
Transport properties of molecular nanostructures - Physics ...
76
UNIVERSIT ` A DEGLI STUDI DI MODENA E REGGIO EMILIA Facolt` a di Scienze Matematiche, Fisiche e Naturali Tesi per il conseguimento del titolo di Dottore di Ricerca in Fisica Transport properties of molecular nanostructures Candidato: Andrea Ferretti Supervisori: Il Coordinatore: Dott.ssa Rosa Di Felice Prof. Virginio Bortolani Prof.ssa Elisa Molinari XVII CICLO - Dicembre 2004
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of Transport properties of molecular nanostructures - Physics ...

UNIVERSITA DEGLI STUDI DI MODENA E REGGIO EMILIA
Facolta di Scienze Matematiche, Fisiche e Naturali
Tesi per il conseguimento del titolo di Dottore di Ricerca in Fisica
Transport properties of molecularnanostructures
Candidato:
Andrea Ferretti
Supervisori: Il Coordinatore:
Dott.ssa Rosa Di Felice Prof. Virginio Bortolani
Prof.ssa Elisa Molinari
XVII CICLO - Dicembre 2004


Contents
Contents 5
1 Introduction 7
2 Electronic structure from a transport point of view 152.1 Intermolecular coupling in polymer crystals by transfer integrals 15
Electronic properties of polymer crystals: the effect ofinterchain interactions . . . . . . . . . . . . . . . . . . 18Ab initio study of transport parameters in polymercrystals . . . . . . . . . . . . . . . . . . . . . . . . . . 22Charge transport and radiative recombination in poly-thiophene crystals: a first-principles study . . . . . . . 32
2.2 The case of organic-metallic hybrid interfaces . . . . . . . . . 35Electron delocalization at the hybrid aromatic-thiol/Cu(100) interface . . . . . . . . . . . . . . . . . 37Surface-science approach to the study of mercapto-benzoxazole on Cu(100) . . . . . . . . . . . . . . . . . 44
3 Transport in a device geometry with the inclusion of corre-lation 51
First principles theory of correlated transport through nano-junctions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 54First principles theoretical description of transport includingelectron-electron correlation . . . . . . . . . . . . . . . . . . . 59
Conclusions and outlook 73
Acknowledgments 75
Full list of publications 77


Chapter 1
Introduction
The search for faster and faster electronic devices is currently driving thecontinuous shrinking of their dimensions. Today’s Silicon technology is nowapproaching a physical limit where classical descriptions break down andquantum effects become dominant. While this represents a formidable chal-lenge in the field, it also offers the exploitation of unusual properties ofmatter, thereby opening important new scenarios. Among these, currentresearch is focusing on the possible use of nanoscale building blocks andmolecular devices to replace present silicon transistors [1]. Molecular de-vices could realize functionalities that are not available in silicon systems(e.g., efficient light emission for high-speed optoelectronics) while, at thesame time, provide an efficient means to selectively self-assemble devicesand circuit components. This would allow for a high integration densitybased on intrinsic intra-molecular features and inter-molecular recognitionproperties (bottom-up approach), rather than on complicated lithographicsteps (top-down approach).
This thesis work focuses on three main issues – namely intermolecularinteractions, organic-inorganic contacts, and correlation effects in transport– that are relevant for molecular electronics. As illustrated below, theyhave great impact in a variety of systems, ranging from organic films andself-assembled monolayers to single-molecule junctions.
Organic thin films. Some of the first attempts to realize optoelectronicelements by means of organic objects can be found in the synthesis and pro-duction of thin films made by π-conjugated polymers or oligomers [2, 3, 4].Among the most studied materials adopted in organic electronics, polyacetylene (PA), poly para-phenylene (PPP), poly para-phenylene-vinylene(PPV), tetracene, pentacene and oligo-thiophenes which can be consideredprototypical examples [5,6,4]. Many working devices such as light emittingdiodes [7, 8], transistors [9, 10] or solar cells [11] have been demonstratedand some of them have already been commercialized. Here the main role

8 Introduction
is played by the interplay between the proper mechanical properties ofplastics, their easy production and doping and the electronic structurearising from the π-conjugation. Moreover large room exists in orderto ad hoc tailor film properties both by means of chemical engineeringon the molecules themselves or by controlled modifications of physicalcharacteristics such as morphology or packing [5, 6, 4]. The latter issueis particularly interesting since, by just modifying deposition conditionsand techniques, it is possible to obtain specific film properties. Currentresearch is addressing both these chemical and physical functionalizations,highlighting that a deeper knowledge of the microscopic mechanisms isneeded. Important open questions addressed in this thesis focus on therelation between intermolecular and intramolecular properties which isfundamental in understanding and controlling the physical and chemicalfunctionalizations of these materials as well as the basic mechanismsunderlying their physics.
Self-assembled monolayers. One of the most promising ideas ofnano-electronics is the specific possibility of reverting the usual top-downapproach starting from suitable nanosized building blocks able to self-organize and statistically reproduce some desired functionality. This is oneof the fundamental issues of the so-called bottom-up nanotechnology, whichshould be able to take advantage of the specific properties of nano-objectsin order to face inadequacies of usual approaches. Recognition capabilityof molecular systems are among the basic keys in order to reach sucha goal. Self-assembled monolayers (SAM’s) have been the subject ofintense research in the last two decades [12, 13, 14, 15] in order to explorethe possibility of exploiting such a reverted approach. Here the usefulproperties of the molecules are connected to their ability to adsorb onmetal or semiconducting surfaces with specific and reproducible geometriesallowing for a long range order of the deposited film. Among the hugevariety of SAM’s, particular attention has been devoted to layers of thiols(-SH) deposited on metallic (especially Gold) surfaces [12]. The action ofthe thiol head-group as a chemical hook able to bind molecules on metallicsurfaces has been studied for different metals and different molecular tails,both aromatic and aliphatic. In spite of such ubiquitous role of thiols,detailed results exists only for few specific molecules and surfaces, e.g.alkanethiols on Au(111) [16]. In the general case, the chemical reactionsinvolved in the adsorption process, the favorite S adsorption site andthe adsorbed molecular geometry are partly or completely unknown. Onthe other hand, the above parameters are of fundamental importance inorder to understand the electronic structure of the SAM and the complexinteractions between molecules and surface. A detailed description of thesystem at the atomic level, as developed in this thesis, is thus of importancein order to account for charge transfer between SAM and surface and finally

