
University of Groningen Processing grammatical evidentiality ...
Upload
khangminh22Category
view
2download
0
University of Groningen
Modulation of lipoxygenase activity and chemistry-based detection of protein nitration ininflamationWisastra, Rosaline
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:2013
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Wisastra, R. (2013). Modulation of lipoxygenase activity and chemistry-based detection of protein nitrationin inflamation. s.n.
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license.More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne-amendment.
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 05-02-2022

Modulation of lipoxygenase activity and
chemistry-based detection of protein
nitration in inflammation

Paranymphs : Jenny Novianty Soetedjo
Thea van den Bosch
The research project described in this thesis was carried out in the division of Pharmaceutical Gene Modulation, Groningen
Research Institute of Pharmacy, according to the requirements of the Graduate School of Science (Faculty of Mathematics
and Natural Sciences, University of Groningen).
This work was financially supported by the University of Groningen
Cover design : Andreas Hidayat Gunawan
Layout : Rosalina Wisastra
Printing : Off Page (www.offpage.nl)
This thesis also available in electronic format at http://dissertations.ub.rug.nl
Copyright © 2013 by Rosalina Wisastra. All rights reserved.

Modulation of lipoxygenase activity and
chemistry-based detection of
protein nitration in inflammation
Proefschrift
ter verkrijging van het doctoraat in de
Wiskunde en Natuurwetenschappen
aan de Rijksuniversiteit Groningen
op gezag van de
Rector Magnificus, dr. E. Sterken,
in het openbaar te verdedigen op
vrijdag 24 mei 2013
om 11.00 uur
door
Rosalina Wisastra
geboren op 17 mei 1985
te Bandung, Indonesië

Promotor : Prof. dr. H.J. Haisma
Copromotor : Prof. dr. F.J. Dekker
Beoordelingscommissie : Prof. dr. A.S.S. Dömling
Prof. dr. R.M.J. Liskamp
Prof. dr. A.P. IJzerman
ISBN : 978-90-367-6182-6 (printed version)
978-90-367-6183-3 (electronic version)

For my dad, mom, brother and my beloved families
I can do all things through Him who strengthens me.
Philippians 4:13


Contents
CHAPTER 1 Introduction and scope of the thesis 11
CHAPTER 2 Anacardic acid derived salicylates are inhibitors or activators of
lipoxygenases 37
CHAPTER 3 Discovery of a novel activator of 5-lipoxygenase from an anacardic acid
derived compound collection 65
CHAPTER 4 Isothiazolones; thiol-reactive inhibitors of cysteine protease cathepsin B
and histone acetyl transferase PCAF 103
CHAPTER 5 Antibody-free detection of protein tyrosine nitration in tissue sections 123
CHAPTER 6 Summary, General Discussion and Future Perspectives 143
Appendix Dutch summary - Nederlandse samenvatting 151
Acknowledgements 161
List of Publications 165


Introduction and
scope of the thesis 1

12
Introduction and scope of the thesis
Initiation and termination of inflammatory responses
Inflammation is a defensive body response to restoring tissue homeostasis upon chemical or
biological invasion. The initial defensive response in acute inflammation is the production of
inflammatory mediators such as chemokines, cytokines, and eicosanoids followed by recruitment of
blood components such as for example neutrophils, to the site of injury or infection.1 The release of
reactive oxygen species (ROS) and reactive nitrogen species (RNS) is an important component of the
defensive strategy to eliminate invading agents.2 If the acute inflammation is resolved, pro-
inflammatory mediators such as prostaglandins and leukotrienes are exchanged for anti-inflammatory
mediator such as lipoxins.3 Lipoxins prevent the neutrophil recruitment and promote the monocyte
recruitment to remove the dead cells and initiate tissue remodeling.4 In the case that acute
inflammatory responses fail to be resolved and the inflammatory processes persist, a chronic
inflammatory state occurs.5 The consequences of chronic inflammation might be detrimental since it
can result in tissue malfunction, damage, fibrosis and even cancer.
Lipoxygenases
Lipoxygenases, which are a group of oxidative enzymes with a non-heme iron atom in their active
site, are involved in the initiation of inflammatory responses by generation of pro-inflammatory
mediators known as leukotrienes or anti-inflammatory mediators, lipoxins. These enzymes catalyze
the insertion of oxygen (O2) into poly-unsaturated fatty acids (PUFAs) such as arachidonic acid and
linoleic acid. It has been described that the catalytic reaction of lipoxygenases involves a single
electron oxidation by the active site iron atom which switches between Fe2+ and Fe3+ redox states.6 In
the catalytic reaction, Fe3+ is reduced to Fe2+ with concomitant oxidation of the lipid substrate by
hydrogen abstraction from a bis-allylic methylene to give a pentadienyl radical, which is re-arranged
to provide a 1-cis,3-trans-conjugated diene moiety. Subsequently, a stereo-specific insertion of
oxygen at the pentadienyl radical takes place to form a oxygen centered fatty acid hydroperoxide
radical. The intermediate hydroperoxide radical is reduced to the corresponding anion with
concomitant re-oxidation of iron to Fe3+ (Figure 1).7
Lipoxygenases catalyze the formation of hydroperoxy eicosatetraenoic acids (HPETEs) from
arachidonic acid. These HPETEs are subsequently reduced and transformed to form so called
eicosanoids, which are signaling molecules that play an important regulatory role in the immune
responses and other physiological processes. In general, lipoxygenases are classified as 5-, 8-, 12, and
15-lipoxygenases according to their selectivity to oxygenate fatty acids in a specific position.8 The
importance of fatty acids oxygenation by lipoxygenase enzymes has been described for many
physiological processes (Table 1).

13
Introduction and scope of the thesis
Lipoxygenases are commonly found in the plant and animal kingdoms. Although the overall
architecture of plant lipoxygenases such as soybean lipoxygenase is similar to mammalian
lipoxygenases, they share little sequence similarity (about 25%).9 In contrast, there are sequence
similarities of about 60% among human 5-, 12- and 15-lipoxygenases.10 Even though these enzymes
show a high sequence similarity, the regulatory mechanism of 5-lipoxygenase (5-LOX) is more
complex compared to the other human lipoxygenases. In general, lipoxygenases are comprised of two
domains; N-terminal and C-terminal domains. The N-terminal domain is a regulatory domain and
consists mostly of β-barrels, while the C-terminal domain is a catalytic domain and consists mostly of
α-helices.9 The non-heme iron atom is located in the catalytic C-terminal domain, whereas the
function of the N-terminal domain is not unambiguously characterized. For 5-LOX, it is clear that the
N-terminal domain is essential for translocation to the nuclear membrane whereas for the other LOXs,
this is still under debate.9, 11
Table 1. Human lipoxygenase types and their most important substrates, products, and functions 12-23
Lipoxygenase Substrate Product Physiological functions Ref.
arachidonic acid 5(S)-HPETE, Leukotriene A4 pro-inflammatory mediator 12
γ-linolenic acidDihomo-γ-linoleic acid
(DGLA)
inhibition of arachidonic acid
conversion13
eicosapentaenoic acid (EPA) Leukotriene A5anti-inflammatory mediator/
inhibitor LTA4 hydrolase14
arachidonic acid 12(S)-HPETE
Dihomo-γ-linoleic acid
(DGLA)12(S)-HPETrE
eicosapentaenoic acid (EPA) 12(S)-HPEPE
α-linoleic acid 13(S)-HPOTrE
arachidonic acid 12(R)-HPETE
linoleyl-ω-hydroxy ceramide9(R)-hydroperoxylinoleoyl-ω-
hydroxy ceramide
epidermis LOX3
(eLOX3)
9(R)-hydroperoxylinoleoyl-ω-
hydroxy ceramide
9(R)-10(R)-trans-epoxy-11E-
13(R)- hydroxylinoleoyl-ω-
hydroxy ceramide
linoleic acid 13(S)-HPODEmodulation of MAP kinase
signaling pathways
arachidonic acid 15(S)-HPETEmodulation of leukotriene B4,
pro-inflammatory mediators
15-lipoxygenase-
2 (15-LOX2)arachidonic acid 15(S)-HPETE
negative cell cycle regulator
and tumor supressor22-23
15-17
18
19-21
5-lipoxygenase
(5-LOX)
Platelet 12-
lipoxygenase
(p12-LOX)
12R-
lipoxygenase
(12R-LOX)
15-lipoxygenase-
1 (15-LOX1)
epidermal barrier acquisition
modulation of platelet
agregation
Human 5-LOX activity is influenced by the presence of Ca2+, which reversibly binds to the
enzyme with maximum binding of two Ca2+ ions per 5-LOX. A Ca2+ binding causes an increase in
hydrophobicity, which promotes membrane association of 5-LOX.24 Furthermore, the presence of
adenosine tri-phosphate (ATP) appears to be important for an optimal 5-LOX activity. It has been
reported that 5-LOX has an ATP binding site, in which both the adenine-base and the phosphate
moieties of ATP are essential for the activation. However, the stoichiometry, the affinity and the

14
Introduction and scope of the thesis
location of ATP binding on 5-LOX remain elusive.25 In addition, the cellular 5-LOX activity is
essentially dependent on a small membrane protein called five-lipoxygenase-activating protein
(FLAP). Although, FLAP shares about 50% sequence similarity with human LTC4 synthase, its
activity is not glutathione dependent. The influence of FLAP on 5-LOX activity is exerted via an
allosteric mechanism. FLAP plays a role in arachidonic acid recruitment to 5-LOX and its conversion
to 5-HPETE and LTA4.26
Two different types of 12-LOX have been identified based on the differences in tissue distribution,
which are respectively named as platelet 12-LOX (p12-LOX) and 12R-LOX. Platelet 12-LOX is
mostly found in platelets as a platelet aggregation modulator, whereas 12R-LOX is mostly found in
skin cells in which it plays a role in epidermal barrier properties.17, 18 There are also two subtypes of
15-LOX, named as15-LOX-1 and 15-LOX-2. 15-LOX-1 is highly expressed in leukocytes and airway
endothelial cells27, 28 while, in contrast, 15-LOX-2 is expressed in prostate, lung, cornea, and many
tissues such as liver, colon, kidney, spleen, ovary, and brain, but not in leukocytes.29 Moreover, cells
induced by interleukin (IL)-4 and IL-13 show selective increase of 15-LOX-1 expression and not 15-
LOX-2.30. A lack of similarity between 15-LOX-1 and 15-LOX-2 at the primary sequence level
contributes to their distinct biological roles.31
Figure 1 – Oxidation reactions of lipoxygenases in the Leukotriene (LT) biosynthesis pathways.
Biosynthesis of Leukotrienes : Initiation of inflammatory responses
Leukotrienes (LTs) received their name because they were found in various types of leukocytes, such
as granulocytes, monocytes and mast cells. The pro-inflammatory leukotrienes are divided into two
classes, dihydroxy acid leukotriene LTB4 and the cysteinyl leukotrienes LTC4, LTD4, and LTE4.32

15
Introduction and scope of the thesis
Biosynthesis of leukotrienes is regulated by the activity of 5-lipoxygenase. Upon inflammatory
stimulation, cytosolic phospholipase A2-α (cPLA2α) releases arachidonic acid from membrane lipids
to start the leukotrienes biosynthesis. 5-lipoxygenase catalyzes the oxidation of arachidonic acid to 5-
HPETE, which is subsequently converted into Leukotriene A4 (LTA4). LTA4, which is a LT
precursor, is hydrolyzed by LTA4 hydrolase to form dihydroxy acid leukotriene LTB4. Another route
is the conversion of LTA4 to cysteinyl leukotriene LTC4 by addition of a glutathione group by LTC4
synthase. Conversion of LTC4 by γ-glutamyl transferase results in LTD4 and glutamic acid release.
Furthermore, dipeptidase (DiP) breaks the amide bond in LTD4 to give LTE4 (Figure 1).
LTB4 has an important function as chemo-attractant and is also involved in the formation of
reactive oxygen species. Binding of LTB4 to the Leukotriene B4 receptor 1 or 2 (LTBR1/2) activates
the phosphatidylinostisol 3-kinase (PI3K) pathways.33 In this way LTB4 is involved in the NF- B
pathway by stimulating the phosphorylation of IκBα, which results in activation of the NF- B
pathway. The cysteinyl leukotrienes LTC4, LTD4, and LTE4 activate two cysteinyl leukotriene
receptors (CysLTR) 1 and 2, which also play a role in the regulation of NF- B pathway.34 LTC4
induces the phosphorylation of NF- B p65 and activates the NF- B complex p50-p65. It also has
been proposed that the LTC4 binding to the CycLT2 receptor will induce the phosphorylation of I Bα
by involving protein kinase C (PKC) family enzymes (Figure 2).35
Nuclear Factor B (NF- B) in inflammation
Among all the lipoxygenase products, leukotrienes have exceptional biological functions. A particular
function is their action as pro-inflammatory mediators in the activation of the NF- B pathway.36 The
nuclear factor -B (NF- B) is an inducible transcription factor comprised of homo- and hetero-
dimers of the NF- B and Rel protein family.9, 37 The NF- B sub-family is comprised of two precursor
proteins, p105 and p100, while the Rel sub-family is comprised of RelA/p65, RelB and c-Rel. p105
and p100 respectively are precursors of p50 and p52, which are transcription factors in the NF- B
pathways. The transcription factors of NF- B are normally present in the cytoplasm in their inactive
state in a complex with the inhibitory proteins of I B family.9, 37 The production of pro- and anti-
inflammatory mediators is highly correlated with gene expression through the NF- B pathway.38
There are two major pathways for NF- B activation, the canonical pathway and the non-canonical
pathway. In addition, an atypical pathway has also been identified. The heterodimer of RelA/p65and
p50 is involved in the canonical pathway, whereas the heterodimer of RelB and p52 is involved in the
non-canonical pathway.39, 40 The activated NF- B pathway is involved in the pathogenesis of
inflammatory diseases such as asthma, arthritis, inflammatory bowel diseases (IBD) and chronic
obstructive pulmonary diseases (COPD).41-43 During inflammatory responses, both pro- and anti-
inflammatory mediators are produced. The regulation of inflammatory responses relies on the careful
orchestration of the expression of mediators that activate or suppress the immune response.

16
Introduction and scope of the thesis
The canonical NF- B activation pathway
Under normal conditions, the activity of the transcription factor complex RelA/p65-p50 is inhibited
by its natural inhibitors, I B proteins. Upon stimulation by pro-inflammatory cytokines such as TNFα
and IL-1, I B kinase (IKK) complex phosphorylates I B proteins that cause the release of the
RelA/p65-p50 dimer, which can subsequently translocate to the nucleus. The IKKs consist of the
subunits IKKα, IKKβ and IKKγ, which is also known as the NF- B essential modulator (NEMO)
protein. In addition, the functions of RelA/p65 are also regulated by two groups of enzymes;
phosphoinositide 3-kinase (PI3K) and protein kinase B (PKB)/Akt kinases.44 Kinases in the PI3K and
PKB/Akt pathways induce the activation of I B kinase to phosphorylate the I B and stimulate the
activation of transcription factors.45, 46 Furthermore, the phosphorylated I Bα protein is ubiquitinated
and subsequently degraded.47 Degradation of I B leads to the translocation of the free p65-p50 dimer
to the nucleus, in which p65-p50 then bind to the B promoter regions and activates gene expression
(Figure 2).39, 47
The non-canonical NF- B activation pathway
RelB in complex with p100 is present in the cytoplasm as inactive form of the transcription factor
RelB-p52. The activation of the NF- B via the non-canonical pathway is mediated by the IKK
complex, which comprises two IKKα sub-units. The activation of the homodimer of IKKα is
involving NF- B activation of inducing kinase (NIK) and tumor necrosis factor receptor-associated
factor (TRAF).9, 44 Upon stimulation, the IKK complex is activated by NIK through a phosphorylation
process, then the activated IKKα phosphorylates the inactive form of p100 subunit. Phosphorylation
of p100 then leads to another post-translational modification; ubiquitination, which induces the
proteolytic processing of p100 to form the active transcription factor p52. The formed heterodimer
RelB-p52 is recruited to the nucleus to initiate the gene transcription (Figure 2). 48

17
Introduction and scope of the thesis
Figure 2 – The roles of leukotrienes and acetylation in the expression of pro-inflammatory mediators through the NF-κB
pathway. The activated cPLA2α produces arachidonic acid, which is further converted to LTA4 by the 5-LOX. LTA4 is then
converted to LTB4 and cys-LTs and their binding to the leukotriene receptors activate the NF- B pathway in leukocytes
during inflammation. cPLA2α - cytosolic phospholipase A2-α; 5-LOX – 5-lipoxygenase; LTA4 – leukotriene A4; LTB4 –
leukotriene B4; Cys-LTs – cysteinyl leukotrienes; LTBR1/2 – leukotriene B receptors 1 or 2; CysLTR1/2 – cysteinyl
leukotriene receptors 1 or 2; PI3K - phosphoinositide 3-kinase; PKC – protein kinase C; NEMO – NF- B essential
modulator; I Bα – inhibitor NF- B; IKK - I B kinase; NIK - NF- B activation of inducing kinase; HAT – histone
acetyltransferase. TNFα – tumor necrosis factor α; MIP-2 - macrophage inflammatory protein-2; COX-2 – cycloxygenas-2;
iNOS – inducible nitric oxide synthase.

18
Introduction and scope of the thesis
Role of leukotrienes in inflammatory diseases
Over-expression of lipoxygenases and their pro-inflammatory products, leukotrienes, has been
implicated in many human acute and chronic inflammatory diseases such as asthma, atherosclerosis,
rheumatoid arthritis, inflammatory bowel diseases, dermatitis, and cancer.
Asthma
Highly increased levels of LTC4, LTD4, and LTE4 have been observed in lung tissues that were
challenged with allergens. Up-regulation of these mediators is considered as the main cause of asthma
since leukotrienes are also potent regulators for smooth muscle contraction in bronchoconstriction. In
addition, cysteinyl leukotrienes can cause plasma leakage from post-capillary venules in respiratory
tissues, which can lead to inflammatory edema.49 These findings indicate that the modulation of the
production of pro-inflammatory leukotrienes using small molecule inhibitors has potential for
treatment of asthma.
Cardiovascular diseases
Lipoxygenase activity has been implicated in the pathogenesis of cardiovascular diseases such as
atherosclerosis. Lipoxygenases, as oxidative enzymes, are believed to have an important role in the
oxidation of low density lipoproteins (LDLs) in the macrophage to form foam cells.50 The formed
foam cells will develop plaques of atheroma and their accumulation in the arteries leads to
atherosclerosis. In addition, an increase of cysteinyl LTE4 levels in urine and LTB4 in the atheroma
were observed in patients with atherosclerosis. Inhibition of lipoxygenase activity can provide a
treatment strategy for this inflammatory disease.
Rheumatoid Arthritis
Since 5-lipoxygenase is the main catalyst for the formation of LTB4, its role in the development of
rheumatoid arthritis becomes apparent with the identification of high LTB4 levels in the synovial
fluid of arthritis patient.51 This leukotriene is produced mainly by neutrophils, which are the most
abundant leukocytes in rheumatoid joints.52 A crucial role of LTB4 in arthritis induction and severity
has been revealed in a mouse serum transfer model of inflammatory arthritis.53 Importantly, the
inflammatory responses are reduced in mice with 5-LOX and leukotriene A4 hydrolase enzyme
deficiency.54 In addition, another lipoxygenase type, namely 15-lipoxygenase, is also involved in the
pathogenesis of rheumatoid arthritis via the NF- B pathway. It has been described that the 15-
lipoxygenase metabolite, 15-(S)-HETE increases the IκBα degradation and the nuclear translocation
of NF-κB subunit.55 It has been observed that the NF- B pathway is activated in the early stage of
joint inflammation56 and NF- B DNA binding activity is increased in rheumatoid arthritis patients.56
These results suggest that inhibition of lipoxygenases could find a therapeutic application also in this
field.

19
Introduction and scope of the thesis
Inflammatory Bowel Disease
The role of leukotrienes in inflammatory bowel disease (IBD) has been explored. A colonic biopsy
test from patients with IBD showed 3-7 fold enhancement of 5-lipoxygenase, FLAP and LTA4
hydrolase expression in the colonic mucosa and the rectal dialysates, which form the cellular basis for
LTB4 synthesis.57 More recently, Cys-leukotiene E4 (LTE4) was considered as a biomarker for IBD
since the urinary excretion of LTE4 was significantly increased in patients with IBD.58 All these data
together suggest that inhibition of lipoxygenase activity and leukotriene bio-synthesis can be a
valuable approach for treatment of such inflammatory diseases.
Lipoxygenase in cancer
Lipoxygenases and their catalysis products are associated with carcinogenic processes such as tumor
cell proliferation, differentiation, and apoptosis.59 Several lines of evidence have proven the crucial
role of lipoxygenases in cancer. In human prostate cancer cells, the overexpression of platelet 12-
lipoxygenase (p12-LOX) has been observed, which is a trigger for angiogenesis and tumor growth.60
The roles of 15-LOX-1 metabolites are reported in the development of breast cancer by promoting the
invasion of tumor cells into the lymphatic vessels and the formation of lymph node metastasis.61 The
increased expression of the 5-LOX enzyme and the LTB4 receptors were observed in pancreatic
cancer. In addition, 5-LOX expression levels were suggested as indicator for early neoplastic
lesions.62 Leukotriene LTB4 is a potential stimulator for cancer cell growth and also plays a role in the
formation of ROS in response to hypoxia.62, 63 These studies indicate that the increase of lipoxygenase
expression and activity is associated with the development of cancer, therefore, small molecule
modulators of these enzymes can provide comprehensive information about lipoxygenase function in
cancer and subsequently lead to development of drugs for this disease.
Biosynthesis of Lipoxins : Termination of inflammatory responses
Within the eicosanoid cascade, lipoxins that are formed by lipoxygenases have potential as counter-
regulator to resolve inflammation and to restore the cellular homeostasis. Lipoxins (LXs) are
generated from arachidonic acid through two lipoxygenase-based synthesis routes. The first route
involves the formation of LTA4 by 5-LOX and the conversion of LTA4 to the intermediate 5(6)-
epoxytetraene, which is subsequently converted into LXA4 and LXB4. The second route for LXs
formation is initiated by 15-LOX activity to oxidize arachidonic acid to 15-HPETE then followed by
5-LOX activity, which convert 15-HPETE to 5(6)-epoxytetraene.64 Both routes, which are involving
5-LOX activity in the lipoxin production, show that 5-LOX activity is important, not only in the
formation of pro-inflammatory mediators, but also in the formation of anti-inflammatory mediators.
Moreover, like the leukotrienes, an addition of glutathione (GSH) by GSH-S-transferase activity
generates cysteinyl lipoxin LXC4. LXD4 and LXE4 are generated in a similar manner as in the
leukotriene biosynthesis pathways (Figure 3).

20
Introduction and scope of the thesis
Only a few explorations on LXC4, LXD4, and LXE4 have been done and their biological roles
have not been investigated in detail. However, it has been reported that LXC4, LXD4, and LXE4 are
selectively generated by eosinophils and not by neutrophils and platelets.64 LXA4 and LXB4, with
LXA4 being the most studied, are emerging as mediators to stop the inflammatory responses and to
switch the cells to normal homeostasis.65 LXA4 and LXB4 actions in cells and tissues are mediated
through their interactions with lipoxin receptors. The lipoxin A receptor (ALXR) transmits stop
signals to reduce the pro-inflammatory signals to terminate neutrophil migration. Furthermore, it
stimulates the activation of monocytes and macrophages, and inhibits the leukotriene B4 formation.66
In addition, LXA4 can also act as a partial agonist for the LTD4 receptor by blocking the LTD4
binding.64 LXA4 stimulated-ALXR is able to block the NF- B-mediated gene expression and inhibits
the degradation of I Bα.65, 67
Figure 3 –Two lipoxygenase-based synthesis routes of lipoxins (LXs).
Lipoxins production, which is related to the activity of 5-, p12-, and 15-LOXs, has been proven to
be important and the alteration of the enzyme activity determines the levels of lipoxin.68, 69 Up-
regulation of arachidonate 15-lipoxygenase genes has been reported in the gene profiling of
glucocorticoid-treated nasal polyps,70 which is also an indication of 15-HPETE production during the

21
Introduction and scope of the thesis
termination of inflammatory process. Another study on the blood polymorphonuclear cells (PMN)
from asthmatic patients shows an increase of lipoxin production together with the activation of 5-
lipoxygenase.71 In addition, aspirin, a non-steroidal anti-inflammatory drug which inhibits the activity
of pro-inflammatory eicosanoids produced by cyclooxygenase (COX), triggers the biosynthesis of
LXA4 and the 15-epimer of LXA4 accompanied by the increase of brain 5-LOX activity in rat
infused with lipopolysaccharide (LPS).72 Taking all these findings together, these studies indicate that
the increase of 5-LOX activity does not solely contribute to the production of leukotrienes but also to
the increase of lipoxin levels. Since the 5-LOX activity is important for both initiation and termination
of inflammation, the modulation of this enzyme is crucial for inflammation therapy. Furthermore, its
dual functions in the inflammatory processes make 5-LOX an interesting enzyme for further
investigation of both inhibitors and activators.
Lipoxygenase inhibitors
Considering the potent pro-inflammatory properties of lipoxygenases and their products, the
modulation of the lipoxygenase pathways using small molecule inhibitors should provide new
therapeutic approaches for numerous inflammatory diseases and cancer. Various approaches have
been developed to inhibit lipoxygenases. Several synthetic small molecules as well as isolated natural
compounds have been tested for the inhibition of lipoxygenases (Figure 4). Recently, it was reported
that Δ9-tetrahydrocannabinol (Δ
9-THC), which is an active component extracted from cannabis, shows
an inhibition of 15-lipoxygenase with an IC50 of 2.42 µM.73 Nordihydroguaiaretic acid (NDGA),
which is a well-known antioxidant, inhibits platelet 12-lipoxygenase and 15-lipoxygenase.74 Another
compound with iron binding properties; 4-(2-oxopentadeca-4-yne)phenyl propanoic acid (OPP)
(Figure 4), shows a mixed type inhibition towards leukocyte 12-lipoxygenase with Ki and Ki’ values
respectively are 0.2 µM and 4.5 µM.75 The natural product curcumin, which is found in turmeric, is a
modulator of arachidonic acid metabolism through the 5-LOX pathway.76
The role of 5-lipoxygenase in inflammation has been intesively investigated. The fact that the
mechanism of 5-LOX activation is more complex compared to the others lipoxygenases, opens
opportunities for alternative strategies of inhibition. Inhibitors for leukotriene biosynthesis via 5-
lipoxygenase can be divided into FLAP inhibitors, redox inhibitors, non-redox inhibitors, and iron-
chelator inhibitors (Figure 4).
FLAP inhibitors
Compound MK-866, which was introduced by Gillard, et.al., is a potent leukotriene biosynthesis
inhibitor.77 This compound selectively inhibits FLAP without affecting 5-LOX, or phospholipase in
the leukotriene biosynthesis pathway.77,78 MK-866 was shown to be safe for consumption and has an
effect on the early and late stages of asthmatic responses to allergens.79 The other compounds that
belong to the FLAP inhibitor class are MK-0591 and Bay-X-1005 (Figure 4). These compounds show

22
Introduction and scope of the thesis
a potent leukotriene inhibition in the nanomolar range.80, 81 However, the presence of arachidonic acid
and other cis-unsaturated fatty acids in blood can compete with those inhibitors for FLAP binding,
causing a low inhibitors efficacy in whole blood assay.82 This results in a 50-200 fold reduction in
potency in whole blood assays in comparison with assays in isolated leukocytes.80, 81 This reduced
efficacy for FLAP inhibition in excess of arachidonic acid indicates that inhibition of FLAP in the
leukotriene biosynthesis pathway might be less effective.83
Redox inhibitors
Redox inhibitors basically act as antioxidants for the oxidation reaction performed by lipoxygenases.
The redox inhibitors Phenidone, BW755C, and AA-861 are well known as reducing agents (Figure
4).84, 85 Structure activity relationships for this class of inhibitors are relatively difficult to describe.
Nevertheless, it has been recognized that, apart from the redox potency,86 lipophilicity is also
important.85 Recently, a new redox inhibitor for 5-LOX has been reported, which is a trimer or
tetramer of caffeoyl clusters (Figure 4) with IC50 values of respectively 0.79 µM and 0.66 µM87.
Furthermore, redox inhibitors have a low selectivity for 5-LOX inhibition compared to COXs
inhibition.84 Although they display a high potency to inhibit leukotriene biosynthesis, an interference
with other biological redox processes has been reported. The formation of methaemoglobin is one of
the problems that were reported upon application of redox inhibitors.88
Iron-chelator Inhibitors
In general a non-heme iron atom in lipoxygenases coordinates with amino acid residues and a water
molecule forming an octahedral complex.89 The coordinated water molecule in the active site is
stabilized by a hydrogen bond with the carboxylate of an Ile residue. The iron atom in the 12-
lipoxygenase active site is more ordered in comparison to 5- or 15-lipoxygenase. The water molecule
in 5-lipoxygenase still coordinates with the iron atom but is slightly off the position to form an
octahedral complex, while in contrast no water molecule is coordinated with the iron atom in the 15-
Iipoxygenase active site. Besides coordinating with a water molecule, in 5-lipoxygenase the iron atom
coordinates with three His residues, and one Asn, whereas in 12- and 15-lipoxygenases four His
residues with one Ile are coordinated with the iron.90 The crystal structure of the enzymes with their
iron complex provides an understanding about the regio- and stereoselectivity of the catalytic
reaction, which is important for the development of inhibitors of the iron-chelator class.
Inhibition of 5-LOX can be achieved by replacing one of the iron ligands with a small molecule
ligand to create a complex. Molecules with iron-chelating functionalities such as hydroxamic acid or
N-hydroxyurea are potent inhibitors for 5-LOX (Figure 4).91 Zileuton is one of the 5-LOX iron-
chelator inhibitors that is already on the market for the treatment of asthma. In a number of clinical
trials, Zileuton has been shown to improve airway function and reduce the asthmatic symptoms as
well as the inflammation in the airway systems. Despite its effectiveness, Zileuton is not the first

23
Introduction and scope of the thesis
choice therapy due to its side effects such as nausea and idiosyncratic effects on the liver.92 Further
development of this class of inhibitors led to the identification of Atreleuton, which inhibits LTB4 and
cys-LTE4 production and has a potency that is about 5 fold enhanced in comparison to Zileuton. 93
Atreleuton, which has entered clinical trials for atherosclerosis and cardiovascular diseases, is one of
the leading 5-LO inhibitors in clinical development.94 Another N-hydroxyurea derivative, CMI-977
(LDP-977)95 showed potency as a new drug for asthma by suppressing 5-LOX activity in blood and
also by inhibition of anti-IgE-induced contractions of the airway tissue.96, 97 These studies suggest that
the development of iron-chelator inhibitors for lipoxygenases could be an interesting concept to be
explored.
Non-redox inhibitors
Non-redox inhibitors do not interfere with the oxidation reaction of lipoxygenases or have apparent
iron-binding properties. Inhibition of the enzyme activity can take effect by competitive binding to the
active site or by binding to an allosteric binding site that regulates the activity of the enzyme. The
(methoxyalkyl)thiazole (ICI211965) (Figure 4) selectively inhibits 5-LOX activity, which reduces
LTC4 and LTB4 synthesis in animal and human blood samples.98 Unfortunately, steady-state kinetic
analyses of this compound for 5-LOX have not been successfully performed and therefore it has not
been possible to determine wheter the inhibition is competitive with the substrate arachidonic acid or
not.99 Although, ICI211965 is a highly potent 5-LOX inhibitor from a novel structural class, it has
been reported to have a low oral potency. The methoxytetrahydropyran compound ZD-2138 (Figure
4) shows an improvement of the oral potency compared to ICI211965 for the treatments of arthritis
and asthma.100 Furthermore, ZD-2138 inhibits antigen-induced leukotriene release at the micromolar
concentration range.101 However, the results from a clinical trial for its application as an anti-arthritis
agent were disappointing and therefore research on this molecule was discontinued.102
Leukotriene antagonist
Recently, leukotriene receptor antagonists have appeared as a class of compounds that have superior
properties for suppression of leukotriene biosynthesis. Pranlukast, Zafirlukast and Montelukast
(Figure 4), three of the leukotriene receptor antagonists, have also shown good efficacy in the
treatment of asthma.103, 104 These drugs block the binding of leukotriene D4 and also LTC4 and LTE4
to the CysLTR1 in the lungs and bronchial tubes, which resulted in the reduction of airway
constriction, and mucus accumulation in the lungs and airways. Interestingly, it has also been reported
that Montelukast possesses secondary anti-inflammatory properties to inhibit the activitiy of 5-LOX
and HATs.105 Montelukast suppresses the leukotriene biosynthesis by selectively inhibition of 5-LOX
and gives no effect on the other enzymes involved in the leukotrienes biosynthesis pathway such as
LTA4 hydrolase and LTC4 synthase.106 Moreover, Montelukast alters the activity of the NF- B
transcription factor p65-associated HAT activity and reduces the TNF-α-stimulated IL-8

24
Introduction and scope of the thesis
expression.107 However, it has been reported that the usage of this leukotriene antagonist gives
neuropsychiatric side effects which is a major concern for its safety.
Different types of compounds have been introduced to modulate the enzyme activity and
ultimately to provide new drugs for inflammation. Despite of their high potency to inhibit leukotriene
production, their limitation in different aspects is still a concern that needs to be resolved. The
development of lipoxygenase modulator with an improve potency and selectivity for new therapeutic
applications becomes a major challenge.
Figure 4 – Lipoxygenases inhibitors
FLAP inhibitors
redox inhibitors iron-chellator inhibitors
non-redox inhibitors
leukotriene antagonist

25
Introduction and scope of the thesis
Histone acetyltrasefases (HATs) are co-activators in the NF- B pathway
Inhibition of leukotriene production or inhibition of receptor binding suppresses the up-stream
activation of the NF- B pathway. In contrast, modulation of inhibitory of activatory posttranslational
modifications of the NF- B transcription factor itself or the histones at B promotor sites is an
alternative strategy for down-stream inhibition and modulation of this pathway.
Acetylation of -amino groups of lysine residues is an activatory protein post-translational
modification in the NF- B pathway that rapidly gains attention. The acetylation of specific lysine
residues in the p65 and p50 NF- B subunits plays an important role in the regulation of gene
expression. The acetylation and de-acetylation of the transcription factor NF- B influence the affinity
of the NF- B transcription factors complex to the B region in the DNA, the selectivity for specific
promoters and also the transcriptional activation or inhibition.108 Furthermore, the B regions in the
DNA are located in the promoter regions of most of the cytokine and chemokine encoding genes. The
NF- B induction activates the transcription of pro-inflammatory mediators such as pro-TNFα, IL-8,
macrophage-inflammatory protein-2 (MIP-2), cyclooxygenase-2 (COX-2), and inducible NO synthase
(iNOS) (Figure 2).109 Therefore, the balance between acetylation and deacetylation processes play a
crucial role in controlling the normal homeostasis and the perturbation of this balance alters the
inflammatory responses for specific stimuli.
Acetylation levels are regulated by two important groups of enzymes, histone acetyltransferases
(HATs) and histone deacetylases (HDACs), which are respectively responsible for the introduction or
the removal of acetyl groups.110 HAT and HDAC activities are required for normal gene regulation,
because they are involved in several physiological processes, including cell-cycle progression,
checkpoint responses to DNA damage, DNA replication and chromosome stability.110, 111 Histone
acetyltransferases can also acetylate non-histone proteins, such as transcription factors and nuclear
receptors to facilitate gene expression. Two main types of HATs have been identified based on their
location in cells, HAT-A and HAT-B, which are respectively nucleosomal and cytoplasmic histone
acetyltransferases.112 However, most of the extensively characterized acetyltransferases are known as
nuclear enzymes. Type B acetyltransferases, which are located in the cytoplasm, acetylate newly
synthesized histones during the process of chromatin assembly.113 Furthermore, HATs are classified
into several families based on their primary sequence analysis, which displays a high sequence
similarity within families but there is a poor or no sequence similarity between families.114 The HAT
families that have been studied extensively are the GNAT family (including Gcn5 and PCAF), the
p300/CBP family, and the MYST family (including Esa1, MOZ and Tip60). In general, HATs activity
is linked to chromatin dynamics and transcriptional activation. Lysine acetylation eliminates the
positive charge of the polybasic histones, which results in a reduction electrostatic interactions with
the DNA.115 Decreasing electrostatic contacts between the core histones and the DNA provides a less
tightly packed chromatin and facilitates the recruitment of co-regulators and RNA polymerase

26
Introduction and scope of the thesis
complexes to the locus of modification and then activates the expression of genes that are involved in
inflammatory responses.116 The shift of the HAT/HDAC balance towards HAT activity can lead to
the increase of pro-inflammatory gene expression, which ultimately leads to the development of
several inflammatory diseases and also cancer.
HATs inhibitors
Modulation of the acetylation levels through HAT and HDAC inhibition are important approaches to
regulate inflammatory NF-κB-mediated gene expression. Several classes of HAT inhibitors have been
described. Bisubstrate inhibitors are constructs in which both the peptide and the CoA substrates are
linked together. Bisubstrate inhibitors (Figure 5) are selective and potent inhibitors for HATs, but
poor in cellular permeability due to their large size, polarity and charge. Therefore, the development
of small molecule HAT inhibitors that are selective and cell-permeable is essentially needed.
Screening of natural products for HAT inhibition has led to several potent inhibitor candidates (Figure
5). Curcumin, a natural compound from turmeric inhibits p300/CBP activity and suppresses NF- B-
mediated gene transcription.117, 118 Another natural product from mangosteen, Garcinol shows a potent
inhibition of p300 and gene transcription, and induces apoptosis in HeLa cells.119, 120 Anacardic acid,
isolated from cashew nutshells, inhibits the HATs Tip60 and p300 and suppresses NF- B activation,
down-regulates NF-κB–dependent gene expression, and induces apoptosis.108, 121 Anacardic acid
association effects on the inhibition of NF-κB are related to the suppression of IκBα degradation, the
inhibition of p65 acetylation and the nuclear translocation.121
Besides the natural product inhibitor class, synthetic inhibitors of HATs have been developed
(Figure 5). Isothiazolones are one of the compound classes that inhibit the HATs PCAF and p300,
which have been identified through a high-throughput screening.122 The heterocyclic structure of the
isothiazolones is reactive towards the cysteine in the HAT active site.123 The formed covalent bond
with the cysteine thiol group gives the isothiazolones compound class a good potential for
development of probes for activity-based protein profiling (ABPP). Recently, inhibitor C646 has been
described as a potent and selective compound for inhibition of p300 and is able to induce apoptosis of
cancer cells.124, 125 The induction of apoptosis is mediated by a down-regulation of the expression of
the NF- B transcription factor subunit p65.125 Taking all these data together, the inhibition of HATs
can be considered as a valuable strategy to suppress the pro-inflammatory gene expression and can be
a potential approach for treatment of inflammatory diseases and cancer.

27
Introduction and scope of the thesis
Figure 5 – Histone acetyltransferase inhibitors

28
Introduction and scope of the thesis
Scope of the thesis
Inflammation has become the global health issue, which poses the challenge to improve the available
treatment. Modulation of lipoxygenase (LOXs) activity is an interesting starting point for drug
discovery because of their involvement in the initiation and termination of inflammatory processes, as
well as carcinogenic processes. Small molecule modulators of LOXs are tools to investigate their
functions in cellular physiology and pathology. Changes in the expression and the activity of
lipoxygenases and their related products, known as the pro-inflammatory mediators; leukotrienes, and
anti-inflammatory mediators; lipoxins, provide chances for restoring normal function using small
molecule modulator of LOXs.
LOXs inhibition represents a potential novel approach for the design of new drugs. Despite their
importance, the development of novel lipoxygenase inhibitors has considerably decreased over the
past years due to disappointing results in clinical trials. These failures may be due to the fact that the
currently described inhibitor classes suffer from low potency, lack of specificity or low cell-
permeability. This argues for the development of novel classes of modulators in order to get a
comprehensive understanding of LOXs and their roles in inflammatory diseases and cancer. It is
expected that the modulation of LOXs activity will result in novel concepts for regulation of
lipoxygenase activity and innovative strategies for drug discovery in these diseases. Therefore, the
development of novel LOXs modulators is needed. The aims of studies described in this thesis are to
develop novel compounds to modulate LOXs activity in association with inflammation. In addition,
HAT activity is also associated with inflammation and therefore, an approach to study HAT activity
and to develop ABPP probes for HATs are included in this thesis.
In chapter 2, we describe the identification of new inhibitors and activators of LOXs from a
compound collection based on the natural product anacardic acid. We investigate the in vitro
inhibitory potency of these compounds for soybean 15-lipoxygenase-1 (SLO-1) and potato 5-
lipoxygenase (potato 5-LOX) which are used as model enzymes of LOXs. We also evaluate the
structure-activity relationship of these compounds for LOXs modulation. Furthermore, we analyze the
steady-state kinetics for these enzymes in order to clarify their inhibition or activation mechanisms.
This study identifies the presence of an allosteric site in the lipoxygenases in which salicylate based
molecules can bind and regulate the enzymes activity.
Chapter 3 extends our studies on anacardic acid derivatives as lipoxygenase modulators. Here, we
focus on investigating the influence of anacardic acid-derived salicylates on human 5-lipoxygenase
(h-5-LOX) in comparison to cyclooxygenase-2. We explore the potency of newly developed
anacardic acid derivatives and analyze the structure activity relationships in order to determine their
influence on LOX activity. Furthermore, we resolve the modulatory mechanism of these compounds
by performing extensive studies on the kinetic behavior of 5-LOX.

29
Introduction and scope of the thesis
A study on cysteine protease cathepsin B and HAT PCAF is described in chapter 4. We aimed to
explore the utility of the isothiazolone class of HAT inhibitors for development of activity-based
protein profiling (ABPP) probes for HATs by assessing their reactivity towards thiolates, which are
also present in the active sites of the HAT PCAF and protease cathepsin B. We investigate the in vitro
potency of isothiazolone and 5-chlorothiazolone derivatives to inhibit the cysteine protease cathepsin
B in comparison with the HAT PCAF. In this chapter we show the structure-activity relationships and
selective inhibition of cathepsin B. Using a combination of organic synthesis, crystal structure
elucidation of isothiazolones and cathepsin B kinetic studies, the reactivity of these compounds
against thiolates in enzyme active sites is now well understood.
We expand our studies on inflammation in chapter 5 by development of a chemistry-based
method to detect nitrotyrosine, which is generated under oxidative stress conditions. For this purpose,
we synthesized a 2-aminophenol-salicylate-aluminium fluorophore and analyzed its chemical and
physical properties. Subsequently, we develop a method to convert nitrotyrosine into this fluorophore
by two reaction steps under mild conditions. Furthermore, we determine the detection limit of this
novel method on blood samples compared with a conventional immunostaining method. We also
investigate the application of this novel method for histochemical staining of nitrotyrosine in different
tissue sections with or without pre-exposure to inflammation inducers. The novel fluorescence method
proved to have advantages in comparison to the conventional method using an anti-nitrotyrosine
antibody. Although we were unable to detect 3-nitrotyrosine from a blood sample due to the very low
endogenous levels of tyrosine nitrated protein in blood, this novel method shows selective staining in
tissue sections.
All results and a general discussion of the studies described in this thesis are summarized in
chapter 6. Future perspectives of these studies are also incorporated in this chapter.
References
1. Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428-435.
2. Nathan, C. Neutrophils and immunity: challenges and opportunities. Nat. Rev. Immunol. 2006, 6, 173-182.
3. Serhan, C. N.; Savill, J. Resolution of inflammation: the beginning programs the end. Nat. Immunol. 2005,
6, 1191-1197.
4. Serhan, C. N. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving
lipid mediators and pathways. Annu. Rev. Immunol. 2007, 25, 101-137.
5. Drayton, D. L.; Liao, S.; Mounzer, R. H.; Ruddle, N. H. Lymphoid organ development: from ontogeny to
neogenesis. Nat. Immunol. 2006, 7, 344-353.
6. Haining, J. L.; Axelrod, B. Induction period in the lipoxidase-catalyzed oxidation of linoleic acid and its
abolition by substrate peroxide. J. Biol. Chem. 1958, 232, 193-202.
7. Solomon, E. I.; Zhou, J.; Neese, F.; Pavel, E. G. New insights from spectroscopy into the structure/function
relationships of lipoxygenases. Chem. Biol. 1997, 4, 795-808.
8. Brash, A. R. Lipoxygenases: Occurrence, Functions, Catalysis, and Acquisition of Substrate. J. Biol.
Chem. 1999, 274, 23679-23682.
9. Gilmore, T. D. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene 2006, 25, 6680-
6684.

30
Introduction and scope of the thesis
10. Sigal, E. The molecular biology of mammalian arachidonic acid metabolism. Am. J. Physiol. 1991, 260,
L13-L28.
11. Chen, X.; Funk, C. D. The N-terminal “β-Barrel” Domain of 5-Lipoxygenase Is Essential for Nuclear
Membrane Translocation. J. Biol. Chem. 2001, 276, 811-818.
12. Samuelsson, B. Leukotrienes: mediators of immediate hypersensitivity reactions and inflammation.
Science 1983, 220, 568-575.
13. Iversen, L.; Fogh, K.; Bojesen, G.; Kragballe, K. Linoleic acid and dihomogammalinolenic acid inhibit
leukotriene B4 formation and stimulate the formation of their 15-lipoxygenase products by human
neutrophils in vitro. Evidence of formation of antiinflammatory compounds. Agents Actions 1991, 33, 286-
291.
14. Nathaniel, D. J.; Evans, J. F.; Leblanc, Y.; Léveillé, C.; Fitzsimmons, B. J.; Ford-Hutchinson, A. W.
Leukotriene A5 is a substrate and an inhibitor of rat and human neutrophil LTA4 hydrolase. Biochem.
Biophys. Res. Commun. 1985, 131, 827-835.
15. Brüne, B.; Ullrich, V. 12-hydroperoxyeicosatetraenoic acid inhibits main platelet functions by activation of
soluble guanylate cyclase. Mol. Pharmacol. 1991, 39, 671-678.
16. Yeung, J.; Holinstat, M. 12-Lipoxygenase: a Potential Target for Novel Anti-Platelet Therapeutics.
Cardiovasc. Hematol. Agents Med. Chem. 2011, 9, 154-164.
17. Ikei, K. N.; Yeung, J.; Apopa, P. L.; Ceja, J.; Vesci, J.; Holman, T. R.; Holinstat, M. Investigations of
human platelet-type 12-lipoxygenase: role of lipoxygenase products in platelet activation. J. Lipid Res.
2012, 53, 2546-2559.
18. Epp, N.; Fürstenberger, G.; Müller, K.; de Juanes, S.; Leitges, M.; Hausser, I.; Thieme, F.; Liebisch, G.;
Schmitz, G.; Krieg, P. 12R-lipoxygenase deficiency disrupts epidermal barrier function. J. Cell Biol. 2007,
177, 173-182.
19. Profita, M.; Sala, A.; Riccobono, L.; Pace, E.; Paternò, A.; Zarini, S.; Siena, L.; Mirabella, A.; Bonsignore,
G.; Vignola, A. M. 15(S)-HETE modulates LTB4 production and neutrophil chemotaxis in chronic
bronchitis. Am. J. Phys. 2000, 279, C1249-C1258.
20. Sordillo, L. M.; Weaver, J. A.; Cao, Y.; Corl, C.; Sylte, M. J.; Mullarky, I. K. Enhanced 15-HPETE
production during oxidant stress induces apoptosis of endothelial cells. Prostaglandins Other Lipid Mediat.
2005, 76, 19-34.
21. Hsi, L. C.; Wilson, L. C.; Eling, T. E. Opposing Effects of 15-Lipoxygenase-1 and -2 Metabolites on
MAPK Signaling in Prostate: alteration in peroxisome proliferator-activated receptor γ. J. Biol. Chem.
2002, 277, 40549-40556.
22. Bhatia, B.; Maldonado, C. J.; Tang, S.; Chandra, D.; Klein, R. D.; Chopra, D.; Shappell, S. B.; Yang, P.;
Newman, R. A.; Tang, D. G. Subcellular Localization and Tumor-suppressive Functions of 15-
Lipoxygenase 2 (15-LOX2) and Its Splice Variants. J. Biol. Chem. 2003, 278, 25091-25100.
23. Tang, S.; Bhatia, B.; Maldonado, C. J.; Yang, P.; Newman, R. A.; Liu, J.; Chandra, D.; Traag, J.; Klein, R.
D.; Fischer, S. M.; Chopra, D.; Shen, J.; Zhau, H. E.; Chung, L. W. K.; Tang, D. G. Evidence That
Arachidonate 15-Lipoxygenase 2 Is a Negative Cell Cycle Regulator in Normal Prostate Epithelial Cells ,
J. Biol. Chem. 2002, 277, 16189-16201.
24. Rådmark, O. The Molecular Biology and Regulation of 5-Lipoxygenase. Am. J. Respir. Crit. Care Med.
2000, 161, S11-S15.
25. Noguchi, M.; Miyano, M.; Matsumoto, T. Physicochemical characterization of ATP binding to human 5-
lipoxygenase. Lipids 1996, 31, 367-371.
26. Peters-Golden, M.; Brock, T. G. 5-Lipoxygenase and FLAP. Prostaglandins Leukot. Essent. Fatty Acids.
2003, 69, 99-109.
27. Hunter, J. A.; Finkbeiner, W. E.; Nadel, J. A.; Goetzl, E. J.; Holtzman, M. J. Predominant generation of
15-lipoxygenase metabolites of arachidonic acid by epithelial cells from human trachea. Proc. Natl. Acad.
Sci. 1985, 82, 4633-4637.
28. Nadel, J. A.; Conrad, D. J.; Ueki, I. F.; Schuster, A.; Sigal, E. Immunocytochemical localization of
arachidonate 15-lipoxygenase in erythrocytes, leukocytes, and airway cells. J. Clin. Invest. 1991, 87, 1139-
1145.

