than starting materials inchemical synthesis.76,77 Arsenic trihalides AsF3, AsCl3 and AsI3 are available but the ra-tio quantity:price is not convenient. Mono- and dialkyl/aryl substituted halogenoarsinesare not commercially produced due to their possible warfare use. Triphenylarsine is theonly trisubstituted arsine available.
2.3. 75As NMR as an analytical toolThe rapid development in phosphorus chemistry was signi�cantly helped by the magneticproperties of the element. 31P NMR enabled virtually immediate product or mixturecharacterisation. Arsenic, similarly to phosphorus, is monoisotopic (75As) but its nu-
∗The commercial availability data were taken from the internet catalogues of Sigma-Aldrich(http://www.sigmaaldrich.com), Alfa Aesar (http://www.alfa-chemcat.com) and Apollo Scienti�c(http://www.apolloscienti�c.co.uk), accessed during my research period.
38

2. Introduction
clear spin of 32and relatively high quadrupolar moment result in broad lines in spectra
and make 75As NMR impossible to use for characterisation of other than symmetricalmolecules.78,79
2.4. Methods of preparation of common startingmaterials
The term �common starting materials� relates to simple inorganic and organoarseniccompounds, which were described in vast majority of the studied literature as startingmaterials for particular syntheses. They can be sorted in 4 classes:
• Arsenic trihalogenides
• Arsonic acids
• Arsinic acids
• Organohalogenoarsines
2.4.1. Arsenic trihalogenidesThe most convenient laboratory preparations of arsenic trihalogenides on a medium scaleuse arsenic trioxide and follow similar procedures. AsF3 and AsI3 are prepared by thetreatment of As2O3 with suitable halogenating agent (CaF2, KI) in concentrated acid:80,81
As2O3 + 3 CaF2 + 3 H2SO4 2 AsF3 + 3 CaSO4 + 3 H2O (2.5)
As2O3 + 6 KI + 6 HCl 2 AsI3 + 6 KCl + 3 H2O (2.6)
The preparation of AsF3 is carried out in a combined apparatus where a still headwith a water condenser is attached to the reaction �ask from the beginning. The mix-ture requires gentle heating in a water bath. The reaction starts at 30 ◦C and at 65 ◦Cthe colourless product distills to the collection �ask. AsF3 can be puri�ed from smallamounts of HF by the addition of dried NaF which is usually placed in the collection�ask. Subsequent distillation to a metal storage cylinder a�ords su�ciently pure AsF3as a colourless, air-sensitive liquid.80,82,83
Solid AsI3 precipitates when mixture of As2O3 and conc. HCl is carefully added to anaqueous solution of KI (eq. 2.6).81 AsI3 is �ltered o� and puri�ed by recrystallisation fromCS2 81 or diethyl ether.84 AsI3 is an orange/red crystalline solid at room temperature.81
39

2. Introduction
AsBr3 can be prepared within a couple of hours when As2O3 is allowed to react withconc. HBr at 120 ◦C in the absence of any solvents (eq. 2.7).85
As2O3 + 6 HBr 4 AsBr3 + 3 H2O (2.7)
HBr is used in excess which shifts the equilibrium to the right. AsBr3 crystallises out ofthe solution after cooling, is isolated and puri�ed by extraction with hexane. At roomtemperature, AsBr3 is a colourless, crystalline and volatile substance.85
AsCl3 is best prepared from As2O3 and SOCl2:86,87
As2O3 + 3 SOCl2 2 AsCl3 + 3 SO2 (2.8)
SOCl2 is used in excess as it ful�lls also the role of a solvent. The reaction proceedssmoothly over the period of 2 days, excess SOCl2 is then separated from AsCl3 byfractionation distillation using a Vigreux column and the crude product is distilled undernitrogen at atm. pressure to give pure AsCl3 as a clear and colourless, air-sensitive liquid.This preparation route gives good yields of AsCl3, leaves minimum waste and the distilledSOCl2 may be used for another preparation.87
2.4.2. Alkylarsonic acidsIn 1883 Meyer observed that iodomethane oxidises sodium arsenite to sodium methylar-sonate at 75 ◦C. Su�cient amount of alcohol had to be added to ensure mixing of theinorganic aqueous phase with CH3I (eq. 2.9 and 2.10).88
As2O3 + 6 NaOH 2 Na3AsO3 + 3 H2O (2.9)
Na3AsO3 + CH3I + NaI
O
AsH3C
ONaONa
EtOH
(2.10)
The alkaline solution is neutralised and acidi�ed with conc. HCl to convert the arsonateto arsonic acid. Meyer's general route was further re�ned in later reports. The reactiontimes, the amount of alcohol added and the acidity of the mixture were described.89,90
Meyer's discovery is of huge signi�cance. The oxidation of arsenite to arsonic acidwith alkyl halide is frequently and deservedly referred to as �Meyer's reaction� in theliterature. Unfortunately, the use of Meyer's reaction is limited for alkylarsonic acidsonly.91 Arylarsonic acids need to be prepared by di�erent routes which will be introducedin the next section.
40

2. Introduction
2.4.3. Arylarsonic acids2.4.3.1. Rosenmund's method
In 1921, Rosenmund attempted to apply Meyer's reaction for the synthesis of arylar-sonic acids. In a reaction analogous to equations 2.9 and 2.10 (page 40), he preparedphenylarsonic and 2-carboxyphenylarsonic acids from K3AsO3 and bromobenzene or2-bromobenzoic acid, respectively. In both reactions copper catalysts were used, butthe conditions of particular reactions were di�erent. While phenylarsonic acid was ob-tained nearly from neat reactants at the boiling point of bromobenzene (180�200 ◦C),2-carboxyphenylarsonic acid was formed in much milder conditions, by a re�ux (90 ◦C)of an aqueous solution mixed with a small amount of ethanol. The yield of phenylarsonicacid was low, that of the latter acid was decent, approximately 50%. Rosenmund ex-plained the low yield of phenylarsonic acid as a result of its thermal decomposition causedby the temperature necessary to maintain the re�ux during the reaction.92 Unfortunately,Rosenmund's method was generally shown to be unsuitable for the preparation of arylar-sonic acids. With the exception of 2-carboxyphenylarsonic acid and o-phenylenediarsonicacid, no other arylarsonic acid has been prepared in good yields.93
2.4.3.2. Bart's reaction
The most convenient method for the preparation of arylarsonic acids thus remainsBart's reaction, a coupling of alkaline arsenites with diazonium salts. Heinrich Bartpatented94�96 and improved97�99 this process in the years 1910�1912 but the publicationof his results was not possible until 1922.100 The original procedure is shown in Scheme2.2 on page 42. An aromatic amine is converted to ammonium salt by aqueous HCl andthe salt is diazotised with sodium nitrite. A solution of sodium arsenite in water is thenadded to the solution of the diazonium salt, the pH of the resulting mixture is madeslightly alkaline and the mixture is heated to help the nitrogen gas release. After the N2evolution is �nished, the mixture is �ltered and the arylarsonic acid is isolated from the�ltrate. The methods of isolation depend on the properties of particular acids. Acids in-soluble in water precipitate from the alkaline �ltrate upon acidi�cation with HCl. Acidswith higher solubility in water are isolated by extraction with alcohol from a dry residueformed after acidi�cation of the �ltrate and evaporation of water. If organic byproductsare formed during the preparation of an arylarsonic acid, the �ltrate is evaporated todryness and the byproducts are extracted with alcohol from the dry residue, i.e. fromthe Na or K salt of the arsonic acid. The remaining solid matter is then redissolved inwater, acidi�ed and the acid is isolated as was mentioned above. Additional puri�cationis possible by a recrystallisation from a suitable solvent, usually methanol, ethanol or
41

2. Introduction
R NH2 R N+ N Cl-R NH3 Cl-NaNO2
HCl
HCl
H2O
HClR As
O
ONaONa
1. + Na3AsO3 + NaOH - N2
2. heating
R As
O
OHOH
Scheme 2.2 The preparation of arylarsonic acids by Bart's reaction
water, with or without the use of activated charcoal.93,100 The outcome of the reactionbetween an arsenite and an aryldiazonium salt strongly depends on pH and dilution ofthe mixture and on the type of substituents on the aromatic ring.
Acidobasic conditions of Bart's reaction Bart described the optimum conditions as�an alkaline solution with not too high concentration of OH � �, where the diazoniumsalt would be converted to diazohydroxide with only marginal or zero formation of syn-diazoate (vide infra).100 He carried out two series of experiments to demonstrate the roleof pH and dilution of the reaction mixture. In acidic as well as in strongly alkaline solu-tions no reaction between arsenite and diazonium salt occured. Similarly, concentratedmixtures of certain alkalinity resulted in zero or low yields of the arsonic acid comparedto more diluted systems with the same OH � concentration.100 Bart used these resultsto contribute to discussions about the acidobasic behaviour of diazonium compounds,which were ongoing already for two decades at that time. He suggested that higher alka-linity is necessary for the transformation of the diazonium salt into a syn-diazoate, whichin diluted conditions undergoes hydrolysis to set up an equilibrium with its conjugatedacid, �diazohydrate�, nowadays named diazohydroxide (eq. 2.11). Bart was convincedthat diazohydroxide was the key reactant during the substitution with alkali arsenites.100
diazonium diazohydroxide diazoate
Aryl N+ N Aryl N N OH Aryl N N O-OH- OH-
+ H2O
(2.11)
Bart's theory deserves huge respect since he had a very correct view on this problemdespite the fact, that in the early 20th century there were little means to study in depthsuch a sensitive system like diazonium ions.
Later it was shown that arenediazonium ions behave like strong Lewis acids, which arestable in acidic aqueous solution in the company of anions of strong acids.101 When anaqueous base (hydroxides, Na2CO3) is added to the diazonium salt, it replaces those acid
42

2. Introduction
anions with hydroxide anions which do not have such a stabilising e�ect. The diazoniumcation becomes less stable and either enters substitution reactions 2.12 and 2.13 or reactswith OH � to form covalent diazo compounds according to eq. 2.11.101,102 The equilib-rium in 2.11 has been studied thoroughly and it was shown that the diazohydroxide istransformed quickly into the diazoate and that in reality �the diazonium salt reacts atonce with 2 equivalents of OH � to form the diazoate�. The concentration of the diazo-hydroxide in the course of the reaction remains negligible. This means that the onlycoupling entity in the whole aqueous diazotation system is the diazonium ion and notdiazohydroxide as was suggested by Bart.101,103
+ NaOH + N2 + NaClH2O
Cl-
NN+
OH
(2.12)
Cl-
+N
N+
OH
H2O
NaOHONa
NN + NaCl + H2O
(2.13)
Furthermore, as predicated by Hantzsch104 and supported by Bart,100 it was con�rmedthat the diazoates indeed form two geometric isomers syn- and anti -. Their structure wasproved by spectroscopic methods, examples of both aliphatic and aromatic syn-diazoateswere successfully analysed by X-ray di�raction. A comparison of bond lengths of the -NNO � unit in syn-diazoates o�ers strong support for partial delocalisation of the negativecharge in aromatic syn-diazoates unlike in the aliphatic ones (Fig. 2.2).105�107 Similarly,localised bonds can be expected in aliphatic anti -diazoates as was demonstrated by theX-ray structure of thallium methyl-anti -diazoate.108 To the best of my knowledge therehas been no report on the crystal structure of an aromatic anti -diazoate.
N N
Aryl O-
N N
Aryl
O-
syn-arenediazoate anti-arenediazoate
N N
Aryl O
Fig. 2.2. The structures of syn- and anti - arenediazoates
Because the syn � anti isomerisation depends on pH of the mixture, it can be incor-porated into the general equilibrium 2.11. The result of such incorporation is the setof equilibria 2.14�2.18, which represent a detailed description of the dependence of the
43

2. Introduction
entire aqueous diazotation system on acidobasic conditions.109
Aryl–N=N+ + OH- syn-Aryl–N=N–OH (2.14)
syn-Aryl–N=N–OH + OH- syn-Aryl–N=N–O- + H2O (2.15)
syn-Aryl–N=N–OH anti-Aryl–N=N–OH (2.16)
syn-Aryl–N=N–O- anti-Aryl–N=N–O- (2.17)
anti-Aryl–N=N–OH + OH- anti-Aryl–N=N–O- + H2O (2.18)
These equilibria were drawn on the basis of a series of spectrophotometric and elec-trochemical measurements.109,110 It can be seen that after the addition of a base to anaqueous solution of a diazonium salt it is the syn-diazoate which is formed �rst andthen it isomerises to anti -diazoate. Thus, diazonium ion is in equilibrium only withsyn-diazoate.†This is the most important outcome, which justi�es the acidobasic con-ditions described in Bart's experimental instructions.100 For a successful preparation ofarylarsonic acids it is necessary to maintain a stable pH of the mixture in the area mostconvenient for the diazonium salt�syn-diazoate equilibrium.93,102
The role of substituents in the aromatic ring. The e�ect of the substituents is car-ried over onto the diazonium cation, where it in�uences the formation of diazoates andtheir stability.102 Thus, for aromatic diazonium salts bearing various substituents on thearomatic ring, the equilibria 2.14�2.18 are established at various pH. Bart found, thatarenediazonium salts bearing electron withdrawing substituents undergo the reaction
†The anti -diazoate is disadvantaged due to kinetic factors. Littler110 compared the diazoates to alkenesand deduced that since the position of the lone pair on the aryl-bound nitrogen in the syn-diazoateis trans to the hydroxyl group, it must facilitate the leaving of the hydroxyl group while a diazoniumsalt is formed. Similarly as alkenes, where a proton situated trans to a leaving group will react fasterwith a base in a bimolecular elimination reaction:27
N N
Aryl OHAryl
N+
N
+ OH-N N
Aryl
OH
Aryl
N+
N
+ OH-is faster than
as well as
R1 Br R1
+ BH+ + Br-
R1
Bris faster than
H R2B
R2
H
R2
B
R1
+ BH+ + Br-
R2
syn- anti-
44

2. Introduction
with alkali arsenite in less alkaline environment in comparison with other arenediazo-nium salts. However, he refused to draw any general conclusions stating that no ruleapplies for the preparation of the acids. He recommended an empirical approach andshared his experience that ortho- and para- substituted aromatics react better thanmeta-isomers.100
In later years measurements of a series of diazonium salts were undertaken and theresulting pH and equilibrium constants were listed alongside the Hammett constant σ ofthe corresponding substituent. It turned out that arenediazonium salts with strong elec-tron withdrawing substituents (�NO2, �CN or �CF3 groups; high positive σ values) reachequilibrium at higher pH and rapid isomerisation from syn- to anti - follows. Arenedia-zonium salts bearing electron donating substituents (�NH2, �O � and �C(O)Me groups;most negative σ values) did not establish an equilibrium within the conditions of themeasurement, the reaction ran too slowly.111 These results con�rmed Bart's observationsand pointed at the problem of side-reactions. Both quick syn � anti isomerisation andvery slow equilibrium allow side-reactions.111 The formation of diazo dyes as products ofside-reactions was observed by Bart, who concluded that side-reactions can be e�cientlysupressed by working in diluted conditions. Such conditions enable hydrolysis of diazoateto syn-diazohydroxide, which Bart considered the main substance that reacts with alkaliarsenite.100
Catalysts in Bart's reaction. In one of supplements to his original patent, Bart sug-gested the use of elemental copper or its salts as catalysts in his reaction. The catalystswere added to the alkali arsenite solution and during the coupling with diazonium saltthey assisted the nitrogen gas evolution so that it proceeded at lower temperatures andthe resulting arylarsonic acid could be obtained in higher purity.99 Interestingly, the useof catalysts is not mentioned in the published version of Bart's discoveries.100 Catalystswould probably do little work in Bart's reaction as it is autocatalysed: in the moleculeof an alkali arsenite the lone pair on arsenic helps the nitrogen release (Scheme 2.3).The role of the metal ions as catalysts is believed to be in preventing side reactions byscavenging aryl radicals converting them into less harmful cations.102
N+ N
Cl
As-O
O-
ONa+
Na+
Na+
As
O
O-Na+
O-Na+ + N2 + NaCl
Scheme 2.3 Coupling of benzenediazonium chloride with sodium arsenite
45

2. Introduction
2.4.3.3. Modi�cations of Bart's method
Over many years, Bart's method was slightly modi�ed, which led to increased yields ofsome arylarsonic acids. The most substantial modi�cation was �rst presented by Földi,who observed formation of arylarsonic acids when arenediazonium salts were coupledwith AsCl3 instead of alkaline arsenite.112 But it was Scheller, who later described de-tailed conditions, under which good yields of arylarsonic acids could be obtained.113,114
The reaction should be carried out in the absence of water or in the presence of onlya necessary minimum of water, depending on the nitrite used for diazotisation. Whenworking in anhydrous conditions, Scheller used glacial acetic acid as solvent and amylnitrite or solid, powdered sodium nitrite. The diazotisation can be successfully achievedalso if sodium nitrite is dissolved in minimum water.114 AsCl3 can be present in the mix-ture either before the diazotation or can be added afterwards to a solution of a diazoniumsalt. CuCl114 or CuBr93 are used as a catalysts, which help the release of nitrogen gasand hence they need to be added to the ��nal� mixture, i.e. the mixture which containsall reactants and was diazotised (Scheme 2.4).93 An intermediate containing pentacoor-dinate arsenic is formed �rst, which is hydrolysed in the second step to give arylarsonicacid.93
R N+ N + AsCl3HSO4- MeOH H2O
O
AsR OH
OHR As
Cl
OSO3HCl
ClCuCl,CuBr
Scheme 2.4 Reaction scheme of Scheller's modi�cation of Bart's reaction
This modi�cation proved suitable for preparing arylarsonic acids with electron withdraw-ing substituents in meta- position to the diazonium group.93 Nowadays its use would belimited by di�cult accessibility to, and the cost of AsCl3.
2.4.3.4. Béchamp's reaction
The last synthetic route leading to arylarsonic acids mentioned in this thesis is themethod by Béchamp. Arylarsonic acids are obtained by arsonation, i.e. substitutionof an aromatic proton by arsono group.93 The source of the arsono group is arsenicacid (H3AsO4), which can be obtained either by hydration of As2O5
115 or by oxidationof As2O3 with 46�52% nitric acid in the presence of iodine.116 In a typical Béchampexperiment a neat aromatic compound is heated/melted and solid arsenic acid is added.The temperature of the mixture is then rapidly increased well above the melting point ofthe aromatic reactant and is later allowed to cool down. The heating period di�ers fromminutes to days, depending on the aromatic reactant. The resulting matter is extracted
46

2. Introduction
with aqueous alkali and the arsonic acid is precipitated from the extracts by addition ofHCl.93,115
The process is analogous to sulfonation. Arsenic acid is, however, a weak arsonatingagent and therefore the aromatic proton needs to be more labile. As a consequence,Béchamp's method gives satisfactory results only for a narrow range of aromatics, usuallywith amino or hydroxo groups attached to the aromatic ring. Aromatic compoundsbearing electron withdrawing substituents o�er very low yields of arsonic acids.93
2.4.4. Dialkyl- and diarylarsinic acidsBoth Meyer's and Bart's reactions can be performed twice to prepare dialkyl- and diary-larsinic acids, respectively. First an arsonic acid is prepared, which is in the next stepreduced to arylarsonous acid.
O
AsROH
OH + SO2 + H2O + H2SO4AsR
OHOH
H+
KI
(2.19)
The reduction of the arsonic acid is best carried out by SO2 or HSO �3 in acidic envi-
ronment in the presence of HI or KI. During the reduction, diluted sulfuric acid100 orglacial acetic acid98 are recommended. Hydrochloric acid is not used, since it convertsthe arsonous acid to alkyl- or aryldichloroarsine.117 The arsonous acid usually precipi-tates out from the solution, may be puri�ed by recrystallisation from suitable solventand is �nally dissolved in aqueous hydroxide solution to form arsonite. The arsonite isthen either oxidised by a new portion of alkyl halide (eq. 2.20) or is coupled with a newarenediazonium salt (eq. 2.21).98,100,118
NaOHR As
O
OHR'
AsROH
OH AsRONa
ONa2. HCl
1. + R'X + EtOH
(2.20)
R' N+ N Cl-
NaOH
+ NaOH
2. heatingR As
O
OHR'
AsROH
OH AsRONa
ONa
1.
- N2
+
3. HCl (2.21)
47

2. Introduction
2.4.5. OrganohalogenoarsinesThis section will concentrate on monoalkyl/aryl substituted halogenoarsines, since theseare most relevant to this thesis.
2.4.5.1. From arsonic and arsinic acids
The most comfortable way leading to substituted halogenoarsines is the reduction of thecorresponding arsonic acids with gaseous SO2 in the presence of concentrated hydrochlo-ric or hydrobromic acid (eq. 2.22). The preparation of RAsCl2 proceeds better if a smallamount of iodide is added as a catalyst.89,117 Similarly, reduction of an arsinic acid withSO2 gives dialkyl-/diarylhalogenoarsines.118
O
AsROH
OH + 2 HX + SO2 + H2SO4 + H2OAsRX
X
X = Cl, Br (2.22)
Another possible way to prepare RAsBr2 is to reduce the parent alkylarsonic acid withexcess PBr3.119 Diiodoarsines RAsI2 are probably the easiest to prepare of all organo-halogenoarsines. The parent arsonic acid can be reduced either by SO2 in the presenceof HCl and KI or only by hydroiodic acid itself.120 The preparation of RAsF2 by thereduction of arsonic acids has not been reported.
If the parent arsonic acid is not commercially available, it can be prepared by eitherMeyer's or Bart's reaction (sections 2.4.2 and 2.4.3.2). Meyer's reaction can be directlyfollowed by the reduction with SO2 without isolating and purifying the alkylarsonic acid.Alkyldihalogenoarsines can thus be prepared in a one-pot-synthesis from As2O3 accordingto eq. 2.9, 2.10 and 2.22.121
The reduction of arsonic acids or arsonates is especially convenient for the preparationof monoalkyl/aryldihalogenoarsines.
2.4.5.2. From AsX3 and organometallic reagents
The posession of arsenic trihalogenides opens the gates to a vast number of substitutedarsines. The alkyl/aryl group is introduced on arsenic by means of Grignard reagentsor organolithium compounds, many of which are commercially available or alternativelythey can be readily prepared from alkyl- and arylbromides (eq. 2.23�2.26).
R Br + Mg R Mgreflux
+ I2, etherBr
(2.23)
48

2. Introduction
AsX3 + n RnAsX(3-n) + n MgBrXR = alkyl, aryl
X = Cl, Br, I
n = 1-3
R Mg Br
(2.24)
R Br + BuLi + BuBrR Liether
low temp. (2.25)
AsX3 + n RLi RnAsX(3-n) + n LiXR = alkyl, aryl
X = F, Cl, Br
n = 1-3 (2.26)
This method is versatile and fast. Both alkyl and aryl halogenoarsines may be preparedin short time periods. A drawback is the impossibility of controlling the number ofhalogens being substituted. This becomes most apparent when preparing monosubsti-tuted halogenoarsines, the preparation of which requires an excess of AsX3 to minimisethe formation of di- and trialkylarsines. However, an entire suppression is not possibleand mixtures of mono-, di- and trialkyl substituted halogenoarsines are always obtained.
In general, AsCl3 is the most suitable for the reactions with both Grignard reagentsand organolithium compounds, which is re�ected in the high number of publications.The published preparations of iPrAsCl2,122 tBuAsCl2,123 MesAsCl2,124 Mes*AsCl2 125 orFcAsCl2 126 are good examples.
Grignard reagents. There is no report on the preparation of RAsF2 using Grignardreagents. AsBr3 was shown to react with MesMgBr to give only Mes2AsBr and magne-sium bromoarsenate with the formula MgAs5Br17 (eq. 2.27), even if the stoichiometricratio of the reactants was 1:1.127
2 + 6 AsBr3 Mes2AsBr + MgAs5Br17 + MgBr2Mes Mg BrTHF / hexane
(2.27)
The reaction between AsI3 and Grignard reagents is usually not adopted, since the con-version of RAsCl2 to RAsI2 is very simple (section 2.4.5.7, page 58). Nevertheless, it hasbeen reported for the preparation of cyclohexyldiiodoarsine.128
Organolithium reagents were successfully used during the syntheses of Cp*AsF2129 and
Mes*AsF2130,131 from AsF3. Also CpAsBr2 132 and Cp*AsBr2 129 were prepared in this
way from AsBr3, but the reaction between AsI3 and organolithium compounds has notbeen reported.
49

2. Introduction
2.4.5.3. Controlled substitution of halogens in AsX3
Since the reaction between organometallic reagents and AsX3 yields a mixture of mono-,di- and trialkylarsines RnAsX3-n (n = 1�3), an e�ort has been put in the investigation ofsyntheses, which would o�er full control over the substitution process and would reliablylead purely to RAsX2.
Halogenation-elimination reaction. One means to control the substitution of halo-gens in AsX3 is to convert AsX3 into R3As with a particular Grignard reagent and tosubstitute one or two of the organic groups back with a halogen in a halogenation-elimination reaction. The process was described in 1928 in a study on the strength ofthe As−C bond. The study revealed that in some arsoranes, sterically demanding or-ganic groups can be substituted by means of thermal cleavage of the As−C bond.128 Forexample tricyclohexylarsine was oxidised with chlorine to dichlorotricyclohexylarsorane,Cy3AsCl2 (eq. 2.29). When heated (200 ◦C) in vacuum, Cy3AsCl2 released chlorocy-clohexane to leave Cy2AsCl (eq. 2.30). Cy2AsCl was once more oxidised with chlorineto Cy2AsCl3 which was heated in vacuum and decomposed already at 80�90 ◦C intochlorocyclohexane and CyAsCl2 (eq. 2.31 and 2.32).
AsCl3 + 3 MgCl + 3 MgCl2As
(2.28)
AsCCl4
+ Cl2 0 ºC
Cl
As
Cl (2.29)
AsCl
vacuum
200 ºC +
ClCl
As
Cl (2.30)
AsCl
+ Cl2 0 ºC
petroleumether
Cl
As
Cl
Cl
(2.31)
50

2. Introduction
AsClCl
vacuum
80-90 ºC+
ClCl
As
Cl
Cl
(2.32)
Neopentyldibromoarsine was prepared by the same method.133 Although halogenation-elimination is only of very limited use, it represents a reliable and controlled way leadingto monoalkyldihalogenoarsines.
Protection by cyclisation. Another possibility is to restrict the number of halogensavailable for the reaction with Grignard reagents. An example is the formation of 10-chloro-5,10-dihydrophenarsazine (4) from AsCl3 and diphenylamine (eq. 2.33).134
As
Cl
ClClN
HN
As
Cl
H
+ + 2 HClo-dichlorobenzene
reflux
4 (2.33)
In 4 there is only one chlorine atom available for the reaction with Grignard reagents (eq.2.34)135,136 and the resulting 10-alkyl-5,10-dihydrophenarsazine can be easily decomposedby gaseous HCl into the required RAsCl2 and diphenylamine hydrochloride (eq. 2.35).136
N
As
Cl
H
+ RMgBr
N
As
R
H
+ MgBrClether
(2.34)
N
As
R
H
+ 3 HCl NH·HCl+110-130 ºC
R
AsClCl
(2.35)
Protecting groups. Suitable protecting groups can be utilised in the same sense asin the case of the phenarsazine formation. The substitution of halogens with dialky-lamino group is frequently employed during preparation of various alkyl derivatives ofhalogenophosphines137,138 and it can be used with similar success in the chemistry ofhalogenoarsines. The most frequently used protecting groups are dimethylamino groups.
51

2. Introduction
The substitution of AsCl3 with two protecting �NMe2 groups leaves only one halogenavailable for the reaction with the Grignard reagent and the resulting alkylbis(dimethyl-amino)arsine is then nearly quantitatively converted to dichloroalkylarsine by gaseousHCl. The method is versatile (eq. 2.36 and 2.37).139
As Cl
R2N
R2N+ R' Mg Cl R'As
R2N
R2N+ MgCl2
Et2O
-10 ºC
(2.36)
R'As
R2N
R2NR'As
Cl
Cl
+ 4 HCl(g) + 2 R2NH·HClEt2O
(2.37)
Mono- and bis(dimethylamino)halogenoarsines can be prepared from stoichiometricamounts of AsCl3 and dimethylamine in ether,140 but more reliable route is o�ered by fullconversion of AsCl3 into As(NMe2)3 followed by a ligand-exchange between As(NMe2)3and AsCl3 as is shown by equations 2.38 � 2.40.141�143
AsCl3 + 6 Me2NH + 3 Me2NH·HCl0-5 ºC
hexaneAs
NMe2
NMe2Me2N (2.38)
As(NMe2)3 + 2 AsCl3 3 Me2NAsCl2 (2.39)
2 As(NMe2)3 + AsCl3 3 (Me2N)2AsCl (2.40)
The equilibria described by equations 2.39 and 2.40 are strongly shifted to the right andare not random, which is the main reason why a de�ned stoichiometric ratio of AsCl3and As(NMe2)3 provides nearly quantitative yields of the corresponding dimethylamino-halogenoarsines (Me2N)nAsCl3-n (n = 1, 2).143,144
2.4.5.4. From AsX3 and organosilicon and organotin reagents
Organosilicon reagents. An alternative to the highly air-sensitive organometallicreagents is the use of alkyl- and aryltrimethylsilanes RSiMe3, which upon reaction withAsX3 yield substituted halogenoarsines. The driving force in this reaction is the faciledissociation of a Si−C bond together with the high a�nity of silicon to form bondswith halogens.132 The usefulness of this method was demonstrated in the preparation ofCpAsX2 (X = F, Cl, Br).132
RSiMe3 + AsX3 RAsX2 + Me3SiX4 ºC
(2.41)
52

2. Introduction
Alkyl-/aryltrimethylsilanes are more stable than lithiated hydrocarbons and their solu-bility in organic solvents is incomparably higher than that of the lithiated hydrocarbons.Moreover, Me3SiF is a gas at room temperature, Me3SiCl and Me3SiBr have relativelylow boiling points (57 and 79 ◦C, respectively)145 and therefore can be easily removedfrom the reaction mixture under reduced pressure. Alkyl/aryltrimethylsilanes thus o�erbetter control over the reaction and higher comfort of work.
Organotin reagents. Organotin reagents, in particular alkyl/aryltributylstannanesmay be applied in an identical way (eq. 2.42 and 2.43).146,147
[Na]+ + Bu3SnCl SnBu3 + NaClr.t., 2 days
benzene
(2.42)
SnBu3 + AsCl3 AsCl2 + Bu3SnCl1. -30 ºC
2. r.t., 10 min. (2.43)
The butyl group C−Sn bond is probably the most chemically stable of all organotinbonds, whereas the cyclopentadienyl C−Sn bond is known to be reactive for both homo-and heterolytic cleavages.148,149 The heterolytic cleavage is believed to proceed through anelectrophilic attack at carbon but is considerably assisted by a nucleophilic attack at thetin centre.148 AsCl3, as a medium strength Lewis acid,150 can attack the cyclopentadienylC−Sn bond. Furthermore, a nucleophilic chlorine atom from AsCl3 can attack the tincentre and as a result the reaction proceeds smoothly giving the product, CpAsCl2, in67% yield. The e�ciency of organotin reagents can be demonstrated by a whole row oforganometallic compounds. For example HfCl4 reacts with Bu3SnCp to give CpHfCl3.151
Other examples are displayed in Scheme 2.5.All the syntheses using the organotin reagents (Scheme 2.5) show one remarkable
feature. Purely by mixing the reactants in 1:1 stoichiometric ratio, they enable thepreparation of monoorganyl metallohalogenides from metal halogenides in 70�80% yields.No mixtures of mono-, di- and multiorganometallohalogenides have been reported. Thereliability of the monosubstitution is most likely a consequence of a bonding changeoccuring during the reaction. In the organotin reagents, the non-butyl organic group isσ bonded to tin, whereas in the products, the organic ligands are π bonded to the metalcentre (Scheme 2.5). In CpAsCl2 prepared from Bu3SnCp, the Cp-ligand is σ boundto arsenic, which was con�rmed by both 1H NMR and IR spectroscopies and furthersupported by theoretical calculations, which identi�ed two stable conformers with theCp-unit σ bound to arsenic.132,147 On the contrary, the crystal structures of all fourbulkier analogues Cp*AsX2 revealed a σ bound Cp*-ring with signi�cant π-interactionsof the neighbouring carbon atoms.129
53
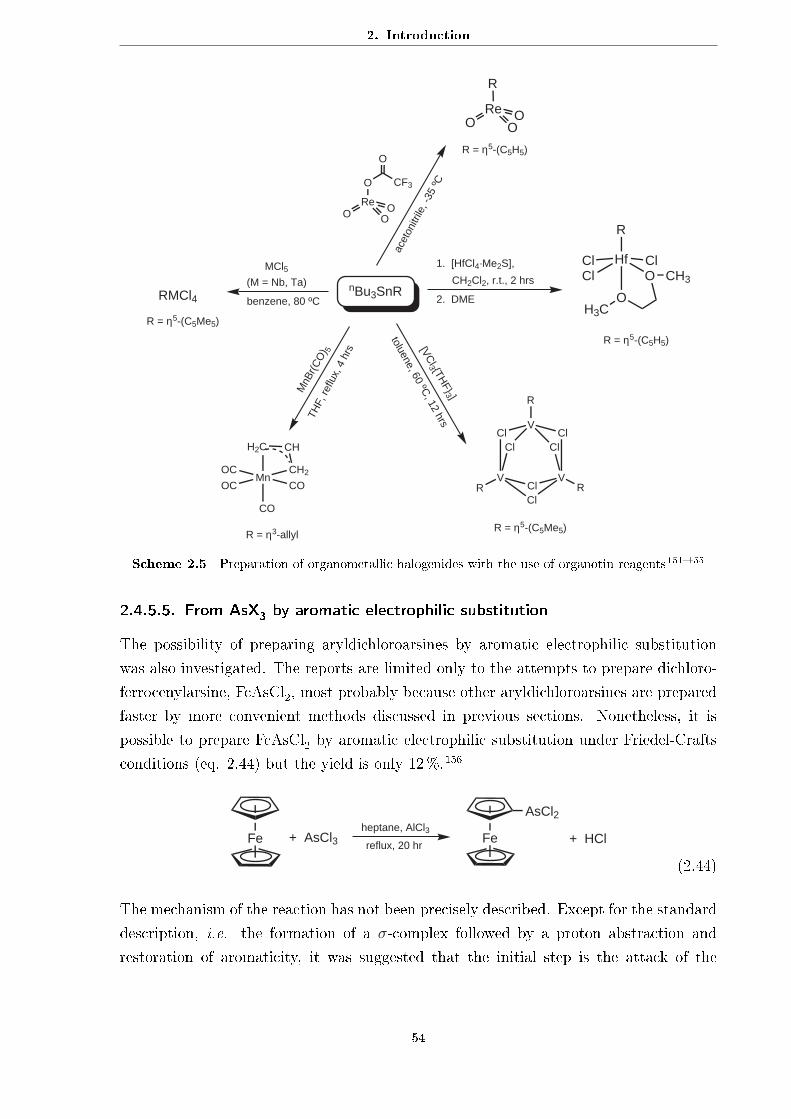
2. Introduction
nBu3SnR
1. [HfCl4·Me2S],
CH2Cl2, r.t., 2 hrs
2. DME
R
Hf
O
ClClClO
H3C
CH3
R = η5-(C5H5)
MCl5(M = Nb, Ta)
benzene, 80 ºCRMCl4
R = η5-(C5Me5)
Re
O
O OO
CF3
O
acet
onitr
ile, -
35 ºC
Re
R
O OO
toluene, 60 ºC, 12 hrs
[VCl3 {TH
F}3 ]
V
ClCl
V
ClCl
VClCl
R
RR
R = η5-(C5Me5)
R = η5-(C5H5)
MnB
r(CO
) 5
THF,
reflu
x, 4
hrs
Mn
H2C
CO
OC
CH2OC
CO
CH
R = η3-allyl
Scheme 2.5 Preparation of organometallic halogenides with the use of organotin reagents151�155
2.4.5.5. From AsX3 by aromatic electrophilic substitution
The possibility of preparing aryldichloroarsines by aromatic electrophilic substitutionwas also investigated. The reports are limited only to the attempts to prepare dichloro-ferrocenylarsine, FcAsCl2, most probably because other aryldichloroarsines are preparedfaster by more convenient methods discussed in previous sections. Nonetheless, it ispossible to prepare FcAsCl2 by aromatic electrophilic substitution under Friedel-Craftsconditions (eq. 2.44) but the yield is only 12%.156
Fe Fe
AsCl2heptane, AlCl3
+ AsCl3 reflux, 20 hr + HCl
(2.44)
The mechanism of the reaction has not been precisely described. Except for the standarddescription, i.e. the formation of a σ-complex followed by a proton abstraction andrestoration of aromaticity, it was suggested that the initial step is the attack of the
54

2. Introduction
electrophile on the iron centre.156 This, however, has not been fully con�rmed yet. A laterstudy only speci�ed that �the metal atom is not an essential participant in electrophilicsubstitution of ferrocene� and that it could not be proved that the participation of ironin the process would �provide some energetic advantage�.157
FcAsCl2 is not obtained directly from the reaction mixture as is described by eq. 2.44but it is treated with aqueous NaOH to prepare disodium ferrocenylarsonite. Acidi�ca-tion with hydrochloric acid results in precipitation of the dimeric ferrocenylarsenic oxide,which is in the next step converted by conc. HCl to ferrocenyldichloroarsine (Scheme2.6).156
Fe
Fe
AsCl2Fe
AsONa
ONa
Fe
AsO
OAs
2 2
Fe
AsCl22
(crude, not isolated)
(isolated) (pure, isolated)
NaClH2ONaOH
NaClH2O
HCl
HCl H2O
Scheme 2.6 Isolation of dichloroferrocenylarsine.
This classical organic synthesis is on one hand very time consuming, on the other ityields reliably monosubstituted halogenoarsine, although the yield is low. For a com-parison, Spang et al.126 prepared FcAsCl2 from AsCl3 and lithioferrocene in yield ofapproximately 20% if the reactants were in 1:1 ratio.‡ However, the impression of ahigher yield is blurred by the fact that unrecoverable 7-fold excess AsCl3 was used tosupport the formation of the monosubstituted haloarsine.
2.4.5.6. Special methods of RAsX2 preparation
In 1955, Knunyants and Pilskaya showed how chemical weapons can be both destroyedand used for the synthesis of useful starting materials. They used Lewisite and bis(2-chlorovinyl)chloroarsine for the preparation of R2AsCl and RAsCl2, respectively.158,159
The method makes use of the 2-chlorovinyl groups as protecting groups during alkylationwith a Grignard reagent in the �rst step (eq. 2.45). The protecting groups then undergo‡The yields for both Friedel-Crafts and lithioferrocene routes are based on AsCl3 used.
55

2. Introduction
an elimination of HCl in the presence of a base to form alkyldiethynyl- or dialkylethynyl-arsine. Water formed during the elimination reaction causes a hydrolysis of the C−Asbond, acetylene is released and alkylarsenic oxides are formed (eq. 2.46 and 2.47).
Cl
AsCl
Cl
Cl
As Cl
Cl
+ 2 RMgX
+ RMgX
Cl
AsR
R
Cl
As R
Cl
ether+ 2 MgClX
+ MgClX
0 ºC
ether
0 ºC
Lewisite
(2.45)
+ 2 KOH
Cl
AsR
R + 2 H2O + 2 KClEtOH
AsRR
H
2 2
AsO
AsR
R
R
R
+ 2 C2H2 + H2O
(2.46)
AsR
+ 4 KOH
Cl
As R
Cl
+ 4 H2O + 4 KClAs R
H
H
2 2EtOH
O
OAs R + 4 C2H2 + 2 H2O
(2.47)
56

2. Introduction
The alkylarsenic oxides are then converted to dialkylchloro- or alkyldichloroarsine byconc. HCl (eq. 2.48).
AsO
AsR
R
R
R
AsRO
OAs R
+ 2 HCl 2 + H2OAs ClR
R
+ 4 HCl As
R
ClCl2 + 2 H2O
(2.48)
At the end of the reaction, the oily crude product is swiftly extracted with a suitableorganic solvent (e.g. chloroform), the organic layer is separated, dried and the solventis evaporated. The alkylhaloarsines are puri�ed by vacuum distillation. The yields aregenerally high (over 70%).
Although smart, this recipe is nowadays virtually of no use for practical preparationsof organohaloarsines RAsCl2 or R2AsCl. The starting materials are, for obvious reasons,not easily available and, moreover, their time consuming preparation in order to convertthem to di�erent compounds of the same type, which can be prepared by much fasterprocedures, is of little sense.
In 1949, Kharasch et al. described an interesting and e�cient synthetis of EtAsCl2from AsCl3 and tetraethyllead.160
3 AsCl3 + PbEt4 3 EtAsCl2 + C2H5Cl + PbCl2100-110 ºC
(2.49)
Solvents can be used but the best yields (95�97%) were obtained in the absence of anysolvents. The authors found that the reaction has a radical mechanism and proceeds intwo steps, of which the �rst (eq. 2.50) starts at room temperature but the second needstemperatures above 90 ◦C to proceed rapidly (eq. 2.51).
2 AsCl3 + PbEt4 2 EtAsCl2 + Et2PbCl2 (2.50)
AsCl3 + Et2PbCl2 EtAsCl2 + C2H5Cl + PbCl2t > 90 ºC
(2.51)
The experimental procedure itself is somewhat adventurous, since PbEt4 is added slowlydropwise to neat AsCl3 preheated to 100 ◦C. Nevertheless, the reaction runs smoothly,the volatile C2H5Cl is collected in an attached trap cooled with acetone/dry ice bathand after the reaction is �nished, the product is distilled under reduced pressure straight
57

2. Introduction
from the reaction �ask leaving the solid PbCl2. The distilled EtAsCl2 may be furtherpuri�ed by fractional distillation to remove residues of unreacted AsCl3.
The advantage of this synthetic route is that no mixtures of mono-, di- and triethy-larsines are formed at prescribed temperatures (i.e. below 120 ◦C).160 The correct stoi-chiometric ratio of the reactants leads to quantitative yields of dichloroethylarsine. Still,this method will �nd only scarce application due to high toxicity of both starting mate-rials.
2.4.5.7. Halogen exchange reactions of RAsX2
RAsF2. So far in section 2.4.5 only little has been written about the preparation of�uoroarsines RAsF2. It is because RAsF2 generally cannot be prepared from simplereactants by the methods described for other RAsX2 systems. Only one route has beenreported - the reaction between organolithium reagents and AsF3,129�131 but this methoddepends on the type of the organic rest R and is applicable for only a few derivatives.RAsF2 are usually prepared by conversion of alkyl- and arylarsine oxides (RAsO) or fromRAsCl2 by halogen exchange.
The preparation of RAsF2 in relatively dilute aqueous conditions (e.g. from alkylarsineoxides in the presence of conc. HF) brings little success as the high solubility of RAsF2 inwater makes their isolation di�cult.161 It is therefore necessary to work in concentratedconditions (CaF2/H2SO4 system).80,161
The halogen exchange is carried out most frequently between RAsCl2 and suitable�uorides. Anhydrous NH4F was found as a universal reagent converting RAsCl2 toRAsF2 (R = Me, Et, Ph) at 90 ◦C in the absence of solvents.161 In case of Cp*AsF2,cobaltocenium �uoride was used.129
Other RAsX2. RAsCl2 can be converted to RAsBr2 by halogen exchange with LiBr.The reaction proceeds smoothly at room temperature in acetone but the yields were notreported.162 The conversion of RAsCl2 to RAsI2 is easily done by mixing with KI in asuitable dry solvent163 or with the mixture KI/HI.164
The reverse process leading from RAsI2 to RAsCl2 is not so simple. RAsI2, dissolvedin an organic solvent, must be hydrolysed to the corresponding alkyl- or arylarsine oxideby Na2CO3,121 aqueous NH3
165 or NaOH.126 The alkylarsine oxide is then suspended inconc. HCl and is treated with gaseous HCl.121
58

2. Introduction
2.5. Arsenic containing main-group ring compoundsThe chemistry of arsenic is well developed and arsenic containing ring compounds havebeen investigated so extensively that the data and experience could easily �ll pages ofa book. Therefore only some examples are picked to illustrate the width of this area ofchemistry.
2.5.1. Organoarsenic heterocycles2.5.1.1. Five-membered heterocycles
The propensity of arsenic to become a part of stable heterocyclic compounds was observedand fully exploited in the years 1939�1940 during intensive works, which led to thediscovery of 2,3-dimercaptopropan-1-ol as a suitable Lewisite antidote (British Anti-Lewisite, BAL).65,166�169 The �ve-membered ring 2-chlorvinyl-4-hydroxymethyl-1,3,2-di-thiarsolane formed after the application of BAL (eq. 2.52) must be named as one of thebest known examples of arsenic containing main-group heterocycles.170
AsCl Cl
Cl
SH
SH
OH
Lewisite BAL
+S
AsS
OH
Cl
+ 2 HCl
(2.52)
Greater attention has been paid to arsole, the analogue of cyclopentadiene. The un-substituted compound, C4H4AsH, has not yet been prepared but discussions about itsaromaticity have been ongoing for a long time. Theoretical calculations of its groundstate structure revealed pyramidalisation at arsenic with an inversion barrier of 33.9kcal/mol, a much lower value than that of AsH3 (54.5 kcal/mol). This lowering wasattributed to the conjugation of the lone pair on arsenic with the other π-electrons de-spite the pyramidal geometry.171 Ab initio calculations produced a molecular geometrywith the C−As single bonds shorter than an isolated C−As single bond, i.e. a furthersupport for delocalisation of the 6 π-electrons.172 The only �vote� against aromaticity ofarsole is o�ered by delocalisation stabilisation energies, which were found to contain zerocontribution when compared with the delocalisation energies within buta-1,3-diene.172
Finally, the calculated aromatic stabilisation energies (ASE), Λ and NICS suggest thatarsole is aromatic, though the extent of aromaticity is very modest.173,174
The 1-alkyl- and 1-arylarsoles C4H4AsR (R = tBu, Ph)175�177 as well as other deriva-tives bearing more alkyl groups are known.178 The compounds are best prepared from a
59

2. Introduction
mixture of E and Z isomers of 1,4-dichlorobuta-1,3-diene according to eq. 2.53.176,177
RAsH2 RAsLi2+ nBuLi
- C4H10
Cl Cl+
- LiClAs
R
R = Ph, tBu (2.53)
Similarly to the cyclopentadienide anion, the arsolide anion [C4H4As] � isaromatic173,174 and forms sandwich complexes with η5-coordination of a metal centre.Since arsole has not been prepared yet, the source of [C4H4As] � is 1-phenylarsole. Thearsenic-carbon bond is e�ciently cleaved with alkali metals (Li or K) and the result-ing arsolide readily reacts with a metal halogenide to give the desired sandwich com-plex.177,179,180 Equation 2.54 shows the preparation of 1,1'-diarsaferrocene.177
As
Ph
Li (excess)
THF, 3 hrs, r. t.- PhLi
AsLi+
1. AlCl3, THF, 0 ºC
2. FeCl2, THF, 24 hrs
As
As
Fe- LiCl
(2.54)
2.5.1.2. Six-membered heterocycles
The most attractive examples of six-membered heterocycles are probably arsabenzene,arsanaphthalene and arsaanthracene. Arsaanthracene181�183 was the �rst to be synthe-sised followed by arsabenzene.184,185 Both compounds were prepared from cyclic organ-otin reagents by substitution of the metal with AsCl3 and subsequent abstraction of HClby a base (eq. 2.55 and 2.56).182,185
Sn
H H
Me Me
AsCl3120 ºC, 3 hrs
As
H H
Cl
DBU
THFAs- Me2SnCl2 - DBU·HCl
(2.55)
SnnBu nBu
AsCl3THF- nBu2SnCl2
As
Cl
DBN
- DBN·HClAs
(2.56)
The synthesis of arsanaphthalene was more complicated. A new route had to be foundand thus it took nearly another ten years before arsanaphthalene was prepared. Ben-zenediazonium carboxylate was allowed to react with arsabenzene to yield 4H-1,2-etheno-
60

2. Introduction
arsinoline, from which ethylene was removed by means of 3,6-di(pyridin-2-yl)-1,2,4,5-tet-razine (eq. 2.57 and 2.58).186 Surprisingly, in the view of the successful application ofcyclic organotin precursors during the syntheses of arsabenzene and -anthracene, a suit-able precursor to arsanaphthalene was not isolated until 2001. Arsanaphthalene is ob-tained from this precursor, 1,4-dihydro-1,1-dimethyl-1-stannanaphthalene, in two steps,which are analogous to those described by eq. 2.55.187
N2+
O-
OAs
As+ - N2
- CO2
CH2Cl2
(2.57)
+As N
N
R
R
AsN
N N
N
R
R
- N2
CDCl3+
R =N (2.58)
All three compounds are air sensitive. Arsabenzene is thermally stable, colourless liq-uid, arsanaphthalene and -anthracene are unstable yellow liquids. Arsanaphthalene and-anthracene show a high tendency to react in Diels-Alder reactions resulting in prod-ucts containing the more stable three-coordinate arsenic.182,186 2-trimethylsilyl-1-arsa-naphthalene is found as a ligand in a half-sandwich complex of the Mo(CO)3 moiety.187
Arsabenzene �gures as a ligand in a number of half-sandwich (M = Cr,188 Mo189) andsandwich (M = Ti,188 V,188 Cr,190 Mo191) metal complexes and can be coordinated alsoas a monodentate ligand (M = Mo, W).189,191
2.5.2. Arsenic homocyclesAnother group of main group heterocycles are multiarsaheterocycles, which in the limit-ing case may become arsahomocycles.
2.5.2.1. Five-membered arsenic homocycles
Arsenomethane, [CH3As]5, was conveniently prepared by reduction of sodium methy-larsonate with H3PO2 in aqueous solution at elevated temperature.192 It was isolated asa yellow oil, which crystallised upon cooling. The compound forms �ve-membered ringsin a distorted envelope conformation. Four methyl groups alternate above and below theAs4 moiety and the �fth methyl group is placed in an equatorial position on the �fth,
61

2. Introduction
envelope-tip As atom. The As−As bond lengths (mean value 2.428Å) are just below theAs−As single bond range (2.44�2.8Å).193,194
The cyclic anion [As5] 4 � can be prepared e.g. from arsenomethane according to eq.2.59.195
AsAs
As
As As+ [CpMo(CO)3]2
190 ºC, 2 days As AsAsH3C
CH3
CH3
CH3H3CMo
As As
Mo
- 5×CH3- 6 CO
5
(2.59)The loss of the �ve methyl groups is believed to be metal-catalysed and is not unusualunder the reaction conditions.195 The three rings in 5 are all eclipsed in the solid state.The As5 ring is much larger than the Cp rings. Due to structural irregularities withinthe As5 ring it was recommended to formulate the anion [As5] 4 � as a bridging anionicligand [µ-(η4-As5)] 4 � rather than [η5-As5] 4 � .195
The aromatic196,197 anion [As5] � is found in pentaarsametallocenes η5-(C5R5)M(η5-As5)(R = H, Me; M = Fe, Ru), where it takes the position of one cyclopentadienide anion.In these compounds the As5 ring is regular and coordinates the metal centre in the η5-mode.198,199 Decaarsametallocenes have been studied only theoretically.200,201 The ions[(η5-As5)2M] � (M = K, Nb) and [(η5-As5)2Nb] 3 � were observed in ES mass spectra butno examples have been isolated so far.202
2.5.2.2. Six-membered arsenic homocycles
Arsenobenzene, [PhAs]6, is a white crystalline solid prepared by reduction of pheny-larsonic acid in a way analogous to arsenomethane.192,203 The molecule consists of theAs6 ring which is in a chair conformation, similarly to cyclohexane. The six phenylgroups are in equatorial positions. The As−As bond lengths are very close to those ofarsenomethane (mean value 2.459Å) indicating As−As single bonds.204
Hexaarsacyclohexa-1,3,5-triene (cyclo-As6) is an analogue of benzene, which has notyet been isolated as a pure chemical species. According to ab initio SCF MO calculationsthe planar structure of cyclo-As6 is stabilised by resonance energy and the As−As bondlength value (2.307Å) is an average between single and double bond values (2.44�2.48Å
62

2. Introduction
and 2.22�2.24Å, respectively).194
The anion [As6] 4 � was prepared by thermal205,206 and photochemical205,207,208 reactionroutes (Scheme 2.7) and its structure was determined by X-ray analysis.205 The anion
Mo
[Cp×Mo(CO)2]2 + As4p-xylene
reflux, 2.5 days
As AsAs
AsAsAs
Me
MeMe
MeMe
MeMe
MeMe Me
Mo
[Cp×Mo(CO)2(η3-As3)]toluenehν ,
reflux, 1 hr
Scheme 2.7 Preparation of the anion [As6] 4 �
[As6] 4 � forms a planar, regular six-membered ring stabilised by two Cp×Mo moieties(Cp× = C5Me4Et). The delocalisation of As−−As double bonds was suggested on thebasis of experimental values of the As−As bond lengths in the ring. The values areequivalent within experimental error and the mean value (2.35Å) �ts well between thoseof a single and a double As−As bonds. Though, aromaticity of the anion was notclaimed.205 Later it was shown, that the shape of the As6 ring depends on the coor-dination mode of the anion. Highly coordinated (e.g. η6-) [As6] 4 � is planar, whereaslow coordination of [As6] 4 � gives a chair conformation of the As6 ring with signi�cantlylonger, yet equal As−As distances.209 Ab initio calculations and analysis of the electronlocalisation function refuted aromaticity of the �chair-anion� [As6] 4 � .209
2.5.2.3. Arsafullerenes
An attractive example of coexistence between �ve- and six-membered rings are fullerenes.The stabilities of their all-arsenic analogues Asn (n = 4, 8, 20, 28, 32, 36, 60) havebeen calculated using DFT methods. The higher clusters with the exception of As20were found to be �unstable against dissociation into As4 units� with the dissociationbeing an exothermic process.210 The cluster As20 is the only crystallographically char-acterised member of the row. It has been obtained as a part of the �onion-skin� anion[As@Ni12@As20] 3 � .211,212 The term �onion-skin� is used since the anion consists of threesurrounding layers. The �rst layer is a sole arsenic atom, which is situated in the centre ofthe icosahedron Ni12. The unit [Ni12(µ12-As)] 3 � is then surrounded by the fullerene-likecage As20, which forms the third layer (Fig. 2.3). The cluster As20 forms a dodecahe-dron consisting only of pentagonal faces. The As−As bond lengths (2.752 ± 0.025Å)
63