9
to describe the lineup of the molecular and surface levels in the adsorbedconfiguration.
Single molecule electronics. Technologically more complex examples ofnano-electronic devices can be achieved considering single objects such asmolecules connected to external electrodes. These devices are also calledsingle-molecule devices and represent one of the ultimate frontiers of cur-rent research. The first proposal of a discrete molecular device dates backto 1974, when Aviram and Ratner envisioned the design of a rectifier basedon a single donor-acceptor molecule short-circuiting a metal-insulator-metaljunction [17]. However, it is only very recently that these seminal ideashave been implemented in a tangible device. Interesting advancements infabrication methods and probes are now leading towards the construction ofprototype devices where individual molecules are connected in a controlledway [18, 19]. Moreover, nano-mechanical controls of samples allow the pro-duction and the study of ultimate nano-contacts made by single atoms oratomic wires [20].
One of the problems in this context is still the reproducibility ofexperiments. Usually in fact not much is known about the nano-junctiongeometries of real samples and large fluctuations are experienced. Sucha degree of uncertainty is a problem both for device production andfor the advance of the knowledge in the field. Moreover, due to thereduced scale and increased confinement, the interplay between differenteffects such as contact resistances, dissipation and electron-ion coupling,electron-electron interactions or transport boundary conditions appearsfar from being understood. The viability of reliable simulations able todescribe these features is therefore of great interest. In the present thesiswe explicitly treat the problem of including electron-electron correlation inthe calculation of transport properties.
Theoretical simulations. In the theoretical description of electronic struc-ture and transport at the nanoscale, the need for a detailed atomistic de-scription of the systems is quite evident. Semiclassical methods for transport(solving the classical Boltzmann equation only using effective mass param-eters and transition rates from quantum descriptions) were found to wellreproduce mesoscale experiments up to the early nineties [21]. These ap-proaches were essentially classical and continuous due to the confinementof quantum effects in the input material properties. With the advent ofconfined systems such as quantum wells, wires and dots, quantum effectscould no longer be neglected and were introduced in the transport formal-ism (for example by means of the quantum Boltzmann equation [21,22]) butno atomistic detail was added to the treatment and effective mass approachremained the state-of-the-art. Reaching the nanosize limit, the atomisticdescription becomes essential in the electronic structure or transport cal-

10 Introduction
culation themselves. The interplay of intermolecular and intramolecularproperties appears to be fundamental in modifying polymer film properties,the very detail of organo-metallic interfaces determines itself the nature ofSAM’s and the possibility to use them in some specific application, thelineup of molecular and electrode levels, their broadening and the chargetransfer at the interfaces are all fundamental parameters for a reliable de-scription of the system. Moreover the energy scales typically associatedwith electron-electron or electron-ion couplings become comparable withother energy scales of the system largely increasing the relative importanceof these effects [5, 23,24].
Many methods from solid state physics and quantum chemistry treatthe full atomistic quantum problem. The latter approaches are usually ableto treat finite systems like molecules while the former ones typically assumeperiodic boundary conditions. Nanosystems are especially complex in thatthey usually retain an extended nature, while translation symmetry islowered (such as in polymer or oligomer crystals) or even absent (molecularconductors). For this reason, appropriate formulations and implementa-tions are needed in order to apply the above described methods. Theseapproximations rely in the description of extended systems by means oflarge clusters in quantum chemical approaches, or in the use of supercelltechniques for non periodic (or low symmetry) systems within solid stateimplementations.
The present work mainly deals with the computational analysis of trans-port properties of nanoscaled systems. It is centered on some general ques-tions along three main guidelines: (i) We numerically studied the effects ofdimensionality and packing on the hopping terms (transfer integrals) in thecase of polymer crystals, stressing the role of interchain versus intrachaintransport. (ii) We address the issue of molecule-metal interfaces studying indetail the electronic structure of a thioled heteroaromatic molecule adsorbedon a metal surface. Here great importance has been given to the analysisof interface effects on the electronic structure. Finally, (iii) we focus on theproblem of introducing electron-electron correlation effects in the calculationof ballistic transport through nano-junctions. Here we revised and adaptedthe formulation of the problem and applied the formalism on a model case.
All the applications presented in this work move from ab initio sim-ulations of total energy and electronic structure within the formalism ofDensity Functional Theory [25, 26, 27, 28] (DFT). Calculations of single-particle quantities have been performed by the PWscf [29] package whichadopt pseudopotentials techniques and plane waves as basis set. Part of thework has been also focused on further code develpments: In particular, thecalculation of intermolecular transfer integrals was performed by a new codefully interfaced with the PWscf package. This code will be soon freelyavailable online within the GNU Generic Public Licence (GPL) [30]. In

