
University of Groningen Pemphigoid diseases
231
University of Groningen Pemphigoid diseases: Insights in the nonbullous variant and disease management Lamberts, Aniek DOI: 10.33612/diss.132159641 IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check the document version below. Document Version Publisher's PDF, also known as Version of record Publication date: 2020 Link to publication in University of Groningen/UMCG research database Citation for published version (APA): Lamberts, A. (2020). Pemphigoid diseases: Insights in the nonbullous variant and disease management. University of Groningen. https://doi.org/10.33612/diss.132159641 Copyright Other than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons). The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license. More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne- amendment. Take-down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons the number of authors shown on this cover page is limited to 10 maximum. Download date: 23-01-2022
-
Upload
khangminh22 -
Category
Documents
-
view
4 -
download
0
Transcript of University of Groningen Pemphigoid diseases

University of Groningen
Pemphigoid diseases: Insights in the nonbullous variant and disease managementLamberts, Aniek
DOI:10.33612/diss.132159641
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:2020
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Lamberts, A. (2020). Pemphigoid diseases: Insights in the nonbullous variant and disease management.University of Groningen. https://doi.org/10.33612/diss.132159641
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license.More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne-amendment.
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 23-01-2022

1
Pemphigoid diseases: insights in the nonbullous variant and disease management
Aniek Lamberts

2
ISBN: 978-94-034-2431-6 ISBN: 978-94-034-2430-9 (e-book) ©Copyright M.A. Lamberts 2020, the Netherlands All rights reserved. No part of this thesis may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, recording, or by any information storage or retrieval system, without written permission of the author. The copyright of previously published chapters of this thesis remains with the publisher or journal. Financial support for the publication of this thesis was provided by: nanoString, Eurocept, Rijksuniversiteit Groningen, Studiefonds Dermatologie, Universitair Medisch Centrum Groningen. Cover design: Martijn O. Wolf – MOTTOW – mottow.nl Interior design: Aniek Lamberts Print: Ridderprint | www.ridderprint.nl

3
Pemphigoid diseases: insights in the nonbullous variant and disease management
Proefschrift
ter verkrijging van de graad van doctor aan de Rijksuniversiteit Groningen
op gezag van de rector magnificus prof. dr. C. Wijmenga
en volgens besluit van het College voor Promoties.
De openbare verdediging zal plaatsvinden op
dinsdag 22 september 2020 om 11.00 uur
door
Marlou Aniek Lamberts
geboren op 17 juli 1990 te Beilen

4
Promotores Dr. B. Horváth Prof. dr. M.F. Jonkman† Copromotor Dr. H.H. Pas Reading committee Prof. dr. P. Heeringa Prof. dr. M.A. de Rie Prof. dr. R. Ludwig

5
Paranimfen Lisette Prens Angelique Rondags

6
Table of contents Chapter 1 Introduction in pemphigoid diseases 9
PART 1 Nonbullous pemphigoid: disease characteristics and immunological aspects
Chapter 2 Significantly higher prevalence of circulating bullous 35 pemphigoid-specific IgG autoantibodies in elderly patients with a nonbullous skin disorder
Chapter 3 Nonbullous pemphigoid: a systematic review 43 Chapter 4 Nonbullous pemphigoid: insights in clinical and 61
diagnostic findings, treatment response and prognosis Chapter 4A Reply to: “Pruritus with pemphigoid autoantibodies 81
is the tip of an iceberg” Chapter 5 Prevalence of pemphigoid as a potentially unrecognized 85
cause of pruritus in nursing home residents Chapter 6 IgE autoantibodies in serum and skin of nonbullous and 93
bullous pemphigoid patients Chapter 7 Gene expression profile of lesional skin in bullous and 111
nonbullous pemphigoid: an explorative pilot study

7
PART 2 Management of pemphigoid diseases Chapter 8 Unmet needs in pemphigoid diseases: an international 133
survey amongst patients, clinicians and researchers Chapter 9 Effectiveness and safety of rituximab in recalcitrant 147
pemphigoid diseases Chapter 10 Determining the incidence of pneumocystis pneumonia 171
in patients with autoimmune blistering diseases not receiving routine prophylaxis
Chapter 11 Discussion and future perspectives 185 Chapter 12 Summary 209 Chapter 13 Samenvatting 217 Appendices Dankwoord 226
List of publications 228 Curriculum vitae 230

8
1

9
CHAPTER 1 Introduction in pemphigoid diseases Aniek Lamberts Center for Blistering Diseases, Department of Dermatology, University Medical Center Groningen, University of Groningen, Groningen, The Netherlands Published in adapted form: Lamberts A, Rashid H, Diercks GFH, Pas HH, Meijer JM, Horváth B. Pemphigoid variants affecting the skin: a review. Clinical and Experimental Dermatology, 2019 Oct;44(7):721-727 Lamberts A*, Rashid H*, Diercks GFH, Pas HH, Meijer JM, Bolling MC, Horváth B. Oral lesions in autoimmune bullous diseases: an overview of clinical characteristics and diagnostic algorithm. American Journal of Clinical Dermatology, 2019 Dec;20(6):847-861 * Authors contributed equally

10
Pemphigoid diseases Pemphigoid diseases are autoimmune skin diseases mediated by autoantibodies targeting structural proteins within, or closely related to the hemidesmosome (figure 1).1,2 Hemidesmosomes are specialized protein-complexes which connect the keratin cytoskeleton of the keratinocytes to the extracellular matrix in the dermis, providing structure and integrity to the skin.3
Many subtypes of pemphigoid diseases exist, and they can be subdivided into pemphigoid diseases predominantly affecting the skin, or mucous membranes. Beside the clinical subdivision, pemphigoid diseases can also be characterized by the targeted antigen. While clinical disease features may overlap, management and prognosis often differs. It is therefore important to differentiate between the various pemphigoid subtypes.
Figure 1. Schematic overview of the skin on the left. The epidermis is attached to the dermis by hemidesmosomes that connect the basal keratinocytes to the extracellular dermal matrix. On the right, an overview is given of proteins within, or closely related to the hemidesmosome. Proteins in white can be targeted by autoantibodies in pemphigoid diseases. Adapted from M.F. Jonkman.

11
Bullous pemphigoid Bullous pemphigoid (BP) is the most common autoimmune blistering disease, and predominantly affects the skin.1 The disease presents with severe pruritus and blisters, and typically has an onset at old age, with a reported median of 77 to 83 years.4–8 The annual incidence of BP is estimated between 2.4 to 21.7 new cases per million inhabitants in the general population of countries worldwide, and exponentially increases to 190-312 cases per million in the elderly population aged above 80 years.5–8 Interestingly, reported incidence numbers show an increasing trend over the last two decades, possibly related to more awareness for atypical BP variants, and the development of better diagnostic tests.5,9 Moreover, several drugs that can trigger BP are more commonly used, such as gliptins, TNF-α inhibitors, and check point inhibitors.10–12 Another possible explanation is the increasing incidence of neurodegenerative diseases, which are associated with BP.5,13 The highest associations were found between the co-occurrence of BP and Alzheimer’s disease, multiple sclerosis, and Parkinson’s disease.13–15 Neurological disorders precede BP in the majority of the cases.15 Interestingly, BP antigens BP180 and BP230 are also expressed in a neuronal isoform in the central and peripheral nervous system, and theories on cross reactivity have been postulated, yet, no strong conclusions could be drawn.14,16 Pathogenesis - Several studies observed a genetic susceptibility to develop pemphigoid diseases in patients carrying the human leucocyte antigen (HLA) allele DQB1*03:01.17–20 This HLA allele presumably contributes in the pathophysiology by presenting pemphigoid-specific antigens to autoreactive T cells.17–20 T cell activation subsequently leads to B cell activation, and ultimately to the production of autoantibodies by plasma cells. In BP, these autoantibodies are directed against BP180 and BP230 (figure 1).1,2 BP230, also termed BP antigen 1, is a member of the plakin family and is located intracellular.2 BP180 is a transmembrane protein, and is also termed type XVII collagen, or BP antigen 2.1,2 The extracellular noncollagenous 16A (NC16A) domain of BP180 is an important immunodominant region, and the pathogenicity of IgG autoantibodies to NC16A is proven in multiple studies, while studies on the relevance of BP230 reactivity showed conflicting results.10,21–24 Circulating IgE autoantibodies against BP180 and BP230 were also detected in BP patients, and anti-NC16 IgE showed a correlation with disease activity as well. 21,25–
31 In the skin, IgE was found in a linear pattern along the basement membrane zone

12
(BMZ)32–35, whereas others reported IgE bound to mast cells and eosinophils in the upper dermis31,36. Yet, the exact role of IgE in the disease pathogenesis of BP is unknown.
The mechanism of blister formation in BP may follow complement dependent and independent pathways.37,38 Complement activation, also termed complement fixation, can be induced through the classical pathway by autoantibody binding, or through the lectin or alternative pathway.39 Upon activation, a cascade of cleavage of complement components is initiated, resulting in stimulation of chemotaxis and phagocytosis of immune cells, inflammation, and direct cell lysis by forming a membrane attack complex.39 Evidence suggests that in BP the complement system is activated by binding of autoantibodies to BP180, which leads to migration of mast cells, eosinophils, and neutrophils towards the skin.40,41 It is hypothesized that, upon activation, immune cells release cytotoxic substances and proteases that degrade extracellular matrix proteins, therefore causing a subepidermal split.10 Other studies suggest complement independent blistering through autoantibody induced internalization of the complete BP180 protein.37,38 Depletion of BP180 from the hemidesmosome weakens its adhesion strength, and may result in blister formation. Clinical presentation - The clinical presentation of a typical BP patient includes symptoms of severe pruritus accompanied by tense blisters on erythematous plaques (figure 2).42–44 In early stages of BP, a prodromal phase may occur in which pruritic symptoms are the sole manifestation, and patients are frequently misdiagnosed.45,46 Lesions are predominantly located on the extremities and trunk, and limited mucosal involvement can be observed in 10-15%.42–44 BP has a chronic disease course with a tendency to relapse, and symptoms negatively influence the patients’ quality of life.47,48 Mortality rates in BP are heightened 3.4- to 6.6-fold compared with the general population49–51, with a pooled 1-year mortality rate in BP patients of 23.5% worldwide49. These findings emphasize the importance of early disease recognition, and timely adequate therapy in patients with BP.
Nonbullous pemphigoid Several studies reported that approximately 20% of BP patients present with atypical clinical features in which blisters are absent (figure 3).9,44,52,53 In these reports patients commonly present with severe itch and skin lesions, which were

13
eczematous-like, prurigo nodularis-like, or consisted of erythematous urticarial plaques. Also, few cases that displayed no primary skin lesions at all were described.53 The diagnosis of pemphigoid was confirmed in all cases by the detection of pemphigoid-specific autoantibodies in serum and skin. Authors have given the nonbullous disease phenotype various descriptive names, such as pruritic pemphigoid, pemphigoid incipiens, pemphigoid nodularis, or prodromal bullous pemphigoid.52,54–56 The shared clinical characteristic in the reported cases is the lack of a blistering phenotype, therefore, we favor the term nonbullous pemphigoid
Figure 2. Bullous pemphigoid. A/B. Tense fluid-filled and hemorrhagic blisters, erosions, erythematous plaques and crusts on the right flank, the chest, and upper arm in a patient with severe bullous pemphigoid. C. Tense blisters, erosions and crusts on erythematous skin on the right upper leg. D. Detail of tense blisters and erosions with remnants of the blister roof.
A
B
C
D

14
(NBP). It is not clear whether NBP patients are diagnosed during an early (prodromal) phase of BP, or whether NBP should be seen as a different disease entity within the pemphigoid spectrum. Moreover, NBP is not well characterized and is understudied. It is unknown why blisters do not develop, while these patients have autoantibodies against antigens that are also targeted in BP patients (BP180 and BP230). Furthermore, important clinical practice data on the management and prognosis of NBP patients are lacking.
Figure 3. Nonbullous pemphigoid as cause of severe pruritus in elderly patients. A. Fixed erythematous urticarial plaques on the left arm. B. Secondary skin lesions by pruritus consisting of excoriations and hypopigmented maculae on the shoulders and back. C. Erythematous papules and urticarial plaques on the chest.
B
A C

15
Mucous membrane pemphigoid Mucous membrane pemphigoid (MMP) is a group of pemphigoids that predominantly affect the mucous membranes.1,57,58 The annual reported incidence is 1.3 to 2.0 newly diagnosed cases per million inhabitants of France and Germany, with an average age of disease onset of 60 years.59–61 Patients present with blisters, erosions and inflammation of mucosal surfaces, and the oral (85%) and ocular (65%) mucosa are most frequently affected.57,58,62 Other sites may include nasal (20-40%), anogenital (20%), pharyngeal (20%), laryngeal (20%), and esophageal mucosa (5-15%). One third of the MMP patients also display mild skin lesions. Most patients have autoantibodies against BP180, mainly targeting the C-terminal domain and/or the NC16A domain.63–66 However, antibodies may also target BP230, type VII collagen, integrin α6β4, p200 or laminin 332 (figure 1). Anti-laminin 332 reactivity is associated with severe disease, scarring, and pharyngeal and laryngeal involvement, with risk of airway obstruction.67–69 Several studies have reported an increased risk of malignancy in patients with anti-laminin 332 reactivity, whereas others did not find such association.68–71
Epidermolysis bullosa acquisita Epidermolysis bullosa acquisita (EBA) comprises approximately 6% of all pemphigoid diseases.72 The targeted antigen is type VII collagen, a major component of anchoring fibrils located below the lamina densa (figure 1).73 EBA can roughly be divided into the mechanobullous subtype characterized by skin fragility, milia formation, nail dystrophy, and scarring, and the inflammatory subtype, clinically resembling other pemphigoid diseases.72 Mortality data are lacking, however, clinical experience learns that mortality rates are lower compared to BP.51 Nevertheless, EBA patients are often treatment resistant, and suffer from a chronic disease course.74,75 Linear IgA disease Linear IgA disease (LAD) is a heterogeneous group of pemphigoids characterized by exclusive IgA class autoantibodies.76 The disease can be drug induced, most commonly by vancomycin.77 The annual incidence ranges between 0.2 and 1.0 cases per million estimated in different regions.78 The majority of patients recognize the 120 kDa (LAD-1), or the 97 kDa (LABD97) antigen, both cleavage products of the extracellular domain of BP180 (figure 1).79 A less common subtype

16
shows IgA reactivity to type VII collagen, and is named sublamina densa-type LAD, or IgA EBA. LAD has a biphasic distribution, affecting young children, and adults above 50 years.76 Childhood LAD presents with blisters on urticarial plaques in a typical circinate or serpiginous configuration forming a ‘crown of jewels’ or a ‘string of pearls’.80 In adults LAD more often resembles BP. LAD is usually self-limiting in childhood within one to five years, whereas adults may have a chronic disease course with poor response to different therapies.80 Other pemphigoid diseases Other rare pemphigoid variants, not further discussed in this thesis, include pemphigoid gestationis81 with disease onset during pregnancy; Brunsting-Perry pemphigoid82 with blisters localized on the scalp, face and neck, leaving atrophic scars; lichen planus pemphigoides83 with clinical features of both BP and lichen planus; anti-p200 pemphigoid, with autoantibodies to a 200 kDa sized protein of yet unknown molecular identity.84–86
Diagnosis of pemphigoid diseases The diagnosis of pemphigoid diseases is based on clinical features and autoantibody detection in skin and/or serum (figure 4).1,87 Histopathology In general, histopathologic features of pemphigoid diseases include a subepithelial split, and a dermal infiltrate with eosinophilic or neutrophilic granulocytes and lymphocytes.88 Histopathology alone is not sufficient to diagnose pemphigoid, but can support diseases in the differential diagnosis. Direct immunofluorescence microscopy Direct immunofluorescence (DIF) microscopy has a highly important role in the diagnosis of pemphigoid diseases. Autoantibodies and complement bound in the skin are visualized by incubation of a fluorescent labeled antibody against human IgG, IgA or complement on a frozen skin section.89 Additional serration pattern analysis differentiates pemphigoid variants with an n-serrated pattern (figure 5A) from EBA with a u-serrated pattern (figure 5B).90

17
Figu
re 4
. Flo
wch
art o
f the
dia
gnos
tic p
athw
ay in
pat
ient
s pr
esen
ting
with
blis
ters
on
the
skin
or m
ucou
s m
embr
anes
. DIF
, dire
ct
imm
unof
luor
esce
nce;
EBM
Z, e
pide
rmal
bas
emen
t mem
bran
e zo
ne; I
IF, i
ndire
ct im
mun
oflu
ores
cenc
e m
icro
scop
y; S
SS, s
alt-
split
ski
n; E
LISA
, enz
yme-
linke
d im
mun
osor
bent
ass
ay; E
BA, e
pide
rmol
ysis
bullo
sa a
cqui
sita
.

18
Serologic tests The detection of circulating autoantibodies against pemphigoid-specific antigens can be valuable to diagnose pemphigoid diseases. Indirect immunofluorescence microscopy - Indirect immunofluorescence (IIF) microscopy is frequently performed using a monkey esophagus or salt-split skin (SSS) substrate. Monkey esophagus is commercially available, while SSS is obtained by incubation of human skin in 1M sodium chloride for 24 hours, resulting in a reproducible artificial split in the lamina lucida. Antigens are located either at the
B
C D
A
Figure 5. Immunofluorescence results compatible with the diagnosis of pemphigoid diseases. A. Direct immunofluorescence (DIF) microscopy shows linear IgG along the basement membrane zone (BMZ) in an n-serrated pattern. B. DIF microscopy shows linear IgG along the BMZ in a u-serrated pattern, compatible with epidermolysis bullosa acquisita. C. Indirect immunofluorescence microscopy on salt-split skin (IIF SSS) shows IgG bound to the epidermal side of the artificial split, compatible with pemphigoid diseases targeting BP180, BP230, LAD-1, LABD97, or integrin α6β4. D. IIF SSS shows IgG bound to the dermal side of the artificial split, compatible with pemphigoid diseases targeting p200, laminin 332, or type VII collagen.

19
epidermal or dermal side of the split. IIF SSS discriminates between pemphigoid diseases targeting BP180 and BP230 located in the lamina lucida (epidermal staining; figure 5C) and those targeting laminin 332, type VII collagen, or p200, all located beneath the lamina lucida (dermal staining; figure 5D). ELISA and immunoblot – Enzyme-linked immunosorbent assay (ELISA) and immunoblot are the most commonly used techniques to specify the targeted antigen. ELISA kits are commercially available to detect and quantify antibodies to specific pemphigoid antigens (BP180 and BP230). By measuring the intensity of an enzyme induced color reaction, an antibody titer can be calculated. The immunoblot technique first sorts denatured skin proteins by molecular size through gel electrophoresis.91,92 The sorted proteins are then transferred onto a membrane. Autoantibodies directed against skin proteins bind the membrane, and are visualized by staining the bound IgG. The molecular size of the stained protein identifies which pemphigoid antigen is targeted. Other serological tests – Additional serological tests can differentiate between anti-p200 pemphigoid, anti-laminin 332 pemphigoid and EBA. The IIF knockout analysis is a technique using skin sections of patients with hereditary epidermolysis bullosa, in which laminin 332, or type VII collagen is absent (‘knocked-out’).86 IIF microscopy is negative if patient serum contains autoantibodies to the knocked-out protein. In anti-p200 pemphigoid IIF remains positive in both laminin 332 and type VII collagen knock-out skin. Another technique to confirm or rule out the presence of anti-laminin 332 autoantibodies is the novel keratinocyte footprint assay.93 This fast and specific assay uses the unique laminin 332 footprints that cultured keratinocytes leave on the bottom of the culture dish when moving. Anti-laminin-332 autoantibodies in the serum of a patient will bind to the footprints, and can be stained by immunofluorescence.
Management of pemphigoid diseases The treatment of pemphigoid diseases mainly relies on immunosuppressive or immunomodulating drugs. In general, there is a lack of randomized placebo controlled trials with a sufficient sample size. Reasons for this are the low incidence

20
of most pemphigoid variants, and, especially in BP, the fragile elderly population with multiple comorbidities in which the diseases mainly occurs. Bullous pemphigoid The consensus guideline for the management of BP provides treatment recommendations for mild limited BP, and extensive generalized BP.87 The first treatment choice for both mild and generalized disease is super potent topical corticosteroids applied on the whole body, except the face.94,95 Secondly, systemic corticosteroids are recommended if topical steroids are insufficient, in a dosage of 0.5-0.75 mg/kg/day.94 Adjuvant therapies that may be considered are tetracyclines, azathioprine, mycophenolate mofetil, methotrexate, dapsone, chlorambucil, and cyclosporine.87 For therapy resistant BP that does not respond to the therapies mentioned above, the guideline advises to consider intravenous immunoglobulins, rituximab (RTX), anti-IgE monoclonal antibodies and plasma exchange.87 Mucous membrane pemphigoid The first international guideline for the treatment of MMP was developed in 2002 by Chan and colleagues, using a consensus based methodology.96 Management recommendations were separated for ‘high risk’ and ‘low risk’ patients. ‘High risk’ patients were defined as those who have disease occurring in ocular, genital, nasopharyngeal, esophageal, and laryngeal mucosa, as they have high likelihood of therapy resistance and scarring, which in case of airway obstruction can be life threatening. First line therapy in ‘high risk’ MMP is prednisone (1-1.5 mg/kg/day), and cyclophosphamide (1-2 mg/kg/day).97,98 Alternative therapeutic options are azathioprine, and dapsone.97,99 ‘Low risk’ patients were defined as those who have disease occurring only in oral mucosa, or in both oral mucosa and skin. Recommendations for initial therapy in low risk patients include topical corticosteroids, or tetracycline hydrochloride with nicotinamide.100,101 Alternatively, dapsone, low doses of prednisolone, or azathioprine are advised.96,97 Epidermolysis bullosa acquisita The management of EBA is challenging, and systemic corticosteroids are widely used as first treatment choice, with dosages ranging from 0.5 to 2.0 mg/kg/day.102 In mild cases the use of colchicine 1 to 2 mg/day is preferred, as it only gives minor side effects compared to other treatment options.103 Other therapies that may be

21
prescribed as monotherapy, or may be combined with systemic corticosteroids, are dapsone, methotrexate, azathioprine, cyclosporine, mycophenolate mofetil, and cyclophosphamide.102 For treatment resistant EBA cases it can be considered to treat with high-dose intravenous immunoglobulin, RTX, plasmapheresis, immunoadsorption, or extracorporeal photochemotherapy.102 Linear IgA disease Dapsone is the first treatment choice in LAD, and on average a dose of 100mg/day is sufficient to induce disease control.104,105 Dapsone is prescribed in a number of diseases that involve the accumulation of neutrophils, and inhibits the adherence of neutrophils to anti-BMZ autoantibodies.106 In some LAD patients dapsone can be ineffective, and systemic corticosteroids, with or without adjuvant immunosuppressants, such as azathioprine, mycophenolate mofetil, cyclosporine or cyclophosphamide may be needed.105 No additional treatment recommendations exist for the sublamina densa type LAD, also called IgA-EBA. In drug induced LAD, first the suspected drug needs to be stopped, which usually resolves the symptoms within four weeks after withdrawal.77,107
Novel and emerging therapies The management of pemphigoid diseases can be challenging, partly due to the frailty of the patients. For many years high doses of systemic corticosteroids have been used to treat pemphigoid diseases, however, they have been associated with high mortality rates.50,108 Conventional immunosuppressive drugs may give insufficient disease control or severe side effects. Therefore, the search for better therapies with more effectiveness and less side effects is ongoing. Several novel and emerging therapies are discussed below. Rituximab RTX is an anti-CD20 monoclonal antibody that has been used for many years in the fields of rheumatology and oncology.109 In 2017 groundbreaking results of a multicenter open label randomized trial illustrated a beneficial effect of RTX 1000mg on day 1 and 15 combined with short term oral corticosteroids, over monotherapy with oral corticosteroids in pemphigus vulgaris.110 Based on these outcomes, RTX was recently registered as therapy for pemphigus vulgaris. Limited data are available on RTX in pemphigoid diseases, however, it is suggested that RTX

22
could be a relatively safe and valuable therapeutic option.111–113 Currently, most clinical guidelines recommend the use of RTX as a 3rd line therapy in pemphigoid diseases. IgE targeting therapy Based on potential pathogenic role of IgE in pemphigoid, targeting IgE can be a novel and interesting approach in the treatment portfolio. Omalizumab is a monoclonal antibody targeting unbound human IgE, and therefore prevents its binding to the high affinity IgE receptor.114 The drug is registered for chronic urticaria. Fairley et al. were the first to publish a BP case successfully treated with omalizumab in 2009, and in the last ten years over 22 more cases were treated.115,116 Meta-analysis of these cases showed a surprisingly high success rate, with complete remission in up to 80% of the cases, however recurrence was seen in 80% after an average duration of 3.8 months, or when the therapy was stopped.116 Anti-complement therapy A novel innovative therapeutic target in inflammatory diseases is the complement system.117 Most experience with anti-complement therapies was gained in renal disease, particularly in anti-neutrophilic cytoplasmic antibody associated vasculitis.118 Several animal studies reported evidence that complement may play an important role in the pathogenesis of pemphigoid diseases, providing a rational for anti-complement therapy in BP.40,41,119 Recently, complement component 1s (C1s) was blocked by BIVV009 (previously termed TNT009) in ten BP patients, intervening in the classical complement activation pathway.120 The drug appeared relatively safe, however, no disease activity measurements were performed. BP180 and BP230 autoantibody levels remained stable throughout the treatment period, while C3 depositions in the skin disappeared in 80%. Studies reporting on effectiveness of anti-complement therapy are expected soon.

23
Outline and aim of this thesis PART 1 Nonbullous pemphigoid: Disease characteristics and
immunological aspects NBP is an understudied disease, and only few case reports have provided limited information on its disease features. Therefore, NBP is easily overlooked as a cause of pruritus in elderly individuals. In part 1 of this thesis, we aim to provide clinicians with more insights in the clinical features of NBP to improve disease recognition, and to gain information on the prognosis and disease management of NBP. Moreover, we intended to learn more about the immunological aspects of NBP, to answer the question ‘why do NBP patients lack blisters’.
Interpretation of serological pemphigoid test results can be challenging. Therefore, we investigated the presence of serum autoantibodies in a population of dermatology patients with nonbullous skin disorders in chapter 2. To give an overview of the available literature on NBP, we systematically reviewed the literature on NBP in chapter 3, and summarized the disease characteristics of all published cases. Chapter 4 describes patient characteristics of our cohort of NBP patients, and provides daily practice data on the treatment and prognosis. In chapter 5 we performed a cross-sectional study to determine the prevalence of pemphigoid as an unrecognized cause of pruritus in the potential high-risk population of nursing home residents. In chapter 6 we attempted to find an answer on the question ‘why do NBP patients lack blisters?’ by assessing the presence of IgE in the serum and skin of BP and NBP patients. A second effort to find the answer was made in chapter 7, where we analyzed and compared the gene expression profile of lesional skin in NBP and BP. PART 2 Management of pemphigoid diseases In part 2 of this thesis we focus on the management of pemphigoid diseases. In chapter 8 we performed an international survey study, in which we explored the unmet needs in pemphigoid diseases from the perspective of patients, researchers and clinicians. The disease management of pemphigoid diseases can be challenging, and off-label drugs may be necessary in treatment resistant

24
pemphigoid cases. The recent success of the CD20 targeting drug RTX for the autoimmune blistering disease pemphigus vulgaris caught our attention and made us question whether it may also be effective in pemphigoid diseases. Therefore, we retrospectively assessed the effectiveness and safety of RTX in recalcitrant pemphigoid diseases in chapter 9. In chapter 10 we assessed the prevalence of pneumocystis pneumonia in patients with autoimmune blistering diseases to answer whether or not routine prophylaxis is advised. References 1 Schmidt E, Zillikens D. Pemphigoid diseases. Lancet (London, England) 2013; 381:320–32. 2 Goletz S, Zillikens D, Schmidt E. Structural proteins of the dermal-epidermal junction targeted
by autoantibodies in pemphigoid diseases. Exp Dermatol 2017; 26:1154–62. 3 Walko G, Castanon MJ, Wiche G. Molecular architecture and function of the hemidesmosome.
Cell Tissue Res 2015; 360:529–44. 4 Hubner F, Recke A, Zillikens D, et al. Prevalence and Age Distribution of Pemphigus and
Pemphigoid Diseases in Germany. J. Invest. Dermatol. 2016; 136:2495–8. 5 Kridin K, Ludwig RJ. The Growing Incidence of Bullous Pemphigoid: Overview and Potential
Explanations. Front Med 2018; 5:220. 6 Joly P. Incidence of bullous pemphigoid and pemphigus vulgaris. BMJ. 2008; 337:a209. 7 Langan SM, Smeeth L, Hubbard R, et al. Bullous pemphigoid and pemphigus vulgaris--incidence
and mortality in the UK: population based cohort study. BMJ 2008; 337:a180. 8 Marazza G, Pham HC, Scharer L, et al. Incidence of bullous pemphigoid and pemphigus in
Switzerland: a 2-year prospective study. Br J Dermatol 2009; 161:861–8. 9 Cozzani E, Gasparini G, Burlando M, et al. Atypical presentations of bullous pemphigoid: Clinical
and immunopathological aspects. Autoimmun Rev 2015; 14:438–45. 10 Lo Schiavo A, Ruocco E, Brancaccio G, et al. Bullous pemphigoid: etiology, pathogenesis, and
inducing factors: facts and controversies. Clin Dermatol 2013; 31:391–9. 11 Lopez AT, Khanna T, Antonov N, et al. A review of bullous pemphigoid associated with PD-1 and
PD-L1 inhibitors. Int J Dermatol 2018; 57:664–9. 12 Nishie W. Dipeptidyl peptidase IV inhibitor-associated bullous pemphigoid: a recently
recognized autoimmune blistering disease with unique clinical, immunological and genetic characteristics. Immunol Med 2019; 42:22–8.
13 Försti AK, Jokelainen J, Ansakorpi H, et al. Psychiatric and neurological disorders are associated with bullous pemphigoid - A nationwide Finnish Care Register study. Sci Rep 2016; 6:1–6.
14 Forsti A-K, Huilaja L, Schmidt E, Tasanen K. Neurological and psychiatric associations in bullous pemphigoid-more than skin deep? Exp Dermatol 2017; 26:1228–34.
15 Milani-Nejad N, Zhang M, Kaffenberger J. The association between bullous pemphigoid and neurological disorders: a systematic review. Eur J Dermatol 2017; 27:472–81.
16 Julio TA, Vernal S, Massaro JD, et al. Biological predictors shared by dementia and bullous pemphigoid patients point out a cross-antigenicity between BP180/BP230 brain and skin isoforms. Immunol Res 2018; 66:567–76.

25
17 Delgado JC, Turbay D, Yunis EJ, et al. A common major histocompatibility complex class II allele HLA-DQB1* 0301 is present in clinical variants of pemphigoid. Proc Natl Acad Sci U S A 1996; 93:8569–71.
18 Setterfield J, Theron J, Vaughan RW, et al. Mucous membrane pemphigoid: HLA-DQB1*0301 is associated with all clinical sites of involvement and may be linked to antibasement membrane IgG production. Br J Dermatol 2001; 145:406–14.
19 Amber KT, Zikry J, Hertl M. A multi-hit hypothesis of bullous pemphigoid and associated neurological disease: Is HLA-DQB1*03:01, a potential link between immune privileged antigen exposure and epitope spreading? HLA 2017; 89:127–34.
20 Budinger L, Borradori L, Yee C, et al. Identification and characterization of autoreactive T cell responses to bullous pemphigoid antigen 2 in patients and healthy controls. J Clin Invest 1998; 102:2082–9.
21 Dopp R, Schmidt E, Chimanovitch I, et al. IgG4 and IgE are the major immunoglobulins targeting the NC16A domain of BP180 in Bullous pemphigoid: serum levels of these immunoglobulins reflect disease activity. J Am Acad Dermatol 2000; 42:577–83.
22 Daneshpazhooh M, Ghiasi M, Lajevardi V, et al. BPDAI and ABSIS correlate with serum anti-BP180 NC16A IgG but not with anti-BP230 IgG in patients with bullous pemphigoid. Arch Dermatol Res 2018; 310:255–9.
23 Patsatsi A, Kyriakou A, Pavlitou-Tsiontsi A, et al. Association of autoantibodies to BP180 with disease activity in Greek patients with bullous pemphigoid. Clin Dev Immunol 2012; 2012:854795.
24 Yoshida M, Hamada T, Amagai M, et al. Enzyme-linked immunosorbent assay using bacterial recombinant proteins of human BP230 as a diagnostic tool for bullous pemphigoid. J Dermatol Sci 2006; 41:21–30.
25 Messingham KAN, Noe MH, Chapman MA, et al. A novel ELISA reveals high frequencies of BP180-specific IgE production in bullous pemphigoid. J Immunol Methods 2009; 346:18–25.
26 van Beek N, Luttmann N, Huebner F, et al. Correlation of Serum Levels of IgE Autoantibodies Against BP180 With Bullous Pemphigoid Disease Activity. JAMA dermatology 2017; 153:30–8.
27 Bing L, Xiping Z, Li L, et al. Levels of anti-BP180 NC16A IgE do not correlate with severity of disease in the early stages of bullous pemphigoid. Arch Dermatol Res 2015; 307:849–54.
28 Ishiura N, Fujimoto M, Watanabe R, et al. Serum levels of IgE anti-BP180 and anti-BP230 autoantibodies in patients with bullous pemphigoid. J Dermatol Sci 2008; 49:153–61.
29 Iwata Y, Komura K, Kodera M, et al. Correlation of IgE autoantibody to BP180 with a severe form of bullous pemphigoid. Arch Dermatol 2008; 144:41–8.
30 Hashimoto T, Ohzono A, Teye K, et al. Detection of IgE autoantibodies to BP180 and BP230 and their relationship to clinical features in bullous pemphigoid. Br J Dermatol 2017; 177:141–51.
31 Dimson OG, Giudice GJ, Fu CL, et al. Identification of a potential effector function for IgE autoantibodies in the organ-specific autoimmune disease bullous pemphigoid. J Invest Dermatol 2003; 120:784–8.
32 Yayli S, Pelivani N, Beltraminelli H, et al. Detection of linear IgE deposits in bullous pemphigoid and mucous membrane pemphigoid: a useful clue for diagnosis. Br J Dermatol 2011; 165:1133–7.
33 Provost TT, Tomasi TBJ. Immunopathology of bullous pemphigoid. Basement membrane

26
deposition of IgE, alternate pathway components and fibrin. Clin Exp Immunol 1974; 18:193–200.
34 Moriuchi R, Nishie W, Ujiie H, et al. In vivo analysis of IgE autoantibodies in bullous pemphigoid: a study of 100 cases. J Dermatol Sci 2015; 78:21–5.
35 Kamata A, Kurihara Y, Funakoshi T, et al. Basement membrane zone IgE deposition is associated with bullous pemphigoid disease severity and treatment results. Br J Dermatol 2019 Jul 22. doi: 10.1111/bjd.18364. [Epub ahead of print]
36 Freire PC, Munoz CH, Stingl G. IgE autoreactivity in bullous pemphigoid: eosinophils and mast cells as major targets of pathogenic immune reactants. Br J Dermatol 2017; 177:1644–53.
37 Natsuga K, Nishie W, Shinkuma S, et al. Antibodies to Pathogenic Epitopes on Type XVII Collagen Cause Skin Fragility in a Complement-Dependent and -Independent Manner. J Immunol 2012; 188:5792–9.
38 Iwata H, Ujiie H. Complement-independent blistering mechanisms in bullous pemphigoid. Exp Dermatol 2017 Dec;26(12):1235-1239. doi:10.1111/exd.13367.
39 Nesargikar PN, Spiller B, Chavez R. The complement system: history, pathways, cascade and inhibitors. Eur J Microbiol Immunol (Bp) 2012; 2:103–11.
40 Nelson KC, Zhao M, Schroeder PR, et al. Role of different pathways of the complement cascade in experimental bullous pemphigoid. J Clin Invest 2006; 116:2892–900.
41 Heimbach L, Li Z, Berkowitz P, et al. The C5a receptor on mast cells is critical for the autoimmune skin-blistering disease bullous pemphigoid. J Biol Chem 2011; 286:15003–9.
42 Di Zenzo G, Della Torre R, Zambruno G, Borradori L. Bullous pemphigoid: from the clinic to the bench. Clin Dermatol 2012; 30:3–16.
43 Tanaka M, Hashimoto T, Dykes PJ, Nishikawa T. Clinical manifestations in 100 Japanese bullous pemphigoid cases in relation to autoantigen profiles. Clin Exp Dermatol 1996; 21:23–7.
44 Della Torre R, Combescure C, Cortés B, et al. Clinical presentation and diagnostic delay in bullous pemphigoid: A prospective nationwide cohort. Br J Dermatol 2012; 167:1111–7.
45 Zhang Y, Luo Y, Han Y, et al. Non-bullous lesions as the first manifestation of bullous pemphigoid: A retrospective analysis of 181 cases. J Dermatol 2017 Jul;44(7):742-746. doi:10.1111/1346-8138.13782.
46 Sun C, Chang B, Gu H. Non-bullous lesions as the first manifestation of bullous pemphigoid: a retrospective analysis of 24 cases. J Dermatolog Treat 2009; 20:233–7.
47 Kalinska-Bienias A, Piotrowski T, Kowalczyk E, et al. Actigraphy-measured nocturnal wrist movements and assessment of sleep quality in patients with bullous pemphigoid: a pilot case-control study. Clin Exp Dermatol 2019 Oct;44(7):759-765. doi:10.1111/ced.13902.
48 Kouris A, Platsidaki E, Christodoulou C, et al. Quality of life, depression, anxiety and loneliness in patients with bullous pemphigoid. A case control study. An Bras Dermatol 2016; 91:601–3.
49 Kridin K, Bergman R. Mortality in Patients with Bullous Pemphigoid: A Retrospective Cohort Study, Systematic Review and Meta-analysis. Acta Derm Venereol 2019 Jan 1;99(1):72-77. doi:10.2340/00015555-2930.
50 Kalinska-Bienias A, Lukowska-Smorawska K, Jagielski P, et al. Mortality in bullous pemphigoid and prognostic factors in 1st and 3rd year of follow-up in specialized centre in Poland. Arch Dermatol Res 2017 Nov;309(9):709-719. doi:10.1007/s00403-017-1772-x.
51 Joly P, Baricault S, Sparsa A, et al. Incidence and mortality of bullous pemphigoid in France. J

27
Invest Dermatol 2012; 132:1998–2004. 52 Lamb PM, Abell E, Tharp M, et al. Prodromal bullous pemphigoid. Int J Dermatol 2006; 45:209–
14. 53 Bakker C V, Terra JB, Pas HH, Jonkman MF. Bullous pemphigoid as pruritus in the elderly: a
common presentation. JAMA dermatology 2013; 149:950–3. 54 Asbrink E, Hovmark A. Clinical variations in bullous pemphigoid with respect to early symptoms.
Acta Derm Venereol 1981; 61:417–21. 55 Powell AM, Albert S, Gratian MJ, et al. Pemphigoid nodularis (non-bullous): a clinicopathological
study of five cases. Br J Dermatol 2002; 147:343–9. 56 Alonso-Llamazares J, Rogers RS 3rd, Oursler JR, Calobrisi SD. Bullous pemphigoid presenting as
generalized pruritus: observations in six patients. Int J Dermatol 1998; 37:508–14. 57 Chan LS, Ahmed AR, Anhalt GJ, et al. The first international consensus on mucous membrane
pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol 2002; 138:370–9.
58 Chan LS, Wojnarowska F, Fine J-D, et al. The First International Consensus on Mucous Membrane Pemphigoid. Arch Dermatol 2004; 138. doi:10.1001/archderm.138.3.370.
59 Bertram F, Bröcker EB, Zillikens D, Schmidt E. Prospektive Untersuchung der Inzidenz blasenbildender Autoimmundermatosen in Unterfranken. JDDG - J Ger Soc Dermatology 2009; 7:434–40.
60 Arduino PG, Broccoletti R, Carbone M, et al. Describing the gingival involvement in a sample of 182 Italian predominantly oral mucous membrane pemphigoid patients: A retrospective series. Med Oral Patol Oral Cir Bucal 2017; 22:e149–52.
61 Bagan J, Jiménez Y, Murillo J, Bagan L. Oral mucous membrane pemphigoid: A clinical study of 100 low-risk cases. Oral Dis 2018; 24:132–4.
62 Schmidt E, Zillikens D. Pemphigoid diseases. Lancet 2013; 381:320–32. 63 Balding SD, Prost C, Diaz LA, et al. Cicatricial Pemphigoid Auto antibodies React with Multiple
Sites on the BP180 Extracellular Domain. J Invest Dermatol 1996; 106:141–6. 64 Oyama N, Setterfield JF, Powell AM, et al. Bullous pemphigoid antigen II (BP180) and its soluble
extracellular domains are major autoantigens in mucous membrane pemphigoid: The pathogenic relevance to HLA class II alleles and disease severity. Br J Dermatol 2006; 154:90–8.
65 Lee JB, Liu Y, Hashimoto T. Cicatricial pemphigoid sera specifically react with the most C-terminal portion of BP180. J Dermatol Sci 2003; 32:59–64.
66 Calabresi V, Arduino P, Tirone F, et al. Oral pemphigoid autoantibodies preferentially target BP180 ectodomain. Clin Immunol 2006; 122:207–13.
67 Chin-Che Hsu R, Lazarova Z, Lee HG, et al. Antiepiligrin cicatricial pemphigoid. J Am Acad Dermatol 2000; 42:841–4.
68 Amber KT, Bloom R, Hertl M. A systematic review with pooled analysis of clinical presentation and immunodiagnostic testing in mucous membrane pemphigoid: Association of anti-laminin-332 IgG with oropharyngeal involvement and the usefulness of ELISA. J Eur Acad Dermatology Venereol 2016; 30:72–7.
69 Bernard P, Antonicelli F, Bedane C, et al. Prevalence and clinical significance of anti-laminin 332 autoantibodies detected by a novel enzyme-linked immunosorbent assay in mucous membrane pemphigoid. JAMA Dermatology 2013; 149:533–40.

28
70 Egan CA, Lazarova Z, Darling TN, et al. Anti-epiligrin cicatricial pemphigoid and relative risk for cancer. Lancet 2001; 357:1850–1.
71 Goletz S, Probst C, Komorowski L, et al. A sensitive and specific assay for the serological diagnosis of antilaminin 332 mucous membrane pemphigoid. Br J Dermatol 2018; :149–56.
72 Buijsrogge JJA, Diercks GFH, Pas HH, Jonkman MF. The many faces of epidermolysis bullosa acquisita after serration pattern analysis by direct immunofluorescence microscopy. Br J Dermatol 2011; 165:92–8.
73 Woodley DT, Briggaman RA, O’Keefe EJ, et al. Identification of the skin basement-membrane autoantigen in epidermolysis bullosa acquisita. N Engl J Med 1984; 310:1007–13.
74 Iwata H, Vorobyev A, Koga H, et al. Meta-analysis of the clinical and immunopathological characteristics and treatment outcomes in epidermolysis bullosa acquisita patients. Orphanet J Rare Dis 2018; 13:153.
75 Kim JH, Kim S-C. Epidermolysis bullosa acquisita. J Eur Acad Dermatol Venereol 2013; 27:1204–13.
76 Chorzelski TP, Jablonska S. IgA linear dermatosis of childhood (chronic bullous disease of childhood). Br J Dermatol 1979; 101:535–42.
77 Chanal J, Ingen-Housz-Oro S, Ortonne N, et al. Linear IgA bullous dermatosis: comparison between the drug-induced and spontaneous forms. Br J Dermatol 2013; 169:1041–8.
78 Kridin K. Subepidermal autoimmune bullous diseases: overview, epidemiology, and associations. Immunol Res 2018; 66:6–17.
79 Pas HH, Kloosterhuis GJ, Heeres K, et al. Bullous pemphigoid and linear IgA dermatosis sera recognize a similar 120-kDa keratinocyte collagenous glycoprotein with antigenic cross-reactivity to BP180. J Invest Dermatol 1997; 108:423–9.
80 Gottlieb J, Ingen-Housz-Oro S, Alexandre M, et al. Idiopathic linear IgA bullous dermatosis: prognostic factors based on a case series of 72 adults. Br J Dermatol 2017; 177:212–22.
81 Jenkins RE, Hern S, Black MM. Clinical features and management of 87 patients with pemphigoid gestationis. Clin Exp Dermatol 1999; 24:255–9.
82 Brunsting LA, Perry HO. Benign pemphigold; a report of seven cases with chronic, scarring, herpetiform plaques about the head and neck. AMA Arch Derm 1957; 75:489–501.
83 Zaraa I, Mahfoudh A, Sellami MK, et al. Lichen planus pemphigoides: four new cases and a review of the literature. Int J Dermatol 2013; 52:406–12.
84 Chen KR, Shimizu S, Miyakawa S, et al. Coexistence of psoriasis and an unusual IgG-mediated subepidermal bullous dermatosis: identification of a novel 200-kDa lower lamina lucida target antigen. Br J Dermatol 1996; 134:340–6.
85 Zillikens D, Kawahara Y, Ishiko A, et al. A novel subepidermal blistering disease with autoantibodies to a 200-kDa antigen of the basement membrane zone. J Invest Dermatol 1996; 106:1333–8.
86 Meijer JM, Diercks GFH, Schmidt E, et al. Laboratory Diagnosis and Clinical Profile of Anti-p200 Pemphigoid. JAMA dermatology 2016; 152:897–904.
87 Feliciani C, Joly P, Jonkman MF, et al. Management of bullous pemphigoid: the European Dermatology Forum consensus in collaboration with the European Academy of Dermatology and Venereology. Br J Dermatol 2015; 172:867–77.
88 Buonavoglia A, Leone P, Dammacco R, et al. Pemphigus and mucous membrane pemphigoid: An

29
update from diagnosis to therapy. Autoimmun Rev 2019 Apr;18(4):349-358. 89 Jordon RE, Beutner EH, Witebsky E, et al. Basement zone antibodies in bullous pemphigoid.
JAMA 1967; 200:751–6. 90 Vodegel RM, Jonkman MF, Pas HH, de Jong MCJM. U-serrated immunodeposition pattern
differentiates type VII collagen targeting bullous diseases from other subepidermal bullous autoimmune diseases. Br J Dermatol 2004; 151:112–8.
91 Pas H. immunoassays. In: Autoimmune bullous diseases. , 2016; 57–62. 92 Pas HH. Immunoblot assay in differential diagnosis of autoimmune blistering skin diseases. Clin
Dermatol 2001; 19:622–30. 93 Giurdanella F, Nijenhuis AM, Diercks GFH, et al. Keratinocyte footprint assay discriminates anti-
laminin-332 pemphigoid from all other forms of pemphigoid diseases. Br J Dermatol 2020 Feb;182(2):373-381. doi:10.1111/bjd.18129.
94 Joly P, Roujeau J-C, Benichou J, et al. A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. N Engl J Med 2002; 346:321–7.
95 Joly P, Roujeau J-C, Benichou J, et al. A comparison of two regimens of topical corticosteroids in the treatment of patients with bullous pemphigoid: a multicenter randomized study. J Invest Dermatol 2009; 129:1681–7.
96 Chan LS, Ahmed AR, Anhalt GJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol 2002; 138:370–9.
97 Mondino BJ, Brown SI. Immunosuppressive therapy in ocular cicatricial pemphigoid. Am J Ophthalmol 1983; 96:453–9.
98 Elder MJ, Lightman S, Dart JK. Role of cyclophosphamide and high dose steroid in ocular cicatricial pemphigoid. Br J Ophthalmol 1995; 79:264–6.
99 Rogers RS 3rd, Seehafer JR, Perry HO. Treatment of cicatricial (benign mucous membrane) pemphigoid with dapsone. J Am Acad Dermatol 1982; 6:215–23.
100 Lozada-Nur F, Miranda C, Maliksi R. Double-blind clinical trial of 0.05% clobetasol propionate (corrected from proprionate) ointment in orabase and 0.05% fluocinonide ointment in orabase in the treatment of patients with oral vesiculoerosive diseases. Oral Surg Oral Med Oral Pathol 1994; 77:598–604.
101 Reiche L, Wojnarowska F, Mallon E. Combination therapy with nicotinamide and tetracyclines for cicatricial pemphigoid: further support for its efficacy. Clin Exp Dermatol 1998; 23:254–7.
102 Koga H, Prost-Squarcioni C, Iwata H, et al. Epidermolysis Bullosa Acquisita: The 2019 Update. Front Med 2019 Jan 10;5:362.
103 Cunningham BB, Kirchmann TT, Woodley D. Colchicine for epidermolysis bullosa acquisita. J Am Acad Dermatol 1996; 34:781–4.
104 Kasperkiewicz M, Zillikens D, Schmidt E. Pemphigoid diseases: pathogenesis, diagnosis, and treatment. Autoimmunity 2012; 45:55–70.
105 Vale ECS do, Dimatos OC, Porro AM, Santi CG. Consensus on the treatment of autoimmune bullous dermatoses: dermatitis herpetiformis and linear IgA bullous dermatosis - Brazilian Society of Dermatology. An Bras Dermatol 2019; 94:48–55.
106 Thuong-Nguyen V, Kadunce DP, Hendrix JD, et al. Inhibition of neutrophil adherence to antibody by dapsone: a possible therapeutic mechanism of dapsone in the treatment of IgA dermatoses.

30
J Invest Dermatol 1993; 100:349–55. 107 Garel B, Ingen-Housz-Oro S, Afriat D, et al. Drug-induced linear immunoglobulin A bullous
dermatosis: A French retrospective pharmacovigilance study of 69 cases. Br J Clin Pharmacol 2019; 85:570–9.
108 Rzany B, Partscht K, Jung M, et al. Risk factors for lethal outcome in patients with bullous pemphigoid: low serum albumin level, high dosage of glucocorticosteroids, and old age. Arch Dermatol 2002; 138:903–8.
109 Gurcan HM, Keskin DB, Stern JNH, et al. A review of the current use of rituximab in autoimmune diseases. Int Immunopharmacol 2009; 9:10–25.
110 Joly P, Maho-Vaillant M, Prost-Squarcioni C, et al. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet (London, England) 2017; 389:2031–40.
111 Lourari S, Herve C, Doffoel-Hantz V, et al. Bullous and mucous membrane pemphigoid show a mixed response to rituximab: experience in seven patients. J. Eur. Acad. Dermatol. Venereol. 2011; 25:1238–40.
112 Kasperkiewicz M, Shimanovich I, Ludwig RJ, et al. Rituximab for treatment-refractory pemphigus and pemphigoid: a case series of 17 patients. J Am Acad Dermatol 2011; 65:552–8.
113 Schmidt E, Seitz CS, Benoit S, et al. Rituximab in autoimmune bullous diseases: mixed responses and adverse effects. Br J Dermatol 2007; 156:352–6.
114 Kawakami T, Blank U. From IgE to Omalizumab. J Immunol 2016; 197:4187–92. 115 Fairley JA, Baum CL, Brandt DS, Messingham KAN. Pathogenicity of IgE in autoimmunity:
successful treatment of bullous pemphigoid with omalizumab. J. Allergy Clin. Immunol. 2009; 123:704–5.
116 Kremer N, Snast I, Cohen ES, et al. Rituximab and Omalizumab for the Treatment of Bullous Pemphigoid: A Systematic Review of the Literature. Am J Clin Dermatol 2019 Apr;20(2):209-216. doi:10.1007/s40257-018-0401-6.
117 Morgan BP, Harris CL. Complement, a target for therapy in inflammatory and degenerative diseases. Nat Rev Drug Discov 2015; 14:857–77.
118 Reddy YN V, Siedlecki AM, Francis JM. Breaking down the complement system: a review and update on novel therapies. Curr Opin Nephrol Hypertens 2017; 26:123–8.
119 Liu Z, Giudice GJ, Swartz SJ, et al. The role of complement in experimental bullous pemphigoid. J Clin Invest 1995; 95:1539–44.
120 Freire PC, Munoz CH, Derhaschnig U, et al. Specific inhibition of the classical complement pathway prevents C3 deposition along the dermal-epidermal junction in bullous pemphigoid. J Invest Dermatol 2019 Dec;139(12):2417-2424.e2. doi:10.1016/j.jid.2019.04.025.

