
Pathophysiology of Cardiorenal Syndrome Type 2 in Stable ...
Upload
khangminh22Category
view
1download
0
University of Groningen
Cardiorenal interaction in heart failureDamman, Kevin
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:2009
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Damman, K. (2009). Cardiorenal interaction in heart failure. s.n.
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license.More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne-amendment.
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 18-03-2022

Cardiorenal Interaction in Heart Failure
Kevin Damman

© copyright 2009 Kevin Damman
Financial support by the Netherlands Heart Foundation (NHS) and the Groningen Institute for Drug Exploration (GUIDE) for publication of this thesis is gratefully acknowledged.
Damman, KevinCardiorenal Interaction in Heart FailureProefschrift Groningen.
ISBN 978-90-367-3787-6ISBN Elektronische versie 978-90-367-3786-9Correspondence [email protected] & cover Arne Heijenga, Riverside, California (www.stukjewebgebeuren.nl)Printed by Gildeprint, EnschedeFonts Garamond Premier Pro (13pt) Helvetica Neue LT Std (10pt)
Text backcover: Reprinted with permission from Elsevier. Full citation: Moxon. Lancet. 1868;91:498-499.
All rights reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means, without permission of the author.

Cardiorenal Interaction in Heart Failure
Proefschrift
ter verkrijging van het doctoraat in deMedische Wetenschappen
aan de Rijksuniversiteit Groningenop gezag van de
Rector Magnificus, dr. F. Zwarts,in het openbaar te verdedigen op
woensdag 22 april 2009om 13.15 uur
door
Kevin Dammangeboren op 24 mei 1980
te Dalfsen

Promotores: Prof. dr. H.L. Hillege Prof. dr. D.J. van Veldhuisen Prof. dr. G. Navis
Copromotor: Dr. A.A. Voors
Beoordelingscomissie: Prof. dr. K. Amann Prof. dr. W.H. van Gilst Prof. dr. P.A. de Graeff

Paranimfen: B. Daan Westenbrink Bart van der Heij
Part of the research described in this thesis was supported by a grant of the Netherlands Heart Foundation (NHF-2006B157).
Additional financial support by the following sponsors for the publication of this thesis is gratefully acknowledged:
Amgen B.V., AstraZeneca B.V., Baxter Nederland B.V., Biotronik Nederland B.V., BMEYE B.V., Bristol-Myers Squibb B.V., Fresenius Medical Care Nederland B.V., Genzyme Nederland, Guide, Interuniversitair Cardiologisch Instituut Nederland, Medtronic Bakken Research Center, Medtronic Trading Nederland B.V., Menarini Farma Nederland, Merck Sharp & Dohme B.V., Novartis Pharma B.V., Pfizer B.V., Rijksuniversiteit Groningen, Roche Diagnostics Nederland B.V., Roche Nederland B.V., Sanofi-Aventis Nederland B.V., Schering-Plough B.V., Servier Nederland Farma B.V., Stichting Edu Cardio Groningen.


Table of Contents
Introduction 9
PART IPathophysiology of reduced glomerular filtration rate in chronic heart failure
Chapter 1 25Differential associations between renal function and “modifiable” risk factors in patients with chronic heart failureClin Res Cardiol. 2009;98:121-129
Chapter 2 43Decreased cardiac output, venous congestion and the association with renal impairment in patients with cardiac dysfunctionEur J Heart Fail, 2007; 9:872-78
Chapter 3 59Increased central venous pressure is associated with impaired renal function and mortality in a broad spectrum of patients with cardiovascular diseaseJ Am Coll Cardiol. 2009;53:582-588
Chapter 4 77Venous congestion in chronic systolic heart failure is related to renal dysfunction and increased mortalitySubmitted
PART IIWorsening renal function in patients with heart failure
Chapter 5 97Worsening renal function and prognosis in heart failure: Systematic review and meta-analysisJ Card Fail, 2007;13:599-608
Chapter 6 115Both in and outhospital worsening of renal function predict outcome in patients with heart failure Results from the Coordinating Study Evaluating Outcome of Advising and Counseling in Heart Failure (COACH)Submitted

PART IIIEmerging pathophysiological pathways of the cardiorenal connection in patients with heart failure
Chapter 7 135Renal function relates to outcome through different pathways of renal perfusion and filtration efficacy, hemodilution and volume overload in patients with chronic heart failureSubmitted
Chapter 8 153Urinary neutrophil gelatinase associated lipocalin (NGAL), a marker of tubular damage, is increased in patients with chronic heart failureEur J Heart Fail, 2008; 10:997-1000
Chapter 9 163Tubular damage is common and associated with reduced survival in patients with chronic systolic heart failureSubmitted
Summary 183
Discussion and Future Perspectives 189
Nederlandse Samenvatting 219
Dankwoord 229
Bibliography 237

Introduction

10

Introduction
11
Heart failure (HF) is a condition characterized by signs and symptoms mainly attributable to reduced forward flow and increased venous congestion. The estimated prevalence in Europe (> 900 million people) is at least 15 million patients according to recent European guidelines on HF [1]. Despite the initiation of new therapies, the prognosis of patients with HF is still incredibly poor: almost half of patients have a life expectancy of less than 4 years [1]. The reason for the persisting poor prognosis of patients with HF is not evident, but the changing characteristics of patients with HF may be an important aspect of the problem. In comparison to patients with HF who were diagnosed in the late 80’s, HF patients now tend to be older (Figure 1) and more often female [2].
Furthermore, with the aging HF population, co-morbidities or other organ dysfunction are much more frequent, which further complicates treatment and may reduce or obscure the effect of HF treatment on mortality and morbidity [3-8]. The life-expectancy of these co-morbidities itself could be a limiting factor, which may indicate the need for a shift in focus of targets of treatment in HF. In particular renal failure has received increasing attention in the last decade [7,9-13]. Interestingly, renal impairment is not only more frequently observed in patients with HF who are older and female [14], but the frequency of patients with chronic kidney disease as defined by different definitions in large HF trials has risen in the last two decades (Figure 2).
Figure 1. Relationship between year of publication of study and mean age of included patients. Shown are HF studies, including registries, clinical trials and observational cohort studies in the period 1985–2008. Included are acute as well as chronic heart failure studies, in combination with studies that included left ventricular dysfunction after myocardial infarction. Mean age is taken, irrespective of inclusion or exclusion criteria. Size of circles is relative to the study size. The solid line represents the fitted regression line, weighted for study size. Dashed lines represent 95% confidence intervals [2].
Publication date trial
1985 1990 1995 2000 2005 2010
Mea
n ag
e of
tria
l pat
ient
s
45
50
55
60
65
70
75
80
85

12
Cardiorenal interactionThe kidney is the main organ responsible for water and salt homeostasis, blood pressure
control and secretion of important hormones for hemodynamic stability. In normal physiologic circumstances, the kidney receives approximately 20-25% of the total cardiac output [15]. It therefore is the organ that receives the highest blood flow per gram of body weight in the human body. The main function of the kidney is usually measured by the glomerular filtration rate (GFR). Normally, GFR is maintained constant by renal autoregulatory mechanisms, which are capable of maintaining renal blood flow (RBF) by changing vasomotor tone in the efferent and afferent renal arteriole, despite changes in systemic blood pressure. In cardiovascular disease however, kidney function or GFR is often compromised [16]. In general, renal impairment is referred to as any decrease in GFR below normal, but the most used definition is CKD, with (estimated) GFR below 60 mL/min/1.73m. The National Kidney Foundation Kidney Disease Outcomes Quality Initiative (KDOQI) further classifies different stages from ≥ 90, 60-89, 30-59, 15-29 and < 15 (or dialysis) mL/min/1.73m. Finally, also the method used to determine GFR may influence the definition of renal impairment, as different formulas estimating GFR may be biased and imprecise in specific conditions, including HF [17]. Main reasons for renal impairment in cardiovascular disease include atherosclerosis, hypertension, endothelial dysfunction and inflammation, but many others have been identified or suggested [18-22]. Although the pathogenesis of reduced GFR in individual patients may differ, the result is the same: reduced GFR is strongly related to increased mortality and morbidity [7,16]. In addition, (micro) albuminuria, as a marker of glomerular damage is frequently observed in patients with cardiovascular disease, and further adds to the impaired prognosis in these patients [23-25].
Figure 2. Relationship between year of conduction of study and percentage of patients with chronic kidney disease. Shown are some key HF studies. The solid line represents the fitted regression line. Image obtained from data from original reports of studies mentioned.
Conduction years of study
1985 1990 1995 2000 2005 2010
Per
cent
age
of p
atie
nts
with
CK
D a
t b
asel
ine
10
20
30
40
50
60
COACHMERIT
DIG
SOLVDTreatment
SOLVDPrevention
CHARM
SAVECIBIS II
ELITE
CORONA
CARE-HF

Introduction
13
Renal impairment in heart failureIn the spectrum of cardiovascular disease, HF is a particularly important disease with respect
to renal function and renal function impairment. Renal impairment is frequently observed in patients with HF, and has consistently been shown to increase the risk for all-cause mortality and HF rehospitalizations [7,11,12,16]. The presence of CKD (as defined as GFR below 60 mL/min/1.73m2) relates to a strongly increased mortality (Figure 3), while the extent of renal impairment as estimated by serum creatinine is also associated with the severity of impaired prognosis (Figure 4).
Importantly, not only renal function in patients with chronic HF (CHF) is an important mediator of outcome, but also inhospital renal impairment in patients with acute HF (AHF) plays an important role [26]. New therapies are emerging specifically targeted at prevention of worsening of renal function, or even improvement of GFR to improve subsequent prognosis [27]. The striking morbidity associated with a combination of renal impairment and HF may mutually influence the disease progression of both diseases.
Figure 3. Forrest plot of relationship between chronic kidney disease and mortality in HF. Shown is risk for all-cause mortality of HF patients with CKD versus without CKD. Odds ratio’s were estimated using event rates presented in individual studies. Overall odds ratio was estimated using random effects meta-analysis.

14
Pathophysiology of renal failure in heart failureRenal impairment as co-existing in and as a consequence of HF has been recognized as
early as 1868 [28]. Historically, hemodynamic alterations in HF have been considered the cornerstone of the pathophysiology of renal impairment [15,29,30]. Indeed, RBF may decrease disproportionate in comparison to reduction in cardiac output [30]. Early studies conducted in the first half of the 20th century have investigated the mechanisms responsible for reduction in GFR and/or preservation of GFR, which resulted in the discovery of the renal autoregulatory mechanisms [31]. Only after the angiotensin II mediated efferent vasoconstriction was discovered, angiotensin converting enzyme inhibitors (ACEi) were developed to specifically target the autoregulatory response [15,32-37]. In studies in patients with CHF without ACEi, a reduction in RBF has been established as the main determinant of a reduction in GFR [29,30]. Interestingly, when cardiac index and subsequently RBF decreased, GFR was preserved to some extent by increasing the filtration fraction. However, eventually GFR decreases when the renal autoregulatory mechanisms are unable to further increase filtration fraction. Whether the introduction of ACEi has had a mediating effect on the relationship between RBF and GFR is one of the focuses of the present thesis as discussed in chapter 1.
HF is not only characterized by a decreased cardiac output and subsequent decreased organ perfusion, but also by increased venous congestion. This was already recognized 100 years
80 100 120 140 160 180
Ann
ual m
orta
lity
0
10
20
30
40
50
60
Baseline serum creatinine (mg/dL)
1,0 1,2 1,4 1,6 1,8 2,0
Baseline serum creatinine (µmol/L)
Figure 4. Relationship between serum creatinine and annual mortality in published HF studies. Shown are HF studies, including registries, clinical trials and observational cohort studies in the period 1985–2008. Included are acute as well as chronic heart failure studies, in combination with studies that included left ventricular dysfunction after myocardial infarction. Size of circles is relative to the study size. The solid line represents the fitted regression line, weighted for study size. Dashed lines represent 95% confidence intervals

Introduction
15
ago, when experimental work in HF animal models was conducted to evaluate the precise pathophysiologic link between HF and renal failure [38-40]. Different models were used to establish renal failure in these models, including artificially increasing renal venous pressure [34,39-41]. In these studies, especially the effect of increasing renal venous pressure (or central venous pressure (CVP)) on reducing renal perfusion pressure was observed. However, also more structural abnormalities with increasing CVP were studied, which may indicate more direct effects on renal function by increased CVP [39,40,42]. In addition, observations made in the abdominal compartment syndrome have more recently re-introduced the concept of an effect of increased CVP on renal function [43,44]. In HF however, little is known about the relationship between CVP and renal function. Therefore, we evaluated the link between CVP and RBF with GFR in patients with cardiac dysfunction in chapter 2, and in a more general cardiovascular population in chapter 3. Measuring CVP invasively in every patient with HF is now considered obsolete, even in patients with AHF [45]. Therefore, we investigated the relationship between non-invasively determined symptoms and signs of venous congestion, renal function and outcome in a large cohort of patients with CHF in chapter 4.
Changes in renal function in patients with heart failureAlthough renal impairment at any point in time has been shown to be related to poor
prognosis in patients with HF, it may be much more important to know the progression of cardiorenal disease. At the present time, little is known about the progression of renal failure in patients with HF, but it has been suggested that the (downward) slope of renal function over time may be similar to patients with CKD. This would indicate a much steeper decline in GFR as compared to the normal population, in which GFR decreases at around 0.5 -1.0 mL/min/1.73m2 every year [46,47]. Considering the already depressed baseline renal function at which patients with HF start of with, this further emphasizes the need for close control of renal function in these patients. In addition, recent evidence is accumulating that not only a great proportion of patients with HF experience an exceptional fast decline in renal function, but also that this occurrence of worsening renal function (WRF) is associated with an unfavorable outcome [48,49]. To further address this issue, we have pooled several of the studies examining WRF and outcome in patients with HF in chapter 5. Renal function is a dynamic process, and GFR may fluctuate over time. Therefore we assessed the relationship between the occurrence of WRF at different points in time and the slope of renal function in patients with HF in chapter 6.
Emerging pathophysiological pathways of the cardiorenal connection in pa-tients with heart failure
While decreased RBF may be the crucial step in the pathophysiology of decreased GFR in HF, the actual mechanisms leading to a reduction in RBF may be more than only a reduction in cardiac output. General endothelial dysfunction may have profound effects on renal perfusion [50]. Renal artery stenosis, especially in patients with atherosclerotic disease, may be found in almost 20% of patients with CHF [51]. A key component is the activation of the renin angiotensin system, together with an increase in sympathetic nervous system activation

16
[50]. Adenosine mediated vasoconstriction may be an important mechanism responsible for reduced RBF, and now emerges as a possible target for therapy in especially AHF [27]. The interrelationship of these different domains and the effect on renal function and outcome is outlined in chapter 7.
Finally, decreasing RBF may not only cause a decrease in GFR, but may also trigger more general renal hypoxia [52]. This may give rise to problems, especially in regions of the kidney that consume the largest amount of oxygen, which are the proximal tubules. In CKD, the final common pathway of renal disease is considered to be renal hypoxia [52,53]. This in turn will lead to increased interstitial fibrosis and glomerular sclerosis. Urinary tubular marker protein concentrations, measured in acute and CKD, have shown a strong increase in these markers of tubular dysfunction in response to (acute) renal dysfunction [54-56]. Additionally, their urinary concentrations correlated with the extent of tubulointerstitial injury, prognosis and response to treatment [54,57]. In CHF however, very little is known about tubulointerstitial damage, or tubular dysfunction. Therefore, we investigated the prevalence of tubular damage in CHF patients in chapter 8, and extended our investigation to include the relationship with prognosis in chapter 9.

Introduction
17
Aims of the thesisIn the last decade, renal impairment in HF has emerged as an important marker for
prognosis, disease progression, pathophysiology of HF and as target for therapy. In this thesis, the underlying pathophysiology of renal impairment in HF, its decline over time, the relationship with prognosis, and the importance of tubular damage and new therapeutic targets, were studied.
In PART I, the pathophysiology of renal impairment in HF was investigated. Chapter 1 focuses on different associations between established risk markers of renal impairment and GFR as well as RBF in patients with HF who underwent invasive determination of renal function and perfusion. In chapter 2, we addressed the effect of reduced RBF and increased CVP on the relationship with GFR in patients with cardiac dysfunction, secondary to pulmonary hypertension. We further explored the relationship between CVP and GFR in a more general cardiovascular population in chapter 3, while signs of venous congestion, and the relationship with prognosis and renal function in patients with HF were the primary focus in chapter 4.
In PART II, we investigated the decline of renal function and WRF, and the relationship with prognosis in patients with HF. Chapter 5 describes a pooled analysis of eight studies investigating the relationship between the occurrence of WRF at a given time point and prognosis in patients with both chronic and acute HF. To further investigate the effect of WRF at different points during and after HF hospitalization, and to investigate the slope of renal function over time, we studied these associations in a substudy of the COACH in chapter 6.
PART III focuses on tubular damage and possible new targets for therapy in HF patients with renal impairment. In chapter 7 different pathways by which renal failure may initiate a worse prognosis are investigated. In the last two chapters, a new entity in patients with HF is studied, which consists of the occurrence of tubular damage. Chapter 8 studied the prevalence of tubular dysfunction, while in chapter 9 the relationship with prognosis was investigated. Finally, in the discussion and future directions the results of the present thesis are discussed and put into clinical context. Future perspectives, especially with regards to therapy in patients with renal impairment and HF are discussed.

18
ReferencesDickstein K, Cohen-Solal A, Filippatos G et al. ESC Guidelines for the diagnosis and treatment of 1. acute and chronic heart failure 2008: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM). Eur Heart J. 2008;29:2388-2442.
Damman K, de Boer RA, and van Veldhuisen DJ. Heart failure, aging and beta-blockers: the need for 2. more data on tolerability and efficacy. Clin Res Cardiol. 2008;97:575-577.
Groenveld HF, Januzzi JL, Damman K et al. Anemia and mortality in heart failure patients a systematic 3. review and meta-analysis. J Am Coll Cardiol. 2008;52:818-827.
Lang CC and Mancini DM. Non-cardiac comorbidities in chronic heart failure. Heart. 2007;93:665-4. 671.
Le Jemtel TH, Padeletti M, and Jelic S. Diagnostic and therapeutic challenges in patients with coexistent 5. chronic obstructive pulmonary disease and chronic heart failure. J Am Coll Cardiol. 2007;49:171-180.
MacDonald MR, Petrie MC, Hawkins NM et al. Diabetes, left ventricular systolic dysfunction, and 6. chronic heart failure. Eur Heart J. 2008;29:1224-1240.
Smith GL, Lichtman JH, Bracken MB et al. Renal impairment and outcomes in heart failure: systematic 7. review and meta-analysis. J Am Coll Cardiol. 2006;47:1987-1996.
Sturm HB, Haaijer-Ruskamp FM, Veeger NJ et al. The relevance of comorbidities for heart failure 8. treatment in primary care: A European survey. Eur J Heart Fail. 2006;8:31-37.
Al Ahmad A, Rand WM, Manjunath G et al. Reduced kidney function and anemia as risk factors for 9. mortality in patients with left ventricular dysfunction. J Am Coll Cardiol. 2001;38:955-962.
Dries DL, Exner DV, Domanski MJ, Greenberg B, and Stevenson LW. The prognostic implications of 10. renal insufficiency in asymptomatic and symptomatic patients with left ventricular systolic dysfunction. J Am Coll Cardiol. 2000;35:681-689.
Hillege HL, Girbes AR, de Kam PJ et al. Renal function, neurohormonal activation, and survival in 11. patients with chronic heart failure. Circulation. 2000;102:203-210.
Hillege HL, Nitsch D, Pfeffer MA et al. Renal function as a predictor of outcome in a broad spectrum 12. of patients with heart failure. Circulation. 2006;113:671-678.
Smilde TD, Hillege HL, Voors AA, Dunselman PH, and van Veldhuisen DJ. Prognostic importance of 13. renal function in patients with early heart failure and mild left ventricular dysfunction. Am J Cardiol. 2004;94:240-243.
Owan TE, Hodge DO, Herges RM et al. Secular trends in renal dysfunction and outcomes in hospitalized 14. heart failure patients. J Card Fail. 2006;12:257-262.
Leithe ME, Margorien RD, Hermiller JB, Unverferth DV, and Leier CV. Relationship Between Central 15. Hemodynamics and Regional Blood-Flow in Normal Subjects and in Patients with Congestive Heart-Failure. Circulation. 1984;69:57-64.
Tonelli M, Wiebe N, Culleton B et al. Chronic kidney disease and mortality risk: a systematic review. J 16. Am Soc Nephrol. 2006;17:2034-2047.

Introduction
19
Smilde TD, van Veldhuisen DJ, Navis G, Voors AA, and Hillege HL. Drawbacks and prognostic value 17. of formulas estimating renal function in patients with chronic heart failure and systolic dysfunction. Circulation. 2006;114:1572-1580.
Go AS, Chertow GM, Fan D, McCulloch CE, and Hsu CY. Chronic kidney disease and the risks of 18. death, cardiovascular events, and hospitalization. N Engl J Med. 2004;351:1296-1305.
London GM, Guerin AP, Marchais SJ et al. Arterial media calcification in end-stage renal disease: 19. impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant. 2003;18:1731-1740.
Muntner P, Hamm LL, Kusek JW et al. The prevalence of nontraditional risk factors for coronary heart 20. disease in patients with chronic kidney disease. Ann Intern Med. 2004;140:9-17.
Raggi P, Boulay A, Chasan-Taber S et al. Cardiac calcification in adult hemodialysis patients. A link 21. between end-stage renal disease and cardiovascular disease? J Am Coll Cardiol. 2002;39:695-701.
Shlipak MG, Fried LF, Crump C et al. Elevations of inflammatory and procoagulant biomarkers in 22. elderly persons with renal insufficiency. Circulation. 2003;107:87-92.
Hillege HL, Fidler V, Diercks GF et al. Urinary albumin excretion predicts cardiovascular and 23. noncardiovascular mortality in general population. Circulation. 2002;106:1777-1782.
Solomon SD, Lin J, Solomon CG et al. Influence of albuminuria on cardiovascular risk in patients with 24. stable coronary artery disease. Circulation. 2007;116:2687-2693.
Wachtell K, Ibsen H, Olsen MH et al. Albuminuria and cardiovascular risk in hypertensive patients 25. with left ventricular hypertrophy: the LIFE study. Ann Intern Med. 2003;139:901-906.
Heywood JT, Fonarow GC, Costanzo MR et al. High prevalence of renal dysfunction and its impact 26. on outcome in 118,465 patients hospitalized with acute decompensated heart failure: a report from the ADHERE database. J Card Fail. 2007;13:422-430.
Cotter G, Dittrich HC, Weatherley BD et al. The PROTECT pilot study: a randomized, placebo-27. controlled, dose-finding study of the adenosine A1 receptor antagonist rolofylline in patients with acute heart failure and renal impairment. J Card Fail. 2008;14:631-640.
Moxon. Guy.s Hospital. (Department of Morbid Anatomy.) : Notes of autopsies of - I. Hemiplegia from 28. embolus; renal disease. II. Pulmonary apoplexy from embolus; phtisis. III. Hypertrophy and dilatation of the heart; thickening of middlesized systemic arteries. Lancet. 1868;91:498-499.
Cody RJ, Ljungman S, Covit AB et al. Regulation of glomerular filtration rate in chronic congestive 29. heart failure patients. Kidney Int. 1988;34:361-367.
Ljungman S, Laragh JH, and Cody RJ. Role of the Kidney in Congestive Heart-Failure - Relationship 30. of Cardiac Index to Kidney-Function. Drugs. 1990;39:10-21.
Selkurt EE, Hall PW, and Spencer MP. Influence of graded arterial pressure decrement on renal clearance 31. of creatinine, p-aminohippurate and sodium. Am J Physiol. 1949;159:369-378.
Creager MA, Halperin JL, Bernard DB et al. Acute regional circulatory and renal hemodynamic effects 32. of converting-enzyme inhibition in patients with congestive heart failure. Circulation. 1981;64:483-489.

20
Levine TB, Olivari MT, Garberg V, Sharkey SW, and Cohn JN. Hemodynamic and clinical response 33. to enalapril, a long-acting converting-enzyme inhibitor, in patients with congestive heart failure. Circulation. 1984;69:548-553.
Maxwell MH, Breed ES, and Schwartz IL. Renal venous pressure in chronic congestive heart failure. J 34. Clin Invest. 1950;29:342-348.
Werko L, Varnauskas E, Ek J et al. Studies on the renal circulation and renal function in mitral valvular 35. disease. II. Effect of apresoline. Circulation. 1954;9:700-705.
Werko L, Varnauskas E, Eliasch H et al. Studies on the renal circulation and renal function in mitral 36. valvular disease. I. Effect of exercise. Circulation. 1954;9:687-699.
Werko L, Ek J, Varnauskas E et al. The relationship between renal blood flow, glomerular filtration rate 37. and sodium excretion, cardiac output and pulmonary and systemic blood pressures in various heart disorders. Am Heart J. 1955;49:823-837.
Kerr WJ. Heart failure: Its underlying causes, clinical manifestations and treatment. California State 38. Journal of Medicine. 1923;21:417-420.
Robinson G. Researches into the connection existing between an unnatural degree of compression of 39. the blood contained in the renal vessels, and the presence of certain abnormal matters in the urine. Med Chir Soc Tr. 1843;26:51-79.
Rowntree LG, Fitz R, and Geraghty JT. The effect of experimental chronic passive congestion on renal 40. function. Arch Int Med. 1913;11:121-147.
Blake WD, Wegria R, Keating RP, and Ward HP. Effect of increased renal venous pressure on renal 41. function. Am J Physiol. 1949;157:1-13.
Wegria R, Capeci NE, Blumenthal MR et al. The pathogenesis of proteinuria in the acutely congested 42. kidney. J Clin Invest. 1955;34:737-743.
Doty JM, Saggi BH, Sugerman HJ et al. Effect of increased renal venous pressure on renal function. J 43. Trauma. 1999;47:1000-1003.
Doty JM, Saggi BH, Blocher CR et al. Effects of increased renal parenchymal pressure on renal function. 44. J Trauma. 2000;48:874-877.
Binanay C, Califf RM, Hasselblad V et al. Evaluation study of congestive heart failure and pulmonary 45. artery catheterization effectiveness: the ESCAPE trial. JAMA. 2005;294:1625-1633.
Hoang K, Tan JC, Derby G et al. Determinants of glomerular hypofiltration in aging humans. Kidney 46. Int. 2003;64:1417-1424.
Lindeman RD, Tobin J, and Shock NW. Longitudinal studies on the rate of decline in renal function 47. with age. J Am Geriatr Soc. 1985;33:278-285.
Khan NA, Ma I, Thompson CR et al. Kidney function and mortality among patients with left ventricular 48. systolic dysfunction. J Am Soc Nephrol. 2006;17:244-253.
Metra M, Nodari S, Parrinello G et al. Worsening renal function in patients hospitalised for acute heart 49. failure: clinical implications and prognostic significance. Eur J Heart Fail. 2008;10:188-195.
Bongartz LG, Cramer MJ, Doevendans PA, Joles JA, and Braam B. The severe cardiorenal syndrome: 50. Guyton revisited. Eur Heart J. 2005;26:11-17.

Introduction
21
de Silva R, Loh H, Rigby AS et al. Epidemiology, associated factors, and prognostic outcomes of renal 51. artery stenosis in chronic heart failure assessed by magnetic resonance angiography. Am J Cardiol. 2007;100:273-279.
Norman JT and Fine LG. Intrarenal oxygenation in chronic renal failure. Clin Exp Pharmacol Physiol. 52. 2006;33:989-996.
Manotham K, Tanaka T, Matsumoto M et al. Evidence of tubular hypoxia in the early phase in the 53. remnant kidney model. J Am Soc Nephrol. 2004;15:1277-1288.
Ding H, He Y, Li K et al. Urinary neutrophil gelatinase-associated lipocalin (NGAL) is an early 54. biomarker for renal tubulointerstitial injury in IgA nephropathy. Clin Immunol. 2007;123:227-234.
Mishra J, Dent C, Tarabishi R et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker 55. for acute renal injury after cardiac surgery. Lancet. 2005;365:1231-1238.
Mori K, Lee HT, Rapoport D et al. Endocytic delivery of lipocalin-siderophore-iron complex rescues the 56. kidney from ischemia-reperfusion injury. J Clin Invest. 2005;115:610-621.
Machiguchi T, Yoshida H, Yonemoto S et al. Does circulating erythropoietin reflect progression of IgA 57. nephropathy? Comparison with urinary N-acetyl-beta-D-glucosaminidase. Nephrol Dial Transplant. 1999;14:635-640.

22

23
PART IPathophysiology of reduced glomerular filtration rate
in chronic heart failure

24

Chapter 1Differential associations between renal function and “modifiable” risk
factors in patients with chronic heart failure
Tom D.J. Smilde, Kevin Damman, Pim van der Harst, Gerjan Navis, B. Daan Westenbrink, Adriaan A. Voors, Frans Boomsma,
Dirk J. van Veldhuisen, Hans L. Hillege
Clin Res Cardiol. 2009;98:121-129

26
Abstract
Background.Reduced glomerular filtration rate (GFR) is strongly associated with reduced survival in patients
with chronic heart failure (CHF). Our aim was to determine different pathophysiologic markers that are associated with reduced renal function in CHF.
Methods and Results.We studied 86 patients with CHF (58±12 years, 78% male). GFR and renal blood flow (RBF) were
determined by 125I-Iothalamate and 131I-Hippuran clearances. Filtration fraction (FF) was calculated. We determined haemoglobin levels, endothelial function, inflammatory status, plasma renin activity (PRA) and N-terminal pro brain natriuretic peptide (NT-proBNP). Urinary albumin excretion (UAE) was measured in 24 hours urine. Mean GFR was 74±28 ml/min/1.73m2. GFR was strongly related to RBF (r = 0.915, P < 0.001), FF (r = 0.546, P < 0.001), but only weakly to endothelial function and PRA. In multivariate analysis, RBF (r = 0.938, P < 0.001), FF (r = 0.786, P < 0.001) and hemoglobin levels (r = -0.520, P < 0.001) were independently associated with GFR. UAE was mainly dependent on RBF (r = -0.401, P < 0.001) and increased exponentially with decreasing RBF. RBF was mainly associated with NT-proBNP (r = -0.561, P < 0.001) and PRA (r = -0.422, P < 0.001).
Conclusion.Reduced GFR is mainly dependent of decreased RBF in patients with CHF. Endothelial function
and neurohormonal activation showed only mild associations with GFR. NT-proBNP showed a strong relationship with RBF, and may be used as a marker of reduced renal perfusion.

Determinants of renal function in CHF
27
IntroductionRenal dysfunction has consistently been found to be a strong and independent prognostic
factor in patients with chronic heart failure (CHF) [1-4]. Almost two decades ago, Ljungman et al showed that renal blood flow (RBF), due to a decreased cardiac output, is the most important determinant of renal function as estimated by reduced glomerular filtration rate (GFR) in CHF patients not on angiotensin converting enzyme (ACE) inhibitors [5]. Generally, in these CHF patients GFR was maintained in spite of reduced RBF by predominant post-glomerular vasoconstriction, as apparent from an elevated filtration fraction (FF).
In the last two decades, drug treatment for CHF has changed with the introduction of ACE-inhibitor, angiotensin II receptor blocker (ARB), and beta-blocker therapy [6,7]. Considering the effects of ACE-inhibition and ARB therapy on renal afferent and particularly efferent vasomotor tone, it is questionable whether the findings of Ljungman on glomerular hemodynamics still apply to the current CHF population, where all patients are on RAAS-blockade. Evaluation of emerging risk factors show that CHF is not only characterised by impaired hemodynamics, but also endothelial activation [8], atherosclerosis and inflammation [9], neurohormonal activation [10], sympathetic nervous system activation (SNS) [10,11]. Furthermore, we recently showed that not only reduced renal perfusion but also venous congestion is an important determinant of renal impairment [12,13].
The aim of the current study was therefore, first, to determine the relationship of GFR with RBF and FF in chronic heart failure patients on current standard therapy, including ACE-inhibitor and/or ARB therapy. Second, we investigate the relative contribution of a number of domain specific biomarkers on indices of renal impairment, to determine easy obtainable markers of renal impairment and potential targets for risk profiling and therapy.
Methods
Patient populationOutpatient CHF patients, aged ≥ 18 years and clinically stable, were asked to participate in
this study. Patients were recruited from the outpatient CHF-clinic of the University Medical Center Groningen, The Netherlands. CHF was defined as a left ventricular ejection fraction (LVEF) < 45%. Patients were required to be on ACE-inhibitor and/or ARB therapy, and all medication had to be stable for at least one month. Administration of medication was not allowed during renal function measurement. Exclusion criteria included stroke or myocardial infarction within the last three months, cardiac surgery or angioplasty within the last 3 months or scheduled to undergo these procedures, unstable angina pectoris, primary renal disease, patients with prior organ transplant, or chronic use of renal function compromising medication. The study protocol was approved by the institutional ethics committee. All patients gave written informed consent. In this study 110 patients were included. Twenty four patients

28
Chapter 1
were excluded for this analysis, because of missing hematocrit or urinalysis data. In total 86 patients were eligible for the current analysis.
Study designOn the first day, GFR and RBF were measured by the clearances of Iothalamate and
Hippuran. Body weight and length were determined just before renal function measurement started. In addition, during renal measurements, blood pressure and heart rate were determined. Systolic and diastolic blood pressure measurements were calculated as the mean of the last two out of ten consecutive measurements during ten minutes in sitting position with an automatic Dinamap XL Model 9300 series device (Johnson-Johnson Medical INC, Tampa, Florida). MAP was calculated as ⅓ • systolic pressure + ⅔ • diastolic pressure.
Renal function measurement by iothalamate clearanceGFR and effective renal plasma flow (ERPF) were measured by constant infusion of
radiolabelled tracers, 125I-Iothalamate and 131I-Hippuran as described before [14]. The body surface area (BSA) was calculated as 0.007184·weight0.425 ·length0.725, and GFR and ERPF were expressed per 1.73 m2 of BSA. Renal blood flow (RBF) was calculated as ERPF/1-haematocrit. The filtration fraction (FF) was calculated as the ratio of GFR and ERPF and expressed as percentage. GFR and RBF were expressed per 1.73 m2 of BSA. Renal vascular resistance (RVR) was calculated as (MAP/ERPF) x (1-hematocrit) and expressed in mmHg/mL/min.
Cardio-renal (hemodynamic) parametersRBF, FF, LVEF, MAP and N terminal pro brain natriuretic peptide (NT-proBNP) were
used as markers for the cardio-renal hemodynamic status of the patients. LVEF was determined by nuclear ventriculography or echocardiography using Simpsons rule. NT-proBNP was measured by electrochemiluminescence immunoassay on the Roche Elecsys (Roche diagnostics, Netherlands). Peak oxygen consumption (peakVO2 (ml/min/kg) were extracted from medical records if available .
Renin angiotensin system parametersPlasma renin activity (PRA) and Angiotensin II (Ang II) were used as markers for renin
angiotensin system (RAS) activity. PRA was measured by an immunoradiometric assay (Nichols Institute Diagnostics, Middlesex, United Kingdom). Ang II was measured by specific radioimmunoassays after SepPak extraction of plasma. Analyses were performed in a routine setting according to the guidelines of the manufacturer.
Endothelial function parameters and inflammationVon Willebrand factor (vWf), plasma nitrite/nitrate (NOx) and asymmetric di-methyl
arganine (ADMA), soluble vascular adhesion molecule 1 (sVCAM-1), and soluble E-selectin (sES) were used as markers for endothelial damage and activation. vWF was determined using a validated in-house ELISA, as described previously. NOx was determined in plasma after

Determinants of renal function in CHF
29
ultrafiltration through a 10 kDa molecular weight cut-off filter (Millipore BV). A colorimetric assay was used according to the instructions of the manufacturer (Cayman Chemical Company, Ann Abor, MI). ADMA was determined in plasma using a commercially available ELISA kit (DLD diagnostika GmbH, Hamburg, Germany) according to the instructions as supplied by the manufacturer. Serum levels of sVCAM-1 and sES were determined by commercially available ELISA kits (R&D Systems, Abingdon, UK and Bender Med Systems, Vienna, Austria, respectively) according to the manufacturer’s instructions.
High sensitive CRP was (hs-CRP) determined by nephelometry with a threshold of 0.156 mg/L and intra- and inter-assay coefficients of less than 4.4% and 5.7%, respectively (BNII N, Dade Behring, Marburg, Germany). CRP levels below the detection level were scored as 0.156 mg/L.
UrinalysisUrinary albumin concentrations were determined by nephelometry (Dade Behring
Diagnostics, Marburg, Germany). Serum and urine creatinine was determined by Kodak Ektachem dry chemistry (Eastman Kodak, Rochester, NY, U.S.A.). Urinary albumin excretion was determined as the mean of two 24-h urine collections.
The investigation conforms to the principles outlined in the Declaration of Helsinki.
Statistical analysesData are given as mean ± standard deviation when normally distributed, as median and
interquartile range when skewed distributed, and as frequencies and percentages for categorical variables. Correlations between GFR, RBF, FF and various variables were performed using partial correlation coefficients, adjusted for age and gender. In multivariate analysis, beta coefficients are shown. Non-normally distributed continuous variables were log-transformed. Multivariate stepwise linear regression analysis, including all univariate associated variables (P < 0.10), was used to investigate independent contributions of different variables. In our primary analysis, we investigated the relationships between FF, RBF and GFR. Furthermore, we assessed the relationship between other pathophysiologic variables and GFR. Fractional polynomial modelling was used to visualize the relationship between RBF, RVR, FF, and GFR. In secondary analysis, we investigated the pathophysiologic factors related to the closest determinants of GFR, namely RBF and FF. Finally, again fractional polynomial modelling was used to investigate the possibility of a curvilinear effect between NT-proBNP and RBF. A P value < 0.05 was considered statistically significant. Statistical analyses were performed using SPSS, Chicago version 12.0 and STATA, College Station, Texas, version 9.0.

30
Chapter 1
Table 1. Baseline characteristics for total study population
Variables Total Population (n=86)
Age (years) 58 ± 12
Sex (n, % male) 67 (78)
NYHA class I/II/III/IV (%) 16 / 41 / 31 / 12
Diabetes (n, %) 7 (8)
Current smoking (n, %) 14 (18)
Ischemic etiology (n, %) 43 (50)
Cardio-renal hemodynamic parameters
Serum creatinine (µmol/L) 104 (92 – 121)
Serum BUN (mmol/L) 7.1 (6.0 – 9.8)
GFR (mL/min/1.73m2) 74 ± 28
RBF (mL/min/1.73m2) 465 ± 161
FF (%) 27 ± 5
RVR (mmHg/mL/min) 0.18 (0.15 – 0.23)
LVEF (%) 27 ± 9
MAP (mmHg) 85 ± 14
UAE (mg/day)* 8.7 (4.5 - 21.4)
NT-proBNP (pg/ml)* 786 (303 - 1930)
Hemoglobin (mmol/l) 8.7 ± 0.86
Peak VO2 (L/min/kg)# 18.4 ± 6.3
Renin Angiotensin System parameters
PRA (ng/mL/h)* 24.3 (5.5 - 60.3)
Ang II (pmol/l)* 9.8 (4.2 - 14.0)
Endothelial function parameters
ADMA (μmol/l) 0.68 ± 0.19
vWf (%)* 78 (48 - 174)
sVCAM-1 (ng/ml)* 348 (273 - 374)
Plasma NOx (μmol/l)* 29 (18 – 39)
sES (ng/ml)* 62 (47 - 88)
Inflammation parameter
hs-CRP (mg/l) * 2.5 (1.2-4.3)
Therapy
ACEi/ARB use (n (%)) 86 (100)
Beta-blocker use (n (%)) 74 (86)
Diuretic use (n (%)) 56 (65)
Aldosterone Antagonist use (n (%)) 33 (38)
Statin use (n (%)) 45 (52)
All continuous variables are presented with mean ± SD. If * is present median value with (25th – 75th percentile) are presented. # N = 48. Abbreviations: BUN: Blood urea nitrogen, NYHA; new york heart association functional class, GFR; glomerular filtration rate, RBF; renal blood flow, FF; filtration fraction, LVEF; left ventricular ejec-tion fraction, MAP; mean arterial pressure, NT-proBNP; N terminal pro brain natriuretic peptide, PRA; plasma renin activity, Ang II; angiotensin II, UAE; urinary excretion rate, ADMA; asymmetric dimethyl arganine, vWF; von Willebrand factor, sVCAM-1; soluble vascular adhesion molecule-1, NOx; nitrate/nitrate, sES; soluble E-selectine, CRP; C-reactive protein, ACEi; Angiotensin converting enzyme inhibitors, ARB; Angiotensin II recep-tor blockers.

Determinants of renal function in CHF
31
ResultsThe baseline characteristics of the 86 patients are presented in Table 1. The studied
population consisted of predominantly male patients (76%) with a mean age of 58 ± 12 years. The severity of CHF ranged from NYHA I to IV with an average of 2.3 ± 0.8. All patients received RAS-inhibition (85% ACE-inhibitor). The majority was treated with beta-blockers (86%) and diuretics (65%). Mean LVEF was 27 ± 9% and mean GFR was slightly impaired (74 ± 28 ml/min/1.73m2).
Relationship of cardiorenal (hemodynamic), endothelial dysfunction and in-flammation parameters with GFR
In univariate regression analysis, RBF was the main determinant of GFR (r = 0.888, P < 0.001), accounting for over 80% of the variance in GFR. Also FF (r = 0.573, P < 0.001), RVR (r = -0.707, P < 0.001), and UAE (r = -0.306, P = 0.005) were related to GFR. Curvilinear fitting of RBF, RVR and FF with GFR revealed that the relationship between FF and GFR showed a drop-off in the lower ranges of GFR and RBF (Figure 1). No early increase in FF was observed with decreasing RBF. FF started to decrease when RBF dropped below approximately 350 ml/min/1.73m2. RVR showed an exponential increase with decreasing RBF.
Table 2 shows the relationship between GFR and other parameters. Hemodynamic parameters and markers related to GFR included NT-proBNP, MAP and LVEF. Of RAS activity parameters, only PRA showed a strong and significant association with GFR. Endothelial function parameters, including sVCAM-1, vWF and plasma NOx, were moderately associated with GFR. High sensitive-CRP, as a marker of inflammation, did not show any relationship with GFR. In multivariate stepwise linear regression analysis, including all univariate associated variables, only RBF (r = 0.938, P < 0.001), FF (r = 0.786, P < 0.001) and hemoglobin levels
Figure 1. Relationship between RBF, RVR, FF and GFR. Abbreviations: GFR: Glomerular filtration rate, FF: Filtration Fraction, RBF: Renal blood flow, RVR: Renal vascular resistance.
RBF (mL/min/1.73m2)
100200300400500600700
GFR
(mL/
min
/1.7
3m2 )
0
20
40
60
80
100
120
FF (%
)
28
24
20
FF
GFR
RV
R (m
mH
g/m
L/m
in)
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
RVR

32
Chapter 1
(r = -0.520, P < 0.001) remained significant and independent predictors of GFR (adjusted R2 = 0.953).
RVR was significantly associated with age (r = -0.420, P < 0.001) and gender (r = -0.296, P = 0.006), and by definition with MAP and RBF. In addition, RVR related to VCAM-1 (r = 0.347 P = 0.001), NT-proBNP (r = 0.556, P < 0.001), UAE (r = 0.472, P < 0.001) and haemoglobin levels(r = -0.245, P =0.025). In multivariate analysis, only UAE (r = 0.375,
Table 2. Regression analysis for GFR.
GFR
Variable Partial R P-value
Age -0.338 0.001
Gender -0.312 0.003
Cardio-renal hemodynamic parameters
RBF 0.888 < 0.001
FF# 0.573 < 0.001
RVR# -0.707 < 0.001
NT-proBNP# -0.533 < 0.001
MAP 0.386 < 0.001
UAE# -0.306 0.005
LVEF 0.297 0.006
Hemoglobin 0.312 0.004
Peak VO2* 0.552 < 0.001
Renin Angiotensin System parameters
PRA# -0.501 < 0.001
Ang 2# 0.189 0.089
Endothelial function parameters
sVCAM-1# -0.279 0.010
vWf# -0.283 0.009
NOx# -0.276 0.011
ADMA -0.168 0.126
sES# -0.056 0.616
Inflammation parameter
CRP# -0.016 0.883
* N = 48. #Data logarithmically transformed. Shown are partial correlation coefficients adjusted for age and gen-der where appropriate. Abbreviations: GFR; glomerular filtration rate, RBF; Renal blood flow, FF; Filtration frac-tion, RVR: renal vascular resistance, UAE; urinary excretion rate, NT-proBNP; N terminal pro brain natriuretic peptide, MAP; mean arterial pressure, LVEF; left ventricular ejection fraction, PRA; plasma renin activity, Ang II; angiotensin II, sVCAM-1; soluble vascular adhesion molecule-1, vWF; von Willebrand factor, NOx; nitrate/nitrate, ADMA; asymmetric dimethyl arganine, sES; soluble E-selectin, CRP; C-reactive protein.
Determinants of renal function in CHF
33
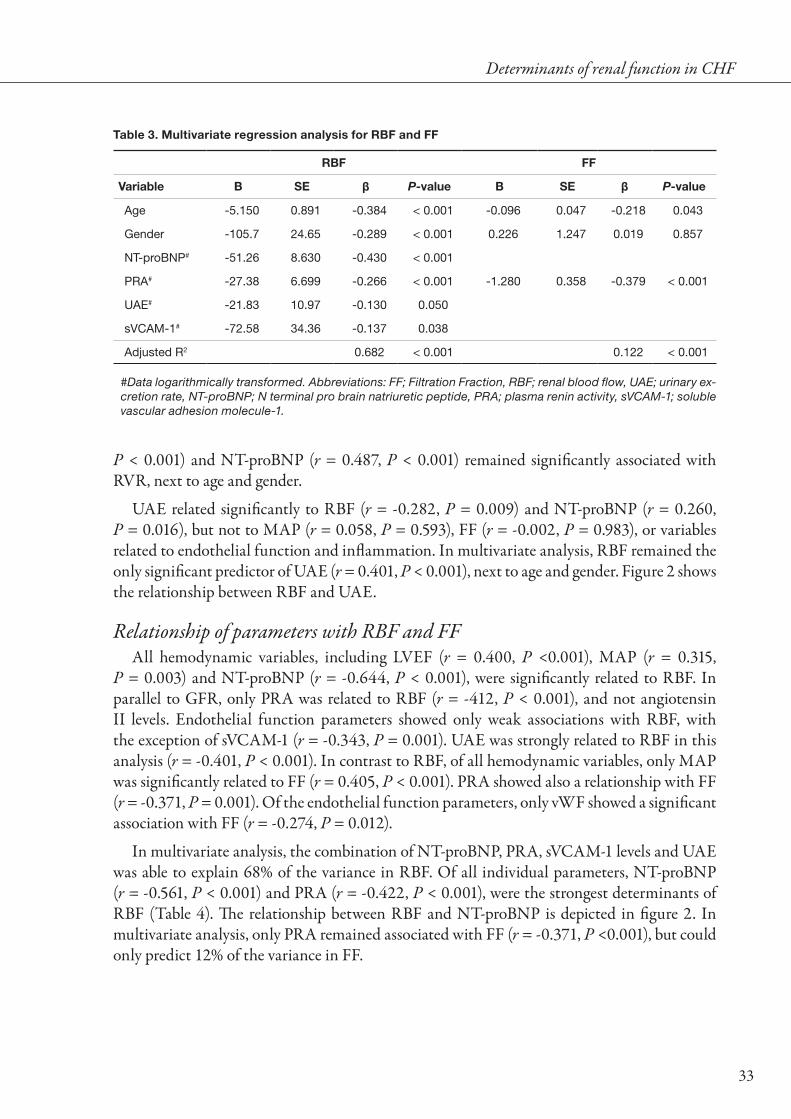
P < 0.001) and NT-proBNP (r = 0.487, P < 0.001) remained significantly associated with RVR, next to age and gender.
UAE related significantly to RBF (r = -0.282, P = 0.009) and NT-proBNP (r = 0.260, P = 0.016), but not to MAP (r = 0.058, P = 0.593), FF (r = -0.002, P = 0.983), or variables related to endothelial function and inflammation. In multivariate analysis, RBF remained the only significant predictor of UAE (r = 0.401, P < 0.001), next to age and gender. Figure 2 shows the relationship between RBF and UAE.
Relationship of parameters with RBF and FFAll hemodynamic variables, including LVEF (r = 0.400, P <0.001), MAP (r = 0.315,
P = 0.003) and NT-proBNP (r = -0.644, P < 0.001), were significantly related to RBF. In parallel to GFR, only PRA was related to RBF (r = -412, P < 0.001), and not angiotensin II levels. Endothelial function parameters showed only weak associations with RBF, with the exception of sVCAM-1 (r = -0.343, P = 0.001). UAE was strongly related to RBF in this analysis (r = -0.401, P < 0.001). In contrast to RBF, of all hemodynamic variables, only MAP was significantly related to FF (r = 0.405, P < 0.001). PRA showed also a relationship with FF (r = -0.371, P = 0.001). Of the endothelial function parameters, only vWF showed a significant association with FF (r = -0.274, P = 0.012).
In multivariate analysis, the combination of NT-proBNP, PRA, sVCAM-1 levels and UAE was able to explain 68% of the variance in RBF. Of all individual parameters, NT-proBNP (r = -0.561, P < 0.001) and PRA (r = -0.422, P < 0.001), were the strongest determinants of RBF (Table 4). The relationship between RBF and NT-proBNP is depicted in figure 2. In multivariate analysis, only PRA remained associated with FF (r = -0.371, P <0.001), but could only predict 12% of the variance in FF.
Table 3. Multivariate regression analysis for RBF and FF
RBF FF
Variable B SE β P-value B SE β P-value
Age -5.150 0.891 -0.384 < 0.001 -0.096 0.047 -0.218 0.043
Gender -105.7 24.65 -0.289 < 0.001 0.226 1.247 0.019 0.857
NT-proBNP# -51.26 8.630 -0.430 < 0.001
PRA# -27.38 6.699 -0.266 < 0.001 -1.280 0.358 -0.379 < 0.001
UAE# -21.83 10.97 -0.130 0.050
sVCAM-1# -72.58 34.36 -0.137 0.038
Adjusted R2 0.682 < 0.001 0.122 < 0.001
#Data logarithmically transformed. Abbreviations: FF; Filtration Fraction, RBF; renal blood flow, UAE; urinary ex-cretion rate, NT-proBNP; N terminal pro brain natriuretic peptide, PRA; plasma renin activity, sVCAM-1; soluble vascular adhesion molecule-1.

34
Chapter 1
DiscussionThe present study shows that RBF is the most important contributor of GFR in patients
with CHF on ACE-inhibition or ARB therapy. Markers of neurohormonal activation and endothelial function showed a less pronounced relationship with GFR. Furthermore, we observed strong relationships between NT-proBNP, PRA and RBF.
HemodynamicsWe observed a parallel decline in GFR with declining RBF. This relationship was the result
of a stable FF over almost the full range of RBF. Only in the extreme ranges of hypoperfusion, FF decreased in parallel with decreasing RBF. In contrast to findings by Cody and Ljungman in 34 patients with CHF not on ACE-inhibitor therapy, FF did not increase with reducing RBF [5,11]. Apparently, this phenomenon represents the effect of ACE-inhibition which blocks the action of angiotensin II on the efferent arteriole. The finding of a disproportionate decrease in FF when RBF is severely reduced is in fact in agreement with hypofiltration that occurs in the absence of ACE-inhibition [5].
The inability to adequately increase or preserve FF can be explained by a combination of three factors: 1) low glomerular plasma flow, 2) low transcappillary hydraulic pressure and 3) reduced ultrafiltration coefficient [11]. We were able to show that reduced RBF is the main contributing factor in the condition of hypofiltration and impaired GFR in CHF patients. SNS activity, which stimulates pre-glomerular vasomotor tone, decreases RBF [15]. However, we did not measure sympathetic activity in our cohort. MAP was related to both RBF and FF
Figure 2. Relationship between RBF and UAE. Box plots for UAE levels are shown. Boxes display median (horizontal bars), interquartile ranges (lower and upper limits of boxes) and 5th and 95th percentiles (error bars). Abbreviations: RBF: Renal blood flow, UAE: urinary albumin excretion.
RBF (mL/min/1.73m2)
> 550 400 - 550 250 - 400 < 250
UA
E (m
g/d
ay)
0
20
40
60
80
100

Determinants of renal function in CHF
35
in univariate analysis. Especially the latter indicates that with the decrease of FF in the lower regions of RBF, MAP decreased. This suggests that in these patients, hydraulic pressure is in fact reduced, which may be one of the mechanisms by which GFR is reduced.
Furthermore, remarkably we observed that FF decreased in the lower regions of RBF, despite an exponential increase in RVR. Total RVR is determined by both afferent and efferent vascular resistance, whereas FF is predominantly determined by efferent vascular resistance. A higher RVR due to efferent vasoconstriction would be expected to lead to an increase in FF as well, but, apparently, at the higher extreme of RVR, this is not the case. Accordingly, this part of the curve is explained by a predominant increase in afferent vascular tone. A very high afferent tone, with consequently a low glomerular perfusion pressure, would require a correspondingly high efferent vascular tone to maintain filtration pressure. The divergence between RVR and FF in patients with severe renal function impairment indicates that in these patients postglomerular tone can no longer be maintained, which might be related to the use of RAS-blockade that is known to blunt efferent vasoconstriction. Thus, the striking increase in RVR is likely to represent strongly increased afferent vasoconstriction. In the light of new therapies in acute heart failure which are targeted at blocking adenosine mediated afferent vasoconstriction, this may indicate a similar pathophysiologic mechanism for reduced GFR in CHF [16]. This is further supported by similar effects of adenosine blockade in the presence of RAS blockade in either animal models of renal hypoxia or patients with CHF [17,18], although in the latter only 59% were on RAS blocking therapy. We recently showed that also venous congestion as estimated by increased central venous pressure, may be an important determinant of GFR, especially in the condition of an impaired RBF [12,13]. The observed decrease in GFR with higher levels of NT-proBNP in the current study might be a reflection of this finding.
Figure 3. Relationship of RBF with NT-proBNP. Abbreviations: NT-proBNP: N terminal pro brain natriuretic peptide, RBF: Renal blood Flow. Solid lines represent regression lines, while dotted lines represent 95% confidence intervals of the regression line.
NT-proBNP (pg/mL)
10 100 1000 10000
RB
F (m
L/m
in/1
.73m
2 )
0
100
200
300
400
500
600
700
800
900r = -0.698Y = 1016 - 83.3 Ln(x)P < 0.001

36
Chapter 1
Endothelial function and inflammation.Increased levels of markers of endothelial dysfunction and inflammation have been found
to correlate with renal dysfunction in non-CHF patient populations [19,20]. Interestingly, endothelial dependent vasodilatation was not associated with estimated GFR in non-CHF patients [21]. We showed that next to RBF and FF, parameters of endothelial function were related to GFR in univariate analysis. However, in multivariate analysis, no independent relationship could be established with GFR. This may suggest that the univariate relationships are attributable to the inter-relationship between endothelial function and RBF, which was confirmed in regression analysis for RBF. Of those factors measured, vWF showed the most consistent relationship with GFR and FF, while sVCAM-1 was related to RBF. Both vWF and sVCAM-1 have been shown to correlate with estimated GFR, but also to prognosis in CHF and other patient populations [8,22]. We did not find any relationship between hs-CRP with GFR, RBF or FF. This suggests that inflammation may not be a strong mediator of renal impairment in CHF.
NT-proBNP NT-proBNP showed an inverse relationship with both GFR and RBF. This is to our
knowledge the first study that showed a strong correlation between endogenous NT-proBNP levels and RBF. In normal subjects, the active brain natriuretic peptide (BNP) may preferably increase RBF by afferent vasodilatation [23]. However, a decrease [24] or no effect on RBF, due to efferent and afferent vasomotor tone unbalance, has also been reported [25]. However, in CHF, the renal actions of BNP are known to be blunted [26]. Therefore, the absence of BNP mediated afferent vasodilatation in the lower regions of RBF may be partly responsible for the markedly reduced GFR in these patients. On the other hand, increased levels of NT-proBNP in parallel to decreased RBF may also be a consequence of more severe cardiac dysfunction. The strong relationship between RBF and NT-proBNP suggests that NT-proBNP may serve as an easily obtainable marker of renal perfusion, which is an important clinical finding because the measurement of RBF is invasive, patient-unfriendly, time-consuming and expensive. Finally, in agreement with our earlier findings, NT-proBNP levels may be a reflection of increased venous congestion, and the relationship with RBF may be a result of the effect of congestion on renal perfusion [12,13]. The ability of NT-proBNP levels for profiling cardiorenal risk and potential target for reno-protective therapy needs to be investigated in future studies.
Renin angiotensin system activityPRA showed a linear relationship with decreasing RBF and GFR. PRA levels are increased
in CHF due to activation of renin secretion in response to different stimuli [27]. In addition, renin is especially secreted from outer cortical glomeruli in response to decreased RBF [28]. This is important, while in CHF renal blood flow is especially diminished in the cortial regions and relatively preserved in the medulla [29]. Via these pathways PRA does not only influence RBF, but RBF also has profound effects on PRA secretion. Therefore, the relationship between PRA and RBF is probably mainly driven by a marked reduction in forward flow. PRA has

Determinants of renal function in CHF
37
been shown to correlate with estimated GFR in other CHF populations, even in patients using ACE-inhibitors [30]. Higher PRA levels might also reflect more severe ACE-inhibition and/or ARB therapy. However, it is plausible, that with more severe renal impairment, ACE escape occurs, which may result in continuing efferent vasoconstriction [31]. Probably, because of a parallel decline in RBF, this is not expressed in an increase in FF.
Albumin excretion Albuminuria is highly prevalent in CHF, and predisposes to CHF in patients with
hypertension [32,33]. We found that UAE was inversely related to GFR in our CHF population, and we observed that UAE was primarily associated with RBF. We were unable to demonstrate any relationship between UAE and markers of endothelial function and inflammation. This suggests that increased UAE in CHF reflects intrinsic renal damage, possibly due to chronic hypoperfusion. In other chronic conditions, intrarenal hypoperfusion injury progressively compromises the entire kidney [34]. Chronic renal hypoperfusion might ultimately result in reduced tubular reabsorption of albumin, suggesting also tubular dysfunction [35].
Clinical implications.Our findings have important clinical implications. First, impaired hemodynamics are
the key determinants of reduced GFR. Therapies aimed at preserving GFR in CHF patients should most likely focus on preservation of RBF. It is however difficult to measure RBF in every heart failure patient, especially in those with compromised hemodynamics. Our present findings suggest that NT-proBNP levels may be used as a marker of RBF, and may therefore be a target for therapy to indirectly improve RBF and subsequent GFR. Therapy targeted at PRA levels seems to be of less interest, since the slope between PRA levels and RBF was much less pronounced. The decrease in FF, together with decrease in GFR and increase in RVR suggests that renal afferent vasoconstriction may be an important target for therapy, even in CHF. Finally, therapy should focus on preventing renal impairment below a GFR of 40 ml/min/1.73m2 as these patients not only experience functional impairment, but also structural renal damage.
LimitationsThis study is of cross-sectional design and thus only can be hypothesis generating. Similarly,
cause-effect relationships cannot be distilled from cross-sectional studies. Future studies are therefore needed to intervene in the pathophysiologic mechanisms we have explored to establish the cause-effect relationships. Regretfully we were not able to determine the relationship of SNS-activation and the ultrafiltration coefficient with renal function in our patients. Both may play an important role in renal function impairment in CHF. PRA levels will be altered by ACE-inhibitor and ARB therapy, thereby limiting interpretability. Although our study was rather small, this is to our knowledge by far the largest cohort of patients comparing the results of true renal function measurements with a large number of parameters linked to the biology of impaired renal function.