31
Introduction and scope of the thesis
29. Brash, A. R.; Boeglin, W. E.; Chang, M. S. Discovery of a second 15S-lipoxygenase in humans. Proc.
Natl. Acad. Sci. 1997, 94, 6148-6152.
30. Brown, C. D.; Kilty, I.; Yeadon, M.; Jenkinson, S. Regulation of 15-lipoxygenase isozymes and mucin
secretion by cytokines in cultured normal human bronchial epithelial cells. Inflammation Res. 2001, 50,
321-326.
31. Chanez, P.; Bonnans, C.; Chavis, C.; Vachier, I. 15-Lipoxygenase. Am. J. Respir. Cell Mol. Biol. 2002, 27,
655-658.
32. Haeggstrom, J. Z.; Wetterholm, A. Enzymes and receptors in the leukotriene cascade. Cell Mol. Life Sci.
2002, 59, 742-753.
33. Okamoto, F.; Saeki, K.; Sumimoto, H.; Yamasaki, S.; Yokomizo, T. Leukotriene B4 Augments and
Restores FcγRs-dependent Phagocytosis in Macrophages. J. Biol. Chem. 2010, 285, 41113-41121.
34. Lee, K. S.; Kim, S. R.; Park, H. S.; Park, S. J.; Min, K. H.; Lee, K. Y.; Jin, S. M.; Lee, Y. C. Cysteinyl
leukotriene upregulates IL-11 expression in allergic airway disease of mice. J. Allergy Clin. Immunol.
2007, 119, 141-149.
35. Thompson, C.; Cloutier, A.; Bossé, Y.; Poisson, C.; Larivée, P.; McDonald, P. P.; Stankova, J.; Rola-
Pleszczynski, M. Signaling by the Cysteinyl-Leukotriene Receptor 2: involvement in chemokine gene
transcription. J. Biol. Chem. 2008, 283, 1974-1984.
36. Kawano, T.; Matsuse, H.; Kondo, Y.; Machida, I.; Saeki, S.; Tomari, S.; Mitsuta, K.; Obase, Y.;
Fukushima, C.; Shimoda, T.; Kohno, S. Cysteinyl leukotrienes induce nuclear factor κb activation and
rantes production in a murine model of asthma. J. Allergy Clin. Immunol. 2003, 112, 369-374.
37. Hoffmann, A.; Natoli, G.; Ghosh, G. Transcriptional regulation via the NF-kappaB signaling module.
Oncogene 2006, 25, 6706-6716.
38. Lawrence, T. The Nuclear Factor NF-κB Pathway in Inflammation. Cold Spring Harb. Perspect. Biol.
2009, 1, .
39. Zheng, C.; Yin, Q.; Wu, H. Structural studies of NF-kappaB signaling. Cell Res. 2011, 21, 183-195.
40. Karin, M.; Ben-Neriah, Y. Phosphorylation Meets Ubiquitination: The Control of NF- B Activity. Annu.
Rev. Immunol. 2000, 18, 621-663.
41. Holgate, S. T. Cytokine and anti-cytokine therapy for the treatment of asthma and allergic disease.
Cytokine , 28, 152-157.
42. Williams, R. O.; Paleolog, E.; Feldmann, M. Cytokine inhibitors in rheumatoid arthritis and other
autoimmune diseases. Curr. Opin. Pharmacol. 2007, 7, 412-417.
43. Chung, K. F. Cytokines as targets in chronic obstructive pulmonary disease. Curr. Drug Targets 2006, 7,
675-681.
44. Perkins, N. D.; Gilmore, T. D. Good cop, bad cop: the different faces of NF-kappaB. Cell Death Differ.
2006, 13, 759-772.
45. Haller, D.; Russo, M. P.; Sartor, R. B.; Jobin, C. IKKβ and Phosphatidylinositol 3-Kinase/Akt Participate
in Non-pathogenic Gram-negative Enteric Bacteria-induced RelA Phosphorylation and NF-κB Activation
in Both Primary and Intestinal Epithelial Cell Lines. J. Biol. Chem. 2002, 277, 38168-38178.
46. Madrid, L. V.; Mayo, M. W.; Reuther, J. Y.; Baldwin, A. S. Akt Stimulates the Transactivation Potential
of the RelA/p65 Subunit of NF-κB through Utilization of the IκB Kinase and Activation of the Mitogen-
activated Protein Kinase p38. J. Biol. Chem. 2001, 276, 18934-18940.
47. Perkins, N. D. Post-translational modifications regulating the activity and function of the nuclear factor
kappa B pathway. Oncogene 2006, 25, 6717-6730.
48. Bonizzi, G.; Karin, M. The two NF-κB activation pathways and their role in innate and adaptive immunity.
Trends Immunol. 2004, 25, 280-288.
49. Dahlen, S. E.; Bjork, J.; Hedqvist, P.; Arfors, K. E.; Hammarstrom, S.; Lindgren, J. A.; Samuelsson, B.
Leukotrienes promote plasma leakage and leukocyte adhesion in postcapillary venules: in vivo effects with
relevance to the acute inflammatory response. Proc. Natl. Acad. Sci. U. S. A. 1981, 78, 3887-3891.
50. Ylä-Herttuala, S.; Rosenfeld, M. E.; Parthasarathy, S.; Sigal, E.; Sarkioja, T.; Witztum, J. L.; Steinberg, D.
Gene expression in macrophage-rich human atherosclerotic lesions. 15-lipoxygenase and acetyl low
density lipoprotein receptor messenger RNA colocalize with oxidation specific lipid-protein adducts. J.
Clin. Invest. 1991, 87, 1146-1152.

32
Introduction and scope of the thesis
51. Hui, A. Y.; McCarty, W. J.; Masuda, K.; Firestein, G. S.; Sah, R. L. A systems biology approach to
synovial joint lubrication in health, injury, and disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2012, 4, 15-
37.
52. Serhan, C. N. Novel Lipid Mediators and Resolution Mechanisms in Acute Inflammation: To Resolve or
Not? Am. J. Pathol. 2010, 177, 1576-1591.
53. Chen, M.; Lam, B. K.; Kanaoka, Y.; Nigrovic, P. A.; Audoly, L. P.; Austen, K. F.; Lee, D. M. Neutrophil-
derived leukotriene B4 is required for inflammatory arthritis. J. Exp. Med. 2006, 203, 837-842.
54. Gheorghe, K. R.; Korotkova, M.; Catrina, A. I.; Backman, L.; af Klint, E.; Claesson, H.; Radmark, O.;
Jakobsson, P. Expression of 5-lipoxygenase and 15-lipoxygenase in rheumatoid arthritis synovium and
effects of intraarticular glucocorticoids. Arthritis Res. Ther. 2009, 11, R83.
55. Wu, M.; Lin, T.; Chiu, Y.; Liou, H.; Yang, R.; Fu, W. Involvement of 15-lipoxygenase in the
inflammatory arthritis. J. Cell. Biochem. 2012, 113, 2279-2289.
56. Asahara, H.; Asanuma, M.; Ogawa, N.; Nishibayashi, S.; Inoue, H. High DNA-binding activity of
transcription factor NF-kappa B in synovial membranes of patients with rheumatoid arthritis. Biochem.
Mol. Biol. Int. 1995, 37, 827-832.
57. Jupp, J.; Hillier, K.; Elliott, D. H.; Fine, D. R.; Bateman, A. C.; Johnson, P. A.; Cazaly, A. M.; Penrose, J.
F.; Sampson, A. P. Colonic expression of leukotriene-pathway enzymes in inflammatory bowel diseases.
Inflamm. Bowel Dis. 2007, 13, 537-546.
58. Stanke-Labesque, F.; Pofelski, J.; Moreau-Gaudry, A.; Bessard, G.; Bonaz, B. Urinary leukotriene E4
excretion: a biomarker of inflammatory bowel disease activity. Inflamm. Bowel Dis. 2008, 14, 769-774.
59. Wang, D.; Dubois, R. N. Eicosanoids and cancer. Nat. Rev. Cancer. 2010, 10, 181-193.
60. Pidgeon, G. P.; Tang, K.; Cai, Y. L.; Piasentin, E.; Honn, K. V. Overexpression of Platelet-type 12-
Lipoxygenase Promotes Tumor Cell Survival by Enhancing αvβ3 and αvβ5 Integrin Expression. Cancer
Res. 2003, 63, 4258-4267.
61. Kerjaschki, D.; Bago-Horvath, Z.; Rudas, M.; Sexl, V.; Schneckenleithner, C.; Wolbank, S.; Bartel, G.;
Krieger, S.; Kalt, R.; Hantusch, B.; Keller, T.; Nagy-Bojarszky, K.; Huttary, N.; Raab, I.; Lackner, K.;
Krautgasser, K.; Schachner, H.; Kaserer, K.; Rezar, S.; Madlener, S.; Vonach, C.; Davidovits, A.; Nosaka,
H.; Hämmerle, M.; Viola, K.; Dolznig, H.; Schreiber, M.; Nader, A.; Mikulits, W.; Gnant, M.; Hirakawa,
S.; Detmar, M.; Alitalo, K.; Nijman, S.; Offner, F.; Maier, T. J.; Steinhilber, D.; Krupitza, G. Lipoxygenase
mediates invasion of intrametastatic lymphatic vessels and propagates lymph node metastasis of human
mammary carcinoma xenografts in mouse. J. Clin. Invest. 2011, 121, 2000-2012.
62. Hennig, R.; Grippo, P.; Ding, X.; Rao, S. M.; Buchler, M. W.; Friess, H.; Talamonti, M. S.; Bell, R. H.;
Adrian, T. E. 5-Lipoxygenase, a Marker for Early Pancreatic Intraepithelial Neoplastic Lesions. Cancer
Res. 2005, 65, 6011-6016.
63. Steiner, D. R. S.; Gonzalez, N. C.; Wood, J. G. Leukotriene B4 promotes reactive oxidant generation and
leukocyte adherence during acute hypoxia. J. Appl. Physiol. 2001, 91, 1160-1167.
64. Serhan, C. N. Lipoxin biosynthesis and its impact in inflammatory and vascular events. Biochim. Biophys.
Acta. 1994, 1212, 1.
65. Chiang, N.; Arita, M.; Serhan, C. N. Anti-inflammatory circuitry: Lipoxin, aspirin-triggered lipoxins and
their receptor ALX. Prostaglandins Leukot. Essent. Fatty Acids 2005, 73, 163-177.
66. Serhan, C. N. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nature
rev. Immunol. 2008, 8, 349.
67. Fierro, I. M.; Colgan, S. P.; Bernasconi, G.; Petasis, N. A.; Clish, C. B.; Arita, M.; Serhan, C. N. Lipoxin
A4 and Aspirin-Triggered 15-epi-Lipoxin A4 Inhibit Human Neutrophil Migration: Comparisons Between
Synthetic 15 Epimers in Chemotaxis and Transmigration with Microvessel Endothelial Cells and Epithelial
Cells. .J. Immunol. 2003, 170, 2688-2694.
68. Clària, J.; Titos, E.; Jiménez, W.; Ros, J.; Ginès, P.; Arroyo, V.; Rivera, F.; Rodés, J. Altered biosynthesis
of leukotrienes and lipoxins and host defense disorders in patients with cirrhosis and ascites.
Gastroenterology 1998, 115, 147-156.
69. Karp, C. L. Defective lipoxin-mediated anti-inflammatory activity in the cystic fibrosis airway. Nat.
Immunol. 2004, 5, 388.

33
Introduction and scope of the thesis
70. Benson, M.; Carlsson, L.; Adner, M.; Jernås, M.; Rudemo, M.; Sjögren, A.; Svensson, P. A.; Uddman, R.;
Cardell, L. O. Gene profiling reveals increased expression of uteroglobin and other anti-inflammatory
genes in glucocorticoid-treated nasal polyps. J. Allergy Clin. Immunol. 2004, 113, 1137-1143.
71. Chavis, C.; Vachier, I.; Chanez, P.; Bousquet, J.; Godard, P. 5(S),15(S)-dihydroxyeicosatetraenoic acid
and lipoxin generation in human polymorphonuclear cells: dual specificity of 5-lipoxygenase towards
endogenous and exogenous precursors. J. Exp. Med.1996, 183, 1633-1643.
72. Basselin, M. Anti-inflammatory effects of chronic aspirin on brain arachidonic acid metabolites.
Neurochem. Res. 2011, 36, 139.
73. Takeda, S.; Jiang, R.; Aramaki, H.; Imoto, M.; Toda, A.; Eyanagi, R.; Amamoto, T.; Yamamoto, I.;
Watanabe, K. ?9-tetrahydrocannabinol and its major metabolite ?9-tetrahydrocannabinol-11-oic acid as 15-
lipoxygenase inhibitors. J. Pharm. Sci. 2011, 100, 1206-1211.
74. Whitman, S. Structure-activity relationship studies of nordihydroguaiaretic acid inhibitors toward soybean,
12-human, and 15-human lipoxygenase. J. Med. Chem. 2002, 45, 2659.
75. Moody, J. S.; Marnett, L. J. Kinetics of Inhibition of Leukocyte 12-Lipoxygenase by the Isoform-Specific
Inhibitor 4-(2-Oxapentadeca-4-yne)phenylpropanoic Acid . Biochemistry 2002, 41, 10297-10303.
76. Hong, J.; Bose, M.; Ju, J.; Ryu, J.; Chen, X.; Sang, S.; Lee, M.; Yang, C. S. Modulation of arachidonic
acid metabolism by curcumin and related β-diketone derivatives: effects on cytosolic phospholipase A2,
cyclooxygenases and 5-lipoxygenase. Carcinogenesis 2004, 25, 1671-1679.
77. Gillard, J.; Ford-Hutchinson, A. W.; Chan, C.; Charleson, S.; Denis, D.; Foster, A.; Fortin, R.; Leger, S.;
McFarlane, C. S.; Morton, H. L-663,536 (MK-886) (3-[1-(4-chlorobenzyl)-3-t-butyl-thio-5-isopropylindol-
2-yl]-2,2 - dimethylpropanoic acid), a novel, orally active leukotriene biosynthesis inhibitor. Can. J.
Physiol. Pharmacol. 1989, 67, 456-464.
78. Evans, J. F.; Ferguson, A. D.; Mosley, R. T.; Hutchinson, J. H. What's all the FLAP about?: 5-
lipoxygenase-activating protein inhibitors for inflammatory diseases. Trends Pharmacol. Sci. 2008, 29, 72-
78.
79. Friedman, B. S.; Bel, E. H.; Buntinx, A.; Tanaka, W.; Han, Y. R.; Shingo, S.; Spector, R.; Sterk, P. Oral
Leukotriene Inhibitor (MK-886) Blocks Allergen-induced Airway Responses. Am. Rev. Respir. Dis. 1993,
147, 839-844.
80. Fruchtmann, R.; Mohrs, K. H.; Hatzelmann, A.; Raddatz, S.; Fugmann, B.; Junge, B.; Horstmann, H.;
Muller-Peddinghaus, R. In vitro pharmacology of BAY X1005, a new inhibitor of leukotriene synthesis.
Agents Actions 1993, 38, 188-195.
81. Mancini, J. A.; Prasit, P.; Coppolino, M. G.; Charleson, P.; Leger, S.; Evans, J. F.; Gillard, J. W.; Vickers,
P. J. 5-Lipoxygenase-activating protein is the target of a novel hybrid of two classes of leukotriene
biosynthesis inhibitors. Mol. Pharmacol. 1992, 41, 267-272.
82. Charleson, S.; Evans, J. F.; Leger, S.; Perrier, H.; Prasit, P.; Wang, Z.; Vickers, P. J. Structural
requirements for the binding of fatty acids to 5-lipoxygenase-activating protein. Eur. J. Pharmacol. 1994,
267, 275-280.
83. Werz, O. 5-Lipoxygenase: Cellular Biology and Molecular Pharmacology. Curr. Drug Targets Inflamm.
Allergy 2002, 1, 23-44.
84. McMillan, R. M.; Walker, E. R. H. Designing therapeutically effective 5-lipoxygenase inhibitors. Trends
Pharmacol. Sci. 1992, 13, 323-330.
85. Batt, D. G.; Maynard, G. D.; Petraitis, J. J.; Shaw, J. E.; Galbraith, W.; Harris, R. R. 2-Substituted-1-
Naphthols as Potent 5-Lipoxygenase Inhibitors with Topical Antiinflammatory Activity. J. Med. Chem.
1990, 33, 360-370.
86. Corey, E. J.; Wright, S. W.; Matsuda, S. P. T. Stereochemistry and mechanism of the biosynthesis of
leukotriene A4 from 5(S)-hydroperoxy-6(E),8,11,14(Z)-eicosatetraenoic acid. Evidence for an organoiron
intermediate. J. Am. Chem. Soc. 1989, 111, 1452-1455.
87. Doiron, J.; Boudreau, L. H.; Picot, N.; Villebonet, B.; Surette, M. E.; Touaibia, M. Synthesis and 5-
lipoxygenase inhibitory activity of new cinnamoyl and caffeoylclusters. Bioorg. Med. Chem. Lett. 2009,
19, 1118-1121.
88. Ford-Hutchinson, A.; Gresser, M.; Young, R. N. 5-Lipoxygenase. Annu. Rev. Biochem. 1994, 63, 383-417.

34
Introduction and scope of the thesis
89. Gillmor, S. A.; Villasenor, A.; Fletterick, R.; Sigal, E.; Browner, M. F. The structure of mammalian 15-
lipoxygenase reveals similarity to the lipases and the determinants of substrate specificity. Nat. Struct.
Biol. 1997, 4, 1003-1009.
90. Xu, S.; Mueser, T.; Marnett, L.; Funk Jr., M. Crystal Structure of 12-Lipoxygenase Catalytic-Domain-
Inhibitor Complex Identifies a Substrate-Binding Channel for Catalysis. Structure 2012, 20, 1490-1497.
91. Connolly, P. J.; Wetter, S. K.; Beers, K. N.; Hamel, S. C.; Chen, R. H. K.; Wachter, M. P.; Ansell, J.;
Singer, M. M.; Steber, M.; Ritchie, D. M.; Argentieri, D. C. N-Hydroxyurea and hydroxamic acid
inhibitors of cyclooxygenase and 5-lipoxygenase. Bioorg. Med. Chem. Lett. 1999, 9, 979-984.
92. Nelson, H.; Kemp, J.; Berger, W.; Corren, J.; Casale, T.; Dube, L.; Walton-Bowen, K.; LaVallee, N.;
Stepanians, M. Efficacy of zileuton controlled-release tablets administered twice daily in the treatment of
moderate persistent asthma: a 3-month randomized controlled study. Ann. Allergy Asthma Immunol. 2007,
99, 178-184.
93. Brooks, C. D.; Stewart, A. O.; Basha, A.; Bhatia, P.; Ratajczyk, J. D.; Martin, J. G.; Craig, R. A.; Kolasa,
T.; Bouska, J. B.; Lanni, C. (R)-(+)-N-[3-[5-[(4-fluorophenyl)methyl]-2-thienyl]-1-methyl- 2-propynyl]-N-
hydroxyurea (ABT-761), a second-generation 5-lipoxygenase inhibitor. J. Med. Chem. 1995, 38, 4768-
4775.
94. Back, M. Inhibitors of the 5-lipoxygenase pathway in atherosclerosis. Curr. Pharm. Des. 2009, 15, 3116-
3132.
95. Gurjar, M. K.; Murugaiah, A. M. S.; Radhakrishna, P.; Ramana, C. V.; Chorghade, M. S. A novel and
simple asymmetric synthesis of CMI-977 (LDP-977): a potent anti-asthmatic drug lead. Tetrahedron:
Asymmetry 2003, 14, 1363-1370.
96. Pergola, C.; Werz, O. 5-Lipoxygenase inhibitors: a review of recent developments and patents. Expert
Opin. Ther. Patents 2010, 20, 355-375.
97. Werz, O. Pharmacological intervention with 5-lipoxygenase: new insights and novel compounds. Expert
Opin. Ther. Patents 2005, 15, 505.
98. Bird; Bruneau, P.; Crawley, G. C.; Edwards, M. P.; Foster, S. J.; Girodeau, J. M.; Kingston, J. F.;
McMillan, R. M. (Methoxyalkyl)thiazoles: a new series of potent, selective, and orally active 5-
lipoxygenase inhibitors displaying high enantioselectivity. J. Med. Chem. 1991, 34, 2176-2186.
99. Falgueyret, J.; Hutchinson, J. H.; Riendeau, D. Criteria for the identification of non-redox inhibitors of 5-
lipoxygenase. Biochem. Pharmacol. 1993, 45, 978-981.
100. Crawley, G. C.; Dowell, R. I.; Edwards, P. N.; Foster, S. J.; McMillan, R. M.; Walker, E. R. H.; Waterson,
D.; Bird, T. G.; Bruneau, P.; Girodeau, J. M. Methoxytetrahydropyrans. A new series of selective and
orally potent 5-lipoxygenase inhibitors. J. Med. Chem. 1992, 35, 2600-2609.
101. Kusner, E. J.; Buckner, C. K.; Dea, D. M.; DeHaas, C. J.; Marks, R. L.; Krell, R. D. The 5-lipoxygenase
inhibitors ZD2138 and ZM230487 are potent and selective inhibitors of several antigen-induced guinea-pig
pulmonary responses. Eur. J. Pharmacol. 1994, 257, 285-292.
102. Young, R. N. Inhibitors of 5-lipoxygenase: a therapeutic potential yet to be fully realized? Eur. J. Med.
Chem. 1999, 34, 671-685.
103. Sorkness, C. A. The Use of 5-Lipoxygenase Inhibitors and Leukotriene Receptor Antagonists in the
Treatment of Chronic Asthma. Pharmacotherapy 1997, 17, 50S-54S.
104. Renzi, P. M. Antileukotriene agents in asthma: The dart that kills the elephant? CMAJ. 1999, 160, 217-
223.
105. Tintinger, G. R. Montelukast: more than a cysteinyl leukotriene receptor antagonist?
TheScientificWorldJournal 2010, 10, 2403.
106. Ramires, R.; Caiaffa, M. F.; Tursi, A.; Haeggström, J. Z.; Macchia, L. Novel inhibitory effect on 5-
lipoxygenase activity by the anti-asthma drug montelukast. Biochem. Biophys. Res. Commun. 2004, 324,
815-821.
107. Tahan, F. Montelukast inhibits tumour necrosis factor-a-mediated interleukin-8 expression through
inhibition of nuclear factor- B p65-associated histone acetyltransferase activity. Clin. Exp. Allergy 2008,
38, 805.
108. Ghizzoni, M.; Haisma, H. J.; Maarsingh, H.; Dekker, F. J. Histone acetyltransferases are crucial regulators
in NF-κB mediated inflammation. Drug Discov. Today 2011, 16, 504-511.

35
Introduction and scope of the thesis
109. Nam, N. H. Naturally occurring NF-kappaB inhibitors. Mini Rev. Med. Chem. 2006, 6, 945.
110. Wang, C.; Fu, M.; Pestell, R. Histone Acetylation/Deacetylation As a Regulator of Cell Cycle Gene
Expression. Methods Mol. Biol. 2004, 241, 207-216.
111. Moore, J. D.; Krebs, J. E. Histone modifications and DNA double-strand break repair. Biochem. Cell Biol.
2004, 82, 446-452.
112. Roth, S. Y.; Denu, J. M.; Allis, C. D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81-120.
113. Parthun, M. R. Hat1: the emerging cellular roles of a type B histone acetyltransferase. Oncogene 2007, 26,
5319-5328.
114. Marmorstein, R. Structure of histone acetyltransferases. J. Mol. Biol. 2001, 311, 433-444.
115. Allan, J.; Harborne, N.; Rau, D. C.; Gould, H. Participation of core histone "tails" in the stabilization of the
chromatin solenoid. J. Cell Biol. 1982, 93, 285-297.
116. Yang, X. Lysine acetylation and the bromodomain: a new partnership for signaling. Bioessays 2004, 26,
1076-1087.
117. Balasubramanyam, K.; Varier, R. A.; Altaf, M.; Swaminathan, V.; Siddappa, N. B.; Ranga, U.; Kundu, T.
K. Curcumin, a Novel p300/CREB-binding Protein-specific Inhibitor of Acetyltransferase, Represses the
Acetylation of Histone/Nonhistone Proteins and Histone Acetyltransferase-dependent Chromatin
Transcription. J. Biol. Chem. 2004, 279, 51163-51171.
118. Taher, M. M. Curcumin inhibits ultraviolet light induced human immunodeficiency virus gene expression.
Mol. Cell. Biochem. 2003, 254, 289.
119. Arif, M. Mechanism of p300 specific histone acetyltransferase inhibition by small molecules. J. Med.
Chem. 2009, 52, 267.
120. Prasad, S. Garcinol potentiates TRAIL-induced apoptosis through modulation of death receptors and
antiapoptotic proteins. Mol. Cancer Ther. 2010, 9, 856.
121. Sung, B.; Pandey, M. K.; Ahn, K. S.; Yi, T.; Chaturvedi, M. M.; Liu, M.; Aggarwal, B. B. Anacardic acid
(6-nonadecyl salicylic acid), an inhibitor of histone acetyltransferase, suppresses expression of nuclear
factor-κB–regulated gene products involved in cell survival, proliferation, invasion, and inflammation
through inhibition of the inhibitory subunit of nuclear factor-κBα kinase, leading to potentiation of
apoptosis. Blood 2008, 111, 4880-4891.
122. Stimson, L. Isothiazolones as inhibitors of PCAF and p300 histone acetyltransferase activity. Mol. Cancer
Ther. 2005, 4, 1521.
123. Morley, J. O.; Oliver Kapur, A. J.; Charlton, M. H. Structure-activity relationships in 3-isothiazolones.
Org. Biomol. Chem. 2005, 3, 3713-3719.
124. Bowers, E. M.; Yan, G.; Mukherjee, C.; Orry, A.; Wang, L.; Holbert, M. A.; Crump, N. T.; Hazzalin, C.
A.; Liszczak, G.; Yuan, H.; Larocca, C.; Saldanha, S. A.; Abagyan, R.; Sun, Y.; Meyers, D. J.;
Marmorstein, R.; Mahadevan, L. C.; Alani, R. M.; Cole, P. A. Virtual Ligand Screening of the p300/CBP
Histone Acetyltransferase: Identification of a Selective Small Molecule Inhibitor. Chem. Biol. 2010, 17,
471-482.
125. Santer, F. R.; Höschele, P. P. S.; Oh, S. J.; Erb, H. H. H.; Bouchal, J.; Cavarretta, I. T.; Parson, W.;
Meyers, D. J.; Cole, P. A.; Culig, Z. Inhibition of the Acetyltransferases p300 and CBP Reveals a
Targetable Function for p300 in the Survival and Invasion Pathways of Prostate Cancer Cell Lines. Mol.
Cancer Ther. 2011, 10, 1644-1655.

Anacardic acid derived
salicylates are inhibitors or
activators of lipoxygenases
Part of this chapter has been published:
Rosalina Wisastra, Massimo Ghizzoni, Andre Boltjes,
Hidde J. Haisma, and Frank J. Dekker
Bioorganic & Medicinal Chemistry, 2012, 20(16), 5027-5032
Department of Pharmaceutical Gene Modulation, Groningen Research Institute of Pharmacy,
University of Groningen, Antonius Deusinglaan 1, 9713 AV Groningen, The Netherlands.
2

38
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
Abstract
Lipoxygenases catalyse the oxidation of unsaturated fatty acids, such as linoleic acid, which play
a crucial role in inflammatory responses. Selective inhibitors may provide a new therapeutic
approach for inflammatory diseases. In this study, we describe the identification of a novel
soybean lipoxygenase-1 (SLO-1) inhibitor and a potato 5-lipoxygenase (5-LOX) activator from a
screening of a focused compound collection around the natural product anacardic acid. The
natural product anacardic acid inhibits SLO-1 with an IC50 of 52 µM, whereas the inhibitory
potency of the novel mixed type inhibitor 23 is five-fold enhanced. In addition, another
derivative (21) caused non-essential activation of potato 5-LOX. This suggests the presence of an
allosteric binding site that regulates the lipoxygenase activity.

39
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
Introduction
Lipoxygenases (LOXs) are a family of non-heme iron-containing enzymes that catalyse the
oxygenation of cis,cis-1,4-pentadiene moieties in lipids. In general, lipoxygenases (LOXs), which
are widely distributed in both the plant and animal kingdom, are categorized into 5-LOX, 8-LOX,
12-LOX and 15-LOX based on the position of oxygenation of their substrates. Lipoxygenases
convert their natural substrates, arachidonic acid, to hydroperoxy eicosatetraenoic acids
(HPETEs) by a radical mechanism.1 Lipoxygenases found in plant oxygenate linoleic acid to
generate 13-hydroperoxy-9(Z),11(E)-octadecadienoic acid (13-HPOD). 5-HPETE is transformed
into Leukotriene A4, which contains an unstable epoxide. Subsequently, Leukotriene A4 is
converted either into Leukotriene B4 by an enzymatic hydrolysis or into Leukotriene C4 by
glutathione S-transferase. The resulting Leukotriene C4 is further metabolized to Leukotriene D4
and Leukotriene E4.2 It has been reported that activities of lipoxygenases and their related
products play an important role in numerous inflammatory and proliferative diseases such as
asthma, atherosclerosis, and cancer.3-5 Leukotriene B4 activates inflammatory cells such as
neutrophils and macrophages.6 This broad range of biological effects demonstrates the
importance of lipoxygenases as a therapeutic target.
6-pentadecyl salicylic acid, commonly known as anacardic acid, is a natural product found in
cashew nut shells. This compound is often associated with anti-inflammatory, anti-tumor,
molluscicidal, and anti-microbial activities.7-9 Previous studies reported that anacardic acid
inhibits peroxidation of linoleic acid by soybean lipoxygenase-1 (SLO-1) with an IC50 of 85
µM.10 In addition, anacardic acid and its derivatives inhibit histone acetyl transferases (HATs)
and cyclooxygenases, which are also involved in inflammation and cancer.11-13
In this study, we investigate the affinity and selectivity of anacardic acid derived inhibitors for
SLO-1 and potato 5-LOX as representatives of the lipoxygenase family. Although lipoxygenases
are well-known drug targets, relatively little inhibitors with high selectivity and potency have
been described (reviewed by Pergola and Werz14). Currently, the selective 5-LOX inhibitor
Zileuton (IC50 = 0.5 µM) is marketed for treatment of asthma.15 Although there are no 15-LOX
inhibitors available for the clinic, recent studies identified inhibitors with nanomolar affinity. 16, 17
Nevertheless, their efficacy in cell-based studies remains limited. Therefore, development of
inhibitors of new-structural classes remains necessary in order to explore these enzymes further
as drug targets.
Here, we describe the identification of lipoxygenase inhibitors and activators from a focused
compound collection based on anacardic acid. Anacardic acid and its derivatives were
synthesized following previous research18 and inhibition of SLO-1 and potato 5-LOX were
investigated. We found that derivative 23 inhibits SLO-1 selectively to potato 5-LOX, whereas
derivative 21 shows a three-fold activation on potato 5-LOX. The enzyme kinetic studies of these

40
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
modulators indicate the presence of an allosteric site that influences the activity of the
lipoxygenase active site.
Results and discussion
Design
The fact that lipoxygenases are iron-containing enzymes combined with the fact that salicylates are
known iron-binding compounds justifies the hypothesis that the iron-binding properties of salicylates
are keys to their inhibitory potency. Based on this hypothesis a molecular modeling study was
performed in order to propose a binding configuration of anacardic acid 18 to the soybean
lipoxygenase-1 active site. A docking simulation shows that the salicylate from 18 can coordinate to
the iron and that its hydrophobic tail fits well in the hydrophobic cavity in the active site of soybean
lipoxygenase-1 (Figure 1). The hydrophobic interactions between the amino acid residues and the
ligand are probably important for the interaction in the enzyme active site. Based on our proposed
binding model we expect that the different 6-alkyl substituents in our focused compound collection
fit, in principle, in the lipoxygenase active site. Therefore, we screened this focused compound
collection for lipoxygenase inhibition.
Figure 1. Binding pose of anacardic acid 18 (A) and inhibitor 23 (B) in a crystal structure of soybean
lipoxygenase-1 (PDB files 1F8N). The iron atom is shown as a ball, the binding residues are shown as
wireframes, and the ligand is shown as sticks. The hydrogen bonds are shown as cyan dashed lines. The iron
coordination with the ligand and enzyme are illustrated with green lines.
Synthesis
A compound collection was assembled around the natural product anacardic acid 18. This collection
comprises the commercially available salicylates 12, 13 and 14, the previously published salicylates
15-23, 26-30, and 32 and the newly synthesized salicylates 24, 25, 31, and 33. The synthesis of
anacardic acid and its derivatives 15-23, 26-30, and 32 were described previously by Ghizzoni et al.18,
19 and compounds 24, 25, 31, and 33 were synthesized using similar procedures (Scheme 1). Triflate 1
(A) (B)

41
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
was synthesized as described by Uchiyama et al.20 Alkynes 2 21 and 5 were prepared from triflate 1 or
3-iodobenzoic acid 3 by Sonogashira coupling with trimethylsilyl (TMS) acetylene and subsequent
cleavage of the TMS protective group to yield the corresponding products (Scheme 1). Benzoxazoles
7a and 7b were prepared from 2-amino-5-chlorophenol (6) and benzoic acid through amidation and
cyclization.22 Subsequently, 7a was attached to 2 and 7b was attached to 5 to give the corresponding
alkynyls, which were reduced using catalytic hydrogenation to give 24, and 31 (Scheme 1).
Sonogashira coupling of aryl chlorides has been described to proceed with PdCl2(PPh3)2, P(tBu)3,
and 1,8-Diazabicyclo [5.4.0]undec-7-ene (DBU) as catalysts, and Cs2CO3 as a base to give >80%
yields.23 However, our attempt to synthesize compounds 24, and 31 under these conditions yielded
20-32% of the desired products. These relatively low yields are possibly due to electronic properties
of the benzoxazole and/or the limited stability of the ester functionality in 2 or 5 under the basic
coupling conditions.
Scheme 1. Reagent and conditions : a) Trimethylsilylacetylene, CuI, PdCl2(PPh3)2, Et2NH, PPh3, CH3CN, (MW,
120 ⁰C, 95 W), b) TBAF, THF, 0 ⁰C; c) CuI, PdCl2(PPh3)2, Et2NH, HC≡CR, CH3CN (MW, 100 ⁰C, 70 W); d)
H2, Pd/C, MeOH, 40 ⁰C ; e) aqueous KOH 5 N, THF, 55 ⁰C; f) benzyl bromide, K2CO3, DMF, R.T. g) benzoic
acid anhydride, pyridine, DMF, R.T.; h) SOCl2, 60 ⁰C followed by 2-aminophenol, 0 ⁰C; i) p-toluensulfonic
acid, toluene, reflux; j) SOCl2, DMAP, DME, acetone, R.T; k) Bn-Br, K2CO3, DMF, R.T.; l) Bn-O-Na+, THF,
R.T.; m) Tf2O, pyridine, CH2Cl2, 0 ⁰C ; n) HC≡CR, aryl chloride, Cs2CO3, PdCl2(PPh3)2, P(tBu)3, DBU, DMF
(MW, 150 ⁰C, 95 W)
Benzoxazole 9 was prepared from commercially available 2-(4-bromophenyl)acetic acid (8) and 2-
aminophenol following two reaction steps. Firstly, 2-(4-bromophenyl)acetic acid (8) was converted
into the acyl chloride using thionyl chloride followed by amide bond formation with 2-aminophenol
to give the amide with high yields (70%). Secondly, this amide was cyclized using p-toluenesulfonic
acid in toluene at reflux for 4 hours to give the product 9 with high yields (78-85%). Subsequently,

42
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
the arylbromide (9) was coupled to trimethylsilyl acetylene by a Sonogashira coupling. The
trimethylsilyl protective group was cleaved to give the corresponding alkyne 10. This alkyne was
coupled to triflate 11 24 using a Sonogashira coupling to provide 55% yield of the corresponding
alkyne (Scheme 1), which was hydrogenated to yield 25 in a single step.
Acetonide 13 was synthesized from 2,4-dihydroxybenzoic acid 12 as described by Tranchimand
et.al.24 Furthermore, acetonide 13 was converted to triflate 14 as described previously.20 Sonogashira
reaction, a coupling of 1-ethynyl-4- heptyl benzene with triflate 14, was performed to give the alkyne
with high yield (79%), which was hydrogenated to give compound 33.
Table 1. Compounds collection of anacardic acid and its derivatives
R1 R2 R3
12 H H H
13 H OH H
14 C2H5 OH CH3
15 H H
16 H H
17 H OH
18 H H
19 CH3 H
20 H OH
21 H H
22 H H
23 H OH
24 H H
25 H OH
26 H H
27 H H
28 H H
29 H H
30 H OH
31 H H
32 H H
33 H OH
Compounds
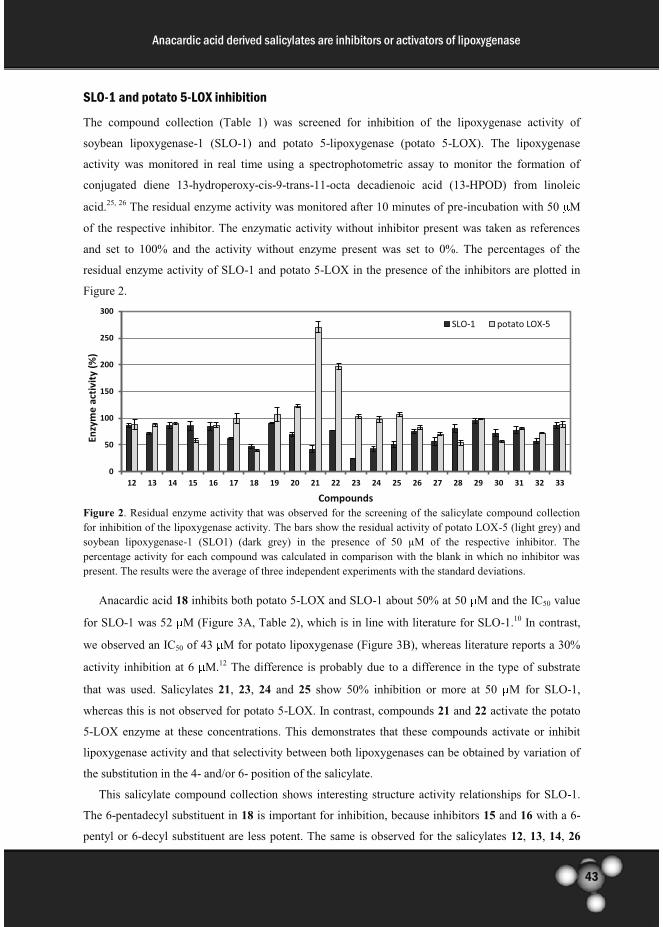
43
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
SLO-1 and potato 5-LOX inhibition
The compound collection (Table 1) was screened for inhibition of the lipoxygenase activity of
soybean lipoxygenase-1 (SLO-1) and potato 5-lipoxygenase (potato 5-LOX). The lipoxygenase
activity was monitored in real time using a spectrophotometric assay to monitor the formation of
conjugated diene 13-hydroperoxy-cis-9-trans-11-octa decadienoic acid (13-HPOD) from linoleic
acid.25, 26 The residual enzyme activity was monitored after 10 minutes of pre-incubation with 50 M
of the respective inhibitor. The enzymatic activity without inhibitor present was taken as references
and set to 100% and the activity without enzyme present was set to 0%. The percentages of the
residual enzyme activity of SLO-1 and potato 5-LOX in the presence of the inhibitors are plotted in
Figure 2.
Figure 2. Residual enzyme activity that was observed for the screening of the salicylate compound collection
for inhibition of the lipoxygenase activity. The bars show the residual activity of potato LOX-5 (light grey) and
soybean lipoxygenase-1 (SLO1) (dark grey) in the presence of 50 µM of the respective inhibitor. The
percentage activity for each compound was calculated in comparison with the blank in which no inhibitor was
present. The results were the average of three independent experiments with the standard deviations.
Anacardic acid 18 inhibits both potato 5-LOX and SLO-1 about 50% at 50 M and the IC50 value
for SLO-1 was 52 M (Figure 3A, Table 2), which is in line with literature for SLO-1.10 In contrast,
we observed an IC50 of 43 M for potato lipoxygenase (Figure 3B), whereas literature reports a 30%
activity inhibition at 6 M.12 The difference is probably due to a difference in the type of substrate
that was used. Salicylates 21, 23, 24 and 25 show 50% inhibition or more at 50 M for SLO-1,
whereas this is not observed for potato 5-LOX. In contrast, compounds 21 and 22 activate the potato
5-LOX enzyme at these concentrations. This demonstrates that these compounds activate or inhibit
lipoxygenase activity and that selectivity between both lipoxygenases can be obtained by variation of
the substitution in the 4- and/or 6- position of the salicylate.
This salicylate compound collection shows interesting structure activity relationships for SLO-1.
The 6-pentadecyl substituent in 18 is important for inhibition, because inhibitors 15 and 16 with a 6-
pentyl or 6-decyl substituent are less potent. The same is observed for the salicylates 12, 13, 14, 26
0
50
100
150
200
250
300
12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33
En
zym
e a
ctiv
ity
(%
)
Compounds
SLO-1 potato LOX-5

44
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
and 27 without an aliphatic substituent in the 6-position. These results demonstrate that the side chain
length of the 6-alkyl chain is important for the SLO-1 inhibitory potency. This is possibly due to the
hydrophobic interactions between the inhibitor and the binding pocket of the enzyme. In addition, a
complete loss of inhibition was observed if the carboxylate of anacardic acid 18 was converted into a
methyl ester 19, which demonstrates that the free carboxylate is also important for binding.
Furthermore, the importance of the 2-hydroxyl functionality of the salicylate is demonstrated by the
inactivity of the inhibitors 31-33, which show less than 50% inhibition 50 µM. These results
demonstrate that the carboxylate and the hydroxyl of the salicylate core are both crucial for inhibition
of SLO-1.
Salicylates 24 and 25 with a benzoxazole functionality in their side chain maintain their inhibition
of SLO-1, whereas no inhibition of potato 5-LOX was observed. This demonstrates that selectivity
between potato 5-LOX and SLO-1 can be obtained by variation of the 6-alkyl substituent in anacardic
acid.
Figure 3. The IC50 value for inhibition of (A) SLO-1 and (B) potato 5-LOX by anacardic acid (18). The results
were the average of three independent experiments with error bars (±S.D.)
Inhibitor 23 shows an IC50 of 11 M (Table 2) for SLO-1, which is five-fold improved compared to
anacardic acid 18, whereas no inhibition of potato 5-LOX was observed. Furthermore, inhibitor 23
also shows selectivity towards HATs, in which inhibitor 23 give no inhibitory effect on the activity of
p300, and PCAF and modest effect on Tip60 at 200 µM.19 In comparison, the lower SLO-1 inhibition
of 22 and 33 demonstrate that both hydroxyl groups of 23 are important for inhibition. In addition, the
reduced potency of 20 shows that also the 1-ethyl-4-heptylbenzene substituent in 23 is important for
inhibition.
Table 2. IC50 values for inhibition of SLO-1 by anacardic acid derivatives
Compounds IC50 (µM)
18 51.9 ± 5.0
21 55.4 ± 6.3
23 11.1 ± 0.8
25 58.5 ± 3.6
(B) (A)

45
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
In an attempt to improve the potency of inhibitor 23, we designed a novel compound 25 in which
the aliphatic chain was replaced for a heterocycle. However, this derivative did not provide a better
inhibition of SLO-1 (Table 2).
The influence of 23 on the Michaelis-Menten kinetics for conversion of the substrate linoleic acid
was determined in order to establish the mechanism of SLO-1 inhibition. Inhibitor 23 causes both a
reduction of Vmaxapp and an increase of Km
app (Table 3), which indicates a mixed type of inhibition
(Figure 4). The enzyme activity is expected to obey the model in Scheme 2, in which the inhibitor can
bind to the free enzyme as well as to the substrate bound enzyme.
The values for Vmax and Km were determined from the Lineweaver-Burk plot using equation 1
(Figure 5) in the absence of inhibitor 23, respectively from the y-intercept and x-intercept. Vmaxapp and
Kmapp for each inhibitor 23 concentration were calculated in the same manner as in the absence of
inhibitor. α’ and α/α’ values, which are the change in Vmax and Km, are derived respectively from
Vmaxapp divided by Vmax and from Km
app divided by Km. The inhibitor binding constant, Ki and Ki’
values, were calculated from the equation 2 and 3 (Figure 5) as described in the experimental sections.
The binding constant for inhibition of the free enzyme (Ki) is 9.8 µM and the binding constant to the
substrate bound enzyme (Ki’) is 15.7 µM. The fact that inhibitor 23 binds to the free as well as the
substrate bound enzyme as a mixed type inhibitor suggests the presence of an allosteric binding site.28-
30
Figure 4. Conversion of the substrate linoleic acid by the enzyme SLO-1 with no inhibitor present (♦), after
preincubation with 6 µM inhibitor 23 (▲), and with 11 µM inhibitor 23 (■). The results were the average of
three independent experiments with error bars (±S.D.)
Table 3. Enzyme kinetics parameter for SLO-1 inhibition
-5,0E+06
0,0E+00
5,0E+06
1,0E+07
1,5E+07
2,0E+07
-1,5E+04 -5,0E+03 5,0E+03 1,5E+04
1/V
o (
sec/
M)
1/[S] (1/M)
no Inh 11 µM 23 6µM 23
(A)
[23] (µM) VmaxApp (µM/s) Km
App (µM) R2
0 0.437 12.4 0.963
6 0.321 14.3 0.967
12 0.251 15.5 0.979

46
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
Scheme 2. Kinetic model for mixed enzyme inhibition.
equation 1
equation 2 equation 3
Figure 5. Equations for the enzyme kinetics according to the model in Scheme 2. v is the reaction velocity, Vmax
is the maximal reaction velocity, [S] is the substrate concentration and Km is the Michaelis-Menten constant. α
and α’, respectively, are the parameters to describe the change of substrate binding affinity to the enzyme and the change of the maximum velocities. Ki is the dissociation constant of inhibitor to the free enzyme and Ki’is the dissociation constant of inhibitor to the enzyme-substrate complex.
Interestingly, inhibitor 21, in which the 6-aliphatic side chain is replaced by an ether functionality,
shows equal inhibition compared to anacardic acid 18 on SLO-1 (Table 2), whereas this inhibitor (and
also inhibitor 22) shows about three-fold activation of potato 5-LOX. Compound 21 provided a
concentration dependent activation with a maximal activation that is about 3 fold higher than the
control activity at concentrations higher than 50 M (Figure 6).
We decided to study the enzyme kinetics for the activation of potato 5-LOX by 21 in order to
investigate the binding mechanism. The concentration dependent effect of 21 on the Lineweaver-Burk
plot of potato 5-LOX activity was investigated (Figure 7A). Activator 21 causes an increase of the
Vmax and a decrease of the Km values. This behavior, in which the reaction can occur in the presence
or in the absence of the activator, indicates non-essential activation of potato 5-LOX 31 (Table 4). The
enzyme kinetics for the activation of potato 5-LOX obeys the model shown in Scheme 3. The
activator dissociation constants (KA), the change in the affinity of substrate binding (α value) and the
change in the catalytic constant (β value) were determined from the re-plot of 1/Δslope and the 1/Δy-
intercept (Figure 7B) according to equation 4 (Figure 8) as described in the experimental sections.
The kinetic analysis shows that is 0.013, is 1.4 and KA is 1.8 mM. The value indicates that
the substrate and the activator mutually enhance their binding by close to 100-fold. Consequently, the
affinity of the activator for the substrate bound enzyme is 24 M, which explains the observed
activation at concentrations of 25 M and higher. In addition, activator binding increases the catalysis
rate by 1.4 fold. Taking this together, activator 21 shows non-essential activation and binds with
relatively high affinity to substrate bound lipoxygenase and enhances the enzymatic conversion of the
substrate.