31

32

33
PART 1
Nonbullous pemphigoid: Disease characteristics and immunological aspects

34
2

35
CHAPTER 2 Significantly higher prevalence of circulating bullous pemphigoid-specific IgG autoantibodies in elderly patients with a nonbullous skin disorder Aniek Lamberts*, Joost M. Meijer*, Hendri H. Pas, Marcel F. Jonkman * Authors contributed equally Center for Blistering Diseases, Department of Dermatology, University Medical Center Groningen, University of Groningen, Groningen, The Netherlands Published in the British Journal of Dermatology, 2015 Nov; 173(5), 1274-6

36
Dear editor, we read with interest the recent article of Van Beek et al. on “Serum autoantibodies against the dermal–epidermal junction in patients with chronic pruritic disorders, elderly individuals and blood donors prospectively recruited” and the recent review article of Schmidt et al. on “BP180- and BP230-specific IgG autoantibodies in pruritic disorders of the elderly: a preclinical stage of bullous pemphigoid?” about the association between pruritus in the elderly and the presence of bullous pemphigoid (BP)-specific IgG autoantibodies.1,2 Van Beek et al. studied autoantibody reactivity against the epidermal basement membrane zone (EBMZ) by indirect immunofluorescence microscopy (IIF), enzyme-linked immunosorbent assay (ELISA) and immunoblot. Positive reactivity in any test was found in 31.2% of the sera of elderly individuals (≥ 70 years; n=93), 16.7% of the sera of patients with chronic pruritic disorders (n=78) and in 26% of the sera of healthy blood donors of all ages (n=50), respectively. In our opinion these are remarkably high percentages, probably due to false-positive rates of immuno-assays, as mentioned in the discussion of their article. Van Beek et al. concluded that neither advanced age nor chronic pruritus have been verified as risk factors for autoantibodies against the EBMZ.1 Recently Schmidt et al. reviewed clinical and experimental studies about the possible association between senile pruritus and BP IgG autoantibodies, and question whether this could be a preclinical stage of BP.2 Prior studies by Rieckhoff-Cantoni et al. (1992), Hofmann et al. (2003) and Feliciani et al. (2009) on the presence of circulating BP autoantibodies in elderly patients with pruritic disorders, but without blistering, reported IgG reactivity against BP180 or BP230 in 10 of 43 patients (23%), 3 of 25 patients (12%) and 5 of 15 patients (33%), respectively.3-5 The question remains whether circulating autoantibodies against BP antigens in the elderly and patients with pruritic disorders indicate the presence of a BP subtype, may identify patients with an increased risk of developing BP, or may have no clinical relevance at all. As an extension to these studies we present our results of a retrospective database study, which included 374 patients who consulted our department for a skin disorder, without blistering. Data was collected from patients in our dermatology database in whom direct immunofluorescence (DIF) and serological testing were performed at the University Medical Center Groningen (UMCG). Patients were excluded if DIF was positive or if they clinically presented with blisters or erosions on skin or mucous membranes, to exclude those with an

37
evident diagnosis of autoimmune blistering disease. Patient characteristics are shown in table 1. The following serological test results were studied: IIF on monkey esophagus, IIF on salt-split skin (SSS), immunoblot testing on BP180 and BP230 antibodies, and BP180 NC16A- and BP230-specific enzyme-linked immunosorbent assay (ELISA, MBL, Nagoya, Japan, cut-off <9 U/ml) (Fig. 1). In our study, we found at least one positive serological test in 13.6% (n=51) of the dermatology patients with a non-bullous skin disorder. The median age of these patients (71.1 years, n=51) was significantly higher (p=0.009, Mann-Whitney U-test) than the median age of patients with no positive test results (64.0 years, n=323). Moreover, logistic regression showed that age above 75 years had a significant predictive value for positive reactivity for at least one serological test (p=0.025; odds ratio 3.8) compared to patients aged < 45 years. The higher prevalence of BP IgG autoantibodies in patients aged above 75 years was confirmed with X2-test (p=0.012). In contrast, no relation was found between the presence of pruritus and BP IgG autoantibodies. Furthermore, no relation was found between the combined presence of pruritus and older age (>65 years) and BP IgG autoantibodies.
Table 1. Patient characteristics with sex, pruritus and age vs. serological test results
Total No reactivity At least one positive test result P-valuea
Total population 374 323 (86.4) 51 (13.6) - Sex
Female 218 (58.3) 190 (87.2) 28 (12.8) 0.60 Male 156 (41.7) 133 (85.3) 23 (14.7)
Pruritus No 67 (17.9) 59 (88) 8 (12) 0.66
Yes 307 (82.1) 264 (86.0) 43 (14.0) Age (years)
Mean (range) 62.2 (5.5-96.0) ≤ 75 years 268 (71.7) 239 (89.2) 29 (10.8) 0.012*
> 75 years 106 (28.3) 84 (79.2) 22 (20.8)
Values are n (%) unless stated otherwise. a P-value by X2-test. *Significant (p < 0.05).

38
Van Beek et al. stated that positive ELISA results could not be confirmed with ELISA tests of other manufacturers, nevertheless, these values were considered positive. In our study ELISA results were only considered positive if the result remained positive after replication of the tests. This does not exclude nonspecific binding of antibodies, but minimizes false-positive results due to technical errors. Therefore another 15 former positive ELISA results were negative after replicated testing. In 13 of 374 patients (3.5%) multiple positive reactivity was seen in tests of different methodology, of which seven patients had positive reactivity in two different tests, three patients in three tests, two patients in four tests and one patient with reactivity in five different tests. Although detection of circulating BP IgG antibodies has been reported previously in patients with various (chronic) pruritic dermatoses and in elderly individuals, the mechanism and relevance are not yet fully understood. Both Hofmann et al. and Schmidt et al. discussed the mechanism of repeated cell injury to the EBMZ due to itching in pruritic dermatoses, which may lead to exposure of hidden epitopes. In combination with a loss of self-tolerance related to the ageing process, these patients could be at risk of developing anti-EBMZ antibodies and, eventually, BP.2,4,6 Conversely, anti-BP230 IgG autoantibodies may trigger pruritus, leading to the development of pruritic skin lesions and possibly anti-BP180 IgG antibodies, by the process of epitope spreading.2,6 Our findings did not show a relation between pruritus and circulating BP IgG autoantibodies in our non-bullous dermatology population. A possible association might have been obscured due to the selection bias, since our study was based on a population that consulted a dermatologist for a dermatosis that may be pruritic due to various causes. The non-bullous clinical variant of BP and the minimal criteria to diagnose BP are a topic of recent discussion.7-9 Positive epidermal binding of IIF on SSS may play an important diagnostic role, with a reported high specificity and positive predictive value in typical BP patients.10 The finding in our study of a significantly higher prevalence of BP-specific IgG autoantibodies in elderly dermatology patients with non-bullous skin disorders raises the question whether a diagnosis of pemphigoid in elderly with pruritus and a negative DIF is often missed. However, the results of testing sera of elderly patients with pruritic disorders should be interpreted with caution, and additional studies are needed in the general

39
population (as control) and in a high-risk population for developing BP (elderly persons in nursing homes).
Figure 1. Bullous pemphigoid-specific IgG autoantibody reactivity in our study population of dermatology patients with nonbullous skin disorders, divided into age >75 years and ≤75 years. Sera were tested with six different serological tests, including indirect immunofluorescence (IIF) microscopy on monkey oesophagus (n = 308), IIF on human salt-split skin (SSS, n = 359), immunoblot (n = 306) and BP180 and BP230 enzyme-linked immunosorbent assay (ELISA, n = 239 and 227, respectively).

40
References 1. van Beek N, Dohse A, Riechert F, et al. Serum autoantibodies against the dermal-epidermal
junction in patients with chronic pruritic disorders, elderly individuals and blood donors prospectively recruited. Br J Dermatol 2014 Apr;170(4):943-947.
2. Schmidt T, Sitaru C, Amber K, Hertl M. BP180- and BP230-specific IgG autoantibodies in pruritic disorders of the elderly: a preclinical stage of bullous pemphigoid? Br J Dermatol 2014 Aug;171(2):212-219.
3. Rieckhoff-Cantoni L, Bernard P, Didierjean L, Imhof K, Kinloch-de Loes S, Saurat JH. Frequency of bullous pemphigoid-like antibodies as detected by western immunoblot analysis in pruritic dermatoses. Arch Dermatol 1992 Jun;128(6):791-794.
4. Hofmann SC, Tamm K, Hertl M, Borradori L. Diagnostic value of an enzyme-linked immunosorbent assay using BP180 recombinant proteins in elderly patients with pruritic skin disorders. Br J Dermatol 2003 Oct;149(4):910-912.
5. Feliciani C, Caldarola G, Kneisel A, et al. IgG autoantibody reactivity against bullous pemphigoid (BP) 180 and BP230 in elderly patients with pruritic dermatoses. Br J Dermatol 2009 Aug;161(2):306-312.
6. Chan LS, Vanderlugt CJ, Hashimoto T, et al. Epitope spreading: lessons from autoimmune skin diseases. J Invest Dermatol 1998 Feb;110(2):103-109.
7. Bakker CV, Terra JB, Pas HH, Jonkman MF. Bullous pemphigoid as pruritus in the elderly: a common presentation. JAMA Dermatol 2013 Aug;149(8):950-953.
8. Borradori L, Joly P. Toward a practical renaming of bullous pemphigoid and all its variants: cutaneous pemphigoid. JAMA Dermatol 2014 Apr;150(4):459.
9. Bakker CV, Terra JB, Jonkman MF. Toward a practical renaming of bullous pemphigoid and all its variants-reply. JAMA Dermatol 2014 Apr;150(4):459-460.
10. Sardy M, Kostaki D, Varga R, Peris K, Ruzicka T. Comparative study of direct and indirect immunofluorescence and of bullous pemphigoid 180 and 230 enzyme-linked immunosorbent assays for diagnosis of bullous pemphigoid. J Am Acad Dermatol 2013 Nov;69(5):748-753.

41

42
3

43
CHAPTER 3 Nonbullous pemphigoid: a systematic review Aniek Lamberts, Joost M. Meijer, Marcel F. Jonkman Center for Blistering Diseases, Department of Dermatology, University Medical Center Groningen, University of Groningen, Groningen, The Netherlands Published in the Journal of the American Academy of Dermatology, 2018 May;78(5):989-995

44
Abstract
Background Bullous pemphigoid is an autoimmune disease that typically presents with tense bullae and severe pruritus. However, bullae may be lacking, a subtype termed nonbullous pemphigoid. Objective To summarize the reported characteristics of nonbullous pemphigoid. Methods The EMBASE and MEDLINE databases were searched using ‘nonbullous pemphigoid’ and various synonyms. Case reports and series describing nonbullous pemphigoid were included. Results The search identified 133 articles. After selection 39 articles were included, presenting 132 cases. Erythematous, urticarial plaques (52.3%) and papules/nodules (20.5%) were the most reported clinical features. The mean age at presentation was 74.9 years. Histopathology was commonly nonspecific. Linear depositions of IgG/C3 along the basement membrane zone were found by direct immunofluorescence microscopy in 93.2%. Indirect immunofluorescence on salt-split skin was positive in 90.2%. The mean diagnostic delay was 22.6 months. The minority of patients (9.8%) developed bullae during the reported follow-up. Limitations Results are mainly based on case reports/small case series. Conclusion Nonbullous pemphigoid is an underdiagnosed variant of pemphigoid that most often does not evolve to bullous lesions, and mimics other pruritic skin diseases. Greater awareness among doctors is needed to avoid delay in diagnosis.

45
Introduction Bullous pemphigoid (BP) is the most common autoimmune bullous disease affecting the skin and mucous membranes, with autoantibodies directed against the 180-kDa BP antigen (BP180) and the 230-kDa BP antigen (BP230) located in the basement membrane zone.1 The disease commonly affects older patients and is associated with an increased risk of mortality, as well as a significant decline in quality of life and psychological well-being.2-6 The clinical phenotype of pemphigoid is polymorphic. The typical presentation consists of tense blisters that arise on erythematous, urticarial plaques, and is accompanied by severe pruritus.1,3 Prior to blister formation, pruritus can occur as a prodrome, with or without primary skin manifestations.7 In contrast to the typical bullous presentation, various atypical variants have been reported with terms such as papular pemphigoid, pemphigoid nodularis, pemphigoid vegetans, erythrodermic pemphigoid, pruritic nonbullous pemphigoid and erythema multiforme-like pemphigoid.8-11 The nonbullous variant of pemphigoid presents with pruritus and various nonbullous findings on the skin, such as erythematous patches, urticarial plaques, papules, nodules, excoriations, eczema, and erythroderma. Moreover, this variant can even present without primary skin lesions, called ‘pruritus on primary, non-diseased, non-inflamed skin’ according to the International Clinical Classification of Itch.11,12 Cohort studies show that at least 20% of all pemphigoid patients do not have blisters at the time of diagnosis.3,13 Thus, nonbullous pemphigoid is not that uncommon or atypical as may be assumed.14 Bullous and nonbullous pemphigoid are immunologically indistinguishable. The diagnosis is usually based on the combination of clinical presentation, histopathological findings, direct immunofluorescence (DIF) microscopy, and immunoserology.13 One of the main obstacles currently is the lack of consensus on the minimal diagnostic criteria of pemphigoid.8,14-17 The absence of blistering in nonbullous pemphigoid can make the recognition of this disease difficult for clinicians and might result in a delay of diagnosis.18,19 The aim of our study is to characterize and define nonbullous pemphigoid by systematic review, which has not been performed previously. Our study lists reported clinical presentations, histopathologic findings, laboratory findings, and prognosis regarding patients with nonbullous pemphigoid.

46
Methods Search strategy The literature search for this review was conducted in the EMBASE and MEDLINE databases on the 4th of November 2016. Various terms and synonyms for ‘nonbullous pemphigoid’ were used (supplementary appendix). There were no limitations on article type. After the selection procedure the references of all included articles were checked for missing articles. Selection of articles Language was limited to Dutch, German or English. Independent screening of the titles and abstracts was carried out by Drs Lamberts and Meijer. Discrepancies between the researchers were resolved through discussion. All articles reporting on one or multiple cases of nonbullous pemphigoid were included. Nonbullous pemphigoid was defined as all symptomatic cases with a nonbullous phenotype, that lacked a previous history of bullae, and fulfilled the following diagnostic criteria of pemphigoid: a positive DIF with linear IgG and/or C3c along the basement membrane zone and/or positive indirect immunofluorescence (IIF), in combination with compatible clinical presentation, histopathologic findings, or other immunoserologic tests. If the full text was not available online it was ordered at the national library. Poster abstracts were only included if sufficient individual patient data was presented. Data collection The following variables were gathered: age at diagnosis, gender, duration of symptoms before diagnosis, clinical presentation, results of diagnostic tests, histopathologic findings, total follow-up time, and blister development during follow-up. Statistical analyses were done in IBM SPSS statistics 23. Results Systematic search results A total of 39 articles presenting a total of 132 cases of nonbullous pemphigoid were identified (supplemental table 1). Figure 1 displays the selection procedure. The first case of nonbullous pemphigoid was reported in 1983 by Barker et al.20 The largest case series was from Lamb et al.21, who described the clinical presentation of 53 patients diagnosed with ‘prodromal bullous pemphigoid’. This large case

47
series did not present individual patient characteristics concerning age, gender, duration of symptoms, histopathological findings and total duration of follow-up. However, we were able to include the reported clinical presentation and the number of cases that developed blisters during follow-up. Clinical presentation Table 1 shows the demographics of the reported patients with nonbullous pemphigoid. The mean age at presentation was 74.9 years. The reported efflorescences and configurations of skin lesions seen at dermatological examination are displayed in table 2. Table 3 presents the location of skin lesions, reported in 64 of the 132 cases.
Articles identified by the literature search, n=208 - MEDLINE, n=84 - EMBASE, n=124
Unique articles screened on titles and abstracts, n=133
75 duplicates
Total of articles excluded, n=99 • Exclusion of articles reporting cases with blistering at the time of diagnosis n=51 - reports on patients with undefined symptoms prior to blistering, n=28 - reports on patients without undefined symptoms prior to blistering, n=23 • Exclusion due to language restriction, n=15 • Exclusion of articles not relevant to the question, n=33
Relevant articles, n=34 Inclusion after screening of the references, n=5 Total of articles included in the study, n=39
Figure 1. Study selection flow diagram
48
Histopathology The histopathological findings were described in 53 individual cases. A perivascular infiltrate was seen most frequently (n=32; 60.4%), which is a non-specific finding. Additionally, non-specific findings not further specified were reported in 14 cases (26.4%). Eosinophils were present in the biopsies of 25 cases (47.2%) and neutrophils in 7 cases (13.2%). Spongiosis without eosinophils was reported in 10 cases (18.9%), while eosinophilic spongiosis was seen in 4 (7.5%). The presence of dermal edema was reported in 8 cases (15.1%). The presence of a microscopic subepidermal split was reported in 8 cases (15.1%). Laboratory findings Table 4 shows the reported laboratory findings of patients with nonbullous pemphigoid. In all cases DIF microscopy was performed. In cases with a negative DIF result, the diagnosis was based on positive IIF with additional serological tests that specified the targeted antigen. IIF was the most commonly performed immunoserologic test (55 cases).
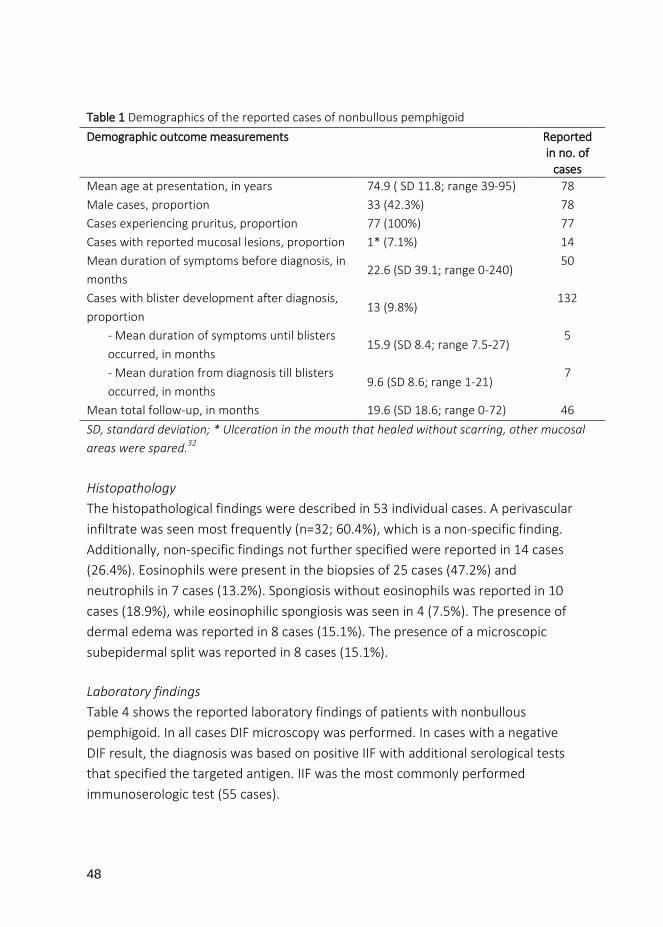
Table 1 Demographics of the reported cases of nonbullous pemphigoid Demographic outcome measurements Reported
in no. of cases
Mean age at presentation, in years 74.9 ( SD 11.8; range 39-95) 78 Male cases, proportion 33 (42.3%) 78 Cases experiencing pruritus, proportion 77 (100%) 77 Cases with reported mucosal lesions, proportion 1* (7.1%) 14 Mean duration of symptoms before diagnosis, in months
22.6 (SD 39.1; range 0-240) 50
Cases with blister development after diagnosis, proportion
13 (9.8%) 132
- Mean duration of symptoms until blisters occurred, in months
15.9 (SD 8.4; range 7.5-27) 5
- Mean duration from diagnosis till blisters occurred, in months
9.6 (SD 8.6; range 1-21) 7
Mean total follow-up, in months 19.6 (SD 18.6; range 0-72) 46 SD, standard deviation; * Ulceration in the mouth that healed without scarring, other mucosal areas were spared.32

49
Table 2. Skin findings and configurations reported in 132 cases of nonbullous pemphigoid Skin findings reported n (%) Erythematous, urticarial papules and plaques
69 (52.3)
Papules/nodules 27 (20.5) Eczematous lesions 16 (12.1) No primary lesions reported* 6 (4.5) Dermatitis herpetiformis-like lesions 5 (3.8) Ulcerations 3 (2.3) Erythroderma 3 (2.3) Other: Scarring alopecia 1 (0.8) Vegetations 1 (0.8) Solitary macule 1 (0.8) Excoriations 30 (22.7) Configuration reported Annular configuration** 8 (6.1) Figurated configuration 2 (1.5) Gyrated configuration 1 (0.8) * All 6 cases presented with secondary lesions in the form of excoriations. ** Two cases presented with erythema multiformis-like lesions
Table 3. Localization of skin lesions reported in 64 cases of nonbullous pemphigoid Localization n (%) Extremities 43 (67.2) Trunk 42 (65.6) Generalized 14 (21.9) Head and/or neck 7 (10.9) Scalp 6 (9.4) Hands and/or feet 5 (7.8)
The substrate used in IIF was not specified in 15 cases. In the other cases monkey esophagus (n=27) or human skin (n=13) were used as substrate. The BP230 ELISA was the least performed immunoserologic test (n=19). Additionally in four cases

50
immunoprecipitation was used to identify antigens, resulting in a positive reaction to both BP180 and BP230 in one case and only a positive reaction to BP230 in three cases. Eosinophilia in peripheral blood was reported in 13 of 15 cases (86.7%).
Table 4. Laboratory findings in reported cases of nonbullous pemphigoid
Diagnostic test Cases with positive test results, n (%)
Total no. reported
cases DIF microscopy, linear IgG and/or C3c depositions along the BMZ
123 (93.2) 132
IIF, IgG* 42 (76.4) 55 IIF on SSS, IgG, epidermal binding 46 (90.2) 51 Nc16a ELISA, IgG 15 (57.7) 26 BP230 ELISA, IgG 10 (52.6) 19 Immunoblot BP180, IgG 11 (32.4) 34 Immunoblot BP230, IgG 20 (55.6) 36 BMZ, basement membrane zone; BP, bullous pemphigoid; DIF, direct immunofluorescence; ELISA, enzyme-linked immunosorbent assay; IIF, indirect immunofluorescence; SSS, salt-split skin; Nc16a, non-collagen 16a. * different substrates were used by different authors.
Discussion This systematic review summarizes the reported characteristics of nonbullous pemphigoid. The most frequently reported skin efflorescences were erythematous, urticarial plaques (52.3%). Pruritus was reported in 100% of the cases. Overall, the duration between the start of symptoms and the correct diagnosis was very long (mean 22.6 months). Only 13 patients (9.8%) developed bullae during the reported follow-up, thus were actually prodromal to bullous pemphigoid. However, in the majority of the cases (90.2%) bullae never occurred. The findings of this review show that although the clinical presentation of nonbullous pemphigoid is various, pruritus at high age may be a clinical clue. Our study identified several similarities in clinical characteristics of nonbullous and bullous pemphigoid. Both present at older age (mean 74.9 years in nonbullous pemphigoid versus 77.2 – 82.6 years in bullous pemphigoid).4-6 Furthermore, in both variants lesions are most frequently located on the trunk and extremities.18,22 Most of the skin efflorescences reported in nonbullous pemphigoid

51
cases can also be found in patients with bullous pemphigoid.1,13 On the other hand, mucosal involvement was rarely reported in nonbullous pemphigoid, while reported in 10-30% of patients with bullous pemphigoid.3,13,22 In 14 cases the configurations of the skin lesions were reported to be annular, gyrate, figurate or herpetiform.21,23-30 Two of these patients presented with targetoid lesions.21,25 We also found three case reports that were possibly drug induced due to nifedipine, lisinopril and the combination of allopurinol plus colchicine.25,31,32 Nifedipine and lisinopril were previously associated with bullous pemphigoid, however it is not shown that these drugs actually cause a higher risk to develop bullous pemphigoid.33,34 Studies did show that the use of spironolactone and neuroleptics are independent risk factors for the development of bullous pemphigoid.35,36 The reported histopathologic findings in nonbullous pemphigoid differ from bullous pemphigoid with typical bullae in several aspects. Histopathologic findings were commonly nonspecific in nonbullous pemphigoid and resembled eczema or prurigo nodularis. While bullous pemphigoid is usually characterized by the presence of eosinophilic spongiosis (>50%) and a subepidermal split (± 80%), in the cases with nonbullous pemphigoid histopathologic findings only described eosinophilic spongiosis in 7.5% and a subepidermal split in 15.1%.1,37 These findings emphasize the need to always perform DIF microscopy and immunoserology in addition to histopathology in patients in which nonbullous pemphigoid is suspected. In nonbullous pemphigoid DIF microscopy was the most reported positive diagnostic test (positive in 93.2%) followed by IIF on salt-split skin (SSS) (90.2%). Both DIF microscopy and IIF on SSS have a high specificity (98% and 100% respectively).38 Yet, the reported percentage of positive findings in DIF microscopy in nonbullous pemphigoid might be an overestimation, since this test is regarded as the reference standard for diagnosis of pemphigoid and commonly the only performed immunopathologic test.39 Consequently the diagnosis of pemphigoid might be rejected when DIF microscopy is negative and immunoserologic analysis might not have been performed. The mean duration of symptoms of nonbullous pemphigoid until the correct diagnosis of pemphigoid was 22.6 months. These results seem to be consistent with other research that also found long diagnostic delays in pemphigoid cases that lack bullae. Previously, we reported a mean delay in diagnosis of 33.6 months in 15 patients with nonbullous pemphigoid.16 The studies of Zhang et al. and Sun et al. reported misdiagnosis with eczema, nodular prurigo

52
or other dermatologic diseases in all pemphigoid patients that initially presented without bullae, 181 and 24 patients respectively.18,40 In both studies the correct diagnosis was made when bullae appeared, which was after a mean duration of 15.9 months and 20.75 months (range 1 month to 19 years). Although these studies only identified misdiagnosis in prodromal bullous pemphigoid patients, they also illustrate the importance of more awareness and better knowledge regarding the characteristics of nonbullous pemphigoid. In contrast, Della Torre et al. did not find a significant difference in delay of diagnosis between patients with bullous (n=97) and nonbullous (n=20) pemphigoid in their cohort.3 Whether early recognition and immunosuppressive treatment of nonbullous pemphigoid can prevent later blister development is unknown. A much debated question is whether patients diagnosed with nonbullous pemphigoid are prodromal or have a distinct pemphigoid variant.10,21 The finding that the majority of nonbullous pemphigoid patients did not develop blisters during follow-up supports the hypothesis that nonbullous pemphigoid is not a prodromal stage but merely a variant within the clinical spectrum of pemphigoid diseases. We can conclude that ‘prodromal pemphigoid’ is an incorrect term and that there is a need for consensus regarding the terminology to describe this disease variant. We strongly argue for insertion of the term nonbullous pemphigoid in the EMTREE. During our literature search we identified a number of other subepidermal autoimmune blistering diseases with nonbullous clinical presentations: nonbullous epidermolysis bullosa acquisita41, nonbullous linear IgA dermatosis42 and nonbullous pemphigoid gestationis43. Furthermore we came across reports of pemphigoid patients that first presented with bullae and later experienced a nonbullous flare-up of the disease.44-49 These cases strengthen the idea that nonbullous pemphigoid should be seen as a disease variant within the spectrum of pemphigoid diseases. Previous publications reported a higher prevalence of BP-specific autoantibodies in older dermatology patients (>75 years) without blisters, healthy blood donors, and elderly individuals with pruritus.50-52 How these patients fit the pemphigoid spectrum has not been clarified. Our systematic review provides insight on reported literature on nonbullous pemphigoid so far. A limitation of this review is that the results are mainly based on single case reports and small case series. Consequently missing values were present in the summarized data. Moreover, in some publications the

53
clinical picture was described very briefly. A second limitation of this review is the risk of reporting bias, since cases with unusual atypical presentations are more likely to be reported in the literature. Furthermore, the finding that the majority (90.2%) of nonbullous pemphigoid patients did not develop blisters during the reported follow-up (mean 19.8 months;range 0-72) might be slightly biased by selection, since we excluded cases of pemphigoid that were diagnosed after bullae appeared, even though authors retrospectively described pruritic symptoms prior to blistering. However, it is uncertain whether these symptoms prior to diagnosis were caused by pemphigoid, or by other pruritic dermatoses, such as prurigo nodularis or eczema. This study therefore highlights the importance of larger observational studies with longer follow-up for a better representation of nonbullous pemphigoid Another interesting focus for future research is why patients with nonbullous pemphigoid do not develop bullae. Several factors have been suggested to influence blister formation, such as autoantibody titers,53 the antigens or epitopes targeted by autoantibodies,22,54 complement involvement,55,56 and eosinophils.57 More knowledge of the underlying pathophysiology of this subtype of pemphigoid might lead to more awareness and less delay in diagnosis of nonbullous pemphigoid. In conclusion, our review showed that the reported clinical presentation of nonbullous pemphigoid can be heterogeneous. The reported long duration of symptoms until correct diagnosis (mean 22.6 months) illustrates that nonbullous pemphigoid can be difficult to recognize for clinicians. Pruritus in elderly is a common denominator in patients with nonbullous pemphigoid and in our opinion the most important clue for recognition. Clinicians should therefore perform DIF on a skin biopsy and immunoserologic analysis on a blood sample in elderly with unexplained or refractory chronic pruritus and erythematous, urticarial papules and plaques. Further study is needed to evaluate the prevalence of nonbullous pemphigoid.
References 1. Schmidt E., Zillikens D. Pemphigoid diseases. - Lancet.2013 Jan 26;381(9863):320-32.doi:
10.1016/S0140-6736(12)61140-4.Epub 2012 Dec 11. 2. Kouris A, Platsidaki E, Christodoulou C, et al. Quality of life, depression, anxiety and loneliness in
patients with bullous pemphigoid. A case control study. - An Bras Dermatol.2016 Sep-Oct;91(5):601-603.doi: 10.1590/abd1806-4841.20164935.

54
3. Della Torre R, Combescure C, Cortes B, et al. Clinical presentation and diagnostic delay in bullous pemphigoid: A prospective nationwide cohort. - Br J Dermatol.2012 Nov;167(5):1111-7.doi: 10.1111/j.1365-2133.2012.11108.x.
4. Marazza G, Pham H, Scharer L, et al. Incidence of bullous pemphigoid and pemphigus in switzerland: A 2-year prospective study. - Br J Dermatol.2009 Oct;161(4):861-8.doi: 10.1111/j.1365-2133.2009.09300.x.Epub 2009 May 8.
5. Langan S, Smeeth L, Hubbard R, Fleming K, Smith C, West J. Bullous pemphigoid and pemphigus vulgaris--incidence and mortality in the UK: Population based cohort study. - BMJ.2008 Jul 9;337:a180.doi: 10.1136/bmj.a180.
6. Joly P, Baricault S, Sparsa A, et al. Incidence and mortality of bullous pemphigoid in france. - J Invest Dermatol.2012 Aug;132(8):1998-2004.doi: 10.1038/jid.2012.35.Epub 2012 Mar 15.
7. Asbrink E, Hovmark A. Clinical variations in bullous pemphigoid with respect to early symptoms. - Acta Derm Venereol.1981;61(5):417-21.
8. Cozzani E, Gasparini G, Burlando M, Drago F, Parodi A. Atypical presentations of bullous pemphigoid: Clinical and immunopathological aspects. Autoimmun Rev. 2015;14(5):438-445.
9. Bernard P, Antonicelli F. Bullous pemphigoid: A review of its diagnosis, associations and treatment. Am J Clin Dermatol. 2017 Aug;18(4):513-528.
10. Strohal R, Rappersberger K, Pehamberger H, Wolff K. Nonbullous pemphigoid: Prodrome of bullous pemphigoid or a distinct pemphigoid variant? J Am Acad Dermatol. 1993;29(2 II):293-299.
11. Bakker CV, Terra JB, Pas HH, Jonkman MF. Bullous pemphigoid as pruritus in the elderly a common presentation. JAMA Dermatol. 2013;149(8):950-953.
12. Stander S, Weisshaar E, Mettang T, et al. Clinical classification of itch: A position paper of the international forum for the study of itch. Acta Derm Venereol. 2007;87(4):291-294.
13. Di Zenzo G, Della Torre R, Zambruno G, Borradori L. Bullous pemphigoid: From the clinic to the bench. - Clin Dermatol.2012 Jan-Feb;30(1):3-16.doi: 10.1016/j.clindermatol.2011.03.005.
14. Lipsker D, Borradori L. 'Bullous' pemphigoid: What are you? urgent need of definitions and diagnostic criteria. Dermatology. 2010;221(2):131-134.
15. Borradori L, Joly P. Toward a practical renaming of bullous pemphigoid and all its variants: Cutaneous pemphigoid. - JAMA Dermatol.2014 Apr;150(4):459.doi: 10.1001/jamadermatol.2014.51.
16. Bakker C, Terra J, Jonkman M. Toward a practical renaming of bullous pemphigoid and all its variants-reply. - JAMA Dermatol.2014 Apr;150(4):459-60.doi: 10.1001/jamadermatol.2014.54.
17. Feliciani C, Joly P, Jonkman M, et al. Management of bullous pemphigoid: The european dermatology forum consensus in collaboration with the european academy of dermatology and venereology. - Br J Dermatol.2015 Apr;172(4):867-77.doi: 10.1111/bjd.13717.
18. Zhang Y, Luo Y, Han Y, Tian R, Li W, Yao X. Non-bullous lesions as the first manifestation of bullous pemphigoid: A retrospective analysis of 181 cases. J Dermatol. 2017 Jul;44(7):742-746.
19. Hertl M, Schmidt T. Underrecognition of the heterogeneous clinical spectrum of bullous pemphigoid. JAMA Dermatol. 2013;149(8):954-955.
20. Barker DJ. Generalized pruritus as the presenting feature of bullous pemphigoid. Br J Dermatol. 1983;109(2):237-239. 21. Lamb PM, Abell E, Tharp M, Frye R, Deng J-. Prodromal bullous pemphigoid. Int J Dermatol.

55
2006;45(3):209-214. 22. Tanaka M, Hashimoto T, Dykes PJ, Nishikawa T. Clinical manifestations in 100 japanese bullous
pemphigoid cases in relation to autoantigen profiles. Clin Exp Dermatol. 1996;21(1):23-27. 23. Altman K, Lloyd R, Longley J. Granuloma annulare-like presentation of bullous pemphigoid. J Am
Acad Dermatol. 2015;72(5):AB119. 24. Amato DA, Silverstein J, Zitelli J. The prodrome of bullous pemphigoid. Int J Dermatol.
1988;27(8):560-563. 25. Axelrod S, Davis-Lorton MA. Erythema multiforme-like bullous pemphigoid drug eruption. Ann
Allergy Asthma Immunol. 2010;105(5):A57-A58. 26. Ise Y, Suga Y, Okumura K, Negi O, Ishii N, Hashimoto T. Erythematous variety of bullous
pemphigoid: Case report and literature review. Acta Derm -Venereol. 2016;96(3):412-413. 27. Kabuto M, Fujimoto N, Tanaka T. Two cases of annular erythema without bullous lesions by
autoimmune blistering diseases. Mod Rheumatol. 2018 Jan;28(1):200-202. 28. Park KY, Hyun MY, Yeo IK, Seo SJ, Hong CK. An eczema-like, pruritic, nonbullous form of bullous
pemphigoid. J Dermatol Case Rep. 2015;9(2):55-57. 29. Patel M, Gilbert E, Tzu J. Case report: Bullous pemphigoid associated with renal cell carcinoma
and invasive squamous cell carcinoma. J Am Acad Dermatol. 2012;66(4):AB44. 30. Tashiro H, Arai H, Hashimoto T, Takezaki S, Kawana S. Pemphigoid nodularis: Two case studies
and analysis of autoantibodies before and after the development of generalized blistering. J Nippon Med Sch. 2005;72(1):60-65.
31. Liu VT, Cheng KY, Yu P. Where are the blisters? A case of bullous pemphigoid without bullae. J Gen Intern Med. 2014;29:S470.
32. Ameen M, Harman KE, Black MM. Pemphigoid nodularis associated with nifedipine [11]. Br J Dermatol. 2000;142(3):575-577.
33. Shachar E, Bialy-Golan A, Srebrnik A, Brenner S. "Two-step" drug-induced bullous pemphigoid. Int J Dermatol. 1998;37(12):938-939.
34. Kalinska-Bienias A, Rogozinski TT, Wozniak K, Kowalewski C. Can pemphigoid be provoked by lisinopril? Br J Dermatol. 2006;155(4):854-855.
35. Bastuji-Garin S, Joly P, Lemordant P, et al. Risk factors for bullous pemphigoid in the elderly: A prospective case-control study. J Invest Dermatol. 2011;131(3):637-643.
36. Bastuji-Garin S, Joly P, Picard-Dahan C, et al. Drugs associated with bullous pemphigoid. A case-control study. Arch Dermatol. 1996;132(3):272-276.
37. Chan YC, Sun YJ, Ng PP, Tan SH. Comparison of immunofluorescence microscopy, immunoblotting and enzyme-linked immunosorbent assay methods in the laboratory diagnosis of bullous pemphigoid. Clin Exp Dermatol. 2003;28(6):651-656.
38. Sardy M, Kostaki D, Varga R, Peris K, Ruzicka T. Comparative study of direct and indirect immunofluorescence and of bullous pemphigoid 180 and 230 enzyme-linked immunosorbent assays for diagnosis of bullous pemphigoid. J Am Acad Dermatol. 2013;69(5):748-753.
39. Bagci IS, Horvath ON, Ruzicka T, Sardy M. Bullous pemphigoid. Autoimmun Rev. 2017;16(5):445-455.
40. Sun C, Chang B, Gu H. Non-bullous lesions as the first manifestation of bullous pemphigoid: A retrospective analysis of 24 cases. J Dermatolog Treat. 2009;20(4):233-237.

56
41. Furukita K, Ansai S, Hida Y, Kubo Y, Arase S, Hashimoto T. A case of epidermolysis bullosa acquisita with unusual clinical features. Clin Exp Dermatol. 2009;34(8):e702; e704.
42. Chaudhari S, Mobini N. Linear IgA bullous dermatosis: A rare clinicopathologic entity with an unusual presentation. J Clin Aesthetic Dermatol. 2015;8(10):43-46.
43. Ogilvie P, Trautmann A, Dummer W, Rose C, Bröcker EB, Zillikens D. Pemphigoid gestationis without blisters. Hautarzt. 2000;51(1):25-30.
44. Borradori L, Prost C, Wolkenstein P, Bernard P, Baccard M, Morel P. Localized pretibial pemphigoid and pemphigoid nodularis. J Am Acad Dermatol. 1992;27(5 II):863-867.
45. Bourke JF, Berth-Jones J, Gawkrodger DJ, Burns DA. Pemphigoid nodularis: A report of two cases. Clin Exp Dermatol. 1994;19(6):496-499.
46. Gallo R, Parodi A, Rebora A. Pemphigoid nodularis [1]. Br J Dermatol. 1993;129(6):744-745. 47. Mochizuki M, Fujine E, Tawada C, Kanoh H, Seishima M. Pemphigoid nodularis possibly induced
by etanercept. J Dermatol. 2013;40(7):578-579. 48. Yung CW, Soltani K, Lorincz AL. Pemphigoid nodularis. J Am Acad Dermatol. 1981;5(1):54-60. 49. Ross JS, McKee PH, Smith NP, et al. Unusual variants of pemphigoid: From pruritus to
pemphigoid nodularis. J Cutaneous Pathol. 1992;19(3):212-216. 50. Meijer JM, Lamberts A, Pas HH, Jonkman MF. Significantly higher prevalence of circulating
bullous pemphigoid-specific IgG autoantibodies in elderly patients with a nonbullous skin disorder. Br J Dermatol. 2015;173(5):1274-1276.
51. van Beek N, Dohse A, Riechert F, et al. Serum autoantibodies against the dermal-epidermal junction in patients with chronic pruritic disorders, elderly individuals and blood donors prospectively recruited. Br J Dermatol. 2014;170(4):943-947.
52. Schmidt T, Sitaru C, Amber K, Hertl M. BP180- and BP230-specific IgG autoantibodies in pruritic disorders of the elderly: A preclinical stage of bullous pemphigoid? Br J Dermatol. 2014;171(2):212-219.
53. Kawahara Y, Matsumura K, Hashimoto T, Nishikawa T. Immunoblot analysis of autoantigens in localized pemphigoid and pemphigoid nodularis. Acta Derm Venereol. 1997;77(3):187-190.
54. Thoma-Uszynski S, Uter W, Schwietzke S, et al. BP230- and BP180-specific auto-antibodies in bullous pemphigoid. J Invest Dermatol. 2004;122(6):1413-1422.
55. Romeijn TR, Jonkman MF, Knoppers C, Pas HH, Diercks GF. Complement in bullous pemphigoid: Results from a large observational study. Br J Dermatol. 2017;176(2):517-519.
56. Nelson KC, Zhao M, Schroeder PR, et al. Role of different pathways of the complement cascade in experimental bullous pemphigoid. J Clin Invest. 2006;116(11):2892-2900.
57. de Graauw E, Sitaru C, Horn M, et al. Evidence for a role of eosinophils in blister formation in bullous pemphigoid. Allergy. 2017 Jul;72(7):1105-1113.
58. Bingham EA, Burrows D, Sandford JC. Prolonged pruritus and bullous pemphigoid. Clin Exp Dermatol. 1984;9(6):564-570.
59. Borradori L, Rybojad M, Verola O, Flageul B, Puissant A, Morel P. Pemphigoid nodularis. Arch Dermatol. 1990;126(11):1522-1523.
60. Wolf R, Ophir J, Dechner E. Nonbullous bullous pemphigoid. Int J Dermatol. 1992;31(7):498-500.
61. Wever S, Rank C, Hornschuh B, et al. Bullous pemphigoid simulation subacute simple prurigo. Hautarzt. 1995;46(11):789-795.

57
62. Jeong SJ, Lee CW. Bullous pemphigoid: Persistent lesions of eczematous/urticarial erythemas. Cutis. 1995;56(4):225-226.
63. Cliff S, Holden CA. Pemphigoid nodularis: A report of three cases and review of the literature. Br J Dermatol. 1997;136(3):398-401.
64. Alonso-Llamazares J, Dietrich SM, Gibson LE. Bullous pemphigoid presenting as exfoliative erythroderma. J Am Acad Dermatol. 1998;39(5 Pt 2):827-830.
65. Alonso-Llamazares J, Rogers III RS, Oursler JR, Calobrisi SD. Bullous pemphigoid presenting as generalized pruritus: Observations in six patients. Int J Dermatol. 1998;37(7):508-514.
66. Scrivener Y, Heid E, Grosshans E, Cribier B, Gibson LE. Erythrodermic bullous pemphigoid [4] (multiple letters). J Am Acad Dermatol. 1999;41(4):658-659.
67. Schmidt E, Sitaru C, Schubert B, et al. Subacute prurigo variant of bullous pemphigoid: Autoantibodies show the same specificity compared with classic bullous pemphigoid. J Am Acad Dermatol. 2002;47(1):133-136.
68. Powell AM, Albert S, Gratian MJ, Bittencourt R, Bhogal BS, Black MM. Pemphigoid nodularis (non-bullous): A clinicopathological study of five cases. Br J Dermatol. 2002;147(2):343-349.
69. Goel A, Balchandran C, Shenoi SD, Pai B. S. Non-bullous variant of bullous pemphigoid: Role of immunofluorescence in diagnosis. Indian J Dermatol Venereol Leprol. 2003;69(4):294-295.
70. Mechtel D, Prugovecki V, Knopf B. Pemphigoid nodularis (non bullous) - A case report. Aktuel Dermatol. 2003;29(7):296-299.
71. Von Felbert V, Simon D, Braathen L, Hunziker T. Pemphigoid nodularis triggered by hypereosinophilic syndrome? Hautarzt. 2006;57(5):434-436.
72. Yesudian PD, Shah D, Giles M, Williamson D. Nonbullous variant of pemphigoid in a patient with absent C4 allotype: A lesson in immunology for dermatologists. Br J Dermatol. 2009;161:97.
73. Matsudate Y, Ansai S-, Hirose K, Kubo Y, Arase S. Pemphigoid nodularis: The importance of ELISA for diagnosis. Eur J Dermatol. 2009;19(1):83-84.
74. Safa G, Darrieux L. Nonbullous pemphigoid treated with doxycycline monotherapy: Report of 4 cases. J Am Acad Dermatol. 2011;64(6):e116; e118.
75. McCourt C, Corry A, Walsh M, McMillan C. Generalized pruritic eruption in a patient with chronic kidney disease. Dermatol Online J. 2010;16(4).
76. Geiss Steiner J, Trüeb RM, Mühleisen B, Kerl K, French LE, Hofbauer GF. Ecthyma gangrenosum-like bullous pemphigoid. J Invest Dermatol. 2009;129:S20.
77. Lehman JS, Kalaaji AN, Rogers 3rd. RS, Stone RA. Pemphigoid nodularis: A case report. Cutis. 2011;88(5):224-226.
78. Balakirski G, Merk HF, Megahed M. Bullous pemphigoid: A new look at a well-known disease. Hautarzt. 2014;65(12):1013-1016.
79. Huet F, Karam A, Lemasson G, et al. Image gallery: Erythroderma revealing a nonbullous bullous pemphigoid. Br J Dermatol. 2016;175(5):e136-e137.

58
Supplementary appendix Keywords used in the systematic search (performed in EMBASE & MEDLINE): ('non*bullous' AND 'pemphigoid') OR ‘non*bullous pemphigoid' OR non*bullous bullous pemphigoid’ OR ‘non*bullous BP’ OR 'pruritic pemphigoid' OR 'pruritic non*bullous pemphigoid' OR 'pemphigoid nodularis' OR 'nodular pemphigoid' OR 'prurigo nodularis-like pemphigoid' OR 'papular pemphigoid' OR 'prodromal BP' OR 'prodromal bullous pemphigoid' OR 'prodromal pemphigoid' OR 'prodrome of bullous pemphigoid' OR ‘non bullous variant' NEAR/10 'pemphigoid' OR 'nonbullous variant' NEAR/10 'pemphigoid' OR 'bullous pemphigoid mimicking' OR '-like bullous pemphigoid' OR 'erythrodermic bullous pemphigoid' OR ('bullous pemphigoid' AND 'without blister*') OR ('bullous pemphigoid'/exp AND 'without blister*') OR ('bullous pemphigoid' AND 'without bullae') OR ('bullous pemphigoid' AND 'without bullous lesions')

59
Supplemental table
Supplemental table 1. All included articles presenting cases of nonbullous pemphigoid First author Publication year Barker et al.20 1983 Bingham et al.58 1984 Amato et al.24 1988 Borradori et al.59 1990 Wolf et al.60 1992 Ross et al.49 1992 Strohal et al.10 1993 Bourke et al.45 1994 Wever et al.61 1995 Jeong et al.62 1995 Cliff et al.63 1996 Kawahara et al.53 1997 Alonso-Llamazares et al.64 1998 Alonso-Llamazares et al.65 1998 Scrivener et al.66 1999 Ameen et al.32 2000 Schmidt et al.67 2002 Powell et al.68 2002 Goel et al.69 2003 Mechtel et al.70 2003 Tashiro et al.30 2005 von Felbert et al.71 2005 Lamb et al.21 2006 Yesudian et al.72 2009 Matsudate et al.73 2009 Axelrod et al.25 2010 Safa et al.74 2010 McCourt et al.75 2010 Geiss Steiner et al.76 2010 Lehman et al.77 2011 Patel et al.29 2012 Bakker et al.11 2013 Balakirski et al.78 2014 Liu et al.31 2014 Kabuto et al.27 2015 Altman et al.23 2015 Park et al.28 2015 Huet et al.79 2016 Ise et al.26 2016

60
4

61
CHAPTER 4 Nonbullous pemphigoid: Insights in clinical and diagnostic findings, treatment responses, and prognosis Aniek Lamberts1, Joost M. Meijer1, Hendri H. Pas1, Gilles F.H. Diercks1,2, Barbara Horváth1, Marcel F. Jonkman1
Center for Blistering Diseases, Department of Dermatology1, and the Department of Pathology2, University Medical Center Groningen, University of Groningen, Groningen, The Netherlands Published in the Journal of the American Academy of Dermatology, 2019 Aug;81(2):355-363

62
Abstract
Background Nonbullous pemphigoid is an under-recognized phenotype of the autoimmune bullous disease pemphigoid, characterized by the absence of blisters. Several disease aspects have not been studied previously. Objective To describe the characteristics of nonbullous pemphigoid. Methods A retrospective chart review study. The diagnosis of pemphigoid was based on meeting two of the following three criteria; 1) pruritus, 2) positive direct immunofluorescence microscopy (DIF), 3) positive indirect immunofluorescence microscopy (IIF) on salt-split skin. Results Sixty-nine patients were included. The mean delay in diagnosis was 29 months. Skin examination most often showed pruritic papules/nodules (31%) or pruritus without primary skin lesions (22%). Histopathological findings were mainly nonspecific. DIF and IIF were positive in 60% and 69%. During follow-up blisters formed in 17%, which was associated with a positive IIF (p=0.014), and positive BP180 immunoblot result (p=0.032). The Kaplan-Meier estimates of 1-, 2- and 3-year mortality were 14%, 34%, and 46%, with an 8.6 fold increased all-cause mortality risk. Limitations The retrospective study design. Conclusion Nonbullous pemphigoid presented with heterogeneous pruritic skin lesions, resulting in delayed diagnosis. DIF and IIF are essential to diagnose nonbullous pemphigoid, in contrast to histopathology, mainly showing nonspecific findings. During follow-up, an increased all-cause mortality risk was observed.

63
Capsule summary
• Nonbullous pemphigoid is an under-recognized variant of pemphigoid with long diagnostic delays. Patients present with symptoms of pruritus, with or without skin lesions.
• Clinicians should realize that histopathology is not useful for diagnosing nonbullous pemphigoid; direct- and indirect immunofluorescence are required. Patients have an increased all-cause mortality risk.
List of abbreviations
BP bullous pemphigoid BMZ basement membrane zone CR complete remission DC disease control DIF direct immunofluorescence microscopy ELISA enzyme-linked immunosorbent assay HR hazard ratio IIF indirect immunofluorescence microscopy MO monkey oesophagus NC16A noncollagenous 16A domain of BP180 PR partial remission SMR standardized mortality ratio SSS salt-split skin

64
Introduction
Pemphigoid is an autoantibody mediated skin disease mainly affecting elderly patients.1 Autoantibodies target structural proteins BP180 and BP230 located in the basement membrane zone (BMZ), inducing an eosinophilic inflammatory response in the skin.2 Interestingly, the immunological disease mechanism in pemphigoid can lead to two distinct clinical phenotypes, termed bullous and nonbullous pemphigoid. Bullous pemphigoid (BP) classically presents with severe pruritus and tense blisters on urticarial plaques.1 There is a high co-occurrence of psychiatric- and neurodegenerative diseases, and patients have an increased mortality risk compared to the age-matched general population.3–6 One in five patients lack typical blisters, termed nonbullous pemphigoid.7 Patients present with pruritus and a wide spectrum of skin manifestations that may resemble other pruritic skin diseases.8–12 Consequently, patients often have a long diagnostic delay.13,14 Urticarial plaques and papules/nodules are reported most frequently.8–12 Blister development during disease course is reported in ten percent.8 The diagnosis of nonbullous pemphigoid is based on the detection of either skin-bound IgG or complement C3 in a linear deposition along the BMZ by direct immunofluorescence microscopy (DIF), and/or circulating antibodies by indirect immunofluorescence microscopy (IIIF) on salt-split skin (SSS).7 Histopathology is often nonspecific.8 To date, little is known about the management and prognosis of nonbullous pemphigoid. Treatment recommendations of BP are often followed, recommending whole body application of superpotent topical corticosteroids as initial therapy.15 The aim of this study was to describe the clinical and diagnostic findings, treatment responses, and follow-up of patients with nonbullous pemphigoid, to support early recognition and improve patient care. Materials and methods
Selection of cases and data collection This retrospective study included patients diagnosed with nonbullous pemphigoid between 2002-2017 at the dermatology department of the University Medical Center Groningen, the Netherlands. Inclusion criteria were based on recently established diagnostic criteria by Meijer et al. with two positive out of the following three criteria: 1) pruritus, 2) linear IgG and/or C3 depositions along the BMZ by DIF,

65
3) positive staining of IgG on the epidermal side of SSS substrate by IIF.7 Exclusion criteria were blisters or vesicles prior to diagnosis, or at initial presentation objectified by a physician. Cases were excluded if blisters occurred within two months after the onset of pruritus, and considered prodromal BP. Clinical characteristics were assessed by reviewing patient charts. Data was collected anonymously in electronic case report forms using Open Clinica software. The following variables were collected: age, date of first symptoms and diagnosis, clinical presentation, skin manifestations, diagnostic findings, treatment response, side effects, blister development and death during follow-up. The study was approved by the Institutional Review Board at our local ethics committee. Laboratory tests for pemphigoid Laboratory techniques DIF, IIF on SSS and monkey oesophagus (MO), and immunoblot were performed at our Immuno-dermatology Laboratory as reported previously.7 Autoantibodies against the noncollagenous 16A domain of BP180 (NC16A) and BP230 were detected with commercially available enzyme-linked immunosorbent assays (ELISA) according to the manufacturer’s protocol (MBL Japan, cut-off index ≥ 9 u/mL). Treatment response and safety Treatment response was assessed by using outcome measurements defined by international consensus, consisting of disease control (DC), partial remission (PR) on minimal/off therapy, complete remission (CR) on minimal/off therapy, and relapse.16 We deviated from the consensus definition by allowing a dose of 7.5 mg methotrexate per week to count for minimal therapy. Reported side effects were registered. Uncertainties during retrospective assessment were resolved through discussion with the study team. Statistical analysis Correlations between bivariate outcomes were analyzed with the Pearson Chi-Square test, or Fisher’s exact test when appropriate. Comparisons of means for non-normally distributed data were done with the Mann-Whitney U test. Estimated cumulative survival after 1-, 2-, and 3-year follow-up was assessed by Kaplan-Meier analysis. Univariate cox regression was performed to investigate the effect of selected variables on the 3-year survival. Age-adjusted standardized

66
mortality ratios (SMR) were calculated by comparing the objectified 1-year all-cause mortality rates in nonbullous pemphigoid with the expected 1-year all-cause mortality rates per age group, using mortality data of the Dutch population over the year 2017, provided by Statistics Netherlands (CBS, www.cbs.nl). The 95% confidence interval for SMR was calculated using Poisson distribution.17 A p-value <0.05 was defined as statistical significance. Statistical analyses were performed using IBM SPSS statistics version 23. Results
Patient characteristics and clinical findings Patients’ characteristics are shown in table 1. Patients were relatively old (mean 76.1 years), and had several comorbidities, such as hypertension (34%), diabetes mellitus (24%), atrial fibrillation (13%), and stroke (12%). ACE inhibitors and loop diuretics were used in 26% and 20%. Six individual patients reported a time relation with onset of symptoms and the use of acenocoumarol, simvastatin, candesartan, metoprolol, perindopril and acitretin. Observed skin lesions are displayed in figure 1. Fifteen patients (22%) presented with pruritus on primary, nondiseased, noninflamed skin, and showed a significantly longer delay in diagnosis compared to patients with primary skin lesions (49.9 vs. 22.6 months; p=0.018). The average duration of follow-up was 22 months, but varied per patient (0-218 months). Factors influencing the follow-up duration include death during follow-up, response to therapy, and transfer of care of elderly patients with physical limitations or long travel distance. Follow-up was longer in patients with systemic treatment. Diagnostic findings Diagnostic findings are summarized in table 2. Histopathology most often showed a dermal perivascular infiltrate (98%) with eosinophils (69%), and a subepidermal split in only one case. Pathologists most frequently reported nonspecific findings (n=21; 39%), or findings compatible with cutaneous drug reactions (n=18; 33%), eczema (n=12; 22%), urticaria (n=6; 11%), chronic scratching (n=5; 9%), insect bites (n=3; 6%), or a psoriasiform dermatitis (n=3; 6%).