38
Chapter 1
Conclusion.Reduced RBF is the main determinant of impaired GFR in patients with CHF.
Neurohormonal activation and endothelial function were only moderately associated with impaired GFR. NT-proBNP levels may be a non-invasive marker of RBF, and may serve as a target for therapy in future studies.

Determinants of renal function in CHF
39
ReferencesHillege HL, van Gilst WH, van Veldhuisen DJ et al. Accelerated decline and prognostic impact of renal 1. function after myocardial infarction and the benefits of ACE inhibition: the CATS randomized trial. Eur Heart J. 2003;24:412-420.
Hillege HL, Nitsch D, Pfeffer MA et al. Renal function as a predictor of outcome in a broad spectrum 2. of patients with heart failure. Circulation. 2006;113:671-678.
Ruilope LM, van Veldhuisen DJ, Ritz E, and Luscher TF. Renal function: The Cinderella of cardiovascular 3. risk profile. J Am Coll Cardiol. 2001;38:1782-1787.
Smilde TDJ, Hillege HL, Voors AA, Dunselman PHJ, and van Veldhuisen DJ. Prognostic importance 4. of renal function in patients with early heart failure and mild left ventricular dysfunction. Am J Cardiol. 2004;94:240-243.
Ljungman S, Laragh JH, and Cody RJ. Role of the Kidney in Congestive Heart-Failure - Relationship 5. of Cardiac Index to Kidney-Function. Drugs. 1990;39:10-21.
Bohm M, Werner N, and Kindermann M. Drug treatment of chronic heart failure. Clin Res Cardiol. 6. 2006;95 Suppl 4:36-54.
van der Horst I, Voors AA, and van Veldhuisen DJ. Treatment of heart failure with ACE inhibitors and 7. beta-blockers: what is next? Aldosterone receptor antagonists? Clin Res Cardiol. 2007;96:193-195.
Chong AY, Freestone B, Patel J et al. Endothelial activation, dysfunction, and damage in congestive heart 8. failure and the relation to brain natriuretic peptide and outcomes. Am J Cardiol. 2006;97:671-675.
Torre-Amione G, Kapadia S, Benedict C et al. Proinflammatory cytokine levels in patients with 9. depressed left ventricular ejection fraction: a report from the Studies of Left Ventricular Dysfunction (SOLVD). J Am Coll Cardiol. 1996;27:1201-1206.
Bongartz LG, Cramer MJ, Doevendans PA, Joles JA, and Braam B. The severe cardiorenal syndrome: 10. ‘Guyton revisited’. Eur Heart J. 2005;26:11-17.
Cody RJ, Ljungman S, Covit AB et al. Regulation of glomerular filtration rate in chronic congestive 11. heart failure patients. Kidney Int. 1988;34:361-367.
Damman K, Navis G, Smilde TD et al. Decreased cardiac output, venous congestion and the association 12. with renal impairment in patients with cardiac dysfunction. Eur J Heart Fail. 2007;9:872-878.
Damman K, van Deursen VM, Navis G et al. Increased central venous pressure is associated with 13. impaired renal function and mortality in a broad spectrum of patients with cardiovascular disease. J Am Coll Cardiol. 2009;53:582-588.
Donker AJM, Vanderhem GK, Sluiter WJ, and Beekhuis H. Radioisotope Method for Simultaneous 14. Determination of Glomerular-Filtration Rate and Effective Renal Plasma-Flow. Netherlands Journal of Medicine. 1977;20:97-103.
Evans RG, Eppel GA, Anderson WP, and Denton KM. Mechanisms underlying the differential control 15. of blood flow in the renal medulla and cortex. J Hypertens. 2004;22:1439-1451.
Givertz MM, Massie BM, Fields TK, Pearson LL, and Dittrich HC. The effects of KW-3902, an 16. adenosine A1-receptor antagonist,on diuresis and renal function in patients with acute decompensated heart failure and renal impairment or diuretic resistance. J Am Coll Cardiol. 2007;50:1551-1560.

40
Chapter 1
Prevot A, Huet F, Semama DS, Gouyon JB, and Guignard JP. Complementary effects of adenosine and 17. angiotensin II in hypoxemia-induced renal dysfunction in the rabbit. Life Sci. 2002;71:779-787.
Dittrich HC, Gupta DK, Hack TC et al. The effect of KW-3902, an adenosine A1 receptor antagonist, 18. on renal function and renal plasma flow in ambulatory patients with heart failure and renal impairment. J Card Fail. 2007;13:609-617.
Bonomini M, Reale M, Santarelli P et al. Serum levels of soluble adhesion molecules in chronic renal 19. failure and dialysis patients. Nephron. 1998;79:399-407.
Stam F, van GC, Becker A et al. Endothelial dysfunction contributes to renal function-associated 20. cardiovascular mortality in a population with mild renal insufficiency: the Hoorn study. J Am Soc Nephrol. 2006;17:537-545.
van der Harst P., Smilde TD, Buikema H et al. Vascular function and mild renal impairment in stable 21. coronary artery disease. Arterioscler Thromb Vasc Biol. 2006;26:379-384.
Wannamethee SG, Shaper AG, Lowe GD et al. Renal function and cardiovascular mortality in elderly 22. men: the role of inflammatory, procoagulant, and endothelial biomarkers. Eur Heart J. 2006;27:2975-2981.
La Villa G, Fronzaroli C, Lazzeri C et al. Cardiovascular and renal effects of low dose brain natriuretic 23. peptide infusion in man. J Clin Endocrinol Metab. 1994;78:1166-1171.
Jensen KT, Carstens J, and Pedersen EB. Effect of BNP on renal hemodynamics, tubular function and 24. vasoactive hormones in humans. Am J Physiol. 1998;274:F63-F72.
van der Zander K, Houben AJ, Hofstra L, Kroon AA, and de Leeuw PW. Hemodynamic and renal 25. effects of low-dose brain natriuretic peptide infusion in humans: a randomized, placebo-controlled crossover study. Am J Physiol Heart Circ Physiol. 2003;285:H1206-H1212.
Marcus LS, Hart D, Packer M et al. Hemodynamic and renal excretory effects of human brain 26. natriuretic peptide infusion in patients with congestive heart failure. A double-blind, placebo-controlled, randomized crossover trial. Circulation. 1996;94:3184-3189.
Brown MJ. Renin: friend or foe? Heart. 2007;93:1026-1033.27.
Nushiro N, Ito S, and Carretero OA. Renin release from microdissected superficial, midcortical, and 28. juxtamedullary afferent arterioles in rabbits. Kidney Int. 1990;38:426-431.
Kilcoyne MM, Schmidt DH, and Cannon PJ. Intrarenal blood flow in congestive heart failure. 29. Circulation. 1973;47:786-797.
Hillege HL, Girbes AR, de Kam PJ et al. Renal function, neurohormonal activation, and survival in 30. patients with chronic heart failure. Circulation. 2000;102:203-210.
van de Wal RM, Plokker HW, Lok DJ et al. Determinants of increased angiotensin II levels in severe 31. chronic heart failure patients despite ACE inhibition. Int J Cardiol. 2006;106:367-372.
Ingelsson E, Sundstrom J, Lind L et al. Low-grade albuminuria and the incidence of heart failure in a 32. community-based cohort of elderly men. Eur Heart J. 2007;28:1739-1745.
van de Wal RM, Asselbergs FW, Plokker HW et al. High prevalence of microalbuminuria in chronic 33. heart failure patients. J Card Fail. 2005;11:602-606.

Determinants of renal function in CHF
41
Brezis M and Rosen S. Hypoxia of the renal medulla--its implications for disease. N Engl J Med. 34. 1995;332:647-655.
Pollock CA and Poronnik P. Albumin transport and processing by the proximal tubule: physiology and 35. pathophysiology. Curr Opin Nephrol Hypertens. 2007;16:359-364.

42

Chapter 2Decreased cardiac output, venous congestion and the association with
renal impairment in patients with cardiac dysfunction
Kevin Damman, Gerjan Navis, Tom D. J. Smilde, Adriaan A. Voors, Wim van der Bij, Dirk J. van Veldhuisen, Hans L. Hillege
Eur J Heart Fail, 2007; 9:872-78

44
Abstract
Background.Renal failure in heart failure is related to decreased cardiac output. However, little is known about its
association with venous congestion. We aimed to investigate the relationship between venous congestion and glomerular filtration rate (GFR) in patients with cardiac dysfunction.
Methods and Results.Right atrial pressure (RAP) and cardiac index (CI) were determined by right heart catheterization
in 51 patients with cardiac dysfunction, secondary to pulmonary hypertension. GFR and renal blood flow (RBF) were measured as 125I-Iothalamate and 131I-Hippuran clearances, respectively. Mean age was 40 ± 11 years and 69% of patients were female. GFR was 73 ± 19 ml/min/1.73m² with a CI of 2.1 ± 0.7 l/min/m². In multivariate analysis, RBF (r = 0.664, P < 0.001) and RAP (r = - 0.367, P = 0.020) were independently associated with GFR. In patients in the lower ranges of RBF, venous congestion was an important determinant of renal function.
Conclusion.RBF is the main factor determining GFR in patients with cardiac dysfunction. Venous congestion,
characterised by an increased RAP, adjusted for RBF is also related to GFR. Treatment to preserve GFR should not only focus on improvement of renal perfusion, but also on decreasing venous congestion.

CVP and renal function
45
IntroductionImpaired renal function independently increases the risk of death, cardiovascular death and
hospitalisation for worsening heart failure in patients with chronic heart failure (CHF) [1-3]. The main determinant of renal function in CHF is renal blood flow (RBF) [4]. Reduction in cardiac output (CO) results in a disproportionate reduction in renal perfusion, which consequently leads to a diminished glomerular filtration rate (GFR).
CHF is not only characterised by decreased cardiac output and subsequent decreased organ perfusion, but also by increased venous congestion. However, most previous reports have studied the interrelationship between reduced cardiac output, renal function and prognosis in CHF of different aetiologies, including the impact of right ventricular function on mortality [5,6].
There are very few data on the association between venous congestion and indices of renal function. One very small study suggested a relationship between venous congestion and RBF in CHF [7]. The authors showed an inverse relationship between RBF and venous pressure. However, the association between venous congestion and GFR, remains to be elucidated.
Patients with pulmonary hypertension often have decreased cardiac output in combination with elevated right sided filling pressures and signs of congestion. Therefore, from a haemodynamic point of view, these patients are a suitable cohort to investigate the relationship between venous congestion and GFR in patients with cardiac dysfunction.
The aim of the present study was therefore to investigate the relative contribution of decreased renal perfusion and determinants of venous congestion on renal function in patients with cardiac dysfunction secondary to pulmonary hypertension.
Methods
Patients.The study enrolled consecutive patients diagnosed with pulmonary hypertension who were
potential candidates for lung transplantation. All patients underwent right heart catheterization and clearance measurements of renal haemodynamic parameters as a part of the work up for transplantation. Patients with idiopathic pulmonary arterial hypertension, as well as secondary pulmonary hypertension, were included. The study protocol was approved by the institutional ethics committee. All patients gave written informed consent.
Right heart catheterization.Right sided cardiac catheterization data included measurements of mean arterial pressure
(MAP, mmHg), right atrial pressure (RAP, mmHg), mean pulmonary artery pressure (MPAP, mmHg), pulmonary capillary wedge pressure (PCWP, mmHg), systemic vascular resistance (SVR, dyne·sec·cm-5) and pulmonary vascular resistance (PVR, dyne·sec·cm-5). Cardiac output

46
Chapter 2
(CO, l/min) was determined using the method of thermodilution. Systemic blood flow was used as measurement of cardiac output in patients with intracardiac shunting. Cardiac index (CI, l/min/m2) was calculated as CO divided by body surface area (BSA). Cardiac catheterization measurements were obtained from the patient at rest.
Renal function measurement by 125I-Iothalamate and 131I-Hippuran clear-ances.
GFR and effective renal plasma flow (ERPF) were measured by constant infusion of radiolabelled tracers, 125I-Iothalamate and 131I-Hippuran. The clearances were calculated as (U·V)/P and (I·V)/P, respectively. U·V represents the urinary excretion of the tracer, I·V represents the infusion rate of the tracer; P represents the tracer value in plasma at the end of each clearance period. GFR was measured as U·V/P for 125I-Iothalamate, and corrected for voiding errors by the ratio of (I·V/P)/(U·V/P)131I-Hippuran as described previously. RBF was calculated as ERPF / 1-haematocrit. The filtration fraction (FF) was calculated as the ratio of GFR and ERPF and expressed as a percentage. GFR and ERPF were expressed per 1.73m2 of BSA.
Relative contributions of RAP and RBF.In order to assess and visualise relative contributions of RBF and RAP, RBF was dichotomised
(≤ 400 vs > 400 ml/min/1.73m2) and subsequently ranked to low and high RAP within both subgroups. This stratification is based on the haemodynamic renal response in a setting of reduced perfusion when RBF declines below approximately 400 ml/min/1.73m2 [4,8]. It considers a potential relationship between RBF and venous congestion and allows a within-group comparison of patients with and without venous congestion, who have comparable RBF [4].
Statistical analysis.Results are expressed as means ± SD unless otherwise indicated. All variables were normally
distributed. Pearson correlation coefficients were calculated to determine which variables had a significant univariate association with GFR. Stepwise multivariate linear regression analysis was used to determine the independent relationships between univariately associated variables with GFR. Subjects with missing data were excluded from multivariate analysis. In a secondary analysis, the multivariate regression analysis was repeated after imputing missing values using expectation maximization as estimation method. To examine all possible interactions of the effects of various variables, a secondary analysis was performed including interaction terms. Differences between the different groups of low vs high RBF or RAP were carried out using Student’s T-tests. All reported probability values are 2-tailed, and a P value <0.05 was considered statistically significant. Statistical analyses were performed using SPSS, Chicago version 12.
The investigation conforms with the principles outlined in the Declaration of Helsinki

CVP and renal function
47
Results
Patient characteristics. In total 51 consecutive patients were included (Table 1). Twenty eight patients (55%) had
idiopathic pulmonary arterial hypertension. Secondary pulmonary hypertension was mostly due to pulmonary embolism (n=9) and atrial septum defect (n=7). Both left ventricular ejection fraction (52 ± 14%) and right ventricular ejection fraction (42 ± 11%) were mildly impaired. Mean MPAP (60 ± 16 mmHg) and RAP (11 ± 6 mmHg) were elevated and the cardiac index was decreased (2.1 ± 0.7 l/min/m2, normal range 2.5-4.0 l/min/m2). GFR was
Table 1. Patient characteristics
Variable Total
Age (years) 40 ± 11
Gender (% male) 31
Hemoglobin level (g/dL) 15.5 ± 1.9
Hematocrit (%) 44 ± 6
Cardiac Output (l/min) * 3.6 ± 1.3
Cardiac Index (l/min/m2) * 2.1 ± 0.7
Left Ventricular Ejection Fraction (%) † 52 ± 14
Right Ventricular Ejection Fraction (%) † 42 ± 11
Mean Arterial Pressure (mmHg) 90 ± 12
Mean Pulmonary Arterial Pressure (mmHg) 60 ± 16
Right Atrial Pressure (mmHg) ‡ 11 ± 6
Pulmonary Wedge Pressure (mmHg) # 15 ± 10
Pulmonary Vascular Resistance (dyne·sec·cm-5) 1160 ± 560
Systemic Vascular Resistance (dyne·sec·cm-5) 1886 ± 652
Renal Function Parameters
Serum Creatinine (µmol/l) 93 ± 18
Glomerular Filtration Rate (ml/min/1.73m2) 73 ± 19
Renal Blood Flow (ml/min/1.73m2) 384 ± 133
Effective Renal Plasma Flow (ml/min/1.73m2) 212 ± 73
Filtration Fraction (%) 37 ± 6
Therapy
Diuretic use (n, (%)) 26 (51)
ACE-inhibitor use (n, (%)) 6 (12)
* n = 50, † n = 34, ‡ n = 41, # n = 49. Values are presented as mean (SD) unless indicated otherwise.

48
Chapter 2
mildly impaired (73 ± 19 ml/min/1.73m2) in combination with an increased filtration fraction. No differences in baseline characteristics were found between patients with idiopathic and secondary pulmonary hypertension.
Relationship between haemodynamic parameters and GFR.Univariate analysis showed that RBF was strongly associated with GFR (r = 0.797, P <
0.001). Next to RBF, RAP was inversely related to GFR (r = - 0.616, P < 0.001). Other variables related to renal function were CI (r = 0.404, P = 0.007) and PVR (r = -0.298, P = 0.040). In
Table 2. Regression analysis for GFR
Variable Univariate correlation coefficient
Univariate β Multivariate correlation coefficient
Multivariate β MultivariateP-value
Age -0.072 0.023
Gender -0.218 -0.028
RBF 0.797 0.782 0.664 0.621 <0.001
RAP -0.616 -0.579 -0.367 -0.276 0.020
CI 0.404 0.396
PVR -0.298 -0.297
Adjusted R2 0.609 <0.001
CI, Cardiac Index; PVR, Pulmonary Vascular Resistance; RAP, Right Atrial Pressure; RBF, Renal Blood Flow.
Figure 1. Relative contributions of right atrial pressure (RAP) and renal blood flow (RBF) to glomerular filtration rate (GFR). Error bars represent 95% Confidence Interval. * P < 0.001 for difference with High RAP, Low RBF. † P < 0.01 for difference with Low RAP, Low RBF.
High RAPLow RBF
Low RAPLow RBF
High RAPHigh RBF
Low RAPHigh RBF
*
* *
GFR
(ml/m
in/1
.73m
2 )
20
40
60
80
100
120
††

CVP and renal function
49
multivariate regression analysis, including all univariately associated variables (P < 0.1), only RBF and RAP remained significant and independent predictors of GFR (Table 2). Addition of diuretic treatment or ACE-inhibition to the model did not interfere with these results. Also beta-blocker therapy was not a mediator of the relationship with GFR.
Analysis of interaction terms between the different variables revealed no significant interaction on a continuous scale. We further analyzed possible interactions between the two variables in the multivariate model, when both were dichotomised by the median. The interaction term of these dichotomised variables showed a trend toward significance (P 0.095). The multivariate analysis showed, after imputing of missing values, similar results.
Figure 2. Pathophysiology of the relation between venous congestion and reduced glomerular filtration rate (GFR). ANP; Atrial Natriuretic Peptide, SNS; Sympathetic Nervous System, RAAS; rennin-angiotensin aldosterone system. Numbers in circles represent the targets for specific therapies, as follows:1. Ultrafiltration, diuretics, sodium and water restriction and arginine vasopressin receptor antagonists [36,37,39] .2. Ultrafiltration, diuretics and sodium and water restriction [36,37].3. ACE-inhibitors and angiotensin II receptor blockers.4. Statin therapy [38].5. Beta-blocker therapy.6. Angiotensin II receptor blockers.7. Neutral endopeptidase inhibitors [41].8. Urodilatin [40].
HypoxicTrigger
High Central Venous Pressure
Reduced GFR
Renal Venous Pressure
Renal InterstitialPressure
Intrarenal Angiotensin II
Hydrostaticpressure in Bowman ’s
capsule
SNS Activation
Angiotensin II Filtration
Coef�cient
ANP
Hypo-responsiveness to
ANP in CHF
RAAS
[13]
[16,17]
[18]
[20,21][21]
[14] [13]
[19]
[23]
[15]
[11]
[22]
22
1
7
6
4
3
5
8
[24]

50
Chapter 2
Figure 1 illustrates the relative contributions of RBF and RAP to GFR when dichotomised to high versus low values. The effect of increased RAP appears to reside solely among patients with already reduced RBF. Characteristics of these groups are shown in Table 3. Lowest GFR was seen in patients with a relatively high RAP and low RBF. The highest GFR was present in patients with high RBF and low RAP; in these patients GFR was almost similar when compared to the group of patients with a high RAP (P = 0.736). In patients with a low RBF, a high RAP was associated with a significantly lower GFR when compared to patients with normal RBF and/or normal RAP (P < 0.001 and P < 0.01, respectively). Both RBF and RAP differed significantly between high and low subgroups. Both groups with low RBF had higher RAP than the groups with high RBF.
Figure 3. Different haemodynamic profiles. Schematic representation of haemodynamic profiles, as adapted from Stevenson et al [25].
+--
+
--
AD
EQ
UA
TE
PE
RFU
SIO
N
CONGESTION
Dry - Warm Wet - Warm
Dry - Cold Wet - Cold
Low RAP,
High RBF
High RAP,
High RBF
Low RAP,Low RBF
High RAP,High RBF
Table 3. Characteristics of the different subgroups
High RAP,Low RBF
Low RAP,Low RBF
High RAP,High RBF
Low RAP,High RBF
N 11 10 10 10
RAP (mmHg) 19 ± 4 9 ± 2 12 ± 3 5 ± 2
RBF (ml/min/1.73m2) 256 ± 62 291 ± 50 489 ± 74 474 ± 65
GFR (ml/min/1.73m2) 52 ± 9 67 ± 9 83 ± 12 85 ± 17
CI (l/min/m2) 1.7 ± 0.5 1.8 ± 0.6 2.2 ± 0.6 2.5 ± 0.7
CI, Cardiac Index; GFR, Glomerular Filtration Rate; RAP, Right Atrial Pressure; RBF, Renal Blood Flow.

CVP and renal function
51
DiscussionIn the present study we show that GFR is not only determined by reduced cardiac output,
but also by venous congestion, characterised by an increased RAP in patients with cardiac dysfunction, secondary to pulmonary hypertension. The association between RAP and renal function was primarily present in patients with a reduced RBF.
Relationship between venous congestion and renal function in CHFIn a substudy of the SOLVD, Drazner et al established the prognostic implication of jugular
venous pressure in patients with CHF [9]. Elevated jugular venous pressure was associated with adverse outcomes. Furthermore, patients with increased venous pressure had a significantly higher serum creatinine level (115 ± 27 vs 106 ± 27 µmol/l). This suggests an important relationship between renal dysfunction and venous congestion in patients with cardiac dysfunction.
The main determinant of GFR in CHF is RBF [4]. However, we recently showed that increased levels of atrial and brain natriuretic peptides (ANP, BNP respectively) are associated with renal dysfunction in patients with CHF [10]. Both peptides are secreted when volume overload causes ventricular stretch, while ANP is also released in response to volume overload in the atria. This could indicate that not only reduced renal perfusion but also volume overload is related to renal dysfunction, again suggesting a relation between venous congestion and renal impairment.
Independent component of venous congestion in pathophysiology of reduced GFRIn a multivariate analysis we found that both RAP and RBF were independent determinants
of GFR. Both parameters are reflections of cardiac status and are mutually associated. Renal venous pressure is closely related to RBF in certain pathophysiological states, such as CHF [11]. Therefore, it is to be expected that it influences GFR. Ljungman showed that even during decreased RBF, the kidney can preserve GFR by increasing filtration fraction in CHF [4]. Below a certain threshold value (400 ml/min/1.73m2) of RBF, GFR declines. To address this in our population we dichotomised RBF to low and high values, and then assessed the relationship between RAP in the patients with low and high RBF. We observed an association between venous pressure and GFR independent of RBF. Furthermore, we showed that in the lower regions of RBF, there is an additive effect on GFR between increased RAP and decreased RBF, which is absent in the higher regions of RBF. This suggests that the independent contribution of RAP is mainly relevant in the low ranges of renal perfusion. While RAP is less elevated in the high RBF group with high RAP, this could also imply that the effect of venous congestion on GFR is limited to higher levels of RAP.
The specific circulatory physiology underlying this direct link between increased venous pressure and GFR is unclear, but there are a number of possibilities as shown in Figure 2. Renal venous pressure rises, in response to increased central venous pressure and causes an increase in renal interstitial pressure [11-13]. This may lead to impairment of GFR induced by an hypoxic

52
Chapter 2
state in analogy to hepatic congestion that leads to liver failure in CHF [14], or by an increase in hydrostatic pressure in Bowman’s capsule [13]. Furthermore, with increasing renal venous pressure, not only intrarenal but also systemic, angiotensin II concentrations increase [13,15]. This will lead to a further fall in GFR [16,17], either directly or by modulation of the sympathetic nervous system (SNS) [18,19]. Increased SNS activity will influence GFR by changing the filtration-coefficient [20,21]. Furthermore, increased SNS activity triggers angiotensin II release, increasing the effect on GFR [21]. Finally, the effect of ANP on preserving GFR by decreasing sensitivity of the tubuloglomerular feedback mechanism is blunted in CHF, thereby compromising GFR [22]. In addition, both SNS-activation and angiotensin II are mediators of the blunted response to ANP observed in CHF [23,24].
The combination of both decreased cardiac output and increased venous congestion in CHF was introduced in a model of four clinical profiles by Stevenson et al [25,26]. These profiles were defined as the presence or absence of venous congestion (wet or dry) in combination with adequacy of perfusion (warm or cold). These clinical profiles were shown to correlate strongly with outcome [27]. Those patients who not only suffered from hypoperfusion but also had clinical signs of venous congestion had the worst prognosis. The recent ESCAPE trial investigated whether treatment directed at lowering invasively measured RAP and PCWP, was superior to treatment guided by clinical assessment [28]. No difference was observed in clinical outcome between the treatment groups. However, in the group tailored to filling pressures, renal function did not worsen when RAP was actively lowered, while it did worsen in the group tailored by clinical assessment. More recently, Stevenson argued that haemodynamic goals are (still) important in the treatment of CHF and that worsening of renal function is not solely attributable to a fall in cardiac output [29]. The current study indicates that venous congestion could be one of these additional factors.
Clinical profiles and relation to renal functionThe profiles provided by Stevenson can now be extended, in light of our findings of a
combined effect of decreased perfusion and venous congestion on renal function (Figures 1 and 3). In the ‘warm’ profiles, GFR is mainly characterised by a relatively preserved RBF. However, there are patients with preserved RBF who experience increased RAP. Possibly, in these patients increased RAP reflects adaptation to reduced cardiac function by the Frank-Starling mechanism [30]. The dependency on renal perfusion rather than RAP indicates that the contribution of increased venous pressure on GFR is limited. We therefore hypothesize that therapy to reduce venous congestion and/or filling pressures in these patients will have little impact on GFR. This profile might reflect the haemodynamic state of patients with heart failure and preserved systolic function. Interestingly, in this patient group, renal impairment is associated with greater mortality than observed in patients with systolic dysfunction [31]. In addition, these patients (compared to the dry-warm profile) are at risk for accelerated renal function loss when renal perfusion is compromised, shifting to the ‘wet-cold’ profile. Therefore, reducing venous congestion in this patient group will result in a shift towards the more favourable ‘dry-warm’ profile, with subsequent improved survival [27].

CVP and renal function
53
In contrast, when renal perfusion is compromised (‘cold’), especially patients in the ‘wet’ profile have trouble preserving GFR. While RBF is closely related to reduced cardiac output, these profiles represent more advanced stages of cardiac dysfunction [4,32]. The observation that in the presence of reduced RBF, increased RAP can also be absent, indicates that at least a subgroup of patients is able to adapt to reduced renal perfusion and reduced cardiac output, without increasing filling pressures.
Clinical implications for therapy.Therapy to preserve GFR is especially warranted in profiles with reduced RBF. First
line therapy to improve RBF consists of ACE inhibitors and ARBs [33,34]. In addition to improved renal perfusion, this will also lead to improvement of GFR by interaction with venous congestion. This interaction and the targets for other therapies are shown in figure 2. Diuretics are prescribed frequently in patients with CHF who are fluid overloaded, mainly to relieve symptoms. Concerns have been raised about whether aggressive diuresis in patients in the ‘wet-cold’ profile leads to further renal impairment [35]. However, our present data suggest that when diuretic therapy is tailored to decrease venous congestion, it may benefit GFR and subsequently improve prognosis. Future therapeutic options include ultrafiltration which is a promising therapy to reduce venous congestion [36,37]. In addition, beta-blockers can impact on the relationship between venous congestion and renal function, by blocking SNS activation. Also, as observed in a study of experimental heart failure [38], statins can indirectly influence SNS activation by down-regulation of angiotensin II receptor expression. Other new therapies to preserve renal function and to reduce venous congestion have effects on the cardiorenal interaction. Arginine vasopressin antagonists inhibit water reabsorption in the distal tubule [39]. Urodilatin is a recombinant ANP which increases natriuresis and improves symptoms in acute heart failure [40], while neutral endopeptidase inhibitors reduce breakdown of ANP, thereby increasing the bioavailability of ANP resulting in delayed onset of sodium retention [41]. The safety and efficacy of these therapies is still under investigation, but they may be potential candidates for renal-protective therapy in patients with reduced renal perfusion and venous congestion.
Study limitationsThe present analysis was conducted in a cross sectional design. Therefore, a cause and effect
relationship from venous congestion to renal dysfunction can not be demonstrated, while decreased renal function leads to increased water and salt retention, thereby increasing venous congestion. Furthermore, although patients with pulmonary hypertension provide unique data on the relationship between cardiac dysfunction, venous congestion and renal haemodynamics, they have secondary heart problems due to increased pulmonary pressure, instead of primary cardiac illness. On the other hand, in this population use of ACE-inhibitors, that can confound the interrelationships between systemic and renal haemodynamics, was limited. The current analysis was conducted in a small cohort with a specific cause for cardiac dysfunction, future research to assess the relationship between venous congestion and renal function in a broader cohort of patients with different aetiologies of cardiac dysfunction, is now requires.

54
Chapter 2
ConclusionRenal function is not only related to decreased renal perfusion, but also to increased venous
congestion in patients with cardiac dysfunction secondary to pulmonary hypertension. Differentiation between haemodynamic profiles may be useful in tailoring treatment for preservation of GFR in CHF, by not only focussing on improvement of RBF, but also on alleviation of venous congestion.

CVP and renal function
55
ReferencesDries DL, Exner DV, Domanski MJ, Greenberg B, and Stevenson LW. The prognostic implications of 1. renal insufficiency in asymptomatic and symptomatic patients with left ventricular systolic dysfunction. J Am Coll Cardiol. 2000;35:681-689.
Hillege HL, Nitsch D, Pfeffer MA et al. Renal function as a predictor of outcome in a broad spectrum 2. of patients with heart failure. Circulation. 2006;113:671-678.
Al Ahmad A, Rand WM, Manjunath G et al. Reduced kidney function and anemia as risk factors for 3. mortality in patients with left ventricular dysfunction. J Am Coll Cardiol. 2001;38:955-962.
Ljungman S, Laragh JH, and Cody RJ. Role of the Kidney in Congestive Heart-Failure - Relationship 4. of Cardiac Index to Kidney-Function. Drugs. 1990;39:10-21.
Ghio S, Gavazzi A, Campana C et al. Independent and additive prognostic value of right ventricular 5. systolic function and pulmonary artery pressure in patients with chronic heart failure. J Am Coll Cardiol. 2001;37:183-188.
Zornoff LAM, Skali H, Pfeffer MA et al. Right ventricular dysfunction and risk of heart failure and 6. mortality after myocardial infarction. J Am Coll Cardiol. 2002;39:1450-1455.
Kos T, Pacher R, Wimmer A et al. Relationship between kidney function, hemodynamic variables and 7. circulating big endothelin levels in patients with severe refractory heart failure. Wien Klin Wochenschr. 1998;110:89-95.
Smilde TDJ, van der Harst P, van Veldhuisen DJ et al. Reduced renal blood flow is the main determinant 8. of GFR in heart failure. Meeting Abstract. 2005.
Drazner MH, Rame JE, Stevenson LW, and Dries DL. Prognostic importance of elevated jugular venous 9. pressure and a third heart sound in patients with heart failure. N Eng J Med. 2001;345:574-581.
Smilde TD, Hillege HL, Navis G et al. Impaired renal function in patients with ischemic and 10. nonischemic chronic heart failure: association with neurohormonal activation and survival. Am Heart J. 2004;148:165-172.
Maxwell MH, Breed ES, and Schwartz IL. Renal Venous Pressure in Chronic Congestive Heart Failure. 11. J Clin Invest. 1950;29:342-348.
Fiksen-Olsen MJ and Romero JC. Renal effects of prostaglandin inhibition during increases in renal 12. venous pressure. Am J Physiol. 1991;260:525-529.
Fiksen-Olsen MJ, Strick DM, Hawley H, and Romero JC. Renal effects of angiotensin II inhibition 13. during increases in renal venous pressure. Hypertension. 1992;19:137-141.
Seeto RK, Fenn B, and Rockey DC. Ischemic hepatitis: clinical presentation and pathogenesis. Am J 14. Med. 2000;109:109-113.
Kastner PR, Hall JE, and Guyton AC. Renal hemodynamic responses to increased renal venous pressure: 15. role of angiotensin II. Am J Physiol. 1982;243:F260-F264.
Blantz RC, Konnen KS, and Tucker BJ. Angiotensin II effects upon the glomerular microcirculation 16. and ultrafiltration coefficient of the rat. J Clin Invest. 1976;57:419-434.

56
Chapter 2
Tucker BJ, Mundy CA, Maciejewski AR et al. Changes in glomerular hemodynamic response to 17. angiotensin II after subacute renal denervation in rats. J Clin Invest. 1986;78:680-688.
Taddei S, Favilla S, Duranti P, Simonini N, and Salvetti A. Vascular renin-angiotensin system and 18. neurotransmission in hypertensive persons. Hypertension. 1991;18:266-277.
DiBona GF. Nervous kidney. Interaction between renal sympathetic nerves and the renin-angiotensin 19. system in the control of renal function. Hypertension. 2000;36:1083-1088.
DiBona GF and Kopp UC. Neural control of renal function. Physiol Rev. 1997;77:75-197.20.
Kon V, Yared A, and Ichikawa I. Role of renal sympathetic nerves in mediating hypoperfusion of renal 21. cortical microcirculation in experimental congestive heart failure and acute extracellular fluid volume depletion. J Clin Invest. 1985;76:1913-1920.
Morsing P, Stenberg A, Casellas D et al. Renal interstitial pressure and tubuloglomerular feedback 22. control in rats during infusion of atrial natriuretic peptide (ANP). Acta Physiol Scand. 1992;146:393-398.
De Leon H. and Garcia R. Regulation of glomerular atrial natriuretic factor receptor subtypes by renal 23. sympathetic nerves. Am J Physiol. 1991;260:R1043-R1050.
Charloux A, Piquard F, Doutreleau S, Brandenberger G, and Geny B. Mechanisms of renal 24. hyporesponsiveness to ANP in heart failure. Eur J Clin Invest. 2003;33:769-778.
Stevenson LW and Perloff JK. The limited reliability of physical signs for estimating hemodynamics in 25. chronic heart failure. JAMA. 1989;261:884-888.
Stevenson LW. Design of therapy for advanced heart failure. Eur J Heart Fail. 2005;7:323-331.26.
Nohria A, Tsang SW, Fang JC et al. Clinical assessment identifies hemodynamic profiles that predict 27. outcomes in patients admitted with heart failure. J Am Coll Cardiol. 2003;41:1797-1804.
Binanay C, Califf RM, Hasselblad V et al. Evaluation study of congestive heart failure and pulmonary 28. artery catheterization effectiveness: the ESCAPE trial. JAMA. 2005;294:1625-1633.
Stevenson LW. Are hemodynamic goals viable in tailoring heart failure therapy? Hemodynamic goals 29. are relevant. Circulation. 2006;113:1020-1027.
Reddi BA and Carpenter RH. Venous excess: a new approach to cardiovascular control and its teaching. 30. J Appl Physiol. 2005;98:356-364.
Ahmed A, Rich MW, Sanders PW et al. Chronic kidney disease associated mortality in diastolic versus 31. systolic heart failure: a propensity matched study. Am J Cardiol. 2007;99:393-398.
Leithe ME, Margorien RD, Hermiller JB, Unverferth DV, and Leier CV. Relationship between central 32. hemodynamics and regional blood flow in normal subjects and in patients with congestive heart failure. Circulation. 1984;69:57-64.
Creager MA, Halperin JL, Bernard DB et al. Acute Regional Circulatory and Renal Hemodynamic-33. Effects of Converting-Enzyme Inhibition in Patients with Congestive Heart-Failure. Circulation. 1981;64:483-489.
Hillege HL, van Gilst WH, van Veldhuisen DJ et al. Accelerated decline and prognostic impact of renal 34. function after myocardial infarction and the benefits of ACE inhibition: the CATS randomized trial. Eur Heart J. 2003;24:412-420.

CVP and renal function
57
Weinfeld MS, Chertow GM, and Stevenson LW. Aggravated renal dysfunction during intensive therapy 35. for advanced chronic heart failure. Am Heart J. 1999;138:285-290.
Costanzo MR, Guglin ME, Saltzberg MT et al. Ultrafiltration versus intravenous diuretics for patients 36. hospitalized for acute decompensated heart failure. J Am Coll Cardiol. 2007;49:675-683.
Bart BA, Boyle A, Bank AJ et al. Ultrafiltration versus usual care for hospitalized patients with heart 37. failure: the Relief for Acutely Fluid-Overloaded Patients With Decompensated Congestive Heart Failure (RAPID-CHF) trial. J Am Coll Cardiol. 2005;46:2043-2046.
Gao L, Wang W, Li YL et al. Simvastatin therapy normalizes sympathetic neural control in experimental 38. heart failure: roles of angiotensin II type 1 receptors and NAD(P)H oxidase. Circulation. 2005;112:1763-1770.
Costello-Boerrigter LC, Smith WB, Boerrigter G et al. Vasopressin-2-receptor antagonism augments 39. water excretion without changes in renal hemodynamics or sodium and potassium excretion in human heart failure. Am J Physiol Renal Physiol. 2006;290:F273-F278.
Mitrovic V, Seferovic PM, Simeunovic D et al. Haemodynamic and clinical effects of ularitide in 40. decompensated heart failure. Eur Heart J. 2006;27:2823-2832.
Martin FL, Stevens TL, Cataliotti A et al. Natriuretic and antialdosterone actions of chronic oral NEP 41. inhibition during progressive congestive heart failure. Kidney Int. 2005;67:1723-1730.

58

Chapter 3Increased central venous pressure is associated with impaired
renal function and mortality in a broad spectrum of patients with cardiovascular disease
Kevin Damman, Vincent M. van Deursen, Gerjan Navis, Adriaan A. Voors, Dirk J. van Veldhuisen, Hans L. Hillege
J Am Coll Cardiol. 2009;53:582-588

60
Abstract
BackgroundThe pathophysiology of impaired renal function in cardiovascular disease is multifactorial. The relative
importance of increased CVP has not been addressed before. We aimed to investigate the relationship between increased central venous pressure (CVP), renal function and mortality in a broad spectrum of cardiovascular patients.
Methods and ResultsA total of 2557 patients who underwent right heart catheterization in the University Medical Center
Groningen, The Netherlands between January 1, 1989, and December 31, 2006, were identified and data extracted from electronic databases. Estimated glomerular filtration rate (eGFR) was assessed using the simplified modification of diet in renal disease formula. Mean age was 59 ± 15 years, and 57% was male. Mean eGFR was 65 ± 24 mL/min/1.73m2, with a cardiac index of 2.9 ± 0.8 L/min/m2 and CVP of 5.9 ± 4.3 mmHg. CVP was associated with cardiac index (r = -0.259, P < 0.0001) and eGFR (r = -0.147, P < 0.0001). Also cardiac index was associated with eGFR (r = 0.123, P < 0.0001). In multivariate analysis CVP remained associated with eGFR (r = -0.108, P < 0.0001). In a median follow up time of 10.7 years 741 (29%) of patients died. CVP was an independent predictor of reduced survival (Hazard ratio 1.03 per mmHg increase, 95% confidence interval 1.01 to 1.05, P = 0.0032).
ConclusionsIncreased CVP is associated with impaired renal function and independently related to all cause
mortality in a broad spectrum of patients with cardiovascular disease.

CVP, eGFR and prognosis
61
IntroductionRenal dysfunction is a strong and independent predictor of prognosis in the general
population, but also in patients with diabetes, hypertension, coronary artery disease, and heart failure [1-7]. The pathophysiology is multifactorial and associated with decreased renal perfusion, atherosclerosis and inflammation, endothelial dysfunction and neurohormonal activation [8-10]. We recently showed that in patients with cardiac dysfunction secondary to pulmonary hypertension, not only renal perfusion was a strongly associated with renal function impairment, but also venous congestion [11]. It is however unclear whether this observation is limited to those patients with reduced cardiac function and pulmonary hypertension, or may also be present in patients with a mixture of cardiovascular diseases with varying aetiologies and treatments. In addition, there is only limited data on the relationship between venous congestion, as estimated by central venous pressure (CVP) and the impact on prognosis, even in patients with and without heart failure. The studies that have been conducted are either small or include only non-invasive assessment of increased venous congestion, such as jugular venous pressure [12-15].
In the present study we therefore aimed to investigate the relationship between CVP, renal function and mortality in patients with a mixture of cardiovascular diseases with varying aetiologies and treatments.
Methods
Case IdentificationUsing the patient registration system of the University Medical Center Groningen, The
Netherlands, all patients that underwent right heart catheterization between January 1, 1989, and December 31, 2006, were identified.
Data ExtractionRetrospective chart review was done to analyze characteristics of all patients that were
identified during the electronic search. For each patient, date of birth, gender, race, and weight and height were collected. Comorbid conditions including hypertension, coronary artery disease, cardiac valve disease, congenital heart disease, history of stroke, hypercholesterolemia, and diabetes, medical treatment at the time of catheterization were also extracted for each patient. Furthermore, the reason for performing right heart catheterization was identified. The study was approved by the institutional review board.
Heart catheterizationHemodynamic variables obtained during catheterization included systolic blood pressure
(SBP, mmHg), diastolic blood pressure (DBP, mmHg), cardiac output (thermodilution, L/min) and right atrial pressure as indicator of CVP (CVP, mmHg). Cardiac index (L/min/m2)

62
Chapter 3
was determined as cardiac output divided by the body surface area (BSA). BSA was calculated as 0.007184·weight0.425 ·length0.725. Measurements obtained from cardiac catheterization were obtained from the patient during a resting state.
Renal function measurementSerum creatinine at the day of catheterization was extracted. For the patients who did not
have laboratory measurements on the day of catheterization, measurements obtained within 3 days prior to catheterization were taken as the baseline value. Of patients included in the study, 2282 (89%) had at least one serial creatinine measurement within 3 days of catheterization. Renal function was estimated as glomerular filtration rate by using the simplified modification of diet in renal disease equation (eGFR (mL/min/1.73m2) = 186.3 x (serum creatinine)-1.154 x age-0.203 x (0.742 if female)) [16]. Estimated GFR values over 200 mL/min/1.73 m2 were set equal to 200 mL/min/1.73 m2, according to Coresh et al [17].
Mortality dataSurvival status was determined using the electronic patient registration database of
the University Medical Center Groningen. Follow up started directly after right heart catheterization. The primary endpoint consisted of death from any cause.
Statistical analysisData are given as mean ± standard deviation when normally distributed, as median and
interquartile range when skewed distributed, and as frequencies and percentages for categorical variables. Associations between baseline variables were evaluated by means of 1-way ANOVA, the Kruskal-Wallis test, and χ2 or Fisher exact tests, when appropriate. Two-sided P values were used, taking P < 0.05 to be statistically significant. CVP was divided into tertiles to assess relationships between baseline characteristics and CVP. A fractional polynomial parameterization of exposure was used to explore nonlinearity between different predictors and renal function. In this technique, each exposure value is expressed as a polynomial of degree greater than one (eg. quadratic, cubic, etc), yielding an estimated model with multiple predictors (ie – separate predictors for the linear, quadratic, etc terms respectively). We used a Cox proportional hazards model to estimate hazard ratios (HR) with 95% CI. At first, in multivariate analysis, CVP was fitted into a stepwise multivariate Cox regression analysis on a continuous scale. In secondary analysis, CVP was fitted into the model and in multiple steps, the model was adjusted for other variables and parameters. The internal validity of the regression model was assessed by the bootstrap re-sampling technique [18]. For each of 100 bootstrap samples, the model was re-fitted and tested on the original sample to obtain a bias-corrected estimate of predictive accuracy. Statistical analyses were performed using SPSS, Chicago version 12.0 and STATA, College Station, Texas, version 9.0.

CVP, eGFR and prognosis
63
Results
Baseline characteristicsA total of 3757 right heart catheterizations were carried out between 1989 and 2006. Of
these, 2557 (68%) were first or only right heart catheterization of unique patients and formed the study population. Main reasons for right heart catheterization are shown in table 1. Aortic and mitral valve disorders accounted for 44% of indications, while in 16% acute or chronic heart failure was the predominant reason. Mean age was 59 ± 15 years, and 57% was male (Table 2). In the total study population, both mean cardiac index (2.9 ± 0.8 L/min/m2) and mean CVP (5.9 ± 4.3 mmHg) were within the normal range. The distribution of CVP among the study population is shown in figure 1. Mean eGFR was moderately impaired: 65 ± 24 mL/min/1.73m2.
The distribution of different factors over tertiles of CVP is shown in table 2. Most of the characteristics were equally distributed across tertiles of CVP, except for the highest tertile (CVP > 6 mmHg). Both cardiac output and cardiac index were significantly lower in the highest tertile compared to lower tertiles (P < 0.0001), corresponding to r = -0.259 (P < 0.0001) for the association between CVP and cardiac index. Furthermore, patients in the highest tertile were more frequently treated with ACEi/ARB, beta-blockers, diuretics and aldosterone antagonists. Prevalence of heart failure showed a trend towards increasing with higher tertiles of CVP (P = 0.0781), while also congenital heart disease was more prevalent in the highest tertile. Finally,
Figure 1. Distribution of CVP and curvilinear relationship between CVP and eGFR in the study population. Adjusted for age, gender and cardiac index. Abbreviations: CVP: central venous pressure, eGFR: estimated glomerular filtration rate. The curvilinear model had the following individual polynomial components for the relationship between CVP and eGFR: First order: Y = -25.8 * (CVP+1)/10, (Wald 28.2, P < 0.0001) and second order: Y = 35.7* ((CVP+1)/10)0.5, (Wald 17.4, P < 0.0001).
CVP (mmHg)
0 10 15 20 25 30
eGFR
(mL/
min
/1.7
3m2 )
(95%
CI)
0
10
20
30
40
50
60
70
80
Num
ber
of p
atie
nts
0
100
200
300
400
500
5

64
Chapter 3
Figure 2. Curvilinear relationship between CVP and eGFR according to different values of CI. P = 0.0217 for interaction between CI and CVP on the relationship with eGFR. Abbreviations: CI: cardiac index, CVP: central venous pressure, eGFR: estimated glomerular filtration rate
CVP (mmHg)
0 10 15 20 25 30
eGFR
(mL/
min
/1.7
3m2 )
0
30
40
50
60
70
80
CI < 2.5 L/min/m2
CI 2.5 - 3.2 L/min/m2
CI > 3.2 L/min/m2
5
Table 1. Primary indication for heart catheterization.
Percentage of Patients
Aortic valve disorders 29
Aortic valve stenosis 23
Aortic valve insufficiency 6
Mitral valve disorders 15
Mitral valve insufficiency 14
Mitral valve stenosis 1
Pulmonary valve disorders 1
Pulmonary valve insufficiency 0.8
Pulmonary valve stenosis 0.2
Heart failure 16
Coronary artery disease 13
Pre transplantation (non-cardiac) 11
Rhythm disorders 5
Pulmonary hypertension 3
Congenital heart disease 2
Post heart transplantation 2
Other 6

CVP, eGFR and prognosis
65
eGFR was significantly lower in the highest tertile of CVP, compared to both lower tertiles (P < 0.001).
Curvilinear fitting and the relationship between CVP and eGFRFigure 1 shows the curvilinear relationship between CVP and eGFR in the total study
population as obtained by fractional polynomial modelling. Estimated GFR showed a small increase when CVP increased from 1 to 6 mmHg. However, in CVP values above 6 mmHg, a steep decline is observed with increasing CVP values. This resulted in a partial correlation of r = 0.064, P =0.0218 in patients with CVP ≤ 6 mmHg, and r = -0.212, P < 0.0001 in patients
Table 2. Baseline characteristics according to tertiles of CVP.
Total Tertile 1(0 - 3 mmHg)
Tertile 2(4 - 6 mmHg)
Tertile 3(> 6 mmHg)
P for trend
N 2557 911 855 791
Age (yr) 59 ± 15 60 ± 15 59 ± 15 58 ± 15 0.0032
Gender (% male) 57 59 58 54 NS
SBP (mmHg) 133 ± 29 133 ± 28 134± 27 129 ± 31 0.0100
DBP (mmHg) 68 ± 13 66 ± 12 68 ± 12 69 ± 13 0.0010
CO (L/min) 5.5 ± 1.6 5.7 ± 1.6 5.5 ± 1.5 5.0 ± 1.5 < 0.0001
CI (L/min/m2) 2.9 ± 0.8 3.1 ± 0.7 3.0 ± 0.7 2.7 ± 0.8 < 0.0001
CVP (mmHg) 5.9 ± 4.3 2 ± 1 5 ± 1 11 ± 4 < 0.0001
eGFR (ml/min/1.73 m2) 65 ± 24 65 ± 23 67 ± 24 62 ± 24 0.0001
Medical History (%)
Heart Failure 16 15 15 19 NS
Coronary artery disease 24 24 25 24 NS
Congenital heart disease 5 4 5 7 0.0189
Valve disease 51 50 55 49 NS
Hypercholesterolemia 6 7 5 6 NS
Diabetes mellitus 9 8 8 10 NS
Hypertension 20 21 20 18 NS
Stroke 5 4 5 6 NS
Medication (%)
Diuretics 42 37 38 53 0.0001
Beta-Blocker 28 25 29 31 0.0388
ACEi or ARB 38 36 32 45 0.0001
Aldosterone-antagonist 9 5 6 15 0.0001
Abbreviations: ACEi: angiotensin converting enzyme inhibitor, ARB: angiotensin II receptor blocker, CI: cardiac index, CO: cardiac output, CVP: central venous pressure, DBP: diastolic blood pressure, eGFR: estimated glomerular filtration rate, NS: Not significant, SBP: systolic blood pressure.
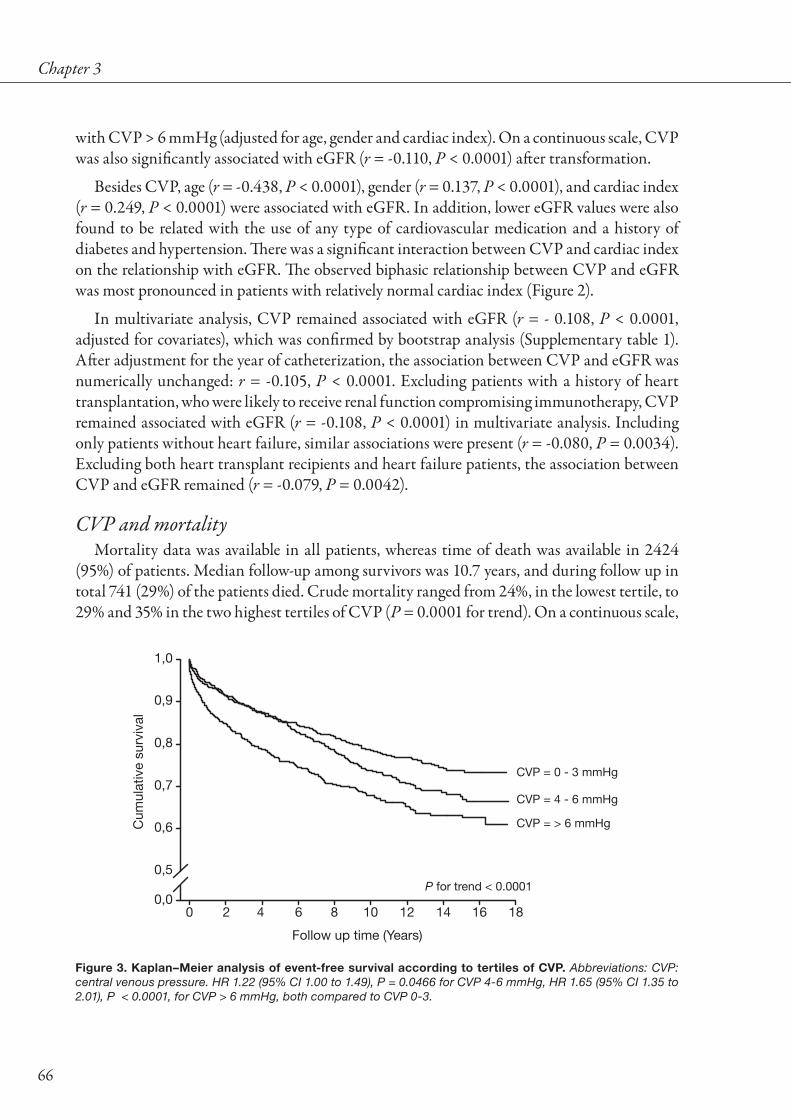
66
Chapter 3
with CVP > 6 mmHg (adjusted for age, gender and cardiac index). On a continuous scale, CVP was also significantly associated with eGFR (r = -0.110, P < 0.0001) after transformation.
Besides CVP, age (r = -0.438, P < 0.0001), gender (r = 0.137, P < 0.0001), and cardiac index (r = 0.249, P < 0.0001) were associated with eGFR. In addition, lower eGFR values were also found to be related with the use of any type of cardiovascular medication and a history of diabetes and hypertension. There was a significant interaction between CVP and cardiac index on the relationship with eGFR. The observed biphasic relationship between CVP and eGFR was most pronounced in patients with relatively normal cardiac index (Figure 2).
In multivariate analysis, CVP remained associated with eGFR (r = - 0.108, P < 0.0001, adjusted for covariates), which was confirmed by bootstrap analysis (Supplementary table 1). After adjustment for the year of catheterization, the association between CVP and eGFR was numerically unchanged: r = -0.105, P < 0.0001. Excluding patients with a history of heart transplantation, who were likely to receive renal function compromising immunotherapy, CVP remained associated with eGFR (r = -0.108, P < 0.0001) in multivariate analysis. Including only patients without heart failure, similar associations were present (r = -0.080, P = 0.0034). Excluding both heart transplant recipients and heart failure patients, the association between CVP and eGFR remained (r = -0.079, P = 0.0042).
CVP and mortalityMortality data was available in all patients, whereas time of death was available in 2424
(95%) of patients. Median follow-up among survivors was 10.7 years, and during follow up in total 741 (29%) of the patients died. Crude mortality ranged from 24%, in the lowest tertile, to 29% and 35% in the two highest tertiles of CVP (P = 0.0001 for trend). On a continuous scale,
Figure 3. Kaplan–Meier analysis of event-free survival according to tertiles of CVP. Abbreviations: CVP: central venous pressure. HR 1.22 (95% CI 1.00 to 1.49), P = 0.0466 for CVP 4-6 mmHg, HR 1.65 (95% CI 1.35 to 2.01), P < 0.0001, for CVP > 6 mmHg, both compared to CVP 0-3.
Follow up time (Years)
0 4 8 10 12 14 16 18
Cum
ulat
ive
surv
ival
0,0
0,5
0,6
0,7
0,8
0,9
1,0
P for trend < 0.0001
CVP = 0 - 3 mmHg
CVP = 4 - 6 mmHg
CVP = > 6 mmHg
2 6

CVP, eGFR and prognosis
67
higher CVP levels were associated with impaired survival (HR = 1.05 per mmHg increase, 95% CI 1.04 to 1.07, P < 0.0001). Kaplan Meier survival curves for tertiles of CVP are shown in figure 3, showing that especially patients with the highest CVP were at risk for increased mortality (HR for CVP > 6 mmHg versus ≤ 6 mmHg 1.49, 95% CI 1.26 to 1.76, P < 0.0001). Baseline eGFR (HR = 1.09 per 10 mL/min/1.73m2 decrease, 95% CI 1.05 to 1.13, P < 0.0001) and cardiac index (HR = 0.74 per L/min/m2, 95% CI 0.66 to 0.84, P < 0.0001) were also strong predictors of mortality. Other factors associated with reduced survival are shown in table 3. In stepwise multivariate Cox regression analysis, CVP remained significantly associated with reduced survival (HR = 1.03 per mmHg increase, 95% CI 1.01 to 1.05, P = 0.0032) (table 3). Finally, we fitted a second model, adjusting CVP for other covariates (Supplementary Table 2). CVP remained an independent predictor of impaired survival (HR = 1.03 per mmHg increase, 95% CI 1.01 to 1.05, P = 0.0144). To account for the effects of changing therapy during the study inclusion time, we adjusted for the year of catheterization. This secondary analysis yielded similar results: HR = 1.03 per mmHg increase, 95% CI 1.00 to 1.05, P = 0.0207). Excluding patients with known heart failure and heart transplant recipients, CVP was still associated with mortality (HR = 1.03 per 5 mmHg increase, 95% CI 1.00 to 1.06, P = 0.0369).
Table 3. Multivariate stepwise Cox regression model.
Univariate Multivariate
Variables Hazard ratio (95% CI) P-value Hazard ratio (95% CI) Wald statistic P-value
Age (per 10 year increase) 1.05 (1.00 – 1.10) 0.0880 1.21 (1.12 – 1.31) 25.4 < 0.0001
Gender 1.03 (0.88 – 1.20) 0.713
Cardiac Index (per L/min/m2 increase) 0.74 (0.66 – 0.84) < 0.0001 0.81 (0.71 – 0.93) 9.1 0.0026
CVP (per mmHg increase) 1.05 (1.04 – 1.07 < 0.0001 1.03 (1.01 – 1.05) 8.7 0.0032
eGFR (per 10 mL/min/1.73m2 decrease) 1.09 (1.05 – 1.13) < 0.0001
Medication
Diuretic use 1.43 (1.22 – 1.67) < 0.0001
ACEi or ARB use 1.29 (1.10 – 1.52) 0.0017
Aldosterone Antagonist use 1.92 (1.50 – 2.45) < 0.0001 1.50 (1.10 – 2.02) 6.9 0.0087
Medical History (%)
Coronary artery disease 1.26 (1.05 – 1.50) 0.0112
Diabetes mellitus 1.83 (1.44 – 2.31) < 0.0001 1.76 (1.34 – 2.31) 16.6 < 0.0001
Indication for catheterization
Aorta insufficiency 0.63 (0.43 – 0.93) 0.0203
Congenital heart disease 0.36 (0.17 – 0.77) 0.0078
Pre transplantation (non-cardiac) 1.50 (1.21 – 1.85) < 0.0001 2.54 (1.90 – 3.40) 39.2 < 0.0001
Heart failure 1.48 (1.21 – 1.85) 0.0001 1.71 (1.34 – 2.20) 18.1 < 0.0001
Rhythm disorders 0.49 (0.29 – 0.82) 0.0062
Abbreviations: ACEi: angiotensin converting enzyme inhibitor, ARB: angiotensin II receptor blocker, CVP: cen-tral venous pressure, eGFR: estimated glomerular filtration rate.