47
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
Figure 6. Compound 21 activates potato 5-LOX. The experiments were performed in the absence (control) or
the presence of compound 21 with various concentrations. The results were the average of three independent
experiments with error bars (±S.D.)
Figure 7. (A) Lineweaver-Burk plot of the potato 5-LOX activity with no activator present (♦), after
preincubation with 12.5 µM activator 21 (▲), with 25 µM activator 21 (■), and with 50 µM activator 21 (●).
The results were the average of three independent experiments with error bars (±S.D.) (B) Re-plot of 1/Δslope
(●) and the 1/Δy-intercept (♦)
Table 4. Enzyme kinetics parameter for potato 5-LOX activation
0
50
100
150
200
250
300
control 12.5 25 50 100 200
LOX
-5 a
ctiv
ity
(%
)
Conc. of Compound 21 (µM)
-1,0E+07
0,0E+00
1,0E+07
2,0E+07
3,0E+07
-1,0E+04 0,0E+00 1,0E+04 2,0E+04 3,0E+04 4,0E+04
1/V
o (
sec/
M)
1/[S] (1/M)
no Actvtr 25 µM 21 12.5 µM 21 50 µM 21(A)
(B)
[21] (µM) VmaxApp (µM/s) Km
App (µM) R2
0 1.597 1.160 0.998
12.5 1.754 0.733 0.999
25 1.852 0.537 0.995
50 1.949 0.370 0.992

48
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
Scheme 3. Kinetic model for non-essential activation.
equation 4
Figure 8. Equation for the enzyme kinetics according to the model in Scheme 3.31 v is the reaction velocity,
Vmax is the maximal reaction velocity, [S] is the substrate concentration and Km is the Michaelis-Menten
constant, [A] is the activator concentration. α and β, respectively, are the parameters to describe the the change in the affinity of substrate binding and the change in the catalytic constant.
Furthermore, the critical micelle concentrations (CMCs) for both anacardic acid (18) and 23 at the
assay conditions (0.2 M borate buffer, pH 9.0, R.T) are, respectively, 390 µM and 436 µM (Figure
9B,D) whereas for compound 21 (in 0.1 M phosphate buffer pH 6.3, R.T), the CMC value is 208 µM
(Figure 9C). These results indicate that micelle formation does not occur at concentrations employed
for inhibition or activation. In addition, we found a CMC value for linoleic acid of 163 µM (Figure
9A)27, which demonstrates that the substrate concentration in the inhibition studies (100 µM) was
below the CMC. Thus the enzyme kinetics was evaluated in a homogeneous system.
The kinetic studies on both potato 5-LOX and SLO-1 indicate the presence of an allosteric binding
site that influences substrate binding and conversely is influenced by binding of the substrate. For
potato 5-LOX we demonstrated activation of the enzyme activity and for SLO-1 we demonstrated
inhibition. This behavior means that the described docking simulation as in the initial design (Figure
1) is not in line with the allosteric mechanism indicated from the kinetic studies of the modulator.
These indications for an allosteric binding pocket are in line with previous studies. The observation of
substrate inhibition of soybean lipoxygenase by linoleic acid indicates the presence of an allosteric
binding site.32 It can be presumed that the salicylate based inhibitors resemble the lipid substrate
linoleic acid and binds in a similar way to the enzyme, thereby inhibiting the enzyme activity via the
allosteric binding pocket. In addition, mixed inhibition has been described previously for soybean
lipoxygenase by the inhibitor oleyl sulfate, which also indicates the presence of an allosteric binding
site.28 Non-essential activation has been described previously for 5-LOX by compound (R,S)-2-
hydroxy-2-trifluoromethyl-trans-n-octadec-4-enoic acid (HTFOA) and phosphatidic acid.33,34
These findings also indicate the existence of allosteric site as enzyme activity regulator. Nevertheless,

49
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
the location and structural properties of the allosteric binding pocket in these enzymes remain
elusive.35 This demonstrates that structural characterization of the allosteric binding pocket remains a
major challenge in lipoxygenase research.
Figure 9. Surface tensions of (A) Linoleic acid, (B) Anacardic acid 18, (C) compound 21, and (D) compound
23 against the logarithm of concentration. CMC values for A and C were measured in SLO-1 assay conditions,
0.2 M borate buffer, pH 9.0, R.T.(t = 19°C), whereas CMC value for B was measured in potato 5-LOX assay
conditions, 0.1M phosphate buffer, pH 6.3,R.T. (t = 25°C).
This study identifies salicylate based molecules as allosteric regulators of lipoxygenase enzyme
activity. Such inhibitors might find applications as starting points for development of therapeutic
agents for asthma and inflammations. Nevertheless, the potency and selectivity of this compound
class for human lipoxygenases remains to be investigated. It is encouraging that the anacardic acid
derived salicylates show clear structure activity relationships, which indicates that further
optimization of the affinity and selectivity of this compound class might be feasible.
Conclusion
This study demonstrates that anacardic acid 18 and its derivatives are lipoxygenase inhibitors and that
their potency depends on the substitution pattern in the 4- and 6-position of the salicylate core. A
novel inhibitor 23 was identified for soybean lipoxygenase-1 with a five-fold improved IC50 value
compared to anacardic acid 18. Enzyme kinetics reveal a mixed type inhibition pattern for 23 with a
Ki of 9.8 M and a Ki’ of 15.7 M (Scheme 3). In addition, an activator 21 of potato 5-lipoxygenase
(A)
(C)
(B)
(D)

50
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
was identified for which KA is 1.8 mM, is 0.013 and is 1.4 (Scheme 4). The affinity of 21 for the
substrate bound enzyme ( KA) is 24 M. To our knowledge, this is the first activator with micromolar
potency for potato lipoxygenase. These inhibitors might provide valuable starting points for
development of inhibitors that target lipoxygenases, which is relevant for inflammatory diseases.
Experimental
General
All reagent and solvent were purchased from commercial suppliers (Fluka, Sigma-Aldrich, Acros
Organics) and were used without further purification unless stated otherwise. Dichloromethane was
distilled over CaH2 before use. Merck silica gel 60 F254 plates were used for analytical thin layer
chromatography (TLC) and spots were detected by UV light, or stained using KMNO4 or ninhydrin
solution. Column chromatography was performed with MP Ecochrom silica gel 32-63, 60Ǻ using the
flash chromatography technique. 1H (200 MHz) and 13C (50 MHz) NMR spectra were recorded on a
Varian Gemini 200 spectrometer. 13C NMR spectra were recorded using the attached proton test
(APT). Chemical shifts are reported in ppm (δ) relative to the solvent signals. Electrospray ionization
mass spectra (ESI-MS) were recorded on an Applied Biosystems/SCIEX API3000-triple quadrupole
mass spectrometer. High-resolution mass spectra (HR-MS) were recorded using a flow injection
method on a LTQ-Orbitrap XL mass spectrometer (Thermo Electron, Bremen, Germany) with a
resolution of 60,000 at m/z 400. Protonated testosterone (lock mass m/z = 289.2162) was used for
internal recalibration in real time.
Organic Synthesis of the focused compound collection of salicylates
The synthesis of anacardic acid and its derivatives 15-23, 26-30, and 32 was described previously by
Ghizzoni et.al.18, 19 and compounds 24, 25, 31, and 33 were synthesized using similar procedures
according to Scheme 1.
Synthetic procedure 1; Sonogashira coupling of aryl iodides or aryl triflates
Freshly distilled diethylamine (0.15 mL, 1.5 mmol) and the alkyne (1.1 mmol) were subsequently
added to a solution of the aryl iodide or aryl triflate (1.0 mmol), CuI (9.5 mg, 0.05 mmol), and
PdCl2(PPh3)2 (35 mg, 0.05 mmol) in degassed anhydrous acetonitrile (1.0 mL) under nitrogen
atmosphere. The mixture was subjected to microwave irradiation for 20 min at 100 °C (70 W). The
reaction mixture was diluted with ethyl acetate (50 mL) and extracted with demi water (2 × 75 mL).
The water phase was separated and washed with EtOAc (2 × 50 mL). The combined organic phases
were washed with brine (2 × 75 mL), dried over Na2SO4 and filtered. The solvent was evaporated
under reduced pressure, and the residue was purified by column chromatography.

51
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
Synthetic procedure 2; Sonogashira coupling of aryl chlorides
The alkyne (1.2 mmol), the aryl chloride (1.3 mmol), and Cs2CO3 (0.47 g, 1.4 mmol) were dissolved
in dry DMF (2 mL) under nitrogen atmosphere. Subsequently, 2 mol % PdCl2(PPh3)2 (17 mg, 0.024
mmol), 4 mol % P(tBu)3 (10 mg, 0.05 mmol), and 10% mol 1,8-Diazabicyclo[5.4.0]undec-7-ene
(DBU) (18 mg, 0.12 mmol) were added to the reaction mixture. The mixture was subjected to
microwave irradiation for 15 min at 150 °C (95 W). The mixture was diluted with 50 mL EtOAc and
filtered over Celite. Then the filtrate was washed with brine (2 × 50 mL), and dried over MgSO4 and
the solvent was evaporated under reduced pressure. The product was obtained after purification using
column chromatography.
Synthetic procedure 3; reductive hydrogenation and or hydrogenolysis
The starting material (0.5 mmol) was dissolved in MeOH and 10 mol% Pd/C (10%) (53 mg, 0.05
mmol) was added. The compound precursor of 25 named benzyl 2-((4-(benzo[d]oxazol-2-
ylmethyl)phenyl)ethynyl)-4,6-bis(benzyloxy)benzoate was dissolved in EtOAc due to its low
solubility in MeOH. The suspension was shaken with 3 atm H2-pressure in a Parr apparatus at 40 oC
overnight. Subsequently, the mixture was filtered through Celite. The solvent was evaporated under
reduced pressure and the residue was purified by column chromatography.
Synthetic procedure 4; saponification of the acetonide
The starting material (1.0 mmol) was dissolved in THF (10 mL) and an aqueous 5 N KOH solution (2
mL, 10 mmol) was added. The solution was stirred overnight at 55 °C. Subsequently, the reaction
mixture was diluted with EtOAc (10 mL) and neutralized with 1 N HCl (20 mL). The mixture was
extracted with EtOAc (3 × 30 mL). The organic phases were collected, washed with brine (1 × 50
mL), dried over Mg2SO4 and filtered. The solvent was evaporated and the product was purified by
column chromatography.
Synthetic procedure 5; benzoxazole synthesis
2-amino-5-chlorophenol (1.4 g, 10 mmol) was dissolved in DMF (15 mL). Subsequently, pyridine
(4.0 mL, 50 mmol) and (p-methoxy) benzoic acid anhydride (10 mmol) were added and the reaction
mixture was stirred for 3 hours at room temperature. The reaction mixture was diluted with 100 mL
water and extracted with EtOAc (2 × 100 mL). The organic layer was combined and extracted with
1 N HCl (2 × 150 mL) and saturated Na2CO3 (2 × 150 mL). The organic layer was washed with brine
(1 × 150 mL), dried over Mg2SO4 and filtered. The solvent was evaporated to give a corresponding
amide.
The amide (1 mmol) was dissolved in toluene (10 mL) and p-toluensulfonic acid (2 mmol) was
added. The solution was heated to reflux for 4 hours. The reaction mixture was diluted with EtOAc

52
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
(40 mL) and extracted with saturated NaHCO3 (2 × 50 mL). The organic phases were collected,
washed with brine (1 × 50 mL), dried over Mg2SO4 and filtered. The solvent was evaporated and the
product was purified by column chromatography.
2-(4-bromobenzyl)benzo[d]oxazole (9)
4-bromophenyl acetic acid (2.15 g, 10 mmol) was dissolved in 10 mL SOCl2 and heated to 60 °C for
30 minutes. The solvent was distilled off to obtain the acyl chloride as a yellow liquid. 2-aminophenol
(1.09 g, 10 mmol) was suspended in dichloromethane (20 mL), Et3N (2.77 mL, 20 mmol) was added
and the mixture was cooled at 0 °C. The acyl chloride was added drop-wise to the mixture, which was
stirred overnight while it warmed to room temperature. The reaction mixture was diluted with EtOAc
(30 mL) and extracted with a saturated aqueous Na2CO3 solution (3 × 50 mL), 1 N aqueous HCl (3 ×
50 mL) and brine (1 × 50 mL). The organic phase was dried with Mg2SO4 and the solvent was
evaporated to obtain the corresponding amide.
The amide (1.78 g, 5.8 mmol) was dissolved in toluene (40 mL) and p-toluensulfonic acid (2.21 g,
11.6 mmol) was added. The solution was heated to reflux for 4 hours. Subsequently, the reaction
mixture was diluted with EtOAc (60 mL) and extracted with saturated aqueous NaHCO3 (2 × 100
mL). The organic phase was collected, washed with brine (1 × 100 mL), dried over Mg2SO4 and
filtered. The solvent was evaporated and the TLC confirmed product purity after the work up and
therefore no further purification was performed. Yield 85%. Brown solid. Rf = 0.55 (heptane:EtOAc
1:1). 1H NMR (200 MHz, DMSO) δ 4.32 (s, 2H); 7.31-7.36 (m, 4H); 7.55 (d, J = 8.3Hz, 2H); 7.62-
7.70 (m, 2H) .13C NMR (50 MHz DMSO) δ 33.69; 110.86; 119.69; 120.57; 124.67; 125.23; 131.64;
131.78; 134.83; 141.04; 150.61; 165.19. MS ESI m/z 288.2; 290.2 [M+H]+.
2-(4-((trimethylsilyl)ethynyl)benzyl)benzo[d]oxazole
Into a dried 10-20 mL microwave vial, PdCl2(PPh3) (133 mg, 0.2 mmol), CuI (36 mg, 0.2 mmol), and
PPh3 (200 mg, 0.8 mmol) were suspended in CH3CN (5 mL) under nitrogen atmosphere.
Subsequently, the benzoxazole 9 (1.10 g, 3.8 mmol), diethyl amine (Et2NH) (5.9 mL, 57 mmol), and
trimethylsilyl acetylene (TMSA) (402 mg, 4.2 mmol), were added to the mixture. The mixture was
heated at 120 °C for 35 minutes via microwave irradiation (95 W). The reaction mixture was diluted
with EtOAc (50 mL) and washed with water (3 × 50 mL). The water layer was washed with EtOAc (2
× 50 mL). The combined organic phases were extracted with brine (1 × 50 mL) and dried over

53
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
Mg2SO4. The solvent was evaporated under reduced pressure and the residue was purified by column
chromatography with heptane/EtOAc 12:1 (v/v) as eluent. Yield 75%. Yellow to orange solid. Rf =
0.76 (heptane:EtOAc 1:1). 1H NMR (200 MHz, CDCl3) δ 0.09 (s, 9H); 4.08 (s, 2H); 7.05-7.12 (m,
4H); 7.21-7.27 (m, 2H); 7.47-7.52 (m, 2H) .13C NMR (50 MHz CDCl3) δ 0.16; 35.25; 104.84;
105.21; 110.79; 119.92; 122.60; 124.71; 125.27; 129.14; 132.63; 135.05; 151.15; 165.08. MS ESI m/z
306.2 [M+H]+.
2-(4-ethynylbenzyl)benzo[d]oxazole (10)
2-(4-((trimethylsilyl)ethynyl)benzyl)benzo[d]oxazole (611 mg, 2.0 mmol) was dissolved in THF (5
mL) and the solution was cooled to 0 oC. Tetra butyl ammonium fluoride (TBAF) (2.6 mmol) in THF
(1 M, 2.6 mL) was added and the reaction mixture was stirred for 10 minutes. The reaction mixture
was diluted with EtOAc (50 mL), extracted with water (4 × 50 mL) and washed with brine (2 × 50
mL). The organic phase was dried over MgSO4 and filtered. The solvent was evaporated and the
product was used without further purification. Yield 99%. Brown solid. Rf = 0.59 (heptane:EtOAc
1:1). 1H NMR (200 MHz, CDCl3) δ 3.07 (s, 1H); 7.29-7.36 (m, 4H); 7.44-7.50 (m, 2H); 7.68-7.72 (m,
2H).13C NMR (50 MHz CDCl3) δ 35.07; 76.41; 104.09; 110.48; 119.83; 121.24; 124.31; 124.87;
129.02; 132.56; 135.45; 151.00; 164.57. MS ESI m/z 234.1 [M+H]+.
Benzyl 2-((4-(benzo[d]oxazol-2-ylmethyl)phenyl)ethynyl)-4,6-bis(benzyloxy)benzoate
The product was obtained using sonogashira coupling of benzyl 2,4-bis(benzyloxy)-6-
(((trifluoromethyl)sulfonyl)oxy)benzoate 11 and 2-(4-ethynylbenzyl)benzo[d]oxazole 10 using
synthetic procedure 1. The product was purified by column chromatography with heptane/EtOAc 4:1
(v/v) as eluent. Yield 51%. Yellow gum. Rf = 0.52 (heptane:EtOAc 1:1). 1H NMR (200 MHz, CDCl3)
δ 4.29 (s, 2H); 5.02 (s, 2H); 5.05 (s, 2H); 5.34 (s, 2H); 6.56 (d, J=2.1 Hz, 1H); 6.74 (d, J= 2.1 Hz,
1H); 7.17-7.22 (m, 4H); 7.32-7.36 (m, 15H); 7.46-7.50 (m, 2H); 7.68-7.73 (m, 2H) .13C NMR (50
MHz CDCl3) δ 35.25; 67.31; 70.54; 70.81; 87.23; 92.67; 102.19; 109.73; 110.81; 119.90; 122.18;
123.51; 124.76; 125.31; 127.30; 127.74; 128.14; 128.20; 128.45; 128.61; 128.76; 128.89; 129.25;
132.25; 132.42; 133.18; 135.15; 136.01; 136.32; 136.41; 157.28; 160.56; 166.80.

54
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
2-(4-(benzo[d]oxazol-2-ylmethyl)phenethyl)-4,6-dihydroxybenzoic acid (25)
The product was obtained from benzyl 2-((4-(benzo[d]oxazol-2-ylmethyl)phenyl)ethynyl)-4,6-
bis(benzyloxy)benzoate using synthetic procedure 3. The product was purified by column
chromatography with heptane/EtOAc 1:2 (v/v) as eluent. Yield 86%. White solid. Rf = 0.45
(heptane:EtOAc 1:1). 1H NMR (200 MHz, DMSO) δ 2.67-2.75 (m, 2H); 2.94-3.02 (m, 2H); 4.26 (s,
2H); 6.14 (d, J=2.4 Hz, 1H); 6.20 (d, J= 2.4 Hz, 1H); 7.16-7.35 (m, 6H); 7.60-7.68(m, 2H). 13C NMR
(50 MHz DMSO) δ 34.18; 37.53; 38.08; 101.26; 105.63; 110.62; 111.01; 119.77; 124.88; 125.40;
128.97; 129.39; 132.97; 141.12; 146.70; 150.73; 162.03; 164.16; 166.01; 173.06. HRMS: m/z
390.1336 [M+H]+, calcd for C23H20O5N1 390.1331.
5-chloro-2-phenylbenzo[d]oxazole (7a)
The product was obtained from 2-amino-4-chlorophenol and benzoic acid using synthetic procedure
5. The product was purified by column chromatography with hexane/EtOAc 10:1 (v/v) as eluent.
Yield 80%. Pink solid. Rf = 0.44 (hexane:EtOAc 3:1). 1H NMR (200 MHz, CDCl3) δ 7.25 (dd, J= 8.5;
1.9 Hz, 1H); 7.43-7.61 (m, 5H); 8.12-8.16 (m, 2H). 13C NMR (50 MHz CDCl3) δ 104.98; 111.22;
120.45; 125.26; 126.69; 127.64; 128.95; 130.65; 131.78; 140.88; 150.90; 163.68. MS ESI m/z 230.1;
232 [M+H]+.
2,2-dimethyl-5-((2-phenylbenzo[d]oxazol-5-yl)ethynyl)-4H-benzo[d][1,3]dioxin-4-one
The product was obtained from 5-ethynyl-2,2-dimethyl-4H-benzo[d][1,3]dioxin-4-one 2 and 5-
chloro-2-phenylbenzo[d]oxazole 7a using synthetic procedure 2. The product was purified by column
chromatography with hexane/EtOAc 5:1 (v/v) as eluent. Yield 20%. Yellow solid. Rf = 0.27
(hexane:EtOAc 3:1). 1H NMR (200 MHz, CDCl3) δ 1.7 (s, 6H); 6.96-7.01 (m, 3H); 7.53-7.61 (m,
5H); 7.82 (s, 1H); 8.24-8.29 (m, 2H). TOF MS ES+: m/z 396.1233 [M+H]+, calcd for C25H22O4N1
396.1236.

55
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
2,2-dimethyl-5-(2-(2-phenylbenzo[d]oxazol-5-yl)ethyl)-4H-benzo[d][1,3]dioxin-4-one
The product was obtained from 2,2-dimethyl-5-((2-phenylbenzo[d]oxazol-5-yl)ethynyl)-4H-
benzo[d][1,3]dioxin-4-one using synthetic procedure 3. This intermediate was used for the next
reaction without any further purification. Yield 81%. Yellow oil. Rf = 0.44 (hexane:EtOAc 3:1). 1H
NMR (200 MHz, CDCl3) δ 1.63 (s, 6H); 2.95-3.03 (m, 2H); 3.34-3.42 (m, 2H); 6.75-6.83 (m, 2H);
7.28-7.36 (m, 2H); 7.44-7.48 (m, 5H); 8.17-8.22 (m, 2H). TOF MS ES+: m/z 340.1540 [M+H]+, calcd
for C25H22O4N1 340.1549.
2-hydroxy-6-(2-(2-phenylbenzo[d]oxazol-6-yl)ethyl)benzoic acid (24)
The product was obtained from 2,2-dimethyl-5-(2-(2-phenylbenzo[d]oxazol-5-yl)ethyl)-4H-
benzo[d][1,3]dioxin-4-one using synthetic procedure 4. The product was purified by column
chromatography with hexane/EtOAc 4:1 (v/v) as eluent. Yield 50%. White solid. Rf = 0.31 (EtOAc +
0.1% AcOH). 1H NMR (200 MHz, DMSO) δ 2.93-3.04 (m, 4H); 6.72-6.78 (m, 2H); 7.15-7.28 (m,
2H); 7.61-7.64 (m, 5H); 8.17-8.22 (m, 2H) . 13C NMR (50 MHz DMSO) δ 35.93; 40.21; 110.70;
114.48; 119.82; 120.57; 120.76; 125.80; 126.99; 127.57; 129.75; 131.29; 132.24; 140.15; 140.31;
140.97; 150.89; 156.80; 162.38; 170.94. HRMS: m/z 344.1280 [M+H]+, calcd for C22H18O3N1
344.1281
5-chloro-2-(4-methoxyphenyl)benzo[d]oxazole (7b)
The product was obtained from 2-amino-4-chlorophenol and 4-methoxybenzoic acid using synthetic
procedure 5. The product was purified by column chromatography with hexane/EtOAc 8:1 (v/v) as
eluent. Yield 48%. Pink solid. Rf = 0.62 (hexane:EtOAc 1:1). 1H NMR (200 MHz, CDCl3) δ 3.87 (s,
3H); 7.00 (d, J= 8.9 Hz, 2H); 7.28 (dd, J= 8.5; 1.9 Hz, 1H); 7.52-7.62 (m, 2H); 8.13 (d, J= 8.9 Hz,
2H).13C NMR (50 MHz CDCl3) δ 55.68; 111.25; 114.64; 119.40; 120.24; 125.28; 129.67; 130.29;
141.30; 151.06; 162.75; 164.11. MS ESI m/z 260.1; 262.1 [M+H]+.

56
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
Benzyl 3-((2-(4-methoxyphenyl)benzo[d]oxazol-5-yl)ethynyl)benzoate
The product was obtained from benzyl 3-ethynylbenzoate 5 and 5-chloro-2-(4-
methoxyphenyl)benzo[d]oxazole 7b using synthetic procedure 2. The product was purified by column
chromatography with hexane/EtOAc 6:1 (v/v) as eluent. Yield 32%. Yellow solid. Rf = 0.43
(hexane:EtOAc 1:1). 1H NMR (200 MHz, CDCl3) δ 3.81 (s, 3H); 5.31 (s, 2H); 6.95 (d, J=9.0 Hz, 2H);
7.30-7.46 (m, 7H); 7.58-7.67 (m, 3H); 7.95-8.17 (m, 4H). 13C NMR (50 MHz CDCl3) δ 55.72; 67.22;
90.59; 113.78; 114.69; 119.25; 119.48; 119.70; 128.55; 128.60; 128.79; 128.88; 129.66; 129.84;
130.73; 133.00; 136.03; 143.05; 150.61; 154.37; 162.87; 164.64; 165.94. MS ESI m/z 460.3 [M+H]+.
3-(2-(2-(4-methoxyphenyl)benzo[d]oxazol-6-yl)ethyl)benzoic acid (31)
The product was obtained from benzyl 3-((2-(4-methoxyphenyl)benzo[d]oxazol-5-
yl)ethynyl)benzoate using synthetic procedure 3. The product was obtained in high purity and no
further purification was required. Yield 98%. White solid. Rf = 0.46 (EtOAc + 0.1% AcOH). 1H NMR
(200 MHz, CD3OD) δ 3.00-3.06 (m, 4H); 3.85 (s, 3H); 7.05 (d, J=9.0 Hz 2H); 7.14-7.29 (m, 3H);
7.38 (s, 1H); 7.52 (d, J=8.1Hz, 1H); 7.72-7.81 (m, 1H); 7.84 (s, 1H); 8.08 (d, J=9.0 Hz 2H). 13C NMR
(50 MHz CD3OD) δ 39.24; 39.98; 56.22; 111.57; 115.79; 119.68; 126.46; 126.92; 128.17; 128.85;
130.45; 130.58; 131.71; 139.36; 141.02; 142.27; 152.18; 164.34; 164.37; 166.32. HRMS: m/z
374.1385 [M+H]+, calcd for C23H20O4N1 374.1387.
7-(benzyloxy)-2,2-dimethyl-4H-benzo[d][1,3]dioxin-4-one (13)
The acetonide 7-hydroxy-2,2-dimethyl-4H-benzo[d][1,3]dioxin-4-one (558 mg, 2.9 mmol) was
dissolved in DMF (10 mL) under nitrogen atmosphere. K2CO3 (955 mg, 6.9 mmol) and benzyl
bromide (0.41 mL, 3. 5 mmol) were added and the reaction mixture was stirred at room temperature
overnight. The reaction mixture was diluted with EtOAc (50 mL). The organic phase was washed
with water (4 × 50 mL) and brine (3 × 20 mL). The organic phase was dried over MgSO4 and the

57
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
solvent was evaporated. The residue was purified using column chromatography with heptane/EtOAc
4:1 (v/v) as eluent. Yield 84%. Colorless oil. Rf = 0.46 (heptane:EtOAc 1:1). 1H NMR (200 MHz,
DMSO) δ 1.68 (s, 6H); 5.20 (s, 2H); 6.75 (d, J=2.3 Hz, 1H); 6.84 (dd, J=8.7, 2.4 Hz, 1H); 7.31-7.48
(m,5H); 7.78 (d, J=8.7 Hz, 1H). 13C NMR (50 MHz DMSO) δ 25.26; 70.02; 102.01; 105.75; 106.23;
111.20; 127.98; 128.18; 128.52; 130.76; 136.00; 157.39; 159.87; 165.16. MS ESI m/z 285.2 [M+H]+.
Benzyl 4-(benzyloxy)-2-(((trifluoromethyl)sulfonyl)oxy)benzoate (14)
Sodium (0.2 g, 9.1 mmol) was added to a solution of benzyl alcohol (1.2 mL, 11.4 mmol) in THF (10
mL) under nitrogen atmosphere. The suspension was stirred until the sodium disappeared.
Subsequently, the 7-(benzyloxy)-2,2-dimethyl-4H-benzo[d][1,3]dioxin-4-one 13 was added (0.65 g,
2.28 mmol) and the reaction mixture was stirred for 30 minutes at room temperature. The reaction
mixture was diluted with EtOAc (50 mL) and washed with water (3 × 50 mL) and brine (3 × 20 mL).
The organic phase was dried over MgSO4 and the solvent was evaporated. The product was purified
using column chromatography to obtain benzyl 4-(benzyloxy)-2-hydroxybenzoate as a product.
Benzyl 4-(benzyloxy)-2-hydroxybenzoate (56 mg, 1.79 mmol) and pyridine (0.71 mL, 8.97 mmol)
were dissolved in CH2Cl2 (10 mL) and cooled to 0°C under nitrogen atmosphere. Triflic anhydride
(0.52 mL, 1.97 mmol) was added drop wise to the mixture and stirred for 1 hour. The mixture was
poured onto solid NaHCO3 and washed with 0.1 N HCl (3 × 30 mL), brine (2 × 15 mL), and dried
over MgSO4. The solvent was evaporated and the product was used without further purification. Yield
90%. Colorless solid. Rf = 0.57 (heptane:EtOAc 1:1). 1H NMR (200 MHz, CDCl3) δ 5.12 (s, 2H);
5.38 (s, 2H); 6.86 (d, J=2.4Hz, 1H); 7.0 (dd, J=8.9, 2.5 Hz, 1H); 7.31-7.49 (m, 10H); 8.05 (d, J=8.9
Hz, 1H).13C NMR (50 MHz, CDCl3) δ 63.37; 71.09; 109.97; 114.40; 116.77; 117.31; 120.50; 127.75;
128.61; 128.76; 128.82; 128.84; 129.04; 134.29; 135.31; 135.68; 149.92; 163.27; 163.34. MS ESI m/z
467.1 [M+H]+.
Benzyl 4-(benzyloxy)-2-((4-heptylphenyl)ethynyl)benzoate
The product was obtained from benzyl 4-(benzyloxy)-2-(((trifluoromethyl)sulfonyl)oxy)benzoate 14
and 1-ethynyl-4-heptylbenzene using synthetic procedure 1. The product was purified by column
chromatography with heptane/EtOAc 21:4 (v/v) as eluent. Yield 79%. Orange oil. Rf = 0.64

58
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
(heptane:EtOAc 1:1). 1H NMR (200 MHz, CDCl3) δ 0.90 (t, J=6.8Hz, 3H); 1.30-1.33 (m, 8H); 1.60-
1.64 (m, 2H); 2.62 (t, J=7.6Hz, 2H); 5.13 (s, 2H); 5.39 (s, 2H); 6.95 (dd, J=8.8, 2.6 Hz, 1H); 7.13 (d,
J=8.0 Hz, 2H); 7.23 (d, J=2.6 Hz, 1H); 7.28-7.55 (m, 12H); 8.02 (d, J=8.8 Hz, 1H). 13C NMR (50
MHz CDCl3) δ 14.31; 22.86; 29.36; 29.40; 31.46; 32.02; 36.14; 66.92; 70.40; 88.01; 95.06; 115.08;
119.66; 120.49; 124.24; 126.31; 127.72; 128.25; 128.46; 128.59; 128.72; 128.88; 131.88; 133.10;
136.23; 136.34; 143.92; 161.42; 165.86. MS ESI m/z 517.3 [M+H]+.
2-(4-heptylphenethyl)-4-hydroxybenzoic acid (33)
The product was obtained from benzyl 4-(benzyloxy)-2-((4-heptylphenyl)ethynyl)benzoate using
synthetic procedure 3. No purification of the crude product was required. Yield 100%. White solid. Rf
= 0.41 (heptane:EtOAc 1:1). 1H NMR (200 MHz, CDCl3) δ 0.86 (t, J= 6.3 Hz, 3H); 1.26-1.30 (m,
8H); 1.54-1.61 (m, 2H); 2.55 (t, J= 7.7 Hz, 2H); 2.83-2.91 (m, 2H); 3.23-3.31 (m, 2H); 6.66-6.73 (m,
2H); 7.05-7.16 (m, 4H); 8.05 (d, J= 8.3 Hz, 1H).13C NMR (50 MHz, CDCl3) δ 14.34; 22.91; 29.44;
29.60; 31.84; 32.07; 35.84; 37.66; 113.32; 118.20; 120.63; 128.59; 134.93; 139.27; 140.71; 148.55;
159.50; 171.78. HRMS: m/z [M+H]+, calcd for C22H29O3 341.2111, found 341.2112; m/z [M+Na]+,
calcd for C22H28O3Na 363.1931, found 363.1931; m/z 379.1670 [M+K]+, calcd for C22H28O3K
379.1670.
Inhibition screening UV Assay
The enzyme potato 5-lipoxygenase (potato 5-LOX) and soybean lipoxygenase-1 (SLO-1) were
obtained from Cayman Chemicals. Linoleic acid was obtained from Sigma.
Enzyme inhibition was measured by the residual enzyme activity after 10 minutes incubation with
the inhibitor at room temperature. The enzyme activity was determined by conversion of the
lipoxygenase substrate linoleic acid into hydroperoxy eicosatetraenoic acid (HPETE). The conversion
rate was followed by UV absorbance of the conjugated diene at 234 nm (ε = 25000 M-1cm-1) over a
period of 20 minutes. The UV absorbance increase over time was used to determine the enzyme
activity.
Phosphate buffer (NaH2PO4-Na2HPO4 0.1 M) pH 6.3 was used as an assay buffer for all potato 5-
LOX inhibitory experiments. The potato 5-LOX enzyme was diluted 1:2000 with the assay buffer.
The inhibitor (100 mM in DMSO) was diluted with the assay buffer to 100.5 µM. The substrate,
linoleic acid was diluted with EtOH to 20 mM. Subsequently, 1 mL of enzyme solution (1:2000) was
mixed with 1 mL inhibitor solution (100.5 µM), which was incubated for 10 minutes. Subsequently,
the linoleic acid solution (10 µL, 20 mM) was added to give a mixture with 1:4000 enzyme dilution,

59
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
50 µM inhibitor and 100 µM linoleic acid. The conversion rate of the substrate was measured after 10
second reaction of the enzyme with the substrate. The reaction rate in absence of the inhibitor was
used as positive control. In the positive control experiments the assay buffer was supplemented with a
little amount of DMSO (3.0 μL of DMSO in 1.5 mL) in order to replace the DMSO inhibitor solution,
which was also pre-incubeted for 10 minutes.
Sodium borate buffer (H3BO3 0.2 M) pH 9.0 was used as an assay buffer for all SLO-1 inhibition
experiments. The enzyme SLO-1 was diluted 1:4000 using the SLO-1 assay buffer and the inhibitor
(100 mM in DMSO) was diluted using the same buffer to 200 µM. The linoleic acid (20 mM in
EtOH) was diluted with SLO-1 assay buffer to 200 µM. The enzyme (400 µL, 1:4000) was mixed
with the inhibitor (400 µL, 200 µM) and the mixture was incubated for 10 minutes. Subsequently, the
linoleic acid (800 µL, 200 µM) was added to give a mixture with 50 µM inhibitor and 100 µM
linoleic acid. The UV absorbance at 234 nm was immediately measured after 10 seconds reaction of
the enzyme with the substrate. The measurement in absence of inhibitor was used as a positive
control. The assay buffer with a little amount of DMSO (3.0 μL of DMSO in 1.5 mL) was used to
replace the inhibitor solution. This solution was also pre-incubated for 10 minutes.
Lipoxygenase IC50 determination
Inhibitory concentration 50% (IC50) for both potato 5-LOX and SLO-1 were determined using the
same assay setup as the inhibitor screening. Various concentrations of inhibitor were added to the
enzyme and the residual enzyme activity was determined (Table 2, Figure 2 and 10). These
experiments were performed in triplicate. The calculation was performed with Excel 2010 and the
non-linear curve fitting was performed with the Origin 8 software.
Figure 10. The IC50 values for inhibition of SLO-1 by inhibitor 21 (A), inhibitor 23 (B) and inhibitor 25 (C).
The results were the average of three independent experiments with error bars (±S.D.)
Michaelis Menten Enzyme Kinetics
The enzyme kinetics of potato 5-LOX and SLO-1 were also studied by the formation of the
conjugated diene product at 234 nm (ε = 25000 M-1cm-1) using the same experimental setup as for the
IC50 determination and the inhibitor screening. The substrate concentration was varied between 62.5 –
500 µM for SLO-1 kinetics analyses in absence or presence of fixed concentrations of inhibitor 23 (6
A B C

60
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
µM, and 11 µM). The reaction velocities (v), which are the concentration change over time, were
plotted against substrate concentration in Lineweaver-Burk plot (Figure 4) and the Km and Vmax and
the apparent values (Kmapp and Vmax
app) in presence of the inhibitor were derived (Table 3).
The Vmax and Vmaxapp were derived from the y-interception of Lineweaver-Burk plot for each
inhibitor consentration, whereas Km and Kmapp from the x-interception. α’ which is the change in the
Vmax, was calculated from the Vmaxapp divided by Vmax. The change in the affinity of substrate binding
represented by α/α’ was calculated from the Kmapp divided by Km. Furthermore, the Ki and Ki’ were
derived using equation 1.3 by substituting the α and α’ values for each inhibitor concentration. The
reported Ki and Ki’ values are the averages of two inhibitor concentration (6 µM, and 11 µM).
Kinetic analyses of potato 5-LOX activation were performed in absence or presence of fixed
concentrations of activator 21 (12.5 µM; 25 µM; 50 µM) and various concentration of substrate,
ranging from 25-200 µM. Lineweaver-Burk plots (Figure 7A) were used to determine Km and Vmax
and the apparent values (Kmapp and Vmax
app) in presence of the inhibitor were derived (Table 4). The
slopes and y-intercepts were derived and re-plotted as 1/Δslope or 1/Δy-intercept versus 1/[activator]
(Figure 7B). From these plots α, β and KA values were derived as described by Leskovac3 for non-
essential activation. According to this method in the re-plot of 1/Δy-intercept vesus 1/[activator] the y
intercept correspond to β Vmax/(β-1), whereas in the re-plot of 1/Δslope vesus 1/[activator] the y
intercept correspond to β Vmax/Km(β-α). The x intersection point of two lines from the plot of
1/Δintercept and 1/Δslope is –β/αKA, which can be employed to derive the KA value if α and β are
known. All the experiments were performed in triplicate and the average triplicate values and their
standard deviations are plotted. Calculations were performed with Excel 2010.
Concentration-Dependent potato 5-LOX Activity
Concentration-dependent potato 5-LOX activation profile was determined by comparing the enzyme
activity in the presence of various concentration of activator. The experiment was performed using the
same assay setup as for the inhibitor screening of potato 5-LOX. The potato 5-LOX enzyme was
diluted 1:2000 with phosphate buffer (NaH2PO4-Na2HPO4 0.1 M) pH 6.3. Various concentrations of
activator, ranged from 12.5–200 µM, were used in the assay. The enzyme (1.0 mL) was pre-incubated
with the activator (1.0 mL) for 10 minutes before the diluted substrate (10 µL, 20 mM) was added.
The formation of HPETE was measured by the UV absorption at 234 nm. The experiments were
performed in triplicate and the calculations were performed with Excel 2010.
Critical Micelle Concentration (CMC) Determination
The Critical Micelle Concentration (CMC) value is the upper limit of concentration range in which
the substrate system is still homogenous. The CMC values were determined by measuring surface
tension of the solutions as a function of the concentrations (1-1000 μM). The CMC values for
anacardic acid, inhibitor 23, and linoleic acid were determined in a buffer containing 0.2 M H3BO3 at

61
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
pH 9.0, whereas for activator 21, the CMC value was determined in 0.1 M phosphate buffer
(NaH2PO4-Na2HPO4) at pH 6.3. The surface tension of the solutions was measured using Du Noüy
ring method with Pt-Ir ring of diameter 0.8 cm and Krüss tensiometer. The CMC was defined as the
intersection of two linear lines extrapolating the surface tension as function of logarithm of the
compounds concentration (Figure 9). The experiments were performed in triplicate and the
calculations were performed with Excel 2010. We found that under these experimental conditions, the
CMCs are 163 µM for linoleic acid, 390 µM for anacardic acid, 208 µM for 21 and 436 µM for 23.
Molecular Docking
The crystal structure of soybean lipoxygenase-1 was downloaded from Protein Data Bank (code
1F8N). All small molecules were drawn using Chemaxon MarvinSketch (www.chemaxon.com) and
exported as mol2 files. The enzyme and small molecules were prepared for docking (structure
recognition and protonation) using SPORES (www.tcd.uni-konstanz.de/research/spores.php).
Molecular docking simulations were performed using PLANTS v1.6.4, 5 The detailed of preparation
using SPORES and the docking using PLANTS was done following the software instruction. The Fe2+
in the SLO-1 active site was set as a docking site center. Fifteen poses were generated for each
compound. The docking results were analyzed using Molegro Virtual Docker (www.molegro.com).
The fitness of a pose is evaluated in terms of intermolecular interaction energy between the ligand and
the enzyme, and the intramolecular interaction energy of the enzyme, which is represent by MolDock
score (Table 5).
Table 5. The docking scores for the 15 poses of 18 and 23 in the SLO-1 active site
Pose 1 Pose 2 Pose 1 Pose 2
-126,891 -112,037 -111,305 -125,933
-124,409 -113,931 -111,416 -119,462
-111,028 -113,773 -101,914 -107,51
-103,624 -107,062 -81,462 -102,518
-98,273 -100,513 -73,637 -90,812
-106,888 -97,789
-106,125 -88,03
-110,794 -84,374
-101,798 -90,111
-101,435 -77,108
Compound 18 Compound 23MolDock
Score*

62
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
*Pose 1 is the pose in which the salicylate group occupies the the position shown in Figure 1 (A); Pose 2 is the
pose in which the salicylate group flips to the position shown Figure 1(B); The MolDock score is a calculated
total energy for each pose (in arbitrary energy units)
References
1. McGinley, C. M.; van der Donk, W. A. Enzymatic hydrogen atom abstraction from polyunsaturated fatty
acids. Chem. Commun. (Camb) 2003, (23), 2843-2846.
2. De Caterina, R.; Zampolli, A. From Asthma to Atherosclerosis - 5-Lipoxygenase, Leukotrienes, and
Inflammation. N. Engl. J. Med. 2004, 350, 4-7.
3. Cathcart, M. K.; Folcik, V. A. Lipoxygenases and atherosclerosis: protection versus pathogenesis. Free
Radic. Biol. Med. 2000, 28, 1726-1734.
4. Holroyde, M. C.; Cole, M.; Altounyan, R. E. C.; Dixon, M.; Elliott, E. V. Bronchoconstriction Produces in
Man by Leukotrienes C and D. The Lancet 1981, 318, 17-18.
5. Greene, E. R.; Huang, S.; Serhan, C. N.; Panigrahy, D. Regulation of inflammation in cancer by
eicosanoids. Prostaglandins Other Lipid Mediat. 2011, 96, 27-36.
6. Chen, M.; Lam, B. K.; Kanaoka, Y.; Nigrovic, P. A.; Audoly, L. P.; Austen, K. F.; Lee, D. M. Neutrophil-
derived leukotriene B4 is required for inflammatory arthritis. J. Exp. Med. 2006, 203, 837-842.
7. Rea, A. I.; Schmidt, J. M.; Setzer, W. N.; Sibanda, S.; Taylor, C.; Gwebu, E. T. Cytotoxic activity of
Ozoroa insignis from Zimbabwe. Fitoterapia 2003, 74, 732-735.
8. Sullivan, J. T.; Richards, C. S.; Lloyd, H. A.; Krishna, G. Anacardic acid: molluscicide in cashew nut shell
liquid. Planta Med. 1982, 44, 175-177.
9. Trevisan, M. T. S.; Pfundstein, B.; Haubner, R.; Würtele, G.; Spiegelhalder, B.; Bartsch, H.; Owen, R. W.
Characterization of alkyl phenols in cashew (Anacardium occidentale) products and assay of their
antioxidant capacity. Chem. Food Toxicol. 2006, 44, 188-197.
10. Shobha, S. V.; Ramadoss, C. S.; Ravindranath, B. Inhibition of Soybean Lipoxygenase-1 by Anacardic
Acids, Cardols, and Cardanols. J. Nat. Prod. 1994, 57, 1755-1757.
11. Paramashivappa, R.; Phani Kumar, P.; Subba Rao, P. V.; Srinivasa Rao, A. Design, synthesis and
biological evaluation of benzimidazole/benzothiazole and benzoxazole derivatives as cyclooxygenase
inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 657-660.
12. Grazzini, R.; Hesk, D.; Heininger, E.; Hildenbrandt, G.; Reddy, C. C.; Cox-Foster, D.; Medford, J.; Craig,
R.; Mumma, R. O. Inhibition of lipoxygenase and prostaglandin endoperoxide synthase by anacardic acids.
Biochem. Biophys. Res. Commun. 1991, 176, 775-780.
13. Ghizzoni, M.; Haisma, H. J.; Maarsingh, H.; Dekker, F. J. Histone acetyltransferases are crucial regulators
in NF-κB mediated inflammation. Drug Discov. Today 2011, 16, 504-511.
14. Pergola, C.; Werz, O. 5-Lipoxygenase inhibitors: a review of recent developments and patents. Expert
Opin. Ther. Patents 2010, 20, 355-375.
15. Carter, G. W.; Young, P. R.; Albert, D. H.; Bouska, J.; Dyer, R.; Bell, R. L.; Summers, J. B.; Brooks, D.
W. 5-lipoxygenase inhibitory activity of zileuton. J. Pharm. Exp. Ther 1991, 256, 929-937.
16. Rai, G.; Kenyon, V.; Jadhav, A.; Schultz, L.; Armstrong, M.; Jameson, J. B.; Hoobler, E.; Leister, W.;
Simeonov, A.; Holman, T. R.; Maloney, D. J. Discovery of Potent and Selective Inhibitors of Human
Reticulocyte 15-Lipoxygenase-1. J. Med. Chem. 2010, 53, 7392-7404.
17. Ngu, K.; Weinstein, D. S.; Liu, W.; Langevine, C.; Combs, D. W.; Zhuang, S.; Chen, X.; Madsen, C. S.;
Harper, T. W.; Ahmad, S.; Robl, J. A. Pyrazole-based sulfonamide and sulfamides as potent inhibitors of
mammalian 15-lipoxygenase. Bioorg. Med. Chem. Lett. 2011, 21, 4141-4145.
18. Ghizzoni, M.; Boltjes, A.; Graaf, C. d.; Haisma, H. J.; Dekker, F. J. Improved inhibition of the histone
acetyltransferase PCAF by an anacardic acid derivative. Bioorg. Med. Chem. 2010, 18, 5826-5834.
19. Ghizzoni, M.; Wu, J.; Gao, T.; Haisma, H. J.; Dekker, F. J.; George Zheng, Y. 6-alkylsalicylates are
selective Tip60 inhibitors and target the acetyl-CoA binding site. Eur. J. Med. Chem. 2012, 47, 337-344.
20. Uchiyama, M.; Ozawa, H.; Takuma, K.; Matsumoto, Y.; Yonehara, M.; Hiroya, K.; Sakamoto, T.
Regiocontrolled Intramolecular Cyclizations of Carboxylic Acids to Carbon-Carbon Triple Bonds
Promoted by Acid or Base Catalyst. Org. Lett. 2006, 8, 5517-5520.

63
Anacardic acid derived salicylates are inhibitors or activators of lipoxygenase
21. Molander, G. A.; Dehmel, F. Formal Total Synthesis of Oximidine II via a Suzuki-Type Cross-Coupling
Macrocyclization Employing Potassium Organotrifluoroborates. J. Am. Chem. Soc. 2004, 126, 10313-
10318.
22. DeLuca, M. R.; Kerwin, S. M. The para-Toluenesulfonic Acid-Promoted Synthesis of 2-Substituted
Benzoxazoles and Benzimidazoles from Diacylated Precursors. Tetrahedron 1997, 53, 457-464.
23. Huang, H.; Liu, H.; Jiang, H.; Chen, K. Rapid and Efficient Pd-Catalyzed Sonogashira Coupling of Aryl
Chlorides. J. Org. Chem. 2008, 73, 6037-6040.
24. Tranchimand, S.; Tron, T.; Gaudin, C.; Iacazio, G. First Chemical Synthesis of Three Natural Depsides
Involved in Flavonol Catabolism and Related to Quercetinase Catalysis. Synth. Commun. 2006, 36, 587-
597.
25. Gibian, M. J.; Galaway, R. A. Steady-state kinetics of lipoxygenase oxygenation of unsaturated fatty acids.
Biochemistry 1976, 15, 4209-4214.
26. Ha, T. J.; Kubo, I. Lipoxygenase Inhibitory Activity of Anacardic Acids. J. Agric. Food Chem. 2005, 53,
4350-4354.
27. Mogul, R.; Johansen, E.; Holman, T. R. Oleyl Sulfate Reveals Allosteric Inhibition of Soybean
Lipoxygenase-1 and Human 15-Lipoxygenase . Biochemistry 2000, 39, 4801-4807.
28. Ruddat, V. C.; Whitman, S.; Holman, T. R.; Bernasconi, C. F. Stopped-Flow Kinetic Investigations of the
Activation of Soybean Lipoxygenase-1 and the Influence of Inhibitors on the Allosteric Site. Biochemistry
2003, 42, 4172-4178.
29. Wecksler, A. T.; Kenyon, V.; Garcia, N. K.; Deschamps, J. D.; van der Donk, W. A.; Holman, T. R.
Kinetic and structural investigations of the allosteric site in human epithelial 15-lipoxygenase-2.
Biochemistry 2009, 48, 8721-8730.
30. Leskovac, V. Comprehensive Enzyme Kinetics. 2004; 111-116.
31. Verhagen, J.; Vliegenthart, J. F.; Boldingh, J. Micelle and acid-soap formation of linoleic acid and 13-L-
hydroperoxylinoleic acid being substrates of lipoxygenase-1. Chem. Phys. Lipids 1978, 22, 255-259.
32. Egmond, M. R.; Brunori, M.; Fasella, P. M. The Steady-state kinetics of the Oxygenation of Linoleic Acid
Catalysed by Soybean Lipoxygenase. Eur. J. Biochem. 1976, 61, 93-100.
33. Butovich, I. A.; Soloshonok, V. A.; Kukhar, V. P. The unusual action of (R,S)-2-hydroxy-2-
trifluoromethyl-trans-n-octadec-4-enoic acid on 5-lipoxygenase from potato tubers. Eur. J. Biochem. 1991,
199, 153-155.
34. Skaterna, T. D.; Kharytonenko, H. I.; Kharchenko, O. V. Thermoinactivation of potato 5-lipoxygenase and
effect of phosphatidic acid on activation energy of denaturation. Ukr. Biokhim. Zh. 2010, 82, 22-28.
35. Ivanov, I.; Heydeck, D.; Hofheinz, K.; Roffeis, J.; O’Donnell, V. B.; Kuhn, H.; Walther, M. Molecular
enzymology of lipoxygenases. Arch. Biochem. Biophys. 2010, 503, 161-174.
36. Korb, ,O.; Stützle, ,Thomas; Exner, ,Thomas An ant colony optimization approach to flexible protein-
ligand docking. Swarm Intelligence 2007, 115-134.
37. Korb, O.; Stützle, T.; Exner, T. E. Empirical Scoring Functions for Advanced Protein-Ligand Docking
with PLANTS. J. Chem. Inf. Model. 2009, 49, 84-96.

Rosalina Wisastra,a Petra A.M. Kok,a Matthew P. Baumgartner,b
Carlos J. Camacho,b Hidde J. Haisma,a and Frank J. Dekkera
submitted for publication 2013
Discovery of a novel activator
of 5-lipoxygenase from
an anacardic acid-derived
compound collection
3
a Department of Pharmaceutical Gene Modulation, Groningen Research Institute of Pharmacy,
University of Groningen, Antonius Deusinglaan 1, 9713 AV Groningen, The Netherlands.
b Department of Computational and Systems Biology, University of Pittsburgh, Pittsburgh,
Pennsylvania 15260, United States
.

66
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
Abstract
Lipoxygenases (LOXs) and cyclooxygenases (COXs) metabolize poly-unsaturated fatty acids into
inflammatory signaling molecules. Modulation of the activity of these enzymes may provide new
approaches for therapy of inflammatory diseases. In this study, we screened novel anacardic acid
derivatives as modulators of human 5-LOX and COX-2 activity. Interestingly, a novel salicylate
derivative 23a was identified as a surprisingly potent activator of human 5-LOX. This compound
showed both non-competitive activation towards the human 5-LOX activator ATP and non-essential
mixed type activation against the substrate linoleic acid, while having no effect on the conversion of
the substrate arachidonic acid. The kinetic analysis demonstrated a non-essential activation of the
linoleic acid conversion with a KA of 8.65 µM, αKA of 0.38 µM and a β value of 1.76. It is also of
interest that a comparable derivative 23d showed a mixed type inhibition for linoleic acid conversion.
These observations indicate the presence of an allosteric binding site in human 5-LOX distinct from
the ATP binding site. The activatory and inhibitory behavior of 23a and 23d on the conversion of
linoleic compared to arachidonic acid are rationalized by docking studies, which indicate that the
activator 23a stabilizes linoleic acid, whereas the larger inhibitor 23d blocks the enzyme active site.
Finally, it has been shown in a cellular assay for activation of the NF- B pathway as a model for
inflammation that 23a was able to stimulate this pathway.