11
the context of topic (iii) we performed the transport calculations adoptingmaximally localized Wannier functions [31,32] (MLWF’s) as real space basisset and treating the many body corrections within the three body scattering(3BS) method [33] already adopted in the calculation of photoemissionspectra in the case of strong short range correlation [34]. I contributed tothe online release (within GPL licencing as well) of the WanT [35] packagefor the calculation of mean field transport properties within MLWFs. Ontop of this code, I also implemented the treatment of electron electroncorrelation.
The thesis is organized as a collection of some of the papers producedduring the PhD activity. Chapter 2 deals with the results more directly con-nected to electronic structure studies such as (i) the calculation of transferintegrals for polymers (Sec. 2.1) and (ii) the analysis of MBO adsorption onCu(100) surface (Sec. 2.2). In chapter 3 we discuss about (iii) the inclusionof interactions and their effects in the treatment of nano-junction transportproperties. Bibliography is reported at the end of each chapter, and the fulllist of the published and submitted papers is given in the last section of thethesis.
Bibliography
[1] C. Joachim, J. K. Gimzewski, and A. Aviram, Electronic using hybrid-molecular and mon-molecular devices, Nature 408, 541–548 (Nov.2000).
[2] J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks,K. Mackay, R. H. Friend, P. L. Burns, and A. B. Holmes, Light-emitingdiodes based on conjugated polymers, Nature 347, 539–541 (1990).
[3] J. C. Scott, Conducting Polymers: From Novel Science to New Tech-nology, Science 278(5346), 2071–2072 (Dec. 1997).
[4] R. Friend, R. Gymer, A. Holmes, J. Burroughes, R. Marks, C. Tal-iani, D. Bradley, D. D. Santos, J. Bredas, M. Logdlung, and W. Sala-neck, Electroluminescence in conjugated polymers, Nature 397, 121–128 (Jan. 1999).
[5] N. Greenham and R. Friend, Semiconductor Device Physics of Conju-gated Polymers, in Solid State Physics, edited by H. Ehrenreich andF. Spaepen, pages 2–150, Academic San Diego USA, 1995.
[6] C. Taliani, F. Biscarini, and M. Muccini, Intermolecular interactionsand energy transfer in solid α-sexithienyl, in Conjugated Oligomers,Polymers, and Dendrimers: from polyacetylene to DNA, edited by J.-L.

12 Introduction
Bredas, volume 4 of Bibliotheque Scientifique Francqui, pages 163–203,De Boeck Universite, 1998.
[7] R. F. Service, Plastics May Add New Colors To Lasers’ Light Show,Science 273(5283), 1800–1801 (Sept. 1996).
[8] H. Sirringhaus, N. Tessler, and R. H. Friend, Integrated OptoelectronicDevices Based on Conjugated Polymers, Science 280(5370), 1741–1744(June 1998).
[9] R. F. Service, Transistors and Diodes Link and Light Up, Science280(5370), 1691 (June 1998).
[10] Z. Bao, Materials and Fabrication Needs for Low-Cost Organic Tran-sistor Circuits, Adv. Mater. 12(3), 227–230 (Feb. 2000).
[11] J. Nelson, Solar Cells by Self-Assembly, Science 293(5532), 1059–1060(Aug. 2001).
[12] A. Ulman, Formation and structure of self-assembled monolayers,Chem. Rev. 96(4), 1533–1554 (June 1996).
[13] F. Schreiber, Structure and growth of self-assembling monolayers,Prog. Surf. Sci. 65, 151–256 (2000).
[14] S. F. Bent, Organic functionalization of group IV semiconductor sur-faces: principles, examples, applications, and prospects, Surf. Sci. 500,879–903 (2002).
[15] J. M. Buriak, Organometallic Chemistry on Silicon and GermaniumSurfaces, Chem. Rev. 102(5), 1271–1308 (May 2002).
[16] M. C. Vargas, P. Giannozzi, A. Selloni, and G. Scoles, Coverage-Dependent Adsorption of CH3S and (CH3S)2 on Au(111): a DensityFunctional Theory Study, J. Phys. Chem. B 105, 9509–9513 (2001).
[17] A. Aviram and M. A. Ratner, Molecular rectifier,Chem. Phys. Lett. 29(2), 277–283 (Nov. 1974).
[18] A. Aviram and M. Ratner, editors, Molecular electronics: science andtechnology, volume 852 of Annals of the New York Academy of Sciences,New York Academy of Sciences, 1998.
[19] A. Aviram, M. Ratner, and V. Mujica, editors, Molecular electronicsII, volume 960 of Annals of the New York Academy of Sciences, NewYork Academy of Sciences, 2002.
[20] N. Agraıt, A. L. Yeyati, and J. M. van Ruitenbeek, Quantum propertiesof atomic-sized conductors, Phys. Rep. 377(2–3), 81–279 (2003).

