
University of Groningen Osteoprotegerin in organ fibrosis: biomarker ...
185
University of Groningen Osteoprotegerin in organ fibrosis: biomarker, actor, and target of therapy? Putri, Kurnia Sari Setio IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check the document version below. Document Version Publisher's PDF, also known as Version of record Publication date: 2019 Link to publication in University of Groningen/UMCG research database Citation for published version (APA): Putri, K. S. S. (2019). Osteoprotegerin in organ fibrosis: biomarker, actor, and target of therapy?. University of Groningen. Copyright Other than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons). The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license. More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne- amendment. Take-down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons the number of authors shown on this cover page is limited to 10 maximum. Download date: 31-01-2022
-
Upload
khangminh22 -
Category
Documents
-
view
2 -
download
0
Transcript of University of Groningen Osteoprotegerin in organ fibrosis: biomarker ...

University of Groningen
Osteoprotegerin in organ fibrosis: biomarker, actor, and target of therapy?Putri, Kurnia Sari Setio
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:2019
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Putri, K. S. S. (2019). Osteoprotegerin in organ fibrosis: biomarker, actor, and target of therapy?. Universityof Groningen.
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license.More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne-amendment.
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 31-01-2022

OSTEOPROTEGERIN IN ORGAN FIBROSIS:BIOMARKER, ACTOR AND
TARGET OF THERAPY?
Kurnia Sari Setio Putri

ParanymphsDorenda OosterhuisFransien van Dijk
The research presented in this PhD thesis was performed at the Department ofPharmaceutical Technology and Biopharmacy, University of Groningen, theNetherlands. Printing of this thesis was financially supported by University ofGroningen, Faculty of Science and Engineering and the University Library.Kurnia Sari Setio Putri received a PhD grant from Ubbo Emmius sandwich scholarshipbetween University of Groningen, The Netherlands and Universitas Indonesia,Indonesia.Cover design : Suryadi (addieadie, cubucubu.id)Layout : Kurnia Sari Setio PutriPrinted by : ProefschriftMaken (www.proefschriftmaken.nl)ISBN (printed version) : 978-94-034-1663-2ISBN (digital version) : 978-94-034-1662-5© Kurnia Sari Setio Putri, 2019All rights reserved. Copyright of the published articles is with the correspondingjournal or otherwise with the author. No part of this publication may be reproduced,stored in a retrieval system, or transmitted, in any form or by any means, without theprior permission in writing from author or the copyright-owning journal.

Osteoprotegerin in OrganFibrosis: Biomarker, Actor, and
Target of Therapy?
PhD thesis
to obtain the degree of PhD at theUniversity of Groningenon the authority of the
Rector Magnificus Prof. E. Sterkenand in accordance with
the decision by the College of Deans.
This thesis will be defended in public on
Monday 17 June 2019 at 11.00 hours
by
Kurnia Sari Setio Putri
born on 29 August 1985in Jakarta, Indonesia

SupervisorsProf. P. OlingaProf. B.N. Melgert
Co-supervisorDr. W.L.J. Hinrichs
Assessment CommitteeProf. J.K. BurgessProf. R. GosensProf. S. Meiners

BismillahirrahmanirrahimIn the name of Allah, the Most Gracious, the Most Merciful
Minazh zhulumaati ilannuur (Al-Quran, Surah Al- Hadid (57), verse 9)Door Duisternis tot Licht - Dari Kegelapan menuju Cahaya


CONTENTS
General Introduction 9
Chapter 1 The elusive antifibrotic macrophage 25
Chapter 2 The RANK/RANKL/OPG axis has a role in regulatingtissue repair processes in lung 51
Chapter 3 Osteoprotegerin is an early marker of the fibroticprocess and of antifibrotic treatment responses in ex vivolung fibrosis85
Chapter 4 Osteoprotegerin as a new marker to study early fibrosisin various organs 107
Chapter 5 Osteoprotegerin expression in liver tissue is induced byIL13 through TGFβ 139
General Discussion 157
Summary, Samenvatting, Ringkasan 167
Acknowledgements 177
Author Affiliations 183


General Introduction

General Introduction
10
FIBROSISFibrosis is associated with many diseases and is characterized by excessivedeposition of extracellular matrix (ECM) in tissue. Production of ECM is actually partof a normal repair response after tissue damage and includes a clotting phase, aninflammatory phase, a remodelling phase and a resolution phase, which will ultimatelylead to resolution of the damage and restoration of normal tissue architecture1.However, persistent injury and/or inflammation of a tissue may lead to an imbalancedregulation of ECM formation and resolution in this tissue.Fibrosis can occur in many organs, including vital organs like liver2, kidney3,lung4, intestine5, heart6,7, and bone marrow8. The development of fibrosis in each organis associated with several persistent triggers and damage, as described in Table 1.Table 1. Triggers of organ fibrosis
Triggers of fibrosis development Organ affectedViral andbacterialinfections Hepatitis B9 and hepatitis C10 LiverTuberculosis11, diphtheria12 LungMicrobiota13 Liver & intestineEnvironmentalfactors Cigarette smoke14, metal and silica dust4 LungFat and alcohol15 LiverChronicdiseases Ischemic/hypertensive and diabetic nephropathy3 KidneyHypertension and cardiomyophaty16,17 HeartAdverse effectsof radiation andchemotherapy4Lung
An advanced stage of fibrosis often leads to organ failure and thus malfunctionof these vital organs and can ultimately cause the death of patients. To date there areno possibilities to stop or reverse fibrosis, besides transplantation, and only a fewtherapies are available that slow down the fibrotic process7,18,19.Late detection of fibrosis is one of the main reasons for the high mortality inpatients. In most patients, fibrosis is only detected when the organ is already severelydamaged and the fibrotic organ is not able to properly perform its normal functionsanymore. Several diagnostic tools have been applied to assess the stage of fibrosis,including non-invasive imaging methods like magnetic resonance and transientelastography9,20 and high-resolution computed tomography21,22 to assess fibrosis stage

Osteoprotegerin in Organ Fibrosis: Biomarker, Actor and Target of Therapy?
11
in liver and lung, respectively. However, it is still of paramount importance to havereliable and easy-to-assess diagnostic methods and markers to detect the slow andlong-term progression of fibrosis in the earliest phase possible in order to prevent theincurable end-stage of the disease. Furthermore, the markers may also be applied toassess the response of fibrotic organs towards antifibrotic therapy, to determinetherapeutic effectiveness of antifibrotic drug candidates, and eventually be used assurrogate endpoints in drug studies. In order to determine the reliability of candidatefibrosis markers that can accurately diagnose fibrosis and therapeutic effectiveness, itis necessary to understand how mechanisms, pathways, cell types, growth factors, andcytokines interact in fibrosis development and resolution.FIBROSIS-ASSOCIATED CELLSFibrosis is a complex condition, which involves many different cell types,growth factors, and cytokines. Therefore, it is of utmost importance to understandintercellular mechanisms and signalling pathways of fibrosis, to identify specifictargets for therapy and to establish reliable markers for detecting fibrosis andevaluating therapy effectiveness. Extensive studies on those subjects will give moreinsights on the progression of fibrosis, which will facilitate the development of effectiveantifibrotic drugs.Fibroblasts, including tissue resident (like hepatic stellate cells in liver) typesand circulating precursors i.e. fibrocytes, play key-role in fibrosis development.Injury/damage of tissue leads to activation of fibroblasts into myofibroblasts. Thesemyofibroblasts secrete profibrotic cytokines and chemokines, initiate migration ofcirculating fibrocytes and other profibrotic cells into the injured tissue/organ, induceproliferation and differentiation of fibrosis-associated cells, promote tissuecontraction, and produce ECM1,2. Due to the central role of myofibroblasts in thedevelopment of fibrosis, many studies aim to inhibit activation of fibroblasts intomyofibroblasts, e.g. by inhibiting the transforming growth factor beta (TGFβ)-pathway23,24, the Wnt-pathway25 and the PI3K/Akt pathway26.Beside fibroblasts, smooth muscle cells exhibit similar properties as fibroblaststowards fibrosis stimulation27,28. Several studies have shown that smooth muscle cells

General Introduction
12
can differentiate into myofibroblasts and secrete TGFβ29 and platelet-derived growthfactor (PDGF) in lung30, and PDGF in intestine31.Myofibroblasts, being the major producers of extracellular matrix, have beenthe focus of fibrosis research for many years. However, in recent years, there isincreasing evidence that several other cell types also play an important role in fibrosisdevelopment and resolution. Other cells that are involved in the development offibrosis, are described in Table 2.Table 2. Cells involved in fibrosis
Fibrosis-associatedcells
Role in fibrosisFibroblasts1,32,33 - produce profibrotic cytokines and chemokines- initiate migration of circulating fibrocytes and other profibroticcells into injured tissues/organs- induce proliferation and differentiation of fibrosis-associatedcells- promote tissue contraction and produce ECM(Profibrotic)macrophages34,35 - produce profibrotic mediators like TGF and PDGF that induceproliferation and activation of fibroblasts- produce CC chemokines that can attract profibrotic cells(Antifibrotic)macrophages36–39 - produce specific matrix metalloproteinases (MMPs) and otherproteolytic enzymes like cathepsins to degrade ECM.- phagocytose pieces of degraded ECM- produce tissue inhibitor of metalloproteinases (TIMPs)Fibrocytes 28,40,41 - can develop into (myo)fibroblasts and produce connective tissueproteins such as vimentin and collagens I and IIITh2 (Type 2 Thelper) cells 42 - produce profibrotic cytokines including interleukin-4 (IL4),interleukin-10 (IL10), and interleukin-13 (IL13)- produce growth factors (TGFβ, PDGF)B cells43 - produce IL-6, IL10, and TGFSmooth musclecells29–31,44 - produce TGF and PDGF- produce cytokines and chemokines- produce matrix proteins, MMPs and TIMPs- express integrinsDendritic cells45–47 - produce MMPs, including MMP2 and MMP7, and produce IL10and TGFβStudies showed that Th2 cells42 and B cells43 also play role in producingprofibrotic cytokines (IL4, IL10 and IL13) and growth factors (TGFβ, PDGF) that can

Osteoprotegerin in Organ Fibrosis: Biomarker, Actor and Target of Therapy?
13
activate fibroblasts to produce ECM. Dendritic cells contributes to remodeling of tissueby secreting MMPs, including MMP2 and MMP746 and producing IL10 and TGFβ47.Macrophages play an important role in controlling ECM homeostasis, which isdysregulated during fibrosis42,48. Several studies have shown that macrophagespromote fibrosis by producing profibrotic mediators like TGF and PDGF that induceproliferation and activation of fibroblasts35,42 . However, other studies have shown thatmacrophages also facilitate resolution of fibrosis by producing specific MMPs andother proteolytic enzymes like cathepsins that degrade fibrotic ECM49,50. In addition,macrophages have also been shown to express receptors that can phagocytose piecesof degraded ECM48,51,52. Macrophages can also express/ produce membrane-type MMP(MT-MMP) and TIMPs39 which can activate proteolytic activity of MMPs via proteinasecleavage53,54. These studies thus reveal that macrophages behaviour is highly plastic.The interaction and roles of fibrosis-associated cells are schematicallysummarized in Figure 1. This scheme illustrates key players (including mechanisms,pathways, cell types, growth factors, and cytokines) in fibrosis development andresolution that may be studied in more detail for development of fibrosis markers andtargets of therapy.
Figure 1. The interactions and roles of fibrosis-associated cells

General Introduction
14
FIBROSIS MARKERSIn several studies, possible fibrosis markers have been identified in variousorgans. These markers can be classified as follows:1. Fibrosis-associated cells, including fibroblasts32,33, fibrocytes28,40,macrophages34,37,38, monocytes35, dendritic cells45–47, Th2 cells42, and B cells43 asdescribed in Table 22. Fibrogenesis-related cytokines, including TGFβ, connective-tissue growth factor(CTGF), PDGF , IL13, tumor necrosis factor alpha (TNFα), vascular endothelialgrowth factor (VEGF), epidermal growth factor (EGF) and their receptors553. Profibrotic chemokines, including ligand of CXC chemokine-13 (CXCL13)56 andligand of CC chemokine-8 (CCL18)41,57,584. Fibrosis-associated proteins, including galectin-359,60 and klotho615. Markers of myofibroblast activation/ differentiation: α-smooth muscle actin(α-SMA)626. Markers of ECM formation, including collagen, glycoprotein, hyaluronan, pro-peptide of collagen type III (PIIINP), pro-peptide of collagen type I (PINP), type IVcollagen, hydroxyproline, fibronectin, and plasminogen activator inhibitor 1(PAI-1)63.7. Collagen chaperones, including heat shock protein 47 (Hsp47)64, FK506-bindingprotein 10 (FKBP10)658. Markers of fibrolytic processes, including MMP-2, MMP-9, MMP-13639. ECM degradation products, including collagen type III, VI, I fragments generated byMMP-2, 9, 1355,66,6710. Epithelium-specific markers, including αvβ6 integrin68, integrin alpha 1169,surfactant protein A, C, and D70, MMP-771–73 and MMP-374Among the above-mentioned markers, fibrosis markers that can be detected inserum/plasma (blood-based biomarkers) offer advantages in diagnosing fibrosis dueto their easier sampling procedure. Therefore, several blood-based (serum)biomarkers, have been further developed to be applied in the clinical field. Maher etal.70 for instance have shown that four serum biomarkers (surfactant protein D,

Osteoprotegerin in Organ Fibrosis: Biomarker, Actor and Target of Therapy?
15
MMP-7, carbohydrate antigen 19-9, and carbohydrate antigen 125) predict diseaseprogression in a cohort of patients with idiopathic pulmonary fibrosis (IPF).In the case of liver fibrosis, several serum tests have been developed to diagnoseliver fibrosis, i.e. the European Liver Fibrosis test (ELF)™ using hyaluronic acid,procollagen III N-terminal peptide and TIMP120, the FibroTest/Fibrosure using α-2-macroglobulin, apolipoprotein A1, haptoglobin, L-glutamyltranspeptidase, andbilirubin75, and the Coopscore using α-2-macroglobulin, apolipoprotein A1, AST,collagen IV and osteoprotegerin. For the latter one, osteoprotegerin (OPG) wasincluded as an additional biomarker to increase the accuracy of liver fibrosisdiagnosis76.A few other studies have shown that higher OPG serum levels are associatedwith having liver fibrosis 77,78. However, it is unclear whether higher OPG levels areonly associated with fibrosis of the liver or also of other organs. Moreover, the role ofOPG in fibrotic processes and how OPG is regulated during fibrosis are still unclear.Therefore, the aim of this thesis is to elucidate the role of OPG in fibrosis andinvestigate whether it is a general phenomenon or only associated with liver fibrosis.OSTEOPROTEGERIN IN FIBROSISOPG is a secretory protein that belongs to the tumor necrosis factor (TNF)receptor superfamily. It functions as a decoy receptor for several ligands includingreceptor of nuclear factor B ligand (RANKL), TNF-related apoptosis-inducing ligand(TRAIL) and glycosaminoglycan79,80. OPG is best known for its regulation of bone tissueECM by binding RANKL and blocking RANKL-RANK interactions, thus inhibitingosteoclast activation and preventing bone ECM degradation81. However, recent studieshave shown that higher OPG levels were not only detected in bone but also in fibroticlung82, heart83, and vasculature84of murine models as well as in fibrotic liver77,78,epidural fibrosis85, and chronic kidney disease due to vascular damage86, andinflammatory bowel disease87 in humans.Unlike markers that can only be detected in fibrotic organs, OPG is a solubleprotein and can be detected in blood and urine. Several studies have shown that higherOPG serum levels correlate with fibrosis of the liver76,78, kidney88,89 and colon90. These

General Introduction
16
results suggest that OPG could be a biomarker to assess organ fibrosis and maypossibly also be used to assess the effectiveness of antifibrotic therapy. Furthermore,having a better understanding of the role of OPG in fibrogenesis in specificcells/tissues/organs may even lead to new opportunities for OPG as a target forantifibrotic therapy.OPG was first discovered to be produced by osteoblasts81 but recent studieshave shown that OPG is also produced by fibroblasts91, smooth muscle cells92 andepithelial cells93. OPG production was found to be stimulated by TGFβ, IL4, and IL17and inhibited by interferon-γ (IFNγ)94. OPG itself could also induce the expression offibronectin, collagen type I, III, IV, and TGFβ184. The association with several types offibrosis and its production by key cells in fibrogenesis indicate that OPG may play rolein fibrosis, however, how OPG contributes to the development of organ fibrosis needsto be further studied.There are several hypotheses that could explain the role of OPG in fibrosis.Firstly, OPG may bind TRAIL and avert TRAIL-induced apoptosis of myofibroblasts andtherefore myofibroblasts will continue to produce ECM84,95,96. Secondly, OPG may bindRANKL thereby preventing interaction of RANKL with RANK-expressing macrophages,which may lead to inactivation of MMP-producing macrophages and consequently toinhibition of ECM degradation97. Key to these hypotheses is the assumption OPG isproduced locally in the fibrotic organ of study. However, in patients and animal modelsof fibrosis OPG protein in serum or even the organ itself may originate from multiplesources. Therefore, beside using patient material or whole animals, we need additionalmethods to be able to study OPG regulation on the organ level. One such method is theex vivo method of precision-cut tissue slices. This technique offers advantages toinvestigate OPG production and regulation in in specific organs without interference ofother organs in more details.PRECISION-CUT TISSUE SLICESTo study fibrosis, many different models are being used, including in vitro andin vivo models98,99. In vitro studies on fibrosis focus on how specific cell types respondto fibrotic stimulation. However, fibrosis is a complex disease involving interactions

Osteoprotegerin in Organ Fibrosis: Biomarker, Actor and Target of Therapy?
17
between various types of cells and in vitro studies with only a single cell type cannotcomprehensively explain these complex mechanisms. On the other hand, in vivo studieswith animals provide better insight into the complex mechanisms between cells in thefibrotic organ, but are sometimes overly complex and often require large numbers ofanimals. In addition, animal models often do not accurately mimic human disease.Over the years our lab has specialized precision-cut tissue slices as an inbetween, alternative model to study chronic diseases such as fibrosis in lung, liver,intestine and kidney100–104. Intercellular and cell-matrix interactions remain intact inthese tissue slices102 and therefore tissue slices allow study of multicellular processesas the contain all the different cells in their original environment and their tissuearchitecture. Precision-cut human tissue slices can also provide better prediction oftherapy success in clinical research since precision-cut human tissue slices enablemore accurate translation of preclinical studies into clinical studies because there areno species differences. Therefore, an important part of this thesis has been generatedusing the model of precision-cut tissue slices to study different aspects of fibrogenesis.SCOPE OF THE THESISSeveral studies have shown that macrophages exhibit a “dual role” in fibrosis35,38,42,105–107. Therefore, in Chapter 1, we discuss this elusive behaviour ofmacrophages during the development of fibrosis in various organs and identify pro-and antifibrotic characteristics of macrophages to design strategies to suppress theirfibrotic nature and to stimulate the antifibrotic nature of macrophages.We further continued our studies in Chapter 2, investigating whether OPGplays a role in pulmonary fibrosis and to test our hypothesis that OPG has an effect onfibrosis development through interactions with RANKL. To test this we treated micewith silica-induced pulmonary fibrosis with RANKL to possibly activate antifibroticmacrophages and we assessed fibrosis development after RANKL treatment.In Chapter 3, we used precision-cut lung slices to study the regulation of OPGin lung tissue during fibrogenesis in more detail using TGFβ-stimulated murine lungslices and slices from human fibrotic lung tissue. In this chapter, we also investigatedwhether pirfenidone and nintedanib, currently the only approved treatments for IPF,

General Introduction
18
affect OPG production to assess whether OPG could potentially be used as a marker fortreatment effects.One of the challenges of using precision cut tissue slices is that they can only beused for short-term (2 days) experiments, while fibrosis is a chronic disease thatdevelops over several months/years. Therefore, it is important to have a marker thatcan detect development of fibrosis in early stages as well as detect early changes afterantifibrotic therapy. In Chapter 4, we further explore OPG as marker of both early- andend-stage fibrosis in lung, liver, kidney and intestine using murine and human tissueslices. We further evaluated OPG as marker to assess treatment effect using a new drugcandidate: galunisertib, a TGFβ-receptor type I kinase inhibitor, which was previouslyapplied as an anticancer drug108–110.To gain deeper understanding of OPG regulation in fibrotic organs, in Chapter
5, we further studied regulation of OPG in liver tissue after TGFβ- and IL13-stimulation.Finally, in the General Discussion, we summarize our findings and discuss theperspectives of the use of OPG as biomarker to detect fibrosis in early stages, to assesstherapy effectiveness of new drug candidates, and as target for antifibrotic therapy.REFERENCES
1. Wynn, T. Cellular and molecular mechanisms of fibrosis. J. Pathol. 214, 199–210 (2008).2. Pellicoro, A., Ramachandran, P., Iredale, J. P. & Fallowfield, J. A. Liver fibrosis and repair: immuneregulation of wound healing in a solid organ. Nat. Rev. Immunol. 14, 181–94 (2014).3. Duffield, J. S. & Duffield, J. S. Cellular and molecular mechanisms in kidney fibrosis Find the latestversion : Review series Cellular and molecular mechanisms in kidney fibrosis. 124, 2299–2306(2014).4. Wilson, M. S. & Wynn, T. A. Pulmonary fibrosis: Pathogenesis, etiology and regulation. MucosalImmunol. 2, 103–121 (2009).5. Curciarello, R., Docena, G. H. & MacDonald, T. T. The Role of Cytokines in the Fibrotic Responsesin Crohn’s Disease. Front. Med. 4, 1–9 (2017).6. Kong, P., Panagiota, ·, Nikolaos, C. · & Frangogiannis, G. The pathogenesis of cardiac fibrosis. Cell.Mol. Life Sci 71, 549–574 (2014).7. Fan, Z. & Guan, J. Antifibrotic therapies to control cardiac fibrosis. Biomater. Res. 20, 1–13 (2016).8. Zahr, A. A. et al. Bone marrow fibrosis in myelofibrosis: pathogenesis, prognosis and targetedstrategies. Haematologica 101, 660–671 (2016).9. Chon, Y. E. et al. Performance of Transient Elastography for the Staging of Liver Fibrosis inPatients with Chronic Hepatitis B: A Meta-Analysis. PLoS One 7, 1–7 (2012).10. Rizzo, L. et al. Comparison of transient elastography and acoustic radiation force impulse for non-invasive staging of liver fibrosis in patients with chronic hepatitis C. Am. J. Gastroenterol. 106,2112–2120 (2011).

Osteoprotegerin in Organ Fibrosis: Biomarker, Actor and Target of Therapy?
19
11. Jarvela, J. R., Tuscano, L., Lee, H. & Silver, R. F. Pulmonary responses to pathogen-specific antigensin latent Mycobacterium tuberculosis infection. Tuberculosis 96, (2016).12. Sisson, T. H. et al. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis.Am. J. Respir. Crit. Care Med. 181, 254–263 (2010).13. Seki, E. & Schnabl, B. Role of innate immunity and the microbiota in liver fibrosis: Crosstalkbetween the liver and gut. J. Physiol. 590, 447–458 (2012).14. King, T. E., Pardo, A. & Selman, M. Idiopathic pulmonary fibrosis. Lancet 378, 1949–1961 (2011).15. Ellis, E. L. & Mann, D. A. Clinical evidence for the regression of liver fibrosis. J. Hepatol. 56, 1171–1180 (2012).16. Díez, J. Mechanisms of cardiac fibrosis in hypertension. J. Clin. Hypertens. (Greenwich). 9, 546–550 (2007).17. Khan, R. & Sheppard, R. Fibrosis in heart disease: Understanding the role of transforming growthfactor-β1 in cardiomyopathy, valvular disease and arrhythmia. Immunology 118, 10–24 (2006).18. Caminati, A., Cassandro, R., Torre, O. & Harari, S. Severe idiopathic pulmonary fibrosis: What canbe done? Eur. Respir. Rev. 26, (2017).19. Arakeri, G. et al. Current protocols in the management of oral submucous fibrosis: An update. J.Oral Pathol. Med. 46, 418–423 (2017).20. Fitzpatrick, E. & Dhawan, A. Noninvasive biomarkers in non-alcoholic fatty liver disease: Currentstatus and a glimpse of the future. World J. Gastroenterol. 20, 10851–10863 (2014).21. Hunninghake, G. W. et al. Utility of a Lung Biopsy for the Diagnosis of Idiopathic PulmonaryFibrosis. 2–5 (2001).22. Raghu, G., Weycker, D., Edelsberg, J., Bradford, W. Z. & Oster, G. Incidence and prevalence ofidiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 174, 810–816 (2006).23. Fernandez, I. E. & Eickelberg, O. The Impact of TGF-β on Lung Fibrosis. Proc. Am. Thorac. Soc. 9,111–116 (2012).24. Meng, X., Nikolic-Paterson, D. J. & Lan, H. Y. TGF-β: the master regulator of fibrosis. Nat. Rev.Nephrol. 12, 325–338 (2016).25. Akhmetshina, A. et al. Activation of canonical Wnt signalling is required for TGF-β-mediatedfibrosis. Nat. Commun. 3, (2012).26. Kulkarni, A. A. et al. PPAR-γ Ligands Repress TGFβ-Induced Myofibroblast Differentiation byTargeting the PI3K/Akt Pathway: Implications for Therapy of Fibrosis. PLoS One 6, e15909(2011).27. Maharaj, S. S. et al. Increased Fibrocytes In Patients Newly Diagnosed With Idiopathic PulmonaryFibrosis. in B28. EMERGING BIOMARKERS OF COMPLEX LUNG DISEASES A2655–A2655(American Thoracic Society, 2012). doi:10.1164/ajrccm-conference.2012.185.1_MeetingAbstracts.A265528. Borie, R. et al. Alveolar Fibrocytes Are A Markers A Lung Fibrosis Severity. in B28. EMERGINGBIOMARKERS OF COMPLEX LUNG DISEASES A2656–A2656 (American Thoracic Society, 2012).doi:10.1164/ajrccm-conference.2012.185.1_MeetingAbstracts.A265629. Xie, S. et al. Regulation of TGF-β1-induced connective tissue growth factor expression in airwaysmooth muscle cells. Am. J. Physiol. Cell. Mol. Physiol. 288, L68–L76 (2005).30. Chung, K. F. Airway smooth muscle cells: Contributing to and regulating airway mucosalinflammation? Eur. Respir. J. 15, 961–968 (2000).31. Speca, S., Giusti, I., Rieder, F. & Latella, G. Cellular and molecular mechanisms of intestinal fibrosis.World J. Gastroenterol. 18, 3635–3661 (2012).32. McAnulty, R. J. Fibroblasts and myofibroblasts: Their source, function and role in disease. Int. J.Biochem. Cell Biol. 39, 666–671 (2007).33. Lawrance, I. C. et al. Cellular and Molecular Mediators of Intestinal Fibrosis. J. Crohn’s Colitisj.crohns.2014.09.008 (2015). doi:10.1016/j.crohns.2014.09.00834. Barron, L. & Wynn, T. A. Macrophage activation governs schistosomiasis-induced inflammationand fibrosis. Eur. J. Immunol. 41, 2509–2514 (2011).

General Introduction
20
35. Gibbons, M. A. et al. Ly6Chimonocytes direct alternatively activated profibrotic macrophageregulation of lung fibrosis. Am. J. Respir. Crit. Care Med. 184, 569–581 (2011).36. Brancato, S. K. & Albina, J. E. Wound macrophages as key regulators of repair: Origin, phenotype,and function. Am. J. Pathol. 178, 19–25 (2011).37. Huang, W. C., Sala-Newby, G. B., Susana, A., Johnson, J. L. & Newby, A. C. Classical macrophageactivation up-regulates several matrix metalloproteinases through mitogen activated proteinkinases and nuclear factor-κB. PLoS One 7, (2012).38. Boorsma, C. E., Draijer, C. & Melgert, B. N. Macrophage Heterogeneity in Respiratory Diseases.Mediators Inflamm. 2013, 1–19 (2013).39. Laquerriere, P. et al. MMP-2, MMP-9 and their inhibitors TIMP-2 and TIMP-1 production byhuman monocytes in vitro in the presence of different forms of hydroxyapatite particles.Biomaterials 25, 2515–2524 (2004).40. Quan, T. E., Cowper, S. E. & Bucala, R. The role of circulating fibrocytes in fibrosis. Curr.Rheumatol. Rep. 8, 145–150 (2006).41. Prasse, A. & Müller-Quernheim, J. Non-invasive biomarkers in pulmonary fibrosis. Respirology14, 788–795 (2009).42. Barron, L. & Wynn, T. A. Fibrosis is regulated by Th2 and Th17 responses and by dynamicinteractions between fibroblasts and macrophages. Am. J. Physiol. Gastrointest. Liver Physiol. 300,G723-8 (2011).43. Hasegawa, M., Fujimoto, M., Takehara, K. & Sato, S. Pathogenesis of systemic sclerosis: Altered Bcell function is the key linking systemic autoimmunity and tissue fibrosis. J. Dermatol. Sci. 39, 1–7 (2005).44. Black, J. L., Burgess, J. K. & Johnson, P. R. A. Airway smooth muscle—its relationship to theextracellular matrix. Respir. Physiol. Neurobiol. 137, 339–346 (2003).45. Rahman, A. H. & Aloman, C. Dendritic cells and liver fibrosis. BBA - Mol. Basis Dis. 1832, 998–1004 (2013).46. Tort Tarrés, M. et al. Role of dendritic cells in pulmonary fibrosis in mice. in 1.5 DiffuseParenchymal Lung Disease 48, PA3891 (European Respiratory Society, 2016).47. Lu, T. T. Dendritic Cells: Novel Players in Fibrosis and Scleroderma. doi:10.1007/s11926-011-0215-548. McKleroy, W., Lee, T.-H. & Atabai, K. Always cleave up your mess: targeting collagen degradationto treat tissue fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 304, L709–L721 (2013).49. Cabrera, S. et al. Overexpression of MMP9 in macrophages attenuates pulmonary fibrosisinduced by bleomycin. Int. J. Biochem. Cell Biol. 39, 2324–2338 (2007).50. Fonović, M. & Turk, B. Cysteine cathepsins and extracellular matrix degradation. Biochim.Biophys. Acta - Gen. Subj. 1840, 2560–2570 (2014).51. Atabai, K. et al. Mfge8 diminishes the severity of tissue fibrosis in mice by binding and targetingcollagen for uptake by macrophages. J. Clin. Invest. 119, 3713–22 (2009).52. Popov, Y. et al. Macrophage-mediated phagocytosis of apoptotic cholangiocytes contributes toreversal of experimental biliary fibrosis. Am. J. Physiol. Liver Physiol. 298, G323–G334 (2010).53. Ra, H.-J. & Parks, W. C. Control of matrix metalloproteinase catalytic activity. Matrix Biol. 26, 587–596 (2007).54. Morrison, C. J. et al. Cellular activation of MMP-2 (gelatinase A) by MT2-MMP occurs via a TIMP-2-independent pathway. J. Biol. Chem. 276, 47402–10 (2001).55. Liu, T., Wang, X., Karsdal, M. A., Leeming, D. J. & Genovese, F. Molecular Serum Markers of LiverFibrosis. Biomark. Insights 7, BMI.S10009 (2012).56. Vuga, L. J. et al. C-X-C motif chemokine 13 (CXCL13) is a prognostic biomarker of idiopathicpulmonary fibrosis. Am. J. Respir. Crit. Care Med. 189, 966–974 (2014).57. Zhang, Y., Kaminski, N. & Simmons, R. P. Biomarkers in idiopathic pulmonary fibrosis. (2012).doi:10.1097/MCP.0b013e328356d03c58. Zissel, G. et al. Serum CC-Chemokine Ligand 18 Concentration Predicts Outcome in IdiopathicPulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 179, 717–723 (2009).

Osteoprotegerin in Organ Fibrosis: Biomarker, Actor and Target of Therapy?
21
59. de Boer, R. A. et al. The fibrosis marker galectin-3 and outcome in the general population. J. Intern.Med. 272, 55–64 (2012).60. Darabantiu, D., Pilat, L., Lala, R. I., Puschita, M. & Lungeanu, D. Galectin-3 as a marker for clinicalprognosis and cardiac remodeling in acute heart failure. Herz 43, 146–155 (2017).61. Nitta, K. et al. Reduced Klotho expression level in kidney aggravates renal interstitial fibrosis.Am. J. Physiol. Physiol. 302, F1252–F1264 (2012).62. Brenner, D. A. et al. Origin of myofibroblasts in liver fibrosis. Fibrogenes. Tissue Repair 5, 2–5(2012).63. Liu, T., Wang, X., Karsdal, M. A., Leeming, D. J. & Genovese, F. Molecular serum markers of liverfibrosis. Biomark. Insights 7, 105–117 (2012).64. Ishida, Y. & Nagata, K. Chapter 9 - Hsp47 as a Collagen-Specific Molecular Chaperone. Biol. Serpins499, 167–182 (2011).65. Staab-Weijnitz, C. A. et al. FK506-binding protein 10, a potential novel drug target for idiopathicpulmonary fibrosis. Am. J. Respir. Crit. Care Med. 192, 455–467 (2015).66. Jenkins, R. G. et al. Longitudinal change in collagen degradation biomarkers in idiopathicpulmonary fibrosis: an analysis from the prospective, multicentre PROFILE study. Lancet Respir.Med. 3, 462–472 (2015).67. Crooks, M. G. & Hart, S. P. Biomarkers in idiopathic pulmonary fibrosis: picking the winners fortrials. Lancet Respir. Med. 3, 421–422 (2015).68. Saini, G. et al. Αvβ6 Integrin May Be a Potential Prognostic Biomarker in Interstitial Lung Disease.Eur. Respir. J. 46, 486–494 (2015).69. Bansal, R. et al. Integrin alpha 11 in the regulation of the myofibroblast phenotype: implicationsfor fibrotic diseases. Exp. Mol. Med. 49, e396 (2017).70. Maher, T. M. et al. An epithelial biomarker signature for idiopathic pulmonary fibrosis: ananalysis from the multicentre PROFILE cohort study. Lancet Respir. Med. 5, 946–955 (2017).71. Rosas, I. O. et al. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathicpulmonary fibrosis. PLoS Med. 5, 0623–0633 (2008).72. Richards, T. J., Kaminski, N. & Gibson, K. F. Plasma proteins for risk prediction in idiopathicpulmonary fibrosis. Am. J. Respir. Crit. Care Med. 185, 1329–1330 (2012).73. Chu, J. et al. Validation of the prognostic value of MMP-7 in idiopathic pulmonary fibrosis.Respirology 22, 486–493 (2016).74. Yamashita, C. M. et al. Matrix metalloproteinase 3 is a mediator of pulmonary fibrosis. Am. J.Pathol. 179, 1733–1745 (2011).75. Salkic, N. N., Jovanovic, P., Hauser, G. & Brcic, M. Fibro test/fibrosure for significant liver fibrosisand cirrhosis in chronic hepatitis B: A meta-analysis. Am. J. Gastroenterol. 109, 796–809 (2014).76. Bosselut, N. et al. Including osteoprotegerin and collagen IV in a score-based blood test for liverfibrosis increases diagnostic accuracy. Clin. Chim. Acta J.L. Bosson; A. Paris; ANRS L. Allain, Paris.Methodol. Hospices Civ. Lyon. M-C. Gelineau, B. Poggi 415, 63–68 (2013).77. Yilmaz, Y. et al. Serum levels of osteoprotegerin in the spectrum of nonalcoholic fatty liverdisease. Scand. J. Clin. Lab. Invest. 70, 541–546 (2010).78. García-Valdecasas-Campelo, E. et al. Serum osteoprotegerin and rankl levels in chronic alcoholicliver disease. Alcohol Alcohol. 41, 261–266 (2006).79. Baud’huin, M. et al. Osteoprotegerin: Multiple partners for multiple functions. Cytokine GrowthFactor Rev. 24, 401–409 (2013).80. Kuźniewski, M. et al. Osteoprotegerin and osteoprotegerin/TRAIL ratio are associated withcardiovascular dysfunction and mortality among patients with renal failure. Adv. Med. Sci. 61,269–275 (2016).81. Boyce, B. F. & Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch.Biochem. Biophys. 473, 139–146 (2008).82. Brass, D. M. et al. Fibroproliferation in LPS-induced airway remodeling and bleomycin-inducedfibrosis share common patterns of gene expression. Immunogenetics 60, 353–369 (2008).

General Introduction
22
83. Liu, W. et al. Osteoprotegerin/RANK/RANKL axis in cardiac remodeling due to immuno-inflammatory myocardial disease. Exp. Mol. Pathol. 84, 213–217 (2008).84. Toffoli, B. et al. Osteoprotegerin promotes vascular fibrosis via a TGF-β1 autocrine loop.Atherosclerosis 218, 61–68 (2011).85. Sen, O. et al. The relation between serum levels of osteoprotegerin and postoperative epiduralfibrosis in patients who underwent surgery for lumbar disc herniation. Neurol. Res. 27, 452–455(2005).86. Yilmaz, M. I. et al. Osteoprotegerin in Chronic Kidney Disease: Associations with VascularDamage and Cardiovascular Events. Calcif. Tissue Int. 99, 121–130 (2016).87. Sylvester, F. A., Draghi, A., Bausero, M. A., Fernandez, M. L. & Vella, A. T. 1149 OsteoprotegerinExpression is Upregulated in the Colon of Children With Active Inflammatory Bowel Disease.Gastroenterology 142, S-209 (2012).88. Montañez-Barragán, A. et al. Osteoprotegerin and kidney disease. J. Nephrol. 27, 607–617 (2014).89. Bernardi, S. et al. Circulating osteoprotegerin is associated with chronic kidney disease inhypertensive patients. BMC Nephrol. 18, 1–9 (2017).90. Draghi, A., Ramanarasimhaiah, R., Fernandez, M. L., Vella, A. T. & Sylvester, F. A. Sa1165 ColonicOsteoprotegerin: Higher Expression in Children With Ulcerative Colitis Than Crohn’s Disease.Gastroenterology 146, S-217 (2014).91. Adhyatmika, A. et al. Osteoprotegerin is more than a serum marker in liver fibrosis. J. Hepatol.66, S147–S148 (2017).92. Zhang, J. et al. PDGF induces osteoprotegerin expression in vascular smooth muscle cells bymultiple signal pathways. FEBS Lett. 521, 180–184 (2002).93. Vidal, K. et al. Osteoprotegerin production by human intestinal epithelial cells: a potentialregulator of mucosal immune responses. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G836-44(2004).94. Tunyogi-Csapo, M. et al. Cytokine-controlled RANKL and osteoprotegerin expression by humanand mouse synovial fibroblasts: Fibroblast-mediated pathologic bone resorption. ArthritisRheum. 58, 2397–2408 (2008).95. McGrath, E. E. et al. Deficiency of tumour necrosis factor-related apoptosis-inducing ligandexacerbates lung injury and fibrosis. Thorax 67, 796–803 (2012).96. Vitovski, S., Phillips, J. S., Sayers, J. & Croucher, P. I. Investigating the Interaction betweenOsteoprotegerin and Receptor Activator of NF-κB or Tumor Necrosis Factor-related Apoptosis-inducing Ligand. J. Biol. Chem. 282, 31601–31609 (2007).97. Meng, H., Bai, X., Yu, H., Wang, Z. & Guo, A. Osteoprotegerin Promotes Cell Growth by RegulatingMatrix Metalloprotease-13 in Chondrocytes. J. Biomater. Tissue Eng. 7, 257–260 (2017).98. Chua, F., Gauldie, J. & Laurent, G. J. Pulmonary fibrosis: Searching for model answers. Am. J. Respir.Cell Mol. Biol. 33, 9–13 (2005).99. Molina-Molina, M., Pereda, J. & Xaubet, A. Experimental models for the study of pulmonaryfibrosis: current usefulness and future promise. Arch. Bronconeumol. 43, 501–507 (2007).100. Hansen, N. U. B. et al. Tissue turnover of collagen type I, III and elastin is elevated in the PCLSmodel of IPF and can be restored back to vehicle levels using a phosphodiesterase inhibitor.Respir. Res. 17, 1–10 (2016).101. de Graaf, I. A. M. et al. Preparation and incubation of precision-cut liver and intestinal slices forapplication in drug metabolism and toxicity studies. Nat. Protoc. 5, 1540–1551 (2010).102. Westra, I. M., Oosterhuis, D., Groothuis, G. M. M. & Olinga, P. Precision-cut liver slices as a modelfor the early onset of liver fibrosis to test antifibrotic drugs. Toxicol. Appl. Pharmacol. 274, 328–338 (2014).103. Iswandana, R. et al. Organ- and species-specific biological activity of rosmarinic acid. Toxicol. Vitr.32, 261–268 (2016).104. Poosti, F. et al. Precision-cut kidney slices (PCKS) to study development of renal fibrosis andefficacy of drug targeting ex vivo. Dis. Model. Mech. 8, 1227–1236 (2015).

Osteoprotegerin in Organ Fibrosis: Biomarker, Actor and Target of Therapy?
23
105. Stout, R. D. et al. Macrophages sequentially change their functional phenotype in response tochanges in microenvironmental influences. J. Immunol. 175, 342–349 (2005).106. Duffield, J. S. et al. Selective depletion of macrophages reveals distinct, opposing roles duringliver injury and repair. J. Clin. Invest. 115, 56–65 (2005).107. Pesce, J. T. et al. Arginase-1–Expressing Macrophages Suppress Th2 Cytokine–DrivenInflammation and Fibrosis. PLoS Pathog. 5, e1000371 (2009).108. Fujiwara, Y. et al. Phase 1 study of galunisertib, a TGF-beta receptor I kinase inhibitor, in Japanesepatients with advanced solid tumors. Cancer Chemother. Pharmacol. 76, 1143–1152 (2015).109. Herbertz, S. et al. Clinical development of galunisertib (LY2157299 monohydrate), a smallmolecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Devel. Ther.9, 4479–99 (2015).110. Yingling, J. M. et al. Preclinical assessment of galunisertib (LY2157299 monohydrate), a first-in-class transforming growth factor-β receptor type I inhibitor. Oncotarget 9, 6659–6677 (2018).

General Introduction
24

CHAPTER 1
The elusive antifibrotic macrophage
Adhyatmika* | Kurnia S.S. Putri* | Leonie Beljaars | Barbro N. Melgert
*) equal contributionspublished in Frontiers in Medicine, 2015, 2: 81, 1-11

Chapter 1
26
ABSTRACTFibrotic diseases, especially of the liver, the cardiovascular system, the kidneys, andthe lungs, account for approximately 45% of deaths in Western societies. Fibrosis is aserious complication associated with aging and/or chronic inflammation or injury andcannot be treated effectively yet. It is characterized by excessive deposition ofextracellular matrix (ECM) proteins by myofibroblasts and impaired degradation bymacrophages. This ultimately destroys the normal structure of an organ, which leadsto loss of function. Most efforts to develop drugs have focused on inhibiting ECMproduction by myofibroblasts and have not yielded many effective drugs yet. Anotheroption is to stimulate the cells that are responsible for degradation and uptake ofexcess ECM, i.e., antifibrotic macrophages. However, macrophages are plastic cells thathave many faces in fibrosis, including profibrotic behavior-stimulating ECMproduction. This can be dependent on their origin, as the different organs have tissue-resident macrophages with different origins and a various influx of incomingmonocytes in steady-state conditions and during fibrosis. To be able topharmacologically stimulate the right kind of behavior in fibrosis, a thoroughcharacterization of antifibrotic macrophages is necessary, as well as an understandingof the signals they need to degrade ECM. In this review, we will summarize the currentstate of the art regarding the antifibrotic macrophage phenotype and the signals thatstimulate its behavior.Keywords: macrophages, antifibrotic, fibrosis, resolution, monocytes, MMP,cathepsin K, polarization

The elusive antifibrotic macrophage
27
INTRODUCTIONFibrosis is a serious complication associated with aging and with chronic injuryand inflammation within an organ. It is characterized by progressive and irreversibledestruction of normal architecture of an organ by excessive deposition of extracellularmatrix (ECM). The excess ECM ultimately leads to organ malfunction and deathbecause there are no effective therapies to stop or reverse fibrosis development. Amechanistic understanding of how ECM homeostasis is maintained in healthysituations, the similarities and differences between the various organs, and how itbecomes dysregulated in fibrosis is of vital importance for defining novel targets fortherapy. More insight into these processes will help the development of novelantifibrotic drugs.Production of ECM is part of a normal repair response after tissue damage.Tissue repair has distinct stages including a clotting phase, an inflammatory phase, a(myo)fibroblast proliferation phase and a remodeling phase in which normal tissuearchitecture is restored1. During the remodeling phase, myofibroblasts produce ECMand promote tissue contraction, which will ultimately lead to resolution of the damage.Current dogma is that ongoing micro injury within an organ induces an imbalance inECM homeostasis and subsequently leads to fibrosis2,3. In most organs, extracellularmatrix-producing myofibroblasts are found in close proximity with macrophages, andthere is increasing evidence that suggests that normally these two cell types interact inmany ways to control ECM homeostasis and that these interactions may bedysregulated in fibrosis3-6. Myofibroblasts, as the major producers of extracellularmatrix, have been the focus of fibrosis research for many years. Unfortunately, this hasnot yielded many successful drugs yet. Therefore, the role macrophages have incontrolling extracellular matrix production in fibrosis is getting more attentionrecently.Macrophages are important cells in all stages of the fibrotic process7. On the onehand they have been found to promote fibrosis by secreting profibrotic mediators liketransforming growth factor beta (TGFβ) and platelet-derived growth factor (PDGF)that induce proliferation and activation of myofibroblast7-9. On the other hand, theyalso facilitate the resolution of fibrosis by producing specific matrixmetalloproteinases (MMPs) and other proteolytic enzymes like cathepsins that

Chapter 1
28
degrade fibrotic ECM and they express receptors that can phagocytose pieces ofdegraded ECM10. Studies in models of pulmonary and liver fibrosis have shown thatwhen macrophages are depleted during the early inflammatory phase of fibrosis, ECMdeposition was reduced but when they are depleted during the remodeling phase ECMdeposition was aggravated8-11. These studies elegantly showed that the behavior ofmacrophages is highly plastic, but it remains unclear how the pro- and antifibroticactivities of macrophages are regulated. Knowing which signals induce antifibroticbehavior of macrophages is particularly important because restoration of normaltissue architecture can only proceed if the deposited excess ECM is removed. Thesesignals may subsequently be used for the development of a whole new class ofantifibrotic drugs. However, discerning antifibrotic macrophages from othermacrophages is difficult, since characteristic markers are unclear, as are the signalsthat induce antifibrotic macrophages.In this review we will discuss evidence currently present in literature thatenables us to identify antifibrotic macrophages and the signals that are needed toinduce them in order to design macrophage-directed antifibrotic therapeutics. Studiesused for this review were gathered by a systematic search of Pubmed using thekeywords “macrophages” and “fibrosis” and “(resolution OR antifibrotic)”. Only studiesdiscussing pro-or antifibrotic activities of macrophages or phenotypical markers ofthese macrophages were included.MACROPHAGE PLASTICITYMacrophages have many roles in the immune system and are strongly involvedin fighting microbial threats, inflammation, repair and resolution to return tohomeostasis. For years, researchers have tried to define distinct macrophagepolarization states or phenotypes that are responsible for these different tasks12. Theyhave been classified in several different ways, mostly into two main groups with M1macrophages as the classically activated macrophages and M2 macrophages as thealternatively activated macrophages13-14. Broadly speaking M1-activated macrophagesare associated with inflammatory responses and are involved in fighting infections.This phenotype develops after exposure to microbial products, and pro-inflammatorycytokines like tumor necrosis factor alpha (TNFα) and interferon gamma (IFNγ). M2-

The elusive antifibrotic macrophage
29
activated macrophages are more difficult to capture into one phenotype and this hasled to the suggestion to group them into the different subsets M2a, M2b, and M2c15.These subsets are associated with repair processes and resolution of inflammation andare induced by a variety of signals such as interleukin-4/interleukin-13 (IL-4/IL-13)for M2a, immune complexes and lipopolysaccharides (LPS) for M2b and IL-10/TGFβ/glucocorticosteroids for M2c. This classification had its uses for well-controlled in vitroexperiments but could not capture the multitude or spectrum of polarization statespresent in vivo leading to much confusion in the field. This has led to the suggestion toidentify macrophages through their origin, the polarizing substance and/or throughmarkers they do or do not express16.The confusion about macrophage polarization is also apparent in the field offibrosis. The widespread use of the M1/M2 classification has led to the suggestion thatM1 macrophages promote inflammation in the inflammatory stages of wound repairand subsequently polarize to or are being replaced by M2 macrophages that promotefibrosis. However, the complex microenvironment macrophages are exposed to in vivohas many stimuli that induce different functions that cannot be captured in M1 and M2.Furthermore, the M2 phenotype is a complex collection of divergent activities that aresometimes even contradictory. For example, in mice M2 macrophages have beendescribed by their expression of arginase-1 and these macrophages were consideredto be profibrotic. However, Pesce et al. showed, using macrophage-specific arginase-1(Arg-1) knockout mice that these arginase-1 expressing macrophages were actuallyresponsible for suppressing fibrosis development17. This intriguing result shows theplasticity of profibrotic and antifibrotic behavior within the M2 macrophage subset ina complex tissue environment.Other studies have circumvented the M1/M2 dichotomy by namingmacrophages after their roles in inflammation and tissue remodeling: i.e. pro-inflammatory, pro-fibrotic, pro-resolution, resolving or scar-associatedmacrophages4,10,18-20. For the purpose of this review we will be specifically addressingthe macrophages that are associated with areas of existing fibrosis and are responsiblefor clearing away excess extracellular matrix, also known as pro-resolution orantifibrotic macrophages.

