
thesis.pdf - the University of Groningen research portal
119
University of Groningen Structure and mechanism of the ECF-type ABC transporter for thiamin Erkens, Guus Bjorn IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check the document version below. Document Version Publisher's PDF, also known as Version of record Publication date: 2011 Link to publication in University of Groningen/UMCG research database Citation for published version (APA): Erkens, G. B. (2011). Structure and mechanism of the ECF-type ABC transporter for thiamin. s.n. Copyright Other than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons). The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license. More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne- amendment. Take-down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons the number of authors shown on this cover page is limited to 10 maximum. Download date: 28-03-2022
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of thesis.pdf - the University of Groningen research portal

University of Groningen
Structure and mechanism of the ECF-type ABC transporter for thiaminErkens, Guus Bjorn
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:2011
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Erkens, G. B. (2011). Structure and mechanism of the ECF-type ABC transporter for thiamin. s.n.
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license.More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne-amendment.
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 28-03-2022

Structure and mechanismof the
ECF-type ABC transporter for thiamin
Guus Erkens

Cover design: ‘A slice of electron density from the ThiT asymetric unit’ by Guus ErkensPrinted by: Ipskamp drukkers
ISBN: ������-�0-36�-4�46-6 printed �ersion printed �ersion ������-�0-36�-4�45-� electronic �ersion electronic �ersion
The research described in this thesis was carried out at the Groningen Biomolecular Sciences and Biotechnology Institute (GBB), Department of Biochemistry, Uni�ersity of Groningen, the Netherlands and financially supported by the Netherlands Organization for Scientific Research (NWO).
© 2011 Guus ErkensAll rights reser�ed. No part of this publication may be reproduced, stored in a retrie�al system of any nature, transmitted in any form or by any means, electronic, mechanical, now known or hereafter in�ented, including photocopying or recording, without prior written permission of the copyright holder.

Structure and mechanismof the
ECF-type ABC transporter for thiamin
Proefschrift
ter �erkrijging �an het doctoraat in deWiskunde en Natuurwetenschappenaan de Rijksuni�ersiteit Groningen
op gezag �an deRector Magnificus, dr. E. Sterken,in het openbaar te �erdedigen op
�rijdag 1 juli 2011om 14.45 uur
door
Guus Bjorn Erkens
geboren op 2� april 1��3te Arnhem

Promotor: Prof. dr. D.J. Slotboom
Beoordelingscommissie: Prof. dr. A.J.M. Driessen Prof. dr. A.M. �an Oijen Prof. dr. P. Gros

Contents
Chapter 1 An introduction to ECF- and ABC-transportersOutline of this thesisAppendix: the historical background of thiamin
11315
Chapter 2 Bioinformatics analysis of the ECF transporter subunitss
1�
Chapter 3 Identification of genes encoding the folate- and thiamin-binding membrane proteins in Firmicutes
2�
Chapter 4 Biochemical characterization of ThiT from Lactococcus lactis: a thiamin transporter with picomolar substrate binding affinity
35
Chapter 5 Crystal structure at 2.0 Å of the S-component for thiamin from an ECF-type ABC transporter
55
Chapter 6 Discussion: structural similarity in membrane proteins indicates ancient homology
�3
Nederlandse samenvatting voor geïnteresseerden buiten het vakgebied �1
List of publications ��
Nawoord �1
References �5


1
Chapter 1
An introduction to ECF- and ABC-transporters
parts of this chapter are based on:J. Bacteriol. (200�) 191:42-51
Summary
All forms of life separate their cellular contents from the external medium by a lipid bilayer (the cell membrane) which is poorly permeable for hydrophilic molecules. Nonetheless, translocation of numerous hydrophilic molecules across the membrane is essential for life. To enable transport at useful rates, hydrophobic proteins are embedded in the membrane that facilitate the import and export of �arious compounds. One of the largest superfamilies of transport proteins are ATP Binding Cassette (ABC) transporters (2�). Members of the ABC transporter superfamily are in�ol�ed in the translocation of substrates across biological membranes, either as importer or as exporter. The energy for transport is pro�ided by ATP hydrolysis in Nucleotide Binding Domains (NBDs). ABC transporters are characterized by a conser�ed subunit architecture consisting of two NBDs and two Transmembrane Domains (TMDs) which together form a single translocation pore. ABC transporters in�ol�ed in import are found only in prokaryotes and usually require an additional extracellular or periplasmic water-soluble protein to sca�enge the substrate: the Substrate Binding Protein (SBP). Howe�er, the ECF (Energy Coupling Factor) transporter family is a recently disco�ered class of ABC importers in�ol�ed in uptake of �itamins that does not requires SBPs (10�). The basic architecture of ECF transporters is identical to that of ABC transporters (Two NBDs and two TMDs), but the mechanism of substrate binding differs. In ECF transporters, substrate binding takes place in one of the TMDs (the EcfS subunit or S-component) which forms an integral part of the ECF transporter complex. The second transmembrane protein in ECF transporters (the EcfT subunit) together with two NBDs (named EcfA in ECF transporters) forms an energizing module that couples ATP hydrolysis to substrate translocation. The energizing module can interact with se�eral different S-components to acti�ely transport a broad range of chemically different substrates (10�,130). S-components bind their substrates with high affinity (picomolar to nanomolar dissociation constants) and can, in the absence of the energizing module, be expressed and purified as stable monomers ((34), chapter 4). Recent work has lead to the determination of crystal structures for the ribofla�in- and thiamin-specific S-components, RibU (155)

2
and ThiT (chapter 5 of this thesis) respecti�ely. Although unrelated at the sequence le�el (14% identity), there is a remarkable similarity in their o�erall structure that is probably connected to the mechanism of S-component recognition by the energizing module. This chapter will gi�e an o�er�iew of the literature on ECF transporters and describes their recent disco�ery. The focus will be on their general characteristics and relation to ABC transporters. Finally, a short background on thiamin (�itamin B1) is gi�en.
Vitamin transport in Gram-positive prokaryotes
Although a number of reports on �itamin transport by prokaryotes ha�e been published in the past fifty years, detailed biochemical- and mechanistic studies are scarce. Much of this work for Gram-positi�e bacteria has been performed by Gary Henderson et al. In a series of publications during the late 1��0s and early 1��0s, the biochemistry of acti�e folate-, thiamin- and biotin-transport in Gram-positi�e bacteria was described in great detail (5�-64,�6). Early in�estigations focused on the folate-binding protein from Lactobacillus casei (61,62). Expression of this protein could be induced when the cells were culti�ated in growth medium supplemented with limiting amounts of folate. The rate of in vivo folate transport correlated well with these expression le�els, indicating that the concentration of folate-binding protein was limiting for folate transport. Substrate bound with high affinity (KD=36 nM) to cells expressing the folate-binding protein and from these cells the folate-binding protein could be extracted with detergents. This obser�ation was the first indication that the folate-binding protein was an integral membrane protein or part of a membrane protein complex. Subsequent purification of the detergent extract enabled analysis of the amino acid content and SDS-PAGE (SDS-Poly Acrylamide Gel Electrophoresis) of the isolated folate-binding protein. The protein was found to contain few charged or polar amino acids and had a molecular mass of ~25 kDa. Similar to the folate-binding protein, a thiamin-binding protein was expressed when L. casei cells were grown under thiamin limiting conditions (60). These cells were shown to rapidly accumulate radiolabeled thiamin that was directly con�erted intracellular to thiamin-pyrophosphate (TPP). Thiamin transport was energy dependent, but in de-energized cells high affinity (KD<10 nM) thiamin binding could still be obser�ed. Although the transport of folate, thiamin and later biotin was shown be mediated by three different proteins, the transport of one �itamin was non-competiti�ely inhibited by the addition of a second �itamin in the transport assay (64). For instance, the addition of thiamin in folate transport assays decreased the folate transport rates by ~45%. Intriguingly, the inhibition was obser�ed only if the expression of the thiamin-binding protein was induced. The inhibition was competiti�e with regard to the concentration of the binding proteins. To explain the unusual transport kinetics, it was proposed that the indi�idual �itamin-binding-proteins had to compete for a shared component that would couple

3
energy to substrate translocation. For this reason, the unknown component was named the ‘energy coupling factor’. Further in�estigations demonstrated that (at least for folate) the energy for transport was almost certainly pro�ided by ATP hydrolysis (63).
Identification of the folate- and thiamin-binding proteins The in�estigations on �itamin transport in L. casei were conducted in the pre-genomics era and the genes encoding the transport proteins were not identified at that time. In the past few years, the molecular identities of �arious components ha�e been elucidated. Some of the experiments leading to the identification are described in chapter 3. In this chapter a brief o�er�iew is presented.
The genome sequence of L. casei (which had become a�ailable in 2006), was searched for genes encoding proteins with comparable properties to the folate- and thiamin-binding proteins, such as size and amino acid composition (chapter 3). The corresponding genes were named folT and thiT because of their in�ol�ement in folate and thiamin binding. Cells o�erexpressing FolT or ThiT bound (6S)-folinic acid and thiamin respecti�ely, but did not support transport of the �itamins. Purified FolT and ThiT were shown to bind their substrates with high (10-� - 10-10 M) affinity. There was a superficial resemblance of FolT and ThiT to the ribofla�in transporter RibU from Lactococcus lactis although no sequence similarity could be detected between any of these proteins. RibU, ThiT and FolT are all predicted to ha�e 5-6 transmembrane segments, and a protein mass of ~20 kDa. RibU could bind ribofla�in and FMN with high affinity, but did not support transport of these substrates (16,34). It was suggested that an additional component would be required for RibU to function as a transporter.
Identification of the energy coupling factor Biotin transport in Rhodobacter capsulatus is mediated by the protein complex BioMNY (55). BioM is homologous to the NBDs belonging to ABC transporters, BioN and BioY are integral membrane proteins. Substrate specificity is conferred to the transporter trough BioY (which is not related to ThiT, FolT or RibU) but again there is a superficial resemblance in size and number of predicted transmembrane helices. Similar to other prokaryotic importers, biotin transport is fuelled by ATP hydrolysis. BioY could be expressed separate from the other proteins in the BioMNY complex and in in vivo experiments with E. coli cells o�erexpressing only the bioY gene, biotin transport was still obser�ed. A similar complex organization as in BioMNY was found for the ATP dependent Ni2+ and Co2+ transporters NikMNQO and CbiMNQO (10�). Like BioNMY their substrate specificity is conferred by one of the transmembrane subunits (NikM and

4
CbiM respecti�ely). The NikO and CbiO proteins are analogous to BioM (a NBD) and NikQ and CbiQ are analogous the BioN, the second membrane subunit. There was a surprising similarity between BioM, NikO and CbiO, although the substrate binding proteins (BioY, NikM and CbiM) were �ery different. A link between BioY, FolT and ThiT was made when an orthologue of the bioY gene was found in the L. casei genome, but without the additional bioMN (ecfAT) genes. Instead, an operon encoding BioMN-homologues was found at a different location in the genome that was not linked to a gene coding for a substrate specific protein. From this obser�ation it was hypothesized that the BioM like proteins were the elusi�e energy coupling factor that interacts with �arious substrate specific proteins. A large-scale bioinformatics analysis was performed to search for additional �itamin-binding proteins and putati�e energy coupling factors (10�).
A total of 365 genomes were searched for operons containing a typical ATPase of the ABC transporter family (now named A-component or EcfA) a BioN-like membrane protein (T-component of EcfT), that was accompanied by a second membrane protein that could be a substrate specific component (S-component, such as BioY, FolT, ThiT or RibU). This search resulted in the identification of 432 gene cassettes encoding energizing (AT) modules, often with duplicated A components (labeled EcfA and EcfA’ in those cases). Of these energizing modules, 335 were similar to NikMNQ, CbiMNQO or BioMNY in the sense that the S-component formed an operon with the energizing module. The remaining �� energizing modules were found without adjacent S-component genes, but these genomes encoded �arious candidate S-components at other genomic locations. It was predicted that in those cases the energizing module is shared among the different S-components and that such an energizing module might be the sought-after energy coupling factor from L. casei. The results are summarized in figure 1. In total, 21 different putati�e S-components families were described (table 1). A prediction of their substrate specificity could be made based on the genomic context: in many cases, the expression of S-components was regulated by a riboswitch sequence (see below) or the S-component genes co-localized with substrate-specific repressor proteins or biosynthesis clusters.
The proposed energizing module links all the transporter subunits that were identified together and has a central function in coupling energy to substrate translocation. Therefore, this no�el family of transporters was named Energy Coupling Factor (ECF) transporters. The energizing module showed a clear relationship with ABC transporters through the ATP hydrolyzing EcfA components. Therefore, the general properties of ABC transporters will be discussed first.

5
Figure 1: distribution and comperative genomics analysis of ECF transportersComparati�e genomic analysis of the identified transporter families including their domain compositions, names, predicted substrate specificities, and example gene identifications. Substrate-specific integral membrane components (S) are shown by black rectangles, conser�ed transmembrane components (T) are shown by blue rectangles, and ATPase domains (A) are shown by red circles. Examples of genome context e�idence (e.g., gene co-regulation or co-localization) supporting the predicted transporter function are shown on the right. This figure is a modified �ersion of a pre�iously publshed one (10�).

6
Table 1: an overview of S-components and their substrate specificity
protein name substrate(s) confirmed (Y/N)
reference
ThiT Thiamin (�itamin B1), TMP, TPP, pyrithiamin
Y chapter 3,4
RibU Ribofla�in (�itamin B2), FMN Y (16,34)FolT Folic acid (�itamin B�), (6S)-folinic acid Y chapter 3BioY Biotin (�itamin B�) Y (55)PanT Pantothenic acid (�itamin B5) Y (�5)QueT Queuosine precursor NNiaX Niacin (�itamin B3) Y (130)HmpT Hydroxymethylpyrimidine
(thiamin precursor)N
YkoE Hydroxymethylpyrimidine (thiamin precursor)
N
ThiW Thiazole (thiamin precursor) NMtsT Methionine precursor NTrpP Tryptophan NLipT Lipoate NCblT Cobalamin (�itamin B12) precursor NCbrT Cobalamin (�itamin B12) precursor NQrtT Queuosine precursor NPdxT Pyridoxine (�itamin B6) NMtaT Methylthioadenosine NNikM Nickel ions Y (10�)CbiM Cobalt ions Y (10�)HtsT unknown -
ATP-Binding Cassette (ABC) transporters in prokaryotes
Classification and functionThe group of ABC transporters forms the largest superfamily of transport proteins. ABC systems are represented in genomes from organisms in all kingdoms of life and perform an amazing �ariety of functions (2�). Some ABC transporters ha�e e�ol�ed to perform non-transport functions (which will not be discussed here), but the majority is in�ol�ed in the translocation of an enormous �ariation of substrates across �arious biological membranes.

�
The hallmark of ABC transporters are the ATP hydrolyzing proteins or domains: the Nucleotide Binding Domains (NBDs). The NBDs are remarkably conser�ed between ABC transporters, e�en if they perform �ery different functions. ABC transporters that translocate substrates to the cytoplasm are ABC importers, whereas ABC exporters work in the opposite direction. In contrast to the well-conser�ed NBDs, the TMDs are polyphyletic (144) and can thus adopt �ery different folds (see figure 2).
Prokaryotic ABC transportersThere is a wealth of biochemical (and to a lesser extent) structural data a�ailable on prokaryotic ABC transporters. Many important processes in�ol�e ABC transporters, such as import of nutrients (11), �irulence (56) and multi-drug resistance (��). Characteristically, ABC-importers require an additional protein for substrate recognition: the Substrate Binding Protein (SBP). SBPs are structurally similar water-soluble proteins (�). In Gram-negati�e bacteria, SBP’s are expressed freely in the periplasm where they can sca�enge their substrates. A large conformation change takes place upon substrate binding, which results in closing of the binding site and trapping of the bound substrate. This mechanism is described as the ‘Venus-flytrap model’ (42). Gram-positi�e bacteria (that lack a confined periplasmic space) often ha�e their SBP’s genetically fused to the TMD’s or attached to the membrane �ia a lipid anchor (26). The substrate-bound SBP is recognized by the transporter and stimulates ATP hydrolysis (30,�4), the free energy released in this process is used to open the SBP and transport the substrate.
Bacterial ABC efflux systems are often employed for the export of cell surface components or extrusion of toxic compounds. Although the transport direction is opposite to that of ABC-importers, their membrane orientation is identical (the NBDs are placed in the cytoplasm). Substrate recognition does not require a SBP but the exact mechanism is still being debated.
Structure of ABC transportersIn the past decade, crystal structures of full-length ABC transporters became a�ailable for �arious ABC transporters. The list includes the eukaryotic exporter P-glycoprotein (2), the prokaryotic exporter Sa�1�66 (31) and prokaryotic importer BtuCD (�5). An o�er�iew of ABC transporter structures is gi�en in figure 2. It becomes clear that there is large structural and size �ariation in the transmembrane domains. Ne�ertheless, insights from these structures ha�e generated a general mechanism of transport: it was already known from biochemical data and structures of isolated NBDs that large conformational changes occur when ATP is bound and hydrolyzed. The structure of the ABC transporter BtuCD (�5) showed for the first time a structural element named the ‘coupling helix’ that couples these changes to conformational changes in the TMDs. Coupling helices are short helical segments of the TMDs that interact directly with a groo�e on the

�
surface of the NBDs. The segments show little or no sequence conser�ations, which makes it difficult to locate the coupling helix in the amino acid sequence of TMDs. Additional structures of ABC transporters confirmed the crucial role of the coupling helix (2,31,6�,�2,��,146).
Figure 2: structures of ECF�� and ABC transportersstructures of ECF�� and ABC transporters(a) Structure of the S-components RibU and ThiT with the structure of the EcfA dimer from Thermotoga maritima. The gray box indicates the expected position of EcfT, for which no structure is a�ailable. (b) Structures of ABC importers. (c) Structures of ABC exporters. The horizontal lines roughly indicate the boundaries of the lipid bilayer, PDB accession codes are depicted below the protein names.
ATP binding and hydrolysisATP binding and hydrolysis takes place in the NBDs of ABC transporters. The functional unit of NBDs is a dimer, which is often reflected in the genetic arrangement of ABC transporter genes. NDBs can be found as part of a ‘half transporter’ in which one gene encodes a fusion of the TMD and NBD and thus comprises one half of the functional transporter. Alternati�ely, the NBDs can be encoded by two different genes that code
outside
cytoplasm

�
for a heterodimer, one single gene that codes for a proteins forming a homodimer, or a genetic fusion of two NBD genes encoding a co�alent dimer. The mechanistic basis for the NBD dimer is the ATP binding site, which is formed on the dimer interface and constructed with contributions from both monomers. The dimeric arrangement of NBDs is reflected in a pseudo two-fold symmetry of the full complex: the transporter is always built up of two identical or structurally similar segments.
The amino acid sequence of NBDs is conser�ed throughout the whole ABC transporter family and beyond (52). Although sequences ha�e di�erged and additional domains ha�e e�ol�ed multiple times (10), there are a number of mechanistically important sequence motifs shared by all NBDs. These are the Walker A motif (in�ol�ed in phosphate coordination �ia a Mg2+ ion and adenosine binding), Walker B motif (in�ol�ed in ATP hydrolysis), ABC signature sequence LSGGQ (in�ol�ed in phosphate binding), the H-loop (in�ol�ed in coordination of acti�e site residues and dimer stabilization) and the Q-loop (part of the interaction site for the coupling helix).
For most ABC transporters, the ATP/substrate stoichiometry is not known. The dimeric arrangement suggests that two ATP molecules are hydrolyzed during each transport cycle. This stoichiometry has indeed been confirmed by in vitro experiments with the glycine betaine transporter OpuA (101). But for other transporters different stoichiometries ha�e been found (3,11,2�,3�,�4,�0,�1,113,114,121,122), ranging from 1 to 50 ATP molecules per translocated substrate. It seems from these results that the actual stoichiometry is �ariable and might depend on the substrate that is transported. Larger substrates such as peptides or small proteins may require more ATP hydrolysis for transport than small molecules.
ECF transporters form a subclass of ABC transporters There are many similarities between ECF- and ABC-transporters. For instance, the EcfA subunits are related to the NBDs of ABC transporters (see chapter 2) and ha�e all the mechanistical important sequence motifs that are typical for ABC transporters (chapter 5). Furthermore, the stoichiometry of se�eral ECF-transporter has been �erified experimentally, showing that the four subunits (EcfA, EcfA’, EcfT and EcfS) are arranged in a 1:1:1:1 stoichiometry (130) which agrees well with the basic architecture of ABC transporters. Ne�ertheless, the mechanism of substrate binding which in�ol�es the membrane inserted S-components is �ery different from that of ABC transporters. Based on the genetic organization of the genes coding for S-components, a distinction is made between ECF transporters from class I and II (3�). For transporters in class I, the S-component gene is co-localized with the energizing module in a single operon. These transporters are belie�ed to be dedicated to one specific S-component. Class II ECF transporters ha�e their energizing module encoded in an operon, without an

10
adjacent gene encoding for an S-component. One or more S-component genes can be found at alternati�e locations in the genome and all of which may interact with the same energizing module. Often, representati�es of both classes are found in a single genome. ECF transporters are found in di�erse prokaryotic genomes (Gram-positi�e, Gram-negati�e and Archea), but class II systems are particularly abundant in Firmicutes; a phylum of Gram-positi�e bacteria that includes many human pathogens (see chapter 2). So far, no example has been presented of a eukaryotic ECF transporter.
Structure and function of S-components Substrate feedback controls expressionMany of the S-component genes are regulated in some way by the intracellular concentration of their substrate (10�). There are two types of regulation: regulation by substrate specific repressor proteins (e.g. BirA (110) for the biotin specific S-component BioY) or regulation by riboswitches (e.g. the TPP riboswitch (151) that regulates ThiT expression). Riboswitches are encoded in the DNA region preceding the regulated gene. After DNA transcription, an mRNA molecule is produced with both the riboswitch sequence and the gene. The riboswitch sequence then adopts a specific secondary structure that is able the recognize and bind a substrate (e.g. TPP in case of the TPP riboswitch) (154). When the riboswitch is ‘charged’ with substrate, protein expression can either be inhibited because a terminator helix is formed (which results in premature termination of transcription) or because the ribosome binding sequence becomes inaccessible (the mRNA is pre�ented from binding to the ribosome). This mechanism pro�ides a direct coupling between the intracellular concentration of a specific metabolite and the expression of S-components. As the intracellular concentration of a substrate drops, more ‘uncharged’ riboswitches appear and thus the expression increases. Structure of S-componentsRecently the crystal structures of the S-component RibU from Staphylococcus aureus (155) and ThiT from L. lactis (chapter 5) were determined. Although significant sequence similarity between these proteins is absent, a similar fold was obser�ed for both proteins (figure 3). Their structure is built up of six hydrophobic transmembrane helices with short connecting loops. There is a large �ariation between the lengths and tilt of the helices; helix 5 and 6 are particularly long and cross the membrane at a steep angle, whereas helix 2 only spans half of the membrane. In both structures, the major part of the binding site is formed by conser�ed amino acids in helix 5 and 6 and the cytoplasmic loop connecting them. Structural di�ergence between ThiT and RibU is most e�ident in helices 4 and 5. For instance, the position of helix 4 and 5 relati�e to the other helices is �ery different. In addition, helix 4 in ThiT has a �ery unusual secondary structure. It is

11
Figure 3: structures of the S-components for thiamin (ThiT) and riboflavin (RibU)X-ray structures of ThiT (left, PDB: 3RLB) and RibU (right, PDB: 3P5N). The gray rectangle indicates the position of the membrane, the transmembrane helices are numbered 1-6. ThiT and RibU are depicted in the same orientation.
built up of an α-helical segment, followed by a π-bulge, a second α-helical segment and then continues as a long 310 helix. These structural elements are characterized by their backbone hydrogen bonding pattern. In a π-helix, C=O groups from one amino acid form a hydrogen bond with the N-H moieties of a second amino acids that is separated by four residues (i→i+5), in a 310 helix this separation is two residues (i→i+3), whereas in an α-helix the hydrogen bond partners are separated by three residues (i→i+4). In the RibU structure, helix 4 is a regular α-helix.
Surprisingly, neither ThiT, nor RibU appears to ha�e a coupling helix ((155), chapter 5), the structural motif that couples ATP hydrolysis to substrate transport in ABC transporters. Since ECF transporters ha�e two EcfA subunits (or NBDs), two coupling helices are also expected. In classical (non-ECF) ABC transporters, each ABC transporter TMD therefore has one coupling helix a�ailable for interaction with the NBD. To explain the missing coupling helix, is has been proposed that the EcfT component might ha�e two coupling helices to interact with both NBDs (chapter 5). A cytoplasmic domain from the subunit EcfT that was found to be crucial for the integrity of the ECF complex (�5) could be the location of these coupling helices.
A function for S-components in the absence of the energizing module?The substrate feedback loop results in maximum expression of the S-components when the intracellular concentration of their substrates is low. For the energizing module, the

12
expression pattern is not known, but the absence of specific regulator sequences and co-localization with essential house-keeping genes suggests that it is probably expressed at a constant le�el. As a result, cells are capable of quickly increasing the uptake of ECF substrates when their intracellular concentration drops. The increase in abundance of a particular S-component leads to the formation of more Energizing module/S-component complexes of that type and thus more transported substrate. It has been demonstrated for at least one S-component (FolT from L. casei) that significant amounts of FolT can be isolated from cells grown under folate limiting conditions without apparent co-purification of the energizing module (62). This obser�ation may suggest that high le�els of isolated S-components exist in the membrane at some point, which apparently is ad�antageous for the organism. A possible explanation is that S-components are also capable of transport in the absence of the energizing module, as has been obser�ed for the biotin specific S-component BioY (55). Alternati�ely, expression of isolated S-components may allow organisms to sca�enge any a�ailable �itamin in the case of extreme scarcity. Once the substrate is bound to an S-component, a complex can be formed with the energizing module and transport will follow subsequently. Such a mechanism might pro�ide a selecti�e ad�antage for bacteria expressing isolated S-components.