67
Table 1. Clinical characteristics of patients with nonbullous pemphigoid General characteristics Mean (SD; Range) or n (%) Mean age at diagnosis, years 76.1 (13.5; 39-101) Gender, n / n 29 M / 40 F Mean delay in diagnosis, months 28.9 (53.7; 0-385) Mean time of follow-up, months 21.9 (38.2; 0-218) Living in a nursing home, n (%) 7 (11.5) Location of symptoms n (%)* Extremities 58 (92.1) Back 46 (79.3) Abdomen 29 (55.8) Scalp 18 (34.0) Hands/feet 16 (32.0) Neck 17 (31.5) Face 6 (12.0) Mucosa 0 (0.0) Findings during skin examination n (%)* Pruritus** 68 (98.5) generalized pruritus 40 (58.0) excoriations 52 (76.5) Localized disease*** 6 (8.7) Xerosis cutis 8 (11.8) Papules/nodules 21 (30.9) Pruritus on primary nondiseased, noninflamed skin 15 (22.1) 3 sensu stricto
Urticarial papules/plaques 8 (11.8) 1 with pustules Eczematous lesions 3 (4.4) Mixed skin findings
Urticarial papules/plaques + papules/nodules 10 (14.7) Papules/nodules + eczematous lesions 1 (1.5) Papules/nodules + erythematous macules 3 (4.4) Other 7 (10.3) 4 Erythematous plaques with squamous borders 2 pityriasis rubra pilaris-like
1 Suberythrodermia
1 Ulcerations
1 Localized livid, erythematous macules M, male; F, female; SD, standard deviation; R, range. * percentages were calculated after exclusion of cases of which data was unknown. ** Data extracted from anamnesis. ***Localized disease was defined as the presence of localized lesions involving one body site, conform the European consensus on the management of bullous pemphigoid 2015.15

68
DIF results were positive in 41 of 69 cases (60%), of which 20 cases (29%) had a positive IIF on SSS result, and 21 cases (31%) a negative result. In ten of these 21 cases circulating antibodies against BP180 or BP230 were demonstrated by immunoblot or ELISA. In 27 of 69 cases (40%) DIF was negative and the diagnosis based on a positive IIF on SSS and compatible pruritic symptoms. Additional positive results by IIF on MO, ELISA and immunoblot were found in 26 (96%), 21 (78%) and 17 (63%) cases. In one case DIF was not performed, and the diagnosis based on IIF on SSS positivity.
Figure 1. Nonbullous pemphigoid presenting with pruritus and various skin lesions. A. Papules and nodules. B. Pruritus on primary nondiseased, noninflamed skin with secondary excoriations. C. Urticarial papules and plaques. D. Eczematous lesions.

69
Table 2. Diagnostic findings in nonbullous pemphigoid Histopathology (n=54) n (%)* Subepidermal split 1 (1.9) Spongiosis 22 (40.7) without inflammatory cells 12 (22.2) Eosinophilic spongiosis 3 (5.6) Lymphocytic spongiosis 7 (13.0) Dermal lymphocytic infiltrate 53 (98.1) located perivascular 49 (90.7) presence of eosinophils 37 (68.5) DIF on a skin biopsy (n=68) n (%)* Positive DIF result 41 (60.3) IgG 41 (60.3) C3c 14 (20.6) IgA 10 (14.7) IgM 5 (7.4) n-serrated pattern 19 of 41 (46.3) indeterminable serration pattern 22 of 41 (53.7) Immunoserological findings (n=68) n (%)* Positive IIF result 47 (69.1) IIF on MO, IgG 42 (61.8) IIF on SSS, IgG 45 (66.2) IIF on SSS, IgA 5 (7.4) Positive immunoblot result 37 (54.4) BP180 9 (13.2) BP230 28 (41.2) BP230 doubtful 5 (7.4) Positive ELISA result 41 (60.3) NC16A, n (%); mean titer u/mL 21 (30.9); 44.6 (SD 31.4; R 11-146) BP230, n (%); mean titer u/mL 30 (46.9); 39.7 (SD 29.5; R 11-122) Eosinophilia in peripheral blood (n=57) 26 (44.8) Mean titer, 10E9/L 1.02 (SD 0.61; R 0.4-2.4) DIF, direct immunofluorescence microscopy; IIF, indirect immunofluorescence microscopy; MO, monkey oesophagus; SSS, salt split skin; LAD-1, linear IgA disease-1; ELISA, enzyme-linked immunosorbent assay; NC16A, noncollagenous 16a; SD, standard deviation; R, range. * Percentages were calculated after exclusion of cases of which data was unknown.

70
Immunoblot and ELISA showed that autoantibodies were predominantly directed against BP230. BP230 reactivity correlated with negative DIF (p=0.019). Conversely, BP180 reactivity correlated with positive DIF (p=0.048). In cases with only BP230 reactivity and no BP180 autoantibodies, a stronger association was seen (p=0.001). In 16 cases ELISA titers of IgG against NC16A and BP230 were repeated during follow-up, and changes corresponded to clinical symptoms in seven, and did not corresponded in nine cases. Treatment response Treatment strategy could vary for individual patients, due to ineffectiveness of prescribed topical or systemic therapies prior to diagnosis, or individual patient characteristics and comorbidities. Treatment response to initial and second prescribed therapies are displayed in table 3. Topical corticosteroids were often prescribed awaiting diagnostic test results. Twenty-two patients reported side effects during the complete follow-up period. Side effects were experienced by 47% of the patients treated with methotrexate (n=32), and treatment needed to be discontinued in 28%. Azathioprine (n=10) and dapsone (n=6) gave side effects in 50% of the cases. Disease course Twelve of 69 cases (17%) developed blisters during follow-up, after a mean disease duration of 41.4 months (SD 65.9; R 5-242). Four patients were in remission off therapy at the time blisters formed, and six patients in remission on minimal systemic therapy. Two patients developed blisters during initially prescribed whole body application of superpotent topical corticosteroids. The mean follow-up time of cases with blister formation was significantly longer (41.4 months (SD 58.2; R 2-218)) compared to patients without blister formation (17.7 months (SD 31.7; R 0-172; p=0.008)).

71
Tabl
e 3
- par
t 1. T
reat
men
t res
pons
e on
firs
t and
sec
ond
pres
crib
ed th
erap
ies
in lo
caliz
eda n
onbu
llous
pem
phig
oid
(n=6
).
Resp
onse
un
know
n N
o re
spon
se
DC
time
till
DC,
wee
ks
Rem
issio
n,
eith
er
PR/C
R PR
time
till
PR,
wee
ks
CR
time
till
CR,
wee
ks
rela
pse
time
till
rela
pse,
w
eeks
Firs
t (n=
6) a
nd se
cond
(n=2
) th
erap
ies
n n
n (%
)*
n (%
)*
mea
n
(SD;
R)
n (%
)*
n (%
)*
mea
n (S
D;R)
n
(%)*
m
ean
(SD;
R)
n (%
)**
mea
n
(SD;
R)
Lesio
nal c
lobe
taso
l cre
am
2 -
2 (1
00)
- -
- -
- -
- -
-
Doxy
cylin
e 2
- 1
(50)
-
- 1
(50)
-
- 1
off (
50)
5.0
- -
Who
le b
ody
appl
icat
ion
of
supe
rpot
ent t
opic
al
cort
icos
tero
ids
1 1
- -
- -
- -
- -
- -
Tria
mci
nolo
n le
sion
al
1 -
- -
- 1
(100
) -
- 1
on (1
00)
5.0
- -
Met
hotr
exat
eb 1
- -
1(10
0)
11.0
-
- -
- -
- -
Azat
hiop
rinec
1 -
1 (1
00)
- -
- -
- -
- -
-
DC, d
isea
se c
ontr
ol; P
R, p
artia
l rem
issio
n; C
R, c
ompl
ete
rem
issio
n; o
n/of
f, on
min
imal
/off
ther
apy,
as
defin
ed b
y in
tern
atio
nal c
onse
nsus
1 ; SD
, sta
ndar
d de
viat
ion;
R, r
ange
. * P
erce
ntag
es w
ere
calc
ulat
ed w
ithou
t tak
ing
unkn
own
resp
onse
s in
to a
ccou
nt. *
* Pe
rcen
tage
s of
pat
ient
s re
laps
ing
wer
e ca
lcul
ated
ove
r the
num
ber o
f cas
es th
at a
chie
ved
DC, P
R or
CR.
a. L
ocal
ized
dise
ase
was
def
ined
as
the
pres
ence
of l
ocal
ized
lesio
ns in
volv
ing
one
body
sit
e, c
onfo
rm th
e Eu
rope
an c
onse
nsus
on
the
man
agem
ent o
f bul
lous
pem
phig
oid
2015
15; b
. Thi
s pa
tient
had
pso
riasi
s, m
etho
trex
ate
was
cho
sen
as
trea
tmen
t for
bot
h sk
in d
isea
ses;
c. T
his
patie
nt u
sed
low
dos
e az
athi
oprin
e fo
r Cro
hn’s
dis
ease
, the
dos
e w
as h
eigh
tene
d w
hen
pem
phig
oid
was
di
agno
sed.

72
Tabl
e 3
– pa
rt 2
. Tre
atm
ent r
espo
nse
on fi
rst a
nd s
econ
d pr
escr
ibed
ther
apie
s in
gen
eral
ized
non
bullo
us p
emph
igoi
d (n
=61)
.*
Resp
onse
un
know
n N
o re
spon
se
DC
time
till D
C,
wee
ks
Rem
issio
nei
ther
PR
/CR
PR
time
till P
R,
wee
ks
CR
time
till
CR, w
eeks
re
laps
e
time
till
rela
pse,
w
eeks
Fi
rst (
n=61
) and
seco
nd
(n=4
2) th
erap
ies
n n
n (%
)**
n (%
)**
mea
n
(SD;
R)
n (%
)**
n (%
)**
mea
n
(SD;
R)
n (%
)**
mea
n
(SD;
R)
n (%
)***
m
ean
(SD;
R)
W
hole
bod
y ap
plic
atio
n of
sup
erpo
tent
topi
cal
cort
icos
tero
ids
41
2 11
(28)
18
(46)
7.
4 (8
; 2-3
7)
10 (2
6)
4 on
(10)
; 1
off (
2)
17.8
(7
; 9-2
8)
1 on
(3);
4
off(1
0)
17.0
(1
1; 5
-31
) 12
(43)
41
.8
(80;
2-2
75)
Met
hotr
exat
e (+
sho
rt-
term
pre
dnis
olon
e in
3)
15
1 3
(21)
5
(36)
11
.8
(11;
3-2
9)
6 (4
3)
4 on
(29)
; 1
off (
7)
38.8
(3
2; 8
-87)
1
on (7
) 19
.0
5 (4
6)
94.6
(1
00; 8
-254
)
Pred
niso
lone
13
1
3 (2
5)
7 (5
8)
4.3
(6; 1
-18)
2
(17)
1
on (8
) 17
.0
1 on
(8)
12.0
9
(100
) 9.
9 (5
; 1-2
0)
Lesi
onal
clo
beta
sol
crea
m
10
1 4
(44)
1
(11)
7.
0 4
(44)
2
on (2
2);
1 of
f (11
) 8.
7 (8
; 1-1
7)
1 of
f(11)
20
.0
1 (2
0)
9.0
Dox
ycyl
ine
(with
or
with
out n
icot
inam
ide)
5
- 5
(100
) -
- -
- -
- -
- -
Pred
niso
lone
+
doxy
cycl
ine
3 -
1 (3
3)
2 (6
7)
1.0
(0; 1
-1)
- -
- -
- 1
(50)
5.
0
Who
le b
ody
appl
icat
ion
of s
uper
pote
nt to
pica
l co
rtic
oste
roid
s +
do
xycy
clin
e
2 -
1 (5
0)
1 (5
0)
4.0
- -
- -
- 1
(100
) 3.
0
Pred
niso
lone
+
azat
hiop
rine
3 -
1 (3
3)
2 (6
7)
3.0
(2; 2
-5)
- -
- -
- 1
(50)
14
.0
Dap
son
3 1
1 (5
0)
1 (5
0)
8.0
- -
- -
- 1
(100
) 1.
0
Oth
er th
erap
ies a
8 -
4 (5
0)
3 (3
8)
3.0
(2; 2
-5)
1 (1
3)
1 of
f (13
) 2.
0 -
- -
-
DC,
dis
ease
con
trol
; PR,
par
tial r
emis
sion
; CR,
com
plet
e re
mis
sion;
on/
off,
on m
inim
al/o
ff th
erap
y, a
s de
fined
by
inte
rnat
iona
l con
sens
us1 ; S
D, s
tand
ard
devi
atio
n;
R, ra
nge.
* T
wo
case
s di
d no
t rec
eive
ther
apy,
one
pat
ient
had
min
imal
com
plai
nts
and
was
lost
to fo
llow
-up,
and
one
firs
t sto
pped
sus
pect
ed re
late
d m
edic
atio
n an
d di
ed s
hort
ly a
fter
. **
Perc
enta
ges
wer
e ca
lcul
ated
with
out t
akin
g un
know
n re
spon
ses
into
acc
ount
. ***
Per
cent
ages
of p
atie
nts
rela
psin
g w
ere
calc
ulat
ed
over
the
num
ber o
f cas
es th
at a
chie
ved
DC,
PR
or C
R. a
. Mom
etas
on n
=3 (l
esio
nal,
+ ta
crol
imus
in 2
), Tr
iam
cino
lon
n=3
(lesi
onal
), te
rbin
afin
e n=
1 (s
yste
mic
), ta
crol
imus
n=1
(les
iona
l, to
pica
l)

73
Blister development during follow-up was associated with positive IIF on MO/SSS (p=0.014), and positive BP180 immunoblot (p=0.032). Immunoserology tests and DIF were repeated in three of 12 cases with blister formation. Increased autoantibody titers against both NC16A and BP230 were detected by ELISA in two of three cases. DIF was already positive at diagnosis in two cases; in the third case DIF turned out positive after blisters occurred. Nevertheless, the alteration from negative to positive DIF during follow-up was also observed in four cases without blister development. Mortality rates Twenty-five patients (36%) died during follow-up, after mean disease duration of 51.2 months (SD40.6; R16-153). The mean time between diagnosis and death was 24.1 months (SD26.3; R0-127). Causes of death were lung cancer (n=1), sepsis after a surgical procedure (n=2), heart failure (n=2), and most often unknown (n=20). Ten patients were lost to follow-up within the 1-year follow-up period, and four additional patients within the 3-year follow-up period, mainly due to referral to a peripheral hospital after the diagnosis was made. The Kaplan-Meier estimates of 1-, 2-, and 3-year all-cause mortality in nonbullous pemphigoid were 14%, 34%, and 46%. Univariate Cox Regression analysis showed a significant effect of age on the 3-year survival (HR 1.04; p=0.028). No other factors significantly influenced the 3-year mortality risk in our population. The SMR per age group is displayed in table 4, showing an 8.6-fold increased all-cause mortality risk in the overall nonbullous pemphigoid population.
Table 4. Standardized mortality ratio's (SMR) in nonbullous pemphigoid
Age groups Total,
n Mortality,
n
Expected 1-year mortality*
Observed 1-year mortality SMR 95% CI
30-59 7 0 0.0018 0.0000 0.0 0.0 - 2.1 60-69 10 1 0.0089 0.1000 11.2 9.1 - 13.7 70-79 19 2 0.0243 0.1053 4.3 3.5 - 5.2 80-89 26 5 0.0806 0.1923 2.4 2.1 - 2.7 90 or older 7 0 0.2643 0.0000 0.0 0.0 - 0.1 Total group (aged ≥30) 69 8 0.0134 0.1159 8.6 7.1 - 10.3 SMR, standardized mortality ratio; CI, confidence interval. * Expected deaths in the Dutch population are based on population wide data of the year 2017.

74
Discussion
Patients with nonbullous pemphigoid endured symptoms for an average duration of 29 months before the correct diagnosis was made. Our study confirmed that histopathological findings in nonbullous pemphigoid are nonspecific, and DIF and IIF should be performed to establish the diagnosis of nonbullous pemphigoid. Methotrexate was most successful in achieving remission, though side effects were reported by almost half of the patients. Of importance, an increased all-cause mortality risk was demonstrated, indicating that a lack of blisters is not equivalent to a mild prognosis. We found a considerable longer diagnostic delay in nonbullous pemphigoid, compared to BP (29 vs. 6 months).18 Dermatologists should perform DIF and IIF on SSS even in the absence of blisters when considering pemphigoid. The low-hanging fruit of unrecognized nonbullous pemphigoid can easily be harvested by the aforementioned tests in elderly patients with refractory chronic pruritus. The diagnostic value of routine histopathology for the diagnosis of pemphigoid is poor. Eosinophilic spongiosis and a subepidermal split are considered histopathological hallmarks of BP.1 In fact, these findings are less typical than implied, as a recent study could only confirm eosinophilic spongiosis in 50% and a subepidermal split in 54% of BP cases.19 We observed that only in 6% of the nonbullous pemphigoid cases eosinophils infiltrated the epidermis. Furthermore, eosinophils were found in a perivascular infiltrate (69%), and in the peripheral blood (45%). Eosinophils are hypothesized to mediate blister formation through secretion of toxic granule proteins.20,21 In nonbullous pemphigoid eosinophils may be activated and attracted towards the skin, but may be unable to infiltrate the epidermis and induce blistering. A notable observation is the predominant reactivity against BP230 in nonbullous pemphigoid, correlating with a negative DIF result, also described by previous studies.8,22,23 It is suggested that the intracellular localization of BP230 might hinder binding of autoantibodies in skin, resulting in negative staining by DIF.24 In contradiction, we found ten nonbullous pemphigoid cases with positive DIF, and only circulating BP230 autoantibodies. Meijer et al. previously showed that autoantibodies against BP180 NC16A are more often present in BP compared to nonbullous pemphigoid, and serum titers appear higher.7 The pathogenicity of BP180 autoantibodies was repeatedly confirmed in vitro and in vivo.7,25 In contrast, BP230 autoantibodies failed to spontaneously induce blisters in several animal

75
studies.25,26 Several studies suggest that symptoms and binding capacity of BP230 autoantibodies depend on coinciding intracellular epitope exposure, for instance by ultraviolet irradiation, epithelial injury, or the transient presence of BP180 autoantibodies.27,28 Recently, BP230 autoantibodies were found to bind in the skin and induce blisters in scurfy mice lacking regulatory T cells.29 Based on these findings we suggest that loss of regulatory T cell function, seen in the aging process, might also influence the pathogenic ability of circulating BP230 antibodies. Other studies linked epitope recognition to BP phenotype, with less inflammation when antibodies recognized the middomain of BP180, or the C-terminal domain of BP230 in cases with BP230 antibodies only.24,30 Future studies are needed to illuminate the pathophysiology of nonbullous pemphigoid. Interestingly, five patients with initial negative DIF results turned positive when DIF was repeated. This demonstrates that the used minimal diagnostic criteria truly support early diagnosis in pemphigoid.7 The changed DIF result coincided with blister development in one case, and in general no trend of altered antigen recognition, or relation with biopsy site was observed in these patients. Our study provides data concerning treatment responses in nonbullous pemphigoid, with only limited data available on the treatment of localized cases. In generalized cases, the highest effectiveness was seen of methotrexate, followed by lesional clobetasol cream and whole body application of superpotent topical corticosteroids. Caution is advised for treatment with methotrexate in elderly patients, as many side effects were reported. Prednisolone often led to DC (58%), though all patients relapsed over time, suggesting it is useful for short-term disease control only. Lesional corticosteroids were ineffective in most cases, however, remission was still seen in 28%. Williams et al. showed non-inferiority of a treatment strategy starting doxycycline over prednisolone in BP.31,32 Our data showed that doxycycline was not effective in the majority (86%) of nonbullous pemphigoid patients. Prognostic data of our study showed that 36% of the study population died after an average disease duration of 51.2 months. Compared to BP we found a lower 1-year all-cause mortality rate in nonbullous pemphigoid, and similar to higher 2- and 3-year all-cause mortality rates.5,6,33 Moreover, an overall SMR of 8.6 was found in nonbullous pemphigoid compared to reported SMR of 3.4, 3.6 and 6.6 in BP.5,6,33 Our mortality data might be influenced by the low sample size, and limited follow-up data of cases that were censored in the Kaplan-Meier analysis.

76
Furthermore, it can be hypothesized that the long delay in diagnosis, and therefore prolonged disease exposure without adequate treatment, might influence the prognosis. A limitation of this study was the retrospective design. Consequently, disease severity measurements such as the BPDAI and autoantibody titers during follow-up were not available. Moreover, the cause of death was unknown to the authors in 20 of 25 deceased cases. A selection bias might affected epidemiology, treatment and prognostic results, since patients visiting an academic hospital are more likely to have severe complaints. Furthermore, our cohort misses patients residing in nursing homes who are not be able to visit a hospital, which could explaining the low co-occurrence of neurodegenerative diseases in our cohort.3,4 Another limitation was the significant shorter follow-up in cases without blister formation, not allowing us to draw hard conclusions on the number of patients with late blister development. This study brought insight in unrevealed disease aspects of nonbullous pemphigoid. Most important, pathologists and dermatologists should be aware that nonbullous pemphigoid cannot be excluded by histopathology and performance of DIF and IIF are required for diagnosis. Once the diagnosis is established, the best therapeutic effect was seen with methotrexate. The mortality rates in nonbullous pemphigoid are increased, indicating that a lack of blisters is not equivalent to having a better prognosis.

77
References
1. Schmidt E, Zillikens D. Pemphigoid diseases. Lancet (London, England). 2013;381(9863):320-332. doi:10.1016/S0140-6736(12)61140-4
2. Hammers CM, Stanley JR. Mechanisms of Disease: Pemphigus and Bullous Pemphigoid. Annu Rev Pathol. 2016;11:175-197. doi:10.1146/annurev-pathol-012615-044313
3. Chen YJ, Wu CY, Lin MW, et al. Comorbidity profiles among patients with bullous pemphigoid: A nationwide population-based study. Br J Dermatol. 2011;165(3):593-599. doi:10.1111/j.1365-2133.2011.10386.x
4. Försti AK, Jokelainen J, Ansakorpi H, et al. Psychiatric and neurological disorders are associated with bullous pemphigoid - A nationwide Finnish Care Register study. Sci Rep. 2016;6(September):1-6. doi:10.1038/srep37125
5. Kalinska-Bienias A, Lukowska-Smorawska K, Jagielski P, Kowalewski C, Wozniak K. Mortality in bullous pemphigoid and prognostic factors in 1st and 3rd year of follow-up in specialized centre in Poland. Arch Dermatol Res. 2017 Nov;309(9):709-719. doi:10.1007/s00403-017-1772-x
6. Kridin K, Bergman R. Mortality in Patients with Bullous Pemphigoid: A Retrospective Cohort Study, Systematic Review and Meta-analysis. Acta Derm Venereol. 2019 Jan 1;99(1):72-77. doi:10.2340/00015555-2930
7. Meijer JM, Diercks GFH, de Lang EWG, Pas HH, Jonkman MF. Assessment of Diagnostic Strategy for Early Recognition of Bullous and Nonbullous Variants of Pemphigoid. JAMA dermatology. 2019 Feb 1;155(2):158-165. doi:10.1001/jamadermatol.2018.4390
8. Lamberts A, Meijer JM, Jonkman MF. Nonbullous pemphigoid: A systematic review. J Am Acad Dermatol. 2018;78(5). doi:10.1016/j.jaad.2017.10.035
9. Bakker C V, Terra JB, Pas HH, Jonkman MF. Bullous pemphigoid as pruritus in the elderly: a common presentation. JAMA dermatology. 2013;149(8):950-953. doi:10.1001/jamadermatol.2013.756
10. Lamb PM, Abell E, Tharp M, Frye R, Deng JS. Prodromal bullous pemphigoid. Int J Dermatol. 2006;45(3):209-214. doi:10.1111/j.1365-4632.2004.02457.x
11. Cozzani E, Gasparini G, Burlando M, Drago F, Parodi A. Atypical presentations of bullous pemphigoid: Clinical and immunopathological aspects. Autoimmun Rev. 2015;14(5):438-445. doi:10.1016/j.autrev.2015.01.006
12. Alonso-Llamazares J, Rogers RS 3rd, Oursler JR, Calobrisi SD. Bullous pemphigoid presenting as generalized pruritus: observations in six patients. Int J Dermatol. 1998;37(7):508-514.
13. Zhang Y, Luo Y, Han Y, Tian R, Li W, Yao X. Non-bullous lesions as the first manifestation of bullous pemphigoid: A retrospective analysis of 181 cases. J Dermatol. 2017 Jul;44(7):742-746 . doi:10.1111/1346-8138.13782
14. Sun C, Chang B, Gu H. Non-bullous lesions as the first manifestation of bullous pemphigoid: a retrospective analysis of 24 cases. J Dermatolog Treat. 2009;20(4):233-237. doi:10.1080/09546630802683876
15. Feliciani C, Joly P, Jonkman MF, et al. Management of bullous pemphigoid: the European Dermatology Forum consensus in collaboration with the European Academy of Dermatology and Venereology. Br J Dermatol. 2015;172(4):867-877. doi:10.1111/bjd.13717
16. Murrell DF, Daniel BS, Joly P, et al. Definitions and outcome measures for bullous pemphigoid: recommendations by an international panel of experts. J Am Acad Dermatol. 2012;66(3):479-485. doi:10.1016/j.jaad.2011.06.032
17. Altman DG, Machin D, Bryant TN, Gardner MJ. Statistics with Confidence. 2nd edition. BMJ Books; 2000. doi:10.4135/9781446218525
18. Della Torre R, Combescure C, Cortés B, et al. Clinical presentation and diagnostic delay in bullous pemphigoid: A prospective nationwide cohort. Br J Dermatol. 2012;167(5):1111-1117. doi:10.1111/j.1365-2133.2012.11108.x

78
19. Hodge B, Roach J, Reserva JL, et al. The Spectrum of Histopathologic Findings in Pemphigoid: Avoiding Diagnostic Pitfalls. J Cutan Pathol. 2018 Nov;45(11):831-838. doi:10.1111/cup.13343
20. de Graauw E, Sitaru C, Horn M, et al. Evidence for a role of eosinophils in blister formation in bullous pemphigoid. Allergy Eur J Allergy Clin Immunol. 2017;72(7):1105-1113. doi:10.1111/all.13131
21. Messingham KN, Holahan HM, Frydman AS, Fullenkamp C, Srikantha R, Fairley JA. Human Eosinophils Express the High Affinity IgE Receptor, FcεRI, in Bullous Pemphigoid. PLoS One. 2014;9(9):e107725. doi:10.1371/journal.pone.0107725
22. Gammon WR, Briggaman RA, Inman AO 3rd, Queen LL, Wheeler CE. Differentiating anti-lamina lucida and anti-sublamina densa anti-BMZ antibodies by indirect immunofluorescence on 1.0 M sodium chloride-separated skin. J Invest Dermatol. 1984;82(2):139-144.
23. Sardy M, Kostaki D, Varga R, et al. Comparative study of direct and indirect immunofluorescence and of bullous pemphigoid 180 and 230 enzyme-linked immunosorbent assays for diagnosis of bullous pemphigoid. J Am Acad Dermatol. 2013;69(5):748-753. doi:https://dx.doi.org/10.1016/j.jaad.2013.07.009
24. Hayakawa T, Teye K, Hachiya T, et al. Clinical and immunological profiles of anti-BP230-type bullous pemphigoid: Restriction of epitopes to the C-terminal domain of BP230, shown by novel ELISAs of BP230-domain specific recombinant proteins. Eur J Dermatol. 2016;26(2):155-163. doi:10.1684/ejd.2015.2719
25. Bieber K, Sun S, Ishii N, et al. Animal models for autoimmune bullous dermatoses. Exp Dermatol. 2010;19(1):2-11. doi:10.1111/j.1600-0625.2009.00948.x
26. Feldrihan V, Licarete E, Florea F, et al. IgG antibodies against immunodominant C-terminal epitopes of BP230 do not induce skin blistering in mice. Hum Immunol. 2014;75(4):354-363. doi:10.1016/j.humimm.2014.01.005
27. Pas HH, de Jong MC, Jonkman MF, Heeres K, Slijper-Pal IJ, van der Meer JB. Bullous pemphigoid: serum antibody titre and antigen specificity. Exp Dermatol. 1995;4(6):372-376.
28. Hall RP 3rd, Murray JC, McCord MM, Rico MJ, Streilein RD. Rabbits immunized with a peptide encoded for by the 230-kD bullous pemphigoid antigen cDNA develop an enhanced inflammatory response to UVB irradiation: a potential animal model for bullous pemphigoid. J Invest Dermatol. 1993;101(1):9-14.
29. Haeberle S, Wei X, Bieber K, et al. Regulatory T-cell deficiency leads to pathogenic bullous pemphigoid antigen 230 autoantibody and autoimmune bullous disease. J Allergy Clin Immunol. 2018 Dec;142(6):1831-1842.e7. doi:10.1016/j.jaci.2018.04.006
30. Izumi K, Nishie W, Mai Y, et al. Autoantibody Profile Differentiates between Inflammatory and Noninflammatory Bullous Pemphigoid. J Invest Dermatol. 2016;136(11):2201-2210. doi:10.1016/j.jid.2016.06.622
31. Sapadin AN, Fleischmajer R. Tetracyclines: nonantibiotic properties and their clinical implications. J Am Acad Dermatol. 2006;54(2):258-265. doi:10.1016/j.jaad.2005.10.004
32. Williams HC, Wojnarowska F, Kirtschig G, et al. Doxycycline versus prednisolone as an initial treatment strategy for bullous pemphigoid: a pragmatic, non-inferiority, randomised controlled trial. Lancet (London, England). 2017;389(10079):1630-1638. doi:10.1016/S0140-6736(17)30560-3
33. Joly P, Baricault S, Sparsa A, et al. Incidence and mortality of bullous pemphigoid in France. J Invest Dermatol. 2012;132(8):1998-2004. doi:10.1038/jid.2012.35

79

80
4A

81
CHAPTER 4A Reply to: “Pruritus with pemphigoid autoantibodies is the tip of an iceberg” Aniek Lamberts*1, Joost M. Meijer*1, Gilles F.H. Diercks1,2, Hendri H. Pas1, Barbara Horváth1
* Authors contributed equally Center for Blistering Diseases, Department of Dermatology1, and the Department of Pathology2, University Medical Center Groningen, University of Groningen, Groningen, The Netherlands In reply to: Byth LA. Pruritus with pemphigoid autoantibodies is the tip of an iceberg. Journal of the American Academy of Dermatology, 2019 Nov;81(5):e151. Published in the Journal of the American Academy of Dermatology, 2019 Nov;81(5): e153-e154.

82
To the editor: We appreciate the response of dr. Byth to our article1, and agree that nonbullous pemphigoid (NBP) deserves more attention in the clinical practice guidelines for chronic pruritus, as a rather unknown cause of chronic pruritus in elderly patients.2,3 We politely disagree with dr. Byth that NBP patients with pruritus without rash should be referred to as ‘pruritus with pemphigoid autoantibodies (PPA)’. Firstly, we advocate for NBP as umbrella term for all pemphigoid variants without blisters, and believe that introducing the term PPA is needless and confusing. Secondly, PPA does not accurately describe the intended population of NBP patients without primary skin lesions, since all patients with bullous pemphigoid (BP) and NBP experience pruritus and have pemphigoid autoantibodies.
Dr. Byth questioned whether testing for pemphigoid autoantibodies in elderly patients with pruritus would be cost-effective. In our opinion, the burden of disease in these patients with chronic pruritus is too high to deny them a possible diagnosis of NBP and adequate therapy. Therefore, we included pemphigoid in the standard diagnostic workup of elderly patients with chronic pruritus. We like to emphasize that caution is needed if only ELISA is used as screening method, as these have frequent false positive results. The recently published article of Wang et al.4 reports positive BP180 and BP230 autoantibodies by ELISA in 208 patients with a negative skin biopsy for direct immunofluorescence (DIF). Various lesion morphologies were described in these patients, most commonly dermatitis, but also essential pruritus. The authors conclude that low positive levels of BP180 and BP230 autoantibodies should not be overinterpreted as evidence for BP in the setting of a negative DIF, and still consider DIF positivity to be the golden standard for diagnosis of NBP.
Recent work of our group assessed this clinical dilemma with a diagnostic accuracy study in 1125 patients suspected of NBP or BP, providing minimal diagnostic criteria.5 IIF on salt-split skin (SSS) showed a positive predictive value for diagnosis of pemphigoid of 99.6%, and therefore plays an essential role for the serological diagnosis of pemphigoid. The BP180 NC16A ELISA showed frequent false-positivity (11,3%) and is not recommended for initial diagnosis, but only for disease monitoring in confirmed patients. The established minimal diagnostic criteria consists of a 2 out of 3 rule: (1) pruritus and/or predominant cutaneous blisters, (2) linear (n-serrated) IgG and/or C3c deposits by DIF on a skin biopsy specimen, and (3) positive epidermal side staining of IgG by IIF SSS on a serum

83
sample. Thereby, extending the spectrum of pemphigoid with the unrecognized nonbullous variant, and allowing a diagnosis with negative DIF.
Our article complements the study of Wang et al., demonstrating the use of the minimal diagnostic criteria in the broad spectrum of NBP. In conclusion, not all patients with ‘pruritus with pemphigoid autoantibodies’ with ELISA positivity have pemphigoid, and IIF SSS positivity is essential for diagnosis in DIF negative cases. References 1. Lamberts A, Meijer JM, Pas HH, Diercks GFH, Horvath B, Jonkman MF. Nonbullous pemphigoid:
insights in clinical and diagnostic findings, treatment responses and prognosis. J Am Acad Dermatol. 2019 Aug;81(2):355-363. doi:10.1016/j.jaad.2019.04.029
2. Weisshaar E, Szepietowski JC, Dalgard FJ, et al. European S2k Guideline on Chronic Pruritus. Acta Derm Venereol. 2019;99(5):469-506. doi:10.2340/00015555-3164
3. Millington GWM, Collins A, Lovell CR, et al. British Association of Dermatologists’ guidelines for the investigation and management of generalized pruritus in adults without an underlying dermatosis, 2018. Br J Dermatol. 2018;178(1):34-60. doi:10.1111/bjd.16117
4. Wang M, Lehman JS, Camilleri MJ, Drage LA, Wieland CN. Circulating bullous pemphigoid autoantibodies in the setting of negative direct immunofluorescence findings for bullous pemphigoid: A single-center retrospective review. J Am Acad Dermatol. 2019;81(2):472-479. doi:10.1016/j.jaad.2019.03.062
5. Meijer JM, Diercks GFH, de Lang EWG, Pas HH, Jonkman MF. Assessment of Diagnostic Strategy for Early Recognition of Bullous and Nonbullous Variants of Pemphigoid. JAMA dermatology. 2019 Feb 1;155(2):158-165. doi:10.1001/jamadermatol.2018.4390

84
5

85
CHAPTER 5 Prevalence of pemphigoid as a potentially unrecognized cause of pruritus in nursing home residents Aniek Lamberts*1, Joost M. Meijer*1, Hendrika J. Luijendijk2, Gilles F.H. Diercks1, Hendri H. Pas1, Sytse U. Zuidema2, Marcel F. Jonkman1† * Authors contributed equally. 1 University of Groningen, University Medical Center Groningen, Department of Dermatology, Center for Blistering Diseases, Groningen, The Netherlands 2 University of Groningen, University Medical Center Groningen, Department of General Practice and Elderly Care Medicine, Groningen, The Netherlands
Published in the Journal of the American Medical Association Dermatology. 2019 Nov 6. doi: 10.1001/jamadermatol.2019.3308. [Epub ahead of print]

86
Bullous pemphigoid may commonly present as pruritus in elderly patients.1 Approximately one in five patients with pemphigoid has nonbullous skin manifestations that resemble other pruritic skin diseases, termed nonbullous pemphigoid.1,2 According to recently established minimal diagnostic criteria the diagnosis of bullous and nonbullous pemphigoid can be based on either a skin biopsy, or a serum sample.2 The prevalence of bullous pemphigoid strongly increases with age and the disease is associated with neurodegenerative diseases, such as dementia and Parkinson’s disease.3,4 Our aim was to assess the prevalence of pemphigoid in a high-risk population of nursing home residents, with nonbullous pemphigoid a potential unrecognized cause of pruritus. Methods This prospective cross-sectional study was conducted in seven nursing homes affiliated with the University Network for Elderly Care of the UMCG in the Netherlands between July 2016 and December 2017. Participants aged 65 years or older were eligible when a routine vena punction was scheduled and one extra blood sample could be withdrawn. Exclusion criteria were systemic immunosuppressive therapy or life expectancy of less than four weeks. Written informed consent was required of cognitively capable participants, or legal representatives of cognitively impaired participants. Pruritus was assessed by skin examination using the Bullous Pemphigoid Disease Area Index (BPDAI) excoriation score. Cognitively competent participants rated pruritus from 0 to 10, and for cognitively incompetent participants nursing staff was interviewed about signs of pruritus. Diagnostic criteria for pemphigoid were: 1) pruritus and/or skin blisters, and 2) positive epidermal side staining of IgG by indirect immunofluorescence on salt-split skin (IIF SSS).2 A skin biopsy for direct immunofluorescence (DIF) was only performed on voluntary basis due to ethical considerations. Results We enrolled 125 nursing home residents (figure 1) on average 84.1 years [SD 6.9], with a history of neurodegenerative disease in 75%. Pruritus was present in 59 of 125 participants (47%) and of chronic duration (>6 weeks) in 48 participants (81%). Pemphigoid was diagnosed in seven of 125 participants, yielding an overall prevalence of 6%. Table 1 describes the clinical and serological findings. Three

87
participants with bullous pemphigoid had already been diagnosed. Nonbullous pemphigoid was unrecognized and newly diagnosed in four participants, all of whom had a history of chronic pruritus. Nonbullous skin manifestations consisted of erythematous papules/nodules, urticarial plaques and excoriations, mainly distributed on the back and extremities (figure 2). Non-specific serological findings in controls were single detection of IgA by IIF SSS (two cases), and low titers of IgG autoantibodies by BP180 NC16A (ten cases) or BP230 ELISA (three cases). Discussion We observed a prevalence of pemphigoid of 6% among nursing home residents. More than half of the cases did not show blisters and had not yet been diagnosed with pemphigoid.
Figure 1. Flowchart of the study.

88
Only two previous studies assessed bullous pemphigoid in nursing home residents, and reported a prevalence of 1% by survey5, and an annual incidence of 5% by skin biopsy.6
Our study confirms that the prevalence of pemphigoid is substantially higher in nursing home residents than in the general population, which was estimated at 0.026% in total and 0.3% in persons aged ≥85 years (data provided by the authors).3 Possibly this finding might be explained by neurodegenerative diseases preceding development of pemphigoid as a result of cross-reactivity between neuronal and epithelial isoforms of the pemphigoid antigens.4 While the confirmative IIF SSS test is highly specific, the sensitivity is approximately 80% and a skin biopsy for DIF required to exclude pemphigoid in negative cases.2
The striking finding of this study that nonbullous pemphigoid was more frequent than bullous pemphigoid merits attention from clinicians. We therefore recommend to include pemphigoid in the diagnostic work-up of chronic pruritus in elderly patients. References 1 Lamberts A, Meijer JM, Diercks GFH, et al. Nonbullous pemphigoid: Insights in clinical and
diagnostic findings, treatment responses, and prognosis. J Am Acad Dermatol 2019; 81(2):355-363. doi:10.1016/j.jaad.2019.04.029
2 Meijer JM, Diercks GFH, de Lang EWG, et al. Assessment of Diagnostic Strategy for Early Recognition of Bullous and Nonbullous Variants of Pemphigoid. JAMA Dermatol. 2019;155(2):158-165 doi:10.1001/jamadermatol.2018.4390.
3 Hubner F, Recke A, Zillikens D, et al. Prevalence and Age Distribution of Pemphigus and Pemphigoid Diseases in Germany. J. Invest. Dermatol. 2016; 136:2495–8.
4 Forsti AK, Jokelainen J, Ansakorpi H, et al. Psychiatric and neurological disorders are associated with bullous pemphigoid - a nationwide Finnish Care Register study. Sci Rep 2016; 6:37125.
5 Tseng HW, Lam HC, Ger LP, et al. A survey of dermatological diseases among older male adults of a Veterans Home in Southern Taiwan. Aging Clin Exp Res 2015; 27:227–33.
6 Fernandez-Viadero C, Arce Mateos F, Verduga Velez R, Crespo Santiago D. Blisters in a nursing home: bullous pemphigoid more often than we think? J. Am. Geriatr. Soc. 2004; 52:1405–6.

89
Figure 2. Clinical features of a 89 year old female nursing home resident with nonbullous pemphigoid. A) Excoriated erythematous papules and excoriations on the back. B) Complete remission achieved after 6 weeks of treatment with methotrexate 7.5mg per week monotherapy. C) Excoriated papules and urticarial plaques on the right flank. D) Excoriated erythematous papules and nodules on the left arm.
A B
C D

90
Tabl
e 1.
Cha
ract
eris
tics
of n
ursi
ng h
ome
resi
dent
s di
agno
sed
with
pem
phig
oid
Sex
/ age
(y
ears
),
pem
phig
oid
phen
otyp
e
Clin
ical
find
ings
, ski
n le
sions
and
di
strib
utio
n IIF
SSS
(r
oof
stai
ning
)
IIF M
E An
tigen
by
ELI
SA
BP18
0 N
C16A
, BP
230,
im
mun
oblo
t
DIF*
an
ti-BM
Z Tr
eatm
ent a
nd fo
llow
-up
(dur
atio
n)
F / 8
3,
bullo
us
mild
pru
ritus
, mod
erat
e bl
iste
rs,
eryt
hem
a, e
xcor
iatio
ns a
nd h
yper
-pi
gmen
tatio
n on
abd
omen
, bac
k an
d ex
trem
ities
IgG
3+
IgG
+
BP18
0, B
P230
n.
a.
Lesi
onal
sup
er p
oten
t cor
ticos
tero
ids
Part
ial r
emis
sion
(9 m
onth
s), u
ntil
deat
h du
e to
na
tura
l cau
ses.
M /
81,
bullo
us
seve
re p
rurit
us, g
ener
aliz
ed b
liste
rs,
eros
ions
, ery
them
a, p
apul
es a
nd
urtic
aria
IgG
3+
IgA
1+
IgG
+/-
BP
180
IgG
, C3c
lin
ear
Ora
l cor
ticos
tero
ids,
dox
ycyc
line,
lesi
onal
sup
er
pote
nt c
ortic
oste
roid
s D
eath
due
to n
atur
al c
ause
s sh
ortly
aft
er s
tudy
ex
amin
atio
n
F / 7
3,
bullo
us
mild
pru
ritus
, loc
aliz
ed b
liste
rs o
n le
gs
with
exc
oria
tions
, hyp
er-p
igm
enta
tion
and
hem
atom
a Ig
G 1
+ Ig
G +
BP
180,
BP2
30
IgG
, C3c
n-
serr
ated
Ora
l cor
ticos
tero
ids,
met
hotr
exat
e (1
5 m
g/w
k) a
nd
lesi
onal
sup
er p
oten
t cor
ticos
tero
ids
Com
plet
e re
mis
sion
(18
mon
ths)
M
/ 84
, no
nbul
lous
se
vere
gen
eral
ized
pru
ritus
with
ex
coria
tions
, pap
ules
, ery
them
a Ig
G 1
+ Ig
G +
BP
230
IgA
n-
serr
ated
D
oxyc
yclin
e, w
hole
-bod
y su
per p
oten
t co
rtic
oste
roid
s. P
artia
l rem
issio
n (1
7 m
onth
s)
F / 8
9,
nonb
ullo
us
seve
re p
rurit
us w
ith e
xcor
iatio
ns,
eryt
hem
a, p
apul
es a
nd u
rtic
aria
on
back
and
ext
rem
ities
Ig
G 3
+ Ig
G +
BP
230
nega
tive
Met
hotr
exat
e (7
.5m
g/w
k) le
d to
co
mpl
ete
rem
issi
on (1
1 m
onth
s), f
lare
aft
er
tape
ring
and
com
plet
e re
mis
sion
(9 m
onth
s) a
fter
re
trea
tmen
t
F / 8
1,
nonb
ullo
us
seve
re p
rurit
us w
ith e
xcor
iatio
ns,
eryt
hem
a an
d ur
ticar
ia o
n ba
ck a
nd
extr
emiti
es
IgG
1+
IgG
+
BP23
0 ne
gativ
e
Met
hotr
exat
e (7
.5m
g/w
k) le
d to
com
plet
e re
mis
sion
(7 m
onth
s), f
lare
aft
er ta
perin
g an
d co
mpl
ete
rem
issi
on (1
mon
th) a
fter
retr
eatm
ent,
until
dea
th d
ue to
nat
ural
cau
ses.
F / 8
3,
nonb
ullo
us
mild
pru
ritus
with
ery
them
a, p
apul
es
and
exco
riatio
ns o
n ex
trem
ities
Ig
G 2
+ Ig
G +
BP
230
nega
tive
Topi
cal s
tero
id o
intm
ents
Co
mpl
ete
rem
issi
on (1
8 m
onth
s)
F, fe
mal
e; M
, mal
e; Ig
G, i
mm
unog
lobu
lin G
; IgA
, im
mun
oglo
bulin
e A;
C3c
, com
plem
ent C
3; +
, pos
itive
; -, n
egat
ive;
+/-
, dou
btfu
l pos
itive
; IIF
, ind
irect
im
mun
oflu
ores
cenc
e; S
SS, s
alt-
split
ski
n; M
E, m
onke
y es
opha
gus;
ELI
SA, e
nzym
e lin
ked
imm
unos
orbe
nt a
ssay
; NC1
6A, n
on-c
olla
geno
us 1
6A d
omai
n of
BP1
80;
DIF
, dire
ct im
mun
oflu
ores
cenc
e; B
MZ,
bas
emen
t mem
bran
e zo
ne. *
No
stud
y re
quire
men
t, pe
rfor
med
at v
olun
tary
bas
is

91
Acknowledgements The authors wish to thank the participating care organisations and staff of the SSENIOR Study Group*, and Janny Zuiderveen, Gonnie Meijer, Marije van der Molen and Laura Nijen-Vos (Immunodermatology Laboratory, Department of Dermatology, University Medical Center Groningen, the Netherlands) for technical assistance and performing laboratory tests. *SSENIOR Study Group: University Medical Center Groningen: Alet Leus, Nanda Hommes Patyna Bloemkamp Bolsward: Erica Gerbrandy, Mariëlle Gaster Dignis De Omloop Norg, De Enk Zuidlaren: Carolien van Bruggen Interzorg Anholt Assen: Sifra van Rest, Arenda Krol Dignis Blauwbörgje Groningen: Margreet Schaaf ZINN De Es/De Dilgt Haren: Arnold Bisschop CERTE Department of Primary Care Diagnostics and Trial Coordination Centre

92
6

93
CHAPTER 6 IgE autoantibodies in serum and skin of nonbullous and bullous pemphigoid patients Aniek Lamberts1, Nika Kotnik2, Gilles F.H. Diercks1,3, Joost M. Meijer1, Giovanni Di Zenzo4, Hendri H. Pas1, Marcel F. Jonkman1, Bernhard F. Gibbs2, Ulrike Raap2, Barbara Horváth1
Affiliations 1 Department of Dermatology, University Medical Center Groningen, University of Groningen, Groningen, the Netherlands 2 Department of Experimental Dermatology and Allergology, University of Oldenburg, Oldenburg, Germany
3 Department of Pathology, University Medical Center Groningen, University of Groningen, Groningen, the Netherlands 4 Molecular and Cell Biology Laboratory, Istituto Dermopatico dell'Immacolata, IDI-IRCCS, Rome, Italy Submitted

94
Abstract Background Nonbullous pemphigoid (NBP) is a pemphigoid variant which frequently resembles other pruritic skin diseases. In contrast to bullous pemphigoid (BP), blisters are absent. In BP, previous studies showed that IgE autoantibodies may be involved in its pathogenesis. IgE-activated mast cells, basophils and eosinophils may participate in BP by inducing pruritus and possibly blister formation, although the differential role of IgE in NBP compared to BP has not yet been described. Objective To assess IgE in serum and skin of NBP and BP patients. Methods We examined total IgE and pemphigoid-specific IgE in the serum of 68 NBP and 50 BP patients by enzyme-linked immunosorbent assay (ELISA). Sera of 25 pemphigus patients, and 25 elderly patients with pruritus were included as controls. Skin biopsies of 14 NBP and 14 BP patients with the highest IgE titers to NC16A were stained for IgE by immunofluorescence techniques. Results Total IgE was elevated in 63% of NBP and 60% of BP patients, and in 20% of pemphigus controls, as well as 60% of elderly controls. IgE ELISAs were more frequently positive in BP than in NBP (NC16A 18% vs. 9%, p=0.139; BP230 34% vs. 22%, p=0.149). IgE ELISAs for NC16A and BP230 were positive in 8% and 20% of elderly controls, respectively, while all pemphigus controls were negative. Two of 28 biopsies (7%; 1 NBP, 1 BP) showed linear IgE along the basement membrane zone, while in most biopsies (71% NBP; 86% BP) IgE was bound to dermal cells. Conclusion Since IgE was present in the serum and skin of both NBP and BP patients this supports IgE-dependent mechanisms common to both diseases, such as pruritus. However, it remains to be elucidated whether IgE contributes to blister formation in BP.