67
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
Introduction
The lipoxygenases (LOXs) and the cyclooxygenases (COXs) are enzymes that play key roles in the
major metabolic pathways of poly-unsaturated fatty acids (PUFAs). These pathways convert PUFAs
into inflammatory mediators such as lipoxins, leukotrienes, and prostaglandins, which play a
regulatory role in numerous inflammatory and proliferative diseases including asthma, arthritis and
cancer.1 The biological roles of LOXs and COXs in metabolic pathways towards inflammatory
mediators and signaling molecules demonstrate the utility of these enzymes as therapeutic targets.
Lipoxygenases are non-heme iron containing enzymes. There are four types of lipoxygenases with
different positional specificities for arachidonic acid oxidation present in mammalian tissues; 5-LOX,
8-LOX, 12-LOX and 15-LOX. In general, lipoxygenases comprise of two domains in which the N-
terminal domain is the regulatory domain and the C-terminal domain is the catalytic domain.2
Although, human lipoxygenases show about 60% sequence similarities,3 their regulatory mechanisms
are variable. Unlike other LOXs, 5-LOX activity requires the presence of activators such as Ca2+ and
ATP. In addition, different types of LOXs have been identified based on their distribution over
different tissues. For example, 12-LOX is subdivided in platelet 12-LOX (p12-LOX) and 12R-LOX.
As its name implies, platelet 12-LOX (p12-LOX) is mostly found in platelets, whereas 12R-LOX is
most frequently found in skin cells4, 5. Similarly, 15-LOX is subdivided in 15-LOX-1 and 15-LOX-2.
15-LOX-1 is highly expressed in leukocytes and airways endothelial cells,6, 7 whereas 15-LOX-2 is
expressed in multiple tissues such as prostate, lung, cornea, liver, colon, kidney, and brain but not in
leukocytes.8 Moreover, stimulation of cells by interleukin (IL)-4 and IL-13 shows selective
enhancement of the 15-LOX-1 expression and not 15-LOX-2 expression.9 In addition, 15-LOX-1 and
15-LOX-2 lack similarity at the primary sequence level, which also hints at distinct biological roles
and the possibility to develop selective inhibitors or activators.10
The overexpression of certain lipoxygenases has been reported in numerous inflammatory
diseases. For example, increased 15-LOX-1 expression in bronchial epithelium, which is
accompanied by elevated concentrations of 15- hydroxyeicosatetraenoic acid (15-HETE), has been
observed in patients with asthma and chronic bronchitis.11, 12 In addition, activity of other LOXs
isoenzymes has been reported to be significantly increased in disease conditions. For example,
upregulation of 12R-LOX activity has been observed in the development of skin tumor and
melanoma.13, 14 In addition, overexpression of 5-LOX has been reported to contribute to airway
inflammation in asthma and to the growth of some tumors in, for example, lung, prostate, brain and
colon.15-19 In contrast, lipoxins, another type of arachidonic acid metabolites, have been reported to
have important roles as anti-inflammatory and pro-resolution mediators.20 Production of lipoxins upon
inflammatory stimulation involves two main lipoxygenase-mediated pathways. The first pathway
involving 15-LOX and 5-LOX occurs in mucosal tissues, such as the respiratory tract, gastrointestinal
tract and the oral cavity. The second pathway involves 5-LOX and p12-LOX, in which 5-LOX

68
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
produces the Leukotriene A4 (LTA4) and p12-LOX converts the LTA4 to lipoxin A4 (LXA4).21
Lipoxin A4 has important roles in stopping neutrophil migration, stimulation of monocyte and
macrophage activation, and inhibition of leukotriene B4 formation.20 Induction of 15-LOX-1 by IL-13
in synovial tissues of patients with rheumatoid arthritis enhances the level of LX4 in a negative
feedback loop to limit inflammatory responses induced by pro-inflammatory mediators such as
Leukotriene B4.22 Another study showed that prior to induction with peroxisome proliferator-
activated receptors gamma (PPARγ) agonists in brain, the 5-LOX expression was enhanced in
association with the increase of cerebral LX4 production and inhibition of Leukotriene B4
production.23 The association of 5-lipoxygenase activity in the production of both pro- and anti-
inflammatory mediators indicates its crucial role in pathophysiological processes of inflammation.
Based on these findings we speculate that activators of 5-lipoxygenase might also have beneficial
effects in inflammation by triggering the termination of immune responses. We stress, however, that
lipoxygenase activation by small molecules with the aim to limit inflammation is a novel concept and
lacks experimental proof.
Prostaglandin endoperoxide H synthases (PGHSs) are also known as cyclooxygenases (COXs) and
catalyze the formation of prostaglandins (PGs). COXs are comprised of two main sites, a heme site
with peroxidase activity and a bis-oxygenase site. COXs convert arachidonic acid into prostaglandin
endoperoxide H2 (PGH2) in a two steps process. The first step is enzymatic catalysis of the insertion
of two molecules O2 into arachidonic acid to generate prostaglandin G2 (PGG2). The second step is
the peroxidase reaction in which the PGG2 is oxidized to PGH2 by a two electron abstraction. The
peroxidase reaction occurs at a heme-containing active site located near the protein surface, whereas
the oxygenation reaction occurs in a hydrophobic channel in the core of the enzyme.24 The COX
isoenzymes COX-1 and COX-2 are differentially expressed in various tissues. Under normal
conditions, COX-1 is expressed in most mammalian cells, whereas COX-2 is not expressed. The
COX-2 expression is increased under inflammatory conditions. Animal models of inflammatory
arthritis indicate that overexpression of COX-2 is responsible for the increase of PGs in joint tissue
and cause acute and chronic inflammation.25 Elevated levels of PGs were observed in inflammatory
bowel diseases (IBD).26-28 A similar result was observed in cancers. For example, the expression of
COX-2, but not COX-1, was observed in various regions of the colon in human colorectal cancer.29, 30
Therefore, COX-2 inhibition is considered to be a relevant therapeutic strategy for treatment of cancer
and inflammatory diseases.
Anacardic acid is a naturally occurring 6-alkyl substituted salicylate that is identified from cashew
nutshells. Previously, we found that this salicylate and its derivatives are allosteric regulators of
lipoxygenase activity. We identified an allosteric inhibitor of soybean lipoxygenase-1 (SLO-1) (19)
and an allosteric activator of potato 5-lipoxygenase (5-LOXs) (21).31 It should, however, be noted that
the lack of similarity between the human and plant enzymes prohibits extrapolation or these data
towards biological effects in cell-based studies.32 Therefore, we continue our previous studies by

69
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
exploring the inhibitory potency of a salicylate-based compound collection for inhibition of human 5-
lipoxygenase and cyclooxygenase-2. In this study, we describe the synthesis of a novel focused
collection of salicylate-based compounds and we investigate their influence on the enzyme activity of
h-5-LOX and COX-2. This enabled the identification of a novel activator 23a of the conversion of the
linoleic acid conversion by h-5-LOX. Interestingly, no effect on the arachidonic acid conversion was
observed. Enzyme kinetic investigations demonstrated that this activator binds to an allosteric binding
site on h-5-LOX that is different from the ATP regulatory site. Modeling studies suggest that 23a
binds close to the substrate linoleic acid, thereby enhancing the Km and kcat for its conversion.
Furthermore, cell-based studies on 23a demonstrated an increase of NF- B expression, which is in
line with cellular h-5-LOX activation.
Result and Discussion
Synthesis
Our compound collection that is inspired by anacardic acid (14) comprises of the previously published
salicylates 14-19, and 21 and the newly synthesized salicylates 20, 22, 23a-d, 24b-c and benzoxazole
25. This collection includes the previously described compounds that were reported as inhibitors or
activator of soybean lipoxygenase-1 (SLO1) (19) and potato 5-LOX (21).31 The synthesis of
anacardic acid (14) and its derivatives 15-19, and 21 were described previously by Ghizzoni et.al.33, 34
The starting materials, acetonide 1 and triflate 10, were synthesized as described by Uchiyama et
al.35 (Scheme 1). 2-hydroxy-6-(nonyloxy)benzoic acid (22) was synthesized from commercially
available 1-bromononane and acetonide 1 following synthetic procedure described by Ghizzoni
et.al.33 (Scheme 1). p-alkyloxybenzylbromides (3a-d) and m-alkyloxybenzylbromides (5b-c) were
prepared in three steps from p-hydroxybenzaldehyde or m-hydroxybenzaldehyde. The first step
involves the coupling of alkylbromides with the hydroxybenzaldehydes to give the corresponding
ethers. Subsequently, the benzaldehydes were reduced to the corresponding benzyl alcohols 2a-d and
4b-c36 in high yields. Direct coupling of benzyl alcohols to phenol 1 by Mitsunobu reaction did not
provide the desired ethers. Therefore, the benzylalcohols 2a-d and 4b-c were brominated using PBr3
to give the corresponding benzylbromides 3a-d and 4b-c. The benzylbromides were coupled to
acetonide 1 to give the corresponding products 12a-d and 13b-c in high yields (>70%). Subsequently,
the acetonide protecting group was cleaved using potassium hydroxide in THF to provide the final
products 23a-d as potassium salts (Scheme 1). Isolation of salicylates 23a-d as salicylate salts proved
to be necessary to avoid degradation of the p-alkoxybenzylethers by the strongly acidic salicylic acids.
Acidification results a slow hydrolysis of the product to give 2,6-dihydroxybenzoate and the
corresponding (4-(alkyloxy)benzylium cation, which is stabilized by the electrondonating alkyloxy
substituent on the para- postion. As expected, for the meta-substituted benzyl substituents 24b-c this
problem was not observed.

70
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
Scheme 1. Reagents and conditions: a) SOCl2, DMAP, DME, Acetone, 0 ⁰C for 1 h, then R.T. overnight; b) 1-
bromo alkane, K2CO3, DMF, R.T. overnight; c) NaBH4, MeOH, 0 ⁰C for 1 h, then R.T. for 30 min; d) PBr3,
CH2Cl2, 0 ⁰C for 1.5 h; e) 1-bromononane, K2CO3, DMF, R.T. overnight; f) compound 1, K2CO3, DMF, R.T.
overnight; g) 5 M KOH, THF, 60 ⁰C overnight.
p-decylphenyl bromide (8) was prepared from bromobenzene 6 and decanoylchloride following two
reaction steps. The first step was the formation of 1-(4-bromophenyl)decan-1-one 7 through Friedel-
Craft acylation using AlCl3.37 The second step was the Wolff-Kishner reduction of 1-(4-
bromophenyl)decan-1-one 7 using hydrazine monohydrate and potassium hydroxide to give product
8.37 Subsequently, arylbromide 8 was converted to alkyne 9 by coupling with trimethyl-silyl (TMS)
acetylene by a Sonogashira reaction and subsequent removal of the TMS protection using tetra butyl
ammonium fluoride (TBAF). The resulted alkyne 9 was coupled to triflate 10 38 to give the product in
high yield (80%), which was subsequently converted into product 20 by catalytic hydrogenation
(Scheme 2).
Benzoxazole 25 was designed based on the presumption that iron-binding is a key property for the
inhibition of lipoxygenases and cyclooxygenases. Benzoxazole 25 was synthesized from salicylic acid
and 2-amino phenol through direct cyclization using polyphosphoric acid (Scheme 2).39 Compound 25
did not inhibit h-5-LOX, however it gives 50% COX-2 inhibition at 50 µM inhibitor concentration.
This indicates that 25 is a potential starting point for development of selective inhibitors of COX-2
(Figure 1).

71
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
Scheme 2. Reagents and conditions: a) Decanoylchloride, AlCl3, CH2Cl2, 60 ⁰C for 1 h; b) NH2NH2.H2O,
KOH, 1-octanol, reflux, 3h; c) Trimethylsilyl-acetylene, CuI, PdCl2(PPh3)2, Et2NH, PPh3, CH3CN, (MW, 120
⁰C, 95 W, 35 min), d) TBAF, THF, 0 ⁰C, 10 min; e) CuI, PdCl2(PPh3)2, Et2NH, triflate 10, CH3CN (MW, 120
⁰C, 70 W, 35 min); f) H2, Pd/C, MeOH, 45 ⁰C, 24 h; g) 2-aminophenol, polyphosphatic acid, 180 ⁰C for 6.5 h.
The influence of the salicylates (Table 1) on the enzyme activity of human 5-lipoxygenase and/or
cyclooxygenase-2 was investigated. The human 5-lipoxygenase (h-5-LOX) activity was monitored in
real time by detection of the formation of the UV absorbance of the conjugated diene 13-
hydroperoxy-cis-9-trans-11-octa decanoic acid (13-HPOD) from linoleic acid. 40, 41 The residual h-5-
LOX activity was monitored after 10 minutes pre-incubation with 50 µM of the compound of interest.
The COX-2 activity was determined by measuring the peroxidase activity using a colorimetric assay42
to monitor the oxidation of N,N,N’,N’-tetramethyl-p-phenylenediamine (TMPD). The residual
enzyme activity was measured after 5 minutes pre-incubation in the presence of 50 µM of the
compound of interest. The enzyme activity without inhibitor present was taken as control and set to
100% and the activity without enzyme present was set to 0%. The residual enzyme activities of h-5-
LOX and COX-2 in the presence of different salicylates are shown in Figure 1.
Figure 1. Residual enzyme activity that was observed for the screening of a salicylate-based compound
collection for inhibition of h-5-LOX and COX-2 activity in the presence of 50 µM the respective compounds.
The results presented were the average of three independent experiments and the standard deviations are shown.
0
50
100
150
200
control 14 15 16 17 18 19 20 21 22 23a 23b 23c 23d 24b 24c 25
En
zym
e a
ctiv
ity
(%
)
Compounds
h-5-LOX COX-2

72
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
Anacardic acid 14, 15, and 16 show little inhibition of both h-5-LOX and COX-2 at 50 µM. In
comparison to anacardic acid 14, a slight improvement in the inhibitory potency of h-5-LOX was
observed for compound 17. Importantly, neither inhibition nor activation was observed from
compound 21, which in our previous report31 shows a strong activation on potato 5-LOX, which
demonstrates the difference between plant and human enzymes.
Compounds 23a-d with alkoxy substituents in the para- positions of the benzylethers, show an
interesting structure activity relationship for modulation of h-5-LOX activity. Compound 23a shows
strong activation of h-5-LOX at 50 µM, whereas compound 23b and 23c with a longer side chain
show no or little inhibition of h-5-LOX. Furthermore, compound 23d with a branched side chain
provides almost 50% inhibitions of h-5-LOX at the same concentration.
Table 1. Focused compound collections
The data in Figure 1 demonstrate that the salicylate-based compounds do not inhibit COX-2
activity more than 50% at 50 µM, which indicates that these types of compounds have a tendency for
R1 R2 R3
14 H H
15 H H
16 H H
17 H OH
18 H H
19 H OH
20 H OH
21 H H
22 H H
23a K H
23b K H
23c K H
23d K H
24b H H
24c H H
25
Compounds

73
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
selective modulation of h-5-LOX compared to COX-2. Interestingly, benzoxazole 25 inhibits the
COX-2 activity by about 50%, whereas no inhibition was observed on h-5-LOX, which indicates that
further optimization of this class of compound may be feasible.
The critical micelle concentration (CMCs) values for 23a and 23d at the assay conditions (Tris
buffer (50 mM), 2 mM EDTA and 2 mM CaCl2, pH 7.5, R.T.) are, respectively, 628 µM and 395 µM
(Figure 2B,C) which indicate that micelle formation does not occur at concentrations employed for
inhibition or activation. In addition, we found a CMC value for linoleic acid of 185 µM (Figure 2A)43,
which demonstrates that the substrate concentration in the inhibition studies (100 µM) was below the
CMC. Thus the enzyme kinetics was evaluated in a homogeneous system.
Figure 2. Surface tensions of (A) Anacardic acid 14 and (B) compound 23a (C) compound 23d against the
logarithm of concentration. CMC values (A–C) were measured in human 5-LOX assay conditions, Tris buffer
(50 mM), 2 mM EDTA and 2 mM CaCl2, pH 7.5, R.T.( t = 19°C)
Enzyme kinetic studies for human 5-lipoxygenase
The steady-state enzyme kinetic analysis of h-5-LOX in presence of the activator 23a was performed
in order to unravel the activation mechanism. The kinetic characterization of h-5-LOX activity versus
linoleic acid in the presence of activator 23a shows a concentration dependent activation. (Figure 3).
The activation was determined in presence of various concentrations of the natural substrate linoleic
acid or the h-5-LOX activator ATP. The initial velocities of h-5-LOX were determined at different
concentration of ATP, different concentrations 23a (0, 12.5 and 25 µM) and a fixed concentration of
linoleic acid (100 µM) The velocities were plotted in the Michaelis-Menten plot and the Lineweaver-
Burk plot as shown in Figure 4.
Figure 3. Concentration dependent activation of the linoleic acid conversion by h-5-LOX in the presence of
various concentrations of compound 23a and in its absence (control). The results were the average of three
independent experiments with error bars (±S.D)
0
50
100
150
200
250
control 1 μM 5 μM 12.5 μM 25 μM 50 μM 100 μM 200 μM
h-5
-LO
X a
ctiv
ity
(%
)
Conc. of Compound 23a (µM)
(A) (C) (B)

74
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
Figure 4. Steady-state kinetic characterization of the linoleic acid conversion versus the ATP concentration of
h-5-LOX activator 23a. (a) Michaelis-Menten plots and (b) Lineweaver-Burk plots show relation of h-5-LOX
activity versus ATP concentration at three selected concentration of 23a (¡) 0 µM, (●) 12.5 µM, and (�) 25 µM
in the presence of 100 µM linoleic acid substrate.
Table 2. Enzyme kinetic parameter for activation of h-5-LOX by activator 23a versus ATP
23a (µM) Kmapp (µM) Vmax
app (nM/s) R2
0 27.9 ± 1.7 230.9 ± 4.2 0.998
12.5 28.3 ± 1.7 386.1 ± 7.3 0.999
25 28.6 ± 2.3 523.6 ± 13.7 0.999
The Michaelis-Menten data show that activator 23a causes an increase in Vmax, whereas the Km
remains constant (Table 2). Lineweaver-Burk analysis provided similar values. The values
demonstrate non-competitive activation of linoleic acid conversion by h-5-LOX by compound 23a
compared to ATP. The fact that binding of 23a does not influence the binding constant of ATP to h-5-
LOX indicates that 23a does not compete for binding in the ATP binding pocket that allosterically
regulates h-5-LOX activity. This indicates the presence of an allosteric binding pocket that is distinct
from the ATP binding pocket and regulates the h-5-LOX enzyme activity.
The enzyme kinetics of 23a were further investigated through Michaelis-Menten and Lineweaver-
Burk plots (Figure 5). The initial velocities of h-5-LOX at different concentration of linoleic acid and
a fixed concentration of ATP (100 µM), and at several selected concentrations of 23a (0, 12.5 and 25
µM) were measured. Activator 23a causes an increase in Vmax and a decrease in Km (Table 3), which
indicates non-essential mixed type activation. This indicates that the activator can bind to the free
enzyme (E, Scheme 3) and also to the substrate bound enzyme (ES, Scheme 3). In this model binding
of the substrate and 23a mutually influence each other. According to this model the activation of h-5-
LOX by 23a can be described by equation 1 (Figure 7).
-4,0E+06
0,0E+00
4,0E+06
8,0E+06
1,2E+07
1,6E+07
-1,0E+05 -5,0E+04 0,0E+00 5,0E+04 1,0E+05
1/V
o (
sec/M
)
1/[ATP] (1/M)
(B) (A)

75
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
The enzyme kinetic parameters were derived as described in the material and methods using
methods from Leskovac.44 The activator dissociation constant (KA), the change in the affinity of
substrate binding (α) and the change in the catalytic constant (β) were determined by re-plotting the
slopes and the y-interceptions derived from the Lineweaver-Burk plot as a function of activator 23a
concentration (Figure 6).44 The KA value was determined from the intersection of 1/Δslopes plot and
1/Δy-intercept plot. The intersection of 1/Δy-intercept plot with the ordinate is defined as β.Vmax/(β-1)
from which the β value was derived. Subsequently, the resulting β value was substituted to the
equation β.Vmax/Km(β-α), which is the intersection of 1/ Δslopes plot with the ordinate, to acquire the
α value.
The analysis shows that α is 0.0438, β is 1.76 and KA is 8.65 µM. The α value, which indicates the
change in substrate binding, gives about 50-fold affinity enhancement of the substrate binding to the
activator bound enzyme compared to the free enzyme. Compound 23a shows a high potency as an
activator of h-5-LOX with the KA value in the micromolar range. Furthermore, the affinity of 23a to
the substrate bound enzyme is about 50-fold enhanced, which indicates that activation of h-5-LOX at
high substrate concentrations (> 10 µM) takes place at low activator 23a (<10 µM) concentrations
(Figure 3). The catalytic constant for substrate conversion reaction was enhanced by 1.76 fold in the
presence of the activator 23a compared to the normal reaction conditions. In conclusion, compound
23a gives a non-essential mixed-type activation of h-5-LOX and binds with high affinity (αKA =
390nM) to the substrate bound enzyme and enhances the conversion of the substrate linoleic acid.
The observed results from the enzyme kinetic studies that give non-essential mixed-type activation
versus linoleic acid and non-essential non-competitive activation versus ATP demonstrate that there is
an allosteric regulatory site in h-5-LOX that is distinct from the ATP binding site. In order to get a
better understanding of this allosteric site in the regulation of the enzyme activity, we also studied the
inhibition mechanism of h-5-LOX by compound 23d.
Figure 5. Steady-state kinetic characterization of h-5-LOX activation by activator 23a. (a) Michaelis-Menten
plots and (b) Lineweaver-Burk plots show the relation of h-5-LOX activity versus linoleic acid concentrations at
three selected concentration of 23a (¡) 0 µM, (●) 12.5 µM, and (�) 25 µM.
-1,0E+06
0,0E+00
1,0E+06
2,0E+06
3,0E+06
4,0E+06
5,0E+06
-5,0E+03 0,0E+00 5,0E+03 1,0E+04 1,5E+04
1/V
o (
sec/M
)
1/[S] (1/M)
(B) (A)

76
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
Figure 6. Re-plot of 1/Δslopes and 1/Δy-intercept versus concentration of 23a.
Table 3. Enzyme kinetic parameter for activation of h-5-LOX by activator 23a versus linoleic acid
23a (µM) Kmapp (mM) Vmax
app (µM/s) R2
0 0.590 ± 0.080 2.14 ± 0.21 0.999
12.5 0.438 ± 0.020 2.39 ± 0.07 0.999
25 0.383 ± 0.011 2.70 ± 0.05 0.999
Scheme 3. Kinetic model for non-essential activation.
equation 1
Figure 7. Equation for the enzyme kinetics according to the model in Scheme 2.44 v is the reaction velocity,
Vmax is the maximal reaction velocity, [S] is the substrate concentration and Km is the Michaelis-Menten
constant, [A] is the activator concentration. α and β, respectively, are the parameters to describe the the change
in the affinity of substrate binding and the change in the catalytic constant.
Interestingly, compound 23d has a structure that is closely related to 23a and this compound
inhibits h-5-LOX at 50 µM. We analyzed the underlying enzyme kinetic parameters. The Michaelis-
Menten plots and the Lineweaver-Burk plots show an increase of the Kmapp and reduction of the
Vmaxapp (Table 4), which is a mixed-type inhibition of h-5-LOX (Figure 8). This indicates that
inhibitor 23d can bind to the free enzyme as well as to the substrate bound enzyme following the
inhibition model in Scheme 4 from which the kinetic parameters Vmax and Km can be derived by non-
-5.00 104 0 5.00 104 1.00 105
2.50 10-3
5.00 10-3
7.50 10-3
1.00 10-2
1.25 10-2
5.00 10-6
1.00 10-5
1.50 10-5
2.00 10-5
2.50 10-5
1/ slope 1/ intercept
1/[23a]
1/
slo
pe
1/
inte
rcep
t

77
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
linear curve fitting of equation 2 (Figure 9). Furthermore, the changes in the Km and Vmax are α/α’. Km
and Vmax/α’ are used to determine the Ki and Ki’ values for inhibitor 23d using equation 3 and 4
(Figure 9). Ki and Ki’ values for inhibitor 23d respectively are 70.3 µM and 140 µM. The fact that
inhibitor 23d shows a mixed type inhibition also indicate the presence of an allosteric site in human 5-
lipoxygenase. We presume that compound 23d binds to the same allosteric binding pocket as 23a but
that the difference in the alkyl chain causes a difference in affinity as well as a change from activation
to inhibition.
Figure 8. Steady-state kinetic characterization of h-5-LOX inhibition by 23d. (a) Michaelis-Menten plots and
(b) Lineweaver-Burk plots show relation of h-5-LOX activity versus linoleic acid concentration at three selected
concentration of 23d (¡) 0 µM, (●) 25 µM, and (�) 50 µM.
Table 4. Enzyme kinetic parameter for inhibition of h-5-LOX by inhibitor 23d versus linoleic acid
23d (µM) Kmapp (mM) Vmax
app (µM/s) R2
0 0.624 ± 0.085 2.24 ± 0.23 0.999
25 0.666 ± 0.121 1.84 ± 0.25 0.998
50 0.690 ± 0.101 1.47 ± 0.16 0.999
Scheme 4. Kinetic model for mixed enzyme inhibition.
-4,0E+06
0,0E+00
4,0E+06
8,0E+06
1,2E+07
1,6E+07
-1,0E+04 0,0E+00 1,0E+04 2,0E+04 3,0E+04
1/V
o (
sec/M
)
1/[S] (1/M)
(B) (A)

78
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
equation 2
equation 3 equation 4
Figure 9. Equations for the enzyme kinetics according to the model in Scheme 2. v is the reaction velocity, Vmax is the
maximal reaction velocity, [S] is the substrate concentration and Km is the Michaelis-Menten constant. α and α’,
respectively, are the parameters to describe the change of substrate binding affinity to the enzyme and the change of the
maximum velocities. Ki is the dissociation constant of the inhibitor to the free enzyme and Ki’ is the dissociation constant of
the inhibitor to the enzyme-substrate complex.
Taking this together, the kinetic studies on activation or inhibition of human 5-lipoxygenase by
either 23a or 23d indicate the presence of an allosteric binding site that influences the enzyme
activity. In addition, both activation and inhibition obey a comparable kinetic model, which suggests
that both effects originate from the same binding site in the enzyme. Presumably, depending on the
structure of the compounds that bind to this allosteric site the enzyme would either be activated or
inhibited. We conclude that anacardic acid derivatives can be used as tools to modulate the h-5-LOX
activity toward linoleic acid and that these compounds modulate the activity of this enzyme by
binding to an allosteric binding site.
We continued our studies with investigation of the modulation of 23a and 23d of the h-5-LOX
activity for arachidonic acid (Figure 10). Interestingly, no modulation of the h-5-LOX activity toward
arachidonic acid was observed in presence of 23a or 23d. These results demonstrate that 23a and 23d
are selective modulators of linoleic acid conversion and not arachidonic acid conversion by h-5-LOX.
Figure 10. Residual enzyme activity that was observed for compounds 23a and 23d for inhibition of h-5-LOX
in the presence of 50 µM of the respective compounds with linoleic acid or arachidonic acid as a substrate. The
results presented were the average of three independent experiments and the standard deviations are shown.
Molecular modeling
These results justify the hypothesis that the allosteric regulators 23a and 23d bind to an allosteric
regulatory site in h-5-LOX that is close to the active site. The hypothesis is supported by a previous
report, which stated that h-5-LOX has a wide active site compared to the others LOXs, which could
harbor two substrate molecules.45 The observation that 23a and 23d modulate the conversion of
linoleic acid, which has 18 carbon atoms, and not arachidonic acid, which has 20 carbon atoms, is in
line with this hypothesis, because it is an indication for steric interactions between the substrate and
23a or 23d. Therefore, we hypothesize that the substrates together with either 23a or 23d bind in the
0
50
100
150
200
control 23a 23dEn
zym
e a
ctiv
ity
(%
)
Compounds
Linoleic acid
Arachidonic acid

79
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
active site, thus resulting in a different binding constant and a different turnover rate of the substrate
linoleic acid. We employed this presumption as a basis for the modeling studies.
The binding models of the activator 23a and the inhibitor 23d in the linoleic acid bound as well as
the arachidonic acid bound h-5-LOX active site were made based on the recently published crystal
structure of h-5-LOX (PDB code 3V99)46. This crystal structure contains an arachidonic acid substrate
molecule in the active site. The arachidonic acid substrate molecule lacks visible specific interactions
except for the interaction of the cis double bond at carbon 11 to the iron. This lack of visible specific
interactions prohibits the re-docking of arachidonic acid and de novo docking of linoleic acid in the h-
5-LOX enzyme active site. Therefore, we modeled a bound structure of linoleic acid by threading the
lipid into the crystal structure of arachidonic acid using PyMOL.47 The threaded linoleic acid was then
minimized using UCSF Chimera48 to produce the final model of linoleic acid bound to the h-5-LOX
active site as shown in Figure 11a. This model is justified by the high similarity of linoleic acid to
arachidonic acid, which is present in the crystal structure.
Using the model of linoleic acid bound to the active site of h-5-LOX, compounds 23a and 23d
were docked using smina and the poses were reranked using the vina scoring function. Most of the
top 10 docked structures are held bound by a strong pi-pi stacking interaction between the phenyl ring
of the compounds 23a and 23d (as well as 23b and 23c) and Phe177 on one side and the hydrophobic
side chain of Ala410 on the other (Figure 11b). The buried polar functionalities of the salicylate head
group may also be stabilized by a hydrogen bond between the carboxylic -OH group of the head
group and the side chain of His372. Also of importance is the hydrogen bond between the backbone
nitrogen of Phe177 and the ether oxygen of the hydrophobic tail (Figure 11c). The dominant docking
poses of compounds 23a-d share a similar geometry, with the exception that in some docking poses
the salicylate head group is rotated by 180 degrees. Compounds 23a-d are identical except for the
length of their alkoxy tails (Table 1). The only differences in the full series of docked conformations
is in the flexible alkoxy tails of 23a-d that can fit in a variety of conformations with almost identical
energy scores (Figure 11d).
The models of 23a-d (Figure 11d) are fully consistent with our experimental observations. Figure
1 and Table 1 demonstrate that the h-5-LOX enzymatic activity decreases with an increase in the
alkoxy tail length. In addition, it was observed that 23a and 23d influence the linoleic acid conversion
and not the arachidonic acid conversion by h-5-LOX (Figure 10). Figure 11a demonstrates that the
most prominent difference between linoleic acid (C18) and arachidonic acid (C20) is that linoleic acid
protrudes less far in the active site than arachidonic acid. This indicates that linoleic acid as a
substrate leaves space for the alkoxy tails of 23a and 23d (Figure 11c). On the other hand, arachidonic
acid fills the binding pocket blocking the access of 23d but not necessarily of compound 23a. The
space that is left unfilled by the shorter linoleic (relative to arachidonic) acid is occupied by the
alkoxy tails of 23a (C5), thereby filling the binding pocket in a manner similar to arachidonic acid.
This model explains the high affinity of 23a for the linoleic acid bound enzyme (αKA 0.38 µM)
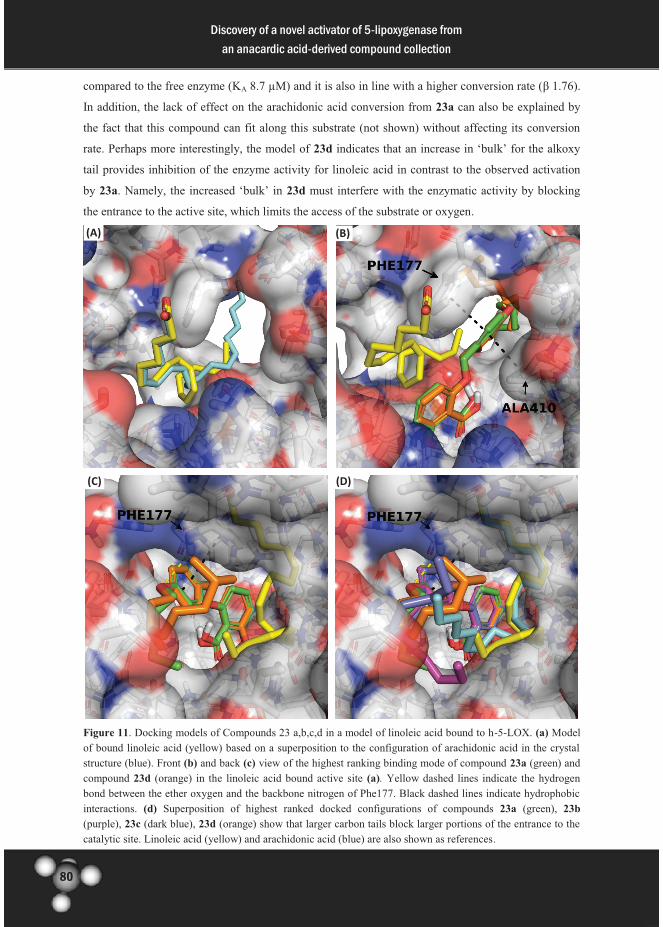
80
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
compared to the free enzyme (KA 8.7 µM) and it is also in line with a higher conversion rate (β 1.76).
In addition, the lack of effect on the arachidonic acid conversion from 23a can also be explained by
the fact that this compound can fit along this substrate (not shown) without affecting its conversion
rate. Perhaps more interestingly, the model of 23d indicates that an increase in ‘bulk’ for the alkoxy
tail provides inhibition of the enzyme activity for linoleic acid in contrast to the observed activation
by 23a. Namely, the increased ‘bulk’ in 23d must interfere with the enzymatic activity by blocking
the entrance to the active site, which limits the access of the substrate or oxygen.
Figure 11. Docking models of Compounds 23 a,b,c,d in a model of linoleic acid bound to h-5-LOX. (a) Model
of bound linoleic acid (yellow) based on a superposition to the configuration of arachidonic acid in the crystal
structure (blue). Front (b) and back (c) view of the highest ranking binding mode of compound 23a (green) and
compound 23d (orange) in the linoleic acid bound active site (a). Yellow dashed lines indicate the hydrogen
bond between the ether oxygen and the backbone nitrogen of Phe177. Black dashed lines indicate hydrophobic
interactions. (d) Superposition of highest ranked docked configurations of compounds 23a (green), 23b
(purple), 23c (dark blue), 23d (orange) show that larger carbon tails block larger portions of the entrance to the
catalytic site. Linoleic acid (yellow) and arachidonic acid (blue) are also shown as references.
(A)
(C)
(B)
(D)

81
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
Taking all these data together we have a plausible model to explain our observations. We conclude
that compound 23a is an activator for linoleic acid conversion by h-5-LOX, presumably because it
stabilizes the substrate-enzyme complex without blocking the entrance to the active site. On the other
hand, compound 23d could bind in a comparable configuration but its larger carbon tail, presumably,
blocks one end of the active site which interferes with the diffusion of the substrate and molecular
oxygen thus overriding the stabilization effect. Although these models provide plausible explanations
for the observed behavior of this compound class, we realize the crystallography studies would
provide the ultimate experimental proof for the mode of action of these molecules.
Modulation of NF- B pathway
The endogenous h-5-LOX metabolites can activate the NF- B pathway by stimulating the
phosphorylation of I Bα via phosphatidylinostisol 3-kinase (PI3K) pathway or protein kinase C
(PKC) pathway.49-51 The relevance of these modulators in cell-based studies on NF- B activation was
investigated using the commercially available RAW-Blue NF- B reporter cell line. First, the
cytotoxicity of the compound on this cell line was investigated and the cells showed more than 75%
viability at concentration below 3 µM. (Figure 12). Modulation of NF- B gene expression was
determined by measuring the concentration of secreted embryonic alkaline phosphatase (SEAP) in the
medium using a quanti-blue assay. The SEAP secretion of the untreated cells was plotted as
background and set to 100% and the absorbance of empty medium was set to 0%. The changes of the
SEAP concentrations in the presence of different modulator compounds are shown in Figure 13.
Interestingly, the addition of 3 µM of compound 23a gives about 7% elevation of the NF- B gene
expression, whereas inhibitor 23d shows no effect at this concentration. Furthermore, stimulation of
the cells with IFN- (10 ng/mL) gives a 20% increase of SEAP concentrations and an addition of
compound 23a increases the gene expression to 27%, whereas no significant change in the SEAP
concentrations were observed from the addition of 23d to IFN- treated cells. These results
demonstrates that activator 23a gives an additive effect to the activation of NF- B gene expression by
IFN- stimulation, which indicates that h-5-LOX activate the NF- B pathway via a different
mechanism than IFN- by interferon- induced protein kinase R (PKR) via Janus kinase (JAK)-Signal
Transducer and Activator of Transcription (STAT) pathway, which has also been described in the
literature.52

82
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
Figure 12. Cytotoxicity assay of RAW-Blue cells in the presence of modulator 23a and 23d at different
concentration.
Figure 13. NF- B SEAP assay of the untreated cells (control) and in the presence of 3 µM compounds 23a and
23d, 10 ng/mL IFN- , and the combination of modulators and IFN- .
Conclusion
Anacardic acid derivatives show interesting structure activity relationships that demonstrate that these
compounds can either activate or inhibit the activity of human-5-LOX. Compound 23a and 23d either
activate or inhibit the activity of human 5-LOX toward linoleic acid as a substrate depending on the
substitution in para- position of the 2-(benzyloxy)-6-hydroxybenzoic acid core. Compound 23a was
identified as a non-essential activator of human 5-lipoxygenase with KA is 8.65 µM, α is 0.0438, and
β is 1.76. The affinity of activator 23a to the substrate bound enzyme (αKA) is 0.38 µM, which is 50
times higher than to the free enzyme. Furthermore, activator 23a acts as non-competitive activator
against ATP, which indicates the presence of an allosteric site that is different from the ATP binding
site. Kinetic studies on inhibitor 23d show a mixed type inhibition, which indicates the presence of an
allosteric regulatory site for human 5-LOX activity. Both modulators 23a and 23d demonstrated no
effect on the conversion of the substrate arachidonic acid by h-5-LOX. Molecular modeling studies
indicate that 23a and 23d bind close to the substrate linoleic acid in the active site of h-5-LOX, but
that these modulators do not fit simultaneously in the active site with the arachidonic acid, which is
0
20
40
60
80
100
120
100 50 25 13 6,25 3,13 1,56 0,78 0
% c
ell
s v
iab
ilit
y
Conc. of modulator (µM)
23a 23d
-5
0
5
10
15
20
25
30
Δ %
SE
AP

83
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
slightly larger than linoleic acid. Cell-based studies on the utility of the modulators in inflammatory
models demonstrated that activator 23a is able to activate the NF- B pathway in presence of
inflammatory stimuli such as IFN- . This provides a novel compound class that modulates human 5-
LOX activity via a novel mechanism, which can be explored further in cell-based studies and drug
discovery projects.
Experimental
General
All reagents and solvents were purchased from commercial suppliers (Fluka, Sigma-Aldrich, Acros
Organics) and were used without further purification. Dichloromethane was distilled over CaH2
before use. Merck silica gel 60 F254 plates were used for analytical thin layer chromatography (TLC)
and spots were detected by UV light, or stained using KMNO4 solution. Column chromatography was
performed with MP Ecochrom silica gel 32-63, 60Ǻ using the flash chromatography technique. 1H
(200 MHz) and 13C (50 MHz) NMR spectra were recorded on a Varian Gemini 200 spectrometer. 13C
NMR spectra were recorded using the attached proton test (APT). Chemical shifts are reported in ppm
(δ) relative to the solvent. Atmospheric Pressure Photoionization mass spectra (APPI-MS) were
recorded on an Applied Biosystems/SCIEX API3000-triple quadrupole mass spectrometer. High-
resolution mass spectra (HR-MS) were recorded using a flow injection method on a LTQ-Orbitrap XL
mass spectrometer (Thermo Electron, Bremen, Germany) with a resolution of 60,000 at m/z 400.
Protonated testosterone (lock mass m/z = 289.2162) was used for internal recalibration in real time.
N,N,N',N'-tetramethyl-p-phenylenediamine (TMPD) hydrochloride, human 5-lipoxygenase
enzyme, and COX-2 (human recombinant) were obtained from Cayman Chemicals. Adenosine tri-
phosphate (ATP), arachidonic acid, and linoleic acid were obtained from Sigma-Aldrich. Raw Blue
cells assay was obtained from InvivoGen. The MTS assay kit was obtained from Promega and
interferon- (IFN- ) was obtained from PeproTech.
Synthesis
Synthetic Procedure 1: Synthesis of alkoxy benzyl alcohol
1-bromo alkane (13 mmol) was added to a light yellow suspension of hydroxybenzaldehyde (1.1 g,
9.0 mmol) and K2CO3 (5.2 g, 37 mmol) in 45 mL DMF under nitrogen atmosphere. The suspension
was stirred overnight at room temperature. The reaction mixture was diluted with demiwater (100
mL) and extracted with ethylacetate (3 x 80 mL). The combined organic layers were washed with
demiwater (4 x 80 mL), 0.1 M aqueous HCl (100 mL), brine (2 x 30 mL), dried with MgSO4, filtered
and concentrated in vacuo.
NaBH4 (0.17 g, 4.5 mmol) was added to a solution of benzaldehyde (8.0 mmol) in 30 mL methanol
at 0 °C under nitrogen atmosphere. The reaction mixture was stirred for one hour at 0 °C and then

84
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
warmed to room temperature. The reaction mixture was diluted with demiwater (80 mL) and extracted
with ethylacetate (2 x 60 mL). The combined organic layers were washed with demiwater (2 x 40
mL), brine (2 x 40mL), dried with MgSO4, filtered and concentrated in vacuo. The product was
obtained after crystallization or purification using column chromatography.
Synthetic procedure 2: Bromination of alkyloxy benzyl alcohol
Phosphorus tribromide (PBr3) (0.34 mL, 3.6 mmol) was added slowly to a suspension of alkyloxy
benzyl alcohol (2.5 mmol) in 10 mL CH2Cl2 under nitrogen atmosphere at 0 °C to give an orange
suspension. The reaction mixture was stirred for 1.5 hours at 0 °C. The reaction mixture was poured
into 80 mL ice water, and extracted with ethylacetate (3 x 50 mL). The combined organic layers were
washed with demiwater (50 mL), 0.1 M aqueous HCl (2 x 30 mL), brine (50 mL), dried with MgSO4,
filtered and concentrated in vacuo. The aryl bromide product was obtained after purification using
column chromatography.
Synthetic Procedure 3: Williamson ether synthesis coupling of aryl bromide
5-hydroxy-2,2-dimethyl-4H-benzo[d][1,3]dioxin-4-one 1 (0.22 g, 1.1 mmol) and K2CO3 (0.63 g, 4.6
mmol) were suspended in 8 mL DMF under nitrogen atmosphere. A suspension of aryl bromide in 7
mL DMF was added to the suspension. The mixture was stirred overnight at room temperature and
was diluted with demiwater (60 mL) and extracted with ethylacetate (3 x 45 mL). The combined
organic layers were washed with demiwater (2 x 40 mL), 0.1 M aqueous HCl (40 mL), brine (2 x 20
mL), dried over MgSO4, filtered and concentrated in vacuo. The product was obtained after
purification using column chromatography.
Synthetic procedure 4: saponification of the acetonide
5 M KOH (0.16 mL, 0.80 mmol) was added to a solution of 2,2-dimethyl-5-((4-
(alkyloxy)benzyl)oxy)-4H-benzo[d][1,3]dioxin-4-one (0.15 g, 0.40 mmol) in 10 mL THF. The
reaction mixture was heated till 60 °C and stirred overnight affording an orange suspension. The
reaction mixture was concentrated in vacuo yielding the product as potassium salt.
Synthetic procedure 5: hydrolysis of acetonide
5 M KOH (1.2 mL, 6 mmol) was added to a colorless solution of acetonide (0.60 mmol) in 10 mL
THF. The reaction mixture was stirred for 24 hours at 62 °C. The reaction mixture was diluted and
acidified with 1 N aqueous HCl (2.6 mL) and extracted with ethylacetate (2 x 40 mL). The combined
organic layers were washed with water (50 mL), brine (2 x 30 mL), dried over MgSO4, filtered and
concentrated in vacuo.

85
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
(4-(pentyloxy)phenyl)methanol (2a)
The product was obtained using Williamson ether synthesis followed by aldehyde reduction of 1-
bromopentane, and 4-hydroxybenzaldehyde using synthetic procedure 1. The product was purified
using column chromatography with heptane:EtOAc 5:1 (v/v) as an eluent. Yield 73%. Light orange
solid. Rf=0.41 (heptane:EtOAc 1:1). 1H NMR (200 MHz, CDCl3) δ 7.40 – 7.18 (m, 2H), 6.97 – 6.79
(m, 2H), 4.59 (s, 2H), 3.95 (t, J = 6.6, 2H), 1.87 (s, 1H), 1.86 – 1.72 (m, 2H), 1.46 – 1.34 (m, 4H),
0.94 (t, J = 7.0, 3H). 13C NMR (50 MHz, CDCl3) δ 158.47, 133.10, 128.80, 114.73, 68.24, 65.20,
29.14, 28.38, 22.65, 14.20. MS (APPI): m/z 194.2 [M+.].
1-(bromomethyl)-4-(pentyloxy)benzene (3a)
The product was obtained from (4-(pentyloxy)phenyl)methanol 2a using synthetic procedure 2. The
product was obtained in high purity and no further purification was required. Yield 97%. Yellow
solid. Rf = 0.72 (heptane:EtOAc 1:1).1H NMR (200 MHz, CDCl3) δ 7.38 – 7.22 (m, 2H), 6.94 – 6.74
(m, 2H), 4.51 (s, 2H), 3.94 (t, J = 6.2, 2H), 1.91 – 1.65 (m, 2H), 1.59 – 1.21 (m, 4H), 0.94 (t, J = 7.1,
3H). 13C NMR (50 MHz, CDCl3) δ 159.47, 130.60, 129.88, 114.94, 68.26, 34.30, 29.11, 28.38, 22.65,
14.22.
2,2-dimethyl-5-((4-(pentyloxy)benzyl)oxy)-4H-benzo[d][1,3]dioxin-4-one (12a)
The product was obtained from 5-hydroxy-2,2-dimethyl-4H-benzo[d][1,3]dioxin-4-one 1, and 1-
(bromomethyl)-4-(pentyloxy)benzene 3a using synthetic procedure 3. The product was crystallized
from iso-propanol. Yield 70%. White solid. Rf = 0.6 (heptane:EtOAc 1:1).1H NMR (200 MHz,
CDCl3) δ 7.50 – 7.32 (m, 3H), 6.90 (d, J = 8.3, 2H), 6.65 (d, J = 8.5, 1H), 6.54 (d, J = 8.2, 1H), 5.18
(s, 2H), 3.95 (t, J = 6.6, 2H), 1.81 – 1.70 (m, 8H), 1.46 - 1.40 (m, 4H), 0.93 (t, J = 7.1, 3H). 13C NMR
(50 MHz, CDCl3) δ 160.69, 159.10, 157.99, 136.40, 128.53, 128.26, 114.82, 109.57, 107.62, 105.41,
105.22, 104.17, 70.89, 68.23, 29.17, 28.41, 25.86, 22.67, 14.23. MS (APPI): m/z 370.2 [M+.].

86
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
potassium 2-hydroxy-6-((4-(pentyloxy)benzyl)oxy)benzoate (23a)
The product was obtained from 2,2-dimethyl-5-((4-(pentyloxy)benzyl)oxy)-4H-benzo[d][1,3]dioxin-
4-one 12a using synthetic procedure 4. Yield quantitatively determined from the final yield of product
in excess of KOH. Orange gum. Rf = 0.16 (heptane:EtOAc 1:1 containing 1% acetic acid). 1H NMR
(200 MHz, CD3OD) δ 7.49 – 7.22 (m, 2H), 7.15 – 6.82 (m, 3H), 6.51 – 6.08 (m, 2H), 5.03 (s, 2H),
3.95 (t, J = 6.4, 2H), 1.93 – 1.63 (m, 2H), 1.55 – 1.26 (m, 4H), 0.94 (t, J = 7.0, 3H).13C NMR (50
MHz, CD3OD) δ 164.21, 161.49, 160.78, 158.80, 133.04, 131.92, 130.70, , 116.19, 111.57, 106.31,
99.93, 72.43, 69.88, 46.09, 30.89, 30.17, 24.32, 15.24. HRMS: m/z [M−H]−calcd for C19H21O5
329.13945, found 329.13995.
(4-(heptyloxy)phenyl)methanol (2b)
The product was obtained using Williamson ether synthesis followed by aldehyde reduction of 1-
bromoheptane, and 4-hydroxybenzaldehyde using synthetic procedure 1. The product was crystallized
from heptane. Yield 87%. Colorless to white crystal. Rf = 0.44 (heptane:EtOAc 1:1). 1H NMR (200
MHz, CDCl3) δ 7.49 – 7.16 (m, 2H), 6.99 – 6.79 (m, 2H), 4.59 (s, 2H), 3.95 (t, J = 6.6, 2H), 1.83 (s,
1H), 1.81 – 1.71 (m, 2H), 1.55 – 1.19 (m, 8H), 0.89 (t, J = 6.4, 3H).13C NMR (50 MHz, CDCl3) δ
158.97, 133.09, 128.81, 114.75, 68.27, 65.22, 31.98, 29.46, 29.26, 26.20, 22.80, 14.28. MS (APPI):
m/z 222.2 [M+.].
1-(bromomethyl)-4-(heptyloxy)benzene (3b)
The product was obtained from (4-(heptyloxy)phenyl)methanol 2b using synthetic procedure 2. The
product was obtained in high purity and no further purification was required. Yield 97%. Yellow oil.
Rf = 0.71 (heptane:EtOAc 1:1).1H NMR (200 MHz, CDCl3) δ 7.37 – 7.27 (m, 2H), 6.91 – 6.75 (m,
2H), 4.51 (s, 2H), 3.95 (t, J = 6.5, 2H), 1.91 – 1.68 (m, 2H), 1.57 – 1.18 (m, 8H), 0.91 (t, J = 6.5,
3H).13C NMR (50 MHz, CDCl3) δ 159.47, 130.60, 129.87, 114.94, 68.27, 34.30, 31.98, 29.42, 29.25,
26.19, 22.81, 14.29.