Chapter 1
30
MURINE VERSUS HUMAN MACROPHAGESThe discovery of macrophages phenotypes has largely been driven by murinemodels. Translation to human steady-state conditions or diseases is scarce andhampered by the fact that many phenotypical and functional markers are murine-specific and the human counterparts are unknown12,21. For instance, the widely-usedM2 markers Ym1 (chitinase 3–like protein 3) and FIZZ1 (resistin-like molecule alpha1/found in inflammatory zone 1) are only expressed on murine IL-4/IL-13 activatedmacrophages and not in their human counterparts. Though firmly associated withdevelopment of fibrosis in mouse models, how these markers themselves play a role isunclear22-24, making it even more difficult find their human equivalents. Most of theinformation on antifibrotic macrophages will therefore be derived from murinestudies. Whenever possible we will try to make the translation to the human situation.THE ORIGIN OF TISSUE MACROPHAGESMature macrophages in adult tissues can originate from two different sources:either from circulating blood monocytes that infiltrate the tissues after birth or fromembryonic macrophages infiltrating tissues before birth and that self-maintainthroughout life25-32. The distinction between hematopoetic versus embryonic originmay be important because this may determine their functionality33. For instance, liver-resident alternatively activated macrophages were found to be phenotypically andfunctionally distinct from monocyte-derived alternatively activated macrophages. Thefirst were found to be key in suppressing schistosomiasis-induced chronicinflammation, while the latter monocyte-derived ones could slow the progression offibrosis34.Recent experiments have shown that during steady state conditions, in mostorgans, tissue macrophages are of embryonic origin25-32. These embryonicmacrophages can develop from yolk sac macrophages directly or, through erythro-myeloid progenitors in the fetal liver25,30,35,36. In the developing embryo, hematopoiesisbegins in the yolk sac with primitive erythrocytes and macrophages developing in theabsence of hematopoietic stem cells and spreading into developing peripheraltissues37. This primitive hematopoiesis is not sufficient to support the developingembryo until hematopoietic stem cells are functional. Therefore, a second wave of
The elusive antifibrotic macrophage
31
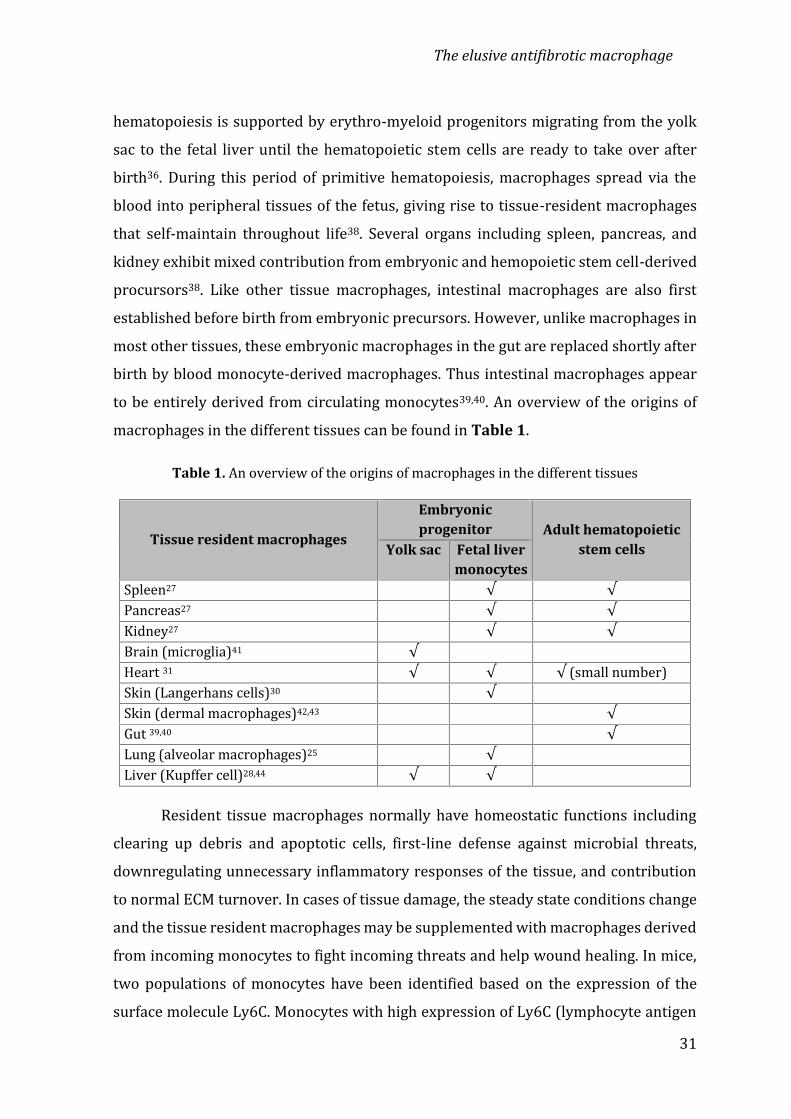
hematopoiesis is supported by erythro-myeloid progenitors migrating from the yolksac to the fetal liver until the hematopoietic stem cells are ready to take over afterbirth36. During this period of primitive hematopoiesis, macrophages spread via theblood into peripheral tissues of the fetus, giving rise to tissue-resident macrophagesthat self-maintain throughout life38. Several organs including spleen, pancreas, andkidney exhibit mixed contribution from embryonic and hemopoietic stem cell-derivedprocursors38. Like other tissue macrophages, intestinal macrophages are also firstestablished before birth from embryonic precursors. However, unlike macrophages inmost other tissues, these embryonic macrophages in the gut are replaced shortly afterbirth by blood monocyte-derived macrophages. Thus intestinal macrophages appearto be entirely derived from circulating monocytes39,40. An overview of the origins ofmacrophages in the different tissues can be found in Table 1.Table 1. An overview of the origins of macrophages in the different tissues
Tissue resident macrophages
Embryonicprogenitor Adult hematopoietic
stem cellsYolk sac Fetal livermonocytesSpleen27 √ √Pancreas27 √ √Kidney27 √ √Brain (microglia)41 √Heart 31 √ √ √ (small number)Skin (Langerhans cells)30 √Skin (dermal macrophages)42,43 √Gut 39,40 √Lung (alveolar macrophages)25 √Liver (Kupffer cell)28,44 √ √Resident tissue macrophages normally have homeostatic functions includingclearing up debris and apoptotic cells, first-line defense against microbial threats,downregulating unnecessary inflammatory responses of the tissue, and contributionto normal ECM turnover. In cases of tissue damage, the steady state conditions changeand the tissue resident macrophages may be supplemented with macrophages derivedfrom incoming monocytes to fight incoming threats and help wound healing. In mice,two populations of monocytes have been identified based on the expression of thesurface molecule Ly6C. Monocytes with high expression of Ly6C (lymphocyte antigen

Chapter 1
32
6C) are generally called classical or inflammatory monocytes and these patrol theextravascular tissues in homeostatic conditions29. During this patrolling function theyremain monocytic and do not commit to being macrophages. During inflammation,however, they respond with rapid extravasion into the affected tissues and they canreadily transform into macrophages with limited potential for migration29. Monocyteswith low expression of Ly6C are called nonclassical monocytes and patrol the bloodvessels to monitor endothelial cell homeostasis45,46. They develop from the Ly6C-hisubset26,47,48 and this can also take place in injured or inflamed tissue with subsequentconversion to wound-healing macrophages that can proliferate locally49,50.In humans, similar monocytes subsets are found based on expression of CD14and CD1651. Classical monocytes express high levels of CD14 and no CD16, whilenonclassical monocytes express high levels of CD16 and low levels of CD14. Both inhumans and mice, an intermediate third subset is suggested to exist characterized inhumans by high levels of CD14 and intermediate levels of CD16. The functions of thissubset are not well understood, although they have been found to preferentiallyaccumulate in inflamed human livers and have been postulated to play a role infibrogenesis52.Unfortunately, there are no reliable markers to distinguish betweenmacrophages from embryonic or hematopoietic/monocytic origin, which makes itdifficult to study the contributions of the two types of macrophages to changes inhomeostatic conditions, especially in humans. In mice, some lineage tracing studieshave been performed with special mouse models in the context of fibrosis to get someinsight into the origin of macrophages in fibrotic tissues and these studies arediscussed below.THE ORIGIN OF MACROPHAGES DURING FIBROSISSeveral papers have investigated the various origins of macrophages in thecontext of fibrosis. There is a clear role for infiltrating Ly6C-hi monocytes in fibrosis.These monocytes have high expression of the CCR2 (C-C motif chemokine receptortype 2) and have been shown to CCR2-dependently infiltrate the kidney, liver, heartand lung after acute injury8,53-56. Less fibrosis is found when this migration is preventedeither by specific depletion of the Ly6C-hi subset or when interfering with CCR2

The elusive antifibrotic macrophage
33
function53,57. In liver and lung it was shown that Ly6C-hi monocytes clearly facilitatethe progression of fibrosis, but without obviously engrafting into the tissue asmacrophages, which may indicate their patrolling behavior of extravascular tissues isnot restricted to steady state conditions8,53.Many models of fibrosis consist of toxic injury (e.g. carbon tetrachloride,bleomycin) with an acute inflammatory phase followed by a fibrotic phase and aresolution phase with a return to fairly normal tissue structure. In these models it wasshown that depletion of macrophages in the resolution phase slowed down the processof resolution8,18,57-62. These restorative macrophages appear to be derived from therecruited Ly6C-hi monocytes that undergo a phenotypic switch to a Ly6C-lophenotype18,57. However, in a study by Baeck et al. inhibiting a transient CCR2-dependent accumulation of Ly6C-hi monocytes in the resolution phase acceleratedscar resolution in two models of hepatic fibrosis62. Therefore, contributions of bothrecruited Ly6C-lo monocytes and tissue-resident macrophages are also likely8,59-61.Corroboration for involvement of Ly6C-lo monocytes comes from a study showing thatdeletion of the fractalkine receptor CX3CR1 (C-X3-C motif chemokine receptor 1),which is highly expressed on Ly6C-lo monocytes, inhibits resolution of hepaticfibrosis60. Gibbons et al. showed that ablation of tissue-resident macrophages in thelung during the resolution phase of bleomycin-induced injury also slowed downresolution8.In conclusion, macrophages of various origins, hematopoietic and embryonic,contribute to fibrosis and its resolution. The evidence available points at antifibroticmacrophages being either derived from CX3CR1-expressing Ly6C-lo monocytesand/or embryonically derived tissue-resident macrophages, while ly6C-hi monocytesappear to be profibrotic. For a summary of the available data also see Table 2.Table 2. Origins of antifibrotic macrophages
OrganAntifibrotic macrophages
Tissue-resident Ly6C-lo recruited monocytePeritoneal √ 63 √ 63Lung √ 8,64,65 √ 59,66Liver (Kupffer cell) √ 60

Chapter 1
34
ANTIFIBROTIC MACROPHAGES:
HOW TO INDENTIFY AND INDUCE OR RECRUIT THEM?Within fibrotic parts of tissues higher numbers of macrophages were shown tobe present as compared to the healthy parts and these were shown to be important forfibrosis resolution61,67,68. One of the main tasks of these antifibrotic macrophages isclearance of fibrotic ECM, in particular of fibrillar types of collagen. Macrophages areimportant sources of various matrix-degrading enzymes and they can take up partiallydegraded collagen fragments6. The expression of these matrix-degrading enzymes andof the receptors for uptake of collagen fragments could therefore potentially bemarkers of antifibrotic macrophages in vivo.Collagen fibers are cleaved extracellularly by proteases such as matrixmetalloproteinases (MMPs) and cathepsins. Intact fibrillar collagen can only be cleavedby a subset of MMPs (MMP1, MMP8, MMP13, MMP14) and by other proteases such ascathepsin K69-71. Subsequently, collagen pieces are further degraded by other membersof the MMP family like MMP2 and MMP96. The main cellular source of matrix-degrading enzymes is macrophages. Huang et al. showed expression of different MMPsin the various macrophage phenotypes in vitro72. Therefore, MMP expression bymacrophages might serve as a functional marker to identify antifibrotic macrophagesin vivo. Scar-associated macrophages were shown to be a source of MMP13 and astrong correlation between the presence of MMP13-positive macrophages andenhanced regression was shown in fibrotic carbon tetrachloride mouse livers68. Notonly MMP13, but also other members of the MMP family (MMP3, MMP8, MMP9,MMP12, MMP14) were identified in scar-associated macrophages and associated withresolution activities in liver73,74. The presence of MMP-expressing macrophages in scartissue was also seen in other fibrotic tissues such as in the lung, kidneys, heart, andspinal cord. Shechter showed MMP13-expressing macrophages in glial scar tissue andrelated this to a resolving macrophage phenotype58. Cabrera showed increased MMP9expression in alveolar macrophages that appear in the regression phase of thebleomycin-induced lung fibrosis75. Also, Popov showed that MMP9, in contrast toMMP12 and MMP13, was particularly induced during resolution and higher expressedthan during fibrogenesis74. Within lung and liver, MMP9 expression is particularly

The elusive antifibrotic macrophage
35
observed in macrophages as can be checked in immunohistochemical stainingsprovided by the human protein atlas76.In addition to MMPs, macrophages also express other ECM-degrading enzymessuch as the cysteine proteases, i.e. cathepsins71. MMPs are traditionally considered tobe the main agents of ECM degradation, but the lysosomal cathepsins, can also besecreted into the extracellular space where they can remain proteolyticaly active anddegrade various components of the extracellular matrix71. Cathepsin K is the onlyprotease with the ability to degrade intact fibrillar collagen, both at the ends of the fibriland at multiple sites within the triple helix. Overexpression of cathepsin K protectedanimals from developing bleomycine or silica-induced pulmonary fibrosis, whiledeleting it accelerated the development of fibrosis66,77,78. These findings all suggesthigh antifibrotic activity of cathepsin K and therefore of macrophages in the lung.Alveolar macrophages in the resolution phase are also reported to produce plasmin, aprotease associated with reducing TGFβ1 levels and thus with reduced stimulation ofcollagen synthesis64.Matrix metalloproteinases can also contribute to other activities, such ascellular migration79 and activation of cytokines and growth factors80,81. Theexpressions and activities of MMPs are therefore not limited to the resolution phase.Certain subtypes are more enhanced during fibrogenesis as compared to resolution,e.g. MMP2 in liver fibrosis74. This might hamper the use of certain MMPs as markersfor antifibrotic macrophages. Based on the current knowledge about the expressionpatterns of matrix-degrading enzymes in macrophages in fibrosis and resolution, inparticular MMP9, MMP13, and cathepsin K seem suitable markers to discernantifibrotic macrophages in vivo from other macrophage phenotypes.In addition to the matrix degrading activities of antifibrotic macrophages,candidate markers of antifibrotic macrophages could also be proteins involved ininduction of proteolytic enzymes and proteins involved in clearance of degraded ECMproteins. After extracellular degradation, further processing of collagen fragmentsoccurs intracellularly, predominantly in the lysosomal compartments of the cell. Tothat end, collagen fragments are internalized via phagocytosis, macropinocytosis orreceptor-mediated endocytosis6.

Chapter 1
36
Phagocytosis for instance is mediated by binding of collagen fragments tocellular membrane integrin α2β1. For receptor-mediated endocytosis binding totransmembrane mannose receptor CD206 or mannose receptor 2 (Mrc2; also calledEndo180) is required6,82-84. Lopez-Guisa showed upregulation of Mrc2 in a subset ofmacrophages at sites of renal fibrosis directing the process of repair. Renal fibrosis wassignificantly worse in Mrc2-deficient mice, which was related to lower collagenturnover. In addition, treatment of wild-type mice with a cathepsin inhibitor, whichblocks the proteases implicated in Mrc2-mediated collagen degradation, worsenedUUO-induced renal fibrosis83.The extracellular bridging glycoprotein Mfge8 (Milk fat globule-EGF factor 8)has also been described to be involved in the cellular uptake of collagenfragments6,65,85. Atabai showed that Mfge8 decreased the severity of tissue fibrosis in amouse model of pulmonary fibrosis by binding and targeting collagen for cellularuptake through its discoidin domains85. Reddy et al. showed that nitrated fatty acidsregulated the expression of Mfge8 in alveolar macrophages and thus stimulatedcollagen uptake and its further degradation65. The usefulness of these receptors,involved in the cellular uptake of collagen, in identifying antifibrotic macrophages hasnot been investigated in great detail and will require more studies.Other proteins expressed by macrophages that have been shown to contributeto the antifibrotic phenotype of macrophages are arginase-117 and FIZZ122. Both wereshown to limit Th2-dependent responses that are required for the development offibrosis.As is clear from the previous sections, production of matrix-degrading enzymesis one of the key characteristics of antifibrotic macrophages. Therefore, to induce thistype of macrophage, it will be helpful to understand the signals involved in attractingthese macrophages to the fibrotic areas and/or the signals that induce the expressionof matrix-degrading enzymes and collagen uptake receptors. These could be cytokineslike TNFα, IL-1β, IFNα/β and IL-4, growth factors, chemokines or even processes58,81,86-88. Popov et al. showed that the enhanced proteolytic activity of macrophages wasinduced after phagocytosis of apoptotic cholangiocytes that were increasingly presentin the resolution phase of biliary fibrosis74. The receptor involved in this phagocytosis-

The elusive antifibrotic macrophage
37
induced proteolytic activity was most probably the Tyrosine-protein Kinase Merreceptor (MERTK), which is highly expressed on macrophages74,89,90. Gene variants ofMERTK have been shown to be risk factors for progression of hepatitis C-induced liverfibrosis91,92. Through no functional data of the gene variants of MERTK were shown,making it hard to interpret this data. Similar phagocytosis-induced proteolytic activitywas reported in the lung, in which apoptotic cell instillation induced peroxisomeproliferator-activated receptor-γ (PPARγ) expression in macrophages andsubsequently stimulated resolution of bleomycin-induced fibrosis93. Whether MERTKand PPARγ are useful markers for antifibrotic macrophages needs to be investigated infurther detail. PPARγ seems to be a promising candidate as agonists of PPARγ havebeen investigated as a possible antifibrotic therapy in multiple settings65,94-100.Some of the cytokines or their receptors that induce antifibrotic behavior areexpressed by macrophages themselves, therefore these cytokines or their receptorscould potentially also be markers of antifibrotic macrophages. However, theirubiquitous expression by various other cells may hamper their use in vivo.Tumor necrosis factor alpha receptor (TNFαR) or the production of TNFα maybe potential inducers and/or markers of antifibrotic macrophages, though thisdepends on the stage of the disease limiting their use. Macrophages are importantproducers of TNFα and thereby contribute to inflammation after injury. InhibitingTNFα at this point has been shown to lead to less fibrosis in models of kidney, liver,heart, and lung fibrosis101-105. However, TNFα has also been shown to have antifibroticactivities, especially in the resolution stage of fibrosis. Recent research showed thatintratracheal delivery of TNFα reduced lung collagen levels and improved lungarchitecture. In addition, mice deficient in TNFα exhibited delayed resolution ofbleomycin-induced pulmonary fibrosis, further showing that TNFα may be importantin the resolution phase of fibrosis by inducing antifibrotic macrophages106. A study inpatients with pulmonary fibrosis showed that release of TNFα by macrophages andmonocytes of these patients was higher than of controls, which may be a sign that thelung is trying to degrade excess collagen or a sign that inflammation is still importantin patients diagnosed with pulmonary fibrosis107. The fact that anti-inflammatorydrugs like corticosteroids are harmful to pulmonary fibrosis patients indicates thatTNFα is probably involved in attempted resolution108. Production of TNFα by

Chapter 1
38
antifibrotic macrophages may have an effect on macrophages themselves throughTNFα type 1 and/or 2 receptors or affect other cells. Both in the heart and in the kidneyTNFα type 2 receptor expression on macrophages was found to essential foraccelerating fibrosis resolution109,110. A recent publication by Lemos et al. showed thatthe effect of TNFα in muscle fibrosis was through induction of apoptosis ofmyofibroblast progenitors111.Treatment of liver macrophages with Interferon-a2b induced a higher MMP13expression and these macrophages also showed a higher expression of IL-1088. Similarfindings were reported in glial scars by Shechter et al.58. The effect of IL-10 on fibrosis,however, is not clear since increased levels of IL-10 were accompanied by reducedfibrosis in one study73, while other studies have reported that IL-10 actsprofibrotic112,113.Cytokines and chemokines that are involved in recruitment antifibroticmacrophages are macrophage migration inhibitory factor (MIF), CX3C ligand 1(fractalkine), and vascular endothelial growth factor (VEGF)60,61,114. CD74, CXCR2, andCXCR4 are receptors for MIF and their expressions appear to be associated withrecruitment of resolving macrophages61,115,116. This also is the case for chemokinereceptor CX3CR1 and this receptor may also be helpful in the detection of antifibroticmacrophages60. Another chemokine involved in the recruitment of resolution-promoting monocytes appears to be VEGF. Treatment with a neutralizing antibodyagainst VEGF during fibrosis resolution delayed resolution and this was shown to bedependent on CXCL9 and MMP13114. In addition, enhanced expression of CXCL10 inmacrophages has been shown to accelerate resolution of pulmonary fibrosis59,117,118.Interestingly, in the study by Tighe et al., IFNγ was found to be able to stimulateproduction of CXCL10 in macrophages and this may therefore contribute to the knownantifibrotic effects of IFNγ59,119.In conclusion, various studies indicate the existence of antifibrotic macrophagesthat play a key role in resolving fibrotic ECM and therefore these macrophages may bea target for therapeutic intervention. Identification of this subset in vivo is not easy, butvarious options can be explored. One of most obvious is the expression of matrix-degrading enzymes in macrophages, in particular MMP9, MMP13 and cathepsin K.Other options include the chemokines CXCL10 and CXCL9, chemokine receptor

The elusive antifibrotic macrophage
39
CX3CR1, M2-markers arginase-1, FIZZ1, and PPARγ, collagen-uptake receptors MRC1,MRC2, and Mfge8, and cytokines like TNFα (see also Table 3). However, most of theseproteins are not specific to macrophages and even the different phenotypes ofmacrophages in the pro-inflammatory/fibrotic phase and in the resolution phase seemto use them.Table 3. Markers of antifibrotic macrophages and potential therapeutic approaches inducingor attracting antifibrotic macrophages or inhibiting the recruitment of profibrotic monocytes.
Markers Prospective DrugTNF receptor 109,110CX3CR160TNFα106,107,111CXCL1059,117,118CXCL9117,118MMP973-75MMP1368,118Cathepsin K66,71,77,78MERTK89,90PPARγ93-100MRC1120MRC2121Mfge86,65,85Arg-117FIZZ122
TNFα106RANKL122-124PPARγ agonist93-100IFNγ59,125IFNα58,88Asprin-triggered lipoxin A analogues126CCL2 inhibitors62
Induction or recruitment of antifibrotic macrophages is even less well defined.Monocytes that turn into antifibrotic macrophages appear to be recruited by CX3Cligand 1 or VEGF. Cytokines that can induce antifibrotic behaviour of macrophages inwell-defined circumstances are TNFα, IFNα or IFNγ.FROM CONCEPT TO MARKET: THERAPEUTIC APPLICATION AND CHALLENGESAs antifibrotic macrophages can be crucial in the resolution of fibrosis invarious organs, they constitute a valid novel target for therapeutic intervention.Therefore, understanding of how to specifically induce their beneficial activities maylead to a generation of new antifibrotic compounds.In addition to the aforementioned TNFα, IFNγ, and IFNα, only a few potentialtherapeutic compounds affecting antifibrotic macrophages have been described in

Chapter 1
40
literature. One of few examples is the use of PPARγ agonists that can induce antifibroticproperties in macrophages. Experimental studies in kidney, liver, heart and lung haveshown that various PPARγ agonists can alleviate fibrosis, though not all haveinvestigated macrophages specifically65,94-100. There is even phase 1 safety study inclinicaltrials.gov describing the use of PPARγ -agonist rosiglitazone for the treatmentof focal glomerulosclerosis. This study ended in 2007 but no results have been postedyet. A currently unexplored option is the possible use of receptor activator ofnuclear factor-κB ligand (RANKL). Many tissue macrophages express the receptorRANK for this ligand and there are several studies showing that RANKL stimulationinduces the release of proteases, which can degrade ECM76. Wittrant et al. showed thatRANKL stimulated MMP9 and cathepsin K expression122 and Matsumoto et al. alsoshowed that RANKL induced cathepsin K gene expression123. Another study showedthat RANKL, through binding to RANK, activated the nuclear factor-κB pathway andinduced MMP9 expression. They also suggested that by costimulating with IL-1β orTNFα it was possible to synergize with RANKL to further enhance MMP9 expression124.We are currently investigating whether RANKL can indeed induce antifibroticmacrophages in settings of established fibrosis.Another option described was the use of a Spiegelmer-based inhibitor of CCL2,named mNOX-E36, that was found to inhibit recruitment of Ly6C-hi monocytes andthereby accelerated resolution of liver fibrosis62. The last of the few examples was asynthetic analog of asprin-triggered lipoxin A4. Lipoxins have potent proresolutioneffects and this synthetic analog called ATLa reversed collagen deposition by inducingarginase-1-positive macrophages in a bleomycin model of pulmonary fibrosis126. Asummary of the origin and all characteristics of antifibrotic macrophages is depicted inFigure 1.

The elusive antifibrotic macrophage
41
Figure 1. Antifibrotic macrophages, derived from either embryonic tissue macrophages and/or Ly6C-lomonocytes, contribute to fibrosis resolution by expressing extracellular matrix (ECM)-degradingenzymes and receptors to take up pieces of degraded ECM and by expression of proteins thatdownregulate Th2-associated inflammation. These antifibrotic macrophages can be induced or attractedby a number of signals such as cytokines, chemokines and growth factors.Abbreviations: MIF: Macrophages migration inhibitory factor; CX3CR ligand 1: ligand for C-X3-C motifchemokine receptor 1; VEGF: vascular endothelial growth factor; CXCL-9 and -10: C-X-C motifchemokine ligand -9 and -10; RANKL: receptor activator of nuclear factor-κB ligand; TNFα: tumornecrosis factor α; TNFαR1/2: tumor necrosis factor receptor type 1 or 2; IFNγ: interferon γ; IFNγ:interferon γ; MMP9 and MMP13: matrix metalloproteinase 9 and 13; Mfge8: Milk fat globule-EGF factor8; MERTK: Tyrosine-protein Kinase Mer receptor; Mrc1 and Mrc2: mannose receptor 1 and 2; PPARγ:Peroxisome proliferator-activated receptor-γ; Arg-1: Arginase-1; FIZZ1: resistin-like molecule alpha 1;Th2: T helper 2 lymphocyted-mediated.

Chapter 1
42
One factor worth considering is the translation of these results in rodents to thehuman situation. As said before, an obstacle in this translation is that most knowledgeso far is obtained with mouse models and the markers and effector molecules ofantifibrotic macrophages in humans are largely unexplored127.In addition, several fibrosis-inducing agents such as carbon tetrachloride,bleomycine, silica, or nutritional interventions are highly effective in establishingadvanced fibrosis in mice, but they do not represent key elements of human diseasecompletely.CONCLUSIONSThe flurry in new studies investigating antifibrotic behavior of macrophages inrecent years has made the elusive antifibrotic macrophage slightly more tangible. Thissubset of macrophages appears to be derived from embryonic tissue-residentmacrophage or recruited Ly6C-lo monocytes and expresses a variety of markerstraditionally assigned to both M1 and M2 macrophages, including: MMP9, MMP13,cathepsin K, CXCL10, CXCL9, CX3CR1, arginase-1, FIZZ1, PPARγ, MRC1, MRC2, andMfge8, and TNFα. Although therapy aimed at the antifibrotic macrophage is still in itsinfancy, it is expected that more targets for therapeutic entities will appear whenantifibrotic macrophages are better understood.REFERENCES
1. Wynn T. Cellular and molecular mechanisms of fibrosis. J Pathol (2008) 214:199–210.doi:10.1002/path.22772. Noble PW, Homer RJ. Back to the future: historical perspective on the pathogenesis of idiopathicpulmonary fibrosis. Am J Respir Cell Mol Biol (2005) 33:113–20. doi:10.1165/rcmb.F3013. Wynn T, Ramalingam T. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. NatMed (2012) 18:1028–40. doi:10.1038/nm.28074. Pellicoro A, Ramachandran P, Iredale J, Fallowfield J. Liver fibrosis and repair: immune regulationof wound healing in a solid organ. Nat Rev Immunol (2014) 14:181–94. doi:10.1038/nri36235. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci (2014)71:549–74. doi:10.1007/s00018-013-1349-66. McKleroy W, Lee T-H, Atabai K. Always cleave up your mess: targeting collagen degradation to treattissue fibrosis. Am J Physiol Lung Cell Mol Physiol (2013) 304:L709–21.doi:10.1152/ajplung.00418.2012

The elusive antifibrotic macrophage
43
7. Barron L, Wynn TA. Fibrosis is regulated by Th2 and Th17 responses and by dynamic interactionsbetween fibroblasts and macrophages. Am J Physiol Gastrointest Liver Physiol (2011) 300:G723–8. doi:10.1152/ajpgi.00414.20108. Gibbons MA, MacKinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, et al.Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis.Am J Respir Crit Care Med (2011) 184:569–81. doi:10.1164/rccm.201010-1719oc9. Song E, Ouyang N, Hörbelt M, Antus B, Wang M, Exton MS. Influence of alternatively and classicallyactivated macrophages on fibrogenic activities of human fibroblasts. Cell Immunol (2000) 204:19–28. doi:10.1006/cimm.2000.168710. Boorsma CE, Draijer C, Melgert BN. Macrophage heterogeneity in respiratory diseases. MediatorsInflamm (2013) 2013:769214. doi:10.1155/2013/76921411. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, VuthooriS, et al. Selective depletion ofmacrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest (2005)115:56–65. doi:10.1172/ JCI2267512. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol (2005) 5:953–64.doi:10.1038/nri173313. Mills CD. Anatomy of a discovery: m1 and m2 macrophages. Front Immunol (2015) 6:212.doi:10.3389/fimmu.2015.0021214. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment.F1000Prime Rep (2014) 6:13. doi:10.12703/P6-1315. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci(2008) 13:453–61. doi:10.2741/269216. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation andpolarization: nomenclature and experimental guidelines. Immunity (2014) 41:14–20.doi:10.1016/j.immuni.2014.06.00817. Pesce JT, Ramalingam TR, Mentink-Kane MM, Wilson MS, El Kasmi KC, Smith AM, et al. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog(2009) 5:e1000371. doi:10.1371/journal.ppat.100037118. Ramachandran P, Pellicoro A, Vernon M, Boulter L, Aucott R, Ali A, et al. Differential Ly-6C expressionidentifies the recruited macrophage phenotype, which orchestrates the regression of murine liverfibrosis. Proc Natl Acad Sci U S A (2012) 109:E3186–95. doi:10.1073/pnas.111996410919. Wynn T, Chawla A, Pollard J. Macrophage biology in development, homeostasis and disease. Nature(2013) 496:445–55. doi:10.1038/nature1203420. Anders H-J, Ryu M. Renal microenvironments and macrophage phenotypes determine progressionor resolution of renal inflammation and fibrosis. Kidney Int (2011) 80:915–25.doi:10.1038/ki.2011.21721. Wermuth PJ, Jimenez SA. The significance of macrophage polarization subtypes for animal modelsof tissue fibrosis and human fibrotic diseases. Clin Transl Med (2015) 4:2. doi:10.1186/s40169-015-0047-422. Pesce JT, Ramalingam TR, Wilson MS, Mentink-Kane MM, Thompson RW, Cheever AW, et al. Retnla(relmalpha/fizz1) suppresses helminth-induced Th2-type immunity. PLoS Pathog (2009)5:e1000393. doi:10.1371/journal. ppat.100039323. Madala SK, Edukulla R, Davis KR, Schmidt S, Davidson C, Kitzmiller JA, et al. Resistin-like moleculeα1 (Fizz1) recruits lung dendritic cells without causing pulmonary fibrosis. Respir Res (2012)13:51. doi:10.1186/1465-9921-13-5124. Liu T, Yu H, Ullenbruch M, Jin H, Ito T, Wu Z, et al. The in vivo fibrotic role of FIZZ1 in pulmonaryfibrosis. PLoS One (2014) 9:e88362. doi:10.1371/ journal.pone.008836225. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages developfrom fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. JExp Med (2013) 210:1977–92. doi:10.1084/jem.2013119926. Yona S, Kim K-W, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins anddynamics of monocytes and tissue macrophages under homeostasis. Immunity (2013) 38:79–91.doi:10.1016/j.immuni.2012.12.001

Chapter 1
44
27. Hashimoto D, Chow A, Noizat C, Teo P, Beasley M, Leboeuf M, et al. Tissueresident macrophagesself-maintain locally throughout adult life with minimal contribution from circulating monocytes.Immunity (2013) 38:792–804. doi:10.1016/j.immuni.2013.04.00428. Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage ofmyeloid cells independent of Myb and hematopoietic stem cells. Science (2012) 336:86–90.doi:10.1126/science.121917929. Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, et al. Minimal differentiationof classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes.Immunity (2013) 39:599–610. doi:10.1016/j.immuni.2013.08.00730. Hoeffel G, Wang Y, Greter M, See P, Teo P, Malleret B, et al. Adult Langerhans cells derivepredominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derivedmacrophages. J Exp Med (2012) 209:1167–81. doi:10.1084/jem.2012034031. Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady stateand during inflammation. Immunity (2014) 40:91–104. doi:10.1016/j.immuni.2013.11.01932. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity (2014)41:21–35. doi:10.1016/j.immuni.2014.06.01333. Gundra UM, Girgis NM, Ruckerl D, Jenkins S, Ward LN, Kurtz ZD, et al. Alternatively activatedmacrophages derived from monocytes and tissue macrophages are phenotypically and functionallydistinct. Blood (2014) 123:e110–22. doi:10.1182/blood-2013-08-52061934. Vannella KM, Barron L, Borthwick LA, Kindrachuk KN, Narasimhan PB, Hart KM, et al. Incompletedeletion of IL-4Rα by LysM(Cre) reveals distinct subsets of M2 macrophages controllinginflammation and fibrosis in chronic schistosomiasis. PLoS Pathog (2014) 10:e1004372.doi:10.1371/journal.ppat.100437235. Hoeffel G, Chen J, Lavin Y, Low D, Almeida F, See P, et al. C-Myb+ erythro-myeloid progenitor-derivedfetal monocytes give rise to adult tissue-resident macrophages. Immunity (2015) 42(4):665–78.doi:10.1016/j.immuni.2015.03.01136. McGrath KE, Frame JM, Fegan KH, Bowen JR, Conway SJ, Catherman SC, et al. Distinct sources ofhematopoietic progenitors emerge before HSCs and provide functional blood cells in themammalian embryo. Cell Rep (2015) 11:1892–904. doi:10.1016/j.celrep.2015.05.03637. Palis J. Primitive and definitive erythropoiesis in mammals. Front Physiol (2014) 5:3.doi:10.3389/fphys.2014.0000338. Hashimoto D, Miller J, Merad M. Dendritic cell and macrophage heterogeneity in vivo. Immunity(2011) 35(3):323–35. doi:10.1016/j.immuni.2011.09.00739. Bogunovic M, Ginhoux F, Helft J, Shang L, Hashimoto D, Greter M, et al. Origin of the lamina propriadendritic cell network. Immunity (2009) 31:513–25. doi:10.1016/j.immuni.2009.08.01040. Gross M, Salame T-M, Jung S. Guardians of the gut – murine intestinal macrophages and dendriticcells. Front Immunol (2015) 6:254. doi:10.3389/fimmu.2015.0025441. Ginhoux F, Lim S, Hoeffel G, Low D, Huber T. Origin and differentiation of microglia. Front CellNeurosci (2013) 7:45. doi:10.3389/fncel.2013.0004542. Haniffa M, Ginhoux F, Wang X-N, Bigley V, Abel M, Dimmick I, et al. Differential rates of replacementof human dermal dendritic cells and macrophages during hematopoietic stem cell transplantation.J Exp Med (2009) 206:371–85. doi:10.1084/jem.2008163343. Tamoutounour S, Guilliams M, Montanana Sanchis F, Liu H, Terhorst D, Malosse C, et al. Origins andfunctional specialization of macrophages and of conventional and monocyte-derived dendritic cellsin mouse skin. Immunity (2013) 39:925–38. doi:10.1016/j.immuni.2013.10.00444. Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophagesoriginate from yolk-sac-derived erythro-myeloid progenitors. Nature (2015) 518:547–51.doi:10.1038/nature1398945. Carlin LM, Stamatiades EG, Auffray C, Hanna RN, Glover L, Vizcay-Barrena G, et al. Nr4a1-dependentLy6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell (2013)153:362–75. doi:10.1016/j.cell.2013.03.010

The elusive antifibrotic macrophage
45
46. Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, et al. Monitoring of blood vessels andtissues by a population of monocytes with patrolling behavior. Science (2007) 317:666–70.doi:10.1126/science.114288347. Hettinger J, Richards DM, Hansson J, Barra MM, Joschko A-C, Krijgsveld J, et al. Origin of monocytesand macrophages in a committed progenitor. Nat Immunol (2013) 14:821–30.doi:10.1038/ni.263848. Hanna RN, Carlin LM, Hubbeling HG, Nackiewicz D, Green AM, Punt JA, et al. The transcription factorNR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C- monocytes. NatImmunol (2011) 12:778–85. doi:10.1038/ni.206349. Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, et al. Inflammatory monocytesrecruited after skeletal muscle injury switch into antiinflammatory macrophages to supportmyogenesis. J Exp Med (2007) 204:1057–69. doi:10.1084/jem.2007007550. Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG, et al. Ly-6Chighmonocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarctedmyocardium. Circ Res (2014) 114:1611–22. doi:10.1161/CIRCRESAHA.114.30320451. Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, et al. Comparisonof gene expression profiles between human and mouse monocyte subsets. Blood (2010) 115:e10–9. doi:10.1182/blood-2009-07-23502852. Liaskou E, Zimmermann HW, Li K-K, Oo YH, Suresh S, Stamataki Z, et al. Monocyte subsets in humanliver disease show distinct phenotypic and functional characteristics. Hepatology (2013) 57:385–98. doi:10.1002/hep.2601653. Karlmark KR, Weiskirchen R, Zimmermann HW, Gassler N, Ginhoux F, Weber C, et al. Hepaticrecruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis.Hepatology (2009) 50:261–74. doi:10.1002/hep.2295054. Lin SL, Castaño AP, Nowlin BT, Lupher ML, Duffield JS. Bone marrow Ly6Chigh monocytes areselectively recruited to injured kidney and differentiate into functionally distinct populations. JImmunol (2009) 183:6733–43. doi:10.4049/jimmunol.090147355. Falkenham A, de Antueno R, Rosin N, Betsch D, Lee TD, Duncan R, et al. Nonclassical residentmacrophages are important determinants in the development of myocardial fibrosis. Am J Pathol(2015) 185:927–42. doi:10.1016/j.ajpath.2014.11.02756. Ji W-J, Ma Y-Q, Zhou X, Zhang Y-D, Lu R-Y, Sun H-Y, et al. Temporal and spatial characterization ofmononuclear phagocytes in circulating, lung alveolar and interstitial compartments in a mousemodel of bleomycin-induced pulmonary injury. J Immunol Methods (2014) 403:7–16.doi:10.1016/j.jim.2013.11.01257. Mitchell C, Couton D, Couty J-P, Anson M, Crain A-M, Bizet V, et al. Dual role of CCR2 in theconstitution and the resolution of liver fibrosis in mice. Am J Pathol (2009) 174:1766–75.doi:10.2353/ajpath.2009.08063258. Shechter R, Raposo C, London A, Sagi I, Schwartz M. The glial scar-monocyte interplay: a pivotalresolution phase in spinal cord repair. PLoS One (2011) 6:e27969.doi:10.1371/journal.pone.002796959. Tighe RM, Liang J, Liu N, Jung Y, Jiang D, Gunn MD, et al. Recruited exudative macrophages selectivelyproduce CXCL10 after noninfectious lung injury. Am J Respir Cell Mol Biol (2011) 45:781–8.doi:10.1165/rcmb.2010-0471OC60. Karlmark KR, Zimmermann HW, Roderburg C, Gassler N, Wasmuth HE, Luedde T, et al. Thefractalkine receptor CX-CR1 protects against liver fibrosis by controlling differentiation andsurvival of infiltrating hepatic monocytes. Hepatology (2010) 52:1769–82. doi:10.1002/hep.2389461. Barnes MA, McMullen MR, Roychowdhury S, Madhun NZ, Niese K, Olman MA, et al. Macrophagemigration inhibitory factor is required for recruitment of scar-associated macrophages during liverfibrosis. J Leukoc Biol (2015) 97:161–9. doi:10.1189/jlb.3A0614-280R62. Baeck C, Wei X, Bartneck M, Fech V, Heymann F, Gassler N, et al. Pharmacological inhibition of thechemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liverfibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice. Hepatology (2014)59:1060–72. doi:10.1002/hep.26783

Chapter 1
46
63. Stout R, Jiang C, Matta B, Tietzel I, Watkins S, Suttles J. Macrophages sequentially change theirfunctional phenotype in response to changes in microenvironmental influences. J Immunol (2005)175:342–9. doi:10.4049/jimmunol.175.1.34264. Khalil N, Corne S, Whitman C, Yacyshyn H. Plasmin regulates the activation of cell-associated latentTGF-beta 1 secreted by rat alveolar macrophages after in vivo bleomycin injury. Am J Respir CellMol Biol (1996) 15:252–9. doi:10.1165/ajrcmb.15.2.870348265. Reddy AT, Lakshmi SP, Zhang Y, Reddy RC. Nitrated fatty acids reverse pulmonary fibrosis bydedifferentiating myofibroblasts and promoting collagen uptake by alveolar macrophages. FASEBJ (2014) 28:5299–310. doi:10.1096/fj.14-25626366. Srivastava M, Steinwede K, Kiviranta R, Morko J, Hoymann H-G, Länger F, et al. Overexpression ofcathepsin K in mice decreases collagen deposition and lung resistance in response to bleomycin-induced pulmonary fibrosis. Respir Res (2008) 9:54. doi:10.1186/1465-9921-9-5467. Beljaars L, Schippers M, Reker-Smit C, Martinez FO, Helming L, Poelstra K, et al. Hepatic localizationof macrophage phenotypes during fibrogenesis and resolution of fibrosis in mice and humans.Front Immunol (2014) 5:430. doi:10.3389/fimmu.2014.0043068. Fallowfield JA, Mizuno M, Kendall TJ, Constandinou CM, Benyon RC, Duffield JS, et al. Scar-associatedmacrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate theresolution of murine hepatic fibrosis. J Immunol (2007) 178:5288–95.doi:10.4049/jimmunol.178.8.528869. Borel O, Gineyts E, Bertholon C, Garnero P. Cathepsin K preferentially solubilizes matured bonematrix. Calcif Tissue Int (2012) 91:32–9. doi:10.1007/s00223-012-9604-770. Costa AG, Cusano NE, Silva BC, Cremers S, Bilezikian JP. Cathepsin K: its skeletal actions and role asa therapeutic target in osteoporosis. Nat Rev Rheumatol (2011) 7:447–56.doi:10.1038/nrrheum.2011.7771. Fonović M, Turk B. Cysteine cathepsins and extracellular matrix degradation. Biochim Biophys Acta(2014) 1840:2560–70. doi:10.1016/j.bbagen.2014.03.01772. Huang W-C, Sala-Newby GB, Susana A, Johnson JL, Newby AC. Classical macrophage activation up-regulates several matrix metalloproteinases through mitogen activated protein kinases andnuclear factor-κB. PLoS One (2012) 7:e42507. doi:10.1371/journal.pone.004250773. Thomas JA, Pope C, Wojtacha D, Robson AJ, Gordon-Walker TT, Hartland S, et al. Macrophage therapyfor murine liver fibrosis recruits host effector cells improving fibrosis, regeneration, and function.Hepatology (2011) 53:2003–15. doi:10.1002/hep.2431574. Popov Y, Sverdlov DY, Bhaskar KR, Sharma AK, Millonig G, Patsenker E, et al. Macrophage-mediatedphagocytosis of apoptotic cholangiocytes contributes to reversal of experimental biliary fibrosis.Am J Physiol Gastrointest Liver Physiol (2010) 298:G323–34. doi:10.1152/ajpgi.00394.200975. Cabrera S, Gaxiola M, Arreola JL, Ramírez R, Jara P, D’Armiento J, et al. Overexpression of MMP9 inmacrophages attenuates pulmonary fibrosis induced by bleomycin. Int J Biochem Cell Biol (2007)39:2324–38. doi:10.1016/j.biocel.2007.06.02276. Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science (2015) 347:1260419. doi:10.1126/science.126041977. Van den Brûle S, Misson P, Bühling F, Lison D, Huaux F. Overexpression of cathepsin K during silica-induced lung fibrosis and control by TGF-beta. Respir Res (2005) 6:84. doi:10.1186/1465-9921-6-8478. Bühling F, Röcken C, Brasch F, Hartig R, Yasuda Y, Saftig P, et al. Pivotal role of cathepsin K in lungfibrosis. Am J Pathol (2004) 164:2203–16. doi:10.1016/ S0002-9440(10)63777-779. Gong Y, Hart E, Shchurin A, Hoover-Plow J. Inflammatory macrophage migration requires MMP-9activation by plasminogen in mice. J Clin Invest (2008) 118:3012–24. doi:10.1172/JCI3275080. Rohani MG, Parks WC. Matrix remodeling by MMPs during wound repair. Matrix Biol (2015) 44-46C:113–21. doi:10.1016/j.matbio.2015.03.00281. Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell DevBiol (2001) 17:463–516. doi:10.1146/annurev.cellbio.17.1.463

