
University of Groningen The multiple faces of the human ...
333
University of Groningen The multiple faces of the human immune system Pruimboom, Leo IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check the document version below. Document Version Publisher's PDF, also known as Version of record Publication date: 2017 Link to publication in University of Groningen/UMCG research database Citation for published version (APA): Pruimboom, L. (2017). The multiple faces of the human immune system: Modern life causes low-grade inflammation and thereby provokes conflict between the selfish immune system and the selfish brain. [Groningen]: Rijksuniversiteit Groningen. Copyright Other than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons). Take-down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons the number of authors shown on this cover page is limited to 10 maximum. Download date: 12-11-2019
-
Upload
khangminh22 -
Category
Documents
-
view
3 -
download
0
Transcript of University of Groningen The multiple faces of the human ...

University of Groningen
The multiple faces of the human immune systemPruimboom, Leo
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:2017
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Pruimboom, L. (2017). The multiple faces of the human immune system: Modern life causes low-gradeinflammation and thereby provokes conflict between the selfish immune system and the selfish brain.[Groningen]: Rijksuniversiteit Groningen.
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 12-11-2019

THE MULTIPLE FACES
OF THE HUMAN IMMUNE SYSTEM
Modern life causes low-grade inflammation and thereby provokes conflict
between the selfish immune system and the selfish brain
Proefschrift
ter verkrijging van de graad van doctor aan de Rijksuniversiteit Groningen op gezag van de
rector magnificus prof. dr. E. Sterken en volgens besluit van het College voor Promoties.
De openbare verdediging zal plaatsvinden op maandag 30 January 2017 om 14.30 uur
Door
Leo Pruimboom
geboren op 12 mei 1961 te Amsterdam
Promotores Prof. Dr. F.A.J. Muskiet
Prof. Dr. C.L. Raison (University of Arizona)

2
Beoordelingscommissie Prof.dr G. Feixas
Prof.dr. Marja van Luyn
Prof.dr H.J. Wichers
Having a human immune system is like having a life insurance.

3
If it pays out it is too late.

4
Table of contents
Introduction 7
Aim of the thesis 11
Chapter 1. Chronic systemic low-grade inflammation 16
Paragraph 1.1. The Selfish Immune System. When the Immune System Overrides the “Selfish”
Brain. Pruimboom L, Raison CL, Muskiet FA. Submitted Journal of Evolution and Health
Paragraph 1.2. Lifestyle and nutritional imbalances associated with Western diseases: causes
and consequences of chronic systemic low-grade inflammation in an evolutionary context.
Ruiz-Núñez B, Pruimboom L, Dijck-Brouwer DA, Muskiet FA. J Nutr Biochem 2013;24:1183-
201.
Paragraph 1.3. Chronic inflammatory diseases are stimulated by current lifestyle: how diet,
stress levels and medication prevent our body from recovering. Bosma-den Boer MM, van
Wetten ML, Pruimboom L. Nutr Metab (Lond) 2012;9:32.
Paragraph 1.4. Stress induces endotoxemia and low-grade inflammation by increasing barrier
permeability. de Punder K, Pruimboom L. Front Immunol 2015;6:223. Epub 2015 May 15.
Paragraph 1.5. The dietary intake of wheat and other cereal grains and their role in
inflammation. de Punder K, Pruimboom L. Nutrients 2013;5:771-87.
Paragraph 1.6. Lactase persistence and augmented salivary alpha-amylase gene copy
numbers might have been selected by the combined toxic effects of gluten and (food born)
pathogens. Pruimboom L, Fox T, Muskiet FA. Med Hypotheses 2014;82:326-34.
Chapter 2. Physical inactivity 238
Paragraph 2.1. Physical Activity Protects the Human Brain against Metabolic Stress Induced
by a Postprandial and Chronic Inflammation. Pruimboom L, Raison CL, Muskiet FA. Behav
Neurol. 2015. Epub 2015 May 5.
Paragraph 2.2. Physical inactivity is a disease synonymous for a non-permissive brain
disorder. Pruimboom L. Med Hypotheses. 2011;77:708-13.

5
Chapter 3. Lifestyle intervention 316
Paragraph 3.1. Influence of a 10 days mimic of our ancient lifestyle on anthropometrics and
parameters of metabolism and inflammation. The ‘Study of Origin’. Pruimboom L1,2, Ruiz-
Núñez B2, Raison RL3, Muskiet FA2, on behalf of the ‘Study of Origin” Consortium. Biomed
Research International 2016
Summary and future perspectives 317
Samenvatting en toekomstperspectieven 319
Dankwoord 327
Curriculum Vitae 328

6
Introduction
This thesis focuses on the human immune system as the ultimate organ explaining the
development of most, if not all, ‘typically Western’ chronic diseases. Patho-physiologically,
the emphasis is on the development of ‘systemic low-grade inflammation’ (LGI). A red thread
throughout this thesis is Charles Darwin’s (1809-1882) ‘adaptation to the conditions of
existence’. We explain our physiology and functioning in the light of evolution, being the
only approach that really ‘makes sense in biology’ (Theodosius Dobzhansky, 1900-1975).
During millions years of evolution the immune system has saved simple organisms, such as
the lamprey, and later on much more complex types of life, including humans and plants,
although the latter make use of a much less complicated innate-like immune system that e.g.
does not rely on mobile defence cells. Analogous to all other systems, the immune system has
been adapted by evolutionary pressure to the benefit of survival and reproduction. There is
good evidence to show that infection should be considered as the ‘roller coaster’ of evolution
that shaped all forms of life, and humans in particular. Even today it are still infections by
parasites, bacteria, fungi and viruses causing the most devastating conditions in humans.
Contemporary examples are Zika in the America’s, ebola outbreak in Africa, HIV affecting
millions of humans, salmonella infections worldwide and Aspergillus in intensive care units
(Bellet 2013, Tyghe 2011, Wingard 2008).
For example, about half of the world’s population is at risk of malaria infection, causing about
438,000 deaths on a yearly basis (WHO http://www.who.int/features/factfiles/malaria/en/).
The malaria parasite might be at least 200 million years old, as compared to the about 160,000
years of homo sapiens. The battle between the two species has become intensified from
about 80,000 years ago, i.e. long after homo sapiens started to leave Africa some 100,000
years ago (Cserti 2007). According to Darwin’s ‘adaptation to the conditions of existence’ this
battle has shaped both the genomes of Plasmodium and modern humans. Some examples of
human adaptations by positive selection of polymorphisms are given in the Table 1
(Kwiatkowsi 2005). At least some of these adaptations, like sickle haemoglobin, confer
heterozygote advantage, also named balancing selection, with homozygotes exhibiting
(serious) disease, while heterozygotes are not even fully protected. The existence of
haplotypes witness the independence of the occurrence and preservation of the sickle
haemoglobin mutation in at least 4 malaria-endemic regions in the world, adding to the
notion of the very strong selective pressure that has and is still exerted by Plasmodium.

7
Table 1. Anti-malarial strategies related to malaria survival.
Membrane proteins Duffy antigen (chemokine receptor functioning as
recognition site, confers resistance to P. vivax). Glycophorin.
Chloride/HCO3- exchanger (ovalocytosis).
Blood group resistance: 0>B>A (South America 100% type 0).
(Energy) metabolism Glucose-6-phosphate dehydrogenase (G6PD; higher RBC
oxidative stress).
Hemoglobin Hb structural variants (examples HbS, HbC, HbE).
Thalassemias (notably alpha, beta).
Haptoglobulin Binding of free Hb.
Immunity,
inflammation, adhesion
Ag presentation (HLA), cytokines (ILs, interferon, TNF-
alpha), NO-synthase, adhesion molecules (ICAM1).
Humans have conquered the world and survived their continuing search of novelty
(Pruimboom 2015). Encountered challenges included e.g. climate, famine, thirst, but most of
all infections (Nunn 2014). The corresponding evolutionary pressure triggered many
phenotypic adjustments by either genetic or epigenetic modifications. As an example we
mention 3 major strategies:
1. A shift from strong to smart (Watve 2007), needed to ‘outthink’ much stronger animals
and to guarantee food, water, shelter from cold and heat, protection from violence, and
solutions for infections (Nunn 2014)
2. A shift from strong to smart leading to the highest encephalization quotient among all
animals (Roth 2005) and a change from present to future thinking through the
development of a more dopamine-oriented neuroanatomy (Vernier 2004)
3. A highly adequate-functioning immune system, though expensive, needed to survive
pathogenic load and novel environmental challenges, prior to the institution of hygiene
and modern medicine (e.g. antibiotics).
A highly effective ‘somatic immune system’ has provided humans with many fitness benefits
that are nevertheless endowed with important limitations and costs. A reactive pro-
inflammatory immune response is metabolically expensive; not only from a caloric point of
view, but also from the view of nutrients, such as calcium and magnesium. Calcium, as signal
transducer, is essential for adequate functioning of the immune system, muscles, brain and
others. The immune system’s activation in general (Vig 2009), and more specifically of T-
cells, induces profound increases of intracellular calcium, needed to initiate the production of

8
cytotoxic substances and T-cell antigen receptor ligation. Sickness syndrome may
accompany infection and the concomitant lack of mobilization causes loss of calcium from
bones and augmented urinary calcium excretion, illustrating ‘use it or lose it’. The
concomitant anorexia of severe infection causes long-term reduced intestinal calcium and
magnesium intakes, and may, aggravated by vitamin D and K deficiencies, add to the
negative calcium balance that ultimately results in osteopenia (Straub, J Intern Med 2010).
Thus, the immune system’s actions can be highly debilitating and damaging in the face of its
causation of sickness behaviour, cytotoxicity, fever, fatigue and micronutrient depletion, all
of which may ultimately lead to multiple organ failure, including damage to the immune
system itself. Prevention of immune responses are therefore of paramount importance. A
unique part of the human immune system is the ‘behavioural immune system’ (Schaller 2011),
since it aims at the predictable benefits of prevention. This system developed to avoid
pathogen contact, thereby preventing an energy-costly reactive response and saving energy
and resources for other essential organs, including the brain and the heart (Schaller 2011, Park
2007).
The human brain and the immune system have become so important for human physiology
and evolutionary fitness that both systems have developed selfish behaviours by their
capabilities to pull on energy and other resources, with the solemn purpose to maintain their
own anatomies and optimal functioning (see for the selfish brain concept: Kubera 2012,
Peters 2011, Peters 2009, Peters 2007, Peters 2004 and for the selfish immune system: Straub
2014, Pruimboom 2015). Upon challenge, an optimal human response therefore depends on
the flexibility to (re)distribute energy, and thereby in first instance prevent energy conflicts
between the brain and the immune system. The circadian rhythm (Figure 1), allowing the
brain to dominate daytimes and the immune system to exploit night times, illustrates how
evolution shaped collaboration between these two highly selfish systems, while avoiding
conflict (Straub 2011; Pacheco-Lopez 2011). The selfish behaviour of the immune system may,
from a pathophysiological point of view, explain why LGI disturbs the functioning of many
other organs, including the brain. LGI puts the immune system on top of hierarchic priorities
as e.g. evidenced by the observation that the system even endorses breakdown of an essential
muscle like the diaphragm (Hafner 2014).

9
Figure 1. Circadian fuel allocation.
At daytime (A): the immunosuppressive stress hormones, cortisol and adrenalin, together with anti-inflammatory myokines,
shut-off the immune system. At night (B): growth hormone, prolactin and melatonin, secreted shortly after sleep onset,
activate the immune system. Modified from Pacheco-Lopez (Philos Trans R Soc Lond Sci 2011).
Known risk factors for LGI and chronic disease are age, smoking, socioeconomic status,
obesity, chronic psychosocial stress, sedentary lifestyle, toxins, insufficient sleep, nutritional
factors (nutrient composition, time, frequency) and abuse of legal and illegal drugs, alcohol
included (Schmid 2015, Henson 2015, Ruiz 2014, Ruiz 2013, Egger 2012a, Egger 2012b, Egger
2011). These, mostly environmentally-driven, risk factors seem inevitable in current Western
societies and their shares and intensities are most likely destined to further increase in the
future. Importantly, many of these risk factors exhibit interaction, while contemporary
humans are likely to suffer from them in concert. This current ‘condition of existence’, occurs
as opposed to the stress factors experienced by humans living in the environment of our
ancestors. In that environment, they had to cope with short-term mono-metabolic danger
factors (e.g. hunger, thirst, cold, heat), whereas modern humans are exposed to multi-
metabolic risk factors that stimulate an energy conflict between organs and major systems
(Wells 2012). The ensuing conflict between what we currently experience and to what our
genes are adapted is the basis of the so-called ‘mismatch hypothesis’ of ‘typically Western’
diseases.
Mono-metabolic stress factors have shaped mechanisms for survival and reproduction, such
as short-lasting infections, insulin resistance, activation of the sympathetic nervous system
and others. All of these responses emerged with the purpose to first protect the brain from
damage and energy deficits (Pruimboom 2011). Mono-metabolic challenge increases basal
metabolic rate and frequent challenges in a resource-restricted (thrifty) environment prevent
a pathological increase of adipose tissue size. A sizeable fat-mass in concert with other risk
factors, is not unlikely to become an inflammatory compartment (Exley 2015). Multi-
metabolic risk factors may, on the other hand, cause LGI and a hypo-metabolic state (Wells
2012). The latter is probably the main reason for the deleterious effects of LGI, since a hypo-

10
metabolic state causes energy deficits in multiple organs, and consequently multi-organ
damage (Straub 2010).
While among our ancestors, cold, heat, starvation and repetitive infections were major causes
of death, exposure to mild cold, mild heat, short fasting periods and the regular consumption
of small amounts of ‘toxic’ nutrients provided hormetic triggers. Mildly toxic insults, for
example, derive from plant secondary metabolites, many with bitter tastes. The discovery of
the nrf2 receptor has revolutionized toxicology by unveiling the benefits of low amounts of
toxins (Bryan 2013, Hayes 2014; Forman 2014). Ultimately, it is all about hormesis: every dose-
response curve is U-shaped (Calabrese 2014), as opposed to the saturation curves that are
usually depicted in textbooks and lectures, and is the first to pop-up when entered into
Google-pictures. Establishment of ‘dietary reference intakes’ (e.g. AIs, RDAs, ULs) have for
long used dose-response curves showing a range starting from deficiency, via adequacy, to
toxicity, but the step to ‘what does not kill you makes you stronger’ has only recently become
appreciated. This notion deserves rethinking of the definition of ‘essential nutrients’.
Mild triggers might at least in part recover physiologic and metabolic dysfunctioning in
patients suffering from ‘typically Western’ diseases (Mattson 2015). In other words: they may
provide low-cost opportunities for secondary prevention. Conversely, the chronic absence of
mild stress factors may have rendered modern 21st century humans less resistant to major
toxic insults and susceptible to the development of many, ‘typically Western’, chronic
diseases of affluence, including metabolic disorders, some types of cancer, depression and
cardiovascular diseases (Mattson 2015, Calabrese 2015, Mattson 2014). Re-introduction of
exposure provides low-cost opportunities for primary prevention with huge favourable
potential for the society as a whole. Many changes in lifestyle are involved and their adoption
is not necessarily unpleasant, as is frequently claimed. For instance, a recent study suggested
that men taking sauna bathing sessions at a frequency of 4-7 times/week have 63% lower risk
of all-cause and CVD mortality, compared with those having one sauna session/week. There
was also a significant trend of lower fatal CVD mortality of 19 minutes sessions, compared
with sessions lasting less than 11 minutes (Laukkanen 2015). A sauna session may be regarded
as a mild, heat-based, stress factor with hormetic actions and broad protecting ability from
the insults of the 21st century environment (Sarup 2014, Rattan 2009).
Aim of the thesis
The purpose of this thesis was to contribute to the notion that the human immune system
may be selfish and perhaps even more selfish than the human brain. The exploitable part of
this knowledge is that the triggering of the immune system’s selfish behaviour might be
responsible for most, if not all, current chronic non-communicable diseases (CNCD), and that

11
a chronically activated immune system produces a long-term hypo-metabolic state.
Along these lines, the aim of our field study (Chapter 3) was to show that metabolism and
normal immunological functioning of contemporary humans can be recovered by ‘suffering’
from ancient mild stress factors, analogous to what our ancestors have experienced in the
past, and as opposed to the LGI ensuing from modern life. We definitely do not propose to
return to the ‘caveman’s life’, like popular ‘counterarguments’ tend to posit, but to exploit the
positive effects of evolutionary-based lifestyle challenges to the benefit of our current health.
The only realistic and affordable road to ‘healthy aging’ is comprised in the ‘adaptations to the
past conditions of existence’ according to culture of the 21st century (Cordain 2005). For this
we hypothesize that living a life with short-term mild stress factors could prevent metabolic
diseases and LGI (primary prevention) and even cure patients suffering from CNCD, at least to
some extent (secondary prevention). In other words: short-term mild stress factors may
protect and even cure modern humans from the deleterious effects of Western lifestyle.
Chapter 1 reviews LGI as the major cause of most, if not all, CNCD. Paragraph 1.1 describes the
‘selfish immune system’ concept, as compiled from the existing scientific literature.
Paragraphs 1.2 and 1.3 review the current lifestyle factors held responsible for the
development of LGI and CNCD, respectively. In paragraph 1.4 we describe several pathways
leading to LGI and ensuing from modern lifestyle factors. Paragraph 1.5 discusses the gluten
in grains as a specific, uniformly present, nutritional factor that is likely to be involved in the
development of LGI. Paragraph 1.6 discusses the background of genetic adaptations triggered
by the consumption of non-human milk and starchy-rich foods, which are two recently
introduced foods into homo sapiens’ diet. Our hypothesis in this paragraph may illustrate the
widely misunderstood distinction between ultimate and proximate explanations of our
physiology, that nevertheless in both explanations ensue from evolutionary pressure with the
aim to survive and reproduce. The question here is not ‘how’ we adapted (proximate
explanation) but ‘to what’ and ‘why’ (evolutionary approach). Chapter 2 deals with physical
activity. Paragraphs 2.1 and 2.2 discuss the impact of sedentary lifestyle and a lack of physical
activity on the developments of LGI and CNCD. Finally, in Chapter 3, we present our field
study aiming to show that a concerted lifestyle ‘intervention’, based on what made us
humans, may favourably affect both our metabolism and immune functioning.

12
References
1. Bellet, M. M., Deriu, E., Liu, J. Z., Grimaldi, B., et al. Circadian clock regulates the host
response to Salmonella. Proceedings of the National Academy of Sciences 110, 9897-
9902 (2013).
2. Bryan, H. K., Olayanju, A., Goldring, C. E. & Park, B. K. The Nrf2 cell defence pathway:
Keap1-dependent and -independent mechanisms of regulation. Biochemical
Pharmacology 85, 705-717 (2013).
3. Calabrese, E. J. Model Uncertainty via the Integration of Hormesis and LNT as the
Default in Cancer Risk Assessment. Dose-Response 13, (2015).
4. Cserti, C. M. & Dzik, W. H. The ABO blood group system and Plasmodium falciparum
malaria. Blood 110, 2250-2258 (2007).
5. Egger, G. Obesity, Chronic Disease, and Economic Growth: A Case for “Big Picture”
Prevention. Advances in Preventive Medicine 2011, 1-6 (2011).
6. Egger, G., Swinburn, B. & Amirul Islam, M. Economic growth and obesity: An interesting
relationship with world-wide implications. Economics & Human Biology 10, 147-153
(2012).
7. Egger, G. In Search of a Germ Theory Equivalent for Chronic Disease. Preventing
Chronic Disease (2012).
8. Exley, M. A., Hand, L., O'Shea, D. & Lynch, L. Interplay between the immune system and
adipose tissue in obesity. Journal of Endocrinology 223, R41-R48 (2014).
9. Feske, S. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol 7,
690-702 (2007).
10. Hafner, S., Radermacher, P., Frick, M., Dietl, P. & Calzia, E. Hyperglycemia, oxidative
stress, and the diaphragm: a link between chronic co-morbidity and acute stress?
Critical Care 18, 149 (2014).
11. Hanson, L. A., Zahn, E. A., Wild, S. R., Döpfer, D., et al. Estimating global mortality from
potentially foodborne diseases: an analysis using vital registration data. Population
Health Metrics 10, 5 (2012).
12. Hayes, J. D. & Dinkova-Kostova, A. T. The Nrf2 regulatory network provides an interface
between redox and intermediary metabolism. Trends in Biochemical Sciences 39, 199-
218 (2014).
13. HENSON, J., EDWARDSON, C. L., MORGAN, B., HORSFIELD, M. A., et al. Associations of
Sedentary Time with Fat Distribution in a High-Risk Population. Medicine & Science in
Sports & Exercise 47, 1727-1734 (2015).
14. Joseph, N., Reicher, B. & Barda-Saad, M. The calcium feedback loop and T cell
activation: How cytoskeleton networks control intracellular calcium flux. Biochimica et
Biophysica Acta (BBA) - Biomembranes 1838, 557-568 (2014).

13
15. Kubera, B., Hubold, C., Zug, S., Wischnath, H., et al. The brain's supply and demand in
obesity. Frontiers in Neuroenergetics 4, (2012).
16. Kwiatkowski, D. P. How malaria has affected the human genome and what human
genetics can teach us about malaria. The American Journal of Human Genetics 77, 171-
192 (2005).
17. Laukkanen, T., Khan, H., Zaccardi, F. & Laukkanen, J. A. Association Between Sauna
Bathing and Fatal Cardiovascular and All-Cause Mortality Events. JAMA Internal
Medicine 175, 542 (2015).
18. Mattson, M. P. Challenging Oneself Intermittently to Improve Health. Dose-Response
12, 600-618 (2014).
19. Mattson, M. P. Lifelong brain health is a lifelong challenge: from evolutionary principles
to empirical evidence. Ageing research reviews 20, 37-45 (2015).
20. Nunn, A. V. W., Guy, G. W. & Bell, J. D. The intelligence paradox; will ET get the
metabolic syndrome? Lessons from and for Earth. Nutrition & Metabolism 11, 34 (2014).
21. Pacheco-Lopez, G. & Bermudez-Rattoni, F. Brain-immune interactions and the neural
basis of disease-avoidant ingestive behaviour. Philosophical Transactions of the Royal
Society B: Biological Sciences 366, 3389-3405 (2011).
22. Park, J. H., Schaller, M. & Crandall, C. S. Pathogen-avoidance mechanisms and the
stigmatization of obese people. Evolution and Human Behavior 28, 410-414 (2007).
23. Peters, A., Schweiger, U., Pellerin, L., Hubold, C., et al. The selfish brain: competition for
energy resources. Neuroscience & Biobehavioral Reviews 28, 143-180 (2004).
24. Peters, A., Pellerin, L., Dallman, M. F., Oltmanns, K. M., et al. Causes of obesity: looking
beyond the hypothalamus. Progress in neurobiology 81, 61-88 (2007).
25. Peters, A. Build-ups in the supply chain of the brain: on the neuroenergetic cause of
obesity and type 2 diabetes. Frontiers in Neuroenergetics 1, (2009).
26. Peters, A. The selfish brain: Competition for energy resources. Am. J. Hum. Biol. 23, 29-
34 (2010).
27. Peters, A. & McEwen, B. S. Stress habituation, body shape and cardiovascular mortality.
Neuroscience & Biobehavioral Reviews 56, 139-150 (2015).
28. Pruimboom, L. Physical inactivity is a disease synonymous for a non-permissive brain
disorder. Medical Hypotheses 77, 708-713 (2011).
29. Rattan, S. I. S., Fernandes, R. A., Demirovic, D., Dymek, B. & Lima, C. F. Heat Stress and
Hormetin-Induced Hormesis in Human Cells: Effects on Aging, Wound Healing,
Angiogenesis, and Differentiation. Dose-Response 7, 90-103 (2008).
30. Roth, G. & Dicke, U. Evolution of the brain and intelligence. Trends in cognitive sciences
9, 250-257 (2005).
31. Ruiz-Núñez, B., Pruimboom, L., Dijck-Brouwer, D. J. & Muskiet, F. A. Lifestyle and
nutritional imbalances associated with Western diseases: causes and consequences of

14
chronic systemic low-grade inflammation in an evolutionary context. The Journal of
Nutritional Biochemistry 24, 1183-1201 (2013).
32. Ruiz-Núñez, B., Kuipers, R. S., Luxwolda, M. F., De Graaf, D. J., et al. Saturated fatty acid
(SFA) status and SFA intake exhibit different relations with serum total cholesterol and
lipoprotein cholesterol: a mechanistic explanation centered around lifestyle-induced
low-grade inflammation. The Journal of Nutritional Biochemistry 25, 304-312 (2014).
33. Sarup, P., Sørensen, P. & Loeschcke, V. The long-term effects of a life-prolonging heat
treatment on the Drosophila melanogaster transcriptome suggest that heat shock
proteins extend lifespan. Experimental Gerontology 50, 34-39 (2014).
34. Schaller, M. The behavioural immune system and the psychology of human sociality.
Philosophical Transactions of the Royal Society of London B: Biological Sciences 366,
3418-3426 (2011).
35. Schmid, D., Ricci, C. & Leitzmann, M. F. Associations of Objectively Assessed Physical
Activity and Sedentary Time with All-Cause Mortality in US Adults: The NHANES Study.
PLOS ONE 10, e0119591 (2015).
36. Schwarz, E. C., Qu, B. & Hoth, M. Calcium, cancer and killing: The role of calcium in
killing cancer cells by cytotoxic T lymphocytes and natural killer cells. Biochimica et
Biophysica Acta (BBA) - Molecular Cell Research 1833, 1603-1611 (2013).
37. Straub, R. H., Cutolo, M., Buttgereit, F. & Pongratz, G. Energy regulation and
neuroendocrine-immune control in chronic inflammatory diseases. J Intern Med 267,
543-560 (2010).
38. Straub, R. H. Neuroendokrinimmunologie: Neue Aspekte zur Pathogenese und
klinischen Anwendung (Leitthema). Zeitschrift für Rheumatologie 70, 767-774 (2011).
39. Straub, R. H. Systemic disease sequelae in chronic inflammatory diseases and chronic
psychological stress: comparison and pathophysiological model. Ann. N.Y. Acad. Sci.
1318, 7-17 (2014).
40. VERNIER, P. The Degeneration of Dopamine Neurons in Parkinson's Disease: Insights
from Embryology and Evolution of the Mesostriatocortical System. Annals of the New
York Academy of Sciences 1035, 231-249 (2004).
41. Vig, M. & Kinet, J. -P. Calcium signaling in immune cells. Nature immunology 10, 21-27
(2009).
42. Watve, M. G. & Yajnik, C. S. Evolutionary origins of insulin resistance: a behavioral
switch hypothesis. BMC Evolutionary Biology 7, 61 (2007).
43. Wells, J. C. K. The evolution of human adiposity and obesity: where did it all go wrong?
Disease Models & Mechanisms 5, 595-607 (2012).
44. Wingard, J. R., Ribaud, P., Schlamm, H. T. & Herbrecht, R. Changes in causes of death
over time after treatment for invasive aspergillosis. Cancer 112, 2309-2312 (2008).

15
Chapter 1. Chronic systemic low-grade inflammation
Paragraph 1.1. The Selfish Immune System. When the Immune System Overrides the “Selfish”
Brain. Pruimboom L, Raison CL, Muskiet FA. Submitted.
The selfish immune system
When the Immune System Overrides the ‘Selfish’ Brain
Leo Pruimboom1,2, Charles L Raison3, Frits A.J. Muskiet1
1Laboratory Medicine, University Medical Center Groningen (UMCG), University of
Groningen, P.O. Box 30.001 Groningen, The Netherlands; 2Natura Foundation, Edisonstraat 66, 3281 NC Numansdorp, The Netherlands; 3School of Human Ecology and School of Medicine and Public Health, University of
Wisconsin Madison, Madison, WI, USA
Corresponding author
L. Pruimboom
Burgemeester Marijnenlaan 98
2585 DR Den Haag, The Netherlands
Email: [email protected]

16
Review
The Selfish Immune System
When the Immune System Overrides the ‘Selfish’ Brain
Leo Pruimboom 1,2,*, Charles L Raison3, Frits A.J. Muskiet1
1. Laboratory Medicine, University Medical Center Groningen (UMCG), University of
Groningen, P.O. Box 30.001 Groningen, the Netherlands; [email protected]
2. Natura Foundation, Edisonstraat 66, 3281 NC Numansdorp, the Netherlands;
3. School of Human Ecology and School of Medicine and Public Health, University of
Wisconsin Madison, Madison, WI, USA; [email protected]
* Correspondence: [email protected]
Abstract
The brain and the immune system are the only two systems of the human body that can
dominate all others by extracting resources, including glucose. The brain dominates during
daytime hours and stressful situations, whereas the immune system protects us principally at
night, during periods of infection and when wounds are healing. Both systems are similarly
capable of drawing on energy and other essential resources using strategies beneficial to their
own function and anatomy. Human evolution has made the brain the most important of the
body’s systems, resulting in a shift from strong to smart. However, the immune system is very
old and robust; when necessary it is activated by a variety of non-specific immune challenges
such as psychoemotional stress and most often when immune activating risk factors
(including endotoxemia) are not solved in an appropriate timeframe. When chronically
activated, the immune system demonstrates even more selfish behaviour than the selfish
brain, inducing chronic low-grade inflammation and multiple related diseases. But before
castigating the immune system for this behaviour, it is crucial to recognise that it is only
doing what it is made for: trying to protect us.
Keywords
immune system; selfish brain; inflammation; evolution; stress; chronic disease; Alzheimer;
fibromyalgia syndrome; insulin resistance

17
1. Introduction
1.1. The selfish brain and immune system in evolution
Low-grade inflammation; the cause of causes
Chronic inflammatory diseases (CID) are increasing in frequency, while treatments for these
conditions are still in their infancy (1). Many of these diseases hardly existed 200 years ago (2)
and proximate interventions addressing their genuine aetiologies have not been very
successful (3). Chronic inflammation involves the innate and adaptive immune system (4),
which can be considered very costly at the level of the use of resources, including energy (5),
proteins and certain minerals, such as calcium (6) and magnesium (7-9). Long-term
activation of the innate and adaptive immune system causes further maturation of antigen
presenting cells (10). It is therefore plausible that low-grade inflammation is a direct cause of
multiple diseases related with increased activity of the innate and adapted immune system
including multiple autoimmune disorders and cancer. A recent meta analysis showed a linear
association between C reactive protein (CRP), a marker for low grade inflammation and risk
for breast cancer (11)).
Physiological processes in all living organisms are direct consequences of evolutionary
pressure promoting overall evolutionary fitness, defined as survival taking precedence over
reproduction and direct survival/reproduction taking precedence over long-term
survival/growth (12). Because of this precedence being set, injuries or atrophy of tissues such
as skin, bone and tendons occur when resources needed for survival are scarce, including
situations of starvation (13), and in times of energy depletion due to acute and even chronic
inflammation (1). Many CIDs manifest at older age and therefore exert little selection pressure.
Our ancestors had much lower life expectancies and rarely suffered from CIDs or the
resulting adverse health consequences, and when they did this would hardly have affected
survival and reproduction (14). Nevertheless, although inflammation can affect important
organs such as the liver and liver inflammatory mechanisms are essential for the
maintenance of liver health, it is important to note that even in these situations, hepatic
gluconeogenesis is maintained during immune activation, providing the energy required for
survival and reproduction (15, 16).
During the preparation of this review, which started in 2008, other authors published the term
“selfish immune system” and so we refere to those publications (17, 18). Nevertheless this
paper has been written as original and focuses on total different insides of the selfish
character of the immune system. The purpose of this review is to give evidence that the
immune system overrides the brain in situations of low grade inflammation and that the
dominance of the immune system is the major reason for the development of most if nota ll

18
chronic diseases. It is obvious that the comparance between a solid organ such as the brain
and a circulatory organ such as the immune system is difficult. Nevertheless, if we do not
consider anatomy, both Systems are perfectly comparable at the level of behaviour, health and
disease
1.2. Protective role of the immune system during human evolution
Robust adaption to new environmental challenges involves epigenetic changes that
influence rapid (epigenetic; individuals, some generations) and long-term (genetic;
generations) adjustment of the phenotype, for instance by epimutation, single nucleotide
polymorphisms and gene copy number variation (19, 20). Numerous environmental factors
have shaped the human genome, including climate, food and microbial load (21). Although
the first two challenges certainly show selective pressure in humans, the main selective
pressure seems to derive from pathogens because of their high degree of potential lethality
(22).
When hominins began exploring new environments looking for food and scavenging, they
were exposed to new pathogens. For example, dead meat, when spoiled, is a perfect source of
pathogens such as Escherichia coli, Salmonella and other possible lethal microbes (23). The
struggle to survive in new situations led to the development of an incredibly effective and
robust immune system. The survival mechanisms evolved at least four times and entailed:
upregulation of anti-inflammatory and anti-pathogenic strategies (24), spontaneous physical
activity (25, 26), the development of a highly sophisticated behavioural immune system (27),
and higher immunological reactivity, when compared with our evolutionary closest
counterpart, the chimpanzee (28, 29).
The higher reactivity of the immune system enabled the exploration of new environments
and, when necessary, the ability to mount a massive innate immune response to prevent
lethal infection (30). This response is extremely costly and would have hardly permitted the
further brain growth observed in later hominins if the pro-inflammatory reaction to
pathogens had prevailed over the needs of the brain on a chronic basis. The use of the first
three strategies might have been necessary because of a much lower energetic cost, thus
protecting against pathogenic load, without suffering from the possible secondary damaging
effects of a pro-inflammatory strategy (31). It is therefore conceivable that the combination of
these three strategies ‘liberated’ energy for larger brains and an expansion of brain functions.
Failure of these three strategies (i.e. upregulation of anti-inflammatory and anti-pathogenic
strategies, spontaneous physical activity and the development of a highly sophisticated
behavioural immune system) necessitates a protective high-cost pro-inflammatory response

19
and the entire body is then at the disposal of the immune system; “prima vivere e dopo
filosofare” (first live and then philosophise). This selfish behaviour of the immune system is
observed not only in acute inflammation but also in chronic inflammation, the major
difference between these two states being a shift from a hypermetabolic state to a
hypometabolic state. Figures 1 and 2 show how the immune system puts the body at its
disposal in acute inflammatory (Figure 1) and chronic inflammatory (Figure 2) states.
Observing the actual pandemic increase of non-communicable diseases, we consider that it
is this selfish behaviour of the immune system that causes the majority, if not all, of these
diseases. Although the selfish immune system gave humans the ability to explore the entire
world, it now seems to be responsible for most modern diseases, including cardiovascular
disorders, autoimmune and neurodegenerative diseases.
Figure 1. The immune/metabolic response during acute inflammation.
The increased need for energy during acute inflammation causes a hypermetabolic state and allocation of resources,
including proteins and minerals, to the immune system. Insulin levels are down-regulated by inhibition of pancreatic B-cells
and glucose can be used by the immune system through development of adaptive insulin resistance of competing organs
such as muscles, fat and liver. Short-term use of neurotransmitters by the immune system increases the activity level of the
innate immune inflammatory response, ‘helping’ the need to mount an intense but optimal reaction that will resolve in a
maximum of 4 to 7 days. Sickness behaviour induced by hyperleptinaemia and pro-inflammatory cytokines further saves
energy, induces sleep and adaptive cachexia. The optimal IIS response is short-term and will moderately activate antigen-
presenting cells. The adaptive immune system will produce anti-inflammatory memory cells that can be recruited when the
host encounters a different immune challenge of the same type. This adaptive immune response generally ends after a
maximum of 27 days, leaving sufficient energy to maintain health of the organs disposed of during the start of the immune
response. Resolving substances such as protectins and resolvins finish immune activation and homeostasis of the whole
body is recovered through normalised energy distribution. Ach, acetylcholine; APC, antigen presenting cell; IIS, innate
immune system.

20
Figure 2. Chronic low-grade inflammation and hypometabolism.
In the modern world, long-term pro-inflammatory activity of the immune system is frequently caused by anthropogenic
factors and other conditions that challenge the immune system only weakly, but chronically. In normal situations any
inflammation would produce a compensatory immune suppression, which is why low-grade inflammation needs a logical
explanation. To maintain pro-inflammatory activity, the immune system itself puts the entire body at its disposal, but at the
same time protects the body against multiple organ failure by inducing a hypometabolic state. Cortisol and noradrenaline
induce gluconeogenesis and this extra glucose is allocated to the immune system by increasing insulin resistance of
competing organs. At the same time, leptin reactivates the immune system, whereas brain regions associated with satiety
develop leptin resistance, inducing increased food craving. Low thyroid hormone (rT3>T3) decreases total energy
expenditure (protective hypometabolism) and rT3 is needed to fight pathogens. Nerve-driven immunity provides the
immune system software (serotonin and dopamine) to maintain pro-inflammatory activity, whereas immune-suppressive
mediators are down-regulated (cannabinoids and acetylcholine). Retraction of sympathetic fibres of inflamed/immune
tissue and increase of sensory fibres are hardware strategies of nerve-driven immunity. Chronic pro-inflammatory activity is
further maintained by a shift from an anti-inflammatory androgenic to a pro-inflammatory oestrogenic state in both males
and females. The total picture of this ‘selfish immune system behaviour’ might be considered protective when it does not
last too long; it is however highly deleterious when the human body starts developing all kinds of modern disorders such as
autoimmune diseases, neurodegeneration and other maladies. Nevertheless, even today it is usually better to develop a
CNCD than to die of cancer or infection.

21
1.3. Acute inflammation resolves itself; controlled resolution
The immune system is self-regulating through negative biofeedback mechanisms, just like
any other system in the human body. Acute inflammation induces the production of several
substances responsible for finishing the immune reaction, including arachidonic acid
derived lipoxins, EPA-derived protectins and DHA-derived resolvins (1), but only if the
aforementioned fatty acids, notably the fish-derived fatty acids EPA and DHA, are available in
sufficient amounts (32-34). These substances decrease the activity of pro-inflammatory cells
of the innate immune system and, at the same time, stimulate migration of phagocytizing
macrophages to the danger zone/battlefield (35-37)
Another more intrinsic negative biofeedback signal is lactic acid. The activated immune
system uses (cytoplasmic) glycolysis as energy metabolism, in which 90% of glucose
molecules are converted into lactic acid, a metabolic shift from mitochondrial oxidative
phosphorylation (MOP) to cytoplasmic substrate level phosphorylation (SLP) (38). This
metabolic shift seems counterintuitive when considering that only 2 molecules of ATP are
generated from 1 molecule of glucose during SLP, whereas MOP yields 36 molecules of ATP.
This is, however, conceivable in the light of the velocity of SLP, which is a hundred times
faster than MOP (38, 39).
A second benefit of SLP is that the glucose molecule is only partially used for ATP generation.
Lactic acid and other macromolecules are metabolites of SLP that confer several favourable
positive conditions during acute inflammation that guarantee cell division and cytokine
production and render the immune system to a state independent of food and oxygen. The
immune system’s capacity to engage in SLP is also known as the ‘Warburg effect’, which not
only provides the immune system with fast energy, but also the precursor (glucose) needed
for the synthesis of structural elements for the production of all DNA, RNA, organelles and the
majority of cell membranes (for an excellent review, see 40). The oxygen-independent
fermentation of glucose in the cytoplasm thus leads to the production of amino acids as
precursors of proteins, (deoxy)ribose for DNA and RNA, glycerol for lipids and NADPH
through the pentose phosphate pathway, needed for the production of phospholipids and
glutathione (40). The activated immune system is now capable of growth and proliferation,
largely without the need to uptake oxygen and building blocks. Considering the sickness
behaviour associated with acute infectious disorders, characterised by cachexia {food
absence} and even diaphragmatic breakdown {low oxygen} (41, 42), this makes sense.
Lactic acid supports the role of lipoxins, resolvins and protectins in finishing the
inflammatory response in a maximum of 4 to 7 days. Finishing the inflammatory response in
time protects the body against possible deleterious secondary effects caused by the immune

22
system itself. Not only have intrinsic mechanisms emerged to end an acute inflammatory
response, but brain-coordinated strategies, including the production of substances such as
cortisol (43), certain cannabinoids (44), acetylcholine (45) and catecholamines (46, 47), are able
to switch off the immune system. These inhibiting mechanisms are activated through
coordinated processes during acute inflammation and the same holds true for the serotonin
and dopamine pathways (see chapter ‘behaviour at the disposal of the immune system’).
Observing the multiple mechanisms responsible for inhibiting the pro-inflammatory activity
of the immune system, it can only be concluded that the inflammatory response should be
finalised in time, leading to controlled resolution even after infection (48).
Therefore, why is immune-inhibition absent when people suffer from weak inflammation
caused by anthropogenic factors (AF)? AF activate the immune system through indirect
pathways and causes a weak, ‘cold’ inflammation, without any of the typical signs of ‘hot’
inflammation (49). Usually adipocytes are activated by AF, such as high caloric food intake
and psycho-emotional stress. Although this type of inflammation is not as strong, it still
produces a metabolic shift of the immune system, giving rise to the production of substantial
amounts of lactic acid, which serves as a potent immune suppressor through the creation of
significant acidosis (50, 51). The question therefore remains how the immune system
‘manages’ its activity for years and years, in spite of intrinsic and extrinsic mechanisms that
inhibit the immune system and generally protect the body, but especially the selfish brain,
against secondary damaging effects.
We suggest that the immune system manages long-term activity by pursuing at least two
strategies, firstly to achieve energy (glucose) and secondly to reactivate itself. The first strategy
has to induce constant gluconeogenesis and energy allocation to the immune system and the
second strategy has to reactivate the immune system. Pro-inflammatory immune system
activity depends on several conditions. 1. Pro-inflammatory cytokines have to be produced
through activation of the key regulator of the immune system being nuclear factor-κB (NFkB),
2. Immune cell growth and cell proliferation depends on activation of the mammalian target
of rhapamacin (mTOR), 3. cytoplasmic glycolysis is needed for the production of
macromolecules and energy during immune activation and this requires the activation of
hypoxia-induced factor-1 (HIF-1) and 4. The activated immune system demands large
amounts of glucose and therefore a higher number of glucose transporters type 1, achieved
by upregulation of c-myc (52-54).

23
Strategies used by the immune system to reactivate itself include:
• Insulin resistance and high insulin levels
• Leptin resistance and hyperleptinaemia
• Low thyroid hormone syndrome
• Catecholamine resistance of the immune system
• Cortisol resistance of the immune system
• Systemic androgens to oestrogens shift
• Peripheral serotonin recruitment
• Peripheral dopamine recruitment
The majority of these different strategies are associated with sickness behaviour exhibited
during acute infection (55). This behaviour is characterised by symptoms such as cachexia,
fatigue, increased sleep and fever. It suggests that the activated immune system itself is
responsible for fasting during infection and ‘senses’ that food will not be available under these
conditions. The outcome is the mentioned shift from MOP to SLP, resulting in immune cell
proliferation and activity becoming independent from food intake, all to enable the infection
to be cured (56). Chronic activity of the immune system, during which the same metabolic
shift occurs, would require a large amount of glucose, which is the basic energy fuel of the
activated immune system and, of which, the limited availability constitutes the basic problem
in chronic non-communicable diseases (CNCD) (56). This ‘nutrition-independent’ state may
be responsible for the recently evidenced decrease in basal metabolic rate in chronic
inflammatory disorders, rendering the subject vulnerable to the development of multiple
metabolic disorders, including metabolic syndrome and diabetes mellitus type 2 (57).
In summary, the metabolic shift observed in conditions of an activated immune system
renders the system glucose-dependent and will therefore activate all strategies to maintain
glucose homeostasis through systemic gluconeogenesis (43). The immune system can apply
multiple strategies to maintain activity, although at a low level and puts the whole body at its
disposal, including the brain, if necessary. This selfish behaviour has a protective effect
during acute infection, but may have a dramatically deleterious effect in the long run,
evidenced by the number of people suffering from CNCD in our society (58, 59). Low-grade
inflammation should therefore be regarded as the cause for the majority, if not all, cases of
CNCD. Treatment should therefore target the strategies used by the immune system to
maintain its activity over a prolonged period of time. The only way to provide the correct
treatment is to understand the ways the immune system puts the whole body at its disposal,
which is the aim of this review.

24
1.4. The selfish immune system; brain damage leads to more brain damage
caused by the immune system and overriding the brain as most dominant
organ
The immune system is one of the systems with a high level of biological robustness (60).
Some diseases and their effects produced by the robust character of the immune system will
not necessarily benefit the host, but that is the price to pay. We provide evidence that the
mechanisms leading to pathologies affecting the whole body, including the brain, should be
considered robust and part of the survival strategies developed during hominin evolution (61).
The interactive neuro-endocrine-immune system evolved to cope with acute immunological
challenges such as infection and wound healing, but is also activated by non-immunological
danger for which it was not designed (2, 62). Theories explaining biological priorities
consistently put the human brain in first place (63, 64). However, brain functions, blood
circulation and anatomy are disrupted in those suffering from Alzheimer’s disease,
Parkinson’s disease, fibromyalgia syndrome (FMS), chronic fatigue syndrome (CFS) and
depression (65-68), which suggests that certain pathophysiological processes override the
protective behaviour of the selfish brain.
FMS depression and Alzheimer’s disease (AD) are obviously not diseases of choice, but may
rather reflect the involuntary alternative of suffering from a low energetic state leading to a
non-permissive brain disorder (e.g. AD, depression and FMS), rather than dying from multiple
organ failure (69-71) caused by a chronic activated pro-inflammatory immune system.
Rogers (72) stated that “Inflammation seems useful when controlled, but deadly when it is not.
For example, ‘head trauma may kill hundreds of thousands of neurons, but the secondary
inflammatory response to head trauma may kill millions of neurons or the patient” and the
same holds true for people who suffer a stroke (73, 74). The inflammatory response of the
immune system causing severe secondary damage to the brain after traumatic brain injury or
ischaemic stroke seems maladaptive in the face of the ‘selfish’ brain hypothesis. It would,
however, correspond with the ‘selfish’ immune system hypothesis, which states that danger
gives precedence to the immune system, and thereby overriding the selfish brain.
It may even be the case that dramatic inflammatory response following brain injury is not
caused by the injury itself, but by the presence of infectious pathogens. Pre-existing infection
is present in one third of clinical ischaemic stroke patients (75). Clinical data suggest that
stroke risk peaks at three days after infection onset and that this risk remains high for three
months (76). Almost three out of every four people in the world who suffer a fatal stroke live in
developing countries and malaria, Chagas disease and Gnathostomiasis seem to be the major

25
causes for this surprising fact (77). Children and young adults are less susceptible to all-cause
strokes with the exception of infectious stroke and diseases such as sickle cell disease. A
recent case-control study revealed that pre-existing infection is an independent risk factor for
stroke in 33% of affected children also in developed countries, such as the USA where 2,400
children suffer from strokes every year (78). These data suggest that a possible pathogenic
presence ‘programmed’ a severe pro-inflammatory immune response when the brain or the
heart muscle are damaged. The selfish behaviour of the immune in these cases should be
considered as adaptive but also possibly deleterious. Nevertheless the fact that people
suffering from immune suppression after stroke are highly susceptible for sepsis and dead
(79) suggests that the selfish behaviour of the immune system should be considered as
needed when damage to the heart or the brain has been done.

26
2. It’s all about energy – The selfish immune system
2.1. Robustness and energy reallocation between visceral organs, muscles,
the immune system and the brain
The immune system is one of the energetically most costly systems in humans when
‘activated’. Consequently, immune metabolism has a profound effect on the functioning of
the body. Metabolic conflicts between organs seem to explain the emergence of several
disorders and, more specifically, modern non-communicable diseases such as autoimmune
disorders (80).
Anthropogenic danger signals, such as psychoemotional stress and sleep deprivation, are
capable of activating the immune system and a chronically activated immune system
demands high energy, protein and immune-specific minerals such as calcium (2). Such high
costs would never have permitted the development of the phylogenetic newer metabolic
expensive brain in general and, more specifically, the neocortex during evolution. A recent
review describes how exercise (searching for food, water and shelter) in primates and early
hominids produced a shift from a pro-inflammatory immune reaction with a high metabolic
demand to an anti-inflammatory response with a low metabolic demand (31). This shift made
it possible to allocate energy to other organs e.g. the brain without large amounts of energy,
proteins and minerals having to be invested in the immune system, whilst at the same time
maintaining protection against microbes.
2.2. Energy and energetic conflict as the driving force behind evolution
Changes in energy allocation between organs are a consequence of energy conflicts that
affect all animals, but non-human primates and humans in particular (81, 82). Several
scenarios have been proposed, with the brain being the organ to benefit from loss of colon
length and high caloric dense food [the expensive tissue hypothesis] (81, 82)], a decrease in
muscle mass, an increase in fat mass (49), cooking (83), human locomotion costing less
energy (84) and, very recently, a change in the expression of glucose transporters beneficial to
the brain (85). To our knowledge, as yet it has not been suggested that immune function may
also have benefitted from a smaller gut, a lower energy demand for locomotion, an increase
in fat mass and tissue-specific differences in the expression of glucose transporters. The latter
entails a higher expression of GLUT1 in activated immune cells, when energy is needed to
protect against pathogens and other immune challenges (86), at the expense of GLUT4
expression in muscles and adipocytes (87-90).

27
The work of Fedrigo et al. (85) shows that glucose transport capacity has been essential for
brain growth and function during human evolution. They demonstrated that human brain
cells express more activity of the SLC2A1 gene, which is the genetic code for the production
of GLUT-1 glucose transporters compared with the chimpanzee and macaque (human >
chimpanzee > macaque). At the same time, SLC2A4 expression in muscle is significantly
higher in chimpanzee > human > macaque.
Logical reasoning makes it plausible that a concomitant per gram tissue reduction of SLC2A4
in skeletal muscle and an increase of SLC2A1 in the brain will lead to higher glucose uptake by
the brain at a given plasma glucose concentration. This would result in a shift of glucose
allocation away from the body (strong) and towards the brain (smart).
2.3. Glucose to the immune system – prioritising energy guidance
Along a similar vein, the same holds true for glucose allocation to the immune system. The
energy demand of lymphocytes and leukocytes increases dramatically upon activation (2, 4)
and all activated immune cells express GLUT1 glucose transporters (86, 91). Higher expression
of GLUT1 will promote energy allocation to the immune system, which could be considered
to be an ‘energy demand reaction’ (92). Glucose allocation to the immune system maintains
its function even under strong energy restriction (80).
The foregoing demonstrates that activation of the immune system through danger signs will
attract and redistribute energy, favouring the brain and the immune system. Prolonged
activation of the immune system (as has been observed in people with CIDs) would allocate
glucose chronically to the immune system through immune-controlled down-regulation of
GLUT1 transporters at the level of the blood-brain barrier and would decrease GLUT4
transporters at the level of muscle and adipose tissue (93-96).

28
3. Evolution shaped the selfish immune system
3.1. Evolution and the human selfish immune system - over-reactivity of the
human immune system when compared with chimpanzees
It has been shown that the human immune system is relatively over-reactive when compared
with our closest evolutionary relative, the chimpanzee (97). The increased activity of the
human immune system holds true for both the innate and the adaptive immune systems (98).
It seems that all major cell groups of the human immune system show lower levels of
mediators capable of down-regulation of the immune response against pathogens and
phytohaemagglutinin (28). These mediators, called inhibitory sialic acid-recognising Ig-
superfamily lectins (SIGLEC), are expressed on most immune cells including B lymphocytes
(99). The difference between chimpanzees and humans is three-fold. Firstly, humans express
different SIGLECs; secondly, humans exhibit lower SIGLEC numbers and little or none are
present on T lymphocytes, and thirdly, they show a lower production rate of inhibitory
SIGLECs when challenged with pathogens or other immune stimuli (99, 100).
The observed development of a more reactive immune response in humans is probably a
consequence of being faced by unique immune challenges to numerous pathogens through
scavenging, increased population density, hunting and migration (101, 102).
3.2. The selfish brain is less selfish than the much more ancient immune
system
The brain is selfish in almost every situation, including mild and severe stress (103) and
multiple studies support the ‘selfish brain’ hypothesis. Acute mild mental stress requires 12%
additional energy from the human brain (104) and the same holds when humans are
challenged by intense exercise (105). The group of Peters showed that social stress also
augments the brain’s energy need (106) and that the brain switches to the use of ketone
bodies (107) and lactic acid (107) when glucose is unavailable. Therefore, several lines of
research give evidence for the ‘selfish brain’ hypothesis in which it is stated that brain energy
is maintained through multiple pathways, including activation of central stress axes and the
use of multiple energy sources (glucose, ketone bodies, lactic acid). Immune activation also
produces activity of central stress axes and a state of high arousal of the central nervous
system, the purpose being to sense and avoid further danger (108). An acute inflammatory
response produces energy allocation to the immune system until this is resolved, and only
when the system is challenged by mono-metabolic danger signals (108). However, multi-
metabolic risk factors produce an energetic conflict between the immune system and the
brain. The combined need for resources (energy-producing macronutrients, blood, oxygen)

29
of the stressed brain, of the activated immune system and of other organs responsible for
maintaining organ functions during multiple metabolic signalling challenges, caused by
psychogenic, psychosocial and physical factors at the same time, would probably override the
maximum capacity of energy uptake by the gut and, although speculative, would demand a
maintained heart rate of around 180/minute and a chronically increased blood pressure at
around 160/120 mm Hg. This would lead to severe damage to the heart and probably the
brain, which is contrary to the evolutionary drive of maintaining brain function and anatomy
against at any cost (106). The only feasible response to maintain life throughout chronic
situations of high energy demands of ‘conflicting’ organs, is the ‘creation’ of an organ-
specific low thyroid hormone syndrome and other adaptations with the purpose of lowering
the activity of all organs and puts the body at the disposal of the immune system. This is
evidenced by the development of immunological sickness behaviour, immunologically
induced secondary damage of vital organs, protective depression, gluconeogenesis by the
liver and kidneys, and the use of metabolic hormones and neurotransmitters by the immune
system to benefit its pro-inflammatory activity and thereby protect against possible lethal
pathogens (109, 110, 111, 112).
It is definitely true that the brain ‘behaves’ selfishly in almost every situation, including an
energy deficient intra-uterine environment. Nevertheless, although it seems that one of the
most fundamental biological drivers in humans is supplying the brain with nutrients and
energy, how is it possible for people to suffer from diseases related with lack of brain energy?’
Something has to be so wrong that it may even cause a reaction that overrides this interest
and ‘accept’ the collateral damage to the selfish brain. Acute sepsis, severe burn wounds,
multiple traumata and major surgery are known to allocate up to 100% of resting energy
expenditure to the immune system, but these are acute situations which often lead to instant
death, even in children (3, 4, 113-116). Neurodegenerative disease, fibromyalgia, and chronic
fatigue disorders develop slowly and should therefore be caused by factors that demand long-
term energy allocation to systems other than the brain, e.g. the immune system, ultimately
affecting the transport of resources to the selfish brain. Sedentary lifestyle, overeating,
childhood abuse, oral sepsis, chronic life stress, leaky barriers, perceived social stress,
environmental toxins, social jetlag, meal frequency and even father-daughter conflict all
activate the immune system and an energy demand response based on activation of the SAM
and HPA axes (117-121). This latter response should provide energy for, primarily, the brain
and secondarily, the immune system. When brain allocation fails, brain functions and
possibly anatomy will be disturbed. The latter occurs in people suffering from acute infection,
but also in those suffering from Alzheimer’s disease, FMS, CFS, depression and other diseases
affecting the central nervous system. It seems that multiple metabolic danger signals produce
a state mimicking acute life threatening danger, allocating energy to the immune system and
disposing of energy from the rest of the body, including the brain.

30
4. Genetic and environmental evidence supporting the
hypothesis
4.1. Genetic evidence for the selfish immune system hypothesis
Depression and other maladies, including FMS and neurodegenerative disorders, such as
Parkinson’s and Alzheimer’s diseases, are related to increased immune activity (122, 123, 124).
All of these disorders seem to have a genetic predisposition and the majority of the genes
related with neuro-degenerative disorders and depression influence the immune system.
Raison hypothesised that if depression is related to certain polymorphisms, then these genes
are primarily protective in the face of infection (for review see 125). The same holds for genes
related to Alzheimer’s disease and FMS.
The gene most widely accepted to be associated with Alzheimer’s disease susceptibility is
ApoE 4 (apolipoprotein E4), although many others have been proposed as Alzgenes (126).
Classical functions of apolipoprotein E relate to metabolism (transporter of lipids in the
periphery and in the central nervous system (127)). More recently, it was found that ApoE
influences the innate and adaptive immune systems (128). The overall influence of ApoE on
the innate immune system is complex and depends on ApoE polymorphism. ApoE4 induces
an inflammatory response, whereas ApoE3 inhibits inflammation and enhances repair (see
references in 128). The pro-inflammatory activity of ApoE4 is classically seen as deleterious,
but can also be considered protective in the light of the pathogen-host defence (PATHOS-D)
hypothesis proposed by Raison and Miller (125). In our recent evolution, the share of the
ApoE3 allele appears to have increased at the expense of the ancestral ApoE4 (129). ApoE3 has
spread significantly in first world populations, whereas the prevalence of ApoE4 is still very
high in cultures historically exposed to disease-causing pathogens (116, 130-132). The
observation seems consistent with the protective role of ApoE4 against infection. The
Yoruban population in Nigeria has a 70% lower incidence of dementia when compared to
African Americans. This difference is probably caused by the lifelong low-fat diet of the
Yorubans (133).
Candidate genes possibly associated with increased susceptibility to fibromyalgia syndrome
have been reported, but no definite conclusions can be drawn as yet (134). The serotonin
transporter gene SLC6A4 is perhaps the strongest candidate in terms of its relation to FMS (51,
135, 136). Two major SLC6A4 polymorphisms have been identified. The short allele carrier
produces a protein, which is less efficient in the reuptake of serotonin and carriers show an
increased risk for the development of depression when facing psychosocial challenges.
Carriers of the short allele further show higher levels of circulating pro-inflammatory
cytokines compared with anti-inflammatory cytokines (IL-6/IL-10 rate) when challenged

31
with a psychosocial stressor (137).
Another disease with an immunological genetic background is celiac disease. Celiac disease
is caused by the intake of gluten and it’s interaction with a great number of genes, of which
most are related with an increased reactivity of the immune system (138). More specific, it are
alleles related with IL18, IL23, IL2 and IL12 that increase the susceptibility for celiac disease,
but at the same time people expressing these genes are probably better protected against
pathogens, including virus and bacteria (139)
4.2. Pathogens shaped genetics to the benefit of the immune system and
pathogen load prioritises the immune system over the brain
If the PATHOS-D hypothesis is correct, that the microbial world is co-responsible for the
chronically increased activity of the immune system and disposal of expensive brain
functions, then pathogenic load should not only affect brain function and anatomy of older
people, but also younger individuals. Evidence for this supporting the PATHOS-D hypothesis
comes from studies investigating the development of intelligence during human evolution in
general and, more recently, the past two hundred years. A recent study of Eppig (140) showed
that infectious disease and the consequent immune activity is the most important predictor
of lower intelligence in almost every population on earth. Nutrition was also correlated with
IQ, but became insignificant when corrected for infection. The connection between
pathogen load, infection and intelligence seems plausible considering the high energetic cost
of infectious disease. (141).

32
5. Long -term immune activity: the need for reactivation and
fuelling strategies
5.1. Evolutionary stored energy limits the timescale of immune activity
Both the innate and adaptive immune responses are normally self-limiting (142). The self-
limiting timeframe of 28 days is probably based on the energy-resource model. A (much)
longer massive activity of both systems would lead to severe secondary damage and even
death because of sustained energy deficit of vital organs including the liver, kidneys and heart
muscle (143). Recruitment of the adaptive immune system, although very expensive at first
exposure time (141), and the generation of immune memory, should be considered beneficial
from an evolutionary point of view, because of the shortening of the immune response and
protection against energy depletion, when the host is newly exposed to the pathogen (64).
Chronic low-grade inflammation literally means long-term low activity of the innate immune
system and lack of the production of self-limiting substances, such as resolvins and
protectins (142). Chronic low-grade inflammation probably starts after a non-optimal acute
inflammatory response (supranormal or subnormal) and when self-limiting mechanisms or
strategies fail (143). Sterile wounds, low pathogenic load and non-specific immunological
challenges such as psychogenic stress activate the immune system, but lack the strength of
optimal immune activation which would normally lead to complete resolution (144).
Nevertheless if the factors that activate the immune system in a subnormal manner are not
resolved, the capacity to maintain immune activity is essential for survival, as evidenced in
people suffering from inflammation-associated immune suppression. People suffering from
inflammation-associated immune suppression (IAIS) are highly susceptible for the
development of different types of cancer and secondary infections, and IAIS significantly
increases the mortality rate (145, 146). IAIS is induced by immunological intrinsic (e.g lactic
acid)., but also multiple brain derived strategies including the activation of the
parasympathetic nervous system. This is where the immune system has to put the body at its
disposal, thereby overriding the selfish brain.
5.2. The consequence: the whole body at the disposal of the selfish immune
system
The way in which the immune system puts the body at its disposal might follow a
coordinated sequence. The initial activation of energy demanding central stress axes allocates
resources to the immune system through induction of gluconeogenesis and insulin
resistance of competing organs, such as, bone, muscle, adipose tissue and the liver (1, 40).
Once the innate immune system (maximum 4-7 days) and, when necessary, the adaptive
immune system (27–42 days) has/have triggered the immune response, problems should

33
have been resolved. If immune activation is required for a longer period of time, this induces
further insulin resistance/hyperinsulinemia, hyperleptinaemia/leptin resistance, and
hypercortisolism/glucocorticoid resistance. The possible hypermetabolic state produced by
the constantly activated immune system could cause multiple organ disorders, failure and
even death (MODFD). The development of a low thyroid hormone state (rT3>T3) protects the
body against MODFD, putting homeostatic regulation at the disposal of the immune system.
The pro-inflammatory activity can also be maintained by higher aromatase activity and the
production of pro-inflammatory oestrogens (see below): a further step in putting the whole
body, including the reproductive system, at the disposal of the immune system.

34
6. Fuelling and reactivation strategies of the immune system
6.1. Thyroid hormone prevents multiple organ failure during
hypermetabolism and maintains immune homeostasis; thyroid hormone
at the disposal of the selfish immune system
Immune activity depends on aerobic glycolysis (147). It is only possible to maintain aerobic
glycolysis when the intrinsic inhibitory pathways of the immune system can be overruled.
Low T3 and high rT3 can maintain aerobic glycolysis in immune cells. Thyroid hormone T3
induces mitochondrial activity in all kinds of cells, including immune cells (148). T3 can even
strongly activate mitochondrial oxidation in cancer cells and render them more sensitive for
chemotherapy (149). Intracellular T3 would therefore inhibit the inflammatory activity of the
immune system, which would be highly deleterious during severe infection or other
immunological challenges. Extracellular T4 and T3 are necessary for immune activation (150),
but intracellular T4 is converted by deiodinase 3 (D3) into rT3 and T3 is rapidly downregulated
by the same enzyme, preventing mitochondrial activation and maintenance of cytoplasmic
substrate level phosphorylation through upregulation of D3 by the pro-inflammatory
cytokine IL-6 (151). The final state is that of a low thyroid hormone syndrome (LTHS).
LTHS does not only benefit the immune system, but is also protective against the possible
secondary damage of chronic immune system activation. These rather deleterious effects of
immune activation on other organs and tissues is prevented by down-regulation of the
conversion of T4 into T3 both systemically and tissue specifically, causing the reduction of
overall metabolic rate, but especially the activity of organs less important for direct survival
during immune activity, such as muscle tissue, liver, kidneys, the heart muscle and the
digestive system (152-154). The lower activity of these organs protects them against acute
organ failure and sudden death of the host. The protection and lower metabolic rate is a
product of low thyroid syndrome and high reverse T3 (rT3) (151). This state is protective
initially, but can be deleterious in the long run (155).
A recent discovery by the group of Klein and Schaefer has shed new light on the interaction
between the immune system and metabolism. Their group proposed a new model of
controlled metabolic regulation by an activated immune system (156-158). They showed that
TSH can be produced by several tissues other than the thyroid gland. Dendritic cells (DCs) and
several cells from the small intestine are capable of producing TSH and this happens mostly
during bacterial or viral infection (159). Certain leukocytes also produce TSH, but with a
slightly different structure and this hormone has been named TSHbeta splice variant (160).
The TSHbeta splice variant seems to change the thyroid phenotype inducing a shift from T3
to rT3. This shift decreases total body metabolism because of lower systemic T3 (159) and

35
initially saves the brain by continued conversion of T4 to T3 in the brain itself (161).
The possible function of D3 expression in activated innate immune cells is intriguing.
Thyroid hormones (TH) play a role in differentiation and proliferation of cells, with high T3
inducing cell differentiation and low T3 inducing cell proliferation. Granulocytes are short-
lived, fully differentiated cells that migrate to the site of infection and do not proliferate,
which may argue against a role for D3 induction in differentiation or proliferation of activated
granulocytes. Studies in the 1960s suggested a role for thyroid hormone in the bacterial killing
capacity of leukocytes. Iodide in combination with hydrogen peroxide (H2O2) provides one
of the most effective antibacterial substances of the immune system. Thyroid hormones are
an important source of iodide, and leukocytes generate inorganic iodide by the uptake of
iodide and by de-iodinating T4, (162). In combination with the recent demonstration of D3
induction in infiltrating leukocytes during infection, we suggest that D3 induction helps to
generate iodide as part of the innate immune response (150). Studies in S. pneumonia-
infected D3 knockout mice indeed showed a defective bacterial clearance compared with
wild-type mice, which supports this hypothesis (163). Further evidence is given by the work of
Kwakkel et al, showing a dramatic increase of D3 production by neutrophils when challenged
with bacterial LPS (164).
The resulting state of immune activation, low T3, high rT3, combined with the energy
demand reaction of the HPA axis and the sympathetic nervous system maintains
immunological homeostasis during prolonged stress. The brain will maintain anatomy and
function as long as brain metabolism can be guaranteed. The same holds for the immune
system, although long-term stress and inflammation suppress immune activity (46). The
latter situation could expose the host to possible infection and death. Protection of the host
integrity will now depend on the use of alternative mechanisms to postpone this dangerous
state whilst maintaining pro-inflammatory immune activity. This would be the time at which
the immune system 1. puts every possible organ at its disposal to guarantee its own metabolic
homeostasis, 2. induces resistance to hormones with pleiotropic immunological functions
(leptin, insulin, cortisol) and 3. produces a state of nerve-driven immunity, putting almost all
neurotransmitters, including dopamine, serotonin, acetylcholine, glutamate and GABA at its
disposal (165-169).
6.2. Gluconeogenesis and glucocorticoid resistance at the disposal of the
immune system
Endogenous cortisol has several effects on the immune system, including suppression
through activation of the inhibiting factor kappa B (IkB) (170), apoptosis of immune cells that
are no longer needed (171) and migration of immune cells to the so-called ‘battlefield’

36
(dangerous zone) or back into the ‘barracks’ (lymph knots, bone marrow, thymus) (172) and
through activation of macrophage migration activating factor (173). Intact cortisol signalling
in the immune system would lead to suppression of the immune system, which is why
immune cells show an intrinsic mechanism to develop cortisol resistance, which is essential
during acute infection but at the same time co-responsible for low-grade inflammation (174).
Glucocorticoid resistance (GR) of the immune system leads to hypercortisolaemia and
constant gluconeogenesis. The glucose produced by GR-gluconeogenesis can cover the
energetic needs of the selfish immune system. GR-gluconeogenesis can be induced in
muscle, the liver, kidneys and perhaps even the pancreas (175-178). So GR serves two basic
strategies to maintain immune activity: immunological GR prevents inhibition of the
immune system and GR-induced hypercortisolaemia increases glucose production,
necessary for the constant nourishment of chronic active selfish immune cells.
Glucocorticoid resistance itself seems to protect the host against possible viral infection,
including HIV, by maintaining high activity of the anti-viral Th1 component of the adapted
immune system (179, 180), although GR can be highly deleterious (181). The GR observed
during chronic inflammation is universal and mostly occurs along with another ancient
protective mechanism: insulin resistance (182). Cells of the immune system show inherent
genetically imprinted resistance mechanisms, which protect the body against the immune-
suppressive effects of glucocorticoids, although side-effects can be severe, including chronic
leukaemia (174).
GR is observed in rheumatoid arthritis, inflammatory bowel disease and COPD and is mostly
considered deleterious (183). Treatment of these diseases normally focuses on increasing
cortisol sensitivity (184) with contrasting results (1). Increasing GC-sensitivity can even lead to
higher mortality when animals are challenged with pathogens such as E. Coli (185). If intrinsic
or acquired GR conveys protection against pathogenic load, than asthma, rheumatoid
arthritis and inflammatory bowel disease should be associated with increased pathogenic
microbial load. Indeed, the group of Siala showed that reactive and undifferentiated
oligoarthritis is associated with the presence of a high number of bacteria in the synovial fluid
(186, 187). Patients with arthritis also show a high incidence of glucocorticoid resistance (188).
Those with chronic asthma present higher bacterial colonisation of the lower airways, linked
to the severity and duration of asthma (189), whilst GR is also a characteristic of asthmatic
patients (190). Inflammatory bowel disease (IBD) normally evolves with pathogenic bacteria
(20) and, as mentioned above, patients suffering from IBD also show a high prevalence of GR
(191).

37
It therefore appears that GR prevents suppression of the innate immune system and the
glucocorticoids-induced shift from Th1 to Th2 activity of the adapted immune system (192),
thus maintaining protection against microbial infiltration and infection. GR serves the selfish
immune system to maintain activity, nourish itself with glucose, but with just one purpose,
which is to protect the individual from lethal infection.
6.3. Leptin and insulin at the disposal of the selfish immune system
Leptin and insulin are needed to maintain long-term activity of the immune system and the
immune system itself increases the production of leptin by adipocytes via TNFα signalling
(193). Leptin is highly inflammogenic (194) and hyperleptinaemia, together with central leptin
resistance, maintains pro-inflammatory activity and energy allocation to the immune system
(195, 196). Proinflammatory cytokines induce leptin production by adipocytes, as does food
intake.
Adipose tissue is present in immune-cell-harbouring tissues, such as lymphoid organs, bone
marrow and adipocytes that infiltrate wounds (34) and so adipocyte derived leptin can have
direct influence on immune cell functioning.
Leptin activates all types of immune cells and increases glucose uptake during
immunological activity. The principal target of leptin-induced immune cell reactivation is the
key immune response regulator nuclear factor-κB, responsible for transcription of genes
encoding for IL1, IL6 and TNFα (197). In summary, leptin activates the immune system
through different pathways with a focus on the innate immune system and Th1. Under
physiological circumstances, this leads to increased protection against infection and
pathogenic growth. During low-grade inflammation, leptin should be considered to be a re-
activator, which can perpetuate immune activity.
The strategies used by the immune system to maintain its activity and guarantee glucose
availability could merely have evolved for their beneficial effects; a basic rule in evolutionary
biology. This also holds true for the leptin and insulin responses observed during acute and
chronic inflammation. The leptin response during inflammation supports different protective
traits. Hyperleptinaemia during immune activity informs the brain about the adequacy of
long-term energy stores in adipose tissue, asking for/demanding permission to produce a
costly fever reaction and a short-term hypermetabolic state, following immune activation
(198, 91). The hyper-leptinaemic state will also produce inflammatory cachexic behaviour,
which is protective at the start when facing an acute inflammatory response, but could be
deleterious when chronic, as is observed in patients and animals with chronic kidney
inflammation (199).

38
Long-term hyperleptinaemia leads to central leptin resistance. Several researchers noted that
hyperleptinaemia is required for the development of leptin resistance (200). Central leptin
resistance is responsible for an increased risk of overeating (201) and overeating rapidly
produces leptin resistance (202). Central leptin resistance can be considered to be an
evolutionary advantage when energy availability is low, or when the need for energy is
chronically increased as observed during prolonged immune activity (203). The beneficial
effect of hyperleptinaemia and LR is observed in different situations. Hyperleptinaemia and
LR protect against cardiovascular disorders by preventing lipid deposition in the heart muscle
itself (204, 205), although recent publications have challenged this view (206). The influence
of leptin on the anti-pathogenic function of the immune system has recently been
demonstrated in two new studies from the same group (207, 208). Children with low leptin
levels are more susceptible to infection (209). The required pro-inflammatory effect of leptin
to fight against pathogens has also been demonstrated in a recent in vitro study (210). The
overall effect of leptin on the immune system seems to be permissive, which implies that
intact leptin-signalling towards the immune system maintains Th1-Th2 functioning and, if
necessary, ‘permits’ pro-inflammatory activity (211).
Like leptin, insulin is also recognised as a pleiotropic hormone. Energy demands of the brain,
or the immune system during starvation, infection or stress are covered by gluconeogenesis
and the temporary development of insulin resistance of various organs, caused by
proinflammatory cytokines and stress hormones (212, 213). Energy allocation to the immune
system is achieved by activating the energy-demand stress systems (sympatho-
adrenomedullary, axis, SAM and hypothalamic-pituitary-adrenocortical axis, HPA) and stress
systems-induced gluconeogenesis. Hyperinsulinaemia precedes stress-induced and
inflammation-induced insulin resistance (214). Hyperinsulinaemia is seen immediately after
a stress challenge and/or direct immunological activators, such as injuries and pathogen
invasion (215). Low insulin levels increase the susceptibility to develop infections, suggesting
that insulin protects against pathogens (216). Acute inflammation produces down-regulation
of insulin levels through inhibition of pancreatic β-cells (217) and also insulin resistance in
competing organs for glucose uptake (such as liver, muscles and adipose tissue (212). In this
way, glucose becomes available for the energy-demanding immune system. Chronic
inflammation maintains the state of insulin resistance and enhances insulin production,
leading to hyperinsulinemia (218). In the latter situation, glucose remains available for the
immune system and insulin can now be used as reactivator through the mTOR pathway in
immune cells, protecting against possible infections which is, however, deleterious in the
long run (219). Insulin can also upregulate the specific glucose transporters on immune cells,
including GLUT1, GLUT3 and GLUT4, thereby increasing glucose uptake by leukocytes and
lymphocytes (220). Insulin signalling through insulin receptors on immune cells stimulate
the IRS-1/PI3K/AKT pathway that activates mTOR1 and mTOR2 (221). mTOR signalling

39
recruits c-myc, NFkB and HIF1, facilitating further glucose uptake, production of pro-
inflammatory cytokines and maintenance of cytoplasmic aerobic glycolysis, respectively
(222-224).
It seems clear that leptin and insulin pathways are capable of fuelling and reactivating the
immune system, not only during acute infection, but also to maintain long-term activation.
The capacity of redistributing glucose to the immune system and away from peripheral
tissues mediates the immune response and has been crucial to human survival. In other
words: leptin and insulin signalling beneficial to immune system activity are vital for survival,
and are meanwhile also responsible for chronic low-grade inflammation and its associated
diseases.
6.4. The reproductive system at the disposal of the selfish immune system
The combined metabolic shift produced at the disposal of the pro-inflammatory activity of
the immune system is directly responsible for the pro-inflammatory systemic
hypoandrogenic state observed in individuals suffering from low- grade inflammatory
disorders (225). This systemic hypoandrogenic state is not produced because of lower
testosterone production in the sex organs. To the contrary, obese males, characterised by
increased plasma leptin and low-grade inflammation, exhibit a higher testosterone-
dependent risk of prostate cancer (226), while the same holds true for the polycystic ovaria
syndrome in females (227).
The testosterone boost produced by metabolic hormones precedes the increased systemic
and tissue-specific conversion of testosterone into pro-inflammatory oestrogens. This shift
has been observed in different diseases, including obesity and inflammation-related breast
cancer (228).
The systemic shift from testosterone to oestrogens could benefit the anti-pathogenic pro-
inflammatory activity of the immune system. Males with higher testosterone levels are more
susceptible to parasite infection, microbial transmission (229) and have decreased resistance
against tick infection (230). Conversely, low testosterone protects against bacterial infection
in general and specifically against prostate infection (231). It is therefore conceivable, but at
the same time striking, that both males and females exhibit higher aromatase activities during
inflammation as evidenced in patients with rheumatic diseases, characterised by high pro-
inflammatory oestrogens and low testosterone levels (232). The protective effect of oestrogens
against microbial infiltration and infection is supported by epidemiological data showing that
postmenopausal women are disproportionately susceptible to recurrent urinary tract
infections (233). Urinary tract infections (UTI) in elderly patients are more treatment resistant

40
and oestrogen replacement diminishes UTI frequency (234). Chronic inflammation and stress
lead to low testosterone and high oestrogens in men (235) and high oestrogen levels protect
against possible infection.
A possible negative effect of this shift from testosterone to oestrogen is the loss of fertility (41).
Obese men, characterised by high aromatase activity in adipocytes (236) and low-grade
inflammation (237), have lower fertility (238). Individuals engaged in a chronic struggle
against pathogens rather not reproduce, preventing damage to offspring, which is beneficial
to overall reproductive success (125). The septic danger posed by non-sterile wounds is
included in the selective pressure factors shaping human behaviour (phenotype) and genome
(genotype). It has been shown that the shift from testosterone to oestrogen in the skin is
highly protective against pathogenic infection, prolonged infection, wound healing and
overall cutaneous repair (239). It is consequently conceivable that the immune system also
puts the reproductive system at its disposal. Immediate survival overrules reproduction. Once
again, the immune system dominates the whole body, including the timing of reproduction
and, if necessary, protecting genetically-related individuals against possible pathogenic
damage, or damage to the immune system itself (240). Oestrogens activate the immune
system through several mechanisms including stimulation of NFkB, c-myc and mTOR,
facilitating immune cell proliferation and inflammatory activity (241).
6.5. Behaviour at the disposal of the immune system: serotonin-dependent
reactivation of the immune system
The observed changes in behaviour during inflammation suggest that the immune system
actively affects neurophysiological function, putting behaviour at its own disposal. Pro-
inflammatory cytokines such as TNFα, IL-1 beta, and IL-6 produce adaptive behavioural
effects when entering the brain (55). Sickness behaviour includes social withdrawal, increased
sleeping time, fatigue and exercise avoidance. This reduces energy uptake by muscles and
the brain and this is reallocated to the immune response.
Several pathways explaining inflammation-induced sickness behaviour have been proposed
and all of them probably contribute to this state (120). Sickness behaviour not only benefits
the host’s immune system in terms of energy/resource reallocation, but also helps the
immune system to fight pathogens and restore homeostasis when immune activity is no
longer needed (55). Pro-inflammatory cytokines (IL1β, IL6, TNF-α and IFNy) and stress
hormones, produced during inflammation, activate tryptophan 2,3-dioxygenase (TDO) and
indoleamine 2,3-dioxygenase (IDO), affecting serotonin production from its precursor
tryptophan and favouring the production of kynurenine and quinolinic acid (242) The
resulting serotonin depletion is considered to be one of the factors causing sickness

41
behaviour (243), which, when considered from a proximate prospective, could be considered
a maladaptive response. The latter is supported by showing that quinolinic acid, produced by
cells in the central nervous system, is highly neurotoxic and associated with the development
of numerous neurodegenerative conditions, including Parkinson’s and Alzheimer’s diseases
(215).
An evolutionary explanation for the underlying mechanism considers that upregulation of
IDO and TDO during acute inflammation protects the host significantly by depleting
tryptophan and efficiently suppressing the growth of pathogens and malignant cells (120).
Serotonin further inhibits activation of the sympathetic nervous system (244), while SNS is
needed for energy production and its allocation to the brain and the immune system during
inflammation. Inhibition of serotonin production during inflammation will therefore favour
SNS activity and energy production/allocation to the immune system.
Serotonin is present in high concentrations at the sites of inflammation and is used by
activated immune cells as co-stimulator through reuptake via the serotonin transporter
protein (245). This is beneficial, considering the need to mount an optimal immune response
during inflammation, but could be deleterious in the long run and cause several disorders
including autoimmune diseases (245). IDO will not only deplete tryptophan but also serotonin
and both pathways will inhibit the immune system activity when it is no longer needed,
thereby recovering tissue homeostasis and facilitating tissue repair. A feeling of sickness and
even pain are common consequences of this highly effective neuroimmunological reaction
but that is the price to be paid (246).
The total picture of inflammation-caused sickness behaviour should be considered beneficial
to the host. Only when inflammation is supramaximal, such as in sepsis, or when
inflammation lasts too long, sickness behaviour has more of a negative impact because of the
possible damage caused by immune system dependent pathways. These deleterious effects to
the brain show that, if necessary, the immune system will take over and override the interests
of the selfish brain, supporting the ‘selfish immune system’ hypothesis.
6.6. Behaviour and immune system co-evolution: dopamine-dependent
reactivation of the immune system
The use of dopamine as an immunological co-stimulator has been studied extensively and is
of high clinical importance (247). Dopamine recruitment by the immune system has
profound effects on inflammatory behaviour. Humans have engaged in exploring new
environments and this demands several traits, including curiosity (25), a large brain and
immune protection.

42
Dopamine is considered to be the main neurotransmitter responsible for curiosity (248),
novelty seeking (249), motivation and aggressiveness (250). A polymorphism of the dopamine
receptor D4 (DRD4) is associated with novelty seeking, risk-taking and increased exploratory
behaviour (251). Novel environments produce new immunological challenges, including
climate, food availability and pathogens (252). The long allele of the DRD4 receptor is related
to the migratory distance from Africa. Matthews and Butler (251) suggested that this allele
been positively selected, as opposed to genetic drift.
Other behavioural traits, in addition to the association of 7R DRD4 polymorphism with
environmental exploration and novelty seeking, are increased anger and a decreased feeling
of disgust (253). Disgust is amongst the most intensively investigated emotions belonging to
the behavioural immune system (248). Immune defence is usually a reaction following tissue
damage or some pathogenic infection. It is highly costly and intense and long-term activity
could result in secondary lesions and even multiple organ failure. Humans have explored
new environments with constant new immunological challenges throughout evolution. The
development of a pro-reactive behavioural immune system, preventing contact with possible
pathogens could have been beneficial to save energy and guide them to important other
physiological functions, including those of the brain and skeletal muscles (254, 27). Disgust as
a proactive strategy to avoid disease, produces aversion to a wide range of factors. High levels
of disgust, i.e. increased activity of the behavioural immune system, produce neophobia (255),
rejection of other individuals, decrease in mating behaviour (256), food neophobia (257),
prejudicial attitudes to old people (258) and even discrimination (259).
The behavioural immune system (BIS) can be very sensitive and dominate free will. However,
individuals, carrying the longer allele of the DRD4 gene exhibit a higher level of novelty
seeking, less disgust and more spontaneous activity (236). This implies that people with
increased exploratory behaviour through DRD4 polymorphism would be at a higher risk of
pathogenic infection, because of less aversion and disgust. This combination argues against
the current opinion about pathogens dominating selective pressure in human evolution
(260). The only feasible explanation would be that the longer allele of the DRD4 gene should
have some immune function, protecting the ‘seeking’ carrier against pathogens.
The evidence for an immune function of the long allele of the DRD4 comes from different
investigations studying the influence of the expression of dopamine receptors on innate
immune cells and lymphocytes of the adaptive immune system. The various immune cells
express different dopamine receptors (DRD1-DRD5) (165). The net function of dopamine
receptor activation is an increase in the pro-inflammatory activity of the immune system,
with the exception of the wild type DRD4 (the short allele’) (261). Activation of the wild type
DRD4 receptor leads to the production of the immune-suppressing cytokine IL10 (247, 254).

43
The long 7R allele, on the contrary, is associated with diminished cAMP production and
reduced intracellular response (262). Reduced response will lead to lower immune quiescence
(the normal function of wild type DRD4 (247, 263) and increase the pro-inflammatory effects
of dopamine by activating other dopamine receptors (165). It is therefore conceivable that
migration out of Africa selected the longer allele of the D4 dopamine receptor by inducing
novelty seeking, while increasing protective inflammatory activity. Dopamine can stimulate
the production of NFkB and pro-inflammatory cytokines such as TNFα and IL1, although the
opposite, production of anti-inflammatory IL10, is also possible (165). This probably depends
on the individual’s genotype, implying that not every individual will be capable of using the
dopamine mechanism as reactivation strategy.
It seems clear that dopamine (DA) plays an important role in the immune system. Dopamine
is not only produced in neurons, but also in several immune cells, including T lymphocytes
(247). Dopamine seems to be recruited by the immune system to protect the host against
acute infiltration by pathogens. The protective effect of dopamine signalling in the immune
system against new infections is evidenced by the fact that dopamine activates resting T cells,
but inhibits activated T cells (165) even in the absence of other danger signals (261). This is in
line with the effects of other neurotransmitters on the immune system, increasing protection
against new invaders (evolutionary beneficial) but having a negative effect on the
immunological memory (264). The immunological ‘use’ of the whole body to fight infection
makes sense in an evolutionary framework, considering that humans have had to fight
infections as the main cause of death for thousands of generations and, as stated before,
almost all humans died because of infection before the start of the 20th century.

44
7. Summary and conclusion
It can be concluded that the activated immune system puts the whole body at its disposal, by
reversing the functions of metabolic hormones, organs, and even the nervous system to the
energetic and pro-inflammatory benefit of the immune system (Figure 3). This response is
highly protective during acute inflammation/infection and even at the start of a chronic
process. The longer the inflammatory response lasts, the more it contributes to (severe) loss of
lean body mass (265, 266), organ dysfunction (4), brain damage and neurodegenerative
diseases (77). The protective pro-inflammatory activity of the selfish immune system is no
longer beneficial once severe secondary damage to organs has been caused by the immune
system.
Life would not have been possible without an immune system, and the development of
complex organisms needed an even more complex immune system. The human immune
system belongs to the most complex among all living organisms and serves as the blueprint
for the development of antivirus software in computer programming (267). Newer systems
and organs normally dominate older systems as the most basic phylogenetical law in
evolution. However, this sequence may change in the face of severe or long-term danger,
known as evo-devo1 mechanisms (268, 269). Evo-devo1 can reach so far back in time that
inflamed lung and kidney tissue literally resembles a swim bladder (270). Chronic disease is
characterised by chronic inflammation (and vice versa) and gradual loss of functions and
even anatomy. It affects the whole body, including the brain. The evidence brought together
in this review shows that the immune system captures a major part of energy and resources
during acute inflammation, putting the whole body at its disposal. This state relates to
disposal of muscles (muscle wasting), the cardiovascular system (high blood pressure,
atherosclerosis), the gut (digestive problems and food intolerance) and even the brain (loss of
memory and concentration in, for instance, individuals suffering from FMS). Long-term pro-
inflammatory activation of the immune system would not be possible without putting the
whole body at the disposal of the immune system. Because of the immune system’s capability
of recruiting metabolic hormones and neurotransmitters and using them for its own benefit,
it is the most selfish organ in human beings. The body at the disposal of the immune system
protects the host during acute inflammation by mounting an optimal response and during
chronic stress to maintain pro-inflammatory activity and to protect against possible
infectious pathogens. This situation is initially protective, but becomes severely deleterious
when the secondary damage to organs and tissues overrides the benefit of infectious
protection. The environment in which current human beings live constantly challenges the
body with multiple new metabolic signalling factors. The only organ capable of
1 Evolutionary developmental biology, or evo-devo, broadly investigates how body plan diversity and morphological novelties have arisen and persisted in nature (132).

45
communicating with all organs involved in the energetic conflict because of these multiple
metabolic signalling is the immune system. To prevent further conflict, the immune system
takes over, using its robust power to put the whole body at its disposal and showing its selfish
behaviour. This selfish behaviour of the immune system has saved hominins for millions of
years. A slow-changing environment to which the immune system could gradually adapt,
characterised these years. This selfish behaviour of the immune system has to be considered
to be the main cause of the majority, if not all, modern diseases. The reason lies in the
interaction between the evolutionary background of immune function, genetic development
and notably, the current environment as the primary cause. Genes and functions are old; the
environment is brand new and this conflict underlies modern disease. It should, however, be
noted that the immune system is only doing what it is made for: trying to protect us.
Figure 3. The total picture of the body at the disposal of the selfish immune system.
If the immune system succeeds in doing so, the host is protected against inflammation-induced immune suppression,
which would lead to cancer and possibly lethal infections (bottom right), but at the expense of the development of modern
low-grade inflammatory diseases (bottom left). GLUT 1, glucose transporter 1: GLUT 4, glucose transporter 4; IS, immune
system; SAM, sympathetic adrenal medular system; HPA, hypothalamus-pituitary-adrenal axis; GR, glucocorticoid receptor;
BH4, tetrahydrobiopterin; IDO, indoleamine 2,3-deoxigenase: TDO, tryptophan 2,3-deoxigenase; CVD, cardiovascular
diseases; FMS, fibromyalgia syndrome; CFS, chronic fatigue syndrome.

46
References
1. Bosma-den Boer MM, van Wetten ML, Pruimboom L. Chronic inflammatory diseases
are stimulated by current lifestyle: How diet, stress levels and medication prevent our
body from recovering. Nutr Metab (Lond) 2012;9(1):32.
2. Straub RH. How energy shifts lead to systemic illness. 2011. The Rheumatologist, July
2011, www.the-rheumatologist.org
3. Straub RH. Concepts of evolutionary medicine and energy regulation contribute to the
etiology of systemic chronic inflammatory diseases. Brain Behav Immun 2011,
Jan;25(1):1-5.
4. Ghigliotti, G., Barisione, C., Garibaldi, S., Fabbi, P., et al. Adipose tissue immune
response: novel triggers and consequences for chronic inflammatory conditions.
Inflammation 37, 1337-1353 (2014).
5. Bajgar, A., Kucerova, K., Jonatova, L., Tomcala, A., et al. Extracellular adenosine mediates
a systemic metabolic switch during immune response. PLoS Biol 13, e1002135 (2015).
6. Straub RH, Cutolo M, Buttgereit F, Pongratz G. Energy regulation and neuroendocrine-
immune control in chronic inflammatory diseases. J Intern Med 2010, Jun;267(6):543-
60.
7. Cavicchia PP, Steck SE, Hurley TG, Hussey JR, Ma Y, Ockene IS, Hébert JR. A new
dietary inflammatory index predicts interval changes in serum high-sensitivity c-
reactive protein. J Nutr 2009, Dec;139(12):2365-72.
8. Laires MJ, Monteiro CP, Bicho M. Role of cellular magnesium in health and human
disease. Front Biosci 2004;9:262-76.
9. Tam, M., Gomez, S., Gonzalez-Gross, M. & Marcos, A. Possible roles of magnesium on
the immune system. European journal of clinical nutrition 57, 1193-1197 (2003).
10. Montserrat-de la Paz, S., Naranjo, M. C., Lopez, S., Abia, R., et al. Niacin and olive oil
promote skewing to the M2 phenotype in bone marrow-derived macrophages of mice
with metabolic syndrome. Food Funct (2016).
11. Chan, D. S., Bandera, E. V., Greenwood, D. C. & Norat, T. Circulating C-Reactive Protein
and Breast Cancer Risk-Systematic Literature Review and Meta-analysis of Prospective
Cohort Studies. Cancer Epidemiol Biomarkers Prev 24, 1439-1449 (2015).
12. Speakman JR, Mitchell SE. Caloric restriction. Mol Aspects Med 2011, Jun;32(3):159-221.
13. Kirkwood TB, Shanley DP. Food restriction, evolution and ageing. Mech Ageing Dev
2005, Sep;126(9):1011-6.
14. Finch CE. Evolution of the human lifespan, past, present, and future: Phases in the
evolution of human life expectancy in relation to the inflammatory load. Proceedings of
the American Philosophical Society 2012;156(1):9.

47
15. McCann JC, Ames BN. Adaptive dysfunction of selenoproteins from the perspective of
the triage theory: Why modest selenium deficiency may increase risk of diseases of
aging. FASEB J 2011, Jun;25(6):1793-814.
16. Nalam RL, Pletcher SD, Matzuk MM. Appetite for reproduction: Dietary restriction, aging
and the mammalian gonad. J Biol 2008;7(7):23.
17. Straub, R. H. Systemic disease sequelae in chronic inflammatory diseases and chronic
psychological stress: comparison and pathophysiological model. Ann N Y Acad Sci 1318,
7-17 (2014).
18. Straub, R. H. Insulin resistance, selfish brain, and selfish immune system: an
evolutionarily positively selected program used in chronic inflammatory diseases.
Arthritis Res Ther 16 Suppl 2, S4 (2014).
19. Hancock AM, Witonsky DB, Alkorta-Aranburu G, Beall CM, Gebremedhin A, Sukernik R,
et al. Adaptations to climate-mediated selective pressures in humans. PLoS Genet 2011,
Apr;7(4):e1001375.
20. Novembre J, Han E. Human population structure and the adaptive response to
pathogen-induced selection pressures. Philos Trans R Soc Lond B Biol Sci 2012, Mar
19;367(1590):878-86.
21. Fumagalli M, Sironi M, Pozzoli U, Ferrer-Admetlla A, Ferrer-Admettla A, Pattini L,
Nielsen R. Signatures of environmental genetic adaptation pinpoint pathogens as the
main selective pressure through human evolution. PLoS Genet 2011,
Nov;7(11):e1002355.
22. Kwiatowski DP. How Malaria Has Affected the Human Genome and What Human
Genetics Can Teach Us about Malaria. Am. J. Hum. Genet. , 2005, 77:171–192
23. Oviasogie FE, Ajuzie CU, Ighodaro UG. Bacterial analysis of soil from waste dumpsite.
Arch Appl Sci Res 2010;2:161–7.
24. Belloni V, Faivre B, xGuerreiro R, Arnoux E, Bellenger J, Sorci G. Suppressing an anti-
inflammatory cytokine reveals a strong age-dependent survival cost in mice. PLoS ONE
2010;5(9):e12940.
25. Garland T, Schutz H, Chappell MA, Keeney BK, Meek TH, Copes LE, et al. The biological
control of voluntary exercise, spontaneous physical activity and daily energy
expenditure in relation to obesity: Human and rodent perspectives. J Exp Biol 2011, Jan
15;214(Pt 2):206-29.
26. Stranahan AM, Mattson MP. Bidirectional metabolic regulation of neurocognitive
function. Neurobiol Learn Mem 2011, Nov;96(4):507-16.
27. Schaller M, Neuberg SL. Danger, disease, and the nature of prejudice(s). In: Danger,
Disease, and the Nature of Prejudice(s); 2012dv. p. 1-54.
28. Varki A. Nothing in medicine makes sense, except in the light of evolution. J Mol Med
(Berl) 2012, May;90(5):481-94.