2. Introduction
are longer than As−As single bonds (2.42�2.44Å)195 but lie within a very close rangecon�rming the regularity of the cluster.211
Fig. 2.3. Ball and stick model of the anion [As@Ni12@As20] 3 � : the dodecahedron As20 (A), theanionic subunit [Ni12(µ12-As)] 3 � (B) and the whole anion (C). The bonds between particularsubunits are omitted for clarity.§, 211
2.5.3. Arsenic containing analogues of S4N4
By exchanging atoms of sulfur or nitrogen in an S4N4 molecule for arsenic a row ofcompounds with general formulae AsnS4-nN4 and S4AsnN4-n (n = 1�4) can be obtained.In this section the known types of compounds and their reactivity are described and theirstructural relationship to S4N4 is highlighted. The heterocycles are sorted according toelemental composition of the eight membered ring, which can be binary (As4S4 andAs4N4) as well as mixed of all three elements - As, S and N - in various ratios.
2.5.3.1. Cyclotetrathiatetrarsocane
As4S4 is the most known example - the mineral realgar. It is an analogue of S4N4, whereall nitrogen atoms are exchanged for arsenic. The structure of As4S4 is analogous toS4N4 but the positions of the S and As atoms are reversed, as is the case in P4S4.213 TheS atoms are coplanar and form a square, the As atoms - two above and two below thesquare - are arranged into vertices of a tetrahedron. The atoms in the As4S4 moleculeare bound by 10 single bonds, 8 As−S and 2 transannular As−As bonds.214,215
The chemistry of As4S4 is quite established. Particular attention has been paid toits role as a ligand in complexes with d-block metals, where the As4S4 cage structurefeatures in various degrees of distortion. For example, the cage structure is fully pre-served in adducts with HgX2 (X = Br, I). This is quite surprising taking into accountthat (HgBr2)3(As4S4)2 is prepared by melting the reactants in vacuum at 400 ◦C with asubsequent annealing period of two weeks at 190 ◦C.216 The adduct [HgI2 ·As4S4]2 was
§Fig. 2.3 was copied from the Science magazine website accessed on 09/12/2008(http://www.sciencemag.org/cgi/content/full/300/5620/778/FIG1).
64

2. Introduction
prepared under identical conditions from HgI2, As and S in molar ratio 1:4:4.217 Analternative way leading to better yields of (HgBr2)3(As4S4)2 is a solvothermal reactionbetween HgBr2 and As4S4 in CS2 at 160 ◦C.216 Both adducts contain an undistortedmolecule of As4S4, which is coordinated to mercury by very weak Hg−S bonds.216,217
On the contrary, total decomposition of the As4S4 cage occurs upon interaction with ag-gressive reagents such as chlorine. In the presence of [PPh4]Cl compound (PPh4)2As4Cl14is formed according to eq. 2.60.218
As-
Cl
As4S4 + 2 [PPh4]Cl + 10 Cl2CH2Cl2 As
Cl
ClAs
ClCl
Cl
Cl
Cl
ClCl
Cl
Cl Cl
As- Cl- 4 SCl2
PPh4+
PPh4+ (2.60)
The [As4Cl14] 2 � anion consists of two tetrachloroarsenite units and two AsCl3 molecules.The weak interactions between particular entities (displayed by dashed bonds in eq. 2.60)make the spatial coordination of the As atoms that of a distorted pseudooctahedron.218
When As4S4 reacts with d-block metal carbonyls at elevated temperatures the cagestructure is broken into fragments, which are coordinated to the metal centre. No pat-tern was observed, according to which the As4S4 cage structure would fragment. How-ever, for complexes containing larger As−S fragments it is believed, that the As4S4molecule �rst breaks into smaller fragments which may later recombine to form biggerfragments.219,220 The fragmentation was demonstrated by a series of reactions betweenAs4S4 and Cp×2M2(CO)4 (M = Fe, Ru) or Cp×2Co2(CO)2 (Cp× = C5Me4Et). The ironcomplexes contained smaller fragments As2S2 and As2S3 220 whereas in the rutheniumcomplexes larger fragments As4S2, As2S5 and As3S5 were found.219 The cobalt carbonylyielded complexes bearing a wider range of As−S fragments.221
Brunner et al. also demonstrated, that the presence of organometallic species asadducts of As4S4 strongly in�uences the outcome of the cage fragmentation. Whilepure As4S4 reacts with Cp×2Co2(CO)2 breaking into various AsS fragments, the adductAs4S4 ·Cr(CO)5 yielded complexes of low sulfur- and high arsenic content. Brunner orig-inally thought the Cr(CO)5 moiety could ful�l a role of a fragment-stabilising group buthe found out that it acts rather as a sulfur-abstracting reagent.222
Despite the fragmentation process has been shown to occur in vast majority of reac-tions between As4S4 and metal carbonyls, it cannot be taken for granted. Surprisingly,complexes with fully preserved, albeit distorted, As4S4 cage were isolated from the re-action mixtures with the Co and Ru carbonyls mentioned above. In the cobalt complex6, the metal centre is inserted into the transannular As−As bond.221 The structure ofthe ruthenium complex 7 is derived from the geometrical isomer of realgar, As4S4(II),223
65

2. Introduction
RuCp×
S
As
As
S S
As
As
S
realgar(α-As4S4)
realgar As4S4(II)
SAs
As
S S
As
As
S
Co CO
Et
MeMe
MeMe
As
S
S
Cp×Ru
As
S
As
As
As
S As
S
S
As
S
As
S
76
Fig. 2.4. The structures of realgar (α-As4S4), realgar As4S4(II) and their metal complexes 6 and 7
and there are two metal centres inserted into As−As and As−S bonds (Fig. 2.4).219
2.5.3.2. Cyclotetraarsazenes
While there are no reports on other S4N4 analogues with the general formula AsnS4N4-n,more species with sulfur exchanged for arsenic are known.
Tetraarsenic tetranitride, As4N4, is not known as a binary compound, only deriva-tives bearing substituents have been prepared. Begley et al. obtained Ph8As4N4 fromdiphenyltrichloroarsorane and ammonia (eq. 2.61�2.62).224,225
2 Ph3As + AsCl3 3 Ph2AsCl (2.61)
As
Ph
Ph2AsClCl2
Ph2AsCl3CH2Cl2, -78 ºC
CHCl3, 20 ºC
NH3 (g)
CH2Cl2, -78 ºC
NH3 (l)
+ NH4Cl
N N
AsN
As
N
AsPh
Ph Ph
Ph
Ph Ph
Ph
+
(2.62)
The As4N4 ring in Ph8As4N4 is puckered and the molecule has an approximate S 4 symme-try. The As−N bonds are nearly equal in length and are considerably shorter than As−Nsingle bond (1.87Å).11 The authors expected �some degree of π bonding� on the basis ofvalence considerations, but they did not wish to describe the nature of the As−N bonduntil other As4N4 derivatives could be characterised, especially those bearing electron-withdrawing substituents.225 This was achieved nine years later, when (CF3)8As4N4 was
66

2. Introduction
prepared by thermal condensation of compound 8 (eq. 2.63�2.65).226,227
(CF3)3-nAsCln + 3 LiN(SiMe3)2 (CF3)2AsN(SiMe3)2 + (CF3)As[N(SiMe3)2]2 +1. hexane, -20 ºC
2. r.t., 4 hrs+ 3 LiCl(n = 1, 2)
(2.63)
As NF3C
F3C SiMe3
SiMe3
As N
F3C
F3CSiMe3
Clr.t.
NAs
NAs
SiMe3
SiMe3
Cl
F3CF3C Cl
CF3
CF3
Cl2 (excess),CH2Cl2, -189 ºC
- Me3SiCl 24 hrs
8
(2.64)
reflux, 8 hrs
heptaneN
AsN
As
SiMe3
SiMe3
Cl
F3CF3C Cl
CF3
CF3 + 4 Me3SiCl
As
N
AsN
As
N
AsNF3C
F3C CF3
CF3
CF3
CF3F3CF3C
2
8 (2.65)
The structure of (CF3)8As4N4 was found to be nearly identical to that of Ph8As4N4. Themolecule has also an approximate S 4 symmetry, the N atoms are more deviated fromthe mean ring plane than the As atoms and the As−N bonds are of nearly equal length.However, no explanation of bonding within the As4N4 cage was provided.227
2.5.3.3. Cyclotetraarsazanes
Cyclotetraarsazanes contain an As4N4 ring with As−N single bonds. The �rst reportedexample was the tetramer [As(C6H5N)I]4, which can be prepared in two steps from AsI3and aniline.228
4 I As
HN
HN
AsI3 +Et2O
reflux+ 2 C6H5NH2·HI
NH2
(2.66)
67

2. Introduction
N
As
NAs
N
As
NAs
IPh
I
PhI
Ph
I
Ph
3 I As
HN
HN
+ AsI3Et2O
reflux+ 2 C6H5NH2·HI
(2.67)
The structure of [As(C6H5N)I]4 was unfortunately not determined by X-ray analysis, itstetrameric constitution was suggested on the basis of the results of microanalysis andcryoscopic determination of molecular mass.228 However, the crystal structure of anothercyclotetraarsazane, [Cp*AsNH]4, was successfully determined. The molecule has a C i
symmetry and the As4N4 ring is puckered with a � long chair conformation� (eq. 2.68).The As−N bond lengths correspond to As−N single bonds.229
4 Cp*AsCl2 + 12 NH3NH3 / Et2O
- 40 ºC
HN
AsAs
HN AsNH
As
NH
Cp*
Cp* Cp*
Cp*
+ 8 NH4Cl
(2.68)
Tetraarsahexaazaadamantanes. Another class of compounds containing the As4N4cage with As−N single bonds takes up an adamantane-like cage structure.230 They canbe prepared by the reaction between AsX3 (X = Cl, I) and primary aliphatic231 oraromatic228 amines. Same products are obtained when As(NMe2)3 reacts with aliphaticprimary amines (eq. 2.69).231
4 AsX3 + 18 RNH2
(X = Cl, I)
(R = Me, iPr, nBu)
4 As(NMe2)3 + 6 RNH2
(R = Me, nBu)
As
RNAs
NR
AsNR
RN
RN
NRAs- 12 RNH2·HX
- 12 Me2NH
benzene,elev. temp.
(2.69)
X-ray analysis of the derivative As4(NMe)6 230 con�rmed the adamantane-like structureof the molecule with all As atoms connected by alkylimino bridges. When observingthe As4(NR)4 moiety highlighted in bold in eq. 2.69, it can be seen that unlike cyclote-trarsazanes, tetraarsahexaazaadamantanes are structurally related to S4N4: the atomsof As are situated in vertices of a tetrahedron, which is bisected by a square de�ned byfour �NR groups.
68

2. Introduction
2.5.3.4. Dithiatetrazadiarsocines
More accurately, this section describes 3,7-Dialkyl/diaryl-3H,7H -1λ4,5λ4,2,4,6,8,3,7-di-thiatetrazadiarsocines, but for practical reasons only �dithiatetrazadiarsocines� will beused.
In 1971 Scherer and Wies prepared Me2As2S2N4, the �rst S4N4 derivative with twosulfur atoms substituted by arsenic.232 Later also the Ph, Mes,124 tBu233 and ferrocenyl126
analogues were reported. The general synthetic route towards these AsSN heterocycles isthe reaction between the corresponding alkyl/aryldichloroarsine and bis(trimethylsilyl)-sulfurdiimide124,126,232 eventually dipotassiumsulfurdiimide233,234 (eq. 2.70).
2 RAsCl2 + 2- 4 Me3SiCl
N
As
NS
N
As
NS
RR
SN
NMe3Si
Me3Sir. t.
R = Me, Ph,
2 RAsCl2 + 2 KNSNK - 4 KCl
R = tBu
MeCN, - 30 ºC
Mes, Fc
(2.70)
Me2As2S2N4 is an oil which does not crystallise at low temperature but the structures ofthe remaining compounds were determined. The As2S2N4 cage resembles that of S4N4but there are signi�cant di�erences between the two structures. The As−N−−S−−N−Asfragments are coplanar, unlike the S−N−−S−−N−S fragments in S4N4 (Fig. 2.5).233 Themolecules of the dithiatetrazadiarsocines thus have an approximate C 2v symmetry incomparison with D2d for S4N4. Furthermore, there are no transannular S−S interactionsin R2As2S2N4 and the As−As distances are on the edge of the sum of Van der Waalsradii.235 Finally, the S−N bond lengths in R2As2S2N4 are approximately 0.1Å shorterand the NSN angles 20◦ wider than in the molecule of S4N4.233,236
On the basis of bond lengths analysis Alcock proposed delocalisation of the �N−−S−−N�bonds in R2As2S2N4 (R = Ph, Mes) but his suggestion was not supported by eitherGieren or Spang who prepared the tert-butyl- and ferrocenyl- derivatives, respectively.Indeed, the S−N bond lengths in all dithiatetrazadiarsocines are very similar and Gierenet al. concluded, that the presence of an aromatic versus aliphatic substituent on As haslittle e�ect on bonding within the heterocycle.126,233
69

2. Introduction
N2
S4
S2
N4 N1
S1
S3
N3
90 º
N2
S1
As2
N4 N1
As1
S2
N3
S2
N3 N2
S3 S4
(S1)
(N4) (N1)
90 º
N3
As2
N2
S2 S1
(As1)
(N4) (N1)
R2R1 R2
(R1)
Fig. 2.5. A schematic demonstration of the structural di�erence between S4N4 and dithiatetrazadiar-socines. The atoms and alkyl groups in parentheses occupy positions eclipsed by the atomswithout parentheses.
Reactivity. The two arsenic atoms each have a lone electron pair and dithiatetraza-diarsocines can thus be employed as ligands. Scheme 2.8 on page 71 shows complexesprepared in reactions with some d-metal carbonyls.126,237�240 Monodentate coordinationto the metal centre (compounds 9, 13a and 13b) has little e�ect on the structure of theAs2S2N4 ring. Only slight increase of the As−As distance is observed in the bidentatecomplexes 10a and 10b.237�239 The biggest changes were observed in the chelates 12a�12c, where the As2S2N4 ring is considerably compressed. The As−As distances lie in therange 3.132�3.236Å and the As−N−S angles have mean values around 125◦ (the values inthe free R2As2S2N4 rings lie in the range 3.47�3.68Å and 130◦).124,233,239,241,242 The AsSNeight-membered ring undergoes a cleavage during the reaction with Fe3(CO)12 to formcomplex 11. The course of the reaction was not monitored, nor the byproducts analysedand therefore the mechanism of the loss of one NSN unit from the eight-membered ringremains unknown.238 In any case, the fragmentation of the As2S2N4 ring into smallfragments upon a reaction with iron carbonyls is reminiscent of similar behaviour ofAs4S4 (section 2.5.3.1).
Dithiatetrazadiarsocines are prepared in good yields from alkyl/aryldichloroarsinesaccording to eq. 2.70. It was anticipated that if unsubstituted AsX3 would be usedinstead of RAsCl2, a cage compound 14 would be obtained. However, the reaction gaveproduct 15, with two dithiatetrazadiarsocines connected by a diaminosulfane bridge (eq.
70

2. Introduction
S
N
NS
As
N N
As
S
N
RR
R2As2S2N4
: M = Cr; R = tBu, Ph, Fc: M = Mo; R = tBu, Ph, Fc: M = W; R = tBu
+ M(CO)4 (η 4-C7H
8 )
(CO)4M
(OC)3Os Os(CO)4
(CO)4Os
+ O
s 3(C
O) 1
1(M
eCN
)
NS
As
NN
AsR RM(CO)3(OC)3M
(CO)4M
N
S
As
N N
As
S
N
RR+ Me3NO·2 H2O
+ R
u 3(CO) 12
+ Fe3(CO)12
(OC)3Fe Fe(CO)3
AsAs
N NS
R R
: M = Ru; R = tBu: M = Os; R = tBu
- 2 C
O
- M
eCN
- Me3N- CO2- H2O
: R = tBu
- ( )
+ M
(CO
)5 (thf)
NS
As
N N
As
S
N
RR
M(CO)5
- thf
hν or thermolysis
: M = Cr; R = tBu: M = W; R = tBu
- CO
910a
11
: R = tBu
10b
12a12b12c
13a13b
Scheme 2.8 Dithiatetraazadiarsocines as ligands
71

2. Introduction
2.71).243
4 AsBr3 + 6 KNSNKMeCN/Et2O
- 40 ºC
N
S
As
NN
As
S
N
N
NS
As
N N
As
S
N
NS
N
N
S
As
NN
As
S
N
NS- 12 KBr
- [NSN]
2
14
15 (2.71)
The structures of the eight-membered AsSN rings are very similar to the previouslydescribed ones, i.e. coplanar AsNSNAs units with S−−N double bonds. The As−Asdistance in 15 is, however, shorter and all angles are slightly more acute. The bridgingS−N bonds are slightly shorter than S−N single bonds.243
2.5.3.5. 1,5,2,4,6,8,3,7-dithiatetrazadiarsocanes
Only two members containing an As2S2N4 ring with all bonds being single have been re-ported so far. Sommer and Lauer prepared compounds 16a and 16b in a transaminationreaction between As(NMe2)3 and sulfamide:244
As(NMe2)3 + SO2(NHR)2
RN
S
RNAs
NR
S
NRAs
NR
S
NR
O
O
O
O
O
O
benzene
reflux+ NHMe2
R = H, Me
16a16b
: R = H: R = Me (2.72)
The X-ray structures of 16a and 16b have not been determined, the authors suggestedthe shape of the molecule on the basis of elemental analysis and IR spectra of the prod-ucts.244
2.5.3.6. Other AsSN eight-membered heterocycles
Alcock tried to prepare the trithiatetrazaarsocines RAsS3N4 from RAsCl2 andMe3SiNSN−S−NSNSiMe3 (R = Ph, Mes). Although he detected the intermediate 17
72

2. Introduction
and the desired product 18 (eq. 2.73) by 1H NMR, all he could isolate were the dithi-atetrazadiarsocines R2As2S2N4.124
SN
N
S
S
NSiMe3
NSiMe3
SN
N
S
S
N
NSiMe3
As
R
Cl
N
S
NAsR
N
S
NS
+ RAsCl2- Me3SiCl - Me3SiCl
17 18 (2.73)
No other trithiatetrazaarsocines are known. Also, no reports have been published onthiatetrazatriarsocines, i.e. compounds containing the As3SN4 heterocycle.
2.5.4. Arsenic containing analogues of S2N2
The arsenic-containing derivatives of S2N2 form only two types of compounds with theprincipal structural motives being the rings As2S2 or As2N2. Four-membered rings con-taining all three elements As, S and N have not yet been described in the literature.
2.5.4.1. As2S2
Unlike S2N2, diarsenic disul�de is not known as an isolated molecule. Its structure hasbeen predicted by theoretical calculations, the results of which showed that a butter�yconformation (C 2v) is prefered to the planar one (D2h) (Fig. 2.6). The calculated en-ergetical di�erence was very small though (3.28 kcal/mol), and the authors pointed outthat higher level of theory might reverse the order of stability.245
SAs
SAsS
As
SAs
butterfly (C2v) planar (D2h)
Fig. 2.6. Possible conformations of the As2S2 ring
A planar As2S2 ring was found in a one-dimensional polymeric anion [NiAs4S8] 2n �nsynthesised under hydrothermal conditions from NiCl2 and K3AsS3 in the presence ofPh4PBr. A part of the anion is displayed in Fig. 2.7.246 Arsenic and sulfur atoms forma tetradentate anionic bridging ligand [As4S8] 4 � , which is coordinated on both sides toNi 2+ cations. In the centre of the ligand is the planar ring As2S2, which can be regarded asthe common part of two edge-sharing pyramids AsS3. These edge-sharing pyramids sharevertices with another two AsS3 pyramids, which are in an anti position with respect to
73

2. Introduction
AsS As
S
S
S
As
AsS-
Ni
S-
S
S S-
Ni
S-
S
SAs
SAsS
S
S
As
AsAs
S AsS
S
S
As
As
Fig. 2.7. A part cut out from the polymeric anion [NiAs4S8] 2n �n . The building unit [NiAs4S8] 2 � ishighlighted.
the plane of the As2S2 ring. This structure was suggested to be a result of a condensationof four [AsS3] 3 � anions with sul�dic anions S 2 � being eliminated as byproducts.246,247
The As2S2 ring is planar and nearly of a square shape. The opposite As−S bonds havethe same lengths and the two pairs of bonds di�er only by 0.06Å. All internal angles arevery close to 90◦. The As−S bonds (mean 2.282Å) are slightly elongated in comparisonwith the As−S single bonds observed in realgar (2.243Å).214 The Ni 2+ cation is foundin an approximately square planar coordination environment.246
An As2S2 ring with additional S atoms bound to each arsenic in a syn con�gurationwith respect to the ring plane has also been reported, e.g. in the salt PbTlAs3S6. Thering is planar and its shape inclines more towards a rhombus than square, since theinternal angles di�er slightly more from 90◦. The mean value of the pairs of oppositeAs−S bond lenghts (2.313Å) is even longer than in case of the [NiAs4S8] 2n �n anion.248
A planar As2S2 ring is also formed by dimerisation of unstable monomeric As−Sspecies. For example abstraction of chloride from 2-chloro-1,3,2-dithiarsolane by a Lewisacid gives a salt 19, the cation of which is stabilised by dimer formation (eq. 2.74).249
SAs
SCl + 2 GaCl3
[GaCl4]-
2CH2Cl2 S
As+S
As+
S'
S'
[GaCl4]-
19 (2.74)
The dication consists of two monomeric [C2H4S2As]+ ions connected by a pair of As−Sbonds to form a central four membered As2S2 ring. The rests of the monomeric cations arein an anti con�guration with respect to the ring and they are in an envelope conformation.The central As2S2 ring is planar and has the shape of a parallelogram with the anglesat As being more acute than those at S and their values di�ering by ± 3◦ from 90◦.249
The As−S bonds connecting the two monomers (2.423Å) are longer than single As−Sbonds in realgar. The As−S bonds, which belong to the monomers and also participate
74

2. Introduction
in the As2S2 ring are shorter (2.326Å) and the As−S′ bonds are very short (2.181Å)suggesting multiple bonding. It was the short As−S′ bond length which prompted theauthors to suggest, that the connection of the two monomers can be explained by meansof mesomeric structures 19a and 19b (Fig. 2.8). As a result, the bonding within theAs2S2 ring was described as �an intermediate between fully σ bonded As environmentand As−S π bonding.� The X-ray structure of the dication showed that dimerisationand thus stabilisation of the σ bonded alternative is preferred rather than π bonding ofthe S−As−S moiety.249
S
As
S S+
As
S+
S+
As
S+S
As
S
19a 19b
Fig. 2.8. Mesomeric structures of bis(1,3,2-dithiarsolylium) dication 19
2.5.4.2. As2N2
Diarsenic dinitride is not known as an isolated molecule. The As2N2 ring is a commonstructural motive observed frequently in arsenic-nitrogen chemistry. In most cases itis formed by dimerisation of iminoarsines, i.e. compounds containing a As−−N doublebond. Iminoarsines are formed as intermediates in reactions of arsenic compounds withprimary amines (eq. 2.75 and 2.76).250�252
AsCl3 + 3 RNH2 As N
Cl
R
- 2 RNH2·HCl
AsRN As NR
Cl
Cl (2.75)
As(NMe2)3 + RNH2 As N
Me2N
R
- 2 Me2NH
AsRN As NR
NMe2
NMe2 (2.76)
The dimerisation is thermodynamically very convenient and has been observed for anumber of compounds bearing both small and bulky, and both electron donating andwithdrawing substituents on As and N. Further evidence for the propensity to dimerisa-tion is, that the only acyclic compound with an As−−N double bond remains compound
75

2. Introduction
20 prepared in 1986 according to eq. 2.77.253
AsCl3 + 3 LiNHMes*Et2O, reflux
- 3 LiCl NH
Mes* AsN
Mes*
NMes* As
NH
Mes*
- Mes*NH2
20 (2.77)
The conformation of the As2N2 ring can be either planar or butter�y. In case of theplanar geometry, the substituents on As take up either syn or anti positions with respectto the plane of the ring. Vetter et al. suggested that the dimers, cyclodiarsazanes, containthe As2N2 ring in the butter�y conformation with the substituents anti with respect tothe ring plane.252 This hypothesis was shown to be incorrect by later investigations, whichcon�rmed, that even cyclodiarsazanes bearing bulky substituents on nitrogen (Mes*) orarsenic (C i
5Pr4H) contain a planar As2N2 ring. The con�guration of the substituents onarsenic is indeed anti.229,254 Butter�y conformation of the four-membered AsN ring wasobserved in cyclodiarsazanes bearing sterically demanding substituents on arsenic (Cp*).However, butter�y conformation implies syn con�guration on As atoms, since then boththe substituents are situated in pseudo-equatorial positions.229
Planar As2N2 ring with syn con�guration of substituents on arsenic was observedin compounds bearing sterically less demanding substituents such as [tBuNAsCl]2 orcompound 21 (eq.2.78).255,256
NMe NH
SiMe3BuLi
NMe N- SiMe3
Li+
AsCl3- LiCl- C4H10 NMe N
SiMe3
AsClCl
- Me3SiCl80 ºC, 4 hrs
NMe NSiMe3
AsCl
NMe
NAs
NAs N
MeCl
Cl
21 (2.78)
In the case of sterically less demanding substituents, the geometry of the As2N2 ring
76

2. Introduction
and the con�guration of the substituents on arsenic is in full agreement with theoreti-cal calculations (PM3), which showed that the syn con�guration is thermodynamicallypreferred in a planar [RAsNH]2 ring.257
2.5.5. Five- and six-membered arsenic-sulfur-nitrogen ringsSurprisingly, only one representative from each type mentioned in the title is known.5-methyl-1,3λ4δ2,2,4,5-dithiadiazarsole, MeAs(S2N2), was �rst prepared in 1972 by Sche-rer and Wies from MeAsCl2 and N,N' -bis(trimethylsilyl)sulfurdiimide (eq. 2.79).258
As
CH3
ClCl Me3Si
NS
N
SiMe3
+
S N
SAs
N
CH3
- 2 Me3SiCl2
(2.79)
The reaction was carried out in the absence of solvents and the product was isolated ina low yield as an orange, volatile and air sensitive oil which did not crystallise at lowtemperatures.
The six-membered heterocycle 2H,5λ4-3-tertbutyl-1,5,2,4,6,3-dithiatriazaarsinine wasreported as a ligand in a chromium complex 22 (eq. 2.80). The complex was formed as aminor product. Nevertheless, neither its characterisation was o�ered nor an informationwhy the ligand should have such a structure.239,242 Thus, the existence of the complexremains in doubt despite the fact that the analogous phosphorus compound was preparedand its X-ray structure determined.259
[(CO)5Cr(tBuAsCl2)]
K2[NSN],MeCN, -40 ºC
NS
As
N N
As
S
N
tButBu
Cr(CO)5
Cr(CO)5
As
tBuN
N
S N
S
H
- KCl+
(40%) (10%)13a 22 (2.80)
2.5.6. AsEN heterocycles (E = Se, Te)2.5.6.1. Eight-membered As4E4 heterocycles (E = Se, Te)
Cyclotetraselenatetrarsocane, As4Se4, is best prepared from the stoichiometric amountsof elements in vacuum at 500 ◦C followed by 2 days annealing at 255 ◦C.260 As4Se4 hasbeen extensively studied. Its molecular structure is isomorphous with As4S4, i.e. thefour in-plane Se atoms and two pairs of As atoms situated above and below the plane andlying in the vertices of an As4 tetrahedron. The As atoms within each pair are connected
77

2. Introduction
by transannular As−As interactions.261 As4Se4 was frequently used as a starting materialfor the preparations of numerous AsSe clusters.262�264 However, the chemistry of As4Se4as a ligand is not well developed. An interesting example is the ionic complex 23, wherethe Mn atom displaces one of the As atoms with the shape of the original As4Se4 cagebeing preserved (eq. 2.81).265,266 It is interesting to note that while As4S4 enters reactionswith metal centres readily, As4Se4 needs a strong support such as reduction with alkalimetals, as was the case during the preparation of 23.266
As4Se4 + KDMF
r. t., 12 hrs SeAs
As
Se Se
As
As
Se
Se-Se1. Mn2(CO)10,
3. Ph4PBr SeAs
As
Se Se
Mn
As
Se
Se-C C
C
O O
O [PPh4]+r. t., 1 hr
2. 100 ºC, 12 hrs
23
(2.81)
The chemistry of As4S4 is the most developed from the heavier stable chalcogens. As4Se4falls short behind and tellurium con�rms the trend with no information on As4Te4 pub-lished so far. No eight-membered AsNE, AsSE or AsSNE heterocycles have been reported(E = Se, Te) and neither were six- and �ve-membered AsEN or AsESN heterocycles(E = Se, Te).
2.5.6.2. Four-membered As2E2 heterocycles (E = Se, Te)
The unsubstituted, electroneutral rings have not been prepared yet. Ab initio calcula-tions predict higher stability of butter�y structures of these rings.245 A few examplescontaining the As2E2 (E = Se, Te) moiety were reported.
As2Se2. As2Se2 was found in complex 24 prepared according to eq. 2.82.267
SeSe
Mn(CO)2Cp'
Cp'(CO)2Mn
+ RAsH2As Se AsSe
Cp'(CO)2Mn
R
R
Mn(CO)2Cp'
CH2Cl2reflux
(R = tBu, Ph, Cy;
- H2
24Cp' = C5H4Me) (2.82)
The reaction involves a rearrangement, in which the organomanganese unit is transferredfrom Se to As. The description of the product is very limited due to low quality X-raydi�raction data. Still, the main features could be observed, i.e. the planarity of the four-
78

2. Introduction
membered ring and an anti arrangement of the substituents on the As centres.267 Theplanarity of the As2Se2 ring is in contradiction to the results of the previously mentionedab initio calculations,245 which indicates that the ring formation may be a result of somestabilisation process such as dimerisation. Slightly puckered As2Se2 ring was observed inBa2As2Se5 synthesised at high temperatures (267 and 697 ◦C).268
As2Te2. The �rst four-membered As2Te2 ring was described recently. The anion[As2Te2] 2 � was prepared from elemental K, As and from As2Te3 in a two-step procedureat room temperature (eq. 2.83).269
As + 3 Kethylenediamine
12 hrs, r. t.K3As
1. + As2Te3,
[K(18-crown-6)]2[As2Te2]2 hrs, r. t.
2. + 18-crown-6,toluene (2.83)
The anion forms a parallelogram, with two uneven As−Te bond lengths (2.69 and 2.81Å)and with the angles 107.2◦ (at As) and 72.8◦ (at Te). The negative charge of the anionis counterballanced by two [K(18-crown-6)]+ cations situated above and below the planeof the anion so that the molecule has the shape of a distorted octahedron.269
DFT calculations performed in the same study showed that the geometry of the anionis strongly in�uenced by electron spin e�ects, with the triplet structure being slightlymore stable and in better agreement with the real structure of the anion than the sin-glet structure, for which a square planar geometry resulted. NICS calculations indicatearomatic character of the anion [As2Te2] 2 � .269
2.6. ConclusionThis lengthy introduction was intended to o�er an overview of selected arsenic analoguesof the famous sulfur-nitrogen compounds S4N4 and S2N2 and the wide range of diversechemical reactions they can enter. Before that it was necessary to review the preparationmethods of basic arsenicals as suitable starting materials. It became clear that the areaof small size arsenic-sulfur-nitrogen ring compounds has not been properly investigated,which set the target of the author's research and became the subject of the followingchapter.
79

3. Results and Discussion
Scherer and Wies prepared the �rst �ve-membered As−S−N ring, MeAs(S2N2) (25),from bis(trimethylsilyl)sulfur diimide (26) and MeAsCl2 according to eq. 2.79 (page77).258 25 was formed in a poor yield as an air-sensitive, extremely volatile liquid andthe authors characterised it by 1H NMR, MS, IR and UV spectroscopies. The fact that25 has remained the only prepared 1,3λ4δ2,2,4,5-dithiadiazarsole for nearly 40 years issurprising.
The aims. The primary aim was to repeat Scherer and Wies' synthesis of 25 and totry to prepare more analogues by a rational route with the hope, that some of themmight be crystalline substances. A low temperature single crystal X-ray analysis of theserings was the desired target. Little is known about the reactivity of 25 and therefore thereactivity of the novel heterocycles RAs(S2N2) was also to be investigated.
Strategy. The original route brings several complications: First, 26 is not a stoichio-metric source of the (S2N2) 2 � moiety. The formation of (S2N2) 2 � thus appears as aresult of a complex rearrangement within the reaction system, which is not understood.To date, 25 remains the only reported example of a �ve-membered heterocycle contain-ing the S2N2 moiety being prepared from 26 suggesting that this method may not bethe most suitable one. Second, MeAsCl2 is nowadays not commercially available. Theeasiest synthetic route leading to MeAsCl2 is the reduction of methylarsonic acid, whichis not commercially available either and must be prepared by Meyer reaction from As2O3and iodomethane (section 2.4.2, page 40). The preparation of MeAsCl2 is not di�cultbut it requires isolation and puri�cation of the acid. The preparation of MeAsI2 by thesame method is faster, as it is a one-pot-synthesis starting from As2O3 and iodomethaneand �nishing with MeAsI2.90
For these two reasons it was desirable to �nd a method which would o�er higheryields of the products and which could utilise RAsX2 other than chlorides. The lig-and exchange reactions between an alkyl- or aryldihalogenoarsine and the dimer of5,5-dibutyl-1,3λ4δ2,2,4,5-dithiadiazastannole, [nBu2Sn(S2N2)]2 (27), was the most con-venient option (eq. 3.1). 27 can be readily prepared on a medium-to-large scale and itwas previously successfully used as the source of the (S2N2) 2 � ligand in reactions with
80
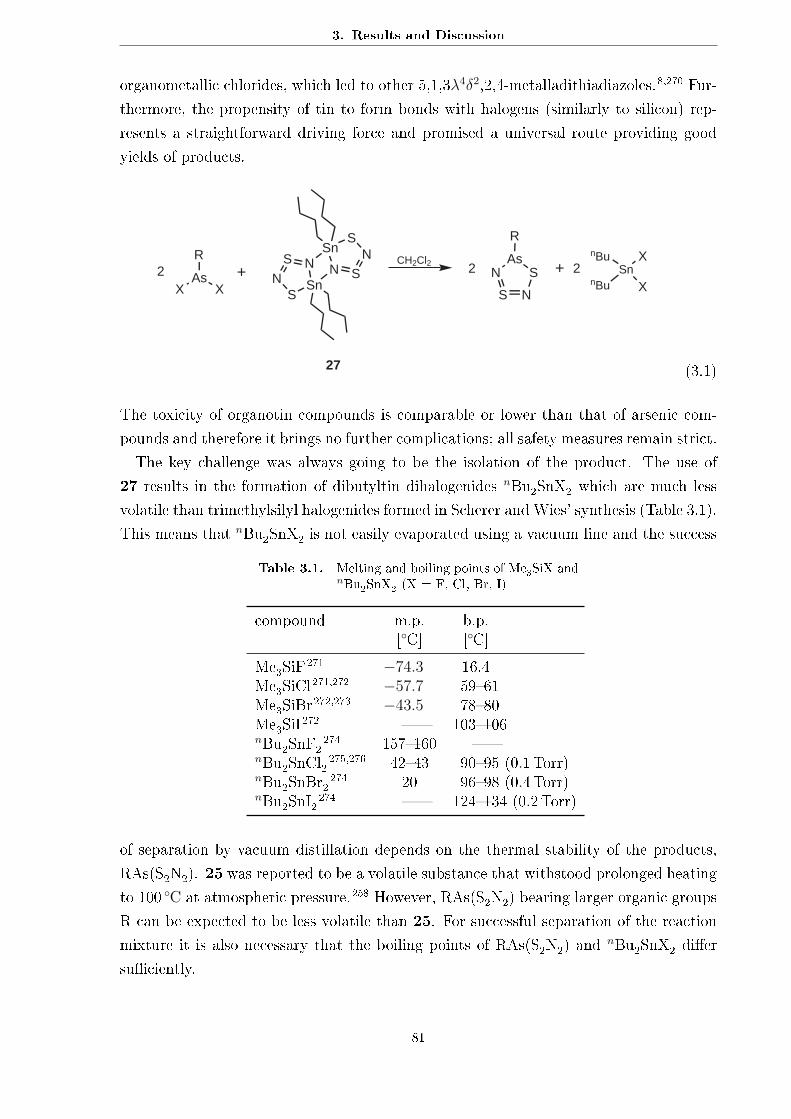
3. Results and Discussion
organometallic chlorides, which led to other 5,1,3λ4δ2,2,4-metalladithiadiazoles.8,270 Fur-thermore, the propensity of tin to form bonds with halogens (similarly to silicon) rep-resents a straightforward driving force and promised a universal route providing goodyields of products.
+CH2Cl2 2 2
X
XSn
nBu
nBu+
R
AsXX
2
S N
SAs
N
R
SnS
N
S NSn
SN
SN
27 (3.1)
The toxicity of organotin compounds is comparable or lower than that of arsenic com-pounds and therefore it brings no further complications; all safety measures remain strict.
The key challenge was always going to be the isolation of the product. The use of27 results in the formation of dibutyltin dihalogenides nBu2SnX2 which are much lessvolatile than trimethylsilyl halogenides formed in Scherer and Wies' synthesis (Table 3.1).This means that nBu2SnX2 is not easily evaporated using a vacuum line and the success
Table 3.1. Melting and boiling points of Me3SiX andnBu2SnX2 (X = F, Cl, Br, I)
compound m.p. b.p.[◦C] [◦C]
Me3SiF271 −74.3 16.4Me3SiCl271,272 −57.7 59�61Me3SiBr272,273 −43.5 78�80Me3SiI272 � 103�106nBu2SnF2
274 157�160 �nBu2SnCl2 275,276 42�43 90�95 (0.1Torr)nBu2SnBr2 274 20 96�98 (0.4Torr)nBu2SnI2 274 � 124�134 (0.2Torr)
of separation by vacuum distillation depends on the thermal stability of the products,RAs(S2N2). 25 was reported to be a volatile substance that withstood prolonged heatingto 100 ◦C at atmospheric pressure.258 However, RAs(S2N2) bearing larger organic groupsR can be expected to be less volatile than 25. For successful separation of the reactionmixture it is also necessary that the boiling points of RAs(S2N2) and nBu2SnX2 di�ersu�ciently.
81

3. Results and Discussion
The chemical stability of RAs(S2N2) would determine whether they may be puri�edby column chromatography (on silica, alumina or Bio-Beads) or not. Unfortunately thereported air-sensitivity of 25 suggested that other RAs(S2N2) would also be air-sensitive.
3.1. Comments on preparative procedures
3.1.1. Alkyl/aryldihalogenoarsinesFor practical reasons (section 2.4.5), the most convenient starting materials are theorganodichloroarsines. The exceptions were MeAsI2 and EtAsI2, the preparation of whichis faster than that of the corresponding dichlorides.
All organodihalogenoarsines RAsX2 (R = Me, Et, iPr, tBu, Ph, Mes; X = Cl, I) wereprepared according to published procedures. MeAsI2 90 was prepared in only 16% yield,which was unexpected. The instructions were followed and nothing was observed whichwould indicate an unusual course of the reaction. The instructions, however, contain afew inaccuracies. First, the reported yield (78%) is incorrect, 39% is the correct value.And second, MeAsI2 separates as a dense oil at the bottom of the reaction �ask; if theoil is not present, the authors suggest to heat the mixture to 50 ◦C. This suggestion is,in fact, quite necessary. Before the isolation of the product, the mixture needs to be�ltered and the oil separated in a separating funnel. Thus, the mixture must be liquidduring the work-up. MeAsI2 melts at 30 ◦C and if the mixture is not su�ciently warm,the product will crystallise spontaneously causing unnecessary delays in work.EtAsI2 121 was prepared according to the instructions published in 1920 by McKenzie
and Wood (who worked at the universities of Dundee and St Andrews). Since their workconcentrated mainly on the preparation of EtAsCl2, it was necessary to combine theirinstructions with those for the preparation of MeAsI2.90 The general synthesis of RAsI2by Meyer's reaction followed by the reduction of the alkylarsonate proceeds according toeq. 3.2�3.4.
As2O3 + 6 NaOH 2 Na3AsO3 + 3 H2O (3.2)
Na3AsO3 + RI + NaI
O
AsR
ONaONa
EtOH
(3.3)
+ 2 HCl + 2 NaI + SO2 + 2 NaCl +
O
AsR
ONaONa
R
AsII
+ Na2SO4 + H2O (3.4)
In reaction 3.3 1mol of NaI is formed, whereas to a full conversion of an alkylarsonate
82

3. Results and Discussion
to RAsI2 2mol of NaI are needed (eq. 3.4). Thus, when performing a one-pot-synthesis,additional NaI must be added during the reduction of the alkylarsonate, so that the molarratios in reaction 3.4 are correct. This has been complied with during the preparationof MeAsI2 but not during the preparation of EtAsI2. McKenzie and Wood added conc.HCl instead, which, however, must have led to the formation of ethylchloroiodoarsine,EtAsClI, rather than EtAsI2 (eq. 3.5).
+ 3 HCl + NaI + SO2 + 2 NaCl +
O
AsR
ONaONa
R
AsICl
+ NaHSO4 + H2O (3.5)
For the purpose of their work this did not matter, as they did not need to obtain apure substance after the reduction of sodium ethylarsonate. They needed any ethyl-dihalogenoarsine, which they subsequently converted into pure EtAsCl2 (eq. 3.6 and3.7).
R
AsII
2 + 4 NaOH + 4 NaI + 2 H2OR AsO
OAs R
(3.6)
R
AsClCl
2+ 4 HCl(conc.) + 2 H2OR AsO
OAs R
HCl(g)
(3.7)
In this work, pure EtAsI2 had to be prepared for the purpose of analysis and there-fore both conc. HCl and aqueous NaI were added. The reduction with SO2 proceededsmoothly but the mixture soon became oversaturated and a white solid started to precip-itate with every portion of HCl and NaI added. Such a precipitation was not reported,presumably because McKenzie and Wood worked with a more diluted mixture. Sincethe amount of the precipitate was quite signi�cant, the addition of HCl was stoppedafter 10ml were added and only the addition of NaI was continued. Without any furthercomplications EtAsI2 was isolated in 30% yield (reported was 47%).
It is important to note that both alkyldiiodoarsines cannot be stored for unlimitedtime periods. After 6 months they start to darken, even if they are stored under nitrogenin the dark. A comparison of 1H NMR spectra of freshly prepared and the darkenedproducts suggests slow decomposition of the diiodoarsines.
iPrAsCl2 and tBuAsCl2 were prepared in diethyl ether from AsCl3 and the cor-responding Grignard reagent RMgCl. So far, this preparation of iPrAsCl2 has beendescribed only very brie�y in a patent.122 However, the route described for tBuAsCl2 123
is applicable also for iPrAsCl2. Both preparations were repeated several times to �nd
83

3. Results and Discussion
the optimum reaction conditions giving the best yields.To enhance the formation of monoalkyldichloroarsine, the Grignard reagent must be
added to the solution of AsCl3, the speed of the addition should be rather medium-slow,the concentration of AsCl3 should be higher than the concentration of the Grignardreagent and the mixture must be stirred vigorously. The mixture does not need to becooled to -78 ◦C, good yields were obtained when the reactants were mixed at -40 ◦C. Theconcentrations of the reactants are important. The mixture must be suitably diluted,otherwise it soon becomes a thick slurry of the precipitated MgCl2 in ether and magneticstirring is not possible. The solutions of the reactants should not be more than 1.5m. Astraightforward procedure with good yields was observed when 0.5m Grignard reagentwas added to 1.0m AsCl3 in diethyl ether.
After the reaction is �nished the mixture needs to be worked up and maintained cold.The �ltration (-20 ◦C) must be done through a celite plug, otherwise the sinter becomesclogged with MgCl2 particles. It is better to collect the �ltrate into a �ask immersedin a cooling bath (below -20 ◦C). If allowed to heat up to around 0 ◦C, crude tBuAsCl2deteriorates very quickly, which is apparent by formation of a sand-coloured precipitate,which quickly reappears after �ltration. iPrAsCl2 does not deteriorate so dramaticallybut a concentrated solution also starts to darken at room temperature.
The crude products should be puri�ed as soon as possible. iPrAsCl2 was obtained asa colourless oily liquid by distillation at atmospheric pressure. During the distillation,three fractions were collected at three distinct oil bath temperatures. The highest boilingfraction was collected with the oil bath temperature on 204 ◦C, a value expected forpure iPrAsCl2 boiling at 169 ◦C.136 Such procedure was believed to su�ce given thatthe di�erence between the boiling points of AsCl3 and iPrAsCl2 is nearly 40 ◦C.87,136
Nevertheless, Raman spectroscopy revealed the presence of AsCl3 in the distillate withthe lines being of lesser intensity than those of iPrAsCl2. Since the total amount of thedistillate was only 2.80ml, further distillation was not attempted and the mixture wasused for further synthesis.
According to the original instructions, tBuAsCl2 should be distilled at 50�54 ◦C un-der reduced pressure (11Torr).123 The attempted distillation of the product under highvacuum (0.3Torr, toil = 40�45 ◦C) resulted in sublimation of the product and formationof well shaped colourless crystals in the condenser. Gentle heating of the condenser didnot cause melting of the crystals, it moved the condensation spot more towards a coldtrap connected between the distillation apparatus and the vacuum line. At the end ofthe distillation only negligible amount of the product collected in the distillate-�ask andmajority of the product condensed in the cold trap. The content of the cold trap (i.e. thepreviously evaporated solvent, volatile impurities and tBuAsCl2) was therefore allowed tomelt under nitrogen and the solution was transferred to a predried 250 ml Schlenk �ask.
84

3. Results and Discussion
tBuAsCl2 was eventually isolated from this solution as colourless crystalline solid. Itsmelting point was found to lie in between 42 and 45 ◦C but these are the temperatures atwhich the crystals started and �nished melting rapidly. The �rst wetting of the crystalsstarted as soon as the substance reached room temperature after being removed from afridge. tBuAsCl2 seems to exist in a transition phase between a solid and a liquid, whichwas re�ected predominantly in the absence of di�raction when crystals were submittedfor X-ray analysis.
In spite of the complications during its isolation, the purity of tBuAsCl2 was good.Raman spectroscopy proved the presence of small amounts of AsCl3 but the intensitiesof its bands were weaker than those of tBuAsCl2. 1H and 13C NMR showed only signalsof the product.
The propensity of tBuAsCl2 to sublime was surprising and it shows evidence of highvolatility of the substance. Also, tBuAsCl2 has the most penetrating odour of all thehalogenoarsines mentioned in this thesis. Whether it is a special property of tBuAsCl2or if it is a consequence of the volatility of the compound cannot be stated.PhAsCl2 was obtained by reduction of phenylarsonic acid in conc. HCl without any
problems.117
MesAsCl2 was prepared by the reaction of AsCl3 and MesLi in molar ratio 1:1. MesLiwas prepared by lithium-halogen exchange from MesBr and nBuLi. MesLi precipitatedas a white solid, the mixture was evaporated to dryness and the solid was kept under highvacuum for 1 hour to remove volatile impurities. However, this was not enough to removeall bromine-containing byproducts, as emerged after the reaction of MesLi and AsCl3.The crude product was distilled under high vacuum and the highest boiling fraction wasanalysed by NMR and MS spectroscopies. Both 1H and 13C NMR and MS revealed thatthe product consisted of a mixture of MesAsCl2, MesAsClBr and MesAsBr2. To savetime, the mixture was not converted to pure MesAsCl2, it was used for further synthesesas it was. Since it was impossible to determine the ratio of particular components, thestoichiometric calculations assumed the mixture contained only the lighter MesAsCl2.
The synthesis of 1,4-bis(dichloroarsino)benzene (28) by reduction of p-phenylene-diarsonic acid (29) was described by Goldsworthy et al.277 The attempted preparationof 29 by the Bart reaction failed.
The failure of Bart's method was caused by use of the wrong reaction conditions. In1922, after the publication of his results became possible, Bart wrote a crucial 50 pageslong report in which he described in detail the reaction conditions for the preparations ofarsonic acids.100 Unfortunately, this article was not known to the author of this thesis atthe time when the preparation of 29 was performed. Thus, the Bart reaction was carriedout according to the instructions of Lieb published in 1921.278 Obviously, Lieb could notdraw from Bart's experience and the reaction conditions he chose were unsuitable for the
85

3. Results and Discussion
As
As
O
OOH
OH
OHOHAs
AsClCl
ClClNH2
AsO
ONaOH
302928
Fig. 3.1. 1,4-bis(dichloroarsino)benzene (28), p-phenylenediarsonic acid(29) and Atoxyl (sodium hydrogen-4-aminophenylarsonate, 30)
formation of 29.Lieb diazotised atoxyl (30) and instead of using Na3AsO3, he allowed to couple the
diazonium salt with an arsenite bu�er consisting of Na2HAsO3 and NaH2AsO3. Headded the hydrogenarsenites into �signi�cantly acidic� solution of the diazonium salt,after which he observed a �tempestuous nitrogen evolution�. Lieb eventually obtained 29in 11% yield.278
One year later in his publication, Bart commented directly on Lieb's unsuccess sayingthat Lieb's conditions were far too acidic for the formation of 29, which requires ratherbasic pH during the coupling. Especially in case of 29 it was recommended to add thesolution of the diazonium salt to the alkaline solution of Na3AsO3 and keep the overallpH basic by adding hydroxide when necessary.100 Bart reported 27% yield of 29.
In this work, the preparation of 29 under Lieb's conditions was repeated but only verymild nitrogen evolution was observed indicating that only small amount of the diazoniumsalt was converted into 29. The rest recombined to form unidenti�ed byproducts ofred/brown colour. The mixture was worked up as is described in the experimental sectionand at the end a fair bulk of a cream-coloured solid dispersed in a dark red/brown mudwas obtained, from which it was not possible to obtain 29 in a pure form.
A part of the crude product was boiled with decolourising charcoal in water (as advisedby Lieb) but this caused only a deterioration. The mixture darkened signi�cantly, thecream-coloured solid was lost and after the mixture was �ltered and water evaporated,no signals in the aromatic region were detected by 1H NMR.
Another part of the crude product was treated with more care. The mixture wasdiluted with a small amount of ethanol and the cream-coloured solid was �ltered o� bysuction (sinter). Only a small amount of the solid remained on the sinter and it quicklystarted turning dark red. However, a 1H NMR spectrum of the solid was recorded and29 could be detected (singlet at 7.95 ppm).
Since it was obvious that the yield of 29 would be very low and since it was nearlyimpossible to isolate it as a pure solid substance, the decision was made to test the
86

3. Results and Discussion
possibility to reduce the crude product with gaseous SO2 in the presence of HCl and totry to isolate 28. This attempt was unsuccessful since the reduction with SO2 yieldedonly As2O3 as was shown by mass spectrommetry (EI).
It is noteworthy, that following the course of the reaction by 1H NMR was of littleassistance, as 29 is insoluble in common organic solvents and water. Satisfactory spectrawere recorded in DMSO-d6 using the small quantities of partially puri�ed 29.
3.1.2. 5-alkyl/aryl-1,3λ4δ2,2,4,5-dithiadiazarsolesThe chosen method, i.e. the reaction between RAsX2 and 27, was successful and a seriesof 5-alkyl/aryl-1,3λ4δ2,2,4,5-dithiadiazarsoles were obtained in good yields. All reactionswere carried out in CH2Cl2 because of its convenient properties when working under mildreaction conditions.
In general, the reactants were mixed as their CH2Cl2 solutions in 2:1 (RAsX2 : 27)stoichiometric ratio, RAsX2 being added to 27 (eq. 3.1, page 81). A colour changefrom lemon yellow to orange or red was observed shortly after the �rst drops of RAsX2were added and the colour deepened as more RAsX2 was added. The reactions werenot exothermic and proceeded smoothly at room temperature. The end of a reactionwas indicated visually (no further colour change) with 1H NMR spectrum providing thecon�rmation. The �nal reaction mixture was usually a clear orange solution, though inthe case of the preparation of 25 and especially 32 an o�-white solid precipitated, whichwas later �ltered o� and was not analysed.
Pure MeAs(S2N2) (25) was obtained by vacuum distillation. Its boiling point(toil = 90 ◦C at 0.3Torr) is su�ciently lower than that of nBu2SnI2 (Table 3.1, page81) and thus its separation was perfect.EtAs(S2N2) (31) was also puri�ed by vacuum distillation. It distilled at toil = 120 ◦C
(0.3Torr), which is much closer to the boiling point of nBu2SnI2 and as a result thedistillate was contaminated with nBu2SnI2. In contrast to 25, 31 did not show signs ofdecomposition during a TLC test. After silica column chromatography (hexane followedby toluene) pure 31 was obtained as an orange oil which did not crystallise upon cooling.Despite su�ering no damage during the column chromatography, an exposure to air foran hour resulted in deterioration of 31 and therefore it had to be stored under nitrogen.
With the boiling points of RAs(S2N2) increasing when going from R = Me to theheavier rests R and with the boiling point of the byproducts dereasing when goingfrom nBu2SnI2 to nBu2SnCl2, there was a high probability that iPrAs(S2N2) (32),tBuAs(S2N2) (33) and nBu2SnCl2 would have similar boiling points, which wouldmake the reaction mixtures inseparable by vacuum distillation. The distillation of 33was attempted but the compound decomposed before its vapours reached the condenser,
87