87
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
5-((4-(heptyloxy)benzyl)oxy)-2,2-dimethyl-4H-benzo[d][1,3]dioxin-4-one (12b)
The product was obtained from 5-hydroxy-2,2-dimethyl-4H-benzo[d][1,3]dioxin-4-one 1, and 1-
(bromomethyl)-4-(heptyloxy)benzene 3b using synthetic procedure 3. The product was purified using
column chromatography with heptane:EtOAc 11:1 (v/v) as an eluent. Yield 78%. White solid. Rf =
0.41 (heptane:EtOAc 3:1).1H NMR (200 MHz, CDCl3) δ 7.50 – 7.32 (m, 3H), 6.90 (d, J = 8.6, 2H),
6.90 (d, J = 8.4, 1H), 6.65 (d, J = 8.4, 1H), 5.18 (s, 2H), 3.94 (t, J = 6.5, 2H), 1.85 – 1.65 (m, 8H),
1.40 – 1.30 (m, 8H), 0.89 (t, J = 6.6, 3H). 13C NMR (50 MHz, CDCl3) δ 160.68, 159.11, 158.23,
157.99, 136.40, 128.53, 128.25, 114.82, 109.57, 107.61, 105.41, 104.17, 70.88, 68.25, 31.99, 29.48,
29.27, 26.21, 25.85, 22.82, 14.29. MS (APPI): m/z398.2 [M+.].
Potassium 2-((4-(heptyloxy)benzyl)oxy)-6-hydroxybenzoate (23b)
The product was obtained from 5-((4-(heptyloxy)benzyl)oxy)-2,2-dimethyl-4H-benzo[d][1,3]dioxin-
4-one 12b using synthetic procedure 4. Yield quantitatively determined from the final yield of product
in excess of KOH. Orange solid. Rf = 0.49 (heptane:EtOAc 2:1 containing 1% acetic acid).1H NMR
(200 MHz, CD3OD) δ 7.42 (d, J = 8.7, 1H), 7.32 (d, J = 8.7, 1H), 7.08 (t, J = 8.2, 1H), 6.97 – 6.81 (m,
2H), 6.48 – 6.38 (m, 1H), 6.37 – 6.13 (m, 1H), 5.06 (s, 2H), 3.99 – 3.92 (m, 2H), 1.80 - 1.70 (m, 2H),
1.59 – 1.19 (m, 8H), 0.91 (t, J = 6.5, 3H). 13C NMR (50 MHz, CD3OD) δ 179.61, 161.89, 161.16,
160.88, 133.19, 131.99, 130.65, 116.15, 111.26, 106.62, 105.29, 72.48, 69.85, 33.85, 31.30, 31.08,
28.01, 24.53, 15.27. HRMS: m/z [M−H]−calcd for C21H25O5357.17075, found 357.17109.
(4-(nonyloxy)phenyl)methanol (2c)
The product was obtained using Williamson ether synthesis followed by aldehyde reduction of 1-
bromononane, and 4-hydroxybenzaldehyde using synthetic procedure 1. The product was crystallized
from heptane. Yield 73%. White crystal. Rf = 0.44 (heptane:ethylacetate 1:1).1H NMR (200 MHz,
CDCl3) δ 7.35 – 7.20 (m, 2H), 6.95 – 6.82 (m, 2H), 4.60 (s, 2H), 3.95 (t, J = 6.6, 2H), 1.85 – 1.74 (m,
2H), 1.71 (s, 1H), 1.42 – 1.29 (m, 12H), 0.89 (t, J = 7.0, 3H).13C NMR (50 MHz, CDCl3) δ 158.99,

88
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
133.09, 128.82, 114.76, 68.28, 65.27, 32.08, 29.74, 29.61, 29.47, 26.24, 22.87, 14.31. MS (APPI): m/z
250.2 [M+.].
1-(bromomethyl)-4-(nonyloxy)benzene (3c)
The product was obtained from (4-(nonyloxy)phenyl)methanol 2c using synthetic procedure 2. The
product was obtained in high purity and no further purification was required. Yield 94%. Light yellow
oil. Rf = 0.74 (heptane:EtOAc 1:1).1H NMR (200 MHz, CDCl3) δ 7.39 – 7.20 (m, 2H), 6.94 – 6.76
(m, 2H), 4.50 (s, 2H), 3.95 (t, J = 6.5, 2H), 1.88 – 1.66 (m, 2H), 1.54 – 1.14 (m, 12H), 0.89 (t, J = 6.4,
3H).13C NMR (50 MHz, CDCl3) δ 159.48, 130.61, 129.88, 114.96, 68.29, 34.31, 32.09, 29.74, 29.59,
29.47, 29.42, 26.23, 22.88, 14.32.
2,2-dimethyl-5-((4-(nonyloxy)benzyl)oxy)-4H-benzo[d][1,3]dioxin-4-one (12c)
The product was obtained from 5, -hydroxy-2,2-dimethyl-4H-benzo[d][1,3]dioxin-4-one 1, and 1-
(bromomethyl)-4-(nonyloxy)benzene 3c using synthetic procedure 3. The product was purified using
column chromatography with heptane:EtOAc 15:1 (v/v) as an eluent. Yield 85%. White solid. Rf =
0.48 (heptane:EtOAc 3:1). 1H NMR (200 MHz, CDCl3) δ 7.53 – 7.29 (m, 3H), 6.90 (d, J = 8.4, 2H),
6.65 (d, J = 8.4, 1H), 6.54 (d, J = 8.4, 1H), 5.18 (s, 2H), 3.94 (t, J = 6.5, 2H), 1.88 – 1.64 (m, 8H), 1.36
(d, J = 30.5, 12H), 0.88 (t, J = 6.2, 3H).13C NMR (50 MHz, CDCl3) δ 160.67, 159.09, 158.20, 157.98,
136.39, 128.52, 128.24, 114.81, 109.56, 107.60, 105.40, 102.26, 70.87, 68.24, 32.08, 29.74, 29.61,
29.47, 26.25, 25.85, 22.87, 14.35, 14.31. MS (APPI): m/z 426.2 [M+.].
Potassium 2-hydroxy-6-((4-(nonyloxy)benzyl)oxy)benzoate (23c)
The product was obtained from 2,2-dimethyl-5-((4-(nonyloxy)benzyl)oxy)-4H-benzo[d][1,3]dioxin-4-
one 12c using synthetic procedure 4. Yield quantitatively determined from the final yield of product
in excess of KOH. Rf = 0.18 (heptane:EtOAc 1:1 containing 1% acetic acid). 1H NMR (200 MHz,
CD3OD) δ 7.43 (d, J = 8.7, 1H), 7.31 (d, J = 8.7, 1H), 7.08 (t, J = 8.2, 1H), 7.04 – 6.86 (m, 2H), 6.43

89
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
(d, J = 8.3, 2H), 4.98 (s, 2H), 3.96 (t, J = 6.4, 2H), 1.80 - 1.69 (m, 2H), 1.41 – 1.14 (m, 12H), 0.91 (t,
J = 7.0, 3H).13C NMR (50 MHz, CD3OD) δ 172.3, 164.24, 160.76, 156.88, 133.10, 131.98, 130.67,
116.19, 111.48, 109.33, 106.36, 72.41, 69.89, 61.77, 33.83, 31.47, 31.31, 31.18, 28.10, 27.95, 24.52,
14.22. HRMS: m/z [M−H]−calcd for C23H29O5 385.20205, found 385.20248.
(4-((3,7-dimethyloctyl)oxy)phenyl)methanol (2d)
The product was obtained using Williamson ether synthesis followed by aldehyde reduction of 1-1-
bromo-3,7-dimethyloctane, and 4-hydroxybenzaldehyde using synthetic procedure 1. The product was
used for the next reaction step without further purification. Yield 92%. Yellow oil. Rf=0.48
(heptane:EtOAc 1:1). 1H NMR (200 MHz, CDCl3) δ 7.27 (d, J = 8.6, 2H), 6.88 (d, J = 8.6, 2H), 4.59
(s, 2H), 3.98 (t, J = 6.5, 2H), 1.88 – 1.46 (m, 5H), 1.32 – 1.14 (m, 6H), 0.93 (d, J = 6.2, 3H),0.87 (d, J
= 6.5, 6H).13C NMR (50 MHz, CDCl3) δ 158.98, 133.10, 128.82, 114.77, 66.59, 65.27, 39.45, 37.49,
36.40, 30.06, 28.17, 24.86, 22.91, 22.81, 19.86. MS (APPI): m/z 264.1 [M+.].
1-(bromomethyl)-4-((3,7-dimethyloctyl)oxy)benzene (3d)
The product was obtained from (4-((3,7-dimethyloctyl)oxy)phenyl)methanol 2d using synthetic
procedure 2. The product was used for the next reaction step without further purification. Yield
quantitative. Light yellow oil. Rf = 0.75 (heptane:EtOAc 1:1). 1H NMR (200 MHz, CDCl3) δ 7.30 (d,
J = 8.6, 2H), 6.85 (d, J = 8.6, 2H), 4.49 (s, 2H), 3.98 (t, J = 6.5, 2H), 1.87 – 1.46 (m, 4H), 1.32 – 1.10
(m, 6H), 0.93 (d, J = 6.2, 3H), 0.87 (d, J = 6.4, 6H).13C NMR (50 MHz, CDCl3) δ 159.46, 130.60,
129.88, 114.95, 66.59, 39.45, 37.47, 36.35, 34.30, 30.04, 28.18, 24.86, 22.92, 22.82, 19.85.
5-((4-((3,7-dimethyloctyl)oxy)benzyl)oxy)-2,2-dimethyl-4H-benzo[d][1,3]dioxin-4-one (12d)
The product was obtained from 5-hydroxy-2,2-dimethyl-4H-benzo[d][1,3]dioxin-4-one 1, and 1-
(bromomethyl)-4-((3,7-dimethyloctyl)oxy)benzene 3d using synthetic procedure 3. The product was
purified using column chromatography with heptane:EtOAc 19:1 (v/v) as eluent. Yield 68%. White
solid. Rf = 0.35 (heptane:EtOAc 4:1). 1H NMR (200 MHz, CDCl3) δ 7.46 – 7.26 (m, 3H), 6.92 (d, J =
8.6, 2H), 6.65 (d, J = 8.5, 1H), 6.54 (d, J = 8.5, 1 H), 5.18 (s, 2H), 3.98 (t, J = 6.5, 2H), 1.84 – 1.5 (m,
10H), 1.32 – 1.14 (m, 6H), 0.93 (d, J = 6.2, 3H), 0.87 (d, J = 6.4, 6H).13C NMR (50 MHz, CDCl3) δ

90
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
160.69, 159.10, 158.23, 157.99, 136.40, 128.53, 128.25, 114.82, 109.57, 107.61, 105.41, 70.89, 66.56,
39.45, 37.49, 36.40, 30.05, 28.17, 25.85, 24.85, 22.91, 22.81, 19.86. MS (APPI): m/z 440.4 [M+.].
Potassium 2-((4-((3,7-dimethyloctyl)oxy)benzyl)oxy)-6-hydroxybenzoate (23d)
The product was obtained from 5-((4-((3,7-dimethyloctyl)oxy)benzyl)oxy)-2,2-dimethyl-4H-
benzo[d][1,3]dioxin-4-one 12d using synthetic procedure 4. Yield quantitatively determined from the
final yield of product in excess of KOH. Orange solid. Rf = 0.54 (heptane:EtOAc 1:1 containing 1%
acetic acid). 1H NMR (200 MHz, CD3OD) δ 7.42 (d, J = 8.7, 2H), 7.05 (t, J = 8.2, 1H), 6.95 – 6.85
(m, 2H), 6.43 – 6.38 (m, 2H), 5.03 (s, 2H), 3.99 (t, J = 6.7 2H), 1.92 – 1.48 (m, 4H), 1.40 – 1.16 (m,
6H), 0.95 (d, J = 6.2, 3H), 0.89 (d, J = 6.5, 6H).13C NMR (50 MHz, CD3OD) δ 192.95, 164.42,
161.42, 160.86, 132.74, 132.06, 130.69, 116.17, 111.76, 105.95, 72.47, 68.11, 41.28, 39.28, 38.27,
31.89, 29.99, 26.67, 23.95, 23.86, 20.90. HRMS: m/z [M−H]−calcd for C24H31O5 399.2177, found
399.21844.
(3-(heptyloxy)phenyl)methanol (4b)
The product was obtained using Williamson ether synthesis followed by aldehyde reduction of 1-
bromoheptane, and 3-hydroxybenzaldehyde using synthetic procedure 1. The product was used for the
next reaction step without further purification. Yield 94%. Brown solid. Rf = 0.48 (heptane:EtOAc
1:1). 1H NMR (200 MHz, CDCl3) δ 7.26 – 7.18 (m, 1H), 6.96 – 6.75 (m, 3H), 4.64 (s, 2H), 3.95 (t, J =
6.5, 2H), 1.88 – 1.67 (m, 3H), 1.55 – 1.17 (m, 8H), 0.89 (t, J = 6.5, 3H).13C NMR (50 MHz, CDCl3) δ
159.62, 142.67, 129.74, 119.09, 114.01, 113.09, 68.18, 65.48, 31.99, 29.49, 29.27, 26.22, 22.82,
14.34. MS (APPI): m/z 222.2 [M+.].
1-(bromomethyl)-3-(heptyloxy)benzene (5b)
The product was obtained from (3-(heptyloxy)phenyl)methanol 4b using synthetic procedure 2.
Therefore product was used for the next reaction step without further purification. Yield 86%. Brown
solid. Rf = 0.71 (heptane:EtOAc 1:1). 1H NMR (200 MHz, CDCl3)7.32 – 6.27 (m, 4H), 4.76 (s, 2H),
3.92 (t, J = 6.4, 2H), 1.72-1.65(m, 2H), 1.34 – 1.15 (m, 8H), 0.89 (t, J = 6.5, 3H).13C NMR (50 MHz,
CDCl3) δ 159.41, 129.63, 119.81, 114.36, 105.23, 68.05, 32.65, 32.01, 29.53, 29.36, 26.24, 22.84,
14.31.

91
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
5-((3-(heptyloxy)benzyl)oxy)-2,2-dimethyl-4H-benzo[d][1,3]dioxin-4-one (13b)
The product was obtained from 5-hydroxy-2,2-dimethyl-4H-benzo[d][1,3]dioxin-4-one 1, and 1-
(bromomethyl)-3-(heptyloxy)benzene 5b using synthetic procedure 3. The product was purified using
column chromatography with heptane:EtOAc 16:1 (v/v) as eluent. Yield 58%. White solid. Rf = 0.29
(heptane:EtOAc 5:1).1H NMR (200 MHz, CDCl3) δ 7.42 – 7.35 (m, 2H), 7.25 (d, J = 7.5, 1H), 7.04
(d, J = 7.5, 1H), 6.83 (d, J = 7.9, 1H), 6.70 – 6.49 (m, 2H), 5.22 (s, 2H), 3.99 (t, J = 6.4, 2H), 1.87 –
1.68 (m, 2H) 1.56 (s, 6H), 1.42– 1.12 (m, 8H), 0.89 (t, J = 6.5, 3H). 13C NMR (50 MHz, CDCl3) δ
160.53, 159.77, 158.23, 158.01, 138.02, 136.48, 129.73, 118.57, 114.61, 112.43, 109.67, 107.36,
105.46, 70.74, 68.19, 32.00, 29.50, 29.29, 26.26, 25.85, 22.82, 14.30. MS (APPI): m/z 399.2 [M+H]+.
2-((3-(heptyloxy)benzyl)oxy)-6-hydroxybenzoic acid (24b)
The product was obtained from 5-((3-(heptyloxy)benzyl)oxy)-2,2-dimethyl-4H-benzo[d][1,3]dioxin-
4-one 13b using synthetic procedure 5. Yield quantitative. Yellow oil. Rf = 0.42 (heptane:EtOAc 1:1
containing 1% acetic acid).1H NMR (200 MHz, DMSO-d6) δ 7.26 – 7.19 (m, 2H), 7.08 (d, J = 7.5,
1H), 7.04 (d, J = 7.5, 1H), 6.79 (d, J = 8.11H), 6.31 (d, J = 8.2, 2H), 5.04 (s, 2H), 3.95 (t, J = 6.4, 2H),
1.73 - 1.63 (m, 2H), 1.36 – 1.18 (m, 8H), 0.87 (t, J = 7.0, 3H).13C NMR (50 MHz, DMSO-d6) δ
170.28, 164.46, 159.34, 158.66, 139.68, 130.82, 129.05, 118.77, 113.03, 110.05, 109.85, 102.77,
69.61, 67.26, 31.26, 28.75, 28.48, 25.55, 22.07, 13.96. HRMS: m/z [M−H]−calcd for C21H25O5
357.17075, found 357.17114.
(3-(nonyloxy)phenyl)methanol (4c)
The product was obtained using Williamson ether synthesis followed by aldehyde reduction of 1-
bromoheptane, and 3-hydroxybenzaldehyde using synthetic procedure 1. The product was used for the
next reaction step without further purification. Yield 89%. Rf = 0.54 (heptane:EtOAc 1:1). 1H NMR
(200 MHz, CDCl3) δ 7.22 (t, J = 7.8, 1H), 6.89 (s, 1H), 6.85 – 6.72 (m, 2H), 4.59 (s, 2H), 3.92 (t, J =
6.5, 2H), 2.32 (s, 1H), 1.79 – 1.69 (m, 2H), 1.42 – 1.28 (m, 12H), 0.88 (t, J = 6.3, 3H).13C NMR (50

92
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
MHz, CDCl3) δ 159.52, 142.67, 129.62, 119.04, 113.88, 113.02, 68.11, 65.25, 32.05, 29.72, 29.58,
29.44, 26.22, 22.84, 14.26. MS (APPI): m/z250.2 [M+].
1-(bromomethyl)-3-(nonyloxy)benzene (5c)
The product was obtained from (3-(nonyloxy)phenyl)methanol 4c using synthetic procedure 2.
Therefore the product was used for the next reaction step without further purification. Yield 78%.
Yellow oil. Rf = 0.71 (heptane:EtOAc 1:1).1H NMR (500 MHz, CDCl3) δ 7.22 (t, J = 7.8, 1H), 6.96 –
6.92 (m, 2H), 6.84 – 6.81 (m, 1H), 4.46 (s, 2H), 3.95 (t, J = 7.0, 2H), 1.87 - 1.75 (m, 2H), 1.47 - 1.28
(m, 12H), 0.89 (t, J = 6.8, 3H). 13C NMR (50 MHz, CDCl3) δ 159.33, 139.04, 129.73, 121,03, 115.03,
114.67, 68.02, 33.55, 31.86, 29.52, 29.38, 29.24, 26.02, 22.65, 14.09.
2,2-dimethyl-5-((3-(nonyloxy)benzyl)oxy)-4H-benzo[d][1,3]dioxin-4-one (13c)
The product was obtained from 5-hydroxy-2,2-dimethyl-4H-benzo[d][1,3]dioxin-4-one 1, and 1-
(bromomethyl)-3-(nonyloxy)benzene 5c using synthetic procedure 3. The product was purified using
column chromatography with heptane:EtOAc 16:1 (v/v) as eluent. Yield 88%. Yellow oil. Rf = 0.29
(heptane:EtOAc 5:1). 1H NMR (500 MHz, CDCl3) δ 7.41-7.35 (m, 2H), 7.25 - 7.20 (m, 2H), 7.04 (d,
J = 7.0, 1H), 6.82 (d, J = 6.9, 1H), 6.59 (d, J = 8.0, 1H), 5.21 (s, 2H), 3.99 (t, J = 6.0, 2H), 1.89 – 1.62
(m, 8H), 1.51 – 1.13 (m, 12H), 0.87 (d, J = 6.3, 3H).13C NMR (50 MHz, CDCl3) δ 160.46, 159.71,
157.95, 137.97, 136.45, 129.66, 118.51, 114.55, 112.37, 109.62, 107.30, 105.39, 103.97, 70.68, 68.13,
32.04, 29.71, 29.60, 29.43, 26.26, 25.79, 22.83, 14.28. MS (ESI): m/z 427.3 [M+H]+.
2-hydroxy-6-((3-(nonyloxy)benzyl)oxy)benzoic acid (24c)
The product was obtained from 5-((3-(heptyloxy)benzyl)oxy)-2,2-dimethyl-4H-benzo[d][1,3]dioxin-
4-one 13c using synthetic procedure 5. The product was obtained in high purity and no further
purification was required. Yield 58%. Yellow oil. Rf = 0.60 (heptane:EtOAc 1:1 containing 1% acetic
acid). 1H NMR (200 MHz, CD3OD) δ 7.27 - 7.20 (m, 2H), 7.11 - 7.01 (m, 2H), 6.81 (d, J = 7.8, 1H),
6.49 (d, J = 7.5, 2H), 5.16 (s, 2H), 3.97 (t, J = 6.2, 2H), 1.75 – 1.71 (m, 2H), 1.41 – 1.23 (m, 12H),

93
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
0.90 (t, J = 6.5, 3H).13C NMR (50 MHz, CD3OD) δ 185.96, 161.65, 156.96, 141.16, 131.25, 121.03,
115.76, 115.03, 107.29, 94.05, 72.58, 69.81, 33.92, 31.56, 31.43, 31.30, 28.07, 24.59, 15.30. HRMS:
m/z [M−H]−calcd for C23H29O5 385.20205, found 385.20261.
2,2-dimethyl-5-(nonyloxy)-4H-benzo[d][1,3]dioxin-4-one (11)
The product was obtained from 5-hydroxy-2,2-dimethyl-4H-benzo[d][1,3]dioxin-4-one 1, and 1-
bromononane using synthetic procedure 3. The product was purified using column chromatography
with heptane as eluent followed by heptane:EtOAc 50:1 (v/v) as eluent until the starting material was
completely removed from the column and the product was obtained by eluting the column with
EtOAc as eluent. Yield 53%. Light yellow solid. Rf = 0.64 (heptane:EtOAc 1:1). 1H NMR (200 MHz,
CDCl3) δ 7.40 (t, J = 8.4, 1H), 6.59 (d, J =, 8.1, 1H), 6.51 (d, J =, 8.2, 1H) 4.06 (t, J = 6.7, 2H), 1.95
– 1.81 (m, 2H), 1.69 (s, 6H), 1.50 – 1.26 (m, 12H), 0.85 (t, J = 6.6, 3H).13C NMR (50 MHz, CDCl3) δ
161.30, 158.20, 157.96, 136.41, 109.03, 106.62, 105.28, 103.67, 69.60, 32.07, 29.69, 29.55, 29.46,
29.14, 26.03, 25.84, 22.86, 14.31. MS (APPI): m/z 321.0 [M+H]+.
2-hydroxy-6-(nonyloxy)benzoic acid (22)
The product was obtained from 5-((3-(heptyloxy)benzyl)oxy)-2,2-dimethyl-4H-benzo[d][1,3]dioxin-
4-one 11 using synthetic procedure 5. The product was obtained in high purity and no further
purification was required. Yield quantitative. Light brown solid. Rf = 0.60 (heptane:EtOAc 1:1
containing 1% acetic acid). 1H NMR (200 MHz, DMSO-d6) δ 7.02 (t, J = 8.2, 1H), 6.28 (d, J = 8.0,
1H), 6.24 (d, J = 8.1, 1H), 3.87 (t, J = 6.5, 2H), 1.68 – 1.58 (m, 2H), 1.40 – 1.17 (m, 12H), 0.86 (t, J =
6.5, 3H).13C NMR (50 MHz, DMSO-d6) δ 164.31, 160.21, 131.19, 110.51, 109.82, 103.07, 69.00,
31.74, 29.48, 29.34, 29.14, 25.92, 22.54, 14.40. HRMS: m/z [M−H]−calcd for C16H23O4 279.16018,
found 279.16061.
1-(4-bromophenyl)decan-1-one (7)
The product was obtained using Friedel-Craft acylation of bromobenzene 6 with decanoylchloride.
AlCl3 (0.81 g, 6.1 mmol) was added slowly to a solution of bromobenzene (0.54 mL, 5.1 mmol) and

94
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
decanoylchloride (1.2 mL, 5.8 mmol) in 20 mL CH2Cl2 in a 50 mL flask at 0 °C. AlCl3 slowly
dissolved in the mixture and the reaction mixture was stirred for 1 hour at 60 °C. The reaction mixture
was poured into ice-water (25 mL) and extracted with CH2Cl2 (2 x 30 mL). The combined organic
layers were dried over brine and concentrated in vacuo. The product was dissolved in CH2Cl2 and
washed with 2 N aqueous HCl (2 x 15 mL) and dried over brine (15 mL) and concentrated in vacuo.
The product was crystallized from heptane in a yield 62% (0.49 mg, 1.5 mmol) as a white solid. Rf =
0.69 (heptane:EtOAc 1:1). 1H NMR (200 MHz, CDCl3) δ 7.89 – 7.75 (m, 2H), 7.65 – 7.53 (m, 2H),
2.92 (t, J = 7.5, 2H), 1.75 – 1.64 (m, 2H), 1.32 – 1.27 (m, 12H), 0.87 (t, J = 6.4, 3H).13C NMR (50
MHz, CDCl3) δ 199.72, 136.01, 132.06, 129.82, 128.19, 38.81, 32.08, 29.67, 29.54, 29.49, 24.50,
22.88, 14.32. MS (APPI): m/z 311.2 [M+].
1-bromo-4-decylbenzene (8)
The product was obtained using a Wolff-Kishner reaction to reduce 1-(4-bromophenyl)decan-1-one 7.
1-(4-bromophenyl)decan-1-one 7 (1.3 g, 4.2 mmol), hydrazine monohydrate (1.2 mL, 16 mmol) and
KOH (1.8 g, 32 mmol) were dissolved in 25 mL of 1-octanol to yield a green suspension in a two-
necked 50 mL flask. The reaction mixture was stirred at reflux at 135 °C for 2¾ hours to give an
almost colorless solution. The reaction mixture was then cooled to room temperature and diluted with
ether (50 mL), washed with 1 N aqueous HCl (50 mL), 2 N aqueous HCl (20 mL), brine (2 times, 30
mL), dried over MgSO4, filtered, concentrated in vacuo. 1-octanol was removed by vacuum
distillation to yield about 2.7 g of the crude product. The product was purified using column
chromatography with heptane as eluent. Yield 40%. Colorless oil. Rf = 0.61 (100% heptane). 1H NMR
(500 MHz, CDCl3) δ 7.37 (d, J = 8.3, 2H), 7.03 (d, J = 8.1, 2H), 2.54 (t, J = 7.7, 2H), 1.60 – 1.54 (m,
2H), 1.28 – 1.25 (m, 14H), 0.88 (t, J = 6.8, 3H).13C NMR (50 MHz, CDCl3) δ 142.05, 131.46, 130.37,
119.47, 35.59, 32.14, 31.56, 29.85, 29.82, 29.71, 29.57, 29.43, 22.93, 14.34. MS (APPI): m/z 296.2
[M+].
((4-decylphenyl)ethynyl)trimethylsilane
Into a dried 10-20 mL microwave vial, PdCl2(PPh3) (52 mg, 74 µmol), CuI (18 mg, 95 µmol), and
PPh3 (80 mg, 0.30 mmol) were suspended in CH3CN (2 mL) under nitrogen atmosphere.
Subsequently, 1-bromo-4-decylbenzene 8 (0.46 g, 1.5 mmol), diethyl amine (Et2NH) (2.4 mL, 23
mmol), and trimethylsilyl acetylene (TMSA) (0.24 mL, 1.7 mmol), were added to the mixture. The
mixture was heated at 120 °C for 35 minutes via microwave irradiation (95 W). The reaction mixture

95
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
was diluted with EtOAc (50 mL) and washed with water (3 x 50 mL). The water layer was washed
with EtOAc (2 x 50 mL). The combined organic phases were extracted with brine (1 x 50 mL) and
dried over Mg2SO4. The solvent was evaporated under reduced pressure and the residue was purified
by column chromatography with heptane as eluent. Yield 65%. Brown oil. Rf = 0.41 (100% heptane). 1H NMR (500 MHz, CDCl3) δ 7.37 (d, J = 8.0, 2H), 7.10 (d, J = 7.9, 2H), 2.58 (t, J = 7.6, 2H), 1.58 –
1.54 (m, 2H), 1.28 – 1.25 (m, 14H), 0.88 (t, J = 6.8, 3H), 0.24 (s, 9H);13C NMR (50 MHz, CDCl3) δ
143.89, 132.08, 128.52, 120.44, 105.63, 93.45, 36.11, 32.12, 31.42, 29.82, 29.69, 29.55, 29.42, 22.91,
14.34, 0.26. MS (APPI): m/z 314.2 [M+].
1-decyl-4-ethynylbenzene (9)
((4-decylphenyl)ethynyl)trimethylsilane (0.29 g, 0.91 mmol) was dissolved in THF (5 mL) and the
solution was cooled to 0 oC. Tetra butyl ammonium fluoride (TBAF) (1.4 mL, 1.4 mmol) in THF (1
M, 2.6 mL) was added dropwise and the reaction mixture was stirred for 10 minutes. The reaction
mixture was diluted with EtOAc (50 mL), extracted with water (4 x 50 mL) and washed with brine (2
x 50 mL). The organic phase was dried over MgSO4 and filtered. The solvent was evaporated and the
product was used without further purification. Yield 93%. Brown oil. Rf = 0.45 (100% heptane). 1H
NMR (500 MHz, CDCl3) δ 7.40 (d, J = 8.0, 2H), 7.13 (d, J = 7.8, 2H), 3.02 (s, 1H), 2.59 (t, J = 7.6,
2H), 1.59 – 1.55 (m, 2H), 1.29 – 1.26 (m, 14H), 0.88 (t, J = 6.8, 2H).13C NMR (50 MHz, CDCl3) δ
144.22, 132.25, 128.63, 119.41, 105.22, 92.67, 36.12, 32.12, 31.45, 29.82, 29.79, 29.69, 29.55, 29.46,
22.91, 14.34. MS (APPI): m/z 242.2 [M+.].
Benzyl 2,4-bis(benzyloxy)-6-((4-decylphenyl)ethynyl)benzoate
The product was obtained using Sonogashira coupling reaction of 2,4-bis(benzyloxy)-6-
(((trifluoromethyl)sulfonyl)oxy)benzoate 10, and 1-decyl-4-ethynylbenzene 9. Distilled diethylamine
(0.11 mL, 1.05 mmol) and 1-decyl-4-ethynylbenzene 9 (0.18 g, 0.74 mmol) were subsequently added
to a solution of the triflate 10 (0.36 g, 0.63 mmol), CuI (9.0 mg, 47 µmol), and PdCl2(PPh3)2 (18 mg,
0.026 mmol) in degassed anhydrous acetonitrile (1.0 mL) under nitrogen atmosphere. The mixture
was subjected to microwave irradiation for 35 min at 120 °C (70 W). The reaction mixture was
diluted with ether (50 mL) and washed with demi water (2 x 30 mL), brine (2 x 50 mL), dried over
MgSO4 and filtered. The solvent was evaporated under reduced pressure, and the residue was purified

96
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
by column chromatography with heptanes/EtOAc 5:1 (v/v) as eluent. Yield 80%. Brown oil. Rf = 0.40
(heptane:EtOAc 1:1). 1H NMR (500 MHz, CDCl3) δ 7.43 – 7.27 (m, 15H), 7.22 (d, J = 8.0, 2H), 7.12
(d, J = 7.9, 2H), 6.75 (s, 1H), 6.56 (s, 1H), 5.37 (s, 2H), 5.06 (s, 2H), 5.04 (s, 2H), 2.60 (t, J = 7.5,
2H), 1.60- 1.55 (m, 2H), 1.30- 1.26 (m, 14H), 0.88 (t, J = 6.6, 3H).13C NMR (50 MHz, CDCl3) δ
166.91, 160.54, 157.24, 144.06, 136.47, 136.38, 136.09, 131.89, 128.89, 128.76, 128.61, 128.48,
128.44, 128.17, 128.12, 127.75, 127.30, 123.86, 120.06, 119.89, 109.67, 101.97, 93.47, 86.28, 70.80,
70.53, 67.30, 36.15, 32.12, 31.47, 29.81, 29.71, 29.55, 29.46, 22.91, 14.34. MS (APPI): m/z 665.2
[M+H+].
2-(4-decylphenethyl)-4,6-dihydroxybenzoic acid (20)
Benzyl 2,4-bis(benzyloxy)-6-((4-decylphenyl)ethynyl)benzoate (0.28 g, 0.42 mmol) was dissolved in
EtOAc (due to low solubility in MeOH) and added to 10 mol% Pd/C (10%) (45 mg, 42 nmol). The
suspension was shaken with 3 atm H2-pressure in a Parr apparatus at 45 oC for 24 hours.
Subsequently, the mixture was filtered through Celite. The solvent was evaporated under reduced
pressure and the residue was purified by column chromatography with heptane/EtOAc 9:1 (v/v) as
eluent, followed by EtOAc as eluent to obtain the desired product. Yield 72%. White solid. Rf = 0.40
(heptane:EtOAc 1:1 + acetic acid). 1H NMR (200 MHz, CD3OD) δ 7.12 – 7.02 (m, 4H), 6.18 (d,
J=2.4 Hz, 1H); 6.16 (d, J= 2.4 Hz, 1H), 3.18 – 3.10 (m, 2H), 2.83 – 2.75 (m, 2H), 2.62 – 2.48 (m,
2H), 1.60 – 1.50 (m, 2H), 1.28 – 1.20 (m, 14H), 0.89 (t, J = 6.5, 3H).13C NMR (50 MHz, CD3OD) δ
167.82, 164.57, 150.02, 142.23, 141.60, 130.15, 112.86, 106.44, 102.82, 41.15, 40.02, 37.41, 33.93,
33.67, 31.59, 31.47, 31.31, 31.19, 24.59, 15.30. HRMS: m/z [M−H]−calcd for C25H33O4 397.23843,
found 397.2388.
2-(benzo[d]oxazol-2-yl)phenol (26)
Salicylic acid (0.138 g, 1.0 mmol) was mixed with 2-aminophenol (0.11 g, 1.0 mmol) and 3 mL of
polyphosphoric acid was added to the mixture. The mixture was heated in oil bath at 180 °C under N2
for 6.5 hours. The reaction mixture was cooled to room temperature and subsequently poured into 150
mL demi water. The mixture was extracted with ethylacetate (2 x 30 mL). The combined organic
layers were washed with demiwater (2 x 40 mL), brine (1 x 40mL), dried with MgSO4, filtered and
concentrated in vacuo. The product was obtained after crystallization or purification using column

97
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
chromatography with heptane/EtOAc 50:1 (v/v) as eluent. Yield 47%. Colorless to white crystals. Rf
= 0.69 (heptane:EtOAc 1:1). 1H NMR (200 MHz, DMSO) δ 8.04 (dd, J = 7.9, 1.7, 1H), 7.83-7.89 (m,
2H), 7.53-7.58 (m, 1H), 7.45-7.50 (m, 2H), 7.06-7.16 (m, 2H).13C NMR (50 MHz, DMSO) 158.17,
151.40, 149.24, 139.90, 134.41, 128.04, 126.31, 125.77, 120.43, 119.64, 117.63, 111.50, 110.84.
HRMS: m/z [M+H]+ calcd for C13H10O2N1 212.07061, found 212.07051.
Human 5-LOX inhibition screening UV Assay
Enzyme inhibition was measured by the residual enzyme activity after 10 minutes incubation with the
inhibitor at room temperature. The enzyme activity was determined by conversion of lipoxygenase
substrate linoleic acid into hydroperoxy-octadecadienoate (HPOD). The conversion rate was followed
by UV absorbance of the conjugated diene at 234 nm (ε = 25000 M-1cm-1) over a period of 20
minutes. The UV absorbance increase over time was used to determine the enzyme activity.
Tris buffer (50 mM) pH 7.5 containing 2 mM EDTA and 2 mM CaCl2 was used as an assay buffer
for human 5-LOX experiments. The human 5-LOX enzyme was diluted 1:4000 with the assay buffer.
The inhibitor (100 mM in DMSO) was diluted with the assay buffer to 1 mM. The substrate, linoleic
acid was diluted with EtOH to 20 mM. Subsequently, 1 mL of enzyme solution (1:4000) was mixed
with 100 µL ATP (2 mM), 100 µL inhibitor (1 mM) and 790 µL Tris buffer and incubated for 10
minutes. Subsequently, the linoleic acid solution (10 µL, 20 mM) was added to a mixture and the
conversion rate of the substrate was measured after 10 second mixing of the enzyme with the
substrate. The reaction rate in the absence of the inhibitor was used as positive control. In the positive
control experiment, the assay buffer was supplemented with a little amount of DMSO (3 μL of DMSO
in 1.5 mL) in order to replace the DMSO inhibitor solution, which was also pre-incubated for 10
minutes.
COX-2 inhibition screening assay
The COX-2 activity was measured spectrophotometrically by measuring the formation of oxidized
N,N,N',N'-tetramethyl-p-phenylenediamine (TMPD). Tris buffer (0.1M) pH 8.0 was used as an assay
buffer for COX-2 experiments. The COX-2 enzyme was diluted 1:1000 with the assay buffer.
Hematin (7.5 mM in DMSO) was diluted with assay buffer to 330 µM. The inhibitor was diluted to
1.1 mM in DMSO. The substrate, arachidonic acid was diluted by adding 50 µL KOH 0.1 M and 800
µL H2O into 150 µL of a 22 mM arachidonic acid solution in EtOH. The colorimetric substrate
TMPD (2.64 mM in H2O) was prepared freshly prior to the experiment. The substrate, linoleic acid
was diluted with EtOH to 20 mM. Subsequently, in a 96 wells plate, 150 µL assay buffer, 10 µL
hematin 330 µM, 10 µL COX-2 enzyme (1:1000) was added. 10 µL inhibitor solution (1.1 mM in
DMSO) was added to the inhibitor well and 10 µL of DMSO was added to the positive control wells.
The plate was incubated for 5 minutes at room temperature. 20 µL of freshly prepared TMPD was
added followed by the addition of 20 µL arachidonic acid. Arachidonic acid addition was excluded for

98
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
the negative control wells. The plate was incubated for another 5 mintes at room temperature. The
absorbance was measured at 550 nm, and the calculations were performed with Excel 2010. The
experiments were performed in triplicate, and the presented results were the average of three
measurements with the standard deviation.
Michaelis Menten Enzyme Kinetics
The enzyme kinetics of human 5-LOX were also studied by the formation of the conjugated diene
product at 234 nm (ε = 25000 M-1cm-1) using the same experimental setup as for the concentration
dependent and the inhibitor screening. Enzyme kinetics were performed against linoleic acid and ATP
to get a complete indication about the regulation of human 5-LOX enzyme activity.
In the kinetic study of the human 5-LOX against ATP, the ATP concentration was varied between
12.5-200 µM while the concentration of linoleic acid was fixed (100 µM). In the kinetic study of the
human 5-LOX against the linoleic acid, substrate concentration was varied between 37.5–300 µM.
The enzyme activity was measured in the absence or presence of fixed concentrations of activator 23a
(0 µM, 12.5 µM, and 25 µM) or inhibitor 23d (0 µM, 25 µM, and 50 µM). The reaction velocities (v),
which are the concentration changes over time, were plotted against the substrate concentrations in
Michaelis-Menten plots and the Km and Vmax and the apparent values (Kmapp and Vmax
app) in the
presence of the activator were derived. The results from the non-linear curve fitting were in line with
the results from Lineweaver-Burk plot.
The slopes and y-intercepts from the Lineweaver-Burk plot of h-5-LOX activation was derived and
then re-plotted as 1/Δslope or 1/Δy-intercept versus 1/[activator]. From these plots α, β and KA values
were derived as described by Leskovac44 for non-essential activation. According to this method in the
re-plot of 1/Δy-intercept versus 1/[activator] the y intercept corresponds to β (Vmax/(β-1), whereas in
the re-plot of 1/Δslope versus 1/[activator] the y intercept correspond to β Vmax/Km(β-α). The x
intersection point of two lines from the plot of 1/Δy-intercept and 1/Δslope is –β/αKA, which can be
employed to derive the KA value if α and β are known. All the experiments were performed in
triplicate and the average triplicate values and their standard deviations are plotted. Calculations were
performed with Excel 2010.
Docking Studies
Protein and ligand structures
Our docking models were predicted based on the co-crystal structure between stable-5-LOX and
arachidonic acid (PDB 3V99). The model of stable-5-LOX was further refined using the related PDB
structures 3V98 and 3V92 to model the carboxylic acid of the C-terminal Ile673 (missing in 3V99),
which coordinates the catalytic iron in the active site.

99
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
Structures of compounds 23a, 23b, 23c and 23d were built using Omega2 from OpenEye. 53 We
fixed between the benzyl ring and the carboxylic acid group on all molecules using AutoDockTools to
dock structures that kept a planar geometry for the benzyl ring and carboxylic acid.54
Docking and scoring
The receptor structure was prepared for docking using the prepare_receptor4.py script from
AutoDockTools. Compounds were docked using smina55 with the default vina scoring function. The
docked poses were rescored using the '--score_only' command in smina and then reranked. The top
ranked poses were used for further analysis.
Cytotoxicity assay
The Raw Blue cells (~7,500) were seeded in DMEM (100 µL) in 96 wells plate. For the blank, no
cells were seeded. After 24 hours incubation at 37°C, the medium was removed and replaced by 100
µL of fresh medium containing different concentrations of the compounds (1.6 – 200 µM). Then after
the plate was incubated overnigth at 37°C, 20 µL per well of filter-sterilized MTS solution containing
5% v/v phenazine methosulphate (PMS) (obtained from Promega) was added to give the final
concentration of MTS and PMS of respectively 333 µg/mL and 25 µM. The absorbance was measured
using a ThermoMax microplate reader at 490 nm after 6 hours of incubation at 37°C.
Modulation of NF-kB pathway
The influences of the compounds on the NF- B pathway were studied using the Raw Blue Cells assay
from the InvivoGen. Raw Blue cells, which are derived from the macrophages, stably express a NF-
B induced gene, secreting embryonic alkaline phosphatase (SEAP). The change in the NF- B
pathway was determined by measuring the concentration of secreted SEAP in the medium using
Quanti-Blue. The measurements were performed following the provided protocols with some
adjustments.
The cells were seeded in the 96 wells plate with ~100,000 cells per well in 200 µL Dulbecco's
Modified Eagle Medium (DMEM) containing 0.1% (v/v) zeocin and the plate was incubate overnight
at 37°C. For the blank, no cells were seeded. The medium was removed and replaced by 180 µL of
fresh medium containing 3 µM of the modulator compounds 23a or 23d. After 6 hour incubation at
37°C, 20 µL of NF- B stimulant (IFN- ) was added. Empty medium was used as the control. The
plate was incubated overnight at 37°C. 150 µL of the incubated medium were transferred into a flat-
bottom 96 wells plate containing 50 µL Quati-blue suspension. The plate was incubated for another 6
hours at 37°C before the absorbance was measured using a ThermoMax microplate reader at 650 nm.
The experiments were performed in triplicate, and the presented results were the average of three
independent experiments with the standard deviation.

100
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
References
1. Martel-Pelletier, J.; Lajeunesse, D.; Reboul, P.; Pelletier, J. Therapeutic role of dual inhibitors of 5-LOX
and COX, selective and non-selective non-steroidal anti-inflammatory drugs. Ann. Rheum. Dis. 2003, 62,
501-509.
2. Gillmor, S. A.; Villasenor, A.; Fletterick, R.; Sigal, E.; Browner, M. F. The structure of mammalian 15-
lipoxygenase reveals similarity to the lipases and the determinants of substrate specificity. Nat. Struct.
Biol. 1997, 4, 1003-1009.
3. Sigal, E. The molecular biology of mammalian arachidonic acid metabolism. Am. J. Physiol. 1991, 260,
L13-L28.
4. Epp, N.; Fürstenberger, G.; Müller, K.; de Juanes, S.; Leitges, M.; Hausser, I.; Thieme, F.; Liebisch, G.;
Schmitz, G.; Krieg, P. 12R-lipoxygenase deficiency disrupts epidermal barrier function. J. Cell Biol. 2007,
177, 173-182.
5. Ikei, K. N.; Yeung, J.; Apopa, P. L.; Ceja, J.; Vesci, J.; Holman, T. R.; Holinstat, M. Investigations of
human platelet-type 12-lipoxygenase: role of lipoxygenase products in platelet activation. J. Lipid Res.
2012, 53, 2546-2559.
6. Hunter, J. A.; Finkbeiner, W. E.; Nadel, J. A.; Goetzl, E. J.; Holtzman, M. J. Predominant generation of
15-lipoxygenase metabolites of arachidonic acid by epithelial cells from human trachea. Proc. Natl. Acad.
Sci. 1985, 82, 4633-4637.
7. Nadel, J. A.; Conrad, D. J.; Ueki, I. F.; Schuster, A.; Sigal, E. Immunocytochemical localization of
arachidonate 15-lipoxygenase in erythrocytes, leukocytes, and airway cells. J. Clin. Invest. 1991, 87, 1139-
1145.
8. Brash, A. R.; Boeglin, W. E.; Chang, M. S. Discovery of a second 15S-lipoxygenase in humans. Proc.
Natl. Acad. Sci. 1997, 94, 6148-6152.
9. Brown, C. D.; Kilty, I.; Yeadon, M.; Jenkinson, S. Regulation of 15-lipoxygenase isozymes and mucin
secretion by cytokines in cultured normal human bronchial epithelial cells. Inflamm. Res. 2001, 50, 321-
326.
10. Chanez, P.; Bonnans, C.; Chavis, C.; Vachier, I. 15-Lipoxygenase: a Janus enzyme? Am. J. Respir. Cell
Mol. Biol. 2002, 27, 655-658.
11. Chu, H. W.; Balzar, S.; Westcott, J. Y.; Trudeau, J. B.; Sun, Y.; Conrad, D. J.; Wenzel, S. E. Expression
and activation of 15-lipoxygenase pathway in severe asthma: relationship to eosinophilic phenotype and
collagen deposition. Clin. Exp. Allergy 2002, 32, 1558-1565.
12. Shannon, V. R.; Chanez, P.; Bousquet, J.; Holtzman, M. J. Histochemical Evidence for Induction of
Arachidonate 15-Lipoxygenase in Airway Disease. Am. Rev. Respir. Dis. 1993, 147, 1024-1028.
13. Piao, Y.; Du, Y.; Oshima, H.; Jin, J.; Nomura, M.; Yoshimoto, T.; Oshima, M. Platelet-type 12-
lipoxygenase accelerates tumor promotion of mouse epidermal cells through enhancement of cloning
efficiency. Carcinogenesis 2008, 29, 440-447.
14. Rásó, E.; Döme, B.; Somlai, B.; Zacharek, A.; Hagmann, W.; Honn, K.V.; Tímár, J.; Molecular
identification, localization and function of platelet-type 12-lipoxygenase in human melanoma progression,
under experimental and clinical conditions. Melanoma Res. 2004, 14, 245-250.
15. Gupta, S.; Srivastava, M.; Ahmad, N.; Sakamoto, K.; Bostwick, D. G.; Mukhtar, H. Lipoxygenase-5 is
overexpressed in prostate adenocarcinoma. Cancer 2001, 91, 737-743.
16. Melstrom, L. G.; Bentrem, D. J.; Salabat, M. R.; Kennedy, T. J.; Ding, X.; Strouch, M.; Rao, S. M.; Witt,
R. C.; Ternent, C. A.; Talamonti, M. S.; Bell, R. H.; Adrian, T. A. Overexpression of 5-Lipoxygenase in
Colon Polyps and Cancer and the Effect of 5-LOX Inhibitors In vitro and in a Murine Model. Clin. Cancer
Res. 2008, 14, 6525-6530.
17. Nathoo, N.; Prayson, R.A.; Bondar, J.; Vargo, L.; Arrigain, S.; Mascha, E.J.; Suh J.H.; Barnett,
G.H.; Golubic, M. Increased Expression of 5-Lipoxygenase in High-Grade Astrocytomas. Neurosurgery
2006, 58, 347-354.
18. Wilborn, J.; Bailie, M.; Coffey, M.; Burdick, M.; Strieter, R.; Peters-Golden, M. Constitutive activation of 5-
lipoxygenase in the lungs of patients with idiopathic pulmonary fibrosis. J. Clin. Invest. 1996, 97, 1827-
1836.