48
29. Wang M, Jiang YY, Kim KM, Qu G, Ji HF, Mittenthal JE, et al. A universal molecular clock
of protein folds and its power in tracing the early history of aerobic metabolism and
planet oxygenation. Mol Biol Evol 2011, Jan;28(1):567-82.
30. O'Bleness M, Searles VB, Varki A, Gagneux P, Sikela JM. Evolution of genetic and
genomic features unique to the human lineage. Nat Rev Genet 2012, Dec;13(12):853-66.
31. Pruimboom L, Raison CL, Muskiet FA. Physical activity protects the human brain
against metabolic stress induced by a postprandial and chronic inflammation. Behav
Neurol 2015;2015:569869.
32. Filep JG. Resolution pathways in inflammation: The devil in the adipose tissues and in
the details. Focus on "diversity of lipid mediators in human adipose tissue depots". Am J
Physiol Cell Physiol 2013, Jun 15;304(12):C1127-8.
33. Clària J, Nguyen BT, Madenci AL, Ozaki CK, Serhan CN. Diversity of lipid mediators in
human adipose tissue depots. Am J Physiol Cell Physiol 2013, Jun 15;304(12):C1141-9.
34. Pond CM. Adipose tissue and the immune system. Prostaglandins Leukot Essent Fatty
Acids 2005, Jul;73(1):17-30.
35. Zhang MJ, Spite M. Resolvins: Anti-inflammatory and proresolving mediators derived
from omega-3 polyunsaturated fatty acids. Annu Rev Nutr 2012, Aug 21;32:203-27.
36. Pixley FJ. Macrophage migration and its regulation by CSF-1. Int J Cell Biol
2012;2012:501962.
37. Bannenberg GL, Chiang N, Ariel A, Arita M, Tjonahen E, Gotlinger KH, et al. Molecular
circuits of resolution: Formation and actions of resolvins and protectins. The Journal of
Immunology 2005;174(7):4345-55.
38. Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and competition in the evolution of
atp-producing pathways. Science 2001, Apr 20;292(5516):504-7.244.
39. Seyfried TN, Shelton LM. Cancer as a metabolic disease. Nutr Metab (Lond) 2010;7:7.
40. Lunt SY, Vander Heiden MG. Aerobic Glycolysis: Meeting the Metabolic Requirements
of Cell Proliferation. Annu. Rev. Cell Dev. Biol. 2011. 27:441–64
41. Straub RH, Schradin C. Chronic inflammatory systemic diseases. Evolution, Medicine,
and Public Health 2016, pp. 37–51
42. Hafner S, Radermacher P, Frick M, Dietl P and Calzia E. Hyperglycemia, oxidative stress,
and the diaphragm: a link between chronic co-morbidity and acute stress?. Critical Care
2014, 18:149
43. Chrousos GP. Stress and disorders of the stress system. Nat Rev Endocrinol 2009,
Jul;5(7):374-81.
44. Zogopoulos P, Vasileiou I, Patsouris E, Theocharis SE. The role of endocannabinoids in
pain modulation. Fundam Clin Pharmacol 2013, Feb;27(1):64-80.
45. Mavroudis PD, Scheff JD, Calvano SE, Androulakis IP. Systems biology of circadian-
immune interactions. J Innate Immun 2013;5(2):153-62.

49
46. Dhabhar FS. A hassle a day may keep the doctor away: Stress and the augmentation of
immune function. Integr Comp Biol 2002;42(3):556-64.
47. Pongratz G, Straub RH. Role of peripheral nerve fibres in acute and chronic
inflammation in arthritis. Nat Rev Rheumatol 2013, Feb;9(2):117-26. (
48. Russell CD, Schwarze J. The role of pro-resolution lipid mediators in infectious disease.
Immunology 2014, Feb;141(2):166-73.
49. Calay ES, Hotamisligil GS. Turning off the inflammatory, but not the metabolic, flames.
Nat Med 2013;19(3):265-7.
50. Kraut JA, Madias NE. Treatment of acute metabolic acidosis: A pathophysiologic
approach. Nat Rev Nephrol 2012, Oct;8(10):589-601.
51. Kellum JA, Song M, Li J. Science review: Extracellular acidosis and the immune
response: Clinical and physiologic implications. Critical Care 2004;8(5):331.
52. Delmastro-Greenwood MM, Piganelli JD. Changing the energy of an immune response.
Am J Clin Exp Immunol 2013;2(1):30-54.
53. Delmastro-Greenwood MM, Votyakova T, Goetzman E, Marre ML, Previte DM,
Tovmasyan A, et al. Mn porphyrin regulation of aerobic glycolysis: Implications on the
activation of diabetogenic immune cells. Antioxid Redox Signal 2013, Dec 1;19(16):1902-
15.
54. Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat
Rev Immunol 2009, Sep;9(9):609-17.
55. Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to
sickness and depression: When the immune system subjugates the brain. Nat Rev
Neurosci 2008, Jan;9(1):46-56.
56. Fitzpatrick M, Young SP. Metabolomics--a novel window into inflammatory disease.
Swiss Med Wkly 2013;143:w13743.
57. Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol
2011;29:415-45.
58. Ruiz-Núñez B, Kuipers RS, Luxwolda MF, De Graaf DJ, Breeuwsma BB, Dijck-Brouwer
DA, Muskiet FA. Saturated fatty acid (SFA) status and SFA intake exhibit different
relations with serum total cholesterol and lipoprotein cholesterol: A mechanistic
explanation centered around lifestyle-induced low-grade inflammation. J Nutr
Biochem 2014, Mar;25(3):304-12.
59. Ruiz-Nunez B, Pruimboom L, Dijck-Brouwer J. Lifestyle and nutritional imbalances
associated with western diseases: Causes and 3 consequences of chronic systemic low-
grade inflammation in an evolutionary context. Journal of Nutritional Biochemistry
2013.
60. Kitano H. Towards a theory of biological robustness. Mol Syst Biol 2007;3:137.
61. Pruimboom L. Physical inactivity is a disease synonymous for a non-permissive brain
disorder. Med Hypotheses 2011, Nov;77(5):708-13.

50
62. Straub RH, Besedovsky HO. Integrated evolutionary, immunological, and
neuroendocrine framework for the pathogenesis of chronic disabling inflammatory
diseases. FASEB J 2003, Dec;17(15):2176-83.
63. Ames BN. Low micronutrient intake may accelerate the degenerative diseases of aging
through allocation of scarce micronutrients by triage. Proceedings of the National
Academy of Sciences 2006;103(47):17589-94.
64. Kirkwood TB. Understanding ageing from an evolutionary perspective. J Intern Med
2008, Feb;263(2):117-27.
65. Cunnane SC, Crawford MA. Survival of the fattest: Fat babies were the key to evolution
of the large human brain. Comparative Biochemistry and Physiology-Part A: Molecular
& Integrative Physiology 2003;136(1):17-26.
66. Maes M, Twisk FN, Kubera M, Ringel K, Leunis JC, Geffard M. Increased iga responses to
the LPS of commensal bacteria is associated with inflammation and activation of cell-
mediated immunity in chronic fatigue syndrome. J Affect Disord 2012, Feb;136(3):909-
17.
67. Maes M, Ringel K, Kubera M, Berk M, Rybakowski J. Increased autoimmune activity
against 5-HT: A key component of depression that is associated with inflammation and
activation of cell-mediated immunity, and with severity and staging of depression. J
Affect Disord 2012, Feb;136(3):386-92.189.
68. Mosconi L, Tsui WH, De Santi S, Li J, Rusinek H, Convit A, et al. Reduced hippocampal
metabolism in MCI and AD: Automated FDG-PET image analysis. Neurology 2005, Jun
14;64(11):1860-7.
69. Gutierrez T, Hornigold R, Pearce A. The systemic response to surgery. Surgery (Oxford)
2011;29(2):93-6.
70. Castellheim A, Brekke OL, Espevik T, Harboe M, Mollnes TE. Innate immune responses
to danger signals in systemic inflammatory response syndrome and sepsis. Scand J
Immunol 2009, Jun;69(6):479-91.
71. Reser JE. Alzheimer's disease and natural cognitive aging may represent adaptive
metabolism reduction programs. Behav Brain Funct 2009;5:13.
72. Rogers J. The inflammatory response in alzheimer's disease. J Periodontol 2008,
Aug;79(8 Suppl):1535-43.
73. Shichita T, Ago T, Kamouchi M, Kitazono T, Yoshimura A, Ooboshi H. Novel therapeutic
strategies targeting innate immune responses and early inflammation after stroke. J
Neurochem 2012, Nov;123 Suppl 2:29-38.
74. Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T. Inflammatory response in
acute traumatic brain injury: A double-edged sword. Current Opinion in Critical Care
2002;8(2):101-5.
75. Murray KN, Buggey HF, Denes A, Allan SM. Systemic immune activation shapes stroke
outcome. Mol Cell Neurosci 2013, Mar;53:14-25.

51
76. Clayton TC, Thompson M, Meade TW. Recent respiratory infection and risk of
cardiovascular disease: Case-control study through a general practice database. Eur
Heart J 2008, Jan;29(1):96-103.
77. Carod-Artal FJ. Strokes caused by infection in the tropics. Rev Neurol 2007;44(12):755.
78. Hills NK, Johnston SC, Sidney S, Zielinski BA, Fullerton HJ. Recent trauma and acute
infection as risk factors for childhood arterial ischemic stroke. Ann Neurol 2012,
Dec;72(6):850-8.
79. Roquilly A, Lejus C, Asehnoune K. Brain injury, immunity and infections. Ann Fr Anesth
Reanim 2012, Jun;31(6):e97-100.
80. Spies CM, Straub RH, Buttgereit F. Energy metabolism and rheumatic diseases: From
cell to organism. Arthritis Res Ther 2012;14(3):216.
81. Aiello LC, Wheeler P. The expensive-tissue hypthesis: The brain and the digestive
system in human and primate evolution. Curr Anthropol 1995, Apr;36(2):199-221.
82. Milton K. A hypothesis to explain the role of meat-eating in human evolution.
Evolutionary Anthropology Issues News and Reviews 1999;8.1(9)(11-21).
83. Carmody RN, Wrangham RW. The energetic significance of cooking. J Hum Evol 2009,
Oct;57(4):379-91.
84. Navarrete A, van Schaik CP, Isler K. Energetics and the evolution of human brain size.
Nature 2011, Dec 1;480(7375):91-3.
85. Fedrigo O, Pfefferle AD, Babbitt CC, Haygood R, Wall CE, Wray GA. A potential role for
glucose transporters in the evolution of human brain size. Brain Behav Evol
2011;78(4):315-26.
86. Maciver NJ, Jacobs SR, Wieman HL, Wofford JA, Coloff JL, Rathmell JC. Glucose
metabolism in lymphocytes is a regulated process with significant effects on immune
cell function and survival. J Leukoc Biol 2008, Oct;84(4):949-57.
87. Herman MA, Kahn BB. Glucose transport and sensing in the maintenance of glucose
homeostasis and metabolic harmony. J Clin Invest 2006, Jul;116(7):1767-75.127.
88. Huang S, Czech MP. The GLUT4 glucose transporter. Cell Metab 2007, Apr;5(4):237-52.
89. Uldry M, Thorens B. The SLC2 family of facilitated hexose and polyol transporters.
Pflugers Arch 2004, Feb;447(5):480-9.
90. Woods JA, Ceddia MA, Zack MD, Lowder TW, Lu Q. Exercise training increases the
näive to memory T cell ratio in old mice. Brain Behav Immun 2003;17(5):384-92.
91. Wolowczuk I, Verwaerde C, Viltart O, Delanoye A, Delacre M, Pot B, Grangette C.
Feeding our immune system: Impact on metabolism. Clin Dev Immunol
2008;2008:639803.
92. Pacheco-López G, Bermúdez-Rattoni F. Brain-immune interactions and the neural
basis of disease-avoidant ingestive behaviour. Philos Trans R Soc Lond B Biol Sci 2011,
Dec 12;366(1583):3389-405.

52
93. Abdul Muneer PM, Alikunju S, Szlachetka AM, Murrin LC, Haorah J. Impairment of brain
endothelial glucose transporter by methamphetamine causes blood-brain barrier
dysfunction. Mol Neurodegener 2011;6:23.
94. Deng Y, Li B, Liu Y, Iqbal K, Grundke-Iqbal I, Gong CX. Dysregulation of insulin
signaling, glucose transporters, o-glcnacylation, and phosphorylation of tau and
neurofilaments in the brain: Implication for alzheimer's disease. Am J Pathol 2009,
Nov;175(5):2089-98.
95. Peters A. The energy request of inflammation. Endocrinology 2006, Oct;147(10):4550-2.
96. Plomgaard P, Bouzakri K, Krogh-Madsen R, Mittendorfer B, Zierath JR, Pedersen BK.
Tumor necrosis factor-α induces skeletal muscle insulin resistance in healthy human
subjects via inhibition of akt substrate 160 phosphorylation. Diabetes 2005;54(10):2939-
45.
97. Soto PC, Stein LL, Hurtado-Ziola N, Hedrick SM, Varki A. Relative over-reactivity of
human versus chimpanzee lymphocytes: Implications for the human diseases
associated with immune activation. J Immunol 2010, Apr 15;184(8):4185-95.
98. O'Bleness M, Searles VB, Varki A, Gagneux P, Sikela JM. Evolution of genetic and
genomic features unique to the human lineage. Nat Rev Genet 2012, Dec;13(12):853-66.
99. Nguyen DH, Hurtado-Ziola N, Gagneux P, Varki A. Loss of siglec expression on T
lymphocytes during human evolution. Proceedings of the National Academy of
Sciences 2006;103(20):7765-70.
100. Finch CE. Evolution of the human lifespan, past, present, and future: Phases in the
evolution of human life expectancy in relation to the inflammatory load. Proceedings of
the American Philosophical Society 2012;156(1):9.
101. Wang X, Mitra N, Secundino I, Banda K, Cruz P, Padler-Karavani V, et al. Specific
inactivation of two immunomodulatory SIGLEC genes during human evolution. Proc
Natl Acad Sci U S A 2012, Jun 19;109(25):9935-40.
102. Ricart W, Fernández-Real JM. Insulin resistance as a mechanism of adaptation during
human evolution. Endocrinol Nutr 2010, Oct;57(8):381-90.
103. Peters A, Schweiger U, Pellerin L, Hubold C, Oltmanns KM, Conrad M, et al. The selfish
brain: Competition for energy resources. Neurosci Biobehav Rev 2004, Apr;28(2):143-
80.
104. Madsen PL, Hasselbalch SG, Hagemann LP, Olsen KS, Bülow J, Holm S, et al. Persistent
resetting of the cerebral oxygen/glucose uptake ratio by brain activation: Evidence
obtained with the kety-schmidt technique. Journal of Cerebral Blood Flow &
Metabolism 1995;15(3):485-91.
105. Hitze B, Hubold C, van Dyken R, Schlichting K, Lehnert H, Entringer S, Peters A. How
the selfish brain organizes its supply and demand. Front Neuroenergetics 2010;2:7.
106. Kubera B, Hubold C, Zug S, Wischnath H, Wilhelm I, Hallschmid M, et al. The brain's
supply and demand in obesity. Front Neuroenergetics 2012, Jan 10;4:4.

53
107. Patchev VK, Patchev AV. Experimental models of stress. Dialogues in Clinical
Neuroscience 2006;8(4):417.
108. Wells JC. The evolution of human adiposity and obesity: Where did it all go wrong? Dis
Model Mech 2012, Sep;5(5):595-607.
109. Hale MW, Raison CL, Lowry CA. Integrative physiology of depression and
antidepressant drug action: Implications for serotonergic mechanisms of action and
novel therapeutic strategies for treatment of depression. Pharmacol Ther 2013,
Jan;137(1):108-18.
110. Fidalgo S, Ivanov DK, Wood SH. Serotonin: From top to bottom. Biogerontology 2012,
Oct 26.
111. Idova GV, Alperina EL, Cheido MA. Contribution of brain dopamine, serotonin and
opioid receptors in the mechanisms of neuroimmunomodulation: Evidence from
pharmacological analysis. Int Immunopharmacol 2012, Apr;12(4):618-25.
112. Sperner-Unterweger B, Kohl C, Fuchs D. Immune changes and neurotransmitters:
Possible interactions in depression? Prog Neuropsychopharmacol Biol Psychiatry 2012,
Oct 17.
113. Muehlenbein MP, Hirschtick JL, Bonner JZ, Swartz AM. Toward quantifying the usage
costs of human immunity: Altered metabolic rates and hormone levels during acute
immune activation in men. Am J Hum Biol 2010;22(4):546-56.
114. Atiyeh BS, Gunn SW, Dibo SA. Metabolic implications of severe burn injuries and their
management: A systematic review of the literature. World J Surg 2008, Aug;32(8):1857-
69.
115. Wetzel RC, Burns RC. Multiple trauma in children: Critical care overview. Crit Care Med
2002;30(11):S468-77.
116. Briegel J, Jochum M, Gippner-Steppert C, Thiel M. Immunomodulation in septic shock:
Hydrocortisone differentially regulates cytokine responses. Journal of the American
Society of Nephrology 2001;12(suppl 1):S70-4.
117. Roenneberg T, Allebrandt KV, Merrow M, Vetter C. Social jetlag and obesity. Curr Biol
2012, May 22;22(10):939-43.
118. Byrd-Craven J, Auer BJ, Granger DA, Massey AR. The father-daughter dance: The
relationship between father-daughter relationship quality and daughters' stress
response. J Fam Psychol 2012, Feb;26(1):87-94.
119. Dixit VD, Yang H, Sayeed KS, Stote KS, Rumpler WV, Baer DJ, et al. Controlled meal
frequency without caloric restriction alters peripheral blood mononuclear cell cytokine
production. J Inflamm (Lond) 2011;8:6.
120. Miller AH, Maletic V, Raison CL. Inflammation and its discontents: The role of cytokines
in the pathophysiology of major depression. Biol Psychiatry 2009, May 1;65(9):732-41.

54
121. Way BM, Taylor SE, Eisenberger NI. Variation in the µ-opioid receptor gene (OPRM1) is
associated with dispositional and neural sensitivity to social rejection. Proceedings of
the National Academy of Sciences 2009;106(35):15079-84.
122. Taga, M., Minett, T., Classey, J., Matthews, F. E., et al. Metaflammasome components in
the human brain: a role in dementia with alzheimer's pathology? Brain Pathol (2016).
123. Vazquez-Roque, M. I., Camilleri, M., Smyrk, T., Murray, J. A., et al. A controlled trial of
gluten-free diet in patients with irritable bowel syndrome-diarrhea: effects on bowel
frequency and intestinal function. Gastroenterology 144, 903-911.e3 (2013).
124. Littlejohn, G. Neurogenic neuroinflammation in fibromyalgia and complex regional
pain syndrome. Nat Rev Rheumatol 11, 639-648 (2015).
125. Raison CL, Miller AH. The evolutionary significance of depression in pathogen host
defense (PATHOS-D). Mol Psychiatry 2013, Jan;18(1):15-37
126. Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of
alzheimer disease genetic association studies: The alzgene database. Nat Genet 2007,
Jan;39(1):17-23.
127. Leduc V, Domenger D, De Beaumont L, Lalonde D, Bélanger-Jasmin S, Poirier J.
Function and comorbidities of apolipoprotein e in alzheimer's disease. Int J Alzheimers
Dis 2011;2011:974361.
128. Vitek MP, Brown CM, Colton CA. APOE genotype-specific differences in the innate
immune response. Neurobiol Aging 2009, Sep;30(9):1350-60.
129. Fullerton SM, Clark AG, Weiss KM, Nickerson DA, Taylor SL, Stengård JH, et al.
Apolipoprotein E variation at the sequence haplotype level: Implications for the origin
and maintenance of a major human polymorphism. The American Journal of Human
Genetics 2000;67(4):881-900.
130. Eisenberg DT, Kuzawa CW, Hayes MG. Worldwide allele frequencies of the human
apolipoprotein E gene: Climate, local adaptations, and evolutionary history. Am J Phys
Anthropol 2010, Sep;143(1):100-11.
131. Zekraoui L, Lagarde JP, Raisonnier A, Gerard N, Aouizerate A, Lucotte G. High
frequency of the apolipoprotein E* 4 allele in african pygmies and most of the african
populations in sub-saharan africa. Human Biology 1997:575-81.
132. Sandholzer C, Delport R, Vermaak H, Utermann G. High frequency of the apo epsilon 4
allele in khoi san from south africa. Hum Genet 1995, Jan;95(1):46-8.
133. Hendrie C. Incidence of dementia and alzheimer disease in 2 communities: Yoruba
residing in ibadan, nigeria, and african americans residing in indianapolis, indiana.
JAMA: The Journal of the American Medical Association 2001, Feb 14;285(6):739-47.
134. Light KC, White AT, Tadler S, Iacob E, Light AR. Genetics and gene expression involving
stress and distress pathways in fibromyalgia with and without comorbid chronic fatigue
syndrome. Pain Res Treat 2012;2012:427869.

55
135. Low LA, Schweinhardt P. Early life adversity as a risk factor for fibromyalgia in later life.
Pain Res Treat 2012;2012:140832.
136. Offenbaecher M, Bondy B, De Jonge S, Glatzeder K, Krueger M, Shoeps P, Ackenheil M.
Possible association of fibromyalgia with a polymorphism in the serotonin transporter
gene regulatory region. Arthritis & Rheumatism 1999;42(11):2482-8.
137. Fredericks CA, Drabant EM, Edge MD, Tillie JM, Hallmayer J, Ramel W, et al. Healthy
young women with serotonin transporter SS polymorphism show a pro-inflammatory
bias under resting and stress conditions. Brain Behav Immun 2010, Mar;24(3):350-7.
138. Qiao, S. W., Iversen, R., Ráki, M. & Sollid, L. M. The adaptive immune response in celiac
disease. Semin Immunopathol 34, 523-540 (2012).
139. Sironi, M. & Clerici, M. The hygiene hypothesis: an evolutionary perspective. Microbes
Infect 12, 421-427 (2010).
140. Eppig C, Fincher CL, Thornhill R. Parasite prevalence and the worldwide distribution of
cognitive ability. Proc Biol Sci 2010, Dec 22;277(1701):3801-8.
141. Martin LB, Han P, Kwong J, Hau M. Cutaneous immune activity varies with
physiological state in female house sparrows (passer domesticus). Physiological and
Biochemical Zoology 2006;79(4):775-83.
142. Fredman G, Li Y, Dalli J, Chiang N, Serhan CN. Self-limited versus delayed resolution of
acute inflammation: Temporal regulation of pro-resolving mediators and microrna. Sci
Rep 2012;2:639.
143. Spite M, Serhan CN. Novel lipid mediators promote resolution of acute inflammation:
Impact of aspirin and statins. Circ Res 2010, Nov 12;107(10):1170-84.
144. Nathan C, Ding A. Nonresolving inflammation. Cell 2010, Mar 19;140(6):871-82.
145. Hoetzenecker W, Echtenacher B, Guenova E, Hoetzenecker K, Woelbing F, Brück J, et al.
ROS-induced ATF3 causes susceptibility to secondary infections during sepsis-
associated immunosuppression. Nat Med 2012, Jan;18(1):128-34.
146. Sevko A, Sade-Feldman M, Kanterman J, Michels T, Falk CS, Umansky L, et al.
Cyclophosphamide promotes chronic inflammation-dependent immunosuppression
and prevents antitumor response in melanoma. J Invest Dermatol 2012, Dec 6.
147. Dandapani M, Hardie DG. AMPK: Opposing the metabolic changes in both tumour cells
and inflammatory cells? Biochem Soc Trans 2013, Apr;41(2):687-93.
148. Harvey CB, Williams GR. Mechanism of thyroid hormone action. Thyroid
2002;12(6):441-6.
149. Suhane S, Ramanujan VK. Thyroid hormone differentially modulates warburg
phenotype in breast cancer cells. Biochem Biophys Res Commun 2011, Oct 14;414(1):73-
8.
150. Hodkinson CF, Simpson EE, Beattie JH, O'Connor JM, Campbell DJ, Strain JJ, Wallace
JM. Preliminary evidence of immune function modulation by thyroid hormones in
healthy men and women aged 55-70 years. J Endocrinol 2009, Jul;202(1):55-63.

56
151. Wajner SM, Maia AL. New insights toward the acute non-thyroidal illness syndrome.
Front Endocrinol (Lausanne) 2012;3:8.
152. Pappa TA, Vagenakis AG, Alevizaki M. The nonthyroidal illness syndrome in the non-
critically ill patient. Eur J Clin Invest 2011, Feb;41(2):212-20.
153. Iglesias P, Díez JJ. Thyroid dysfunction and kidney disease. Eur J Endocrinol 2009,
Apr;160(4):503-15.
154. Rodriguez-Perez A, Palos-Paz F, Kaptein E, Visser TJ, Dominguez-Gerpe L, Alvarez-
Escudero J, Lado-Abeal J. Identification of molecular mechanisms related to
nonthyroidal illness syndrome in skeletal muscle and adipose tissue from patients with
septic shock. Clin Endocrinol (Oxf) 2008, May;68(5):821-7.
155. Galli E, Marchini M, Saba A, Berti S, Tonacchera M, Vitti P, et al. Detection of 3-
iodothyronamine in human patients: A preliminary study. J Clin Endocrinol Metab
2012, Jan;97(1):E69-74.
156. Schaefer JS, Klein JR. A novel thyroid stimulating hormone beta-subunit isoform in
human pituitary, peripheral blood leukocytes, and thyroid. Gen Comp Endocrinol 2009,
Jul;162(3):241-4.
157. Schaefer JS, Klein JR. Immunological regulation of metabolism--a novel quintessential
role for the immune system in health and disease. FASEB J 2011, Jan;25(1):29-34.
158. Klein JR. The immune system as a regulator of thyroid hormone activity. Experimental
Biology and Medicine 2006;231(3):229-36.
159. Kishida Y, Imaizumi N, Tanimura H, Haruna Y, Kashiwamura S, Kashiwagi T.
Restoration of innate and adaptive immune responses by HCV viral inhibition with an
induction approach using natural interferon-beta in chronic hepatitis C. Clin Dev
Immunol 2012;2012:582716.
160. Grossmann M, Szkudlinski MW, Wong R, Dias JA, Ji TH, Weintraub BD. Substitution of
the seat-belt region of the thyroid-stimulating hormone (TSH) beta-subunit with the
corresponding regions of choriogonadotropin or follitropin confers luteotropic but not
follitropic activity to chimeric TSH. J Biol Chem 1997, Jun 13;272(24):15532-40.
161. Boelen A, Kwakkel J, Fliers E. Beyond low plasma T3: Local thyroid hormone
metabolism during inflammation and infection. Endocr Rev 2011, Oct;32(5):670-93.
162. Siegel E, Sachs BA. In vitro leukocyte uptake of 131I labeled iodide, thyroxine and
triiodothyronine, and its relation to thyroid function. Journal of Clinical Endocrinology
& Metabolism 1964;24(4):313-8.
163. Boelen A, Kwakkel J, Wieland CW, St Germain DL, Fliers E, Hernandez A. Impaired
bacterial clearance in type 3 deiodinase-deficient mice infected with streptococcus
pneumoniae. Endocrinology 2009, Apr;150(4):1984-90.
164. Kwakkel J, Fliers E, Boelen A. Illness-induced changes in thyroid hormone metabolism:
Focus on the tissue level. Hepatitis C: Many Small Steps 209 2011:224.

57
165. Toth BE, Vecsernyes M, Zelles T, Kadar K, Nagy GM. Role of peripheral and brain-
derived dopamine (DA) in immune regulation. Advances in Neuroimmune Biology
2012(3):111-55.
166. Gaskill PJ, Carvallo L, Eugenin EA, Berman JW. Characterization and function of the
human macrophage dopaminergic system: Implications for CNS disease and drug
abuse. J Neuroinflammation 2012;9:203.
167. Levite M, Chowers Y. Nerve-driven immunity: Neuropeptides regulate cytokine
secretion of T cells and intestinal epithelial cells in a direct, powerful and contextual
manner. Annals of Oncology 2001;12(suppl 2):S19-25.
168. Carlton ED, Demas GE, French SS. Leptin, a neuroendocrine mediator of immune
responses, inflammation, and sickness behaviors. Horm Behav 2012, Aug;62(3):272-9.
169. Ottaviani E, Malagoli D, Capri M, Franceschi C. Ecoimmunology: Is there any room for
the neuroendocrine system? Bioessays 2008, Sep;30(9):868-74.
170. Van Bogaert T, De Bosscher K, Libert C. Crosstalk between TNF and glucocorticoid
receptor signaling pathways. Cytokine Growth Factor Rev 2010, Aug;21(4):275-86.
171. Chinenov Y, Rogatsky I. Glucocorticoids and the innate immune system: Crosstalk with
the toll-like receptor signaling network. Mol Cell Endocrinol 2007, Sep 15;275(1-2):30-
42.
172. Dhabhar FS. Effects of stress on immune function: The good, the bad, and the beautiful.
Immunol Res 2014, May;58(2-3):193-210.
173. Dhabhar FS, Malarkey WB, Neri E, McEwen BS. Stress-induced redistribution of immune
cells-from barracks to boulevards to battlefields: A tale of three hormones--curt richter
award winner. Psychoneuroendocrinology 2012, Sep;37(9):1345-68.
174. Sifakis EG, Lambrou GI, Prentza A, Vlahopoulos S, Koutsouris D, Tzortzatou-
Stathopoulou F, Chatziioannou AA. Elucidating the identity of resistance mechanisms
to prednisolone exposure in acute lymphoblastic leukemia cells through transcriptomic
analysis: A computational approach. J Clin Bioinforma 2011;1:36.
175. Stanya KJ, Jacobi D, Liu S, Bhargava P, Dai L, Gangl MR, et al. Direct control of hepatic
glucose production by interleukin-13 in mice. J Clin Invest 2013, Jan 2;123(1):261-71.
176. Machado MV, Yang Y, Diehl AM. The benefits of restraint: A pivotal role for IL-13 in
hepatic glucose homeostasis. J Clin Invest 2013, Jan 2;123(1):115-7.
177. Mitrakou A. Kidney: Its impact on glucose homeostasis and hormonal regulation.
Diabetes Res Clin Pract 2011;93:S66-72.
178. Gerich JE, Meyer C, Woerle HJ, Stumvoll M. Renal gluconeogenesis its importance in
human glucose homeostasis. Diabetes Care 2001;24(2):382-91.
179. Norbiato G, Bevilacqua M, Vago T, Taddei A, Clerici M. Glucocorticoids and the immune
function in the human immunodeficiency virus infection: A study in hypercortisolemic
and cortisol-resistant patients. Journal of Clinical Endocrinology & Metabolism
1997;82(10):3260-3.

58
180. Norbiato G. Endocrine, metabolic, and immunologic components of HIV infection. Ann
N Y Acad Sci 2012, Jul;1262:51-5.
181. Yang N, Ray DW, Matthews LC. Current concepts in glucocorticoid resistance. Steroids
2012, Sep;77(11):1041-9.
182. Teelucksingh S, Jaimungal S, Pinto Pereira L, Seemungal T, Nayak S. Does insulin
resistance co-exist with glucocorticoid resistance in the metabolic syndrome? Studies
comparing skin sensitivity to glucocorticoids in individuals with and without
acanthosis nigricans. Cardiovasc Diabetol 2012;11:31.
183. Barnes PJ, Adcock IM. Glucocorticoid resistance in inflammatory diseases. The Lancet
2009;373(9678):1905-17.
184. Barnes PJ. Mechanisms and resistance in glucocorticoid control of inflammation. J
Steroid Biochem Mol Biol 2010, May 31;120(2-3):76-85.
185. Huff GR, Huff WE, Balog JM, Rath NC. The effects of behavior and environmental
enrichment on disease resistance of turkeys. Brain Behav Immun 2003;17(5):339-49.
186. Siala M, Jaulhac B, Gdoura R, Sibilia J, Fourati H, Younes M, et al. Analysis of bacterial
DNA in synovial tissue of tunisian patients with reactive and undifferentiated arthritis
by broad-range PCR, cloning and sequencing. Arthritis Res Ther 2008;10(2):R40.
187. Siala M, Gdoura R, Fourati H, Rihl M, Jaulhac B, Younes M, et al. Broad-range PCR,
cloning and sequencing of the full 16S rrna gene for detection of bacterial DNA in
synovial fluid samples of tunisian patients with reactive and undifferentiated arthritis.
Arthritis Res Ther 2009;11(4):R102.
188. Quax, R. A., Koper, J. W., Huisman, A. M., Weel, A., et al. Polymorphisms in the
glucocorticoid receptor gene and in the glucocorticoid-induced transcript 1 gene are
associated with disease activity and response to glucocorticoid bridging therapy in
rheumatoid arthritis. Rheumatol Int 35, 1325-1333 (2015).
189. Zhang, Q., Illing, R., Hui, C. K., Downey, K., et al. Bacteria in sputum of stable severe
asthma and increased airway wall thickness. Respir Res 13, 35 (2012).
190. Keenan CR, Salem S, Fietz ER, Gualano RC, Stewart AG. Glucocorticoid-resistant asthma
and novel anti-inflammatory drugs. Drug Discov Today 2012, Sep;17(17-18):1031-8.
191. Chen HL, Li LR. Glucocorticoid receptor gene polymorphisms and glucocorticoid
resistance in inflammatory bowel disease: A meta-analysis. Dig Dis Sci 2012,
Dec;57(12):3065-75.
192. Korte SM, Koolhaas JM, Wingfield JC, McEwen BS. The darwinian concept of stress:
Benefits of allostasis and costs of allostatic load and the trade-offs in health and disease.
Neurosci Biobehav Rev 2005, Feb;29(1):3-38.
193. Finck BN, Johnson RW. Tumor necrosis factor (TNF)-α induces leptin production
through the p55 TNF receptor. American Journal of Physiology-Regulatory, Integrative
and Comparative Physiology 2000;278(2):R537-43.

59
194. Paz-Filho G, Mastronardi C, Franco CB, Wang KB, Wong M-L, Licinio J. Leptin:
Molecular mechanisms, systemic pro-inflammatory effects, and clinical implications.
Arquivos Brasileiros De Endocrinologia & Metabologia 2012;56(9):597-607.
195. Carlton ED, Demas GE, French SS. Leptin, a neuroendocrine mediator of immune
responses, inflammation, and sickness behaviors. Horm Behav 2012, Aug;62(3):272-9.
196. Steiner AA, Dogan MD, Ivanov AI, Patel S, Rudaya AY, Jennings DH, et al. A new
function of the leptin receptor: Mediation of the recovery from lipopolysaccharide-
induced hypothermia. FASEB J 2004, Dec;18(15):1949-51.
197. Nanjappa V. A comprehensive curated reaction map of leptin signaling pathway.
Journal of Proteomics & Bioinformatics 2011;04(09).
198. Steiner AA, Romanovsky AA. Leptin: At the crossroads of energy balance and systemic
inflammation. Prog Lipid Res 2007;46(2):89-107.
199. Mak RH, Cheung W, Cone RD, Marks DL. Leptin and inflammation-associated cachexia
in chronic kidney disease. Kidney Int 2006, Mar;69(5):794-7.
200. Knight ZA, Hannan KS, Greenberg ML, Friedman JM. Hyperleptinemia is required for
the development of leptin resistance. PLoS ONE 2010;5(6):e11376.
201. Myers MG, Cowley MA, Münzberg H. Mechanisms of leptin action and leptin resistance.
Annu Rev Physiol 2008;70:537-56.
202. Wang J, Obici S, Morgan K, Barzilai N, Feng Z, Rossetti L. Overfeeding rapidly induces
leptin and insulin resistance. Diabetes 2001;50(12):2786-91.
203. Obici S, Rossetti L. Minireview: Nutrient sensing and the regulation of insulin action
and energy balance. Endocrinology 2003, Dec;144(12):5172-8.
204. Unger RH. Hyperleptinemia: Protecting the heart from lipid overload. Hypertension
2005, Jun;45(6):1031-4.
205. Lee Y, Naseem RH, Park BH, Garry DJ, Richardson JA, Schaffer JE, Unger RH. Alpha-
lipoic acid prevents lipotoxic cardiomyopathy in acyl coa-synthase transgenic mice.
Biochem Biophys Res Commun 2006, May 26;344(1):446-52.
206. Koh KK, Park SM, Quon MJ. Leptin and cardiovascular disease: Response to therapeutic
interventions. Circulation 2008, Jun 24;117(25):3238-49.
207. Duggal P, Guo X, Haque R, Peterson KM, Ricklefs S, Mondal D, et al. A mutation in the
leptin receptor is associated with entamoeba histolytica infection in children. J Clin
Invest 2011;121(3):1191.
208. Guo X, Roberts MR, Becker SM, Podd B, Zhang Y, Chua SC, et al. Leptin signaling in
intestinal epithelium mediates resistance to enteric infection by entamoeba histolytica.
Mucosal Immunol 2011, May;4(3):294-303.
209. Vedantam G, Viswanathan VK. Leptin signaling protects the gut from entamoeba
histolytica infection. Gut Microbes 2012;3(1):2-3.

60
210. Marie CS, Verkerke HP, Paul SN, Mackey AJ, Petri WA. Leptin protects host cells from
entamoeba histolytica cytotoxicity by a stat3-dependent mechanism. Infect Immun
2012, May;80(5):1934-43.
211. Mantzoros CS, Magkos F, Brinkoetter M, Sienkiewicz E, Dardeno TA, Kim SY, et al. Leptin
in human physiology and pathophysiology. Am J Physiol Endocrinol Metab 2011,
Oct;301(4):E567-84.
212. Tsatsoulis A, Mantzaris MD, Bellou S, Andrikoula M. Insulin resistance: An adaptive
mechanism becomes maladaptive in the current environment - an evolutionary
perspective. Metabolism 2012, Dec 18.
213. Johnson AR, Justin Milner J, Makowski L. The inflammation highway: Metabolism
accelerates inflammatory traffic in obesity. Immunol Rev 2012;249(1):218-38.
214. Patil P, Patil P, Watve M. Hyperinsulinemia and insulin resistance: What comes first?
Nature Precedings 2010, Dec 20.
215. Shelton RC, Miller AH. Eating ourselves to death (and despair): The contribution of
adiposity and inflammation to depression. Prog Neurobiol 2010, Aug;91(4):275-99.
216. Gauglitz GG, Toliver-Kinsky TE, Williams FN, Song J, Cui W, Herndon DN, Jeschke MG.
Insulin increases resistance to burn wound infection-associated sepsis. Crit Care Med
2010, Jan;38(1):202-8.
217. Newsholme P, Keane K, De Bittencourt P, Krause M. The impact of inflammation on
pancreatic beta-cell metabolism, function and failure in T1DM and T2DM:
Commonalities and difference. In: Type 1 Diabetes. 2013bi.
218. Hamasaki H, Mishima S, Yanai H. Hyperinsulinemia and insulin resistance in a patient
with type 2 diabetes complicated with myelofibrosis. World J Diabetes 2012, Aug
15;3(8):156-7.
219. Calder PC, Dimitriadis G, Newsholme P. Glucose metabolism in lymphoid and
inflammatory cells and tissues. Current Opinion in Clinical Nutrition & Metabolic Care
2007;10(4):531-40.
220. Maratou E, Dimitriadis G, Kollias A, Boutati E, Lambadiari V, Mitrou P, Raptis SA. Glucose
transporter expression on the plasma membrane of resting and activated white blood
cells. Eur J Clin Invest 2007, Apr;37(4):282-90.
221. Van der Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mtor
mediated by the akt/PKB substrate PRAS40. Nat Cell Biol 2007, Mar;9(3):316-23.
222. Cheng SC, Joosten LA, Netea MG. The interplay between central metabolism and innate
immune responses. Cytokine Growth Factor Rev 2014, Jun 23.
223. Jóźwiak P, Forma E, Bryś M, Krześlak A. O-GlcNAcylation and metabolic reprograming
in cancer. Front Endocrinol (Lausanne) 2014;5:145.
224. Heikamp EB, Powell JD. Sensing the immune microenvironment to coordinate T cell
metabolism, differentiation & function. Semin Immunol 2012, Dec;24(6):414-20.

61
225. Härle P, Pongratz G, Weidler C, Büttner R, Schölmerich J, Straub RH. Possible role of
leptin in hypoandrogenicity in patients with systemic lupus erythematosus and
rheumatoid arthritis. Ann Rheum Dis 2004, Jul;63(7):809-16.
226. Allott EH, Masko EM, Freedland SJ. Obesity and prostate cancer: Weighing the evidence.
Eur Urol 2012, Nov 15.
227. Olszanecka-Glinianowicz M, Madej P, Nylec M, Owczarek A, Szanecki W, Skałba P,
Chudek J. Circulating apelin level in relation to nutritional status in polycystic ovary
syndrome and its association with metabolic and hormonal disturbances. Clin
Endocrinol (Oxf) 2012, Dec 1.
228. Subbaramaiah K, Morris PG, Zhou XK, Morrow M, Du B, Giri D, et al. Increased levels of
COX-2 and prostaglandin E2 contribute to elevated aromatase expression in inflamed
breast tissue of obese women. Cancer Discov 2012, Apr;2(4):356-65.
229. Grear DA, Perkins SE, Hudson PJ. Does elevated testosterone result in increased
exposure and transmission of parasites? Ecol Lett 2009, Jun;12(6):528-37.
230. Hughes VL, Randolph SE. Testosterone depresses innate and acquired resistance to
ticks in natural rodent hosts: A force for aggregated distributions of parasites. J Parasitol
2001, Feb;87(1):49-54.
231. Quintar AA, Leimgruber C, Pessah OA, Doll A, Maldonado CA. Androgen depletion
augments antibacterial prostate host defences in rats. Int J Androl 2012, Dec;35(6):845-
59.
232. Cutolo M, Sulli A, Straub RH. Estrogen metabolism and autoimmunity. Autoimmun Rev
2012, May;11(6-7):A460-4.
233. Raz R. Urinary tract infection in postmenopausal women. Korean J Urol 2011,
Dec;52(12):801-8.
234. Arinzon Z, Shabat S, Peisakh A, Berner Y. Clinical presentation of urinary tract infection
(UTI) differs with aging in women. Arch Gerontol Geriatr 2012;55(1):145-7.
235. Gautier A, Bonnet F, Dubois S, Massart C, Grosheny C, Bachelot A, et al. Associations
between visceral adipose tissue, inflammation and sex steroid concentrations in men.
Clin Endocrinol (Oxf) 2013, Mar;78(3):373-8.
236. Cohen PG. Obesity in men: The hypogonadal-estrogen receptor relationship and its
effect on glucose homeostasis. Med Hypotheses 2008;70(2):358-60.
237. Dulloo AG, Montani JP. Body composition, inflammation and thermogenesis in
pathways to obesity and the metabolic syndrome: An overview. Obes Rev 2012, Dec;13
Suppl 2:1-5.
238. Ghanayem BI, Bai R, Kissling GE, Travlos G, Hoffler U. Diet-induced obesity in male
mice is associated with reduced fertility and potentiation of acrylamide-induced
reproductive toxicity. Biol Reprod 2010, Jan;82(1):96-104.
239. Gilliver SC, Ashworth JJ, Ashcroft GS. The hormonal regulation of cutaneous wound
healing. Clin Dermatol 2007;25(1):56-62.

62
240. Abrams ET, Miller EM. The roles of the immune system in women's reproduction:
Evolutionary constraints and life history trade-offs. Am J Phys Anthropol 2011;146
Suppl 53:134-54.
241. Cutolo M. Sex and rheumatoid arthritis: Mouse model versus human disease. Arthritis
Rheum 2007, Jan;56(1):1-3.
242. Capuron L, Miller AH. Immune system to brain signaling:
Neuropsychopharmacological implications. Pharmacol Ther 2011, May;130(2):226-38.
243. Hayley S. Toward an anti-inflammatory strategy for depression. Front Behav Neurosci
2011;5:19.
244. Motyl KJ, Rosen CJ. Understanding leptin-dependent regulation of skeletal
homeostasis. Biochimie 2012, Oct;94(10):2089-96.
245. Baganz NL, Blakely RD. A dialogue between the immune system and brain, spoken in
the language of serotonin. ACS Chem Neurosci 2013, Jan 16;4(1):48-63.
246. Thorslund K, Amatya B, Dufva AE, Nordlind K. The expression of serotonin transporter
protein correlates with the severity of psoriasis and chronic stress. Arch Dermatol Res
2013, Mar;305(2):99-104.
247. Sarkar C, Basu B, Chakroborty D, Dasgupta PS, Basu S. The immunoregulatory role of
dopamine: An update. Brain Behav Immun 2010, May;24(4):525-8.
248. Schaller M. The behavioural immune system and the psychology of human sociality.
Philos Trans R Soc Lond B Biol Sci 2011, Dec 12;366(1583):3418-26.
249. Alcaro A, Huber R, Panksepp J. Behavioral functions of the mesolimbic dopaminergic
system: An affective neuroethological perspective. Brain Res Rev 2007, Dec;56(2):283-
321.
250. Dishman RK. Gene-Physical activity interactions in the etiology of obesity: Behavioral
considerations. Obesity 2008;16(S3):S60-5.
251. Ptacek, Kuzelova H, Stefano G. Dopamine D4 receptor gene DRD4 and its association
with psychiatric disorders. Med Sci Monit 2011;17(9):RA215-20.
252. Matthews LJ, Butler PM. Novelty-seeking DRD4 polymorphisms are associated with
human migration distance out-of-africa after controlling for neutral population gene
structure. Am J Phys Anthropol 2011, Jul;145(3):382-9.
253. McDade TW, Worthman CM. Evolutionary process and the ecology of human immune
function. American Journal of Human Biology 1999;11(6):705-17.
254. Kang JI, Kim SJ, Namkoong K, An SK. Association of DRD4 and COMT polymorphisms
with disgust sensitivity in healthy volunteers. Neuropsychobiology 2010;61(2):105-12.
255. Fessler DM. Shame in two cultures: Implications for evolutionary approaches. Journal
of Cognition and Culture 2004;4(2):207-62.
256. Cavigelli SA, McClintock MK. Fear of novelty in infant rats predicts adult corticosterone
dynamics and an early death. Proc Natl Acad Sci U S A 2003, Dec 23;100(26):16131-6.

63
257. Stevenson RJ, Case TI, Oaten MJ. Proactive strategies to avoid infectious disease. Philos
Trans R Soc Lond B Biol Sci 2011, Dec 12;366(1583):3361-3.
258. Wright F, Eberhard R, Hobson A, Avery L, Russello A. Behavioral flexibility and species
invasions: The adaptive flexibility hypothesis. Ethology Ecology & Evolution 2010, Nov
10;22(4):393-404.
259. Duncan LA, Schaller M. Prejudicial attitudes toward older adults may be exaggerated
when people feel vulnerable to infectious disease: Evidence and implications. Analyses
of Social Issues and Public Policy 2009, Dec;9(1):97-115.
260. Faulkner J, Schaller M, Park H, Duncan A. Evolved disease-avoidance mechanisms and
contemporary xenophobic attitudes. Group Processes & Intergroup Relations 2004, Oct
1;7(4):333-53.
261. Altmann DM, Balloux F, Boyton RJ. Diverse approaches to analysing the history of
human and pathogen evolution: How to tell the story of the past 70 000 years. Philos
Trans R Soc Lond B Biol Sci 2012, Mar 19;367(1590):765-9.
262. Besser MJ, Ganor Y, Levite M. Dopamine by itself activates either D2, D3 or D1/D5
dopaminergic receptors in normal human t-cells and triggers the selective secretion of
either IL-10, tnfalpha or both. J Neuroimmunol 2005, Dec;169(1-2):161-71.
263. Poston WS, Ericsson M, Linder J, Haddock CK, Hanis CL, Nilsson T, et al. D4 dopamine
receptor gene exon III polymorphism and obesity risk. Eating and Weight Disorders:
EWD 1998;3(2):71.
264. Sarkar C, Das S, Chakroborty D, Chowdhury UR, Basu B, Dasgupta PS, Basu S. Cutting
edge: Stimulation of dopamine D4 receptors induce T cell quiescence by up-regulating
krüppel-like factor-2 expression through inhibition of ERK1/ERK2 phosphorylation. The
Journal of Immunology 2006;177(11):7525-9.
265. Levite M. The 1st international meeting on nerve-driven immunity: Neurotransmitters
and neuropeptides in the immune system. Future Neurology 2012;7(3):247-53.
266. Ferreira IM, Brooks D, Lacasse Y, Goldstein RS, White J. Nutritional supplementation for
stable chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2005;2.
267. Kim J, Bentley PJ, Aickelin U, Greensmith J, Tedesco G, Twycross J. Immune system
approaches to intrusion detection – a review. Natural Computing 2007, Oct 18;6(4):413-
66.
268. Torday JS, Rehan VK. A cell-molecular approach predicts vertebrate evolution. Mol Biol
Evol 2011, Nov;28(11):2973-81.
269. Vestbo J. Body mass, fat free body mass and prognosis in COPD patients from a random
population sample. Am J Respir Crit Care Med 2005, Sep 15.
270. Torday JS, Rehan VK. Cell-cell signaling drives the evolution of complex traits:
Introduction-lung evo-devo. Integr Comp Biol 2009, Aug;49(2):142-54.

64
Paragraph 1.2. Lifestyle and nutritional imbalances associated with Western diseases: causes
and consequences of chronic systemic low-grade inflammation in an evolutionary context.
Ruiz-Núñez B, Pruimboom L, Dijck-Brouwer DA, Muskiet FA. J Nutr Biochem 2013;24:1183-
201.
Lifestyle and nutritional imbalances associated with Western diseases: causes
and consequences of chronic systemic low grade inflammation in an
evolutionary context
Begoña Ruiz-Núñez1, Leo Pruimboom2, D.A. Janneke Dijck-Brouwer1,
Frits A.J. Muskiet1
1Laboratory Medicine, University Medical Center Groningen (UMCG),
The Netherlands; 2University of Gerona, Faculty of Sciences, Spain and University of Graz, Unit
for Life, Austria

65
Contents
Abstract
Keywords
Introduction
Our brain growth rendered us sensitive to glucose deficits
Reallocation of energy-rich nutrients by insulin resistance
Changes in serum lipoproteins
Lifestyle-induced chronic systemic low grade inflammation
Mechanisms of lifestyle-induced inflammation
Fatty acids and inflammation
Role of the antioxidant network
Evolutionary nutrition vs. randomized controlled trials
Nurture, not nature
Conclusions
References

66
Abstract
In this review, we focus on lifestyle changes, especially dietary habits, that are at the basis of
chronic systemic low grade inflammation, insulin resistance and Western diseases. Our
sensitivity to develop insulin resistance traces back to our rapid brain growth in the past 2.5
million years. An inflammatory reaction jeopardices the high glucose needs of our brain,
causing various adaptations, including insulin resistance, functional reallocation of energy-
rich nutrients and changing serum lipoprotein composition. The latter aims at redistribution
of lipids, modulation of the immune reaction, and active inhibition of reverse cholesterol
transport for damage repair. With the advent of the agricultural and industrial revolutions, we
have introduced numerous false inflammatory triggers in our lifestyle, driving us to a state of
chronic systemic low grade inflammation that eventually leads to typically Western diseases
via an evolutionary conserved interaction between our immune system and metabolism. The
underlying triggers are an abnormal dietary composition and microbial flora, insufficient
physical activity and sleep, chronic stress and environmental pollution. The disturbance of
our inflammatory/anti-inflammatory balance is illustrated by dietary fatty acids and
antioxidants. The current decrease in years without chronic disease is rather due to ‘nurture’
than ‘nature’, since less than 5% of the typically Western diseases are primary attributable to
genetic factors. Resolution of the conflict between environment and our ancient genome
might be the only effective manner for ‘healthy aging’, and to achieve this we might have to
return to the lifestyle of the Paleolithic era as translated to the 21st century culture.
Keywords
Chronic systemic low grade inflammation, evolution, brain, encephalization quotient,
immune system, diet, fatty acids, fish oil, fruits, vegetables, antioxidant network, metabolic
syndrome, glucose, homeostasis, insulin resistance, cholesterol, lifestyle, antioxidants,
resoleomics, pro-inflammatory nutrients, anti-inflammatory nutrients.
List of abbreviations
AA, arachidonic acid; ADHD, attention deficit hyperactivity disorder; AHA, American Heart
Association; ALA, alpha-linolenic acid; BMI, body mass index; CARS, compensatory anti-
inflammatory response syndrome; CAT, catalase; COX-2, cyclo-oxygenase-2; CRP, C-reactive
protein; CVD, cardiovascular disease; DHA, docosahexaenoic acid; EBM, evidence based
medicine; EBN, evidence based nutrition, EPA, eicosapentaenoic acid; EQ, encephalization
quotient; FADS2, Δ-6-desaturase; FDB, familial defective apo-B100; FH, familial
hypercholesterolemia; GPR120, G-protein-coupled receptor 120; GPx, glutathione peroxidase;
GWAS, genome wide association studies; HDL, high density lipoprotein; HPA-axis,
hypothalamus-pituitary-adrenal gland axis; HPG-axis, hypothalamus-pituitary-gonadal
gland axis; HPL, human placental lactogen; HPT-axis, hypothalamic-pituitary-thyroid axis;

67
IGF-1, insulin-like-growth factor-1; LA, linoleic acid; LCP, long-chain polyunsaturated fatty
acids; LDL, low density lipoprotein; LOX-12, lipoxygenase-12; LOX-15, lipoxygenase-15; LOX-
5, lipoxygenase-5; LPS, lipopolysaccharides; LTB4, leukotrienes-B4; LX, lipoxin; NFκB, nuclear
factor kappa B; NTIS, non-thyroidal illness syndrome; PGD2, prostagladins- D2; PGE2,
prostagladins-E2; PLA2, phospholipase A2; PPAR; peroxisome proliferator activated receptor;
RCT, randomized controlled trial; ROS, reactive oxygen species; RR, relative risk; SAA, serum
amyloid A; SIRS, systemic inflammatory response syndrome; TNFα, tumor necrosis factor
alpha; TSH, thyroid stimulating hormone; VLDL, very low density lipoprotein.
Introduction
In recent years, it has become clear that chronic systemic low grade inflammation is at the
basis of many, if not all, typically Western diseases centered on the metabolic syndrome. The
latter is the combination of an excessive body weight, impaired glucose homeostasis,
hypertension and atherogenic dyslipidemia (the ‘deadly quartet’), that constitutes a risk for
diabetes mellitus type 2, cardiovascular disease (CVD), certain cancers (breast, colorectal,
pancreas), neurodegenerative diseases (e.g. Alzheimer's disease), pregnancy complications
(gestational diabetes, preeclampsia), fertility problems (polycystic ovarian syndrome) and
other diseases (1). Systemic inflammation causes insulin resistance and a compensatory
hyperinsulinemia that strives to keep glucose homeostasis in balance. Our glucose
homeostasis ranks high in the hierarchy of energy equilibrium, but becomes ultimately
compromised under continuous inflammatory conditions via glucotoxicity, lipotoxicity, or
both, leading to the development of beta-cell dysfunction and eventually type 2 diabetes
mellitus (2).
Insulin resistance has a bad name. The ultimate aim of this survival strategy is, however,
deeply anchored in our evolution, during which our brain has grown tremendously. The goal
of reduced insulin sensitivity is, among others, the reallocation of energy-rich nutrients
because of an activated immune system (3, 4), limitation of the immune response, and the
repair of the inflicted damage. To that end, serum lipoproteins adopt a pattern that bears
resemblance with the ‘hyperlipidemia of sepsis’, accompanied by seemingly inconsistent
changes in serum cholesterol, increased triglycerides, decreased HDL-cholesterol, and an
increase of ‘small dense’ LDL-particles, of which the latter three constitute the triad of
atherogenic dyslipidemia that is part of the metabolic syndrome (5-10).
From the perspective of our brain growth during evolution, we address the question of why
homo sapiens is so sensitive to the development of insulin resistance. The purpose and the
underlying mechanisms leading to insulin resistance and the associated dyslipidemia are
subsequently discussed in more detail. We argue that our current Western lifestyle is the

68
cause of many false inflammatory triggers which successively lead to a state of chronic
systemic low grade inflammation, insulin resistance, the metabolic syndrome, and eventually
to the development of the above mentioned typically Western diseases of affluence. To find a
solution for the underlying conflict between our environment and our ancient genome, we
also go back in time. With the reconstruction of our Paleolithic diet, we might be able to
obtain information on the nutritional balance that was at the basis of our genome. We argue
that insight into this balance bears greater potential for healthy aging than the information
from the currently reigning paradigm of ‘Evidence Based Medicine’ (EBM) and ‘randomized
controlled trials’ (RCTs) with single nutrients.
Our brain growth rendered us sensitive to glucose deficits
Homo sapiens and the current chimpanzees and bonobos share a common ancestor, who
lived in Africa around 6 million years ago. Since about 2.5 million years ago, our brain has
strongly grown from an estimated volume of 400 mL to the current volume of approximately
1,400 mL (Figure 1). This growth was enabled by the finding of a high-quality dietary source2,
that was easy to digest and contained an ample amount of nutrients, necessary for the
building and maintenance of a larger brain. The nutritional quality of primate food correlates
positively with relative brain size and inversely with body weight, suggesting that a larger
brain requires a higher dietary quality (11). The necessary so-called ‘brain selective nutrients’
include, among others, iodine, selenium, iron, vitamins A and D, and the fish oil fatty acids
eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), that jointly are abundantly
available in the land-water ecosystem. There are compelling arguments that a sizeable part of
our evolution occurred at places where the land meets the water (12-15), but also that we have
changed our lifestyle in a too short period of time. These changes started from the
agricultural revolution (around 10,000 years ago) and became accelerated since the industrial
revolution (about 100-200 years ago). They created a conflict between our current lifestyle,
including our diet, and our ancient genome, that, with an average effective mutation rate of
0.5% per million years, still resides for the greater part in the Paleolithic era (16, 17). It is not by
chance that the above mentioned brain selective nutrients are among those of which we
currently exhibit the largest deficits worldwide. These deficits are masked by enrichment and
fortification of our current diet with iodine (in salt), vitamins A and D (e.g. in margarines and
milk) and iron (flour, cereals).
2 Food quality refers to the energy content and/or the nutrient content of a diet. An increase in food quality may derive from the consumption of a diet with another composition or the modification of the diet by e.g. cooking or genetic manipulation (11).

69
Figure 1. Evolution of our brain size within the past 3.5 million years.
Our brain has grown fast since the homo erectus (1.7-2.0 million years ago). The newborn homo sapiens, the adult
chimpanzee and the homo floresiensis (18) have brain volumes of around 400 mL. Adapted from Aiello and Wheeler (19)
with permission from The University of Chicago Press.
Figure 2. Relationship between body weight and basal metabolism in 51 land mammals (20 non-
primates, 30 primates, and humans).
Adapted from Leonard et al.(11) with permission from Elsevier.

70
Our brain consumes 20-25%3 of our basal metabolism (11-17, 20) and is thereby together with
the liver (19%2), our gastrointestinal tract (15%2), and skeletal musculature (15%2) among the
quantitatively most important organs in energy consumption (19). The infant brain consumes
as much as 74% of the basal metabolism (11, 21). In contrast to most other organs, the brain
uses mostly glucose as an energy source. There is no other primate equipped with such a
large, glucose-consuming, luxury organ as our brain. For example, our closest relative, the
chimpanzee, has a brain volume of 400 mL, which consumes about 8-9% of the basal
metabolism. Because of the high energy expenditure of a large brain, it was necessary to
make various adjustments in the sizes of some other organs. There is a linear relationship
between body weight and basal metabolism among terrestrial mammals (Figure 2). This
apparently dogmatic relationship predicts that, due to the growth of our brain, other organs
with high energy consumption had to be reduced in size, what in evolution is known as a
‘trade-off’4. As a consequence of this ‘expensive tissue hypothesis’ of Aiello and Wheeler (19)
our intestines, amongst others, had to become reduced in size. However, this exchange of
expensive tissue probably occurred prior to, or simultaneous with, our brain growth, in which
the trigger was the consumption of the easily digestible high-quality food (20) that contains
the above-mentioned ‘brain selective nutrients’ from the land-water ecosystem. Under these
‘conditions of existence’ (Darwin), a single mutation in a growth regulatory gene is likely to
have been sufficient for the brain to grow. This notion derives from the existence of
genetically-determined micro- (22) and macrocephaly (23) and it is as a ‘proof of principle’
demonstrated by the differences in the beak lengths of Darwins’ legendary Galapagos finches
(24-26). Compared with our close (vegetarian) relatives in the primate world, we possess a
relatively long small intestine and a relatively short large intestine, which corresponds with
the digestion of high quality food (such as meat and fish) in the small intestine, and the lesser
need of a long colon for the digestion of complex carbohydrates (e.g. fiber) from a typically
vegetarian diet (19). Unlike our near primates, such as the gorilla, our teeth and the
attachments of our jaw muscles are not specialized for the processing of tough vegetarian
food. Also our muscle mass became adapted, since its current size is relatively small
compared to our body weight. For instance, when compared with the chimpanzee, we are
definitely weak. On the other hand, we have a relatively sizeable fat mass, which probably
serves as a guarantee for the high energy requirement of our brain.
Our brain’s energy consumption is quite stable. Unlike other organs, the energy consumption
of the brain can not be downregulated at times of a negative energy balance or fasting (11, 20).
Our brain also gets spared during prolonged fasting, while other organs such as the liver,
spleen, kidneys and even the heart, are sacrificed for energy generation (27). This hierarchy
3 These estimates derive from various publications and therefore do not add to 100%. They should be regarded as indications. 4 The beneficial exchange of a certain property into another one

71
also applies to the prenatal brain, whose development is conserved during intrauterine
growth restriction (28). An example is the Indian 'thin fat baby’, with a birth weight of 2,700 g.
Compared with its 3,500 g counterpart from the UK, this infant has a similar brain size and a
relatively large fat compartment, at the expense of the somatic growth of the skeletal muscle,
kidneys, liver and the pancreas (28). Our brain ranks high in the functional hierarchy and
should be provided with the necessary energy at all times.
Apart from its large size, there is nothing special about our brain within the primate world.
Compared with other species, primates have a very economical space-saving brain, but
among the primates, brain weight correlates with the number of neurons (29-32) and
intelligence (33). Actually, our brain is no more than an oversized primate brain (29). What
does distinguish us from other species is the high ratio between our brain size and our body
weight, which is also named encephalization quotient (EQ) (Figure 3). Toothed whales (brain
weight 9,000 g) and African elephants (4,200 g) have much larger brains than humans, but
they have lower EQs (34). Among the primates, EQ does not correlate with intelligence (33).
Our high EQ has major implications for our energy management, particularly at times of
‘glucose shortage’. Under normal circumstances, our brain functions almost entirely on
glucose, consuming up to 130 g/day (27). Compared with the apparently unlimited storage
capacity for fat, we only dispose of a small reserve of glucose that is stored as glycogen in the
liver (up to 100-120 g; mobilizable) and muscles (360 g; for local usage), while some glycogen
can even be found in brain’s astrocytes (35). With the exception of the glycerol moiety, we can
not convert fat into glucose. The reduced carbohydrate intake that came along during
evolution with the transition from vegetarians to omnivores rendered us strongly dependent
on gluconeogenesis from (glucogenic) amino acids. This was possible because we
simultaneously consumed more protein from meat and fish, which is also referred to as the
‘carnivore connection’ (36). After the depletion of our glycogen reserves, for instance after an
overnight fast, we obtain the necessary glucose for our brain via gluconeogenesis from
glycerol and amino acids. Under normal conditions, these amino acids derive from our
dietary proteins after a meal, but during starvation, they become extracted from our tissues by
catabolism of functional proteins, at the expense of our lean body mass. Under such
circumstances of severe glucose deficit, the energetic need of our brain becomes increasingly
covered by ketone bodies from fat (37, 38).

72
Figure 3. Encephalization quotient (EQ) of selected mammals.
The EQ has been normalized with the cat as a reference. Data adapted from Roth and Dicke (34).
A glucose deficit leads to competition between organs for the available glucose. As previously
mentioned, this occurs during fasting, but also during pregnancy and
infection/inflammation. Fasting is characterized by a generalized shortage of glucose (and
other macronutrients), but in case of pregnancy and inflammation we deal with active
compartments competing with the brain for the available glucose, i.e. the growing child and
the activated immune system, respectively. During competition between organs for glucose,
we fulfill the high glucose needs of the brain by a reallocation of the energy-rich nutrients,
and to that end, we need to become insulin resistant.
Reallocation of energy-rich nutrients by insulin resistance
The developing child grows fast in the third trimester of pregnancy. In this period, the supply
of the necessary building blocks like glucose and fatty acids should be independent of the
maternal metabolic status, which is known as the state of ‘accelerated starvation’ and
‘facilitated anabolism’ (38). Glucose crosses the placenta without restriction. Fetal needs are
directive, since the developing fetus is high in the evolutionary hierarchy. If necessary, the
fetal needs become covered at the expense of the mother, which is known as the ‘depletion
syndrome’.

73
During infection/inflammation we deal with the metabolic needs of an activated immune
system for acute survival. The inactive immune system consumes about 23%2 of our basal
metabolism, of which as much as 69% derives from glucose (47%) and the glycogenic amino
acid glutamine (22%). Upon activation, the energy requirement of our immune system may
increase with about 9-30% of our basal metabolic rate. In multiple fractures, sepsis and
extensive burns, we deal with increases up to 15-30, 50, and 100% of our basal metabolism,
respectively (3, 4, 39).
The way we save glucose for our brain during starvation, for the brain and the fetus during
pregnancy, and for the brain and immune system during infection/inflammation, is by
causing insulin resistance in selected insulin-dependent tissues. These tissues are thereby
forced to switch to the burning of fat. Due to insulin resistance, the adipose tissue
compartment will be encouraged to distribute free fatty acids, while the liver will be
encouraged to produce glucose via gluconeogenesis and to distribute triglycerides via VLDL.
The aforementioned (asymmetric) ‘thin fat baby’ with its spared brain, relatively high adipose
tissue compartment, and the growth restricted body (islets of Langerhans included), has
relatively high cord plasma insulin and glucose concentrations at birth (28). These
characteristics of insulin resistance and diabetes mellitus are probably necessary for the
postpartum, saving of as much as possible of the available glucose for the brain, whereas the
other organs are provided with fatty acids from the sizeable adipose tissue stores. This
intrauterine 'programming', that follows the prediction of a thrifty postnatal life comes along
with health risks, notably when the prediction proves false (40, 41). According to the ‘Barker
hypothesis’, at adult age, these children have a higher chance of diseases related to the
metabolic syndrome, especially when they are raised in our current obesogenic society. The
unfavorable interaction of their high EQ with a high body weight is already demonstrable at
the age of 8 years (42). Essentially, their postnatal risk is attributable to a (probably epigenetic)
‘intrauterine programming’, that traces back to the high hierarchical ranking of our brain in
both growth and energy needs, also referred to as ‘the selfish brain’ (43).
Glucose intolerance (26) and insulin resistance have been reported in calorie restriction,
extreme fasting and anorexia nervosa, and may even cause, under these circumstances,
diabetes mellitus type 2, notably in those subjects sensitive to its development (44). According
to textbooks, insulin resistance during the third trimester of pregnancy is caused by the
hormonal environment, among which HPL, progesterone, estrogens, prolactin and cortisol
are mentioned. However, placental tumor necrosis factor alpha (TNFα) correlates best with
measures of maternal insulin resistance (45, 46). Pregnancy is therefore sometimes referred to
as a physiological state of systemic low grade inflammation (47). As a consequence of
reduced insulin sensitivity, maternal circulating concentrations of energy-rich nutrients,
such as glucose and fat, tend to increase, promoting their transport across the placenta.

74
Under non-pregnant conditions, this situation would resemble pathology, but is tolerable
during the 9 months of a pregnancy, while the largest changes occur during the third
trimester.
Figure 4. Mechanistic connection between inflammation and insulin resistance.
The NFκB and AP-1 Fos/June inflammatory pathways inhibit the PI3K/AKT signal transduction pathway for nutrient
metabolism and the Ras/MAPK pathway for gene expression, both part of the insulin signaling. CAP, Cbl associated protein;
Cbl, Proto-oncogene product; ER, endoplasmic reticulum; FFAs, Free fatty acids; Gqα/11, heterotrimeric G protein; Ikkb, I
kappa B kinase Beta; IRS, insulin receptor substrate; JNK, C-jun N-terminal kinase; NFkB, nuclear factor kappa B; NO, nitric
oxide; Ras/MAPK; PI3K, phosphatidylinositol 3-kinase; Ras-mitogen activated protein kinase; Shc, Src homology 2
containing protein; SOCS, supressor of citokyne signaling; TLRs, Toll-like receptors. Adapted from de Luca and Olefsky (48)
with permission from Elsevier.
During infection and inflammation, the signals for metabolic adaptation become transmitted
by pro-inflammatory cytokines. The resulting insulin resistance causes reallocation of energy
(i.e. the aim of the process; see above), which illustrates that inflammation and metabolism
are highly integrated (49-51). At the molecular level, the interaction takes place through the
influences of the nuclear factor kappa B (NFκB) and the AP-1 Fos/June inflammatory
pathways on the PI3K/Akt signal transduction pathway for nutrient metabolism and the
Ras/MAPK pathway for gene expression, which are both part of the insulin signal
transduction (48, 52). To put it simply: the activated inflammatory signal transduction

75
pathway causes inhibition of the postreceptor insulin signaling pathway, which becomes
noticeable by what we know as insulin resistance (Figure 4). Insulin resistance especially
refers to a grossly diminished reduction of the circulating glucose concentration by insulin.
However, insulin has many functions, and thereby exerts different effects in the various
organs carrying the insulin receptor. Consequently, the ‘resistance’ affects the many insulin
signal transduction pathways at various degrees, and thereby works out differently with
respect to the various insulin functions (1, 53). Some processes are impaired (i.e. are genuinely
‘resistant’), while others remain intact and become excessively stimulated by the
compensatory hyperinsulinemia. This compensatory increase of the circulating insulin levels
aims at the prevention of a disturbed glucose homeostasis and thereby the onset of type 2
diabetes mellitus. The persistence of compensatory hyperinsulinism is responsible for most, if
not all, of the abnormalities that belong to the metabolic syndrome (1).
In muscle and fat cells, insulin resistance induces a diminished glucose uptake and therefore
a reduced storage of glucose as glycogen and triglycerides. In fat cells, it causes decreased
uptake of circulating lipids, increased hydrolysis of stored triglycerides and their mobilization
as free fatty acids and glycerol. In liver cells, insulin resistance induces the inability to
suppress glucose production and secretion, in addition to decreased glycogen synthesis and
storage. The hereby promoted reallocation of energy-rich substrates (glucose to the brain,
fetus and immune system; fat to the fetus and the organs that became insulin resistant) and
the compensatory hyperinsulinemia, are meant for short-term survival, and their persistence
as a chronic state are at the basis of the ultimate changes that we recognize as the symptoms
of the metabolic syndrome, including the changes in glucose and lipid homeostasis (3, 4) and
the increasing blood pressure. For example, the concomitant hypertension has been
explained by a disbalance between the effects of insulin on renal sodium reabsorption and
NO-mediated vasodilatation, in which the latter effect, but not the first, becomes
compromised by insulin resistance, causing salt sensitivity and hypertension (54).
Reaven coined the term ‘metabolic syndrome’ and subsequently renamed it the ‘insulin
resistance syndrome’ (1). However, it becomes increasingly clear that we could better refer to
it as the ‘chronic systemic low-grade inflammation induced energy reallocation syndrome’.
The reason for this broader name derives from the recognition that insulin resistance is only
part of the many simultaneously occurring adaptations. To their currently known extent,
these adaptations and consequences are composed of: i) reduced insulin sensitivity (glucose
and lipid redistribution, hypertension), ii) increased sympathetic nervous system activity
(stimulation of lipolysis, gluconeogenesis and glycogenolysis), iii) increased activity of the
HPA-axis [hypothalamus-pituitary-adrenal gland (stress) axis, mild cortisol increase,
gluconeogenesis, with cortisol resistance in the immune system], iv) decreased activity of the
HPG-axis (hypothalamus-pituitary-gonadal gland axis; decreased androgens for

76
gluconeogenesis from muscle proteins, sarcopenia, androgen/estrogen disbalance, inhibition
of sexual activity and reproduction), v) IGF-1 resistance (insulin-like growth factor-1; no
investment in growth) and vi) the occurrence of ‘sickness behavior’ (energy-saving, sleep,
anorexia, minimal activity of muscles, brain, and gut) (3).
The HPT-axis (hypothalamic-pituitary-thyroid axis) has a central role in our energy
management. The adaptation of thyroid function in subjects with the metabolic syndrome is
yet unclear, possibly due to the many concerted changes, such as an altered thyroid hormone
binding capacity, tissue uptake, conversion of T4 into T3, and tissue-specific receptor
expression and function. For example, T4 may become converted into the highly active T3
within the target cell and thereby, without visible changes of circulating hormone
concentrations, bind to the intracellular thyroid hormone receptor (55). Whether intracellular
T4 is converted into T3 or the inactive reverse T3 (rT3), or is used as a source of iodine to kill
bacteria, depends on several factors, including cytokines, that determine the expression
pattern of the three involved deiodinases (55-57). In euthyroid subjects, free T4 (FT4) is
associated with insulin resistance, inversely related to total- and LDL-cholesterol, while also a
positive relationship between TSH and triglycerides has been documented (58). The reported
changes during metabolic syndrome (59), low-grade inflammation and insulin resistance (60)
are inconsistent, but do bear great resemblance with subclinical hypothyroidism, with high-
normal or slightly elevated TSH, and normal FT4 concentrations (61, 62). Insulin resistance has
recently been associated with an increased T3/rT3 ratio, which is a measure of peripheral
thyroid hormone metabolism and suggests increased thyroid hormone activity (63). In
contrast, during fasting, energy expenditure becomes downregulated, resulting in a normal
or decreased TSH and decreased serum thyroid hormone concentrations (64).
Downregulation of the HPT-axis with reductions of T3, T4 and TSH, and an increase of rT3
(and thus a decrease of the T3/rT3 ratio) occurs progressively with the severity of the ‘non-
thyroidal illness syndrome’ (NTIS, also called the ‘Low T3 syndrome’ and ‘euthyroid sick
syndrome’) (55) which is explained as an adaptation of the body to prevent excessive (protein)
catabolism as part of the acute phase response (56).
All of the above mentioned adaptations of our metabolism are associated with changes in the
serum lipoprotein profile, which are part of the metabolic syndrome. The purpose of these
changes will be explored in more detail below.
Changes in serum lipoproteins
The quantitative and qualitative changes in the composition of serum lipoproteins resulting
from an inflammatory trigger have, in addition to the reallocation of energy-rich nutrients
(fatty acids to the insulin resistant organs), at least two other goals (5-10, 65). These are: i) the

77
modulation of the immune response by which we protect ourselves from the harmful effects
of invading bacteria, viruses and parasites, and ii) the restoration of the hereby inflicted
damage. However, if the subsequent changes in structure and function of lipoproteins persist,
they contribute to the development of atherosclerosis (66). These long term complications
have not exerted selection pressure during evolution and, consequently, no solution has
come into existence via the habitual process of spontaneous mutation and natural selection.
The inflammatory trigger during an infection with Gram-negative bacteria is initiated by
lipopolysaccharides (LPS). Circulating lipoproteins aid in the clearance of this LPS. Hence,
lipoproteins do not only have functions in transporting lipids to and from tissues, but also
play important roles in limiting the inflammatory response (67). The ability of lipoproteins to
bind LPS is proportional to the cholesterol content of the lipoprotein (68), but the
phospholipids/cholesterol ratio of the lipoprotein is the principal determinant of the LPS-
binding capacity (69). The available phospholipid surface is thus of special importance and is,
under normal circumstances, the largest for the circulating HDL. However, critically ill
patients exhibit decreases of both esterified cholesterol and HDL (see below) and in those
patients, LPS is mainly taken up in the phospholipid layers of LDL and VLDL. Binding of LPS
to lipoproteins prevents activation of LPS-responsive cells and encourages LPS clearance via
the liver to the bile. In line with this mechanism, it has been observed that a decrease in
plasma lipoproteins in experimental models increases LPS-induced lethality (69).
The protective role of LDL is already known for some time, and this process has probably
been exploited during evolution. Currently, there are over one thousand LDL-receptor
mutations, many of which lead to a reduced or absent hepatic uptake of LDL particles, and
consequently, to an elevated serum LDL-cholesterol (70). The carriers of these mutations have
‘familial hypercholesterolemia’ (FH; incidence about 1/400 in The Netherlands) or ‘defective
apo-B100’ (FDB), if the mutation is located in the LDL-receptor ligand. They constitute
autosomal dominant disorders with a high risk of premature atherosclerosis and mortality
from CVD (71). The arising question is why evolution has preserved so many apparently
detrimental mutations in the LDL-receptor. Research with data from the population registry
office in The Netherlands showed that subjects with FH lived longer until 1800, which turned
into a shorter lifespan than the general population after 1800 (72). Important support for an
explanation came from studies with LDL-receptor knockout mice, and also with transgenic
mice overexpressing apo-A1, the structural protein of HDL. These mutants have a high LDL-
and HDL-cholesterol, respectively, are resistant to LPS-induced mortality, and have better
survival of severe Gram-negative infection compared with the wild type (66, 73). In other
words, FH might have become widespread during evolution due to the modulating effect of a
high LDL (i.e. ‘a high cholesterol’) during Gram-negative infections, that were much more
common in the past. This benefit might have become a risk following the introduction of a

78
typically Western lifestyle (see below), to which subjects with FH seem particularly sensitive
(72).
Figure 5. Changes in reverse cholesterol transport during the acute phase response.
Lipopolysaccharides (LPS) and cytokines reduce the ABCA1 (ATP binding cassette transporter A1) and the cholesterol efflux
from peripheral cells to HDL. LPS reduces the activities of various proteins involved in HDL metabolism, such as lecithin-
cholesterol acyltransferarse (LCAT), cholesterol ester transfer protein (CETP) and hepatic lipase (HL). LPS and cytokines also
down-regulate hepatic scavenger receptor class B type 1 (SRB1), resulting in a decreased cholesterol ester (CE) uptake in
the liver. FC, free cholesterol; LDL-R, LDL receptor; LRP, LDL receptor-related protein; PLTP, phospholipid transfer protein.
Adapted from Khovidhunkit et al. (66) with permission from The American Society for Biochemistry and Molecular Biology.
As mentioned above, among the lipoproteins, notably HDL has the capacity to bind LPS and
thereby to prevent an LPS-induced activation of monocytes and the subsequent secretion of
proinflammatory cytokines (5). However, during the ‘lipidemia of sepsis’, the HDL
concentration decreases while also the HDL particles decrease in size (6). Their function
changes as part of the acute phase response: the immunomodulatory properties vanish to a
high extent and HDL even becomes proinflammatory. The apo-A1 and cholesterol esters are
lost from the HDL particle, the activities of HDL-associated enzymes and exchange proteins
decrease, and these proteins are, among others, replaced by serum amyloid A (SAA) (5, 6). Like
CRP, SAA is produced in the liver as part of the acute phase response. SAA is 90% located in
HDL, prevents the uptake of cholesterol by the liver and directs it to other cells such as
macrophages (8, 66). Both the decreasing HDL-cholesterol and the concomitantly reduced
‘cholesterol reverse transport’, promote the accumulation of cholesterol in the tissues, where
it is needed for the synthesis of steroid hormones (e.g. cortisol) in the adrenal glands, the
immune system and for the synthesis of cellular membranes that became damaged by the
infection (66). Also the formation of small dense LDL (74) might be functional because these

79
particles are poorly cleared by the LDL-receptor, easily penetrate the subendothelial space
and by their binding to the subendothelial matrix, take their cholesterol cargo to the sites of
damage in a highly efficient manner. It appears that there are numerous mechanisms that
jointly cause the active inhibition of the reverse cholesterol transport in response to an acute
phase response (Figure 5) (66, 75).
Summarizing thus far, we humans are extremely sensitive to glucose deficits, because our
large brain functions mainly on glucose. During starvation, pregnancy and
infection/inflammation, we become insulin resistant, along with many other adaptations.
The goal is the reallocation of energy-rich substrates to spare glucose for the brain, the
rapidly growing infant during the third trimester of pregnancy, and our activated immune
system that also functions mainly on glucose. Under these conditions, the insulin resistant
tissues are supplied with fatty acids. Other goals of the changes in the serum lipoprotein
composition are their role in the modulation of the immune response by the clearance of LPS
during infection/inflammation and the redirection of cholesterol to tissues for local damage
repair. The metabolic adaptations caused by inflammation illustrate the intimate relationship
between our immune system and metabolism. This relation is designed for the short term. In
a chronic state it eventually causes the metabolic syndrome and its sequelae. We are
ourselves the cause of the chronicity. Our current Western lifestyle contains many false
inflammatory triggers and is also characterized by a lack of inflammation suppressing factors.
These will be described in more detail below.
Lifestyle-induced chronic systemic low grade inflammation
An inflammatory reaction is the reflection of an activated immune system that aims to
protect us from invading pathogens or reacts to a sterile infection. If an activated immune
system is uncontrolled, the resulting secondary reactions have the ability to kill us. Rogers (76)
expresses it as follows: ‘...inflammation may be useful when controlled, but deadly when it is
not. For example, head trauma may kill hundreds of thousands of neurons, but the secondary
inflammatory response to head trauma may kill millions of neurons or the patient’. It is clear
that an inflammatory reaction that has started should subsequently be ended.

80
Table 1. Environmental factors that may cause chronic systemic low grade inflammation.
Adapted from Egger and Dixon (77).
There are many factors in our current Western lifestyle that jointly cause a state of chronic
systemic low grade inflammation, which in turn leads to chronically compromised insulin
sensitivity, compensatory hyperinsulinemia and, eventually, the diseases related to the
metabolic syndrome. Lifestyle factors that cause inflammation can be subdivided into an
unbalanced composition of the diet (usually referred to as ‘malnutrition’) (78-80) and non-
food related factors (77), which partly exert their influence via obesity (81) (Table 1). Among the
pro-inflammatory factors in our current diet, we find: the consumption of saturated fatty
acids (82) and industrially produced trans fatty acids (83, 84), a high ω6/ω3 fatty acid ratio (85-
87), a low intake of long-chain polyunsaturated fatty acids (LCP) of the ω3 series (LCPω3)
from fish (88, 89), a low status of vitamin D (90-92), vitamin K (93) and magnesium (94-96),
the ‘endotoxemia’ of a high-fat low-fiber diet (97, 98), the consumption of carbohydrates with
a high glycemic index and a diet with a high glycemic load (99, 100), a disbalance between the
many micronutrients that make up our antioxidant/pro-oxidant network (101-103), and a low
intake of fruit and vegetables (103, 104). The ‘dietary inflammation index’ of the University of
North Carolina is composed of 42 anti- and proinflammatory food products and nutrients. In
Lifestyle Exercise too little (inactivity) Lifestyle Exercise/physical activity/fitnesstoo much
Nutrition alcohol (excessive) Nutrition alcoholexcessive energy intake energy intake (restricted)starvation 'fast food'/ Western style diet Mediterranean dietfat high-fat diet fat fish/fish oil
saturated fats mono-unsaturated fatstrans fatty acids olive oilhigh ω6/ω3 ratio low ω6/ω3 ratio
fiber (low intake) fiber (high intake)fructose nutsglucose high glucose/GI foods low GI foods
glycemic load grapes/raisinsglycemic status dairy calcium sugar-sweetened drinks eggs
meat (domesticated) lean meats (wild)salt soy protein
fruits/vegetablescocoa/chocolate (dark)herbs and spicestea/green teacapsaicin (pepper)garlicpepper
Obesity 'Healthy obesity'Weight gain Weight lossSmoking Smoking cessation 'Unhealthy lifestyle' Intensive lifestyle changeStress/anxiety/depression/burn outSleep deprivation
Age
Environment Socioeconomic status (low)Perceived organizational injusticeAir pollution (indoor/outdoor)Second-hand smoking 'Sick building syndrome'AtmosphericCO2
Pro-Inflammatory Anti-Inflammatory

81
this index, a magnesium deficit scores high in the list of pro-inflammatory stimuli (105).
Magnesium has many functions, some of them, not surprisingly, related to our energy
metabolism and immune system, e.g., it is the cation most intimately connected to ATP (95).
Indirect diet-related factors are an abnormal composition of the bacterial flora in the mouth
(106), gut (106, 107), and gingivae (108-110). Chronic stress (111, 112), (passive) smoking and
environmental pollution (77), insufficient physical activity (113-118) and insufficient sleep
(119-123) are also involved.
All of the above listed lifestyle factors exhibit interaction and are therefore difficult to study in
isolation. As an example, the bacterial flora may change secondary to the composition of our
diet. An inflammatory reaction might be at the basis of the observed relation between the
abnormal bacterial species in both our oral cavity and intestine and our serum HDL- and
LDL-cholesterol (106). Saturated fats may cause an inflammatory reaction especially when
they are combined with a carbohydrate-rich diet, notably carbohydrates with a high glycemic
index, and especially in subjects with the insulin resistance syndrome (124-128).
Mechanisms of lifestyle-induced inflammation
Diets high in refined starches, sugar, saturated and trans fats, and low in LCPω3, natural
antioxidants, and fiber from fruits and vegetables, have been shown to promote inflammation
(82-84, 129-131) (Table 1). As most chronic (inflammatory) diseases have been linked to diet,
modifying it could prevent, delay or even heal these diseases. Obviously. inflammation is an
essential process for survival, but our immune system should be carefully controlled to limit
the unavoidably associated collateral damage (132). For instance, wound healing and other
immune challenges become controlled in our body by a process coined by Serhan et al. (133-
135) as resoleomics, using metabolites produced from the LCP arachidonic acid (AA), EPA and
DHA (85, 133-136). However, our inflammatory and resolution genes operate nowadays in a
completely different environment than the one to which they became adapted through
mutation and natural selection. In most (if not all) chronic diseases typical of Western
societies, the inflammatory response is not concluded because of suboptimal or
supramaximal responses (137, 138).
It has been estimated that 10% of all deaths in the Netherlands are attributable to unfavorable
dietary composition and 5% to overweight. In this scenario, the major contributors to diet-
associated death were insufficient intakes of fish, vegetables and fruits, with less important
roles for too high intakes of saturated and trans fatty acids (139). The consumption of fish,
fruit and vegetables is considered too low in most Western countries (139-143). In the USA,
low dietary ω3 fatty acids and high dietary trans fatty acids may have accounted for up to
84,000 and 82,000 deaths, respectively, in 2005, while a low intake of fruit and vegetables

82
might have been responsible for 58,000 deaths (144). The Dutch (145) and the American Heart
Association (AHA) (146) dietary guidelines recommend to consume at least two servings of
fish per week (particularly fatty fish), but in 1998, the average fish consumption in The
Netherlands amounted to hardly 3 times per month (139). Only about 7% of the 9-13 year-old
Dutch children eat fish twice or more per week and 10% never eat fish (147). In the USA, the
estimated intake of fish in 2007 was about 0.7 kg per month, per person. More preoccupying
is the fact that the USA is considered the third largest consumer of seafood in the world (148,
149). Despite improvements of the fatty acid contents of food products, only 5% of the Dutch
population follows a diet with the recommended fatty acid pattern (139). Eating fish once
weekly was associated with a 15% lower risk of CVD death compared with a consumption of
less than once per month (150), while each 20 g/day increase in fish consumption was related
to a 7% lower risk of CVD mortality (151).
The current Dutch recommendation for adults is 200 g fruits and 200 g vegetables per day
(139), while in the USA, 4-5 servings of fruits and 4-5 servings of vegetables are recommended
in a 2,000 kcal diet (152). Between 1988 and 1998, the consumption of fruit and vegetables in
The Netherlands declined 15-20% and currently, less than 25% of the Dutch population follows
the recommendations regarding the consumption of fruit, vegetables and dietary fiber (139).
As an example, currently 99% and 95% of the 9-13 year old Dutch do not adhere to the advice
of consuming 150 g/day vegetables and 200 g/day fruits, respectively (147). Meta analyses of
prospective studies indicated that <3 vs. >5 servings of fruits and vegetable per day
correspond with a 17% reduction in coronary heart disease (153) and 26% reduction in stroke
(154), while the relation of low intakes with mouth, pharynx, esophagus, lung, stomach, colon
and rectum cancer is considered substantially convincing (155).
In view of the numerous nutrients present in our food and their many mechanisms of action
in the inflammatory response, we selected two nutrient classes, i.e. the LCP from fish (LCPω3;
notably EPA and DHA), and the antioxidants in fruit and vegetables, to illustrate the many
dietary components involved in our pro-inflammatory/anti-inflammatory balance. However,
before embarking into these nutrient classes, it should be emphasized that our food is in
reality composed of biological systems, such as meat, fish, vegetables and fruits, in which
nutrients obey to the balance that comes along with living material. Therefore, focusing on
specific, presently known mechanisms without sufficient knowledge of the many possible
interactions between the numerous nutrients in our food should be regarded as a serious
limitation. This is a reductionist approach, whereas system dynamics and holistic
approximations would be more appropriate.

83
Fatty acids and inflammation
The media are consistently reporting on advises to reduce fat consumption to avoid risks
associated with obesity, CVD, diabetes and other chronic diseases and conditions. Among the
macronutrients, fat does indeed contain the highest amount of energy per gram. However,
from a thermodynamic point of view, a ‘calorie is a calorie’ (156), implying that any
macronutrient consumed in disbalance with energy expenditure and thermogenesis might
cause obesity. A recent in-depth study revealed that ‘a calorie is not a calorie’ in a metabolic
sense, showing that isocaloric diets with different macronutrient compositions have different
effects on resting and total energy expenditure with decreasing energy expenditures in the
sequence low-fat diet<low-glycemic diet<very low-carbohydrate diet (157), and thereby
suggesting that the diet with the highest protein and fat content gives rise to the lowest
weight gain. However, whether the intake of fat per se and, as a matter of fact, any isolated
nutrient (158), can be held responsible for the epidemics of obesity, remains controversial and
counter intuitive (159-161). Moreover, it is becoming increasingly clear that about 10-25% of
obese subjects have little CVD and type 2 diabetes mellitus risk (a condition coined ‘healthy
obesity’) (162, 163), that lean physically unfit subjects have higher risk of CVD mortality than
obese, but fit, subjects (164), and that it is the quality and not the quantity of fat that conveys a
major health hazard (165). The type of dietary fat affects vital functions of the cell and its
ability to resist disfunction e.g. by influencing the interaction with receptors, by determining
basic membrane characteristics and by producing highly active lipid mediators (166, 167).
Saturated fat intake has been associated with inflammation (168, 169). However, the widely
promoted reduction of saturated fatty acids is increasingly criticized (170) and also the AHA
advisory to replace saturated fatty acids in favor of linoleic acid (LA) to 5-10 en% (171).
Insufficient intake of particular fatty acids is, on the other hand, likely to contribute to health
hazards, including increased risk of infection (172), dysregulated chronobiological activity
and impaired cognitive and sensory functions (especially in infants) (173). Among these
important fatty acids are the LCPω3 derived from fish, of which EPA and DHA are the most
important members. In 2003, the intake of EPA+DHA by adults in The Netherlands amounted
to approximately 90 mg/day (women 84 mg/day and men 103 mg/day) (174), while the
recommendation is 450 mg/day (175). This recommendation is based on an optimal effect in
preventing CVD (anti-arrhythmic effect), but there is good evidence that higher intakes may
convey additional favorable effects because of their anti-thrombotic properties and their
ability to reduce blood pressure, heart rate and triglyceride levels (131). It was calculated that
our Paleolithic ancestors living in the water-land ecosystem had daily intakes of 6-14 g
EPA+DHA (176), which correspond with the intakes by traditionally living Greenland Eskimos
(177), who, because of their low incidence of CVD, were at the basis of the research on the
beneficial effects of fish oil that started in the seventies (178-180).

84
Both EPA and DHA must be in balance with AA, which is the major LCPω6 derived from
meat, poultry, eggs (181-183) and also lean fish (184, 185). Each of these LCP may be
synthesized by desaturation, chain elongation and chain shortening from the parent
‘essential fatty acids’ LA (converted to AA) and alpha-linolenic acid (ALA) (converted to EPA
and DHA) (186), even though the production of EPA, and notably DHA, occurs with difficulty
in humans (187). Included among the symptoms of LA, LCPω3 and LCPω6 deficiencies are
fatigue, dermatological problems, immune problems, weakness, gastrointestinal disorders,
heart and circulatory problems, growth retardation, development or aggravation of breast and
prostate cancer, rheumatoid arthritis, asthma, preeclampsia, depression, schizophrenia and
ADHD (173, 188-190).
LCPω3 are implicated in many diseases and conditions, including CVD, psychiatric diseases,
pregnancy complications and suboptimal (neuro) development (86, 191-196). Moreover, a
growing number of studies indicate the protective effects of dietary LCPω3 on mood
symptoms, cognitive decline, depression (197, 198), Alzheimer’s disease (199) and, more
generally, impaired quality of life both in the elderly (200, 201) and younger (202) populations.
LCPω3 are involved in numerous processes including energy generation, growth, cell
division, transfer of oxygen from the air to the bloodstream, hemoglobin synthesis, normal
nerve impulse transmission and brain function. Many different mechanisms are operational:
LCPω3 mediate potent anti-inflammatory and insulin sensitizing effects through their
interaction with a membrane receptor named G-protein-coupled receptor 120 (GPR120) (203,
204); they act at the gene expressional level by binding to nuclear receptors, such as the
peroxisome proliferator activated receptors (PPARs) (205-207); and they modulate physical
and metabolic properties of membranes through their incorporation into phospholipids and
thereby impact on the formation of lipid rafts (134, 208, 209). Important common
denominators in each of these interactions seem to be their anti-inflammatory and metabolic
effects, again illustrating the intimate connection between the immune system and
metabolism (50, 51).
The modernization of food manufacturing, preservation processes and food choices have
dramatically altered the balance between LCPω3 and LCPω6 in the Western diet, notably by
increasing the intake of LA from refined vegetable oils and a concomitant decrease in the
intake of LCPω3 from fish (210, 211). It is gaining acceptance that it is not the amount of fat
but the balance between the different types of fatty acids that is important (211, 212). A high
ω6/ω3 fatty acid ratio has been demonstrated to have an inflammatory effect (86, 212, 213),
while a higher intake of LCPω3 in the form of EPA and DHA regulates the production of
inflammatory and resolving cytokines and decreases LA levels in both plasma phospholipids

85
and cell membranes (183, 214). The conversions of LA and ALA to AA and to EPA+DHA,
respectively, depend on the same enzymes in the desaturase and elongase cascade, with Δ6-
desaturase (FADS2) as a rate-limiting enzyme (215) that functions twice in the biosynthesis of
DHA (216). Increased consumption of ALA gives rise to an increased ALA/LA ratio and
EPA+DHA content in cell membranes that comes together with a reduction of the AA content
(216, 217), and thereby influences the balance between inflammation and its subsequent
resolution (Figure 6) (218-220). Conversely, a higher LA level in plasma phospholipids and cell
membranes emerges as a major factor responsible for incomplete resoleomics reactions and
the associated immune paralysis (214, 220, 221) (Figure 6), which is attributed to the
competitive inhibition of LA in the conversion of ALA to EPA and DHA and also to the
competition of LA in the incorporation of EPA and DHA into cellular phospholipids (183, 214,
216).
Figure 6. LCPω6 and LCPω3 postulated involvement in the inflammatory reaction in sepsis and
its subsequent resolution.
Sepsis causes a systemic inflammatory response giving rise to the ‘systemic inflammatory
response syndrome’ (SIRS). The inflammatory response is followed by a compensatory anti-
inflammatory response, which results in the ‘e’ (CARS), characterized by a weakened host
defense and augmented susceptibility to secondary infections. An inflammatory response
should not only be initiated, but also stopped to limit collateral damage produced by the
immune system and to prevent immune paralysis. LCPω6 (AA) are involved in the initiation
of the inflammatory reaction, while LCPω3 (EPA and DHA) are involved in its resolution (see
also Figure 7). a) A high LCPω6/LCPω3 ratio, e.g. low fish intake, intensifies the SIRS reaching
a state of hyper-inflammation, while the CARS leads to a state of immune paralysis. b) A low
LCPω6/LCPω3 ratio dampens both the SIRS and CARS, resulting in a more balanced immune
response and preventing hyper-inflammation and immune-paralysis. SIRS, systemic
inflammatory response syndrome; CARS, compensatory anti-inflammatory response
syndrome. Adapted from Mayer et al. (220) with permission from Wolters Kluwer Health.
LCPω3 and LCPω6 have distinct functions in the inflammatory reaction and its resolution. In
the first phase of the inflammatory process, the pro-inflammatory eicosanoids leukotrienes-

86
B4 (LTB4) and prostagladins-E2 and D2 (PGE2 and PGD2) (222, 223) are generated by
macrophages from their precursor AA with the help of the lipid-oxidizing enzyme
lipoxygenase-5 (LOX-5) and cyclo-oxygenase-2 (COX-2) (224-226). At the same time, PGE2
and/or PGD2, although initially pro-inflammatory, determine the switch to the next phase: the
resolution of the inflammation (227) via the so-called ‘eicosanoid-switch’. The production of
the LOX-5 enzyme becomes limited, while anti-inflammatory lipoxins (LXs) are produced
from AA through the activation of lipoxygenase-12 (LOX-12), lipoxygenase-15 (LOX-15) and
acetylated COX-2 (228). At the site of inflammation, LOX-12 produced by platelets converts
LTA4 to LXA4 and LXB4. Along with AA, both LOX-12 and -15 are involved in the biosynthesis
of specialized bioactive lipid mediators, coined resolvins, (neuro)protectins (135) and
maresins (229), which derive from EPA and DHA (Figure 7) (85, 134, 172). Several studies have
illustrated the involvement of these lipid mediators in vascular inflammation and
atherosclerosis (85, 228, 230, 231). They possess potent anti-inflammatory and pro-resolving
actions that stimulate the resolution of acute inflammation by reducing and/or limiting the
production of a large proportion of the pro-inflammatory cytokines produced by
macrophages. Furthermore, LXA4, protectin D1 and resolvin D1 stimulate the phagocytic
activity of macrophages toward apoptotic cells and inhibit inflammatory cell recruitment
(232, 233) thereby protecting tissues from excessive damage by the oxidative stress that
comes along with immune defense mechanisms and others. By their inhibitory actions on
the recruitment of inflammatory cells, they allow the resolution phase to set in (234) and
finish the inflammatory process with the return to homeostasis (136, 227).
Accordingly, LCPω3 given at doses of hundreds of milligrams to grams per day, exhibits
beneficial actions in many inflammatory diseases (88, 190, 194, 235, 236). For example, DHA
has been shown to suppress NFκB activation and COX-2 expression in a macrophage cell line
(168, 237). Different studies demonstrated the nutrigenetic modulation of the 12/15-LOX by
providing endogenous anti-inflammatory signals and protection during the progression of
atherogenesis (231, 238, 239), which seem to be totally annulled in the presence of Western
diet induced hyperlipidemia. As some eicosanoids regulate the production of inflammatory
cytokines (85, 134, 135) an LCPω3-induced decrease in pro-inflammatory eicosanoid
production might affect the production of pro-inflammatory cytokines. Equally important is
the observation that LCPω3 also modulate the activation of transcription factors involved in
the expression of inflammatory genes (e.g. NFκB, phosphatidylinositol 3-kinase (PI3K)) (240).
Hence, a high fish consumption, and especially fatty fish, rich in EPA and DHA, seems of
crucial importance in the primary and secondary prevention of (Western) chronic diseases
(241, 242), although it should be emphasized that fish is not a synonym of fish oil and also that
insufficient fish consumption is certainly not the only factor involved in the pro-
inflammatory Western lifestyle (Table 1).