The elusive antifibrotic macrophage
47
82. Engelholm LH, List K, Netzel-Arnett S, Cukierman E, Mitola DJ, Aaronson H, et al. uPARAP/Endo180is essential for cellular uptake of collagen and promotes fibroblast collagen adhesion. J Cell Biol(2003) 160:1009–15. doi:10.1083/jcb.20021109183. López-Guisa JM, Cai X, Collins SJ, Yamaguchi I, Okamura DM, Bugge TH, et al. Mannose receptor 2attenuates renal fibrosis. J Am Soc Nephrol (2012) 23:236–51. doi:10.1681/ASN.201103031084. Boskovic J, Arnold JN, Stilion R, Gordon S, Sim RB, Rivera-Calzada A, et al. Structural model for themannose receptor family uncovered by electron microscopy of Endo180 and the mannose receptor.J Biol Chem (2006) 281:8780–7. doi:10.1074/jbc.M51327720085. Atabai K, Jame S, Azhar N, Kuo A, Lam M, McKleroy W, et al. Mfge8 diminishes the severity of tissuefibrosis in mice by binding and targeting collagen for uptake by macrophages. J Clin Invest (2009)119:3713–22. doi:10.1172/JCI4005386. Creasy BM, McCoy KL. Cytokines regulate cysteine cathepsins during TLR responses. Cell Immunol(2011) 267:56–66. doi:10.1016/j.cellimm.2010.11.00487. Balce DR, Li B, Allan ER, Rybicka JM, Krohn RM, Yates RM. Alternative activation of macrophages byIL-4 enhances the proteolytic capacity of their phagosomes through synergistic mechanisms. Blood(2011) 118:4199–208. doi:10.1182/blood-2011-01-32890688. Yu Z, Xie M, Fan X, Jia J. Interferon α2b increases MMP-13 and IL-10 expression in Kupffer cellsthrough MAPK signaling pathways. Hepatogastroenterology (2015) 62:350–4.89. Sather S, Kenyon KD, Lefkowitz JB, Liang X, Varnum BC, Henson PM, et al. A soluble form of the Merreceptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation.Blood (2007) 109:1026–33. doi:10.1182/blood-2006-05-02163490. Zagórska A, Través PG, Lew ED, Dransfield I, Lemke G. Diversification of TAM receptor tyrosinekinase function. Nat Immunol (2014) 15:920–8. doi:10.1038/ni.298691. Rüeger S, Bochud P-Y, Dufour J-F, Müllhaupt B, Semela D, Heim MH, et al. Impact of common riskfactors of fibrosis progression in chronic hepatitis C. Gut (2014) 64(10):1605–15.doi:10.1136/gutjnl-2014-30699792. Patin E, Kutalik Z, Guergnon J, Bibert S, Nalpas B, Jouanguy E, et al. Genomewide association studyidentifies variants associated with progression of liver fibrosis from HCV infection.Gastroenterology (2012) 143:1244–52.e1_12. doi:10.1053/j.gastro.2012.07.09793. Yoon Y-S, Kim S-Y, Kim M-J, Lim J-H, Cho M-S, Kang JL. PPARγ activation following apoptotic cellinstillation promotes resolution of lung inflammation and fibrosis via regulation of efferocytosisand proresolving cytokines. Mucosal Immunol (2015) 8(5):1031–46. doi:10.1038/mi.2014.13094. Kawai T, Masaki T, Doi S, Arakawa T, Yokoyama Y, Doi T, et al. PPAR-gamma agonist attenuatesrenal interstitial fibrosis and inflammation through reduction of TGF-beta. Lab Invest (2009)89:47–58. doi:10.1038/labinvest.2008.10495. Uto H, Nakanishi C, Ido A, Hasuike S, Kusumoto K, Abe H, et al. The peroxisome proliferator-activatedreceptor-gamma agonist, pioglitazone, inhibits fat accumulation and fibrosis in the livers of rats feda choline-deficient, l-amino acid-defined diet. Hepatol Res (2005) 32:235–42. doi:10.1016/j.hepres.2005.05.00896. Ihm S-H, Chang K, Kim H-Y, Baek SH, Youn H-J, Seung K-B, et al. Peroxisome proliferator-activatedreceptor-gamma activation attenuates cardiac fibrosis in type 2 diabetic rats: the effect ofrosiglitazone on myocardial expression of receptor for advanced glycation end products and ofconnective tissue growth factor. Basic Res Cardiol (2010) 105:399–407. doi:10.1007/ s00395-009-0071-x97. Higashi K, Oda T, Kushiyama T, Hyodo T, Yamada M, Suzuki S, et al. Additive antifibrotic effects ofpioglitazone and candesartan on experimental renal fibrosis in mice. Nephrology (Carlton) (2010)15:327–35. doi:10.1111/j.1440-1797.2009.01253.x98. Yang L, Stimpson SA, Chen L, Wallace Harrington W, Rockey DC. Effectiveness of the PPARγ agonist,GW570, in liver fibrosis. Inflamm Res (2010) 59:1061–71. doi:10.1007/s00011-010-0226-099. Wei J, Zhu H, Komura K, Lord G, Tomcik M, Wang W, et al. A synthetic PPAR-γ agonist triterpenoidameliorates experimental fibrosis: PPAR- γ-independent suppression of fibrotic responses. AnnRheum Dis (2014) 73:446–54. doi:10.1136/annrheumdis-2012-202716

Chapter 1
48
100. Attia YM, Elalkamy EF, Hammam OA, Mahmoud SS, El-Khatib AS. Telmisartan, an AT1 receptorblocker and a PPAR gamma activator, alleviates liver fibrosis induced experimentally bySchistosoma mansoni infection. Parasit Vectors (2013) 6:199. doi:10.1186/1756-3305-6-199101. Piguet PF, Vesin C. Treatment by human recombinant soluble TNF receptor of pulmonary fibrosisinduced by bleomycin or silica in mice. Eur Respir J (1994) 7:515–8.doi:10.1183/09031936.94.07030515102. Kitamura K, Nakamoto Y, Akiyama M, Fujii C, Kondo T, Kobayashi K, et al. Pathogenic roles of tumornecrosis factor receptor p55-mediated signals in dimethylnitrosamine-induced murine liverfibrosis. Lab Invest (2002) 82:571–83. doi:10.1038/labinvest.3780452103. Rojanasakul Y, Weissman DN, Shi X, Castranova V, Ma JK, Liang W. Antisense inhibition of silica-induced tumor necrosis factor in alveolar macrophages. J Biol Chem (1997) 272:3910–4.doi:10.1074/jbc.272.7.3910104. Gurantz D, Cowling RT, Varki N, Frikovsky E, Moore CD, Greenberg BH. IL-1beta and TNF-alphaupregulate angiotensin II type 1 (AT1) receptors on cardiac fibroblasts and are associated withincreased AT1 density in the post-MI heart. J Mol Cell Cardiol (2005) 38:505–15.doi:10.1016/j.yjmcc.2004.12.015105. Meldrum KK, Misseri R, Metcalfe P, Dinarello CA, Hile KL, Meldrum DR. TNF-alpha neutralizationameliorates obstruction-induced renal fibrosis and dysfunction. Am J Physiol Regul Integr CompPhysiol (2007) 292:R1456–64. doi:10.1152/ajpregu.00620.2005106. Redente EF, Keith RC, Janssen W, Henson PM, Ortiz LA, Downey GP, et al. Tumor necrosis factor-αaccelerates the resolution of established pulmonary fibrosis in mice by targeting profibrotic lungmacrophages. Am J Respir Cell Mol Biol (2014) 50:825–37. doi:10.1165/rcmb.2013-0386oc107. Zhang Y, Lee TC, Guillemin B, Yu MC, Rom WN. Enhanced IL-1 beta and tumor necrosis factor-alpharelease and messenger RNA expression in macrophages from idiopathic pulmonary fibrosis or afterasbestos exposure. J Immunol (1993) 150:4188–96.108. Woodcock HV, Maher TM. The treatment of idiopathic pulmonary fibrosis. F1000Prime Rep (2014)6:16. doi:10.12703/P6-16109. Hamid T, Gu Y, Ortines RV, Bhattacharya C, Wang G, Xuan Y-T, et al. Divergent tumor necrosis factorreceptor-related remodeling responses in heart failure: role of nuclear factor-kappaB andinflammatory activation. Circulation (2009) 119:1386–97. doi:10.1161/CIRCULATIONAHA.108.802918110. Morimoto Y, Gai Z, Tanishima H, Kawakatsu M, Itoh S, Hatamura I, et al. TNF-alpha deficiencyaccelerates renal tubular interstitial fibrosis in the late stage of ureteral obstruction. Exp Mol Pathol(2008) 85:207–13. doi:10.1016/j.yexmp.2008.08.003111. Lemos DR, Babaeijandaghi F, Low M, Chang C-K, Lee ST, Fiore D, et al. Nilotinib reduces musclefibrosis in chronic muscle injury by promoting TNF-mediated apoptosis of fibro/adipogenicprogenitors. Nat Med (2015) 21:786–94. doi:10.1038/nm.3869112. Sun L, Louie MC, Vannella KM, Wilke CA, LeVine AM, Moore BB, et al. New concepts of IL-10-inducedlung fibrosis: fibrocyte recruitment and M2 activation in a CCL2/CCR2 axis. Am J Physiol Lung CellMol Physiol (2011) 300:L341–53. doi:10.1152/ajplung.00122.2010113. Barbarin V, Arras M, Misson P, Delos M, McGarry B, Phan SH, et al. Characterization of the effect ofinterleukin-10 on silica-induced lung fibrosis in mice. Am J Respir Cell Mol Biol (2004) 31:78–85.doi:10.1165/rcmb.2003-0299OC114. Yang L, Kwon J, Popov Y, Gajdos GB, Ordog T, Brekken RA, et al. Vascular endothelial growth factorpromotes fibrosis resolution and repair in mice. Gastroenterology (2014) 146:1339–50.e1.doi:10.1053/j.gastro.2014.01.061115. Heinrichs D, Knauel M, Offermanns C, Berres M-L, Nellen A, Leng L, et al. Macrophage migrationinhibitory factor (MIF) exerts antifibrotic effects in experimental liver fibrosis via CD74. Proc NatlAcad Sci U S A (2011) 108:17444–9. doi:10.1073/pnas.1107023108116. Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, et al. MIF is a noncognate ligandof CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med (2007)13:587–96. doi:10.1038/nm1567

The elusive antifibrotic macrophage
49
117. Liang Y, Luo J, Lu Q, Zhou Y, Wu H, Zheng D, et al. Gene profile of chemokines on hepatic stellatecells of schistosome-infected mice and antifibrotic roles of CXCL9/10 on liver non-parenchymalcells. PLoS One (2012) 7:e42490. doi:10.1371/journal.pone.0042490118. Tager A, Kradin R, LaCamera P, Bercury S, Campanella G, Leary C, et al. Inhibition of pulmonaryfibrosis by the chemokine IP-10/CXCL10. Am J Respir Cell Mol Biol (2004) 31:395404.doi:10.1165/rcmb.2004-0175OC119. Poelstra K, Beljaars L, Melgert BN. Cell-specific delivery of biologicals: problems, pitfalls andpossibilities of antifibrotic compounds in the liver. Drug Discov Today (2013) 18:1237–42.doi:10.1016/j.drudis.2013.05.013120. Madsen DH, Jürgensen HJ, Ingvarsen S, Melander MC, Vainer B, Egerod KL, et al. Endocytic collagendegradation: a novel mechanism involved in protection against liver fibrosis. J Pathol (2012)227:94–105. doi:10.1002/path.3981121. Madsen DH, Leonard D, Masedunskas A, Moyer A, Jürgensen HJ, Peters DE, et al. M2-likemacrophages are responsible for collagen degradation through a mannose receptor-mediatedpathway. J Cell Biol (2013) 202:951–66. doi:10.1083/jcb.201301081122. Wittrant Y, Theoleyre S, Couillaud S, Dunstan C, Heymann D, Redini F. Regulation of osteoclastprotease expression by RANKL. Biochem Biophys Res Commun (2003) 310:774–8.doi:10.1016/j.bbrc.2003.09.084123. Matsumoto M, Kogawa M, Wada S, Takayanagi H, Tsujimoto M, Katayama S, et al. Essential role ofp38 mitogen-activated protein kinase in cathepsin K gene expression during osteoclastogenesisthrough association of NFATc1 and PU.1. J Biol Chem (2004) 279:45969–79.doi:10.1074/jbc.M408795200124. Sundaram K, Nishimura R, Senn J, Youssef R, London S, Reddy S. RANK ligand signaling modulatesthe matrix metalloproteinase-9 gene expression during osteoclast differentiation. Exp Cell Res(2006) 313:168–78. doi:10.1016/j.yexcr.2006.10.001125. Jiang D, Liang J, Hodge J, Lu B, Zhu Z, Yu S, et al. Regulation of pulmonary fibrosis by chemokinereceptor CXCR3. J Clin Investig (2004) 114:291299. doi:10.1172/JCI16861126. Guilherme RF, Xisto DG, Kunkel SL, Freire-de-Lima CG, Rocco PR, Neves JS, et al. Pulmonaryantifibrotic mechanisms aspirin-triggered lipoxin A(4) synthetic analog. Am J Respir Cell Mol Biol(2013) 49:1029–37. doi:10.1165/rcmb.2012-0462OC127. Alagesan S, Griffin MD. Alternatively activated macrophages as therapeutic agents for kidneydisease: in vivo stability is a key factor. Kidney Int (2014) 85:730–3. doi:10.1038/ki.2013.405

Chapter 1
50

CHAPTER 2
The RANK/RANKL/OPG axishas a role in regulating
tissue repair processes in lung
Carian E. Boorsma | Kurnia S.S. Putri | Christina Draijer |Peter Heukels | Bernt van den Blink | Robbert H. Cool |
Corry-Anke Brandsma | George Nossent | David M. Brass |Peter Olinga | Wim Timens | Barbro N. Melgert

Chapter 2
52
ABSTRACTOsteoprotegerin (OPG) is associated with fibrotic processes, but its role in pulmonaryfibrosis (PF) is unknown. OPG is a decoy receptor for Receptor-Activator-of-NF-BLigand (RANKL). RANKL can bind to RANK on macrophages, inducing degradation ofextracellular matrix (ECM) in bone and OPG prevents this. Pulmonary macrophagesalso express RANK. We hypothesized that RANKL similarly induces ECM degradationin lung, while high levels of OPG dampen ECM degradation by macrophages andcontribute to fibrosis development.OPG levels were higher in fibrotic human and mouse lung tissue and correlated withhigher collagen-1 content in mice. Expression was found in fibroblasts and isolatedfibroblasts of PF patients had higher OPG production as compared to controlfibroblasts. TGF stimulated OPG production in fibroblasts and in precision-cut-lungslices. RANKL-treatment of mice with fibrosis did not result in resolution but greatlyinduced OPG-production and epithelial cell numbers in lung.In conclusion, the induction of OPG and the increase in epithelial cell numbers in lungfollowing RANKL-treatment of fibrosis suggest that the RANK/RANKL/OPG-axis is notonly active in bone, but also has a role in regulating tissue repair processes in lung. AsOPG production is closely linked to fibrosis it is interesting new avenue to explore fordrug and biomarker development.Keywords: Pulmonary fibrosis, osteoprotegerin, resolution, macrophage and RANKL

The RANK/RANKL/OPG axis has a role in regulating tissue repair processes in lung
53
INTRODUCTIONPulmonary fibrosis is characterized by relentless scarring of the lung withextracellular matrix (ECM) deposition and progressive destruction of lung architecturethat leads to impaired lung function with high mortality rates1,2. Fibroblasts andmyofibroblasts are abundantly present in fibrotic lung tissue and are considered themain producers of excess ECM. Macrophages are also abundantly present in fibroticlung tissue and can contribute to fibrosis by producing profibrotic cytokines3,4.However, macrophages have also been shown to be antifibrotic by virtue of their abilityto degrade ECM with matrix metalloproteinases (MMPs), cathepsins and their abilityto internalize degraded collagens5-8. Gibbons et al. showed that resolution ofexperimental lung fibrosis in mice was attenuated when lung macrophages weredepleted during the resolution phase of the disease9. However, it has not yet beenelucidated whether this is a distinct population of macrophages and how their behavioris regulated.OPG is a decoy receptor for receptor activator of nuclear factor κb ligand(RANKL), which is best known for its role in the regulation of bone ECM10. RANKL canstimulate macrophages in bone via its receptor RANK towards osteoclastdifferentiation and these can degrade bone ECM. As the decoy receptor for RANKL, OPGblocks the interaction of RANKL with RANK thereby inhibiting osteoclastdifferentiation and bone ECM degradation. Increased osteoprotegerin (OPG)expression has recently been associated with fibrotic processes in liver10-12, heart13 andin the vasculature14. Increased OPG expression has also been observed in a mousemodel of chronic lipopolysaccharide-induced airway remodelling15 and in silica-16 andbleomycin-induced lung fibrosis15. OPG levels increase early after bleomycin treatmentand wane in time accompanied by a decrease in collagen deposition, suggesting thatOPG may be important in active fibrotic processes. However, how OPG plays a role infibrosis and whether it is involved in human pulmonary fibrosis remains to bedetermined.As pulmonary macrophages were shown to express RANK as well(www.proteinatlas.com), we hypothesized that RANK activation could initiate a matrixresolving phenotype in the lung in the presence of RANKL, which may be hampered byhigh levels of OPG in fibrotic conditions. We therefore characterized the expression of

Chapter 2
54
RANK, RANKL and OPG in lung tissue samples of patients with pulmonary fibrosis andof mice with experimental silica-induced lung fibrosis. To address the role of OPG inlung fibrosis, mice with silica-induced fibrosis were treated therapeutically withsoluble RANKL (sRANKL) to neutralize OPG and we examined the progression offibrosis in lung tissue.MATERIALS AND METHODS
Human tissueThe study protocol was consistent with the Research Code of the University MedicalCenter Groningen, Dutch national ethical and professional guidelines, and the MedicalEthical Committee of Rotterdam. Patient characteristics are displayed in Table 1.Table 1. Characteristics of patients whose lung tissue was used for determining RANK,RANKL and OPG levels (medians with range are presented).
Controls Pulmonary Fibrosis
Cancer (n=15) COPD (n=9) (n=11)Sex, male/female 1/5 9/0 10/1Age, years 63 (57-81) 58 (50-62) 56 (37-64)Smoking status 4 non-smokers/2 ex-smokers 1 non-smokers/8 ex-smokers 4 non-smokers/6 ex-smokers/1 unknownFEV1 % predicted 103 (85-125) 32 (10-72) 49 (45-89)Diagnosis 3 lung cancer/3 lung metastases 1 GOLD stage 3/8 GOLD stage 4 11 IPF/UIPDefinition of abbreviations: PF = pulmonary fibrosis; FEV1 = forced exhaled volume; COPD = chronicobstructive pulmonary disease; IPF = idiopathic pulmonary fibrosis; UIP = usual interstitial pneumonia.Isolation and culture of primary fibroblastsPrimary lung fibroblasts were isolated from lung explants using a techniquedescribed previously17,18. Patient characteristics are summarized in Table 2. Cells usedin the experiments were maximally passaged three times. Plated cells were grown toconfluence and transferred to low-serum medium (0.5% FCS). After 72 h, supernatantswere collected for analysis.

The RANK/RANKL/OPG axis has a role in regulating tissue repair processes in lung
55
Table 2. Characteristics of patients whose lung tissue was used for the isolation of primarylung fibroblasts (medians with range are presented)Controls Pulmonary Fibrosis
Cancer (n=3) COPD (n=2) (n=6)Sex, male/female 2/1 1/1 4/2Age, years 65 (45-74) 55 (53-57) 64 (58-66)Smoking status 1 non-smoker/2 ex-smoker 2 ex-smoker 2 non-smoker/4 ex-smokerFEV1 % predicted 98 (90-130) 22.5 (20-25) 62.5 (52-75)Diagnosis Lung cancer COPD 2 IPF/UIP, 1 ILD/EAA,1 NSIP/dust-exposed,1 FNSIP/UIPDefinition of abbreviations: PF = pulmonary fibrosis; FEV1 = forced exhaled volume; COPD = chronicobstructive pulmonary disease; IPF = idiopathic pulmonary fibrosis; UIP = usual interstitial pneumonia;ILD = interstitial lung disease; EAA = extrinsic allergic alveolitis; NSIP = non-specific interstitialpneumonia; FNSIP = fibrosing NSIP.Animal experimentsMale C57BL/6 mice were obtained from Harlan (Zeist, The Netherlands). Allanimal experiments were approved by the Institutional Animal Care and UseCommittee, (DEC6064) and (DEC6416AA). Animal experiments were performed in theanimal facility of the University of Groningen according to strict governmental andinternational guidelines on animal experimentation.Silica-induced lung fibrosis in miceFibrosis was induced using a single intratracheal dose of Min-U-Sil 5 crystallinesilica (0.2 g/kg in 50 l 0.9% saline, a kind gift from Dr. Andy Ghio, US EPA, Chapel Hill,NC) following isoflurane anesthesia. Control animals received an equivalent volume of0.9% saline. Mice were sacrificed after 28 days and serum and lung tissue werecollected (n=6-8).In mice treated with sRANKL, silica was administered on day zero and sRANKLtreatment began after 28 days and was continued for an additional 2 weeks. sRANKLwas administered intranasally in 40 l saline or 40 l saline (vehicle) three times perweek (n=12). On day 43, mice were sacrificed and lungs were collected for flow

Chapter 2
56
cytometry, western blot analysis, ELISA, or histology. See Figure 1 for an overview ofthe experimental setup.
Figure 1. Experimental setup of the animal experimens: pulmonary fibrosis was induced intratracheal(i.t.) instillation of silica on day 0. After 28 days, mice were either sacrificed to assess pulmonary fibrosisdevelopment or treated intranasally (i.n.) with sRANKL or vehicle for two additional weeks.Mouse lung precision-cut lung slicesLungs of six male C57BL/6 mice (20-30 gr, age 6-8 weeks) were used to makeprecision-cut lung slices. This method was previously described by Oenema et.al.19.Lung slices were incubated in triplicate for 48h with TGF (5 ng/ml) or 48h with TGFfollowed by 24 h of either vehicle or RANKL (200 ng/ml) stimulation. Three slices fromeach condition were pooled for further analyses and tissue slice supernatants werecollected separately.Mouse cell culturesNIH 3T3 fibroblasts and RAW264.7 macrophages were purchased at theAmerican Type Culture Collection. Self-propagating mouse alveolar-like macrophages(MPI cells) were a kind gift from Dr. G. Fejer, Plymouth University, Devon, UK.Before TGF stimulation of 3T3 fibroblasts, cells were incubated in 0.5% FCSmedium. After 24h, cells were stimulated with TGF (5 ng/ml, Peprotech, Rocky Hill,NJ) for 48h after which the supernatant was collected.To induce a proteolytic phenotype, RAW264.7 macrophages (until max. passage12) or MPI macrophages were stimulated with 200 ng/ml soluble RANKL (sRANKL,

The RANK/RANKL/OPG axis has a role in regulating tissue repair processes in lung
57
produced at our own facility). For each experiment, the data presented arerepresentative of at least three independent replicates.Protein isolation from lung tissueProtein was isolated from frozen human or mouse lung tissue (30-40 mg) inbuffer (0.25 M Tris/HCl buffer, 2.5 % Igepal, 0.5% SDS, Protease Inhibitor Cocktail(Boehringer Mannheim, Germany), pH 7.5). Overall protein concentration wasdetermined using an RC DCTM Protein Assay based on the Lowry method(cat#5000111, Bio-Rad, Hercules, CA).Western blot analysisOne hundred micrograms of tissue lysate per sample was used for western blotanalysis. The following primary antibodies were used: mouse RANK (1:200, Acris,Herford, Germany), human RANK (1:100, R&D Systems, Minneapolis, MN), RANKL(human and mouse, 1:100, Acris) and β-actin (1:20,000 Sigma-Aldrich). RANK andRANKL expression levels were normalized to β-actin expression.ELISA Human and mouse OPG levels in lung tissue and culture supernatants wereassessed using ELISA (cat#DY805 (human), cat#DY459 (mouse), R&D Systems)according to the instructions provided by the manufacturer. One hundred microgramsof tissue lysate total protein in a total of 100 μl was analyzed per sample. Cell culturemedia was analyzed undiluted while lung slice culture medium was diluted 1:5 beforeanalysis.ImmunohistochemistryImmunohistochemical analysis of collagen I, RANK, and OPG was performed on3 m paraffin sections of mouse lung tissue using anti-mouse collagen I (SouthernBiotech, Birmingham, AL), anti-human RANK (R&D Systems), and anti-mouse OPG(Antibodies-online, Atlanta, GA). The amount of collagen deposition in the lung wasdetermined using ImageScope software (Aperio, Burlingame, USA).

Chapter 2
58
Cathepsin K activityCathepsin K activity of RAW264.7 macrophages was measured using aCathepsin K Activity Kit (Cat#KA0769, Abnova, Taipei, Taiwan) following instructionsprovided by the manufacturer. Cathepsin K activity was normalized against the activityobserved in unstimulated cells.Quantitative Real-time PCRTotal mRNA was isolated from cells or tissues using a Maxwell® LEV simply RNACells/Tissue kit (cat# AS1280, Promega, Madison, WI) according to the instructionsprovided by the manufacturer. The following primers were used: collagen IαI,fibronectin, MMP9, OPG, β-actin, YWHAZ and 18s (Sigma-Aldrich, Zwijndrecht, TheNetherlands). mRNA expression was normalized to β-actin for cell culture samples, toYWHAZ for tissue samples, and to 18S for precision-cut lung slices and expressed asfold-change of healthy controls or control conditions.Production and purification of sRANKL proteinRecombinant sRANKL was produced by R. Cool following standard proceduresat our facility.Flow cytometryMouse-left lungs were used for the isolation of a single cell suspension for flowcytometry. Total cell numbers were determined using a FACS Array cell counter (BDBiosciences) and used to calculate total numbers of specific cell types. Single lung cellsuspensions were stained for T cells, epithelial cells, neutrophils, interstitial andalveolar macrophage, and RANKL expression by flow cytometry. See supplementalTable 1 and 2 for antibody details. Samples were analyzed using an LSRII flowcytometer (BD Biosciences). Data was analyzed using FlowJo software (Tree Start,Ashland, USA). See supplemental data Figure 3 and 4 for the gating strategy.

The RANK/RANKL/OPG axis has a role in regulating tissue repair processes in lung
59
Statistical analysisAll groups were considered non-normally distributed due to small samplessizes. Statistical differences between two groups were assessed using Mann-WhitneyU tests. Multiple-group comparison was performed with Kruskal Wallis. Whensignificant, a Mann-Whitney-U test with a Bonferroni adjustment for multiplecomparisons was used as a post-test. Correlations were assessed by calculating theSpearman correlation coefficient (R). Significance was considered when p<0.05. Thedata were analyzed using GraphPad Prism 6.RESULTS
OPG levels are higher in fibrotic lung tissueWe observed higher OPG levels in human lung tissue with end stage fibrotic lungdisease than in control lung tissue (Figure 2A). OPG levels in the lungs of silica-treatedmice were also significantly higher when compared with control lungs (Figure 2B).Fibrosis in our mouse model was confirmed by significantly higher collagen expressionand deposition in lungs of mice exposed to silica (Supplemental Figures 1A and 1B).Staining for OPG in mouse lung tissue showed that OPG expression localized in thesmooth muscle layers around vessels and airways in control lung tissue (Figures 2C-
D). In mouse lungs exposed to silica, OPG expression was also observed within areasof active fibrosis development, which was difficult to pinpoint to a specific cell type(Figures 2E-F, positive staining is indicated by the arrows). To assess whether OPGcould be produced by fibroblasts in the fibrotic areas, human primary lung fibroblastswere tested for their ability to produce OPG. We found they produced copious amountsof OPG and lung fibroblasts from fibrotic lungs produced significantly more OPG thanlung fibroblasts isolated from lung tissue of patients with lung carcinoma or COPD(Figure 2G). We also assessed whether bronchial or alveolar epithelial cells andmacrophages could produce OPG and found that cultures of these cells did not produceany OPG (data not shown).

Chapter 2
60

The RANK/RANKL/OPG axis has a role in regulating tissue repair processes in lung
61
Figure 2. ELISA analysis of OPG levels on lung tissue lysates showed that OPG protein levels were higherin fibrotic conditions in both human (A) and mouse lung tissue (B). OPG levels were measured in controlhuman lung tissue obtained from 15 patients undergoing surgical resection for carcinoma or COPD andpulmonary fibrosis tissue obtained from 11 patients undergoing lung transplantation. Mouse lung tissuewas collected from mice 28 days after receiving silica (experimental fibrosis, n=7) or saline (healthycontrol, n=8). (C-D). Immunohistochemical staining for OPG in paraffin-embedded mouse lung tissuerevealed expression in smooth muscle cells underneath endothelial cells and bronchial epithelial cells(E-F). Under fibrotic condition, OPG expression was also observed in fibrotic areas as indicated by thearrows, but it was unclear which cells were responsible for this staining. (G). Cell cultures of humanprimary fibroblasts isolated from lung tissue of patients with pulmonary fibrosis (n=6) or patientsundergoing surgical resection for carcinoma or COPD (control patients, n=5) revealed that fibroblastsfrom fibrotic lung tissue produced significantly higher levels of OPG in culture supernatant thanfibroblasts from control lung tissue. Groups were compared using a Mann-Whitney U test and p<0.05was considered significant.
Figure 3. (A) Western blot analysis of RANKL shows expression of this protein in human lung tissue.RANKL levels were similar in lung tissue lysates from control patients undergoing surgical resection forcarcinoma or COPD (n=15) and pulmonary fibrosis patients (n=11). (B) Western blot analysis of RANKLshows expression of this protein in murine lung tissue. Mouse lung tissue of healthy controls and micewith silica-induced pulmonary fibrosis showed similar RANKL protein expression levels. (C-D) Flowcytometric analysis of cells from murine lung suspensions showed RANKL expression on T cells (C) andepithelial cells (D). More T cells positive for RANKL were found in fibrotic as compared to healthyconditions, while the number of RANKL+ epithelial cells was similar between fibrotic and controlconditions. Groups were compared using a Mann-Whitney U test and p<0.05 was considered significant.

Chapter 2
62
RANKL is expressed by epithelial cells and T cells in lung tissueAs OPG is the decoy receptor of RANKL, we assessed RANKL expressionpatterns in human and mouse lung tissue. We found clear expression of RANKL in bothhuman and mouse lung tissue (Figure 3). Overall RANKL protein levels were similarin fibrotic lung tissue compared with control lung tissue in both humans (Figure 3A)and mice (Figure 3B).To assess which cells in mouse lung tissue express RANKL, we analyzed RANKLexpression in by flow cytometry. We found clear RANKL+ expression on CD3+ T cellsand EpCAM+ epithelial cells (Figure 3C and D) and no significant expression onmacrophages. Induction of fibrosis by silica resulted in higher number of RANKL+ Tcells (Figure 3C) and no change in the number of RANKL+ epithelial cells (Figure 3D).RANK expression on lung macrophages in decreased in fibrotic conditionsRANK, the membrane-bound receptor of RANKL, was also expressed in bothhuman and mouse lung tissue and levels were similar between fibrotic and control lungtissue in both humans and mice (Figures 4A and 4B). When staining for RANK wefound expression by bronchial epithelial cells and interstitial as well as alveolarmacrophages (Figures 4C-F).OPG levels correlated with collagen I levels and are induced by TGF-βTo assess its relationship with fibrosis, we studied whether OPG levels inmurine lung tissue correlated with markers of fibrosis. Indeed, OPG levels in the lungcorrelated with the collagen-I content of the lung (Figure 5A) and serum OPG levelsreflected the OPG levels in the lung (Figure 5B). To confirm OPG is produced by lungtissue under the control of a profibrotic cytokine, we incubated murine precision-cutmouse lung slices with TGF, the hallmark cytokine of fibrosis. We found thatprecision-cut mouse lung slices produced more OPG following TGF stimulation thanunstimulated slices (Figure 5C). Moreover, 3T3 murine fibroblasts produceddetectable levels of OPG at baseline and stimulation with TGF resulted in even higherOPG levels in culture supernatant (Figure 5D).

The RANK/RANKL/OPG axis has a role in regulating tissue repair processes in lung
63
Figure 4. Western blot analysis of RANK shows expression of this protein in human and murine lungtissue (A-B). No differences were found in RANK expression between control and fibrotic lung tissue inhumans (A) and mice (B). Lung tissue lysates were collected from 15 control patients undergoingsurgical resection for carcinoma or COPD and 11 samples were collected from pulmonary fibrosispatients undergoing lung transplantation. Mice were either silica-exposed to induce pulmonary fibrosis(n=4) or vehicle-exposed (n=3) for 28 days. (C-D) Immunohistochemical staining for RANK in paraffin-embedded control mouse lung tissue revealed expression by alveolar and interstitial macrophages andbronchial epithelial cells. (E-F) Immunohistochemical staining of fibrotic murine lung tissue showed asimilar expression pattern of macrophages and bronchial epithelial cells as for control lung tissue.

Chapter 2
64
Figure 5. (A) OPG levels in lysates of lung tissue from mice exposed to silica correlated with collagen Ilevels in that same lung tissue. (B) OPG serum levels correlated with OPG levels in murine fibrotic lungtissue. A Spearman correlation coefficient was calculated for each correlation and p<0.05 wasconsidered significant. (C) OPG production was higher in murine precision-cut lung slices following 48hours of TGF stimulation compared to vehicle-treated controls (n=7 independent experiments). (D)OPG levels in culture supernatant of murine 3T3 fibroblasts were higher after 24 hours of TGFstimulation than in supernatant from vehicle-stimulated fibroblasts (n=7 independent experiments).Groups were compared using a Mann-Whitney U test and p<0.05 was considered significant.RANKL induces ECM-degrading enzymes in macrophages with a hematopoietic
origin We tested whether RANKL is able to induce a proteolytic phenotype in eithermacrophages with an embryonic origin (alveolar macrophages) or with ahematopoietic origin (infiltrating monocytes)20. We found that 48 hours of RANKLstimulation induced significant expression of MMP9 mRNA (Figure 6A) and activity ofcathepsin K (Figure 6B) in monocyte-derived RAW264.7 macrophages as comparedto unstimulated cultures. No polynuclear cells were seen, which would be an indicationof osteoclast formation. In contrast, we found that RANKL did not induce MMP9 mRNAexpression in the embryonic alveolar-like MPI macrophages (Figure 6C).

The RANK/RANKL/OPG axis has a role in regulating tissue repair processes in lung
65
Figure 6. In RAW264.7 monocytic macrophages, MMP9 mRNA (A) and cathepsin K activity (B) werehigher following RANKL stimulation (24h and 72h respectively) as compared to unstimulated controls.(C) sRANKL stimulation of MPI alveolar-like macrophages did not result in higher MMP9 expression ascompared to unstimulated controls. Groups were compared using a Mann-Whitney U test and p<0.05was considered significant.RANKL treatment of mice with silica-induced pulmonary fibrosis does not reverse
fibrosis, but does result in more OPG and epithelial cellsBy treating mice with silica-induced pulmonary fibrosis with recombinantsRANKL, we aimed to overcome the high levels of OPG and induce the development ofECM-degrading macrophages. Induction of fibrosis by silica resulted in higher collagenI deposition and more neutrophils, more macrophages (both interstitial and alveolar)and, as expected, more production of OPG as compared to nonexposed healthy mice(Figures 7A-K). Exposure to silica did not change the number of infiltrating T cells(Figure 7B) or the number of epithelial cells (Figure 7D), but did lead to a highernumber of RANKL+ T cells in lung tissue (Figure 7C) as compared to nonexposedhealthy mice.

66
Chapter 2

The RANK/RANKL/OPG axis has a role in regulating tissue repair processes in lung
67
Figure 7. Mice were treated with sRANKL (3x per week for two weeks, 5 or 10 g permouse/administration) four weeks after inducing pulmonary fibrosis with silica. Collagen I deposition(A), numbers of CD3+ T cells (B), RANKL+CD3+ T cells (C), Epcam+ epithelial cells (D), RANKL+Epcam+epithelial cells (E), neutrophils (F), total macrophages (G), alveolar macrophages (H), interstitialmacrophages (I), and levels of OPD mRNA expression (J) and protein expression (K) were assessed.Silica-exposure led to higher collagen I deposition in lung tissue, more RANKL+CD3+ T cells, neutrophils,total, interstitial and alveolar macrophages, and higher expression of OPG mRNA and protein in lungtissue as compared to nonexposed animals. sRANKL treatment did not change collagen I deposition butdid dose-dependently increase the number of (RANKL+) epithelial cells and also resulted in higher OPGmRNA and protein levels in lung tissue as compared to mice with untreated fibrosis. Groups werecompared using a Kruskall Wallis test for multiple testing and p<0.05 was considered significant. (L)OPG mRNA expression did not change in TGF + sRANKL-treated murine precision-cut lung slicescompared to TGF + vehicle-treated controls (n=6 independent experiments).
Treatment with 5 or 10 g sRANKL had no effect on collagen I deposition andthe number of (RANKL+) T cells, neutrophils, and (alveolar and interstitial)macrophages as compared to silica-exposed nontreated animals. Collagen IαI, MMP9and fibronectin mRNA expressions also increased after silica exposure and remainedunchanged following sRANKL treatment (Supplemental Figures 2A, 2B and 2C).sRANKL treatment did result in a strong induction of OPG expression (Figure
7J) and this also resulted in higher OPG levels in lung tissue as compared to silica-exposed nontreated animals (Figure 7K). In addition, sRANKL treatment resulted in adose-dependent significant increase in the number of epithelial cells in lung tissue ascompared to silica-exposed nontreated animals and this was paralleled by a similarpattern for the number of RANKL+ epithelial cells (Figures 7D and E).To investigate whether sRANKL could directly induce the expression of OPG inlung tissue, we incubated precision-cut murine lung slices with TGF for 72 hours andadded sRANKL during the last 24 hours to mimic our in vivo experiment. sRANKLtreatment for 24 hours did not lead to higher expression of OPG mRNA in lung slices ascompared to nontreated slices (Figure 7L).

Chapter 2
68
DISCUSSIONOur study shows that OPG protein levels are higher in lung tissue of bothhumans and mice with (silica-induced) pulmonary fibrosis and that in addition to OPG,its ligand RANKL and the membrane-bound receptor RANK are all present in lungtissue. In mice with silica-induced fibrosis, these lung tissue levels of OPG correlatepositively with the amount of collagen in the lung. The findings that sRANKL treatmentincreases epithelial cell numbers and OPG expression in lung tissue suggests that theRANK/RANKL/OPG axis is not only active in bone, but also has a role in regulatingtissue repair processes in the lung.OPG has previously been found to associate with fibrosis-related diseases likevascular fibrosis21, cystic fibrosis22 and liver fibrosis11,12. In addition, high OPG levelswere found bronchoalveolar lavage fluid of mice with silica- and bleomycin-inducedfibrosis15,16 and in a model of endotoxin-induced airway remodeling15. We now showthat OPG expression is also high in human fibrotic lung tissue and that it is producedby fibroblasts isolated from this lung tissue. The higher OPG production resulted fromproduction in lung tissue itself, as OPG mRNA expression in lung tissue was also higherin experimental fibrosis. Some have suggested that OPG levels are higher to counteractincreased RANKL levels that may be elevated due to the actions of IL-6 and TNF-23.However, we showed that TGF, the hallmark cytokine of fibrosis, is capable ofincreasing OPG production. Therefore, our findings strongly support the concept thatOPG is associated with the fibrotic process itself and is produced locally in the lung.In bone, OPG prevents the development of osteoclasts by binding RANKL toreduce bone ECM degradation. We hypothesized a similar role for OPG in the lungs.However, when treating with sRANKL to overcome the excess OPG in fibrosis, we foundno differences in collagen I deposition in lung tissue, our outcome measure forpulmonary fibrosis. In contrast, sRANKL treatment even further induced OPG levels,potentially still preventing the induction of ECM degradation. Possibly, the sRANKLadministered binds to free OPG, triggering a pulmonary or extrapulmonary feedbackmechanism to compensate for the lower levels of free OPG. To explain thisphenomenon, we studied whether RANKL could directly induce OPG in precision-cutlung slices and found it did not change OPG mRNA expression. It therefore seems likelythat RANKL somehow indirectly induces the expression of OPG in lung tissue. Clearly,

The RANK/RANKL/OPG axis has a role in regulating tissue repair processes in lung
69
our findings have provided interesting leads to further investigate the regulation ofOPG in the lung.Interestingly, the treatment with sRANKL clearly was able to affect cells withinlung tissue as we found a strong dose-dependent increase in the number of Epcam+cells, which is indicative of an increase in the number of epithelial cells or anupregulation of the Epcam protein by these cells. Immunohistochemical staining forRANK showed expression of this receptor of RANKL on bronchial epithelial cells andthese cells could therefore potentially be responsive to the effects of RANKL. To thebest of our knowledge no reports on RANK expression on lung epithelial cells havebeen published nor on the effects of stimulating this receptor with RANKL in lungtissue. However, both mammary epithelial cells as well as a subset of thymic epithelialcells were found to expand after exposure to RANKL24,25 which may suggest RANKL isa more general epithelial growth factor. Further studies investigating proliferationeffects of RANKL on bronchial and alveolar epithelial cells are needed to address thisquestion. In combination with the clear pulmonary regulation of OPG during fibrosis,these data do suggest a RANK/RANKL/OPG axis is operating in lung tissue duringtissue repair.RANKL could theoretically still play a role in the resolution of fibrosis bystimulating the expression of ECM-degrading enzymes in macrophages. However, itsusefulness seems limited because of its ability to upregulate OPG production. In vitrostimulation of embryonic alveolar-like macrophages with sRANKL did not induceMMP9 expression, but it did induce a proteolytic phenotype in monocyte-derivedRAW264.7 macrophages as shown by our data and those of others previously20,26-32.MMP9 and cathepsin K can both contribute to the ECM degradation process6,7,33 andthese monocyte-derived macrophages may be therapeutically more relevant asGibbons et al. previously showed that infiltrating monocyte-derived macrophagescontributed largely to the development of fibrosis, whereas resident alveolarmacrophages did not9. The development of ligands that induce this proteolytic state inmonocte-derived macrophages without inducing OPG production may be a novel wayof inducing ECM degradation and treating advanced fibrotic disease34.Based on our findings that serum OPG levels correlated significantly with OPGlevels in murine lung tissue, OPG may be considered as a biomarker of disease severity

Chapter 2
70
or treatment effects. OPG serum levels in patients with liver cirrhosis have beenincluded into a panel of serum biomarkers to improved diagnostic accuracy of anoninvasive test to assess liver fibrosis severity35,36. OPG may be similarly useful inpulmonary fibrosis based on our in vivo data, even though McGrath et al. found nodifferences between controls and IPF patients OPG serum levels and no associationwith lung function parameters37. Further studies could aim at investigating whetherOPG correlates with non-invasive diagnostic parameters for (early) detection of thedisease or with disease progression.A limitation of our findings is the fact that our mouse model of silica-inducedpulmonary fibrosis only represents one cause of pulmonary fibrosis; while in humansthe causes are diverse and usually not known. The patients from whom the lung tissuesamples were obtained, however, represented many types of pulmonary fibrosis(idiopathic pulmonary fibrosis, fibrotic non-specific interstitial pneumonia andprogressed chronic extrinsic allergic alveolitis). All samples showed high levels of OPG,while we found low levels of OPG in COPD and in control lung tissue obtained frompatients undergoing surgical resection for carcinoma with normal lung function. Thissuggests that the induction of OPG production is specific for fibrotic lung diseases,though other lung diseases should be considered to confirm this assumption.RANKL is just one of the ligands for OPG and we focused on in relation to fibrosissince RANKL possesses the highest affinity for OPG out of all other possible ligands38.OPG is expressed in various organs and can also function as a decoy receptor of TNF-related apoptosis-inducing ligand (TRAIL), heparin and glycosaminoglycans35,36.Involvement of one of these ligands may also explain possible actions of OPG in thelung or generally in fibrosis39,40. For example, by binding TRAIL, OPG may be able toprevent TRAIL-induced apoptosis of lung myofibroblasts and thereby allowingmyofibroblasts to continue producing ECM37,41. To investigate all ligands togetherinstead of investigating them separately, a lung-specific conditional OPG knockoutmouse could be considered in combination with models of experimental pulmonaryfibrosis to determine the impact of OPG on the development and onset of fibrosis.In conclusion, OPG expression is higher in fibrotic lung tissue as compared tocontrol lung tissue and the profibrotic mediator TGF induces its production by

The RANK/RANKL/OPG axis has a role in regulating tissue repair processes in lung
71
fibroblasts. The induction of OPG and the increase in epithelial cell numbers in lungtissue following sRANKL treatment of pulmonary fibrosis suggest that theRANK/RANKL/OPG axis is not only active in bone, but also has a role in regulatingtissue repair processes in the lung. Even though the exact role of OPG in fibrosisremains undetermined, the fact that OPG production is closely linked to fibrosis makesit an interesting new avenue to explore for drug and biomarker development.ACKNOWLEDGEMENTSThe Pender Foundation is gratefully acknowledged for their support of theexchange of fibrotic lung tissue between the Groningen and Rotterdam centers (BMand BB). A Young Investigator Award of the Netherlands Respiratory Society for ourresearch on OPG in pulmonary fibrosis is gratefully acknowledged (CB). Barbro NMelgert is an active member of COST action BM1201. We would like to thank thefollowing Bachelor students of Pharmacy and Master students of Pharmacy for theirpractical contributions to the manuscript: Burak Güney, Axel Haak, AnnemarieBroesder, Thirsa Tuin and Marcelina Stel.

Chapter 2
72
REFERENCES1. Bois du RM. Idiopathic pulmonary fibrosis: present understanding and future options. Eur RespirRev 2011;20:132–3. doi:10.1183/09059180.000015112. Strieter RM. What Differentiates Normal Lung Repair and Fibrosis?: Inflammation, Resolution ofRepair, and Fibrosis. Proc Am Thorac Soc 2008;5:305–10. doi:10.1513/pats.200710-160DR3. Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis2010;30:245–57. doi:10.1055/s-0030-12553544. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol2008;8:958–69. doi:10.1038/nri24485. Atabai K, Jame S, Azhar N, et al. Mfge8 diminishes the severity of tissue fibrosis in mice by bindingand targeting collagen for uptake by macrophages. J Clin Invest 2009;119:3713–22.doi:10.1172/JCI40053DS16. Ishii T, Abboud RT, Wallace AM, et al. Alveolar macrophage proteinase/antiproteinase expressionin lung function and emphysema. Eur Respir J 2014;43:82–91. doi:10.1183/09031936.001746127. Srivastava M, Steinwede K, Kiviranta R, et al. Overexpression of cathepsin K in mice decreasescollagen deposition and lung resistance in response to bleomycin-induced pulmonary fibrosis.Respir Res 2008;9:54. doi:10.1186/1465-9921-9-548. JS. Selective depletion of macrophages reveals distinct, opposing roles during liver injury andrepair. J Clin Invest 2005;115:56–65. doi:10.1172/JCI2005226759. Gibbons MA, Mackinnon AC, Ramachandran P, et al. Ly6Chi Monocytes Direct AlternativelyActivated Pro-fibrotic Macrophage Regulation of Lung Fibrosis. Am J Respir Crit Care Med PublishedOnline First: 23 July 2011. doi:10.1164/rccm.201010-1719OC10. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature 2003;423:337–42.11. Szalay F, Hegedus D, Lakatos PL, et al. High serum osteoprotegerin and low RANKL in primarybiliary cirrhosis. J Hepatol 2003;38:395–400.12. Garcia-Valdecasas-Campelo E, Gonzalez-Reimers E, Santolaria-Fernandez F, et al. Serumosteoprotegerin and RANKL levels in chronic alcoholic liver disease. Alcohol Alcohol 2006;41:261–6. doi:10.1093/alcalc/agl00413. Shen A, Hou X, Yang D, et al. Role of osteoprotegerin and its gene polymorphisms in the occurrenceof left ventricular hypertrophy in essential hypertensive patients. Medicine 2014;93:e154.14. Toffoli B, Pickering RJ, Tsorotes D, et al. Osteoprotegerin promotes vascular fibrosis via a TGF-beta1 autocrine loop. Atherosclerosis 2011;218:61–8. doi:10.1016/j.atherosclerosis.2011.05.01915. Brass DM, Yang IV, Kennedy MP, et al. Fibroproliferation in LPS-induced airway remodeling andbleomycin-induced fibrosis share common patterns of gene expression. Immunogenetics2008;60:353–69. doi:10.1007/s00251-008-0293-316. Brass DM, McGee SP, Dunkel MK, et al. Gender influences the response to experimental silica-induced lung fibrosis in mice. Am J Physiol Lung Cell Mol Physiol 2010;299:L664–71.doi:10.1152/ajplung.00389.200917. Noordhoek JA, Postma DS, Chong LL, et al. Different modulation of decorin production by lungfibroblasts from patients with mild and severe emphysema. COPD 2005;2:17–25.18. Noordhoek JA, Postma DS, Chong LL, et al. Different proliferative capacity of lung fibroblastsobtained from control subjects and patients with emphysema. Exp Lung Res 2003;29:291–302.19. Oenema TA, Maarsingh H, Smit M, Groothuis GMM, Meurs H, et al. Bronchoconstriction InducesTGF-b Release and Airway Remodelling in Guinea Pig Lung Slices. PLoS ONE 2013. 8(6): e6558020. Gomez Perdiguero E, Klapproth K, Schulz C, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015;518:547–51. doi:10.1038/nature1398921. Toffoli B, Bernardi S, Candido R, et al. Osteoprotegerin induces morphological and functionalalterations in mouse pancreatic islets. Mol Cell Endocrinol 2011;331:136–42.doi:10.1016/j.mce.2010.08.01922. Ambroszkiewicz J, Sands D, Gajewska J, et al. Bone turnover markers, osteoprotegerin and RANKLcytokines in children with cystic fibrosis. Adv Med Sci 2013;58:338–43.