3. Results and Discussion
forming a pale yellow solid on the walls of the microdistillation kit.TLC tests showed on one hand that 32 and 33 did not decompose on silica but on the
other no separation from nBu2SnCl2 could be observed. The insu�cient separation wasmost probably due to the larger alkyl chains on arsenic in 32 and 33, which decreasedthe overall polarity of the molecules to the level of nBu2SnCl2. As a result, columnchromatography would have to be carried out at least twice, which was to be avoidedbecause of the questionnable stability of 32 and 33. An attempted puri�cation of 33by a silica column chromatography con�rmed the presumption. First run showed that33 is less polar than nBu2SnCl2. 33 was eluted �rst and although it formed an isolatedcoloured band in the column, it was contaminated with nBu2SnCl2. A second run wasrequired, during which 33 decomposed (its 1H NMR signal could not be seen in any ofthe collected fractions). Similar results could be expected for compound 32 and thereforeits puri�cation by either vacuum distillation or silica column chromatography was notattempted.
The most successful isolation method turned out to be size-exclusion chromatographyusing Bio-Beads. The bulky iPr or tBu groups enabled satisfactory separation of theproducts from nBu2SnCl2, which contains the sterically much less demanding nBu chains.Generally, nBu2SnCl2 was eluted �rst followed by the coloured wide band of the product.Since the product was contaminated with residual nBu2SnCl2, it had to be passed twicemore through the column. During the �nal elution the coloured band was divided in twoparts, which were collected separately. While the �rst part (approx. 1
3of the length of the
band) was still contaminated with nBu2SnCl2, the other was pure. Attempts to isolatemore product from the fractions contaminated with nBu2SnCl2 were not successful. Theeluates from the repeated runs remained contaminated and from the 1H NMR spectra itcould be seen that the amount of the product decreased with every run.It is important to note that Bio-Beads were soaked in CH2Cl2 (not dry) that has greaterdensity than Bio-Beads. To ensure a good contact of the beads with the solvent, theslurry needs to be stirred frequently. If left standing in CH2Cl2, Bio-Beads �oat andswell only locally. To prevent �oating of the packed column a glass wool plug was puton top.
Both 32 and 33 are very volatile substances with a characteristic old-sweat odour.Similarly to tBuAsCl2, the odour of 33 is particularly pungent.
The preparations of PhAs(S2N2) (34) and MesAs(S2N2) (35) were probably theeasiest of all. The aromatic organic substituents signi�cantly stabilise the molecules, asa result of which they could be puri�ed with minimum losses in yields. 35 was preparedwithout complications from the mixture of halogenoarsines, MesAsXX′ (X = Cl, Br; X′
= Cl, Br). The stoichiometric amounts of the reactants were calculated for X = X′ = Cland an excess of 27 was used.
88

3. Results and Discussion
In general, the resulting reaction mixture was evaporated to dryness and the residuewas distilled under high vacuum. The orange/red distillate crystallised spontaneouslyin the distillation �ask. If, during the distillation, the distillate solidi�ed already inthe condenser, the condenser was heated gently with a heating gun to enforce meltingand the distillation was resumed. An inspection under a microscope revealed that thesolid distillate consisted of colourless needles contaminated with an orange/red oil and1H NMR con�rmed that the crystals were nBu2SnCl2. The oil consisted probably of amixture of 34 and nBu2SnCl2, because an additional amount of 34 could be isolatedfrom the whole distillate bulk. However, the amount was only 10mg, which made it notworth the extra time spent.
The distillation residue was an orange/brown solid (in case of 34) and a red/browntarry matter (in case of 35). A TLC suggested both substances to be stable on silicaand good separation from residual nBu2SnCl2 was shown. Silica column chromatography(mixture petroleum ether�toluene) removed the residual impurities and pure productswere eluted. After evaporation of solvents, 34 was obtained as a yellow oil, 35 was ared oil, both were stored under nitrogen. Unlike the alkyldithiadiazarsoles, both thearyldithiadiazarsoles eventually solidi�ed at room temperature into non-glassy solids.34 solidi�ed in a freezer overnight and could be recrystallised as follows: the solid was
heated in a �ask under the hot water tap and when the substance melted, the �ask wasswirled smoothly at room temperature. The melt was allowed to roll over the inner wallsof the �ask and to cool down gradually leaving well shaped yellow prisms of the producton the walls. This procedure was repeated several times to obtain crystals with a bettershape.
After 35 had not solidi�ed in a freezer overnight, it was placed in a fume cupboard,where it eventually solidi�ed after two days. However, its recrystallisation by the methoddescribed for 34 was less successful. After the substance was melted and once it solidi�edtwo days later, the crystals were of the same or worse shape than before the recrystalli-sation. Attempts to recrystallise 35 by a gas phase di�usion of hexane into a CH2Cl2solution of 35 failed and as a result the best crystals chosen for single crystal X-rayanalysis were still of only average quality.
3.2. NMR spectroscopy
3.2.1. 1H and 13C NMR dataIn both 1H and 13C NMR spectra, RAs(S2N2) gave signals shifted from those of thestarting materials RAsX2 and these two substances could be therefore easily distinguishedwithin the reaction mixture. The identi�cation of 31 and especially 32 was more di�cult,
89

3. Results and Discussion
since their signals overlapped with the much more intense ones of nBu2SnCl2.
Table 3.2. 1H NMR data of RAsX2a
RAsX2 δ multi- assignment[ppm] plet
MeAsI2 3.11 s 3H, CH3
EtAsI2 1.42 t 3H, CH32.81 qrt 2H, CH2
iPrAsCl2 1.38 d 6H, 2×CH32.46 sept. 1H, CH
tBuAsCl2 1.32 s 9H, tBuPhAsCl2 7.40�8.00 m 5H, Ph-groupMesAsCl2 124 2.30 s 3H, para-CH3
2.70 s 6H, 2×ortho-CH36.93 s 2H, 2×meta-Harom.
a All spectra measured in CDCl3 (270.2MHz,298K).
A molecule of RAsX2 has a plane of symmetry and therefore the alkyl- and aryldihalo-genoarsines produced simple spectra (Table 3.2). The 1H NMR data of MesAsCl2 givenin the table are those of a pure substance prepared by Alcock et al.124 The 1H NMRspectrum of the mixture of MesAsXX′ (X = Cl, Br; X′ = Cl, Br) corresponded well withthat published for MesAsCl2 with the chemical shifts di�ering only slightly. However, acloser look at the peaks revealed shoulders indicating an overlap of signals. Much betterproof of the presence of a mixture of compounds came from the 13C NMR spectrum ofthe MesAsXX′ mixture, where the chemical shifts of the signals di�ered more markedly(see the Experimental section).
The introduction of the (S2N2) 2 � moiety brings chirality to the molecules ofRAs(S2N2), which could be observed in the 1H NMR spectra. The 1H and 13C NMRspectra of 25 and 33 measured at 298K showed simple patterns as a result of chemicalequivalency of the 3 protons in the Me group and all 9 protons and 3 carbons in the tBugroup.
The 1H NMR spectrum of 31 showed an ABX3 spin system, which consists of a doubletof doublet (CH3 group) and a more deshielded complex multiplet (the two inequivalentprotons in the CH2 group). The spectrum was successfully simulated to determine theexact experimental values of the chemical shifts and coupling constants. The recordedand simulated spectrum are shown on Fig. 3.2, data are listed in Table 3.3. 32 gave aspectrum containing two overlapping doublets (the two inequivalent CH3 groups) and amultiplet (the sole CH proton), which on the �rst glance looked like a septet. However,
90

3. Results and Discussion
Table 3.3. 1H NMR data of RAs(S2N2) a
RAs(S2N2) δ multi- assignment notes[ppm] plet
MeAs(S2N2) (25) 1.17 s 3H, CH3
EtAs(S2N2) (31) 0.99 dd 3HX in CH32JHA−HB = 13.42Hz
1.36 m HA in CH23JHA−HX = 7.59Hz
1.52 m HB in CH23JHB−HX = 7.78Hz
iPrAs(S2N2) (32) 0.942 d 3H, CH30.960 d 3H, CH31.62 qqrt 1H, CH
tBuAs(S2N2) (33) 0.91 s 9H, tBuPhAs(S2N2) (34) 7.27�7.41 m 5H, phenylMesAs(S2N2) (35) b 2.25 s 3H, para-CH3
2.45 s 6H, 2×ortho-CH36.82 s 2H, 2×meta-Harom.
a All spectra measured in CDCl3 (270.2MHz, 298K). Chemical shifts are calibrated to the peakof the residual CHCl3 (7.26 ppm).279
b 499.9MHz spectrometer (CDCl3, 298K).
Fig. 3.2. Simulated (A) and recorded (B) 1H NMR spectrum of 31(270.2MHz, CDCl3, 298K)
91

3. Results and Discussion
integration analysis proved that the multiplet is a quartet of quartet formed by couplingof the CH signal by two chemically inequivalent CH3 groups.34 produced 1H NMR spectrum with a characteristic multiplet in the aromatic region.In the 1H NMR spectrum of 35, the presence of a chiral centre did not result in
anisochronicity of the diastereotopic ortho-Me groups of the mesityl moiety. The spec-trum was recorded at both 270.2 and 499.9MHz but all the proton signals appeared asisochronic. Similarly, no anisochronicity was observed in the 13C NMR of 35.
Table 3.4. 13C NMR data of RAsX2a
RAsX2 δ multi- assignment[ppm] plet
MeAsI2 20.6 s 1C, CH3
EtAsI2 15.1 s 1C, CH328.3 s 1C, CH2
iPrAsCl2 16.5 s 2C, 2×CH342.5 s 1C, CH
tBuAsCl2 24.0 s 3C, 3×CH344.7 s 1C, Cquart.
PhAsCl2 129.4 s 2C, 2×meta-C b
130.1 s 2C, 2×ortho-C b
132.3 s 1C, para-C b
145.3 s 1C, C−As b
a All spectra measured in CDCl3 (67.9MHz, 298K).b Assignments according to Bodner et al.280
The 13C NMR spectra showed only the expected simple patterns. The 13C NMR dataare listed in Table 3.4 and Table 3.5. The 13C NMR chemical shifts of PhAsCl2 come fromthe experimental work presented in this thesis, their assignment is based on literaturedata.280 Further details of the NMR experiments are given in the Experimental section.
3.2.2. 14N NMR dataAll dithiadiazarsoles gave 14N NMR spectra with two well de�ned peaks. In Table 3.6it can be seen that the di�erence between the chemical shifts of the two peaks decreaseswith the decreasing size of the organic group. This suggests that in molecules withsterically less demanding alkyl group on arsenic the two nitrogen atoms are chemicallymore equivalent than in the molecules with bulky groups. Whether this is a consequenceof a delocalisation of π-electrons within the As(S2N2) ring cannot be stated at present.
A suggestion about the assignment of the 14N NMR peaks is usually obtained from the-oretical calculations, although the exact values of the theoretical chemical shifts are not
92

3. Results and Discussion
Table 3.5. 13C NMR data of RAs(S2N2) a
RAs(S2N2) δ multi- assignment[ppm] plet
MeAs(S2N2) (25) 20.6 s 1C, CH3
EtAs(S2N2) (31) 15.1 s 1C, CH328.3 s 1C, CH2
iPrAs(S2N2) (32) 16.5 s 2C, 2×CH342.5 s 1C, CH
tBuAs(S2N2) (33) 24.0 s 3C, 3×CH344.7 s 1C, Cquart.
PhAs(S2N2) (34) 129.4 s 2C, 2×meta-Carom.130.1 s 2C, 2×ortho-Carom.132.3 s 1C, para-Carom.145.3 s 1C, C−As
MesAs(S2N2) (35) 21.0 s 1C, para-CH321.5 s 2C, 2×ortho-CH3
130.3 s 2C, 2×meta-Carom.140.5 s 1C, C−As140.8 s 1C, para-Carom.141.7 s 2C, 2×ortho-Carom.
a All spectra measured in CDCl3 (67.9MHz, 298K).
Table 3.6. 14N NMR data of RAs(S2N2) a
RAs(S2N2) δ assignment[ppm]
MeAs(S2N2) (25) 274.2 SNS301.4 As−N
EtAs(S2N2) (31) 271.6 SNS303.3 As−N
iPrAs(S2N2) (32) 272.3 SNS303.4 As−N
tBuAs(S2N2) (33) 272.6 SNS303.7 As−N
PhAs(S2N2) (34) 269.2 SNS304.8 As−N
MesAs(S2N2) (35) 265.9 SNS309.8 As−N
a All spectra measured in CDCl3 (28.9MHz, 298K).
93

3. Results and Discussion
always in a good agreement with the experimental ones.40,281 A detailed theoretical studydescribing �ve-membered arsenic-sulfur-nitrogen heterocycles has not been published yetand is currently in progress.
Meanwhile, a decision which set of compounds to choose for a comparison is not aneasy one. In (S2N2)CO and S3N2O the lower chemical shift was attributed to the nitrogenbound directly to the keto or sulfoxo group respectively,40,281 while in the organometalliccomplexes CpCo(S2N2) and Cp*M(S2N2) (M = Co, Rh, Ir) the trend was the opposite(Part III of this thesis). Arsenic, being a main group element, is often referred to asa metalloid as a re�ection of its properties between non-metals and metals.78 For thisreason the assignment suggested in Table 3.6 is based on the trend observed for theorganometallic complexes, i.e. the lower chemical shift is attributed to the nitrogen boundto the two sulfur atoms whereas the higher chemical shift is attributed to the arsenic-bound nitrogen.
3.3. Mass spectrometryElectrospray ionisation turned out to be too mild and produced spectra containing oneor two peaks, which could not be assigned either to any possible product of reactionor solvolysis, or to any fragment or product of recombination. Without any observablefragmentation paths, these spectra were of no use. The only peaks, which could beassigned were those of I � and [I3] � anions observed in the ES � spectra of MeAsI2 andEtAsI2.
On the contrary, electron impact provided high quality spectra, in which molecularpeaks could be identi�ed and fragmentation paths followed. For technical reasons chem-ical ionisation was used for 31 but the spectra obtained were of comparable quality withthe other ones. The m/z values and their assignments are listed in the Experimentalsection.
Except for iPrAsCl2 and tBuAsCl2, all compounds gave molecular peaks and frag-mented into easily characterisable units of lower molecular masses. The spectra of thechloroarsines and of the mixtures of chloro- and bromoarsines showed characteristic iso-topic patterns, which come as a result of the natural abundancies of the isotopes of Cland Br. Chlorine and bromine have each two naturally abundant isotopes (35Cl, 78.78%;37Cl, 24.22%; 79Br, 50.69%; 81Br, 49.31%), iodine is monoisotopic (127I).282
The spectra of iPrAsCl2 and tBuAsCl2 showed predominantly oxygen-containing frag-ments suggesting high air-sensitivity of the compounds. The spectrum of the mixtureMesAsXX′ contained the peaks of all the possible chloro- and bromoarsines and theirfragments.
94

3. Results and Discussion
The As(S2N2) moiety gave base peaks in the spectra of all the alkyldithiadiazarsoleswith the exception of 31. Thanks to the milder ionisation method (CI), 31 gave aspectrum with a [MH]+ base peak and with the As(S2N2) fragment being the secondmost abundant (65%). Nevertheless, these results suggest relatively high stability of theAs(S2N2) ring.
The spectra of the aryldithiadiazarsoles 34 and 35 contained molecular peaks with theabundance above 60% while the abundance of the As(S2N2) unit did not exceed 40%.The stabilising e�ect of the aromatic hydrocarbon rests is thus apparent.
3.4. IR and Raman spectroscopyIn a full multi-step vibrational analysis, the actual assignment of vibrational frequencies isonly a minor task. The spectra of new compounds are interpreted using as much advice aspossible from known spectra of suitable reference molecules. Characteristic vibrations areoften obtained from the spectra of simple molecules containing the same type of chemicalbonds as the investigated molecule. Further guidance is obtained from the spectra ofmore complex reference molecules. Such molecules need to be as close as possible to theinvestigated molecule in terms of molecular symmetry, chemical composition and massesof participating elements. This approach was adopted in the case of the spectra of thestarting materials RAsX2, the interpretation of which had to be carried out in order too�er a justi�ed assignment of the series of the novel dithiadiazarsoles RAs(S2N2) and toidentify peaks due to vibrations of the AsS2N2 ring.
The molecules of RAsX2 belong to the point group of symmetry Cs. EtAsI2 andiPrAsCl2 can form two conformers - trans (Cs) and gauche (C1). The dithiadiazarsolesRAs(S2N2) belong to the point group of symmetry C1 with 31 and 32 able to adopt threepossible conformations (Fig. 3.3). As a non-linear unit, the As(S2N2) moiety is expectedto exhibit 9 internal vibrational modes. However, since it is known that molecules withlow symmetry give spectra with higher amount of bands due to complicated interactionsof the vibrational modes,283 only the assignments for the least doubtful frequencies areproposed. The data are summarised in Table 3.7�Table 3.12. Each table contains data fora pair of RAsX2 and the corresponding RAs(S2N2). The tables also contain frequenciesdue to dominant features in particular spectra such as intense peaks. A full vibrationalanalysis has not been performed, therefore all the proposed assignments need to betreated as tentative.
95

3. Results and Discussion
RB
RARAAs
XXRA
RBRAAs
XXRA
RB RAAs
X X
RB
RARAAs
NS
N S
RA
RBRAAs
NS
N S
RA
RARBAs
NS
N S
trans gauche gauche
trans to the lone pair trans to N trans to S
(Cs) (C1) (C1)
(C1) (C1) (C1)
Fig. 3.3. A general presentation of conformers for EtAsI2,iPrAsCl2 (top) and EtAs(S2N2) (31) andiPrAs(S2N2) (32) (bottom). The two gaucheconformers of RAsX2 (top) are enantiomers with re-spect to each other and will give identical vibrationalspectra.
3.4.1. The assignments and their discussionScherer and Wies recorded the IR spectra of 25, but con�ned themselves only to aprovisional suggestion to attribute two bands at 1050 and 930 cm−1 to the NSN sys-tem.258 More information was obtained after comparison of the spectra of the RAsX2and RAs(S2N2) series with the following results.
Irrespective of the organic rest, the peaks at 1050, 932, 680, 602, 500 and 365 cm−1
could be easily recognised in the spectra of the dithiadiazarsoles 25�35 and were observedin none of the spectra of the starting materials. The bands strong in IR are those at1050, 680, 602 and 365 cm−1, the remaining peaks appear as strong Raman lines. Thesepeaks are most likely due to the As(S2N2) ring vibrational modes. An additional pairof peaks could be identi�ed in the low frequency region: the IR band at 297 cm−1 andthe corresponding Raman line at 305 cm−1 regularly appeared only in the RAs(S2N2)compounds, but their intensities were rather inconsistent.
An unequivocal assignment of the peaks is di�cult. The range of stretch frequenciesof a cyclic S−−N bond is 1161�911 cm−1, while those of a cyclic S−N bond lie in theinterval 780�670 cm−1. The deformation vibrations frequencies cover the remaining low-frequency area.40,281,284,285 Ring stretches within the molecule of As4S4 range from 384 to
96

3. Results and Discussion
329 cm−1 and the deformation vibrations are scattered over the remaining low-frequencyregion.286,287 The two As−S stretching frequencies in 2-chloro- and 2-iodo-1,3,2-dithiarso-lanes were reported at 390 and 360 cm−1, respectively.288 The As−N stretch gives riseto bands around 585 cm−1.289 However, it is unlikely that stretches of particular bondscould be identi�ed, since the ring will most probably have its own vibrational motions,similarly to e.g. the phenyl ring. Since a detailed analysis leading to description ofparticular vibrational modes of the As(S2N2) ring was not the primary subject of thisthesis, a general assignment was used.
The chemical propinquity of the alkyl rests is re�ected in similar appearance of thespectra throughout the series of both RAsX2 and RAs(S2N2). The spectra of the deriva-tives bearing aromatic groups have also common features.
The IR spectra of MeAsI2, iPrAsCl2 and tBuAsCl2 were measured as thin pressed KBrdiscs whereas the spectrum of EtAsI2 was measured as a liquid �lm between two CsI discs.The spectra of the three compounds measured in KBr discs contain six intense bandsdue to high vacuum silicon grease. The bands appear at unchanged positions at 2963,1261, 1090, 1020, 866 and 804 cm−1, with only the band at 2963 cm−1 having a Ramancounterpart. The bands at 1090 and 1020 cm−1 form a broad �fork� which completelyovershadows the area from 1100 to 1010 cm−1. The shapes and intensities of the bands arein a perfect agreement with those in the spectrum of pure grease. Although utmost carewas taken to avoid contact of the halogenoarsines with grease, the six bands appearedrepeatedly when new spectra were recorded with a new set of samples. The IR spectrumof EtAsI2 also contains these bands but their intensities and widths are insigni�cant.The spectra of the dithiadiazarsoles RAs(S2N2) do not contain these bands.
MeAsI2 and 25 The vibrational frequencies and assignments are listed in Table 3.7.The IR and Raman spectra of MeAsI2 presented in this thesis are in a good agreementwith the published ones and hence their assignment was not di�cult.290 The most intenseband observed in the solid state IR spectrum of MeAsI2 is found at 820 cm−1. Duriget al. recorded the IR spectrum of MeAsI2 in a liquid state and observed one strongIR band at 832 cm−1 with a single very weak Raman counterpart. They assigned theband to degenerate symmetric and antisymmetric CH3 rocking modes.290 The shift of thefrequency of the IR band at 820 cm−1 towards lower values is most probably a consequenceof di�erent state of the sample and thus it can be assigned to rocking of the CH3 group.
The IR spectrum of 25 contains no band around the area 830�820 cm−1. Purely on thebasis of vibrational frequency, the closest candidate is a medium-strong, slightly broadband at 792 cm−1, which was not observed in MeAsI2 nor in any other RAs(S2N2). ItsRaman counterpart, however, was not observed.
In the high frequency region of the IR spectrum of 25 two bands are observed at 3145
97

3. Results and Discussion
and 3050 cm−1. The bands are either combinational vibrations or overtones, since theirfrequencies are signi�cantly higher than a typical frequency of an antisymmetric C−Hstretch observed for the alkyl groups throughout the RAsX2 series. By coincidence, thehighest νas C−H frequency within the series was observed for MeAsI2 (3000 cm−1 in IR).
Table 3.7. Selected IR and Raman wavenumbers (in cm−1) of MeAsI2 and MeAs(S2N2)
MeAsI2 MeAs(S2N2) (25) AssignmentIR Raman IR Raman
3000 vvw 3005 vvw ν C-H2984 sh, vvw 2991 vvw 2992 w 2997 w ν C-H2907 vvw 2910 vvw 2906 w 2902 m ν C-H1396 mw 1408 vvw 1402 m 1403 vvw δCH31374 sh, vw 1382 vvw 1372 sh, vvw � δCH31231 ms 1231 vvw 1235 w 1233 vw δCH3
1054 vs 1057 vw AsS2N2 ring vibr.926 w 929 vvs AsS2N2 ring vibr.
820 vvs 830 vvw ρCH3804 vvs � �
677 vvs 679 vw AsS2N2 ring vibr.605 s � AsS2N2 ring vibr.
556 s 563 vw 561 mw 562 m ν As-C503 mw 506 s AsS2N2 ring vibr.363 ms � AsS2N2 ring vibr.
225 w � ν As-I203 vvs � ν As-I180 w 183 w γ As−CH3; ωCH3AsI2
EtAsI2 and 31 The spectra of EtAsI2 were published on several occasions but o�eredonly incomplete information.291,292 A complete listings of IR and Raman frequencies werepublished by Shagidullin et al.293 whose brief report concentrates on conformational ana-lysis and, unfortunately, lacks a broader discussion about the spectra. Their assignmentsof CH3 and CH2 deformation vibrations are in disagreement with the assignments byEllermann et al.292 as well as with those for a series of ethyldihalogenoarsines by Revittand Sowerby.294 In this thesis the assignment of the area 1450�1210 cm−1 in the spectraof both EtAsI2 and 31 is based on the reports by Ellermann et al. and Revitt andSowerby.
EtAsI2 takes up two conformations, trans- and gauche, which can be identi�ed byvibrational spectroscopy. The only evidence of the presence of two conformers in thespectra of EtAsI2 presented here were two As−C stretches. Revitt and Sowerby ob-served more evidence in the spectra of EtAsCl2 and EtAsBr2,294 but none of the featuresdescribed by them was identi�ed in the present spectrum of EtAsI2.
98

3. Results and Discussion
Table 3.8. Selected IR and Raman wavenumbers (in cm−1) of EtAsI2 and EtAs(S2N2)
EtAsI2 EtAs(S2N2) (31) AssignmentIR Raman IR Raman
2967 sh, s 2975 sh, vvw ν C-H (CH2)2955 vs 2957 vvw 2959 s 2955 vw ν C-H (CH3)2917 vs 2918 vw 2922 ms 2923 w ν C-H (CH2)2888 mw 2890 vvw 2900 sh, mw 2898 sh, w ν C-H (CH3)2861 ms 2863 vvw 2866 m 2867 vw �2815 vw � 2819 sh, vvw � �1446 vvs 1450 vvw 1451 m 1450 vvw δCH31398 m 1401 vvw 1406 w 1404 vvw δCH21374 s 1378 vvw 1373 w 1373 vvw δCH3� 1221 vvw 1215 sh, vw 1217 vw ωCH2
1209 ms 1209 vvw 1200 w 1208 vw τ CH21059 vvs 1061 vvw AsS2N2 ring vibr.
1018 ms 1019 vvw 1021 mw � ν C-C970 mw 970 vvw 964 vw � ρCH3
931 vw 932 vvs AsS2N2 ring vibr.712 ms � 720 sh, vw 716 vvw ρCH2
675 s 679 vw AsS2N2 ring vibr.611 m 612 vvw AsS2N2 ring vibr.
547 m 548 vw 546 vw 548 mw ν As-C519 mw 521 vvw 522 vw 523 m ν As-C
503 w 506 vvs AsS2N2 ring vibr.370 mw 367 vvw AsS2N2 ring vibr.
286 mw 289 vvw 286 vw 289 m δAsCC215 vvs � ν As-I
31 should exist in three conformers - a trans and two inequivalent gauche (Fig. 3.3).Two As−C stretches show up also in the spectra of 31 indicating the presence of at leasttwo conformers. On the basis of the data published for EtAsX2 it is possible to suggestthat the As−C stretch with higher frequency belongs to the trans-to-lone-pair conformerand the other to one of the remaining conformers (Fig. 3.3, page 96). The evidence forthe third conformer could not be found.
Similarly to 25, the IR spectrum of 31 contains two weak high-frequency bands at3145 and 3050 cm−1 without Raman counterparts, which must be either combinationalvibrations or overtones. Both the IR and Raman spectra of EtAsI2 and 31 containidentical patterns of bands corresponding to the vibrations of the ethyl group (Table 3.8).The antisymmetrical C−H stretch of the CH2 group was not observed in the IR spectrumof 31. The band forms a thin shoulder in the spectrum of EtAsI2 and it is possible thatin the spectrum of 31 the stretch became accidentally degenerate with the C−H stretchof the CH3 group.
iPrAsCl2 and 32 A full assignments of the vibrational spectra of iPrAsCl2 have notbeen published yet. The assignments presented in this thesis are derived from those of
99
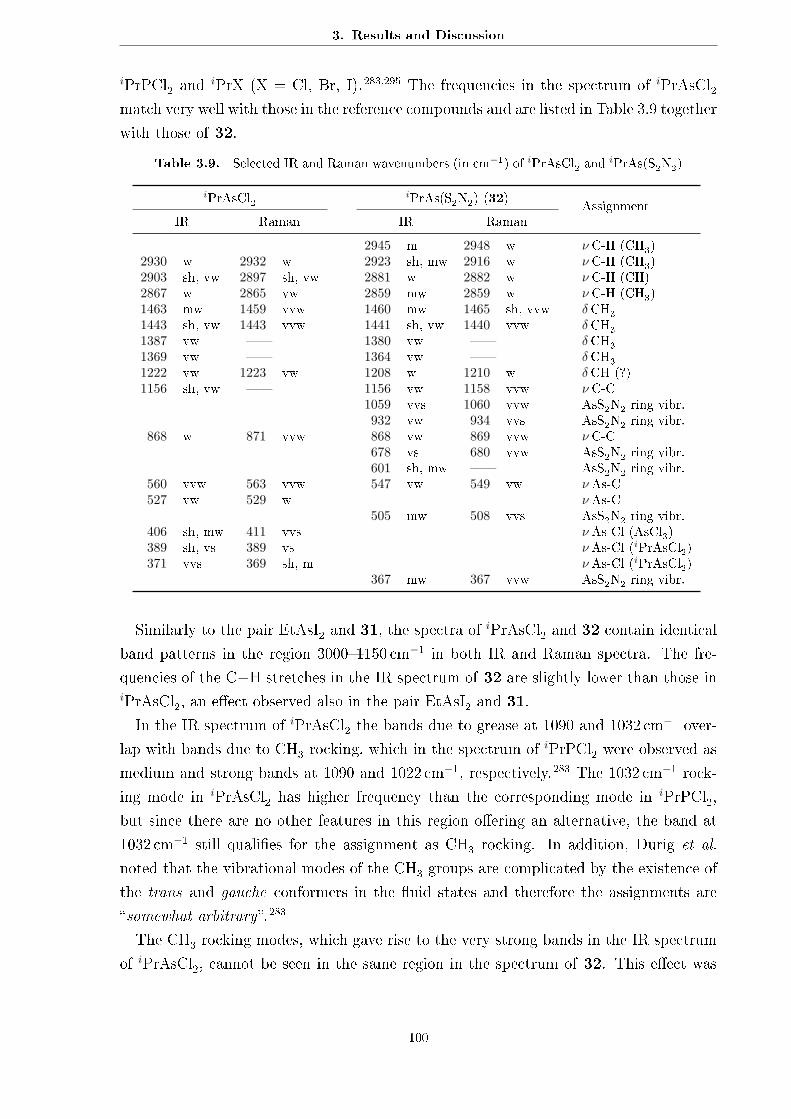
3. Results and Discussion
iPrPCl2 and iPrX (X = Cl, Br, I).283,295 The frequencies in the spectrum of iPrAsCl2match very well with those in the reference compounds and are listed in Table 3.9 togetherwith those of 32.
Table 3.9. Selected IR and Raman wavenumbers (in cm−1) of iPrAsCl2 and iPrAs(S2N2)
iPrAsCl2 iPrAs(S2N2) (32) AssignmentIR Raman IR Raman
2945 m 2948 w ν C-H (CH3)2930 w 2932 w 2923 sh, mw 2916 w ν C-H (CH3)2903 sh, vw 2897 sh, vw 2881 w 2882 w ν C-H (CH)2867 w 2865 vw 2859 mw 2859 w ν C-H (CH3)1463 mw 1459 vvw 1460 mw 1465 sh, vvw δCH31443 sh, vw 1443 vvw 1441 sh, vw 1440 vvw δCH31387 vw � 1380 vw � δCH31369 vw � 1364 vw � δCH31222 vw 1223 vw 1208 w 1210 w δCH (?)1156 sh, vw � 1156 vw 1158 vvw ν C-C
1059 vvs 1060 vvw AsS2N2 ring vibr.932 vw 934 vvs AsS2N2 ring vibr.
868 w 871 vvw 868 vw 869 vvw ν C-C678 vs 680 vvw AsS2N2 ring vibr.601 sh, mw � AsS2N2 ring vibr.
560 vvw 563 vvw 547 vw 549 vw ν As-C527 vw 529 w ν As-C
505 mw 508 vvs AsS2N2 ring vibr.406 sh, mw 411 vvs ν As-Cl (AsCl3)389 sh, vs 389 vs ν As-Cl (iPrAsCl2)371 vvs 369 sh, m ν As-Cl (iPrAsCl2)
367 mw 367 vvw AsS2N2 ring vibr.
Similarly to the pair EtAsI2 and 31, the spectra of iPrAsCl2 and 32 contain identicalband patterns in the region 3000�1150 cm−1 in both IR and Raman spectra. The fre-quencies of the C−H stretches in the IR spectrum of 32 are slightly lower than those iniPrAsCl2, an e�ect observed also in the pair EtAsI2 and 31.
In the IR spectrum of iPrAsCl2 the bands due to grease at 1090 and 1032 cm−1 over-lap with bands due to CH3 rocking, which in the spectrum of iPrPCl2 were observed asmedium and strong bands at 1090 and 1022 cm−1, respectively.283 The 1032 cm−1 rock-ing mode in iPrAsCl2 has higher frequency than the corresponding mode in iPrPCl2,but since there are no other features in this region o�ering an alternative, the band at1032 cm−1 still quali�es for the assignment as CH3 rocking. In addition, Durig et al.noted that the vibrational modes of the CH3 groups are complicated by the existence ofthe trans and gauche conformers in the �uid states and therefore the assignments are�somewhat arbitrary�.283
The CH3 rocking modes, which gave rise to the very strong bands in the IR spectrumof iPrAsCl2, cannot be seen in the same region in the spectrum of 32. This e�ect was
100

3. Results and Discussion
observed also in the spectrum of 25. Purely on the basis of the vibrational frequencies,only one candidate can be suggested - a weak band at 997 cm−1. The band was notobserved in the spectrum of iPrAsCl2 or any other RAs(S2N2) and can correspond to thelower-frequency rocking mode observed at 1032 cm−1 in the IR spectrum of iPrAsCl2. ItsRaman counterpart was not observed. No option could be found for the higher-frequencyrocking mode. It is possible that, if active, this band became accidentally degeneratewith the strong As(S2N2) ring mode at 1059 cm−1.
In the Raman spectrum of iPrAsCl2 lines at 411, 195 and 178 cm−1 showed up. After adetailed inspection also the tip of a line at 374 cm−1 could be spotted. These four listedsignals form a complete Raman spectrum of AsCl3 which, contrary to expectation, wasnot completely separated from iPrAsCl2 (section 3.1.1).
tBuAsCl2 and 33 Full assignments of the vibrational spectra of tBuAsCl2 have not beenpublished yet. The interpretation proposed in this thesis is based on the assignmentsof tBuPCl2 and tBuX (X = Cl, Br)296�298 and was used during the assignments of thevibrational spectra of 33 (Table 3.10).
Table 3.10. Selected IR and Raman wavenumbers (in cm−1) of tBuAsCl2 andtBuAs(S2N2)
tBuAsCl2 tBuAs(S2N2) (33) AssignmentIR Raman IR Raman
2959 sh, mw 2961 sh, w ν C-H2923 vw 2923 m 2929 m 2930 mw ν C-H2853 vw 2861 mw 2855 mw 2855 mw �1460 vvw 1464 vw 1462 mw 1463 vvw δCH3� 1443 w 1441 sh, vvw 1437 vw δCH3� � 1389 vvw 1395 vvw δCH3
1367 vvw � 1362 mw � δCH31202 sh, vvw 1198 vw 1203 sh, vvw 1204 vvw ν C-C1165 sh, vw 1170 mw 1164 w 1165 mw ρCH3
1060 vvs 1061 vvw AsS2N2 ring vibr.933 vw 935 vvs AsS2N2 ring vibr.
� vvs 792 mw 791 vw 792 w ν C-C679 vs 681 vvw AsS2N2 ring vibr.611 m 616 vvw AsS2N2 ring vibr.
� 526 s � 518 sh, ms ν As-C507 w 511 vvs AsS2N2 ring vibr.
404 vw 403 s 400 vvw 401 vvw �381 vw 385 vvs ν As-Cl368 vw 371 ms ν As-Cl
362 w 366 vvw AsS2N2 ring vibr.
The quality of both the IR and Raman spectra of tBuAsCl2 is low, which can bethe consequence of the phase of the compound (section 3.1.1). The sample for IR wasprepared as a thin pressed KBr disc. With the exception of the C−H stretches and
101

3. Results and Discussion
the �grease bands� at 1262, 1096, 1023 and 803 cm−1, the IR bands are badly de�nedand their localisation is based mainly on the presence of the Raman counterparts. TheRaman spectrum is in general statisfactory, though the tops of the lines are often splitinto two or three tips separated by no more than 7 cm−1.
The IR band due to C−C stretch was found at 799 cm−1 in tBuPCl2 296 and at 804 cm−1
in tBuBr,298 which is exactly in the region of the strong �grease band� at 803 cm−1. Inthe IR spectrum of tBuAsCl2, this �grease band� is slightly broadened most probably asa result of an overlap with the C−C stretch band. The C−C stretch gave reasonablystrong Raman counterparts in tBuPCl2, tBuBr and tBuCl and on this basis the line at792 cm−1 in the spectrum of tBuAsCl2 was assigned to this mode. It is in excellentagreement with the frequencies observed for tBuPCl2.296
Due to the low quality of the IR spectrum, the CH3 deformation vibrations and the twousually strong bands due to As−Cl stretches are supressed and appear as insigni�cantmembers within a huddle of bands badly de�ned in the 1480�1350 and 400�360 cm−1
regions, respectively. For the same reason there is no IR band corresponding to theAs−C stretch.
Taking into consideration the complications that accompanied the preparation oftBuAsCl2, a presence of AsCl3 was expected. The Raman spectrum of tBuAsCl2 in-deed showed two lines due to AsCl3, at 403 and 176 cm−1. The AsCl3 line expectedaround 374 cm−1 was overlapped by the strong As−Cl stretches of tBuAsCl2, while thelast AsCl3 band expected around 195 cm−1 surprisingly did not show up at all, probablyas a result of the solid/liquid phase of the sample.
Both IR and Raman spectra of 33 are of much better quality. The CH3 deformationvibrations region as well as the low frequency region are well de�ned and all the peaksare sharp. The As−C stretch was not observed in the IR spectrum but there is a veryslim shoulder at 518 cm−1 in Raman, the frequency of which is in a good agreement withthat of a C−Br stretch in tBuBr.298
PhAsCl2 and 34 In addition to the C−H stretches, the normal vibrational modesof a phenyl ring are characterised by various deformations of the cyclic skelet. Whi�enprovided a graphic outline of the complete set of vibrational modes of a phenyl group in aseries of halogenobenzenes. He also created an annotation system using the letters of theLatin alphabet.299 Other annotation systems used for the phenyl ring vibrational modesare based on Herzberg's or Wilson's annotations of the vibrations of benzene.300,301 Allthree systems were listed alongside each other for conversion purposes by Stenzenbergerand Schindlbauer.302 The vibrational modes listed in Table 3.11 are assigned accordingto Herzberg.302,303 The corresponding Whi�en's annotation is given in parentheses.299
Where possible, the type of vibration is described using the greek letters.
102

3. Results and Discussion
Table 3.11. Selected IR and Raman wavenumbers (in cm−1) of PhAsCl2 and PhAs(S2N2)
PhAsCl2 PhAs(S2N2) (34) AssignmentIR a Raman IR Raman
� 3147 vvw � 3146 vvw �3067 s � � 3067 sh, vw �3055 m 3059 mw 3055 vvw 3048 w ν15
′ (z3); ν C-H3008 w � 3014 vvw � ν1 (z1) or ν5 (z2); ν C-H1575 mw 1579 vw 1563 vvw 1579 vw ν16
′andν16 (k and l)1482 ms 1482 vvw 1475 vw 1479 vvw ν13 (m)1304 m 1310 vvw 1302 vw � �1182 mw 1184 vvw 1180 vvw 1183 vvw ν17
′ (a); β C-H1160 w 1162 vvw 1154 vvw 1159 vvw ν17 (c); β C-H1067 sh 1068 sh, vvw 1068 sh, m 1073 vvw ν10 (d); β C-H
1052 vvs 1055 vvw AsS2N2 ring vibr.1023 mw 1025 w 1023 sh, s 1024 vw ν14 (b); β C-H999 s 1000 vvs 995 mw 999 vvs ν6 (p); trigonal
�ring-breathing�932 w 931 s AsS2N2 ring vibr.
914 w � 916 vw 912 sh, vvw ν11′ (i); γ C-H
736 vs � 743 s � ν4 (f); γ C-H688 vs � 689 sh, m 692 sh, vvw ν8 (v); ring-torsion
(chair)678 s 684 vvw AsS2N2 ring vibr.
672 mw 674 vw 667 sh, m 669 vw ν2 (r); X-sensitive(in-plane ring-def.)
615 vw 616 vvw � 617 vvw ν18 (s);602 m � AsS2N2 ring vibr.499 mw 505 ms AsS2N2 ring vibr.
460 s � 459 m � ν19′ (y); X-sensitive
(out-of-plane ring-def.)361 ms 368 vvw AsS2N2 ring vibr.
a The wavenumbers 3067�3008 cm−1 are taken from Schindlbauer and Stenzenberger,303 the re-maining wavenumbers are from Revitt and Sowerby.304
Vibrational spectra of monosubstituted benzenes show two characteristic features.First, there is not one characteristic vibrational mode due to a C−X stretch as wasthe case for monosubstituted alkanes. The substituent X is involved in several modesthat are usually referred to as �X-sensitive�. Second, majority of the phenyl ring vibra-tions are independent of the mass of the substituent.299 It is thus possible to observea high number of identical frequencies in a series of diverse compounds, such as thehalogenobenzenes,299 PhPH2, PhAsH2, PhPCl2 or PhAsCl2.302�304 It was believed thatthis trend can be successfully extended to the spectra of 34.
The IR spectrum of PhAsCl2 was not recorded, since the colourless substance reactedquickly with both the KBr and the CsI disc to give the corresponding orange/brownbromide or iodide, respectively. The IR frequencies listed in Table 3.11 are taken fromliterature.303,304 The experimental Raman frequencies are in a good agreement with thepublished IR and Raman values.303,304
103

3. Results and Discussion
The vibrational frequencies of 34 were derived from those of PhAsCl2. With theexception of a weak line at 1055 cm−1 which is due to the As(S2N2) ring mode, theRaman spectra of both PhAsCl2 and 34 contain identical line patterns in the region 3150�950 cm−1. In the IR spectrum of 34 a medium band at 1261 cm−1 is observed, which inthe spectra of all the alkyl derivatives �gures as a very weak band. The same band withmedium intensity was found also in the spectrum of 35 suggesting that the enhancedintensity is connected with the vibrational modes of the aromatic ring. Similar conclusioncan be drawn about a medium IR band at 801 cm−1 with a shoulder at 817 cm−1. Thebands do not have a Raman counterpart and do not �gure in the published IR spectra ofPhAsCl2. None of these bands was found in the spectra of any of the alkyl derivatives,but they do appear in the IR spectrum of 35. These bands also seem to be connectedwith the vibrational modes of the phenyl ring.
MesAsX2 and 35 Neither IR nor Raman spectra were recorded for the mixture ofhalogenoarsines MesAsXX′, since without an exact data on the composition of the mix-ture such spectra would be of little use. As a reference compound for the assignment ofthe spectra of 35 was therefore used 2-bromomesitylene, the spectra of which togetherwith the three remaining 2-halogenomesitylenes were studied by Kainz and Schmidt.305
The authors also brought a graphic outline of the complete set of the normal vibra-tions for the molecules. The frequencies of both 2-bromomesitylene and 35 are listed inTable 3.12.
The C−CH3 stretching mode gives rise to a weak IR band at 1307 cm−1 with a mediumRaman counterpart at 1303 cm−1 in the spectra of MesBr. No band was observed between1290 and 1350 cm−1 in the spectra of 35. However, at lower frequencies a weak IR bandat 1288 cm−1 with a sharp medium-weak Raman counterpart at 1291 cm−1 can be found,which are strong candidates for this vibrational mode.
The IR bands at 1262, 816 and 803 cm−1 were not observed either in MesBr, PhAsCl2or any of the previously discussed alkyl derivatives, but they were observed in 34 (videsupra). The enhanced intensity of the 1262 cm−1 band as well as the presence of the tworemaining bands in the IR spectrum of 35 thus can be attributed to the mesityl ringvibrational modes.
The IR band due to the As(S2N2) ring vibration at 932 cm−1 with its very strongRaman counterpart could be observed in all the spectra throughout the series. In thespectra of 35 these bands could overlap with the peaks due to CH3 group rocking mode,which in the spectra of 2-halogenomesitylenes was found around 935 cm−1.
104

3. Results and Discussion
Table 3.12. Selected IR and Raman wavenumbers (in cm−1) of MesBr and MesAs(S2N2)
MesBr305 MesAs(S2N2) (35) AssignmentIR Raman IR Raman
3025 mw � 3016 vvw 3021 w Q1; ν C-H (arom.)2925 ms � 2910 vw 2918 ms ν C-H (CH3)1600 1602 1597 w 1601 mw Q2
1588 w 1590 sh 1583 sh, vw 1587 sh, vvw Q22
1466 vs 1469 vvw 1463 sh, vw 1467 vvw Q3
1437 sh 1443 vvw 1440 mw 1437 sh, vvw �1412 sh 1406 sh 1405 vw � �1379 m 1383 w 1372 sh, vw 1379 w δCH3� 1275 sh � 1276 sh, vvw Q24
� 1250 vvw 1243 sh, vvw 1241 vvw Q25; δC-H (arom.)1178 mw 1180 vvw 1176 vvw 1178 vvw Q5; δC-H (arom.)
1057 vvs 1060 vvw AsS2N2 ring vibr.1028 vs 1030 vw 1028 ms 1038 vvw Q6; X-sensitive952 m 951 vvw 954 vvw 953 sh, vvw Q8; ν C-CH3
932 vw 932 vvs AsS2N2 ring vibr.,ρCH3
846 vvs � 851 m 852 vvw Q15; γ C-H (arom.)681 s 684 vw AsS2N2 ring vibr.602 mw 607 vvw AsS2N2 ring vibr.
589 s 593 vvw 583 w 585 vw Q9; X-sensitive564 mw 570 vvs 558 vw 561 s Q10; �ring-breathing�542 mw 542 vvw 543 vw 544 sh, vw Q17; ring-torsion (boat)
494 w 503 vvs AsS2N2 ring vibr.363 mw 369 vw AsS2N2 ring vibr.
� 343 vw 340 sh, vvw 339 mw Q29; δC-X240 sh 238 vvw 242 sh, vw Q19; γ C-CH3
3.5. X-ray structures
3.5.1. PhAs(S2N2)Molecular structure. The X-ray structure of 34 is displayed on Fig. 3.4. The centralmotif of the molecule is the �ve-membered As(S2N2) ring, which is slightly puckered.Arsenic is in a pseudotetrahedral coordination and posesses a stereochemically activelone pair. It is hinged approx. 0.246Å above the least squares plane of the S2N2 moiety;the S2N2 and S(2)−As(1)−N(1) planes are inclined by 9.6◦. The phenyl group is attachedto the As atom by an As−C single bond and is inclined by 99.4◦ to the least squares planeof the As(S2N2) ring. The plane of the phenyl ring is nearly coplanar with the As(1)−S(2)
bond; the dihedral angle between the phenyl group and the S(2)−As(1)−C(1) planes isonly 11.8◦.
Selected bond lengths and angles are listed in Table 3.13. The As(1)−S(2) bond iselongated (2.303(19)Å) with respect to the As−S single bond in As4S4 (2.243Å meanvalue).214 For a comparison, Pauling's value calculated with Schomaker and Stevenson's
105

3. Results and Discussion
Fig. 3.4. The X-ray structure of 34: a view along the plane of the S2N2 moiety toshow the puckering of the As(S2N2) ring and coordination environmentof the As atom (A); a view along the C(1)−As(1) bond showing theorientation of the phenyl group with respect to the As(1)−S(2) bond (B).Hydrogen atoms are omitted for clarity.
electronegativity correction is 2.23Å. The elongation in 34 is, however, not too drastic.Longer As−S single bonds were observed e.g. in the dimeric cation 19 (2.326(2)Å, page74).249 Elongation of the bond between the terminal sulfur of the S2N2 moiety and the�fth element bound to this moiety has been observed in other �ve-membered main groupsulfur-nitrogen heterocycles, for example in Roesky's ketone ((S2N2)CO) or Roesky'ssulfoxide (S3N2O) (Part IV of this thesis).40,45
Table 3.13. Selected bond lengths (Å) and angles (◦) of PhAs(S2N2) (34)
As(1)−S(2) 2.3028(19) S(2)−As(1)−N(1) 90.01(15)As(1)−N(1) 1.899(5) As(1)−N(1)−S(1) 116.6(3)N(1)−S(1) 1.540(5) N(1)−S(1)−N(2) 115.1(3)S(1)−N(2) 1.573(5) S(1)−N(2)−S(2) 116.9(3)N(2)−S(2) 1.671(5) N(2)−S(2)−As(1) 100.23(18)As(1)−C(1) 1.965(6) S(2)−As(1)−C(1) 101.07(17)
N(1)−As(1)−C(1) 96.5(2)
The As(1)−N(1) bond (1.899(5)Å) is slightly longer than Pauling's As−N single bondcalculated with the electronegativity correction (1.87Å). It is, however, in a good agree-ment with As−N single bonds found in the eight-membered As−N and As−S−N ringcompounds mentioned in the previous chapter (pages 67 and 69).124,126,229,230,233 Thelength of the As(1)−C(1) bond is 1.965(6)Å which is nearly identical to the mean As−Cbond length in arsenobenzene.306,307 Pauling's corrected As−C single bond length is1.942Å.
The alternating sulfur and nitrogen atoms are connected by two shorter (1.540(5) and
106

3. Results and Discussion
1.573(5)Å) and one longer (1.671(5)Å) bonds. A comparison with Pauling's values ofS−−N double bonds justi�es the formal description of 34 by a Lewis formula with twodouble bonds coming out from S(1). On the other hand, the S−−N bonds in 34 arelonger than those in dithiatetrazadiarsocines and acyclic sulfurdiimides.124,126,237,308 Theelongation suggests there is some degree of π-electrons delocalisation in 34, which isfurther supported by the shortened length of the N(2)−S(2) bond single bond.
Delocalisation of π-electrons is closely related to aromaticity. Whether 34 is aromaticor not is the subject of an ongoing computational study. The length di�erence of theformal S−−N bonds in 34 is 0.033Å, which is more than in Roesky's ketone and sulfoxide(0.005 and 0.014Å, respectively) which were both suggested to be aromatic.40,45
The distribution of the bond angles in 34 is comparable to other known �ve-memberedsulfur-nitrogen rings. The most open angles are found at the N atoms. Their mean valuetogether with that of the N(1)−S(1)−N(2) angle (116.7◦ and 115.1(3)◦, respectively) isclose to the optimum 120◦ expected for a trigonal planar coordination, which supportsthe formal Lewis formula of 34. The S(2)−As(1)−N(1) right angle corresponds wellwith the trend of inverse proportionality between van der Waals' radius of an atom anda bond angle value at this atom in �ve-membered sulfur-nitrogen rings.
Crystal structure. The unit cell contains in total two molecules of 34, of which only oneforms the asymmetric unit. It was mentioned that the phenyl ring is oriented in suchway that its plane is eclipsed with the As−S single bond. The surprising orientationseems to be thoroughly �xed by a system of intra- and intermolecular contacts withinthe crystal. Fig. 3.4 shows stacking arrangement of the melocules with the S(1B)−S(2)
and S(1)−S(2A) distances (3.614(5)Å) being just on the edge of the sum of van derWaals' radii.
Fig. 3.5. The stacking of PhAs(S2N2) molecules in the crystal
107

3. Results and Discussion
3.5.2. MesAs(S2N2)The molecular structure of 35 di�ers to that of 34. The As(S2N2) ring in 35 is essentiallyplanar and the orientation of the mesityl group also changes - its plane bisects theAs(S2N2) ring. The two planes are nearly perpendicular. One of the ortho methylgroups is situated right above the �ve-membered ring (Fig. 3.6). Due to bigger stericdemand of mesityl compared to phenyl group, the molecule of 35 is more �open� thanthat of 34: the mesityl group is bound to arsenic with a wider angle with respect to theleast squares plane of the As(S2N2) ring (approx. 110◦ and 111◦ for the two independentmolecules). Selected bond lengths and angles are listed in Table 3.14. The bonding
Fig. 3.6. The X-ray structure of 35: a view on the asymmetric unit of the unit cell(A); a view showing the planarity of the As(S2N2) ring (B); a view alongthe C(1)−As(1) bond showing the orientation of the mesityl group withrespect to the As(S2N2) ring (C). Hydrogen atoms are omitted for clarity.
within the molecule can be formally described by a Lewis formula with two double bondscoming from the NSN sulfur atoms. The As−S, As−N and As−C bonds are single andare slightly longer than those in 34, which can be explained as a consequence of thesteric e�ect of the mesityl group and its better electron-donating ability. The S−N bondlengths can be regarded as intermediate between a single and a double S−N bonds and
108