101
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
19. Zou, L. Y. Tumor 5-Lipoxygenase Expression Correlates with Gastric Cancer Metastasis and Its Selective
Inhibitor Induces Cancer Cell Apoptosis. J. Cancer Mol. 2006, 2, 227.
20. Serhan, C. N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: dual anti-inflammatory and pro-
resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349.
21. Edenius, C.; Haeggström, J.; Lindgren, J. Å. Transcellular conversion of endogenous arachidonic acid to
lipoxins in mixed human platelet-granulocyte suspensions. Biochem. Biophys. Res. Commun. 1988, 157,
801-807.
22. Hashimoto, A.; Hayashi, I.; Murakami, Y.; Sato, Y.; Kitasato, H.; Matsushita, R.; Iizuka, N.; Urabe, K.;
Itoman, M.; Hirohata, S.; Endo, H. Antiinflammatory mediator lipoxin A4 and its receptor in synovitis of
patients with rheumatoid arthritis. J. Rheumatol. 2007, 34, 2144-2153.
23. Sobrado, M.; Pereira, M.P.; Ballesteros, I.; Hurtado, O.; Fernández-López, D.; Pradillo, J.M.; Caso,
J.R.; Vivancos, J.; Nombela, F.; Serena, J.; Lizasoain, I.; Moro, M.A. Synthesis of lipoxin A4 by 5-
lipoxygenase mediates PPARγ-dependent, neuroprotective effects of rosiglitazone in experimental stroke.
J. Neurosci. 2009, 29, 3875.
24. Smith, W. L.; DeWitt, D.L., Garavito, R.M. Cyclooxygenases: structural, cellular, and molecular biology.
Annu. Rev. Biochem. 2000, 69, 145.
25. Anderson, G. D.; Hauser, S. D.; McGarity, K. L.; Bremer, M. E.; Isakson, P. C.; Gregory, S. A. Selective
inhibition of cyclooxygenase (COX)-2 reverses inflammation and expression of COX-2 and interleukin 6
in rat adjuvant arthritis. J. Clin. Invest. 1996, 97, 2672-2679.
26. Nielsen, O. H.; Ahnfelt-Ronne, I.; Elmgreen, J. Abnormal metabolism of arachidonic acid in chronic
inflammatory bowel disease: enhanced release of leucotriene B4 from activated neutrophils. Gut 1987, 28,
181-185.
27. Reuter, B. K.; Asfaha, S.; Buret, A.; Sharkey, K. A.; Wallace, J. L. Exacerbation of inflammation-
associated colonic injury in rat through inhibition of cyclooxygenase-2. J. Clin. Invest. 1996, 98, 2076-
2085.
28. Hendel, J.; Nielsen, O. H. Expression of cyclooxygenase-2 mRNA in active inflammatory bowel disease.
Am. J. Gastroenterol. 1997, 92, 1170-1173.
29. Kargman, S. L.; O'Neill, G. P.; Vickers, P. J.; Evans, J. F.; Mancini, J. A.; Jothy, S. Expression of
prostaglandin G/H synthase-1 and -2 protein in human colon cancer. Cancer Res. 1995, 55, 2556-2559.
30. Cianchi, F.; Cortesini, C.; Bechi, P.; Fantappiè, O.; Messerini, L.; Vannacci, A.; Sardi, I.; Baroni, G.;
Boddi, V.; Mazzanti, R.; Masini, E. Up-regulation of cyclooxygenase 2 gene expression correlates with
tumor angiogenesis in human colorectal cancer. Gastroenterology 2001, 121, 1339-1347.
31. Wisastra, R.; Ghizzoni, M.; Boltjes, A.; Haisma, H. J.; Dekker, F. J. Anacardic acid derived salicylates are
inhibitors or activators of lipoxygenases. Bioorg. Med. Chem. 2012, 20, 5027-5032.
32. Brash, A. R. Lipoxygenases: Occurrence, Functions, Catalysis, and Acquisition of Substrate. J. Biol.
Chem. 1999, 274, 23679-23682.
33. Ghizzoni, M.; Boltjes, A.; Graaf, C. d.; Haisma, H. J.; Dekker, F. J. Improved inhibition of the histone
acetyltransferase PCAF by an anacardic acid derivative. Bioorg. Med. Chem. 2010, 18, 5826-5834.
34. Ghizzoni, M.; Wu, J.; Gao, T.; Haisma, H. J.; Dekker, F. J.; George Zheng, Y. 6-alkylsalicylates are
selective Tip60 inhibitors and target the acetyl-CoA binding site. Eur. J. Med. Chem. 2012, 47, 337-344.
35. Uchiyama, M.; Ozawa, H.; Takuma, K.; Matsumoto, Y.; Yonehara, M.; Hiroya, K.; Sakamoto, T.
Regiocontrolled Intramolecular Cyclizations of Carboxylic Acids to Carbon-Carbon Triple Bonds
Promoted by Acid or Base Catalyst. Org. Lett. 2006, 8, 5517-5520.
36. Harrowven, D. C.; Woodcock, T.; Howes, P. D. Total Synthesis of Cavicularin and Riccardin C:
Addressing the Synthesis of an Arene That Adopts a Boat Configuration. Angew. Chem. Int. Ed. Engl.
2005, 44, 3899-3901.
37. Sista, P.; Nguyen, H.; Murphy, J. W.; Hao, J.; Dei, D. K.; Palaniappan, K.; Servello, J.; Kularatne, R. S.;
Gnade, B. E.; Xue, B.; Dastoor, P. C.; Biewer, M. C.; Stefan, M. C. Synthesis and Electronic Properties of
Semiconducting Polymers Containing Benzodithiophene with Alkyl Phenylethynyl Substituents.
Macromolecules 2010, 43, 8063-8070.
38. Tranchimand, S.; Tron, T.; Gaudin, C.; Iacazio, G. First Chemical Synthesis of Three Natural Depsides
Involved in Flavonol Catabolism and Related to Quercetinase Catalysis. Synth. Comm. 2006, 36, 587-597.

102
Discovery of a novel activator of 5-lipoxygenase from
an anacardic acid-derived compound collection
39. Back, D. F.; Manzoni de Oliveira, G.; Ballin, M. A.; Corbellini, V. A. Complexes of vanadyl and uranyl
ions with a benzoxazole derivative: Synthesis, structural features and remarks on luminescence properties.
Inorg. Chim. Acta 2010, 363, 807-812.
40. Gibian, M. J.; Galaway, R. A. Steady-state kinetics of lipoxygenase oxygenation of unsaturated fatty acids.
Biochemistry. 1976, 15, 4209-4214.
41. Ha, T. J.; Kubo, I. Lipoxygenase Inhibitory Activity of Anacardic Acids. J. Agric. Food Chem. 2005, 53,
4350-4354.
42. Copeland, R. A.; Williams, J. M.; Giannaras, J.; Nurnberg, S.; Covington, M.; Pinto, D.; Pick, S.;
Trzaskos, J. M. Mechanism of selective inhibition of the inducible isoform of prostaglandin G/H synthase.
Proc. Natl. Acad. Sci. 1994, 91, 11202-11206.
43. Verhagen, J.; Vliegenthart, J. F.; Boldingh, J. Micelle and acid-soap formation of linoleic acid and 13-L-
hydroperoxylinoleic acid being substrates of lipoxygenase-1. Chem. Phys. Lipids 1978, 22, 255-259.
44. Leskovac, V. Comprehensive Enzyme Kinetics. 2004; pp 111-116.
45. Gilbert, N. C.; Bartlett, S. G.; Waight, M. T.; Neau, D. B.; Boeglin, W. E.; Brash, A. R.; Newcomer, M. E.
The Structure of Human 5-Lipoxygenase. Science 2011, 331, 217-219.
46. Gilbert, N. C.; Rui, Z.; Neau, D. B.; Waight, M. T.; Bartlett, S. G.; Boeglin, W. E.; Brash, A. R.;
Newcomer, M. E. Conversion of human 5-lipoxygenase to a 15-lipoxygenase by a point mutation to mimic
phosphorylation at Serine-663. FASEB J. 2012, 26, 3222-3229.
47. PyMOL The PyMOL Molecular Graphics System. PyMOL 2010, 1.3, .
48. Pettersen, E. F.; Goddard, T. D.; Huang, C. C.; Couch, G. S.; Greenblatt, D. M.; Meng, E. C.; Ferrin, T. E.
UCSF Chimera? A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25,
1605-1612.
49. Lee, K. S.; Kim, S. R.; Park, H. S.; Park, S. J.; Min, K. H.; Lee, K. Y.; Jin, S. M.; Lee, Y. C. Cysteinyl
leukotriene upregulates IL-11 expression in allergic airway disease of mice. J. Allergy Clin. Immunol.
2007, 119, 141-149.
50. Okamoto, F.; Saeki, K.; Sumimoto, H.; Yamasaki, S.; Yokomizo, T. Leukotriene B4 Augments and
Restores FcγRs-dependent Phagocytosis in Macrophages. J. Biol. Chem. 2010, 285, 41113-41121.
51. Thompson, C.; Cloutier, A.; Bossé, Y.; Poisson, C.; Larivée, P.; McDonald, P. P.; Stankova, J.; Rola-
Pleszczynski, M. Signaling by the Cysteinyl-Leukotriene Receptor 2: Involvement in Chemokine Gene
Transcription. J. Biol. Chem. 2008, 283, 1974-1984.
52. Deb, A.; Haque, S. J.; Mogensen, T.; Silverman, R. H.; Williams, B. R. G. RNA-Dependent Protein Kinase
PKR Is Required for Activation of NF-κB by IFN-γ in a STAT1-Independent Pathway. J. Immunol. 2001,
166, 6170-6180.
53. Boström, J.; Greenwood, J. R.; Gottfries, J. Assessing the performance of OMEGA with respect to
retrieving bioactive conformations. J. Mol. Graph. Model. 2003, 21, 449-462.
54. Morris, G. M.; Huey, R.; Lindstrom, W.; Sanner, M. F.; Belew, R. K.; Goodsell, D. S.; Olson, A. J.
AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput.
Chem. 2009, 30, 2785-2791.
55. Koes, D. R.; Baumgartner, M. P.; Camacho, C. J. Lessons Learned in Empirical Scoring with smina from
the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Model. 2013 .

Isothiazolones; thiol-reactive
inhibitors of cysteine protease
cathepsin B and histone
acetyl transferase PCAF
Part of this chapter has been published:
Rosalina Wisastra,a Massimo Ghizzoni,a Harm Maarsingh,b
Adriaan J. Minnaard,c Hidde J. Haisma,a and Frank J. Dekker a
Organic & Bio-molecular Chemistry, 2011, 9(6), 1817-1822
a Department of Pharmaceutical Gene Modulation, Groningen Research Institute of Pharmacy,
University of Groningen, Antonius Deusinglaan 1, 9713 AV Groningen, The Netherlands.
bDepartment of Molecular Pharmacology, Groningen Research Institute of Pharmacy,
University of Groningen, Antonius Deusinglaan 1, 9713 AV Groningen, The Netherlands.
cDepartment of Bio-organic Chemistry, Stratingh Institute for Chemistry,
University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands
4

104
Isothiazolones; thiol-reactive inhibitors of
cysteine protease cathepsin B and histone acetyl transferase
Abstract
Isothiazolones and 5-chloroisothiazolones chemoselectively react with thiols by cleavage of the
weak nitrogen-sulfur bond to form disulfides. They show selectivity for inhibition of the thiol -
dependent cysteine protease cathepsin B and the histone acetyltransferase p300/CBP associated
factor (PCAF) based on their substitution pattern. Furthermore, enzyme kinetics and mass
spectrometry indicate covalent binding a 5-chloroisothiazolone to cathepsin B, which
demonstrates their potential utility as probes for activity-based protein profiling.

105
Isothiazolones; thiol-reactive inhibitors of
cysteine protease cathepsin B and histone acetyl transferase
Introduction
Functional groups that react covalently with thiolates have been used extensively for activity-
based protein profiling of cysteine-proteases.1 This technology depends on covalent labelling of
active site thiolates of enzymes in cell-lysates. Labelling is often performed with biotinylated
functionalities, which can be employed for purification with immobilized streptavidin. Cleavable
inhibitor-biotin linkers have been developed to improve the recovery of the proteins of interest. 2
Our current study indicates that isothiazolones and 5-chloroisothiazolones are excellent
candidates for this purpose, because the disulfides that are formed upon reaction with active site
thiolates (Scheme 1) can be cleaved under mild conditions.
The activity of isothiazolones and 5-chloroisothiazolones is expected to originate for a major
extend from the reactivity of these heterocycles towards thiolates.3 In a careful study, Alvarez-
Sánchez et al.4 described that N-methyl-5-chloroisothiazolone reacts with oxygen and nitrogen
nucleophiles via an addition-elimination in the 5-position, thereby substituting the chloride.
Treatment of N-methyl-5-chloroisothiazolone with thiol-nucleophiles, however, provided ring-
opened thioester and dithioester products that can not be explained this way. This indicates that
the thiol nucleophiles react through cleavage of the N-S bond (Scheme 1).
Cathepsin B is a biologically relevant enzyme that is implicated in tumor formation,
angiogenesis, tumor invasion and metastasis.5, 6 Furthermore, cathepsin B has be been considered
as a potentical biomarker for prognosis in cancer.7 Biotinylated irreversible inhibitors can be used
as a probe in activity-based protein profiling of cathepsin B.8, 9 In this study we identify
isothiazolones and 5-chloroisothiazolones as covalent inhibitors of cathepsin B. Furthermore, we
evaluated the selectivity of cathepsin B inhibition compared to the histone acetyltransferase
(HAT) p300/CBP associated factor (PCAF). This enzyme plays a role in the Nuclear Factor κ B
inflammation.10 Upon activation, NFκB binds to cofactors that have intrinsic HAT activity, like
PCAF, which in turn acetylate lysines in core histone H4.11 This leads to a less compact
chromatin structure, which enables the transcription of proinflammatory genes, including
Interleukin 8 (IL-8).12, 13 Isothiazolones and 5-chloroisothiazolones (Scheme 1) are potent and
cell-permeable inhibitors of PCAF, and attempts to optimize their inhibitory activity have been
made in several studies.14-17 However, their mode of action, selectivity and activity in cell-based
studies remain to be investigated.
Herein we describe isothiazolones, 5-chloroisothiazolones and 5-chloroisothiazolone-1-oxides
as new classes of cathepsin B inhibitors. Selectivity between cathepsin B and PCAF inhibition
can be obtained by variation of the substitution. 5-chloroisothiazolones are shown to be non-
competitive inhibitors of cathepsin B and mass spectroscopy demonstrates covalent binding. The
reaction products of isothiazolones and 5-chloroisothiazolones that are formed upon thiolate

106
Isothiazolones; thiol-reactive inhibitors of
cysteine protease cathepsin B and histone acetyl transferase
treatment proved to be disulfides, which could be advantageous in activity-based protein
profiling. Futhermore, the most potent PCAF inhibitor is evaluated in a cell -based study for
inhibition of histone acetylation in comparison to the known HAT inhibitor anacardic acid.
Scheme 1. Reaction of isothiazolones and 5-chloroisothiazolones with propane-1-thiolate results in
cleavage of the N-S bond. a) propane-1-thiol, Et3N, CH2Cl2 b) extended reaction times resulted in
hydrolysis of 7.
Results and discussion
Synthesis
Compounds 1, 5 and 11 were prepared by reaction of the corresponding primary amines with
3,3’-dithiobispropionyl chloride to yield the 3,3’-dithiobispropionic amides as described
previously for compounds 9, 10, 12-18 and 19-26.15, 16 These amides were treated with 3 equiv. of
sulfuryl chloride in dichloromethane to yield a mixture of the 5-chloroisothiazolone and the
isothiazolone, which were separated by column chromatography.
Thiolate reactivity of (5-chloro)isothiazolones
The reaction products that are formed upon treatment with thiolates were identified in order to
study the thiol-reactivity of isothiazolones and 5-chloroisothiazolones. Isothiazolones 1 and 2
were treated with 1.0 equiv. of propane-1-thiol and triethylamine in dichloromethane (Scheme 1).
The resulting products were isolated and characterized by NMR and mass analysis. In substrate 1
the observed 3J(H4,H5) is 6.5 Hz, whereas in product 3 it amounts to 14.1 Hz. Comparably, in 2 3J(H4,H5) is 6.5 Hz, whereas it is 9.7 Hz in 4. This is a strong indication for an E-configuration of
the double bond in 3 and 4, which could be caused by a catalytic amount of thiol that reacts via
subsequent addition, rotation and elimination. When 5-chloroisothiazolones 5 and 6 were
subjected to the same reaction conditions, 7 was isolated upon treatment of 5, however we were
not able to isolate a comparable product after treatment of 6. Upon extended reaction times for
the formation of 7, a new product 8 was isolated that turned out to be the hydrolysis product of 7.
The 1H and 13C NMR spectra of 8 show two isomers most likely due to keto-enol tautomerism.

107
Isothiazolones; thiol-reactive inhibitors of
cysteine protease cathepsin B and histone acetyl transferase
Figure 1. Three resonance structures of 1.
Figure 2. Crystal structure of 2-phenylisothiazolone 1 (A) and 5-chloro-2-phenylisothiazolone 5 (B).
CCDC 763228 & 763229 contain the supplementary crystallographic data for this paper. These data can be
obtained free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html.
Table 1. Interatomic distances (Å) in the crystal structures for compounds 1 and 5.
2-phenyl-isothiazolone 1
5-chloro-2-phenyl-isothiazolone 5
Literature values
sp2 a
Literature values
sp3 a
Cl C1 1.7084(18) 1.76
S N 1.715(4) 1.7038(15) 1.56 1.78
1.70 (thiophene)b
1.61
O C3 1.243(6) 1.231(2) 1.23-1.24 (amides) 1.42-1.44 (ethers)
N C3 1.401(7) 1.396(2) 1.35 (imines) 1.46-1.48 (amines)
N C4 1.433(7) 1.433(2) 1.35 (imines) 1.46-1.48 (amines)
C1 C2 1.345(7) 1.346(3) 1.31-1.34 (alkene)
C2 C3 1.452(7) 1.454(3)1.45-1.46
(single conjugated)
C4 C5 1.399(7) 1.396(3) 1.38-1.40 (aromatic)
C4 C9 1.408(7) 1.393(3) 1.38-1.40 (aromatic)
C5 C6 1.383(7) 1.384(3) 1.38-1.40 (aromatic)
C6 C7 1.391(8) 1.395(3) 1.38-1.40 (aromatic)
C7 C8 1.402(8) 1.392(3) 1.38-1.40 (aromatic)
C8 C9 1.374(9) 1.389(3) 1.38-1.40 (aromatic)
1.81S C1 1.702(6) 1.7269(18)
Standard uncertainty in the last decimal place is given in parentheses
a18 , b CCDC 617103
The reactivity of isothiazolones originates from the electronic structure of the heterocycle,
which is supposed to have considerable aromatic character. This is illustrated by resonance

108
Isothiazolones; thiol-reactive inhibitors of
cysteine protease cathepsin B and histone acetyl transferase
structure II and III in Figure 1. To provide experimental evidence for the aromaticity of
isothiazolones, the crystal structures of 1 and 5 were resolved (Figure 2, Table 1, CCDC 763228
and 763229). The numbering of the carbon atoms in the crystal structure deviates from the
IUPAC numbering in Scheme 1. The crystal structure shows a planar isothiazolone ring for both
1 and 5. In comparison with literature data (Table 1) the C1-C2 bond length fits to a double bond,
C2-C3 fits to a single, conjugated bond and C3-O to a double bond. This indicates that resonance
structure I has a major contribution and that the S-N bond length has single bond character.
Nevertheless, the C3-N bond is shorter than expected for a single bond and the C1-S bond is also
shorter than observed for a single bond, which indicates that resonance structures II and III also
contribute to the structure.
The observation that thiol nucleophiles attack isothiazolones on the sulfur can be explained by
the strength of the formed sulfur-sulfur bond and the weakness of the sulfur-nitrogen bond. In
contrast, isothiazolones are non-reactive towards oxygen or nitrogen nucleophiles, whereas 5-
chloroisothiazolones react with these nucleophiles via addition-elimination at the 5-position.4
Cathepsin B and PCAF inhibition
Cathepsin B inhibition was evaluated by the cleavage of 7-amino-4-methoxy coumarin (AMC)
from the substrate Cbz-Arg-Arg-AMC.18 Inhibiting concentration (IC50) values were derived after
15 minutes of pre-incubation with the inhibitors (Table 2). Since these inhibitors are expected to
interact covalently with the enzyme, differences in potency reflect changes in enzyme binding as
well as differences in the reaction rate of covalent binding to the enzyme. The assay for the HAT
PCAF inhibition and the IC50 values of 9, 10, 12-18 and 19-26 have been reported previously.15, 16
Disulfides 3, 4, and 7, isothiazolone 12 and 5-chloroisothiazolone 5 inhibit cathepsin B, whereas
no inhibition of PCAF is observed in the applied concentrations. Remarkably, disulfide 7 showed the
highest potency for cathepsin B (46 nM). Isothiazolone 9 inhibits the HAT PCAF, whereas cathepsin
B is not inhibited, which demonstrates that selective PCAF inhibition can be obtained with N-aryl
substituents. This is not observed for N-aliphatic substituted isothiazolones and 5-
chloroisothiazolones 12-18. Substitution in the isothiazolone 4-position (R2), in 14, 15 and 16,
changed the cathepsin B inhibition modestly. Remarkably, N-aliphatic substituted 5-
chloroisothiazolone 11 was more potent for cathepsin B than the other N-aliphatic substituted 5-
chloroisothiazolones. Furthermore, 5-chloroisothiazolone-1-oxides also inhibit cathepsin B (Table 2)
and their potency also depends on their substitution pattern. Taking these data together it is concluded
that isothiazolones, 5-chloroisothiazolones and 5-chloroisothiazolone-1-oxides are inhibitors of
cathepsin B and the HAT PCAF and that their potency and selectivity depends on the substitution in
the 2-, 4- and 5-position.

109
Isothiazolones; thiol-reactive inhibitors of
cysteine protease cathepsin B and histone acetyl transferase
Table 2. IC50 values for inhibition of cathepsin B and PCAF by isothiazolones, 5-chloroisothiazolones,
disulfides (Scheme 1), and 5-chloroisothiazolone-1-oxides
1 H H phenyl >100 >10
3* H H phenyl 12 ± 1 >10
4* H H -(CH2)5CO2Me 24 ± 1 >10
5 Cl H -phenyl 13 ± 1 >10
7* Cl H -phenyl 0.046 ± 0.002 >10
9 H H 3-Cl-4-F-phenyl >100 4.2 ± 0.6
10 Cl H 3-Cl-4-F-phenyl 42 ± 2 2.5 ± 0.1
12 H H -pentyl 13 ± 1 >10
13 Cl H -pentyl 7.3 ± 0.5 2.9 ± 0.3
14 Cl H -(CH2)2CO2Me 12 ± 1 1.8 ± 0.2
15 Cl Cl -(CH2)2CO2Me 8.5 ± 0.7 2.6 ± 0.6
16 Cl CH3 -(CH2)2CO2Me 11 ± 1 >10
17 Cl H -(CH2)2CO2Et 14 ± 2 2.0 ± 0.2
18 Cl H -(CH2)3CO2Me 11 ± 1 2.5 ± 0.3
19 Cl H -Ethyl 27.4 ± 1.1 >10
20 Cl Cl -Ethyl 39.0 ± 0.5 >10
21 Cl CH3 -Ethyl 17.4 ± 1.3 >10
22 Cl H -(CH2)2CO2Me 21.3 ± 1.5 5.6 ± 0.2
23 Cl Cl -(CH2)2CO2Me 34.5 ± 1.2 >10
24 Cl CH3 -(CH2)2CO2Me 23.1 ± 3.1 >10
R3
4.1 ± 0.2 3.9 ± 0.42-Cl-4,5-dimethoxy
phenethyl11 Cl H
R1 R2CompoundsCat B
a
(µM)
PCAFb
(µM)
a maximal concentration in the assay 100 M. b maximal concentration in the assays 10 M
* Disulfide compounds as shown in Scheme 1

110
Isothiazolones; thiol-reactive inhibitors of
cysteine protease cathepsin B and histone acetyl transferase
Figure 3. Conversion of the substrate Cbz-Arg-Arg-AMC by the cathepsin B enzyme shows substrate
inhibition at high substrate concentrations with (A) Inhibitor 5 : (▲) with no inhibitor present, (●) after
preincubation with 7 M inhibitor 5 and (■) after preincubation with 14 M inhibitor 5; (B) Inhibitor 14 :
(▲) with no inhibitor present, (●) after preincubation with 6 M inhibitor 14 and (■) after preincubation
with 12 M inhibitor 14; (C) Inhibitor 22 : (▲) with no inhibitor present, (●) after preincubation with 11
M inhibitor 22 and (■) after preincubation with 22 M inhibitor 22. The data were fitted to equation 1
and the parameters are shown in Table 3.
Scheme 2. Enzyme kinetics influence by substrate inhibition
equation 1
Figure 4. Equation for non-linear curve fitting of enzyme kinetics that is influenced by substrate inhibition
according to the model in Scheme 220 v is the reaction velocity, Vmax is the maximal reaction velocity, [S]
is the substrate concentration and Km is the Michaelis-Menten constant. Ks is the binding constant of a
second substrate molecule to the enzyme substrate complex resulting in inhibition of the enzyme activity.
(B) (A)
(C)

111
Isothiazolones; thiol-reactive inhibitors of
cysteine protease cathepsin B and histone acetyl transferase
Table 3 Enzyme kinetics parameters for cathepsin B inhibition (n = 3)
To establish the mechanism of cathepsin B inhibition further, the influence of inhibitors 5 and
14 on the Michaelis-Menten kinetics for conversion of the substrate Cbz-Arg-Arg-AMC was
determined. Cathepsin B conversion of Cbz-Arg-Arg-AMC shows a strong substrate inhibition at
higher concentrations (Figures 3A-C). Substrate inhibition of the enzyme activity is expected to
obey the model in Scheme 2 from which the kinetic parameters can be derived by non-linear
curve fitting of equation 1 (Figure 4). Inhibitor 5 causes a reduced Vmax and a constant Km and Ks
(Table 3). The same is observed for 14 (Figure 3B, Table 3), whereas the data for 22 did not fit
well to equation 1 (Figure 3C, Table 3). This result indicates non-competitive inhibition of the
cathepsin B activity by 5-chloroisothiazolones. Inhibition of cathepsin B activity by 5 proved to
be time dependent (Table 4) and dilution experiments did not show reactivation of cathepsin B
(Table 5). Pre-steady state kinetic analysis revealed a very fast inactivation of the enzyme (50%
inhibition after 20 seconds by 14 M 5, Table 6). The IC50 value proved to decrease slightly over
time. This result suggests a fast initial binding event followed by a slow second binding event
(Table 4, 6).
Furthermore, mass-spectrometry experiments showed an increased mass after incubation of
cathepsin B with the inhibitor. The mass spectrometry data showed that the inhibitor can bind one
or two times to the enzyme. The mass spectrum of the non-reacted enzyme (Figure 5A) gives m/z
27960 as the [M+H]+ peak and 13985 as the [M+2H]2+ peak. The mass spectrum of the enzyme
pre-incubated with the inhibitor (Figure 5B) gives m/z 28111 and 28354 as the [M+H]+ and
14082 and 14177 as the [M+H]2+ peaks. The difference between reacted and non-reacted enzyme
is 151 and 394 for the [M+H]+ peaks and 97 and 192 for the [M+H]2+ peaks, This indicates that 5-
chloroisothiazolone 5 binds once or twice to the enzyme to give a product corresponding to 8,
which would mean a mass increase of 194 or 388. Taken these experiments together it is
concluded that the 5-chloroisothiazolone 5 binds covalently to the thiolate in the enyme active
[inhibitor] (µM) Vmax (nmol/s) Km (mM) KS (mM) R2
0 0.60 ± 0.41 1.2 ± 0.92 0.11 ± 0.087 0.996
7 0.36 ± 0.20 1.1 ± 0.68 0.11 ± 0.068 0.996
14 0.22 ± 0.10 1.1 ± 0.60 0.12 ± 0.069 0.997
0 0.65 ± 0.66 1.2 ± 1.3 0.10 ± 0.11 0.989
6 0.51 ± 0.24 1.4 ± 0.7 0.10 ± 0.051 0.998
12 0.23 ± 0.13 1.2 ± 0.7 0.20 ± 0.13 0.995
0 0.56 ± 0.67 1.1 ± 1.4 0.098 ± 0.13 0.982
11 0.52 ± 0.59 1.1 ± 1.2 0.11 ± 0.15 0.972
22 0.50 ± 0.70 1.1 ± 1.9 0.090 ± 0.15 0.972
5
14
22

112
Isothiazolones; thiol-reactive inhibitors of
cysteine protease cathepsin B and histone acetyl transferase
site and/or to another surface exposed cysteine of cathepsin B.
Table 4. Time dependent inhibition of cathepsin B by 5. The IC50 of the residual enzyme activity was
determined after 1, 15, 40 or 120 minutes pre-incubation of the enzyme with the inhibitor (n = 3).
Time (min) IC50 (µM)
1 13.6 ± 1.5
15 12.7 ± 1.3
40 9.3 ± 1.1
120 8.1 ± 1.0
Table 5. Dilution experiments after pre-incubation of Cathepsin B (20 nM) and inhibitor 5 (17 µM) with a
substrate solution (100 µM) demonstrates a reduction of activity that is proportional to the dilution. This
demonstrates covalent binding of the enzyme to the inhibitor (n = 3) 6.
Dilution v (fluorescence units/min) Reduction in activity
1 13.3 ± 0.5 1.00
2 6.9 ± 0.1 1.93
5 2.7 ± 0.1 4.93
Table 6. Reduction rate of cathepsin B by 5. The initial rate of cathepsin B (1.5 nM) activity with or without
inhibitor 5 (14 µM) were determine after 20 second reaction of the enzyme with the substrate (33.3 µM) (n = 3).
The result shows a very quick reduction of the enzyme activity in presence of the inhibitor.
Inhibitor V (fluorescence units/s)
No inhibitor 25.3 ± 1.2
5 (14 µM) 11.9 ± 5.0
Figure 5. MALDI – TOF mass spectrum of the non-reacted cathepsin B (A) and the mass spectrum of the
cathepsin B that was pre-incubated with inhibitor 5 (B).
(B) (A)

113
Isothiazolones; thiol-reactive inhibitors of
cysteine protease cathepsin B and histone acetyl transferase
Inhibition of histone acetylation
The most potent PCAF inhibitor 14 was evaluated in a cell-based assay for inhibition of histone
acetylation in HEP-G2 cells. To study inhibition of acetylation, deacetylation was blocked by
treatment with the HDAC inhibitor suberoylanilide hydroxamic acid (SAHA). Inhibition of
histone deacetylation by treatment with 10 µM of SAHA for 20 h gave a significant increase in
histone acetylation (lane 2 and 5, Figure 6). In a separate experiment, the cells were treated for 1
hour with SAHA before the HAT inhibitors were added and the cells were treated with SAHA
and the HAT inhibitor for 20 h (lane 3 and 6, Figure 6). Treatment with SAHA and 14 did not
decrease the acetylation level compared to SAHA treated cells (lane 3, Figure 6). The acetylation
level decreased in cells that were treated with SAHA and the known HAT inhibitor anacardic
acid21 compared to SAHA treated cells (lane 6, Figure 6). The quantification results show that 5 -
chloroisothiazolone 14 does not effectively down-regulate acetylation of histone H4 in HEP G2
cells under the applied conditions in contrast to the known HAT inhibitor anacardic acid. The
lack of inhibition might be explained by a lack of cellular permeability or metabolic stability.
Alternatively, the inhibition of the HAT PCAF might be compensated by other HATs that are not
affected by 14. Furthermore, the cytotoxicity assay result shows that the treatment with the HAT
inhibitor at concentration employed in the inhibition assay is not toxic to the cell.
Figure 6. Cell-based studies on histone acetylation. HEP G2 cell were treated for 24 h with vehicle or HDAC
and HAT inhibitors. The histones were extracted and blotted with an anti-acetyl H4 antibody (C). The loading
was standardized based on a Coomassie staining on a separate blot (C). The data were quantified and shown in
bar graphs (A and B). The bars represent the average of four independent experiments with their respective
standard deviations. Lane 1 and 4 are histones from vehicle (DMSO dilution) treated cells. Lane 2 and 5 are
cells that are treated with 10 M of the HDAC inhibitor SAHA. Lane 3 is histones from cell that were treated
with 10 M SAHA and 10 M 14. Lane 6 is histones from cell that were treated with 10 M SAHA and 50 M
anacardic acid. The p-value for the difference between lane 5 and 6 in B is 0.006, which indicates a significant
difference.
A
0
100
200
300
400
500
1 2 3
Ac
ety
late
d H
4
B
0
100
200
300
400
500
600
700
4 5 6
Ac
ety
late
d H
4
anacardic acid 14

114
Isothiazolones; thiol-reactive inhibitors of
cysteine protease cathepsin B and histone acetyl transferase
Conclusion
In conclusion, isothiazolones and 5-chloroisothiazolones react with thiolates by disulfide
formation and cleavage of the nitrogen-sulfur bond. The crystal structures illustrate the aromatic
character of isothiazolones. Whereas this aromaticity provides stability to the compounds, the
weak sulfur-nitrogen bond allows a fast reaction with sulfur nucleophiles. Based on their
reactivity, 5-chloroisothiazolones and isothiazolones inhibit thiol containing enzymes, such as the
cysteine protease cathepsin B and the HAT PCAF. Their potency depends on the substitution
pattern. Selectivity for inhibition of the HAT PCAF was observed for N-aryl substituted
isothiazolone 9. The most potent HAT inhibitor 14 did not effectively downregulate histone
acetylation in cell-based studies under the applied conditions. Enzyme kinetics and mass
spectrometry indicate covalent binding of 5-chloroisothiazolone 5 to the active site thiol and/or to
another surface exposed cysteine of cathepsin B. The fact that isothiazolones and 5-
chloroisothiazolones react with thiolates to form disulfides makes them excellent candidates for
activity-based protein profiling of thiol-containing enzymes.
Experimental
General
All reagents and solvents were obtained from commercial suppliers (Sigma-Aldrich, Acros
Organics) and were used without further purification. CH2Cl2 was distilled over CaH2 before use.
Merck silica gel 60 F254 plates were used for analytical thin layer chromatography (TLC) and
spots were detected by UV light, or stained using KMNO4 or ninhydrin solution. Column
chromatography was performed with MP Ecochrom silica gel 32-63, 60Ǻ using the flash
chromatography technique. 1H (200 MHz) and 13C (50 MHz) NMR spectra were recorded on a
Varian Gemini 200 spectrometer in CDCl3 unless otherwise specified. 1H chemical shift are
reported in ppm (δ) downfield from internal standard tetramethylsilane (TMS). 13C NMR spectra
were recorded using the attached proton test (APT) and chemical shifts are relative to the solvent.
Melting points were determined using an Electrocome digital melting point measurement
apparatus. High-resolution mass spectra (HR-MS) were recorded using a flow injection method
on a LTQ-Orbitrap XL mass spectrometer (Thermo Electron, Bremen, Germany) with a
resolution of 60,000 at m/z 400. Protonated testosterone (lock mass m/z = 289.2162) was used for
internal recalibration in real time.
The enzyme cathepsin B (human liver) and cathepsin Substrate III, Fluorogenic (Z-RR-AMC)
was obtained from Calbiochem. 7-amino 4-methyl coumarine was obtained from Sigma. HEP G2
cells were purchased from the American Type Culture Collection and cultured in Dulbecco's modified
Eagle's medium (GIBCO, cat. 41966) containing 10% fetal bovine serum (PAA, cat. A15-151) and

115
Isothiazolones; thiol-reactive inhibitors of
cysteine protease cathepsin B and histone acetyl transferase
1% penicillin and 1% streptomycin (GIBCO, cat. 15140) at 37 ºC, in 5% CO2. Cells were not cultured
beyond 50 passages.
Organic Synthesis
Synthetic procedure 1
Isothiazolones and 5-chloroisothiazolones were synthesized according to the procedure described
by Dekker et al.15, 16. The 3,3’-dithiobis-propionamide was dissolved in dry CH2Cl2 at 0 oC and
SO2Cl2 was added dropwise to the solution. The mixture was stirred at 0 °C for 2 hours. The
mixture was washed with water (2 times) and brine (1 time), dried using Na 2SO4, filtered and
concentrated under reduced pressure. The residue was purified by column chromatography.
5-chloro-2-phenylisothiazol-3(2H)-one (5)
The product was obtained using synthetic procedure 1. Purification was performed using column
chromatography with hexane/EtOAc 10:1 (v/v) as eluent. Yield 43%. Off-white solid. Crystals
(colorless needles) for X-ray diffraction were obtained by crystallization from EtOAc:hexanes 1:1
at 4 oC over 2 days. Mp = 118.4 °C. Rf = 0.52 (hexane:EtOAc 1:1). 1H NMR (200 MHz, CDCl3) δ
6.35 (s, 1H), 7.27-7.55 (m, 5H). 13C NMR (50 MHz CDCl3) δ 114.9, 124.8, 127.8, 129.5, 135.7,
146.4, 165.4. MS (ESI) m/z 211.9 [M+H]+. HRMS (m/z) 211.9931 [M+H]
+, calcd 211.9931
C9H7ClNOS+.
2-phenylisothiazol-3(2H)-one (1)
After elution of product 5 product 1 was eluted from the column with hexane/EtOAc 1:1 (v/v) as
eluent. Yield 29%. Off-white solid. Crystals (colorless platelets) for X-ray diffraction were
obtained by crystallization from EtOAc:hexanes 3:4 at 4oC over 2 days. Mp = 92.8 °C. Rf = 0.16
(hexane:EtOAc 1:1). 1H NMR (200 MHz, CDCl3) δ 6.32 (d, J = 6.2 Hz, 1H), 7.29-7.61 (m, 5H),
8.18 (d, J = 6.5 Hz). 13C NMR (50 MHz CDCl3) δ 114.8, 124.8, 127.5, 129.4, 136.5, 139.8, 167.6.
MS (ESI) m/z 178.1 [M+H]+. HRMS (m/z) 178.0321 [M+H]+
, calcd 178.0321 C9H8NOS+.

116
Isothiazolones; thiol-reactive inhibitors of
cysteine protease cathepsin B and histone acetyl transferase
5-chloro-2-(2-chloro-4,5-dimethoxyphenethyl)isothiazol-3(2H)-one (11)
The product was obtained using synthetic procedure 1. Purification was performed using column
chromatography with hexane/EtOAc 1:1 (v/v) as eluent. Yield 6%. White-yellow solid. Mp =
108.5 °C. Rf = 0.20 (hexane:EtOAc 1:1). 1H NMR (200 MHz, CDCl3) δ 3.04 (t, J = 7.0 Hz, 2H),
3.82 (s, 3H), 3.86 (s, 3H), 3,95 (t, J = 7.0 Hz, 2H), 6.25 (s, 1H), 6.68 (s, 1H), 6.86 (s, 1H). 13C
NMR (50 MHz CDCl3) δ 32.8, 43.3, 56.0, 112.5, 113.2, 114.3, 124.9, 126.4, 145.8, 147.8, 148.5,
166.6. MS (ESI) m/z 334.3 [M+H]+. HRMS (m/z) 334.0065 [M+H]+
, calcd 334.0066
C13H14Cl2NO3S+; (m/z) 119.0521 [M-C3H1ClNO2S]+
, calcd 119.0520 C10H12ClO2+.
Synthetic procedure 2
The 5-chloroisothiazolone (1 mmol) was dissolved in 2 mL CH2Cl2 and cooled to 0 °C. A 0.5 M
propane-1-thiol solution in CH2Cl2 (1 mL) was added, and the progress of reaction was followed
by TLC. A 0.5 M triethylamine (TEA) solution in CH2Cl2 (1 mL) was added to the mixture and
reacted for another 30 minutes. The mixture was diluted with 20 mL of CH2Cl2 and washed with
H2O (2 times 50 mL), brine (1 times 50 mL) and dried using Na2SO4.
N-phenyl-3-(propyldisulfanyl)acrylamide (3)
The product was obtained using synthetic procedure 2. Purification was performed using column
chromatography with hexane/EtOAc 15:1 (v/v) as eluent. Yield 21%. White solid. R f = 0.70
(hexane:EtOAc 1:1). 1H NMR (200 MHz, CD3CN) δ 1.03 (t, J = 7.4 Hz, 3H), 1.72 (m, 2H), 2.80
(t, J = 7.2 Hz, 2H), 6.50 (d, J = 14.1 Hz, 1H), 7.09 (t, J = 7.4 Hz, 1H), 7.32 (t, J = 7.5 Hz, 2H)
7.51-7.64 (m, 3H), 8.50 (br. 1H). 13C NMR (50 MHz CD3CN) δ 13.2, 23.2, 40.4, 120.4, 121.2,
124.7, 129.8, 139.9, 142.9, 163.0. MS (ESI) m/z 254.1 [M+H]+. HRMS (m/z) 254.0668 [M+H]
+,
calcd 254.0668 C12H16NOS2+; (m/z) 178.0323 [M-C3H7S]
+, calcd 178.0321 C9H8NOS+.

117
Isothiazolones; thiol-reactive inhibitors of
cysteine protease cathepsin B and histone acetyl transferase
Methyl 6-(3-(propyldisulfanyl)acrylamido)hexanoate (4)
The product was obtained using synthetic procedure 2. Purification was performed using column
chromatography with hexane/EtOAc 3:1 (v/v) as eluent. Yield 24%. White solid. R f = 0.36
(hexane:EtOAc 1:1). 1H NMR (200 MHz, CD3CN) δ 0.98 (t, J = 7.2 Hz, 3H), 1.25-1.70 (m, 10H),
2.77 (t, J = 7.2 Hz, 2H), 3.10-3.21 (m, 2H), 3.60 (s, 3H), 5.86 (d, J = 9.7 Hz, 1H), 6.59 (br. 1H),
6.99 (d, J = 9.7 Hz, 1H). 13C NMR (50 MHz CD3CN) δ 12.2, 22.1, 24.3, 26.0, 28.9, 33.4, 38.6,
41.0, 50.9, 118.0, 152.1, 165.9, 173.8. MS (ESI) m/z 306.0 [M+H]+. HRMS (m/z) 328.1009
[M+Na]+, calcd 328.1012 C13H23NNaO3S2
+; (m/z) 306.1191 [M+H]+, calcd 306.1192
C13H24NO3S2+; (m/z) 230.0844 [M-C3H7S]
+, calcd 230.0845 C10H16NO3S
+.
N-phenyl-3-(propyldisulfanyl)-3-chloro-acrylamide (7)
The product was obtained using synthetic procedure 2. Purification was performed using column
chromatography with hexane/EtOAc 20:1 (v/v) as eluent. Yield 60%. White solid. R f = 0.67
(hexane:EtOAc 1:1). 1H NMR (200 MHz, CD3CN) δ 0.99 (t, J = 7.4 Hz, 3H), 1.72 (m, 2H), 2.79
(t, J = 7.2 Hz, 2H), 6.56 (s, 1H), 7.10 (t, J = 7.4 Hz, 1H), 7.33 (m, 2H) 7.58 (d, J = 7.6 Hz, 2H),
8.50 (br. 1H). 13C NMR (50 MHz CD3CN) δ 13.2, 22.7, 42.8, 120.3, 122.2, 125.0, 129.8, 139.4,
151.4, 163.3. MS (ESI) m/z 288.1 [M+H]+. HRMS (m/z) 288.0277 [M+H]
+, calcd 288.0278
C12H15ClNOS2+; (m/z) 211.9931 [M- C3H7S]
+, calcd 211.9931 C9H7ClNOS+.
phenyl-3-(propyldisulfanyl)-3-hydroxy-acrylamide (8)
The product was obtained using synthetic procedure 2 with overnight reaction time. Purification
was performed using colomn chromatography with hexane/EtOAc 5:1 (v/v) as eluent. Yield 30%.
Yellow solid. Rf = 0.57 (hexane:EtOAc 1:1). 1H NMR (200 MHz, CDCl3) δ 0.97 (m, 3H), 1.63
(m, 2H), 2.67 (m, 2H), 7.22 (m, 1H), 7.25 (m, 2H), 7.50 (m, 3H), 8.73 (br. 1H). 13C NMR (50
MHz CDCl3) δ 13.7, 23.3, 36.7, 37.1, 119.6, 119.7, 120.6, 120.7, 124.4, 125.4, 127.1, 129.2,
129.3, 130.8, 136.9, 137.1, 138.0, 157.4, 163.5. MS (ESI) m/z 270.1 [M+H]+.

118
Isothiazolones; thiol-reactive inhibitors of
cysteine protease cathepsin B and histone acetyl transferase
Cathepsin B inhibition
Enzyme inhibition was measured by the residual enzyme activity after 15 min incubation with the
inhibitor. The enzyme activity was determined by conversion of cathepsin B substrate Cbz-Arg-
Arg-AMC.18 The conversion rate was determined by fluorescence detection of 7-amino 4-methyl
coumarine with 1 min intervals over 15 min at 25oC. Detection was performed with an excitation
wavelength of 355 nm and an emission wavelength of 450 nm. The cathepsin B enzyme (0.5
mg/ml) was diluted 1:4000 with cathepsin B assay buffer (100 mM Na2HPO4.2H2O; 1.25 mM
EDTA.2Na; pH=6.8) containing 100 mM DTT. The inhibitor (10 mM in CH3CN) was diluted
with cathepsin B assay buffer containing 1% (v/v) Triton-X to the desired concentration.
Calculations were performed with Excel 2003 and non-linear curve fitting was performed with
the Origin 7 software.
Michaelis Menten enzyme kinetics
The enzyme kinetic experiments were performed using the same assay setup as used for the IC50
determinations. Various concentrations of fluorogenic substrate Cbz-Arg-Arg-AMC (7.81-1000
µM) were added to the enzyme. The fluorescence values were converted to concentrations using a
7-amino 4-methyl coumarine calibration curve (0.78-25 µM). The concentration change over time
is the reaction velocity (v) that is plotted against the substrate concentration (s) in Figure 3. The
experiments were performed in triplicate and the average triplicate values and their standard
deviations are plotted. Calculations were performed with Excel 2003 and non-linear curve fitting
with equation 1 was performed with the Origin 7 software. The results are shown in Table 3.
Mass spectroscopy
Inhibitor 5 (50 mM) and cathepsin B (0.25 mg/mL) were incubated in phosphate buffer (100 mM
Na2HPO4.2H2O; 1.25 mM EDTA.2Na; pH = 6.8) for 20 min at room temperature. This mixture
was mixed 1 to 1 with the sinapinic acid matrix and subjected to MALDI TOF mass
spectroscopy.
Western blot on histone acetylation
HEP G2 cells were seeded (10,000 cells) in T-175 flasks and cultured until 50% confluence.
Subsequently, the cells were treated with HDAC and HAT inhibitors or control solutions. The
inhibitors were diluted from stock solutions (10 mM) in DMSO. The residual DMSO at the applied
concentrations did not have an effect on histone acetylation. Cells were treated with phosphate buffer
saline pH 7.4 (PBS) as control or with SAHA (10 µM) for 20 hours. The HAT inhibitors were added
to cells that were pretreated with SAHA (10 µM) for 1 hour after which the cells were cultured for 20
hours in medium with SAHA and the HAT inhibitor. Anacardic acid was applied at 50 µM and 5-

119
Isothiazolones; thiol-reactive inhibitors of
cysteine protease cathepsin B and histone acetyl transferase
chloroisothiazolone 14 was applied at 10 µM. The viability of the cells was more than 70% at the
applied concentrations as determined by a MTT assay.
The histones were extracted using the following procedure. Cells were trypsinized for 10 minutes
at 37 ºC and washed twice with ice-cold PBS. The PBS was replaced with 5 mL ice-cold lysis buffer
(10mM Tris, 50 mM sodium bisulfite, 10 mM MgCl2, 8.6% sucrose, 1% Triton-X, pH 6.5) and the
cells were sonicated at 0 oC (3 times 15 sec with 15 sec intervals) Subsequently the suspension was
centrifuged (10 min 12 000 rcf, 4 oC) and the pellet was washed three times with ice-cold lysis buffer.
The pellet was washed once in washing buffer (10 mM Tris, 10 mM EDTA, pH 7.4) and resuspended
in 400 μL ice-cold water. H2SO4 was added to a final concentration of 0.4 M and samples were
incubated for one hour at 0 oC and centrifuged (10 min, 12 000 rcf, 4 ºC). The supernatant was then
transferred to a vial with five volumes acetone and incubated overnight at -20 ºC. The suspension was
centrifuged (10 min, 16 000 rcf, 4 ºC), the supernatant was discarded and the pellet was air-dried at
room temperature. The pellets were then resuspended in 500 μL demi water.
The protein concentrations of samples were determined using the BioRad Protein Assay, with BSA
as calibration. Samples of 6.25 μg of protein from each extract were loaded on polyacrylamide gels
(SDS-PAGE). Acidic samples were neutralized using 100 mM Tris-buffer pH 8.8 and if necessary,
100 mM sodium hydroxide until the laemmli buffer regained its blue coloration. Samples of 20 μL
were then run on 12.5% polyacrylamide gels at 70 volts through the stacking gel and 110 volts
through the resolving gel before blotting onto PVDF membrane for two hours at 200 mA. The PVDF
membrane was stained using Coomassie blue and destained using destaining solution (40% methanol
and 10% acetic acid in water) for 30 minutes at room temperature. The Coomassie-stained membrane
was used as a loading control for each sample. Another membrane with the same sample was blocked
using 10 mL 5% BSA in PBS. The membrane was washed (3 times, 5 min, room temp) with PBS
containing 0.05% v/v Tween 20 (PBS-T). The membrane was stained with 10 mL 1:2000 anti-acetyl
H4 (Millipore 06-598) in PBS-T. The membrane was washed (3 times, 5 min, room temp.) with PBS-
T. The membrane was incubated with 10 mL 1:1000 HRP-conjugated swine anti-rabbit antibody
(DakoCytomation, cat. P0217) and washed with PBS-T (3 times, 5 min, room temp.) Staining was
performed using AEC (Sigma-Aldrich, cat. AEC101-1KT) according to the manufacturer's
instructions. The membrane was exposed to this solution until bands appeared and the reaction was
stopped by washing the membrane with demi water. The bands were scanned using a Bio-Rad GS-
710 densitometer with the blue filter setting and quantified using the open source software Image J.
The antibody-based quantifications were normalized against the total protein as measured by
Coomassie staining. The average and standard-deviations of four independent experiments are
reported (Figure 6).