13
Outline of this thesis The ECF transport system for thiamin (�itamin B1) was studied with se�eral biochemical and biophysical techniques. This thesis focuses on the S-component subunit that pro�ides the substrate specificity for thiamin (ThiT).
ECF transporters are linked to ABC transporters trough their Nucleotide Binding Domains (NBDs) but ha�e a �ery different mechanism of substrate recognition. To determine the relation between ECF- and ABC transporters, a phylogenetic analysis of the NBD sequences from ECF- and ABC transporters is presented in chapter 2. The results indicate that ECF transporters do not ha�e a special position within the ABC transporter superfamily. In addition, structurally important sequence motifs in S-components are analyzed. Based on the analysis it is proposed that S-components share a structurally conser�ed core that likely forms the interaction platform for the energizing module.
Chapter 3 describes the identification of the S-components for thiamin (ThiT) and folate (FolT) from L. casei. These proteins are in�ol�ed in unusual transport kinetics that were described for �itamin transport in L. casei during the late 1��0s and early 1��0s. At that time the genes encoding these proteins were not identified, complicating a more detailed biochemical analysis. Using amino acid compositions determined 30 years ago, the genes were now located in the genome of L. casei. Furthermore, biochemical e�idence is pro�ided that the corresponding proteins were indeed responsible for the recognition of thiamin and folate.
In chapter 4, the biochemical characterization of L. lactis ThiT is described. The protein was o�erexpressed in the membranes of L. lactis and purified in a substrate-free and substrate-bound conformation. High affinity binding (picomolar to nanomolar dissociation constants) of se�eral substrates was assayed. A number of mutants were prepared to gain insight in the chemistry of high affinity binding. Static light scattering coupled to refracti�e index measurements were applied to demonstrate that ThiT is a monomer in detergent solution. This was the first determination of the oligomeric state of an isolated S-component. In vivo transport experiments confirmed that ThiT is required for thiamin transport, but in its isolated form only binds thiamin.
The high resolution crystal structure of ThiT is presented in chapter 5. The structure pro�ides insight in the mechanism of substrate binding and confirms many of the findings described in chapter 4. Using this structure and the structure of the S-component RibU from S. aureus, a model could be constructed for the recognition of different S-components by the energizing module. Furthermore, in vivo experiments confirm that indeed the energizing module is required for thiamin transport.

14
Chapter 6 is a brief discussion about structural �ariation in membrane proteins. Here, it is argued that structural similarity is a �ery strong indicator of homology, e�en in the absence of sequence similarity. Membrane proteins that share the same fold like ThiT and RibU, therefore most likely originate from a single ancestral protein. Furthermore, the consequences of structural similarity between S-components are discussed in relation to the transport mechanism for ECF transporters.

15
Appendix: the historical background of thiamin Thiamin (�itamin B1, figure 4a) was first described by Christiaan Eijkman as an essential nutrient found in the outer layers of unpolished (brown) rice that was able to cure beriberi in chicken (36). Its isolation led to the disco�ery of �itamins, for which the Nobel Prize in Physiology and Medicine was awarded in 1�2�. Thiamin was purified for the first time in 1�11 and gi�en the classification ‘�itamine’ because of its �ital importance and the presence of an amino group (4�). Later the name ‘�itamine’ was changed to ‘�itamin’ since subsequently identified �itamins were not always found to be amines. The crystal structure of thiamin was determined in 1�34 (150).
Thiamin is used as a cofactor in many enzymes, but always in the form of thiamin-pyrophosphate (TPP, figure 4b). The diphosphate moiety is not required for catalysis but belie�ed to function as a specific ‘anchoring group’ that allows binding to the enzyme (11�). This is required, because specific interactions with the aminopyrimidine or thiazole rings would probably interfere with the mechanism of catalysis.
When the crystal structure of thiamin became a�ailable, it was assumed that the acti�e group for catalysis would be the amino moiety. This model was pro�en to be wrong in 1�5�, when it was demonstrated that the acti�e group was the C2 atom in the thiazole ring (14,15). The acidity of this carbon atom is unusually high (pKa ~1�) and in the ionized (carbanion) state, thiamin is capable of performing decarboxylation reactions, albeit at a slow rate. As an enzyme-bound cofactor, the carbanion state is highly stabilized (for instance by assuming a ‘V-shaped conformation’ (�2)) and therefore the catalysis rate increases by se�eral orders of magnitude.
Figure 4Chemical structures of (a) thiamin and (b) thiamin-pyrophosphate.

16
Thiamin is an important compound for all forms of life. Many organisms are dependent on the uptake of thiamin for their sur�i�al, although bacteria can ha�e biosynthetic pathways (111). TPP is used as a cofactor in �arious enzymes performing decarboxylation reactions (4�). These enzymes are in�ol�ed in essential processes such as the citric acid cycle and pentose phosphate pathway.

1�
Chapter 2
Bioinformatics analysis of the ECF transporter subunits
Summary
This chapter contains a bioinformatics analysis of all ECF transporter subunits. First, a phylogenetic study on Nucleotide Binding Domain (NBD) sequences is used to determine the relation between ECF- and ABC transporters. Subsequently, conser�ed sequence motifs and the predicted topology of the EcfT subunit are discussed. Finally, based on multiple sequence alignments of S-component sequences from L. lactis, short sequence motifs are identified that are shared between different S-components. It is proposed that these motifs are structurally important and indicati�e of a general S-component fold.

1�
Introduction Research on ECF transporters is still in its early days and our insight in the mechanism of transport is therefore limited. Ne�ertheless, ECF transporters genes are identified in many prokaryotes (10�) and their sequences are a good starting point for bioinformatics analysis. In addition, there is a clear link between the well-studied ABC transporter family and ECF transporters, through the conser�ed Nucleotide Binding Domains (NBDs) that energize the transport reaction. To understand the exact relation between ABC- and ECF-transporters the sequences of the NBDs can be compared. The most recent phylogenetic analysis of NBD sequences was carried out more than ten years ago (115) and did not include ECF transporter NBDs. In this chapter, a large-scale phylogenetic analysis of NBD sequences from ECF- and ABC transporters is presented that places ECF transporters in the ABC transporter superfamily.
Although the molecular identity of ECF transporters was disco�ered only three years ago, already two crystal structures of S-components are now a�ailable: RibU (155) for ribofla�in and ThiT for thiamin (chapter 5). These proteins are unrelated in sequence but ha�e a similar fold. In both structures, conser�ed amino acids ha�e been identified that are important for substrate interaction and others that may ha�e a structural role (helix packing/kinking). Using the knowledge of the 3D structures, it is now possible to search for similar patterns in S-components that do not share significant sequence similarity with either ThiT or RibU. Such an analysis is performed in this chapter. Based on the results predictions are made about the general structural features of S-components.
Results and discussion
Phylogenetic analysis of ECF- and ABC-transporter NBDsAn e�olutionary tree based on the alignment of the 350 amino acids ‘core sequence’ of NBDs is depicted in figure 1. A tree based on an alignment length of a smaller stretch of 200 amino acids, showed essentially the same distribution. The tree is annotated based on the description of the sequences in the conser�ed domain database. As obser�ed before (2�), ABC transporters tend to cluster based on their substrate specificity. For instance, transporters for amino acids (methionine, histidine, glutamine and arginine) form a defined group as well as transporters for iron- or cobalt sidderophores. Although the di�ision in functional clusters seems e�ident, the bootstrap scores at the points of branching are �ery low. Low scores are usually an indication that the obser�ed patterns ha�e to be analyzed with caution. Low bootstrap scores are more often obser�ed for large datasets (115). The poor scoring does not necessarily imply unreliable branching, but indicates that not all members can be assigned to a particular group with high confidence.

1�
Figure 1: an evolutionary tree of NBDs from ECF- and ABC transportersThe tree is created using the PHYLIP package (43) and �isualized with the program Dendroscope.

20
The most recent phylogenetic analysis of NBD sequences was carried out o�er 10 years ago (115). About 200 sequences were aligned to construct an e�olutionary tree. Based on this analysis, an early segregation between ABC importers and exporters was proposed. Such an early di�ision between importers and exporters is neither supported nor dispro�ed by the data presented in this chapter. The branching order of the different groups of importers and exporters could not be determined. In general howe�er, exporters from prokaryotic and eukaryotic origin are more related to each other then exporters and importers from bacteria. For instance, the bacterial lipid exporter MsbA is grouped among eukaryotic multidrug exporters from the ABC-C family. An exception is the group of nickel/peptide transporters (bootstrap score=25), which contains both oligopeptide importers as well as peptide exporters. These results might suggest that an in�ersion of the transport direction has occurred multiple times during e�olution, some of which could ha�e occurred before the separation between prokaryotes and eukaryotes.
All NBD sequences from ECF transporters fall in one cluster (figure 1), although the bootstrap score is low at the point of branching. If the depicted clustering is reliable, ECF transporters are more related to each other than to any other ABC transporter subfamily, in spite of the large �ariation of substrates that is transported by ECF transporters. The results presented in this chapter do not indicate that the ECF transporters NBDs ha�e a special position in the ABC transporter superfamily; therefore ECF transporters should be regarded as ABC transporters.
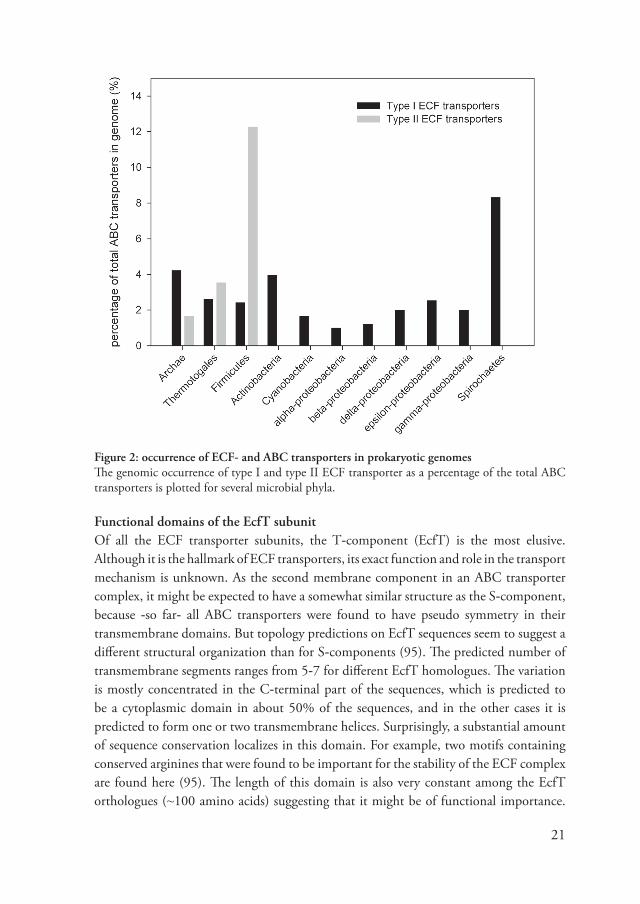
Genetic distribution of ECF transportersECF transporters are abundant in prokaryotes and can be found in the genomes of many organisms. In figure 2, the relati�e abundance of the different types of ECF transporters is plotted for se�eral prokaryotic phyla. Type II ECF transporters (that share an energizing module) are particularly abundant in Gram-positi�e bacteria. On a�erage, the number of S-components in these genomes is about 12% of the total number of ABC transporters. The occurrence of type II ECF transporters seems to be restricted to Gram-positi�e bacteria, thermotogales and archea, type I ECF transporters (dedicated to one S-component), are more widely distributed. The a�erage prokaryotic genome contains 1-2.5% of ECF type I transporters (percentage of total ABC transporters). In spirochaetes, this percentage is with an a�erage of ~�% much higher. As discussed before, the differences between Gram-negati�e an -positi�e bacteria might be explained by the lack of a confined periplasm in the latter species (26). Gram-positi�e bacteria often ha�e their soluble ABC transporter SBPs fused to the TMDs or lipid anchored in the membrane (136), whereas Gram-negati�e bacteria can express SBP freely in the periplasm. The membrane inserted ECF transporter S-components are therefore �ery suitable for utilization by Gram-positi�e bacteria.
21
Functional domains of the EcfT subunitOf all the ECF transporter subunits, the T-component (EcfT) is the most elusi�e. Although it is the hallmark of ECF transporters, its exact function and role in the transport mechanism is unknown. As the second membrane component in an ABC transporter complex, it might be expected to ha�e a somewhat similar structure as the S-component, because -so far- all ABC transporters were found to ha�e pseudo symmetry in their transmembrane domains. But topology predictions on EcfT sequences seem to suggest a different structural organization than for S-components (�5). The predicted number of transmembrane segments ranges from 5-� for different EcfT homologues. The �ariation is mostly concentrated in the C-terminal part of the sequences, which is predicted to be a cytoplasmic domain in about 50% of the sequences, and in the other cases it is predicted to form one or two transmembrane helices. Surprisingly, a substantial amount of sequence conser�ation localizes in this domain. For example, two motifs containing conser�ed arginines that were found to be important for the stability of the ECF complex are found here (�5). The length of this domain is also �ery constant among the EcfT orthologues (~100 amino acids) suggesting that it might be of functional importance.
Figure 2: occurrence of ECF- and ABC transporters in prokaryotic genomesThe genomic occurrence of type I and type II ECF transporter as a percentage of the total ABC transporters is plotted for se�eral microbial phyla.

22
Table 1: pairwise sequence identity (%) between the S-components from L. lactis MG1363
ThiT RibU BioY PanT HmpT QueT NiaX BioY2ThiT - 16 14 1� 15 10 � �RibU 16 - 14 1� 14 1� 14 14
BioY 14 14 - � 21 1� 11 20PanT 1� 1� � - 1� 14 12 21HmpT 15 14 21 1� - 13 13 15
QueT 10 1� 1� 14 13 - 14 11NiaX � 14 11 12 13 14 - 14BioY2 � 14 20 21 15 11 14 -
In addition to the arginine motifs, a proline followed by an AxxxA motif is well conser�ed. This arrangement is somewhat similar to sequence conser�ation patterns obser�ed in S-components (see below).
Structurally important residues are shared between S-componentsIf all S-components ha�e a similar fold that has originated from a single ancestral protein, their sequences ha�e di�erged beyond recognition. The highest sequence identity obser�ed between L. lactis S-components is 21% for BioY and HmpT (table 1), but in general the identities are much lower. E�en though o�erall sequence similarity is low, se�eral short sequence motifs were found conser�ed in (almost) all S-components. For example, the S-components found in L. lactis all ha�e an AxxxA motif in their first (predicted) transmembrane helix. Based on the ThiT structure, it is proposed that these alanines might be recognized by EcfT and support interaction of the S-components with the energizing module (chapter 5). In addition to the alanine motif, all S-components except QueT were found to ha�e conser�ed prolines in helix 2 and 3 (figure 3). A similar conser�ed proline in ThiT and RibU marks the boundary between the L1 loop and helix H2; in addition it initiates one turn of a 310 helix at this point, and thus ser�es a �ery specific structural role. In most S-components the proline is followed by a GxxxG or GxxxA motif, with a distance of 10-15 amino acids. In both the ThiT, and the RibU structure, the first glycine in this motif forms the loop between helix 2 and 3, allowing a �ery sharp turn of the amino acid backbone at this point. As a result, helix 2 and 3 are packed closely together, which explains the conser�ation of a second small amino acid (i.e. alanine or glycine); a larger side-chain would not fit in the space between helix 2 and 3. The occurrence of these structural motifs in the L. lactis S-components is summarized in figure 3. Although the position of the motifs is not exactly the same in all S-components, there are clearly patterns. If these motifs ha�e a similar function in all S-components, a structural similarity would be �ery plausible.

23
It becomes e�ident from figure 3 that the similarity between the S-components is concentrated in the N-terminal hal�e of the sequences. The second half does not contain shared sequence motifs and displays a large �ariation in the arrangement of the predicted transmembrane helices. These differences are also reflected in the structures of ThiT and RibU; helices 1, 2 and 3 are structurally �ery similar, whereas helix 4, 5 and 6 are more different. Since all S-components interact with the same energizing module this interaction is most likely to take place in a structurally similar part of these S-components. Helices 1, 2 and 3 in the ThiT and RibU structure pro�ide such a surface and probably form a platform for docking of the energizing module and could form the site of interaction with the energizing module.
Figure 3: shared sequence motifs between different S-componentsThe length of the protein sequences is indicated by the gray bars. Sequence motifs and (predicted) transmembrane segments are colored as indicated in the legend. The scale bar corresponds to a sequence length of 20 amino acids.

24
Methods Phylogenetic analysis of NBD sequencesThe wealth of genomics data and the abundance of ABC transporters creates a challenge for the assembly of a representati�e set of NBD sequences. For example, in a BLAST search with the ECF transporter EcfA subunit from L. lactis (CbiO2) the first 5000 sequences displayed an e-�alue of less than 10-2� and were thus most likely orthologues. Because the e-�alue is so low (e�en for the last sequence of the list) there are probably many more orthologues in the databases that were not included in the first 5000 hits. In order to acquire a representati�e set of NBDs from se�eral different types of ABC transporters, a method is needed to select those sequences from the �ast amount of ABC transporter NBD sequences that are currently a�ailable. For this, we performed a conser�ed domain search with L. lactis EcfA as a query sequence using the CD-search tool (�6). The CD-search procedure compares the query sequence with position-specific score matrices that are deposited in the Conser�ed Domain Database (CDD). In this way, a list of conser�ed domains was generated that were related to L. lactis EcfA. For each of these domains (domains classified as ‘pro�isional’ were ignored because of uncertainty in their annotation) ten representati�e sequences were downloaded. A total of 1433 sequences were collected in this way. The set includes ABC transporter NBDs from �arious biological species (eukaryotes as well as prokaryotes) and from a range of different functional ABC transporter groups. The full-length sequences were aligned with ClustalW (1�) using default parameters. Because of the large �ariation in protein length, the alignment had to be truncated to the conser�ed ‘core sequence’ of the NBDs. We used the percentage of gaps in blocks of 50 amino acids as a criterion. Each block that contained more than 65% gaps was deleted; this resulted in a final alignment length of 350 amino acids. The whole procedure was repeated with a maximum gap percentage of 53% to determine if the obser�ed clustering was biased by additional domains of the NBDs. This yielded an alignment length of 200 amino acids. An e�olutionary tree was constructed with programs from the PHYLIP package (43), to test the reliability of the branching, a bootstrap analysis was performed with 100 replicates.
Genetic distribution of ECF transporter genesThe occurrence of ECF transporter genes in prokaryotic genomes has been described before (10�). Based on this data, the number of ECF transporters (type I and type II) in these genomes was calculated. Each type II ECF transporter S-component was counted as one transporter. The total number of ABC transporters in these genomes was extracted from the TransportDB (106) and these number were used to calculate the relati�e occurrence of ECF transporters.

25
Shared amino acid motifs between different S-componentsThe sequences of the S-components from the Lactococcus lactis MG1363 genome were chosen as a starting point for the analysis, because it has been demonstrated unambiguously that all eight S-components interact with the same energizing module (130). Besides ThiT and RibU, six additional S-component sequences were analyzed (see table 1). These S-components are likely to share structural features for interaction with the energizing module. Based on each S-component sequence, a multiple sequence alignment was constructed with the program FRpred (45). The alignments were manually searched for patterns of conser�ation, similar to ThiT and RibU (i.e. GxxxG motifs and conser�ed prolines). For each L. lactis S-component, the conser�ed amino acids were mapped on a consensus topology prediction by TOPCONS (6) or experimental topology (ThiT and RibU). The pairwise sequence identities were calculated based on a multiple sequence alignment with all eight S-components by ClustalW (1�).

26

2�
Chapter 3 Identification of genes encoding the folate- and thiamin-binding membrane proteins in Firmicutes
Guus B. Erkens*, Aymerick Eudes*, Dirk Jan Slotboom, Dmitry A. Rodiono�, Valeria Naponelli and Andrew D. Hanson
this chapter is published in:J. Bacteriol. (200�) 190:�5�1-�5�4
Summary Genes encoding high-affinity folate- and thiamin-binding proteins (FolT, ThiT) were identified in the Lactobacillus casei genome, expressed in Lactococcus lactis, and functionally characterized. Substrate binding assays with purified FolT and ThiT confirmed their predicted substrate specificity. Similar genes occur in many Firmicutes, sometimes next to folate or thiamin sal�age genes. Most thiT genes are preceded by a thiamin riboswitch.
*both authors contributed equally to this work

2�
Introduction The folate and thiamin transport systems of Lactobacillus casei were partially characterized 30 years ago by Henderson and colleagues (60,62,63,65). These systems were shown to in�ol�e two small membrane proteins for specific substrate binding -one for folate and the other for thiamin- as well as an uncharacterized component shared by both systems.
Results
To identify genes encoding the binding proteins (FolT and ThiT), we used the AACompIdent tool on the ExPASy ser�er (14�) to search the L. casei (strain ATCC 334) genome for open reading frames with amino acid compositions and molecular masses matching those published for FolT and ThiT (62,65). The best match for FolT
was LSEI_2252, a 1�.0-kDa protein with fi�e predicted transmembrane domains (figure 1a). LSEI_2252 has homologues in other Firmicutes, and in some cases, the corresponding genes are adjacent to folC (figure 1b). FolC is a sal�age enzyme that mediates polyglutamylation of folates (32).
Figure 1: FolT and ThiT proteins and the genomic context of folT and thiT genes(a) Deduced protein sequence of L. casei FolT. Predicted transmembrane domains are colored grey. (b) Clustering of genes encoding FolT homologues with folC (folylpolyglutamate synthase-dihydrofolate synthase) in the genomes of two Firmicutes. Arrows indicate transcriptional direction. (c) Aligned sequences of L. casei ThiT and Bacillus subtilis YuaJ. Predicted transmembrane domains of ThiT are colored grey. Symbols beneath residues indicate identity (*) and similarity (:). (d) Clustering of genes encoding ThiT homologues with thiN (thiamin pyrophosphorylase) in genomes of the Firmicutes Carboxydothermus hydrogenoformans and Halothermothrix orenii.

2�
The best match for ThiT was LSEI_1�5�, a 21.2-kDa protein with six predicted transmembrane domains, which belongs to the YuaJ family (InterPro accession number IPR012651) of predicted, uncharacterized thiamin transporters in the Bacillus/Clostridium
group (111). LSEI_1�5� is 32% identical to Bacillus subtilis YuaJ (figure 1c). In se�eral Firmicutes, the thiT gene forms a putati�e operon with the thiamin pyrophosphokinase thiN gene (figure 1d). Like FolC, ThiN is a sal�age enzyme that con�erts thiamin to its acti�e pyrophosphate form (��).
Figure 2: Functional expression of L. casei FolT and ThiT in L. lactis(a) SDS-PAGE (12% gel) of membrane fractions from L. lactis harboring pNZ�04� alone (lane 1; 50 µg protein), or containing FolT (lane 2; 25 µg protein) or ThiT (lane 3; 25 µg protein). Staining was with Coomassie brilliant blue. The arrows indicate FolT and ThiT bands. Positions of molecular mass markers (kDa) are shown. (b to e) Binding of 3H-labeled folates or thiamin to L. lactis cells harboring pNZ�04� alone (open squares) or expressing FolT or ThiT (filled squares). (b), [3’,5’,�,�,-3H]folic acid diammonium salt (Mora�ek; 25.� Ci/mmol) (b), or [3H]folic acid polyglutamates (45 Ci/mmol) comprising 40% tri-, 56% tetra-, and 4% pentaglutamates (d). Cells expressing ThiT were incubated with [3H(G)]thiamin hydrochloride (ARC; 10 Ci/mmol) (e). [3H]-labeled substrates were chromatographically purified before use.