3. Results and Discussion
π-electrons delocalisation can be expected. The N(1)−S(1) and S(1)−N(2) double bondsdi�er in average only by 0.020Å in length, which is less than in 34.
Table 3.14. Selected bond lengths (Å) and angles (◦) of MesAs(S2N2) (35)
As(1)−S(2) 2.315(3) As(11)−S(12) 2.339(3)As(1)−N(1) 1.927(7) As(11)−N(11) 1.886(7)N(1)−S(1) 1.543(8) N(11)−S(11) 1.537(7)S(1)−N(2) 1.563(7) S(11)−N(12) 1.556(8)N(2)−S(2) 1.665(7) N(12)−S(12) 1.663(9)As(1)−C(1) 1.969(9) As(11)−C(11) 1.973(9)
S(2)−As(1)−N(1) 90.6(2) S(12)−As(11)−N(11) 88.9(2)As(1)−N(1)−S(1) 114.7(4) As(11)−N(11)−S(11) 118.5(4)N(1)−S(1)−N(2) 117.3(4) N(11)−S(11)−N(12) 114.7(4)S(1)−N(2)−S(2) 116.9(5) S(11)−N(12)−S(12) 117.8(5)N(2)−S(2)−As(1) 100.4(3) N(12)−S(12)−As(11) 100.0(3)S(2)−As(1)−C(1) 105.2(3) S(12)−As(11)−C(11) 106.9(2)N(1)−As(1)−C(1) 103.3(3) N(11)−As(11)−C(11) 103.4(3)
Crystal structure. The unit cell contains eight molecules of 35, with two crystallo-graphically independent molecules in the asymmetric unit. The �ve-membered ringsin the asymmetric unit are approx. coplanar but slipped so that the S(1)−N(1) bondlies over the S(12)−N(12) bond of the second independent molecule (Fig. 3.6). TheS(1)−S(12) and N(1)−N(12) distances are 3.713(7)Å and 3.952(8)Å respectively, whichis beyond the sum of van der Waals' radii and thus there are no contacts between the twoindependent molecules. The molecules do not take up a stacking arrangement, presum-ably since the bulky mesityl group precludes an arrangement in closely packed layers.Similarly to 34, the orientation of the mesityl group seems strongly determined by anumber of intra- and intermolecular interactions.
3.6. Cyclic voltammetryThe voltammogram of 34 was recorded at the scan rate of 0.1V s−1 (Fig. 3.7). 34 showedirreversible oxidation and reduction under the experimental conditions. Observed valuesof potentials Ep and currents Ip are presented in Table 3.15. To determine the numberof electrons transferred during the redox reaction at the working electrode, a simulationof a one-electron process under the conditions of the experiment was performed. Thecurrents resulting from the simulated process are approx. 3× higher than the valuesobtained in the experiment for both oxidation and reduction (Table 3.15). The lower
109

3. Results and Discussion
Table 3.15. Experimental and simulated voltammetric data for 34
Ep [V] Ip [µA]experiment simulation experiment simulation
anodic 1.08 1.825 5.49 16.74cathodic −1.71 −1.025 −3.07 −10.24
observed currents are most probably due to passivation of the working electrode causedby a strong adsorption through sulfur, nitrogen or arsenic on its surface.
Fig. 3.7. Cyclic voltammogram of 34
3.7. Reactivity of PhAs(S2N2)It was shown that dialkyl- and diaryldithiatetrazadiarsocines react with metal carbonylsto give complexes, in which the eight-membered rings are coordinated as ligands via thelone pair of electrons on arsenic (page 69). The presence of a lone pair on the arsenicatom in 34 suggests similar behaviour of 34. In order to investigate the ability of 34 toact as a ligand a series of reactions with d-metal complexes was carried out.
Reaction with Pt(COD)Cl2. Pt(COD)Cl2 is known to readily replace the labile CODunder mild reaction conditions with a stronger donor ligands such as phosphines.309,310
Reactions with arsines have also been reported.311,312 The products are obtained after
110

3. Results and Discussion
stirring at room temperature for a couple of hours. They usually precipitate from the re-action mixture but in the opposite case precipitation is induced by addition of a nonpolarsolvent, e.g. hexane.
When the reaction between 34 and Pt(COD)Cl2 was carried out at room tempera-ture, neither precipitation nor a colour change occured. After a gentle re�ux in CH2Cl2the mixture darkened. The dark matter obtained after work-up was separated by sil-ica column chromatography. Several coloured fractions were obtained but only two ofthem contained multiplets in the aromatic region which corresponded very well withpure PhAs(S2N2). Remaining fractions contained signals of neither PhAs(S2N2) nor free1,5-cyclooctadiene. The fraction obtained by elution with methanol gave a 1H NMR spec-trum showing very intense aromatic signals as well as the signals of platinum-coordinatedCOD suggesting that the reaction did not occur. Recrystallisation attempts of some frac-tions were carried out with no success.
Reaction with metal carbonyls. The reaction of 34 with [Mo(CO)4(piperidine)2] atroom temperature in CH2Cl2 resulted in formation of a brown/orange solution, whichupon evaporation of the solvent left a tarry black residue. IR spectrum showed a changein the area of ν(CO) - the bands at 1780, 1837 and 1892 cm−1 due to carbonyl groupsin [Mo(CO)4(piperidine)2] disappeared and a single band appeared at 1927 cm−1. TLCshowed decomposition of the product on silica and therefore the product was precipitatedfrom a concentrated CH2Cl2 solution by addition of diethyl ether (10ml) and petroleumether (40ml). The dark brown precipitate was �ltered o� by suction, washed twice withpetroleum ether and dried under high vacuum. Recrystallisation attempt by gas phasedi�usion of hexane into a CH2Cl2 solution resulted in deposition of orange plates, whichunfortunately did not di�ract X-rays. Mass spectrum (ES−TOF) showed a molecularpeak at m/z 246.83 which could not be assigned to either any expected product, byprod-uct or unreacted starting material.34 was allowed to react with Cp2Ti(CO)2 at room temperature for 2 days at which
point some �ne powder started to precipitate. The stirring was stopped and the mixturewas �ltered through a sinter. TLC of the �ltrate showed several well de�ned spots withno signs of decomposition. The subsequent silica column chromatography of the �ltrateseparated unreacted PhAs(S2N2) and a purple product which in a 1H NMR spectrumgave two very close signals looking like a doublet (6.27 and 6.29 ppm). This suggestedthe presence of two chemically non-equivalent cyclopentadienyl rings. The most probableproduct that could be formed in this reaction system and that could conform to suchan 1H NMR spectrum was Cp2Ti(S2N2). In an attempt to isolate this compound in acrystalline state, the product was dissolved in CH2Cl2 and was treated with hexane in agas phase di�usion recrystallisation system. However, after several days the encouraging
111

3. Results and Discussion
purple colour was lost and a beige precipitate was formed, the 1H NMR spectrum ofwhich was di�erent to that of the purple compound. Attempts to recrystallise this beigeprecipitate unfortunately resulted only in formation of a powder, no crystals were formed.
The reaction of 34 with Cp*Co(CO)2 was carried out in toluene at room temperature.After 2 days a suspension of a black solid in a dark wine-red solution was formed. Themixture was �ltered, the �ltrate was reduced in volume and was subjected to silica columnchromatography. Two coloured fractions were collected, one containing the unreacted 34and a purple band, which was con�rmed by 1H NMR and MS as Cp*Co(S2N2) (Part IIIof this thesis).
Methylation with CH3I. An attempt to prepare the ionic N -methyldithiadiazarsoliumiodide by methylation of 34 with CH3I was not succesful. The reaction was carried outin dry acetone at room temperature overnight. It was possible to identify CH2I2 by 1Hand 13C NMR. The most intense signal of the spectrum was a new singlet at 1.23 ppm,which could be the product. The 13C NMR spectrum also showed an intense singlet at29.3 ppm which could be assigned to the product. Soon, yellow crystals started to growon the walls of the NMR tube which were identi�ed by X-ray analysis as the unreacted34.
Oxidation with sulfur. Inconsistent reports were published concerning the possibleoxidation of As (III) compounds by elementary sulfur to obtain As (V) products with anAs−−S double bond. In spite of thermal lability of the arsenic�chalcogen double bonds,313
the oxidation of bis(diphenylarsino)oxide and -sul�de at high temperatures accompaniedby the formation of As−−S bond was observed.314 Since 34 showed remarkable thermalstability, it was worth trying to oxidise by elementary sulfur at elevated temperature.
By re�uxing 34 and sulfur �owers in toluene a yellow/orange mixture was formed. Af-ter �ltration and after reducing the �ltrate in volume, well shaped yellowish crystals weredeposited on the walls of the Schlenk tube. Unfortunately, the crystals were identi�edby X-ray analysis as unreacted 34.
3.8. ConclusionThis part described a contribution to the little investigated area of �ve-membered AsSNring compounds. The versatile tin reagent 27 was used as the source of the (S2N2) 2 �
ligand in substitution reactions with alkyl- and aryldihalogenoarsines, which led to theisolation of �ve new 5-alkyl/aryl-1,3λ4δ2,2,4,5-dithiadiazarsoles 25�35. All products wereunequivocally characterised and two single crystal structures were determined. The crys-
112

3. Results and Discussion
tal structures of the two relatively stable aryldithiadiazarsoles revealed interesting intra-and intermolecular e�ects in�uencing strongly the geommetry of the dithiadiazarsoles.
The ability of PhAs(S2N2) (34) to act as a ligand was investigated in a series ofreactions with selected d-metals complexes. The results suggest low tendency of 34to serve as donor of its lone pairs of electrons. The reactions with complexes of d-metals from the fourth period (Ti, Co) resulted in a ligand exchange. The existence ofthe complex Cp2Ti(S2N2) was suggested by 1H NMR spectra, whereas the formation ofCp*Co(S2N2) was proved by both 1H NMR and MS. The reactions with �fth and sixthperiod d-metals complexes (Mo, Pt) resulted neither in the (S2N2) 2 � ligand exchangenor in coordination of 34 as a ligand. 34 was always obtained in minor quantities asunreacted material and the products usually formed black tarry matters from which noproduct could be isolated.
The stability of the �ve-membered As(S2N2) ring was con�rmed by unsuccessful methy-lation with CH3I. Finally it was shown that the oxidation of 34 by elemental sulfur atelevated temperatures cannot be achieved.
113

4. Experimental
The �rst sample of AsCl3 was purchased from ABCR and the next was prepared ac-cording to the published procedure.87 Other purchased chemicals were CH3I, C2H5Iand SOCl2 (all Fluka), diethyl ether solutions of iPrMgCl and tBuMgCl (Aldrich),2-bromomesitylene (Alfa-Aesar) and 4-aminobenzenearsonic acid (TCI). Phenylarsonicacid and As2O3 were obtained from the departmental store. Pt(COD)Cl2 315
and Cp2Ti(CO)2 316 were prepared according to literature procedures.
4.1. Preparation of AsCl387
A 500ml three-neck-�ask equipped with a stirring bar was charged with As2O3 (100 g,0.505mol) and the �ask was �tted with a dropping funnel and a water condenser. Theoutlet from the condenser was secured with a drying tube (CaSO4) and was connectedwith an empty washing �ask (backsuction protection) followed by two washing �asks�lled with Ca(OH)2 suspensions. SOCl2 (200ml, 327 g, 2.75mol) was added from thedropping funnel portionwise to As2O3. Since no reaction was observed after 50ml SOCl2were added, the mixture was gently heated with a heating gun to 50�60 ◦C. Slight increasein SO2 bubbles evolution was observed indicating the start of the reaction. The remainingamount of SOCl2 was added in 50ml portions with 20 minutes idle period between everyaddition. The mixture was then stirred at room temperature for 36 hours by which timenearly all As2O3 reacted and a heavy, impure colourless solution was formed. The �askwas �tted with a 25 cm Vigreux column well insulated with glass wool and aluminiumfoil and the mixture was fractionally distilled under nitrogen at atmospheric pressure.The nitrogen gas inlet was introduced at the receiver adapter. The oil bath was heatedto 140 ◦C, the Vigreux column was allowed to warm up slowly and after a longer periodSOCl2 started to distill (tvapours = 70�80 ◦C). At some point the distillation stopped andno progress was observed even when the oil bath temperature was set to 150 ◦C. Thedistillation was stopped, SOCl2 was stored aside and AsCl3 was distilled without theVigreux column under nitrogen at atmospheric pressure (toil = 162 ◦C, tvapours = 127�130 ◦C). Yield 152 g (83%).Raman data: 408 (vs), 377 (m), 195 (m), 175 (sh) cm−1.
114

4. Experimental
4.2. Preparation of CH3AsI290
A 2 litre Erlenmeyer �ask with an overhead stirrer was loaded with water (150ml), As2O3(55.0 g, 0.278mol) and, with stirring, NaOH (97.0 g, 2.43mol). After the solution cooleddown to room temperature, ethanol (800ml) was added. The �ask was immersed intoan ice/water cooling bath, CH3I (53ml, 121 g, 0.850mol) was carefully added and themixture was stirred for 20 hours at room temperature. Afterwards, as much solventas possible was evaporated on a rotary evaporator with the temperature of the heatingbath not exceeding 50 ◦C. Additional evaporation for 15 minutes under high vacuumwas carried out. The resulting white slurry was diluted with water (400ml) and thecloudy mixture was acidi�ed with conc. HCl (approx. 110ml) to pH 3 (indicator paper).After reaching the desired pH, SO2 was bubbled into the mixture and at the same timea solution of NaI (105 g, 0.700mol) in water (120ml) was added from a dropping funnel.The SO2 was bubbled for 2 hours and the addition of NaI was �nished slightly beforethe bubbling was stopped. During the bubbling, heavy oily layer of the product startedto form at the bottom of the �ask and also some white solid precipitated. After thebubbling was �nished, the yellow mixture was heated in a water bath to 50 ◦C and thewarm mixture was �ltered through a sinter. The heavy oily product was separated in aseparating funnel and was collected in a 100ml �ask, where it crystallised spontaneouslyupon cooling. The crude product was cooled in a water/ice bath and was transferred ontoa sinter where it was washed with cold water (0 ◦C, 50ml). The pure, yellow, crystallineproduct was dried under high vacuum for 2.5 hours. Yield 30.5 g (16%).MS(CI+TOF): m/z 344.76 (100%) [MH]+, 343.75 (40%) M+, 328.73 (5%) [AsI2]+,216.85 (85%) [CH3AsI]+.1H NMR (CDCl3, 298 K): δ = 3.11 (s, 3H, CH3).13C NMR (CDCl3, 298 K): δ = 20.6 (s, 1C, CH3).IR data: 3000 (vvw), 2984 (sh, vvw), 2964 (w), 2907 (vvw), 1396 (mw), 1374 (sh, vw),1261 (ms), 1231 (ms), 1094 (ms, br), 1022 (ms, br), 943 (vvw), 866 (vw), 850 (sh, vvw),820 (vvs), 804 (vvs), 733 (sh, vvw), 661(vvw), 593(vw), 556 (s), 512 (sh, w), 388 (vw),368 (sh, vvw), 345 (vvw), 287 (vvw) cm−1.Raman data: 3005 (vvw), 2991 (vvw), 2910 (vw), 1408 (vvw), 1382 (vvw), 1231 (vvw),830 (vvw), 563 (vw), 290 (vvw), 256 (vvw), 225 (w), 203 (vvs), 180 (w) cm−1.
4.3. Preparation of EtAsI290,121
In a beaker, As2O3 (50.0 g, 0.253mol) was allowed to react with a solution of NaOH(60.6 g, 1.52mol) in 200ml of water to form an aqueous solution of Na3AsO3.
A 2 litre round bottom �ask equipped with a suitable stirring bar was charged with
115

4. Experimental
500ml of water and the prepared Na3AsO3 solution. Ethyl iodide (40.4ml, 78.8 g,0.505mol) was carefully added to the stirred solution whereupon two liquid layers wereformed. The mixture was allowed to cool down a bit and ethanol was added in such anamount so that only one liquid phase was formed (850ml). The mixture was then stirredfor 6 days: after the �rst day a white precipitate appeared and over the remaining 5 daysthe mixture gradually became more and more yellow. When the reaction was �nished,the mixture was divided in two parts and ethanol was evaporated on a rotary evaporatorfrom both parts. The alcohol-free residues were collected to a 2 litre �ask equipped witha stirring bar. The mixture was acidi�ed to pH 3 (indicator paper) with conc. HCl(approx. 90ml). After the mixture reached pH 9, a white precipitate appeared. Whenthe desired pH was reached, the mixture was �ltered by suction (Büchner funnel) and theclear �ltrate was placed in a 2 litre three-neck-�ask equipped with a stirring bar. Twonecks of the �ask were �tted with dropping funnels, one charged with conc. HCl (10ml),the other with a solution of NaI (75.8 g, 0.505mol) in water (100ml). A gas inlet glasstube was immersed to the �ltrate through the third neck and SO2 was passed throughthe stirred mixture for 4 hours. NaI was added �uently throughout the bubbling periodand the addition �nished just before the bubbling was terminated. HCl was added inone portion after 30 minutes of SO2 bubbling. A heavy yellow oil kept forming at thebottom of the �ask. After the SO2 inlet was stopped, the heterogeneous mixture was�ltered through a sinter, the �ltrate was collected in a 1 litre �ask and was heated to50 ◦C to help separate the remaining oil from the aqueous mixture. The two liquidswere then separated in a separation funnel, the heavy oily product was collected and theaqueous layer was discarded. The product was puri�ed by vacuum distillation (0.3Torr,toil = 103 ◦C). The clear, orange/red oil was stored in dark under nitrogen. Yield 55.0 g(30%). B.p. 63 ◦C (0.3Torr), approximate density measured by weighing 1ml of theneat product, d = 2.76 g cm−3.MS(EI+TOF): m/z 357.77 (25%) [M]+, 328.74 (5%) [AsI2]+, 230.87 (15%) [EtAsI]+,201.83 (6%) [AsI]+, 126.90 (66%) [I]+, 74.92 (3%) [As]+.1H NMR (CDCl3, 298 K): δ = 1.42 (t, 3H, CH3), 2.81 (qrt, 2H, CH2).13C NMR (CDCl3, 298 K): δ = 15.1 (s, 1C, CH3), 28.28 (s, 1C, CH2).IR data (neat liquid between CsI discs): 2967 (sh, s), 2955 (vs), 2917 (vs), 2888 (mw),2861 (ms), 2815 (vw), 1446 (vvs), 1398 (m), 1374 (s), 1297 (vvw), 1260 (vw), 1209 (ms),1090 (vw), 1044 (w), 1018 (ms), 970 (mw), 874 (vvw), 800 (vw), 712 (ms), 547 (m), 519(mw), 426 (vvw), 400 (sh, vw), 390 (vw), 374 (vw), 361 (vw), 344 (vvw), 321 (vvw), 286(mw), 273 (), 246 (w) cm−1.Raman data: 2975 (sh, vvw), 2957 (vvw), 2918 (vw), 2890(vvw), 2863 (vvw), 1450 (vvw,br), 1401 (vvw, br), 1378 (vvw), 1221 (vvw), 1209 (vvw), 1019 (vvw), 970 (vvw), 548(vw), 521 (vvw), 323 (vvw), 289 (vvw), 215 (vvs), 182 (sh, vvw) cm−1.
116

4. Experimental
4.4. Preparation of iPrAsCl2122,136
Commercial 2.0m iPrMgCl in diethyl ether (26.5ml, 0.053mol) was diluted with dryether (80ml) and was added dropwise to a stirred cold (-40 ◦C) solution of AsCl3 (8.9ml,19.2 g, 0.106mol) in dry ether (100ml). The mixture was stirred for 30 minutes and wasallowed to warm up to -20 ◦C, then it was �ltered under nitrogen through a celite plug(2 cm) and since the �ltrate was still cloudy, the �ltration was repeated. The solvents wereevaporated under high vacuum, the colourless oily residue was transferred to a microdis-tillation kit and was distilled at atmospheric pressure. Three fractions of colourless oilyliquid were collected at the oil bath temperatures 190, 195 and 204 ◦C respectively. Afteranalysis of the 1H NMR spectra, the lowest boiling fraction was discarded. The middlefraction contained the product slightly contaminated with some unidenti�ed byproductsand the highest boiling fraction contained virtually pure iPrAsCl2. Yield 2.31 g (23%).MS(EI+TOF): m/z 144.86 (100%) [AsCl2]+, 109.89 (20%) [AsCl]+, 74.92 (25%) [As]+.1H NMR (CDCl3, 298 K): δ = 1.38 (d, 6H, 2×CH3), 2.46 (sept., 1H, C−H).13C NMR (CDCl3, 298 K): δ = 16.5 (s, 2C, 2×CH3), 42.5 (s, 1C, CH).IR data: 2962 (ms), 2930 (w), 2903 (sh, vw), 2867 (w), 1463 (mw), 1443 (sh, vw), 1411(vw), 1387 (vw), 1369 (vw), 1261 (vs), 1222 (vw), 1211 (sh, vvw), 1156 (sh, vw), 1090(vvs), 1032 (sh, s), 924 (mw), 868 (w), 807 (vvs), 753 (sh, vvw), 700 (vw), 664 (vvw),621 (vw), 560 (vvw), 527 (vw), 499 (sh, vvw), 389 (sh, vs), 371 (vvs), 319 (sh, vvw)cm−1.Raman data: 2959 (vw), 2932 (w), 2897 (sh, vw), 2890 (vw), 2865 (vw), 1459 (vvw),1443 (vvw), 1223 (vvw), 1212 (sh, vvw), 1098 (vvw), 871 (vvw), 563 (vvw), 529 (w), 411(vvs), 389 (vs), 369 (m), 274 (vw), 195 (mw), 178 (vw) cm−1.
4.5. Preparation of tBuAsCl2123,163
A 500ml �ask equipped with a stirring bar and �tted with a dropping funnel with apressure-equalising shoulder were all predried and after cooling down they were wrappedin aluminium foil.
Dry diethyl ether (45ml) was transferred via a syringe through the dropping funnel tothe 500ml �ask and then in the same way AsCl3 (4.2ml, 9.06 g, 0.050mol) was carefullyadded. The dropping funnel was then washed with a little bit of dry ether, the stopcockwas closed and the AsCl3 solution was cooled to -30 ◦C (acetone/dry ice). Dry ether(75ml) was transferred to the dropping funnel and commercial 2.0m tBuMgCl in ether(25ml, 0.050mol) was quickly added. The solution was shortly stirred with a glass rodand was added dropwise to the stirred and cooled solution of AsCl3. The mixture wasstirred at -30 ◦C for 1 hour and then was �ltered through a celite plug (2 cm). The �ltra-
117

4. Experimental
tion was carried out under nitrogen in a Schlenk sinter but the mixture was transferredonto the celite plug quickly through a powder funnel. As soon as all collected, the �ltratewas reduced to ca. 20ml and the concentrated solution was kept at -40 ◦C. When a mi-crodistillation apparatus was assembled and predried, the cold solution was transferred tothe distillation �ask, the remaining solvent was evaporated and the residue was distilledunder high vacuum (0.3Torr, toil = 40�45 ◦C). Under these conditions volatile impuritieswere evaporated and the product sublimed over to a cold trap connected between thedistillation apparatus and the vacuum line. The content of the cold trap was allowed tomelt, was transferred to a predried 250ml Schlenk �ask and was worked up as follows:a portion was taken to a predried Schlenk tube and the solvent was slowly evaporatedunder high vacuum with only gentle hand-heating applied when necessary. The slowevaporation together with the usual temperature decrease caused a gradual crystallisa-tion of the product in the Schlenk tube. When there was no more liquid present, nextportion from the 250ml �ask was added and the procedure was repeated.
tBuAsCl2 of su�cient purity was obtained as a volatile colourless, crystalline substancewith a characteristic old-sweat-odour and was stored under nitrogen in a freezer. Yield4.30 g (42%). M.p. 42�45 ◦C.MS(EI+TOF): m/z 395.66 (100%) [As4O6]+, 288.74 (30%) [As3O4]+, 90.92 (29%)[AsO]+, 73.04 (26%) [tBuO]+, 57.07 (78%) [C4H9]+.1H NMR (CDCl3, 298 K): δ = 1.32 (s, 9H, tBu).13C NMR (CDCl3, 298 K): δ = 24.0 (s, 3C, 3×CH3), 44.7 (s, 1C, Cquart.).IR data: 2963 (mw), 2933 (sh, vw), 2923 (vw), 2903 (sh, vw), 2853 (vw), 1460 (vvw),1401 (vvw), 1379 (vvw), 1367 (vvw), 1262 (vs), 1202 (sh, vvw), 1176 (sh, vvw), 1165(sh, vw), 1096 (vs), 1023 (vs), 866 (vw), 803 (vvs), 671 (vvw), 660 (vvw), 498 (w), 404(vw), 381 (vvw), 368 (vvw), 343 (vvw), 308 (vvw), 286 (vw) cm−1.Raman data: 2964 (mw), 2939 (m), 2923 (m), 2893 (ms), 2861 (mw), 2782 (vw), 2720(vw), 1464 (vw), 1443 (w), 1393 (vvw), 1221 (vw), 1198 (vw), 1187 (sh, vw), 1170 (mw),943 (vw), 792 (mw), 526 (s), 403 (s), 385 (vvs), 371 (ms), 301 (w), 272 (mw), 246 (vw),176 (mw) cm−1.
4.6. Preparation of PhAsCl2117
Phenylarsonic acid (12.0 g, 0.059mol) was mixed with conc. HCl (300ml) in a 1 litre�ask. After the mixture was stirred for 1 hour, SO2 was bubbled into the mixture for6 hours. The mixture was then left standing overnight to allow the heavy oily productseparate from the mixture at the bottom of the �ask. The crude product was isolatedin a separating funnel as an impure colourless liquid which was puri�ed by vacuum
118

4. Experimental
distillation (toil = 130�140 ◦C) to give a clear, colourless liquid. The product was storedunder nitrogen. Yield 6.7 g (50%).MS(EI+TOF): m/z 221.90 (10%) M+, 186.93 (50%) [PhAsCl]+, 150.95 (15%)[C6H4As]+, 109.89 (2%) [AsCl]+, 77.03 (100%) [C6H5]+.1H NMR (CDCl3, 298 K): δ = 7.40�8.00 (m, 5H, Ph-group).13C NMR (CDCl3, 298 K): δ = 129.4 (s, 2C, meta-C), 130.1 (s, 2C, ortho-C), 132.3 (s,1C, para-C), 145.3 (s, 1C, C−As).Raman data: 3147 (vvw), 3059 (mw), 3034 (sh, vvw), 1579 (vw), 1482 (vvw), 1310(vvw), 1184 (vvw), 1162 (vvw), 1077 (vvw), 1068 (sh, vvw), 1025 (w), 1000 (vvs), 674(vw), 616 (vvw), 389 (s), 365 (mw), 309 (vvw), 251 (w), 229(vvw), 223 (vvw), 181 (vvw)cm−1.
4.7. Preparation of mixture ofmesityldihalogenoarsines317
2-Bromomesitylene (3.84ml, 5.0 g, 0.025mol) was dissolved in dry diethyl ether (100ml),the solution was cooled to 0 ◦C (water/ice) and nBuLi (0.030mol, 12.0ml of 2.5m solutionin hexanes) was added dropwise to the stirred solution. The cooling bath was removedand the mixture was stirred at room temperature for 4 hours. The resulting suspensionwas evaporated to dryness, the bright white solid was resuspended in fresh dry ether(70ml) and the suspension was added dropwise (syringe) to a vigorously stirred, cold(-78 ◦C) solution of AsCl3 (2.0ml, 4.31 g, 0.024mol) in dry ether (60ml). The mixturewas allowed to warm up gradually to room temperature and was stirred for 2 hours. Thesolids were �ltered o� (sinter), the solvent was evaporated from the �ltrate and the oilyresidue was distilled under high vacuum. The �rst fraction distilling at toil = 64 ◦C wasunreacted AsCl3, the second fraction (toil = 80 ◦C) was proved by 1H NMR to be unre-acted 2-bromomesitylene and at toil = 135 ◦C the product started to distill and solidi�edin the condenser. Gentle heating with a heating gun caused melting of the crystals andthe distillation was continued. The product was stored under an inert atmosphere. Yield2.40 g (36%).MS(EI+TOF):m/z 353.85 (3%) [MesAsBr2]+, 309.89 (8%) [MesAsClBr]+, 274.92 (13%)[MesAsBr]+, 263.95 (3%) [MesAsCl2]+, 228.97 (62%) [MesAsCl]+, 193.00 (8%)[C9H10As]+, 119.08 (100%) [C9H11]+.1H NMR (CDCl3, 298 K): δ = 2.26�2.33 (singlets, mixture of para-CH3 groups), 2.68�2.73 (singlets, mixture of ortho-CH3 groups), 6.89�6.92 (m, mixture of Harom.).13C NMR (CDCl3, 298 K): δ = 21.3 (s, 1C, ortho-CH3), 21.4 (s, 1C, ortho-CH3), 21.7(s, 1C, ortho-CH3), 130.0 (m, 2C, a part of meta-C multiplet in MesAsClBr), 130.1 (m,
119

4. Experimental
2C, a part of meta-C multiplet in MesAsClBr), 130.4 (m, 2C, a part of meta-C multipletin MesAsClBr), 130.5 (s, 2C, meta-C in MesAsCl2 or MesAsBr2), 130.6 (s, 2C, meta-Cin MesAsCl2 or MesAsBr2), 135.4 (single peak, mixture of Cquart.), 130.1 (overlappingpeaks, mixture of Cquart.), 139.6 (single peak, mixture of Cquart.), 141.7 (single peak,mixture of Cquart.), 141.8 (single peak, mixture of Cquart.), 142.7 (single peak, mixture ofCquart.).
4.8. Preparation of p-phenylenediarsonic acid byBart's reaction278,318
The following solutions were prepared: 2.5m H2SO4 (100ml), 5.0m NaOH (250ml), 2.0mNa2HAsO3 (100ml) and 5.0m HCl (250ml).
A 5 litre �ask equipped with a big te�on stirring bar was charged with p-aminoben-zenearsonic acid (25.0 g, 0.115mol). Water (1 litre) followed by 2.5m H2SO4 (89ml,0.223mol) were added. The mixture was stirred for a while so that all p-aminoben-zenearsonic acid could dissolve. The solution was cooled in an ice/water bath to 8 ◦Cand a solution of NaNO2 (7.95 g, 0.115mol) in water (100ml) was added. The mixtureturned yellow indicating the formation of the diazonium salt. The mixture was kept inthe cooling bath and was cooled to 0 ◦C. 5.0m NaOH (20ml, 0.100mol) was added todecrease the acidity of the mixture.
The copulation with the sodium hydrogenarsenite solution required a pre-treatment ofthe sodium hydrogenarsenite. Because the preparation of p-phenylenediarsonic acid wascarried out with 0.115mol of p-aminobenzenearsonic acid, an equal amount of sodiumhydrogenarsenite (0.115mol) was needed. Therefore, 65ml of the 2.0m Na2HAsO3 solu-tion (0.130mol) was pipetted in a 100ml conical �ask and 2.5m H2SO4 (3ml, 0.0075mol)was added to convert the excessive amount (0.015mol) of Na2HAsO3 into NaH2AsO3 andthus to obtain a bu�er with exactly 0.115mol of Na2HAsO3.
The arsenite bu�er was then poured cautiously in one portion to the cold (0 ◦C) di-azotised mixture. Nitrogen gas was evolved unexpectedly mildly, as if only from thesurface of the mixture, no voluminous foams were formed and the mixture darkenedslightly. Right after the copulation another 5.0m NaOH (20ml, 0.100mol) was addedwhereupon the mixture became slightly basic (pH = 8, indicator paper) and darkenedto red/brown. The coolig bath was removed, the stirring was continued at room tem-perature for 2.5 hours and then the mixture was heated to 50 ◦C but neither more gasevolution nor any colour change were observed. After cooling to room temperature themixture was acidi�ed with 5.0m HCl (20ml, 0.100mol) which resulted in precipitation
120

4. Experimental
of a dark red solid. The solid was �ltered o� by suction (Büchner funnel), water wasevaporated from the �ltrate on a rotary evaporator and the resulting mixture of a cream-coloured solid and a dark red mud was extracted with boiling ethanol (3 × 600ml). Thejoined extracts were �ltered through a Büchner funnel and ethanol was completely re-moved from the �ltrate on a rotary evaporator to leave a dark red/brown mud with bitsof a cream-coloured solid.
A small amount of the cream-coloured solid was obtained by dilution of the crudemixture with a small amount of ethanol and �ltration of the suspension through a sin-ter. 1H NMR spectrum of the solid indicated the presence of p-phenylenediarsonic acid(1H NMR (DMSO−d6, 298 K): δ = 7.95 (s, 4H, arom. H)). Only poor amounts of theacid could be isolated from the crude mixture. Therefore all fractions of the crude acidwere placed in a 2 litre �ask. Water (600ml) and 5m HCl (140ml) were added andSO2 was bubbled through the stirred mixture for 3 hours. A sand-coloured precipitatekept forming, which was then �ltered o� and identi�ed by MS (EI) as As2O3. 1,4-bis(di-chloroarsino)benzene could not be isolated from the �ltrate. MS(EI+TOF): m/z 395.65(100%) [As4O6]+, 288.74 (30%) [As3O4]+, 181.84 (2%)[As2O2]+, 90.91 (15%) [AsO]+.
4.9. Reaction of MeAsI2 with [nBu2Sn(S2N2)]2MeAsI2 (370mg, 1.08mmol) dissolved in dry CH2Cl2 (10ml) was added dropwise to astirred solution of [nBu2Sn(S2N2)]2 (351mg, 0.538mmol) in dry CH2Cl2 (10ml). Themixture was gently re�uxed for 3 hours during which time it gradually darkened toorange. After cooling to room temperature, the mixture was �ltered through a sinter.The solvent was removed from the �ltrate to leave a small amount of a red/brown oilwhich was transferred into a microdistillation apparatus and distilled under high vacuum(0.3Torr, toil = 90 ◦C). Due to the small amount of the crude oil, the necessary insulationof the apparatus with glass wool and aluminium foil was not enough and the �pre-condenser� part of the apparatus had to be gently heated with a heat gun to help thevapours run over to the condenser. The distillate was collected to a �ask immersed in asmall acetone/dry ice cooling bath to avoid loss of the product to the vacuum line coldtrap. MeAs(S2N2) is an orange/brown, air-sensitive, volatile oil which does not crystalliseupon cooling. Yield 54mg (30%). The distillation residue, an impure colourless oil, wasproved by 1H NMR to be nBu2SnI2 and was discarded.MS(EI+TOF): m/z 181.89 (18%) [M]+, 166.87 (100%) [AsS2N2]+, 135.92 (8%)[MeAsSN]+, 120.90 (10%) [AsSN]+, 106.89 (10%) [AsS]+, 89.94 (5%) [MeAs]+, 74.92(2%) [As]+.
121

4. Experimental
1H NMR (CDCl3, 298 K): δ = 1.17 (s, 3H, CH3).13C NMR (CDCl3, 298 K): δ = 24.5 (s, 1C, CH3).14N NMR (CDCl3, 298 K): δ = 274.2 (s, 1N), 301.4 (s, 1N).IR data (neat oil between CsI discs): 3145 (w), 3050 (w), 2993 (w), 2971 (sh, w), 2906(w), 1402 (m), 1261 (w), 1235 (w), 1192 (w), 1130 (mw), 1101 (sh, mw), 1089 (sh, mw),1054 (vs), 1046 (sh, vs), 971 (vvw), 926 (w), 862 (m), 792 (ms), 694 (sh, s), 677 (vvs),605 (s), 561 (mw), 503 (mw), 448 (vvw), 422 (vvw), 363 (ms), 334 (mw), 297 (s), 279(m), 254 (m), 232 (vvw), 224 (w), 209 (w) cm−1.Raman data: 2997 (w), 2902 (m), 1403 (vvw), 1233 (vw), 1057 (vw), 1047(vw), 1038(vw), 929 (vvs), 679 (vw), 585 (sh, w), 562 (m), 524 (sh, w), 506 (s), 477 (mw), 419(vvw), 337 (m), 297 (vs), 220 (m), 183 (w) cm−1.
4.10. Reaction of EtAsI2 with [nBu2Sn(S2N2)]2EtAsI2 (0.627 g, 1.75mmol) dissolved in dry CH2Cl2 (10ml) was added dropwise to astirred solution of [nBu2Sn(S2N2)]2 (0.572 g, 0.877mmol) in dry CH2Cl2 (10ml) andthe resulting orange mixture was gently re�uxed for 5 hours. The mixture was thenreduced to a third of its original volume and was transferred into a microdistillationapparatus. High vacuum was introduced, the rest of solvents was evaporated and theresidue was distilled (toil = 120 ◦C). Similarly to the preparation of MeAs(S2N2), gentleheating with a heating gun was used to help the vapours run over to the condenser. Thedistillation residue, an impure colourless oil was veri�ed by 1H NMR to be nBu2SnI2and was discarded. The distilled product, EtAs(S2N2), was slightly contaminated withnBu2SnI2. The distillate was therefore puri�ed by silica column chromatography (25 ×1.5 cm). Elution with hexane removed majority of nBu2SnI2 and toluene eluted the pureproduct which was collected to a Schlenk tube. The solvent was quickly removed fromthe eluate and the orange oily product was stored under nitrogen. EtAs(S2N2) is anorange, air-sensitive, oil which can be brie�y handled on air but must be stored underan inert atmosphere. It does not crystallise upon cooling. Yield 86mg (25%).MS(CI+TOF): m/z 196.92 (100%) [MH]+, 195.91 (35%) [M]+, 166.87 (65%) [AsS2N2]+,149.94 (6%) [EtAsSN]+, 136.92 (6%) [EtAsSH]+, 117.96 (20%) [EtAsN]+, 106.89 (13%)[AsS]+, 104.97 (4%) [EtAsH]+.1H NMR (CDCl3, 298 K): δ = 0.99 (dd, 3H, 3HX in CH3), 1.36 (m, 1H, HA in CH2),1.52 (m, 1H, HB in CH2); 2JHA−HB = 13.42Hz, 3JHA−HX = 7.59Hz, 3JHB−HX = 7.78Hz.13C NMR (CDCl3, 298 K): δ = 6.5 (s, 1C, CH3), 32.5 (s, 1C, CH2).14N NMR (CDCl3, 298 K): δ = 271.6 (s, 1N), 303.3 (s, 1N).IR data (neat liquid between CsI discs): 3145 (vvw, br), 3049 (vvw, br), 2959 (s), 2922
122

4. Experimental
(ms), 2900 (sh, mw), 2866 (m), 2819 (sh, vvw), 1451 (m), 1406 (w), 1373 (w), 1260(vvw), 1215 (sh, vw), 1200 (w), 1129 (sh, w), 1113 (w), 1081 (sh, mw), 1059 (vvs), 1021(mw), 964 (vw), 931 (vw), 876 (vvw), 799 (vw), 720 (sh, vw), 708 (vw), 675 (s), 611 (m),546 (vw), 522 (vw), 503 (w), 448 (vvw, br), 424 (sh, vvw), 398 (vw), 370 (mw), 346 (sh,vvw), 309 (sh, vvw), 299 (sh, vvw), 286 (vw), 237 (vw) cm−1.Raman data: 2955 (vw), 2923 (w), 2910 (sh, w), 2898 (sh, w), 2867 (vw), 1450 (vvw),1404 (vvw), 1373 (vvw), 1217 (vw), 1208 (vw), 1061 (vvw), 1004 (vvw), 932 (vvs), 787(vvw), 716 (vvw), 679 (vw), 612 (vvw), 548 (mw), 523 (m), 506 (vvs), 446 (vvw), 367(vvw), 342 (w), 317 (m), 302 (mw), 289 (m), 230 (vw), 179 (vvw) cm−1.
4.11. Reaction of iPrAsCl2 with [nBu2Sn(S2N2)]2iPrAsCl2 (0.4ml, 0.660 g, 3.50mmol) dissolved in dry CH2Cl2 (25ml) was added dropwiseat room temperature to a stirred solution of [nBu2Sn(S2N2)]2 (1.90 g, 2.91mmol) in dryCH2Cl2 (40ml). The mixture changed colour gradually from yellow to orange. After halfof the iPrAsCl2 amount was added, some solid started to precipitate. After the additionof iPrAsCl2 was �nished, the mixture was stirred at room temperature overnight. ABio-Beads column (27 × 1.5 cm) soaked in CH2Cl2 (reagent grade) and secured on topwith a glass wool plug was prepared. The resulting cloudy orange reaction mixture wasreduced in volume by 3
4and was �ltered through a short glass pipette with a small celite
plug directly onto the column. Elution with CH2Cl2 (not dry) was repeated three times.The byproduct, nBu2SnCl2, was always removed �rst as a yellowish band followed by awide orange band of the product. During the third elution the orange band was dividedinto two parts (the front 1
3and the remaining 2
3of the length of the band), which were
collected separately. The second part contained pure product. Evaporation of the solventleft the product, iPrAs(S2N2), as an orange, volatile and air-sensitive oil which did notcrystallise upon cooling. Yield 169mg (23%).Microanalysis: Found C, 16.8; H, 3.0; N, 13.3. Calc. for C3H7AsS2N2: C, 17.1; H, 3.3;N, 13.3%.MS(EI+TOF): m/z 209.93 (8%) [M]+, 166.87 (100%) [AsS2N2]+, 120.90 (1%) [AsSN]+,89.93 (3%) [AsN]+.1H NMR (CDCl3, 298 K): δ = 0.942 (d, 3H, CH3), 0.960 (d, 3H, CH3), 1.62 (qqrt, 1H,C−H).13C NMR (CDCl3, 298 K): δ = 15.1 (s, 1C, CH3), 15.7 (s, 1C, CH3), 38.2 (s, 1C, C−H).14N NMR (CDCl3, 298 K): δ = 272.3 (s, 1N), 303.4 (s, 1N).IR data: 2945 (m), 2923 (sh, mw), 2881 (w), 2859 (mw), 1460 (mw), 1453 (sh, w), 1441(sh, vw), 1402 (vvw), 1380 (vw), 1364 (vw), 1294 (vvw), 1261 (vvw), 1208 (w), 1156
123

4. Experimental
(vw), 1112 (sh, vw), 1059 (vvs), 997 (vw), 960 (vvw), 932 (vw), 868 (vw), 803 (vvw),706 (w), 678 (vs), 612 (ms), 601 (sh, mw), 547 (vw), 505 (mw), 439 (vvw), 418 (vvw),367 (mw), 337 (sh, vvw), 305 (vvw), 283 (vvw) cm−1.Raman data: 2948 (w), 2916 (w), 2882 (w), 2859 (w), 1465 (sh, vvw), 1456 (vvw), 1440(vvw), 1295 (), 1210 (w), 1158 (vvw), 1060 (vvw), 1000 (vvw), 934 (vvs), 869 (vvw), 710(vvw), 680 (vvw), 617 (vvw), 549 (vw), 508 (vvs), 447 (vvw), 411 (vvw), 367 (vvw), 345(sh, w), 337 (w), 312 (m), 291 (w), 274 (vw), 253 (w), 232 (vvw), 216 (vvw), 183 (vvw)cm−1.
4.12. Reaction of tBuAsCl2 with [nBu2Sn(S2N2)]2A solution of tBuAsCl2 (400mg, 1.97mmol) in dry CH2Cl2 (25ml) was added dropwiseto a stirred solution of [nBu2Sn(S2N2)]2 (643mg, 0.986mmol) in dry CH2Cl2 (25ml)and the mixture was stirred at room temperature for 7 hours. Meanwhile a Bio-Beadscolumn (27 × 1.5 cm) soaked in CH2Cl2 (reagent grade) and secured with a glass woolplug on top was prepared. The resulting orange and clear reaction mixture was reducedto a quarter of its original volume and was subjected three times to a size-exclusionchromatography using a Bio-Beads column (27 × 1.5 cm) in an identical way as in thecase of iPrAs(S2N2).
tBuAs(S2N2) was obtained as an orange, volatile, air-sensitive oil with a characteristicpungent old-sweat odour. It was stored under nitrogen in a freezer but no crystals wereformed. Yield 88mg (20%).Microanalysis: Found C, 22.7; H, 4.2; N, 10.1. Calc. for C4H9AsS2N2: C, 21.4; H, 4.0;N, 12.5%.MS(EI+TOF): m/z 223.94 (5%) [M]+, 166.86 (100%) [AsS2N2]+, 120.90 (5%) [AsSN]+,106.89 (3%) [AsS]+, 91.95 (2%) [S2N2]+, 57.07 (37%) [C4H9]+.1H NMR (CDCl3, 298 K): δ = 0.91 (s, 9H, tBu).13C NMR (CDCl3, 298 K): δ = 23.3 (s, 3C, 3×CH3), 41.4 (s, 1C, Cquart.).14N NMR (CDCl3, 298 K): δ = 272.6 (s, 1N), 303.7 (s, 1N).IR data: 2959 (sh, mw), 2942 (sh, m), 2929 (m), 2886 (w), 2855 (mw), 2710 (vvw), 1462(mw), 1441 (sh, vvw), 1408 (vvw), 1389 (vvw), 1362 (mw), 1261 (vvw), 1217 (sh, vvw),1203 (sh, vvw), 1164 (w), 1110 (vvw), 1097 (vvw), 1060 (vvs), 1011 (sh, vvw), 933 (vw),791 (vw), 701 (sh, vw), 679 (vs), 611 (m), 507 (w), 400 (vvw), 376 (sh, vvw), 362 (w),343 (vvw), 307 (vw), 297 (sh, vw), 278 (vvw), 260 (vvw) cm−1.Raman data: 2961 (sh, w), 2946 (sh, w), 2930 (mw), 2886 (mw), 2855 (mw), 2772 (vvw),2711 (vvw), 1463 (vvw), 1437 (vw), 1395 (vvw), 1220 (vvw), 1212 (vvw), 1204 (vvw),1165 (mw), 1061 (vvw), 1013 (vvw), 935 (vvs), 792 (w), 681 (vvw), 616 (vvw), 518 (sh,
124

4. Experimental
ms), 511 (vvs), 401 (vvw), 385 (vvw), 366 (vvw), 341 (w), 311 (m), 300 (sh, w), 266(vvw), 240 (w), 221 (sh, vvw), 182 (vvw) cm−1.
4.13. Reaction of PhAsCl2 with [nBu2Sn(S2N2)]2[nBu2Sn(S2N2)]2 (1.32 g, 2.0mmol) was dissolved in dry CH2Cl2 (100ml) in a 250ml �ask.A solution of PhAsCl2 (0.900 g, 4.0mmol) in dry CH2Cl2 (10ml) was added dropwiseupon stirring and the mixture was re�uxed for 8 hours. The solvent was evaporatedin vacuo and the residue was distilled under high vacuum (toil = 125�130 ◦C). Theorange/red distillate quickly solidi�ed forming colourless needles contaminated with anorange/red oil. 1H NMR con�rmed that the crystals were nBu2SnCl2. The distillate wasput aside.
The orange/brown distillation residue was dissolved in toluene and was added to apacked silica column (25 × 2 cm). Elution with toluene separated su�ciently the resid-ual nBu2SnCl2 (at the front) from the product, which was eluted as a yellow band.Evaporation of the solvent left an orange clear oil which solidi�ed in a freezer overnight.Repeated melting in warm water and swirling the �ask while cooling down led to crystalli-sation of the oil into well formed yellow prisms, which were identi�ed by X-ray di�ractionas PhAs(S2N2). Additional work-up of the distillate in an identical way provided up to10mg of pure PhAs(S2N2). Total yield 0.568 g (51%).Microanalysis: Found C, 29.6; H, 2.2; N, 10.9; S, 26.7. Calc. for C6H5AsS2N2: C, 29.5;H, 2.1; N, 11.5; S, 26.2%.MS(EI+TOF): m/z 243.90 (75%) [M]+, 197.93 (100%) [PhAsSN]+, 166.87 (40%)[AsS2N2]+, 165.96 (62%) [PhAsN]+, 106.90 (10%) [AsS]+, 77.04 (5%) [C6H5]+.1H NMR (CDCl3, 298 K): δ = 7.27�7.41 (m, 5H, phenyl group).13C NMR (CDCl3, 298 K): δ = 129.2 (s, 2C, meta-C), 129.5 (s, 2C, ortho-C), 131.0 (s,1C, para-C), 146.4 (s, 1C, C−As).14N NMR (CDCl3, 298 K): δ = 269.2 (s, 1N, ), 304.8 (s, 1N, ).IR data: 3055 (vvw), 3042 (sh, vvw), 3014 (vvw), 2963 (w), 2928 (vvw), 2857 (vvw),1563 (vvw), 1475 (vw), 1426 (mw), 1331 (vvw), 1302 (vw), 1261 (m), 1208 (vvw), 1180(vvw), 1154 (vvw), 1100 (m), 1068 (sh, m), 1052 (vvs), 1023 (sh, s), 995 (mw), 932 (w),916 (vw), 866 (vw), 817 (sh, m), 801 (ms), 743 (s), 689 (sh, m), 678 (s), 667 (sh, m), 602(m), 556 (sh, w), 499 (mw), 459 (m), 403 (vw), 361 (ms), 338 (mw), 308 (vvw), 295 (w)cm−1.Raman data: 3146 (vvw), 3067 (sh, vw), 3048 (w), 3027 (sh, vvw), 2951 (vvw), 1579(vw), 1479 (vvw), 1183 (vvw), 1159 (vvw), 1073 (vvw), 1066 (sh, vvw), 1055 (vvw), 1024(vw), 999 (vvs), 931 (s), 912 (sh, vvw), 692 (sh, vvw), 684 (vvw), 669 (vw), 617 (vvw),
125

4. Experimental
505 (ms), 368 (vvw), 344 (w), 306 (sh, m), 301 (m), 248 (vw), 220 (vw), 182 (vvw) cm−1.
4.14. Reaction of mesityldihalogenoarsines with[nBu2Sn(S2N2)]2
Molar amounts calculated for X = X′ = Cl. Solutions of MesAsXX′ (1.97 g, 7.44mmol) indry CH2Cl2 (60ml) and [nBu2Sn(S2N2)]2 (2.81 g, 4.32mmol) in dry CH2Cl2 (60ml) wereprepared and the solution of the arsine was added �uently, not necessarily dropwise,to the stirred solution of [nBu2Sn(S2N2)]2 at room temperature. The mixture turnedimmediately orange, the stirring was continued for 30 minutes at room temperature andthen the mixture was gently re�uxed for 6 hours. Afterwards, the volume of the mixturewas reduced to ca. 25ml and the concentrate was transferred to a predried 50ml �askattached to a microdistillation kit. The rest of the solvent was evaporated and theoily residue was distilled under high vacuum (0.3Torr, toil = 110�113 ◦C). The orangedistillate crystallised spontaneously into colourless needles contaminated with an orangeoil. The red/brown tarry distillation residue was partially dissolved in a su�ciently smallamount of the mixture toluene : petroleum ether (1:4) and was added to a silica column(27 × 2.5 cm) prepared in the same solvent system. The same solvent system was usedas eluant. The early yellow and orange bands were discarded and the wide red band wascollected to a Schlenk �ask. The solvents were evaporated under high vacuum and thedark red oil was additionally evacuated for 1 hour. The oil was stored under nitrogen andcrystallised spontaneously when allowed to stand at room temperature for 2 days. X-rayanalysis identi�ed the product as MesAs(S2N2). Yield 1.07 g (50%). M.p. 46�48 ◦C.Microanalysis: Found C, 37.3; H, 4.1; N, 9.4; S, 21.4. Calc. for C9H11AsS2N2: C, 37.8;H, 3.9; N, 9.8; S, 22.4%.MS(EI+TOF): m/z 285.96 (65%) [M]+, 239.98 (90%) [MesAsSN]+, 208.01 (60%)[MesAsN]+, 166.87 (35%) [AsS2N2]+, 151.06 (85%) [MesS]+, 119.08 (100%) [C9H11]+.1H NMR (499.9MHz, CDCl3, 298K): δ = 2.25 (s, 3H, CH3 (C 4)), 2.45 (s, 6H, 2×CH3(C 2,6)), 6.82 (s, 2H, Harom. (C 3,5)).13C NMR (CDCl3, 298 K): δ = 21.3 (s, 1C, CH3 (C 4)), 21.7 (s, 2C, 2×CH3 (C 2,6)), 130.5(s, 2C, C 3,5), 140.8 (s, 1C, C 1), 141.0 (s, 1C, C 4), 141.9 (s, 2C, C 2,6).14N NMR (CDCl3, 298 K): δ = 265.9 (s, 1N), 309.8 (s, 1N).IR data: 3016 (vvw), 2962 (w), 2910 (vw), 2859 (vvw), 2726 (vvw), 1597 (w), 1583 (sh,vw), 1545 (vw), 1463 (sh, vw), 1446 (sh, mw), 1440 (mw), 1405 (vw), 1372 (sh, vw),1367 (vw), 1288 (vw), 1262 (mw), 1243 (sh, vvw), 1176 (vvw), 1099 (mw), 1057 (vvs),1028 (ms), 954 (vvw), 932 (vw), 851 (m), 816 (sh, mw), 803 (mw), 707 (vw), 681 (s),626 (vvw), 602 (mw), 583 (w), 558 (vw), 543 (vw), 532 (sh, vvw), 494 (w), 405 (vvw),
126

4. Experimental
363 (mw), 340 (sh, vvw), 321 (vw), 290 (vvw), 277 (vvw) cm−1.Raman data: 3021 (w), 2966 (w), 2918 (ms), 2859 (vw), 2728 (vvw), 1601 (mw), 1587(sh, vvw), 1557 (vvw), 1467 (vvw), 1445 (vvw), 1437 (sh, vvw), 1379 (w), 1291 (mw),1276 (sh, vvw), 1241 (vvw), 1178 (vvw), 1060 (vvw), 1038 (vvw), 1012 (vw), 953 (sh,vvw), 932 (vvs), 852 (vvw), 684 (vw), 607 (vvw), 585 (vw), 561 (s), 544 (sh, vw), 534(vw), 503 (vvs), 369 (vw), 339 (mw), 321 (w), 295 (ms), 262 (vw), 242 (sh, vw), 207 (w),183 (vw) cm−1.
4.15. Reaction of PhAs(S2N2) with Pt(COD)Cl2A solution of PhAs(S2N2) (114mg, 0.470mmol) in dry CH2Cl2 (6ml) was prepared.In another �ask, Pt(COD)Cl2 (88mg, 0.235mmol) was dissolved in dry CH2Cl2 (4ml)and the solution of PhAs(S2N2) was added dropwise upon stirring. Neither a colour ortemperature change were observed, nor the characteristic odour of 1,5-cyclooctadienewhich would indicate its dissociation from the platinum complex. The mixture wasgently re�uxed for 1.5 hours which resulted in darkening of the mixture. The volatileswere evaporated and the black residue was separated on a silica column (20 × 1.5 cm)with CH2Cl2 being the eluant. Several coloured fractions were obtained but only twoof them contained multiplets in the aromatic region which corresponded very well withpure PhAs(S2N2). Remaining fractions contained signals of neither PhAs(S2N2) nor free1,5-cyclooctadiene. The fraction obtained by elution with methanol gave a 1H NMRspectrum showing very intense aromatic signals as well as the signals of coordinated 1,5-cyclooctadiene suggesting that the reaction did not occur. Recrystallisation attempts ofsome fractions were carried out with no success.
4.16. Methylation of PhAs(S2N2) with CH3IPhAs(S2N2) (174mg, 0.710mmol) dissolved in dry acetone (7ml) was treated with asolution of CH3I (101mg, 0.710mmol) in dry acetone (1ml) and the mixture was stirredat room temperature overnight. The 1H NMR spectrum of the mixture showed a newsinglet at 3.77 ppm which corresponds well with the chemical shift of CH2I2 and a singletat 1.23 ppm, the most intense signal of the spectrum, which can be assigned to the prod-uct. The 13C NMR spectrum also showed the signal corresponding to CH2I2 (53.8 ppm)and an intense singlet at 29.3 ppm corresponding to the possible product. Soon, yellowcrystals started to grow on the walls of the NMR tube which were identi�ed by X-rayanalysis as the unreacted starting material, PhAs(S2N2).
127

4. Experimental
4.17. Reaction of PhAs(S2N2) with[Mo(CO)4(piperidine)2]
PhAs(S2N2) (139mg, 0.569mmol) was added in one portion to a suspension of[Mo(CO)4(piperidine)2] (107mg, 0.284mmol) in dry and degassed CH2Cl2 (10ml) andthe mixture was stirred overnight at room temperature. The colour changed from yel-low to brown/orange. The solvent was evaporated under high vacuum to leave a tarryblack residue. TLC showed decomposition of the product on silica and therefore theproduct was precipitated from a concentrated CH2Cl2 solution by addition of diethylether (10ml) and petroleum ether (40ml). The dark brown precipitate was �ltered o�by suction, washed twice with petroleum ether and dried under high vacuum for 30 min-utes. Recrystallisation attempt by gas phase di�usion of hexane into a CH2Cl2 solutionresulted only in reprecipitation of the product, no crystals were formed. Mass spectrum(ES−TOF) showed a molecular peak at m/z 246.83 which could not be assigned to eitherany expected product, byproduct or unreacted starting material.
4.18. Reaction of PhAs(S2N2) with Cp2Ti(CO)2Cp2Ti(CO)2 (96mg, 0.409mmol) was dissolved in dry toluene (10ml) and a solution ofPhAs(S2N2) (200mg, 0.819mmol) in dry toluene (10ml) was added dropwise. With notemperature change or other visible change observed, the mixture was stirred at roomtemperature for 2 days at which point some �ne precipitate started to separate. Thestirring was stopped and the mixture was �ltered through a sinter. TLC of the �ltrateshowed several well de�ned spots and therefore the �ltrate was reduced in volume and wasadded to a silica column (15 × 1.5 cm) packed in toluene. Elution with toluene removedsome unreacted PhAs(S2N2) and the mixture CH2Cl2 : ethyl acetate (20:1) eluted a purpleproduct which in a 1H NMR spectrum gave two very close, doublet-like signals (6.26and 6.28 ppm). The product was dissolved in CH2Cl2 and was treated with hexane(gas phase di�usion) only to form a beige precipitate which gave a di�erent 1H NMRspectrum. Attempts to recrystallise this beige precipitate unfortunately resulted againonly in separation of a powder, no crystals were formed.
4.19. Reaction of PhAs(S2N2) with Cp*Co(CO)2To Cp*Co(CO)2 (77mg, 0.307mmol) dissolved in dry toluene (12ml) a solution ofPhAs(S2N2) (150mg, 0.614mmol) in dry toluene (8ml) was added dropwise and themixture was stirred at room temperature for 2 days. The resulting mixture was �ltered
128

4. Experimental
leaving a black precipitate on the sinter and a dark wine-red �ltrate. The �ltrate wasreduced in volume and was passed through a silica column (20 × 1.5 cm) packed inCH2Cl2. The elution with CH2Cl2 removed unreacted PhAs(S2N2) and the mixture ace-tone : hexane (1:2) eluted a purple band which was collected and evaporated to dryness.Analysis by 1H NMR and MS con�rmed that the product is Cp*Co(S2N2).MS(ES+TOF): m/z 286.96 (100%) [MH]+.1H NMR (CDCl3, 298 K): δ = 1.96 (s, 15H, Cp*).
4.20. Reaction of PhAs(S2N2) with sulfurPhAs(S2N2) (114mg, 0.466mmol) was dissolved in dry toluene (10ml), sulfur �owers(15mg, 0.058mmol) were added in one portion and the mixture was re�uxed for 6 hours(toil = 130 ◦C). Afterwards, the mixture still hot was �ltered through a sinter and theorange/yellow �ltrate was allowed to cool down to room temperature. When no crystalsappeared upon cooling, the �ltrate was reduced to a quarter of its original volume,heated to 50 ◦C, allowed to cool down to room temperature and was placed to a freezer.Overnight well shaped yellowish crystals were formed which were identi�ed by X-rayanalysis as PhAs(S2N2).
129

Part III.
Transition Metal Sulfur-NitrogenRing Compounds
Abstract
This part of the thesis deals with organometallic sulfur-nitrogen complexes with theemphasis on �ve-membered rings, in which a metal centre is coordinated by the [S2N2] 2 �
ligand. The topic is introduced by a short literature survey. The main body of this partcontains a contribution to the chemistry of the popular pentamethylcyclopentadienylhalf-sandwich complexes. The series of the known complexes Cp*M(S2N2) (M = Co, Ir) iscompleted with the preparation of the missing rhodium analogue and with the previouslyunpublished X-ray structure of Cp*Co(S2N2). The experimental characterisation of thetitle compounds is compared to the results of theoretical calculations.