87
Figure 7. Biosynthesis of inflammatory and resolving lipid mediators.
AA is released from membrane phospholipids by phospholipase A2 (PLA2) and metabolized by COXs or 5-LO to form
inflammatory mediators, such as prostaglandins and leukotrienes. During the process of resolution, there is a ‘switch’ from
the biosynthesis of inflammatory mediators to the formation of lipid derivatives with anti-inflammatory and pro-resolving
properties, including lipoxins and 15-d-PGJ2. EPA and DHA are converted to potent anti-inflammatory and pro-resolving
lipid mediators like resolvins (E1 and D1) and protectins. ASA, acetylsalicylic acid, CYP450, cytochrome P450, COX-1, cyclo-
oxygenasa-1, COX-2, cyclo-oxygenasa-2; 5-LO, 5-lipo-oxygenase; 12-LO, 12-lipo-oxygenase; 15-LO, 15-lipo-oxygenase;
PGE2, prostagladin-E2; PGD2, prostaglandin-D2; LTs, leukotrienes; 15d-PGJ2,15-deoxy-delta-12,14-prostaglandin J2; 15-epi-
LXA4 , 15-epi-lipoxin A4 ; LXA4 , lipoxin A4. Adapted from González-Périz and Clària (243) with permission.
Role of the antioxidant network
The largest contributor to mortality and morbidity worldwide is age-related, non
communicable disease, including cancer, CVD, neurodegenerative diseases and diabetes
(244). Even though these are multi-factorial diseases with many pathophysiological
mechanisms, a common finding is oxidation-induced damage through oxidative stress (245,
246). Appropriate antioxidant intake has been proposed as a solution to counteract the
deleterious effects of reactive oxygen species (ROS; e.g. hydrogen peroxide, hypochlorite
anion, superoxide anion and hydroxyl radical), with substantial evidence upholding the
contention that: a diet rich in natural antioxidants supports health (104, 246), is associated
with lower oxidative stress and inflammation (77, 103, 140), and is therefore associated with
lower risk of cancer, CVD, Alzheimer’s disease, cataracts, and some of the functional declines
associated with aging (247-251).

88
Molecular oxygen is essential to aerobic life and, at the same time, an oxidizing agent,
meaning that it can gain electrons from various sources that thereby become ‘oxidized’, while
oxygen itself becomes ‘reduced’ (252, 253). In general terms, an antioxidant is ‘anything that
can prevent or inhibit oxidation’ and these are therefore needed in all biological systems
exposed to oxygen (252). The emergence of oxygenic photosynthesis and subsequent
changes in atmospheric environment (254) forced organisms to develop protective
mechanisms against oxygen’s toxic effects (255). Change is implicit to evolution and
evolution results in adaptation to change (256). As a result, many enzymatic reactions central
to anoxic metabolism were effectively replaced in aerobic organisms and antioxidant defense
mechanisms evolved (257, 258). The continuous exposure to free radicals from a variety of
sources led organisms to develop a series of systems (259) acting as a balanced and
coordinated network where each one relies on the action of the others (260, 261).
Oxidative stress occurs when there is a change in this balance in favor of ROS (262) that may
occur under several circumstances, ranging from malnutrition to disease (263, 264). Damage
by oxidation of lipids (262, 265, 266), nucleic acids and proteins changes the structure and
function of key cellular constituents resulting in the activation of the NFκB pathway,
promoting inflammation, mutation, cell damage and even death (252, 260, 267), and is
thereby believed to underlie the deleterious changes in aging and age-related diseases (102,
244). The prevention and/or inhibition of oxidation can be achieved by several types of
specialized antioxidant mechanisms depicted in Table 2 (260). Our antioxidant system is
composed of two networks (Figure 8), namely, the antioxidant network of non-enzymatic
antioxidants that we obtain mostly via the diet (268), and the antioxidant enzymes that we
synthesize ourselves and that carry metal ions for their appropriate functioning in ROS
clearance. Members of the non-enzymatic antioxidants are e.g. ascorbic acid (vitamin C),
alpha-tocopherol (vitamin E), carotenoids, and the polyphenols (269, 270). For instance,
quercetin, one of the most common flavonoids in the human diet, and resveratrol, a well-
known stilbenoid present mostly in berries and the skin of red grapes, have demonstrated
favorable effects on glucose metabolism by attenuating TNFα-mediated inflammation and
insulin resistance in primary human adipocytes (271). Typical examples of the antioxidant
enzymes are superoxide dismutase (SOD), glutathione peroxidase (GPx) and catalase (CAT)
(252).

89
Table 2. Types of antioxidant action.
Action Examples
Prevention
Protein binding/inactivation of metal
ions
Transferrin, ferritin,
ceruloplasmin, albumin
Enzymatic
Neutralization
Specific channelling of ROS into
harmless products
SOD, catalase, glutathione
peroxidase
Scavenging
Sacrificial interaction with ROS by
expendable (recyclable or replaceble)
substrates
Ascorbic acid, alpha
tocopherol, uric acid,
glutathione
Quenching
Absorption of electrons and/or energy
α-tocopherol, β-carotene,
astaxanthin
ROS, reactive oxygen species; SOD, superoxide dismutase. Adapted from Benzie (260).
While the prevention of oxidative stress by enhancing the antioxidant defense mechanisms
may diminish the production of inflammatory mediators and thereby slow aging and lower
risk of certain diseases (102, 245, 249), it should at the same time be appreciated that ROS also
exert essential metabolic and immune functions. For example, oxidative phosphorylation is
based on electron transport (272), which renders free radicals’ inevitable byproducts of
mitochondrial metabolism (273). Mitochondrial oxidants may function as signaling molecules
in the communication between the mitochondria and the cytosol (273), while TNFα-induced
apoptosis may involve mitochondria-derived ROS (274). The innate immune system kills
microbes by means of the respiratory burst (275). A certain level of ROS may also be essential
to trigger antioxidant responses (276). Repeated exposure to sublethal stress has been
proposed to result in enhanced stress resistance and increased survival rates, which in the
dose-response curve is better known as hormesis (277). Intracellular ROS may stimulate gene
expression of antioxidant and immunoreactive proteins (278), while SOD may become
upregulated in chronic exercise through the binding of NFκB to the SOD promoter (279, 280).
Consequently, certain antioxidants may inhibit mitochondrial biogenesis, interfere with the
hormetic effects of ROS (281, 282) or have other adverse effects. Effective prevention of ROS
formation and their removal may therefore upset energy metabolism, cell signaling pathways
and the immune system, and thereby paradoxically increase the risk of chronic disease (283).
Moreover, any antioxidant is also a potential pro-oxidant because in its scavenging action it
gains an extra electron that can initiate a new radical reaction when transferred to an
acceptor, either spontaneously or upon decomposition (284, 285). Possibly through its

90
prooxidant action or other mechanisms (286), meta-analyses of studies with β-carotene
dosages above 20 mg/day have shown increased risk of lung cancer in the total population,
smokers and asbestos workers; and of stomach cancer in smokers and asbestos workers (287).
Analogously, oral antioxidants to limit muscle damage following exercise training may be
detrimental to health and performance (288), while β-carotene, vitamin A and vitamin E
supplements have been connected with higher risk of all-cause mortality (289), although the
outcome of the latter meta-analysis has been contested (290). Moreover, not all antioxidants
are created equal. Astaxanthin, a carotenoid from the land-water ecosystem, does not appear
to exhibit pro-oxidant properties (291) when supplemented alone, even at high doses (292),
and has been shown to decrease oxidative stress and inflammation in various circumstances
(266, 293).
Chronic inflammation results in the chronic generation of free radicals, which may cause
collateral damage and stimulate signaling and transcription factors associated with chronic
diseases (294, 295). The hypothesis that dietary antioxidants lower the risk of chronic diseases
has been developed from epidemiological studies consistently showing that consumption of
fruit and vegetables is strongly associated with a reduced risk of these diseases (104, 248, 250).
Regular consumption of green tea (296) and red wine (103, 297), both rich in polyphenols,
decreases DNA damage, and the same holds for the kiwifruit (298) and watercress (299), both
harboring high amounts of carotenoids and vitamin C. On a calorie basis, fruits and
vegetables are not only richer in many vitamins and minerals, when compared with cereals,
meat or fish, but also in antioxidants (300). These may collectively be responsible of the
aforementioned protection of fruits and vegetables in chronic diseases, including CVD (248)
and cancer (249). Plants harbor similar defense mechanisms as animals for protection against
ROS (301). Some of their antioxidants are part of their arsenal of ‘secondary metabolites’,
defined as those organic compounds that are not directly involved in normal growth,
development and reproduction, but in long term survival and fecundity (302). The plant
secondary metabolites are largely involved in the chemical defense against herbivores,
microbes, viruses and competing plants, in signaling and in nitrogen storage (303); and some
(e.g. polyphenols, carotenoids) also serve functions in the protection against ROS. The
underlying metabolic pathways towards secondary metabolites lead to a series of related
compounds that are usually composed of few major metabolites and several minor
components differing in the position of their functional groups (303). Animals consuming
fruits and vegetables may employ these plant secondary metabolite networks for their own
purposes, including maintenance of inflammatory/anti-inflammatory balance, cancer
chemoprevention and protection against ROS (303).

91
Figure 8. Antioxidant defense mechanisms.
An overview of the antioxidant system present in the human body. Various types of antioxidant systems have developed
through time, reflecting different selection pressures. Different forms have developed for the same purpose, for example,
SODs, peroxidases and GPx are important members of the antioxidant enzyme capacity group. Tocopherols and ascorbic
acid, as representatives of the antioxidant network, are manufactured only in plants, but are needed by animals. Ascorbic
acid is an essential antioxidant, but cannot be synthesized by homo sapiens. In humans, therefore, antioxidant defense
against toxic oxygen intermediates comprises an intricate network which is heavily influenced by nutrition. GR, glutathione
reductase; GSG, reduced glutathione; GSH-Px, glutathione peroxidase; GSSG, oxidized glutathione; GST, glutathione-S-
transferase; MSR, methionine sulphoxide reductase; PUFA, polyunsaturated fatty acids; S-AA, sulphur amino-acids; SH-
proteins, sulphydryl proteins; SOD, superoxide dismutase; Fe Cu, transition metal-catalysed oxidant damage to
biomolecules. Adapted from Strain (304) with permission from Cambridge University Press.
In view of the yet poorly understood complex antioxidant networks composed of many
compounds, it seems improbable to find a single ‘magic bullet’ to prevent and treat chronic
diseases associated with ROS. Protective effects of fruits and vegetables may originate from
their numerous phytochemicals working in concert (305) and from many different
mechanisms of action that are not solely related to ROS. A purified phytochemical may not
have the same health benefit as that phytochemical present in whole foods or a mixture of
foods (250, 306). In biological systems, toxins may become nutrients, while nutrients may
become toxic in other situations (268), for example when disbalanced with other nutrients.
Rather than translating our food into an assembly of nutrients where each has to prove its

92
health benefits by scientific means, the objective should be to embrace a eucaloric diet that
provides the adequate amount of nutrients from whole foods to maintain our body
homeostasis. ‘Adequacy’ may in this sense be translated into causing an optimal interaction
between our diet (and our lifestyle in general) with our genome, that is: nurture in balance
with nature.
Evolutionary nutrition vs. randomized controlled trials
Coherence between lifestyle factors, including the composition of our diet, is quite obvious
from an evolutionary point of view. After all, there was first an environment, and from this
environment originated a genome that was adapted to that environment: it is the substrate
(environment) that selects the organism, not viceversa. This is exactly what Darwin meant
with ‘conditions of existence’, as the most important driving force in evolution. In other
words, our only slowly changing genome is indissolubly linked to a certain environment and
lifestyle. However, we have changed this environment since the agricultural revolution and
continue to do so with still increasing paste. The resulting conflict does not generate acute
toxicity, but acts as an assassin in the long term. Probably, the conflict does not exert much
selection pressure either, because its associated mortality occurs mainly after reproductive
age.
To solve the conflict, it is virtually impossible to study all of the introduced errors in our
lifestyle (Table 1) in isolation, according to the reigning paradigm of EBM (307). EBM is widely
confused with the results of RCTs and preferably the meta-analysis thereof (308, 309). This
paradigm, originally designed for objective evaluation of medical treatments and drugs in
particular, and named in nutrition research ‘Evidence Based Nutrition’ (EBN); is at present
misused by food scientists and Health and Nutrition advisory boards. In contrast to drugs, this
(expensive) RCT paradigm usually lends itself poorly for the study of single nutrients with
meaningful outcomes (308). For each nutrient, we are dealing with poorly researched dose-
response relationships, multiple mechanisms of action, small effects causing pathology in the
long-term, numerous interactions, ethical limitations regarding the choice of intervention
and control groups, and the inability to patent its outcomes (309). The RCT criteria are
moreover inconsistently applied in the current development of nutritional recommendations.
For example, there is no RCT-supported evidence for the saturated fat hypothesis (170), and
also not for the trans fatty acids, while such an approach is considered mandatory for the
adjustment of the vitamin D nutritional standards (310-312). Incidentally, there was also no
RCT prior to the introduction of trans fatty acids showing that they could be consumed
without adverse effects on the long term. However, there is an RCT on the effects of smoking
cessation, which showed an equal mortality among the quitters (313, 314). The meta-analyses
of RCTs studying the influence of LCP on brain development are negative (315-318). However,

93
recommendations for their addition to infant formulas have been issued (196), probably
because nobody wants to take chances with the brains of our offspring. By applying EBM in a
rigorous manner and by merely taking a view from the ‘precautionary principle’ (i.e. zero risk 5) this well meant concept has become a burden in the nutritional science, that calls for
replacement by appropriate risk-cost-benefit analyses such as e.g. performed for vitamin D
(319).
Our diet is composed of millions of substances that are part of a biological network. In fact,
we eat ‘biological systems’ like a banana, a fish or a piece of meat. There is a connection
between the various nutrients in these systems. In other words,, there is a balance and an
interaction that is part of a living organism. This balance can be found in the reconstruction
of our Paleolithic diet, and various attempts for this reconstruction have already been made
(28, 131, 320-322). Preliminary results of interventions with a Paleolithic diet are utterly
positive (for a review see (323)). For example, in an indeed uncontrolled study with non-obese
sedentary healthy subjects, an eucaloric Paleolithic diet resulted within 10 days in beneficial
effects on three out of the four symptoms of the metabolic syndrome, i.e. blood pressure,
dyslipidemia and glucose homeostasis. The fourth symptom, overweight/obesity, was
deliberately not changed to prevent the attribution of any beneficial changes to weight loss
(324).
Nurture, not nature
Less than 5% of our diseases can primarily be ascribed to heritable genetic factors (325, 326).
‘Genome wide association studies’ (GWAS) will not make this figure change; not even if the
number of patients and controls are further increased. As it could have been predicted from
evolution, these GWAS identify only a few genes that are associated with typically Western
diseases. Moreover, the so far identified genes merely convey low risks. In one of these
disappointing GWAS, where 14,000 patients with seven major typically Western diseases and
3.000 controls were studied, it was concluded that: ‘... for any given trait, there will be few (if
any) large effects, a handful of modest effects and a substantial number of genes generating
small or very small increases in disease risk‘ (327). The differences in genetic susceptibility to
environmental factors is widely confused with a primary genetic origin of Western disease.
Environmental factors may mimic genetic heritability, especially when the exposure has
become widespread, As clearly explained by Rose (328): "If everyone smoked 20 cigarettes a
day, then clinical, case-control and cohort studies alike would lead us to conclude that lung
cancer was a genetic disease; and in one sense that would be true, since if everyone is
5 The precautionary principle is a moral and political principle stating that, if an intervention or policy may cause serious or irreversible damage to society or the environment, the burden of proof lies with the proponents of the intervention or the measure if there is no scientific consensus on the future damage. The precautionary principle is particularly applicable in health care and environment; in both cases we deal with complex systems in which interventions result in unpredictable effects (source: Wikipedia).

94
exposed to the necessary agent, then the distribution of cases is wholly determined by
individual susceptibility”. In other words: ‘disease susceptibility genes’ is a misnomer from an
evolutionary point of view.
Most of the currently demonstrated polymorphisms associated with typically Western
diseases already existed when homo sapiens emerged about 160,000 years ago in East-Africa.
After all, the largest inter-individual genetic variation can be found between individuals
belonging to a single population (93-95% of the total genetic variation), and only little genetic
variation is on the account of differences between populations belonging to a single race (2%)
and between the 5 races (3-5%) (329). The allele that, according to current knowledge, is linked
with the highest penetrance of type 2 diabetes mellitus in the general population (with
Western lifestyle!) conveys 46% higher relative risk (RR=1.46) (330). In contrast, a woman with
a body mass index (BMI) of 35+ kg/m2 has a one hundred-fold higher risk (RR=100) of diabetes
mellitus type 2 (331), which translates into a 9,900% higher relative risk. ‘Genetic’ diseases with
relative risks below 1.5 have no practical value in Public Health. They are only important to
our understanding of the etiology of the concerning disease and for drug development (326),
which is part of Health Care.
Between 70 and 90% of the cases of type 2 diabetes mellitus, CVD and colon cancer can be
prevented by paying more attention to nutrition, smoking, overweight and lack of physical
activity (325). Hemminki et al. (326) stated that ‘if the Western population was to live in the
same conditions as the populations of developing countries, the risk of cancer would
decrease by 90%, provided that viral infections and mycotoxin exposures could be avoided’.
The popular counter argument that people in developing countries have (on average!) shorter
life spans is not valid. The reason that we (on average!) live longer in Western societies, is
mainly due to the strong reduction of infectious diseases (particularly in childhood), famine
and violence (332, 333), and also in part on the account of Health Care. However, together
with our increasing life expectancy, there is a decrease in the number of years without
chronic disease (334).
Conclusions
It has become clear that most, if not all, typically Western chronic illnesses find their primary
cause in an unhealthy lifestyle and that systemic low grade inflammation is a common
denominator. From an evolutionary point of view, the current conflict between environment
and our Paleolithic genome traces back to our brain growth and the ensuing intimate
relationship between inflammation and metabolism. The present disbalance between
inflammatory and anti-inflammatory stimuli does not originate from a single cause and can
consequently also not be solved by a single ‘magic bullet’. Resolution of the conflict between

95
environment and our ancient genome might be the only effective manner to arrive at
‘healthy aging’ and to achieve this objective we might have to return to the lifestyle of the
Paleolithic era according to the culture of the 21st century (16, 322).

96
References 1. Reaven GM. The insulin resistance syndrome: Definition and dietary approaches to
treatment. Annu Rev Nutr. 2005;25:391-406.
2. Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: Principles of pathogenesis
and therapy. Lancet. 2005 04/09;365(9467):1333-46.
3. Straub RH. Concepts of evolutionary medicine and energy regulation contribute to the
etiology of systemic chronic inflammatory diseases. Brain Behav Immun. 2011
01;25(1):1-5.
4. Straub RH, Cutolo M, Buttgereit F, Pongratz G. Energy regulation and neuroendocrine-
immune control in chronic inflammatory diseases. J Intern Med. 2010 06;267(6):543-60.
5. Murch O, Collin M, Hinds CJ, Thiemermann C. Lipoproteins in inflammation and
sepsis. I. Basic science. Intensive Care Med. 2007 01;33(1):13-24.
6. Wendel M, Paul R, Heller AR. Lipoproteins in inflammation and sepsis. II. Clinical
aspects. Intensive Care Med. 2007 01;33(1):25-35.
7. Van Leeuwen HJ, Heezius ECJM, Dallinga GM, van Strijp JAG, Verhoef J, van Kessel
KPM. Lipoprotein metabolism in patients with severe sepsis. Crit Care Med. 2003
05;31(5):1359-66.
8. van Leeuwen HJ, van Beek AP, Dallinga-Thie GM, van Strijp JA, Verhoef J, van Kessel
KP. The role of high density lipoprotein in sepsis. Neth J Med. 2001 09;59(3):102-10.
9. Van Amersfoort ES, Van Berkel TJC, Kuiper J. Receptors, mediators, and mechanisms
involved in bacterial sepsis and septic shock. Clin Microbiol Rev. 2003 07;16(3):379-414.
10. Hudgins LC, Parker TS, Levine DM, Gordon BR, Saal SD, Jiang X, et al. A single
intravenous dose of endotoxin rapidly alters serum lipoproteins and lipid transfer
proteins in normal volunteers. J Lipid Res. 2003 08;44(8):1489-98.
11. Leonard WR, Robertson ML, Snodgrass JJ, Kuzawa CW. Metabolic correlates of hominid
brain evolution. Comparative Biochemistry and Physiology - Part A: Molecular &
Integrative Physiology. 2003 9;136(1):5-15.
12. Broadhurst CL, Cunnane SC, Crawford MA. Rift valley lake fish and shellfish provided
brain-specific nutrition for early homo. Br J Nutr. 1998 01;79(1):3-21.
13. Broadhurst CL, Wang Y, Crawford MA, Cunnane SC, Parkington JE, Schmidt WF. Brain-
specific lipids from marine, lacustrine, or terrestrial food resources: Potential impact on
early African homo sapiens. Comp Biochem Physiol B, Biochem Mol Biol. 2002
04;131(4):653-73.
14. Gibbons A. Becoming human. In search of the first hominids. Science. 2002 Feb
15;295(5558):1214-9.
15. Muskiet FAJ, Kuipers RS. Lessons from shore-based hunter-gatherer diets in east africa.
In: Human Brain Evolution. John Wiley & Sons, Inc.; 2010;. p. 77-104.

97
16. Cordain L, Eaton SB, Sebastian A, Mann N, Lindeberg S, Watkins BA, et al. Origins and
evolution of the Western diet: Health implications for the 21st century. Am J Clin Nutr.
2005 02;81(2):341-54.
17. Muskiet FAJ. Evolutionaire geneeskunde. U bent wat u eet, maar u moet weer worden
wat u at. Ned Tijdschr Klin Chem Labgeneesk. 2005:163-84.
18. Brown P, Sutikna T, Morwood MJ, Soejono RP, Jatmiko , Saptomo EW, et al. A new
small-bodied hominin from the late pleistocene of Flores, Indonesia. Nature. 2004
10/28;431(7012):1055-61.
19. Aiello LC, Wheeler P. The expensive-tissue hypothesis: The brain and the digestive
system in human and primate evolution. Curr Anthropol. 1995 Apr.;36(2):pp. 199-221.
20. Leonard WR, Snodgrass JJ, Robertson ML. Effects of brain evolution on human
nutrition and metabolism. Annu Rev Nutr. 2007;27:311-27.
21. Cunnane SC, Crawford MA. Survival of the fattest: Fat babies were the key to evolution
of the large human brain. Comp Biochem Physiol, Part A Mol Integr Physiol. 2003
09;136(1):17-26.
22. Kaindl AM, Passemard S, Kumar P, Kraemer N, Issa L, Zwirner A, et al. Many roads lead
to primary autosomal recessive microcephaly. Prog Neurobiol. 2010 03;90(3):363-83.
23. Williams CA, Dagli A, Battaglia A. Genetic disorders associated with macrocephaly. Am J
Med Genet A. 2008 08/01;146A(15):2023-37.
24. Abzhanov A, Kuo WP, Hartmann C, Grant BR, Grant PR, Tabin CJ. The calmodulin
pathway and evolution of elongated beak morphology in Darwin's finches. Nature.
2006 08/03;442(7102):563-7.
25. Patel NH. Evolutionary biology: How to build a longer beak. Nature. 2006 Aug
3;442(7102):515-6.
26. Fontana L, Klein S, Holloszy JO. Effects of long-term calorie restriction and endurance
exercise on glucose tolerance, insulin action, and adipokine production. Age (Dordr).
2010 03;32(1):97-9108.
27. Peters A. The selfish brain: Competition for energy resources. Am J Hum Biol.
2011;23(1):29-34.
28. Yajnik CS. Obesity epidemic in india: Intrauterine origins? Proc Nutr Soc. 2004
08;63(3):387-96.
29. Azevedo FAC, Carvalho LRB, Grinberg LT, Farfel JM, Ferretti REL, Leite REP, et al. Equal
numbers of neuronal and nonneuronal cells make the human brain an isometrically
scaled-up primate brain. J Comp Neurol. 2009 04/10;513(5):532-41.
30. Herculano-Houzel S. Coordinated scaling of cortical and cerebellar numbers of
neurons. Front Neuroanat. 2010 Mar 10;4:12.
31. Gabi M, Collins CE, Wong P, Torres LB, Kaas JH, Herculano-Houzel S. Cellular scaling
rules for the brains of an extended number of primate species. Brain Behav Evol.
2010;76(1):32-44.

98
32. Herculano-Houzel S. Scaling of brain metabolism with a fixed energy budget per
neuron: Implications for neuronal activity, plasticity and evolution. PLoS One. 2011 Mar
1;6(3):e17514.
33. Deaner RO, Isler K, Burkart J, van Schaik C. Overall brain size, and not encephalization
quotient, best predicts cognitive ability across non-human primates. Brain Behav Evol.
2007;70(2):115-24.
34. Roth G, Dicke U. Evolution of the brain and intelligence. Trends Cogn Sci (Regul Ed ).
2005 05;9(5):250-7.
35. Dinuzzo M, Mangia S, Maraviglia B, Giove F. The role of astrocytic glycogen in
supporting the energetics of neuronal activity. Neurochem Res. 2012.
36. Colagiuri S, Brand Miller J. The 'carnivore connection'-- Evolutionary aspects of insulin
resistance. Eur J Clin Nutr. 2002 Mar;56 Suppl 1:S30-5.
37. Cahill GF. Fuel metabolism in starvation. Annu Rev Nutr. 2006;26:1-22.
38. Hadden DR, McLaughlin C. Normal and abnormal maternal metabolism during
pregnancy. Semin Fetal Neonatal Med. 2009 04;14(2):66-71.
39. Calder PC, Dimitriadis G, Newsholme P. Glucose metabolism in lymphoid and
inflammatory cells and tissues. Curr Opin Clin Nutr Metab Care. 2007 07;10(4):531-40.
40. Gluckman PD, Hanson MA. The consequences of being born small - An adaptive
perspective. Horm Res. 2006;65 Suppl 3:5-14.
41. Gluckman P, Hanson M, Morton SMB, Pinal C. Life-long echoes--a critical analysis of
the developmental origins of adult disease model. Biol Neonate. 2005;87(2):127-39.
42. Fall CHD. The fetal and early life origins of adult disease. Indian Pediatr. 2003
05;40(5):480-502.
43. Lumbers ER, Yu ZY, Gibson KJ. The selfish brain and the Barker hypothesis. Clin Exp
Pharmacol Physiol. 2001 11;28(11):942-7.
44. Koffler M, Kisch ES. Starvation diet and very-low-calorie diets may induce insulin
resistance and overt diabetes mellitus. J Diabetes Complications. 1996;10(2):109-12.
45. Kirwan JP, Hauguel-De Mouzon S, Lepercq J, Challier J, Huston-Presley L, Friedman
JE, et al. TNF-alpha is a predictor of insulin resistance in human pregnancy. Diabetes.
2002 07;51(7):2207-13.
46. Lain KY, Catalano PM. Metabolic changes in pregnancy. Clin Obstet Gynecol. 2007
12;50(4):938-48.
47. Hauguel-de Mouzon S, Guerre-Millo M. The placenta cytokine network and
inflammatory signals. Placenta. 2006 Aug;27(8):794-8.
48. de Luca C, Olefsky JM. Inflammation and insulin resistance. FEBS Lett. 2008
1/9;582(1):97-105.
49. Horng T, Hotamisligil GS. Linking the inflammasome to obesity-related disease. Nat
Med. 2011 02;17(2):164-5.

99
50. Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006 Dec
14;444(7121):860-7.
51. Hotamisligil GS, Erbay E. Nutrient sensing and inflammation in metabolic diseases. Nat
Rev Immunol. 2008 Dec;8(12):923-34.
52. Lee JY, Zhao L, Hwang DH. Modulation of pattern recognition receptor-mediated
inflammation and risk of chronic diseases by dietary fatty acids. Nutr Rev. 2010
01;68(1):38-61.
53. Biddinger SB, Kahn CR. From mice to men: Insights into the insulin resistance
syndromes. Annu Rev Physiol. 2006;68:123-58.
54. Sarafidis PA, Bakris GL. The antinatriuretic effect of insulin: An unappreciated
mechanism for hypertension associated with insulin resistance? Am J Nephrol.
2007;27(1):44-54.
55. Warner MH, Beckett GJ. Mechanisms behind the non-thyroidal illness syndrome: An
update. J Endocrinol. 2010 04;205(1):1-13.
56. Kwakkel J, Fliers E, Boelen A. Illness-induced changes in thyroid hormone metabolism:
Focus on the tissue level. Neth J Med. 2011 05;69(5):224-8.
57. Dentice M, Salvatore D. Deiodinases: The balance of thyroid hormone: Local impact of
thyroid hormone inactivation. J Endocrinol. 2011 06;209(3):273-82.
58. Roos A, Bakker SJ, Links TP, Gans RO, Wolffenbuttel BH. Thyroid function is associated
with components of the metabolic syndrome in euthyroid subjects. J Clin Endocrinol
Metab. 2007 Feb;92(2):491-6.
59. Ruhla S, Weickert MO, Arafat AM, Osterhoff M, Isken F, Spranger J, et al. A high normal
TSH is associated with the metabolic syndrome. Clin Endocrinol (Oxf). 2010
May;72(5):696-701.
60. Tuzcu A, Bahceci M, Gokalp D, Tuzun Y, Gunes K. Subclinical hypothyroidism may be
associated with elevated high-sensitive c-reactive protein (low grade inflammation)
and fasting hyperinsulinemia. Endocr J. 2005 02;52(1):89-94.
61. Surks MI, Ocampo E. Subclinical thyroid disease. Am J Med. 1996 02;100(2):217-23.
62. Jones DD, May KE, Geraci SA. Subclinical thyroid disease. Am J Med. 2010
06;123(6):502-4.
63. Ruhla S, Arafat AM, Weickert MO, Osterhoff M, Isken F, Spranger J, et al. T3/rT3-ratio is
associated with insulin resistance independent of TSH. Horm Metab Res. 2011
Feb;43(2):130-4.
64. Boelen A, Wiersinga WM, Fliers E. Fasting-induced changes in the hypothalamus-
pituitary-thyroid axis. Thyroid. 2008 Feb;18(2):123-9.
65. Esteve E, Ricart W, Fernandez-Real JM. Dyslipidemia and inflammation: An
evolutionary conserved mechanism. Clin Nutr. 2005 02;24(1):16-31.
66. Khovidhunkit W, Kim M, Memon RA, Shigenaga JK, Moser AH, Feingold KR, et al.
Thematic review series: The pathogenesis of atherosclerosis. effects of infection and

100
inflammation on lipid and lipoprotein metabolism mechanisms and consequences to
the host. Journal of Lipid Research. 2004 July 01;45(7):1169-96.
67. Ravnskov U. High cholesterol may protect against infections and atherosclerosis. QJM.
2003 12;96(12):927-34.
68. Rauchhaus M, Coats AJ, Anker SD. The endotoxin-lipoprotein hypothesis. Lancet. 2000
09/09;356(9233):930-3.
69. Kitchens RL, Thompson PA, Munford RS, O'Keefe GE. Acute inflammation and infection
maintain circulating phospholipid levels and enhance lipopolysaccharide binding to
plasma lipoproteins. J Lipid Res. 2003 12;44(12):2339-48.
70. Leigh SEA, Foster AH, Whittall RA, Hubbart CS, Humphries SE. Update and analysis of
the University College London. Low density lipoprotein receptor familial
hypercholesterolemia database. Ann Hum Genet. 2008 07;72:485-98.
71. Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic causes of monogenic
heterozygous familial hypercholesterolemia: A HuGE prevalence review. Am J
Epidemiol. 2004 Sep 1;160(5):407-20.
72. Sijbrands EJ, Westendorp RG, Defesche JC, de Meier PH, Smelt AH, Kastelein JJ.
Mortality over two centuries in large pedigree with familial hypercholesterolaemia:
Family tree mortality study. BMJ. 2001 04/28;322(7293):1019-23.
73. Netea MG, Demacker PN, Kullberg BJ, Boerman OC, Verschueren I, Stalenhoef AF, et al.
Low-density lipoprotein receptor-deficient mice are protected against lethal
endotoxemia and severe Gram-negative infections. J Clin Invest. 1996 03/15;97(6):1366-
72.
74. Solano MP, Goldberg RB. Management of dyslipidemia in diabetes. Cardiol Rev.
2006;14(3):125-35.
75. Feingold KR, Grunfeld C. The acute phase response inhibits reverse cholesterol
transport. J Lipid Res. 2010 Apr;51(4):682-4.
76. Rogers J. The inflammatory response in Alzheimer's disease. J Periodontol. 2008
08;79(8):1535-43.
77. Egger G, Dixon J. Non-nutrient causes of low-grade, systemic inflammation: Support
for a 'canary in the mineshaft' view of obesity in chronic disease. Obesity Reviews.
2011;12(5):339-45.
78. Galland L. Diet and inflammation. Nutr Clin Pract. 2010 Dec;25(6):634-40.
79. Anand P, Kunnumakkara AB, Kunnumakara AB, Sundaram C, Harikumar KB, Tharakan
ST, et al. Cancer is a preventable disease that requires major lifestyle changes. Pharm
Res. 2008 09;25(9):2097-116.
80. Egger G, Dixon J. Inflammatory effects of nutritional stimuli: Further support for the
need for a big picture approach to tackling obesity and chronic disease. Obesity
Reviews. 2010;11(2):137-49.

101
81. Egger G, Dixon J. Obesity and chronic disease: Always offender or often just
accomplice? Br J Nutr. 2009 10;102(8):1238-42.
82. Jimenez-Gomez Y, Lopez-Miranda J, Blanco-Colio LM, Marin C, Perez-Martinez P,
Ruano J, et al. Olive oil and walnut breakfasts reduce the postprandial inflammatory
response in mononuclear cells compared with a butter breakfast in healthy men.
Atherosclerosis. 2009 Jun;204(2):e70-6.
83. Mozaffarian D. Trans fatty acids - Effects on systemic inflammation and endothelial
function. Atheroscler Suppl. 2006 May;7(2):29-32.
84. Mozaffarian D, Aro A, Willett WC. Health effects of trans-fatty acids: Experimental and
observational evidence. Eur J Clin Nutr. 2009 05;63 Suppl 2:5-21.
85. Serhan CN, Chiang N. Endogenous pro-resolving and anti-inflammatory lipid
mediators: A new pharmacologic genus. Br J Pharmacol. 2008 03;153 Suppl 1:200-15.
86. Simopoulos AP. The importance of the omega-6/omega-3 fatty acid ratio in
cardiovascular disease and other chronic diseases. Exp Biol Med (Maywood). 2008
Jun;233(6):674-88.
87. Calder PC. N-3 polyunsaturated fatty acids, inflammation, and inflammatory diseases.
Am J Clin Nutr. 2006 06;83(6):1519.
88. He K, Liu K, Daviglus ML, Jenny NS, Mayer-Davis E, Jiang R, et al. Associations of
dietary long-chain n-3 polyunsaturated fatty acids and fish with biomarkers of
inflammation and endothelial activation (from the multi-ethnic study of atherosclerosis
[MESA]). Am J Cardiol. 2009 05/01;103(9):1238-43.
89. Din JN, Newby DE, Flapan AD. Omega 3 fatty acids and cardiovascular disease--
Fishing for a natural treatment. BMJ. 2004 01/03;328(7430):30-5.
90. Adorini L, Penna G. Control of autoimmune diseases by the vitamin D endocrine
system. Nat Clin Pract Rheumatol. 2008 08;4(8):404-12.
91. Nagpal S, Na S, Rathnachalam R. Noncalcemic actions of vitamin D receptor ligands.
Endocr Rev. 2005 08;26(5):662-87.
92. Peterson CA, Heffernan ME. Serum tumor necrosis factor-alpha concentrations are
negatively correlated with serum 25(OH)D concentrations in healthy women. J
Inflamm (Lond). 2008 Jul 24;5:10.
93. Shea MK, Booth SL, Massaro JM, Jacques PF, D'Agostino RB S, Dawson-Hughes B, et al.
Vitamin K and vitamin D status: Associations with inflammatory markers in the
Framingham offspring study. Am J Epidemiol. 2008 Feb 1;167(3):313-20.
94. Kim DJ, Xun P, Liu K, Loria C, Yokota K, Jacobs DR, et al. Magnesium intake in relation
to systemic inflammation, insulin resistance, and the incidence of diabetes. Diabetes
Care. 2010 12;33(12):2604-10.
95. Laires MJ, Monteiro CP, Bicho M. Role of cellular magnesium in health and human
disease. Front Biosci. 2004 01/01;9:262-76.

102
96. Laires MJ, Monteiro C. Exercise, magnesium and immune function. Magnes Res. 2008
06;21(2):92-6.
97. Cani PD, Delzenne NM. Involvement of the gut microbiota in the development of low
grade inflammation associated with obesity: Focus on this neglected partner. Acta
Gastroenterol Belg. 2010;73(2):267-9.
98. Cani PD, Delzenne NM. The role of the gut microbiota in energy metabolism and
metabolic disease. Curr Pharm Des. 2009;15(13):1546-58.
99. Liu S, Manson JE, Buring JE, Stampfer MJ, Willett WC, Ridker PM. Relation between a
diet with a high glycemic load and plasma concentrations of high-sensitivity C-reactive
protein in middle-aged women. Am J Clin Nutr. 2002 03;75(3):492-8.
100. Levitan EB, Cook NR, Stampfer MJ, Ridker PM, Rexrode KM, Buring JE, et al. Dietary
glycemic index, dietary glycemic load, blood lipids, and C-reactive protein. Metab Clin
Exp. 2008 03;57(3):437-43.
101. Vertuani S, Angusti A, Manfredini S. The antioxidants and pro-antioxidants network: An
overview. Curr Pharm Des. 2004;10(14):1677-94.
102. Benzie IFF. Evolution of dietary antioxidants. Comp Biochem Physiol , Part A Mol Integr
Physiol. 2003 09;136(1):113-26.
103. Pan M, Lai C, Dushenkov S, Ho C. Modulation of inflammatory genes by natural dietary
bioactive compounds. J Agric Food Chem. 2009 06/10;57(11):4467-77.
104. Holt EM, Steffen LM, Moran A, Basu S, Steinberger J, Ross JA, et al. Fruit and vegetable
consumption and its relation to markers of inflammation and oxidative stress in
adolescents. J Am Diet Assoc. 2009 03;109(3):414-21.
105. Cavicchia PP, Steck SE, Hurley TG, Hussey JR, Ma Y, Ockene IS, et al. A new dietary
inflammatory index predicts interval changes in serum high-sensitivity C-reactive
protein. J Nutr. 2009 12;139(12):2365-72.
106. Koren O, Spor A, Felin J, Fak F, Stombaugh J, Tremaroli V, et al. Human oral, gut, and
plaque microbiota in patients with atherosclerosis. Proc Natl Acad Sci U S A. 2011
03/15;108 Suppl 1:4592-8.
107. De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al. Impact
of diet in shaping gut microbiota revealed by a comparative study in children from
Europe and rural Africa. Proc Natl Acad Sci U S A. 2010 08/17;107(33):14691-6.
108. Humphrey LL, Fu R, Buckley DI, Freeman M, Helfand M. Periodontal disease and
coronary heart disease incidence: A systematic review and meta-analysis. J Gen Intern
Med. 2008 12;23(12):2079-86.
109. Takahashi N, Honda T, Domon H, Nakajima T, Tabeta K, Yamazaki K. Interleukin-1
receptor-associated kinase-M in gingival epithelial cells attenuates the inflammatory
response elicited by porphyromonas gingivalis. J Periodont Res. 2010 08;45(4):512-9.
110. Nakajima T, Yamazaki K. Periodontal disease and risk of atherosclerotic coronary heart
disease. Odontology. 2009 07;97(2):84-91.

103
111. Garcia-Bueno B, Caso JR, Leza JC. Stress as a neuroinflammatory condition in brain:
Damaging and protective mechanisms. Neurosci Biobehav Rev. 2008 08;32(6):1136-51.
112. Black PH, Garbutt LD. Stress, inflammation and cardiovascular disease. J Psychosom
Res. 2002 01;52(1):1-23.
113. Huffman KM, Samsa GP, Slentz CA, Duscha BD, Johnson JL, Bales CW, et al. Response
of high-sensitivity C-reactive protein to exercise training in an at-risk population. Am
Heart J. 2006 10;152(4):793-800.
114. Petersen A,Marie W., Pedersen BK. The anti-inflammatory effect of exercise. J Appl
Physiol. 2005 04;98(4):1154-62.
115. Petersen AMW, Penkowa M, Iversen M, Frydelund-Larsen L, Andersen JL, Mortensen J,
et al. Elevated levels of IL-18 in plasma and skeletal muscle in chronic obstructive
pulmonary disease. Lung. 2007;185(3):161-71.
116. Roubenoff R. Physical activity, inflammation, and muscle loss. Nutr Rev. 2007
12;65(12):208-12.
117. Roubenoff R. Molecular basis of inflammation: Relationships between catabolic
cytokines, hormones, energy balance, and muscle. JPEN J Parenter Enteral Nutr. 2008
Nov-Dec;32(6):630-2.
118. Handschin C, Spiegelman BM. The role of exercise and PGC1alpha in inflammation and
chronic disease. Nature. 2008 07/24;454(7203):463-9.
119. Simpson N, Dinges DF. Sleep and inflammation. Nutr Rev. 2007 12;65(12):244-52.
120. Meier-Ewert HK, Ridker PM, Rifai N, Regan MM, Price NJ, Dinges DF, et al. Effect of
sleep loss on C-reactive protein, an inflammatory marker of cardiovascular risk. J Am
Coll Cardiol. 2004 02/18;43(4):678-83.
121. Irwin MR, Wang M, Ribeiro D, Cho HJ, Olmstead R, Breen EC, et al. Sleep loss activates
cellular inflammatory signaling. Biol Psychiatry. 2008 09/15;64(6):538-40.
122. Irwin MR, Wang M, Campomayor CO, Collado-Hidalgo A, Cole S. Sleep deprivation and
activation of morning levels of cellular and genomic markers of inflammation. Arch
Intern Med. 2006 09/18;166(16):1756-62.
123. Mullington JM, Simpson NS, Meier-Ewert HK, Haack M. Sleep loss and inflammation.
Best Pract Res Clin Endocrinol Metab. 2010 10;24(5):775-84.
124. Forsythe CE, Phinney SD, Fernandez ML, Quann EE, Wood RJ, Bibus DM, et al.
Comparison of low fat and low carbohydrate diets on circulating fatty acid composition
and markers of inflammation. Lipids. 2008 01;43(1):65-77.
125. Forsythe CE, Phinney SD, Feinman RD, Volk BM, Freidenreich D, Quann E, et al. Limited
effect of dietary saturated fat on plasma saturated fat in the context of a low
carbohydrate diet. Lipids. 2010 10;45(10):947-62.
126. Volek JS, Fernandez ML, Feinman RD, Phinney SD. Dietary carbohydrate restriction
induces a unique metabolic state positively affecting atherogenic dyslipidemia, fatty
acid partitioning, and metabolic syndrome. Prog Lipid Res. 2008 09;47(5):307-18.

104
127. Volek JS, Ballard KD, Silvestre R, Judelson DA, Quann EE, Forsythe CE, et al. Effects of
dietary carbohydrate restriction versus low-fat diet on flow-mediated dilation. Metab
Clin Exp. 2009 12;58(12):1769-77.
128. Volek JS, Phinney SD, Forsythe CE, Quann EE, Wood RJ, Puglisi MJ, et al. Carbohydrate
restriction has a more favorable impact on the metabolic syndrome than a low fat diet.
Lipids. 2009 04;44(4):297-309.
129. Esposito K, Giugliano D. Mediterranean diet and the metabolic syndrome: The end of
the beginning. Metab Syndr Relat Disord. 2010 Jun;8(3):197-200.
130. Giugliano D, Ceriello A, Esposito K. The effects of diet on inflammation: Emphasis on
the metabolic syndrome. J Am Coll Cardiol. 2006 Aug 15;48(4):677-85.
131. Mozaffarian D, Rimm EB. Fish intake, contaminants, and human health: Evaluating the
risks and the benefits. JAMA. 2006 10/18;296(15):1885-99.
132. Eisenächer K, Krug A. Regulation of RLR-mediated innate immune signaling – It is all
about keeping the balance. Eur J Cell Biol. 2012 1;91(1):36-47.
133. Serhan CN, Chiang N. Novel endogenous small molecules as the checkpoint controllers
in inflammation and resolution: Entrée for resoleomics. Rheumatic Disease Clinics of
North America. 2004 2;30(1):69-95.
134. Serhan CN. Novel ω − 3-derived local mediators in anti-inflammation and resolution.
Pharmacol Ther. 2005 1;105(1):7-21.
135. Serhan CN, Yacoubian S, Yang R. Anti-inflammatory and proresolving lipid mediators.
Annu Rev Pathol. 2008;3:279-312.
136. Ariel A, Serhan CN. Resolvins and protectins in the termination program of acute
inflammation. Trends Immunol. 2007 4;28(4):176-83.
137. Chiang N, Fredman G, Backhed F, Oh SF, Vickery T, Schmidt BA, et al. Infection
regulates pro-resolving mediators that lower antibiotic requirements. Nature. 2012 Apr
25;484(7395):524-8.
138. Nathan C, Ding A. Nonresolving inflammation. Cell. 2010 3/19;140(6):871-82.
139. National Institute for Public Health and the Environment, The Netherlands (RIVM). Our
food, our heath. healthy diet and safe food in The Netherlands. Bilthoven: National
Institute for Public Health and the Environment; 2006. Report No.: 270555009.
140. Hou JK, Abraham B, El-Serag H. Dietary intake and risk of developing inflammatory
bowel disease: A systematic review of the literature. Am J Gastroenterol. 2011
Apr;106(4):563-73.
141. O'Neil CE, Keast DR, Fulgoni VL3rd, Nicklas TA. Tree nut consumption improves
nutrient intake and diet quality in US adults: An analysis of National Health and
nutrition examination survey (NHANES) 1999-2004. Asia Pac J Clin Nutr.
2010;19(1):142-50.
142. Guenther PM, Dodd KW, Reedy J, Krebs-Smith SM. Most Americans eat much less than
recommended amounts of fruits and vegetables. J Am Diet Assoc. 2006 9;106(9):1371-9.

105
143. Marriott BP, Olsho L, Hadden L, Connor P. Intake of added sugars and selected nutrients
in the United States, National Health and nutrition examination survey (NHANES)
2003-2006. Crit Rev Food Sci Nutr. 2010 Mar;50(3):228-58.
144. Danaei G, Ding EL, Mozaffarian D, Taylor B, Rehm J, Murray CJL, et al. The preventable
causes of death in the United States: Comparative risk assessment of dietary, lifestyle,
and metabolic risk factors. - PLoS Med. (4):e1000058.
145. Health Council of the Netherlands. Dietary reference intakes: Energy, proteins, fats and
digestible carbohydrates. The Hague: Health council of The Netherlands. Publication
no. 2001/19. ISBN 90-5549-384-8. 2001.
146. Kris-Etherton PM, Harris WS, Appel LJ, . Fish consumption, fish oil, omega-3 fatty acids,
and cardiovascular disease. Circulation. 2002 11/19;106(21):2747-57.
147. van Rossum C, Fransen HP, Verkaik-Kloosterman J, Buurma-Rethans E, Ocké MC.
Dutch National food consumption survey 2007-2010. Diet of children and adults aged 7
to 69 years. 2011 2011-10-05. Report No.: RIVM Report 350050006.
148. The NOAA administration. per capita consumption [Internet]. Available from:
http://www.st.nmfs.noaa.gov/st1/fus/fus04/08_perita2004.pdf.
149. Per capita consumption [Internet]. Available from:
http://www.st.nmfs.noaa.gov/st1/fus/fus04/08_perita2004.pdf.
150. He K, Song Y, Daviglus ML, Liu K, Van Horn L, Dyer AR, et al. Accumulated evidence on
fish consumption and coronary heart disease mortality. Circulation. 2004 June
08;109(22):2705-11.
151. de Goede J, Geleijnse JM, Boer JMA, Kromhout D, Verschuren WMM. Marine (n-3) fatty
acids, fish consumption, and the 10-year risk of fatal and nonfatal coronary heart
disease in a large population of Dutch adults with low fish intake. The Journal of
Nutrition. May 2010 May 2010;140(5):1023-8.
152. U.S. Department of Agriculture and U.S. Department of Health and Human Services.
Dietary guidelines for Americans, 2010. 7th edition, Washington, DC: U.S. Government
printing office, december 2010.
153. He FJ, Nowson CA, Lucas M, MacGregor GA. Increased consumption of fruit and
vegetables is related to a reduced risk of coronary heart disease: Meta-analysis of cohort
studies. J Hum Hypertens. 2007;21(9):717-28.
154. He F, Nowson C, MacGregor G. Fruit and vegetable consumption and stroke: Meta-
analysis of cohort studies. Lancet. 2006;367(9507):320-6.
155. Riboli E, Norat T. Epidemiologic evidence of the protective effect of fruit and vegetables
on cancer risk. Am J Clin Nutr. 2003;78(3 Suppl):559S-69S.
156. Buchholz AC, Schoeller DA. Is a calorie a calorie? The American Journal of Clinical
Nutrition. 2004 May 01;79(5):899S-906S.

106
157. Ebbeling C, Swain J, Feldman H, Wong W, Hachey D, Garcia Lago E, et al. Effects of
dietary composition on energy expenditure during weight-loss maintenance. JAMA
(Chicago, Ill.). 2012;307(24):2627-34.
158. Shao Q, Chin K. Survey of American food trends and the growing obesity epidemic.
Nutr Res Pract. 2011 /6/;5(3):253-9.
159. Bray GA, Lovejoy JC, Smith SR, DeLany JP, Lefevre M, Hwang D, et al. The influence of
different fats and fatty acids on obesity, insulin resistance and inflammation. The
Journal of Nutrition. 2002 September 01;132(9):2488-91.
160. Willett WC. Dietary fat and obesity: An unconvincing relation. Am J Clin Nutr. 1998
Dec;68(6):1149-50.
161. Hession M, Rolland C, Kulkarni U, Wise A, Broom J. Systematic review of randomized
controlled trials of low-carbohydrate vs. low-fat/low-calorie diets in the management of
obesity and its comorbidities. Obesity Reviews. 2009;10(1):36-50.
162. Hamer M, Stamatakis E. Metabolically healthy obesity and risk of all-cause and
cardiovascular disease mortality. J Clin Endocrinol Metab. 2012;97(7):2482-8.
163. Blher M. The distinction of metabolically 'healthy' from 'unhealthy' obese individuals.
Curr Opin Lipidol. 2010;21(1):38-43.
164. Lee CD, Blair SN, Jackson AS. Cardiorespiratory fitness, body composition, and all-
cause and cardiovascular disease mortality in men. The American Journal of Clinical
Nutrition. 1999 March 01;69(3):373-80.
165. Moussavi N, Gavino V, Receveur O. Could the quality of dietary fat, and not just its
quantity, be related to risk of obesity? Obesity (Silver Spring). 2008 Jan;16(1):7-15.
166. Schwenk RW, Holloway GP, Luiken JJFP, Bonen A, Glatz JFC. Fatty acid transport across
the cell membrane: Regulation by fatty acid transporters. Prostaglandins, Leukotrienes
and Essential Fatty Acids. 2010 0;82(4–6):149-54.
167. Brookheart RT, Michel CI, Schaffer JE. As a matter of fat. Cell Metabolism. 2009
7/8;10(1):9-12.
168. Lee JY, Zhao L, Youn HS, Weatherill AR, Tapping R, Feng L, et al. Saturated fatty acid
activates but polyunsaturated fatty acid inhibits toll-like receptor 2 dimerized with toll-
like receptor 6 or 1. J Biol Chem. 2004 Apr 23;279(17):16971-9.
169. Huang S, Rutkowsky J, Snodgrass R, Ono Moore K, Schneider D, Newman J, et al.
Saturated fatty acids activate TLR-mediated pro-inflammatory signaling pathways. J
Lipid Res. 2012.
170. Kuipers RS, de Graaf DJ, Luxwolda MF, Muskiet MH, Dijck-Brouwer DA, Muskiet FA.
Saturated fat, carbohydrates and cardiovascular disease. Neth J Med. 2011 Sep;69(9):372-
8.
171. Ramsden CE, Hibbeln JR, Majchrzak SF, Davis JM. N-6 fatty acid-specific and mixed
polyunsaturate dietary interventions have different effects on CHD risk: A meta-
analysis of randomised controlled trials. Br J Nutr. 2010 Dec;104(11):1586-600.

107
172. Ji R, Xu Z, Strichartz G, Serhan CN. Emerging roles of resolvins in the resolution of
inflammation and pain. Trends Neurosci. 2011 11;34(11):599-609.
173. Yehuda, S., Rabinovitz, S., Mostofsky, DI. Effects of essential fatty acids preparation, (SR-
3) on brain biochemistry and on behavioral and cognitive functions. In: Yehuda S,
Rabinovitz S, Mostofsky DI., editor. Handbook of essential fatty acids biology:
biochemistry, physiology and behavioral neurobiology. New York: Humana Press; 1997.
p. 427-52.
174. Kruizinga, A.G., Westenbrink, S., van Bosch, L.M.C., Jansen, M.C.J.F. TNO kwaliteit van
leven. De inneming van omega-3 en -6 vetzuren, van vitamines A en E, bij jong
volwassenen. Aanvullende berekeningen op basis van voedselconsumptiepeiling 2003.
2007. Report No.: V7451.
175. Gezondheidsraad 2006. Richtlijnen goede voeding 2006 - achtergronddocument.
Gezonheidsraad. 2006(A06/08).
176. Kuipers RS, Luxwolda MF, Dijck-Brouwer DA, Eaton SB, Crawford MA, Cordain L, et al.
Estimated macronutrient and fatty acid intakes from an East African Paleolithic diet. Br
J Nutr. 2010 Dec;104(11):1666-87.
177. Feskens EJ, Kromhout D. Epidemiologic studies on eskimos and fish intake. Ann N Y
Acad Sci. 1993 06/14;683:9-15.
178. Bang HO, Dyerberg J, Hjoorne N. The composition of food consumed by greenland
eskimos. Acta Med Scand. 1976;200(1-2):69-73.
179. Dyerberg J, Bang HO, Hjorne N. Fatty acid composition of the plasma lipids in
Greenland eskimos. Am J Clin Nutr. 1975 09;28(9):958-66.
180. Dyerberg J, Bang HO, Stoffersen E, Moncada S, Vane JR. Eicosapentaenoic acid and
prevention of thrombosis and atherosclerosis? Lancet. 1978 07/15;2(8081):117-9.
181. Sinclair AJ, Johnson L, O'Dea K, Holman RT. Diets rich in lean beef increase
arachidonic acid and long-chain omega 3 polyunsaturated fatty acid levels in plasma
phospholipids. Lipids. 1994 May;29(5):337-43.
182. Naughton JM, O'Dea K, Sinclair AJ. Animal foods in traditional Australian aboriginal
diets: Polyunsaturated and low in fat. Lipids. 1986 Nov;21(11):684-90.
183. Nelson GJ, Schmidt PC, Bartolini G, Kelley DS, Phinney SD, Kyle D, et al. The effect of
dietary arachidonic acid on plasma lipoprotein distributions, apoproteins, blood lipid
levels, and tissue fatty acid composition in humans. Lipids. 1997 Apr;32(4):427-33.
184. Kuipers RS, Fokkema MR, Smit EN, van der Meulen J, Boersma ER, Muskiet FA. High
contents of both docosahexaenoic and arachidonic acids in milk of women consuming
fish from Lake Kitangiri (tanzania): Targets for infant formulae close to our ancient diet?
Prostaglandins Leukot Essent Fatty Acids. 2005 Apr;72(4):279-88.
185. O'Dea K, Sinclair AJ. Increased proportion of arachidonic acid in plasma lipids after 2
weeks on a diet of tropical seafood. The American Journal of Clinical Nutrition. 1982
November 01;36(5):868-72.

108
186. Das UN. Essential fatty acids: Biochemistry, physiology and pathology. Biotechnol J.
2006 Apr;1(4):420-39.
187. Muskiet FAJ, Fokkema MR, Schaafsma A, Boersma ER, Crawford MA. Is
docosahexaenoic acid (DHA) essential? Lessons from DHA status regulation, our
ancient diet, epidemiology and randomized controlled trials. The Journal of Nutrition.
2004 January 01;134(1):183-6.
188. Yehuda S, Rabinovitz S, L. Carasso R, I. Mostofsky D. The role of polyunsaturated fatty
acids in restoring the aging neuronal membrane. Neurobiol Aging. 2002 0;23(5):843-53.
189. Freeman LM. Beneficial effects of omega-3 fatty acids in cardiovascular disease. J Small
Anim Pract. 2010 Sep;51(9):462-70.
190. Gerber M. Omega-3 fatty acids and cancers: A systematic update review of
epidemiological studies. Br J Nutr. 2012;107 Suppl 2:S228-39.
191. Muskiet FAJ. Pathophysiology and evolutionary aspects of dietary fats and long-chain
polyunsaturated faty acids across the life cycle. In: Montmayeur JP, le Coutre J, editors.
Fat Detection. Taste, Texture, and Post Ingestive Effects. Boca Raton (FL): Frontiers of
Neuroscience; 2010. p. 19.
192. Freeman MP, Davis M, Sinha P, Wisner KL, Hibbeln JR, Gelenberg AJ. Omega-3 fatty
acids and supportive psychotherapy for perinatal depression: A randomized placebo-
controlled study. J Affect Disord. 2008 Sep;110(1-2):142-8.
193. Freeman MP, Hibbeln JR, Wisner KL, Davis JM, Mischoulon D, Peet M, et al. Omega-3
fatty acids: Evidence basis for treatment and future research in psychiatry. J Clin
Psychiatry. 2006 Dec;67(12):1954-67.
194. Campoy C, Escolano-Margarit MV, Anjos T, Szajewska H, Uauy R. Omega 3 fatty acids
on child growth, visual acuity and neurodevelopment. Br J Nutr. 2012 Jun;107 Suppl
2:S85-S106.
195. Karr JE, Alexander JE, Winningham RG. Omega-3 polyunsaturated fatty acids and
cognition throughout the lifespan: A review. Nutr Neurosci. 2011 Sep;14(5):216-25.
196. Koletzko B, Lien E, Agostoni C, Bohles H, Campoy C, Cetin I, et al. The roles of long-
chain polyunsaturated fatty acids in pregnancy, lactation and infancy: Review of
current knowledge and consensus recommendations. J Perinat Med. 2008;36(1):5-14.
197. Sublette ME, Ellis S, Geant A, Mann JJ. Meta-analysis of the effects of eicosapentaenoic
acid (EPA) in clinical trials in depression. J Clin Psychiatry. 2011;72(12):1577-84.
198. Astorg P, Couthouis A, Bertrais S, Arnault N, Meneton P, Guesnet P, et al. Association of
fish and long-chain n-3 polyunsaturated fatty acid intakes with the occurrence of
depressive episodes in middle-aged french men and women. Prostaglandins Leukot
Essent Fatty Acids. 2008 Mar;78(3):171-82.
199. Cunnane SC, Plourde M, Pifferi F, Begin M, Feart C, Barberger-Gateau P. Fish,
docosahexaenoic acid and Alzheimer's disease. Prog Lipid Res. 2009 Sep;48(5):239-56.

109
200. Feart C, Torres MJ, Samieri C, Jutand MA, Peuchant E, Simopoulos AP, et al. Adherence
to a mediterranean diet and plasma fatty acids: Data from the Bordeaux sample of the
three-city study. Br J Nutr. 2011 Jul;106(1):149-58.
201. Barberger-Gateau P, Letenneur L, Deschamps V, Peres K, Dartigues JF, Renaud S. Fish,
meat, and risk of dementia: Cohort study. BMJ. 2002 Oct 26;325(7370):932-3.
202. Kiecolt-Glaser JK. Stress, food, and inflammation: Psychoneuroimmunology and
nutrition at the cutting edge. Psychosom Med. 2010 May;72(4):365-9.
203. Im DS. Omega-3 fatty acids in anti-inflammation (pro-resolution) and GPCRs. Prog
Lipid Res. 2012 Jul;51(3):232-7.
204. Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, et al. GPR120 is an omega-3
fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects.
Cell. 2010 Sep 3;142(5):687-98.
205. Poudyal H, Panchal SK, Diwan V, Brown L. Omega-3 fatty acids and metabolic
syndrome: Effects and emerging mechanisms of action. Prog Lipid Res. 2011
10;50(4):372-87.
206. Calder P. Omega-3 polyunsaturated fatty acids and inflammatory processes: Nutrition
or pharmacology? Br J Clin Pharmacol. 2012.
207. Dubuquoy L, Rousseaux C, Thuru X, Peyrin Biroulet L, Romano O, Chavatte P, et al.
PPARgamma as a new therapeutic target in inflammatory bowel diseases. Gut.
2006;55(9):1341-9.
208. Calder PC. Polyunsaturated fatty acids and inflammatory processes: New twists in an
old tale. Biochimie. 2009 Jun;91(6):791-5.
209. Rietveld A, Simons K. The differential miscibility of lipids as the basis for the formation
of functional membrane rafts. Biochimica et Biophysica Acta (BBA) - Reviews on
Biomembranes. 1998 11/10;1376(3):467-79.
210. Sanders TA. Polyunsaturated fatty acids in the food chain in Europe 1. Am J Clin Nut.
2000 January 01;71(1):176S-8S.
211. Simopoulos AP. Importance of the omega-6/Omega-3 balance in health and disease:
Evolutionary aspects of diet World Rev Nutr Diet. 2011;102:10-21.
212. Harris WS. The omega-3 index: From biomarker to risk marker to risk factor. Curr
Atheroscler Rep. 2009 11;11(6):411-7.
213. Luxwolda MF, Kuipers RS, Smit EN, Velzing-Aarts FV, Dijck-Brouwer DA, Muskiet FAJ.
The relation between the omega-3 index and arachidonic acid is bell shaped:
Synergistic at low EPA+DHA status and antagonistic at high EPA+DHA status.
Prostaglandins Leukot Essent Fatty Acid. 2011 10;85(3-4):171-8.
214. Liou YA, King DJ, Zibrik D, Innis SM. Decreasing linoleic acid with constant alpha-
linolenic acid in dietary fats increases (n-3) eicosapentaenoic acid in plasma
phospholipids in healthy men. J Nutr. 2007 Apr;137(4):945-52.

110
215. Nakamura MT, Nara TY. Structure, function, and dietary regulation of delta6, delta5, and
delta9 desaturases. Annu Rev Nutr. 2004;24:345-76.
216. Gibson R, Muhlhausler B, Makrides M. Conversion of linoleic acid and alpha-linolenic
acid to long-chain polyunsaturated fatty acids (LCPUFAs), with a focus on pregnancy,
lactation and the first 2 years of life. Maternal and child nutrition. 2011;7 Suppl 2:17-26.
217. Burdge GC, Calder PC. Dietary alpha-linolenic acid and health-related outcomes: A
metabolic perspective. Nutr Res Rev. 2006 Jun;19(1):26-52.
218. Lopez-Garcia E, Schulze MB, Fung TT, Meigs JB, Rifai N, Manson JE, et al. Major dietary
patterns are related to plasma concentrations of markers of inflammation and
endothelial dysfunction. Am J Clin Nutr. 2004 Oct;80(4):1029-35.
219. Lopez-Garcia E, Schulze MB, Manson JE, Meigs JB, Albert CM, Rifai N, et al.
Consumption of (n-3) fatty acids is related to plasma biomarkers of inflammation and
endothelial activation in women. J Nutr. 2004 Jul;134(7):1806-11.
220. Mayer K, Schaefer MB, Seeger W. Fish oil in the critically ill: From experimental to
clinical data. Curr Opin Clin Nutr Metab Care. 2006 Mar;9(2):140-8.
221. Cunnane SC, Guesnet P. Linoleic acid recommendations- A house of cards.
Prostaglandins Leukot Essent Fatty Acids. 2011 Sep 29.
222. Hansson G. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med.
2005 04/21; 2012/01;352(16):1685-95.
223. Libby P. Inflammation in atherosclerosis. Nature. 2002 Dec 19-26;420(6917):868-74.
224. Hamberg M, Samuelsson B. Prostaglandin endoperoxides. novel transformations of
arachidonic acid in human platelets. Proceedings of the National Academy of Sciences.
1974 September 01;71(9):3400-4.
225. Borgeat P, Hamberg M, Samuelsson B. Transformation of arachidonic acid and homo-
gamma-linolenic acid by rabbit polymorphonuclear leukocytes. monohydroxy acids
from novel lipoxygenases. Journal of Biological Chemistry. 1976 December
25;251(24):7816-20.
226. Soberman RJ, Harper TW, Betteridge D, Lewis RA, Austen KF. Characterization and
separation of the arachidonic acid 5-lipoxygenase and linoleic acid omega-6
lipoxygenase (arachidonic acid 15-lipoxygenase) of human polymorphonuclear
leukocytes. Journal of Biological Chemistry. 1985 April 10;260(7):4508-15.
227. Bannenberg GL, Chiang N, Ariel A, Arita M, Tjonahen E, Gotlinger KH, et al. Molecular
circuits of resolution: Formation and actions of resolvins and protectins. J Immunol.
2005 Apr 1;174(7):4345-55.
228. Chiang N, Arita M, Serhan CN. Anti-inflammatory circuitry: Lipoxin, aspirin-triggered
lipoxins and their receptor ALX. Prostaglandins, Leukotrienes and Essential Fatty Acids.
2005 0;73(3–4):163-77.

111
229. Serhan CN, Yang R, Martinod K, Kasuga K, Pillai PS, Porter TF, et al. Maresins: Novel
macrophage mediators with potent antiinflammatory and proresolving actions. The
Journal of Experimental Medicine. 2009 January 16;206(1):15-23.
230. Spite M, Serhan CN. Novel lipid mediators promote resolution of acute inflammation:
Impact of aspirin and statins. Circ Res. 2010 11/12;107(10):1170-84.
231. Merched AJ, Serhan CN, Chan L. Nutrigenetic disruption of inflammation-resolution
homeostasis and atherogenesis. J Nutrigenet Nutrigenomics. 2011;4(1):12-24.
232. Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis.
Nat Rev Immunol. 2010 Jan;10(1):36-46.
233. Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011 Apr
29;145(3):341-55.
234. Merched AJ, Ko K, Gotlinger KH, Serhan CN, Chan L. Atherosclerosis: Evidence for
impairment of resolution of vascular inflammation governed by specific lipid
mediators. FASEB J. 2008 Oct;22(10):3595-606.
235. Lee JH, O'Keefe JH, Lavie CJ, Marchioli R, Harris WS. Omega-3 fatty acids for
cardioprotection. Mayo Clin Proc. 2008 Mar;83(3):324-32.
236. Das UN. Essential fatty acids and their metabolites could function as endogenous HMG-
CoA reductase and ACE enzyme inhibitors, anti-arrhythmic, anti-hypertensive, anti-
atherosclerotic, anti-inflammatory, cytoprotective, and cardioprotective molecules.
Lipids Health Dis. 2008 Oct 15;7:37.
237. Lee JY, Plakidas A, Lee WH, Heikkinen A, Chanmugam P, Bray G, et al. Differential
modulation of toll-like receptors by fatty acids: Preferential inhibition by n-3
polyunsaturated fatty acids. J Lipid Res. 2003 Mar;44(3):479-86.
238. Reilly KB, Srinivasan S, Hatley ME, Patricia MK, Lannigan J, Bolick DT, et al. 12/15-
lipoxygenase activity mediates inflammatory monocyte/endothelial interactions and
atherosclerosis in vivo. J Biol Chem. 2004 Mar 5;279(10):9440-50.
239. Helgadottir A, Manolescu A, Thorleifsson G, Gretarsdottir S, Jonsdottir H,
Thorsteinsdottir U, et al. The gene encoding 5-lipoxygenase activating protein confers
risk of myocardial infarction and stroke. Nat Genet. 2004 Mar;36(3):233-9.
240. Weaver K, Ivester P, Seeds M, Case LD, Arm J, Chilton F. Effect of dietary fatty acids on
inflammatory gene expression in healthy humans. J Biol Chem. 2009;284(23):15400-7.
241. Tavazzi L, Maggioni A, Marchioli R, Barlera S, Franzosi M, Latini R, et al. Effect of n-3
polyunsaturated fatty acids in patients with chronic heart failure (the GISSI-HF trial): A
randomised, double-blind, placebo-controlled trial. Lancet. 2008;372(9645):1223-30.
242. Itakura H, Yokoyama M, Matsuzaki M, Saito Y, Origasa H, Ishikawa Y, et al. Relationships
between plasma fatty acid composition and coronary artery disease. J Atheroscler
Thromb. 2011;18(2):99-107.
243. Gonzalez-Periz A, Claria J. Resolution of adipose tissue inflammation.
ScientificWorldJournal. 2010 May 4;10:832-56.

112
244. Benzie IFF, Wachtel-Galor S. Increasing the antioxidant content of food: A personal
view on whether this is possible or desirable. Int J Food Sci Nutr. 2012 03/01;
2012/06;63:62-70.
245. Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature.
2000;408(6809):239-47.
246. Benzie IF, Wachtel-Galor S. Vegetarian diets and public health: Biomarker and redox
connections. Antioxid Redox Signal. 2010 Nov 15;13(10):1575-91.
247. Ames BN, Wakimoto P. Are vitamin and mineral deficiencies a major cancer risk? Nat
Rev Cancer. 2002 Sep;2(9):694-704.
248. Hertog MG, Feskens EJ, Hollman PC, Katan MB, Kromhout D. Dietary antioxidant
flavonoids and risk of coronary heart disease: The Zutphen elderly study. Lancet.
1993;342(8878):1007-11.
249. Block G, Patterson B, Subar A. Fruit, vegetables, and cancer prevention: A review of the
epidemiological evidence. Nutr Cancer. 1992;18(1):1-29.
250. Liu RH. Health benefits of fruit and vegetables are from additive and synergistic
combinations of phytochemicals. The American Journal of Clinical Nutrition. 2003
September 01;78(3):517S-20S.
251. Engelhart M, Geerlings M, Ruitenberg A, van Swieten J, Hofman A, Witteman JCM, et al.
Dietary intake of antioxidants and risk of Alzheimer disease. JAMA (Chicago, Ill.).
2002;287(24):3223-9.
252. Halliwell B., Gutteridge JMC. Free radicals in biology and medicine. Oxford: Clarendon
Press; 1999.
253. Halliwell B. Free radicals and antioxidants: A personal view. Nutr Rev. 1994;52(8):253-65.
254. Cloud PE. Atmospheric and hydrospheric evolution on the primitive Earth. Both secular
accretion and biological and geochemical processes have affected earth's volatile
envelope. Science. 1968;160(3829):729-36.
255. Raymond J, Segrè D. The effect of oxygen on biochemical networks and the evolution
of complex life. Science. 2006 March 24;311(5768):1764-7.
256. Laland KN, Odling-Smee J, Feldman MW. Niche construction, biological evolution, and
cultural change. Behav Brain Sci. 2000;23(01):131.
257. Walker JC. Atmospheric constraints on the evolution of metabolism. Origins of life and
evolution of the biosphere. 1980;10(2):93-104.
258. Fridovich I. Superoxide and evolution. Horiz Biochem Biophys. 1974;1:1-37.
259. Cadenas E. Basic mechanisms of antioxidant activity. Biofactors. 1997;6(4):391-7.
260. Benzie IF. Evolution of antioxidant defence mechanisms. Eur J Nutr. 2000 Apr;39(2):53-
61.
261. Evans P, Halliwell B. Micronutrients: Oxidant/antioxidant status. Br J Nutr. 2001;85
Suppl 2:S67-74.
262. Sies H. Oxidative stress: Oxidants and antioxidants. Exp Physiol. 1997 Mar;82(2):291-5.

113
263. Klarod K, Hongsprabhas P, Khampitak T, Wirasorn K, Kiertiburanakul S,
Tangrassameeprasert R, et al. Serum antioxidant levels and nutritional status in early
and advanced stage lung cancer patients. Nutrition. 2011 Nov-Dec;27(11-12):1156-60.
264. Ames BN. Low micronutrient intake may accelerate the degenerative diseases of aging
through allocation of scarce micronutrients by triage. Proc Natl Acad Sci U S A. 2006
Nov 21;103(47):17589-94.
265. Calder PC, Albers R, Antoine JM, Blum S, Bourdet-Sicard R, Ferns GA, et al.
Inflammatory disease processes and interactions with nutrition. Br J Nutr. 2009
May;101 Suppl 1:S1-45.
266. Nakagawa K, Kiko T, Miyazawa T, Carpentero Burdeos G, Kimura F, Satoh A, et al.
Antioxidant effect of astaxanthin on phospholipid peroxidation in human erythrocytes
Br J Nutr. 2011 Jan 31:1-9.
267. Martin HD, Ruck C, Schmidt M, Sell S, Beutner S, Mayer B, et al. Chemistry of carotenoid
oxidation and free radical reactions Pure and Applied Chemistry. 1999;71(12):2253-62.
268. Halliwell B. Free radicals and antioxidants: Updating a personal view. Nutr Rev.
2012;70(5):257-65.
269. Valko M, Leibfritz D, Moncol J, Cronin MTD, Mazur M, Telser J. Free radicals and
antioxidants in normal physiological functions and human disease. International
journal of biochemistry cell biology. 2007;39(1):44-84.
270. Avignon A, Hokayem M, Bisbal C, Lambert K. Dietary antioxidants: Do they have a role
to play in the ongoing fight against abnormal glucose metabolism? Nutrition.
2012;28(7-8):715-21.
271. Chuang C, Martinez K, Xie G, Kennedy A, Bumrungpert A, Overman A, et al. Quercetin is
equally or more effective than resveratrol in attenuating tumor necrosis factor-{alpha}-
mediated inflammation and insulin resistance in primary human adipocytes. Am J Clin
Nutr. 2010;92(6):1511-21.
272. Linnane AW, Ziegler DM. Studies on the mechanism of oxidative phosphorylation. V.
the phosphorylating properties of the electron transport particle. Biochim Biophys Acta.
1958;29(3):630-8.
273. Nemoto S, Takeda K, Yu ZX, Ferrans VJ, Finkel T. Role for mitochondrial oxidants as
regulators of cellular metabolism. Mol Cell Biol. 2000;20(19):7311-8.
274. Sidoti-de Fraisse C, Rincheval V, Risler Y, Mignotte B, Vayssire JL. TNF-alpha activates
at least two apoptotic signaling cascades. Oncogene. 1998;17(13):1639-51.
275. Babior BM. The respiratory burst of phagocytes. J Clin Invest. 1984;73(3):599-601.
276. Jones D. Redefining oxidative stress. Antioxidants redox signalling. 2006;8(9-10):1865-
79.
277. Calabrese E, Baldwin L. Hormesis: The dose-response revolution. Annu Rev Pharmacol
Toxicol. 2003;43:175-97.

114
278. Meyer M, Pahl HL, Baeuerle PA. Regulation of the transcription factors NF-kappa B and
AP-1 by redox changes. Chem Biol Interact. 1994;91(2-3):91-100.
279. Ji LL. Exercise and oxidative stress: Role of the cellular antioxidant systems. Exerc Sport
Sci Rev. 1995;23:135-66.
280. Hollander J, Fiebig R, Gore M, Ookawara T, Ohno H, Ji LL. Superoxide dismutase gene
expression is activated by a single bout of exercise in rat skeletal muscle. Pflügers
Archiv. 2001;442(3):426-34.
281. Gomez Cabrera M, Domenech E, Romagnoli M, Arduini A, Borras C, Pallardo F, et al.
Oral administration of vitamin C decreases muscle mitochondrial biogenesis and
hampers training-induced adaptations in endurance performance. Am J Clin Nutr.
2008;87(1):142-9.
282. Via J, Gomez Cabrera M, Borras C. Fostering antioxidant defences: Up-regulation of
antioxidant genes or antioxidant supplementation? Br J Nutr. 2007;98 Suppl 1:S36-40.
283. Finley J, Kong A, Hintze K, Jeffery E, Ji L, Lei X. Antioxidants in foods: State of the
science important to the food industry. J Agric Food Chem. 2011;59(13):6837-46.
284. Barros MP, Pinto E, Colepicolo P, Pedersén M. Astaxanthin and peridinin inhibit
oxidative damage in Fe2+-loaded liposomes: Scavenging oxyradicals or changing
membrane permeability? Biochem Biophys Res Commun. 2001 10/19;288(1):225-32.
285. Halliwell B, Wasil M, Grootveld M. Biologically significant scavenging of the
myeloperoxidase-derived oxidant hypochlorous acid by ascorbic acid. implications for
antioxidant protection in the inflamed rheumatoid joint. FEBS Lett. 1987 Mar 9;213(1):15-
7.
286. Veeramachaneni S, Wang X. Carotenoids and lung cancer prevention. Frontiers in
bioscience (Scholar edition). 2009;1:258-74.
287. Druesne Pecollo N, Latino Martel P, Norat T, Barrandon E, Bertrais S, Galan P, et al. Beta-
carotene supplementation and cancer risk: A systematic review and metaanalysis of
randomized controlled trials. International Journal of Cancer. 2010;127(1):172-84.
288. Peternelj T, Coombes J. Antioxidant supplementation during exercise training:
Beneficial or detrimental? Sports Medicine. 2011;41(12):1043-69.
289. Bjelakovic G, Nikolova D, Gluud L, Simonetti R, Gluud C. Mortality in randomized trials
of antioxidant supplements for primary and secondary prevention: Systematic review
and meta-analysis. JAMA (Chicago, Ill.). 2007;297(8):842-57.
290. Biesalski H, Grune T, Tinz J, Zllner I, Blumberg J. Reexamination of a meta-analysis of
the effect of antioxidant supplementation on mortality and health in randomized trials.
Nutrients. 2010;2(9):929-49.
291. McNulty H, Jacob RF, Mason RP. Biologic activity of carotenoids related to distinct
membrane physicochemical interactions. Am J Cardiol. 2008 5/22;101(10, Supplement
1):S20-9.

115
292. Spiller GA, Dewell A. Safety of an astaxanthin-rich haematococcus pluvialis algal
extract: A randomized clinical trial. J Med Food. 2003;6:51-6.
293. Park JS, Chyun JH, Kim YK, Line L, Chew B. Astaxanthin decreased oxidative stress and
inflammation and enhanced immune response in humans Nutrition & Metabolism.
2010 03-05;7:18.
294. Lavrovsky Y, Chatterjee B, Clark RA, Roy AK. Role of redox-regulated transcription
factors in inflammation, aging and age-related diseases. Exp Gerontol. 2000
Aug;35(5):521-32.
295. Rahman I. Oxidative stress, chromatin remodeling and gene transcription in
inflammation and chronic lung diseases. J Biochem Mol Biol. 2003 Jan 31;36(1):95-109.
296. Han KC, Wong WC, Benzie IFF. Genoprotective effects of green tea (camellia sinensis) in
human subjects: Results of a controlled supplementation trial. Br J Nutr. 2011;105(2):171-
9.
297. Arendt B, Ellinger S, Kekic K, Geus L, Fimmers R, Spengler U, et al. Single and repeated
moderate consumption of native or dealcoholized red wine show different effects on
antioxidant parameters in blood and DNA strand breaks in peripheral leukocytes in
healthy volunteers: A randomized controlled trial (ISRCTN68505294). Nutrition journal.
2005;4:33-.
298. Brevik A, Gaivo I, Medin T, Jrgenesen A, Piasek A, Elilasson J, et al. Supplementation of
a western diet with golden kiwifruits (actinidia chinensis var.'hort 16A':) effects on
biomarkers of oxidation damage and antioxidant protection. Nutrition journal.
2011;10:54-.
299. Gill CIR, Haldar S, Boyd L, Bennett R, Whiteford J, Butler M, et al. Watercress
supplementation in diet reduces lymphocyte DNA damage and alters blood antioxidant
status in healthy adults. Am J Clin Nutr. 2007;85(2):504-10.
300. Cordain L. Cereal grains: Humanity's double-edged sword. World Rev Nutr Diet.
1999;84:19-73.
301. Bartosz G. Oxidative stress in plants. Acta Physiologiae Plantarum. 1997;19:47-64.
302. Hartmann T. From waste products to ecochemicals: Fifty years research of plant
secondary metabolism. Phytochemistry. 2007;68(22-24):2831-46.
303. Wink M, editor. Annual plant reviews volume 39. Functions and biotechnology of
plants' secondary metabolites. Second edition. 2nd ed. Oxford, United Kingdom:
Blackwell Publishing Ltd; 2010.
304. Strain JJ. Disturbances of micronutrient and antioxidant status in diabetes. Proc Nutr
Soc. 1991;50(03):591.
305. Holst B, Williamson G. Nutrients and phytochemicals: From bioavailability to
bioefficacy beyond antioxidants. Curr Opin Biotechnol. 2008 4;19(2):73-82.
306. Norman J. T. Antioxidants and disease: More questions than answers. Nutr Res. 2000
3;20(3):449-59.

116
307. Sackett, D. L., Strauss, S. E., Scott Richardson W., Rosenberg W., Hayness, R. B. Evidence
based medicine. How to practice and teach EBM. Edinburgh: Churchill Livingstone;
2000.
308. Blumberg J, Heaney RP, Huncharek M, Scholl T, Stampfer M, Vieth R, et al. Evidence-
based criteria in the nutritional context. Nutr Rev. 2010 08;68(8):478-84.
309. Mitchell HL, Aggett PJ, Richardson DP, Stowell JD. Food & health forum meeting:
Evidence-based nutrition. Br J Nutr. 2011 01;105(2):322-8.
310. Wielders JPM, Muskiet FAJ, van dW. [Shedding new light on vitamin D--reassessment
of an essential prohormone]. Ned Tijdschr Geneeskd. 2010;154.
311. Muskiet FAJ, van dV, Schuitemaker GE, Wielders JPM. Response to: Towards an
adequate intake of vitamin D. an advisory report of the Health Council of The
Netherlands. Eur J Clin Nutr. 2010 06;64(6):655-.
312. Weggemans RM, Schaafsma G, Kromhout D. Towards an adequate intake of vitamin D.
an advisory report of The Health Council of The Netherlands. Eur J Clin Nutr. 2009
12;63(12):1455-7.
313. Rose G, Hamilton PJ, Colwell L, Shipley MJ. A randomised controlled trial of anti-
smoking advice: 10-year results. J Epidemiol Community Health. 1982 06;36(2):102-8.
314. Rose G, Hamilton PJ. A randomised controlled trial of the effect on middle-aged men of
advice to stop smoking. Journal of Epidemiology and Community Health. 1978
December 01;32(4):275-81.
315. Makrides M, Gibson RA, Udell T, Ried K. Supplementation of infant formula with long-
chain polyunsaturated fatty acids does not influence the growth of term infants. Am J
Clin Nutr. 2005 05;81(5):1094-101.
316. McCann JC, Ames BN. Is docosahexaenoic acid, an n-3 long-chain polyunsaturated
fatty acid, required for development of normal brain function? An overview of evidence
from cognitive and behavioral tests in humans and animals. Am J Clin Nutr. 2005
08;82(2):281-95.
317. Simmer K, Schulzke SM, Patole S. Longchain polyunsaturated fatty acid
supplementation in preterm infants. Cochrane Database Syst Rev. 2008(1).
318. Simmer K, Patole SK, Rao SC. Longchain polyunsaturated fatty acid supplementation in
infants born at term. Cochrane Database Syst Rev. 2008 Jan 23;(1)(1):CD000376.
319. Bischoff Ferrari HA, Shao A, Dawson Hughes B, Hathcock J, Giovannucci E, Willett WC.
Benefit-risk assessment of vitamin D supplementation. Osteoporosis Int.
2010;21(7):1121-32.
320. Cordain L, Miller JB, Eaton SB, Mann N, Holt SH, Speth JD. Plant-animal subsistence
ratios and macronutrient energy estimations in worldwide hunter-gatherer diets. Am J
Clin Nutr. 2000 Mar;71(3):682-92.
321. Eaton SB, Eaton SB, Konner MJ. Paleolithic nutrition revisited: A twelve-year
retrospective on its nature and implications. Eur J Clin Nutr. 1997 04;51(4):207-16.

117
322. Eaton SB, Eaton SB,3rd. Paleolithic vs. modern diets--selected pathophysiological
implications. Eur J Nutr. 2000 Apr;39(2):67-70.
323. Klonoff DC. The beneficial effects of a Paleolithic diet on type 2 diabetes and other risk
factors for cardiovascular disease. J Diabetes Sci Technol. 2009 Nov 1;3(6):1229-32.
324. Frassetto LA, Schloetter M, Mietus-Synder M, Morris RC, Sebastian A. Metabolic and
physiologic improvements from consuming a Paleolithic, hunter-gatherer type diet.
Eur J Clin Nutr. 2009 08;63(8):947-55.
325. Willett WC. Balancing life-style and genomics research for disease prevention. Science.
2002 04/26;296(5568):695-8.
326. Hemminki K, Lorenzo Bermejo J, Forsti A. The balance between heritable and
environmental aetiology of human disease. Nat Rev Genet. 2006 12;7(12):958-65.
327. Genome-wide association study of 14,000 cases of seven common diseases and 3,000
shared controls. Nature. 2007 06/07;447(7145):661-78.
328. Rose G. Sick individuals and sick populations. Int J Epidemiol. 1985;14(1):32-8.
329. Rosenberg NA, Pritchard JK, Weber JL, Cann HM, Kidd KK, Zhivotovsky LA, et al.
Genetic structure of human populations. Science. 2002 12/20;298(5602):2381-5.
330. Cauchi S, El Achhab Y, Choquet H, Dina C, Krempler F, Weitgasser R, et al. TCF7L2 is
reproducibly associated with type 2 diabetes in various ethnic groups: A global meta-
analysis. J Mol Med (Berl). 2007 07;85(7):777-82.
331. Bray GA. Medical consequences of obesity. J Clin Endocrinol Metab. 2004
06;89(6):2583-9.
332. Gurven M, Kaplan H. Longevity among hunter- gatherers: A cross-cultural
examination. Population and Development Review. 2007;33(2):321-65.
333. Hill K, Hurtado AM, Walker RS. High adult mortality among Hiwi hunter-gatherers:
Implications for human evolution. J Hum Evol. 2007 04;52(4):443-54.
334. Bruggink, J-W., Garssen, J., Lodder, B., Kardal, M. Trends in gezonde
levensverwachting. CBS. bevolkingstrends 1e kwartaal 2009.

118
Paragraph 1.3. Chronic inflammatory diseases are stimulated by current lifestyle: how diet,
stress levels and medication prevent our body from recovering. Bosma-den Boer MM, van
Wetten ML, Pruimboom L. Nutr Metab (Lond) 2012;9:32.
Chronic inflammatory diseases are stimulated by current lifestyle: how diet,
stress levels and medication prevent our body from recovering
Margarethe M Bosma-den Boer1* * Corresponding address
Email: [email protected]
Marie-Louise van Wetten1
Email: [email protected]
Leo Pruimboom1
Email: [email protected]
1 University of Gerona, Gerona, Spain

119
Abstract
Serhan and colleagues introduced the term “Resoleomics” in 1996 as the process of
inflammation resolution. The major discovery of Serhan´s work is that onset to conclusion of
an inflammation is a controlled process of the immune system (IS) and not simply the
consequence of an extinguished or “exhausted” immune reaction. Resoleomics can be
considered as the evolutionary mechanism of restoring homeostatic balances after injury,
inflammation and infection. Under normal circumstances, Resoleomics should be able to
conclude inflammatory responses. Considering the modern pandemic increase of chronic
medical and psychiatric illnesses involving chronic inflammation, it has become apparent
that Resoleomics is not fulfilling its potential resolving capacity. We suggest that recent
drastic changes in lifestyle, including diet and psycho-emotional stress, are responsible for
inflammation and for disturbances in Resoleomics. In addition, current interventions, like
chronic use of anti-inflammatory medication, suppress Resoleomics. These new lifestyle
factors, including the use of medication, should be considered health hazards, as they are
capable of long-term or chronic activation of the central stress axes. The IS is designed to
produce solutions for fast, intensive hazards, not to cope with long-term, chronic stimulation.
The never-ending stress factors of recent lifestyle changes have pushed the IS and the central
stress system into a constant state of activity, leading to chronically unresolved inflammation
and increased vulnerability for chronic disease. Our hypothesis is that modern diet, increased
psycho-emotional stress and chronic use of anti-inflammatory medication disrupt the
natural process of inflammation resolution ie Resoleomics.
Keywords
Chronic inflammation, Central stress system, Nutrition, Resoleomics, Sympathetic-adrenal-
medulla axis, Hypothalamus-pituitary-adrenal axis, Anti-inflammatory medication, Insulin
resistance, Polyunsaturated fatty acids, Glycemic index

120
Introduction
The number of people suffering from chronic diseases such as cardiovascular diseases (CVD),
diabetes, respiratory diseases, mental disorders, autoimmune diseases (AID) and cancers has
increased dramatically over the last three decades. The increasing rates of these chronic
systemic illnesses suggest that inflammation [1,2], caused by excessive and inappropriate
innate immune system (IIS) activity, is unable to respond appropriately to danger signals that
are new in the context of evolution. This leads to unresolved or chronic inflammatory
activation in the body.
Inflammation is designed to limit invasions and damage after injury, a process which has
been essential for the survival of Homo sapiens in the absence of medication such as
antibiotics. Recently, it has been discovered that onset to conclusion of an inflammation is a
self-limiting and controlled process of the immune system (IS). This process of inflammation
resolution is defined by Serhan as Resoleomics [3], a term which will be used throughout this
article.
Our genes and physiology, which are still almost identical to those of our hunter-gatherer
ancestors of 100,000 years ago, preserve core regulation and recovery processes [4,5].
Nowadays our genes operate in an environment which is completely different to the one for
which they were designed.
Modern man is exposed to an environment which has changed enormously since the time of
the industrial revolution. In recent decades there has been a tremendous acceleration in
innovations which have changed our lives completely. As a consequence, more than 75 % of
humans do not meet the minimum requirement of the estimated necessary daily physical
activity [6], 72 % of modern food types is new in human evolution [7], psycho-emotional stress
has increased and man is exposed to an overwhelming amount of information on a daily
basis. All these factors combine to produce an environment full of modern danger signals
which continuously activate the IIS and central stress axes. The question is whether the IIS
and its natural inflammatory response, Resoleomics, can still function optimally in this
modern, fast-changing environment, considering that the IIS is designed to produce short,
intensive reactions to acute external danger [8,9]. It would seem that in the bodies of people
who have adopted a Western lifestyle the inflammatory response is not concluded because of
an initial excessive or subnormal onset of the response [10].
This article postulates how triggers from chronic altered diet and psycho-emotional stress
negatively influence Resoleomics, thereby increasing susceptibility to the development of
chronic, low-grade, inflammation-based diseases due to the constant activation of both the
central stress axes and the IIS. In addition, an attempt is made to demonstrate the ways in
which the use of anti-inflammatory medication could influence Resoleomics.