68
Chapter 3
DiscussionThe present study shows that increased CVP is associated with impaired renal function in
a broad spectrum of cardiovascular patients who underwent right heart catheterization. In addition, the slope between CVP and impaired eGFR was steeper with relatively preserved cardiac function. Finally, an increased CVP was a strong and independent determinant of all cause mortality, which was especially observed in patients with a CVP above 6 mmHg.
There is only limited data on the relationship between increased CVP and renal impairment. Studies in animals have shown that raising renal venous pressure leads to a reduction in glomerular filtration, which was probably mediated by a decreased renal perfusion [19]. Renal vein constriction lead to a drop in glomerular filtration rate in rats [20], while renal function decreased when renal vein pressure was increased in dogs, but only when cardiac output was reduced [21]. We recently showed that in patient with reduced cardiac function, secondary to pulmonary hypertension, increased CVP was strongly associated with renal impairment, especially when renal perfusion was already impaired [11]. Early studies on increased renal vein pressure in heart failure patients and animals showed a marked reduction in renal blood flow as well as water and salt excretion [22,23], but the effect on GFR was not uniform. One small report showed a strong relationship between CVP and renal blood flow in advanced heart failure [24].
Drazner et al reported that in patients with elevated jugular venous pressure on examination, serum creatinine was significantly higher [13]. In patients who underwent elective cardiac surgery, preoperative presence of high CVP was a strong predictor of the occurrence of acute renal injury, independent of the presence of low CO [25].
However, especially in patients with preserved cardiac function, data regarding the relationship between CVP and renal function is scarce. Diastolic dysfunction, a disease characterised by elevated filling pressures, often co-exists with renal failure and vise versa [26-28]. Interestingly, a recent study showed that renal dysfunction is even more important in defining mortality risk in patients with preserved cardiac function compared to those with systolic dysfunction. [29,30]. Our present study confirmed that increased CVP is an important risk factor for decreased renal function in patients with preserved and decreased cardiac function.
Curvilinear effect of CVP with eGFRWe observed a biphasic relationship between eGFR and increasing CVP. In the physiologic
ranges of CVP, up to 6 mmHg, eGFR increases gradually. This subtle increase in eGFR may be a reflection of increased cardiac filling to preserve cardiac function by Frank-Starling mechanism (preload), and subsequent renal perfusion [31]. This gradual increase in eGFR was observed across the full spectrum of low to high cardiac index. We observed a decrease in eGFR when CVP rises above 6 mmHg. In these patients, the equilibrium between venous return, CO and CVP may have shifted towards a plateau phase or optimum, where CO is not further increased in response to higher CVP [32]. Higher CVP levels will then decrease renal perfusion pressure

CVP, eGFR and prognosis
69
which will further impair eGFR. However, if higher CVP levels preserve CO, and despite this mechanism eGFR drops with higher CVP, this suggests that increased CVP may also exert an effect on GFR in this group of patients, independent of renal perfusion.
Importantly, the relationship between CVP and GFR is bound to be bidirectional. Not only CVP may influence GFR, but even mildly impaired renal function may initiate salt and water retention, resulting in elevated filling pressures [33]. Due to the cross-sectional nature of our analysis, we were unable to investigate the cause effect associations, and our present analysis must be regarded as hypothesis generating.
CVP and eGFR in patients with and without cardiac dysfunctionOf particular interest is the interaction between cardiac index and CVP on eGFR. Patients
who have a combination of reduced perfusion (cardiac index) and increased venous congestion (CVP) suffer from fluid overload and decreased organ perfusion, leading among other things to renal dysfunction. We showed that patients with high CVP levels often also have decreased cardiac index and reduced eGFR. Remarkably, CVP and cardiac index showed an interaction on the relationship with eGFR, with an even more pronounced relationship in patients with relatively normal cardiac index. This further strengthens the observation of a relationship between CVP and eGFR, which is not exclusively due to reduced cardiac systolic function. It also challenges the intuitive notion that fluid overload, although deleterious from a cardiovascular perspective, will invariably be beneficial from the point of view of preservation of renal function. As our analysis does not allow to dissect cause and effect relationships, however, it might also well be that the relatively normal cardiac index is maintained at the expense of the elevated CVP. In that case, apparently, such a renal hemodynamic profile does not translate into better renal function. Our present findings seem inconsistent with our earlier findings, showing that especially patients with reduced renal perfusion are prone to a detrimental effect of CVP on GFR [11]. However, we did not measure renal hemodynamics or renal function by clearance techniques, while also the population was entirely different. Furthermore, our previous study consisted of patients with much lower cardiac indices, all of which makes comparison difficult. Nevertheless, this inconsistency needs further addressing in future studies.
New therapeutic agents in especially acute heart failure, which are specifically targeted at improvement of cardiac function and reducing venous congestion have recently shown promising results regarding renal function. A substudy of the SURVIVE study, comparing levosimendan with dobutamine, showed that improvement of renal function was more pronounced in the group receiving levosimendan, despite the obvious positive inotropic effect of dobutamine [34]. The specific venodilatory effect observed with Levosimendan, with subsequent reduced central venous pressure may be the pathophysiologic mechanism, supporting a direct pathophysiologic link between CVP and renal impairment.[35]
CVP and mortalityIncreased CVP and jugular venous pressure predispose to the development of heart failure
in patients with cardiac dysfunction and have been associated with reduced survival in patients

70
Chapter 3
with heart failure [12,13,36]. Small studies have shown that invasive assessment of CVP is a predictor of cardiovascular outcome in patients with advanced heart failure [14,15]. In other selected patients populations, such as patients who underwent fontan surgery or lung transplantation, higher central venous pressures were strong predictors of outcome [37,38]. The prognostic importance of raised CVP in patients with normal cardiac function has not been reported before. We show that in a selected patient population, raised CVP remained a determinant of all-cause mortality, independent of cardiac function. This association with mortality was most prominent in patients with severely increased CVP, even after adjustment for other baseline characteristics. This finding was additive to the observation that higher CVP levels predispose to lower eGFR, which may influence survival by different mechanisms.
Clinical implicationsRaised CVP is frequently observed in patients with and without reduced cardiac function,
comprising of almost 20% of patients in the present patient population. Recognition of these patients is essential, while not only renal dysfunction is much more frequently observed, but also mortality risk rises with increasing CVP levels. Treatment to selectively lower CVP may be favourable to reduce symptoms and signs of congestion, improve GFR, and improve prognosis.
LimitationsThe present study comprises of a selected patient population, who all had a specific indication
for right heart catheterization. Patients undergoing right heart catheterization are prone to have higher right sided filling pressures, and the present observations may therefore not represent the general cardiovascular population. However, this is a large cohort study, with invasive cardiac function and CVP measurements across the full range. Second, this is a retrospective analysis, and no invasive data is available on renal blood flow and true GFR in these patients. Furthermore, it should be addressed that our study population had very different catheterization indications, medical history and medication regime, all of which could have influenced our results. In our study, the presence of heart failure was a clinical diagnosis, rather than related to reduced cardiac index on catheterization. Therefore, the prevalence of heart failure in our study is most likely underestimated. The relationship between increased CVP and renal perfusion has been observed in heart failure. However our present study is the first to show an independent effect of CVP on renal function. Finally, the retrospective nature of this study and the cross sectional design limit the ability to investigate the cause-effect relationship between renal impairment and increased CVP, that may actually mutually influence each other.
ConclusionIncreased CVP is associated with impaired renal function and independently related to all
cause mortality in a broad spectrum of patients with cardiovascular disease.

CVP, eGFR and prognosis
71
ReferencesGo AS, Chertow GM, Fan D, McCulloch CE, and Hsu CY. Chronic kidney disease and the risks of 1. death, cardiovascular events, and hospitalization. N Engl J Med. 2004;351:1296-1305.
Ruilope LM, van Veldhuisen DJ, Ritz E, and Luscher TF. Renal function: The Cinderella of cardiovascular 2. risk profile. J Am Coll Cardiol. 2001;38:1782-1787.
Hillege HL, van Gilst WH, van Veldhuisen DJ et al. Accelerated decline and prognostic impact of renal 3. function after myocardial infarction and the benefits of ACE inhibition: the CATS randomized trial. Eur Heart J. 2003;24:412-420.
Nag S, Bilous R, Kelly W et al. All-cause and cardiovascular mortality in diabetic subjects increases 4. significantly with reduced estimated glomerular filtration rate (eGFR): 10 years’ data from the South Tees Diabetes Mortality study. Diabet Med. 2007;24:10-17.
Rahman M, Pressel S, Davis BR et al. Renal outcomes in high-risk hypertensive patients treated with 5. an angiotensin-converting enzyme inhibitor or a calcium channel blocker vs a diuretic: a report from the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). Arch Intern Med. 2005;165:936-946.
Reddan DN, Szczech LA, Tuttle RH et al. Chronic kidney disease, mortality, and treatment strategies 6. among patients with clinically significant coronary artery disease. J Am Soc Nephrol. 2003;14:2373-2380.
Hillege HL, Nitsch D, Pfeffer MA et al. Renal function as a predictor of outcome in a broad spectrum 7. of patients with heart failure. Circulation. 2006;113:671-678.
Ljungman S, Laragh JH, and Cody RJ. Role of the Kidney in Congestive Heart-Failure - Relationship 8. of Cardiac Index to Kidney-Function. Drugs. 1990;39:10-21.
Stam F, van Guldener C, Schalkwijk CG et al. Impaired renal function is associated with markers of 9. endothelial dysfunction and increased inflammatory activity. Nephrol Dial Transplant. 2003;18:892-898.
Zoccali C, Mallamaci F, and Tripepi G. Inflammation and atherosclerosis in end-stage renal disease. 10. Blood Purif. 2003;21:29-36.
Damman K, Navis G, Smilde TD et al. Decreased cardiac output, venous congestion and the association 11. with renal impairment in patients with cardiac dysfunction. Eur J Heart Fail. 2007;9:872-878.
Unverferth DV, Magorien RD, Moeschberger ML et al. Factors influencing the one-year mortality of 12. dilated cardiomyopathy. Am J Cardiol. 1984;54:147-152.
Drazner MH, Rame JE, Stevenson LW, and Dries DL. Prognostic importance of elevated jugular venous 13. pressure and a third heart sound in patients with heart failure. N Engl J Med. 2001;345:574-581.
Gardner RS, Henderson G, and McDonagh TA. The prognostic use of right heart catheterization data 14. in patients with advanced heart failure: how relevant are invasive procedures in the risk stratification of advanced heart failure in the era of neurohormones? J Heart Lung Transplant. 2005;24:303-309.
Morley D and Brozena SC. Assessing risk by hemodynamic profile in patients awaiting cardiac 15. transplantation. Am J Cardiol. 1994;73:379-383.

72
Chapter 3
Smilde TD, van Veldhuisen DJ, Navis G, Voors AA, and Hillege HL. Drawbacks and prognostic value 16. of formulas estimating renal function in patients with chronic heart failure and systolic dysfunction. Circulation. 2006;114:1572-1580.
Coresh J, Selvin E, Stevens LA et al. Prevalence of chronic kidney disease in the United States. JAMA. 17. 2007;298:2038-2047.
Harrell FE, Jr., Lee KL, Califf RM, Pryor DB, and Rosati RA. Regression modelling strategies for 18. improved prognostic prediction. Stat Med. 1984;3:143-152.
Doty JM, Saggi BH, Sugerman HJ et al. Effect of increased renal venous pressure on renal function. J 19. Trauma. 1999;47:1000-1003.
Stolarczyk J and Carone FA. Effects of renal lymphatic occlusion and venous constriction on renal 20. function. Am J Pathol. 1975;78:285-296.
Priebe HJ, Heimann JC, and Hedley-Whyte J. Effects of renal and hepatic venous congestion on renal 21. function in the presence of low and normal cardiac output in dogs. Circ Res. 1980;47:883-890.
Maxwell MH, Breed ES, and Schwartz IL. Renal venous pressure in chronic congestive heart failure. J 22. Clin Invest. 1950;29:342-348.
Blake WD, Wegria R, Keating RP, and Ward HP. Effect of increased renal venous pressure on renal 23. function. Am J Physiol. 1949;157:1-13.
Kos T, Pacher R, Wimmer A et al. Relationship between kidney function, hemodynamic variables 24. and circulating big endothelin levels in patients with severe refractory heart failure. Wiener Klinische Wochenschrift. 1998;110:89-95.
Palomba H, de Castro I, Neto AL, Lage S, and Yu L. Acute kidney injury prediction following elective 25. cardiac surgery: AKICS Score. Kidney Int. 2007;72:624-631.
Bruch C, Rothenburger M, Gotzmann M et al. Chronic kidney disease in patients with chronic 26. heart failure--impact on intracardiac conduction, diastolic function and prognosis. Int J Cardiol. 2007;118:375-380.
Miyazato J, Horio T, Takiuchi S et al. Left ventricular diastolic dysfunction in patients with chronic 27. renal failure: impact of diabetes mellitus. Diabet Med. 2005;22:730-736.
Ogata C, Horio T, Kamide K, Takiuchi S, and Kawano Y. Association between left ventricular diastolic 28. dysfunction and renal hemodynamic change in patients with treated essential hypertension. Hypertens Res. 2003;26:971-978.
Ahmed A, Rich MW, Sanders PW et al. Chronic kidney disease associated mortality in diastolic versus 29. systolic heart failure: a propensity matched study. Am J Cardiol. 2007;99:393-398.
Bruch C, Reinecke H, Rothenburger M et al. Transmitral flow patterns and the presence of chronic 30. kidney disease provide independent and incremental prognostic information in patients with heart failure and systolic dysfunction. J Am Soc Echocardiogr. 2007;20:989-997.
Guyton AC and Jones CE. Central venous pressure: physiological significance and clinical implications. 31. Am Heart J. 1973;86:431-437.
Magder S and Bafaqeeh F. The clinical role of central venous pressure measurements. J Intensive Care 32. Med. 2007;22:44-51.

CVP, eGFR and prognosis
73
Schrier RW. Role of diminished renal function in cardiovascular mortality: marker or pathogenetic 33. factor? J Am Coll Cardiol. 2006;47:1-8.
Yilmaz MB, Yalta K, Yontar C et al. Levosimendan improves renal function in patients with acute 34. decompensated heart failure: comparison with dobutamine. Cardiovasc Drugs Ther. 2007;21:431-435.
Damman K and Voors AA. Levosimendan improves renal function in acute decompensated heart 35. failure: cause and clinical application. Editorial to: „Levosimendan improves renal function in patients with acute decompensated heart failure: comparison with dobutamine by Yilmaz et al.“. Cardiovasc Drugs Ther. 2007;21:403-404.
Drazner MH, Rame JE, and Dries DL. Third heart sound and elevated jugular venous pressure as 36. markers of the subsequent development of heart failure in patients with asymptomatic left ventricular dysfunction. Am J Med. 2003;114:431-437.
Khairy P, Fernandes SM, Mayer JE, Jr. et al. Long-Term Survival, Modes of Death, and Predictors of 37. Mortality in Patients With Fontan Surgery. Circulation. 2008;117:13-15.
Pilcher DV, Scheinkestel CD, Snell GI et al. High central venous pressure is associated with prolonged 38. mechanical ventilation and increased mortality after lung transplantation. J Thorac Cardiovasc Surg. 2005;129:912-918.
74
Chapter 3
Supplementary Table 1. Regression analysis for eGFR.
Regression analysis Bootstrapped analysis
B SE BCorrelation coefficient
P-valueBoot-
strapped SEz-statistic P-value
Model 1 *
Cardiac Index 3.397 0.634 0.123 < 0.0001 0.779 4.36 < 0.0001
CVP† -3.068 0.476 -0.147 < 0.0001 0.484 -6.34 < 0.0001
Adjusted R2 0.249 < 0.0001 0.249 < 0.0001
Model 2 ‡
Cardiac Index 2.140 0.661 0.080 0.0012 0.791 2.71 0.0068
CVP* -2.146 0.496 -0.107 < 0.0001 0.482 -4.45 < 0.0001
Adjusted R2 0.254 < 0.0001 0.254 < 0.0001
Model 3 §
Cardiac Index 2.219 0.661 0.083 0.0008 0.813 2.73 0.0063
CVP* -2.203 0.496 -0.110 < 0.0001 0.476 -4.62 < 0.0001
Adjusted R2 0.260 < 0.0001 0.260 < 0.0001
Model 4 ||
Cardiac Index 1.885 0.662 0.071 0.0045 0.803 2.35 0.0189
CVP* -2.163 0.496 -0.108 < 0.0001 0.489 -4.60 < 0.0001
Adjusted R2 0.279 < 0.0001 0.279 < 0.0001
† CVP transformed as follows: ((CVP+1)/10)2 – 0.457* Adjusted for age and gender‡ Adjusted for age, gender, ACEi/ARB use, diuretic use, aldosterone antagonist use, and beta blocker use.§ Adjusted for age, gender, ACEi/ARB use, diuretic use, aldosterone antagonist use, and beta blocker use his-tory of hypertension, diabetes mellitus, coronary artery disease, congenital heart disease, valve disease, stroke and hypercholesterolemia.|| Adjusted for age, gender, ACEi/ARB use, diuretic use, aldosterone antagonist use, and beta blocker use his-tory of hypertension, diabetes mellitus, coronary artery disease, congenital heart disease, valve disease, stroke and hypercholesterolemia, and indication for right heart catheterization.Abbreviations: CVP: central venous pressure
CVP, eGFR and prognosis
75
Supplementary Table 2. Multivariate Cox regression analysis for all cause mortality.
Model Hazard ratio (95% CI) Wald statistic P-value
CVP (unadjusted)* 1.05 (1.04 – 1.07) 38.1 < 0.0001
Adjusted for age and gender* 1.06 (1.04 – 1.07) 42.1 < 0.0001
Adjusted for preceding and cardiac index* 1.05 (1.03 – 1.07) 20.7 < 0.0001
Adjusted for preceding and eGFR* 1.04 (1.02 – 1.06) 14.0 < 0.0001
Adjusted for preceding and medication* 1.03 (1.00 – 1.05) 5.7 0.0170
Adjusted for preceding and medical history* 1.03 (1.00 – 1.05) 5.6 0.0175
Adjusted for preceding and indication for right heart catheterization* 1.03 (1.01 – 1.05) 6.0 0.0144
* per 1 mmHg increase. Abbreviations: CVP: central venous pressure, eGFR: estimated glomerular filtration rate

76

Chapter 4Venous congestion in chronic systolic heart failure is related to renal
dysfunction and increased mortality
Kevin Damman, Adriaan A. Voors, Hans L. Hillege, Gerjan Navis, Philippe Lechat, Dirk J. van Veldhuisen, Henry J. Dargie
Submitted

78
Abstract
BackgroundIt has been suggested that invasively determined central venous pressure is associated with renal
impairment in patients with cardiac dysfunction. It is however unclear whether signs of venous congestion are also related to renal impairment and whether they are independently related to outcome in chronic heart failure (CHF) patients.
Methods and ResultsThe cardiac insufficiency bisoprolol study II (CIBIS-II) included 2647 NYHA class III/IV CHF
patients. Venous congestion was estimated by the presence of elevated jugular venous pressure, orthopnea, ascites or edema. Glomerular filtration rate (eGFR) was estimated using the sMDRD formula. The primary endpoint was all-cause mortality. Mean age was 61 ± 11 years, 80% was male, and mean eGFR was 77 ± 31 mL/min/1.73m2. A total of 1234 (47%) patients had at least one sign, and 10% of patients had > 3 signs of congestion. Mean eGFR was lower in patients with signs of congestion (75 ± 30 vs 79 ± 31 mL/min/1.73m2, P < 0.001). In multivariate analysis, signs of congestion remained independently related to eGFR (P = 0.016). Mortality rates almost doubled from no signs to ≥ 3 signs of congestion (11% vs 20%, respectively, P < 0.0001). The presence of venous congestion was also independently associated with a worse outcome. Adjusted hazard ratio (HR) was 1.42 (1.14 – 1.77), P < 0.01) for mortality and 1.21 (1.05 – 1.39), P < 0.01 for the composite endpoint.
ConclusionSigns and symptoms of venous congestion are associated with renal impairment and are independent
determinants of prognosis in patients with CHF.

Signs of congestion, eGFR and prognosis
79
IntroductionSymptoms and signs of venous congestion are important targets for therapy in patients with
chronic systolic heart failure (CHF), and have a profound effect on quality of life.[1] However, the association of venous congestion with prognosis in CHF has only been investigated in a limited number of studies. Drazner et al established the independent prognostic value of an elevated jugular venous pressure (JVP) or a third heart sound in a substudy of the SOLVD trial.[2] A simple score of symptoms, including elevated JVP, peripheral edema, and radiologic signs of congestion were independent predictors of mortality in the DIG trial.[3] However, the SOLVD patients were recruited twenty years ago with only half of patients on angiotensin converting enzyme inhibition (ACEi). In addition, only few were on beta-blocker therapy while in the DIG trial beta-blocker use was not registered. Furthermore, the substudy of the DIG did not specifically address signs and symptoms of venous congestion, but also more general symptoms such as angina, fatigue, limited activity and dyspnea.
One of the most important prognostic factors in CHF is renal impairment.[4,5] The pathophysiology of renal dysfunction in CHF is mainly characterised by decreased renal perfusion.[6] However, we recently that increased venous congestion, measured invasively during right heart catheterisation, is an important determinant of renal impairment in patients with and without cardiac dysfunction.[7,8] It is unclear whether the presence of congestive symptoms may predispose to worse renal function, and subsequent prognosis.
The aims of the present study are twofold. First, we aimed to investigate the relationship between the presence of venous congestion and renal impairment and second, we aimed to determine the independent predictive value of signs of venous congestion, in a large modern cohort of CHF patients.
MethodsThe main results of the Cardiac Insufficiency Bisoprolol Study II (CIBIS-II) has previously
been published.[9] A total of 2647 patients with class III or IV New York Heart Association (NYHA) heart failure and left ventricular ejection fraction of ≤ 35% were randomly assigned to receive either bisoprolol or placebo. Patients needed a diagnosis of chronic heart failure, made at least 3 months previously, with clinical stability during the preceding 6 weeks for heart failure or 3 months for acute myocardial infarction or unstable angina. Cardiovascular therapy had to be unchanged in the 2 weeks before randomisation. Treatment had to include a diuretic and an angiotensin-converting-enzyme (ACE) inhibitor, in case of intolerance for ACE inhibition, other vasodilators were accepted. Patients with renal failure as defined by serum creatinine above 3.4 mg/dL (300 µmol/L) were excluded.

80
Chapter 4
Demographic variablesBaseline measurements included height, weight, blood pressure, heart rate, NYHA
classification, left ventricular ejection fraction (LVEF), use of concomitant medication, medical history and serum creatinine. Renal function was estimated as glomerular filtration rate (eGFR) by using the simplified modification of diet in renal disease equation (eGFR (mL/min/1.73m2) = 186.3 x (serum creatinine)-1.154 x age-0.203 x (0.742 if female))[10]. Estimated GFR values over 200 mL/min/1.73 m2 were set equal to 200 mL/min/1.73 m2, according to Coresh et al.[11] Renal function was categorized according to the Kidney Disease Outcomes Quality Initiative (K/DOQI) classification of chronic kidney disease for visual presentation of the effect of renal function on prognosis.
Assessment of signs and symptoms of venous congestionAt baseline visit, investigators evaluated the presence or absence of signs and symptoms of
venous congestion. These included the following: raised JVP, presence of ascites, presence of peripheral edema and presence of orthopnea (all yes versus no). Presence or absence of signs or symptoms of venous congestion was classified either as any sign of venous congestion or number of signs of venous congestion. The latter was divided into no signs, 1 sign, 2 signs or ≥ 3 signs of venous congestion. Via this approach, different combinations of individual signs of congestion are possible; therefore the observed effect estimates represent the overall estimate across different possible combinations of signs of congestion. This approach was chosen for simplicity reasons, rather than pathophysiological reasons. To address the effect on outcome of some of these differences, we carried out secondary analyses.
OutcomeThe primary endpoint of the CIBIS-II trial was all-cause mortality. Secondary endpoints
included all-cause hospital admissions, cardiovascular hospital admissions, a combined endpoint of cardiovascular mortality and cardiovascular hospital admissions, sudden death and death from pump failure.
Statistical analysisData are given as mean ± standard deviation when normally distributed, as median and
interquartile range when skewed distributed, and as frequencies and percentages for categorical variables. Associations between baseline variables were evaluated by means of 1-way ANOVA, the Kruskal-Wallis test, and χ2 or Fisher exact tests, when appropriate. Two-sided P values were used, taking P < 0.05 to be statistically significant. We used a Cox proportional hazards model to estimate hazard ratios with 95% CI. Signs and symptoms of venous congestion were entered into the models as categorical variables. Because of the relatively small number of patients with one specific individual sign of congestion, signs and symptoms of congestion were entered into the model without correction for the presence of other signs or symptoms. The presented effect measurements are therefore a representation of the effect when at least the studied parameter is present. To further address specific combinations of signs and symptoms

Signs of congestion, eGFR and prognosis
81
of congestion on outcome, secondary analyses were carried out to investigate the contribution of specific combinations. Statistical analyses were performed using SPSS, Chicago version 12.0 and STATA, College Station, Texas, version 10.0.
ResultsData on signs and symptoms of venous congestion were available in 2643 (99.8%) of
patients. Mean age of the total study population was 61 ± 11 years, 80% was male, and mean eGFR was 77 ± 31 mL/min/1.73m2. Baseline characteristics of patients with or without signs of venous congestion are shown in table 1. In total 1234 (47%) of patients had at least one sign of venous congestion. Of these, 594 (22%) had an elevated JVP, 79 (3%) had ascites, 687 (26%) had peripheral edema, and 513 (19%) patients experienced orthopnea. Patients with any sign of venous congestion were more often female, and had more co-morbidities, including diabetes, hypertension and previous myocardial infarction. Patients with signs of venous congestion more frequently had symptoms of hemodynamic distress, as indicated by higher NYHA functional class heart failure, higher heart rate, but also lower eGFR and higher serum creatinine. There was no difference in the presence of congestive signs and the assignment to bisoprolol or placebo, while the frequency of diuretic use was higher in patients with congestive signs. In table 2 the range of congestive symptoms in relation to baseline characteristics is depicted. Across patients with higher frequencies of congestive symptoms, patients had more severe heart failure, indicated by more often NYHA class IV and higher heart rate.
Figure 1. Relationship between estimated GFR at baseline and number of signs of congestion. Abbreviations: eGFR: estimated glomerular filtration rate
eGFR (mL/min/1.73m2)
< 30 30 - 60 60 - 90 90 -120 120 - 150 >150
% o
f pat
ient
s
-75
-50
-25
0
25
50
751 sign of congestion
2 signs of congestion
≥3 signs of congestion
no signs of congestion
P = 0.007 for trend
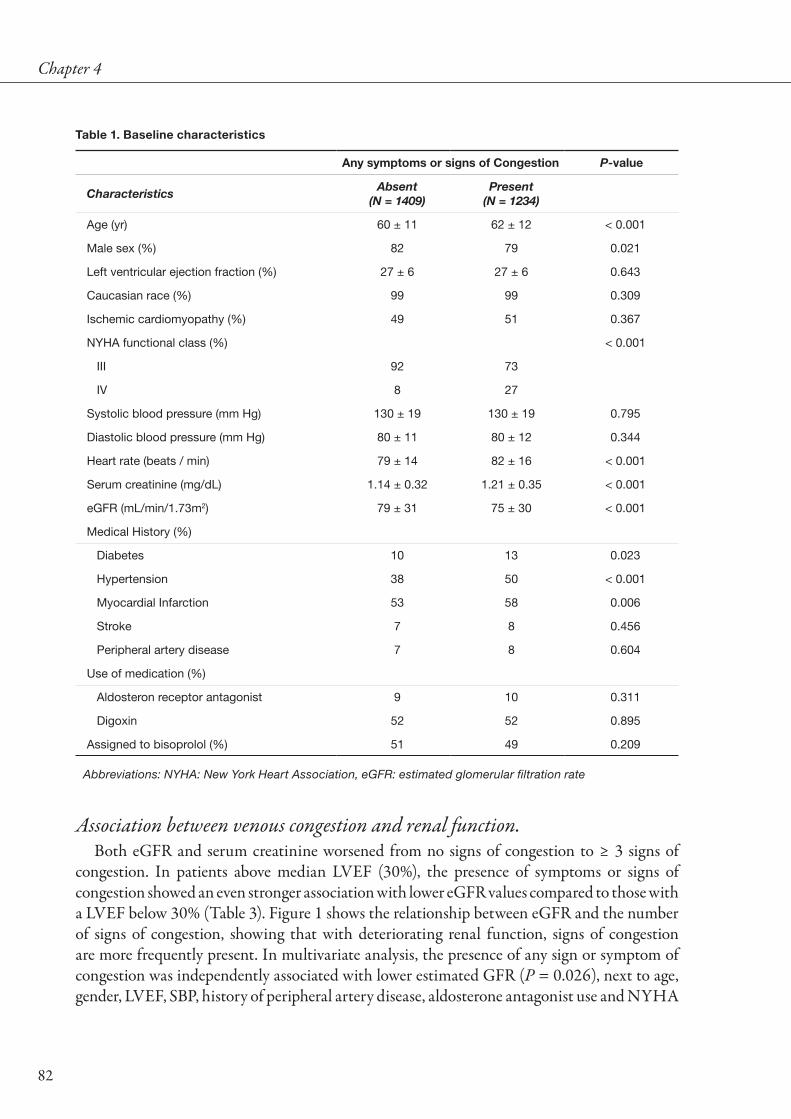
82
Chapter 4
Association between venous congestion and renal function.Both eGFR and serum creatinine worsened from no signs of congestion to ≥ 3 signs of
congestion. In patients above median LVEF (30%), the presence of symptoms or signs of congestion showed an even stronger association with lower eGFR values compared to those with a LVEF below 30% (Table 3). Figure 1 shows the relationship between eGFR and the number of signs of congestion, showing that with deteriorating renal function, signs of congestion are more frequently present. In multivariate analysis, the presence of any sign or symptom of congestion was independently associated with lower estimated GFR (P = 0.026), next to age, gender, LVEF, SBP, history of peripheral artery disease, aldosterone antagonist use and NYHA
Table 1. Baseline characteristics
Any symptoms or signs of Congestion P-value
CharacteristicsAbsent
(N = 1409)Present
(N = 1234)
Age (yr) 60 ± 11 62 ± 12 < 0.001
Male sex (%) 82 79 0.021
Left ventricular ejection fraction (%) 27 ± 6 27 ± 6 0.643
Caucasian race (%) 99 99 0.309
Ischemic cardiomyopathy (%) 49 51 0.367
NYHA functional class (%) < 0.001
III 92 73
IV 8 27
Systolic blood pressure (mm Hg) 130 ± 19 130 ± 19 0.795
Diastolic blood pressure (mm Hg) 80 ± 11 80 ± 12 0.344
Heart rate (beats / min) 79 ± 14 82 ± 16 < 0.001
Serum creatinine (mg/dL) 1.14 ± 0.32 1.21 ± 0.35 < 0.001
eGFR (mL/min/1.73m2) 79 ± 31 75 ± 30 < 0.001
Medical History (%)
Diabetes 10 13 0.023
Hypertension 38 50 < 0.001
Myocardial Infarction 53 58 0.006
Stroke 7 8 0.456
Peripheral artery disease 7 8 0.604
Use of medication (%)
Aldosteron receptor antagonist 9 10 0.311
Digoxin 52 52 0.895
Assigned to bisoprolol (%) 51 49 0.209
Abbreviations: NYHA: New York Heart Association, eGFR: estimated glomerular filtration rate

Signs of congestion, eGFR and prognosis
83
class. Finally, the number of signs present was independently related to lower eGFR values in multivariate analysis (P = 0.009).
Prognosis.Table 4 shows the incidence of endpoints, according to the presence of each sign of congestion,
and number of signs of congestion. The presence of elevated JVP, ascites, peripheral edema, orthopnea and the number of signs present were associated with significantly more deaths, hospitalisations, deaths from pump failure and the occurrence of the combined endpoint. The
Table 2. Baseline characteristics according to number of signs of congestion
Number of signs of congestion P-value
Characteristics0
(N = 1409)1
(N = 774)2
(N = 305)≥3
(N = 155)
Age (yr) 60 ± 11 61 ± 10 62 ± 10 62 ± 10 0.002
Male gender (%) 82 80 76 80 0.067
Left ventricular ejection fraction (%) 27 ± 6 27 ± 6 27 ± 6 28 ± 6 0.556
Caucasian race (%) 99 99 99 100 0.424
Ischemic cardiomyopathy (%) 49 50 52 53 0.720
NYHA functional class (%) < 0.001
III 92 81 69 39
IV 8 19 31 61
Systolic blood pressure (mm Hg) 130 ± 19 130 ± 19 128 ± 19 130 ± 26 0.675
Diastolic blood pressure (mm Hg) 80 ± 11 80 ± 11 79 ± 11 81 ± 13 0.293
Heart rate (beats / min) 79 ± 14 81 ± 15 82 ± 15 85 ± 17 < 0.001
Serum creatinine (µmol/L) 1.14 ± 0.32 1.19 ± 0.34 1.22 ± 0.35 1.24 ± 0.38 < 0.001
eGFR (mL/min/1.73m2) 79 ± 31 76 ± 30 73 ± 29 74 ± 31 0.001
Medical History (%)
Diabetes 10 12 14 16 0.057
Hypertension 38 48 51 56 < 0.001
Myocardial Infarction 53 58 55 65 0.007
Stroke 7 7 11 7 0.125
Peripheral artery disease 7 8 9 6 0.709
Use of medications (%)
Aldosterone receptor antagonist 9 9 14 7 0.011
Digoxin 52 53 55 37 0.001
Assigned to bisoprolol (%) 51 47 51 56 0.091
Abbreviations: NYHA: New York Heart Association, eGFR: estimated glomerular filtration rate

84
Chapter 4
incidence of sudden death did not differ between the presence or absence of symptoms. Kaplan-Meier curves of event free survival for each of the individual symptoms is shown in figure 2.
In univariate analysis, patients with any signs of congestion were at severely increased risk for all cause mortality (Hazard ratio (HR) 1.76, 95% confidence interval (CI) 1.43 – 2.15), P < 0.001). This increased to HR 2.00, 95% CI 1.36 – 2.94, P < 0.001 when ≥ 3 signs of congestion were present (Figure 3). This was mainly attributable to the presence of either increased JVP
Figure 3. Number of signs of congestion and all-cause mortality
Days
0 120 240 360 480 600 720 840
Eve
nt fr
ee s
urvi
val
0,4
0,5
0,6
0,7
0,8
0,9
1,0
No signs of congestion
1 sign of congestion
2 signs of congestion
≥ 3 signs of congestion
P < 0.0001
Table 3. Relationship between presence of different types of signs of congestion and eGFR
Total Population(N = 2643)
LVEF < 30%(N = 1447)
LVEF ≥ 30%(N = 1196)
eGFR (ml/min/1.73m2) eGFR (ml/min/1.73m2) eGFR (ml/min/1.73m2)
Sign or symptom of congestion
Sign or symptom of congestion
Sign or symptom of congestion
Absent Present Absent Present Absent Present
Raised JVP 79 ± 32 73 ± 30† 77 ± 32 73 ± 30* 81 ± 31 73 ± 30†
N 2051 594 1093 342 945 245
Ascites 78 ± 32 71 ± 26 76 ± 32 76 ± 27 80 ± 31 65 ± 23#
N 2549 79 1392 43 1153 36
Peripheral Edema 77 ± 31 78 ± 31 76 ± 32 77 ± 31 79 ± 31 79 ± 31
N 1942 687 1102 333 836 354
Orthopnea 79 ± 31 72 ± 32† 77 ± 31 71 ± 33# 81 ± 31 73 ± 29†
N 2115 513 1150 284 962 228
† P < 0.001, * P < 0.05, # P < 0.01, Abbreviations: eGFR: estimated glomerular filtration rate, JVP: Jugular venous pressure, LVEF: Left ventricular ejection fraction.

Signs of congestion, eGFR and prognosis
85
Figure 4. Risk of the combined endpoint in relation to K/DOQI stages of chronic kidney disease and number of signs of congestion. Three-dimensional bar graph showing risk of combined endpoint in relation to K/DOQI stages of chronic kidney disease and number of signs of congestion. Abbreviations: GFR: estimated glomerular filtration rate.
0
1
2
3
4
5
6
7
0
1
2
3
4
5
6
7
0
1
2
3
> 90
60 - 89
30 - 59< 30
Haz
ard
Rat
io
Haz
ard
Rat
io
Signs of congestionGFR (mL/min/1.73m2)
Figure 2. Kaplan-Meier analysis of event free survival according to the presence of specific signs and symptoms of congestion. All cause mortality (primary endpoint CIBIS-II)
Days0 120 240 360 480 600 720 840
Eve
nt fr
ee s
urvi
val
0,4
0,5
0,6
0,7
0,8
0,9
1,0
No signs of congestion
Raised JVP
Ascites
All P < 0.0001 vs no signs of congestion
OrthopneaPeripheral eadema

86
Chapter 4Ta
ble
4. I
nci
den
ce o
f en
dp
oin
ts a
cco
rdin
g t
o t
he
pre
sen
ce o
f sp
ecifi
c si
gn
s o
f co
ng
esti
on
and
nu
mb
er o
f si
gn
s o
f co
ng
esti
on
.
Ele
vate
d J
VP
(N =
594
)A
scit
es(N
= 7
9)P
erip
hera
l Ed
ema
(N =
687
)O
rtho
pne
a(N
= 5
13)
Num
ber
of
sig
ns p
rese
nt(N
= 1
409
/ 77
4 /
305
/ 15
5)
No
. of
even
tsN
o. o
f ev
ents
No
. of
even
tsN
o. o
f ev
ents
No
. of
even
ts
Ab
sent
Pre
sent
Ab
sent
Pre
sent
Ab
sent
Pre
sent
Ab
sent
Pre
sent
01
2≥
3
All
caus
e m
orta
lity
252
(12)
130
(22)
†36
2 (1
4)20
(25)
*27
0 (1
4)11
2 (1
6)‡
289
(14)
93 (1
8)‡
158
(11)
131
(17)
62 (2
0)31
(20)
†
All
caus
e ho
spita
lisat
ion
706
(34)
247
(42)
†91
9 (3
6)33
(42)
‡69
2 (3
5)26
1 (3
8)‡
730
(34)
222
(43)
†46
7 (3
3)28
5 (3
7)13
3 (4
4)66
(43)
†
CV
hos
pita
lisat
ion
534
(26)
204
(34)
†70
8 (2
8)30
(38)
#52
2 (2
7)21
6 (3
1)#
555
(26)
182
(35)
†34
7 (2
5)22
1 (2
9)10
8 (3
5)61
(39)
†
Dea
th fr
om p
ump
failu
re44
(2)
29 (7
)†76
(3)
7 (9
)*49
(3)
34 (5
)*56
(3)
27 (5
)#27
(2)
24 (3
)18
(6)
14 (9
)†
Sud
den
dea
th96
(5)
35 (6
)12
7 (5
)4
(5)
100
(5)
31 (5
)11
0 (5
)21
(4)
64 (5
)48
(6)
15 (5
)4
(3)
Com
bin
ed e
ndp
oint
613
(30)
238
(40)
†81
4 (3
2)37
(47)
*60
5 (3
1)24
6 (3
5)‡
649
(31)
201
(39)
†40
3 (2
9)25
5 (3
3)12
5 (4
1)67
(43)
†
† P
< 0
.000
1 by
log
rank
tes
t fo
r co
mp
aris
on
with
ab
senc
e o
r nu
mb
er o
f co
nges
tive
sig
ns
* P
< 0
.001
by
log
rank
tes
t fo
r co
mp
aris
on
with
ab
senc
e o
r nu
mb
er o
f co
nges
tive
sig
ns#
P <
0.0
1 by
log
rank
tes
t fo
r co
mp
aris
on
with
ab
senc
e o
r nu
mb
er o
f co
nges
tive
sig
ns‡
P <
0.0
5 by
log
rank
tes
t fo
r co
mp
aris
on
with
ab
senc
e o
r nu
mb
er o
f co
nges
tive
sig
nsA
bb
revi
atio
ns: C
V: C
ard
iova
scul
ar, J
VP
: Jug
ular
ven
ous
pre
ssur
e.

Signs of congestion, eGFR and prognosis
87
Tab
le 5
. Rel
atio
nsh
ip b
etw
een
pre
sen
ce a
nd
nu
mb
er o
f si
gn
s o
f co
ng
esti
on
and
ou
tco
me
in m
ult
ivar
iate
an
alys
is.
All
Cau
seM
ort
alit
yA
ll ca
use
hosp
ital
isat
ion
CV
ho
spit
alis
atio
nsD
eath
fro
m p
ump
fa
ilure
Sud
den
dea
thC
om
bin
ed e
ndp
oin
t
Par
amet
ers
of
Co
nges
tio
n
E
leva
ted
JV
P1.
52 (1
.22
– 1.
91)*
*1.
18 (1
.01
– 1.
37)#
1.21
(1.0
2 –
1.43
)#2.
13 (1
.35
– 3.
37)*
1.14
(0.7
6 –
1.72
)1.
23 (1
.06
– 1.
44)*
A
scite
s1.
70 (1
.06
– 2.
72)#
1.11
(0.7
8 –
1.59
)1.
22 (0
.83
– 1.
78)
2.19
(0.9
7 –
4.97
)1.
20 (0
.43
– 3.
34)
1.33
(0.9
4 –
1.87
)
P
erip
hera
l ed
ema
1.23
(0.9
7 –
1.56
)1.
16 (1
.00
– 1.
35)
1.25
(1.0
6 –
1.48
)*2.
07 (1
.30
– 3.
30)*
1.00
(0.6
5 –
1.52
)1.
23 (1
.05
– 1.
44)#
O
rtho
pne
a1.
04 (0
.81
– 1.
34)
1.19
(1.0
2 –
1.40
)#1.
23 (1
.03
– 1.
47)#
1.17
(0.6
9 –
1.97
)0.
70 (0
.43
– 1.
14)
1.16
(0.9
8 –
1.37
)
Num
ber
of
sig
ns p
rese
nt
A
ny1.
42 (1
.14
– 1.
77)*
1.19
(1.0
4 –
1.36
)#1.
23 (1
.05
– 1.
43)*
1.76
(1.0
8 –
2.89
)#1.
12 (0
.78
– 1.
61)
1.21
(1.0
5 –
1.39
)*
N
one
1.00
‡1.
00†
1.00
‡1.
00‡
1.00
1.00
‡
1
1.35
(1.0
6 –
1.71
)#1.
12 (0
.96
– 1.
30)
1.13
(0.9
5 –
1.35
)1.
40 (0
.79
– 2.
46)
1.24
(0.8
4 –
1.83
)1.
12 (0
.95
– 1.
32)
2
1.59
(1.1
6 –
2.16
)*1.
32 (1
.09
– 1.
62)*
1.37
(1.0
9 –
1.72
)*2.
11 (1
.11
– 4.
01)#
0.98
(0.5
5 –
1.76
)1.
36 (1
.10
– 1.
67)*
≥3
1.55
(1.0
1 –
2.36
)#1.
34 (1
.01
– 1.
77)#
1.57
(1.1
6 –
2.12
)*3.
38 (1
.60
– 7.
17)*
0.60
(0.2
1 –
1.74
)1.
49 (1
.12
– 1.
98)*
LVE
F (p
er 5
% d
ecre
ase)
1.15
(1.0
6 –
1.25
)**
1.08
(1.0
2 –
1.14
)*1.
07 (1
.00
– 1.
14)#
1.10
(0.9
2 –
1.32
)1.
15 (1
.00–
1.3
2)1.
08 (1
.02
– 1.
14)*
eGFR
(per
10
mL/
min
/1.7
3m2 d
ecre
ase)
1.11
(1.0
6 –
1.17
)**
1.07
(1.0
4 –
1.11
)**
1.08
(1.0
5 –
1.12
)**
1.29
(1.1
5 –
1.45
)**
1.08
(0.9
9 –
1.17
)1.
09 (1
.05
– 1.
12)*
*
** P
< 0
.001
,* P
< 0
.01,
# P
< 0
.05,
† P
for
tren
d <
0.0
5, ‡
P fo
r tr
end
< 0
.01.
All
mo
del
s ad
just
ed fo
r ag
e, g
end
er, r
ace,
eje
ctio
n fr
actio
n, e
GFR
, NY
HA
cla
ss, t
reat
-m
ent
assi
gnm
ent,
etio
log
y of
hea
rt fa
ilure
, ele
ctro
card
iog
rafic
ab
norm
aliti
es, a
his
tory
of d
iab
etes
or
hyp
erte
nsio
n, b
asel
ine
med
icat
ion
use,
and
blo
od
pre
ssur
e.
Ab
bre
viat
ions
: CV
: car
dio
vasc
ular
, eG
FR: e
stim
ated
glo
mer
ular
filtr
atio
n ra
te, J
VP
: jug
ular
ven
ous
pre
ssur
e, L
VE
F: le
ft v
entr
icul
ar e
ject
ion
frac
tion.

88
Chapter 4
(HR 1.93, 95% CI 1.56 – 2.38, P < 0.001) or ascites (HR 2.30, 95% CI 1.46 – 3.61, P < 0.001), while also the presence of peripheral edema (HR 1.29, 95% CI 1.03 – 1.61, P < 0.05) and orthopnea (HR 1.34, 95% CI 1.06 – 1.70), P < 0.05 were related to all cause mortality. The risk of all cause hospitalisations or cardiovascular hospitalisations was similar among each type of congestion sign, and rose to HR 1.58 , 95% CI 1.20 – 2.00, P =0.001 (all cause hospitalisations) and HR 1.97, 95% CI 1.50 – 2.59, P < 0.001 (cardiovascular hospitalisations) when ≥ 3 signs of congestion were present. Most congestion parameters were strong predictors of death from pump failure: JVP (HR 1.51, 95% CI 2.15 – 5.10, P < 0.001), ascites (HR 3.82, 95% CI 1.76 – 8.32, P < 0.001), peripheral edema (HR 2.15, 95% CI 1.39 - 3.34, P < 0.01) and orthopnea (HR 2.01, 95% CI 1.27 – 3.18, P < 0.01). None were significant predictors of the occurrence of sudden death. Finally, all symptoms of congestion were significantly related to the combined endpoint , resulting in a HR 1.25, 95% CI 1.07 – 1.46, P < 0.01 when one symptom was present, HR 1.71, 95% CI 1.40 – 2.09, P < 0.001 when two signs were present and HR 1.87, 95% CI 1.45 – 2.43, P < 0.001 when ≥ 3 signs or symptoms were present.
Renal impairment was a strong predictor of all cause mortality (HR 1.13, 95% CI 1.09 - 1.18, P < 0.001), all cause hospitalisations (HR 1.11, 95% CI 1.08 - 1.13, P < 0.001), cardiovascular hospitalisations (HR 1.11, 95% CI 1.08 - 1.14, P < 0.001), death from pump failure (HR 1.11, 95% CI 1.07 - 1.16, P < 0.001), sudden death (HR 1.08, 95% CI 1.01 - 1.15, P < 0.05) and the combined endpoint (HR 1.11, 95% CI 1.08 - 1.14, P < 0.001) (all per 10 mL/min/1.73m2 decrease).
Combining categories of eGFR and numbers of signs of congestion, a stepwise increase in the risk for the combined endpoint was present (Figure 4). No interaction between signs of congestion and eGFR was present, therefore both signs and symptoms of venous congestion and eGFR had an effect that was additive in terms of predicting the combined endpoint.
In multivariate analysis, the presence of an elevated JVP remained a strong and independent predictor of all cause mortality, hospitalisations, death from pump failure and the composite endpoint. Also the presence of ascites was independently related to mortality, while peripheral edema and orthopnea were primarily independent predictors of hospitalisations (Table 5). Table 5 shows that increasing number of signs of congestion was associated with a higher risk for all cause mortality, hospitalisations, death from pump failure and the combined endpoint. In comparison, both LVEF and eGFR were strong and independent predictors of most endpoints, including mortality and hospitalisations, but not sudden death.
Finally, in a secondary analysis, we investigated the relationship between different combinations of individual signs of congestion and outcome (Supplementary Table 1). There was a significant positive interaction between the presence of JVP and ascites on all cause mortality, which persisted in multivariate analysis. The combination of JVP and ascites in this analysis was therefore not associated with increased mortality. The results of the secondary analyses further emphasize the observation that elevated JVP is the most important symptom of venous congestion, showing the most consistent findings in combination with other symptoms.

Signs of congestion, eGFR and prognosis
89
DiscussionThe present study shows that symptoms of venous congestion, including elevated JVP,
ascites, peripheral edema and orthopnea, are frequently found in and associated with decreased renal function in patients with CHF. Furthermore, the extent of venous congestion, as assessed by the number of symptoms and signs present, was also associated with the extent of renal impairment. Finally, the presence of signs and symptoms of congestion was associated with unfavourable outcome, including all-cause mortality, hospitalisations and the combined endpoint of cardiovascular mortality and hospitalisations.
Signs and symptoms of venous congestion and renal impairmentRenal function is regarded as one of the most important determinants of prognosis in
CHF.[4] It is thought to be closely related to decreased renal perfusion as a result of decreased cardiac output.[6] However, we recently showed that also invasively determined central venous pressure is associated with renal impairment in patients with cardiac dysfunction secondary to pulmonary hypertension.[7] Although invasively determined central venous pressure seems to be poorly correlated with the presence of congestive symptoms and signs,[12-14] the latter have been indirectly related to reduced renal function.[2] Our present data suggests that if signs and symptoms of venous congestion are present, renal function is often impaired. Furthermore, renal function seems to be even more deteriorated in the presence of increasing number of symptoms and signs of venous congestion.
However, the observed relationship between renal impairment and the presence of congestion may also have a different etiology. Renal impairment itself may be the initiating factor for salt and water retention as a response to decreased renal perfusion, secondary to reduced cardiac output.[15] In contrast to this hypothesis, we found that in patients who had the relatively best LVEF in our present cohort of CHF patients, renal function was even more deteriorated when signs and symptoms of venous congestion were present. Although LVEF is not the most reliable marker of cardiac output and subsequent renal perfusion, this suggests that the relationship between venous congestion and renal function is at least bidirectional, and includes an effect of congestion on renal function.
Signs and symptoms of venous congestion and prognosisAlthough widely appreciated as marker for the extent of fluid overload, as marker for severity
of heart failure, and now also as marker for renal dysfunction, data regarding venous congestion as marker for prognosis are surprisingly scarce. An elevated JVP or a third heart sound, or a combination of both were strong and independent predictors of outcome in the SOLVD trial.[2] A substudy of the COMET trial investigated the prognostic value of angina, fatigue and breathlessness, showing that only the latter was independently associated with mortality.[16] A substudy of the DIG trial showed that a simple score of symptoms, including elevated JVP, a third heart sound, peripheral edema, dyspnea at rest or exertion, limited activity and radiologic signs of congestion were independent predictors of mortality.[3] First symptoms in idiopathic dilated cardiomyopathy have been associated with poor outcome,[17] while absence

90
Chapter 4
of congestion is associated with improved survival.[18] In acute heart failure, the presence of symptoms and signs of congestion is associated with strongly reduced survival.[19] However, tailoring therapy to reducing central venous pressure was not superior to conventional therapy in these patients, although interestingly, renal function was preserved in these patients.[20] Small studies have investigated the relationship between invasively determined central venous pressure and outcome, showing that right atrial pressures above 12 mmHg are related to poor outcome in patients listed for cardiac transplantation.[21,22] In our present analysis we have shown that independent of established prognostic markers in CHF, such as renal dysfunction, NYHA class and LVEF, not only the presence but also the number of symptoms and signs of venous congestion are important predictors of outcome in these patients. This further emphasizes the need for standard thorough physical examination in these patients, to identify those at risk for impaired renal function and strongly impaired prognosis.
LimitationsOur present analysis is hampered by its retrospective nature. Furthermore, no longitudinal
data was available on either renal function measurements or signs and symptoms of congestion. We were therefore unable to investigate possible cause and effect relationships, which are bound to be bidirectional As a consequence, our results must be regarded as hypothesis generating. The assessment of physical signs and symptoms is of an individual nature, and interobserver variability is high. But even despite these limitations, we found that congestive symptoms were strong independent predictors of prognosis. Renal function was assessed using the simplified MDRD formula, which may have over or underestimated true renal function in CHF patients. Finally, patients with severely impaired renal function (serum creatinine > 300 µmol/L were excluded from the CIBIS-II trial. Therefore our present retrospective analysis does not allow us to investigate the relationship between parameters of venous congestion and severely deteriorated renal function.
ConclusionThe presence of any sign of venous congestion is associated with impaired renal function in
patients with CHF. Renal function is even more deteriorated when more signs and symptoms are present. Furthermore, not only the presence of congestive symptoms, but also the extent of congestion is independently related to poor outcome in these patients. These findings may help to identify those patients at risk for reduced renal function and subsequent poor survival, and may lead to more tailored and effective therapy.

Signs of congestion, eGFR and prognosis
91
AcknowledgementsK. Damman is supported by the Netherlands Heart Foundation (grant 2006B157) A.A.
Voors and D.J. van Veldhuisen are Clinical Established Investigators of the Netherlands Heart Foundation (grants 2006T37 and D97-017, respectively).
DisclosuresNone.
ReferencesLesman-Leegte I, Jaarsma T, Sanderman R, Linssen G, and van Veldhuisen DJ. Depressive symptoms 1. are prominent among elderly hospitalised heart failure patients. Eur J Heart Fail. 2006;8:634-640.
Drazner MH, Rame JE, Stevenson LW, and Dries DL. Prognostic importance of elevated jugular venous 2. pressure and a third heart sound in patients with heart failure. N Engl J Med. 2001;345:574-581.
Brophy JM, Dagenais GR, McSherry F, Williford W, and Yusuf S. A multivariate model for predicting 3. mortality in patients with heart failure and systolic dysfunction. Am J Med. 2004;116:300-304.
Hillege HL, Nitsch D, Pfeffer MA et al. Renal function as a predictor of outcome in a broad spectrum 4. of patients with heart failure. Circulation. 2006;113:671-678.
Ruilope LM, van Veldhuisen DJ, Ritz E, and Luscher TF. Renal function: The Cinderella of cardiovascular 5. risk profile. J Am Coll Cardiol. 2001;38:1782-1787.
Ljungman S, Laragh JH, and Cody RJ. Role of the Kidney in Congestive Heart-Failure - Relationship 6. of Cardiac Index to Kidney-Function. Drugs. 1990;39:10-21.
Damman K, Navis G, Smilde TD et al. Decreased cardiac output, venous congestion and the association 7. with renal impairment in patients with cardiac dysfunction. Eur J Heart Fail. 2007;9:872-878.
Damman K, van Deursen VM, Navis G et al. Increased central venous pressure is associated with 8. impaired renal function and mortality in a broad spectrum of patients with cardiovascular disease. J Am Coll Cardiol. 2009;53:582-588.
CIBIS-II Investigators and Committees. The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a 9. randomised trial. Lancet. 1999;353:9-13.
Smilde TD, van Veldhuisen DJ, Navis G, Voors AA, and Hillege HL. Drawbacks and prognostic value 10. of formulas estimating renal function in patients with chronic heart failure and systolic dysfunction. Circulation. 2006;114:1572-1580.
Coresh J, Selvin E, Stevens LA et al. Prevalence of chronic kidney disease in the United States. JAMA. 11. 2007;298:2038-2047.
Connors AF, Jr., McCaffree DR, and Gray BA. Evaluation of right-heart catheterization in the critically 12. ill patient without acute myocardial infarction. N Engl J Med. 1983;308:263-267.

92
Chapter 4
Eisenberg PR, Jaffe AS, and Schuster DP. Clinical evaluation compared to pulmonary artery 13. catheterization in the hemodynamic assessment of critically ill patients. Crit Care Med. 1984;12:549-553.
Stein JH, Neumann A, and Marcus RH. Comparison of estimates of right atrial pressure by physical 14. examination and echocardiography in patients with congestive heart failure and reasons for discrepancies. Am J Cardiol. 1997;80:1615-1618.
Schrier RW. Role of diminished renal function in cardiovascular mortality: marker or pathogenetic 15. factor? J Am Coll Cardiol. 2006;47:1-8.
Ekman I, Cleland JG, Swedberg K et al. Symptoms in patients with heart failure are prognostic predictors: 16. insights from COMET. J Card Fail. 2005;11:288-292.
Komajda M, Jais JP, Reeves F et al. Factors predicting mortality in idiopathic dilated cardiomyopathy. 17. Eur Heart J. 1990;11:824-831.
Lucas C, Johnson W, Hamilton MA et al. Freedom from congestion predicts good survival despite 18. previous class IV symptoms of heart failure. Am Heart J. 2000;140:840-847.
Nohria A, Tsang SW, Fang JC et al. Clinical assessment identifies hemodynamic profiles that predict 19. outcomes in patients admitted with heart failure. J Am Coll Cardiol. 2003;41:1797-1804.
Binanay C, Califf RM, Hasselblad V et al. Evaluation study of congestive heart failure and pulmonary 20. artery catheterization effectiveness: the ESCAPE trial. JAMA. 2005;294:1625-1633.
Morley D and Brozena SC. Assessing risk by hemodynamic profile in patients awaiting cardiac 21. transplantation. Am J Cardiol. 1994;73:379-383.
Unverferth DV, Magorien RD, Moeschberger ML et al. Factors influencing the one-year mortality of 22. dilated cardiomyopathy. Am J Cardiol. 1984;54:147-152.

Signs of congestion, eGFR and prognosis
93
Su
pp
lemen
tary table 1. R
elation
ship
betw
een differen
t com
bin
ation
s of in
divid
ual sig
ns o
f con
gestio
n and
ou
tcom
e
All C
auseM
ortality
All cause
hosp
italisation
CV
ho
spitalisatio
nsD
eath from
pum
p
failureS
udd
en death
Co
mb
ined end
po
int
Two
Sig
ns
JVP
+ A
scites1.52 (0.82 – 2.84)
1.10 (0.75 – 1.62)1.21 (0.80 – 1.81)
2.45 (0.98 – 6.14)1.34 (0.45 – 4.00)
1.37 (0.95 – 1.98)
JVP
+ E
dem
a1.61 (1.18 – 2.20)*
1.27 (1.03 – 1.56)#1.37 (1.08 – 1.73)*
3.11 (1.66-5.81)**1.15 (0.66 – 2.04)
1.37 (1.10 – 1.70)*
JVP
+ O
rthopnea
1.41 (1.02 – 1.95)#1.30 (1.05 – 1.60)#
1.36 (1.07 – 1.72)#1.76 (0.91 – 3.43)
0.80 (0.43 – 1.50)1.31 (1.05 – 1.63)#
Ascites +
Ed
ema
1.53 (0.93 – 2.52)1.09 (0.75 – 1.59)
1.25 (0.84 – 1.86)2.39 (0.98 – 5.87)
1.13 (0.39 – 3.29)1.34 (0.93 – 1.92)
Ascites +
Orthop
nea1.34 (0.76 – 2.36)
1.11 (0.74 – 1.67)1.24 (0.80 – 1.92)
1.36 (0.48 – 3.83)0.78 (0.24 – 2.54)
1.28 (0.86 – 1.91)
Ed
ema +
Orthop
nea1.47 (0.84 – 2.58)
1.28 (1.04 – 1.59)#1.41 (1.11 – 1.79)*
1.72 (0.85 – 3.47)0.68 (0.36 – 1.27)
1.28 (1.02 – 1.61)#
Three S
igns
JVP
+ A
scites + O
rthopnea
1.96 (1.11 – 3.47)#1.26 (0.84 – 1.90)
1.43 (0.92 – 2.21)2.42 (0.85 – 6.93)
0.92 (0.28 – 3.02)1.51 (1.01 – 2.26)#
JVP
+ E
dem
a + O
rthopnea
1.55 (1.08 – 2.25)#1.45 (1.15 – 1.85)*
1.61 (1.24 – 2.11)*3.07 (1.46 – 6.49)*
0.79 (0.39 – 1.60)1.52 (1.18 – 1.95)#
JVP
+ A
scites + E
dem
a2.24 (1.36 – 3.71)*
1.24 (0.85 – 1.81)1.44 (0.96 – 2.15)
4.27 (1.72 – 10.59)*1.33 (0.45 – 3.88)
1.59 (1.10 – 1,29)#
Ascites +
Ed
ema +
Orthop
nea1.48 (0.84 – 2.58)
1.25 (0.83 – 1.87)1.48 (0.96 – 2.28)
2.36 (0.83 – 6.75)0.77 (0.24 – 2.49)
1.48 (1.00 – 2.20)#
Four S
igns
JVP
+ A
scites + E
dem
a + O
rthopnea
2.16 (1.25 – 3.74)*1.42 (0.95 – 2.11)
1.70 (1.11 – 2.60)#4.22 (1.52 – 11.72)*
0.91 (0.29 – 2.89)1.75 (1.19 – 2.59)*
** P <
0.001,* P <
0.01, # P <
0.05. All m
od
els adjusted
for ag
e, gend
er, race, ejection fractio
n, eGFR
, NY
HA
class, treatment assig
nment, etio
logy of heart failure,
electrocard
iografic ab
norm
alities, a history of d
iabetes o
r hypertensio
n, baseline m
edicatio
n use, and b
loo
d p
ressure. Ab
breviatio
ns: CV
: cardiovascular, eG
FR:
estimated
glo
merular filtratio
n rate, JVP
: jugular veno
us pressure, LV
EF: left ventricular ejectio
n fraction.