The RANK/RANKL/OPG axis has a role in regulating tissue repair processes in lung
73
23. Tanaka Y, Nakayamada S, Okada Y. Osteoblasts and osteoclasts in bone remodeling andinflammation. Curr Drug Targets Inflamm Allergy 2005;4:325–8.24. Danzl NM, Jeong S, Choi Y, Alexandropoulos K. Identification of Novel Thymic Epithelial Cell SubsetsWhose Differentiation Is Regulated by RANKL and Traf6. PLoS ONE 2014; 9(1): e86129.25. Gonzalez-Suarez E Jacob A Jones J Miller R Roudier-Meyer M et. al. RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature 2010; 46:103–10726. Guilliams M, De Kleer I, Henri S, et al. Alveolar macrophages develop from fetal monocytes thatdifferentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med 2013;210:1977–92.doi:10.1084/jem.2013119927. Yona S, Kim K-W, Wolf Y, et al. Fate mapping reveals origins and dynamics of monocytes and tissuemacrophages under homeostasis. Immunity 2013;38:79–91. doi:10.1016/j.immuni.2012.12.00128. Hashimoto D, Chow A, Noizat C, et al. Tissue-resident macrophages self-maintain locallythroughout adult life with minimal contribution from circulating monocytes. Immunity2013;38:792–804. doi:10.1016/j.immuni.2013.04.00429. Schulz C, Gomez Perdiguero E, Chorro L, et al. A lineage of myeloid cells independent of Myb andhematopoietic stem cells. Science 2012;336:86–90. doi:10.1126/science.121917930. Jakubzick C, Gautier EL, Gibbings SL, et al. Minimal differentiation of classical monocytes as theysurvey steady-state tissues and transport antigen to lymph nodes. Immunity 2013;39:599–610.doi:10.1016/j.immuni.2013.08.00731. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity2014;41:21–35. doi:10.1016/j.immuni.2014.06.01332. Vaananen HK, Zhao H, Mulari M, et al. The cell biology of osteoclast function. J Cell Sci 2000;113 (
Pt 3):377–81.33. van den Brule S, Misson P, Buhling F, et al. Overexpression of cathepsin K during silica-induced lungfibrosis and control by TGF-beta. Respir Res 2005;6:84. doi:10.1186/1465-9921-6-8434. Adhyatmika A, Putri KSS, Beljaars L, Melgert BN. The Elusive Antifibrotic Macrophage. Front. Med2015. 2:8135. Boursier J, de Ledinghen V, Zarski J-P, et al. A new combination of blood test and fibroscan foraccurate non-invasive diagnosis of liver fibrosis stages in chronic hepatitis C. Am J Gastroenterol2011;106:1255–63. doi:10.1038/ajg.2011.10036. Bosselut N, Taibi L, Guechot J, et al. Including osteoprotegerin and collagen IV in a score-basedblood test for liver fibrosis increases diagnostic accuracy. Clin Chim Acta 2013;415:63–8.doi:10.1016/j.cca.2012.09.02037. McGrath EE, Lawrie A, Marriott HM, et al. Deficiency of tumour necrosis factor-related apoptosis-inducing ligand exacerbates lung injury and fibrosis. Thorax 2012;67:796–803.doi:10.1136/thoraxjnl-2011-20086338. Baud'huin M, Duplomb L, Teletchea S, et al. Osteoprotegerin: multiple partners for multiplefunctions. Cytokine Growth Factor Rev 2013;24:401–9. doi:10.1016/j.cytogfr.2013.06.00139. Perez de Ciriza C, Lawrie A, Varo N. Osteoprotegerin in Cardiometabolic Disorders. Int J Endocrinol2015;2015:564934. doi:10.1155/2015/56493440. Sheridan JP, Marsters SA, Pitti RM, et al. Control of TRAIL-induced apoptosis by a family of signalingand decoy receptors. Science 1997;277:818–21.41. Emery JG, McDonnell P, Burke MB, et al. Osteoprotegerin is a receptor for the cytotoxic ligandTRAIL. J Biol Chem 1998;273:14363–7.

Chapter 2
74
SUPPLEMENTARY DATA
MATERIALS AND METHODSHuman tissueHuman fibrotic lung tissue was collected with informed consent from patients with end-stagepulmonary fibrosis undergoing lung transplantation at either the University Medical CenterGroningen (UMCG) or at the Erasmus Medical Center Rotterdam. In Groningen, the studyprotocol was consistent with the Research Code of the University Medical Center Groningen(http://www.umcg.nl/EN/Research/Researchers/General/ResearchCode/Paginas/default.aspx (accessed 18 Feb2016)), and Dutch nationalethical and professional guidelines (http://www.federa.org (accessed 18 Feb2016)). InRotterdam, the Medical Ethical Committee approved all protocols followed in that center.Control lung tissue was obtained at the UMCG from patients undergoing surgicalresection for carcinoma or chronic obstructive pulmonary disease (COPD). In the cases oftumor resections, histologically normal lung tissue was taken as far distally as possible fromthe tumor and assessed visually for abnormalities with standard haematoxylin and eosinstaining.Isolation and culture of primary human fibroblastsLung fibroblasts were isolated from lung explants using a technique describedpreviously[18,19]. In short, fresh parenchymal lung tissue, excluding visible vessels andairways, was cut into 1-2 mm pieces that were then cultured for 4-5 weeks in 12-well plates inthe presence of complete Ham’s F12 medium [10% fetal calf serum (FCS, Invitrogen, TheNetherlands), Penicillin/Streptavidin, fungizone, glutamine] at 37°C in an atmosphere of 5%CO2. At 25% confluency, the cells were transferred to T25 flasks. After 1 week, the cells wereplated in 12-wells plates (50,000 cells/well) for experiments or cryopreserved in FCS with10% DMSO under slow cooling conditions and eventually stored at -150°C. Cells used in theexperiments described were maximally passaged three times. Plated cells were grown toconfluence and transferred to low-serum medium (0.5% FCS). After 72h, supernatants werecollected for ELISA analysis of OPG levels and stored at -80°C for later analysis.Animal experimentsMale C57BL/6 mice were obtained from Harlan (Zeist, The Netherlands). Animals weremaintained with permanent access to food and water in a temperature-controlledenvironment with a 12h dark/light cycle regimen. All animal experiments were approved bythe Institutional Animal Care and Use Committee. Mice were used in experiments with silica-induced pulmonary fibrosis (DEC6064) and for the preparation of precision cut lung slices(DEC6416AA). Animal experiments were performed in the animal facility of the University ofGroningen according to strict governmental and international guidelines on animalexperimentation.Silica-induced lung fibrosis in miceFibrosis was induced using Min-U-Sil 5 crystalline silica (a kind gift from Dr. Andy Ghio,US EPA, Chapel Hill, NC). Animals were anesthetized using isoflurane before they received asingle administration of crystalline silica (0.2 g/kg) in 50 l 0.9% saline by intratracheal

The RANK/RANKL/OPG axis has a role in regulating tissue repair processes in lung
75
installation, while control animals received an equivalent volume of 0.9% saline. Mice weresacrificed after 28 days and serum and lung tissue were collected (n=6-8).In mice treated with sRANKL, silica was administrated on day zero and sRANKLtreatment began after 28 days and was continued for an additional 2 weeks. We calculated thatthe administered sRANKL dose should exceed the calculated OPG content (0.8 g OPG/lung) infibrotic lungs to be able to see a biological effect of sRANKL. We therefore used doses of 5 μg(n=12) and 10 μg RANKL (n=12) per mouse three times per week. sRANKL was administeredintranasally in 40 l saline. Control mice with pulmonary fibrosis received 40 l saline(vehicle) three times per week (n=12). On day 43, mice were sacrificed and lungs werecollected for flow cytometry, western blot analysis, ELISA, or histology. See Figure 1 for anoverview of the experimental setup.Mouse lung precision-cut lung slicesAn ex vivo model of early lung fibrosis development was used to investigate the effectof TGF stimulation on OPG production by lung tissue. Lungs of male C57BL/6 mice (20-30gr,age 6-8 weeks) of in total six mice were used to make precision-cut lung slices. After sacrificeby exsanguination via the aorta abdominalis under isoflurane anaesthesia, mouse lungs werefilled with 1.5% low-melting temperature agarose in 0.9% NaCl (Sigma-Aldrich) andtransferred directly into ice-cold University of Wisconsin organ preservation solution (DuPontCritical Care, Waukegab, IL). Lung slices of 5-mm in diameter and 10-15 cell layers thickness,were prepared with a Krumdieck tissue slicer (Alabama Research and Development, AL) usingice-cold Krebs-Henseleit Buffer [25 mM D-glucose (Merck, Darmstadt, Germany), 25 mMNaHCO3 (Merck), 10 mM HEPES (MP Biomedicals, Aurora, OH), saturated with carbogen (95%O2/5% CO2) and adjusted to pH 7.4] as we have previously described20.Lung slices were incubated in 12-well plates in DMEM + Glutamax medium [4.5g/L D-glucose and pyruvate (Gibco) supplemented with a non-essential amino acid mixture (1:100),100U/ml penicillin, 100g/ml streptomycin, 45 µg/ml gentamycin and 10% FCS]. After a 1hpre-incubation at 37C in a 95% O2/5% CO2 atmosphere with continuous shaking at 90 rpm,the slices were transferred into fresh medium and incubated in triplicate for 48h with TGF (5ng/ml) or 48h with TGF followed by either vehicle or RANKL (200ng/ml) stimulation for 24hours. Three slices from each condition were pooled for further analyses and tissue slicesupernatants were collected separately. All samples were stored at -80C for further analyses.Mouse cell culturesNIH 3T3 fibroblasts (American Type Culture Collection) were cultured in Dulbecco’smodified Eagle’s medium (Invitrogen, The Netherlands) [10% FCS, penicillin/streptomycin] at37°C in an atmosphere of 5% CO2.RAW 264.7 macrophages (American Type Culture Collection) were cultured inDulbecco’s modified Eagle’s medium (Invitrogen, The Netherlands) [10% FCS, L-Glutamine,Gentamycin] and self-propagating mouse alveolar-like macrophages (MPI cells, a kind gift fromDr. G. Fejer, Plymouth University, Devon, UK) were cultured in RPMI 1640 medium (Gibco,Bleiswijk, The Netherlands) [10% FCS, 20 ng/ml GM-CSF]17. Macrophages were cultured at37°C in an atmosphere of 5% CO2.

Chapter 2
76
3T3 cells were stimulated with TGF to transform them to myofibroblasts. Before TGFstimulation, plated cells were incubated in 0.5% FCS medium. After 24h, cells were stimulatedwith TGF (5 ng/ml, Peprotech, Rocky Hill, NJ) for 48h. Afterwards, the supernatant wascollected and stored at -80°C until further use.To induce a proteolytic phenotype, RAW macrophages (until max. passage 12) or MPImacrophages were stimulated with 200 ng/ml soluble RANKL (sRANKL, produced at our ownfacility. Technical details are described in “Production and purification of sRANKL protein”).Cells were lysed after 24h for the isolation of mRNA or after 72 hours for protein analysis.Samples were stored at -80°C for later analysis. For each experiment, the data presented arerepresentative of at least three independent replicates.Protein isolation from lung tissueProtein was isolated from frozen human or mouse lung tissue (30-40 mg) in buffer(0.25 M Tris/HCl buffer, 2.5% Igepal, 0.5% SDS, Protease Inhibitor Cocktail (BoehringerMannheim, Germany), pH 7.5) by incubating for 1 hour at 4°C, homogenizing and centrifugingat 13,200 rpm for 30 min. Supernatants were collected and the overall protein concentrationof the tissue lysates was determined using an RC DCTM Protein Assay based on the Lowrymethod (cat#5000111, Bio-Rad, Hercules, CA). Samples were stored at -80°C until furtheranalyses by Western blot or ELISA analysis.Western blot analysisWestern blot analysis was used to compare levels of mouse and human RANK andRANKL in control and fibrotic lung tissue. One hundred micrograms of total protein per samplewas loaded into the well of a 10% SDS-PAGE gel and transferred to a polyvinylidene-fluoridemembrane. Membranes were blocked with 5% nonfat milk in Tris-Buffered saline with 0.05%Tween 20 (TBST) for 2 hours, following incubation with the first antibody in blocking bufferovernight at 4°C. The following primary antibodies were used: rabbit-anti-mouse RANK(1:200, Acris, Herford, Germany), goat-anti-human RANK (1:100, R&D Systems, Minneapolis,MN), rabbit-anti-RANKL (human and mouse, 1:100, Acris) and mouse-anti β-actin (1:20,000Sigma-Aldrich) as a loading control. After washing 30 min with TBST, the membrane wasincubated with an appropriate secondary antibody in blocking buffer for 2 hours at roomtemperature. Subsequently, membranes were washed with TBST and with TBS before thebands were visualized with enhanced chemiluminescence (Perkin-Elmer Life Sciences, Boston,MA). Band intensity was measured and quantified using GeneSnap (SynGene, Synoptics,Cambridge, UK). RANK and RANKL expression levels were normalized to β-actin expression.ELISA Human and mouse OPG levels in lung tissue and culture supernatants were assessedusing ELISA (cat#DY805 (human), cat#DY459 (mouse), R&D Systems) according to theinstructions provided by the manufacturer. One hundred micrograms of tissue lysate totalprotein in a total of 100 μl was analyzed per sample. Cell culture media was analyzed undilutedwhile lung slice culture medium was diluted 1:5 before analysis.

The RANK/RANKL/OPG axis has a role in regulating tissue repair processes in lung
77
ImmunohistochemistryImmunohistochemical analysis of collagen I, RANK, and OPG was performed on 3 mparaffin sections of mouse lung tissue. Tissue sections were deparaffinized in xylene,rehydrated in alcohol, rinsed in milliQ water and placed in PBS. When applicable, sections weresubjected to antigen retrieval (0.1 N Tris-HCl pH 9.0 buffer, overnight at 80C). Prior to theincubation with the OPG antibody, sections were blocked with 1% BSA in 5% nonfat milk(Sigma-Aldrich). Primary antibodies were then incubated for 1h at room temperature in thepresence of 5% normal mouse serum. Antibodies used were goat-anti-mouse collagen I (1:75,Southern Biotech, Birmingham, AL), rabbit-anti-mouse OPG (1:400, Antibodies-online, Atlanta,GA), and goat-anti-human RANK (1:100, R&D Systems). Primary antibody incubation wasfollowed by incubation with species-specific PO-labeled secondary and optional tertiarypolyclonal antisera. PO-labeled antibodies were visualized using ImmPACT NovaRED kit(Vector, Burlingame, USA) with a hematoxylin counterstain when appropriate.The amount of collagen deposition in the lung was determined using ImageScopesoftware (Aperio, Burlingame, USA). After selecting the stained area of the lung sectionsexcluding edges of the tissue, large airways and vessels, a threshold was set to identify positivestaining of the tissue (represented by collagen). The percentage of stained-tissue surface pertotal-tissue surface analyzed was then calculated for each section.Cathepsin K activityCathepsin K activity of RAW264.7 macrophages was measured using a Cathepsin KActivity Kit (Cat#KA0769, Abnova, Taipei, Taiwan) following instructions provided by themanufacturer. Cathepsin K activity was normalized against the activity observed inunstimulated cells.Quantitative Real-time PCRTotal mRNA was isolated from cells or tissues using a Maxwell® LEV simply RNACells/Tissue kit (cat# AS1280, Promega, Madison, WI) according to the instructions providedby the manufacturer. Final mRNA concentrations were determined using a Nanodrop ND-100spectrophotometer (Nanodrop Technologies, Wilmington, DE). All primers were obtainedfrom Sigma-Aldrich (Zwijndrecht, The Netherlands), as described in Supplemental Table 1.Transcription levels of these genes were measured by using 20 ng cDNA per sample in aquantitative real-time PCR (SensiMix™ SYBR kit (Bioline, Taunton, MA)) and an ABI7900HTsequence detection system (Applied Biosystems, Foster City, CA). For each sample, thethreshold cycles (Ct values) were calculated using SDS 2.3 software (Applied Biosystems), andmRNA expression was normalized to β-actin for cell culture mRNA samples, YWHAZ for tissuemRNA samples, and 18S for precision-cut murine lung slices. Results are expressed as fold-change of healthy controls or control conditions.Production and purification of sRANKL proteinPlasmid pET15b-mRANKL, encoding the extracellular domain of murine RANKL(sRANKL; amino acids 160-316), was transformed into Escherichia coli strain BL21(DE3). A 10ml overnight culture was used to inoculate 1L 2xYT [16g Bacto Tryptone, 10g Bacto YeastExtract, 5g NaCl, pH 7.0] medium containing 0.1mg/l ampicillin. Cells were grown at 370C until

Chapter 2
78
the absorbance at 600 nm was 0.5 after which sRANKL production was induced by adding 0.1mM isopropyl β-D-1-thiogalactopyranoside and incubation continued for another 16 hours at20 °C. After harvesting and resuspending wet cells at 1 g/3 ml in buffer A [50 mM MES, pH5.8, 2 mM dithiothreitol, 10 % glycerol], cells were carefully sonicated on ice. Cell debris wasremoved by centrifugation for 60 min at 38,000xg and 4 °C. Supernatants were loaded on an 8ml cation exchange column (Source 30S, GE Healthcare, USA). sRANKL eluted early from alinear 0-500 mM NaCl gradient in buffer A. Pooled fractions at 4 °C were brought to pH 7.5 byadding drops of 1 M NaOH and to 1.3 M (NH4)2SO4 by slowly adding pulverised salt crystals.The protein was loaded on a 5 ml HiTrap phenyl sepharose column (GE Healthcare), pre-equilibrated with buffer B [20 mM sodium phosphate buffer, 2 mM dithiothreitol, 10 %glycerol, pH 7.5] and eluted early from a linear 1.3-0 M (NH4)2SO4 gradient in buffer B. Pooledfractions were run on a HiTrap Superdex75 column (GE Healtcare) in 20 mM sodiumphosphate buffer NaPi, [NaH2PO4, Na2HPO4, 10 % glycerol, pH 7.5]. Protein purity waschecked by SDS-PAGE and sRANKL concentrations were measured with a Coomassie Bradfordprotein assay kit (Pierce, USA) using BSA as reference and with a RANKL ELISA kit (cat#DY462,R&D Systems). Values from both assays were always in the same range and the concentrationobtained from the ELISA analysis was used for further experiments. Purified RANKL isroutinely tested for endotoxin contamination comparing heat-inactivated RANKL with nativeRANKL using RAW264.7 macrophages and no measurable contamination was found.Flow cytometryMouse left lungs were minced and incubated in RPMI containing 10% FCS, 10 μg/mlDNase I (grade II from bovine pancreas, Roche Applied Science, Almere, The Netherlands), and0.7 mg/ml collagenase A (Sigma-Aldrich) in a shaking water bath (37°C) for 45 min. Digestedlung tissue was mashed through a 70 μm nylon strainer (BD Biosciences, Breda, TheNetherlands) resulting in a single cell suspension. To remove contaminating erythrocytes, thecells were incubated with Pharmlyse (BD Biosciences) for 2 min at room temperature. Totalcell numbers were determined using a FACS Array cell counter (BD Biosciences) and used tocalculate total numbers of specific cell types.Single lung cell suspensions were stained for T cells, neutrophils, epithelial cells andmacrophage subsets by flow cytometry (see Supplemental Table 2 and 3 for antibodydetails). A viability dye was included (Fixable Viability Dye eFluor® 506, eBioscience, SanDiego, CA) to exclude dead cells. T cells were identified using anti-CD3-APC/Cy7 and epithelialcells were identified using anti-EpCAM-Alexafluor 647. Interstitial macrophages wereidentified as CD68+CD64negCD11c-low-expressing cells and alveolar macrophages wereidentified as CD68+CD64varCD11C-high-expressing cells using anti-CD68-PerCp/Cy5.5, anti-CD64-PE/Cy7 and anti-CD11-Brilliant Violet 785. Neutrophils were identified asCD68negGR1+ cells. In addition, the expression of RANKL on T cells, epithelial cells, andmacrophages was determined by using anti-RANKL-FITC. See supplemental data Figure 3 and4 for the gating strategy.Approximately 106 cells were incubated with cell surface marker antibodies in thepresence of 1% normal mouse serum (30 min, on ice, protected from light). The cells were thenwashed with PBS containing 2% FCS and 5 mM EDTA and fixed and permeabilized using a

The RANK/RANKL/OPG axis has a role in regulating tissue repair processes in lung
79
fixation and permeabilization buffer (30 min, eBioscience) to stain for intracellular markers.After washing the cells with permeabilization buffer, all samples were resuspended in FACSlysing solution (Biosciences) and stored at 4 °C until analysis. Samples were analyzed using anLSRII flow cytometer (BD Biosciences). The data were analyzed using FlowJo software (TreeStart, Ashland, USA). For each antibody, a Fluorescent Minus One (FMO) control was includedfor proper gating during data analyses.Statistical analysisAll groups were considered non-normally distributed due to small samples sizes.Therefore, statistical differences between two groups were assessed using nonparametricMann-Whitney U tests. For the comparison of multiple groups, we used Kruskal Wallis. Whensignificant, a Mann-Whitney-U test with a Bonferroni adjustment for multiple comparisons wasused as a post-test to calculate statistical differences between specific groups. Correlationsbetween OPG levels in lung and serum, and the correlation with collagen content in the lungswere assessed by calculating a nonparametric Spearman correlation coefficient (R).Significance was considered *= p<0.05. The data were analyzed using GraphPad Prism 6.

Chapter 2
80
Supplemental Table 1. Sequences of primersPrimer Forward sequence Reverse sequenceCol1α1 TGACTGGAAGAGCGGAGAGT ATCCATCGGTCATGCTCTCTFibronectin GCGACTCTGACTGGCCTTAC CCGTGTAAGGGTCAAAGCATMMP9 GTCCAGACCAAGGGTACAGC GCCTTGGGTCAGGCTTAGAGOPG ACAGTTTGCCTGGGACCAAA CTGTGGTGAGGTTCGAGTGGβ-actin ATCGTGCGTGACATCAAAGA ATGCCACAGGATTCCATACCYWHAZ ATCGTGCGTGACATCAAAGA ATGCCACAGGATTCCATACC18s CTTAGAGGGACAAGTGGCG ACGCTGAGCCAGTCAGTGTA
Supplemental Table 2. Overview of flow cytometry antibodies used to stain for T cells andepithelial cells.Antigen Fluorochrome Cataloge # Company
CD3 APC/Cy7 100222 BiolegendRANKL 488 510002 BiolegendEpCAM A647 118211 BiolegendLive/Dead e506 65-0866 eBioscience
Supplemental Table 3. Overview of flow cytometry antibodies used to stain for macrophages.Antigen Fluorochrome Cataloge # Company
CD11c BV785 563735 eBioscienceGR1 A700 108421 BiolegendLive/Dead e506 65-0866 eBioscienceCD68 PerCp/Cy5.5 137009 BiolegendCD64 PE/Cy7 139314 Biolegend

The RANK/RANKL/OPG axis has a role in regulating tissue repair processes in lung
81
Supplemental Figure 1. A. Collagen I mRNA expression was higher in lungs of mice exposed to silica(fibrosis, n=7) as compared to healthy controls (n=8). Levels were corrected for YHWAZ ashousekeeping gene. B. Representative pictures of an immunohistochemical staining for collagen Ideposition, showing that collagen I deposition was higher in the lungs of mice exposed to silica ascompared to saline-exposed controls. Collagen I expression is indicated by the red colour.

Chapter 2
82
Supplemental Figure 2. mRNA expressions of collagen IαI (A), fibronectin (B) and MMP9 (C) werehigher in fibrotic mouse lungs following silica exposure for 28 days (n=6) and vehicle treatment for twoweeks as compared to healthy controls (n=3), but unchanged in fibrotic lungs of mice that receivedsRANKL treatment for two weeks (5μg/mouse, n=6 and 10ug/mouse, n=6) as compared to vehicletreatment. Groups were compared using a Kruskall Wallis test for multiple testing and p<0.05 wasconsidered significant.

The RANK/RANKL/OPG axis has a role in regulating tissue repair processes in lung
83
Supplemental Figure 3. Gating strategy for T cells and epithelial cells.
Supplemental Figure 4. Gating strategy for macrophages and neutrophils.

Chapter 2
84

CHAPTER 3
Osteoprotegerin is an early markerof the fibrotic process and
of antifibrotic treatment responsesin ex vivo lung fibrosis
Kurnia S.S. Putri | Carian E. Boorsma | Adhyatmika |Peter Heukels | Wim Timens | Habibie | Marina H. de Jager |
Wouter L.J. Hinrichs | Peter Olinga | Barbro N. Melgert

Chapter 3
86
ABSTRACTLung fibrosis is a chronic lung disease with a high mortality rate with only twoapproved drugs (pirfenidone and nintedanib) to attenuate its progression. To datethere are no reliable biomarkers that can assess fibrosis development and/ortreatment effect for these two drugs. Osteoprotegerin (OPG) is used as a serum markerto diagnose liver fibrosis and we have found it may be applicable in lung fibrosis aswell. We therefore set out an ex vivo study to investigate the regulation of OPG in lungtissue to elucidate whether it tracks with fibrosis development and responds toantifibrotic treatment to assess its potential use as a biomarker.Control murine and human lung slices were incubated with transforming growth factorbeta-1 (TGFβ1) or interleukin-13 (IL-13) to induce early-stage fibrosis. Pirfenidone ornintedanib were added to evaluate OPG responses towards therapy. mRNAexpressions of OPG, collagen1α1 (Col1α1), fibronectin (Fn1) and plasminogenactivator inhibitor-1 (PAI-1) were measured by qPCR to assess fibrosis developmentand OPG protein production was measured by ELISA.OPG mRNA expression in murine lung slices was higher after 48 hours incubation andwith TGFβ1 or IL-13 stimulation and closely correlated with Fn1 and PAI-1 mRNAexpression. More OPG protein was released from TGFβ1- and IL13-stimulated murinelung slices and from fibrotic human lung slices than from the control slices. OPG releasewas lower from pirfenidone- and nintedanib-treated murine TGFβ-induced fibroticslices, and from pirfenidone-treated human fibrotic lung slices than from theirrespective controls.OPG can already be detected during early stage fibrosis development and responds,both in early- and late-stage fibrosis, to treatment with the two antifibrotic drugscurrently on the market for lung fibrosis. Therefore, OPG should be further investigatedas a potential biomarker for lung fibrosis and a potential surrogate marker fortreatment effect.Keywords: precision-cut lung slices, lung fibrosis, osteoprotegerin, biomarker, woundhealing, fibronectin, PAI-1

Osteoprotegerin is an early marker of the fibrotic process andof antifibrotic treatment responses in ex vivo lung fibrosis
87
INTRODUCTIONLung fibrosis is a chronic lung disease that is characterized by deposition ofexcessive extracellular matrix (ECM) in lung parenchyma leading to organ malfunction,disruption of gas exchange, and death from respiratory failure 1,2. It has a high mortalityrate with an average survival after diagnosis of about 2-3 years 3,4. The etiology of lungfibrosis remains unclear but likely includes repeated injury to and therefore loss ofepithelial cells due to exposure to allergens, chemicals, radiation, and/orenvironmental particles, followed by a dysregulated wound repair response involvingfibroblasts and macrophages 2,3,5. To date there are no effective drugs to stop or reversefibrosis development, but two drugs have been approved by the FDA that can attenuateprogression of the disease: pirfenidone and nintedanib6–8. Unfortunately, thedevelopment of more and better drugs is hampered by the lack of easy-to-assess earlymarkers of fibrosis development and treatment responses.We recently described a novel marker called osteoprotegerin (OPG) that ishighly upregulated in lung tissue of patients with lung fibrosis and appears to beinvolved in regulating alveolar epithelial regeneration9. OPG is also known as tumornecrosis factor receptor superfamily member 11B (TNFRSF11B) and is a decoyreceptor for receptor activator for nuclear factor kappa-B ligand (RANKL) and tumornecrosis factor-related apoptosis-inducing ligand (TRAIL) 10. OPG is a key factor in theregulation of osteogenesis and is produced by osteoblasts to control osteoclastactivity11, but interestingly our recent studies indicate that through its capturing ofRANKL it can also inhibit alveolar epithelial repair9. It is produced and secreted by lungfibroblasts under the influence of the key profibrotic mediator transforming growthfactor beta (TGF) 9,12 . Studies in patients with liver fibrosis have shown that OPG isdetectable in serum and that is correlated with disease severity13–15 but for lungfibrosis the association of serum levels with disease severity are unclear 16. In addition,there is no data available if and how OPG secretion is affected by treatment with thetwo antifibrotic drugs currently on the market. We therefore set out to investigate theregulation of OPG early in the process of fibrosis development to elucidate whether ittracks with fibrosis development and responds to antifibrotic treatments to assess itspotential use as a biomarker.

Chapter 3
88
For these studies we used precision-cut lung slices because this model allowsus to study multicellular processes in lung tissue of both humans and experimentalanimals, without the influence of other organs (e.g. bone) on production of OPG. Lungslices contain all resident lung cells in their original environment and tissuearchitecture and, importantly, intercellular and cell-matrix interactions remain intactin these tissue slices 17. To model early processes of fibrosis development we incubatedmurine lung slices with either transforming growth factor beta-1 (TGFβ1) orinterleukin-13 (IL-13) as overexpression of both cytokines in lung tissue has beenshown to lead to development of lung fibrosis 18,19. To investigate whether OPGsecretion is affected by treatment with antifibrotic drugs pirfenidone and nintedaniband tracks with inhibition of fibrotic processes, we treated murine fibrotic lung slicesand slices from lungs of patients with end-stage lung fibrosis with pirfenidone andnintedanib.MATERIALS AND METHODS
Murine Precision-cut Lung SlicesEight- to twelve-week old C57BL/6 male mice from Harlan (Horst, TheNetherlands) were kept in cages with a 12 hours of light/dark cycle and received foodand water ad libitum. The experiments were approved by the Institutional Animal Careand Use Committee of the University of Groningen (DEC6416AA).Precision-cut lung slices were prepared according to the method of Oenemaand co-workers20, with several modifications. Shortly, mice were anaesthetized withisoflurane/O2 (Nicholas Piramal, London, UK) and then sacrificed by exsanguinationvia the aorta abdominalis. The lungs were filled with low-melting temperature agarose(1.5%) (Sigma-Aldrich, Steinheim, Germany) in 0.9% NaCl through the cannulatedtrachea, and the lungs were directly transferred into ice-cold University of Wisconsinorgan preservation solution (UW-solution). Cores of lung tissue were made by using abiopsy-puncher with a 5-mm diameter. Slices, with a weight of about 5 mg (orthickness of 250-300 µm), were prepared from these cores with a Krumdieck tissueslicer (Alabama Research and Development, USA), which was filled with ice-coldKrebs-Henseleit Buffer supplemented with 25 mM D-glucose (Merck, Darmstadt,

Osteoprotegerin is an early marker of the fibrotic process andof antifibrotic treatment responses in ex vivo lung fibrosis
89
Germany), 25 mM NaHCO3 (Merck), 10 mM HEPES (MP Biomedicals, Aurora, OH),saturated with carbogen (95% O2/5% CO2) and adjusted to pH 7.4.After slicing, slices were incubated in a pre-warmed 12-well plate which wasfilled with 1.3 mL DMEM + Glutamax medium containing 4.5g/L D-glucose andpyruvate (Gibco® by Life Technologies, Grand Island, New York, USA) supplementedwith non-essential amino acid mixture (1:100), penicillin-streptomycin, 45 µg/mlgentamycin (Gibco® by Life Technologies, Grand Island, New York, USA) and 10% fetalcalf serum (FCS). After 1 hour of pre-incubation at 37 C in an O2/CO2-incubator (MCO-18M, Sanyo, USA) which was continuously shaken at a speed of 90 rpm and saturatedwith 80% O2 and 5% CO2, medium was refreshed. Slices were then incubated for 48hours in medium with or without several cytokines to mimic the fibrotic process, andwith or without antifibrotic compounds. Transforming growth factor-1 (TGFβ1, 5ng/ml) and interleukin-13 (IL-13, 10 ng/ml) were added to investigate their influenceon development of fibrosis and production of OPG, while 1 mM pirfenidone and 0.5 µMnintedanib were added as antifibrotic compounds. Medium and treatments wererefreshed after 24 hours. After 48 hours of incubation, culture medium and slices weresnap frozen into liquid nitrogen, and stored at -80 C until analysis.Human Precision-cut Lung SlicesHuman fibrotic lung tissue was collected with informed consent from patientswith end-stage lung fibrosis undergoing lung transplantation at either the UniversityMedical Center Groningen (UMCG) or at the Erasmus Medical Center Rotterdam. InGroningen, the study protocol was consistent with the Research Code of the UniversityMedical Center Groningen and Dutch national ethical and professional guidelines(http://www.federa.org). In Rotterdam, the Medical Ethical Committee approved allprotocols followed in that center. Control lung tissue was obtained at the UMCG frompatients undergoing surgical resection for suspected carcinoma. During preparation,lung tissue was preserved in ice-cold University of Wisconsin organ preservationsolution (UW-solution). Cores of lung tissue of 5 mm were made and slices wereprepared from these cores with a Krumdieck tissue slicer and were incubated insupplemented DMEM medium, as described for murine slices. Pirfenidone (2.5 mM)was added as antifibrotic drug. Medium and treatments were refreshed after 24 hours.

Chapter 3
90
After 48 hours of incubation, culture medium and slices were snap frozen into liquidnitrogen, and stored at -80 C until analysis.ELISA Murine and human OPG levels in slice incubation medium were measured usingELISA (cat #DY459 (murine), cat #DY805 (human), R&D Systems) according to theinstructions provided by the manufacturer. Total OPG in the medium was corrected forthe protein content of the slices, which was measured by Lowry (BIO-rad RC DC ProteinAssay, Bio Rad, Veenendaal, The Netherlands).Quantitative Real-time PCRTotal mRNA was isolated from slices using a Maxwell® LEV simply RNACells/Tissue kit (Promega, Madison, WI). Final RNA concentrations were determinedusing Biotek Synergy HT (Biotek®, Winoosku, Vermont, USA). The conversion of RNAinto cDNA was performed by using a reverse transcriptase kit (Promega, Leiden, TheNetherlands) in a master-cycler gradient (25°C for 10 min, 45°C for 60 min, and 95°Cfor 5 min). Transcription levels of OPG, fibrosis-associated genes (collagen 1α1(Col1α1), fibronectin (Fn1), plasminogen activator inhibitor-1 (PAI-1)) weremeasured using a SensiMix™ SYBR kit (Bioline, Luckenwalde, Germany) or Taqman(Eurogentech, Maastricht, The Netherlands) and a 7900HT Real-Time RT-PCRsequence detection system (Applied Biosystems, Bleiswijk, The Netherlands) with 45cycles of 10 min 95°C, 15 sec at 95°C, and 25 sec at 60°C following with a dissociationstage (SYBR kit) or or with 40 cycles of 10 min at 95 °C, 15 s at 95 °C and 1 min at 60 °C(TaqMan).Gene expression was quantified using Ct values of the genes in the SDS 2.3software program (Applied Biosystems). mRNA expression in murine lung slices wasnormalized against 18s as housekeeping gene, while human lung slices werenormalized against GAPDH as housekeeping gene. Gene expression is shown as the foldinduction (2-ΔCt) in graphs. All primers, as listed in Table 1, were obtained from Sigma-Aldrich (Zwijndrecht, The Netherlands).

Osteoprotegerin is an early marker of the fibrotic process andof antifibrotic treatment responses in ex vivo lung fibrosis
91
Table 1. Sequences of primersPrimer Forward sequence Reverse sequence Probe
Murine18s CTTAGAGGGACAAGTGGCG ACGCTGAGCCAGTCAGTGTACol1α1 TGACTGGAAGAGCGGAGAGT ATCCATCGGTCATGCTCTCTFn1 CGGAGAGAGTGCCCCTACTA CGATATTGGTGAATCGCAGAPAI-1 GCCAGATTTATCATCAATGACTGGG GGAGAGGTGCACATCTTTCTCAAAGOPG ACAGTTTGCCTGGGACCAAA CTGTGGTGAGGTTCGAGTGGTGFβ AGGGCTACCATGCCAACTTC GTTGGACAACTGCTCCACCTIL13Rα2 TGAAAGTGAAGACCTATGCTTT GACAAACTGGTACTATGAAAATHumanGAPDH ACCAGGGCTGCTTTTAACTCT GGTGCCATGGAATTTGCC TGCCATCAATGACCCCTTCACol1α1 CAATCACCTGCGTACAGAACGCC CGGCAGGGCTCGGGTTTC CAGGTACCATGACCGAGACGTGFn1 AGGCTTGAACCAACCTACGGATGA GCCTAAGCACTGGCACAACAGTTT ATGCCGTTGGAGATGAGTGGGAAPAI-1 CACGAGTCTTTCAGACCAAG AGGCAAATGTCTTCTCTTCCOPG CCTGGCACCAAAGTAAACGC TGCTCGAAGGTGAGGTTAGC
StatisticsResults are presented as box-and-whisker plots using the median and min/maxwhiskers including individual data points or as aligned before-after plots. All resultsobtained from more than 8 individual experiments were analyzed for its normality byusing D’Agostino & Pearson omnibus normality test. Datasets that did not have anormal distribution were log-transformed to obtain normality and if data were still notnormally distributed then nonparametric tests were used. All results that werenormally distributed, were analyzed by using a paired or unpaired Student’s t-test. Allresults that were not normally distributed or obtained from less than 8 individualexperiments were analyzed by Wilcoxon or Mann Whitney test for paired or unpaireddata, respectively. When comparing multiple groups, a parametric one-way ANOVAwith Holm-Sidak correction or non-parametric paired Friedman with Dunn’scorrection was performed depending on normality of the data. Correlations wereassessed by calculating the Pearson correlation coefficient. A p-value lower than 0.05(p<0.05) was considered as significant.

Chapter 3
92
RESULTS
More osteoprotegerin as well as other fibrosis-associated markerswere higher
after TGFβ1 stimulation as compared to controlTo study early onset of fibrosis development, we incubated murine lung sliceswith and without TGFβ1 and quantified mRNA expressions levels of several fibrosismarkers after 48 hours of incubation. We found that OPG mRNA levels in murine lungslices (Figure 1a) were significantly higher after 48 hours of incubation, which wasaccompanied by higher Col1α1 (Figure 1c) and Fn1 (Figure 1d) mRNA levels.Furthermore, incubation of slices with TGFβ1 resulted in an even higher OPG mRNAlevel (Figure 1a) and protein excretion (Figure 1b) and corresponded withsignificantly higher expression of Fn1 (Figure 1d) and PAI-1 (Figure 1e).Osteoprotegerin exhibits a more sensitive response towards IL-13 stimulation in
murine lung slices than other fibrosis markersIn addition to TGFβ1, we also investigated the effect of IL-13, anotherprofibrotic cytokine, on murine lung slices. As depicted in Figure 2a and b, OPG mRNAand protein levels were higher after IL-13 stimulation as compared to untreated lungslices, similar to the effect of TGFβ1 on murine lung slices. IL-13 did not induceexpression of the three other fibrosis markers tested in this study, i.e. Col1α1, Fn1 andPAI-1 (Figures 2c, d, e).

Osteoprotegerin is an early marker of the fibrotic process andof antifibrotic treatment responses in ex vivo lung fibrosis
93
Figure 1. Responses of murine lung slices to incubation and treatment with TGFβ1. Osteoprotegerin(OPG) mRNA expression was significantly higher after 48 hours of incubation than freshly cut slices andexpressed at even higher levels with TGFβ1 treatment (a), and this was accompanied by higher OPGprotein excretion (b). Collagen 1α1 (Col1α1) mRNA expression was significantly higher after 48 hoursof incubation as compared to freshly cut slices and showed no extra response towards TGFβ1 treatment(c). The mRNA expression of fibronectin (Fn1) was significantly higher after 48 hours of incubation thanfreshly cut slices and even higher with TGFβ1 treatment (Figure 1d). Plasminogen activator inhibitor-1(PAI-1) expression was not higher after 48 hours of incubation, but significantly higher with TGFβ1stimulation (Figure 1e). Groups were compared using a one-way ANOVA with Holm-Sidak correction formultiple testing, p<0.05 was considered significant.

Chapter 3
94
Figure 2. Responses of murine lung slices to treatment with IL-13. Osteoprotegerin (OPG) mRNA (a) andprotein expression (b) were significantly (or near-significantly) higher after IL-13 stimulation. IL-13 didnot induce expressions of collagen 1a1 (Col1α1, c), fibronectin (Fn1, d) or plasminogen activatorinhibitor-1 (PAI-1, e) mRNA. Groups were compared using a paired Wilcoxon test, p<0.05 wasconsidered significant.To investigate the mechanism behind IL13-stimulated OPG production, we alsomeasured mRNA expression of TGFβ and interleukin 13 receptor alpha 2 (IL13Rα2) inIL-13 stimulated murine lung slices. We found that IL-13 did not lead to higher TGFβmRNA expression (Figure 3a), but did significantly induce IL13Rα2 mRNA expression(Figure 3b). We further treated IL13-stimulated murine lung slices with galunisertib,an inhibitor of TGFβ receptor 1 kinase, and measured OPG mRNA expression and OPGprotein excretion. As depicted in Figure 3c and Figure 3d, galunisertib treatment

Osteoprotegerin is an early marker of the fibrotic process andof antifibrotic treatment responses in ex vivo lung fibrosis
95
resulted in significantly lower OPG mRNA expression and induced a trend towardslower OPG protein excretion by IL13-stimulated murine lung slices.
Figure 3. Responses of murine lung slices to IL-13 stimulation and galunisertib treatment. IL-13 did notlead to higher TGFβ mRNA expression (a) but did result in significantly higher interleukin 13 receptoralpha 2 mRNA expression (IL13Rα2) (b). Galunisertib treatment of IL13-stimulated murine lung slicesresulted in significantly lower osteoprotegerin (OPG) mRNA expression (c) and a trend towards lowerOPG protein excretion (d) compared to IL-13 stimulation alone. Groups were compared using aWilcoxon test, p<0.05 was considered significant.Osteoprotegerin expression strongly correlates with Fn1 and PAI-1 expression in
TGFβ1-stimulated murine lung slices and less so in IL-13-stimulated slicesTo investigate whether or not expression of OPG correlates with other fibrosismarkers, we compared OPG mRNA expression with mRNA expressions of each of theother fibrosis marker from the same experiments. We also compared OPG mRNAexpression with the OPG released in the medium from the same experiments to analyzethe correlation between mRNA and protein expression.For TGFβ1-stimulated lung slices, there were strong positive correlationsbetween OPG mRNA expression and secreted OPG protein in slice medium (Figure 4a),Fn1 mRNA expression (Figure 4e) and PAI-1 mRNA expressions (Figure 4g), but notwith Col1α1 mRNA expression (Figure 4c).

Chapter 3
96Figure 4. Correlations of OPG mRNA expression with OPG protein excretion and with the expression ofother fibrosis markers (Col1α1, Fn1, and PAI-1) in murine lung slices stimulated with TGFβ1 (a, c, e, g)and IL-13 (b, d, f, h). Correlations were tested using a Pearson test and presented as log data. p<0.05 wasconsidered significant

Osteoprotegerin is an early marker of the fibrotic process andof antifibrotic treatment responses in ex vivo lung fibrosis
97
For IL-13-stimulated lung slices, OPG mRNA expression only showed a trendtowards a positive correlation with PAI-1 mRNA expression (Figure 4h) and asignificant positive correlation with OPG secretion in slice medium (Figure 4a).More osteoprotegerin was released from human fibrotic lung than from slices of
control lung tissueSimilar to our findings in murine lung slices, more OPG was released fromcontrol human lung slices that were incubated with TGFβ1 for 48 hours than fromuntreated slices (Figure 5a). In addition, slices made from fibrotic human tissuereleased more OPG in during 48 hours of incubation than slices made from control lungtissue (Figure 5b). We also checked whether or not expression of OPG mRNAcorrelated with expression of fibrosis markers Col1α1, Fn1, and PAI-1 and found onlya trend towards a positive correlation with Col1α1 (r=0.51, p=0.09) and Fn1expression (r=0.53, p=0.07) (data in supplementary data Figures 1a-d).
Figure 5. More osteoprotegerin was released from TGFβ1-stimulated human control lung slices thanfrom the unstimulated slices, n=1 (a). After 48 hours of incubation, human fibrotic lung slices releasedmore OPG than the human control lung slices (b).Osteoprotegerin release responds to antifibrotic treatment in murine and human
fibrotic lung slicesWe investigated whether OPG release would be affected by treatment withpirfenidone or nintedanib using TGFβ1-stimulated murine lung slices and humanfibrotic lung slices, respectively. Incubating human fibrotic lung slices with 2.5 mMpirfenidone or murine lung slices with 0.5 µM nintedanib, did not affect the viability ofslices (no changes in ATP content with treatment, data not shown). However, in the

Chapter 3
98
case of murine lung slices, treatment with 2.5 mM pirfenidone resulted in a significantloss of viability (data not shown). Therefore, for murine lung slices we used pirfenidoneat a concentration of 1 mM, which did not result in significantly lower viability scores.Treatment of TGFβ-stimulated murine lung slices with 1 mM pirfenidoneresulted in a trend towards lower OPG levels in culture medium (Figure 6a). Similarto murine slices, treatment of human fibrotic lung slices with 2.5 mM pirfenidone alsoresulted in a trend towards lower OPG protein excretion into culture medium (Figure
6b). Lower levels of OPG protein excretion from pirfenidone-treated murine lungslices were not accompanied by lower levels of Col1α1 or Fn1 mRNA expression(Figures 6c, 6e), but was accompanied by a trend towards lower mRNA expression ofPAI-1 as compared to lung slices only stimulated with TGFβ1 (Figure 6g). In contrast,lower OPG protein excretion from pirfenidone-treated human lung slices was notaccompanied by lower mRNA expressions of Col1α1, Fn1, and PAI-1 as compared tountreated slices (Figures 6d, 6f, 6h).Similar to the effects of pirfenidone, nintedanib treatment of TGFβ-stimulatedmurine lung slices also resulted in lower OPG mRNA expression (Figure 7a) OPGprotein excretion (Figure 7b) albeit not significantly due to the low number ofreplicates. These lower levels of OPG mRNA and protein excretion after nintedanibtreatment were accompanied by lower mRNA expression of Fn1 (Figure 7d), but notCol1α1 (Figure 7c) and PAI-1 (Figure 7e).
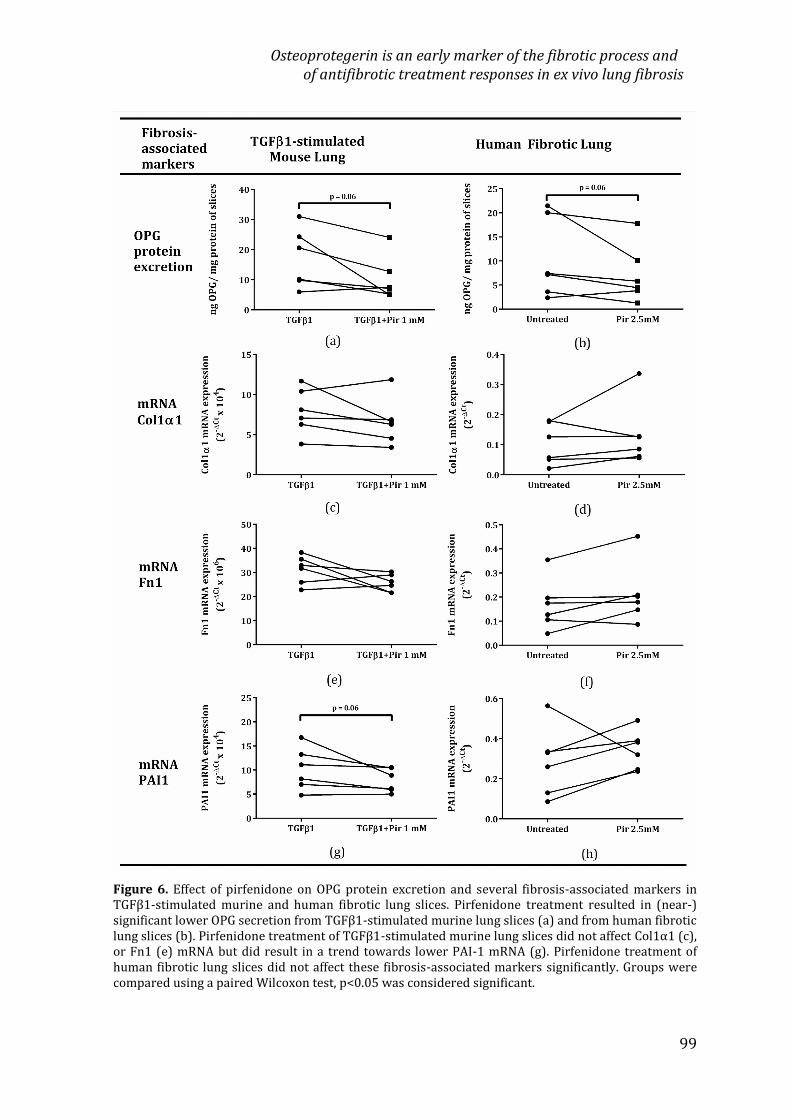
Osteoprotegerin is an early marker of the fibrotic process andof antifibrotic treatment responses in ex vivo lung fibrosis
99
Figure 6. Effect of pirfenidone on OPG protein excretion and several fibrosis-associated markers inTGFβ1-stimulated murine and human fibrotic lung slices. Pirfenidone treatment resulted in (near-)significant lower OPG secretion from TGFβ1-stimulated murine lung slices (a) and from human fibroticlung slices (b). Pirfenidone treatment of TGFβ1-stimulated murine lung slices did not affect Col1α1 (c),or Fn1 (e) mRNA but did result in a trend towards lower PAI-1 mRNA (g). Pirfenidone treatment ofhuman fibrotic lung slices did not affect these fibrosis-associated markers significantly. Groups werecompared using a paired Wilcoxon test, p<0.05 was considered significant.