5. Introduction
Metal complexes of sulfur-nitrogen ligands form a signi�cant part within the vast areaof sulfur-nitrogen chemistry. The adducts S4N4 ·MCl4 (M = Ti, Sn) and S4N4 ·MCl5(M = Mo, Sb) have been known since 1906319,320 but a rapid development was notachieved until the 1980's. A wide range of four- to eight-membered ring compoundshave been prepared. Although the development has steadied in the last two decades,the interest in sulfur-nitrogen ligands persists. The knowledge has been summarised inseveral reviews and books2,3,321,322 and therefore only a brief account is presented in thischapter.
5.1. Four-membered ringsThe famous sulfur-nitrogen heterocycles have been prepared in several forms. For ex-ample in addition to S4N4 also S4N4H4 and S4N4O2 are known and their properties andreactivity are often compared. The same can be said about several metal-sulfur-nitrogenheterocycles, especially the �ve-membered ones (vide infra). An example of an unsubsti-tuted four -membered MSN heterocycle has not been isolated so far, which is probablythe reason why four-membered MSN ring compounds have been mostly omitted fromreviews.2,3,321,322
A range of four-membered MSN heterocycles with exocyclic substituents on nitrogenor on both nitrogen and sulfur were reported. In general, a sulfur-nitrogen ligand canbe coordinated to a metal centre in four ways (Fig. 5.1). Only the coordination mode A
NM
NS
SM
NN
SM
SN
SM
NS
A B C D
Fig. 5.1. A schematic chart of possible coordination modes of the N2S andNS2 ligands onto a metal centre. The bonds demonstrate connec-tivity, not a single bond.
has been described in the literature to date. A large number of rings of this type have
131

5. Introduction
been prepared since the early 1970's and thus the literature background of this area isbroad.
The bidentate S-N ligand can be attached to the metal centre in the form of an anionor as a neutral species. The coordination occurs through the lone pairs on the donornitrogen atom.
The anionic ligands are best prepared by deprotonation of an amido group by a strongbase or with the help of catalysts. In this way metallacycles containing Pt, Al and Tacentres were prepared (Scheme 5.1).323�327
NH
SNH
O O
R'R'
PtNH
S
HN O
O
R3P
R3PPt
NS
N OO
R3P
R3P
TaN
SN O
O
Me2N
Me2N
NS
NAl-
NS
N
Me2N
(PR
3)P
tCl 2
NH
3 (l)
(COD)PtCl2
PR3
Ag2O
CH2Cl2
(COD)PtCl2PR3
KOH
CH2Cl2 / THF
MAlH4 (M = Li, Na)
THF
Ta(N
Me
2 )5
- 2 Me
2 NH
O
O
O
O
Ph
Ph
PtN
SN O
O
R3P
R3P
Ph
Ph
tBu tBu
tButBu
tBu
tBu
[M+]
R' =
H
R' = Ph
R' = Ph
R' = tBu
R' =
tBu
Scheme 5.1 Preparation methods of four-membered ring complexes with anionic NSN ligands
The insertion of SO2 into the N−−N bond of an azobenzene molecule in the iron carbonylcomplex 36 is also interesting. The resulting complex 37 contains a bridging S-N ligand
132

5. Introduction
that forms two FeNSN four-membered rings with the Fe centres (eq. 5.1).328
(CO)3Fe Fe(CO)3
N NPh Ph
+ SO2hexane
r.t.(CO)3Fe Fe(CO)3
N NPh PhS
O O
36 37 (5.1)
The electroneutral molecules of sulfurdiimides R−N−−S−−N−R are found as both mono-and bidentate ligands in a wide range of transition metals complexes. The RNSNRmolecule is attached to the metal either as an adduct or as a substitution for the originalligands. The formation of a four-membered MNSN ring is determined by steric factors.Sulfurdiimides bearing the bulky iPr or tBu groups form the ring whereas those with Meor Et groups coordinate in a monodentate mode.
The preparative routes involve a simple mixing of the reactants in a suitable solventand stirring at room or elevated temperature. Some examples are shown in equations5.2 and 5.3.329,330
ML4 + tBuNSNtBupentane
r.t.L4M
NS
N
tBu
tBuM = Ti, Zr. L = Cl.
ML4 = VOCl3 (5.2)
[M(CO)4(MeCN)2] + tBuNSNtBuhexane
r.t.(CO)4M
NS
N
tBu
tBuM = Cr, Mo, W
+ 2 MeCN
(5.3)
The X-ray structure of [(CO)4W(tBuNSNtBu)] (eq. 5.3) con�rmed the existence of aplanar four-membered MNSN ring in the complex.331
Other metals forming four-membered ring complexes with sulfurdiimides as NSN li-gands are Mn, Re, Rh or Ir.332,333 Noteworthy is the adduct of N,N ′-bis(trimethylsilyl)-sulfurdiimide with SnCl4 (eq. 5.4).334
SnCl4 + Me3SiNSNSiMe3
SO2 (l)Sn
Cl
Cl
NCl
N
Cl
S
SiMe3
SiMe3
(5.4)
133

5. Introduction
On one hand SnCl4 is known to form complexes, of which especially those with thecoordination number 6 on the Sn atom are well established.78 On the other hand, thepropensity of the Me3Si group to couple readily with halogens has been utilised in count-less synthetic procedures and a similar reaction path could be expected when SnCl4 istreated with Me3SiNSNSiMe3. However, suitable reaction conditions - low temperatureand polar aprotic solvent (liquid SO2) - enable the formation of SnCl4 ·Me3SiNSNSiMe3,which exhibits thermal stability at room temperature.334
5.2. Five-membered ringsFive-membered MSN rings attracted a great attention in the past, mainly because it wasbelieved that they might be incorporated in superconductors. The chemistry of the �ve-membered MSN complexes thus developed rapidly and is probably the most explored ofall the MSN ring compounds.
A wide range of complexes are known where the bidentate ligands are formed by theanions (S2N2) 2 � , (S2N2H) � , (S3N) � or by the (NSSN) moiety. A compound containingthe hypothetical (SN3) � anion has not been reported yet. The complexes are preparedby a reaction of simple metal salts or metal complexes with a suitable source of the SNligand. The original preparative procedures utilised the explosive S4N4 but over the yearsmany alternative routes were developed, the most revolutionary of which are without adoubt the reactions of a metal complex with a reactive SN species prepared in situ inliquid ammonia. The following sections will give a brief insight into the great diversityof preparation methods.
5.2.1. M(S2N2) complexes containing the (S2N2) 2 � anion5.2.1.1. Preparation from S4N4 or S2N2
S4N4 in liquid ammonia. Historically the �rst metalladithiadiazole was prepared fromPbI2 and S4N4 in liquid ammonia.335 The equation expressing the exact course of thereaction has not been reported yet but equation 5.5 can be suggested on the basis ofwhat is known about the behaviour of SN species in liquid ammonia.5,6 The product,[Pb(S2N2)] ·NH3, is a stable species but upon removal of the coordinated molecule ofammonia (80�90 ◦C, 10−4Torr) Pb(S2N2) is formed, which is explosive.336
2 PbI2 + 3 S4N4 + 10 NH3NH3 (l)
2 [(NH3)·Pb(S2N2)] + 4 NH4I +
+ 2 [S4N5]- + 2 [NH4]+ (5.5)
134

5. Introduction
Oxidative addition. More often, S4N4 is used at room temperature in reactions withcomplexes containing a low valent metal centre. It was proposed that in such reactionsthe metal centre promotes the cleavage of the S4N4 cage into two reactive S2N2 moieties,which undergo a quick oxidative addition onto the metal centre to form a �ve-memberedM(S2N2) ring.322 In this way Co,337,338 Ir,339 Pd and Pt340�342 or Sn343 complexes wereprepared. Some examples are shown in eq. 5.6 and 5.7.341,343
(PPh3)2MCH2
CH2+ S4N4 [(PPh3)2M(S2N2)]2 + 2 C2H42
M = Pd, Pt (5.6)
2 R2Sn + S4N4 2 R2Sn(S2N2)
CF3
CF3
F3CR =
(5.7)
The metal carbonyls Ni(CO)4 and Co2(CO)8 react with S4N4 only to form dark amor-phous solids of the approximate formula M(NS)4.344,345 The same results from a reactionbetween S2N2 and Raney-Ni, Raney-Co or vapours of elemental nickel (Scheme 5.2).346,347
The solids have not been characterised but it is believed they have a polymeric structure[M(NS)4] x.344,347 On treatment with protic solvents or traces of water in organic solventsthe polymer rearranges into a mixture of complexes M(S2N2H)2, M(S2N2H)(S3N) andM(S3N)2.347�351
[M(SN)4]xS2N2
N
SHN
MS
S
N S
HN
Ni (g)
Raney-M
MeOH
M = Co, Ni
+S
NSM
S
S
N S
HN
+
S
NSM
S
N
S S
S+
Scheme 5.2 Reactivity of metals with S2N2
5.2.1.2. Preparation from other SN rings
S4N4H4 or [S3N2Cl]Cl can also be used in oxidative additions, although the preparationof Ph3Sb(S2N2) is the only example reported for such use of [S3N2Cl]Cl (eq. 5.8).342,352,353
On the contrary to S4N4, S4N4H4 can enter substitution reactions also with complexescontaining a metal in higher oxidation states. Such reactions are enabled by the presence
135

5. Introduction
of a base such as DBU (eq. 5.9).354
5 SbPh3 + 2 [S3N2Cl]Cl Ph3Sb(S2N2) + 2 Ph3SbS +
+ 2 Ph3SbCl2 + 1/2 S4N4
CH2Cl2r.t.
(5.8)
2 (PR3)2MCl2 + S4N4H4 + 4 DBU 2 [(PR3)2M(S2N2)] + 4 DBU·HClCH2Cl2
r.t.
M = Pd, Pt (5.9)
Na[S3N3]. A series of Pd and Pt complexes was obtained by the reaction between[(PR3)2PtCl2] and Na[S3N3] (eq. 5.10).6 Similarly to S4N4, Na[S3N3] is explosive andtherefore has not been used very frequently.
(PR3)2MCl2 + 2 Na(S3N3) [(PR3)2M(S2N2)] + S4N4 + 2 NaClEtOH / CH2Cl2
r.t.
M = Pd, Pt (5.10)
Reactions in liquid ammonia. To avoid handling of explosive materials, reactive SNspecies can be conveniently prepared from sulfur or its compounds in liquid ammonia,which serves as both a reagent and a solvent. The (S2N2) 2 � anion is formed when liquidammonia reacts with [S4N3]Cl, [S3N2Cl]Cl or SCl2.5,6 In a subsequent reaction with ametal complex the (S2N2) 2 � anion replaces two anionic ligands coordinated to the metalcentre. In this method most usually metal chlorides are used as reactants (eq. 1.3�1.5and 5.11�5.12).7,8,323
2 [S3N2Cl]Cl + 8 NH3 (S2N2)2- + S4N4 + 4 NH4Cl + 2 [NH4]+NH3 (l)
(5.11)
(PR3)2PtCl2 + 2 SCl2 + 8 NH3 [(PR3)2Pt(S2N2)] + 6 NH4ClNH3 (l)
(5.12)
The reported examples of the M(S2N2) complexes prepared in liquid ammonia containPt7,323 or Sn8 metal centres. The relatively low variety of products is counterbalancedby the importance of the R2Sn(S2N2) compounds, which are excellent transmetallatingagents leading to a wide range of M(S2N2) complexes (vide infra). The signi�cance ofthe reactions of sulfur compounds in liquid ammonia lies in the possibility to scale upthe quantities of the reactants with virtually no risk of explosion.
136

5. Introduction
5.2.1.3. Transmetallation reactions
The transmetallation reactions using the tin reagents with the general formulaR2Sn(S2N2) are well established. A simple exchange of ligands between the tin reagentand metal halogenocomplexes results in formation of a number of M(S2N2) complexes(eq. 5.13 and 5.14). The driving force of the transmetallation is the readiness of tin toform bonds with halogens. The reported examples are complexes of Sn,355 Ru, Os,270
and Ir.8
[Me2Sn(S2N2)]2 + 2 SnCl4 [Cl2Sn(S2N2)]2 + 2 Me2SnCl2CH2Cl2reflux (5.13)
[Cp*IrCl2]2 + [nBu2Sn(S2N2)]2 2 Cp*Ir(S2N2) + 2 nBu2SnCl2CH2Cl2
r.t. (5.14)
5.2.1.4. Deprotonation of M(S2N2H) complexes
M(S2N2) complexes can be prepared by deprotonation of the M(S2N2H) complexes witha suitable base (eq. 5.15) or by a proton exchange with another M(S2N2) complex (eq.5.16).309,356,357
2 Ni(S2N2H)2 MeOH, r.t.
N S
NS
SN
SNi
N
S N
SN
NS
NNi
SNi[Ph4P]2 + 2 [Ph4P]Cl + 10 H2O
+ NiCl2 · 6H2O
+ 4 [Ph4P][OH]
(5.15)
BrPt
R3P
HN
S
S
N
R3PPd
R3P
N
S
S
N
BrPd
R3P
HN
S
S
N
R3PPt
R3P
N
S
S
N
[BF4]
[BF4]
+
+
CH2Cl2
PR3 = PPhMe2 PR3 = PPhMe2
(5.16)
137

5. Introduction
5.2.1.5. Preparation from acyclic SN compounds
Five-membered M(S2N2) ring compounds are prepared also from metal complexes andsulfurdiimides (eq. 5.17),358,359 S(NSO)2 (eq. 5.18),342 and are formed in minor quantitiesas byproducts of reactions involving other MSN complexes.359
S
NSnMe3
NSnMe3
2 + 4 MeLi
- 4 SnMe4S
NLi
NLi
+ 2 tBu2SnCl2- 4 LiCl2
NSn
N
S
NSn
N
S
tBu tBu
tBu tBu
+ 1/4 S8
[tBu2Sn(S2N2)]2 (5.17)
(PPh3)2PtCH2
CH2+ S(NSO)2 (PPh3)2Pt(S2N2) + C2H4
toluene, r.t.- SO2 (5.18)
The �nal remark in this section is dedicated to a theoretical study on RAl(S2N2) (R= H, Cl, CH3), which was published recently. The calculated geometries describe thecompounds as conventional M(S2N2) �ve-membered rings with one single S−N bondand two S−N bonds with double character. The calcualted bond lengths and bondorders indicate delocalisation of π-electrons of the [S2N2] 2 � moiety and negative valuesof NICS(0) and NICS(1) suggest aromaticity of the compounds, which means a certaindegree of stability once prepared. However, it must be noted that the Al analogueswere the least aromatic of the whole set, which involved also the analogous B, C and Sicompounds.31
5.2.2. M(S2N2) complexes containing the (NSSN) moietyA �ve-membered ring complex of rhenium containing the (NSSN) 4 � anion is the onlypublished example of such a metallacycle. The complex 38 is formed according to eq.5.19.360 It is a structural isomer of the more conventional [S2N2] 2 � ligand with the SNSN
138

5. Introduction
sequence.
+ PPh3CH2Cl2
0 ºCRe
Cl
ClCl
NSClCl3PONSCl
Re
Cl
ClCl
NClN
S
S[PPh3Cl]+
-
+ POCl3
38 (5.19)
The mechanism of this interesting rearrangement has not been elucidated so far. A seriesof analogous complexes were obtained from the salts of the [Cl4Re(NSCl)2] � anion,361
but this area of sulfur-nitrogen metallacycles has not developed further.
Adducts. Adducts of suitable electroneutral compounds with metal complexes or saltsalso contain a �ve-membered M(NSSN) ring. Although the possible range of such com-pounds is wide, only the adducts of N, N ′-dithiodimorpholine with CoCl2 and FeCl2 werereported (eq. 5.20).362
acetoneMCl2 +
ON
ON
S S
ON
ON
S S
MCl Cl
M = Fe, Co (5.20)
5.2.3. M(S2N2H) ring complexesThe majority of the preparation methods leading to the unprotonated M(S2N2) com-pounds can be used also for the synthesis of the protonated M(S2N2H) complexes. Theoutcome of the reaction depends mainly on reaction conditions and the type of the metalatom involved.
5.2.3.1. Preparation from S4N4
The reaction of metal salts with S4N4 results in the formation of a mixture of complexesconsisting of the M(S2N2H) as well as M(S3N) rings (M = Co, Ni, Pd and Pt) (Scheme5.3).348,350,363 Typically, the syntheses were carried out under re�ux in dry methanol.According to Piper349 it is the solvent that provides the protons as the same reactionsperformed in CS2 or C6H6 gave no product (see also sections 5.2.1 and 5.2.4).The risks associated with the use of S4N4 in re�uxing solvents can be avoided by activa-tion of S4N4 by UV radiation at room temperature.364
139

5. Introduction
S4N4
(M = Co, Ni)
MeOH, reflux
MCl2
MeOH, reflux
PdCl2 + NH4Cl
MeOH, reflux
Na2PtCl6 + Na2S2O4
N
SHN
MS
S
N S
HN
+S
NSM
S
S
N S
HN
+S
NSM
S
N
S S
S
Scheme 5.3 Preparation of M(S2N2H)2 complexes from S4N4
5.2.3.2. Protonation of M(S2N2) complexes
The vast majority of Co, Pd and Pt M(S2N2H) complexes was prepared by protonationof the corresponding M(S2N2) complexes with HBF4 or HCl.354,365�367 An interestingprotonation path was observed in the reactions of palladium complexes with [S4N3]Cl inliquid ammonia.7 The primary product is an ammonium salt of Pd(S2N2) complex thatdisproportionates into Pd(S2N2H) complex and free ammonia (eq. 5.21).7
NH3 (l)
[S4N3]Cl / NH3 (l)
N S
NHPd
SN
C ClPd
Cl
N
CPd
CH3
N N
CH3
CH3
N C
N S
NPd
S
N C
[NH4] - NH3
C N =
vacuum
(5.21)
M(S2N2H) complexes were also found as byproducts in reaction mixtures involving otherMSN ring compounds.368,369
5.2.4. M(S3N) ring complexes5.2.4.1. Preparation from [S7N] �
Five-membered M(S3N) rings are most conveniently prepared by the reaction of binarymetal salts and S7NH or its salts. Reactions with S7NH are generally carried out in thepresence of a base, which deprotonates the imido-group and the generated [S7N] � aniondissociates into [S3N] � and S8 (eq. 5.22).366 In this way a wide range of Fe,370 Ru,371,372
Ni,373,374 Pd,7,366,375 Pt,376 Cu374,377,378 and Au379 complexes were prepared.
2 [S7N]- 2 [S3N]- + S8 (5.22)
140

5. Introduction
5.2.4.2. Desulfuration of [S4N] �
The [S3N] � anion can be generated in situ by desulfuration of the [S4N] � anion withPPh3.380�382 [S3N] � prepared in this way reacts with the salts of Co, Ni, Cu and Ag togive the corresponding �ve-membered M(S3N) ring complexes (eq. 5.23).381,383
MeCN, r.t.
MeCN, r.t.
2 [S4N]- + 2 PPh3 2 [S3N]-
M(S3N)2 + 2 X-
- 2 Ph3PS
+ MX
M = Co, Ni; X = BrMeCN, r.t.
+ MX2
[M(S3N)2]- + X-
M = Cu; X = ClM = Ag; X = NO3 (5.23)
5.2.4.3. Preparation from other SN compounds
Possible sources of the [S3N] � chelating anion are also S4N4, S2N2 or [S4N3]Cl. In sections5.2.1 and 5.2.3 it was shown that in reactions of elemental metals with S2N2, metal M 0
complexes with S4N4, as well as metal binary salts with S4N4 a mixture of M(S2N2H)2,M(S2N2H)(S3N) and M(S3N)2 (M = Co, Ni, Pd, Pt) complexes is formed, which canbe easily separated on preparative TLC.348,350,384,385 On silica, the M(S2N2H)(S3N) com-plex undergoes a disproportionation into the complexes M(S2N2H)2 and M(S3N)2 (eq.5.24).348,350,384
2 [M(S2N2H)(S3N)] M(S2N2H)2 + M(S3N)2
M = Co, Ni, Pd, Pt (5.24)
Both Pt(S2N2H) and Pt(S3N) complexes were formed also when (PR3)2PtCl2 was re-�uxed with S4N4 at 160 ◦C in xylene367 or when the two reactants were allowed to reactat room temperature under a UV lamp.364
E�cient formation of [S3N] � from [S4N3]Cl was observed in the presence of a base([Me4N]OH) but details about a possible mechanism were not given.386
5.2.5. Structure of the �ve-membered MSN ringsIn all the above ring compounds the MSN ring moieties were essentially planar. The metalcentres Co, Ni, Pd and Pt in the M(S2N2H)2 complexes are found in an approximatelysquare planar coordination environment as well as palladium in Pd(S3N)2.387�391 The cop-per atom in the [Cu(S3N)2] � anion is in a tetrahedral coordination.383 A few complexesof Pt and Pd showed stacking of the �ve-membered MSN rings in crystals.365,392,393
Crystal structure re�nement of the Re(NSSN) complex 38 resulted in a model contain-ing two separated thionitrosyl ligands with the S−S distance of 2.53(2)Å.360 However,
141

5. Introduction
theoretical calculations suggested that coupling of two thionitrosyl ligands via their sul-fur atoms is possible and that there is a bonding interaction between the two sulfuratoms.394
5.3. Six-membered ringsThe [S3N2] 2 � , [S2N3] � , [S2N3] 3 � and [N2S2N(SO2NH2)] 2 � anions are known to act asbidentate ligands and to form six-membered MSN rings with metal centres.
5.3.1. M(S3N2) ring complexesThe SNSNS sequence. Palladium is the only metal to form complexes with the[SNSNS] 2 � anion. In all of the reported examples the anion forms a planar bridgingligand with each of the two terminal sulfur atoms coordinated to two metal centres. Thecomplexes are prepared predominantly from halogenocomplexes of palladium with S4N4or S(NSNSiMe3)2 (eq. 5.25)359,395,396 but complex 39 was found among the products ofthe reaction between PdCl2 and S4N4 in methanol (eq. 5.26).397
2 [Pd2X6]2-CH2Cl2, r.t.
SPdPd
S
XX
XX
N NS 2-
NS
NS
PdPd
X
X
X
X
X
X
2-
+S4N4
(5.25)
MeOHS
PdPdS
S
S
S
S
N NS
S4N4PdCl2
NS
S
N
+ Pd(S2N2H)2 + Pd(S3N)2 +
+ Pd(S2N2H)(S3N)
39 (5.26)
The SSNSN sequence. When Cp2Ti(CO)2 reacts with S4N4 at room temperaturean eight- and a six-membered ring are formed (eq. 5.27).398 The six-membered ring ispuckered and is organised in an SSNSN sequence with the terminal sulfur and nitrogenchelating the Ti centre. To date no other complex with analogous structure has been
142

5. Introduction
reported.
Cp2Ti(CO)2 + S4N4THFr.t.
SN
S
NS
N
Ti
N
Cp
CpN S
N
SS
TiCp
Cp+
(5.27)
The NSSSN sequence. The only example is the copper complex 40, which was pre-pared by the reaction of CuCl2 and bis(oxamido)trisulfane (eq. 5.28).399 The S-N-Cu-N-Sfragment is planar while the central sulfur atom is situated out of that plane.
CuCl2 +HN S
S
SNH
OH2N
O
O
O
H2N
- 2 HClCu
OO
N
N
S
S
SH2N
H2N
O
O
40 (5.28)
5.3.2. M(S2N3) ring complexes bearing the (S2N3) � anionOnly palladium complexes have been reported to date. The �rst complex was preparedfrom dipalladium hexachlorate and the basket-like compound S5N6 (eq. 5.29).400 Lateranother method was developed using the safer S(NSNSiMe3)2 instead of S5N6 which ismore explosive than S4N4 (eq. 5.30).359
[PPh4]2[Pd2Cl6] + S5N6 2 [PPh4] PdN
Cl
Cl
N
S
SN
CH2Cl2r.t.
(5.29)
[Pd2X6]- + S(NSNSiMe3)2 PdN
X
X
N
S
SN
CH2Cl2r.t.
+
-
SPdPd
S
XX
XX
N NS 2-
X = Cl, Br (5.30)
5.3.3. M(S2N3) ring complexes bearing the (S2N3) 3 � anionThis is the most studied type of six-membered MSN rings. In 1983 the vanadium com-plex Cl2V(S2N3) became the �rst published example401 and a number of V, Mo andW complexes soon followed.402�413 The last metal reported to form a complex with the(S2N3) 3 � anion was rhenium in 2000.414
143

5. Introduction
Most frequently S4N4 or S3N3Cl3 are used for the generation of the (S2N3) 3 � anion withtheir reaction partners being metal halogenides, oxyhalogenides, carbonyls or salts. Thepreparations are usually carried out in halogenated solvents but more extreme conditionssuch as a reaction in liquid bromine407 or in an S3N3Cl3 melt406 were also reported. Someexamples are shown in equations 5.31 and 5.32.401�404
MCln + S4N4CH2Cl2
r.t.[ClnM(S2N3)]2
M = V; n = 4M = Mo; n = 5M = W; n = 6
M = V; n = 2M = Mo; n = 3M = W; n = 3 (5.31)
MECln + S3N3Cl3 [ClnM(S2N3)]2
M = V; E = O; n = 3M = Mo; E = N; n = 3M = W; E = O, S; n = 4
M = V; n = 2M = Mo; n = 3M = W; n = 3 (5.32)
The protonated ligand (S2N3H) 2 � was observed in the [Cl3WO(S2N3H)] � anion, whichresulted from partial hydrolysis of [Cl4W(S2N3)] � .409
The rhenium metallacycle Re(S2N3) forms a part of a complex anion 41 (Fig. 5.2),which was prepared from [Cl4Re(NSCl)2] � and N(SiMe3)3.414 The monomeric units con-sist of the hypothetical [Cl2Re(S2N3)2] � complex, in which one sulfur atom is replacedby ReCl3 moiety.
ReN
NCl
Cl
SN
S NN
S
Re
Cl
N
Cl
N Re N
Cl
Cl Cl
ReN
S
Cl
Cl
N
N
S
SN
Cl
41
2-
Fig. 5.2. The structure of [Cl2Re(S2N3)(Cl3ReSN3)] 2 �2
5.3.4. M(S2N3) ring complexes bearing the [N2S2N(SO2NH2)] 2 �anion
The reported examples are a series of Pt complexes and one Pd compound.324,415 Theywere prepared from chlorocomplexes (PR3)2MCl2 by a substitution of the Cl � anionsfor the bidentate [N2S2N(SO2NH2)] 2 � anion, which was generated from S4N4O2 in liquid
144

5. Introduction
ammonia (eq. 5.33).324
(PR3)2MCl2 + S4N4O2 + 3 NH3NH3 (l)
MN
NR3P
R3P S
SN
SOO
NH2
- NH4Cl
M = PtM = Pd; (PR3)2 = dppe
(5.33)
5.4. Seven-membered ringsAn unsubstituted seven-membered MSN ring complex has not been synthesised yet.However, a series of tin complexes containing the Sn(S2N4) ring was reported. Thecompounds were prepared by oxidation of cyclic bis(amido)stannylenes with N, N ′-di-alkylsulfurdiimides (eq. 5.34).416
tBuN
Me2SiNtBu
Sn
RN SNR
NRSRNtBu
NMe2Si
NtBu
Sn + 2 S
NR
NR
R = Me, Et,nPr, nBu (5.34)
The Sn centre is coordinated only by nitrogen atoms and is in a tetrahedral environment.The M(S2N4) ring is puckered and consists of two sulfurdiamido groups attached to theSn centre on one side and connected with each other through nitrogen atoms on theother.
5.5. Eight-membered rings
5.5.1. Complexes of the (S3N4) 2 � anionAs was mentioned earlier, the reaction between Cp2Ti(CO)2 and S4N4 produces the eight-membered ring complex Cp2Ti(S3N4), in addition to the six-membered Cp2Ti(S3N2) (eq.5.27).398 The eight-membered ring consists of alternating S and N atoms and the Cp2Timoiety takes the position of one sulfur eliminated from S4N4. The ring is only very slightlypuckered. No other complexes bearing the (S3N4) 2 � anion have been synthesised to date.
145

5. Introduction
5.5.2. Complexes of the (S4N3) � anionThe only example in this class of compounds is the complex ClPt(S4N3) prepared bydisplacement of two benzonitrile molecules and a chloride anion from Cl2Pt(NCPh)2 byS4N4 (eq. 5.35).417
Cl2Pt(NCPh)2 + S4N4toluenereflux
SN
PtS
NS
N
S
Cl
+ 2 PhCN + S2+ + Cl-
(5.35)
The planar (S4N3) � anion coordinates the Pt centre as a tridentate ligand, which resultsin the formation of a bicyclic compound. Platinum is in a square planar environment.
5.6. Nine-membered rings
5.6.1. Monocyclic nine-membered ringsOne monocyclic nine-membered ring has been reported - the product of an insertion ofTi centre into the S7NH molecule. Cp2Ti(CO)2 reacts with S7NH at room temperatureto give the complex Cp2Ti(S7NH) (eq. 5.36).418
Cp2Ti(CO)2 + S7NHhexane
r.t.+ 2 CO
NH
SS
S
SSS
Ti
SCp
Cp
(5.36)
It is interesting that only one structural isomer is formed in this reaction. The Cp2Timoiety is inserted only in such position that the Ti and N atoms are bridged by µ-S5 andµ-S2 sulfur chains. No other structural isomer was observed in the mixture. When theproton on nitrogen is substituted with a methyl group, again only one structural isomeris formed, this time in the µ-S4 and µ-S3 arrangement. A rationale for this behaviourhas not been o�ered.418
5.6.2. Insertion of a metal centre in S4N4
The products of metal complexes insertions into S4N4 represent a famous and well stud-ied group of compounds. Hypothetically, an insertion of a metal centre into a moleculeof S4N4 should result in the formation of a nine-membered MSN ring but such productwould be most probably very unstable. Instead, the insertion results in a disruption
146

5. Introduction
of the S4N4 cage and gives rise to the (S4N4) 2 � anion, which coordinates the metalcentre as a tridentate ligand. Thus, the reaction of S4N4 with IrCl(CO)(PPh3)2 leads
S
N
Ir
Ph3P
S S
Cl
N
CO
N NS
S
N
Pt
Cl
S S
Cl
N
Cl
N NS
[PPh4]+
42 43
Fig. 5.3. The structures of complexes 42 and 43
to the formation of complex 42 in which the (S4N4) 2 � anion is in facial coordination(Fig. 5.3).419,420 Platinum can be inserted into S4N4 with the same result as was demon-strated by compound 43 produced by the reaction of K[PtCl3(C2H4)] with S4N4 followedby metathesis with [PPh4]Cl. The structure of 43 was determined by 14N and 15N NMRspectroscopy. The crystal structure has not been solved and therefore the S−−N doublebonds in Fig. 5.3 are drawn in positions suggested according to analogy with 42.421
The dimer [PtCl2(PMe2Ph)]2 reacts with S4N4 to give compound 44 with the (S4N4) 2 �
anion coordinated meridionally (Fig. 5.4).422 44 slowly isomerises in CH2Cl2 solution togive complex 45, the structure of which was suggested after thorough NMR studies.423
In 45 the coordination of the SN anion is facial and occurs through two nitrogens andone sulfur whereas in 42, 43 and 44 the ligand was attached through two sulfurs andone nitrogen. Since the crystal structure of 45 has not been determined, the positionsof the S−−N double bonds in Fig. 5.4 are suggested on the basis of the comparison withthe structure of the bicyclic complex ClPt(S4N3) (page 146).
PtMe2PhP
Cl N
N
S
Cl
SN
S
S N
S N
PtMe2PhP
S S
NCl
NN
S
Cl
44 45
Fig. 5.4. The structures of complexes 44 and 45
147

5. Introduction
5.7. Half-sandwich M(S2N2) complexesHalf-sandwich complexes are popular in synthetic chemistry because they contain a metalatom which is a centre of reactivity from one side and at the same time it is completelyprotected by a stabilising, structurally simple and esthetic ligand from the other.
The number of half-sandwich MSN complexes is remarkably low. The titanoceniumcomplexes listed in the previous sections cannot be counted as they are not half-sandwichcomplexes. This means that currently there are only 7 known half-sandwich complexes,all of which contain �ve-membered MSN rings. The most famous are 5,1,3,2,4-metalladi-thiadiazoles stabilised with cyclopentadienyl (Cp) or pentamethylcyclopentadienyl (Cp*)ligands. CpCo(S2N2) (46) is known and fully characterised, whereas the X-ray struc-ture of Cp*Co(S2N2) (47) has not been reported.337,338,424 The rhodium analogues arenot known and Cp*Ir(S2N2) (51) remains the only fully characterised iridium analogue(Fig. 5.5).8
M
CH3
CH3H3C
H3CCH3
S
N S
NM
H
HH
HH
S
N S
N
M
: Co
M
46 47: Co: Rh: Ir
4850
: Rh: Ir
4951
Fig. 5.5. Numbering of the CpM(S2N2) and Cp*M(S2N2) complexes (M = Co, Rh, Ir)
Four 5,1,2,4,3-ruthenatrithiazoles LRu(S3N) (L = Cp, C6H6, C6H5Me, C6H4Me2) wereprepared and characterised but received much less attention than the metalladithiadi-azoles.372 Several attempts to synthesise both Cp- and Cp*-metalladithiadiazoles usingvarious transition metals were unsuccessful.424
5.8. ConclusionA brief literature survey provided an insight into the high number of synthetic approachesused during preparations of metal-sulfur-nitrogen heterocycles of various sizes. Althoughrich in numbers, particular classes of MSN rings do not span over a broader area withinthe d-block of the Periodic System. In the next chapter a contribution to the �eld of�ve-membered half-sandwich M(S2N2) complexes is presented.
148

6. Results and discussion
The aim was to prepare η5-pentamethylcyclopentadienyl-5,1,3,2,4-metalladithiadiazolesof the group 9 metals (Co, Rh and Ir). The main target was to create a consistent set ofstructural data obtained from low temperature X-ray structure analysis, which could beused for a comparison with geometries calculated for the gas phase structures.
Strategy. To date the Cp* complexes have been prepared with greater success thanthe Cp analogues. The decision was to start with the preparation of Cp*Co(S2N2) (47)and to determine its low temperature X-ray structure. There was a small uncertaintyabout success in this task, since although the preparation of 47 was published on severaloccasions,338,424 its X-ray structure has not been determined.
The next step was a routine synthesis of Cp*Ir(S2N2) in order to remeasure its X-raystructre - this time at low temperature.
The last task was to prepare and characterise the previously unreported Cp*Rh(S2N2)(49). The fact that 49 was the only analogue missing in the Cp* series of the group 9complexes raised a suspicion that also in this case the success may not be granted.
6.1. Syntheses of the complexes
6.1.1. Cp*Co(S2N2) (47)47 was prepared by an oxidative addition of (S2N2) onto a Co I centre.338 This is a well-established procedure generating the product in 50% yield. The entire synthetic routeincluding the synthesis of the starting material is displayed by equations 6.1 and 6.2.The most time consuming procedure of the preparative sequence was the puri�cation ofthe starting material, Cp*Co(CO)2, which had to be performed under nitrogen on analumina column.425
Co
CH3
CH3H3C
H3CCH3
OC CO
Co2(CO)8 + 2 C5Me5H + + 4 COCH2Cl2 2reflux+
(6.1)
149

6. Results and discussion
toluene
12 hrs, r.t.2 Cp*Co(CO)2 + S4N4 + 4 COCo
CH3
CH3H3C
H3CCH3
S
N S
N
47 (6.2)
47 was characterised by 1H and 13C NMR spectroscopy and the chemical shifts wereidentical to the published ones (Table 6.7).338 The ES mass spectrum of 47 showed twopeaks, at m/z 308.99 ([MNa]+) and 287.00 ([MH]+).
6.1.2. Cp*Rh(S2N2) (49)The oxidative addition of (S2N2) onto a Rh I centre leads to the formation of insolubleprecipitates which are thought to be of a polymeric nature.424 The method proved un-suitable for the synthesis of other metalladithiadiazoles.424 However, 49 can be preparedfrom [Cp*RhCl2]2 in two possible routes.
The liquid ammonia route. The reaction of [S4N3]Cl with liquid ammonia is a wellknown and reliable source of the [S2N2] 2 � anion,5,6,426 which reacts with [Cp*RhCl2]2 togive a mixture of products containing 49 (eq. 1.3�1.4 and 6.3�6.4).
[Cp*RhCl2]2 + 2 HCl + 2x H2O2 RhCl3 · xH2O + 2 C5Me5HMeOH
reflux (6.3)
[Cp*RhCl2]2 + 2 [S2N2]2- + 4 Cl-Rh
CH3
CH3H3C
H3CCH3
S
N S
N
49
2
(6.4)
The presence of 49 was determined on the basis of NMR and mass spectrometry of thecrude solid obtained after the evaporation of ammonia. The ES mass spectrum showedtwo peaks corresponding to [Cp*Rh(S2N2H)]+ (330.95) and decamethylrhodioceniumcation [Cp*RhCp*]+ (373.11). The 1H NMR spectrum showed a number of signals witha dominant singlet at 2.07 ppm and a less intense singlet at 1.80 ppm. There was littledoubt that the signals belonged to the products observed in the mass spectrum. Giventhe composition of the liquid ammonia reaction system, the most probable decamethyl-rhodiocenium byproduct was [Cp*RhCp*]Cl (52), which is a known compound427 butits NMR data and crystal structure have not been reported so far. 52 thus had to be
150

6. Results and discussion
prepared to enable an unequivocal assignment of the 1H NMR spectrum of the crudereaction mixture. Its X-ray structure was also determined and will be discussed in aseparate section.
The comparison of the recorded 1H and 13C NMR spectra of the crude mixture of solidsand 52 con�rmed that the singlet at 1.80 ppm belongs to 52 and the one at 2.07 ppm to49. The origin of 52 is not clear but it seems reasonable to suggest that it is formed ina side reaction 6.5.
[Cp*RhCl2]2 [Cp*RhCp*]Cl + RhCl3 (6.5)
In view of these observations the formation of 49 is more accurately described by eq.6.6.
2 [Cp*RhCl2]2 + 2 [S2N2]2- 2 Cp*Rh(S2N2) + [Cp*RhCp*]Cl +
+ RhCl3 + 4 Cl- (6.6)
All attempts to purify 49 obtained in the liquid ammonia route were unsuccessful.Column chromatographies using silica, alumina and size-exclusion chromatography usingBio-Beads were attempted both in air and under a nitrogen atmosphere. Several solventsystems were tested but the 1H NMR spectra of the eluates always showed a large numberof signals due to products of decomposition.
The tin reagent route. An alternative strategy for the synthesis of 49 is the trans-metallation reaction between [nBu2Sn(S2N2)]2 (27) and the dimer [Cp*RhCl2]2, in afashion similar to the preparation of 51 (eq. 6.7).8
[Cp*RhCl2]2 + [nBu2Sn(S2N2)]2 2 Cp*Rh(S2N2) + 2 nBu2SnCl2CH2Cl2
r.t. (6.7)
TLC of the crude mixture suggested decomposition of 49 on both silica and alumina.However, small amounts of pure 49 were obtained by a careful elution with an ace-tone/toluene (1:2) mixture from a silica column. Both the 1H and 13C NMR spectrawere identical to the spectra of 49 prepared by the liquid ammonia route. The ESmass spectrum of pure 49 showed a single peak at m/z 330.99 ([MH]+). To obtainbetter yields, the separation on alumina, size-exclusion chromatography using Bio-Beadsand chromatographies using predried adsorbents and dry and degassed solvents wereattempted. Although the yields were very small the reproducibility of the preparationof 49 was demonstrated. The microanalyses of samples collected from three separatepreparations were satisfactory given the instability of 49.
151

6. Results and discussion
6.1.3. Cp*Ir(S2N2) (51)The transmetallation reaction using the tin reagent 27 is a straightforward route giving51 (eq. 5.14).8 Surprisingly, after the published procedure was carefully followed, it wascompound 54 which was isolated (eq. 6.8).
2 [Cp*IrCl2]2 + [nBu2Sn(S2N2)]2 + 2 nBu2SnCl2CH2Cl2
r.t.
Ir
CH3
CH3H3C
H3CCH3
S
N S
N
IrH3C CH3
CH3CH3
H3CCl
Cl
2
54 (6.8)
Its structure suggests that 54 is formed if [Cp*IrCl2]2 is in excess. This is the case inthe initial stages of the published reaction when 27 is added as a solid in one portion toa solution of [Cp*IrCl2]2.8 Before all 27 dissolves, the conditions favour the formation of54 according to eq. 6.8. To obtain 51, both reactants must be mixed in solutions.
6.2. The X-ray structures
6.2.1. Cp*M(S2N2) (M = Co, Rh, Ir)The X-ray analysis con�rmed the structure of 47 as 5-(η5-pentamethylcyclopentadi-enyl)-5,1,3,2,4-cobaltadithiadiazole with a planar Co(S2N2) ring which is perpendicularto the plane of the Cp* ring (Fig. 6.1). 47 has an approximate non-crystallographicCs symmetry. The solid-state molecular geometry of 47 is quite similar to that of theCp-analogue 46.46 However, the overall size of a molecule of 47 appears to be largerthan that of 46. The distance between the Cp* ring and the Co atom in 47 is greaterthan the Cp−Co distance in 46 and (with the exception of the S(1)−N(1) bond) thebond lengths in the Co(S2N2) ring are approximately 0.05Å longer in 47 than in 46(Table 6.1). The S−Co−N angle is approximately 0.4◦ larger in 47 than in 46. Theresults of the calculations presented in Table 6.2 suggest that this size increase is acrystal e�ect, as the calculated geometry of 47 is almost identical to that of 46. Thebond lengths in the Co(S2N2) ring di�er by no more than 0.011Å. The calculatedCp/Cp*−Co distances show an opposite trend to the experimental ones: the Cp*−Codistance in 47 is shorter by 0.015Å than the Cp−Co distance in 46. The di�erencebetween these distances is considerably smaller than between the two experimental ones.
152

6. Results and discussion
Fig. 6.1. The X-ray structure of Cp*Co(S2N2) (47): front and perspectiveview (A and B) and packing (C).
153

6. Results and discussion
Table 6.1. Selected bond lengths (Å) and angles (◦) of CpCo(S2N2) (46),Cp*Co(S2N2) (47) and Cp*Ir(S2N2) (51)
4646 47 51M(1)−N(1) 1.816(1) 1.8780(19) 1.963(3)N(1)−S(1) 1.556(1) 1.594(2) 1.562(4)S(1)−N(2) 1.597(2) 1.634(2) 1.583(4)N(2)−S(2) 1.657(2) 1.718(2) 1.682(3)S(2)−M(1) 2.0764(6) 2.1371(7) 2.1891(12)Cp/Cp*−M(1) 1.662(2) 1.703(2) 1.819(4)
M(1)−N(1)−S(1) 118.32(8) 117.83(11) 118.59(19)N(1)−S(1)−N(2) 112.04(7) 112.75(11) 113.67(17)S(1)−N(2)−S(2) 111.22(8) 111.27(12) 112.7(2)N(2)−S(2)−M(1) 107.40(5) 106.86(8) 108.13(13)S(2)−M(1)−N(1) 91.02(5) 91.29(6) 86.94(11)
The observation regarding the S−Co−N angle is also found in the calculated data, asthe value in 47 is approximately 0.4◦ larger than in 46.
Table 6.2. Selected calculated (DFT/B1B95/6-311+G*/def-TZPP/ecp) geometrical data forCpM(S2N2) (M = Co (46), Rh (48), Ir (50)) and Cp*M(S2N2) (M = Co (47),Rh (49), Ir (51)) complexes
46 48 50 47 49 51M(1)−N(1) 1.792 1.931 1.933 1.801 1.944 1.944N(1)−S(1) 1.558 1.562 1.573 1.559 1.561 1.571S(1)−N(2) 1.600 1.594 1.592 1.596 1.589 1.589N(2)−S(2) 1.646 1.648 1.651 1.657 1.660 1.661S(2)−M(1) 2.102 2.215 2.216 2.106 2.228 2.225Cp/Cp*−M 1.649 1.810 1.815 1.634 1.792 1.803
M(1)−N(1)−S(1) 119.9 120.0 120.3 119.2 119.4 119.9N(1)−S(1)−N(2) 110.7 112.0 111.6 111.4 112.8 112.2S(1)−N(2)−S(2) 112.2 114.0 114.0 112.3 114.2 114.0N(2)−S(2)−M(1) 106.6 107.4 107.7 106.2 106.8 107.4S(2)−M(1)−N(1) 90.5 86.6 86.3 90.9 86.8 86.4
Attempts to obtain crystals of 49 suitable for single crystal X-ray analysis failed.Recrystallisation by gas-phase di�usion of hexane into a dichloromethane solution of 49resulted only in deposition of a thin red �lm on the walls of a sample vial suggesting a lowtendency of 49 to crystallise. Attempts to recrystallise 49 from a hot dichloromethaneor toluene solutions led to the formation of black tarry globules as a result of a heatdecomposition, while saturated toluene or dichloromethane solutions of 49 stored at
154

6. Results and discussion
−20 ◦C over several weeks resulted only in powdery precipitates. Powder di�ractogramsof several samples of 49 contained broad and badly de�ned peaks raising only slightlyfrom a very noisy baseline suggesting little crystallinity of 49.
Hoping that an ionic compound could be obtained in slightly better yield and moreimportantly that it would be more prone to crystallisation, the protonated derivative[Cp*Rh(S2N2H)][PF6] was prepared from [Cp*RhCl2]2, [S4N3]Cl and [NH4][PF6] in liquidammonia. The 1H NMR proved the presence of Cp*Rh(S2N2) and the 31P NMR showeda septet of the [PF6] � anion. The sample in the NMR tube was carefully layered withdry hexane but no crystals were formed by the liquid phase di�usion. The rest of themixture was extracted with dry CH2Cl2, �ltered, the �ltrate was concentrated to 1
5of its
original volume and was placed in a freezer. After no crystals were formed during severalweeks, the mixture was diluted with CH2Cl2 and the product was precipitated by a slowaddition of diethyl ether followed by petroleum ether. Although the 1H, 13C,31P NMRand mass spectra of the collected precipitate showed that [Cp*Rh(S2N2H)][PF6] wasformed, all recrystallisation attempts were unsuccessful.
The low temperature X-ray structure of 51 (Fig. 6.2) is nearly identical to the structuremeasured at 293K.8 In the low temperature structure, the Ir−S and all N−S bond lengthsappear slightly elongated, though the e�ect is modest.
47 and 51 show very similar crystal packing with stacking of molecules. In the packingof 51 intermolecular contacts between two NSN sulfur atoms are highlighted with thedistance being just on the edge of the sum of van der Waals' radii. Similar contacts werenot observed between two molecules of 47.
155