94

95
PART IIWorsening renal function in patients with heart failure

96

Chapter 5Worsening renal function and prognosis in heart failure
Systematic review and meta-analysis
Kevin Damman, Gerjan Navis, Adriaan A. Voors, Folkert W. Asselbergs, Tom D.J. Smilde, John, G.F. Cleland, Dirk J. van Veldhuisen, Hans L. Hillege
J Card Fail, 2007;13:599-608

98
Abstract
BackgroundRenal impairment is associated with increased mortality in heart failure (HF). Recently, reports suggest
that worsening renal function (WRF) is another predictor of clinical outcome in HF. The present study was designed to establish the proportion of patients with HF that exhibits (WRF) and the associated risk for mortality and hospitalization by conducting a systematic review and meta analysis.
Methods and ResultsA systematic search of MEDLINE revealed 8 studies on the relationship between WRF and mortality
in 18,634 patients with HF. The mortality risk associated with WRF was estimated using random-effects meta-analysis. WRF was defined as an increase in serum creatinine ≥ 0.2 mg/dl or a corresponding decrease in estimated glomerular filtration rate ≥ 5 ml/min/1.73m2. Subgroup analysis included differentiation between in and out-hospital patients, degree of WRF and time until endpoint occurrence. WRF developed in 4,734 (25%) patients and was associated with a higher risk for mortality (odds ratio [OR] = 1.62; 95% confidence interval [CI] 1.45 to 1.82, P < 0.001) and hospitalization (OR = 1.30, 95% CI 1.04 to 1.62, P = 0.022). The severity of WRF was also associated with greater mortality. Patients with impaired renal function at baseline were more prone to progressive renal function loss.
ConclusionWRF predicts substantially higher rates of mortality and hospitalization in patients with HF.

WRF and outcome
99
IntroductionImpaired renal function is an important independent risk factor for all-cause mortality in
patients with heart failure (HF).[1-4] Recently, Smith et al and de Silva et al emphasized the risk associated not only with renal dysfunction but also with change in renal function over time.[5] We conducted a systematic review and meta-analysis of all available relevant clinical studies to determine the proportion of patients with HF that exhibit worsening of renal function (WRF) and to investigate whether WRF was associated with an increase in mortality and hospitalization.
MethodsLiterature Search
MEDLINE was searched to identify eligible studies using search tools provided by Pubmed (http://www.ncbi.nlm.nih.gov/entrez/query/static/clinical.shtml; used January 2007). These search tools have been validated by Haynes et al. as optimizing retrieval.[6] We also used keywords including (worsening) renal (kidney) function, WRF, heart failure, mortality, prognosis, left ventricular dysfunction, creatinine and a combination of these. We included papers limited to the English language. Furthermore, we searched our own files, reviewed reference lists from eligible studies, used the “see related articles” feature for key publications in PubMed, consulted the Cochrane Library, and searched the ISI Web of Knowledge (http://scientific.thomson.com/webofknowledge) for publications that cited key publications.
Study SelectionWe included those studies primarily investigating the association between WRF and
outcome in patients with HF. These included retrospective secondary analyses of randomized controlled trials, cohort studies and observational studies. Articles were excluded if: a) no measurement of WRF was available, b) no data were available for all cause mortality, c) follow up duration for outcome measurements was less than 6 months (in secondary analysis, also studies with short follow up were included), d) data was only published in abstract form and e) no definition for HF was given: either by combination of symptoms and signs (using New York Heart Association functional class or physical examination), imaging (impaired left ventricular ejection fraction) or a combination of both. In secondary analysis, also studies investigating early mortality were included in the analysis.
Definition of Worsening Renal FunctionMost reports included in the analysis used a pre-defined cut-off value of an increase of >
0.3 mg/dl (> 26.5 µmol/l) in serum creatinine as a definition of WRF. Different intervals between serum creatinine measurements were used. In most reports the in-hospital variance in creatinine measurements was reported. The different definitions used are shown in Table 3.
An increase in serum creatinine of < 0.2 mg/dl (< 17.7 µmol/l) or corresponding decline of < 5 ml/min/1.73m2, [7] was considered within the limits of normal physiological variation, and

100
Chapter 5
used as reference decline. This cut off point was chosen for practical, rather than pathophysiologic reasons. WRF was defined as any decline in renal function greater than the above. To examine the effect of WRF severity on outcome, we defined different classes of WRF. Class I WRF was defined as an increase in serum creatinine between 0.2 mg/dl and 0.3 mg/dl (17.7 and 26.5 µmol/l) or a corresponding decrease in eGFR of 5 to 10 ml/min/1.73m2. Class II WRF was defined as an increase in serum creatinine between 0.3 and 0.5 mg/dl (26.5 and 44.2 µmol/l) or a decrease in eGFR between 11 and 15 ml/min/1.73m2. Class III was defined as an increase in serum creatinine of > 0.5 mg/dl or a decrease in eGFR of > 15 ml/min/1.73m2. These values represent the spectrum of WRF definitions used by Smith et al.[8]
OutcomesThe primary outcome measure was defined as all cause mortality. In the main analysis, only
studies reporting outcome after a minimum of 6 months were included. In secondary analysis, early mortality was also taken into account, and difference in risk between the different times to events was analyzed. Secondary outcome variable included all cause hospitalization as reported in the individual reports.
Study QualityThe quality of the individual studies was assessed according to the guidelines provided by the
United States Preventive Task Force [9] and published recommendations.[10] We assessed eleven factors: 1) sufficiently inclusion and exclusion criteria, 2) did the study explain sample selection, 3) were clinical and demographic variables fully specified, 4) is the study sample representative for the mentioned patient population, 5) was the outcome measure fully specified, 6) was the definition of WRF adequately defined, 7) did the study assess dose-response relationship between different values of WRF and outcome, 8) adjustment for possible confounders in the analysis, 9) reporting of lost to follow-up rates, 10) study design and 11) duration of follow up. Grading was as follows; good quality: 8-11 criteria, fair quality: 5-7 criteria and poor quality: < 5 criteria.
Statistical analysisMeta-analysis was performed using a random-effects model to determine risk associated with
WRF and all-cause mortality, as measured by combined crude mortality rates and unadjusted risk ratios. For comparison adjusted hazard ratios were extracted from Cox regression analysis when available. These adjusted risk estimates were corrected for covariates such as: age, gender, race, left ventricular ejection fraction, medication, medical history, baseline serum creatinine, and other known cardiovascular risk factors Among study heterogeneity of risk estimates was examined using a standard chi-square test and I2 statistic for heterogeneity. I2 is the percentage of variance that is due to between-study variance. Reasons for diversity in study results were explored using meta-regression analysis. Results are presented as odds ratios (ORs) with their 95% confidence intervals (CIs) and p values. A P value of < 0.05 was considered statistically significant. Statistical analyses were performed using Stata 9.0, College Station, Texas.

WRF and outcome
101
Results
Study Search and General CharacteristicsOf 1124 citations retrieved by the search, 13 investigated the relationship between WRF and
outcome. Five studies were excluded for reasons shown in figure 1.[11-15] Eight studies remained for our analysis.[7,8,16-21] Of all selected studies, three assessed deteriorating renal function in out-patients, [7,18,20] while the remaining five investigated patients in the in-hospital setting.[8,16,17,19,21] with follow up extending beyond admission. One study described the relationship between WRF and outcome in patients with left ventricular dysfunction after myocardial infarction without overt heart failure at the moment of inclusion in the study.[20] Therefore, results are also shown with this study excluded.
The main characteristics of the individual studies are outlined in Tables 1, 2 and 3. Altogether, eight studies with 18,634 patients (mean age 67 years) were included of whom 4,734 (25%) developed some degree of WRF. Baseline serum creatinine ranged from 1.2 to 1.8 mg/dl (106-
Figure 1. Quality of Reporting of Meta-Analyses (QUOROM) flow diagram for study selection.
1124 Potentially relevant publications identi�ed and
screened for retrieval
95 Full-text articles retrieved for detailed review
13 Studies of worsening renal function and prognosis
8 Studies of worsening renal function and prognosis in HF
1,029 Publications excluded based on title and abstract
83 Studies excluded based on: (no renal function measurements,
no follow up data, etc)
5 Studies excluded (no HF, short follow up)
102
Chapter 5Ta
ble
1. I
ncl
ud
ed S
tud
ies:
Sam
ple
Ch
arac
teri
stic
s
Stu
dy &
pub
licat
ion
date
F/U
nTy
pe
Pat
ient
sE
ndp
oin
tsM
ajo
r E
xclu
sio
n C
rite
ria
Stu
dy
Des
ign
Stu
dy
Qua
lity
Kru
mho
lz e
t al
. 200
0 [1
6]In
-hos
pita
l30
day
s6
mon
ths
1681
Hos
pita
lized
HF
pat
ient
s w
ith
prin
cip
al d
isch
arge
cod
e of
HF
Mor
talit
y,A
dm
issi
on<
65
year
s, s
ever
e A
OS
/MI
maj
or c
omp
licat
ions
, Mor
talit
y <
24h
afte
r ho
spita
lizat
ion
Ret
rosp
ectiv
e m
ulti-
cent
er c
hart
rev
iew
Fair
Sm
ith e
t al
. 200
3 [8
]6
mon
ths
412
Hos
pita
lized
HF
pat
ient
s w
ith
pre
senc
e of
HF
on a
dm
issi
on.
Mor
talit
yA
dm
issi
on
< 5
0 ye
ars
Term
inal
illn
ess
In h
osp
ital m
orta
lity
Pro
spec
tive,
ob
-se
rvat
iona
l, si
ngle
ce
nter
coh
ort
stud
y G
ood
Akh
ter
et a
l. 20
04 [1
7]30
day
s6
mon
ths
480
Hos
pita
lized
HF
pat
ient
s.M
orta
lity
Ad
mis
sion
Hyp
oten
sion
, Sho
ck,
Ant
icip
ated
sur
viva
l < 1
mon
th
Sec
ond
ary
anal
ysis
ra
ndom
ized
clin
ical
tr
ial:
VM
AC
(200
2)Fa
ir
De
Silv
a et
al.
2005
[18]
16.5
mon
ths
1216
HF
outp
atie
nts
Mor
talit
yD
ialy
sis
pat
ient
sR
enal
tra
nsp
lant
atio
n
Pro
spec
tive,
ob
-se
rvat
iona
l, si
ngle
ce
ntre
coh
ort
stud
y G
ood
Kha
n et
al.
2006
[7]
34.2
and
32
.3 m
onth
s66
40H
F ou
tpat
ient
s.M
orta
lity
Cre
atin
ine
> 2
.5 m
g/d
lS
econ
dar
y an
alys
is
rand
omiz
ed c
linic
al
tria
l: S
OLV
D (1
992)
Goo
d
Cow
ie e
t al
. 200
6 [1
9]In
-hos
pita
l30
day
s6
mon
ths
299
Hos
pita
lized
HF
pat
ient
s.M
orta
lity
Ad
mis
sion
< 2
0 ye
ars,
Sev
ere
AO
SD
ialy
sis
pat
ient
sTe
rmin
al il
lnes
s
Pro
spec
tive,
ob
ser-
vatio
nal m
ultic
ente
r co
hort
stu
dy
PO
SH
Goo
d
Jose
et
al. 2
006
[20]
36.8
mon
ths
1854
Pat
ient
s w
ith a
cute
MI a
nd le
ft
vent
ricul
ar d
ysfu
nctio
n (<
40%
)M
orta
lity,
Ad
mis
sion
Cre
atin
ine
> 2
.5 m
g/d
lS
econ
dar
y an
alys
is
rand
omiz
ed c
linic
al
tria
l: S
AV
E (1
992)
Goo
d
Ow
an e
t al
. 200
6 [2
1]
3 m
onth
s4.
8 ye
ars
6052
Hos
pita
lized
HF
pat
ient
s.M
orta
lity
No
HF
pat
ient
s, N
o se
rial s
e-ru
m c
reat
inin
e m
easu
rem
ents
Ret
rosp
ectiv
e si
ngle
ce
nter
cha
rt r
evie
w.
Fair
AO
S: a
ort
ic v
alve
ste
nosi
s, H
F; h
eart
failu
re, M
I; m
yoca
rdia
l inf
arct
ion,
PO
SH
; Pro
spec
tive
Out
com
e S
tud
y in
Hea
rt fa
ilure
, SA
VE
; Sur
viva
l and
Ven
tric
ular
Enl
arg
e-m
ent
stud
y, s
Cr;
ser
um c
reat
inin
e, S
OLV
D;
Stu
die
s O
f Le
ft V
entr
icul
ar D
ysfu
nctio
n, V
MA
C;
Vas
od
ilatio
n in
the
Man
agem
ent
of A
cute
Co
nges
tive
Hea
rt F
ailu
re
stud
y, W
RF;
wo
rsen
ing
rena
l fun
ctio
n

WRF and outcome
103
Tab
le 2
. Ch
arac
teri
stic
s o
f in
clu
ded
stu
die
s.
Stu
dy
Ag
e (S
D)
Men
(%)
DM
(%
)H
TN
(%)
IHD
(%)
LVE
F (%
)sC
r (m
g/d
l)eG
FR /
CrC
l(*)
Kru
mho
lz e
t al
. (2
000)
Tota
lN
o W
RF
WR
F
79 (8
)- -
42 44 37
38 36 44
60 57 68
37 38 34
29%
< 4
051
% <
40
45%
< 4
0
41%
> 1
.537
% >
1.5
49%
> 1
.5
- - -
Sm
ith e
t al
. (20
03)
Tota
lN
o W
RF
WR
F
72 (1
1)- -
51 - -
47 - -
- - -
- - -
39 - -
1.8 - -
- - -
Akh
ter
et a
l. (2
004)
Tota
lN
o W
RF
WR
F
62 (1
4)- -
69 - -
47 - -
70 - -
46 - -
27 - -
1.71 - -
- - -
De
Silv
a et
al.
(200
5)To
tal
No
WR
FW
RF
71(1
1)- -
69 69 66
21 21 24
41 41 46
66 65 69
3468
% <
40
78%
< 4
0
1.39 - -
5756
% <
60
74%
< 6
0
Kha
n et
al.
(200
6)To
tal
No
WR
FW
RF
:
60(1
0)- -
86 - -
19 - -
39 - -
79 - -
27 - -
1.17 - -
70.1 - -
Cow
ie e
t al
. (20
06)
Tota
lN
o W
RF
WR
F
68 (1
2)68 68
74 73 76
33 31 36
47 47 46
51 51 53
28 28 28
1.58
1.
50
1.77
56*
49*
44*
Jose
et
al. (
2006
)To
tal
No
WR
FW
RF
59 (1
1)59 61
83 84 76
21 20 28
42 42 46
35 38 29
31 31 31
1.19
1.
20
1.09
-34
% <
60
27%
< 6
0
Ow
an e
t al
. (20
06)
Tota
lN
o W
RF
WR
F
73 (1
3)74 74
56 56 56
34 33 39
52 52 60
56 56 62
- - -
1.53
1.
50 2.0
57 59 49
CrC
l; cr
eatin
ine
clea
ranc
e (m
l/min
), D
M; D
iab
etes
Mel
litus
, eG
FR; e
stim
ated
Glo
mer
ular
Filt
ratio
n R
ate
(ml/m
in/1
.73m
2 ), H
TN; H
yper
tens
ion,
IHD
; Isc
hem
ic H
eart
d
isea
se /
his
tory
of M
yoca
rdia
l Inf
arct
ion,
LV
EF;
Lef
t Ve
ntric
ular
Eje
ctio
n Fr
actio
n, s
CR
; ser
um c
reat
inin
e.

104
Chapter 5
159 µmol/l). Only four studies also reported estimated GFR by serum creatinine formulae such as the sMDRD or by creatinine clearance. Follow up ranged from duration of 6 months to a median follow up of 4.8 years. Table 2 also shows information on the subgroups of No WRF versus WRF, as reported by the original reports.
All-cause mortalityIn the included studies,
data were available for all-cause mortality with a minimum follow up of at least 6 months. The crude mortality rates of patients with versus those without were 43% and 36%, respectively. This resulted in a combined mortality risk of OR = 1.62, 95% CI 1.45 to 1.82, P < 0.001 in patients with WRF. No significant heterogeneity amongst studies was observed (I2 = 32.8%, P < 0.166).We performed meta-regression analysis to determine factors explaining possible confounding. Variables explored included the presence of hypertension, diabetes and a history of myocardial infarction, baseline serum creatinine, LVEF, age; type of patients, study quality and class of WRF as stratified in our analysis. Patients with more severe WRF, a history of myocardial infarction, of greater age and men showed an increased risk for mortality when experiencing WRF.
When adjusted hazard ratios were used to determine adjusted mortality risk which were published Ta
ble
3. D
efin
itio
ns
of
WR
F as
sp
ecifi
ed in
th
e d
iffe
ren
t st
ud
ies.
Stu
dy
Gen
eral
defi
niti
on
WR
FIn
tim
eS
trat
ified
WR
FN
pat
ient
s W
RF
N p
atie
nts
no W
RF
Incl
uded
in t
he a
naly
sis
as:
Kru
mho
lz>
0.3
mg/
dl i
ncre
ase*
Dur
ing
adm
issi
on46
912
12C
lass
II W
RF
Sm
ithA
ny in
crea
se*
Dur
ing
adm
issi
on>
0.1
mg/
dl i
ncre
ase*
> 0
.2 m
g/d
l inc
reas
e*>
0.3
mg/
dl i
ncre
ase*
> 0
.4 m
g/d
l inc
reas
e*>
0.5
mg/
dl i
ncre
ase*
103
173
227
280
313
309
239
185
132
99
Cla
ss I
WR
FC
lass
I W
RF
Cla
ss II
WR
FC
lass
II W
RF
Cla
ss II
I WR
F
Akh
ter
> 0
.5 m
g/d
l inc
reas
e*D
urin
g ad
mis
sion
119
361
Cla
ss II
I WR
F
De
Silv
a>
0.3
mg/
dl i
ncre
ase*
6 m
onth
s16
110
55C
lass
II W
RF
Kha
nA
dec
reas
e in
eG
FR o
f > 5
ml/m
in/1
.73m
2Ye
ar5-
10 m
l/min
/1.7
3m2
11-1
5 m
l/min
/1.7
3m2
> 1
5 m
l/min
/1.7
3m2
960
344
756
4475
†44
75†
4475
†
Cla
ss I
WR
FC
lass
II W
RF
Cla
ss II
I WR
F
Cow
ie>
0.3
mg/
dl i
ncre
ase*
Dur
ing
adm
issi
on98
201
Cla
ss II
WR
F
Jose
> 0
.3 m
g/d
l inc
reas
e*2
wee
ks22
316
31C
lass
II W
RF
Ow
an>
0.3
mg/
dl i
ncre
ase*
Dur
ing
adm
issi
on>
0.3
mg/
dl i
ncre
ase*
1419
4633
Cla
ss II
WR
F
* in
crea
se in
ser
um c
reat
inin
e, †
Kha
n co
mp
ared
diff
eren
t st
ratifi
catio
ns o
f WR
F w
ith t
he p
atie
nt g
roup
with
a ‘n
orm
al’ d
ecre
ase
of <
5 m
l/min
/1.7
3m2

WRF and outcome
105
in the original reports, WRF was still independently associated with worse outcome (HR 1.65, 95% CI 1.42 to 1.88), although only 4 out of 8 studies reported adjusted hazard ratios. One study also reported a decreased risk of all cause mortality associated with improving renal function (HR 0.8, 95% CI 0.6-1.0).[18]
Secondary outcome: HospitalizationWe assessed the risk for all-cause hospitalization associated with WRF in four reports
addressing this endpoint . WRF was associated with modest increase in the rate of all-cause hospitalization OR = 1.30, 95% CI 1.04 to 1.62, P = 0.022.
Severity of WRFMortality increased with greater severity of WRF. The OR ranged from 1.03, 95% CI 0.65
to 1.64, P = 0.895 in WRF Class I, 1.48, 95% CI 1.35 to 1.63, P < 0.001 in Class II, to 3.22, 95% CI 2.36 to 4.40, P < 0.001 in Class III WRF (Figure 3a). Figure 3b shows the relationship between WRF measured by an increase in serum creatinine or decrease in eGFR and mortality risk on a continuous scale.
A linear trend was observed between the development of WRF and the baseline renal function as estimated by serum creatinine (Figure 4), indicating that the risk for WRF was larger in patients with more renal function impairment at baseline.
Figure 2. Combined adjusted all-cause mortality: any degree of WRF. Test for heterogeneity: Chi2 = 10.42, df = 7 (P = 0.166), I2 = 32.8%. Test for overall effect: Z = 8.35 (P < 0.001). * Total OR = 1.64 (1.45 – 1.86) when Jose removed[20].

106
Chapter 5
Time to endpointSecondary analysis revealed two studies that reported only in-hospital mortality.[14,22]Two
reports reported in-hospital mortality, 30 day mortality and 6 months mortality as well.[16,19] Other reports analyzed mortality at 1 and/or 6 months,[8,17] while four reports assessed mortality rates after more than 6 months of baseline measurements.[7,18,20,21] Figure 5 shows OR related to the time until endpoint occurrence. We observed a declining trend in risk for mortality with increasing time til endpoint.
Hospitalized versus Out-hospital patientsWe analyzed the difference between the prognostic value of WRF in patients who were
admitted to hospital and those who were out-hospital patients. Hospitalized patients with WRF had similar chance to die 6 months after hospitalization OR = 1.61, 95% CI 1.35 to 1.93, P < 0.001 as was observed with out-hospital patients OR = 1.69, 95% CI 1.45 to 1.94, P < 0.001 (Figure 6).
Figure 3a. All-cause mortality: stratification by classes of WRF.Class I WRF: Test for heterogeneity: Chi2 = 2.70 df = 6 (P = 0.100), I2 = 63%.Class II WRF: Test for heterogeneity: Chi2 = 0.73 df = 6 (P = 0.994), I2 = 0%. * Total OR = 1.48 (1.34 – 1.64) when Jose removed [20].Class III WRF: Test for heterogeneity: Chi2 = 4.17 df = 2 (P = 0.124), I2 = 52.0%.

WRF and outcome
107
DiscussionTo our knowledge, this is the first systematic review to address the relation between WRF
and outcome in patients with HF. WRF was common, whether defined by increase in serum creatinine or estimated GFR, especially in patients with impaired renal function at baseline. It was associated with an increase in mortality and all-cause hospitalization especially when WRF was more severe. This was true whether WRF occurred in the setting of acute or chronic HF. On the other hand, improvement in renal function, reported in only one study, was associated with a better prognosis.[18]
Definition of worsening renal functionAn important problem that arises with the use of serial serum creatinine measurements as a
marker of kidney function, is the exponential relationship between serum creatinine and GFR. Therefore, a rise in serum creatinine in patients with higher baseline values will be associated with a less marked decline in GFR than a similar rise in patients with lower baseline values. However, the consequences of a fall in GFR from 25 to 20 ml/min/1.73m2 may be greater than a decline from 60 to 40 ml/min/1.73m2. This emphasizes the need to use estimated GFR to assess changes in renal function. We recently showed that these formulas are also biased in estimating GFR in patients with HF;[23] underestimating GFR in the higher regions and overestimating GFR in the lower regions. We concluded that the (s)MDRD-formula is the
Figure 3b. All-cause mortality: stratification by degree of WRF on a continuous scale. The area of each circle is proportional to the sample size of each cohort. The center line shows the estimated risk for worsening of renal function on a continuous scale. Dotted lines represent the 95% confidence intervals.
0,0 0,1 0,2 0,3 0,4 0,5 0,6
Od
ds
Rat
io
0,0
1,0
2,0
3,0
4,0
5,0
6,0
0,0 2,5 5,0 9,0 13,0 15,0 17,0
Increase in serum creatinine (mg/dl)
Estimated decrease in GFR (ml/min/1.73m2)
Class I Class IIIClass IIReference decline
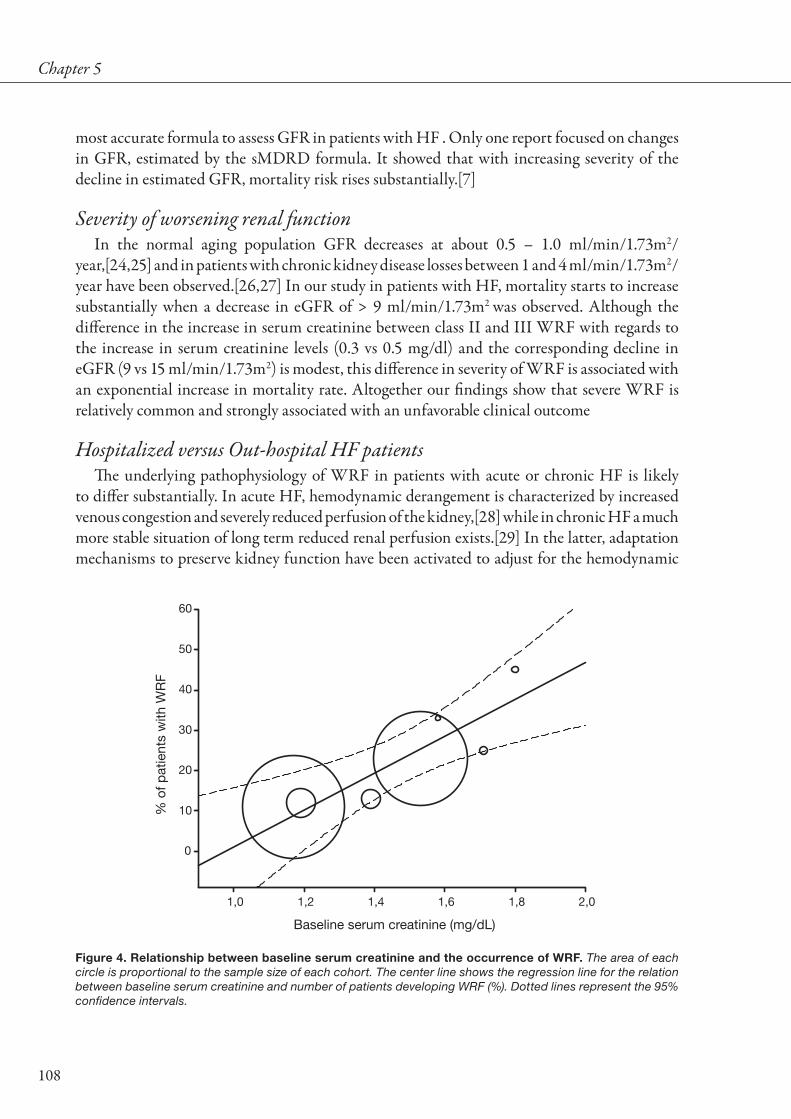
108
Chapter 5
most accurate formula to assess GFR in patients with HF . Only one report focused on changes in GFR, estimated by the sMDRD formula. It showed that with increasing severity of the decline in estimated GFR, mortality risk rises substantially.[7]
Severity of worsening renal functionIn the normal aging population GFR decreases at about 0.5 – 1.0 ml/min/1.73m2/
year,[24,25] and in patients with chronic kidney disease losses between 1 and 4 ml/min/1.73m2/year have been observed.[26,27] In our study in patients with HF, mortality starts to increase substantially when a decrease in eGFR of > 9 ml/min/1.73m2 was observed. Although the difference in the increase in serum creatinine between class II and III WRF with regards to the increase in serum creatinine levels (0.3 vs 0.5 mg/dl) and the corresponding decline in eGFR (9 vs 15 ml/min/1.73m2) is modest, this difference in severity of WRF is associated with an exponential increase in mortality rate. Altogether our findings show that severe WRF is relatively common and strongly associated with an unfavorable clinical outcome
Hospitalized versus Out-hospital HF patientsThe underlying pathophysiology of WRF in patients with acute or chronic HF is likely
to differ substantially. In acute HF, hemodynamic derangement is characterized by increased venous congestion and severely reduced perfusion of the kidney,[28] while in chronic HF a much more stable situation of long term reduced renal perfusion exists.[29] In the latter, adaptation mechanisms to preserve kidney function have been activated to adjust for the hemodynamic
Figure 4. Relationship between baseline serum creatinine and the occurrence of WRF. The area of each circle is proportional to the sample size of each cohort. The center line shows the regression line for the relation between baseline serum creatinine and number of patients developing WRF (%). Dotted lines represent the 95% confidence intervals.
Baseline serum creatinine (mg/dL)
1,0 1,2 1,4 1,6 1,8 2,0
% o
f pat
ient
s w
ith W
RF
0
10
20
30
40
50
60

WRF and outcome
109
state that has developed,[30] while in the acute setting the kidney is much more vulnerable to rapid and considerable changes in hemodynamics. Although we observed no difference in the mortality risk associated with WRF between both groups, caution must be taken to extrapolate our findings to the general HF patient population.
Cause of worsening renal functionThe precise pathophysiology of WRF in patients with HF is unclear. Renal dysfunction
is generally considered as a reflection of decreased kidney perfusion, which decreases disproportionally fast with declining cardiac output.[29] Within this perspective, the kidney can be considered as a sensitive marker of decreased organ perfusion. Multiple reports focused on variables associated with worsening of kidney function. In the presence of comorbid conditions related to mortality, such as impaired baseline renal function, impaired left ventricular function, higher blood pressure, higher age and symptoms of HF, WRF occurs more frequently.[7,14,16,18,31] However, no serial measurements of these variables, and especially variables of left ventricular function, were reported in any of these studies. Other studies focused on worsening of renal function in response to the initiation of treatment, such as ACE-inhibitor therapy, diuretics and nesiritide.[13,32-34] Future research is warranted to elucidate the factors involved in the worsening and improvement of renal function in HF.
ImplicationsThis analysis has several important clinical implications. First, we show that risk appraisal
using renal function in patients with HF is a dynamic process which can be improved using longitudinal information. In addition, it is especially important to monitor the time course of renal function in already compromised patients while predominantly these patients are at
Figure 5. All-cause mortality: stratification by time until endpoint and any degree of WRF.* Krumholz (2000), † Cowie (2006), ‡ Forman (2004), § Chittineni (2007), []Akhter (2004), ¶ Smith (2003), #Jose (2006), ** Owan (2006), †† De Silva (2005), ‡‡ Khan (2006)
Time till all-cause mortality
Inhospital 1 Month 6 Month > 6 Month
Od
ds
Rat
io (9
5% C
I)
1
10
100
*
**#
[]
§
**
†
†
† ††
‡
‡
‡‡
¶

110
Chapter 5
greater risk of developing WRF. Also, renoprotective therapy focusing on the prevention or progression of renal impairment may prove to be a valuable new treatment strategy, not only to preserve renal function but also to improve survival.
Study LimitationsQuality of studies was assessed using an instrument with 11 variables, according to published
guidelines and recommendations.[9,10] All included studies were at least of fair quality and all met at least seven of eleven criteria.
Accurate assessment of changes in serum creatinine may be hampered by the problem of assay variability of laboratory measurements. Inter-laboratory differences in measurements are also likely to exist. Therefore, the presented increases in serum creatinine might be biased and may therefore not reflect actual changes in renal function. However, the possibility of reporting potential biased estimates might be diluted by regression to the mean due to the use of multiple measurements.
Furthermore, a change in predictive performance of estimated GFR from underestimation to overestimation, across the spectrum from mild to severe CHF, has been reported before.[23] This could mean that, although estimated GFR suggests a stable condition over time, it may in fact be declining. If one takes that into account, the results would be even more convincing.
Figure 6 All-cause mortality: stratification by type of patient; in- versus out-hospital patients.In-hospital patients: Test for heterogeneity: Chi2 = 6.20, df = 4 (P = 0.185), I2 = 35.5 %. Test for overall effect: Z = 5.26 (P < 0.001). Out-hospital patients: Test for heterogeneity: Chi2 = 2.32, df = 2 (P = 0.313), I2 = 14.0 %. Test for heterogeneity between subgroups: P = 0.169.Total: Test for heterogeneity: Chi2 = 10.42, df = 7 (P = 0.166), I2 = 32.8%.
Krumholz (2000), n = 1681 235/1212119/4691.41 (1.10 - 1.82)
Smith (2003), n = 412 27/22735/1851.73 (1.00 - 2.98)
Akhter (2004), n = 480 68/36145/1192.62 (1.66 - 4.13)
Cowie (2006), n = 299 35/20126/981.71 (0.96 - 3.05)
Owan (2006), n = 6052 3215/46331095/14191.41 (1.30 - 1.71)
Odds ratio (95% CI)
No. of events
WRF n/N no WRF n/NInhospital patients
Outhospital patients
Subtotal 1.61 (1.35 - 1.93) 1320/2290 3580/6634
Khan (2006), n = 6535
Jose (2006), n = 1854
De Silva (2005), n = 1216
879/4475628/20601.79 (1.59 - 2.02)
316/163158/2231.46 (1.30 - 1.71)
219/105544/1611.44 (0.98 - 2.09)
Subtotal 1.69 (1.48 - 1.94) 730/2444 1414/7161
Overall 1.62 (1.45 - 1.82 2050/4734 4994/13795
.1 .2 .5 12 48
Higher risk for WRFLower risk for WRF

WRF and outcome
111
Several post hoc analyses of prospective randomized controlled trials with or without ACE-inhibitor therapy, were included in the present analysis, which might have biased the findings. ACE-inhibition causes an acute decline in GFR, preserves it on the long term, and ultimately leads to improved prognosis. As a consequence WRF in these groups may actually reflect the initiation of the treatment that will in the long term lead to improved prognosis, indicating that the cause of WRF is not always directly related to poorer outcome.
ConclusionWorsening of renal function is commonly observed in patients with HF, especially in
those who already have renal dysfunction. WRF is associated with an increase in subsequent mortality, especially in patients with more severe WRF, and all cause. On the other hand, improving of kidney function is associated with better survival. Therefore therapy targeted at improving kidney function in HF may be a valuable new strategy to improve prognosis in this high mortality patient group. Further research is needed to investigate the factors associated with WRF.
ReferencesDries DL, Exner DV, Domanski MJ, Greenberg B, and Stevenson LW. The prognostic implications of 1. renal insufficiency in asymptomatic and symptomatic patients with left ventricular systolic dysfunction. J Am Coll Cardiol. 2000;35:681-689.
Hillege HL, Girbes AR, de Kam PJ et al. Renal function, neurohormonal activation, and survival in 2. patients with chronic heart failure. Circulation. 2000;102:203-210.
Al Ahmad A, Rand WM, Manjunath G et al. Reduced kidney function and anemia as risk factors for 3. mortality in patients with left ventricular dysfunction. J Am Coll Cardiol. 2001;38:955-962.
Hillege HL, Nitsch D, Pfeffer MA et al. Renal function as a predictor of outcome in a broad spectrum 4. of patients with heart failure. Circulation. 2006;113:671-678.
Smith GL, Lichtman JH, Bracken MB et al. Renal impairment and outcomes in heart failure: systematic 5. review and meta-analysis. J Am Coll Cardiol. 2006;47:1987-1996.
Haynes RB, McKibbon KA, Wilczynski NL, Walter SD, and Werre SR. Optimal search strategies 6. for retrieving scientifically strong studies of treatment from Medline: analytical survey. BMJ. 2005;330:1179.
Khan NA, Ma I, Thompson CR et al. Kidney function and mortality among patients with left ventricular 7. systolic dysfunction. J Am Soc Nephrol. 2006;17:244-253.
Smith GL, Vaccarino V, Kosiborod M et al. Worsening renal function: what is a clinically meaningful 8. change in creatinine during hospitalization with heart failure? J Card Fail. 2003;9:13-25.
Harris RP, Helfand M, Woolf SH et al. Current methods of the US Preventive Services Task Force: a 9. review of the process. Am J Prev Med. 2001;20:21-35.

112
Chapter 5
Altman DG. Systematic reviews of evaluations of prognostic variables. BMJ. 2001;323:224-228.10.
Goldberg A, Hammerman H, Petcherski S et al. Inhospital and 1-year mortality of patients who 11. develop worsening renal function following acute ST-elevation myocardial infarction. Am Heart J. 2005;150:330-337.
Koreny M, Karth GD, Geppert A et al. Prognosis of patients who develop acute renal failure during the 12. first 24 hours of cardiogenic shock after myocardial infarction. Am J Med. 2002;112:115-119.
Hillege HL, van Gilst WH, van Veldhuisen DJ et al. Accelerated decline and prognostic impact of renal 13. function after myocardial infarction and the benefits of ACE inhibition: the CATS randomized trial. Eur Heart J. 2003;24:412-420.
Forman DE, Butler J, Wang Y et al. Incidence, predictors at admission, and impact of worsening renal 14. function among patients hospitalized with heart failure. J Am Coll Cardiol. 2004;43:61-67.
Gottlieb SS, Abraham W, Butler J et al. The prognostic importance of different definitions of worsening 15. renal function in congestive heart failure. J Card Fail. 2002;8:136-141.
Krumholz HM, Chen YT, Vaccarino V et al. Correlates and impact on outcomes of worsening renal 16. function in patients > or =65 years of age with heart failure. Am J Cardiol. 2000;85:1110-1113.
Akhter MW, Aronson D, Bitar F et al. Effect of elevated admission serum creatinine and its worsening 17. on outcome in hospitalized patients with decompensated heart failure. Am J Cardiol. 2004;94:957-960.
de Silva R, Nikitin NP, Witte KK et al. Incidence of renal dysfunction over 6 months in patients with 18. chronic heart failure due to left ventricular systolic dysfunction: contributing factors and relationship to prognosis. Eur Heart J. 2006;27:569-581.
Cowie MR, Komajda M, Murray-Thomas T, Underwood J, and Ticho B. Prevalence and impact of 19. worsening renal function in patients hospitalized with decompensated heart failure: results of the prospective outcomes study in heart failure (POSH). Eur Heart J. 2006;27:1216-1222.
Jose P, Skali H, Anavekar N et al. Increase in Creatinine and Cardiovascular Risk in Patients with 20. Systolic Dysfunction after Myocardial Infarction. J Am Soc Nephrol. 2006;10:2886-2891.
Owan TE, Hodge DO, Herges RM et al. Secular trends in renal dysfunction and outcomes in hospitalized 21. heart failure patients. J Card Fail. 2006;12:257-262.
Chittineni H, Miyawaki N, Gulipelli S, and Fishbane S. Risk for Acute Renal Failure in Patients 22. Hospitalized for Decompensated Congestive Heart Failure. Am J Nephrol. 2007;27:55-62.
Smilde TD, van Veldhuisen DJ, Navis G, Voors AA, and Hillege HL. Drawbacks and prognostic value 23. of formulas estimating renal function in patients with chronic heart failure and systolic dysfunction. Circulation. 2006;114:1572-1580.
Hoang K, Tan JC, Derby G et al. Determinants of glomerular hypofiltration in aging humans. Kidney 24. Int. 2003;64:1417-1424.
Halbesma N, Kuiken DS, Brantsma AH et al. Macroalbuminuria is a better risk marker than low 25. estimated GFR to identify individuals at risk for accelerated GFR loss in population screening. J Am Soc Nephrol. 2006;17:2582-2590.

WRF and outcome
113
Eriksen BO and Ingebretsen OC. The progression of chronic kidney disease: a 10-year population-based 26. study of the effects of gender and age. Kidney Int. 2006;69:375-382.
Hunsicker LG, Adler S, Caggiula A et al. Predictors of the progression of renal disease in the Modification 27. of Diet in Renal Disease Study. Kidney Int. 1997;51:1908-1919.
Binanay C, Califf RM, Hasselblad V et al. Evaluation study of congestive heart failure and pulmonary 28. artery catheterization effectiveness: the ESCAPE trial. JAMA. 2005;294:1625-1633.
Ljungman S, Laragh JH, and Cody RJ. Role of the Kidney in Congestive Heart-Failure - Relationship 29. of Cardiac Index to Kidney-Function. Drugs. 1990;39:10-21.
Bongartz LG, Cramer MJ, Doevendans PA, Joles JA, and Braam B. The severe cardiorenal syndrome: 30. 'Guyton revisited'. Eur Heart J. 2005;26:11-17.
Butler J, Forman DE, Abraham WT et al. Relationship between heart failure treatment and development 31. of worsening renal function among hospitalized patients. Am Heart J. 2004;147:331-338.
Weinfeld MS, Chertow GM, and Stevenson LW. Aggravated renal dysfunction during intensive therapy 32. for advanced chronic heart failure. Am Heart J. 1999;138:285-290.
Iglesias J, Hom D, Antoniotti M, Ayoub S, and Levine JS. Predictors of worsening renal function in 33. adult patients with congestive heart failure receiving recombinant human B-type brain natriuretic peptide (nesiritide). Nephrol Dial Transplant. 2006;21:3458-3465.
de Silva R, Nikitin NP, Witte KK et al. Effects of applying a standardised management algorithm for 34. moderate to severe renal dysfunction in patients with chronic stable heart failure. Eur J Heart Fail. 2007;9:415-423.

114
Chapter 5

Chapter 6Both in and outhospital worsening of renal function predict outcome in
patients with heart failure Results from the Coordinating Study Evaluating Outcome of Advising
and Counseling in Heart Failure (COACH)
Kevin Damman, Tiny Jaarsma, Adriaan A. Voors, Gerjan Navis, Hans L. Hillege and Dirk J. van Veldhuisen
Submitted

116
Abstract
BackgroundImpaired renal function and inhospital worsening of renal function (WRF) are common in patients
with heart failure and associated with poor outcome. The effect of WRF after discharge on outcome in these patients is unknown.
Methods and resultsThe Coordinating Study Evaluating Outcome of Advising and Counseling in Heart Failure (COACH)
included 1023 heart failure patients. We assessed estimated glomerular filtration rate (eGFR) and serum creatinine at admission, discharge, and 6 and 12 months after discharge. WRF was defined as increase in serum creatinine of > 0.3 mg/dL and >25%. The primary outcome was a composite of all-cause mortality and heart failure admissions. Mean age was 71 ± 11 years, 62% were male. Mean eGFR at admission was 55 ± 21 mL/min/1.73m2 while mean LVEF was 33 ± 14%. Inhospital WRF occurred in 11% of patients, while 16% and 9% experienced WRF from 0 to 6, and 6 to 12 months after discharge, respectively. The occurrence of WRF or lower eGFR over time was associated with lower baseline eGFR, anemia, history of hypertension, type II diabetes, peripheral artery disease, and higher age. In multivariate analysis, WRF at any point in time was associated with worse prognosis: HR 1.41 (1.01–1.97), P = 0.047 for inhospital WRF, HR 1.94 (1.13 – 3.34), P = 0.016 for WRF between 0-6 months, and HR 3.47 (1.36 – 8.87), P = 0.009 for WRF between 6-12 months.
ConclusionBoth in and outhospital worsening of renal function are independently related to poor prognosis in
patients with heart failure, suggesting that renal function in heart failure patients should be monitored long after discharge.

WRF at different timepoints and outcome
117
IntroductionRenal impairment is common in both patients with acute heart failure (AHF), and patients
with chronic heart failure (CHF). In both patient groups, renal impairment is a prominent predictor of poor survival and (re)hospitalizations for heart failure [1-4]. However, not only baseline renal function is important in these patients, but also worsening renal function (WRF) is an independent predictor of prognosis [5-7]. In a pooled analysis of 8 studies, we recently showed that WRF was associated with a significantly higher mortality rate in both patients with AHF and CHF [8]. Results of the AHF studies were based on WRF which was defined as inhospital worsening of renal function [6,7,9-14]. Importantly, WRF is also frequently observed after discharge in both patients with heart failure and after myocardial infarction, but to this date there is no information of the effect of such worsening of renal function on prognosis [5,15-17]. While outhospital WRF is an important predictor of prognosis in patients with stable CHF [5,15], it is important to know whether similar associations are present in patients with AHF, long after they have been discharged from the hospital. In the present study, we therefore set out to investigate the effect of WRF at different time points during hospitalization and after discharge on prognosis in AHF patients who participated in the Coordinating Study Evaluating Outcome of Advising and Counseling in Heart Failure (COACH) [18].
MethodsThis was a retrospective analysis of the Coordinating Study Evaluating Outcome of
Advising and Counseling in Heart Failure (COACH), a multicenter, randomized, open trial, with blinded endpoint evaluation, designed to compare basic support and intensive support in patients with heart failure, after hospitalization for AHF, which was conducted from 2002 to 2007 in The Netherlands. A detailed description of the rationale, design, and results of the COACH has been previously published [18,19]. Patients with both reduced and preserved left ventricular ejection fraction (LVEF) were allowed to participate in the study. Patients were routinely followed every 6 months after discharge in all treatment groups, and additional visits in the intensive support and moderate support groups. For comparison purposes, only the 6 months visits, which were available in all treatments arms were used for the primary analysis.
Renal function and WRFWe estimated the glomerular filtration rate (eGFR) using the simplified Modification of
Diet in Renal Disease formula (186.3 x (serum creatinine (mg/dL))-1.154 x age-0.203 x (0.742 if female) (mL/min/1.73m2)), at admission, discharge, 6 months, 12 months and 18 months after discharge [20]. For patients without serum creatinine at discharge, the last known serum creatinine during hospitalization used. WRF was defined as an absolute increase in serum creatinine >0.3mg/dL in combination with >25% increase in serum creatinine between two time points, the latter chosen to adjust for the exponential relationship between eGFR and

118
Chapter 6
serum creatinine [6]. We evaluated WRF at the following time points: from admission to discharge, from discharge to 6 months follow up, and from 6 months to 12 months follow up. For determination of predictors of the occurrence of WRF, and slope of eGFR over time, also WRF from discharge to 2 months and WRF from 2 months to 6 months follow up were considered in patients that attended those visits according to protocol. Serum creatinine at admission was available in all patients, and a discharge serum creatinine was available in 98% (1000) of patients. Among survivors, 72% (647), 73% (582) and 75% (562) had a serum creatinine at 6, 12 and 18 months follow up, respectively.
Follow-up and prognosisPatients were followed for 18 months after index hospitalization. The present analysis was
conducted using the same endpoints as the main study. There were two primary endpoints of the COACH: a composite endpoint of heart failure hospitalization and all-cause mortality. A second primary endpoint was the number of ‘unfavourable days’. This is the number of days a patient is not alive or not out of hospital, as described earlier.[18,19] Secondary endpoints included the individual endpoints in the primary endpoint, all-cause hospitalizations and the number of all-cause hospitalizations per patient.
Statistical analysisData are given as mean ± standard deviation when normally distributed, as median and
interquartile range when skewed distributed, and as frequencies and percentages for categorical variables. Associations between baseline variables were evaluated by means of 1-way ANOVA, the Kruskal-Wallis test, and χ2 or Fisher exact tests, when appropriate. To establish continuous change of renal function over time, and the association with demographic variables, we used two types of analysis. First, we analyzed the occurrence of WRF (> 0.3 mg/dL and > 25% increase in serum creatinine) during the total follow up. Time to determination of serum creatinine was used as follow up variable. A Cox proportional hazard analysis, with WRF being the failure variable, was used to determine univariate and multivariate predictors of the occurrence of WRF. Second, we constructed a multilevel mixed-effects linear model using the xtmixed command in STATA to investigate the determinants of the slope of eGFR over time. Finally, we used a Cox proportional hazards model to estimate hazard ratios with 95% confidence intervals (CI) for the association between WRF and prognosis, and in a secondary model we estimated the effect of time-varying eGFR on outcome. Missing values were imputed using expectation maximization as estimation method. Two-sided P values were used, taking P < 0.05 to be statistically significant. Statistical analyses were performed using SPSS, Chicago version 12.0 and STATA, College Station, Texas, version 10.0.

WRF at different timepoints and outcome
119
ResultsIn the present analysis, all 1023 patients who were randomized in the COACH were
included. Table 1 shows characteristics at baseline (admission) for the study population and the characteristics for the different subgroups of patients that experienced WRF. Mean age was 71 ± 11 years, while 62% were male. Mean eGFR was 55 ± 21 mL/min/1.73m2, while 59% had chronic kidney disease as defined as eGFR below 60 mL/min/1.73m2.
During index hospitalization, the mean change in serum creatinine showed a normal distribution, with a mean delta serum creatinine of 0.01 ± 0.44 mg/dL (Figure 1A). Changes in serum creatinine from discharge to 6 months, from 6 months to 12 months, and 12 to 18 months follow up were also normally distributed and showed a mean delta serum creatinine of 0.08 ± 0.45 mg/dL, 0.05 ± 0.34 mg/dL, and 0.04 ± 0.44 mg/dL, respectively (Figure 1 B-D). This corresponded to a change in eGFR of -1.1 inhospital, and -2.1, -2.3, and -0.7 mL/min/1.73m2, respectively per 6 month follow up.
Inhospital WRF occurred in 11% of patients, while 16% and 9% of patients experienced WRF from discharge to 6 months, and from 6 to 12 months follow-up, respectively. Of those
Figure 1. Distributions of the change in serum creatinine inhospital, 6 months, 12 months and 18 months.A. Absolute change in serum creatinine inhospitalB. Absolute change in serum creatinine 0 – 6 monthsC. A bsolute change in serum creatinine 6 -12 monthsD. Absolute change in serum creatinine 12 – 18 months
Delta serum creatinine (mg/dL) inhospital-1.5 -1.2 -0.9 -0.6 -0.3 0.0 +0.3 +0.6 +0.9 +1.2 +1.5 +1.8
Per
cent
age
of p
atie
nts
0
5
10
15
20
25
A
Delta serum creatinine (mg/dL) 0 - 6 months-1.5 -1.2 -0.9 -0.6 -0.3 0.0 +0.3 +0.6 +0.9 +1.2 +1.5 +1.8
Per
cent
age
of p
atie
nts
0
5
10
15
20
25
B
Delta serum creatinine (mg/dL) 6 - 12 months-1.5 -1.2 -0.9 -0.6 -0.3 0.0 +0.3 +0.6 +0.9 +1.2 +1.5 +1.8
Per
cent
age
of p
atie
nts
0
5
10
15
20
25
C
Delta serum creatinine (mg/dL) 12 - 18 months-1.5 -1.2 -0.9 -0.6 -0.3 0.0 +0.3 +0.6 +0.9 +1.2 +1.5 +1.8
Per
cent
age
of p
atie
nts
0
5
10
15
20
25
D

120
Chapter 6
Table 1. Baseline characteristics according to the occurrence of WRF
Characteristics No WRF
(N = 763)
WRFInhospital(N = 106)
WRF0 – 6 months
(N = 101)
WRF6 – 12 months
(N = 43)
Age (years) 70 ± 12 73 ± 11* 73 ± 10* 71 ± 9
Male sex (%) 64 59 55 77
LVEF (%) 33 ± 14 36 ± 16 34 ± 14 34 ± 13
NYHA
II 5 7 4 2
III 51 44 47 49
IV 44 49 49 49
Blood pressure
SBP (mmHg) 118 ± 21 119 ± 21 118 ± 22 124 ± 23
DBP (mmHg) 68 ± 12 69 ± 12 69 ± 14 69 ± 12
Heart rate (bpm) 75 ± 13 75 ± 13 74 ±14 74 ± 14
Comorbidities
Hypertension 42 45 52* 54
Atrial fibrillation 44 45 48 40
Diabetes 26 30 38 28
Stroke 9 11 8 14
COPD 26 31 29 30
PAD 15 24 12 21
MI 43 34 52* 51
Renal function
Serum creatinine (mg/dL) 1.41 ± 0.7 1.24 ± 0.4* 1.37 ± 0.6 1.39 ± 0.4
Blood urea nitrogen (mmol/L) 11 ± 6 10 ± 6 11 ± 5 11 ± 6
eGFR (ml/min/1.73m2) 56 ± 22 60 ± 21 57 ± 22 56 ± 21
Hemoglobin levels (g/dL) 13.5 ± 2.0 13.5 ± 2.0 13.1 ± 1.8* 13.9 ± 1.9
Medication (%)
ACEi 50 51 60 63
ARB 13 12 11 12
Beta-blockers 46 43 43 54
Loop Diuretics 75 75 77 74
Digoxin 22 21 26 26
Statins 33 41 33 47
Calcium antagonists 20 23 21 30
* P < 0.05 for difference with no WRF. Abbreviations: LVEF: left ventricular ejection fraction, NYHA: New York heart association, SBP: systolic blood pressure, DBP: diastolic blood pressure, COPD: chronic obstructive pulmonary disease, PAD: peripheral artery disease, MI: history of myocardial infarction, eGFR: estimated glomerular filtration rate, ACEi: angiotensin converting enzyme inhibition, ARB: angiotensin II receptor antagonist

WRF at different timepoints and outcome
121
patients who experienced WRF between discharge and 6 months, only 9% had experienced WRF inhospital. In contrast, 26% of patients that experienced WRF from 6 to 12 months follow-up had experienced WRF during hospitalization or from discharge to 6 months. During 18 months follow up, mean eGFR among survivors decreased from 60 ± 24 mL/min/1.73m2 at admission to 53 ± 23 mL/min/1.73m2 after 18 months (Figure 2).
Predictors of occurrence of WRF and change in eGFR over timeAfter 18 months follow up, in total 260 (25%) of patients had experienced WRF at any point
in time. In univariate analysis, significant predictors of WRF were a history of type II diabetes (Hazard ratio (HR) 1.53, 95% confidence interval (CI) 1.14 to 2.03, P = 0.004), peripheral artery disease (HR 1.44 (95% CI 1.06 to 1.94) P = 0.019), age (HR 1.17 per 10 years increase (95% CI 1.05 to 1.31) , P = 0.007), as well as the presence of anemia (HR 1.42 (95% CI 1.11 to 1.84), P = 0.008). Randomized treatment assignment showed no effect on the occurrence of WRF. Table 2 shows the outcome of the multivariate analysis, showing that age, type II diabetes, and anemia, were independent predictors of the occurrence of WRF.
Figure 2. Change of mean eGFR over time in survivors. Abbreviations: eGFR: estimated glomerular filtration rate
Admiss
ion
Discha
rge
6 m
onth
s
12 m
onth
s
18 m
onth
s
eGFR
(mL/
min
/1.7
3m2 )
0
40
45
50
55
60
65
70
P < 0.001 for trend
Table 2. Multivariate Cox proportional hazard analysis for the occurrence of WRF at any point
Variable Hazard Ratio (95% CI) P-value
Age (per 10 year increase) 1.13 1.01 to 1.27 0.037
History of type II diabetes 1.51 1.13 to 2.01 0.006
Anemia 1.36 1.13 to 1.76 0.022

122
Chapter 6
In a multilevel mixed-effects linear model estimating eGFR change over time, the most important predictor of eGFR change was renal function at admission. Other predictors included a history of hypertension, type II diabetes, atrial fibrillation, pulmonary disease, and
Figure 3. Relationship between WRF at different time points and the combined endpoint. Reference group of No WRF depicts patients who did not have WRF during that particular time period. Abbreviations: WRF: worsening renal function.A. Relationship between WRF inhospital and combined endpointB. Relationship between WRF from 0 to 6 months and combined endpointC. Relationship between WRF from 6 to 12 months and combined endpoint
Follow Up time (days)
600 120 180 240 300 360 420 480 540 600
Cum
ulat
ive
surv
ival
(Dea
th o
r H
F ad
mis
sion
)
0,0
0,4
0,5
0,6
0,7
0,8
0,9
1,0
No WRFWRF
WRF Inhospital A
Follow Up time (days)
0 60 120 180 240 300 360
Cum
ulat
ive
surv
ival
(Dea
th o
r H
F ad
mis
sion
)
0,0
0,4
0,5
0,6
0,7
0,8
0,9
1,0
No WRFWRF
WRF from discharge to 6 months B
Follow Up time (days)
0 60 120 180
Cum
ulat
ive
surv
ival
(Dea
th o
r H
F ad
mis
sion
)
0,0
0,4
0,5
0,6
0,7
0,8
0,9
1,0
No WRFWRF
WRF from 6 to 12 months C

WRF at different timepoints and outcome
123
peripheral artery disease, low sodium diet, gender, age, beta-blocker therapy and hemoglobin levels. The multivariate mixed model results are depicted in table 3. These results indicate that the slope of eGFR over time is particularly dependent on baseline eGFR, a fixed effect, with an extra random effect of baseline eGFR on the individual patient level.
Prognosis.During the 18 months follow up of the study, in total 411 patients reached the primary
endpoint. Both eGFR at admission (HR 1.23 per 10 mL/min/1.73m2 decrease, 95% CI 1.17 to 1.29, P < 0.001), and eGFR at discharge (HR 1.24 per 10 mL/min/1.73m2 decrease, 95% CI 1.17 to 1.30, P < 0.001), were prominent predictors of survival. Inhospital WRF was a not a predictor of the primary endpoint on the long term HR 1.24, 95% CI 1.92 to 1.68, P = 0.157 (figure 3a). Both WRF from discharge to 6 months HR 1.62, 95% CI 1.08 to 2.43, P = 0.019 and WRF from 6 to 12 months HR 3.71, 95% CI 1.84 to 7.49, P < 0.001, were related to a poor outcome (Figure 3b and 3c). In a landmark analysis, WRF at any of the pre-specified time-intervals was significantly related to impaired prognosis (Figure 4). This resulted in a HR 1.55, 95% CI 1.07 to 2.26, P = 0.022, for inhospital WRF, HR 2.02, 95% CI 1.20 to 3.40, P = 0.008 for WRF between 0 and 6 months and HR 3.68, 95% CI 1.83 to 7.41, P < 0.001 for WRF between 6 and 12 months (all 6 months follow up). Table 4 shows the unadjusted and adjusted hazard ratio’s for WRF at the different time points for all endpoints with 6 months follow up for each category. WRF during any time was independently associated with a greater risk for the primary endpoint of all-cause mortality and heart failure hospitalizations. This was mainly attributable to the effect on all-cause mortality, with the effect on heart failure admissions subsiding in multivariate analysis.
In a secondary model, we investigated the relationship of eGFR with the primary endpoint when introduced as a time-dependent covariate. In multivariate analysis, eGFR was an independent predictor of the combined endpoint (HR 1.19 per 10 mL/min/1.73m2 decrease
Table 3. Multilevel longitudinal mixed effects model for eGFR over time
Variable Coefficient (95% CI) Z-score P-value
Fixed effects
Baseline eGFR (per 10 mL/min/1.73m2 decrease) -7.86 -8.17 to -7.54 - 49.2 < 0.0001
Age (per 10 years increase) -1.56 -2.09 to -1.03 - 5.8 < 0.0001
Hemoglobin levels (per g/dl decrease) -0.74 -0.88 to -0.34 - 4.4 < 0.0001
History of hypertension -1.56 -2.69 to -0.44 - 2.72 0.0065
History of type II diabetes -1.41 -2.84 to 0.02 - 1.9 0.0533
History of peripheral artery disease -1.57 -3.04 to -0.10 - 2.1 0.0363
Random effects SD random Coeff
Baseline eGFR (per 10 mL/min/1.73m2 decrease) 1.80 1.43 to 2.26
Abbreviations: eGFR: estimated glomerular filtration rate, SD: standard deviation, Coeff: coefficient