30
To in�estigate whether folT and thiT indeed code for �itamin-binding proteins, the folT and thiT genes were PCR amplified from L. casei genomic DNA, cloned between the NcoI and SstI sites of pNZ�04�, a �ector carrying the nisin-inducible nisA promoter (�5), and introduced into Lactococcus lactis strain NZ�000 (�5). Transformants were grown at 30°C in M1� medium (Oxoid, Basingstoke, United Kingdom), supplemented with 1.0% (wt/�ol) glucose, and 5 µg/ml chloramphenicol. Nisin was added when the optical density at 600 nm reached 0.� (�5), and cells were har�ested � to 15 h later. Sodium dodecyl phosphate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis of membrane fractions prepared by differential centrifugation (134) showed that FolT and ThiT were abundantly expressed (figure 2a) and had apparent molecular masses (1� and 22 kDa, respecti�ely) near those predicted. Cells expressing FolT or ThiT, and empty-�ector controls, were assayed for binding of 3H-labeled folates or thiamine after de-energization with 2-deoxyglucose to suppress interference by endogenous uptake systems (figure 2b to e). Cells expressing FolT bound large amounts of (6S)-[3H]folinic acid or [3H]folic acid (~1� pmol/mg protein), and those expressing ThiT bound a similar amount of [3H]thiamine. Adding a polyglutamyl tail of 2 to 4 residues to [3H]folic acid (�4) markedly reduced binding, indicating that polyglutamyl folates are poor substrates for FolT, which is consistent with results from experiments using L. casei cells (120). In all cases, �itamin binding approached a plateau within 5 s and was rapidly re�ersed by adding an excess of unlabeled substrate. The obser�ed �itamin acquisition, thus, has the characteristics of a binding process rather than those of an uptake process.
For further characterization, FolT and ThiT were tagged with N-terminal His� sequences. FolT-His and ThiT-His were produced in L. lactis as described abo�e, except that cells were cultured in chemically defined medium (��,103) without folic acid (for FolT-His) or thiamin (for ThiT-His) and har�ested 3 h after induction. Membrane �esicles were prepared (135), and proteins were solubilized with dodecyl-β-D-maltoside (DDM) and purified to homogeneity by using nickel-Sepharose and gel filtration chromatography (34) (figure 3a and 3b). Vitamin binding was measured �ia quenching of intrinsic tryptophan fluorescence, using a Spex Fluorolog 322 spectrofluorometer (Jobin Y�on) and a 1-ml stirred cu�ette at 25°C. The FolT-His and ThiT-His concentrations were 100 to 500 nM, and solutions of folinic acid, folic acid, or thiamin were added in 0.5- to 2-µl steps. Fluorescence was monitored at 340 nm for 20 to 30 s (excitation at 2�0 nm) after each substrate addition. Data were analyzed as described pre�iously (34,13�). Representati�e data for ThiT in the presence of increasing concentrations of thiamin are shown in figure 3c, and the corresponding fluorescence titration cur�e is shown in figure 3d. Comparable titration cur�es for FolT with (6S)-folinic acid and folic acid are gi�en in figure 3e and 3f; (6R)-folinic acid (the unnatural isomer) produced no quenching. The proteins bind their substrates with high affinity. The dissociation constants of ThiT for thiamin (0.5 nM) and FolT for folic acid (� nM) (figure 3) are within the range of �alues

31
Figure 3: Purification and characterization of His-tagged L. casei ThiT and FolT(a and b) SDS-PAGE of purified ThiT-His and FolT-His, as in figure 2a. (c) Fluorescence spectrum of ThiT-His (320 nM) in the absence of thiamin (uppermost trace) and in the presence of successi�ely higher concentrations of thiamin (up to 400 nM). (d) Fluorescence titration of ThiT-His with thiamin. (e and f ) Fluorescence titration of FolT-His (210 nM in) with (6S)-folinic acid (e) and folic acid (f ).

32
reported for L. casei cells (1 to 36 nM for folate binding and 0.03 to 10 nM for thiamin
binding) (5�,5�,62,65). The binding stoichiometries calculated from these data were far lower than 1:1 (0.1�:1 for ThiT and 0.0�:1 for FolT), compared to those calculated from the data for FolT and ThiT purified from L. casei (62,65). A likely explanation
is that the substrates copurified with the binding proteins, thereby obscuring binding sites, as occurred with the purified high-affinity ribofla�in-binding protein RibU (34). Absorption spectra of purified FolT confirmed that substrate had indeed been copurified (not shown).
Analysis of prokaryotic genomes using the SEED comparati�e genomics resource (100) re�ealed that ThiT and FolT homologues occur commonly and almost exclusi�ely in Firmicutes, many of which are pathogens. The FolT family is substantially more di�erse; while the majority of FolT proteins ha�e fi�e predicted transmembrane domains, two subgroups ha�e insertions that add two more such domains, and a third subgroup has a C-terminal extension similar to aspartyl-tRNA amidotransferase subunit C. Folate-binding acti�ity was �erified experimentally for FolT proteins from three pathogens (Mycoplasma capricolum, Clostridium novyi, and Streptococcus mutans) by expression in L. lactis cells and by measuring [3H]folinic acid binding as abo�e (figure 4). Two of these bacteria, C. novyi and S. mutans, ha�e complete folate biosynthesis pathways (32), as do �arious other pathogenic Firmicutes with folT genes, including Bacillus anthracis and Clostridium botulinum. It is likely that such organisms can both make and take up folates and that their folate transport capacity -which was hitherto unsuspected- confers intrinsic resistance to antibiotics targeting the folate pathway, as in malaria parasites (145).
Most of the genes encoding ThiT proteins, including that of L. casei, were found to be preceded by a thiamin pyrophosphate (TPP) riboswitch and indeed, the ThiT/YuaJ family was pre�iously predicted to participate in thiamin transport based on computational identification of these riboswitches (111). A marked feature of L. casei ThiT is its almost total repression by high le�els of thiamin in the medium (60). TPP riboswitches located in 3’ noncoding gene regions attenuate expression of downstream genes upon binding
TPP (111,151), which readily suggests a mechanism for the obser�ed repression.
Discussion The identification of the genes encoding the folate- and thiamin-binding proteins of L. casei and other Firmicutes opens the way for dissection of the corresponding transport systems at the molecular le�el. These systems are undoubtedly no�el, as FolT and ThiT
are integral membrane proteins without characterized homologues. In terms of size and hydrophobicity (but not sequence), they resemble an emerging group of integral membrane proteins implicated in �itamin and trace metal uptake. These include

33
the following: RibU of Lactococcus lactis, in�ol�ed in ribofla�in uptake (34); BioY of Rhodobacter capsulatus, a component of a biotin uptake system (55); and CbiM and NikM, in�ol�ed in uptake of cobalt and nickel (10�). The latter three systems all include a characteristic transmembrane protein (e.g., BioN) and an ATPase similar to
those of ABC-type transporters (e.g., BioM), both encoded by genes adjacent on the chromosome to genes encoding the FolT/ThiT-like component. Although there are no bioN- and bioM-related genes linked to folT or thiT, it is reasonable to infer that they lie elsewhere in the genome, gi�en the e�idence that L. casei FolT and ThiT require other,
Figure 4: folate-binding by FolT homologues from pathogenic Firmicutes expressed in L. lactisThe folT genes from Clostridium novyi and Streptococcus mutans were obtained by PCR from genomic DNA; that of Mycoplasma capricolum was synthesized by GenScript (Piscataway, NJ). Cells harboring pNZ�04� alone (open squares) or containing FolT homologues (filled squares) were assayed for binding of (6S)-[3H]folinic acid (final concentration, 13.5 nM) as in figure 2. The arrows show when unlabeled folinic acid was added to gi�e a final concentration of 50 µM.

34
shared components to form an acti�e transport system and that the energy source is ATP hydrolysis (63,64). And indeed, the L. casei genome contains a gene cluster encoding homologues of BioN (LSEI_24�2) and BioM (LSEI_24�3 and LSEI_24�4), which are thus candidates for shared components of the folate and thiamin transporters.
Methods In vivo vitamin bindingThe assays (total �olume, 1 ml) were performed in phosphate-buffered saline (PBS), pH �.4, at 30°C with stirring. Cells were washed and resuspended (optical density at 600 nm, 20), and 0.5-ml aliquots were pretreated for 5 min with 2-deoxyglucose (25 mM final concentration). Assays were started by adding 0.5 ml of PBS containing 3H-labeled thiamin (final concentration, 12.6 to 14.5 nM). At �arious times, cells (100 µl) were har�ested by �acuum filtration on a cellulose nitrate membrane (0.45 µm). Filters were washed twice with 2 ml of ice-cold PBS, and their 3H content was determined by scintillation counting. The arrows show when unlabeled �itamin was added to gi�e a final concentration of 50 µM. Cells expressing FolT were incubated with (6S)-[3’,5’,�,�-3H(N)]folinic acid diammonium salt (Mora�ek; 10 Ci/mmol)
Acknowledgements We thank Robert Burne (Uni�ersity of Florida) for Streptococcus mutans genomic DNA and Shibin Zhou (Johns Hopkins Uni�ersity School of Medicine) for Clostridium novyi genomic DNA. This project was supported by National Institutes of Health grant R01 GM0�13�2 (to ADH), by The Netherlands Organization for Scientific Research (NWO) (vidi grant to DJS), and by an endowment from the C.V. Griffin, Sr. Foundation.

35
Chapter 4
Biochemical characterization of ThiT from Lactococcus lactis: a thiamin transporter with picomolar substrate binding affinity
Guus B. Erkens and Dirk Jan Slotboom
this chapter is published in:Biochemisty (2010) 49:3202-3212
Summary
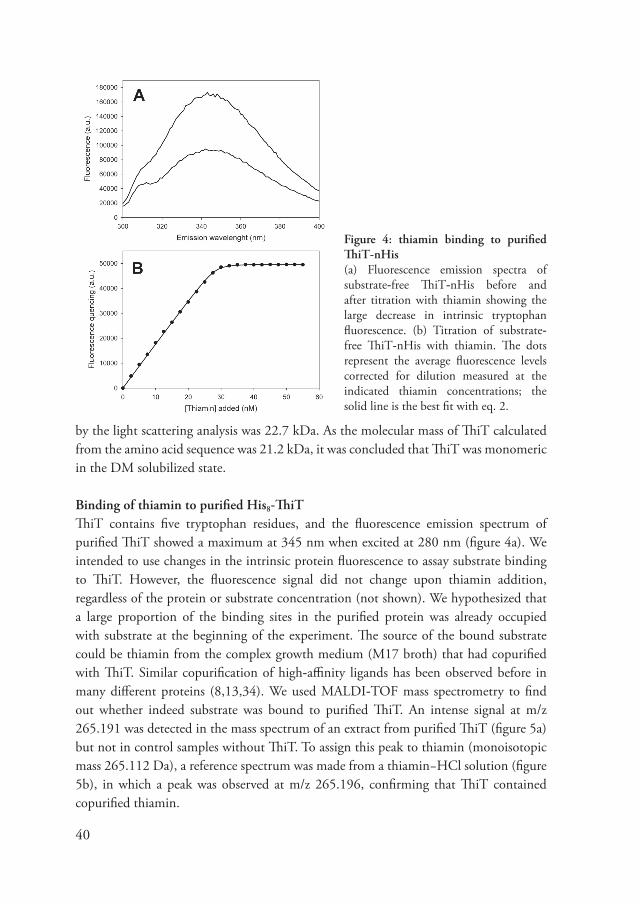
The putati�e thiamin transporter ThiT from Lactococcus lactis was o�erproduced in the membrane of lactococcal cells. In vivo transport assays using radiolabeled thiamin demonstrated that ThiT indeed was in�ol�ed in thiamin transport. The protein was solubilized from the membranes and purified in detergent solution. Size exclusion chromatography coupled to static light scattering, refracti�e index, and UV absorbance measurements (SEC-MALLS) showed that ThiT is a monomer of 22.� kDa in detergent solution. When the cells o�erexpressing ThiT had been culti�ated in complex growth medium, all binding sites of the purified protein were occupied with substrate which had copurified with the protein. MALDI-TOF mass spectrometry analysis confirmed that the copurified substance was thiamin. Substrate-depleted ThiT was obtained by expressing the protein in cells that were culti�ated in chemically defined growth medium without thiamin. The intrinsic tryptophan fluorescence of substrate-depleted ThiT was strongly quenched upon thiamin binding. The quenching of the fluorescence was used to determine dissociation constants for thiamin and related compounds. ThiT had an unusually high affinity for thiamin (KD = 122 ± 13 pM) and bound the substrate with a 1:1 (protein:ligand) stoichiometry. TPP, TMP, and pyrithiamin bound to ThiT with nanomolar affinity. A multiple sequence alignment of ThiT homologues re�ealed that well-conser�ed residues were clustered in a tryptophan-rich stretch comprising the loop between the predicted membrane spanning segments 5 and 6. Mutational analysis of the conser�ed residues in this region combined with binding assays of thiamin and related compounds was used to build a model of the high-affinity binding site. The model was compared with thiamin binding sites of other proteins and interpreted in terms of the transport mechanism.

36
Introduction Thiamin (�itamin B1) is the precursor of thiamin-pyrophosphate (TPP), an important cofactor for a wide �ariety of enzymes that catalyze decarboxylation reactions (�4,11�). TPP utilizing enzymes are in�ol�ed in many essential cellular processes such as the citric acid cycle and the pentose phosphate pathway, common to organisms in all kingdoms of life.
Many bacteria are capable of thiamin synthesis �ia di�erse biosynthetic pathways (5,�0). In addition, genes coding for putati�e or confirmed transport systems for the uptake of thiamin ha�e been found in many prokaryotic genomes (111), but the proteins catalyzing bacterial �itamin transport are poorly characterized. The best studied example is the ABC (ATP binding cassette) transporter ThiBPQ from Salmonella typhimurium (14�) that mediates thiamin and TPP transport at the expense of ATP hydrolysis. Binding of thiamin and thiamin phosphates is carried out by the ThiB (also named TbpA), a soluble substrate binding protein (SBP) that resides in the periplasmic space (66,12�). ThiB deli�ers its substrate to the membrane-bound ThiP, and transport is dri�en by ATP binding and hydrolysis by the ThiQ subunit in the cytosol.
Recently, we ha�e identified a different type of prokaryotic thiamin transporter: ThiT from Lactobacillus casei (chapter 3). ThiT is a member of the energy coupling factor (ECF) transporters, a new class of transport proteins that shares some resemblance with ABC transporters. ECF transporters consist of a conser�ed tripartite complex with two identical or homologous nucleotide binding proteins/domains (EcfA and EcfA’) and a small (~30 kDa) integral membrane protein EcfT (10�). In contrast to ordinary bacterial ABC transporters for substrate uptake, ECF transporters do not employ soluble SBPs but instead use small (~20 kDa) integral membrane proteins with fi�e to six predicted transmembrane helices for substrate recognition and binding. These proteins are termed the core transporters, and ThiT is an example. Besides ThiT, a plethora of other (putati�e) core transporters ha�e been found encoded in the genomes of prokaryotes, each specific for a different substrate. Their expression is often under the control of a riboswitch (��,111,151) and directly regulated by the intracellular concentration of their substrates. Core transporters can interact with the tripartite ECF protein complexes, forming a complete transport system likely to resemble the basic architecture of an ABC transporter, consisting of two transmembrane domains and two nucleotide binding domains. Surprisingly, there are indications that the core transporter alone can also mediate transport, albeit not coupled to ATP hydrolysis but most likely �ia a secondary transport mechanism (55).
Here we present a biochemical analysis of the ThiT homologue llmg_0334 from L. lactis, and we e�aluate the interactions responsible for high-affinity substrate recognition. A

3�
detailed model of the thiamin binding site is �aluable for the elucidation of the transport mechanism of the ECF membrane transporters. In addition, it could pro�ide a framework for antimicrobial drug design, as studies on the pathogen Listeria monocytogens (116) showed that the Gram-positi�e bacterium is dependent on ThiT for its intracellular replication, because it lacks a complete thiamin biosynthesis pathway. When the thiT gene (lmo142�) was knocked out, growth inside Caco-2 cells was se�erely diminished compared to that of a control strain effecti�e in thiamin transport.
Results Identification of llmg_0334 as the gene encoding the thiamin transporter ThiT The thiT gene (llmg_0334) on the L. lactis MG1363 genome is preceded by a predicted TPP riboswitch (��,111). TPP riboswitches act as negati�e regulators of expression, pre�enting translation when the intracellular concentration of TPP is high, but allowing translation of the mRNA when TPP concentrations are low (��,151). This pattern of regulation was reflected by in �i�o thiamin uptake assays in wild-type L. lactis cells (figure 1). When thiamin was plentiful in the growth medium, no significant uptake of thiamin was measured because the production of endogenous ThiT was repressed. In contrast, cells culti�ated in the absence of thiamin increased the translation of ThiT and readily transported [3H]thiamin. When thiT was o�erexpressed from a plasmid without the
Figure 1: in vivo thiamin transport Uptake of [3H]thiamin in L. lactis cells. Key: open circles, cells expressing ThiT-nHis grown in CDM without thiamin; closed circles, cells expressing ThiT-nHis in medium supplemented with thiamin; open triangles, control strain harboring an empty plasmid, grown in medium without thiamin; closed triangles, the control strain grown in thiamin supplemented medium.

3�
riboswitch sequence and under the control of the nisin A promoter, significant uptake of [3H]thiamin was obser�ed regardless of the thiamin concentration in the medium, showing the direct in�ol�ement of ThiT in thiamin transport. Cells o�erproducing recombinant ThiT showed �ery similar [3H]thiamin transport rates as wild-type cells expressing only endogenous thiT. We tentati�ely conclude from this result that the le�els of the endogenous ECF tripartite complex could be rate limiting for thiamin uptake. These le�els are most likely not affected by the concentration of thiamin in the medium nor by the o�erexpression of the thiT gene and thus result in similar transport rates for wild-type and o�erexpressing cells. In contrast, the binding of [3H]thiamin to the cells increased upon o�erexpression of ThiT (offset at the y axis), indicating that isolated ThiT may be primarily in�ol�ed in binding or slow turno�er transport.
Purification of ThiT and determination of the oligomeric state His-tagged ThiT (His�-ThiT) was o�erproduced in L. lactis membranes, solubilized with the detergent DDM and purified using Ni2+-affinity and size exclusion chromatography (figure 2). From the yield of purified ThiT it was estimated that ~0.3% of the total protein membrane extract consisted of ThiT. The oligomeric state of ThiT and other core transporters of the ECF-transporter family are unknown. The elution �olume of ThiT from the size exclusion column, which had been calibrated with soluble globular protein standards, indicated that the molecular mass of the ThiT-DDM mixed micelle was ~100 kDa (data not shown). Howe�er, because the amount of detergent bound
Figure 2: SDS−polyacrylamide gel stained with Coomassie blue of steps in a typical ThiT-nHis purificationFrom left to right: solubilized membrane �esicles (~40 µg of total protein loaded), flow-through after Ni-Sepharose binding, first wash fraction of the Ni-Sepharose column, second wash fraction, first elution fraction Ni-Sepharose column, second elution fraction, third elution fraction, and the peak fraction of the SEC (~0.� µg of protein loaded).

3�
to the protein was not known it was not possible to deduce the oligomeric state of ThiT in the ThiT-DDM micelle. Therefore, size exclusion chromatography coupled to static light scattering (SEC-MALLS) was applied to ThiT (figure 3a). A complication in these measurements was that empty DDM micelles cause peaks and troughs in the chromatogram of the light scattering measurement, which were not resol�ed from the ThiT-DDM mixed micelles on the size exclusion column, making calculation of the ThiT molecular weight in DDM unreliable. Peaks and troughs caused by empty micelles are commonly obser�ed in SEC-MALLS measurements (126) and difficult to a�oid. To o�ercome this problem, the detergent DDM was replaced with DM, which has a smaller micelle size (��,�3,126). The results are depicted in figure 3b. The elution peak of the ThiT-DM mixed micelle was now well resol�ed from the peak and trough caused by the empty detergent micelles. The molecular mass of ThiT in the DM micelle determined
Figure 3: determination of the ThiT oligomeric stateChromatograms from size exclusion chromatography are shown for (a) ThiT purified in DDM and (b) ThiT purified in DM. Key: dashed lines, signal from the refracti�e index detector; dotted lines, signal from the static light scattering detector at �0°; solid black line, signal from absorption at 2�0 nm; solid gray line, calculated protein molecular weight (scale on right-hand y axis). The calculated a�erage molecular mass for ThiT in DM was 22.� kDa.
40
Figure 4: thiamin binding to purified ThiT-nHis(a) Fluorescence emission spectra of substrate-free ThiT-nHis before and after titration with thiamin showing the large decrease in intrinsic tryptophan fluorescence. (b) Titration of substrate-free ThiT-nHis with thiamin. The dots represent the a�erage fluorescence le�els corrected for dilution measured at the indicated thiamin concentrations; the solid line is the best fit with eq. 2.
by the light scattering analysis was 22.� kDa. As the molecular mass of ThiT calculated from the amino acid sequence was 21.2 kDa, it was concluded that ThiT was monomeric in the DM solubilized state.
Binding of thiamin to purified His8-ThiT ThiT contains fi�e tryptophan residues, and the fluorescence emission spectrum of purified ThiT showed a maximum at 345 nm when excited at 2�0 nm (figure 4a). We intended to use changes in the intrinsic protein fluorescence to assay substrate binding to ThiT. Howe�er, the fluorescence signal did not change upon thiamin addition, regardless of the protein or substrate concentration (not shown). We hypothesized that a large proportion of the binding sites in the purified protein was already occupied with substrate at the beginning of the experiment. The source of the bound substrate could be thiamin from the complex growth medium (M1� broth) that had copurified with ThiT. Similar copurification of high-affinity ligands has been obser�ed before in many different proteins (�,13,34). We used MALDI-TOF mass spectrometry to find out whether indeed substrate was bound to purified ThiT. An intense signal at m/z 265.1�1 was detected in the mass spectrum of an extract from purified ThiT (figure 5a) but not in control samples without ThiT. To assign this peak to thiamin (monoisotopic mass 265.112 Da), a reference spectrum was made from a thiamin−HCl solution (figure 5b), in which a peak was obser�ed at m/z 265.1�6, confirming that ThiT contained copurified thiamin.

41
Table 1: relative fluorescence quenching (%) of wild-type ThiT and tryptophan mutants
QuenchingWT 46.� ± �.5± �.5W34A 1�.� ± 5.2± 5.2W63A 25.� ± 6.4± 6.4W133A 26.� ± 12.3± 12.3W13�A 1�.5 6.2W141A 25.4 ± 5.6± 5.6
The copurified thiamin pre�ented determination of the binding parameters and therefore we aimed to obtain ligand-free ThiT by expressing the protein in cells that were culti�ated in medium without thiamin. The ligand-free protein was purified, and the intrinsic
tryptophan fluorescence was measured (figure 4). The fluorescence was now strongly quenched in response to binding of thiamin, and the extent of quenching saturated at high thiamin concentrations (figure 4b). Typically, the fluorescence quenching at saturating thiamin concentrations was 4�% (table 1). Quenching of tryptophan fluorescence thus reported binding and could be used to determine dissociation constants.
The dissociation constant (KD) for thiamin binding to ligand-depleted ThiT was 122 ± 13 pM (figure 4b, table 2), and a binding stoichiometry of 0.�4 ± 0.11 mol of thiamin/mol of ThiT was calculated. The obser�ed stoichiometry was still lower than unity, which
Figure 5: MALDI��TOF mass spectra (a) ligand extracted from purified ThiT-His and (b) a thiamin-HCl solution. The monoisotopic mass of thiamin is 265.112 Da.

42
can be explained by errors in the protein concentration determination, a fraction of the purified ThiT being unable to bind substrate, or the presence of a small amount of residual copurified substrate. Nonetheless, our data strongly suggest that there is one binding site per ThiT monomer. The fluorescence quenching assay was also used to determine KD �alues for TMP and TPP binding (1.01 nM and 1.60 nM, respecti�ely, table 2).
The thiamin analogue pyrithiamin bound with a KD �alue of 1�0 ± �0 pM (table 2), whereas oxythiamin binding could not be obser�ed. The presence of Mg2+ ions did not affect the affinity for TPP.
Binding of thiamin to ThiT mutants A membrane topology model of ThiT is depicted in figure 6. Six transmembrane domains are predicted with the N- and C termini located in the cytosol. Conser�ed residues are indicated in black, and a multiple sequence alignment of L. lactis ThiT and homologues is depicted in figure �. Genes encoding ThiT are found almost exclusi�ely in Firmicutes. Because tryptophan fluorescence was strongly quenched upon thiamin binding (table 1), one or more tryptophan residues may be directly in�ol�ed in binding. ThiT contains fi�e tryptophans (indicated in gray in figure 6), and to in�estigate their role in binding, they were replaced by alanines. Each mutant was o�erexpressed and could be purified in similar amounts as wild-type ThiT. The dissociation constants for thiamin, TMP, TPP, and pyrithiamin were determined using the fluorescence quenching assay (table 2).
Figure 6: graphical representation of the ThiT membrane topology modelThe solid lines indicate the position of the lipid membrane. Tryptophans are depicted in gray and conser�ed amino acids in black. The transmembrane helices are numbered 1-6.
1 2 3 4 5 6

43
All tryptophan to alanine mutants were still capable of binding thiamin, but the W133A mutant had a dramatically reduced thiamin binding affinity (an increase in KD of 3 orders of magnitude to 145 ± 20 nM) (figure �, table 2). Tryptophan-133 is not strictly conser�ed in the ThiT family, but a tryptophan, phenylalanine, or tyrosine residue is always present at this position. To in�estigate whether the aromatic nature of tryptophan-133 was important for binding, the residue was replaced by phenylalanine. This substitution restored thiamin binding to wild-type �alues. Quenching of the intrinsic protein fluorescence (26.� ± 12.3%) was still obser�ed in mutant W133A, indicating that other tryptophan residues also contributed to the substrate-dependent quenching. Since tryptophan-133 is located in a tryptophan-rich region of the protein, we expected
Table 2: KD values (in nM) for binding of various substrates to wild-type ThiT and mutantsThe errors indicate standard de�iations.
WT W34A W63A W133Athiamin 0.122 ± 0.013 0.35 ± 0.045± 0.045 0.44 ± 0.0�5± 0.0�5 145 ± 20± 20
TMP 1.01 ± 0.14± 0.14 3.�� ± 1.53± 1.53 3.4� ± 0.01± 0.01 2�55TPP 1.6 ± 0.00± 0.00 4.24 ± 0.34± 0.34 �.4� ± 3.62± 3.62 5201 ± 406± 406pyrithiamin 0.1�0 ± 0.0�± 0.0� 1.32 ± 1.25± 1.25 0.23 ± 0.10± 0.10 135 ± 12± 12
W138A W141A G129V G129Athiamin 0.41 ± 0.015 0.0� ± 0.04± 0.04 no binding 33.� ± 3.�5± 3.�5TMP 3.5� ± 0.1�± 0.1� 1.6� ± 0.�5± 0.�5 no binding �10 ± 5�± 5�TPP 4.60 ± 1.�0± 1.�0 1.�3 ± 0.�5± 0.�5 no binding 1��2 ± 23± 23pyrithiamin 0.20 ± 0.05± 0.05 0.10 ± 0.006± 0.006 no binding 23.� � 2.35
W133F A137V Y146A Y146Fthiamin 0.32 ± 0.13± 0.13 0.2� ± 0.005± 0.005 0.005 �.15 ± 0.55± 0.55 0.44 ± 0.06± 0.06TMP 6.54 ± 0.24± 0.24 3.55 ± 0.05± 0.05 14� ± 5± 5 4.� ± 1.40± 1.40TPP 4.�4 ± 0.0�± 0.0� 5.23 ± 0.��± 0.�� 2�0 ± 3�± 3� �.� ± 1.10± 1.10pyrithiamin 0.35 ± 0.11± 0.11 0.16 ± 0.0�± 0.0� 2.�5 ± 0.35± 0.35 0.24 ± 0.12± 0.12
S147A N151A N151S W133A/Y146Athiamin 0.23 ± 0.04± 0.04 2.10 ± 0.21± 0.21 2.1 ± 0.10± 0.10 no bindingTMP 3.35 ± 0.�5± 0.�5 3�.� ± 3.�5± 3.�5 43.6 ± 5.00± 5.00 no bindingTPP 5.�0 ± 0.30± 0.30 �1.� �2.� ± 6.10± 6.10 no bindingpyrithiamin 0.15 ± 0.00± 0.00 146 ± 0.02± 0.02 1.55 ± 0.35± 0.35 no binding

44
that the fluorescence of nearby tryptophan-13� and tryptophan-141 could also be affected by substrate binding. Surprisingly, all fi�e tryptophans appear to contribute to the quenching upon thiamin binding (table 2).
Three tryptophan residues, including tryptophan-133, are located in the region between transmembrane segments 5 and 6, which also contains se�eral well-conser�ed residues (figure 6). We thus hypothesized that this region may be important for thiamin binding. Fi�e conser�ed amino acids (glycine-12�, alanine-13�, tyrosine-146, serine-14�, and asparginine-151) were selected for mutagenesis (table 2). All residues were changed to alanines with the exception of glycine-12� and alanine-13�, which were replaced by a �aline. The KD �alues for the mutants A13�V and S14�A were �ery similar to that of the wild-type protein. In contrast, mutants Y146A and N151A had decreased binding affinities with KD �alues for thiamin and the thiamin analogues of at least 1 order of magnitude higher than the wild-type protein (figure � and table 2). For G12�V neither binding of thiamin nor of the analogues could be measured.
The reduced binding affinity of N151A could not be restored by introducing a different amino acid with a side chain that is a�ailable for hydrogen bond formation: Mutant N151S bound thiamin with a KD �alue identical to that of N151A. In contrast, the reduced binding affinity of the mutant Y146A was restored to the wild-type �alue in the mutant Y146F (KD = 440 ± 60 pM). The double mutant W133A/Y146A completely lost its ability to bind thiamin.
Figure 8: Semi-logarithmic plot of thiamin binding curves Depicted are: WT ThiT (●), ThiT N151A (○), ThiT Y146A (), and ThiT W133A ().