95
Introduction Bullous pemphigoid (BP) is the most common autoimmune bullous disease, mainly affecting elderly patients aged over 70.1,2 Most patients present with severe pruritus and skin blisters.1 However, one-in-five patients lack blisters, a subtype termed nonbullous pemphigoid (NBP).3–5 While BP is well-characterized and well-studied, comparatively little is known about NBP. The pathogenesis of BP involves IgG autoantibodies targeting hemidesmosome proteins BP180 and BP230.6,7 BP230 (also BP antigen 1) is a 230kDa intracellular protein.8 BP180 (also BP antigen 2, or type XVII collagen) is a transmembrane protein of 180kDa.8 The extracellular non-collagenous 16A (NC16A) domain of BP180 contains immunodominant epitopes, and anti-NC16A IgG antibodies correlate with disease activity.8–10 Interestingly, IgE autoantibodies targeting pemphigoid antigens were demonstrated in serum and skin of BP patients as well.9 IgE is a key mediator of allergic responses by inducing degranulation of mast cells and basophils through crosslinking of IgEs bound to high-affinity IgE receptors (FcɛRI).11,12 Elevated total IgE levels in BP patients (70%) were first reported in 1974.13 Circulating IgE antibodies to BP180 were observed in 30-77% of BP patients, and correlated with disease activity in several studies.9,14–20 IgE antibodies to BP230, though less extensively studied, were detected in 22-67% of BP patients.17,18,21,22 In the skin, linear IgE deposits were reported along the basement membrane zone (BMZ)23–26, whereas other reports described IgE bound to eosinophils and mast cells in the dermis.20,27 Recently, Ben Mordehai et al. assessed total IgE levels in NBP, and reported significantly higher levels in the serum of NBP compared to BP patients.28 However, pemphigoid-specific IgE levels and skin biopsies were not assessed. Omalizumab, a monoclonal antibody targeting IgE, has been used therapeutically in several BP patients.29,30 A meta-analysis found complete responses in approximately 84% of patients, with decreased pruritus and blister development.30 These results imply an important role for IgE in the disease mechanism of BP, and potentially in blister formation. The aim of this study was to assess whether IgE is implicated in blister formation, by comparing the presence of IgE antibodies in the serum and skin of patients with NBP and BP.

96
Material and Methods Selection of patients and controls Patients diagnosed with pemphigoid at our Center for Blistering Diseases were retrospectively selected. Patients were diagnosed with NBP or BP if two of the following three diagnostic criteria were met; 1) compatible clinical features of pruritus and/or cutaneous blisters, 2) positive linear IgG or C3c staining by direct immunofluorescence (DIF) microscopy, 3) positive IgG staining on the epidermal side of salt-split skin by indirect immunofluorescence microscopy.3 Sixty-eight NBP patients were included, of whom clinical, diagnostic and prognostic features have been published separately.5 Fifty consecutive BP patients diagnosed between 2014 and 2015 were selected retrospectively for comparison. Leftover diagnostic material was used, consisting of serum samples taken for immunoserology and biopsies taken for DIF microscopy, both stored at minus 80 degrees Celsius. Patient characteristics, and data concerning peripheral eosinophilia, and eosinophils in lesional hematoxylin-eosin stained skin biopsies were noted if available. Control sera included 25 consecutive pemphigus patients with high levels of autoantibodies to desmoglein 1 and/or 3, and 25 consecutive elderly patients with pruritus who tested negative for pemphigoid. The study was approved by the local Ethics Committee (University Medical Center Groningen, the Netherlands). Laboratory tests Enzyme-linked immunosorbent assay IgE antibodies against BP180 and BP230 were measured by enzyme-linked immunosorbent assay (ELISA) using NC16A and BP230 coated plates from MBL (Nagoya, Japan). Samples were diluted 1:25 with phosphate buffered saline (PBS) with 0.05% Tween20 and 0.5% bovine serum albumin, and incubated for 1 hour at room temperature. Anti-human IgE-horseradish peroxidase (Acris, Herford, Germany) was diluted 1:250 and used as conjugate with 1 hour incubation time. A substrate was added for 30 minutes after which the enzyme reaction was stopped. Optical density values were measured with a spectrophotometer, set to 450 nm. IgG cross-reactivity was tested by protein G column chromatography, running a patients serum with high titers for anti-NC16A IgE and IgG through a protein G column. The IgG depleted flow-through and IgG rich fraction were both collected. The flow-through showed a complete negative result on the IgG ELISA, and

97
remained equally positive for IgE. Conversely, the IgG rich fraction was positive by IgG ELISA, and completely negative for IgE, showing no cross-reactivity. Previous studies with similar methodologies reported that antigen-specific IgE antibodies in sera of BP and epidermolysis bullosa acquisita could be effectively measured without preadsorption of IgG, and showed no significant difference in OD values between pre- and postadsorption of IgG.19,31 The diagnostic accuracy of the novel IgE ELISA was examined by receiver operating characteristic curve analysis. The area under the curve for the anti-NC16A IgE ELISA was 0.507, and 0.553 for the anti-BP230 IgE ELISA, indicating that they were unsuitable as diagnostic tests for pemphigoid. The ELISA cut-off values were set on the mean plus three times the standard deviation of sera of the pemphigus control population (A450 0.620 for anti-NC16A ELISA; 0.929 for anti-BP230 ELISA). Serum total IgE was measured by Phadia immunoCap technology (cut-off value ≥ 115 kU/L). Immunofluorescent IgE staining of skin biopsies Skin biopsies taken for DIF microscopy of 14 NBP and 14 BP patients with the highest anti-NC16A IgE titers were additionally stained for IgE. According to our standard protocol for DIF microscopy, biopsies were kept in saline overnight at room temperature before storage at minus 80 degrees Celsius. We used polyclonal rabbit anti-human IgE, ɛ-chain specific (Dako, Glostrup, Denmark) diluted 1:200 with PBS containing 1% ovalbumin, and incubated for 30 minutes. Slides were washed for 15 minutes with PBS. In a second step polyclonal donkey anti-rabbit IgG was used with a fluorescein isothiocyanate label (Jackson, Ely, United Kingdom) diluted 1:100 with PBS containing 1% ovalbumin, and incubated for 30 minutes. Biopsies were washed for 15 minutes with PBS, and viewed independently by two authors (AL and GD). Any discrepancies between ratings were discussed. To study IgE staining patterns in a larger sample of control skin, diagnostic biopsies received by our immunodermatology laboratory of patients without an autoimmune blistering disease were additionally stained for IgE during a period of 3 months.

98
Statistical analysis Non-normally distributed data are presented as median with interquartile range (IQR). Correlations between binary data were analyzed by the Chi-square test, or the Fisher’s exact test when required. The Mann-Whitney U test was used for comparing non-normally distributed data. A p-value < 0.05 was considered statistically significant. Statistical analyses were performed using IBM SPSS Statistics 23 software (IBM, Armonk, New York). Results Patient characteristics are described in table 1. The median age of pemphigoid patients and elderly controls corresponded (79 years, IQR=14 vs. 76 years, IQR=10). IgE in serum Total and specific IgE against NC16A and BP230 Total IgE and IgE ELISA results are shown in table 1. Pemphigoid-specific IgE autoantibodies were more commonly detected in BP (NC16A 18%, BP230 34%) than in NBP (NC16A 9%, BP230 22%), however, no significant difference was found (p=0.139 and p=0.149, respectively). Overall, median IgE ELISA titers were significantly higher in BP compared with NBP (p<0.001). In general, elevated total IgE was associated with specific IgE to NC16A (p=0.004) and BP230 (p<0.001). In NBP, elevated total IgE levels were associated with specific IgE to BP230 (p<0.001) but not to NC16A (p=0.339). Conversely, in BP a strong association was found between elevated total IgE and specific IgE to NC16A (p=0.007), but not to BP230 (p=0.058). In elderly controls with pruritus no associations were observed between either elevated total IgE levels or specific IgE to NC16A or BP230 (p=1.000 and p=0.061). Associations between serum IgE and IgG autoantibodies Specific IgE autoantibodies to NC16A did not correlate with IgG autoantibodies to NC16A in serum of NBP and BP patients (p=0.363 and p=0.141). However, when analyzing pemphigoid as one group, we did find an association between specific IgE and IgG autoantibodies to NC16A (p=0.009). Specific IgE antibodies to BP230 were strongly associated with IgG antibodies to BP230 in both NBP and BP patients (p<0.001 and p=0.001), and in pemphigoid patients as one group (p<0.001).

99
Table 1. Patient characteristics and results of IgE ELISA in pemphigoid patients and controls
Study group Control group
NBP n=68
BP n=50
P-value NBP vs. BP
Pemphigus disease, n=25
Elderly subjects with pruritus, n=25
Median age, years (IQR) 79.0 (16) 78.0 (14) 0.794 60.0 (29) 76.0 (10)
Gender, male, n (%) 29 (42) 15 (30) 0.160 17 (68) 12 (48)
Female, n(%) 40 (58) 35 (70) 8 (32) 13 (52)
Eosinophilia, n (%) (> 0.40 109/L) 26 of 58 (45) 10 of 27 (37) 0.499 1 of 13 (8) 6 of 10 (60)
Lesional histopathology biopsy
Eosinophils in dermal infiltrate, n(%)
37 of 54 (69) 18 of 20 (90) 0.123
Eosinophilic spongiosis, n(%) 3 of 54 (6) 6 of 20 (30) 0.014
Diagnostic features
DIF IgG and/or C3c positive, n(%) 40 of 67 (60) 44 (88) 0.001
IIF on SSS IgG positive (roof), n(%) 45 (66.2) 44 (88) 0.007
IgG ELISA
NC16A IgG positive, n(%) 21 (31) 33 (66) < 0.001
Anti-NC16A IgG titer, median (IQR) 32.0 (40) 65.0 (94) 0.016
BP230 IgG positive, n(%) 30 (47) 27 (54) 0.450
Anti-BP230 IgG titer, median (IQR) 29.50 (28) 51.0 (44) 0.067
Study results
Elevated total IgE, (≥ 115 kU/L), n (%) 42 (63) 28 (59.6) 0.737 5 (20) 15 (60)
Median total IgE (IQR), kU/L 243 (970.1) 164 (554.8) 0.350 40 (83.2) 137 (300.7)
IgE ELISA OD value anti-NC16A IgE, median (IQR)
213.0 (120) 354.5 (225) < 0.001 288 (97) 294 (70)
OD value anti-BP230 IgE, median (IQR)
402.5 (567) 559.5 (611) < 0.001 475 (203) 545 (272)
NC16A IgE positive*, n(%) 6 (9) 9 (18) 0.139 0 2 (8)
BP230 IgE positive**, n(%) 15 (22) 17 (34) 0.149 0 5 (20)
NC16A AND BP230 IgE positive, n(%) 2 (3) 3 (6) 0.649 0 0
* Cut-off 620 (3xSD pemphigus). ** Cut-off 929 (3xSD pemphigus). IQR, interquartile range; L, liter; IIF, indirect immunofluorescent microscopy; SSS, salt-split skin; DIF, direct immunofluorescent microscopy; ELISA, enzyme-linked immunosorbent assay; kU, kilo unit; OD, optical density; n.a., not applicable.

100
Specific IgE autoantibodies to NC16A were not associated with linear IgG along the BMZ by DIF in NBP and BP (p=1.000; p=1.000). Specific IgE autoantibodies to BP230 were associated with a negative DIF result in NBP (p=0.013), but not in BP (p=0.162). IgE in the skin Biopsies for DIF microscopy of 14 patients of each pemphigoid phenotype were stained for IgE. NBP biopsies were taken from lesional (n=6), perilesional (n=6), and healthy skin (n=1), and from an unknown location in one patient. BP biopsies were taken from perilesional (n=9), lesional (n=2), or healthy skin (n=2), and from the buccal mucosa in one patient. IgE on the surface of cells in the dermis IgE in skin was mainly observed on the surface of cells in the upper dermis (NBP 71% and BP 86%; Fig. 1abc, Fig 2abc, table 2). Two of 14 BP biopsies lacked IgE-expressing cells; one taken from buccal mucosa, and one from healthy skin. Four of 14 NBP biopsies showed no IgE-expressing cells; three taken from perilesional skin, and one from healthy skin. The latter NBP healthy skin biopsy did, however, show linear IgE staining along the BMZ. The presence of IgE on cells was not associated with circulating specific IgE antibodies to NC16A and BP230 (p=0.648 and p=1.000). However, the cell-surface expression of IgE did correlate with circulating IgG antibodies to NC16A (p=0.038), and with positive linear IgG staining along the BMZ (p=0.021) by DIF microscopy. No correlation was found with circulating IgG antibodies to BP230 (p=0.673). In BP patients, median total IgE levels were significantly higher in patients with IgE-expressing cells in the upper dermis (659 vs. 87; p=0.048). Such a correlation was not found in NBP patients (p=0.777). No significant differences in median titers of specific IgE to NC16A and BP230 were observed in pemphigoid patients with or without IgE-positive cells present in the skin. Linear IgE along the basement membrane zone In total, two biopsies (7%, 1 NBP and 1 BP) showed IgE deposits along the BMZ (Fig. 1d, Fig. 2d, table 2). Both biopsies with linear IgE staining were taken from healthy skin. The NBP patient had no specific IgE against NC16A and BP230, but showed

101
IgG antibodies to NC16A. The BP patient was positive for IgE and IgG antibodies to BP230, but not to NC16A. Both patients had linear IgG along the BMZ by DIF microscopy. Additional IgE staining of diagnostic skin biopsies In total, 122 biopsies of subjects suspected of autoimmune blistering diseases, but with negative routine IgG diagnostic pemphigoid tests, were stained for IgE. None of the biopsies showed IgE in a linear pattern along the BMZ. Binding of IgE on a few cells was seen occasionally, but less frequently and extensively as in either BP or NBP biopsies.
(a) (b)
(c) (d)
Figure 1. Immunofluorescent staining of IgE in the skin of bullous pemphigoid (BP) patients. (a) Many IgE-positive cells in the dermis (++), representative for 5 of 14 BP skin samples. (b) Multiple IgE-positive cells in the dermis (+) representative for 1 of 14 BP skin samples. (c) Few IgE-positive cells in the dermis, representative for 6 of 14 BP skin samples. (d) One BP skin sample showed linear IgE along the basement membrane zone, while 13 other skin samples did not. White bar is 50 μm.

102
Eosinophils and IgE in serum and histopathologic skin biopsies Data on eosinophils in serum and lesional histopathologic skin biopsies of pemphigoid patients are shown in table 1. Peripheral eosinophilia was significantly associated with elevated total IgE levels in NBP (p=0.028), but not in BP (p=0.208). No associations were found between peripheral eosinophilia and specific IgE antibodies to NC16A and BP230 (p=0.516 and p=0.326). Peripheral eosinophilia was also not associated with IgE on cells (p=0.594), or IgE in a linear pattern along the BMZ (p=0.481). No correlations were found between the presence of eosinophils in histopathologic lesional skin biopsies, and results of total IgE, specific IgE to
(a) (b)
(c) (d)
Figure 2. Immunofluorescent staining of IgE in the skin of nonbullous pemphigoid (NBP) patients. (a) Many IgE-positive cells in the dermis (++), representative for 2 of 14 NBP skin samples. (b) Multiple IgE-positive cells in the dermis (+) representative for 4 of 14 NBP skin samples. (c) Few IgE-positive cells in the dermis, representative for 4 of 14 NBP skin samples. (d) One BP skin sample showed linear IgE along the basement membrane zone, while 13 other skin samples did not. White bar is 50 μm.

103
NC16A/BP230, IgE in the skin on cells in the upper dermis, or IgE in the skin in a linear pattern along the BMZ (data not shown).
Table 2. IgE in the skin of nonbullous and bullous pemphigoid
Nonbullous pemphigoid (n=14)
Bullous pemphigoid (n=14)
Immunofluorescence findings n (%) n (%) IgE deposits linear along the BMZ
1 (7.1) 1 (7.1)
Positive, ++ 0 (0) 1 (7.1) Positive, + 1 (7.1) 0 (0) Negative 12 (85.7) 12 (85.7) IgE on cells in the dermis 10 (71.4) 12 (85.7) Positive, ++ many cells 2 (14.3) 5 (35.7) Positive, + multiple cells 4 (28.6) 1 (7.1) Positive, only few cells 4 (28.6) 6 (42.9) Negative 4 (28.6) 2 (14.3) BMZ, basement membrane zone.
Discussion IgE autoantibodies were clearly present in the serum and skin of both NBP and BP patients. Although not statistically significant, circulating specific IgE to NC16A and BP230 was more often detected in BP (18% and 34%) than in NBP (9% and 22%). In the skin, IgE was most frequently observed on dermal cells (NBP 71% and BP 86%), and only two biopsies (7%; 1 NBP, 1 BP) showed linear IgE depositions along the BMZ. These findings imply that IgE autoantibodies could have indirect effects on blistering by binding to immune cells, likely eosinophils and mast cells, but other factors presumably play a more conclusive role in whether pemphigoid patients form blisters or not. In contrast with the findings of Ben Mordehai et al., our study did not detect significantly higher total IgE levels in NBP compared to BP serum.28 Overall, pemphigoid-specific IgE was more frequently directed to BP230 than to NC16A, in line with several BP studies.17,32 Predominant reactivity of IgE to intracellular BP230 might account for the low occurrence of linear IgE staining by DIF. Hashimoto et al. reported an association between specific IgE to BP230 and a pemphigoid nodularis phenotype.19 Interestingly, subanalysis of previously published clinical NBP data5

104
showed a similar trend, with 47% of the NBP patients with specific IgE to BP230 presenting with papules and nodules (p=0.121; data not shown). This suggests that specific IgE to BP230 might be pathognomonic for a prurigo nodularis-like pemphigoid phenotype. In NBP, specific IgE to NC16A or BP230 was not associated with an urticarial phenotype, similar to previous reports on BP.15,19,33 Overall, we found a lower percentage of IgE reactivity to NC16A and BP230 by ELISA in BP patients, compared with other studies.9,14–22 This might be dependent on methodological differences of established IgE assays, such as serum dilutions, or determined cut-off values. Another explanation could be differences between the BP study cohorts, since the percentage of IgG reactivity to BP180 and BP230 in our consecutive BP patients was lower (66% and 44%, respectively) compared with other studies (90% and 60%, respectively) that might have selected their patients differently.34 We observed linear IgE along the BMZ in only 2 of 24 pemphigoid biopsies (7%), while others reported linear IgE in 0%, 3%, 18%, 25%, and 44% of BP biopsies, indicating that linear IgE is not commonly observed in pemphigoid skin.20,23–27 Differences in methodology and biopsy location may account for the discrepancies between these studies. Interestingly, IgE on the surface of dermal cells was associated with IgG reactivity to NC16A (82%; p=0.038), and linear IgG deposits along the BMZ by DIF (86%; p=0.021), but not with pemphigoid-specific IgE. Our study did not specify the cell types expressing IgE. However, previous studies identified IgE on mast cells and eosinophils in perilesional BP skin.20,27 It has been hypothesized that IgE could induce mast cell and eosinophil degranulation, releasing proteolytic enzymes matrix metalloprotease 9 (MMP9) and neutrophil elastase, substances that are known to cleave the extracellular domain of BP180 in vitro.35–37 This has recently been supported by a murine BP model demonstrating eosinophil-dependent blister formation following sensitization with NC16A-specific IgE.38 Furthermore, since omalizumab (anti-IgE) therapy reduces both pruritus and blisters, a role for IgE in blister formation is supported.30 In contrast, our data does not support the hypothesis that IgE plays an essential role in blister formation in pemphigoid, since we observed IgE on cells in the dermis in 71% of NBP patients that clinically lack blisters. Based on our findings, it is likely that factors other than IgE are more important for blister development. Several studies designate eosinophils as the

105
most important regulators of blister formation, since activated eosinophils were shown to accumulate in serum, skin and blister fluids of BP, and were able to induce dermal-epidermal separation upon activation by IL-5.7,39,40 Beside FcɛRI, eosinophils also express receptors for complement anaphylatoxins C3a and C5a.41–
43 These complement anaphylatoxins not only orchestrate eosinophil migration, but were also capable to evoke eosinophil degranulation.43 Previous studies demonstrated that complement activation might be important for blister formation in BP.44–46 Nonetheless, others found evidence for blister induction via complement-independent pathways by IgG and IgE autoantibody induced internalization of the complete BP180 molecule.47–50 Surprisingly, a subset of elderly controls with pruritus showed specific IgE antibodies to NC16A and BP230 in the absence of IgG autoantibodies to these antigens. In contrast, none of the pemphigus controls had pemphigoid-specific IgE. In accordance, Freire et al. reported specific IgE antibodies to BP180 in one-third of healthy control sera.27 Previously, IgG autoantibodies to NC16A and BP230 by ELISA were detected in elderly individuals, chronic pruritus patients, healthy controls, and dermatology patients, all without pemphigoid.51–55 It has been suggested that repeated cell injury, for example by scratching, could expose new and otherwise hidden antigens to the immune system, a phenomenon termed epitope spreading.56 Moreover, aging of the immune system can result in a pro-inflammatory immune status, increasing the risk of autoimmunity.57 Both events could conceivably contribute to the development of IgE and IgG autoantibodies in elderly controls with pruritus. Yet, the relevance of these autoantibodies in individuals without pemphigoid remains unknown. This study has several limitations, of which the most important is its retrospective nature. Consequently, diagnostic skin samples were taken from different locations by different healthcare professionals. Moreover, our study only assessed IgE targeting NC16A, therefore, no conclusions could be drawn on IgE autoantibodies to other BP180 epitopes. Previous epitope mapping studies of BP180 IgE in BP sera mainly detected NC16A reactivity, however, reactivity to other extracellular and intracellular domains of BP180 has been reported.27,58,59 No epitope mapping data of NBP are available. In conclusion, IgE in NBP and BP skin was more often found on the surface of immune cells rather than deposited linear along the BMZ. The IgE ELISAs for NC16A and BP230 were slightly more often positive in BP than in NBP patients, however,

106
are unsuitable as a diagnostic pemphigoid test due to low specificity. Our main finding is that IgE autoantibodies are present in serum and skin of both NBP and BP patients, supporting the notion that IgE potentially modulates pruritus associated with these pemphigoid diseases rather than being centrally involved in blister formation per se. Further studies, however, are needed to determine why blisters are absent in NBP, and to understand the exact role of IgE in the disease mechanism of pemphigoid. References 1 Schmidt E, Zillikens D. Pemphigoid diseases. Lancet (London, England) 2013; 381:320–32. 2 Hubner F, Recke A, Zillikens D, et al. Prevalence and Age Distribution of Pemphigus and
Pemphigoid Diseases in Germany. J. Invest. Dermatol. 2016; 136:2495–8. 3 Meijer JM, Diercks GFH, de Lang EWG, et al. Assessment of Diagnostic Strategy for Early
Recognition of Bullous and Nonbullous Variants of Pemphigoid. JAMA dermatology 2019 Feb 1;155(2):158-165. doi:10.1001/jamadermatol.2018.4390.
4 Lamberts A, Meijer JM, Jonkman MF. Nonbullous pemphigoid: A systematic review. J Am Acad Dermatol 2018 May;78(5):989-995.e2. doi:10.1016/j.jaad.2017.10.035.
5 Lamberts A, Meijer JM, Pas HH, et al. Nonbullous pemphigoid: insights in clinical and diagnostic findings, treatment responses and prognosis. J Am Acad Dermatol 2019 Aug;81(2):355-363. doi:10.1016/j.jaad.2019.04.029.
6 Hammers CM, Stanley JR. Mechanisms of Disease: Pemphigus and Bullous Pemphigoid. Annu Rev Pathol 2016; 11:175–97.
7 Lo Schiavo A, Ruocco E, Brancaccio G, et al. Bullous pemphigoid: etiology, pathogenesis, and inducing factors: facts and controversies. Clin Dermatol 2013; 31:391–9.
8 Goletz S, Zillikens D, Schmidt E. Structural proteins of the dermal-epidermal junction targeted by autoantibodies in pemphigoid diseases. Exp Dermatol 2017; 26:1154–62.
9 Dopp R, Schmidt E, Chimanovitch I, et al. IgG4 and IgE are the major immunoglobulins targeting the NC16A domain of BP180 in Bullous pemphigoid: serum levels of these immunoglobulins reflect disease activity. J Am Acad Dermatol 2000; 42:577–83.
10 Daneshpazhooh M, Ghiasi M, Lajevardi V, et al. BPDAI and ABSIS correlate with serum anti-BP180 NC16A IgG but not with anti-BP230 IgG in patients with bullous pemphigoid. Arch Dermatol Res 2018; 310:255–9.
11 Humbert M, Bousquet J, Bachert C, et al. IgE-Mediated Multimorbidities in Allergic Asthma and the Potential for Omalizumab Therapy. J allergy Clin Immunol Pract 2019 May - Jun;7(5):1418-1429. doi:10.1016/j.jaip.2019.02.030.
12 Gould HJ, Sutton BJ. IgE in allergy and asthma today. Nat Rev Immunol 2008; 8:205–17. 13 Arbesman CE, Wypych JI, Reisman RE, Beutner EH. IgE levels in sera of patients with pemphigus
or bullous pemphigoid. Arch Dermatol 1974; 110:378–81. 14 Messingham KAN, Noe MH, Chapman MA, et al. A novel ELISA reveals high frequencies of
BP180-specific IgE production in bullous pemphigoid. J Immunol Methods 2009; 346:18–25. 15 van Beek N, Luttmann N, Huebner F, et al. Correlation of Serum Levels of IgE Autoantibodies

107
Against BP180 With Bullous Pemphigoid Disease Activity. JAMA dermatology 2017; 153:30–8. 16 Bing L, Xiping Z, Li L, et al. Levels of anti-BP180 NC16A IgE do not correlate with severity of
disease in the early stages of bullous pemphigoid. Arch Dermatol Res 2015; 307:849–54. 17 Ishiura N, Fujimoto M, Watanabe R, et al. Serum levels of IgE anti-BP180 and anti-BP230
autoantibodies in patients with bullous pemphigoid. J Dermatol Sci 2008; 49:153–61. 18 Iwata Y, Komura K, Kodera M, et al. Correlation of IgE autoantibody to BP180 with a severe form
of bullous pemphigoid. Arch Dermatol 2008; 144:41–8. 19 Hashimoto T, Ohzono A, Teye K, et al. Detection of IgE autoantibodies to BP180 and BP230 and
their relationship to clinical features in bullous pemphigoid. Br J Dermatol 2017; 177:141–51. 20 Dimson OG, Giudice GJ, Fu CL, et al. Identification of a potential effector function for IgE
autoantibodies in the organ-specific autoimmune disease bullous pemphigoid. J Invest Dermatol 2003; 120:784–8.
21 Delaporte E, Dubost-Brama A, Ghohestani R, et al. IgE autoantibodies directed against the major bullous pemphigoid antigen in patients with a severe form of pemphigoid. J Immunol 1996; 157:3642–7.
22 Koga H, Ishii N, Hashimoto T, Nakama T. Case of shift from linear immunoglobulin A bullous dermatosis to pemphigus herpetiformis for a short period of time. J Dermatol 2017; 44:189–93.
23 Yayli S, Pelivani N, Beltraminelli H, et al. Detection of linear IgE deposits in bullous pemphigoid and mucous membrane pemphigoid: a useful clue for diagnosis. Br J Dermatol 2011; 165:1133–7.
24 Provost TT, Tomasi TBJ. Immunopathology of bullous pemphigoid. Basement membrane deposition of IgE, alternate pathway components and fibrin. Clin Exp Immunol 1974; 18:193–200.
25 Moriuchi R, Nishie W, Ujiie H, et al. In vivo analysis of IgE autoantibodies in bullous pemphigoid: a study of 100 cases. J Dermatol Sci 2015; 78:21–5.
26 Kamata A, Kurihara Y, Funakoshi T, et al. Basement membrane zone IgE deposition is associated with bullous pemphigoid disease severity and treatment results. Br J Dermatol 2019 Jul 22. doi: 10.1111/bjd.18364. [Epub ahead of print]
27 Freire PC, Munoz CH, Stingl G. IgE autoreactivity in bullous pemphigoid: eosinophils and mast cells as major targets of pathogenic immune reactants. Br J Dermatol 2017; 177:1644–53.
28 Ben Mordehai Y, Faibish H, Astman N, et al. Characteristics of patients with bullous pemphigoid: comparison of classic bullous pemphigoid to non-bullous pemphigoid. J Eur Acad Dermatol Venereol 2020 Jan;34(1):161-165. doi:10.1111/jdv.15883.
29 Fairley JA, Baum CL, Brandt DS, Messingham KAN. Pathogenicity of IgE in autoimmunity: successful treatment of bullous pemphigoid with omalizumab. J. Allergy Clin. Immunol. 2009; 123:704–5.
30 Kremer N, Snast I, Cohen ES, et al. Rituximab and Omalizumab for the Treatment of Bullous Pemphigoid: A Systematic Review of the Literature. Am J Clin Dermatol 2019 Apr;20(2):209-216. doi:10.1007/s40257-018-0401-6.
31 Koga H, Teye K, Yamashita K, et al. Detection of anti-type VII collagen IgE antibodies in epidermolysis bullosa acquisita. Br J Dermatol 2019; 180:1107–13.
32 Ghohestani RF, Cozzani E, Delaporte E, et al. IgE antibodies in sera from patients with bullous pemphigoid are autoantibodies preferentially directed against the 230-kDa epidermal antigen

108
(BP230). J Clin Immunol 1998; 18:202–9. 33 Saniklidou AH, Tighe PJ, Fairclough LC, Todd I. IgE autoantibodies and their association with the
disease activity and phenotype in bullous pemphigoid: a systematic review. Arch Dermatol Res 2018; 310:11–28.
34 Di Zenzo G, Della Torre R, Zambruno G, Borradori L. Bullous pemphigoid: from the clinic to the bench. Clin Dermatol 2012; 30:3–16.
35 Verraes S, Hornebeck W, Polette M, et al. Respective contribution of neutrophil elastase and matrix metalloproteinase 9 in the degradation of BP180 (type XVII collagen) in human bullous pemphigoid. J Invest Dermatol 2001; 117:1091–6.
36 Stahle-Backdahl M, Inoue M, Guidice GJ, Parks WC. 92-kD gelatinase is produced by eosinophils at the site of blister formation in bullous pemphigoid and cleaves the extracellular domain of recombinant 180-kD bullous pemphigoid autoantigen. J Clin Invest 1994; 93:2022–30.
37 Amber KT, Valdebran M, Kridin K, Grando SA. The Role of Eosinophils in Bullous Pemphigoid: A Developing Model of Eosinophil Pathogenicity in Mucocutaneous Disease. Front Med 2018; 5:201.
38 Lin L, Hwang B-J, Culton DA, et al. Eosinophils Mediate Tissue Injury in the Autoimmune Skin Disease Bullous Pemphigoid. J Invest Dermatol 2018; 138:1032–43.
39 Engmann J, Rüdrich U, Behrens G, et al. Increased Activity and Apoptosis of Eosinophils in Blister Fluids, Skin and Peripheral Blood of Patients with Bullous Pemphigoid. Acta Derm Venereol 2017; 97:464–71.
40 de Graauw E, Sitaru C, Horn M, et al. Evidence for a role of eosinophils in blister formation in bullous pemphigoid. Allergy Eur J Allergy Clin Immunol 2017; 72:1105–13.
41 Messingham KN, Wang JW, Holahan HM, et al. Eosinophil localization to the basement membrane zone is autoantibody- and complement-dependent in a human cryosection model of bullous pemphigoid. Exp Dermatol 2016; 25:50–5.
42 Sayed BA, Christy A, Quirion MR, Brown MA. The master switch: the role of mast cells in autoimmunity and tolerance. Annu Rev Immunol 2008; 26:705–39.
43 DiScipio RG, Schraufstatter IU. The role of the complement anaphylatoxins in the recruitment of eosinophils. Int Immunopharmacol 2007; 7:1909–23.
44 Nelson KC, Zhao M, Schroeder PR, et al. Role of different pathways of the complement cascade in experimental bullous pemphigoid. J Clin Invest 2006; 116:2892–900.
45 Heimbach L, Li Z, Berkowitz P, et al. The C5a receptor on mast cells is critical for the autoimmune skin-blistering disease bullous pemphigoid. J Biol Chem 2011; 286:15003–9.
46 Romeijn TR, Jonkman MF, Knoppers C, et al. Complement in bullous pemphigoid: results from a large observational study. Br. J. Dermatol. 2017; 176:517–9.
47 Messingham KN, Srikantha R, DeGueme AM, Fairley JA. FcR-independent effects of IgE and IgG autoantibodies in bullous pemphigoid. J Immunol 2011; 187:553–60.
48 Kitajima Y, Nojiri M, Yamada T, et al. Internalization of the 180 kDa bullous pemphigoid antigen as immune complexes in basal keratinocytes: an important early event in blister formation in bullous pemphigoid. Br J Dermatol 1998; 138:71–6.
49 Iwata H, Kamio N, Aoyama Y, et al. IgG from patients with bullous pemphigoid depletes cultured keratinocytes of the 180-kDa bullous pemphigoid antigen (type XVII collagen) and weakens cell attachment. J Invest Dermatol 2009; 129:919–26.

109
50 Ujiie H, Sasaoka T, Izumi K, et al. Bullous Pemphigoid Autoantibodies Directly Induce Blister Formation without Complement Activation. J Immunol 2014; 193:4415–28.
51 van Beek N, Dohse A, Riechert F, et al. Serum autoantibodies against the dermal-epidermal junction in patients with chronic pruritic disorders, elderly individuals and blood donors prospectively recruited. Br J Dermatol 2014; 170:943–7.
52 Meijer JM, Lamberts A, Pas HH, Jonkman MF. Significantly higher prevalence of circulating bullous pemphigoid-specific IgG autoantibodies in elderly patients with a nonbullous skin disorder. Br J Dermatol 2015 Nov;173(5):1274-6. doi:10.1111/bjd.13874.
53 Rieckhoff-Cantoni L, Bernard P, Didierjean L, et al. Frequency of bullous pemphigoid-like antibodies as detected by western immunoblot analysis in pruritic dermatoses. Arch Dermatol 1992; 128:791–4.
54 Hofmann SC, Tamm K, Hertl M, Borradori L. Diagnostic value of an enzyme-linked immunosorbent assay using BP180 recombinant proteins in elderly patients with pruritic skin disorders. Br. J. Dermatol. 2003; 149:910–2.
55 Feliciani C, Caldarola G, Kneisel A, et al. IgG autoantibody reactivity against bullous pemphigoid (BP) 180 and BP230 in elderly patients with pruritic dermatoses. Br J Dermatol 2009; 161:306–12.
56 Didona D, Di Zenzo G. Humoral Epitope Spreading in Autoimmune Bullous Diseases. Front Immunol 2018; 9:779.
57 Pawelec G. Age and immunity: What is ‘immunosenescence’? Exp Gerontol 2018; 105:4–9. 58 Dresow SK, Sitaru C, Recke A, et al. IgE autoantibodies against the intracellular domain of
BP180. Br J Dermatol 2009; 160:429–32. 59 Fairley JA, Fu CL, Giudice GJ. Mapping the binding sites of anti-BP180 immunoglobulin E
autoantibodies in bullous pemphigoid. J Invest Dermatol 2005; 125:467–72.

110
7

111
CHAPTER 7 Gene expression profile of lesional skin in bullous and nonbullous pemphigoid patients: an explorative pilot study Aniek Lamberts Center for Blistering Diseases, Department of Dermatology, University Medical Center Groningen, University of Groningen, Groningen, The Netherlands Preliminary results

112
Introduction Bullous pemphigoid (BP) is the most common autoimmune bullous disease that typically presents in elderly patients with severe pruritus, and tense blisters on erythematous skin.1–3 One-in-five patients present with severe pruritus and a phenotype without blisters, a disease variant termed nonbullous pemphigoid (NBP).4–6 The clinical presentation of NBP is heterogeneous and may mimic other pruritic dermatological diseases, causing misdiagnosis and long diagnostic delays.7,8 While the pathogenesis of BP is only partly clarified, no studies have assessed the pathogenesis of NBP.
A higher susceptibility for pemphigoid diseases was observed in individuals expressing the major histocompatibility complex class II allele HLA-DQB1*03:01, which likely presents pemphigoid antigens to autoreactive T cells.9–12 Subsequently, autoantibodies against hemidesmosome proteins BP180 and BP230 are formed.13,14 Autoantibodies against BP180 were demonstrated to be pathogenic in animal and human studies.15–19 The role of autoantibodies to the intracellular BP230 is less clear, and single BP230 autoantibody reactivity was associated with a less inflammatory BP phenotype, and with NBP.4,15,20–22
The disease mechanism of BP is not completely elucidated. Most studies support the hypothesis that autoantibodies to BP180 mediate blister formation by activation of the complement system through classical and alternative pathways, attracting inflammatory cells towards the skin.23–25 Studies particularly found evidence for an important role of eosinophils and mast cells, whom upon activation release proteolytic enzymes that break down structural hemidesmosome proteins, causing subepithelial blister formation.13,26–28 Complement independent mechanisms of blistering were also described, involving depletion of the BP180 molecule from the hemidesmosome by autoantibody induced pinocytosis.29 So far, it is unknown why NBP patients do not develop blisters, while the autoantibody profile can be similar to that of BP.
In this study, we assessed the activation of the immune response in lesional skin of BP and NBP patients. By quantification of immune-response related gene transcripts, we aim to find differences in the immune response in BP and NBP.

113
Material and methods Selection of patients and patient material Patients diagnosed with BP (n=12) and NBP (n=12) at the outpatient clinic of our dermatology department were retrospectively selected. Diagnostic inclusion criteria consisted of the 2-out-of-3 rule, meaning patients had to meet two of the following three criteria; 1) pruritus and/or cutaneous blisters, 2) positive linear IgG or C3c staining by direct immunofluorescence (DIF) microscopy, 3) positive IgG staining on the epidermal side of salt-split skin by indirect immunofluorescence microscopy.4 Another inclusion criterion was the performance of a lesional skin biopsy for histopathology for diagnostic purposes. Patients using systemic immunosuppressive drugs at the time of the biopsy were excluded. The use of topical corticosteroids was avoided, but allowed at lowest class if a more suitable biopsy was not available.
Patient characteristics were assessed by reviewing patient charts. Patient material utilized for analyzing gene transcripts consisted of histopathologic skin biopsies that were formalin fixed, and embedded in paraffin before stored at room temperature, according to standard protocol. BP skin biopsies contained one-third of a blister, and NBP skin biopsies were taken from lesional erythematous skin.
NanoString gene expression profiling The nanoString nCounter 30 Myeloid Innate Immunity Panel (nanoString Technologies, Seattle, WA) was used to quantify the expression of 180 genes associated with innate and adaptive immune responses. RNA was isolated from four 5 μm thick formalin-fixed and paraffin-embedded skin sections of 12 confirmed NBP and 12 confirmed BP patients using the RNeasy mini kit (Qiagen) according to suppliers instructions. RNA (100ng as measured by Qubit (ThermoFisher)) was hybridized with the nanoString reporter and capture probes overnight at 65 degrees Celsius. Each probe set (reporter and capture probe) is designed to bind a unique mRNA target, and has a unique color-coded molecular tag. The RNA-probe complexes were loaded on an nCounter cartridge, and washed and read on a SPRINT digital counter, using digital photography. Statistical analysis nSolver Analysis Software (nanoString Technologies, Seattle, WA) was used for normalization of counts using the arithmetic mean of spiked-in reference gene

114
transcripts as well as the geometric mean of the hybridization controls. Gene expression data was visualized with the nSolver advanced analysis module. Heatmaps were created with unsupervised clustering. Binomial regression models were used to estimate the differential expression of genes in BP versus NBP patients. To control the rate of type I errors occurring when conducting multiple comparisons, the Benjamini-Yekutieli procedure was applied controlling for the false discovery rate, and providing an adjusted p-value.31 Results Patients characteristics Two BP samples were excluded from the study, due to a low quality of the transcript data. Gene expression levels were successfully measured in 10 BP, and 12 NBP patients. Patient characteristics, and the results of gene expression involving complement activation, T helper (Th1), and T helper 2 (Th2) responses are presented in table 1. Complement activation High expression of genes related to complement activation was observed in six BP patients (figure 1). The patients with a lower expression of complement activation related genes included four BP patients, and 12 NBP patients. C3c positivity in direct immunofluorescence microscopy Complement C3c in the skin is routinely stained in NBP and BP patients for diagnostic purposes by DIF. Three of the six BP patients with high expression of complement related genes showed linear C3c along the basement membrane zone (BMZ) by DIF (table 1). Moreover, five of the 16 biopsies with lower expression of complement related genes did show linear C3c along the BMZ by DIF.

115
Table 1. Patient characteristics and gene expression results
Patient characteristics Gene expression* NBP vs.
BP Age Gender DIF IgG
DIF C3c
Antigen recognition**
Complement activation
Th1 response
Th2 response
NBP 1 67 female 2+ neg BP180 only Low Low Low
NBP 2 41 male +/2+ neg BP180 only Low Low Low
NBP 3 95 male +/2+ neg BP180 only Low Low Low
NBP 4 101 female 2+ neg BP180 + BP230 Low High High
NBP 5 88 female 2+ neg BP180 + BP230 Low Low Low
NBP 6 78 female dub 3+ BP180 + BP230 Low High High
NBP 7 82 female dub neg BP180 + BP230 Low Low Low
NBP 8 79 male neg neg BP230 only Low Low Low
NBP 9 79 male neg neg BP230 only Low Low Low
NBP 10 91 female neg neg BP230 only Low High Low
NBP 11 92 female neg neg BP230 only Low Low Low
NBP 12 86 female neg neg BP230 only Low High Low
BP 1 76 male 3+ 2+ BP180 + BP230 Low High High
BP 2 53 male 3+ 2+ BP180 only Low High High
BP 3 87 male 2+ 3+ BP180 + BP230 High High High
BP 4 71 female 2+ + BP180 + BP230 High High High
BP 5 80 male + 3+ BP180 only High High High
BP 6 87 female + 2+ BP180 only Low High High
BP 7 74 female + +/- BP180 + BP230 High High High
BP 8 80 female + neg BP230 only High High High
BP 9 89 female neg 2+ BP180 + BP230 Low High High
BP 10 61 male neg neg BP180 + BP230 High High High * Whether gene expression was scored low or high was determined based on unsupervised clustering and visualization of the data by heatmaps (see figure 1, 2 and 3). ** Antigen recognition is based on results of immunoblot and/or enzyme-linked immunosorbent assay. NBP, nonbullous pemphigoid; BP, bullous pemphigoid; DIF, direct immunofluorescence microscopy; IgG, immunoglobulin G; C3c, complement component C3c; Th1, T helper 1; Th2, T helper 2.

116
T helper 1 and 2 responses In all BP specimens a high expression of both Th1 and Th2 related genes was observed (figure 2 and 3). In NBP specimens, genes involved in Th1 responses were highly expressed in one-third of the cases, while a gene expression signature associated with a Th2 response was observed in 2 out of 12 NBP cases.
Figure 1. Complement activation. Heat map of expressed genes involved in complement activation. Unsupervised clustering was performed. Highly expressed genes are depicted in orange, whereas genes with lower expression are blue. The phenotype (BP or NBP) is presented per sample.
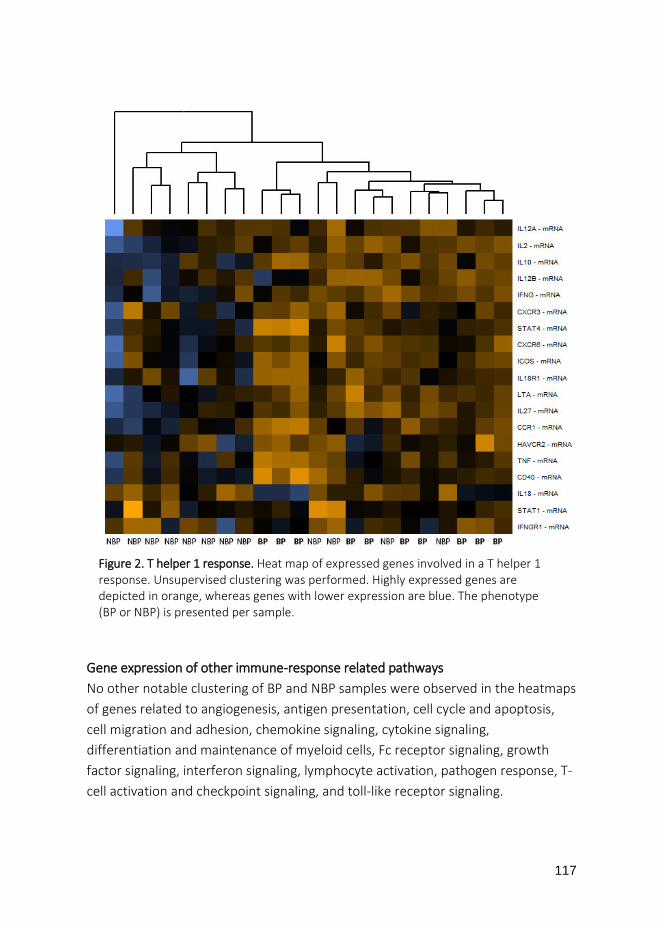
117
Gene expression of other immune-response related pathways No other notable clustering of BP and NBP samples were observed in the heatmaps of genes related to angiogenesis, antigen presentation, cell cycle and apoptosis, cell migration and adhesion, chemokine signaling, cytokine signaling, differentiation and maintenance of myeloid cells, Fc receptor signaling, growth factor signaling, interferon signaling, lymphocyte activation, pathogen response, T-cell activation and checkpoint signaling, and toll-like receptor signaling.
Figure 2. T helper 1 response. Heat map of expressed genes involved in a T helper 1 response. Unsupervised clustering was performed. Highly expressed genes are depicted in orange, whereas genes with lower expression are blue. The phenotype (BP or NBP) is presented per sample.
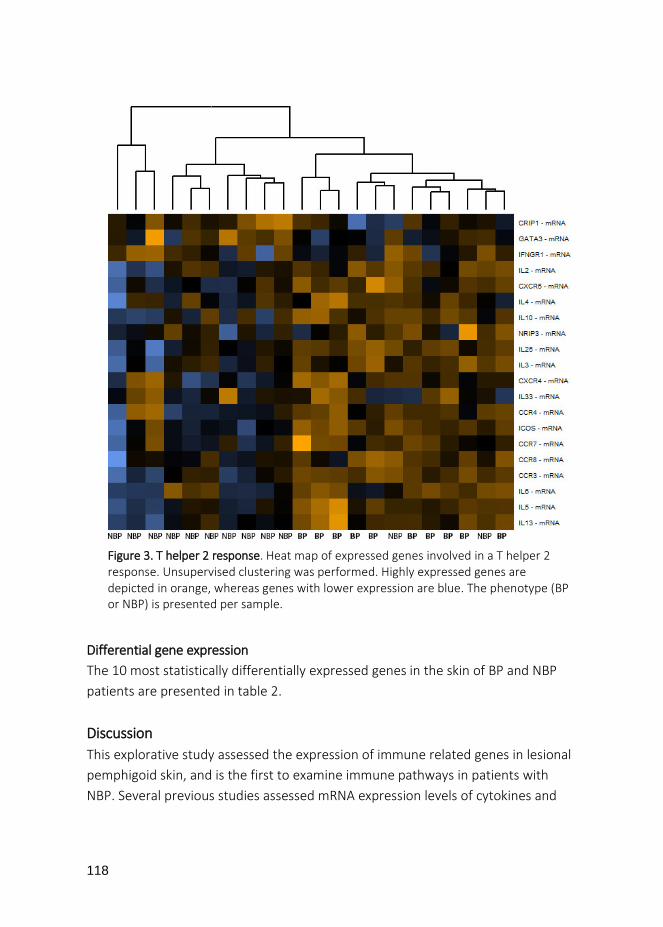
118
Differential gene expression The 10 most statistically differentially expressed genes in the skin of BP and NBP patients are presented in table 2. Discussion This explorative study assessed the expression of immune related genes in lesional pemphigoid skin, and is the first to examine immune pathways in patients with NBP. Several previous studies assessed mRNA expression levels of cytokines and
Figure 3. T helper 2 response. Heat map of expressed genes involved in a T helper 2 response. Unsupervised clustering was performed. Highly expressed genes are depicted in orange, whereas genes with lower expression are blue. The phenotype (BP or NBP) is presented per sample.

119
chemokines in BP, but only in single cell types, such as macrophages, eosinophils, or peripheral blood mononuclear cells.32–35
Table 2. Top 10 of the most statistically differentially expressed genes in BP versus NBP skin (NBP is baseline).
Gene Encodes for Fold
change (log2)
Standard error (log2)
95% Confidence
interval (log2)
P-value Adjusted p-value*
HBEGF
Heparin-binding epidermal growth factor-like growth factor
3.47 0.438 2.61 – 4.33 1.38e-07 0.000683
CXCL8 Chemokine IL-8 3.74 0.533 2.70 – 4.79 8.35e-07 0.00194
PTGS2
Prostaglandin-endoperoxide synthase 2
1.49 0.218 1.07 – 1.92 1.18e-06 0.00194
ADAMTS4
A disintegrin and metalloproteinase with trombospondin motifs
2.38 0.429 1.54 – 3.22 1.95e-05 0.0101
AREG amphiregulin 3.51 0.635 2.27 – 4.76 2.05e-05 0.0101
CCL3 chemokine C-C motif ligand 3
1.45 0.251 0.96 – 1.95 1.14e-05 0.0101
CREM
cAMP (cyclic adenosine monophosphate) responsive element modulator
1.33 0.236 0.87 – 1.79 1.64e-05 0.0101
FOSL1 Fos-related antigen 1 2.1 0.375 1.37 – 2.84 1.78e-05 0.0101
GPR65 G protein-coupled receptor 65 1.1 0.195 0.72 – 1.48 1.6e-05 0.0101
IL1RL1 Interleukin-1 receptor-like 1
1.78 0.307 1.18 – 2.38 1.16e-05 0.0101
* P-values were adjusted for multiple testing by applying the Benjamini-Yekutieli procedure, controlling for the false discovery rate. BP, bullous pemphigoid; NBP, nonbullous pemphigoid.