124
Chapter 6
(95% CI 1.07 to 1.31), P < 0.001). Other independent time dependent predictors were hemoglobin levels (HR 1.13 per g/dL decrease (95% CI 1.04 to 1.23), P = 0.005), age (HR 1.03 per year increase (95% CI 1.01 to 1.06), P = 0.006) and diuretic use (HR 2.64 (95% CI 1.05 to 6.63), P = 0.039).
Figure 5 shows the association between the second primary endpoint of the COACH (unfavourable days) and the occurrence of WRF. In general, the presence of WRF resulted in a higher number of unfavourable days compared to patients without WRF. Especially patients who experienced WRF after discharge showed a significantly higher number of unfavourable days compared to patients with preserved renal function.
Tab
le 4
. Rel
atio
nsh
ip b
etw
een
occ
urr
ence
of
WR
F an
d o
utc
om
e
WR
F in
hosp
ital
(N =
100
0)W
RF
fro
m 0
to
6 m
ont
hs(N
= 6
32)
WR
F fr
om
6 t
o 1
2 m
ont
hs(N
= 4
76)
Eve
nts
Una
dju
sted
H
R (9
5% C
I)A
dju
sted
*H
R (9
5% C
I)U
nad
just
ed
HR
(95%
CI)
Ad
just
ed*
HR
(95%
CI)
Una
dju
sted
HR
(95%
CI)
Ad
just
ed*
HR
(95%
CI)
Prim
ary
end
poi
nt1.
55 (1
.07
– 2.
26)†
1.63
(1.1
0 –
2.40
)†2.
02 (1
.20
– 3.
40)‡
2.06
(1.1
3 –
3.74
)†3.
68 (1
.83
– 7.
41)#
5.03
(2.1
3 –
11.8
8)#
All-
caus
e m
orta
lity
1.86
(1.1
7 –
2.94
)‡1.
73 (1
.16
– 2.
60)‡
2.47
(1.2
9 –
4.74
)‡4.
80 (2
.15
– 10
.70)
#5.
08 (2
.26
– 11
.39)
#8.
44 (2
.79
– 25
.50)
#
Hea
rt fa
ilure
hos
pita
lizat
ions
1.30
(0.7
8 –
2.17
)1.
50 (0
.89
– 2.
53)
1.76
(0.9
5 –
3.27
)1.
65 (0
.81
– 3.
38)
4.88
(2.1
7 –
10.9
8)#
6.56
(2.2
1 –
19.4
3)‡
All-
caus
e ho
spita
lizat
ions
1.47
(1.0
8 –
1.99
)†1.
57 (1
.15
– 2.
15)‡
1.57
(0.9
0 –
2.75
)1.
18 (0
.61
– 2.
29)
1.42
(0.5
6 –
3.56
)1.
89 (0
.64
– 5.
59)
* A
dju
sted
fo
r ag
e, g
end
er, N
YH
A c
lass
, LV
EF,
tre
atm
ent
assi
gnm
ent,
sys
tolic
and
dia
sto
lic b
loo
d p
ress
ure,
hea
rt r
ate,
hem
oglo
bin
leve
ls, e
GFR
at
bas
elin
e, t
he
occ
urre
nce
of W
RF
bef
ore
the
stu
die
d p
erio
d,
med
ical
the
rapy
, his
tory
of m
yoca
rdia
l inf
arct
ion
/ at
rial
fib
rilla
tion
/ d
iab
etes
/ s
tro
ke /
CO
PD
/ H
yper
tens
ion
/ P
e-ri
phe
ral a
rter
y d
isea
se /
tim
e si
nce
dia
gno
sis
of h
eart
fai
lure
† P
< 0
.05
‡ P
< 0
.01
# P
< 0
.001

WRF at different timepoints and outcome
125
DiscussionOur present study is the first to show that in patients with AHF not only WRF during
hospitalization, but also WRF on short and moderate time span after hospitalization is common and associated with impaired survival. WRF after hospitalization is associated with a higher number of unfavourable days compared to patients that had relatively preserved renal function.
Renal impairment at any point in time has been recognized as an important risk factor in both CHF and AHF. When present, decreased renal function predisposes to further worsening of renal function [7,15]. In a recent meta-analysis, we showed that both in AHF and CHF patient populations, the risk associated with WRF for all-cause mortality was similar [8]. Furthermore, we showed that the risk associated with a decline in renal function started to rise when an increase in serum creatinine was observed of > 0.3 mg/dl. This cut-off for WRF has been used by others, and has been suggested as clinically meaningful [5,14]. To address the exponential relationship between serum creatinine, we included also a relative increase in serum creatinine in our definition of WRF, which may give a more reliable estimate of WRF.[7] Using this definition, the incidence of inhospital WRF in the COACH-study was 11%, which was slightly lower compared to other AHF studies, in which percentages of 12-55% have been observed, depending on definition. The outhospital incidence of WRF (16% and 9%), was more or less similar to the incidence observed in populations with CHF patients. Our sub
Figure 4. Landmark analysis of the relationship between WRF and outcome. * P < 0.01, † P < 0.001. Reference group of No WRF depicts patients who did not have WRF during that particular time period. Patients who experience an endpoint in a previous period are excluded from subsequent analyses. Abbreviations: WRF: worsening renal function
Follow Up time (days)
0 12060 180 240 300 360 420 480 540
Cum
ulat
ive
surv
ival
(Dea
th o
r H
F ad
mis
sion
)
0,0
0,6
0,7
0,8
0,9
1,0
No WRFWRF
WRF inhospital* WRF 0-6 months† WRF 6-12 months†

126
Chapter 6
analysis of the COACH study is the first to investigate changes in renal function both during hospitalization and after discharge in the same patient with heart failure. Our results indicate that even long after discharge, these patients are at increased risk for WRF, and should be monitored closely.
WRF and prognosisIn agreement with other studies in both AHF and CHF, WRF in our population was a strong
and independent predictor of the primary endpoint of the COACH trial, which consisted of heart failure (re)hospitalization and all-cause mortality. The relative risk observed with WRF increased with time after index hospitalization, with the highest mortality of re-hospitalization risk observed when WRF occurred between 6 and 12 months post-discharge. The increased risk with WRF was mainly attributable to an increased all-cause mortality risk. This is consistent with findings from our recent meta-analysis, showing only a borderline increase in the risk for heart failure hospitalization [8]. We also analyzed the relationship between WRF and unfavourable days, the second primary endpoint of the COACH study. WRF occurrence was associated with a significant increase in unfavourable days, especially when WRF was observed outhospital. This is the first analysis to show that WRF is not only associated with an increased risk for mortality, but to a higher number of unfavourable days as well.
Pathophysiology of WRFThe pathophysiology of renal impairment in heart failure is mainly attributable to decreased
renal perfusion and venous congestion, while endothelial dysfunction, neurohormonal activation and inflammation play a mediating role [21,22]. The driving force behind WRF is much less established. In a recent study, Metra et al found that baseline renal impairment, reduced ejection fraction, NYHA class and diuretic dose were independent determinants of WRF [7]. Diuretic use and dosing have been established as important risk factors for WRF in
Figure 5. Relationship between occurrence of WRF and unfavourable days in the COACH. * P < 0.05
180
20
40
60
80
100
120
140
160
61 21 8
No.
of u
nfav
orab
le d
ays
0
20
40
60
80
100
120
140
160
12 18
No WRFWRF
Duration of Follow Up (months)
**
*
*
WRFinhospital
WRF0-6 months
WRF6-12 months

WRF at different timepoints and outcome
127
other studies [15,23]. Other factors established as risk factors for WRF include the presence of vascular disease, hemoglobin and blood pressure [5,15,24]. Our present analysis confirmed baseline renal impairment and reduced hemoglobin levels as prominent predictors of WRF or decreasing GFR. In addition, we were able to show in a longitudinal mixed effects multilevel model that the slope of eGFR was also dependent of baseline eGFR and anemia, but also age, low sodium diet on admission, hypertension, peripheral artery disease, and type II diabetes. To our knowledge, we are the first to use this kind of estimation models to predict determinants of the slope of renal function over time.
Our results, and those previously published, seem to indicate that WRF (or lower eGFR over time) is especially observed in those with many comorbidities. The association with diabetes may indicate the occurrence of diabetic nephropathy, while the association with peripheral artery disease may have a relationship with either renal artery stenosis, or general atherosclerosis [25,26]. Low sodium diet as a predictor of WRF may be a representation of treatment bias; especially volume overload patients (sicker patients) are likely to be advised to use a low sodium diet. Finally, the association between lower hemoglobin levels and anemia with lower eGFR may be bidirectional, while also reduced renal function may predispose to the occurrence of anemia [27].
Clinical implicationsOur present analysis has some important clinical implications. First, our study confirms
earlier reports that WRF is frequently observed in patients with heart failure, both in and outhospital. Second, while WRF occurs also shortly after discharge, patients should be monitored closely in the weeks and months following admission: WRF at any time after discharge heralds a worse prognosis. Initiation of adequate therapy may prevent further WRF and eventually also improve renal function. Third, the effect of WRF on mortality and morbidity should be acknowledged. Although an increase in serum creatinine > 0.3 mg/dL may seem insignificant, it increases the risk for all cause mortality substantially. Finally, predisposing and modifiable factors to WRF should be monitored and possibly treated. These may include anemia, baseline renal impairment and diuretic use. Future studies are needed to assess whether treatment targeted at prevention or preservation of renal function will lead to improvement in mortality and morbidity.
LimitationsThis is a retrospective analysis of a randomized controlled trial. We did not measure renal
hemodynamics or GFR by clearance methods, and the used estimated GFR formula is only a surrogate marker of real GFR, but has been shown to be the most accurate in heart failure [20]. The definition used to determine WRF has been used by multiple previous studies, but the cut-off value of > 0.3 mg/dl increase in serum creatinine is arbitrary. We have shown in our recent meta-analysis that the relative risk for mortality rises steeply above >0.3mg/dl increase, and therefore this cut-off seems justifiable.

128
Chapter 6
ConclusionsIn our present retrospective analysis of the COACH, we found that WRF is not only
prevalent inhospital, but also on short and moderate time span after hospitalization in patients with AHF. WRF was associated with impaired survival and morbidity especially when WRF occurred late after discharge.
AcknowledgementsThe COACH study was supported by the Netherlands Heart Foundation (grant 2000Z003).
K. Damman is supported by the Netherlands Heart Foundation (grant 2006B157) A.A. Voors and D.J. van Veldhuisen are Clinical Established Investigators of the Netherlands Heart Foundation (grants 2006T37 and D97-017, respectively).
DisclosuresNone.

WRF at different timepoints and outcome
129
ReferencesDries DL, Exner DV, Domanski MJ, Greenberg B, and Stevenson LW. The prognostic implications of 1. renal insufficiency in asymptomatic and symptomatic patients with left ventricular systolic dysfunction. J Am Coll Cardiol. 2000;35:681-689.
Hillege HL, Nitsch D, Pfeffer MA et al. Renal function as a predictor of outcome in a broad spectrum 2. of patients with heart failure. Circulation. 2006;113:671-678.
Ruilope LM, van Veldhuisen DJ, Ritz E, and Luscher TF. Renal function: The Cinderella of cardiovascular 3. risk profile. J Am Coll Cardiol. 2001;38:1782-1787.
Tonelli M, Wiebe N, Culleton B et al. Chronic kidney disease and mortality risk: a systematic review. J 4. Am Soc Nephrol. 2006;17:2034-2047.
Khan NA, Ma I, Thompson CR et al. Kidney function and mortality among patients with left ventricular 5. systolic dysfunction. J Am Soc Nephrol. 2006;17:244-253.
Krumholz HM, Chen YT, Vaccarino V et al. Correlates and impact on outcomes of worsening renal 6. function in patients > or =65 years of age with heart failure. Am J Cardiol. 2000;85:1110-1113.
Metra M, Nodari S, Parrinello G et al. Worsening renal function in patients hospitalised for acute heart 7. failure: clinical implications and prognostic significance. Eur J Heart Fail. 2008;10:188-195.
Damman K, Navis G, Voors AA et al. Worsening renal function and prognosis in heart failure: systematic 8. review and meta-analysis. J Card Fail. 2007;13:599-608.
Akhter MW, Aronson D, Bitar F et al. Effect of elevated admission serum creatinine and its worsening 9. on outcome in hospitalized patients with decompensated heart failure. Am J Cardiol. 2004;94:957-960.
Cowie MR, Komajda M, Murray-Thomas T, Underwood J, and Ticho B. Prevalence and impact of 10. worsening renal function in patients hospitalized with decompensated heart failure: results of the prospective outcomes study in heart failure (POSH). Eur Heart J. 2006;27:1216-1222.
Logeart D, Tabet JY, Hittinger L et al. Transient worsening of renal function during hospitalization for 11. acute heart failure alters outcome. Int J Cardiol. 2007.
Owan TE, Hodge DO, Herges RM et al. Secular trends in renal dysfunction and outcomes in hospitalized 12. heart failure patients. J Card Fail. 2006;12:257-262.
Smith GL, Lichtman JH, Bracken MB et al. Renal impairment and outcomes in heart failure: systematic 13. review and meta-analysis. J Am Coll Cardiol. 2006;47:1987-1996.
Smith GL, Vaccarino V, Kosiborod M et al. Worsening renal function: what is a clinically meaningful 14. change in creatinine during hospitalization with heart failure? J Card Fail. 2003;9:13-25.
de Silva R, Nikitin NP, Witte KK et al. Incidence of renal dysfunction over 6 months in patients with 15. chronic heart failure due to left ventricular systolic dysfunction: contributing factors and relationship to prognosis. Eur Heart J. 2006;27:569-581.
Hillege HL, van Gilst WH, van Veldhuisen DJ et al. Accelerated decline and prognostic impact of renal 16. function after myocardial infarction and the benefits of ACE inhibition: the CATS randomized trial. Eur Heart J. 2003;24:412-420.

130
Chapter 6
Jose P, Skali H, Anavekar N et al. Increase in Creatinine and Cardiovascular Risk in Patients with 17. Systolic Dysfunction after Myocardial Infarction. J Am Soc Nephrol. 2006;10:2886-2891.
Jaarsma T, van der Wal MH, Lesman-Leegte I et al. Effect of moderate or intensive disease management 18. program on outcome in patients with heart failure: Coordinating Study Evaluating Outcomes of Advising and Counseling in Heart Failure (COACH). Arch Intern Med. 2008;168:316-324.
Jaarsma T, van der Wal MH, Hogenhuis J et al. Design and methodology of the COACH study: a 19. multicenter randomised Coordinating study evaluating Outcomes of Advising and Counselling in Heart failure. Eur J Heart Fail. 2004;6:227-233.
Smilde TD, van Veldhuisen DJ, Navis G, Voors AA, and Hillege HL. Drawbacks and prognostic value 20. of formulas estimating renal function in patients with chronic heart failure and systolic dysfunction. Circulation. 2006;114:1572-1580.
Damman K, Navis G, Smilde TD et al. Decreased cardiac output, venous congestion and the association 21. with renal impairment in patients with cardiac dysfunction. Eur J Heart Fail. 2007;9:872-878.
Bongartz LG, Cramer MJ, Doevendans PA, Joles JA, and Braam B. The severe cardiorenal syndrome: 22. 'Guyton revisited'. Eur Heart J. 2005;26:11-17.
Weinfeld MS, Chertow GM, and Stevenson LW. Aggravated renal dysfunction during intensive therapy 23. for advanced chronic heart failure. Am Heart J. 1999;138:285-290.
Forman DE, Butler J, Wang Y et al. Incidence, predictors at admission, and impact of worsening renal 24. function among patients hospitalized with heart failure. J Am Coll Cardiol. 2004;43:61-67.
Feringa HH, Karagiannis SE, Chonchol M et al. Lower progression rate of end-stage renal disease in 25. patients with peripheral arterial disease using statins or Angiotensin-converting enzyme inhibitors. J Am Soc Nephrol. 2007;18:1872-1879.
Mui KW, Sleeswijk M, van den HH et al. Incidental renal artery stenosis is an independent predictor of 26. mortality in patients with peripheral vascular disease. J Am Soc Nephrol. 2006;17:2069-2074.
Westenbrink BD, Visser FW, Voors AA et al. Anaemia in chronic heart failure is not only related to 27. impaired renal perfusion and blunted erythropoietin production, but to fluid retention as well. Eur Heart J. 2007;28:166-171.

WRF at different timepoints and outcome
131

132

133
PART IIIEmerging pathophysiological pathways
of the cardiorenal connection in patients with heart failure

134

Chapter 7Renal function relates to outcome through different pathways of renal
perfusion and filtration efficacy, hemodilution and volume overload in patients with chronic heart failure
Kevin Damman, Gerjan Navis, Adriaan A. Voors, Daan B. Westenbrink, Tom D.J. Smilde, Dirk J. van Veldhuisen, Hans L. Hillege
Submitted

136
Abstract
IntroductionRenal function is strongly related to prognosis in chronic heart failure (CHF). It is however unclear
which parameters determine the prognostic value of renal function. We therefore set out to determine those factors associated with the prognostic value of renal function.
Methods and ResultsCHF patients underwent invasive measurement of glomerular filtration rate (GFR), effective renal
plasma flow (ERPF) and extracellular volume by 125I-Iothalamate and 131I-Hippuran clearances. The combined primary endpoint consisted of the first occurrence of either all cause mortality, heart transplantation or admission for CHF. In total 102 patients (76% male, 58 ± 12 years) were included, with a median follow up of 26 months. GFR was a strong predictor of survival (hazard ratio(HR) 2.84 per standard deviation (SD) decrease, P < 0.001. Also ERPF was a major predictor of outcome (HR 2.79 per SD decrease, P < 0.001). Only PRA (HR 2.41 per SD increase, P < 0.001) was independently associated with endpoint occurrence, next to ERPF (HR 2.58 per SD decrease, P < 0.001). In pathway analysis, the interrelationship between ERPF, filtration fraction, hemoglobin levels and markers of volume overload with outcome, reflect the prognostic value of GFR.
ConclusionERPF and PRA are the most important factors related to prognosis in this group of patients with CHF.
The risk associated with a combination of impaired renal perfusion, reduced adequacy of filtration, volume overload and, as a complication, hemodilution, reflect the prognostic value observed with impaired GFR.

Pathophysiologic pathways and prognosis
137
IntroductionRenal function is a strong predictor of cardiovascular and all cause mortality in patients with
chronic heart failure (CHF) [1-3]. This observation is independent of functional parameters of left ventricular function. It is unclear which determinants of renal function are responsible for the prognostic value in CHF.
Renal perfusion is the crucial determinant of glomerular filtration rate (GFR). It is mainly influenced by renal vascular resistance, and therefore susceptible to afferent and efferent vasoconstriction of the glomerulus. The kidney is able to cope with reduced renal perfusion, by increasing the ratio of filtration across the glomerular membrane. This filtration fraction (GFR/renal perfusion), is increased at the cost of increased hydrostatic glomerular pressure, but can only preserve GFR to a certain extent. Eventually, GFR will drop as a consequence of reduced perfusion, at the cost of reduced filtration fraction.
Renal perfusion and filtration fraction are therefore the two main parameters determining GFR. Both are influenced by renin-angiotensin activation, sympathetic nervous system activation, but also by increases in central venous pressure and therapy modulating these systems such as angiotensin-converting enzyme inhibitors and beta-blockers. Recently we also found a close relationship between parameters of neurohormonal activation, endothelial dysfunction and inflammation with renal function in CHF [4].
Possibly these parameters are not only causally related to kidney function, but are also responsible for the prognostic value of renal impairment. Other factors associated with kidney function and prognosis in CHF include hemoglobin levels [5,6], etiology of CHF [7,8] and use of medication [9,10]. Therefore, the cause of reduced renal function is multifactorial. However, therapy to modulate renal function itself (GFR) is not yet available. Therapy to improve prognosis associated with impaired renal function should possibly focus on those factors, or a combination of those factors, associated with the prognostic value of GFR.
In the present study we therefore aimed to investigate which parameters are the determinants of the prognostic value of renal function in patients with CHF and which parameters are independently associated with prognosis, next to renal impairment.
Methods
Patient population and study designThe main study design has been published elsewhere [11]. In short, outpatient CHF patients,
aged ≥ 18 years, left ventricular ejection fraction (LVEF) < 45%, and clinically stable, were asked to participate. All patients used renin-angiotensin-system inhibitors, and all medication had to be stable for at least 1 month. Baseline measurements included standard weight, height, blood pressure and assessment of NYHA function class heart failure. Patients underwent clearance measurements of renal function. GFR, effective renal plasma flow (ERPF) and extracellular

138
Chapter 7
volume (ECV) were measured by the clearances of 125I-Iothalamate and 131I-Hippuran as described earlier [12]. The filtration fraction (FF) was calculated as the ratio of GFR and ERPF and expressed as percentage.
Assessment of different parameters of endothelial function, inflammation, neurohormonal activation and volume overload was done by laboratory measurements. These included Von Willebrand factor (vWf), plasma nitrite/nitrate (NOx), asymmetric di-methyl arganine (ADMA), soluble vascular adhesion molecule 1 (sVCAM-1), soluble E-selectin (sES), high sensitive C-reactive protein (hs-CRP), Plasma Renin Activity (PRA), Angiotensin II (Ang II) and N-terminal pro Brain Natriuretic Peptide (NT-proBNP).
Urinary albumin excretion (UAE) was determined as parameter of structural kidney damage.
Follow upPatients follow up started after renal function measurements had taken place. The primary
(combined) endpoint consisted of the first occurrence of either all cause mortality, heart transplantation or admission to hospital for CHF.
StatisticsData of variables are presented as mean ± standard deviation, unless indicated otherwise.
We used Cox-proportional-hazards regression analyses to evaluate the association with baseline parameters and the occurrence of endpoints. P-values for entry and removal of 0.10 were used. Continuous variables were divided by standard deviation to allow comparison of relative contributions of the different variables, except for ADMA and NOx, which showed a non-linear relationship with prognosis and were therefore dichotomized, i.e., upper tertile versus lower tertiles (preliminary analyses with tertiles of the variables showed increase in HR in only the highest tertile). GFR was also divided by the K/DOQI guidelines for chronic kidney disease (< 30, 30-59, 60-89, >90 ml/min/1,73m2, respectively), and ERPF was divided into quartiles to allow comparison with GFR.
In secondary analyses, pathway analysis was done to investigate multilevel relationships between variables and outcome. Pathway analysis allows simultaneous examination of several multiple regression analyses in a hypothetical model. This results in a better understanding of complex interrelationships and possible causal relationships between variables under investigation. To compare cox proportional hazard analysis with analysis obtained with pathway analysis, which basically consists of multilevel multivariate logistic regression analysis, figure 2 shows univariate standardized coefficients obtained with simple pathway analysis.
To identify eligible variables to include in the pathway models, we investigated variables that lost significant relationship with outcome after adjustment for GFR, with the hypothesis that the dependent prognostic implication of the variable was attributable to the prognostic value of GFR. Other variables were added to the hypothetical models because of their (patho)physiologic association with GFR. Those included NT-proBNP, hemoglobin levels, ECV and

Pathophysiologic pathways and prognosis
139
MAP. The relationship between these variables and outcome was investigated in a logistic regression analysis, hypothesizing that a combination of those variables could explain the prognostic value of GFR. We deliberately excluded PRA in this analysis because of the complex nature of its relationship with GFR, and the independent nature of the prognostic value in comparison to ERPF (and GFR). Secondly, a simplified model was constructed to improve likelihood of fit, excluding non-significant pathways. Finally, PRA was added to the model to allow comparison of variables in the full outcome model.
Goodness of fit. Because of the theoretical nature of the pathways constructed, indices of model fitting were used to account for goodness of fit. We used the minimum discrepancy score divided by the degrees of freedom for overall model fit (CMIN/df). A ratio < 3 indicates acceptable fit. The Akaike information criterion (AIC) and expected cross validation index (ECVI) are compared against the satured model and are actual better parameters for ‘badness of fit’, and therefore lower values are desirable. Normal fit index (NFI) varies from 0 to 1. By convention, a NFI < 0.9 can be improved, while a NFI > 0.95 indicates a good fit. The relative fit index (RFI) is compared with an independence model and varies also between 0 and 1, in which higher values represent better fit. Finally, root mean square error of approximation (RMSEA) was used, in which a value of <0.05 indicates a close fit of the model, with 0.08 or less being reasonable for the RMSEA.
The statistical computer packages SPSS, Chicago, version 13.0, AMOS, Chicago, version 6.0 and STATA, College Station, Texas, version 9.0 were used for the statistical analysis. All reported P values are two-tailed, and a P value <0.05 was considered statistically significant.
ResultsThe baseline characteristics of the total study population are shown in table 1. After
a median follow up of 26 months, in total 8 patients had died, 3 patients underwent heart transplantation and 12 were admitted to the hospital for CHF. The differences between patients who experienced endpoints and those who did not are depicted in table 2. Patients who reached an endpoint were more often female, had higher NYHA functional heart class, had lower hemoglobin levels and lower MAP values at baseline. All parameters of renal function were significantly lower in patients who reached an endpoint. Of the neurohormonal parameters investigated, NT-proBNP, angiotensin II and PRA were also higher in these patients.
Outcome related to baseline GFR and ERPFIn the different groups of renal function, patients with the lowest renal function had the
worst prognosis. In patients with baseline GFR ≥ 90 and 60-89 ml/min/1.73m2, 6% and 11% reached an endpoint, respectively. When GFR fell below 60 ml/min/1.73m2, respectively 48% (30-59 ml/min/1.73m2) and 75% (< 30 ml/min/1.73m2) reached an endpoint. This is comparable to a hazard ratio of 2.84 (1.86-4.34), P < 0.001 per SD decrease in GFR. Next to GFR, we also determined the relationship between ERPF and outcome. With decreasing renal

140
Chapter 7
Table 1. Baseline characteristics; total and stratification for outcome.
Endpoint Reached
Variables Total (n=102) Yes (n = 23 ) No (n = 79) P-value
Age (years) 58 ± 12 58 ± 13 58 ± 11 0.887
Gender: Male (N (%)) 78 (77) 14 (61) 64 (81) 0.046
NYHA I or II / III / IV (%) 61 / 31 / 10 17 / 48 / 35 72 / 25 / 3 < 0.001
Ischemic etiology (N (%)) 47 (46) 7 (30) 40 (51) 0.089
LVEF (%) 28 ± 9 25 ± 9 28 ± 9 0.158
MAP (mmHg) 86 ± 14 76 ± 14 88 ± 12 < 0.001
Hemoglobin (g/dL) 14.0 ± 1.5 13.1 ± 1.7 14.3 ± 1.3 < 0.001
ECV (L/kg BW) 0.26 ± 0.05 0.27 ± 0.06 0.26 ± 0.04 0.589
Renal Function Parameters
Serum creatinine (µmol/l) 113 ± 36 144 ± 51 104 ± 23 < 0.001
GFR (ml/min/1.73m2) 75 ± 27 52 ± 27 82 ± 23 < 0.001
ERPF (ml/min/1.73m2) 275 ± 88 204 ± 71 295 ± 82 < 0.001
Filtration Fraction (%) 27 ± 5 24 ± 8 28 ± 4 0.003
UAE (mg/day) 9.3 (6.6 – 18.6) 17.1 (6.6 – 33.4) 8.6 (6.4 – 14.5) 0.060
Neurohormonal, endothelial and inflammation parameters
NT-proBNP (pg/mL) 634 (272 – 1849) 2076 (971 – 4951) 465 (219 – 1230) < 0.001
vWF (%) 85 (48 – 179) 158 (95 – 194) 73 (47 - 149) 0.013
CRP (mg/dL) 2.5 (1.1 – 4.3) 3.6 (0.8 – 5.5) 2.4 (1.1 – 4.2) 0.311
sVCAM-1 (ng/mL) 325 (261 – 374) 347 (308 – 430) 317 (249 – 366) 0.032
E-selectin (ng/mL) 68 (48 – 93) 76 (51 – 96) 64 (47 – 93) 0.199
Plasma NOx (μmol/L) 11.5 (7.3 – 15.5) 16.1 (8.9 – 20.7) 10.9 (7.14 – 13.8) 0.012
ADMA (µmol/L) 0.64 (0.53 – 0.81) 0.63 (0.53 – 0.99) 0.65 (0.52 – 0.74) 0.355
Renin Angiotensin System parameters
PRA (ng/mL/h) 24.3 (5.6 – 72.2) 124 (51.1 – 162.6) 13.8 (4.9 – 42.5) < 0.001
ANG II (pmol/L) 9.9 (4.6 – 15.2) 13.6 (9.7 – 18.2) 9.1 (3.9 – 13.2) 0.010
Medication use (N (%))
ACEi, n (%) 89 (87) 12 (78) 72 (91) 0.093
% recommended dose 114 ± 73 111 ± 56 111 ± 77 0.585
ARB, n (%) 14 (13) 5 (22) 8 (10) 0.144
% recommended dose 64 ± 38 61 ± 40 66 ± 39 0.754
Beta Blocker n (%) 86 (84) 20 (87) 66 (84) 0.694
% recommended dose 50 ± 30 58 ± 34 47 ± 28 0.245
Diuretic, n (%) 71 (70) 20 (87) 51 (65) 0.041
Statin, n (%) 51 (50) 11 (48) 39 (49) 0.814
* continuous and normally distributed variables are expressed as Mean ± SD. Non-normally distributed vari-ables as median (25%-75%). Abbreviations: ACEi: Angiotensin Converting Enzyme Inhibitor, ADMA: Asymmetric dimethyl arganine, ANG II: Angiotensin II, ARB: Angiotensin II Receptor Blocker, CRP: C-reactive Protein, ECV: Extracellular volume, ERPF: Effective Renal Plasma Flow, GFR: Glomerular Filtration Rate, LVEF: Left Ventricular Ejection Fraction, MAP: Mean Arterial Pressure, NOx: Nitrite/Nitrate, NT-proBNP: N-Terminal pro Brain Natri-uretic Peptide, NYHA: New York Heart Association, PRA: Plasma Renin Activity, sE-Selectin: soluble E-selectin, sVCAM-1: soluble Vascular Adhesion Molecule -1, UAE: Urine Albumin Excretion, vWF: von Willebrand Factor

Pathophysiologic pathways and prognosis
141
perfusion, the risk of adverse clinical outcome increased from 4 % in the highest quartile, to 48 % in the lowest quartile. This resulted in a HR of 2.79 (1.76 – 4.43), P <0.001 per SD decrease in ERPF. Figure 1 shows the incremental Hazard ratio’s for both declining GFR and ERPF.
Other variables associated with prognosisIn addition to GFR and ERPF, other variables related to prognosis in univariate analysis
consisted of parameters of endothelial function (VCAM-1, plasma NOx, ADMA), hemodynamics (LVEF, MAP, NT-proBNP), diuretic use, hemoglobin levels and PRA (Table 2). Also FF was associated with prognosis. After adjustment for GFR, only PRA, hemoglobin levels, etiology of CHF, MAP and NT-proBNP were independently associated with prognosis.
Multivariate analysisIn multivariate stepwise cox regression analysis, including all univariate associated variables,
only ERPF (2.58 (1.54 – 4.31), P < 0.001, per SD decrease) and PRA (2.41 (1.75 – 3.32), P < 0.001, per SD increase) were independently related to worse outcome. After adjustment for percentage of recommended dose for ACEi/ARB use, PRA was still associated with prognosis.
Table 2. Univariate and multivariate cox regression analysis and after adjustment for GFR
Variable Univariate HR (95% CI)
Wald P-value HR after adjustment for GFR (95% CI)
P-value Multivariate HR (95% CI)
P-value
Age 1.00 (0.65 – 1.55) 0 0.995
Gender 2.38 (1.03 – 5.50) 4.10 0.043 1.31 (0.54 – 3.16) NS
Etiology 2.23 (0.92 – 5.41) 3.11 0.078 2.70 (1.09 – 6.68) 0.032
LVEF 1.42 (0.94 – 2.15) 2.69 0.101 1.23 (0.79 – 1.92) NS
MAP 0.33 (0.19 – 0.57) 15.93 < 0.001 0.53 (0.32 – 0.88) 0.013
GFR 2.84 (1.86 – 4.34) 23.30 < 0.001 NA NA
ERPF 2.79 (1.76 – 4.43) 18.9 < 0.001 1.24 (0.47 – 3.28) NS 2.58 (1.54 – 4.31) < 0.001
FF 1.89 (1.14 – 2.71) 11.87 0.001 1.14 (0.78 – 1.66) NS
Diuretic use 3.30 (0.98 – 11.10) 3.71 0.054 1.46 (0.40 – 5.36) NS
Hemoglobin 2.51 (1.59 – 3.94) 15.83 < 0.001 1.69 (1.05 – 2.72) 0.031
NT-proBNP 1.70 (1.39 – 2.08) 26.53 < 0.001 1.30 (1.02 – 1.65) 0.035
sVCAM-1 4.43 (1.02 – 2.01) 4.27 0.039 1.16 (0.77 – 1.73) NS
Plasma NOx 2.88 (1.26 – 6.57) 6.30 0.012 1.52 (0.63 – 3.68) NS
ADMA 1.52 (1.02 – 2.27) 4.18 0.041 1.13 (0.49 – 2.61) NS
PRA 2.44 (1.85 – 3.23) 39.08 < 0.001 2.03 (1.47 – 2.79) < 0.001 2.41 (1.75 – 3.32) < 0.001
HRs for continuous variables are shown per SD except for plasma NOx and ADMA (higher tertile versus both lower tertiles). Abbreviations: ADMA: Asymmetric dimethyl arganine, ECV: Extracellular Volume, ERPF: Effective Renal Plasma Flow, FF: Filtration Fraction, GFR: Glomerular Filtration Rate, LVEF: Left Ventricular Ejection Frac-tion, MAP: Mean Arterial Pressure, NOx: Nitrite/Nitrate, NT-proBNP: N-Terminal pro Brain Natriuretic Peptide, PRA: Plasma Renin Activity, sVCAM-1: soluble Vascular Adhesion Molecule

142
Chapter 7
In additional analysis, the HR for both PRA and ERPF was not affected by adjustment for variables associated with prognosis in univariate analysis, or adjustment for mutual confounding between both independent parameters. Even after adjustment for all comorbid variables, including LVEF, MAP, NT-proBNP, hemoglobin levels and endothelial markers, ERPF was associated with prognosis (HR 2.10 (0.97 – 4.58), independent of PRA, although only a strong trend was observed (P = 0.06).
Pathway analysisFigure 3a shows the hypothetical model (Model 1) constructed with pathway analysis for the
relationship between variables related to renal function and outcome. LVEF is an important determinant of both ERPF (r = 0.22) and FF (r = -0.15). Also MAP was related to ERPF (r = -0.14), FF (r = -0.35) and NT-proBNP (r = -0.30). The model shows that both lower levels of FF (r = 0.18 ) and lower levels of ERPF (r = 0.28) are independently related to prognosis. Next to their direct effect on prognosis, an indirect effect exists, which is mediated through hemoglobin (r = 0.26 for ERPF, r = 0.33 for FF) levels and for ERPF also through parameters of volume overload (r = -0.16 for ECV, r = 0.39 for NT-proBNP). Both NT-proBNP (r = 0.19) and hemoglobin levels (r = 0.14) have a direct effect on prognosis. Addition of diuretic use, VCAM, ADMA and plasma NOx did not have significant interaction with the model variables and did not improve model fit.
A simplified model (model 2) excluded MAP and LVEF (no individual relationship with prognosis), to improve model fit (Figure 3b). Most coefficients did not differ between model
Figure 1. Relationship of GFR and ERPF with prognosis.
GFR (mL/min/1.73m2)020406080100120140
Haz
ard
Rat
io (9
5% C
I)
0
1
2
3
4
5
6
7
8
9
10
ERPF (mL/min/1.73m2)
0100200300400500
GFRERPF

Pathophysiologic pathways and prognosis
143
1 and 2, however the relationship between ERPF with NT-proBNP and FF was stronger in model 2.
Figure 4 shows the total model (Model 3) for outcome when PRA was added as a prominent predictor for outcome in this population. PRA was the strongest predictor of adverse clinical outcome (r = 0.53). The addition of PRA to the model diminished the direct effect of both ERPF and FF on outcome. However, an indirect effect, mediated through PRA was revealed with strong correlations with both ERPF(r = 0.27) and FF (0.39).
Table 3. Goodness of fit measures for path models
NFI AIC ECVI (90% CI) RFI RMSEA (90% CI) CMIN/df ratio
Model 1 0.897 82.4 0.816 (0.762 – 0.964) 0.663 0.070 (0.000 – 0.135) 1.49
Model 2 0.920 57.2 0.566 (0.511 – 0.697) 0.440 0.143 (0.043 – 0.253) 3.07
Model 3 0.886 75.1 0.744 (0.645 – 0.918) 0.602 0.127 (0.062 – 0.195) 2.64
Desired > 0.90 0 0 1 < 0.08 < 3.0
Abbreviations: AIC: Akaike information criterion, ECVI: Expected Cross Validation Index, NFI: Normal fit Index, RFI: Relative Fit Index, RMSEA: Root mean square error of approximation.
Figure 2. Univariate pathway analysis. Shown are standardized coefficients.

144
Chapter 7
Goodness of fitFor model 1, CMIN/df was 1.49, and NFI was 0.90 (>0.90 desired) (Table 3). In the
simplified model NFI improved to 0.92, however, RFI decreased while RMSEA increased. The CMIN/DF ratio was still satisfying (3.07), the total model achieving goodness of fit of χ2
= 9.21, p 0.027 (p > 0.05 desired). In model 3, with PRA added, NFI approached desired value of >0.9 (0.89), while RFI increased again. The CMIN/DF ratio decreased below a favourable value of 3.0 (2.64). Both AIC and ECVI had best results in model 2, however model 3 showed better model fit compared to model 1.
DiscussionThe present study shows that not only GFR is related to prognosis in patients with CHF,
but also ERPF, the main determinant of renal function. PRA levels were the strongest markers of outcome in this cohort of patients with CHF, who were all on RAS blocking medication. In pathway analysis, we showed that also hemoglobin levels, in combination with parameters of volume overload add to the pathophysiologic relationship between renal function and prognosis.
Renal perfusion, filtration fraction and prognosisRenal perfusion is the main determinant of GFR in this population [4]. In the present study,
ERPF was a strong predictor of prognosis, and the only variable associated with prognosis independently of PRA. Renal perfusion is a sensitive reflection of decreased cardiac output,
Figure 3. Path analysis diagram for model 1 and 2. Model 1: dotted and solid lines. Model 2: only solid lines. Estimates of standardized coefficients of the hypothesized model.

Pathophysiologic pathways and prognosis
145
therefore this finding may reflect the severity of increased pump failure and subsequent related poorer outcome [13].
While ERPF represents adequacy of renal perfusion, FF resembles efficacy of renal filtration, which tries to preserve GFR in the presence of reduced ERPF by increasing hydrostatic glomerular pressure. In patients with CHF, FF is therefore increased, reflecting the need of the kidney to preserve GFR, despite decreased ERPF [13]. In patients on ACE-inhibitor or ARB therapy, FF is actually decreased to more normal values [14]. We recently showed that in severely underperfused kidneys (ERPF below 180 ml/min/1.73m2), the kidney struggles to preserve GFR, and as a consequence FF decreases disproportionately fast [4]. Possibly, the prognostic implication of decreasing FF is a reflection of this observation.
Hemodilution and prognosisNext to reduced renal perfusion and impaired efficacy of renal function, hemoglobin levels,
or anemia, have also been shown to correlate strongly with prognosis in CHF [15,16]. Recently, de Silva et al [17] and Go et al [18], showed that this prognostic value was independent of and additive to the prognostic value of renal function. Our present findings are in agreement with this observation.
Figure 4. Path analysis diagram for the full model, including PRA. Estimates of standardized coefficients of the hypothesized model.

146
Chapter 7
The etiology of anemia in CHF is multifactorial. It includes impaired erythropoietin production by the kidney, impaired erythropoiesis in the bone marrow and volume retention secondary to salt and water retention, which will lead to hemodilution [19-21]. In our present analysis, we show that at least part of the prognostic value of ERPF (and GFR) is mediated through NT-proBNP and ECV. Both are related to volume overload and are indirectly related to prognosis by a pathway that is mediated by hemoglobin levels, suggesting that hemodilution is an important prognostic marker.
Disturbances in volume regulation is common in CHF. Based on radioisotope measurements Androne et al reported that merely 18% of the ambulatory patients with CHF are normovolemic, whereas 65% are hypervolemic in the absence of congestive symptoms or anemia [22]. These hypervolemic patients displayed an increased risk of death. Recently we revealed that non-symptomatic fluid retention is independently related to anemia in CHF patients, indicating that hemodilution is a common cause of anemia in CHF [20]. This has been corroborated by Androne et al in a small cohort of anemic patients with severe CHF, where patients with anemia caused by hemodilution were at increased risk of death [21]. Interestingly, we recently showed that GFR does not only depend on renal perfusion, but also on venous congestion, indicating the possible pathophysiologic pathway of our present finding [23].
PRA and outcomePharmacologic intervention in CHF has focussed on hemodynamic stability and RAS
blockade in the last two decades [24]. Blocking of the RAS by either ACE-inhibitors and/or ARB’s is now common practice, and with it mortality rates have substantially decreased in CHF [25]. However, in the present analysis, even with all patients using RAS blocking agents, PRA was the most important factor related to prognosis. PRA levels correlate with increasing severity of CHF and with kidney function as well [4,26,27]. In addition, increased levels of PRA were strong predictors of mortality in patients in the VAL-HEFT population [28] and in a small Spanish study in patients with severe CHF [29], in which 93% and 97% of patients were on ACE inhibitor therapy, respectively. In both the present study and in the VAL-HEFT, beta-blocker therapy and ACE-inhibitor therapy did not modulate the prognostic value of PRA.
The link between higher PRA levels and mortality in the presence of RAS blockade is not clear. In absence of RAS blockade, RAS activity has been showed to play a key role in the pathophysiology of CHF and the cardiorenal syndrome [30,31]. However, no studies have investigated the effect of activity of the RAS system on pathophysiology in the presence of ACE inhibitor therapy. Possibly, increased PRA levels merely reflect the severity of the disease, and/or more pronounced impaired renal perfusion [28], although the independent character of the observed relationships are in contrast to this hypothesis. Another possibility is that higher PRA levels represent more severe ACE inhibition, as a consequence of negative feedback inhibition [32], but this would again be in contradiction to the general belief of survival benefit with higher doses of ACE inhibitor therapy.

Pathophysiologic pathways and prognosis
147
Future therapy specifically targeted on renin activity, such as selective renin-inhibitors which are currently under investigation, could therefore proof to be an important additive treatment strategy in CHF, next to ACE inhibition and/or ARB therapy.
Clinical ImplicationsThe prognostic nature of impaired renal function suggests that modulation may result in
a strong survival benefit. However, it is difficult to modulate renal function (GFR) itself. We now show that the components responsible for the prognostic implication may be an alternative target for therapy.
ERPF is mainly dependent on renal perfusion pressure and renal vascular resistance. The latter is largely dependent on afferent and efferent vasotone, and therefore susceptible to ACE-inhibition, improving renal perfusion by efferent vasodilation [14]. Also FF will improve by ACE-inhibitor therapy, by modulation of the ultrafiltration coefficient. (REF) Therefore, ACE-inhibition should be the cornerstone of treatment in combined heart and renal dysfunction.
Renal perfusion pressure is dependent on (mean) arterial and (central) venous blood pressure. It is often decreased when central venous pressure is elevated due to venous congestion. We recently hypothesized that venous congestion does not only influence renal perfusion, but also has direct effects on GFR. Therapy to improve renal perfusion pressure in favour of maintaining GFR should therefore (also) focus on alleviation of venous congestion.
Another reason to improve venous congestion, other than improvement of renal perfusion and function, is the observation of an interrelationship between hemoglobin levels, hemodilution and volume overload with outcome. We and others have demonstrated that hemodilution is relatively common in these patients with CHF, even in the absence of congestive symptoms [20,21]. Since hemodilution is associated with prognosis, meticulous control of fluid retention is warranted, especially when anemia and impaired renal function are present [20].
Finally, therapy should focus on reducing the risk of worse outcome associated with increasing levels of PRA. As suggested, inhibiting the rate-limiting step in the RAS-cascade by renin-inhibition may be a promising additive treatment in especially these patients. ARB therapy was found to be more effective in patients who had high PRA levels in the VAL-HEFT population, suggesting that in these patients dual RAS-blockade could be of benefit for prognosis [28]. Also beta-blockers reduce PRA in the presence of ACE-inhibition [33-35], but this reduction seems to be transient as a result of an escape phenomenon [35]. More insight in the pathophysiology behind the persistent relation between higher PRA and mortality in the presence of RAS-blockade is needed to optimize therapy in patients with CHF and renal dysfunction.
LimitationsThe present study is hampered by its limited study size, and therefore the hypothetical
models may not be representative for the entire CHF population. Furthermore, there may be unmeasured significant contributors to the hypothetical models, which could influence

148
Chapter 7
the results. The findings are observational in nature, and therefore we were not able to show effects of possible treatments or changes in measured variables on our findings. The effect of the suggested therapies is hypothetical and based on our present observations and need further investigation. It is however the first to address the underlying pathophysiology of the prognostic value of GFR in CHF. In addition, this is the first study in cardio-renal dysfunction in which pathway analysis is used to understand multilevel associations, rather than multivariate analysis. Although also this type of analysis has drawbacks, it provides elegant and comprehensive insights in multivariate interaction between pathophysiologic variables.
ConclusionIn conclusion, ERPF and PRA are the most important factors related to prognosis in
this group of patients with CHF. The risk associated with a combination of impaired renal perfusion, reduced adequacy of filtration, volume overload and, as a complication, hemodilution, reflect the prognostic value observed with impaired GFR. The relationship between PRA and prognosis, even in the presence of RAS blockade, suggests that therapy targeted to RAS activation in patients with CHF should be intensified and broadened. Other therapies should focus on preservation of renal perfusion, and reducing venous congestion to improve prognosis associated with renal function.

Pathophysiologic pathways and prognosis
149
ReferencesAl Ahmad A, Rand WM, Manjunath G et al. Reduced kidney function and anemia as risk factors for 1. mortality in patients with left ventricular dysfunction. J Am Coll Cardiol. 2001;38:955-962.
Dries DL, Exner DV, Domanski MJ, Greenberg B, and Stevenson LW. The prognostic implications of 2. renal insufficiency in asymptomatic and symptomatic patients with left ventricular systolic dysfunction. J Am Coll Cardiol. 2000;35:681-689.
Hillege HL, Girbes AR, de Kam PJ et al. Renal function, neurohormonal activation, and survival in 3. patients with chronic heart failure. Circulation. 2000;102:203-210.
Smilde TDJ, Damman K, van der Harst P et al. Differential associations between renal function and 4. "modifiable" risk factors in patients with chronic heart failure. Clin Res Cardiol. 2009;98:121-129.
Anand IS. Pathogenesis of anemia in cardiorenal disease. Rev Cardiovasc Med. 2005;6 Suppl 5. 3:S13-S21.
Iaina A, Silverberg DS, and Wexler D. Therapy insight: congestive heart failure, chronic kidney disease 6. and anemia, the cardio-renal-anemia syndrome. Nat Clin Pract Cardiovasc Med. 2005;2:95-100.
Smilde TD, Hillege HL, Navis G et al. Impaired renal function in patients with ischemic and 7. nonischemic chronic heart failure: association with neurohormonal activation and survival. Am Heart J. 2004;148:165-172.
Smilde TD, Hillege HL, Navis G et al. Difference in long-term prognostic value of renal function 8. between ischemic and non-ischemic mild heart failure. Int J Cardiol. 2006;107:73-77.
Hillege HL, van Gilst WH, van Veldhuisen DJ et al. Accelerated decline and prognostic impact of renal 9. function after myocardial infarction and the benefits of ACE inhibition: the CATS randomized trial. Eur Heart J. 2003;24:412-420.
de Silva R, Nikitin NP, Witte KK et al. Incidence of renal dysfunction over 6 months in patients with 10. chronic heart failure due to left ventricular systolic dysfunction: contributing factors and relationship to prognosis. Eur Heart J. 2006;27:569-581.
Smilde TD, van Veldhuisen DJ, Navis G, Voors AA, and Hillege HL. Drawbacks and prognostic value 11. of formulas estimating renal function in patients with chronic heart failure and systolic dysfunction. Circulation. 2006;114:1572-1580.
Donker AJM, Vanderhem GK, Sluiter WJ, and Beekhuis H. Radioisotope Method for Simultaneous 12. Determination of Glomerular-Filtration Rate and Effective Renal Plasma-Flow. Netherlands Journal of Medicine. 1977;20:97-103.
Ljungman S, Laragh JH, and Cody RJ. Role of the Kidney in Congestive Heart-Failure - Relationship 13. of Cardiac Index to Kidney-Function. Drugs. 1990;39:10-21.
Creager MA, Halperin JL, Bernard DB et al. Acute Regional Circulatory and Renal Hemodynamic-14. Effects of Converting-Enzyme Inhibition in Patients with Congestive Heart-Failure. Circulation. 1981;64:483-489.
O'Meara E, Clayton T, McEntegart MB et al. Clinical correlates and consequences of anemia in a broad 15. spectrum of patients with heart failure: results of the Candesartan in Heart Failure: Assessment of Reduction in Mortality and Morbidity (CHARM) Program. Circulation. 2006;113:986-994.

150
Chapter 7
Anand IS, Kuskowski MA, Rector TS et al. Anemia and change in hemoglobin over time related to 16. mortality and morbidity in patients with chronic heart failure: results from Val-HeFT. Circulation. 2005;112:1121-1127.
de Silva R, Rigby AS, Witte KK et al. Anemia, renal dysfunction, and their interaction in patients with 17. chronic heart failure. Am J Cardiol. 2006;98:391-398.
Go AS, Yang J, Ackerson LM et al. Hemoglobin level, chronic kidney disease, and the risks of death and 18. hospitalization in adults with chronic heart failure: the Anemia in Chronic Heart Failure: Outcomes and Resource Utilization (ANCHOR) Study. Circulation. 2006;113:2713-2723.
Ezekowitz JA, McAlister FA, and Armstrong PW. Anemia is common in heart failure and is associated 19. with poor outcomes: insights from a cohort of 12 065 patients with new-onset heart failure. Circulation. 2003;107:223-225.
Westenbrink BD, Visser FW, Voors AA et al. Anaemia in chronic heart failure is not only related to 20. impaired renal perfusion and blunted erythropoietin production, but to fluid retention as well. Eur Heart J. 2007;28:166-171.
Androne AS, Katz SD, Lund L et al. Hemodilution is common in patients with advanced heart failure. 21. Circulation. 2003;107:226-229.
Androne AS, Hryniewicz K, Hudaihed A et al. Relation of unrecognized hypervolemia in chronic heart 22. failure to clinical status, hemodynamics, and patient outcomes. Am J Cardiol. 2004;93:1254-1259.
Damman K, Navis G, Smilde TD et al. Decreased cardiac output, venous congestion and the association 23. with renal impairment in patients with cardiac dysfunction. Eur J Heart Fail. 2007;9:872-878.
Hunt SA, Abraham WT, Chin MH et al. ACC/AHA 2005 Guideline Update for the Diagnosis and 24. Management of Chronic Heart Failure in the Adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation. 2005;112:e154-e235.
Garg R and Yusuf S. Overview of randomized trials of angiotensin-converting enzyme inhibitors on 25. mortality and morbidity in patients with heart failure. Collaborative Group on ACE Inhibitor Trials. JAMA. 1995;273:1450-1456.
Francis GS, Benedict C, Johnstone DE et al. Comparison of neuroendocrine activation in patients with 26. left ventricular dysfunction with and without congestive heart failure. A substudy of the Studies of Left Ventricular Dysfunction (SOLVD). Circulation. 1990;82:1724-1729.
Dzau VJ, Colucci WS, Hollenberg NK, and Williams GH. Relation of the renin-angiotensin-aldosterone 27. system to clinical state in congestive heart failure. Circulation. 1981;63:645-651.
Latini R, Masson S, Anand I et al. The comparative prognostic value of plasma neurohormones at 28. baseline in patients with heart failure enrolled in Val-HeFT. Eur Heart J. 2004;25:292-299.
Orus J, Roig E, Perez-Villa F et al. Prognostic value of serum cytokines in patients with congestive heart 29. failure. J Heart Lung Transplant. 2000;19:419-425.
Schrier RW. Role of diminished renal function in cardiovascular mortality: marker or pathogenetic 30. factor? J Am Coll Cardiol. 2006;47:1-8.

Pathophysiologic pathways and prognosis
151
Bongartz LG, Cramer MJ, Doevendans PA, Joles JA, and Braam B. The severe cardiorenal syndrome: 31. 'Guyton revisited'. Eur Heart J. 2005;26:11-17.
Benedict CR, Francis GS, Shelton B et al. Effect of long-term enalapril therapy on neurohormones in 32. patients with left ventricular dysfunction. SOLVD Investigators. Am J Cardiol. 1995;75:1151-1157.
Cohen SA, Jondeau G, Beauvais F, and Berdeaux A. Beneficial effects of carvedilol on angiotensin-33. converting enzyme activity and renin plasma levels in patients with chronic heart failure. Eur J Heart Fail. 2004;6:463-466.
The RESOLVD Investigators. Effects of metoprolol CR in patients with ischemic and dilated 34. cardiomyopathy : the randomized evaluation of strategies for left ventricular dysfunction pilot study. Circulation. 2000;101:378-384.
Fung JW, Yu CM, Yip G et al. Effect of beta blockade (carvedilol or metoprolol) on activation of the 35. renin-angiotensin-aldosterone system and natriuretic peptides in chronic heart failure. Am J Cardiol. 2003;92:406-410.

152

Chapter 8Urinary neutrophil gelatinase associated lipocalin (NGAL), a marker
of tubular damage, is increased in patients with chronic heart failure
Kevin Damman, Dirk J. van Veldhuisen, Gerjan Navis, Adriaan A. Voors, Hans L. Hillege
Eur J Heart Fail, 2008; 10:997-1000

154
Abstract
BackgroundRenal impairment as measured by reduced glomerular filtration rate (GFR) and increased urinary
albumin excretion (UAE) is prevalent in patients with chronic heart failure (CHF) and associated with reduced survival. The prevalence of structural tubular damage in CHF is unknown.
Methods and resultsWe investigated 90 CHF patients and 20 age and gender balanced healthy controls, and determined
estimated GFR, UAE, N terminal pro brain natriuretic peptide (NT-proBNP) and urinary neutrophil gelatinase associated lipocalin (NGAL) as marker for tubular damage. CHF patients had significantly lower averaged estimated GFR (64 ± 17 vs 90 ± 12 mL/min/1.73m2, P < 0.0001), but higher NT-proBNP and UAE levels (both P < 0.0001). Median urinary NGAL levels were markedly increased in CHF patients compared to controls (175 (70–346) vs 37 (6–58) μg /gCr, P < 0.0001). Both serum creatinine (r = 0.26, P = 0.006) and eGFR (r = -0.29, P = 0.002) were significantly associated with urinary NGAL levels as were NT-proBNP and UAE but to a lesser extent.
ConclusionRenal impairment in CHF patients is not only characterised by decreased eGFR and increased UAE,
but also by the presence of tubular damage, as measured by increased urinary NGAL concentrations.

Urinary NGAL in CHF
155
BackgroundRenal dysfunction, as measured by decreased glomerular filtration rate (GFR), is common
in chronic systolic heart failure (CHF) and associated with severely increased mortality and morbidity already apparent in the early stages of borderline renal dysfunction [1-3]. Even if GFR is only mildly impaired, increased urinary albumin excretion (UAE) levels, as a marker of early renal damage, are commonly observed in patients with CHF [4]. In primary renal disease, renal impairment is not only associated with decreased GFR and increased UAE, but also with the presence of structural tubular damage, as measured by increased urinary concentrations of specific tubular marker proteins [5-7]. One of these markers is neutrophil gelatinase associated lipocalin (NGAL), which has been shown to be highly increased in patients with acute and chronic renal injury in different clinical stages [5,8].
AimIn the present study, we aimed to 1) investigate the prevalence of structural tubular damage
as measured by urinary NGAL concentrations, 2) establish the relationship between urinary NGAL levels and estimated GFR, and 3) investigate the relationship between urinary NGAL levels and UAE, in patients with CHF.
MethodsIn short, 90 outpatient CHF patients, aged ≥ 18 years, left ventricular ejection fraction
(LVEF) < 45%, and clinically stable, were asked to participate. All patients were on angiotensin converting enzyme inhibitors and/or angiotensin II receptor blockers, and all medication had to be stable for at least 1 month. In addition, 20 healthy, age and gender matched controls were studied. All subjects gave informed consent to participate in the study, which was approved by the ethics review committee of the study centre. Baseline measurements included standard weight, height, systolic and diastolic blood pressure, serum creatinine, hemoglobin levels, N terminal- pro brain natriuretic peptide (NT-proBNP) and assessment of NYHA function class heart failure. LVEF was determined by nuclear ventriculography. Patients and controls collected 24-hours urine and urinary albumin excretion (UAE) was determined. Urinary creatinine was determined to correct for concentration of urine. Urinary NGAL was determined by means of a commercially available ELISA testkit from Antibody Shop (Gentofte, Denmark) and expressed as per gram urinary creatinine (μg/gCr). In brief, a monoclonal antibody against human NGAL, and biotinylated antibody against bound NGAL was used to detect NGAL in the urine samples. Horseradish peroxidise conjugated streptavidin was added, followed by color-forming peroxidise substrate containing tetramethylbenzidine. The color was then measured at 450 nm by a microtiter plate reader and compared with a standard curve. NGAL levels below the detection level were scored as 0.1 μg/gCr. Estimated GFR was calculated using the simplified modification of diet in renal disease formula (186.3 x serum creatinine -1.154 x age -0.203 (x 0.742 if female) (x 1.212 if black)) as validated in CHF patients [9].