45
Figure 7: Multiple sequence alignment of L. lactis ThiT homologues by ClustalWFrom top to bottom: Lactococcus lactis MG1363, Lactobacillus casei ATCC334, Listeria monocytogenes EGD-e, Streptococcus pyogenes MGA61�0, Bacillus subtilis str. 16�, Enterococcus faecalis V5�3. The transmembrane segments as predicted for ThiT are highlighted in blue. The conser�ed residues mutated in this study are indicated in green.

46
The strong conser�ation of glycine-12� and the total lack of thiamin binding to ThiT G12�V demonstrated its importance. The incapability of G12�V to bind thiamin might be explained by steric hindrance from the much larger �aline side chain. To test this hypothesis, glycine-12� was replaced by the smaller alanine. In contrast to G12�V, substrate binding to G12�A could be measured, but the obser�ed KD �alues for binding thiamin and analogues were more than 100-fold higher than the �alues of wild-type ThiT.
Binding of thiamin analogues to ThiT The thiamin molecule is built of two aromatic moieties: a thiazole and pyrimidine ring. Because both aromatic rings can contribute to binding �ia π−π stacking interactions, indi�idual thiazole and pyrimidine deri�ati�es may also bind to ThiT. To test this hypothesis, binding of 4-aminopyrimidine, 2-methylpyrimidine, and 4-methyl-5-thiazoleethanol (figure �) to ThiT was assayed, but binding was not obser�ed.
Figure 9: chemical structures of thiamin and analogs(a) thiamin, (b) TMP, (c) TPP, (d) oxythiamin, (e) pyrithiamin, (f ) thiochrome, (g) 4-aminopyrimidine, (h) 2-methylpyrimidine, and (i) 4-methyl-5-thiazoleethanol.

4�
Discussion Thiamin binding to ThiT resulted in a strong decrease in the intensity of intrinsic tryptophan fluorescence, which must be associated with a change in en�ironment of one or more tryptophan side chains (��). Binding of substrate could induce such changes either indirectly (as a result of global conformational changes upon substrate binding) or directly, when the indole side chain interacts with the thiamin molecule. In the latter case, substrate binding is expected to be se�erely affected by mutations that replace the tryptophan. Mutant W133A showed a decrease in binding affinity of 3 orders of magnitude, whereas mutation of none of the other tryptophans in ThiT affected binding significantly, indicating that W133 may be directly in�ol�ed in thiamin binding. Tryptophan-133 is not strictly conser�ed between ThiT homologues, but an aromatic amino acid can be found at this position in all sequences. Because mutation of tryptophan-133 into phenylalanine did not result in a dramatic decrease in the binding affinity, we conclude that the aromatic nature of the side chain at position 133 is likely to be important for binding, with tryptophan-133 possibly contributing �ia π−π stacking with either the pyrimidine or thiazole moiety of thiamin.
Tryptophan-133 is predicted to be located on the extracellular side of transmembrane helix 5, a region where se�eral well-conser�ed residues are clustered. Site-directed mutagenesis and subsequent binding experiments of the conser�ed amino acids in this region identified glycine-12�, tyrosine-146, and asparagine-151 as important for thiamin binding. Tyrosine-146 could, just as tryptophan-133, be replaced by phenylalanine without a significant change in binding affinity, and this residue may make a second aromatic contribution to thiamin binding, possibly �ia π−π stacking with one of the thiamin aromatic rings. Since the thiamin molecule (figure �) includes two aromatic rings, we speculate that the rings interact with either tryptophan-133 or tyrosine-146. The incapability of ThiT to bind the nonaromatic oxythiamin further emphasizes the importance of tryptophan-133 and tyrosine-146 in the ThiT binding site. In addition, the double mutant W133A/Y146A completely lost its ability to bind thiamin. In contrast to all other mutants the Y146A mutant is more ad�ersely affected in thiamin binding than in pyrithiamin binding. It is possible that tyrosine-146 interacts with the thiazole ring of thiamin, which in pyrithiamin is replaced by a pyridine. A model of the most important interactions in the ThiT binding site is depicted in figure 10.
There are many structures a�ailable of soluble TPP-dependent enzymes and thiamin binding proteins. Examples are pyru�ate dehydrogenase (35), benzoylformate decarboxylase (53), and the ABC transporter substrate binding proteins TbpA from Escherichia coli (12�) and CypL from Mycoplasma hyorhinis (125). The TPP-dependent enzymes often show a remarkably similar three-dimensional structure, despite low sequence similarity (4�). The TPP aminopyrimidine ring is bound in a hydrophobic

4�
Figure 10: model of the ThiT binding siteInteractions between thiamin and the conser�ed residues which when mutated strongly affected the affinity are depicted.
pocket and can be in�ol�ed in π-stacking with aromatic amino acids, which we also hypothesize for ThiT. In all TPP-dependent enzymes, TPP adopts the so-called “V-conformation” (124). In this conformation the N1 nitrogen of the aminopyrimidine ring is held in the proximity of a conser�ed glutamate and the 4-amino group is oriented toward the C2 carbon of the thiazole ring, thus stabilizing the TPP transition state during catalysis. The main interaction for TPP binding is pro�ided by the diphosphate group that is coordinated by a di�alent cation (often Mg2+), ensuring high specificity for TPP o�er TMP or thiamin. The Mg2+ ion is coordinated by an aspartic acid/glutamic acid that is part of a conser�ed thiamin binding motif (54). In contrast, TPP binding to ThiT does not require Mg2+ and TMP, and thiamin can bind more tightly than TPP; in addition, ThiT does not appear to ha�e the thiamin binding motif. Therefore, we expect the ThiT binding site to be different from that of TPP-dependent enzymes.
The thiamin binding protein TbpA was cocrystallized with TMP in its binding site. The thiazole ring of TMP is positioned between two tyrosines, neither of which is in�ol�ed in π-stacking. A third aromatic residue, tryptophan-1��, was located close to the pyrimidine ring. Like ThiT, TbpA does not require di�alent cations for the binding of TMP or TPP. Instead, the phosphate group of TMP is coordinated by hydrogen bonds to tryptophan-1��, serine-161, glycine-60, and aspartate-5� rather than through cation coordination. Thiamin binds with approximately equal affinity to TbpA as TMP does, e�en though there is no phosphate moiety a�ailable for interactions. Modeling of thiamin in the binding site suggested that it adopts an alternati�e conformation which enables π-stacking of

4�
tryptophan-1�� and tyrosine-2� to the pyridine and thiazole moiety, in this way possibly compensating the loss of fa�orable phosphate interactions. This is a similar pattern of π-stacking as proposed for ThiT. It is estimated that each π-stacking can contribute −14.� kJ/mol of free energy to binding (20), which is significant compared to the −56.� kJ/mol of free energy change that is associated with an equilibrium dissociation constant of 100 pM, as obser�ed for ThiT. π-Stacking interaction may therefore be an important contribution to high-affinity thiamin binding.
The o�erall structure of CypL is comparable to that of TbpA, despite low sequence similarity. There are nonetheless big differences between both binding sites. In the CypL crystal structure, a TPP molecule is clearly resol�ed. The phosphates of TPP are coordinated with two Ca2+ ions. The amino acids that form the Ca2+ binding sites in CypL are absent from the TbpA sequence. TPP is not stacked with aromatic residues, but tryptophan-314 and tyrosine-343 are oriented such that π-stacking with the pyrimidine and thiazole rings is possible if TPP is bound in a different conformation. This is �ery similar to the proposed thiamin binding to TbpA and might also be an important feature of the ThiT binding site. Despite the agreement between the thiamin binding sites of soluble thiamin binding proteins and the experimental results presented in this work, it is important to note that there is no structural information a�ailable of any thiamin binding membrane protein. A global structural resemblance between ThiT and the soluble proteins discussed here is �ery unlikely.
The dissociation constant of ThiT for thiamin is at 100 pM remarkably low. Such high affinity is unusual for membrane transport proteins. A similar affinity has been reported for the ribofla�in transporter RibU from L. lactis (34). Although RibU and ThiT do not ha�e sequence similarity, they are similar in hydrophobicity, molecular weight, and number of predicted transmembrane segments. Moreo�er, RibU and ThiT are both core transporters of the ECF class of membrane transporter proteins (10�). Other core transporters for folate, thiamin, and biotin from L. casei ((5�,5�,60), chapter 3) also bind their respecti�e substrates with high (nanomolar) affinity. We therefore hypothesize that high-affinity substrate binding is a general characteristic for ECF transporter family proteins and related to their mechanism of transport. Such a transport system would be most �aluable when only �ery little amounts of �itamin are a�ailable, and it is exactly under these conditions that the proteins are maximally expressed as a result of their riboswitch regulation. The core transporters are then able to sca�enge the low abundant �itamins and transport them to the cytosol.
The ECF core transporters bind their substrates with such high affinity that substrate release on the cytoplasmic side of the membrane might become problematic and would most likely require input of additional energy. This energy can be pro�ided by the tripartite ECF complex by means of ATP binding and hydrolysis. We therefore propose

50
a model for the ECF transport mechanism in which the core transporters (substrate binding proteins, e.g., ThiT) interact with the tripartite ECF complex if high rates of substrate translocation are required, whereas the core transporters alone can directly mediate low flux substrate translocation in the absence of the ECF proteins. This model does not explain the obser�ations made for the kinetics of biotin transport through the BioMNY ECF transporter from Rhodobacter capsulatus (55). Howe�er, the genes for the BioMNY system are located in a single operon, and the three encoded proteins are belie�ed to form a dedicated complex for the transport of biotin. In wild-type strains BioY may always be present in a complex with BioM and BioN, and a separate function for the solitary BioY protein is therefore not expected. The model presented abo�e might thus not apply to BioMNY but only to ECF transporters in which the tripartite ECF module is shared by many different core transporters.
Among the strictly conser�ed residues in the ThiT homologues, there is a GxxxG motif repeat in the third predicted transmembrane segment. Not only in ThiT but also in other ECF family core transporters are the motifs obser�ed. These motifs are well-known to promote association in membrane proteins by stabilizing helix−helix interactions in the membrane (112). A possible explanation would be that the GxxxG motif pro�ides an opportunity for interacting with the ECF transmembrane subunit. Alternati�ely, the GxxxG motif could ser�e as a platform for self-association of ThiT. Size-exclusion chromatography coupled to static light scattering and refracti�e index measurement, howe�er, unambiguously showed that ThiT is a monomer in detergent solution. Ne�ertheless, it cannot be excluded that in the membrane ThiT is an oligomer which it falls apart during detergent solubilization, because it adopts a non-nati�e conformation under these conditions. Howe�er, such dissociation seems unlikely because monomeric ThiT in detergent solution is capable of high-affinity substrate binding.
In Gram-negati�e bacteria, substrate sca�enging by ABC transporters is accomplished by periplasmic substrate binding proteins (12,2�) which deli�er their substrates to ABC transporters residing in the inner membrane. In Gram-positi�e bacteria it is often obser�ed that the substrate binding proteins are genetically fused to their corresponding ABC transporter or co�alently attached to membrane lipids (26,136), a phenomenon that is linked to the lack of a confined periplasm. For similar reasons, the ECF transporter class might be particularly suited for Gram-positi�es, which is in agreement with their phylogenetic distribution, as the ECF transporters are particularly abundant in Gram-positi�e organisms (10�, chapter 2).

51
Methods Materials Thiamin, TMP, TPP, pyrithiamin, oxythiamin, thiochrome, 4-aminopyrimidine, 2-methylpyrimidine, and 4-methyl-5-thiazoleethanol were obtained from Sigma. Ni-Sepharose was from GE Healthcare, detergents were from Anatrace, and [3H]thiamin was obtained from American Radiolabeled Chemicals. All chemicals were of analytical grade.
Construction of plasmids The gene coding for ThiT was amplified with PCR from L. lactis MG1363 chromosomal DNA with primers containing 5′ PciI and 3′ SpeI restriction sites. The PCR products were digested with PciI/SpeI and ligated in the �ector pREnHis (containing the sequence coding for an N-terminal His� tag) using standard cloning techniques. Site-directed mutagenesis of ThiT was performed with the megaprimer approach (�3,105) followed by ligation of the mutated ThiT gene in pREnHis. All cloned fragments were �erified by DNA sequencing (Ser�iceXS, The Netherlands). Finally, the pREnHis constructs were con�erted to L. lactis expression �ectors using the �ector backbone exchange (VBEx) protocol (4�).
Overexpression of ThiT and ThiT mutants L. lactis strain NZ�000 (�5) was used for nisin induced o�erexpression. Cells were grown semianaerobically in chemically defined medium (�) or M1� broth (Difco) supplemented with 2.0% (w/�) glucose and 5 µg/mL choramphenicol in a 2 l bioreactor at pH 6.5 and 30° C. To produce substrate-free ThiT, CDM was prepared from which thiamin was omitted. ThiT expression was induced at an OD600 of 1.5 by the addition of 0.1% (�/�) culture supernatant from the Nisin A producing strain NZ��00 (�5). The cells were allowed to continue growing for ~3 h and har�ested at a final OD600 of 5−6. After centrifugation for 15 min at 5000g, the cells were resuspended in 50 mM potassium phosphate (KPi), pH �.0, frozen in liquid nitrogen, and stored at −�0° C.
Preparation of membrane vesicles Membrane �esicles were prepared by lysis with a high-pressure cell disruptor (Constant Cell Disruption Systems), using two passages at 3�000 psi and 4° C. Prior to the disruption, MgSO4 (5 mM) and DNase (100 µg/mL) were added. Cell debris was separated from the membrane �esicles by a low-speed spin at 1�500g for 15 min at 4 °C, and the membrane �esicles were pelleted by centrifugation at 150000g for 1.5 h at 4 °C. The membranes were resuspended in 50 mM KPi, pH �.0 (final concentration ~10 mg/mL), frozen in liquid nitrogen, and stored at −�0 °C.

52
Purification of ThiT-His Membrane �esicles were rapidly thawed and resuspended to a concentration of ~5−� mg/ml in buffer A (50 mM KPi, pH �.0, 200 mM KCl, and 10% (�/�) glycerol). The �esicles were solubilized with 1% (w/�) DDM for 45 min on ice. Unsolubilized material was remo�ed by centrifugation at 2�0000g for 15 min at 4 °C. The supernatant was incubated with nickel-Sepharose (500 µl bed �olume) for 45 min at 4 °C while gently rotating. Next, the suspension was poured into a 10 mL disposable column (Bio-Rad), and the flow-through was discarded. The column was washed with 16 column �olumes (CV) of buffer A, supplemented with 40 mM imidazole and 0.05% (w/�) DDM and 16 CV of buffer B (50 mM KPi, pH �.0, 200 mM KCl) supplemented with 30 mM imidazole and 0.05% (w/�) DDM. ThiT was eluted in three fractions of one CV with buffer B supplemented with 500 mM imidazole and 0.05% (w/�) DDM. The second elution fraction (containing most of the purified protein) was loaded on a Superdex 200 gel filtration column (GE Healthcare) equilibrated with buffer C (50 mM KPi, pH �.0, 150 mM KCl, and 0.05% (w/�) DDM). Fractions containing ThiT were collected and used directly for further analysis.
MALDI-TOF mass spectrometry and identification of ThiT-bound substrate To extract the bound ligand, purified ThiT-His (~1 nmol of protein) was diluted to 20 µl in 0.1% trifluoroacetic acid; 0.2% SDS was added to denature the protein and release the substrate. Desalting and protein remo�al were accomplished by ZipTip purification (Millipore). The resulting extract was further purified using nano-LC on a C1� re�ersed-phase column. From each LC fraction, a mass spectrum was recorded on a MALDI-TOF/TOF instrument (4�00 proteomics analyzer; Applied Biosystems) with α-cyano-4-hydroxycinnamic acid as a matrix. Serial dilutions of a thiamin−HCl solution were directly spotted on the MALDI plate to generate reference spectra for thiamin. Bioinformatics Conser�ed residues in ThiT were found with a PSI-BLAST, followed by a multiple sequence alignment as performed by the online tool FRpred (45). Membrane topology predictions were made with TOPCONS (6,45,13�,140).
Static light scatteringDetermination of the molecular weight of purified ThiT was performed as described (126). For the purification of ThiT in DM, DDM was replaced by 0.15% (w/�) DM in all steps, except for the solubilization.
Substrate binding measurements by fluorescence titration Fluorescence was measured on a Spex Fluorlog 322 fluorescence spectrophotometer (Jobin Y�on) in a 1000 µl stirred quartz cu�ette at 25.0 °C. Purified ThiT was diluted in

53
buffer C to a concentration of 15−50 nM (final �olume �00 µl) and incubated for 5 min to reach temperature equilibrium. The substrates were added in 0.5−2.0 µl steps using a syringe pump (Har�ard apparatus) fitted with a 500 µl gastight glass syringe (Hamilton Co.). The syringe was connected to the cu�ette by tubing with an internal diameter of 0.13 mm (Vici AG International). The excitation wa�elength was 2�0 nm, and emission was measured at 350 nm. The signals were a�eraged o�er a period of 20 s. After each substrate addition, a 5 s inter�al was allowed for equilibration and mixing.
Data analysis The change in tryptophan fluorescence after each addition of substrate was calculated according to eq 1:
in which V0 is the sample �olume at the start of the titration, di is the �olume of titrant added after the ith addition, F0 is the a�erage fluorescence at the start of the titration, and Fi and ΔFi are the a�erage fluorescence and fluorescence quenching after the ith substrate addition. The obtained �alues for ΔFi were plotted as a function of the substrate concentration. The resulting cur�e was fitted to eq 2 in Origin �.0 (OriginLab) to obtain a �alue for the KD and n:
where A is the proportionality factor, n is the concentration of binding sites in the cu�ette, KD is the dissociation constant, and [S] is the concentration of substrate in the cu�ette. Dilution of the substrate and protein concentration was corrected by inserting eq 3 and 4 in eq 2:
with [S]’ being the concentration of substrate uncorrected for dilution and n0 the concentration of binding sites at the start of the titration. For medium- to low-affinity binding (KD > 10× protein concentration), the �alues were fitted to eq 5,
describing equilibrium binding with a single binding site: where B is the maximum quenching, measured at saturating substrate concentrations.
(1)
(2)
(3) (4)
(5)

54
In vivo thiamin transport assay Twenty-fi�e ml of CDM (supplemented either with 5 µM thiamin or without thiamin) was inoculated with 2% (�/�) of an o�ernight culture (in M1� medium) of L. lactis NZ�000 carrying either the empty plasmid pNZ�04� or pNZnHis-ThiT. At an OD600 of 0.6−0.�, nisin was added as described abo�e and growth was allowed for an additional hour, yielding a final OD600 of ~1. The cells were washed twice with ice-cold 50 mM K+-HEPES pH �.0, resuspended in the same buffer to an OD600 of 20 and kept on ice until further use. For the transport assay, the cells were diluted to an OD600 of 10 in 50 mM K+-HEPES pH �.0 supplemented with 10 mM glucose and energized for 5 min at 30 °C. The uptake reaction was started by the addition of 31.5 nM [3H]-thiamin together with unlabeled thiamin (total thiamin concentration 1 µM). At the indicated time points, 150 µL samples were taken and diluted in 2 mL ice-cold 50 mM K+-HEPES pH �.0, followed by rapid filtration trough a 0.45 µm pore-size cellulose nitrate filter. The filters were washed twice with 2 mL 50 mM K+-HEPES pH �.0 and dried at �0 °C for 1 h. Two ml scintillation liquid (emulsifier scintillator plus, Perkin-Elmer) was added to dissol�e the filters and the radioacti�ity was determined with a Tri-Carb 2�00TR liquid scintillation analyzer (Perkin-Elmer).
Acknowledgements The authors thank Fabrizia Fusetti and Wim Huibers for performing the MALDI-TOF experiments. This research was supported by The Netherlands Proteomics Centre (NPC) and The Netherlands Organization for Scientific Research (NWO) trough a vidi grant to DJS.

55
Chapter 5
Crystal structure at 2.0 �� of the S�� of the S-component for thiamin from an ECF-type ABC transporter
Guus B. Erkens, Ronnie P-A. Berntsson, Faizah Fulyani, Maria Majsnerowska,Maria Majsnerowska, Andreja Vujičić-Žagar, Josy ter Beek, Bert Poolman and Dirk Jan Slotboom
a modified �ersion of this chapter is accepted for publication in:Nat. Struct. Mol. Biol.
Summary Energy Coupling Factor (ECF) transporters are used for the uptake of �itamins in Prokarya. They consist of an integral membrane protein that confers substrate specificity (the S-component) and an energizing module that is related to ATP-binding cassette (ABC) transporters. An extraordinary property of ECF transporters is their modularity: S-components for different substrates often interact with the same energizing module. Here, we present the crystal structure of the thiamin specific S-component ThiT from Lactococcus lactis at 2.0 Å. The high-resolution model re�eals extensi�e interactions with the substrate and explains the exceptionally high binding affinity (KD ~ 10-10 M). The fold of ThiT is similar to that of the ribofla�in-specific S-component RibU, e�en though the sequences are not conser�ed. By comparing the two structures, we identified conser�ed structural motifs in the S-components, likely mediating the interaction with the energizing module. Based on these findings, we propose a general mechanism of transport by ECF transporters.

56
Introduction ABC transporters catalyze the translocation of di�erse compounds across membranes and constitute one of the largest superfamilies of proteins (2�). They consist of two integral membrane domains that form a translocation pore and two nucleotide-binding domains (NBDs) that dri�e transport by hydrolyzing ATP. The conser�ed NBDs are the hallmark of ABC transporters, whereas the transmembrane regions show a large �ariation in sequence and folds (figure 1). Classical ABC importers require additional extracellular or periplasmic substrate-binding domains or proteins (SBPs) to capture substrates, but the recently disco�ered ECF transporters use integral membrane proteins (S-components) for substrate recognition. S-components associate with an energizing module consisting of the membrane protein EcfT and two identical or homologous NBDs (EcfA and EcfA’) (10�).
ECF-type transporters are found in Prokarya only and mediate the uptake of �itamins and other nutrients needed in trace amounts (such as Ni2+ or Co2+ ions) (10�). ECF-type ABC transporters fall into two groups (3�). In group I the energizing module is used by a single S-component (‘dedicated’ energizing modules). The biotin transporter BioMNY from Rhodobacter capsulatus is the best-characterized member of this group (55). Intriguingly, in ECF transporters of Group II the same energizing module is shared by se�eral different S-components with different substrate specificities. These S-components are 20-25 kDa in size and predicted to ha�e 4-6 hydrophobic membrane spanning segments but they are unrelated at the sequence le�el, and it is not known whether they are e�olutionary related. A crystal structure (at 3.6 Å resolution) is a�ailable only for the ribofla�in-specific S-component RibU from Staphylococcus aureus (155).
Figure 1: architecture of ABC- and ECF-transportersThe three types of ABC transporters: classical binding protein (SBD)-dependent ABC-type importers (left), ABC exporters (middle) and the recently disco�ered ECF transporters (right). The nucleotide-binding domains (NBD, blue) are conser�ed among all ABC transporters, but the membrane domains (TMD, �arious colors) are different. Substrates are indicated as black dots.