120
Isothiazolones; thiol-reactive inhibitors of
cysteine protease cathepsin B and histone acetyl transferase
Cytotoxicity assay
Cells (7,500) were seeded in DMEM medium (95 µL) in 96 well plates. In the blank determination no
cells were seeded. After 24 hours different concentrations of the compounds were added in PBS (5
μL). In the positive control no compound was added. Each plate was centrifuged briefly before
returning to incubation. Determinations were performed in 8-fold on each plate. After 48 hours, cells
were treated with filter-sterilized MTT solution (20 μL, 5 mg.ml-1) per well, followed by brief
centrifugation and three hours incubation at 37 ºC, 5% CO2. The medium was removed and 4 mM
HCl, 0.1% v/v Triton-X in isopropanol (150 μL) was added. The plate was shaken for 15 minutes and
absorbance read at 550 nm on a ThermoMax microplate reader.
References
1. Cravatt, B. F.; Wright, A. T.; Kozarich, J. W. Activity-Based Protein Profiling: From Enzyme Chemistry
to Proteomic Chemistry. Annu. Rev. Biochem. 2008, 77, 383-414.
2. Verhelst, S. H. L.; Fonović , M.; Bogyo, M. A Mild Chemically Cleavable Linker System for Functional
Proteomic Applications. Angew. Chem. Int. Ed. 2007, 46, 1284-1286.
3. Morley, J. O.; Oliver Kapur, A. J.; Charlton, M. H. Structure-activity relationships in 3-isothiazolones.
Org. Biomol. Chem. 2005, 3, 3713-3719.
4. Alvarez-Sánchez, R.; Basketter, D.; Pease, C.; Lepoittevin, J. Studies of Chemical Selectivity of Hapten,
Reactivity, and Skin Sensitization Potency. 3. Synthesis and Studies on the Reactivity toward Model
Nucleophiles of the 13C-Labeled Skin Sensitizers, 5-Chloro-2-methylisothiazol-3-one (MCI) and 2-
Methylisothiazol-3-one (MI). Chem. Res. Toxicol. 2003, 16, 627-636.
5. Podgorski, I.; Sloane, B. F. Cathepsin B and its role (s) in cancer progression. Biochem. Soc. Symp. 2003,
70, 263-276.
6. Gocheva, V.; Zeng, W.; Ke, D.; Klimstra, D.; Reinheckel, T.; Peters, C.; Hanahan, D.; Joyce, J. A. Distinct
roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev. 2006, 20, 543-556.
7. Herszenyi, L.; Farinati, F.; Cardin, R.; Istvan, G.; Molnar, L.; Hritz, I.; De Paoli, M.; Plebani, M.;
Tulassay, Z. Tumor marker utility and prognostic relevance of cathepsin B, cathepsin L, urokinase-type
plasminogen activator, plasminogen activator inhibitor type-1, CEA and CA 19-9 in colorectal cancer.
BMC Cancer 2008, 8, 194.
8. Walker, B.; Cullen, B. M.; Kay, G.; Halliday, I. M.; McGinty, A.; Nelson, J. The synthesis, kinetic
characterization and application of a novel biotinylated affinity label for cathepsin B. Biochem. J. 1992,
283 ( Pt 2), 449-453.
9. Cullen, B. M.; Halliday, I. M.; Kay, G.; Nelson, J.; Walker, B. The application of a novel biotinylated
affinity label for the detection of a cathepsin B-like precursor produced by breast-tumour cells in culture.
Biochem. J. 1992, 283 ( Pt 2), 461-465.
10. Lentsch, A. B.; Ward, P. A. Activation and Regulation of NF- B during Acute Inflammation. Clin. Chem.
Lab. Med. 2005, 37, 205.
11. Barnes, P. J. Transcription factors in airway diseases. Lab. Invest. 2006, 86, 867-872.
12. Yao, H.; Yang, S. R.; Kode, A.; Rajendrasozhan, S.; Caito, S.; Adenuga, D.; Henry, R.; Edirisinghe, I.;
Rahman, I. Redox regulation of lung inflammation: role of NADPH oxidase and NF-kappaB signalling.
Biochem. Soc. Trans. 2007, 35, 1151-1155.
13. Stimson, L.; Rowlands, M. G.; Newbatt, Y. M.; Smith, N. F.; Raynaud, F. I.; Rogers, P.; Bavetsias, V.;
Gorsuch, S.; Jarman, M.; Bannister, A.; Kouzarides, T.; McDonald, E.; Workman, P.; Aherne, G. W.
Isothiazolones as inhibitors of PCAF and p300 histone acetyltransferase activity. Mol. Cancer Ther. 2005,
4, 1521-1532.

121
Isothiazolones; thiol-reactive inhibitors of
cysteine protease cathepsin B and histone acetyl transferase
14. Gorsuch, S.; Bavetsias, V.; Rowlands, M. G.; Aherne, G. W.; Workman, P.; Jarman, M.; McDonald, E.
Synthesis of isothiazol-3-one derivatives as inhibitors of histone acetyltransferases (HATs). Bioorg. Med.
Chem. 2009, 17, 467-474.
15. Dekker, F. J.; Ghizzoni, M.; van der Meer, N.; Wisastra, R.; Haisma, H. J. Inhibition of the PCAF histone
acetyl transferase and cell proliferation by isothiazolones. Bioorg. Med. Chem. 2009, 17, 460-466.
16. Ghizzoni, M.; Haisma, H. J.; Dekker, F. J. Reactivity of isothiazolones and isothiazolone-1-oxides in the
inhibition of the PCAF histone acetyltransferase. Eur. J. Med. Chem. 2009, 44, 4855-4861.
17. Dekker, F. J.; Haisma, H. J. Histone acetyl transferases as emerging drug targets. Drug Discov. Today
2009, 14, 942-948.
18. Barrett, A. J.; Kirschke, H. Cathepsin B, Cathepsin H, and cathepsin L. Methods Enzymol. 1981, 80 Pt C,
535-561.
19. Anslyn, E. V.; Dougherty, D. A. Modern Physical Organic Chemistry. In University Science Books: 2006;
pp 22.
20. Copeland, R. A. Enzymes 2nd edition. in Wiley 2000.
21. Balasubramanyam, K.; Swaminathan, V.; Ranganathan, A.; Kundu, T. K. Small Molecule Modulators of
Histone Acetyltransferase p300. J. Biol. Chem. 2003, 278, 19134-19140.

123
123
Part of this chapter has been published:
Rosalina Wisastra,a Klaas Poelstra,b Rainer Bischoff,c
Harm Maarsingh,d Hidde J. Haisma,a and Frank J. Dekker a
ChemBioChem, 2011, 12(13), 2016-2020
a Department of Pharmaceutical Gene Modulation, Groningen Research Institute of Pharmacy,
bDepartment of Pharmacokinetics, Toxicology and Targeting, Groningen Research Institute of Pharmacy,
cDepartment of Analytical Biochemistry, Groningen Research Institute of Pharmacy,
dDepartment of Molecular Pharmacology, Groningen Research Institute of Pharmacy,
University of Groningen, Anthonius Deusinglaan 1, 9713 AV Groningen, The Netherlands.
Antibody-free detection of
protein tyrosine nitration in
tissue sections
5

124
Antibody-free detection of protein tyrosine nitration in tissue sections
Abstract
Tyrosine nitration is a covalent posttranslational protein modification that represents a biomarker for
inflammatory diseases. We introduce a novel methodology to detect nitrotyrosine in biological
samples by chemoselective conversion into a fluorophore. We describe the applicability of this
methodology to detect protein bound nitrotyrosine in tissue sections using fluorescence microscopy
and on western blot membranes.

125
Antibody-free detection of protein tyrosine nitration in tissue sections
Introduction
Tyrosine nitration is a covalent posttranslational protein modification that occurs in vivo as a
consequence of oxidative stress.1 It has been described that peroxynitrite, which is formed by reaction
between superoxide anion (O2-) and nitric oxide (NO), causes tyrosine nitration.2 Moreover, also other
reactive nitrogen species, such as NO2+, NO2Cl, NO2·, can be involved in tyrosine nitration.3, 4 Recent
studies show that 3-nitrotyrosine is a biomarker for several inflammatory diseases such as
inflammatory bowel disease (IBD), rheumatoid arthritis, and asthma.5-7 Tyrosine nitration has often
been regarded as non-selective oxidative damage to proteins; however, recently more specific roles
have been described.8 For example, tyrosine nitration triggers dissociation of IκB from Nuclear
Factor-κB (NF-κB), which results in activation of this pathway that plays an important role in cancer
and inflammation.7, 8
The importance of tyrosine nitration in the pathology of multiple diseases and its potential role in
signal transduction demonstrate the urgent need for reliable analytical methods to study protein
nitration. Several methods have been developed for the detection of nitrotyrosine that apply
antibodies, mass spectrometry, or fluorogenic tagging.9-11 Currently, tyrosine nitrated proteins in
tissue sections are detected by immunohistochemistry using antibodies directed at nitrotyrosine.5, 6, 12
Protein bound nitrotyrosine in tissue homogenates and blood plasma is usually detected by
immunoblot methods, which also rely on nitrotyrosine directed antibodies.13 Nevertheless,
nitrotyrosine antibodies have disadvantages such as a lack of selectivity for nitrotyrosine, high
background signals in certain tissues, high costs and a limited stability upon prolonged storage.
In this study, we introduce a novel methodology that does not rely on antibodies to detect
nitrotyrosine in biological samples. This method implies two reaction steps to convert the 2-
nitrophenol functionality of protein-bound nitrotyrosine chemoselectively into a fluorophore (Figure
1). In the first reaction step the 2-nitrophenol functionality is reduced to a 2-aminophenol
functionality by sodium dithionite (Na2S2O4). In the second reaction step the 2-aminophenol
functionality is converted into a fluorophore by reaction with salicylaldehyde and Al3+.14 This novel
histochemical staining selectively detects 2-nitrophenol as well as 2-nitrosophenol and 2-aminophenol
functionalities. In this study, we describe the applicability of this methodology to detect nitrotyrosine
in tissue sections using fluorescence microscopy. This method is also applicable for staining of
protein bound 2-nitrophenol functionalities on western blot membranes in complex biological samples
such as blood plasma.

126
Antibody-free detection of protein tyrosine nitration in tissue sections
Figure 1. Reduction of a protein bound 2-nitrophenol functionality to a 2-aminophenol functionality and
subsequent conversion into a fluorophore: a) aqueous Na2S2O4; b) aqueous AlCl3 and salicylaldehyde.
Results and discussion
Initially, we studied the fluorescence of the Schiff base of 2-aminophenol and salicylaldehyde
complexed with aluminium. The fluorescence spectra of this complex in solution show an excitation
wavelength (λex) of 412 nm and an emission wavelength (λem) of 520 nm in H2O and in MeOH
(Figure 2). The fluorescence proved to be linear between 0.31 µM and 10 µM in both solvents (Figure
3) and the fluorescence signal in H2O is 8 times stronger than in MeOH. Thus, H2O is the preferred
solvent for detection of nitrotyrosine in biological samples.
Figure 2. (A) The excitation and emission spectra of 2-aminophenol-salicylaldehyde-aluminium complex in
H2O. The amplification was (HV) was 1000 and the bandwidth for λex and λem were 5 nm. (B) The excitation
and emission spectra of 2-aminophenol-salicylaldehyde-aluminium complex in methanol. The amplification was
(HV) was 800 and the bandwidth for λex and λem were 5 nm. The concentration of the fluorophore was 10 µM.
(B) (A)

127
Antibody-free detection of protein tyrosine nitration in tissue sections
Figure 3. Fluorescence intensity plotted against the concentration of the 2-aminophenol-salicylaldehyde-
aluminium complex solution in demi water ( ) (R2 = 0.999), and in methanol ( ) (R2 = 0.994).
The first reaction step for the histochemcial staining according to Figure 1 is reduction of 2-
nitrophenol using a freshly prepared solution of sodium dithionite in demi-water. This reagent
provides fast and complete conversion of 2-nitrophenol in aqueous environment, which demonstrates
that sodium dithionite is an appropriate reducing agent for this reaction. Several control experiments
were performed for the second reaction step for the histochemical staining according to Figure 1,
which leads to the formation of the Schiff base of the 2-aminophenol functionality and
salicylaldehyde in complex with Al3+.
Figure 4. Excitation and emission spectra for salicylaldehyde and aluminium in methanol.
Analysis of the fluorescence spectra of the reagent solution that was used for this reaction step,
which contains salicylaldehyde and aluminium, gives a very weak fluorescence with λex 395 nm and
λem 475 nm (Figure 4). This indicates that the background fluorescence of the reagents will not
interfere with the detection of 2-aminophenol functionalities in proteins. Quantification of 2-
nitrophenol in solution using UV detection is about 100 times less sensitive then the fluorescence of
the Schiff base of 2-aminophenol and salicylaldehyde in complex with aluminium (linear range UV
detection between 20 µM to 1200 µM), which shows that fluorescence detection is preferable. Further
experiments showed that the fluorescence of the aluminium complex disappears upon addition of
buffers such as phosphate buffer (50 mM, pH 7.5) and citric acid buffer (10 mM, pH 4.0) and also
0,00
1,00
2,00
3,00
4,00
5,00
6,00
0,00E+00 4,00E-06 8,00E-06 1,20E-05
flu
ore
sce
nce
(f.
u)
Al-complex (M)

128
Antibody-free detection of protein tyrosine nitration in tissue sections
upon addition of reducing agents such as sodium dithionite (Figure 5A). Furthermore, high
concentrations of bovine serum albumin (BSA) reduced the fluorescence of the aluminium complex
with 2-aminophenol and salicylaldehyde (Figure 5B). This indicates that the formation of the
fluorescent aluminium complex should be performed in demi-water.
Figure 5. Fluorescence of Aluminium complex (A) in the presence (1) and in the absence (2) of sodium
dithionite in aqueous solution, and (B) in the presence of BSA
Fluorescence microscopy on the new histochemical staining of nitrotyrosine has been performed in
tissue sections obtained from animal models on inflammatory diseases and was compared with
immunohistochemical staining using an anti-nitrotyrosine antibody. Figure 2 shows staining of fresh
frozen liver sections from mice that were treated with carbon tetrachloride (CCl4) in olive oil, which
induces chronic inflammation, in comparison with liver sections from vehicle (olive oil) treated mice
as negative control. The anti-nitrotyrosine staining of tissue from olive oil treated mice (Figure 6E)
shows a background signal that cannot be attributed to nitrotyrosine. A section from the same tissue
with the novel histochemical shows little staining (Figure 6F). Intensity measurement of the
background fluorescence shows that the antibody staining gives significantly higher background
intensities (P<0.01, see Figure 10). In contrast tissue sections from CCl4 treated mice (Figure 6A-D),
show a distinct staining pattern with both methods, which confirms that nitrotyrosine is a marker for
inflammation. Figure 6A and 6B show a comparable staining on the same location in a tissue section.
Interestingly, the novel histochemical staining gives much less background fluorescence and shows
clear cellular structures (Figure 6E-F). Some differences were observed between the novel
histochemical staining and the antibody staining, which could possibly be attributed to reduced
variants of the 2-nitrophenol functionality. The histochemical staining in inflamed liver tissue (Figure
6D) shows a non-homogenous distributed around the nucleus in the cytoplasm (Figure 7), which is
consistent with literature data for immunohistochemistry on nitrotyrosine.15,16 The novel
histochemical staining provided no signal if the sodium dithionite reduction was not performed, which
shows that the staining can be attributed to 2-nitrophenol functionalities (Figure 8).
0,0
2000,0
4000,0
6000,0
8000,0
10000,0
12000,0
1 2
flu
ore
sce
nce
un
it
Samples
0,00
20,00
40,00
60,00
80,00
100,00
120,00
0,00E+00 4,00E-06 8,00E-06 1,20E-05 1,60E-05
Flu
ore
sce
nce
pre
sen
tag
e
(%)
BSA concentration (M)
(B) (A)

129
Antibody-free detection of protein tyrosine nitration in tissue sections
Figure 6. Fluorescent staining of nitrotyrosine in fresh frozen mice liver tissue by immunohistochemical
staining using rabbit polyclonal anti-nitrotyrosine - goat-anti rabbit-FITC (A, C, E) and the novel histochemical
staining using Na2S2O4 followed by Al3+ and salicylaldehyde (B, D, F). A, B, C and D are tissue section from
CCl4 treated mice and E and F from vehicle treated mice. Magnification (20X 1.0X) for A, B, E and F and (40X
1.6X) for C and D.
Figure 7. Novel histochemical staining of fresh frozen liver tissue sections from mice that were treated with
CCl4. Picture A1, A2 and A3 is the overlay picture from filter A [UV light: 340-380 nm excitation filter and 425
nm emission filter], Filter +L5 [blue light : 480/40 nm excitation filter and 527/30 nm emission filter], and Filter
+N3 [green light : 546/12 nm excitation filter and 600/40 nm emission filter]. Picture B was the picture obtained
using filter A, Picture C was obtained using filter +L5, whereas Picture D was obtained using filter +N3. In the
overlay picture the picture from every filter was artificially colored (Filter A colored blue, Filter +L5 colored
green and filter +N3 colored red) to distinguish the fluorescence signals. The fluorescences which were
observed with filter +N3 (colored as red or bright purple in the overlay pictures) were not derived from the
Aluminium-complex.

130
Antibody-free detection of protein tyrosine nitration in tissue sections
Vehicle treated mice CCl4 in olive oil treated mice
Figure 8. Control experiment of the novel histochemical staining of 2-nitrophenol functionalities in fresh frozen
liver tissue sections from mice that were challenged with CCl4 in olive oil (B and D) and vehicle (olive oil)
treated mice (A and C). Liver tissue sections from vehicle (olive oil) treated mice with sodium dithionite
reduction (A) and without sodium dithionite reduction (C) showed no fluorescence. Distinct fluorescence in
liver tissue section of CCl4 treated mice (B). Liver tissue sections from a CCl4 treated mice (D) without sodium
dithionite reduction showed modest fluorescence. Magnification for (A) and (B) 40X, 1.0X and for (C) and (D)
20X 1.0X
Figure 9. Fluorescent staining of nitrotyrosine in paraffin embedded inflamed human trans colon tissue (A-D)
by immunohistochemical staining using Rabbit Polyclonal anti-nitrotyrosine - goat-Anti Rabbit FITC
conjugated (A and C) and the novel histochemical staining using Na2S2O4 followed by Al3+ and salicylaldehyde
(B and D). A (area enlargement in C) and B (area enlargement in D) (20X 1.0X) magnification.
C D
A B
with Na2S2O4 reduction
without Na2S2O4 reduction

131
Antibody-free detection of protein tyrosine nitration in tissue sections
Figure 10. Background fluorescence intensity of tissue sections stained with Aluminium complex and with anti-
nitrotyrosine antibody (P<0.01). Statistical significance of different was evaluated with difference at P<0.01 is
considered statistically different.
Figure 11. Immunohistochemical or the novel histochemical staining of paraffin embedded inflamed ileum
tissue sections from a patient with inflammation bowel disease (IBD). The stained was performed with Rabbit
Polyclonal anti-nitrotyrosine – Goat Anti-Rabbit/FITC (A1 and A2) and with salicylate-aluminium complex (B-
D). Picture B1 and B2 is the overlay picture from filter A [UV light : 340-380 nm excitation filter and 425 nm
emission filter], Filter +L5 [blue light : 480/40 nm excitation filter and 527/30 nm emission filter], and Filter
+N3 [green light : 546/12 nm excitation filter and 600/40 nm emission filter]. Picture C was the picture obtained
using filter A, whereas Picture D was obtained using filter +N3. In the overlay picture the picture from every
filter was artificially colored (Filter A colored blue, Filter +L5 colored green and filter +N3 colored red) to
distinguish the fluorescence signals. The fluorescences which were observed with filter +N3 (colored as red or
bright purple in the overlay pictures) were not derived from the Aluminium-complex.
The novel histochemical staining was tested on paraffin embedded human colon tissue sections
from a patient with inflammatory bowel disease (IBD). The material was kindly provided by Gerard
Dijkstra, MD, Ph.D. from University Medical Centre Groningen, Department of Gastroenterology and
Hepatology. (Informed, signed consent was obtained from the patient.) The results show a fluorescent
staining in inflamed transverse colon tissue with the antinitrotyrosine immunostaining as well as with
the novel histochemical staining, which is probably located in lymphoid cells (Figure 9C and D, see
also Figure 11). The colon tissue shows also fluorescence if the histochemical staining was applied
with no sodium dithionite reduction. This shows that the tissue samples contain fluorescent
components that interfere with the nitrotyrosine detection. Nevertheless, distinct differences were
0,00
20,00
40,00
60,00
80,00
Al-complex Antibody
Flu
ore
sce
nce
In
ten
sity
Staining Method

132
Antibody-free detection of protein tyrosine nitration in tissue sections
observed between the histochemical staining with and without reduction (Figure 12A-B). This
demonstrates that control experiments without reduction need to be included in these studies. A
similar result was obtained if the primary antibody was pre-absorbed with the antigen (Figure 12C-D).
Furthermore, this method was also tested in fresh frozen lung sections from guinea pigs that were
repeatedly challenged with ovalbumin as a model for chronic allergic asthma. However, in the
histochemical staining no clear difference was observed between the experiments with sodium
dithionite reduction and the negative controls without sodium dithionite reductions (Figure 13 A and
B). A similar result was obtained for the immunohistochemical staining and the negative experiment
in which the antigen was preabsorbed with the antibody (Figure 13 C and D). Therefore, we conclude
that it is not possible to detect nitrotyrosine in the guinea pig tissue sections using these both methods.
Figure 12. Immunohistochemical or the novel histochemical staining of paraffin embedded inflamed transverse
colon tissue sections from IBD patient. The staining was performed with the novel histochemical staining (A-B)
without sodium dithionite reduction (A) and with sodium dithionite reduction (B). Additional positive signals
(yellow arrow) were observed from the transverse colon tissue section with sodium dithionite reduction.
Immunohistochemical staining (C-D) using Rabbit Polyclonal anti-nitrotyrosine - goat-Anti Rabbit-FITC (D)
and pre-incubated anti-nitrotyrosine antibody with the antigen (C). Magnification is (20X 1.0X) for A-D.
Figure 13. Immunohistochemical or the novel histochemical staining of fresh frozen lung tissue sections from
ovalbumin treated guinea pig. The novel histochemical staining was performed without sodium dithionite
reduction in A and with sodium dithionite reduction in B. Immunohistochemical (C-D) was stained using Rabbit
Polyclonal anti-nitrotyrosine - goat-Anti Rabbit-FITC (D) and pre-incubated anti-nitrotyrosine antibody with the
antigen (C). Magnification is (20X 1.0X) for A-D. There is no distinct signal from a negative control with the
result from novel method as well as from the antibody staining.

133
Antibody-free detection of protein tyrosine nitration in tissue sections
Labeling of proteins by techniques such as activity-based protein profiling 17 is currently often
performed with inhibitors that are directly linked to a fluorophore or inhibitors that contain an alkyne
or azide functionality that can be conjugated to a fluorophore via ’click chemistry’.18 We evaluated
the applicability of the 2-nitrophenol functionality for fluorescent detection of labeled proteins in
biological matrices using SDS-PAGE and electroblotting on PVDF membrane. For this purpose
bovine serum albumin (BSA) was conjugated to 3-nitro-4-hydroxyphenylacetic acid (NHPA). The
NHPA conjugated BSA was subjected to SDS PAGE followed by electrotransfer to PVDF membrane.
Figure 14. Detection of BSA conjugated to NHPA after SDS-PAGE and electrotransfer to PVDF membrane
using either immunostaining with antinitrotyrosine (A) or fluorescent staining by conversion of 2-nitrophenol
functionalities into a fluorophore (B-C). (A) Lane 1 - 7.0 µg unconjugated BSA and Lane 2 to 5 contain
respectively 0.8, 0.4, 0.04 and 0.01 nmol NHPA conjugated-BSA. (B) Lane 1 – 7.0 µg non-conjugated BSA and
Lane 2 to 5 contain respectively 0.4, 0.1, 0.04 and 0.01 nmol NHPA conjugated-BSA. (C) Fluorescent staining
NHPA conjugated BSA in complex matrices guinea pig blood plasma. The guinea pig blood plasma samples
were standardized on 7µg total protein content with NHPA conjugated-BSA addition in various concentrations.
Lane 1 to 4 contain respectively guinea pig blood plasma enriched with 0.4, 0.1, 0.04, 0.01 nmol NHPA
conjugated-BSA
Figure 15. Western blot of NHPA conjugated-BSA with rabbit polyclonal anti-nitrotyrosine – swine ati
rabbit/HRP. Lane 1 – 7.0 µg untreated BSA; Lane 2 - 0.4 nmol of NHPA conjugated-BSA; Lane 3 – 0.01 nmol
of NHPA conjugated-BSA; Lane 4 – 0.5 pmol of NHPA conjugated-BSA;
The PVDF membrane was stained with either an anti-nitrotyrosine antibody (Figure 14A) or by
chemical conversion of 2-nitrophenol functionalities into the fluorescent aluminium complex (Figure
14B). Both staining methods did not detect non-conjugated BSA, whereas both methods showed a
concentration dependent staining of NHPA conjugated to BSA in a concentration range between 0.01
and 0.4 nmol NHPA conjugated to BSA per lane. However, bands with less than 0.01 nmol NHPA

134
Antibody-free detection of protein tyrosine nitration in tissue sections
conjugated to BSA were not visible by the fluorescent staining of 2-nitrophenol functionalities by
aluminium complex formation, in contrast to the antinitrotyrosine immunoblot in which 0.5 pmol
NHPA conjugated to BSA per lane was still visible (Figure 15, lane 4). This indicates a detection limit
for the aluminium complex on blot of 0.01 nmol NHPA conjugated to BSA in 20 µL samples, which
is a concentration of 500 nmol/L. The immunoblot is about a factor 20 more sensitive and can detect
concentration down to 25 nmol/L. As a negative control another PVDF membrane with NHPA
conjugated BSA was stained using the anti-nitrotyrosine antibody preincubated with the antigen or the
novel chemical staining without the sodium dithionite reduction (Figure 16). Both controls did not
show a positive signal (Figure 16B, and Figure 16C). This shows that both the antibody as well as the
novel fluorescent staining selectively detect 2-nitrophenol functionalities on PVDF membranes.
Figure 16. Western blot of NHPA conjugated-BSA with rabbit polyclonal anti-nitrotyrosine – swine ati
rabbit/HRP staining (A-B) and with the novel fluorescent staining (C-D). PVDF membranes stained either by
anti-nitrotyrosine antibody preincubated with the antigen (B) or the novel fluorescent staining sodium dithionite
reduction (D) gives no visible signals. A.) Western blot with anti-nitrotyrosine antibody staining; Lane 1 – 0.01
nmol nmol of NHPA conjugated-BSA; Lane 2 – 7 µg total protein of blood plasma with an addition of 0.4 nmol
of NHPA conjugated-BSA; Lane 3 – 0.4 nmol of NHPA conjugated-BSA; Lane 4 – 7 µg of untreated BSA; B.)
Western blot with anti-nitrotyrosine antibody that was preincubated with the antigen; Lane 5 - 7 µg of untreated
BSA; Lane 6 - 0.4 nmol of NHPA conjugated-BSA; Lane 7 - 7 µg total protein of blood plasma with an addition
of 0.4 nmol of NHPA conjugated-BSA; Lane 8 - 0.01 nmol nmol of NHPA conjugated-BSA. C.) Western blot
with the novel fluorescent staining without sodium dithionite reduction; Lane 1 – 0.01 nmol nmol of NHPA
conjugated-BSA; Lane 2 – 7 µg total protein of blood plasma with an addition of 0.4 nmol of NHPA
conjugated-BSA; Lane 3 – 0.4 nmol of NHPA conjugated-BSA; Lane 4 – 7 µg of untreated BSA; D.) Western
blot with the novel fluorescent staining with sodium dithionite reduction; Lane 5 - 7 µg of untreated BSA; Lane
6 - 0.4 nmol of NHPA conjugated-BSA; Lane 7 - 7 µg total protein of blood plasma with an addition of 0.4
nmol of NHPA conjugated-BSA; Lane 8 - 0.01 nmol nmol of NHPA conjugated-BSA.
In order to evaluate the 2-nitrophenol detection in complex biological matrices blood plasma from
guinea pigs was enriched with NHPA conjugated BSA (Figure 14C and Figure 16D Lane 7). The
novel staining method still detected NHPA conjugated BSA in the concentration range between 0.01
nmol and 0.4 nmol (Figure 14C). The presence of blood plasma did not significantly alter the
fluorescent signal of the 2-nitrophenol staining. This demonstrates the utility of this novel method to
detect 2-nitrophenol functionalities in complex biological matrices on western blot without extensive
sample pre-treatment. The endogenous levels of tyrosine nitrated proteins in tissue homogenates or
blood plasma are usually very low, in contrast to the localized nitrotyrosine concentrations, which

135
Antibody-free detection of protein tyrosine nitration in tissue sections
proved to be relatively high (Figure 6 and 9). Total 3-nitrotyrosine levels of 1 nmol/L to 4 nmol/L in a
human blood plasma have been reported 19. Guinea pig blood plasma samples were subjected to SDS-
PAGE and immunoblot in order to study endogenous tyrosine nitration (Figure 17B, lane 9 and 11).
However, the nitrotyrosine levels were below the detection limit of the immunoblot and therefore the
samples were enriched using immunoprecipitation (IP) (Figure 17B, lane 8 and 10). Nevertheless, the
immunostaining of nitrotyrosine after enrichment with IP indicate nitrotyrosine levels that are close to
the detection limit of the immunoblot and consequently no signal was observed upon application of
the fluorescent 2-nitrophenol staining of the corresponding guinea pig blood samples (Figure 17A).
We conclude that the levels of endogenous tyrosine nitrated proteins in blood plasma samples are too
low for detection with the new 2-nitrophenol staining.
Figure 17. Detection of nitrotyrosine in guinea pig blood plasma samples with the novel fluorescent staining
method (A) compared with antinitrotyrosine immunoblot detection using Rabbit Polyclonal Antinitrotyrosine –
Swine Anti Rabbit/HRP (B). As described in the protocol ‘preparation in blood plasma samples’, 1 mg total
protein content of the guinea pig blood plasma was subjected for immunoprecipitation before it was loaded onto
SDS PAGE gel (lane 1, lane 8 and lane 10) whereas for lane 2, lane 9 and lane 11, the guinea pig blood plasma
samples were standardized on 7 µg total protein content. (A) Lane 1 – plasma blood of OVA challenged guinea
pig with immunoprecipitation; Lane 2 - plasma blood of OVA challenged guinea pig without
immuneprecipitation; Lane 3 - 0.4 nmol NHPA conjugated-BSA; Line 4 – 7.0 µg untreated BSA. (B) Lane 5 –
7.0 µg untreated BSA; Lane 6 - 0.4 nmol NHPA conjugated-BSA; Lane 7 - 0.01 nmol NHPA conjugated-BSA;
Lane 8 – plasma blood of OVA challenged guinea pig with immuneprecipitation; Lane 9 - plasma blood of
OVA challenged guinea pig without immunoprecipitation; Lane 10 – plasma blood of saline challenged guinea
pig blood plasma with immunoprecipitation; Lane 11 – plasma blood of saline challenged guinea pig blood
plasma without immunoprecipitation.
Conclusion
In conclusion, we have developed a novel fluorescent staining method for the detection of tyrosine
nitrated proteins in tissue sections using fluorescence microscopy. The staining is also applicable for
detection of proteins that are labelled with 2-nitrophenol functionalities in complex biological
matrices on western blot. The staining involves reduction of the 2-nitrophenol functionality to a 2-
aminophenol functionality, which is subsequently reacted with salicylaldehyde and aluminium to
form a fluorescent complex. The fluorescent staining is chemoselective for the 2-nitrophenol
functionality of nitrotyrosine and its reduced analogs such as 2-nitrosophenol. The novel fluorescent
staining provides a similar staining pattern as observed for the anti-nitrotyrosine antibody. Moreover,

136
Antibody-free detection of protein tyrosine nitration in tissue sections
the novel staining provides lower background fluorescence in mice liver tissue and shows a more
distinct staining pattern. In addition, the fact that the novel fluorescent staining stains 2-nitrophenol
functionalities and also reduced its analogues such as 2-nitrosophenol provides a broader detection
method towards oxidative nitration of protein bound tyrosine. The newly described fluorescent
staining of 2-nitrophenol functionalities is less labour intensive and employs commercially available
reagents that are cheap and stable upon prolonged storage, which are significant advantages. Thus, the
novel histochemical staining is superior to the commonly used immunohistochemical staining for
nitrotyrosine in tissue sections.
Experimental
Materials
All reagents and solvents were obtained from commercial suppliers (Sigma, Aldrich, Across Organic,
Fluka) and were used without further purification. Bovine serum albumin (Fraction V powder, 96%)
was purchased from Sigma. Anti-Nitrotyrosine, clone 1A6, agarose conjugate (mouse monoclonal
IgG) and anti-nitrotyrosine (Rabbit Polyclonal IgG) were provided by Upstate-Millipore Inc. Swine
anti rabbit/Horseradish Peroxidase antibody was purchased from Dako Cytomation. Goat anti
rabbit/FITC antibody was provided by Abcam.
Fluorescence properties of the aluminium complex with the Schiff base of salicylaldehyde
and 2-aminophenol
A solution of salicylaldehyde (5 µL, 0.1 M) in DMSO and a solution of AlCl3 (5 µL, 0.1 M) in H2O
were added to a solution of 2-aminophenol (5 µL, 0.1 M) in DMSO. The resulting solution was
diluted with either methanol or demi water to get a final concentration of 10 µM. After 90 minutes
incubation at room temperature the emission and excitation spectra were recorded using a
spectrofluorometer (SLM Amico SPF-500C). The amplification factor (HV) for measurement in H2O
was 1000 and for measurement in methanol 800. Subsequently, the fluorescence intensity with λ ex
412 nm and λ em 515 nm was determined over a concentration range of 0.31 µM and 10 µM (Figure
3). Furthermore, a solution of salicylaldehyde (5 µL, 0.1 M) in DMSO and a solution of AlCl3 (5 µL,
0.1 M) in H2O were mixed and diluted with H2O to a concentration of 10 µM. The excitation and
emission spectra were measured in order to estimate the background signal for the combination of
both reagents. The UV absorption of 2-nitrophenol was determined using UV extinction at 405 nm in
a 96 well plate reader. The UV absorption was determined in phosphate buffer pH 7.5 in a
concentration range of from 20 µM to 1200 µM. Reduction of 2-nitrophenol with sodium dithionite
(Na2S2O4) in aqueous environment proved to proceed very quickly as determined by the UV
absorption of 2-nitrophenol at 405 nm.

137
Antibody-free detection of protein tyrosine nitration in tissue sections
Aniline and 1-propanamine in combination with salicylaldehyde and aluminium at 10 µM did not
provide a measureable signal.
Conjugation of NHPA to BSA
A conjugate of bovine serum albumin (BSA) with 3-nitro-4-hydroxyphenylacetic acid (NHPA) was
used as a positive control to mimic tyrosine nitrated proteins (Scheme 1). The conjugate was prepared
by incubation of NHPA (99 mg, 0.50 mmol) in CH3CN (0.5 mL) with ethyl chloroformate (96 µL, 1.0
mmol) and triethylamine (146 µL, 1.00 mmol) with magnetic stirring at room temperature for 2 hours.
This yields {4-[(ethoxycarbonyl)oxy]-3-nitrophenyl} acetyl ethyl carbonate as a brown solution with
a white precipitate. The supernatant (100 µL) was added to 900 µL 5 mg/mL BSA solution in
carbonate buffer (0.1 M, pH 9.6) and the resulting mixture was reacted overnight at room temperature.
Subsequently, the mixture was dialyzed using a membrane cutoff of 10.000 Dalton in order to remove
the non-conjugated NHPA. The dialysis was performed in a Slidelyser with phosphate buffer (0.1 M,
pH 7.5). The buffer was changed 2 times with 2 hour intervals followed by dialysis overnight at 4 oC.
The NHPA conjugated BSA was obtained as a yellow solution. The conjugation of NHPA to BSA
was quantified using UV absorption at 405 nm. The UV extinction was calibrated on 2-nitrophenol
(2.44 µM to 0.625 mM) in Tris buffer (0.1M, pH 7.5). The result shows that about 3.75 NHPA
molecules were conjugated to BSA.
Scheme 1. Synthesis of NHPA labeled BSA.
Preparation of blood plasma samples
Blood plasma samples for analysis were subjected to protein determination using the Bradford protein
assay. The samples were diluted with demiwater to provide 5 mg/mL protein content and 1.4 µL was
mixed with 23.6 µL sample buffer (6 µL Laemmli 4X, 16.6 µL H2O and 1 µL β-mercaptoethanol).
After 3 minutes incubation at 110 °C, 20 µL of sample mixture was loaded into SDS PAGE.
Immunoprecipitation with an antinitrotyrosine antibody was performed to increase the
nitrotyrosine content of the blood plasma samples. For this purpose, blood plasma samples were
subjected to immunoprecipitation using anti-Nitrotyrosine, clone 1A6, agarose conjugate (mouse
monoclonal IgG – Upstate/Millipore). The blood plasma samples were diluted to a protein
concentration of roughly 1 mg/mL, and subsequently 10 µL gel slurry of the anti-nitrotyrosine,
agarose conjugate was added into 1 mL of diluted plasma blood sample. The mixture was gently

138
Antibody-free detection of protein tyrosine nitration in tissue sections
rocked at 4 oC overnight. The agarose beads were collected by centrifugation (10 seconds 14,000
rpm), the supernatant was drained off and then the beads were washed 3-4 times with PBS buffer. The
beads were resuspended in 62 µL Laemmli sample buffer (45 µL H2O, 15 µL Laemmli 4x, and 2 µL
-mercaptoethanol). The beads suspension was heated 3 minute at 110 oC. The agarose beads were
separated from the supernatant by centrifugation and the supernatant (20 µL) was load in SDS-PAGE.
Animal material
Animal material was obtained from animals experiments that were approved by the University of
Groningen Committee for animal experiments (DEC-RUG) for the investigators H. Maarsingh and K.
Poelstra. Liver tissue sections for microscopy were obtained from male balb/c mice (20-22g) that
were treated with olive oil, as control, or increasing doses of carbon tetrachloride CCl4 in olive oil, to
induce inflammation, twice weekly by intra-peritoneal injections for 8 weeks (week 1: 0.5 ml/kg;
week 2: 0.8 ml/kg and week 3-8: 1 ml/kg prepared in olive oil). The livers were cut with a cryostat (4
mm thickness), dried and stored at -20 °C.
Lung tissue slices and blood plasma samples for microscopy were obtained from guinea pigs. The
guinea pigs were challenged with ovalbumin in saline (OVA) (Sigma, St. Louis, MO) or saline 20.
Inhalations of an increasing concentration of OVA in saline (0.05%, 0.1%, 0.3%, 0.5% and 0.7% w/v)
were applied for 3 minutes for each concentration with 7 minutes interval in-between. No
antihistaminic agent was needed to prevent anaphylactic shock. At 24 h after the last challenge, the
guinea pigs were sacrificed, the blood was taken from their hearts and the main bronchi from both left
and right lung lobes were cut with a cryostat (4 μm). The slices were dried and stored at -20°C before
further processing. The blood samples were centrifuged (14,000 rpm) to separate the blood plasma
and aliquots (1 mL) of blood plasma were stored at -20 °C before analysis.
Detection of nitrotyrosine in tissue sections using fluorescence microscopy
Fresh frozen tissue sections (4 µm) were dried using a hair blower (20 minutes, room temperature)
and fixed with acetone for 15 minutes. Subsequently, the tissue sections were dried again using the
hair blower (20 minutes). After fixation, the sections were subjected for immunohistochemical
staining using an antinitrotyrosine antibody or the novel histochemical staining.
For the immunohistochemical staining, 400 µL blocking solution (1% BSA in cold PBS) was
added per slide of tissue sections and incubated for 3 hours. After that period, the blocking solution
was drained from the sections and 100 µL of diluted rabbit IgG polyclonal anti-nitrotyrosine antibody
(3:250) was applied per slide. The sections were then incubated overnight at 4°C. Afterward the
sections were washed 4 times in 1X PBS-T (PBS with 0.05% Tween 20) for 5 minutes and then 100
µL of diluted goat anti-rabbit/FITC conjugated antibody (1:50) was applied. The sections were
incubated at room temperature for 1 hour and washed 2 times with 1X PBS-T and 3 times with PBS
(5 minutes per washing step).

139
Antibody-free detection of protein tyrosine nitration in tissue sections
For the histochemical staining, the tissue sections were incubated with freshly prepared aqueous
sodium dithionite solution (400 µL, 10 mM) for 10 minutes. The sections were washed with demi-
water (3 times, 5 minutes) and subsequently incubated overnight with 500 µL aqueous solution of
AlCl3 (200 µM) and salicylaldehyde (200 µM) at 4 °C. The next day, the AlCl3 and salicylaldehyde
solution was removed and a few drops of Citifluor (glycerol/PBS) solution were added before the
cover slips were applied to prevent fluorescence bleaching. Microscopy was performed using a Leica
DAS Microscope LEITZ DMR and photos were made using a digital camera Leica DC350FX
connected to Leica-QWin software. The filter sets used for fluorescence detection were: Filter A
(UV): 340-380 nm excitation filter and 425 nm emission cutoff filter, Filter +L5 (blue): 480/40 nm
excitation filter and 527/30 nm emission filter, Filter +N3 (green): 546/12 nm excitation filter and
600/40 nm emission filter.
Negative controls for the immunohistochemical staining were performed by substitution of the
primary antibody with the antibody that was preabsorbed with 10 mM NHPA in PBS buffer for 1 hour
at room temperature. For the negative control of the histochemical staining, the tissue sections were
not incubated with the sodium dithionite solution. The histochemical staining in the tissue section was
stable for prolonged period of time. The fluorescence was still visible in the same intensity after the
slides were stored for 1 week at 4 ºC (Figure 18). The background intensity measurement was carried
out digitally using quantification software (ImageJ). Statistical significance of differences was
evaluated using the T-test in which differences at P<0.01 is considered statistically significant.
Figure 18. Immunohistochemical or the novel histochemical staining of fresh frozen liver tissue sections from
mice that were treated with CCl4. The staining was observed at 1 day and 6 days after the immunohistochemical
or the histochemical staining. Rabbit anti-nitrotyrosine – goat anti rabbit/FITC (A and B) and 2-aminophenol,
salicylaldehyde, aluminium complex (C and D) stained liver tissue with enlargement after prolonged storage at
4°C.
Fluorescent staining of 2-nitrophenol on western blot
NHPA-conjugated BSA or blood plasma samples were loaded on SDS PAGE (20 µL samples on 1.5
mm SDS-PAGE). The voltage for electrophoresis was set to 70 V for the stacking gel and was

140
Antibody-free detection of protein tyrosine nitration in tissue sections
increased to 110 V for the resolving gel. Subsequently, the proteins were electroblotted (2 hours, 200
mA) on a PVDF membrane. Subsequently, the PVDF membrane was incubated with 16 mL
phosphate buffer (0.5 M, pH 7.5) containing 10 mM sodium dithionite for 10 minutes at room
temperature. As a control, the PVDF membrane was washed directly with demi water without sodium
dithionite incubation. Subsequently, the PVDF membrane was washed with demi water 2 times for 1
hour. Finally, the membrane was incubated with an aqueous solution (16 mL) containing AlCl3 (200
µM) and salicylaldehyde (200 µM) overnight at room temperature. The membrane was dried with a
tissue paper before measurement. The fluorescent aluminium complex was visualized using the blot
scanner Fuji Film LAS-4000 with λex = 365 nm and λem = 515 nm and the images were recorded using
an Image Reader LAS-4000 ver. 2.0.
Immunostaining of nitrotyrosine on western blot
NHPA conjugated-BSA was used as a reference protein for the immunostaining of nitrotyrosine on
western blot. After electrophoresis with SDS PAGE and electrotransfer to PVDF membrane, the
membrane was blocked with 5% milk powder in PBS overnight at 4°C. The membrane was washed 3
times with PBS-T, incubated 1 hour with Rabbit monoclonal antibody raised against nitrotyrosine
(Upstate, 06-284) in PBS (1:400). The negative control was performed by pre-incubating the anti
nitrotyrosine antibody with 10 mM NHPA for 1 hour at room temperature. The membrane was
washed 3 times with PBS-T for 10 minutes and incubated for 2 hours with swine anti rabbit/HRP
antibody in PBS (1:200). After 2 hours of incubation, the membrane was washed 3 times with PBS
before stained with enhanced chemiluminescence (ECL) (Amersham biosciences).
References
1. Ohshima, H.; Friesen, M.; Brouet, I.; Bartsc, H. Nitrotyrosine as a new marker for endogenous nitrosation
and nitration of proteins. Fd Chem.Toxic. 1990, 28, 647-652.
2. Beckman, J. S.; Ischiropoulos, H.; Ling Zhu; van der Woerd, M.; Smith, C.; Jun Chen; Harrison, J.;
Martin, J. C.; Tsai, M. Kinetics of Superoxide Dismutase- and Iron-Catalyzed Nitration of Phenolics by
Peroxynitrite. Arch.Biochem.Biophys. 1992, 298, 438-445.
3. van der Vliet, A.; Eiserich, J. P.; Oneill, C. A.; Halliwell, B.; Cross, C. E. Tyrosine Modification by
Reactive Nitrogen Species: A Closer Look. Arch.Biochem.Biophys. 1995, 319, 341-349.
4. van der Vliet, A.; Eiserich, J. P.; Shigenaga, M. K.; Cross, C. E. Reactive Nitrogen Species and Tyrosine
Nitration in the Respiratory Tract. Am. J. Respir. Crit. Care Med. 1999, 160, 1-9.
5. Dijkstra, G.; Moshage, H.; Van Dullemen, H. M.; De Jager-Krikken, A.; Tiebosch, A. T. M. G.;
Kleibeuker, J. H.; Jansen, P. L. M.; Van Goor, H. Expression of nitric oxide synthases and formation of
nitrotyrosine and reactive oxygen species in inflammatory bowel disease. J. Pathol. 1998, 186, 416-421.
6. Mapp, P. I.; Klocke, R.; Walsh, D. A.; Chana, J. K.; Stevens, C. R.; Gallagher, P. J.; Blake, D. R.
Localization of 3-Nitrotyrosine to Rheumatoid and Normal Synovium. Arthritis Rheum. 2001, 44, 1534-
1539.
7. Hanazawa, T.; Kharitonov, S. A.; Barnes, P. J. Increased Nitrotyrosine in Exhaled Breath Condensate of
Patients with Asthma. Am. J. Respir. Crit. Care. Med. 2000, 162, 1273–1276.

141
Antibody-free detection of protein tyrosine nitration in tissue sections
8. Abello, N.; Kerstjens, H. A. M.; Postma, D. S.; Bischoff, R. Protein Tyrosine Nitration: Selectivity,
Physicochemical and Biological Consequences, Denitration, and Proteomics Methods for the Identification
of Tyrosine-Nitrated Proteins. J. Proteome Res. 2009, 8, 3222-3238.
9. Nikov, G.; Bhat, V.; Wishnok, J. S.; Tannenbaum, S. R. Analysis of nitrated proteins by nitrotyrosine-
specific affinity probes and mass spectrometry. Anal. Biochem. 2003, 320, 214-222.
10. Söderling, A. S.; Ryberg, H.; Gabrielsson, A.; Lärstad, M.; Torén, K.; Niari, S.; Caidahl, K.
A derivatization assay using gaschromatography/negative chemical ionization tandem mass spectrometry
to quantify 3-nitrotyrosine in human plasma. J. Mass Spectrom. 2003, 38, 1187-1196.
11. Sharov, V. S.; Dremina, E. S.; Galeva, N. A.; Gerstenecker, G. S.; Li, X.; Dobrowsky, R. T.; Stobaugh, J.
F.; Schöneich, C. Fluorogenic Tagging of Peptide and Protein 3-Nitrotyrosine with 4-(Aminomethyl)-
benzenesulfonic Acid for Quantitative Analysis of Protein Tyrosine Nitration. Chromatographia 2010, 71,
37-53.
12. Haddad, Y. I.; G., P.; Hu, P.; Galliani, C.; Beckman, J. S.; Matalon, S. Quantittation of Nitrotyrosine
Levels in Lung Sections of Patients and Animals with Acute Lung Injury. J. Clin. Invest. 1994, 94, 2407-
2413.
13. MacMillan-Crow, L. A.; Thompson, J. A. Immunoprecipitation of nitro-tyrosine containing proteins.
Methods Enzymol. 1999, 301, 135-145.
14. Morisige, K. Metal complexes of aromatic schiff base compounds : Part I. The fluorescence properties of
aluminium and gallium complexes of aromatic schiff bases and their use in fluorimetry. Anal. Chim. Acta
1974, 72, 295-305.
15. Ahn, B.; Han, B. S.; Kim, D. J.; Ohshima, H. Immunohistochemical localization of inducible nitric oxide
synthase and 3-nitrotyrosine in rat liver tumors induced by N-nitrosodiethylamine. Carcinogenesis 1999,
20, 1337-1344.
16. Hinson, J. A.; Michael, S. L.; Ault, S. G.; Pumford, N. R. Western Blot Analysis for Nitrotyrosine Protein
Adducts in Livers of saline-treated and acetaminophen-treated mice. Toxicol. Sci. 2000, 53, 467-473.
17. Cravatt, B. F.; Wright, A. T.; Kozarich, J. W. Activity Based Protein Profiling - From Enzyme Chemistry
to Proteomic Chemistry. Annu. Rev. Biochem. 2008, 77, 383-414.
18. Speers, A. E.; Cravatt, B. F. Profiling Enzyme Activities In Vivo Using Click Chemistry Methods. Chem.
Biol. 2004, 11, 535-5446.
19. Radabaugh, M. R.; Nemirovskiy, O. V.; Misko, T. P.; Aggarwal, P.; Mathews, W. R. Immunoaffinity
liquid chromatography-tandem mass spectrometry detection of nitrotyrosine in biological fluids:
Development of clinically transatable biomarker. Anal. Biochem. 2008 , 380, 68–76.
20. Meurs, H.; Santing, R. E.; Remie, R.; van der Mark, T. W.; Westerhof, F. J.; Zuidhof, A. B.; Bos, I. S. T.;
Zaagsma, J. A guinea pig model of acute and chronic asthma using permanently instrumented and
unrestrained animals. Nature Protocol 2006, 1, 840-847.