6. Results and discussion
Fig. 6.2. The X-ray structure of Cp*Ir(S2N2) (51): the molecule (A) andpacking (B).
6.2.1.1. The in�uence of M in CpM(S2N2) and Cp*M(S2N2)
Comparison of the calculated iridium-containing structures 50 and 51 (Table 6.2) showsthat the structures are very similar, in accordance with what was found for the calculatedstructures of 46 and 47; the maximum di�erence in bond length in the heterocycle isjust 0.011Å. Likewise, the Cp* ring sits closer to the iridium atom than the Cp ring by0.012Å and the S−Ir−N angles are virtually identical. When the calculated data of 48and 49, for which no experimental structures exist, are compared, similar conclusionscan be drawn: the largest di�erence in bond length in the heterocycle is 0.013Å, the
156

6. Results and discussion
Cp* ring sits closer to the rhodium atom by 0.018Å and the S−Rh−N angles di�er byno more than 0.2◦.
Comparison of the experimental structures of 47 and 51 shows the in�uence of di�erentcovalent radii of the metal centre on the structure of the M(S2N2) rings (Table 6.1): thebonds between the metal centre and N(1) and S(2) are considerably shorter in 47. Theremaining bonds within the (S2N2) moiety are longer in 47 than in 51. The di�erencesin the bond lengths M−N(1) and M−S(2) are directly linked to the value of the angleS(2)−M−N(1). In 47, where the bonds coming from the metal centre are shorter, theangle is more obtuse than in 51, where the longer distances between the metal centreand N(1) and S(2) accompany a more acute angle.
Analysis of the calculated geometrical data of 46, 48 and 50, which includes allthree metal complexes, leads to a more detailed description of the in�uence of the metal(Table 6.2). In the series 46 −→ 48 −→ 50 the elongation of the Cp−M, M−S(2) andM−N(1) bonds can be clearly seen, as well as the reduction of the S(2)−M−N(1) angle.In addition, the S(2)−N(2) and S(1)−N(1) bond lengths increase slightly, by 0.005 and0.015Å, respectively. In contrast, the N(2)−S(1) distance becomes slightly shorter, by0.008Å. As a result of this reshu�ing of electron density due to the change in metalatom, the shortest S(1)−N(1) bond has become longer while the medium N(2)−S(1)
bond has become shorter. This is indicative of a slight increase in delocalisation inthe heterocyclic ring upon going from cobalt to iridium. Similar conclusions can bedrawn from the calculated Wiberg bond orders (Table 6.3). Despite the elongation of
Table 6.3. Calculated Wiberg bond orders for the most importantbonds in CpM(S2N2) and Cp*M(S2N2) complexes (M =Co, Rh, Ir)
46 48 50 47 49 51N(2)−S(2) 1.19 1.18 1.16 1.16 1.15 1.14S(1)−N(2) 1.25 1.26 1.28 1.27 1.28 1.29N(1)−S(1) 1.49 1.48 1.42 1.48 1.47 1.42M(1)−N(1) 0.72 0.74 0.81 0.66 0.69 0.76S(2)−M(1) 0.79 0.81 0.87 0.74 0.77 0.84M(1)−C(1) 0.23 0.24 0.27 0.23 0.24 0.27M(1)−C(2) 0.27 0.29 0.34 0.27 0.30 0.35M(1)−C(3) 0.23 0.24 0.27 0.23 0.24 0.26M(1)−C(4) 0.23 0.24 0.27 0.23 0.24 0.28M(1)−C(5) 0.27 0.29 0.34 0.27 0.30 0.33
M−S(2) and M−N(1), the bond order values correspond to strengthening of these bondswhen along the series 46 −→ 48 −→ 50. At the same time, S(2)−N(2) and S(1)−N(1)
become slightly longer and weaker, and N(2)−S(1) becomes slightly shorter and stronger;
157

6. Results and discussion
according to these values the M−S(2) and M−N(1) bonds are somewhat weaker thanthe three sulfur-nitrogen bonds.
According to the calculated geometries the ring-to-metal distance increases when Cp*is replaced by Cp. However, the carbon-metal bond order values, which are a measure ofstrength of the ring-metal bond, suggest that there is no reduction in bonding when Cp*is replaced by Cp; the di�erences between the values of the di�erent pairs are negligible.
6.2.2. Cp*Rh(µ−S3N2)(µ−S2O3)RhCp* (53)and [Cp*RhCp*]Cl (52)
During one of the lengthy recrystallisation attempts (gas-phase di�usion of hexane intoa CH2Cl2 solution of 49) 49 decomposed to give compound 53, which formed rosettesof fragile red needles at the bottom of the sample vial. The quality of the crystalswas below average but the X-ray structure could be solved. 53 consists of two Cp*Rhunits connected by a bridging [S3N2] 2 � ligand and by bridging terminal sulfur of athiosulfate anion (Fig. 6.3). Metal complexes bearing either [S3N2] 2 � or thiosulfateanion in a bridging position are known359,395,397,428�430 but no rhodium analogues havebeen reported. 53 also contains both of these ligands in one molecule, which has notbeen observed before.
Fig. 6.3. The X-ray structure of Cp*Rh(µ−S3N2)(µ−S2O3)RhCp* (53)
The Cp* rings in 53 are in an eclipsed position and the distance between the two Rhatoms is 3.1347(17)Å (Table 6.4). Both the [S3N2] 2 � ligand and the two S atoms of thethiosulfate anion lie in one plane which is parallel to the planes of the Cp* rings and
158

6. Results and discussion
is bisecting the distance between them. The molecule is electroneutral with Rh in theformal oxidation state +III and has approximate non-crystallographic Cs symmetry.
There are three Rh−S−Rh planes in the molecule which can be labelled accordingto the central sulfur atoms as σ1, σ3 and σ4 (Fig. 6.4). The regular distribution ofthe planes is slightly disrupted by steric hindrance of the bridging ligands. The anglesσ1−σ3 and σ3−σ4 are determined by the steric demands of the [S3N2] 2 � ligand and by theinterference of the terminal thiosulfate oxygen O(1) with the bridging S(3), respectively.The two angles are 122◦ wide. There is no feature causing a steric hindrance betweenthe planes σ4 and σ1 which is re�ected by a more acute angle between them (115◦). AllRh−S−Rh angles have values around 83◦. All Rh−S bonds are approximately equal
Table 6.4. Selected bond lengths (Å) and angles (◦) for 53
Rh(1)−S(1) 2.369(4) Rh(1)−S(1)−Rh(2) 82.75(12)Rh(1)−S(3) 2.367(3) Rh(1)−S(3)−Rh(2) 82.79(11)Rh(1)−S(4) 2.376(4) Rh(1)−S(4)−Rh(2) 82.68(12)Rh(2)−S(1) 2.374(4) S(1)−Rh(1)−S(3) 82.38(12)Rh(2)−S(3) 2.374(4) S(3)−Rh(1)−S(4) 82.31(12)Rh(2)−S(4) 2.370(4) S(4)−Rh(1)−S(1) 78.54(13)S(1)−N(1) 1.690(14) S(1)−Rh(2)−S(3) 82.13(13)N(1)−S(2) 1.566(13) S(3)−Rh(2)−S(4) 82.30(13)S(2)−N(2) 1.569(12) S(4)−Rh(2)−S(1) 78.57(13)N(2)−S(3) 1.679(13) S(1)−N(1)−S(2) 125.9(8)S(4)−S(5) 2.163(5) N(1)−S(2)−N(2) 120.0(7)S(5)−O(1) 1.422(10) S(2)−N(2)−S(3) 127.9(7)S(5)−O(2) 1.469(11) Rh(1)−S(1)−N(1) 106.7(5)S(5)−O(3) 1.450(11) Rh(2)−S(1)−N(1) 106.8(5)Rh(1)−Rh(2) 3.1347(17)Cp*(1)−Rh(1) 1.820(15)Cp*(2)−Rh(2) 1.830(15)
in length (2.37Å). The structure of the [S3N2] 2 � ligand is analogous to that in thepalladium compounds prepared earlier.359,395,397 The S(4)−S(5) distance in the bridgingthiosulfate anion is 2.163(5)Å which is in good agreement with the values published forthiosulfate anions in [Cp*M(µ−S2)(µ−S)(µ−S2O3)MCp*] (M = Cr, Mo).428�430
Fig. 6.5 shows the X-ray structure of 52. The molecule is in the form of monohydrate,most probably as a consequence of recrystallisation from non-dried solvents. The rhodiumatom is in the formal oxidation state +III and is coordinated by two coplanar Cp*rings in an eclipsed conformation which imparts the decamethylrhodiocenium cationwith approximate non-crystallographic D5h symmetry.
The most important bond lengths and angles are listed in Table 6.5. The hydrogenatoms in the molecule of water exhibit hydrogen bonding to the chloride anion of the
159

6. Results and discussion
S(3)
S(4)
S(1)
O3SN(2)
N(1)
S(2)σ1
σ4
σ3
Fig. 6.4. A Newman projection of 53 along the Rh−Rh axis
Fig. 6.5. The X-ray structure of [Cp*RhCp*]Cl ·H2O (52): a view of the molecule (A)and a view along the Cp*−Cp* axis to see the orientation of the Cp* rings (B).Hydrogen atoms are omitted for clarity.
parent molecule and to the oxygen of a neighbouring water molecule.
Table 6.5. Selected bond lengths (Å) and angles (◦) for 52
Cp*A−Rh(1) 1.812(4)Cp*B−Rh(1) 1.808(3)Rh(1)−Cl(1) 5.283O(1)−H(1A) 0.980(3)O(1)−H(1B) 0.979(3)H(1A)···Cl(1) 2.158(13)H(1B)···O(1)#2 2.28(5)
H(1A)−O(1)−H(1B) 85(5)
6.2.3. Cp*Ir[S2N2(IrCl2Cp*)] (54)The low temperature single crystal X-ray structure of 54 revealed cocrystallisation ofone molecule of nBu2SnCl2. The correct formula of the system thus must be writtenas Cp*Ir[S2N2(IrCl2Cp*)] · nBu2SnCl2. The crystal structure also shows a disorderedmolecule of toluene from which 54 was recrystallised (Fig. 6.6).
160

6. Results and discussion
Fig. 6.6. The X-ray structure of 54 · nBu2SnCl2. Hydrogen atoms are omitted for clarity.
The molecule of 54 is electroneutral and consists of the Cp*Ir(S2N2) core to which theCp*IrCl2 ligand is bound. It can be seen that the distance of the Cp* ring from the atomIr(1) is 0.054Å longer than the Cp*−Ir(2) distance (Table 6.6). This can be explained bythe presence of the two chlorine atoms on Ir(2) which are withdrawing electron densityfrom the metal centre forcing the Cp* ring to be situated closer to the Ir(2) atom.
The in�uence of the Cp*IrCl2 ligand on the Cp*Ir(S2N2) core can be described by acomparison between 51 and 54. The surroundings of the Ir(1) centre in 54 are virtuallyuna�ected by the presence of the ligand. On the contrary, the geometry of the (S2N2)unit changes considerably which is most apparent from the values of the bond angles.In 54, the angles at both nitrogen atoms become wider and those at both sulfur atomsbecome more acute. Particularly noteworthy is the angle at the donor nitrogen atomN(2) which opens by nearly 2◦, and the angle at S(1) which closes by approximately2.4◦. The bond lengths of the Ir(S2N2) ring in 54 do not change as considerably as theangles. The most signi�cant is the elongation of the S(1)−N(2) bond (1.583(4)Å in 51vs. 1.612(3)Å in 54) which is probably directly linked to the change of the angle at S(1)discussed above.
161

6. Results and discussion
Table 6.6. Selected bond lengths (Å) and angles (◦) for 51, 54 and 55
51 54 55Ir(1)−N(1) 1.963(3) 1.969(3) 1.961(10)N(1)−S(1) 1.562(4) 1.550(3) 1.558(10)S(1)−N(2) 1.583(4) 1.612(3) 1.531(9)N(2)−S(2) 1.682(3) 1.694(3) 1.685(9)S(2)−Ir(1) 2.1891(12) 2.1864(10) 2.179(3)Cp*−Ir(1) 1.819(4) 1.815(4) 1.790(11)Cp*−Ir(2) � 1.761(4) 1.849(11)Ir(2)−N(2) � 2.100(3) 2.178(8)Ir(2)−Cl(1) � 2.4174(9) �Ir(2)−Cl(2) � 2.4222(10) �Ir(2)−P(1) � � 2.336(3)Ir(2)−Cl(1) � � 2.421(3)
Ir(1)−N(1)−S(1) 118.59(19) 119.81(18) 120.6(5)N(1)−S(1)−N(2) 113.67(17) 111.31(16) 110.0(5)S(1)−N(2)−S(2) 112.7(2) 114.68(19) 117.4(5)N(2)−S(2)−Ir(1) 108.13(13) 106.41(11) 105.5(3)S(2)−Ir(1)−N(1) 86.94(11) 87.78(9) 86.5(3)Ir(2)−N(2)−S(1) � 121.66(17) 125.1(5)Ir(2)−N(2)−S(2) � 123.12(16) 117.1(5)N(2)−Ir(2)−Cl(1) � 85.82(8) �N(2)−Ir(2)−Cl(2) � 86.84(9) �Cl(1)−Ir(2)−Cl(2) � 90.39(3) �N(2)−Ir(2)−P(1) � � 90.6(2)N(2)−Ir(2)−Cl(1) � � 86.4(2)P(1)−Ir(2)−Cl(2) � � 90.39(10)
54 is related to the known compound 55 (Fig. 6.7).8 54 is electroneutral whereas 55has a positive charge with [PF6] � as the counterion. The common feature of 54 and55 is the planarity of the Ir(S2N2) ring. Table 6.6 gives an interesting insight into thestructural di�erences between the two compounds.
The X-ray data show that the [Cp*IrCl2] and [Cp*IrCl(PPh3)]+ units have a �xedinternal structure. This is demonstrated not only by the Ir(2)−Cl bond lengths, which areclose to 2.42Å in both compounds, but mainly by the L−Ir(2)−L′ angle (L, L′ = ligands)which is identical in both compounds, despite Ir(2) bearing di�erent pairs of ligands in54 and 55. However, the overall e�ect of the [Cp*IrCl2] and [Cp*IrCl(PPh3)]+ moietieson the Ir(S2N2) ring is noticeable. In 54, the two Cl atoms are withdrawing electronsfrom the Ir(2) atom forcing the N(2) atom to get closer to Ir(2). The Ir(2)−N(2) bondshortening induces closing of the S(1)−N(2)−S(2) angle and elongation of the S(1)−N(2)
bond in the Ir(S2N2) ring in 54.
162

6. Results and discussion
Ir
CH3
CH3H3C
H3CCH3
S
N S
N
Ir
H3C CH3
CH3CH3
H3CCl
Cl
[PF6]-
+
Ir
CH3
CH3H3C
H3CCH3
S
N S
N
Ir
H3C CH3
CH3CH3
H3CPPh3
Cl
54 55
Fig. 6.7. Cp*Ir[S2N2(IrCl2Cp*)] (54) and [Cp*Ir[S2N2(IrCl(PPh3)Cp*)]][PF6] (55)
The steric e�ects of the ligands can be demonstrated on 55. While in 54 the anglesIr(2)−N(2)−S(1) and Ir(2)−N(2)−S(2) are nearly identical, in 55 the presence of thetriphenylphosphino group results in slant of the [Cp*IrCl(PPh3)]+ moiety.
6.3. NMR spectroscopyThe 1H NMR data of all isolated Cp* complexes are summarised in Table 6.7. The 1H sig-nals of the methyl groups of the Cp*M(S2N2) complexes are signi�cantly deshielded withrespect to the signals of the starting materials, which enabled their quick identi�cation inthe spectra of the reaction mixtures. The CH3 signals in all the Cp*M(S2N2) complexes
Table 6.7. 1H NMR data of Cp*M(S2N2) (M = Co (47), Rh (49), Ir (51)),[Cp*RhCp*]Cl (52), Cp*Rh(µ−S3N2)(µ−S2O3)RhCp* (53) andCp*Ir[S2N2(IrCl2Cp*)] (54) a
compound δ multi- assignment[ppm] plet
47 1.98 s 15H, 5×CH3
49 2.07 s 15H, 5×CH3
51 2.22 s 15H, 5×CH3
52 1.79 s 30H, 10×CH3
53 1.66 s 30H, 10×CH3
54 1.57 s 15H, 5×CH3 in Cp*IrCl22.17 s 15H, 5×CH3 in Cp*Ir(S2N2)
a All spectra measured in CDCl3 (270.2MHz, 298K). Chemical shiftsare calibrated to the peak of residual CHCl3 (7.26 ppm).279
are singlets which means that lowering the symmetry when going from Cp*Co(CO)2 or[Cp*MCl2]2 to Cp*M(S2N2) has no e�ect on the chemical equivalency of the CH3 groups.
163

6. Results and discussion
The three title complexes 47, 49 and 51 nicely show a trend of increased deshielding ofthe 1H NMR signals of the CH3 groups when going from Co towards Ir.
In accordance with expectations, the 13C NMR spectra of all the discussed Cp* com-plexes showed two signals for every Cp* ring, one due to the CH3 groups and the otherdue to the aromatic C5 ring (Table 6.8). Since 103Rh has nuclear spin of 1
2, the C5 signals
in the Rh complexes are split into doublets. The values of the interaction constantsindicate only a small 103Rh− 13C interaction.
The 1H and 13C NMR signals due to the Cp* rings in 54 were assigned after a com-parison with the spectra of both 51 and [Cp*IrCl2]2.
Table 6.8. 13C NMR data of Cp*M(S2N2) (M = Co (47), Rh (49), Ir (51)), [Cp*RhCp*]Cl(52), Cp*Rh(µ−S3N2)(µ−S2O3)RhCp* (53) and Cp*Ir[S2N2(IrCl2Cp*)] (54) a
compound δ multi- assignment notes[ppm] plet
47 10.8 s 5C, 5×CH394.8 s 5C, C5
49 9.3 s 5C, 5×CH394.9 d 5C, C5
1J 103Rh−13C = 8.30 Hz51 10.7 s 5C, 5×CH3
95.6 s 5C, C5
52 8.7 s 10C, 10×CH398.5 d 10C, 2×C5
1J 103Rh−13C = 7.27 Hz53 8.7 s 10C, 10×CH3
99.6 d 10C, 2×C51J 103Rh−13C = 7.27 Hz
54 9.1 s 5C, 5×CH3 in Cp*IrCl210.7 s 5C, 5×CH3 in Cp*Ir(S2N2)86.5 s 5C, C5 in Cp*IrCl297.1 s 5C, C5 in Cp*Ir(S2N2)
a All spectra measured in CDCl3 (67.9MHz, 298K).
6.3.1. 14N NMR spectroscopy14N NMR spectroscopy proved unsuitable for the characterisation of 5-(η5-pentamethyl-cyclopentadienyl)-5,1,3,2,4-metalladithiadiazoles. The nitrogen atoms of 47 and 49 gaveonly very weak response in the spectra and the broad signals could not be convincinglyidenti�ed within the rolling baseline. The spectrum of 51 could be recorded and showedtwo broad and deformed peaks at 372.0 and 454.4 ppm. On the basis of previous 15NNMR studies on 46 and 47,424 the peak with the higher chemical shift can be assignedto the iridium-bound nitrogen and that with the lower shift to the SNS nitrogen.
164

6. Results and discussion
6.4. Mass spectrometryElectrospray ionisation turned out to be the most convenient ionisation method. Thespectra were of good quality and contained only the most important peaks which couldbe quickly assigned. The ES+ spectra often contained pairs of peaks corresponding tothe cations [MH]+ and [MNa]+.
Iridium has two naturally abundant isotopes - 191Ir (37.3%) and 193Ir (62.7%)282 -and therefore the spectra of the iridium complexes contained peaks with characteristicisotopic patterns. The molecular peak of 54 could not be observed due to the presenceof two atoms of chlorine in the molecule. The peak with the highest m/z value displayedin the spectrum of 54 was assigned to the protonated fragment without the two chlorineatoms, [(Cp*)2Ir2S2N2H]+. The base peak was produced by the protonated fragment[Cp*Ir(S2N2H)]+.
6.5. IR and Raman spectroscopyThe majority of the IR bands were due to the Cp* rings. Spectra of all the compoundsshowed characteristic C−H stretches in the area 3000�2900 cm−1 and the CH3 deforma-tion vibrations between 1460 and 1370 cm−1.
A few peaks could be tentatively assigned on the basis of comparison with the vibra-tional spectra of the starting materials Cp*Co(CO)2 and [Cp*MCl2]2 (M = Rh, Ir). Inthe infrared spectra the intense IR bands at 724 (M = Co) and 700 cm−1 (M = Rh,Ir) and at 646 (M = Rh) and 635 cm−1 (M = Co, Ir) are the most likely M(S2N2) ringvibrational modes. The very strong Raman line at 919 cm−1 in the spectra of 49 is acertain Rh(S2N2) ring vibrational mode and those at 649, 426 and 403 cm−1 are strongcandidates. The Raman lines in the spectra of 51 are shifted towards lower wavenum-bers. The very strong line at 864 cm−1 must be a Ir(S2N2) ring vibration and listed canbe also the lines at 639, 504, 419 and 385 cm−1. Under the experimental conditions (nearIR Raman laser) no Raman spectrum of 47 could be recorded. However, the frequenciesin the IR spectrum of 47 matched perfectly with the published values.338
6.6. Protonation of Cp*Co(S2N2)A number of Co, Pd and Pt M(S2N2H) complexes were prepared by protonation ofthe corresponding M(S2N2) complexes with HBF4 or HCl.354,365�367 The protonation of47 with both acids was attempted. The �rst experiment was performed with HBF4.Immediately on the contact with the acids a grey/black solid started to precipitate. Its
165

6. Results and discussion
1H and 13C NMR spectra showed only residual unreacted 47. Neither a concentratedNMR sample revealed the presence of an N−H proton.
Once the grey solid matter was �ltered and thoroughly washed from all residual violet47, it was dissolved in CH2Cl2 to form a black solution from which the grey/black solidwas reprecipitated by the addition of diethyl ether and hexane. After its isolation thepuri�ed solid was dissolved in a small amount of CH2Cl2 in an NMR tube and was treatedwith hexane in a liquid phase di�usion, which resulted only in a deposition of the samemicrocrystalline solid.
The black solid was formed also when HBF4 was layered below the Cp*Co(S2N2)solution with no stirring applied and also when both reactants were introduced at lowtemperature (-40 ◦C).
Once the immediate precipitation occured also on the contact with HCl, the mixturewas only allowed to reduce in volume spontaneously in a gentle nitrogen �ow and whenno crystals were formed, the attempts were stopped.
6.7. Conclusion5-(η5-pentamethylcyclopentadienyl)-5,1,3,2,4-rhodiadithiadiazole, Cp*Rh(S2N2) (49),was prepared from [Cp*RhCl2]2 by two substitution reactions. The �rst method usesthe [S2N2] 2 � anion generated in situ in liquid ammonia and yields the product contam-inated with [Cp*RhCp*]Cl. The other method takes advantage of the ligand exchangewith the versatile tin reagent [nBu2Sn(S2N2)]2. 49 is an unstable substance but smallamounts of material of satisfactory purity were obtained after silica column chromatog-raphy after several separate preparations con�rming the reproducibility of the synthesisof 49. 49 was characterised by 1H and 13C NMR spectroscopy, mass spectrometry andits constitution was supported by vibrational spectra. 49 is a microcrystalline substancewhich does not crystallise into well grown single crystals or crystals of su�cient size tobe investigated by powder X-ray di�raction.
The low-temperature crystal structure of Cp*Co(S2N2) (47) was determined. Thecomparison of the solid state structures of both CpCo(S2N2) (46) and 47 showed in-signi�cant di�erences between the two molecules and theoretical calculations suggestedthat the larger size of the molecule of 47 is a result of a crystal e�ect.
The low-temperature crystal structure of Cp*Ir(S2N2) (51) was determined and athorough comparison of the calculated gas-phase structures has been carried out betweenthe non-methylated and methylated pairs of cyclopentadienylmetalladithiadiazoles. Thecrystal structures of 46 and 47 show an increase of the ring-metal distance, while thecalculated gas-phase structures show an opposite trend. The Wiberg bond order analysis
166

6. Results and discussion
demonstrated that substitution of a Cp ring with a Cp* ring has no e�ect on bondingbetween the ring and the metal centre, despite the change of distance. Theoreticalcalculations also indicated increasing degree of π-electron delocalisation in the M(S2N2)heterocycle when going from Co to Ir.
Several Rh and Ir complexes obtained during this work were characterised by multi-nuclear NMR spectroscopy, mass spectrometry and single crystal X-ray analysis. It wasshown that decamethylrhodiocenium cation in [Cp*RhCp*]Cl contains eclipsed Cp* ringsin the crystal. X-ray analysis also revealed that hydrolysis of 49 leads to the formationof complex 53, which contains bridging [S3N2] 2 � and [S2O3] 2 � ligands in one molecule.
167

7. Experimental
HBF4 ·Et2O was purchased from Sigma-Aldrich, RhCl3 · xH2O and IrCl3 · xH2O wereobtained on loan from Johnson Matthey Plc. S4N4,431 [S4N3]Cl4 and [Cp*IrCl2]2,432
were prepared according to published methods.
Recrystallisation of S4N4 CAUTION: S4N4 explodes upon mechanical or heat shock.Its sensitivity increases with purity of the substance. Kevlar gloves and visor were wornwhen it was manipulated with S4N4. Residues of S4N4 were disposed of by decompositionwith aqueous NaOH.433
The crude S4N4/S mixture obtained from the dioxane extract431 contained high amountof sulfur. Signi�cant amount of sulfur was removed by suspending the S4N4/S mixturein su�ciently small amount of CH2Cl2 and subsequent decanting of the orange S4N4-richsolution. The solvent was evaporated on a rotary evaporator and the S4N4-rich crudemixture was recrystallised from toluene. The purity of S4N4 was checked by TLC.
7.1. Preparation of [Cp*RhCl2]2432
RhCl3 · xH2O (7.49 g, 0.036mol) was dissolved in degassed methanol (250ml) in a 500mlSchlenk �ask. C5Me5H (5.82ml, 4.91 g, 0.036mol) was added, the �ask was �tted with awater condenser with an oil-bubbler on top, the apparatus was �ushed with nitrogen andthe stirred mixture was gently re�uxed under nitrogen for 2 days. After cooling downto room temperature, the mixture was �ltered through a sinter on air and the dark redsolid was collected. The �ltrate was reduced in volume to ca. 50ml and the second cropwas collected by suction. Both crops were washed with diethyl ether (3 × 50ml) andwere dried on air overnight. Yield 8.27 g (74%).1H NMR (CDCl3, 298 K): δ = 1.58 (s, 15H, Cp*).13C NMR (CDCl3, 298 K): δ = 9.5 (s, 5C, 5×CH3), 94.2 (d, 5C, C5, 1J 103Rh−13C = 8.30Hz).
168

7. Experimental
7.2. Preparation of Cp*Co(CO)2425
In a three-neck-�ask equipped with a stirring bar, dry CH2Cl2 (70ml) was deoxygenatedby bubbling nitrogen gas through for 40 minutes. The deoxygenating process left 50mlof dry and deoxygenated CH2Cl2. Co2(CO)8 (6.61 g, 19.3mmol) was dissolved in thedeoxygenated CH2Cl2 and C5Me5H (3.55ml, 3.0 g, 22.0mmol) was added followed bycyclohexa-1,3-diene (2.5ml, 2.15 g, 26.8mmol). The mixture was gently re�uxed for45 minutes, then another portion of C5Me5H (3.0ml, 2.53 g, 18.5mmol) was added andthe re�ux was continued for additional 13
4hour. The resulting dark red mixture was
allowed to reach room temperature, solvent and volatiles were distilled o� under highvacuum and the crude solid was puri�ed on an alumina column (25 × 2.5 cm) packedunder nitrogen in deoxygenated hexane. Elution with deoxygenated hexane removed theproduct as an orange/brown band which was collected under nitrogen and evaporatedto dryness. The wine red crystals of the pure product were stored under nitrogen in afreezer. Yield 5.48 g (62%).1H NMR (CDCl3, 298 K): δ = 1.88 (s, 15H, Cp*).
7.3. Preparation of Cp*Co(S2N2) (47)338
Cp*Co(CO)2 (1.71 g, 6.80mmol) was dissolved in dry and deoxygenated toluene (60ml).The solution was vigorously stirred and S4N4 (0.630 g, 3.40mmol) was added carefullyin one portion. Shortly after, bubbles of CO were evolved, the colour changed fromorange/brown to very deep violet and some solid precipitated. The mixture was stirredat room temperature overnight and then was directly passed through a silica column (20× 5 cm) prepared in toluene. Elution with toluene removed unreacted starting materialsand the mixture acetone:toluene (1:2) eluted the product as a black band which wasshown to be a very concentrated purple solution of the product. The band was collectedunder nitrogen and evaporated to dryness. Crystals suitable for X-ray analysis weregrown from a saturated hot hexane solution by slow cooling and keeping in a fridge.Yield 0.978 g (50%). 1H NMR (CDCl3, 298 K): δ = 1.96 (s, 15H, Cp*).IR data: 2980 (mw), 2960 (mw), 2909 (mw), 2854 (sh), 1485 (m), 1465 (m), 1452 (sh),1429 (m), 1404 (sh), 1377 (s), 1360 (sh), 1242 (sh), 1198 (mw), 1158 (mw), 1102 (sh),1076 (m), 1022 (vs), 963 (ms), 913 (sh), 839 (vw), 798 (vw), 767 (vw), 724 (vs), 634(ms), 618 (sh), 590 (w), 541 (vw), 510 (vw), 470 (vw), 453 (w), 444 (w), 399 (w), 384(mw), 328 (vw), 293 (vw) cm−1.
169

7. Experimental
7.4. Cp*Rh(S2N2) (49)The liquid ammonia method Ammonia (20�30ml) was condensed into a predriedthree-neck �ask. [S4N3]Cl (51mg, 0.247mmol) was added in one portion. The mixtureturned immediately red and was stirred for 30 minutes at -78 ◦C. [Cp*RhCl2]2 (153mg,0.247mmol) was added in one portion and the mixture was allowed to warm up to-33 ◦C. Once the temperature was reached, the cooling bath in the ammonia condenserwas regularly supplied with dry ice and the mixture was kept under cold re�ux for 2 hourstill the colour of the mixture changed to dark purple with slight greenish glimpse at therim of the level. After that period, the ammonia was allowed to evaporate spontaneouslyleaving a beige/brown mixture of solids. The mixture was put under high vacuum for15 minutes to evaporate residual ammonia and after reintroduction of nitrogen gas it wasextracted with just one portion of dry CH2Cl2 (10ml). The extract was �ltered througha sinter, solvent was removed in vacuo and the solid dried under high vacuum. Yield114mg (70%), product contaminated with [Cp*RhCp*]Cl.MS(ES+TOF): m/z 373.11 (80%) [Cp*RhCp*]+, 330.95 (100%) [MH]+.1H NMR (CDCl3, 298 K): δ = 2.04 (s, 15H, Cp*), 1.77 (s, 30H, decamethylrhodiocenium).13C NMR identical to the product obtained by the transmetallation route.
The transmetallation method using 27 27 (161mg, 0.247mmol) was dissolved in dryCH2Cl2 (30ml). A solution of [Cp*RhCl2]2 (153mg, 0.247mmol) in dry CH2Cl2 (30ml)was added dropwise and the mixture was stirred for 6 hours at room temperature. Thesolvent was evaporated, the crude product was adsorbed on a small amount of silicaand added to a packed silica column (25 × 2 cm). Unreacted starting material and thebyproduct were removed with toluene as a pale yellow band. Elution with toluene:acetone(2:1) gave several similarly coloured fractions which had to be analysed by 1H NMR sothat the product could be found. The residues were washed out with toluene:acetone(1:1) but they contained only small amount of very contaminated product and were notworth repurifying. Yield 6mg (10%). Compound decomposed without melting at 190 ◦C(blackening).Microanalysis: Found C, 36.4; H, 5.0; N, 6.1. Calc. for C10H15RhS2N2: C, 36.4; H, 4.6;N, 8.5%.MS(ES+TOF): m/z 330.99 (100%) [MH]+.1H NMR (CDCl3, 298 K): δ = 2.04 (s, 15H, Cp*).13C NMR (CDCl3, 298 K): δ = 9.2 (s, 5C, 5×CH3), 94.1 (d, 5C, C5, 1J 103Rh−13C = 8.30Hz).IR data: 2963 (mw), 2912 (mw), 2853 (w), 1477 (sh), 1449 (mw), 1423 (sh), 1377 (m),1357 (sh), 1261 (ms), 1197 (vw), 1153 (sh), 1096 (s), 1079 (s), 1061 (vs), 1023 (vs), 964
170

7. Experimental
(sh), 918 (mw), 871 (vw), 802 (s), 701 (vw), 647 (m), 618 (mw), 538 (vw), 501 (vw), 423(vw), 397 (mw), 387 (sh), 368 (sh), 330 (vw), 295 (vw), 233 (w) cm−1.Raman data: 2962 (w), 2917 (ms), 1483 (vw), 1450 (vw), 1421 (mw), 1387 (vw), 1361(vw), 1158 (vw), 1028 (w), 920 (s), 649 (mw), 620 (m), 592 (mw), 541 (w), 426 (ms),403 (vs), 388 (sh), 370 (w), 327 (w), 300 (vw), 261 (w), 242 (vw), 199 (m) cm−1.
7.5. [Cp*RhCp*]Cl (52)427
[Cp*RhCl2]2 (309mg, 0.500mmol) was dissolved in dry CH2Cl2 (15ml) and AlCl3 (0.5 g,3.70mmol) was added in one portion to the dark red solution. Some solid precipitated,the resulting pale orange suspension was cooled to 0 ◦C (ice/water) and C5Me5H (1ml,0.844 g, 6.20mmol) was added dropwise. The resulting dark red mixture was stirred at0 ◦C for 3 hours, then was heated to 40 ◦C for 15 minutes, cooled again to 0 ◦C and washydrolysed with ice (8.0 g). When all the ice melted, the aqueous layer was separated,the CH2Cl2 layer was extracted twice with 10ml of water and the separated aqueouslayer joined with the two aqueous extracts was extracted with CH2Cl2 (3 × 10ml). Theorganic extracts were dried with MgSO4, �ltered through a sinter and the �ltrate wasconcentrated to approx. 4�5ml. The product was precipitated by a slow addition ofpentane as an o�-white powder which was collected by suction �ltration and was driedunder high vacuum for 30 minutes. Crystals suitable for X-ray analysis were obtained byslow evaporation of CDCl3 from a sample in an NMR tube. Yield 274mg (67%). M.p.182 ◦C.Microanalysis: Found C, 57.3; H, 7.6. Calc. for C20H32RhClO: C, 56.3; H, 7.6%.MS(ES+TOF): m/z 373.05 (100%) [Cp*RhCp*]+.1H NMR (CDCl3, 298 K): δ = 1.77 (s, 30H, 2×Cp*).13C NMR (CDCl3, 298 K): δ = 8.7 (s, 10C, 10×CH3), 98.5 (d, 10C, 2×C5, 1J 103Rh−13C
= 7.27 Hz).
7.6. [Cp*Rh(µ-S3N2)(µ-S2O3)RhCp*] (53)The compound was obtained as a product of hydrolysis of Cp*Rh(S2N2) followed by arearrangement during a long period recrystallisation (gas phase di�usion of hexane intoa CH2Cl2 solution of Cp*Rh(S2N2)). Compound decomposed without melting at 210 ◦C(blackening).Microanalysis: Found C, 35.1; H, 4.3; N, 2.8. Calc. for C20H30Rh2S5N2O3: C, 33.7; H,4.2; N, 3.9%.MS(ES+TOF): m/z 734.62 (100%) [MNa]+, 712.70 (5%) [MH]+.
171

7. Experimental
1H NMR (CDCl3, 298 K): δ = 1.65 (s, 30H, 2×Cp*).13C NMR (CDCl3, 298 K): δ = 8.7 (s, 10C, 10×CH3), 99.5 (d, 10C, 2×C5, 1J 103Rh−13C
= 7.27 Hz).IR data: 2960 (w), 2915 (mw), 2853 (w), 1461 (mw), 1424 (mw), 1378 (m), 1229 (s),1158 (vw), 1105 (vw), 1078 (vw), 1016 (vs), 901 (vw), 800 (vw), 641 (vw), 620 (sh), 599(s), 558 (vw), 534 (w), 403 (mw), 359 (w), 332 (vw), 309 (vw), 283 (vw) cm−1.Raman data: 2918 (m), 1426 (vw), 1015 (w), 902 (m), 625 (w), 590 (w), 540 (w), 440(mw), 412 (vs), 336 (vw), 310 (mw), 255 (vw), 214 (vw) cm−1.
7.7. Preparation of [Cp*Rh(S2N2H)]PF6
Ammonia gas was condensed (30ml) in a 100ml three-neck-�ask (-78 ◦C, aceone/dry icecooling bath). [S4N3]Cl (51mg, 0.247mmol) was added in one portion and the red mix-ture was stirred for 30 minutes. [Cp*RhCl2]2 (153mg, 0.247mmol) followed by [NH4]PF6(60mg, 0.370mmol) were added in one portion and the mixture was stirred for 1.5 hoursat -78 ◦C. It was then re�uxed at -33 ◦C for 2 hours before the ammonia was allowed toevaporate slowly. To remove the residues of ammonia, the mixture was kept for 15 min-utes under high vacuum. After the 1H and 31P NMR proved the presence of the productin the mixture, the sample in the NMR tube was carefully layered with dry hexane. Nocrystals were formed by the liquid phase di�usion.
The rest of the mixture was extracted with dry CH2Cl2 (10ml) and �ltered througha sinter. The �ltrate was concentrated to 1/5 of its original volume and was placed ina freezer but no crystals were formed during several weeks. The following work-up wasdone with no more nitrogen gas protection. The mixture was taken out of the freezer,diluted with CH2Cl2 (7ml) and the product was precipitated by a slow addition ofdiethyl ether (20ml) followed by petroleum ether (200ml). The precipitate was collectedby suction, washed with diethyl ether (3 × 10ml) and dried brie�y under high vacuum.The �ltrate was evaporated to dryness and after the 1H NMR showed no Cp* signals, itwas discarded.
The 1H and 13C NMR spectrum of the precipitate showed the signals of Cp*Rh(S2N2)and [Cp*RhCp*]Cl together with a high number of much more intense signals of uniden-ti�ed byproducts. The 31P NMR spectra showed the septet of the [PF6] � anion butits intensity was very low. The presence of the peak of [Cp*Rh(S2N2H)]+ in the ES+
mass spectrum could not be taken as a serious evidence as the peak appears also in theES+ mass spectrum of the neutral Cp*Rh(S2N2). The mass spectrum also proved thepresence of the [PF6] � anion. Recrystallisation attempts were unsuccessful.MS(ES+TOF): m/z 330.93 (100%) [Cp*RhS2N2H]+, 373.10 (30%) [Cp*RhCp*]+. MS
172

7. Experimental
(ES−TOF): m/z 144.84 (100%) [PF6] � .1H NMR (CDCl3, 298 K): δ = 2.05 (s, 15H, Cp*Rh(S2N2)), 1.77 (s, 30H, [Cp*RhCp*]+).13C NMR (CDCl3, 298 K): δ = 9.2 (s, 5C, 5×CH3 in Cp*Rh(S2N2)), 8.8 (s, 10C, 10×CH3in [Cp*RhCp*]Cl).31P NMR (CDCl3, 298 K): δ = -143.8 (sept., 1P, [PF6] � ).
7.8. Cp*Ir(S2N2) (51)8
27 (161mg, 0.247mmol) was dissolved in dry CH2Cl2 (20ml) and [Cp*IrCl2]2 (197mg,0.247mmol) was dissolved in another �ask in dry CH2Cl2 (20ml). The solution of[Cp*IrCl2]2 was �uently added to the stirred solution of 27 and the mixture was stirredfor 8 hours at room temperature. The product was absorbed on a small amount of silicaand added to a packed silica column (20 × 2 cm) prepared in toluene. Unreacted 27 andthe byproduct were eluted as a yellow band by toluene and the crude product was elutedwith acetone. Pure product was obtained after additional silica column chromatographyusing the mixture acetone:toluene (1:4) as eluant. Yield 91mg (44%). Melting point180 ◦C (decomp.).Microanalysis: Found C, 28.1; H, 3.6; N, 6.4. Calc. for C10H15IrS2N2: C, 28.6; H, 3.6;N, 6.7%.MS(ES+TOF): m/z 442.89 (100%) [MNa]+, 420.93 (80%) [MH]+. The correct isotopicpatterns were observed for each peak.1H NMR (CDCl3, 298 K): δ = 2.18 (s, 15H, Cp*).13C NMR (CDCl3, 298 K): δ = 10.7 (s, 5C, 5×CH3), 95.6 (s, 5C, C5).14N NMR (CDCl3, 298 K): δ = 372.0 (s, 1N, SNS), 454.4 (s, 1N, IrNS).IR data: 2982 (mw), 2962 (mw), 2918 (mw), 2853 (w), 1490 (mw), 1457 (m), 1427 (mw),1402 (sh), 1377 (m), 1361 (sh), 1293 (vw), 1265 (vw), 1158 (vw), 1078 (w), 1026 (ms),956 (m), 862 (vw), 795 (vw), 701 (vs), 680 (sh), 635 (m), 611 (w), 589 (vw), 542 (vw),502 (mw), 452 (w), 420 (vw), 397 (mw), 382 (w), 303 (vw), 277 (vw) cm−1.Raman data: 2991 (mw), 2966 (mw), 2913 (m), 1493 (vw), 1459 (w), 1450 (w), 1431(mw), 1407 (vw), 1382 (vw), 1361 (vw), 1159 (vw), 1033 (w), 959 (w), 864 (vs), 703 (w),639 (w), 614 (vw), 593 (mw), 538 (w), 504 (s), 449 (mw), 420 (ms), 385 (m), 285 (s),202 (mw), 176 (w) cm−1.
7.9. Cp*Ir[S2N2(IrCl2)]Cp* (54)[Cp*IrCl2]2 (197mg, 0.247mmol) was dissolved in dry CH2Cl2 (25ml). 27 (161mg,0.247mmol) was added in one portion and the mixture was stirred for 8 hours at room
173

7. Experimental
temperature. The solvent was evaporated under high vacuum, the mixture was adsorbedon a small amount of silica and added to a packed silica column (20 × 2 cm) preparedin toluene. Unreacted tin reagent and the byproduct were eluted with toluene andthe product with acetone as a yellow/orange band. The acetone fraction was collected,evaporated to dryness and recrystallised from warm toluene (60�70 ◦C). The product wasobtained as orange/red crystals. Yield 230mg (79%). Melting point 166 ◦C (decomp.).Microanalysis: Found C, 32.1; H, 4.4; N, 2.4. Calc. for C31.5H52Ir2S2N2Cl4Sn: C, 32.4;H, 4.5; N, 2.4%.MS(ES+TOF): m/z 749.08 (5%) [M(2×193Ir)H - Cl2]+, 747.07 (10%) [M(193,191Ir)H - Cl2]+,420.95 (100%) [(M(Cp∗IrS2N2)+2)H]+, 418.96 (30%) [M(Cp∗IrS2N2)H]+.1H NMR (CDCl3, 298 K): δ = 1.55 (s, 15H, Cp*Ir(2)), 2.16 (s, 15H, Cp*Ir(1)).13C NMR (CDCl3, 298 K): δ = 9.1 (s, 5C, 5×CH3 in Cp*Ir(2)), 10.7 (s, 5C, 5×CH3 inCp*Ir(1)), 86.5 (s, 5C, C5 in Cp*Ir(2)), 97.1 (s, 5C, C5 in Cp*Ir(1)).IR data: 3145 (vw), 3050 (vw), 2951 (s), 2924 (vs), 2866 (ms), 1492 (sh), 1448 (s), 1412(m), 1383 (s), 1361 (sh), 1261 (w), 1193 (vw), 1147 (mw), 1098 (sh), 1079 (m), 1023(vs), 969 (ms), 875 (w), 856 (w), 802 (w), 780 (ms), 735 (m), 688 (m), 615 (w), 582 (w),536 (vw), 497 (m), 463 (w), 398 (w), 277 (mw) cm−1.Raman data: 2970 (w), 2927 (ms), 2872 (w), 1448 (vw), 1420 (w), 1388 (vw), 1156 (w),859 (vs), 784 (w), 627 (mw), 587 (m), 539 (w), 500 (m), 472 (vw), 441 (m), 429 (m), 401(vw), 343 (sh), 306 (ms), 277 (vw), 211 (mw), 181 (ms) cm−1.
7.10. Protonation of Cp*Co(S2N2) with HBF4
Cp*Co(S2N2) (191mg, 0.667mmol) was dissolved in dry diethyl ether (10ml) andHBF4 ·Et2O (108mg, 0.667mmol) was added slowly dropwise to the stirred mixture.A grey/black solid started to precipitate immediately. The mixture was �ltered on airthrough a sinter and the orange/pink �ltrate was brought to dryness leaving a negligibleamount of an impure colourless solid which was discarded. The black solid was collectedfrom the sinter, dissolved in 5ml of CH2Cl2 and precipitated with the addition of diethylether (10ml) and hexane (50ml). The precipitate was collected on a sinter, washed oncewith diethyl ether (10ml) and brie�y dried under high vacuum. A recrystallisation bya liquid phase di�usion in an NMR tube was unsuccessful, only a �ne black precipitatewas formed.
174

7. Experimental
7.11. Protonation of Cp*Co(S2N2) with HClA solution of anhydrous HCl in dry CH2Cl2 was prepared by the following way: GaseousHCl - prepared by dropping conc. H2SO4 into conc. HCl placed in a �ask with an outlet- was dried by passing through a tube �lled with P2O5 and was directly bubbled intodry CH2Cl2 (20ml) in a Schlenk �ask immersed in an ice/water cooling bath. HCl wasbubbled for approx. 15 minutes by which time the volume of CH2Cl2 decreased to 10ml.
Ten drops of the HCl solution were added to Cp*Co(S2N2) (120mg, 0.420mmol) dis-solved at 0 ◦C in dry CH2Cl2 (10ml). An unidenti�ed grey substance formed immediatelyon the contact of Cp*Co(S2N2) with HCl but the overall colour remained violet. The�ask was �tted with an oil-bubbler and the mixture was gradually reduced in volume ina gentle nitrogen �ow. Unfortunately, no crystals were formed and the mixture deteri-orated with time which was indicated by blackening and precipitation of a black solidknown from previous attempts.
7.12. Computational detailsAll calculations were performed by Dr. Karla Tersago under the supervision of Prof.Frank Blockhuys at the University of Antwerp, Belgium. The calculations were per-formed on isolated molecules using Gaussian 03434 at the Density Functional (DFT)level of theory using the B1B95435 functional, in combination with the 6-311+G* ba-sis set as it is implemented in Gaussian 03 for hydrogen, carbon, nitrogen and sulfur.For cobalt, rhodium and iridium the def-TZVPP basis sets436�438 and Stuttgart ECPsfrom 1990 were used.439,440 The geometries of 46, 47, 48, 49 and 50 were calculatedin Cs symmetry; the geometry of 51 was calculated in C1 symmetry. Force �eld cal-culations were used to ascertain whether the resulting structures were energy minima.All subsequent calculations of molecular properties were performed using the optimisedgeometries. Wiberg bond orders were calculated using the NBO 3.1 program441 as it isimplemented in the Gaussian 03 program package.
175

Part IV.
Roesky's Sulfoxide, S3N2O
Abstract
This part of the thesis describes the structure and reactivity of 1-oxo-1,2,4,3,5-trithiadi-azole, S3N2O, also known as Roesky's sulfoxide. The compound was prepared and lowtemperature single crystal X-ray structure was determined. The structural data werecompared to the results of a detailed theoretical study. Reactivity of S3N2O is alsodescribed.
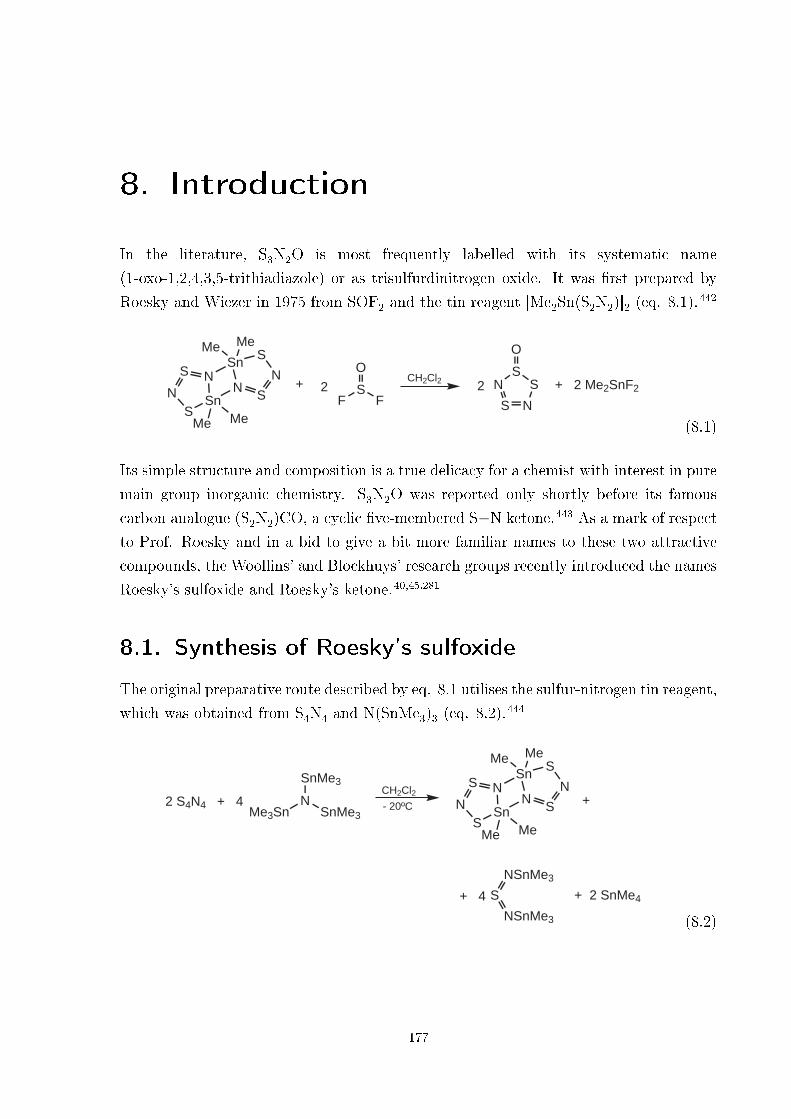
8. Introduction
In the literature, S3N2O is most frequently labelled with its systematic name(1-oxo-1,2,4,3,5-trithiadiazole) or as trisulfurdinitrogen oxide. It was �rst prepared byRoesky and Wiezer in 1975 from SOF2 and the tin reagent [Me2Sn(S2N2)]2 (eq. 8.1).442
+ 2 Me2SnF2CH2Cl2
S
O
F F+ N
S N
SS
OSn S
N
SN
SnS
N
S N
Me Me
MeMe
2 2
(8.1)
Its simple structure and composition is a true delicacy for a chemist with interest in puremain group inorganic chemistry. S3N2O was reported only shortly before its famouscarbon analogue (S2N2)CO, a cyclic �ve-membered S−N ketone.443 As a mark of respectto Prof. Roesky and in a bid to give a bit more familiar names to these two attractivecompounds, the Woollins' and Blockhuys' research groups recently introduced the namesRoesky's sulfoxide and Roesky's ketone.40,45,281
8.1. Synthesis of Roesky's sulfoxideThe original preparative route described by eq. 8.1 utilises the sulfur-nitrogen tin reagent,which was obtained from S4N4 and N(SnMe3)3 (eq. 8.2).444
2 S4N4 + 4 N
SnMe3
SnMe3Me3Sn
CH2Cl2- 20ºC
Sn S
N
SN
SnS
N
S N
Me Me
MeMe
+ 4 + 2 SnMe4S
NSnMe3
NSnMe3
+
(8.2)
177

8. Introduction
A safer method with no participation of both the toxic organotin reagents and the explo-sive S4N4 was introduced later. By hydrolysis of 1-chloro-1,2,4,3,5-trithiadiazolyl chlo-ride, [S3N2Cl]Cl, with anhydrous formic acid or acetic anhydride S3N2O can be obtainedin a good yield (eq. 8.3).445
N
S N
SS
O
+ HCOOHCH2Cl2, reflux
1 week+ 2 HCl + CON
S N
SS+
Cl
Cl-
(8.3)
S3N2O is obtained also in other reactions, of which none is su�ciently convenient tobecome a practical preparative route. For example the reaction of (Me2PhP)2Pt(S2N2)with SOCl2 gives S3N2O in a decent yield (35%, eq. 8.4).354 Platinum can be recoveredfrom the reaction mixture in the form of (Me2PhP)2PtCl2, which can be converted backto (Me2PhP)2Pt(S2N2) e.g. by reaction with Na(S3N3) (eq. 8.5).6 However, due to thehigh cost of primary platinum precursors this preparation of S3N2O cannot be a�ordedon bigger scales.
PR3 = PPhMe2
+ (PR3)2PtCl2CH2Cl2
S
O
Cl Cl+ N
S N
SS
O
N
S N
SPt
R3P PR3
r.t.
(8.4)
(PR3)2PtCl2 + 2 Na(S3N3) [(PR3)2Pt(S2N2)] + S4N4 + 2 NaClEtOH / CH2Cl2
r.t.
PR3 = PPhMe2 (8.5)
A cheaper alternative o�ers the reaction between Ni(S2N2H)2 and SOCl2 but this methodhas not been used frequently either.446 The last example is the formation of S3N2O ona non-preparative scale as one of �ve products when Na[S4N5] reacts with SOCl2 (eq.8.6). S3N2O was found only when the Na[S4N5] : SOCl2 ratio was greater than 1. In theopposite case S3N2O is converted to S(NSO)2 by the excess SOCl2. The mechanism ofthis complex reaction course has not been suggested.447
x Na[S4N5] + SOCl2 CH2Cl2, 0ºCS4N4 + S5N6 + S(NSO)2 + S3N2O + NaCl
x > 1
(8.6)
178