120
By using a broad explorative approach, our results indicated involvement of complement activation in 6 out of 10 BP patients but not in NBP patients (0 out of 12). Moreover, all BP patients showed strong Th1 and Th2 responses, whereas NBP patients did not (high expression of Th1 in 33%, Th2 in 17%). These observations support the hypothesis that blister formation involves Th1 and Th2 responses, and complement activation. Genes related to complement activation Six BP patients showed higher expression of genes related to complement activation compared to 4 BP and all 12 NBP patients. In line with these findings, mice studies on the pathogenesis of BP demonstrated that complement activation via the classical pathway and alternative pathway might be essential for blister development.25,36,37 Complement independent disease mechanisms might have played an more important role in the pemphigoid patients with a lower expression of complement related genes.29 Surprisingly, expression of genes related to complement activation did not correspond with positivity of C3c linear along the BMZ by DIF in several patients. C3c deposits in perilesional skin apparently do not reflect on lesional complement activation.
Interestingly, several patients (4 BP, and 7 NBP) had IgG deposits along the BMZ by DIF, but did not show an high expression of complement related genes. We hypothesize that IgG visualized by DIF in these patients might consist of IgG2 or IgG4 subclasses, whom have low complement activating capacity.38 In line with this hypothesis, it was previously shown that NBP patients were IgG4 subclass dominant, and showed less complement deposits in the skin compared to BP patients.4,39,40 Our observations underscore the complexity of the complement system, and its various activation routes. Genes involved in T helper 1 and 2 responses High expression of both Th1 and Th2 related genes was found in skin of all BP patients, but in a minority of NBP patients. Previous studies in BP detected autoreactive T cells producing both Th1 and Th2 cytokines, and found elevated levels of Th1 and Th2 chemokines in serum.12,41–43 More evidence for a Th2 response in BP includes increased serum levels of total IgE, and IgE against the NC16A domain of BP180, which correlates with disease activity.17,44–46 IgE was also observed bound to the cell surface of mast cells and eosinophils in the dermis of BP

121
skin, while others found IgE deposits in a linear pattern along the BMZ.28,47–49 Peripheral eosinophilia was present in approximately half of BP and NBP patients, and dermal infiltrates commonly include eosinophils in both disease phenotypes.20,50,51 All data above point towards a strong Th2 footprint in BP. Only limited data are published about the immune response in NBP. In chapter 6 of this thesis we detected IgE in serum and skin of NBP patients, suggesting a role for a Th2 immune response in its pathogenesis (unpublished data). However, based on our gene expression data, we may conclude that Th2 responses likely play a less prominent role in the disease pathogenesis of NBP compared to BP. Differential gene expression Several genes that were most differentially expressed in BP compared to NBP skin are discussed below. Epidermal growth factors The HBEGF gene encodes for heparin-binding epidermal growth factor-like growth factor, and was most differentially expressed, with high expression in BP. HBEGF is an epidermal growth factor produced by monocytes and macrophages, and plays a role in many physiological and pathological processes. No publications link HBEGF gene expression to pemphigoid so far. In rheumatoid arthritis (RA), single cell sequencing identified a population of HBEGF+ inflammatory macrophages in inflamed synovial tissues.52 These macrophages produced a defined subset of inflammatory products, such as IL-1, HBEGF, and epiregulin, and promoted pathologic fibroblast mediated joint destruction. In a lupus nephritis mouse model, high expression of the HBEGF gene was found in kidney tissue and kidney macrophages, suggesting that HBEGF might be important in the SLE pathogenesis.53
A second epithelial growth factor gene that was highly expressed in BP compared to NBP skin is amphiregulin (AREG). Interestingly, AREG is an important paralog of the HBEGF gene, meaning the genes descent from the same ancestral gene.54 AREG is a transmembrane protein, and in RA its expression seems to be related to fibroblasts proliferation, and increased levels of IL-8.54,55 AREG and HBEGF were significantly upregulated in bone marrow mononuclear cells, and peripheral blood mononuclear cells of RA patients, but only AREG was significantly upregulated in synovial fluids.55

122
HBEGF and AREG expression is dependent on ADAM17 activation.56 ADAM17 belongs to the family of disintegrins and metalloproteases, and is best known for processing tumor necrosis factor α (TNF-α). While HBEGF and AREG expression were not previously studied in BP, ADAM17 was highly expressed in the epidermis of BP patients, and was suggested to deplete BP180 from keratinocytes.56,57
Pro-inflammatory chemokines, cytokines and enzymes The CXCL8 gene encodes for the pro-inflammatory chemokine IL-8 and is highly expressed in BP. IL-8 is a potent chemoattractant and activator of leukocytes. It was shown that keratinocytes in vitro release IL-8 and IL-6, after binding with anti-BP180 IgG.58 Moreover, multiple studies found elevated IL-8 levels in blister fluid and serum of BP patients compared to controls.59–62
The PTGS2 gene encodes for the enzyme prostaglandin-endoperoxide synthase 2, more often referred to as cyclooxygenase-2 (COX-2). COX-2 catalyzes arachidonic acid to prostaglandin H2, and is highly expressed during inflammation. In literature, the only link between BP and COX-2 is a report describing a pemphigoid case possibly induced by selective COX-2 inhibitor celecoxib.64 Interestingly, COX-2 is naturally inhibited by calcitriol, the active form of vitamin D.65 Several studies found an association between low levels of vitamin D and BP.66–
69 In vitro, calcitriol showed anti-inflammatory effects in keratinocytes that were treated with BP specific autoantibodies.70
The CCL3 gene encodes for chemokine C-C motif ligand 3, also termed macrophage inflammatory protein-1-α (MIP-1- α), and is produced by macrophages. CCL3 has a function in recruitment and activation of granulocytes.74 Two previous studies observed elevated expression of CCL3 in BP serum and blister fluid.75,76 However, one of these studies did report that Th2 associated chemokines (eotaxin and MCP-4) were higher in the blister fluid of BP patients, than Th1 associated chemokines (CXCL10 and CCL3).76
Transcription factors CREM is a gene encoding for transcription factor cAMP (cyclic adenosine monophosphate) responsive element modulator, and is an important component of the cAMP mediated signal transduction.77 No data is available on the expression of CREM and its possible function in pemphigoid. However, in T lymphocytes of patients with systemic lupus erythematosus (SLE) a high expression of CREM is

123
reported.78 In SLE, CREM binds to the IL-2 promotor to repress IL-2 transcription by T lymphocytes, while enhancing the expression of IL-17 by direct transcriptional mechanisms.77–79 IL-2 is important for proliferation of T and B lymphocytes, while IL-17 is a pro-inflammatory cytokine described to have a potential role in the pathogenesis of several autoimmune diseases, such as RA, SLE, multiple sclerosis, inflammatory bowel diseases, and also in BP.60,77–80
A second gene that influences gene transcription is the FOSL1 gene, encoding for the fos-related antigen 1 (FRA1). FOSL1 is member of the FOS gene family, that encode leucine zipper proteins that dimerize with proteins of the JUN family to form the transcription factor complex ‘activator protein 1’ (AP-1).81 The AP-1 complex regulates a variety of mitogen activated protein kinase (MAPK) pathways. No articles have previously described FOSL1 or AP-1 expression in pemphigoid. Interestingly, CREM expression is regulated by two promotors named P1 and P2, of which the promotor P2 is under tight transcriptional control of AP-1, linking the high CREM and FOSL1 expression found in this study.77
Receptor genes The Interleukin-1 receptor-like 1 (IL1RL1) gene encodes for a receptor that is highly expressed on Th2 cells and mast cells. Binding of IL-33 to IL1RL1 induces the production of Th2 associated cytokines, and IL-8.83 In BP, IL1RL1 expression and IL-33 levels were measured in serum and blister fluid, but in contrast to our results, IL1RL1 expression and IL-33 protein levels were below the detection limit.84 Possibly, the difference in patient material en testing method by which IL1RL1 expression was measured could account for these conflicting results.
GPR65 encodes for a G protein-coupled receptor 65, also termed TDAG8 (T cell death associated gene 8). The GPR65 receptor is expressed in several tissue types, as well as in lymphocytes, macrophages and leucocytes.87 The GPR65 receptor is able to sense the extracellular pH, and in acidic environments its activation results in intracellular cAMP accumulation.87 GPR65 receptor activation in T cells and macrophages showed a reduced production of pro-inflammatory cytokines IL-6 and TNF-α, and increased production of anti-inflammatory cytokine IL-10 in vitro.88 In line, a GPR65 knockout mice model showed a significant exacerbation of antibody induced arthritis and delayed hypersensitivity.89 These findings suggest anti-inflammatory effects of GPR65 expression but its function in BP remains elusive

124
Proteinase The ADAMTS4 gene encodes for a proteinase that is a member of the “a disintegrin and metalloproteinase with trombospondin motifs (ADAMTS)” family, and is known for degradation of proteoglycans in articular cartilage in osteoarthritis.90 The role of ADAMTS4 in inflammation is unclear and is not linked to pemphigoid or other autoimmune diseases so far. Summary differential gene expression All genes discussed above showed a significant higher expression in BP than in NBP, and therefore might be important in the pathogenesis of BP, and less important in the pathogenesis of NBP. In summary, we may carefully conclude that chemoattractants IL-8 and CCL3 possibly play a role in migration of leukocytes towards the site of inflammation. High expression of epidermal growth factors HBEGF and AREG suggest a role of macrophages in BPs pathogenesis. The high expression of transcription factors CREM and FOSL1 were previously described in T lymphocytes, and possibly enhance the production of pro-inflammatory IL-17 in BP skin. The ILR1RL1 receptor was previously found on Th2 cells and mast cells, and after activation might contribute to a pro-inflammatory environment by releasing Th2 related cytokines and IL-8. In contrast, activation of the GPR65 receptor expressed on macrophages and T cells may have anti-inflammatory effects. The expression of COX-2 is known to be upregulated in inflammation, but its function in BP is not clear. Also the relevance of the observed high expression of the proteinase ADAMTS4 in BP is unknown. Limitations of the study The greatest limitations of this study is the limited sample size, combined with performing multiple comparisons, causing a multiple testing problem. By correcting for false discovery rates, the number of type I errors is only partially controlled. Therefore, our explorative genetic data needs further validation.
Another limitation is that a preselected panel of genes was analyzed, which could be seen as a limitation opposed to RNA sequencing. By only focusing on preselected genes, we might have missed differences in genes outside of the tested panel. Besides this limitation, the nanoString technique has some advantages over RNA-sequencing. The nanoString avoids reversed transcription of RNA, and cDNA amplification, two steps in RNA-sequencing that are prone to bias.

125
Moreover, in RNA-sequencing the use of formalin-fixed paraffin-embedded tissue samples that contain degraded RNA remains a challenge, while nanoString is able to still generate high-quality data from this tissue. Conclusions Based on the data of this explorative study, we may carefully conclude that genes related to complement activation, Th1 and Th2 responses showed a higher expression in BP skin compared to NBP skin, suggesting they may be important in blister formation. In BP skin, T-helper responses showed a strong dual character, with both Th1 and Th2 related genes involved, whereas in NBP only few biopsies showed high expression of Th1 and Th2 genes. Further studies for validation of the genetic data are needed, and should assess specific gene and protein expression in a larger patient population so that multiple testing problems are avoided. Moreover, an important next step is to include a series of healthy skin biopsies as control samples to analyze the possible more subtle changes in NBP gene expression. References 1 Schmidt E, Zillikens D. Pemphigoid diseases. Lancet (London, England) 2013; 381:320–32. 2 Hubner F, Recke A, Zillikens D, et al. Prevalence and Age Distribution of Pemphigus and
Pemphigoid Diseases in Germany. J. Invest. Dermatol. 2016; 136:2495–8. 3 Joly P. Incidence of bullous pemphigoid and pemphigus vulgaris. BMJ. 2008; 337:a209. 4 Meijer JM, Diercks GFH, de Lang EWG, et al. Assessment of Diagnostic Strategy for Early
Recognition of Bullous and Nonbullous Variants of Pemphigoid. JAMA dermatology 2019 Feb 1;155(2):158-165. doi:10.1001/jamadermatol.2018.4390.
5 Della Torre R, Combescure C, Cortés B, et al. Clinical presentation and diagnostic delay in bullous pemphigoid: A prospective nationwide cohort. Br J Dermatol 2012; 167:1111–7.
6 Di Zenzo G, Della Torre R, Zambruno G, Borradori L. Bullous pemphigoid: from the clinic to the bench. Clin Dermatol 2012; 30:3–16.
7 Zhang Y, Luo Y, Han Y, et al. Non-bullous lesions as the first manifestation of bullous pemphigoid: A retrospective analysis of 181 cases. J Dermatol 2017 Jul;44(7):742-746. doi:10.1111/1346-8138.13782.
8 Sun C, Chang B, Gu H. Non-bullous lesions as the first manifestation of bullous pemphigoid: a retrospective analysis of 24 cases. J Dermatolog Treat 2009; 20:233–7.
9 Delgado JC, Turbay D, Yunis EJ, et al. A common major histocompatibility complex class II allele HLA-DQB1* 0301 is present in clinical variants of pemphigoid. Proc Natl Acad Sci U S A 1996; 93:8569–71.
10 Setterfield J, Theron J, Vaughan RW, et al. Mucous membrane pemphigoid: HLA-DQB1*0301 is associated with all clinical sites of involvement and may be linked to antibasement membrane IgG production. Br J Dermatol 2001; 145:406–14.
11 Amber KT, Zikry J, Hertl M. A multi-hit hypothesis of bullous pemphigoid and associated neurological disease: Is HLA-DQB1*03:01, a potential link between immune privileged antigen

126
exposure and epitope spreading? HLA 2017; 89:127–34. 12 Budinger L, Borradori L, Yee C, et al. Identification and characterization of autoreactive T cell
responses to bullous pemphigoid antigen 2 in patients and healthy controls. J Clin Invest 1998; 102:2082–9.
13 Lo Schiavo A, Ruocco E, Brancaccio G, et al. Bullous pemphigoid: etiology, pathogenesis, and inducing factors: facts and controversies. Clin Dermatol 2013; 31:391–9.
14 van Beek N, Schulze FS, Zillikens D, Schmidt E. IgE-mediated mechanisms in bullous pemphigoid and other autoimmune bullous diseases. Expert Rev Clin Immunol 2016; 12:267–77.
15 Goletz S, Zillikens D, Schmidt E. Structural proteins of the dermal-epidermal junction targeted by autoantibodies in pemphigoid diseases. Exp Dermatol 2017; 26:1154–62.
16 Dopp R, Schmidt E, Chimanovitch I, et al. IgG4 and IgE are the major immunoglobulins targeting the NC16A domain of BP180 in Bullous pemphigoid: serum levels of these immunoglobulins reflect disease activity. J Am Acad Dermatol 2000; 42:577–83.
17 van Beek N, Luttmann N, Huebner F, et al. Correlation of Serum Levels of IgE Autoantibodies Against BP180 With Bullous Pemphigoid Disease Activity. JAMA dermatology 2017; 153:30–8.
18 Ishiura N, Fujimoto M, Watanabe R, et al. Serum levels of IgE anti-BP180 and anti-BP230 autoantibodies in patients with bullous pemphigoid. J Dermatol Sci 2008; 49:153–61.
19 Iwata Y, Komura K, Kodera M, et al. Correlation of IgE autoantibody to BP180 with a severe form of bullous pemphigoid. Arch Dermatol 2008; 144:41–8.
20 Lamberts A, Meijer JM, Pas HH, et al. Nonbullous pemphigoid: insights in clinical and diagnostic findings, treatment responses and prognosis. J Am Acad Dermatol 2019 Aug;81(2):355-363. doi:10.1016/j.jaad.2019.04.029.
21 Tanaka M, Hashimoto T, Dykes PJ, Nishikawa T. Clinical manifestations in 100 Japanese bullous pemphigoid cases in relation to autoantigen profiles. Clin Exp Dermatol 1996; 21:23–7.
22 Hayakawa T, Teye K, Hachiya T, et al. Clinical and immunological profiles of anti-BP230-type bullous pemphigoid: Restriction of epitopes to the C-terminal domain of BP230, shown by novel ELISAs of BP230-domain specific recombinant proteins. Eur J Dermatol 2016; 26:155–63.
23 Dainichi T, Chow Z, Kabashima K. IgG4, complement, and the mechanisms of blister formation in pemphigus and bullous pemphigoid. J Dermatol Sci 2017; 88:265–70.
24 Edwards G, Diercks GFH, Seelen MAJ, et al. Complement Activation in Autoimmune Bullous Dermatoses: A Comprehensive Review. Front Immunol 2019; 10:1477.
25 Nelson KC, Zhao M, Schroeder PR, et al. Role of different pathways of the complement cascade in experimental bullous pemphigoid. J Clin Invest 2006; 116:2892–900.
26 Hammers CM, Stanley JR. Mechanisms of Disease: Pemphigus and Bullous Pemphigoid. Annu Rev Pathol 2016; 11:175–97.
27 de Graauw E, Sitaru C, Horn M, et al. Evidence for a role of eosinophils in blister formation in bullous pemphigoid. Allergy Eur J Allergy Clin Immunol 2017; 72:1105–13.
28 Dimson OG, Giudice GJ, Fu CL, et al. Identification of a potential effector function for IgE autoantibodies in the organ-specific autoimmune disease bullous pemphigoid. J Invest Dermatol 2003; 120:784–8.
29 Iwata H, Ujiie H. Complement-independent blistering mechanisms in bullous pemphigoid. Exp Dermatol 2017 Dec;26(12):1235-1239. doi:10.1111/exd.13367.
30 Tsang H-F, Xue VW, Koh S-P, et al. NanoString, a novel digital color-coded barcode technology: current and future applications in molecular diagnostics. Expert Rev Mol Diagn 2017; 17:95–103.
31 Benjamini BY, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann statitistics 2001; 29:1165–88.
32 Fang H, Shao S, Cao T, et al. Increased expression of NLRP3 inflammasome components and

127
interleukin-18 in patients with bullous pemphigoid. J Dermatol Sci 2016; 83:116–23. 33 Furudate S, Fujimura T, Kambayashi Y, et al. Comparison of CD163+ CD206+ M2 macrophages in
the lesional skin of bullous pemphigoid and pemphigus vulgaris: the possible pathogenesis of bullous pemphigoid. Dermatology 2014; 229:369–78.
34 Messingham KN, Holahan HM, Frydman AS, et al. Human Eosinophils Express the High Affinity IgE Receptor, FcεRI, in Bullous Pemphigoid. PLoS One 2014 Sep 25;9(9):e107725.
35 Gambichler T, Tsitlakidon A, Skrygan M, et al. T regulatory cells and other lymphocyte subsets in patients with bullous pemphigoid. Clin Exp Dermatol 2017; 42:632–7.
36 Heimbach L, Li Z, Berkowitz P, et al. The C5a receptor on mast cells is critical for the autoimmune skin-blistering disease bullous pemphigoid. J Biol Chem 2011; 286:15003–9.
37 Liu Z, Giudice GJ, Swartz SJ, et al. The role of complement in experimental bullous pemphigoid. J Clin Invest 1995; 95:1539–44.
38 Tao MH, Smith RI, Morrison SL. Structural features of human immunoglobulin G that determine isotype-specific differences in complement activation. J Exp Med 1993; 178:661–7.
39 Lamb PM, Patton T, Deng JS. The predominance of IgG4 in prodromal bullous pemphigoid. Int J Dermatol 2008; 47:150–3.
40 Romeijn TR, Jonkman MF, Knoppers C, et al. Complement in bullous pemphigoid: results from a large observational study. Br. J. Dermatol. 2017; 176:517–9.
41 Lin MS, Fu CL, Giudice GJ, et al. Epitopes targeted by bullous pemphigoid T lymphocytes and autoantibodies map to the same sites on the bullous pemphigoid 180 ectodomain. J Invest Dermatol 2000; 115:955–61.
42 Echigo T, Hasegawa M, Shimada Y, et al. Both Th1 and Th2 chemokines are elevated in sera of patients with autoimmune blistering diseases. Arch Dermatol Res 2006; 298:38–45.
43 Eming R, Budinger L, Riechers R, et al. Frequency analysis of autoreactive T-helper 1 and 2 cells in bullous pemphigoid and pemphigus vulgaris by enzyme-linked immunospot assay. Br J Dermatol 2000; 143:1279–82.
44 Asbrink E, Hovmark A. Serum IgE levels in patients with bullous pemphigoid and its correlation to the activity of the disease and anti-basement membrane zone antibodies. Acta Derm Venereol 1984; 64:243–6.
45 Arbesman CE, Wypych JI, Reisman RE, Beutner EH. IgE levels in sera of patients with pemphigus or bullous pemphigoid. Arch Dermatol 1974; 110:378–81.
46 Saniklidou AH, Tighe PJ, Fairclough LC, Todd I. IgE autoantibodies and their association with the disease activity and phenotype in bullous pemphigoid: a systematic review. Arch Dermatol Res 2018; 310:11–28.
47 Freire PC, Munoz CH, Stingl G. IgE autoreactivity in bullous pemphigoid: eosinophils and mast cells as major targets of pathogenic immune reactants. Br J Dermatol 2017; 177:1644–53.
48 Kamata A, Kurihara Y, Funakoshi T, et al. Basement membrane zone IgE deposition is associated with bullous pemphigoid disease severity and treatment results. Br J Dermatol 2019 Jul 22. doi: 10.1111/bjd.18364. [Epub ahead of print]
49 Yayli S, Pelivani N, Beltraminelli H, et al. Detection of linear IgE deposits in bullous pemphigoid and mucous membrane pemphigoid: A useful clue for diagnosis. Br J Dermatol 2011; 165:1133–7.
50 Kridin K. Peripheral eosinophilia in bullous pemphigoid: Prevalence and influence on the clinical manifestation. Br J Dermatol 2018 Nov;179(5):1141-1147.
51 Lamberts A, Meijer JM, Jonkman MF. Nonbullous pemphigoid: A systematic review. J Am Acad Dermatol 2018 May;78(5):989-995.e2.
52 Kuo D, Ding J, Cohn IS, et al. HBEGF(+) macrophages in rheumatoid arthritis induce fibroblast invasiveness. Sci Transl Med 2019 May 8;11(491). pii: eaau8587.

128
doi:10.1126/scitranslmed.aau8587. 53 Qing X, Chinenov Y, Redecha P, et al. iRhom2 promotes lupus nephritis through TNF-alpha and
EGFR signaling. J Clin Invest 2018; 128:1397–412. 54 Singh B, Carpenter G, Coffey RJ. EGF receptor ligands: recent advances. 2016 Sep 8;5. pii: F1000
Faculty Rev-2270. doi:10.12688/f1000research.9025.1. 55 Yamane S, Ishida S, Hanamoto Y, et al. Proinflammatory role of amphiregulin, an epidermal
growth factor family member whose expression is augmented in rheumatoid arthritis patients. J Inflamm (Lond) 2008; 5:5.
56 Zebrowska A, Wagrowska-Danilewicz M, Danilewicz M, et al. Does Adam17 cause the destruction of anchoring fibers via shedding tumor necrosis factor alpha in bullous pemphigoid and dermatitis herpetiformis? J. Cutan. Med. Surg. 2012; 16:149–50.
57 Liu Y, Peng L, Li L, et al. TWEAK/Fn14 Activation Contributes to the Pathogenesis of Bullous Pemphigoid. J Invest Dermatol 2017; 137:1512–22.
58 Schmidt E, Reimer S, Kruse N, et al. Autoantibodies to BP180 associated with bullous pemphigoid release interleukin-6 and interleukin-8 from cultured human keratinocytes. J Invest Dermatol 2000; 115:842–8.
59 Tukaj S, Gruner D, Zillikens D, Kasperkiewicz M. Hsp90 blockade modulates bullous pemphigoid IgG-induced IL-8 production by keratinocytes. Cell Stress Chaperones 2014; 19:887–94.
60 Kowalski EH, Kneibner D, Kridin K, Amber KT. Serum and blister fluid levels of cytokines and chemokines in pemphigus and bullous pemphigoid. Autoimmun Rev 2019; 18:526–34.
61 Engmann J, Rüdrich U, Behrens G, et al. Increased Activity and Apoptosis of Eosinophils in Blister Fluids, Skin and Peripheral Blood of Patients with Bullous Pemphigoid. Acta Derm Venereol 2017; 97:464–71.
62 Tabatabaei-Panah P-S, Moravvej H, Sadaf Z, et al. Proinflammatory Cytokine Gene Polymorphisms in Bullous Pemphigoid. Front Immunol 2019; 10:636.
63 Mitchell JA, Warner TD. COX isoforms in the cardiovascular system: understanding the activities of non-steroidal anti-inflammatory drugs. Nat Rev Drug Discov 2006; 5:75–86.
64 Apap C, Boffa MJ, Cordina J. Celecoxib-induced bullous pemphigoid. Skinmed. 2013; 11:190–1. 65 Wang Q, He Y, Shen Y, et al. Vitamin D inhibits COX-2 expression and inflammatory response by
targeting thioesterase superfamily member 4. J Biol Chem 2014; 289:11681–94. 66 Marzano A V, Trevisan V, Eller-Vainicher C, et al. Evidence for vitamin D deficiency and
increased prevalence of fractures in autoimmune bullous skin diseases. Br J Dermatol 2012; 167:688–91.
67 Marzano AV, Trevisan V, Cairoli E, et al. Vitamin D and skeletal health in autoimmune bullous skin diseases: a case control study. Orphanet J Rare Dis 2015; 10:8.
68 Sarre ME, Annweiler C, Legrand E, et al. Association between bullous pemphigoid and hypovitaminosis D in older inpatients: Results from a case-control study. Eur J Intern Med 2016; 31:25–8.
69 Sarre ME, Riou J, Duval GT, et al. Association between serum 25-hydroxyvitamin D concentration and severity of first-diagnosed bullous pemphigoid in older adults. Arch Gerontol Geriatr 2019; 83:28–30.
70 Tukaj S, Gruner D, Tukaj C, et al. Calcitriol exerts anti-inflammatory effects in keratinocytes treated with autoantibodies from a patient with bullous pemphigoid. J Eur Acad Dermatol Venereol 2016; 30:288–92.
71 Agrawal U, Kumari N, Vasudeva P, et al. Overexpression of COX2 indicates poor survival in urothelial bladder cancer. Ann Diagn Pathol 2018; 34:50–5.
72 Tong D, Liu Q, Wang L-A, et al. The roles of the COX2/PGE2/EP axis in therapeutic resistance. Cancer Metastasis Rev 2018; 37:355–68.

129
73 Friedrich M, Reichert K, Woeste A, et al. Effects of Combined Treatment with Vitamin D and COX2 Inhibitors on Breast Cancer Cell Lines. Anticancer Res 2018; 38:1201–7.
74 Hatano Y, Katagiri K, Arakawa S, et al. Successful treatment by double-filtration plasmapheresis of a patient with bullous pemphigoid: effects in vivo on transcripts of several genes for chemokines and cytokines in peripheral blood mononuclear cells. Br J Dermatol 2003; 148:573–9.
75 Nakashima H, Fujimoto M, Asashima N, et al. Serum chemokine profile in patients with bullous pemphigoid. Br J Dermatol 2007; 156:454–9.
76 Gounni Abdelilah S, Wellemans V, Agouli M, et al. Increased expression of Th2-associated chemokines in bullous pemphigoid disease. Role of eosinophils in the production and release of these chemokines. Clin Immunol 2006; 120:220–31.
77 Rauen T, Hedrich CM, Tenbrock K, Tsokos GC. cAMP responsive element modulator: a critical regulator of cytokine production. Trends Mol Med 2013; 19:262–9.
78 Juang Y-T, Wang Y, Solomou EE, et al. Systemic lupus erythematosus serum IgG increases CREM binding to the IL-2 promoter and suppresses IL-2 production through CaMKIV. J Clin Invest 2005; 115:996–1005.
79 Ohl K, Wiener A, Lippe R, et al. CREM Alpha Enhances IL-21 Production in T Cells In Vivo and In Vitro. Front Immunol 2016; 7:618.
80 Arakawa M, Dainichi T, Ishii N, et al. Lesional Th17 cells and regulatory T cells in bullous pemphigoid. Exp. Dermatol. 2011; 20:1022–4.
81 Vesely PW, Staber PB, Hoefler G, Kenner L. Translational regulation mechanisms of AP-1 proteins. Mutat Res 2009; 682:7–12.
82 Trop-Steinberg S, Azar Y. AP-1 Expression and its Clinical Relevance in Immune Disorders and Cancer. Am J Med Sci 2017; 353:474–83.
83 Yagami A, Orihara K, Morita H, et al. IL-33 mediates inflammatory responses in human lung tissue cells. J Immunol 2010; 185:5743–50.
84 Wakatabi K, Komine M, Meephansan J, et al. The levels of soluble ST2 in sera and bullous fluid from patients with bullous pemphigoid. Eur J Dermatol 2012; 22:333–6.
85 Margiotta DPE, Navarini L, Vadacca M, et al. The IL33/ST2 axis in Sjogren syndrome in relation to disease activity. Eur Rev Med Pharmacol Sci 2016; 20:1295–9.
86 Mok MY, Huang FP, Ip WK, et al. Serum levels of IL-33 and soluble ST2 and their association with disease activity in systemic lupus erythematosus. Rheumatology (Oxford) 2010; 49:520–7.
87 Okajima F. Regulation of inflammation by extracellular acidification and proton-sensing GPCRs. Cell Signal 2013; 25:2263–71.
88 Onozawa Y, Fujita Y, Kuwabara H, et al. Activation of T cell death-associated gene 8 regulates the cytokine production of T cells and macrophages in vitro. Eur J Pharmacol 2012; 683:325–31.
89 Onozawa Y, Komai T, Oda T. Activation of T cell death-associated gene 8 attenuates inflammation by negatively regulating the function of inflammatory cells. Eur J Pharmacol 2011; 654:315–9.
90 Kelwick R, Desanlis I, Wheeler GN, Edwards DR. The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin motifs) family. Genome Biol 2015; 16:113.

130

131
PART 2
Management of pemphigoid diseases

132
8

133
CHAPTER 8 Unmet needs in pemphigoid diseases: an international survey amongst patients, clinicians and researchers Aniek Lamberts1, Marc Yale2, Sergei A. Grando3, Barbara Horváth1, Detlef Zillikens4, Marcel F. Jonkman1
Affiliations 1 Center for Blistering Diseases, Department of Dermatology, University Medical Center Groningen, University of Groningen, Groningen, The Netherlands 2 International Pemphigus and Pemphigoid Foundation, Sacramento, United States of America 3 Department of Dermatology, University of California, UC Irvine Medical Center, Irvine, United States of America 4 Department of Dermatology, University of Lübeck, University Medical Center Schleswig-Holstein, Lübeck, Germany Published in Acta Dermato-Venereology, 2019 Feb 1;99(2):224-225

134
Introduction Pemphigoid diseases are subepidermal autoimmune bullous diseases, characterized by autoantibodies against structural proteins of the dermal-epidermal junction.1 Symptoms of severe pruritus, with or without tense blistering of the skin or mucosa, cause a high disease burden.2 Basic and clinical research led to better understanding of disease mechanisms, and novel therapies emerged.3 Nonetheless, gaps in knowledge exist, and several disease areas are understudied. These unmet needs have not been well-characterized. While in research available time and resources are often limited, it is essential to address research topics relevant to both patients and health care professionals.4 Nowadays, it is widely recognized that patients play an important role in setting the research agenda.5 This study sought to explore and prioritize unmet needs in pemphigoid diseases from the perspective of patients, clinicians and researchers, with the intent to guide future research towards important research topics. A secondary aim was to identify points of improvement in patient care.
Methods A steering group was established in February 2017, consisting of a project coordinator (AL), experts on pemphigoid diseases (SAG, BH, DZ, MFJ) and a patient representative (MY; director of the International Pemphigus and Pemphigoid Foundation (IPPF)). A preliminary list of unmet needs was composed and discussed by the steering group during a kick-off meeting in June 2017 that took place at the IPPF conference in Lübeck. An online anonymous survey was developed using Qualtrics survey software (supplement 1), containing questions on participants characteristics, and unmet needs in pemphigoid diseases. Seven or eight pre-listed needs were provided and participants were asked whether they recognized the needs as unmet, to designate a top three of the most urgent unmet needs, and to complement the list. In addition, patients received questions about patient care satisfaction, and their reasons for (dis)satisfaction.
The survey was distributed internationally between October 2017 and April 2018. Patients were invited by email via the International Pemphigus and Pemphigoid Foundation, and national German and Dutch patient organizations. Clinicians and researchers were invited by email via pemphigoid research groups, and via the European Academy of Dermatology and Venereology. Data was exported from Qualtrics directly into SPSS Statistics version 23 (IBM, Chicago, USA).

135
Descriptive and qualitative statistics were used for data analysis. An overall ranking score was calculated by awarding three points every time unmet needs were ranked highest, two points if ranked second, and one point if ranked third.
Results The in- and exclusion process is shown in figure 1. The clinicians and researchers response rate was 36/99 (36%). The patients response rate was unknown.
175 people visited the survey
26 people discontinued before answering the first question
42 participants were excluded 21 pemphigus patients 2 patients filled in the survey twice 19 participants ended the survey before answering an unmet need related question
107 participants completed 134 surveys 71 patients:
- 52 BP - 17 MMP - 2 EBA
35 clinicians 28 researchers
149 participants
Figure 1. Flowchart of in- and exclusion of study participants. BP, bullous pemphigoid; MMP, mucous membrane pemphigoid; EBA, epidermolysis bullosa acquisita.

136
Participants characteristics and the top three of most urgent needs are displayed in table 1. Patients, clinicians and researchers agreed that the most urgent need is the improvement of therapeutic options for pemphigoid diseases (table 1). Additionally, patients expressed the need for more public information most frequently (n=9) (supplemental table 1).
Data on patient satisfaction showed that half of the patients were unsatisfied with patient care during the diagnostic process, mainly due to misdiagnosis and long doctors delay (mentioned by 88% of unsatisfied patients; table 2). Six patients visited more than five doctors before a correct diagnosis was made. Patients with epidermolysis bullosa acquisita and mucous membrane pemphigoid reported a longer diagnostic delay (mean 90.3 ±127 and 19.7 ±23 months), compared to patients with bullous pemphigoid (9.0 ±22 months). Most patients (76%) were satisfied with current patient care, especially due to successful treatment (mentioned by 41% of satisfied patients) in centers of expertise (mentioned by 37% of satisfied patients) (table 2). Treatment side effects, insurance issues, and poor disease knowledge by doctors were main reasons for unsatisfactory current patient care.
Discussion Our survey data confirmed that a long diagnostic delay and suboptimal treatment are important concerns in pemphigoid diseases. Though our results were not surprising, this study is the first to explore patients priorities in the field of pemphigoid diseases. Our method has some resemblance to the James Lind Alliance (JLA) methodology of prioritisation of research topics.5 The greatest difference is the lack of a finalisation workshop, where patients and health professionals discuss the final prioritisation of uncertainties face-to-face. In our study we choose for prioritisation by survey, considering that pemphigoid diseases are rare, and therefore a low attendance and a high geographic selection bias would be expected. Still, the risk of selection bias was not completely prevented, as participants from only three continents were included. Other limitations of this study include a relatively low sample size and missing values. Geographical differences might have caused small deviations in the ranked needs (supplemental tables 2, 3, 4).

137
Table 1. Participants characteristics and top three ranking of unmet needs in pemphigoid diseases. Participants characteristics (n=107) Patients (n=71) Clinicians (n=35) Researchers (n=28) Mean age: 66.6 years (R 34-94) Mean diagnostic delay: 14.1 months (R 1 day-15 years) Mean disease duration: 4.7 years (R 0-20)
Mean age 51.9 (R 28-65) Setting: Academic 34 (97%) Peripheral 1 (3%) Consider yourself AIBD expert? Yes: 35 (100%)
Mean age 51.6 (R 28-65) Research experience: >10 years: 20 (71%) AIBD main research topic? Yes: 21 (75%), No: 7 (25%)
Continent of origin: Europe: 22 (31%) Northern America: 48 (68%) Asia: 1 (1%)
Continent of origin: Europe: 21 (60%) Northern America: 7 (20%) Asia: 4 (11%)
Continent of origin: Europe: 18 (64%) Northern America: 6 (21%) Asia: 4 (14%)
Overall top three ranking of unmet needs
Unmet need recognized? Ranking score*
Yes, n (%)
No, n (%)
Missing, n (%)
Patients (n=71)
1. Need for better treatment options
48 (68) 5 (7) 18 (25) 92 2. Need for quicker diagnosis
47 (66) 9 (13) 15 (21) 84
3. Need for more disease awareness
46 (65) 8 (11) 17 (24) 81 Clinicians (n=35)
1. Need for labeling of new drugs for the indication pemphigoid (anti- CD20, anti-complement, anti-FcRn, anti-neutrophil activating pathways)
30 (86) 4 (11) 1 (3) 53
2. Need for easy laboratory tests to diagnose pemphigoid diseases
27 (77) 5 (14) 3 (9) 38
3. Need for better recognition of nonbullous pemphigoid
30 (86) 3 (9) 2 (6) 37
Researchers (n=28)
1. Need for more head-to-head randomized controlled trials comparing the effectiveness and safety of current treatments
25 (89) 1 (4) 2 (7) 51
2. Need for understanding of the pathophysiology of pemphigoid for drug development
27 (96) 0 (0) 1 (4) 38
3. Need for understanding trigger mechanism (e.g., infections, drugs) in addition to genetic predisposition
27 (96) 0 (0) 1 (4) 30
R, range; AIBD, autoimmune blistering diseases; * Ranking score was calculated by awarding three points for every time an unmet needs was ranked highest, two points if ranked second highest and one point if ranked third highest.

138
Table 2. Satisfaction of patients (n=71) with patient care during the diagnostic process, and their current patient care.
Patients satisfied with diagnostic process (n=36, 51%)
Patients unsatisfied with diagnostic process (n=35, 49%)
Reasons categorized Times mentioned
Reasons categorized
Times mentioned
Quick diagnosis 10 Misdiagnosis/mistreatment - lack of recognition of nonbullous variant (n=2)
31 Good disease recognition 5
Good awareness 3 High number of doctors seen before diagnosis
6
Disease information provided
3 Lack of disease knowledge 5
Good treatment 3 Symptoms were not taken seriously 2 Good disease knowledge 2 Side effects of steroids 2 Doctor was determinate after unspecific results of biopsy
2 Long waiting time for referral to specialist
2
Lack of information 2
Doctor induced bullous pemphigoid by drug prescription
1
Poor treatment 1 Many referrals, thereby high costs of
care 1
No biopsy was taken 1 Patients satisfied with current patient care (n=54, 76%)
Patients unsatisfied with current patient care (n=17, 24%)
Reasons categorized
Times mentioned
Reasons categorized
Times mentioned
Satisfied with therapy/ in remission
22 Side effects - steroids (n=3)/ other therapy (n=1)
5
Experienced/professional dermatologist/care center
20 Little disease knowledge by doctor 5
Happy with correct diagnosis
3 Insurance issues - insurance does not cover therapy
(n=3) - specialist did not accept the
medical insurance (n=1)
4
Disease information provided
2
Not satisfied with interaction with specialist
2
No information/support for patients in remission still dealing with skin issues
1
No satisfying treatment for itching 1 BP, bullous pemphigoid.

139
Patients from Northern America ranked the need for better treatment availability fourth, in contrast to European patients that rated the urgency of this need second last. This might be explained by lower health care availability in Northern America than in Europe (supplemental table 2).
Another interesting finding is the higher need for easy diagnostic laboratory tests expressed by clinicians in Northern America and Asia, in comparison with European clinicians (supplemental table 3). Whether this difference is caused by approachability, laboratory equipment, or by the use of different diagnostic techniques cannot be concluded based on our survey data.
In conclusion, our data shows that future studies are needed to improve and widen the currently available treatment options for pemphigoid diseases. Moreover, patients point out a high need for shortening the diagnostic delay. Therefore, more awareness for pemphigoid diseases should be pursued. Data on patient satisfaction showed that after the correct diagnosis was established, patients were most satisfied with care in centers of expertise. Focus group sessions might be useful to provide extended information in order to formulate concrete interventions for patient care improvement. References 1. Schmidt E, Zillikens D. Pemphigoid diseases. Lancet 2013; 381: 320-332. 2. Kouris A, Platsidaki E, Christodoulou C, Armyra K, Korkoliakou P, Stefanaki C, et al. Quality of life,
depression, anxiety and loneliness in patients with bullous pemphigoid. A case control study. An Bras Dermatol 2016; 91: 601-603.
3. Antonicelli F, Ludwig RJ. New insights into pemphigoid diseases. Exp Dermatol 2017; 26: 1151-1153.
4. Tallon D, Chard J, Dieppe P. Relation between agendas of the research community and the research consumer. Lancet 2000; 355: 2037-2040.
5. NETSCC & Cowan K. The James Lind Alliance Guidebook 2016; Available from: URL: http://www.jla.nihr.ac.uk/jla-guidebook/downloads/JLA-Guidebook-Version-6-February-2016.pdf
Acknowledgements The authors would like to thank all participants that completed the survey.

140
Supplemental tables Supplemental table 1. Additional unmet needs in pemphigoid diseases by patients, clinicians and researchers
Categorized additional unmet needs in pemphigoid diseases of patients Times
mentioned, n Need for more (public) information 9
Need for more awareness amongst doctors/medical staff 7
Need for better disease knowledge by doctors 7
Need for better health care organization 5
Need for attention and care for intense itching post BP (after blistering stage; after remission)
4
Need for better medicine 4
Need for more information/guidance 4
Need for research 4
Need for more local support groups 2
Need for guidance of frail elderly population (caregiver to help with nutrition/self-care)
1
Categorized additional unmet needs in pemphigoid diseases of clinicians Times
mentioned, n Need for better therapies: 11
Need for resolving issues with coverage of medication by insurance companies 4
Need for better diagnostics: 3
Need for a centralized national serum database/DNA bank and need for registries 2
Need for nosology for linear IgA disease and pemphigoid (not a synonym for bullous pemphigoid)
1
Need for guideline for diagnosis/treatment of MMP and treatment of BP 1
Need for better education of patients on their disease 1
Need for transfer of knowledge to GP’s on the nonbullous stages of disease 1
Need for access to nursing support in outpatient setting 1
Categorized additional unmet needs in pemphigoid diseases of researchers Times
mentioned, n Need for animal/ ex-vivo model of human pemphigoid 2
Need for research on pemphigoid diseases pathogenesis 2
Need for research on brain and skin associations in BP 2
Need for research; why is BP a self-limiting disorder 1
Need for research on cytokine network in amplifying blistering development 1
BP, bullous pemphigoid; RCT, randomized controlled trial; DNA, deoxyribonucleic acid; MMP, mucous membrane pemphigoid; GP, general practitioner.

141
Supplemental table 2. Patients’ ranking of unmet needs in pemphigoid diseases per continent
Unmet need recognized? Ranking score* Yes,
n (%) No,
n (%) Missing,
n (%) Europe (n=22)
1. Need for better treatment options 16 (73) 3 (14) 3 (14) 34
2. Need for awareness 15 (68) 6 (27) 1 (5) 30
3. Need for quicker diagnosis 15 (68) 6 (27) 1 (5) 29
4. Need for guidance of psychological impact 14 (64) 7 (32) 1 (5) 24
5. Need for easy assessable information 12 (55) 9 (41) 1 (5) 21
6. Need for knowledge on impact on comorbidities 15 (68) 5 (23) 2 (9) 16
7. Need better treatment availability 8 (36) 13 (59) 1 (5) 8
8. Need for self-assessment tool 9 (41) 12 (55) 1 (5) 7
Northern America (n=48)
1. Need for better treatment options 31 (65) 2 (4) 15 (31) 54
2. Need for quicker diagnosis 31 (65) 3 (6) 14 (29) 52
3. Need for awareness 30 (63) 2 (4) 16 (33) 45
4. Need better treatment availability 19 (40) 4 (8) 25 (52) 33
5. Need for easy assessable information 22 (46) 5 (10) 21 (44) 32
6. Need for self-assessment tool 19 (40) 4 (8) 25 (52) 22
7. Need for guidance of psychological impact 18 (38) 5 (10) 25 (52) 21
8. Need for knowledge on impact on comorbidities 21 (44) 3 (6) 24 (50) 16
Asia (n=1)
No preference noted
* Ranking score was calculated by awarding three points for every time an unmet needs was ranked highest, two points if ranked second highest and one point if ranked third highest.

142
Supplemental table 3. Clinicians’ ranking of unmet needs in pemphigoid diseases per continent
Unmet need recognized? Ranking
score* Yes, n (%)
No, n (%)
Missing, n (%)
Europe (n=21) 1. Need for labeling of new drugs for the indication pemphigoid 16 (76) 4 (19) 1 (5) 31 2. Need for better recognition of nonbullous cutaneous pemphigoid 17 (81) 3 (14) 1 (5) 24 3. Need for a multidisciplinary approach: building multidisciplinary teams 15 (71) 4 (19) 2 (10) 17 3. Need for easy laboratory tests to diagnose pemphigoid diseases 13 (62) 5 (24) 3 (14) 17 4.Need for consensus on minimal requirements for diagnosis of pemphigoid 16 (76) 4 (19) 1 (5) 16 5. Need for definition of the treatment goal at certain time points during treatment (e.g. at 3 months)
14 (67) 5 (24) 2 (10) 13
6. Need for standardized use of PROMS during treatment 15 (71) 4 (19) 2 (10) 11 7. Need for low threshold for detection of autoantibodies in referral laboratories
12 (57) 7 (33) 2 (10) 0
Asia (n=4)
1. Need for easy laboratory tests to diagnose pemphigoid diseases 4 (100) 0 (0) 0 (0) 7
1. Need for a multidisciplinary approach: building multidisciplinary teams 4 (100) 0 (0) 0 (0) 7
2. Need for labeling of new drugs for the indication pemphigoid 4 (100) 0 (0) 0 (0) 6
3.Need for consensus on minimal requirements for diagnosis of pemphigoid 4 (100) 0 (0) 0 (0) 5
4. Need for better recognition of nonbullous cutaneous pemphigoid 4 (100) 0 (0) 0 (0) 4 5. Need for definition of the treatment goal at certain time points during treatment (e.g. at 3 months) 4 (100) 0 (0) 0 (0) 3
6. Need for standardized use of PROMS during treatment 4 (100) 0 (0) 0 (0) 3 7. Need for low threshold for detection of autoantibodies in referral laboratories
3 (75) 0 (0) 1 (25) 2
Northern America (n=7)
1. Need for labeling of new drugs for the indication pemphigoid 7 (100) 0 (0) 0 (0) 14
2. Need for easy laboratory tests to diagnose pemphigoid diseases 7 (100) 0 (0) 0 (0) 9
3. Need for better recognition of nonbullous cutaneous pemphigoid 6 (86) 0 (0) 1 (14) 7 4. Need for low threshold for detection of autoantibodies in referral laboratories
5 (71) 2 (29) 0 (0) 4
4.Need for consensus on minimal requirements for diagnosis of pemphigoid 5 (71) 2 (29) 0 (0) 4 4. Need for definition of the treatment goal at certain time points during treatment (e.g. at 3 months) 5 (71) 2 (29) 0 (0) 4
5. Need for a multidisciplinary approach: building multidisciplinary teams 3 (43) 4 (57) 0 (0) 3
6. Need for standardized use of PROMS during treatment 5 (71) 2 (29) 0 (0) 0 PROMS, patient reported outcome measurements.* Ranking score was calculated by awarding three points for every time an unmet needs was ranked highest, two points if ranked second highest and one point if ranked third highest.

143
Supplemental table 4 – part 1. Researchers’ ranking of unmet needs in pemphigoid diseases per continent
Unmet need recognized? Ranking
score* Yes, n (%)
No, n (%)
Missing, n (%)
Europe (n=18) 1. Need for more head-to-head randomized controlled trials comparing the effectiveness and safety of current treatments
15 (83) 1 (6) 2 (11) 33
2. Need for understanding of the pathophysiology of pemphigoid for drug development
17 (94) 0 (0) 1 (6) 26
3.Need for understanding trigger mechanism (e.g., infections, drugs) in addition to genetic predisposition 17 (94) 0 (0) 1 (6) 20
4. Need for studies on personalized treatment based on patients characteristics
15 (83) 1 (6) 2 (11) 16
5. Need for humanized animal models that simulate the human eosinophilic pathogenesis and the interactions between human IgG/IgA/IgE with human Fc receptors
11 (73) 3 (17) 4 (22) 10
6. Need for animal models for studying the break of tolerance to desmosomal and hemidesmosomal constituents
12 (67) 3 (17) 3 (17) 5
7. Need for consensus on BPDAI cut-off values for staging disease severity 13 (72) 3 (17) 2 (11) 4
Asia (n=4)
1. Need for more head-to-head randomized controlled trials comparing the effectiveness and safety of current treatments
4 (100) 0 (0) 0 (0) 11
2. Need for studies on personalized treatment based on patients characteristics 4 (100) 0 (0) 0 (0) 6
2. Need for animal models for studying the break of tolerance to desmosomal and hemidesmosomal constituents
4 (100) 0 (0) 0 (0) 6
3. Need for consensus on BPDAI cut-off values for staging disease severity 4 (100) 0 (0) 0 (0) 4 4.Need for understanding trigger mechanism (e.g., infections, drugs) in addition to genetic predisposition
4 (100) 0 (0) 0 (0) 3
4. Need for understanding of the pathophysiology of pemphigoid for drug development
4 (100) 0 (0) 0 (0) 3
4. Need for humanized animal models that simulate the human eosinophilic pathogenesis and the interactions between human IgG/IgA/IgE with human Fc receptors
3 (75) 0 (0) 1 (25) 3
BPDAI, bullous pemphigoid disease activity index. * Ranking score was calculated by awarding three points for every time an unmet needs was ranked highest, two points if ranked second highest and one point if ranked third highest.
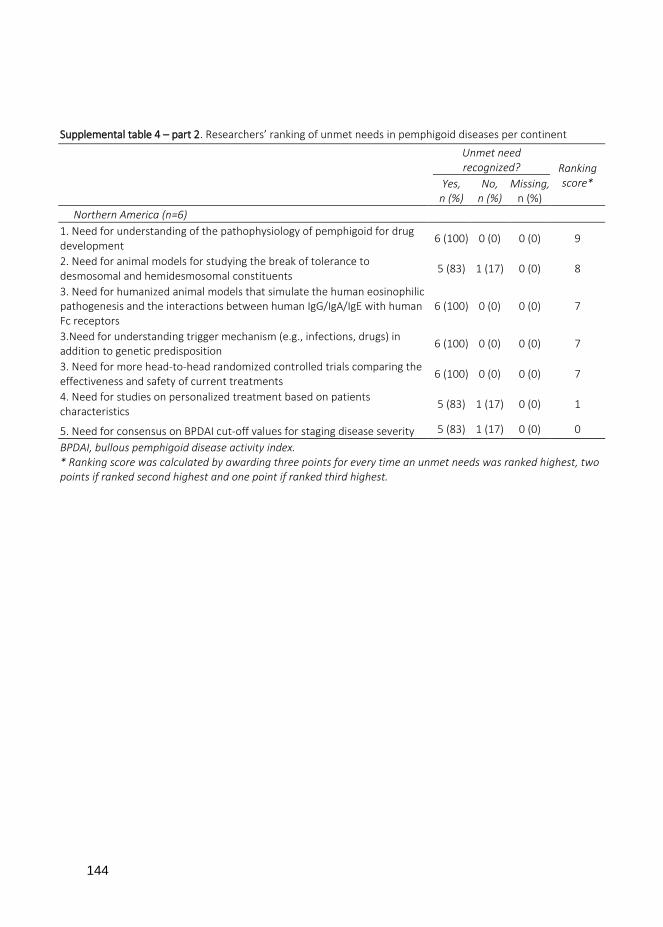
144
Supplemental table 4 – part 2. Researchers’ ranking of unmet needs in pemphigoid diseases per continent
Unmet need recognized? Ranking
score* Yes, n (%)
No, n (%)
Missing, n (%)
Northern America (n=6) 1. Need for understanding of the pathophysiology of pemphigoid for drug development
6 (100) 0 (0) 0 (0) 9
2. Need for animal models for studying the break of tolerance to desmosomal and hemidesmosomal constituents
5 (83) 1 (17) 0 (0) 8
3. Need for humanized animal models that simulate the human eosinophilic pathogenesis and the interactions between human IgG/IgA/IgE with human Fc receptors
6 (100) 0 (0) 0 (0) 7
3.Need for understanding trigger mechanism (e.g., infections, drugs) in addition to genetic predisposition
6 (100) 0 (0) 0 (0) 7
3. Need for more head-to-head randomized controlled trials comparing the effectiveness and safety of current treatments
6 (100) 0 (0) 0 (0) 7
4. Need for studies on personalized treatment based on patients characteristics
5 (83) 1 (17) 0 (0) 1
5. Need for consensus on BPDAI cut-off values for staging disease severity 5 (83) 1 (17) 0 (0) 0
BPDAI, bullous pemphigoid disease activity index. * Ranking score was calculated by awarding three points for every time an unmet needs was ranked highest, two points if ranked second highest and one point if ranked third highest.

145

146
9

147
CHAPTER 9 Effectiveness and safety of rituximab in recalcitrant pemphigoid diseases Aniek Lamberts, H. Ilona Euverman, Jorrit B. Terra, Marcel F. Jonkman, Barbara Horváth Center for Blistering Diseases, Department of Dermatology, University Medical Center Groningen, University of Groningen, Groningen, The Netherlands Published in the Frontiers in Immunology, 2018 Feb 19;9:248

148
Abstract Introduction Rituximab (RTX) is a monoclonal antibody targeting CD20, a transmembrane protein expressed on B cells, causing B cell depletion. RTX has shown great efficacy in studies of pemphigus vulgaris, but data of pemphigoid diseases are limited. Objective To assess the effectiveness and safety of RTX in pemphigoid diseases. Methods The medical records of 28 patients with pemphigoid diseases that were treated with RTX were reviewed retrospectively. Early and late endpoints, defined according to international consensus, were disease control (DC), partial remission (PR), complete remission (CR) and relapses. Safety was measured by reported adverse events. Results Patients with bullous pemphigoid (n=8), mucous membrane pemphigoid (n=14), epidermolysis bullosa acquisita (n=5) and linear IgA disease (n=1) were included. Treatment with 500mg RTX (n=6) or 1000mg RTX (n=22) was administered on day 1 and 15. Eight patients received additional 500mg RTX at month 6 and 12. Overall, DC was achieved in 67.9%, PR in 57.1%, and CR in 21.4% of the cases. During follow-up 66.7% patients relapsed. Repeated treatment with RTX led to remission (PR or CR) in 85.7% of the retreated cases. No significant difference in response between pemphigoid subtypes was found. IgA-dominant cases (n=5) achieved less DC (20% vs. 81.3%; p=0.007), less PR (20% vs. 62.5%; p=0.149) and less CR (0% vs. 18.8%; p =0.549) compared to IgG-dominant cases (n=16). Five severe adverse events and three deaths were reported. One death was possibly related to RTX and one death was disease related. Conclusion RTX can be effective in recalcitrant IgG-dominant pemphigoid diseases, however not in those where IgA is dominant.