13
[21] D. Ferry and S. M. Goodnik, Transport in nanosctructures, CambridgeUniversity press, 1997.
[22] H. Haug and A.-P. Jauho, Quantum kinetics in transport and optics ofsemiconductors, Springer Berlin, 1996.
[23] J. Park, A. N. Pasupathy, J. I. Goldsmith, C. Chang, Y. Yaish, J. R.P. M. Rinkoski, J. P. Sethna, H. D. Abrunna, P. L. McEuen, and D. C.Ralph, Coulomb blockade andthe Kondo effect in single-atom transis-tors, Nature 417, 722–725 (June 2002).
[24] W. Liang, M. P. Shores, M. Bockrath, J. R. Long, and H. Park, Kondoresonance in a single-molecule transistor, Nature 417, 725–729 (June2002).
[25] R. O. Jones and O. Gunnarsson, The density functional formalism,its applications and prospects, Rev. Mod. Phys. 61(3), 689–746 (July1989).
[26] R. M. Dreizler and E. K. U. Gross, Density Functional Theory: AnApproach to the Quantum Many-Body Problem, Springer, 1990.
[27] M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias, and J. D.Joannopoulos, Iterative minimization techniques for ab initio total-energy calculations: molecular dynamics and conjucate gradients,Rev. Mod. Phys. 64(4), 1045–1097 (Oct. 1992).
[28] W. Kohn, Nobel Lecture: Electronic structure of matterwave functionsand density functionals, Rev. Mod. Phys. 71(5), 1253–1266 (Oct.1999).
[29] S. Baroni, A. Dal Corso, S. de Gironcoli, and P. Giannozzi, 2001,http://www.pwscf.org.
[30] GNU General Public Licence (GPL), seehttp://www.gnu.org/licenses/gpl.txt.
[31] N. Marzari and D. Vanderbilt, Maximally localized generalized Wannierfunctions for composite energy bands, Phys. Rev. B 56(20), 12847–12865 (Nov. 1997).
[32] A. Calzolari, N. Marzari, I. Souza, and M. Buongiorno Nardelli, Abinitio transport properties of nanostructures from maximally-localizedWannier functions, Phys. Rev. B 69(3), 035108 (2004).
[33] C. Calandra and F. Manghi, Three-body scattering theory of correlatedhole and electron states, Phys. Rev. B 50(4), 2061–2073 (July 1994).

14 Introduction
[34] F. Manghi, V. Bellini, and C. Arcangeli, On-site correlation in valenceand core states of ferromagnetic nickel, Phys. Rev. B 56(12), 7149–7161(Sept. 1997).
[35] A. Calzolari, C. Cavazzoni, N. Marzari, and M. Buongiorno Nardelli,2004, http://www.wannier-transport.org.

Chapter 2
Electronic structure from atransport point of view
In this chapter I present papers that are mainly focused on the analysis ofelectronic properties of interest for the study of transport. In a first applica-tion (Sec. 2.1) we extract the transfer integral (TI) parameters, which enterthe calculation of transport properties both in the band-like and hoppingregimes, from the electronic structure of different polymer crystals. Sec-ondly, we apply an electronic structure analysis to the study of a molecule-metal interface (Sec. 2.2). Here the powerful combination of clear-cut sur-face science and ab initio techniques allows to directly focus on the detailedstructure of interface hybrid states which are of fundamental importance fortransport.
2.1 Intermolecular coupling in polymer crystalsby transfer integrals
Polymer and oligomer systems have demonstrated electronic properties rang-ing from highly insulating to semiconducting and metallic behaviors. Inview of possible technological advantages [1] with respect to silicon-basedelectronics, large effort has been devoted to implement basic devices (tran-sistors, LED’s, solar cells) using organic layers as active materials [1,2], andsome of the resulting devices are indeed already in production. Due to theworse intrinsic transport properties of organic thin films wrt silicon, thefirst and more diffused applications concern optoelectronics: here the goodoptical properties, their large tunability and the ease of fabrication makeapplications interesting. The possibility to apply these materials in lightemission devices (LED’s) or in optical charge injection devices (e.g. solarcells) depends dramatically on the transport and charge localization proper-ties of the films. While first studies mainly referred these properties to thoseof the isolated molecules, it was later understood that important effects arise

16 Electronic structure from a transport point of view
from crystal packing [3], which further controls the intrinsic properties ofthe films.
In this scenario we applied solid state techniques, which are able to ac-count for interchain interactions, to analyze packing effects among differentpolymers and crystal structures. Quantum chemical approaches are insteadmainly focused on intramolecular properties. In order to compare trans-port properties of these systems, we chose a tight-binding point of view andadopted the interchain transfer integrals (TI’s) as key parameters describ-ing electron transfer between chains. TI’s in fact enter both the descriptionof band-like transport, essentially giving the band width, as well as that ofhopping mechanisms through the Marcus formalism [4] for electron transfer.These two regimes were found to well describe transport in organic materialsat low and high temperatures respectively, thus making TI’s significant ina wide range of physical conditions. Our simulations are performed withinthe Density Functional Theory (DFT) framework. Once the electronic struc-ture has been computed we calculate TI’s parameters both by means of atight-binding interpolation of the obtained band structure and by a directcalculation.
In the first work of this section (Ref. [5]) we address the computation ofTI’s for two different structures of poly para-phenylene-vinylene (PPV), thenative herring-bone (HB) packing [6] and the π-stack structure of some ofits derivative (e.g. MEH-PPV [7]). In spite of the reduced dimensionalityof the latter crystal, suggesting a higher degree of localization, our resultsshow a much larger interchain coupling in the stacked structure than in thenative HB one. The origin of this phenomena is discussed in the paper.In the second work of the series (Ref. [8]) we present in detail the formal-ism used in the tight-binding interpolation of interchain TI’s and a directway to compute them based on the knowledge of the Kohn and Sham [9]Hamiltonian for the crystal. We compare the results of the two methodsapplying them to different structures of PPV and poly-thiophene (PT). Theinterpolation method is in very good accordance with the numerical directcalculations, allowing us to check the approximations done in the first case.The last example presented (Ref. [10]) compares the above-mentioned resultsfor PT crystals with ab initio results on optical properties. Here my maincontribution is related to the TI’s calculation. Further details on this topicwere presented at international conferences, with proceedings published inRefs. [11,12].
Bibliography
[1] N. Greenham and R. Friend, Semiconductor Device Physics of Conju-gated Polymers, in Solid State Physics, edited by H. Ehrenreich andF. Spaepen, pages 2–150, Academic San Diego USA, 1995.