156
Chapter 8
Statistic analysisData are given as mean ± standard deviation when normally distributed, as median and
interquartile range when skewed distributed. Differences between patients and controls were tested using Mann-Whitney U or students T testing where appropriate. Correlations were performed using Spearman’s correlation coefficients. Multivariate regression analysis was used to correct for eGFR when comparing patients with controls. All reported probability values are 2-tailed, and a P value <0.05 was considered statistically significant. Statistical analyses were performed using SPSS, Chicago version 12.0.
ResultsClinical characteristics of patients and controls are summarized in Table 1. NGAL levels
were highly elevated in patients with CHF (175(70–346) μg/gCr) compared to healthy controls (37 (6–58) μg/gCr, P < 0.0001) (Figure 1). Combining findings in CHF patients and controls, urinary NGAL levels were significantly related to eGFR(r =-0.29, P = 0.002) (Figure 2), serum creatinine (r = 0.26, P = 0.006), UAE levels (r =0.33, P = 0.001) and NT-proBNP (r = 0.26, P = 0.007) (Figure 3). When adjusted for eGFR in multivariate analysis, CHF patients still had significantly higher urinary NGAL levels (P = 0.0004). Urinary NGAL levels were similar among different etiologies of heart failure, NYHA class and hemoglobin levels.
Figure 1. NGAL and UAE levels in CHF patients versus controls. Shown are boxplots for urinary NGAL and UAE levels. Boxes display median (horizontal bars), interquartile ranges (lower and upper limits of boxes) and 5th and 95th percentiles (error bars). Abbreviations: CHF: chronic heart failure, NGAL: neutrophil gelatinase associated lipocalin, UAE: urinary albumin excretion.
Controls CHF Controls CHF
UA
E (m
g/d
ay)
0
5
10
15
20
25
30
0
100
200
300
400
500
600 UAEUrinary NGAL
P < 0.0001 P < 0.0001
NG
AL
(μg/
gCr)

Urinary NGAL in CHF
157
ConclusionIn the present study we show for the first time that structural tubular damage, as measured
by increased urinary concentrations of NGAL, is highly prevalent in patients with CHF. Furthermore, across the spectrum of heart failure and healthy controls, urinary NGAL levels are not only associated with different indices of renal dysfunction, but also positively associated with increased levels of NT-proBNP.
NGAL is a 21 kD protein of the lipocalin family and is normally secreted in low amounts in lung, kidney, trachea, stomach and colon tissue [10]. NGAL levels are elevated in various
Table 1. Baseline characteristics
VariableCHF
(n = 90)Controls(n = 20) P-value
Age (years) 59 ± 11 58 ± 4 0.91
Gender, n (% male) 71 (79) 16 (80) 0.91
NYHA (I /II / III or IV (%)) 16 / 47 / 38 NA NA
Ischemic etiology, n (%) 43 (48) NA NA
LVEF (%) 28 ± 9 NA NA
Systolic BP (mmHg) 121 ± 20 120 ± 11 0.67
Diastolic BP (mmHg) 69 ± 11 71 ± 8 0.30
Hemoglobin (g/dL)* 14.2 ± 1.3 14.2 ± 1.0 0.99
NT-proBNP (pg/mL) 515 (219 – 1283) 84 (57 – 108) < 0.0001
Renal indices
Serum creatinine (μmol/L) 112 ± 36 79 ± 13 < 0.001
eGFR (mL/min/1.732) 64 ± 17 90 ± 12 < 0.0001
UAE (mg/24h) 5.1 (3.4 – 9.8) 1.5 (1.2 – 1.9) < 0.0001
Tubular marker
Urinary NGAL (µg/L) 106 (56 – 217) 27 (6 – 53) < 0.0001
Urinary NGAL (µg/gCr) 175 (70 – 346) 37 (6 – 58) < 0.0001
Use of medications (%)
RAS inhibitor 100 0 NA
Beta Blocker 87 0 NA
Diuretic 70 0 NA
Aldosteron receptor blocker 32 0 NA
Presented are mean ± standard deviations or median with interquartile ranges. * for conversion to mmol/L, multiply by 0.6206. Abbreviations: NYHA: New York Heart Association, LVEF: left ventricular ejection fraction, BP: blood pressure, NT-proBNP: N terminal pro brain natriuretic peptide, eGFR: estimated glomerular filtration rate, UAE: urinary albumin excretion, NGAL: neutrophil gelatinase associated lipocalin, RAS: renin angiotensin system.

158
Chapter 8
Figure 2. Relationship between eGFR and urinary NGAL levels. Abbreviations: CHF: chronic heart failure, eGFR: estimated glomerular filtration rate, NGAL: neutrophil gelatinase associated lipocalin.
eGFR (mL/min/1.73m2)02 04 06 08 0 100 120
0,1
1
10
100
1000
10000 ControlsCHF
r = -0.29, P = 0.002
NG
AL
(μg/
gCr)
Figure 3. Relationship between plasma NT-proBNP and urinary NGAL levels. Abbreviations: CHF: chronic heart failure, NGAL: neutrophil gelatinase associated lipocalin, NT-proBNP: N terminal-pro brain natriuretic peptide.
NT-proBNP (pg/mL)
10 100 1000 10000
0,1
1
10
100
1000
10000ControlsCHF
r = 0.26, P = 0.007
NG
AL
(μg/
gCr)

Urinary NGAL in CHF
159
pathologic states, and possess bacteriostatic effects, by inhibition of iron-binding molecules that are important to certain bacteria. In renal failure both serum and urinary concentrations rise massively, and therefore (urinary) NGAL is thought to be a marker of tubular injury, an effect independent of its bacteriostatic properties [10].
NGAL has been associated with morphological changes and albuminuria in patients with primary renal disease, but never in patients with heart failure [5]. In CHF, reduced GFR is mainly dependent of reduced renal perfusion, which may serve as a hypoxic trigger for tubular damage [11,12]. In the chronic setting, patients with renal insufficiency due to IgA nephropathy had higher urinary NGAL levels compared to controls, and the urinary NGAL concentration also correlated strongly with the extent of tubulointerstitial injury [5].
Chronic renal hypoxia has not only been proposed as the final common pathway to end stage renal disease [12], but may also be the initiating trigger for a vicious circle between tubulointerstitial injury and chronic renal insufficiency [13]. This hypothesis may be one of the pathways by which chronic renal insufficiency may develop in patients with CHF.
Recently we hypothesized that also venous congestion might be a determinant of renal damage [14]. Plasma NT-proBNP levels were indeed correlated to NGAL levels, fitting this hypothesis, but may also be an expression of impaired cardiac systolic function leading to renal impairment.
NGAL has mainly been studied in the setting of acute renal failure. During cardiopulmonary bypass operation, patients who experienced acute renal dysfunction showed a marked in urinary NGAL levels, which preceded the increase in serum creatinine by a day [15,16]. In one single case of acute tubular necrosis due to heart failure induced hypotension, NGAL tubular expression was strongly increased [8].
Hence, measurements of NGAL may serve as a very early marker of worsening renal function, even preceding plasma serum creatinine rise. Urinary (or plasma) NGAL levels could therefore be used to adjust therapy, to anticipate and possibly prevent expected renal injury, even before a peak in serum creatinine occurs.
In conclusion, patients with CHF frequently suffer from a combination of reduced GFR, increased UAE and structural tubular damage. NGAL may serve as a novel non-invasive marker for (worsening) renal function in heart failure.

160
Chapter 8
AcknowledgementsK. Damman is supported by the Netherlands Heart Foundation (grant 2006B157) A.A.
Voors and D.J. van Veldhuisen are Clinical Established Investigators of the Netherlands Heart Foundation (grants 2006T37 and D97-017, respectively).
DisclosuresNone.

Urinary NGAL in CHF
161
ReferencesHillege HL, Nitsch D, Pfeffer MA et al. Renal function as a predictor of outcome in a broad spectrum 1. of patients with heart failure. Circulation. 2006;113:671-678.
Ruilope LM, van Veldhuisen DJ, Ritz E, and Luscher TF. Renal function: The Cinderella of cardiovascular 2. risk profile. J Am Coll Cardiol. 2001;38:1782-1787.
Hillege HL, van Gilst WH, van Veldhuisen DJ et al. Accelerated decline and prognostic impact of renal 3. function after myocardial infarction and the benefits of ACE inhibition: the CATS randomized trial. Eur Heart J. 2003;24:412-420.
van de Wal RM, Asselbergs FW, Plokker HW et al. High prevalence of microalbuminuria in chronic 4. heart failure patients. J Card Fail. 2005;11:602-606.
Ding H, He Y, Li K et al. Urinary neutrophil gelatinase-associated lipocalin (NGAL) is an early 5. biomarker for renal tubulointerstitial injury in IgA nephropathy. Clin Immunol. 2007;123:227-234.
Liangos O, Perianayagam MC, Vaidya VS et al. Urinary N-acetyl-beta-(D)-glucosaminidase activity and 6. kidney injury molecule-1 level are associated with adverse outcomes in acute renal failure. J Am Soc Nephrol. 2007;18:904-912.
Trachtman H, Christen E, Cnaan A et al. Urinary neutrophil gelatinase-associated lipocalcin in 7. D+HUS: a novel marker of renal injury. Pediatr Nephrol. 2006;21:989-994.
Mori K, Lee HT, Rapoport D et al. Endocytic delivery of lipocalin-siderophore-iron complex rescues the 8. kidney from ischemia-reperfusion injury. J Clin Invest. 2005;115:610-621.
Smilde TD, van Veldhuisen DJ, Navis G, Voors AA, and Hillege HL. Drawbacks and prognostic value 9. of formulas estimating renal function in patients with chronic heart failure and systolic dysfunction. Circulation. 2006;114:1572-1580.
Schmidt-Ott KM, Mori K, Li JY et al. Dual action of neutrophil gelatinase-associated lipocalin. J Am 10. Soc Nephrol. 2007;18:407-413.
Ljungman S, Laragh JH, and Cody RJ. Role of the Kidney in Congestive Heart-Failure - Relationship 11. of Cardiac Index to Kidney-Function. Drugs. 1990;39:10-21.
Nangaku M. Chronic hypoxia and tubulointerstitial injury: a final common pathway to end-stage renal 12. failure. J Am Soc Nephrol. 2006;17:17-25.
Norman JT and Fine LG. Intrarenal oxygenation in chronic renal failure. Clin Exp Pharmacol Physiol. 13. 2006;33:989-996.
Damman K, Navis G, Smilde TD et al. Decreased cardiac output, venous congestion and the association 14. with renal impairment in patients with cardiac dysfunction. Eur J Heart Fail. 2007;9:872-878.
Mishra J, Dent C, Tarabishi R et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker 15. for acute renal injury after cardiac surgery. Lancet. 2005;365:1231-1238.
Wagener G, Jan M, Kim M et al. Association between increases in urinary neutrophil gelatinase-associated 16. lipocalin and acute renal dysfunction after adult cardiac surgery. Anesthesiology. 2006;105:485-491.

162

Chapter 9Tubular damage is common and associated with reduced survival in
patients with chronic systolic heart failure
Kevin Damman, Dirk J. van Veldhuisen, Gerjan Navis, Vishal S. Vaidya, Tom D.J. Smilde, B. Daan Westenbrink,
Joseph V. Bonventre, Adriaan A. Voors, Hans L. Hillege
Submitted

164
Abstract
BackgroundThere is little information on the prevalence of tubular damage, and the association with renal function
and prognosis in patients with chronic heart failure (CHF).
Methods and ResultsCHF patients underwent accurate invasive measurement of glomerular filtration rate (GFR) and
effective renal plasma flow (ERPF) by 125I-Iothalamate and 131I-Hippuran clearances. Neutrophil gelatinase associated lipocalin (NGAL), N-acetyl-beta-D-glucosaminidase (NAG) and kidney injury molecule 1 (KIM-1) as markers of tubular damage, as well as urinary albumin excretion (UAE) were determined in 24 hours urine collections. In total 90 patients (79% male, mean age 59±11 years) were included. Mean GFR was 78±26 ml/min/1.73m2 and mean LVEF was 28±9%. Urinary NGAL (175 (70-346) µg/gram creatinine (gCr)), NAG (12 (6-17 U/gCr)) and KIM-1 (277 (188-537) ng/gCr) levels were increased compared to 20 healthy controls (all P < 0.001). Urinary NAG, but not NGAL or KIM-1 correlated with GFR (r = -0.34, P = 0.001) and ERPF (r = -0.29, P = 0.006). Both NAG (r = 0.21, P = 0.048) and KIM-1 (r = 0.23, P = 0.033) correlated with plasma N terminal-pro brain natriuretic peptide levels. Both urinary KIM-1 (Hazard ratio (HR) 1.15 (1.02-1.30) per 100 ng/gCr increase, P = 0.025) and NAG (HR 1.42 (1.02–1.04) per 5 U/gCr increase, P = 0.039), were associated with an increased risk of death or heart failure hospitalizations, independent of GFR.
ConclusionStructural tubular damage is prevalent in patients with CHF, as measured by increased urinary
concentrations of NGAL, NAG and KIM-1. Urinary NAG levels relate to the severity of reduced GFR and ERPF. Both urinary KIM-1 and NAG showed prognostic information additional to GFR.

Tubular damage and prognosis in CHF
165
IntroductionImpaired renal function is an important independent risk factor for all cause mortality
and morbidity in patients with chronic heart failure (CHF) [1-3]. The pathogenesis of renal impairment in CHF is multifactorial, but the main determinant is a disproportionate decrease in renal perfusion as a consequence of decreased cardiac output [4,5]. Theoretically, decreased renal perfusion will not only result in decreased renal filtration function, but possibly also in structural glomerular and tissue pathology.
Tubular pathology is present in patients with chronic and acute renal failure and is associated with prognosis in these patients [6-9]. These reports showed that with different etiologies of renal failure, tubular dysfunction develops, as characterized by increases in the urinary concentrations of tubular damage proteins, such as N-acetyl-beta-D-glucosaminidase (NAG), kidney injury molecule 1 (KIM-1), and neutrophil gelatinase associated lipocalin (NGAL). In a recent proof of concept study in patients with CHF, we showed that tubular dysfunction, as measured by increased urinary NGAL levels is highly prevalent, and it is not only associated with different indices of renal dysfunction (estimated glomerular filtration rate (eGFR) and urinary albumin excretion (UAE)), but also positively associated with increased levels of N-terminal pro brain natriuretic peptide (NT-proBNP) [10].
In the present study we further analyzed the prevalence of tubular damage as estimated by different tubular marker proteins, the relationship with renal function in patients with CHF, and the association with different tubular marker proteins and prognosis.
Methods
Patient population and study designThe main study design has been published elsewhere [11]. A total of 90 patients were available
for this analysis. In addition 20 age- and gender- balanced healthy controls were studied.
In short, outpatient CHF patients, aged ≥ 18 years, left ventricular ejection fraction (LVEF) < 45%, and clinically stable, were asked to participate. All patients were on angiotensin converting enzyme inhibitors and/or angiotensin II receptor blockers, and all medication had to be stable for at least 1 month. All subjects gave informed consent to participate in the study, which was approved by the ethics review committees of the study centre. The study was conducted in accordance with the guidelines of the Declaration of Helsinki.
Baseline measurements included standard weight, height, systolic and diastolic blood pressure and assessment of NYHA function class heart failure. Patients underwent clearance measurements of renal function. GFR and effective renal plasma flow (ERPF) were measured by the clearances of 125I-Iothalamate and 131I-Hippuran as described earlier. The filtration fraction (FF) was calculated as the ratio of GFR and ERPF and expressed as percentage. Estimated GFR (eGFR) was additionally determined using the simplified modification of diet in renal

166
Chapter 9
disease formula (186.3 x serum creatinine -1.154 x age -0.203 (x 0.742 if female) (x 1.212 if black)), for comparison between CHF patients and controls, since the latter did not undergo invasive renal function measurements.
Laboratory measurements consisted of serum creatinine, haemoglobin levels and NT-proBNP.
Urinalysis and markers of tubular damagePatients and controls collected 24-hours urine and UAE was determined. Urinary creatinine
was determined to correct for concentration of urine. NGAL was determined by means of a commercially available ELISA test kit from Antibody Shop (Gentofte, Denmark). Samples were diluted 500 times in dilution buffer supplied with the test kit. NGAL was expressed in ng/ml and values are also shown corrected for urinary creatinine concentration. The enzyme NAG was evaluated using the substrate p-nitrophenyl N-acetyl-β-D-glucosaminide (Sigma, St Louis, MO) in citrate buffer at pH 4.5. After 60 minutes at 37 oC, 1 M Na2CO3 was added to the mixture to terminate the reaction and to develop a yellow color released from the converted substrate. Controls were obtained from each sample by addition of Na2CO3 at time=0. The color was measured at 405 nm by a microtiter plate reader and controls were subtracted. A standard curve was made with N-acetyl-β-D-glucosaminidase. Urinary NAG activity was expressed as U/gram urinary creatinine (U/gCr). Urinary KIM-1 measurements were performed using microsphere-based Luminex xMAP technology with polyclonal antibodies raised against the human KIM-1 ectodomain. For measurements, 30 µl of urine samples were analyzed in duplicate. The lowest limit of detection for this assay is 0.125 ng/ml. The inter and intra-assay variability was < 20%. The urinary KIM-1 level was normalized to the urinary creatinine concentration.
Follow up and prognosisPatients follow-up started after the 125I-Iothalamate and 131I-Hippuran clearances were
performed. A combined clinical outcome parameter was defined consisting of death, heart transplantation, cardiovascular event (myocardial infarction or primary PTCA), and first hospitalization for heart failure with a maximum 30 months follow up.
StatisticsData are given as mean ± standard deviation when normally distributed, as median and
interquartile range when skewed distributed, and as frequencies and percentages for categorical variables. Differences between patients and controls were tested using Mann-Whitney U or students T testing where appropriate. One-sided ANOVA or Kruskal-Wallis were used for multiple comparisons where appropriate. Spearman’s correlation coefficients were calculated between NAG, NGAL and other variables. Testing of equality of correlations was carried out using Fisher’s Z transformation for the correlation coefficients. Survival analysis was carried out using Cox proportional hazard analysis. In the multivariate analyses, we first constructed a model with the individual urinary markers and GFR. In a second model, GFR was replaced

Tubular damage and prognosis in CHF
167
by serum creatinine. In the final model, we adjusted for numerous confounding factors (age, gender, LVEF, blood pressure, haemoglobin levels, GFR, NT-proBNP and UAE). Testing for interaction revealed no significant (P < 0.1) interactions between the investigated variables. All reported probability values are 2-tailed, and a P value <0.05 was considered statistically significant. Statistical analyses were performed using SPSS, Chicago version 12.0 and STATA, College Station, Texas, version 10.0. The authors had full access to the data and take responsibility for its integrity. All authors have read and agree to the manuscript as written.
ResultsClinical characteristics of patients and controls are summarized in Table 1. The majority of
patients were in NYHA functional class heart failure II and III, with a mean LVEF of 28 ± 9 %. Mean GFR was mildly impaired (78 ± 26 ml/min/1.73m2), with a corresponding decreased ERPF (282 ± 83 ml/min/1.73m2). The urinary concentration of markers of tubular damage, including NAG (12 (6.2 – 17) U/gCr), NGAL (175 (70 – 346) µg/gCr) and KIM-1 (277 (188 – 537) ng/gCr)) were significantly elevated in CHF patients compared to healthy controls (NAG 1.6 (0.7 – 2.2) U/gCr, NGAL 37 (6 – 58) µg/gCr, and KIM-1 136 (63 – 195) ng/gCr, respectively (all P < 0.001) (figure 1). Even after adjustment for the difference in eGFR, urinary NGAL, NAG and KIM-1 levels were significantly higher in CHF patients (all P < 0.001) compared to controls.
Correlations with markers of tubular damage. Urinary NAG, NGAL and KIM-1 levels only showed weak, non-significant associations
with each other (Table 2). Urinary NAG levels were significantly lower in men than in women, while both urinary NGAL and KIM-1 levels increased with advancing age. Table 2 shows relationships between urinary NAG, NGAL and KIM-1 and different cardiorenal functional
Figure 1. Urinary NGAL, NAG and KIM-1 levels in CHF patients versus controls. Shown are boxplots with median and interquartile ranges (box) and 5-95% ranges (error bars) of urinary NAG, NGAL and KIM-1 levels. * P < 0.0001 versus controls. Abbreviations: NAG: N-acetyl-β-D-glucosaminidase, NGAL: Neutrophil gelatinase associated lipocalin, KIM-1: kidney injury molecule 1.
Controls CHF Controls CHF Controls CHF
NA
G (U
/gC
r)
0
5
10
15
20
25
30
0
100
200
300
400
500
600
KIM
-1 (n
g/gC
r)
0
200
400
600
800
1000
1200NAGNGALKIM-1
* *
NG
AL
(μg/
gCr)
*
168
Chapter 9
parameters including GFR, plasma NT-proBNP and UAE. The relationship between urinary NAG and GFR (r = -0.34, P = 0.001) and ERPF (r = -0.29, P = 0.006), was stronger compared to the relationship of urinary KIM-1 and NGAL with these renal indices. Both urinary NAG (r = 0.21, P = 0.048) and urinary KIM-1 levels (r = 0.23, P = 0.033) showed a significant association with NT-proBNP levels. Urinary NAG, NGAL or KIM-1 did not show any significant correlation with blood pressure, haemoglobin levels or LVEF.
Table 1. Baseline Characteristics and relationship between quartiles of NAG.
Variable Total Population(n = 90)
Controls(n = 20)
P-value
Age (years) 59 ± 11 58 ± 4 0.91
Gender, n (% male) 71 (79) 16(80) 0.91
NYHA (I /II / III or IV (%)) 16 / 47 / 37 NA NA
Ischemic etiology, n (%) 43 (48) NA NA
LVEF (%) 28 ± 9 NA NA
Systolic BP (mmHg) 121 ± 20 120 ± 11 0.67
Diastolic BP (mmHg) 69 ± 11 71 ± 8 0.30
Hemoglobin (g/dl) 14 ± 1.3 14 ± 1.0 0.99
NT-proBNP (pg/mL) 515 (219 – 1283) 84 (57 - 108) <0.0001
Renal indices
Serum creatinine (mg/dL) 1.27 ± 0.41 0.89 ± 0.15 <0.001
eGFR (mL/min/1.73m2) 64 ± 17 90 ± 12 <0.0001
GFR (mL/min/1.73m2) 78 ± 26 NA NA
ERPF (mL/min/1.73m2) 282 ± 83 NA NA
Filtration Fraction (%) 27 ± 5 NA NA
UAE (mg/day) 5.1 (3.4 – 10) 1.5 (1.2 - 1.9) <0.0001
Tubular markers
NGAL (µg/gCr) 175 (70 – 346) 37 (6 – 58) <0.0001
NAG (U/gCr) 12 (6.2 – 17) 1.6 (0.7 – 2.2) <0.0001
KIM-1 (ng/gCr) 277 (188 – 537) 136 (63 – 195) <0.0001
Therapy (n (% use))
RAS inhibitor 90 (100) 0 NA
Beta Blocker 78 (87) 0 NA
Diuretic 63 (70) 0 NA
Aldosterone receptor blocker 29 (32) 0 NA
Abbreviations: BP: blood pressure, ERPF: effective renal plasma flow, GFR: glomerular filtration rate, LVEF: left ventricular ejection fraction, NAG: N-acetyl-β-D-glucosaminidase, NGAL: Neutrophil gelatinase associated lipocalin, NT-proBNP: N Terminal pro-brain natriuretic peptide, RAS: Renin angiotensin system, UAE: urinary albumin excretion.

Tubular damage and prognosis in CHF
169
Data on tubular markers, stratified for eGFR according to the definition of chronic kidney disease (CKD) (</≥ 60 mL/min/1.73m2) are given in figure 2. These show that tubular markers are elevated in CHF, even when renal function is preserved. Yet, with a decrease in renal function a further elevation of NAG is observed, while urinary NGAL and KIM-1 levels did not differ between CHF patients with or without CKD.
Table 2. Correlation between markers of tubular damage and renal function parameters
Variable Univariate correlation coefficient (95% CI)
NGAL NAG KIM-1
Tubular markers
NGAL NA 0.15 (-0.06 to 0.35) 0.16 (-0.05 to 0.35)
NAG 0.15 (-0.06 to 0.35) NA 0.13 (-0.08 to 0.33)
KIM-1 0.16 (-0.05 to 0.35) 0.13 (-0.08 to 0.33) NA
Renal Function Measurements
GFR -0.07 (-0.27 to 0.14) -0.34 (-0.51 to -0.14)† -0.01 (-0.22 to 0.20)
ERPF -0.06 (-0.26 to 0.15) -0.29 (-0.47 to -0.09)† -0.11 (-0.31 to 0.10)
Filtration Fraction 0.04 (-0.17 to 0.25) -0.16 (-0.36 to 0.05) 0.17 (-0.04 to 0.36)
UAE 0.10 (-0.11 to 0.30) 0.16 (-0.05 to 0.35) 0.21 (-0.01 to 0.40)
NT-proBNP -0.03 (-0.24 to 0.18) 0.21 (0.00 to 0.40)* 0.23 (0.02 to 0.42)*
† P < 0.01, *P < 0.05. Abbreviations: ERPF: Effective renal plasma flow, GFR: Glomerular filtration rate, NAG: N-acetyl-β-D-glucosaminidase, NGAL: Neutrophil gelatinase associated lipocalin, NT-proBNP: N-terminal pro brain natriuretic peptide, UAE: Urinary albumin excretion.
Table 3. Univariate and adjusted cox proportional hazard analysis for urinary NAG, NGAL and KIM-1as predictors for the combined endpoint.
Predictor Variable
Urinary NGAL (per 100 µg/gCr)
(HR [95% CI]) P-value
Urinary NAG (per 5 U/gCr) (HR [95% CI]) P-value
Urinary KIM-1 (per 100 ng/gCr)
(HR [95% CI]) P-value
Unadjusted 1.02 (0.87 – 1.20) NS 1.43 (1.10 – 1.84) 0.007 1.13 (1.00 – 1.28) 0.047
Adjusted for GFR 1.01 (0.88 – 1.16) NS 1.42 (1.02 – 1.94) 0.039 1.15 (1.02 – 1.30) 0.025
Adjusted for serum creatinine 1.03 (0.89 – 1.19) NS 1.49 (1.11 – 2.00) 0.008 1.18 (1.04 – 1.33) 0.009
Adjusted for Age, gender, LVEF, SBP, DBP, Hb, GFR, NT-proBNP and UAE
1.01 (0.85 – 1.19) NS 1.46 (0.98 – 2.17) 0.066 1.16 (1.00 – 1.35) 0.046
Abbreviations: GFR: glomerular filtration rate, NGAL: neutrophil gelatinase associated lipocalin, NAG: N-acetyl-β-D-glucosaminidase, KIM-1: kidney injury molecule 1, gCr: gram urinary creatinine, HR: hazard ratio, CI: con-fidence interval, LVEF: left ventricular ejection fraction, SBP: systolic blood pressure, DBP: diastolic blood pressure, Hb: hemoglobin levels, NT-proBNP: N terminal pro brain natriuretic peptide, UAE: urinary albumin excretion.
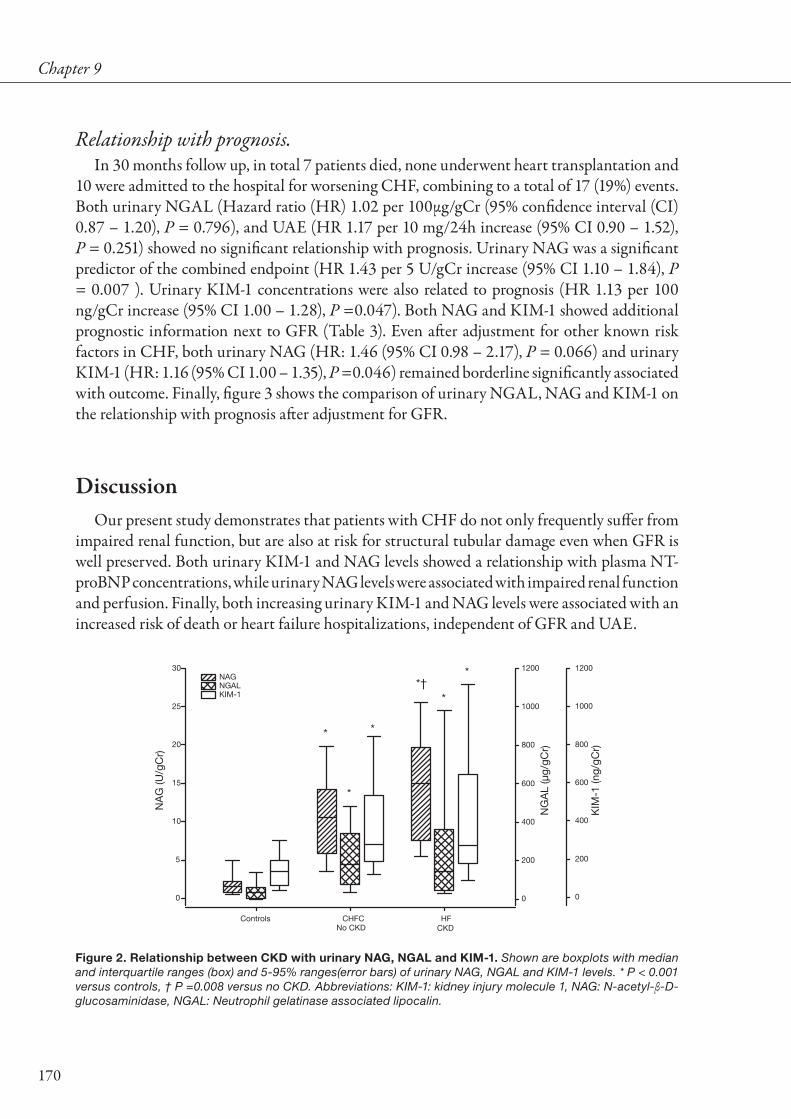
170
Chapter 9
Relationship with prognosis.In 30 months follow up, in total 7 patients died, none underwent heart transplantation and
10 were admitted to the hospital for worsening CHF, combining to a total of 17 (19%) events. Both urinary NGAL (Hazard ratio (HR) 1.02 per 100µg/gCr (95% confidence interval (CI) 0.87 – 1.20), P = 0.796), and UAE (HR 1.17 per 10 mg/24h increase (95% CI 0.90 – 1.52), P = 0.251) showed no significant relationship with prognosis. Urinary NAG was a significant predictor of the combined endpoint (HR 1.43 per 5 U/gCr increase (95% CI 1.10 – 1.84), P = 0.007 ). Urinary KIM-1 concentrations were also related to prognosis (HR 1.13 per 100 ng/gCr increase (95% CI 1.00 – 1.28), P =0.047). Both NAG and KIM-1 showed additional prognostic information next to GFR (Table 3). Even after adjustment for other known risk factors in CHF, both urinary NAG (HR: 1.46 (95% CI 0.98 – 2.17), P = 0.066) and urinary KIM-1 (HR: 1.16 (95% CI 1.00 – 1.35), P =0.046) remained borderline significantly associated with outcome. Finally, figure 3 shows the comparison of urinary NGAL, NAG and KIM-1 on the relationship with prognosis after adjustment for GFR.
DiscussionOur present study demonstrates that patients with CHF do not only frequently suffer from
impaired renal function, but are also at risk for structural tubular damage even when GFR is well preserved. Both urinary KIM-1 and NAG levels showed a relationship with plasma NT-proBNP concentrations, while urinary NAG levels were associated with impaired renal function and perfusion. Finally, both increasing urinary KIM-1 and NAG levels were associated with an increased risk of death or heart failure hospitalizations, independent of GFR and UAE.
Figure 2. Relationship between CKD with urinary NAG, NGAL and KIM-1. Shown are boxplots with median and interquartile ranges (box) and 5-95% ranges(error bars) of urinary NAG, NGAL and KIM-1 levels. * P < 0.001 versus controls, † P =0.008 versus no CKD. Abbreviations: KIM-1: kidney injury molecule 1, NAG: N-acetyl-β-D-glucosaminidase, NGAL: Neutrophil gelatinase associated lipocalin.
Controls CHFC HF
NA
G (U
/gC
r)
0
5
10
15
20
25
30
0
200
400
600
800
1000
1200
KIM
-1 (n
g/gC
r)
0
200
400
600
800
1000
1200NAGNGALKIM-1
No CKD CKD
*
*
*
*†*
*
NG
AL
(μg/
gCr)

Tubular damage and prognosis in CHF
171
Tubular marker proteins in CHF patientsIn the present analysis, we showed that urinary NGAL, NAG and KIM-1 levels were
highly increased in patients with CHF compared to controls, even after adjustment for lower (e)GFR levels. In the normal population, or in control samples of other patient populations, urinary NGAL, NAG and KIM-1 levels were much lower than in our present CHF cohort (Supplementary Table 1a-c).
NGAL is a 21 kD protein of the lipocalin family and is normally secreted in low amounts in lung, kidney, trachea, stomach and colon tissue [12,13]. It is thought to possess bacteriostatic properties [13]. In patients with acute kidney injury, both serum and urinary concentrations rise massively in response to tubular ischemia, and therefore NGAL is thought to be a marker of ongoing tubular injury [13]. Furthermore, acute tubular necrosis caused by heart failure-induced hypotension was shown to be associated with higher expression of renal NGAL [9]. NGAL levels in our present CHF cohort were comparable to those in children with cyanotic heart disease and chronic kidney disease, but higher than observed in elective PCI patients or IgA nephropathy (Supplementary Table 1a).
Figure 3. Relationship of tubular markers, UAE and GFR with prognosis. GFR is shown as decreasing
GFR. Abbreviations: GFR: glomerular filtration rate, KIM-1: kidney injury molecule 1, NAG: N-acetyl-β -D-glucosaminidase, NGAL: Neutrophil gelatinase associated lipocalin.
GFR (mL/min/1.73m2)020406080100120140
Haz
ard
rat
io fo
r co
mb
ined
end
poi
nt
0
2
4
6
8
10
12
NAG (U/gCr)05 10 15 20 25 30 35 40
0 250 500 750 1000 1250 1500 1750 2000
KIM-1 (ng/gCr)0 200 400 600 800 1000 1200 1400 1600
GFRNAGNGALKIM-1
Adjusted for GFR
NGAL (μg/gCr)

172
Chapter 9
NAG is a lysosomal brush border enzyme found in proximal tubular cells. It is relatively large (>130kD), and is therefore not filtered through the glomerular membrane [7,14,15]. It is found at elevated urinary concentrations in acute and chronic kidney disease, as well as diabetic patients [16,17] and in patients with essential hypertension [18]. The elevated NAG excretion in renal disease is correlated to proteinuria, and is reduced by antiproteinuric treatment by RAAS-blockade [19]. In selected patient populations, patients with higher NAG levels which are produced by the nephron, also more often experience albuminuria and more severe structural renal damage [8]. Compared to other patient populations, urinary NAG values in our present study were comparable to those found in diabetes, hypertension and acute MI. (Supplementary Table 1b). In CHF patients without overt renal impairment, urinary NAG levels were already higher than in the normal population, but not as high as in the present study [20].
KIM-1 is a transmembrane protein which is believed to play a role in tubulo-interstitial damage [20,21]. In response to acute kidney injury, the KIM-1 ectodomain is cleaved and shedded into the urine, where it is undetectable in normal circumstances. Urinary KIM-1 levels are elevated in acute renal disease of various origin [20,22], in diabetes [20,23], and various other causes of chronic renal failure in humans [24,25]. Furthermore, the extent of tubulo-interstitial damage and fibrosis has been associated with urinary KIM-1 concentrations, and KIM-1 mRNA levels correlate strongly with urinary KIM-1 concentration in rats exposed to bilateral renal ischemia [21]. Urinary KIM-1 concentrations in our present cohort were somewhat lower compared to values reported in patients with diabetes, acute renal dysfunction, and ischemic ATN, but comparable to subjects with contrast induced nephropathy or chronic kidney disease (Supplementary Table 1c). The elevation of urinary KIM-1 in acute renal damage is reversible upon restoration of renal function, as well as during reduction of proteinuria by RAS-blockade [19]. This reduction corresponds to the reduction in intrarenal expression [26].
The sensitivity and specificity for (histological) tubulointerstitial damage of NAG, NGAL and KIM-1 has mainly been studied in patients with renal disease, showing strong sensitivity and reasonable specificity for the presence of tubular damage [6,7,21,27]. In addition, urinary concentrations of NAG, NGAL and KIM-1 showed strong correlations with the extent of histological presence of tubulointerstitial damage in different studies [6,8,24]. In our present study, the interrelationships between urinary NAG, NGAL and KIM-1 were modest, and non-significant. This is in disagreement with findings in chronic kidney disease. Reasons for this discrepancy may be the small sample size, but also differences in the pathophysiology of renal damage in CHF as compared to primary renal parenchymal disease, as well as the extent of tubular injury in CHF patients. In addition, we found strong differences between associations of NAG, NGAL and KIM-1 with clinical and cardiorenal parameters. Only urinary NAG showed a relationship with estimates of glomerular function, which might suggest partly dependency of glomerular filtration, limiting sensitivity for actual tubular damage. In contrast, neither KIM-1, nor NGAL showed a relationship with glomerular indices, suggesting a better specificity for tubulointerstitial damage.

Tubular damage and prognosis in CHF
173
Pathophysiology of tubular damage in CHFImpaired GFR in CHF is mainly dependent on renal perfusion and is thought to be a
reversible upon restoration of renal perfusion, as estimated GFR has been shown to improve after LVAD implantation in pre-transplant CHF patients [28]. Also in the general CHF population impaired GFR may improve over time [29]. However, reports in experimental animal settings suggest that hypoperfused kidneys may in fact suffer from structural damage in addition to, and contributing to the belief of functionally decreased filtration function [30]. Furthermore, chronic renal hypoxia is not only considered as a common final pathway in end-stage renal disease, but is also related to tubulointerstitial damage [31]. Additionally, tubulointerstitial damage itself may predispose to a vicious circle of kidney injury and hypoxia, leading to chronic renal insufficiency [32]. Therefore, CHF patients, who have relatively poor renal perfusion, may be especially at increased risk for development of hypoxic tubulo(interstitial) damage. Indeed, we were able to show that urinary NAG showed the strongest relationship with ERPF and GFR, supporting this hypothesis. However, the association between NGAL and KIM-1 with either the degree of renal impairment or reduced ERPF were weak and non-significant. One reason for this contrast may be that urinary concentrations of NGAL not only rises massively in response to acute (tubular) renal injury, but also very quickly decreases when the initiating trigger has vanished [33,34]. The discrepancy with urinary KIM-1 levels is much less clear and should be the focus of larger studies.
Interestingly, we recently showed that venous congestion is also an important determinant of GFR, possibly by increasing renal interstitial pressure and subsequently causing damage [35]. In agreement with this hypothesis, we observed a significant relationship between both urinary NAG and KIM-1 levels with NT-proBNP in our present analysis.
Next to reduced GFR and now also tubular damage, albuminuria is frequently observed in CHF [36]. Albuminuria is assumed to be caused by glomerular leakage or damage, which often precedes reduced GFR, but these two do not always co-exist. In addition, albuminuria may reflect impairment of tubular reabsorption of proteins. In our present cohort, we were unable to establish a relationship between markers of tubular damage and UAE. This is in disagreement with findings in renal failure, in which both NGAL and NAG correlated well with UAE [6,14,15]. In addition, all of our patients were on RAS-blocking therapy, which will have limited albuminuria and could have influenced the associations between tubular damage and UAE.
Tubular damage and prognosisThere is only limited data available on the relationship between the presence of tubulo-
interstitial injury as measured by histological abnormalities or increased levels of urinary marker proteins and prognosis, even in primary kidney disease. In patients with acute kidney injury, both urinary NAG and KIM-1 predicted the occurrence of hospital death or need for dialysis, although only urinary NAG remained an independent predictor after adjustment [7]. In primary glomerulonephritis, urinary NAG levels were significant predictors of functional outcome [14]. Urinary KIM-1 levels were independent predictors of graft loss in renal transplant

174
Chapter 9
recipients [37]. Our present cohort of CHF patients further emphasizes the prognostic importance of both urinary NAG and KIM-1. Both markers of tubular damage were predictors of the combined endpoint of death, heart failure hospitalisation and heart transplantation. In addition, both showed prognostic importance, independent of GFR. This might implicate that, in terms of risk profiling, the presence of tubular damage may be an additive component related to prognosis in CHF patients, which is independent of generally impaired GFR. The absence of any relationship with prognosis of urinary NGAL warrants further investigation, especially in patients who frequently develop acute worsening or renal function.
LimitationsThis study is hampered by its small size, therefore observed associations may not represent
the general CHF population. Urinary NGAL, NAG and KIM-1 are markers of tubular dysfunction, and, although they correlate with histological abnormalities in renal disease, we did not perform renal biopsies in the present study. Therefore, actual histological renal damage in these patients cannot be assessed. Furthermore, all patients in the present study were on renin angiotensin system blocking medication, which has been shown to reduce urinary excretion of both KIM-1 and NAG, along with proteinuria [19]. The present study shows cross-sectional data with follow up, but no serial measurements are available. We did not investigate the effect of treatment on the observed relationships. Finally, the interrelationships between these urinary markers of tubular damage, including the specificity and sensitivity for both decreased renal function and outcome should be the focus of larger prospective studies in both acute and chronic heart failure.
ConclusionStructural tubular damage, as measured by increased levels of urinary tubular proteins levels,
is prevalent in patients with CHF. The prognostic impact of these tubular markers, especially urinary KIM-1 and NAG, is additive to that of impaired GFR, and seems independent of the prevalence of albuminuria. These proteins may be new non-invasive markers of renal dysfunction and prognosis in these patients.

Tubular damage and prognosis in CHF
175
AcknowledgementsK. Damman is supported by the Netherlands Heart Foundation (grant 2006B157) A.A.
Voors and D.J. van Veldhuisen are Clinical Established Investigators of the Netherlands Heart Foundation (grants 2006T37 and D97-017, respectively). Joseph V. Bonventre is supported by the National Institute of Health (grants DK39773 and DK74099).
DisclosuresJoseph V. Bonventre is co-inventor on KIM-1 patents.

176
Chapter 9
ReferencesDries DL, Exner DV, Domanski MJ, Greenberg B, and Stevenson LW. The prognostic implications of 1. renal insufficiency in asymptomatic and symptomatic patients with left ventricular systolic dysfunction. J Am Coll Cardiol. 2000;35:681-689.
Ruilope LM, van Veldhuisen DJ, Ritz E, and Luscher TF. Renal function: The Cinderella of cardiovascular 2. risk profile. J Am Coll Cardiol. 2001;38:1782-1787.
Hillege HL, Nitsch D, Pfeffer MA et al. Renal function as a predictor of outcome in a broad spectrum 3. of patients with heart failure. Circulation. 2006;113:671-678.
Ljungman S, Laragh JH, and Cody RJ. Role of the Kidney in Congestive Heart-Failure - Relationship 4. of Cardiac Index to Kidney-Function. Drugs. 1990;39:10-21.
Smilde TDJ, Damman K, van der Harst P et al. Differential associations between renal function and 5. "modifiable" risk factors in patients with chronic heart failure. Clin Res Cardiol. 2009;98:121-129.
Ding H, He Y, Li K et al. Urinary neutrophil gelatinase-associated lipocalin (NGAL) is an early 6. biomarker for renal tubulointerstitial injury in IgA nephropathy. Clin Immunol. 2007;123:227-234.
Liangos O, Perianayagam MC, Vaidya VS et al. Urinary N-acetyl-beta-(D)-glucosaminidase activity and 7. kidney injury molecule-1 level are associated with adverse outcomes in acute renal failure. J Am Soc Nephrol. 2007;18:904-912.
Machiguchi T, Yoshida H, Yonemoto S et al. Does circulating erythropoietin reflect progression of IgA 8. nephropathy? Comparison with urinary N-acetyl-beta-D-glucosaminidase. Nephrol Dial Transplant. 1999;14:635-640.
Mori K, Lee HT, Rapoport D et al. Endocytic delivery of lipocalin-siderophore-iron complex rescues the 9. kidney from ischemia-reperfusion injury. J Clin Invest. 2005;115:610-621.
Damman K, van Veldhuisen DJ, Navis G, Voors AA, and Hillege HL. Urinary neutrophil gelatinase 10. associated lipocalin (NGAL), a marker of tubular damage, is increased in patients with chronic heart failure. Eur J Heart Fail. 2008;10:997-1000.
Smilde TD, van Veldhuisen DJ, Navis G, Voors AA, and Hillege HL. Drawbacks and prognostic value 11. of formulas estimating renal function in patients with chronic heart failure and systolic dysfunction. Circulation. 2006;114:1572-1580.
Cowland JB and Borregaard N. Molecular characterization and pattern of tissue expression of the gene 12. for neutrophil gelatinase-associated lipocalin from humans. Genomics. 1997;45:17-23.
Schmidt-Ott KM, Mori K, Li JY et al. Dual action of neutrophil gelatinase-associated lipocalin. J Am 13. Soc Nephrol. 2007;18:407-413.
Bazzi C, Petrini C, Rizza V et al. Urinary N-acetyl-beta-glucosaminidase excretion is a marker of tubular 14. cell dysfunction and a predictor of outcome in primary glomerulonephritis. Nephrol Dial Transplant. 2002;17:1890-1896.
Wellwood JM, Ellis BG, Price RG et al. Urinary N-acetyl- beta-D-glucosaminidase activities in patients 15. with renal disease. Br Med J. 1975;3:408-411.
Uslu S, Efe B, Alatas O et al. Serum cystatin C and urinary enzymes as screening markers of renal 16. dysfunction in diabetic patients. J Nephrol. 2005;18:559-567.

Tubular damage and prognosis in CHF
177
Weitgasser R, Schnoell F, Gappmayer B, and Kartnig I. Prospective evaluation of urinary N-acetyl-beta-17. D-glucosaminidase with respect to macrovascular disease in elderly type 2 diabetic patients. Diabetes Care. 1999;22:1882-1886.
Harmankaya O, Ozturk Y, Basturk T, and Obek A. Urinary excretion of N-acetyl-beta-D-glucosaminidase 18. in newly diagnosed essential hypertensive patients and its changes with effective antihypertensive therapy. Int Urol Nephrol. 2001;32:583-584.
Waanders F, Vaidya VS, Goor HV et al. Effect of Renin-Angiotensin-Aldosterone System Inhibition, 19. Dietary Sodium Restriction, and/or Diuretics on Urinary Kidney Injury Molecule 1 Excretion in Nondiabetic Proteinuric Kidney Disease: A Post Hoc Analysis of a Randomized Controlled Trial. Am J Kidney Dis. 2009;53:16-25.
Matsushima H, Yoshida H, Machiguchi T et al. Urinary albumin and TGF 1 levels as renal damage 20. indices in patients with congestive heart failure. Clin Exp Nephrol. 2002;6:21-29.
Vaidya VS, Ramirez V, Ichimura T, Bobadilla NA, and Bonventre JV. Urinary kidney injury molecule-1: 21. a sensitive quantitative biomarker for early detection of kidney tubular injury. Am J Physiol Renal Physiol. 2006;290:F517-F529.
Prozialeck WC, Vaidya VS, Liu J et al. Kidney injury molecule-1 is an early biomarker of cadmium 22. nephrotoxicity. Kidney Int. 2007;72:985-993.
Nosadini R, Carboni A, Manconi A et al. The decline of glomerular function is not always associated 23. with the development of micro- and macroalbuminuria in hypertensive patients with type 2 diabetes. Diabetologia. 2008;epub ahead of print.
van Timmeren MM, van den Heuvel MC, Bailly V et al. Tubular kidney injury molecule-1 (KIM-1) in 24. human renal disease. J Pathol. 2007;212:209-217.
Waikar SS and Bonventre JV. Biomarkers for the diagnosis of acute kidney injury. Curr Opin Nephrol 25. Hypertens. 2007;16:557-564.
de Borst MH, van Timmeren MM, Vaidya VS et al. Induction of kidney injury molecule-1 in homozygous 26. Ren2 rats is attenuated by blockade of the renin-angiotensin system or p38 MAP kinase. Am J Physiol Renal Physiol. 2007;292:F313-F320.
Zhou Y, Vaidya VS, Brown RP et al. Comparison of kidney injury molecule-1 and other nephrotoxicity 27. biomarkers in urine and kidney following acute exposure to gentamicin, mercury, and chromium. Toxicol Sci. 2008;101:159-170.
Butler J, Geisberg C, Howser R et al. Relationship between renal function and left ventricular assist 28. device use. Ann Thorac Surg. 2006;81:1745-1751.
de Silva R, Nikitin NP, Witte KK et al. Incidence of renal dysfunction over 6 months in patients with 29. chronic heart failure due to left ventricular systolic dysfunction: contributing factors and relationship to prognosis. Eur Heart J. 2006;27:569-581.
Chade AR, Rodriguez-Porcel M, Grande JP et al. Distinct renal injury in early atherosclerosis and 30. renovascular disease. Circulation. 2002;106:1165-1171.
Nangaku M. Chronic hypoxia and tubulointerstitial injury: a final common pathway to end-stage renal 31. failure. J Am Soc Nephrol. 2006;17:17-25.

178
Chapter 9
Norman JT and Fine LG. Intrarenal oxygenation in chronic renal failure. Clin Exp Pharmacol Physiol. 32. 2006;33:989-996.
Coca SG, Yalavarthy R, Concato J, and Parikh CR. Biomarkers for the diagnosis and risk stratification 33. of acute kidney injury: a systematic review. Kidney Int. 2008;73:1008-1016.
Mishra J, Dent C, Tarabishi R et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker 34. for acute renal injury after cardiac surgery. Lancet. 2005;365:1231-1238.
Damman K, Navis G, Smilde TD et al. Decreased cardiac output, venous congestion and the association 35. with renal impairment in patients with cardiac dysfunction. Eur J Heart Fail. 2007;9:872-878.
van de Wal RM, Asselbergs FW, Plokker HW et al. High prevalence of microalbuminuria in chronic 36. heart failure patients. J Card Fail. 2005;11:602-606.
van Timmeren MM, Vaidya VS, van Ree RM et al. High urinary excretion of kidney injury molecule-1 37. is an independent predictor of graft loss in renal transplant recipients. Transplantation. 2007;84:1625-1630.

Tubular damage and prognosis in CHF
179
Su
pp
lemen
tary Table 1a. N
GA
L values in co
ntro
ls and
oth
er patien
t po
pu
lation
s
Stud
yN
C
ontro
lN
GA
L value contro
lN
d
isease g
roup
Disease g
roup
NG
AL value
Type p
atients
Bachorzew
ska-Gajew
ska et al. 2006
3510.9 (0.2-60.2) μg/L b
aseline35
14.8 (1.4 – 117.4) μg/L 24 h after PC
I12.2 (0.2 – 98.2) μg/L 48 h after P
CI
Patients und
ergoing elective PC
I
Ding et al. 2007
401.5 ±
0.3 μg/L70
4.1 ± 1.6 μg/L G
rade II nep
hropathy*
21.3 ± 4.6 μg/L G
rade III nep
hropathy*
Ad
justed for urine concentration
4.0 ± 1.5 ng/gC
r Grad
e II nephrop
athy*25 ±
3 ng/gCr G
rade III nep
hropathy*
Patients w
ith IgA nep
hropathy
Mishra et al. 2005
102.2 ±
0.5 μg/L (adult)
1.6 ± 0.3 μg/L (child
)71
Max 175 ±
40 μg/L*A
djusted
for urine concentrationM
ax 150 ± 40 ng/gC
r*
Child
ren with congenital heart d
is-ease, N
GA
L after cardiop
ulmonary
byp
ass
Wagener et al. 2006
None
NA
815994 ±
7616 μg/L AR
D1760 ±
3527 μg/L no AR
DP
atient undergoing card
iac surgery, w
ith or without A
RD
Bachorzew
ska-Gajew
ska et al. 2007
601.5 (0-58) μg/L b
aseline60
10.5 (0.55 – 117) μg/L 4 h after PC
I14.8 (0.5 – 237) μg/L 8 h after P
CI
13.3 (0.5 – 251) μg/L 24 h after PC
I
Patients und
ergoing PC
I
Parikh et al. 2006
711.7 ±
0.5 μg/L without A
RD
1.8 ± 0.5 μg/L w
ith AR
D71
5.0 ± 1.1 μg/L w
ithout AR
D (m
ax)178 ±
36.2 μg/L with A
RD
(max)
Child
ren undergoing card
iopulm
onary b
ypass surgery, w
ith or without A
RD
Mori et al. 2005
1022 μg/L
39557 μg/L in ATN119 μg/L in C
KD
Patients w
ith either acute or chronic renal failure
Hirsch et al. 2007
800.5 ±
0.2 μg/gCr
(6 h after CA
G, no C
IN)
1113.6 ±
4.2 μg/gCr
Ad
olescents who und
erwent C
AG
, w
ith and w
ithout CIN
Bolignano et al. 2007
267 ±
6 ng/mL
26119 ±
42 ng/mL
Autosom
al-dom
inant polycystic kid
-ney d
isease
Current stud
y20
37 (6 – 58) µg/gCr
90175 (70 – 346) µg/gC
rC
HF p
atients
* from
figure. A
bb
reviations: A
RD
: acute renal dysfunctio
n, ATN
: acute tubulus necro
sis, CA
G: co
ronary ang
iograp
hy, CIN
: contrast ind
uced nep
hrop
athy, CK
D:
chronic kid
ney disease, P
CI: p
ercutaneous co
ronary interventio
n, NG
AL: N
eutrop
hil gelatinase asso
ciated lip
ocalin.

180
Chapter 9S
up
ple
men
tary
Tab
le 1
b. N
AG
val
ues
in c
on
tro
ls a
nd
oth
er p
atie
nt
po
pu
lati
on
s
Stu
dy
N
cont
rol
NA
G v
alue
cont
rol
N d
isea
se
gro
upD
isea
se g
roup
NA
G v
alue
Typ
e p
atie
nts
Aw
ad e
t al
. 200
3 14
8.94
± 7
.63
U /
Cr
(nm
ol/m
g)72
31.6
8 –
244.
77 (n
mol
/mg)
Chi
ldre
n su
fferin
g fr
om c
yano
tic c
onge
nita
l hea
rt
dis
ease
.
Baz
zi e
t al
. 200
2 N
one
NA
136
17.4
-25.
4 U
/g C
rP
rimar
y gl
omer
ulon
ephr
itis
Mac
higu
chi e
t al
. 199
9 20
1.9
± 0
.5 U
/g
Cr
386.
7 ±
6.2
U /
g C
rIg
A N
ephr
opat
hy
Usl
u et
al.
2005
20
1.74
(0.9
4 –
2.50
) U /
g C
r56
4.41
(1.4
3 –
16.8
0) U
/g
Cr
Dia
bet
ic p
atie
nts
Wei
tgas
ser
et a
l. 19
99
Non
eN
A12
45.
4 –
7.7
U /
g C
rD
iab
etic
pat
ient
s
Har
man
kaya
et
al. 2
001
365.
0 ±
1.2
U /
L38
7.8
± 0
.5 U
/ L
New
ly d
iagn
osed
pat
ient
s w
ith e
ssen
tial h
yper
ten-
sion
Lian
gos
et a
l. 20
07
Non
eN
A20
10
– 43
0 U
/g C
r (e
stim
ated
from
figu
re)
Pat
ient
s w
ith a
cute
ren
al fa
ilure
Loef
et
al. 2
004
200
(bef
ore
CP
B)
(est
imat
ed fr
om fi
gure
)20
Max
9.0
U/g
Cr/
h (e
stim
ated
from
figu
re)
Pat
ient
s w
ith C
AD
, und
ergo
ing
CA
BG
. Mea
sure
-m
ents
bef
ore,
dur
ing
and
aft
er C
AB
G. N
orm
al L
VE
F.
Din
g et
al.
2007
40
1.5
± 0
.3 U
/g
Cr
Tota
l: 70
Gra
de
II*G
rad
e III
* 2.
3 ±
0.8
U /
g C
r 5.
1 ±
2.3
U /
g C
r
Pat
ient
s w
ith Ig
A n
ephr
opat
hy
Tayl
or e
t al
. 199
7 26
01.
38 ±
0.7
3 U
/g
Cr
71 63 112
2.53
U /
g C
r1.
33 U
/g
Cr
2.23
U /
g C
r
Cad
miu
m e
xpos
ure
Mer
cury
exp
osur
eLe
ad e
xpos
ure
Mat
sush
ima
et a
l. 20
02
200.
17 ±
0.1
7 ng
/ml
6.8
± 4
.8 U
/g
Cr
P
atie
nts
with
CH
F, w
ithou
t re
nal i
mp
airm
ent.
No
optim
al m
edic
al t
hera
py.
Waa
nder
s et
al.
2008
20
2.7
± 0
.5 U
/ D
3412
.8 ±
1.2
U /
D
Pat
ient
s w
ith C
KD
with
out
pro
tein
uria
or
DM
Cur
rent
stu
dy
201.
6 (0
.7 –
2.2
) U/g
Cr
9012
(6.2
-17)
U/g
Cr)
CH
F p
atie
nts
Ab
bre
viat
ions
: CA
D: c
oro
nary
art
ery
dis
ease
, CH
F: C
hro
nic
Hea
rt F
ailu
re, C
KD
: chr
oni
c ki
dne
y d
isea
se, D
M: d
iab
etes
mel
litus
, NA
G: N
-ace
tyl-
β-D
-glu
cosa
min
idas
e,
g C
r: g
ram
urin
ary
crea
tinin
e.

Tubular damage and prognosis in CHF
181
Su
pp
lem
enta
ry T
able
1c.
KIM
-1 v
alu
es in
co
ntr
ols
an
d o
ther
pat
ien
t p
op
ula
tio
ns
Stu
dy
N
Co
ntro
lK
IM-1
val
ue c
ont
rol
N
dis
ease
gro
upD
isea
se g
roup
KIM
-1 v
alue
Typ
e p
atie
nts
Lian
gos
et a
l. 20
07N
one
NA
201
0 –
3500
0 ng
/g C
r (e
stim
ated
from
figu
re)
Pat
ient
s w
ith a
cute
ren
al fa
ilure
Han
et
al. 2
002
8<
0.1
ng/m
L7 7 9 9
2.9
± 0
.61
μg/g
Cr
0.85
± 0
.30
μg/g
Cr
0.48
± 0
.19
μg/g
Cr
0.72
± 0
.37
μg/g
Cr
Isch
emic
ATN
Con
tras
t N
ephr
opat
hyO
ther
AR
DC
KD
Van
Tim
mer
en e
t al
. 200
711
0 (0
– 0
.37)
μg/
g C
r53
1.42
(0.3
3 –
14.4
7) μ
g/g
Cr
Pat
ient
s w
ith v
ario
us c
ause
s of
ren
al im
pai
rmen
t
Nos
adin
i et
al. 2
008
Non
eN
A30 27 15 17
4.1
(0.6
– 1
0)3.
8 (0
.7 –
9.7
)4.
4 (0
.7 –
11)
3.9
(0.7
– 1
0.7)
Typ
e II
DM
, eG
FR 6
0-75
, mic
roal
bum
inur
iaTy
pe
II D
M, e
GFR
60-
75, n
orm
oalb
umin
uria
Typ
e II
DM
, eG
FR 7
6-90
, mic
roal
bum
inur
iaTy
pe
II D
M, e
GFR
76-
90, n
orm
oalb
umin
uria
Han
et
al.
2008
300.
1 (0
.0-0
.2) μ
g/g
Cr
29 103.
3 (2
.1-5
.5) μ
g/g
Cr
0.4
(0.2
-2.0
) μg/
g C
rA
RD
Urin
ary
trac
t in
fect
ion
Waa
nder
s et
al.
2008
3558
± 8
ng
/ D
3417
06 ±
498
ng/
DP
atie
nts
with
CK
D w
ithou
t p
rote
inur
ia o
r D
M
Cur
rent
stu
dy
2013
6 (6
3 –
195)
ng/
gCr
9027
7 (6
3 –
195)
ng/
gCr
CH
F p
atie
nts
Ab
bre
viat
ions
: ATN
: acu
te tu
bul
us n
ecro
sis,
AR
D: a
cute
ren
al d
ysfu
nctio
n, C
HF:
chr
oni
c he
art f
ailu
re, C
KD
: chr
oni
c ki
dne
y d
isea
se, D
M: d
iab
etes
mel
litus
, KIM
-1:
Kid
ney
inju
ry m
ole
cule
1,
g C
r: g
ram
urin
ary
crea
tinin
e.