Chapter 3
100
Figure 7. Effects of nintedanib on TGFβ1-stimulated murine lung slices. Nintedanib treatment of TGFβ1-stimulated murine lung slices resulted in trends towards lower mRNA expression of OPG (a) and Fn1(d), lower OPG protein secretion (b), and exhibited no effects on mRNA expression of Col1α1 (c) andPAI-1 (e). Groups were compared using a Wilcoxon test, p<0.05 was considered significant.DISCUSSIONOur study has shown that the slicing procedure itself triggers a repair orregenerative response, which was indicated by higher Col1α1 and Fn1 mRNAexpressions and was accompanied by higher production of OPG. In addition, promotingfibrosis by incubating with TGFβ1 aggravates this repair response with even higherproduction of OPG. These data suggest that OPG is part of normal wound repair and

Osteoprotegerin is an early marker of the fibrotic process andof antifibrotic treatment responses in ex vivo lung fibrosis
101
also tracks with ‘wound repair gone wrong’ when developing into fibrosis.Interestingly, OPG expression also responded to pirfenidone and nintedanib treatment(the two antifibrotic drugs currently on the market for treatment of IPF), even whenother fibrosis-associated markers did not respond (yet), making it a potential sensitivesurrogate marker for treatment effect.OPG was initially studied for its role in bone turnover in which it prevents boneresorption and stimulates production of extracellular matrix in cartilage21. However,in recent years, an increasing number of studies have shown correlations between OPGand several fibrotic conditions including liver, vascular, cardiac, kidney and intestinalfibrosis 22-299,22–29. This study, and our own previous study 9, have now shown this alsoappears the case for wound healing and fibrosis in lung tissue. How OPG actuallyinfluences wound repair and fibrosis is still an open question, but a study by Hao et al.suggests an interaction between TRAIL, OPG, and collagen-producing cells 30. UsingOPG -/- mice, they showed activation of matrix metalloproteinases without acompensatory increase in collagen synthesis leading to lower collagen deposition inheart tissue and development of left ventricle dilatation. This was accompanied byincreased expression of TRAIL and a higher level of apoptosis in heart tissue,suggesting an association between TRAIL, OPG, and collagen-producing cells inphysiological tissue remodeling. We have previously shown fibroblasts andmyofibroblasts to be important producers of OPG 9,12 and these cells are also key cellsin collagen production 31,32. Combined these findings indicate that OPG may protect(myo)fibroblasts from TRAIL-induced apoptosis and can thereby contribute to bothphysiological wound healing and fibrosis.Interestingly, there appears to be no direct interaction of OPG with collagenproduction as OPG mRNA expression did not correlate with Col1α1 mRNA expression,while it did correlate with Fn1 and PAI-1. Little is known about interactions betweenFn1, PAI-1 and OPG, however, a study by Vial et al showed that PAI-1 stimulates Fn1matrix assembly by disrupting the interaction between αvβ5 and vitronectin that thenstimulates activation of α5β1 integrin, which increases the rate of Fn1polymerization33. OPG has been shown to bind to αv integrins as well and maytherefore have a stimulating effect on these interactions 34.

Chapter 3
102
As we have shown before, OPG expression in lung tissue is clearly controlled byTGFβ, the master regulator of fibrotic responses 9,12,23. To get more insight into theregulation of OPG expression, we also studied the effect of IL-13, a fibrosis-associatedcytokine, on murine lung slices. Previous studies have shown that IL-13 plays animportant role in the development of lung fibrosis 35–37. However, unlike TGFβ1, IL-13stimulation of murine lung slices did not induce mRNA expression of Col1α1, Fn1 andPAI-1, while it did induce production of OPG mRNA and protein. This inductionappeared to be solely dependent on TGFβ1 as co-treatment with galunisertib, a TGFβreceptor-I kinase inhibitor, could completely block the OPG-inducing effect of IL-13.Our previous studies in liver have shown that IL-13 can induce OPG through IL-13receptor α2 (IL13Rα2)-induced TGFβ production 12. In these lung studies we did findhigher expression of IL13Rα2 after treatment with IL-13, but not a concomitantincrease in TGFβ expression, which is in contrast with the clear inhibitory effect ofgalunisertib on IL-13-induced OPG production. This may be caused by a difference inkinetics of the different mRNA transcripts studied as we did find a trend towards acorrelation between PAI-1 and OPG mRNA expression after IL-13 stimulation.Importantly, we found that OPG was also released by human lung slices. Bothslices from lung tissue of a patient with normal lung function as well as slices from lungtissue of patients with lung fibrosis secreted OPG, with the latter releasing far higheramounts of it than slices of control lung tissue. The OPG production by control lungtissue could also be increased by stimulating with TGFβ1, suggesting similar pathwaysin mice and men. Due to limited availability of lung tissue from patients with normallung function, we only obtained one sample for this study and these studies shouldtherefore be extended for definite conclusions. Our results, however, do confirm thepossibility of studying OPG as a marker of lung fibrosis in a clinical setting. Theadvantage of using OPG as marker of remodeling and fibrosis over other extracellularmatrix proteins is that OPG is a soluble protein that can easily be measured in blood orculture media. Our results show a strong correlation between mRNA and protein levelsof OPG. Higher OPG mRNA expression was always accompanied by higher OPGreleased from lung tissue slices. Serum OPG could therefore mirror what happens inlung tissue and should be investigated as a marker for lung fibrosis or for progressionof fibrotic disease in clinical practice.

Osteoprotegerin is an early marker of the fibrotic process andof antifibrotic treatment responses in ex vivo lung fibrosis
103
In the interest of clinical applicability of OPG as a marker for treatment effects,we further investigated whether OPG production is affected by treatment withantifibrotic drugs. Pirfenidone and nintedanib were used in this study since they havebeen proven to slow down lung function decline due to fibrosis and were thusapproved by the FDA for IPF treatment 6–8. We found that OPG mRNA and proteinproduction were indeed inhibited by both drugs in TGFβ1-stimulated murine andfibrotic human lung slices, even though expressions of fibrosis-associated ECMmarkers were not (yet) affected. The reason for this discrepancy is unclear but may berelated to different kinetics of mRNA transcripts. The half-life of OPG mRNA wasdescribed to be around 4 hours 38, whereas Col1a1 mRNA was shown to have a half-life between 12-24 hours depending on the type of fibroblast studied39. As many in
vitro, in vivo, and clinical studies have shown pirfenidone and nintendanib can inhibitextracellular matrix production 40–46, our results reinforce the notion that OPG may bean early marker of treatment effect as OPG production was already inhibited beforeother markers are affected both in experimental early and late fibrotic disease.CONCLUSIONOPG seems to be part of normal wound repair and also tracks with ‘wound repairgone wrong’ when developing into fibrosis. Its expression closely correlates with Fn1and PAI-1 mRNA suggesting interactions between these genes during fibrogenesis. AsOPG can easily be measured in serum it is an interesting candidate to furtherinvestigate as a potential biomarker for fibrotic disorders of the lung. OPG expressionalso responds to pirfenidone and nintedanib, therefore also making it a potentialsurrogate marker for treatment effect.REFERENCES1. Wilson, M. S. & Wynn, T. A. Pulmonary fibrosis: Pathogenesis, etiology and regulation. Mucosal
Immunol. 2, 103–121 (2009).2. Wynn, T. A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 208, 1339–1350 (2011).3. Rafii, R., Juarez, M. M., Albertson, T. E. & Chan, A. L. A review of current and novel therapies foridiopathic pulmonary fibrosis. J. Thorac. Dis. 5, 48–73 (2013).4. Raghu, G., Weycker, D., Edelsberg, J., Bradford, W. Z. & Oster, G. Incidence and prevalence ofidiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 174, 810–816 (2006).

Chapter 3
104
5. Chua, F., Gauldie, J. & Laurent, G. J. Pulmonary fibrosis: Searching for model answers. Am. J. Respir.Cell Mol. Biol. 33, 9–13 (2005).6. Karimi-Shah, B. A. & Chowdhury, B. A. Forced Vital Capacity in Idiopathic Pulmonary Fibrosis —FDA Review of Pirfenidone and Nintedanib. N. Engl. J. Med. 372, 1189–1191 (2015).7. Costabel, U. et al. Efficacy of nintedanib in idiopathic pulmonary fibrosis across prespecifiedsubgroups in INPULSIS. Am. J. Respir. Crit. Care Med. 193, 178–185 (2016).8. Keating, G. M. Nintedanib: A Review of Its Use in Patients with Idiopathic Pulmonary Fibrosis.Drugs 75, 1131–1140 (2015).9. Boorsma, C. et al. The RANKL-OPG balance in pulmonary fibrosis. in 1.5 Diffuse Parenchymal LungDisease 46, PA3809 (European Respiratory Society, 2015).10. Reid, P. & Holen, I. Pathophysiological roles of osteoprotegerin (OPG). Eur. J. Cell Biol. 88, 1–17(2009).11. Harada, S. & Rodan, G. A. Control of osteoblast function and regulation of bone mass. Nature 423,349–355 (2003).12. Adhyatmika, A. et al. Osteoprotegerin production and profibrotic activity in liver fibrosis areTGFβ dependent. J. Hepatol. 66, S147 (2017).13. Yilmaz, Y. et al. Serum levels of osteoprotegerin in the spectrum of nonalcoholic fatty liverdisease. Scand. J. Clin. Lab. Invest. 70, 541–546 (2010).14. García-Valdecasas-Campelo, E. et al. Serum osteoprotegerin and rankl levels in chronic alcoholicliver disease. Alcohol Alcohol. 41, 261–266 (2006).15. Bosselut, N. et al. Including osteoprotegerin and collagen IV in a score-based blood test for liverfibrosis increases diagnostic accuracy. Clin. Chim. Acta J.L. Bosson; A. Paris; ANRS L. Allain, Paris.Methodol. Hospices Civ. Lyon. M-C. Gelineau, B. Poggi 415, 63–68 (2013).16. McGrath, E. E. et al. Deficiency of tumour necrosis factor-related apoptosis-inducing ligandexacerbates lung injury and fibrosis. Thorax 67, 796–803 (2012).17. Westra, I. M. Precision-cut liver slices: an ex vivo model for the early onset and end-stage of liverfibrosis. 97 (2014).18. Sime, P. J., Xing, Z., Graham, F. L., Csaky, K. G. & Gauldie, J. Adenovector-mediated gene transfer toactivate transforming growth factor beta1 induces prolonged severe fibrosis in lung. J.Clin.Invest.100, 768–776 (1997).19. Lee, C. G. et al. Interleukin-13 Induces Tissue Fibrosis by Selectively Stimulating and ActivatingTransforming Growth Factor β 1. J. Exp. Med. 194, 809–822 (2001).20. Oenema, T. A. et al. Bronchoconstriction Induces TGF-β Release and Airway Remodelling inGuinea Pig Lung Slices. PLoS One 8, (2013).21. Kostenuik, P. & Shalhoub, victoria. Osteoprotegerin A Physiological and PharmacologicalInhibitor of Bone Resorption. Curr. Pharm. Des. 7, 613–635 (2001).22. Sen, O. et al. The relation between serum levels of osteoprotegerin and postoperative epiduralfibrosis in patients who underwent surgery for lumbar disc herniation. Neurol. Res. 27, 452–455(2005).23. Toffoli, B. et al. Osteoprotegerin promotes vascular fibrosis via a TGF-β1 autocrine loop.Atherosclerosis 218, 61–68 (2011).24. Ambroszkiewicz, J., Sands, D., Gajewska, J., Chelchowska, M. & Laskowska-Klita, T. Bone turnovermarkers, osteoprotegerin and RANKL cytokines in children with cystic fibrosis. Adv. Med. Sci. 58,338–343 (2013).25. Jiang, J. Q., Lin, S., Xu, P. C., Zheng, Z. F. & Jia, J. Y. Serum osteoprotegerin measurement for earlydiagnosis of chronic kidney disease-mineral and bone disorder. Nephrology 16, 588–594 (2011).26. Montañez-Barragán, A. et al. Osteoprotegerin and kidney disease. J. Nephrol. 27, 607–617 (2014).27. Sylvester, F. A., Draghi, A., Bausero, M. A., Fernandez, M. L. & Vella, A. T. 1149 OsteoprotegerinExpression is Upregulated in the Colon of Children With Active Inflammatory Bowel Disease.Gastroenterology 142, S-209 (2012).28. Adhyatmika, A. et al. Osteoprotegerin is more than a serum marker in liver fibrosis. J. Hepatol.66, S147–S148 (2017).

Osteoprotegerin is an early marker of the fibrotic process andof antifibrotic treatment responses in ex vivo lung fibrosis
105
29. Bosselut, N. et al. Including osteoprotegerin and collagen IV in a score-based blood test for liverfibrosis increases diagnostic accuracy. Clin. Chim. Acta 415, 63–68 (2013).30. Hao, Y. et al. Cardiac hypertrophy is exacerbated in aged mice lacking the Osteoprotegerin gene.Cardiovasc. Res. 110, 62–72 (2016).31. McAnulty, R. J. Fibroblasts and myofibroblasts: Their source, function and role in disease. Int. J.Biochem. Cell Biol. 39, 666–671 (2007).32. Wynn, T. Cellular and molecular mechanisms of fibrosis. J. Pathol. 214, 199–210 (2008).33. Vial, D. & KcKeown-Longo, P. J. PAI1 stimulates assembly of the fibronectin matrix inosteosarcoma cells through crosstalk between the αvβ5 and α5β1 integrins. J. Cell Sci. 121,1661–1670 (2008).34. Jia, D. et al. Osteoprotegerin Disruption Attenuates HySu-Induced Pulmonary HypertensionThrough Integrinvβ3/FAK/AKT Pathway Suppression. Circ. Cardiovasc. Genet. 10, 1–10 (2017).35. Belperio, J. A. et al. Interaction of IL-13 and C10 in the Pathogenesis of Bleomycin-InducedPulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 27, 419–427 (2002).36. Jakubzick, C. et al. Therapeutic Attenuation of Pulmonary Fibrosis Via Targeting of IL-4- and IL-13-Responsive Cells. J. Immunol. 171, 2684–2693 (2003).37. Jakubzick, C., Kunkel, S. L., Puri, R. K. & Hogaboam, C. M. of IL-4- and IL-13-Responsive Cells inPulmonary Fibrosis. Immunol. Res. 339–349 (2004).38. Rubin, J. et al. IGF-I regulates osteoprotegerin (OPG) and receptor activator of nuclear factor-κBligand in vitro and OPG in vivo. J. Clin. Endocrinol. Metab. 87, 4273–4279 (2002).39. Stefanovic, B. RNA protein interactions governing expression of the most abundant protein inhuman body, type I collagen. Wiley Interdiscip. Rev. RNA 4, 535–545 (2013).40. Inomata, M. et al. Pirfenidone inhibits fibrocyte accumulation in the lungs in bleomycin-inducedmurine pulmonary fibrosis. Respir. Res. 15, 1–14 (2014).41. Hisatomi, K. et al. Pirfenidone inhibits TGF-β1-induced over-expression of collagen type I andheat shock protein 47 in A549 cells. BMC Pulm. Med. 12, 1–9 (2012).42. Schaefer, C. J., Ruhrmund, D. W., Pan, L., Seiwert, S. D. & Kossen, K. Antifibrotic activities ofpirfenidone in animal models. Eur. Respir. Rev. 20, 85–97 (2011).43. A., A. et al. Exploratory analysis of a phase III trial of pirfenidone identifies a subpopulation ofpatients with idiopathic pulmonary fibrosis as benefiting from treatment. Respir. Res. 12, 143(2011).44. Conte, E. et al. Effect of pirfenidone on proliferation, TGF-β-induced myofibroblast differentiationand fibrogenic activity of primary human lung fibroblasts. Eur. J. Pharm. Sci. 58, 13–19 (2014).45. Wollin, L., Maillet, I., Quesniaux, V., Holweg, A. & Ryffel, B. Antifibrotic and Anti-inflammatoryActivity of the Tyrosine Kinase Inhibitor Nintedanib in Experimental Models of Lung Fibrosis. J.Pharmacol. Exp. Ther. 349, 209–220 (2014).46. Wollin, L. et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosisREVIEW INTERSTITIAL LUNG DISEASES |. Eur Respir J 45, 1434–1445 (2015).

Chapter 3
106
SUPPLEMENTARY DATA
Supplemental Figure 1. Correlations of OPG mRNA expression with the expression of other fibrosismarkers: Col1α1 (a), Fn1 (b), PAI-1 (c) in fibrotic human lung slices. Correlations were tested using aPearson test and presented as log data. p<0.05 was considered significant

CHAPTER 4
Osteoprotegerin as a new markerto study early fibrosis in various organs
Kurnia S.S. Putri | Adhyatmika | Suriguga | Theerut Luangmonkong |Emilia Bigaeva | Emilia Gore | Fransien van Dijk | Habibie |Dorenda Oosterhuis | Detlef Schuppan | Hendrik S. Hofker |
Koert P. de Jong | Igle J. de Jong | Peter Heukels | Wim Timens |Wouter L.J. Hinrichs | Barbro N. Melgert | Peter Olinga

Chapter 4
108
ABSTRACTFibrosis is a chronic disease that is characterized by excessive extracellular matrixdeposition and is usually only detected at a late stage when the organ is alreadyseverely damaged. Therefore, it is highly important to have reliable and easy-to-assessearly biomarkers. Osteoprotegerin (OPG) has been postulated to be a serum marker ofadvanced fibrosis in lung, liver and kidney. Little is known about the regulation of OPGin early fibrosis in these organs and its response to treatment.Therefore, we aimed at studying expression and production of OPG in early- and late-stage fibrosis in various organs and its response to treatment with an antifibrotic drugusing murine and human precision-cut tissue slices.Tissue slices of murine lung, liver, kidney and colon were incubated with and withoutTGFβ1 and/or galunisertib, a TGFβ receptor 1 kinase inhibitor, to study OPGproduction and expression in relation to fibrotic responses. OPG levels were measuredin plasma of mice with unilateral ureteral obstruction (UUO) and in mice deficient forMDR2, and slices of UUO-kidney and MDR2-/- -liver were treated with galunisertib.OPG levels were also measured in incubation medium of healthy and fibrotic humanlung, liver and kidney slices, as well as in incubation medium of galunisertib-treatedhuman tissue slices.Murine lung, liver, kidney and colon tissue slices all expressed OPG mRNA and this washigher after 48 h of incubation than directly after slicing. TGFβ1 treatment resulted inmore OPG mRNA expression in all organs and more OPG production in lung, liver andkidney but not colon slices as compared to untreated slices. This higher OPGproduction could be inhibited with galunisertib in all organ slices. OPG plasma levelswere higher in UUO and MDR2-/- mice than in control mice. OPG protein levels werelower after galunisertib treatment of both UUO fibrotic kidney and MDR2-/- fibroticliver slices. More OPG was also secreted from human fibrotic tissue slices than fromcontrols, and galunisertib-treated human fibrotic tissue slices released less OPG thanuntreated slices. These results indicate that OPG production is upregulated both inearly- and late-stage fibrosis and is responsive to TGFβ1-inhibition treatment offibrosis. The next steps should include testing OPG as a blood-based biomarker forearly-onset organ fibrosis and/or as a marker of treatment effect in patients.Keywords: early-stage fibrosis, osteoprotegerin, marker, precision-cut tissue slices

Osteoprotegerin as a new marker to study early fibrosis in various organs
109
INTRODUCTIONFibrosis is characterized by excessive deposition of extracellular matrix (ECM)in tissue, leading to organ malfunction and even death1. To date, besidestransplantation, there are only few therapies to slow the fibrotic process and nopossibilities to stop or reverse fibrosis2–5.One of the challenges in treating and studying chronic diseases such as fibrosisis the lack of suitable (early) diagnostic methods and markers to detect the slow andlong-term progression of this disease. Therefore, most fibrotic conditions in patientsare detected at a late-stage when the organ is already severely damaged. Until nowbiopsies and/or high resolution computed tomography are the only reliable diagnosticmethods to accurately stage organ fibrosis6. However, these have a high burden and ittherefore important to have reliable and easy-to-assess biomarkers to detect organfibrosis preferably also in an earlier stage.Osteoprotegerin (OPG) is a secreted protein that belongs to the tumor necrosisfactor (TNF) receptor superfamily and is expressed in various organs. It functions as adecoy receptor for several ligands including nuclear factor B ligand (RANKL), TNF-related apoptosis-inducing ligand (TRAIL), heparin, and glycosaminoglycan7–9. OPG isbest known for its regulation of bone extracellular matrix10. However, our recentstudies showed that OPG levels in fibrotic lung11 and cirrhotic liver tissue12 weresignificantly higher than in control lung and liver tissue, respectively. OPG has alsobeen found to be associated with chronic kidney disease in hypertensive patients andwith kidney damage13. Another study has shown that OPG is overexpressed by colonicepithelium during active colonic inflammatory bowel disease14, which is of interestbecause inflammation appears to be the central driving force behind colonic fibrosis15.These results suggested that OPG is linked to fibrosis in multiple organs.OPG is a soluble protein and can be detected in blood and urine as was shownin patients with cirrhotic livers16 and chronic kidney disease9,17–19, respectively. OPGserum levels also correlated with the severity of liver and kidney fibrosis. In addition,urinary OPG from chronic kidney disease patients was higher than from healthyvolunteers19. These results support the study of OPG as an easy access biomarker fororgan fibrosis and further studies into the applicability of OPG as a biomarker for organfibrosis seem warranted.

Chapter 4
110
In this study we, therefore, used murine precision-cut tissue slices of lung, liver,kidney and colon incubated with TGFβ1 to investigate OPG in relation to onset offibrosis in different organs. Furthermore, we used galunisertib, a TGFβ-receptor type Ikinase inhibitor, to investigate OPG as marker of treatment success in both human andmurine slices.MATERIALS AND METHODS
Animal experimentsMale C57BL/6 mice and FVB mice (both 8-12 weeks old weighing 20 - 30 grams)were obtained from Harlan (Horst, The Netherlands) and Jackson Laboratory (JacksonLaboratory, Bar Harbor, ME, USA), respectively. All mice were housed with permanentaccess to water and food in a temperature-controlled room with a 12 h dark/light cycleregimen. The Institutional Animal Care and Use Committee of the University ofGroningen (DEC6416AA and DEC6427A) and the Animal Ethical Committee of theRhineland Palatinate approved the use of all animals in these studies.To induce kidney fibrosis, C57BL/6 mice were subjected to unilateral ureteralobstruction. Mice were anesthetized with isoflurane/O2 (Nicholas Piramal, London,UK) and the left ureter was ligated. After 3 days, mice were sacrificed and the ligated(UUO) kidney was collected20,21.MDR2-/- mice are genetically modified FVB mice (which were bred inhomozygosity at the Institute of Translational Immunology at Mainz UniversityMedical Center) that spontaneously develop liver fibrosis. These were used to studyOPG in relation to liver fibrosis. Genetically unmodified FVB mice were used ascontrols. Seven days before sacrifice the MDR2-/- mice were injected subcutaneouslywith 500 μl of 20 wt-% control microspheres (prepared from multi-block co-polymers[PCL-PEG-PCL]/[PLLA]) dispersed in 0.4% carboxymethyl cellulose (CMC, Aqualonhigh Mw, Ashland, pH 7.0-7.4) as a control group for another experiment22. Thistreatment was not expected to influence OPG levels.Murine precision-cut tissue slicesMice were anaesthetized with isoflurane/O2 (Nicholas Piramal, London, UK) andthen sacrificed by exsanguination via the aorta abdominalis. Precision-cut tissue slices

Osteoprotegerin as a new marker to study early fibrosis in various organs
111
of the lung, liver, kidney and colon were prepared according to the method of Oenemaet al.23, Westra et al.24, Stribos et al.25 and Iswandana et al.26, respectively. Corepreparation, preservation medium and incubation medium of each organ are describedin Table 1. The tissue and cores of each organ were incubated in preservation mediumbefore the slicing process. Slices were prepared from these cores with a Krumdiecktissue slicer (Alabama Research and Development, USA), which was filled with ice-coldKrebs-Henseleit Buffer supplemented with 25 mM D-glucose (Merck, Darmstadt,Germany), 25 mM NaHCO3 (Merck), 10 mM HEPES (MP Biomedicals, Aurora, OH),saturated with carbogen (95% O2/5% CO2) and adjusted to pH 7.4.Immediately after preparation, slices were incubated in 1.3 mL (or 500 μL forcolon) pre-warmed incubation medium (Table 1) in 12-wells plates (or 24-wells platefor colon). Tissue slices were incubated at 37 ⁰C in an incubator (MCO-18M, Sanyo,USA) with an atmosphere of 80% O2 and 5% CO2, which was continuously shaken at aspeed of 90 rpm25. The slices were incubated for 48 h in medium with or without 5ng/mL TGFβ1 and with or without 10 µM galunisertib (Selleckchem, Munich,Germany). Culture medium was refreshed after 24 h. The concentration of TGFβ124,25or galunisertib27 applied in this study has previously been found optimal for inducingfibrosis or inhibiting TGFβ1-induced induction of fibrosis-associated markers26,respectively. After 48 h of incubation, slices were collected, snap frozen into liquidnitrogen and stored at -80 ⁰C until analysis. Incubation medium was collected andstored in -20 ⁰C until analysis.Human precision-cut tissue slicesThe experimental protocols of human tissues were approved by the MedicalEthical Committee of the University Medical Center Groningen and Erasmus MedicalCenter Rotterdam according to Dutch legislation and the Code of Conduct for dealingresponsibly with human tissue in the context of health research (www.federa.org),refraining the need of written consent for ‘further use’ of coded-anonymous humantissue. The source of human tissue is described in Table 2 and the demographic ofhuman tissue donor is describe in Table 3.
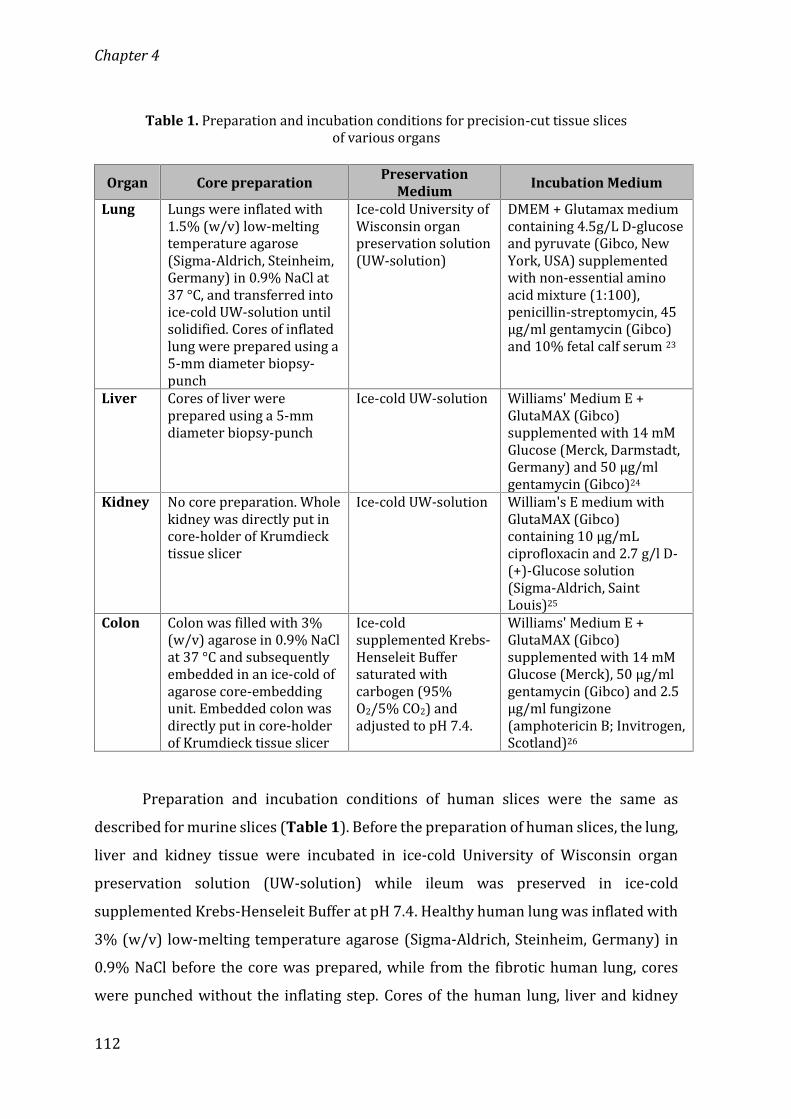
Chapter 4
112
Table 1. Preparation and incubation conditions for precision-cut tissue slicesof various organsOrgan Core preparation Preservation
Medium Incubation Medium
Lung Lungs were inflated with1.5% (w/v) low-meltingtemperature agarose(Sigma-Aldrich, Steinheim,Germany) in 0.9% NaCl at37 °C, and transferred intoice-cold UW-solution untilsolidified. Cores of inflatedlung were prepared using a5-mm diameter biopsy-punch
Ice-cold University ofWisconsin organpreservation solution(UW-solution)DMEM + Glutamax mediumcontaining 4.5g/L D-glucoseand pyruvate (Gibco, NewYork, USA) supplementedwith non-essential aminoacid mixture (1:100),penicillin-streptomycin, 45µg/ml gentamycin (Gibco)and 10% fetal calf serum 23
Liver Cores of liver wereprepared using a 5-mmdiameter biopsy-punch Ice-cold UW-solution Williams' Medium E +GlutaMAX (Gibco)supplemented with 14 mMGlucose (Merck, Darmstadt,Germany) and 50 μg/mlgentamycin (Gibco)24Kidney No core preparation. Wholekidney was directly put incore-holder of Krumdiecktissue slicer
Ice-cold UW-solution William's E medium withGlutaMAX (Gibco)containing 10 μg/mLciprofloxacin and 2.7 g/l D-(+)-Glucose solution(Sigma-Aldrich, SaintLouis)25Colon Colon was filled with 3%(w/v) agarose in 0.9% NaClat 37 °C and subsequentlyembedded in an ice-cold ofagarose core-embeddingunit. Embedded colon wasdirectly put in core-holderof Krumdieck tissue slicer
Ice-coldsupplemented Krebs-Henseleit Buffersaturated withcarbogen (95%O2/5% CO2) andadjusted to pH 7.4.
Williams' Medium E +GlutaMAX (Gibco)supplemented with 14 mMGlucose (Merck), 50 μg/mlgentamycin (Gibco) and 2.5μg/ml fungizone(amphotericin B; Invitrogen,Scotland)26Preparation and incubation conditions of human slices were the same asdescribed for murine slices (Table 1). Before the preparation of human slices, the lung,liver and kidney tissue were incubated in ice-cold University of Wisconsin organpreservation solution (UW-solution) while ileum was preserved in ice-coldsupplemented Krebs-Henseleit Buffer at pH 7.4. Healthy human lung was inflated with3% (w/v) low-melting temperature agarose (Sigma-Aldrich, Steinheim, Germany) in0.9% NaCl before the core was prepared, while from the fibrotic human lung, coreswere punched without the inflating step. Cores of the human lung, liver and kidney

Osteoprotegerin as a new marker to study early fibrosis in various organs
113
were made by using a 5-mm diameter biopsy-punch. Human ileum was cleaned byflushing Krebs-Henseleit Buffer through the lumen and the muscle layer was removedbefore cores were prepared using 3 % (w/v) agarose in 0.9 % NaCl at 37°C andembedded in an agarose core-embedding unit.Table 2. The surgical waste material source of human tissue for precision-cut tissueslices of various organs
Organ Fibrotic tissue Control tissueLung Explanted end-stage fibroticlung Unaffected part of surgical resectionfor suspected carcinomaLiver Explanted cirrhotic liver Surgical excess material from thereduced-size donor liver fortransplantationKidney Explanted fibrotic kidneys Unaffected part of kidney of anexplanted kidneysIleum Fibrotic part from ileocecalresection procedure Unaffected part of ileum fromileocecal resection procedureTable 3. Demographic of human tissue donor
Organ Information Fibrotic tissue Control tissueLung Gender 5 males, 1 female maleAge 57 – 64 years old unknownReason of surgicalprocedure Idiophatic pulmonaryfibrosis; fibrotic nonspecificinterstitial pneumonia Lung carcinomaKidney Gender 3 females, 1 male 4 malesAge 27 – 43 years old 56 – 80 years oldNephrectomy side 2 right, 2 left side 3 right, 1 left sideCreatinine beforenephrectomy(μmol/L) 124 – 627 81 - 100
Reason of surgicalprocedure Rejected afunctional kidney Renal cell carcinomaIleum Gender 3 females 3 femalesAge 32 – 64 years old 32 – 64 years oldReason of surgicalprocedure Colitis (stenosis) Colitis (stenosis)

Chapter 4
114
ELISA Murine and human OPG levels in slice incubation medium and mouse plasmawere measured using ELISA (cat #DY459 (mouse), cat #DY805 (human), R&DSystems) according to the instructions provided by the manufacturer. Total OPG in themedium was corrected for the protein content of the slices, which was measured byLowry (BIO-rad RC DC Protein Assay, Bio Rad, Veenendaal, The Netherlands).Quantitative real-time PCRTotal mRNA was isolated from slices using a Maxwell® LEV simply RNACells/Tissue kit (Promega, Madison, WI). Final RNA concentrations were determinedusing Biotek Synergy HT (Biotek®, Winoosku, Vermont, USA). Conversion of RNA intocDNA was performed by using a reverse transcriptase kit (Promega, Leiden, TheNetherlands), in a master-cycler gradient (25 °C for 10 min, 45 °C for 60 min, and 95 °Cfor 5 min). For murine slices, transcription levels of OPG and fibrosis-associatedmarkers collagen 1α1 (Col1α1), α-smooth muscle actin (αSMA), fibronectin (Fn1),plasminogen activator inhibitor-1 (PAI-1) were measured by using a SensiMix™ SYBRkit (Bioline, Taunton, MA) and the 7900HT Real-Time RT-PCR sequence detectionsystem (Applied Biosystems, Bleiswijk, The Netherlands) with 45 cycles of 10 min 95°C, 15 sec at 95 °C, and 25 sec at 60 °C following with a dissociation stage.
Table 4. Mouse primers of fibrosis-associated markersPrimer Forward sequence Reverse sequence18s CTTAGAGGGACAAGTGGCG ACGCTGAGCCAGTCAGTGTAGAPDH ACAGTCCATGCCATCACTGC GATCCACGACGGACACATTGβ-actin ATCGTGCGTGACATCAAAGA ATGCCACAGGATTCCATACCCol1α1 TGACTGGAAGAGCGGAGAGT ATCCATCGGTCATGCTCTCTαSMA ACTACTGCCGAGCGTGAGAT CCAATGAAAGATGGCTGGAAFn1 CGGAGAGAGTGCCCCTACTA CGATATTGGTGAATCGCAGAPAI-1 GCCAGATTTATCATCAATGACTGGG GGAGAGGTGCACATCTTTCTCAAAGOPG ACAGTTTGCCTGGGACCAAA CTGTGGTGAGGTTCGAGTGG
Output data were analyzed using SDS 2.3 software (Applied Biosystems) and Ctvalues were normalized to housekeeping gene 18s RNA (lung and kidney), β-actin(liver), and GAPDH (colon) and relative gene expression was calculated as 2-ΔCt. In the
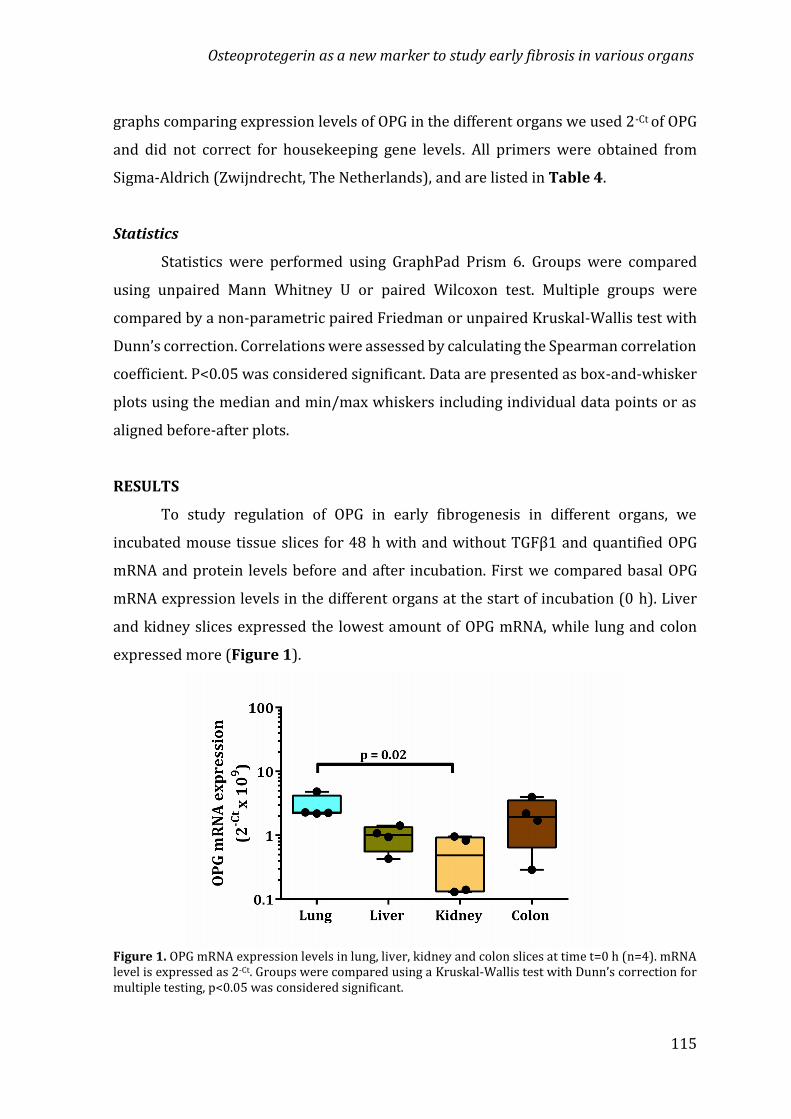
Osteoprotegerin as a new marker to study early fibrosis in various organs
115
graphs comparing expression levels of OPG in the different organs we used 2-Ct of OPGand did not correct for housekeeping gene levels. All primers were obtained fromSigma-Aldrich (Zwijndrecht, The Netherlands), and are listed in Table 4.StatisticsStatistics were performed using GraphPad Prism 6. Groups were comparedusing unpaired Mann Whitney U or paired Wilcoxon test. Multiple groups werecompared by a non-parametric paired Friedman or unpaired Kruskal-Wallis test withDunn’s correction. Correlations were assessed by calculating the Spearman correlationcoefficient. P<0.05 was considered significant. Data are presented as box-and-whiskerplots using the median and min/max whiskers including individual data points or asaligned before-after plots.RESULTSTo study regulation of OPG in early fibrogenesis in different organs, weincubated mouse tissue slices for 48 h with and without TGFβ1 and quantified OPGmRNA and protein levels before and after incubation. First we compared basal OPGmRNA expression levels in the different organs at the start of incubation (0 h). Liverand kidney slices expressed the lowest amount of OPG mRNA, while lung and colonexpressed more (Figure 1).
Figure 1. OPG mRNA expression levels in lung, liver, kidney and colon slices at time t=0 h (n=4). mRNAlevel is expressed as 2-Ct. Groups were compared using a Kruskal-Wallis test with Dunn’s correction formultiple testing, p<0.05 was considered significant.
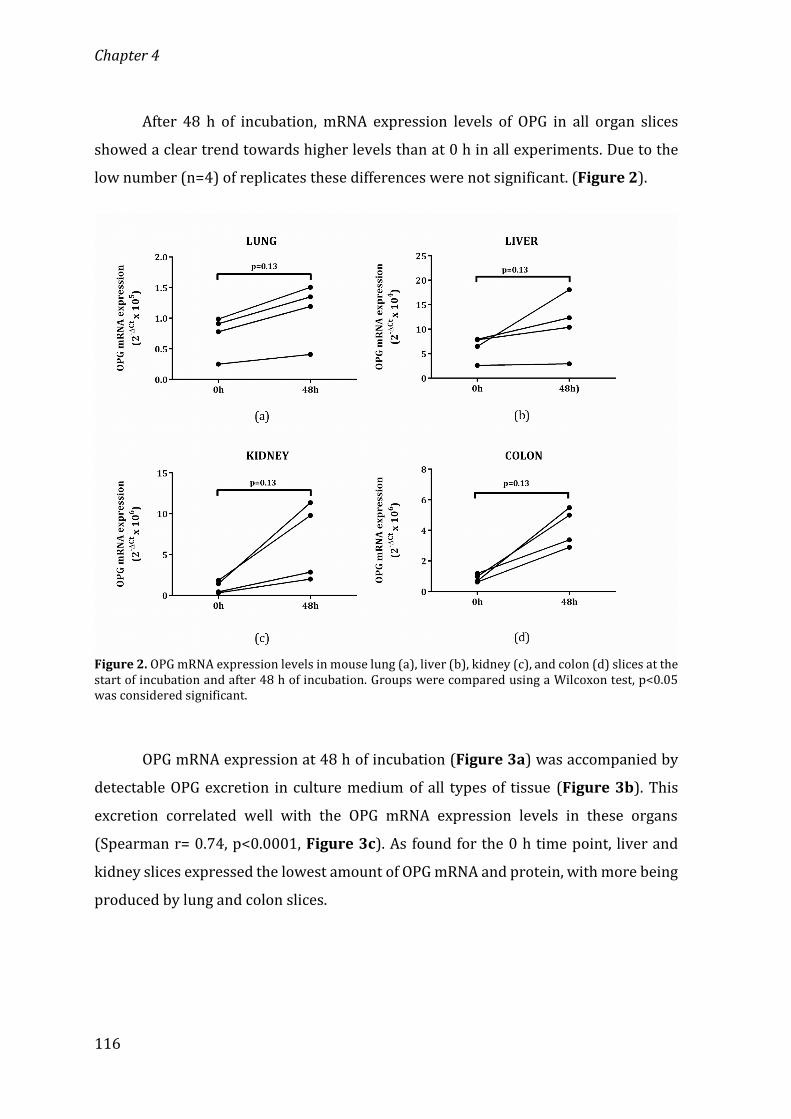
Chapter 4
116
After 48 h of incubation, mRNA expression levels of OPG in all organ slicesshowed a clear trend towards higher levels than at 0 h in all experiments. Due to thelow number (n=4) of replicates these differences were not significant. (Figure 2).
Figure 2. OPG mRNA expression levels in mouse lung (a), liver (b), kidney (c), and colon (d) slices at thestart of incubation and after 48 h of incubation. Groups were compared using a Wilcoxon test, p<0.05was considered significant.OPG mRNA expression at 48 h of incubation (Figure 3a) was accompanied bydetectable OPG excretion in culture medium of all types of tissue (Figure 3b). Thisexcretion correlated well with the OPG mRNA expression levels in these organs(Spearman r= 0.74, p<0.0001, Figure 3c). As found for the 0 h time point, liver andkidney slices expressed the lowest amount of OPG mRNA and protein, with more beingproduced by lung and colon slices.

Osteoprotegerin as a new marker to study early fibrosis in various organs
117
Figure 3. OPG mRNA expression in mouse lung, liver, kidney, and colon slices after 48 h of incubation.mRNA level is expressed as 2-Ct (a). OPG protein release from mouse lung, liver, kidney, and colon slicesinto incubation medium after 48 h of incubation (b). OPG mRNA and protein production correlate wellwhen all organs were combined (Spearman r = 0.74 and p < 0.0001) (c). Groups were compared using aKruskal-Wallis test with Dunn’s correction for multiple testing. Correlation was tested using a Spearmantest. p<0.05 was considered significant.We subsequently induced the onset of fibrosis in these murine organ slices byadding profibrotic stimulus of TGFβ1 during incubation, which resulted in higherexpression of OPG mRNA in lung, liver, kidney and colon slices than in the control slices(Figures 4a, 4c, 4e, 4g). This higher mRNA expression after TGFβ1 treatment wasmatched with higher OPG protein excretion by lung, liver and kidney slices (Figures
4b, 4d, 4f). Due to the low n of the experiments for lung, kidney and colon thisdifference was not significant, but all experiments showed the similar trend of moreOPG after TGFβ1. In addition to OPG mRNA expression and protein excretion, severalfibrosis-associated markers such as Fn1 and PAI-1 were also expressed at higher levelsin TGFβ1-stimulated lung (Figures 5c, 5d), liver (Figures 5g, 5h) and kidney slices(Figures 5k, 5l). Col1α1 mRNA expression was relatively higher in TGFβ1-stimulated
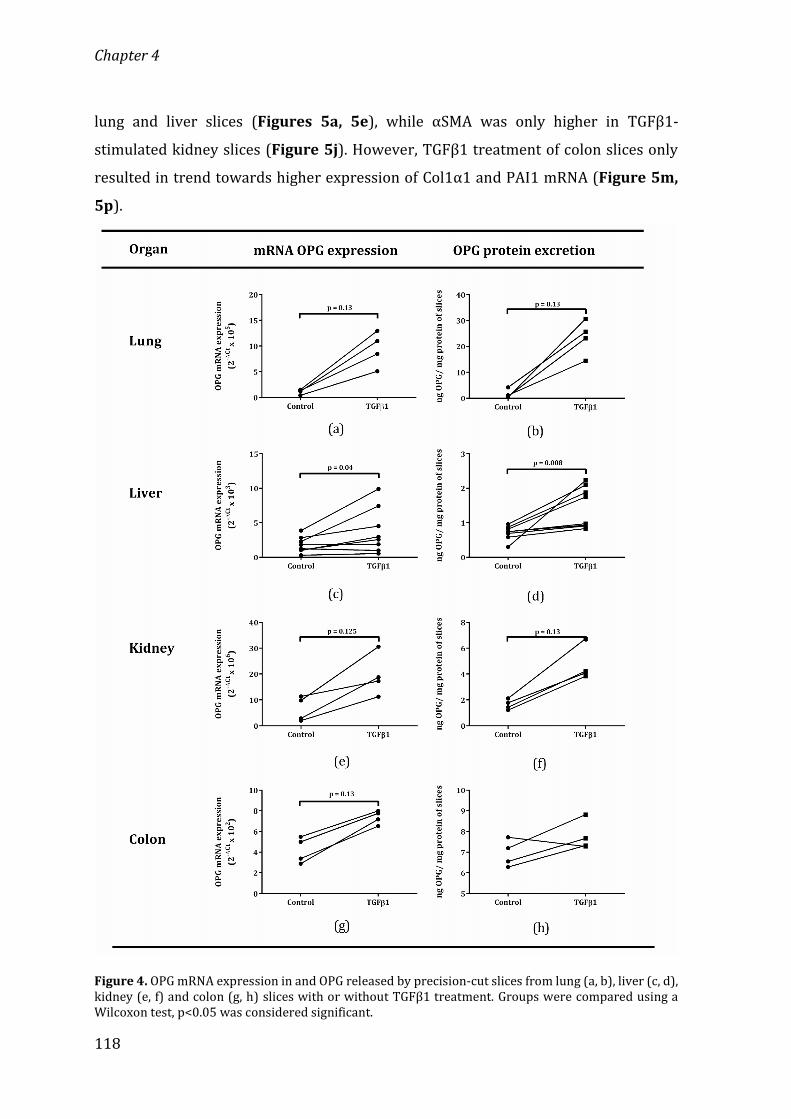
Chapter 4
118
lung and liver slices (Figures 5a, 5e), while αSMA was only higher in TGFβ1-stimulated kidney slices (Figure 5j). However, TGFβ1 treatment of colon slices onlyresulted in trend towards higher expression of Col1α1 and PAI1 mRNA (Figure 5m,
5p).
Figure 4. OPG mRNA expression in and OPG released by precision-cut slices from lung (a, b), liver (c, d),kidney (e, f) and colon (g, h) slices with or without TGFβ1 treatment. Groups were compared using aWilcoxon test, p<0.05 was considered significant.