2.1 Intermolecular coupling in polymer crystals by transfer integrals 17
[2] R. Friend, R. Gymer, A. Holmes, J. Burroughes, R. Marks, C. Tal-iani, D. Bradley, D. D. Santos, J. Bredas, M. Logdlung, and W. Sala-neck, Electroluminescence in conjugated polymers, Nature 397, 121–128 (Jan. 1999).
[3] T. Q. Nguyen, R. C. Kwong, M. E. Thompson, and B. J. Schwartz,Improving the performance of conjugated polymer-based devicesby control of interchain interactions and polymer film morphology,Appl. Phys. Lett. 76, 2454–2456 (2000).
[4] R. A. Marcus and N. Sutin, Electron transfers in chemistry and biology,Biochim. Biophys. Acta 811, 265–322 (1985).
[5] A. Ferretti, A. Ruini, E. Molinari, and M. J. Caldas, ElectronicProperties of Polymer Crystals: The Effect of Interchain Interactions,Phys. Rev. Lett. 90(8), 086401 (Feb. 2003).
[6] D. Chen, M. J. Winokur, M. A. Masse, and F. E. Karasz, Structuralphases of sodium-doped polyparaphenylene vinylene, Phys. Rev. B41(10), 6759–6767 (Apr. 1990).
[7] C. Y. Yang, F. Hide, M. A. Dıaz-Garcıa, A. J. Heeger, and Y. Cao,Microstructure of thin films of photoluminescent semiconducting poly-mers, Polymer 39(11), 2299–2304 (1998).
[8] A. Ferretti, A. Ruini, G. Bussi, E. Molinari, and M. J. Caldas, Abinitio study of transport parameters in polymer crystals, Phys. Rev. B69(20), 205205 (May 2004).
[9] W. Kohn and L. J. Sham, Self-Consistent Equations Including Ex-change and Correlation Effects, Phys. Rev. 140(4A), A1133–A1138(Nov. 1965).
[10] A. Ruini, G. Bussi, A. Ferretti, M. J. Caldas, and E. Molinari, Chargetransport and radiative recombination in polythiophene crystals: afirst-principles study, Synth. Met. 139(3), 755–757 (Oct. 2003).
[11] G. Bussi, A. Ferretti, A. Ruini, M. J. Caldas, and E. Molinari, Opticsand Transport in Conjugated Polymer Crystals: Interchain InteractionEffects, Adv. Solid State Phys. 43, 313– 326 (Sept. 2003).
[12] A. Ruini, A. Ferretti, G. Bussi, E. Molinari, and M. J. Caldas, Rela-tionship between structural and optoelectronic properties in semicon-ducting polymers, Semicond. Sci. Technol. 19(4), S362–S364 (Mar.2004).