121
Resoleomics, a self-limiting process of inflammation
Serhan and his colleagues [3] introduced the term Resoleomics to describe a self-limiting
process of inflammation, executed and controlled by the innate immune system (IIS) and
regulated by the sympathetic nervous system (SNS) and the hypothalamus-pituitary-adrenal
(HPA) axis. This process controls inflammation using metabolites produced from arachidonic
acid (AA), eicosapentaenoic acid (EPA) and docosahexenoic acid (DHA). Resoleomics operates
locally when polymorphonuclear neutrophils (PMNs) are attracted by increased pro-
inflammatory cytokine and eicosanoids production during microbial invasion, wound
healing or chemical injury. The function is to limit the inflammation response. The central
control system of the inflammatory reaction is very complex. Local and central processes
influence each other and both are responsible for an optimal resolving response (Figure 1).
The local process can be divided into three phases [11] (Figure 2):
Figure 1 Start and finish of a physiological inflammatory reaction in wound healing and
situations of microbial challenge.
Cellular damage and leakage of alarmins attract neutrophils to the damaged area (PMN’s). Sympathetic afferents activate
the locus coeruleus (central nucleus of the sympathetic nervous system, SNS) and Noradrenaline (Norepinephrine, NE) is
released. The released NE activates the adrenal medulla inducing the production of systemic catecholamins that supports
the activation of the PMN. Damaged blood vessels are a source of an omega 3 rich edema (EPA and DHA). DHA and EPA
inhibit LOX-5 directly and through conversion into resolvins and protectins. Both PGE2 and PGD2, produced by the
breakdown of AA by COX-2 activity, will now override the strong chemotaxic effect of LTB4. The combined action of
protectins, resolvins and lipoxins produced out of AA will put a hold on the pro-inflammatory activity of PMN’s, which is
supported by the increased production of systemic cortisol. Cortisol further activates macrophages (M-Ph) to phagocytose

122
issue debris and quiet PMN by releasing substances such as LXA4, resolvin E1 (RvE1), prostanoid D1 (PD1), fibroblast growth
factor (FGF), vascular endothelial growth factor (VEGF) and epithelial growth factor (EGF) at the same time. Further edema
leakage will be stopped, whereas angiogenesis and production of connective tissue will take place, finishing the
inflammatory reaction and starting the production of new tissue.
Figure 2 Inflammation is a controlled process with an initiation, resolution and termination
phase. After microbial invasion, lesion or chemical injury, the initiation phase starts with the production of pro-inflammatory
mediators like LTB4 and PG2. These mediators increase inflammation until the Eicosanoid Switch, the end of the initiation
phase, takes place. This occurs when the level of PGE2 plus PGD2 is equal to the LTB4 level. The resolution phase is
entered, triggering the generation of anti-inflammatory mediators like LK, resolvins, protectins, maresins, PGD2 and PGF2a.
When the total level of anti-inflammatory mediators exceeds the level of LTB4 the Stop Signal takes place. This is the last
phase, the inflammation will be terminated by clearing the affected area [11]. The stress hormones produced by the
systemic stress axes have a direct effect on the inflammation phases. A microbial invasion, lesion or injury sends off an
alarm in the body, setting off the systemic stress system which produces NE as response and tunes the system to insulin
and cortisol resistance [12]. The Eicosanoids Switch to resolution can only take place when NE is equal to the level of
cortisol plus insulin and when cortisol sensitivity is recovered. The Stop Signal requires a low level of NE and normalized
cortisol sensitivity. The termination phase is entered when the stress axes are switched off

123
1. Initiation phase
2. Resolution phase
3. Termination phase
Initiation phase
Pro-inflammatory eicosanoids, like leukotrienes B4 (LTB4) and prostaglandins (PGs) initiate
the inflammatory response. PMNs generate LTB4 and PGE2 from precursor AA with the use
of lipoxygenase-5 (LOX-5) and cyclo-oxygenase 2 (COX-2). Both eicosanoids enhance
inflammation, LTB4 being the strongest chemotoxic compound of cytotoxic neutrophils.
PGE2 and/or PGD2, although initially pro-inflammatory, determine the switch to the next
phase, the resolution of the inflammation.
Resolution phase
This phase starts with the Eicosanoid Switch to resolution. When the PGE2 and/or PGD2 level
is equal to the level of LTB4, the PMNs activate the switch from pro-inflammatory to anti-
inflammatory eicosanoids production by limiting the production of LOX-5. This switch is
responsible for the production of anti-inflammatory lipoxins (LXs) from AA through
activation of lipoxygenase −12 (LOX-12), lipoxygenase-15 (LOX-15) and acetylated COX-2
[13,14]. This last mechanism has been found to be responsible for the production of more
stable aspirin-triggered LXs (ATLs) with a longer half-value period [15]. Other resolving
metabolites that support LXs are resolvins, (neuro)protectins and maresins produced from
respectively EPA and DHA [11,16]. A second substantial increase of COX-2 activity will
produce anti-inflammatory PGs (PGD2 and PGF2a) during this phase [17].
Termination phase
This phase starts when the Stop Signal takes place. This happens when sufficient anti-
inflammatory mediators such as LXs are available to stop the pro-inflammatory process
[13,14]. LXs are capable of inhibiting both PMN infiltration and the activity of cytotoxic cells of
the ISS, inducing phagocytosis to clear debris by non-cytoxic macrophages and attenuating
an accumulation of the pro-inflammatory transcription factors, ie nuclear factor-kappaB (NF-
kB) and activator protein 1 (AP-1) [18,19].

124
Central stress axes and Resoleomics
This section deals solely with the effect of the sympathetic, parasympathetic and the HPA axis
on Resoleomics. The systemic stress system is closely linked to the IIS via the stress axes of
our body. Anything that can activate the sympathetic-adrenal-medulla (SAM) and HPA axes
will have its effect on the IIS [20] and therefore on Resoleomics. Seen in reverse, it is precisely
the IIS that can trigger stress axes, inducing a systemic stress reaction in the body [21]. In the
SNS, which initially activates the IIS, inhibition of the IIS is provided by the strong anti-
inflammatory neurotransmitter acetylcholine (ACh), produced by the parasympathetic
nervous system [22].
The systemic stress reaction follows a two-wave pattern. Activation of the SAM axis is
considered the first wave, giving rise to the excretion of brain norepinephrine (NE) by the
Locus Coeruleus (LC). The descending pathway activates sympathetic motor neurons in the
medulla oblongata, which stimulate the adrenal glands (through sympathetic efferent nerves).
The adrenal gland will now excrete catecholamines, which activate and induce proliferation
of ISS cells. NF-kB increases pro-inflammatory cytokines production, such as interleukin 1-
beta (IL1-β), interleukin 6 (IL-6) and tumor necrosis factor (TNF). Both the IIS and Th1 of the
adaptive IS contain receptors sensitive to catecholamines. Cerebral catecholamines affect the
activity of spleen, thymus, bone marrow and lymphoid nodes [23]. NE has been shown to
activate the IIS at the onset of inflammation, while long-term activation of the SNS induces
IIS inhibition [24].
The second wave of the systemic stress reaction corresponds with the activation of the HPA
axis, with glucocorticoids (GCs) as end product. Cortisol is capable of inhibiting the IIS
through the upward regulation of inhibiting factor kappa B (IkB), while informing the
immunological cortex through the migration of different immune cells to the brain [25,26].
Cortisol, the regulator of the IIS response, can guide the inflammation into resolution phase.
Termination is instigated when cortisol “overrules” the NE effect on NF-kB signalling through
genetic influence and reduction of transcription of the NF-kB sensitive pro-inflammatory
gene, resulting in the finalization of the inflammatory response (Figure 2).
This “termination” effect of cortisol is normally supported by a compensatory anti-
inflammatory response through activation of the vagal anti-inflammatory loop [27]. The
resulting production of ACh inhibits the IS through the alpha-7-nicotin-Acetylcholinergic
Receptor (α7nAChR) [28] (Figure 1).
The SNS (NE) increases the initial pro-inflammatory immune response in the initiation phase,
whereas delayed cortisol response, induced by the HPA axis, inhibits the pro-inflammatory

125
response [29]. Integrity of the SAM axis with its NE response/ reaction is necessary for an
adequate initial inflammatory response [30]. At the beginning of the initiation phase, there is
resistance to both cortisol and insulin in order to allow for the activation of the IIS [12]. At the
end of this phase, cortisol sensitivity and insulin sensitivity should be recovered to facilitate
the Eicosanoid Switch to the resolution phase.
Chronic stress exposure reduces the capacity to mount an acute stress response [31], resulting
in an inadequate pro-inflammatory response. Chronic (psycho-emotional) stress situations
can be responsible for the continuous production of catecholamines by the SAM axis. People
suffering from “perpetual stress”, for example the parents of a child with cancer, showed
chronic, increased levels of circulating pro-inflammatory cytokines [26]. This situation
requires a high level of energy expenditure. The metabolic rate is increased to provide extra
energy for the brain (arousal of all senses), the heart muscle and the locomotive system. The
existing cells from the IIS are activated and will proliferate (relatively low energy expenditure),
whereas proliferation of new immune cells (much more costly energy expenditure) will be
blocked. Further consequences of chronic SAM activity are narrowing of the cell spectrum of
the IIS and complete loss of activity of the Th1 section of the adaptive IS, leading to an
insufficient capability to fight viruses, (pre)neoplastic cells and intracellularly presented
pathogens [31].
An inflammatory response leading to solution depends on the sensitivity of glucocorticoid
receptors (GR) and catecholamine receptors of the IIS [32]. Factors such as stress endured
early in life, trauma and polymorphisms are possible risk factors for loss of GR and
catecholamine sensitivity [33-35].
Suboptimal inflammatory response as a consequence of chronic stress prevents the
Eicosanoid Switch from functioning, since the switch to the resolution phase requires
recovered cortisol and insulin sensitivity. The initiation phase should have a maximum
duration of 8 to 12 hrs. PMN number and activation levels should reach their maximum
during this phase; longer duration caused by chronic stress could produce secondary damage
to neighbouring tissues due to the strong cytotoxic effects of activated PMNs [11].
Supramaximal activation of PMNs could sensitize the adapted IS if contact time between self-
antigens and the IS is significantly increased [11,29].
The crosstalk between the IS and stress axes is further evidenced by the fact that acute
production of high levels of catecholamines activate the IIS strongly [23], whereas
eicosanoids produced from AA induce the production of local and systemic catecholamines
[36]. Long-term activation may lead to catecholamine resistance and lack of eicosanoid
production. This situation, combined with the aforementioned possibility of resistance to

126
insulin and cortisol, provokes a suboptimal inflammatory response and consequently the
perpetuation and development of low-grade inflammation [26,37].
Nutritional factors and Resoleomics
Several dietary factors influence the activity of the IIS and the function of a wide range of
hormones, including cortisol, insulin and catecholamines. The dramatic changes in dietary
composition since the agricultural revolution (some 10,000 years ago) and, to a greater extent,
since the industrial revolution (some 200 years ago) have turned the intake of food into a
common daily danger and therefore a cause of continuous systemic stress. Some of these
changes include an increase in the omega 6/omega 3 fatty acid ratio, a high intake of
saturated fatty acids (SFA) and refined carbohydrates, the introduction of industrially
produced trans fatty acids, a lower intake of vitamins D and K, imbalanced intake of
antioxidants, high intake of anti-nutrients (eg lectines, saponins) and an altered intake of
dietary fibre [38].
The following section will discuss the impact of the changed ratio of polyunsaturated fatty
acids (PUFAs) and the intake of food with a high glycemic load on Resoleomics. The pro-
inflammatory effects of anti-nutrients present in cereals [39], potatoes [40], legumes [41], and
tomato have previously been extensively reviewed [7].
Role of PUFAs in inflammation
The intake ratio of α-linoleic acid (LA) (omega 6), α-linolenic acid (ALA) (omega 3),
docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) in the Western diet has
changed dramatically compared to the estimated intake ratio of hunter-gatherer diets from
2–3:1 to 10–20:1 in the contemporary diet [42,43]. All of these PUFAs are essential for normal
Resoleomics response, as they function as precursors for the special small mediators
responsible for the instigation and conclusion of the inflammatory response. One of the toxic
changes in fatty acid composition of food corresponds to the increased intake of LA since the
production of vegetable oils in 1913. Increased LA levels affect the inflammation process in
three ways (Figure 3):

127
Figure 3 Summary of the effects of high LA intake on Resoleomics 1. Increase of the omega 6 / omega 3 fatty acid ratio
2. Altered AA level
3. Increases of inflammatory compounds, leukotoxins (LK) production
Increased omega 6 / omega 3 fatty acid ratio
The inflammatory effect of a high omega 6/ omega 3 fatty acid ratio during inflammation has
been demonstrated in recent human studies [44,45], in vitro studies [46,47] and animal studies
[48,49]. The higher LA levels in phospholipids in plasma and cell membranes seem to be a
major factor responsible for incomplete Resoleomics reactions. Higher intake of omega 3
fatty acids in the form of DHA and EPA regulate the production of pro-inflammatory
cytokines and decrease LA levels in phospholipids in plasma and cell membranes [46,48]. The
conversion of LA and ALA into respectively AA, DHA and EPA depend on the same enzymes
in the desaturase and elongase cascade, with δ-6-desaturase as the rate-limiting enzyme
(Figure 4) [50].

128
Figure 4 Synthesis of unsaturated fatty acids in mammals by Desaturase and Elongase
Human trials investigating the effects of omega 3 dietary supplements showed significant improvements of symptoms in
patients suffering from diseases such as RA, inflammatory bowel disease, asthma, psoriasis, breast cancer and CVD.
However, full remission of symptoms was not achieved [43,51]. Our conclusion is that an increased intake of omega 3 alone
is not enough to restore Resoleomics; the intake of LA must be decreased as well.
LA effect on AA level
Higher AA levels in plasma result in more adequate inflammatory reactions, since AA is a
precursor of pro- and anti-inflammatory substances within the self-limiting inflammatory
process [52]. LA is the precursor for AA in the desaturase/ elongase conversion (Figure 4).
Theoretically, LA could be the source of a sufficient level of endogenous AA. However, higher
intake of LA does not deliver increased levels of AA in comparison to low intake [53,54]. To
achieve the required AA level, AA should be present in the regular diet [45]. The combined
situation of AA deficiency together with a reduced intake of omega 3 fatty acids such as DHA
and EPA (necessary for the flip flop reaction of LOX-5 and the Eicosanoid Switch [3]), enable a
perpetuation of the pro-inflammatory initiation phase and therefore of chronic
inflammation.

129
Increased production of leukotoxin
The third harmful effect of high LA intake is the possible production of so-called leukotoxins
(LK). High LA levels are metabolized by CYP2C9 in the liver into biologically active oxidation
products known as LK and leukotoxin diol (LTD). These metabolites promote oxidative stress
responses and the activation of NFkB and AP-1, increasing the systemic release of pro-
inflammatory cytokines [55]. LK and LTD are toxic for T cells, and can kill these cells with
pathways resembling necrosis and programmed cell death [56].
Role of high glycemic food in Inflammation
An abundant intake of high glycemic food appears to be related to an increased susceptibility
to the development of chronic inflammation, as has been demonstrated by several research
groups [57-59]. The consequences of a high carbohydrate diet are complex and multiple. The
pathways leading to disturbances of normal inflammation are:
1. High glycemic food intake increases inflammation markers
2. High glycemic food intake causes hyperglycemia and hyperinsulinemia leading to
disturbed balances in insulin growth factor-1 (IGF-1) and androgens
3. Chronic intake of high glycemic food causes hypoglycemia, which triggers central
stress axes
High Glycemic food increases inflammation markers
Various clinical trials have shown that an abundant intake of high glycemic food increases
inflammatory markers and markers of metabolic syndrome such as postprandial NFkB in
mononuclear cells [57], high sensitive-C-Reactive Protein (hs-CRP)[58], interleukin (IL)-6, IL-
7, IL-18 [60], levels of free radicals [59], cholesterol, triglycerides [61] and even blood pressure
[62]. Changes incurred by following a low glycemic diet include improved insulin sensitivity,
lower blood pressure and total cholesterol, which are all key markers of the metabolic
syndrome [58,60,61]. The high glucose-induced inflammatory response is accompanied by
hyperinsulinemia and insulin resistance, characteristic for people suffering from obesity
[57,59]. Increased hsCRP values, hyperinsulinemia and insulin resistance are strongly related
to CVD risk [60]. Glycemic index (GI) and glycemic load (GL) have therefore been proposed as
biomarkers and predictors for (chronic) inflammation [63].
Hyperglycemia and hyperinsulinemia
Cordain demonstrated that high glycemic food is a potential risk factor for inflammation
through disturbed signalling of mechanisms as a result of hyperglycemia and
hyperinsulinemia [64] (Figure 5b). Long exposure to high glucose levels in blood, which leads
to a slow recovery of the homeostasis, makes tissues vulnerable to disease [65]. High plasma

130
insulin can increase the production of IGF-1 and androgens. Both hormones are related to
disorders such as polycystic ovarian syndrome (PCOS) [66], epithelial cell cancer (breast,
prostate, colon) [67,68], acne [69], androgenic alopecia [70], and acanthosis nigricans [71].
Several pathways in this respect have been previously described in medical literature, but
these go beyond the scope of this article.
Figure 5 High glycemic food intake could cause inflammation and diseases as a result of
hyperinsulinemia. The pathways in the shaded area have been extensively described by Cordain [64] (part B). Part A: The consequential
reactive hyperglycemia is another deleterious pathway. Hyperglycemia is a danger signal, which activates the systemic
stress system. Chronic activation will suppress the IIS, resulting in low grade inflammation and an increased vulnerability for
excessive inflammation
Hypoglycemia triggers the systemic stress system
As previously mentioned, intake of a high glycemic diet can cause hyperglycemia and
hyperinsulinemia. Hyperglycemia will push abundant glucose via insulin into muscle and
adipocytes at the instigation of the inflammatory process. However, continuous intake of
high glycemic food results in reactive hypoglycemia, ie an energy-deficient situation which

131
threatens the homeostasis of the body. As a consequence, the brain will maintain its own
energy supply aimed at the survival of the organism (the selfish brain) [25]. To ensure
sufficient energy supply, the brain activates its systemic stress system to induce
gluconeogenesis (Figure 5a). Excreted catecholamines and cortisol will mobilize extra energy,
which is allocated with priority to the brain and to the activated IS, at the expense of other
body tissues [72].
On the basis of the above information and other referenced data, it seems plausible to state
that aspects of the Western diet, of the modern industrialised environment and of their
resultant lifestyles form a chronic danger to the body, triggering both the central stress axes
and the IIS into a state of chronic activity. This state seems to be a direct cause of the
development of low-grade inflammation and consequently of chronic inflammatory diseases
(Figure 5a).
Impact of current medication on Resoleomics
The role of the IIS is to limit the damage of inflammation in acute situations. Anti-
inflammatory medication can be used to dampen the immune response. Nowadays, as a
result of lifestyle changes, man is exposed to chronic inflammation and consequently to the
chronic use of anti-inflammatory medication, much of which in fact suppresses
Resoleomics. Current medication used to treat chronic inflammatory diseases does suppress
the symptoms of inflammation, but complete remission of the disease is seldom realized [73].
Resoleomics is hindered and complete resolution of the inflammation does not take place.
Modern chronic inflammatory diseases are treated by several groups of medication. In this
article we focus on rheumatoid arthritis (RA) medication as an example. Four groups of anti-
inflammatory RA medication are taken into account: the prostaglandin inhibitors
[Nonsteroidal anti-inflammatory drugs [NSAIDs: Aspirin (ASA) and COX-inhibitors], the
Glucocorticoids (GCs), the Disease Modifying Drugs [DMARDs: Methotrexate (MTX) and
Sulfasalazine (SSZ)] and the cytokine blockers [Biological agents: anti TNF-αlpha and IL-1
blockers]. The mechanisms of action and possible effects on the IIS and Resoleomics are
summarized from literature (see Table 1). Most current therapies target the IIS in an attempt to
inhibit the production of pro-inflammatory chemical mediators (Table 1). However, an
equally important target is the active induction of pro-resolution programs by stromal cells
such as fibroblasts within the inflamed tissues [74]. Inhibition of MIF [75] and production of
NO [76] are not addressed in this article.

132
Tab
le 1 Cu
rrent R
A treatm
ents an
d th
eir effect on
imm
un
e system cells an
d p
redicted
effect on
Reso
leom
ics
Med
ication
M
echan
ism o
f action
C
urren
t RA
treatmen
t effects on
Imm
un
e System
Cells
Pred
icted effects o
n
Reso
leom
ics Ph
ase 1:
initiatio
n, P
hase 2
: resolu
tion
,
Ph
ase 3: termin
ation
NSA
IDs: A
spirin
(ASA
)
[11,14,32
,77-83]
CO
X-1 in
hib
ition
, CO
X-2
acetylation
; PG
E2 ↓
; AT
Ls (15-ep
i-
LX
) ↑; A
ctivation
AL
X/FP
R2
recepto
r ↑; P
LA
2 ↓
: free AA
; PG
E
& L
T ↓
PM
N in
filtration
↓, P
GE
s ↓, ch
emo
kines ↓
;
Leu
cocyte accu
mu
lation
↓; N
eutro
ph
il recruitm
ent
↓; V
ascular p
ermeab
ility ↓; N
on
ph
log
istic
ph
ago
cytosis o
f apo
pto
tic neu
trop
hils ↑
Neg
ative: PG
< L
T levels: p
hase
1 ↑; P
ositive: P
G n
ot
com
pletely ↓
: switch
from
ph
ase 1 to p
hase 2
↑; P
ositive:
AT
Ls ↑
: ph
ase 2 ↑
, ph
ase 3 ↑
NSA
IDs: C
OX
-
inh
ibito
rs [84
,85]
CO
X-2
inh
ibitio
n >
CO
X-1
inh
ibitio
n; P
GE
2 ↓
, LT
B4
↑, P
GF2α
↓, P
GD
2 ↓
CO
X-2
expressio
n m
acrop
hag
es ↓: C
hem
otaxis
neu
trop
hils, eo
sino
ph
ils & m
on
ocytes in
to syn
oviu
m
↓
Neg
ative: PG
< L
TB
4 levels:
Ph
ase 1 ↑; sw
itch fro
m p
hase 1
to 2
↓; sw
itch fro
m p
hase 2
to
3 ↓
Glu
coco
rticostero
id
(GC
s) [32,80
,86
,87]
Tran
scriptio
n o
f IKB
↑: N
FkB ↓
;
Tran
scriptio
n b
y GC
R &
CR
EB
-
bin
din
g p
rotein
(CB
P) ↓
; PL
A2
↓:
free AA
, PG
E &
LT
↓; A
nn
exin-1 ↑
;
Activatio
n o
f the A
LX
/FPR
2
recepto
r ↑
PM
N in
filtration
↓, P
GE
s ↓, ch
emo
kines ↓
;
Leu
cocyte accu
mu
lation
↓; N
eutro
ph
il recruitm
ent
↓; N
FkB –
transcrip
tion
↓; E
xpressio
n
inflam
mato
ry gen
es ↓; M
acrop
hag
e mig
ration
&
ph
ago
cytosis ↑
Neg
ative: PG
E, L
T ↓
: switch
from
ph
ase 1 to 2
↓; C
ortiso
l
resistance: sw
itch fro
m p
hase
1 to p
hase 2
↓ o
r no
switch
;
Lip
oxin
s ↓: sw
itch fro
m p
hase
2 to
3 ↓

133
DM
AR
Ds:
Meth
otrexate (M
TX
)
[88
-102
]
Folate an
alog
s: 1. Folate-d
epen
den
t
enzym
es ↓: 1a. T
hym
idylate
synth
etase; 1b. A
ICA
R
transfo
rmylase; 1c. D
ihyd
rofo
late
redu
ctase; 2. C
ytoso
l pero
xide
(RO
S) ↑
Ad
1a. Synth
esis of D
NA
& R
NA
↓; T
-cell
pro
liferation
& p
rotein
- & cyto
kine-exp
ression
↓; L
T
& IL
-1 ↓; A
d 1b
. Ad
eno
sine ↑
: NK
-cell, mo
no
cytes &
macro
ph
ages fu
nctio
nin
g ↓
; Cyto
kine syn
thesis
TN
F-α, IL
-1, IL-6
& IL
-8↓; 1c. T
HF ↓
: pu
rine &
pyrim
idin
e ↓; A
d 2
. T-cell ap
op
tosis ↑
Neg
ative: cytokin
es ↓,
T-cell activity ↓
, LT
↓: sw
itch
from
ph
ase 1 to 2
↓ o
r no
switch
DM
AR
Ds:
Sulp
hasalazin
e (SSZ)
[86
]
SSZ: stro
ng
& p
oten
t inh
ibito
r of
NFkB
-activation
; 5-amin
o
acytelate (5-ASA
): PG
↓; su
lph
a-
pyrid
ine m
etabo
lites
NFkB
activation
↓: IL
-2 activated
T-cells ↓
, TN
F-α &
IL-1 m
acrop
hag
es ↓; A
ntib
od
y plasm
a cells ↓;
Neu
trop
hils, m
on
ocytes, m
acroh
ages, g
ranu
locyte
activation
↓, IK
B ↓
: NFkB
translo
cation
↓ &
transcrip
tion
of cyto
kines, ad
hesio
n m
olecu
les,
chem
okin
es ↓: C
OX
-2 &
PG
↓
Neg
ative: Imm
un
e cell activity
↓: sw
itch fro
m p
hase 1 to
2 ↓
Bio
log
ical agen
ts: An
ti
TN
F-alph
a [54,10
3-105] T
NF-alp
ha sig
nallin
g o
f
mo
no
cytes, PM
N’s, T
-cells,
end
oth
elial cells, syno
vial
fibro
blasts &
adip
ocytes ↓
; CO
X-2
ind
uctio
n ↓
Mo
no
cyte activation
, cytokin
e & P
G release ↓
; PM
N
prim
ing
, apo
pto
sis and
oxid
ative bu
rst; T-cell
apo
pto
sis, clon
al regu
lation
& T
-cell recepto
r ↓;
En
do
thelial-cell ad
hesio
n m
olecu
le expressio
n,
cytokin
e release ↓, syn
ovial fib
rob
last pro
liferation
,
collag
en syn
thesis, M
MP
& cyto
kine release ↓
;
Ad
ipo
cyte FFA release ↑
Neg
ative: Imm
un
e cell activity
↓: sw
itch fro
m p
hase 1 to
2 ↓

134
Bio
log
ical agen
ts: IL-1
blo
cker [54,10
3-105]
IL-1 sig
nallin
g m
on
ocytes, B
-cells,
end
oth
elial cell, syno
vial
fibro
blasts, ch
ron
dro
cytes ↓; C
OX
-
2 in
du
ction
↓
Syno
vial fibro
blast cyto
kine, ch
emo
kine, M
MP
, iNO
S
& P
G release ↓
; Mo
no
cytes cytokin
e, RO
I & P
G
release ↓; O
steoclast activity ↑
; GA
G syn
thesis ↓
,
iNO
S ↑, M
MP
& ag
grecan
ase; En
do
thelial-cell
adh
esion
mo
lecule exp
ression
↓
Neg
ative: Imm
un
e cell activity
↓: sw
itch fro
m p
hase 1 to
2 ↓

135
Positive effect of ASA and GCs on Resoleomics
Medical intervention should stimulate the endogenous pathways of resolution and two drugs
already known to possess these qualities are central to contemporary medicine:
glucocorticoids (GCs) [77] and aspirin (ASA) [106,107]. It is apparent that ASA and GCs have a
positive effect on Resoleomics, while other medications prolong the initiation phase,
tempering and/ or blocking the resolution and termination phase of Resoleomics in various
ways (Table 1). The positive effect of ASA on Resoleomics can be ascribed to its ability to
produce ASA-triggered lipoxins (ATLs) through acetylation (and not through an irreversible
inhibition) of the COX-2 enzymes [78]. These ATLs show many pro-resolving properties,
which are essential in the resolution and termination phase of the inflammation process
[79,108]. Long-term intake of high doses of ASA blocks PGE2 production and initiates the
resolution phase without affecting the biosynthesis of other pro-resolving mediators [108].
Low and high doses of ASA increase the production of lipoxin A4 (LXA4) and 15-epi-LXA4 in
the rat brain, suggesting that ASA could protect against neuroinflammation [109]. However,
because of its side effects, ASA is no longer the treatment of choice for RA. In high doses,
inhibition of the COX-1 enzyme by ASA is responsible for damage to the stomach lining.
ASA and also GCs activate the ALX/FRP2 receptor, making them the ideal collaborator in the
resolution process [77]. GCs-induced annexin-1 protein (ANXA1) [110,111] as well as ASA-
induced ATLs act on the same ALX/FPR2 receptor and dampen PMN infiltration [77,80].
ANXA1 also inhibits the phospholipids A2 enzyme (PLA2). Reduced PLA2 activity appears to
reduce AA release from the cell membrane [32,112], which possibly leads to decreased levels of
both PGs and LTs and to the delay of resolution. Besides their anti-inflammatory effects, GCs
have a positive influence on resolution by enhancing macrophage migration and
phagocytosis [11,113].
Adverse effects of medication on Resoleomics
The use of anti-inflammatory medication without the capacity to induce (complete)
resolution should be considered solution-toxic, ie hindering Resoleomics. NSAIDs are strong
inhibitors of COX-2 and less of COX-1 enzymes [114]. Almost complete COX-2 inhibition
decreases the PGs synthesis, and consequently leads to a higher production of LTs via LOX-5
in PMNs [115]. PGE2 and PgD2 decrease the activity of LOX-5, decreasing neutrophil activity
and facilitating the end of the inflammatory phase and the instigation of resolution.
Immune-suppressors such as SSZ (and less powerful GCs) almost completely block NF-kB
transcription, leading to insufficient cytokine production and suboptimal inflammation [86].
Again the resolution process will not be completed, with perpetuation of inflammation as the
logical consequence.

136
Perhaps the most deleterious drugs, interfering negatively with resolution, are TNF-alpha
inhibitors such as anti TNF-alpha and MTX. MTX inhibits the proliferation of the IIS cells,
decreasing the production and accumulation of adenosine within the IS cells [88,116]. These
effects lead to rapid anti-inflammatory effects and symptom release. However, because of its
side effects and incomplete resolution, this medication is qualified as solution-toxic. This
conclusion is supported by many patients who have discontinued this treatment [73].
Another group of possible solution-toxic drugs are biological agents with an inhibiting effect
on TNF-alpha and IL-1. Biological agents together with DMARDS (Table 1) are strong anti-
inflammatory compounds, decreasing the production of pro-inflammatory cytokines. The
absence or insufficient activity of pro-inflammatory cytokines decreases cell communication
and induction of COX-2 in activated neutrophils. This can lead to less production of
resolution substances such as PgE2, PgD2 and lipoxins [54,103]. Furthermore, DMARDs and
biological agents appear to reduce the functioning and number of IIS cells, causing
suboptimal inflammation and possibly inflammation perpetuation [104].
Discussion
Long-term activity of the IIS results in low-grade inflammation and chronic disease. Over the
past years, ideas regarding the treatment of inflammation have started to change as evidence
accumulates which shows that, although the targeting of infiltrating immune cells can control
the inflammatory response, it does not lead to its complete resolution and a return to
homeostasis, which is essential for healthy tissue and good health in general.
Hotamisligil describes how low-grade, chronic inflammation (‘meta-inflammation’) induced
by a nutritional and metabolic surplus, is accompanied by disturbed metabolic pathways and
chronic metabolic disorders. He states that this inflammatory response differs from the
classical inflammation response caused by injury [117]. However, others have shown that the
classical response of the IIS dealing with injuries can be linked to activation of the central
stress axes [26,28]. This article specifically discusses the relationship between the over-
activated systemic stress system and the self-limited process of inflammation, known as
Resoleomics, executed and controlled by the innate immune system (IIS).
Changes in lifestyle which are new to our evolutionary process should be considered a major
trigger in causing chronic activation of the IS and consequently of the central stress axes and
vice versa, thereby leading to chronic diseases such as cardiovascular diseases (CVD),
diabetes, respiratory diseases, mental disorders, auto-immune diseases (AID) and cancers.
This article evaluates two of the lifestyle changes which contribute to long-term activity of the
ISS, namely, nutrition and continuous psycho-emotional stress. Other risk factors such as

137
physical inactivity [6], genetic susceptibility [118], smoking, environmental toxicity and shift
work [119] fall beyond the scope of this article but should not be ruled out.
Nutrition is an important factor in understanding the development of chronic inflammation.
The current Western diet can disturb the resolution response in various ways (Figure 6). In the
Ancestral human diet, foodstuffs with an increased risk of inflammation were virtually
unknown, while nutrients able to activate the IIS are now abundant in our diet [38,120].
Cordain’s research has focused on relating these anti-nutrients in food (eg lectines,
saponines) to the development of chronic inflammation and autoimmune diseases (AID)
[7,39]. Fortunately, it seems that the human body possesses a strong capacity to recover from
illness. If our genes are exposed to their ‘original’ environment by intake of an ancestral
human diet, their function can recover rapidly. Research has shown that obese persons
improve their blood markers after just 10 days following a paleolithic diet consisting of fish,
lean meat, fruit, vegetables and nuts [121]. Similar results have been found in a study with
aboriginals suffering from Diabetes II, who showed normalized blood markers after returning
to their traditional lifestyle for seven weeks [122].
Figure 6 Reflection of the working mechanism demonstrating how several nutritional factors
could induce and inhibit inflammation

138
People suffering from chronic inflammatory disease demonstrate over-activated central stress
axes, which then lead to catecholamines, cortisol and insulin resistance. McGowan et al [123]
show the impact of childhood abuse on the epigenetic pattern of different genes including the
gene for GR in the hippocampus. They found a decreased level of GR and an increased
methylation pattern of the GR gene, giving rise to a situation of lower cortisol sensibility and
altered HPA stress responses. This could make people more vulnerable to developing diseases.
An altered sensitivity to cortisol has been linked to diseases such as rheumatoid arthritis (RA)
[124], post-traumatic stress syndrome [125], chronic fatigue syndrome [126], inflammatory
diseases and AID in general [127].
The key priority in the treatment of people with chronic inflammation is to induce the
Eicosanoid Switch to the anti-inflammatory resolution phase. Long-lasting cortisol resistance
and insulin resistance will definitely delay or block complete resolution. The combination of
local factors (ie DHA deficiency, low levels of protectins) disturbing the process of complete
resolution (ie Resoleomics) and the absence of adequate NE and cortisol signalling can be
responsible for perpetuatual inflammation by delaying the resolution phase of the
inflammatory response (Figure 7).
Figure 7 Chronic over-activation of the systemic stress system as a result of external stressors
plays a central role in the development of chronic inflammatory diseases. Current intervention with anti-inflammatory medication suppresses Resoleomics and the IIS and so enhance the over-
activation of the systemic stress system

139
Current anti-inflammatory medication used in RA treatment is aimed at the suppression of
the IIS and its inflammatory response and thus hinders Resoleomics. In addition, these
medication interventions do not solve underlying catecholamine, cortisol and insulin
resistance, and consequently making it impossible to achieve full recovery of the chronic
inflammation. This suggests that chronic use of anti-inflammatory medication in fact
impedes the body from making a full recovery. Furthermore, the ongoing low-grade
inflammation will continuously trigger the activity of the systemic stress system [28].
Health care should focus on early detection of silent, ongoing and low-grade inflammation in
order to avoid the development of many chronic diseases. Further research is needed to
validate a questionnaire which addresses early symptoms of chronic low-grade inflammation,
ie avoidance of exercise, fatigue, emotional flatness, social isolation, decreased libido, hyper or
hyposomnia, obsessive behaviour or sensitivity to addiction [6,128].
We have made an effort to demonstrate that the science of Resoleomics can help to find new
ways to treat people suffering from diseases based on chronic inflammation. Since over-
activated central stress axes directly delay Resoleomics, and thereby delay the resolution of
inflammation, treatment should focus on restoring the central stress system to its default,
healthy homeostasis. Dietary changes, psycho-emotional stress release and physical activity
should always be included in treatment of all chronic inflammatory diseases.

140
Abbreviations
AA, Arachidonic acid; ACh, Acetylcholine; AID, Autoimmune diseases; ALA, α-linolenic acid;
ALX/FPR2, Lipoxin A(4) receptor; ANXA 1, Annexin 1 protein; AP-1, Activator protein 1; ASA,
Aspirin; ATLs, Stable aspirin-triggered lipoxin; COX, Cyclo-oygenase; CRP, High sensitive-C-
Reactive Protein; CVD, Cardiovascular diseases; DHA, Docosahexaenoic acid; DMARDs,
Disease Modifying Drugs; EPA, Eicosapentaenoic acid; GI, Glycemic index; GL, Glycemic load;
GCs, Glucocorticoids; HPA, Hypothalamus-pituitary-adrenal; IGF-1, Insulin growth factor-1;
IS, Immune system; IIS, Innate immune system; IL, Interleukin; LA, α-linoleic acid; LC, Locus
Coeruleus; LOX, Lipoxygenase; LK, Leukotoxins; LTs, Leukotrienes; LTD, Leukotoxin diol; LXs,
Lipoxins; MTX, Methotrexate; NE, Norepinephrine (ie noradrenaline); NF-kB, Nuclear factor-
KappaB; NSAIDs, Nonsteroidal anti-inflammatory drugs; PCOS, Polycystic ovarian syndrome;
PGs/ PGE2/ PGD2/ PGF2a, Prostaglandins/ prostaglandin E2, D2, F2a; PLA2, Phospholipase A2
enzyme; PMNs, Polymorphonuclear leukocytes; PUFAs, Polyunsaturated fatty acids; RA,
Rheumatoid arthritis; SAM, Sympathetic-adrenal-medulla; SFA, Saturated fatty acids; SNS,
Sympathetic nervous system; TNF, Tumour necrosis factor.
Competing interests
The author(s) declare that they have no competing interests.
Authors’ contributions
MMB executed an analysis and review of the relationship between chronic inflammatory
pathways and the central stress systems, Resoleomics and nutrition. MMB also drafted the
manuscript. MLvW reviewed the MOA of currently used anti-inflammatory medication and its
effect on Resoleomics. LP played a central role in integrating the results of various stressors on
chronic inflammation pathways and also acted as lead reviewer. All authors have approved
the final manuscript.
Authors’ information
MMB and MLvW, MD treat patients with chronic diseases in a private practice. LP, a practising
psychoneuroimmunologist and director of the master in CPNI at the University of Gerona,
Spain, has developed valuable insights into the metabolic pathways of chronic diseases, which
he has applied in the treatment of numerous patients.

141
References
4. Kolb H, Mandrup-Poulsen T: The global diabetes epidemic as a consequence of lifestyle-
induced low-grade inflammation. Diabetologia 2010, 53:10–20.
5. Miller AH, Maletic V, Raison CL: Inflammation and its discontents: the role of cytokines
in the pathophysiology of major depression. Biol Psychiatry 2009, 65:732–741.
6. Serhan CN, Chiang N: Novel endogenous small molecules as the checkpoint controllers
in inflammation and resolution: entree for resoleomics. Rheum Dis Clin North Am 2004,
30:69–95.
7. Macaulay V, Richards M, Hickey E, Vega E, Cruciani F, Guida V, Scozzari R, Bonne-Tamir
B, Sykes B, Torroni A: The emerging tree of West Eurasian mtDNAs: a synthesis of
control-region sequences and RFLPs. Am J Hum Genet 1999, 64:232–249.
8. Smith E, Morowitz HJ: Universality in intermediary metabolism. Proc Natl Acad Sci U S
A 2004, 101:13168–13173.
9. Pruimboom L: Physical inactivity is a disease synonymous for a non-permissive brain
disorder. Med Hypothesis 2011, 77:708–713.
10. Cordain L, Toohey L, Smith MJ, Hickey MS: Modulation of immune function by dietary
lectins in rheumatoid arthritis. Br J Nutr 2000, 83:207–217.
11. Straub RH, Cutolo M, Buttgereit F, Pongratz G: Energy regulation and neuroendocrine-
immune control in chronic inflammatory diseases. J Intern Med 2010, 267:543–560.
12. Peters A, Hitze B, Langemann D, Bosy-Westphal A, Muller MJ: Brain size, body size and
longevity. Int J Obes 2005, 34:1349–1352.
13. Nathan C, Ding A: Nonresolving inflammation. Cell 2010, 140:871–882.
14. Serhan CN: Resolution phase of inflammation: novel endogenous anti-inflammatory
and proresolving lipid mediators and pathways. Annu Rev Immunol 2007, 25:101–137.
15. Muskiet FAJ: The evolutionairy background, cause and consequences of chronic system
low grade inflammation. Significance for clinical chemistry. Ned Tijdschr Klin Chem
Labgeneesk 2011, 36:199–214.
16. Bannenberg GL, Chiang N, Ariel A, Arita M, Tjonahen E, Gotlinger KH, Hong S, Serhan
CN: Molecular circuits of resolution: formation and actions of resolvins and protectins. J
Immunol 2005, 174:4345–4355.
17. Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA:
Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med 1999,
5:698–701.
18. Serhan CN, Maddox JF, Petasis NA, Akritopoulou-Zanze I, Papayianni A, Brady HR,
Colgan SP, Madara JL: Design of lipoxin A4 stable analogs that block transmigration and
adhesion of human neutrophils. Biochemistry 1995, 34:14609–14615.

142
19. Serhan CN, Yang R, Martinod K, Kasuga K, Pillai PS, Porter TF, Oh SF, Spite M: Maresins:
novel macrophage mediators with potent antiinflammatory and proresolving actions. J
Exp Med 2009, 206:15–23.
20. Willoughby DA, Moore AR, Colville-Nash PR: COX-1, COX-2, and COX-3 and the future
treatment of chronic inflammatory disease. Lancet 2000, 355:646–648.
21. Maddox JF, Serhan CN: Lipoxin A4 and B4 are potent stimuli for human monocyte
migration and adhesion: selective inactivation by dehydrogenation and reduction. J
Exp Med 1996, 183:137–146.
22. Maderna P, Godson C: Taking insult from injury: lipoxins and lipoxin receptor agonists
and phagocytosis of apoptotic cells. Prostaglandins Leukot Essent Fatty Acids 2005,
73:179–187.
23. Miller GE, Chen E, Sze J, Marin T, Arevalo JM, Doll R, Ma R, Cole SW: A functional
genomic fingerprint of chronic stress in humans: blunted glucocorticoid and increased
NF-kappaB signaling. Biol Psychiatry 2008, 64:266–272.
24. Rosas-Ballina M, Tracey KJ: The neurology of the immune system: neural reflexes
regulate immunity. Neuron 2009, 64:28–32.
25. Elenkov IJ: Neurohormonal-cytokine interactions: implications for inflammation,
common human diseases and well-being. Neurochem Int 2008, 52:40–51.
26. Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES: The sympathetic nerve–an integrative
interface between two supersystems: the brain and the immune system. Pharmacol Rev
2000, 52:595–638.
27. Kin NW, Sanders VM: It takes nerve to tell T and B cells what to do. J Leukoc Biol 2006,
79:1093–1104.
28. Peters A, Schweiger U, Pellerin L, Hubold C, Oltmanns KM, Conrad M, Schultes B, Born J,
Fehm HL: The selfish brain: competition for energy resources. Neurosci Biobehav Rev
2004, 28:143–180.
29. Miller GE, Cohen S, Ritchey AK: Chronic psychological stress and the regulation of pro-
inflammatory cytokines: a glucocorticoid-resistance model. Health Psychol 2002,
21:531–541.
30. Guarini S, Cainazzo MM, Giuliani D, Mioni C, Altavilla D, Marini H, Bigiani A, Ghiaroni V,
Passaniti M, Leone S, et al: Adrenocorticotropin reverses hemorrhagic shock in
anesthetized rats through the rapid activation of a vagal anti-inflammatory pathway.
Cardiovasc Res 2004, 63:357–365.
31. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H,
Ulloa L, et al: Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of
inflammation. Nature 2003, 421:384–388.
32. Liezmann C, Klapp B, Peters EM: Stress, atopy and allergy: A re-evaluation from a
psychoneuroimmunologic persepective. Dermatol Endocrinol 2011, 3:37–40.

143
33. Whitaker AM, Sulzer J, Walker E, Mathis K, Molina PE: Sympathetic modulation of the
host defense response to infectious challenge during recovery from hemorrhage.
Neuroimmunomodulation 2010, 17:349–358.
34. Straub RH: Physiologische Grundlagen. Lehrbuch der klinischen Pathophysiologie
komplexer chronischer Erkrankungen. Vandenhoe & Ruprecht 2006.
35. Rhen T, Cidlowski JA: Antiinflammatory action of glucocorticoids–new mechanisms
for old drugs. N Engl J Med 2005, 353:1711–1723.
36. Heim C, Newport DJ, Bonsall R, Miller AH, Nemeroff CB: Altered pituitary-adrenal axis
responses to provocative challenge tests in adult survivors of childhood abuse. Am J
Psychiatry 2001, 158:575–581.
37. Danese A, Moffitt TE, Pariante CM, Ambler A, Poulton R, Caspi A: Elevated inflammation
levels in depressed adults with a history of childhood maltreatment. Arch Gen Psychiatry
2008, 65:409–415.
38. Simmons RA: Role of metabolic programming in the pathogenesis of beta-cell failure in
postnatal life. Rev Endocr Metab Disord 2007, 8:95–104.
39. Malcher-Lopes R, Buzzi M: Glucocorticoid-regulated crosstalk between arachidonic acid
and endocannabinoid biochemical pathways coordinates cognitive-, neuroimmune-,
and energy homeostasis-related adaptations to stress. Vitam Horm 2009, 81:263–313.
40. Pace TW, Hu F, Miller AH: Cytokine-effects on glucocorticoid receptor function:
relevance to glucocorticoid resistance and the pathophysiology and treatment of major
depression. Brain Behav Immun 2007, 21:9–19.
41. Cordain L, Eaton SB, Sebastian A, Mann N, Lindeberg S, Watkins BA, O'Keefe JH, Brand-
Miller J: Origins and evolution of the Western diet: health implications for the 21st
century. Am J Clin Nutr 2005, 81:341–354.
42. Cordain L: Cereal grains: humanity's double-edged sword. World Rev Nutr Diet 1999,
84:19–73.
43. Patel B, Schutte R, Sporns P, Doyle J, Jewel L, Fedorak RN: Potato glycoalkaloids
adversely affect intestinal permeability and aggravate inflammatory bowel disease.
Inflamm Bowel Dis 2002, 8:340–346.
44. Vasconcelos IM, Oliveira JT: Antinutritional properties of plant lectins. Toxicon 2004,
44:385–403.
45. Simopoulos AP: Overview of evolutionary aspects of omega 3 fatty acids in the diet.
World Rev Nutr Diet 1998, 83:1–11.
46. Simopoulos AP: Evolutionary aspects of diet, the omega-6/omega-3 ratio and genetic
variation: nutritional implications for chronic diseases. Biomed Pharmacother 2006,
60:502–507.
47. Guebre-Egziabher F, Rabasa-Lhoret R, Bonnet F, Bastard JP, Desage M, Skilton MR, Vidal
H, Laville M: Nutritional intervention to reduce the n-6/n-3 fatty acid ratio increases

144
adiponectin concentration and fatty acid oxidation in healthy subjects. Eur J Clin Nutr
2008, 62:1287–1293.
48. Liou YA, King DJ, Zibrik D, Innis SM: Decreasing linoleic acid with constant alpha-
linolenic acid in dietary fats increases (n-3) eicosapentaenoic acid in plasma
phospholipids in healthy men. J Nutr 2007, 137:945–952.
49. Wang L, Reiterer G, Toborek M, Hennig B: Changing ratios of omega-6 to omega-3 fatty
acids can differentially modulate polychlorinated biphenyl toxicity in endothelial cells.
Chem Biol Interact 2008, 172:27–38.
50. Chene G, Dubourdeau M, Balard P, Escoubet-Lozach L, Orfila C, Berry A, Bernad J, Aries
MF, Charveron M, Pipy B: n-3 and n-6 polyunsaturated fatty acids induce the expression
of COX-2 via PPARgamma activation in human keratinocyte HaCaT cells. Biochim
Biophys Acta 2007, 1771:576–589.
51. Ghosh S, Novak EM, Innis SM: Cardiac proinflammatory pathways are altered with
different dietary n-6 linoleic to n-3 alpha-linolenic acid ratios in normal, fat-fed pigs.
Am J Physiol Heart Circ Physiol 2007, 293:H2919–H2927.
52. Riediger ND, Azordegan N, Harris-Janz S, Ma DW, Suh M, Moghadasian MH: 'Designer
oils' low in n-6:n-3 fatty acid ratio beneficially modifies cardiovascular risks in mice. Eur
J Nutr 2009, 48:307–314.
53. Nakamura MT, Nara TY: Structure, function, and dietary regulation of delta6, delta5, and
delta9 desaturases. Annu Rev Nutr 2004, 24:345–376.
54. Calder PC: Session 3: Joint Nutrition Society and Irish Nutrition and Dietetic Institute
Symposium on 'Nutrition and autoimmune disease' PUFA, inflammatory processes and
rheumatoid arthritis. Proc Nutr Soc 2008, 67:409–418.
55. Ferrucci L, Cherubini A, Bandinelli S, Bartali B, Corsi A, Lauretani F, Martin A, Andres-
Lacueva C, Senin U, Guralnik JM: Relationship of plasma polyunsaturated fatty acids to
circulating inflammatory markers. J Clin Endocrinol Metab 2006, 91:439–446.
56. Adam O, Tesche A, Wolfram G: Impact of linoleic acid intake on arachidonic acid
formation and eicosanoid biosynthesis in humans. Prostaglandins Leukot Essent Fatty
Acids 2008, 79:177–181.
57. McInnes IB, Schett G: Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev
Immunol 2007, 7:429–442.
58. Viswanathan S, Hammock BD, Newman JW, Meerarani P, Toborek M, Hennig B:
Involvement of CYP 2 C9 in mediating the proinflammatory effects of linoleic acid in
vascular endothelial cells. J Am Coll Nutr 2003, 22:502–510.
59. Mangan DF, Taichman NS, Lally ET, Wahl SM: Lethal effects of Actinobacillus
actinomycetemcomitans leukotoxin on human T lymphocytes. Infect Immun 1991,
59:3267–3272.

145
60. Dickinson S, Hancock DP, Petocz P, Ceriello A, Brand-Miller J: High-glycemic index
carbohydrate increases nuclear factor-kappaB activation in mononuclear cells of young,
lean healthy subjects. Am J Clin Nutr 2008, 87:1188–1193.
61. Liu S, Manson JE, Buring JE, Stampfer MJ, Willett WC, Ridker PM: Relation between a
diet with a high glycemic load and plasma concentrations of high-sensitivity C-reactive
protein in middle-aged women. Am J Clin Nutr 2002, 75:492–498.
62. Hu Y, Block G, Norkus EP, Morrow JD, Dietrich M, Hudes M: Relations of glycemic index
and glycemic load with plasma oxidative stress markers. Am J Clin Nutr 2006, 84:70–76.
quiz:266–267.
63. Esposito K, Marfella R, Ciotola M, Di Palo C, Giugliano F, Giugliano G, D'Armiento M,
D'Andrea F, Giugliano D: Effect of a mediterranean-style diet on endothelial dysfunction
and markers of vascular inflammation in the metabolic syndrome: a randomized trial.
JAMA 2004, 292:1440–1446.
64. Du H, der AD van, van Bakel MM, van der Kallen CJ, Blaak EE, van Greevenbroek MM,
Jansen EH, Nijpels G, Stehouwer CD, Dekker JM, Feskens EJ: Glycemic index and
glycemic load in relation to food and nutrient intake and metabolic risk factors in a
Dutch population. Am J Clin Nutr 2008, 87:655–661.
65. Pereira MA, Swain J, Goldfine AB, Rifai N, Ludwig DS: Effects of a low-glycemic load diet
on resting energy expenditure and heart disease risk factors during weight loss. JAMA
2004, 292:2482–2490.
66. Halton TL, Willett WC, Liu S, Manson JE, Albert CM, Rexrode K, Hu FB: Low-
carbohydrate-diet score and the risk of coronary heart disease in women. N Engl J Med
2006, 355:1991–2002.
67. Cordain L, Eades MR, Eades MD: Hyperinsulinemic diseases of civilization: more than
just Syndrome X. Comp Biochem Physiol A Mol Integr Physiol 2003, 136:95–112.
68. Nappo F, Esposito K, Cioffi M, Giugliano G, Molinari AM, Paolisso G, Marfella R,
Giugliano D: Postprandial endothelial activation in healthy subjects and in type 2
diabetic patients: role of fat and carbohydrate meals. J Am Coll Cardiol 2002, 39:1145–
1150.
69. Ehrmann DA: Polycystic ovary syndrome. N Engl J Med 2005, 352:1223–1236.
70. Goodwin PJ, Ennis M, Bahl M, Fantus IG, Pritchard KI, Trudeau ME, Koo J, Hood N: High
insulin levels in newly diagnosed breast cancer patients reflect underlying insulin
resistance and are associated with components of the insulin resistance syndrome.
Breast Cancer Res Treat 2009, 114:517–525.
71. Tran TT, Naigamwalla D, Oprescu AI, Lam L, McKeown-Eyssen G, Bruce WR, Giacca A:
Hyperinsulinemia, but not other factors associated with insulin resistance, acutely
enhances colorectal epithelial proliferation in vivo. Endocrinology 2006, 147:1830–1837.

146
72. Smith RN, Mann NJ, Braue A, Makelainen H, Varigos GA: A low-glycemic-load diet
improves symptoms in acne vulgaris patients: a randomized controlled trial. Am J Clin
Nutr 2007, 86:107–115.
73. Matilainen V, Laakso M, Hirsso P, Koskela P, Rajala U, Keinanen-Kiukaanniemi S: Hair
loss, insulin resistance, and heredity in middle-aged women. A population-based study.
J Cardiovasc Risk 2003, 10:227–231.
74. Barth JH, Ng LL, Wojnarowska F, Dawber RP: Acanthosis nigricans, insulin resistance
and cutaneous virilism. Br J Dermatol 1988, 118:613–619.
75. Peters A, Langemann D: Build-ups in the supply chain of the brain: on the
neuroenergetic cause of obesity and type 2 diabetes mellitus. Front Neuroenergetics
2009, 1:2.
76. Gaujoux-Viala C, Smolen JS, Landewe R, Dougados M, Kvien TK, Mola EM, Scholte-
Voshaar M, van Riel P, Gossec L: Current evidence for the management of rheumatoid
arthritis with synthetic disease-modifying antirheumatic drugs: a systematic literature
review informing the EULAR recommendations for the management of rheumatoid
arthritis. Ann Rheum Dis 2010, 69:1004–1009.
77. Filer A, Pitzalis C, Buckley CD: Targeting the stromal microenvironment in chronic
inflammation. Curr Opin Pharmacol 2006, 6:393–400.
78. Das UN: Vagal nerve stimulation in prevention and management of coronary heart
disease. World J Cardiol 2010, 3:105–110.
79. Desneves KJ, Todorovic BE, Cassar A, Crowe TC: Treatment with supplementary
arginine, vitamin C and zinc in patients with pressure ulcers: a randomised controlled
trial. Clin Nutr (Edinburgh, Scotland) 2005, 24:979–987.
80. Perretti M, Chiang N, La M, Fierro IM, Marullo S, Getting SJ, Solito E, Serhan CN:
Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with
glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat Med 2002,
8:1296–1302.
81. Roth GJ, Stanford N, Majerus PW: Acetylation of prostaglandin synthase by aspirin. Proc
Natl Acad Sci U S A 1975, 72:3073–3076.
82. Serhan CN: Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of
endogenous anti-inflammation and resolution. Prostaglandins Leukot Essent Fatty
Acids 2005, 73:141–162.
83. Gilroy DW: The role of aspirin-triggered lipoxins in the mechanism of action of aspirin.
Prostaglandins Leukot Essent Fatty Acids 2005, 73:203–210.
84. Claria J, Serhan CN: Aspirin triggers previously undescribed bioactive eicosanoids by
human endothelial cell-leukocyte interactions. Proc Natl Acad Sci U S A 1995, 92:9475–
9479.

147
85. Paul-Clark MJ, Van Cao T, Moradi-Bidhendi N, Cooper D, Gilroy DW: 15-epi-lipoxin A4-
mediated induction of nitric oxide explains how aspirin inhibits acute inflammation. J
Exp Med 2004, 200:69–78.
86. Serhan CN, Takano T, Chiang N, Gronert K, Clish CB: Formation of endogenous
“antiinflammatory” lipid mediators by transcellular biosynthesis: Lipoxins and aspirin-
triggered lipoxins inhibit neutrophil recruitment and vascular permeability. Am J Respir
Crit Care Med 2000, 161:S95–S101.
87. Bertolini A, Ottani A, Sandrini M: Dual acting anti-inflammatory drugs: a reappraisal.
Pharmacol Res 2001, 44:437–450.
88. Warner TD, Mitchell JA: Cyclooxygenases: new forms, new inhibitors, and lessons from
the clinic. FASEB J 2004, 18:790–804.
89. Wahl C, Liptay S, Adler G, Schmid RM: Sulfasalazine: a potent and specific inhibitor of
nuclear factor kappa B. J Clin Invest 1998, 101:1163–1174.
90. Barnes PJ: Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin
Sci (Lond) 1998, 94:557–572.
91. Genestier L, Paillot R, Quemeneur L, Izeradjene K, Revillard JP: Mechanisms of action of
methotrexate. Immunopharmacology 2000, 47:247–257.
92. Baggott JE, Morgan SL, Ha TS, Alarcon GS, Koopman WJ, Krumdieck CL: Antifolates in
rheumatoid arthritis: a hypothetical mechanism of action. Clin Exp Rheumatol 1993,
11(Suppl 8):S101–S105.
93. Chan ES, Cronstein BN: Methotrexate–how does it really work?. Nat Rev Rheumatol
2010, 6:175–178.
94. Cronstein BN: Molecular therapeutics. Methotrexate and its mechanism of action.
Arthritis Rheum 1996, 39:1951–1960.
95. Cronstein BN, Naime D, Ostad E: The antiinflammatory mechanism of methotrexate.
Increased adenosine release at inflamed sites diminishes leukocyte accumulation in an
in vivo model of inflammation. J Clin Invest 1993, 92:2675–2682.
96. Herman S, Zurgil N, Deutsch M: Low dose methotrexate induces apoptosis with reactive
oxygen species involvement in T lymphocytic cell lines to a greater extent than in
monocytic lines. Inflamm Res 2005, 54:273–280.
97. Herman S, Zurgil N, Langevitz P, Ehrenfeld M, Deutsch M: he immunosuppressive effect
of methotrexate in active rheumatoid arthritis patients vs. its stimulatory effect in
nonactive patients, as indicated by cytometric measurements of CD4+ T cell
subpopulations. Immunol Invest 2004, 33:351–362.
98. Kim YJ, Song M, Ryu JC: Mechanisms underlying methotrexate-induced pulmonary
toxicity. Expert Opin Drug Saf 2009, 8:451–458.

148
99. Nesher G, Moore TL: The in vitro effects of methotrexate on peripheral blood
mononuclear cells. Modulation by methyl donors and spermidine. Arthritis Rheum 1990,
33:954–959.
100. Olsen NJ, Callahan LF, Pincus T: Immunologic studies of rheumatoid arthritis patients
treated with methotrexate. Arthritis Rheum 1987, 30:481–488.
101. Olsen NJ, Murray LM: Antiproliferative effects of methotrexate on peripheral blood
mononuclear cells. Arthritis Rheum 1989, 32:378–385.
102. Phillips DC, Woollard KJ, Griffiths HR: The anti-inflammatory actions of methotrexate
are critically dependent upon the production of reactive oxygen species. Br J Pharmacol
2003, 138:501–511.
103. Sperling RI, Coblyn JS, Larkin JK, Benincaso AI, Austen KF, Weinblatt ME: Inhibition of
leukotriene B4 synthesis in neutrophils from patients with rheumatoid arthritis by a
single oral dose of methotrexate. Arthritis Rheum 1990, 33:1149–1155.
104. Thomas R, Carroll GJ: Reduction of leukocyte and interleukin-1 beta concentrations in
the synovial fluid of rheumatoid arthritis patients treated with methotrexate. Arthritis
Rheum 1993, 36:1244–1252.
105. van Ede AE, Laan RF, Blom HJ, De Abreu RA, van de Putte LB: Methotrexate in
rheumatoid arthritis: an update with focus on mechanisms involved in toxicity. Semin
Arthritis Rheum 1998, 27:277–292.
106. McInnes IB, Gracie JA: Targeting cytokines beyond tumor necrosis factor-alpha and
interleukin-1 in rheumatoid arthritis. Curr Pain Headache Rep 2005, 9:405–411.
107. Scott DL, Kingsley GH: Tumor necrosis factor inhibitors for rheumatoid arthritis. N Engl
J Med 2006, 355:704–712.
108. Raza K, Falciani F, Curnow SJ, Ross EJ, Lee CY, Akbar AN, Lord JM, Gordon C, Buckley
CD, Salmon M: Early rheumatoid arthritis is characterized by a distinct and transient
synovial fluid cytokine profile of T cell and stromal cell origin. Arthritis Res Ther 2005,
7:R784–R795.
109. Planaguma A, Titos E, Lopez-Parra M, Gaya J, Pueyo G, Arroyo V, Claria J: Aspirin (ASA)
regulates 5-lipoxygenase activity and peroxisome proliferator-activated receptor alpha-
mediated CINC-1 release in rat liver cells: novel actions of lipoxin A4 (LXA4) and ASA-
triggered 15-epi-LXA4. FASEB J 2002, 16:1937–1939.
110. Bannenberg GL: Therapeutic applicability of anti-inflammatory and proresolving
polyunsaturated fatty acid-derived lipid mediators. Sci World J 2010, 10:676–712.
111. Yacoubian S, Serhan CN: New endogenous anti-inflammatory and proresolving lipid
mediators: implications for rheumatic diseases. Nat Clin Pract Rheumatol 2007, 3:570–
579. quiz 571 p following 589.
112. Basselin M, Ramadan E, Chen M, Rapoport SI: Anti-inflammatory effects of chronic
aspirin on brain arachidonic acid metabolites. Neurochem Res 2011, 36:139–145.

149
113. Scheinman RI, Cogswell PC, Lofquist AK, Baldwin AS Jr: Role of transcriptional
activation of I kappa B alpha in mediation of immunosuppression by glucocorticoids.
Science 1995, 270:283–286.
114. MacDermott RP, Schloemann SR, Bertovich MJ, Nash GS, Peters M, Stenson WF:
Inhibition of antibody secretion by 5-aminosalicylic acid. Gastroenterology 1989,
96:442–448.
115. Tanabe T, Tohnai N: Cyclooxygenase isozymes and their gene structures and
expression. Prostaglandins Other Lipid Mediat 2002, 68–69:95–114.
116. Miller AH: Depression and immunity: a role for T cells?. Brain Behav Immun 2010, 24:1–
8.
117. Vane JR: Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like
drugs. Nat New Biol 1971, 231:232–235.
118. Fiorucci S, Distrutti E, de Lima OM, Romano M, Mencarelli A, Barbanti M, Palazzini E,
Morelli A, Wallace JL: Relative contribution of acetylated cyclo-oxygenase (COX)-2 and
5-lipooxygenase (LOX) in regulating gastric mucosal integrity and adaptation to aspirin.
FASEB J 2003, 17:1171–1173.
119. Tabor CW, Tabor H: Polyamines. Annu Rev Biochem 1984, 53:749–790.
120. Hotamisligil GS: Inflammation and metabolic disorders. Nature 2006, 444:860–867.
121. Fall CHD: Developmental origins of cardiovascular disease, type 2 diabetes and obesity
in humans. Early Life Orig Health Dis 2006, 2:8–28.
122. Egger G, Dixon J: Inflammatory effects of nutritional stimuli: further support for the
need for a big picture approach to tackling obesity and chronic disease. Obes Rev 2010,
11:137–149.
123. Eaton SB, Cordain L, Sparling PB: Evolution, body composition, insulin receptor
competition, and insulin resistance. Prev Med 2009, 49:283–285.
124. Frassetto LA, Schloetter M, Mietus-Synder M, Morris RC Jr: Sebastian A: Metabolic and
physiologic improvements from consuming a paleolithic, hunter-gatherer type diet. Eur
J Clin Nutr 2009, 63:947–955.
125. O'Dea K: Marked improvement in carbohydrate and lipid metabolism in diabetic
Australian aborigines after temporary reversion to traditional lifestyle. Diabetes 1984,
33:596–603.
126. McGowan PO, Sasaki A, D'Alessio AC, Dymov S, Labonte B, Szyf M, Turecki G, Meaney
MJ: Epigenetic regulation of the glucocorticoid receptor in human brain associates with
childhood abuse. Nat Neurosci 2009, 12:342–348.
127. Cutolo M, Foppiani L, Minuto F: Hypothalamic-pituitary-adrenal axis impairment in the
pathogenesis of rheumatoid arthritis and polymyalgia rheumatica. J Endocrinol Invest
2002, 25:19–23.

150
128. Strohle A, Scheel M, Modell S, Holsboer F: Blunted ACTH response to dexamethasone
suppression-CRH stimulation in posttraumatic stress disorder. J Psychiatr Res 2008,
42:1185–1188.
129. Van Den Eede F, Moorkens G, Van Houdenhove B, Cosyns P, Claes SJ: Hypothalamic-
pituitary-adrenal axis function in chronic fatigue syndrome. Neuropsychobiology 2007,
55:112–120.
130. Cobb JM, Steptoe A: Psychosocial stress and susceptibility to upper respiratory tract
illness in an adult population sample. Psychosom Med 1996, 58:404–412.
131. Segerstrom SC: Social networks and immunosuppression during stress: relationship
conflict or energy conservation?. Brain Behav Immun 2008, 22:279–284.

151
Paragraph 1.4. Stress induces endotoxemia and low-grade inflammation by increasing barrier
permeability. de Punder K, Pruimboom L. Front Immunol 2015;6:223. Epub 2015 May 15.
Stress induces endotoxemia and low grade inflammation by increasing barrier
permeability
Karin de Punder1,2,*, Leo Pruimboom1, 3
1. Natura Foundation, Numansdorp, The Netherlands
2. Institute of Medical Psychology, Charité University Medicine, Berlin, Germany
3. University of Groningen, Groningen, Holland
Correspondence: Karin de Punder, Institute of Medical Psychology, Charité University
Medicine, Berlin, Germany.
Keywords: endotoxemia, endotoxin, inflammation, intestinal permeability,
lipopolysaccharide, LPS, stress, tight junction

152
Abstract
Chronic non-communicable diseases (NCDs) are the leading causes of work absence,
disability and mortality worldwide. Most of these diseases are associated with low-grade
inflammation. Here we hypothesize that stresses (defined as homeostatic disturbances) can
induce low-grade inflammation by increasing the availability of water, sodium and energy-
rich substances to meet the increased metabolic demand induced by the stressor. One way of
triggering low-grade inflammation is by increasing intestinal barrier permeability through
activation of various components of the stress system. Although beneficial to meet the
demands necessary during stress, increased intestinal barrier permeability also raises the
possibility of the translocation of bacteria and their toxins across the intestinal lumen into the
blood circulation. In combination with modern life-style factors, the increase in
bacteria/bacterial toxin translocation arising from a more permeable intestinal wall causes a
low-grade inflammatory state. We support this hypothesis with numerous studies finding
associations with NCDs and markers of endotoxemia, suggesting that this process plays a
pivotal and perhaps even a causal role in the development of low-grade inflammation and its
related diseases.
Introduction
Inflammation is the response of the innate immune system triggered by stimuli like microbial
pathogens and injury. Acute systemic inflammation such as in sepsis, trauma, burns, and
surgery is characterized by a quick increase in plasma-levels (up to 100 fold) of pro-
inflammatory cytokines and acute phase proteins, while in low-grade inflammation there is a
sustained but only two to three fold increase in circulation pro-inflammatory mediators (1).
Chronic low-grade inflammation is characteristic for many non-communicable diseases
(NCD) including diabetes type II, cardiovascular disorders, autoimmune diseases, chronic
fatigue syndrome, depression and neurodegenerative pathologies, but until now the exact
mechanism behind the elevated levels of inflammatory mediators found in these conditions is
not well understood (2-5).
Inflammation can be induced by the binding of pathogen-associated molecular patterns
(PAMP) to toll-like receptors (TLR), which are expressed on different cells types including
immune cells, adipocytes and endothelial cells. The most extensively studied PAMP is
lipopolysaccharide (LPS) or endotoxin (the terms LPS and endotoxin will be used
interchangeably throughout the rest of the article), a major cell wall component of Gram-
negative bacteria, which is normally present in the human circulation in very low
concentrations. It has been hypothesized that most of this circulating LPS is derived from the
gut, since the gut-microbiota is the biggest source of Gram-negative bacteria-derived LPS.
However, LPS found in the circulation could also be derived from Gram-negative bacteria

153
residing in the oral cavity, respiratory and genitourinary tracts, or can be food derived (6-8).
Under certain circumstances there can be an increase of endotoxin translocation across the
intestinal barrier, leading to mildly increased concentrations in blood circulation. This process
has been associated with several NCDs, like depression (9), chronic fatigue syndrome (10),
chronic heart failure (11), type 2 diabetes (12), autism (13), non-alcoholic fatty liver disease
(NAFLD) (14) and inflammatory bowel disease (IBD) (15), diseases that are all linked to chronic
systemic low-grade inflammation, indicating that endotoxemia could be an important
contributor in the development of these conditions.
Here we hypothesize that stress-induction leads to a more permeable intestinal wall intended
to facilitate an increase in the availability of water, sodium and energy-rich substances
necessary to meet the increased metabolic demand induced by the stressor. Modern life-style
factors, such as long-term psychosocial stress and components of our “Western” diet
constantly challenge the stress-axis and further compromise intestinal barrier function,
resulting in endotoxemia, low grade inflammation and its related diseases. We support our
hypothesis by describing literature surrounding stress- and immune system activation
processes and their relation to gut barrier function and explain how lifestyle choices impact all
these systems. In addition we present a vast amount of literature describing associations with
NCDs and markers of endotoxemia. Overall we conclude that stress-induced disrupted barrier
function in parallel with elevated circulating endotoxin levels may underlie disease onset and
progression and should be considered much more than just a risk factor for chronic disease; it
could be a cause.
1. Bacterial toxins activate the immune system via TLR
LPS, the major cell wall component of Gram-negative bacteria, is characterized by its capacity
to induce inflammation, fever, shock and death (1). Additionally in recent years, other cell wall
components of Gram-negative and -positive bacteria, have been recognized to have
endotoxic properties (16), but these will not be further addressed in the rest of the paper.
Endotoxins are released from bacteria during infection or as a consequence of bacterial lysis.
Although both whole bacteria and bacterial toxins can translocate transcellular or paracellular
into the lymph, blood and mesenteric lymph nodes, it is still not precisely clear if the presence
of endotoxin in the blood circulation (endotoxemia) also presents whole bacteria translocation
across the intestinal wall (17).
Inflammation can be induced by the binding of LPS to TLR4. The lipid-A moiety of LPS
interacts with the LPS-sensing machinery composed of TLR4, myeloid differential protein 2,
CD14 and LPS-binding protein (LBP). LBP transports and delivers circulating aggregates of LPS
to lipoproteins, resulting in hepatic clearance, or delivers LPS to CD14 (the membrane bound

154
or secreted, soluble form of this molecule), leading to TLR4 activation. TLR4 activation
activates two transcription factors, activator protein (AP)-1 and nuclear factor κB (NF-κB) (18,
19), and stimulates the production of pro-inflammatory mediators such as prostaglandin 2
(PgE2) (20), tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, Interferon (IFN)-γ and the
acute phase protein, C-reactive protein (CRP) (19). Simultaneously, an uncontrolled pro-
inflammatory reaction is prevented by the induction of TLR4, NF-κB and AP-1 signaling
inhibitors, which are probably involved in creating endotoxin tolerance (21). LPS tolerance is
defined as a reduced responsiveness to a LPS challenge following a first encounter of
endotoxin (22). It has been suggested that the dose of LPS exposure is important for
determining the switch between LPS tolerance and priming. For example, in macrophages,
high LPS concentrations induced a robust pro-inflammatory response in parallel with the
activation of inhibitory feedback mechanisms. Lower concentrations of LPS, like those
observed in NCDs, removed transcriptional suppressors on the promotors of pro-
inflammatory genes and induced a mild but persistent expression of pro-inflammatory
mediators (21, 23).
2. Intestinal barrier function
2.1. The paracellular pathway is important for water, mineral and nutrient
uptake
The intestinal barrier allows for the regulated uptake of water, minerals and nutrients and
protects the gut lumen from damage due to harmful substances. Components can cross the
epithelial barrier by active transport and endocytosis (transcellular) or via the paracellular
route. Because hydrophilic solutes are limited to cross lipid membranes of epithelial cells, the
paracellular route is an important and major route for the transport of water, solutes and
minerals across the intestinal barrier (24, 25). Active glucose, sodium and water uptake is
mediated by the activity of sodium-dependent glucose co-transporters (SGLT) (26). The
transcellular absorption of glucose and sodium and the resulting basolateral disposition of
glucose and sodium by these transporters opens up the paracellular pathway structure and
allowing the passive flow of water and small nutrients by creating an osmotic gradient (27).
Intestinal permeability is a measure of the barrier function of the gut and relates to the
paracellular space surrounding the brush border surface of the enterocytes and the junctional
complexes (28). The junctional complex, containing tight junctions, adherens junctions and
desmosomes is an important regulator of the paracellular pathway and allows the passage of
water, solutes and ions, but under normal conditions provides a barrier to larger molecules
(28, 29). The claudin family of junctional transmembrane proteins have a substantial effect on
paracellular permeability. While one group of sealing claudins makes the paracellular barrier
less permeable, the other group of claudins is known to increase paracellular permeability by

155
the formation of pores that increase permeability for small solutes (30, 31). The expression of
claudin proteins varies between tissues, explaining the variances in permeability of tight
junctions among tissues (27). The paracellular pathway can be divided into the pore and non-
pore pathway. The pore pathway is mainly controlled by the expression of claudins, while the
non-pore pathway is more sensitive to cytoskeletal disruptions (30). Cytoskeletal
rearrangements can be induced by phosphorylation of the regulatory myosin light chain
(MLC), induced by MLC-kinase (MLCK). Phosphorylation of the MLC facilitates myosin
binding to actin and therefore aids in cytoskeletal contractility. MLCK can be activated by
cytokines such as TNF-α, causing increases in tight junction permeability by actomyosin
contraction and reorganization of the tight junction (32, 33). In addition, SGLT1 activation and
associated increases in tight junction permeability are also paralleled with phosphorylation of
MLC, indicating that MLCK is an important mediator in tight junction and paracellular
permeability regulation (25, 34) (Figure 1).
Increased intestinal permeability has been associated with autoimmune diseases, such as type
1 diabetes (35), rheumatoid arthritis, multiple sclerosis (36), and diseases related to chronic
inflammation-like IBD (36, 37), asthma (38), chronic fatigue syndrome and depression (10, 39).
It has been hypothesized that chronic intestinal hyper-permeability results in a pro-
inflammatory phenotype induced by the enhanced paracellular translocation of microbial
(and dietary) antigens across the gut barrier (40).
3. Intestinal barrier function
3.1. Stress increases permeability of the intestinal barrier
Stressful stimuli activate the sympathetic nervous system (SNS) and hypothalamic-pituitary-
adrenal (HPA)-axis. Activation of both systems increases the availability of water, minerals and
energy-rich substances in order to meet with the body’s metabolic demand (41, 42). The SNS
responds instantly to physical and psychological stress by reallocating energy into different
organs by neuronal regulation of heart rate, blood flow, release of catecholamines (adrenalin
and noradrenalin) from the adrenal medulla (43) and stimulation of the renin-angiotensin-
aldosterone system (44), involved in retention of water and sodium from the kidneys. In
addition to the kidneys, water and sodium reabsorption can also be achieved at the level of the
intestine. The intestinal wall is innervated by adrenergic sympathetic nerve fibers that upon
stimulation increase water and sodium absorption (45, 46), which is paralleled by increases in
intestinal permeability. The SNS-induced increase in permeability is likely mediated by β2-
adrenergic receptors expressed on epithelial cells (47). Activation of the β2-adrenergic-
receptors stimulated SGLT1-mediated glucose absorption from the gut (48, 49) and the
resulting basolateral disposition of glucose and sodium by these transporters opens up the

156
paracellular pathway (27) (Figure 1). Not surprisingly, blockage of the SNS by means of thoracic
epidural anesthesia resulted in the blockage of the endotoxin-induced increase in intestinal
permeability in rats (50).
Activation of the HPA-axis leads to the release of glucocorticoids that potentiate some of the
actions of catecholamines. Essential to this response are the neurons in the paraventricular
nucleus of the hypothalamus expressing corticotropin-releasing hormone (CRH) and other
co-secretagogues, such as arginine vasopressin and oxytocin, both involved in the regulation
of water homeostasis. Arginine vasopressin and CRH trigger the immediate release of
adrenocorticotropic hormone (ACTH) from the anterior pituitary, which in turn induces the
release of glucocorticoids and to some extend mineralocorticoids from the adrenal cortex,
stimulating gluconeogenesis and increasing sodium and water retention respectively (51, 52).
Intestinal permeability is regulated by several components of the HPA-axis.
In epithelial HT-29 monolayers, exposure to CRH resulted in an increased response to LPS as
reflected by a decrease in transepithelial resistance and a significant increase in the
expression of the pore forming protein, claudin 2 (27). Interestingly enough, these effects were
mediated by an increase in TLR4 expression, an observation that could be repeated in mice
treated with the water-avoid stressor (53). TLR4 activation resulted in the activation of the
transcription factor NF-κB, which has specific binding sites in the claudin 2 gene promoter
(54), indicating that in epithelial cells CRH affects both intestinal permeability and
inflammatory pathways.
In rats, exposure to restricted stress or swimming stress increased intestinal permeability
throughout the whole intestinal tract as measured by the fractional secretion of the urinary
recovery of sucrose (reflecting gastric permeability), the lactulose-mannitol ratio (as a marker
for small intestinal permeability) and sucralose (reflecting both small intestinal and colonic
permeability) (55). Other experimental animal stress models such as thermal injury or early
maternal deprivation induced the development of gastric ulcers, altered gastrointestinal
motility and ion secretion, and increased intestinal permeability (reviewed by Caso et al. 2008
(56)). SGLT1 expression was markedly increased in the rat jejunum and ileum after 8 weeks of
restraint stress. These findings were paralleled with an increase in intestinal lymphocytic
infiltration and adrenal gland weight gain (26). The up-regulation of the SGLT1 is probably
necessary to meet with the increased water, sodium and nutrient demand, induced by
chronic stress (42).
The effect of acute stress on intestinal permeability was also investigated in humans (57). In
healthy volunteers subjected to a public speech test, high cortisol-responders displayed

157
increased intestinal permeability as measured by the lactulose-mannitol ratio. Exogenous
CRH administration also increased intestinal permeability, yet the CRH-induced hyper-
permeability could be suppressed by the mast cell stabilizer disodium cromoglycate. Mast cell
stabilization before the public speech test also did not alter intestinal permeability, however, it
should be noted that in this experiment a control group was not included. Nevertheless, these
results identify CRH as an important factor in the stress-induced alterations of the intestinal
barrier function. These alterations seemed to be mediated by intestinal mast cells that upon
activation secrete pro-inflammatory mediators like IFN-γ and TNF-α. A variety of pro-
inflammatory cytokines increases epithelial and endothelial paracellular permeability by
modulating the structure of the tight junction and by inducing cytoskeletal disruptions via
activation of MLCK (32, 34, 58) (Figure 1). For example, IFN-γ increased epithelial permeability
of T84 monolayers to large molecules (10 kDa). Interestingly, the IFN-γ-induced increase in
permeability also up-regulated the passage of FITC-labeled-endotoxin by ten-fold (59).
4. Neuroendocrine- immune interactions
The complex neuroendocrine-immune interactions are evidenced by the fact that emotional
stressors influence the immune response and that pure immunological stimuli impact on
cognitive performance (60). Inflammatory mediators activate the HPA-axis with the purpose
to provoke disease behavior and redirect energy-rich nutrients towards the immune system
(61). Cytokines have been shown to increase nutrient availability to meet with the
inflammation-dependent increased metabolic demand. For example, the cytokine Il-1α
increased whole body glucose metabolism on a central level (62) and cytokines like Il-6, TNF-
α, Il-1 and IFN independently evoke a HPA-axis response (63-65). Immune mediators can
communicate with the brain via several pathways. By stimulating afferent sensory nerve
fibers, entering the brain via the circumventricular organs or by binding to cerebral blood
vessel endothelium immune mediators effectively redirect energy-rich substrates towards the
immune system (41, 42).
Besides inflammatory cytokines, prostaglandins synthesized via the cyclooxygenase system
play a central role in inflammation and HPA-axis activation. Zimomra et al. (65) demonstrated
that in rats the initial activation of the HPA-axis by LPS is mediated by prostaglandins, like
PgE2, while inflammatory cytokines maintain corticosterone levels at later time-points. In this
study it was suggested that prostaglandins stimulated corticosterone release in a direct
manner, since the peak in circulating corticosterone levels was observed long before the peak
in circulating ACTH. This idea was confirmed by a study in rodents, showing that PgE2
directly stimulated the release of glucocorticoids from the adrenal gland (66). In human
adrenal cells expressing TLR2 and TLR4, LPS stimulation resulted in the release of cortisol.
This effect was mediated by PgE2, since inhibition of cyclooxygenase-2 attenuated cortisol

158
release (67).
As indicated, TLR4 activation stimulates the release of PgE2 by immune cells, adipocytes,
endothelial, epithelial and probably also adrenal cells (68), inducing the peripheral release of
glucocorticoids from the adrenal gland (66). PgE2 also activates glucocorticoid production
through activation of the HPA-axis at the level of the hypothalamus and the pituitary (69).
Macrophages, homing in blood vessels in the cranium, are directly activated by danger signals
such as LPS. Activation of these special macrophages induces the production of PgE2 which
directly stimulates the paraventricular nucleus of the hypothalamus, leading to higher
production of glucocorticoids which should probably protect against possible inflammation
of the brain (69).
5. Acute stress increases pro-inflammatory pathways by
increasing intestinal permeability
Acute stress modulates the immune response and changes immune cell distribution. These
neuroendocrine effects on the immune system are mediated by stress-hormones released
from the adrenal gland, by direct innervation of sympathetic nerve fibers into lymphoid
organs and by stress hormone receptors expressed on immune cells, like glucocorticoid
receptors (GR) and α- and β-adrenergic receptors (70-72). It has been suggested that by
mobilizing immune cells, the stress response, also known as the “fight-flight reaction”,
prepares the immune system for oncoming challenges (70).
In addition, acute stress increases circulating pro-inflammatory mediators (73-75). In subjects
exposed to acute stress, NF-κB was up-regulated in peripheral blood mononuclear cells in
parallel with elevated levels of circulating catecholamines and glucocorticoids (76). Until now
it is not completely understood what causes this pro-inflammatory response. Glucocorticoids
mostly have an inhibitory effect on inflammatory pathways and catecholamines a rather
modulating than activating influence on the immune system (71, 72, 77), however, it has been
shown that activation of the β-adrenergic receptor by noradrenalin (but not adrenalin)
increased NF-κB binding to DNA in monocytes in vitro (76). A recent study in rodents showed
that acute stress-induced neuro-inflammation could be prevented by a pre-stress treatment
with antibiotics or an inhibitor of MLCK. In addition, these treatments prevented stress-
induced hyper-permeability and endotoxemia, indicating that it is not the stress-factor itself
producing a pro-inflammatory response of the immune system, but the fact that stress
increases barrier permeability and the translocation of endotoxin into the circulation. Pre-
stress probiotic treatment with Lactobacillus farciminis had similar effects, which could be
explained by its ability to enhance intestinal barrier function (78). In agreement with these
results, it could be hypothesized that the (short lasting) pro-inflammatory activity in humans

159
observed during acute stress is initiated by a stress-induced increase in intestinal
permeability, mediated by the SNS and components of the HPA-axis, and resulting in higher
levels of translocating endotoxin interacting with TLR on immune cells, adipocytes and
epithelial cells. A schematic overview of the complex neuroendocrine-immune interactions
and their relation to gut barrier function are displayed in Figure 2.
6. Chronic stress dysregulates the HPA-axis and changes immune
function
Chronic psychological stress is known to dysregulate the immune system. These alterations
are accompanied by low-grade inflammation, delayed wound healing and increased
susceptibility to infectious diseases (79). Chronic stress leads to hypercortisolemia (77), long
term permeability of barriers, endotoxemia and low-grade inflammation (our hypothesis and
theory). Normally, the release of glucocorticoids puts a limit on the maximum activity of the
immune system, however, chronic HPA-axis stimulation can result in glucocorticoid
resistance at the level of the immune system, making it insensitive to its inhibitory and
modulatory actions (2). This process is observed in several conditions (including conditions
related to psychosocial stress), whereby immune cells from patients are less responsive to the
inhibitory actions of glucocorticoids on cytokine release and cell proliferation after
stimulation in vitro (80-83). In addition, chronic stress induces a shift in the production of
type 1 cytokines towards type 2 cytokine production. It can be deducted that by this
mechanism, the part of the immune system involved in the clearance of extracellular bacteria
and bacterial toxins (the type 2 response) is prevented from being suppressed, protection
against ongoing microbial infiltration (endotoxemia) is guaranteed, while the type 1 response,
involved in clearance of intracellular pathogens (like viruses) is inhibited (71, 84).
7. Life style-related factors induce endotoxemia
The fact that stress increases barrier permeability and thereby enhances the availability of
water, sodium and nutrients, makes sense from an evolutionary perspective. However, the
question arises if the accompanied translocation of bacteria and their toxins should also be
considered beneficial for the host. We speculate that when the composition of the microbiota
is physiological, and barrier opening is short-lasting, acute stress will not produce low-grade
inflammation. However, modern people suffer from new multi-factorial stressors, such as
chronic psychosocial stress and the consumption of a “Western diet”, which constantly
challenges the stress-axis, alters microbiota composition and thereby compromises intestinal
barrier function. This next section discusses how modern lifestyle factors impact the gut-
brain-immune-axis and promote endotoxemia, low-grade inflammation and its related
diseases.

160
7.1. Gut microbiota modulate stress-axis and influence gut barrier function
Large differences in the composition of the gut-microbiota and an overall reduction in
microbial diversity are observed in Western populations when compared to traditional
Hunter-gatherers or people from rural Africa (85, 86). These environment and diet-induced
changes in gut microbiota have been connected to an increased susceptibility to chronic
diseases, like IBD, obesity, type 1 and type 2 diabetes (87). The gut microbiota influences
inflammatory (88) and metabolic processes (89) and has been shown to influence the
development of the HPA-axis and immune system (90, 91). For example, exposure to LPS
during developmental periods can exaggerate the HPA-axis and immune response to stress
(92, 93), but also the absence of bacteria can induce these effects. Animals raised in germ-free
environments showed an exaggerated HPA-axis response, which was normalized by
colonization with fecal matter from specifically germ free animals or by the administration of
the Gram-positive Bifidobacterium infantis (94). Vice versa, exposure to social stress changed
the composition of the gut microbiota in mice (95, 96) and prenatal stress altered the
microbiome in rhesus monkeys by reducing the overall numbers of the Gram-positive
Bifidobacteria and Lactobacilli (97), indicating that chronic stress affected the composition of
the gut microbiome. Stress influences gut motility, secretions, and mucin production, thereby
altering the habitat of resident bacteria, promoting changes in the composition of the gut
microbiome (98) and allowing the growth of pathogenic bacteria (99).
Increasing evidence supports an important role for microbiota on the homeostasis of the
intestinal barrier. Certain strains of the Gram-positive Lactobacilli decreased intestinal
permeability in several animal and human disease models (78, 100). Bifidobacterium infantis
reduced intestinal permeability (as assessed by 70-kDa fluorescein isothiocyanate–dextran
transmucosal flux) and ameliorated symptoms in a neonatal necrotizing enterocolitis mouse
model (101). Further evidence indicating the influence of the gut-microbiota on intestinal
permeability was presented in detoxifying alcoholic-dependent subjects: Lower levels of
Ruminococcaceae and higher abundance of Lachnospiraceae (Dorea) and Blautia were
associated with increased intestinal permeability (102). In addition, higher levels of certain
pathogenic bacteria can increase intestinal permeability by disrupting the epithelial barrier
and triggering cell death and inflammation. These bacteria have the ability to bind and/or
translocate through endothelial and microfold cells and have been shown to secrete toxins or
other effector molecules via specialized secretion systems. Although the exact mechanisms
are not well described, most pathogenic gut bacteria including Escherichia coli, Helicobacter
pylori, Staphylococcus aureus, Cholera Pseudomanas fluorescens, Pseudomanas eruginosa,
Yersinia enterocolitica, Campylobacter jejuni and Salmonella typhimurium alter paracellular
permeability by disassembling tight junctions and generating cytoskeleton changes by
increasing inflammation (reviewed by Barreau et al. (103)). As an example, a strain of

161
Escherichia coli, normally present in the human gut, induced focal leaks in colonic epithelial
monolayers and in rat distal colon by using α-hemolysin, allowing for their paracellular
translocation across the epithelial layer (104).
7.2. High-caloric and high-fat diets induce inflammation and increase
circulating endotoxin levels
Compared to ealthy individuals, patients suffering from obesity have higher circulating
endotoxin levels together with greater levels of circulating pro-inflammatory cytokines and
insulin resistance (105). Food intake can produce postprandial immune activation and elevate
endotoxin levels when a meal is high in calories (106) or has a high fat content (6, 107-109).
Rodents fed a 4-week high-fat diet (72% fat) showed a constant elevation in circulating
endotoxin levels, while in control animals endotoxin levels only increased during feeding
hours. Furthermore, a high-fat diet produced fasting glycaemia, insulin resistance, general
weight gain and weight gain of the liver and visceral and subcutaneous adipose tissue. In
addition, adipose tissue F4/80-positive cells (indicating the infiltration of macrophages),
markers of inflammation and liver triglyceride content were increased. Interestingly, almost
similar effects were observed in mice subcutaneously infused with LPS (resulting in similar
circulating LPS levels as observed in the high-fat fed mice). These effects were mediated by
TLR4, since mice lacking CD14, which is important for the recognition of LPS to this receptor,
showed a delayed response to a high-fat diet or LPS injections (107).
In healthy humans a 910 calories high-fat and high carbohydrate meal resulted in increased
circulating endotoxin levels and elevated levels of LBP in parallel with higher inflammatory
markers and increased protein expression of TLR2 and TLR4 in isolated leukocytes. A meal
high in fruits and fibre did not induce these effects (108). Plasma endotoxin levels, pro-
inflammatory markers and leukocyte TLR4 expression increased after the intake of cream (300
calories), while the intake of 300 calories of glucose resulted only in a pro-inflammatory
response and the intake of orange juice and water showed none of these effects (110). In
healthy individuals, plasma endotoxin levels increased about 50% after the intake of a high-fat
meal (900 calories) (6) and 4 weeks consumption of a Western-style diet raised plasma
endotoxin activity levels by 71% (111).
How exactly the intake of a high-caloric meal increases circulating endotoxin levels is still
unclear but has been explained by several mechanisms (reviewed by Kelly et al., 2012 (112).
One of these suggested mechanisms is that the introduction of a high-fat diet modulates the
expression of genes involved in the barrier function in epithelial cells, thereby directly
compromising the integrity of the tight junction (113). Another explanation could be that a

162
high-caloric/high-fat meal induces high levels of insulin and leptin, hormones that directly
activate the SNS (114, 115). Moreover insulin enhances SGLT1 mediated glucose absorption
(116). Activation of the SGLT1 and the SNS leads to increased permeability of the gut barrier,
which may induce the observed post-prandial endotoxemia (our hypothesis and theory).
7.3. Gliadin compromises the integrity of tight junctions
The intake of wheat and other cereal grains has been implicated in the development of
inflammation-related diseases, by inducing inflammation and increasing intestinal
permeability (40). Gliadin, a component of gluten, has been demonstrated to increase
permeability in human Caco-2 intestinal epithelial cells by reorganizing actin filaments and
altering expression of junctional complex proteins (117). Several studies by the group of
Fasano et al. show that the binding of gliadin to the chemokine receptor CXCR3 on epithelial
IEC-6 and Caco2 cells releases zonulin, a protein that directly compromises the integrity of
the junctional complex (118, 119).
7.4. Alcohol consumption increases intestinal permeability
Alcohol consumption is an important risk factor for disease and is one of the major causes of
chronic liver disease. Increased intestinal permeability has been observed during chronic
alcohol consumption. In an animal model of chronic alcoholic liver disease, alcohol feeding
for 8 weeks increased intestinal permeability (120). In humans, alcohol dependence induced
changes in the gut microbiota composition that were associated with increased intestinal
permeability (102). Furthermore, increased intestinal permeability and higher circulating
endotoxin levels were observed in patients with chronic alcohol abuse (121-123).
7.5. Exercise induced heat-stress increases intestinal permeability
Exercise increases body temperature, reduces intestinal blood flow (reallocated to the muscles
and cardiac system) and increases intestinal permeability by activating the SNS and HPA-axis.
Already in 1992, Oktedalen et al. (124) showed that marathon runners displayed a significant
increase in intestinal permeability. In addition, studies have indicated that strenuous exercise
induced higher circulating endotoxin levels and activated the immune system (125-128).
Further evidence of exercise- and heat-induced increased intestinal permeability, leading to
gastrointestinal complaints in people engaging in physical activity, has been recently
reviewed (129).

163
8 Endotoxemia is associated with diseases related to chronic
inflammation
Multiple human studies have emerged that find associations with NCDs and markers of
endotoxemia. Even aging, associated with higher sympathetic nerve activity (130) and higher
circulating inflammatory mediators like Il-6, has been linked to higher plasma concentration
of LPS and LBP (131). In further support of our theory, in this section an overview is given of
human studies finding changes in levels of endotoxin or endotoxin-related markers in NCDs
(Table 1).
8.1. Metabolic syndrome
Metabolic syndrome is accompanied by an increased risk for NAFLD, obesity, type 2 diabetes
and cardiovascular diseases. All of these conditions are related to and even predicted by an
increased sympathetic activity (132) and a dysregulated HPA-axis (133). Higher circulating
endotoxin and LBP levels are associated with risk factors of the metabolic syndrome, like
insulin resistance, obesity, dyslipidemia and chronic inflammation (134-137). Patients
suffering from NAFLD exhibited significantly higher serum endotoxin levels in contrast to
healthy controls (14). Farhardi et al. (138) indicated that elevated plasma endotoxin levels in
these patients were related to an impaired intestinal barrier function, because, only in the
patient group was the intake of a permeability stressor (aspirin) shown to increase the 0–24 h
urinary excretion of sucralose (a marker of whole-gut permeability). Furthermore, augmented
plasma LBP levels in concert with increased plasma levels of TNF-α were observed in obese
NAFLD patients compared to healthy controls (139).
Elevated circulating levels of endotoxin and LBP were detected in type 2 diabetics (12, 135,
140-142). Compared to healthy controls, obese individuals and type 2 diabetics showed higher
endotoxin levels after the intake of a high-fat meal. Increased endotoxin levels were observed
in all challenged individuals, yet higher endotoxin levels were seen in individuals suffering
from metabolic illnesses, suggesting an increased intestinal permeability in these patients
(143). This was further indicated by a recent study showing that increased serum levels of
endotoxin, Il-6 and TNF-α are found in type 2 diabetic patients compared to healthy
individuals. The level of endotoxin was positively related to zonulin, a marker for intestinal
permeability (12).
A large cohort of patients with coronary artery disease identified increased serum LBP levels
to be associated with total and cardiovascular mortality (144). Moreover, circulating LBP levels
were associated with carotid intima media thickness (a marker of atherosclerosis), obesity,
insulin resistance and high sensitive CRP (145).