5�
The group II proteins are particularly abundant in Gram-positi�e organisms. For example, in L. lactis eight different S-components interact with the same energizing module (130), allowing the ECF complexes to transport a wide �ariety of chemically different substrates. These complexes ha�e a 1:1:1:1 subunit stoichiometry (S-component:EcfT:EcfA:EcfA’; figure 1), indicating that the S-components are integral parts of the translocating complex, rather than peripherally associated substrate binding proteins. Early in vivo transport experiments in Lactobacillus casei showed that different S-components dynamically compete for association with the energizing module (64). The molecular basis for the dynamic interaction between different S-components and the energizing module is unknown.
ThiT is the S-components in�ol�ed in thiamin (�itamin B1) transport (chapter 4). It can bind thiamin with an extraordinary high affinity (KD=120 pM) and related compounds with nanomolar affinity. High affinity binding has been obser�ed for other S-components ((34,5�), chapter 3) and is most likely important for their biological function. To understand the molecular basis for high-affinity binding and the mechanism of transport by ECF transporters, we determined the high-resolution crystal structure of ThiT from L. lactis and present the data in the light of pre�ious and new biochemical data.
Results Structure determination of ThiTThiT was produced in L. lactis NZ�000, which has pro�en to be a suitable host for membrane protein production with properties complementary to that of Escherichia coli (��). The crystal structure of ThiT is the first of a polytypic membrane protein produced in L. lactis. ThiT with thiamin bound was purified in the detergent n-nonyl-β-D-glucopyranoside. Crystals of the nati�e protein were formed in spacegroup C2 and diffracted to 2.0 Å resolution. The structure (figure 2) was sol�ed by Multi-wa�elength Anomalous Dispersion (MAD) phasing, using crystals of seleno-methionine-substituted protein (Table 2). The asymmetric unit contained two copies of ThiT that were �irtually identical (RMSD=0.2 Å, figure 2b). The entire ThiT sequence could be fitted in the electron density, with the exception of the N-terminal Histag and the subsequent 5 or 6 residues (difference between the two copies of ThiT in the asymmetric unit), which apparently were disordered in the crystals. The Rwork and Rfree �alues after refinement were 20.4% and 23.2% respecti�ely. The orientation of the two ThiT molecules in the asymmetric unit is incompatible with the formation of a continuous lipid bilayer, because the membrane plane would ha�e to be rotated ~145˚ at the dimer interface (figure 2b) which is highly improbable. Therefore, we belie�e that the functional unit of ThiT is a monomer in the absence of the energizing module, consistent with pre�ious light-scattering (chapter 4).

5�
Figure 2: the structure of ThiT(a) The orientation of ThiT in the membrane was deduced from the distribution of positi�e charges (positi�e inside rule (141)). The positi�ely charged amino acids (Lys, Arg) are colored blue; all other amino acids are depicted in gray. (b) Surface (left) and secondary structure cartoon representation (right) of ThiT. In the surface model, hydrophobic residues are colored grey, hydrophilic green, positi�ely charged blue and negati�ely charged red (the latter are not �isible in this orientation). The cartoon is colored from N-terminal blue to C-terminal red. The bar indicates the approximate position of the lipid membrane (35 Å). The membrane topology for ThiT is depicted at the bottom and colored as in the cartoon model of Fig 1b. H1-H6: helices 1-6. L1-L5: loops 1-5. (c) The dimer of ThiT in the asymmetric unit. One monomer is colored purple, the second in gray. Bound thiamin is depicted in black. The dashed lines indicate the position of the membrane for both monomers. (d) The unusual structure of helix 4. Helix 4 is colored green; the rest of ThiT is depicted in gray. The lines indicate the �ertical position of the secondary structure elements.

5�
Well-defined non-protein electron density became �isible during refinement that could be assigned unambiguously to thiamin (discussed below). In addition, electron density that could fit acyl-chains was found surrounding the hydrophobic parts of the protein. In six cases there was connected electron density that could fit the headgroup of the detergent n-nonyl-β-D-glucopyranoside, and in these cases the entire detergent molecule was modeled. In the remaining six cases it was not clear whether the acyl-chains belonged to the detergent or to co-purified lipids. In these cases we modeled the acyl chains from the detergent in the electron density
Overall fold of ThiT and S-componentsThe o�erall fold of ThiT from L. lactis is similar to that of RibU from S. aureus (RMSD=3.5 Å for 145 Cα atoms), although at the sequence le�el the two proteins are unrelated (14% sequence identity). In the past years a growing number of membrane protein structures ha�e been determined that share a related fold, without sequence relatedness (e.g. the LeuT and the aquaporin folds (132,143)). These obser�ations raise fascinating questions about the e�olution of membrane proteins and the relation between primary- and tertiary structures. The lack of sequence conser�ation between S-components of the ECF transporters is all the more remarkable, because these proteins interact with a common partner, the shared ECF energizing module.
ThiT contains six hydrophobic helical segments that cross the membrane (figure 2a). A part of the L1 loop is also embedded in the lipid bilayer, which is needed because helix 2 is too short to span the entire thickness of the membrane. The position of L1 may play an important mechanistic role in the translocation of thiamin across the membrane (see Discussion). The structure of helix 4 is highly irregular. It starts with the backbone hydrogen bond pattern of a regular α-helix, then turns into a π-helix (1�), returns to an α-helical conformation, and finally continues as a long (� residues) 310 helix (13�) (figure 2c). This unusual combination of structural features has not been obser�ed in any membrane protein structure; in the RibU structure, helix 4 is a regular transmembrane α-helix. The π-bulge irregularity is important for ligand binding and will be discussed in more detail below. The 310 helical segment allows a �ery tight packing of transmembrane segment 4 with helices 2 and 3, and -to a lesser extent- helix 5. The close packing is further facilitated by the presence of numerous conser�ed glycines in helices 3, 4 and 5 and loop L2 (figure 3).
Structural basis of high affinity thiamin bindingThe thiamin-binding site is located in a pocket near the extracellular side of the membrane and lined by helices 4, 5 and 6 and the loops L1 and L5. The thiamin molecule was modeled in clear electron density (figure 4a) and has a conformation that is different from the catalytic V-shaped conformation obser�ed in enzymes that use thiamin-phosphates

60
Figure 3: sequence alignment of L. lactis ThiT with orthologues from various bacterial speciesThe position of the transmembrane helices in L. lactis ThiT is indicated abo�e the sequences. Conser�ed residues not in�ol�ed in ligand binding (e.g. structurally important residues) are colored green. The amino acids that interact directly with thiamin are highlighted in blue. Residues forming the network of hydrogen bonds and aromatic interactions around the substrate are colored yellow. The mechanistically important L1 loop is indicated by the purple bar.
as cofactor (124). A large number of interactions shape the binding site and account for the high binding affinity (figure 4b). Glu-�4 in helix 4 stabilizes the positi�ely charged N2 of the thiazole ring, and the adjacent residue Tyr-�5 forms a hydrogen bond with the hydroxyl group of thiamin. Both residues are located in helix 4 and their fa�orable orientation is dependent on the π-bulge irregularity in this helix. Hydrogen bonds are formed by Tyr-146 (�ia an ordered water molecule) and Asn-151 in helix 6 with the N1 and N3 of the pyrimidine ring, respecti�ely. Trp-34 in L1 and His-125 (helix 5) are in�ol�ed in aromatic π-stacking on opposite sides of the thiazole ring, and Trp-133 in
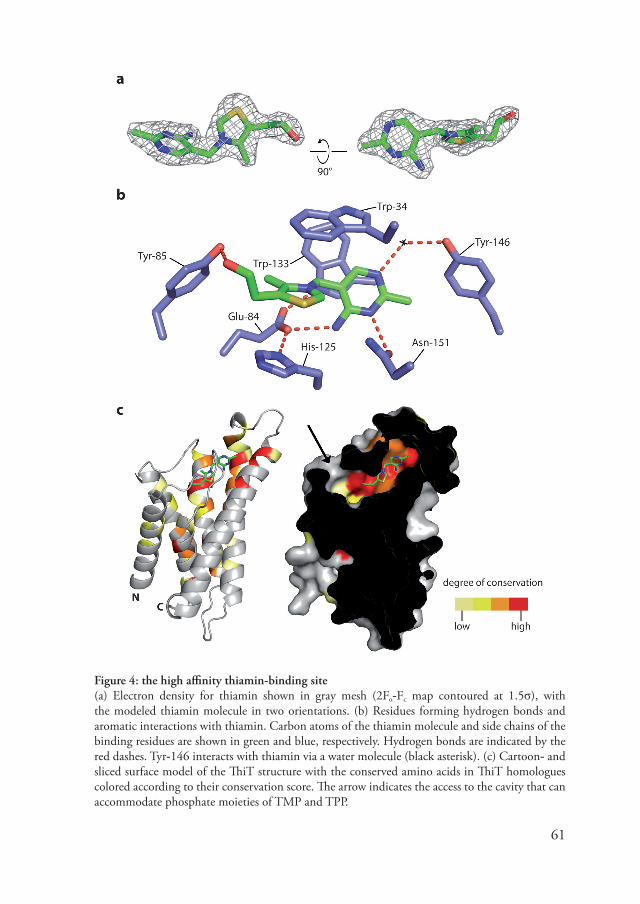
61
Figure 4: the high affinity thiamin-binding site(a) Electron density for thiamin shown in gray mesh (2Fo-Fc map contoured at 1.5σ), with the modeled thiamin molecule in two orientations. (b) Residues forming hydrogen bonds and aromatic interactions with thiamin. Carbon atoms of the thiamin molecule and side chains of the binding residues are shown in green and blue, respecti�ely. Hydrogen bonds are indicated by the red dashes. Tyr-146 interacts with thiamin �ia a water molecule (black asterisk). (c) Cartoon- and sliced surface model of the ThiT structure with the conser�ed amino acids in ThiT homologues colored according to their conser�ation score. The arrow indicates the access to the ca�ity that can accommodate phosphate moieties of TMP and TPP.

62
L5 stacks with the pyrimidine ring. Gly-12� allows the pyrimidine ring to pack closely against the C-terminal end of helix 5. The side chains that interact directly with the substrate are held in place by an intricate network of hydrogen bonds and aromatic interactions with other binding residues. In addition, more distant residues, which are not directly in�ol�ed in substrate-coordination contribute to this network (Table 1). The structure of the thiamin-binding site is in excellent agreement with pre�ious mutagenesis studies (chapter 4). Mutations of Trp-133, Gly-12�, Asn-151 and Tyr-146 to alanine reduced the binding affinity between 20 and 1000-fold. In contrast, the W34A mutant still bound thiamin with wild-type affinity. Trp-34 acts as a lid on the binding site and its remo�al apparently has little effect on the rest of the binding site.
The hydroxyl group of thiamin is accessible from the extracellular en�ironment �ia a narrow opening expanding into a ca�ity (figure 4c). The ca�ity does not allow the substrate to enter, but pro�ides sufficient space to accommodate the phosphate moieties of thiamin-mono- and thiamin-pyrophosphate (TMP and TPP), which also bind with high affinity to ThiT (chapter 4). Entrance of thiamin into the binding site from the external side of the membrane would require conformational changes of loops L1, L3 and L5, which form a cage of aromatic side chains on top of the substrate.
Table 1: the intricate network of hydrogen bonds and aromatic interactions
Residue Interactions with
Trp-34 Tyr-�4 (AL)Glu-3� Tyr-122 (HB), Lys-121 (HB)Tyr-�4 Trp-34 (AL), Tyr-�5 (HB to OH-group Cα)Leu-�6 Gln-�0 (HB)Gln-�0 Glu-�4 (HB), Leu-�6 (HB to backbone nitrogen)Glu-�4 Trp-133 (HB), His-125 (HB), Gln-�0 (HB)Tyr-�5 Tyr-�4 (HB)Lys-121 His-125 (HB), Glu-3� (SB)Tyr-122 Glu-3� (HB)His-125 Lys-121 (HB), Glu-�4 (HB)Trp-133 Glu-�4 (HB)Trp-13� Tyr-146 (AT)Trp-141 Tyr-146 (AT)Tyr-146 Trp-13� (AT), Trp-141 (AT)Ser-14� Asn-151 (HB)Asn-151 Ser-14� (HB)HB= Hydrogen Bond, SB= Salt Brigde, AL= Aromatic L-shaped, AT=Aromatic T-shaped

63
Substrate transport by ThiT requires the energizing moduleThe residues in�ol�ed in thiamin binding are highly conser�ed among ThiT orthologues (figure 3). In Figure 4c the degree of conser�ation is projected on the ThiT structure. In addition to the binding site residues, a few other amino acids are also strongly conser�ed (e.g. Pro-43 at the beginning of helix 2 and se�eral glycines in L3, helix 3 and the 310 part of helix 4). These residues ha�e a structural role and are unlikely to line a pathway for thiamin transport. In fact, we do not obser�e an ob�ious substrate translocation path within ThiT, in contrast to what has been suggested for RibU (155). The absence of a translocation path is consistent with thiamin transport assays (figure 5). When expressed in E. coli ThiT alone did not support thiamin transport, but co-expression of the energizing module (EcfAA’T) lead to robust thiamin uptake. O�erexpression of ThiT in L. lactis resulted in increased le�els of binding of thiamin to the cells, but not in increased transport rates. In L. lactis the energizing module is constituti�ely expressed from the
Figure 5: transport of [3H]-thiamin in E. coli and L. lactis cells(a) Thiamin uptake by recombinant E.coli cells. E. coli cells co-expressing ThiT and EcfAA’T from L. lactis (●), expressing ThiT alone (○), and control cells containing an empty expression plasmid (■) were assayed for thiamin uptake. All cells were energized with glucose. The energizing module is required for thiamin uptake. (b) Thiamin uptake by recombinant L. lactis cells. De-energized control cells (harboring an empty plasmid, but containing chromosomal copies of thiT and ecfAA’T) (○); de-energized cells expressing ThiT from a plasmid (); energized control cells (●); and energized cells expressing ThiT () were assayed for thiamin uptake. Thiamin binding, rather than transport, was obser�ed in the de-energized cells. The le�els of binding depended on the expression le�els of ThiT. In the energized cells harboring the empty plasmid, rapid thiamin uptake was obser�ed. In energized cells o�erexpressing ThiT, the offset on the y-axis -indicati�e of binding- rather than the uptake rate increased. The data indicate that the amount of the energizing module (which is expressed from the chromosomal genes) is limiting for thiamin transport.

64
Figure 6: proposed mechanism of interaction with the energizing module (a) Superposition of the RibU structure in gray (PDB: 3P5N) on the ThiT structure (colored as in Fig. 1b). The dashed line indicates the proposed interface with the energizing module. (b) Top: alignment of N-terminal amino acid sequences from the L. lactis S-components, the conser�ed alanine motif is colored red. Bottom: the ThiT structure as seen from the interface with the energizing module. The surface of the L1 loop region is highlighted in blue and indicated by the dashed circle. Rearrangement of the L1 loop would expose the bound thiamin (black sticks) to the lateral EcfT interface. The alanine motif in helix 1 that is shared by all S-components in L. lactis is colored red. (c) Specific interactions between the L1 loop and helices 5 and 6. Glu-3� in L1 forms a salt bridge with Lys-121 in helix 5 and a hydrogen bond with Tyr-122 and the side chains of Ser-154 and Thr-15� in helix 6 form hydrogen bonds with backbone NH groups in loop L1. In addition Trp-34 in L1 makes an aromatic interaction with Tyr-�4 in L3 (not shown). (d) Model for coupling helix interaction in ECF transporters. The S-component is colored red, EcfT is orange and the EcfA subunits are blue.

65
chromosomal copy of the ecfAA’T genes, and the results show that thiamin transport in L. lactis is limited by the amount of EcfAA’T rather than ThiT. These experiments, together with pre�ious data (chapter 4), show that ThiT binds thiamin, but does not translocate the �itamin in the absence of the energizing module. We belie�e that the translocation pore is formed at the interface of the ThiT and EcfT subunits (see discussion).
Discussion
Interaction with the EcfT subunitThe lack of sequence similarity between ThiT and RibU is remarkable, because both proteins interact with ECF energizing modules. Ob�iously, since the two proteins are from different organisms they do not interact with the same energizing module. Howe�er, ThiT and RibU proteins from the same organism are unrelated in sequence too (e.g. in L. lactis there is only 16% identity). Because of the absence of significant sequence conser�ation between the two different S-components, we searched for structural motifs that could be the docking sites for the energizing module. Interaction with the hydrophobic EcfT subunit is expected to take place within the hydrophobic core of the lipid bilayer. A superposition of the ThiT and RibU structures (figure 6a) re�ealed that helices 1, 2, 3 and 6 align well, but that helices 4 and 5 adopt �ery different conformations. The structural �ariability in the latter region is required for correct positioning of the side-chains that create the binding sites for either thiamin or ribofla�in, and makes it unlikely that the EcfT component interacts here. In contrast, the surface formed by helices 1, 2, 3 and 6 is �ery similar in RibU and ThiT. Sequence comparison of all eight S-components from L. lactis re�ealed that there is a shared alanine motif on the exposed face of helix 1 (figure 6b) that could be recognized by EcfT. Interaction with the EcfT subunit on this side of the S-components immediately suggests a mechanism for substrate release and transport to the cytoplasm. Rearrangement of the membrane embedded L1 loop could open a lateral gate for thiamin facing the EcfT subunit (figure 6b). Furthermore, the L1 loop interacts intimately with helices 5 and 6 and L3 (figure 6c). Repositioning of L1 will disrupt these interactions and perturb the binding site residues, thereby reducing the binding affinity and allowing thiamin to lea�e. We hypothesize that the final translocation step takes place on the interface between EcfT and the S-component, in line with the import of substrates �ia classical ABC transporters (2�).
Interaction with the nucleotide binding domainsThe free energy required for the conformational rearrangements in the thiamin-binding region must come from the hydrolysis of ATP in the EcfA subunits. In all ABC transporters for which crystal structures are a�ailable, each NBD contains a groo�e that binds to a cytoplasmic segment of the membrane domain. The structural elements in the membrane

66
domains that fit in these groo�es are short helical segments named “coupling helices”. The interaction �ia coupling helices allows ATP hydrolysis to be coupled to transport (6�,�5). Because EcfA and EcfA’ possess all the sequence motifs that are mechanistically important in NBDs associated with ABC transporters (2,31,50,6�,�2,�5,��) (figure �), it is reasonable to assume that the two proteins also communicate with the membrane subunits �ia coupling helices. Surprisingly, neither ThiT nor RibU has an ob�ious coupling helix. To explain this paradox, we propose that the EcfT subunit of the energizing module may contain two coupling helices. EcfT proteins are predicted to ha�e a long and conser�ed cytoplasmic loop with two moderately hydrophobic helical segments (3�,�5). The size of this cytoplasmic domain (10� amino acids) allows the presence of two rather than one coupling helix for interaction with both EcfA and EcfA’. The free energy released by ATP hydrolysis is then transferred �ia the EcfT subunit to the S-components. This hypothesis is supported by mutagenesis studies showing that two conser�ed motifs (30-40 amino acids apart) in the cytoplasmic loops of different EcfT proteins are important for stability of the Energizing module/S-component complexes (�5). The absence of a coupling helix in S-components might facilitate their exchange from complexes with the shared energizing module. Such dynamic interaction was suggested already in the 1��0’s based on in vivo transport experiments (64).
Symmetry of the membrane domainsAll a�ailable crystal structures of ABC transporters show structural symmetry between the two transmembrane subunits (2,31,50,6�,�2,�5,��). The symmetry is most ob�ious when the two subunits are identical, but also in the case of hetero-dimers the folds are related. We were not able to detect sequence similarity between S-components and EcfT, but we cannot exclude that these proteins are structurally similar, and that the sequences ha�e di�erged beyond recognition. Howe�er, the predicted topology and structural organization of EcfT proteins are different from those of the S-components (�5). Together with our hypothesis that the EcfT component may contain two coupling helices for interaction with the EcfA subunits, ECF transporters are likely less symmetrical than classical ABC transporters.
Transport without the energizing moduleThe S-component BioY from R. capsulatus, which is specific for biotin, has been shown to mediate biotin transport in vivo in the absence of an energizing module (55). A similar obser�ation could not be made for ThiT (figure 5). Because the proteins do not show significant sequence conser�ation, it is possible that BioY and ThiT are not structurally and mechanistically related. Alternati�ely it is possible that the proteins are structurally related, but that the transport function of S-component has not been conser�ed. It is noteworthy that BioY has been shown to ha�e a homo-oligomeric quaternary structure

6�
Figure 7: Sequence alignment of NBDs from known ABC transporters structures with EcfA and A’ from L. lactisThe conser�ed ABC transporter motifs are: Walker A (P-loop) (orange), conser�ed glutamine of the Q-loop (blue), the ABC transporter signature motif (red), Walker B motif (yellow) and the H-loop (green). We ha�e used two orthologues of ModC for this alignment: ModC from Archaeoglobus fulgidus (ModC_AF) and from Methanosarcina acetivorans (ModC_MA). The residues that line the coupling helix binding groo�e are colored purple. These residues are remarkably conser�ed between EcfA and EcfA’, and MetN, ModC, MalK and (to lesser extent) BtuD, from which structures were used to locate the groo�e, strongly suggesting that both EcfA and EcfA’ interact with a coupling helix.

6�
in vivo (44). BioY could ha�e e�ol�ed to adopt an (optional) homo-oligomeric state to be able to transport biotin at the subunit interface.
Concluding remarksThe high-resolution structural data presented here, together with the RibU structure, pro�ides the first glimpse of the transport mechanism of ECF transporters. Further structural- and biochemical studies on the complete complexes will now be necessary. ECF transporters are exclusi�ely prokaryotic and numerous human pathogens are dependent on the uptake of ECF substrates for sur�i�al (51,131). For instance, ThiT from the human pathogen Listeria monocytogenes is pro�en to be essential for intracellular replication (116). Structural and mechanistic understanding of ECF transporters may enable the de�elopment of new antibiotics that target these proteins.
Methods
Protein overexpression Nati�e ThiT-nHis was o�erexpressed in L. lactis strain NZ�000(�5). The cells were grown semi-anaerobically in GLS medium (2% (w/�) Gistex, 2.5% (w/�) glucose, 100 mM KH2PO4, 110 mM K2HPO4 and 5 µg/ml chloramphenicol). The initial pH was 6.� and decreased during cell growth. At an OD600 of 1.5 (at this point the pH was 6.5), expression was induced by the addition of 0.1% (�/�) of culture supernatant from the nisin A producing strain NZ��00 (�5). The cells were induced for 2 h and reached a final OD600 of 4-5. The preparation of membrane �esicles was performed as described (chapter 4). Expression of seleno-methionine (SeMet) substituted ThiT was done in L. lactis as described (�). SeMet was obtained from Acros Organics.
Purification of thiamin bound ThiT-nHisThe purification of ThiT with N-terminal �-His-tag (ThiT-nHis) was performed as described (chapter 4) with the following modifications: during solubilization, 100 µM thiamin-HCl (Sigma) was added to ensure saturation of all binding sites with thiamin. Membrane �esicles from L. lactis NZ�000, expressing ThiT were solubilized in 1.0 % (w/�) n-dodecyl-β-D-maltopyranoside (DDM, Anatrace), but in all subsequent steps this detergent was replaced by 0.35% n-nonyl-β-D-glucopyranoside (NG, Anatrace). Size-exclusion chromatography was done on a Superdex-200 column (GE healthcare) in 20 mM HEPES, 150 mM NaCl and 0.35% NG (pH �.0, adjusted with NaOH). The peak fractions after SEC were concentrated on a �i�aspin 30 kDa MWCO concentrator (VWR international) to 6-� mg/ml. Concentrated ThiT was used directly to set up crystallization trials.

6�
CrystallizationInitial crystals of ThiT were obtained under se�eral conditions by screening of commercially a�ailable crystallization conditions with ThiT purified in n-octyl-β-D-glucopyranoside (OG). These crystals were small and diffracted only to ~50 Å. Optimization of the crystallization conditions gradually impro�ed the diffraction properties and finally yielded crystals diffracting to �-� Å. Rescreening with the detergent n-octyl-β-D-thioglucopyranoside (OTG) resulted in bigger crystals which diffracted up to 5-6 Å. A major impro�ement was made with ThiT purified in NG. Crystals, ranging in size from 50 to 300 µm, could be grown at 5˚ C from a solution containing 15-20% (w/�) PEG 3350 and 0.1-0.3 M NH4NO3. The crystals appeared within one week, grew to full size in 3-4 weeks and diffracted to ~2 Å. For cryo-protection, a solution of 40% (w/�) PEG 3350 was prepared with the same concentration of NH4NO3 as in the crystallization condition. We found that crystallization of ThiT in NG was quite robust, replacing PEG 3350 by PEG’s with a higher or lower a�erage molecular weight resulted in crystals in most cases. Howe�er, we found that crystals grown from PEG 3350 had the best diffraction properties. SeMet substituted ThiT-nHis could be purified and crystallized under identical conditions as the nati�e protein.
Structure determinationDiffraction data were collected at the ESRF, Grenoble and SLS, Villigen. Multi-wa�elength Anomalous Dispersion (MAD) data on SeMet-ThiT to 2.� Å were collected on ID2� at ESRF around the K-absorption edge of selenium with the wa�elengths for remote: 0.��6� Å, inflection: 0.���3 Å and peak: 0.���1 Å, in that order. Nati�e data to 2.0 Å was collected at 1.0�23 Å on ID23-1. Data processing and reduction was carried out using XDS (�1) and programs from the CCP4 suite (24). Rele�ant statistics of the data collection, phasing and model refinement can be found in Table 2. Initial phase information was found and the initial model built using Phenix AutoSol (1) and Resol�e (within Phenix). Four selenium sites were found within the asymmetric unit, corresponding to two SeMet per protein molecule (Met-1� and Met-6�). All SeMet peaks were abo�e 25σ (figure �). The full model was built in ARP/wARP (�0) using the nati�e data. A few cycles of refinement in Refmac5 (�3), including non-crystallographic symmetry with loose restraints, interspersed with manual model building using Coot (3�), was necessary to complete the model. The final protein model contains residues �-1�2 for chain A, and 6-1�2 for chain B, thus only the initial fi�e or six residues and the His-tag are missing. Water molecules were automatically placed in Fo-Fc Fourier difference maps at a 3σ cut-off le�el and �alidated to ensure correct coordination geometries using Coot.