149
Introduction Pemphigoid diseases are a heterogeneous group of autoantibody mediated subepidermal blistering diseases.1 IgG, IgA or IgM autoantibodies target distinct antigens located in the basement membrane zone (BMZ) inducing different pemphigoid subtypes. Cutaneous pemphigoid is the subgroup of pemphigoid diseases that predominantly affect the skin.1,2 Pemphigoid, be it nonbullous or bullous (BP), is the most prevalent disease within this subgroup and mainly presents at older age.3 Mucous membrane pemphigoid (MMP) opposes cutaneous pemphigoid in the spectrum of pemphigoid diseases, and is characterized by primary involvement of the mucosa.2,4 Beside the classification based on body localization, pemphigoid diseases may be classified based on targeted auto-antigens, such as 180 kDa BP antigen (BP180) and the 230 kDa BP antigen (BP230) in pemphigoid, laminin-332 in anti-laminin-332 MMP, the p200 protein in anti-p200 pemphigoid (anti-laminin γ1 pemphigoid), and type VII collagen in epidermolysis bullosa acquisita (EBA).2 Last, classification may be based on predominant class of autoantibodies IgG or IgA. Pemphigoid diseases with the exclusive IgA involvement are named linear IgA disease (LAD), regardless of the targeted antigen or clinical presentation.5 IgA-mediated pemphigoid diseases are difficult to treat if dapsone is contraindicated, and mostly show high resistance to usual immunosuppressants.6
The 2014 European consensus guideline for the management of BP recommends transcutaneous systemic clobetasol therapy as initial treatment.7,8 The alternative is oral systemic prednisolon therapy (0.5-1.0 mg/kg/day), which was associated with adverse events and higher mortality.8,9 Recently, doxycycline was found to be non-inferior to and safer than prednisolone for short-term blister control10, although the statistical margins were wide.11 As third line rituximab (RTX) is recommended in cases in which conventional immunosuppressive drugs were not effective, were contraindicated, or showed unacceptable side effects.12-14
RTX is a chimeric monoclonal antibody targeting CD20, a transmembrane protein expressed by all B cells in the pre-plasma cell lineage.15 Binding of RTX to CD20 leads to B cell depletion in the peripheral circulation by various mechanisms.16 RTX is registered for treatment of B cell lymphoma’s, rheumatoid arthritis (RA) and granulomatosis with polyangiitis.16,17 Recently RTX was shown to be effective as first line therapy in pemphigus.18 However, the position of RTX on the therapeutically ladder of pemphigoid diseases is unknown. Data regarding the

150
effectiveness and safety of RTX in pemphigoid diseases are limited and are mainly of retrospective nature.19-27 Furthermore, it is unclear which treatment regime is most beneficial and which factors might be predictive for treatment response.28,29 Therefore the aim of our study was to retrospectively analyze our daily practice experience with RTX in pemphigoid diseases by evaluating the effectiveness and safety, and to identify clinical or serological factors that might be associated with treatment response. Materials and Methods All patients with pemphigoid diseases treated with RTX between 2010 and September 2017 at the Center for Blistering Diseases at the University Medical Center Groningen were included in the study. Pemphigoid diseases were diagnosed based on the following criteria: linear depositions of IgG, IgA, IgM or C3c along the BMZ by direct immunofluorescence microscopy (DIF) and/or positive indirect immunofluorescence microscopy (IIF) on monkey esophagus (MO) or salt-split skin (SSS), in combination with clinical presentation, histopathological findings, or other immunoserological tests compatible with the diagnosis of a pemphigoid disease. Patients with a linear u-serrated immunodeposition pattern seen by DIF were diagnosed with EBA. Patients with exclusive involvement of IgA were diagnosed with LAD.
Patients charts were reviewed retrospectively by the first (AL) and second (HIE) authors. Response outcomes were defined according to international consensus and measured by the early endpoint disease control (DC), and the late endpoints partial remission (PR), complete remission (CR) and the number of relapses.30,31 Safety was measured by reported adverse events. Discrepancies in the assessment by AL and HIE were resolved through discussion with the other authors (BH, MJ). Treatment regimes There were two treatment protocols administered. In the period of 2010-2012 patients were treated with RTX 500mg at day 1 and 15 (low dose RA protocol), since this dose was effective in pemphigus patients (Horvath et al. 2011).32,33 Additional 500mg RTX at month 6 and/or 12 was only administered on indication.34 From 2012 the protocol was adjusted to 1000mg RTX at day 1 and 15 (high dose RA protocol – published in 2011).29 Since 2014 patients standardly received additional

151
500mg RTX at month 6 and 12, and if indicated at month 18. Patients that relapsed within one year after the last RTX infusion received re-treatment with a single infusion of 500mg RTX. Relapsed patients beyond one year after the last RTX infusion were retreated with a new cycle of 1000mg RTX at day 1 and 15. Statistical analysis The Kolmogorov–Smirnov test was used to test for normal distributions. Correlations between bivariate outcome measures were analyzed with Fisher’s exact test. Comparing means of non-normally distributed data was done with the Mann-Whitney U test. Statistical significance was defined by a p-value <0.05. Statistical analyses were performed in IBM SPSS statistics version 23. Results Patient population A total of 28 patients were included. The patient characteristics are listed in table 1. The mean delay in diagnosis was 10.5 months in BP (range 1-19), 24.3 months in MMP (range 4-60) and 19.0 months in EBA patients (range 3-47). One MMP outlier with an exceptional long delay in diagnosis of 285 months was not taken into account. This patient showed severe laryngeal, oral, genital and ocular (foster stage 4) involvement. The mean time between diagnosis and RTX treatment was longer for BP patients (64.3 months; range 1-272), EBA (29.1 months; range 0.5-84) and LAD patients (49.0 months) compared to MMP (13.8 months; range 2-63). Prior to RTX all patients received one or more immunosuppressants (supplement 1) with suboptimal effect or with unacceptable side effects. Therefore, RTX was administered as last resort in several cases. Six patients received low dose RTX (500mg) and 22 patients high dose (1000mg), of which eight patients also received repeated RTX doses (500mg) at month 6 and 12. All patients also received a local steroid and/or one or two systemic drugs (supplement 1). Effectiveness of first course of RTX DC was achieved in 21 of 28 patients (75%) at a mean time of 14.5 weeks (range 1-36; sd 9.1). Remission (partial or complete) was achieved by 57.1% (n=16) of the treatment resistant pemphigoid cases (figure 1 and 2). PR was achieved by 16 patients (57.1%) at a mean time of 34.2 weeks (range 9-71; sd 18.1). Six of 28

152
patients (21.4%) also achieved CR at a mean time of 59.2 weeks (range 24-85; sd 22.1). Table 1. Demographics of pemphigoid patients treated with rituximab (RTX)
Mean age at first cycle RTX BP (n=8)a 67.13 years sd 9; R 53-78
MMP (n=14) Ocular involvement (n=7) b Oral involvement (n=11) Laryngeal involvement (n=4) Genital involvement (n=2)
64.9 years sd 12; R 45-84
EBA, all inflammatory subtype (n=5) 54.0 years sd 23; R 25-87
LAD (n=1) 48.0 years -
Total (n=28) 63.0 years sd 14; R 25-87
Dominant immunoglobuline in DIF and IIF on SSS
IgG-dominant IgA-dominant IgM-dominant IgG/IgA equally dominant
16 patients 5 patients 1 patient 6 patients
Gender Male 13 (46.4%)
Female 15 (53.6%)
First cycle of 2x500mg 6 patients
Additional cycle 2x1000mg 3 patients
Additional cycle 2x500mg 1 patient
First cycle of 2x1000mg 22 patientsc
Additional cycle 2x1000mg 1 patients
Additional cycle 2x500mg 1 patient
Additional gifts of RTX
500mg at M6 and/or M12 500mg at M6 and M12
15 patientsd
8 patients
Mean total follow-up time (first RTX cycle till last contact)
30.3 months sd 23; R 2-79
RTX, rituximab; BP, bullous pemphigoid; MMP, mucous membrane pemphigoid; EBA, epidermolysis bullosa acquisita; LAD, linear IgA disease; sd, standard deviation; R, range; DIF, direct immunofluorescence microscopy; IIF, indirect immunofluorescence microscopy; SSS, salt-split skin; M6, month 6; M12, month 12. a All patients with pemphigoid presented with the bullous subtype (BP). b Two patients had exclusive ocular involvement, also known as pure ocular MMP. c One patient only received 1x1000mg due to the development of pneumocystis pneumonia. d Five patients only received 500mg RTX at M6, 2 patients only received 500mg RTX at M12.

153
Figure 1. Bullous pemphigoid in a 69-year old male. (A,C) Erythematous plaques and papules on both legs before rituximab treatment. (B,D) Remission with minimal therapy after rituximab treatment.

154
Figures 3 and 4 display a flowchart and bar chart of the achieved early and late endpoints during follow-up. A complete overview of the outcome measurements of all included patients can be found in supplement 1.
Figure 2. Epidermolysis bullosa acquisita in a 59-year old female. (A) Nummular erythematous plaques, papules and circinate configurated crustae, vesicles and bullae on the trunk, before rituximab treatment. (B) Remission off therapy after rituximab treatment.
Figure 3. Flowchart of the effectiveness of RTX in pemphigoid patients, showing the highest endpoint reached after the first RTX cycle. RTX, rituximab; DC, disease control ; PR, partial remission ; CR, complete remission. a Two patients already achieved DC before RTX was administered.

155
Figu
re 4
. Bar
cha
rt s
how
ing
the
achi
eved
end
poin
ts a
nd re
peat
ed tr
eatm
ent o
f RTX
of e
ach
indi
vidu
al p
emph
igoi
d pa
tient
. Bar
s re
pres
ent
patie
nts
until
the
end
of fo
llow
-up.
The
pem
phig
oid
subt
ypes
are
indi
cate
d on
the
y-ax
is an
d th
e x-
axis
disp
lays
tim
e in
yea
rs. R
TX,
ritux
imab
; BP,
bul
lous
pem
phig
oid;
MM
P, m
ucou
s m
embr
ane
pem
phig
oid;
EBA
, epi
derm
olys
is bu
llosa
acq
uisi
ta; L
AD, l
inea
r IgA
dise
ase.

156
We analyzed whether early administration of RTX was more beneficial. We compared the mean time between onset of symptoms and RTX treatment of patients with PR or CR (52.2 months; n=16) and patients without PR or CR (64.9 months; n=12). No significant difference was found (Mann Whitney test (p=0.642)). Comparison of treatment regimes Significantly more patients achieved DC with 1000mg RTX at day 1 and 15 (85.0%) compared to 500 mg RTX (33.3%; p=0.028). Furthermore, patients more often achieved PR (63.6% vs. 16.7%; p=0.057) and CR (27.3% vs. 0%; p=0.289). Relapses were seen in both two cases receiving 500mg RTX and 12 out of 19 cases (63.2%) with 1000mg RTX (p=0.533).
Patients receiving repeated RTX infusions (n=8) achieved DC (100% vs. 63.2%; p=0.134) and PR (87.5% vs. 45.0%; p=0.088) more frequently than patients without additional RTX infusions. A similar number of patients achieved CR (25.0% vs. 20%; p=1.000). The relapse rate in the group with the additional gifts was lower (50.0% vs. 76.9%; p=0.346), and the mean time untill relapse was longer (81.3 weeks (range 32-170; sd 62.3) vs. 69.0 weeks (range 12-238; sd 66.6); p=0.572). Response in the different pemphigoid subtypes MMP patients showed the most benefit of RTX with DC in 85.7%, PR in 64.3% and CR in 28.6% patients. During follow-up 75% of the MMP patients relapsed. BP patients achieved DC in 83.3%, PR in 62.5% and CR in 12.5% with a relapse rate of 71.4%. In EBA patients DC was found in 40%, PR in 40% and CR in 20% without relapse. The patient with LAD was unresponsive to RTX treatment. No significant difference was found in the effectiveness of RTX between the different pemphigoid subtypes by Fisher’s exact test. Immunological findings The dominant immunoglobulin class prior to RTX treatment assessed by staining intensity in DIF and IIF on SSS was IgG in the majority of the cases (57.1%; n=16), IgA in 17.9% (n=5), and IgM in 3.6% (n=1). Equal intensity of IgA and IgG staining was observed in 21.4% (n=6) of the cases. IgA-dominant pemphigoid cases (n=5) showed significantly less DC (20% vs. 81.3%; p=0.007) compared to IgG-dominant cases (n=16). The proportion of patients achieving PR (20% vs. 62.5%; p=0.149) and CR (0% vs. 18.8%; p=0.549) did not differ significantly, however showed a clear

157
trend of ineffectiveness of RTX in IgA-dominant cases. Post treatment analysis was not performed.
Deposition of C3c along the BMZ was seen in 67.9% (n=19) of DIF biopsies. No differences in effectiveness of RTX were found between patients with or without complement depositions.
Relapses Fourteen of 21 patients (66.7%) relapsed after a mean time of 72.5 weeks (range 12-238; sd 63.2). Four of 14 relapsed patients showed B cell repopulation; in one case repopulation preceded relapse and in three cases repopulation was objectified after the relapse. Five of 14 relapsed patients showed maintained B cell depletion and in five patients B cells were not followed up. Seven of 14 relapsed patients were retreated with RTX, which led to PR or CR in six out of seven patients (85.7%). Safety Table 2 provides an overview of reported adverse events and deaths after RTX treatment. One patient died four months after RTX infusion due to sepsis which led to multi-organ failure. This death was interpreted as possibly related to RTX, since neutropenia (possibly RTX induced late onset neutropenia) might have contributed to a higher infection risk. All cases that reported grade 3 or grade 4 adverse events fully recovered. Five infusion reactions were observed in three patients during RTX administration: dyspnea with chest pain, tired feeling of the legs, and dizziness plus a burning sensation in the groins. All infusions could be successfully continued at lower infusion rate. One patient was accidently shortly infused with RTX subcutaneously, causing temporary pain and swelling of the arm. B cell depletion B cells were undetectable in the peripheral blood within two weeks in all patients after a single RTX infusion. In 13 patients B cell levels remained undetectable during a mean follow-up time of 77.5 weeks (range 24-269; sd 64.6). All 13 patients received repeated RTX infusions at month 6, 12 or both. Repopulation of B cells was seen in six patients after a mean time of 95.2 weeks (range 36-250; sd 82.4). B cell levels were not followed-up in nine patients. B cell levels showed no clear relation with response to RTX. All data on B cell levels should be interpreted with

158
Table 2. Adverse events and deaths reported in pemphigoid patients treated with RTX
Reported adverse events GRADE* Concomitant immunosuppressive drugs
Pt 1 Erysipelas right arm 3 prednisolone 30mg/day Herpes simplex labialis (confirmed
HSV-1) 2 prednisolone 30mg/day
Pt 2,3 Upper respiratory infection probably viral (not confirmed)
1 patient 2: prednisolone 10mg/day patient 3: none
Pt 4 PCP twice (no prophylaxis) - after first gift of 1000 mg RTX
4 prednisolone 60mg/day + cyclophosphamide 150mg
- after second cycle of 2x1000mg RTX
4 prednisolone 20mg
Pt 5 Urticaria e.c.i., self-limiting 1 prednisolone 15mg/day
Pt 6 Flare-up of concomitant psoriasis 2 prednisolone 10mg/day + dapsone 100mg/day
Pt 7 Polyarthritis and fever, possibly caused by serum sickness (not confirmed)
3 prednisolone 7.5mg/day
Pt 8 Diarrhoea and loss of consciousness, followed by hospitalization
3 prednisolone 40mg/day
Pt 9 Generalized pain e.c.i., self-limiting 2 prednisolone 35mg/day Urinary tract infection (female) 2 prednisolone 35mg/day
Pt 10 Upper respiratory infection probably viral (not confirmed)
2 prednisolone 5mg/day + cyclophosphamide 50mg/day
Urinary tract infection(male) 1 prednisolone 5mg/day + cyclophosphamide 50mg/day
Pt 11 Myalgia e.c.i., self-limiting 1 prednisolone 5mg/day
Deaths that occurred after RTX administration
Male, 78 years old, BP Cognitive and physical decline. Exact cause of death unknown.
Female, 73 years old, BP
Sepsis due to neglected urinary tract infection and neutropenia/leukopenia (possibly late onset neutropenia due to RTX), multi-organ failure eventually led to death.
Female, 87 years old, EBA
Active disease with severe mucosal involvement, weight loss and physical decline, exact cause of death unknown (possibly disease-related).
RTX, rituximab; pt, patient; HSV-1, Herpes Simplex Virus type 1; PCP, pneumocystis pneumonia; e.c.i., e causa ingnota (of unknown cause); BP, bullous pemphigoid; MMP, mucous membrane pemphigoid; EBA, epidermolysis bullosa acquisita; LAD, linear IgA disease. * Adverse events were graded according to the Common Terminology Criteria for Adverse Events v4.0 (CTCAE).48

159
caution, since B cells were not measured at standard time points in our study population. Discussion Our study showed partial or complete remission with RTX in 57.1% of the cases with a pemphigoid disease, that previously failed on a variety of immunosuppressants. RTX was most beneficial in refractory MMP and BP patients with partial or complete remission in 64.3% and 62.5% of the cases. Interestingly, IgA-dominant pemphigoid diseases responded poorly on RTX.
Only two other studies described RTX treatment of MMP patients (n=14; 11 cases with isolated ocular involvement) according to the RA protocol, and showed PR or CR in all cases.20,21 Both studies reported high relapse rates (83.3% and 100%), however repeated treatment led to remission in all cases. These findings are in accordance with our observed relapse rate (66.7%) and remission rate after repeated RTX treatment (85.7%).
Previous studies on RTX therapy in MMP and BP reported either mixed responses with serious infectious adverse events and death,25-27 or high remission rates with limited non-serious adverse events19-24. Studies on RTX in combination with immunoadsorption (protein A) or HIVIg found high response rates in ocular MMP, resistant EBA and recalcitrant BP.35-38 Interestingly, our study showed lower remission rates compared to most reports in literature; DC in 100%22, PR and/or CR in 66%25, 86%26, 88%27, 92%19 and 100%24. These differences in the results can be explained by the clinical heterogeneity of the previous studies, caused by using different RTX regimes22,23,25-27, by assessing different populations (multiple pemphigoid subtypes in a tertiary referral center in our study, MMP patients in most studies)20-22,27,36, or by using different definitions for the outcome measurements20,23,24,39. Studies prior to the pemphigoid consensus of 2012 either used definitions of the pemphigus consensus of 2008, in which minimal adjuvant therapy was less well defined, or other definitions for treatment response.20,23,24,39
An important result is the observation of significantly more DC (p=0.028), and more PR (p=0.057) and CR (p=0.289) in patients treated with a high dosage regime compared to a low dosage regime. Moreover, we noticed a beneficial effect of repeated RTX infusions with less relapses.
A major finding of our study is that four out of five (80%) IgA-dominant pemphigoid diseases were completely unresponsive to RTX treatment. Previously

160
He et al. demonstrated persistent IgA-secreting plasma cells in a MMP patient not responding to RTX treatment.40 They speculated that plasma cells could be derived from a tissue resident memory B cell population that is resistant to anti-CD20 therapy. Mei et al. described the continuous presence of IgA+ plasma cells in the peripheral circulation and the gastrointestinal mucosa of RA patients during successful B cell depletion by RTX.41 Further characterization of the circulating plasma cells revealed a mucosal phenotype, indicating that their precursor B cells are mucosal resident, and not depleted by RTX. All these findings could explain the unresponsiveness in our IgA-dominant cases.
Fourteen adverse events were reported in 11 (39.3%) pemphigoid cases treated with RTX. The majority of adverse events were infectious (n=8). Five adverse event were severe (grade 3 of 4), and one reported death was possibly related to RTX. Noteworthy is one disease-related death in an older EBA patient, demonstrating that pemphigoid diseases can be life-threatening in therapy resistant cases and underlining the urgent need for effective treatment options. Our safety data is comparable with the reports of Maley et al. and Cho et al. who found adverse events in 33% and 31% of the MMP patients treated with RTX.19,22 Yet, both studies observed a significantly higher adverse event rate in patients treated with conventional therapies (48% and 53%). Other studies have reported less adverse events compared to our data.24,35-37 This might be explained by the frequent use of concomitant immunosuppressive drugs in our population with high disease severity, causing a high risk of infection.
Pemphigoid diseases appear to respond less on RTX than pemphigus, despite successful B cell depletion in the peripheral circulation in both diseases.18,33 Furthermore, our data also showed that it takes almost 4 months (mean time till DC is 14.4 weeks) until RTX has effect, whereas in pemphigus effect is noticed within two months (DC at 4.0-9.3 weeks).42-46 Possibly, B cell depletion stops pathogenic autoantibody production in both diseases, though other ongoing pathophysiological mechanisms that are not interrupted by B cell depletion might play a more substantial role in the pathogenesis of pemphigoid.28,47 Nevertheless, PR and CR in responding pemphigoid disease patients was reached after the same time or slightly later than in pemphigus patients.33,45,46 The greatest limitation of this study is its retrospective nature, which led to incomplete and/or missing data, such as objective disease scores (BPDAI and MMPDAI), and laboratory measurements of B cells and pathogenic autoantibodies at standard time points.

161
Furthermore, a major limitation is the small sample size, especially when comparing the different pemphigoid subtypes, and the heterogeneity of our study population. The use of concomitant immunosuppressants in almost all patients (supplement 1) did not allow us to draw conclusions regarding rituximab monotherapy. Nonetheless, the immunosuppressants alone did not succeed to establish disease control or remission prior to rituximab treatment. Moreover, the use of co-medication in severe pemphigoid diseases does reflect upon our daily practice. Last, it is important to emphasize that consensus late endpoints PR and CR imply to define two contrasting outcomes, but in clinical setting the difference can be minimal (one insignificant lesion once weekly versus no lesions), therefore patients on PR and CR might be equally satisfied with treatment result. Prospective studies with a greater sample size are needed to provide an higher level of evidence on the effectiveness of RTX in pemphigoid diseases. In conclusion, this study demonstrated that RTX was effective in 57.1% of recalcitrant pemphigoid diseases and that the high dose regime of twice 1000mg was more effective than the low dose. Although relapse rates were high (66.7%), repeated RTX therapy led to remission in the majority of the relapsed cases (85.7%). An important finding is that most pemphigoid patients with IgA-dominant disease showed poor response to RTX. This finding suggests that RTX can be eliminated from the clinicians’ arsenal when encountering IgA-dominant pemphigoid patients, however, future studies are required for confirmation. RTX showed to be relatively safe. Prospective comparative studies are needed to further determine the position of RTX in the therapeutic algorithm for pemphigoid diseases. References 1. Schmidt E, Zillikens D. Pemphigoid diseases. Lancet. 2013;381(9863):320-332. 2. Amber KT, Murrell DF, Schmidt E, Joly P, Borradori L. Autoimmune subepidermal bullous
diseases of the skin and mucosae: Clinical features, diagnosis, and management. Clin Rev Allergy Immunol. 2018 Feb;54(1):26-51.
3. Joly P, Baricault S, Sparsa A, et al. Incidence and mortality of bullous pemphigoid in france. J Invest Dermatol. 2012;132(8):1998-2004.
4. Chan LS, Ahmed AR, Anhalt GJ, et al. The first international consensus on mucous membrane pemphigoid: Definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol. 2002;138(3):370-379.
5. Guide SV, Marinkovich MP. Linear IgA bullous dermatosis. Clin Dermatol. 2001;19(6):719-727.

162
6. Gottlieb J, Ingen-Housz-Oro S, Alexandre M, et al. Idiopathic linear IgA bullous dermatosis: Prognostic factors based on a case series of 72 adults. Br J Dermatol. 2017;177(1):212-222.
7. Feliciani C, Joly P, Jonkman MF, et al. Management of bullous pemphigoid: The european dermatology forum consensus in collaboration with the european academy of dermatology and venereology. Br J Dermatol. 2015;172(4):867-877.
8. Joly P, Roujeau JC, Benichou J, et al. A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. N Engl J Med. 2002;346(5):321-327.
9. Kirtschig G, Middleton P, Bennett C, Murrell DF, Wojnarowska F, Khumalo NP. Interventions for bullous pemphigoid. Cochrane Database Syst Rev. 2010;(10):CD002292. doi(10):CD002292.
10. Williams HC, Wojnarowska F, Kirtschig G, et al. Doxycycline versus prednisolone as an initial treatment strategy for bullous pemphigoid: A pragmatic, non-inferiority, randomised controlled trial. Lancet. 2017;389(10079):1630-1638.
11. Grantham HJ, Stocken DD, Reynolds NJ. Doxycycline: A first-line treatment for bullous pemphigoid? Lancet. 2017;389(10079):1586-1588.
12. Joly P, Roujeau JC, Benichou J, et al. A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. N Engl J Med. 2002;346(5):321-327.
13. Kalinska-Bienias A, Lukowska-Smorawska K, Jagielski P, Kowalewski C, Wozniak K. Mortality in bullous pemphigoid and prognostic factors in 1st and 3rd year of follow-up in specialized centre in poland. Arch Dermatol Res. 2017 Nov;309(9):709-719.
14. Sadik CD, Zillikens D. Current treatments and developments in pemphigoid diseases as paradigm diseases for autoantibody-driven, organ-specific autoimmune diseases. Semin Hematol. 2016;53 Suppl 1:S51-3.
15. Reff ME, Carner K, Chambers KS, et al. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood. 1994;83(2):435-445.
16. Gurcan HM, Keskin DB, Stern JN, Nitzberg MA, Shekhani H, Ahmed AR. A review of the current use of rituximab in autoimmune diseases. Int Immunopharmacol. 2009;9(1):10-25.
17. Pescovitz MD. Rituximab, an anti-cd20 monoclonal antibody: History and mechanism of action. Am J Transplant. 2006;6(5 Pt 1):859-866.
18. Joly P, Maho-Vaillant M, Prost-Squarcioni C, et al. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (ritux 3): A prospective, multicentre, parallel-group, open-label randomised trial. Lancet. 2017;389(10083):2031-2040.
19. Cho YT, Chu CY, Wang LF. First-line combination therapy with rituximab and corticosteroids provides a high complete remission rate in moderate-to-severe bullous pemphigoid. Br J Dermatol. 2015;173(1):302-304.
20. Rubsam A, Stefaniak R, Worm M, Pleyer U. Rituximab preserves vision in ocular mucous membrane pemphigoid. Expert Opin Biol Ther. 2015;15(7):927-933.
21. Heelan K, Walsh S, Shear NH. Treatment of mucous membrane pemphigoid with rituximab. J Am Acad Dermatol. 2013;69(2):310-311.
22. Maley A, Warren M, Haberman I, Swerlick R, Kharod-Dholakia B, Feldman R. Rituximab combined with conventional therapy versus conventional therapy alone for the treatment of mucous membrane pemphigoid (MMP). J Am Acad Dermatol. 2016;74(5):835-840.
23. Shetty S, Ahmed AR. Critical analysis of the use of rituximab in mucous membrane pemphigoid: A review of the literature. J Am Acad Dermatol. 2013;68(3):499-506.

163
24. Kasperkiewicz M, Shimanovich I, Ludwig RJ, Rose C, Zillikens D, Schmidt E. Rituximab for treatment-refractory pemphigus and pemphigoid: A case series of 17 patients. J Am Acad Dermatol. 2011;65(3):552-558.
25. Schmidt E, Seitz CS, Benoit S, Brocker EB, Goebeler M. Rituximab in autoimmune bullous diseases: Mixed responses and adverse effects. Br J Dermatol. 2007;156(2):352-356.
26. Lourari S, Herve C, Doffoel-Hantz V, et al. Bullous and mucous membrane pemphigoid show a mixed response to rituximab: Experience in seven patients. J Eur Acad Dermatol Venereol. 2011;25(10):1238-1240.
27. Le Roux-Villet C, Prost-Squarcioni C, Alexandre M, et al. Rituximab for patients with refractory mucous membrane pemphigoid. Arch Dermatol. 2011;147(7):843-849.
28. Sitaru C, Thiel J. The need for markers and predictors of rituximab treatment resistance. Exp Dermatol. 2014;23(4):236-237.
29. Buch MH, Smolen JS, Betteridge N, et al. Updated consensus statement on the use of rituximab in patients with rheumatoid arthritis. Ann Rheum Dis. 2011;70(6):909-920.
30. Murrell DF, Marinovic B, Caux F, et al. Definitions and outcome measures for mucous membrane pemphigoid: Recommendations of an international panel of experts. J Am Acad Dermatol. 2015;72(1):168-174.
31. Murrell DF, Daniel BS, Joly P, et al. Definitions and outcome measures for bullous pemphigoid: Recommendations by an international panel of experts. J Am Acad Dermatol. 2012;66(3):479-485.
32. Smolen JS, Keystone EC, Emery P, et al. Consensus statement on the use of rituximab in patients with rheumatoid arthritis. Ann Rheum Dis. 2007;66(2):143-150.
33. Horvath B, Huizinga J, Pas HH, Mulder AB, Jonkman MF. Low-dose rituximab is effective in pemphigus. Br J Dermatol. 2012;166(2):405-412.
34. Cianchini G, Lupi F, Masini C, Corona R, Puddu P, De Pita O. Therapy with rituximab for autoimmune pemphigus: Results from a single-center observational study on 42 cases with long-term follow-up. J Am Acad Dermatol. 2012;67(4):617-622.
35. Kolesnik M, Becker E, Reinhold D, et al. Treatment of severe autoimmune blistering skin diseases with combination of protein A immunoadsorption and rituximab: A protocol without initial high dose or pulse steroid medication. J Eur Acad Dermatol Venereol. 2014;28(6):771-780.
36. Foster CS, Chang PY, Ahmed AR. Combination of rituximab and intravenous immunoglobulin for recalcitrant ocular cicatricial pemphigoid: A preliminary report. Ophthalmology. 2010;117(5):861-869.
37. Ahmed AR, Shetty S, Kaveri S, Spigelman ZS. Treatment of recalcitrant bullous pemphigoid (BP) with a novel protocol: A retrospective study with a 6-year follow-up. J Am Acad Dermatol. 2016;74(4):700-8.e3.
38. Oktem A, Akay BN, Boyvat A, et al. Long-term results of rituximab-intravenous immunoglobulin combination therapy in patients with epidermolysis bullosa acquisita resistant to conventional therapy. J Dermatolog Treat. 2017;28(1):50-54.
39. Murrell DF, Dick S, Ahmed AR, et al. Consensus statement on definitions of disease, end points, and therapeutic response for pemphigus. J Am Acad Dermatol. 2008;58(6):1043-1046.

164
40. He Y, Shimoda M, Ono Y, et al. Persistence of autoreactive IgA-secreting B cells despite multiple immunosuppressive medications including rituximab. JAMA Dermatol. 2015;151(6):646-650.
41. Mei HE, Frolich D, Giesecke C, et al. Steady-state generation of mucosal IgA+ plasmablasts is not abrogated by B-cell depletion therapy with rituximab. Blood. 2010;116(24):5181-5190.
42. Sharma VK, Bhari N, Gupta S, et al. Clinical efficacy of rituximab in the treatment of pemphigus: A retrospective study. Indian J Dermatol Venereol Leprol. 2016;82(4):389-394.
43. Bhattacharjee R, De D, Handa S, Minz RW, Saikia B, Joshi N. Assessment of the effects of rituximab monotherapy on different subsets of circulating T-regulatory cells and clinical disease severity in severe pemphigus vulgaris. Dermatology. 2016;232(5):572-577.
44. Leshem YA, David M, Hodak E, et al. A prospective study on clinical response and cell-mediated immunity of pemphigus patients treated with rituximab. Arch Dermatol Res. 2014;306(1):67-74.
45. Kim JH, Kim YH, Kim MR, Kim SC. Clinical efficacy of different doses of rituximab in the treatment of pemphigus: A retrospective study of 27 patients. Br J Dermatol. 2011;165(3):646-651.
46. Wang HH, Liu CW, Li YC, Huang YC. Efficacy of rituximab for pemphigus: A systematic review and meta-analysis of different regimens. Acta Derm Venereol. 2015;95(8):928-932.
47. Hammers CM, Stanley JR. Mechanisms of disease: Pemphigus and bullous pemphigoid. Annu Rev Pathol. 2016;11:175-197.
48. National cancer institute, common terminology criteria for adverse events v4.0. NCI, NIH, DHHS. ;NIH publication # 09-7473.

165
List of abbreviations RTX rituximab BMZ basement membrane zone BP bullous pemphigoid MMP mucous membrane pemphigoid EBA epidermolysis bullosa acquisita LAD linear IgA disease RA rheumatoid arthritis DIF direct immunofluorescence microscopy IIF indirect immunofluorescence microscopy SSS salt-split skin DC disease control PR partial remission CR complete remission HIVIg human intravenous immunoglobulin MTX methotrexate AZA azathioprine cyclo cyclophosphamide MMF mycophenolate mofetil HIVIg human intravenous immunoglobulin IV intravenous sd standard deviation

166
Supp
lem
ent 1
. Pa
rt 1
: Ove
rvie
w o
f all
incl
uded
pat
ient
s w
ith p
emph
igoi
d di
seas
es tr
eate
d w
ith R
TX
Case
no/
G
ende
r/
age
Pem
phig
oid
subt
ype
Ig in
DI
F +
IIF o
n SS
Sa
Med
icat
ion
prio
r to
RTX
RT
X do
se
first
cyc
le
500m
g RT
X at
M
6&12
Med
icat
ion
conc
omita
nt
with
RTX
DC
PR
CR
Rela
pse
Follo
w-u
p
1/f/
77
BP
IgG
+
IgA
pred
, MTX
, dox
y 2x
1000
mg
yes
loca
l st
eroi
ds,
pred
yes
min
imal
th
erap
y no
no
Su
stai
ned
PR
2/m
/78
BP
IgG
+
IgA
pred
2x
1000
mg
no
loca
l st
eroi
ds,
pred
yes
no
no
yes
Dea
th, n
ot R
TX
rela
ted
3/m
/65
BP
IgG
+
IgA
+ Ig
M
pred
, dox
y,
daps
one,
IV
ster
oids
2x10
00m
g ye
s pr
ed
yes
min
imal
th
erap
y no
Ye
s PR
on
retr
eatm
ent w
ith
RTX
4/m
/72
BP
IgG
+
IgA
pred
, MTX
, dox
y,
daps
one,
AZA
2x
1000
mg
yes
pred
, AZA
be
fore
RT
X m
inim
al
ther
apy
no
no
Sust
aine
d PR
5/f/
56
BP
IgG
+
IgA
pred
, MTX
, dox
y,
daps
one,
AZA
, IV
ster
oids
, MM
F,
HIV
Ig
2x10
00m
g no
lo
cal
ster
oid,
pr
ed
yes
min
imal
th
erap
y m
inim
al
ther
apy
yes
Mul
tiple
tim
es
retr
eatm
ent w
ith
RTX
PR/C
R/re
laps
e in
term
itten
t 6/
m/6
3 BP
Ig
G
pred
, MTX
, dox
y,
daps
one,
AZA
, M
MF,
HIV
Ig
2x 5
00m
g no
lo
cal
ster
oid,
pr
ed
yes
min
imal
th
erap
y no
ye
s M
ultip
le ti
mes
re
trea
tmen
t with
RT
X: in
term
itten
t PR
/CR/
rela
pse
7/
f/53
BP
Ig
G +
Ig
M
pred
, MTX
dox
y,
AZA,
cyc
lo, M
MF,
H
IVIg
2x10
00m
g no
on
e gi
ft o
f H
IVIg
be
fore
RT
X no
no
ye
s Re
spon
se o
n H
IVIg
m
aint
enan
ce
ther
apy
8/f/
73
BP
IgG
+
IgA
pred
, dox
y, A
ZA,
MM
F 2x
500
mg
no
loca
l st
eroi
d,
MM
F
no
no
no
n.a.
D
eath
, pos
sibly
RT
X re
late
d
9/f/
73
MM
P Ig
G +
Ig
A pr
ed, d
oxy,
da
pson
e, A
ZA,
cycl
o
2x 5
00m
g no
lo
cal
ster
oid,
pr
ed, A
ZA
yes
no
no
yes
CR a
fter
sec
ond
cycl
e of
2x
1000
mg
RTX
10/m
/81
MM
P Ig
G +
Ig
A pr
ed, d
apso
ne,
cycl
o 2x
500
mg
no
pred
, AZA
no
no
no
n.
a.
Lost
to fo
llow
-up
RTX,
ritu
xim
ab; D
IF, d
irect
imm
unof
luor
esce
nce;
IIF,
indi
rect
imm
unof
luor
esce
nce;
SSS
, sal
t spl
it sk
in; D
C, d
isea
se c
ontr
ol; P
R, p
artia
l rem
issio
n; C
R, c
ompl
ete
rem
issio
n; f,
fem
ale;
m, m
ale;
BP,
bul
lous
pem
phig
oid;
MM
P, m
ucou
s mem
bran
e pe
mph
igoi
d; E
BA, e
pide
rmol
ysis
bullo
sa a
cqui
sita
; LAD
, lin
ear I
gA d
isea
se; p
red,
pr
edni
solo
ne; d
oxy,
dox
ycyc
line;
MTX
, met
hotr
exat
e; A
ZA, a
zath
iopr
ine;
cyc
lo, c
yclo
phos
pham
ide;
MM
F, m
ycop
heno
late
mof
etil;
HIV
Ig, H
uman
Intr
aven
ous
Imm
unog
lobu
lin; I
V st
eroi
ds, i
ntra
veno
us s
tero
ids;
n.a
., no
t app
licab
le. a T
he d
omin
ant i
mm
unog
lobu
line
clas
s is
und
erlin
ed.
Supplement 1

167
Supp
lem
ent 1
. Pa
rt 2
: Ove
rvie
w o
f all
incl
uded
pat
ient
s w
ith p
emph
igoi
d di
seas
es tr
eate
d w
ith R
TX
Case
no/
G
ende
r/
age
Pem
phig
oid
subt
ype
Ig in
DIF
+
IIF o
n SS
Sa M
edic
atio
n pr
ior t
o RT
X RT
X do
se
first
cyc
le
500m
g RT
X at
M
6&12
Med
icat
ion
conc
omita
nt
with
RTX
DC
PR
CR
Rela
pse
Follo
w-u
p
11/m
/75
MM
P Ig
G +
IgA
Pred
, MTX
, da
pson
e, c
yclo
2x
1000
mg
yes
loca
l ste
roid
, cy
clo
yes
off
ther
apy
no
yes
CR o
n re
trea
tmen
t w
ith R
TX
12/f
/63
MM
P Ig
G +
IgA
+ Ig
M
pred
, dap
sone
, AZ
A, c
yclo
, MM
F 2x
1000
mg
no
loca
l ste
roid
ye
s of
f th
erap
y of
f th
erap
y ye
s CR
by
loca
l ste
roid
13/m
/62
MM
P Ig
G +
IgA
pred
, dox
y,
daps
one,
cyc
lo,
MM
F
2x10
00m
g no
pr
ed, c
yclo
ye
s no
no
ye
s D
C on
cyc
lo +
pre
d
14/f
/84
MM
P Ig
G +
IgA
pred
, dap
sone
, AZ
A, c
yclo
2x
1000
mg
no
pred
ye
s m
inim
al
ther
apy
no
no
Sust
aine
d PR
15/m
/46
MM
P Ig
G
daps
one,
cyc
lo
2x10
00m
g no
lo
cal s
tero
id
yes
off
ther
apy
no
yes
PR/C
R on
re
trea
tmen
t with
RT
X 16
/m/6
8 M
MP
IgG
+ Ig
A pr
ed, c
yclo
2x
1000
mg
yes
pred
, cyc
lo
yes
no
no
yes
DC
on p
red
17/m
/69
MM
P Ig
G +
IgA
pred
, dap
sone
, cy
lco
2x10
00m
g ye
s lo
cal s
tero
id,
pred
, cyc
lo
yes
min
imal
th
erap
y of
f th
erap
y no
Su
stai
ned
CR
18/f
/49
MM
P Ig
G
pred
, cyc
lo, M
MF
2x10
00m
g no
lo
cal s
tero
id
yes
off
ther
apy
no
yes
RTX
retr
eatm
ent i
s pl
anne
d 19
/m/7
4 M
MP
IgG
+ Ig
A pr
ed, d
apso
ne,
cycl
o 2x
1000
mg
no
loca
l ste
roid
, pr
ed, c
yclo
ye
s m
inim
al
ther
apy
no
yes
Retr
eatm
ent w
ith
RTX:
PR/
rela
pse
inte
rmitt
ent
20/f
/58
MM
P Ig
G +
IgA
pred
, dap
sone
, cy
clo,
MM
F 2x
1000
mg
no
loca
l ste
roid
, pr
ed
no
no
no
n.a.
N
o re
miss
ion
on
kena
cort
inje
ctio
ns
21/f
/45
MM
P Ig
G +
IgA
pred
, dap
sone
, cy
clo.
2x
1000
mg
no
pred
, dap
sone
ye
s m
inim
al
ther
apy
min
imal
th
erap
y no
Su
stai
ned
CR
RTX,
ritu
xim
ab; D
IF, d
irect
imm
unof
luor
esce
nce;
IIF,
indi
rect
imm
unof
luor
esce
nce;
SSS
, sal
t spl
it sk
in; D
C, d
isea
se c
ontr
ol; P
R, p
artia
l rem
issio
n; C
R, c
ompl
ete
rem
issio
n; f,
fem
ale;
m
, mal
e; B
P, b
ullo
us p
emph
igoi
d; M
MP,
muc
ous m
embr
ane
pem
phig
oid;
EBA
, epi
derm
olys
is bu
llosa
acq
uisit
a; L
AD, l
inea
r IgA
dis
ease
; pre
d, p
redn
isol
one;
dox
y, d
oxyc
yclin
e;
MTX
, met
hotr
exat
e; A
ZA, a
zath
iopr
ine;
cyc
lo, c
yclo
phos
pham
ide;
MM
F, m
ycop
heno
late
mof
etil;
HIV
Ig, H
uman
Intr
aven
ous
Imm
unog
lobu
lin; I
V st
eroi
ds, i
ntra
veno
us s
tero
ids;
n.
a., n
ot a
pplic
able
. a The
dom
inan
t im
mun
oglo
bulin
e cl
ass
is u
nder
lined
.

168
Supp
lem
ent 1
. Pa
rt 3
: Ove
rvie
w o
f all
incl
uded
pat
ient
s w
ith p
emph
igoi
d di
seas
es tr
eate
d w
ith R
TX
Case
no/
G
ende
r/
age
Pem
phig
oid
subt
ype
Ig in
DIF
+
IIF o
n SS
Sa
Med
icat
ion
prio
r to
RTX
RT
X do
se
first
cyc
le
500m
g RT
X at
M
6&12
Med
icat
ion
conc
omita
nt
with
RTX
DC
PR
CR
Rela
pse
Follo
w-u
p
22/m
/62
MM
P Ig
G +
IgA
pred
, MTX
, da
pson
e, c
yclo
2x
1000
mg
yes
pred
, dap
sone
ye
s m
inim
al
ther
apy
min
imal
th
erap
y ye
s Re
trea
tmen
t with
RT
X pl
anne
d 23
/f/5
9 EB
A Ig
G +
IgA
pred
, dap
sone
, AZ
A, c
yclo
, MM
F,
HIV
Ig
2x 5
00m
g no
lo
cal s
tero
id,
pred
no
no
no
n.
a.
CR o
n pr
ed
24/f
/87
EBA
IgG
+ Ig
A +
IgM
pr
ed, A
ZA, I
V st
eroi
ds
2x 5
00m
g no
pr
ed, A
ZA
no
no
no
n.a.
D
eath
, dis
ease
re
late
d 25
/f/5
6 EB
A Ig
G +
IgA
pred
, dox
y,
daps
one,
AZA
2x
1000
mg
yes
loca
l ste
roid
, pr
ed, A
ZA
yes
off
ther
apy
no
no
Sust
aine
d PR
26/m
/25
EBA
IgG
+ Ig
A pr
ed, d
oxy,
AZA
2x
1000
mg
no
loca
l ste
roid
, pr
ed, d
apso
ne
yes
min
imal
th
erap
y m
inim
al
ther
apy
no
Sust
aine
d CR
27/f
/43
EBA
IgG
+ Ig
A pr
ed, d
apso
ne,
AZA,
MM
F
2x10
00m
g no
pr
ed, d
apso
ne
no
no
no
n.a.
N
o re
miss
ion
on:
- H
IVIg
+ d
apso
ne
+ pr
ed +
col
chic
ine
- sul
fasa
lazi
ne +
da
pson
e +
pred
28
/f/4
8 LA
D Ig
A pr
ed, d
apso
ne
2x10
00m
g no
pr
ed
no
no
no
n.a.
N
o re
miss
ion
on:
colc
hici
ne +
pre
d,
MM
F +
pred
, MTX
, do
xy +
ni
acin
amid
e, A
ZA
RTX,
ritu
xim
ab; D
IF, d
irect
imm
unof
luor
esce
nce;
IIF,
indi
rect
imm
unof
luor
esce
nce;
SSS
, sal
t spl
it sk
in; D
C, d
isea
se c
ontr
ol; P
R, p
artia
l rem
issio
n; C
R, c
ompl
ete
rem
issio
n; f,
fem
ale;
m
, mal
e; B
P, b
ullo
us p
emph
igoi
d; M
MP,
muc
ous m
embr
ane
pem
phig
oid;
EBA
, epi
derm
olys
is bu
llosa
acq
uisit
a; L
AD, l
inea
r IgA
dis
ease
; pre
d, p
redn
isol
one;
dox
y, d
oxyc
yclin
e;
MTX
, met
hotr
exat
e; A
ZA, a
zath
iopr
ine;
cyc
lo, c
yclo
phos
pham
ide;
MM
F, m
ycop
heno
late
mof
etil;
HIV
Ig, H
uman
Intr
aven
ous
Imm
unog
lobu
lin; I
V st
eroi
ds, i
ntra
veno
us s
tero
ids;
n.
a., n
ot a
pplic
able
. a The
dom
inan
t im
mun
oglo
bulin
e cl
ass
is u
nder
lined
.

169

170
10

171
CHAPTER 10 Determining the incidence of Pneumocystis Pneumonia in patients with autoimmune blistering diseases not receiving routine prophylaxis
Kyle Amber1, Aniek Lamberts2, Farzan Solimani3, Arianna Agnoletti1,4, Daria Didona5, Ilona Euverman2, Emanuele Cozzani4, Lee Haur Yueh6, Giovanni Di Zenzo7, Yael Anne Leshem8,9, Daniel Mimouni8,9, Michael Hertl3, Barbara Horváth2
Affiliations 1 Department of Dermatology, University of California, Irvine, United States of America 2 Center for Blistering Diseases, Department of Dermatology, University Medical Center Groningen, University of Groningen, Groningen, the Netherlands 3 Department of Dermatology and Allergology, Philipps University, Marburg, Germany 4 DISSAL Section of Dermatology, IRCCS Azienda Universitaria Ospedaliera San Martino-IST, Genoa, Italy 5 Dermatology Division, Istituto Dermopatico Dell’Immacolata, IDI-IRCCS, FLMM, Rome, Italy 6 Department of Dermatology, Singapore General Hospital, Singapore 7 Laboratory of Molecular and Cell Biology, Istituto Dermopatico Dell’Immacolata, IDI- IRCCS, FLMM, Rome, Italy 8 Department of Dermatology, Rabin Medical Center, Petah Tiqva, Israel 9 Sackler School of Medicine, Tel Aviv University, Tel Aviv, Israel Published in the JAMA Dermatology, 2017 Nov 1;153(11):1137-1141

172
Abstract Importance Pneumocystis pneumonia (PCP) is a potentially lethal opportunistic infection that primary prophylaxis can help prevent. The risk of prophylactic therapy must be weighed against the incidence of PCP in the patient population. Prophylaxis most frequently involves trimethoprim-sulfamethoxazole, with second-line therapies, including atovaquone, dapsone, and pentamide. The indication for prophylaxis in immunocompromised patients without HIV is less well defined. Previously, an incidence of at least 3.5% has been proposed as a cutoff to justify prophylaxis. Objective To assess the incidence of PCP in patients with autoimmune blistering diseases receiving no routine prophylaxis. Design, setting, and participants This was a retrospective analysis of patient medical records to determine the incidence of PCP infections. The multicenter study was performed at tertiary care centers that provide care for patients with autoimmune blistering diseases in Germany, Italy, Singapore, Israel, and the Netherlands. Patients had a confirmed diagnosis of pemphigus vulgaris, pemphigus foliaceus, bullous pemphigoid, epidermolysis bullosa acquisita, mucous membrane pemphigoid, or anti-p200 pemphigoid. Main outcomes and measures To determine the incidence of PCP defined as patients with the International Classification of Diseases, Ninth Revision (ICD-9), code 136.3, for PCP, or free text documentation of PCP occurring based on characteristic radiographic findings with elevated lactate dehydrogenase, or hospitalization for pneumonia with bronchioalveolar lavage demonstrating Pneumocystis jiroveci on confirmatory stains. Results A total of 801 patients with autoimmune blistering diseases were included in this study; their mean (SD) age was 66.5 (17.6) years, and a total of 465 (58%) were female. Only 1 patient developed PCP, resulting in an incidence rate of 0.1%. This incidence significantly fell below the recommended threshold of 3.5% (0.1% vs. 3.5%, χ2
1 = 27.0; P < .001). This incidence was significantly lower than the previously reported incidence of PCP in all immunosuppressed dermatologic patients (0.1% vs. 1.3%; χ2
1 1 = 8.2; P = .004). Conclusions and relevance Routine PCP prophylaxis for patients with autoimmune blistering diseases does not seem to be warranted. Patients with autoimmune blistering diseases seem to have a lower risk of PCP than the general population of immunosuppressed dermatology patients. Risks of routine prophylaxis include hyperkalemia, hypoglycemia, photosensitivity, thrombocytopenia, and more rare adverse reactions.

173
Introduction Pneumocystis pneumonia (PCP) is an opportunistic fungal infection caused by Pneumocystis jiroveci, formerly named Pneumocystis carinii.1 PCP can occur in the setting of human immunodeficiency virus (HIV), as well as in the setting of congenital or iatrogenic immunosuppression. Its incidence in patients with HIV has been significantly decreased with the use of routine prophylaxis in patients with CD4+ T lymphocyte counts of less than 2000. Prophylaxis most frequently involves trimethoprim sulfamethoxazole, with second-line therapies, including atovaquone, dapsone, and pentamide. The indication for prophylaxis in immunocompromised patients without HIV is less well defined. Meta-analysis of immunocompromised patients with HIV has suggested a PCP incidence of at least 3.5% to outweigh the risks of therapy.2 These risks include hyperkalemia, hypoglycemia, photosensitivity, thrombocytopenia, and more rare adverse reactions, such as Stevens-Johnson syndrome, agranulocytosis, aplastic anemia, drug reaction with eosinophilia and systemic symptoms, and fulminant hepatic necrosis. Other Cochrane meta-analyses of prophylactic trimethoprim sulfamethoxazole in immunocompromised patients without HIV demonstrated that adverse events necessitating the cessation of prophylaxis occurred in 13.8% of patients compared with 5.9% in patients receiving either placebo or alternative prophylactic antibiotics.3-5 This results in a number needed to harm of 12.7, although this does not specify severe adverse events vs. minor adverse events.3 While primary PCP prophylaxis with trimethoprim-sulfamethoxazole was found to improve survival in these patients, it is notable that the incidence of PCP in this cohort was estimated at 6.2%.4 This patient population included afebrile neutropenic patients, children with leukemia, and both solid and bone marrow transplant patients, but notably did not include studies of patients with dermatologic diseases. The use of prophylactic treatment in the iatrogenically immunosuppressed patient is controversial. Some have suggested use or primary prophylaxis for PCP for patients receiving and equivalent of at least 20 mg of prednisone daily for more than 4 weeks, particularly if a second risk factor exists, including malignant neoplasm, interstitial lung disease, or additional immunosuppressive therapies.6,7 The disease in question, however, plays a significant role in the decision for PCP prophylaxis. Few studies have assessed the incidence of PCP in dermatologic patients. Lehman and Kalaaji8 assessed 150 dermatology patients receiving

174
immunosuppressive therapy for more than a month, finding that PCP occurred in 0.5% of patients. A larger study of 334 patients with immunobullous and connective tissue disease receiving immunosuppressive therapies showed that 7 patients (2%) developed PCP, with a 1-month mortality rate of 43% in those patients.9 Of the patients developing PCP, only 1 had an immunobullous disease. A Chinese study10 of 202 patients with immunobullous disease demonstrated an incidence of PCP in 1.9%. In contrast, an Israeli study11 of 172 patients following individuals newly diagnosed as having pemphigus failed to demonstrate any patients with PCP. Based on these studies, the incidence of PCP in the dermatologic immunosuppressed population can be estimated at 1.3%.3 PCP carries a significant mortality in these patients, estimated at 47%.12 Patients with certain diseases carry a greater innate risk for PCP. For example, granulomatosis with polyangiitis (formerly Wegener granulomatosis) is associated with a PCP incidence of 6%. Therefore, it would be indicated to use primary PCP prophylaxis in these patients.13 Thus, evidence-based guidelines must be based on the disease in question rather than a generalized immunosuppressed state. Autoimmune blistering diseases (AIBDs) are characterized by circulating autoantibodies targeting epidermal antigens located at the basement membrane zone or in the epidermis, but sparing of the vasculature and other organs as would been involved in connective tissue disease. Patients with AIBD often require prolonged use of often multiple immunosuppressive therapies, putting them at risk for opportunistic infections.11 Among experts in the treatment of immune bullous disease, there is significant discord in regard to use of opportunistic infection prophylaxis.14 As such, we sought to characterize the incidence of PCP in a large cohort of patients with AIBD to generate evidence-based recommendations regarding routine PCP prophylaxis in these patients. We hypothesized that patients with AIBDs not receiving routine prophylaxis fail to reach a PCP incidence of 3.5% and that the current estimation of 1.3% in all dermatologic patients overestimates the incidence of PCP in patients with AIBDs. Methods Study design A retrospective multicenter study was performed in 6 tertiary referral centers for AIBD. Study populations included Israel, Germany, the Netherlands, Italy, and Singapore. Routine use of PCP prophylaxis was not used at these institutions.