164
Patients suffering from chronic heart failure with aggravated renal function showed increased
circulating endotoxin levels and an impairment of the intestinal barrier (11). Wiedermann et al.
(146), showed that subjects with the highest levels of circulating endotoxin (90th percentile)
had a 3-fold increased risk of incident atherosclerosis. Higher serum endotoxin and pro-
inflammatory cytokine concentrations were seen in patients with edematous chronic heart
disease compared to stable patients and healthy controls. Intriguingly, after short-term
diuretic treatment, circulating endotoxin concentrations decreased in edematous patients
(147). Diuretic treatment (like angiotensin-converting enzyme (ACE) inhibitors) ameliorated
intestinal inflammation, perhaps by impacting on intestinal permeability through interference
with the renin-angiotensin-aldosterone system. Several components of this system (renin,
ACE and angiotensin II) have been shown to stimulate pro-inflammatory pathways (44, 148).
8.2. IBD
Ulcerative colitis and Crohn’s disease are intestinal inflammatory disorders, also known as
IBD, which have been causally linked to chronic psychological stress (149), altered immune
function, changes in the gut microbiota, increased intestinal permeability and endotoxemia
(150). For example, increased plasma endotoxin and LBP levels were measured in both patient
groups, but were more pronounced in patients with active disease compared to inactive
disease and were associated with disease severity (151). In addition, detectable plasma
endotoxin levels and higher plasma levels of LBP were more frequently observed in IBD
patients compared to controls (15, 152) and were correlated with disease severity and
circulating TNF-α levels (153).
8.3. Psychiatric diseases
Over the last decade, the role of the gut-brain axis has emerged as an important mediator in
the development of psychiatric and mood disorders (154). Moreover, higher endotoxin levels
and intestinal barrier dysfunction are observed in several of these conditions. For example,
Parkinson’s patients exhibited increased total intestinal permeability and a more intense
staining for Escherichia coli LPS and oxidative stress markers in intestinal sigmoid mucosa
samples. However, in these patients endotoxin levels resembled control samples and serum
LBP concentrations were lower compared to healthy individuals (155). Higher serum
endotoxin levels are associated with severe autism, sporadic amyotrophic lateral sclerosis and
Alzheimer’s disease (13, 156). Furthermore, increased IgA and IgM responses against LPS of
commensal bacteria were seen in chronic fatigue syndrome (10) and depression (9).
Intriguingly, chronic oral infection of periodontitis was associated with Alzheimer’s disease
where higher antibody levels against oral pathogens were observed years before the onset of
symptoms in people suffering from Alzheimer’s disease (157), suggesting there was an

165
increased translocation of bacteria and/or bacterial toxins from the mouth into the
bloodstream.
Table 1. Associations found between markers of endotoxemia and disease.

166
9 Conclusion
Chronic low-grade inflammation is an eminent feature of NCDs. In addition, many studies
report increased circulating endotoxin levels and increased gut permeability in patients
suffering from these conditions. As reviewed in this paper, stress-induced increases in
intestinal permeability, in combination with modern life-style factors, raise the possibility of
translocation of bacteria and/or their toxins across the more permeable gut barrier. The
resulting, long lasting, endotoxemia should be considered much more than just a risk factor
for chronic disease; it could be a cause. Notwithstanding the fact that the exact origin and
sequence of events involved in development of NCDs remain to be unsolved, evidence
indicates that a disrupted barrier function in parallel with elevated circulating endotoxin levels
may underlie disease onset and progression. For this reason, applying therapies aimed at
restoring intestinal barrier function, lifestyle changes and stress management should be
considered as an important strategy in preventing and attenuating the pro-inflammatory state
observed in NCDs.
Conflict of interest statement
The authors declare no conflict of interest.
Acknowledgments
We would like to thank Alicia Lammerts van Bueren for the editorial work on the manuscript.

167
References
1. Andreasen AS, Krabbe KS, Krogh-Madsen R, Taudorf S, Pedersen BK, Moller K. Human
endotoxemia as a model of systemic inflammation. Current medicinal chemistry (2008)
15(17):1697-705. Epub 2008/08/05. PubMed PMID: 18673219.
2. Bosma-den Boer MM, van Wetten ML, Pruimboom L. Chronic inflammatory diseases are
stimulated by current lifestyle: how diet, stress levels and medication prevent our body
from recovering. Nutrition & metabolism (2012) 9(1):32. doi: 10.1186/1743-7075-9-32.
PubMed PMID: 22510431; PubMed Central PMCID: PMC3372428.
3. Eckel RH, Alberti KG, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet (2010)
375(9710):181-3. doi: 10.1016/S0140-6736(09)61794-3. PubMed PMID: 20109902.
4. Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the
pathogenesis of depression. Trends in immunology (2006) 27(1):24-31. doi:
10.1016/j.it.2005.11.006. PubMed PMID: 16316783; PubMed Central PMCID: PMC3392963.
5. Ruiz-Nunez B, Pruimboom L, Dijck-Brouwer DA, Muskiet FA. Lifestyle and nutritional
imbalances associated with Western diseases: causes and consequences of chronic
systemic low-grade inflammation in an evolutionary context. The Journal of nutritional
biochemistry (2013) 24(7):1183-201. Epub 2013/05/10. doi: 10.1016/j.jnutbio.2013.02.009.
PubMed PMID: 23657158.
6. Erridge C, Attina T, Spickett CM, Webb DJ. A high-fat meal induces low-grade
endotoxemia: evidence of a novel mechanism of postprandial inflammation. The
American journal of clinical nutrition (2007) 86(5):1286-92. PubMed PMID: 17991637.
7. Moreira AP, Texeira TF, Ferreira AB, Peluzio Mdo C, Alfenas Rde C. Influence of a high-
fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. The
British journal of nutrition (2012) 108(5):801-9. Epub 2012/06/22. doi:
10.1017/S0007114512001213. PubMed PMID: 22717075.
8. Vaishnavi C. Translocation of gut flora and its role in sepsis. Indian journal of medical
microbiology (2013) 31(4):334-42. Epub 2013/09/26. doi: 10.4103/0255-0857.118870.
PubMed PMID: 24064638.
9. Maes M, Mihaylova I, Leunis JC. Increased serum IgM antibodies directed against
phosphatidyl inositol (Pi) in chronic fatigue syndrome (CFS) and major depression:
evidence that an IgM-mediated immune response against Pi is one factor underpinning
the comorbidity between both CFS and depression. Neuro endocrinology letters (2007)
28(6):861-7. Epub 2007/12/08. PubMed PMID: 18063934.
10. Maes M, Mihaylova I, Leunis JC. Increased serum IgA and IgM against LPS of
enterobacteria in chronic fatigue syndrome (CFS): indication for the involvement of
gram-negative enterobacteria in the etiology of CFS and for the presence of an
increased gut-intestinal permeability. Journal of affective disorders (2007) 99(1-3):237-
40. Epub 2006/09/30. doi: 10.1016/j.jad.2006.08.021. PubMed PMID: 17007934.

168
11. Sandek A, Bjarnason I, Volk HD, Crane R, Meddings JB, Niebauer J, et al. Studies on
bacterial endotoxin and intestinal absorption function in patients with chronic heart
failure. International journal of cardiology (2012) 157(1):80-5. Epub 2010/12/31. doi:
10.1016/j.ijcard.2010.12.016. PubMed PMID: 21190739.
12. Jayashree B, Bibin YS, Prabhu D, Shanthirani CS, Gokulakrishnan K, Lakshmi BS, et al.
Increased circulatory levels of lipopolysaccharide (LPS) and zonulin signify novel
biomarkers of proinflammation in patients with type 2 diabetes. Molecular and cellular
biochemistry (2014) 388(1-2):203-10. doi: 10.1007/s11010-013-1911-4. PubMed PMID:
24347174.
13. Emanuele E, Orsi P, Boso M, Broglia D, Brondino N, Barale F, et al. Low-grade
endotoxemia in patients with severe autism. Neuroscience letters (2010) 471(3):162-5.
doi: 10.1016/j.neulet.2010.01.033. PubMed PMID: 20097267.
14. Harte AL, da Silva NF, Creely SJ, McGee KC, Billyard T, Youssef-Elabd EM, et al. Elevated
endotoxin levels in non-alcoholic fatty liver disease. Journal of inflammation (2010)
7:15. doi: 10.1186/1476-9255-7-15. PubMed PMID: 20353583; PubMed Central PMCID:
PMC2873499.
15. Caradonna L, Amati L, Lella P, Jirillo E, Caccavo D. Phagocytosis, killing, lymphocyte-
mediated antibacterial activity, serum autoantibodies, and plasma endotoxins in
inflammatory bowel disease. The American journal of gastroenterology (2000)
95(6):1495-502. doi: 10.1111/j.1572-0241.2000.02085.x. PubMed PMID: 10894586.
16. Wang JE, Dahle MK, McDonald M, Foster SJ, Aasen AO, Thiemermann C. Peptidoglycan
and lipoteichoic acid in gram-positive bacterial sepsis: receptors, signal transduction,
biological effects, and synergism. Shock (2003) 20(5):402-14. doi:
10.1097/01.shk.0000092268.01859.0d. PubMed PMID: 14560103.
17. Giannelli V, Di Gregorio V, Iebba V, Giusto M, Schippa S, Merli M, et al. Microbiota and
the gut-liver axis: bacterial translocation, inflammation and infection in cirrhosis. World
journal of gastroenterology : WJG (2014) 20(45):16795-810. doi:
10.3748/wjg.v20.i45.16795. PubMed PMID: 25492994; PubMed Central PMCID:
PMC4258550.
18. Bohannon JK, Hernandez A, Enkhbaatar P, Adams WL, Sherwood ER. The
immunobiology of toll-like receptor 4 agonists: from endotoxin tolerance to
immunoadjuvants. Shock (2013) 40(6):451-62. doi: 10.1097/SHK.0000000000000042.
PubMed PMID: 23989337; PubMed Central PMCID: PMC3919163.
19. Neves AL, Coelho J, Couto L, Leite-Moreira A, Roncon-Albuquerque R, Jr. Metabolic
endotoxemia: a molecular link between obesity and cardiovascular risk. Journal of
molecular endocrinology (2013) 51(2):R51-64. Epub 2013/08/15. doi: 10.1530/JME-13-
0079. PubMed PMID: 23943858.

169
20. Weinlich R, Bortoluci KR, Chehab CF, Serezani CH, Ulbrich AG, Peters-Golden M, et al.
TLR4/MYD88-dependent, LPS-induced synthesis of PGE2 by macrophages or dendritic
cells prevents anti-CD3-mediated CD95L upregulation in T cells. Cell death and
differentiation (2008) 15(12):1901-9. doi: 10.1038/cdd.2008.128. PubMed PMID: 18820644.
21. Maitra U, Gan L, Chang S, Li L. Low-dose endotoxin induces inflammation by selectively
removing nuclear receptors and activating CCAAT/enhancer-binding protein delta.
Journal of immunology (2011) 186(7):4467-73. doi: 10.4049/jimmunol.1003300. PubMed
PMID: 21357541.
22. Cavaillon JM, Adib-Conquy M. Bench-to-bedside review: endotoxin tolerance as a
model of leukocyte reprogramming in sepsis. Critical care (2006) 10(5):233. doi:
10.1186/cc5055. PubMed PMID: 17044947; PubMed Central PMCID: PMC1751079.
23. Deng H, Maitra U, Morris M, Li L. Molecular mechanism responsible for the priming of
macrophage activation. The Journal of biological chemistry (2013) 288(6):3897-906. doi:
10.1074/jbc.M112.424390. PubMed PMID: 23264622; PubMed Central PMCID:
PMC3567643.
24. Kapus A, Szaszi K. Coupling between apical and paracellular transport processes.
Biochemistry and cell biology = Biochimie et biologie cellulaire (2006) 84(6):870-80. doi:
10.1139/o06-202. PubMed PMID: 17215874.
25. Turner JR, Rill BK, Carlson SL, Carnes D, Kerner R, Mrsny RJ, et al. Physiological
regulation of epithelial tight junctions is associated with myosin light-chain
phosphorylation. The American journal of physiology (1997) 273(4 Pt 1):C1378-85.
PubMed PMID: 9357784.
26. Lee CY. Chronic restraint stress induces intestinal inflammation and alters the
expression of hexose and lipid transporters. Clinical and experimental pharmacology &
physiology (2013) 40(6):385-91. doi: 10.1111/1440-1681.12096. PubMed PMID: 23586523.
27. Turner JR. Show me the pathway! Regulation of paracellular permeability by Na(+)-
glucose cotransport. Advanced drug delivery reviews (2000) 41(3):265-81. PubMed PMID:
10854686.
28. Turner JR. Intestinal mucosal barrier function in health and disease. Nature reviews
Immunology (2009) 9(11):799-809. Epub 2009/10/27. doi: 10.1038/nri2653. PubMed
PMID: 19855405.
29. Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. American
journal of physiology Cell physiology (2004) 286(6):C1213-28. doi:
10.1152/ajpcell.00558.2003. PubMed PMID: 15151915.
30. Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring
Harbor perspectives in biology (2009) 1(2):a002584. doi: 10.1101/cshperspect.a002584.
PubMed PMID: 20066090; PubMed Central PMCID: PMC2742087.

170
31. Overgaard CE, Daugherty BL, Mitchell LA, Koval M. Claudins: control of barrier function
and regulation in response to oxidant stress. Antioxidants & redox signaling (2011)
15(5):1179-93. doi: 10.1089/ars.2011.3893. PubMed PMID: 21275791; PubMed Central
PMCID: PMC3144428.
32. Graham WV, Wang F, Clayburgh DR, Cheng JX, Yoon B, Wang Y, et al. Tumor necrosis
factor-induced long myosin light chain kinase transcription is regulated by
differentiation-dependent signaling events. Characterization of the human long myosin
light chain kinase promoter. The Journal of biological chemistry (2006) 281(36):26205-
15. doi: 10.1074/jbc.M602164200. PubMed PMID: 16835238.
33. Shen L, Black ED, Witkowski ED, Lencer WI, Guerriero V, Schneeberger EE, et al. Myosin
light chain phosphorylation regulates barrier function by remodeling tight junction
structure. Journal of cell science (2006) 119(Pt 10):2095-106. doi: 10.1242/jcs.02915.
PubMed PMID: 16638813.
34. Cunningham KE, Turner JR. Myosin light chain kinase: pulling the strings of epithelial
tight junction function. Annals of the New York Academy of Sciences (2012) 1258:34-42.
doi: 10.1111/j.1749-6632.2012.06526.x. PubMed PMID: 22731713; PubMed Central PMCID:
PMC3384706.
35. Secondulfo M, Iafusco D, Carratu R, deMagistris L, Sapone A, Generoso M, et al.
Ultrastructural mucosal alterations and increased intestinal permeability in non-celiac,
type I diabetic patients. Digestive and liver disease : official journal of the Italian Society
of Gastroenterology and the Italian Association for the Study of the Liver (2004) 36(1):35-
45. PubMed PMID: 14971814.
36. Fasano A. Leaky gut and autoimmune diseases. Clinical reviews in allergy &
immunology (2012) 42(1):71-8. doi: 10.1007/s12016-011-8291-x. PubMed PMID:
22109896.
37. Keita AV, Soderholm JD. The intestinal barrier and its regulation by neuroimmune
factors. Neurogastroenterology and motility : the official journal of the European
Gastrointestinal Motility Society (2010) 22(7):718-33. Epub 2010/04/10. doi: 10.1111/j.1365-
2982.2010.01498.x. PubMed PMID: 20377785.
38. Hijazi Z, Molla AM, Al-Habashi H, Muawad WM, Molla AM, Sharma PN. Intestinal
permeability is increased in bronchial asthma. Archives of disease in childhood (2004)
89(3):227-9. PubMed PMID: 14977697; PubMed Central PMCID: PMC1719843.
39. Maes M, Kubera M, Leunis JC, Berk M, Geffard M, Bosmans E. In depression, bacterial
translocation may drive inflammatory responses, oxidative and nitrosative stress
(O&NS), and autoimmune responses directed against O&NS-damaged neoepitopes. Acta
psychiatrica Scandinavica (2013) 127(5):344-54. doi: 10.1111/j.1600-0447.2012.01908.x.
PubMed PMID: 22900942.

171
40. de Punder K, Pruimboom L. The dietary intake of wheat and other cereal grains and
their role in inflammation. Nutrients (2013) 5(3):771-87. doi: 10.3390/nu5030771. PubMed
PMID: 23482055; PubMed Central PMCID: PMC3705319.
41. Straub RH. Evolutionary medicine and chronic inflammatory state--known and new
concepts in pathophysiology. Journal of molecular medicine (2012) 90(5):523-34. doi:
10.1007/s00109-012-0861-8. PubMed PMID: 22271169; PubMed Central PMCID:
PMC3354326.
42. Straub RH. TRPV1, TRPA1, and TRPM8 channels in inflammation, energy redirection,
and water retention: role in chronic inflammatory diseases with an evolutionary
perspective. Journal of molecular medicine (2014). doi: 10.1007/s00109-014-1175-9.
PubMed PMID: 24871046.
43. Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J. The
sympathetic nervous system in heart failure physiology, pathophysiology, and clinical
implications. Journal of the American College of Cardiology (2009) 54(19):1747-62. doi:
10.1016/j.jacc.2009.05.015. PubMed PMID: 19874988.
44. Groeschel M, Braam B. Connecting chronic and recurrent stress to vascular dysfunction:
no relaxed role for the renin-angiotensin system. American journal of physiology Renal
physiology (2011) 300(1):F1-10. doi: 10.1152/ajprenal.00208.2010. PubMed PMID:
20980410.
45. Brunsson I, Eklund S, Jodal M, Lundgren O, Sjovall H. The effect of vasodilatation and
sympathetic nerve activation on net water absorption in the cat's small intestine. Acta
physiologica Scandinavica (1979) 106(1):61-8. doi: 10.1111/j.1748-1716.1979.tb06370.x.
PubMed PMID: 463580.
46. Levens NR, Peach MJ, Carey RM. Interactions between angiotensin peptides and the
sympathetic nervous system mediating intestinal sodium and water absorption in the
rat. The Journal of clinical investigation (1981) 67(4):1197-207. PubMed PMID: 7204574;
PubMed Central PMCID: PMC370682.
47. Lange S, Delbro DS. Adrenoceptor-mediated modulation of Evans blue dye permeation
of rat small intestine. Digestive diseases and sciences (1995) 40(12):2623-9. PubMed
PMID: 8536522.
48. Aschenbach JR, Borau T, Gabel G. Glucose uptake via SGLT-1 is stimulated by beta(2)-
adrenoceptors in the ruminal epithelium of sheep. The Journal of nutrition (2002)
132(6):1254-7. PubMed PMID: 12042442.
49. Ishikawa Y, Eguchi T, Ishida H. Mechanism of beta-adrenergic agonist-induced
transmural transport of glucose in rat small intestine. Regulation of phosphorylation of
SGLT1 controls the function. Biochimica et biophysica acta (1997) 1357(3):306-18.
PubMed PMID: 9268055.

172
50. Schaper J, Wagner A, Enigk F, Brell B, Mousa SA, Habazettl H, et al. Regional sympathetic
blockade attenuates activation of intestinal macrophages and reduces gut barrier failure.
Anesthesiology (2013) 118(1):134-42. Epub 2012/12/12. doi:
10.1097/ALN.0b013e3182784c93. PubMed PMID: 23221864.
51. de Kloet ER, Joels M, Holsboer F. Stress and the brain: from adaptation to disease. Nature
reviews Neuroscience (2005) 6(6):463-75. doi: 10.1038/nrn1683. PubMed PMID: 15891777.
52. Hattangady NG, Olala LO, Bollag WB, Rainey WE. Acute and chronic regulation of
aldosterone production. Molecular and cellular endocrinology (2012) 350(2):151-62. doi:
10.1016/j.mce.2011.07.034. PubMed PMID: 21839803; PubMed Central PMCID:
PMC3253327.
53. Yu Y, Liu ZQ, Liu XY, Yang L, Geng XR, Yang G, et al. Stress-Derived Corticotropin
Releasing Factor Breaches Epithelial Endotoxin Tolerance. PloS one (2013) 8(6):e65760.
doi: 10.1371/journal.pone.0065760. PubMed PMID: 23840363; PubMed Central PMCID:
PMC3686760.
54. Suzuki T, Yoshinaga N, Tanabe S. Interleukin-6 (IL-6) regulates claudin-2 expression
and tight junction permeability in intestinal epithelium. The Journal of biological
chemistry (2011) 286(36):31263-71. doi: 10.1074/jbc.M111.238147. PubMed PMID:
21771795; PubMed Central PMCID: PMC3173073.
55. Meddings JB, Swain MG. Environmental stress-induced gastrointestinal permeability is
mediated by endogenous glucocorticoids in the rat. Gastroenterology (2000)
119(4):1019-28. PubMed PMID: 11040188.
56. Caso JR, Leza JC, Menchen L. The effects of physical and psychological stress on the
gastro-intestinal tract: lessons from animal models. Current molecular medicine (2008)
8(4):299-312. Epub 2008/06/10. PubMed PMID: 18537637.
57. Vanuytsel T, van Wanrooy S, Vanheel H, Vanormelingen C, Verschueren S, Houben E, et
al. Psychological stress and corticotropin-releasing hormone increase intestinal
permeability in humans by a mast cell-dependent mechanism. Gut (2013). Epub
2013/10/25. doi: 10.1136/gutjnl-2013-305690. PubMed PMID: 24153250.
58. Capaldo CT, Nusrat A. Cytokine regulation of tight junctions. Biochimica et biophysica
acta (2009) 1788(4):864-71. doi: 10.1016/j.bbamem.2008.08.027. PubMed PMID:
18952050; PubMed Central PMCID: PMC2699410.
59. Watson CJ, Hoare CJ, Garrod DR, Carlson GL, Warhurst G. Interferon-gamma selectively
increases epithelial permeability to large molecules by activating different populations
of paracellular pores. Journal of cell science (2005) 118(Pt 22):5221-30. doi:
10.1242/jcs.02630. PubMed PMID: 16249235.
60. Wersching H, Duning T, Lohmann H, Mohammadi S, Stehling C, Fobker M, et al. Serum
C-reactive protein is linked to cerebral microstructural integrity and cognitive function.

173
Neurology (2010) 74(13):1022-9. doi: 10.1212/WNL.0b013e3181d7b45b. PubMed PMID:
20350977.
61. Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to
sickness and depression: when the immune system subjugates the brain. Nature
reviews Neuroscience (2008) 9(1):46-56. doi: 10.1038/nrn2297. PubMed PMID: 18073775;
PubMed Central PMCID: PMC2919277.
62. Lang CH, Molina PE, Yousef KA, Tepper PG, Abumrad NN. Role of IL-1 alpha in central
nervous system immunomodulation of glucoregulation. Brain research (1993) 624(1-
2):53-60. PubMed PMID: 8252416.
63. Dunn AJ. Cytokine activation of the HPA axis. Annals of the New York Academy of
Sciences (2000) 917:608-17. PubMed PMID: 11268389.
64. Goebel MU, Baase J, Pithan V, Exton M, Saller B, Schedlowski M, et al. Acute interferon
beta-1b administration alters hypothalamic-pituitary-adrenal axis activity, plasma
cytokines and leukocyte distribution in healthy subjects. Psychoneuroendocrinology
(2002) 27(8):881-92. PubMed PMID: 12383450.
65. Zimomra ZR, Porterfield VM, Camp RM, Johnson JD. Time-dependent mediators of
HPA axis activation following live Escherichia coli. American journal of physiology
Regulatory, integrative and comparative physiology (2011) 301(6):R1648-57. doi:
10.1152/ajpregu.00301.2011. PubMed PMID: 21917906; PubMed Central PMCID:
PMC3233844.
66. Mohn CE, Fernandez-Solari J, De Laurentiis A, Prestifilippo JP, de la Cal C, Funk R, et al.
The rapid release of corticosterone from the adrenal induced by ACTH is mediated by
nitric oxide acting by prostaglandin E2. Proceedings of the National Academy of
Sciences of the United States of America (2005) 102(17):6213-8. doi:
10.1073/pnas.0502136102. PubMed PMID: 15837925; PubMed Central PMCID:
PMC1087960.
67. Vakharia K, Hinson JP. Lipopolysaccharide directly stimulates cortisol secretion by
human adrenal cells by a cyclooxygenase-dependent mechanism. Endocrinology (2005)
146(3):1398-402. doi: 10.1210/en.2004-0882. PubMed PMID: 15564329.
68. Zacharowski K, Zacharowski PA, Koch A, Baban A, Tran N, Berkels R, et al. Toll-like
receptor 4 plays a crucial role in the immune-adrenal response to systemic
inflammatory response syndrome. Proceedings of the National Academy of Sciences of
the United States of America (2006) 103(16):6392-7. doi: 10.1073/pnas.0601527103.
PubMed PMID: 16606831; PubMed Central PMCID: PMC1458888.
69. Serrats J, Schiltz JC, Garcia-Bueno B, van Rooijen N, Reyes TM, Sawchenko PE. Dual
roles for perivascular macrophages in immune-to-brain signaling. Neuron (2010)
65(1):94-106. doi: 10.1016/j.neuron.2009.11.032. PubMed PMID: 20152116; PubMed
Central PMCID: PMC2873837.

174
70. Dhabhar FS, Malarkey WB, Neri E, McEwen BS. Stress-induced redistribution of immune
cells--from barracks to boulevards to battlefields: a tale of three hormones--Curt Richter
Award winner. Psychoneuroendocrinology (2012) 37(9):1345-68. Epub 2012/06/26. doi:
10.1016/j.psyneuen.2012.05.008. PubMed PMID: 22727761; PubMed Central PMCID:
PMC3412918.
71. Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve--an integrative
interface between two supersystems: the brain and the immune system.
Pharmacological reviews (2000) 52(4):595-638. PubMed PMID: 11121511.
72. Kohm AP, Sanders VM. Norepinephrine and beta 2-adrenergic receptor stimulation
regulate CD4+ T and B lymphocyte function in vitro and in vivo. Pharmacological
reviews (2001) 53(4):487-525. PubMed PMID: 11734616.
73. Prather AA, Carroll JE, Fury JM, McDade KK, Ross D, Marsland AL. Gender differences in
stimulated cytokine production following acute psychological stress. Brain, behavior,
and immunity (2009) 23(5):622-8. doi: 10.1016/j.bbi.2008.11.004. PubMed PMID:
19070658; PubMed Central PMCID: PMC2694847.
74. Steptoe A, Hamer M, Chida Y. The effects of acute psychological stress on circulating
inflammatory factors in humans: a review and meta-analysis. Brain, behavior, and
immunity (2007) 21(7):901-12. doi: 10.1016/j.bbi.2007.03.011. PubMed PMID: 17475444.
75. Yamakawa K, Matsunaga M, Isowa T, Kimura K, Kasugai K, Yoneda M, et al. Transient
responses of inflammatory cytokines in acute stress. Biological psychology (2009)
82(1):25-32. doi: 10.1016/j.biopsycho.2009.05.001. PubMed PMID: 19446599.
76. Bierhaus A, Wolf J, Andrassy M, Rohleder N, Humpert PM, Petrov D, et al. A mechanism
converting psychosocial stress into mononuclear cell activation. Proceedings of the
National Academy of Sciences of the United States of America (2003) 100(4):1920-5. doi:
10.1073/pnas.0438019100. PubMed PMID: 12578963; PubMed Central PMCID:
PMC149934.
77. Barnes PJ, Adcock IM. Glucocorticoid resistance in inflammatory diseases. Lancet
(2009) 373(9678):1905-17. doi: 10.1016/S0140-6736(09)60326-3. PubMed PMID: 19482216.
78. Ait-Belgnaoui A, Durand H, Cartier C, Chaumaz G, Eutamene H, Ferrier L, et al.
Prevention of gut leakiness by a probiotic treatment leads to attenuated HPA response to
an acute psychological stress in rats. Psychoneuroendocrinology (2012) 37(11):1885-95.
Epub 2012/05/01. doi: 10.1016/j.psyneuen.2012.03.024. PubMed PMID: 22541937.
79. Glaser R, Kiecolt-Glaser JK. Stress-induced immune dysfunction: implications for
health. Nature reviews Immunology (2005) 5(3):243-51. doi: 10.1038/nri1571. PubMed
PMID: 15738954.
80. Huang CJ, Acevedo EO, Mari DC, Randazzo C, Shibata Y. Glucocorticoid inhibition of
leptin- and lipopolysaccharide-induced interlukin-6 production in obesity. Brain,
behavior, and immunity (2013). doi: 10.1016/j.bbi.2013.10.004. PubMed PMID: 24126150.

175
81. Maranville JC, Micic D, Hanauer SB, Di Rienzo A, Kupfer SS. In vitro sensitivity assays
and clinical response to glucocorticoids in patients with inflammatory bowel disease.
Journal of Crohn's & colitis (2014). doi: 10.1016/j.crohns.2014.06.013. PubMed PMID:
25052346.
82. Pariante CM. Glucocorticoid receptor function in vitro in patients with major
depression. Stress (2004) 7(4):209-19. doi: 10.1080/10253890500069650. PubMed PMID:
16019586.
83. Sauer J, Polack E, Wikinski S, Holsboer F, Stalla GK, Arzt E. The glucocorticoid sensitivity
of lymphocytes changes according to the activity of the hypothalamic-pituitary-
adrenocortical system. Psychoneuroendocrinology (1995) 20(3):269-80. PubMed PMID:
7777655.
84. Glaser R. Stress-associated immune dysregulation and its importance for human health:
a personal history of psychoneuroimmunology. Brain, behavior, and immunity (2005)
19(1):3-11. doi: 10.1016/j.bbi.2004.06.003. PubMed PMID: 15581732.
85. De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al. Impact of
diet in shaping gut microbiota revealed by a comparative study in children from Europe
and rural Africa. Proceedings of the National Academy of Sciences of the United States
of America (2010) 107(33):14691-6. doi: 10.1073/pnas.1005963107. PubMed PMID:
20679230; PubMed Central PMCID: PMC2930426.
86. Schnorr SL, Candela M, Rampelli S, Centanni M, Consolandi C, Basaglia G, et al. Gut
microbiome of the Hadza hunter-gatherers. Nature communications (2014) 5:3654. doi:
10.1038/ncomms4654. PubMed PMID: 24736369; PubMed Central PMCID: PMC3996546.
87. Brown K, DeCoffe D, Molcan E, Gibson DL. Diet-induced dysbiosis of the intestinal
microbiota and the effects on immunity and disease. Nutrients (2012) 4(8):1095-119. doi:
10.3390/nu4081095. PubMed PMID: 23016134; PubMed Central PMCID: PMC3448089.
88. van Baarlen P, Troost FJ, van Hemert S, van der Meer C, de Vos WM, de Groot PJ, et al.
Differential NF-kappaB pathways induction by Lactobacillus plantarum in the
duodenum of healthy humans correlating with immune tolerance. Proceedings of the
National Academy of Sciences of the United States of America (2009) 106(7):2371-6. doi:
10.1073/pnas.0809919106. PubMed PMID: 19190178; PubMed Central PMCID:
PMC2650163.
89. Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, et al. Gut microbiota from
twins discordant for obesity modulate metabolism in mice. Science (2013)
341(6150):1241214. doi: 10.1126/science.1241214. PubMed PMID: 24009397; PubMed
Central PMCID: PMC3829625.
90. Dinan TG, Cryan JF. Regulation of the stress response by the gut microbiota:
implications for psychoneuroendocrinology. Psychoneuroendocrinology (2012)

176
37(9):1369-78. Epub 2012/04/10. doi: 10.1016/j.psyneuen.2012.03.007. PubMed PMID:
22483040.
91. Karrow NA. Activation of the hypothalamic-pituitary-adrenal axis and autonomic
nervous system during inflammation and altered programming of the neuroendocrine-
immune axis during fetal and neonatal development: lessons learned from the model
inflammagen, lipopolysaccharide. Brain, behavior, and immunity (2006) 20(2):144-58.
doi: 10.1016/j.bbi.2005.05.003. PubMed PMID: 16023324.
92. Sominsky L, Fuller EA, Bondarenko E, Ong LK, Averell L, Nalivaiko E, et al. Functional
programming of the autonomic nervous system by early life immune exposure:
implications for anxiety. PloS one (2013) 8(3):e57700. doi: 10.1371/journal.pone.0057700.
PubMed PMID: 23483921; PubMed Central PMCID: PMC3590226.
93. Walker AK, Nakamura T, Hodgson DM. Neonatal lipopolysaccharide exposure alters
central cytokine responses to stress in adulthood in Wistar rats. Stress (2010) 13(6):506-
15. doi: 10.3109/10253890.2010.489977. PubMed PMID: 20666652.
94. Sudo N, Chida Y, Aiba Y, Sonoda J, Oyama N, Yu XN, et al. Postnatal microbial
colonization programs the hypothalamic-pituitary-adrenal system for stress response in
mice. The Journal of physiology (2004) 558(Pt 1):263-75. doi:
10.1113/jphysiol.2004.063388. PubMed PMID: 15133062; PubMed Central PMCID:
PMC1664925.
95. Bailey MT, Dowd SE, Galley JD, Hufnagle AR, Allen RG, Lyte M. Exposure to a social
stressor alters the structure of the intestinal microbiota: implications for stressor-
induced immunomodulation. Brain, behavior, and immunity (2011) 25(3):397-407. doi:
10.1016/j.bbi.2010.10.023. PubMed PMID: 21040780; PubMed Central PMCID:
PMC3039072.
96. Galley JD, Nelson MC, Yu Z, Dowd SE, Walter J, Kumar PS, et al. Exposure to a social
stressor disrupts the community structure of the colonic mucosa-associated microbiota.
BMC microbiology (2014) 14:189. doi: 10.1186/1471-2180-14-189. PubMed PMID:
25028050; PubMed Central PMCID: PMC4105248.
97. Bailey MT, Coe CL. Maternal separation disrupts the integrity of the intestinal microflora
in infant rhesus monkeys. Developmental psychobiology (1999) 35(2):146-55. PubMed
PMID: 10461128.
98. de Jonge WJ. The Gut's Little Brain in Control of Intestinal Immunity. ISRN
gastroenterology (2013) 2013:630159. doi: 10.1155/2013/630159. PubMed PMID: 23691339;
PubMed Central PMCID: PMC3649343.
99. Bien J, Palagani V, Bozko P. The intestinal microbiota dysbiosis and Clostridium difficile
infection: is there a relationship with inflammatory bowel disease? Therapeutic
advances in gastroenterology (2013) 6(1):53-68. doi: 10.1177/1756283X12454590. PubMed
PMID: 23320050; PubMed Central PMCID: PMC3539291.

177
100. Ahrne S, Hagslatt ML. Effect of lactobacilli on paracellular permeability in the gut.
Nutrients (2011) 3(1):104-17. Epub 2012/01/19. doi: 10.3390/nu3010104. PubMed PMID:
22254077; PubMed Central PMCID: PMC3257727.
101. Bergmann KR, Liu SX, Tian R, Kushnir A, Turner JR, Li HL, et al. Bifidobacteria stabilize
claudins at tight junctions and prevent intestinal barrier dysfunction in mouse
necrotizing enterocolitis. The American journal of pathology (2013) 182(5):1595-606. doi:
10.1016/j.ajpath.2013.01.013. PubMed PMID: 23470164; PubMed Central PMCID:
PMC3644725.
102. Leclercq S, Matamoros S, Cani PD, Neyrinck AM, Jamar F, Starkel P, et al. Intestinal
permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence
severity. Proceedings of the National Academy of Sciences of the United States of
America (2014) 111(42):E4485-93. doi: 10.1073/pnas.1415174111. PubMed PMID: 25288760;
PubMed Central PMCID: PMC4210345.
103. Barreau F, Hugot JP. Intestinal barrier dysfunction triggered by invasive bacteria.
Current opinion in microbiology (2014) 17:91-8. doi: 10.1016/j.mib.2013.12.003. PubMed
PMID: 24440560.
104. Troeger H, Richter JF, Beutin L, Gunzel D, Dobrindt U, Epple HJ, et al. Escherichia coli
alpha-haemolysin induces focal leaks in colonic epithelium: a novel mechanism of
bacterial translocation. Cellular microbiology (2007) 9(10):2530-40. doi: 10.1111/j.1462-
5822.2007.00978.x. PubMed PMID: 17587334.
105. Moreno-Navarrete JM, Manco M, Ibanez J, Garcia-Fuentes E, Ortega F, Gorostiaga E, et
al. Metabolic endotoxemia and saturated fat contribute to circulating NGAL
concentrations in subjects with insulin resistance. International journal of obesity (2010)
34(2):240-9. doi: 10.1038/ijo.2009.242. PubMed PMID: 19949414.
106. Amar J, Burcelin R, Ruidavets JB, Cani PD, Fauvel J, Alessi MC, et al. Energy intake is
associated with endotoxemia in apparently healthy men. The American journal of
clinical nutrition (2008) 87(5):1219-23. PubMed PMID: 18469242.
107. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic
endotoxemia initiates obesity and insulin resistance. Diabetes (2007) 56(7):1761-72. Epub
2007/04/26. doi: 10.2337/db06-1491. PubMed PMID: 17456850.
108. Ghanim H, Abuaysheh S, Sia CL, Korzeniewski K, Chaudhuri A, Fernandez-Real JM, et al.
Increase in plasma endotoxin concentrations and the expression of Toll-like receptors
and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-
carbohydrate meal: implications for insulin resistance. Diabetes care (2009) 32(12):2281-
7. doi: 10.2337/dc09-0979. PubMed PMID: 19755625; PubMed Central PMCID:
PMC2782991.

178
109. Herieka M, Erridge C. High-fat meal induced postprandial inflammation. Molecular
nutrition & food research (2014) 58(1):136-46. doi: 10.1002/mnfr.201300104. PubMed
PMID: 23847095.
110. Deopurkar R, Ghanim H, Friedman J, Abuaysheh S, Sia CL, Mohanty P, et al. Differential
effects of cream, glucose, and orange juice on inflammation, endotoxin, and the
expression of Toll-like receptor-4 and suppressor of cytokine signaling-3. Diabetes care
(2010) 33(5):991-7. doi: 10.2337/dc09-1630. PubMed PMID: 20067961; PubMed Central
PMCID: PMC2858203.
111. Pendyala S, Walker JM, Holt PR. A high-fat diet is associated with endotoxemia that
originates from the gut. Gastroenterology (2012) 142(5):1100-1 e2. doi:
10.1053/j.gastro.2012.01.034. PubMed PMID: 22326433; PubMed Central PMCID:
PMC3978718.
112. Kelly CJ, Colgan SP, Frank DN. Of microbes and meals: the health consequences of
dietary endotoxemia. Nutrition in clinical practice : official publication of the American
Society for Parenteral and Enteral Nutrition (2012) 27(2):215-25. doi:
10.1177/0884533611434934. PubMed PMID: 22378797; PubMed Central PMCID:
PMC4046172.
113. Cani PD, Delzenne NM, Amar J, Burcelin R. Role of gut microflora in the development of
obesity and insulin resistance following high-fat diet feeding. Pathologie-biologie
(2008) 56(5):305-9. doi: 10.1016/j.patbio.2007.09.008. PubMed PMID: 18178333.
114. Cassaglia PA, Hermes SM, Aicher SA, Brooks VL. Insulin acts in the arcuate nucleus to
increase lumbar sympathetic nerve activity and baroreflex function in rats. The Journal
of physiology (2011) 589(Pt 7):1643-62. doi: 10.1113/jphysiol.2011.205575. PubMed PMID:
21300750; PubMed Central PMCID: PMC3099021.
115. Simonds SE, Cowley MA. Hypertension in obesity: is leptin the culprit? Trends in
neurosciences (2013) 36(2):121-32. doi: 10.1016/j.tins.2013.01.004. PubMed PMID:
23333346.
116. Stumpel F, Scholtka B, Jungermann K. Stimulation by portal insulin of intestinal glucose
absorption via hepatoenteral nerves and prostaglandin E2 in the isolated, jointly
perfused small intestine and liver of the rat. Annals of the New York Academy of
Sciences (2000) 915:111-6. PubMed PMID: 11193565.
117. Sander GR, Cummins AG, Henshall T, Powell BC. Rapid disruption of intestinal barrier
function by gliadin involves altered expression of apical junctional proteins. FEBS letters
(2005) 579(21):4851-5. PubMed PMID: 16099460.
118. Drago S, El Asmar R, Di Pierro M, Grazia Clemente M, Tripathi A, Sapone A, et al. Gliadin,
zonulin and gut permeability: Effects on celiac and non-celiac intestinal mucosa and
intestinal cell lines. Scandinavian journal of gastroenterology (2006) 41(4):408-19.
PubMed PMID: 16635908.

179
119. Lammers KM, Lu R, Brownley J, Lu B, Gerard C, Thomas K, et al. Gliadin induces an
increase in intestinal permeability and zonulin release by binding to the chemokine
receptor CXCR3. Gastroenterology (2008) 135(1):194-204 e3. PubMed PMID: 18485912.
120. Chen P, Starkel P, Turner JR, Ho SB, Schnabl B. Dysbiosis-induced intestinal
inflammation activates TNFRI and mediates alcoholic liver disease in mice. Hepatology
(2014). doi: 10.1002/hep.27489. PubMed PMID: 25251280.
121. Bode C, Kugler V, Bode JC. Endotoxemia in patients with alcoholic and non-alcoholic
cirrhosis and in subjects with no evidence of chronic liver disease following acute
alcohol excess. Journal of hepatology (1987) 4(1):8-14. PubMed PMID: 3571935.
122. Fukui H, Brauner B, Bode JC, Bode C. Plasma endotoxin concentrations in patients with
alcoholic and non-alcoholic liver disease: reevaluation with an improved chromogenic
assay. Journal of hepatology (1991) 12(2):162-9. PubMed PMID: 2050995.
123. Parlesak A, Schafer C, Schutz T, Bode JC, Bode C. Increased intestinal permeability to
macromolecules and endotoxemia in patients with chronic alcohol abuse in different
stages of alcohol-induced liver disease. Journal of hepatology (2000) 32(5):742-7.
PubMed PMID: 10845660.
124. Oktedalen O, Lunde OC, Opstad PK, Aabakken L, Kvernebo K. Changes in the
gastrointestinal mucosa after long-distance running. Scandinavian journal of
gastroenterology (1992) 27(4):270-4. PubMed PMID: 1589703.
125. Ashton T, Young IS, Davison GW, Rowlands CC, McEneny J, Van Blerk C, et al. Exercise-
induced endotoxemia: the effect of ascorbic acid supplementation. Free radical biology
& medicine (2003) 35(3):284-91. PubMed PMID: 12885590.
126. Jeukendrup AE, Vet-Joop K, Sturk A, Stegen JH, Senden J, Saris WH, et al. Relationship
between gastro-intestinal complaints and endotoxaemia, cytokine release and the
acute-phase reaction during and after a long-distance triathlon in highly trained men.
Clinical science (2000) 98(1):47-55. PubMed PMID: 10600658.
127. Pals KL, Chang RT, Ryan AJ, Gisolfi CV. Effect of running intensity on intestinal
permeability. Journal of applied physiology (1997) 82(2):571-6. PubMed PMID: 9049739.
128. Selkirk GA, McLellan TM, Wright HE, Rhind SG. Mild endotoxemia, NF-kappaB
translocation, and cytokine increase during exertional heat stress in trained and
untrained individuals. American journal of physiology Regulatory, integrative and
comparative physiology (2008) 295(2):R611-23. doi: 10.1152/ajpregu.00917.2007. PubMed
PMID: 18565834.
129. de Oliveira EP, Burini RC, Jeukendrup A. Gastrointestinal complaints during exercise:
prevalence, etiology, and nutritional recommendations. Sports medicine (2014) 44 Suppl
1:S79-85. doi: 10.1007/s40279-014-0153-2. PubMed PMID: 24791919; PubMed Central
PMCID: PMC4008808.

180
130. Hart EC, Charkoudian N. Sympathetic neural regulation of blood pressure: influences of
sex and aging. Physiology (2014) 29(1):8-15. doi: 10.1152/physiol.00031.2013. PubMed
PMID: 24382867.
131. Ghosh S, Lertwattanarak R, Garduno JD, Galeana JJ, Li J, Zamarripa F, et al. Elevated
Muscle TLR4 Expression and Metabolic Endotoxemia in Human Aging. The journals of
gerontology Series A, Biological sciences and medical sciences (2014). doi:
10.1093/gerona/glu067. PubMed PMID: 24846769.
132. Licht CM, de Geus EJ, Penninx BW. Dysregulation of the autonomic nervous system
predicts the development of the metabolic syndrome. The Journal of clinical
endocrinology and metabolism (2013) 98(6):2484-93. doi: 10.1210/jc.2012-3104. PubMed
PMID: 23553857.
133. Kazakou P, Kyriazopoulou V, Michalaki M, Ierodiakonou V, Psyrogiannis A, Habeos I.
Activated hypothalamic pituitary adrenal axis in patients with metabolic syndrome.
Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones
et metabolisme (2012) 44(11):839-44. doi: 10.1055/s-0032-1311632. PubMed PMID:
22549399.
134. Lassenius MI, Pietilainen KH, Kaartinen K, Pussinen PJ, Syrjanen J, Forsblom C, et al.
Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin
resistance, obesity, and chronic inflammation. Diabetes care (2011) 34(8):1809-15. doi:
10.2337/dc10-2197. PubMed PMID: 21636801; PubMed Central PMCID: PMC3142060.
135. Moreno-Navarrete JM, Ortega F, Serino M, Luche E, Waget A, Pardo G, et al. Circulating
lipopolysaccharide-binding protein (LBP) as a marker of obesity-related insulin
resistance. International journal of obesity (2012) 36(11):1442-9. doi: 10.1038/ijo.2011.256.
PubMed PMID: 22184060.
136. Romani J, Caixas A, Escote X, Carrascosa JM, Ribera M, Rigla M, et al.
Lipopolysaccharide-binding protein is increased in patients with psoriasis with
metabolic syndrome, and correlates with C-reactive protein. Clinical and experimental
dermatology (2013) 38(1):81-4. doi: 10.1111/ced.12007. PubMed PMID: 23082944.
137. Sun L, Yu Z, Ye X, Zou S, Li H, Yu D, et al. A marker of endotoxemia is associated with
obesity and related metabolic disorders in apparently healthy Chinese. Diabetes care
(2010) 33(9):1925-32. doi: 10.2337/dc10-0340. PubMed PMID: 20530747; PubMed Central
PMCID: PMC2928335.
138. Farhadi A, Gundlapalli S, Shaikh M, Frantzides C, Harrell L, Kwasny MM, et al.
Susceptibility to gut leakiness: a possible mechanism for endotoxaemia in non-
alcoholic steatohepatitis. Liver international : official journal of the International
Association for the Study of the Liver (2008) 28(7):1026-33. Epub 2008/04/10. doi:
10.1111/j.1478-3231.2008.01723.x. PubMed PMID: 18397235.

181
139. Ruiz AG, Casafont F, Crespo J, Cayon A, Mayorga M, Estebanez A, et al.
Lipopolysaccharide-binding protein plasma levels and liver TNF-alpha gene expression
in obese patients: evidence for the potential role of endotoxin in the pathogenesis of
non-alcoholic steatohepatitis. Obesity surgery (2007) 17(10):1374-80. doi:
10.1007/s11695-007-9243-7. PubMed PMID: 18000721.
140. Al-Attas OS, Al-Daghri NM, Al-Rubeaan K, da Silva NF, Sabico SL, Kumar S, et al.
Changes in endotoxin levels in T2DM subjects on anti-diabetic therapies.
Cardiovascular diabetology (2009) 8:20. doi: 10.1186/1475-2840-8-20. PubMed PMID:
19368716; PubMed Central PMCID: PMC2674418.
141. Creely SJ, McTernan PG, Kusminski CM, Fisher f M, Da Silva NF, Khanolkar M, et al.
Lipopolysaccharide activates an innate immune system response in human adipose
tissue in obesity and type 2 diabetes. American journal of physiology Endocrinology and
metabolism (2007) 292(3):E740-7. doi: 10.1152/ajpendo.00302.2006. PubMed PMID:
17090751.
142. Pussinen PJ, Havulinna AS, Lehto M, Sundvall J, Salomaa V. Endotoxemia is associated
with an increased risk of incident diabetes. Diabetes care (2011) 34(2):392-7. doi:
10.2337/dc10-1676. PubMed PMID: 21270197; PubMed Central PMCID: PMC3024355.
143. Harte AL, Varma MC, Tripathi G, McGee KC, Al-Daghri NM, Al-Attas OS, et al. High fat
intake leads to acute postprandial exposure to circulating endotoxin in type 2 diabetic
subjects. Diabetes care (2012) 35(2):375-82. doi: 10.2337/dc11-1593. PubMed PMID:
22210577; PubMed Central PMCID: PMC3263907.
144. Lepper PM, Kleber ME, Grammer TB, Hoffmann K, Dietz S, Winkelmann BR, et al.
Lipopolysaccharide-binding protein (LBP) is associated with total and cardiovascular
mortality in individuals with or without stable coronary artery disease--results from the
Ludwigshafen Risk and Cardiovascular Health Study (LURIC). Atherosclerosis (2011)
219(1):291-7. doi: 10.1016/j.atherosclerosis.2011.06.001. PubMed PMID: 21722903.
145. Serrano M, Moreno-Navarrete JM, Puig J, Moreno M, Guerra E, Ortega F, et al. Serum
lipopolysaccharide-binding protein as a marker of atherosclerosis. Atherosclerosis
(2013) 230(2):223-7. doi: 10.1016/j.atherosclerosis.2013.07.004. PubMed PMID: 24075748.
146. Wiedermann CJ, Kiechl S, Dunzendorfer S, Schratzberger P, Egger G, Oberhollenzer F, et
al. Association of endotoxemia with carotid atherosclerosis and cardiovascular disease:
prospective results from the Bruneck Study. Journal of the American College of
Cardiology (1999) 34(7):1975-81. PubMed PMID: 10588212.
147. Niebauer J, Volk HD, Kemp M, Dominguez M, Schumann RR, Rauchhaus M, et al.
Endotoxin and immune activation in chronic heart failure: a prospective cohort study.
Lancet (1999) 353(9167):1838-42. doi: 10.1016/S0140-6736(98)09286-1. PubMed PMID:
10359409.

182
148. Garg M, Angus PW, Burrell LM, Herath C, Gibson PR, Lubel JS. Review article: the
pathophysiological roles of the renin-angiotensin system in the gastrointestinal tract.
Alimentary pharmacology & therapeutics (2012) 35(4):414-28. doi: 10.1111/j.1365-
2036.2011.04971.x. PubMed PMID: 22221317.
149. Mawdsley JE, Rampton DS. Psychological stress in IBD: new insights into pathogenic
and therapeutic implications. Gut (2005) 54(10):1481-91. doi: 10.1136/gut.2005.064261.
PubMed PMID: 16162953; PubMed Central PMCID: PMC1774724.
150. Caradonna L, Amati L, Magrone T, Pellegrino NM, Jirillo E, Caccavo D. Enteric bacteria,
lipopolysaccharides and related cytokines in inflammatory bowel disease: biological and
clinical significance. Journal of endotoxin research (2000) 6(3):205-14. PubMed PMID:
11052175.
151. Pastor Rojo O, Lopez San Roman A, Albeniz Arbizu E, de la Hera Martinez A, Ripoll
Sevillano E, Albillos Martinez A. Serum lipopolysaccharide-binding protein in
endotoxemic patients with inflammatory bowel disease. Inflammatory bowel diseases
(2007) 13(3):269-77. doi: 10.1002/ibd.20019. PubMed PMID: 17206721.
152. Funderburg NT, Stubblefield Park SR, Sung HC, Hardy G, Clagett B, Ignatz-Hoover J, et
al. Circulating CD4(+) and CD8(+) T cells are activated in inflammatory bowel disease
and are associated with plasma markers of inflammation. Immunology (2013) 140(1):87-
97. doi: 10.1111/imm.12114. PubMed PMID: 23600521; PubMed Central PMCID:
PMC3809709.
153. Gardiner KR, Halliday MI, Barclay GR, Milne L, Brown D, Stephens S, et al. Significance of
systemic endotoxaemia in inflammatory bowel disease. Gut (1995) 36(6):897-901.
PubMed PMID: 7615280; PubMed Central PMCID: PMC1382629.
154. Dinan TG, Stanton C, Cryan JF. Psychobiotics: a novel class of psychotropic. Biological
psychiatry (2013) 74(10):720-6. Epub 2013/06/14. doi: 10.1016/j.biopsych.2013.05.001.
PubMed PMID: 23759244.
155. Forsyth CB, Shannon KM, Kordower JH, Voigt RM, Shaikh M, Jaglin JA, et al. Increased
intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and
endotoxin exposure markers in early Parkinson's disease. PloS one (2011) 6(12):e28032.
doi: 10.1371/journal.pone.0028032. PubMed PMID: 22145021; PubMed Central PMCID:
PMC3228722.
156. Zhang R, Miller RG, Gascon R, Champion S, Katz J, Lancero M, et al. Circulating
endotoxin and systemic immune activation in sporadic amyotrophic lateral sclerosis
(sALS). Journal of neuroimmunology (2009) 206(1-2):121-4. doi:
10.1016/j.jneuroim.2008.09.017. PubMed PMID: 19013651; PubMed Central PMCID:
PMC2995297.
157. Sparks Stein P, Steffen MJ, Smith C, Jicha G, Ebersole JL, Abner E, et al. Serum
antibodies to periodontal pathogens are a risk factor for Alzheimer's disease. Alzheimer's

183
& dementia : the journal of the Alzheimer's Association (2012) 8(3):196-203. doi:
10.1016/j.jalz.2011.04.006. PubMed PMID: 22546352; PubMed Central PMCID:
PMC3712346.
158. Pussinen PJ, Tuomisto K, Jousilahti P, Havulinna AS, Sundvall J, Salomaa V.
Endotoxemia, immune response to periodontal pathogens, and systemic inflammation
associate with incident cardiovascular disease events. Arteriosclerosis, thrombosis, and
vascular biology (2007) 27(6):1433-9. doi: 10.1161/ATVBAHA.106.138743. PubMed PMID:
17363692.
159. Lepper PM, Schumann C, Triantafilou K, Rasche FM, Schuster T, Frank H, et al.
Association of lipopolysaccharide-binding protein and coronary artery disease in men.
Journal of the American College of Cardiology (2007) 50(1):25-31. doi:
10.1016/j.jacc.2007.02.070. PubMed PMID: 17601541.

184
Figure 1. MLC phosphorylation increases intestinal permeability.
Activation of the SNS stimulates intestinal permeability by increasing the activity of SGLT1 on epithelial cells. Activation of
SGLT1 is paralleled by MLC phosphorylation by MLCK, inducing actomyosin contraction and reorganization of the tight
junction. The resulting increase in paracellular permeability raises the possibility of translocation of bacteria and/or their
toxins across the more permeable gut barrier. Pro-inflammatory cytokines produced by activated immune cells residing in
the lamina propria further increase intestinal permeability by activating MLCK. JC, junctional complex.

185
Figure 2. The complex neuroendocrine-immune interactions and their relation to gut barrier
function.
Stressors, including inflammatory mediators, activate the SNS and HPA-axis. Activation of the HPA-axis stimulates the
neurons in the paraventricular nucleus of the hypothalamus to secrete CRH and AVP that trigger the release of ACTH from
the anterior pituitary, resulting in the secretion of corticosteroids from the adrenal cortex. CRH has been shown to affect
intestinal permeability. SNS activation results in the release of catecholamines from the adrenal medulla. The intestinal wall
is innervated by adrenergic sympathetic nerve fibers that upon stimulation increase water, sodium and glucose absorption,
paralleled by increased intestinal permeability. The resulting increase in translocation of endotoxin across the intestinal
barrier can stimulate immune cells in the underlying lamina propria to secrete pro-inflammatory cytokines and
prostaglandins like PgE2. Inflammatory mediators communicate with the brain by stimulating afferent sensory nerve fibers,
by entering the brain via the circumventricular organs or by binding to cerebral blood vessel endothelium. Continuous
stress-induced impairment of the intestinal barrier creates a vicious circle whereby inflammatory cytokines will persistently
activate the SNS and HPA-axis resulting in barrier disruption, increased endotoxin translocation and a pro-inflammatory
state.

186
Paragraph 1.5. The dietary intake of wheat and other cereal grains and their role in
inflammation. de Punder K, Pruimboom L. Nutrients 2013;5:771-87.
The Dietary Intake of Wheat and other Cereal Grains and their Role in
Inflammation
Karin de Punder 1, Leo Pruimboom1, 2,*
1. University of Girona, Plaça Sant Domènec, 3 Edifici Les Àligues, 17071 Girona, Spain; E-
Mail: [email protected]
2. Uni for Life, Universität Graz, Beethovenstraße 9, 8010, Graz, Austria; E-Mail:
Abstract
Wheat is one of the most consumed cereal grains worldwide and makes up a substantial part
of the human diet. Although government supported dietary guidelines in Europe and the
U.S.A advise individuals to eat adequate amounts of (whole) grain products per day, cereal
grains contain “anti-nutrients”, such as wheat gluten and wheat lectin, that in humans can
elicit dysfunction and disease. In this review we discuss evidence from in vitro, in vivo and
human intervention studies that describe how the consumption of wheat, but also other
cereal grains, can contribute to the manifestation of chronic inflammation and auto-immune
diseases by increasing intestinal permeability and initiating a pro-inflammatory immune
response.
Keywords cereal grains; coeliac disease; gluten; gliadin; inflammation; intestinal permeability; lectins;
wheat; wheat germ agglutinin

187
1. Introduction
Inflammation is the response of the innate immune system triggered by noxious stimuli,
microbial pathogens and injury. When a trigger remains or when immune cells are
continuously activated an inflammatory response may become self-sustainable and chronic.
Chronic inflammation has been associated with many medical and psychiatric disorders,
including cardiovascular disease, metabolic syndrome, cancer, auto-immune diseases,
schizophrenia and depression [1,2,3]. Furthermore, it is usually associated with elevated levels
of pro-inflammatory cytokines and acute phase proteins, such as interferons (IFNs),
interleukin (Il)-1, Il-6, tumor necrosis factor-α (TNF-α), and C-reactive protein (CRP). While
clear peripheral sources for this chronic inflammation are apparent in some conditions (i.e. fat
production of cytokines in the metabolic syndrome), in other disorders, such as major
depression, the inflammatory source is not completely understood. Genetic vulnerability,
psychological stress and poor dietary patterns have all been repeatedly implicated as being of
significant importance in the development of an inflammatory phenotype [3,4,5]. Dietary
factors associated with inflammation include a shift towards a higher n-6:n-3 fatty acid
ratio [5] and a high intake of simple sugars [6]. Other substances in our daily food, like those
found in wheat and other cereal grains, are also capable of activating pro-inflammatory
pathways.
2. Wheat grain, gluten and disease
2.1. Wheat allergy and intolerance
The ingestion of wheat products has been reported to be responsible for IgE-mediated allergic
reactions. Wheat-dependent exercise-induced anaphylaxis is a syndrome in which the
ingestion of a product containing wheat followed by physical exercise can result in an
anaphylactic response. Several proteins present in wheat, most notably gluten proteins have
been shown to react with IgE in patients [7]. Other allergic responses that appear to be related
to a range of wheat proteins include Bakers asthma, rhinitis and contact urticaria [7,8].
More common than wheat allergies are conditions involving wheat intolerance, including
coeliac disease (CD), which is estimated to affect 1% of the population of Western Europe, and
dermatitis herpetiformis, which has an incidence between about 2-fold and 5-fold lower than
CD [9]. The close association between type 1 diabetes and CD [10] and the observation that
auto-immune diseases seem to be more prevalent in coeliac patients and their relatives [11]
associate the intake of wheat with several other conditions.

188
2.2. Wheat grain and gluten
Gluten is the main structural protein complex of wheat consisting of glutenins and gliadins.
When wheat flour is mixed with water to form dough, the gluten proteins form a continuous
network which provides the cohesiveness and viscoelasticity that allows dough to be
processed into bread, noodles and other foods. The protein contents of wheat varies between
7-22% with gluten constituting about 80% of the total protein of the seed [9]. Glutenins are the
fraction of wheat proteins that are soluble in dilute acids and are polymers of individual
proteins. Prolamins are the alcohol soluble proteins of cereal grains and are specifically
named gliadins in wheat. Gliadins are monomeric proteins and are classified into three
groups: these are α/β-gliadins, γ-gliadins and ω-gliadins [7].
2.3. Gluten, gliadin and CD
Gliadin epitopes from wheat gluten and related prolamins from other gluten-containing
cereal grains, including rye and barley, can trigger CD in genetically susceptible people. The
symptoms of this disease are mucosal inflammation, small intestine villous atrophy, increased
intestinal permeability and malabsorption of macro- and micronutrients. CD, a chronic
inflammatory disorder mediated by T-cells, is preceded by changes in intestinal permeability
and pro-inflammatory activity of the innate immune system. Gliadin immunomodulatory
peptides can be recognized by specific T-cells, a process that can be enhanced by the
deamidation of gliadin epitopes by tissue transglutaminases that convert particular glutamine
residues into glutamic acid resulting in a higher affinity for HLA-DQ2 or DQ8 expressed on
antigen presenting cells (APC) [10]. Serum antibodies, among which are antibodies against
tissue transglutaminases, are also found in CD. The HLA-DQ2 or HLA-DQ8 is expressed in
99.4% of the patients suffering from CD [10], but interestingly enough there is a group HLA-
DQ2/DQ8 negative patients suffering from gastrointestinal symptoms that respond well to a
gluten-free diet. This group of “gluten-sensitive” patients does not have the CD serology and
histopathology, but does present the same symptoms and shows improvements when
following a gluten-free diet [12,13].
2.4. Gliadin and immunity
There are at least 50 gliadin epitopes that exert immunomodulatory, cytotoxic and gut
permeating activities that can be partially traced back to different domains of α-gliadin. Where
some immunomodulatory gliadin peptides activate specific T-cells, others are able to induce a
pro-inflammatory innate immune response [10]. Stimulation of immune cells by gliadin is not
only restricted to CD patients; the incubation of peripheral blood mononuclear cells (PBMC)
from healthy HLA-DQ2 positive controls and CD patients with gliadin peptides stimulated the
production of IL-23, IL-1β and TNF-α in all donors tested. Still the production of cytokines was
significantly higher in PBMC derived from CD patients [14]. Similar results were obtained by

189
Lammers et al. [15] showing that gliadin induced an inflammatory immune response in both
CD patients and healthy controls, yet IL-6, Il-13 and IFN-γ were expressed at significantly
higher levels in CD patients. IL-8 production was only expressed in a subset of healthy and CD
individuals after stimulation with a specific gliadin peptide and appeared to dependent on the
CXCR3 chemokine receptor only in CD patients. Sapone et al. [16] showed that in a subset of
CD patients, but not in gluten-sensitive patients (with 36% of the studied individuals in this
group being HLA-DQ2/DQ8 positive), there is an increased IL-17 mRNA expression in the
small-intestinal mucosa compared to healthy controls. The same group showed that in a
subset of gluten sensitive patients (with 50% of the studied individuals being HLA-DQ2/DQ8
positive) there is a prevailing stimulation of cells of the innate immune system, while in CD
both the innate and adaptive immune system are involved [13].
2.5. Gliadin and intestinal permeability
In order for gliadin to interact with cells of the immune system it has to overcome the
intestinal barrier. Gliadin peptides cross the epithelial layer by transcytosis or paracellular
transport. Paracellular transport occurs when intestinal permeability is increased, a feature
that is characteristic for CD [17]. It is indicated by several studies that increased intestinal
permeability precedes the onset of CD and is not just a consequence of chronic intestinal
inflammation [18,19]. Gliadin has been demonstrated to increase permeability in human
Caco-2 intestinal epithelial cells by reorganizing actin filaments and alter expression of
junctional complex proteins [20]. Several studies by the group of Fasano et al. show that the
binding of gliadin to the chemokine receptor CXCR3 on epithelial IEC-6 and Caco2 cells
releases zonulin, a protein that directly compromises the integrity of the junctional
complex [21,22]. Although zonulin levels were more up-regulated in CD patients, zonulin was
activated by gliadin in all intestinal biopsies from both CD and non-CD patients [21,22],
suggesting that gliadin can increase intestinal permeability also in non-CD patients, yet
increased intestinal permeability was not observed in a group of gluten-sensitive patients [13].
3. Increased intestinal permeability
3.1. Increased intestinal permeability is associated with disease
Chronically increased intestinal permeability (or leaky gut syndrome) allows for the increased
translocation of both microbial and dietary antigens to the periphery which can then interact
with cells of the immune system. Shared amino acid motifs among exogenous peptides (HLA-
derived peptides and self tissue) may produce cross reactivity through immunological
mimicry, thereby disturbing immune tolerance in genetically susceptible individuals [23]. Not
surprisingly, increased intestinal permeability has been associated with auto-immune
diseases, such as type 1 diabetes [24], rheumatoid arthritis, multiple sclerosis [18], but also with

190
diseases related to chronic inflammation like inflammatory bowel disease [18,25], asthma [26],
chronic fatigue syndrome and depression. The latter two conditions see patients with
significantly greater values of serum IgA and IgM to LPS of gram-negative enterobacteria
compared to controls, implying intestinal permeability is increased in these
patients [27,28,29].
3.2. Intestinal barrier function and inflammation
The intestinal barrier allows the uptake of nutrients and protects from damage of harmful
substances from the gut lumen. Macromolecules that can be immunogenic like proteins, large
peptides but also bacteria and lectins can be endocytosed or phagocytosed by enterocytes
forming the epithelial layer of the gut. Absorbed proteins generally will be entering the
lysosomal route and will be degraded to small peptides. Normally, only small amounts of
antigen pass the barrier by transcytosis and interact with the innate and adaptive immune
system situated in the lamina propria. Highly specialized epithelial microfold (M) cells
function as active transporters of dietary and microbial antigens from the gut lumen to the
immune system where either a pro-inflammatory or tolerogenic immune response can be
generated. The paracellular route is regulated by the junctional complex that allows the
passage of water, solutes and ions, but under normal conditions provides a barrier to larger
peptides and protein-sized molecules. When the barrier function is disrupted there is an
increased passage of dietary and microbial antigens interacting with cells of the immune
system [25,30] (Figure 1).
3.3. The role of zonulin signaling on intestinal permeability
Intestinal permeability is a measure of the barrier function of the gut which relates to the
paracellular space surrounding the brush border surface of the enterocytes and the junctional
complexes consisting of tight junctions, adherent junctions, desmosomes and gap
junctions [31]. The junctional complexes are regulated in response to physiological and
immunological stimuli, like stress, cytokines, dietary antigens and microbial products [31]. As
mentioned before, zonulin, a protein identified as prehaptoglobulin-2 (the precursor of
haptoglobin-2) is also a regulator of intestinal permeability. Haptoglobin-2, together with
haptoglobin-1, is one of the two gene variants of the multifunctional protein haptoglobin and
is associated with an increased risk for CD (homozygotes and heterozygotes) and severe
malabsorption (homozygotes) [32,33]. The haptoglobulin-2/zonulin allele has a frequency of
about 0.6 in Europe and the U.S.A, but varies throughout the world depending on racial
origin [34].

191
Figure 1. Increased intestinal permeability allows for the passage of microbial and dietary
antigens across the epithelial layer into the lamina propria where these antigens can be taken up
by APC and presented to T-cells. JC, junctional complex.
3.4. High zonulin levels are observed in auto-immune and inflammatory
diseases
Zonulin signalling is proposed to cause rearrangements of actin filaments and induces the
displacement of proteins from the junctional complex, thereby increasing
permeability [18,32,35]. Gliadin peptides initiate intestinal permeability through the release of
zonulin, thereby enabling paracellular translocation of gliadin and other dietary and microbial
antigens, which by interacting with the immune system give rise to inflammation. In this
manner a vicious cycle is created in which, as a consequence of the persistent presence of
pro-inflammatory mediators, intestinal permeability will increase even further. High zonulin
levels (together with increased intestinal permeability) have been observed in auto-immune
and inflammatory diseases like CD, multiple sclerosis, asthma and inflammatory bowel
disease and the haptoglobin polymorphism is associated with rheumatoid arthritis,
ankylosing spondylitis, schizophrenia and certain types of cancer [32].
The zonulin inhibitor Larozotide acetate was tested in an inpatient, double-blind randomized
placebo controlled trial. The group of CD patients in the placebo group that were exposed to

192
gluten showed a 70% increase in intestinal permeability, while no changes were seen in the
group exposed to Larazotide acetate. Also gastrointestinal symptoms were significant more
frequent in the placebo group [32]. These results suggest that in CD patients, when intestinal
barrier function is restored, autoimmunity will disappear while the trigger (gluten) is still there.
Besides gliadin from wheat gluten, the lectin wheat germ agglutinin (WGA) has also been
shown to stimulate cells of the immune system and increase intestinal permeability, as we will
now further discuss.
4. WGA
4.1. Dietary WGA
Lectins are present in a variety of plants, especially in seeds, where they serve as defense
mechanisms against other plants and fungi. Because of their ability to bind to virtually all cell
types and cause damage to several organs, lectins are widely recognized as anti-nutrients
within food [36]. Most lectins are resistant to heat and the effects of digestive enzymes and are
able to bind to several tissues and organs in vitro and in vivo (reviewed by Freed 1991 [37]). The
administration of the lectin WGA to experimental animals caused hyperplastic and
hypertrophic growth of the small intestine, hypertrophic growth of the pancreas and thymus
atrophy [36]. Lectin activity has been demonstrated in wheat, rye, barley, oats, corn and rice,
however the best studied of the cereal grain lectins is WGA [38].
The highest WGA concentrations are found in wheat germ (up to 0.5 g/kg [39]). Although
unprocessed wheat germ, like muesli, contains far higher amounts of active WGA than do
processed wheat germ products, WGA activity is still apparent in several processed breakfast
cereals as assessed by hemagglutination and bacterial agglutination assays [40,41]. A summary
of the amount of active WGA in commonly consumed wheat derived products is listed in
Table 1.
4.2. WGA binds to cell surface glycoconjugates
WGA binds to N-glycolylneuraminic acid (Neu5Ac), the sialic acid predominantly found in
humans [42], allowing it to adhere to cell surfaces like the epithelial layer of the gut. The
surface of many prokaryotic and eukaryotic cells are covered with a dense coating of
glycoconjugates, also named glycocalyx. Sialic acids are a wide family of nine-carbon sugars
that are typically found at the terminal positions of many surface exposed glycoconjugates
and function for self recognition in the vertebrate immune system, but they can also be used
as binding target for pathogenic extrinsic receptors and molecular toxins [43,44,45]. WGA
binding to Neu5Ac of the glycocalyx of human cells (and pathogens expressing Neu5Ac)
allows for cell entry and could disturb immune tolerance by evoking a pro-inflammatory
immune response (discussed below).

193
4.3. WGA and immunity
WGA induces inflammatory responses by immune cells. For example, WGA has been shown
to trigger histamine secretion and granule extrusion from non-stimulated rat peritoneal mast
cells [46], induce NADP-oxidase activity in human neutrophils [47] and stimulate the release
of the cytokines IL-4 and IL-13 from human basophils [48]. In human PBMC, WGA induced
the production of IL-2, while simultaneously inhibiting the proliferation of activated
lymphocytes [49]. WGA stimulated the secretion of IL-12, in a T and B-cell independent
manner in murine spleen cells. IL-12, in turn, activated the secretion of IFN-γ by T or natural
killer cells [50]. In murine peritoneal macrophages WGA induced the production of the pro-
inflammatory cytokines TNF-α, IL-1β, IL-12 and IFN-γ [51]. Similar results have been observed
in isolated human PBMC, given that nanomolar concentrations of WGA stimulated the release
of several pro-inflammatory cytokines. In the same study a significant increase in the
intracellular accumulation of IL-1β was measured in monocytes after WGA exposure [52].
These results indicate that when delivered in vitro WGA is capable of directly stimulating
monocytes and macrophages, cells that have the ability to initiate and maintain inflammatory
responses. Monocytic cells have been shown to engulf WGA via receptor-mediated
endocytosis or by binding to non-receptor glycoproteins [53].
Human data showing the influence of WGA intake on inflammatory markers are lacking,
however, antibodies to WGA have been detected in the serum of healthy individuals [54].
Significantly higher antibody levels to WGA were measured in patients with CD compared to
patients with other intestinal disorders. These antibodies did not cross-react with gluten
antigens and could therefore play an important role in the pathogenesis of this disease [55].

194
Table 1. Amount of active WGA in wheat derived products
Wheat derived products WGA µg/g (±SD) Reference source
Wheat germ 300 (±35) Vincenzi et al., 2000 [56]
Wheat germ 100 - 500 Peumans and Van Damme, 1996
[39]
Semolinaa 4.0 (±1.0) – 10.7 (±1.5) Matucci et al., 2004 [57]
Floura 4.3 (±0.7) – 4.4 (±1.0)
Wholemeal flour a 29.5 (±2.5) – 50 (±5.5)
Pastaa ≤ 0.4 (±0.2) – 3.2
(±0,2)
Pasta cookeda ≤ 0.3 (±0.2)
Wholemeal pasta (enriched
with wheat germ)
40 (±2.7)
Wholemeal pasta (enriched
with wheat germ) cooked
Not detectable
Wholemeal pastaa 0 – 5.7 (±0.2)
Wholemeal pasta cookeda Not detectable
Breakfast cerealsa 13 - 53 Ortega-Barria et al., 1994 [41]
a Values are obtained from more than one product and from different manufacturers.
4.4. WGA and intestinal permeability
After ingestion, WGA is capable of crossing the intestinal barrier. In animal models, WGA has
been shown to reach the basolateral membrane and walls of the small blood vessels in the
subepithelium of the small intestine [36]. WGA can be phagocytosed by binding to membrane
non-receptor glycoproteins, a process that has been observed in Caco-2 cells [58]. WGA can
also be endocytosed by antigen sampling M-cells [59,60] or by enterocytes via binding to
epidermal growth factor receptors [61]. Another possible route for lectin entry into the
periphery is by paracellular transport, a process that can be further aggravated by the binding
of gliadin to the chemokine receptor CXR3 on enterocytes.
WGA itself has been found to affect enterocyte permeability. Investigations by Dalla Pellegrina
et al. [52] showed in vitro that exposure to micromolar concentrations of WGA impairs the
integrity of the intestinal epithelial layer, allowing passage of small molecules, like lectins. At
the basolateral side of the epithelium, WGA concentrations in the nanomolar range induced
the secretion of pro-inflammatory cytokines by immune cells [52]. This may further affect the
integrity of the epithelial layer, heightening the potential for a positive feedback loop between
WGA, epithelial cells and immune cells. When combined, these mechanisms are likely able to
significantly increase the percentage of consumed WGA that can cross the epithelial layer

195
compared to the low percentage of WGA crossing by means of transcytosis (0.1%) alone [52].
This suggests that together with gliadin, WGA can increase intestinal permeability, resulting
in an increase of translocating microbial and dietary antigens interacting with cells of the
immune system.
5. Animal data on cereal grain intake
There are two rodent models of spontaneous type 1 diabetes: the non-obese-diabetic (NOD)
mouse and the diabetes-prone BioBreeding (BBdp) rat. In these animals a cereal-based diet
containing wheat induced the development of type 1 diabetes, while animals fed a
hypoallergenic diet (gluten free) or a hypoallergenic diet supplemented with casein showed a
decreased incidence and a delayed onset of this disease. BBdp rats fed a cereal-based diet
showed increased intestinal permeability and a significant increase in the percentage of IFN-γ
producing Th1 lymphocytes in the mesenteric lymph nodes in the gut [30]. Compared to
animals fed a hypoallergenic diet, NOD mice fed a wheat-based diet expressed higher mRNA
levels of the pro-inflammatory cytokines IFN-γ and TNF-α and the inflammatory marker
inducible NO synthase in the small intestine. While these diet-induced changes in gut-wall
inflammatory activity did not translate to increased cytokine mRNA in Peyers patches,
structures that contribute to immune regulation to exogenous antigens, it is possible that the
gut-signal may promote systemic inflammation via other mechanisms, such as activating
intraepithelial lymphocytes and mesenteric lymph node cells [62]. These in vivo results show
that in two rodent models of spontaneous type 1 diabetes a cereal containing diet induces the
(early) onset of disease and increases markers of inflammation. In addition, Chignola et al. [63]
have shown in rats that a WGA-depleted diet was associated with reduced responsiveness of
lymphocytes from primary and secondary lymphoid organs after in vitro stimulation and
attenuated spontaneous proliferation when compared to lymphocytes from rats fed a WGA-
containing diet, indicating the stimulatory effect of WGA on cells of the immune system.
6. Human studies on cereal grain intake and inflammation
6.1. Human epidemiological data on cereal grain intake and inflammation
Observational prospective and cross-sectional studies show that the intake of whole grain
products is associated with reduced risks for developing type 2 diabetes, cardiovascular
diseases, obesity and some types of cancer [64]. Inflammation is associated with these
conditions and some studies have shown that associations between the intake of whole grains
and decreased inflammatory markers (CRP, Il-6) are found [65]. Intervention studies, however,
do not demonstrate a clear effect of the intake of whole grains on
inflammation [66,67,68,69,70,71] and it could therefore be that other components in the diet
modulate the immune response.

196
It has been shown that the intake of whole grains is associated with healthier dietary factors
and a healthier lifestyle in general. In a Scandinavian cross sectional study, the intake of whole
grains was directly associated with the length of education, the intake of vegetables, fruits,
dairy products, fish, shellfish, coffee, tea and margarine and inversely associated with
smoking, BMI and the intake of red meat, white bread, alcohol, cakes and biscuits [72]. Good
quality epidemiological studies attempt to control for these confounding factors, but with the
consequence that associations are attenuated or become insignificant.
6.2. Human intervention trials on cereal grain intake and inflammation
To really estimate the causal relationship of cereal grain intake and inflammation,
intervention trials provide us with better evidence. Wolever et al. [71] showed that a diet with a
low glycemic index (containing whole grains) compared to high (containing refined grain
products), resulted in sustained reductions in postprandial glucose and CRP levels on the
long-term in patients with type 2 diabetes treated with diet alone. A refined grain is a whole
grain that has been stripped of its outer shell (fiber) and its germ, leaving only the endosperm,
resulting in lower levels of macro- and micronutrients and a higher dietary glycemic index for
refined grains compared to whole grains. Refined wheat products contain less WGA, but still
contain a substantial amount of gluten. It should be noted that whole grains contain
phytochemicals, like polyphenols, that can exert anti-inflammatory effects which could
possibly offset any potentially pro-inflammatory effects of gluten and lectins [73].
The substitution of whole grain (mainly based on milled wheat) for refined grains products in
the daily diet of healthy moderately overweight adults for 6 weeks did not affect insulin
sensitivity or markers of lipid peroxidation and inflammation [66]. Consistent with these
finding are the results of Brownlee et al. [67], who showed that infrequent whole-grain
consumers, when increasing whole grain consumption (including whole wheat products),
responded with no improvements of the studied biomarkers of cardiovascular health,
including insulin sensitivity, plasma lipid profile and markers of inflammation. The
substitution of refined cereal grains and white bread with 3 portions of whole wheat food or 1
portions of whole wheat food combined with 2 servings of oats significantly decreased the
systolic blood pressure and pulse pressure in middle-aged, healthy, overweight men and
women, yet none of the interventions significantly affected systemic markers of
inflammation [70]. In obese adults suffering from metabolic syndrome there were
significantly greater decreases in CRP and the percentage of body fat in the abdominal region
in participants consuming whole grains compared to those consuming refined grains. It must
be noted that both diets were hypocaloric (reduced by 500 kcal/d) [69]. Most of the
intervention studies mentioned above attempted to increase whole-grain intake and were

197
using refined grain diets as controls, thereby making it very difficult to draw any conclusions
on the independent role of cereal grains in disease and inflammation.
6.3. Health effects of the paleolithic diet
There are few studies that investigate the influence of a paleolithic type diet comprising lean
meat, fruits, vegetables and nuts, and excluding food types, such as dairy, legumes and cereal
grains, on health. In domestic pigs the paleolithic diet conferred higher insulin sensitivity,
lower CRP and lower blood pressure when compared to a cereal based diet [74]. In healthy
sedentary humans the short-term consumption of a paleolithic type diet improved blood
pressure and glucose tolerance, decreased insulin secretion, increased insulin sensitivity and
improved lipid profiles [75]. Glucose tolerance also improved in patients suffering from a
combination of ischemic heart disease and either glucose intolerance or type 2 diabetes that
were advised to follow a paleolithic diet. Control subjects who were advised to follow a
Mediterranean-like diet based on whole grains, low-fat dairy products, fish, fruits and
vegetables did not significantly improve their glucose tolerance despite decreases in weight
and waist circumference [76]. Similar positive results on glycemic control were obtained in
diabetic patients when the paleolithic diet was compared with the diabetes diet. Participants
were on each diet for 3 months where the paleolithic diet resulted in a lower BMI, weight and
waist circumference, higher mean HDL, lower mean levels of hemoglobin A1c, triacylglycerol
and diastolic blood pressure, yet levels of CRP were not significantly different [77]. Although
the paleolithic diet studies are small, these results suggest that, together with other dietary
changes, the withdrawal of cereal grains from the diet has a positive effect on health.
Nevertheless, because these studies are confounded by the presence or absence of other
dietary substances and by differences in energy and macronutrient intake, factors that could
all affect markers of inflammation, it is difficult to make a concise statement on the impact of
cereal grains on these health outcomes.
6.4. Rechallenge trial of effects of dietary gluten
One human intervention study specifically focused on the effects of dietary gluten on
inflammation. Biesiekierski et al. [12] undertook a double-blind randomized, placebo-
controlled rechallenge trial to investigate the influence of gluten in individuals with irritable
bowel syndrome but without clinical features of CD, who reached satisfactory levels of
symptom control with a gluten-free diet. After screening the participants, about 50% of the
individuals in both the gluten and placebo group were HLA-DQ2 and/or HLA-DQ8 positive.
Participants received either gluten or placebo together with a gluten-free diet for 6 weeks.
End-points in the study were symptom assessments and biomarkers of inflammation and
intestinal permeability. The patients receiving gluten reported significantly more symptoms
compared to the placebo group. The most striking outcome of this study was that for all the

198
endpoints measured there were no differences in individuals with or without HLA-DQ2/DQ8,
indicating that the intake of gluten can cause symptoms also in individuals without this
specific HLA-profile. No differences in biomarkers for inflammation and intestinal
permeability were found between both groups, however, inflammatory mediators have been
implicated in the development of symptoms in patients with irritable bowel syndrome [78]. It
could therefore be that the markers used to measure inflammation and intestinal permeability
were not sensitive enough to detect subtle changes on the tissue level.
7. Conclusion
In the present review we describe how the daily consumption of wheat products and other
related cereal grains could contribute to the manifestation of chronic inflammation and auto-
immune diseases. Both in vitro and in vivo studies demonstrate that gliadin and WGA can
both increase intestinal permeability and activate the immune system. The effects of gliadin
on intestinal permeability and the immune system have also been confirmed in humans.
Other cereal grains containing related prolamins and lectins have not been so extensively
studied and therefore more research investigating their impact on intestinal permeability and
inflammation is required. It would be interesting to further elucidate the role of other
prolamins on zonulin release and intestinal permeability.
In CD and gluten-sensitive individuals adverse reactions to the intake of wheat, rye and barley
are clinically apparent, however, it is important to gain better insights on the effects of the
consumption of these cereal grains in other groups of patients and in healthy individuals. It
would be of high interest to investigate the effects of the withdrawal of cereal grain products
from the diet on inflammatory markers and intestinal permeability in healthy subjects and
patients suffering from inflammation-related diseases and measure the same parameters in a
rechallenge trial. Ideally, in such an intervention study, the diet must be completely controlled
and combined with the appropriate substitution of foods in the cereal grain-deprived diet so
that small dietary variations and alterations in energy intake can be avoided and cannot
potentially influence inflammatory markers.
Until now, human epidemiological and intervention studies investigating the health-effects
of whole grain intake were confounded by other dietary and lifestyle factors and therefore well
designed intervention studies investigating the effects of cereal grains and their individual
components on intestinal permeability and inflammation are warranted.

199
Acknowledgments
We would like to thank Charles L. Raison and Alicia Lammerts van Bueren for the editorial
work on the manuscript
Conflict of Interest
The authors declare no conflict of interest.

200
References
1. Barnes, P.J.; Adcock, I.M. Glucocorticoid resistance in inflammatory diseases. Lancet
2009, 373, 1905-1917.
2. Libby, P. Role of inflammation in atherosclerosis associated with rheumatoid arthritis.
Am J Med 2008, 121, S21-31.
3. Raison, C.L.; Capuron, L.; Miller, A.H. Cytokines sing the blues: inflammation and the
pathogenesis of depression. Trends Immunol 2006, 27, 24-31.
4. Bosma-den Boer, M.M.; van Wetten, M.L.; Pruimboom, L. Chronic inflammatory
diseases are stimulated by current lifestyle: how diet, stress levels and medication
prevent our body from recovering. Nutr Metab (Lond) 2012, 9, 32.
5. Shelton, R.C.; Miller, A.H. Eating ourselves to death (and despair): the contribution of
adiposity and inflammation to depression. Prog Neurobiol 2010, 91, 275-299.
6. Brown, C.M.; Dulloo, A.G.; Montani, J.P. Sugary drinks in the pathogenesis of obesity
and cardiovascular diseases. Int J Obes (Lond) 2008, 32 Suppl 6, S28-34.
7. Tatham, A.S.; Shewry, P.R. Allergens to wheat and related cereals. Clin Exp Allergy 2008,
38, 1712-1726.
8. Sapone, A.; Bai, J.C.; Ciacci, C.; Dolinsek, J.; Green, P.H.; Hadjivassiliou, M.; Kaukinen, K.;
Rostami, K.; Sanders, D.S.; Schumann, M.; Ullrich, R.; Villalta, D.; Volta, U.; Catassi, C.;
Fasano, A. Spectrum of gluten-related disorders: consensus on new nomenclature and
classification. BMC Med 2012, 10, 13.
9. Shewry, P.R. Wheat. J Exp Bot 2009, 60, 1537-1553.
10. Troncone, R.; Jabri, B. Coeliac disease and gluten sensitivity. J Intern Med 2011, 269,
582-590.
11. Neuhausen, S.L.; Steele, L.; Ryan, S.; Mousavi, M.; Pinto, M.; Osann, K.E.; Flodman, P.;
Zone, J.J. Co-occurrence of celiac disease and other autoimmune diseases in celiacs
and their first-degree relatives. J Autoimmun 2008, 31, 160-165.
12. Biesiekierski, J.R.; Newnham, E.D.; Irving, P.M.; Barrett, J.S.; Haines, M.; Doecke, J.D.;
Shepherd, S.J.; Muir, J.G.; Gibson, P.R. Gluten causes gastrointestinal symptoms in
subjects without celiac disease: a double-blind randomized placebo-controlled trial. Am
J Gastroenterol 2011, 106, 508-514; quiz 515.
13. Sapone, A.; Lammers, K.M.; Casolaro, V.; Cammarota, M.; Giuliano, M.T.; De Rosa, M.;
Stefanile, R.; Mazzarella, G.; Tolone, C.; Russo, M.I.; Esposito, P.; Ferraraccio, F.; Carteni,
M.; Riegler, G.; de Magistris, L.; Fasano, A. Divergence of gut permeability and mucosal
immune gene expression in two gluten-associated conditions: celiac disease and gluten
sensitivity. BMC Med 2011, 9, 23.
14. Harris, K.M.; Fasano, A.; Mann, D.L. Cutting edge: IL-1 controls the IL-23 response
induced by gliadin, the etiologic agent in celiac disease. J Immunol 2008, 181, 4457-
4460.

201
15. Lammers, K.M.; Khandelwal, S.; Chaudhry, F.; Kryszak, D.; Puppa, E.L.; Casolaro, V.;
Fasano, A. Identification of a novel immunomodulatory gliadin peptide that causes
interleukin-8 release in a chemokine receptor CXCR3-dependent manner only in
patients with coeliac disease. Immunology 2011, 132, 432-440.
16. Sapone, A.; Lammers, K.M.; Mazzarella, G.; Mikhailenko, I.; Carteni, M.; Casolaro, V.;
Fasano, A. Differential mucosal IL-17 expression in two gliadin-induced disorders:
gluten sensitivity and the autoimmune enteropathy celiac disease. Int Arch Allergy
Immunol 2010, 152, 75-80.
17. Catassi, C.; Pierani, P.; Natalini, G.; Gabrielli, O.; Coppa, G.V.; Giorgi, P.L. Clinical
application of a simple HPLC method for the sugar intestinal permeability test. J Pediatr
Gastroenterol Nutr 1991, 12, 209-212.
18. Fasano, A. Leaky gut and autoimmune diseases. Clin Rev Allergy Immunol 2012, 42, 71-
78.
19. van Elburg, R.M.; Uil, J.J.; Mulder, C.J.; Heymans, H.S. Intestinal permeability in patients
with coeliac disease and relatives of patients with coeliac disease. Gut 1993, 34, 354-357.
20. Sander, G.R.; Cummins, A.G.; Henshall, T.; Powell, B.C. Rapid disruption of intestinal
barrier function by gliadin involves altered expression of apical junctional proteins.
FEBS Lett 2005, 579, 4851-4855.
21. Drago, S.; El Asmar, R.; Di Pierro, M.; Grazia Clemente, M.; Tripathi, A.; Sapone, A.;
Thakar, M.; Iacono, G.; Carroccio, A.; D'Agate, C.; Not, T.; Zampini, L.; Catassi, C.; Fasano,
A. Gliadin, zonulin and gut permeability: Effects on celiac and non-celiac intestinal
mucosa and intestinal cell lines. Scand J Gastroenterol 2006, 41, 408-419.
22. Lammers, K.M.; Lu, R.; Brownley, J.; Lu, B.; Gerard, C.; Thomas, K.; Rallabhandi, P.; Shea-
Donohue, T.; Tamiz, A.; Alkan, S.; Netzel-Arnett, S.; Antalis, T.; Vogel, S.N.; Fasano, A.
Gliadin induces an increase in intestinal permeability and zonulin release by binding to
the chemokine receptor CXCR3. Gastroenterology 2008, 135, 194-204 e193.
23. Cordain, L.; Toohey, L.; Smith, M.J.; Hickey, M.S. Modulation of immune function by
dietary lectins in rheumatoid arthritis. Br J Nutr 2000, 83, 207-217.
24. Secondulfo, M.; Iafusco, D.; Carratu, R.; deMagistris, L.; Sapone, A.; Generoso, M.;
Mezzogiomo, A.; Sasso, F.C.; Carteni, M.; De Rosa, R.; Prisco, F.; Esposito, V.
Ultrastructural mucosal alterations and increased intestinal permeability in non-celiac,
type I diabetic patients. Dig Liver Dis 2004, 36, 35-45.
25. Keita, A.V.; Soderholm, J.D. The intestinal barrier and its regulation by neuroimmune
factors. Neurogastroenterol Motil 2010, 22, 718-733.
26. Hijazi, Z.; Molla, A.M.; Al-Habashi, H.; Muawad, W.M.; Molla, A.M.; Sharma, P.N.
Intestinal permeability is increased in bronchial asthma. Arch Dis Child 2004, 89, 227-
229.

202
27. Maes, M. An intriguing and hitherto unexplained co-occurrence: Depression and
chronic fatigue syndrome are manifestations of shared inflammatory, oxidative and
nitrosative (IO&NS) pathways. Prog Neuropsychopharmacol Biol Psychiatry 2011, 35,
784-794.
28. Maes, M.; Kubera, M.; Leunis, J.C.; Berk, M. Increased IgA and IgM responses against gut
commensals in chronic depression: Further evidence for increased bacterial
translocation or leaky gut. J Affect Disord 2012, 141, 55-62.
29. Maes, M.; Mihaylova, I.; Leunis, J.C. Increased serum IgA and IgM against LPS of
enterobacteria in chronic fatigue syndrome (CFS): indication for the involvement of
gram-negative enterobacteria in the etiology of CFS and for the presence of an
increased gut-intestinal permeability. J Affect Disord 2007, 99, 237-240.
30. Sonier, B.; Patrick, C.; Ajjikuttira, P.; Scott, F.W. Intestinal immune regulation as a
potential diet-modifiable feature of gut inflammation and autoimmunity. Int Rev
Immunol 2009, 28, 414-445.
31. Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol
2009, 9, 799-809.
32. Fasano, A. Zonulin, regulation of tight junctions, and autoimmune diseases. Ann N Y
Acad Sci 2012, 1258, 25-33.
33. Papp, M.; Foldi, I.; Nemes, E.; Udvardy, M.; Harsfalvi, J.; Altorjay, I.; Mate, I.; Dinya, T.;
Varvolgyi, C.; Barta, Z.; Veres, G.; Lakatos, P.L.; Tumpek, J.; Toth, L.; Szathmari, E.;
Kapitany, A.; Gyetvai, A.; Korponay-Szabo, I.R. Haptoglobin polymorphism: a novel
genetic risk factor for celiac disease development and its clinical manifestations. Clin
Chem 2008, 54, 697-704.
34. Carter, K.; Worwood, M. Haptoglobin: a review of the major allele frequencies worldwide
and their association with diseases. Int J Lab Hematol 2007, 29, 92-110.
35. Fasano, A. Zonulin and its regulation of intestinal barrier function: the biological door to
inflammation, autoimmunity, and cancer. Physiol Rev 2011, 91, 151-175.
36. Pusztai, A.; Ewen, S.W.; Grant, G.; Brown, D.S.; Stewart, J.C.; Peumans, W.J.; Van Damme,
E.J.; Bardocz, S. Antinutritive effects of wheat-germ agglutinin and other N-
acetylglucosamine-specific lectins. Br J Nutr 1993, 70, 313-321.
37. Freed, D.L.J. Lectins in food: Their importance in health and disease. Journal of
Nutritional Medicine 1991, 2.
38. Cordain, L. Cereal grains: humanity's double-edged sword. World Rev Nutr Diet 1999,
84, 19-73.
39. Peumans, W.J.; Van Damme, E.J. Prevalence, biological activity and genetic
manipulation of lectins in foods. Trends in Food Science & Technology 1996, 7, 132-138.