8. Introduction
8.2. Structure and reactivityS3N2O was isolated as a dense, red liquid which did not wet glass. Roesky and Wiezerdetermined its structure on the basis of elemental analysis and IR and mass spectra,442
later also 15N and 14N NMR data were published.426,448 However, the crystal structureof S3N2O was not reported. It is possible that the e�orts to perform X-ray analysison S3N2O were stopped after Roesky and coworkers noted in one of their papers thatS3N2O - being a red oily liquid - solidi�ed into a glassy material upon cooling.448 Thecrystal structure remained unresolved even after a later statement that S3N2O formsyellow needles with the melting point of 18 ◦C.445
The chemical behaviour of S3N2O has been investigated brie�y. It was shown that theoxygen atom can be substituted with a nitrogen bearing a suitable electron withdrawinggroup. Typically, the electron withdrawing group is a �uorinated derivative of sulfuric orsulfonic acid. The nitrogen atom is a part of an N -sul�nyl- (R−N−−S−−O) or an isocyano-(R−N−−C−−O) group. The S3N2O oxygen reacts with the sulfur or carbon atoms in theR−NSO or R−NCO groups, the groups undergo a cleavage, a molecule of a gas (SO2or CO2) is liberated and the nitrogen bearing the electron withdrawing substituent isattached to the �vemembered ring (eq. 8.7�8.9).355
S3N2O + + SO2SNF
O OS
ONS
NS
SS
N F
OO
50ºC
(8.7)
S3N2O + + CO2SNF3C
O O
C ONS
NS
SS
N CF3
OO
50ºC
(8.8)
S3N2O + + SO2SNC4F9
O OS
ONS
NS
SS
N C4F9
OO
50ºC
(8.9)
The presence of the electron withdrawing substituents together with the central sulfurin a high oxidation state was shown to be an important condition, since when the sameexperiments were carried out with CH3SO2NSO, CF3S(O)NSO or Me3SiNSO, only sulfurand S4N4 were obtained.355
The adducts of S3N2O with SnCl4, TiCl4, AsF5 and SbF5 were described, in whichS3N2O was coordinated to the metal exclusively through the oxygen atom.448 The samecoordination mode was observed in the complexes [M(S3N2O)6][AsF6]2 (M = Zn, Cd)prepared from the elemental metals, AsF5 and S3N2O in liquid SO2 at room temperature
179

8. Introduction
(eq. 8.10 and 8.11).
M + 3 AsF51. SO2 (l), -78ºC
[M(SO2)x][AsF6]2 + AsF3
M = Zn, Cd
2. r.t., 12 hrs
(8.10)
[M(SO2)x][AsF6]2
1. SO2 (l), -78ºC;
- x SO2
M2+
O
O O
O
O
O
N
S N
SS N
S
N
SS
N
S
NS
S
N
SN
SS
NS
N
SS
N
S
N S
S
[AsF6]22. + 6 S3N2O
3. r.t., 16 hrs
(8.11)
The number of the SO2 ligands (x ) in eq. 8.10 depends on the pressure of SO2 and variesfrom 2 to 4.449
The infrared spectra of both the adducts and the Zn and Cd complexes showed theS−−O stretch bands shifted towards lower frequencies in comparison with the free S3N2O,which indicates a weakening of the S−−O bond.449
A coordination via the nitrogen atom adjacent to the S−−O group in S3N2O was ob-served in the silver complex [Ag(S3N2O)2][AsF6]. The Ag centre is coordinated in alinear arrangement by the nitrogens of two S3N2O molecules. The entire coordinationenvironment around silver is, however, octahedral generated by additional two more dis-tant molecules of S3N2O coordinated through oxygens and by two [AsF6] � anions (eq.8.12).450
Ag+
O O N
S
NS
S
NS
N
SS
AgAsF6 + 2 S3N2OSO2 (l)
ON
S
N S
S
ON
S
N S
S
As-
F
F F
F
F
F
As-
F
F F
F
F
F (8.12)
At this point it must be noted that the structures of both the adducts with Lewis acidsand the Ag, Zn and Cd complexes were determined by X-ray analysis and therefore someinferred facts about the real structure of S3N2O were known, such as puckering of theS3N2 ring or approximate values of bond lengths and angles.
180

8. Introduction
S3N2O is also a potential source of the [S2N2] 2 � anion as was demonstrated by thereaction between chlorocomplexes of platinum and S3N2O in liquid ammonia (eq. 8.15).It was suggested that the [S2N2] 2 � anion is formed from NH4[S3N3], which results fromthe reaction between ammonia and S3N2O (eq. 8.13 and 8.14).324
2 S3N2O + 4 NH3 2 NH4[S3N3] + 2 H2O (8.13)
[NH4]2[S2N2] + S4N42 NH4[S3N3] (8.14)
[NH4]2[S2N2] + (PR3)2PtCl2
N S
NPt
S
R3P PR3
+ 2 NH4Cl
(8.15)
8.3. ConclusionRoesky's sulfoxide has been syntesised by several methods. Its structure has been deter-mined by non-crystallographic techniques and the crystal structures of transition metalcomplexes in which S3N2O �gures as a ligand revealed the shape of the molecule. How-ever, exact structural data of free S3N2O were not available until the publication of theresults of this work,40 which are presented in the next chapter.
181

9. Results and Discussion
9.1. The crystal structureWhen S3N2O in the form of the dark red oil obtained from reaction 8.3 was placed ina freezer, it solidi�ed into a brown and opaque solid. The fact that the solid was nottransparent indicated a non-glassy nature of the solid. Recrystallisation attempts fromsolvents at low temperatures lead only to separation of S3N2O in the form of solid bulks,not even a powder. Sublimation under static vacuum resulted in a formation of brightyellow and well shaped needles on the cooling �nger, which melted as soon as the puresubstance was transferred into a predried Schlenk �ask kept at room temperature. Thusit became clear that if single crystals were to be grown and subjected to X-ray di�raction,all procedures would have to be carried out at low temperatures.
Single crystals suitable for X-ray analysis sublimed over a bulk of pure S3N2O stored ina Schlenk �ask in a freezer for three weeks. The needles were very thin and had to be keptat low temperature throughout the selection and mounting process. The cooling devicewas a wide polystyrene box �lled with dry ice, which enabled a reasonably comfortablemanipulation with both the Schlenk �ask and the microscope glass plate.
The needle with the crystal had to be transported onto the di�ractometer while thewarm air protection layer around the low temperature device was switched o�, as evensuch a short contact of the crystal with a warm air caused melting of the crystal.
The crystal structure of S3N2O at 93K is shown in Fig. 9.1. The �ve-membered SNring is puckered with S(1) approx. 0.48Å above the S(2)−N(3)−S(4)−N(5) plane. Thetwo SN planes of the �ve-membered ring are inclined by 22◦ while the exocyclic oxygenis bound to the ring with the angle of 114◦ with respect to the S(1)−S(2)−N(5) plane.
The bond lengths and angles are listed in Table 9.1. The sulfur-nitrogen ring is char-acterised by two longer (about 1.64Å) and two shorter (about 1.57Å) S−N bonds anda S−S single bond (2.2158(9)Å). The S−S bond is rather long in comparison with thebonds in Sx rings (x = 6�12), in which the sulfur-sulfur single bonds are on average2.05Å, within the very narrow range between 1.998 and 2.113Å.451 The oxygen O(1) isbound to S(1) by a localised double bond (1.4769(17)Å).
The nature of the S−S single bond was investigated by theoretical calculations which
182

9. Results and Discussion
Fig. 9.1. The crystal structure of Roesky's sulfoxide (S3N2O)
con�rmed the presence of bonding between the atoms S(1) and S(2), though the bond isweak (bond order 0.7).40
Table 9.1. The bond lengths (Å) and angles (◦) for S3N2O
S(1)−O(1) 1.4769(17) S(1)−S(2)−N(3) 97.49(7)S(1)−S(2) 2.2158(9) S(2)−N(3)−S(4) 116.63(9)S(2)−N(3) 1.6372(18) N(3)−S(4)−N(5) 108.85(8)N(3)−S(4) 1.5689(19) S(4)−N(5)−S(1) 119.03(10)S(4)−N(5) 1.5827(18) N(5)−S(1)−S(2) 92.43(7)N(5)−S(1) 1.6447(16) N(5)−S(1)−O(1) 109.05(10)
O(1)−S(1)−S(2) 105.30(7)
The packing of S3N2O molecules in the crystal is shown in Fig. 9.2. The view alongthe crystallographic c axis reveals a stacked arrangement of molecules. Even though itis common in sulfur-nitrogen chemistry to observe intermolecular S···S interactions, anexamination of the structure of S3N2O shows that S···O contacts are the predominantpacking interactions (Fig. 9.2). All the S···O contacts are well within the sum of the vander Waals radii of sulfur and oxygen (3.20Å).
Fig. 9.2. The packing of S3N2O molecules in the crystal with highlighted intermolecularcontacts (distances in Å)
183

9. Results and Discussion
9.1.1. Aromaticity of Roesky's sulfoxideAromaticity of Roesky's sulfoxide was investigated by evaluation of geometrical andmagnetic criteria.
Taking into account the lengths of a single and a double S−N bond de�ned by Pauling(1.74 and 1.56Å, respectively),11 the S−N bond lengths in Roesky's sulfoxide (Table 9.1)can be classi�ed as intermediate between a single and a double S−N bond, which speaksin favour of π-electrons delocalisation and aromaticity. The calculated bond orders forthe S−N bonds re�ect the situation with the values being all well above 1.40
The values of NICS(0), NICS(1) and NICS(-1) obtained for Roesky's sulfoxide are allconvincingly negative suggesting that Roesky's sulfoxide is aromatic.
It must be noted that the calculated properties such as bond orders or NICS are basedon the calculated geometries for a gas phase molecule. However, since the agreementbetween the calculated and solid state geometries is perfect,40 the calculated propertiescan be extended also onto the structure obtained from X-ray di�raction.
9.2. Vibrational spectraA molecule of Roesky's sulfoxide is not symmetrical and therefore it belongs to thesymmetry point group C1. A molecule of S3N2O should give rise to 12 normal vibrationalmodes, of which not all were observed in the spectra presented here. Table 9.2 containsthe list of the IR and Raman frequencies and thanks to theoretical calculations also theirassignments. The assignments are based on the Potential Energy Distribution calculatedfor each of the normal modes. In case of mixing of symmetry coordinates only the majorcontribution is listed.
Table 9.2. Selected IR and Raman wavenumbers (in cm−1) of S3N2O
IR (KBr) Raman (l) Assignment1130 vs 1125 w ν S=O981 m � ν N(3)-S(4)911 m 914 m ν S(4)-N(5)735 s 737 w ν S(2)-N(3)670 m 676 w ν S(1)-N(5)582 w 583 vs ring def.503 s 505 w γ S=O384 m 390 w ring torsion287 w 301 vs ν S-S
245 m β S=O178 w ring torsion
In addition to the Raman lines listed in Table 9.2 an intense line at 1040 cm−1 appearedin the Raman spectrum, which cannot be assigned to any of the normal modes. The line
184

9. Results and Discussion
is probably a combination vibration. Quite a number of possible combinations producewavenumbers close to this value. The wavenumber assigned to ν S-S is somewhat higherthan those reported in the literature for a number of weak S−S bonds in sulfur-nitrogencages and bicyclic compounds, for which the values lie between 186 and 269 cm−1.452
This can be directly related to the fact that in Roesky's sulfoxide the S−S distance issomewhat shorter than those in the mentioned compounds (between 2.43 and 2.71Å).
9.3. 14N NMR spectroscopyThe 14N NMR spectrum of S3N2O contains two well de�ned, broad singlets at 254.8 and324.3 ppm. The theoretical calculations suggest that the deshielded signal is due to N(3).
9.4. Cyclic voltammetryThe voltammogram of S3N2O was recorded at several scan rates. At the scan rateof 0.1V s−1, S3N2O showed irreversible oxidation and reduction (Fig. 9.3), which is inaccord with previous polarographic measurements carried out by Chivers and Hojo.453
At higher rates (5�10V s−1) the oxidation was still irreversible but the reduction showedreversibility. Observed values of potentials Ep and currents Ip are presented in Table 9.3.To determine the number of electrons transferred during the redox reaction at the working
Table 9.3. Experimental and simulated voltammetric data for S3N2O
Ep [V] Ip [µA]experiment simulation experiment simulation
anodic 1.75 1.825 2.54 16.74cathodic −1.01 −1.025 −0.85 −10.24
electrode, a simulation of a one-electron process under the conditions of the experimentwas performed. The anodic current resulting from the simulated process is approx. 6×higher and the cathodic current approx. 12× higher than the currents observed in theexperiment (Table 9.3). The lower observed currents are most probably a consequenceof passivation of the working electrode caused by a strong adsorption through sulfur ornitrogen on its surface.
9.5. Reactivity of Roesky's sulfoxideThe reported reactions with Roesky's sulfoxide were summarised in the introductorypart. A molecule of S3N2O either remained preserved, or the exocyclic oxygen was
185

9. Results and Discussion
Fig. 9.3. Cyclic voltammogram of S3N2O
substituted, or the �ve-membered ring was fragmented and only certain part was presentin the product.
Reactions with metal carbonyls. The reaction of S3N2O with Cp*Co(CO)2 was car-ried out in the molar ratio 2:1 at room temperature in toluene with the intention ofreplacing the two CO ligands with S3N2O (eq. 9.1).
Cp*Co(CO)2 + 2 S3N2O Cp*Co(S3N2O)2 + 2 COtoluene
r.t. (9.1)
Under these conditions the reaction occured instantly forming a dark solution, from whichmixtures of products were separated by silica column chromatography. Their 1H NMRspectra contained a number of signals. The only product that could be safely identi�edwas Cp*Co(S2N2) (singlet at 1.96 ppm), which was eluted as a short violet band withthe mixture CH2Cl2 : ethyl acetate (1:1).
An analogous reaction with Cp2Ti(CO)2 in toluene at room temperature gave a di�erentresult. After two days the mixture stopped changing colour and gave a 1H NMR spectrumwith an intense singlet at 6.18 ppm, among others. Two products were separated bysilica column chromatography. Elution with CH2Cl2 removed the �rst product, whichwas collected as a red solution (Fraction A). The second product was eluted as a yellowband with the mixture CH2Cl2 : acetone (20:1) (Fraction B).
Fraction A was evaporated to dryness and the solid was analysed by 1H NMR. Thespectrum showed two singlets of equal intensity at 6.05 and 6.34 ppm. Mass spectrum(ES) of this product contained �ve peaks, the base peak was at m/z 685. Since elec-
186

9. Results and Discussion
trospray was used as ionisation method fragmentation of molecules was less likely andtherefore the origin of the remaining four peaks was not certain. It was not possibleto assign the peaks to any fragments or products of hydrolysis, solvolysis or fragmentrecombination. During an attempted recrystallisation by a slow evaporation of solventfrom a CH2Cl2 solution the product decomposed: a beige solid deposited in the samplevial which did not dissolve into a red solution again.
Fraction B was similarly analysed by 1H NMR and its spectrum showed a sharp singletat 6.18 ppm which was strong already in the spectrum of the crude mixture. Recrystalli-sation by a gas phase di�usion of hexane into a CH2Cl2 solution of the product ledto formation of orange crystals which were identi�ed by X-ray analysis as the knowncomplex Cp2Ti(N3)−O−Ti(N3)Cp2 (Fig. 9.4).454
Ti O Ti
NCp
Cp
NCp
Cp
N+
N-
N+
N-
Fig. 9.4. The structure of the complex Cp2Ti(N3)−O−Ti(N3)Cp2
N-metallation. Aucott et al. reported an N -metallation of the metalladithiadiazolesCpCo(S2N2) and Cp*Ir(S2N2) with a Au I electrophile. The electrophile is generatedfrom ClAu(PPh3) by chloride abstraction with AgClO4 and the metallation of the �ve-membered M(S2N2) ring occurs on the metal-bound nitrogen.455 Roesky's sulfoxideshowed similar reactivity pattern in the silver complex [Ag(S3N2O)2][AsF6] (eq. 8.12,page 180).450 It was therefore possible that Roesky's sulfoxide could form complexes alsowith the gold(I) electrophile. However, the experiment carried out in the dark at roomtemperature in THF resulted only in precipitation of a grey/black solid, presumably goldsul�de, which after �ltration reappeared in the �ltrate. No products could be obtainedfrom the reaction mixture.
Protonation. Attempts to protonate S3N2O with HBF4 resulted in formation of yellowplatelets of a possible product which unfortunately did not di�ract. Recrystallisationattempts resulted only in precipitation of yellow powders.
Reaction with Woollins' Reagent. During his systematic work withWoollins' Reagent(WR), Hua found that ketones react with WR upon C−C bond formation.456 It wastherefore in our interest to �nd out whether this reactivity pattern could be extended
187

9. Results and Discussion
also to Roesky's sulfoxide and ketone. Both reactions were unsuccessful, most proba-bly due to the conditions necessary for the reaction mixture to become homogeneous.Woollins' Reagent is only poorly soluble at room temperature and reactions with it aregenerally carried out in re�uxing toluene. 31P NMR showed that neither Roesky's sul-foxide nor ketone reacted with WR in toluene at room temperature within two days.Both compounds decomposed under the re�ux conditions.
The reaction mixture involving Roesky's sulfoxide showed decomposition on TLC.Attempts to isolate at least some products by crystallisation from concentrated hotsolution resulted only in formation of yellow precipitates.
The mixture containing Roesky's ketone was �rst concentrated till the �rst solid par-ticles appeared, then was heated to 60 ◦C and was allowed to cool slowly. Overnight,yellow needle-like crystals were formed which were identi�ed by X-ray analysis as sulfur.TLC showed no decomposition on silica and therefore the solution was puri�ed by silicacolumn chromatography. Elution with hexane and subsequently toluene removed Se3S5which was identi�ed by MS and Raman spectroscopy and later also by X-ray analysis.It was the only product that could be removed from the column. Final elution withmethanol removed colourless phosphorus containing decomposition products which wasshown by the 31P NMR.
9.6. ConclusionThe low temperature single crystal X-ray structure of Roesky's sulfoxide S3N2O wasdetermined. The data for a gas-phase structure obtained from theoretical calculationsare in a perfect agreement with the solid state data. Evaluation of S−N bond lengthstogether with the calculated values of bond orders and NICS indicate delocalisation ofπ-electrons over the S−N framework and aromaticity of Roesky's sulfoxide. With thehelp of calculations it was possible to perform vibrational analysis of S3N2O and toassign 14N NMR chemical shifts to particular nitrogen atoms. Roesky's sulfoxide showedinstability in chemical reactions with metal complexes performed at room temperaturein common organic solvents. In all cases the ring was disrupted. Reaction with Woollins'Reagent led to formation of powdery precipitates which could not be recrystallised.
188

10. Experimental
NH4Cl and sulfur �owers were purchased from Fisher and S2Cl2, AgClO4 and (Ph3P)AuClfrom Aldrich. Anhydrous formic acid was prepared from commercially available 95%formic acid (Aldrich) by re�uxing with phthalic anhydride for 8.5 hours followed by dis-tillation. The distillate was then re�uxed for 5 hours with anhydrous CuCl2 and distilledfrom CuCl2. Woollins' Reagent was prepared according to the published procedure.457
10.1. Preparation of S3N2O445
[S3N2Cl]Cl (15.5 g, 0.079mol) was placed in a 500ml �ask equipped with a stirring bar.Dry CH2Cl2 (300ml) was added to form a suspension. Anhydrous formic acid (6ml,7.32 g, 0.159mol) was added dropwise within 2 hours to the stirred suspension. The �askwith the mixture was �tted with a water re�ux condenser with an oil bubbler on the topand was gently re�uxed for 7 days. By then, the evolution of HCl ceased. The resultingorange mixture was concentrated to a quarter of its original volume, �ltered through asinter and the �ltrate was placed in a freezer. Overnight, orange needle-like crystals ofS4N4 were formed and some dark byproduct precipitated. Both were �ltered o� (sinter),the solvent was evaporated and the oily residue was distilled under high vacuum (toil =55�60 ◦C). The distillate was additionally puri�ed by vacuum sublimation in vacuum.Yield 2.45 g (22%).14N NMR (CDCl3, 298 K): δ = 324.3 (s, 1N, SNS), 254.8 (s, 1N, NSO).IR data: 1199 (sh), 1181 (m), 1130 (vs), 1041 (vw), 981 (ms), 911 (ms), 800 (vw), 735(s), 670 (ms), 582 (w), 556 (vw), 503 (s), 384 (ms), 287 (sh) cm−1.Raman data: 1205 (vw), 1183 (vw), 1125 (w), 1040 (s), 914 (m), 737 (sh), 701 (sh), 676(mw), 583 (vs), 566 (sh), 505 (w), 387 (sh), 374 (mw), 301 (s), 245 (m), 220 (sh), 178(w) cm−1.
10.2. Reaction of S3N2O with Cp*Co(CO)2Cp*Co(CO)2 (178mg, 0.713mmol) was dissolved in dry toluene (13ml). S3N2O (200mg,1.43mmol) was also dissolved in dry toluene and was added dropwise to the stirred
189

10. Experimental
solution of Cp*Co(CO)2. The colour of the mixture changed immediately to dark redwith some black precipitate formed. The mixture was stirred overnight, then �lteredthrough a sinter and to the dark brown/red �trate a small amount of silica was added.The solvent was evaporated and the products absorbed on silica were added to a packedcolumn of silica (20 × 1.5 cm) prepared in the mixture CH2Cl2 : ethyl acetate (2:1).Elutions were carried out with mixtures of these two solvents (2:1, then 1:1) followed byacetone. Analysis of the coloured eluates by 1H NMR spectroscopy revealed mixtures ofproducts with a number of signals, often broad and overlapping. The peak at 1.96 ppmfound in the 1H NMR spectrum of a short violet band removed with CH2Cl2 : ethylacetate (1:1) corresponded to Cp*Co(S2N2), other products could not be identi�ed.
10.3. Reaction of S3N2O with Cp2Ti(CO)2Cp2Ti(CO)2 (220mg, 0.940mmol) was dissolved in dry toluene (15ml). Solution ofS3N2O (263mg, 1.88mmol) in dry toluene (5ml) was added dropwise and the mixturewas stirred at room temperature for 2 days. Over that period, the mixture darkenedsigni�cantly and then turned orange/brown. The crude mixture was dissolved in a smallamount of toluene and was added to a packed silica column (20 × 1.5 cm). Elution withtoluene removed some impurities and elution with CH2Cl2 removed a pale pink bandcollected as a red solution (Fraction A). The mixture CH2Cl2 : acetone (20:1) eluted ayellow band (Fraction B).
Fraction A was evaporated to dryness. The solid gave 1H NMR signals at 6.05 and6.34 ppm. Mass spectrum (ES+TOF) of this byproduct contained peaks at m/z 702(1%), 686 (2.5%), 685 (100%), 413 (0.5%) and 265 (3%). The product decomposedduring recrystallisation.
Fraction B gave a 1H NMR spectrum with a sharp singlet at 6.18 ppm. Recrystalli-sation by a gas phase di�usion of hexane into a CH2Cl2 solution of the product led toformation of orange crystals which were identi�ed by X-ray analysis asCp2Ti(N3)−O−Ti(N3)Cp2.MS(ES+TOF): m/z 478.91 (100%) [[MNa]+].1H NMR (CDCl3, 298 K): δ = 6.18 (s, 20H, 4×Cp).
10.4. Reaction of S3N2O and[(Ph3P)Au(CH3CN)][ClO4]
Preparation of [(Ph3P)Au(CH3CN)][ClO4] AgClO4 (58mg, 0.279mmol) dissolvedin dry CH3CN (3ml) was added to a solution of [(Ph3P)AuCl] (138mg, 0.279mmol)
190

10. Experimental
in dry CH2Cl2 (3ml). A shiny white precipitate (AgCl) was formed immediately. Themixture was stirred in dark for 2 hours, the precipitate was �ltered o� (�lter paper) andthe clear �ltrate was evaporated to dryness to give a colourless solid.31P NMR (CDCl3, 298 K): δ = 29.3 (s, 1P, PPh3).
Reaction of [(Ph3P)Au(CH3CN)]ClO4 with S3N2O In a �ask wrapped in aluminiumfoil, [(Ph3P)Au(CH3CN)]ClO4 was redissolved in dry THF (5ml) and S3N2O (39mg,0.279mmol) in dry diethyl ether (1ml) was added dropwise from a syringe. The mixturewas stirred in the dark at room temperature overnight. 31P NMR spectrum of the crudemixture indicated no reaction happening. The resulting dark grey mixture was �lteredthrough a cotton wool plug and the clear pale yellow �ltrate was slowly concentratedunder a gentle �ow of a nitrogen gas. During this procedure more grey/black solid keptprecipitating signalling decomposition of S3N2O and formation of Au2S.
10.5. Protonation of S3N2O with HBF4
S3N2O (249mg, 1.77mmol) was placed in a Schlenk tube and was dissolved in dry Et2O(4ml). HBF4 ·Et2O (287mg, 1.77mmol) was added dropwise upon stirring and themixture was stirred for 1.5 hours at room temperature. No changes were observed. Thesolvents were evaporated in vacuo leaving pale yellow plates and a beige precipitate. Theplates did not di�ract and following recrystallisation attempts were unsuccessful.
The attempt was repeated in more diluted conditions with HBF4 ·Et2O being layeredover the S3N2O solution with no stirring applied. After 2 days of standing, the solventwas allowed to evaporate slowly in a very gentle nitrogen �ow through an oil bubbler.The concentrated mixture was placed in a freezer with no results.
10.6. Reaction of S3N2O with Woollins' ReagentWoollins' Reagent (0.517 g, 0.970mmol) was suspended in dry toluene (15ml). S3N2O(0.300 g, 2.14mmol) dissolved in dry toluene (5ml) was added from a syringe in oneportion and the mixture was stirred at room temperature for 2 days. After 31P NMRshowed no signal of a possible product, the mixture was re�uxed for 2 hours to give asuspension of a black solid in a yellow/orange solution. 31P NMR changed but the numberof signals was high. TLC showed that the product decomposed on silica. The mixturewas therefore �ltered through a sinter and the clear orange �ltrate was concentrated untilthe �rst solid particles appeared. The mixture was then heated to 70 ◦C and was left tocool down gradually to room temperature. No crystals were formed, only powdery solid
191

10. Experimental
precipitated.
10.7. Reaction of (S2N2)CO with Woollins' ReagentWoollins' Reagent (0.690 g, 1.29mmol) was put in a 100ml �ask and dry toluene (20ml)was added. Solid (S2N2)CO (0.343 g, 2.85mmol) was added to the stirred mixture inone portion and the mixture was stirred for 2 days. After 31P NMR showed no signal ofa possible product the mixture was re�uxed for 2 hours to give a suspension of a blacksolid in a yellow solution. The mixture was �ltered through a sinter and the clear yellow�ltrate was concentrated until the �rst solid particles appeared. The mixture was thenheated to 60 ◦C and was left to cool down gradually to room temperature. Overnight,yellow needle-like crystals were formed which were identi�ed by X-ray analysis as sulfur.TLC showed no decomposition on silica and therefore the solution was concentrated andwas added to a silica column (20 × 1.5 cm) packed in hexane. Elution with hexane andsubsequently toluene removed Se3S5.
10.8. Computational detailsThe calculations were performed by Dr. Karla Tersago under the supervision of prof.Frank Blockhuys at the University of Antwerp, Belgium. All calculations were performedon isolated molecules using the Gaussian 03434 and BRABO458 suites of programs ap-plying the Density Functional (DFT) level of theory, using the B1B95435 functional andthe aug-cc-pVTZ basis set, as it is implemented in Gaussian 03. Force �eld calcula-tions were used to ascertain whether the resulting structure was an energy minimum.All subsequent calculations of molecular properties were performed using the optimizedB1B95/aug-cc-pVTZ geometry.
Nucleus-Independent Chemical Shifts38 were calculated for the B1B95/aug-cc-pVTZgeometry at the B3LYP/aug-cc-pVTZ level of theory using the Gauge IndependentAtomic Orbitals (GIAO) method as it is implemented in Gaussian 03.
Chemical shifts for the nitrogen atoms were obtained by subtracting the chemicalshielding values of the two atoms from that calculated for ammonia at the B3LYP/aug-cc-pVTZ level of theory (260.9437 ppm) using the geometry in C3v symmetry optimisedat the same level of theory. For further details see the work by Tersago et al.40
192

Conclusion
The contribution of the presented thesis to the �eld of synthetic chemistry can be sum-marised in the following points.
Five 1,3,2,4,5-dithiadiazarsoles RAs(S2N2) were prepared and characterised to providesupplementary information to that published for MeAs(S2N2). The analogues bearingaromatic substituents (R = Ph, Mes) showed a remarkable thermal stability. The X-raystructures of these two analogues were determined showing a certain extent of �exibilityof the �ve-membered As(S2N2) ring, which was found to be both puckered and planar.The crystal structures revealed a network of intermolecular contacts. The series of thedithiadiazarsoles o�ered a good opportunity for a range of comparisons, among othersthe comparisons of vibrational spectra, the assignments of which are thus reinforced.
PhAs(S2N2) showed chemical stability in reactions with selected metal complexes andCH3I.
The e�ort to prepare Cp*Rh(S2N2) was crowned with success, although the X-raystructure could not be determined. The low temperature X-ray structures of Cp*M(S2N2)(M = Co, Ir) were determined and were compared to the calculated geometries. Both setsof data were found to be in a good agreement. It was found that the di�erences betweenparticular Cp*M(S2N2) structures are directly linked to the van der Waals' radius ofthe metal centre. Calculated bond orders suggested increasing π-electrons delocalisationwhen proceeding from Co to Ir.
The product of hydrolysis of Cp*Rh(S2N2) was identi�ed by X-ray analysis asCp*Rh(µ-S3N2)(µ-S2O3)RhCp*, which is a rare example of both the [S3N2] 2 � and[S2O3] � anions acting as bridging ligands in one molecule.
The in�uence of ligands on the structure of the Cp*Ir(S2N2L) complexes(L = [IrCl2Cp*] and [IrCl(PPh3)Cp*]) was examined.
The X-ray structure of Roesky's sulfoxide at 93K was determined and revealed apuckered ring with an unexpectedly long S−S single bond, the length of which is re�ectedin its relative weakness. An evaluation of geometrical data, calculated bond orders andNICS values suggests aromaticity of Roesky's sulfoxide.
Reactions of Roesky's sulfoxide carried out at room temperature resulted in general in
193

Conclusion
disruption of the �ve-membered ring.
194

Further work
The projects elaborated in this thesis still o�er challenging ideas.
RAs(S2N2). From the point of view of synthesis, it would be interesting to prepare1,4-bis-(1,3,2,4,5-dithiadiarsol-5-yl)benzene, because the connection of electron-rich sul-fur-nitrogen species with an aromatic hydrocarbon could exhibit interesting properties.For the same reason the syntheses of the same durene (1,2,4,5-tetramethylbenzene) andanthracene derivatives would be interesting.
Since PhAs(S2N2) showed chemical stability in reactions with metal complexes carriedout in common organic solvents, liquid SO2 or NH3 could prove a better environment.Should a suitable high pressure device be at hand, the reactions could be carried out alsoat room temperature.
Finally, thanks to its remarkable thermal stability, both PhAs(S2N2) and MesAs(S2N2)are excellent candidates for reactions with Woollins' Reagent.
Cp*M(S2N2). Given that Cp*Rh(S2N2) is prepared only with very small yields andthat its X-ray structure could not be determined, a search for a more e�cient preparativeroute is still on the list. A suitable derivatives could also bring success. It seems worthto try starting materials such as [NS2]+ or [NS]+. The syntheses of the Cp analogues ofboth Rh and Ir centres are also to be attempted.
Roesky's sulfoxide. The reactivity of Roesky's sulfoxide can still be expanded to awider range of metal complexes. Since experiments in common organic solvents at roomtemperature do not yield the desired products, liquid SO2 seems a better option. Thepreparation of an analogue of Roesky's sulfoxide bearing an oxygen on both sulfurs isalso of interest.
195

References[1] T. Chivers, Chem. Rev., 1985, 85, 341�365.[2] P. F. Kelly, A. M. Z. Slawin, D. J. Williams and J. D. Woollins, Chem. Soc. Rev., 1992, 21,
245�252.[3] T. Chivers, A Guide to Chalcogen-Nitrogen Chemistry, World Scienti�c Publishing Co. Pte. Ltd.,
Singapore, 2005.[4] W. L. Jolly and K. D. Maguire, Inorg. Synth., 1967, 9, 102�111.[5] P. S. Belton, I. P. Parkin, D. J. Williams and J. D. Woollins, J. Chem. Soc., Chem. Commun.,
1988, 1479�1480.[6] P. A. Bates, M. B. Hursthouse, P. F. Kelly and J. D. Woollins, J. Chem. Soc., Dalton Trans.,
1986, 2367�2370.[7] C. A. O'Mahoney, I. P. Parkin, D. J. Williams and J. D. Woollins, J. Chem. Soc., Dalton Trans.,
1989, 1179�1185.[8] S. M. Aucott, A. M. Z. Slawin and J. D. Woollins, Can. J. Chem., 2002, 80, 1481�1487.[9] D. F. Shriver, P. W. Atkins, T. L. Overton, J. P. Rourke, M. T. Weller and F. A. Armstrong,
Inorganic Chemistry, Oxford University Press, Oxford, UK, 4th edn., 2006.[10] V. Schomaker and D. P. Stevenson, J. Am. Chem. Soc., 1941, 63, 37�40.[11] L. Pauling, The Nature of the Chemical Bond and the Structure of Molecules and Crystals: An
Introduction to Modern Structural Chemistry, Cornell University Press, Ithaca, N.Y., USA, 3rdedn., 1960.
[12] B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragánand S. Alvarez, Dalton Trans., 2008, 2832�2838.
[13] P. Pyykkö and M. Atsumi, Chem. Eur. J., 2009, 15, 186�197.[14] J. C. van de Grampel and A. Vos, Acta Crystallogr. Sect. B-Struct. Crystallogr. Crystal Chem.,
1965, 25, 611�617.[15] P. G. Jones, W. Pinkert and G. M. Sheldrick, Acta Crystallogr. Sect. C-Cryst. Struct. Commun.,
1983, 39, 827�829.[16] M. Rock, P. Bravin and K. Seppelt, Z. Anorg. Allg. Chem., 1992, 618, 89�92.[17] R. Faggiani, R. J. Gillespie, C. J. L. Lock and J. D. Tyrer, Inorg. Chem., 1978, 17, 2975�2978.[18] T. Yoshimura, K. Hamada, M. Imado, K. Hamata, K. Tomoda, T. Fujii, H. Morita, C. Shimasaki,
S. Ono, E. Tsukurimichi, N. Furukawa and T. Kimura, J. Org. Chem., 1997, 62, 3802�3803.[19] W. H. Kirchho� and E. B. Wilson, Jr., J. Am. Chem. Soc., 1962, 84, 334�336.[20] T. Borrmann, E. Lork, R. Mews, S. Parsons, J. Petersen, W.-D. Stohrer and P. G. Watson, Inorg.
Chim. Acta, 2008, 361, 479�486.[21] M. L. DeLucia and P. Coppens, Inorg. Chem., 1978, 17, 2336�2338.[22] U. Müller, E. Conradi, U. Demant and K. Dehnicke, Angew. Chem.-Int. Edit. Engl., 1984, 23,
237�238.[23] R. J. Gillespie, J. P. Kent, J. F. Sawyer, D. R. Slim and J. D. Tyrer, Inorg. Chem., 1981, 20,
196

References
3799�3812.[24] S. Parsons and J. Passmore, Accounts Chem. Res., 1994, 27, 101�108.[25] W. J. Hehre, J. Yu, P. E. Klunzinger and L. Lou, A Brief Guide to Molecular Mechanics and
Quantum Chemical Calculations, Wavefunction, Inc., Irvine, CA, USA, 1998.[26] K. Tersago, personal communication.[27] J. Clayden, N. Greeves, S. Warren and P. Wothers, Organic Chemistry, Oxford University Press,
Oxford, UK, 2001.[28] P. von Ragué Schleyer and H. Jiao, Pure Appl. Chem., 1996, 68, 209�218.[29] A. Stanger, Chem. Commun., 2009, 1939�1947.[30] B. M. Gimarc and N. Trinajstic, Pure Appl. Chem., 1980, 52, 1443�1458.[31] J. Van Droogenbroeck, C. Van Alsenoy and F. Blockhuys, J. Phys. Chem. A, 2005, 109, 4847�
4851.[32] J. A. Pople, J. Chem. Phys., 1956, 24, 1111.[33] J. A. Pople and K. G. Untch, J. Am. Chem. Soc., 1966, 88, 4811�4815.[34] H. J. Dauben, Jr., J. D. Wilson and J. L. Laity, J. Am. Chem. Soc., 1968, 90, 811�813.[35] L. Pauling, J. Chem. Phys., 1936, 4, 673�677.[36] F. London, J. Phys. Radium, 1937, 8, 397�409.[37] P. von Ragué Shleyer, P. K. Freeman, H. Jiao and B. Goldfuss, Angew. Chem.-Int. Edit. Engl.,
1995, 34, 337�340.[38] P. von Ragué Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J.
Am. Chem. Soc., 1996, 118, 6317�6318.[39] P. von Ragué Schleyer, M. Manoharan, Z.-X. Wang, B. Kiran, H. Jiao, R. Puchta and N. J. R.
van Eikema Hommes, Org. Lett., 2001, 3, 2465�2468.[40] K. Tersago, V. Matuska, C. Van Alsenoy, A. M. Z. Slawin, J. D. Woollins and F. Blockhuys,
Dalton Trans., 2007, 4529�4535.[41] A. J. Banister, Nature, Phys. Sci., 1972, 237, 92�93.[42] R. Gleiter, Angew. Chem., 1981, 93, 442�450.[43] B. M. Gimarc, A. Juric and N. Trinajstic, Inorg. Chim. Acta, 1985, 102, 105�112.[44] A. Yu. Makarov, I. Yu. Bagryanskaya, F. Blockhuys, C. Van Alsenoy, Yu. V. Gatilov, V. V.
Knyazev, A. M. Maksimov, T. V. Mikhalina, V. E. Platonov, M. M. Shakirov and A. V. Zibarev,Eur. J. Inorg. Chem., 2003, 77�88.
[45] J. Van Droogenbroeck, K. Tersago, C. Van Alsenoy, S. M. Aucott, H. L. Milton, J. D. Woollinsand F. Blockhuys, Eur. J. Inorg. Chem., 2004, 3798�3805.
[46] J. Van Droogenbroeck, C. Van Alsenoy, S. M. Aucott, J. D. Woollins, A. D. Hunter and F. Block-huys, Organometallics, 2005, 24, 1004�1011.
[47] T. Chivers, Accounts Chem. Res., 1984, 17, 166�171.[48] D. D. Perrin and W. L. F. Armarego, Puri�cation of Laboratory Chemicals, Pergammon Press,
Oxford, UK, 3rd edn., 1988.[49] W. R. Cullen and K. J. Reimer, Chem. Rev., 1989, 89, 713�764.[50] C. Thom and K. B. Raper, Science, 1932, 76, 548�550.[51] W. R. Cullen, B. C. McBride, A. W. Pickett and J. Reglinski, Appl. Environ. Microbiol., 1984,
47, 443�444.[52] A. C. Anderson, A. A. Abdelghani and D. McDonell, Sci. Total Environ., 1980, 16, 95�98.[53] H. V. Warren, R. E. Delavault and J. Barakso, Can. Min. Metall. Bull., 1968, 61, 860�866.[54] J. S. Edmonds, K. A. Francesconi, J. R. Cannon, C. L. Raston, B. W. Skelton and A. H. White,
197

References
Tetrahedron Lett., 1977, 18, 1543�1546.[55] R. Xie, W. Johnson, S. Spayd, G. S. Hall and B. Buckley, J. Anal. At. Spectrom., 2007, 22,
553�560.[56] R. Zakharyan, Y. Wu, G. M. Bogdan and H. V. Aposhian, Chem. Res. Toxicol., 1995, 8, 1029�
1038.[57] K. T. Suzuki, T. Tomita, Y. Ogra and M. Ohmichi, Chem. Res. Toxicol., 2001, 14, 1604�1611.[58] J. U. Lakso and S. A. Peoples, J. Agric. Food Chem., 1975, 23, 674�676.[59] M. C. Brumm, A. L. Sutton, V. B. Mayrose and J. L. Krider, J. Anim. Sci., 1979, 48, 1305�1311.[60] B. Adam, Ph.D. thesis, Fakultät für Medizin, Technische Universität München, 2004.[61] M. N. Bates, A. H. Smith and C. Hopenhayn-Rich, Am. J. Epidemiol., 1992, 135, 462�476.[62] H. Tinwell, S. C. Stephens and J. Ashby, Environ. Health Perspect., 1991, 95, 205�210.[63] M. Krishnamohan, L. Qi, P. K. S. Lam, M. R. Moore and J. C. Ng, Toxicol. Appl. Pharmacol.,
2007, 224, 89�97.[64] W. R. Cullen, B. C. McBride and J. Reglinski, J. Inorg. Biochem., 1984, 21, 179�194.[65] V. P. Whittaker, Biochem. J., 1947, 41, 56�62.[66] H. V. Aposhian and M. M. Aposhian, Chem. Res. Toxicol., 2006, 19, 1�15.[67] A. Barchowsky, L. R. Klei, E. J. Dudek, H. M. Swartz and P. E. James, Free Radic. Biol. Med.,
1999, 27, 1405�1412.[68] M. W. G. De Bolster, Pure Appl. Chem., 1997, 69, 1251�1303, p. 1274.[69] T. Samikkannu, C.-H. Chen, L.-H. Yih, A. S. S. Wang, S.-Y. Lin, T.-C. Chen and K.-Y. Jan,
Chem. Res. Toxicol., 2003, 16, 409�414.[70] L. Stryer, Biochemistry, W. H. Freeman and company, New York, USA, 4th edn., 1995.[71] B. K. Mandal, Y. Ogra and K. T. Suzuki, Toxicol. Appl. Pharmacol., 2003, 189, 73�83.[72] A. Shraim, X. Cui, S. Li, J. C. Ng, J. Wang, Y. Jin, Y. Liu, L. Guo, D. Li, S. Wang, R. Zhang
and S. Hirano, Toxicol. Lett., 2003, 137, 35�48.[73] J. S. Petrick, F. Ayala-Fierro, W. R. Cullen, D. E. Carter and H. V. Aposhian, Toxicol. Appl.
Pharmacol., 2000, 163, 203�207.[74] H. V. Aposhian, R. Zakharyan, M. D. Avram, A. Sampayo-Reyes and M. L. Wollenberg, Toxicol.
Appl. Pharmacol., 2004, 198, 327�335.[75] Z. Drobna, S. B. Waters, V. Devesa, A. W. Harmon, D. J. Thomas and M. Styblo, Toxicol. Appl.
Pharmacol., 2005, 207, 147�159.[76] M. Frumar, T. Wagner and M. Vlcek, Eur. J. Solid State Inorg. Chem., 1991, 28, 1193�1203.[77] J. Kristo�k, J. J. Mares and V. Smid, Phys. Status Solidi A-Appl. Res., 1985, 89, 333�345.[78] N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Elsevier Butterworth-Heinemann,
Oxford, UK, 2nd edn., 1998.[79] J. Mason, Multinuclear NMR, Plenum Press, New York, USA, 1987.[80] J. C. Ho�man, Inorg. Synth., 1953, 4, 150�152.[81] J. C. Bailar, Jr., Inorg. Synth., 1939, 1, 103�104.[82] D. R. Aris, C. Knapp, J. Passmore and X. Wang, J. Fluor. Chem., 2005, 126, 1368�1372.[83] A. A. A. Emara, J. F. Lehmann and G. J. Schrobilgen, J. Fluor. Chem., 2005, 126, 1373�1376.[84] H. L. Sievers, H.-F. Grützmacher, H. Grützmacher and S. Pitter, J. Am. Chem. Soc., 1995, 117,
2313�2320.[85] F. J. Arnaiz and M. J. Miranda, Inorg. Synth., 2002, 33, 203�204.[86] H. B. North and A. M. Hageman, J. Am. Chem. Soc., 1913, 35, 352�356.[87] S. K. Pandey, A. Steiner and H. W. Roesky, Inorg. Synth., 1997, 31, 148�150.
198

References
[88] G. Meyer, Ber. Dtsch. Chem. Ges., 1883, 16, 1439�1443.[89] C. K. Banks, J. F. Morgan, R. L. Clark, E. B. Hatlelid, F. H. Kahler, H. W. Paxton, E. J. Cragoe,
R. J. Andres, B. Elpern, R. F. Coles, J. Lawhead and C. S. Hamilton, J. Am. Chem. Soc., 1947,69, 927�930.
[90] I. T. Millar, H. Heaney, D. M. Heinekey and W. C. Fernelius, Inorg. Synth., 1960, 6, 113�115.[91] A. J. Quick and R. Adams, J. Am. Chem. Soc., 1922, 44, 805�816.[92] K. W. Rosenmund, Ber. Dtsch. Chem. Ges., 1921, 54B, 438�440.[93] C. S. Hamilton and J. F. Morgan, Org. Reactions, 1944, 2, 415�454.[94] H. Bart, Verfahren zur Darstellung von organischen Arsenverbindungen, Ger. Pat. # DE 250264,
1910.[95] H. Bart, Improvements in the Manufacture of Organic Derivatives of Arsenic Acid, UK Pat. #
GB 191100568, 1911.[96] H. Bart, Process of Production of Organic Derivatives of Arsenic Acid, US Pat. # US 1061587,
1913.[97] H. Bart, Verfahren zur Darstellung von organischen Arsenverbindungen, Ger. Pat. # DE 254092,
1910.[98] H. Bart, Verfahren zur Darstellung von organischen Arsenverbindungen; Zusatz zum Patent
250264, Ger. Pat. # DE 254345, 1910.[99] H. Bart, Verfahren zur Darstellung von organischen Arsenverbindungen; Zusatz zum Patent
250264, Ger. Pat. # DE 268172, 1912.[100] H. Bart, Justus Liebigs Ann. Chem., 1922, 429, 55�103.[101] V. �t¥rba, in The Chemistry of Diazonium and Diazo Groups, Part 1, ed. S. Patai, John Wiley
& Sons, Ltd, Chichester, UK, 1978, pp. 71�77.[102] K. H. Saunders and R. L. M. Allen, Aromatic Diazo Compounds, Edward Arnold (Publishers)
Ltd, London, UK, 3rd edn., 1985.[103] C. Wittwer and H. Zollinger, Helv. Chim. Acta, 1954, 37, 1954�1968.[104] A. Hantzsch, Ber. Dtsch. Chem. Ges., 1894, 27, 1702�1725.[105] R. Huber, R. Langer and W. Hoppe, Acta Crystallogr., 1965, 18, 467�473.[106] S. Sorriso, in The Chemistry of Diazonium and Diazo Groups, Part 1, ed. S. Patai, John Wiley
& Sons, Ltd, Chichester, UK, 1978, pp. 105�106.[107] N. W. Alcock, P. D. Goodman and T. J. Kemp, J. Chem. Soc., Perkin Trans. 2, 1980, 1093�1095.[108] L. K. Keefer, S. Wang, T. Anjo, J. C. Fanning and C. S. Day, J. Am. Chem. Soc., 1988, 110,
2800�2806.[109] C. D. Ritchie and D. J. Wright, J. Am. Chem. Soc., 1971, 93, 2425�2428.[110] J. S. Littler, Trans. Faraday Soc., 1963, 59, 2296�2304.[111] E. S. Lewis and H. Suhr, Chem. Ber., 1958, 91, 2350�2358.[112] Z. Földi, Ber. Dtsch. Chem. Ges., 1923, 56B, 2489�2498.[113] E. Scheller, A Process for Arsenising Organic Compounds, UK Pat. # GB 261026, 1925.[114] E. Scheller, Verfahren zum Arsenieren organischer Verbindungen, Ger. Pat. # DE 522892, 1926.[115] F. E. Ray and R. J. Garascia, J. Org. Chem., 1950, 15, 1233�1240.[116] J. N. Latimer, Manufacture of Arsenic Acid, US Pat. # US 1974747, 1934.[117] E. Adams, D. Jeter, A. W. Cordes and J. W. Kolis, Inorg. Chem., 1990, 29, 1500�1503.[118] I. T. Millar, H. Heaney, D. M. Heinekey and W. C. Fernelius, Inorg. Synth., 1960, 6, 116�118.[119] R. J. Garascia, G. W. Batzis and J. O. Kroeger, J. Org. Chem., 1960, 25, 1271�1272.[120] K. J. Irgolic, R. A. Zingaro and L. J. Edmonson, Jr., Phosphorus Relat. Group V Elem., 1975, 5,
199

References
183�187.[121] A. McKenzie and J. K. Wood, J. Chem. Soc., 1920, 117, 406�415.[122] R. L. Dicarlo Jr., D. V. Shenai-Khatkhate, M. B. Power and A. Amamchyan, Preparation of
monoalkyl group 15 dihalides and dihydrides for use as chemical vapour deposition precursors,Eur. Pat. Appl. # EP 1335416, 2003.
[123] A. Tzschach and W. Deylig, Z. Anorg. Allg. Chem., 1965, 336, 36�41.[124] N. W. Alcock, E. M. Holt, J. Kuyper, J. J. Mayerle and G. B. Street, Inorg. Chem., 1979, 18,
2235�2239.[125] L. Weber, D. Bungardt, U. Sonnenberg and R. Boese, Angew. Chem.-Int. Edit. Engl., 1988, 27,
1537�1538.[126] C. Spang, F. T. Edelmann, M. Noltemeyer and H. W. Roesky, Chem. Ber., 1989, 122, 1247�1254.[127] H. Nöth and R. Waldhör, Z. Naturforsch. (B), 1999, 54, 603�608.[128] W. Steinkopf, H. Dudek and S. Schmidt, Ber. Dtsch. Chem. Ges., 1928, 61B, 1911�1918.[129] E. V. Avtomonov, K. Megges, S. Wocadlo and J. Lorberth, J. Organomet. Chem., 1996, 524,
253�261.[130] A. H. Cowley, J. G. Lasch, N. C. Norman and M. Pakulski, J. Am. Chem. Soc., 1983, 105,
5506�5507.[131] A. H. Cowley, J. E. Kildu�, J. G. Lasch, S. K. Mehrotra, N. C. Norman, M. Pakulski, B. R.
Whittlesey, J. L. Atwood and W. E. Hunter, Inorg. Chem., 1984, 23, 2582�2593.[132] P. Jutzi and M. Kuhn, Chem. Ber., 1974, 107, 1228�1234.[133] J. C. Pazik and C. George, Organometallics, 1989, 8, 482�487.[134] H. Burton and C. S. Gibson, J. Chem. Soc., 1926, 450�464.[135] J. A. Aeschlimann, J. Chem. Soc., 1927, 413�417.[136] C. S. Gibson and J. D. A. Johnson, J. Chem. Soc., 1931, 2518�2523.[137] P. Kilian, A. M. Z. Slawin and J. D. Woollins, Dalton Trans., 2006, 2175�2183.[138] P. Kilian and A. M. Z. Slawin, Dalton Trans., 2007, 3289�3296.[139] A. Tzschach and V. Kiesel, J. Prakt. Chem., 1971, 313, 259�264.[140] G. Kamai and Z. L. Khisamova, Dokl. Akad. Nauk SSSR, 1955, 105, 489�491.[141] K. Mödritzer, Chem. Ber., 1959, 92, 2637�2640.[142] K. Moedritzer, Inorg. Synth., 1967, 10, 131�135.[143] H.-J. Vetter and H. Nöth, Z. Anorg. Allg. Chem., 1964, 330, 233�244.[144] K. Moedritzer and J. R. Van Wazer, Inorg. Chem., 1964, 3, 139�144.[145] B. O. Pray, L. H. Sommer, G. M. Goldberg, G. T. Kerr, P. A. Di Giorgio and F. C. Whitmore, J.
Am. Chem. Soc., 1948, 70, 433�434.[146] H. P. Fritz and C. G. Kreiter, J. Organomet. Chem., 1964, 1, 323�327.[147] S. El Chaouch, J.-C. Guillemin, T. Kárpáti and T. Veszprémi, Organometallics, 2001, 20, 5405�
5412.[148] A. G. Davies and P. J. Smith, in Comprehensive Organometallic Chemistry; Volume 2, ed.
G. Wilkinson, F. G. A. Stone and E. W. Abel, Pergamon Press Ltd., Oxford, UK, 1st edn.,1982, pp. 536�542.
[149] A. G. Davies, in Comprehensive Organometallic Chemistry II; Volume 2, ed. E. W. Abel, F. G. A.Stone and G. Wilkinson, Elsevier Science Ltd., Oxford, UK, 1st edn., 1995, p. 249.
[150] J.-C. Guillemin and K. Malagu, Organometallics, 1999, 18, 5259�5263.[151] E. C. Lund and T. Livinghouse, Organometallics, 1990, 9, 2426�2427.[152] C. D. Abernethy, F. Bottomley, A. Decken and R. C. Thompson, Organometallics, 1997, 16,
200