119
Osteoprotegerin as a new marker to study early fibrosis in various organs

Chapter 4
120
Figure 5. mRNA levels of fibrosis-associated markers Col1α1, αSMA, Fn1, and PAI-1 expressed in lung(a-d), liver (e-h), kidney (i-l) and colon (m-p) with or without TGFβ1. Groups were compared using aWilcoxon test, p<0.05 was considered significant.To investigate if higher or lower OPG mRNA expression was associated withhigher or lower expression of fibrosis-associated markers, correlations werecalculated using combined results from the multiple experiments on lung, liver, kidneyand colon slices. As depicted in Figure 6, OPG mRNA expression correlatedsignificantly with fibrosis-associated markers Col1α1, αSMA, Fn1, and PAI-1. Inaddition, OPG protein excretion from those organs also significantly correlated withαSMA, Fn1, and PAI-1, but not with Col1α1.To further investigate whether or not these correlations were similar for eachtype of organ, we also investigated correlations of OPG mRNA expression and fibrosis-associated markers per organ. Table 5 and Supplemental Figure 1 show that, inmouse lung slices OPG mRNA expression was only significantly correlated with Fn1and PAI-1 mRNA expression but not with Col1α1 and αSMA mRNA expression. In liverand kidney slices, OPG mRNA expression was significantly correlated with mRNAexpression of all fibrosis-associated markers (Col1α1, αSMA, Fn1, and PAI-1) (Table 5,
Supplemental Figure 2 and Supplemental Figure 3). In colon slices, OPG mRNAexpression was significantly correlated with PAI-1 mRNA expression only and not withCol1α1, aSMA, and Fn1 mRNA expression. (Table 5, Supplemental Figure 4).

Osteoprotegerin as a new marker to study early fibrosis in various organs
121Figure 6. Correlations between mRNA level (2-Ct) of OPG and fibrosis-associated markers expression ofCol1α1 (a), αSMA (c), Fn1 (e), PAI-1 (g) in tissue slices of lung, liver, kidney and colon. Correlationsbetween OPG protein excretion and fibrosis-associated markers expression of Col1α1 (b), αSMA (d), Fn1(f), PAI-1 (h) in tissue slices of lung, liver, kidney and colon. Correlations were tested using a Spearmantest and presented as log data. p<0.05 was considered significant.

Chapter 4
122
Table 5. Correlation between OPG mRNA expression from tissue slices withfibrosis-associated markers in various organsOrgans Fibrosis-
associatedmarkers
mRNA OPG expressionSpearmancorrelationcoefficient (r)
Correlationp-value
Lung Col1α1 0.24 0.36αSMA -0.33 0.21Fn1 0.81 0.0002PAI1 0.78 0.0006
Liver Col1α1 0.75 < 0.0001αSMA 0.64 0.0002Fn1 0.61 0.0006PAI1 0.65 0.0002
Kidney Col1α1 0.92 < 0.0001αSMA 0.71 0.003Fn1 0.93 < 0.0001PAI1 0.94 < 0.0001
Colon Col1α1 0.17 0.59αSMA - 0.52 0.16Fn1 0.43 0.17PAI1 0.91 0.0001
To investigate whether or not antifibrotic treatment is accompanied by a lowerOPG mRNA expression and protein excretion, we treated TGFβ1-induced early fibrosisin tissue slices with galunisertib, a TGFβ-receptor type I kinase inhibitor. Galunisertibmitigated the effects of TGFβ1 in liver, lung and kidney for most fibrosis-associatedgenes, although the inhibitory effect on αSMA mRNA expression seemed lesspronounced (Figure 7). Inhibition of TGFβ1-induced early fibrogenesis wasaccompanied by lower mRNA expression of OPG and lower excretion of OPG in mediumof lung, liver, and kidney slices (Figure 8). We did not use colon slices for thisexperiment since TGFβ1 treatment did not induce clear OPG release nor expression ofother fibrosis-associated markers in colon slices. This decision was supported by otherexperiments from our lab that showed that TGFβ1 treatment did not consistentlystimulate expression of fibrosis-associated mRNA markers in mouse colon slices27.

Osteoprotegerin as a new marker to study early fibrosis in various organs
123
Figure 7. OPG mRNA in and protein excretion from lung (a, b), liver (c, d) and kidney (e, f) slices afterincubation with TGFβ1, with or without 10 µM galunisertib. Groups were compared using a Wilcoxontest, p<0.05 was considered significant.
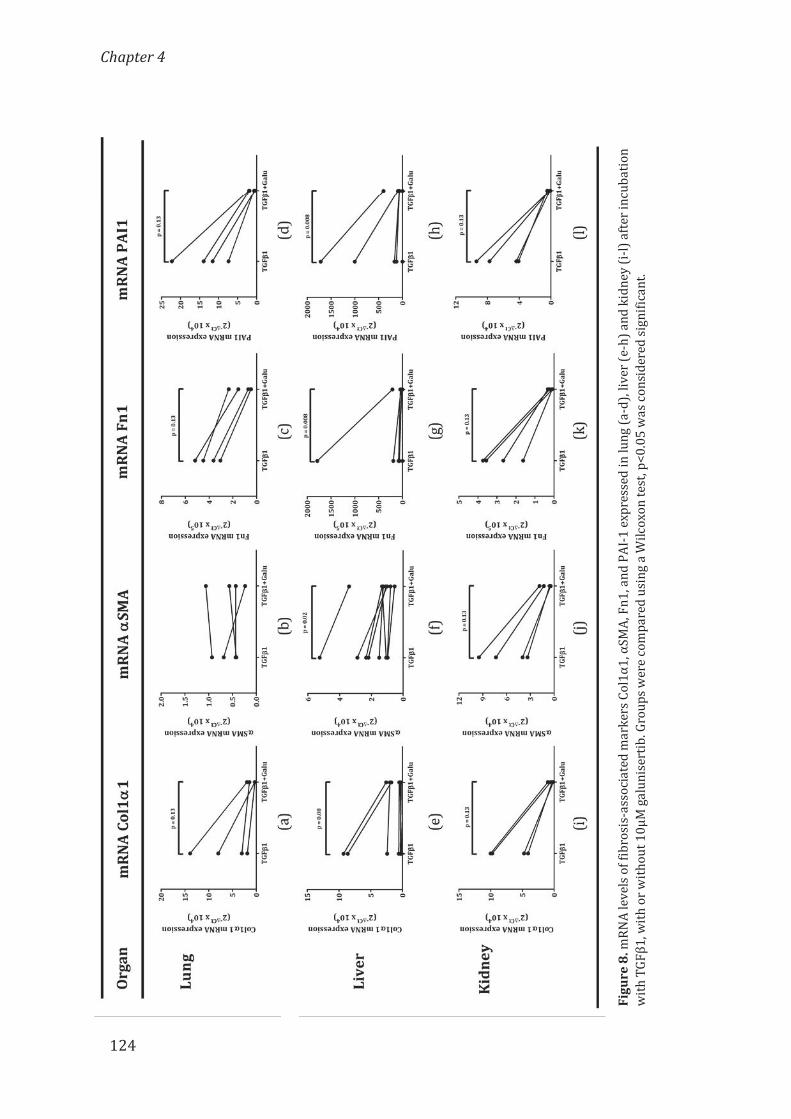
Fig
ur
e 8
. m
RN
A l
ev
els
of
fib
ro
sis
-1
--
--
wit
h
124
Chapter 4

Osteoprotegerin as a new marker to study early fibrosis in various organs
125
To translate our results to in vivo situations we also investigated OPG plasmalevels in mice with either kidney or liver fibrosis. Mice that suffered from kidneyfibrosis induced by UUO had higher OPG levels in plasma 3 days after the obstructionthan healthy control mice (Figure 9a). A similar finding was shown for mice deficientin MDR2 that spontaneously develop liver fibrosis. These mice also had higher OPGplasma levels than healthy control mice (Figure 9b).After sacrificing the animals, slices of the UUO-kidneys and MDR2-/--liverswere prepared and they were treated with or without galunisertib for 48 h. As depictedin Figure 9, galunisertib induced clear inhibition of OPG excretion from both UUO-kidney (c) and MDR2-/--liver slices (d).
Figure 9. Plasma OPG levels in healthy control mice and mice with UUO-induced kidney fibrosis (a) andMDR2-/- mice (b). Groups were compared using unpaired Mann-Whitney test, p<0.05 was consideredsignificant. Kidney slices of mice 3 days after being subjected to UUO (c) and liver slices from MDR2-/-mice (d) produce less OPG after slices were treated with 10 μM galunisertib for 48 h. Groups werecompared using a paired Wilcoxon test, p<0.05 was considered significant

Chapter 4
126
To further investigate if OPG can also be applied as marker to study fibrosis inman, we measured OPG protein excretion from human tissue slices after 48 h ofincubation. We found that more OPG protein was excreted from human fibrotic lungand liver slices (Figures 10a, c) than from the control slices. There was no significantdifference between OPG secretion from human fibrotic and control kidney and ileumslices (Figures 10e, g). Furthermore, we found that human fibrotic lung slices treatedwith galunisertib excreted less OPG than untreated slices (Figure 10b). Surprisingly,we found higher OPG secretion from galunisertib-treated human fibrotic liver slicesthan from untreated slices (Figure 10d). Galunisertib-treated human fibrotic kidneyslices also tended to secrete less OPG than from the untreated kidney slices (Figure
10f). Finally, no significant difference was seen from galunisertib-treated humanfibrotic ileum slices (Figure 10h), as compared to the untreated slices.
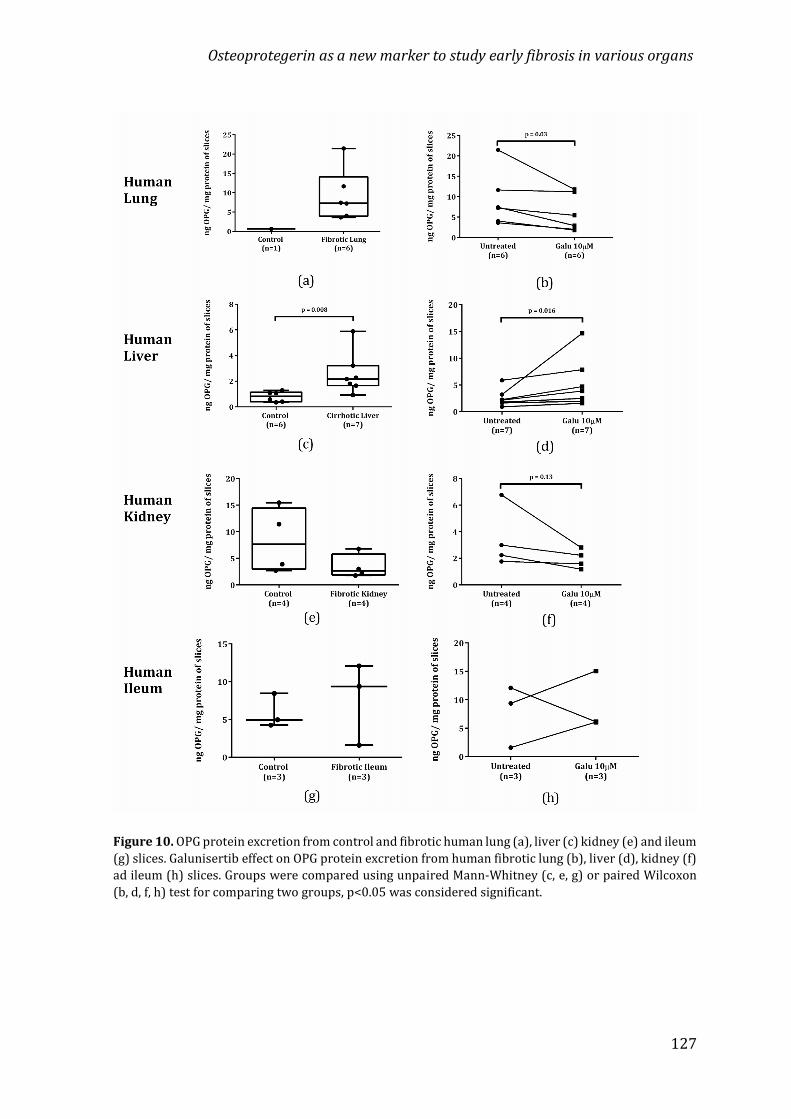
Osteoprotegerin as a new marker to study early fibrosis in various organs
127
Figure 10. OPG protein excretion from control and fibrotic human lung (a), liver (c) kidney (e) and ileum(g) slices. Galunisertib effect on OPG protein excretion from human fibrotic lung (b), liver (d), kidney (f)ad ileum (h) slices. Groups were compared using unpaired Mann-Whitney (c, e, g) or paired Wilcoxon(b, d, f, h) test for comparing two groups, p<0.05 was considered significant.

Chapter 4
128
DISCUSSIONOPG was previously only associated with bone disease and its production innonbone-related pathologies was assumed to be a consequence of some feedbackmechanisms from bone29. Recent studies by us and others, however, indicate that it isalso associated with fibrosis of the lung11, liver12,16, kidney17,30 and colon31. Previousstudies in liver and lung and this study comparing various organs, clearly show thateach of the organs investigated can produce OPG itself. In addition, in all organs, exceptmaybe colon, OPG production is responsive to TGFβ1-stimulation and to inhibition ofits signaling, opening avenues for the use of OPG as a biomarker for detection of fibrosisin the early stages, as well as for the efficacy of the fibrosis treatment.We first compared basal gene expression of OPG in different murine organsand found that OPG mRNA is expressed under basal conditions in lung, liver, kidney,and colon, albeit not to the same extent. As we could not use the same housekeepinggene for all organs, these result should be interpreted with caution, even if all organshad the same cDNA concentration during the OPG gene expression measurements. OPGmRNA expression in lung was similar to colon and higher than in liver and kidney. Thereason for this discrepancy is unclear but may be related to the specific functions ofeach tissue, with both lung and colon being exposed to the outside world on a regularbasis, while kidney and liver do not have this direct interaction. This finding may be aclue towards the function of OPG outside bone tissue. With both tissues being moreexposed to external factors and therefore more prone to damage, the level of OPGexpression may be related to the level of repair needed in steady state conditions. Thisidea is reinforced by our findings that OPG expression is strongly dependent onTGFβ110,32, the master regulator of wound healing, and the finding that incubation ofprecision-cut slices from these organs for 48 h also results in higher expression of OPGin all organs. The damage that is inherent to the slicing procedure will induce a repairprocess during incubation, which may lead to the higher expression of OPG.TGFβ1 is not only important in wound healing, it is also the most importantprofibrotic stimulus in the lung10,33, liver24,27,32,34, kidney19,35,36 and colon4,37,38.Therefore, in this study we incubated mouse organ slices with TGFβ1 to alter theprocess of wound healing in these slices towards onset of fibrosis and to study the OPGresponse. As a read-out for fibrosis development we used four markers commonly

Osteoprotegerin as a new marker to study early fibrosis in various organs
129
associated with fibrosis: Col1α1, αSMA, Fn1, and PAI-1. Col1α1 is one of the buildingblocks of collagen-1, which is deposited excessively as ECM during fibrosisdevelopment39. αSMA is widely known as a marker for myofibroblasts, the cellsresponsible for collagen and ECM production. Its expression levels therefore are oftenassociates with the number of myofibroblasts present in a tissue and the degree offibrosis40. Fibronectin is an ubiquitous ECM glycoprotein that plays an important roleduring tissue repair by providing a scaffold for collagen fibrils to attach to and formmatrix41–43. Increased Fn1 expression was found during wound healing processes inlung, liver and kidney after injury and its expression increases even further duringpathological fibrosis in these organs43. PAI1 is one of proteins responsible formaintaining a balance between production and degradation of ECM during tissuerepair processes by inhibiting urokinase/tissue type plasminogen activator. PAI1expression is almost undetectable in normal tissues by immunohistochemistry,however, its expression immediately increases during wound healing processes and isexcessively induced during chronic injury44.TGFβ1 treatment indeed resulted in significantly higher gene expression ofmost of these different fibrosis-associated markers in all organs though there weresome individual differences. Overall αSMA was the marker least responsive, which onlyincreased in kidney slices treated with TGFβ1. Sun et al. already showed that αSMAexpression is quite variable in organs exposed to TGFβ145. Many other resident cellscan express αSMA, such as smooth muscle cells around vessels46, pericytes47 and alsoLy6Chi circulating monocytes48, which may dilute the effects of TGFβ1 inducing αSMAexpression in transforming fibroblasts in these organs. Consistent with our previousresults, we also found that TGFβ1 treatment did not convincingly upregulateexpression of several fibrosis-associated markers (notably αSMA and Fn1) in mousecolon slices as compared to the other organs28. The reason for this lack of TGFβ1response is unclear but may be related to the role of the peripheral immune system.Rieder et al. showed that intestinal fibrosis is mainly facilitated by infiltration ofimmune cells unlike other organs in which fibrosis is mainly caused by activation ofresident cells15. Therefore, due to absence of infiltrating peripheral immune cells (e.g.monocytes, T cells, neutrophils) in this ex vivo study, fibrosis may not have developedproperly in colon.

Chapter 4
130
Interestingly, in all organs taken together for the expression of fibrosis-associated markers, higher expression was also associated with higher OPG mRNAexpression and this correlation did not improve when we left out the colon data. Theseresults suggest that fibrosis is closely associated with OPG in all organs studied, eventhough the individual colon data are not as convincing on their own.Furthermore, in all organs taken together OPG protein excretion after TGFβ1treatment also correlated with expression of the fibrosis-associated markers except forCol1α1. This lack of correlation of Col1α1 and OPG also did not improve when leavingout the colon data. Apparently the cellular pathways from OPG mRNA expression toprotein excretion contain steps that are less associated with Col1α1 mRNA expressionthan with the other markers that we used.When looking at the separate organs for correlations between mRNAexpression of OPG and fibrosis-associated markers, liver and kidney are again clearlydifferent from lung and colon. In lung OPG mRNA expression only correlates with Fn1and PAI1 and in colon only with PAI1. Therefore, PAI1 appears to be the marker bestlinked to OPG expression, closely followed by Fn1, but how these proteins are linkedto OPG is unclear. Further studies are needed to investigate which cellular pathwaysinteract in the production of OPG, Fn1 and PAI1. In colon slices, the higher OPG mRNAexpression was also not convincingly matched with protein excretion. This may beexplained by the fact that control colon slices incubated for 48 h released the highestamount of OPG protein as compared to the other organs and that a further increasewas therefore more difficult to achieve.We also investigated whether OPG can be used as a biomarker to measureantifibrotic treatment efficacy by treating mouse lung, liver and kidney slices withTGFβ1 in combination with galunisertib, a TGFβ-receptor type I kinase inhibitor.Previous studies have shown that galunisertib exhibited potent antifibrotic activityboth in rat and human tissue slices27. In line with these results, this study also showedthat galunisertib successfully downregulated Col1α1, Fn1 and PAI1 expression inmouse lung, liver and kidney slices and this was accompanied by lower OPG mRNA andprotein expression. Interestingly, galunisertib and TGFβ1-treated mouse lung, liverand kidney slices released similar amounts of OPG as untreated control slices, showingthat galunisertib can completely inhibit the effects of TGFβ1. This confirms our

Osteoprotegerin as a new marker to study early fibrosis in various organs
131
previous results in which we showed that TGFβ1 is the central regulator of OPGproduction in fibroblasts and liver32.Higher OPG mRNA expression and protein production after only 48 h of TGFβ1induction in the murine lung, liver, kidney and colon slices indicates that OPG rapidlyresponds to the profibrotic stimulation. Therefore, developing OPG as biomarker todetect early-stage fibrosis condition in patients may be beneficial in a clinical setting.Furthermore, our results also suggest that OPG can be used as a marker toscreen candidate antifibrotic drugs. To study the value of OPG as a treatment efficacymarker in more clinically relevant conditions of end-stage fibrosis, we also treatedfibrotic kidney slices and liver slices with galunisertib. These fibrotic organs weretaken from animals treated (UUO) or bred (MDR2-/-) to develop fibrosis49–51.Importantly, this fibrosis development was accompanied by higher levels of plasmaOPG, in both the UUO and MDR2-/- mice than in healthy control mice. These resultsindicate that the OPG level in blood may be a prospective marker to diagnose fibrosis,not only ex vivo, but also in vivo. As was found for the TGFβ1-treated slices, OPGexcretion was also lower from galunisertib-treated fibrotic slices, showing OPG mayact as a biomarker for treatment efficacy in more clinically relevant settings too.Therefore, we also studied OPG release by human organ slice, to investigate if OPGcould be a promising tool to be applied in a clinical setting.Our study showed that OPG excretion is higher from human fibrotic lung andcirrhotic liver slices compared to slices of control tissue. This result is supported byprevious studies which showed that OPG levels in lung tissue of pulmonary fibrosis11and cirrhotic liver patients12,52 were significantly higher than the controls.Surprisingly, we found that OPG was not elevated in the medium of human fibrotickidney slices compared to healthy kidney slices. This result differs from previousstudies which showed that serum OPG levels was significantly higher in patients withchronic kidney disease 53,54. This discrepancy may be an effect of age of the donors.Previous studies have shown that OPG production significantly increased with age55–57. Therefore, it is conceivable that OPG secreted from fibrotic kidney slices, derivedfrom donors aged 27-43 years, was not higher than from control kidney slices, derivedfrom donors aged 56-80 years, (see Table 3).

Chapter 4
132
Furthermore, our study also showed that in galunisertib-treated human lungand kidney slices tended to release less OPG than untreated slices. This result is also inagreement with previous studies of our group that showed that galunisertibsuccessfully downregulated fibrosis-associated marker in human kidney slices (datanot shown). However, our study also showed that galunisertib-treated human liverslices secreted more OPG than the untreated slices, even though other fibrosis-associated markers were downregulated in these samples27. The reason for thissurprising result is unclear. Since galunisertib is a TGFβ receptor 1 kinase inhibitor, itsfailure to inhibit OPG production in cirrhotic human liver may indicate that TGFβ is notthe only regulator of OPG production in human liver.Furthermore, there was no difference between OPG production in healthy andfibrotic human ileum slices. Galunisertib treatment also showed no response onfibrotic human ileum slices. This result was similar to the result in mouse colon slicesmodel. We speculate that in intestinal fibrosis OPG may be produced by infiltratingimmune cells15. Thus, due to the absence of these cells in this ex vivo study, fibrosis maynot develop in these slices as it would in vivo. In addition, since the muscle layer ofhuman intestines, which contains OPG+-cells, including dendritic cells andmacrophages58,59, was removed before the preparation of slices, less OPG is producedby the human intestinal slices.Based on our results of human tissue slices, we found that OPG may be apromising marker of lung, liver and kidney fibrosis in a clinical setting and itsapplication to assess efficacy of antifibrotic therapy should be further validated.CONCLUSIONWe have shown that OPG associates with early stages of fibrosis in lung, liverand kidney slices and should be investigated further as a tool to assess fibrosisprogression and to evaluate the effectiveness of antifibrotic therapies. A first stepwould be to investigate levels in patient sera during disease progression and treatmentto reveal its use in clinical practice.

Osteoprotegerin as a new marker to study early fibrosis in various organs
133
REFERENCES1. Wynn, T. Cellular and molecular mechanisms of fibrosis. J. Pathol. 214, 199–210 (2008).2. Lu, Y. T., Tingskov, S. J., Djurhuus, J. C., Nørregaard, R. & Olsen, L. H. Can bladder fibrosis incongenital urinary tract obstruction be reversed? J. Pediatr. Urol. 13, 574–580 (2017).3. Caminati, A., Cassandro, R., Torre, O. & Harari, S. Severe idiopathic pulmonary fibrosis: What canbe done? Eur. Respir. Rev. 26, (2017).4. Curciarello, R., Docena, G. H. & MacDonald, T. T. The Role of Cytokines in the Fibrotic Responsesin Crohn’s Disease. Front. Med. 4, 1–9 (2017).5. Arakeri, G. et al. Current protocols in the management of oral submucous fibrosis: An update. J.Oral Pathol. Med. 46, 418–423 (2017).6. Hunninghake, G. W. et al. Utility of a Lung Biopsy for the Diagnosis of Idiopathic PulmonaryFibrosis. 2–5 (2001).7. Baud’huin, M. et al. Osteoprotegerin: Multiple partners for multiple functions. Cytokine GrowthFactor Rev. 24, 401–409 (2013).8. Kuźniewski, M. et al. Osteoprotegerin and osteoprotegerin/TRAIL ratio are associated withcardiovascular dysfunction and mortality among patients with renal failure. Adv. Med. Sci. 61,269–275 (2016).9. Yilmaz, M. I. et al. Osteoprotegerin in Chronic Kidney Disease: Associations with VascularDamage and Cardiovascular Events. Calcif. Tissue Int. 99, 121–130 (2016).10. Boyce, B. F. & Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch.Biochem. Biophys. 473, 139–146 (2008).11. Boorsma, C. et al. The RANKL-OPG balance in pulmonary fibrosis. in 1.5 Diffuse Parenchymal LungDisease 46, PA3809 (European Respiratory Society, 2015).12. Adhyatmika, A. et al. Osteoprotegerin is more than a serum marker in liver fibrosis. J. Hepatol.66, S147–S148 (2017).13. Bernardi, S. et al. Circulating osteoprotegerin is associated with chronic kidney disease inhypertensive patients. BMC Nephrol. 18, 1–9 (2017).14. Sylvester, F. A., Draghi, A., Bausero, M. A., Fernandez, M. L. & Vella, A. T. 1149 OsteoprotegerinExpression is Upregulated in the Colon of Children With Active Inflammatory Bowel Disease.Gastroenterology 142, S-209 (2012).15. Rieder, F. et al. Inflammation-induced endothelial-to-mesenchymal transition: A novelmechanism of intestinal fibrosis. Am. J. Pathol. 179, 2660–2673 (2011).16. Bosselut, N. et al. Including osteoprotegerin and collagen IV in a score-based blood test for liverfibrosis increases diagnostic accuracy. Clin. Chim. Acta J.L. Bosson; A. Paris; ANRS L. Allain, Paris.Methodol. Hospices Civ. Lyon. M-C. Gelineau, B. Poggi 415, 63–68 (2013).17. Graciolli, F. G. et al. The complexity of chronic kidney disease–mineral and bone disorder acrossstages of chronic kidney disease. Kidney Int. 91, 1436–1446 (2017).18. Benito-Martin, A. et al. Osteoprotegerin in Exosome-Like Vesicles from Human Cultured TubularCells and Urine. PLoS One 8, (2013).19. Montañez-Barragán, A. et al. Osteoprotegerin and kidney disease. J. Nephrol. 27, 607–617 (2014).20. Poosti, F. et al. Selective delivery of IFN-γ to renal interstitial myofibroblasts: A novel strategyfor the treatment of renal fibrosis. FASEB J. 29, 1029–1042 (2015).21. Teekamp, N. et al. Polymeric microspheres for the sustained release of a protein-based drugcarrier targeting the PDGFβ-receptor in the fibrotic kidney. Int. J. Pharm. 534, 229–236 (2017).22. van Dijk, F. et al. Pharmacokinetics of a sustained release formulation of PDGFβ-receptordirected carrier proteins to target the fibrotic liver. J. Control. Release 269, 258–265 (2018).23. Oenema, T. A. et al. Bronchoconstriction Induces TGF-β Release and Airway Remodelling inGuinea Pig Lung Slices. PLoS One 8, (2013).24. Westra, I. M. Precision-cut liver slices: an ex vivo model for the early onset and end-stage of liverfibrosis. 97 (2014).

Chapter 4
134
25. Stribos, E. G. D. et al. Precision-cut human kidney slices as a model to elucidate the process ofrenal fibrosis. Transl. Res. 170, 8–16e1 (2016).26. Iswandana, R. et al. Organ- and species-specific biological activity of rosmarinic acid. Toxicol. Vitr.32, 261–268 (2016).27. Luangmonkong, T. et al. Evaluating the antifibrotic potency of galunisertib in a human ex vivomodel of liver fibrosis. Br. J. Pharmacol. 174, 3107–3117 (2017).28. Pham, B. T., van Haaften, W. T., Iswandana, R., Oosterhuis, D. & Olinga, P. P024 Regionaldifferences in effect of TGF-beta1 and PDGF on the early onset of intestinal fibrosis. J. Crohn’sColitis 8, S74 (2014).29. Fadda, S., Hamdy, A., Abulkhair, E., Mahmoud Elsify, H. & Mostafa, A. Serum levels ofosteoprotegerin and RANKL in patients with rheumatoid arthritis and their relation to bonemineral density and disease activity. Egypt. Rheumatol. 37, 1–6 (2015).30. Znorko, B., Oksztulska-Kolanek, E., Michałowska, M., Kamiński, T. & Pawlak, K. Does theOPG/RANKL system contribute to the bone-vascular axis in chronic kidney disease? A systematicreview. Adv. Med. Sci. 62, 52–64 (2017).31. Draghi, A., Ramanarasimhaiah, R., Fernandez, M. L., Vella, A. T. & Sylvester, F. A. Sa1165 ColonicOsteoprotegerin: Higher Expression in Children With Ulcerative Colitis Than Crohn’s Disease.Gastroenterology 146, S-217 (2014).32. Adhyatmika, A. et al. Osteoprotegerin production and profibrotic activity in liver fibrosis areTGFβ dependent. J. Hepatol. 66, S147 (2017).33. Wei, Y. et al. Fibroblast-specific inhibition of TGF-β1 signaling attenuates lung and tumor fibrosis.J. Clin. Invest. 127, 3675–3688 (2017).34. Karkampouna, S., ten Dijke, P., Dooley, S. & Kruithof-de Julio, M. TGFβ Signaling in LiverRegeneration. Curr. Pharm. Des. 18, 4103–4113 (2012).35. Vega, G., Alarcón, S. & San Martín, R. The cellular and signalling alterations conducted by TGF-βcontributing to renal fibrosis. Cytokine 88, 115–125 (2016).36. Meng, X., Nikolic-Paterson, D. J. & Lan, H. Y. TGF-β: the master regulator of fibrosis. Nat. Rev.Nephrol. 12, 325–338 (2016).37. Ma, Y. et al. Targeting TGF-β1 by employing a vaccine ameliorates fibrosis in a mouse model ofchronic colitis. Inflamm. Bowel Dis. 16, 1040–1050 (2010).38. Lawrance, I. C. et al. Cellular and Molecular Mediators of Intestinal Fibrosis. J. Crohn’s Colitisj.crohns.2014.09.008 (2015). doi:10.1016/j.crohns.2014.09.00839. Bonnans, C., Chou, J. & Werb, Z. Remodelling the extracellular matrix in development and disease.Nat. Rev. Mol. Cell Biol. 15, 786–801 (2014).40. Evans, R. A., Tian, Y. . C., Steadman, R. & Phillips, A. O. TGF-β1-mediated fibroblast–myofibroblastterminal differentiation—the role of smad proteins. Exp. Cell Res. 282, 90–100 (2003).41. Altrock, E. et al. Inhibition of fibronectin deposition improves experimental liver fibrosis. J.Hepatol. 62, 625–633 (2015).42. Sottile, J. & Hocking, D. C. Fibronectin Polymerization Regulates the Composition and Stability ofExtracellular Matrix Fibrils and Cell-Matrix Adhesions. Mol. Biol. Cell 13, 3546–3559 (2002).43. To, W. S. et al. Plasma and cellular fibronectin: distinct and independent functions during tissuerepair. Fibrogenesis Tissue Repair 4, 21 (2011).44. Ghosh, A. K. & Vaughan, D. E. PAI-1 in tissue fibrosis. J. Cell. Physiol. 227, 493–507 (2012).45. Sun, K.-H., Chang, Y., Reed, N. I. & Sheppard, D. α-Smooth muscle actin is an inconsistent markerof fibroblasts responsible for force-dependent TGFβ activation or collagen production acrossmultiple models of organ fibrosis. Am. J. Physiol. - Lung Cell. Mol. Physiol. 310, L824–L836 (2016).46. Moiseenko, A. et al. Origin and characterization of alpha smooth muscle actin-positive cellsduring murine lung development. Stem Cells 35, 1566–1578 (2017).47. Hung, C. et al. Role of lung Pericytes and resident fibroblasts in the pathogenesis of pulmonaryfibrosis. Am. J. Respir. Crit. Care Med. 188, 820–830 (2013).48. Gibbons, M. A. et al. Ly6Chimonocytes direct alternatively activated profibrotic macrophageregulation of lung fibrosis. Am. J. Respir. Crit. Care Med. 184, 569–581 (2011).

Osteoprotegerin as a new marker to study early fibrosis in various organs
135
49. Ucero, A. C. et al. Unilateral ureteral obstruction: beyond obstruction. Int. Urol. Nephrol. 46, 765–776 (2014).50. Chevalier, R. L., Forbes, M. S. & Thornhill, B. A. Ureteral obstruction as a model of renal interstitialfibrosis and obstructive nephropathy. Kidney Int. 75, 1145–1152 (2009).51. Popov, Y., Patsenker, E., Fickert, P., Trauner, M. & Schuppan, D. Mdr2 (Abcb4)-/- micespontaneously develop severe biliary fibrosis via massive dysregulation of pro- andantifibrogenic genes. J. Hepatol. 43, 1045–1054 (2005).52. Bosselut, N. et al. Including osteoprotegerin and collagen IV in a score-based blood test for liverfibrosis increases diagnostic accuracy. Clin. Chim. Acta 415, 63–68 (2013).53. Siomou, E. et al. Serum osteoprotegerin, RANKL and fibroblast growth factor-23 in children withchronic kidney disease. Pediatr. Nephrol. 26, 1105–1114 (2011).54. Jiang, J. Q., Lin, S., Xu, P. C., Zheng, Z. F. & Jia, J. Y. Serum osteoprotegerin measurement for earlydiagnosis of chronic kidney disease-mineral and bone disorder. Nephrology 16, 588–594 (2011).55. Szulc, P., Hofbauer, L. C., Heufelder, A. E., Roth, S. & Delmas, P. D. Osteoprotegerin serum levels inmen: Correlation with age, estrogen, and testosterone status. J. Clin. Endocrinol. Metab. 86, 3162–3165 (2001).56. Khosla, S. et al. International Original Article Correlates of Osteoprotegerin Levels in Women andMen. Metra 394–399 (2002).57. Kudlacek, S., Schneider, B., Woloszczuk, W., Pietschmann, P. & Willvonseder, R. Serum levels ofosteoprotegerin increase with age in a healthy adult population. Bone 32, 681–686 (2003).58. Moschen, A. R. et al. The RANKL/OPG system is activated in inflammatory bowel diseases andrelates to the state or bone loss. Gut 54, 479–487 (2005).59. Mikkelsen, H. B. & Thuneberg, L. Op/op mice defective in production of functional colony-stimulating factor-1 lack macrophages in muscularis externa of the small intestine. Cell TissueRes. 295, 485–493 (1999).

Chapter 4
136
SUPPLEMENTARY DATA
Supplemental Figure 1. Correlation of gene expression (2−ΔCt) of OPG and Col1α1(a), αSMA (b), Fn1 (c)and PAI1 (d) of mouse lung slices. Correlations were tested using Spearman test on 2−ΔCt and presentedas log(2−ΔCt). p<0.05 was considered significant.
Supplemental Figure 2. Correlation of gene expression (2−ΔCt) of OPG and Col1α1(a), αSMA (b), Fn1 (c)and PAI1 (d) of mouse liver slices. Correlations were tested using Spearman test on 2−ΔCt and presentedas log(2−ΔCt). p<0.05 was considered significant.
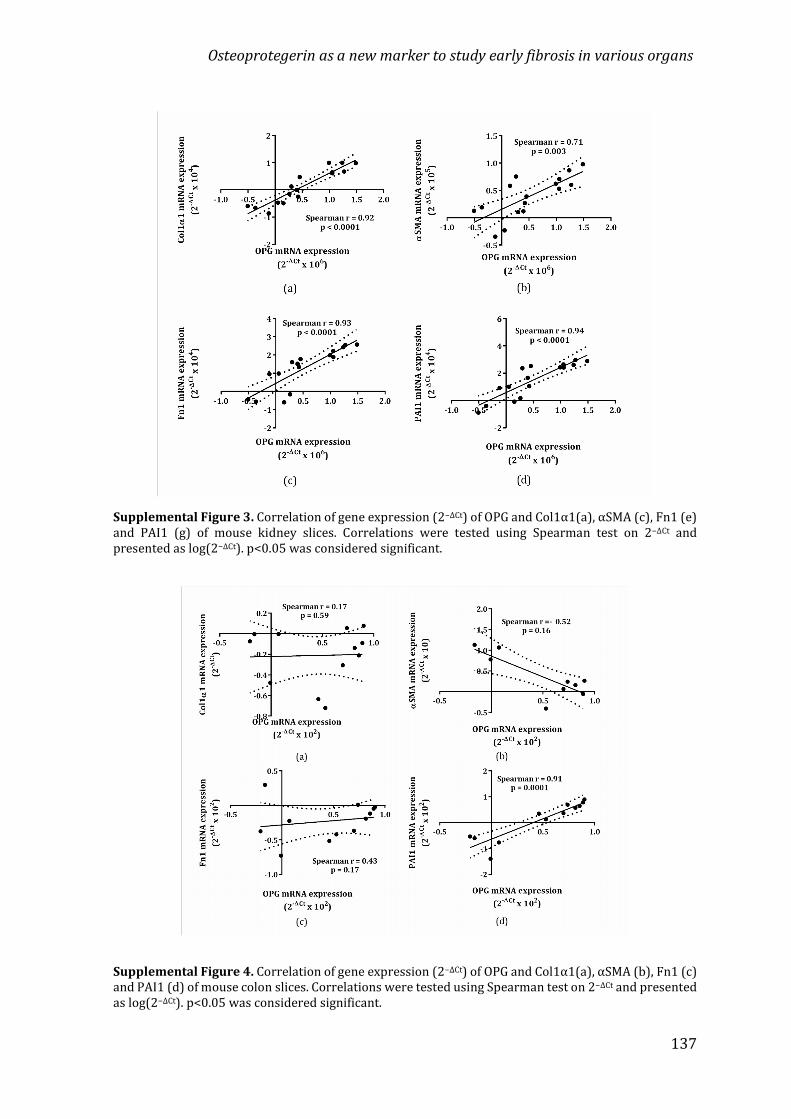
Osteoprotegerin as a new marker to study early fibrosis in various organs
137
Supplemental Figure 3. Correlation of gene expression (2−ΔCt) of OPG and Col1α1(a), αSMA (c), Fn1 (e)and PAI1 (g) of mouse kidney slices. Correlations were tested using Spearman test on 2−ΔCt andpresented as log(2−ΔCt). p<0.05 was considered significant.
Supplemental Figure 4. Correlation of gene expression (2−ΔCt) of OPG and Col1α1(a), αSMA (b), Fn1 (c)and PAI1 (d) of mouse colon slices. Correlations were tested using Spearman test on 2−ΔCt and presentedas log(2−ΔCt). p<0.05 was considered significant.

Chapter 4
138

Osteoprotegerin expression in liver
tissue is induced by IL13 through TGFβ
Adhyatmika | Kurnia S.S. Putri | Emilia Gore | Keri A. MangnusCatharina Reker-Smit | Detlef Schuppan | Leonie Beljaars
Peter Olinga | Barbro N. Melgert

Chapter 5
140
ABSTRACTOur previous studies have shown that osteoprotegerin (OPG) is a profibrotic mediator,produced by myofibroblasts, under influence of Transforming Growth Factor β (TGFβ).Its expression in experimental models of liver fibrosis correlates well with diseaseseverity and treatment responses. The regulation of OPG in liver tissue is largelyunknown and we therefore set out to elucidate which cytokines associated withfibrosis induce OPG and through which pathways.Precision-cut liver slices of wild type and STAT6-deficient mice and 3T3 fibroblastswere used to investigate the effects of TGFβ, IL13, IL1β, and PDGF-BB on expression ofOPG. In addition to TGFβ, only IL13 and not PDGF-BB or IL1β could induce OPGexpression in 3T3 fibroblasts and liver slices. This IL13-dependent induction was notshown in liver slices of STAT6-deficient mice and when wild type slices were cotreatedwith TGFβ receptor 1 kinase inhibitor galunisertib, STAT6 inhibitor AS1517499, orAP1 inhibitor T5224. This suggests that the OPG-inducing effect of IL13 is mediatedthrough IL13 receptor α1-activation and subsequent STAT6-dependent upregulationof IL13 receptor α2, which in turn activates AP1 and induces production of TGFβ andsubsequent production of OPG.We have shown that IL13 induces OPG release by liver tissue through a TGFβ-dependent pathway involving both the α1 and the α2 receptor of IL13 andtranscription factors STAT6 and AP1. OPG may therefore be a novel target for thetreatment liver fibrosis as it is mechanistically linked to two important regulators offibrosis in liver, namely IL13 and TGFβ1.Keywords: IL13, IL13 receptors, STAT6, AP1, murine liver slices, 3T3

Osteoprotegerin expression in liver tissue is induced by IL13 through TGFβ
141
INTRODUCTIONLiver fibrosis is a chronic disease induced by long term injury and/orinflammation initiated by virus infections or chemical-induced injury, for exampledrugs or alcohol1. The main pathological characteristic of liver fibrosis is persistentextracellular matrix formation by hepatic stellate cells, which in turn prevents theregrowth of functional hepatocytes2. The disease has a high burden as there is nopossible therapy to reverse the process when it has fully developed and thereforetransplantation is the only option3.Transforming Growth Factor β (TGFβ) has been widely studied for many yearsas one of the central players in liver fibrosis, but this has not yielded any effective newdrugs yet4,5. It is therefore likely that the process of fibrosis development is far morecomplicated than just the actions of TGFβ alone and that we need to understand thedifferent players and interactions better to develop potential drug candidates.We recently became interested in the actions of osteoprotegerin (OPG, genename TNFRSF11B) after finding that OPG is produced in high quantities by (liver)fibroblasts, especially after stimulation with TGFβ and that OPG itself can induceexpression of TGFβ, indicating a feed-forward loop6. Several clinical studies haveshown that higher serum levels of OPG are associated with having liverfibrosis/cirrhosis7–13. In addition, OPG serum levels are part of a novel diagnostic scorecalled Coopscore® that has better diagnostic performance than Fibrometer®,Fibrotest®, Hepascore® and Fibroscan™ in chronic hepatitis C-associated fibrosis8.Moreover, in our previous studies, we have demonstrated high hepatic OPG productionin liver tissue of patients transplanted for liver cirrhosis and in murine models of liverfibrosis.Osteoprotegerin is well known for its role in protecting bone matrixdegradation14, but little is known about its function in nonbone tissues. In that respect,its role in vascular calcifications is probably best studied showing that OPG protectsagainst vascular calcification15. This contrasts with its known functional influence inbone metabolism in which it induces calcification of bone14. This suggests that OPG hasmore possible functions unrelated to bone and our previous data show its firmassociations with fibrotic processes and TGFβ signalling in (myofibroblasts). However,

Chapter 5
142
little is known about the regulation of OPG production in (liver) fibroblasts16 by othermediators involved in fibrosis. In this study we therefore aimed to further investigateOPG regulation in the liver by studying the effects of several fibrosis-related cytokinesand their downstream signalling pathways, most notably IL13.MATERIALS AND METHODS
AnimalsMale and female wild-type C57BL/6 mice were obtained from Harlan (Horst,The Netherlands) and male STAT6(-/-) C57BL/6 mice were bred in the Institute ofTranslational Immunology, University Medical Center of the Johannes GutenbergUniversity Mainz, Germany. Animals were kept in cages with a 12 hour of light/darkcycle and received food and water ad libitum. The use of C57BL/6 mice in this studywas approved by the Institutional Animal Care and Use Committee of the University ofGroningen (DEC 6416 AA) and the use of STAT6(-/-) mice by the Institutional AnimalCare and Use Committee of the Government of Rhineland Palatinate under thereference number 2317707/G12-1-007.Precision-cut liver slicesMurine precision-cut liver slices were prepared as described before by De Graafet al.17. Slices were treated with 5 ng/mL TGFβ (Peprotech, Rocky Hill, US), 10 ng/mLIL13 (Peprotech), 10 ng/mL IL1β (Peprotech), 10 ng/mL PDGF-BB (Peprotech), 10ng/mL OPG (R&D Systems, Minneapolis, US), 10 mM Galunisertib (Selleckchem,Munich, Germany), 21 nM AS1517499 (Axon MedChem, Groningen, The Netherlands),and/or 10 μM T5224 (ApexBio, Houston, US) in triplicate for a total of 48 hours andculture medium was refreshed every 24 hours.In vitro cell lines50,000/well 3T3 murine fibroblasts (The American Type Culture Collection,ATCC® CRL-1658) were cultured in standard medium of Gibco® Dulbecco’s ModifiedEagle Medium (Thermo Scientific, Waltham, Massachussets, US) containing 4.5 g/L D-Glucose (Sigma-Aldrich, Missouri, US), 2 mM L-Glutamine (Thermo Scientific,

Osteoprotegerin expression in liver tissue is induced by IL13 through TGFβ
143
Waltham, Massachussets, US), and 10 % of fetal calf serum (Biowest, Nuaillé, France).Cells were starved with medium containing 0.5 % serum 24 hours prior to othertreatments. Treatments with TGFβ, IL13, IL1β, PDGF-BB, and OPG were done at similarconcentrations as described for the experiments with slices.Generation of tissue lysateTissue slices were lysed with extraction buffer containing 25 mM Tris (Sigma-Aldrich, Missouri, US), 10 mM sodium phosphate (Sigma-Aldrich), 150 mM NaCl(Sigma-Aldrich, Missouri, US), 0.1 % SDS (Sigma-Aldrich, Missouri, US), 1 % Triton-X100 (Sigma-Aldrich, Missouri, US), and protease inhibitor (Thermo Scientific, Waltham,Massachussets, US) and incubated for 5 minutes at room temperature before snap-freezing and stored at -80oC until analysis.Osteoprotegerin analysisOsteoprotegerin was measured in culture supernatants of cells and slices usinga murine OPG DuoSet® ELISA kit (R&D Systems, Minneapolis, US) according to theinstructions provided by the manufacturer.Messenger RNA analysisMessenger RNA was isolated from cells or slices (three slices per sample,pooled, homogenized prior to extraction) using Maxwell® LEV Simply RNACells/Tissue kit (Promega, Madison, Wisconsin, US). A NanoDrop® ND-1000Spectrophotometer (Thermo Scientific) was used to measure total mRNAconcentration in samples. cDNA synthesis from the mRNA was performed using aMoloney Murine Leukemia Virus Reverse Transcriptase (M-MLV RT) kit (Promega,Madison, Wisconsin, USA) in a Mastercycler® Gradient (Eppendorf, Hamburg,Germany) programmed for 10 minutes at 20 °C, 30 minutes at 42 °C, 12 minutes at 20°C, 5 minutes at 99 °C, and 5 minutes at 20 °C. Transforming growth factor beta 1(TGFβ1), IL13 receptor α2 (IL13Rα2), pro-collagen 1 subunit α1 (Col1α1), α-smoothmuscle actin (αSMA), heat shock protein 47 (HSP47), plasminogen activator inhibitor1 (PAI1), and fibronectin 1 (Fn1) genes were quantified using quantitative real timePCR (RT qPCR) from the synthesized cDNA, using SensiMixTM SYBR® Green (Bioline,