175
Following appropriate ethical approval for medical record review, medical records and/or databases were reviewed within each institution. Inclusion and exclusion criteria Enrollment time was dependent on the availability of accurate medical records (ie, searchable electronic health records) or patient databases at each individual institution. Patients with a confirmed diagnosis of pemphigus vulgaris, pemphigus foliaceus, bullous pemphigoid, epidermolysis bullosa acquisita, mucous membrane pemphigoid, or anti-p200 pemphigoid were included in the study. Diagnosis was based on each individual institutions’ protocol for diagnosing AIBDs, which at a minimum required clinical suspicion and immunofluorescence studies confirming the disease in question, with most patients having histologic and additional serologic confirmation of disease subtype. Patients without a confirmed disease subtype or paraneoplastic pemphigus were excluded. In addition, patients who had received dapsone at any point during their treatment course, had received primary PCP prophylaxis, or had less than 3 months of follow-up available were excluded. All patients, regardless of whether they received systemic therapies, were included to minimize selection bias of more severe presentations, and to account for patients receiving variable doses of topical steroids, which may have systemic immunosuppressive effects. The cohort in the study by Leshem et al11 as well as their method for data extraction has been described previously. Power analysis Sample size was calculated to be greater than 429 to ensure an ability to detect an incidence of 1.3%, the reported incidence of PCP in dermatologic patients, compared with the proposed 3.5%, with an α error of 0.05 and power of 80%. A secondary goal of a sample greater than 718 to determine whether the incidence of PCP in patients with AIBDs was significantly lower than in all immunosuppressed dermatologic patients, with an α error of 0.05 and power of 80%. The enrollment period at each institution is detailed in table 1. End points Enrollment was considered at the time of the first note written in the patient’s medical record in the immunobullous disease clinic. Thus, outside referrals for poorly controlled disease or new diagnoses were treated the same, and both were

176
considered the starting time for enrollment. Follow-up was defined as the time from the first encounter within the clinic, up to the most recent note in the medical record and/or encounter or death, if recorded. In the case of patients receiving trimethoprim-sulfamethoxazole for non-PCP infections, follow-up was stopped at this point. Table 1. A summary of participating institutions’ enrollment
Institution Enrolment years
Singapore General Hospital, Singapore 2005-2014
University of Genoa, Italy 2001-2016
Rabin Medical Center, Israel 2003-2012
Istituto Dermopatico dell’immacolata, Italy 1985-2016
University Medical Center Groningen, the Netherlands 2002-2016
Philipps University, Germany 2004-2016
Patient demographics were extracted, including age, sex, immunobullous disease subtype, systemic medications used for treating the immunobullous disease, associated chronic comorbidities, follow-up time, and the occurrence of PCP. Information on race or ethnicity was not routinely available. Comorbidities evaluated included diabetes, psoriasis, malignant neoplasm, and autoimmune diseases, with hypertension and osteoporosis serving as nonimmunosuppressive comorbidity controls. These were defined as either International Classification of Diseases, Ninth Revision (ICD-9), codes or free text recorded chronic comorbidities. The incidence of PCP was defined as patients with the ICD-9 code 136.3 for Pneumocystis pneumonia or free text documentation of PCP occurring based on characteristic radiographic findings with elevated lactate dehydrogenase or hospitalization for pneumonia with bronchioalveolar lavage demonstrating Pneumocystis jiroveci on confirmatory stains. Statistical analysis Demographic characteristics were summarized descriptively. To determine the incidence of comorbidities, only cases with available information regarding comorbidities were taken into account. Thus, the incidence of each comorbidity was reported as incidence of cases in which comorbidities were available. χ2 Tests

177
were used to compare the incidence of PCP in the study group compared with the proposed cut-off of 3.5% used to justify prophylaxis, as well as 1.3%, which was the mean incidence of PCP in dermatologic patients from the previously discussed literature review. Pertinent subgroup analysis of patients with PCP were additionally performed using a χ2 test to compare subgroup incidence with the proposed 3.5% cut-off. All tests were 2-tailed and performed using the IBM SPSS statistical software, version 20. P < .05 was considered statistically significant. Results In total, 801 patients met the inclusion and exclusion criteria; their mean age (SD) was 66.47 (17.62) years, and a total of 465 (58%) were women. The mean follow-up time was 2.94 years, resulting in 2354 patient-years. Reasons for exclusion included use of dapsone (258 patients), insufficient follow-up (187 patients), and PCP prophylaxis given (6 patients). Additional demographic information is provided in table 2. Of these 801 patients, 1 developed PCP. This patient, a man in his 40s with recalcitrant mucocutaneous pemphigus vulgaris and no reported comorbidities, was treated with high dose of oral prednisolone in combination with rituximab (1000 mg given once on day 1 and repeated on day 15) according to the rheumatology dosing regimen. Because the patient developed erythema multiforme, he was switched to dexamethasone pulse therapy followed by oral dexamethasone, 4 mg per day, as described by Kardaun and Jonkman.15 On day 57 after the first rituximab dosage he developed PCP. The patient subsequently required mechanical ventilation and treatment with trimethoprim-sulfamethoxazole and to date is making a full recovery. Based on the sample size of 801 patients, an estimated 28 patients (3.5%) would need to develop PCP to justify prophylaxis. Comparison of the predicted incidence cut-off (3.5%) to the actual incidence (0.1%) showed χ2
1 (n = 801) = 27.0 (P < .001). To determine whether our sample was significantly lower than that previously reported in the literature for all dermatologic patients, the actual incidence (0.1%) was compared with the previously reported incidence (1.3%) demonstrating χ2
1 (n = 801) = 8.2 (P = .004). A subgroup analysis of the incidence of PCP in patients receiving rituximab demonstrated an incidence of 1 of 140 (0.7%), which compared with the predicted incidence cut-off (3.5%) showed χ2
1 (n = 140) = 3.3 (P = .07).

178
Table 2. Demographics of 801 Patients With Autoimmune Blistering Disease Included in the Present Study Total sample size No. (%)
Follow-up, mean (SD), y 2.94 (3.25)
Age, mean (SD), y 66.47 (17.62)
Female sex 465 (58.0)
Disease subtype
Pemphigus foliaceus 51 (6.4)
Pemphigus vulgaris 360 (44.9)
Bullous pemphigoid 322 (40.2)
Epidermolysis bullosa acquisita 13 (1.6)
Mucous membrane pemphigoid 54 (6.7)
Anti-p200 pemphigoid 1 (0.1)
Medications reviewed
Oral corticosteroid 651 (81.2)
Azathioprine 270 (33.7)
Mycophenolate 113 (14.1)
Methotrexate 66 (8.2)
Cyclosporine 5 (0.6)
Rituximab 140 (17.5)
Intravenous immunoglobulin 34 (4.2)
Oral tetracyclines 120 (15.0)
Cyclophosphamide 28 (3.5)
Comorbidities a
Hypertension 244 (39.4)
Diabetes mellitus 175 (22.9)
Osteoporosis 49 (7.9)
Psoriasis 10 (1.6)
Malignant neoplasm 59 (9.7)
Autoimmune disease 45 (7.3)
Pneumocystis pneumonia incidence
3 months from initial presentation 1 (0.1) a Percentage based on medical records in which comorbidities were available.

179
Because patients with pemphigus often require more significant immunosuppression than patients with pemphigoid, we performed a subgroup analysis of the 1 of 411 patients with PCP and pemphigus (0.2%), which compared with the predicted incidence cut-off (3.5%) showed χ2
1 (n = 411) = 9.7(P = .001). An additional subgroup analysis excluding patients receiving topical steroids, oral tetracyclines, and intravenous immunoglobulin demonstrated an incidence of 1 of 686 (0.14%), which was also significantly smaller than the proposed 3.5% incidence cut-off for prophylaxis use χ2
1 (n = 686) = 21.6 (P = .001). Discussion Patients with AIBD might represent a unique group of iatrogenically immunosuppressed patients. While these patients typically require prolonged use of often multiple immunosuppressive therapies, they may have a lower risk of PCP compared with other dermatologic conditions requiring iatrogenic immunosuppression. Because determining the utility of PCP prophylaxis requires a knowledge of the incidence of PCP in patients not receiving routine prophylaxis, it is essential to characterize this incidence by disease type. Our study of the largest cohort of patients with AIBD highlights the relatively low risk of PCP, with the incidence falling significantly below that of the 3.5% recommended for initiating PCP prophylaxis.2 In addition, our study was sufficiently powered to demonstrate that the incidence of PCP in all dermatologic patients (1.3%) significantly overestimated the incidence in patients with only immunobullous diseases. Thus, the use of routine prophylaxis against PCP in patients with AIBD could not be supported by our data. Because only 1 patient developed PCP, we could not define clear risk factors from our study. This patient developed PCP while receiving both high-dose oral glucocorticoids and after receiving rituximab. He did not have any underlying pulmonary abnormalities, lymphopenia, or neutropenia. In a study of Chinese patients with AIBD, those who developed PCP had absolute lymphocyte counts ranging from 330 to 1200/μL.10 This might indicate that routine laboratory monitoring could identify patients with lymphopenia, prompting either a switch in immunosuppressive therapy, or temporary PCP prophylaxis. Likewise, in a review of all reported cases of PCP developing in dermatology patients, Gonzalez Santiago et al12 described 7 patients who developed PCP, 6 of whom had either lymphopenia, malignant neoplasm, or pulmonary fibrosis and 1 without a

180
description of comorbidities. All of these are known risk factors for PCP, particularly lymphopenia. Limitations Our study has several limitations owing to its retrospective nature. Identification of diagnoses was based on medical records and database review. Multiple criteria to confirm the diagnosis of PCP were chosen to increase the sensitivity for identifying this diagnosis. The medical records of patients who received treatment prior to their referral to tertiary AIBD centers were reviewed for history of pneumonias to avoid underestimation of PCP cases. Cases of pneumonia and atypical pneumonia were all analyzed to ensure that patients did not receive treatment for PCP but rather received antibiotics, such as macrolides or cephalosporins. Still, given that the study captures patients from an initial visit to a tertiary care center with a minimum of 3 months of follow-up, there is a potential for underestimation. This may, however, be balanced by the more severe cases treated in a tertiary care center. Determining duration, treatment courses, or extent of concomitant use of different medication doses could not be performed to further stratify the level of immunosuppression in our population because information from previous medical record systems was summarized in binary form when integrated into newer electronic medical records or databases. Thus, a patient who received simultaneous prednisone and mycophenolate could not be routinely differentiated from a patient receiving prednisone and then later requiring mycophenolate. Further prospective evaluation accounting for the degree of immunosuppression (eg, treatment courses, concomitant therapies) would be beneficial for comparison with other immunosuppressed states. This particularly is true for the use of topical corticosteroids because a patient who received daily whole-body clobetasol would not be considered to be on systemic immunosuppression, despite the significant level of systemic immunosuppression that occurs with this protocol.16 To counter this, all patients with immunobullous disease, whether receiving systemic immunosuppression, were included in the study. Still, a subgroup analysis was performed of patients receiving only systemic immunosuppressive therapy, and our findings of low PCP incidence were not changed even in this high-risk cohort. In addition, the exclusion of patients prescribed dapsone decreased the number of patients assessed. Our study was underpowered to assess the incidence of PCP in patients with AIBDs receiving rituximab. Thus, while the PCP incidence in this group

181
was not significant below the proposed threshold of 3.5%, a larger study would be required to verify this. Finally, our study was performed at tertiary and quaternary care centers, where more aggressive therapies may be used than in community practice. The inclusion of patients from 2 continents and 6 centers, however, improves the generalizability to the larger cohort of patients with AIBDs. Conclusions The high mortality of PCP warrants significant discussion in regard to prophylaxis; however, the incidence of PCP in the disease population must surpass the risks of prophylactic therapy. We demonstrate in a large, multinational cohort of patients with AIBDs that the incidence of PCP does not pass muster. Thus, even in patients with immunobullous disorders receiving various systemic immunosuppressive therapies in the routine clinical setting, lack of prophylaxis was not associated with a sufficient incidence of PCP to warrant prophylaxis. References 1. Thomas CF Jr, Limper AH. Pneumocystis pneumonia. N Engl J Med. 2004;350(24):2487-2498. 2. Green H, Paul M, Vidal L, Leibovici L. Prophylaxis of Pneumocystis pneumonia in
immunocompromised non-HIV-infected patients: systematic review and meta-analysis of randomized controlled trials. Mayo Clin Proc. 2007;82(9):1052-1059.
3. Amber KT. Balancing the risks and benefits of prophylaxis: a reply to “Pneumocystis jiroveci pneumonia in patients treated with systemic immunosuppressive agents for dermatologic conditions”. Int J Dermatol. 2017;56(1):e4-e5.
4. Stern A, Green H, Paul M, Vidal L, Leibovici L. Prophylaxis for Pneumocystis pneumonia (PCP) in non-HIV immunocompromised patients. Cochrane Database Syst Rev. 2014;(10):CD005590.
5. Gafter-Gvili A, Fraser A, Paul M, et al. Antibiotic prophylaxis for bacterial infections in afebrile neutropenic patients following chemotherapy. Cochrane Database Syst Rev. 2012;1:CD004386.
6. Yale SH, Limper AH. Pneumocystis carinii pneumonia in patients without acquired immunodeficiency syndrome: associated illness and prior corticosteroid therapy. Mayo Clin Proc. 1996;71(1):5-13.
7. Caplan A, Fett N, Rosenbach M,Werth VP, Micheletti RG. Prevention and management of glucocorticoid-induced side effects: a comprehensive review: infectious complications and vaccination recommendations. J AmAcad Dermatol. 2017;76(2): 191-198.
8. Lehman JS, Kalaaji AN. Role of primary prophylaxis for Pneumocystis pneumonia in patients treated with systemic corticosteroids or other immunosuppressive agents for immune-mediated dermatologic conditions. J Am Acad Dermatol. 2010;63(5):815-823.
9. Gerhart JL, Kalaaji AN. Development of Pneumocystis carinii pneumonia in patients with immunobullous and connective tissue disease receiving immunosuppressive medications. J Am Acad Dermatol. 2010;62(6):957-961.
10. Li F, Jin HZ, Su F, Jia L, Sun QN. Pneumocystis pneumonia in patients with immunobullous dermatoses. Int J Dermatol. 2011;50(9):1144-1149.
11. Leshem YA, Gdalevich M, Ziv M, David M, Hodak E, Mimouni D. Opportunistic infections in patients with pemphigus. J AmAcad Dermatol. 2014;71 (2):284-292.

182
12. Gonzalez Santiago TM,Wetter DA, Kalaaji AN, Limper AH, Lehman JS. Pneumocystis jiroveci pneumonia in patients treated with systemic immunosuppressive agents for dermatologic conditions: a systematic review with recommendations for prophylaxis. Int J Dermatol. 2016;55(8):823-830.
13. Ognibene FP, Shelhamer JH, Hoffman GS, et al. Pneumocystis carinii pneumonia: a major complication of immunosuppressive therapy in patients withWegener’s granulomatosis. Am J Respir Crit Care Med. 1995;151(3, pt 1):795-799.
14. Leshem YA, Snast I, Friedland R, et al. Is there a role for opportunistic infection prophylaxis in pemphigus? an expert survey. Am J Clin Dermatol. 2017;18(1):127-132.
15. Kardaun SH, Jonkman MF. Dexamethasone pulse therapy for Stevens-Johnson syndrome/toxic epidermal necrolysis. Acta Derm Venereol. 2007;87 (2):144-148.
16. Joly P, Roujeau JC, Benichou J, et al. A comparison of two regimens of topical corticosteroids in the treatment of patients with bullous pemphigoid: a multicenter randomized study. J Invest Dermatol. 2009;129(7):1681-1687.

183

184
11

185
CHAPTER 11 Discussion and future perspectives Aniek Lamberts Center for Blistering Diseases, Department of Dermatology, University Medical Center Groningen, University of Groningen, Groningen, the Netherlands

186
PART 1 : Nonbullous pemphigoid: disease characteristics and immunological aspects Pemphigoid-specific autoantibodies in healthy individuals Several studies have detected pemphigoid-specific IgG autoantibodies in the serum of nondiseased people.1–3 In chapter 2 we assessed the presence of circulating pemphigoid-specific autoantibodies in dermatology patients with a nonbullous skin disorder. A diagnosis of pemphigoid was excluded based on skin biopsy with negative direct immunofluorescence microscopy. Single serological test positivity was found in 14% of the patients, whom had a higher median age compared to patients with negative results. In ten patients (4%), multiple serologic tests were positive. Interestingly, in chapter 6 we also observed circulating IgE autoantibodies to NC16A (8%) and BP230 (20%) in elderly controls with pruritus by enzyme-linked immunosorbent assay (ELISA).
The detection of pemphigoid-specific autoantibodies by ELISA might partly be a result of nonspecific binding, however, it is also hypothesized that such autoantibodies can develop due to a combination of epitope spreading and aging of the immune system. Epitope spreading is previously defined as a specific autoreactive immune response to endogenous epitopes on proteins, secondary to the release of such self-protein during a chronic autoimmune, or inflammatory response.3 Epitope spreading occurs in several autoimmune diseases, including early in BP.3–5 Possibly, immunologically “hidden” epitopes are “revealed” due to cell injury in patients with chronic pruritus by scratching. A second potentially contributing factor is aging of the immune system, which is associated with elevated levels of pro-inflammatory cytokines, and a decline of naive regulatory T cells (Tregs).6,7 Interestingly, a decline of Tregs in blood and skin of BP patients was reported previously.8 Moreover, scurfy mice and patients with IPEX (immunodysregulation, polyendocrinopathy, enteropathy, X-linked) syndrome, whom both lack functional Tregs, spontaneously produced autoantibodies against several antigens located in the BMZ.9–11 This suggests that functional Tregs are essential to prevent self-reactivity to BMZ proteins.
Based on chapter 2, an important question arose; “What are the minimal requirements for a diagnosis of bullous and nonbullous forms of pemphigoid?”. In 2019, Meijer et al. addressed this clinical dilemma in a diagnostic accuracy study, and provided minimal diagnostic criteria for BP and NBP.12 In line with previous

187
findings, IIF SSS showed highest specificity (99.9%), and a positive predictive value of 99.6% for the diagnosis of pemphigoid.12,13 ELISA had no diagnostic value, but was recommended for monitoring disease activity in confirmed pemphigoid patients. Their findings were translated into a 2-out-of-3 rule for the diagnosis of pemphigoid, meaning patients need to fulfill two of the three following criteria: 1) compatible clinical presentation with pruritus and/or predominant skin blisters, 2) positive linear IgG and/or C3c staining along the BMZ by DIF, 3) positive IgG staining on the epidermal side of SSS by IIF. When applying the 2-out-of-3 rule in retrospect, ten patients in chapter 2 fulfilled the diagnostic criteria, and thus received a delayed diagnosis of NBP. Unpublished data regarding these ten patients revealed pruritus in all, and multiple positive serologic tests with predominant BP230 reactivity. It is unknown whether nondiseased patients with pemphigoid-specific autoantibodies detected by ELISA in chapter 2 and 6 are at risk to develop pemphigoid in the future. Long-term follow-up of these patients could answer this question. Disease characteristics of nonbullous pemphigoid The prevalence of nonbullous pemphigoid The NBP phenotype was reported in approximately 20% of the BP cases, but the prevalence of NBP was not studied before.12,14,15 In chapter 5, we assessed the prevalence of pemphigoid, bullous and nonbullous, in a high-risk population of nursing home residents. We observed an exceptional high pemphigoid prevalence of 6%. In 2014, an estimated BP prevalence of 0.026% was reported in the general population of Germany, and 0.3% in elderly people aged above 85 years.16 One previous study assessed the incidence of BP in nursing home residents, and observed a crude incidence of 5% per year, by using clinical skin blisters combined with histopathology findings of a subepithelial split, or positive DIF as diagnostic criteria for BP.17 Our study was the first to assess the full spectrum of pemphigoid in a nursing home population by including NBP. Surprisingly, NBP was more prevalent than BP (4 vs. 3 cases), implying that the nonbullous phenotype of pemphigoid may be more common as previously assumed.18 Importantly, all four NBP cases were misdiagnosed by the nursing home physician with other pruritic skin diseases. This highlights the need for more awareness for NBP, especially amongst healthcare specialists whom care for elderly populations at risk to develop pemphigoid.

188
Interestingly, all seven identified pemphigoid cases had neurodegenerative diseases: dementia in five, Parkinson’s disease in one, and cognitive decline in one patient. In line, several studies observed that neurodegenerative diseases often precede pemphigoid.19–21 The expression of BP180 and BP230 in both skin and brain tissue suggests that cross-reactivity could initiate pemphigoid.21,22 Recently, autoantibodies to BP180 were detected in a large number of patients with Alzheimer’s disease and multiple sclerosis (20% and 54%), however, the autoantibodies were directed against nonpathogenic intracellular and mid-extracellular domains of BP180.23,24 Further studies need to identify factors that trigger intra- and intermolecular epitope spreading, and subsequently initiate pemphigoid disease. Clinical presentation of nonbullous pemphigoid In chapter 3 and 4, we explored the disease characteristics of NBP by systematic review, and a retrospective case study. Corresponding findings were disease onset at old age (75 vs. 76 years), and the heterogeneous clinical presentation with a variety of skin lesions. Both chapters reported nonspecific histopathologic findings in NBP, and blister development during follow-up in the minority of patients (10% and 17%). In line, a recent case series reporting on 36 NBP patients observed blister formation in only 23%.25 In chapter 4, NBP patients with late blister formation had a significant longer follow-up period compared to patients that did not develop blisters. Possibly, blisters might have occurred in more NBP patients if their follow-up period was longer. However, the longer follow-up could also be related to the event of developing blisters, since patients with late blister development likely return for additional treatment advices. Nonetheless, multiple NBP cases remained nonbullous during long follow-up periods, up to 14 years. These findings suggest that NBP should be seen as a phenotypic variant within the pemphigoid spectrum, rather than a prodromal phase of BP.
Several results of chapter 3 and 4 did not correspond. First, published NBP cases most often presented with urticarial papules and plaques (52%), while in our NBP cohort papules and nodules were most common (31%). The high number of published NBP patients with urticarial lesions might be explained by a higher resemblance to the typical clinical picture of BP, triggering clinicians to perform pemphigoid diagnostics more easily. Second, published NBP cases were reasonably equally reactive to BP230 (ELISA 53%; immunoblot 56%), and BP180 (ELISA 58%;

189
immunoblot 32%), while NBP patients in our cohort were predominantly reactive to BP230 (ELISA 47%; immunoblot 41%), and less common to BP180 (ELISA 31%; immunoblot 13%). Possibly, less BP230 reactive NBP cases are published because BP230 reactivity is associated with DIF negativity, and current consensus based guidelines appoint DIF positivity as golden standard for pemphigoid diagnosis.12,26,27 Recently, the need for DIF positivity is refuted by Meijer et al., as was discussed above.12 Presumably, the less NBP patients resemble the typical BP phenotype and immune profile, the less likely clinicians think of pemphigoid. This could explain the subtle differences between published NBP cases, and our cohort of patients diagnosed in a center with high awareness for NBP, and routine pemphigoid diagnostics performed in elderly patients with itch.
Interestingly, Ben Mordehai et al. reported mucosal involvement in 6 of 36 (16.7%) NBP cases.25 In contrast, in chapter 3 and 4 we observed mucosal involvement in only one NBP case published by Ameen et al.28 We question whether patients with bullous or erosive mucosal lesions should receive the diagnosis NBP, as per definition they are not nonbullous. Clinicians must be cautious, and should consider a diagnosis of MMP in patients with nonbullous pruritic skin lesions and mucosal involvement. Immunological aspects of nonbullous pemphigoid An important question is: “Why do NBP patients not develop blisters?”. Although the data in this thesis does not provide the answer, we highlight and discuss potential clues below. Predominant BP230 reactivity in nonbullous pemphigoid In chapter 4, autoantibodies in NBP were predominantly BP230 reactive, opposed to autoantibodies in BP that commonly target BP180, previously described by Meijer et al.12 The pathogenicity of anti-NC16A autoantibodies was proven repeatedly in animal models, and correlated with disease activity in humans.29–33 The pathogenicity of autoantibodies to intracellular BP230 was initially debated, as autoantibodies to BP230 did not lead to skin blistering in mouse studies,34,35 and did not correlate with disease activity in BP patients.30,31,36 Therefore, authors concluded that anti-BP230 autoantibodies must be nonpathogenic.
Nevertheless, other studies did find evidence for a pathogenic role of anti-BP230 autoantibodies. Hall III et al. were one of the first, and induced an anti-

190
BP230 autoantibody response in rabbits, that showed no clinical evidence of skin disease at first.37 However, epithelial injury by ultraviolet B radiation led to an enhanced immune response with epidermal necrosis and linear IgG and C3 deposits at the BMZ, suggesting the intracellular BP230 antigen must be exposed before disease initiates.37 A second study by Kiss et al. subcutaneously injected anti-BP230 autoantibodies into the dorsal skin of seven mice, which induced macroscopic blistering in only one.38 However, all seven mice had erythematous skin, clinically resembling NBP, with histological subepithelial blisters, IgG and C3 depositions along the BMZ, and an intradermal inflammatory reaction. More recently, two studies reported spontaneous anti-BP230 autoantibodies in scurfy mice that lack Tregs.9,10 The mice displayed a blistering pemphigoid phenotype in one study, and eczematous lesions in the other. Haeberle et al. proved the pathogenicity of anti-BP230 autoantibodies by transferring them into neonatal mice, where they induced subepithelial blisters.9 The animal studies discussed above showed evidence that anti-BP230 autoantibodies have pathogenic potential in animals. But what about humans?
In epidermolysis bullosa simplex, homozygous nonsense mutations in the BP230 gene resulted in an absent or truncated BP230 protein, with a clinical phenotype consisting of generalized skin fragility, skin blistering, and in one case prurigo papules.39–41 Moreover, Tanaka et al. studied the relation between clinical data and antigen profile in 100 BP patients, of which 44 patients were reactive to BP230 only.42 These patients needed lower steroid dosages compared with those reactive to BP180 only, however, no differences in disease severity, lesion extent, and steroid responsiveness were observed. Hayawaka et al. performed a similar study, and also found a lower need for systemic corticosteroids in patients with anti-BP230 autoantibodies only (anti-BP230 type BP), but also reported a lower disease severity.43 Moreover, they described ‘unique skin lesions’ compatible with eczematous pemphigoid, pemphigoid nodularis, vesicular pemphigoid, dyshidrosiform pemphigoid, and pretibial localized pemphigoid, implying some of the clinical phenotypes were nonbullous. In contrast, in chapter 4 we did not observe mild disease in NBP patients, whom were often BP230 reactive, but found a frequent necessity of systemic therapy, and increased mortality rates.
Hayakawa et al. additionally mapped the epitopes of patients with anti-BP230 type BP, and showed specific recognition of the C-terminal.43 In contrast, all three domains of BP230 were targeted in BP patients recognizing both BP180 and BP230,

191
suggesting these BP230 autoantibodies might develop secondary to intermolecular epitope spreading.4,43 The authors proposed that anti-BP230 type BP might be a different disease entity, with possibly a different pathogenesis, resulting in a different clinical phenotype.43 We suggest that NBP can be such a phenotype. Complement activation in nonbullous pemphigoid In BP the activation of complement through classical and alternative pathways presumably contributes in blister formation by attraction and activation of immune cells by anaphylatoxins C3a and C5a.44–46 Two studies reported significantly less complement C3c deposits along the BMZ in NBP patients (39% and 52%), compared to BP patients (83% and 77%).12,47 In line, in chapter 4 we observed complement deposits along the BMZ in only 21% of NBP cases, and in chapter 7 a lower expression of genes related to complement activation was found in all NBP biopsies, compared to high expression in 60% of BP biopsies. These findings suggest that complement activation could be the missing link to develop blisters in a subset of NBP patients.
Complement activation through autoantibody binding (classical pathway) can be induced by IgG1 and IgG3 subclasses that have high complement activating abilities, whereas IgG2 has low, and IgG4 has no complement activating abilities.48 In BP, autoantibodies commonly exist of IgG1 and IgG4 subclasses.33,49–51 The lower prevalence of complement deposits in NBP skin suggests that IgG2 or IgG4 might be involved. In line, Lamb et al. found predominant IgG4 subclass positivity in 30 patients with ‘prodromal BP’.52 Zheng et al. assessed IgG subclasses of six patients with anti-BP230 type BP, and found IgG4 positivity in all, while IgG1 and IgG3 were faint or negative.53 Eleven patients with autoantibodies against both BP180 and BP230 also showed IgG4 subclasses (82%), however, together with IgG1 (91%) and IgG3 (64%). As expected, complement C3 deposits were weaker in anti-BP230 type BP compared to BP180+BP230 positive BP.
Interestingly, Buschman et al. linked IgG4 subclass predominance with false-negative DIF results in twelve BP patients.54 The authors demonstrated that BP patients with an initial negative DIF actually did have IgG4 deposits along the BMZ, and suggested that IgG4 subclasses might be below the detection threshold of commercial IgG conjugates used in conventional DIF microscopy.54 However, in this study isotonic salt was not used as biopsy medium, which could have significantly improved the visibility of low IgG4 signals by reducing the overall IgG background

192
staining. The IgG subclasses of autoantibodies in NBP were not studied in this thesis, but will be topic of future research. T helper 2 responses in nonbullous pemphigoid T lymphocytes have a role in initiating and regulating immune responses. T cell responses in NBP were not studied so far, while elevated levels of T helper 1, 2 and 17 (Th1, Th2, Th17) cells and their related cytokines were reported in serum and skin of BP patients.55–58 Moreover, studies on Th2 responses in BP revealed potential pathogenic roles of eosinophils and IgE, which led to targeted therapies that are currently explored.59–63
In chapter 3 and 4 eosinophils were found elevated in the peripheral circulation of NBP (87% and 45%) in similar and higher rates as in BP (50%).64 In line, Ben Mordehai et al. reported a significantly higher blood eosinophil count in NBP compared with BP.25 Eosinophils were detected in histopathologic skin biopsies in 47% of published NBP cases in chapter 3, and in 69% cases of our NBP cohort in chapter 4. Interestingly, eosinophilic spongiosis was less common in NBP (8% and 6%) compared to reports in BP (50%), implying that eosinophils seldom infiltrate the epidermis in NBP.65
We further assessed the presence of pemphigoid-specific IgE autoantibodies in serum and skin of NBP and BP patients in chapter 6, and observed no significant differences. In contrast, Ben Mordehai et al. found significantly higher total IgE levels in serum of NBP compared to BP, but did not assess pemphigoid-specific antibody titers.25 Interestingly, in chapter 6 both NBP and BP skin showed IgE autoantibodies attached to the surface of cells in the dermis, likely eosinophils and mast cells, while IgE deposits in a linear pattern along the BMZ were only found in 2 out of 28 skin biopsies (7%; 1 NBP, 1 BP). The high number of studies that do find linear IgE along the BMZ (in up to 44% of BP biopsies) is surprising, and the conflicting findings must be caused by methodological differences.66–71 Our results suggest that IgE does not directly mediate blister formation by mast cell and eosinophil degranulation in pemphigoid, but could indirectly influence the disease pathogenesis.
Surprisingly, further assessment of the immunological profile of NBP in chapter 7 revealed a lower expression of Th2 related genes compared with BP. Combining our findings, we may conclude that chapter 3, 4 and 6 provide evidence for Th2 responses in both NBP and BP, however, data of chapter 7 implies that Th2

193
responses are more extensive in BP than in NBP. Future studies need to examine Th2 responses in NBP, and need to validate the genetic data presented in chapter 7.
Several immunological pathways and immune players that were thought to play a role in the pathogenesis of NBP and BP are summarized in figure 1. Moreover, an overview of diagnostic immunological findings is provided in table 1.
Management and prognosis of nonbullous pemphigoid Little is published about the management and prognosis of NBP. Lamb et al. treated ‘prodromal BP’ patients with high potent topical steroids (disease resolution in 55%), oral corticosteroids (disease resolution in 27%), tetracycline (disease resolution in 0%), a combination of these three therapies (disease resolution in 50%), or oral corticosteroids combined with an immunosuppressive
Table 1. Diagnostic and immunological findings in bullous and nonbullous pemphigoid
Diagnostic/immunological tests Bullous
pemphigoid* Nonbullous pemphigoid
Direct immune fluorescence microscopy
Positive staining of IgG, % 88% 60%
Positive staining of C3c, % 83% 21%
Positive staining of IgE, % 7% 7%
Serological tests
Positive IgG NC16A ELISA (cut-off index >9), % 70% 31%
Positive IgG BP230 ELISA (cut-off index >9), % 45% 47%
Positive IgE NC16A ELISA** (cut-off OD value 620), % 18% 9%
Positive IgE BP230 ELISA** (cut-off OD value 929), % 34% 22%
Peripheral blood eosinophilia (> 0.40 109/L), % 50% 45%
Elevated total IgE (>115kU/L), % 60% 63%
Predominant IgG subclass IgG1 and IgG4 IgG4 ELISA, enzyme-linked immunosorbent assay. *Percentages on diagnostic findings concerning BP were based on literature.12,64 Percentages concerning diagnostic findings in NBP were based on chapter 4 and 6 of this thesis. Subclass predominance in bullous pemphigoid and nonbullous pemphigoid was based on literature.50-52 ** The methodology of the IgE ELISAs is described in chapter 6 of this thesis.

194
drug (unknown which; disease resolution in 75%).72 Limited follow-up data described relapses within 1 year in 10%, and one death. In line with the study of Lamb et al., chapter 4 showed similar response rates for topical and oral steroids in NBP patients. Moreover, doxycycline monotherapy was also ineffective in our NBP cohort. However, differences in outcome measures in the study of Lamb et al. and chapter 4 does not allow us to draw hard conclusions when comparing results. The study of Ben Mordehai et al. used consensus defined outcome measures73 similar to chapter 4, however, they only focused on achievement of disease control, and did not report remission rates on/off therapy.25 They found sufficient disease control by topical steroids in only 8% and 9% of NBP and BP patients, while all other patients were in need of systemic therapy. Overall, 88% of NBP and 86% of BP patients achieved disease control, but by which treatment was not specified.
Interestingly, our data revealed that NBP responded well to methotrexate (MTX) in a low dose, with long term remission in 43% of the treated patients. Recently, Delaumenie et al. analyzed data of BP patients treated with MTX, and observed a higher response in patients with anti-BP230 autoantibodies compared to patients with anti-BP180 autoantibodies (1 year response rate of 75% vs. 35%).74 Possibly, a difference in disease mechanism in anti-BP230 autoantibody mediated pemphigoid could explain these findings, as was also discussed above.
No previous studies assessed the mortality risk in NBP. In chapter 4, over one-third of the patients in our cohort died within a mean time of 2 years after receiving the diagnosis NBP. The all-cause standardized mortality ratio in the NBP population was 8.6, which is surprisingly higher than reports on BP (3.4 to 6.6).75–77 However, no hard conclusions could be drawn, since our data was limited by a low sample size. It can be hypothesized that the academic setting, in which it is more likely to have severe symptoms, and the long diagnostic delay without adequate therapy influenced the mortality rate. Nonetheless, our findings imply that NBP could not be set aside as a milder disease variant of BP.

195
Figu
re 1
. Sch
emat
ic o
verv
iew
of i
mm
unol
ogic
al fi
ndin
gs in
bul
lous
and
non
bullo
us p
emph
igoi
d. B
ullo
us p
emph
igoi
d: 1
. Aut
oant
ibod
y bi
ndin
g le
ads
to c
ompl
emen
t ac
tivat
ion
by th
e cl
assi
cal p
athw
ay. 2
. Ana
phyl
atox
ins
C3a
and
C5a
bind
rece
ptor
s at
the
endo
thel
ium
, allo
win
g im
mun
e ce
lls to
mig
rate
into
the
derm
is. 3
. Mas
t ce
lls s
ecre
te IL
-5, a
pot
ent a
ctiv
ator
of e
osin
ophi
ls. C
3a, C
5a a
nd Ig
E bi
nd to
mas
t cel
ls a
nd e
osin
ophi
ls c
ould
lead
to a
ctiv
atio
n. A
ctiv
atio
n le
ads
to d
egra
nula
tion
of
prot
eoly
tic e
nzym
es th
at d
egra
de p
rote
ins
of th
e he
mid
esm
osom
e, c
ausi
ng b
liste
r for
mat
ion.
4. C
ompl
emen
t ind
epen
dent
blis
ter f
orm
atio
n m
ay ta
ke p
lace
by
auto
antib
ody
depe
nden
t pin
ocyt
osis
, dep
letin
g BP
180
from
the
hem
ides
mos
ome,
sub
sequ
ently
cau
sing
blis
ter f
orm
atio
n. N
onbu
llous
pem
phig
oid:
1. I
n a
subs
et o
f pa
tient
s co
mpl
emen
t act
ivat
ion
mig
ht o
ccur
thro
ugh
the
clas
sica
l pat
hway
, as
in 2
1% C
3c c
an b
e de
tect
ed li
near
alo
ng th
e ba
sem
ent m
embr
ane
zone
by
dire
ct
imm
unof
luor
esce
nce
mic
rosc
opy.
2. I
n ch
apte
r 4, e
osin
ophi
ls a
re d
etec
ted
in h
isto
path
olog
y bi
opsi
es in
69%
. 3. I
n ch
apte
r 6, I
gE w
as d
etec
ted
on c
ells
in th
e de
rmis
in
71%
, lik
ely
eosin
ophi
ls or
mas
t cel
ls. M
Cs, m
ast c
ells
; eos
, eos
inop
hils
; IL-
5, in
terle
ukin
5.

196
Future perspectives Part 1 of this thesis bundles the first studies that focus on characterizing NBP. In general, NBP is understudied, and many gaps in knowledge exist. The long diagnostic delays, observed in chapter 3 and 4, and frequent misdiagnosis shown in chapter 5, illustrate the high need for more awareness for NBP amongst clinicians. Interestingly, this need was also expressed by others. In chapter 4A, a letter to the editor remarked that NBP was unmentioned in clinical guidelines for chronic pruritus.78,79 In chapter 8, more awareness for NBP was prioritized as third most important unmet need by 30 of 35 clinicians participating in our survey study. This need was also spontaneously mentioned by 2 of 71 participating patients. How to achieve better disease recognition of NBP by clinicians should be the focus and topic of future discussions. Important steps include more research on the prevalence of NBP, and its disease pathogenesis, which may contribute to more acknowledgement of this disease entity by the medical community, and better therapies. PART 2: Management of pemphigoid diseases Unmet needs in pemphigoid diseases Over the last decades, the role of patients to set the research agenda became more important.80,81 The James Lind Alliance (JLA) is an organization that supports Research Priority Setting Partnerships, to prevent a mismatch in studies desired by patients and clinicians, and studies actually performed by researchers.82 In chapter 8, we explored and prioritized unmet needs in pemphigoid diseases by online survey distributed among patients, clinicians and researchers, using a JLA-like methodology. We found that the need for better therapeutic options was ranked most urgent by all interest groups. Patients expressed the need for ‘better treatment options’, clinicians for ‘labeling of new drugs for the indication of pemphigoid’, and researchers for ‘more head-to-head randomized controlled trials comparing effectiveness and safety of current treatments’. Conventional systemic therapies for pemphigoid diseases include oral corticosteroids, methotrexate, dapsone, mycophenolate mofetil, cyclosporine, cyclophosphamide, azathioprine, and all may give serious short- and long-term side effects. Novel treatments for pemphigoid are currently being evaluated, and we will discuss several below.

197
B cell targeting therapies Rituximab Limited data is available on RTX in pemphigoid diseases, therefore, we analyzed our daily practice data of RTX therapy in 28 patients with recalcitrant pemphigoid diseases in chapter 9. We found an overall remission rate of 57%, with best effects in MMP and BP (remission in 64% and 63%). A recent case series of 20 BP patients treated with RTX (19 with 1000mg on day 1 and 15; 1 with 375mg/m2 weekly for four weeks) even reported a higher remission rate (75%).83 Prospective clinical trials need to determine the effectiveness of RTX and its biosimilars in pemphigoid diseases with more certainty. Moreover, further studies are required to evaluate the best dosing schedule.
Only little is known about the changes that B cells undergo during RTX therapy. A recent study analyzed the immunological phenotype of the B cell population in 17 BP patients prior to RTX, and after B cell repopulation.84 BP180-IgG-positive and BP180-IgM-positive B cells were decreased fivefold by RTX, and only BP180-IgM-positive B cells reappeared in a low frequency at month 24. Interestingly, in these B cells a shift in cytokine pattern was noticed with decreased expression of inflammatory cytokines IL-15 and IL-6. Moreover, two patients with long term complete remission off therapy showed elevated expression of anti-inflammatory cytokines IL-10 and IL-1RA on their B cells, suggesting the switch in cytokine pattern reflects on whether or not patients responded well to RTX. Future studies with similar methodology performed in a larger sample size should aim to identify factors that predict treatment success prior to RTX therapy, so that candidates for this treatment can be preselected.
The safety profile of RTX appeared similar, or more favorable than of conventional therapies.83,85,86 In chapter 9, one patient developed a pneumocystis pneumonia (PCP), which is a potentially life threatening opportunistic fungal infection caused by Pneumocystis jiroveci. In chapter 10 we assessed the incidence of PCP infections in patients with autoimmune bullous diseases without PCP prophylaxis, to determine whether or not standard prophylaxis is warranted. Our observed PCP incidence rate was 0.1%, which was below the threshold of 3.5%, calculated by Green et al., by which the number needed to treat significantly outweighs the number needed to harm.87 In 2019, Rekhtman et al. assessed the PCP incidence in 27 health care organizations, and observed the highest PCP

198
incidence (0.2%) in patients using an immunosuppressant combined with systemic corticosteroids.88 By concluding that PCP prophylaxis is warranted in these patients, despite not reaching the 3.5% threshold, the authors launched a discussion wherein it was argued that the calculated threshold of 3.5% did not take severity of the harm into account.87,89,90 Authors debated that the harm of a PCP infection (mortality rates between 30-60%) is higher compared with trimethoprim-sulfamethoxazole use (mostly laboratory abnormalities resolving with discontinuation, and very low percentage of Stevens-Johnson syndrome). Siscos et al. discussed the use of dapsone instead of trimethoprim-sulfamethoxazole for PCP prophylaxis, which provides an additional therapeutic benefit at a low cost.91 In a response letter, it was commented that trimethoprim-sulfamethoxazole is significantly more effective than dapsone in preventing PCP, limiting its use for the proposed dual purpose.92 While our data in chapter 10 made it clear that standard prophylaxis is not necessary in patients with autoimmune bullous diseases, the data of Rekhtman et al. shows that the level of immunosuppression strongly influences the PCP risk. Larger cohort studies on autoimmune bullous patients that take the amount of immunosuppression into account, may reveal which specific drug combinations are an indicator for PCP prophylaxis. Other B cells targeting therapies In chapter 9, RTX appears to be a relatively effective and safe treatment option for pemphigoid diseases, but still, the response rates were lower as in pemphigus diseases.93 Other drugs targeting B cells are currently tested, including ofatumumab, which also targets CD20, belimumab targeting BAFF (B cell activating factor), and ibrutinib, which is a BTK (Bruton’s tyrosine kinase) inhibitor.94 Another interesting attempt to more specifically deplete autoreactive B cells was proposed, using chimeric antigen receptor therapy to target desmoglein-3 specific B cells in pemphigus vulgaris, leaving the nonautoreactive B cell populations intact.95 If this strategy is successful, it might also be possible to engineer this drug into a therapy for pemphigoid diseases. Future perspectives In the upcoming years, there lies an important task for clinicians and researchers to address the need for better therapeutic options for pemphigoid diseases. This need can be addressed by different approaches. Drugs that have shown beneficial

199
effects in other autoimmune diseases with common immunological pathophysiology can be tested. Moreover, a better understanding of the pathophysiology of pemphigoid diseases may identify potential targets to develop novel therapies. A number of promising drugs are currently under investigation.
Complement activation has been designated as an important event in the disease mechanism of pemphigoid diseases. Currently, several clinical trials on anti-complement drugs in pemphigoid diseases are ongoing, and their results are expected soon. Beside complement, IgE and eosinophils were also suggested to play an important role in the pathophysiology. The anti-IgE drug omalizumab, registered for chronic urticaria, has been successful in several preselected BP patients, however a suboptimal long term effectiveness was observed.63 Recent studies demonstrated IgE autoantibodies in serum and skin of EBA and MMP patients, suggesting that omalizumab could be effective in other pemphigoid diseases as well.96–99 Other drugs of interest are mepolizumab, and bertilimumab. Mepolizumab inhibits IL-5, an important mediator of eosinophil activation. The drug was prescribed to BP patients as adjuvant treatment to oral corticosteroids in a placebo controlled, double blind phase 2 pilot study.61 While eosinophils were effectively reduced in skin and blood, mepolizumab showed no additive therapeutic value. Bertilimumab targets eotaxin-1, a potent chemoattractant and regulator of eosinophilic activity. No publications on bertilimumab for BP exist yet, however, ongoing studies are promising, and the drug was designated as orphan drug by the United States Food and Drug Administration for the treatment of BP.100 A last drug of interest is apremilast, an phosphodiesterase 4 (PDE4) inhibitor registered for treatment of psoriasis. PDE4 inhibition reduced blistering in an experimental EBA mouse model, and our Center for Blistering Diseases currently performs a pilot study to examine the effectiveness and safety of apremilast in pemphigoid.101
Over the coming years, novel drugs need to demonstrate whether they have potential as therapeutic option for pemphigoid diseases. The frailty of the elderly pemphigoid population makes it challenging to include large numbers of patients in clinical trials, and emphasizes the need for multicenter collaborations. Beside the necessity for more therapeutic options it would be of great value to patientcare if future research could identify factors that can predict which treatment will be best suitable for individual patients. The ultimate goal for the future would be to realize a more personalized tailored disease management.

200
References 1. Schmidt T, Sitaru C, Amber K, Hertl M. BP180- and BP230-specific IgG autoantibodies in pruritic
disorders of the elderly: a preclinical stage of bullous pemphigoid? Br J Dermatol. 2014;171(2):212-219. doi:10.1111/bjd.12936
2. Hofmann SC, Tamm K, Hertl M, Borradori L. Diagnostic value of an enzyme-linked immunosorbent assay using BP180 recombinant proteins in elderly patients with pruritic skin disorders. Br J Dermatol. 2003;149(4):910-912.
3. Chan LS, Vanderlugt CJ, Hashimoto T, et al. Epitope spreading: lessons from autoimmune skin diseases. J Invest Dermatol. 1998;110(2):103-109. doi:10.1046/j.1523-1747.1998.00107.x
4. Di Zenzo G, Thoma-Uszynski S, Calabresi V, et al. Demonstration of epitope-spreading phenomena in bullous pemphigoid: results of a prospective multicenter study. J Invest Dermatol. 2011;131(11):2271-2280. doi:10.1038/jid.2011.180
5. Cornaby C, Gibbons L, Mayhew V, Sloan CS, Welling A, Poole BD. B cell epitope spreading: mechanisms and contribution to autoimmune diseases. Immunol Lett. 2015;163(1):56-68. doi:10.1016/j.imlet.2014.11.001
6. Pawelec G. Age and immunity: What is “immunosenescence”? Exp Gerontol. 2018;105:4-9. doi:10.1016/j.exger.2017.10.024
7. van der Geest KSM, Abdulahad WH, Tete SM, et al. Aging disturbs the balance between effector and regulatory CD4+ T cells. Exp Gerontol. 2014;60:190-196. doi:10.1016/j.exger.2014.11.005
8. Antiga E, Quaglino P, Volpi W, et al. Regulatory T cells in skin lesions and blood of patients with bullous pemphigoid. J Eur Acad Dermatol Venereol. 2014;28(2):222-230. doi:10.1111/jdv.12091
9. Haeberle S, Wei X, Bieber K, et al. Regulatory T-cell deficiency leads to pathogenic bullous pemphigoid antigen 230 autoantibody and autoimmune bullous disease. J Allergy Clin Immunol. April 2018. doi:10.1016/j.jaci.2018.04.006
10. Muramatsu K, Ujiie H, Kobayashi I, et al. Regulatory T-cell dysfunction induces autoantibodies to bullous pemphigoid antigens in mice and human subjects. J Allergy Clin Immunol. 2018;142(6):1818-1830.e6. doi:10.1016/j.jaci.2018.03.014
11. Hashimoto T, Takahashi H, Sakaguchi S. Regulatory T-cell deficiency and autoimmune skin disease: Beyond the scurfy mouse and immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. J Allergy Clin Immunol. 2018;142(6):1754-1756. doi:10.1016/j.jaci.2018.08.028
12. Meijer JM, Diercks GFH, de Lang EWG, Pas HH, Jonkman MF. Assessment of Diagnostic Strategy for Early Recognition of Bullous and Nonbullous Variants of Pemphigoid. JAMA dermatology. 2019 Feb 1;155(2):158-165. doi:10.1001/jamadermatol.2018.4390
13. Sardy M, Kostaki D, Varga R, et al. Comparative study of direct and indirect immunofluorescence and of bullous pemphigoid 180 and 230 enzyme-linked immunosorbent assays for diagnosis of bullous pemphigoid. J Am Acad Dermatol. 2013;69(5):748-753. doi:https://dx.doi.org/10.1016/j.jaad.2013.07.009
14. Di Zenzo G, Della Torre R, Zambruno G, Borradori L. Bullous pemphigoid: from the clinic to the bench. Clin Dermatol. 2012;30(1):3-16. doi:10.1016/j.clindermatol.2011.03.005
15. Della Torre R, Combescure C, Cortés B, et al. Clinical presentation and diagnostic delay in bullous pemphigoid: A prospective nationwide cohort. Br J Dermatol. 2012;167(5):1111-1117. doi:10.1111/j.1365-2133.2012.11108.x

201
16. Hubner F, Recke A, Zillikens D, Linder R, Schmidt E. Prevalence and Age Distribution of Pemphigus and Pemphigoid Diseases in Germany. J Invest Dermatol. 2016;136(12):2495-2498. doi:10.1016/j.jid.2016.07.013
17. Fernandez-Viadero C, Arce Mateos F, Verduga Velez R, Crespo Santiago D. Blisters in a nursing home: bullous pemphigoid more often than we think? J Am Geriatr Soc. 2004;52(8):1405-1406. doi:10.1111/j.1532-5415.2004.52379_5.x
18. Cozzani E, Gasparini G, Burlando M, Drago F, Parodi A. Atypical presentations of bullous pemphigoid: Clinical and immunopathological aspects. Autoimmun Rev. 2015;14(5):438-445. doi:10.1016/j.autrev.2015.01.006
19. Försti AK, Jokelainen J, Ansakorpi H, et al. Psychiatric and neurological disorders are associated with bullous pemphigoid - A nationwide Finnish Care Register study. Sci Rep. 2016;6(September):1-6. doi:10.1038/srep37125
20. Milani-Nejad N, Zhang M, Kaffenberger J. The association between bullous pemphigoid and neurological disorders: a systematic review. Eur J Dermatol. 2017;27(5):472-481. doi:10.1684/ejd.2017.3066
21. Forsti A-K, Huilaja L, Schmidt E, Tasanen K. Neurological and psychiatric associations in bullous pemphigoid-more than skin deep? Exp Dermatol. 2017;26(12):1228-1234. doi:10.1111/exd.13401
22. Julio TA, Vernal S, Massaro JD, et al. Biological predictors shared by dementia and bullous pemphigoid patients point out a cross-antigenicity between BP180/BP230 brain and skin isoforms. Immunol Res. 2018;66(5):567-576. doi:10.1007/s12026-018-9028-1
23. Tuusa J, Lindgren O, Tertsunen H-M, et al. BP180 Autoantibodies Target Different Epitopes in Multiple Sclerosis or Alzheimer’s Disease than in Bullous Pemphigoid. J Invest Dermatol. 2019;139(2):293-299. doi:10.1016/j.jid.2018.09.010
24. Kokkonen N, Herukka S-K, Huilaja L, et al. Increased Levels of the Bullous Pemphigoid BP180 Autoantibody Are Associated with More Severe Dementia in Alzheimer’s Disease. J Invest Dermatol. 2017;137(1):71-76. doi:10.1016/j.jid.2016.09.010
25. Ben Mordehai Y, Faibish H, Astman N, Greenberger S, Barzilai A, Baum S. Characteristics of patients with bullous pemphigoid: comparison of classic bullous pemphigoid to non-bullous pemphigoid. J Eur Acad Dermatol Venereol. 2020 Jan;34(1):161-165. doi:10.1111/jdv.15883
26. Feliciani C, Joly P, Jonkman MF, et al. Management of bullous pemphigoid: the European Dermatology Forum consensus in collaboration with the European Academy of Dermatology and Venereology. Br J Dermatol. 2015;172(4):867-877. doi:10.1111/bjd.13717
27. Wang M, Lehman JS, Camilleri MJ, Drage LA, Wieland CN. Circulating bullous pemphigoid autoantibodies in the setting of negative direct immunofluorescence findings for bullous pemphigoid: A single-center retrospective review. J Am Acad Dermatol. 2019;81(2):472-479. doi:10.1016/j.jaad.2019.03.062
28. Ameen M, Harman KE, Black MM. Pemphigoid nodularis associated with nifedipine. Br J Dermatol. 2000;142(3):575-577. doi:10.1046/j.1365-2133.2000.03389.x
29. Lo Schiavo A, Ruocco E, Brancaccio G, Caccavale S, Ruocco V, Wolf R. Bullous pemphigoid: etiology, pathogenesis, and inducing factors: facts and controversies. Clin Dermatol. 2013;31(4):391-399. doi:10.1016/j.clindermatol.2013.01.006
30. Schmidt E, Obe K, Brocker EB, Zillikens D. Serum levels of autoantibodies to BP180 correlate

202
with disease activity in patients with bullous pemphigoid. Arch Dermatol. 2000;136(2):174-178. doi:10.1001/archderm.136.2.174
31. Patsatsi A, Kyriakou A, Pavlitou-Tsiontsi A, Giannakou A, Sotiriadis D. Association of autoantibodies to BP180 with disease activity in Greek patients with bullous pemphigoid. Clin Dev Immunol. 2012;2012:854795. doi:10.1155/2012/854795
32. Daneshpazhooh M, Ghiasi M, Lajevardi V, et al. BPDAI and ABSIS correlate with serum anti-BP180 NC16A IgG but not with anti-BP230 IgG in patients with bullous pemphigoid. Arch Dermatol Res. 2018;310(3):255-259. doi:10.1007/s00403-018-1817-9
33. Dopp R, Schmidt E, Chimanovitch I, Leverkus M, Brocker EB, Zillikens D. IgG4 and IgE are the major immunoglobulins targeting the NC16A domain of BP180 in Bullous pemphigoid: serum levels of these immunoglobulins reflect disease activity. J Am Acad Dermatol. 2000;42(4):577-583.
34. Feldrihan V, Licarete E, Florea F, et al. IgG antibodies against immunodominant C-terminal epitopes of BP230 do not induce skin blistering in mice. Hum Immunol. 2014;75(4):354-363. doi:10.1016/j.humimm.2014.01.005
35. Ujiie H, Yoshimoto N, Natsuga K, et al. Immune Reaction to Type XVII Collagen Induces Intramolecular and Intermolecular Epitope Spreading in Experimental Bullous Pemphigoid Models. Front Immunol. 2019;10:1410. doi:10.3389/fimmu.2019.01410
36. Yoshida M, Hamada T, Amagai M, et al. Enzyme-linked immunosorbent assay using bacterial recombinant proteins of human BP230 as a diagnostic tool for bullous pemphigoid. J Dermatol Sci. 2006;41(1):21-30. doi:10.1016/j.jdermsci.2005.11.002
37. Hall RP 3rd, Murray JC, McCord MM, Rico MJ, Streilein RD. Rabbits immunized with a peptide encoded for by the 230-kD bullous pemphigoid antigen cDNA develop an enhanced inflammatory response to UVB irradiation: a potential animal model for bullous pemphigoid. J Invest Dermatol. 1993;101(1):9-14.
38. Kiss M, Husz S, Janossy T, et al. Experimental bullous pemphigoid generated in mice with an antigenic epitope of the human hemidesmosomal protein BP230. J Autoimmun. 2005;24(1):1-10. doi:10.1016/j.jaut.2004.09.007
39. Groves RW, Liu L, Dopping-Hepenstal PJ, et al. A homozygous nonsense mutation within the dystonin gene coding for the coiled-coil domain of the epithelial isoform of BPAG1 underlies a new subtype of autosomal recessive epidermolysis bullosa simplex. J Invest Dermatol. 2010;130(6):1551-1557. doi:10.1038/jid.2010.19
40. He Y, Leppert J, Steinke H, Has C. Homozygous Nonsense Mutation and Additional Deletion of an Amino Acid in BPAG1e Causing Mild Localized Epidermolysis Bullosa Simplex. Acta Derm Venereol. 2017;97(5):657-659. doi:10.2340/00015555-2618
41. Turcan I, Pasmooij AMG, Gostynski A, et al. Epidermolysis Bullosa Simplex Caused by Distal Truncation of BPAG1-e: An Intermediate Generalized Phenotype with Prurigo Papules. J Invest Dermatol. 2017;137(10):2227-2230. doi:10.1016/j.jid.2017.04.041
42. Tanaka M, Hashimoto T, Dykes PJ, Nishikawa T. Clinical manifestations in 100 Japanese bullous pemphigoid cases in relation to autoantigen profiles. Clin Exp Dermatol. 1996;21(1):23-27.
43. Hayakawa T, Teye K, Hachiya T, et al. Clinical and immunological profiles of anti-BP230-type bullous pemphigoid: Restriction of epitopes to the C-terminal domain of BP230, shown by novel ELISAs of BP230-domain specific recombinant proteins. Eur J Dermatol. 2016;26(2):155-163.