182

Summary

184

Summary
185
In the FIRST PART of the thesis, we investigated potential pathophysiologic mechanisms associated with reduced GFR in patients with heart failure (HF). In chapter 1, we studied mechanisms involved in cardiorenal failure in a cross sectional study, investigating GFR and renal blood flow (RBF) by radiolabeled tracer techniques in 110 chronic HF patients, who were all on angiotensin converting enzyme inhibitor and/or angiotensin II receptor blocker therapy. The main finding of this study was that when RBF decreased, a similar reduction in GFR was found, while the filtration fraction remained stable until only very low values of RBF are observed. These observations have consolidated decreased RBF as the most important determinant of GFR in patients with HF, and should therefore be considered as the most important cornerstone of the cardiorenal syndrome. In chapter 2 and 3, we studied the relationship between central venous pressure (CVP) and (estimated) GFR in patients with and without HF. In chapter 2, we showed that in patients with pulmonary hypertension, cardiac dysfunction, and a low RBF, a higher CVP was associated with a lower GFR. Importantly, this effect seemed independent of RBF. We further explored this relationship between CVP and estimated GFR in a large cohort of patients with cardiovascular disease in chapter 3, and found similar results. Independent of cardiac output, patients with higher CVP levels showed significantly lower estimated GFR levels, compared to those with relatively normal CVP. In chapter 4, we investigated the relationship between non-invasive signs of venous congestion, and the relationship with renal function and prognosis in patients who participated in the cardiac insufficiency bisoprolol study II (CIBIS-II) study. Signs of congestion were frequently observed in this high risk chronic HF patient population, and patients with more signs of congestion had more severe renal impairment. We additionally showed that signs of venous congestion assessed by physical examination were a strong, independent predictor of mortality.
In the SECOND PART of the thesis, we investigated the occurrence of worsening renal function (WRF) and the association with outcome in HF. In chapter 5, we showed that WRF was associated with a 60% increase risk for all cause mortality, and 30% increase in the risk for HF hospitalization. In addition, patients who already have impaired renal function are at greatest risk for development of WRF. In chapter 6, we investigated the relationship between the occurrence of WRF at different points in time, and the relationship with prognosis in a substudy of the Coordinating Study Evaluating Outcomes of Advising and Counseling in Heart Failure (COACH). We showed that WRF at any moment in time is associated with a strong increase in the occurrence of the combined endpoint of mortality and heart failure readmissions.
In the THIRD PART of the thesis, we investigated emerging pathophysiological pathways of the cardiorenal connection and the importance of tubular damage in patients with heart failure. In chapter 7, we investigated known and emerging factors associated with prognosis, and the dependency of their prognostic information with GFR. Using pathway analysis, we showed that a combination of decreased perfusion, filtration efficacy, volume overload, and subsequently hemodilution and anemia, were responsible for the combined effect of decreased

186
Summary
GFR on prognosis. This further emphasizes the importance of both reduced perfusion and congestion, not only in the pathophysiology of renal impairment, but also in the effect on clinical outcome. In chapter 8 we showed that urinary neutrophil gelatinase associated lipocalin (NGAL) levels, as markers of tubular damage, are increased in chronic HF patients. In addition, across the severity of eGFR, from HF patients to controls, higher urinary NGAL levels were found in patients with lower eGFR, and higher N terminal pro brain natriuretic peptide (NT-proBNP) levels. In this proof of concept study, urinary NGAL levels remained higher in HF patients compared to controls, even after adjustment for eGFR. This could indicate that tubular dysfunction may progressively worsen, resulting in sustained elevation of urinary NGAL concentrations. In chapter 9, we further explored the prevalence, relationship with renal function and prognosis of different markers of tubular damage in patients with chronic HF. We observed similar results obtained with NGAL for urinary N-acetyl-β-D-glucosaminidase (NAG) and kidney injury molecule 1 (KIM-1) levels, showing that their levels are increased in patients with chronic HF. However, only urinary NAG related to invasively determined GFR and renal perfusion. Finally, both urinary NAG and KIM-1 were related to prognosis, independent of GFR.

Summary
187

188

Discussion and Future Perspectives

190

Discussion and future perspectives
191
Heart failure (HF) is not only a syndrome characterized by its clinical symptoms, signs and poor prognosis, but also by its high degree of co-morbid organ dysfunction and co-morbidities [1-7]. At least part of the extreme mortality and morbidity of HF has been considered attributable to the existence of these co-morbid conditions [3,8]. Perhaps one of the most important co-morbidities in terms of pathophysiology, prognosis and treatment of HF is the co-existence of renal failure [6,8-15]. Although several terms to address this frequently occurring condition have been proposed, the term ‘cardiorenal syndrome’ is favored by many [16-20]. However, the specific definition of this syndrome still needs consensus. Recent reports have put forward at least five types of the cardiorenal syndrome, differentiating between acute and chronic, as well as cardiorenal and renocardiac, reflecting underlying pathophysiology, although also on these subtypes definitions are still being optimalised (Table 1) [18,19].
Several key aspects have been suggested as the cornerstone of this syndrome including: a) the observation that the co-occurrence of heart and renal failure seems to be related to more severe deteriorated prognosis, b) the observation that both entities may worsen each other and c) the presence of pathophysiologic mechanisms relating to each other such as poor hemodynamics, endothelial dysfunction, inflammation and renin-angiotensin system activation [8,17,21-25]. Bongartz et al put forward a model based on Guyton’s model of hemodynamics and fluids control in cardiorenal interaction, introducing ‘cardiorenal connectors’ as specific modifiers of the relationship between hemodynamics and cardiorenal function and failure [17]. However, this model has never been formally tested, and does not include new insights recently observed in the cardiorenal interaction in HF. In the present thesis we have further investigated the contribution of different pathophysiologic determinants of renal failure in patients with heart failure, providing new information on the characteristics and pathophysiology of the cardiorenal syndrome. These and others are listed in Table 2.
Decreased renal functionThe hallmark of HF is the inability of the heart to preserve cardiac output, resulting in
decreased perfusion of peripheral organ systems [24]. However, the most prominent symptoms in HF are not primarily the result of decreased organ perfusion, but consist of the inability of the body to excrete sodium, and secondary water. This is the main cause of the classical symptoms of HF, including dyspnea, peripheral edema, orthopnea, ascites and classical physical signs such as jugular venous distention, rales and weight gain. The reason for excessive salt and water retention is however not primarily related to the cardiac dysfunction itself, but to the co-existing renal failure. The exact mechanism of excessive salt and water retention is in its turn primarily related to decreased glomerular filtration rate (GFR), resulting in the most important question why GFR is reduced in HF.

192
Hemodynamics: Decreased renal perfusionIn the first part of the 20th century, a great deal of work has been done on the relationship
between reduced cardiac function and renal failure, which eventually lead to the discovery of renal autoregulation. In these studies, the main driving force of a reduction in GFR was thought to be attributable to a decrease in renal blood flow (RBF), which was the result of severely increased renal vascular resistance (RVR) as a consequence of increased efferent arteriolar vasoconstriction [26-30]. This concept was re-introduced in the cardiology in the last part of the 20th century when Ljungman et al showed that with decreasing cardiac index, RBF decreased disproportionately fast [25,31]. However, GFR was preserved to some extent by increasing filtration fraction, suggesting increased efferent vasoconstriction. Finally, RBF decreases further, and GFR could no longer be maintained, and dropped along the same line as RBF.
However, with the introduction of angiotensin converting enzyme inhibitors (ACEi) and angiotensin II receptor blockers (ARB), this angiotensin II mediated predominantly efferent vasoconstriction is counteracted [32,33]. This has two possible important consequences. First, RBF is increased by decreasing RVR which, after an initial drop in GFR, may preserve GFR over a longer period of time [25]. However, second, it is also possible that by counteracting the GFR preserving mechanism of efferent vasoconstriction a further decline in RBF cannot be counteracted to preserve GFR. Recently, we investigated this last possibility in a cross sectional study, investigating GFR, RBF by radiolabeled tracer techniques in 110 chronic HF patients, who were all on ACEi and/or ARB therapy [34]. We were able to show that with lower RBF values, we could not establish a preservation of GFR as published by Ljungman. Instead, GFR dropped in a similar way compared to RBF, with filtration fraction remaining stable until only very low values of RBF are observed [34]. In this last patient group, although filtration fraction decreased, RVR increased, which may suggest afferent arteriolar vasoconstriction in these patients, which we will discuss later. The combination of the findings in HF patients with and without renin angiotensin system (RAS) blocking therapy seem to suggest that a) early GFR preservation with mildly impaired RBF is indeed mainly dependent on efferent
Table 1. Definition of subtypes of the Cardiorenal syndrome
Type Title Description
Type 1 Acute Cardiorenal Syndrome Acute worsening cardiac function leading to acute renal injury
Type II Chronic Cardiorenal Syndrome Chronic reduced cardiac function leading to progressive renal injury
Type III Acute Renocardiac Syndrome Acute worsening renal function leading to acute cardiac failure
Type IV Chronic Renocardiac Syndrome Chronic renal failure leading to progressive HF
Type V Secondary Cardiorenal Syndrome Systemic condition causing cardiorenal dysfunction
Adjusted from Ronco et al.[18,19]

Discussion and future perspectives
193
vasoconstriction, that b) the introduction of ACEi and ARB preserves GFR to some extent, but significantly hampers the intrinsic counterregulatory mechanism of the kidney itself to preserve GFR and c) deterioration of GFR with more severe RBF deterioration in the presence of ACEi or ARB may be at least partly attributable to increased afferent vasoconstriction. Together, these observations have consolidated decreased RBF as the most important determinant of GFR in patients with HF, and should be considered as the most important cornerstone of the cardiorenal syndrome.
Hemodynamics: Increased central venous pressureIn the same experimental studies investigating the effect of reduced RBF in HF, different
models were used to initiate decreased renal perfusion pressure. One of these models consisted of increasing renal venous pressure [26,27]. By inducing higher renal venous pressures, the pressure gradient across the glomerulus is decreased when systemic arterial pressure is unchanged, causing a decreased RBF and subsequent decreased GFR. There were however suggestions that increased (central or renal) venous pressure itself may have impact on decreased GFR, independent of the induction of decreased perfusion [26,35,36]. Although these studies could not give a definite answer to whether this was actual the case, more recent studies on this subject showed more convincing results. Higher central venous pressure (CVP) may lead to higher pressures in the encapsulated kidney, causing hypoxia or increase in hydrostatic pressure in Bowman’s capsule, thereby decreasing GFR [37,38]. Other aspects may include an effect of CVP on intrarenal angiotensin II levels, which may have detrimental effect on single nephron GFR [37-39]. Higher CVP may even be linked to proteinuria [40]. We have recently provided more clinical evidence on a direct link between increased CVP and (estimated)
Table 2. Summary of characteristics of the Cardiorenal Syndrome
Characteristics of the Cardiorenal Syndrome
Reduced RBF and GFR
Increased Venous Congestion
Albuminuria
Tubular Damage
Worsening Renal Function
Diuretic Resistance
Activation of the TGF
Anemia
Increased mortality
Abbreviations: GFR: glomerular filtration rate, RBF: renal blood flow, TGF: Tubuloglomerular feedback mechanism

194
Tab
le 3
. Ris
k fa
cto
rs f
or
dev
elo
pm
ent
of
wo
rsen
ing
ren
al f
un
cti
on
Ris
k fa
cto
rs f
or
WR
FS
tud
ies
inve
stig
atin
g W
RF
in H
F#
Kru
mho
lzFo
rman
De
Silv
aK
han
Ow
anLo
gear
tC
owie
Jose
Akh
ter
Met
raW
einf
eld
Chi
ttin
eni
CO
AC
H
Red
uced
bas
elin
e G
FRX
XX
XX
XX
XX
XX
XX
13
Hig
h S
BP
/ H
yper
tens
ion
XX
XX
XX
6
Dia
bet
es
X
X
X
X
XX
6
Ane
mia
/ h
emog
lob
in
X
XX
X
X5
Age
X
X
X
X
X5
Diu
retic
use
*
X
X
XX
4
LVE
F
X
X
X
3
Wom
enX
X
X
3
Vasc
ular
dis
ease
X
XX
3
NY
HA
cla
ss
X
X
2
Sig
ns o
f con
gest
ion
X
X
2
Sm
okin
g
X
X
2
Hig
her
hear
t ra
teX
1
BM
I
X
1
AR
A u
se
X
1
Whi
te e
thni
city
X
1
Sin
us r
hyth
m
X
1
Atr
ial F
ibril
latio
n
X
1
Hyp
onat
rem
ia
X1
* T
hiaz
ides
and
/or
loo
p d
iure
tics.
Ab
bre
viat
ions
: AR
A: a
ldo
ster
one
rec
epto
r an
tag
oni
st, B
MI:
bo
dy
mas
s in
dex
, GFR
: glo
mer
ular
filtr
atio
n ra
te, L
VE
F: le
ft v
entr
icul
ar
ejec
tion
frac
tion,
NY
HA
: New
Yo
rk h
eart
ass
oci
atio
n, S
BP
: sys
tolic
blo
od
pre
ssur
e, W
RF:
wo
rsen
ing
rena
l fun
ctio
n.

Discussion and future perspectives
195
GFR in patients with and without HF. In patients with pulmonary hypertension and cardiac dysfunction, a disease very prone to high right sided filling pressures, we showed that in the presence of relatively preserved RBF, higher CVP has no influence on GFR [41]. However, in the presence of reduced RBF, when GFR is already depressed, the presence of higher CVP was associated with further reduced GFR. Importantly, RBF values were similar in those patients with higher and lower GFR, suggesting that CVP had an effect on GFR which was independent of RBF. We further explored this relationship between CVP and estimated GFR in a large cohort of patients with cardiovascular disease and found similar results [42]. Independent of cardiac output, patients with higher CVP levels showed significantly lower estimated GFR levels, compared to those with relatively normal CVP. We are supported on this finding by evidence from other studies, including a substudy of the ESCAPE trial, showing that the single (invasively determined) parameter associated with reduced renal function was in fact right atrial pressure [43]. Furthermore, specific therapy by levosimendan, aimed at lowering CVP showed improvement in renal function [44,45]. Finally, also tricuspid regurgitation, causing increased CVP, was found to be associated with impaired renal function in patients with HF, strengthening the observation of a relationship between CVP and GFR [46]. While invasively determined CVP is probably not feasible in patients with HF, we investigated the relationship between non-invasive signs of venous congestion, and the relationship with renal function and prognosis in patients who participated in the cardiac insufficiency bisoprolol study II (CIBIS-II) study [47]. Signs of congestion were frequently observed in this high risk chronic HF patient population, and patients with more signs of congestion had more severe renal impairment. This was in agreement with our findings of invasively determined CVP. However, also this relationship may also be partly attributable to excessive salt and water retention in response to decreased GFR, although the relationship was even stronger in patients with the relatively highest left ventricular ejection fraction. Finally, we were able to show that, despite the probability that the interobserver variability is very high, signs of venous congestion assessed by physical examination, was a strong, independent predictor of outcome, which increased the risk for all-cause mortality with the presence of more congestive symptoms.
In addition to our findings of the relationship between increased intravasal CVP and GFR, there is also a different pathophysiologic mechanism by which renal venous pressure may rise. This was already shown in the abdominal compartment syndrome, where patients have very high intra-abdominal pressures, which resulted in high renal venous pressures as mechanical complication [48,49]. Indeed, these patients show lower RBF and lower GFR levels. Recently, Mullens et al argued that this situation is not specific to the abdominal compartment syndrome, showing that elevated intra-abdominal pressure in patients with HF may be an important pathophysiologic mechanism of reduced renal function in these patients [50]. In addition, they showed that reducing the intra-abdominal pressure lead to an improvement in renal function, further supporting this hypothesis [51].
In summary, increased CVP of different origins and the presence of congestive symptoms seem to be important pathophysiologic factors in the cardiorenal syndrome, and deserve future attention.

196
Signs of renal damage: AlbuminuriaIncreased urinary albumin excretion (UAE) is a strong predictor of cardiovascular events in
the general population and in patients with hypertension and diabetes [52-55]. Furthermore, its occurrence in primary renal disease is a predictor of outcome and the first sign of glomerular injury, but is also the main target of therapy in these patients [56]. In addition, numerous reports have showed that the presence of (micro/macro) albuminuria severely increases the risk for the development of HF or risk for HF admissions [53,57-59]. However, in HF itself there is little evidence of the prevalence, pathophysiology and relationship with prognosis of increased UAE, although albuminuria in HF has long been recognized [40]. Van de Wal et al showed that close to one third of patients with HF may have microalbuminuria [60]. Reasons for albuminuria in HF are unclear. In non-HF conditions, albuminuria is considered either as early glomerular injury, or as marker of endothelial dysfunction and leakage [61,62]. Higher blood pressures, and therefore higher hydraulic glomerular pressures have been shown to be correlated to albuminuria in hypertension, but this does not seem to reflect the pathophysiology in HF [63]. On the contrary, lower filtration fraction and the lack of association with higher blood pressures in HF suggest the pathophysiology of albuminuria in HF does not include high hydraulic pressures [34]. Albuminuria may be associated with decreased RBF, which may suggest that albuminuria in HF is related to hypoxic damage [34]. Other mechanisms may also include higher CVP, independent of decreased RBF [40]. And although albuminuria is a strong predictor of events in patient populations other than HF, the relationship between (micro) albuminuria and prognosis in HF is yet to be established.
Worsening renal functionAlthough the normal course of renal function in HF is yet to be established, studies suggest
that up to one third of HF patients experience worsening renal function (WRF) at any point in time [64]. WRF in this regard is defined as an increase in serum creatinine ≥ 26.5 µmol/L (≥ 0.3 mg/dL) with or without a threshold of 25% increase in serum creatinine, roughly corresponding to a decrease in eGFR ≥ 9.0 mL/min/1.73m2 [64-66]. In a meta-analysis, WRF was associated with a 60% increase in risk for all cause mortality after > 6 months follow up time, and 30% increase in the risk for HF hospitalization [64]. In addition, patients who already have impaired renal function are at greatest risk for development of WRF. With more severe WRF, we showed a striking increase in the risk for all-cause mortality, rising up to 300% increase when serum creatinine increases > 44.2 µmol/L. Interestingly, only one report assessed the effect of an improvement in serum creatinine (decrease ≥ 26.5 µmol/L), showing favorable outcome in these patients [9]. We investigated the relationship between the occurrence of WRF at different points in time, the relationship with prognosis, and the factors associated with decreasing renal function or WRF in a substudy of the Coordinating Study Evaluating Outcomes of Advising and Counseling in Heart Failure (COACH) [67]. In our analysis, we showed that WRF during hospitalization, shortly after discharge, or between 6 and 12 months

Discussion and future perspectives
197
after discharge does not only occur frequently, but is associated with a strong increase in the occurrence of the combined endpoint of mortality and heart failure readmissions. In addition, in a landmark analysis, those patients that did not experience an endpoint in the early follow up phase but still experienced WRF, showed strong decreased survival rates. The pathophysiologic mechanisms behind WRF are not yet clear, but the main studies that have investigated the predictors of WRF are listed in Table 3, including the results from the COACH . This summary shows that next to baseline GFR which is the most important factor, (high dose) diuretics, age, anemia and diabetes are a prominent mediators of the development of WRF [9,65,66,68-76]. This may be true inhospital in patients with acute HF, as well as outhospital in patients with chronic HF. In our secondary analysis, we also have investigated the relationship of different factors and the slope of renal function over time (instead of the occurrence of WRF). The factors involved in renal function changes (decline in GFR) were strikingly similar to those observed in WRF, showing that baseline GFR, age, and co-morbidities (anemia, diabetes, peripheral artery disease, and hypertension) predisposed to a more pronounced GFR decline over time. Prevention and treatment of these factors may possibly prevent the occurrence of either WRF or decreasing GFR, which may eventually lead to improved prognosis in these patients with HF. WRF may be an important aspect in the cardiorenal syndrome, as it may connect the occurrence of tubular damage and decreasing GFR with prognosis and cardiorenal disease progression.
Hemodynamics and renin angiotensin system activityHemodynamic derangements seem to be the most important pathophysiologic mechanisms
of renal impairment in HF [25,31,34,41]. While decreased GFR relates to impaired prognosis in HF, it is important to know which aspects of the pathophysiology are responsible for the poor survival of these patients, to establish possible targets for therapy. We investigated known and emerging factors associated with prognosis, and the dependency of their prognostic information with GFR [77]. In pathway analysis, we showed that a combination of decreased perfusion, filtration efficacy (Filtration fraction), volume overload, and subsequently hemodilution and anemia, were responsible for the combined effect of decreased GFR on prognosis. This further emphasizes the importance of both reduced perfusion and congestion, not only in the pathophysiology of renal impairment, but also in the effect on clinical outcome.
In additions to pathways that are dependent on GFR, we found that the sole factor associated with cardiovascular outcome, next to reduced renal perfusion, was plasma renin activity (PRA). This was observed, despite (or in spite) the fact that all patients were on either ACEi and/or ARB therapy. Both therapies may increase PRA [78-80], but the effect of elevated PRA in the presence of these agents has so far not been seriously recognized. Only one study, a substudy of the Valsartan Heart Failure Trial (VALHEFT), showed comparable results, showing sustained prognostic effect of elevated PRA, even in the presence of ACEi or beta-blocker therapy [81]. Reasons for this effect are difficult to unravel, since the negative feedback loop of lower angiotensin II levels, and consequently higher renin levels, should be counteracted by
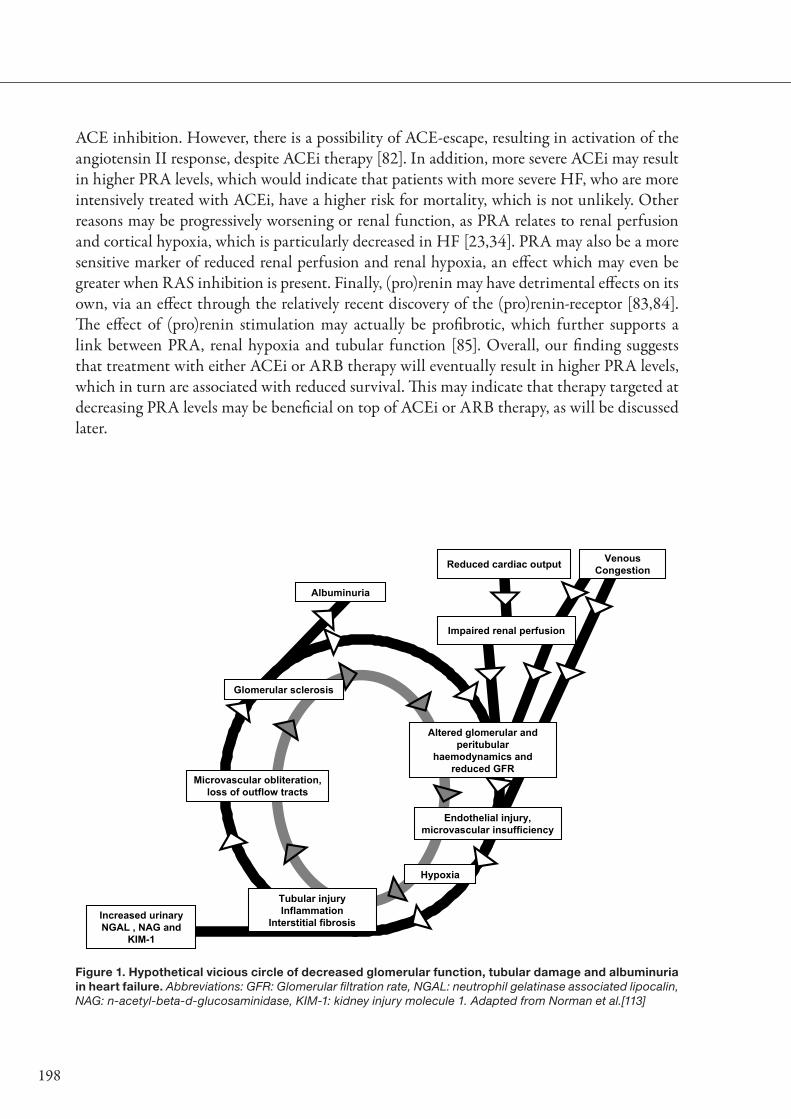
198
ACE inhibition. However, there is a possibility of ACE-escape, resulting in activation of the angiotensin II response, despite ACEi therapy [82]. In addition, more severe ACEi may result in higher PRA levels, which would indicate that patients with more severe HF, who are more intensively treated with ACEi, have a higher risk for mortality, which is not unlikely. Other reasons may be progressively worsening or renal function, as PRA relates to renal perfusion and cortical hypoxia, which is particularly decreased in HF [23,34]. PRA may also be a more sensitive marker of reduced renal perfusion and renal hypoxia, an effect which may even be greater when RAS inhibition is present. Finally, (pro)renin may have detrimental effects on its own, via an effect through the relatively recent discovery of the (pro)renin-receptor [83,84]. The effect of (pro)renin stimulation may actually be profibrotic, which further supports a link between PRA, renal hypoxia and tubular function [85]. Overall, our finding suggests that treatment with either ACEi or ARB therapy will eventually result in higher PRA levels, which in turn are associated with reduced survival. This may indicate that therapy targeted at decreasing PRA levels may be beneficial on top of ACEi or ARB therapy, as will be discussed later.
Figure 1. Hypothetical vicious circle of decreased glomerular function, tubular damage and albuminuria in heart failure. Abbreviations: GFR: Glomerular filtration rate, NGAL: neutrophil gelatinase associated lipocalin, NAG: n-acetyl-beta-d-glucosaminidase, KIM-1: kidney injury molecule 1. Adapted from Norman et al.[113]
Altered glomerular and peritubular
haemodynamics and reduced GFR
Endothelial injury, microvascular insufficiency
Hypoxia
Tubular injuryInflammation
Interstitial fibrosis
Microvascular obliteration, loss of outflow tracts
Glomerular sclerosis
Impaired renal perfusion
Albuminuria
Increased urinaryNGAL , NAG and
KIM-1
Reduced cardiac output VenousCongestion

Discussion and future perspectives
199
AnemiaThe relationship between cardiac and renal failure should not be considered as a simple
straight line across which different pathophysiologic mechanism influence each other. There seem to be different mediators involved in this relationship, but these mediators most of the time only have limited effects [17]. There is however one important mediator of the relationship between heart and renal failure, which is anemia. To emphasize it importance, it is often combined with the cardiorenal syndrome, resulting in the cardio-renal-anemia syndrome [86-88]. Although the pathogenesis of anemia in HF is still under debate, it includes erythropoietin (EPO) resistance, iron deficiency, hemodilution and decreased RBF [89-91]. The co-occurrence of renal failure and anemia in HF seems associated with a severely increased mortality risk, which is much higher than could be expected from the presence of either anemia or renal failure alone [92-95]. This might suggest that both entities may indeed worsen each other, with striking effects on outcome. Both renal failure and anemia relate to decreased perfusion and hemodilution, which all individually relate to outcome [77]. The combination of anemia and renal failure may therefore increase the risk of poor outcome via similar pathways, which may increase the strength of these associations with outcome. In addition, the similarities between the pathophysiology of renal failure and anemia may indicate that therapy to improve renal function, which is focused on improvement of these pathophysiologic pathways, may indirectly also improve hemoglobin levels or anemia.
Arb
itrar
y un
its
0
10
20
30
40
50
60
70
80
90
100
Arbritrary time units
WRF WRF
RBF
GFR
Tubular Dysfunction
Mortality
Albuminuria
Figure 2. Hypothetical time course of renal impairment and associated pathophysiologic entities in heart failure. Abbreviations: GFR: glomerular filtration rate, RBF: renal blood flow, WRF: worsening renal function. RBF may progressively worsen over time. As a consequence, GFR decreases. WRF may develop and may be associated with tubular damage and albuminuria, both worsening over time. The combination of reduced RBF, GFR, and increased tubular dysfunction and albuminuria will lead to strongly increased mortality.

200
Signs of renal damage: Tubular damageIn renal disease, not only albuminuria is frequently observed, but also tubular damage (or
dysfunction) is prevalent [96-101]. However, in HF, there is exceptionally sparse data on the prevalence and pathophysiologic importance of tubular damage [102,103]. In kidney disease, tubular damage is apparent from histological evaluations of renal biopsies in patients with different etiologies of renal failure, and from increased urinary and/or plasma concentrations of specific tubular marker proteins [97,99,101]. There are a number of specific markers of tubular damage, including beta-2-microglobulin, N-acetyl-beta-D-glucosaminidase (NAG), kidney injury molecule 1 (KIM-1), and neutrophil gelatinase associated lipocalin (NGAL). These markers have been widely described and investigated in both acute and chronic kidney disease [96-101,104-107]. In patients with HF without renal impairment, urinary NAG levels have been shown to be elevated compared to the normal population. In a recent study we showed that urinary NGAL levels are increased in chronic HF patients [108]. In addition, across the severity of eGFR, from HF patients to controls, higher urinary NGAL levels were found in patients with lower eGFR, and higher NT-proBNP levels [108]. In this proof of concept study, NGAL levels remained higher in HF patients compared to controls, even after adjustment for eGFR. This could indicate that tubular dysfunction may progressively worsen, resulting in sustained elevation of urinary NGAL concentrations. In a follow up study, we further explored the prevalence, relationship with renal function and prognosis of different markers of tubular damage in patients with chronic HF [109]. We observed similar results obtained with NGAL for urinary NAG and KIM-1 levels, showing that their levels are increased in patients with chronic HF. However, only urinary NAG related to invasively determined GFR
Figure 3. Hemodynamic pathways and detrimental effects of diuretics in the cardiorenal syndrome. Abbreviations: GFR: glomerular filtration rate, RBF: renal blood flow, TGF: tubulo glomerular feedback mechanism.

Discussion and future perspectives
201
and renal perfusion. In addition, urinary NAG, NGAL and KIM-1 levels did not seem to correlate with each other, which would suggest at least some partly different pathophysiology of these markers. Although only NAG was related to renal perfusion, this observation further supports the hypothesis of tubulointerstitial damage occurring when renal (regional) hypoxia develops. In renal disease, tubular damage may arise when high albumin concentrations trigger the tubules to shed these proteins into the urine, but considering the low amount of albumin in HF patients, and the lack of association between urinary NAG, NGAL or KIM-1, a different mechanism may be involved [96,98]. Other studies suggest that also diuretic therapy may initiate renal impairment, possibly also negatively influencing tubular function [76]. Figure 1 shows the hypothetical vicious circle of renal impairment, tubular damage and albuminuria in heart failure. Both decreased perfusion and increased congestion lead to impaired renal function, renal hypoxia, interstitial and tubular damage, albuminuria and subsequently feeds the vicious circle again, to cause sustained renal failure.
The reason why both NGAL and KIM-1 did not correlate to the extent of renal impairment or decreased renal perfusion needs to be investigated further. A reason may be that both markers are more specific for tubular, rather than glomerular damage and dysfunction. Another reason may be that especially NGAL rises quickly in response to tubular damage, but also decrease quickly when the initiating factor has disappeared [110,111]. The striking increase in both urinary NGAL and KIM-1 levels are also observed in patients with acute renal failure, and precedes the rise in serum creatinine by over a day [111,112]. Therefore, these markers may be useful in predicting the occurrence of WRF. This potential of these tubular marker proteins may be useful especially in acute HF, and should be explored in future studies.
In addition to the finding that the concentrations of these tubular marker proteins are elevated in patients with chronic HF, we found that both urinary NAG and KIM-1 levels were predictors of prognosis, independent of GFR. This finding may reflect a different pathophysiologic mechanism through which general renal function, or reduced renal perfusion may ultimately lead to increased mortality. Therefore, the role of these tubular markers as a potential target for therapy for reno-protective treatment may be the focus of new studies.
Finally, the role of tubular damage in the progression of renal failure, cardiorenal disease and prognosis should be the focus of new prospective studies in HF. We have hypothesized on the factors involved in the cardiorenal disease progression in Figure 2, showing that with progressively reduced RBF, GFR decreases, with frequent occurring of WRF, increasing tubular dysfunction and albuminuria, which will eventually lead to increased mortality risk.

202
Tab
le 4
. Tre
atm
ent
in t
he
card
iore
nal
syn
dro
me
/ H
F p
atie
nts
wit
h re
nal
imp
airm
ent
The
rap
yE
SC
Gui
del
ines
AH
A/A
CC
gui
del
ines
Sug
ges
tio
ns in
lite
ratu
re
AC
Ei
Cre
atin
ine
> 2
.5 m
g/d
L co
ntra
ind
icat
ion
for
initi
atio
nC
reat
inin
e ris
e af
ter
initi
atio
n to
3 m
g/d
L ac
cep
tab
leC
reat
inin
e ris
e b
etw
een
3 an
d 3
.5 m
g/d
L: h
alve
dos
eC
reat
inin
e ris
e ab
ove
3.5
mg/
dL:
sto
p A
CE
i
Mai
ntai
n on
AC
Ei a
s lo
ng a
s p
ossi
ble
Cau
tious
initi
atio
n w
hen
crea
tinin
e >
3
mg/
dL
AC
Ei p
rese
rves
ren
al fu
nctio
n on
the
long
te
rm.[1
33]
Defi
nite
sur
viva
l ben
efit
with
GFR
> 3
0 m
L/m
in/1
.73m
2 , p
ossi
ble
bel
ow 3
0 m
L/m
in/1
.73m
2 . [1
24,1
26]
Ob
serv
atio
nal l
ack
of s
urvi
val b
enefi
t in
p
atie
nts
with
HF,
CA
D a
nd C
rCl <
60m
L/m
in
[123
]S
tart
low
, car
eful
eva
luat
ion
rena
l fun
ctio
n
AR
BC
reat
inin
e >
2.5
mg/
dL
cont
rain
dic
atio
n fo
r in
itiat
ion
Cre
atin
ine
rise
afte
r in
itiat
ion
to 3
mg/
dL
acce
pta
ble
Cre
atin
ine
rise
bet
wee
n 3
and
3.5
mg/
dL:
hal
ve d
ose
Cre
atin
ine
rise
abov
e 3.
5 m
g/d
L: s
top
AR
B
Cau
tious
tre
atm
ent
in p
atie
nts
with
se
vere
ren
al im
pai
rmen
t, e
spec
ially
in
com
bin
atio
n w
ith A
CE
i or
Ald
oste
ron
anta
goni
sts
Defi
nite
sur
viva
l ben
efit
with
GFR
> 3
0 m
L/m
in/1
.73m
2 , p
ossi
ble
bel
ow 3
0 m
L/m
in/1
.73m
2 . [1
22,1
25]
Car
eful
eva
luat
ion
rena
l fun
ctio
n.
Bet
a-b
lock
ers
Pre
scrib
e in
pat
ient
s w
ith r
enal
imp
airm
ent
Pre
scrib
e in
pat
ient
s w
ith r
enal
imp
air-
men
tO
bse
rvat
iona
l sur
viva
l ben
efit
in p
atie
nts
with
HF,
CA
D a
nd C
rCl <
60m
L/m
in [1
23]
Sub
stud
y C
IBIS
II s
how
ed b
enefi
t of
bis
o-p
rolo
l in
CrC
l > 3
0 m
L/m
in/1
.73m
2 [1
27]
Ald
ost
ero
n R
ecp
tor
A
ntag
oni
sts
Cre
atin
ine
> 2
.5 m
g/d
L co
ntra
ind
icat
ion
for
initi
atio
nC
reat
inin
e ris
e to
> 2
.5 m
g/d
L: h
alve
dos
eC
reat
inin
e ris
e to
> 3
.5 m
g/d
L: s
top
AR
A
Pre
scrib
e on
ly if
:C
reat
inin
e <
2.5
mg/
dL
in m
enC
reat
inin
e <
2.0
mg/
dL
in w
omen
Defi
nite
sur
viva
l ben
efit
with
GFR
> 6
0 m
L/m
in/1
.73m
2 , p
ossi
ble
bet
wee
n 30
and
60
mL/
min
/1.7
3m2 [1
28]
Diu
reti
csW
RF
with
diu
retic
s: c
heck
for
hyp
ovol
emia
/deh
ydra
-tio
n, e
xclu
de
NS
AID
s, w
ithho
ld A
RA
, sto
p c
on-
com
itant
Thi
azid
es, c
onsi
der
red
uctio
n A
CE
i/AR
B,
cons
ider
ultr
afiltr
atio
nLo
op d
iure
tics
pre
ferr
ed w
hen
CrC
l < 3
0 m
L/m
in
If W
RF
dev
elop
s, d
ecre
ase
diu
retic
d
ose.
If no
t p
ossi
ble
due
to
cong
estio
n: a
c-ce
pt
WR
F to
mai
ntai
n A
CE
i/AR
B.
If re
frac
tory
to
diu
retic
s, c
onsi
der
ul
trafi
ltrat
ion
Diu
retic
s in
crea
se r
isk
for
WR
F [6
8,76
]D
iure
tics
may
initi
ate
rena
l dam
age
Hig
her
dos
es o
f diu
retic
s ne
eded
in p
atie
nts
with
ren
al im
pai
rmen
t. [1
16]
Com
bin
e w
ith s
alt
and
wat
er r
estr
ictio
n, w
ith
care
ful e
valu
atio
n of
ren
al fu
nctio
n. [1
34]
Ult
rafi
ltra
tio
nC
onsi
der
whe
n W
RF
dev
elop
s, r
esis
tanc
e to
tre
at-
men
t or
tre
atm
ent
with
dra
wal
Sev
ere
rena
l im
pai
rmen
t an
d/o
r re
sis-
tanc
e to
tre
atm
ent
No
ben
efici
al e
ffect
of u
ltrafi
ltrat
ion
on r
enal
fu
nctio
n in
UN
LOA
D. U
ltrafi
ltrat
ion
may
eve
n re
duc
e eG
FR in
sim
ilar
fash
ion
as fu
rose
mi-
de
[141
]

Discussion and future perspectives
203
New considerations and future directions in the cardiorenal syndrome
Diuretic resistance and tubuloglom-erular feedback
Diuretic therapy is an important therapeutic tool to relief symptoms and signs of congestion in HF, but there is no proof of a randomized clinical trial showing mortality benefit of diuretics over placebo. Instead, diuretic therapy is associated with WRF, and possibly tubular dysfunction, but does improve quality of life in patients with HF [9,68,76,114]. Therefore, it seems diuretic therapy improves signs and symptoms and quality of life, at the expense of deteriorating renal function. There is however another problem with chronic diuretic use, which is termed ‘diuretic resistance’ [115,116]. The mechanism behind diuretic resistance is based on the fact that the distal tubule and collecting ducts will reabsorb more sodium when, because of the promotion of sodium excretion by diuretics, more sodium reaches the distal tubules. Via this way, the netto sodium excretion and therefore, water excretion, is only limited and higher doses of diuretics are necessary to achieve clinical relevant sodium and water excretion [116]. Adding to this problem is the need for loop diuretics to be filtered before they reach the site of action, which is decreased when GFR is reduced [116]. In addition, high distal sodium load will initiate the tubulo-glomerular feedback mechanism (TGF). The TGF mechanism protects the kidney from losing too much salt by increasing afferent arteriolar glomerular resistance by the action of adenosine, in response to high sodium levels in the distal tubules [117]. This will result in decreased RBF, and therefore
The
rap
yE
SC
Gui
del
ines
AH
A/A
CC
gui
del
ines
Sug
ges
tio
ns in
lite
ratu
re
Dig
oxi
nIf
pre
scrib
ed: l
ow d
oses
0.0
625
/ 0.
125
mg/
day
If p
resc
ribed
: low
dos
es 0
.125
mg/
day
Defi
nite
sur
viva
l ben
efit
with
GFR
> 6
0 m
L/m
in/1
.73m
2 , p
ossi
ble
bet
wee
n 30
and
60
m
L/m
in/1
.73m
2 [1
35]
H-I
SD
NS
ever
e re
nal i
mp
airm
ent
is c
ontr
aind
icat
ion
(dos
e re
duc
tion
may
be
need
ed)
Pre
scrib
e w
hen
no A
CE
i or
AR
B p
os-
sib
le b
ecau
se o
f ren
al fa
ilure
Defi
nite
sur
viva
l ben
efit
with
GFR
> 6
0 m
L/m
in/1
.73m
2 , p
ossi
ble
bet
wee
n 30
and
60
m
L/m
in/1
.732
[136
,137
]
Nes
irit
ide
Not
men
tione
d w
ith r
esp
ect
to r
enal
func
tion.
Not
rec
omm
end
edC
onfli
ctin
g re
sults
of m
eta-
anal
yses
, sho
w-
ing
eith
er n
o ef
fect
or
det
rimen
tal e
ffect
of
Nes
iritid
e on
ren
al fu
nctio
n. [1
38-1
40]
Ult
rafi
ltra
tio
nC
onsi
der
whe
n W
RF
dev
elop
s, r
esis
tanc
e to
tre
at-
men
t or
tre
atm
ent
with
dra
wal
Sev
ere
rena
l im
pai
rmen
t an
d/o
r re
sis-
tanc
e to
tre
atm
ent
No
ben
efici
al e
ffect
of u
ltrafi
ltrat
ion
on r
enal
fu
nctio
n in
UN
LOA
D. U
ltrafi
ltrat
ion
may
eve
n re
duc
e eG
FR in
sim
ilar
fash
ion
as fu
rose
mi-
de
[141
]

204
decreased GFR, which will lead to even more salt and water retention, lower RBF, and lower GFR, eventually feeding a negative vicious circle (Figure 3). Mediation of this mechanism in HF on top of standard HF therapy has been shown to be beneficial for RBF and GFR, and currently trials are underway to determine the effect of an intravenous selective adenosine A1 receptor antagonist (AARA) on clinical outcome in acute decompensated HF patients [118,119]. Interestingly, one small report addressed the effect of AARA on RBF and GFR in chronic HF, showing improvement in both [120]. This further supports our finding that in chronic HF patients on ACEi or ARB therapy, when RBF is very low, GFR is not solely dependent of efferent vasoconstriction, but also on afferent vasoconstriction [34]. This may further emphasize a possible therapeutic role of this new class ‘reno-protective’ therapies, even in chronic HF.
Table 5. Unconventional, new or device therapy
Therapy Suggestions in literature
CRT Substudy of MIRACLE trial showed improvement in estimated GFR in patients receiving CRT with reduced baseline GFR [142]
LVAD LVAD implantation has been shown to improve CrCl in selected pa-tients, especially those with lowest Cardiac index, diabetics and low BMI. [143]
Heart Transplantation ESC guidelines: renal failure (CrCl < 50 mL/min) contraindication for heart transplantation. [1]
Statins Observational survival benefit in patients with HF, CAD and CrCl < 60mL/min [123]No significant interaction between treatment effect and eGFR in CO-RONA [129]
Adenosine A1 receptor antagonists (Rolofylline)
AARAs improve RBF and GFR in chronic HF [120]AARAs improve CrCl and decreased the need for intravenous diuretics in patients with acute HF [144]AARAs protect against the worsening of renal function with diuretic therapy [118]AARA therapy was associated with improved trichotomous endpoint in the PROTECT II pilot trial.[118]
Direct renin inhibition (Aliskiren) Renin inhibition resulted in significant reduction in (NT-pro) BNP and PRA levels in patients with chronic HF. No significant increase in renal dysfunction and/or hyperkalaemia. [145]
Levosimendan Levosimendan improves renal function in acute and advanced chronic HF, despite blood pressure lowering. [45,146]
Vasopressin Antagonists Tolvaptan showed no improvement in renal function over placebo in the EVEREST trial. No significant interaction between treatment effect and creatinine in EVEREST. [147]
Dopamine Low dose dopamine in combination with diuretics may improve renal function in decompensated HF. [148]Dopamine improves RBF in patients with chronic heart failure [149]
Dobutamine No short term effect of dobutamine on eGFR in acute HF [45]

Discussion and future perspectives
205
Treatment of the cardiorenal syndromeThere is no data on effective and safe treatment of patients who present with the cardiorenal
syndrome. In the present thesis, we have put forward pathophysiologic links between cardiac and renal failure, and the relationship with outcome. Therapies in the cardiorenal syndrome should focus on these targets. These include reduced RBF, increased venous congestion, but also anemia, hypertension, tubular dysfunction and sustained RAS-activation. In clinical practice, symptomatic HF therapy is often prescribed, which would normally include RAS-blocking agents, diuretics and beta-blockers [1]. There is however no randomized controlled trial that has evaluated the effectiveness of standard HF care, or specific developed ‘reno-protective’ therapy in patients with combined cardiac and renal failure. Instead, large clinical HF trials have excluded patients with severe renal impairment. Table 4 summarizes key HF therapies as mentioned in the European Society of Cardiology (ESC) and American Heart Association (AHA) guidelines on HF [1,121]. In the recently published guidelines ESC guidelines, increased serum creatinine (above 150 µmol/L (1.7mg/dL)) is acknowledged as a risk factor, but an even higher serum creatinine level (above 220 µmol/L (2.5 mg/dL)) is considered a contra-indication for both ACEi and aldosteron antagonists initiation, while ARBs should only be prescribed in patients with ‘adequate renal function’. When deterioration of renal function occurs, the guidelines suggest halving of ACEi or ARB therapy, even if this occurs after initiation of diuretic therapy. This recommendation however fails to acknowledge that diuretic therapy itself may be the trigger for WRF, although, as discussed, also congestion may predispose to renal failure [9,41,68,121] This is different from the belief in chronic kidney disease in which some decrease in GFR after diuretic therapy is considered favorable. However, considering the low glomerular pressures and low filtration fraction, and the association of diuretic therapy with WRF and secondary outcome, this may not be similar in HF. The AHA guidelines show distinct differences with the ESC guidelines. ACEi and ARB therapy should be continued as long as possible, even if WRF occurs. In agreement with ESC guidelines, beta-blockers should not be withheld, while ultrafiltration may only be indicated in patients who fail to respond to therapy [121]. The last column in table 4 summarizes some findings from observational studies and substudies of clinical trials. Interestingly, ACEi and ARB seem to be effective and safe in patients with mild to moderate renal failure [122-126]. The evidence for aldosteron receptor antagonism seems much less robust in these patients, while beta-blockers show similar results compared to ACEi and ARB [127,128]. Diuretics however have frequently been linked to more severe renal impairment, WRF and patients with renal impairment require higher doses to achieve comparable diuresis [68,76,116,117].
Table 5 shows some unconventional, new or device therapies and their effect in patients with cardiorenal impairment. Levosimendan was shown to improve renal function, an effect which may attributable to its venodilatory effects in combination with the inotropic characteristics of the drug [44,45]. It therefore may be an effective strategy in patients who are congested, but have adequate blood pressures, as levosimendan has been shown to cause some degree of hypotension. Statin therapy seems associated with improved survival in observational studies, irrespective of renal function, but the CORONA and GISSI-HF trial failed to provide evidence

206
of survival benefit of rosuvastatin over placebo, showing no interaction between eGFR and treatment in the CORONA [129,130]. As discussed, new AARAs may counteract the TGF mechanism, which is currently being studied in the PROTECT-II studies, which particularly focuses on patients with cardiac failure and moderate to severe renal failure (creatinine clearance 20-80 mL/min) [118]. A new study, the DIURETICS-HF, is underway to establish the effect of diuretics withdrawal on clinical outcome in patients with chronic HF, which may show interesting results with regards to renal function [131]. If diuretic therapy is considered, this should always be combined with a low sodium diet, as it limits the possibility of the kidney to bypass the diuretic action of the drug by initiating vigorous salt and water retention in the distal tubulus [132]. Furthermore, dietary sodium restriction itself, especially when GFR is relatively preserved, may help reduce congestion via similar mechanisms, thereby decreasing the likelihood of renal failure as a consequence of venous congestion. As discussed earlier, we found that despite ACEi or ARB therapy, PRA was a strong predictor of outcome, suggesting that further blockade of the RAS (other than combining ACEi and ARBs) could be beneficial in patients with chronic HF.
Currently, a large number of trials with a direct renin inhibitor, aliskiren, is being conducted, and has so far resulted in the completion of one chronic HF study. The Aliskiren Observation of heart Failure Treatment trial (ALOFT), showed that with the addition of aliskiren, a significant reduction in (NT-pro)BNP and PRA levels could be achieved, without increasing the incidence of renal dysfunction and hyperkalaemia [145]. This has prompted the planning and design of a large double blind, placebo controlled trial with aliskiren in chronic HF, the Aliskiren Trial to Minimize Outcomes in Patients with Heart Failure (ATMOSPHERE), which will evaluate the effect of aliskiren on clinical outcome. Furthermore, a small safety and renal efficacy study on aliskiren in patients with chronic HF in combination with renal dysfunction (eGFR 30-60mL/min/1.73m2), the effect of Additive Renin Inhibition with Aliskiren on renal blood flow and Neurohormonal Activation in patients with Chronic Heart Failure and Renal Dysfunction (ARIANA-CHF-RD) study, is underway to investigate the effect of aliskiren on RBF, when added to standard HF therapy. Finally, while decreased RBF as a result of decreased cardiac output is the main determinant of decreased GFR, improvement of cardiac function may be an indirect way to improve GFR. A method to establish this may be cardiac resynchronization therapy, which was recently shown to improve estimated GFR in a substudy of the Multicenter InSync Randomized Clinical Evaluation (MIRACLE) trial . Although these new therapies shed some new light on therapeutic potentials in patients with HF and renal impairment, solid evidence based medicine in the cardiorenal syndrome is still lacking.
In the present thesis we have investigated and discussed the cornerstones of the cardiorenal syndrome in HF. We have provided evidence on new pathophysiologic links; we have described new potential targets for therapy, and discussed promising pharmacological new therapies. However, the pathophysiology and treatment of the cardiorenal syndrome should be the focus of experimental and mechanistic studies as well as large randomized trials to eventually improve prognosis in this high mortality patient group.

Discussion and future perspectives
207
ReferencesDickstein K, Cohen-Solal A, Filippatos G et al. ESC Guidelines for the diagnosis and treatment of 1. acute and chronic heart failure 2008: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM). Eur Heart J. 2008;29:2388-2442.
Groenveld HF, Januzzi JL, Damman K et al. Anemia and mortality in heart failure patients a systematic 2. review and meta-analysis. J Am Coll Cardiol. 2008;52:818-827.
Lang CC and Mancini DM. Non-cardiac comorbidities in chronic heart failure. Heart. 2007;93:665-3. 671.
Le Jemtel TH, Padeletti M, and Jelic S. Diagnostic and therapeutic challenges in patients with coexistent 4. chronic obstructive pulmonary disease and chronic heart failure. J Am Coll Cardiol. 2007;49:171-180.
MacDonald MR, Petrie MC, Hawkins NM et al. Diabetes, left ventricular systolic dysfunction, and 5. chronic heart failure. Eur Heart J. 2008;29:1224-1240.
Smith GL, Lichtman JH, Bracken MB et al. Renal impairment and outcomes in heart failure: systematic 6. review and meta-analysis. J Am Coll Cardiol. 2006;47:1987-1996.
Sturm HB, Haaijer-Ruskamp FM, Veeger NJ et al. The relevance of comorbidities for heart failure 7. treatment in primary care: A European survey. Eur J Heart Fail. 2006;8:31-37.
Hillege HL, Girbes AR, de Kam PJ et al. Renal function, neurohormonal activation, and survival in 8. patients with chronic heart failure. Circulation. 2000;102:203-210.
de Silva R, Nikitin NP, Witte KK et al. Incidence of renal dysfunction over 6 months in patients with 9. chronic heart failure due to left ventricular systolic dysfunction: contributing factors and relationship to prognosis. Eur Heart J. 2006;27:569-581.
Dries DL, Exner DV, Domanski MJ, Greenberg B, and Stevenson LW. The prognostic implications of 10. renal insufficiency in asymptomatic and symptomatic patients with left ventricular systolic dysfunction. J Am Coll Cardiol. 2000;35:681-689.
Hillege HL, Nitsch D, Pfeffer MA et al. Renal function as a predictor of outcome in a broad spectrum 11. of patients with heart failure. Circulation. 2006;113:671-678.
Ruilope LM, van Veldhuisen DJ, Ritz E, and Luscher TF. Renal function: The Cinderella of cardiovascular 12. risk profile. J Am Coll Cardiol. 2001;38:1782-1787.
Smilde TD, Hillege HL, Voors AA, Dunselman PH, and van Veldhuisen DJ. Prognostic importance of 13. renal function in patients with early heart failure and mild left ventricular dysfunction. Am J Cardiol. 2004;94:240-243.
Smilde TD, Hillege HL, Navis G et al. Impaired renal function in patients with ischemic and 14. nonischemic chronic heart failure: association with neurohormonal activation and survival. Am Heart J. 2004;148:165-172.
Tonelli M, Wiebe N, Culleton B et al. Chronic kidney disease and mortality risk: a systematic review. J 15. Am Soc Nephrol. 2006;17:2034-2047.

208
Bart BA, Walsh MM, Blake D, and Goldsmith SR. Ultrafiltration for cardiorenal syndrome. J Card 16. Fail. 2008;14:531-532.
Bongartz LG, Cramer MJ, Doevendans PA, Joles JA, and Braam B. The severe cardiorenal syndrome: 17. 'Guyton revisited'. Eur Heart J. 2005;26:11-17.
Ronco C, Haapio M, House AA, Anavekar N, and Bellomo R. Cardiorenal syndrome. J Am Coll 18. Cardiol. 2008;52:1527-1539.
Ronco C, House AA, and Haapio M. Cardiorenal and renocardiac syndromes: the need for a 19. comprehensive classification and consensus. Nat Clin Pract Nephrol. 2008;4:310-311.
Shlipak MG and Massie BM. The clinical challenge of cardiorenal syndrome. Circulation. 2004;110:1514-20. 1517.
DiBona GF. Nervous kidney. Interaction between renal sympathetic nerves and the renin-angiotensin 21. system in the control of renal function. Hypertension. 2000;36:1083-1088.
Fischer D, Rossa S, Landmesser U et al. Endothelial dysfunction in patients with chronic heart failure is 22. independently associated with increased incidence of hospitalization, cardiac transplantation, or death. Eur Heart J. 2005;26:65-69.
Kilcoyne MM, Schmidt DH, and Cannon PJ. Intrarenal blood flow in congestive heart failure. 23. Circulation. 1973;47:786-797.
Leithe ME, Margorien RD, Hermiller JB, Unverferth DV, and Leier CV. Relationship Between Central 24. Hemodynamics and Regional Blood-Flow in Normal Subjects and in Patients with Congestive Heart-Failure. Circulation. 1984;69:57-64.
Ljungman S, Laragh JH, and Cody RJ. Role of the Kidney in Congestive Heart-Failure - Relationship 25. of Cardiac Index to Kidney-Function. Drugs. 1990;39:10-21.
Blake WD, Wegria R, Keating RP, and Ward HP. Effect of increased renal venous pressure on renal 26. function. Am J Physiol. 1949;157:1-13.
Maxwell MH, Breed ES, and Schwartz IL. Renal venous pressure in chronic congestive heart failure. J 27. Clin Invest. 1950;29:342-348.
Werko L, Varnauskas E, Ek J et al. Studies on the renal circulation and renal function in mitral valvular 28. disease. II. Effect of apresoline. Circulation. 1954;9:700-705.
Werko L, Varnauskas E, Eliasch H et al. Studies on the renal circulation and renal function in mitral 29. valvular disease. I. Effect of exercise. Circulation. 1954;9:687-699.
Werko L, Ek J, Varnauskas E et al. The relationship between renal blood flow, glomerular filtration rate 30. and sodium excretion, cardiac output and pulmonary and systemic blood pressures in various heart disorders. Am Heart J. 1955;49:823-837.
Cody RJ, Ljungman S, Covit AB et al. Regulation of glomerular filtration rate in chronic congestive 31. heart failure patients. Kidney Int. 1988;34:361-367.
Creager MA, Halperin JL, Bernard DB et al. Acute regional circulatory and renal hemodynamic effects 32. of converting-enzyme inhibition in patients with congestive heart failure. Circulation. 1981;64:483-489.

Discussion and future perspectives
209
Levine TB, Olivari MT, Garberg V, Sharkey SW, and Cohn JN. Hemodynamic and clinical response 33. to enalapril, a long-acting converting-enzyme inhibitor, in patients with congestive heart failure. Circulation. 1984;69:548-553.
Smilde TDJ, Damman K, van der Harst P et al. Differential associations between renal function and 34. "modifiable" risk factors in patients with chronic heart failure. Clin Res Cardiol. 2009;98:121-129.
Friedman B, Clark G, Resnik H, and Harrison TR. Effect of digitalis on the cardiac output of persons 35. with congestive heart failure. Arch Int Med. 1935;56:710-723.
Seymour WB, Pritchard WH, Longley LP, and Hayman JM. Cardiac output, blood and interstitial 36. fluid volumes, total circulating serum protein, and kidney function during cardiac failure and after improvement. J Clin Invest. 1942;21:229-240.
Fiksen-Olsen MJ and Romero JC. Renal effects of prostaglandin inhibition during increases in renal 37. venous pressure. Am J Physiol. 1991;260:525-529.
Fiksen-Olsen MJ, Strick DM, Hawley H, and Romero JC. Renal effects of angiotensin II inhibition 38. during increases in renal venous pressure. Hypertension. 1992;19:137-141.
Kastner PR, Hall JE, and Guyton AC. Renal hemodynamic responses to increased renal venous pressure: 39. role of angiotensin II. Am J Physiol. 1982;243:F260-F264.
Wegria R, Capeci NE, Blumenthal MR et al. The pathogenesis of proteinuria in the acutely congested 40. kidney. J Clin Invest. 1955;34:737-743.
Damman K, Navis G, Smilde TD et al. Decreased cardiac output, venous congestion and the association 41. with renal impairment in patients with cardiac dysfunction. Eur J Heart Fail. 2007;9:872-878.
Damman K, van Deursen VM, Navis G et al. Increased central venous pressure is associated with 42. impaired renal function and mortality in a broad spectrum of patients with cardiovascular disease. J Am Coll Cardiol. 2009;53:582-588.
Nohria A, Hasselblad V, Stebbins A et al. Cardiorenal interactions: insights from the ESCAPE trial. J 43. Am Coll Cardiol. 2008;51:1268-1274.
Damman K and Voors AA. Levosimendan improves renal function in acute decompensated heart 44. failure: cause and clinical application. Editorial to: "Levosimendan improves renal function in patients with acute decompensated heart failure: comparison with dobutamine by Yilmaz et al.". Cardiovasc Drugs Ther. 2007;21:403-404.
Yilmaz MB, Yalta K, Yontar C et al. Levosimendan improves renal function in patients with acute 45. decompensated heart failure: comparison with dobutamine. Cardiovasc Drugs Ther. 2007;21:431-435.
Maeder MT, Holst DP, and Kaye DM. Tricuspid Regurgitation Contributes to Renal Dysfunction in 46. Patients With Heart Failure. J Card Fail. 2008;14:824-830.
Damman K, Voors AA, Hillege HL et al. Venous congestion in chronic systolic heart failure is related 47. to renal dysfunction and increased mortality. Submitted. 2009.
Doty JM, Saggi BH, Sugerman HJ et al. Effect of increased renal venous pressure on renal function. J 48. Trauma. 1999;47:1000-1003.
Doty JM, Saggi BH, Blocher CR et al. Effects of increased renal parenchymal pressure on renal function. 49. J Trauma. 2000;48:874-877.