Electronic Properties of Polymer Crystals: The Effect of Interchain Interactions
Andrea Ferretti, Alice Ruini, and Elisa MolinariINFM National Center on nanoStructures and bioSystems at Surfaces (S3) and Dipartimento di Fisica,
Universita di Modena e Reggio Emilia, Via Campi 213/A, 41100 Modena, Italy
Marilia J. CaldasInstituto de Fısica, Universidade de Sao Paulo, Cidade Universitaria, 05508-900 Sao Paulo, Brazil and INFM-S3, Italy
(Received 24 July 2002; published 25 February 2003)
We present a theoretical study of the transport parameters in a prototype conjugated-polymer, poly-para-phenylenevinylene, in two different possible crystalline packings. Our analysis is performedthrough density-functional electronic structure calculations, and allows one to obtain the fundamentalparameters describing charge transport. The transfer integrals are found to be a crucial quantity toappreciate the effects of crystalline aggregation on conduction properties: our results indicate thatinterchain interactions can be viewed as a tunable parameter for the design of efficient electronicdevices based on organic materials.
DOI: 10.1103/PhysRevLett.90.086401 PACS numbers: 71.20.Rv, 71.70.–d
The investigation of charge transport in complex or-ganic materials is a huge challenge for both theory andexperiment. This is the case, for example, for the activeconjugated-polymer films in electronic and optoelec-tronic devices [1], and for electron transfer between donorand acceptor sites in proteins [2,3].
Control of carrier mobility is indeed a basic need fororganic devices design, and one would expect clear ex-perimental data and interpretation to be available for themost studied materials, such as poly-para-phenylenevi-nylene (PPV). In the case of long conjugated polymers,with delocalized -electron structure, the main transportpath is known to be intramolecular: this is reflected in thehigh electrical anisotropy seen for stretched material [4].This is not the case for films made from oligomers, wheretransport orthogonal to the long molecular axis coulddominate; it has been shown that for PPV the film mor-phology strongly influences the transport properties, in-dicating that intramolecular interactions are not sufficientto explain experiments [5]. As most polymer films grownby coating or casting from solution are disordered [6],realistic models of transport properties in such configu-rations would be essential. In polymeric systems thisrequires inclusion of two essential components usuallydescribed as ‘‘intrachain’’ and ‘‘interchain’’ charge trans-port. A similar situation exists in the case of proteins [3],where the existence of intrinsically different paths forelectron transfer has been described as ‘‘through bonds’’or ‘‘through space.’’
The main quantity of interest is the transfer integral(TI) Eij between ‘‘sites’’ i; j, to be inserted, e.g., in aButtiker-Landauer [7–9] expression for conductivity[10,11] or in the Marcus [2] formulation for electrontransfer. In order to predict the impact of a specific chainpacking we must obtain reliable values for the relevant TI.
This task can best be performed for simple modelsystems that can be tested against experiment while
retaining the main properties of the samples of interest,namely, the three-dimensional (3D) character of one-dimensional (1D) weakly interacting systems. Veryrecently, extremely ordered crystalline films of someoligomeric materials have been achieved [12], openingthe way to the comparison with accurate theoretical stud-ies. These can now be performed through ab initiodensity-functional theory (DFT) methods, using thesolid-state physics implementations that provide reliablemicroscopic information on structural and electronicproperties of extended systems [13].
In this Letter we investigate and compare interchain TIfor two typical morphologies shown by PPV: herringboneand displaced stacks (HB andS; see Fig. 1). The first isattained in crystalline pure PPV [16], while the stack isassumed by several derivatives of the polymer, for ex-ample, by poly[2-methoxy, 5-(2’-ethyl-hexyloxy) phenyl-ene vinylene] (MEH-PPV) [17]. Our results are based onfirst-principles calculations for the band structure ofmodel crystalline systems, from which we directly ex-tract effective masses and TI for transport orthogonal tothe chain direction. We find that although the transverseeffective masses are similar for both crystal structures,the TI are much more sensitive to the different interchainenvironments. The TI is 1 order of magnitude larger for stacks in the stack direction than for herringbone pack-ing; this indicates that 3D arrangement can be indeed acrucial element for the design of materials with efficienttransport properties.
We first obtain the full band structure of the modelcrystal through first-principles DFT calculations and thenconvert the problem to a tight-binding formulation, in aSlater-Koster spirit [18], by exact discrete Fourier trans-forms. Our ansatz is that the evolution of the highestoccupied states in a k plane (i.e., a plane in reciprocalspace) orthogonal to the chain direction zz can be treatedas coming from one (highest occupied) molecular orbital
P H Y S I C A L R E V I E W L E T T E R S week ending28 FEBRUARY 2003VOLUME 90, NUMBER 8
086401-1 0031-9007=03=90(8)=086401(4)$20.00 2003 The American Physical Society 086401-1
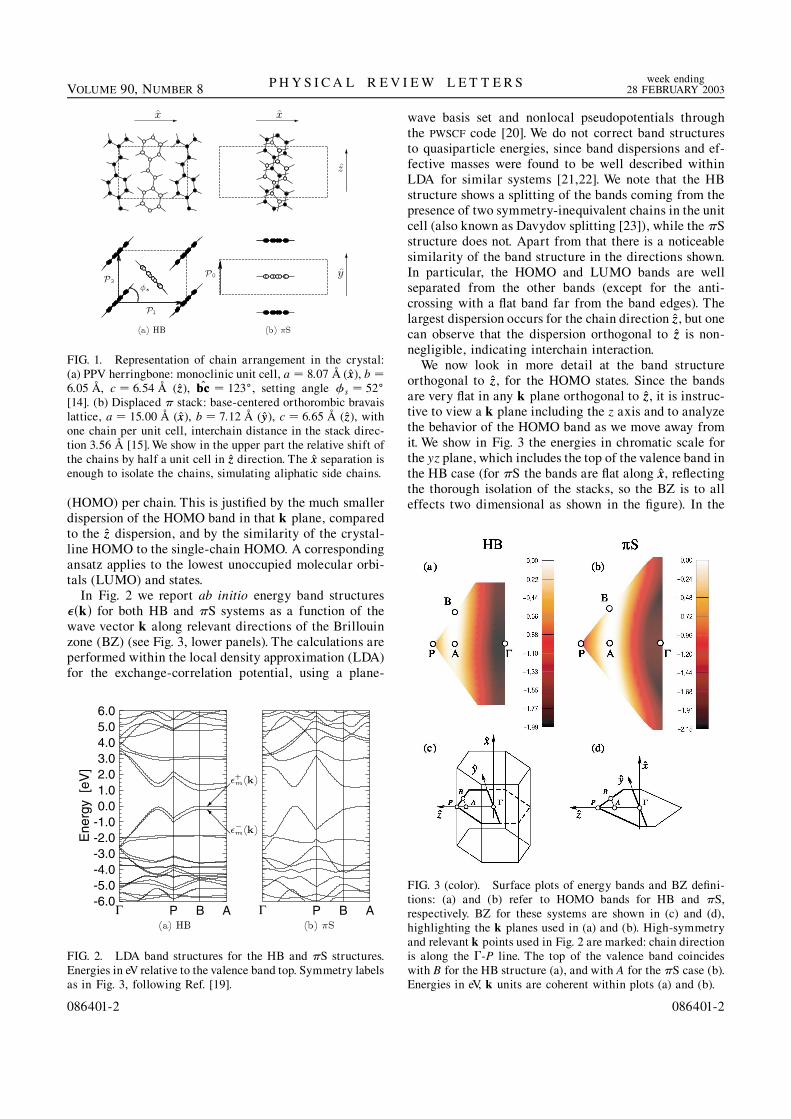
(HOMO) per chain. This is justified by the much smallerdispersion of the HOMO band in that k plane, comparedto the zz dispersion, and by the similarity of the crystal-line HOMO to the single-chain HOMO. A correspondingansatz applies to the lowest unoccupied molecular orbi-tals (LUMO) and states.
In Fig. 2 we report ab initio energy band structuresk for both HB and S systems as a function of thewave vector k along relevant directions of the Brillouinzone (BZ) (see Fig. 3, lower panels). The calculations areperformed within the local density approximation (LDA)for the exchange-correlation potential, using a plane-
wave basis set and nonlocal pseudopotentials throughthe PWSCF code [20]. We do not correct band structuresto quasiparticle energies, since band dispersions and ef-fective masses were found to be well described withinLDA for similar systems [21,22]. We note that the HBstructure shows a splitting of the bands coming from thepresence of two symmetry-inequivalent chains in the unitcell (also known as Davydov splitting [23]), while the Sstructure does not. Apart from that there is a noticeablesimilarity of the band structure in the directions shown.In particular, the HOMO and LUMO bands are wellseparated from the other bands (except for the anti-crossing with a flat band far from the band edges). Thelargest dispersion occurs for the chain direction zz, but onecan observe that the dispersion orthogonal to zz is non-negligible, indicating interchain interaction.
We now look in more detail at the band structureorthogonal to zz, for the HOMO states. Since the bandsare very flat in any k plane orthogonal to zz, it is instruc-tive to view a k plane including the z axis and to analyzethe behavior of the HOMO band as we move away fromit. We show in Fig. 3 the energies in chromatic scale forthe yz plane, which includes the top of the valence band inthe HB case (for S the bands are flat along xx, reflectingthe thorough isolation of the stacks, so the BZ is to alleffects two dimensional as shown in the figure). In the
Γ P B A-6.0-5.0-4.0-3.0-2.0-1.00.01.02.03.04.05.06.0
Ene
rgy
[eV
]
Γ P B A
FIG. 2. LDA band structures for the HB and S structures.Energies in eV relative to the valence band top. Symmetry labelsas in Fig. 3, following Ref. [19].
FIG. 3 (color). Surface plots of energy bands and BZ defini-tions: (a) and (b) refer to HOMO bands for HB and S,respectively. BZ for these systems are shown in (c) and (d),highlighting the k planes used in (a) and (b). High-symmetryand relevant k points used in Fig. 2 are marked: chain directionis along the -P line. The top of the valence band coincideswith B for the HB structure (a), and with A for the S case (b).Energies in eV, k units are coherent within plots (a) and (b).
FIG. 1. Representation of chain arrangement in the crystal:(a) PPV herringbone: monoclinic unit cell, a 8:07 A (xx), b 6:05 A, c 6:54 A (zz), bcbc 123, setting angle s 52
[14]. (b) Displaced stack: base-centered orthorombic bravaislattice, a 15:00 A (xx), b 7:12 A (yy), c 6:65 A (zz), withone chain per unit cell, interchain distance in the stack direc-tion 3:56 A [15]. We show in the upper part the relative shift ofthe chains by half a unit cell in zz direction. The xx separation isenough to isolate the chains, simulating aliphatic side chains.
P H Y S I C A L R E V I E W L E T T E R S week ending28 FEBRUARY 2003VOLUME 90, NUMBER 8
086401-2 086401-2