203
40. Nachbar, M.S.; Oppenheim, J.D.; Thomas, J.O. Lectins in the U.S. Diet. Isolation and
characterization of a lectin from the tomato (Lycopersicon esculentum). J Biol Chem
1980, 255, 2056-2061.
41. Ortega-Barria, E.; Ward, H.D.; Keusch, G.T.; Pereira, M.E. Growth inhibition of the
intestinal parasite Giardia lamblia by a dietary lectin is associated with arrest of the cell
cycle. J Clin Invest 1994, 94, 2283-2288.
42. Shaw, L.; Yousefi, S.; Dennis, J.W.; Schauer, R. CMP-N-acetylneuraminic acid
hydroxylase activity determines the wheat germ agglutinin-binding phenotype in two
mutants of the lymphoma cell line MDAY-D2. Glycoconj J 1991, 8, 434-441.
43. Severi, E.; Hood, D.W.; Thomas, G.H. Sialic acid utilization by bacterial pathogens.
Microbiology 2007, 153, 2817-2822.
44. Varki, A. Multiple changes in sialic acid biology during human evolution. Glycoconj J
2009, 26, 231-245.
45. Varki, A. Colloquium paper: uniquely human evolution of sialic acid genetics and
biology. Proc Natl Acad Sci U S A 2010, 107 Suppl 2, 8939-8946.
46. Lansman, J.B.; Cochrane, D.E. Wheat germ agglutinin stimulates exocytotic histamine
secretion from rat mast cells in the absence of extracellular calcium. Biochem
Pharmacol 1980, 29, 455-458.
47. Karlsson, A. Wheat germ agglutinin induces NADPH-oxidase activity in human
neutrophils by interaction with mobilizable receptors. Infect Immun 1999, 67, 3461-
3468.
48. Haas, H.; Falcone, F.H.; Schramm, G.; Haisch, K.; Gibbs, B.F.; Klaucke, J.; Poppelmann,
M.; Becker, W.M.; Gabius, H.J.; Schlaak, M. Dietary lectins can induce in vitro release of
IL-4 and IL-13 from human basophils. Eur J Immunol 1999, 29, 918-927.
49. Reed, J.C.; Robb, R.J.; Greene, W.C.; Nowell, P.C. Effect of wheat germ agglutinin on the
interleukin pathway of human T lymphocyte activation. J Immunol 1985, 134, 314-323.
50. Muraille, E.; Pajak, B.; Urbain, J.; Leo, O. Carbohydrate-bearing cell surface receptors
involved in innate immunity: interleukin-12 induction by mitogenic and nonmitogenic
lectins. Cell Immunol 1999, 191, 1-9.
51. Sodhi, A.; Kesherwani, V. Production of TNF-alpha, IL-1beta, IL-12 and IFN-gamma in
murine peritoneal macrophages on treatment with wheat germ agglutinin in vitro:
involvement of tyrosine kinase pathways. Glycoconj J 2007, 24, 573-582.
52. Dalla Pellegrina, C.; Perbellini, O.; Scupoli, M.T.; Tomelleri, C.; Zanetti, C.; Zoccatelli, G.;
Fusi, M.; Peruffo, A.; Rizzi, C.; Chignola, R. Effects of wheat germ agglutinin on human
gastrointestinal epithelium: insights from an experimental model of immune/epithelial
cell interaction. Toxicol Appl Pharmacol 2009, 237, 146-153.

204
53. Schumacher, U.; Grafin von Armansperg, N.; Kreipe, H.; Welsch, U. Lectin binding and
uptake in human (myelo)monocytic cell lines: HL60 and U937. Ultrastruct Pathol 1996,
20, 463-471.
54. Tchernychev, B.; Wilchek, M. Natural human antibodies to dietary lectins. FEBS Lett
1996, 397, 139-142.
55. Sollid, L.M.; Kolberg, J.; Scott, H.; Ek, J.; Fausa, O.; Brandtzaeg, P. Antibodies to wheat
germ agglutinin in coeliac disease. Clin Exp Immunol 1986, 63, 95-100.
56. Vincenzi, S.; Zoccatelli, G.; Perbellini, F.; Rizzi, C.; Chignola, R.; Curioni, A.; Peruffo, A.D.
Quantitative determination of dietary lectin activities by enzyme-linked
immunosorbent assay using specific glycoproteins immobilized on microtiter plates. J
Agric Food Chem 2002, 50, 6266-6270.
57. Matucci, A.; Veneri, G.; Dalla Pellegrina, C.; Zoccatelli, G.; Vincenzi, S.; Chignola, R.;
Peruffo, A.; Rizzi, C. Temperature-dependent decay of wheat germ agglutinin activity
and its implications for food processing and analysis. Food control 2004, 15, 391-395.
58. Gabor, F.; Bogner, E.; Weissenboeck, A.; Wirth, M. The lectin-cell interaction and its
implications to intestinal lectin-mediated drug delivery. Adv Drug Deliv Rev 2004, 56,
459-480.
59. Clark, M.A.; Jepson, M.A.; Simmons, N.L.; Booth, T.A.; Hirst, B.H. Differential expression
of lectin-binding sites defines mouse intestinal M-cells. J Histochem Cytochem 1993,
41, 1679-1687.
60. Giannasca, P.J.; Giannasca, K.T.; Leichtner, A.M.; Neutra, M.R. Human intestinal M cells
display the sialyl Lewis A antigen. Infect Immun 1999, 67, 946-953.
61. Lochner, N.; Pittner, F.; Wirth, M.; Gabor, F. Wheat germ agglutinin binds to the
epidermal growth factor receptor of artificial Caco-2 membranes as detected by silver
nanoparticle enhanced fluorescence. Pharm Res 2003, 20, 833-839.
62. Flohe, S.B.; Wasmuth, H.E.; Kerad, J.B.; Beales, P.E.; Pozzilli, P.; Elliott, R.B.; Hill, J.P.;
Scott, F.W.; Kolb, H. A wheat-based, diabetes-promoting diet induces a Th1-type
cytokine bias in the gut of NOD mice. Cytokine 2003, 21, 149-154.
63. Chignola, R.; Rizzi, C.; Vincenzi, S.; Cestari, T.; Brutti, N.; Riviera, A.P.; Sartoris, S.;
Peruffo, A.D.; Andrighetto, G. Effects of dietary wheat germ deprivation on the immune
system in Wistar rats: a pilot study. Int Immunopharmacol 2002, 2, 1495-1501.
64. Jonnalagadda, S.S.; Harnack, L.; Liu, R.H.; McKeown, N.; Seal, C.; Liu, S.; Fahey, G.C.
Putting the whole grain puzzle together: health benefits associated with whole grains--
summary of American Society for Nutrition 2010 Satellite Symposium. J Nutr 2011, 141,
1011S-1022S.
65. Lefevre, M.; Jonnalagadda, S. Effect of whole grains on markers of subclinical
inflammation. Nutr Rev 2012, 70, 387-396.

205
66. Andersson, A.; Tengblad, S.; Karlstrom, B.; Kamal-Eldin, A.; Landberg, R.; Basu, S.; Aman,
P.; Vessby, B. Whole-grain foods do not affect insulin sensitivity or markers of lipid
peroxidation and inflammation in healthy, moderately overweight subjects. J Nutr 2007,
137, 1401-1407.
67. Brownlee, I.A.; Moore, C.; Chatfield, M.; Richardson, D.P.; Ashby, P.; Kuznesof, S.A.; Jebb,
S.A.; Seal, C.J. Markers of cardiovascular risk are not changed by increased whole-grain
intake: the WHOLEheart study, a randomised, controlled dietary intervention. Br J Nutr
2010, 104, 125-134.
68. Giacco, R.; Clemente, G.; Cipriano, D.; Luongo, D.; Viscovo, D.; Patti, L.; Di Marino, L.;
Giacco, A.; Naviglio, D.; Bianchi, M.A.; Ciati, R.; Brighenti, F.; Rivellese, A.A.; Riccardi, G.
Effects of the regular consumption of wholemeal wheat foods on cardiovascular risk
factors in healthy people. Nutr Metab Cardiovasc Dis 2010, 20, 186-194.
69. Katcher, H.I.; Legro, R.S.; Kunselman, A.R.; Gillies, P.J.; Demers, L.M.; Bagshaw, D.M.;
Kris-Etherton, P.M. The effects of a whole grain-enriched hypocaloric diet on
cardiovascular disease risk factors in men and women with metabolic syndrome. Am J
Clin Nutr 2008, 87, 79-90.
70. Tighe, P.; Duthie, G.; Vaughan, N.; Brittenden, J.; Simpson, W.G.; Duthie, S.; Mutch, W.;
Wahle, K.; Horgan, G.; Thies, F. Effect of increased consumption of whole-grain foods
on blood pressure and other cardiovascular risk markers in healthy middle-aged
persons: a randomized controlled trial. Am J Clin Nutr 2010, 92, 733-740.
71. Wolever, T.M.; Gibbs, A.L.; Mehling, C.; Chiasson, J.L.; Connelly, P.W.; Josse, R.G.; Leiter,
L.A.; Maheux, P.; Rabasa-Lhoret, R.; Rodger, N.W.; Ryan, E.A. The Canadian Trial of
Carbohydrates in Diabetes (CCD), a 1-y controlled trial of low-glycemic-index dietary
carbohydrate in type 2 diabetes: no effect on glycated hemoglobin but reduction in C-
reactive protein. Am J Clin Nutr 2008, 87, 114-125.
72. Kyro, C.; Skeie, G.; Dragsted, L.O.; Christensen, J.; Overvad, K.; Hallmans, G.; Johansson,
I.; Lund, E.; Slimani, N.; Johnsen, N.F.; Halkjaer, J.; Tjonneland, A.; Olsen, A. Intake of
whole grain in Scandinavia: intake, sources and compliance with new national
recommendations. Scand J Public Health 2012, 40, 76-84.
73. Fardet, A. New hypotheses for the health-protective mechanisms of whole-grain
cereals: what is beyond fibre? Nutr Res Rev 2010, 23, 65-134.
74. Jonsson, T.; Ahren, B.; Pacini, G.; Sundler, F.; Wierup, N.; Steen, S.; Sjoberg, T.; Ugander,
M.; Frostegard, J.; Goransson, L.; Lindeberg, S. A Paleolithic diet confers higher insulin
sensitivity, lower C-reactive protein and lower blood pressure than a cereal-based diet in
domestic pigs. Nutr Metab (Lond) 2006, 3, 39.
75. Frassetto, L.A.; Schloetter, M.; Mietus-Synder, M.; Morris, R.C., Jr.; Sebastian, A. Metabolic
and physiologic improvements from consuming a paleolithic, hunter-gatherer type
diet. Eur J Clin Nutr 2009, 63, 947-955.

206
76. Lindeberg, S.; Jonsson, T.; Granfeldt, Y.; Borgstrand, E.; Soffman, J.; Sjostrom, K.; Ahren,
B. A Palaeolithic diet improves glucose tolerance more than a Mediterranean-like diet in
individuals with ischaemic heart disease. Diabetologia 2007, 50, 1795-1807.
77. Jonsson, T.; Granfeldt, Y.; Ahren, B.; Branell, U.C.; Palsson, G.; Hansson, A.; Soderstrom,
M.; Lindeberg, S. Beneficial effects of a Paleolithic diet on cardiovascular risk factors in
type 2 diabetes: a randomized cross-over pilot study. Cardiovasc Diabetol 2009, 8, 35.
78. Matricon, J.; Meleine, M.; Gelot, A.; Piche, T.; Dapoigny, M.; Muller, E.; Ardid, D. Review
article: Associations between immune activation, intestinal permeability and the
irritable bowel syndrome. Aliment Pharmacol Ther 2012, 36, 1009-1031.

207
Paragraph 1.6. Lactase persistence and augmented salivary alpha-amylase gene copy numbers
might have been selected by the combined toxic effects of gluten and (food born) pathogens.
Pruimboom L, Fox T, Muskiet FA.Med Hypotheses 2014;82:326-34.
Lactase persistence and augmented salivary alpha-amylase gene copy numbers
might have been selected by the combined toxic effects of gluten and (food
born) pathogens
Leo Pruimboom MSc1, Tom Fox1, Frits A.J. Muskiet PhD2
1. University of Gerona (Spain), Passeig Devesa 41, Gerona, Spain;
2. Laboratory Medicine, University of Groningen, University Medical Center Groningen
(UMCG), P.O. Box 30.001 Groningen, The Netherlands.
Keywords
Lactose, amylase, gene copy number, lactase, starch, Rotavirus, mycotoxins, Aspergillus,
Tuberculosis, glucose transporter, evolution, gluten, dairy, milk, cereals, fungus
Corresponding author
Leo Pruimboom
Avd del Mar Residencial Las Caletas G4
35508 Costa Teguise, Spain

208
Contents
Abstract
Introduction
1. Lactase persistence, existing hypotheses
1.1 The UVB-vitamin D-calcium-rickets hypothesis
1.2 The influence of cereals
1.3 Our hypothesis: Lactase and the immune system
1.4 Conclusions so far
2. Salivary AMY1 gene copy number
2.1 AMY1 copy number and starch consumption
2.2 Existing hypotheses
2.3 AMY1 expression relates to stress and pancreatic amylase activity is huge
2.4 AMY1 and the immune system: domestic dog adaptations as introduction to our
hypotheses
2.5 Evolutionary selective pressure aspects of the consumption of starch-rich
foods; our hypotheses
2.5.1 Toxins in starch-rich foods
2.5.2 Did gluten-toxicity exert sufficient selective pressure to cause
augmented AMY1-GCN?
2.5.3 Can starch itself be sufficiently toxic to produce important selective
pressure?
2.5.4 Fungi growing on starch rich food; a largely ignored global health issue
now and definitely in the past
2.6 Conclusions so far
3. Rotavirus, lactose intolerance and AMY1-GCN
4. Discussion and final conclusions
References

209
Abstract
Various positively selected adaptations to new nutrients have been identified. Lactase
persistence is among the best known, conferring the ability for drinking milk at post weaning
age. An augmented number of amylase gene (AMY1) copies, giving rise to higher salivary
amylase activity, has been implicated in the consumption of starch-rich foods. Higher AMY1
copy numbers have been demonstrated in populations with recent histories of starchy-rich
diets. It is however questionable whether the resulting polymorphisms have exerted positive
selection only by providing easily available sources of macro and miconutrients. Humans
have explored new environments more than any other animal. Novel environments challenge
the host, but especially its immune system with new climatic conditions, food and especially
pathogens. With the advent of the agricultural revolution and the concurrent domestication of
cattle came new pathogens. We contend that specific new food ingredients (e.g. gluten) and
novel pathogens drove selection for lactase persistence and higher AMY gene copy numbers.
Both adaptations provide ample glucose for activating the sodium glucose-dependent co-
transporter 1 (SGLT1), which is the principal glucose, sodium and water transporter in the
gastro-intestinal tract. Their rapid uptake confers protection against potentially lethal
dehydration, hyponatraemia and ultimately multiple organ failure. Oral rehydration therapy
aims at SGLT1 activity and is the current treatment of choice for chronic diarrhoea and
vomiting. We hypothesize that lifelong lactase activity and rapid starch digestion should be
looked at as the evolutionary covalent of oral rehydration therapy.

210
Introduction
Adaption to new environmental challenges aims at phenotypic adjustments either by
epigenetic (rapid, but labile) and genetic (slow, but robust) changes through processes such as
epimutation and de novo mutation, the latter e.g. by variation of gene copy number (GCN) (1,
2). Many environmental factors have shaped the human genome, including climate, diet and
microbial load (3). Although the first two certainly excreted selective pressure on humans, the
main selective pressure seems to derive from pathogens because of their high lethality (4).
Fumagalli et al. (4) identified a surprisingly high number of more than one hundred genes
carrying signatures of a pathogenic environment and presenting as an increase in allele
frequency (GCN). Conversely, for dietary regimes and climatic conditions, no gene shows a
similar correlation between an environmental factor and GCN (4). Other authors have shown
that climate and diet do act as selective pressure factors in humans (5, 6), but the majority of
the literature indicates that it are pathogens and the pathogenic load that should be
considered as the principal environmental factors causing selective pressure in humans (7,8).
Two recent changes in the human diet i.e. the inclusion of dairy products and the increased
intake of starch, have been related with genetic adaptations, i.e. lactase (also known as lactase-
phlorizin hydrolase; LPH) persistence (9) and salivary amylase (AMY1) GCN (10), respectively.
Both adaptations are ascribed to toxicosis as the possible causes of the observed high positive
selection pressure of these alleles, but there have to our knowledge as yet been no suggestions
that they might confer protection against an increased pathogenic load since the rise of
agriculture. Pathogens such as Mycobacterium tuberculosis, Rotavirus, E. coli, Mycoplasma
pneumoniae and fungi producing highly toxic mycotoxins, together with gluten as a novel
food ingredient, may cause devastating effects in humans through pathways ending up in
dehydration, hyponatraemia and lack of energy, ending up in multiple organ failure (MOF).
We suggest (our hypotheses) that improved lactose and starch digestion through lactase
persistence and augmented AMY1-GCN provided a survival advantage by facilitating the
activity of the immune system and the most important glucose, water and sodium transporter
in the gut and thereby conferred protection against infections and infection-driven
dehydration, hyponatremia and multiple organ failure.

211
1. Lactase persistence, existing hypotheses
1.1. The UVB-vitamin D-calcium-rickets hypothesis
Swallow et al. (9) related the higher and longer intake of milk and milk products to lactase
persistence after infant age through the emergence of polymorphisms in the regulation of the
lactase gene (11, 12). The hypothesis was subsequently supported by others (13). Several
explanations have been given for the positive selection of these polymorphisms leading to
lifelong lactase persistence. One of them highlights the advantage of the improved calcium
intake if lactose could be digested (14). The low UV-B levels at high latitudes are associated
with an increased risk of developing rickets and osteomalacia due to the lack of cutaneous
vitamin D production during the long winter period. In the gut, the vitamin D hormone, 1,25-
dihydroxyvitamin D, stimulates the expression of proteins involved in the absorption of
calcium, which is itself an essential mineral required for bone health, signal transduction and
others. Milk born calcium may also help to prevent rickets by impairing the breakdown of
vitamin D in the liver (15).
Rickets narrow the female birth channel and thereby increases the chance of mortality of both
mother and child during labour (16). The only successful preventive measure is by caesarean
section, which has been part of human anthropology probably since millennia (17) and seems
to have saved thousands of people also in recent times (16). However, Itan and Swallow (11),
using a flexible demic computer simulation model to explore the spread of lactase persistence,
challenged the vitamin D-rickets model by showing the absence of a relationship between
lactase persistence and the requirement of more vitamin D in people living at Northern
latitudes.
The vitamin D-calcium-rickets hypothesis based on the low ultraviolet-B (UVB) radiation at
high latitudes does not hold in the light of the very low prevalence of lactase persistence
among other individuals living in northern regions such as those living in Siberia or the
Amerindians living in the north of America (18, 12). Lactase persistence reaches the highest
prevalence in countries around the Baltic- and North Sea (19, 17), which coincides with
extreme dermal depigmentation, unique for this part of the world population (20). It is widely
accepted that depigmentation is a consequence of living at high latitude and lack of vitamin D
producing UVB radiation. UVB radiation (280-340 nm) is necessary for the conversion of pre-
vitamin D3 into vitamin D, which prevents the development of rickets. Pale skin obviously
captures more sunlight because of the reduced absorption by melanocytes (21).
Although there is a trend for lighter skin colour with increasing latitude, no other population
exhibits the extreme dermal depigmentation encountered in Europeans living in the circum
Baltic/North Sea region, even not populations living at higher latitudes, which suggests an

212
additional geographic selection pressure. It is possible that populations in the North of Europe
experienced another threat to their calcium homeostasis due to the spread of agriculture in
general, and in particular because of the consumption of wheat and barley.
1.2. The influence of cereals
Cordain et al. (20) stated that ‘Whole cereals are rachitogenic because their high fibre content
impairs the entero-hepatic circulation of vitamin D, leading to increased faecal elimination of
25-hydroxyvitamin D3, thereby progressively lowering plasma 25-hydroxyvitamin D3
concentrations in humans’. Moreover, whole grains are poor sources of calcium, and their
high phytate content reduces calcium bioavailability and thereby additionally contributes to
their rachitogenicity. The region around the Baltic/North Seas is one of the few high-latitude
regions in the world where cereal grains can be successfully grown without intensive modern
agricultural procedures. Usually, at high latitudes, extreme temperatures render the growing
season too fleeting for cereal crops to effectively compete with animal foods as a staple.
Because the warm Gulf Stream flows into the North and Baltic Seas and because of the
nearness to a maritime heat sink, temperatures in the surrounding landscapes were
sufficiently high to allow Neolithic farmers to successfully grow cereals as staples, primarily
wheat and barley (20).
1.3. Our hypotheses: Lactase and the immune system
To our knowledge, there has been no suggestion of a relation between lactase persistence and
the immune system in which lactase confers protection against potentially lethal pathogens.
Apart from the digestion of lactose, lactase is important for the intestinal absorption of
quercetin. By the hydrolysis of the naturally occurring quercetin-glucosides, quercetin
absorption may increase drastically in the presence of lactase (22). Quercetin is one of the
most abundant flavonols in edible plants (USDA database 2011) and is considered vital to
maintain immune function (23). Epidemiological studies indicate that higher compared to
lower quercetin intake is associated with reduced risk for ischemic heart disease, type 2
diabetes mellitus, asthma, and various types of cancer including lung cancer, colorectal
cancer, prostate cancer (24) and has widespread antimicrobial effects against viruses, bacteria
and fungi (25).
As stated in the introduction, pathogens have exerted strong selective pressure in human
evolution (4). One of the most lethal infectious diseases in humans is and has been
pneumonia (26). Every year 65,000 people in the USA die because of influenza and
pneumonia (27). Pneumococcal load has been a strong selective pressure factor shaping the
human immune system and genome (28). Another microbe producing pneumonia atypical in
humans and pneumonia in cattle is Mycoplasma mycoides (29). Mycoplasma pneumoniae in

213
cattle and probably humans became important with the rise of domestication of cattle,
including cows and goats, some 10,000 years ago (30). Domestication of cattle was the starting
point of introducing milk and milk products into the human diet and also the time point when
the polymorphism for lactase persistence typical for Europeans arose (12). Even now farmers
seem to be at surprisingly high risk (10 to 50 times more than expected) for the development
of pneumonitis (31).
Quercetin has effective antibiotic effects against bacteria producing pneumonia but only at
high doses (32). These might be provided by lactase (32) suggesting benefits in immune
function by lifelong lactase-facilitated quercetin absorption. We suggest that positive
selection of lactase persistence is related with increased protection against potentially lethal
pneumonia-causing pathogens.
1.4. Conclusions so far
Taken together, lactase persistence is a very recent polymorphism and its positive selective
pressure has been extremely strong (33). It is hard to conceive that such a high selective
pressure was caused by diseases with low lethality such as osteomalacia, and osteoporosis, or
because of the adaptation to a novel food with nevertheless high caloric and micronutrient
contents. On the other hand, rickets and pathogenic load are strong selective pressure factors.
Pathogenic load has been the leading roller coaster of human evolution and it is therefore
conceivable that subjects with lactase persistence are better protected against the mentioned
pathogens. This hypothesis is easily testable by measuring the difference in infection
susceptibility between lactase persistent and non-persistent individuals.

214
2. Salivary AMY1 gene copy number
2.1. AMY1 copy number and starch consumption
In 2007 Perry et al. (10) published a seminal paper relating the number of amylase (AMY1) gene
copies to the amount of dietary starch. They found that the number of the salivary amylase
gene copies correlates positively with salivary amylase protein levels and that individuals from
populations with high-starch diets have, on the average, more AMY1 gene copies than those
with traditionally low-starch diets. Since the publication of Perry et al., several hypotheses
have been proposed to explain positive selection of augmented AMY1-GCN. More AMY1
copies and amylase protein could ‘buffer against the fitness-reducing effects of intestinal
disease and toxicosis’ (10), although they did not specify what environmental factor(s) would
cause intestinal disease.
2.2. Existing hypotheses
One hypothesis relates salivary amylase activity to satiety (34). This effect could buffer against
overeating, although higher amylase levels during stressful situations may produce the
opposite. For instance, emotional overeating in response to psychosocial stress is a
behavioural trait related to childhood obesity and psychosocial stress factors increase salivary
amylase production (35). Our society is characterized by food abundance, overeating
increases the risk of obesity, and obesity is a risk factor for diabetes mellitus type 2,
cardiovascular diseases (CVD) and others (36). A buffering effect against overeating could
therefore decrease the incidence of CVD and related disorders. CVD and related disorders
affect a wide range of people, CVD are still the major causes of mortality worldwide (37) and
mortality is an important driving force in evolution by exerting selective pressure (38). It could
therefore theoretically be possible that augmented AMY1-GCN developed through selective
pressure of early mortality caused by increased obesity and CVD.
The above hypothesis, however, does not hold in the light of the estimated time of augmented
AMY1-GCN occurrence and also not when considering that the capacity of overeating
probably saved people from starvation at times when food was not available (39). The
estimated time of appearance of augmented AMY1-GCN is around 200,000 years ago,
although this estimate needs confirmation by the generation of AMY1 sequences from
multiple humans (10). Obesity and CVD are widely considered modern diseases that arose
very recently (40), while CVD mortality is highest among elderly people (41). At the time of the
occurrence of the increase of AMY1-GCN, humans hardly reached an average age of 35 years,
while even 200 years ago the average lifespan was only 30-40 years (42). It is therefore
questionable whether obesity, CVD and mortality caused by overeating would have exerted
the necessary high selective pressure on ancient populations in which the positive selection
for augmented AMY1-GCN occurred.

215
A second hypothesis relates higher AMY1-GCN and increased amylase protein in saliva with
certain sensory properties of starchy foods (43). Taste and viscosity could perhaps be detected
earlier in individuals with higher AMY1-GCN, although the benefit of this trait is unclear (35,
43), making this hypothesis weak while lacking sufficient scientific strength to suggest
positive selection.
Recently it has been shown by Mandel et al. (44) that, following the intake of starch-rich foods,
individuals with high endogenous salivary amylase activity regulate glucose homeostasis
better than individuals with lower salivary amylase activity (44). They state that efficient starch
digestion could have had “immense benefits” and relate this benefit with protection against
toxicosis and lower gastrointestinal malaise. This is exactly in line with our hypothesis which
states that through more efficient starch digestion people were and are protected against
multiple organ failure, dehydration and hyponatremia by upregulating the activity of SGLT1
(see our hypotheses)
It seems clear that no conceivable mono-hypothesis has as yet been generated for the
augmentation of AMY1-GCN in human populations with high starch intakes. Adaptation to
changes in food intake without evolutionary advantages, such as mortality before or during
reproductive age or significantly reduced fitness, is unlikely to provide a sufficiently strong
platform to explain accelerated positive selection. Before stressing our hypothesis, two other
factors speak against the hypothesis that certain human populations have adapted to a
nutrient, merely because it was incorporated in the diet.
2.3. AMY1 expression relates to stress and pancreatic amylase activity is huge
If humans adapted to starch rich food through increased expression of the amylase protein
(related with higher AMY1-GCN), then why would amylase protein production be highest
when salivary glands are (co)activated by the sympathetic nervous system (45)? It is widely
recognized that the sympathetic nervous is the first wave of the acute stress responses. This
would imply that starch intake produces stress, while stress is a reaction to danger (46). The
logical consequence is that starch-rich food is dangerous: something living on starch-rich
food is dangerous, or something within starch-rich food protects the starch-producing plant
against something dangerous and the resulting compounds might be toxic to humans. The
second factor that speaks against a merely nutrient intake driven genetic adaptation refers to
the starch digestive activity of pancreatic amylase. As early as 1995 it was shown that
pancreatic alpha-amylase extensively covers the digestive needs of starch intake (47).
Pancreatic postprandial amylase enzyme output varies from 50 U/min to 2,000 U/min (69)
with amylase having a digesting efficiency of 96% (48).

216
The significant difference in AMY1-GCN between populations with low and high starch
intakes and between humans and our nearest evolutionary counterpart, the chimpanzee, is
likely related to factors in or on starch-rich foods. High alpha-amylase facilitates breakdown
of starch, liberating various monosaccharides, including glucose. The latter is present in roots
and tubers eaten by the people belonging to the high AMY1-GCN tribes in Africa (49). Recent
research with the Hadza, the last authentic hunter-gatherer population in Tanzania, shows
that tubers and roots are an important part of daily food, varying from 18-38 en% of total
caloric intake through the year (50). It is nevertheless their non-preferred food (50) and
should be considered part of the fallback nutritional sources. Fallback nutritional sources
normally provide energy when food is scarce. Given the importance of fallback food, it might
be beneficial to optimize starch digestion efficiency. However, even if starch rich foods would
exceed 38 en% of total intake, it would make no sense to increase salivary amylase level.
Salivary and pancreatic amylase in combination with four other starch-digesting enzymes in
the small gut would cover the starch digestion needs extensively (51).
2.4. AMY1 and the immune system: domestic dog adaptations as introduction
to our hypothesis
It was recently found that domestic dogs exhibit several genetic changes suggesting
adaptation to the intake of starch (52). The three genes showing intense selective pressure
related with starch digestion in this study, are AMY2B, MGAM and SGLT1, which are
responsible for the production of respectively amylase, maltase-glucoamylase and the
sodium-dependent glucose co-transporter 1 (SGLT1). The difference in gene copy number,
enzyme production and enzyme activity in dogs is highly significant compared with their
most closely related counterpart, the wolf (52). The authors concluded that dogs have adapted
to starch intake during domestication, suggesting that a change of ecological niche was the
driving force behind that domestication and the novel niche could have induced scavenging
behaviour in waste dumps. However, waste dumps do not only contain starch, but also other
food wastes like meat, while all waste products come together with microbes, including
Escherichia coli, Salmonella (both meat born microbes) and fungi such as Aspergillus growing
on foul starch (53). These pathogens produce severe symptoms like vomiting, entero-
hemorrhagic enteritis, renal damage and failure, and death in humans, but especially among
children, although adults are not spared (54).
The three genetic changes found in domesticated dogs and related with starch digestion, also
have important effects on metabolism, systemic homeostasis and especially the immune
system. We contend that the effects on the immune system have been the genuine
background for the very high selective pressure on the above-mentioned genes in dogs and

217
that the same could hold for the AMY1-GCN in humans.
2.5. Evolutionary selective pressure aspects of the consumption of starch-rich
foods; our hypotheses
2.5.1. Toxins in starch-rich foods
Starch-rich foods (SRF) contain, like most plants, a series of proteins and anti-nutrients with
certain toxic effects. The mostly consumed SRF in non-African populations are cereals and
legumes, whereas African tribes eat substantial amounts of tubers and roots, although cereals
such as corn are rapidly replacing these ancient food sources (55). Whole grain cereals and
legumes contain both toxic and non-toxic compounds and all of these substances contribute
to the possible net toxic (nettox) effect of these SRF. It is important to consider only
constituents present in whole grains as possible selective factor, because refined cereal
products entered our diet only very recently. Candidate substances are gluten and its main
component gliadin, certain digestive enzyme inhibitors such as amylase-inhibitors in wheat
and lectins. A review was recently published on the potentially toxic impact of grains on
human health (56).
2.5.2. Did gluten-toxicity exert sufficient selective pressure to cause augmented AMY1-GCN?
Gluten can cause a wide range of disorders, varying from celiac disease (57), autoimmune
disorders (58), increased intestinal permeability syndrome (59) and types 1 and 2 diabetes
mellitus (60) and gluten burden is not restricted to gluten intolerant individuals (61).
All of these disorders usually evolve with gastro-intestinal problems and often diarrhoea (62)
The majority of mentioned disorders are negatively correlated with fertility and reproductive
success (63). Diarrhoea can cause severe dehydration and sodium deficiency and both lead to
increased mortality in both genders and at any age, but they affect children the most. Thus, at
least theoretically, it seems possible that gluten intake affected reproduction and mortality by
dehydration and severe sodium deficiency, causing selective pressure on cereal-eating
populations. Improved digestion of starch might increase glucose levels in the gut, facilitating
water, sodium and glucose transport by the sodium-dependent-glucose co transporter
(SGLT1) and thereby protect against lethal dehydration, hyponatraemia and multiple organ
failure because of energy deficiency, supporting our basic hypothesis and knowing that
higher glucose levels in the gut activate SGLT1, whereas normal levels do not (64).
Identification of gluten as a trigger of severe gastro-intestinal complaints, including
uncontrollable diarrhoea, vomiting and abdominal pain, occurred after World War II, when
the Dutch paediatrician Willem-Karel Dicke noticed that the war-related shortage of bread in
The Netherlands caused a significant drop in death rate among children affected by celiac
disease—from greater than 35% to essentially zero. He also reported that once wheat was again

218
available after the war, mortality soared to previous levels. Following up on Dicke’s
observation, other scientists looked at the different components of wheat, discovering that the
major protein in that grain, gluten, was the culprit (65).
Celiac disease is the best-known disorder related with gluten intake and is caused by a
combined impact of environmental factors on perhaps more than one hundred genes (66).
Several genes seem to play major roles in subjects who are susceptible to celiac disease. They
include genes for the production of human lymphocyte antigen (HLA) molecules and non-
HLA genes for the production of interleukins and their receptors (66). The whole group of
genes relates to the immune system and therefore it could well be that these genes originally
protected against pathogens and that celiac disease only occurred after the incorporation of
cereals into the human diet. In this direction, it has recently been shown that several
interleukin/interleukin receptor genes involved in the pathogenesis of celiac disease have
been subjected to pathogen-driven selective pressure. Specifically, celiac disease alleles of
IL18RAP, IL18R1, IL23, IL18R1 and the intergenic region between IL2 and IL21 display higher
frequencies in populations exposed to high microbial/viral loads, suggesting that these
variants play a role in the response to these organisms (67). People with these genotypes are
probably better protected against pathogens, but at the expense of celiac and other
autoimmune diseases,
Gluten intolerance further produces secondary lactose intolerance, because of its damaging
effect on enterocytes, which form the outermost layer of the gut (68). The combined effects of
damage inflicted by the immunological response against gluten and the secondary lactose
intolerance only increases the possibility of developing severe intestinal trouble including the
typical lactose malabsorption symptoms, abdominal pain, diarrhoea, nausea, bloating, and/or
flatulence (69).
The only plausible explanation for the positive selection or the preservation of genes that
confer higher susceptibility to toxic-gluten, has to be related with something even more
dangerous. People living in a highly infectious environment maintained or developed the
need for increased immune reactivity against pathogens (4). When they incorporated gluten
rich foods into their diets, the consequences of diarrhoea and vomiting could and
undoubtedly have killed a countless number of people, demanding another
phenotypical/genotypical adaptation to survive this novel selective pressure factor and this
risk only increases when facing secondary lactose intolerance. And for this, why not employ
another nutrient present in the same food to overcome the deleterious effect of the toxic part
of this altogether important energy-providing nutritional source? Increased breakdown of
starch in gluten-sensitive people would provide sufficient glucose to activate SGLT1 and

219
facilitate glucose, sodium and water transport and thereby prevent the severe burden of
gluten intake. Increased amylase production through higher AMY1-GCN could serve this
purpose.
The previously mentioned study of Axelsson et al. (52) shows that domesticated dogs carry an
increased number of pancreatic amylase genes (AMY2B) and a special SGLT1 haplotype with
structural (better function but not more enzyme) benefits compared with wolves. Dogs suffer
from food allergies, just as humans, with almost identical symptomatology, including
gastroenteritis-caused vomiting and diarrhoea, while canine gastroenteritis seems related
with intestinal bacterial overgrowth with E. coli as main pathogen (70) and gluten intolerance.
It has even been shown that gliadin, the main protein in gluten, produces the most severe
allergic response in dogs (71), while dogs are used to investigate the pathways leading to
clinical celiac disease because of the spontaneous development of celiac disease when fed
with gluten-rich foods (72). This provides evidence for a parallel evolution of humans and
domesticated dogs through increased starch digestion by amylase and improved glucose
transport, protecting the host against possible toxicosis by a novel nutrient, such as gluten.
and/or certain pathogens, such as enterotoxic E. coli. Higher AMY1-GCN and amylase
expression can serve this purpose perfectly and it were both Perry et al. (10) in their first
publication on augmented AMY1-GCN and Mandel et al. in 2012 (44) who already pointed at
the possible beneficial effects of augmented AMY1-GCN for the protection against intestinal
toxicity.
2.5.3. Can starch itself be sufficiently toxic to produce important selective pressure?
Starch is the storable form of energy produced by all green plants.
A part of dietary starch escapes digestion by all enzymes, reaching the colon as resistant
starch, ranging from approximately 1% following the consumption of white rice and 6% after
eating beans (73). Incompletely digested starch has significant impact on the gut microflora in
cattle and humans. Cows fed cereals can have a 1,000 times higher level of Escherichia coli in
their gut and gut E. coli corresponds with higher detectable E. coli levels in their meat (74).
Escherichia coli O157:H7, one of its most toxic mutants, can live undetected in the gut of food
animals and can be spread to humans directly and indirectly. E. coli is a normal inhabitant of
the gastrointestinal tract of mammals. Most E. coli strains do not cause disease, but can release
lipopolysaccharide complexes from their cell walls (including lipid A) upon disintegration.
These endotoxins can cause fever, and even death, but mostly if E. coli translocates from the
gut into the blood. Traditional models of E. coli pathogenesis were based on the ability of
certain strains to attach to mucosal surfaces, but the invasion process itself was poorly
understood.

220
Cattle born pathogens are considered zoonoses (transferable to humans) and they include E.
coli, but also Salmonella, Campylobacter and Clostridium dificile (75). Salmonella can be
highly toxic as evidenced by various severe Salmonella outbreaks through ancient and recent
history (76). Salmonella grows, just like E. coli on meat, and Salmonella/E. Coli infected meat
and poultry are still considered the most important sources of food born illness (76). Survival
of the pathogenic form of these microbes is facilitated by the intake of resistant starch that
escapes total digestion in cattle (74). Recent research shows that this also holds for humans
and that pathogenic growth of gut damaging microbes is substantially enhanced by
maltodextrin, a derivate of incomplete digested starch (77).
It seems obvious that the combination of direct exposure to food born pathogens (E. Coli,
Salmonella) and the nourishing effect of digestion-resistant starch has benefitted the growth
of these microbes and thereby caused severe gastro-intestinal disorders in humans
consuming infected meat and starch rich food. Macronutrient uptake is greatly dependent on
gut surface, passage time and hydrolysis. Complete digestion of starch could have prevented
bacterial overgrowth of pathogenic bacteria, offered protection against possibly severe
gastroenteritis and even death. Augmented AMY1-GCN could have served this purpose, by
causing a more rapid and thereby more complete digestion and uptake in combination with
the other starch-digesting sacharidases.
2.5.4. Fungi growing on starch rich food; a largely ignored global health issue at present and
definitely in the past
Starch provides the major food source for a wide range of fungi, including Fusarium species,
Aspergillus flavus, Penicillum viridicatum and Acremonium coenophialum (78), whereas
another fungus, Claviceps purpurea, parasites on starch rich cereals such as barley, rye and
wheat (79). All of these fungi produce various types of highly toxic mycotoxins, including
aflatoxins, tricothecenes, fumonisins, T-2 toxin, zearalenone, deoxynivalenol and ochratocin
A (80). Among these, Claviceps purpurea is a special fungus, because it has historically often
been looked at as a part of the cereal plant although it produces the most severe mycotoxins
named ergopeptines, including ergometrine (81).
When present in foods in sufficiently high quantities, mycotoxins can produce symptoms
ranging from acute liver or kidney deterioration, severe gastroenteritis, vomiting, anorexia,
reduced weight gain, neuroendocrine changes, immunological effects, diarrhoea,
leukocytosis, haemorrhage or circulatory shock, and acute death, to chronic liver cancer,
mutagenic, and teratogenic effects, skin irritation, immunosuppression, birth defects,
neurotoxicity, and ‘slow’ death (82, 83).

221
Acute mycotoxicosis belongs to the most devastating infections, affecting human being as
evidenced by several recent outbreaks including the one in Kenya in 2004, that affected 317
individuals, killed 125, and was caused by the ingestion of aflatoxin-infected maize (84).
Another fatal outbreak in 1974 killed 100 persons in India because of the consumption of
aflatoxin-infected corn (83). Not only cereals are a rich source of mycotoxins. Other starch
rich foods such as dried fruits (83,), beans, peanuts (83) and underground bulbs (84) are also
frequently affected by fungi, producing different types and amounts of mycotoxins. The
actual economic burden of affected crops is enormous, because of the difficulties to control
fungal growth on SRF (85) and the estimated economic losses are $1.4 billion every year only
in the USA (83). In the European Union, regulations limit the amount of total aflatoxins to 4
ng/g, whereas guidelines in a few developing countries and the US limit total aflatoxins to no
more than 20 ng/g in foodstuffs intended for human consumption (86). In Nigeria, the
National Agency for Food, Drug Administration and Control has set 20 ng/g as the maximum
permissible limit for total aflatoxin in foodstuff (86).
Even though preventive methods are very well defined, mycotoxins keep affecting 25% of
world crops (87). When people started eating SRF thousands of years ago, logical reasoning
informs us that those crops must have been affected by fungal infections and that the
consumers are likely to have suffered from acute and/or chronic mycotoxicosis. The group of
Lesley investigated how mycotoxin growth and fatal doses of mycotoxins could be prevented
in non-industrialized countries, which were basically all countries of the world 10-30,000
years ago (88).
These methods are nowadays likely to be well known in the majority of SRF eating
populations, but it is unlikely that ancient populations knew how to deal with fungal growth
on SRF, while even if they did, it does not imply that they avoided consumption of the affected
foods. ‘Even today situations of relative scarcity of food often forces consumers in many
regions to distressing decisions, such as ‘to eat contaminated grain today and worry about the
consequences tomorrow (or some other time in the future) or starve today and perhaps not
even have a tomorrow’ as stated by Bandyopadhyay (89). This behaviour is not a unique
‘characteristic’ of modern humans, it is the way most individuals think and have thought
when starving to death.
Several data suggest that people have suffered from more or less fatal mycotoxicosis for
hundreds of generations and mycotoxin intoxication has without doubt killed thousands of
people. The mortality rate ranges from 10-60% of the infected people and embryos and
children are among the most affected individuals (90). Important proof for the devastating
effects of mycotoxins comes from data related with ergotism. Ergotamine and other

222
ergopeptines from the fungus Claviceps purpurea are highly toxic to humans and can cause
different types of ‘ergotism’. Convulsive, gangrenous and entero/hyperthermic ergotism
cause respectively epileptic convulsions, confusion and death (convulsive), vasoconstriction,
cold/hot feeling, abortion and gangrene (gangrenous) and nausea, vomiting, diarrhoea,
increased metabolic rate and excessive salivation (entero/hyperthermic, 80). Convincing
historical data for the existence of epidemics of mycotoxicosis are as old as 600 BC, when
people in Assyria suffered from ‘madness’ caused by ‘the noxious pustule in the ear of grain’
(91) and this seems to be the same disease that affected many parts of Europe in the tenth
century referred to as St. Anthony’s or Holy fire, and is considered to have been caused by the
consumption of rye contaminated with ergot alkaloids from Claviceps purpurea. Further
accounts suggest that other contaminated grains have been responsible for major outbreaks
of disease (e.g., the Ten Plagues of Egypt, 83).
Mycotoxins can produce chronic diarrhoea and vomiting. At the same time they highly
influence the functionality of SGLT1 and GLUT5. Very low doses of mycotoxins inhibit
nutrient transport in the gut, especially affecting SGLT1 (50% inhibition with a 10 µmol/L
solution in vitro) and the fructose transporter GLUT5 (42% inhibition, 92). Later studies in
animals confirmed these findings (93). Mycotoxins produced by Aspergillus further inhibit
alpha-amylase activity, which has been evidenced several times in vivo, although mostly in
chickens (94). The combination of these targets and their effects can be considered the perfect
cocktail to die because of dehydration, hyponatraemia and MOF.
Aspergillus and its toxins are definitely highly toxic and have killed thousands of individuals,
including children and adolescents (95).
The described pathways, by which mycotoxins can have affected human health and even
mortality rate, could, and probably have, served as important selective pressure factors for the
observed augmentation of oral AMY1-GCN in populations with high starch intakes. Complete
starch digestion would have provided enough glucose to augment SGLT1 activity, improving
the transport of glucose, water and sodium, preventing dehydration, multiple organ failure
and possibly lethal hyponatraemia in infected individuals.
Of all the pathogens described in this paper and considered as possible cause of selective
pressure on the number of AMY1 gene copies, fungi producing mycotoxins seem to be the
most appropriate candidates.
2.6. Conclusions so far
The entrance of SRF in the human diet introduced several positive and negative factors.
Gluten in SRF can be highly toxic and the incomplete digestion of resistant starch in SRF

223
provides an optimal substrate for the growth of possibly lethal pathogens including food
borne toxic E. coli strains and Salmonella. Dehydration, hyponatraemia and MOF are the most
damaging effect of gluten-toxicity and food borne pathogens due to chronic diarrhoea and
vomiting. The same holds for fungi living on SRF, producing highly toxic mycotoxins that can
be fatal. Higher salivary alpha-amylase activity through augented AMY1-GCN can increase
glucose level in the gut, facilitating water, sodium and glucose uptake and protect against the
above mentioned effects. The observation that people with higher AMY1-GCN have lower
blood glucose levels after starch intake supports the latter conclusion, suggesting that this
polymorphism has been positively selected for higher glucose level in the gut.

224
3. Rotavirus, lactose intolerance and AMY1-GCN
Rotavirus is one of the most important causes of gastro-enteritis, malnutrition, and diarrhoea
in young children and animals. Rotavirus can be extremely dangerous, evidenced by the fact
that more than 550,000 children die every year from this infectious disease (96). Rotavirus
induces diarrhoea and rotavirus-diarrhoea can be lethal because of reduced uptake of sodium
and subsequent systemic hyponatraemia through a direct inhibiting effect of rotavirus on the
SGLT1 mechanism (97). Rotavirus further induces lactose intolerance by inhibiting lactase
production (98) and dairy intake in people suffering from rotavirus diarrhoea seems to
augment the diarrhoea duration (99). As mentioned before, rotavirus is extremely dangerous
for children and newborn and is the mayor death cause in children younger than 5 years old
(96). Newborns produce lactase to break down the lactose in their mother’s own milk,
converting it into glucose and galactose, providing energy and enough glucose to transport
water, sodium and glucose through activation of SGLT1. Newborns hardly produce
endogenous amylase up to 6 months and even after 2 years still show some dependency on
breast-milk amylase to support normal starch breakdown (100). Nothing in breast-milk needs
amylase to become digested, so why would breast-milk contain substantial amylase activity?
It has been proposed that breast-milk amylase could serve as a compensation for low salivary
and pancreatic amylase activities in newborns and aid in the digestion of complex
carbohydrates from the time that complementary foods are introduced in close proximity to
breastfeeding (101). We suggest that the substantial amylase level in breast-milk has become
needed to digest complex starch rich food, at times that pathogens, but especially rotavirus,
cause diarrhea through their combined disturbance of lactase activity, damage to enterocytes
and inhibition of sodium transport (102).
Rotavirus might be considered an important selective pressure factor (103). It has probably
entered the human environment as a zoonose from domestic animals (104). The majority of
individuals dying from rotavirus infections live in developing countries; countries where
domestic animals and novel zoonoses are relatively new. We suggest that augmented AMY1-
GCN has been selected for the protection against lactose intolerance in the newborn caused
by pathogens in general but more specifically by Rotavirus. High amylase level in breast-milk
facilitates starch digesting in newborns that suffer from Rotavirus-induced lactose-
intolerance. Improved starch break down increases glucose level in the newborn gut, activates
SGLT1 and guarantees a minimum of sodium, water and glucose transport into the
bloodstream and thereby protects against possibly lethal hyponatraemia, dehydration and
MOF. This is exactly the rationale behind oral rehydration in individuals suffering from
Rotavirus diarrhoea (105). Rotavirus is nowadays hardly fatal in North European countries.
Augmentation of AMY1-GCN could be at the basis of the lack of virulence of Rotavirus in this

225
part of the world, whereas Rotavirus in Africa is still a major cause of childhood and overall
mortality (99, 105).

226
4. Discussion of our hypotheses and final conclusions
Table 1 shows an overview of the different environmental factors that might have caused
selection for lactase persistence and augmented AMY1-GCN. We suggest that these factors are
not mutually exclusive, but should be viewed upon as complementary and logical in the scene
of evolutionary biology. Lactase persistence and augmented AMY1-GCN are two genetic
adaptations showing intense positive selection (lactase persistence > AMY1-GCN) in humans.
Lactose and starch have deleterious effects on human health when digesting is incomplete
and this occurs when lactase and amylase do not reach sufficiently high activities. Lactose
intolerance (not treated in this review) itself can be detrimental to human health, through
causing symptoms like irritable bowel syndrome, watery stool and excessive flatus (106).
Lactose intolerance can nevertheless hardly have served as the only factor in the observed
highly positive selection of lactase persistence, because of the lack of sufficient influence on
mortality (107), while lactose intolerance also does not seem to affect fertility (108). A recent
publication (109) also challenges the rather simple ‘fresh milk intake’ hypothesis as the driving
force behind lactase persistence. .
Incomplete starch digestion also affects human health, but without bacterial overgrowth of for
instance Escherichia coli, symptoms would be troubling, but mortality rate and reproduction
are less affected. Looking at both genetic adaptations in a broader perspective suggests that
they might protect against the severe deleterious effects of several pathogens and the
damaging effects of gluten. Pathogens like Rotavirus affect lactose digestion directly, whereas
incomplete starch digestion facilitates the growth of possibly lethal cholera like E. Coli strains.
Humans have explored new environments more than any other animal and novel
environments challenge the immune system with new climatological conditions, food and,
most of all, pathogens.
The last big revolution in the human evolutionary history has been the development of
agriculture: somewhat like 10 thousand years ago someone started to exploit the observation
that a plant growths from a seed and agriculture started. With agriculture came the
domestication of cattle and with cattle came new pathogens. The evidence we have given in
this article supports our hypotheses that it are these factors that have driven the highly
positive selection of lactase persistence and augmented AMY1-GCN: both adaptations provide
enough glucose to activate SGLT1, which is the most important glucose, sodium and water
transporter in the gastro-intestinal tract. The increased capacity of water, sodium and glucose
transport from the gut into the blood stream, has probably saved thousand of individuals
infected with pathogenic E. Coli, Rotavirus and TB, but most of all from highly lethal fungi
living on starch rich food. The adaptations also protected them against the highly damaging
effects of gluten intake. We therefore contend that lactase persistence and augmented AMY-

227
GCN are not merely adaptations to nutrients in novel foods; they were needed to protect
humans against novel pathogens that arose with agriculture and the domestication of cattle.
Food born pathogens, living on meat and starch rich foods are still serious threats to human
health and it is therefore necessary to further investigate how we can limit the presence of
possibly lethal pathogens in human and animal foods.

228
Table 1. Overview of the proposed mechanisms for the strong positive selection of lactase
persistence and augmented AMY1-GCN.
Selective pressure
factor
Evolutionary
target
Consequences Contemporary
solution
Evolutionary
adaptation
Benefit
Milk Lactose
intolerance
Diarrhoea, Rickets.
Energy and
calcium deficiency,
Lactose free dairy
products
Vitamin D
Calcium
Lactase
persistence
Availability of important
macro- and
micronutrients including
vitamin D and Calcium
Starch Incomplete
digestion
Intestinal malaise.
Energy deficiency
Higher salivary
amylase
production
and activity by
higher AMY1-
GCN
Availability of important
macro- and
micronutrients
Mycoplasma
Pneumonia (MP)
Immune
system
Pneumonitis death Antibiotics Lactase
persistence
Availability of quercetin
and increased defence
against MP
Gluten in SRF Gut barrier
SGLT1
IIPS, chronic
diarrhoea,
vomiting, loss of
fertility, death
Gluten free cereals Higher salivary
amylase
production
and activity by
higher AMY1-
GCN
Higher gut glucose and
activation of SGLT1.
Protection against
dehydration,
hyponatraemia and MOF
Starch Incomplete
digestion
E. coli overgrowth
syndrome.
Diarrhoea, bladder
infection
Antibiotics
Oral rehydration
Higher salivary
amylase
production
and activity by
higher AMY1-
GCN
Higher gut glucose,
activation of SGLT1.
Protection against
dehydration,
hyponatraemia and MOF.
Availability of mannose;
effective against E. coli
infections
Agriculture and food
born pathogens
(zoonoses),
especially Fungi,
but also E. Coli,
Salmonella,
Campylobacter,
Clostridium
Immune
system.
SGLT1
Mycotoxins.
Diarrhoea,
vomiting,
gangrene, abortion,
epilepsy, infant
death by DH, HN
and/or MOF
Antibiotics
Oral rehydration
Antifungal treatment
Higher salivary
amylase
production
and activity by
higher AMY1-
GCN
Higher gut glucose,
activation of SGLT1.
Protection against
dehydration,
hyponatraemia and MOF.
Rotavirus Lactase gene
Immune
system
Lactose
intolerance, lethal
diarrhoea mostly in
newborn
Oral rehydration
Alternative feeding
Higher salivary
amylase
production
and activity by
higher AMY1-
GCN
Increased amylase in
breast milk, improving
digestion of SRF when
the newborn developed
lactose intolerance and
needed complex food to
survive

229
References
1. Novembre, J. & Han, E. Human population structure and the adaptive response to
pathogen-induced selection pressures. Philos Trans R Soc Lond B Biol Sci 367, 878-886
(2012).
2. Hancock, A. M., Witonsky, D. B., Ehler, E., Alkorta-Aranburu, G., et al. Colloquium paper:
human adaptations to diet, subsistence, and ecoregion are due to subtle shifts in allele
frequency. Proc Natl Acad Sci U S A 107 Suppl 2, 8924-8930 (2010).
3. Hancock, A. M., Witonsky, D. B., Alkorta-Aranburu, G., Beall, C. M., et al. Adaptations to
climate-mediated selective pressures in humans. PLoS Genet 7, e1001375 (2011).
4. Fumagalli, M., Sironi, M., Pozzoli, U., Ferrer-Admetlla, A., et al. Signatures of
environmental genetic adaptation pinpoint pathogens as the main selective pressure
through human evolution. PLoS Genet 7, e1002355 (2011).
5. Weng, K., Hu, H., Xu, A. G., Khaitovich, P. & Somel, M. Mechanisms of dietary response in
mice and primates: a role for EGR1 in regulating the reaction to human-specific
nutritional content. PLoS One 7, e43915 (2012).
6. Babbitt, C. C., Warner, L. R., Fedrigo, O., Wall, C. E. & Wray, G. A. Genomic signatures of
diet-related shifts during human origins. Proc Biol Sci 278, 961-969 (2011).
7. Stevenson, RJ. TI Case, MJ Oaten, Proactive strategies to avoid infectious disease., in
Philos Trans R Soc Lond B Biol Sci
8. Barreiro, L. B. & Quintana-Murci, L. From evolutionary genetics to human immunology:
how selection shapes host defence genes. Nat Rev Genet 11, 17-30 (2010).
9. Swallow, D. M. Genetics of lactase persistence and lactose intolerance. Annu Rev Genet
37, 197-219 (2003).
10. Perry, G. H., Dominy, N. J., Claw, K. G., Lee, A. S., et al. Diet and the evolution of human
amylase gene copy number variation. Nat Genet 39, 1256-1260 (2007).
11. Itan, Y., Jones, B. L., Ingram, C. J., Swallow, D. M. & Thomas, M. G. A worldwide
correlation of lactase persistence phenotype and genotypes. BMC Evol Biol 10, 36 (2010).
12. Ingram, C. J., Mulcare, C. A., Itan, Y., Thomas, M. G. & Swallow, D. M. Lactose digestion
and the evolutionary genetics of lactase persistence. Hum Genet 124, 579-591 (2009).
13. Enattah, N. S., Trudeau, A., Pimenoff, V., Maiuri, L., et al. Evidence of still-ongoing
convergence evolution of the lactase persistence T-13910 alleles in humans. Am J Hum
Genet 81, 615-625 (2007).
14. Flatz, G. & Rotthauwe, H. W. Lactose nutrition and natural selection. Lancet 2, 76-77
(1973).
15. Thacher, T. D., Fischer, P. R., Pettifor, J. M., Lawson, J. O., et al. A comparison of calcium,
vitamin D, or both for nutritional rickets in Nigerian children. New England Journal of
Medicine 341, 563-568 (1999).

230
16. Loudon, I. Deaths in childbed from the eighteenth century to 1935. Medical History 30, 1
(1986).
17. Rosenberg, K. & Trevathan, W. The evolution of human birth. Special Editions (2003).
18. Quillen, E. E., Bauchet, M., Bigham, A. W., Delgado-Burbano, M. E., et al. OPRM1 and
EGFR contribute to skin pigmentation differences between Indigenous Americans and
Europeans. Hum Genet 131, 1073-1080 (2012).
19. Beja-Pereira, A., Luikart, G., England, P. R., Bradley, D. G., et al. Gene-culture coevolution
between cattle milk protein genes and human lactase genes. Nat Genet 35, 311-313
(2003).
20. Cordain, L., Hickey, M. S. & Kim, K. 12 Malaria and Rickets Represent Selective Forces for
the Convergent Evolution of Adult Lactase Persistence. Biodiversity in Agriculture:
Domestication, Evolution, and Sustainability 299 (2012).
21. Jablonski, N. G. & Chaplin, G. Colloquium paper: human skin pigmentation as an
adaptation to UV radiation. Proc Natl Acad Sci U S A 107 Suppl 2, 8962-8968 (2010).
22. Day, A. J., Gee, J. M., DuPont, M. S., Johnson, I. T. & Williamson, G. Absorption of
quercetin-3-glucoside and quercetin-4'-glucoside in the rat small intestine: the role of
lactase phlorizin hydrolase and the sodium-dependent glucose transporter. Biochem
Pharmacol 65, 1199-1206 (2003).
23. Bugianesi, R., Serafini, M., Simone, F., Wu, D., et al. High-performance liquid
chromatography with coulometric electrode array detector for the determination of
quercetin levels in cells of the immune system. Anal Biochem 284, 296-300 (2000).
24. Knekt, P., Kumpulainen, J., Järvinen, R., Rissanen, H., et al. Flavonoid intake and risk of
chronic diseases. Am J Clin Nutr 76, 560-568 (2002).
25. Cushnie, T. T. & Lamb, A. J. Antimicrobial activity of flavonoids. International Journal of
Antimicrobial Agents 26, 343-356 (2005).
26. Ma, Y., Cowling, B. J., Wen, X., Peng, J., et al. Systematic Review and Meta-Analysis on
the Association between Outpatient Statins Use and Infectious Disease-Related
Mortality. PLoS ONE 7, e51548 (2012).
27. Chertow, D. S. & Memoli, M. J. Bacterial Coinfection in InfluenzaA Grand Rounds
ReviewBacterial Coinfection in Influenza. JAMA 309, 275-282 (2013).
28. Hill, A. V. S. Evolution, revolution and heresy in the genetics of infectious disease
susceptibility. Philosophical Transactions of the Royal Society B: Biological Sciences
367, 840-849 (2012).
29. Waites, K. B. & Talkington, D. F. Mycoplasma pneumoniae and its role as a human
pathogen. Clin Microbiol Rev 17, 697-728, table of contents (2004).
30. Fischer, A., Shapiro, B., Muriuki, C., Heller, M., et al. The origin of the 'Mycoplasma
mycoides cluster' coincides with domestication of ruminants. PLoS One 7, e36150
(2012).

231
31. Greskevitch, M., Kullman, G., Bang, K. M. & Mazurek, J. M. Respiratory Disease in
Agricultural Workers: Mortality and Morbidity Statistics. Journal of Agromedicine 12, 5-
10 (2008).
32. Hossion, A. M., Zamami, Y., Kandahary, R. K., Tsuchiya, T., et al. Quercetin
diacylglycoside analogues showing dual inhibition of DNA gyrase and topoisomerase IV
as novel antibacterial agents. Journal of medicinal chemistry 54, 3686-3703 (2011).
33. Gerbault, P., Liebert, A., Itan, Y., Powell, A., et al. Evolution of lactase persistence: an
example of human niche construction. Philos Trans R Soc Lond B Biol Sci 366, 863-877
(2011).
34. Harthoorn, L. F. & Dransfield, E. Periprandial changes of the sympathetic-
parasympathetic balance related to perceived satiety in humans. Eur J Appl Physiol 102,
601-608 (2008).
35. Santos, J. L., Saus, E., Smalley, S. V., Cataldo, L. R., et al. Copy number
polymorphism of the salivary amylase gene: implications in human nutrition
research. J Nutrigenet Nutrigenomics 5, 117-131 (2012).
36. Staiano, A. E., Harrington, D. M., Broyles, S. T., Gupta, A. K. & Katzmarzyk, P. T. Television,
adiposity, and cardiometabolic risk in children and adolescents. Am J Prev Med 44, 40-
47 (2013).
37. Fuster, V., Narula, J. & Kelly, B. B. Promoting global cardiovascular and cerebrovascular
health. Mt Sinai J Med 79, 625-631 (2012).
38. Wells, J. C. The evolution of human fatness and susceptibility to obesity: an ethological
approach. Biol Rev Camb Philos Soc 81, 183-205 (2006).
39. Södersten, P., Bergh, C., Zandian, M. & Ioakimidis, I. Obesity and the brain. Med
Hypotheses 77, 371-373 (2011).
40. Straub, RH. Concepts of evolutionary medicine and energy regulation contribute to the
etiology of systemic chronic inflammatory diseases. Brain, Behavior, and Immunity 25
1–5 (2011)
41. Wells, J. C. The evolution of human adiposity and obesity: where did it all go wrong? Dis
Model Mech 5, 595-607 (2012).
42. Finch, C.E. Evolution of the Human Lifespan, Past, Present, and Future: Phases in the
Evolution of Human Life Expectancy in Relation to the Infl ammatory Load.
Proceedings of the american Philosophical Society vol. 156, no. 1 (2012)
43. Mandel, A. L., Peyrot des Gachons, C., Plank, K. L., Alarcon, S. & Breslin, P. A. Individual
differences in AMY1 gene copy number, salivary α-amylase levels, and the perception of
oral starch. PLoS One 5, e13352 (2010).
44. Mandel, A. L. & Breslin, P. A. High endogenous salivary amylase activity is associated
with improved glycemic homeostasis following starch ingestion in adults. J Nutr 142,
853-858 (2012).

232
45. Rohleder, N. & Nater, U. M. Determinants of salivary alpha-amylase in humans and
methodological considerations. Psychoneuroendocrinology 34, 469-485 (2009).
46. Cohen, S., Janicki-Deverts, D., Doyle, W. J., Miller, G. E., et al. Chronic stress,
glucocorticoid receptor resistance, inflammation, and disease risk. Proc Natl Acad Sci U
S A 109, 5995-5999 (2012).
47. Southgate, D. A. Digestion and metabolism of sugars. The American journal of clinical
nutrition 62, 203S-210S (1995).
48. Keller, J. & Layer, P. Human pancreatic exocrine response to nutrients in health and
disease. Gut 54 Suppl 6, vi1-v28 (2005).
49. Vincent, A. S. Plant foods in savanna environments: a preliminary report of tubers eaten
by the Hadza of northern Tanzania. World Archaeology 17, 131-148 (1985).
50. Marlowe, F. W. & Berbesque, J. C. Tubers as fallback foods and their impact on Hadza
hunter-gatherers. Am J Phys Anthropol 140, 751-758 (2009).
51. Lin, A. H., Nichols, B. L., Quezada-Calvillo, R., Avery, S. E., et al. Unexpected high
digestion rate of cooked starch by the Ct-maltase-glucoamylase small intestine mucosal
α-glucosidase subunit. PLoS One 7, e35473 (2012).
52. Axelsson, E., Ratnakumar, A., Arendt, M. L., Maqbool, K., et al. The genomic signature of
dog domestication reveals adaptation to a starch-rich diet. Nature 495, 360-364 (2013).
53. Oviasogie, F. E., Ajuzie, C. U. & Ighodaro, U. G. Bacterial analysis of soil from waste
dumpsite. Arch Appl Sci Res 2, 161-167 (2010).
54. Scallan, E., Griffin, P. M., Angulo, F. J., Tauxe, R. V. & Hoekstra, R. M. Foodborne Illness
Acquired in the United States—Unspecified Agents. Emerging Infectious Diseases 17, 16-
22 (2011).
55. Westhuizen van der Liana. Fumonisin exposure biomarkers in humans consuming
maize staple diets. PhD Dissertation. University of Stellenbosch (2011)
56. Punder, de K. & Pruimboom, L. The dietary intake of wheat and other cereal grains and
their role in inflammation. Nutrients 5, 771-787 (2013).
57. Fasano, A. & Shea-Donohue, T. Mechanisms of disease: the role of intestinal barrier
function in the pathogenesis of gastrointestinal autoimmune diseases. Nat Clin Pract
Gastroenterol Hepatol 2, 416-422 (2005).
58. Fasano, A. Leaky gut and autoimmune diseases. Clin Rev Allergy Immunol 42, 71-78
(2012).
59. Lammers, K. M., Lu, R., Brownley, J., Lu, B., et al. Gliadin induces an increase in intestinal
permeability and zonulin release by binding to the chemokine receptor CXCR3.
Gastroenterology 135, 194-204.e3 (2008).
60. Kort, de S., Keszthelyi, D. & Masclee, A. A. Leaky gut and diabetes mellitus: what is the
link? Obes Rev 12, 449-458 (2011).

233
61. Ruuskanen, A., Luostarinen, L., Collin, P., Krekelä, I., et al. Persistently positive gliadin
antibodies without transglutaminase antibodies in the elderly: gluten intolerance
beyond coeliac disease. Dig Liver Dis 43, 772-778 (2011).
62. Nussinovitch, U. & Shoenfeld, Y. The role of gender and organ specific autoimmunity.
Autoimmun Rev 11, A377-A385 (2012).
63. Carp, H. J., Selmi, C. & Shoenfeld, Y. The autoimmune bases of infertility and pregnancy
loss. J Autoimmun 38, J266-J274 (2012).
64. Yu, L. C., Flynn, A. N., Turner, J. R. & Buret, A. G. SGLT-1-mediated glucose uptake
protects intestinal epithelial cells against LPS-induced apoptosis and barrier defects: a
novel cellular rescue mechanism? FASEB J 19, 1822-1835 (2005).
65. Fasano, A. Surprises from celiac disease. Scientific American Magazine 301, 54-61 (2009).
66. Qiao, S. W., Iversen, R., Ráki, M. & Sollid, L. M. The adaptive immune response in celiac
disease. Semin Immunopathol 34, 523-540 (2012).
67. Sironi, M. & Clerici, M. The hygiene hypothesis: an evolutionary perspective. Microbes
Infect 12, 421-427 (2010).
68. Brüssow, H. Nutrition, population growth and disease: a short history of l actose.
Environ Microbiol (2013).
69. Hammer, H. F. & Hammer, J. Diarrhea caused by carbohydrate malabsorption.
Gastroenterol Clin North Am 41, 611-627 (2012).
70. German, A. J., Hall, E. J. & Day, M. J. Chronic intestinal inflammation and intestinal
disease in dogs. Journal of veterinary internal medicine 17, 8-20 (2003).
71. Buchanan, B. B. & Frick, O. L. The dog as a model for food allergy. Annals of the New
York Academy of Sciences 964, 173-183 (2002).
72. Marietta, E. V. & Murray, J. A. Animal models to study gluten sensitivity. Semin
Immunopathol 34, 497-511 (2012).
73. Muir, J. G. & O'Dea, K. Validation of an in vitro assay for predicting the amount of starch
that escapes digestion in the small intestine of humans. The American journal of clinical
nutrition 57, 540-546 (1993).
74. Callaway, T. R., Carr, M. A., Edrington, T. S., Anderson, R. C. & Nisbet, D. J. Diet,
Escherichia coli O157: H7, and cattle: a review after 10 years. Current issues in
molecular biology 11, 67 (2009).
75. Marks, S. L., Rankin, S. C., Byrne, B. A. & Weese, J. S. Enteropathogenic bacteria in dogs
and cats: diagnosis, epidemiology, treatment, and control. J Vet Intern Med 25, 1195-
1208 (2011).
76. Wang, L., Wu, C. S., Fan, X. & Mustapha, A. Detection of Escherichia coli O157:H7 and
Salmonella in ground beef by a bead-free quantum dot-facilitated isolation method. Int
J Food Microbiol 156, 83-87 (2012).

234
77. Nickerson, K. P. & McDonald, C. Crohn's disease-associated adherent-invasive
Escherichia coli adhesion is enhanced by exposure to the ubiquitous dietary
polysaccharide maltodextrin. PLoS One 7, e52132 (2012).
78. Yang, F., Jensen, J. D., Svensson, B., Jørgensen, H. J., et al. Secretomics identifies
Fusarium graminearum proteins involved in the interaction with barley and wheat. Mol
Plant Pathol 13, 445-453 (2012).
79. Belser-Ehrlich, S., Harper, A., Hussey, J. & Hallock, R. Human and cattle ergotism since
1900: symptoms, outbreaks, and regulations. Toxicol Ind Health 29, 307-316 (2013).
80. Zaki, M. M., El-Midany, S. A., Shaheen, H. M. & Rizzi, L. Mycotoxins in animals:
Occurrence, effects, prevention and management. J. Toxicol Environ. Health 4, 13-28
(2012).
81. Hulvová, H., Galuszka, P., Frébortová, J. & Frébort, I. Parasitic fungus Claviceps as a
source for biotechnological production of ergot alkaloids. Biotechnol Adv 31, 79-89
(2013).
82. Bonnet, M. S., Roux, J., Mounien, L., Dallaporta, M. & Troadec, J. D. Advances in
deoxynivalenol toxicity mechanisms: the brain as a target. Toxins (Basel) 4, 1120-1138
(2012).
83. Paterson, R.R.M. & Lima, N. Toxicology of mycotoxins. Molecular, Clinical and
Environmental Toxicology. Volume 2: Clinical Toxicology. 31-63 (2010)
84. Lewis, L., Onsongo, M., Njapau, H., Schurz-Rogers, H., et al. Aflatoxin Contamination of
Commercial Maize Products during an Outbreak of Acute Aflatoxicosis in Eastern and
Central Kenya. Environmental Health Perspectives 113, 1763-1767 (2005).
85. Hussein, H. S. & Brasel, J. M. Toxicity, metabolism, and impact of mycotoxins on
humans and animals. Toxicology 167, 101-134 (2001).
86. Egmond, H. P. & Jonker, M. A. Worldwide regulations for mycotoxins in food and feed in
2003 (Food and Agriculture Organization of the United Nations, 2004).
87. Turner, P. C., Flannery, B., Isitt, C., Ali, M. & Pestka, J. The role of biomarkers in
evaluating human health concerns from fungal contaminants in food. Nutr Res Rev 25,
162-179 (2012).
88. Leslie John F, Bandyopadhyay R, Visconti A. Mycotoxins. Detection Methods,
Management, Public Health and Agricultural Trade, in Mycotoxins ISBN-13: 978 1 84593
082 0 (2008)
89. Bandyopadhyay, R., Kumar, M. & Leslie, J. F. Relative severity of aflatoxin contamination
of cereal crops in West Africa. Food Addit Contam 24, 1109-1114 (2007).
90. Peraica, M., Radic, B., Lucic, A. & Pavlovic, M. Toxic effects of mycotoxins in humans.
Bulletin of the World Health Organization 77, 754-766 (1999).
91. De Costa, C. St Anthony's fire and living ligatures: a short history of ergometrine. The
Lancet 359, 1768-1770 (2002).

235
92. Maresca, M., Mahfoud, R., Garmy, N. & Fantini, J. The mycotoxin deoxynivalenol affects
nutrient absorption in human intestinal epithelial cells. J Nutr 132, 2723-2731 (2002).
93. Awad, W. A., Vahjen, W., Aschenbach, J. R. & Zentek, J. A diet naturally contaminated
with the Fusarium mycotoxin deoxynivalenol (DON) downregulates gene expression of
glucose transporters in the intestine of broiler chickens. Livestock Science 140, 72-79
(2011).
94. Richard, J. L., Payne, G. A., eds, Desjardins, A. E., et al. Mycotoxins: risks in plant, animal
and human systems. Council of Food Protective 71, (2003).
95. Fisher, B. T. The Role of Biomarkers for Diagnosis of and Therapeutic Decisions Related
to Invasive Aspergillosis in Children. Curr Fungal Infect Rep 7, 7-14 (2013).
96. Dennehy, P. H. Treatment and prevention of rotavirus infection in children. Curr Infect
Dis Rep 15, 242-250 (2013).
97. Halaihel, N., Liévin, V., Ball, J. M., Estes, M. K., et al. Direct inhibitory effect of rotavirus
NSP4(114-135) peptide on the Na(+)-D-glucose symporter of rabbit intestinal brush
border membrane. J Virol 74, 9464-9470 (2000).
98. Matthews, S. B., Waud, J. P., Roberts, A. G. & Campbell, A. K. Systemic lactose intolerance:
a new perspective on an old problem. Postgrad Med J 81, 167-173 (2005).
99. Dalby-Payne, J. & Elliott, E. Gastroenteritis in children. Clincial Evidence 12, (2004).
100. Hamosh, M. Introduction: Should Infant Formulas Be Supplemented with Bioactive
Components and Conditionally Essential Nutrients Present in Human Milk? The Journal
of nutrition 127, 971S-974S (1997).
101. Heitlinger, L. A., Lee, P. C., Dillon, W. P. & Lebenthal, E. Mammary amylase: a possible
alternate pathway of carbohydrate digestion in infancy. Pediatric research 17, 15-18
(1983).
102. Lorrot, M. & Vasseur, M. How do the rotavirus NSP4 and bacterial enterotoxins lead
differently to diarrhea? Virol J 4, 31 (2007).
103. Usonis, V., Ivaskeviciene, I., Desselberger, U., Rodrigo, C. & Pediatric ROTavirus
European CommiTtee (PROTECT) The unpredictable diversity of co-circulating
rotavirus types in Europe and the possible impact of universal mass vaccination
programmes on rotavirus genotype incidence. Vaccine 30, 4596-4605 (2012).
104. Wolfe, N. D., Dunavan, C. P. & Diamond, J. Origins of major human infectious diseases.
Nature 447, 279-283 (2007).
105. Hagbom, M., Sharma, S., Lundgren, O. & Svensson, L. Towards a human rotavirus
disease model. Curr Opin Virol 2, 408-418 (2012).
106. Swagerty, D. L., Walling, A. D. & Klein, R. M. Lactose intolerance. American family
physician 65, 1845-1860 (2002).
107. Turck, D. Prévention et traitement de la diarrhée aiguë du nourrisson. Archives de
pédiatrie 14, 1375-1378 (2007).

236
108. Chavarro, J. E., Rich-Edwards, J. W., Rosner, B. & Willett, W. C. A prospective study of
dairy foods intake and anovulatory infertility. Hum Reprod 22, 1340-1347 (2007).
109. Vuorisalo, T., Arjamaa, O., Vasemägi, A., Taavitsainen, J. P., et al. High lactose tolerance
in North Europeans: a result of migration, not in situ milk consumption. Perspect Biol
Med 55, 163-174 (2012).

237
Chapter 2. Physical inactivity
Paragraph 2.1. Physical Activity Protects the Human Brain against Metabolic Stress Induced by
a Postprandial and Chronic Inflammation. Pruimboom L, Raison CL, Muskiet FA. Behav
Neurol. 2015. Epub 2015 May 5.
Physical Activity Protects the Human Brain against Metabolic Stress Induced by a
Postprandial and Chronic Inflammation
Leo Pruimboom 1,2, Charles L. Raison 3, and Frits A. J. Muskiet 2
1. Natura Foundation, Edisonstraat 66, 3281 NC Numansdorp, Netherlands
2. Department of Laboratory Medicine, University Medical Center Groningen (UMCG),
University of Groningen, P.O. Box 30.001, 9700 RB Groningen, Netherlands
3. Department of Psychiatry, College of Medicine and Norton School of Family and
Consumer Sciences, College of Agriculture and Life Sciences,University of Arizona,
Tucson, AZ,USA

238
Abstract
In recent years, it has become clear that chronic systemic low-grade inflammation is at the
root of many, if not all, typically Western diseases associated with the metabolic syndrome.
While much focus has been given to sedentary lifestyle as a cause of chronic inflammation, it
is less often appreciated that chronic inflammation may also promote a sedentary lifestyle,
which in turn causes chronic inflammation. Given that even minor increases in chronic
inflammation reduce brain volume in otherwise healthy individuals, the bidirectional
relationship between inflammation and sedentary behaviourmay explain why humans have
lost brain volume in the last 30,000 years and also intelligence in the last 30 years. We review
evidence that lack of physical activity induces chronic low-grade inflammation and,
consequently, an energy conflict between the selfish immune system and the selfish brain.
Although the notion that increased physical activity would improve health in the modern
world is widespread, here we provide a novel perspective on this truism by providing
evidence that recovery of normal human behaviour, such as spontaneous physical activity,
would calm proinflammatory activity, thereby allocating more energy to the brain and other
organs, and by doing so would improve human health.

239
1. Introduction
Chronic inflammatory diseases are a major cause of morbidity and impaired work and social
functioning and are responsable for 35 million to 52 million annual deaths Worldwide (WHO
2014) [1]. A state of low-grade inflammation might be considered the “cause of causes” for
these deadly and disparate conditions. In contrast to inflammatory patterns observed in
hunter-gatherer groups living more in accord with lifestyles that were prototypical across
human evolution and in which inflammatory responses are brisk and time limited (i.e.,
resolving within a maximum of 42 days), in the modern world chronic proinflammatory
activity can last for weeks, months, or even years [2]. Many of the inflammatory diseases that
plague modern societies were uncommon until some 200 years ago [3] but are nowadays
increasingly prevalent. However, treatment is still in its infancy [4], and interventions
addressing the genuine etiologies of the diseases have typically been less than fully
satisfactory [5].
Although the abnormalities that promote allergy, asthma, autoimmunity, and othermore
systemic inflammatory states, such as cardiovascular disease, are usually characterized in
terms of their immune effects, it is less appreciated that the underlying activation of both
innate and adaptive immune inflammatory pathways has costly consequences in terms of
resource use of energy, proteins, and minerals such as calcium [6] andmagnesium [7–9]. In
addition, chronic activation of the immune system produces a constant flux of energy and
blood to the immune system itself, which leads to metabolic stress on other organs and, in
certain circumstances, also the brain. Stress on the brain is unexpected in light of the brain’s
selfish character and humans’ high encephalization quotient, which is the highest of all
mammals and provides evidence of how evolution prioritized energy allocation to the human
brain [10].
Thus, as we discuss in this paper, these metabolic and nutrient imbalances have wide ranging
effects on health-relevant physiological functioning that go beyond the straightforward costs
of immune activation per se. One of the major risk factors for chronic low-grade activity of
the immune systemis frequent and abundant food intake. Postprandial inflammation can put
a high burden on energy availability for the whole body, including the brain, as shown by the
typical “post-lunch dip” in energy and mental activity observed after high-fat and high-
glycemic load meals [11]. Postprandial inflammation increases withmeal size, meal frequency,
and consumption of foul food, and these factors reduce brain growth in mammals, illustrating
the metabolic conflict between the immune system and the brain [12].
Our ancestors experienced rapid and consistent brain growth for 2.5 million years, despite the
likelihood that they frequently consumed spoiled food in the absence of food preservation

240
technology [13]. This pattern of rapid and sustained brain growth despite consumptive
patterns known to activate postprandial inflammation suggests that other mechanisms
inhibited chronic inflammation—and by extension also attenuated postprandial immune
activation. While a number of factors may have contributed to this inhibition, in this review
we will focus on evidence suggesting that preprandial physical activity attenuates
postprandial inflammatory activity while simultaneously protecting from infection via the
induction of immunoglobulins, including lysozymes, and especially, lactoferrin.
In human adults, lactoferrin is produced during and after exercise independent of sex, age,
and menstrual cycle [14]. In infants, lactoferrin is obtained through breast milk, consumed 6 to
7 times daily by newborns. Newborns allocate 74%of their energy intake to brain growth and
differentiation [15–17]; an energy allocation that would be impossible if they experienced a
high-cost bout of postprandial inflammation after every meal. Fortunately, constituents found
in breast milk are capable of downregulating the newborn’s immune system, while they also
protect against possible pathogens. Lactoferrin is probably the most abundant
immunologically active constituent of breast milk that reaches concentrations as high as 8
mg/g in colostrum and 1.5 to 4 mg/g in mature milk [18]. A breastfed newborn who consumes
600 mL of breast milk daily can ingest up to 2.4 grams of lactoferrin daily, an amount that is
probably more than enough to protect against infection, while simultaneously inhibiting
postprandial inflammatory activity of adipocytes and the immune system, thereby saving
energy for constant brain growth.
The hominin brain experienced unprecedented growth despite the challenges that the
immune system faced as our ancestors sought novel experiences and colonized the world.
However, in the last 30,000 years, brain size has shrunk from 1,490mL to 1,350 mL, a loss of
11% [19]. Here we suggest that this reduction in brain size happened because of new
environmental inflammatory factors such as novel pathogens,
high meal frequency, food abundance, and lack of solar radiation but most of all because of
the absence of regular physical activity.

241
2. Energy and Energy Conflict as the Driving Force behind
Evolution
In all animals, energy conflicts shaped physiology during evolution. These conflicts seem to
have caused changes in energy allocation among organs. Nowhere have these conflicts been
more relevant than in the rapid expansion of the brain during hominin evolution [20, 21]. In
principle, a proportionally larger and metabolically expensive brain could have been
supported through an increase in the basal metabolic rate, but no evidence of such a
relationship has been found in humans or other primates [22]. Because of this, several ideas
have been advanced to explain how this larger brain might have been metabolically
supported. For example, the expensive tissue hypothesis [20] and, more recently, various
energy-allocation scenarios [16, 22] propose that the development of the metabolically
expensive human brain must have had consequences for other organs, leading to lower
energy allocation to those organs in favour of the brain.
Navarrete et al. [23] examined evidence for the expensive tissue hypothesis in 100 different
mammalian species, including 23 primates. They found that, controlling for fat free body
mass, brain size is not inversely correlated with the mass of the digestive tract or any other
expensive organ, thus refuting the expensive tissue hypothesis. However, they did find
evidence for a negative correlation between brain size and fat depots in mammals, raising the
intriguing possibility that encephalization and fat storage both evolved as compensatory
strategies to buffer against starvation.When these two strategies are combined, fat might
provide extra energy for the brain during starvation, but only if this fat storage did not
negatively impact locomotor efficiency. Central fat storage (which does not hamper
locomotion efficiency) has probably favoured encephalization, redirecting energy allocation
from growth, reproduction, and high-energy locomotion to brain development and more
efficient locomotion, including during starvation [23].
The observation that human bipedal locomotion and foot anatomy lead to less energy use
than the bipedal or quadrupedal locomotion of chimpanzees is supporting this theory [24].
Bipedal locomotion in humans also facilitates higher central fat depots without major energy
demands during movement [23].
A recent study provides new evidence for the expensive tissue hypothesis [25]. The study
shows that guppies with a bigger brain have smaller guts and produce fewer offspring. This is
in line with the expensive tissue hypothesis and adds a second factor to this hypothesis
relating brain size, gut length, and reproductive capacity [25]. Humans are “under-muscled”
compared with other primates, although this difference is too small to provide sufficient
energy for brain growth [22]. Nevertheless, it is the energy consumed by human muscles
during locomotion which is almost twice as low compared with, for instance, in chimpanzees

242
(332 versus 564 kcal/day) [22] providing enough energy for brain development and
function.The consequence is that humans’ brains are three times bigger than the brains of
chimpanzees, and brain metabolism accounts for 25% of the basal metabolic rate in humans
and only 7 to 8% in other primate species [26].
Overall, there seems to be sufficient scientific support to suggest that the increase of human
brain size and metabolism has been possible because of a change of locomotion, higher
central fat depot storage [27], and (although not addressed in this review) a change of food
intake (see review [24]). In addition, the human brain may have benefitted from a change in
the expression of glucose transporters [28] and the same holds for the immune system. Higher
expression of GLUT1, supporting high and constant glucose uptake, is seen in activated
immune cells, where energy is needed to protect against pathogens and other immune
challenges [29]. This occurs at the expense of GLUT4 that needs insulin for glucose uptake and
is notably expressed in muscles and adipocytes [30–33]. Thus, immune function may have
benefitted from a smaller gut, reduced energy needs for locomotion, increased fat mass, and
tissue specific differences in the expression of glucose transporters.
The work of Fedrigo et al. [28] provides a new explanation for this intriguing “mystery,” that is,
the evidence that during human evolution energy allocation has been directed to brain and
immune system development. They showed that human brain cells express more activity of
the SLC2A1 gene responsible for the production of GLUT1 glucose transporters compared with
the chimpanzee and macaque (human > chimpanzee > macaque). At the same time, SLC2A4
expression (i.e., GLUT4) in muscle is significantly higher in chimpanzee > human >macaque.
Given a certain circulating glucose concentration, an increase in the amount of SLC2A1 or
SLC2A4 protein per gram tissue results in more glucose being captured by that tissue and less
by others. It is important to note that GLUT1 transporters are insulin-independent whereas
GLUT4 in muscles depends on insulin for glucose uptake. An increase in SLC2A1 expression
in the brain and a decrease in SLC2A4 in skeletal muscle would work synergistically to change
the distribution of glucose between these two most energy-demanding tissues in the human
body, in a manner allowing more energy to become available for the brain.

243
3. Glucose to the Immune System: Prioritizing Energy Guidance
A similar line of reasoning explains glucose allocation to the immune system. The energy
demands of lymphocytes and leukocytes increase dramatically upon activation [3, 5] and all
activated immune cells express GLUT1 glucose transporters [29, 34]. Not surprisingly, several
signals and stimuli such as pathogen associated molecular patterns (PAMP), including
lipopolysaccharide (LPS), can promote GLUT1 expression, giving rise to an increase of insulin-
independent glucose transport into all immune cells that supports T-cell receptor stimulation,
immune cell migration, and inflammation [35, 36]. Thus, higher expression of GLUT1
promotes energy allocation to the immune system, which could be considered an “energy
demand reaction,”mobilizing fuel stocks and suppressing the resource demand of other
organs/systems with the overall purpose of preferentially allocating fuels to activated immune
cells [37]. Glucose allocation to the immune system maintains its function even under strong
energy restriction [38].
Metabolic mechanisms and immune control coevolved, conceivably because both processes
are interrelated and essential to survival [39], and both seem to have originated in a single fat
body organ, as can still be seen in Drosophila melanogaster [40, 41]. The integration of
metabolism and immunology persists in higher organisms, in which lymph nodes are
embedded in perinodal adipose tissue that may influence immune responses. In humans,
adipose tissue is well infiltrated with macrophages, and the production of inflammatory
cytokines by both adipocytes and macrophages contributes to systemic inflammation [42].
Activation of the immune system through danger signals utilizes and redistributes energy in a
manner that favours the brain and the immune system. However, prolonged activation of the
immune system, as observed in people with chronic inflammatory states, allocates glucose
chronically to the immune system through immune-controlled down regulation of GLUT1
transporters at the level of the blood-brain barrier and a reduction of GLUT4 transporters at
the level of muscle and adipose tissue [43–45]. This selfish behaviour of the immune system is
responsible for the majority of chronic diseases, if not all [46].
Our hypothesis is that chronic exercise reallocates energy primarily to the brain and muscles,
reducing energy distribution to the immune system, and by doing so recovers metabolic
homeostasis.

244
4. Muscle as a Defence Mechanism in Humans
Muscle is a “forgotten” organ of the immune system. Indeed, the physical activity permitted by
muscles changes the phenotype of immune function from proinflammatory to
antiinflammatory, while maintaining protection against possible lethal pathogens.
Interestingly, chronic exercise is only seen in humans and allowed us to discover and inhabit
widely divergent habitats around the world, while remaining able to survive and reproduce.
Hominins in general and humans in particular tended to seek novelty in their environments,
and this practice would have led them to encounter new pathogens, climatological
challenges, and food scarcity, all of which would have threatened survival and prompted
activation of the immune system. If proinflammatory activity had dominated the biology of
our ancestors when they were challenged with pathogens such as Plasmodium falciparum
malaria, tuberculosis, Salmonella, or pathogenic Escherichia coli, brain growth from 450mL
in early Homo erectus to the current 1,350mL in modern humans would unlikely have been
possible. The energy costs of a consistently active immune system would have been
enormous and would probably have impeded the growth of the brain as well as the body. An
overly active immune system would also have harmed reproduction [47], as evidenced in
modern fertility issues among autoimmune patients [48].
Hominins are not the only organisms in which such a trade-off occurs. Green plants and
hominins probably share one and the same ancestor [49]. The difference is that plants have
chosen an evolutionary path in which locomotion had low or no priority, which implies that
they have not learned to escape from danger in a physical manner [50]. Animals’ ability to
physically flee prevents them from being wounded and thereby from experiencing the
ensuing cytotoxic and self-damaging reaction of the highly expensive innate immune system
[51]. Without the means to physically escape from predators or other direct threats, plants
employ toxins to defend themselves against predators. These toxins {e.g., perforin} are defence
substances similar to those used by animals to kill invading pathogens and are part of the
immune system of the plant [52]. Plants do not have the “migrating” immune cells found in
animals and are therefore dependent on an immune system in each individual cell [53], which
has the potential to be very robust. As a result, in plants a chronic pathogenic load can lead to
overactivity in their equivalent of an immune system causing a change in energy distribution
that favours immune functions at the expense of functions related to growth and
reproduction [54].
Similar to plants, humans and other mammals may experience damage following a robust
reaction of their immune system, such as in sepsis. Long-term and very intense activation of
the immune system in individuals with sepsis can cause loss of lean body mass, tissue
breakdown for use as a source of amino acids and energy, growth impairment, and

245
reproductive disturbances [48, 55]. Even motor and mental functions in later life may be
affected [55, 56]. In addition, a robust inflammatory reaction following acute trauma, such as a
stroke or any other neuro-trauma, often produces severe secondary damage [57]. The strength
of the immune reaction results from the complexity of the human immune system, which has
high receptor diversity that greatly enhances the efficiency of microbial detection. However,
these same qualities also make humans uniquely vulnerable to autoimmune disease (AID)
[58], as high receptor diversity increases the risk of misinterpretation of self-molecules as
foreign, potentially provoking autoimmune responses [59].
4.1. The Sedentary Death Syndrome: The Immune System Takes Over.
The prevalence of autoimmunity is markedly increased in sedentary people [60], who, by
definition, infrequently engage in physical activity. It is interesting to consider whether the
expensive immune reaction seen in sedentary individuals can be considered adaptive in a
manner similar to the strategy plants use to defend against invaders and danger, given that—
like plants—sedentary individuals are less able than others to flee danger.
Exercise has powerful immune effects. Even nonstrenuous exercise, when engaged in
regularly, promotes better health and increases life expectancy [61]. Nonstrenuous exercise
also induces the expression of anti-inflammatory molecules such as lactoferrin and
lysozymes, which provide defence against a large number of invaders, including bacteria,
viruses, and other microbes [62]. A bout of forty five minutes of running at 75% of VO2 max
increases the production of lysozyme and lactoferrin in saliva significantly, independent of
sex and menstrual phase [14]. Serum lactoferrin concentrations also increase immediately
after strenuous exercise, such as running, and may play an antibacterial role in host defences
prior to the mobilization of neutrophils into the circulating pool, thereby attenuating the need
for a possible inflammatory response with high-energy demand [63, 64]. Sedentary
individuals, obese people, and patients with diabetes mellitus type 2 exhibit lower lactoferrin
levels [65, 66], while suffering froman energy-demanding, chronic low-grade inflammation
[10]. All organisms share a need for food intake, and food intake produces postprandial
immune activation as a result of the possible presence of danger signals. This immune
reaction is normally based on the production of inflammation-preventing defence molecules
such as lactoferrin, immunoglobulin A (IgA), and lysozyme, which prevent the high-energy
demands incurred from activation of innate immune cells [65]. However, when this
mechanism fails or is inadequate, as often happens in obese/sedentary individuals, an
inflammatory response ensues [67]. Increasing evidence suggests that this type of
postprandial inflammatory response may contribute to the development of metabolic
syndrome,endothelial dysfunction, cardiovascular diseases, obesity, insulin resistance, and
chronic low-grade inflammation [60, 65].

246
Just as postprandial inflammation is more common in those who are sedentary or obese,
postprandial inflammation is also observed after consumption of a meal that is high in fat,
consists of refined carbohydrates, or contains sugar, fructose, linoleic acid, or other high-
caloric nutrients. Interestingly, a postprandial inflammatory reaction may occur after an even
modestly sized meal that contains cereal-fed meat which should be considered “new” on an
evolutionary time scale [68–70]. The same holds for consumption of a new form of hybridized
beef. The postprandial immune response after ingestion of “new” wagyu beef is significantly
higher than when humans ingest “old” kangaroo meat [70]. All these data raise the intriguing
possibility that the increased immune activity against new nutrients reflects a “borrowed”
ancient immune reaction against evolutionary unknown danger signals, causing low-grade,
systemic, inflammation [70, 71]. Another factor increasing inflammatory activity of the
immune system is chronic food availability.The relationship between food availability and
development of the metabolic syndrome has been investigated in mice [72, 73] and more
recently in humans.
Hatori et al. [72] showed that obesity and metabolic syndrome are caused not only by caloric
content, but also by food being constantly available. Koopman et al. [73] showed that meal
frequency impacts development of fatty liver. Using a 40% hypercaloric diet, they showed that
a high meal frequency (6/day) as compared to a lower frequency (3/day) increases
intrahepatic triglycerides and abdominal fat, independent of caloric content and body weight
gain. Constant food availability further decreases overall physical activity and especially
spontaneous physical activity [74]. In contrast, starvation upregulates glucose transport
through the blood-brain barrier by increasing the number of insulin-independent glucose
transporters
1 (GLUT1) [75], whereas starvation leads to spontaneous physical activity and even
hyperactivity [76].

247
5. Exercise Protects the Body against the Expensive and Damaging
Postprandial Inflammatory Response: Our Hypothesis and Its
Putative Mechanism
Interestingly, exercise prior to meal consumption markedly lowers the postprandial
inflammatory reaction [77]. Evidence suggests that this effect may result from muscle
contraction inducing anti-inflammatory myocytokines (i.e., myokines), including IL-6, IL-15,
and IL-8, that subsequently stimulate production and release of anti-inflammatory molecules
such as lysozyme and lactoferrin by the immune system [64, 78]. The previously mentioned
anti-inflammatory myokines are increased up to 100-fold during exercise [79] and thereby are
responsible for the production of anti-inflammatory molecules such as lactoferrin during and
after physical activity [80]. Elevated concentrations of lactoferrin in serum have been reported
30 minutes after intense running and are associated with elevated serum antibacterial activity
to viable Micrococcus luteus [62]. Furthermore, higher levels of serum lactoferrin have also
been observed two hours after submaximal cycling followed by a bout of eccentric resistance
exercise [81]. The lactoferrin increase appears to be related to intense exercise and eccentric
contraction, which has also been observed in relation to the production of anti-inflammatory
myokines [82]. There seems to be a direct relationship between glycogen depletion and the
production of muscle-derived IL-6, which, despite its inflammatory activities, also
demonstrates anti-inflammatory and metabolic-sensing and regulating properties when
expressed as a muscle myokine [82]. Lactoferrin stimulates the expression of a protein in
adipocytes and in newly identified cells of the innate immune system called six-
transmembrane protein of prostate 2 (STAMP-2) [83].
STAMP-2 links obesity, inflammation, and insulin resistance. When STAMP-2 is activated, the
production of proinflammatory cytokines is inhibited through down regulation of nuclear
factor kappa B {NFkB}, the key transcription factor for activation of genes responsible for the
production of proinflammatory cytokines and enzymes such as IL-1-beta, TNF-alpha, COX2,
and LOX5 [84]. STAMP-2 deficiency can lead to several disorders, including atherosclerosis,
metabolic syndrome, and diabetes [85]. NFkB contributes to the development of insulin
resistance, and STAMP-2 restores insulin sensitivity by inhibiting NFkB [86]. All members of
the STAMP family possess both ferric and cupric reducing activities, which indicate that
STAMP-2 might regulate iron or copper entry into cells [87]. STAMP-2 requires iron or copper
as cofactors, suggesting that STAMP-2 is needed to maintain metabolic homeostasis. STAMP-
2 further protects against atherosclerosis and stabilizes plaques in diabetic mice [88].
Decreased activity of STAMP-2 and similar proteins creates a signalling bottleneck that leads
to insulin resistance [86, 88]. Lactoferrin is an “iron” carrying protein, which could explain its
activating function on STAMP-2 [89]. Considering all these factors, it seems plausible that

248
preprandial (before meal intake) exercise induces the production of anti-inflammatory
myokines and that these myokines stimulate neutrophils to produce lactoferrin [89].
The release of lactoferrin into the circulation activates visceral adipocytes, which
subsequently react with increased expression of STAMP-2 [83]. STAMP-2, in turn, inhibits
NFkB and Janus kinase (JNK) activation and prevents the inhibition of insulin signalling.