203
doi:10.1684/ejd.2015.2719 44. Dainichi T, Chow Z, Kabashima K. IgG4, complement, and the mechanisms of blister formation in
pemphigus and bullous pemphigoid. J Dermatol Sci. 2017;88(3):265-270. doi:10.1016/j.jdermsci.2017.07.012
45. Edwards G, Diercks GFH, Seelen MAJ, Horvath B, van Doorn MBA, Damman J. Complement Activation in Autoimmune Bullous Dermatoses: A Comprehensive Review. Front Immunol. 2019;10:1477. doi:10.3389/fimmu.2019.01477
46. Sadik CD, Miyabe Y, Sezin T, Luster AD. The critical role of C5a as an initiator of neutrophil-mediated autoimmune inflammation of the joint and skin. Semin Immunol. 2018;37:21-29. doi:10.1016/j.smim.2018.03.002
47. Romeijn TR, Jonkman MF, Knoppers C, Pas HH, Diercks GFH. Complement in bullous pemphigoid: results from a large observational study. Br J Dermatol. 2017;176(2):517-519. doi:10.1111/bjd.14822
48. Tao MH, Smith RI, Morrison SL. Structural features of human immunoglobulin G that determine isotype-specific differences in complement activation. J Exp Med. 1993;178(2):661-667. doi:10.1084/jem.178.2.661
49. Jankaskova J, Horvath ON, Varga R, et al. Increased sensitivity and high specificity of indirect immunofluorescence in detecting IgG subclasses for diagnosis of bullous pemphigoid. Clin Exp Dermatol. 2018;43(3):248-253. doi:10.1111/ced.13371
50. Bernard P, Aucouturier P, Denis F, Bonnetblanc JM. Immunoblot analysis of IgG subclasses of circulating antibodies in bullous pemphigoid. Clin Immunol Immunopathol. 1990;54(3):484-494.
51. Al-Karawi KS. Immunoglobulin G subclass distribution of bullous pemphigoid autoantibodies and complement fixation studies. Saudi Med J. 2002;23(12):1492-1495.
52. Lamb PM, Patton T, Deng JS. The predominance of IgG4 in prodromal bullous pemphigoid. Int J Dermatol. 2008;47(2):150-153. doi:10.1111/j.1365-4632.2008.03361.x
53. Zheng M, Ujiie H, Iwata H, et al. Characteristics of IgG subclasses and complement deposition in BP230-type bullous pemphigoid. J Eur Acad Dermatol Venereol. 2019 Mar;33(3):595-600. doi:10.1111/jdv.15325
54. Buschman KE, Seraly M, Thong HY, Deng J-S, Draviam RP, Abernethy JL. A predominant IgG4 subclass may be responsible for false-negative direct immunofluorescence in bullous pemphigoid. J Cutan Pathol. 2002;29(5):282-286.
55. Budinger L, Borradori L, Yee C, et al. Identification and characterization of autoreactive T cell responses to bullous pemphigoid antigen 2 in patients and healthy controls. J Clin Invest. 1998;102(12):2082-2089. doi:10.1172/JCI3335
56. Echigo T, Hasegawa M, Shimada Y, Inaoki M, Takehara K, Sato S. Both Th1 and Th2 chemokines are elevated in sera of patients with autoimmune blistering diseases. Arch Dermatol Res. 2006;298(1):38-45. doi:10.1007/s00403-006-0661-5
57. Eming R, Budinger L, Riechers R, et al. Frequency analysis of autoreactive T-helper 1 and 2 cells in bullous pemphigoid and pemphigus vulgaris by enzyme-linked immunospot assay. Br J Dermatol. 2000;143(6):1279-1282.
58. Arakawa M, Dainichi T, Ishii N, et al. Lesional Th17 cells and regulatory T cells in bullous pemphigoid. Exp Dermatol. 2011;20(12):1022-1024. doi:10.1111/j.1600-0625.2011.01378.x
59. de Graauw E, Sitaru C, Horn M, et al. Evidence for a role of eosinophils in blister formation in

204
bullous pemphigoid. Allergy Eur J Allergy Clin Immunol. 2017;72(7):1105-1113. doi:10.1111/all.13131
60. van Beek N, Schulze FS, Zillikens D, Schmidt E. IgE-mediated mechanisms in bullous pemphigoid and other autoimmune bullous diseases. Expert Rev Clin Immunol. 2016;12(3):267-277. doi:10.1586/1744666X.2016.1123092
61. Simon D, Yousefi S, Cazzaniga S, et al. Mepolizumab failed to affect bullous pemphigoid: a randomized, placebo-controlled, double-blind phase 2 pilot study. Allergy. 2019 Jun 22. doi: 10.1111/all.13950. [Epub ahead of print]
62. Fairley JA, Baum CL, Brandt DS, Messingham KAN. Pathogenicity of IgE in autoimmunity: successful treatment of bullous pemphigoid with omalizumab. J Allergy Clin Immunol. 2009;123(3):704-705. doi:10.1016/j.jaci.2008.11.035
63. Kremer N, Snast I, Cohen ES, et al. Rituximab and Omalizumab for the Treatment of Bullous Pemphigoid: A Systematic Review of the Literature. Am J Clin Dermatol. 2019 Apr;20(2):209-216. doi:10.1007/s40257-018-0401-6
64. Kridin K. Peripheral eosinophilia in bullous pemphigoid: Prevalence and influence on the clinical manifestation. Br J Dermatol. 2018:1-7. doi:10.1111/bjd.16679
65. Hodge B, Roach J, Reserva JL, et al. The Spectrum of Histopathologic Findings in Pemphigoid: Avoiding Diagnostic Pitfalls. J Cutan Pathol. 2018;(August):1-8. doi:10.1111/cup.13343
66. Kamata A, Kurihara Y, Funakoshi T, et al. Basement membrane zone IgE deposition is associated with bullous pemphigoid disease severity and treatment results. Br J Dermatol. 2019 Jul 22. doi: 10.1111/bjd.18364. [Epub ahead of print]
67. Dimson OG, Giudice GJ, Fu CL, et al. Identification of a potential effector function for IgE autoantibodies in the organ-specific autoimmune disease bullous pemphigoid. J Invest Dermatol. 2003;120(5):784-788. doi:10.1046/j.1523-1747.2003.12146.x
68. Freire PC, Munoz CH, Stingl G. IgE autoreactivity in bullous pemphigoid: eosinophils and mast cells as major targets of pathogenic immune reactants. Br J Dermatol. 2017;177(6):1644-1653. doi:10.1111/bjd.15924
69. Moriuchi R, Nishie W, Ujiie H, Natsuga K, Shimizu H. In vivo analysis of IgE autoantibodies in bullous pemphigoid: a study of 100 cases. J Dermatol Sci. 2015;78(1):21-25. doi:10.1016/j.jdermsci.2015.01.013
70. Provost TT, Tomasi TBJ. Immunopathology of bullous pemphigoid. Basement membrane deposition of IgE, alternate pathway components and fibrin. Clin Exp Immunol. 1974;18(2):193-200.
71. Yayli S, Pelivani N, Beltraminelli H, et al. Detection of linear IgE deposits in bullous pemphigoid and mucous membrane pemphigoid: a useful clue for diagnosis. Br J Dermatol. 2011;165(5):1133-1137. doi:https://dx.doi.org/10.1111/j.1365-2133.2011.10481.x
72. Lamb PM, Abell E, Tharp M, Frye R, Deng JS. Prodromal bullous pemphigoid. Int J Dermatol. 2006;45(3):209-214. doi:10.1111/j.1365-4632.2004.02457.x
73. Murrell DF, Daniel BS, Joly P, et al. Definitions and outcome measures for bullous pemphigoid: recommendations by an international panel of experts. J Am Acad Dermatol. 2012;66(3):479-485. doi:10.1016/j.jaad.2011.06.032
74. Delaumenie S, Assikar S, Prudhomme R, et al. Methotrexate is safe and efficient as long-term treatment for bullous pemphigoid. Eur J Dermatol. 2019;29(2):217-218.

205
doi:10.1684/ejd.2019.3501 75. Kridin K, Bergman R. Mortality in Patients with Bullous Pemphigoid: A Retrospective Cohort
Study, Systematic Review and Meta-analysis. Acta Derm Venereol. 2019 Jan 1;99(1):72-77. doi:10.2340/00015555-2930
76. Kalinska-Bienias A, Lukowska-Smorawska K, Jagielski P, Kowalewski C, Wozniak K. Mortality in bullous pemphigoid and prognostic factors in 1st and 3rd year of follow-up in specialized centre in Poland. Arch Dermatol Res. 2017 Nov;309(9):709-719. doi:10.1007/s00403-017-1772-x
77. Joly P, Baricault S, Sparsa A, et al. Incidence and mortality of bullous pemphigoid in France. J Invest Dermatol. 2012;132(8):1998-2004. doi:10.1038/jid.2012.35
78. Weisshaar E, Szepietowski JC, Dalgard FJ, et al. European S2k Guideline on Chronic Pruritus. Acta Derm Venereol. 2019;99(5):469-506. doi:10.2340/00015555-3164
79. Millington GWM, Collins A, Lovell CR, et al. British Association of Dermatologists’ guidelines for the investigation and management of generalized pruritus in adults without an underlying dermatosis, 2018. Br J Dermatol. 2018;178(1):34-60. doi:10.1111/bjd.16117
80. Crowe S, Fenton M, Hall M, Cowan K, Chalmers I. Patients’, clinicians’ and the research communities’ priorities for treatment research: there is an important mismatch. Res Involv Engagem. 2015;1:2. doi:10.1186/s40900-015-0003-x
81. Tallon D, Chard J, Dieppe P. Relation between agendas of the research community and the research consumer. Lancet (London, England). 2000;355(9220):2037-2040. doi:10.1016/S0140-6736(00)02351-5
82. NETSCC, Cowan K. The James Lind Alliance Guidebook 2006. http://www.jla.nihr.ac.uk/jla-guidebook/downloads/JLA-Guidebook-Version-6-February-2016.pdf.
83. Polansky M, Eisenstadt R, DeGrazia T, Zhao X, Liu Y, Feldman R. Rituximab therapy in patients with bullous pemphigoid: A retrospective study of 20 patients. J Am Acad Dermatol. 2019;81(1):179-186. doi:10.1016/j.jaad.2019.03.049
84. Nicolas B, Pascal J, Marie-Laure G, et al. B-cell depletion induces a shift in self antigen specific B-cell repertoire and cytokine pattern in patients with bullous pemphigoid. Sci Rep. 2019;9(1):3525. doi:10.1038/s41598-019-40203-7
85. Maley A, Warren M, Haberman I, Swerlick R, Kharod-Dholakia B, Feldman R. Rituximab combined with conventional therapy versus conventional therapy alone for the treatment of mucous membrane pemphigoid (MMP). J Am Acad Dermatol. 2016;74(5):835-840. doi:10.1016/j.jaad.2016.01.020
86. Cho YT, Chu CY, Wang LF. First-line combination therapy with rituximab and corticosteroids provides a high complete remission rate in moderate-to-severe bullous pemphigoid. Br J Dermatol. 2015;173(1):302-304. doi:10.1111/bjd.13633
87. Green H, Paul M, Vidal L, Leibovici L. Prophylaxis of Pneumocystis pneumonia in immunocompromised non-HIV-infected patients: systematic review and meta-analysis of randomized controlled trials. Mayo Clin Proc. 2007;82(9):1052-1059. doi:10.4065/82.9.1052
88. Rekhtman S, Strunk A, Garg A. Incidence of pneumocystosis among patients exposed to immunosuppression. J Am Acad Dermatol. 2019;80(6):1602-1607. doi:10.1016/j.jaad.2018.12.052
89. Kowalski EH, Kneiber D, Kridin K, Amber KT. Comment on “Incidence of pneumocystosis among patients exposed to immunosuppression”. J Am Acad Dermatol. 2019;81(2):e47.

206
doi:10.1016/j.jaad.2019.02.071 90. Rekhtman S, Strunk A, Garg A. Reply to: “Comment on ‘Incidence of pneumocystosis among
patients exposed to immunosuppression’”. J Am Acad Dermatol. 2019;81(2):e49-e50. doi:10.1016/j.jaad.2019.03.072
91. Siscos SM, Neill BC, Tarantino IS, Aires DJ, Rajpara A. Dapsone advantages over trimethoprim-sulfamethoxazole for Pneumocystis pneumonia prophylaxis in immunobullous patients. J Am Acad Dermatol 2019 Feb 27. pii: S0190-9622(19)30346-9. doi:10.1016/j.jaad.2019.01.091
92. Kneiber D, Kowalski EH, Amber KT. Dapsone, two birds with one stone: a response to “Dapsone advantages over trimethoprim-sulfamethoxazole for Pneumocystis pneumonia prophylaxis in immunobullous patients”. J Am Acad Dermatol. 2019 Apr 19. pii: S0190-9622(19)30631-0. doi:10.1016/j.jaad.2019.03.093
93. Joly P, Maho-Vaillant M, Prost-Squarcioni C, et al. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet (London, England). 2017;389(10083):2031-2040. doi:10.1016/S0140-6736(17)30070-3
94. Izumi K, Bieber K, Ludwig RJ. Current Clinical Trials in Pemphigus and Pemphigoid. Front Immunol. 2019;10:978. doi:10.3389/fimmu.2019.00978
95. Ellebrecht CT, Bhoj VG, Nace A, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. 2016;353(6295):179-184. doi:10.1126/science.aaf6756
96. Yayli S, Pelivani N, Beltraminelli H, et al. Detection of linear IgE deposits in bullous pemphigoid and mucous membrane pemphigoid: A useful clue for diagnosis. Br J Dermatol. 2011;165(5):1133-1137. doi:https://dx.doi.org/10.1111/j.1365-2133.2011.10481.x
97. Corti L, Fanoni D, Venegoni L, Muratori S, Recalcati S, Berti E. Detection of IgE autoantibodies in mucous membrane pemphigoid and their association with disease severity. G Ital Dermatol Venereol. 2018 Oct 4. doi:10.23736/S0392-0488.18.06167-9. [Epub ahead of print]
98. Koga H, Teye K, Yamashita K, Ishii N, Tsuruta D, Nakama T. Detection of anti-type VII collagen IgE antibodies in epidermolysis bullosa acquisita. Br J Dermatol. 2019;180(5):1107-1113. doi:10.1111/bjd.17310
99. Oya K, Watanabe R, Konishi R, et al. Possible involvement of IgE antibody in epidermolysis bullosa acquisita: detection and correlation. Eur J Dermatol. 2019;29(2):210-212. doi:10.1684/ejd.2019.3497
100. Kridin K, Ahn C, Huang WC, Ansari A, Sami N. Treatment Update of Autoimmune Blistering Diseases. Dermatol Clin. 2019;37(2):215-228. doi:10.1016/j.det.2018.12.003
101. Koga H, Recke A, Vidarsson G, et al. PDE4 Inhibition as Potential Treatment of Epidermolysis Bullosa Acquisita. J Invest Dermatol. 2016;136(11):2211-2220. doi:10.1016/j.jid.2016.06.619

207

208
12

209
CHAPTER 12 Summary Aniek Lamberts Center for Blistering diseases, Department of Dermatology University Medical Center Groningen, University of Groningen Groningen the Netherlands

210
This thesis presents studies that are focused on pemphigoid diseases. Pemphigoid diseases are autoantibody mediated bullous diseases that may affect the skin and/or mucous membranes. We further introduce the clinical presentation, diagnostics, and management of different pemphigoid variants in chapter 1. The thesis is divided into two parts. Part 1 of the thesis consists of studies performed to characterize the pemphigoid variant named nonbullous pemphigoid (NBP). Part 2 presents studies that focus on the management of pemphigoid diseases. PART 1 – Nonbullous pemphigoid: diseases characteristics and immunological aspects. Bullous pemphigoid (BP) is the most prevalent pemphigoid subtype, and typically presents with severe pruritus and tense blisters mainly located on the skin. Interestingly, a less known phenotypic variant of BP clinically lacks blisters, a disease termed NBP. NBP is understudied, as only few case reports and small case series exist in literature. Therefore, patients with NBP often have a long diagnostic delay. Diagnostic and immunological findings can be similar to BP, and it is unknown which underlying mechanism prevents blisters to form in NBP. The aim of part 1 of this thesis was to study the disease characteristics of NBP, and to learn more about its underlying immunological disease mechanism. Pemphigoid-specific autoantibodies in healthy individuals In chapter 2 we first assessed the prevalence of pemphigoid-specific IgG autoantibodies in sera of dermatology patients with a nonbullous skin disorder. A diagnosis of pemphigoid was excluded based on skin biopsy with negative direct immunofluorescence microscopy. We found single serological test positivity in 14%. Interestingly, single test positivity was related to a significant higher median age. In chapter 6 we also found circulating pemphigoid-specific IgE autoantibodies in 28% of elderly controls with pruritus. The relevance of these IgG and IgE serum autoantibodies against BP180 and BP230 in ‘healthy’ elderly persons is unknown. It was hypothesized that ELISA positivity might rely on nonspecific binding, or that the immune response could be related to a phenomenon termed ‘epitope spreading’. Moreover, autoantibody formation could be related to a decrease in functional regulatory T cells, due to aging of the immune system. In 2019, Meijer et al. studied the minimal criteria for the diagnosis of NBP and BP in a diagnostic accuracy study, and showed that DIF positivity is not

211
mandatory for the diagnosis of pemphigoid. Based on their findings and proposed diagnostic criteria, ten patients in chapter 2 received a delayed diagnosis of NBP. Disease characteristics of nonbullous pemphigoid Clinical findings In chapter 3 we systematically reviewed the literature on NBP and identified 132 published cases. Overall, patients had long diagnostic delays (mean 23 months), and presented with heterogeneous skin manifestations. Erythematous urticarial plaques (52%), and papules and nodules (21%) were most commonly reported. Reported histopathologic findings were mainly nonspecific. Blister formation during follow-up was observed in 10% of the patients only. In chapter 4 we described the disease characteristics of our cohort of 68 NBP patients. Again, long diagnostic delays (mean 29 months) were observed. Skin examination most frequently showed papules and nodules (31%). Pruritus on primary nondiseased, noninflamed skin was found in 22%. In line with chapter 3, we observed that blisters occurred in the minority of patients (17%) during follow-up. Interestingly, low dose methotrexate showed high remission rates (43%) of long duration (mean 95 weeks). The all-cause mortality rate was high, as 36% of the patients died after a mean follow-up time of 2 years. The standardized mortality ratio was higher (8.6) compared to previous reports on BP, but should be interpreted with caution because of our low sample size. Both chapter 3 and chapter 4 plead for more awareness amongst clinicians for NBP as cause of pruritus in elderly patients. Moreover, our data indicates that NBP has no mild disease course, and shows a frequent necessity for systemic treatment. Prevalence of nonbullous pemphigoid In chapter 5 we assessed the prevalence of pemphigoid in the assumed high-risk population of nursing home residents. An extra blood sample for serologic diagnostic pemphigoid tests was taken of 125 nursing home residents during a routine venous punction. Seven nursing home residents were diagnosed with pemphigoid (3 bullous, 4 nonbullous). The overall pemphigoid prevalence in nursing home residents was 6%, which is substantially higher than the prevalence in the general population (0.3% in persons aged ≥ 85 years). All four NBP cases were newly diagnosed, and had inappropriately treated pruritic symptoms. Our

212
findings illustrate that NBP can be a cause of pruritus in nursing home residents, and merits more attention from nursing home physicians. Immunological aspects of nonbullous pemphigoid In chapter 6 we investigated the presence of IgE autoantibodies in serum and skin of NBP and BP patients, using ELISA and immunofluorescent staining. The percentage of positive anti-BP180 and anti-BP230 IgE ELISAs did not differ significantly between both pemphigoid phenotypes. In the skin of NBP and BP patients, we mainly detected IgE bound to the surface of cells in the dermis (71% and 86%). Only two skin samples (7%; 1 NBP, 1 BP) showed linear IgE along the basement membrane zone. Based on our findings we concluded that IgE might play a role in the pathogenesis of both NBP and BP, but is presumably not centrally involved in blister formation.
In chapter 7 we aimed to learn more about the disease pathogenesis of NBP and BP by assessing the gene expression profile of lesional skin of NBP and BP patients, using the nanoString technology. Six of ten BP patients showed high expression of complement activation related genes, while a low expression was found in four BP, and all 12 NBP skin samples. A dual high expression of Th1 and Th2 related genes was seen in all BP skin samples, while only the minority of NBP samples showed elevated Th1 or Th2 related gene expression. Moreover, several genes were significantly higher expressed in BP compared to NBP skin, including genes encoding for chemoattractants IL-8 and CCL3, epidermal growth factors HBEGF and AREG, transcription factors CREM and FOSL1, receptors ILR1RL1 and GPR65, the gene encoding for COX2, and the proteinase ADAMTS4. These activated genes may be relevant in blister formation, however, further studies are needed to validate this genetic data.
In chapter 11, discussion and future perspectives, we further elaborated on several other immunological aspects that could have a role in the disease pathogenesis of NBP. This includes antigen- and epitope recognition, complement activation, IgG subclass profile, and the activation of eosinophilic granulocytes. PART 2 – Management of pemphigoid diseases Part 2 of this thesis presents studies concerning the management of pemphigoid diseases. In general, the treatment of pemphigoid diseases consists of immunosuppressive- or immunomodulating drugs. These conventional therapies

213
are not always successful in achieving disease control, and may have serious side effects, especially in the vulnerable elderly population that often has multiple comorbidities. Hence, the ongoing search for drugs for pemphigoid diseases with a high effectiveness and a better safety profile. In chapter 8 we performed an international survey study to explore unmet needs in pemphigoid diseases from the perspective of patients, clinicians, and researchers. In total, 107 participants were included, of which most were patients (66%). All three interest groups expressed a high need for improvement and widening of therapeutic options for pemphigoid diseases. Moreover, patients with pemphigoid diseases strongly carried out the need for more disease awareness amongst clinicians, to prevent long diagnostic delays, and misdiagnosis. In chapter 9 we investigated the effectiveness and safety of rituximab infusions in 28 patients with recalcitrant pemphigoid diseases in a retrospective case series. Overall, remission was achieved in 57% of the patients. Mucous membrane pemphigoid patients responded best (remission in 64%), followed by BP patients (63%). Patients with epidermolysis bullosa acquisita showed lower remission rates (40%), and one patient with linear IgA disease showed no response. Overall, two-third of the pemphigoid patients that achieved remission eventually relapsed, and repeated rituximab infusions again led to remission in 86%. Moreover, our data implied that 1000 mg rituximab on day 0 and 15 was more effective than 500 mg, although this difference was not statistically significant. Also, we observed a poor response on rituximab in IgA-dominant pemphigoid cases, suggesting that clinicians should rather prescribe an alternative therapy in such patients. In chapter 10 we assessed the prevalence of pneumocystis pneumonia (PCP) in patients with autoimmune blistering diseases without routine PCP prophylaxis in a multicenter retrospective patient chart study. Data of 801 patients with autoimmune blistering diseases showed an incidence of PCP of 0.1%. This incidence was below the previously determined threshold of 3.5% to justify PCP prophylaxis, and we therefore concluded that routine PCP prophylaxis is not warranted in all patients with autoimmune blistering diseases. Future research should study individual patient characteristics, and the level of immunosuppression

214
in patients that developed a PCP, to provide more guidance in which patients do benefit of PCP prophylaxis. In chapter 11, discussion and future perspectives, we shortly discussed several novel therapies that are currently being researched. These drugs include anti-complement therapies, IgE blockage, eosinophilic granulocyte targeting drugs (anti-IL-5 and anti-eotaxin-1), and phosphodiesterase 4 inhibitors.

215

216
13

217
HOOFDSTUK 13 Samenvatting Aniek Lamberts Centrum voor Blaarziekten, afdeling Dermatologie, Universitair Medisch Centrum Groningen, Universiteit van Groningen Groningen, Nederland

218
Dit proefschrift beschrijft studies gericht op pemfigoïd ziekten. Pemfigoïd ziekten zijn auto-antilichaam gemedieerde blaarziekten van de huid en/of de slijmvliezen. We geven een uitgebreide introductie over de klinische presentatie, diagnostiek en behandeling van verschillende pemfigoïd ziekten in hoofdstuk 1. Dit proefschrift is opgedeeld in twee delen. Deel 1 bestaat uit verschillende studies met als doel de pemfigoïd variant genaamd nonbulleus pemfigoïd (NBP) te karakteriseren. Deel 2 presenteert studies gericht op de behandeling van pemfigoïd ziekten. Deel 1 – Nonbulleus pemfigoïd: ziekte karakteristieken en immunologische aspecten. Bulleus pemfigoïd (BP) is de meest voorkomende pemfigoïd ziekte, en kent een typische presentatie met ernstige jeuk en pral gespannen blaren op de huid. Een minder bekende fenotypische variant van BP kent een klinische presentatie zonder blaren, een ziekte genaamd NBP. NBP is weinig onderzocht, en de literatuur beschrijft slechts enkele casuïstiek. Patiënten met NBP hebben hierdoor vaak een lange diagnostische vertraging. De diagnostische en immunologische bevindingen bij NBP kunnen overeenkomen met BP, en het is onbekend welk onderliggend immunologisch mechanisme voorkomt dat er bij NBP blaren ontstaan. Het doel van deel 1 van dit proefschrift is om de ziekte NBP te karakteriseren, en meer te leren over het onderliggende immunologische ziektemechanisme. Pemfigoïd-specifieke auto-antilichamen in gezonde individuen In hoofdstuk 2 onderzochten wij allereerst de prevalentie van pemfigoïd-specifieke IgG autoantilichamen in het serum van dermatologie patiënten met een niet-bulleuze huidziekte. De diagnose pemfigoïd was bij deze patiënten verworpen op basis van negatieve directe immunofluorescentie microscopie. Bij 14% van deze patiënten was tenminste één serologische test positief. Opvallend was dat serologische test positiviteit gerelateerd bleek aan een significant hogere mediane leeftijd. In hoofdstuk 6 vonden we eveneens circulerende pemfigoïd-specifieke IgE autoantilichamen in 28% van de controle populatie ouderen met jeuk. De relevantie van deze pemfigoïd-specifieke IgG en IgE autoantilichamen in ‘gezonde’ oudere personen is onbekend. Verondersteld werd dat ELISA positiviteit mogelijk kan berusten op aspecifieke antilichaam binding, of dat de immunologische reactie gerelateerd kan zijn aan een fenomeen genaamd ‘epitope spreading’. Daarnaast

219
kan het ontstaan van autoantilichamen gerelateerd zijn aan een vermindering van functionele regulatoire T cellen door veroudering van het immuunsysteem. In 2019 onderzochten Meijer et al. de minimale criteria voor de diagnose NBP en BP, en toonden aan dat DIF positiviteit niet noodzakelijk is voor de diagnose pemfigoïd. Op basis van deze bevindingen en de door hen voorgestelde diagnostische criteria stelden we bij tien patiënten uit hoofdstuk 2 verlaat de diagnose NBP. Ziekte kenmerken van nonbulleus pemfigoïd Klinische bevindingen In hoofdstuk 3 onderzochten wij op systematische wijze de bestaande literatuur over NBP, en identificeerden 132 gepubliceerde casus. Doorgaans was er sprake van een vertraagde diagnose (gemiddelde duur van 23 maanden) en presenteerden patiënten zich met heterogene huidafwijkingen. ‘Erythemateuze urticariële plaques’ (52%) en ‘papels en noduli’ (21%) werden het vaakst beschreven. De beschreven histopathologische bevindingen waren voornamelijk aspecifiek. Blaarvorming tijdens het ziekte beloop werd slechts in 10% van de patiënten gerapporteerd. In hoofdstuk 4 beschreven wij de ziekte karakteristieken van ons eigen cohort van 68 patiënten met NBP. Opnieuw was de tijd tot het stellen van de juiste diagnose lang (gemiddelde 29 maanden). Dermatologisch onderzoek toonde voornamelijk ‘papels en noduli’ (31%). Jeuk zonder primaire huidafwijkingen werd gezien in 22% van de NBP patiënten. In overeenstemming met hoofdstuk 3 ontwikkelde de minderheid van de patiënten blaren tijdens follow-up (17%). Een interessante bevinding was de goede response op een lage dosering methotrexaat (remissie in 43%) met lange remissie duur (gemiddeld 95 weken). Het sterftepercentage, alle oorzaken meegerekend, was hoog: 36% van de patiënten met NBP overleed na een gemiddelde follow-up tijd van 2 jaar. De gestandaardiseerde mortaliteit ratio was opvallend hoog in NBP (8.6) ten opzichte van gepubliceerde mortaliteit ratio’s in BP, echter moet deze bevinding weloverwogen worden geïnterpreteerd vanwege onze lage steekproefomvang. Zowel hoofdstuk 3 en 4 pleiten voor meer aandacht en alertheid onder clinici voor NBP als oorzaak van jeuk in oudere patiënten. Onze data laat tevens zien dat NBP geen mild ziekte beloop heeft, en systemische therapie frequent noodzakelijk is.

220
Prevalentie van nonbulleus pemfigoïd In hoofdstuk 5 onderzochten we de prevalentie van pemfigoïd in de veronderstelde hoog-risico populatie van verpleeghuis bewoners. Van 125 verpleeghuis bewoners werd tijdens een routine vena punctie een extra bloedmonster afgenomen voor serologische diagnostische testen naar pemfigoïd. Zeven verpleeghuis bewoners werden gediagnosticeerd met pemfigoïd (3 bulleus, 4 nonbulleus). De prevalentie van pemfigoïd onder verpleeghuis bewoners was 6%, opvallend hoger dan de prevalentie in de algemene populatie (0.3% onder personen met een leeftijd ≥ 85 jaar). De vier NBP patiënten werden allen nieuw gediagnosticeerd, en hadden inadequaat behandelde jeuk klachten. Onze bevindingen laten zien dat NBP meer aandacht verdient van specialisten ouderengeneeskunde als oorzaak van jeuk onder verpleeghuis bewoners. Immunologische aspecten van nonbulleus pemfigoïd In hoofdstuk 6 onderzochten we de aanwezigheid van IgE autoantilichamen in het serum en in de huid van NBP en BP patiënten middels ELISA en immunofluorescentie. Het percentage van positieve anti-BP180 en anti-BP230 IgE ELISA’s verschilde niet significant tussen beide pemfigoïd fenotypen. In de huid van NBP en BP patiënten vonden we IgE voornamelijk gebonden aan de oppervlakte van cellen in de dermis (71% en 86%). Slechts twee huidmonsters (7%; 1 NBP, 1 BP) lieten IgE in een lineair patroon langs de basaalmembraan zien. Gebaseerd op deze bevindingen concludeerden we dat IgE mogelijk een rol speelt in de ziekte pathogenese van zowel NBP en BP, echter is het minder aannemelijk dat IgE een centrale rol speelt in de blaarvorming. In hoofdstuk 7 trachtten we meer te leren over de ziekte pathogenese van NBP en BP door het gen expressie profiel van lesionale huid te analyseren middels de nanoString technologie. Zes van de tien BP patiënten toonden een hogere expressie van genen betrokken bij complement activatie, terwijl een lage expressie werd gedetecteerd in vier BP en alle 12 NBP huidmonsters. Een hoge expressie van Th1 en Th2 genen werd gezien in alle BP huidmonsters, terwijl de minderheid van de NBP huidmonsters een verhoogde Th1 of Th2 gen expressie liet zien. We identificeerden tevens genen die significant hoger tot expressie kwamen in BP ten opzichte van NBP huid, waaronder chemoattractants IL-8 en CCL3, epidermale groeifactoren HBEGF en AREG, transcriptie factoren CREM en FOSL1, receptoren ILR1RL1 en GPR65, het gen coderend voor COX2, en de protease ADAMTS4. Deze

221
geactiveerde genen kunnen mogelijk relevant zijn in blaarvorming, echter zijn vervolg studies nodig om deze genetische data te valideren. In hoofdstuk 11, discussie en toekomstperspectieven, bediscussiëren we enkele andere immunologische aspecten die mogelijk een rol kunnen hebben in de ziekte pathogenese van NBP. Dit betreft onder andere antigeen- en epitoop herkenning, complement activatie, IgG subklasse profiel en de activatie van eosinofiele granulocyten. Deel 2 – Management van pemfigoïd ziekten Deel 2 van dit proefschrift presenteert studies gericht op management van pemfigoïd ziekten. Over het algemeen bestaat de behandeling van pemfigoïd ziekten uit immunosuppressieve- of immunomodulerende medicijnen. Deze conventionele therapieën zijn niet altijd succesvol in het bereiken van remissie, en hebben mogelijk ernstige bijwerkingen, met name in de populatie kwetsbare ouderen met veelal meerdere comorbiditeiten. Hierdoor is er sprake van een voortdurende zoektocht naar medicatie met een hoge effectiviteit en een beter veiligheidsprofiel. In hoofdstuk 8 hebben we middels een internationale enquête studie de unmet needs (onvervulde behoeften) in pemfigoïd ziekten geëxploreerd, vanuit het perspectief van patiënten, clinici en onderzoekers. In totaal werden 107 deelnemers geïncludeerd, grotendeels bestaande uit patiënten (66%). Alle drie belangengroepen uitte een hoge noodzaak voor verbetering en verbreding van de therapeutische opties voor pemfigoïd ziekten. Daarnaast droegen pemfigoïd patiënten aan dat er een grote noodzaak is voor meer ziekte bewustzijn onder clinici, om vertragingen en het stellen van een foutieve diagnose te voorkomen. In hoofdstuk 9 onderzochten we retrospectief de effectiviteit en veiligheid van rituximab in 28 patiënten met recalcitrante pemfigoïd ziekten. Remissie werd bereikt in 57% van de patiënten. Patiënten met slijmvlies pemfigoïd toonden de beste response (remissie in 64%), gevolgd door BP patiënten (63%). Patiënten met epidermolysis bullosa acquisita behaalden minder vaak remissie (40%), en één patiënt met lineaire IgA dermatose toonde geen response. Twee-derde van de patiënten met remissie kreeg uiteindelijk een opvlamming van de ziekte, waarna herhaalde rituximab behandeling opnieuw tot remissie leidde in 86% van de

222
patiënten. Tevens liet onze data zien dat de dosering van 1000 mg rituximab op dag 0 en 15 effectiever was dan 500 mg, echter was dit verschil niet statistisch significant. Een andere interessante bevinding is de slechte response op rituximab in IgA dominante pemfigoïd patiënten, wat suggereert dat clinici voor deze patiënten beter een alternatieve therapie kunnen kiezen. In hoofdstuk 10 onderzochten we in een multicenter retrospectieve studie de prevalentie van pneumocystis pneumonia (PCP) onder patiënten met auto-immuun blaarziekten bij wie geen PCP profylaxe werd voorgeschreven. Data van 801 patiënten met een auto-immuun blaarziekte toonde een PCP incidentie van 0.1%. Voorgaande studies bepaalden dat de drempel prevalentie waarbij PCP profylaxe gerechtvaardigd is op 3.5% ligt. Daarom concludeerden we dat routine PCP profylaxe niet in elke patiënt met een auto-immuun blaarziekte geïndiceerd is. Toekomstige studies naar individuele patiënten karakteristieken, en de mate van immunosuppressie bij patiënten die een PCP ontwikkelden kunnen mogelijk meer handvaten bieden om te beslissen wanneer PCP profylaxe wel geïndiceerd is. In hoofdstuk 11, discussie en toekomstperspectieven, bediscussiëren we enkele potentiële nieuwe therapieën voor pemfigoïd die momenteel worden onderzocht. Deze therapieën betreffen onder andere anti-complement en anti-IgE medicatie, medicatie gericht tegen eosinofiele granulocyten (anti-IL-5 en anti-eotaxin-1), en phosphodiesterase 4 remmers.

223

224
APPENDICES

225
APPENDICES Dankwoord List of publications Curriculum vitae

226
Dankwoord Na een lange weg vol pieken en dalen, is daar het einde in zicht: dit proefschrift is voltooid! Er zijn een aantal personen die belangrijk zijn geweest in de totstandkoming van dit proefschrift die ik graag wil bedanken.
Eén van deze belangrijke personen is prof. dr. Marcel Jonkman, onder wiens supervisie ik in 2016 startte met mijn promotieonderzoek. In de zomer van 2018 ontvingen wij het treurige nieuws dat prof. Jonkman ernstig ziek was, en na een kort ziekbed overleed hij op 14 januari 2019. Onze korte samenwerking heeft een grote indruk op mij achtergelaten. Ik ben dankbaar voor de kans die ik heb gekregen om met prof. Jonkman samen te werken. Daarnaast ben ik trots en tevreden over de weg die wij hebben afgelegd, en over het eindresultaat: dit boekje, dat hier zonder prof. Jonkman niet in deze vorm zou liggen.
De tweede persoon die ik graag wil bedanken is dr. Barbara Horváth. Barbara, op een geweldige manier ben jij opgestaan in een tijd van onzekerheid en chaos. Zonder enige twijfel nam je mij onder je hoede na het wegvallen van prof. Jonkman. En dat niet alleen, als een ware powervrouw en met een bewonderenswaardige onuitputtelijke bron van energie gaf je leiding aan de afdeling. Ik kan je niet genoeg bedanken voor je steun en hulp met het afronden van mijn proefschrift.
Tevens bedank ik graag mijn copromotor dr. Hendri Pas. Hendri, ik heb genoten van onze samenwerking. Je enthousiasme over de wetenschap werkt aanstekelijk. Discussies vonden plaats in de onderzoekskamer, op het lab, tijdens congressen, en in de kroeg tijdens de tweemaandelijkse labborrels. Door deze discussies ben ik tot interessante inzichten gekomen. Daarnaast was het ook gewoon erg gezellig!
Dank aan mijn collega blaaronderzoekers, Joost en Hanan. Joost, je rol veranderde van stage wetenschap begeleider naar collega onderzoeker. Wij zitten al een tijdje samen op de blaren EN op de niet-blaren. Ik kijk met trots terug op onze samenwerking met mooie resultaten, mede mogelijk gemaakt door vele koppen koffie, vaak met wat lekkers. Dank voor al je hulp en steun. Hanan, ik heb met jou geboft als opvolger in de onderzoekslijn auto-immuun blaarziekten. We hebben mooie stedentripjes gemaakt om congressen en werkgroep vergaderingen te bezoeken. Iets wat ik heb geleerd: reisjes met Hanan gaan altijd gepaard met het eten van ijsjes. Blaarkamergenoten Daniela en Rosalie wil ik ook bedanken voor de gezelligheid en steun. De extra zuurstof van al jullie plantjes hebben zeker bijgedragen aan dit proefschrift.
Mijn paranimfen Angelique en Lisette wil ik graag bedanken. Angelique, in mijn beginfase als onderzoeker was het fijn dat jij mij op weg hielp. Tijdens onze reis naar Orlando heb jij

227
bewezen dat er een correlatie kan bestaan tussen koffie drinken en blaren. Gelukkig verliep de rest van de reis nonbulleus. Lisette, naast de werkvloer komen wij elkaar tegenwoordig ook regelmatig tegen op het volleybalveld, heel leuk! Ons reisje naar Parijs zal ik niet snel vergeten, we hebben ons daar goed vermaakt op het congres, en tot in de late uurtjes. Lieve paranimfen, bedankt voor alle leuke en waardevolle onderzoek gerelateerde herinneringen en voor jullie vriendschap!
Tevens wil ik het immunodermatologie laboratorium bedanken voor jullie belangrijke bijdrage aan dit proefschrift. Ik heb met plezier in jullie lab gewerkt. Daarnaast wil ik Gilles Diercks graag bedanken, met wie ik aan vele onderzoeksprojecten heb samen gewerkt, altijd op een prettige manier.
Andere collega-onderzoekers, Alet, Angelique, Cynthia, Klaziena, en Laura, bedankt voor de gezellige lunchpauzes, koffie breaks en spelletjes avonden. Een waar lichtpuntje tijdens de soms donkere onderzoeksdagen!
De AIOS wil ik bedanken voor jullie steun bij poliklinische of epische vragen, jullie zijn toppers! Ook wil ik de stafleden van de afdeling dermatologie bedanken, en daarnaast het ondersteund personeel: de doktersassistenten, de medische administratie, en de beroemdste persoon van de afdeling: fotograaf Piet Toonder. Last but not least: Alberta en Katarina, dank voor jullie organisatorische talenten, de steun en gezellige praatjes tussen alle bedrijven door. Er hangt een leuke sfeer op de afdeling dermatologie. Ik heb genoten van de leuke afdelingsuitjes, de jaarlijkse kerstborrel met baby/katten quiz, AIOS (en onderzoekers) weekendjes, en de afdelingswintersport, es war supertoll! Ik kan niet wachten meer van de sfeer te proeven!
Dank aan lieve vrienden en vriendinnen binnen en buiten het medische wereldje, voor alle momenten van ontspanning buiten werktijden. Door jullie zijn mijn avonden en weekenden altijd weer snel gevuld met etentjes, borrels, spelletjes, pubquizen, pizza-BBQ avondjes, stoofpot-zondag, volleybal, reisjes, en andere gekkigheid. Dank aan mijn lieve vriendinnen uit Beilen met (en om) wie ik altijd hard kan lachen en ook veel lief & leed heb gedeeld.
Lieve Joke, Michiel en Nynke, dank voor alle steun en jullie luisterende oren. We kennen elkaar inmiddels een lange tijd en ik heb mij bij jullie altijd zeer welkom gevoeld.
Lieve pap, mam en broerlief, dank dat ik bij jullie zonder zorgen heb mogen opgroeien. Ik kan me geen betere jeugd en warmer gezin wensen. Gita, ik ben erg blij met jou als lieve schoonzus. Door jullie voel ik me een rijk mens!
Als laatst een woord aan Steven. Lieverd, na een lange werkdag is er niets fijners dan thuis te komen bij jou. Ik kan niet wachten om met jou de route voor de rest van ons leven verder uit te stippelen.

228
List of publications
1. Lamberts A*, Meijer JM*, Luijendijk HJ, Diercks GFH, Pas HH, Zuidema SU, Jonkman MF. Prevalence of pemphigoid as a potentially unrecognized cause of pruritus in nursing home residents. JAMA Dermatol. 2019 Nov 6. doi: 10.1001/jamadermatol.2019.3308. [Epub ahead of print] * equal contribution
2. Lamberts A, Meijer JM, Pas HH, Diercks GFH, Horváth B. Response to letter to the editor: “pruritus with pemphigoid autoantibodies is the tip of an iceberg”. J Am Acad Dermatol 2019 Nov;81(5):e153-e154. Doi:10.1016/j.jaad.2019.07.078
* equal contribution
3. Lamberts A*, Rashid H*, Diercks GFH, Pas HH, Meijer JM, Bolling MC, Horváth B. Oral lesions in autoimmune bullous diseases: an overview of clinical characteristics and diagnostic algorithm. Am J Clin Dermatol. 2019 Dec;20(6):847-861. Doi: 10.1007/s40257-019-00461-7
* equal contribution
4. Lamberts A, Rashid H, Pas HH, Diercks GFH, Meijer JM, Horváth B. Pemphigoid variants affecting the skin: a review. Clin Exp Dermatol. 2019 Oct;44(7):721-727. doi:10.1111/ced.13984
5. Lamberts A, Meijer JM, Pas HH, Diercks GFH, Horváth B, Jonkman MF. Nonbullous pemphigoid: insights in clinical and diagnostic findings, treatment responses and prognosis. J Am Acad Dermatol 2019 Aug;81(2):355-363. doi:10.1016/j.jaad.2019.04.029
6. Kraaij vd GE, Balak DMW, Busad CI et al (Lamberts A; 12th of 25 coauthors). Highlights of the updated Dutch evidence- and consensus based guideline on psoriasis 2017. Br J Dermatol. 2019 Jan;180(1):31-42.
7. Lamberts A, Yale M, Grando SA, Horváth B, Zillikens D, Jonkman MF. Unmet needs in pemphigoid diseases: an international survey amongst patients, clinicians and researchers. Acta Derm Venereol. 2019 Feb 1;99(2):224-225. doi: 10.2340/00015555-3052.

229
8. Lamberts A, Euverman HI, Terra JB, Jonkman MF, Horváth B. Effectiveness and safety of rituximab in recalcitrant pemphigoid diseases. Front. Immunol. 2018 Feb 19; 9:248. doi: 10.3389/fimmu.2018.00248
9. Lamberts A, Meijer JM, Jonkman MF. Nonbullous pemphigoid: a systematic review. J Am Acad Dermatol 2018 May;78(5):989-995.e2.
10. Amber KT, Lamberts A, Solimani F et al. Determining the incidence of pneumocystis pneumonia in patients with autoimmune blistering diseases not receiving routine prophylaxis. JAMA Dermatol. 2017 Nov 1;153(11):1137-1141. doi:10.1001/jamadermatol.2017.2808.
11. Lamberts A, Jonkman MF. Pruritus in nursing home residents: scabies or nonbullous pemphigoid? NTvDV 2017 Oct; 27(9):490-493
12. Lamberts A, Sande vd A, Horváth B. Abstract of the national guideline of hidradenitis suppurativa 2017. NTvDV. 2017 Aug;27(7): 360-363
13. Lamberts A, Meijer JM, Jonkman MF. An unusual variant of pemphigoid (anti-p200). Journal: NTvDV 2017 Jun; 27(6): 289-292
14. Lamberts A, Meijer JM, Luiendijk D, Zuidema S, Jonkman MF. Pruritus and cutaneous pemphigoid in nursing home residents. Tijdschrift voor ouderengeneeskunde. 2017 Apr; 2: https://www.verenso.nl/magazine-april-2017
15. Lamberts A, Karsch SAT, Kelleners-Smeets NWJ. Abstract of the national guideline of basal cell carcinoma 2015. Nederlands tijdschrift voor dermatologie en venereologie. 2016;26(7): 396-399
16. Lamberts A*, Meijer JM*, Pas HH, Jonkman MF. Significant higher prevalence of circulating bullous pemphigoid (BP)-specific IgG autoantibodies in elderly patients with a non-bullous skin disorder, Br J Dermatol. 2015 Nov;173(5):1274-6. Epub 27.
* equal contribution
17. Lamberts A, Diercks GFH, Terra JB, A child with a S-shaped skin condition at his abdomen, Ned Tijdschr Geneeskd. 2015;159:A8960.

230
Curriculum vitae Marlou Aniek Lamberts, given name Aniek, was born on the 17th of July 1990 in Beilen, the Netherlands. After graduating high school at the Dr. Nassau College in Assen, she started studying medicine at the University of Groningen in September 2008. During her masters, she did one year of internships in the Deventer Ziekenhuis. In her final master year she chose for a senior internship and research internship at the dermatology department of the University Medical Center Groningen, under the supervision of prof. dr. Marcel Jonkman. At the 31st of December 2014 she received her Master of Science degree, and immediately after, Aniek started working at the internal medicine ward in the Scheper Ziekenhuis in Emmen. In January 2016, she moved to Utrecht to work and support national guideline development at the Dutch National Association of Dermatology (NVDV). In October 2016, she started as PhD candidate under the supervision of prof. dr. Marcel Jonkman as promotor, and dr. Hendri Pas as copromotor. Sadly, prof. dr. Marcel Jonkman passed away in January 2019, whereafter the supervision was taken over by dr. Barbara Horváth. In January 2020, Aniek resumed her work as a medical doctor at the outpatient clinic of the department of dermatology at the University Medical Center Groningen.




