Chapter 5
144
London, UK) in a 7900HT Real-Time PCR sequence detection system (AppliedBiosystems, Waltham, Massachussets, US) with primer sequences as presented inTable 1. PCR analysis consisted of 45 cycles of 10 min at 95 °C, 15 seconds at 95 °C,and 25 seconds at 60 °C (repeated for 40 times) followed by a dissociation stage of95oC for 15 seconds, 60 °C for 15 seconds, and 95 °C for 15 seconds. Output data wereanalyzed using SDS 2.4 software (Applied Biosystems) and ΔCt values were calculatedafter β-actin normalization with two to the power of -ΔCt (2-ΔCt) was used as finalvalues to be statistically analyzed.
Table 1. Primers sequencesGene Forward Reverse
β-actin ATCGTGCGTGACATCAAAGA ATGCCACAGGATTCCATACCTGFβ AGGGCTACCATGCCAACTTC GTTGGACAACTGCTCCACCT
IL13Rα2 TGAAAGTGAAGACCTATGCTTT GACAAACTGGTACTATGAAAATCol1α1 TGACTGGAAGAGCGGAGAGT ATCCATCGGTCATGCTCTCTαSMA ACTACTGCCGAGCGTGAGAT CCAATGAAAGATGGCTGGAAHSP47 AGGTCACCAAGGATGTGGAG CAGCTTCTCCTTCTCGTCGTPAI1 GCCAGATTTATCATCAATGACTGGG GGAGAGGTGCACATCTTTCTCAAAGFn1 CGGAGAGAGTGCCCCTACTA CGATATTGGTGAATCGCAGAOPG ACAGTTTGCCTGGGACCAAA CTGTGGTGAGGTTCGAGTGG
Viability assayViability of the slices was assessed by measuring the ATP content per milligramtissue using a bioluminescence assay kit (Sigma-Aldrich) as previously reported byHadi et al.18. For each sample, three slices were collected separately in 1 ml sonificationoptimization (SONOP) solution pH 10.9 containing 70% ethanol and 2 nM EDTA.StatisticsAll statistics were performed using GraphPad Prism 6. As datasets were all n<8,nonparametric tests were used. When comparing 2 groups a Mann Whitney U orWilcoxon test was used depending on the data being paired or not. When comparing

Osteoprotegerin expression in liver tissue is induced by IL13 through TGFβ
145
multiple groups, a Friedman or Kruskall-Wallis with Dunn’s correction was used. Dataare presented as min-to-max box-and-whisker plots with individual data points. Forthe time course experiment using 3T3 fibroblasts, the areas under the curve from 0.5-12 hours and 12-36 hours were calculated and these were compared between groups.Data in this experiment are presented a median + the interquartile range. For allexperiments, p<0.05 was considered significant.RESULTS
IL13 induces fibroblast and hepatic OPG productionTo study possible factors that can induce OPG production by fibroblasts, wetreated 3T3 fibroblasts with several cytokines associated with fibrosis. In this study weused IL1β, IL13, and PDGF-BB with TGFβ as our positive control as we have shownhigher OPG expression with TGFβ in our previous study. In addition to TGFβ, only IL13resulted in higher OPG production as compared to control (Figure 1A). To confirm thatIL13 can have a similar effect in liver tissue, we treated murine precision-cut liver sliceswith IL13 using TGFβ again as a positive control and found that IL-13 also resulted insignificantly higher OPG release from liver tissue as compared to control (Figure 1B).This higher OPG release was accompanied by near-significant higher OPG mRNAexpression and significant higher expression of fibrosis-associated genes col1α1,HSP47, and Fn1, but not aSMA and PAI1 (Figure 1C). None of the treatments affectedthe viability of the slices (Supplemental Figure S1).IL13 induces OPG production at a slower rate than TGFβTo check whether induction of OPG production followed similar kineticsbetween TGFβ and IL13, we followed OPG release in time in culture medium of 3T3fibroblasts after stimulation with TGFβ and IL13. We found that after 36 hours ofincubation IL13 and TGFβ both induced a similar release in OPG although the inductionby TGFβ occurred somewhat faster. When comparing the area under the curvebetween stimulated cells and untreated control cells in the first 12 hours, we found asignificant increase in OPG release by TGFβ, while IL13 was not significantly different

Chapter 5
146
from control. In the time interval from 12 to 36 hours both TGFβ and IL13 significantlyinduced OPG release as compared to control (Figure 2).
FIGURE 1. IL13 induces OPG release. When 3T3 fibroblasts were treated with IL1β, IL13, and PDGFBBfor 24 hours of incubation with TGFβ as a positive control, only IL13 treatment resulted in significantlyhigher OPG release as compared to control (A). Murine precision-cut livers slices also releasedsignificantly more OPG in culture medium after 48 hours of incubation with IL13 as compared to control(B). IL13 incubation also resulted in (near)significant higher expression of the fibrosis-associated genesOPG, Col1α1, HSP47, and Fn1, though not of αSMA and PAI1 (C). Groups were compared using aFriedman test with Dunn’s correction or a Wilcoxon test and p<0.05 was considered significant.
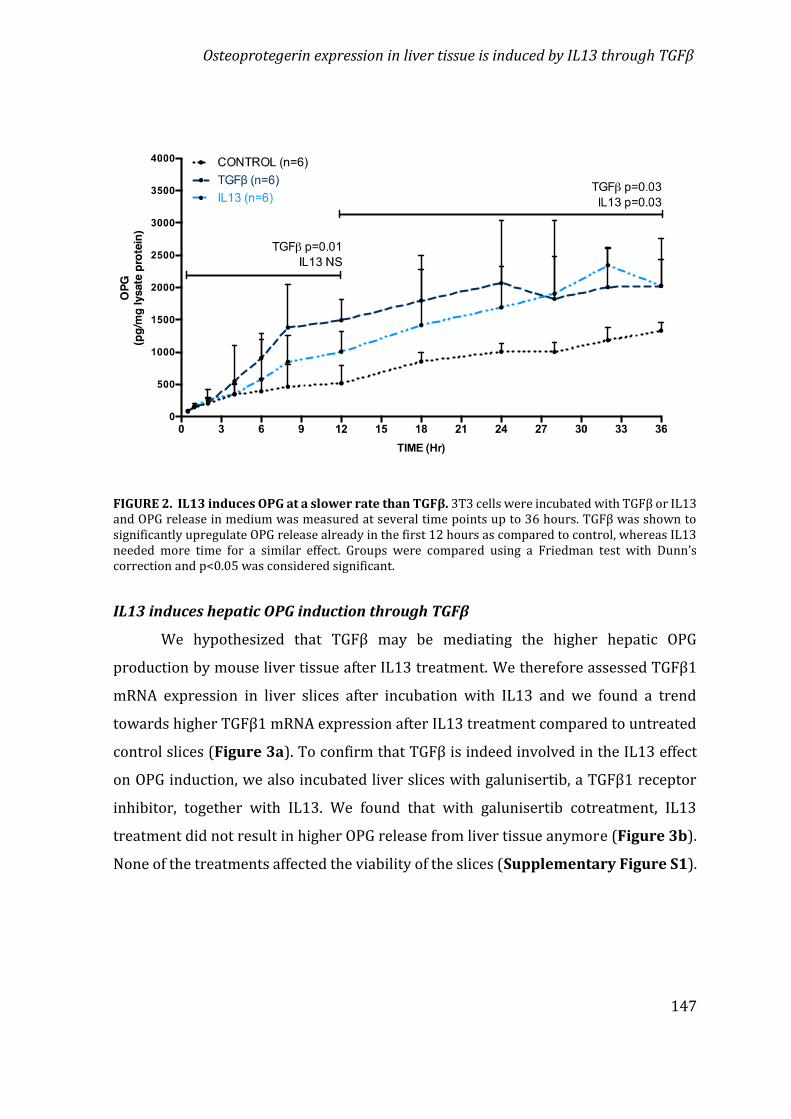
Osteoprotegerin expression in liver tissue is induced by IL13 through TGFβ
147
FIGURE 2. IL13 induces OPG at a slower rate than TGFβ. 3T3 cells were incubated with TGFβ or IL13and OPG release in medium was measured at several time points up to 36 hours. TGFβ was shown tosignificantly upregulate OPG release already in the first 12 hours as compared to control, whereas IL13needed more time for a similar effect. Groups were compared using a Friedman test with Dunn’scorrection and p<0.05 was considered significant.IL13 induces hepatic OPG induction through TGFβWe hypothesized that TGFβ may be mediating the higher hepatic OPGproduction by mouse liver tissue after IL13 treatment. We therefore assessed TGFβ1mRNA expression in liver slices after incubation with IL13 and we found a trendtowards higher TGFβ1 mRNA expression after IL13 treatment compared to untreatedcontrol slices (Figure 3a). To confirm that TGFβ is indeed involved in the IL13 effecton OPG induction, we also incubated liver slices with galunisertib, a TGFβ1 receptorinhibitor, together with IL13. We found that with galunisertib cotreatment, IL13treatment did not result in higher OPG release from liver tissue anymore (Figure 3b).None of the treatments affected the viability of the slices (Supplementary Figure S1).

Chapter 5
148
FIGURE 3. IL13 induces OPG through TGFβ. When mouse liver slices were treated with IL13, we founda trend towards more TGFβ mRNA expression (Wilcoxon test, p<0.05 was considered significant) (A).Moreover, when IL13 was given together with galunisertib, a TGFβ1-receptor inhibitor, the IL13-induced higher OPG release was not found anymore (Friedman test with Dunn’s correction, p<0.05 wasconsidered significant) (B).STAT6 is involved in IL13-induced release of OPGIL13 has been reported to signal through 2 receptors: receptor IL13Rα1 andIL13Rα219. The downstream activation pathway of IL13Rα1 is via transcription factorSTAT620. To study whether the activation of IL13Rα1 and subsequently STAT6 isinvolved in the IL13-induced release of OPG, we treated liver slices of STAT6-deficientmouse with IL13 or TGFβ and measured OPG released in medium. We found that IL13failed to induce OPG release by liver slices of STAT6-deficient mice as compared tountreated controls, whereas TGFβ could still induce OPG release as we found before inwildtype mice (Figure 4a). To confirm our finding, we used AS1517499, a chemicalcompound blocking STAT6 activity, in our wild-type mouse liver slices21 and similarlyfound that IL13 did not induce OPG release anymore when slices were co-incubatedwith this inhibitor as compared to slices only treated with IL13 (Figure 4b). None ofthe treatments affected the viability of the slices (Supplementary Figure S1).

Osteoprotegerin expression in liver tissue is induced by IL13 through TGFβ
149
FIGURE 4. STAT6 is involved in IL13-induced release of OPG. When liver slices of the STAT6 knockout (KO) mice were incubated for 24 hours with IL13 or TGFβ, IL13 failed to induce OPG release, whilethe TGFβ-induced release was not affected by the deficiency in STAT6 (Friedman test with Dunn’scorrection, p<0.05 was considered significant) (A). IL13-induced OPG release in wild type liver slicescould also be blocked with AS1517499, an inhibitor of STAT6 activity (Friedman test with Dunn’scorrection, p<0.05 was considered significant) (B).IL13 receptor α2 is also involved in IL13-induced OPG releaseFichtner-Feigl et al. (2005) reported that IL13Rα2 is involved in induction ofTGFβ expression and fibrosis through transcription factor AP122. However, inhomeostatic conditions, the expression of this receptor is low23, while activation ofIL13Rα1 and subsequently STAT6 can induce IL13Rα2 expression24. In order to checkwhether these findings are also relevant in our system, we assessed IL13Rα2 mRNAexpression in liver slices upon IL13 treatment. We found that IL13Rα2 mRNAexpression level was significantly higher upon IL13 treatment as compared tountreated controls (Figure 5a). We then used T5224, a chemical inhibitor of AP125 tostudy whether AP1 in involved in IL13-induced OPG release and we found that indeedchemical inhibition of AP1 completely abolished the IL13-induced release of OPG(Figure 5b). None of the treatments affected the viability of the slices (Supplementary
Figure S1).
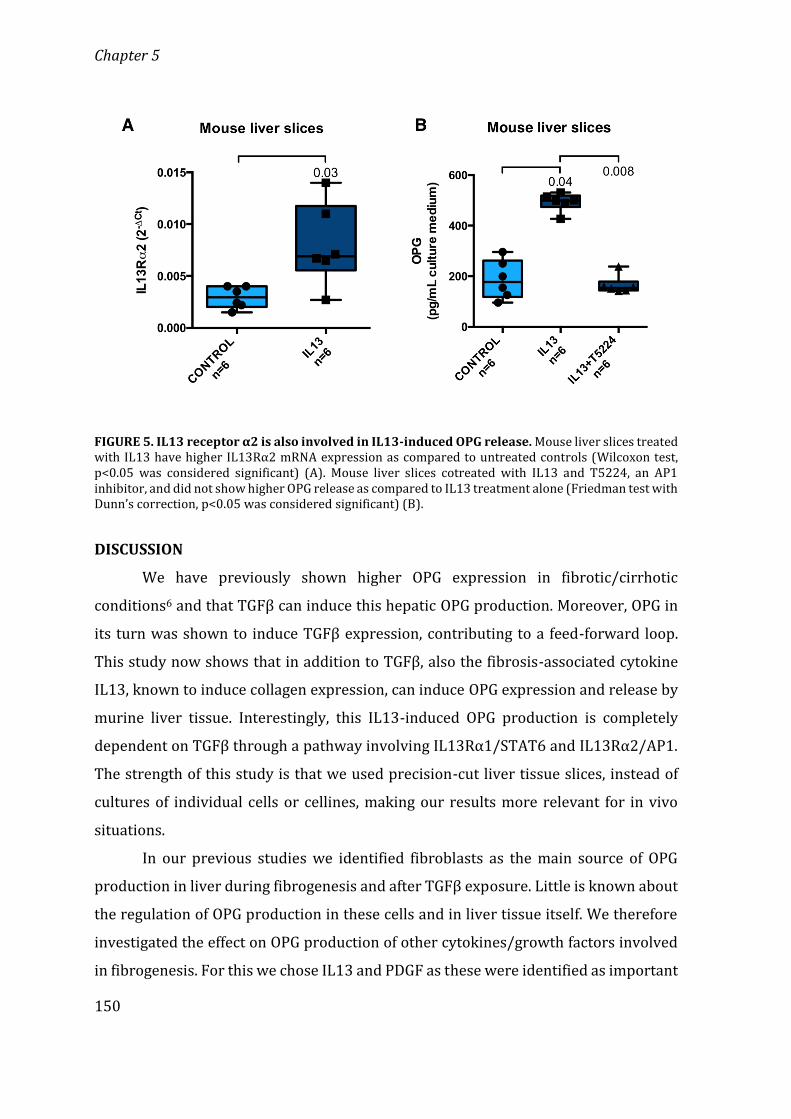
Chapter 5
150
FIGURE 5. IL13 receptor α2 is also involved in IL13-induced OPG release. Mouse liver slices treatedwith IL13 have higher IL13Rα2 mRNA expression as compared to untreated controls (Wilcoxon test,p<0.05 was considered significant) (A). Mouse liver slices cotreated with IL13 and T5224, an AP1inhibitor, and did not show higher OPG release as compared to IL13 treatment alone (Friedman test withDunn’s correction, p<0.05 was considered significant) (B).DISCUSSIONWe have previously shown higher OPG expression in fibrotic/cirrhoticconditions6 and that TGFβ can induce this hepatic OPG production. Moreover, OPG inits turn was shown to induce TGFβ expression, contributing to a feed-forward loop.This study now shows that in addition to TGFβ, also the fibrosis-associated cytokineIL13, known to induce collagen expression, can induce OPG expression and release bymurine liver tissue. Interestingly, this IL13-induced OPG production is completelydependent on TGFβ through a pathway involving IL13Rα1/STAT6 and IL13Rα2/AP1.The strength of this study is that we used precision-cut liver tissue slices, instead ofcultures of individual cells or cellines, making our results more relevant for in vivosituations.In our previous studies we identified fibroblasts as the main source of OPGproduction in liver during fibrogenesis and after TGFβ exposure. Little is known aboutthe regulation of OPG production in these cells and in liver tissue itself. We thereforeinvestigated the effect on OPG production of other cytokines/growth factors involvedin fibrogenesis. For this we chose IL13 and PDGF as these were identified as important

Osteoprotegerin expression in liver tissue is induced by IL13 through TGFβ
151
regulators of fibrosis and are being investigated as potential targets of antifibroticdrugs26–28. We also chose IL1β because this key cytokine is involved in chronic liverinflammation and subsequent development of fibrosis29. Our results showed noinfluence of IL1β on the production of OPG by fibroblasts. This finding suggests thatOPG is mostly produced in connection to fibrogenesis and not during inflammation thatmay precede the development of fibrosis. Furthermore, PDGF-BB did not induce OPGproduction in fibroblasts either. PDGF-BB is the mitogenic agent for fibroblasts andtriggers proliferation and migration of these cells. The pathways leading to stimulationof proliferation by PDGF-BB are apparently not linked to stimulation of OPGproduction.IL13 stimulation on the other hand, did lead to higher production of OPG infibroblasts. This effect was confirmed using precision-cut liver slices of murine liversand this experiment also showed us that the higher OPG expression and productionafter IL13 stimulation was accompanied by higher expression of fibrosis-associatedmarkers Col1α1, HSP47, and Fn1. These profibrotic results of IL13 are in line withpreviously published results by Sugimoto et al.30 and Gieseck et al.27, who showed thatIL13 can induce collagen production and fibrogenesis in hepatic stellate cells and livertissue respectively. Induction of fibrogenesis by IL13 has been suggested to occur viaupregulation of TGFβ1 via IL13Rα1 and IL13Rα2 signaling22,30–32, although otherstudies have suggested that IL13 can also induce fibrosis independently from TGFβ35.Our data show that the IL13-induced production of OPG is completely dependent onTGFβ1, as inhibition of TGFβ1-signalling with galunisertib completely abrogated theeffect of IL13 on OPG production. We also confirmed that both IL13 receptors areinvolved in this TGFβ1-mediated OPG production in liver tissue22,30–32. Blocking or theabsence of STAT6 fully blocked IL13-induced OPG production by liver tissue, showingthat IL13Rα1-signalling through STAT6 is necessary to upregulate TGFβ1 andsubsequently OPG. Furthermore, we showed that IL13 stimulation leads to higherexpression of IL13Rα2 in liver tissue and that blocking signalling of this receptor withan AP1 inhibitor also completely abrogated IL13-induced OPG production in livertissue. A scheme explaining these main findings is depicted in Figure 6.

Chapter 5
152
The signaling through both IL13 receptors and the need for TGFβ1 upregulationbefore OPG can be produced, probably also explains why IL13-induced OPG productionwas lagging in comparison to TGFβ1-induced OPG production. In 36 hours, bothcytokines can induce production of similar levels of OPG, but we found that TGFβ1 canachieve these levels faster. IL13Rα2 is expressed at low levels in normal liver tissueand can therefore not induce TGFβ1 expression immediately when liver tissue isexposed to IL13. The fact that IL13 first needs to upregulate IL13Rα2 through IL13Rα1and STAT6 to be able to stimulate TGFβ1 expression probably accounts for the delayedproduction of OPG after IL13 stimulation.Our results point at an interesting feed-forward mechanism in liver tissueinvolving TGFβ1 as a central player. Both TGFβ1 and IL13 can induce OPG and OPG caninduce expression of TGFβ1 again. In most cases, liver tissue damage will result innormal repair and restoration of fully functional liver tissue. Therefore, there must alsobe brakes in this process to prevent that any type of damage will always end in fibrosis.Our current studies focus on several microRNAs that are induced by TGFβ1 and thatmay serve as the brakes in this feed-forward loop. Another interesting aspect of ourfindings is the possibility of using OPG as a target for therapy. Both TGFβ1 and IL13 aretargets for inhibition of fibrogenesis that are currently being explored in clinicaltrials33-35. As OPG is linking both pathways it seems to be another promising target fortherapy.

Osteoprotegerin expression in liver tissue is induced by IL13 through TGFβ
153
FIGURE 6. IL13 induces liver OPG production via activation of both IL13 receptors andsubsequent induction of TGFβ. A schematic overview of IL13-induced OPG production based on theresults presented in this study. (1) IL13 binds to receptor α1 followed by activation of this receptortriggering (2) STAT6 activation resulting in (3) the increased expression of IL13 receptor, α2, which isinitially expressed at low levels. (4) IL13 then binds to receptor α2, triggering (5) activation oftranscription factor AP1, which induces (6) expression of TGFβ. Finally, as we have reported in ourprevious study, (7) TGFβ can induce OPG protein production by the liver6.CONCLUSIONWe have shown that IL13 induces OPG release by liver tissue through a TGFβ-dependent pathway involving both the α1 and the α2 receptor of IL13 andtranscription factors STAT6 and AP1. OPG may therefore be a novel target for thetreatment liver fibrosis as it is mechanistically linked to two important regulators offibrosis in liver, namely IL13 and TGFβ1.

Chapter 5
154
REFERENCES1. Friedman, S. L. Liver fibrosis – from bench to bedside. J. Hepatol. 38, 38–53 (2003).2. Bataller, R. & Brenner, D. A. Liver fibrosis. J. Clin. Invest. 115, 209–18 (2005).3. Ismail, M. H. & Pinzani, M. Reversal of liver fibrosis. Saudi J. Gastroenterol. 15, 72–9 (2009).4. Xu, F., Liu, C., Zhou, D. & Zhang, L. TGF-β/SMAD Pathway and Its Regulation in Hepatic Fibrosis.J. Histochem. Cytochem. 64, 157–167 (2016).5. Fabregat, I. et al. TGF-β signalling and liver disease. FEBS J. 283, 2219–2232 (2016).6. Adhyatmika, A. et al. Osteoprotegerin is more than a serum marker in liver fibrosis. J. Hepatol.66, S147–S148 (2017).7. García-Valdecasas-Campelo, E. et al. Serum osteoprotegerin and rankl levels in chronic alcoholicliver disease. Alcohol Alcohol. 41, 261–266 (2006).8. Bosselut, N. et al. Including osteoprotegerin and collagen IV in a score-based blood test for liverfibrosis increases diagnostic accuracy. Clin. Chim. Acta J.L. Bosson; A. Paris; ANRS L. Allain, Paris.Methodol. Hospices Civ. Lyon. M-C. Gelineau, B. Poggi 415, 63–68 (2013).9. Prystupa, A. et al. Concentrations of fetuin-A, osteoprotegerin and α-Klotho in patients withalcoholic liver cirrhosis. Exp. Ther. Med. 12, 3464–3470 (2016).10. Monegal, A. et al. Serum osteoprotegerin and its ligand in cirrhotic patients referred fororthotopic liver transplantation: relationship with metabolic bone disease. Liver Int. 27, 492–497 (2007).11. Fabrega, E. et al. Osteoprotegerin and RANKL in alcoholic liver cirrhosis. Liver Int. 25, 305–310(2005).12. Szalay, F. et al. High serum osteoprotegerin and low RANKL in primary biliary cirrhosis. J.Hepatol. 38, 395–400 (2003).13. Moschen, A. R. et al. The RANKL/OPG system and bone mineral density in patients with chronicliver disease. J. Hepatol. 43, 973–983 (2005).14. Boyce, B. F. & Xing, L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res. Ther. 9, S1(2007).15. Harper, E. et al. Vascular calcification in type-2 diabetes and cardiovascular disease: Integrativeroles for OPG, RANKL and TRAIL. Vascul. Pharmacol. 82, 30–40 (2016).16. Wallace, K., Burt, A. D. & Wright, M. C. Liver fibrosis. Biochem. J. 411, 1–18 (2008).17. de Graaf, I. A. M. et al. Preparation and incubation of precision-cut liver and intestinal slices forapplication in drug metabolism and toxicity studies. Nat. Protoc. 5, 1540–1551 (2010).18. Hadi, M., Chen, Y., Starokozhko, V., Merema, M. T. & Groothuis, G. M. M. Mouse Precision-Cut LiverSlices as an ex Vivo Model To Study Idiosyncratic Drug-Induced Liver Injury. Chem. Res. Toxicol.25, 1938–1947 (2012).19. Chomarat, P. & Banchereau, J. Interleukin-4 and lnterleukin-13: Their Similarities andDiscrepancies. Int. Rev. Immunol. 17, 1–52 (1998).20. Murata, T., Husain, S. R., Mohri, H. & Puri, R. K. Two different IL-13 receptor chains are expressedin normal human skin fibroblasts, and IL-4 and IL-13 mediate signal transduction through acommon pathway. Int. Immunol. 10, 1103–1110 (1998).21. Chiba, Y., Todoroki, M., Nishida, Y., Tanabe, M. & Misawa, M. A Novel STAT6 Inhibitor AS1517499Ameliorates Antigen-Induced Bronchial Hypercontractility in Mice. Am. J. Respir. Cell Mol. Biol.41, 516–524 (2009).22. Fichtner-Feigl, S., Strober, W., Kawakami, K., Puri, R. K. & Kitani, A. IL-13 signaling through theIL-13α2 receptor is involved in induction of TGF-β1 production and fibrosis. Nat. Med. 12, 99–106 (2006).23. Daines, M. O. et al. The Journal of Immunology. J. Immunol. 162, 920–930 (2006).24. David, M., Ford, D., Bertoglio, J., Maizel, A. L. & Pierre, J. Induction of the IL-13 receptor α2-chainby IL-4 and IL-13 in human keratinocytes: involvement of STAT6, ERK and p38 MAPK pathways.Oncogene 20, 6660–6668 (2001).

Osteoprotegerin expression in liver tissue is induced by IL13 through TGFβ
155
25. Aikawa, Y. et al. Treatment of arthritis with a selective inhibitor of c-Fos/activator protein-1. Nat.Biotechnol. 26, 817–823 (2008).26. Wynn, T. Cellular and molecular mechanisms of fibrosis. J. Pathol. 214, 199–210 (2008).27. Gieseck, R. L. et al. Interleukin-13 Activates Distinct Cellular Pathways Leading to DuctularReaction, Steatosis, and Fibrosis. Immunity 45, 145–158 (2016).28. Bonner, J. C. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth FactorRev. 15, 255–273 (2004).29. Szabo, G. & Csak, T. Inflammasomes in liver diseases. J. Hepatol. 57, 642–654 (2012).30. Sugimoto, R. et al. Effect of IL-4 and IL-13 on collagen production in cultured LI90 human hepaticstellate cells. Liver Int. 25, 420–428 (2005).31. Lin, J. et al. Assessment of liver steatosis and fibrosis in rats using integrated coherent anti-StokesRaman scattering and multiphoton imaging technique. J. Biomed. Opt. 16, 116024 (2011).32. Shimamura, T. et al. Novel Role of IL-13 in Fibrosis Induced by Nonalcoholic Steatohepatitis andIts Amelioration by IL-13R-Directed Cytotoxin in a Rat Model. J. Immunol. 181, 4656–4665(2008).33. Akhurst, R. J. & Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov.11, 790–811 (2012).34. Schuppan, D. & Kim, Y. O. Evolving therapies for liver fibrosis. J. Clin. Invest. 123, 1887–1901(2013).35. Hoffman-La Roche, completed 2013 (updated 2018), A study of Lebrikizumab in participantswith idiopathic pulmonary fibrosis (IPF), clinical study no. NCT01872689

Chapter 5
156
SUPPLEMENTARY DATA
Supplementary Figure 1. Treatments did not compromise viability mouse liver slices. Treatmentsfor 48 hours of compounds in our experiments did not significantly compromise the viability of themouse liver slices used in our study (n=6, Kruskal-Wallis test corrected for multiple testing).

General Discussion

General Discussion
158
OSTEOPROTEGERIN IN ORGAN FIBROSIS:BIOMARKER, ACTOR AND TARGET OF THERAPY?
Osteoprotegerin as marker of fibrosisFibrosis is a chronic disease, which develops slowly for years and can eventuallycause death of patients1. Therefore, it is highly important to have reliable biomarkersto detect fibrosis in the earliest phase possible in order to prevent an incurable stageof the disease. An easily measurable and sensitive biomarker, which can monitorfibrosis development is crucial to efficiently track antifibrotic efficacy of candidatedrugs in clinical trials2. In this thesis, we have investigated osteoprotegerin (OPG) as apotential marker for fibrosis in different organs and for assessing antifibrotictreatment efficacy.As OPG is already used clinically to diagnoses and stage liver fibrosis, weinvestigated in chapter 2 and chapter 3 whether OPG also plays a role in pulmonaryfibrosis and compared fibrotic responses in lung, liver, kidney and intestine in chapter
4. We found higher OPG levels in lung tissue of IPF patients (chapter 2) as comparedto the control lung tissue. We also showed that pulmonary fibrosis induced by silica inmice was associated with higher OPG mRNA expression and protein production in thelung tissue. In addition, we demonstrated that OPG mRNA expression was higher aftertransforming growth factor β1 (TGFβ1) stimulation and significantly correlated withother fibrosis markers, including collagen1α1 (Col1α1), fibronectin (Fn1), α-smoothmuscle actin (αSMA), and plasminogen activator inhibitor-1 (PAI-1), in vitro in 3T3fibroblasts and ex vivo in murine liver (chapter 4 and 5), lung (chapter 2, 3 and 4),and kidney slices (chapter 4), thus showing that OPG is strongly associated withfibrosis development in those organs. As OPG was strongly correlated with otherfibrosis markers, it should be investigated as a biomarker of fibrosis. Furthermore,higher production of OPG after only 48 hours of TGFβ1 stimulation of healthy tissuesindicate that OPG is upregulated early in the process of wound healing and fibrosis.This idea was reinforced in chapter 3, in which we found that OPG production bymurine lung slices was also higher after stimulation with profibrotic cytokineinterleukin-13 (IL13), while other regular fibrosis-associated markers (Col1α1, αSMA,Fn1 and PAI1 mRNA) did not yet respond.

Osteoprotegerin in Organ Fibrosis: Biomarker, Actor and Target of Therapy?
159
The fact that OPG is a soluble protein, which is secreted into in serum/plasmaand urine of animals and patients, and can therefore be easily detected, may make it anattractive biomarker. In this thesis, we demonstrated that OPG can be detected inmedium of studies with primary lung fibroblasts and 3T3 fibroblasts (chapter 2 andchapter 5), medium of precision-cut tissue slices (chapter 2, 3, 4 and 5), as well as inserum of mice in various models of fibrosis (chapter 4). We also showed that TGFβ1can induce OPG protein secretion from murine liver (chapter 4 and 5), lung (chapter
2, 3 and 4), and kidney slices (chapter 4). Our studies are in line with other studiesshowing higher levels of OPG protein in blood and urine of patients with cirrhoticlivers3 and chronic kidney disease4,5. We also investigated expression of OPG inintestines. Previously, OPG was found to be overexpressed in the colon of patients withinflammatory bowel disease (IBD)6, ulcerative colitis (UC) and Crohn’s disease (CD)7.However, we could not demonstrate OPG was associated with the induction of fibrosisin intestine, nor did we see OPG change with antifibrotic treatment of murine colon orhuman ileum slices (chapter 4). This may be due to the absence of infiltratingperipheral immune cells (e.g. monocytes, T cells, neutrophils) in this study with murinecolon slices8 and/or or due to absence of OPG+-cells in muscle layer of human ileumslices9. In addition to being a possible marker of fibrosis progression, our study alsoshowed that OPG may be useful to assess treatment efficacy of antifibrotic drugs.Lower OPG mRNA expression and protein secretion was associated with decreasedmRNA expression of other fibrosis-associated markers in murine lung slices aftertreatment with two FDA-approved antifibrotic drugs: pirfenidone, and nintedanib(Chapter 3). These result is in line with other studies which showed that pirfenidoneand nintedanib can inhibit production of extracellular matrix (ECM) in vitro10–12, in
vivo13,14 and in clinical studies15. Moreover, lower OPG mRNA and protein levels werealso accompanied with lower mRNA level of other fibrosis-associated markers inmurine lung, liver and kidney slices after treatment with galunisertib, a TGFβ receptortype I kinase inhibitor which was previously applied as an anticancer drug (chapter
4).

General Discussion
160
Osteoprotegerin role in fibrosis developmentIt is an underestimation to think that OPG is only a biomarker of fibrosis. In arecent study by us (manuscript in preparation)16 we demonstrated that OPG itselfcan induce TGFβ1 mRNA expression in murine liver slices and thereby also otherfibrosis-associated markers (Col1α1, αSMA, Fn1 and PAI1 mRNA). Therefore, OPG inliver appears to be involved in a positive feed-forward loop with TGFβ. In lung slicesstimulated with OPG, however, we did not find significant induction of TGFβ mRNAexpression (results not shown) but there seemed to be a trend towards induction thatwe need to investigate in more detail. These results indicate that OPG may have a rolein fibrosis development by stimulating TGFβ1 production.As OPG is a soluble receptor we tried to identify how a receptor can have aneffect on fibrosis development. The most logical explanation being by inhibiting theeffects of its ligands, the most important being RANKL and TRAIL. It is well known thatOPG binding to RANKL prevents RANKL to bind to RANK-expressed macrophages17,18,and OPG binding to TRAIL (TNF-related apoptosis-inducing ligand) prevents TRAIL tobind to DR4 and DR5 on myofibroblasts19,20. We hypothesized that the higherconcentration of OPG in fibrotic lung tissue inhibits activation of ECM-degrading(antifibrotic) macrophages21 and/or inhibits apoptosis of myofibroblasts20,22, whichthus eventually leads to fibrosis progression. In chapter 2, we therefore tried toneutralize excess levels of OPG by treating with soluble RANKL (sRANKL) to amelioratefibrosis. By treating with sRANKL, we expected this excessive RANKL to bind tomacrophages and thus activate an antifibrotic phenotype of macrophages. From thisstudy, we expected RANKL treatment could be considered as a novel potentialtherapeutic for lung fibrosis.Unfortunately, RANKL-treatment did not affect collagen-1 content and Col1α1and Fn1 mRNA expressions in lung tissue of mice with silica-induced pulmonaryfibrosis. RANKL-treatment also did not activate antifibrotic macrophages and did notinduce ECM-degrading proteins, as assessed by MMP-9 mRNA expression, in lungtissue of mice with silica-induced pulmonary fibrosis (chapter 2). The explanationmay have been that RANK expression on interstitial and alveolar macrophages waslower in fibrotic lung tissue than in healthy lungs. Therefore, due to lower RANKexpression on macrophages, additional RANKL may not have had the opportunity to

Osteoprotegerin in Organ Fibrosis: Biomarker, Actor and Target of Therapy?
161
induce effects on macrophages. These results prompted us to do a literature study toget better ideas about how to induce antifibrotic macrophages and the results of thisendeavour are described in chapter 1.The results in chapter 2 did show a tight balance between RANKL and OPG inpulmonary fibrosis. Instead of activating antifibrotic macrophages, administration ofsRANKL to mice with pulmonary fibrosis even stimulated OPG production andsurprisingly resulted in higher numbers of epithelial cells. This suggests that the roleof RANKL may be related to epithelial proliferation and that OPG is regulating thisprocess. Interestingly, in non-small cell lung carcinoma, which develops from epithelialcells, higher RANKL expression and addition of RANKL were associated with highermetastatic potential and this could be inhibited by adding OPG. This suggests that OPGand RANKL may have a role in turnover of epithelial cells in lung23Transforming growth factor β (TGFβ) is a cytokine, which not only plays animportant role in wound healing processes, but is also widely known as key factor infibrosis24. In chapter 4, we show that TGFβ1 stimulation induced mRNA expression ofseveral fibrosis-associated markers like Col1α1, αSMA, Fn1, and PAI-1 in murine lung,liver, kidney and colon slices. In this study we also showed that galunisertib treatment,a TGFβ-receptor type I kinase inhibitor that acts via the SMAD1/SMAD2phosphorylation pathway, of murine lung, liver and kidney slices successfully inhibitedexpression of those fibrosis-associated mRNA markers. In addition, we showed thatOPG mRNA and protein expression in lung, liver and kidney slices after TGFβ1stimulation went up and that it could almost be completely inhibited by Galunisertib,thus indicating that OPG production is mainly regulated by TGFβ1 via the SMADpathway.TGFβ1, however, was not the only cytokine being able to induce expression ofOPG. Our study in chapter 5 revealed that IL13 also stimulated OPG mRNA and proteinexpression in murine liver slices and stimulated TGFβ1 mRNA expression as well.Moreover, the effect of IL13 stimulation on murine liver slices could also be completelyinhibited by galunisertib. This suggests that IL13-induced production of OPG ispredominantly dependent on TGFβ1. Mechanistically, we showed that IL13-inducedOPG production was dependent on IL13Rα1-signalling through STAT6 and subsequentincreased expression of IL13Rα2 and signalling through AP1 as inhibiting both

General Discussion
162
pathways also completely abrogated OPG production. These results indicate that, IL13-induced OPG production is completely dependent on TGFβ through a pathwayinvolving both IL13Rα1/STAT6 and IL13Rα2/AP1.As a soluble protein, OPG in serum or even in the organ itself, may originatefrom multiple sources, not limited to the fibrotic organ itself. Therefore, it is importantto have suitable methods to be able to study OPG regulation on the organ level. Onesuch method is the ex vivo method of precision-cut tissue slices. This technique offersadvantages to investigate OPG production and regulation in specific organs withoutinterference of other organs in more details.PRECISION-CUT TISSUE SLICES: A MODEL TO STUDY FIBROSISFibrosis is a pathological process with multiple pathways and cell typesinvolved. It is important to have an appropriate model to mimic this complexity. In vitrostudies with cells, only provide information about these specific cells involved in aprocess. Therefore, due to the lack of complexity, in vitro studies cannotcomprehensively explain complex mechanisms occurring during fibrosis, and thusextrapolation of in vitro drug effects to the patient is still a big challenge25. In vivostudies with animal models provide models that have the complexity. However, severalpathologic animal models cannot perfectly mimic the pathology and characteristics ofthe disease in humans, for example: unilateral ureteral obstruction (UUO) kidneymodel vs human kidney fibrosis26,27 and bleomycin-induced lung fibrosis vs IPF28.In order to overcome these above challenges, the ex vivo model of precision-cut tissue slices provides a model in which all different cells remain in their originalenvironment, thus serve as an adequate model to study complex mechanisms betweencells and cell-matrix during fibrogenesis29. Precision-cut human tissue slices can alsoprovide better prediction of therapy success in clinical research since precision-cuthuman tissue slices enable more accurate translation of preclinical studies into clinicalstudies because there are no species differences.The model of tissue slices, however, probably cannot mimic in vivo crosstalkbetween organs29, yet this feature offered us the advantage of studying fibrosisregulation in specific organs without interference of other organs. In chapter 2, we

Osteoprotegerin in Organ Fibrosis: Biomarker, Actor and Target of Therapy?
163
combined in vitro and in vivo experiments with a study using murine precision-cut lungslices to gain detailed insight into OPG regulation. For example, even though RANKL-treatment led to higher OPG production in fibrotic murine lung tissue in vivo, RANKL-treatment of TGFβ-treated murine lung slices did not induce OPG mRNA expression.This result implies that OPG was probably produced by infiltrating (immune) cellsrather than tissue-resident cells or was produced in lung tissue under the influence ofa factor produced elsewhere in the body. This result also suggests that the model ofmurine precision-cut tissue slices is beneficial to deliver additional information thatcannot be provided by other models.PERSPECTIVEOPG is used clinically as serum marker to diagnose liver fibrosis and in thisthesis we have shown that OPG is clearly associated with other types of fibrosis too(lung, kidney) and that TGFβ1 is its main inducer. Moreover, OPG responded towardsantifibrotic therapies of pirfenidone, nintedanib (chapter 3), and galunisertib(chapter 4). Therefore, OPG may be a potential marker to diagnose fibrosis and assesstreatment efficacy in patients with lung or kidney fibrosis in clinical practice and it willbe important to validate using serum OPG in relevant patient groups. This also holdstrue for patients with intestinal fibrosis. Even though we could not convincingly showOPG is associated with intestinal fibrosis, previous publications and the fact that inclinical practice intestinal tissue is studied in completeness with infiltrating immunecells present indicate that OPG levels in serum of patients with IBD, UC and CD may stillbe worth to be investigated.OPG production is higher in fibrotic organs in experimental and human fibrosis.Our lung data and a concomitant study by us (not in this thesis, manuscript in
preparation)12 show that OPG is highly produced by (myo)fibroblasts and in liverstimulates fibrosis by inducing TGFβ1 mRNA expression, the key player infibrogenesis. In chapter 3 and chapter 5 we further showed that OPG production inliver and lung can be induced by two profibrotic cytokines, TGFβ and IL13, but bothare completely TGFβ-dependent. These features of OPG regulation offer new insightsindicating that OPG is more than merely a biomarker, OPG may also play role in fibrosis.

General Discussion
164
Our results thus provide evidence for further investigation of OPG as a novel target forthe treatment liver fibrosis. It is important to explore more of how OPG is regulatedand plays a role during fibrosis in various organs to determine whether strategies toinhibit OPG production would be a valid approach.Despite the severity of fibrosis, there is currently no optimal treatment inparticular at the end-stage of the disease. Considering the mechanism of fibrosisdevelopment, there are currently two approaches for improving fibrosis therapy: 1)slowing down fibrosis progression by inhibiting ECM production; 2) reverse fibrosisor promote resolution by inducing ECM degradation. In the context of OPG, we triedthe latter approach in chapter 2 by treating with sRANKL to induce antifibroticmacrophages, but this unexpectedly taught us another possible function of RANKL inlung, namely epithelial regeneration: a mechanism that could potentially be inhibitedby excessive OPG in fibrosis adding further to dysregulated repair. In addition, severalstudies30–32 have suggested that inhibiting excessive OPG production during fibrosismay facilitate apoptosis of myofibroblasts. This hypothesis, however, needs furtherinvestigation.Several issues should be considered when using OPG as a target of therapy.Firstly, ECM production and degradation are part of the normal physiological woundhealing process, thus interfering with this process can cause adverse effect33. Secondly,OPG is not only produced by (healing) tissues but also by healthy bone, thus inhibitionof OPG production or OPG function could affect ECM regulation in bone leading toosteoporosis. Therefore, it may be also be important to design carriers that canspecifically target OPG and deliver antifibrotic drugs to the affected organs or cells,thereby avoiding severe adverse effects.Considering all results presented in this thesis, we suggest to further investigateOPG in the serum of patients with lung, kidney and intestinal fibrosis to investigate itsuse as a biomarker of fibrosis stage, disease progression and therapy success. Furtherstudies should be performed to gain deeper understanding about the role of OPG infibrosis and how OPG is regulated, in order to verify its possibility as a target forantifibrotic therapy.

Osteoprotegerin in Organ Fibrosis: Biomarker, Actor and Target of Therapy?
165
REFERENCES
1. Wynn, T. Cellular and molecular mechanisms of fibrosis. J. Pathol. 214, 199–210 (2008).2. Chin, J. L., Pavlides, M., Moolla, A., Ryan, J. D. & Ryan, J. D. Non-invasive Markers of LiverFibrosis : Adjuncts or Alternatives to Liver Biopsy ? 7, (2016).3. Bosselut, N. et al. Including osteoprotegerin and collagen IV in a score-based blood test for liverfibrosis increases diagnostic accuracy. Clin. Chim. Acta 415, 63–68 (2013).4. Yilmaz, M. I. et al. Osteoprotegerin in Chronic Kidney Disease: Associations with VascularDamage and Cardiovascular Events. Calcif. Tissue Int. 99, 121–130 (2016).5. Benito-Martin, A. et al. Osteoprotegerin in Exosome-Like Vesicles from Human CulturedTubular Cells and Urine. PLoS One 8, (2013).6. Sylvester, F. A., Draghi, A., Bausero, M. A., Fernandez, M. L. & Vella, A. T. 1149 OsteoprotegerinExpression is Upregulated in the Colon of Children With Active Inflammatory Bowel Disease.Gastroenterology 142, S-209 (2012).7. Draghi, A., Ramanarasimhaiah, R., Fernandez, M. L., Vella, A. T. & Sylvester, F. A. Sa1165 ColonicOsteoprotegerin: Higher Expression in Children With Ulcerative Colitis Than Crohn’s Disease.Gastroenterology 146, S-217 (2014).8. Rieder, F. et al. Inflammation-induced endothelial-to-mesenchymal transition: A novelmechanism of intestinal fibrosis. Am. J. Pathol. 179, 2660–2673 (2011).9. Mikkelsen, H. B. & Thuneberg, L. Op/op mice defective in production of functional colony-stimulating factor-1 lack macrophages in muscularis externa of the small intestine. Cell TissueRes. 295, 485–493 (1999).10. Inomata, M. et al. Pirfenidone inhibits fibrocyte accumulation in the lungs in bleomycin-inducedmurine pulmonary fibrosis. Respir. Res. 15, 1–14 (2014).11. Hisatomi, K. et al. Pirfenidone inhibits TGF-β1-induced over-expression of collagen type I andheat shock protein 47 in A549 cells. BMC Pulm. Med. 12, 1–9 (2012).12. Conte, E. et al. Effect of pirfenidone on proliferation, TGF-β-induced myofibroblastdifferentiation and fibrogenic activity of primary human lung fibroblasts. Eur. J. Pharm. Sci. 58,13–19 (2014).13. Schaefer, C. J., Ruhrmund, D. W., Pan, L., Seiwert, S. D. & Kossen, K. Antifibrotic activities ofpirfenidone in animal models. Eur. Respir. Rev. 20, 85–97 (2011).14. Wollin, L., Maillet, I., Quesniaux, V., Holweg, A. & Ryffel, B. Antifibrotic and Anti-inflammatoryActivity of the Tyrosine Kinase Inhibitor Nintedanib in Experimental Models of Lung Fibrosis. J.Pharmacol. Exp. Ther. 349, 209–220 (2014).15. A., A. et al. Exploratory analysis of a phase III trial of pirfenidone identifies a subpopulation ofpatients with idiopathic pulmonary fibrosis as benefiting from treatment. Respir. Res. 12, 143(2011).16. Adhyatmika, A. et al. Osteoprotegerin is more than a serum marker in liver fibrosis. J. Hepatol.66, S147–S148 (2017).17. Liu, W. et al. Osteoprotegerin/RANK/RANKL axis in cardiac remodeling due to immuno-inflammatory myocardial disease. Exp. Mol. Pathol. 84, 213–217 (2008).18. Munasinghe, A., Lin, P. & Colina, C. M. Unraveling Binding Interactions between Human RANKLand Its Decoy Receptor Osteoprotegerin. (2017). doi:10.1021/acs.jpcb.7b0668719. Schneider, P. et al. TRAIL Receptors 1 (DR4) and 2 (DR5) Signal FADD-Dependent Apoptosis andActivate NF-B suggesting that strong regulatory mechanisms control TRAIL receptor signals.TRAIL-R3 (DcR1/TRID) (Pan et al. Immunity 7, (1997).20. Miyashita, T. et al. Osteoprotegerin (OPG) acts as an endogenous decoy receptor in tumournecrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis of fibroblast-likesynovial cells. Clin. Exp. Immunol. 137, 430–436 (2004).21. Jeganathan, S., Fiorino, C., Naik, U., Sun, H. song & Harrison, R. E. Modulation ofOsteoclastogenesis with Macrophage M1- and M2-Inducing Stimuli. PLoS One 9, e104498(2014).