249
6. Physical Activity Was Spontaneous during Evolution and Causes
a Phenotypical Shift of the Immune Response during Immune
Challenges
That exercise prevents postprandial inflammation makes good sense from an evolutionary
perspective. From 2 million years ago until approximately 200 years ago, common threats to
human health included starvation, dehydration, predation, climate, accidents, violence, and
infectious disease [90]. With the exception of infectious disease, all these threats induced
spontaneous activity (SPA) through activation of dopaminergic neuroanatomic nuclei in the
brain, including the ventral tegmentum and the striatum [75]. The major neuropeptide
systems that have been studied relative to spontaneous physical activity include
cholecystokinin, corticotropin-releasing hormone, neuromedin U, neuropeptide Y, leptin,
agouti-related protein, orexins, and ghrelin. All these systems influence dopaminergic
signalling [91]. SPA stimulates the production of anti-inflammatory myokines by energy
depletion of the contracting muscles. As mentioned earlier, these myokines drive neutrophils
to produce antimicrobial/anti-inflammatory molecules {e.g., lactoferrin} and STAMP-2 in
adipocytes and cells of the innate immune system, thus preventing over-activation of NFkB
and the subsequent proinflammatory/insulin resistance response. The cascade of
neurochemical reactions in the brain, when faced with old danger factors such as starvation,
prompted patterns of SPA that reflected the daily lives of our ancestors.
To starve off hunger and obtain food, both men and women often engaged in fishing, while
men also engaged in leg-based long-term hunting and women employed arm based
gathering. Our ancestor’s use of the upper body during physical activity is an important factor
in stimulating an anti-inflammatory reaction. Upper body muscles are energetically more
efficient than lower body muscles and are normally conserved even during severe metabolic
conflicts, as observed in people suffering from chronic obstructive pulmonary disorders
(COPD) [92, 93]. Preservation of upper body muscles may seem counterintuitive considering
that scientists unequivocally recognize horizontal running as the evolutionarily conserved
flight direction and considering that the maximum running speed of predators such as lions,
tigers, and jaguars is by far much higher than the maximum speed of the fastest human.
Conceivably, conserving upper body muscles during metabolically stressful situations may
have occurred because humans could more easily escape threats by climbing/fighting than by
running, making maintenance of the upper body muscles a priority. This view is supported by
the observation that energy is first allocated to arm muscles during fear situations,
manifesting in warm, sweating hands and an instantaneous increase of circulation [94–96].
Palm sweating has a surprising benefit; hands are important parts of the body to eliminate

250
excessive body heat during exercise and increased heat elimination through palm sweating
augments exercise resistance and prevents fatigue. Use of the upper body is also likely to have
led to optimal levels of IL-6, which is anti-inflammatory only when produced in small
amounts. These amounts depend on the amount of metabolic stress on contracting muscles
during exercise, which is significantly less when using the upper body for fishing, digging,
and other gathering functions. Our ancestors were merely gatherers/fishers and part-time
hunters, and they therefore often relied on their upper body, thus producing the optimal anti-
inflammatory cocktail of myokines to cause an anti-inflammatory postprandial response.
As a result, although our ancestors suffered from stress, these patterns of activity made them
less likely than most modern humans to experience a metabolically expensive inflammatory
response of the innate immune system and subsequent low-grade inflammation-based
disorders. The energy thereby conserved would have been used to feed the metabolically
expensive brain, and the absence of immune stress induced insulin resistance would prevent
the development of neurodegenerative diseases and other maladies affecting the central
nervous system in general, but especially the brain [97].

251
7. Spontaneous Physical Activity and the Immune System
Evidence that SPA is caused by ancient stress factors such as food scarceness comes from
research with people suffering from anorexia nervosa and individuals with obesity, that is, the
opposite phenotype [74]. However, SPA and even hyperactivity in periods of reduced food
availability are not unique to humans: animals need to be active to search for more food if the
food supply is limited, as is commonly the case in the natural habitat. Already in 1922, the
psychologist Curt Richter observed that if food is served for a limited period of time, the meal
is preceded by an increase in physical activity [98]. This phenomenon is referred to as “food
anticipatory activity” {FAA}, and the biological explanation for FAA preceding meals is similar
to the above-mentioned evolutionary approach to human SPA in starvation.
The SPA pathway, activated through food restriction, seems to be dependent on orexin and
dopamine [74]. Activation of orexin receptors leads to an increase in physical activity, while
orexin is released in response to food restriction [99]. Orexin-producing hypothalamic
neurons project on dopaminergic neurons in the ventral tegmentum, which are highly
involved in SPA [100]. The same is true for the gut-derived orexigenic hormone ghrelin, which
also activates dopaminergic pathways in the ventral tegmentum and induces SPA [101].
Dopaminergic pathways responsible for spontaneous physical activity have gained
importance during evolution. These dopaminergic neurons belong to the group of the so-
called emotional motor neurons [102–104] and these neurons are part of the behavioural
column.
The behavioural column and its emotional motor neurons literally facilitate muscle
contraction through “gain setting” pathways and motivation. It can be concluded that
motivated activity is energetically less costly (“cheap”) than voluntary exercise and motivated
activity is dopamine dependent [105]. Comparison of the human substantia nigra and ventral
tegmentum with those of other mammals or vertebrates revealed tremendous differences in
the number of dopaminergic neurons.
For example, lizards { Gekko} have about 2,000 dopaminergic neurons and turtles (
Pseudemys) about 5,500, while rats have 45,000 dopaminergic neurons, macaques 165,000,
and humans 590,000 in the first four decades of their lives [106]. Vernier states: “The large
number of dopaminergic neurons in humans is remarkable given that the human brain is
only three times larger than that of the macaque. Normally, the number of dopaminergic
neurons correlates closely with body weight, but humans are a clear exception. This
peculiarity is even more striking because a single dopaminergic neuron of the substantia
nigra or ventral tegmental area (VTA) sends many collaterals with large arborization trees to
several areas of the forebrain. The projections of these neurons are essentially similar among
the different vertebrate species, though they tend to abundantly innervate the striatal areas

252
and, less abundantly, pallial {cortical} areas, under-scoring their role in the control of incentive
and sensorimotor behaviours. Details of these connections are well described only in
mammals and especially in humans [107]”. As mentioned before, the dopaminergic motor
system belongs to the behavioural column defined by Swanson [103], and this system
facilitates and permits behaviour [51].
The opposite response is observed in people with a variety of diseases such as fibromyalgia
syndrome, multiple sclerosis, panic disorder, obesity, and Parkinson’s disease, all of whom
show non-permissive behaviour such as exercise avoidance [108]. Dopamine-induced
preprandial SPA firstly saves energy for brain function and secondly helps to induce a
protective anti-inflammatory response of the immune system after food intake through
upregulation of the production of lactoferrin and its accumulation in dopaminergic neurons
might protect against neurodegeneration and Parkinson’s disease [109]. As mentioned before,
several specific aspects of human locomotion have made it possible to save energy for the
brain [23], but this is not the only way the brain gained access to more resources to grow and
develop during hominin evolution. Chronic exercise shaped the immune system, creating a
highly protective anti-inflammatory phenotype that maintains protection against possibly
lethal invaders while saving energy [82].

253
8. Physical Activity: Part of the Combined and Coordinated
Neuroendocrine-Immunological Stress Response
Another intriguing effect of spontaneous or voluntary exercise is related to its ability to
convert dopamine to codeine and morphine, although in low amounts [110]. Codeine and
morphine are known for their analgesic effects, but much less is known about their influence
on the immune system. Endogenous codeine and morphine induce constitutive nitric oxide
synthetase (cNOS) in immune cells. The resulting NO inhibits mitochondrial energy (ATP)
production in white blood cells and thereby immune inflammatory activity [111]. Voluntary
motivated exercise also produces the immunoregulatory/anti-inflammatory cytokine IL-10,
which may further elicit anti-inflammatory effects through dopamine-regulated physical
activity [112, 113]. This anti-inflammatory response might be dangerous when the bacterial
load is so high that the host would be in serious infectious danger when the immune system
would maintain an anti-inflammatory activity. It is in this context not surprising that recent
research showed that when the pathogenic load is high, the immune response will maintain
proinflammatory capacity and drive sickness behaviour and thereby outweigh the anti-
inflammatory potential of voluntary exercise [114]. This observation in mice further supports
the selfish character of the immune system.
Taken together, both the emergence and function of dopamine, morphine, and nitric oxide
seem to be related to evolutionary pressures to maintain chronic physical activity (for an
excellent review see [115]). The combined production of lactoferrin, anti-inflammatory
myokines, and IL-10 and also the dopamine/morphine/NO triad through motivated and
spontaneous exercise make it at least plausible that SPA has driven the immune system to an
anti-inflammatory phenotype, thereby saving energy for the brain and the muscles, which is
in support of our hypothesis and observed mechanisms.

254
9. Physical Activity Protects against the Damaging Effect of the
Proinflammatory Activity of the Selfish Immune System: Clinical
Evidence
Clinical research in patients suffering from different disorders of the central nervous system
supports the notion that physical activity protects against proinflammatory activity. Patients
with neuroinflammatory diseases such as amyotrophic lateral sclerosis (ALS) react positively
to exercise [116], probably through down regulation of the immune response directed against
brain tissue. Patients with Parkinson’s disease who engage in “forced” exercise show
significant improvement of typical Parkinson symptomatology and less progression of the
disease [117]. These and other neurodegenerative and many other diseases seem to be related
to disturbances in energy metabolism in the brain [118] and also to energy distribution
between the central nervous system and the peripheral organs [119], all caused, at least in part,
by a chronically activated immune system [10, 120].
In summary, SPA and recreational voluntary exercise seems to override the chronically active
immune system [121] and reduce its selfish behaviour. Strenuous obligatory exercise produces
immune suppression, possibly leading to increased susceptibility to infection [114, 122],
although recent human research shows that the immune system reactivates when challenged
with a high load of pathogens in people engaging in strenuous exercise. Physical activity
{ancestors = SPA, contemporary = voluntary} induces anti-inflammatory myokines which
inform the immune system and literally relax it. A relaxed immune system (which is distinct
from “suppressed”) only reacts to the danger signals it was designed to respond to, such as
bacteria and viruses, which conserves energy for the benefit of phylogenetically younger
organs such as the liver, kidneys, and brain and gives rise to normal behaviour and a so-called
permissive brain phenotype. A subject’s permissive brain literally facilitates free will, as
opposed to those suffering from a non-permissive brain syndrome [51, 123–126].
Behavioural changes have high energetic cost and lack of brain energy prevents flexibility and
loss of free will. Spontaneous and voluntary activity can also be considered a strategy
belonging to the proactive behavioural immune system. Avoiding danger, psycho-emotional
problems, social contact with infected individuals, dirt, and other unknown, possibly
dangerous, triggers relax the expensive reactive immune system, once again providing
energy for the human brain. Recent research in insects shows that an acute flight/fight
reconfigures the type of immune reaction from a pro- to an anti-inflammatory phenotype,
just as in humans. Once more, it is this low-cost anti-inflammatory immunological response
that protects against infection and saves energy for brain development and function. In
contrast, the sedentary lifestyle, a disease in itself, demands a proinflammatory response of

255
the immune system and chronic allocation of energy/resources to this system. Low-grade
inflammation is the ultimate consequence and the cause of causes of most, if not all,
noncommunicable chronic diseases [46].

256
10. Conclusions
Because regular muscle contraction diminishes the need for proinflammatory activity of the
innate immune system [127], we suggest that muscles should be considered part of the
immune system [128].The phylogenetic development of muscles, together with that of human
locomotion, increased fat storage. Differences in glucose transporters between muscle and
the brain have been responsible for patterns of energy allocation that enabled the brain to
grow to its evolutionary maximum of 1,490mL approximately 30,000 years ago.
Throughout hominin evolution, our ancestors encountered dangerous situations, such as
violence and dehydration, that threatened the survival of the individual or the species and
usually triggered a survival response in which more ancient systems tend to dominate
younger ones [129], although brain functions and anatomy seem to maintain the highest
priority.
The normal response to survival threats is SPA, and SPA is capable of controlling the immune
system by saving energy for the brain during stressful situations [130]. The absence of SPA or
voluntary exercise, as is occurring in sedentary people living modern lifestyles, provides a
framework in which the immune system overrides the interests of the normally selfish brain,
which could be the reason why brain size shrank in the last 30,000 years from the maximum
of 1,490mL to the current average of 1,350 mL.
Conflict of Interests
Dr. Raison reports the following activities in the prior 12 months: consulting for
Pamlab,Merck, and Otsuka; speaker’s bureau for Pamlab and Sunovion; delivery of nondisease
state presentations for Merck and Otsuka; steering committee membership for North
American Center for Continuing Medical Education (NACCME); preparation and delivery of
continuing medical education material for NACCME, Haymarket, and Medscape.

257
References
1. O.Matheson,M. Kl¨ugl, L. Engebretsen et al., “Prevention and management of non-
communicable disease: the IOC consensus statement, Lausanne 2013,” British Journal
of Sports Medicine, vol. 47, no. 16, pp. 1003–1011, 2013.
2. R. H. Straub, “Evolutionary medicine and chronic inflammatory state—known and new
concepts in pathophysiology,” Journal of Molecular Medicine, vol. 90, no. 5, pp. 523–
534, 2012.
3. R. H. Straub, “Concepts of evolutionary medicine and energy regulation contribute to
the etiology of systemic chronic inflammatory diseases,” Brain, Behavior, and
Immunity, vol. 25, no. 1, pp. 1–5, 2011.
4. M.M. Bosma-Den Boer,M.-L. vanWetten, and L. Pruimboom, “Chronic inflammatory
diseases are stimulated by current lifestyle: howdiet, stress levels and medication
prevent our body from recovering,” Nutrition and Metabolism, vol. 9, no. 1, article 32,
2012.
5. R. H. Straub, F. Buttgereit, and M. Cutolo, “Alterations of the hypothalamic-pituitary-
adrenal axis in systemic immune diseases—a role for misguided energy regulation,”
Clinical and Experimental Rheumatology, vol. 29, no. 5, pp. S23–S31, 2011.
6. R. H. Straub,M.Cutolo, F. Buttgereit, andG. Pongratz, “Energy regulation and
neuroendocrine-immune control in chronic inflammatory diseases,” Journal of Internal
Medicine, vol. 267, no. 6, pp. 543–560, 2010.
7. P. P. Cavicchia, S. E. Steck, T. G. Hurley et al., “A new dietary inflammatory index predicts
interval changes in serum high sensitivity C-reactive protein,” Journal of Nutrition, vol.
139, no. 12, pp. 2365–2372, 2009.
8. M. Otto, “Bacterial sensing of antimicrobial peptides,” Contributions to Microbiology,
vol. 16, pp. 136–149, 2009.
9. M. J. Laires, C. P. Monteiro, and M. Bicho, “Role of cellular magnesium in health and
human disease,” Frontiers in Bioscience, vol. 9, pp. 262–276, 2004.
10. B. Ruiz-Nunez, L. Pruimboom, D. A. J. Dijck-Brouwer, and F.A.J. Muskiet, “Lifestyle and
nutritional imbalances associated with Western diseases: causes and consequences of
chronic systemic low-grade inflammation in an evolutionary context,” Journal of
Nutritional Biochemistry, vol. 24, no. 7, pp. 1183–1201, 2013.
11. T. H. Monk, “The post-lunch dip in performance,” Clinics in Sports Medicine, vol. 24, no.
2, pp. e15–e23, 2005.
12. K. Fonseca-Azevedo and S. Herculano-Houzel, “Metabolic constraint imposes trade off
between body size and number of brain neurons in human evolution,” Proceedings of
the National Academy of Sciences of the United States of America, vol. 109, no. 45, pp.
18571–18576, 2012.

258
13. L. Pruimboom, T. Fox, and F. A. J.Muskiet, “Lactase persistence and augmented salivary
alpha-amylase gene copy numbers might have been selected by the combined toxic
effects of gluten and (food born) pathogens,” Medical Hypotheses, vol. 82, no. 3, pp. 326–
334, 2014.
14. T. Gillum, M. Kuennen, T. Miller, and L. Riley, “The effects of exercise, sex, and menstrual
phase on salivary antimicrobial proteins,” Exercise Immunology Review, vol. 20, pp. 23–
38, 2014.
15. S. M. Innis, “Impact of maternal diet on human milk composition and neurological
development of infants,” American Journal of Clinical Nutrition, vol. 99, no. 3, pp. 34S–
41S, 2014.
16. W. R. Leonard, M. L. Robertson, J. J. Snodgrass, and C. W. Kuzawa, “Metabolic correlates
of hominid brain evolution,” Comparative Biochemistry and Physiology A: Molecular &
Integrative Physiology, vol. 136, no. 1, pp. 5–15, 2003.
17. S. C. Cunnane and M. A. Crawford, “Survival of the fattest: fat babies were the key to
evolution of the large human brain,” Comparative Biochemistry and Physiology Part A:
Molecular & Integrative Physiology, vol. 136, no. 1, pp. 17–26, 2003.
18. F. Berlutti,A. Pilloni,M. Pietropaoli,A. Polimeni, and P.Valenti, “Lactoferrin and oral
diseases: current status and perspective in periodontitis,” Annali Di Stomatologia, vol. 2,
no. 3-4, p. 10, 2011.
19. J. Hawks, “Selection for smaller brains in holocene human evolution,”
http://arxiv.org/abs/1102.5604v1.
20. L. C. Aiello and P. Wheeler, “The expensive-tissue hypothesis: the brain and the digestive
system in human and primate evolution,” Current Anthropology, vol. 36, no. 2, pp. 199–
221, 1995.
21. K. Milton, “A hypothesis to explain the role of meat-eating in human evolution,”
Evolutionary Anthropology, vol. 8, no. 1, pp. 11–21, 1999.
22. K. Isler and C. van Schaik, “Costs of encephalization: the energy trade-off hypothesis
tested on birds,” Journal of Human Evolution, vol. 51, no. 3, pp. 228–243, 2006.
23. A. Navarrete, C. P. van Schaik, and K. Isler, “Energetics and the evolution of human brain
size,” Nature, vol. 480, no. 7375, pp. 91–93, 2011.
24. H. Pontzer and J. M. Kamilar, “Great ranging associated with greater reproductive
investment in mammals,” Proceedings of the National Academy of Sciences of the
United States of America, vol. 106, no. 1, pp. 192–196, 2009.
25. A. Kotrschal, B. Rogell, A. Bundsen et al., “Artificial selection on relative brain size in the
guppy reveals costs and benefits of evolving a larger brain,” Current Biology, vol. 23, no.
2, pp. 168– 171, 2013.

259
26. W. R. Leonard, J. J. Snodgrass, and M. L. Robertson, “Effects of brain evolution on
human nutrition and metabolism,” Annual Review of Nutrition, vol. 27, pp. 311–327,
2007.
27. S. Cunnane, S. Nugent, M. Roy et al., “Brain fuel metabolism, aging, and Alzheimer’s
disease,” Nutrition, vol. 27, no. 1, pp. 3 – 20, 2011.
28. O. Fedrigo, A.D. Pfefferle, C. C. Babbitt, R.Haygood, C. E.Wall, and G. A. Wray, “Apotential
role for glucose transporters in the evolution of human brain size,” Brain, Behavior and
Evolution, vol. 78, no. 4, pp. 315–326, 2011.
29. N. J. MacIver, S. R. Jacobs, H. L. Wieman, J. A. Wofford, J. L. Coloff, and J. C. Rathmell,
“Glucose metabolism in lymphocytes is a regulated process with significant effects on
immune cell function and survival,” Journal of Leukocyte Biology, vol. 84, no. 4, pp.
949–957, 2008.
30. J. A.Woods, “Physical activity, exercise, and immune function,” Brain, Behavior, and
Immunity, vol. 19, no. 5, pp. 369–370, 2005.
31. M.Uldry and B.Thorens, “TheSLC2 family of facilitated hexose and polyol transporters,”
Pfl¨ugersArchiv, vol. 447, no. 5,pp. 480– 489, 2004.
32. M. A. Herman and B. B. Kahn, “Glucose transport and sensing in the maintenance of
glucose homeostasis and metabolic harmony,” The Journal of Clinical Investigation, vol.
116, no. 7, pp. 1767–1775, 2006.
33. S. Huang and M. P. Czech, “The GLUT4 glucose transporter,” Cell Metabolism, vol. 5, no.
4, pp. 237–252, 2007.
34. I. Wolowczuk, C. Verwaerde, O. Viltart et al., “Feeding our immune system: impact on
metabolism,” Clinical and Developmental Immunology, vol. 2008, Article ID 639803, 19
pages, 2008.
35. V. A. Gerriets and J. C. Rathmell, “Metabolic pathways in T cell fate and function,” Trends
in Immunology, vol. 33,no. 4, pp. 168– 172, 2012.
36. J. C. Rathmell, C. J. Fox, D. R. Plas, P. S. Hammerman, R. M. Cinalli, and C. B.Thompson,
“Akt-directed glucose metabolism can prevent Bax conformation change and promote
growth factor-independent survival,” Molecular and Cellular Biology, vol. 23, no. 20, pp.
7315–7328, 2003.
37. G. Pacheco-L´opez and F. Berm´udez-Rattoni, “Brain-immune interactions and the
neural basis of disease-avoidant ingestive behaviour,” Philosophical Transactions of the
Royal Society B: Biological Sciences, vol. 366, no. 1583, pp. 3389–3405, 2011.
38. C. M. Spies, R. H. Straub, and F. Buttgereit, “Energy metabolism and rheumatic diseases:
from cell to organism,” Arthritis Research &Therapy, vol. 14, no. 3, article 216, 2012.
39. G. S. Hotamisligil and E. Erbay, “Nutrient sensing and inflammation in metabolic
diseases,” Nature Reviews Immunology, vol. 8, no. 12, pp. 923–934, 2008.

260
40. G. S. Hotamisligil, “Inflammation and metabolic disorders,” Nature, vol. 444, no. 7121, pp.
860–867, 2006.
41. V. Leclerc and J.-M. Reichhart, “The immune response of Drosophila melanogaster,”
Immunological Reviews, vol. 198, no. 1, pp. 59–71, 2004.
42. M. A. Fitzpatrick and S. P. Young, “Metabolomics—a novel window into inflammatory
disease,” Swiss Medical Weekly, vol. 143, Article ID e13743, 2013.
43. P. M. Abdul Muneer, S. Alikunju, A. M. Szlachetka, L. C. Murrin, and J. Haorah,
“Impairment of brain endothelial glucose transporter by metham-phetamine causes
blood-brain barrier dysfunction,”Molecular Neurodegeneration, vol. 6, no. 1, article 23,
2011.
44. W. Wang, J.-T. Yu, W. Zhang et al., “Genetic association of SLC2A14 polymorphism with
Alzheimer’s disease in a Han Chinese population,” Journal of Molecular Neuroscience,
vol. 47, no. 3, pp. 481–484, 2012.
45. A. Peters, “The energy request of inflammation,” Endocrinology, vol. 147, no. 10, pp.
4550–4552, 2006.
46. B. Ruiz-Nunez, R. S. Kuipers, M. F. Luxwolda et al., “Saturated fatty acid (SFA) status and
SFA intake exhibit different relations with serum total cholesterol and lipoprotein
cholesterol: a mechanistic explanation centered around lifestyle-induced low-grade
inflammation,” Journal of Nutritional Biochemistry, vol. 25, no. 3, pp. 304–312, 2014.
47. E. T. Abrams and E.M.Miller, “The roles of the immune system in Women’s reproduction:
evolutionary constraints and life history trade-offs,” American Journal of Physical
Anthropology, vol. 146, supplement 53, pp. 134–154, 2011.
48. H. J. A. Carp, C. Selmi, and Y. Shoenfeld, “The autoimmune bases of infertility and
pregnancy loss,” Journal of Autoimmunity, vol. 38, no. 2-3, pp. J266–J274, 2012.
49. N. H. Bishopric, “Evolution of the heart from bacteria to man,” Annals of the New York
Academy of Sciences, vol. 1047, pp. 13–29, 2005.
50. T.Hunt, K. Nasmyth, and B.Nov´ak, “The cell cycle,” Philosophical Transactions of the
Royal Society B: Biological Sciences, vol. 366, no. 1584, pp. 3494–3497, 2011.
51. L. Pruimboom, “Physical inactivity is a disease synonymous for a non-permissive brain
disorder,” Medical Hypotheses, vol. 77, no. 5, pp. 708–713, 2011.
52. J. D. G. Jones and J. L. Dangl, “The plant immune system,” Nature, vol. 444, no. 7117, pp.
323–329, 2006.
53. L. Cordain, “Cereal grains: humanity’s double-edged sword,” World Review of Nutrition
and Dietetics, vol. 84, pp. 19–73, 1999.
54. S. I. Kwon, S. H. Kim, S. Bhattacharjee, J.-J. Noh, and W. Gassmann, “SRFR1, a suppressor
of effector-triggered immunity, encodes a conserved tetratricopeptide repeat protein
with similarity to transcriptional repressors,” Plant Journal, vol. 57, no. 1, pp. 109–119,
2009.

261
55. R. Dantzer, J. C. O’Connor, G. G. Freund, R. W. Johnson, and K. W. Kelley, “From
inflammation to sickness and depression: when the immune system subjugates the
brain,” Nature Reviews Neuroscience, vol. 9, no. 1, pp. 46–56, 2008.
56. M. van der Ree, J. C. Tanis, K. N. J. A. Van Braeckel, A. F. Bos, and E. Roze, “Functional
impairments at school age of preterm born children with late-onset sepsis,” Early
Human Development, vol. 87, no. 12, pp. 821–826, 2011.
57. D. J. Loane and K. R. Byrnes, “Role of microglia in neurotrauma,”Neurotherapeutics, vol.
7, no. 4, pp. 366–377, 2010.
58. R. M. Nesse, S. C. Stearns, and G. S. Omenn, “Medicine needs evolution,” Science, vol.
311, no. 5764, p. 1071, 2006.
59. H. Schulenburg, J. Kurtz, Y.Moret, andM. T. Siva-Jothy, “Introduction. Ecological
immunology,” Philosophical Transactions of the Royal Society B: Biological Sciences,
vol. 364, no. 1513, pp. 3– 14, 2009.
60. A. M. Cuevas and A. M. Germain, “Diet and endothelial function,” Biological Research,
vol. 37, no. 2, pp. 225–230, 2004.
61. L. Ferrucci, G. Izmirlian, S. Leveille et al., “Smoking, physical activity, and active life
expectancy,” American Journal of Epidemiology, vol. 149, no. 7, pp. 645–653, 1999.
62. H. Inoue, M. Sakai, Y. Kaida, and K. Kaibara, “Blood lactoferrin release induced by
running exercise in normal volunteers: antibacterial activity,” Clinica Chimica Acta, vol.
341, no. 1-2, pp. 165–172, 2004.
63. N. P. West, D. B. Pyne, J. M. Kyd, G. M. Renshaw, P. A. Fricker, and A. W. Cripps, “The
effect of exercise on innate mucosal immunity,” British Journal of Sports Medicine, vol.
44, no. 4, pp. 227–231, 2010.
64. N. P. Walsh, M. Gleeson, D. B. Pyne et al., “Position statement part two: maintaining
immune health,” Exercise Immunology Review, vol. 17, pp. 64–103, 2011.
65. J. M. Fernandez-Real, E. Garcıa-Fuentes, J. M. Moreno- Navarrete et al., “Fat overload
induces changes in circulating lactoferrin that are associated with postprandial lipemia
and oxidative stress in severely obese subjects,” Obesity, vol. 18, no. 3, pp. 482–488, 2010.
66. J. M. Moreno-Navarrete, F. J. Ortega, J. Bassols, W. Ricart, and J. M. Fernandez-Real,
“Decreased circulating lactoferrin in insulin resistance and altered glucose tolerance as a
possible marker of neutrophil dysfunction in type 2 diabetes,” Journal of Clinical
Endocrinology andMetabolism, vol. 94,no. 10,pp. 4036–4044, 2009.
67. J. Holmer-Jensen, T. Karhu, L. S.Mortensen, S. B. Pedersen, K.- H. Herzig, and K.
Hermansen, “Differential effects of dietary protein sources on postprandial low-grade
inflammation after a single high fat meal in obese non-diabetic subjects,” Nutrition
Journal, vol. 10, no. 1, article 115, 2011.

262
68. J. H. O’Keefe, N. M. Gheewala, and J. O. O’Keefe, “Dietary strategies for improving post-
prandial glucose, lipids, inflammation, and cardiovascular health,” Journal of the
American College of Cardiology, vol. 51, no. 3, pp. 249–255, 2008.
69. L. Dossus and R. Kaaks, “Nutrition, metabolic factors and cancer risk,” Best Practice and
Research: Clinical Endocrinology and Metabolism, vol. 22, no. 4, pp. 551–571, 2008.
70. F. Arya, S. Egger, D. Colquhoun, D. Sullivan, S. Pal, and G. Egger, “Differences in
postprandial inflammatory responses to a ‘modern’ v. traditional meat meal: a
preliminary study,” British Journal of Nutrition, vol. 104, no. 5, pp. 724–728, 2010.
71. G. J. Egger, A. F. Binns, and S. R. Rossner, “The emergence of ‘lifestyle medicine’ as a
structured approach for management of chronic disease,” Medical Journal of Australia,
vol. 190, no. 3, pp. 143–145, 2009.
72. M. Hatori, C. Vollmers, A. Zarrinpar et al., “Time-restricted feeding without reducing
caloric intake prevents metabolic diseases in mice fed a high-fat diet,” CellMetabolism,
vol. 15, no. 6, pp. 848–860, 2012.
73. K. E. Koopman, M. W. Caan, A. J. Nederveen et al., “Hypercaloric diets with increased
meal frequency, but not meal size, increase intra-hepatic triglycerides: a randomized
controlled trial,” Hepatology, vol. 60, no. 2, pp. 545–553, 2014.
74. A. J.W. Scheurink, G. J. Boersma, R.Nerg˚ardh, and P. S¨odersten, “Neurobiology of
hyperactivity and reward: agreeable restlessness in anorexia nervosa,” Physiology &
Behavior, vol. 100, no. 5, pp. 490–495, 2010.
75. N. J. Abbott, L. R¨onnb¨ack, and E. Hansson, “Astrocyte endothelial interactions at the
blood-brain barrier,” Nature Reviews Neuroscience, vol. 7, no. 1, pp. 41–53, 2006.
76. P. Sodersten, R. Nergardh, C. Bergh, M. Zandian, and A. Scheurink, “Behavioral
neuroendocrinology and treatment of anorexia nervosa,” Frontiers in
Neuroendocrinology, vol. 29, no. 4, pp. 445–462, 2008.
77. O. J. MacEneaney, M. Harrison, D. J. O’Gorman, E. V. Pankratieva, P. L. O’Connor, and N.
M. Moyna, “Effect of prior exercise on postprandial lipemia and markers of inflammation
and endothelial activation in normal weight and overweight adolescent boys,” European
Journal of Applied Physiology, vol. 106, no. 5, pp. 721–729, 2009.
78. B. K. Pedersen, “Muscles and their myokines,” Journal of Experimental Biology, vol. 214,
no. 2, pp. 337–346, 2011.
79. B. K. Pedersen and C. P. Fischer, “Beneficial health effects of exercise—the role of IL-6 as
a myokine,” Trends in Pharmacological Sciences, vol. 28, no. 4, pp. 152–156, 2007.
80. N. C. Bishop and M. Gleeson, “Acute and chronic effects of exercise on markers of
mucosal immunity,” Frontiers in Bioscience, vol. 14, no. 12, pp. 4444–4456, 2009.
81. R. A. Fielding, M. A. Violan, L. Svetkey et al., “Effects of prior exercise on eccentric
exercise-induced neutrophilia and enzyme release,” Medicine and Science in Sports and
Exercise, vol. 32, no. 2, pp. 359–364, 2000.

263
82. B. K. Pedersen and M. A. Febbraio, “Muscles, exercise and obesity: skeletal muscle as a
secretory organ,” Nature Reviews Endocrinology, vol. 8, no. 8, pp. 457–465, 2012.
83. J. M.Moreno-Navarrete, F.Ortega, M. Serrano et al., “Decreased STAMP2 expression in
association with visceral adipose tissue dysfunction,” Journal of Clinical Endocrinology
and Metabolism, vol. 96, no. 11, pp. E1816–E1825, 2011.
84. J.Hirosumi,G. Tuncman, L. Chang et al., “Acentral role for JNK in obesity and insulin
resistance,” Nature, vol. 420, no. 6913, pp. 333–336, 2002.
85. H. ten Freyhaus, E. S. Calay, A. Yalcin et al., “Stamp2 controls macrophage inflammation
through nicotinamide adenine dinucleotide phosphate homeostasis and protects
against atherosclerosis,” Cell Metabolism, vol. 16, no. 1, pp. 81–89, 2012.
86. A. Abedini and S. E. Shoelson, “STAMPing out insulin resistance?” Immunology and Cell
Biology, vol. 85,pp. 399–400, 2007.
87. R. S. Ohgami, D. R. Campagna, A. McDonald, and M. D. Fleming, “The Steap proteins
aremetalloreductases,” Blood, vol. 108, no. 4, pp. 1388–1394, 2006.
88. J.Wang, L.Han, Z.-H.Wang et al., “Overexpression of STAMP2 suppresses atherosclerosis
and stabilizes plaques in diabetic mice,” Journal of Cellular and Molecular Medicine, vol.
18, no. 4, pp. 735–748, 2014.
89. J. M. Moreno-Navarrete, F. Ortega, M. Sabater, W. Ricart, and J. M. Fernandez-Real,
“Proadipogenic effects of lactoferrin in human subcutaneous and visceral
preadipocytes,” Journal of Nutritional Biochemistry, vol. 22, no. 12, pp. 1143–1149, 2011.
90. G. P. Chrousos, “Stress and disorders of the stress system,” Nature Reviews
Endocrinology, vol. 5, no. 7, pp. 374–381, 2009.
91. T. Garland Jr., H. Schutz, M. A. Chappell et al., “The biological control of voluntary
exercise, spontaneous physical activity and daily energy expenditure in relation to
obesity: human and rodent perspectives,” The Journal of Experimental Biology, vol. 214,
part 2, pp. 206–229, 2011.
92. E. F. Porto, A. A. M. Castro, M. Velloso, O. Nascimento, F. Dal Maso, and J. R. Jardim,
“Exercises using the upper limbs hyperinflate COPD patients more than exercises using
the lower limbs at the same metabolic demand,” Monaldi Archives for Chest Disease—
Pulmonary Series, vol. 71, no. 1, pp. 21–26, 2009.
93. C. J. Clark, L. M. Cochrane, E. Mackay, and B. Paton, “Skeletal muscle strength and
endurance in patients with mild COPD and the effects of weight training,” European
Respiratory Journal, vol. 15, no. 1, pp. 92–97, 2000.
94. G. A. Khomenok, A. Hadid, O. Preiss-Bloom et al., “Hand immersion in cold water
alleviating physiological strain and increasing tolerance to uncompensable heat stress,”
European Journal of Applied Physiology, vol. 104, no. 2, pp. 303–309, 2008.

264
95. Y. S. Kwon, R. A. Robergs, C. M. Mermier, S. M. Schneider, and A. B. Gurney, “Palm
cooling and heating delays fatigue during resistance exercise in women,” The Journal of
Strength & Conditioning Research. In press.
96. D. A. Grahn, V. H. Cao, C. M. Nguyen, M. T. Liu, and H. C. Heller, “Work volume and
strength training responses to resistive exercise improve with periodic heat extraction
from the palm,” Journal of Strength & Conditioning Research, vol. 26, no. 9, pp. 2558–
2569, 2012.
97. A. Kleinridders, H. A. Ferris, W. Cai, and C. R. Kahn, “Insulin action in brain regulates
systemic metabolism and brain function,” Diabetes, vol. 63, no. 7, pp. 2232–2243, 2014.
98. T. H.Moran and K. L. K. Tamashiro, “Curt richter: spontaneous activity and food intake,”
Appetite, vol. 49, no. 2, pp. 368–375, 2007.
99. J. J. Balcita-Pedicino and S. R. Sesack, “Orexin axons in the rat ventral tegmental area
synapse infrequently onto dopamine and !-aminobutyric acid neurons,” Journal of
Comparative Neurology, vol. 503, no. 5, pp. 668–684, 2007.
100. R. A. Depue and P. F. Collins, “Neurobiology of the structure of personality: dopamine,
facilitation of incentivemotivation, and extraversion,” Behavioral and Brain Sciences,
vol. 22, no. 3, pp. 491–569, 1999.
101. A. Abizaid, Z.-W. Liu, Z. B. Andrews et al., “Ghrelin modulates the activity and synaptic
input organization of midbrain dopamine neurons while promoting appetite,” Journal
of Clinical Investigation, vol. 116, no. 12, pp. 3229–3239, 2006.
102. G. Holstege and J. R. Georgiadis, “The emotional brain: neural correlates of cat sexual
behavior and human male ejaculation,” Progress in Brain Research, vol. 143, pp. 39–45,
2004.
103. L. W. Swanson, “Cerebral hemisphere regulation of motivated behavior,” Brain Research,
vol. 886, no. 1-2, pp. 113–164, 2000.
104. G. Holstege and H. K. Huynh, “Brain circuits for mating behavior in cats and brain
activations and de-activations during sexual stimulation and ejaculation and orgasm in
humans,” Hormones and Behavior, vol. 59, no. 5, pp. 702–707, 2011.
105. M. W. Howe, P. L.Tierney, S.G. Sandberg, P. E. M. Phillips, and A. M. Graybiel, “Prolonged
dopamine signalling in striatum signals proximity and value of distant rewards,” Nature,
vol. 500, no. 7464, pp. 575–579, 2013.
106. P. Vernier, F. Moret, S. Callier, M. Snapyan, C. Wersinger, and A. Sidhu, “The
degeneration of dopamine neurons in Parkinson’s disease: insights from embryology
and evolution of the mesostriatocortical system,” Annals of the New York Academy of
Sciences, vol. 1035, pp. 231–249, 2004.
107. W. J. A. J. Smeets and A. Gonz´alez, “Catecholamine systems in the brain of vertebrates:
new perspectives through a comparative approach,” Brain Research Reviews, vol. 33, no.
2-3, pp. 308– 379, 2000.

265
108. S. T. de Bruijn, A. J. M. vanWijck, R. Geenen et al., “Relevance of physical fitness levels
and exercise-related beliefs for selfreported and experimental pain in fibromyalgia: an
explorative study,” Journal of Clinical Rheumatology, vol. 17, no. 6, pp. 295– 301, 2011.
109. K. Rousseau, Z. Atcha, F. R. A. Cagampang et al., “Photoperiodic regulation of leptin
resistance in the seasonally breeding Siberian hamster (Phodopus sungorus),”
Endocrinology, vol. 143, no. 8, pp. 3083–3095, 2002.
110. G. B. Stefano, R. Pt´acek, H. Kuzelov´a, and R. M. Kream, “Endogenous morphine: up-to-
date review 2011,” Folia Biologica, vol. 58, no. 2, pp. 49–56, 2012.
111. R. M. Kream and G. B. Stefano, “Endogenous morphine and nitric oxide coupled
regulation of mitochondrial processes,” Medical Science Monitor, vol. 15, no. 12, pp.
RA263–RA268, 2009.
112. M. S. Conceic¸˜ao, C. A. Libardi, F. R. D. Nogueira et al., “Effects of eccentric exercise on
systemic concentrations of pro- and anti-inflammatory cytokines and prostaglandin
(E2): comparison between young and postmenopausal women,” European Journal of
Applied Physiology, vol. 112, no. 9, pp. 3205–3213, 2012.
113. P. Cadet, W. Zhu, K. Mantione et al., “Cyclic exercise induces anti-inflammatory signal
molecule increases in the plasma of Parkinson’s patients,” International Journal of
Molecular Medicine, vol. 12, no. 4, pp. 485–492, 2003.
114. S. A. Martin, B. D. Pence, R. M. Greene et al., “Effects of voluntary wheel running on LPS-
induced sickness behavior in aged mice,” Brain, Behavior, and Immunity, vol. 29, pp.
113–123, 2013.
115. G. B. Stefano and R. M. Kream, “Endogenous morphine synthetic pathway preceded and
gave rise to catecholamine synthesis in evolution (review),” International Journal of
Molecular Medicine, vol. 20, no. 6, pp. 837–841, 2007.
116. I.Carreras, S. Yuruker,N. Aytanet al., “Moderate exercisedelays the motor performance
decline in a transgenic model of ALS,” Brain Research, vol. 1313, pp. 192–201, 2010.
117. A. L. Ridgel, J. L. Vitek, and J. L. Alberts, “Forced, not voluntary, exercise improves motor
function in Parkinson’s disease patients,” Neurorehabilitation and Neural Repair, vol. 23,
no. 6, pp. 600–608, 2009.
118. D. Kapogiannis and M. P. Mattson, “Disrupted energy metabolism and neuronal circuit
dysfunction in cognitive impairment and Alzheimer’s disease,” The Lancet Neurology,
vol. 10, no. 2, pp. 187–198, 2011.
119. A. Peters, B. Hitze, D. Langemann, A. Bosy-Westphal, and M. J. Muller, “Brain size, body
size and longevity,” International Journal of Obesity, vol. 34, no. 8, pp. 1349–1352, 2010.
120. C. L. Raison and A. H.Miller, “The evolutionary significance of depression in Pathogen
Host Defense (PATHOS-D),”Molecular Psychiatry, vol. 18, no. 1, pp. 15–37, 2013.

266
121. S. Nehlsen-Cannarella, O. Fagoaga, J. Folz, S. Grinde, C. Hisey, and R. Thorpe, “Fighting,
fleeing, and having fun: the immunology of physical activity,” International Journal of
Sports Medicine, vol. 18, supplement 1, pp. S8–S21, 1997.
122. L. S. McAnulty, D. C. Nieman, C. L. Dumke et al., “Effect of blueberry ingestion on natural
killer cell counts, oxidative stress, and inflammation prior to and after 2.5 h of running,”
Applied Physiology, Nutrition andMetabolism, vol. 36, no. 6, pp. 976–984, 2011.
123. D. De Ridder, J. Verplaetse, and S. Vanneste, “The predictive brain and the ‘free will’
illusion,” Frontiers in Psychology, vol. 4, article 131, 2013.
124. M. Schaller, “The behavioural immune systemand the psychology of human sociality,”
Philosophical Transactions of the Royal Society of London B: Biological Sciences, vol.
366, no. 1583, pp. 3418–3426, 2011.
125. M. Schaller and S. L. Neuberg, “Danger, disease, and the nature of prejudice(s),” in
Advances in Experimental Social Psychology, vol. 46, pp. 1–54, Academic Press, 2012.
126. S. A. Adamo, “The effects of stress hormones on immune function may be vital for the
adaptive reconfiguration of the immune systemduring fight-or-flight behavior,”
Integrative and Comparative Biology, vol. 54, no. 3, pp. 419–426, 2014.
127. A. M.W. Petersen and B. K. Pedersen, “The anti-inflammatory effect of exercise,” Journal
of Applied Physiology, vol. 98, no. 4, pp. 1154–1162, 2005.
128. A. M. Knab, R. S. Bowen, A. T. Hamilton, A. A. Gulledge, and J. T. Lightfoot, “Altered
dopaminergic profiles: implications for the regulation of voluntary physical activity,”
Behavioural Brain Research, vol. 204, no. 1, pp. 147–152, 2009.
129. A. Peters, A. Bosy-Westphal, B. Kubera et al., “Why doesn’t the brain lose weight, when
obese people diet?” Obesity Facts, vol. 4, no. 2, pp. 151–157, 2011.
130. B. K. Pedersen and C. Brandt, “The role of exercise-induced myokines in muscle
homeostasis and the defense against chronic diseases,” Journal of Biomedicine and
Biotechnology, vol. 2010, Article ID 520258, 6 pages, 2010.

267
Paragraph 2.2. Physical inactivity is a disease synonymous for a non-permissive brain
disorder.
Pruimboom L. Med Hypotheses. 2011;77:708-13.
Physical inactivity is a disease
Synonymous for a non-permissive brain disorder
Leo Pruimboom; University of Gerona (Spain), faculty of human Sciences,
University of Graz (Austria), Uni for Life.
Running Title: Physical inactivity; protective?

268
Abstract
The evolution of human kind has taken millions of years in which environmental factors
gradually shaped the actual genome adapted to those circumstances. One of the most vital
behavioural adaptations of mammals in general and especially humans is their capability of
self-sufficiency through physical activity. Physical activity abilities, including long distance
running, jumping, climbing and carrying things have probably been necessary to outrun wild
animals, search for food and hide for danger. In contrast, individuals physically or
psychologically unable to “take care of themselves” were more susceptible for early death and
therefore for genetic extinction.
The actual society is characterized by sedentary instead of “moving” individuals. Physical
inactivity is not just a possible factor related with chronic disease, but should be considered
the actual cause of the majority of human illness. Individuals know that exercise is necessary
and beneficial. Nevertheless almost 75% of the actual population doesn’t reach the estimated
minimum of necessary activity. Physical inactivity belongs to the characteristics of sickness
behaviour; the latter which probably is protective for the organism. Sickness behaviour,
including depressive mood, seems to protect against infection, injury, social conflict and
facilitates energy conservation. Sickness behaviour is based on immune-brain mechanisms
and can be defined as non-permissive behaviour. Long-term non-permissive behaviour can
lead to chronic disease because of reduction of physical activity and self-defeating coping
styles, converting non-permissive behaviour in a non-permissive brain disorder. We propose
that physical inactivity disease is synonymous for a non-permissive brain disorder and that
NPBD produces a so called “reptile phenotype”, characterized by hypothermia, poor hair
growth, decreased fertility and low basal metabolic rate.
Keywords
Physical inactivity, depression, non-permissive, brain, evolution, self defeating behaviour

269
Introduction
Physical inactivity is related to almost every type of chronic disease including heart
insufficiency, diabetes type II, metabolic syndrome, obesity, gall stones depression, early
aging, neurodegeneration and even early death (1). Clinical research about the effect of
exercise on parameters such as blood flow, energy metabolism and immune system
activation is not conclusive because of a basic mistake; the use of “healthy sedentary”
individuals as the control group (2). The human genome has been shaped through several
evolutionary pressure factors including the need of self-sufficiency. Self-sufficiency is
defined sociologically as each human or small group of humans being responsible for their
own food, water, defence, and shelter (3, 4). The self-sufficient phase has dominated human
behaviour for almost 2 million years and only perhaps the last 100 years individuals are
“served” dinner, warmth, drinks and shelter. This non-self-sufficiency is defined here as
humans obtaining a constant supply of food until reproductive age through a distribution
network composed generally of farmers to transporters to sellers to buyers to consumers.
Homo sapiens is the most resistant being to long term exercise, capable of outrunning
animals such as wildebeest and other big mammals, which have been part of the natural
nutritional sources of humans till very recently (5, 6). The capability of outrunning other
animals demands the following unique characteristics of human physiology and morphology
(7):
• Thermoregulation in homo sapiens is unique through the absence of body hair and the
presence of more than 2 million eccrine sweat glands (8, 9)
• Short toes, reducing the energetic cost of running (10, 11)
• The capability of producing great amounts of glucose out glycogen, amino acids and
glycerol; gluconeogenesis
• Expensive organs such as the heart muscle and the brain can use different energy sources
when necessary; the so-called metabolic flexibility (12). This metabolic flexibility
facilitates endurance running, climbing, jumping, swimming, throwing, crawling,
carrying and sprinting (13).

270
Table 1 The relationship between evolutionary based life threatening danger factors, the
developed survival reaction, the benefit and the contemporary consequence of disease.
A = adrenalin; AVP = arginine vasopressin; HIF-1 = hypoxia inducible factor 1; HSR = hypoxia stress reaction; IIS = innate
immune system; IR = insulin resistance; LR = leptin resistance; NA = noradrenalin; SER = serotonin (adapted from 4)
Homo sapiens “is made for walking”, so inactivity is a disease
As it seems that homo sapiens is “made for walking”, it is very strange that so few people
engage in physical activity nowadays. Troiano et al (14) using an accelerometer showed that
only 5% of the adults and 8% of the adolescents reach the minimal amount of recommended
physical activity (figure 1). Nevertheless, looking at Haile Gebreselassi finishing the Berlin
marathon and breaking the world record with a big smile on his face makes it plausible that
exercise, although intense, has to be fun. So knowing why people fail to engage in regular
exercise, leading to possibly severe damage to their body, is of mayor clinical importance. We
state that the background of choosing for a sedentary life is based on a so-called non-
permissive brain syndrome (NPBS, our hypothesis, see further, 17). This syndrome protects the
individual for a certain period (a disorder), while long lasting NPBS is a disease itself (defined
as such by Heskha and Allison, 18), related with increased mortality risk. NPBS is characterized
by low basal metabolism, impaired hair growth, thermoregulation disorders and, naturally,
inactivity.
Figure 1 The actual percentage of individuals reaching the minimal physical activity level
(adapted from 14)

271
The burden of physical inactivity
The systemic changes caused by physical inactivity occur fast and are specific for a disease. In
response to excessive inactivity (continuous bed rest or go into the microgravity environment
of space) individuals exhibit a rapid loss of tissue mass and disruption of normal function in
many cells and tissues. Within weeks of the onset of continuous bed rest by humans,
numerous adaptations occur, including 25% decreases in maximal stroke volume, maximal
cardiac output, and peak consumption of O2 during maximal aerobic exercise. Orthostatic
intolerance occurs due to attenuated baroreflex-mediated sympatho-excitation in response to
incremental declines in arterial blood pressures. Bones lose mass at 10 times their normal rate.
Skeletal muscles become weaker with less endurance for light physical effort. In addition,
metabolic pathways adapt to inactivity by decreasing mitochondrial concentration in skeletal
muscles, lowering the capacity to oxidize fatty acids, affecting metabolic flexibility. Whole
body insulin sensitivity declines within the first 3 days of inactivity, whether it is sedentary or
active individuals stopping daily exercise. Constipation ensues. Deep vein thrombosis can
occur within a time frame as short as an international flight with possible pulmonary
thrombo-embolism in susceptible individuals. In summary, the body’s reaction to physical
inactivity result in a loss of many structural and physiological processes, which often lead to
unhealthy, in some cases life-threatening, conditions (3, 19, 20, 21). Another consequence of
inactivity is the loss of strength of respiratory muscles (22). This effect can cause severe loss of
muscle strength of peripheral muscles through more severe inactivity, oxygen deficiency and
higher insulin resistance (23). On cellular level, changes are even more dramatic. Inactivity
decreases neurogenesis in specific parts of the brain responsible for memory, motor learning
and programs and need-catering factors (food, water, no-drugs, no-alcohol abuse, etc.). Parts
included are the cerebellum (motor programs), the olfactory bulb (need – catering) and the
hippocampus (24). On the contrary, exercise combined with enriched environment is able to
recover earlier lost neurons also in elder animals and humans (25, 26). Decision-making can
be considered the most important trait of Homo sapiens. The actual environment demands
self-control and discipline to make the right decision regarding food choice, physical activity,
smoking, alcohol and other factors that affect hedonic choices. The hippocampus is essential
for decision-making together with other parts of the brain such as the striatum and the frontal
cortex (27, 28). Loss of hippocampus volume (and the subsequence increased susceptibility for
developing inactive behaviour) can further be produced by stress, sleep disruption, smoking,
alcohol abuse and inflammation (26, 29, 30). Another consequence of physical inactivity is its
resistance-inducing effect on insulin, leptin, serotonin, dopamine, thyroxin and cortisol
signalling, affecting the function of the immune system, the endocrinological system and
distribution of energy (50, 53, 54, 55, 56). Resistance to the mentioned hormones is part of the
development of sterile chronic inflammatory diseases (65). Inactivity can also affect the
expression of glucose transporter 4 (GLUT4). GLUT4 is responsible for muscular glucose

272
trafficking in rest (insulin dependent) and activity (insulin independent, 19, 20, 62). GLUT4 is
decreased in sedentary individuals, increased in active ones and persons with high GLUT4 are
more engaged in exercise (62, 63, 64, 65, 66). Increase of GLUT4 expression enables muscle to
uptake more than 80% of the daily glucose load. This would make our high glycemic society
less toxic and therefore less pathological. In resume, the great amount of literature supports
the overall necessity of increase of physical activity as a normal way of living and in one way
or another people have to get conscious about the severe deleterious effects of sedentary
lifestyle on health and life expectation (67, 68).
Possible explanations for the “lazy phenotype”
Chronic intake of food (high meal frequency and abundant meals) can have an important
influence on various forms of physical activity (32), including spontaneous and voluntary
exercise. Spontaneous physical activity, the activity related with daily life, plays a mayor role
in maintaining body weight, caloric intake and overall health (33, 34, 35, 36). Spontaneous
physical activity (SPA) is not chosen and bypasses higher cortical regions while voluntary
exercise is a choice and therefore dependent on neocortical activity and “free will” (36, 37, 38,
39). Brain stem and spinal centres responsible for the maintenance and recovery of
homeostatic balances regulate SPA. Hunger, thirst, cold, warmth, itch, libido, curiosity and
pain are homeostatic feelings inducing movement beyond any doubt. The problem in
modern society is just that the refrigerator, air conditioner and heating are very nearby and
eliminate the SPA stimulus within seconds, whereas our ancestors were obliged to move till
they would find food, water, a place to hide or would have produced so much body heat that
they could survive in the cold (the self-sufficiency period). Homeostatic feelings are related
with the production of certain neuromodulators, including orexin and cholecystokinine (CCK,
for a excellent review see 34). Orexin, related with appetite and environmental food and liquid
exploration, favours SPA (40,41, 42). On the contrary, CCK, produces postprandial satiety,
inhibits SPA and induces sleep (43). Postprandial CCK production influences the body arousal
system probably by inhibiting the sympathetic nervous system and by activating the non-
myelinated vagus nerve (44, 45, 46, 47). This appetite-satiety/exercise-rest rhythm has
probably been developed through evolutionary mechanisms, giving raise to the so-called
feast-rest/famine-exercise hypothesis of Chakravarthy and Booth (3, 47, 48). Several
interventions to promote SPA have failed so far; although something so simple as less
sedentary behaviour and reduction of television hours could already be effective (49).
Human evolution has shaped the genome in such a way that the majority of genes are
selected against physical inactivity (3, 70, 71, 72, 73). Current scientific research shows that a
few genes seem to play a mayor role in engagement in physical activity, including dopamine
receptor 1 (Drd1) and the so-called nescient helix loop helix (Nhlh2, 82). The involvement of a

273
dopamine-signalling pathway in physical activity should not be surprising. Dopamine is
involved in several locomotor disorders such as Parkinson, attention deficit hyperactive
disorder, Tourette syndrome and restless legs. Quite a few studies have been carried out
relating Drd1 expression with activity-level in animals and humans (see review 74). Dopamine
is the key-player in functions such as drive, motivation, “cheap movement”, decision-making,
future thinking, reward searching and control of emotional-obsessive behaviour (7).
Dopamine is fun, but an overload can be responsible for hyperkinetic disorders, paranoia,
fanatic religious behaviour and even uncontrollable aggressive behaviour (75, 76). Dopamine
receptors 1and 2 are the most studied in humans. Theoretically, Drd2 receptor binding signals
the presence of important (often reward-based) information and allows the prefrontal cortex
(PFC) network to respond to this new information by updating its working memory system.
Drd1 receptor stimulation, on the other hand, plays a gating role by controlling the threshold
of significance above which information may pass before it can be admitted to working
memory and processed by the PFC. Drd1 receptor activation stabilizes the pattern of activity
within the PFC neural network and protects the system from distracters until the appropriate
behavioural response is generated (77). Different research groups have found that animals and
humans with a lower expression of Drd1 in the nucleus accumbens belong to the high-active
sample while high expression of Drd1 produces a more inactive phenotype (77, 78, 79, 80, 81).
Dopamine signalling in the brain is very much dependent on the intra-uterine environment
and especially of the presence of enough iodine, L-tyrosine and omega 3 fatty acids (82, 83,
84, 85, 86, 8, 88, 89, 90). Chronic inflammation is especially important in dopamine signalling
and therefore will be considered separately proposing our hypothesis.
Our Hypothesis: “The non-permissive brain disorder; laziness as a
protection?”
We hypothesize that physical inactivity is part of a non-permissive brain syndrome caused by
(adaptive?) disorders of dopamine signalling; not only genes will be involved but also
environmental factors including a lack of brain nutrients during brain development and
maturation (iodine, DHA, L-tyrosine, Cunnane 2005), vitamin D deficiency, increased protein
turn-over and energy distribution disturbances (17). The pathways leading to insufficient
dopamine signalling include hyper activation of indoleamine 2,3 dioxygenase pathway in
chronic inflammation (for an excellent review see 61) and decrease of the conversion of the
amino acid l-tyrosine in dopamine (75). Chronic disease is characterized by higher energy
demand of the immune system and dramatically increased protein turnover (60). The
increased protein/energy demand can be so severe that multiple organs could suffer
functional disorders and even failure, leading to acute death (59). Chronic immune activation
leads to sickness behaviour resembling the non-permissive brain syndrome (91, 92, 93).

274
• Exercise avoidance
• Thermoregulation disorders
• Social isolation
• Reduction of libido
• Gluttony or
• Lack of appetite
• Increased pain sensitivity
• Fatigue
• Concentration deficiency
• Attention disorders
• Memory problems
• Change of body composition (muscle atrophy and increased fat deposits)
• Decreased fertility
• Impaired hair growth and boldness
• Decrease of top-down decision making
• Self-defeating behaviour
• Depressive mood
The physical characteristics lined up belong to a species with low basal metabolism such as a
reptile (51). Low activity belonging to sickness behaviour and NPBS could help to defend the
organism against infection, conflict, injury, exhaustion and perhaps the most important
factor, low activity conserves energy for the immune system to combat infection, although it
would be a sterile one (96, 97, 98). Certain depressive states are part of the non-permissive
brain disorder (NPBS) and therefore depression and low activity should probably be
considered protective at start. Long-term inactivity is deleterious per sé because of the
tremendous damage at physical and mental level. We define NPBS as a behavioural strategy
to prevent more severe damage, conserve energy and proteins to support immune function
and provide protection of the most expensive organs of the human body at start. NPBS can
also protect people against repeated physical abuse (social withdrawal in people who have
suffered childhood abuse, 119) and severe loss of lean body mass during chronic immune
system activity (52, 58, 59). Long lasting NPBS is severely deleterious to the host and should be
treated as one of the most important clinical identities in modern medicine.Humans have the
highest encephalisation quotient of all living animals on earth. Big brains need more energy.
The development of bigger brains, mostly in mammals, has changed the morphology of these
mammals in such a way that more central energy allocation was needed in stead of high

275
allocation to “dismissible” organs such as guts and muscles (99, 100). Possible threat to brain
energy supply will lead to NPBS.
The “selfish brain” will continuously scan energy status in the peripheral body through
neurological and endocrinological communication, including sympathetic interoceptors and
hormones such as insulin (informing about muscle energy) and leptin (informing about the
amount of stored fat. Peters et al (101, 102) showed that the brain would always give priority to
its own energy need although others organs could suffer. Even in insulin resistant states, in
which energy allocation is disturbed, the brain will cover its needs through up regulation of
alternative substrate usage such as creatine and fatty acids (103, 104). NPBS will therefore
protect primarily the brain on behalf of muscle tissue. Muscle tissue uses an average of 13
kcal/24 hours in rest and up to 10 times more during exercise (table 2, 107). Muscle waisting
and physical inactivity conserves a substantial amount of energy and provide proteins to be
used by the immune system.
Chronic immune system activation is just one possibility of energy and protein deficiency
(105). Hepatic overload (through chronic fructose intake, environmental toxins, hormonal
stress) demands a higher energy turnover in the liver. Situations in which an expensive organ
demands more energy and protein, other organs will have to reduce their metabolic rate,
conserving energy and proteins for the demanding organ (103, 106).
Table 2 The energy rate/24 hours of different human organs and tissues.
Muscle, contributing to more than 40% of the body mass, uses approximately 13 kcal/24 hours. Fat tissue and bones belong
to the cheapest tissues and demand 4,6 kcal/24 hours and 2,2 kcal/24 hours respectively (107)
An excellent example of a NPBS is chronic fatigue. The majority of people suffering from
chronic fatigue syndrome (CFS) show symptoms, which should be considered as
consequence of the huge energy and protein demand of the chronic activated immune
system. Another topic in CFS is the high incidence of people suffering from fatty liver disease,
low thyroid hormone function and insulin resistance (108, 109, 110, 111). Impaired thyroid
hormone function induces low basal metabolism and inactivity (¿??).

276
Resuming, it seems plausible to state that physical inactivity as part of NPBD could actually be
a protective strategy against severe or more damage and infection, an energy conservation
strategy and an avoidance strategy against possible psycho-emotional conflicts. If so,
treatment for people with physical inactivity disease (PID) should be based upon inhibition of
the immune system, improvement of energy distribution, recovery of insulin sensitivity and
reduction of psychosocial stress. Even other factors such as shame coping style (117), socio-
demographic conditions (118), childhood abuse (119) and lack of employment (120), causing an
inactive phenotype should be considered just part of the etiological factors leading to NPBS.
NPBS and engaging in physical activity; discussion
As it is stated in this article, physical inactivity disease should be considered a non-permissive
brain syndrome, based on multiple factors but all related with a possible threat for brain
energy deficiency. Our hypothesis can be easily tested through measurement of biomarkers
such as basal metabolic rate, acute phase proteins, the use of validated questionnaires and the
influence of integrative “motivational” treatment and the measurement of engaging in
spontaneous and voluntary exercise after intervention. Basic part of the treatment of people
suffering from NPBS is in depth information about the pathways leading to it and the
subsequence physical inactivity.
Physical inactivity is a disease and should be considered one of the most damaging
behavioural traits not only in developed but also in developing countries. Multiple etiological
factors have been identified to explain this rather strange way of living (without exercise).
Strange because of the fact that exercise has saved Homo sapiens during thousands of
generations and is therefore programmed against inactivity. The impact of physical inactivity
on systemic, metabolic, cellular, molecular en genetic level is so diverse and severe that the
effort to find a so called “exercise pill” will probably will be too good to be true (121). Engaging
people in physical activity needs an integrative intervention based upon deep learning,
immune system regulation, recovery of insulin sensitivity and normalizing of energy and
energy distribution (50). Part of the optimal treatment is based on motivation, increase of
inevitable spontaneous exercise (absence of elevators) and on promoting evolutionary
biorhythm, including a feast/famine rest/exercise rhythm (51). Education in the need of
engagement in physical activity should probably begin when the next generation starts; at
school (69).

277
References
1. Booth, FW. Sedentary death syndrome is what researchers now call America’s second
largest threat to public health. Interview with Frank Booth (2001)
2. McCully, K. K., Smith, S., Rajaei, S., Leigh, J. S. & Natelson, B. H. Blood flow and muscle
metabolism in chronic fatigue syndrome. Clin Sci (Lond) 104, 641-647 (2003).
3. Booth, F. W. & Lees, S. J. Fundamental questions about genes, inactivity, and chronic
diseases. Physiol Genomics 28, 146-157 (2007).
4. Chrousos, G. P. Stress and disorders of the stress system. Nat Rev Endocrinol 5, 374-381
(2009).
5. Liebenberg, L. Persistence hunting by modern hunter-gatherers. Current Anthropology
47, 1017 (2006).
6. Liebenberg, L. The relevance of persistence hunting to human evolution. Journal of
human evolution 55, 1156 (2008).
7. Pruimboom L, Raison C. Homo sapiens sapiens, myelinated vagal controlled exercise
and fun; A new hypotheses considering an unique human feature. In prep. (2011)
8. Perry, G. H. & Dominy, N. J. Evolution of the human pygmy phenotype. Trends Ecol Evol
24, 218-225 (2009).
9. Marino, F. E. The evolutionary basis of thermoregulation and exercise performance. Med
Sport Sci 53, 1-13 (2008).
10. Steudel-Numbers, K. L. & Wall-Scheffler, C. M. Optimal running speed and the evolution
of hominin hunting strategies. Journal of human evolution 56, 355-360 (2009).
11. Rolian, C., Lieberman, D. E., Hamill, J., Scott, J. W. & Werbel, W. Walking, running and
the evolution of short toes in humans. J Exp Biol 212, 713-721 (2009).
12. Corpeleijn, E., Saris, W. H. & Blaak, E. E. Metabolic flexibility in the development of
insulin resistance and type 2 diabetes: effects of lifestyle. Obes Rev 10, 178-193 (2009).
13. Storlien, L., Oakes, N. D. & Kelley, D. E. Metabolic flexibility. Proc Nutr Soc 63, 363-368
(2004).
14. Troiano, R. P., Berrigan, D., Dodd, K. W., Mâsse, L. C., et al. Physical activity in the United
States measured by accelerometer. Medicine & Science in Sports & Exercise 40, 181
(2008).
15. Carducci, B. J. What shy individuals do to cope with their shyness: a content analysis
and evaluation of self-selected coping strategies. Isr J Psychiatry Relat Sci 46, 45-52
(2009).
16. Sirois, F. M. Procrastination and counterfactual thinking: avoiding what might have
been. Br J Soc Psychol 43, 269-286 (2004).
17. Pruimboom L. The non-permissive brain. Van Nature 10, 39 – 42 (2008)
18. Heshka S, Allison DB. Is obesity a disease? Int J Obes Relat Metab Disord;25:1401–1404
(2001)

278
19. Lees, S. J. & Booth, F. W. Physical inactivity is a disease. World Rev Nutr Diet 95, 73-79
(2005).
20. Lees, S. J. & Booth, F. W. Sedentary death syndrome. Applied Physiology, Nutrition, and
Metabolism 29, 447-460 (2004).
21. Booth, F. W., Chakravarthy, M. V., Gordon, S. E. & Spangenburg, E. E. Waging war on
physical inactivity: using modern molecular ammunition against an ancient enemy. J
Appl Physiol 93, 3-30 (2002).
22. Simões, R. P., Castello, V., Auad, M. A., Dionísio, J. & Mazzonetto, M. Prevalence of
reduced respiratory muscle strength in institutionalized elderly people. Sao Paulo Med J
127, 78-83 (2009).
23. Vogiatzis, I., S Zakynthinos, Physical inactivity: common pathway to peripheral muscle
weakness in chronic respiratory diseases?, in Eur Respir J 34 (6); 1213-1214 (2009)
24. Lazarov, O., Mattson, M. P., Peterson, D. A., Pimplikar, S. W. & van Praag, H. When
neurogenesis encounters aging and disease. Trends Neurosci 33, 569-579 (2010).
25. Kerr, A. L. & Swain, R. A. Rapid cellular genesis and apoptosis: Effects of exercise in the
adult rat. Behav Neurosci 125, 1-9 (2011).
26. Lucassen, P. J., Meerlo, P., Naylor, A. S., van Dam, A. M., et al. Regulation of adult
neurogenesis by stress, sleep disruption, exercise and inflammation: Implications for
depression and antidepressant action. Eur Neuropsychopharmacol 20, 1-17 (2010).
27. Johnson, A., van der Meer, M. A. & Redish, A. D. Integrating hippocampus and striatum
in decision-making. Curr Opin Neurobiol 17, 692-697 (2007).
28. Bechara, A. Decision making, impulse control and loss of willpower to resist drugs: a
neurocognitive perspective. Nat Neurosci 8, 1458-1463 (2005). Behav. Sci. Law 25: 295–
308 (2007)
29. Crews, F. T. & Nixon, K. Mechanisms of neurodegeneration and regeneration in
alcoholism. Alcohol Alcohol 44, 115-127 (2009).
30. Gallinat, J., Lang, U. E., Jacobsen, L. K., Bajbouj, M., et al. Abnormal hippocampal
neurochemistry in smokers: evidence from proton magnetic resonance spectroscopy at
3 T. J Clin Psychopharmacol 27, 80-84 (2007).
31. Fasano, A. Zonulin and its regulation of intestinal barrier function: the biological door to
inflammation, autoimmunity, and cancer. Physiol Rev 91, 151-175 (2011).
32. Holmstrup, M. E., Owens, C. M., Fairchild, T. J. & Kanaley, J. A. Effect of meal frequency
on glucose and insulin excursions over the course of a day. e-SPEN, the European e-
Journal of Clinical Nutrition and Metabolism 5, e277-e280 (2010).
33. Thorburn, A W, and J Proietto. "Biological Determinants of Spontaneous Physical
Activity." Obesity reviews : an official journal of the International Association for the
Study of Obesity 1, no. 2 (2000): 87-94.

279
34. Teske, J A, C J Billington, and C M Kotz. "Neuropeptidergic Mediators of Spontaneous
Physical Activity and Non-Exercise Activity Thermogenesis." Neuroendocrinology 87,
no. 2 (2008): 71-90
35. Dishman, Rod K. "Gene-Physical Activity Interactions in the Etiology of Obesity:
Behavioral Considerations." Obesity (Silver Spring, Md.) 16 Suppl 3 (2008):60-65
36. Kotz, Catherine M, Jennifer A Teske, and Charles J Billington. "Neuroregulation of
Nonexercise Activity Thermogenesis and Obesity Resistance." American journal of
physiology. Regulatory, integrative and comparative physiology 294, no. 3 (2008):R699-
710
37. Stranahan, Alexis M, Kim Lee, Bronwen Martin, Stuart Maudsley, Erin Golden, Roy G
Cutler, and Mark P Mattson. "Voluntary Exercise and Caloric Restriction Enhance
Hippocampal Dendritic Spine Density and BDNF Levels in Diabetic Mice." Hippocampus
19, no. 10 (2009):118-127
38. Stranahan, Alexis M, Kim Lee, and Mark P Mattson. "Central Mechanisms of HPAAxis
Regulation by Voluntary Exercise." Neuromolecular medicine 10, no. 2 (2008): 951-961
39. Tancredi, Laurence. The Neuroscience of ‘‘Free Will’’
40. Kotz, C M, C Wang, J A Teske, A J Thorpe, C M Novak, K Kiwaki, and J A Levine. "Orexin
A Mediation of Time Spent Moving in Rats: Neural Mechanisms." Neuroscience 142, no.
1 (2006): 29-36
41. Kotz, C. M. Integration of feeding and spontaneous physical activity: role for orexin.
Physiol Behav 88, 294-301 (2006).
42. Sakurai, T. The neural circuit of orexin (hypocretin): maintaining sleep and wakefulness.
Nat Rev Neurosci 8, 171-181 (2007).
43. Kapas, L. Metabolic signals in sleep regulation: the role of cholecystokinin. Doctor
Thesis 2010
44. Hand, K.V., Bruen, C.M., O'Halloran, F., Giblin, L., and Green, B.D.. Acute and chronic
effects of dietary fatty acids on cholecystokinin expression, storage and secretion in
enteroendocrine STC-1 cells. Mol Nutr Food Res. May;54 Suppl 1:S93-S103 (2010)
45. Maljaars, J., Romeyn, E.A., Haddeman, E., Peters, H.P., and Masclee, A.A. Effect of fat
saturation on satiety, hormone release, and food intake. Am J Clin Nutr (2009) 89: 1019-
024.
46. Tracey, K.J.. Reflex control of immunity. Nat Rev Immunol (2009)9: 418-428.
47. Booth, F. W. & Roberts, C. K. Linking performance and chronic disease risk: indices of
physical performance are surrogates for health. Br J Sports Med 42, 950-952 (2008).
48. Chakravarthy, M. V. & Booth, F. W. Eating, exercise, and "thrifty" genotypes: connecting
the dots toward an evolutionary understanding of modern chronic diseases. J Appl
Physiol 96, 3-10 (2004).

280
49. Garland, T., Schutz, H., Chappell, M. A., Keeney, B. K., et al. The biological control of
voluntary exercise, spontaneous physical activity and daily energy expenditure in
relation to obesity: human and rodent perspectives. J Exp Biol 214, 206-229 (2011).
50. Pruimboom, L. & van Dam, A. C. Chronic pain: a non-use disease. Med Hypotheses 68,
506-511 (2007).
51. Rivera S, Lock B. The reptilian thyroid and parathyroid glands. Vet Clin North Am Exot
Anim Pract. 2008 Jan;11(1):163-75, viii.
52. Ord, H. Nutritional support for patients with infected wounds. Br J Nurs 16:1346-8,
1350-2
53. Sidorenkov, O., Nilssen, O. & Grjibovski, A. M. Metabolic syndrome in Russian adults:
associated factors and mortality from cardiovascular diseases and all causes. BMC Public
Health 10, 582 (2010).
54. Tremblay, M. S. & Willms, J. D. Is the Canadian childhood obesity epidemic related to
physical inactivity? Int J Obes Relat Metab Disord 27, 1100-1105 (2003).
55. Martínez-González, M. A., Martínez, J. A., Hu, F. B., Gibney, M. J. & Kearney, J. Physical
inactivity, sedentary lifestyle and obesity in the European Union. Int J Obes Relat Metab
Disord 23, 1192-1201 (1999).
56. Mayer-Davis, E. J., D'Agostino, R., Karter, A. J., Haffner, S. M., et al. Intensity and amount
of physical activity in relation to insulin sensitivity: the Insulin Resistance
Atherosclerosis Study. JAMA 279, 669-674 (1998).
57. Pruimboom L, Stark M, Fox T, Kool M. De pandemie van chronische aandoeningen. Van
Nature 15, 22 – 28 (2009)
58. Lochmiller, RL., Deerenberg C. Trade-offs in evolutionary immunology: just what is the
cost of immunity?. OIKOS 88: 87–98. Copenhagen (2000)
59. Peters, A. The selfish brain: Competition for energy resources. Am J Hum Biol 23, 29-34
(2011).
60. Straub, R. H., Cutolo, M., Buttgereit, F. & Pongratz, G. Energy regulation and
neuroendocrine-immune control in chronic inflammatory diseases. J Intern Med 267,
543-560 (2010).
61. Miller, A. H. Mechanisms of cytokine-induced behavioral changes:
Psychoneuroimmunology at the translational interface. Brain, behavior, and immunity
23, 149-158 (2009).
62. Bonen, A., Dohm, G. L. & van Loon, L. J. C. Lipid metabolism, exercise and insulin action.
Essays in biochemistry 42, 47 (2006).
63. Stuart, C. A., Howell, M. E., Baker, J. D., Dykes, R. J., et al. Cycle training increased GLUT4
and activation of mammalian target of rapamycin in fast twitch muscle fibers. Med Sci
Sports Exerc 42, 96-106 (2010).

281
64. Tsao TS, Li J, Chang KS, Stenbit AE, Galuska D, Anderson JE, Zierath JR, McCarter RJ,
Charron MJ: Metabolic adaptations in skeletal muscle overexpressing GLUT4: Effects on
muscle and physical activity. FASEB J 2001;15:958–969.
65. Fried LP, Walston J: Frailty and failure to thrive; in Hazzard WR, et al (eds): Principles of
Geriatric Medicine and Gerontology. New York, McGraw Hill, 2003, pp 1487–1492.
66. Daugaard JR, Nielsen JN, Kristiansen S, Andersen JL, Hargreaves M, Richter EA: Fiber
typespecific expression of GLUT4 in human skeletal muscle: Influence of exercise
training. Diabetes 2000;49:1092–1095.
67. Chakravarthy, M. V., Joyner, M. J. & Booth, F. W. An obligation for primary care
physicians to prescribe physical activity to sedentary patients to reduce the risk of
chronic health conditions. Mayo Clin Proc 77, 165-173 (2002).
68. Brownson, R. C., Eyler, A. A., King, A. C., Shyu, Y. L., et al. Reliability of information on
physical activity and other chronic disease risk factors among US women aged 40 years
or older. American journal of epidemiology 149, 379 (1999).
69. Katz, D. L., Cushman, D., Reynolds, J., Njike, V., et al. Putting physical activity where it
fits in the school day: preliminary results of the ABC (Activity Bursts in the Classroom)
for fitness program. Prev Chronic Dis 7, A82 (2010).
70. McCarthy, M. I. Genomics, type 2 diabetes, and obesity. N Engl J Med 363, 2339-2350
(2010).
71. Joosen, A. M. C. P., Gielen, M., Vlietinck, R. & Westerterp, K. R. Genetic analysis of
physical activity in twins. American Journal of Clinical Nutrition 82, 1253 (2005).
72. Hauser, E. R., Mooser, V., Crossman, D. C., Haines, J. L., et al. Design of the Genetics of
Early Onset Cardiovascular Disease (GENECARD) study. Am Heart J 145, 602-613 (2003).
73. Booth, F. W., Chakravarthy, M. V. & Spangenburg, E. E. Exercise and gene expression:
physiological regulation of the human genome through physical activity. J Physiol 543,
399-411 (2002).
74. Lightfoot, J. T. Current understanding of the genetic basis for physical activity. J Nutr
141, 526-530 (2011).
75. Previc, F. H. Dopamine and the Origins of Human Intelligence* 1. Brain and cognition
41, 299-350 (1999).
76. Previc, F. H. The role of the extrapersonal brain systems in religious activity. Conscious
Cogn 15, 500-539 (2006).
77. Savitz, J., Solms, M. & Ramesar, R. The molecular genetics of cognition: dopamine,
COMT and BDNF. Genes Brain Behav 5, 311-328 (2006).
78. Garland, Theodore, Heidi Schutz, Mark A Chappell, Brooke K Keeney, Thomas H Meek,
Lynn E Copes, Wendy Acosta, and others. "The Biological Control of Voluntary Exercise,
Spontaneous Physical Activity and Daily Energy Expenditure in Relation to Obesity:

282
Human and Rodent Perspectives." The Journal of experimental biology 214, no. Pt 2
(2011): 206-229
79. Knab, A. M. & Lightfoot, J. T. Does the difference between physically active and couch
potato lie in the dopamine system? Int J Biol Sci 6, 133-150 (2010).
80. Nehrenberg, D. L., Wang, S., Hannon, R. M., Garland, T. & Pomp, D. QTL underlying
voluntary exercise in mice: interactions with the "mini muscle" locus and sex. J Hered
101, 42-53 (2010).
81. Rhodes, J. S., Gammie, S. C. & Garland, T. Neurobiology of mice selected for high
voluntary wheel-running activity. Integrative and Comparative Biology 45, 438 (2005).
82. Kuipers, R. S., Luxwolda, M. F., Dijck-Brouwer, D. A., Eaton, S. B., et al. Estimated
macronutrient and fatty acid intakes from an East African Paleolithic diet. Br J Nutr 104,
1666-1687 (2010).
83. Mulrine, H. M., Skeaff, S. A., Ferguson, E. L., Gray, A. R. & Valeix, P. Breast-milk iodine
concentration declines over the first 6 mo postpartum in iodine-deficient women. Am J
Clin Nutr 92, 849-856 (2010).
84. Helland, I. B., Smith, L., Blomén, B., Saarem, K., et al. Effect of supplementing pregnant
and lactating mothers with n-3 very-long-chain fatty acids on children's IQ and body
mass index at 7 years of age. Pediatrics 122, e472-e479 (2008).
85. Bernal, J. Thyroid hormone receptors in brain development and function. Nat Clin Pract
Endocrinol Metab 3, 249-259 (2007).
86. Román, G. C. Autism: transient in utero hypothyroxinemia related to maternal flavonoid
ingestion during pregnancy and to other environmental antithyroid agents. J Neurol Sci
262, 15-26 (2007).
87. Kooistra, L., Crawford, S., van Baar, A. L., Brouwers, E. P. & Pop, V. J. Neonatal effects of
maternal hypothyroxinemia during early pregnancy. Pediatrics 117, 161-167 (2006).
88. Singh, M. Essential fatty acids, DHA and human brain. Indian journal of pediatrics 72,
239-242 (2005).
89. Zoeller, T. R., Dowling, A. L. S., Herzig, C. T. A., Iannacone, E. A., et al. Thyroid hormone,
brain development, and the environment. Environmental Health Perspectives 110, 355
(2002).
90. Porterfield, S. P. & Hendrich, C. E. The role of thyroid hormones in prenatal and neonatal
neurological development--current perspectives. Endocr Rev 14, 94-106 (1993).
91. Segerstrom, S. C. Resources, stress, and immunity: an ecological perspective on human
psychoneuroimmunology. Ann Behav Med 40, 114-125 (2010).
92. Segerstrom, S. C. Stress, Energy, and Immunity. Current Directions in Psychological
Science 16, 326 (2007).

283
93. Dantzer, R., O'Connor, J. C., Freund, G. G., Johnson, R. W. & Kelley, K. W. From
inflammation to sickness and depression: when the immune system subjugates the
brain. Nat Rev Neurosci 9, 46-56 (2008).
94. Rosen, C. J. & Bouxsein, M. L. Mechanisms of disease: is osteoporosis the obesity of
bone? Nat Clin Pract Rheumatol 2, 35-43 (2006).
95. Woo, J., Leung, J. & Kwok, T. BMI, Body Composition, and Physical Functioning in Older
Adults&ast. Obesity 15, 1886-1894 (2007).
96. Kinney, D. K. & Tanaka, M. An evolutionary hypothesis of depression and its symptoms,
adaptive value, and risk factors. J Nerv Ment Dis 197, 561-567 (2009).
97. Nesse, R. M. & Williams, G. C. Evolution and the origins of disease. Scientific American
279, 86-93 (1998).
98. Dantzer, R. & Kelley, K. W. Twenty years of research on cytokine-induced sickness
behaviour. Brain Behav Immun 21, 153-160 (2007).
99. Isler, K. & van Schaik, C. P. The Expensive Brain: a framework for explaining
evolutionary changes in brain size. J Hum Evol 57, 392-400 (2009).
100. Crawford, M. A., Bloom, M., Broadhurst, C. L., Schmidt, W. F., et al. Evidence for the
unique function of docosahexaenoic acid during the evolution of the modern hominid
brain. Lipids 34 Suppl, S39-S47 (1999).
101. Peters, A. & Langemann, D. Build-ups in the supply chain of the brain: on the
neuroenergetic cause of obesity and type 2 diabetes mellitus. Front Neuroenergetics 1, 2
(2009).
102. Peters, A., Schweiger, U., Pellerin, L., Hubold, C., et al. The selfish brain: competition for
energy resources. Neurosci Biobehav Rev 28, 143-180 (2004).
103. Hitze, B., Hubold, C., van Dyken, R., Schlichting, K., et al. How the selfish brain organizes
its supply and demand. Front Neuroenergetics 2, 7 (2010).
104. Oltmanns, K. M., Melchert, U. H., Scholand-Engler, H. G., Howitz, M. C., et al. Differential
energetic response of brain vs. skeletal muscle upon glycemic variations in healthy
humans. Am J Physiol Regul Integr Comp Physiol 294, R12-R16 (2008).
105. Maciver, N. J., Jacobs, S. R., Wieman, H. L., Wofford, J. A., et al. Glucose metabolism in
lymphocytes is a regulated process with significant effects on immune cell function and
survival. J Leukoc Biol 84, 949-957 (2008).
106. Peters, A., Pellerin, L., Dallman, M. F., Oltmanns, K. M., et al. Causes of obesity: looking
beyond the hypothalamus. Prog Neurobiol 81, 61-88 (2007).
107. Pruimboom L, Fox T. Nahrung und Bewegung – ein erfahrenes Team. In Bant:
Sportphysiotherapie, Thieme Verlag; 337 – 377 (2011)
108. Newton, J. L. Systemic symptoms in non-alcoholic fatty liver disease. Dig Dis 28, 214-
219 (2010).

284
109. Newton, J. L., Jones, D. E., Henderson, E., Kane, L., et al. Fatigue in non-alcoholic fatty
liver disease (NAFLD) is significant and associates with inactivity and excessive daytime
sleepiness but not with liver disease severity or insulin resistance. Gut 57, 807-813
(2008).
110. Newton, J. L., Pairman, J., Wilton, K., Jones, D. E. & Day, C. Fatigue and autonomic
dysfunction in non-alcoholic fatty liver disease. Clin Auton Res 19, 319-326 (2009).
111. Raszeja-Wyszomirska, J., Lawniczak, M., Marlicz, W., Miezyńska-Kurtycz, J. &
Milkiewicz, P. [Non-alcoholic fatty liver disease--new view]. Pol Merkur Lekarski 24,
568-571 (2008).
112. Kelley DE, Goodpaster B, Wing RR, Simoneau JA: Skeletal muscle fatty acid metabolism
in association with insulin resistance, obesity, and weight loss. Am J Physiol Endocrinol
Metab 277:E1130–E1141, 1999
113. Bruce, C. R., Hoy, A. J., Turner, N., Watt, M. J., et al. Overexpression of carnitine
palmitoyltransferase-1 in skeletal muscle is sufficient to enhance fatty acid oxidation
and improve high-fat diet-induced insulin resistance. Diabetes 58, 550-558 (2009).
114. Dodd, S. L., Johnson, C. A., Fernholz, K. & St Cyr, J. A. The role of ribose in human
skeletal muscle metabolism. Med Hypotheses 62, 819-824 (2004).
115. Hellsten, Y., Skadhauge, L. & Bangsbo, J. Effect of ribose supplementation on resynthesis
of adenine nucleotides after intense intermittent training in humans. Am J Physiol
Regul Integr Comp Physiol 286, R182-R188 (2004).
116. Lazzer, S., Bedogni, G., Lafortuna, C. L., Marazzi, N., et al. Relationship between basal
metabolic rate, gender, age, and body composition in 8,780 white obese subjects.
Obesity (Silver Spring) 18, 71-78 (2010).
117. Campbell, M. C. & Tishkoff, S. A. The evolution of human genetic and phenotypic
variation in Africa. Curr Biol 20, R166-R173 (2010).
118. Cramer, V., Torgersen, S. & Kringlen, E. Socio-demographic conditions, subjective
somatic health, Axis I disorders and personality disorders in the common population:
the relationship to quality of life. J Pers Disord 21, 552-567 (2007).
119. Gladstone, G. L., Parker, G. B., Mitchell, P. B., Malhi, G. S., et al. Implications of childhood
trauma for depressed women: an analysis of pathways from childhood sexual abuse to
deliberate self-harm and revictimization. Am J Psychiatry 161, 1417-1425 (2004).
120. Mitchell, D. P., Betts, A. & Epling, M. Youth employment, mental health and substance
misuse: a challenge to mental health services. J Psychiatr Ment Health Nurs 9, 191-198
(2002).
121. Goodyear, L. J. The exercise pill--too good to be true? N Engl J Med 359, 1842-1844
(2008).

285
Paragraph 3.1. Chronic diseases are caused by the absence of old stress factors – our dearest
friends; The study of origin. Pruimboom L, Muskiet FAJ”. Short Version in Biomed Research
International 2016.
Influence of a 10 days mimic of our ancient lifestyle on anthropometrics and
parameters of metabolism and inflammation.
The ‘Study of Origin’.
Leo Pruimboom1,2, Begoña Ruiz-Núñez2, Jens Freese4, Charles L. Raison3, Frits A.J. Muskiet2
on behalf of the ‘Study of Origin” Consortium
1. Natura Foundation, Numansdorp, The Netherlands
2. Laboratory Medicine, University Medical Center Groningen (UMCG) and University of
Groningen, The Netherlands.
3. Department of Psychiatry, College of Medicine, John and Doris Norton School of Family
and Consumer Sciences, Tucson, USA.
4. German Sport University Cologne, Germany
Corresponding author
L. Pruimboom
Burgemeester Marijnenlaan 98
2585 DR Den Haag, The Netherlands
Email: [email protected]

286
Abstract
Background: Chronic low-grade inflammation and insulin resistance are intimately related
entities that are common to most, if not all, chronic diseases of affluence. We hypothesized
that a short-term intervention based on ‘ancient stress factors’ may improve anthropometrics
and clinical chemical indices.
Objective
Study whether a 10-day-mimic of a hunter-gatherer lifestyle favorably affects
anthropometrics and clinical chemical indices.
Methods
Fifty-three apparently healthy subjects and two patients with fibromyalgia (22–69 years, 28
women), in 5 groups, engaged in a 10-day trip through the Pyrenees. They walked 14 km/day
on average, carrying an 8-kilo backpack. Raw food was provided, self-prepared and water was
obtained from waterholes. They slept outside in sleeping bags and were exposed to
temperatures ranging from 12-42 °C. Anthropometric data and fasting blood samples were
collected at baseline and the study end.
Results
We observed median decreases (p≤0.002) of body weight (-3.5 kg), BMI (-1.1 kg/m2), hip (-3
cm) and waist (-5 cm) circumferences, glucose (-0.6 mmol/L), HbA1c (-0.1%), insulin
(-4.7 pmol/L), HOMA-IR (-1.2 mmol*mU/L2), triglycerides (-0.14 mmol/L), total cholesterol (-
0.7 mmol/L), LDL-cholesterol (-0.6 mmol/L), triglycerides/HDL-cholesterol (-0.55 mol/mol)
and FT3 (-0.8 pmol/L). Changes in anthropometrics were unrelated to changes in clinical
chemical indices, except for FT4 and FT3. ASAT (11 IU/L), ALAT (6 IU/L), ASAT/ALAT ratio
(0.14) and CRP (0.56 mg/L) increased, and their changes were interrelated.
Conclusion
Coping with ‘ancient mild stress factors’, including physical exercise, thirst, hunger and
climate, may influence immune status and improve anthropometrics and metabolic indices
in healthy subjects and patients with fibromyalgia.

287
1. Introduction
Chronic non-communicable diseases (CNCD), including diabetes type 2, cardiovascular
diseases, autoimmune diseases, chronic fatigue, depression and neurodegenerative diseases,
are the major causes of morbidity, work absence and invalidity. They may be responsible for
35 million out of 52 million annual deaths worldwide (1). CNCD has recently become the
major topic for the World Health Assembly. In 2013, the Lancet issued a special on CNCD (2, 3).
Treating patients with CNDC is complex and of limited availability in many countries because
of costs, while its proximate treatment has many side effects. Compliance is often low (e.g.
lipid lowering drugs, anti-hypertensives) and many interventions have proven unsuccessful
(4).
The vast majority of CNCD are caused by unhealthy lifestyle and other anthropogenic factors.
It seems that none of us is immune for the damaging effects of modern lifestyle (5, 6). Not
surprisingly, many of these diseases are preventable by changes in behavior, including
nutrition, physical activity and coping strategies (7, 8, 9, 10). The anthropogenic factors
responsible for the CNCD pandemic include physical inactivity, unhealthy diet (e.g. high
energy density refined food, too low vegetables, fruits and fish), chronic psycho-emotional
stress, insufficient sleep (loss of biorhythm) and environmental toxins, including smoking (5,
11, 12, 13, 14, 15, 16, 17, 18). All of these may be considered ‘danger signals’ that activate central
stress axes [sympathetic nervous system (SNS) and hypothalamus-pituitary-adrenal gland axis
(HPA)] and the immune system (19, 20).
Inflammation is characterized by the five classical symptoms of rubor, dolor, calor, tumor and
torpor. Inflammation requires metabolic adaptations (12). Overt injury and infection bring
about short-term allostatic responses aiming at the removal of the infectious agent, engaging
in repair, and, ultimately, recovering homeostasis in a highly coordinated fashion (21). While
ancient infectious challenges induce optimal responses with self-resolving capacity,
anthropogenic inflammatory stimuli deriving from modern society provide us with weak and
long-lasting immunological responses that take us to a condition of chronic systemic low-
grade inflammation with accompanying chronic hypometabolic adaptations (22, 12, 23). It has
become clear that many, if not all, CNCD are characterized by a state of low-grade
inflammation (LGI, 24, 25) that comes along with (e.g.) insulin resistance, hyperleptinemia,
cortisol resistance, subclinical hypothyroidism and nerve-driven immunity. Jointly, they are
responsible for the gradual development of multiple comorbidities together referred to as the
typically Western diseases of affluence (26, 11, 27, 28, 29, 30, 31).

288
The absence of ancient immune challenges in current Western societies inspired us to
hypothesize that acute stress from ancient danger signals causes redistribution of the
immune system towards its evolutionary preferred locations, and thereby favorably affects the
state of chronic systemic low-grade inflammation, normalizes stress axes activities, recovers
rhythmic function and restores insulin-insensitive pathways. Mild stress factors may activate
resolution responses based on survival mechanisms that originate from millions of years of
evolutionary pressure. In this study we investigated whether such ‘ancient stressors’, provided
by a 10-day trip through the Pyrenees, improved anthropometrics and various clinical
chemical parameters of low-grade inflammation, stress and metabolic control in 53
apparently healthy adults and two patients with fibromyalgia. The objective was to provide
proof of principle that humans can influence their immune and metabolic systems by
exposure to ancient mild acute stress factors. Our intervention mimicked, to some extent, the
‘conditions of existence’ of ancestral and current hunting/fishing-gathering populations.
2. Subjects and methods
Study group
The participants were students, scientists, physicians and other health professionals who were
engaged in clinical Psycho-Neuro-Immunology (PNI) courses throughout Europe. They were
interested to experience the impact of ancient lifestyle on their own health and well-being
and therefore jointly decided to engage in this study. No consent from a medical ethical
committee was deemed necessary, for which we refer to the constitutional law of self-
determination, in which people have a basic right to decide what they want to do with their
health (included in the patients self-determination act, United States 1991 and the Council of
Europe 2009). The participants were part of their own team, united in a research consortium
(see acknowledgments). They covered their own expenses and there were no grants. They
appointed one of us (LP) as the study coordinator. All volunteers signed an informed consent
and all were informed about the trip details. The Catalan Government and the local
government of Tremp (Spain) gave permission to execute our study without any restrictions.
We included apparently healthy adults and two patients with fibromyalgia. The fibromyalgia
syndrome was diagnosed by medical specialists. Exclusion criteria were cardiovascular
diseases, psychiatric diseases and chronic use of medication for serious illnesses.
Study design
Groups of 11 subjects, at most, participated in this 10-day trip through the Spanish Pyrenees
during the summers of 2011 (n=10), 2012 (n=32) and 2013 (n=11). The participants lived
outdoors and walked from one water-source to another. Food was provided by the
organization and with help of forest-guards from official institutes of the Catalan county.