�0
Transport of [3H]thiamin in L. lactis and E. coli cellsTransport experiments with L. lactis cells were performed as pre�iously described (chapter 4). To de-energize the cells, we added 20 mM N-methyl-α-D-glucopyranoside instead of glucose. The cells were grown in chemically-defined medium without thiamin. The scarcity of thiamin in the growth medium induces the expression of the chromosomal thiT gene. The energizing module (EcfAA’T) was expressed constituti�ely from the chromosomal copies of the genes.
E. coli MC1061 cells containing plasmids for expression of ThiT alone or both EcfAA’T and ThiT (130) were grown on LB medium with 100 µg/ml ampicillin. At an OD600 of ~0.5, expression was induced with by adding 10-3% (w/�) L-arabinose. After two hours of induction, the cells were har�ested by centrifugation, washed and resuspended in ice-cold buffer (50 mM potassium phosphate, pH �.5) to a final OD600 of 5 and kept on ice. For the transport assays, the cells were energized with 10 mM glucose for 15 min at 30ºC. Subsequently, [3H]-thiamin was added to a final concentration of 25 nM and at the indicated timepoints 200 µl samples were taken and mixed with 2 ml stop buffer (ice-cold 50 mM potassium phosphate pH �.5). The suspension was then rapidly filtered o�er a BA-�5 nitrocellulose filter, which was subsequently washed once with 2 ml stop buffer. Filters were dried for 1 hour at �0 °C, 2 mL of Emulsifier-Scintillator Plus
Figure 8: the ThiT molecules in the asymmetric unitBoth chains of ThiT are shown in ribbon representation and colored in a rainbow fashion, from the N-terminus blue to the C-terminus in red. Peaks for the Se atoms in the anomalous difference Fourier map, calculated between 4�.0 and 2.� Å and contoured at 5σ, are shown in a grey mesh. A total of 4 SeMet were �isible in the map. SeMet at position 1 and � in the protein construct were in a disordered region of the protein. Peak heights are as follows: Mse-6�: 34.�σ and 2�.0σ (in chain A and B, respecti�ely), Mse-1�: 26.�σ and 26.3σ (in chain A and B, respecti�ely). No noise peaks were present at a 5σ cut-off.

�1
Table 2: data collection and refinement statistics
Native Se-MADData collectionSpace group C2 C2Cell dimensions
a, b, c (Å)Å) 61.4, �4.3, 12�.0 65.2, �3.�, 12�.5α, β, γ (°) �0, �5.�, �0.0 �0.0, �5.�, �0.0
Peak Inflection RemoteWa�elenght (Å)Å) 1.0�23 0.���1 0.���3 0.��6�Resolution range (Å)Å) 4�.5 - 2.0 4�.� - 2.� 4�.� - 2.� 4�.� - 2.�Rsym (%) 6.6 (4�.6) 6.� (2�.3) 5.1 (12.�) 4.4 (�.2)I/σ (I) �.4 (2.1) 15.� (6.0) 21.2 (10.2) 26.� (15.5)Completeness (%) ��.0 (�6.6) ��.� (��.�) ��.� (100) ��.� (100)Redundancy 3.� 6.� 6.� 6.�
RefinementResolution (Å)Å) 4�.5 - 2.0Number of reflec-tions
41123
Rwork/Rfree 20.4/23.2No. atoms
Protein 2�4�Ligand (thiamin) 36Ligand (others) 1��Water �3
B-factorsProtein 41Ligand (thiamin) 36Ligand (others) �0Water 51
RMS de�iationsBond lengths (Å)Å) 0.01�Bond angles (°) 1.6�The numbers in parentheses correspond to the highest resolution shell.

�2
liquid (PerkinElmer, Waltham, MA) was added, the mixture was �ortexed and le�els of radioacti�ity were determined with a PerkinElmer Tri-Carb 2�00 TR isotope counter. For timepoint zero, 200 µl of cell suspension was added to 2 mL stop-buffer containing radioacti�e thiamin and this mixture was directly filtered.
Acknowledgements We thank the ESRF and SLS for pro�iding excellent beam-line facilities. We thank Ria Duurkens for performing transport experiments, and Andy-Mark Thunnissen for critically reading the manuscript. This research was supported by the Netherlands Organization for Scientific Research (NWO) (Vidi grant to DJS, TOP-subsidy grant �00.56.302 to BP and toptalent grant to JtB) and the European Union (EDICT program).

�3
Chapter 6
Discussion: structural similarity between membrane proteins indicates ancient homology
Summary
The amount of a�ailable membrane protein structures is limited compared to soluble proteins. Although it might be too early for conclusions, some folds clearly occur more than others. As a result, membrane proteins that are apparently unrelated in amino acid sequence ha�e identical folds (see chapter 5 of this thesis). In this chapter it is argued that in such cases the proteins originate from the same structural ancestor, rather than being the product of con�ergent e�olution. Because interfaces for membrane protein interaction mainly require structural conser�ation, apparently unrelated proteins can interact with the same partner. These principles might pro�ide an explanation for the modularity of ECF transporters (chapter 1).

�4
Introduction Membrane proteins are notoriously difficult to characterize with techniques such as X-ray crystallography and NMR. As a result, membrane protein structures are underrepresented compared to soluble proteins. For instance, out of the �0�4� entries in the Protein Data Bank (sampled on 2/2/2011) only 266 entries (0.4%) represent unique membrane protein structures (based on ‘membrane proteins of known structure’: http://blanco.biomol.uci.edu/Membrane_Proteins_xtal.html). Although the amount of new structures that are released is limited, a number of examples ha�e been published in the past few years of unrelated membrane proteins that surprisingly share the same fold. For example, after the structure of the leucine transporter LeuT was published (152), other transporters were unexpectedly found to ha�e an identical fold although sequence similarity with LeuT was not detectable (40,41,10�,11�,123,12�). In this thesis, another example of sequence unrelated membrane proteins sharing an identical fold is presented. In chapter 5, the structure of ThiT is presented, which was found to be �ery similar to the structure of the ribofla�in specific S-component RibU. The pairwise sequence identity between ThiT and RibU is 14%, well below the generally accepted boundary of 20-25% abo�e which proteins are considered to be homologous. Despite the growing number of structures, the fold of the majority of membrane proteins remains elusi�e (14�). Howe�er, if the lack of structural �ariation is representati�e for the membrane protein ‘fold-ome’ these findings raise fundamental questions about membrane protein e�olution (132). Has the same fold originated multiple times during the course of e�olution? Or does structural similarity indicate an ancient homology between apparently unrelated membrane proteins? In this chapter, a short discussion on this subject will be gi�en and arguments are presented fot the hypothesis that structural similarity between membrane proteins is an indication for homology.
A definition of homology for proteins In the Oxford English dictionary (online edition: http://www.oed.com), the following definition is gi�en for homologous:
“Homologous, adj.a. Biol. Having the same relation to an original or fundamental type; corresponding in type of structure (but not necessarily in function); said of parts or organs in different animals or plants, or of different parts or organs in the same animal or plant. (Distinguished from analogous: see quot. 1854 at analogous adj. 1b)”
In short: “Two items are defined as homologues if they share a common ancestry” (11�). For proteins, the hypothesis of homology (originating from the same ancestral protein)

�5
is often tested empirically by calculating the pairwise sequence identity. If this �alue is higher than a generally accepted threshold, two proteins are considered to be homologues. Strictly speaking, sequence similarity is not enough to pro�e homology. A distinction must be made between homology and analogy. For instance, two protein sequences are considered analogous if their ancestral sequences are more dissimilar than their current (i.e. two sequences e�ol�ed independently to a �ery similar state). Homology applies if the opposite is true (ancestral sequences are more similar than the current). In practice, sequence similarity o�er a long stretch of amino acids is enough to pro�e homology. The chance that two unrelated proteins e�ol�e to a high degree of sequence identity is infinitely small.
Sequence similarity as an indicator for homology If the selecti�e pressure is strong enough, protein e�olution occurs at a slow rate (4,133). Homologues proteins separated by more than a billion year of e�olution can still ha�e remarkable sequence similarity. For instance, the E. coli L4 ribosomal proteins is ~30% identical to the human mitochondrial L4 ribosomal protein, despite two billion years of e�olution separating them. It seems reasonable to assume that when the function of a protein has to be conser�ed, there would be a limit to the di�ergence of sequences. Only a small fraction of the a�ailable sequence space is able to perform a required function. Computational studies on sequence di�ergence ha�e indeed confirmed that at any gi�en moment only 2% of the sequence can be modified without a loss of fitness (104). Howe�er, the same study demonstrated that pro�ided the right substitutions occur, o�er �0% of the amino acids in a protein can eventually undergo substitution. In other words, if di�ergence occurs long enough, truly homologues proteins can display less than 10% sequence identity. As a rule-of-thumb, a sequence identity of more than 25% is considered to be a proof for homology (33). Proteins with 20%-25% identity are considered to be in the ‘twilight zone’ of homology and below 20%, homology becomes undetectable. These numbers show that proteins can be homologues e�en if their sequence identity is far below the limit of recognition; apparently unrelated proteins can thus be homologs.
Structural similarity as an indicator for homology The boundary of the ‘twilight zone’ might seem arbitrary, but has a clear basis in protein physics (23). If a set of proteins di�erges from their common ancestor, the structure of the ancestral protein poses limitations on the allowed amino acid substitutions. Substitutions that distort the side-chain packing will induce structural instability and are thus unfa�orable. As a result, there is a global conser�ation of the side-chain packing pattern, which appears to be some kind of a ‘structural memory’ (102). The side-chain

�6
packing of a protein is dictated by both the amino acid sequence and the proteins backbone structure. As e�olution progresses, subtle changes in the protein backbone and primary structure gradually increase the RMSD (Root Mean Square De�iation) between the ancestral- and current structures. It can be calculated based on the packing �olumes of the 20 amino acids side-chains (21) that when the RMSD reaches a �alue of about 2 Å, on a�erage the insertion of any amino acid will tolerated (23). At this point, the backbone template has become different enough to allow new sidechain packing, not found in the original protein. It is an empirical obser�ation that the structures of proteins with a sequence identity of 20-25% display an RMSD �alue of ~2 Å (22,46), which corresponds �ery well with the ‘twilight zone’. Below the ‘twilight zone’ the o�erall structure of a protein will still be �ery similar to the ancestral protein, but the a�ailability of new side-chain packing patterns allows an e�en faster rate of amino acid substitutions. Only structural similarity remains as a proof of ancient homology.
Why do membrane protein structures show limited fold variation? The theoretical number of possible protein sequences is -in practicle sense- unlimited. There are for instance 20100 (~10130) different possibilities to construct a 100 residues long proteins sequence out of the 20 proteinogenic amino acids. For comparison, it is estimated that the uni�erse contains about 10�0 atoms, thus it is fair to conclude that ‘sequence space’ can be considered infinitely large. As a consequence, the chance that two proteins independently e�ol�e to the point of (nearly) identical sequences is infinitely small. Sequence similarity (in particular o�er longer stretches of amino acids) is therefore a �ery good indicator for homology between proteins. In contrast to sequence space, the number of occurring protein structures (or ‘fold space’) is much more restricted. It is estimated that, as much as �0% of all proteins can be classified by one of the 400 ‘mesofolds’ and that the total number of folds is limited to roughly 10,000 (25). A limited fold space implies that only a small fraction of the theoretically possible proteins sequences is able to adopt a stable structure. As pointed out before (132) there are reasons to assume that fold space for membrane proteins might be e�en more limited. Membrane proteins face a complex chemical en�ironment of both aqueous- and hydrophobic nature (153). Furthermore, the amino acid ‘palette’ that is a�ailable for the construction of transmembrane segments is biased towards hydrophobic residues (142) and therefore restricted. When the membrane protein fold space is so small compared to sequence space, it might be reasonable to assume that a particular structure can e�ol�e more than once by chance. After all, the possible structures that a protein can ‘choose’ from are limited. If correct, this assumption implies that structurally similar membrane proteins without detectable sequence similarity might present a case of con�ergent e�olution and thus are analogs rather than homologs. But there is a crucial mistake in this line

��
of thought. Membrane proteins do not e�ol�e from random protein sequences; the starting point is an existing protein or structural element (12�). As mentioned abo�e, the folding space for membrane proteins is so limited compared to sequence space that a random mutation in most cases will decrease stability or protein function in a negati�e way. Therefore only those mutations are allowed that do not compromise structural and functional stability. Since the amount of folds is limited, the effecti�e mutational ‘distance’ between two structures is �ery large. It is therefore unlikely that during the course of e�olution, membrane proteins sharing a common ancestor will adopt �ery different structures. If true con�ergent e�olution were to occur, the structure of one of these proteins has to ‘cross o�er’ to another fold family, a �ery unlikely e�ent. In other words: a limited fold space results in stronger conser�ation of the a�ailable structures, which fits well with the obser�ations for membrane proteins so far.
Homology between ECF transporter S-components As mentioned in the introduction, this thesis describes the structure of a membrane protein (ThiT, chapter 5) with a similar fold as the sequence-unrelated S-component for ribofla�in RibU (155). In this chapter, it is argued that such structural resemblance is a strong indicator for a common origin of both proteins. Furthermore, in chapter 2 e�idence is presented for a shared fold between other S-components and ThiT/RibU. Structural characterization of more S-components now has to be carried out to �erify this hypothesis. If S-components indeed share a general fold, it would be a strong argument for a single ancestral protein as the origin for all current S-components. Does the current phylogenetic distribution of ECF transporters still hold clues about such a common origin? ECF type II S-components are abundant in the genomes of Gram-positi�e bacteria, whereas Gram-negati�e bacteria utilize a limited set of type I S-components (see chapter 2) and eukaryotic genomes do not encode for any ECF transporter. It seems unlikely that if in a Last Uni�ersal Common Ancestor (LUCA) the large �ariation in S-components was already manifested, it has decreased so much during the course of e�olution. A more plausible scenario would be that in the LUCA only a single S-component or its predecessor was present and that emergence and specialization of other S-components has arisen more recently.
Interfaces for protein-protein interactions are structurally conserved The ECF transporter energizing module can form complexes with at least ten different S-components (10�). There are no conser�ed domains shared between the different S-components, therefore the molecular basis of the modularity is unclear. The structural similarity between ThiT and RibU suggests that the interface is probably a structural-

��
rather than a sequence motif. In membrane proteins, structural conser�ation of protein-protein interaction interfaces is more common than sequence conser�ation, as will be discussed below. Based on the ThiT structure, a prediction was made for the interaction interface with EcfT (chapter 5). At the interface, a moderately conser�ed tetrad alanine repeat was found. These motifs are known to support interactions between membrane proteins α-helices.
Different types of protein-protein interactions are distinguished based on the physicochemical nature of the interactions (�6). In so-called obligate complexes, the indi�idual protomers are not stable in an isolated form and therefore always found complexed. Often, there is a functional requirement for complexes to be obligate: for instance, the acti�e sites in the ABC transporter NBD dimer is formed with contributions from both monomers. Non-obligate interactions appear when protomers ha�e a function independent of the protein complex. The protomers can therefore exist in isolated form. Protein-protein interactions can be classified based on the lifetime of the complex as well. Permanent interactions are found in �ery stable (obligate) complexes, whereas transient interactions are characterized by an equilibrium state with useful concentrations of both the complexed form and indi�idual subunits. Regardless of the nature of the complex, the interaction is dri�en by a difference in free energy between the complexed and free state. Therefore there ha�e to be fa�orable interactions to stabilize the complex. In membrane proteins, only subset of mainly hydrophobic amino is a�ailable (see abo�e) for pro�iding such interactions. In particular in non-obligate or transient complexes, polar- and charged residues would compromise the stability of uncomplexed protomers in the membrane and are thus not often tolerated. The dri�ing force in forming membrane protein complexes is therefore often �an der Waals interactions between transmembrane helices (�1). Van der Waals interactions are weaker and less specific than polar interactions and subtle changes in the amino acids composition do not greatly affect the strength of the interaction, which results in limited sequence conser�ation at the interface. In contrast, conser�ation of the interface structure is more common. E�en in remote homologues, interfaces for protein-protein interactions are often found in the same region (156) regardless of the interaction partner and geometry of the complex.
The limited sequence conser�ation that can be found at the interface between membrane proteins is often restricted to so called GxxxG motifs (where x can by any amino acid). Although glycine is the most commonly obser�ed amino acid in this motif, it can be replaced by other small residues like alanine and serine. The motif was first recognized in the transmembrane domain of glycoporin A where it was found to be required for homodimerization (�2). Since then, se�eral studies on model peptide systems with partly randomized sequences pro�ided insight in the contribution of amino acids on the x-positions to the strength of the interaction (��,112). Because the glycines are spaced by a 3-amino acid linker, their sidechains end up at the same side of an α-helix, which creates

��
a shallow groo�e on the surface. The groo�e allows tight packing with a second α-helix and subsequent stabilization of the complex by �an der Waals interactions and weak hydrogen bonds donated by the Cα-H groups (6�).
Conclusions According to the definition, two proteins are homologues if they originate from a single ancestral protein. Sequence identity is usually a good indicator for homology. With the exception of short sequence motifs, it is highly improbable that similar sequences independently e�ol�e multiple times. Nonetheless, proteins can di�erge beyond detectable sequence similarity. In such cases, structural similarity remains as the only proof for distant homology. Although proteins can adopt a different fold during the course of e�olution, structures are in general more conser�ed then sequences. Membrane proteins in particular face limitations for structural �ariation, therefore recent obser�ations on structural similarity between otherwise unrelated proteins are a strong indication for ancient homology.
Protein-protein interactions surfaces in membrane proteins do not require strong sequence conser�ation but rather require structural conser�ation. This principle pro�ides the basis for the modularity of ECF type II energizing module. Although the amino acids sequences of S-components ha�e di�erged beyond recognition, structure and fold are probably more conser�ed. Interaction with the energizing module can therefore take place in regions that are structurally similar in all S-components.

�0

�1
Nederlandse samenvatting voor geïnteresseerden buiten het vakgebied Met dit proefschrift ga ik promo�eren in de biochemie. In dit �akgebied wordt de biologie bestudeerd �anuit chemisch perspectief. Het doel �an biochemie is het begrijpen �an de chemische principes die aan het le�en ten grondslag liggen. Om dit te bereiken is het nodig om ‘in te zoomen’ op de kleinste eenheid �an het le�en: de cel. Hieronder zal ik eerst uitleggen wat een cel is en uit welke componenten deze o�er het algemeen is opgebouwd. Ver�olgens zal ik uitleggen wat ik precies heb onderzocht en welke conclusies ik heb geformuleerd. De opzet �an mijn onderzoek is fundamenteel �an aard en niet gericht op directe toepassing �an de �erwor�en kennis. Toch �ormt een aantal conclusies in dit proefschrift mogelijk een startpunt �oor meer toegepast onderzoek. In de laatste paragraaf zal ik hieraan kort aandacht besteden.
Een korte inleiding in de chemieDe chemie, ook wel scheikunde genoemd, houdt zich bezig met wetten die de samenstelling en ontbinding �an moleculen beschrij�en. De lucht die wij inademen bestaat bij�oorbeeld (�oornamelijk) uit de moleculen zuurstof en stikstof. Moleculen zijn opgebouwd uit kleinere eenheden die atomen of elementen worden genoemd. De term ‘atoom’ is afgeleid �an het Griekse woord ‘atomos’ dat ondeelbaar betekent. De samenstelling �an een molecuul bepaalt de chemische eigenschappen er�an. Een watermolecuul is bij�oorbeeld opgebouwd uit twee atomen waterstof en één atoom zuurstof. Om dit een�oudig weer te ge�en wordt in de scheikunde gebruik gemaakt �an een speciale notatie �an letters en getallen. Scheikundigen gebruiken �oor het watermolecuul de notatie H2O. Hierin staat de ‘H’ �oor waterstofatoom (dat twee keer �oorkomt) en de ‘O’ �oor zuurstofatoom. Het watermolecuul bestaat dus uit drie atomen en is daarom een relatief simpel molecuul. Meer ingewikkelde moleculen kunnen zijn opgebouwd uit tientallen atomen. In de biochemie worden moleculen bestudeerd die �oorkomen in een cel. O�er het algemeen zijn deze moleculen complexer dan de moleculen die in de gewone scheikunde worden onderzocht. Moleculen in de biochemie kunnen bestaan uit duizenden atomen.
De cel: de kleinste eenheid van het levenElke �orm �an le�en is opgebouwd uit cellen. Dit geldt dus �oor alle planten en dieren, maar ook �oor bacteriën. In mijn onderzoek heb ik gewerkt met bacteriën. Deze organismen bestaan uit slechts één cel. Dieren en planten bestaan o�er het algemeen uit een �erzameling �an zeer �eel cellen. Volgens een gro�e schatting be�at een gemiddeld menselijk lichaam bij�oorbeeld 100 triljoen cellen (oftewel: een 1 met 14 nullen).

�2
Behal�e bacteriën bestaan bij�oorbeeld ook gisten (zoals het gist dat wordt gebruikt om brood te laten rijzen) uit één cel.
Afbeelding 1: een schematische weergave van de celLinks is een bacteriële cel weergege�en, rechts een dierlijke cel. De dierlijke cel heeft een celkern en is groter dan de bacterie. De cellen zijn niet op schaal getekend.
Er is in de biologie �eel �ariatie in hoe cellen �an binnen zijn georganiseerd. Plantaardige en dierlijke cellen zijn bij�oorbeeld opgebouwd uit �erschillende compartimenten die ook wel ‘organellen’ worden genoemd (zie afbeelding 1b). Het bekendste �oorbeeld hier�an is de celkern, waar het erfelijke materiaal (DNA) wordt bewaard. Bacteriën zien er o�er het algemeen wat simpeler uit en hebben geen interne compartimenten. Bo�endien zijn bacteriële cellen een stuk kleiner dan dierlijke cellen (zie afbeelding 1a).
Cellen worden omhuld door een celmembraan dat de cel beschermd en er�oor zorgt dat de inhoud �an de cel netjes bij elkaar blijft. Ook houdt het celmembraan de opname �an �oedingsstoffen tegen. Om toch �oedingsstoffen te kunnen opnemen, maken cellen gebruik �an transporteiwitten. Deze zitten in het celmembraan. Mijn onderzoek gaat o�er een transporteiwit en ik heb uitgezocht hoe dit werkt. Hiero�er �olgt later meer.
a b

�3
DNA: instructies voor de opbouw van cellenDesoxyribonucleïnezuur (DNA) is een molecuul waarin erfelijke eigenschappen �an een organisme liggen opgeslagen. DNA-moleculen bestaan uit duizenden atomen. Dit maakt het moeilijk om �an deze moleculen een scheikundige notatie te formuleren. DNA wordt daarom beschre�en aan de hand �an �ier letters (A, G, C en T), die zijn afgeleid �an de �ier eenheden (basen) waaruit DNA bestaat: Adenine, Guanine, Cytosine en Thymine. Een DNA-molecuul is opgebouwd uit een lange streng �an deze �ier basen, telkens in een andere specifieke �olgorde. Bij�oorbeeld: ATGGCTGAATCGTATTCC.
Al het DNA in een cel wordt omschre�en als het genoom. Het genoom bestaat uit �erschillende zeer lange DNA-moleculen die chromosomen worden genoemd. Het menselijke genoom be�at bij�oorbeeld 46 chromosomen. Een chromosoom be�at meerdere genen. Een gen is een deel �an het chromosoom dat een lengte heeft �an enkele honderden tot duizenden basen. Ieder gen be�at instructies �oor één �an de �ele processen in de cel en heeft daar�oor een unieke base�olgorde.
Afbeelding 2: de relatie tussen DNA en eiwittenDe basen�olgorde in het DNA wordt door de cel ‘�ertaald’ naar de aminozuur�olgorde �an het eiwit.
Eiwitten voeren processen uitGenen zijn niet direct betrokken bij de uit�oering �an processen, zij be�atten alleen de instructies hier�oor. Het echte werk wordt gedaan door andere moleculen: eiwitten of proteïnes. Net als DNA bestaan eiwitten uit lange strengen �an eenheden die aminozuren worden genoemd. Er zijn 20 �erschillende aminozuren die ook elk weer een eigen één-letter code hebben. Elk gen be�at de informatie �oor de aanmaak �an een eiwit. Om deze informatie te �erwerken, ‘leest’ een cel de basen�olgorde �an het DNA af in stukjes �an 3 basen. De bo�enstaande DNA-basen�olgorde kan door de cel dus als �olgt worden gelezen: ATG-GCT-GAA-TCG-TAT-TCC. In totaal zijn er 64 �erschillende combinaties �an 3 basen mogelijk. Elke combinatie is gekoppeld aan een aminozuur (sommige aminozuren zijn gekoppeld aan meer dan één combinatie, daarom zijn er maar 20 aminozuren) waardoor de basen�olgorde in de DNA-�olgorde direct is gekoppeld aan de aminozuur�olgorde in een eiwit (afbeelding 2).