noninteracting limit, we should obtain completelyplanar energy fronts, orthogonal to zz. However, we see adistinct curvature of the high-energy ridge for S, notseen for HB.
The effective masses are calculated at the top ofthe valence (HOMO) and bottom of the conduction(LUMO) bands, accounting for the full 3D tensor. Theprincipal axes of the inverse mass tensor numericallycoincide with our xx, yy, zz axes for both systems: theresulting effective mass parameters are reported inTable I. These data show the general tendency of LUMObands to be more dispersive in the yy direction; electronshave lower effective masses than holes. Masses along thechain backbones are quite similar for both systems,which indicates at this level that the details of crystallinepacking do not play a relevant role in this direction.Nevertheless, effective masses in directions orthogonalto the chains are only between 1 and 2 orders of magni-tude larger, thus indicating an important contributionof solid-state effects.
An analysis based only on the effective masses is notable to account for differences between packing struc-tures (HB and S), such as those shown in Fig. 3: effec-tive masses are local parameters in k, pertaining to theabsolute maximum (minimum) of the band, and relevantto the low-doping, low-temperature limit. In the caseof polymer or oligomer films, we are usually in anintermediate-to-high doping regime, and we shouldmore properly work with the whole band structure.
We now move on to the study of TI that reflect thewhole band curvature, and can give us information onspecific neighborhood interactions. There are two funda-mentally different periodicities for polymer crystals: theperiodicity of each single chain along the chain axis,described by the lattice vector c, and the (interchain)two-dimensional periodicity, which we denote by P . Wecan explicitly acknowledge the weak interaction on P andwrite the generic eigenstate lk of the crystalHamiltonian as
lkr Xs;m;j
Clm;j eikP sj
mkzr P s j; (1)
where the sum is over cells (s), inequivalent chains in theunit cell (j), and, if necessary, isolated chain band index(m). The ’s are the solutions of the single-chain eigen-value problem: the noteworthy feature in Eq. (1) is thatthe only k state of the isolated chain structure contribu-ting to the expansion is that corresponding to the com-
ponent of k along the chain direction kz k zz. Onecan thus arrive at the following definition of TI:
Ekzmi;njP s Zd3r
mkzr iHnkzr P s j:
(2)
As seen from the band structures in Fig. 2, we canconsider only one state per chain in the unit cell, theHOMO or the LUMO. Here we will focus on the holetransport and the HOMO states. For the simple case ofone chain per unit cell (S), assuming zero overlapbetween the states of different chains, the interchain TIcan be obtained for an arbitrary number of neighborsthrough exact discrete Fourier transforms of the bandalong the orthogonal direction:
Ekzm P s 1
Q
XQq1
eikqP s mkq; (3)
where Q is defined by the number of neighbors at P staken into account.
When by crystal symmetry there are two inequivalent(j 1; 2) chains per unit cell, the TI must be obtainedfrom a 2 2 matrix; taking into account the improperrotation bringing one chain into the other in the HB case,the following relations hold:
Xs
eikP sEkzm1;m1P s 1
2mk mk; (4)
Xs
eikP sEkzm1;m2P s
1
2mk mk; (5)
where mk [mk] is the maximum (minimum) diago-nal element of the 2 2 matrix. From Eq. (4) it ispossible to obtain the TI for equivalent chains through asimple Fourier transform. In order to calculate TI be-tween inequivalent chains [Eq. (5)] we make the furthersimplifying assumption of retaining nearest neighborsonly (the next interaction would be between thirdneighbors).
Our results are reported in Table II, and we now see alarge difference emerging between the structures, withthe nearest-neighbor coupling in the S structure morethat 4 times larger than any coupling in the HB crystal.Surprisingly, the stronger coupling in the HB structurearises for the (nearest-neighbor) nonequivalent chains, in
TABLE I. Effective masses in units of the free-electron mass.
HB SHOMO LUMO HOMO LUMO
xx 3.54 2.52 1 1
yy 5.24 1.31 5.56 2.74zz 0.11 0.11 0.10 0.10
TABLE II. Absolute values of transfer integrals of HOMOstates in meV. For clarity we use EijP s in place of Ekzmi;mjP s,since m refers always to the top valence band, and kz is set tothe z component of the HOMO k vector.
HB S
E120 27.42 EP 0 120.64E11P 2 13.96 E2P 0 10.49
E11P 1 P 2 1.49 E3P 0 2.86E11P 1 0.27
P H Y S I C A L R E V I E W L E T T E R S week ending28 FEBRUARY 2003VOLUME 90, NUMBER 8
086401-3 086401-3