289
Food intake was planned before the trip, based on the average daily food intake by the
traditionally living Hadzabe people in Tanzania (Supplementary Tables 1 and 2). The use of
mobile phones or other electronic devices was not allowed.
The detailed activities and condition during the 10-day study were as follows:
• First day, arrival at the hospital of Tremp (Catalonia, Spain) with an air-conditioned bus
from Barcelona (2.5 h drive). Participants were once again informed about the trip.
Anthropometric data and blood samples were collected in the fasting state.
• Daily walking trips from waterhole to waterhole, with an average walking distance of
about 14 km/day, including altitude differences up to approximately 1,000 m. The
participants carried their own backpacks with an average weight of 8 kg. The trip took
place in the part of the pre-Pyrenees with a maximum altitude of 1,900 meters above sea
level.
• Participants consumed two meals daily. The first was provided by the organization
halfway and the second on arrival at the camping site. Animals, including ducks,
chickens, turkeys, rabbits and fish, were delivered alive and killed by the participants. Fish
were caught with nets in the Noguera river. All foods were prepared on the spot by the
participants. Nutritional details are in Supplemental Table 2.
• The participants slept outside in sleeping bags on small inflatable mattresses. Outside
temperatures varied from 22 to 42 °C during daylight, whereas night temperatures varied
from 12 to 21°C. One group experienced a day of snow in the middle of July, which
prompted the organization to provide hotel accommodations for a single night.
• Bulk (intermittent) drinking behavior was recommended by drinking as much as possible
(up to satiety) after reaching a waterhole. The waterholes contained non-chloritized
drinking water.
• Some manual work was done to clean mountain trails as agreed upon with the Catalan
Government.
Anthropometric data, sample collection and analyses
Sixteen anthropometric and clinical chemical parameters were measured before departure to
the Pyrenees and after return. Anthropometric measurements were measured by one of us
(LP) at the hospital of Tremp. The subgroup of two participants with fibromyalgia syndrome
was followed for more than two years. Fasting heparin-, oxalate- and EDTA- anti-coagulated
blood samples were collected by venipuncture in the first morning and the morning of the
10th day at the study end. All samples were transported at 5 °C and processed within 1 h.
Analyses were done in the clinical laboratory of the hospital of Tremp.

290
HbA1c, total-cholesterol, HDL-cholesterol, aspartate aminotransferase (ASAT), alanine
aminotransferase (ALAT) and glucose were determined with a Cobas c-501 analyzer (Roche,
Madrid, Spain). Serum insulin was measured by chemo-luminescent assays, using Dxi-600
(Beckman, Barcelona, Spain) and Liaison XL (Diasorin, Madrid, Spain), respectively. High
sensitivity C-reactive protein (CRP) was measured with a Behring Nephelometer II Analyzer
System. HOMA-IR (mmol*mU/L2) was calculated by glucose (in mmol/L)*insulin/22.5 (in
mU/L) (Hill 2013)
Statistics
Statistical analyses were performed with IBM SPSS statistics version 23.0 (IBM Corp). Changes
during the intervention were analyzed with the Wilcoxon signed rank test at p<0.05.
Interrelations between variables were analyzed by Spearman´s correlation coefficient at
p<0.05.
Results
Study group
There were 55 participants. Their median age was 38 years (range 22-67). The number of
women was 28 (50.9%). Baseline anthropometrics are in Table 1.
Course of the trip
The 10-day trip went through the low and middle-high part of the Pyrenees. The majority of
participants did very well. If one of them got too tired, LP or JA carried his/her backpack for as
long needed. Trips were made from one waterhole to another, with a consistent midday pause
of 1-1.5 h. The Mountainside of the Pyrenees is composed of hard soil and many of its
vegetation carries thorns. Most participants suffered from small wounds on arms and legs,
caused by the thorns of the aliaga, a plant belonging to the original vegetation of the Pyrenees.
None of them suffered from infected wounds and most appeared fully cured at the end of the
trip. Participants suffered from hunger feelings during the first three days, but gradually got
used to eating only twice daily. Only one participant discontinued the trip, because she had
different expectations. The local organization arranged transport and she left. The other
participants enjoyed the trip for the full 10 days, without exceptions. Interestingly, the
majority, including LP, wanted to go home after 7 days (see discussion).
Feelings of thirst were moderate at the beginning, but ameliorated after 3 days. Participants
noticed that they could drink increasingly more water upon arrival at a waterhole and
gradually experienced less needs for ‘in-between water stops’. Three participants suffered
from what might have been neuroglycopenic periods, feeling weak, hungry, cold, dizzy and

291
trembling. However, measurement of their blood glucose level by finger prick did not reveal
hypoglycemia. One of these participants was grossly overweight and exhibited fasting
glucoses highly surpassing the normal range. He was not able to carry his backpack during
the first three days, but managed surprisingly well during the last. At night, some participants
were affected by the cold. They needed a thicker sleeping bag, which was supplied.
Participants went asleep at sunset and rose at sunrise. Overall, participants felt good,
occasionally tired, but not at all overloaded. Mosquitoes were a nuisance at night and
therefore a liquid mosquito repellent was provided by the organization. Several participants
suffered from diarrhea at the trip’s end, probably from drinking water from the non-
chloritized waterholes. Taken together, the vast majority of the participants enjoyed the trip
and recognized the benefits by feeling more healthy and recovered from Western stressful life.
Anthropometrics and clinical chemical indices
Anthropometric and clinical chemical data collected before and after the excursion were
available from 23-53 participants (Table 1). The missing values were attributable to the 2012
groups. Probably because of procedural imperfections in the Tremp lab, the outcome of the
insulin assay could not be performed in various series.
We found (Table 1) that body weight decreased with a median (range) of -3.4 kg (-7.5 to -0.7),
BMI with -1.2 kg/m2 (-4.4 to -0.2), hip circumference with -3 cm (-17 to +5), waist
circumference with -5 cm (-18 to +9) and waist/hip ratio with -0.02 (-0.14 to +0.10).
We also observed decreases (median; range; Table 1) of: glucose (-0.6; -1.7 to +0.5 mmol/L),
HbA1c (-0.1; -0,4 to +0.2 %), insulin (-4.7; -31.4 to -0.2 pmol/L), HOMA-IR (-1.2; -7.0 to -0.4
mmol*mU/L2), triglycerides (-0.14; -6.12 to +2.18 mmol/L), total cholesterol (-0.7; -2.8 to +0.4
mmol/L), LDL-cholesterol (-0.6; -3.1 to +0.6 mmol/L), triglycerides/HDL-cholesterol ratio (-
0.55; -8.98 to 1.34 mol/mol), and FT3 (-0.8; -3.4 to +3.1 pmol/L).
On the other hand we found that ASAT and ALAT activities increased with 11 IU/L (-8 to 54)
and 6 IU/L (-13 to 52), respectively, while CRP increased with 0.56 mg/L (-15.72 to +41.07).
Figure 1 shows the median and individual changes of ASAT (panel A), ALAT (panel B) and CRP
(panel C). The ASAT/ALAT ratio before the intervention was 1.23 (0.68–2.00) and increased
with 0.08 to 1.31 (0.48–2.06).
Figure 2 shows the medians of the percentage change for the anthropometric and clinical
chemical parameters that were found to change significantly during the 10 days trip through
the Pyrenees.

292
Two subjects with fibromyalgia
The two female participants (ages 37 and 38 years) were part of the 2011 group. Both managed
surprisingly well. The first three days they still suffered from overall pain, but these gradually
disappeared after 7 days. From the start, they preferred to carry their own backpacks but were
in too much pain. LP and one of the others carried their backpacks most of time during the
first three days. They were able to carry their own backpacks from then. Fatigue symptoms
were severe during the first 4 days but diminished substantially after the fifth. Sleeping
problems vanished almost immediately. Bowel movement was compromised at start, but
improved during the trip to become almost normal in one of them at the end. The second
patient kept complaining about abdominal pain, but stool consistency and frequency were
normal. No notable differences were observed in the changes of their clinical chemical
indices when compared with other participants. Both showed increases of ASAT (from 26 to
48; and 36 to 53 IU/L), ALAT (18 to 30; 25 to 39 IU/L) and CRP (0.37 to 2.71; 0.17 to 0.64 mg/L).
while their HOMA-IR decreased (0.48 to 0.31; 1.04 to 0.52 mmol*mU/L2). Their ASAT, ALAT
and CRP values decreased during the months after the trip. Three months after the trip her
ASAT had decreased from 48 to 22 IU/L in one of them, while ALAT decreased from 30 to 19
IU/L, and CRP from 2.71 to 0.5 mg/L. The other showed changes from 53 to 18 IU/L for ASAT,
39 to 16 IU/L for ALAT and 0.64 to 0.2 mg/L for CRP. Follow-up of both patients occurred
during three years. At present both women are healthy. One became pregnant and mother of
a healthy child. She started her own clinical PNI institute, while the other is completing an
University master.
One subject with arrhythmia.
One of the participants suffered from cardiac arrhythmia since 2000, following a sternum
fracture in a car accident. He was on medication since then. During the trip he stopped taking
treatment and did not suffer from arrhythmic periods during the 36-month-period that
passed since the study end.
Interrelationships Relationships between changes of the anthropometric and clinical chemical indices are
presented in Supplemental Table 3. Importantly, we found that the changes in weight, BMI,
hip circumference, waist circumference and waist/hip ratio were generally unrelated to the
changes of clinical chemical indices. Exceptions were the negative associations (p<0.05)
between waist circumference and FT3 (r= -0.359), and waist/hip ratio and FT4 (r= -0.360). The
change in HOMA-IR was negatively related to changes in total cholesterol (r= -0.457) and
LDL-cholesterol (r= -0.436). Finally, we found that the changes in ASAT, ALAT and CRP were
positively interrelated (ASAT vs. ALAT r= 0.777; ASAT vs. CRP r=0.508; and ALAT vs. CRP

293
r=0.440). Age proved positively related to the change of ASAT (r= 0.371), but was unrelated to
the changes of ALAT and CRP.
Discussion
The aim of this study was to investigate whether a 10-day trip through the Pyrenees favorably
affects anthropometric-, metabolic- and inflammatory-parameters in apparently healthy
subjects and two patients with the fibromyalgia syndrome. The trip mimicked to some extent
the ‘conditions of existence’ of ancient and contemporary hunting-gathering populations. We
found that the intervention was well tolerated and that all participants, except for one dropout,
experienced a better subjective feeling of health following its completion. Also the two
patients with fibromyalgia experienced improvements: they were able to meet the needed
physical activity after 7 days. The pain vanished and favorable effects seemed to remain
during a subsequent follow up of three years. The trip caused decreases in body weight and
BMI (median change 4.8%), hip circumference (3%), waist circumference (5.6%) and waist/hip
ratio (2.5%)(Table 1; Figure 2). Among the clinical chemical indices we found decreases of
glucose (12.5%), insulin (55%), HOMA-IR (58.1%), HbA1c (1.8%), triglycerides (20%), total-
cholesterol (13.7%), LDL-cholesterol (21.9%) and triglycerides/HDL-cholesterol ratio (19.3%). On
the other hand, the medians of ASAT and ALAT increased with 48.0% and 35.7%, respectively,
while CRP increased with 110.1%.
Favorable effects
Altogether we found that 3 features of the metabolic syndrome improved, i.e. body mass,
glucose homeostasis and circulating lipids. The fourth, i.e. blood pressure, was not recorded.
The metabolic syndrome, also named the insulin resistance syndrome, is a well-established
risk factor for various diseases of affluence, including type 2 diabetes, cardiovascular disease,
essential hypertension, polycystic ovary syndrome, non-alcoholic fatty liver disease, certain
types of cancer (colon, breast, pancreas), sleep apnea and pregnancy complications, such as
preeclampsia and gestational diabetes (32). Although we did not aim at hard endpoints, our
results suggest that the 10-day intervention could be of value to both primary and secondary
prevention of the ‘typically Western diseases of affluence’.
Potential mechanisms
Our intervention is based on causing ‘mild acute stress’ in humans who in their usual daily
lives are exposed to the chronic stress commensurate with our modern lifestyle. Acute stress
promotes release of stress hormones, including adrenaline, noradrenaline and cortisol, that
each cause profound metabolic and immunologic adaptations (33). For instance, a recent
study by the group of Pickkers (34), showed that extreme cold exposure, combined with
breathing exercise (producing intermittent hypoxia), profoundly increases adrenaline
secretion. This study and others (35, 33) show that acute stress factors increase autonomic

294
activity, accelerate immune cell proliferation and differentiation, and also stimulate the anti-
inflammatory component of the immune system (i.e. production of IL10, lactoferrin,
lysozymes) (26). Nevertheless, mild stress initially produces a pro-inflammatory response,
which may subsequently give rise to recovery from the reigning state of chronic low grade
inflammation and the return to homeostasis (36, 37).
In line with the above, we found that the observed changes in HOMA-IR and lipids were
independent of weight loss, suggesting that a combination of lifestyle factors might be at
stake. Major, highly interacting, lifestyle factors contributing to typically Western diseases are
poor diet, insufficient physical activity, chronic stress, insufficient sleep, abnormal microbial
flora and environmental pollution (smoking included) (38, 11). However, other mismatches
with our ancient lifestyle are less appreciated. For instance, the participants suffered from
thirst. Thirst relates to oxytocin production and the inhibition of stress axis activity (39, 40).
The participants were also disconnected from daily trouble and ‘self-made’ stress, which
reduced the number of anthropogenic stress factors and possibly other inflammatory ‘danger
signals’ (16). Another factor might be the prohibited use of mobile telephones and other
electronic devices. Although controversial, chronic mobile telephone usage may activate
stress systems as evidenced by a recent study of Hamzany et al. (41). Chronic use of mobile
telephones also negatively affects the production of anti-inflammatory substances in saliva
(42). Absence of artificial light might be another factor. The trip forced the participants to
adopt a ’natural day-night rhythm’ in which the sleep-wake cycle was not dominated by social
life, but rather by sunlight (43, 44, 45).
Spontaneous physical activity prior to food and water intake might be another beneficial
factor. The postprandial inflammatory response has been identified as an independent risk
factor for cardiovascular, metabolic and other non-communicable disorders (26, 46, 47, 48).
Pre-prandial exercise did not only blunt the pro-inflammatory response after food intake, but
also produced a shift towards the production of anti-inflammatory mediators by the immune
system and adipose tissue, conferring protection against possible pathogens present in food
(49, 50). Another important difference between Western life and the 10-day trip in the
Pyrenees might be the presence of ‘cutaneous- and other body surface- directed danger
signals’. All participants suffered from little wounds on hands, arm and legs because of small
injuries inflicted by sharp thorns and other natural obstacles. In addition, several participants
suffered from mild gastrointestinal disorders and diarrhea. Fiuza-Luces et al. (51) attributed
positive health effects to so-called hormetic triggers, including small external wounds and
light muscle damage. Although speculative, the immune system might have migrated to sites
that have been most susceptible to the damaging effects of the environment during evolution,
including those affecting the skin, the gastrointestinal tract and the lungs, jointly being the
sites with the highest need of immune surveillance (33).

295
Taken together, we propose that we are dealing with complex interacting lifestyle factors (5,
11) and that the current tendency to perform interventions aiming at single components,
whether or not designed as RCTs, might suffer from a reductionist approach.
Possible adverse effects
We found increases of ASAT, ALAT and CRP, which at first glance might be regarded as
genuine adverse effects. Increases of ASAT and the ASAT/ALAT ratio are related to cardiac
muscle damage (52), while those of ALAT (53) and CRP (54) are intimately related to liver
damage and infection, respectively.
Extreme sports, such as marathon and triathlon, are well known to elicit increases of lactate
dehydrogenase, creatine kinase, ALAT, ASAT, ASAT/ALAT ratio, CRP and cardiac troponins
(55, 56, 57, 58). The responses may easily exceed the upper limit of the reference range into the
myocardial infarction area. The increases of cardiac troponins were similar when exercise was
performed under controlled normoxic or hypoxic conditions, but proved dependent on
exercise duration and intensity, possibly aggravated by hypoxic conditions (56). Noakes et al.
(59) also observed that novice runners had much higher responses than experienced runners
and these results where confirmed in a later study (60). We found that the augmentation of
ASAT increased with age, as previously reported by Jastrzebski et al. (57). Increases of cardiac
markers under these conditions have not been firmly associated with irreversible cardiac
muscle damage and are at present considered benign, at least in well-trained subjects. On the
other hand, there are currently no data from long-term follow-ups (61).
The increases of both ALAT and CRP might also point at moderate liver damage and
inflammation due to environmental exposure, the latter causing mild gastrointestinal
infections from drinking water from waterholes, and small injuries on legs and arms inflicted
by thorns and falls. Although the aforementioned plausible adverse effects will definitely
require more attention in future interventions, they are not unexpected in the light of hunter-
gatherer populations. For instance, the Pygmies exhibit huge gamma-globulin bands in the
classical electrophoretic profiling of serum proteins, pointing at exposure to a host of different
microorganisms and parasites (62). Therefore, apart from the effects of intensive physical
activity, the current findings remind us of the super-hygienic conditions of our current
lifestyle. Hygiene is a major factor in longevity, but may, as a trade-off, also be at the basis of
e.g. autoimmune disease; the so-called ´hygiene hypothesis´ (63, 64, 65). For instance, a
recent study in pregnant and newborn mice revealed that helminth colonization exerts
beneficial effects on the infectious response of the offspring brain and also on microglial

296
sensitization and cognitive dysfunction at adult life (66).
Comparison with previous studies
Our study is not the first to suggest that mimicking the lifestyles of traditional hunter-gatherer
populations may be beneficial to our health. Already in 1984, O’Dea et al. showed that
overweight Australian Aborigines with type 2 diabetes who reverted to their original lifestyle
for 7 weeks, were able to improve, or even normalize, the characteristic abnormalities of
diabetes, including improvements of body weight and fasting glucose, insulin and
triglycerides. The favorable changes were attributed to weight loss, low-fat diet and increased
physical activity (67). Another example of the protective effects of our ‘ancient conditions of
existence’ against modern lifestyle and ‘Western diseases of affluence’, may be compiled from
the studies of the Kitava people in Papua New Guinea (68, 69). Lindeberg et al. showed that
these people, living a traditional lifestyle (e.g. consuming wild foods with profound physical
activity) showed low rates of cardiovascular disease, obesity and other modern diseases,
probably because of higher insulin sensitivity and lower levels of insulin, uric acid and leptin
(70).
Limitations
Our study has many limitations. There was no control group and we also did not employ a
cross-over or RCT-like design. The investigated group was relatively small and we only
measured a small number of ‘soft’ parameters. Hard endpoints can obviously not be expected
in short-term interventions in small groups with good general health. The findings of the two
patients with fibromyalgia and the single subject with arrhythmia should be regarded as
‘anecdotal’. However, whether a placebo effect or not, the subjective feeling of better health is
certainly important and so are the observed changes in anthropometric and clinical chemical
parameters. Our primary objective was to provide a proof of principle. More studies with more
participants are certainly needed, including the recording of more parameters to objectivize
e.g. the feeling of improved health. Also the minimum duration and intensity needed to
demonstrate favorable effects are uncertain as yet.
Conclusions
The outcome of this 10-day ‘Study of Origin’ suggests that a short period of return to the
‘conditions of existence’ similar to those on which our genome is based may improve
anthropometrics and metabolism by favorably challenging the immune system in apparently
healthy subjects and possibly patients with fibromyalgia. The ‘return’ may come with some
costs in more active infection, as a trade-off for the ‘chronic systemic low grade inflammation’
typical of our current lifestyle of affluence. We may increasingly appreciate that we cannot
have it all, while the evolutionary lessons of Darwin and intervention studies (71) teach us that

297
prevention might be more rewarding and affordable than the current culture of medical
treatment.

298
Acknowledgments We thank the Catalan Government and the local government of Tremp for permission to
execute the study. Mr. José Ángel Alert Rafel, head of environmental issues of the city of
Tremp, is gratefully acknowledged for arranging all licenses needed for fishing, hunting,
outdoor sleeping and fire making. We further thank all members of the consortium: Laura
Alapont, Estefanía Alayón, Cristina Alayon Rocio, Josep Angel Alert, Sheila Backus, Timo
Bartel, Rita Beldman, Azemina Bennet, Jeroen Bierman, Jacoline Bogaerds, Dick van Bolhuis,
Mike Budd, Jens Johannes Freese, Anja Gehring, Michaela Gostner, Thomas D´Have, Jan
Willem Den Hollander, Sabine de Hooghe , Javier de la Hoz, Daniel Königer, Richard de Leth,
Nicolai Loboda, Jorge López, Tjaliing Kramer, Anouk Leek, Bianca Manzano, Ivan Maria,
Nennie van der Matten Pablo Medina, Willemijn Melle, Raymond Niemeyer, Willeke v.d. Oord,
Sarai Perez Martinez, Leo Pruimboom, Ralph Ree, Claire Rother, Sandra Rossi, Irine Schmid ,
Carolina Sigalat, Maike Slikker , Michael Stary, Monika Barbara Stary, Lucy Stephens, Fernando
Luca De Tena, Wilma Tuk, Johannes Ullrich, Jerome Jacques Verbeek, Vet-Gos (Proj. Soto),
Boukje Vogel, Robin Witte.

299
References
1. Di Cesare, M., Khang, Y., Asaria, P., Blakely, T., et al. Inequalities in non-communicable
diseases and effective responses. The Lancet 381, 585-597 (2013).
2. Horton, R. Non-communicable diseases: 2015 to 2025. The Lancet 381, 509-510 (2013).
3. Bonita, R., Magnusson, R., Bovet, P., Zhao, D., et al. Country actions to meet UN
commitments on non-communicable diseases: a stepwise approach. The Lancet 381,
575-584 (2013).
4. Hogerzeil, H. V., Liberman, J., Wirtz, V. J., Kishore, S. P., et al. Promotion of access to
essential medicines for non-communicable diseases: practical implications of the UN
political declaration. The Lancet 381, 680-689 (2013).
5. Egger, G. In Search of a Germ Theory Equivalent for Chronic Disease. Preventing
Chronic Disease (2012).
6. Rook, G. A., Raison, C. L. & Lowry, C. A. Childhood microbial experience,
immunoregulation, inflammation and adult susceptibility to psychosocial stressors and
depression in rich and poor countries. Evol Med Public Health 2013, 14-17 (2013).
7. Lenoir-Wijnkoop, I., Jones, P. J., Uauy, R., Segal, L. & Milner, J. Nutrition economics -
food as an ally of public health. Br J Nutr 109, 777-784 (2013).
8. Moore, S. C., Patel, A. V., Matthews, C. E., Berrington de Gonzalez, A., et al. Leisure time
physical activity of moderate to vigorous intensity and mortality: a large pooled cohort
analysis. PLoS Med 9, e1001335 (2012).
9. Fuster V, J Narula, BB Kelly, Promoting global cardiovascular and cerebrovascular
health., in Mt Sinai J Med. Nov-Dec;79(6):625-31 (2012)
10. Amuna, P. & Zotor, F. B. Epidemiological and nutrition transition in developing
countries: impact on human health and development. Proc Nutr Soc 67, 82-90 (2008)
11. Ruiz-Núñez, B., Kuipers, R. S., Luxwolda, M. F., De Graaf, D. J., et al. Saturated fatty acid
(SFA) status and SFA intake exhibit different relations with serum total cholesterol and
lipoprotein cholesterol: a mechanistic explanation centered around lifestyle-induced
low-grade inflammation. J Nutr Biochem 25, 304-312 (2014).
12. Calay, E. S. & Hotamisligil, G. S. Turning off the inflammatory, but not the metabolic,
flames. Nature medicine 19, 265-267 (2013).
13. Goldade, K., Burgess, D., Olayinka, A., Whembolua, G. L. & Okuyemi, K. S. Applying
anthropology to eliminate tobacco-related health disparities. Nicotine Tob Res 14, 631-
638 (2012).
14. Egger, G. Obesity, chronic disease, and economic growth: a case for "big picture"
prevention. Adv Prev Med 2011, 149158 (2011).
15. Egger, G. In Search of a Germ Theory Equivalent for Chronic Disease. Preventing
Chronic Disease (2012).

300
16. Esch, T. & Stefano, G. B. The neurobiology of stress management. Neuroendocrinology
Letters 31, 19-39 (2010).
17. Dampney, R. A., Horiuchi, J. & McDowall, L. M. Hypothalamic mechanisms coordinating
cardiorespiratory function during exercise and defensive behaviour. Auton Neurosci
142, 3-10 (2008).
18. Kudielka, B. M., Federenko, I. S., Hellhammer, D. H. & Wüst, S. Morningness and
eveningness: the free cortisol rise after awakening in "early birds" and "night owls". Biol
Psychol 72, 141-146 (2006).
19. Raison, C. L. & Miller, A. H. The evolutionary significance of depression in Pathogen
Host Defense (PATHOS-D). Mol Psychiatry 18, 15-37 (2013).
20. Straub RH, Concepts of evolutionary medicine and energy regulation contribute to the
etiology of systemic chronic inflammatory diseases., in Brain Behav Immun. 25 (2011) 1–
5 (2011)
21. Buckley, C. D., Gilroy, D. W., Serhan, C. N., Stockinger, B. & Tak, P. P. The resolution of
inflammation. Nat Rev Immunol 13, 59-66 (2013).
22. Dhabhar, F. S. Effects of stress on immune function: the good, the bad, and the beautiful.
Immunol Res 58, 193-210 (2014).
23. Reilly, S. M., Chiang, S. H., Decker, S. J., Chang, L., et al. An inhibitor of the protein
kinases TBK1 and IKK-ɛ improves obesity-related metabolic dysfunctions in mice. Nat
Med 19, 313-321 (2013).
24. Huang, Z. & Kraus, V. B. Does lipopolysaccharide-mediated inflammation have a role in
OA? Nat Rev Rheumatol 12, 123-129 (2016).
25. Wärnberg, J., Cunningham, K., Romeo, J. & Marcos, A. Physical activity, exercise and
low-grade systemic inflammation. Proc Nutr Soc 69, 400-406 (2010).
26. Pruimboom, L., Raison, C. L. & Muskiet, F. A. Physical Activity Protects the Human Brain
against Metabolic Stress Induced by a Postprandial and Chronic Inflammation.
Behavioural neurology 2015, (2015).
27. Vanfleteren, L. E., Spruit, M. A., Groenen, M., Gaffron, S., et al. Clusters of comorbidities
based on validated objective measurements and systemic inflammation in patients with
chronic obstructive pulmonary disease. Am J Respir Crit Care Med 187, 728-735 (2013).
28. Levite, M. The 1st International Meeting on Nerve-Driven Immunity: Neurotransmitters
and Neuropeptides in the Immune System. Future Neurology 7, 247-253 (2012).
29. Carlton, E. D., Demas, G. E. & French, S. S. Leptin, a neuroendocrine mediator of immune
responses, inflammation, and sickness behaviors. Horm Behav 62, 272-279 (2012).
30. Tsatsoulis, A., Mantzaris, M. D., Bellou, S. & Andrikoula, M. Insulin resistance: an adaptive
mechanism becomes maladaptive in the current environment - an evolutionary
perspective. Metabolism 62, 622-633 (2013).

301
31. Steiner, A. A. & Romanovsky, A. A. Leptin: at the crossroads of energy balance and
systemic inflammation. Progress in lipid research 46, 89-107 (2007).
32. Reaven, G.M. THE INSULIN RESISTANCE SYNDROME: Definition and Dietary
Approaches to Treatment. Annu. Rev. Nutr. 2005. 25:391–406 (2005)
33. Dhabhar, F. S., Malarkey, W. B., Neri, E. & McEwen, B. S. Stress-induced redistribution of
immune cells--from barracks to boulevards to battlefields: a tale of three hormones--
Curt Richter Award winner. Psychoneuroendocrinology 37, 1345-1368 (2012).
34. Kox, M., van Eijk, L. T., Zwaag, J., van den Wildenberg, J., et al. Voluntary activation of
the sympathetic nervous system and attenuation of the innate immune response in
humans. Proc Natl Acad Sci U S A 111, 7379-7384 (2014).
35. Kox, M., Stoffels, M., Smeekens, S. P., van Alfen, N., et al. The influence of
concentration/meditation on autonomic nervous system activity and the innate
immune response: a case study. Psychosom Med 74, 489-494 (2012).
36. Dhabhar, F. S. Psychological stress and immunoprotection versus immunopathology in
the skin. Clin Dermatol 31, 18-30 (2013).
37. Dhabhar, F. S. & McEwen, B. S. Acute stress enhances while chronic stress suppresses
cell-mediated immunity in vivo: a potential role for leukocyte trafficking. Brain Behav
Immun 11, 286-306 (1997).
38. Ruiz-Núñez, B., Pruimboom, L., Dijck-Brouwer, D. A. & Muskiet, F. A. Lifestyle and
nutritional imbalances associated with Western diseases: causes and consequences of
chronic systemic low-grade inflammation in an evolutionary context. J Nutr Biochem
24, 1183-1201 (2013).
39. McGregor, I. S. & Bowen, M. T. Breaking the loop: oxytocin as a potential treatment for
drug addiction. Horm Behav 61, 331-339 (2012).
40. Krause, E. G., de Kloet, A. D., Flak, J. N., Smeltzer, M. D., et al. Hydration state controls
stress responsiveness and social behavior. J Neurosci 31, 5470-5476 (2011).
41. Hamzany, Y., Feinmesser, R., Shpitzer, T., Mizrachi, A., et al. Is human saliva an indicator
of the adverse health effects of using mobile phones? Antioxid Redox Signal 18, 622-627
(2013).
42. Hashemipour, M. S., Yarbakht, M., Gholamhosseinian, A. & Famori, H. Effect of mobile
phone use on salivary concentrations of protein, amylase, lipase, immunoglobulin A,
lysozyme, lactoferrin, peroxidase and C-reactive protein of the parotid gland. J Laryngol
Otol 128, 454-462 (2014).
43. Kantermann, T., Duboutay, F., Haubruge, D., Kerkhofs, M., et al. Atherosclerotic risk and
social jetlag in rotating shift-workers: first evidence from a pilot study. Work 46, 273-282
(2013).
44. Roenneberg, T., Allebrandt, K. V., Merrow, M. & Vetter, C. Social jetlag and obesity. Curr
Biol 22, 939-943 (2012).

302
45. Levandovski, R., Dantas, G., Fernandes, L. C., Caumo, W., et al. Depression scores
associate with chronotype and social jetlag in a rural population. Chronobiol Int 28, 771-
778 (2011).
46. Klop, B., Proctor, S. D., Mamo, J. C., Botham, K. M. & Castro Cabezas, M. Understanding
postprandial inflammation and its relationship to lifestyle behaviour and metabolic
diseases. Int J Vasc Med 2012, 947417 (2012).
47. Holmer-Jensen, J., Karhu, T., Mortensen, L. S., Pedersen, S. B., et al. Differential effects of
dietary protein sources on postprandial low-grade inflammation after a single high fat
meal in obese non-diabetic subjects. Nutr J 10, 115 (2011).
48. Peairs, A. D., Rankin, J. W. & Lee, Y. W. Effects of acute ingestion of different fats on
oxidative stress and inflammation in overweight and obese adults. Nutr J 10, 122 (2011).
49. MacEneaney, O. J., Harrison, M., O'Gorman, D. J., Pankratieva, E. V., et al. Effect of prior
exercise on postprandial lipemia and markers of inflammation and endothelial
activation in normal weight and overweight adolescent boys. Eur J Appl Physiol 106,
721-729 (2009).
50. Fielding, R. A., Violan, M. A., Svetkey, L., Abad, L. W., et al. Effects of prior exercise on
eccentric exercise-induced neutrophilia and enzyme release. Med Sci Sports Exerc 32,
359-364 (2000).
51. Fiuza-Luces, C., Garatachea, N., Berger, N. A. & Lucia, A. Exercise is the real polypill.
Physiology (Bethesda) 28, 330-358 (2013).
52. Alvarez, A. & Mukherjee, D. Liver Abnormalities in Cardiac Diseases and Heart Failure.
Int J Angiol 20, 135-142 (2011).
53. Dufour, D. R., Lott, J. A., Nolte, F. S., Gretch, D. R., et al. Diagnosis and monitoring of
hepatic injury. II. Recommendations for use of laboratory tests in screening, diagnosis,
and monitoring. Clinical chemistry 46, 2050-2068 (2000).
54. Han, J. H., Nachamkin, I., Coffin, S. E., Gerber, J. S., et al. Use of a Combination
Biomarker Algorithm To Identify Medical Intensive Care Unit Patients with Suspected
Sepsis at Very Low Likelihood of Bacterial Infection. Antimicrob Agents Chemother 59,
6494-6500 (2015).
55. Sedaghat-Hamedani, F., Kayvanpour, E., Frankenstein, L., Mereles, D., et al. Biomarker
changes after strenuous exercise can mimic pulmonary embolism and cardiac injury--a
metaanalysis of 45 studies. Clin Chem 61, 1246-1255 (2015).
56. Li, F., Hu, Y., Nie, J. & Fu, F. H. Effects of acute, intermittent exercise in hypoxic
environments on the release of cardiac troponin. Scand J Med Sci Sports (2015).
57. Jastrzębski, Z., Żychowska, M., Radzimiński, Ł., Konieczna, A. & Kortas, J. Damage to
Liver and Skeletal Muscles in Marathon Runners During a 100 km Run With Regard to
Age and Running Speed. J Hum Kinet 45, 93-102 (2015).

303
58. Shave, R. & Oxborough, D. Exercise-induced cardiac injury: evidence from novel
imaging techniques and highly sensitive cardiac troponin assays. Prog Cardiovasc Dis
54, 407-415 (2012).
59. Noakes, T. D. & Carter, J. W. The responses of plasma biochemical parameters to a 56-
km race in novice and experienced ultra-marathon runners. European journal of
applied physiology and occupational physiology 49, 179-186 (1982).
60. Smith, J. E. Effects of prolonged strenuous exercise (marathon running) on biochemical
and haematological markers used in the investigation of patients in the emergency
department. British Journal of Sports Medicine 38, 292-294 (2004).
61. Wedin, J. O. & Henriksson, A. E. Postgame elevation of cardiac markers among elite
floorball players. Scand J Med Sci Sports 25, 495-500 (2015).
62. Mann, G. V., Roels, O. A., Price, D. L. & Merrill, J. M. Cardiovascular disease in African
Pygmies: a survey of the health status, serum lipids and diet of Pygmies in Congo.
Journal of chronic diseases 15, 341-371 (1962).
63. Rook, G. A., Lowry, C. A. & Raison, C. L. Hygiene and other early childhood influences on
the subsequent function of the immune system. Brain Res 1617, 47-62 (2015).
64. Rook, G. A. 99th Dahlem conference on infection, inflammation and chronic
inflammatory disorders: darwinian medicine and the 'hygiene' or 'old friends'
hypothesis. Clin Exp Immunol 160, 70-79 (2010).
65. Sironi, M. & Clerici, M. The hygiene hypothesis: an evolutionary perspective. Microbes
Infect 12, 421-427 (2010).
66. Williamson, L. L., McKenney, E. A., Holzknecht, Z. E., Belliveau, C., et al. Got worms?
Perinatal exposure to helminths prevents persistent immune sensitization and cognitive
dysfunction induced by early-life infection. Brain Behav Immun 51, 14-28 (2016).
67. O'dea, K. Marked improvement in carbohydrate and lipid metabolism in diabetic
Australian Aborigines after temporary reversion to traditional lifestyle. Diabetes 33, 596-
603 (1984).
68. Lindeberg, S., NILSSON-EHLE, P., Terent, A., Vessby, B. & Schersten, B. Cardiovascular
risk factors in a Melanesian population apparently free from stroke and ischaemic heart
disease: the Kitava study. Journal of internal medicine 236, 331-340 (1994).
69. Lindeberg, S., Eliasson, M., Lindahl, B. & Ahrén, B. Low serum insulin in traditional
Pacific Islanders—the Kitava Study. Metabolism 48, 1216-1219 (1999).
70. Lindeberg, S., Söderberg, S., Ahren, B. & Olsson, T. Large differences in serum leptin
levels between nonwesternized and westernized populations: the Kitava study. Journal
of internal medicine 249, 553-558 (2001).
71. Manheimer, E. W., van Zuuren, E. J., Fedorowicz, Z. & Pijl, H. Paleolithic nutrition for
metabolic syndrome: systematic review and meta-analysis. Am J Clin Nutr 102, 922-932
(2015).
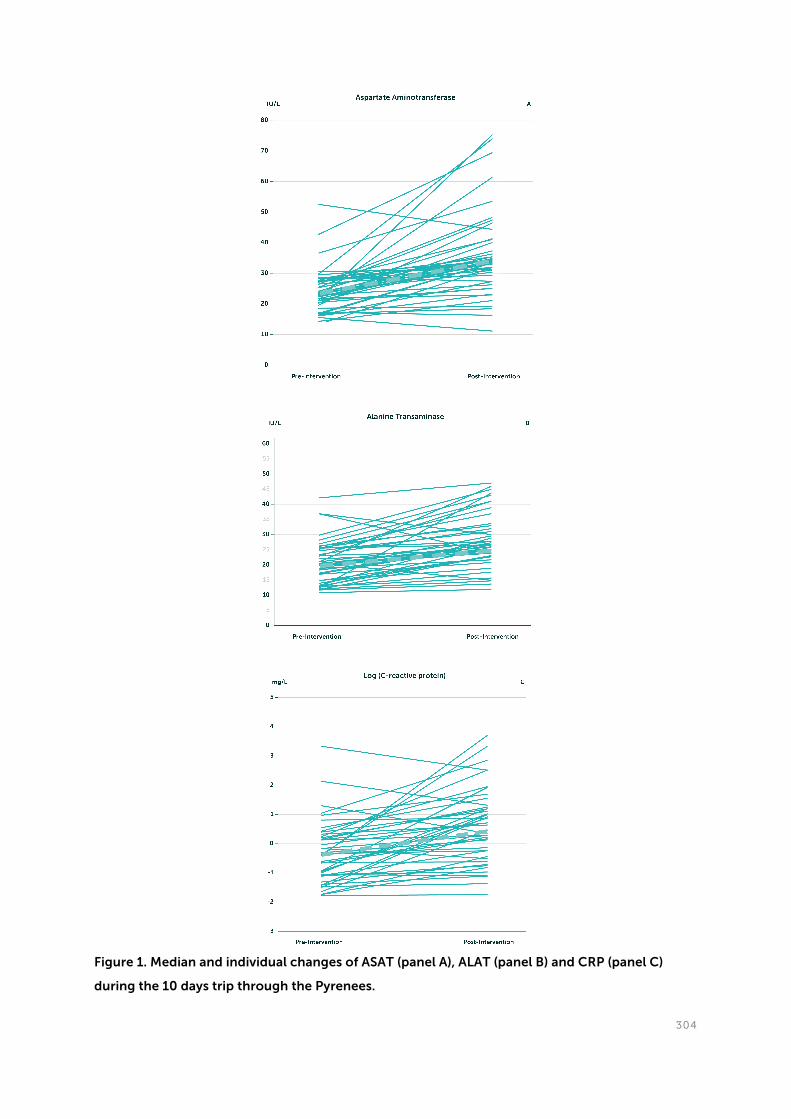
304
Figure 1. Median and individual changes of ASAT (panel A), ALAT (panel B) and CRP (panel C)
during the 10 days trip through the Pyrenees.

305
Figure 2. Medians of the percentage changes of anthropometric and clinical chemical
parameters during the 10 days trip through the Pyrenees.
Only significant changes are shown (see Table 1). For abbreviations see legend of Table 1.

306
Table 1. Anthropometrics and clinical chemical indices at baseline and at the study end
Data are medians (range). Abbreviations: ALAT, alanine aminotransferase; ASAT, aspartate aminotransferase; BMI, body mass
index; CRP, C-reactive protein; FT3, free triiodothyronine; FT4, free thyroxine; HbA1c, hemoglobin A1c; HDL, high-density-
lipoprotein; HOMA-IR, homeostasis model assessment-estimated insulin resistance; LDL, low-density lipoprotein; N.M. Not
measured; TG, triglycerides; TSH, thyroid-stimulating hormone. *, Significant difference between the values before and after
the intervention by Wilcoxon signed rank test at p<0.05.

307
Food consumed Hadza en% kcal
Meat 25 625
Tubers 17 425
Wild Berries 25 625
Fruits
Eggs
Fish
Nuts
Green
Vegetables
Baobab 11 275
Honey 22 550
Total 100 2500
Supplemental Table 1. Average food intake of Hadzabe hunter-gatherers
Data derive from Pontzer 2012, Murray 2001, IDF 2012, www.philosophy.dept.shef.ac.uk/culture&mind/people/crittendena/
1. Murray, S. S., Schoeninger, M. J., Bunn, H. T., Pickering, T. R. & Marlett, J. A. Nutritional Composition of Some
Wild Plant Foods and Honey Used by Hadza Foragers of Tanzania. Journal of Food Composition and Analysis
14, 3-13 (2001).
2. Pontzer, H., Raichlen, D. A., Wood, B. M., Mabulla, A. Z., et al. Hunter-gatherer energetics and human obesity.
PLoS One 7, e40503 (2012).

308
Nutrient Average amount kcal /unit total kcal kcal/part/10 days
Banana 80 105 8400 840
Apple 70 20 1400 140
Melon 10 kilo 300 3000 300
Mangos 4,5 kilo 645 2900 290
Pineapple 3 kilo 460 1380 138
Watermelon 8 kilo 280 2260 226
Dates 1,1 kilo 2760 3036 304
Onions 12 kilo 400 4800 480
Lettuce (6 types) 3 kilo 160 480 48
Cucumber 3 kilo 112 336 33
Garlic 2 kilo 132 264 26
Olives 1,5 kilo 1660 2500 250
Carrots 2,4 kilo 410 985 99
Mushrooms 1,1 kilo 225 250 25
Zucchini 1,5 kilo 160 240 24
Asparagus 1 kilo 220 220 22
Avocado 6 kilo 1650 10100 1010
Pumpkin 2 kilo 200 400 40
Leek 2 kilo 550 1100 110
Sweet potato 1,5 kilo 720 1080 108
Coconut 5 kilo 3180 15900 159
Raisins 1 kilo 2860 2860 286
Berries 3 kilo 480 1440 144
Subtotal 6532
Mayonaise 1 kilo 6300 6300 630
Olive oil 3 liters 8400 25200 2520
Honey 2 kilo 7000 14000 140
Subtotal 3290
Deer 11 kilo 1480 26280 2628
Chicken 9 kilo 2140 19260 1920
Rabbit 2 kilo 2764 5528 553
Sweat water fish 8 kilo 1590 12720 1272
Tuna in olive oil 5 kilo 4030 20150 2015
Duck 6 kilo 2016 12096 1209
Eggs 5,5 kilo 1400 7700 770
Nuts 3,7 kilo 575 20275 2027
Subtotal 12394
Total/pp/trip 22216
Total/pp/day 2222
Supplemental Table 2. Average food intake of participants during the ten days trip through the
Pyrenees (amount/10 persons)

309
Supplemental Table 3. Spearman's correlation coefficients for the relations between the
changes in anthropometric and clinical chemical indices during the study
Changes refer to the difference between pre- and post-intervention data. Abbreviations: ALAT, alanine aminotransferase;
ASAT, aspartate aminotransferase; CRP, C-reactive protein; FT3, free triiodothyronine; FT4, free thyroxine; HBA1c,
hemoglobin A1c; HDL, high-density-lipoprotein; HDL-C, high-density-lipoprotein-cholesterol; LDL, low-density lipoprotein;
; N.M. Not measured; TG, triglycerides; TSH, thyroid-stimulating hormone. *, Significant at p<0.05; ** , significant at p<0.01.

310
Summary and Future perspectives
This thesis describes the multiple ways by which the human immune system can react upon
direct and indirect challenges, such as infection and wounds on the one hand, and chronic
multi metabolic stress factors on the other. The immune system exhibits a type of selfish
behaviour during both acute- and chronic activity states. Acute activation of the immune
system is normally finalized in a short time through multiple mechanisms exerting immune
inhibiting effects. Several of these mechanisms are intrinsic to the immune system itself, such
as the negative biofeedback function of lactic acid produced by the anaerobic metabolism of
an activated immune system (Hoque 2014, Fischer 2007). Systemic inhibitors include cortisol,
testosterone and adrenalin and all these have protective short-lasting immune suppressive
roles. An immune suppressive period serves four purposes. First of all it facilitates the recovery
of energy sources and energy distribution to those organs and systems disposed-of during the
inflammatory phase. Second, wound healing can only start when the inflammation has been
finalized. The third purpose is to prevent the development of lymphocytes with sensitivity
against auto-antigens, which could cause autoimmune disease. Finally, since the opposite
also occurs, increased self-tolerance through long-term contact between activated
lymphocytes and auto-antigens, the immune suppressive state also decreases the chance of
developing cancer. Short-term immune suppression prevents self-tolerance against cancer
cells, which implies that when tumour cells develop they will be recognized as non-self.
A state of chronic activity of the immune system can only be achieved when this state
becomes supported by reactivation and gluconeogenic strategies, of which the development
of insulin resistance with compensatory hyperinsulinemia is an example. Chronic activity of
the immune system is caused by the total number of so-called anthropogenes, which include
proximal lifestyle factors, such as overeating, sitting time, and insufficient sleep, but also
economy, environmental toxins and politics, the latter as examples of distal factors. The sum
of risk factors produces chronic activation of stress axes and endotoxaemia, causing a chronic
infectious state that is referred to as chronic low-grade inflammation. This low-grade
inflammation should be considered as the cause of most, if not all, chronic non-
communicable diseases (CNCD).

311
Chapter 1. Chronic systemic low-grade inflammation
Paragraph 1.1
The brain and the immune system are the only two systems of the human body capable of
dominating all other organs and systems with respect to the use of resources, including
glucose. The brain dominates during daytime hours and stressful situations, whereas the
immune system protects us during the night, infectious periods and wound healing. Both
systems exhibit similar capacities to pull on energy and other essential resources, using
strategies favouring their own functional and anatomical benefits. Human evolution has put
the brain on top of the body’s systems, resulting in a shift from strong to smart. However, the
immune system is very old and robust. If necessary, it is activated through a variety of non-
specific factors and most often when problems are not solved in an appropriate time frame. If
chronically activated the immune system demonstrates an even more selfish behaviour than
the selfish brain, inducing chronic low-grade inflammation and multiple related diseases. But
before castigating the immune system for this behaviour it is crucial to recognize that it is
only doing what it is made for: trying to protect us.
Paragraph 1.2
In this paragraph, we focus on lifestyle changes, especially dietary habits, that are at the basis
of chronic systemic low-grade inflammation, insulin resistance and ‘typically Western’
diseases. Our sensitivity to develop insulin resistance traces back to our rapid brain growth in
the past 2.5 million years. An inflammatory reaction jeopardizes the high glucose needs of our
brain, causing various adaptations, including insulin resistance, functional reallocation of
energy-rich nutrients and by changing the serum lipoprotein composition. The latter aims at
redistribution of lipids, modulation of the immune reaction, and active inhibition of reverse
cholesterol transport for damage repair. With the advent of the agricultural and industrial
revolutions, we have introduced numerous false inflammatory triggers in our lifestyle, driving
us to a state of chronic systemic low grade inflammation that eventually leads to ‘typically
Western’ diseases via an evolutionary-conserved interaction between our immune system
and metabolism. The underlying triggers are an abnormal dietary composition and microbial
flora, insufficient physical activity and sleep, chronic stress and environmental pollution. The
disturbance of our inflammatory/anti-inflammatory balance is illustrated by dietary fatty acids
and antioxidants. The current decrease in years without chronic disease is rather due to
‘nurture’ than ‘nature’, since less than 5% of the ‘typically Western’ diseases are primary
attributable to genetic factors. Resolution of the conflict between environment and our
ancient genome might be the only effective manner for ‘healthy aging’, and to achieve this we
might have to return to the lifestyle of the Palaeolithic era, as translated to culture of the 21st
century.

312
Paragraph 1.3
In 1996, Serhan and colleagues introduced the term ‘Resoleomics’ as the mechanistic process
of inflammation resolution. Their major discovery is that the onset to the conclusion of an
inflammatory reaction is a highly controlled process orchestrated by the immune system
itself, and not simply the consequence of an ‘extinguished’ or ‘exhausted’ immune reaction.
Resoleomics can be considered as the evolutionary conserved mechanism of restoring
homeostatic balances after injury, inflammation and infection. Under normal circumstances,
Resoleomics should be able to conclude inflammatory responses. However, considering the
modern pandemic increase of chronic somatic and psychiatric illnesses involving chronic
inflammation, it has become apparent that Resoleomics is currently not fulfilling its
potentially resolving capacity. We suggest that recent drastic changes in lifestyle, including
diet and psycho-emotional stress, are responsible for inflammation and for disturbances in
Resoleomics. In addition, current treatments, chronic use of anti-inflammatory medication
included, suppress Resoleomics. These, from an evolutionary point of view, new lifestyle
factors should be considered health hazards, as they are capable of causing long-term or
chronic activation of the central stress axes. The immune system is designed to provide
solutions for fast, intensive, hazards and not to cope with long-term, chronic, stimulation. The
never-ending stress factors of recent lifestyle changes have pushed the immune system and
the central stress system into a constant state of activity, leading to chronically unresolved
inflammation and increased vulnerability for chronic disease. Our hypothesis is that modern
diet, increased psycho-emotional stress and chronic use of anti-inflammatory medication
disrupt the natural process of inflammation resolution, i.e. Resoleomics.
Paragraph 1.4
Nowadays, CNCDs are the leading causes of work absence, disability and mortality worldwide.
Most of these diseases are associated with low-grade inflammation. Here we hypothesize that
stresses (defined as homeostatic disturbances) can induce low-grade inflammation, which
augments the need of water, sodium and energy-rich substances to meet the increased
metabolic demand induced by the stressor. One way of triggering low-grade inflammation is
by augmented intestinal barrier permeability through activation of various components of the
stress system. Although beneficial to meet the demands necessary during stress, increased
intestinal barrier permeability also raises the chance of translocation of bacteria and their
toxins, i.e. their passage from the intestinal lumen into the blood circulation. In combination
with modern life-style factors, increased bacteria/bacterial toxin translocation, arising from a
more permeable intestinal wall, causes a low-grade inflammatory state. We support this
hypothesis with numerous studies, showing associations with CNCDs and markers of
endotoxaemia, and thereby suggest that this mechanism plays a pivotal, and perhaps even
causal, role in the development of low-grade inflammation and its related diseases.

313
Paragraph 1.5
Wheat is one of the mostly consumed cereal grains worldwide and makes up a substantial part
of the human diet. Although government-supported dietary guidelines in Europe and USA
advise individuals to eat ample amounts of (whole) grain products per day, cereal grains
contain ‘anti-nutrients’, such as wheat gluten and wheat lectin, that in humans can elicit
dysfunction and disease. In this review we discuss evidence from in vitro, in vivo and human
intervention studies that describe how the consumption of wheat, but also other cereal grains,
may contribute to the manifestation of chronic inflammation and auto-immune diseases by
increasing intestinal permeability and initiating a pro-inflammatory immune response.
Paragraph 1.6
Various recent, positively selected, adaptations to new nutrients have been identified. Lactase
persistence is among the best known, enabling exposed populations to drink milk, notably to
digest lactose, at post-weaning age. An augmented number of amylase gene (AMY1) copies,
giving rise to higher salivary amylase activity, has been implicated in the consumption of
starch-rich foods. Higher AMY1 copy numbers has been demonstrated in populations with
recent histories of starchy diets. It is, however, questionable whether the resulting
polymorphisms have merely exerted positive selection pressure by providing easily-available
sources of macro- and micronutrients. Humans have explored new environments more than
any other animal. Novel environments challenge the host, but especially its immune system
with new climatic conditions, foods and especially pathogens. With the advent of the
agricultural revolution and the concurrent domestication of cattle came new pathogens and
diseases (zoonoses). We contend that specific new food ingredients (e.g. gluten) and novel
pathogens drove selection for lactase persistence and higher AMY gene copy numbers. Both
adaptations provide ample glucose for activating the sodium glucose-dependent co-
transporter 1 (SGLT1), which is the principal transporter of glucose, together with sodium and
water, in the small intestine and to a lesser extent the stomach (Chen 2010). Following starch
ingestion, people with higher AMY1 copy numbers have more rapid increases of insulin and
concomitantly lower plasma glucose, compared with counterparts with lower AMY1 copies
(Mandel 2012). Rapid uptake of glucose, sodium and water confers protection against
potentially lethal dehydration, hyponatraemia and ultimately multiple organ failure, especially
during infection accompanied by diarrhoea and vomiting. Also oral rehydration therapy aims
at SGLT1 activity and is the current treatment of choice for these conditions. We hypothesize
that lifelong lactase activity and rapid starch digestion should be looked at as the evolutionary
precursor of the current oral rehydration therapy.

314
Chapter 2. Physical inactivity
Paragraph 2.1
In recent years, it has become clear that chronic systemic low-grade inflammation is at the
root of many, if not all, ‘typically Western’ diseases associated with the metabolic syndrome.
While much focus has been given to sedentary lifestyle as a cause of chronic inflammation, it
is less often appreciated that chronic inflammation may also promote a sedentary lifestyle,
which in turn causes chronic inflammation. Given that even minor increases in chronic
inflammation reduce brain volume in otherwise healthy individuals, the bidirectional
relationship between inflammation and sedentary behaviour may explain why humans have
lost brain volume in the last 30,000 years and also intelligence in the last 30 years. We review
evidence that lack of physical activity induces chronic low-grade inflammation and,
consequently, an energy conflict between the selfish immune system and the selfish brain.
Although the notion is widespread that increased physical activity would improve health in
the modern world, we here provide a novel perspective on this truism by providing evidence
that recovery of normal human behaviour, such as spontaneous physical activity, would calm
pro-inflammatory immune activity, thereby allocating more energy to the brain and other
organs, and by doing so would improve human health.
Paragraph 2.2
The evolution of humankind has taken millions of years, during which environmental
factors gradually shaped its genome with the aim to adapt to the reigning circumstances
(Darwin). One of the most vital behavioural adaptations of mammals in general, and humans
in particular, is their capacity to be self-sufficient through physical activity. Physical activity
abilities, including long distance running, jumping, climbing and carrying things have
probably been necessary to outrun wild animals, to search for food and to hide from danger.
In contrast, individuals physically or psychologically unable to ‘take care of themselves’, were
more susceptible to early death and therefore genetic extinction.
The current society is characterized by sedentary, instead of ‘moving’, individuals. Physical
inactivity is not just a possible factor related with chronic disease, but should be considered
the actual cause of the majority of human illness. The majority of people know that exercise is
necessary and beneficial. Nevertheless, almost 75% of the current population doesn’t reach the
estimated minimum of the necessary activity. Physical inactivity belongs to the
characteristics of sickness behaviour; which is probably protective for the organism. Sickness
behaviour, including depressive mood, seems to protect against infection, injury, social
conflict and facilitates energy conservation. Sickness behaviour is based on immune-brain
mechanisms and can be defined as non-permissive behaviour. Long-term non-permissive
behaviour can lead to chronic disease because of reduced physical activity and self-defeating

315
coping styles, converting non-permissive behaviour into a non-permissive brain disorder. We
propose that physical inactivity disease is synonymous with a non-permissive brain disorder
and that this disorder produces a so called ‘reptile phenotype’, characterized by hypothermia,
poor hair growth, decreased fertility and low basal metabolic rate.

316
Chapter 3. Lifestyle intervention
Paragraph 3.1
Chronic low-grade inflammation and insulin resistance are intimately related entities that are
common to most, if not all, chronic diseases of affluence. We hypothesized that a short-term
intervention based on ‘ancient stress factors’ may improve anthropometrics and clinical
chemical indices. In this paragraph we investigated whether a 10-days-mimic of a hunter-
gatherer lifestyle favourably affects anthropometrics and clinical chemical indices. Fifty-three
apparently healthy subjects and two patients with fibromyalgia (22–69 years, 28 women), in 5
groups, engaged in a 10-day trip through the Pyrenees. They walked 14 km/day on average,
carrying an 8-kilo backpack. Raw food was provided and self-prepared. while water was
obtained from waterholes. They slept outside in sleeping bags and were exposed to
temperatures ranging from 12-42 °C. Anthropometric data and fasting blood samples were
collected at baseline and the study end. We observed median decreases (p≤0.002) of body
weight (-3.5 kg), BMI (-1.1 kg/m2), hip (-3 cm) and waist (-5 cm) circumferences, glucose (-0.6
mmol/L), HbA1c (-0.1%), insulin (-4.7 pmol/L), HOMA-IR (-1.2 mmol*mU/L2), triglycerides (-
0.14 mmol/L), total cholesterol (-0.7 mmol/L), LDL-cholesterol (-0.6 mmol/L),
triglycerides/HDL-cholesterol ratio (-0.55 mol/mol) and FT3 (-0.8 pmol/L). Changes in
anthropometrics were unrelated to changes in clinical chemical indices, except for FT4 and
FT3. ASAT (11 IU/L), ALAT (6 IU/L), ASAT/ALAT ratio (0.14) and CRP (0.56 mg/L) increased, and
their changes were interrelated. We conclude that coping with ‘ancient mild stress factors’,
including physical exercise, thirst, hunger and climate, may influence immune status and
improve anthropometrics and metabolic indices in healthy subjects and patients with
fibromyalgia.

317
Future perspectives
Many CNCDs are considered ‘incurable’ because of a reigning ‘one cause’ dogma. This way of
thinking was inspired by Louis Pasteur (1822–1895), who showed that an infectious disease
originates from a single causal factor. This factor can be demonstrated and eliminated by
specific treatments. However, the ‘germ theory of disease’ merely states that some diseases are
caused by infectious agents. It is our believe that CNCDs will not become cured if we keep
insisting on mono-etiological backgrounds. We suggest that CNCDs are caused by multi-
metabolic anthropogenic risk factors, which not only include individual lifestyle factors such
as smoking, lack of exercise and high calorie diets, but also more distant political, economic,
social, cultural and even evolutionary risk factors. Our view is largely in line with the papers of
Egger et al. (Egger 2012, Egger 2014a, Egger 2014b). With the exception of diseases with
primary causes in genetics (‘inborn errors’) it seems theoretically possible that each of us is
able to enjoy ‘healthy aging’ and to ultimately die from old age. However, the future does not
seem favourable: the number of anthropogenic factors is likely to increase. Several of these
adverse factors seem inevitable and most of us have little ability to influence them in an
effective manner. We nevertheless believe that prevention remains the most effective manner
to ‘solve’ CNCDs. These preventive measures may aim at resilience against the toxic triggers
of modern life. The best choice would be regular exposure to hormetic triggers, as shown in
our pilot intervention named the ‘Study of origin’ (Chapter 3). It is not difficult, and may even
be enjoyable, to include some of these intermittent hormetic triggers in daily life. There is
increasing support for the health effects of ‘intermittent living’, including intermittent fasting
(Barnosky 2014), intermittent hypoxia (Duennwald 2013), intermittent sitting (Henson 2015)
and intermittent heat (Laukkanen 2015). We suggest that future studies may aim at such
interventions, and that funds will be made available for these aims. There is, however, one
hesitation: the outcomes of these interventions may at most demonstrate that Darwin was
right when he coined his view that ‘organisms adapt to the conditions of existence’. Thus
what is the need to await their outcomes: the future starts today.

318
References
1. Barnosky, A. R., Hoddy, K. K., Unterman, T. G. & Varady, K. A. Intermittent fasting vs daily
calorie restriction for type 2 diabetes prevention: a review of human findings. Transl Res
164, 302-311 (2014).
2. Chen, J., Williams, S., Ho, S., Loraine, H., et al. Quantitative PCR tissue expression
profiling of the human SGLT2 gene and related family members. Diabetes Ther 1, 57-92
(2010).
3. Duennwald, T., Gatterer, H., Groop, P. H., Burtscher, M. & Bernardi, L. Effects of a single
bout of interval hypoxia on cardiorespiratory control and blood glucose in patients with
type 2 diabetes. Diabetes Care 36, 2183-2189 (2013).
4. Egger, G. In Search of a Germ Theory Equivalent for Chronic Disease. Preventing
Chronic Disease (2012).
5. Egger, G. & Dixon, J. Beyond obesity and lifestyle: a review of 21st century chronic
disease determinants. Biomed Res Int 2014, 731685 (2014a).
6. Egger, G., Katz, D., Sagner, M., Dixon, J. & Stevens, J. The art and science of chronic
disease management come together in a lifestyle-focused approach to primary care. Int
J Clin Pract 68, 1406-1409 (2014b).
7. Fischer, K., Hoffmann, P., Voelkl, S., Meidenbauer, N., Ammer, J., Edinger, M.,
8. Gottfried, E., Schwarz, S., Rothe, G., Hoves, S., et al. (2007). Inhibitory effect of tumor cell-
derived lactic acid on human T cells. Blood 109, 3812– 3819. (2007)
9. Hoque, R., Farooq, A., Ghani, A., Gorelick, F., & Mehal, W. Z. Lactate Reduces Organ Injury
in Toll-like Receptor- and Inflammasome-Mediated Inflammation via GPR81-mediated
Suppression of Innate Immunity. Gastroenterology, 1–37. (2014).
10. Laukkanen, T., Khan, H., Zaccardi, F. & Laukkanen, J. A. Association Between Sauna
Bathing and Fatal Cardiovascular and All-Cause Mortality Events. JAMA Internal
Medicine 175, 542 (2015).
11. Mandel, A. L. & Breslin, P. A. High endogenous salivary amylase activity is associated
with improved glycemic homeostasis following starch ingestion in adults. J Nutr 142,
853-858 (2012).

319
Samenvatting en toekomstperspectieven
In dit proefschrift worden de verschillende wegen beschreven waarlangs het menselijk
immuunsysteem kan reageren op directe en indirecte uitlokkers, zoals infectie en verwonding
enerzijds en chronische metabole stressfactoren anderzijds. Het immuunsysteem vertoont
een soort zelfzuchtig gedrag, zowel gedurende acute, als chronische activiteit. Een acuut
geactiveerd immuunsysteem wordt normaal gesproken binnen korte tijd weer uitgezet via
diverse mechanismen met immuunremmende effecten. Verschillende van deze
mechanismen zijn intrinsiek aan het immuunsysteem zelf, zoals de negatieve
feedbackfunctie van melkzuur dat geproduceerd wordt door de anaerobe stofwisseling van
een geactiveerd immuunsysteem (Hoque 2014, Fischer 2007). Systemische remmers zijn
onder meer cortisol, testosteron en adrenaline; deze hebben allemaal een beschermende,
kortdurende, immuunonderdrukkende rol.
Een periode van immuunonderdrukking heeft vier doelen. Ten eerste faciliteert het de
wedertoevoer van energiebronnen en het herstel van de energiedistributie naar de organen
en systemen die tijdens de ontstekingsreactie ‘verwaarloosd’ zijn. Ten tweede kan wondheling
slechts dan van start gaan, wanneer de ontstekingsfase is afgerond. Het derde doel is het
voorkómen van de ontwikkeling van lymfocyten die gevoelig zijn voor auto-antigenen,
waardoor auto-immuunziekten zouden kunnen ontstaan. Tot slot, aangezien het
tegenovergestelde ook voorkomt – i.e. verhoogde zelftolerantie door langdurig contact tussen
geactiveerde lymfocyten en auto-antigenen – verlaagt de toestand van
immuunonderdrukking ook de kans op het ontwikkelen van kanker. Kortdurende
immuunonderdrukking voorkòmt zelftolerantie tegen kankercellen, hetgeen impliceert dat,
wanneer tumorcellen zich ontwikkelen, ze worden herkend als niet eigen.
Een toestand van chronische immuunactiviteit kan alleen worden bereikt indien deze
toestand ondersteuning krijgt middels reactivatie- en gluconeogenesestrategieën. De
ontwikkeling van insulineresistentie met compenserende hyperinsulinemie is daarvan een
voorbeeld. Chronische activatie van het immuunsysteem wordt veroorzaakt door het totaal
van zogenaamde antropogene factoren, waaronder proximale leefstijlfactoren, zoals overeten,
langdurig zitten en onvoldoende slaap, maar ook economie, toxinen in het milieu en politiek.
Deze laatste zijn voorbeelden van distale factoren. De som van de risicofactoren leidt tot
chronische activatie van de stressassen en endotoxemie, waardoor een chronische toestand
van infectie ontstaat waarnaar verwezen wordt als chronische laaggradige ontsteking. Deze
chronische laaggradige ontsteking zou gezien moeten worden als de oorzaak van de meeste,
zo niet alle, chronische niet-overdraagbare aandoeningen (CNCD’s – chronic non-
communicable diseases).

320
Hoofdstuk 1. Chronische systemische lage graad ontsteking
Paragraaf 1.1
De hersenen en het immuunsysteem zijn de enige twee systemen van het menselijk lichaam
die in staat zijn alle andere organen en systemen te domineren op het gebied van
bronnengebruik, waaronder glucose. De hersenen domineren overdag en in stressvolle
situaties, terwijl het immuunsysteem ons vooral beschermt tijdens de nacht, bij infecties en bij
het wondhelingsproces. Beide systemen vertonen een vergelijkbare capaciteit om energie en
andere essentiële bronnen aan te trekken, waarbij ze strategieën gebruiken ten voordele van
hun eigen anatomie en functie. De menselijke evolutie heeft de hersenen in de koppositie
geplaatst ten opzichte van de andere lichaamssystemen, waardoor er een verschuiving heeft
plaatsgevonden van sterk naar slim. Het immuunsysteem is echter zeer oud en robuust.
Indien nodig, wordt het geactiveerd door een keur aan aspecifieke risico factoren, zoals
psychoemotionele stress en meestal wanneer risico factoren die het immuunsysteem
activeren niet binnen een passend tijdsbestek zijn opgelost. Indien chronisch geactiveerd,
laat het immuunsysteem zelfs nog zelfzuchtiger gedrag zien dan het zelfzuchtige brein,
waarbij het chronische laaggradige ontsteking en verschillende daaraan gerelateerde ziekten
veroorzaakt. Maar voordat we het immuunsysteem alle schuld in de schoenen schuiven, is het
van het grootste belang om te erkennen dat het alleen maar doet waar het voor gemaakt is:
proberen ons te beschermen.
Paragraaf 1.2
In deze paragraaf richten we ons op veranderingen in de leefstijl, met name eetgewoonten,
die aan de basis staan van chronische laaggradige ontsteking, insulineresistentie en ‘typisch
westerse’ ziekten. Onze gevoeligheid voor de ontwikkeling van insulineresistentie is terug te
voeren op de snelle hersengroei die in de afgelopen 2,5 miljoen jaar bij onze soort heeft
plaatsgevonden. Een ontstekingsreactie brengt de grote glucosevraag van onze hersenen in
gevaar, waardoor verschillende aanpassingen ontstaan, waaronder insulineresistentie,
functionele herverdeling van energierijke nutriënten en een veranderde samenstelling van
het serumlipoproteïne. Dit laatste heeft als doel het herverdelen van vetten, modulatie van de
immuunreactie en actieve remming van omgekeerd cholesteroltransport om schade te
herstellen.
Met de komst van de landbouw- en industriële revolutie, hebben we verschillende valse
ontstekingsuitlokkers in onze leefstijl geïntroduceerd, waardoor we in de richting worden
geduwd van een toestand van chronische laaggradige ontsteking die uiteindelijk leidt tot
‘typisch westerse’ ziekten via een evolutionair geconserveerde interactie tussen ons
immuunsysteem en onze stofwisseling. De onderliggende uitlokkers zijn een abnormale
voedingssamenstelling en microbiële flora, onvoldoende lichaamsbeweging en slaap,

321
chronische stress en milieuvervuiling. De verstoring van de ontstekings-/anti-
ontstekingsbalans wordt geïllustreerd door voedingsvetzuren en antioxidanten. De huidige
afname in jaren dat men vrij is van chronische ziekte, wordt eerder veroorzaakt door ‘nurture’
dan ‘nature’, aangezien minder dan 5% van de ‘typisch westerse’ ziektebeelden primair zijn toe
te wijzen aan genetische factoren. De oplossing van het conflict tussen de huidige omgeving
en ons oude genoom zou wel eens de enige effectieve manier kunnen zijn om gezond oud te
worden. Om dit te bereiken, zouden we wellicht terug moeten naar de leefstijl van het
paleolithicum, vertaald naar de cultuur van de 21e eeuw.
Paragraaf 1.3
In 1996 introduceerden Serhan en zijn collega’s de term ‘resoleomics’ als benaming voor het
mechanistische proces waarlangs ontstekingen opgelost worden. Hun grootste ontdekking is
dat de beëindiging van een ontstekingsreactie een hoogst gecontroleerd proces is dat door
het immuunsysteem zelf geleid wordt, en dus niet simpelweg het gevolg is van een
‘uitgedoofde’ of ‘uitgeputte’ immuunreactie. Resoleomics kan worden gezien als het
evolutionair bewaard gebleven mechanisme waarmee homeostatische balansen worden
hersteld na verwonding, ontsteking en infectie. Onder normale omstandigheden zou
resoleomics in staat moeten zijn om ontstekingsreacties te beëindigen. Echter, met de
pandemische toename van chronische somatische en psychiatrische ziekten waarbij
chronische ontsteking een rol speelt, is het duidelijk geworden dat resoleomics zijn oplossend
potentieel momenteel niet vervult. We stellen daarom dat recente drastische veranderingen in
leefstijl, waaronder in de voeding en psycho-emotionele stress, verantwoordelijk zijn voor
ontstekingen en verstoringen van resoleomics. Daarbij wordt resoleomics door de huidige
behandelingswijzen onderdrukt, waaronder door chronisch gebruik van ontstekingsremmers.
Deze, vanuit een evolutionair oogpunt bezien, nieuwe leefstijlfactoren zouden gezien moeten
worden als bedreigingen voor de gezondheid, aangezien ze in staat zijn om de stressassen
langdurig of chronisch te activeren. Het immuunsysteem is ontworpen om oplossingen aan
te dragen bij snel optredend en intensief gevaar en niet om opgewassen te zijn tegen
langdurige, chronische stimulatie. De stressfactoren die inherent zijn aan onze recente leefstijl
hebben het immuunsysteem en de centrale stresssystemen in een toestand van voortdurende
activiteit gebracht, waardoor chronisch onopgeloste ontstekingen ontstaan, alsmede een
verhoogde bevattelijkheid voor chronische ziekte. Het is onze hypothese dat de moderne
voeding, verhoogde psycho-emotionele stress en chronisch gebruik van ontstekingsremmers
het natuurlijke proces van ontstekingsremming, ofwel resoleomics, verstoren.
Paragraaf 1.4
Tegenwoordig zijn CNCD’s wereldwijd de voornaamste oorzaak van werkverzuim, invaliditeit
en sterfte. De meeste van deze ziekten worden in verband gebracht met laaggradige

322
ontsteking. Hier presenteren we de hypothese dat stress (gedefinieerd als homeostatische
verstoringen) laaggradige ontsteking kan induceren, waardoor de behoefte aan water,
natrium en energierijke stoffen toeneemt, zodat voldaan kan worden aan de verhoogde
metabole vraag die veroorzaakt wordt door de stressor. Eén manier om een laaggradige
ontsteking uit te lokken is door een verhoogde permeabiliteit van de darmwand langs
activatie van verschillende componenten van het stresssysteem. Hoewel het een voordeel
oplevert bij het voorzien in de vraag tijdens stress, verhoogt een toegenomen permeabiliteit
van de darmwand ook de kans op translocatie van bacteriën en hun toxinen van het
darmlumen naar de bloedsomloop. Samen met moderne leefstijlfactoren veroorzaakt een
verhoogde translocatie van bacteriën en bacteriële toxinen, als gevolg van een meer
permeabele darmwand, een toestand van laaggradige ontsteking. We ondersteunen deze
hypothese met talloze studies, en tonen verschillende verbanden tussen CNCD’s en markers
van endotoxemie, waarmee we suggereren dat dit mechanisme een cruciale – misschien zelfs
een oorzakelijke – rol speelt bij de ontwikkeling van laagradige ontsteking en aanverwante
aandoeningen.
Paragraaf 1.5
Tarwe is één van de meest gegeten granen ter wereld en maakt een substantieel onderdeel uit
van de menselijke voeding. Hoewel door de overheid gesteunde voedingsrichtlijnen in
Europa en de VS adviseren om dagelijks voldoende (volkoren) graanproducten te eten,
bevatten granen ‘antinutriënten’, zoals gluten en lectinen, die bij mensen tot disfunctie en
ziekte kunnen leiden. In deze review behandelen we bewijs uit in vitro, in vivo en menselijke
interventiestudies die beschrijven hoe de consumptie van tarwe, maar ook andere granen,
kunnen bijdragen aan het manifesteren van chronische ontsteking en auto-immuunziekten
door de permeabiliteit van de darm te verhogen en door een ontstekingsbevorderende
immuunreactie te initiëren.
Paragraaf 1.6
Diverse recente, positief geselecteerde aanpassingen aan nieuwe nutriënten zijn
geïdentificeerd. Lactasepersistentie is één van de bekendste, hetgeen populaties die eraan
worden blootgesteld in staat stelt om melk te drinken, in het bijzonder om lactose te verteren
na de zoogtijd. Een verhoogd aantal kopieën van amylasegenen (AMY1), welke zorgen voor
een grotere activiteit van amylase in het speeksel, is in verband gebracht met de consumptie
van zetmeelrijke voedingsmiddelen. Hogere aantallen AMY1 genen zijn aangetoond in
populaties met een recente geschiedenis van zetmeelrijke voeding. Het is echter twijfelachtig
of de resulterende polymorfismen positieve selectiedruk hebben uitgeoefend door louter de
aanwezigheid van eenvoudig verkrijgbare bronnen van macro- en micronutriënten.

323
De mens heeft meer nieuwe omgevingen verkend dan welk ander dier dan ook. Nieuwe
omgevingen bieden nieuwe uitdagingen aan de gastheer, maar confronteren in het bijzonder
het immuunsysteem met nieuwe klimaatomstandigheden, voeding en in het bijzonder
pathogenen. Met de komst van de landbouwrevolutie en de daarop volgende domesticatie van
vee werden nieuwe pathogenen en ziekten geïntroduceerd (zoönosen). We beweren dat
specifieke nieuwe voedingsingrediënten (bijvoorbeeld gluten) en nieuwe pathogenen de
selectie hebben gestuurd richting lactasepersistentie en een groter aantal kopieën van het
AMY-gen. Beide aanpassingen leveren ruim voldoende glucose voor het activeren van de
natrium/glucose-afhankelijke cotransporters 1 (SGLT1), welke de voornaamste transporter is
voor glucose, samen met natrium en water, in de dunne darm en in mindere mate de maag
(Chen 2010). Na inname van zetmeel, hebben mensen met hogere aantallen AMY1-kopieën
een snellere toename van insuline en daarmee gepaard gaande lagere plasmaglucose,
vergeleken met personen met lagere aantallen AMY1-kopieën (Mandel 2012). Snelle opname
van glucose, natrium en water levert bescherming tegen potentieel dodelijke uitdroging,
hyponatremie en uiteindelijk meervoudig orgaanfalen, in het bijzonder tijdens infectie
gecombineerd met diarree en braken. Ook orale rehydratietherapie is gericht op SGLT1-
activiteit en is momenteel de eerste keus behandeling bij deze aandoeningen. Het is onze
hypothese dat levenslange lactase-activiteit en snelle zetmeelvertering gezien moeten worden
als de evolutionaire voorloper van de huidige orale rehydratietherapie.
Hoofdstuk 2. Fysieke inactiviteit
Paragraaf 2.1
De afgelopen jaren is het duidelijk geworden dat chronische systemische laaggradige
ontsteking aan de basis staat van veel, zo niet alle ‘typisch westerse’ aandoeningen die
samenvallen met het metabool syndroom. Hoewel er veel nadruk is gelegd op de zittende
leefstijl als oorzaak van chronische ontsteking, wordt het minder vaak ingezien dat
chronische ontsteking ook een zittende leefstijl kan bevorderen, wat op zijn beurt chronische
ontsteking veroorzaakt. Gegeven dat zelfs kleine toenames in chronische ontsteking het
hersenvolume verkleinen bij anderszins gezonde personen, kan de tweezijdige relatie tussen
ontsteking en een zittende leefstijl wellicht verklaren waarom mensen hersenvolume hebben
verloren in de afgelopen 30.000 jaar en ook intelligentie in de afgelopen 30 jaar. We geven een
overzicht van het bewijs dat gebrek aan lichaamsbeweging chronische laaggradige ontsteking
veroorzaakt en, ten gevolge daarvan, een energieconflict veroorzaakt tussen het zelfzuchtige
immuunsysteem en het zelfzuchtige brein. Hoewel de notie in de hedendaagse wereld
wijdverbreid is dat meer lichaamsbeweging goed is voor de gezondheid, geven we hier een
nieuw perspectief op deze gemeenplaats door bewijs te leveren dat het herstel van normaal
menselijk gedrag, zoals spontane lichaamsbeweging, de ontstekingsbevorderende
immuunactiviteit zou kalmeren, waardoor er meer energie aan de hersenen en andere

324
organen wordt toebedeeld en op deze manier de menselijke gezondheid bevordert.
Paragraaf 2.2
De evolutie van de mens heeft miljoenen jaren in beslag genomen, gedurende welke periode
omgevingsfactoren gaandeweg het genoom hebben gevormd met als doel zich aan te passen
aan de heersende omstandigheden (Darwin). Eén van de meest vitale gedragsmatige
aanpassingen van zoogdieren in het algemeen – en mensen in het bijzonder – is hun
vermogen om in hun levensonderhoud te voorzien middels lichaamsbeweging.
Lichaamsbeweging, zoals rennen, springen, klimmen en dragen, zijn waarschijnlijk nodig
geweest om wilde dieren te snel af te zijn, om naar voedsel te zoeken en om voor gevaar te
schuilen. Aan de andere kant waren individuen die lichamelijk of psychologisch niet in staat
waren om voor zichzelf te zorgen, vatbaarder voor een vroegtijdige dood en daardoor
genetisch uitsterven.
De huidige maatschappij wordt gekenmerkt door locatiegebonden individuen, in plaats van
individuen die ‘in beweging’ zijn. Een gebrek aan lichaamsbeweging is niet slechts een
mogelijke factor die gerelateerd is aan chronische ziekte, maar moet gezien worden als de
eigenlijke oorzaak van vrijwel de gehele menselijke ziektelast. Het overgrote deel van de
mensen weet dat lichaamsbeweging nodig is en voordeel oplevert. Desalniettemin haalt
nagenoeg 75 procent van de huidige populatie niet het geschatte minimale niveau van
lichaamsbeweging. Lichamelijke inactiviteit behoort tot de kenmerken van ziektegedrag,
hetgeen waarschijnlijk een beschermende functie heeft voor het organisme. Ziektegedrag,
waaronder een depressieve stemming, lijkt te beschermen tegen infectie, letsel en sociaal
conflict en bevordert het behoud van energie. Ziektegedrag is gebaseerd op immuun-
hersenmechanismen en kan worden gedefinieerd als niet-permissief gedrag. Langdurig niet-
permissief gedrag kan leiden tot chronische ziekte vanwege verminderde lichaamsbeweging
en zichzelf hinderende copingstrategieën, waarmee niet-permissief gedrag wordt omgezet in
een niet-permissieve hersenaandoening. We stellen voor dat de lichamelijke
inactiviteitsziekte een synoniem is van een niet-permissieve hersenaandoening en dat deze
aandoening leidt tot een zogenaamd ‘reptielenfenotype’, dat wordt gekenmerkt door
hypothermie, slechte haargroei, afgenomen vruchtbaarheid en een laag basaalmetabolisme.
Hoofdstuk 3. Leefstijl interventie
Paragraaf 3.1
Chronische laaggradige ontsteking en insulineresistentie zijn nauw aan elkaar verwante
entiteiten die de meeste, zo niet alle, chronische welvaartsziekten met elkaar gemeen hebben.
Het is onze hypothese dat een kortermijn interventie gebaseerd op ‘oude stressfactoren’ een
verbetering kan geven op antropometrische resultaten en klinisch-chemische indices. In deze

325
paragraaf onderzochten we of een tiendaagse nabootsing van een jager-verzamelaarsleefstijl
een gunstig effect heeft op antropometrische resultaten en klinisch-chemische indices. In
totaal 53 op het oog gezonde deelnemers en twee patiënten met fibromyalgie (22–69 jaar, 28
vrouwen), verdeeld in vijf groepen, deden mee aan een tiendaagse reis door de Pyreneeën.
Gemiddeld wandelden ze 14 kilometer per dag, waarbij een 8 kilogram zware rugzak werd
gedragen. Men werd voorzien van natuurlijke voedingsbronnen en bereidde dit zelf tot
voeding, terwijl water werd verkregen uit waterputten. Ze sliepen buiten in slaapzakken en
stonden bloot aan temperaturen tussen de 12°C en 42°C. Antropometrische gegevens en
bloedmonsters tijdens vasten werden verzameld aan het begin en eind van het onderzoek. We
zagen mediaandalingen (p≤0.002) in lichaamsgewicht (-3,5 kg), BMI (-1,1 kg/m2), heup- (-3
cm) en middelomtrek (-5 cm), glucose (-0,6 mmol/L), HbA1c (-0,1%), insuline (-4,7 pmol/L),
HOMA-IR (-1,2 mmol*mU/L2), triglyceriden (-0,14 mmol/L), totaalcholesterol (-0,7 mmol/L),
LDL-cholesterol (-0,6 mmol/L), verhouding triglyceriden/HDL-cholesterol (-0,55 mol/mol) en
FT3 (-0,8 pmol/L). Veranderingen in antropometrische resultaten hielden geen verband met
veranderingen in klinisch-chemische indices, behalve voor FT4 en FT3. ASAT (11 IU/L), ALAT
(6 IU/L), ASAT/ALAT-verhouding (0,14) en CRP (0,56 mg/L) gingen omhoog en hun
veranderingen stonden met elkaar in onderling verband. We concluderen dat de omgang met
‘oude, milde stressfactoren’, waaronder lichaamsbeweging, dorst, honger en klimaat, invloed
kan hebben op de immuunstatus en antropometrische- , en metabole- indices kunnen
verbeteren bij gezonde personen en patiënten met fibromyalgie.

326
Toekomstperspectieven
Veel CNCD’s worden gezien als ‘ongeneeslijk’ vanwege het heersende dogma dat een
bepaalde ziekte altijd maar één oorzaak heeft. Deze manier van denken is geïnspireerd op het
werk van Louis Pasteur (1822–1895), die aantoonde dat een infectieziekte zijn oorsprong vindt
in één causale factor. Deze factor kan worden aangetoond en bestreden met specifieke
behandelingen. Echter, de ‘kiemtheorie van ziekte’ zegt alleen maar dat sommige ziekten
worden veroorzaakt door ziekteverwekkers. Wij geloven dat CNCD’s onbehandelbaar blijven
als we de nadruk blijven leggen op een enkelvoudige etiologie. We stellen daarom dat CNCD’s
worden veroorzaakt door multi-metabole antropogene risicofactoren, waaronder niet alleen
leefstijlfactoren zoals roken, bewegingsarmoede en calorierijke voeding, maar ook
risicofactoren die iets verder weg staan, zoals politieke, economische, sociale, culturele en
zelfs evolutionaire factoren. Onze visie komt in grote lijnen overeen met de publicaties van
Egger et al. (Egger 2012, Egger 2014a, Egger 2014b). Met uitzondering van ziekten met een
primaire oorzaak gelegen in de genetica (‘aangeboren stofwisselingsziekten’) lijkt het
theoretisch mogelijk dat elk van ons gezond ouder kan worden en kan sterven van ouderdom.
De toekomst lijkt er echter niet gunstig uit te zien: het lijkt aannemelijk dat het aantal
antropogene factoren zal toenemen. Diverse van deze nadelige invloeden lijken
onvermijdelijk en de meesten van ons zijn niet goed in staat om deze factoren op een
effectieve manier te beïnvloeden.
Desalniettemin geloven we dat preventie de meest effectieve manier blijft om CNCD’s op te
lossen. Deze preventieve maatregelen kunnen gericht zijn op het bevorderen van de
weerbaarheid tegen toxische uitlokkers van het moderne leven. De beste optie zou
regelmatige blootstelling aan hormetische uitlokkers zijn, zoals we hebben aangetoond in
onze pilot-interventie de ‘Study of origin’ (Hoofdstuk 3). Het is niet moeilijk, en zou zelfs
aangenaam kunnen zijn, om enkele van deze intermitterende hormetische uitlokkers te
integreren in het dagelijks leven. Er komst steeds meer bewijs voor de gezondheidseffecten
van ‘intermittent living’, waaronder intermittent fasting (Barnosky 2014), intermittent hypoxia
(Duennwald 2013), intermittent sitting (Henson 2015) en intermittent heat (Laukkanen 2015).
We stellen voor dat toekomstige onderzoeken zich op dit soort interventies richten en dat
daarvoor geld beschikbaar komt. Er is echter een enkele aarzeling: de uitkomsten van deze
interventies kunnen op hun hoogst aantonen dat Darwin gelijk had toen hij stelde dat
“organismen zich aanpassen aan de leefomstomstandigheden”. Dus wat is de zin van het
wachten op hun uitkomsten? De toekomst begint vandaag.

327
Dankwoord
When life would vanish
And the apocalypsis would become true
Only you are able to recover life
Just like you did with me
You, stands for my wife Fany and children
Thanks my life
All the other people I have to thank for everything are all the persons who feel connected to
me and especially my friends and my family; Kees, late Bram, Rik, Marjorie, Garda, Tom, Dany,
Elena, Pablo, Gonzalo, Sebastian, Ans, Arnold, Ellie, Rob, Sangeles, Manolo, Lijia, Cristina,
Diana, Gus, my late father and my late mother.
A special thanks for two of the best scientists in recent history. Whereas my brain is crazy and
chaotic since birth, they managed to bring structure in it; without them this work would have
been unreadable for you, the reader of this manuscript.

328
Curriculum Vitae
Name
Leo Pruimboom
Place and date of birth
Amsterdam, The Netherlands, May 12, 1961
Citizenship
Dutch
Current titles
1983 Licensed in Physiotherapy SAFA (Amsterdam)
1988 Study of Biochemistry and Physiology (Amsterdam)
1989 Postgraduate in Sports-physiotherapy (Mainz)
Affiliations
2008 - 2011 Director University of Graz (Master in CPNI)
2006 - 2012 Director Master in PNIC, University of Gerona
2006 - 2007 Inivited Instituto de Empresa (International Business School)
Madrid – Segovia
2004 - 2005 Teacher University of Lisbon (Master in Sportphysiotherapy
– Clinical PNI)
2002 - 2009 Teacher University of Santiago de Compostela
(Master in natural medicine – Clinical PNI)
1987 - AEP Postgraduate director and teacher in Holland,Germany, Spain,
Austria, Belgium, Switzerland, Brasil, Turkey, England
2012 - Teacher University of Middlesex, Wockingham;
Master in nutritional therapy
1999 - Head of Education Natura Foundation
Clinical Affiliations
1984 – 2005 Owner of private clinic in PNIC
2005 - Advicer of multiple private and public clinics in CPNI in Europe

329
Public Projects
2006 – 2009 Director of the First European Proyect in Clinical PNI and health
of a whole city (Chipiona, Andaluzia – Spain)
Private Projects
2015 – Consultant of several professional football teams throughout
Europe
1999 – 2002 Director of the development of “Panticosa Resort”, a PNI health
promoting wellness center
1999 – 2001 Scientific Director of the “Villarreal CF” project of Clinical PNI in
professional soccer
1991 – 1997 Scientific Director of the “Pamesa CB” project of Clinical PNI in
professional Basketbal
1989 – 1992 Physiotherapist and advisor of the “RFEF” project of Clinical PNI
in professional female athletes in sprint
Scientific Invitations
More than 200 congresses in the last 20 years.
Publications
Indexed in Pubmed
1. Pruimboom L, de Punder K. Gluten is Morphine: Asymptomatic Celiac Disease. Journal
of Health, Population and Nutrition (2015) 33:24
2. Pruimboom, L. & Reheis, D. Intermittent drinking, oxytocin and human health. Medical
Hypotheses 92, 80-83 (2016).
3. Kuipers R, Pruimboom L. Short comment on “A review of potential metabolic etiologies
of the observed association between red meat consumption and development of type 2
diabetes mellitus”, by Yoona Kim, Jennifer Keogh, Peter Clifton. Metabolism 2016
4. Pruimboom L, Raison CL, Muskiet FAJ . When the immune system overrides the selfish
brain. (submitted Journal of Evolution and Health 2016)
5. Punder K, Pruimboom L. Stress induces endotoxemia and low-grade inflammation by
increasing barrier permeability. Front. Immunol. 6:223. (2015)
6. Pruimboom L, Raison C, Muskiet FAJ. Physical Activity Protects the Human Brain
against Metabolic Stress Induced by a Postprandial and Chronic Inflammati Hindawi
Publishing Corporation. Behavioural Neurology Volume 2015, Article ID 569869, 11
pages
7. Ruiz-Núñez, B., Pruimboom, L., Dijck-Brouwer, D. A. & Muskiet, F. A. Lifestyle and
nutritional imbalances associated with Western diseases: causes and consequences of

330
chronic systemic low-grade inflammation in an evolutionary context. J Nutr Biochem
24, 1183-1201 (2013)”.
8. Bosma-den Boer MM, van Wetten ML, Pruimboom L. Chronic inflammatory diseases are
stimulated by current lifestyle: How diet, stress levels and medication prevent our body
from recovering. Nutr Metab (Lond) 2012;9(1):32.
9. Pruimboom, L. Physical inactivity is a disease synonymous for a non-permissive brain
disorder. Med Hypotheses 77, 708-713 (2011)”.
10. Punder, K. & Pruimboom, L. The dietary intake of wheat and other cereal grains and
their role in inflammation. Nutrients 5, 771-787 (2013)”
11. Pruimboom, L., Fox, T. & Muskiet, F. A. Lactase persistence and augmented salivary
alpha-amylase gene copy numbers might have been selected by the combined toxic
effects of gluten and (food born) pathogens. Med Hypotheses (2014)”
12. Intervention study: Chronic diseases are caused by the absence of old stress factors –
our dearest friends; The study of origin, based on: ”The study of origin. Pruimboom L,
Consortium, Muskiet FAJ”. Biomed Research International (2016)
13. Pruimboom L, Dam B van. Chronic Pain: “A non-use disease”. Medical Hypotheses
(2007) 68, 506–511.
14. Pruimboom L. Dam B van. Treatment of osteoporosis by activation of endogenous
physiological regenerative capacities. Sportverletz Sportschaden. 1997 Mar;11(1):XVII-
XVIII.
Not indexed in Pubmed
1. Pruimboom L. De pijnneuromatrix. Van Nature nr. 0, okt. 2005, pag. 30-33
2. Pruimboom L. Curcumine en de strijd tegen anorexia. Van Nature nr. 0, okt. 2005, pag.
35
3. Pruimboom L. Vitamine D, vitamine A en DHA, to restore health we have to go back to
the future. Van Nature nr. 1, 2006; pag. 30-32
4. Pruimboom L. U bent niet moe, u heeft dorst. Van Nature nr. 2, 2006, pag. 40-45
5. Dam van B, Pruimboom L. Pijn en het maagdarmkanaal, chronische pijn als
consequentie van een functionele darmstoornis. Van Nature nr. 3, 2006, pag. 51-54
6. Pruimboom L. Neurotraining, een holistische kijk op sport. Van Nature nr. 4, 2007, pag.
26-29
7. Pruimboom L. The origin of species; enzymatische veroudering, het ultieme concept.
Van Nature nr. 5, 2007, pag. 41-45
8. Dam van B, Pruimboom L, Vargas D, et al. Foetale programmering, stoornissen van de
HPA-as en ziekte bij de volwassene. Van Nature nr. 6, 2007, pag. 41-44
9. Pruimboom L. Voedingsfilofosie: het zoekende kind. Van Nature nr. 7, 2007/2008, pag.
34-37

331
10. Pruimboom L. De ‘brain-heart-brain’verbinding. Van Nature nr 8., 2008, pag. 47-51
11. Pruimboom L. Het overleven van de dikste. Van Nature nr. 9, 2008, pag. 52-54
12. Pruimboom L. The non-permissive brain; een klinisch concept voor mentale
stoornissen. Van Nature nr. 10, 2008, pag. 39-42
13. Pruimboom L. De evolutionaire oorsprong van ontsteking. Van Nature nr.12, 2009, pag.
35-37
14. Pruimboom L. Decision making in uncertain times. Van Nature nr. 13, 2009, pag. 39-43.
15. Van Dam B. y Pruimboom L. Un nuevo test en tenis. Revista de Entrenamiento
Deportivo. 1992; Vol. VI.
16. Pruimboom L. La necesidad del entrenamiento contra-lateral. RED VII-1 (1993)
17. Pruimboom L. El pie y su lugar en el deporte. Nuevo sistema de diagnostico y
entrenamiento. Revista de entrenamiento deportivo, 5 (3), 7-12 (1991)
Books and Book chapters
• Pruimboom, Leo. Por y para medicina y nutrición deportiva. Atres Ediciones Deportivas /
978-84-605-8738-5 /
• Pruimboom L, Dam B van. Orthomolekular medicine. In; Van den Berg. Angewandte
Physiologie, Teil 5: Komplementäre Therapien verstehen und integrieren. Thieme Verlag.
ISBN 3-13-131121-5. Pages 81 – 162
• Dam, B van, Pruimboom L. Fibromyalgie. In: Van den Berg. Angewandte Physiologie, Teil
4: Schmerzen verstehen und beeinflussen. Thieme Verlag. 2008
• Pruimboom L, Fox T. Nahrung, Bewegung und Immunology; In Sportphysiotherapy.
Herausg. Hans Josef Haas. Thieme Verlag 2012
• Pruimboom L, Rinderer M, Reheis D. Wirk-Koch-Buch. BUCHER Verlag und den Autoren
ISBN 978-3-99018-177-5. 2014
University teacher and head of education (director) in masters
1995 Master in Topsport, INEF de la Coruña, España
2006 - 2010 Postgraduate in psico-neuro-inmunologia, Universidad Europea
de Madrid, España
2004 - 2006 Master in sports-phisioterapia, Facultade de Motricidade
Humana, Lisboa, Portugal
2004 - 2012 Posgraduate in psico-neuro-inmunologia,
Universidad de Gerona
2006 - 2011 Master in Natural Medicine, Facultad de Medicina de la
Universidad de Santiago de Compostela, España

332
2006 – 2012 Director of Master in Clinical PNI, Universidad de Gerona and
Graz
1987 - Head of Education, and Head Teacher of CPNI, Natura
Foundation, AEP Europe
Mentor of master thesis (32 master dissertations since 2006)
Most important ones
1. Freese J. The role of personality in clinical Psychoneuroimmunology. Cologne (2011)
2. Punder K. Punder, K. & Pruimboom, L. The dietary intake of wheat and other cereal
grains and their role in inflammation. Nutrients 5, 771-787 (2013)
3. Hahn D. The Barker hypothesis revised, part 1. A clinical study. (2012)
4. Biedermann V. The Barker hypothesis revised, part 2. A clinical study. (2012)
5. Hanusch Kay. Hyperthermia in patients suffering from depresión (2012)
Journal reviewer
Medical Hypothesis, Plant Foods for Human Nutrition, Vitamins and Trace Elements, Annual
Research & Review in Biology, BioMed Research International, International Neuropsychiatric
Disease Journal
Membership
Psychoneuroimmunology research society
New York Academy of Science




