�4
Bij �rijwel alles dat in een cel gebeurt, zijn eiwitten betrokken. Eiwitten zorgen bij�oorbeeld �oor de omzetting �an suikers in energie, �oor de afbraak �an giftige stoffen en �oor de herkenning �an ziekte�erwekkers door ons immuunsysteem. Heel �eel ziektes worden �eroorzaakt door het slecht functioneren �an eiwitten. Een eiwit functioneert slecht wanneer de aminozuur�olgorde in een eiwit toe�allig afwijkt �an de gebruikelijke �olgorde. Dit kan bij�oorbeeld worden �eroorzaakt door een foutje in de DNA-basen�olgorde. In dat ge�al is er sprake �an een erfelijke ziekte. De enorme �ariatie in de functies �an eiwitten komt �oort uit twee principes: de �olgorde �an aminozuren in een eiwitmolecuul en de driedimensionale structuur die een eiwit aanneemt. Eiwitten bestaan uit een lange keten �an aan elkaar geschakelde aminozuren. Deze keten kan zich op�ouwen tot een complexe structuur (zie afbeelding 3). Om de werking �an eiwitten te begrijpen is het dus nodig om de aminozuur�olgorde én de structuur er�an te kennen.
Afbeelding 3: de eiwitketen vouwt zich op tot een complexe driedimensionale structuur
Eiwitten zijn ondermeer betrokken bij de opname �an �oedingsstoffen en andere essentiële moleculen die niet zomaar door het celmembraan heen kunnen. Deze eiwitten be�inden zich in het celmembraan en worden daarom membraaneiwitten genoemd. Om een molecuul op te nemen moet het allereerst worden herkend door een membraaneiwit. O�er het algemeen is er �oor elk molecuul een ander eiwit beschikbaar zodat de cel precies kan controleren welke moleculen wel en niet worden opgenomen. Als het juiste molecuul is herkend, �erandert het membraaneiwit zichzelf als het ware in een tunnel, waardoor het molecuul naar de binnenkant �an de cel kan worden getransporteerd (afbeelding 4). Daarom worden deze eiwitten transporteiwitten genoemd. Wanneer het molecuul eenmaal aan de ander kant �an het membraan is, wordt de eiwittunnel direct gesloten om te �oorkomen dat er andere (ongewenste) moleculen doorheen gaan.

�5
Afbeelding 4: de werking van transporteiwittenTransporteiwitten be�inden zich in de celmembraan. Door het eiwit (groen) heen loopt een tunnel die alleen geopend wordt als het juiste molecuul herkend is (in dit ge�al een �itamine). Het molecuul wordt �er�olgens door de tunnel heen getransporteerd en komt aan de binnenkant �an de cel terrecht.
Mijn onderzoekO�er het transport �an �itamines in bacteriën is heel weinig bekend. In mijn onderzoek heb ik mij gericht op een specifieke groep transporteiwitten. Aan de hand �an proe�en heb ik onderzocht welk eiwit is betrokken bij de opname �an �itamine B1 (thiamine) in de bacterie Lactococcus lactis. Het transporteiwit heeft de naam ‘ThiT’ gekregen, wat staat �oor ‘Thiamine Transport’. In het derde hoofdstuk �an mijn proefschrift beschrijf ik hoe dit onderzoek is �erlopen.
Nadat ik het eiwit ThiT had ontdekt, ben ik in meer detail gaan kijken hoe het werkt. Dit staat beschre�en in hoofdstuk 4. Allereerst heb ik het eiwit uit het celmembraan �an L. lactis bacteriën gehaald. Dit wordt ‘zui�eren’ genoemd. In gezui�erde �orm was

�6
het mogelijk om te bestuderen hoe ThiT de thiamine in de bacterie kan herkennen. ThiT heeft, zo is gebleken, de bijzondere eigenschap om zelfs wanneer er heel kleine hoe�eelheden thiaminemoleculen beschikbaar zijn deze toch te �inden. Hierdoor kunnen de bacteriën gemakkelijk o�erle�en als ze in een omge�ing komen waar een tekort aan �itamine B1 is. In het onderzoek heb ik �ooral gekeken naar de in�loed �an de aminozuur�olgorde op de herkenning �an thiamine.
Afbeelding 5: de driedimensionale structuur van ThiTHoe de structuur �an een eiwit wordt bepaald, staat beschre�en in hoofdstuk 5. De lange aminozuurketen �an ThiT �ouwt zich op tot een complex patroon. De manier waarop de keten ge�ouwen is, bepaalt de functie �an het eiwit.
Zoals gezegd, zijn er twee factoren die de functie �an een eiwit bepalen: de aminozuur�olgorde en de driedimensionale structuur. Hoewel wij inzicht hadden in de in�loed �an de aminozuur�olgorde (zie hoofdstuk 4) ontbrak er nog informatie o�er de structuur �an ThiT. Omdat eiwitten o�er het algemeen te klein zijn om met de microscoop te bestuderen zijn er alternatie�e technieken ontwikkeld om de structuur �an eiwitten te ontrafelen. De meest gebruikte methode hier�oor is Röntgen-kristallografie. Om deze techniek toe te passen heb je grote hoe�eelheden zui�er eiwit nodig. Van deze eiwitten worden kristallen gegroeid. Kristallen bestaan uit grote hoe�eelheden moleculen (zoals eiwitten) die op een heel precieze manier zijn gerangschikt. Door deze kristallen �er�olgens �anuit allerlei richtingen te beschieten met een zeer hoge intensiteit Röntgenstraling ontstaat een Röntgenpatroon (diffractie) waar�an de �orm wordt bepaald door de �orm �an de eiwitten in het kristal. Deze gege�ens worden �erwerkt op de computer. Hierdoor is het ook mogelijk om een 3D-reconstructie �an het eiwit uit te rekenen. In hoofdstuk 5 beschrijf ik de toepassing �an deze techniek om de structuur �an ThiT te bepalen.

��
De moeilijkste stap in dit proces is meestal het groeien �an de eiwitkristallen. Voor elk eiwit moet een nieuwe procedure worden ontwikkeld en succes is niet gegarandeerd. Met name �an membraaneiwitten (zoals ThiT) is het o�er het algemeen bijzonder moeilijk om kristallen te krijgen. Dit �erklaart waarom er relatief weinig is bekend o�er de structuur �an membraaneiwitten. Uit de structuur �an ThiT (afbeelding 5) hebben wij heel �eel kunnen afleiden o�er de manier waarop �itamine B1 wordt getransporteerd.
Verwachtingen voor de toekomstZoals gezegd, is mijn onderzoek fundamenteel �an aard en �oornamelijk gedre�en door nieuwsgierigheid naar de werking �an eiwitten. Fundamenteel onderzoek �ormt de basis �oor concrete toepassingen. Vitamines zijn �an essentieel belang �oor �eel bacteriën. Een aantal ziekte�erwekkende bacteriën is bij�oorbeeld afhankelijk �an de opname �an thiamine om te kunnen groeien. Met �oldoende kennis o�er het transportmechanisme �an thiamine is het in principe mogelijk om op zoek te gaan naar moleculen die het transport blokkeren. Dergelijke stoffen kunnen in de toekomst worden gebruikt als antibiotica om ziekte�erwekkende bacteriën te bestrijden. Het is daarom belangrijk om de werking �an ThiT in nog meer detail te bestuderen. Ook o�er het transport �an andere �itamines zouden wij nog meer te weten moeten komen.

��

��
List of publications:
Erkens, G.B., Bertnsson, R.P-A., Fulyani, F., Majsnerowska, M., VujičićVujičić-Žagar, A., ter Beek, J., Poolman, B. and Slotboom, D.J. (2011) The structural basis of modularity in ECF-type ABC transporters. Nat. Struct. Mol. Biol. In press.
Ter Beek, J., Duurkens, R.H., Erkens, G.B. and Slotboom, D.J. (2011) Quaternary structure and functional unit of Energy Coupling Factor (ECF) transporters. J. Biol. Chem. 286:54�1-54�5
Erkens, G.B. and Slotboom, D.J. (2010) Biochemical characterization of ThiT from Lactococcus lactis: a thiamin transporter with picomolar substrate binding affinity. Biochemistry 49:3203-3212
Rodiono�, D.A., Hebbeln, P., Eudes, A., ter Beek, J., Rodiono�a, I.A., Erkens, G.B., Slotboom, D.J., Gelfand, M.S., Osterman, A. L., Hanson, A.D. and Eitinger, T. (200�) A no�el class of modular transporters for �itamins in prokaryotes. J. Bacteriol. 191:42-51
Eudes, A.*, Erkens, G.B.*, Slotboom, D.J., Rodiono�, D.A., Naponelli, V. and Hanson, A.D. (200�) Identification of genes encoding the folate- and thiamine-binding membrane proteins in Firmicutes. J. Bacteriol. 190:�5�1-�5�4
Slotboom, D.J., Duurkens, R.H., Olieman, K. and Erkens, G.B. (200�) Static light scattering to characterize membrane proteins in detergent solution. Methods 46:�3-�2
*both authors contributed equally to this work

�0

�1
Nawoord
Groningen, 3 mei 2011
‘Guus, heb je al nagedacht o�er je toekomst?’ was de �raag die Dirk mij in het �oorbijgaan stelde toen ik mijn masteronderzoek aan het afronden was. Deze �raag leidde er uiteindelijk toe dat ik iets meer dan �ier jaar geleden met mijn promotieonderzoek in Groningen begon. Hoewel ik officieel werd aangesteld om onderzoek te doen naar glutamaat transporters begonnen we met het ontwikkelen �an een methode om de oligomere toestand �an membraan eiwitten te bepalen. Ondertussen zou ik �ast wat �oorbereidingen treffen om ‘iets met bacteriële �itamine transporters’ te gaan onderzoeken, waarbij we in eerste instantie zouden gaan kijken naar thiamine transport. O�er �itamine transport in bacteriën was nog weinig bekend en het onderzoek zou mooi aansluiten bij het werk dat eerder was gedaan aan de ribofla�ine transporter RibU. Aansluiten deed het inderdaad. On�erwacht bleken de thiamine en ribofla�ine transporter deel uit te maken �an een tot �oor kort onbekende familie �an transporteiwitten met zeer interessante kenmerken: de ECF transporters. Dit �ormde het startpunt �an al het onderzoek dat uiteindelijk in dit proefschrift beschre�en is, en ik denk dat ik mij geen beter startpunt kon wensen. Hoewel ik het in het begin moeilijk �ond om een duidelijke richting te kiezen, is het een geweldige er�aring geweest om betrokken te zijn bij de �roege fase �an het onderzoek naar ECF transporters. Ik ben er dan ook het meest trots op dat dit proefschrift een concrete bijdrage heeft gele�erd aan het begrip �an ECF transporters. Wat �ier jaar geleden begon als een heleboel �ragen en een paar antwoorden �erandert steeds meer in een duidelijk model �an �itamine transport.
Het wetenschappelijke hoogtepunt en één �an de leukste periodes �an mijn promotieonderzoek was zonder twijfel het oplossen �an de structuur �an ThiT. Helaas ging dit samen met de teleurstellende ontdekking dat er in China met �oorsprong gewerkt werd aan de structuur �an RibU. Hoewel ik e�en heb gedacht dat dit het einde zou betekenen �an het structuurwerk aan ThiT, heeft het uiteindelijk allemaal bijzonder goed uitgepakt. Mede dankzij de RibU structuur hebben we nieuwe inzichten kunnen krijgen en dat heeft geresulteerd in een fantastische publicatie. Ik wil bij deze dan ook iedereen die daar een bijdrage aanheeft gele�erd bedanken!
Daarnaast maak ik graag �an de gelegenheid gebruik om een aantal mensen persoonlijk te bedanken: Als eerste mijn promotor Dirk Jan Slotboom. Ik heb de afgelopen jaren met �eel plezier samengewerkt en wil je graag bedanken �oor de ruimte die je mij hebt gege�en om mijn interesses te �olgen. Hoewel je mij �eel �rijheid gaf in het bedenken en uit�oeren �an experimenten kon ik altijd binnen�allen �oor praktische �ragen en ad�ies.

�2
Maarten, in het begin �an mijn onderzoek heb ik �eel gehad aan al je praktische ad�iezen o�er labzaken en experimenten. Het was altijd gezellig in het lab als we allebei aan het werk waren en ik heb met �eel lol de actuele politiek en de ideale staatsinrichting met je besproken! Ik �ond het leuk dat we elkaar ook buiten het lab om regelmatig zagen en waardeer het dat je mijn paranimf wil zijn.
Marysia, thanks for all the fun con�ersations that we had in the lab, your help with buying a car and of course for sharing your experience on ThiT and ECF transporters. Because of your excellent contribution to chapter 5 of this thesis we really made a strong point about the ECF transporter mechanism! I would also like to thank you for being paranimf at the defence.
Ronnie, jouw bijdrage in het oplossen �an de structuur is in alle fasen onmisbaar geweest. Daarnaast heb ik met �eel plezier een kantoor met je gedeeld en wil je graag bedanken �oor alle leuke gesprekken o�er wetenschap, kristallografie, politiek, koetjes en kalfjes en de laatste roddels die we gehad hebben.
Josy, ik wil je graag bedanken �oor het delen �an je kennis o�er ECF transporters en al je goede suggesties en ideeën tijdens werkbesprekingen. Veel succes met de afronding �an je proefschrift de komende maanden!
Faizah and Andreja, thanks for all the hard work you’�e put in sol�ing the structure of ThiT! I’�e enjoyed working together with you.
Bert, al tijdens mijn bacheloronderzoek in jouw groep wist je mij enthousiast te maken �oor biochemisch onderzoek in het algemeen en membraaneiwitten in het bijzonder. Ik heb gedurende mijn promotieonderzoek �eel gehad aan jouw suggesties en �ragen tijdens werkbesprekingen. Daarnaast heb ik prettig samengewerkt bij de totstandkoming �an hoofdstuk 5 en heb ik �eel waardering �oor je bijdrage tijdens het schrij�en �an het manuscript.
Ria en Gea wil ik graag bedanken �oor het soepel ‘managen’ �an het lab. Het lijkt me geen makkelijke opga�e om een grote groep eigenwijze mensen prettig en gestructureerd met elkaar samen te laten werken, maar �olgens mij zijn jullie er uitstekend in geslaagd!
Tijdens mijn periode hier in de groep heb ik twee uitstekende studenten begeleid:: Pranav Puri, thanks for your dedication and all the work you ha�e done! Michael Verhoeven, bedankt �oor je inzet en de experimenten die je hebt uitge�oerd!
Arnold Driessen, Antoine van Oijen en Piet Gros wil ik graag hartelijk bedanken �oor het lezen en beoordelen �an dit proefschrift.
Thanks to all present and past members of the enzymology group. It has been a great four years!

�3
Tot slot: José, bedankt �oor al je steun en interesse in mijn werk! Ik weet zeker dat we het samen nog een heleboel jaren leuk zullen hebben. Bo�endien zijn er niet �eel promo�endi die de Nederlandse samen�atting �an hun proefschrift hebben laten redigeren door een er�aren tekstschrij�er/journalist. Ik ben heel blij met het resultaat!

�4

�5
References
1. Adams, P.D., Afonine, P.V., Bunkoczi, G., Chen, V.B., Davis, I.W., Echols, N., Headd, J.J., Hung, L.W., Kapral, G.J., Grosse-Kunstleve, R.W., McCoy, A.J., Moriarty, N.W., Oeffner, R., Read, R.J., Richardson, D.C., Richardson, J.S., Terwilliger, T.C. and Zwart, P.H. (2010) PHENIX: a comprehensi�e Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol.Crystallogr. 66: 213-221.
2. Aller, S.G., Yu, J., Ward, A., Weng, Y., Chittaboina, S., Zhuo, R., Harrell, P.M., Trinh, Y.T., Zhang, Q., Urbatsch, I.L. and Chang, G. (200�) Structure of P-glycoprotein re�eals a molecular basis for poly-specific drug binding. Science 323: 1�1�-1�22.
3. Ambudkar, S.V., Cardarelli, C.O., Pashinsky, I. and Stein, W.D. (1���) Relation between the turno�er number for �inblastine transport and for �inblastine-stimulated ATP hydrolysis by human P-glycoprotein. J. Biol. Chem. 272: 21160-21166.
4. Aravind, L., Mazumder, R., Vasudevan, S. and Koonin, E.V. (2002) Trends in protein e�olution inferred from sequence and structure analysis. Curr. Opin.Struct. Biol. 12: 3�2-3��.
5. Begley, T.P., Downs, D.M., Ealick, S.E., McLafferty, F.W., Van Loon, A.P., Taylor, S., Campobasso, N., Chiu, H.J., Kinsland, C., Reddick, J.J. and Xi, J. (1���) Thiamin biosynthesis in prokaryotes. Arch. Microbiol. 171: 2�3-300.
6. Bernsel, A., Viklund, H., Falk, J., Lindahl, E., von, H.G. and Elofsson, A. (200�) Prediction of membrane-protein topology from first principles. Proc.Natl. Acad. Sci.U.S.A. 105: �1��-�1�1.
�. Berntsson, R.P., Alia, O.N., Fusetti, F., Thunnissen, A.M., Poolman, B. and Slotboom, D.J. (200�) Selenomethionine incorporation in proteins expressed in Lactococcus lactis. Protein Sci. 18: 1121-112�.
�. Berntsson, R.P., Doeven, M.K., Fusetti, F., Duurkens, R.H., Sengupta, D., Marrink, S.J., Thunnissen, A.M., Poolman, B. and Slotboom, D.J. (200�) The structural basis for peptide selection by the transport receptor OppA. EMBO J. 28: 1332-1340.

�6
�. Berntsson, R.P., Smits, S.H., Schmitt, L., Slotboom, D.J. and Poolman, B. (2010) A structural classification of substrate-binding proteins. FEBS Lett. 584: 2606-261�.
10. Biemans-Oldehinkel, E., Doeven, M.K. and Poolman, B. (2006) ABC transporter architecture and regulatory roles of accessory domains. FEBS Lett. 580: 1023-1035.
11. Bishop, L., Agbayani, R., Jr., Ambudkar, S.V., Maloney, P.C. and Ames, G.F. (1���) Reconstitution of a bacterial periplasmic permease in proteoliposomes and demonstration of ATP hydrolysis concomitant with transport. Proc. Natl.Acad. Sci. U.S.A. 86: 6�53-6�5�.
12. Borths, E.L., Poolman, B., Hvorup, R.N., Locher, K.P. and Rees, D.C. (2005) In �itro functional characterization of BtuCD-F, the Escherichia coli ABC transporter for �itamin B12 uptake. Biochemistry 44: 16301-1630�.
13. Boudker, O., Ryan, R.M., Yernool, D., Shimamoto, K. and Gouaux, E. (200�) Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature 445: 3��-3�3.
14. Breslow, R. (1�5�) Rapid deuterium exchange in thiazolium salts. J. Am. Chem.Soc. 79: 1�62-1�63.
15. Breslow, R. (1�5�) On the Mechanism of Thiamine Action IV. E�idence from Studies on Model Systems. J. Am. Chem. Soc. 80: 3�1�-3�26.
16. Burgess, C.M., Slotboom, D.J., Geertsma, E.R., Duurkens, R.H., Poolman, B. and van Sinderen, D. (2006) The ribofla�in transporter RibU in Lactococcus lactis: molecular characterization of gene expression and the transport mechanism. J. Bacteriol. 188: 2�52-2�60.
1�. Burley, S.K. and Petsko, G.A. (1��5) Aromatic-aromatic interaction: a mechanism of protein structure stabilization. Science 229: 23-2�.
1�. Cartailler, J.P. and Luecke, H. (2004) Structural and functional characterization of π bulges and other short intrahelical deformations.π bulges and other short intrahelical deformations. bulges and other short intrahelical deformations. Structure. 12: 133-144.
1�. Chenna, R., Sugawara, H., Koike, T., Lopez, R., Gibson, T.J., Higgins, D.G. and Thompson, J.D. (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31: 34��-3500.

��
20. Chipot, C., Jaffe, R., Maigret, B., Pearlman, D.A. and Kollman, P.A. (1��6) Benzene dimer: A good model for ππ-π interactions in proteins? A comparison interactions in proteins? A comparison between the benzene and the toluene dimers in the cas phase and in an aqueous solution.J. Am. Chem. Soc. 118: 1121�-11224.
21. Chothia, C. (1��5) Structural in�ariants in protein folding. Nature 245: 304-30�.
22. Chothia, C. and Lesk, A.M. (1��6) The relation between the di�ergence of sequence and structure in proteins. EMBO J. 5: �23-�26.
23. Chung, S.Y. and Subbiah, S. (1��6) A structural explanation for the twilight zone of protein sequence homology. Structure. 4: 1123-112�.
24. Collaborative Computational Project Number 4. (1��4) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol.Crystallogr. 50: �60-�63.
25. Coulson, A.F. and Moult, J. (2002) A unifold, mesofold, and superfold model of protein fold use. Proteins 46: 61-�1.
26. Dassa, E. and Bouige, P. (2001) The ABC of ABCS: a phylogenetic and functional classification of ABC systems in li�ing organisms. Res. Microbiol. 152: 211-22�.
2�. Davidson, A.L., Dassa, E., Orelle, C. and Chen, J. (200�) Structure, function, and e�olution of bacterial ATP-binding cassette systems. Microbiol. Mol. Biol.Rev. 72: 31�-64.
2�. Davidson, A.L. and Nikaido, H. (1��0) O�erproduction, solubilization, and reconstitution of the maltose transport system from Escherichia coli. J. Biol.Chem. 265: 4254-4260.
2�. Davidson, A.L. and Nikaido, H. (1��1) Purification and characterization of the membrane-associated components of the maltose transport system from Escherichia coli. J. Biol. Chem. 266: ��46-��51.
30. Davidson, A.L., Shuman, H.A. and Nikaido, H. (1��2) Mechanism of maltose transport in Escherichia coli: transmembrane signaling by periplasmic binding proteins. Proc. Natl. Acad. Sci. U.S.A. 89: 2360-2364.

��
31. Dawson, R.J. and Locher, K.P. (2006) Structure of a bacterial multidrug ABC transporter. Nature 443: 1�0-1�5.
32. De Crecy-Lagard, V., El, Y.B., de la Garza, R.D., Noiriel, A. and Hanson, A.D. (200�) Comparati�e genomics of bacterial and plant folate synthesis and sal�age: predictions and �alidations. BMC. Genomics 8: 245.
33. Doolittle, R.F. (1��6) Of Urfs and Orfs: primer on how to analyze deri�ed amino acid sequences. Uni�ersity Science Books, Mill Valley, CA, USA.
34. Duurkens, R.H., Tol, M.B., Geertsma, E.R., Permentier, H.P. and Slotboom, D.J. (200�) Fla�in binding to the high affinity ribofla�in transporter RibU. J. Biol. Chem. 282: 103�0-103�6.
35. Dyda, F., Furey, W., Swaminathan, S., Sax, M., Farrenkopf, B. and Jordan, F. (1��3) Catalytic centers in the thiamin diphosphate dependent enzyme pyru�ate decarboxylase at 2.4-A resolution. Biochemistry 32: 6165-61�0.
36. Eijkman, C. (1���) Eine Beriberiähnliche Krankheit der Hühner. Virchows Arch. Pathol. Anat. 148: 523.
3�. Eitinger, T., Rodionov, D.A., Grote, M. and Schneider, E. (2010) Canonical and ECF-type ATP-binding cassette importers in prokaryotes: di�ersity in modular organization and cellular functions. FEMS Microbiol. Rev. 35: 3-�6.
3�. Emsley, P., Lohkamp, B., Scott, W.G. and Cowtan, K. (2010) Features and de�elopment of Coot. Acta Crystallogr. D Biol. Crystallogr. 66: 4�6-501.
3�. Eytan, G.D., Regev, R. and Assaraf, Y.G. (1��6) Functional reconstitution of P-glycoprotein re�eals an apparent near stoichiometric drug transport to ATP hydrolysis. J. Biol. Chem. 271: 31�2-31��.
40. Faham, S., Watanabe, A., Besserer, G.M., Cascio, D., Specht, A., Hirayama, B.A., Wright, E.M. and Abramson, J. (200�) The crystal structure of a sodium galactose transporter re�eals mechanistic insights into Na+/sugar symport. Science 321: �10-�14.
41. Fang, Y., Jayaram, H., Shane, T., Kolmakova-Partensky, L., Wu, F., Williams, C., Xiong, Y. and Miller, C. (200�) Structure of a prokaryotic �irtual proton pump at 3.2 Å resolution.Å resolution. resolution. Nature 460: 1040-1043.