210
Mullens W, Abrahams Z, Skouri HN et al. Elevated intra-abdominal pressure in acute decompensated 50. heart failure: a potential contributor to worsening renal function? J Am Coll Cardiol. 2008;51:300-306.
Mullens W, Abrahams Z, Francis GS et al. Prompt reduction in intra-abdominal pressure following 51. large-volume mechanical fluid removal improves renal insufficiency in refractory decompensated heart failure. J Card Fail. 2008;14:508-514.
Farbom P, Wahlstrand B, Almgren P et al. Interaction between renal function and microalbuminuria 52. for cardiovascular risk in hypertension: the nordic diltiazem study. Hypertension. 2008;52:115-122.
Gerstein HC, Mann JF, Yi Q et al. Albuminuria and risk of cardiovascular events, death, and heart 53. failure in diabetic and nondiabetic individuals. JAMA. 2001;286:421-426.
Hillege HL, Fidler V, Diercks GF et al. Urinary albumin excretion predicts cardiovascular and 54. noncardiovascular mortality in general population. Circulation. 2002;106:1777-1782.
Kramer H, Jacobs DR, Jr., Bild D et al. Urine albumin excretion and subclinical cardiovascular disease. 55. The Multi-Ethnic Study of Atherosclerosis. Hypertension. 2005;46:38-43.
KDOQI. KDOQI Clinical Practice Guidelines and Clinical Practice Recommendations for Diabetes 56. and Chronic Kidney Disease. Am J Kidney Dis. 2007;49:S12-154.
Bahrami H, Bluemke DA, Kronmal R et al. Novel metabolic risk factors for incident heart failure and 57. their relationship with obesity: the MESA (Multi-Ethnic Study of Atherosclerosis) study. J Am Coll Cardiol. 2008;51:1775-1783.
Ingelsson E, Sundstrom J, Lind L et al. Low-grade albuminuria and the incidence of heart failure in a 58. community-based cohort of elderly men. Eur Heart J. 2007;28:1739-1745.
Hockensmith ML, Estacio RO, Mehler P et al. Albuminuria as a predictor of heart failure hospitalizations 59. in patients with type 2 diabetes. J Card Fail. 2004;10:126-131.
van de Wal RM, Asselbergs FW, Plokker HW et al. High prevalence of microalbuminuria in chronic 60. heart failure patients. J Card Fail. 2005;11:602-606.
Stehouwer CD, Henry RM, Dekker JM et al. Microalbuminuria is associated with impaired brachial 61. artery, flow-mediated vasodilation in elderly individuals without and with diabetes: further evidence for a link between microalbuminuria and endothelial dysfunction--the Hoorn Study. Kidney Int Suppl. 2004;S42-S44.
Stehouwer CD and Smulders YM. Microalbuminuria and risk for cardiovascular disease: Analysis of 62. potential mechanisms. J Am Soc Nephrol. 2006;17:2106-2111.
Higashikuni Y, Ishizaka N, Ishizaka Y et al. Relationship between blood pressure and chronic kidney 63. disease in the Japanese population: the lower the better even in individuals without hypertension? Hypertens Res. 2008;31:213-219.
Damman K, Navis G, Voors AA, van Veldhuisen DJ, and Hillege HL. Worsening renal function and 64. prognosis in heart failure; a meta-analysis of published studies. Eur J Heart Fail. 207;6:17-18.
Khan NA, Ma I, Thompson CR et al. Kidney function and mortality among patients with left ventricular 65. systolic dysfunction. J Am Soc Nephrol. 2006;17:244-253.

Discussion and future perspectives
211
Krumholz HM, Chen YT, Vaccarino V et al. Correlates and impact on outcomes of worsening renal 66. function in patients > or =65 years of age with heart failure. Am J Cardiol. 2000;85:1110-1113.
Damman K, Jaarsma T, Voors AA et al. Both in and outhospital worsening of renal function predict 67. outcome in patients with heart failure. Submitted. 2009.
Metra M, Nodari S, Parrinello G et al. Worsening renal function in patients hospitalised for acute heart 68. failure: clinical implications and prognostic significance. Eur J Heart Fail. 2008;10:188-195.
Logeart D, Tabet JY, Hittinger L et al. Transient worsening of renal function during hospitalization for 69. acute heart failure alters outcome. Int J Cardiol. 2008;127:228-232.
Akhter MW, Aronson D, Bitar F et al. Effect of elevated admission serum creatinine and its worsening 70. on outcome in hospitalized patients with decompensated heart failure. Am J Cardiol. 2004;94:957-960.
Chittineni H, Miyawaki N, Gulipelli S, and Fishbane S. Risk for Acute Renal Failure in Patients 71. Hospitalized for Decompensated Congestive Heart Failure. Am J Nephrol. 2007;27:55-62.
Cowie MR, Komajda M, Murray-Thomas T, Underwood J, and Ticho B. Prevalence and impact of 72. worsening renal function in patients hospitalized with decompensated heart failure: results of the prospective outcomes study in heart failure (POSH). Eur Heart J. 2006;27:1216-1222.
Forman DE, Butler J, Wang Y et al. Incidence, predictors at admission, and impact of worsening renal 73. function among patients hospitalized with heart failure. J Am Coll Cardiol. 2004;43:61-67.
Jose P, Skali H, Anavekar N et al. Increase in Creatinine and Cardiovascular Risk in Patients with 74. Systolic Dysfunction after Myocardial Infarction. J Am Soc Nephrol. 2006;10:2886-2891.
Owan TE, Hodge DO, Herges RM et al. Secular trends in renal dysfunction and outcomes in hospitalized 75. heart failure patients. J Card Fail. 2006;12:257-262.
Weinfeld MS, Chertow GM, and Stevenson LW. Aggravated renal dysfunction during intensive therapy 76. for advanced chronic heart failure. Am Heart J. 1999;138:285-290.
Damman K, Navis G, Voors AA et al. Renal function relates to outcome through different pathways of 77. renal perfusion and filtration efficacy, hemodilution and volume overload in patients with chronic heart failure. Submitted. 2009.
Benedict CR, Francis GS, Shelton B et al. Effect of long-term enalapril therapy on neurohormones in 78. patients with left ventricular dysfunction. SOLVD Investigators. Am J Cardiol. 1995;75:1151-1157.
Gottlieb SS, Dickstein K, Fleck E et al. Hemodynamic and neurohormonal effects of the angiotensin II 79. antagonist losartan in patients with congestive heart failure. Circulation. 1993;88:1602-1609.
Tsutamoto T, Wada A, Maeda K et al. Angiotensin II type 1 receptor antagonist decreases plasma levels 80. of tumor necrosis factor alpha, interleukin-6 and soluble adhesion molecules in patients with chronic heart failure. J Am Coll Cardiol. 2000;35:714-721.
Latini R, Masson S, Anand I et al. The comparative prognostic value of plasma neurohormones at 81. baseline in patients with heart failure enrolled in Val-HeFT. Eur Heart J. 2004;25:292-299.
Petrie MC, Padmanabhan N, McDonald JE et al. Angiotensin converting enzyme (ACE) and non-ACE 82. dependent angiotensin II generation in resistance arteries from patients with heart failure and coronary heart disease. J Am Coll Cardiol. 2001;37:1056-1061.

212
Danser AH, Batenburg WW, and van Esch JH. Prorenin and the (pro)renin receptor--an update. 83. Nephrol Dial Transplant. 2007;22:1288-1292.
Nguyen G, Delarue F, Burckle C et al. Pivotal role of the renin/prorenin receptor in angiotensin II 84. production and cellular responses to renin. J Clin Invest. 2002;109:1417-1427.
Krebs C, Hamming I, Sadaghiani S et al. Antihypertensive therapy upregulates renin and (pro)renin 85. receptor in the clipped kidney of Goldblatt hypertensive rats. Kidney Int. 2007;72:725-730.
Siems W, Quast S, Carluccio F et al. Oxidative stress in cardio renal anemia syndrome: correlations and 86. therapeutic possibilities. Clin Nephrol. 2003;60 Suppl 1:S22-S30.
Silverberg DS, Wexler D, and Iaina A. The importance of anemia and its correction in the management 87. of severe congestive heart failure. Eur J Heart Fail. 2002;4:681-686.
Silverberg DS, Wexler D, Blum M, and Iaina A. The cardio renal anemia syndrome: correcting anemia in 88. patients with resistant congestive heart failure can improve both cardiac and renal function and reduce hospitalizations. Clin Nephrol. 2003;60 Suppl 1:S93-102.
Anand IS. Pathogenesis of anemia in cardiorenal disease. Rev Cardiovasc Med. 2005;6 Suppl 89. 3:S13-S21.
Androne AS, Katz SD, Lund L et al. Hemodilution is common in patients with advanced heart failure. 90. Circulation. 2003;107:226-229.
Westenbrink BD, Visser FW, Voors AA et al. Anaemia in chronic heart failure is not only related to 91. impaired renal perfusion and blunted erythropoietin production, but to fluid retention as well. Eur Heart J. 2007;28:166-171.
Anand IS, Kuskowski MA, Rector TS et al. Anemia and change in hemoglobin over time related to 92. mortality and morbidity in patients with chronic heart failure: results from Val-HeFT. Circulation. 2005;112:1121-1127.
Al Ahmad A, Rand WM, Manjunath G et al. Reduced kidney function and anemia as risk factors for 93. mortality in patients with left ventricular dysfunction. J Am Coll Cardiol. 2001;38:955-962.
de Silva R, Rigby AS, Witte KK et al. Anemia, renal dysfunction, and their interaction in patients with 94. chronic heart failure. Am J Cardiol. 2006;98:391-398.
Ezekowitz JA, McAlister FA, and Armstrong PW. Anemia is common in heart failure and is associated 95. with poor outcomes: insights from a cohort of 12 065 patients with new-onset heart failure. Circulation. 2003;107:223-225.
Ding H, He Y, Li K et al. Urinary neutrophil gelatinase-associated lipocalin (NGAL) is an early 96. biomarker for renal tubulointerstitial injury in IgA nephropathy. Clin Immunol. 2007;123:227-234.
Liangos O, Perianayagam MC, Vaidya VS et al. Urinary N-acetyl-beta-(D)-glucosaminidase activity and 97. kidney injury molecule-1 level are associated with adverse outcomes in acute renal failure. J Am Soc Nephrol. 2007;18:904-912.
Machiguchi T, Yoshida H, Yonemoto S et al. Does circulating erythropoietin reflect progression of IgA 98. nephropathy? Comparison with urinary N-acetyl-beta-D-glucosaminidase. Nephrol Dial Transplant. 1999;14:635-640.

Discussion and future perspectives
213
Mori K, Lee HT, Rapoport D et al. Endocytic delivery of lipocalin-siderophore-iron complex rescues the 99. kidney from ischemia-reperfusion injury. J Clin Invest. 2005;115:610-621.
Bazzi C, Petrini C, Rizza V et al. Urinary N-acetyl-beta-glucosaminidase excretion is a marker of tubular 100. cell dysfunction and a predictor of outcome in primary glomerulonephritis. Nephrol Dial Transplant. 2002;17:1890-1896.
Wellwood JM, Ellis BG, Price RG et al. Urinary N-acetyl- beta-D-glucosaminidase activities in patients 101. with renal disease. Br Med J. 1975;3:408-411.
Goldfarb M, Abassi Z, Rosen S et al. Compensated heart failure predisposes to outer medullary tubular 102. injury: studies in rats. Kidney Int. 2001;60:607-613.
Matsushima H, Yoshida H, Machiguchi T et al. Urinary albumin and TGF 1 levels as renal damage 103. indices in patients with congestive heart failure. Clin Exp Nephrol. 2002;6:21-29.
Bolignano D, Coppolino G, Campo S et al. Neutrophil gelatinase-associated lipocalin in patients with 104. autosomal-dominant polycystic kidney disease. Am J Nephrol. 2007;27:373-378.
Han WK, Bailly V, Abichandani R, Thadhani R, and Bonventre JV. Kidney Injury Molecule-1 (KIM-105. 1): a novel biomarker for human renal proximal tubule injury. Kidney Int. 2002;62:237-244.
Han WK, Waikar SS, Johnson A et al. Urinary biomarkers in the early diagnosis of acute kidney injury. 106. Kidney Int. 2008;73:863-869.
Waanders F, Vaidya VS, Goor HV et al. Effect of Renin-Angiotensin-Aldosterone System Inhibition, 107. Dietary Sodium Restriction, and/or Diuretics on Urinary Kidney Injury Molecule 1 Excretion in Nondiabetic Proteinuric Kidney Disease: A Post Hoc Analysis of a Randomized Controlled Trial. Am J Kidney Dis. 2008;epub ahead of print.
Damman K, van Veldhuisen DJ, Navis G, Voors AA, and Hillege HL. Urinary neutrophil gelatinase 108. associated lipocalin (NGAL), a marker of tubular damage, is increased in patients with chronic heart failure. Eur J Heart Fail. 2008;10:997-1000.
Damman K, van Veldhuisen DJ, Navis G et al. Tubular damage is common and associated with reduced 109. survival in patients with chronic systolic heart failure. Submitted. 2009.
Coca SG, Yalavarthy R, Concato J, and Parikh CR. Biomarkers for the diagnosis and risk stratification 110. of acute kidney injury: a systematic review. Kidney Int. 2008;73:1008-1016.
Mishra J, Dent C, Tarabishi R et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker 111. for acute renal injury after cardiac surgery. Lancet. 2005;365:1231-1238.
Zhou Y, Vaidya VS, Brown RP et al. Comparison of kidney injury molecule-1 and other nephrotoxicity 112. biomarkers in urine and kidney following acute exposure to gentamicin, mercury, and chromium. Toxicol Sci. 2008;101:159-170.
Norman JT and Fine LG. Intrarenal oxygenation in chronic renal failure. Clin Exp Pharmacol Physiol. 113. 2006;33:989-996.
Prasun MA, Kocheril AG, Klass PH, Dunlap SH, and Piano MR. The effects of a sliding scale diuretic 114. titration protocol in patients with heart failure. J Cardiovasc Nurs. 2005;20:62-70.
Costanzo MR, Saltzberg M, O'Sullivan J, and Sobotka P. Early ultrafiltration in patients with 115. decompensated heart failure and diuretic resistance. J Am Coll Cardiol. 2005;46:2047-2051.

214
Ellison DH. Diuretic therapy and resistance in congestive heart failure. Cardiology. 2001;96:132-143.116.
Vallon V, Miracle C, and Thomson S. Adenosine and kidney function: potential implications in patients 117. with heart failure. Eur J Heart Fail. 2008;10:176-187.
Cotter G, Dittrich HC, Weatherley BD et al. The PROTECT pilot study: a randomized, placebo-118. controlled, dose-finding study of the adenosine A1 receptor antagonist rolofylline in patients with acute heart failure and renal impairment. J Card Fail. 2008;14:631-640.
Givertz MM, Massie BM, Fields TK, Pearson LL, and Dittrich HC. The effects of KW-3902, an 119. adenosine A1-receptor antagonist,on diuresis and renal function in patients with acute decompensated heart failure and renal impairment or diuretic resistance. J Am Coll Cardiol. 2007;50:1551-1560.
Dittrich HC, Gupta DK, Hack TC et al. The effect of KW-3902, an adenosine A1 receptor antagonist, 120. on renal function and renal plasma flow in ambulatory patients with heart failure and renal impairment. J Card Fail. 2007;13:609-617.
Hunt SA, Abraham WT, Chin MH et al. ACC/AHA 2005 Guideline Update for the Diagnosis and 121. Management of Chronic Heart Failure in the Adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation. 2005;112:e154-e235.
Cohn JN and Tognoni G. A randomized trial of the angiotensin-receptor blocker valsartan in chronic 122. heart failure. N Engl J Med. 2001;345:1667-1675.
Ezekowitz J, McAlister FA, Humphries KH et al. The association among renal insufficiency, 123. pharmacotherapy, and outcomes in 6,427 patients with heart failure and coronary artery disease. J Am Coll Cardiol. 2004;44:1587-1592.
Hackam DG, Duong-Hua ML, Mamdani M et al. Angiotensin inhibition in renovascular disease: a 124. population-based cohort study. Am Heart J. 2008;156:549-555.
Pitt B, Poole-Wilson PA, Segal R et al. Effect of losartan compared with captopril on mortality in 125. patients with symptomatic heart failure: randomised trial--the Losartan Heart Failure Survival Study ELITE II. Lancet. 2000;355:1582-1587.
Shlipak MG. Pharmacotherapy for heart failure in patients with renal insufficiency. Ann Intern Med. 126. 2003;138:917-924.
Erdmann E, Lechat P, Verkenne P, and Wiemann H. Results from post-hoc analyses of the CIBIS II trial: 127. effect of bisoprolol in high-risk patient groups with chronic heart failure. Eur J Heart Fail. 2001;3:469-479.
Pitt B, Zannad F, Remme WJ et al. The effect of spironolactone on morbidity and mortality in patients 128. with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341:709-717.
Kjekshus J, Apetrei E, Barrios V et al. Rosuvastatin in older patients with systolic heart failure. N Engl 129. J Med. 2007;357:2248-2261.
Gissi-HF. Effect of rosuvastatin in patients with chronic heart failure (the GISSI-HF trial): a randomised, 130. double-blind, placebo-controlled trial. Lancet. 2008.

Discussion and future perspectives
215
Rauchhaus M. Diuretic use in patients with systolic chronic heart failure: a German competence network 131. heart failure experience. Eur J Heart Fail Suppl. 2008;7:105-105.
Cody RJ, Kubo SH, and Pickworth KK. Diuretic treatment for the sodium retention of congestive heart 132. failure. Arch Intern Med. 1994;154:1905-1914.
Hillege HL, van Gilst WH, van Veldhuisen DJ et al. Accelerated decline and prognostic impact of renal 133. function after myocardial infarction and the benefits of ACE inhibition: the CATS randomized trial. Eur Heart J. 2003;24:412-420.
Maxwell AP, Ong HY, and Nicholls DP. Influence of progressive renal dysfunction in chronic heart 134. failure. Eur J Heart Fail. 2002;4:125-130.
Shlipak MG, Smith GL, Rathore SS, Massie BM, and Krumholz HM. Renal function, digoxin therapy, 135. and heart failure outcomes: evidence from the digoxin intervention group trial. J Am Soc Nephrol. 2004;15:2195-2203.
Taylor AL, Ziesche S, Yancy C et al. Combination of isosorbide dinitrate and hydralazine in blacks with 136. heart failure. N Engl J Med. 2004;351:2049-2057.
Cohn JN, Archibald DG, Francis GS et al. Veterans Administration Cooperative Study on Vasodilator 137. Therapy of Heart Failure: influence of prerandomization variables on the reduction of mortality by treatment with hydralazine and isosorbide dinitrate. Circulation. 1987;75:IV49-IV54.
Owan TE, Chen HH, Frantz RP et al. The effects of nesiritide on renal function and diuretic 138. responsiveness in acutely decompensated heart failure patients with renal dysfunction. J Card Fail. 2008;14:267-275.
Aaronson KD and Sackner-Bernstein J. Risk of death associated with nesiritide in patients with acutely 139. decompensated heart failure. JAMA. 2006;296:1465-1466.
Sackner-Bernstein JD, Skopicki HA, and Aaronson KD. Risk of worsening renal function with nesiritide 140. in patients with acutely decompensated heart failure. Circulation. 2005;111:1487-1491.
Rogers HL, Marshall J, Bock J et al. A randomized, controlled trial of the renal effects of ultrafiltration 141. as compared to furosemide in patients with acute decompensated heart failure. J Card Fail. 2008;14:1-5.
Boerrigter G, Costello-Boerrigter LC, Abraham WT et al. Cardiac resynchronization therapy improves 142. renal function in human heart failure with reduced glomerular filtration rate. J Card Fail. 2008;14:539-546.
Butler J, Geisberg C, Howser R et al. Relationship between renal function and left ventricular assist 143. device use. Ann Thorac Surg. 2006;81:1745-1751.
Greenberg B, Thomas I, Banish D et al. Effects of multiple oral doses of an A1 adenosine antagonist, 144. BG9928, in patients with heart failure: results of a placebo-controlled, dose-escalation study. J Am Coll Cardiol. 2007;50:600-606.
McMurray JJ, Pitt B, Latini R et al. Effects of the Oral Direct Renin Inhibitor Aliskiren in Patients 145. With Symptomatic Heart Failure. Circ Heart Fail. 2008;1:17-24.
Zemljic G, Bunc M, Yazdanbakhsh AP, and Vrtovec B. Levosimendan improves renal function in 146. patients with advanced chronic heart failure awaiting cardiac transplantation. J Card Fail. 2007;13:417-421.

216
Konstam MA, Gheorghiade M, Burnett JC, Jr. et al. Effects of oral tolvaptan in patients hospitalized for 147. worsening heart failure: the EVEREST Outcome Trial. JAMA. 2007;297:1319-1331.
Varriale P. Role of dopamine in congestive heart failure: a contemporary appraisal. Congest Heart Fail. 148. 1999;5:120-124.
Elkayam U, Ng TM, Hatamizadeh P, Janmohamed M, and Mehra A. Renal Vasodilatory Action 149. of Dopamine in Patients With Heart Failure: Magnitude of Effect and Site of Action. Circulation. 2008;117:200-205.

Discussion and future perspectives
217

218

Nederlandse Samenvatting

220

Popular summary in Dutch
221
Hartfalen (HF) is niet alleen een syndroom dat wordt gekarakteriseerd door symptomen en een slechte prognose, maar ook door het veelvuldige voorkomen van co-morbiditeiten. Een deel van de mortaliteit en morbiditeit in HF wordt dan ook toegeschreven aan deze nevendiagnoses. Van deze co-morbiditeiten is nierfalen mogelijk de belangrijkste, en de combinatie van hart en nierfalen samen wordt dan ook vaak het ‘cardiorenaal syndroom’ genoemd, ook als is de definitie van dit syndroom nog niet onomstotelijk vastgelegd. Er zijn een aantal belangrijke aspecten gesuggereerd als de hoeksteen van het syndroom, namelijk: a) de observatie dat het gezamenlijke voorkomen van hart en nierfalen gerelateerd lijkt te zijn aan een ernstig verslechterde prognose, b) de observatie dat beide entiteiten elkaar kunnen verergeren en c) het voorkomen van pathofysiologische processen die zowel aan hart als nierfalen gerelateerd lijken te zijn. In het huidige proefschrift hebben we de specifieke bijdrage van verschillende pathofysiologische mechanismen van nierfalen in hartfalen onderzocht, de relatie met prognose en nieuwe aspecten in cardiorenale interactie, resulterend in nieuwe inzichten in de pathofysiologie van het cardiorenaal syndroom (Tabel 1).
In DEEL 1 van het proefschrift hebben we de bijdrage van verschillende pathofysiologische processen op het ontstaan van verminderde glomerulaire filtratie snelheid (GFR) in patiënten met HF onderzocht.
Hemodynamiek: Verminderde renale bloeddoorstromingIn het laatste deel van de 20ste eeuw werd door Ljungman et al aangetoond dat met
verminderde cardiale functie, renale bloeddoorstroming (RBF) disproportioneel snel afnam. Echter, GFR bleef tot een zekere hoogte gepreserveerd door toename in de filtratiefractie. Dit suggereerde dat efferente vasoconstrictie van het glomerulaire apparaat de nierfunctie behield. Met steeds lagere RBF kon GFR niet meer behouden worden, en daalde parallel met RBF.
Met de introductie van angiotensine converting enzyme inhibitors (ACEi) en angiotensine II receptor blockers (ARB), wordt deze angiotensine II gemedieerde (met name) efferente vasoconstrictie tegengegaan. Dit heeft als consequenties dat RBF toeneemt ten gevolge van verminderde renale vasculaire weerstand (RVR), en dat mogelijk dat door het tegengaan van het GFR preserverende mechanisme van efferente vasoconstrictie, een verdere achteruitgang in RBF niet kan worden tegengegaan. In hoofdstuk 1 onderzochten we deze laatste mogelijkheid. We toonden aan dat in patiënten met lagere RBF waarden geen preservatie van GFR optrad. In tegendeel, GFR daalde proportioneel met verminderde RBF, waarbij filtratie fractie stabiel bleef totdat zeer lage RBF waarden werden geobserveerd. In deze laatste patiëntengroep zagen we een toename van RVR, ondanks afname van filtratie fractie, suggererend dat er afferente vasoconstrictie optreedt in deze patiënten. RBF was in ieder geval de belangrijkste bepalende factor van GFR, en zou daarom moeten worden gezien als de belangrijkste hoeksteen van het cardiorenaal syndroom.
Hemodynamiek: centraal veneuze drukIn het eerste deel van de 20ste eeuw waren er aanwijzingen dat verhoogde centraal veneuze
druk (CVP) een effect op GFR zou kunnen uitoefenen dat onafhankelijk is van het verminderen

222
Popular summary in Dutch
van de perfusiedruk over het glomerulair apparaat. Hoge CVP kan leiden tot hogere drukken in de ingekapselde nier, leidend tot hypoxie of verhoging van de hydrostatische druk in het kapsel van Bowman, en kan daardoor GFR reduceren. Hogere CVP kan zelfs worden gelinked aan proteïnurie. In hoofdstuk 2 en 3 hebben we meer bewijs geleverd voor een directe link tussen CVP en (geschatte) GFR. In hoofdstuk 2 hebben we aangetoond dat in patiënten met pulmonale hypertensie en cardiale dysfunctie, hogere CVP leidt tot een verminderde GFR als RBF al afgenomen is. Ondanks gelijke RBF was een hoge CVP in deze groep patiënten gerelateerd aan een lage GFR. Dit kan mogelijk impliceren dat het effect van CVP op GFR onafhankelijk is van RBF. In hoofdstuk 3 hebben we deze associatie verder onderzocht in een groot cohort patiënten met cardiovasculaire aandoeningen. In deze populatie vonden we dat patiënten met hogere CVP waarden significant lagere GFR hadden vergeleken met patiënten die normale CVP waarden hadden, onafhankelijk van hartminuutvolume. Aangezien invasieve bepaling van CVP niet mogelijk is in alle patiënten met HF hebben we in hoofdstuk 4 de relatie tussen niet-invasieve symptomen van veneuze congestie met nierfunctie en prognose onderzocht. In deze studie vonden we dat patiënten met meer symptomen van congestie een significant slechtere nierfunctie hadden. Hoewel deze associatie ook een uiting zou kunnen zijn van excessieve zout en water retentie als respons op verminderde renale bloeddoorstroming, was het effect zelfs sterker in patiënten met de relatief beste linker ventrikel ejectie fracties. Als laatste hebben we aangetoond dat, onafhankelijk van hoge inter-onderzoeker variabiliteit, symptomen van veneuze congestie sterke en onafhankelijke voorspellers zijn van mortaliteit.
Tekenen van nierschade: AlbuminurieMeerdere studies hebben laten zien dat de aanwezigheid van micro/macro albuminurie de
ontwikkeling van HF en het risico op HF hospitalisaties voorspelt. Er is echter zeer weinig bekend over de prevalentie, pathofysiologie en relatie met prognose van verhoogde albumine uitscheiding
Tabel 1.Samenvatting van de eigenschappen van het Cardiorenaal Syndroom.
Eigenschappen van het Cardiorenaal Syndroom
Afgenomen RBF en GFR
Toegenomen veneuze congestie
Albuminurie
Tubulaire Schade
Verslechterende nierfunctie
Diuretica Resistentie
Activatie van het TGF
Anemie
Toegenomen mortaliteit
Afkortingen: GFR: glomerulaire filtratie snelheid, RBF: renale bloed doorstroming, TGF: Tubuloglomerulair feedback mechanisme

Popular summary in Dutch
223
in patiënten met HF. Tot een derde van alle patiënten met HF lijken microalbuminurie hebben, maar de pathofysiologie van albuminurie in HF is onduidelijk. In aandoeningen anders dan HF wordt albuminurie gezien als marker van vroege glomerulaire schade, of als marker van endotheeldysfunctie. Hogere bloeddruk, en daardoor hogere hydraulische glomerulaire drukken zijn geassocieerd met albuminurie in hypertensie, maar dit lijkt niet het mechanisme in patiënten met HF aangezien deze patiëntengroep eerder een lage glomerulaire druk ervaart. In hoofdstuk 1 toonden we al aan dat albuminurie beperkt geassocieerd was met verminderde RBF. Dit kan suggereren dat albuminurie in HF geassocieerd is met hypoxische schade. Een ander mechanisme zou een verhoogde CVD kunnen zijn, onafhankelijk van verminderde RBF. Hoewel albuminurie een sterke voorspeller van mortaliteit is in andere patiënten populaties dan HF, moet de relatie tussen albuminurie en prognose in patiënten met HF nog aangetoond worden.
In DEEL 2 van dit proefschrift hebben we de veranderingen in nierfunctie, en specifiek het voorkomen van verslechtering van nierfunctie (worsening renal function, WRF) in patiënten met HF onderzocht. Hoewel het normale verloop van nierfunctie in HF nog steeds moet worden vastgesteld, suggereren diverse studies dat bijna een derde van alle HF patiënten op enig moment in hun ziekte WRF ervaren. WRF wordt in dit verband gedefinieerd als een toename in serum creatinine van ≥ 26.5 µmol/L (≥ 0.3 mg/dL) wel of niet in combinatie met een relatieve toename van serum creatinine van > 25%. In hoofdstuk 5 hebben we aangetoond dat, wanneer WRF voorkomt, het geassocieerd is met een 60% toename van risico voor mortaliteit, en 30% toename van het risico op HF opnames. Daarnaast hadden patiënten met de slechtste nierfunctie het hoogste risico voor het ontstaan van WRF. Interessant genoeg was er slechts één studie die het effect van verbetering van nierfunctie heeft onderzocht, en toonde aan dat dit geassocieerd is met een meer gunstige prognose.
In hoofdstuk 6 onderzochten we de relatie tussen het voorkomen van WRF op verschillende punten in tijd, de relatie met prognose en de factoren die geassocieerd zijn met WRF of afnemende GFR in een substudie van de Coordinating Study Evaluating Outcomes of Advising and Counseling in Heart Failure (COACH). WRF kwam niet alleen vaak voor, maar was ook geassocieerd met een sterke toename in voorkomen van het primaire eindpunt van mortaliteit en HF heropnames. De pathofysiologische mechanismen van het ontstaan van WRF zijn nog niet geheel opgehelderd, maar meerdere studies suggereren dat een verminderde GFR op baseline de belangrijkste voorspeller van het ontstaan van WRF is. Daarnaast worden genoemd: (hoge dosis) diuretica, leeftijd, anemie en diabetes, waarvan de laatste drie ook in onze analyses werden gevonden als belangrijke voorspellers voor het ontstaan van WRF.
In DEEL 3 van dit proefschrift onderzochten we de aanwezigheid van tubulaire schade als nieuwe entiteit in cardiorenale interactie in HF en onderzochten de verschillende mechanistische en pathofysiologische pathways waarlangs een verminderde nierfunctie leidt tot verslechterde prognose.

224
Popular summary in Dutch
Hemodynamiek en renine angiotensine systeem activiteitIn HF is een verminderde GFR sterk gerelateerd aan een verhoogde mortaliteit. Het is
daarom belangrijk te weten welke aspecten van de pathofysiologie van verminderde GFR in HF verantwoordelijk zijn voor de slechte overleving van deze patiënten, om zodoende mogelijke specifieke behandelingsdoelen te identificeren. In hoofdstuk 7 onderzochten we daarom alle factoren geassocieerd met prognose, en hoezeer deze associatie afhankelijk was van de relatie met GFR. In pathway analyse toonden we aan dat een combinatie van verslechterde renale perfusie, filtratie efficiëntie (filtratie fractie), toegenomen extracellulair volume en als consequentie hemodilutie en anemie, verantwoordelijk waren voor het totale effect van GFR op prognose. Deze bevindingen bevestigen wederom het belang van zowel vermindere renale perfusie als veneuze congestie, niet alleen in de pathofysiologie van verminderde GFR in HF, maar ook in de relatie met prognose.
Naast pathways afhankelijk van GFR vonden we dat plasma renine activiteit (PRA) de enige factor was, die onafhankelijk van renale bloeddoorstroming geassocieerd was met prognose. Deze observatie werd gedaan ondanks het feit dat alle patiënten op ACEi of ARB therapie stonden. De onderliggende pathofysiologie van dit effect is moeilijk te doorgronden. Redenen zouden kunnen zijn: ACE-escape, hogere dosis ACEi voor patiënten met de slechtste prognose, verslechterende nierfunctie en een direct (pro) fibrotisch effect van renine zelf via de (pro) renine receptor. Samenvattend lijken onze resultaten te suggereren dat behandeling met ACEi of ARB resulteert in hogere PRA spiegels, die geassocieerd zijn met verminderde overleving. Dit kan betekenen dat therapie die specifiek gericht is op PRA effectief zou kunnen zijn bovenop ACEi of ARB therapie, zoals later zal worden toegelicht.
AnemieNaast nierfalen komt anemie veelvuldig voor in patiënten met HF. Hoewel er nog steeds
discussie is over de precieze pathofysiologie van anemie in HF, bestaat deze ten minste uit erythropoietine (EPO) resistentie, ijzerdeficiëntie, en zoals we hebben aangetoond in hoofdstuk 7, hemodilutie en verminderde RBF. Het samen voorkomen van nierfalen en anemie in hartfalen is geassocieerd met een sterk verhoogd mortaliteitsrisico. Deze is veel hoger is dan mag worden verwacht aan de hand van de individuele bijdrage van zowel nierfalen als anemie. Deze interactie suggereert dat beide entiteiten elkaar kunnen verergeren, resulterend in een extreem hoge mortaliteit. De pathway analyse in hoofdstuk 7 geeft hierin enig inzicht. Zowel anemie als nierfalen zijn gerelateerd aan verminderde renale perfusie en hemodilutie, die beide individueel relateren aan prognose. De combinatie van anemie en nierfalen kan daarom via vergelijkbare pathways leiden tot sterk verminderde overleving, die de sterkte van deze associaties met prognose kunnen vergroten.
Tekenen van nierschade: Tubulaire schadeIn nierziekten wordt niet alleen albuminurie vaak geobserveerd, maar ook tubulaire
schade is prevalent. Echter, in HF zijn er weinig tot geen gegevens over de prevalentie en het pathofysiologisch belang van tubulaire schade. Er zijn diverse specifieke urine markers van

Popular summary in Dutch
225
tubulaire schade, waaronder N-acetyl-beta-D-glucosaminidase (NAG), kidney injury molecule 1 (KIM-1), en neutrophil gelatinase associated lipocalin (NGAL). Deze markers zijn uitvoerig beschreven en onderzocht in zowel acuut als chronisch nierfalen.
In hoofdstuk 8 hebben we aangetoond dat urine NGAL concentraties verhoogd zijn in patiënten met chronisch HF. In deze proof of concept studie waren NGAL concentraties hoger in HF patiënten dan in controles, onafhankelijk van GFR. Dit zou kunnen betekenen dat tubulaire schade kan verergeren, en daardoor hogere concentraties van NGAL over een langere periode kan veroorzaken. In hoofdstuk 9 onderzochten we de prevalentie en de relatie met nierfunctie en prognose van verschillende tubulaire schade eiwitten in patiënten met HF in meer detail. In vergelijking met urine NGAL concentraties vonden we vergelijkbare patronen met urine NAG en KIM-1 concentraties. Zowel NAG als KIM-1 urine concentraties waren verhoogd in patiënten met HF. Alleen NAG was gerelateerd aan renale perfusie, maar deze observatie versterkt de hypothese dat tubulo-interstitiële schade optreedt wanneer (regionale) renale hypoxie zich ontwikkelt.
De reden waarom zowel NGAL en KIM-1 niet correleerden aan verminderde GFR of RBF moet onderzocht worden in nieuwe studies. Een reden kan zijn dat zowel NGAL en KIM-1 meer specifiek zijn voor tubulaire dan voor glomerulaire disfunctie. Daarnaast stijgen NGAL spiegels snel in respons op tubulaire schade, maar dalen ook weer snel als de initiërende trigger verdwenen is. Deze snelle toename van zowel NGAL als KIM-1 urine concentraties worden ook gezien bij patiënten met acuut nierfalen, en de stijging treedt bijna één dag eerder op dan de
Figuur 1. Hypothetisch verloop van nierfalen en geassocieerde pathofysiologie in hartfalen. Afkortingen: GFR: glomerulair filtratie snelheid, RBF: renale bloed doorstroming, WRF: verslechtering van nierfunctie. RBF kan sterk verslechteren over een periode. Als consequentie daalt GFR. WRF kan zich ontwikkelen, en zou geassocieerd kunnen zijn met tubulaire schade en albuminuria, die beiden verslechteren over tijd. De combinatie van verminderde RBF, GFR, tubulaire dysfunctie en albuminurie zal leiden tot sterk toegenomen mortaliteitsrisico.
Arb
itrai
re e
enhe
den
0
10
20
30
40
50
60
70
80
90
100
Arbitrair tijdsinterval
WRF WRF
RBF
GFR
Tubulaire Schade
Mortaliteit
Albuminurie

226
Popular summary in Dutch
stijging in serum creatinine. Deze markers kunnen daarom nuttig zijn in het voorspellen van WRF. Deze potentie van deze markers zou vooral in acuut HF belangrijk zijn, en moet verder onderzocht worden.
Naast de bevinding dat de concentraties van tubulaire schade eiwitten verhoogd zijn in patiënten met HF, vonden we dat zowel urine NAG als KIM-1 spiegels significante voorspellers van prognose waren, onafhankelijk van GFR. Dit kan betekenen dat via een heel ander pathofysiologisch mechanisme dan met verminderde GFR, tubulaire schade kan leiden tot een verslechterde prognose. De potentiële rol van deze markers als aangrijpingspunt voor ‘reno-protectieve’ therapie moet en zal de focus zijn van nieuwe studies.
Als laatste zou ook de specifieke plaats van tubulaire schade in de progressie van nierfalen, cardiorenale ziekte en prognose in HF, onderwerp moeten zijn van nieuwe prospectieve studies. In figuur 1 is geïllustreerd hoe de belangrijkste factoren in het cardiorenaal syndroom over de loop van de tijd veranderen. Progressief verslechterende RBF resulteert in afnemende GFR. Daarnaast komt WRF frequent voor, er is toenemende tubulaire schade en albuminurie, en uiteindelijk een hoge mortaliteit.
ToekomstperspectievenEr bestaat geen informatie over effectieve en veilige behandeling van patiënten met het
cardiorenaal syndroom. In het huidige proefschrift hebben we verschillende pathofysiologische interacties tussen hart en nierfalen en het effect op prognose behandeld. Therapieën voor het cardiorenaal syndroom zouden gefocust moeten worden op deze interacties: verminderde RBF, veneuze congestie, maar ook anemie, hypertensie, tubulaire schade en blijvende RAS-activatie. In de praktijk wordt voornamelijk standaard HF therapie voorgeschreven in patiënten met het cardiorenaal syndroom, zoals RAS-blokkers, diuretica en beta-blokkers. Er is echter geen gerandomiseerde studie die de effectiviteit van standaard HF behandeling, of specifiek daarvoor ontwikkelde nier-beschermende therapie in patiënten met gecombineerd hart en nierfalen heeft onderzocht. In tegendeel, grote klinische HF studies hebben patiënten met ernstig nierfalen geëxcludeerd.
Er zijn echter wel nieuwe therapieën in ontwikkeling die (ook) specifiek voor deze patiëntenpopulatie geschikt zou kunnen zijn. Selectieve adenosine A1 receptor antagonisten (AARAs) blokkeren de afferente glomerulaire vasoconstrictie die optreedt wanneer, als respons op meer zoutuitscheiding secundair aan diuretica, het tubuloglomerulaire feedback mechanisme water en zout excretie probeert te minimaliseren. Proof of concept studies met deze AARAs hebben veelbelovende resultaten laten zien in HF en grote klinische trials zijn onderweg om onder andere het effect op nierfunctie en mortaliteit te onderzoeken. Verdere blokkade van het RAS met directe renine remming lijkt een goede kandidaat voor additieve therapie in patiënten met cardiorenaal falen. Eén recente studie in patiënten met HF liet zien dat directe renine remming verdere daling van neurohormonale activatie gaf, zonder ernstige verslechtering van nierfunctie. Ook dit medicijn wordt op dit moment in klinische trials onderzocht, met zowel mortaliteit als nierfunctie als uitkomstmaat. Als laatste kan verbetering van hartfunctie zelf een indirecte manier zijn om GFR te verbeteren. Een methode

Popular summary in Dutch
227
om dit te bewerkstelligen in geselecteerde patiënten zou cardiale resynchronisatie therapie (CRT) kunnen zijn. Recente studies hebben CRT inderdaad geassocieerd met verbetering van geschatte GFR. Hoewel de observaties met deze nieuwe behandelingen nieuw licht werpen op nieuwe effectieve therapieën in patiënten met HF en nierfalen, bestaat er tot op heden nog geen evidence based behandeling voor patiënten met het cardiorenaal syndroom.
In het huidige proefschrift hebben we de hoekstenen van het cardiorenaal syndroom in HF onderzocht en becommentarieerd. We hebben bewijs geleverd voor nieuwe pathofysiologische interacties, we hebben nieuwe potentiële aangrijpingspunten voor therapie beschreven en hebben een overzicht gegeven van nieuwe veelbelovende farmacologische therapieën. Echter, de pathofysiologie en behandeling van het cardiorenaal syndroom in HF moet de focus zijn van experimentele, mechanistische en grote, gerandomiseerde onderzoeken om uiteindelijk de prognose te verbeteren in deze groep patiënten met een uitzonderlijk hoge mortaliteit.

228

Dankwoord

230

Dankwoord
231
Nu mijn promotietraject dan ten einde is gelopen wil ik hierbij alle mensen die op enige wijze hebben bijgedragen aan het tot stand komen van mijn proefschrift hartelijk danken. In het bijzonder wil ik een aantal mensen persoonlijk bedanken.
Mijn promotores prof. dr. H.L. Hillege, prof. dr. D.J. van Veldhuisen en prof. dr. G. Navis.Beste Hans, er kan er maar één zijn die hier als eerste genoemd moet worden, en dat
ben jij uiteraard. Met je staat van dienst op cardiorenaal onderzoeksgebied was je eerst mijn copromotor, maar toen je tot professor werd benoemd, kon het niet anders dan dat je mijn eerste promotor werd, overigens op voorspraak van Dirk Jan. Hans, ik waardeer je enorm als begeleider: met je liefde voor het onderzoek op cardiorenaal gebied en onze gezamenlijke passie op epidemiologische en statistische vlakken hebben we inmiddels samen een aantal mooie artikelen geschreven. Jouw manier van werken leent zich tot open, maar wel stevige discussies, iets wat ook niet kon uitblijven met onze gezamenlijke eigenwijsheid. Hoewel je het ontzettend druk hebt gehad in mijn promotietijd, had je altijd tijd voor overleg, en als het niet uitkwam overdag, dan spraken we elkaar wel ’s avonds of in het weekend. Beste Hans, ik hoop dat we nog lang samen onderzoek kunnen blijven doen op cardiorenaal gebied en ik vind het een eer dat ik je eerste promovendus mag zijn.
Beste Dirk Jan, zoals aangegeven heb jij op het moment dat Hans professor werd gezegd dat je vanaf dat moment meer vanaf de kantlijn zou begeleiden. Uiteraard is niets minder waar. Jouw gedrevenheid in onderzoek, en in het bijzonder onderzoek naar anemie en nierfunctiestoornissen in hartfalen (“de gouden bergen”), hebben geleid tot een vruchtbaar promotieonderzoek. Daar waar Hans en ikzelf af en toe het cardiologische pad verlieten voor een meer nefrologisch georiënteerd onderzoek, was jij altijd degene die ons weer op het juiste spoor bracht. Ik waardeer je kunst van het weglaten, het to-the-point komen, maar ook je sociale belangstelling als je ons weer eens tegen kwam op één of ander internationaal congres.
Beste Gerjan, als enige vrouw in het cardiorenale mannengezelschap weet jij je mannetje prima te staan. Jouw eindeloze kennis over nefrologische onderzoeken die soms al meer dan een eeuw geleden zijn uitgevoerd, kwamen –ongelooflijk maar waar – meermalen van pas in mijn promotietijd. Als ik terug kijk naar onze (eigenlijk te schaarse) besprekingen denk ik vooral aan de hoeveelheid informatie die ik aanbracht, en de hoeveelheid die ik terug kreeg. Meestal kwam ik met meer theorieën de deur uit dan eigenlijk de bedoeling was. Daarnaast hebben jouw nefrologische interpretaties van de nierfunctie-data van onze patiënten enorm geholpen bij het tot stand komen van dit proefschrift. Beste Gerjan, dank voor je inzet in mijn promotie, en ik hoop dat we – samen met Hans – nog vele projecten op cardio-reno-cardiaal gebied kunnen doen.

232
Dankwoord
Mijn copromotor dr. A.A. Voors.Beste Adriaan, eigenlijk heb je een groot deel van mijn promotietijd niet in Groningen
gezeten, maar op talloze internationale congressen en meetingen. Jouw plaats in de internationale wereld, en dan metname in de cardiorenale trials en studies, heeft geleid tot internatonale samenwerking en exposure van onze cardiorenale onderzoeksresultaten. Je bent een echte levensgenieter, en weet werk te combineren met je liefde voor eten en auto’s, iets dat soms zelf alleen al moeilijk genoeg bleek. Ik ben benieuwd hoe het uiteindelijk afloopt met hoofdstuk 6.
I would like to express great gratitude to the members of the examining committee. Prof. dr. K. Amann, the experience you have in your field of interest is remarkable, and I thank you for judging my thesis. Prof. dr. P.A. de Graeff, dank voor de kritische en vlotte beoordeling van mijn proefschrift. Prof. dr. W.H. van Gilst, beste Wiek, ik wil je graag bedanken voor je rol in de beoordelingscommissie en het daarmee gepaard gaande doorspitten van het ‘pak papier’ zoals je het zelf omschreef. Daarnaast wil ik je bedanken voor de goede discussies tijdens brainstorm sessies en metname de sociaal getinte avonden tijdens internationale congressen.
Dr. M.P. van den Berg, beste Maarten, nu mijn promotietraject is afgerond staat een nieuwe uitdaging voor de deur. Sinds ik me kan herinneren wilde ik niet alleen arts worden, maar ook specifiek Cardioloog. Dank dat je me de mogelijkheid geeft om dit te verwezenlijken. Dr. C. Halma, dank voor de mogelijkheid de interne vooropleiding te volgen in het Medisch Centrum Leeuwarden.
Misschien wel het belangrijkste voor een geslaagd promotietraject is een goede sfeer onder de directe collega’s. De sfeer in de Greenhouse en Greenhouse versie 2.1.1 “The unhappy Triad: Ritmisch Falen” was te karakteriseren als geweldig en geweldig chaotisch, maar ook ontzettend relaxed en vriendelijk. Tussen het werken door was er tijd voor sociale refereeravonden, dagjes golf, uitstapjes naar Formule 1, taart – en heel veel – taart, champagne, liters koffie, grootscheepse (onaangekondigde) verhuizingen, af en toe een goede scheldpartij, al dan niet gedwongen ontslagen, en nog veel meer. Trots ben ik op de naar mij genoemde lunch-filmpjes-bespreking: “De Damman-show”, hoewel deze helaas het laatste half jaar minder frequent gegeven kon worden. Als laatste moet natuurlijk de wekelijkse vrijdagmiddagborrel genoemd worden. Echte vrienden volgen je tot aan de bar, en verder, en zo was het ook in het Feithhuis. Hoewel er bijna teveel collega’s zijn om op te noemen wil ik Sheba hierbij apart noemen. Beste Sheba, no-nonsense en niet te veel gezeur is je op je buik geschreven, en ik heb genoten van onze samenwerking en borreluren. Ik ben blij dat we samen twee jaar naar Leeuwarden gaan. Ook de anderen: Pieter, Jasper, Tom, Pim, Erik, Michiel, Martin, Christiane, Tone, Wim, Jessica, Johan, Lucas, Mathijs, Rik, Patrick, Anne, Willem Peter, Sandra, Marcelle, Hessel, Liza, Jan Pieter, Walter, Jardi, Pieter Jan, Lieuwe, Youlan, Suzan, Marieke, Ali, Marthe, Bart en Lennaert, wil ik ontzettend bedanken voor de gezellige tijd. De collega promovendi op de

Dankwoord
233
cardio-renale as, Mariusz en Silvana: ontzettend veel succes verder! Ook promovendi van de afdeling Nefrologie wil ik hierbij hartelijk danken voor de gezellige momenten bij de centrifuge op de nierfunctiekamer en op de spaarzame Kidney Center besprekingen; Folkert, Femke, Titia, Mieneke, Charlotte, Inge, Jan, Steef, Ferdau, Maartje, Else, Marije, Esther en Nynke. Vincent van Deursen wil bedanken voor het monnikenwerk van hoofdstuk 3: het heeft zich uitbetaald! Beste Tiny, dank voor de gezellige momenten tijdens congressen en je bijdrage aan de substudie van de COACH in hoofdstuk 6. Ook dank aan de andere COACH-onderzoekers en hartfalenverpleegkundigen, en in het bijzonder drs. G.C.M. Linssen voor onze samenwerking op gebied van urine NT-proBNP. Ook dank aan de medewerkers van de functieafdeling van de cardiologie. In het bijzonder dank aan de ‘VO2max-dames’: Gean, Jolanda, Monique, Ina, Lineke, Marian, Marijke en Astrid. De dames en studenten van de PREVEND poli wil ik danken voor de gezelligheid en hun bijdrage aan het verzamelen van controle-urines voor hoofdstuk 8 en 9.
Also I would like to thank prof. dr. J.V. Bonventre and dr. V.S. Vaidya for our collaboration and the contribution they have made to chapter 9. I thank prof. dr. J.G.F. Cleland for his critical commentary to chapter 5, and prof. dr. L.M. Ruilope for coming to Groningen to attend the mini-symposium and thesis defense.
In Greenhouse 1.0 waren de cardiologische onderzoekers nog gescheiden door twee vloeren met de cardioresearch, maar in de huidige opzet zijn we buren geworden. Het hele team van de Cardioresearch, te weten: Anja, Peter, Geert, Karin, Carolien, Trienke, Zazza, en Margriet wil ik bedanken. Greetje wil ik in het bijzonder bedanken voor de ervaringen tijdens het korte reisje naar Londen en het onvergetelijke sneeuw-avontuur in Milaan.
Achter de schermen wordt meestal meer uitgevoerd dan iemand kan vermoeden. Beste Alma en Audrey, hoe druk jullie het ook hebben, jullie blijven altijd vriendelijk en bereid zaken voor ons te regelen. Ontzettend bedankt voor jullie gezellige ondersteuning. Ook de secretaresses van de TCC moet ik hier speciaal noemen, beste Brechtel en Diane, dank voor jullie oeverloze geduld als ik weer op zoek was naar Hans.
Een bijzonder woord van dank in dit proefschrift moet uitgaan naar de verpleegkundigen van de nierfunctiekamer. Beste Marian, Roelie en Dirkina, zonder jullie was een groot deel van dit proefschrift niet mogelijk geweest. Wat veel mensen niet weten is dat jullie per jaar meer dan 1000 patiënten ‘aan de nierpomp hangen’, een enorm karwei. Jullie inzet en gezelligheid op de nierfunctiekamer maakte samenwerking met de Nefrologie nog een stuk leuker. Dank voor jullie hulp bij alle nierfunctieonderzoeken en meedenken als weer eens een patiënt uitviel.

234
Dankwoord
Naast onderzoek en studie moet er ook genoeg tijd zijn voor ontspanning. Een groot deel van mijn studenten- en promotietijd heb ik doorgebracht met leden van het Medisch Heerendispuut “Gilde der Priapisten”. Heren leden, ik dank ieder van jullie voor de mooie tijd die ik voor en tijdens mijn promotietijd met eenieder van jullie heb doorgebracht. Ik vind het mooi om te zien dat we zo zijn gegroeid, en dat onze vereniging bloeiende is. Beste Reinoud, als lichtingsgenoot en later huisgenoot hebben we een groot deel van onze studententijd samen doorgebracht. Ik vond het een mooie tijd. Nu je in Utrecht woont, zien we elkaar eigenlijk te weinig, maar de momenten dat we een biertje kunnen drinken zijn des te mooier, net zoals de vakantie naar Frankrijk die je nog bijna gemist had…
Beste Jurrijn, samen met Bart en andere leden van de ‘Champions League’, heb je samen met mij vele vrijdag- en zaterdagavonden doorgebracht. Ook al ken ik je eigenlijk nog niet eens zo ontzettend lang, je bent een goede vriend geworden. Waarschijnlijk zullen we nog wel wat ritjes zuidwaarts gaan maken!
Mijn paranimfen Daan Westenbrink en Bart van der Heij.Beste Daan, vandaag mag jij jouw deel van de afspraak invullen, nadat ik voor jou hetzelfde
heb mogen doen. In de eerste maanden van mijn promotie probeerde je me als ‘knor’ en ‘jongste’ een aantal onderzoekers-mores bij te leren, maar gelukkig hadden we daar samen na een aantal maanden in Jasper een vervanger voor gevonden. Of de geneugten van het leven jou opzoeken of andersom: het maakt niet uit, je geniet ervan en ik heb er een aantal jaar van mogen meegenieten. Op de tee, aan de bar of op congres (of alledrie tegelijk), ik heb me ontzettend vermaakt. Ook ik kijk uit naar de dagen dat we samen op de 1e rij zitten, als het goed is over twee jaar. Ik ben blij dat je ook míjn paranimf wilde zijn.
Beste Bart, als lichtingsgenoot zag je het in eerste instantie helemaal niet zo met mij zitten, maar 9 jaar later ben ik blij dat het allemaal anders is gelopen. Vanaf het moment dat ik meer naar het centrum van de stad ben verhuisd zijn we steeds meer met elkaar opgetrokken. Hoewel jij je talisman hebt moeten inleveren in deze periode, hebben we vooral vele leuke dingen meegemaakt in de vorm van feesten, partijen, squashpartijtjes en bowlingavondjes. Onze gezamenlijke passie voor gadgets heeft onze bankrekening geen goed gedaan, maar heeft ons wel veel plezier gebracht. Helaas moest je door overmacht dit jaar verhuizen naar het zuiden van het land. Ik hoop echter dat we in de spaarzame weekenden die we samen vrij kunnen zijn een ‘pintke’ kunnen drinken op onze vriendschap.
Mijn peetoom en peettante, Hans en Ria, wil ik bedanken voor de enorme gezelligheid en vrolijkheid die jullie altijd weer meebrengen als we elkaar zien. De familiedag op de Universiteit van Twente in 1998 zal ik nooit vergeten, en jullie waarschijnlijk ook niet! Vandaag nog iets formeler, maar ik hoop dat jullie het kunnen waarderen.

Dankwoord
235
Beste Henk en Ineke, ik dank jullie ontzettend voor al jullie goede zorgen voor ons tweeën. Ik wens jullie goede gezondheid toe. Beste Guido, Yvonne, Hans, Wendy, Marc, Ellen, Martijn, Famke en alle kinderen, ook jullie bedank ik voor de gezelligheid!
Mijn Ouders wil ik bedanken voor hun steun en vertrouwen, in het bijzonder tijdens zowel mijn studie als mijn promotieperiode. Hoewel jullie het hele studentenleven in eerste instantie met enige aarzelingen bekeken, kregen Jeffrey en ik al snel het volste vertrouwen. Door jullie steun kan ik vandaag mijn proefschrift verdedigen, ik dank jullie voor alles. Mijn broer, Jeffrey, wil ik ontzettend bedanken voor de gezelligheid en tegenwoordig ook vak-inhoudelijke gesprekken. Beste Jeffrey, ik ben er trots op dat je mijn broer bent en uiteindelijk ook arts zult worden. Ik vind het knap dat je gelijktijdig twee studies hebt kunnen afronden, en straks ook nog in rap tempo gaat promoveren. Ik hoop dat we de komende jaren elkaar vaker zullen zien en spreken!
Lieve Merel, zonder jou had ik deze promotie nooit kunnen volbrengen. Bij jou vind ik liefde en rust, en ik kan me geen leven meer zonder je voorstellen. Ik heb zin in de komende tijd in ons nieuwe huis!

236

Bibliography

238

Bibliography
239
PublicationsHageman J, Eggen BJ, Rozema T, 1. Damman K, Kampinga HH, Coppes RP. Radiation and transforming growth factor-beta cooperate in transcriptional activation of the profibrotic plasminogen activator inhibitor-1 gene. Clin Cancer Res. 2005;11:5956-64.
Damman K2. , Navis G, Smilde TD, Voors AA, van der Bij W, van Veldhuisen DJ, Hillege HL. Decreased cardiac output, venous congestion and the association with renal impairment in patients with cardiac dysfunction. Eur J Heart Fail. 2007;9:872-8.
Damman K3. , Navis G, Voors AA, Asselbergs FW, Smilde TD, Cleland JG, van Veldhuisen DJ, Hillege HL. Worsening renal function and prognosis in heart failure: systematic review and meta-analysis. J Card Fail. 2007;13:599-608.
Hartog JW, Voors AA, Schalkwijk CG, Scheijen J, Smilde TD, 4. Damman K, Bakker SJ, Smit AJ, van Veldhuisen DJ. Clinical and prognostic value of advanced glycation end-products in chronic heart failure. Eur Heart J. 2007;28:2879-85.
Damman K5. , Voors AA. Levosimendan improves renal function in acute decompensated heart failure: cause and clinical application. Editorial to: "Levosimendan improves renal function in patients with acute decompensated heart failure: comparison with dobutamine by Yilmaz et al.". Cardiovasc Drugs Ther. 2007;21:403-4.
Damman K6. , de Boer RA, van Veldhuisen DJ. Heart failure, aging and beta-blockers: the need for more data on tolerability and efficacy. Clin Res Cardiol. 2008;97:575-7.
Groenveld HF, Januzzi JL, 7. Damman K, van Wijngaarden J, Hillege HL, van Veldhuisen DJ, van der Meer P. Anemia and mortality in heart failure patients a systematic review and meta-analysis. J Am Coll Cardiol. 2008;52:818-27.
Damman K8. , van Veldhuisen DJ, Navis G, Voors AA, Hillege HL. Urinary neutrophil gelatinase associated lipocalin (NGAL), a marker of tubular damage, is increased in patients with chronic heart failure. Eur J Heart Fail. 2008;10:997-1000.
Waanders F, Vaidya VS, Goor HV, Leuvenink H, 9. Damman K, Hamming I, Bonventre JV, Vogt L, Navis G. Effect of Renin-Angiotensin-Aldosterone System Inhibition, Dietary Sodium Restriction, and/or Diuretics on Urinary Kidney Injury Molecule 1 Excretion in Nondiabetic Proteinuric Kidney Disease: A Post Hoc Analysis of a Randomized Controlled Trial. Am J Kidney Dis. 2009;53:16-25.
Vlaar PJ, Svilaas T, 10. Damman K, de Smet BJ, Tijssen JG, Hillege HL, Zijlstra F. Impact of pretreatment with clopidogrel on initial patency and outcome in patients treated with primary percutaneous coronary intervention for ST-segment elevation myocardial infarction: a systematic review. Circulation. 2008;118:1828-36.
Smilde TD, 11. Damman K, van der Harst P, Navis G, Westenbrink BD, Voors AA, Boomsma F, van Veldhuisen DJ, Hillege HL. Differential associations between renal function and "modifiable" risk factors in patients with chronic heart failure. Clin Res Cardiol. 2009;98:121-129.
Damman K12. , van Deursen VM, Navis G, Voors AA, van Veldhuisen DJ, Hillege HL. Increased central venous pressure is associated with impaired renal function and mortality in a broad spectrum of patients with cardiovascular disease. J Am Coll Cardiol. 2009;53:582-588.
Damman K13. , van Veldhuisen DJ, Hillege HL. Cardiac Resynchronization Therapy improves Renal Function. Importance of forward ánd backward failure. J Card Fail. 2009;15:78-79.

240
Linssen GCM, 14. Damman K, Hillege HL, Navis G, van Veldhuisen DJ, Voors AA. Urinary NT-proBNP Excretion in Patients With Chronic Heart Failure. Submitted.
van Deursen, 15. Damman K, Hillege HL, van Beek AP, van Veldhuisen DJ, Voors AA. Prevalence, pathophysiology and prognosis of abnormal liver function in heart failure patients. Submitted.
Damman K16. , Voors AA, Hillege HL, Navis G, Lechat P, van Veldhuisen DJ, Dargie HJ. Venous congestion in chronic systolic heart failure is related to renal dysfunction and increased mortality. Submitted.
Damman K,17. Jaarsma T, Voors AA, Navis G, Hillege HL, van Veldhuisen DJ. Both in and outhospital worsening of renal function predict outcome in patients with heart failure. Submitted.
Damman K18. , Navis G, Voors AA, Westenbrink BD, Smilde TDJ, van Veldhuisen DJ, Hillege HL. Renal function relates to outcome through different pathways of renal perfusion and filtration efficacy, hemodilution and volume overload in patients with chronic heart failure. Submitted.
Damman K19. , van Veldhuisen DJ, Navis G, Vaidya V, Smilde TDJ, Westenbrink BD, Bonventre JV, Voors AA, Hillege HL. Tubular damage is common and associated with reduced survival in patients with chronic systolic heart failure. Submitted.
Copyright © 2022 FDOKUMEN




