General Discussion
166
22. Crowder, R. N., Dicker, D. T. & El-Deiry, W. S. The Deubiquitinase Inhibitor PR-619 SensitizesNormal Human Fibroblasts to Tumor Necrosis Factor-related Apoptosis-inducing Ligand(TRAIL)-mediated Cell Death * Downloaded from. J. Biol. Chem. 291, 5960–5970 (2016).23. Peng, X. et al. Differential Expression of the RANKL/RANK/OPG System Is Associated with BoneMetastasis in Human Non-Small Cell Lung Cancer. PLoS One 8, (2013).24. Meng, X., Nikolic-Paterson, D. J. & Lan, H. Y. TGF-β: the master regulator of fibrosis. Nat. Rev.Nephrol. 12, 325–338 (2016).25. Scotcher, D., Jones, C., Posada, M., Rostami-Hodjegan, A. & Galetin, A. Key to Opening Kidney forIn Vitro–In Vivo Extrapolation Entrance in Health and Disease: Part I: In Vitro Systems andPhysiological Data. AAPS J. 18, 1067–1081 (2016).26. Yang, H.-C., Zuo, Y. & Fogo, A. B. Models of chronic kidney disease. Drug Discov. Today Dis.Model. 7, 13–19 (2010).27. Chevalier, R. L., Forbes, M. S. & Thornhill, B. A. Ureteral obstruction as a model of renalinterstitial fibrosis and obstructive nephropathy. Kidney Int. 75, 1145–1152 (2009).28. Mouratis, M. A. & Aidinis, V. Modeling pulmonary fibrosis with bleomycin. Curr. Opin. Pulm.Med. 17, 355–361 (2011).29. Westra, I. M., Oosterhuis, D., Groothuis, G. M. M. & Olinga, P. Precision-cut liver slices as a modelfor the early onset of liver fibrosis to test antifibrotic drugs. Toxicol. Appl. Pharmacol. 274, 328–338 (2014).30. McGrath, E. E. et al. Deficiency of tumour necrosis factor-related apoptosis-inducing ligandexacerbates lung injury and fibrosis. Thorax 67, 796–803 (2012).31. Vitovski, S., Phillips, J. S., Sayers, J. & Croucher, P. I. Investigating the Interaction betweenOsteoprotegerin and Receptor Activator of NF-κB or Tumor Necrosis Factor-related Apoptosis-inducing Ligand. J. Biol. Chem. 282, 31601–31609 (2007).32. Meng, H., Bai, X., Yu, H., Wang, Z. & Guo, A. Osteoprotegerin Promotes Cell Growth by RegulatingMatrix Metalloprotease-13 in Chondrocytes. J. Biomater. Tissue Eng. 7, 257–260 (2017).33. Rockey, D. C., Bell, P. D. & Hill, J. A. Fibrosis — A Common Pathway to Organ Injury and Failure.N. Engl. J. Med. 372, 1138–1149 (2015).

Summary – Samenvatting – Ringkasan

Summary – Samenvatting – Ringkasan
168
SUMMARYFibrosis is a pathological condition caused by chronic injury in an organ, due tovarious external triggers like viral and bacterial infections, exposure to metal dust,habit (smoking, fat and alcohol consumption), chronic disease, radiation andchemotherapy. This chronic and persistent injury causes an excessive production ofextracellular matrix (ECM) in the organ, which leads to organ failure and death. In mostpatients, fibrosis is only detected when the organ is already severely and irreversiblydamaged, and this is one of the main reasons for the high mortality. Therefore, it is veryimportant to have reliable, easily measurable and sensitive biomarkers to detectfibrosis in the earliest phase possible in order to prevent an incurable stage of thedisease, as well as to efficiently track antifibrotic efficacy of candidate drugs in clinicaltrials. Osteoprotegerin (OPG) is best known for its role in regulating of ECM in bonetissue. However, recently, OPG was also included in a panel of biomarkers to increasethe accuracy of the diagnosis of liver fibrosis. Therefore, in this thesis, we investigatedOPG as a potential marker for the diagnoses of fibrosis, not only in liver, but also inother organs, and for assessing the efficacy antifibrotic therapy. We also investigatedthe role and regulation of OPG in fibrosis, to explore its possibility as a target forantifibrotic therapy.Fibrosis is a complex condition, which involves many different cell types. Foryears, (myo)fibroblasts have been the focus of fibrosis research for being the majorproducers of ECM. However, in recent years, there is increasing evidence thatmacrophages exhibit a “dual role” in fibrosis development and resolution. Therefore,in Chapter 1, we discuss this elusive behaviour of macrophages during thedevelopment of fibrosis in various organs and antifibrotic characteristics ofmacrophages to design strategies to stimulate the antifibrotic nature of macrophages.In this chapter, we described that stimulating macrophages in such a way that theyexpress receptor activator of nuclear factor-κB (RANK) with its ligand (RANKL) isconsidered as one of strategies to activate antifibrotic macrophages to secrete ECM-degrading matrix-metalloproteinase, thus induce fibrosis resolution.Considering OPG’s role in ECM regulation in bone, we assumed that excessiveOPG in fibrotic organ will bind to RANKL, thereby inhibiting RANKL-RANK

Osteoprotegerin in Organ Fibrosis: Biomarker, Actor and Target of Therapy?
169
interactions, thus inhibiting the activation of antifibrotic macrophages, and preventingdegradation of excessive ECM. Therefore, in Chapter 2, we investigated whether or notadministration of soluble RANKL to mice with silica-induced lung fibrosis was able toneutralize excessive OPG in fibrotic lung, activate antifibrotic macrophages, and inducefibrosis resolution. In this chapter we confirmed that OPG levels are increased in lungtissue of patients with pulmonary fibrosis and in lung tissue of mice with silica-inducedlung fibrosis. Surprisingly, RANKL treatment did not reduce collagen-1 deposition inlung tissue in this model, instead, it stimulated higher OPG levels and numbers ofepithelial cells. This results suggest that the combination RANK/RANKL/OPG has a rolein regulating tissue repair processes and epithelial proliferation in the lung.Furthermore, our study with precision-cut lung slices implies that OPG productionmight also be influenced by infiltrating cells or other external factors.Due to the fact that higher OPG levels are correlated with fibrosis in lung, wecontinued our studies about the regulation of OPG in lung tissue during fibrogenesis inChapter 3. We found that more OPG was secreted from human fibrotic lung slices andTGFβ1-stimulated murine lung slices than from the controls, and these elevated OPGlevels correlated with other fibrosis-associated markers, including collagen1α1,fibronectin, and plasminogen activator inhibitor-1. In this chapter, OPG mRNAexpression and protein secretion were also induced in murine lung slices afterstimulation with profibrotic cytokine interleukin-13 (IL13). Interestingly, OPG geneand protein expression were down-regulated after treatment with two FDA-approvedantifibrotic drugs: pirfenidone and nintedanib, while other fibrosis-associated markersdid not respond (yet), indicating that OPG may be a potential sensitive marker fordetecting early fibrosis development and assessing antifibrotic treatment efficacy.We further explored OPG as fibrosis marker in murine and human tissue slicesfrom lung, liver, kidney and intestine in Chapter 4. In this chapter, OPG secretion waselevated in ex vivo fibrosis-induced murine lung, liver and kidney slices and fibrotichuman lung and liver slices, suggesting that OPG is associated with early- and end-stage fibrosis in those organs. This chapter also showed elevated OPG concentrationsin plasma of murine models of liver and kidney fibrosis, suggesting that OPG maypotentially be a blood-based biomarker in a clinical setting. Meanwhile, OPG seemedless associated with fibrosis in murine and human intestinal slices, but it may still be

Summary – Samenvatting – Ringkasan
170
worthwhile to investigate OPG levels in serum of patients with intestinal fibrosis inmore detail. Moreover, our results with TGFβ1 stimulation and galunisertib (a TGFβ-receptor type I kinase inhibitor) treatment of slices indicated that OPG production inlung, liver and kidney is regulated by TGFβ via the SMAD pathway.In Chapter 5, we further studied regulation of OPG in liver tissue. We found thatOPG production in liver can be induced by two profibrotic cytokines. IL13-induced OPGproduction appeared also to be completely dependent on TGFβ through a pathwayinvolving both IL13Rα1/STAT6 and IL13Rα2/AP1.In this thesis, we discuss that OPG levels are strongly associated with organfibrosis and OPG can be detected in plasma of murine models of fibrosis, and thereforeit is worth to measure OPG concentration in the serum/plasma of patients with lung,kidney and intestinal fibrosis to further investigate its use as a biomarker of fibrosisstage, disease progression and therapy success. Better understanding the role andregulation of OPG in fibrosis in various organs will be beneficial to determine whetherstrategies to inhibit OPG production or neutralize OPG would be a valid approach forantifibrotic therapy. Furthermore, in this thesis we also discussed that precision-cutmurine and human tissue slices are suitable ex vivo models to explore the role andregulation of OPG in fibrotic organs as well as its features as a biomarker and that itcomplements other in vitro and in vivo models.

Osteoprotegerin in Organ Fibrosis: Biomarker, Actor and Target of Therapy?
171
SAMENVATTINGFibrose is een pathologische aandoening, die veroorzaakt wordt door diverseexterne factoren, zoals virale en bacteriële infecties, blootstelling aan metaalstof,chronische ziektes, bestraling en chemotherapie. Bij deze aandoening wordtextracellulaire matrix (ECM) overmatig geproduceerd wat kan leiden tot chronischeschade van het orgaan en uiteindelijk tot orgaanfalen. Defecten aan vitale organenzoals hart, lever, nier, long, darmen en beenmerg kunnen uiteindelijk leiden tot de doodvan patiënten. Bij de meeste patiënten wordt de diagnose fibrose pas vastgesteldwanneer het orgaan al ernstig en onherstelbaar beschadigd is en dit is de belangrijksteoorzaak voor de hoge sterfte. Het zou daarom van onschatbare waarde zijn als er eengemakkelijk meetbare en gevoelige biomarker zou worden gevonden waarmee fibrosein een zo vroeg mogelijk stadium gedetecteerd kan worden en waarmee de efficiëntievan nieuwe antifibrotische geneesmiddelen in klinisch studies vastgesteld kan worden.Osteoprotegerin (OPG) is vooral bekend van zijn rol in het reguleren van ECM inbotweefsel. Recentelijk werd echter gevonden dat OPG ook gebruikt kan worden alsaanvullende biomarker waarmee de nauwkeurigheid van de diagnose leverfibrosevergroot kan worden. We hebben daarom in dit proefschrift onderzocht of OPGgebruikt kan worden als marker voor het diagnosticeren van fibrose, niet alleen in delever, maar ook in verschillende andere organen. Daarnaast hebben we onderzocht ofOPG gebruikt kan worden als marker voor het al dan niet aanslaan van eenantifibrotische behandeling. Ook onderzochten we de rol en regulatie van OPG bijfibrose. Het doel hiervan was om te onderzoeken of het remmen van de productie ofde neutralisatie van OPG een antifibrotische therapie zou kunnen zijn.Fibrose is een complexe aandoening, waarbij veel verschillende celtypenbetrokken zijn. Jarenlang waren (myo)fibroblasten het onderwerp vanfibroseonderzoek omdat zij de belangrijkste producenten van ECM zijn. De laatstejaren is er echter steeds meer bewijs gekomen dat macrofagen een "dubbele rol" spelenin de ontwikkeling en het verdwijnen van fibrose. Daarom bespreken we in Hoofdstuk1 de pro- en antifibrotische eigenschappen van macrofagen tijdens de ontwikkelingvan fibrose in verschillende organen met als doel strategieën te ontwikkelen om hetantifibrotische karakter van macrofagen te stimuleren. In dit hoofdstuk beschrijven wedat het zodanig stimuleren van macrofagen dat de receptor activator of nuclear factor-κB (RANK) met zijn ligand (RANKL) tot expressie worden gebracht een strategie zoukunnen zijn om het antifibrotische karakter van macrofagen te activeren omdat deze

Summary – Samenvatting – Ringkasan
172
cellen hierdoor ECM-afbrekende matrix-metalloproteinase zouden kunnen gaanuitscheiden, hetgeen leidt tot het verminderen van fibrose.Vanwege de rol van OPG in de regulatie ECM in botweefsel, gingen we ervan uitdat overmatig OPG in een fibrotisch orgaan zal binden aan RANKL waardoor RANKL-RANK-interacties geremd worden. Hierdoor wordt de activatie van antifibrotischemacrofagen geremd en wordt daarmee de degradatie van overmatig ECM voorkomen.We hebben daarom in Hoofdstuk 2 onderzocht of de toediening van oplosbaar RANKLaan muizen met silica-geïnduceerde longfibrose leidt tot de neutralisatie van OPG in defibrotische long, de activatie van antifibrotische macrofagen en daarmee tot hetverminderen van fibrose. In dit hoofdstuk hebben we bevestigd dat de OPG-concentraties in longweefsel van patiënten met longfibrose en muizen met door silicageïnduceerde longfibrose hoger zijn dan in gezonde longen. Verrassend genoeg leiddede RANKL-behandeling niet tot een verminderde afzetting van collageen-1 inlongweefsel van het door silica geïnduceerde fibrose maar tot een hogere OPG-concentratie en een toegenomen aantal epitheelcellen. Deze resultaten suggereren datde combinatie RANK/RANKL/OPG een rol speelt bij het reguleren vanweefselregeneratie en de proliferatie van epitheelcellen in de longen. Bovendienimpliceren onze resultaten in hoofdstuk 2 dat OPG-productie ook zou kunnen wordenbeïnvloed door infiltrerende cellen of externe factoren.Doordat een hogere OPG-concentratie gecorreleerd is met fibrose in de long,hebben we in Hoofdstuk 3 onze studies naar regulatie van OPG in longweefsel tijdensfibrogenese voortgezet. We vonden dat er meer OPG werd uitgescheiden door humanefibrotische weefselplakjes en TGFβ1-gestimuleerde longweefselplakjes van muizendan door het gezonde longweefsel. Bovendien waren de hogere OPG-concentratiesgecorreleerd met andere met fibrose geassocieerde markers, waaronder collageen1α1,fibronectine, en plasminogeen activator inhibitor-1. Daarnaast werd in dit hoofdstukgevonden dat OPG-mRNA-expressie en eiwitsecretie in longweefselplakjes van muizenhoger was na stimulatie met profibrotisch cytokine interleukine-13 (IL13) en lager wasna behandeling met twee door de FDA goedgekeurde antifibrotische geneesmiddelen:pirfenidon en nintedanib. Dit werd zelfs gevonden wanneer de therapie (nog) geeneffect had op andere met fibrose geassocieerde markers, wat aangeeft dat OPGpotentieel een gevoelige marker is voor het detecteren van de efficiëntie van eenantifibrotische behandeling.We onderzochten OPG als fibrose-marker in long, lever, nier en darm met behulpvan weefselplakjes van muizen en humane weefsels in Hoofdstuk 4. In dit hoofdstuk

Osteoprotegerin in Organ Fibrosis: Biomarker, Actor and Target of Therapy?
173
werden hogere OPG-concentraties gevonden in TGFβ1 behandelde long-, lever- ennierplakjes van muizen en humane fibrotische long- en leverplakjes, wat suggereertdat OPG geassocieerd is met fibrose in deze organen in een vroeg- en in eeneindstadium. In dit hoofdstuk werd ook aangetoond dat OPG-concentraties in plasmavan muizen met lever- en nierfibrose hoger waren dan in plasma van gezonde muizen,wat suggereert dat OPG ontwikkeld zou kunnen worden als biomarker in bloed.Ofschoon de plasma OPG-concentraties minder geassocieerd waren met fibrose inmuizen en humane darmplakjes, is het toch de moeite waard om OPG-spiegels inplasma van patiënten met darmfibrose verder te onderzoeken. Voorts gavenexperimenten waarbij weefselplakjes werden gestimuleerd met TGFβ1-stimulatie ofbehandeld met galunisertib (een TGFβ-receptor type I-kinase-remmer) aan dat OPG-productie in long, lever en nier door TGFβ via de SMAD-route wordt gereguleerd.In het onderzoek beschreven in Hoofdstuk 5, hebben we de regulatie van OPG inleverweefsel nader bestudeerd. Gevonden werd dat OPG-productie in de lever kanworden geïnduceerd door twee profibrotische cytokinen, TGFβ1 en IL13. Daarnaastbleek dat IL13-geïnduceerde OPG-productie ook volledig afhankelijk te zijn van TGFbvia een route waarbij zowel IL13Rα1/STAT6 als IL13Rα2/AP1 betrokken zijn.In dit proefschrift is aangetoond dat hogere OPG-concentraties sterkgeassocieerd zijn met orgaanfibrose en dat OPG kan worden gedetecteerd in hetplasma van muizen met lever- en nierfibrose. Het is daarom de moeite waard om deOPG-concentratie in het plasma van patiënten met verschillende vormen vanorgaanfibrose te bestuderen. Hierbij kan verder onderzocht worden of OPG gebruiktkan worden als een biomarker voor fibrose, voor ziekteprogressie en voortherapiesucces. Een beter begrip van de rol en regulatie van OPG bij fibrose inverschillende organen zal noodzakelijk zijn om te bepalen of strategieën om OPG-productie te remmen of OPG te neutraliseren kan leiden tot een effectieveantifibrotische therapie. Daarnaast wordt in dit proefschrift aangetoond datweefselplakjes van muizen en humane weefsels een geschikt ex vivo model zijn om derol en regulatie van OPG in fibrotisch organen te onderzoeken en dat het modelcomplementair is met andere in vitro en in vivo modellen.

Summary – Samenvatting – Ringkasan
174
RINGKASANFibrosis adalah kondisi patologis yang disebabkan oleh cedera kronis pada organakibat berbagai pemicu eksternal seperti infeksi virus dan bakteri, paparan debu logam,kebiasaan (merokok, konsumsi lemak dan alkohol), penyakit kronis, radiasi dankemoterapi. Cedera kronis yang terjadi terus menerus ini menyebabkan produksi matriksekstraselular yang berlebihan di organ, yang menyebabkan kerusakan organ dan kematianpasien. Pada kebanyakan pasien, fibrosis baru terdeteksi ketika organ telah rusak parahdan tidak dapat disembuhkan lagi, dan ini adalah alasan utama tingginya angka kematianakibat fibrosis. Oleh karena itu, sangat penting untuk memiliki biomarker yang mudahdiukur, sensitif dan dapat dipercaya untuk mendeteksi kondisi fibrosis sedini mungkin,untuk mencegah tahap penyakit yang tidak dapat disembuhkan, serta untuk memantauefektivitas antifibrotik pada kandidat obat dalam uji klinis.Osteoprotegerin (OPG) dikenal luas karena perannya dalam mengatur matriksekstraselular dalam jaringan tulang. Namun, baru-baru ini, OPG juga disertakan sebagaipenanda/biomarker untuk meningkatkan akurasi diagnosis fibrosis hati/liver. Olehkarena itu, dalam tesis ini, kami meneliti OPG sebagai penanda/biomarker potensial untukmendiagnosis fibrosis, tidak hanya di hati, tetapi juga di berbagai organ, dan untuk menilaiefektivitas pengobatan antifibrosis. Kami juga menyelidiki peran dan regulasi OPG padaorgan yang mengalami fibrosis, untuk mengeksplorasi kemungkinannya sebagai targetterapi antifibrotik.Fibrosis adalah kondisi kompleks, yang melibatkan banyak jenis sel yang berbeda.Selama bertahun-tahun, (myo)fibroblast telah menjadi fokus penelitian fibrosis karenamerupakan produsen utama matriks ekstraselular. Namun, dalam beberapa tahunterakhir, semakin banyak penelitian yang membuktikan bahwa makrofag menunjukkan"peran ganda" dalam perkembangan dan pemulihan fibrosis. Oleh karena itu, pada Bab 1,kami membahas perilaku unik makrofag selama perkembangan dan pemulihan fibrosis diberbagai organ serta mengidentifikasi karakteristik makrofag yang bersifat antifibrotikuntuk merancang strategi menstimulasi sifat antifibrotik makrofag. Dalam bab ini, kamimenjelaskan bahwa stimulasi terhadap makrofag yang mengekspresikan receptoractivator of nuclear factor-κB (RANK) dengan menggunakan ligannya (RANKL) dianggapsebagai salah satu strategi untuk mengaktifkan sifat antifibrotik dari makrofag sehinggadapat mensekresikan matriks-metaloproteinase yang dapat mendegradasi matriksekstraseluler, sehingga mempercepat pemulihan dari kondisi fibrosis.Dengan mempertimbangkan peran OPG dalam regulasi matriks ekstraselular padatulang, kami menduga bahwa kadar OPG yang berlebihan di dalam organ yang mengalamifibrosis akan berikatan dengan RANKL, menghambat interaksi RANKL-RANK, sehingga

Osteoprotegerin in Organ Fibrosis: Biomarker, Actor and Target of Therapy?
175
menghambat aktivasi makrofag antifibrotik, dan mencegah degradasi matriksekstraseluler yang berlebihan. Oleh karena itu, dalam Bab 2, kami menyelidiki apakahpengobatan dengan menggunakan RANKL mampu menetralkan OPG yang berlebihandalam paru-paru yang mengalami fibrosis, mengaktifkan makrofag antifibrotik, danmempercepat pemulihan fibrosis pada mencit dengan fibrosis paru akibat diinduksiserbuk silika. Bab ini mengkonfirmasi adanya kadar OPG yang lebih tinggi dalam jaringanparu-paru pasien yang menderita fibrosis paru idiopatik dan pada mencit dengan fibrosisparu akibat diinduksi serbuk silika. Di luar dugaan, pengobatan RANKL ternyata tidakmampu mengurangi deposisi kolagen-1 yang berlebih dalam jaringan paru pada mencityang diinduksi silika, dan sebaliknya justru menstimulasi produksi OPG dan jumlah selepitel yang lebih banyak. Hasil ini menunjukkan bahwa RANK/RANKL/OPG memilikiperan dalam mengatur proses perbaikan jaringan dan proliferasi epitel di paru-paru. Lebihlanjut, penelitian kami dengan menggunakan irisan presisi paru menunjukkan bahwaproduksi OPG tidak hanya diproduksi atau dipengaruhi oleh sel-sel di paru, namunmungkin juga dipengaruhi oleh infiltrasi sel atau faktor eksternal dari luar jaringan paru.Dengan fakta bahwa kadar OPG yang lebih tinggi berkorelasi dengan kondisifibrosis di paru-paru, kami melanjutkan penelitian tentang regulasi OPG dalam jaringanparu selama proses fibrogenesis pada Bab 3. Kami menemukan OPG disekresikan lebihbanyak dari irisan presisi paru-paru manusia yang menderita fibrosis paru idiopatik danirisan presisi paru-paru mencit yang distimulasi oleh transforming growth factor β1(TGFβ1), dan sekresi OPG yang lebih banyak ini berkorelasi dengan meningkatnyaekspresi penanda/ biomarker fibrosis lainnya, termasuk collagen1α1, fibronectin, andplasminogen activator inhibitor-1. Dalam bab ini, ekspresi mRNA OPG dan sekresi proteinOPG ditemukan lebih tinggi pada irisan paru-paru mencit setelah stimulasi dengan sitokinprofibrosis interleukin-13 (IL13), dan kadarnya lebih rendah setelah pengobatan dengandua obat antifibrotik yang disetujui FDA: pirfenidone dan nintedanib, bahkan meskipunpenanda/biomarker fibrosis lainnya belum merespon, mengindikasikan bahwa OPGmerupakan penanda yang sensitif dan potensial untuk mendeteksi fibrosis sedini mungkindan untuk menilai efektivitas pengobatan antifibrosis.Kami lebih lanjut mengeksplorasi OPG sebagai penanda fibrosis di paru-paru, hati,ginjal dan usus menggunakan irisan presisi jaringan mencit dan jaringan manusia di Bab4. Dalam bab ini, OPG disekresikan pada konsentrasi yang lebih tinggi dari model ex vivoirisan presisi paru-paru, hati dan ginjal mencit yang diinduksi fibrosis, serta dari irisanpresisi paru-paru dan hati manusia yang menderita fibrosis, menunjukkan kadar OPGterkait dengan kondisi fibrosis pada organ-organ tersebut, baik tahap awal maupun tahapakhir fibrosis. Bab ini juga menunjukkan konsentrasi OPG yang lebih tinggi dalam plasma

Summary – Samenvatting – Ringkasan
176
mencit yang menderita fibrosis hati dan ginjal, menunjukkan bahwa OPG berpotensi untukdikembangkan sebagai biomarker fibrosis dalam bidang klinis karena konsentrasinyadapat diukur dalam sampel darah. Sementara itu, OPG tampaknya kurang terkait denganfibrosis pada irisan presisi usus mencit dan manusia, namun diperlukan penelitian lebihlanjut untuk menyelidiki lebih dalam tentang kadar OPG dalam serum pasien yangmenderita fibrosis usus. Selain itu, hasil kami yang melibatkan stimulasi TGFβ danpengobatan dengan galunisertib (suatu inhibitor kinase reseptor TGFβ- tipe I) pada irisanpresisi jaringan mencit mengindikasikan bahwa produksi OPG di paru-paru, hati dan ginjaldiregulasi oleh TGFβ1 melalui jalur SMAD.Pada Bab 5, kami mempelajari lebih lanjut regulasi OPG dalam jaringan hati. Kamimenemukan bahwa produksi OPG dalam hati dapat diinduksi oleh dua sitokin profibrotik.Produksi OPG yang diinduksi IL13 sepenuhnya tergantung pada TGFβ melalui jalur yangmelibatkan IL13Rα1/STAT6 dan IL13Rα2/AP1.Sebagai kesimpulan, tesis ini membahas tentang kadar OPG yang berhubungan eratdengan kondisi fibrosis suatu organ dan OPG dapat dideteksi dalam plasma mencit yangmengalami fibrosis, sehingga perlu penelitian lebih lanjut untuk mengukur konsentrasiOPG dalam serum/plasma pasien yang menderita fibrosis paru, ginjal dan usus, untukmemvalidasi penggunaannya sebagai biomarker penanda/biomarker untuk mendeteksiderajat keparahan fibrosis, perkembangan penyakit dan keberhasilan terapi. Pemahamanyang lebih mendalam tentang peran dan regulasi OPG dalam fibrosis di berbagai organakan bermanfaat untuk menentukan apakah strategi untuk menghambat produksi OPGatau menetralisir OPG merupakan pendekatan yang valid serta untuk terapi antifibrotik.Lebih lanjut, tesis ini juga membahas bahwa model ex vivo irisan presisi jaringan darimencit dan manusia adalah model yang cocok untuk mengeksplorasi peran dan regulasiOPG dalam organ fibrotik serta menginvestigasi fungsinya sebagai biomarker, daninformasi yang diperoleh dari eksperimen dengan model ini dapat melengkapi informasiyang diperoleh dari eksperimen dengan model in vitro dan in vivo.

Acknowledgements

Acknowledgements
178
After 8 chapters, finally, it is the time to write the hardest chapter of my thesis. I write thischapter with teary eyes as I remembered all immeasurable supports and helps I received duringmy PhD journey. First of all, Alhamdulillahhirrabbil’alamin. All the praises and thanks be to AllahAlmighty, the Lord of the Universe, for all blessings and for sending wonderful people into my life.I definitely would never started this PhD journey without a recommendation from mypromoters and co-promoter to receive Ubbo Emmius Scholarship. Moreover, I also could notcomplete this thesis without their guidance. I am a very lucky student to have them as mysupervisors. I started my project with optimism, yet I faced some unexpected results. But when Ithought that I failed, they said “In research, there is no failure. It is a success when provingsomething acts in practice differently than in theory.” I probably disappointed them many times,yet they continue to support and encourage me endlessly. Therefore, I would like to express mydeepest gratitude Prof. Peter Olinga, Prof. Barbro N. Melgert and Dr. Wouter L.J Hinrichs forsupervising me with all the patience in the world and for all things they have done for me.Dear Peter, thank you very much for giving me the chance to be part of your lab. Your easy-going yet disciplined personality allowed me to explore and express my ideas within a structuredscientific way. You constantly encouraged me to say ”no” (even to yourself) when I disagreed withsomething or unable to help others. This is a priceless lesson for me. Furthermore, being back inIndonesia and having a new-born baby while finishing the thesis and working in UI was challengingfor me. But during this time, you always supported me. I am deeply touched by your sincere caretowards me. Thank you very much, Peter. Dear Barbro, being your student alongside the smart-tough-hard working Carian, Christina, Adhy, Laura and Anienke sometimes made me feel inferior,yet, as a wise mother you always find ways to encourage me. Your point of view, which differedfrom Peter most of the time, enriched my understanding about my project. Your dedication towardsyour work was amazing. Your head injury did not stop you from publish articles and review mythesis. Thank you very much, Barbro. One day you told me that you teach your kids about Borneoand Sulawesi. If you plan to visit Habibi in Sulawesi, please let me know so I can sabotage you duringyour transit in JakartaDear Peter and Barbro, even though it was awkward to see you both arguefor the first time, but thank to you, I learned how to argue professionally (instead of taking itpersonally). That was an important lesson for me to continue my career as an academician.Furthermore, your professionalism at work and dedication to family, make you as my role modelin managing the balance between work and family. Dear Wouter, thank you for picking me up at theairport and helping me with the very impressive thirty-kilograms-red-monster. You are the onlysupervisor who ever saw the worst and the other side of me. You saw me crying, and you watchedme playing Angklung. Thank you very much for your warmth which always succeeded to comfortme. I still owe you an article with you as the last author, therefore, I hope we still can work togetherfor a very long time in the future.Not only I learned how to manage the research, I also learned many things from my threesupervisors. And above all, the way they managed their students is something I value most. I alwaysadmire the way they manage (challenging) students (like me). The way they appreciate every singleachievement of their students always succeeded to encourage me when I was at my lowest.Moreover, not only paying attention to research-related aspects, they also consider the personalaspect of their students. They even considered my physical condition when we decide to have mydefense in summer. Thank you very much, Peter, Barbro, and Wouter. I wish you and your family ahappy, successful and prosperous life. Radit and I are looking forward welcoming you in Indonesia!Furthermore, I would like to extend my sincere thanks to Prof. Janette K. Burgess, Prof.
Reinoud Gosens and Prof. Silke Meiners as the assessment committee, for providing time toassess my thesis and for giving valuable and constructive suggestions to this thesis.

Osteoprotegerin in Organ Fibrosis: Biomarker, Actor and Target of Therapy?
179
Since I now live in Indonesia, I definitely cannot prepare the defense properly without thesetwo amazing and beautiful ladies. Therefore, thank you very much, Dorenda Oosterhuis andFransien van Dijk for all kindness and help you gave me, not only for my defense, but also foreverything you did for all this time. It is such an honor to have you both as my paranymphs. DearDorenda, I always call you fairy godmother, and that was definitely a compliment. Not due to theage, I call you mother because you always take care of us, of our shopping lists, of our requests andeverything with smile and patience. Your cheerful personality never failed to boost up my mood.Thank you for being a mother that I can always rely on. Please send my love to Lois and Nila. I hopesomeday we can play again together. Dear Fransien, your wide smile and kindness always impressme. Your message via WhatsApp or e-mail always succeeded to warm my heart, and I always smileat our conversation about Josefien and Salman. I do hope that one day Salman can meet you, Jakkoand Josefien. I wish you success for your post-doc and future career. Moreover, I would like to thankMas Adjie, for being paranymph for Radit and giving so much help in preparing our defense. I wishyou good luck for your post-doc and future career, and hopefully we can travel again with you aswhat we had in Istanbul and Amsterdam.Most of the time, I performed my experiments at Department of PharmaceuticalTechnology and Biopharmacy (PTB). Therefore, I am extremely grateful to Prof. H.W. (Erik)Frijlink for giving a chance to do research at his department. Dear Erik, I always enjoy the jokesyou shared during tea time, and your encouraging words during R&O meeting never failed toincrease my self-confidence. Thank you very much, Erik. I wish you and your family all thehappiness and good health. Furthermore, beside PTB, I also worked a lot at Department ofPharmacokinetics, Toxicology, and Targeting (PTT). Therefore, I would also like to extend mygratitude to Prof. Geny Groothuis and Prof. Klaas Poelstra for allowing me to work in PTT andjoin PTT’s Friday meeting. Thank you for suggestions during my presentation in PTT’s meeting.Through many experiments and meetings, I received many help, supports, and suggestionsfrom members of the Slicing Group. It such a great pleasure to work with amazing people like them,thus I would like to express my sincere thanks to them. Dear Rick and Miriam, more than orderingand sacrificing the mice, I thank you for every help you gave me in the lab. I wish you all the bestfor your career and family. Dear Naomi, even though you did not work with the slices, we workedside-by-side in the lab quite often. I always admire your good management on your research andstudent while arranging PhD Day and scheduling lab cleaning and presentation for the weeklymeeting. Thank you for your friendliness. I wish you and Frank a successful and happy life. DearTobias, you are such a multi-talented friendly and helpful handsome man! No wonder that myjuniors (Cyntia and Pony) admired you a lot. If this thesis is not read by Radit, I would also admitthat I am admiring you. Thank you for your friendliness, I appreciated a lot when you were beingpolite by incorrectly guessing my age, that made me happy . I wish you success with your PhDand future career. Dear Isabel, I was always impressed by your intelligence, your efficient work,and straight-forward personality. Good luck with your career. Dear Jasper, even though we did notwork together much, you were always very friendly and helpful to me. We also once shared thecakes. Thank you, Jasper. Good luck with your MD-PhD!Our hectic work in the lab, as well as our relaxing time in the canteen, were always cheerful;thanks to duo-Emilia (Emilia Bigaeva and Emilia Gore). Dear Emilia, since our first encounter, youhave been very nice and friendly to me. You even invited me to join you and Emma to the festival inNoorderplantsoen. It was a shame that I could not join you and Emma at that time due to cold. Astime goes by, you continuously showed your friendliness and gave me many help. I thank you a lotfor that. Dear Emma, your active, energetic and friendly personality never fails to cheer up yoursurrounding. Together with Emilia, you always shared exciting stories of Game of Thrones and

Acknowledgements
180
many fascinating-and-updated information. I always enjoyed our conversation. Furthermore, Ispecially thank the both of you to allow me to study osteoprotegerin in your samples. Good luckwith your defenses in July, and I wish you both success with your future careers. Let’s keep in touch!I appreciate Peter a lot that he took many Asian students in his lab. This way, hopefully, wecan build a good network between universities in Asia and visit each other more often in the future.In my first day at university, Wouter introduced me to BT, one of the senior PhD students in Peter’sLab. In Indonesia, we say BT when we feel bored. However, never a single time I felt bored havingconversation with you. Thank you for guiding and collaborating with my husband so that he canfinish his study. I wish you and your family success and a happy life. At this final moment, I wouldlike to specially thank and apologize (three times, hahaha) to my lovely roommates: Jo and Su.Thank you for every help you give, every support you provide, every story, moment and foods weshared together, and above of all, for our friendship. Also, sorry for every inconvenience (to Jo,probably it was my perfume and my tears) and every annoying distraction (“Su, can I ask yousomething?”). Thank you for always being patient towards me I was frustrated when you bothstopped talking to each other, but I can be now relieved that your friendship grow stronger.Congratulation! I wish Jo and Gift, as well as Su, Naqin and the baby, success and a happy life ahead!Let us travel together again someday!Besides the member of Slicing Group, I also received many help, kindness, and warmth fromPTB staff. Therefore, I greatly thank the all PTB members: Herman, Anne de Boer, Sonja, Anko,Jan, Paul, Ed, Marinella, Caroline, Doetie, Lida. Dear Herman, we discussed several times aboutthe review article, which unfortunately Radit and I could not finish. Thank you very much and sorryabout that. Hopefully, we have a chance to collaborate again in the future. I would also like to thankmany generations of PhD’s: Wouter Tonnis, Anne, Marcel, Philip, Gaurav, Imco, Max,Annemarie, and Gerian, and wish you all success with your PhD and future career. My specialthanks go to Niels, Floris, and Maarten for training me when I adjusted myself with the project infirst months of my PhD. Dear Jasmine, thank you for your kindness and warmth to me, Radit andSalman. I was so happy to hear that you got a post-doc position. I wish you, Tushar and Karnik asuccessful and happy life! Dear Mitchel, I was happy when you joined the group and work withprecision-cut lung slices. With your qualities, no wonder that you have published many articles.Thank you for your warmth and kindness. Good luck with your PhD!Being Barbro’s student and Adhy’s partner in crime, I also worked a lot at PTT. Therefore Iwould like to thank all PTT member for their warm welcome: Dr. Leonie Beljaars, Dr. Inge deGraaf, Dr. Anna Salvati, and Prof. Angela Casini. Dear Leonie, thank you very much for yourvaluable input to the review article we wrote and for osteoprotegerin project.During my work in PTT, I often disturbed and asked questions to Eduard, Catharina,Marina, Marjolijn, Gillian and Jan. However, they always answer, explain and help patiently. Forevery answered question, every assistance and suggestion I received, I would like to thank you verymuch. I specially thank Eduard, Catharina, and Marjolijn for their assistance during my work in PTTlab, and to Marina for her assistance when I was in Indonesia.I would also like to thank all colleagues in PTT for their help and friendliness: Carian,Christina, Amirah, Viktoriia, Suresh, Habibie, Anienke, Laura, Gwenda, Roberta, Sylvia,Valentina, Natalia, Andreia, Ming, and Kaisa. Dear Carian, as my senior on lung andosteoprotegerin research, I owe you many things. Thank you very much for teaching me many skillsabout human and mouse lung and allowing me to join your experiments and articles. I wish you,Siegard and the beautiful triplets a wonderful life! Dear Viktoriia, more than a researcher, you alsoshowed amazing talent as a painter. As the senior in slicing lab, thank you for always helping meevery time I need it. Dear Mbak Amirah, thank you for all your help and chat we had in the lab. Good

Osteoprotegerin in Organ Fibrosis: Biomarker, Actor and Target of Therapy?
181
luck with your career in ITB, hopefully, we can meet more often. Dear Laura, Anienke and Gwenda,good luck with your PhDs! Dear Mas Habibie, we only met each other once, via Skype, yet youalready helped me with many things. Mas Adhyat and I were really grateful to have you. Thank youvery much, Mas. I wish you success with your PhD!This thesis is a result of collaboration with many researchers. Therefore, I would like toacknowledge all collaborators and co-authors for their valuable contributions. I am greatly thankfulto Dr. Robbert H. Cool for providing RANKL for our experiments; to Dr. Bernt van den Blink, Dr.Peter Heukels, Prof. Wim Timens, Dr. Corry-Anke Brandsma, Dr. George Nossent, Dr. KoertP. de Jong, Prof. Igle J. de Jong and Dr. Hendrik S. Hofker for cooperation in providing humantissue from the donors; to Prof. Detlef Schuppan for the support in providing genetically modifiedanimals; and Dr. David M. Brass for valuable input to the research.This thesis would never exist without the consent of the organ donors and the sacrifice ofthe mice. Therefore, I would like to express my deep thanks to all mice for their sacrifice, as well asto all donors for their willingness to donate their organs. Thank you very much for not taking yourorgans to heaven because we need them here.During the hectic time of my experiments, I found happiness during lunch and praying time.Thank you for all Indonesian friends in UMCG for all those happy moments we had together inUMCG: Mbak Navessa, Mbak Putri, Mas Zaenal, Pak Tjie, Mbak Niar, Mas Yayok, Mbak Lia,Mbak Astri, Mbak Ferro, Mas Aul, Mbak Neily, Mbak Doti, Mas Hegar, Mbak Icha, Mas Ury,Mbak Monik, Mbak Niken, Mbak Tia, Mbak Ira, Mbak Lusi, Mas Joko, Mas Akbar, Mas Ivan,Mas Pandji, Pak Tjie, Laras, Mbak Pretty, Bhimo, Ujang, Mas Khairul, Mas Riswandy, Aldi.Living far away from home sometimes is hard. However, living together with Mas Didikand his family made it bearable. Bapak Kos, thank you very much for allowing me to stay at yourhouse, helping me with the bike, reminding me to eat fruits, taking me cycling to Delfzijl, and forSaturday night with Capsa, Werewolf, Ticket-to-ride game, and Keramat. Good luck with yourcareer ahead! Ibu Kos, Mbak Rosel, thank you very much for taking care of me when I was ill, fordelicious and nutritious foods you prepared every day, for support during my deadlines and exams,for nasi kuning on my birthday, and for your daily love. I enjoyed every time washing the dishes(and gossiping in the morning) with you and playing with Najwa and Nafi (and Maryam). Let usmeet more often in Indonesia!Living in Planetenlaan 264 with mas Didik’s family open chance for me to meet manyamazing, fun and inspiring people. Their warmth never failed to warm my heart through thefreezing winter and not-so-sunny summer. Thus, I would like to thank them for all the joy andhappiness they have shared: Kak Fean, Mbak Nur, Didin-Anis, Mas Almas, Chacha, Kak Reren,Guntur, Shidiq, Hadar, Mas Zaenal-Mbak Ayu, Ali-Liany, Mbak Icha–Mas Krisna, Insan,Erlangga, Mas Anggar–Mbak Fanny, Mas Windi–Mbak Fitri, Mas Azka, Mas Uchon, MbakErlin, Mas Ilmi, Mas Udin, Mas Insanu, Mas Wahono, May, Dina, Aisyah, Nadia, and Shanti. Wealso thank to Mas Zaki for allowing us to stay at his house when we back to Groningen for defense.I also feel very thankful for having engaged with all Indonesian students in Groningen.Through weekly meetings of deGromiest to learn Al Qur’an, Idul Fitri, Idul Adha and many eventssuch as Indonesian Day&Dinner and GroensCup, they made me enjoy my life in Groningen. Thankyou very much for being my family in Groningen: Mas Kus-Mbak Fitri, Mbak Ira, Mas Asrofi, MasEdy, Mbak Inung, Mas Fikri, Mas Iging-Mbak Desti, Mbak Yosay-Mas Ali, Mbak Monik-MasFajar, Mas Rully-Mbak Intan, Mas Bino-Mbak Susan, Mbak Adel, Mbak Frita, Mbak Rasyida,Mbak Keisia, Mbak Sasha, Mas Muzakki, Mbak Hellen, Mbak Angela, Alfian, Mas Lutfi, MasOky, Mas Zaky-Mbak Sella, Mas Azis-Mbak Nna, Mbak Tiur, Mas Kadek-Mbak Laksmi, Mas

Acknowledgements
182
Ronny, Adit, Mas Doni-Mbak Citra, Mbak Pamela, Mbak Indri, Mbak Delphine, Mas Bintoro-Mbak Sofi, Mbak Fia, Mbak Morita, Mbak Ria-Martijn, Mas Amak, Mbak Vera, Bu Ima, and MasDimas. Moreover, we also acknowledge Mr. Tim Zwaagstra for his support to all PPIG’s events.For all seniors and families in Groningen: Budhe Ari-Oom Herman, Pak Asmoro-Bu Rini,Uwak Asiyah-Oom Meno, Oom Archie-Tante Mary, Budhe Nunung, Pak Tatang-Bu Rohmah,Pak Taufik-Bu Tina, Bu Elvira, thank you for being my parents during my stay in Groningen, andmaking my stay in Groningen feel like home. May God always bless you and your family!Starting a new life in Groningen surely would have been much harder for me without Radit,Mas Didik, and Mas Adhyat. Dear Mas Adhyatmika, I owe you many things which I cannot repay.You helped me a lot with many things, more than just lab things and thesis. You are like Viktor Krumfor Radit and me. We thank you very much for everything. Dear Nuri, even though I alwaysbothered your husband, you always welcomed me in your house with your warm smile and warmfoods. Thank you very much for all your kindness. May Allah always bless you, mas Adhyat, Zidni,Ufi (and probably more? Hehe).I would not have received the scholarship without a recommendation from my supervisorsand support from Dr. Joshita Djajadisastra and Prof. Yahdiana Harahap. Therefore, I would liketo extend my gratitude to Dr. Joshita and Prof. Yahdiana for supporting me to pursue my PhD. Iwould also like to thank all academic staff in Faculty of Pharmacy, Universitas Indonesia for theirsupport during finishing my thesis in Indonesia, especially for the heads of Faculty: Dr. Mahdi Jufri,Dr. Arry Yanuar, and Prof. Abdul Mun’im, as well as all members of Pharmaceutics andPharmaceutical Technology Group: Prof. Effionora Anwar, Dr. Iskandarsyah, Dr. Sutriyo, Dr.Silvia Surini, Ibu Juheini Amin, Ibu Rosmala Dewi, Delly Ramadon, Erny Sagita, AyunArifianti, Pak Imih, Indra dan Wisnu. I realize that this journey would have been more stressfulwithout mental support from the Dosmud; thus, thank you for every FUNtastic, inspiring, andinformative sharing all this time. I also would like to thank my colleagues in Sanofi Aventis (PakSumarno, Bu Rica, Mbak Ika, Mbak Nina, Kak Leni, Kak Resty, Mas Bambang, Kathie, Rina,Stefi) for their continuous support before and during my PhD journey. Furthermore, I and Raditwould like to acknowledge Mas Adi for such beautiful and philosophical cover design of our theses.I would never jump this high without having such a wonderful supporting system: mybeloved family. Therefore, I dedicate this thesis to them, for their unconditional love. Terima kasihsebesar-besarnya untuk Ibu, Bapak dan Etty untuk semua cinta dan doa untuk Nia. Nia nggakmungkin bisa sekolah setinggi ini tanpa doa dan pendidikan yang direncanakan dengan sangat baikoleh Ibu dan Bapak. Nia juga nggak mungkin bisa menyelesaikan tesis ini sambil bekerja di kampusdan mengasuh Salman tanpa dukungan Ibu, Bapak dan Etty. Semoga semua kebaikan Ibu, Bapak danEtty dibalas Allah dengan berlipat. Nia juga berterima kasih kepada Mama, Papa, dan Rara untuksemua dukungan dan doa untuk Adit dan Nia.Finally, much love and thanks go to my little family. Radit, Chagya, as represented in thedesign of our thesis cover; we are black-and-white, yin-and-yang, the role-model and the rule-breaker. Rather than in a romantic way, our relationship developed with daily arguments. We area contradiction in every way. We are different and we argue a lot, yet you are a complement for me.Thank you very much for being the best friend of my life and walking this journey together withme. May Allah bless our family forever. Salman sholeh, thank you for writing this thesis along withme, ever since you were in my womb. Thank you for being a strong boy and being our motivationthrough-out this journey. There will be a time when people make a comparison between you andyour parents. When that time comes, please remember that you should not follow this path, instead,follow your own dream. But if one day you dream of this path, you should know that even thoughthis is not an easy path to walk on, it is always worth to fight for.

Author affiliations
183
AUTHOR AFFILIATIONS
AdhyatmikaCatharina Reker-SmitMarina H. de JagerFransien van DijkLeonie BeljaarsKeri A MangnusDepartment of Pharmacokinetics, Toxicology and Targeting (PTT), Groningen ResearchInstitute for Pharmacy (GRIP), University of Groningen, Groningen, The NetherlandsBarbro N. MelgertCarian E. BoorsmaChristina DraijerDepartment of Pharmacokinetics, Toxicology and Targeting (PTT), Groningen ResearchInstitute for Pharmacy (GRIP), University of Groningen, Groningen, The NetherlandsGRIAC Research Institute, University Medical Center Groningen, University of Groningen,Groningen, The NetherlandsBernt van den BlinkPeter HeukelsErasmus MC, Department of Pulmonary Medicine, Rotterdam, The Netherlands.Corry-Anke BrandsmaDepartment of Pathology, University of Groningen, University Medical Center Groningen,Groningen, The Netherlands.GRIAC Research Institute, University Medical Center Groningen, University of Groningen,Groningen, The NetherlandsDavid M. BrassDepartment of Medicine, Pulmonary Division, Duke University Medical Center, Durham, NC,USA.Dorenda OosterhuisEmilia GoreEmilia BigaevaPeter OlingaS. SurigugaTheerut LuangmonkongWouter L.J. HinrichsDepartment of Pharmaceutical Technology and Biopharmacy (PTB), Groningen ResearchInstitute for Pharmacy (GRIP), University of Groningen, Groningen, The Netherlands

Author affiliations
184
Detlef SchuppanInstitute of Translational Immunology and Research Center for Immunotherapy, University ofMainz Medical Center, Mainz, Germany.Division of Gastroenterology, Beth Israel Deaconess Medical Center, Harvard Medical School,Boston, Massachusetts, US.George NossentDepartment of Pulmonary Diseases, University of Groningen, University Medical CenterGroningen, Groningen, The Netherlands.HabibieDepartment of Pharmacokinetics, Toxicology and Targeting (PTT), Groningen ResearchInstitute for Pharmacy (GRIP), University of Groningen, Groningen, The NetherlandsFaculty of Pharmacy, Universitas Hasanuddin, Makasar, IndonesiaHendrik S. HofkerDepartment of Surgery, University Medical Center Groningen, The NetherlandsKoert P. de JongDepartment of Hepato-Pancreato-Biliary Surgery and Liver Transplantation, UniversityMedical Center Groningen, The NetherlandsIgle J. de JongDepartment of Urology, University Medical Center Groningen, The NetherlandsKurnia S.S PutriDepartment of Pharmaceutical Technology and Biopharmacy (PTB), Groningen ResearchInstitute for Pharmacy (GRIP), University of Groningen, Groningen, The NetherlandsFaculty of Pharmacy, Universitas Indonesia, Depok, IndonesiaRobbert H CoolDepartment of Pharmaceutical Gene Modulation and Department of Pharmaceutical Biology,Groningen Research Institute for Pharmacy, University of Groningen, The Netherlands.Wim TimensDepartment of Pathology, University of Groningen, University Medical Center Groningen,Groningen, The Netherlands.




