Summary,
General Discussion and
Future Perspectives
6

144
Summary, General Discussion and Future Perspectives
SUMMARY
Modulation of Lipoxygenase activity by Anacardic acid Derivatives
Inflammation is a major health problem in the western world. This disease is influenced by numerous
signaling molecules and signal transduction pathways. The NF- B signaling pathway is one of the
main pathways that is involved in the regulation of gene transcription in inflammation and cancer.
Furthermore, the activation and suppression of the NF- B signaling pathway are related to the activity
of inflammatory enzymes such as for example lipoxygenases.
Lipoxygenases (LOXs) are non-heme iron containing enzymes that catalyze the oxidation of cis-
unsaturated fatty acids. LOXs are divided into several sub-families such as 5-LOX, 8-LOX, 12-LOX
and 15-LOX based on their selectivity to oxidize cis-unsaturated fatty acids. These oxidized fatty
acids are mediators that are crucial for the initiation and also the termination of inflammatory
processes. In addition, increasing evidence shows that the development of different types of cancer is
associated with the activity of LOXs and the presence of their metabolites. Due to their relevance in
diseases, the development of small molecule modulators of LOX enzymes is important. These
molecules can be employed as tools to investigate these enzymes in their physiological and
pathological context. Moreover, small molecule modulators of enzyme activity can be employed as
starting points to develop new therapeutic approaches.
In chapter 2 we describe the synthesis of a set of anacardic derivatives in which the substituents in
the 4- and 6- position of the salicylate core were varied. The compound collection was screened in
vitro for inhibition of soybean 15-lipoxygenase-1 (SLO-1) and potato 5-lipoxygenase (potato 5-LOX)
to determine structure-activity relationships. We identified compound 2.23, with a hydroxyl group and
a 4-heptylphenethyl substituent, respectively in the 4- and 6- position of the salicylate core, as the
most potent inhibitor for SLO-1. This compound gives a fivefold improvement of the inhibitory
potency compared to anacardic acid. Kinetic studies of this compound on SLO-1 were performed in
order to resolve its binding mechanism. Compound 2.23 shows a mixed type of inhibition, which
indicates that this inhibitor binds to the free enzyme as well as to the substrate-bound enzyme. Most
likely this inhibitor binds to an allosteric binding pocket, which regulates the catalysis in the active
site of the enzyme.
Interestingly, another derivative 2.21 shows a strong activation of potato 5-LOX with about three
fold activation at concentrations higher than 50 µM. A kinetic study on potato 5-LOX indicates that
this compound is a non-essential activator of the enzymatic reaction, which can occur in the presence
or in the absence of activator. A comparable kinetic model as used for the mixed type inhibition of
SLO-1 can be employed to derive the binding constants. Activator 2.21 shows a relatively high
affinity for the substrate bound enzyme with the αKA of 24 µM and enhances the enzymatic
conversion of the substrate. The kinetic analysis for SLO-1 and potato 5-LOX indicates the presence

145
Summary, General Discussion and Future Perspectives
of a regulatory site in the lipoxygenases that is targeted by anacardic acid and its derivatives. This
provides a novel concept for modulation of lipoxygenase activity.
Figure 1 – Inhibitor and activator of SLO-1 and potato LOX-5 as described in chapter 2.
The anacardic acid structure consists of a hydrophilic salicylate core and a hydrophobic aliphatic
chain and is thus expected to have surfactant characteristics, which can influence the biological
potency by micelle formation. Hence, we investigated the critical micelle concentration (CMC) for
anacardic acid, compound 2.21, and compound 2.23. The CMC values proved to be higher than 100
µM, which indicates that micelle formation does not play a role at concentrations that were used to
investigate enzyme inhibition or activation.
We continued our investigations on anacardic acid and its derivatives as lipoxygenase modulator
on human enzymes. In chapter 3, we screened a collection of compounds based on anacardic acid for
inhibition of human 5-lipoxygenase (h-5-LOX) and compared it to cyclooxygenase-2 (COX-2)
inhibition, which is another oxidative enzyme that plays an important role in inflammation and
cancer. Anacardic acid derivatives show selective modulatory activity against h-5-LOX compared to
COX-2. Compound 2.23 and compound 2.21 which in this chapter were labeled as compound 3.19
and compound 3.21 show no activation and/or inhibition on h-5-LOX and COX-2. These results
demonstrate a different behavior between human lipoxygenase and plant lipoxygenase, which is most
likely due to a lack of similarities in their primary structure. However, activation of h-5-LOX was
observed from another anacardic acid derivative, compound 3.23a. This compound gives 150%
activation of h-5-LOX at a concentration of 12.5 µM. Kinetic studies for this compound on h-5-LOX
reveal that compound 3.23a is a non-competitive activator against the h-5-LOX co-activator ATP and
a mixed non-essential activator against the h-5-LOX substrate linoleic acid. The mixed non-essential
type of activation indicates the presence of an allosteric binding site that regulates the enzyme
activity. The non-competitive behavior with respect to ATP demonstrates that this regulatory site is
distinct from the ATP binding site. Remarkably, the h-5-LOX activator binding constant for 3.23a is
in the nano-molar range (381 nM).
Interestingly, a comparable compound (3.23d) shows inhibition of 5-h-LOX activity. The enzyme
kinetics demonstrates mixed-type inhibition, which is comparable to the mechanism of action of
activator 3.23a. This supports the idea that these compounds bind to a regulatory site in h-5-LOX,
which influences the catalytic processes in the active site. This study indicates the presence of an
allosteric regulatory site that either inhibits or activates enzymatic catalysis based on the structure of
the compound that binds to it.

146
Summary, General Discussion and Future Perspectives
Figure 2 – Inhibitor and activator of human 5-lipoxygenase as described in chapter 3.
Human 5-lipoxygenase has important roles both in the initiation and termination of inflammatory
responses; therefore the modulation of this enzyme is an interesting target for the development of
novel therapeutic approaches. Due to the broad regulatory scope of h-5-LOX metabolites in cancer
and inflammation both inhibitors and activators of this enzyme could provide interesting biological
effects in vivo, which might ultimately be employed for therapeutic purposes.
Several inhibitors for lipoxygenases have been identified and some of them are under investigation
in clinical trial as drugs for treatment of inflammatory diseases. Several inhibitors for the 5-
lipoxygenase sub-family have been clinically used for asthma and arthritis. However, their use is still
limited by a lack of efficacy, selectivity and cell permeability. Considering LOXs important role in
inflammation and cancer, the development of novel classes of small molecule inhibitors of LOXs with
higher selectivity profiles and alternative sub-types selectivities remains necessary.
The studies described in this thesis are focused on the development of chemical tools that can be
used in order to apprehend the regulation of inflammatory processes in association with the NF- B
signaling pathway. In the first part of this thesis, we develop derivatives of the natural product
anacardic acid as novel modulators of lipoxygenase activity. In our first study (chapter 2), we
identify an anacardic acid derivative that selectively inhibits soybean lipoxygenase-1 (SLO-1) and
another derivative that activates potato 5-lipoxygenase (potato 5-LOX). Our subsequent study
(chapter 3) on human 5-lipoxygenase (h-5-LOX) shows that plant enzymes and human enzymes are
modulated by different derivatives. This study led to the identification of a novel inhibitor for 5-
lipoxygenase, which is selective in comparison with cyclooxygenase-2 (COX-2). In addition, we
identified a strong activator for h-5-LOX. Investigations of the effects on the h-5-LOX inhibitors and
activators on signaling via the NF- B pathway are in progress.
We anticipate that both inhibitors and activators of h-5-LOX may find applications in distinct
therapeutic areas. Inhibition of h-5-LOX has been association with the suppression of immune
responses. In contrast, activation of h-5-LOX would be associated with the activation of immune
responses. We anticipate, however, that activation of h-5-LOX might also have beneficial effects in
inflammation since the production of lipoxins by lipoxygenases contributes to the termination of
immune responses. This argues for a more comprehensive understanding of the signaling properties of
lipoxygenase metabolites in their physiological context.

147
Summary, General Discussion and Future Perspectives
Isothiazolones as novel ABPP probes for thiol-dependent enzymes
It has been shown that histone acetylation plays a crucial role in the activation of inflammatory gene
transcription through the NF- B pathway. In previous studies it has been shown that isothiazolones
are small molecule inhibitors of the HAT PCAF and also induce apoptosis in different cancer cell
lines. In chapter 4 of this thesis, we explore the reactivity of isothiazolones towards thiolates and the
selectivity of this compound class for non-related thiol-containing enzymes. In this case we
investigated the selectivity between the HAT PCAF and the cysteine-protease cathepsin B. These
studies provide a starting point to develop this compound class further for the application as probes
for activity based protein profiling (ABPP) for thiol-containing enzymes.
In this study we observed that N-aliphatic substituted 5-chloroisothiazolone 4.11 is the most potent
inhibitor for cathepsin B. Also of interest is that the disulfide 4.7 had a remarkably high potency for
cathepsin B, whereas it did not inhibit the HAT PCAF. In addition, we observed that a selective HAT
PCAF inhibition compared to cathepsin B can be obtained with N-aryl substituent 3-Cl-4-F-phenyl
4.9. In a conclusion, isothiazolones are inhibitors for cysteinyl cathepsin B and HAT PCAF and their
potency and selectivity depend on their substitution pattern.
Figure 3 – Inhibitor of cysteine protease cathepsin B as described in chapter 4.
The reactivity of isothiazolones and 5-chloroisothiazolones towards thiolate originates from the
electronic character of the isothiazolones heterocycle. Crystal structures of isothiazolone and 5-
chloroizothiazolone show an aromatic character of the heterocycle, which provides stability to the
compounds; in contrast the weak sulfur-nitrogen bond allows the reaction with the thiolate to form a
strong disulfide bond.
We analyzed cathepsin B inhibitory mechanism of 5-chloroisothiazolones 4.5 and 4.14 through the
enzyme kinetics. These studies show non-competitive inhibition of cathepsin B by 5-
chloroisothiazolones and a strong substrate inhibition occurs at a higher concentration. Inhibition of
cathepsin B by 5-chloroisothiazolone 4.5 proved to be time dependent and show a decreasing IC50
values overtime, which indicate a fast binding event followed by a slow second binding event. These
data indicate a covalent binding of 5-chloroisothiazolones to cathepsin B. Furthermore, mass
spectroscopy analysis shows that 5-chloroisothiazolone 4.5 binds once or twice to the enzyme, which

148
Summary, General Discussion and Future Perspectives
λex 420 nm λem 520 nm
indicates that 5-chloroisothiazolone 4.5 binds covalently to the thiolate in the enzyme active site
and/or to another surface exposed cysteine of cathepsin B. This study sets the stage to develop the
isothiazolones further for applications as probes for activity-based protein profiling of HATs and
cysteine proteases.
Detection of nitrotyrosine as inflammatory biomarker
Activation of inflammatory pathways leads to oxidative stress and subsequent damage of proteins by
reactive oxygen species (ROS). An important oxidative protein modification is nitration of tyrosine
residues, which has been identified as a biomarker for several inflammatory diseases. In the last part
of this thesis (chapter 5), we describe the development of a novel method to detect nitrotyrosine
containing proteins in biological samples. This novel method involves the conversion of nitrotyrosine
into a fluorophore under mild conditions. This novel method is easy to perform, highly selective for
nitrotyrosine and relies on cheap reagents. It proved to be very efficient in histochemical staining of
inflamed tissue and can also be employed as alternative detection for immunoblotting on PVDF
membranes. As expected, the sensitivity of this novel fluorescence based detection method is lower
than for enzyme linked detection methods, which include enzyme-catalyzed signal amplification.
The method implies two reaction steps; the first step is a conversion of the 2-nitrophenol
functionality into a 2-aminophenol functionality using sodium dithionite, and the second step is a
reaction of the 2-aminophenol functionality with Al3+ and salicylaldehyde to form a fluorophore.
Figure 4 – Fluorescence detection of nitrotyrosine in a tissue slice; reduction of a protein bound 2-nitrophenol functionality
to a 2-aminophenol functionality: a) aqueous Na2S2O4; subsequent conversion into a fluorophore: b) aqueous AlCl3 and
salicylaldehyde.
The fluorescence of the 2-aminophenol - Al3+ - salicylaldehyde complex in demiwater is 8 times
higher than in methanol. Detection in water is the most favorable condition for detection of protein
nitration in biological samples. Furthermore, we investigated the application of this novel method for
histochemical staining in comparison with immunohistochemical staining in different tissue samples.
Remarkably, this novel method shows a clear staining in liver tissue section from CCl4 treated mice,
which is a model for inflammation. The novel histochemical staining was compared to the classical
immunohistochemical staining. Both staining methods showed high intensities in the same locations
of tissue samples; however the classical histochemical staining shows a much higher background
staining. This clearly demonstrates that our newly developed histochemical staining method is, in this
respect, superior to the classical immunohistochemical staining. In addition, the formed fluorophore in
tissue sample was stable for several days with proper storage conditions. These results indicate that

149
Summary, General Discussion and Future Perspectives
this novel histochemical staining method can provide a valuable alternative for detection of protein
tyrosine nitration in tissue samples with high stability upon prolonged storage and lower background
fluorescence signals.
We also evaluated the application of this novel method for fluorescence detection in biological
matrices on western blot. 3-nitro-4-hydroxy phenyl acetic acid (NHPA) was coupled to bovine serum
albumin (BSA) as a model for tyrosine nitrated proteins. This NHPA-conjugated BSA was used as a
standard to optimize the detection method. Our novel staining method enabled the detection of
NHPA-conjugated BSA on western blot. Control experiments with the classically used anti-
nitrotyrosine antibody provided comparable results. The presence of blood plasma did not
significantly alter the fluorescence signal, which demonstrates the applicability of this novel method
to detect 2-nitrophenol in the complex biological matrices. The detection limit for this novel method
is 500 nmol/L, which is about 20 times less sensitive compare to antibody detection method, which
relies on enzymatic amplification. Due to the lower sensitivity no signal was detected from blood
samples before and after immunoprecipitation enrichment using the novel fluorescence staining,
whereas bands from nitrotyrosine enriched blood samples were observed from the antibody staining
method. We conclude that the novel fluorescence method can be employed as an alternative staining
method for 2-nitrophenol functionalities in the complex biological matrices.
Overall, this thesis provides a contribution to a better understanding of inflammatory responses by
the development of novel chemistry-based tools for inhibition and detection of enzyme activity.
Ultimately, these methods could be employed in diagnosis and therapy.

150
Summary, General Discussion and Future Perspectives
FUTURE PERSPECTIVES
Future studies should be focused on the improvement of inhibitors of lipoxygenases in terms of
potency, selectivity, efficacy, and cell permeability. We identified anacardic acid derivatives as
modulators of lipoxygenases with medium to high potency. Here, we revealed a regulatory
mechanism of lipoxygenase activity that is influenced by salicylate-based molecules that bind to the
allosteric site of these enzymes. This allosteric site that is targeted in h-5-LOX is different from the
ATP binding site in The next challenge is to elucidate the location and the structure of this regulatory
site in order to get a model for designing new modulators for this enzyme. The crystal structure of
lipoxygenase in complex with the allosteric modulators may provide the necessary information on this
allosteric site. Based on the crystal structure of the enzyme, the configuration of salicylate based
modulators can be resolved and this information can be further used to design new compounds with
improved inhibitory potency. In our studies, anacardic acid derivatives were shown to selectively
inhibit h-5-LOX in comparison to COX-2 which provides a good basis for a new therapeutic
approach. Furthermore, screening and kinetic studies against other LOXs and COXs enzymes will
provide a more complete understanding of the selectivity of this set of compounds
The roles of lipoxygenase in inflammation and cancer are associated with the activation of NF- B
pathway and the expression of pro-inflammatory genes. The identified modulators of LOX activity
described in this thesis can be employed as tools to explore the connection between the lipoxygenase
activities and activation of the NF- B pathway. Experiments on the potency of lipoxygenase
activators and inhibitors to regulate the NF- B pathway are currently ongoing. Such studies can
further confirm the importance of LOXs inhibitors as a therapeutic approach in inflammation and
cancer.
We also briefly described the role of COXs in inflammation. Inhibition of COX-2 activity has
frequently been employed for the development of anti-inflammatory drugs. Non-selective COXs
inhibitors such as aspirin can provide moderate to severe side effects by inhibition of COX-1.
Therefore the development of selective inhibitors became a major concern for this enzyme. In chapter
3 we describe the benzoxazole core scaffold as a potential starting point for development of a new
class of inhibitors. Further studies on the selectivity of these compounds against COX-1 and the other
LOXs are needed for further development of this compound class.
Compounds that covalently bind with thiolates have been employed frequently as starting points to
develop ABPP probes for a cysteine containing enzymes such as HATs and cysteine proteases. We
revealed the reaction mechanism of isothiazolones with thiolates, and our enzyme kinetic studies in
combination with mass spectroscopy analysis showed that isothiazolones and 5-choloroisothiazolones
bind covalently to the active site thiol and/or to another surface exposed cysteine of cysteine protease
cathepsin B. This sets the stage for development of ABPP probes based on isothiazolones that can be
targeted either to HATs, to cysteine proteases and potentially also other thiol-containing enzymes.

151
151
APPENDIX
Dutch Summary
Acknowledgements
List of Publications

152
Samenvatting en Toekomstperspectieven
Samenvatting
Modulatie van de lipoxygenase enzymactiviteit door stoffen die afgeleid zijn van het
natuurproduct anacardic acid
Ontstekingen zijn een belangrijk gezondheidsprobleem in de westerse wereld. Deze ziektes wordt
gereguleerd door talrijke signaalmoleculen en signaaltransductie routes. De NF- B signaleringsroute
is één van de belangrijkste routes die betrokken is bij de regulatie van genexpressie in ontstekingen en
kanker. Verder zijn de activering en onderdrukking van de NF- B signaalroute gerelateerd aan de
activiteit van inflammatoire enzymen bijvoorbeeld lipoxygenases.
Lipoxygenases (LOXs) zijn enzymen met een ijzeratoom in de ‘active site’ dat niet aan heem
gebonden is. Deze enzymen katalyseren de oxidatie van cis-onverzadigde vetzuren. LOXs zijn
onderverdeeld in verschillende sub-families zoals 5-LOX, 8-LOX, 12-LOX en 15 LOX op basis van
hun selectiviteit voor de oxidatie van specifieke cis-onverzadigde bindingen in vetzuren. Deze
geoxideerde vetzuren zijn signaalmoleculen die cruciaal zijn voor het beginnen en beëindigen van
ontstekingsprocessen. Bovendien blijkt steeds duidelijker dat de ontwikkeling van verschillende typen
kanker wordt geassocieerd met de activiteit van LOXs en de aanwezigheid van metabolieten die
ontstaan door LOXs activiteit. Vanwege hun belang in ziekten is het ontwikkelen van moleculen die
de activiteit van LOX enzymen activeren of remmen belangrijk. Deze moleculen kunnen gebruikt
worden als hulpmiddelen om deze enzymen te onderzoeken in hun fysiologische en
pathofysiologische context. Bovendien kunnen kleine moleculen die enzymactiviteit remmen gebruikt
worden als uitgangspunten om nieuwe therapieën te ontwikkelen.
In hoofdstuk 2 wordt de synthese van een reeks anacardic acid derivaten beschreven waarin de
substituenten in de 4- en 6-positie van de salicylaat werden gevarieerd. De verzameling verbinding
werd gescreend in vitro voor de remming van 15-lipoxygenase uit sojabonen (SLO-1) en 5-
lipoxygenase uit aardappelen (potato 5-LOX) om structuur-activiteitsrelaties te bepalen. De meest
active verbinding voor SLO-1 is 2.23 met een hydroxylgroep in the 4-positie en een 4-
heptylphenethyl substituent in de 6-positie van de salicylaat. Deze verbinding geeft remming in
concentraties die vijf keer zo laag zijn. De enzymekinetiek voor de remming van SLO-1 door 2.23
laat zien dat deze stoffen een ‘mixed type’ remming geeft. Dit geeft aan dat deze stof waarschijnlijk
aan een allostere bindingsplaats bindt, die de katalyse in het actieve centrum van het enzym reguleert.
Een ander derivaat 2.21 liet een sterke activering van aardappel 5-LOX zien wat een opmerkelijke
waarneming is. Deze stof gaf ongeveer een drievoudige activatie bij concentraties hoger dan 50 µM.
De enzymkinetiek van 2.21 met aardappel 5-LOX laat zien dat deze verbinding een niet-essentiële
activator van de enzymatische reactie is. Het type activatie wordt aangeduid als niet-essentieel omdat
de enzymatische reactie zowel in de aanwezigheid als ook in de afwezigheid van de activator kan
verlopen. Door binding van de activator neemt de affiniteit van het enzym voor het substraat en/of de

153
Samenvatting en Toekomstperspectieven
snelheid van de katalytische reactie toe. Met behulp van een model dat vergelijkbaar is aan het ‘mixed
type’ remming kunnen de kinetische parameters voor de niet-essentiële activatie afgeleid worden. De
activator 2.21 heeft een relatief hoge affiniteit voor het substraat gebonden enzym (αKA is 24 µM) en
verhoogt de omzettingssnelheid van het substraat met een factor 1.4 (β). De kinetische analyse laat
zien dat er een allostere bindingsite is die de katalyse in de ‘active site van het enzym reguleert en dat
derivaten van salicylzuur daaraan binden. Dit biedt een nieuw perspectief voor de modulatie van
lipoxygenase activiteit.
Figuur 1 – Een remmer van SLO-1 en een activator van aardappel LOX-5 zoals beschreven in hoofdstuk 2.
De structuur van anacardic acid laat zien dat er een hydrofiele kop en een hydrofobe alifatische
staart is. Het is dus te verwachten dat deze stof en stoffen die hiervan afgeleid zijn oppervlakte-actieve
eigenschappen hebben, die de biologische activiteit kunnen beïnvloeden door micelvorming. Daarom
hebben we de kritische micelconcentratie (CMC) voor de verbinding 2.21, en 2.23 onderzocht. De
CMC waardes bleken hoger te zijn dan 100 µM, wat aangeeft dat micelvorming geen rol speelt bij de
concentraties die werden gebruikt om enzymremming of activering te onderzoeken.
De volgende stap in ons onderzoek was het bestuderen van de activiteit van de anacardic acid
derivaten op menselijke lipoxygenases. In hoofdstuk 3 is een verzameling van verbindingen
gescreend voor remming van menselijk 5-lipoxygenase (h-5-LOX) in vergelijking met
cyclooxygenase-2 (COX-2). Dit zijn beide oxidatieve enzymen die een belangrijke rol spelen bij
ontstekingen en kanker. Een van de anacardic acid derivaten laat een selectieve activerende werking
zien tegen h-5-LOX ten opzichte van COX-2. De verbinding 2.23 en 2.21 die in dit hoofdstuk zijn
aangeduid als verbinding 3.19 en 3.21 vertonen geen activeerde en / of remmende werking voor h-5-
LOX en COX-2. Deze resultaten laten zien dat de menselijke lipoxygenases zich anders gedragen dan
de plantaardige lipoxygenases, wat waarschijnlijk te wijten is aan een gebrek aan overeenkomsten in
hun primaire structuur. Een ander anacardic acid derivaat, verbinding 3.23a, liet activatie van h-5-
LOX zien. Deze verbinding geeft 150% activatie van de h-5-LOX activiteit bij een concentratie van
12,5 µM. Uit kinetische studies op h-5-LOX blijkt dat verbinding 3.23a een niet-competitieve
activator is ten opzichte van co-activator ATP en een gemengde niet-essentiële activator tegen het
substraat linolzuur. De gemengde niet-essentiële activatie wijst op de aanwezigheid van een allostere
bindingsplaats die de enzymactiviteit reguleert. Het niet-competitieve gedrag ten opzichte van ATP
laat zien dat deze regulerende site verschilt van de ATP bindingsplaats. Het is opmerkelijk dat de h-5-
LOX activator 3.23a een bindingsconstante heeft in het nano-molaire gebied (381 nM).

154
Samenvatting en Toekomstperspectieven
Het is interessant dat een vergelijkbare verbinding (3.23d) remming geeft van h-5-LOX activiteit.
De enzymkinetiek toont ‘mixed-type’ remming, wat vergelijkbaar is met het werkingsmechanisme
van activator 3.23a. Dit ondersteunt het idee dat deze verbindingen binden aan een allostere
regulerende site in h-5-LOX, die de katalytische processen in de ‘active site’ beïnvloedt. Deze studie
wijst op de aanwezigheid van een allostere regulerende bindingsplaats die de enzymatische katalyse
remt of activeert op basis van de structuur van de verbinding die aan deze plaats bindt.
Figuur 2 – Een remmer en een activator van humaan 5-lipoxygenase, zoals beschreven in hoofdstuk 3.
Humaan 5-lipoxygenase heeft een belangrijke rol in zowel de start en het beëindigen van
ontstekingsreacties. Daarom is de modulatie van de activiteit van dit enzym een interessant doel voor
de ontwikkeling van nieuwe therapeutische benaderingen. Vanwege de brede regulerende activiteit
van h-5-LOX metabolieten in kanker en ontsteking biedt zowel remming alsook activatie van dit
enzym interessante mogelijkheden om biologische effecten in vivo te beïnvloeden. Deze effecten
zouden uiteindelijk gebruikt kunnen worden voor therapeutische doeleinden.
Er zijn verscheidene remmers voor lipoxygenases geïdentificeerd en sommigen daarvan worden
onderzocht in klinische studies als geneesmiddelen voor de behandeling van ontstekingsziekten. Er
zijn remmers voor 5-lipoxygenase die klinisch worden bestudeerd en toegepast voor ziekten zoals
astma en artritis. Echter de toepassingsmogelijkheden van deze stoffen wordt nog beperkt door een
gebrek aan werkzaamheid, selectiviteit en membraanpermeabiliteit. Gezien de belangrijke rol van
LOXs bij ontstekingen en kanker is de ontwikkeling van nieuwe klassen van remmers met hogere
selectiviteit en voor alternatieve LOX subtypen erg belangrijk.
De studies die beschreven zijn in dit proefschrift zijn gericht op de ontwikkeling van chemische
methodes die gebruikt kunnen worden om de regulatie van de NF- B signaaltransductieroute in
ontstekingsprocessen te begrijpen. In het eerste deel van dit proefschrift wordt de ontwikkeling van
nieuwe derivaten van het natuurproduct anacardic acid die de activiteit van lipoxygenase verhogen of
verlagen beschreven. In onze eerste studie (hoofdstuk 2), identificeren we een anacardic acid
derivaat dat soja lipoxygenase-1 (SLO-1) selectief remt en een ander derivaat dat de aardappel 5-
lipoxygenase (aardappel 5-LOX) activeert. Vervolgens (hoofdstuk 3) laten we zien dat humaan 5-
lipoxygenase (h-5-LOX) door andere stoffen wordt gereguleerd dan de plantaardige enzymen. Dit
onderzoek leidde tot de identificatie van een nieuwe remmer van 5-lipoxygenase, die selectief is ten
opzichte van cyclooxygenase-2 (COX-2). Bovendien identificeren we een sterke activator voor h-5-

155
Samenvatting en Toekomstperspectieven
LOX. Experimenten om de effecten van de h-5-LOX-remmers en activatoren op signalering via de
NF- B signaaltransductieroute te onderzoeken zijn in volle gang.
We verwachten dat zowel remmers en activatoren van h-5-LOX hun toepassingen in verschillende
therapeutische gebieden zullen vinden. Remming van h-5-LOX is betrokken bij de onderdrukking van
immuunreacties en activatie van h-5-LOX is betrokken bij de activering van immuunreacties. We
verwachten echter dat activatie van h-5-LOX ook gunstige effecten kan hebben op ontstekingen
omdat de productie van lipoxines door lipoxygenases bijdraagt aan de beëindiging van
immuunreacties. Dit pleit voor meer onderzoek aan de functies van lipoxygenase metabolieten in hun
fysiologische context. Hierbij kunnen nieuwe remmers en activatoren een belangrijke rol spelen.
Isothiazolonen als nieuwe ABPP probes voor thiol-afhankelijke enzymen
Het is aangetoond dat histon acetylering een cruciale rol speelt bij de activatie van pro-inflammatoire
gentranscriptie door de NF- B signaaltransductie route. In eerdere studies is aangetoond dat
isothiazolonen remmers zijn van de HAT PCAF en dat ze ook apoptose induceren in verschillende
cellijnen. In hoofdstuk 4 van dit proefschrift onderzoeken we de reactiviteit van isothiazolonen met
thiolaten en de selectiviteit van deze klasse van verbindingen voor diverse thiol-bevattende enzymen.
In dit geval hebben we de selectiviteit tussen de HAT PCAF en het cysteïne-protease cathepsine B
bestudeerd. Deze studies geven een startpunt om deze klasse van verbindingen verder te ontwikkelen
voor de toepassing als probes voor activiteits-gebaseerde profilering van voor thiol-bevattende
enzymen.
In deze studie hebben we vastgesteld dat het N-alifatisch gesubstitueerde 5-chloroisothiazolon 4.11
de meest krachtige remmer voor cathepsine B is. Verder is het van belang is dat het disulfide 4.7 een
opmerkelijk hoge potentie voor cathepsine B had, terwijl het de HAT PCAF niet remt. Daarnaast
zagen we een selectieve remming van de HAT PCAF in vergelijking met cathepsine B het met N-aryl
gesubstitueerde isothiazolon 4.9. Hieruit wordt geconcludeerd dat isothiazolonen remmers zijn voor
de cysteïne protease cathepsine B en de HAT PCAF en dat hun potentie en selectiviteit afhangt van
hun substitutiepatroon.
Figuur 3 – Remmers van het cysteïne protease cathepsine B zoals beschreven in hoofdstuk 4.

156
Samenvatting en Toekomstperspectieven
De reactiviteit van isothiazolonen en 5-chloroisothiazolonen voor thiolaten hangt af van de
elektronische eigenschappen van de isothiazolon heterocyclus. Kristalstructuren van N-phenyl-
isothiazolon en N-phenyl-5-chloroizothiazolon laten zien dat deze heterocyclus een aromatisch
karakter heeft die stabiliteit biedt aan de verbindingen. In tegenstelling hiermee geeft de relatief
zwakke zwavel-stikstofbinding een hoge reactiviteit voor thiolaten wat resulteert in een sterke
disulfide binding.
We hebben het remmingsmechanisme van 5-chloroisothiazolonen 4.5 en 4.14 voor cathepsine B
geanalyseerd met enzymkinetiek. Deze studies laten een niet-competitieve remming van cathepsine B
door 5-chloroisothiazolonen zien. Verder werd een sterke substraatremming waargenomen bij hogere
substraatconcentraties. De remming van cathepsine B door 5-chloroisothiazolon 4.5 bleek
tijdsafhankelijk te zijn met IC50 waarden die na verloop van tijd steeds verder afnamen. Dit suggereert
een snelle binding die gevolgd wordt door een langzamere tweede binding. Deze gegevens wijzen op
een covalente binding van 5-chloroisothiazolones aan cathepsine B. Met massaspectrometrie wordt
aangetoond dat 5-chloroisothiazolon 4.5 tweemaal bindt aan het enzym. Dit kan verklaard worden
doordat het 5-chloroisothiazolone 4.5 covalent bindt aan de thiolaat in de active site van het enzym en
/ of aan een andere cysteïne aan het oppervlak van cathepsine B. Dit onderzoek biedt een goede basis
om isothiazolonen verder te ontwikkelen voor toepassingen als probes voor activiteit-gebaseerde
eiwitprofilering van HATs en cysteïneproteasen.
Detectie van inflammatoire nitrotyrosine als biomarker
Ontstekingen leiden tot oxidatieve stress en schade van eiwitten door reactieve vormen van zuurstof.
Een belangrijke oxidatieve eiwitmodificatie is nitrering van tyrosine residuen. Deze modificatie is
geïdentificeerd als een biomarker voor verschillende ontstekingsziekten. In het laatste deel van dit
proefschrift (hoofdstuk 5), beschrijven we de ontwikkeling van een nieuwe methode om nitrotyrosine
in eiwitten te detecteren in biologische monsters. Deze nieuwe methode houdt in dat nitrotyrosine
wordt omgezet in een fluorofoor onder milde omstandigheden. Deze methode is eenvoudig uit te
voeren, zeer selectief voor nitrotyrosine en maakt gebruik van goedkope reagentia. Deze methode
bleek zeer efficiënt te zijn in histochemische kleuring van ontstoken weefsel en kan ook worden
toegepast als alternatieve detectiemethode voor immunoblotting op PVDF-membranen. Zoals
verwacht is de gevoeligheid van deze nieuwe detectiemethode die gebaseerd is op fluorescentie lager
dan detectiemethoden die gebruik maken van enzymgekatalyseerde signaalamplificatie.
Deze nieuwe detectiemethode impliceert twee reactiestappen; de eerste stap is de omzetting van de
2-nitrofenol functionaliteit van nitrotyrosine in een 2-aminofenol functionaliteit met
natriumdithioniet, en in de tweede reactiestap wordt de 2-aminofenol functionaliteit met Al3+ en
salicylaldehyde omgezet in een fluorofoor.

157
Samenvatting en Toekomstperspectieven
λex 420 nm λem 520 nm
Figuur 4 - Fluorescentiedetectie van nitrotyrosine in een weefselmonster in twee reactiestappen: a) behandeling met een
waterige Na2S2O4 oplossing en de daaropvolgende omzetting in een fluorofoor door b) een waterige AlCl3 en
salicylaldehyde oplossing.
Uit ons onderzoek bleek dat de fluorescentie van het 2-aminofenol - Al3+ - salicylaldehyde
complex in demiwater 8 keer hoger is dan in methanol. Detectie in water is het meest gunstige voor
detectie van tyrosine nitrering van eiwitten in biologische monsters. Verder hebben we de
toepasbaarheid van deze nieuwe histochemische kleuringsmethode in vergelijking met klassieke
immunohistochemische kleuringsmethode in verschillende weefselmonsters bestudeerd. Opvallend is
dat deze nieuwe detectiemethode een duidelijke kleuring laat zien in levermonsters van muizen die
met CCl4 zijn behandeld. Als de nieuwe histochemische kleuringsmethode wordt vergeleken met de
klassieke immunohistochemische kleuringsmethode dan blijkt dat beide kleuringsmethoden hoge
kleuringsintensiteiten vertonen op dezelfde plaatsen in weefselmonsters, maar dat de klassieke
histochemische kleuringsmethode een veel hogere achtergrondkleuring heeft. Hieruit blijkt duidelijk
dat de nieuw ontwikkelde histochemische kleuringsmethode in dit opzicht superieur is aan de
klassieke immunohistochemische kleuringsmethode. Bovendien is de gevormde fluorofoor in de
weefselmonsters stabiel gedurende meerdere dagen. Deze resultaten geven aan dat deze nieuwe
histochemische kleuringsmethode een waardevol alternatief kan zijn voor detectie van tyrosine
nitrering in eiwitten in weefselmonsters.
Wij hebben ook de toepassing van deze nieuwe fluorescentiedetectie methode uitgetest in
biologische matrices op western blot. Voor dit doeleind werd 3-nitro-4-hydroxyfenyl azijnzuur
(NHPA) gekoppeld aan runderserumalbumine (BSA) als model voor nitrotyrosine bevattende
eiwitten. Dit NHPA geconjugeerd BSA werd toegepast als standaard om de detectiemethode te
optimaliseren. Met onze nieuwe kleuringsmethode kon NHPA-geconjugeerd BSA gedetecteerd
worden op western blot. Controle-experimenten met het klassiek gebruikte anti-nitrotyrosine
antilichaam gaven vergelijkbare resultaten. De aanwezigheid van bloedplasma had geen significant
effect op het fluorescentie signaal, wat laat zien dat deze nieuwe methode om 2-nitrofenol detecteren
toepasbaar is in complexe biologische matrices. Helaas, is de detectiegrens van deze nieuwe methode
500 nmol/L, wat ongeveer 20 maal minder gevoelig is in vergelijking met antilichamen die gekoppeld
zijn aan enzymen die het detectiesignaal versterken. Door de lagere gevoeligheid van onze nieuwe
methode kon er geen signaal worden gedetecteerd in bloedmonsters voor of na verrijking met
immunoprecipitatie, terwijl in de nitrotyrosine verrijkte bloedmonsters zwakke signalen werken
waargenomen met de antilichaamkleuring. We concluderen dat de nieuwe fluorescentiemethode kan

158
Samenvatting en Toekomstperspectieven
worden gebruikt als een alternatieve methode voor kleuring 2-nitrofenol functionaliteiten en
nitrotyrosine in de complexe biologische matrices.
Over het geheel genomen levert dit proefschrift een bijdrage aan een beter begrip van
ontstekingsreacties door de ontwikkeling van nieuwe chemie-gebaseerde methodes voor remming en
detectie van enzymactiviteit. Uiteindelijk kunnen deze methodes worden toegepast in diagnose en
therapie.

159
Samenvatting en Toekomstperspectieven
TOEKOMSTPERSPECTIEVEN
Toekomstige studies moeten worden gericht op de verbetering van de remmers van lipoxygenasen wat
betreft hun potentie, selectiviteit, werkzaamheid en celdoorlaatbaarheid. We hebben anacardic acid
derivaten geïdentificeerd als modulatoren van lipoxygenases met gemiddelde tot hoge potentie.
Hierbij hebben we een nieuw regulatiemechanisme van lipoxygenase-activiteit geïdentificeerd,
waarbij salicylaat gebaseerde moleculen binden aan de allostere bindingsplaats van deze enzymen.
Deze allostere bindingsplaats in h-5-LOX is een andere bindingsplaats dan de ATP bindingsplaats.
Een belangrijke volgende uitdaging is het identificeren van de plaats en de structuur van deze
bindingsplaats om zo meer inzicht te krijgen voor het ontwerpen van nieuwe modulatoren. De
kristalstructuur van lipoxygenase in complex met deze allostere modulatoren kan hiervoor de
benodigde informatie verschaffen. Met Gebaseerd op de kristalstructuur van het enzym kan de
configuratie van salicylaat gebaseerde modulatoren worden opgehelderd en deze informatie kan
verder gebruikt worden om nieuwe verbindingen te ontwerpen met verbeterde inhibitie of activatie. In
onze studies werd aangetoond dat anacardic acid derivaten selectief h-5-LOX remmen in vergelijking
met COX-2 wat een goede basis biedt voor verdere ontwikkelen van moleculen die mogelijk
therapeutisch kunnen worden toegepast. Bovendien zullen screening en kinetische studies op andere
LOXs en COXs enzymen een vollediger begrip van de selectiviteit van dit soort verbindingen geven.
De rol van LOXs in ontstekingen en kanker is geassocieerd met de activatie van de NF- B
signaaltransductieroute en de expressie van pro-inflammatoire genen. De geïdentificeerde
modulatoren van LOX-activiteit die worden beschreven in dit proefschrift kunnen worden toegepast
als instrumenten om het verband tussen de activiteit van LOXs en de activering van de NF- B
signaaltransductieroute te verkennen. Experimenten om de potentie van de lipoxygenase activatoren
en remmers op de NF- B signaaltransductieroute te bestuderen zijn op dit moment in volle gang.
Verder kan dit onderzoek inzicht geven in het belang van LOXs-remmers voor de mogelijke
behandeling van kanker.
Verder hebben we kort beschreven wat de rol is van COXs bij ontstekingen. Remming van COX-2
activiteit wordt vaak toegepast voor de ontwikkeling van anti-inflammatoire geneesmiddelen. Niet-
selectieve remmers van COXs, zoals aspirine, kunnen matige tot ernstige bijwerkingen geven door
remming van COX-1. Daarom is de ontwikkeling van selectieve remmers voor dit enzym van groot
belang. In hoofdstuk 3 beschrijven we het benzoxazool scaffold als een potentieel startpunt voor de
ontwikkeling van een nieuwe klasse van remmers voor dit enzym. Verdere studies aan de selectiviteit
van deze verbindingen tegen COX-1 en de andere LOXs zijn van belang voor de verdere
ontwikkeling van deze klasse van verbindingen.
Verbindingen die covalent kunnen binden met thiolaten worden vaak gebruikt als uitgangspunten
voor de ontwikkeling van probes voor activiteit-gebaseerde profilering van cysteine-bevattende
enzymen zoals HATs en cysteïneproteasen. Wij hebben het reactiemechanisme van isothiazolonen

160
Samenvatting en Toekomstperspectieven
met thiolaten opgehelderd door enzym kinetische studies in combinatie met massaspectroscopie. Deze
studies toonden aan dat isothiazolonen en 5-choloroisothiazolonen covalent binden aan de thiolaten in
de active site van cathepsine B. Dit laat zien isothiazolonen kunnen worden toegepast voor het
ontwikkelen van ABPP probes voor activiteit-gebaseerde profilering die gericht zijn op HATs,
cysteine-proteasen en mogelijk ook andere thiol-bevattende enzymen.

161
Acknowledgements
Acknowledgements
The view outside my office window is usually very boring, just a rooftop of the next building. But
today it looks different, with the very bright sunshine, it brings back the old memories of my
childhood when I was still dreaming to be a medical doctor. Now here I am on my final step to be a
doctor, not a medical doctor, but a doctor in science. So this is it, the end of my journey in the PhD
tunnel which I have explored for the last 4 years. I feel so blessed with all wonderful people around
me for their continuous support, love, care and encouragement during this journey, and I would like to
thank all of them.
First of all, I would like to express my most gratitude to my promotors, Prof. Dr. Hidde J. Haisma
and Prof. Dr. Frank J. Dekker, who welcomed me as a PhD in Pharmaceutical Gene Modulation
(PGM) group 4 years ago. Hidde, thank you for your help, advice and guidance during these 4 years.
Thank you for making these 4 years of my PhD become more colorful and a lot more interesting than
just the numbers of days and months. I really enjoy all the parties that our group ever had once in a
while and thank for letting me become a captain of your boat once J. Thank you that within your
tight schedule, you always found the time to help me anytime I come to you. Frank, thank you for
being a great supervisor. You really helped me to improve myself during my PhD period. I remember
when I started my PhD, I was very impressed by your passion in science and when I stated that, you
gave me a motivation to become a good scientist. Thank you for your countinuous support and
supervision, without that I would have never been able to finish my PhD on time.
I also would like to thank my reading committee, Prof. Rob Liskamp, Prof. Alexander Dömling,
and Prof. Ad IJzerman for your willingness to read and to assess my thesis. I hope that you really
enjoy reading it.
And for my first paranymph, Jenny N. Soetedjo, thank you for your willingness to be my
paranymph. Even though you have already stated before that you did not want to be a paranymph
again, but you gladly accepted it when I asked you to become one of my paranymphs. Thanks for
always listening for my very long stories and for staying with me and helping me with many tasks that
need to be done, even until past midnight. Many thanks also for my other paranymph, Thea van den
Bosch. Thank you also for your willingness to be my paranymph. Although we have met just for a
couple of months, I am really enjoying all the moment that we have, from a small chit chat at the
office and all the experimental works that we did together in the lab until the ice skating fun session.
Good luck for your PhD project and enjoy as much as possible your PhD time.
I would like to thank all the members of PGM group. Petra Ettema, our best technician, thank
you for all the help you have given to me in the lab and in the LOX expression project that has really
taken a lot of your time. To our cheerful secretary, Janine Buitendijk, who is always helping me
with all the administration things. For all fellow PhDs i.e. Mohammad Arabpour, Maria-Eleni
Ourailidou, Nikolaos Eleftheriadis, Thea van den Bosch, thank you for your warmth and kindness,

162
Acknowledgements
I am going to miss our discussion sessions that we always have in our big office. Good luck and all
the best for the finishing of your PhDs. Our new lecture, Bert-Jan Baas, welcome to the group and
good luck with your own thesis.
My thanks also for all the ex-members of PGM group, with whom I shared a lot of fun and
togetherness. Massimo Ghizzoni, with whom I shared a lot the PhD experiences, thank you for being
a nice office roommate for 2.5 years. I enjoy all our discussions, from works related discussion to
some unimportant issues, such as who is speaking faster: me when I speak Indonesian or you when
you speak Italian (Now I know, no one can beat Maria-Eleni and Nikolaos, they are the fastest when
they speak Greek) J. To Marije Scholte, thanks for answering all my questions about the lab and for
being a nice stairs climbing partner. You are really a good trainer, you have never failed to make me
joining the stairs climbing program with Petra. I thank André Boltjes for his guidance and help
during my work in the synthesis lab. Thanks for all cool tips and tricks, and demonstrations that you
showed me, those are awesome. Even though it was just a short moment, I also would like to thank
Elena Melilo, for making a cheerful atmosphere in the lab.
To my students, Inan Bakhan, Samira Baghban, and Petra Kok, thank you very much for your
hard work and contribution in my projects. It has been a pleasure to work with you and I hope what
you have learned in the lab will be useful for your future.
I would like to express my gratitude to our neighbourhood group (PTT) members, thank you for all
your help. I thank Jan Visser to let me ‘invading’ the HPLC lab very often and for all nice
conversation we had in the lab. Many thanks also to Dr. Harm Maarsingh for our great collaboration.
I also would like to thank Dr. Megawati Santoso, my supervisor during my bachelor and master
project, from whom I learned the passion in bio-organic chemistry. I would like to thank the members
of Medicinal Chemistry Department, with whom I shared synthesis lab with all the synthesis
problems, and also advices, Prof. Durk Dijkstra, Dr. Cor Grol, Jeroen de Graan, Ulrike Dijkman.
I also would like to thank also many Indonesian people for their loves and friendships during my
stay in Groningen. Mackenzie Hadi, with whom I share my happiness and hardness during my PhD
journey, you have introduced me to many beautiful cities in Europe, now I really love to travel and
sometimes I also apply ‘A famous Mackenzie’s traveling type’ too. :p You also never failed to give a
hand and advices anytime I need it. Thank you for being such a good friend, I hope that our friendship
will last forever, despite of the time and the distance. I sincerely thank Tante Hanna de Jong, Om
Fred and Tante Tini Berg, Tante Elly Felten, Tante Ray ‘schatje’ Eman, Om Hendrik Lie and all
oms and tantes, for all love, care, and warmth that make me feel at home eventhough I am far away
from my parents, you are my big family here. Many thanks for Mbak Ika Widyariani and Mas Paul
Hoogland, K’Roga F. Kembaren, K’Laura Junistia and Jerome Wirawan, Mbak Dewi Setyaningsih,
who I consider as my big sisters and brothers. Thank you for always being there for me and helping
me in many circumstances. Big thanks for all my friends, Samuel Sugandhi, who is my first
Indonesian friend both in the lab and in Groningen; geng nonton dan hang-out (Angela Kumalaputri,

163
Acknowledgements
Louis Daniel, Yohanes Siem); trio-Pharma (Amirah Adlia, Doti Parameswari, Putri Dwi Utari);
birthday-party mate (Daniel ‘Daniela’); Double J couple (Jenny Soetedjo and K’ Jeffrey Tan); board
game geng (Mackenzie Hadi, Rieza Aprianto, Dani Setiawan, Lydia Metasari, Widianta Gomulya,
Emeraldo Arias Huvat, Nisa Purwanto, Widyoseno Muhsin); gabut partner (Oktavia Hendrawati);
many thanks also for Yanti, Winarto, Harison ‘lele’ Citrawan, Devi ‘buntal’ Herlianty; Elsye
Agustina, Pak Sandy & Bu Sandra Suharsa, Mbak Gloriani Novita, Navessa Padma Tania, Tita
Alissa Listyowardojo, Kang Handiyana Ariephin, Bang Roysat Alfons, Bang Yohanes Sondang,
Bang Mangarah Silalahi, Mas Prasetyo Adhi Chrisnamoko, Mbak Tata Yunita, Asteria Devy, Mbak
Ria Raraswati, Mbak Hennie Halma, Mbak Rosbina and Bernard Majoor, Mbak Tiurma Pitta
Tallagan, K’Sahat Tinambunan, Laura Gultom, geng UI (Ruby Theofelea, Christal Setyobudi,
Lidia Gabriella, Leidya Dairolouro). Thanks for fellow pharmacy and UMCG people (Rosaline
Sitorus, Pak Adhi Wibawa, Pandji Triadiyaksa, trio-Astri (Rizkiya Astri Hapsari, Astri Handayani,
Astri Ferdiana), Mbak Awalia Febriana & Mas Surahyo Sumarsono, Kadek Yota, Auliya Suwantika,
Neily Zakiyah; KI 105 (Mohammad Iqbal & Erythrina Stavila, Robby Roswanda, Lia ATWA, etc).
I am really grateful for all my friends who still and always support and encourage me in pursuing
my PhD. My friends from my high school period, Silvia Wahyudin, Yessica Cahyadi, Febby Michelle
Bhuana, Devy Natalia, Hanna Romanti, I feel so blessed to have friends like you. My gratitude also
for Anggi Maria Gunita, Felicia Soedradjat, Anastasia Riany, Paramita Jayaratri, MB 2003, and
all KI’03. To my Paskal friends, Valentine Rosadi Sinaga, Septi Ayu Megasiwi, Handy Sugianto,
Budiani Christina, Cornelius Nathanael & Sherly Rivkyan, K’Arnold Nathanael & Ci Levina
Arifin, Tiara Anisa, Elizabeth Hamdani, etc. with whom I shared a lot of enjoyable moments through
Sunday services, fellowship community, and many other occasions, thank you.
Special thanks to my aunt, Ii Heny, thank you for all the support and care, I really appreciate what
you have done for me all these years. For all my uncles, aunts, cousins, thanks for all the support. To
my beloved family, for papih, mamih, oo, thank you for everything. My brother, Ronaldi, thank you
for always supporting me and being the best brother. You are my role model whom I always want to
be. I dedicate this thesis for my beloved parents. I know that you have done everything to give the
very best for me, and thousand words of thank you are still not enough to express how grateful I am
for all what you have done for me. Without your prayers, love, care and support, I will not be able to
be standing at the point where I am right now. I love you pap and mam and I hope that I can make you
both proud with my achievement.
Rosalina


165
List of Publications
List of Publications
Wisastra R, Kok PAM, Baumgartner MP, Camacho CJ, Haisma HJ, Dekker FJ. Discovery of a
novel activator of 5-lipoxygenase from an anacardic acid-derived compound collection.
Submitted for publication 2013
Wisastra R, Ghizzoni M, Boltjes A, Haisma HJ, Dekker FJ. Anacardic acid derived salicylates
are inhibitors or activators of lipoxygenases. Bioorg. Med. Chem., 2012, 20(16), 5027-5032
Dekker F, Abello N, Wisastra R, Bischoff R. Enrichment and detection of tyrosine-nitrated
proteins. Curr. Protoc. Protein Sci. 2012, Chapter 14:Unit 14.13
Wisastra R, Poelstra K, Bischoff R, Haisma HJ, Dekker FJ. Antibody-Free Detection of
Protein Tyrosine Nitration in Tissue Sections, ChemBioChem, 2011, 12(13), 2016-2020
Wisastra R, Ghizzoni M, Maarsingh H, Haisma HJ, Dekker FJ. Isothiazolones; thiol-reactive
inhibitors of cysteine protease cathepsin B and histone acetyltransferase PCAF, Org.
Biomol. Chem., 2011, 9(6), 1817-1822
Dekker FJ, Ghizzoni M, Van der Meer N, Wisastra R. Haisma H.J. Inhibition of the PCAF
histone acetyl transferase and cell proliferation by isothiazolones, Bioorg. Med. Chem., 2009,
17(2), 460-466
Copyright © 2022 FDOKUMEN




