References
1865�1869.[153] W. A. Herrmann, W. Kalcher, H. Biersack, I. Bernal and M. Creswick, Chem. Ber., 1981, 114,
3558�3571.[154] F. E. Kühn, W. A. Herrmann, R. Hahn, M. Elison, J. Blümel and E. Herdtweck, Organometallics,
1994, 13, 1601�1606.[155] E. W. Abel and S. Moorhouse, J. Chem. Soc., Dalton Trans., 1973, 1706�1711.[156] G. P. Sollott and W. R. Peterson, Jr., J. Org. Chem., 1965, 30, 389�393.[157] M. Rosenblum and F. W. Abbate, J. Am. Chem. Soc., 1966, 88, 4811�4815.[158] I. L. Knunyants and V. Y. Pilskaya, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1955, 417�422.[159] I. L. Knunyants and V. Y. Pilskaya, Izv. Akad. Nauk SSSR, Ser. Khim., 1955, 472�479.[160] M. S. Kharasch, E. V. Jensen and S. Weinhouse, J. Org. Chem., 1949, 14, 429�432.[161] L. H. Long, H. J. Emeléus and H. V. A. Briscoe, J. Chem. Soc., 1946, 1123�1126.[162] K. Sommer, Z. Anorg. Allg. Chem., 1970, 377, 128�134.[163] K. Megges, E. V. Avtomonov and J. Lorberth, Z. Naturforsch. (B), 1997, 52, 790�794.[164] W. L. Lewis and H. W. Stiegler, J. Am. Chem. Soc., 1925, 47, 2546�2556.[165] F. F. Blicke and G. L. Webster, J. Am. Chem. Soc., 1937, 59, 534�537.[166] L. A. Stocken and R. H. S. Thompson, Biochem. J., 1946, 40, 529�535.[167] L. A. Stocken and R. H. S. Thompson, Biochem. J., 1946, 40, 535�548.[168] L. A. Stocken and R. H. S. Thompson, Biochem. J., 1946, 40, 548�554.[169] M. G. Ord and L. A. Stocken, Trends Biochem. Sci., 2000, 25, 253�256.[170] L. A. Stocken, J. Chem. Soc., 1947, 592�595.[171] J. D. Andose, A. Rauk and K. Mislow, J. Am. Chem. Soc., 1974, 96, 6904�6907.[172] K. K. Baldridge and M. S. Gordon, J. Am. Chem. Soc., 1988, 110, 4204�4208.[173] M. K. Cyra«ski, T. M. Krygowski, A. R. Katritzky and P. von Ragué Schleyer, J. Org. Chem.,
2002, 67, 1333�1338.[174] H. Fallah-Bagher-Shaidaei, C. S. Wannere, C. Corminboeuf, R. Puchta and P. von Ragué Schleyer,
Org. Lett., 2006, 8, 863�866; Supporting Information, p. S2 and S4.[175] A. J. Ashe III and F. J. Drone, Organometallics, 1985, 4, 1478�1480.[176] L. S. Sunderlin, D. Panu, D. B. Puranik, A. J. Ashe, III and R. R. Squires, Organometallics, 1994,
13, 4732�4740.[177] A. J. Ashe III, S. Mahmoud, C. Elschenbroich and M. Wünsch, Angew. Chem., 1987, 99, 249�250.[178] K. C. Caster, in Comprehensive Heterocyclic Chemistry II, Volume 2, ed. A. R. Katritzky, C. W.
Rees and E. F. V. Scriven, Elsevier Science Ltd., Oxford, UK, 1st edn., 1996, pp. 857�902.[179] F. Nief, L. Ricard and F. Mathey, Polyhedron, 1993, 12, 19�26.[180] A. J. Ashe, III, S. Al-Ahmad, S. Pilotek, D. B. Puranik, C. Elschenbroich and A. Behrendt,
Organometallics, 1995, 14, 2689�2698.[181] P. Jutzi, J. Organomet. Chem., 1969, 19, P1�P2.[182] P. Jutzi and K. Deuchert, Angew. Chem.-Int. Edit. Engl., 1969, 8, 991�992.[183] H. Vermeer and F. Bickelhaupt, Angew. Chem.-Int. Edit. Engl., 1969, 8, 992.[184] A. J. Ashe, III and P. Shu, J. Am. Chem. Soc., 1971, 93, 1804�1805.[185] A. J. Ashe, III, J. Am. Chem. Soc., 1971, 93, 3293�3295.[186] A. J. Ashe, III, D. J. Bellville and H. S. Friedman, J. Chem. Soc., Chem. Commun., 1979, 880�881.[187] A. J. Ashe, III, X. Fang and J. W. Kampf, Organometallics, 2001, 20, 2109�2113.[188] C. Elschenbroich, J. Kroker, M. Nowotny, A. Behrendt, B. Metz and K. Harms, Organometallics,
1999, 18, 1495�1503.
201

References
[189] A. J. Ashe, III and J. C. Colburn, J. Am. Chem. Soc., 1977, 99, 8099�8100.[190] C. Elschenbroich, J. Kroker, W. Massa, M. Wünsch and A. J. Ashe, III, Angew. Chem., 1986, 98,
562�563.[191] C. Elschenbroich, J. Six and K. Harms, Dalton Trans., 2008, 92�95.[192] C. S. Palmer and A. B. Scott, J. Am. Chem. Soc., 1928, 50, 536�541.[193] J. H. Burns and J. Waser, J. Am. Chem. Soc., 1957, 79, 859�864.[194] D. S. Warren, B. M. Gimarc and M. Zhao, Inorg. Chem., 1994, 33, 710�715.[195] A. L. Rheingold, M. J. Foley and P. J. Sullivan, J. Am. Chem. Soc., 1982, 104, 4727�4729.[196] F. De Proft, P. W. Fowler, R. W. A. Havenith, P. von Ragué Schleyer, G. Van Lier and P. Geerlings,
Chem. Eur. J., 2004, 10, 940�950.[197] W. G. Xu and B. Jin, Theochem-J. Mol. Struct., 2006, 759, 101�107.[198] O. J. Scherer, C. Blath and G. Wolmershäuser, J. Organomet. Chem., 1990, 387, C21�C24.[199] B. Rink, O. J. Scherer and G. Wolmershäuser, Chem. Ber., 1995, 128, 71�73.[200] J. Frunzke, M. Lein and G. Frenking, Organometallics, 2002, 21, 3351�3359.[201] M. Lein, J. Frunzke and G. Frenking, Inorg. Chem., 2003, 42, 2504�2511.[202] B. Kesanli, J. Fettinger, B. Scott and B. Eichhorn, Inorg. Chem., 2004, 43, 3840�3846.[203] J. W. B. Reesor and G. F. Wright, J. Org. Chem., 1957, 22, 382�385.[204] A. L. Rheingold and P. J. Sullivan, Organometallics, 1983, 2, 327�331.[205] O. J. Scherer, H. Sitzmann and G. Wolmershäuser, Angew. Chem., 1989, 101, 214�215.[206] O. J. Scherer, H. Sitzmann and G. Wolmershäuser, J. Organomet. Chem., 1986, 309, 77�86.[207] P. Jutzi and T. Wippermann, J. Organomet. Chem., 1985, 287, C5�C7.[208] P. Jutzi and R. Kroos, Chem. Ber., 1988, 121, 1399�1401.[209] F. Kraus, T. Hanauer and N. Korber, Angew. Chem.-Int. Edit., 2005, 44, 7200�7204.[210] T. Baruah, M. R. Pederson, R. R. Zope and M. R. Beltrán, Chem. Phys. Lett., 2004, 387, 476�480.[211] M. J. Moses, J. C. Fettinger and B. W. Eichhorn, Science, 2003, 300, 778�780.[212] B. Kesanli, S. Charles, Y.-F. Lam, S. G. Bott, J. Fettinger and B. Eichhorn, J. Am. Chem. Soc.,
2000, 122, 11101�11107.[213] C.-C. Chang, R. C. Haltiwanger and A. D. Norman, Inorg. Chem., 1978, 17, 2056�2062.[214] E. J. Porter and G. M. Sheldrick, J. Chem. Soc., Dalton Trans., 1972, 1347�1349.[215] A. J. Banister, Nature, Phys. Sci., 1972, 239, 69�71.[216] M. F. Bräu and A. P�tzner, Z. Anorg. Allg. Chem., 2007, 633, 935�937.[217] M. F. Bräu and A. P�tzner, Angew. Chem., 2006, 118, 4576�4578.[218] H. Gruber and U. Müller, Z. Anorg. Allg. Chem., 1997, 623, 1033�1034.[219] H. Brunner, B. Nuber, L. Poll, G. Roidl and J. Wachter, Chem. Eur. J., 1997, 3, 57�61.[220] H. Brunner, L. Poll and J. Wachter, Polyhedron, 1996, 15, 573�576.[221] H. Brunner, H. Kauermann, L. Poll, B. Nuber and J. Wachter, Chem. Ber., 1996, 129, 657�662.[222] H. Brunner, F. Leis, J. Wachter and B. Nuber, Organometallics, 1997, 16, 4954�4955.[223] A. Kutoglu, Z. Anorg. Allg. Chem., 1976, 419, 176�184.[224] A. G. Evans and E. Warhurst, Trans. Faraday Soc., 1948, 44, 189�195.[225] M. J. Begley, D. B. Sowerby and R. J. Tillott, J. Chem. Soc., Dalton Trans., 1974, 2527�2530.[226] H. W. Roesky, R. Bohra and W. S. Sheldrick, J. Fluor. Chem., 1983, 22, 199�203.[227] R. Bohra, H. W. Roesky, J. Lucas, M. Noltemeyer and G. M. Sheldrick, J. Chem. Soc., Dalton
Trans., 1983, 1011�1014.[228] D. Hass, Z. Anorg. Allg. Chem., 1963, 325, 139�148.[229] E. V. Avtomonov, K. Megges, X. Li, J. Lorberth, S. Wocadlo, W. Massa, K. Harms, A. V.
202

References
Churakov and J. A. K. Howard, J. Organomet. Chem., 1997, 544, 79�89.[230] J. Weiss and W. Eisenhuth, Z. Anorg. Allg. Chem., 1967, 350, 9�17.[231] H.-J. Vetter, H. Nöth and W. Jahn, Z. Anorg. Allg. Chem., 1964, 328, 144�153.[232] O. J. Scherer and R. Wies, Angew. Chem.-Int. Edit. Engl., 1971, 10, 812�813.[233] A. Gieren, H. Betz, T. Hübner, V. Lamm, M. Herberhold and K. Guldner, Z. Anorg. Allg. Chem.,
1984, 513, 160�174.[234] M. Herberhold and W. Ehrenreich, Angew. Chem., 1982, 94, 637�638.[235] A. Bondi, J. Phys. Chem., 1964, 68, 441�451.[236] D. Clark, J. Chem. Soc., 1952, 1615�1620.[237] A. Gieren, T. Hübner, M. Herberhold, K. Guldner and G. Süss-Fink, Z. Anorg. Allg. Chem., 1987,
544, 137�148.[238] M. Herberhold, K. Schamel, G. Herrmann, A. Gieren, C. Ruiz-Pérez and T. Hübner, Z. Anorg.
Allg. Chem., 1988, 562, 49�61.[239] M. Herberhold and K. Schamel, J. Organomet. Chem., 1988, 346, 13�22.[240] T. Chivers, K. S. Dhathathreyan, C. Lensink and J. F. Richardson, Inorg. Chem., 1988, 27,
1570�1574.[241] F. Edelmann, C. Spang, M. Noltemeyer, G. M. Sheldrick, N. Keweloh and H. W. Roesky, Z.
Naturforsch. (B), 1987, 42, 1107�1109.[242] M. Herberhold, K. Schamel, A. Gieren and T. Hübner, Phosphorus Sulfur Silicon Relat. Elem.,
1989, 41, 355�360.[243] M. Herberhold, K. Guldner, A. Gieren, C. Ruiz-Pérez and T. Hübner, Angew. Chem., 1987, 99,
81�82.[244] K. Sommer and W. Lauer, Z. Anorg. Allg. Chem., 1970, 378, 310�314.[245] J. M. Mercero, X. Lopez, J. E. Fowler and J. M. Ugalde, J. Phys. Chem. A, 1997, 101, 5574�5579.[246] J.-H. Chou, J. A. Hanko and M. G. Kanatzidis, Inorg. Chem., 1997, 36, 4�9.[247] J.-H. Chou and M. G. Kanatzidis, Chem. Mater., 1995, 7, 5�8.[248] T. Bali¢-�uni¢ and P. Engel, Z. Kristallogr., 1983, 165, 261�269.[249] N. Burford, B. W. Royan, J. M. Whalen, J. F. Richardson and R. D. Rogers, J. Chem. Soc.,
Chem. Commun., 1990, 1273�1275.[250] G. Olah and A. Oswald, Can. J. Chem., 1960, 38, 1428�1430.[251] H.-J. Vetter, H. Strametz and H. Nöth, Angew. Chem., 1963, 75, 417�418.[252] H.-J. Vetter, H. Nöth and U. Hayduk, Z. Anorg. Allg. Chem., 1964, 331, 35�41.[253] P. B. Hitchcock, M. F. Lappert, A. K. Rai and H. D. Williams, J. Chem. Soc., Chem. Commun.,
1986, 1633�1634.[254] N. Burford, T. S. Cameron, C. L. B. Macdonald, K. N. Robertson, R. Schurko, D. Walsh, R. Mc-
Donald and R. E. Wasylishen, Inorg. Chem., 2005, 44, 8058�8064.[255] R. Bohra, H. W. Roesky, M. Noltemeyer and G. M. Sheldrick, Acta Crystallogr. Sect. C-Cryst.
Struct. Commun., 1984, 40, 1150�1152.[256] P. C. Andrews, C. L. Raston, B. W. Skelton, V.-A. Tolhurst and A. H. White, Chem. Commun.,
1998, 575�576.[257] I. Silaghi-Dumitrescu, A. Horea, S. Pascu, L. Silaghi-Dumitrescu and I. Haiduc, Phosphorus Sulfur
Silicon Relat. Elem., 1997, 124&125, 441�444.[258] O. J. Scherer and R. Wies, Angew. Chem.-Int. Edit. Engl., 1972, 11, 529.[259] A. Gieren, C. Ruiz-Pérez, T. Hübner, M. Herberhold, K. Schamel and K. Guldner, J. Organomet.
Chem., 1989, 366, 105�120.
203

References
[260] T. J. Bastow and H. J. Whit�eld, J. Chem. Soc., Dalton Trans., 1973, 1739�1740.[261] P. Goldstein and A. Paton, Acta Crystallogr. Sect. B-Struct. Crystallogr. Crystal Chem., 1974,
30, 915�920.[262] V. Angilella, H. Mercier and C. Belin, J. Chem. Soc., Chem. Commun., 1989, 1654�1655.[263] M. A. Ansari, J. A. Ibers, S. C. O'Neal, W. T. Pennington and J. W. Kolis, Polyhedron, 1992, 11,
1877�1881.[264] M. Grothe and N. Korber, Z. Anorg. Allg. Chem., 2003, 629, 399�402.[265] T. M. Martin, P. T. Wood, G. L. Schimek, W. T. Pennington and J. W. Kolis, Inorg. Chem.,
1995, 34, 4385�4391.[266] S. C. O'Neal, W. T. Pennington and J. W. Kolis, Inorg. Chem., 1992, 31, 888�894.[267] L.-R. Frank, K. Evertz, L. Zsolnai and G. Huttner, J. Organomet. Chem., 1987, 335, 179�188.[268] G. Cordier, C. Schwidetzky and H. Schäfer, Rev. Chim. Miner., 1985, 22, 93�100.[269] A. Ugrinov, A. Sen, A. C. Reber, M. Qian and S. N. Khanna, J. Am. Chem. Soc., 2008, 130,
782�783.[270] S. M. Aucott, A. M. Z. Slawin and J. D. Woollins, Phosphorus Sulfur Silicon Relat. Elem., 2001,
168&169, 559�562.[271] H. S. Booth and J. F. Suttle, J. Am. Chem. Soc., 1946, 68, 2658�2660.[272] M. R. Detty and M. D. Seidler, J. Org. Chem., 1981, 46, 1283�1292.[273] P. A. McCusker and E. L. Reilly, J. Am. Chem. Soc., 1953, 75, 1583�1585.[274] D. L. Alleston and A. G. Davies, J. Chem. Soc., 1962, 2050�2054.[275] R. C. Paul, K. K. Soni and S. P. Narula, J. Organomet. Chem., 1972, 40, 355�357.[276] W. P. Neumann and G. Burkhardt, Justus Liebigs Ann. Chem., 1963, 663, 11�21.[277] L. J. Goldsworthy, W. H. Hook, J. A. John, S. G. P. Plant, J. Rushton and L. M. Smith, J. Chem.
Soc., 1948, 2208�2212.[278] H. Lieb, Ber. Dtsch. Chem. Ges., 1921, 54B, 1511�1519.[279] H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512�7515.[280] G. M. Bodner, C. Gagnon and D. N. Whittern, J. Organomet. Chem., 1983, 243, 305�314.[281] K. Tersago, J. Van Droogenbroeck, C. Van Alsenoy, W. A. Herrebout, B. J. van der Veken, S. M.
Aucott, J. D. Woollins and F. Blockhuys, Phys. Chem. Chem. Phys., 2004, 6, 5140�5144.[282] K. J. R. Rosman and P. D. P. Taylor, Pure Appl. Chem., 1998, 70, 217�235.[283] J. R. Durig, M.-S. Cheng, M. E. Harris and T. J. Hizer, J. Mol. Struct., 1989, 192, 47�62.[284] I. Nevitt, H. S. Rzepa and J. D. Woollins, Spectroc. Acta Pt. A-Molec. Biomolec. Spectr., 1989,
45, 367�369.[285] A. Turowski, R. Appel, W. Sawodny and K. Molt, J. Mol. Struct., 1978, 48, 313�323.[286] W. Bues, M. Somer and W. Brockner, Z. Anorg. Allg. Chem., 1983, 499, 7�14.[287] M. Muniz-Miranda, G. Sbrana, P. Bonazzi, S. Menchetti and G. Pratesi, Spectroc. Acta Pt. A-
Molec. Biomolec. Spectr., 1996, 52, 1391�1401.[288] G. Davidson and K. P. Ewer, Spectroc. Acta Pt. A-Molec. Biomolec. Spectr., 1983, 39, 419�428.[289] G. Davidson and S. Phillips, Spectroc. Acta Pt. A-Molec. Biomolec. Spectr., 1979, 35, 141�146.[290] J. R. Durig, C. F. Jumper and J. N. Willis, Jr., Appl. Spectrosc., 1971, 25, 218�225.[291] L. D. Pettit and D. Turner, Spectroc. Acta Pt. A-Molec. Biomolec. Spectr., 1968, 24, 999�1006.[292] J. Ellermann, H. Schössner, A. Haag and H. Schödel, J. Organomet. Chem., 1974, 65, 33�49.[293] R. R. Shagidullin, F. G. Khalitov, G. M. Doroshkina, A. K. Plyamovatyi, D. F. Fazliev and B. E.
Abolonin, J. Struct. Chem., 1982, 22, 792�794.[294] D. M. Revitt and D. B. Sowerby, J. Chem. Soc. A, 1970, 1218�1222.
204

References
[295] P. Klaboe, Spectroc. Acta Pt. A-Molec. Biomolec. Spectr., 1970, 26, 87�108.[296] R. R. Holmes and M. Fild, Spectroc. Acta Pt. A-Molec. Biomolec. Spectr., 1971, 27, 1537�1547.[297] J. C. Evans and G. Y.-S. Lo, J. Am. Chem. Soc., 1966, 88, 2118�2122.[298] J. E. Bertie and S. Sunder, Can. J. Chem., 1973, 51, 3344�3353.[299] D. H. Whi�en, J. Chem. Soc., 1956, 1350�1356.[300] G. Herzberg, Molecular Spectra and Molecular Structure. Vol. II, Infrared and Raman Spectra of
Polyatomic Molecules, D. Van Nostrand Company, Inc., Princeton, New Jersey, USA, 1945, p.362.
[301] E. B. Wilson, Phys. Rev., 1934, 45, 706�714.[302] H. Stenzenberger and H. Schindlbauer, Spectroc. Acta Pt. A-Molec. Biomolec. Spectr., 1970, 26,
1713�1721.[303] H. Schindlbauer and H. Stenzenberger, Spectroc. Acta Pt. A-Molec. Biomolec. Spectr., 1970, 26,
1707�1712.[304] D. M. Revitt and D. B. Sowerby, Spectroc. Acta Pt. A-Molec. Biomolec. Spectr., 1970, 26, 1581�
1593.[305] B. Kainz and A. Schmidt, Spectroc. Acta Pt. A-Molec. Biomolec. Spectr., 1990, 46, 1361�1370.[306] M. Y. Kraft, G. M. Borodina, I. N. Stre©tsova and Y. T. Struchkov, Dokl. Akad. Nauk SSSR,
1960, 131, 1074�1076.[307] K. Hedberg, E. W. Hughes and J. Waser, Acta Crystallogr., 1961, 14, 369�374.[308] M. Herberhold, S. Gerstmann, W. Milius, B. Wrackmeyer and H. Borrmann, Phosphorus Sulfur
Silicon Relat. Elem., 1996, 112, 261�279.[309] P. F. Kelly, A. M. Z. Slawin, D. J. Williams and J. D. Woollins, Polyhedron, 1988, 7, 1925�1930.[310] P. Bhattacharyya, A. M. Z. Slawin, M. B. Smith and J. D. Woollins, Inorg. Chem., 1996, 35,
3675�3682.[311] F. Novio, R. Mas-Ballesté, I. Gallardo, P. González-Duarte, A. Lledós and N. Vila, Dalton Trans.,
2005, 2742�2753.[312] N. Moldovan, P. Lönnecke, I. Silaghi-Dumitrescu, L. Silaghi-Dumitrescu and E. Hey-Hawkins,
Inorg. Chem., 2008, 47, 1524�1531.[313] G. G. Briand, T. Chivers and M. Parvez, J. Chem. Soc., Dalton Trans., 2002, 3785�3786.[314] L. Silaghi-Dumitrescu, M. N. Gibbons, I. Silaghi-Dumitrescu, J. Zukerman-Schpector, I. Haiduc
and D. B. Sowerby, J. Organomet. Chem., 1996, 517, 101�106.[315] J. X. McDermott, J. F. White and G. M. Whitesides, J. Am. Chem. Soc., 1976, 98, 6521�6528.[316] D. J. Sikora, K. J. Moriarty and M. D. Rausch, Inorg. Synth., 1990, 28, 248�256.[317] E. Vedejs and N. Lee, J. Am. Chem. Soc., 1995, 117, 891�900.[318] H. Schmidt, Justus Liebigs Ann. Chem., 1920, 421, 159�174.[319] O. C. M. Davis, J. Chem. Soc., Trans., 1906, 89, 1575�1578.[320] H. Wölbling, Z. Anorg. Chem., 1908, 57, 281�289.[321] P. F. Kelly and J. D. Woollins, Polyhedron, 1986, 5, 607�632.[322] T. Chivers and F. Edelmann, Polyhedron, 1986, 5, 1661�1699.[323] I. P. Parkin, A. M. Z. Slawin, D. J. Williams and J. D. Woollins, J. Chem. Soc., Chem. Commun.,
1989, 1060�1061.[324] I. P. Parkin and J. D. Woollins, J. Chem. Soc., Dalton Trans., 1990, 519�523.[325] R. D. W. Kemmitt, S. Mason, M. R. Moore and D. R. Russell, J. Chem. Soc., Dalton Trans.,
1992, 409�415.[326] P. Blais, J. K. Brask, T. Chivers and G. Schatte, Inorg. Chem., 2001, 40, 384�388.
205

References
[327] R. C. Mills, P. Doufou, K. A. Abboud and J. M. Boncella, Polyhedron, 2002, 21, 1051�1055.[328] S. E. Kabir, M. Ruf and H. Vahrenkamp, J. Organomet. Chem., 1996, 512, 261�263.[329] J. D. Wilkins, J. Inorg. Nucl. Chem., 1976, 38, 673�675.[330] W. E. Lindsell and G. R. Faulds, J. Chem. Soc., Dalton Trans., 1975, 40�43.[331] R. Meij and K. Olie, Cryst. Struct. Commun., 1975, 4, 515�520.[332] C. Mahabiersing, W. G. J. de Lange, K. Goubitz and D. J. Stufkens, J. Organomet. Chem., 1993,
461, 127�139.[333] J. Kuyper, L. G. Hubert-Pfalzgraf, P. C. Keijzer and K. Vrieze, J. Organomet. Chem., 1976, 108,
271�280.[334] H. W. Roesky, H.-G. Schmidt, M. Noltemeyer and G. M. Sheldrick, Chem. Ber., 1983, 116,
1411�1414.[335] O. Ru� and E. Geisel, Ber. Dtsch. Chem. Ges., 1904, 37, 1573�1595.[336] M. Goehring, J. Weiss and G. Zirker, Z. Anorg. Allg. Chem., 1955, 278, 1�11.[337] F. Edelmann, J. Organomet. Chem., 1982, 228, C47�C50.[338] G. Granozzi, M. Casarin, F. Edelmann, T. A. Albright and E. D. Jemmis, Organometallics, 1987,
6, 2223�2227.[339] F. Cecconi, C. A. Ghilardi, S. Midollini, S. Moneti and A. Orlandini, J. Organomet. Chem., 1984,
275, C22�C24.[340] R. Jones, P. F. Kelly, D. J. Williams and J. D. Woollins, J. Chem. Soc., Chem. Commun., 1985,
1325�1326.[341] C. A. Ghilardi, S. Midollini, S. Moneti and A. Orlandini, J. Organomet. Chem., 1985, 286, 419�
426.[342] T. Chivers, F. Edelmann, U. Behrens and R. Drews, Inorg. Chim. Acta, 1986, 116, 145�151.[343] F. T. Edelmann and M. Belay, J. Fluor. Chem., 1997, 84, 29�33.[344] M. Goehring and A. Debo, Z. Anorg. Allg. Chem., 1953, 273, 319�324.[345] K.-W. Daum, M. Goehring and J. Weiss, Z. Anorg. Allg. Chem., 1955, 278, 260�263.[346] M. Goehring and D. Voigt, Z. Anorg. Allg. Chem., 1956, 285, 181�190.[347] J. D. Woollins, Polyhedron, 1987, 6, 939�941.[348] T. S. Piper, J. Am. Chem. Soc., 1958, 80, 30�32.[349] T. S. Piper, Chem. Ind., 1957, 1101�1102.[350] J. D. Woollins, R. Grinter, M. K. Johnson and A. J. Thomson, J. Chem. Soc., Dalton Trans.,
1980, 1910�1916.[351] M. L. Calatayud, J. A. Ramirez and J. Faus, J. Chem. Soc., Dalton Trans., 1991, 2995�3001.[352] R. Jones, P. F. Kelly, D. J. Williams and J. D. Woollins, Polyhedron, 1985, 4, 1947�1950.[353] F. Kunkel, K. Harms, H.-C. Kang, W. Massa and K. Dehnicke, Z. Naturforsch. (B), 1997, 52,
193�198.[354] R. Jones, P. F. Kelly, D. J. Williams and J. D. Woollins, Polyhedron, 1987, 6, 1541�1545.[355] H. W. Roesky, G. Holtschneider, H. Wiezer and B. Krebs, Chem. Ber., 1976, 109, 1358�1361.[356] J. Weiss, Angew. Chem., 1984, 96, 232.[357] P. F. Kelly and J. D. Woollins, Polyhedron, 1988, 7, 419�420.[358] D. Hänssgen, H. Salz, S. Rheindorf and C. Schrage, J. Organomet. Chem., 1993, 443, 61�65.[359] P. F. Kelly, A. M. Z. Slawin and A. Soriano-Rama, J. Chem. Soc., Dalton Trans., 1996, 53�59.[360] W. Hiller, J. Mohyla, J. Strähle, H. G. Hauck and K. Dehnicke, Z. Anorg. Allg. Chem., 1984, 514,
72�78.[361] E. Conradi, H. G. Hauck, U. Müller and K. Dehnicke, Z. Anorg. Allg. Chem., 1986, 539, 39�49.
206

References
[362] T. F. Gudimovich, A. I. Prisyazhnyuk, G. S. Vilkova, L. M. Tikhonenko and R. I. Makordei, Zh.Neorg. Khim., 1986, 31, 1309�1312.
[363] E. Fluck, M. Goehring and J. Weiss, Z. Anorg. Allg. Chem., 1956, 287, 51�53.[364] C. W. Allen, P. F. Kelly and J. D. Woollins, J. Chem. Soc., Dalton Trans., 1991, 1343�1345.[365] R. Jones, C. P. Warrens, D. J. Williams and J. D. Woollins, J. Chem. Soc., Dalton Trans., 1987,
907�914.[366] R. Jones, T. G. Purcell, D. J. Williams and J. D. Woollins, Polyhedron, 1988, 7, 647�650.[367] J. M. Jolli�e, P. F. Kelly and J. D. Woollins, J. Chem. Soc., Dalton Trans., 1989, 2179�2182.[368] V. C. Ginn, P. F. Kelly, A. M. Z. Slawin, D. J. Williams and J. D. Woollins, Polyhedron, 1993,
12, 1135�1139.[369] U. Swarnalatha, A. Sivaramakrishna, C. S. Venkatachalam, M. N. S. Rao, T. Inoue, T. Ueda and
M. Hojo, Can. J. Chem., 2002, 80, 1428�1434.[370] K. K. Pandey and M. Massoudipour, Inorg. Chim. Acta, 1987, 138, 169�170.[371] K. K. Pandey, D. T. Nehete and M. Massoudipour, Inorg. Chim. Acta, 1987, 129, 253�256.[372] S. Kumar and R. K. Mehta, J. Indian Chem. Soc., 1999, 76, 153�154.[373] J. Weiss, Angew. Chem., 1982, 94, 719�720.[374] J. Weiss, Z. Anorg. Allg. Chem., 1985, 521, 44�50.[375] J. Weiss, Z. Anorg. Allg. Chem., 1986, 542, 137�143.[376] R. Jones, T. G. Purcell, D. J. Williams and J. D. Woollins, Polyhedron, 1987, 6, 2165�2168.[377] H. W. Roesky, K. K. Pandey, M. Noltemeyer and G. M. Sheldrick, Acta Crystallogr. Sect. C-Cryst.
Struct. Commun., 1984, 40, 1555�1556.[378] J. Weiss, Z. Anorg. Allg. Chem., 1986, 532, 184�192.[379] J. D. Woollins, Polyhedron, 1984, 3, 1365�1367.[380] J. Bojes and T. Chivers, J. Chem. Soc., Chem. Commun., 1980, 1023�1024.[381] J. Bojes, T. Chivers, W. G. Laidlaw and M. Trsic, J. Am. Chem. Soc., 1982, 104, 4837�4841.[382] T. Chivers, W. G. Laidlaw, R. T. Oakley and M. Trsic, J. Am. Chem. Soc., 1980, 102, 5773�5781.[383] J. Bojes, T. Chivers and P. W. Codding, J. Chem. Soc., Chem. Commun., 1981, 1171�1173.[384] J. Weiss and U. Thewalt, Z. Anorg. Allg. Chem., 1966, 346, 234�240.[385] D. T. Haworth and G. Y. Lin, J. Inorg. Nucl. Chem., 1977, 39, 1838�1839.[386] J. Weiss, Z. Anorg. Allg. Chem., 1988, 559, 106�110.[387] I. Lindqvist and J. Weiss, J. Inorg. Nucl. Chem., 1958, 6, 184�186.[388] J. Weiss and H. S. Neubert, Z. Naturforsch. (B), 1966, 21, 286�287.[389] J. Weiss and U. Thewalt, Z. Anorg. Allg. Chem., 1968, 363, 159�165.[390] K. F. Mayer and J. Weiss, Acta Crystallogr. Sect. B-Struct. Crystallogr. Crystal Chem., 1978, 34,
1999�2000.[391] I. Kamenicek, A. S. Antsyshkina, M. A. Porai-Koshits and V. I. Sokol, Koord. Khim., 1989, 15,
814�817.[392] R. Jones, P. F. Kelly, C. P. Warrens, D. J. Williams and J. D. Woollins, J. Chem. Soc., Chem.
Commun., 1986, 711�713.[393] R. Jones, P. F. Kelly, D. J. Williams and J. D. Woollins, J. Chem. Soc., Dalton Trans., 1988,
803�807.[394] M. Kersting and R. Ho�mann, Inorg. Chem., 1990, 29, 279�284.[395] P. F. Kelly, A. M. Z. Slawin, D. J. Williams and J. D. Woollins, Polyhedron, 1991, 10, 2337�2340.[396] V. C. Ginn, P. F. Kelly, A. M. Z. Slawin, D. J. Williams and J. D. Woollins, J. Chem. Soc., Dalton
Trans., 1992, 963�968.
207

References
[397] U. Thewalt, Z. Naturforsch. (B), 1982, 37, 276�280.[398] C. G. Marcellus, R. T. Oakley, W. T. Pennington and A. W. Cordes, Organometallics, 1986, 5,
1395�1400.[399] K. Brunn, H. Endres and J. Weiss, Z. Naturforsch. (B), 1988, 43, 113�116.[400] P. F. Kelly, A. M. Z. Slawin, D. J. Williams and J. D. Woollins, Angew. Chem.-Int. Edit. Engl.,
1992, 31, 616�618.[401] H. W. Roesky, J. Anhaus, H. G. Schmidt, G. M. Sheldrick and M. Noltemeyer, J. Chem. Soc.,
Dalton Trans., 1983, 1207�1209.[402] U. Kynast, E. Conradi, U. Müller and K. Dehnicke, Z. Naturforsch. (B), 1984, 39, 1680�1685.[403] J. Hanich, M. Krestel, U. Müller, K. Dehnicke and D. Rehder, Z. Naturforsch. (B), 1984, 39,
1686�1695.[404] J. Anhaus, Z. A. Siddiqui, J. Schimkowiak, H. W. Roesky and H. Lueken, Z. Naturforsch. (B),
1984, 39, 1722�1728.[405] J. Hanich, W. Willing, U. Müller, K. Dehnicke and D. Rehder, Z. Naturforsch. (B), 1985, 40,
1457�1462.[406] H. Wadle, E. Conradi, U. Müller and K. Dehnicke, Z. Naturforsch. (B), 1985, 40, 1626�1630.[407] A. Berg, E. Conradi, U. Müller and K. Dehnicke, Z. Anorg. Allg. Chem., 1985, 529, 74�84.[408] J. Anhaus, Z. A. Siddiqui, H. W. Roesky and J. W. Bats, J. Chem. Soc., Dalton Trans., 1985,
2453�2455.[409] E. Conradi, H. Wadle, U. Müller and K. Dehnicke, Z. Naturforsch. (B), 1986, 41, 48�52.[410] H. W. Roesky, J. Schimkowiak, M. Noltemeyer and G. M. Sheldrick, Z. Naturforsch. (B), 1986,
41, 175�178.[411] H. W. Roesky, J. Schimkowiak and F. Walther, Z. Naturforsch. (B), 1986, 41, 393�394.[412] H. Wadle, E. Conradi, U. Müller and K. Dehnicke, Z. Naturforsch. (B), 1986, 41, 429�435.[413] T. Lübben, M. Witt, H. W. Roesky, M. Noltemeyer and H.-G. Schmidt, Inorg. Chem., 1995, 34,
4275�4277.[414] A. Dietrich, B. Neumüller and K. Dehnicke, Z. Anorg. Allg. Chem., 2000, 626, 315�317.[415] I. P. Parkin, A. M. Z. Slawin, D. J. Williams and J. D. Woollins, J. Chem. Soc., Chem. Commun.,
1989, 58�59.[416] M. Herberhold, C. Köhler, W. Milius and B. Wrackmeyer, Z. Naturforsch. (B), 1995, 50, 1811�
1817.[417] H. Endres and E. Galantai, Angew. Chem., 1980, 92, 644�645.[418] K. Bergemann, M. Kustos, P. Krüger and R. Steudel, Angew. Chem.-Int. Edit. Engl., 1995, 34,
1330�1331.[419] B. J. McCormick and B. M. Anderson, J. Inorg. Nucl. Chem., 1970, 32, 3414�3416.[420] F. Edelmann, H. W. Roesky, C. Spang, M. Noltemeyer and G. M. Sheldrick, Angew. Chem., 1986,
98, 908�909.[421] P. S. Belton, V. C. Ginn, P. F. Kelly and J. D. Woollins, J. Chem. Soc., Dalton Trans., 1992,
1135�1138.[422] M. B. Hursthouse, M. Motevalli, P. F. Kelly and J. D. Woollins, Polyhedron, 1989, 8, 997�998.[423] P. F. Kelly, R. N. Sheppard and J. D. Woollins, Polyhedron, 1992, 11, 2605�2609.[424] R. T. Boeré, B. Klassen and K. H. Moock, J. Organomet. Chem., 1994, 467, 127�133.[425] S. A. Frith and J. L. Spencer, Inorg. Synth., 1990, 28, 273�280.[426] I. P. Parkin, J. D. Woollins and P. S. Belton, J. Chem. Soc., Dalton Trans., 1990, 511�517.[427] U. Kölle and W. Kläui, Z. Naturforsch. (B), 1991, 46, 75�83.
208

References
[428] A. Toupadakis, G. J. Kubas and C. J. Burns, Inorg. Chem., 1992, 31, 3810�3817.[429] H. Brunner, U. Klement, J. Pfauntsch and J. Wachter, Angew. Chem., 1987, 99, 268�269.[430] G. J. Kubas, R. R. Ryan and K. A. Kubat-Martin, J. Am. Chem. Soc., 1989, 111, 7823�7832.[431] M. Villena-Blanco and W. L. Jolly, Inorg. Synth., 1967, 9, 98�102.[432] C. White, A. Yates and P. M. Maitlis, Inorg. Synth., 1992, 29, 228�234.[433] Gmelin Handbook of Inorganic Chemistry, Sulfur-Nitrogen Compounds, Part 2, Springer Verlag,
Berlin, Germany, 8th edn., 1985.[434] M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman,
J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar,J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji,M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda,O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo,J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli,J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G.Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck,K. Raghavachari, J. B. Foresman, J. V. Ortiz, A. Cui, A. G. Baboul, S. Cli�ord, J. Cioslowski,B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith,M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson,W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision C.02, Gaussian, Inc.,Wallingford CT, 2004.
[435] A. D. Becke, J. Chem. Phys., 1996, 104, 1040�1046.[436] A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829�5835.[437] A. J. H. Wachters, J. Chem. Phys., 1970, 52, 1033�1036.[438] F. Weigend, M. Haser, H. Patzelt and R. Ahlrichs, Chem. Phys. Lett., 1998, 294, 143�152.[439] D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77,
123�141.[440] K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119�124.[441] E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO, version 3.1.[442] H. W. Roesky and H. Wiezer, Angew. Chem.-Int. Edit. Engl., 1975, 14, 258�259.[443] H. W. Roesky and E. Wehner, Angew. Chem.-Int. Edit. Engl., 1975, 14, 498.[444] H. W. Roesky and H. Wiezer, Angew. Chem.-Int. Edit. Engl., 1973, 12, 674.[445] H. W. Roesky and M. Witt, Inorg. Synth., 1989, 25, 49�55.[446] A. Heitmann and F. Edelmann, Z. Naturforsch. (B), 1983, 38, 521�522.[447] T. Chivers and J. Proctor, Can. J. Chem., 1979, 57, 1286�1293.[448] H. W. Roesky, M. Kuhn and J. W. Bats, Chem. Ber., 1982, 115, 3025�3031.[449] H. W. Roesky, M. Thomas, J. W. Bats and H. Fuess, J. Chem. Soc., Dalton Trans., 1983, 1891�
1893.[450] H. W. Roesky, M. Thomas, J. Schimkowiak, M. Schmidt, M. Noltemeyer and G. M. Sheldrick, J.
Chem. Soc., Chem. Commun., 1982, 790�791.[451] R. Steudel, in The Chemistry of Inorganic Homo- and Heterocycles, Vol. 2, ed. I. Haiduc and
D. B. Sowerby, Academic Press, London, 1987.[452] N. Burford, T. Chivers, P. W. Codding and R. T. Oakley, Inorg. Chem., 1982, 21, 982�986.[453] T. Chivers and M. Hojo, Inorg. Chem., 1984, 23, 4088�4093.[454] J. Honzí£ek, J. Vinklárek, M. Erben and I. Císa°ová, Acta Crystallogr. Sect. E.-Struct. Rep. Online,
2004, 60, m1090�m1091.
209

References
[455] S. M. Aucott, P. Bhattacharyya, H. L. Milton, A. M. Z. Slawin and J. D. Woollins, New J. Chem.,2003, 27, 1466�1469.
[456] G. Hua and J. D. Woollins, Angew. Chem.-Int. Edit., 2009, 48, 1368�1377.[457] I. P. Gray, P. Bhattacharyya, A. M. Z. Slawin and J. D. Woollins, Chem. Eur. J., 2005, 11,
6221�6227.[458] C. Van Alsenoy and A. Peeters, Theochem-J. Mol. Struct., 1993, 105, 19�34.
210

Appendices
211

A. Crystallographic data∗
Table A.1. Crystal data and structure re�nement for PhAs(S2N2) (34)
Identi�cation code vmdw24Empirical formula C6 H5 As N2 S2Formula weight 244.16Temperature 93(2)KWavelength 0.710 73ÅCrystal system TriclinicSpace group P-1Unit cell dimensions a = 5.046(2)Å α = 103.427(13)◦
b = 8.330(4)Å β = 91.802(15)◦c = 10.155(6)Å γ = 93.833(13)◦
Volume 413.8(4)Å3
Z 2Density (calculated) 1.960Mg/m3
Absorption coe�cient 4.541mm−1
F(000) 240Crystal size 0.2000× 0.0500× 0.0300 mm3
Theta range for data collection 2.06 to 25.34◦Index ranges −6 ≤ h ≤ 5,−10 ≤ k ≤ 8,−9 ≤ l ≤ 12Re�ections collected 2456Independent re�ections 1367 [R(int) = 0.0313]Completeness to theta = 25.00◦ 91.5%Absorption correction MultiscanMax. and min. transmission 1.0000 and 0.6345Re�nement method Full-matrix least-squares on F 2
Data / restraints / parameters 1367 / 0 / 101Goodness-of-�t on F 2 1.118Final R indices [I>2sigma(I)] R1 = 0.0477, wR2 = 0.1047R indices (all data) R1 = 0.0546, wR2 = 0.1114Largest di�. peak and hole 0.883 and −1.042 e·Å=3
∗Full data and the CIF �les are included in the electronic version.
212

A. Crystallographic data
Table A.2. Crystal data and structure re�nement for MesAs(S2N2) (35)
Identi�cation code vmdw42Empirical formula C9 H11 As N2 S2Formula weight 286.24Temperature 93(2)KWavelength 0.710 73ÅCrystal system MonoclinicSpace group P2(1)/cUnit cell dimensions a = 14.367(6)Å α = 90◦
b = 11.372(5)Å β = 91.685(10)◦c = 13.911(5)Å γ = 90◦
Volume 2271.8(16)Å3
Z 8Density (calculated) 1.674Mg/m3
Absorption coe�cient 3.322mm−1
F(000) 1152Crystal size 0.1000× 0.0300× 0.0300 mm3
Theta range for data collection 1.42 to 25.35◦.Index ranges −17 ≤ h ≤ 13,−13 ≤ k ≤ 12,−15 ≤ l ≤ 16Re�ections collected 14163Independent re�ections 4133 [R(int) = 0.1453]Completeness to theta = 25.00◦ 99.4%Absorption correction MultiscanMax. and min. transmission 1.0000 and 0.9268Re�nement method Full-matrix least-squares on F 2
Data / restraints / parameters 4133 / 0 / 261Goodness-of-�t on F 2 1.028Final R indices [I>2sigma(I)] R1 = 0.0901, wR2 = 0.2210R indices (all data) R1 = 0.1388, wR2 = 0.2572Extinction coe�cient 0.0056(12)Largest di�. peak and hole 0.854 and −0.750 e·Å−3
213

A. Crystallographic data
Table A.3. Crystal data and structure re�nement for Cp*Co(S2N2) (47)
Identi�cation code vmdw13Empirical formula C10 H15 Co N2 S2Formula weight 286.29Temperature 93(2)KWavelength 0.710 73ÅCrystal system MonoclinicSpace group P2(1)/nUnit cell dimensions a = 8.1372(10)Å α = 90◦
b = 13.1558(16)Å β = 104.008(7)◦c = 12.9834(16)Å γ = 90◦
Volume 1348.6(3)Å3
Z 4Density (calculated) 1.410Mg/m3
Absorption coe�cient 1.554mm−1
F(000) 592Crystal size 0.100× 0.030× 0.010 mm3
Theta range for data collection 2.69 to 25.35◦.Index ranges −9 ≤ h ≤ 9,−13 ≤ k ≤ 15,−15 ≤ l ≤ 15Re�ections collected 11599Independent re�ections 2372 [R(int) = 0.0675]Completeness to theta = 25.00◦ 96.6%Absorption correction MultiscanMax. and min. transmission 1.0000 and 0.2258Re�nement method Full-matrix least-squares on F 2
Data / restraints / parameters 2372 / 0 / 142Goodness-of-�t on F 2 1.03Final R indices [I>2sigma(I)] R1 = 0.0342, wR2 = 0.0864R indices (all data) R1 = 0.0357, wR2 = 0.0878Largest di�. peak and hole 0.401 and −0.406 e·Å−3
214

A. Crystallographic data
Table A.4. Crystal data and structure re�nement for Cp*Ir(S2N2) (51)
Identi�cation code vmdw6Empirical formula C10 H15 Ir N2 S2Formula weight 419.56Temperature 93(2)KWavelength 0.710 73ÅCrystal system MonoclinicSpace group P2(1)/nUnit cell dimensions a = 7.856(3)Å α = 90◦
b = 13.129(5)Å β = 103.39(3)◦c = 12.860(13)Å γ = 90◦
Volume 1290.4(14)Å3
Z 4Density (calculated) 2.160Mg/m3
Absorption coe�cient 10.641mm−1
F(000) 792Crystal size 0.1000× 0.1000× 0.0100 mm3
Theta range for data collection 3.08 to 25.43◦Index ranges −9 ≤ h ≤ 5,−15 ≤ k ≤ 15,−15 ≤ l ≤ 13Re�ections collected 7725Independent re�ections 2296 [R(int) = 0.0426]Completeness to theta = 25.00◦ 96.9%Absorption correction MultiscanMax. and min. transmission 1.0000 and 0.1902Re�nement method Full-matrix least-squares on F 2
Data / restraints / parameters 2296 / 0 / 142Goodness-of-�t on F 2 0.853Final R indices [I>2sigma(I)] R1 = 0.0217, wR2 = 0.0416R indices (all data) R1 = 0.0236, wR2 = 0.0421Largest di�. peak and hole 1.051 and −0.825 e·Å−3
215

A. Crystallographic data
Table A.5. Crystal data and structure re�nement for[Cp*Rh(µ−S3N2)(µ−SSO3)RhCp*]·CH2Cl2 (53)
Identi�cation code vmdw12Empirical formula C21 H32 Cl2 N2 O3 Rh2 S5Formula weight 797.51Temperature 93(2)KWavelength 0.710 73ÅCrystal system MonoclinicSpace group P2(1)/cUnit cell dimensions a = 10.615(3)Å α = 90◦
b = 23.415(6)Å β = 90.089(4)◦c = 23.361(6)Å γ = 90◦
Volume 5806(2)Å3
Z 8Density (calculated) 1.825Mg/m3
Absorption coe�cient 1.708mm−1
F(000) 3200Crystal size 0.1300× 0.0100× 0.0100 mm3
Theta range for data collection 1.95 to 25.32◦Index ranges −12 ≤ h ≤ 9,−28 ≤ k ≤ 27,−28 ≤ l ≤ 25Re�ections collected 43104Independent re�ections 9933 [R(int) = 0.1725]Completeness to theta = 25.00◦ 94.3%Absorption correction MultiscanMax. and min. transmission 1.0000 and 0.8438Re�nement method Full-matrix least-squares on F 2
Data / restraints / parameters 9933 / 54 / 649Goodness-of-�t on F 2 1.13Final R indices [I>2sigma(I)] R1 = 0.1107, wR2 = 0.2688R indices (all data) R1 = 0.1485, wR2 = 0.2972Largest di�. peak and hole 3.386 and −2.291 e·Å−3
216

A. Crystallographic data
Table A.6. Crystal data and structure re�nement for [Cp*RhCp*]Cl ·H2O (52)
Identi�cation code vmdw39Empirical formula C20 H32 Cl O RhFormula weight 426.82Temperature 93(2)KWavelength 0.710 73ÅCrystal system MonoclinicSpace group C2/mUnit cell dimensions a = 16.909(3)Å α = 90◦
b = 10.1450(17)Å β = 124.929(9)◦c = 13.854(3)Å γ = 90◦
Volume 1948.4(6)Å3
Z 4Density (calculated) 1.455Mg/m3
Absorption coe�cient 1.016mm−1
F(000) 888Crystal size 0.150× 0.100× 0.030 mm3
Theta range for data collection 2.41 to 25.35◦Index ranges −18 ≤ h ≤ 20,−12 ≤ k ≤ 11,−15 ≤ l ≤ 15Re�ections collected 5759Independent re�ections 1646 [R(int) = 0.0703]Completeness to theta = 25.00◦ 87.2%Absorption correction MultiscanMax. and min. transmission 1.0000 and 0.9821Re�nement method Full-matrix least-squares on F 2
Data / restraints / parameters 1646 / 2 / 127Goodness-of-�t on F 2 1.07Final R indices [I>2sigma(I)] R1 = 0.0347, wR2 = 0.0870R indices (all data) R1 = 0.0363, wR2 = 0.0878Extinction coe�cient 0.007(3)Largest di�. peak and hole 0.606 and −0.582 e·Å−3
217

A. Crystallographic data
Table A.7. Crystal data and structure re�nement for Cp*Ir[S2N2(IrCl2Cp*)] · nBu2SnCl2(54 · nBu2SnCl2)
Identi�cation code vmdw3Empirical formula C31.50 H52 Cl4 Ir2 N2 S2 SnFormula weight 1167.76Temperature 93(2)KWavelength 0.710 73ÅCrystal system TriclinicSpace group P-1Unit cell dimensions a = 10.1044(8)Å α = 78.220(8)◦
b = 12.1259(13)Å β = 80.344(8)◦c = 16.8451(18)Å γ = 77.688(9)◦
Volume 1957.3(3)Å3
Z 2Density (calculated) 1.981Mg/m3
Absorption coe�cient 7.819mm−1
F(000) 1118Crystal size 0.1000× 0.0300× 0.0300 mm3
Theta range for data collection 2.28 to 25.35◦Index ranges −10 ≤ h ≤ 12,−14 ≤ k ≤ 14,−17 ≤ l ≤ 20Re�ections collected 12710Independent re�ections 6883 [R(int) = 0.0184]Completeness to theta = 25.00◦ 96.1%Absorption correction MultiscanMax. and min. transmission 1.0000 and 0.7567Re�nement method Full-matrix least-squares on F 2
Data / restraints / parameters 6883 / 0 / 389Goodness-of-�t on F 2 1.054Final R indices [I>2sigma(I)] R1 = 0.0220, wR2 = 0.0445R indices (all data) R1 = 0.0256, wR2 = 0.0459Largest di�. peak and hole 1.145 and −1.017 e·Å−3
218

A. Crystallographic data
Table A.8. Crystal data and structure re�nement for S3N2O
Identi�cation code vmdw2Empirical formula N2 O S3Formula weight 140.2Temperature 93(2)KWavelength 0.710 73ÅCrystal system MonoclinicSpace group CcUnit cell dimensions a = 7.617(5)Å α = 90◦
b = 9.230(6)Å β = 115.920(6)◦c = 7.050(5)Å γ = 90◦
Volume 445.7(5)Å3
Z 4Density (calculated) 2.089Mg/m3
Absorption coe�cient 1.497mm−1
F(000) 280Crystal size 0.1000× 0.0300× 0.0300 mm3
Theta range for data collection 6.98 to 36.62◦Index ranges −12 ≤ h ≤ 6,−15 ≤ k ≤ 15,−6 ≤ l ≤ 11Re�ections collected 3792Independent re�ections 993 [R(int) = 0.0324]Completeness to theta = 36.62◦ 70.4%Absorption correction MultiscanMax. and min. transmission 1.0000 and 0.3400Re�nement method Full-matrix least-squares on F 2
Data / restraints / parameters 993 / 2 / 56Goodness-of-�t on F 2 1.054Final R indices [I>2sigma(I)] R1 = 0.0215, wR2 = 0.0533R indices (all data) R1 = 0.0229, wR2 = 0.0537Absolute structure parameter 0.45(10)Largest di�. peak and hole 0.336 and −0.258 e·Å−3
219




