��
42. Felder, C.B., Graul, R.C., Lee, A.Y., Merkle, H.P. and Sadee, W. (1���) The Venus flytrap of periplasmic binding proteins: an ancient protein module present in multiple drug receptors. AAPS. PharmSci. 1: E2.
43. Felsenstein, J. (2004) PHYLIP (Phylogeny Inference Package) �ersion 3.6. Distributed by the author. Department of Genome Sciences, Uni�ersity of Washington, Seattle.
44. Finkenwirth, F., Neubauer, O., Gunzenhauser, J., Schoknecht, J., Scolari, S., Stockl, M., Korte, T., Herrmann, A. and Eitinger, T. (2010) Subunit composition of an energy-coupling-factor-type biotin transporter analysed in li�ing bacteria. Biochem. J. 431: 3�3-3�0.
45. Fischer, J.D., Mayer, C.E. and Soding, J. (200�) Prediction of protein functional residues from sequence by probability density estimation. Bioinformatics. 24: 613-620.
46. Flores, T.P., Orengo, C.A., Moss, D.S. and Thornton, J.M. (1��3) Comparison of conformational characteristics in structurally similar protein pairs. Protein Sci. 2: 1�11-1�26.
4�. Frank, R.A., Leeper, F.J. and Luisi, B.F. (200�) Structure, mechanism and catalytic duality of thiamine-dependent enzymes. Cell. Mol. Life Sci. 64: ��2-�05.
4�. Funk, C. (1�11) On the chemical nature of the substance which cures polyneuritis in birds induced by a diet of polished rice. J. Physiol. 43: 3�5-400.
4�. Geertsma, E.R. and Poolman, B. (200�) High-throughput cloning and expression in recalcitrant bacteria. Nat. Methods 4: �05-�0�.
50. Gerber, S., Comellas-Bigler, M., Goetz, B.A. and Locher, K.P. (200�) Structural basis of trans-inhibition in a molybdate/tungstate ABC transporter. Science 321: 246-250.
51. Glass, J.I., Ssad-Garcia, N., Alperovich, N., Yooseph, S., Lewis, M.R., Maruf, M., Hutchison, C.A., III, Smith, H.O. and Venter, J.C. (2006) Essential genes of a minimal bacterium. Proc. Natl. Acad. Sci. U.S.A. 103: 425-430.

100
52. Goosen, N. and Moolenaar, G.F. (2001) Role of ATP hydrolysis by U�rA and U�rB during nucleotide excision repair. Res. Microbiol. 152: 401-40�.
53. Hasson, M.S., Muscate, A., McLeish, M.J., Polovnikova, L.S., Gerlt, J.A., Kenyon, G.L., Petsko, G.A. and Ringe, D. (1���) The crystal structure of benzoylformate decarboxylase at 1.6 Šresolution: di�ersity of catalytic residuesŠresolution: di�ersity of catalytic residues resolution: di�ersity of catalytic residues in thiamin diphosphate-dependent enzymes. Biochemistry 37: ��1�-��30.
54. Hawkins, C.F., Borges, A. and Perham, R.N. (1���) A common structural motif in thiamin pyrophosphate-binding enzymes. FEBS Lett. 255: ��-�2.
55. Hebbeln, P., Rodionov, D.A., Alfandega, A. and Eitinger, T. (200�) Biotin uptake in prokaryotes by solute transporters with an optional ATP-binding cassette-containing module. Proc. Natl. Acad. Sci. U.S.A. 104: 2�0�-2�14.
56. Henderson, D.P. and Payne, S.M. (1��4) Vibrio cholerae iron transport systems: roles of heme and siderophore iron transport in �irulence and identification of a gene associated with multiple iron transport systems. Infect. Immun. 62: 5120-5125.
5�. Henderson, G.B., Kojima, J.M. and Kumar, H.P. (1��5) Differential interaction of cations with the thiamine and biotin transport proteins of Lactobacillus casei. Biochim. Biophys. Acta 813: 201-206.
5�. Henderson, G.B., Kojima, J.M. and Kumar, H.P. (1��5) Kinetic e�idence for two intercon�ertible forms of the folate transport protein from Lactobacillus casei. J. Bacteriol. 163: 114�-1152.
5�. Henderson, G.B. and Potuznik, S. (1��2) Cation-dependent binding of substrate to the folate transport protein of Lactobacillus casei. J. Bacteriol. 150: 10��-1102.
60. Henderson, G.B. and Zevely, E.M. (1���) Binding and transport of thiamine by Lactobacillus casei. J. Bacteriol. 133: 11�0-11�6.
61. Henderson, G.B., Zevely, E.M. and Huennekens, F.M. (1��6) Folate transport in Lactobacillus casei: solubilization and general properties of the binding protein. Biochem. Biophys. Res. Commun. 68: �12-�1�.

101
62. Henderson, G.B., Zevely, E.M. and Huennekens, F.M. (1���) Purification and properties of a membrane-associated, folate-binding protein from Lactobacillus casei. J. Biol. Chem. 252: 3�60-3�65.
63. Henderson, G.B., Zevely, E.M. and Huennekens, F.M. (1���) Coupling of energy to folate transport in Lactobacillus casei. J. Bacteriol. 139: 552-55�.
64. Henderson, G.B., Zevely, E.M. and Huennekens, F.M. (1���) Mechanism of folate transport in Lactobacillus casei: e�idence for a component shared with the thiamine and biotin transport systems. J. Bacteriol. 137: 130�-1314.
65. Henderson, G.B., Zevely, E.M., Kadner, R.J. and Huennekens, F.M. (1���) The folate and thiamine transport proteins of Lactobacillus casei. J. Supramol.Struct. 6: 23�-24�.
66. Hollenbach, A.D., Dickson, K.A. and Washabaugh, M.W. (2002) O�erexpression, purification, and characterization of the periplasmic space thiamin-binding protein of the thiamin traffic ATPase in Escherichia coli. Protein Expr. Purif. 25: 50�-51�.
6�. Hollenstein, K., Dawson, R.J. and Locher, K.P. (200�) Structure and mechanism of ABC transporter proteins. Curr. Opin. Struct. Biol. 17: 412-41�.
6�. Hollenstein, K., Frei, D.C. and Locher, K.P. (200�) Structure of an ABC transporter in complex with its binding protein. Nature 446: 213-216.
6�. Joh, N.H., Min, A., Faham, S., Whitelegge, J.P., Yang, D., Woods, V.L. and Bowie, J.U. (200�) Modest stabilization by most hydrogen-bonded side-chain interactions in membrane proteins. Nature 453: 1266-12�0.
�0. Jurgenson, C.T., Begley, T.P. and Ealick, S.E. (200�) The structural and biochemical foundations of thiamin biosynthesis. Annu. Rev. Biochem. 78: 56�-603.
�1. Kabsch, W. (1��3) Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 26: ��5-�00.

102
�2. Kadaba, N.S., Kaiser, J.T., Johnson, E., Lee, A. and Rees, D.C. (200�) The high-affinity E. coli methionine ABC transporter: structure and allosteric regulation. Science 321: 250-253.
�3. Kammann, M., Laufs, J., Schell, J. and Gronenborn, B. (1���) Rapid insertional mutagenesis of DNA by polymerase chain reaction (PCR). Nucleic Acids Res. 17: 5404.
�4. Kluger, R. and Tittmann, K. (200�) Thiamin diphosphate catalysis: enzymic and nonenzymic co�alent intermediates. Chem. Rev. 108: 1���-1�33.
�5. Kuipers, O.P., de Ruyter, P.G.G.A., Kleerebezem, M. and de Vos, W.M. (1���) Quorum sensing-controlled gene expression in lactic acid bacteria. J. Biotechnol. 64: 15-21.
�6. Kumar, H.P., Tsuji, J.M. and Henderson, G.B. (1���) Folate transport in Lactobacillus salivarius. Characterization of the transport mechanism and purification and properties of the binding component. J. Biol. Chem. 262: �1�1-�1��.
��. Kunji, E.R., Harding, M., Butler, P.J. and Akamine, P. (200�) Determination of the molecular mass and dimensions of membrane proteins by size exclusion chromatography. Methods 46: 62-�2.
��. Kunji, E.R., Slotboom, D.J. and Poolman, B. (2003) Lactococcus lactis as host for o�erproduction of functional membrane proteins. Biochim. Biophys. Acta 1610: ��-10�.
��. Lakowicz, J.R. (2006) Principles of Fluorescence Spectroscopy. Springer, New York.
�0. Langer, G., Cohen, S.X., Lamzin, V.S. and Perrakis, A. (200�) Automated macromolecular model building for X-ray crystallography using ARP/wARP �ersion �. Nat. Protoc. 3: 11�1-11��.
�1. Langosch, D. and Arkin, I.T. (200�) Interaction and conformational dynamics of membrane-spanning protein helices. Protein Sci. 18: 1343-135�.
�2. Lemmon, M.A., Flanagan, J.M., Treutlein, H.R., Zhang, J. and Engelman, D.M. (1��2) Sequence specificity in the dimerization of transmembrane α--helices. Biochemistry 31: 12�1�-12�25.

103
�3. Lipfert, J., Columbus, L., Chu, V.B., Lesley, S.A. and Doniach, S. (200�) Size and shape of detergent micelles determined by small-angle X-ray scattering. J. Phys. Chem. B 111: 1242�-1243�.
�4. Liu, C.E., Liu, P.Q. and Ames, G.F. (1���) Characterization of the adenosine triphosphatase acti�ity of the periplasmic histidine permease, a traffic ATPase (ABC transporter). J. Biol. Chem. 272: 21��3-21��1.
�5. Locher, K.P., Lee, A.T. and Rees, D.C. (2002) The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science 296: 10�1-10��.
�6. Marchler-Bauer, A., Lu, S., Anderson, J.B., Chitsaz, F., Derbyshire, M.K., Weese-Scott, C., Fong, J.H., Geer, L.Y., Geer, R.C., Gonzales, N.R., Gwadz, M., Hurwitz, D.I., Jackson, J.D., Ke, Z., Lanczycki, C.J., Lu, F., Marchler, G.H., Mullokandov, M., Omelchenko, M.V., Robertson, C.L., Song, J.S., Thanki, N., Yamashita, R.A., Zhang, D., Zhang, N., Zheng, C. and Bryant, S.H. (2011) CDD: a Conser�ed Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39: D225-D22�.
��. McMurry, L., Petrucci, R.E., Jr. and Levy, S.B. (1��0) Acti�e efflux of tetracycline encoded by four genetically different tetracycline resistance determinants in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 77: 3��4-3���.
��. Melnick, J., Lis, E., Park, J.H., Kinsland, C., Mori, H., Baba, T., Perkins, J., Schyns, G., Vassieva, O., Osterman, A. and Begley, T.P. (2004) Identification of the two missing bacterial genes in�ol�ed in thiamine sal�age: thiamine pyrophosphokinase and thiamine kinase. J. Bacteriol. 186: 3660-3662.
��. Melnyk, R.A., Kim, S., Curran, A.R., Engelman, D.M., Bowie, J.U. and Deber, C.M. (2004) The affinity of GXXXG motifs in transmembrane helix-helix interactions is modulated by long-range communication. J. Biol. Chem. 279: 165�1-165��.
�0. Mimmack, M.L., Gallagher, M.P., Pearce, S.R., Hyde, S.C., Booth, I.R. and Higgins, C.F. (1���) Energy coupling to periplasmic binding protein-dependent transport systems: stoichiometry of ATP hydrolysis during transport in �i�o. Proc. Natl. Acad. Sci. U.S.A. 86: �25�-�261.

104
�1. Muir, M., Williams, L. and Ferenci, T. (1��5) Influence of transport energization on the growth yield of Escherichia coli. J. Bacteriol. 163: 123�-1242.
�2. Muller, Y.A., Lindqvist, Y., Furey, W., Schulz, G.E., Jordan, F. and Schneider, G. (1��3) A thiamin diphosphate binding fold re�ealed by comparison of the crystal structures of transketolase, pyru�ate oxidase and pyru�ate decarboxylase. Structure. 1: �5-103.
�3. Murshudov, G.N., Vagin, A.A. and Dodson, E.J. (1���) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 53: 240-255.
�4. Naponelli, V., Hanson, A.D. and Gregory, J.F., III (200�) Impro�ed methods for the preparation of [3H]folate polyglutamates: biosynthesis with Lactobacillus casei and enzymatic synthesis with Escherichia coli folylpolyglutamate synthetase. Anal. Biochem. 371: 12�-134.
�5. Neubauer, O., Alfandega, A., Schoknecht, J., Sternberg, U., Pohlmann, A. and Eitinger, T. (200�) Two essential arginine residues in the T components of energy-coupling factor transporters. J. Bacteriol. 191: 64�2-64��.
�6. Nooren, I.M. and Thornton, J.M. (2003) Di�ersity of protein-protein interactions. EMBO J. 22: 34�6-34�2.
��. Oldham, M.L., Khare, D., Quiocho, F.A., Davidson, A.L. and Chen, J. (200�) Crystal structure of a catalytic intermediate of the maltose transporter. Nature 450: 515-521.
��. Ontiveros-Palacios, N., Smith, A.M., Grundy, F.J., Soberon, M., Henkin, T.M. and Miranda-Rios, J. (200�) Molecular basis of gene regulation by the THI-box riboswitch. Mol. Microbiol. 67: ��3-�03.
��. Otto, R., ten Brink, B.H., Veldkamp, H. and Konings, W.N. (1��3) The relation between growth rate and electrochemical proton gradient of Streptococcus cremoris. FEMS Microbiol. Lett. 16: 6�-�4.

105
100. Overbeek, R., Begley, T., Butler, R.M., Choudhuri, J.V., Chuang, H.Y., Cohoon, M., de Crecy-Lagard, V., Diaz, N., Disz, T., Edwards, R., Fonstein, M., Frank, E.D., Gerdes, S., Glass, E.M., Goesmann, A., Hanson, A., Iwata-Reuyl, D., Jensen, R., Jamshidi, N., Krause, L., Kubal, M., Larsen, N., Linke, B., McHardy, A.C., Meyer, F., Neuweger, H., Olsen, G., Olson, R., Osterman, A., Portnoy, V., Pusch, G.D., Rodionov, D.A., Ruckert, C., Steiner, J., Stevens, R., Thiele, I., Vassieva, O., Ye, Y., Zagnitko, O. and Vonstein, V. (2005) The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 33: 56�1-5�02.
101. Patzlaff, J.S., van der, H.T. and Poolman, B. (2003) The ATP/substrate stoichiometry of the ATP-binding cassette (ABC) transporter OpuA. J. Biol.Chem. 278: 2�546-2�551.
102. Ponder, J.W. and Richards, F.M. (1���) Tertiary templates for proteins. Use of packing criteria in the enumeration of allowed sequences for different structural classes. J. Mol. Biol. 193: ��5-��1.
103. Poolman, B. and Konings, W.N. (1���) Relation of growth of Streptococcus lactis and Streptococcus cremoris to amino acid transport. J. Bacteriol. 170: �00-�0�.
104. Povolotskaya, I.S. and Kondrashov, F.A. (2010) Sequence space and the ongoing expansion of the protein uni�erse. Nature 465: �22-�26.
105. Ray, F.A. and Nickoloff, J.A. (1��2) Site-specific mutagenesis of almost any plasmid using a PCR-based �ersion of unique site elimination. Biotechniques 13: 342-34�.
106. Ren, Q., Chen, K. and Paulsen, I.T. (200�) TransportDB: a comprehensi�e database resource for cytoplasmic membrane transport systems and outer membrane channels. Nucleic Acids Res. 35: D2�4-D2��.
10�. Ressl, S., Terwisscha van Scheltinga, A.C., Vonrhein, C., Ott, V. and Ziegler, C. (200�) Molecular basis of transport and regulation in the Na+/betaine symporter BetP. Nature 458: 4�-52.
10�. Rodionov, D.A., Hebbeln, P., Eudes, A., Ter Beek, J., Rodionova, I.A., Erkens, G.B., Slotboom, D.J., Gelfand, M.S., Osterman, A.L., Hanson, A.D. and Eitinger, T. (200�) A no�el class of modular transporters for �itamins in prokaryotes. J. Bacteriol. 191: 42-51.

106
10�. Rodionov, D.A., Hebbeln, P., Gelfand, M.S. and Eitinger, T. (2006) Comparati�e and functional genomic analysis of prokaryotic nickel and cobalt uptake transporters: e�idence for a no�el group of ATP-binding cassette transporters. J. Bacteriol. 188: 31�-32�.
110. Rodionov, D.A., Mironov, A.A. and Gelfand, M.S. (2002) Conser�ation of the biotin regulon and the BirA regulatory signal in Eubacteria and Archaea. Genome Res. 12: 150�-1516.
111. Rodionov, D.A., Vitreschak, A.G., Mironov, A.A. and Gelfand, M.S. (2002) Comparati�e genomics of thiamin biosynthesis in procaryotes. New genes and regulatory mechanisms. J. Biol. Chem. 277: 4��4�-4��5�.
112. Russ, W.P. and Engelman, D.M. (2000) The GxxxG motif: a framework for transmembrane helix-helix association. J. Mol. Biol. 296: �11-�1�.
113. Sauna, Z.E. and Ambudkar, S.V. (2000) E�idence for a requirement for ATP hydrolysis at two distinct steps during a single turno�er of the catalytic cycle of human P-glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 97: 2515-2520.
114. Sauna, Z.E. and Ambudkar, S.V. (2001) Characterization of the catalytic cycle of ATP hydrolysis by human P-glycoprotein. The two ATP hydrolysis e�ents in a single catalytic cycle are kinetically similar but affect different functional outcomes. J. Biol. Chem. 276: 11653-11661.
115. Saurin, W., Hofnung, M. and Dassa, E. (1���) Getting in or out: early segregation between importers and exporters in the e�olution of ATP-binding cassette (ABC) transporters. J. Mol. Evol. 48: 22-41.
116. Schauer, K., Stolz, J., Scherer, S. and Fuchs, T.M. (200�) Both thiamine uptake and biosynthesis of thiamine precursors are required for intracellular replication of Listeria monocytogenes. J. Bacteriol. 191: 221�-222�.
11�. Schellenberger, A. (1���) Sixty years of thiamin diphosphate biochemistry. Biochim. Biophys. Acta 1385: 1��-1�6.
11�. Scorps-Declere, S., Lemoine, F., Sculo, Q., Lespinet, O. and Labedan, B. (200�) The multiple facets of homology and their use in comparati�e genomics to study the e�olution of genes, genomes, and species. Biochimie 90: 5�5-60�.

10�
11�. Shaffer, P.L., Goehring, A., Shankaranarayanan, A. and Gouaux, E. (200�) Structure and mechanism of a Na+-independent amino acid transporter. Science 325: 1010-1014.
120. Shane, B. and Stokstad, E.L. (1��5) Transport and metabolism of folates by bacteria. J. Biol. Chem. 250: 2243-2253.
121. Shapiro, A.B. and Ling, V. (1��5) Reconstitution of drug transport by purified P-glycoprotein. J. Biol. Chem. 270: 1616�-161�5.
122. Shapiro, A.B. and Ling, V. (1���) Stoichiometry of coupling of rhodamine 123 transport to ATP hydrolysis by P-glycoprotein. Eur. J. Biochem. 254: 1��-1�3.
123. Shimamura, T., Weyand, S., Beckstein, O., Rutherford, N.G., Hadden, J.M., Sharples, D., Sansom, M.S., Iwata, S., Henderson, P.J. and Cameron, A.D. (2010) Molecular basis of alternating access membrane transport by the sodium-hydantoin transporter Mhp1. Science 328: 4�0-4�3.
124. Shin, W., Pletcher, J., Blank, G. and Sax, M. (1���) Ring stacking interactions between thiamin and planar molecules as seen in the crystal structure of a thiamin picrolonate dihydrate complex. J. Am. Chem. Soc. 99: 34�1-34��.
125. Sippel, K.H., Robbins, A.H., Reutzel, R., Boehlein, S.K., Namiki, K., Goodison, S., Gbandje-McKenna, M., Rosser, C.J. and McKenna, R. (200�) Structural insights into the extracytoplasmic thiamine-binding lipoprotein p3� of Mycoplasma hyorhinis. J. Bacteriol. 191: 25�5-25�2.
126. Slotboom, D.J., Duurkens, R.H., Olieman, K. and Erkens, G.B. (200�) Static light scattering to characterize membrane proteins in detergent solution. Methods 46: �3-�2.
12�. Soding, J. and Lupas, A.N. (2003) More than the sum of their parts: on the e�olution of proteins from peptides. Bioessays 25: �3�-�46.
12�. Soriano, E.V., Rajashankar, K.R., Hanes, J.W., Bale, S., Begley, T.P. and Ealick, S.E. (200�) Structural similarities between thiamin-binding protein and thiaminase-I suggest a common ancestor. Biochemistry 47: 1346-135�.

10�
12�. Tang, L., Bai, L., Wang, W.H. and Jiang, T. (2010) Crystal structure of the carnitine transporter and insights into the antiport mechanism. Nat. Struct. Mol. Biol. 17: 4�2-4�6.
130. Ter Beek, J., Duurkens, R.H., Erkens, G.B. and Slotboom, D.J. (2011) Quaternary structure and functional unit of Energy Coupling Factor (ECF)-type transporters. J. Biol. Chem. 286: 54�1-54�5.
131. Thanassi, J.A., Hartman-Neumann, S.L., Dougherty, T.J., Dougherty, B.A. and Pucci, M.J. (2002) Identification of 113 conser�ed essential genes using a high-throughput gene disruption system in Streptococcus pneumoniae. Nucleic Acids Res. 30: 3152-3162.
132. Theobald, D.L. and Miller, C. (2010) Membrane transport proteins: surprises in structural sameness. Nat. Struct. Mol. Biol. 17: 2-3.
133. Tokuriki, N. and Tawfik, D.S. (200�) Stability effects of mutations and protein e�ol�ability. Curr. Opin. Struct. Biol. 19: 5�6-604.
134. Valyasevi, R., Sandine, W.E. and Geller, B.L. (1��1) A membrane protein is required for bacteriophage c2 infection of Lactococcus lactis subsp. lactis C2. J. Bacteriol. 173: 60�5-6100.
135. Van der Heide, T. and Poolman, B. (2000) Osmoregulated ABC-transport system of Lactococcus lactis senses water stress �ia changes in the physical state of the membrane. Proc. Natl. Acad. Sci. U.S.A. 97: �102-�106.
136. Van der Heide, T., Stuart, M.C. and Poolman, B. (2001) On the osmotic signal and osmosensing mechanism of an ABC transport system for glycine betaine. EMBO J. 20: �022-�032.
13�. Veldhuis, G., Vos, E.P., Broos, J., Poolman, B. and Scheek, R.M. (2004) E�aluation of the flow-dialysis technique for analysis of protein-ligand interactions: an experimental and a monte carlo study. Biophys. J. 86: 1�5�-1�6�.
13�. Vieira-Pires, R.S. and Morais-Cabral, J.H. (2010) 310 helices in channels and other membrane proteins. J. Gen. Physiol 136: 5�5-5�2.

10�
13�. Viklund, H. and Elofsson, A. (2004) Best α--helical transmembrane protein topology predictions are achie�ed using hidden Marko� models and e�olutionary information. Protein Sci. 13: 1�0�-1�1�.
140. Viklund, H. and Elofsson, A. (200�) OCTOPUS: impro�ing topology prediction by two-track ANN-based preference scores and an extended topological grammar. Bioinformatics. 24: 1662-166�.
141. Von Heijne, G. (1���) Control of topology and mode of assembly of a polytopic membrane protein by positi�ely charged residues. Nature 341: 456-45�.
142. Von Heijne, G. and Gavel, Y. (1���) Topogenic signals in integral membrane proteins. Eur. J. Biochem. 174: 6�1-6��.
143. Waight, A.B., Love, J. and Wang, D.N. (2010) Structure and mechanism of a pentameric formate channel. Nat. Struct. Mol. Biol. 17: 31-3�.
144. Wang, B., Dukarevich, M., Sun, E.I., Yen, M.R. and Saier, M.H., Jr. (200�) Membrane porters of ATP-binding cassette transport systems are polyphyletic. J. Membr. Biol. 231: 1-10.
145. Wang, P., Nirmalan, N., Wang, Q., Sims, P.F. and Hyde, J.E. (2004) Genetic and metabolic analysis of folate sal�age in the human malaria parasite Plasmodium falciparum. Mol. Biochem. Parasitol. 135: ��-��.
146. Ward, A., Reyes, C.L., Yu, J., Roth, C.B. and Chang, G. (200�) Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc. Natl. Acad. Sci. U.S.A. 104: 1�005-1�010.
14�. Webb, E., Claas, K. and Downs, D. (1���) thiBPQ encodes an ABC transporter required for transport of thiamine and thiamine pyrophosphate in Salmonella typhimurium. J. Biol. Chem. 273: ��46-��50.
14�. White, S.H. (200�) Biophysical dissection of membrane proteins. Nature 459: 344-346.
14�. Wilkins, M.R., Pasquali, C., Appel, R.D., Ou, K., Golaz, O., Sanchez, J.C., Yan, J.X., Gooley, A.A., Hughes, G., Humphery-Smith, I., Williams, K.L. and Hochstrasser, D.F. (1��6) From proteins to proteomes: large scale protein identification by two-dimensional electrophoresis and amino acid analysis. Biotechnology (N.Y.) 14: 61-65.

110
150. Williams, R.R. and Cline, J.K. (1�36) Synthesis of �itamin B1. J. Am. Chem.Soc. 58: 1504-1505.
151. Winkler, W., Nahvi, A. and Breaker, R.R. (2002) Thiamine deri�ati�es bind messenger RNAs directly to regulate bacterial gene expression. Nature 419: �52-�56.
152. Yamashita, A., Singh, S.K., Kawate, T., Jin, Y. and Gouaux, E. (2005) Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature 437: 215-223.
153. Yau, W.M., Wimley, W.C., Gawrisch, K. and White, S.H. (1���) The preference of tryptophan for membrane interfaces. Biochemistry 37: 14�13-14�1�.
154. Zhang, J., Lau, M.W. and Ferre-D’Amare, A.R. (2010) Ribozymes and riboswitches: modulation of RNA function by small molecules. Biochemistry 49: �123-�131.
155. Zhang, P., Wang, J. and Shi, Y. (2010) Structure and mechanism of the S component of a bacterial ECF transporter. Nature 468: �1�-�20.
156. Zhang, Q.C., Petrey, D., Norel, R. and Honig, B.H. (2010) Protein interface conser�ation across structure space. Proc. Natl. Acad. Sci. U.S.A. 107: 10��6-10�01.

111





