spite of the large interchain angle. The ‘‘stacklike’’ cou-pling along P 1 is prevented by the increased distance, andalong P 2 by the large lateral displacement of one chainrelative to the neighbors. This latter finding is in goodagreement with ab initio results for other organic systems[24] that highlight the relevance of the precise stackinggeometry. It is interesting to compare results for the TIalong the chain direction, for the case of an isolatedchain. Now we consider the HOMO states of the mono-mer as generating orbitals for the band, in a similar spiritas above; we are again justified by the clear origin ofthe HOMO band in these systems. We find that the TIfor nearest neighbors is in that case Ec 523 meV, andthus transport in the stack direction for S films willhave efficiencies of the same order of magnitude as forlong stretched polymer chains along the chain direction.
Recently, semiempirical calculations for selected ag-gregates of similar oligomers were performed [25],assuming ad hoc that the nearest-neighbors-only approxi-mation could be adopted. Our results show that for the HBstructure this hypothesis is quite reasonable; however,this is not the general case, as shown for the S structure.This could be investigated only because we modeled afully periodic system.
We learn from this analysis that, for the HB structure,coupling orthogonal to the chain direction is muchweaker than along the chain, supporting the wealth ofmodels that completely neglect interchain-hopping con-tributions to transport in long-polymer films. However, ithas been accepted that the long aliphatic side chains of,e.g., MEH-PPV were responsible mainly for preventinginteraction between chains, thereby preserving the prop-erties of the isolated chain. What we see is that theorganization in stacks instead promotes interchaininteractions, and on the whole should increase the holeconductivity of the material. This feature, together withthe absence of Davydov splitting (responsible for lumi-nescence quenching [26] in HB-PPV), sums up to betterdevice performance expected for highly organized Sfilms. We did not take into account polaron relaxationwhich should occur in real samples. However, for PPV, therelaxation is associated with slight quinoid distortion ofthe rings, and charge localization [25]. Since the distor-tion does not affect interchain angles and general point-symmetry properties, the values we obtain for TI shouldbe appropriate to the closely packed regions of realsamples.
In conclusion, we have shown that interchain interac-tions are very sensitive to the specific 3D structure, andcan thus be used to tailor the transport properties ofconjugated-polymer films.
We are grateful to G. Bussi and A. Calzolari for stim-ulating discussions. Calculations partly done at CINECAunder an INFM grant.We gratefully acknowledge supportby the EU Network ‘‘Exciting’’ and by the ItalianMinistry for Foreign Affairs (MAE). M. J. C. acknowl-edges support from FAPESP and CNPq (Brazil).
[1] J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N.Marks, K. Mackay, R. H. Friend, P. L. Burns, and A. B.Holmes, Nature (London) 347, 539 (1990).
[2] R. A. Marcus and N. Sutin, Biochim. Biophys. Acta811, 265 (1985).
[3] V. S. Pande and J. N. Onuchic, Phys. Rev. Lett. 78, 146(1997).
[4] M. Ahlskog, M. Reghu, A. J. Heeger, T. Noguchi, andT. Ohnishi, Phys. Rev. B 53, 15 529 (1996).
[5] T. Q. Nguyen, R. C. Kwong, M. E. Thompson, and B. J.Schwartz, Appl. Phys. Lett. 76, 2454–2456 (2000).
[6] N. Greenham and R. Friend, in Solid State Physics,edited by H. Ehrenreich and F. Spaepen (Academic,San Diego, CA, 1995), pp. 2–150.
[7] R. Landauer, Philos. Mag. 21, 863 (1970).[8] M. Buttiker, Y. Imry, R. Landauer, and S. Pinhas, Phys.
Rev. B 31, 6207 (1985).[9] M. Buttiker, Phys. Rev. Lett. 57, 1761 (1986).
[10] P. A. Schulz, D. S. Galvao, and M. J. Caldas, Phys.Rev. B 44, 6073 (1991).
[11] R. Hey, F. Gagel, M. Schreiber, and K. Maschke,Phys. Rev. B 55, 4231 (1997).
[12] See, e.g., F.-J. M. zu Heringdorf, M. C. Reuter, and R. M.Tromp, Nature (London) 412, 517 (2001); C. Kloc, P. G.Simpkins, T. Siegrist, and R. A. Laudise, J. Cryst. Growth182, 416 (1997), and references therein.
[13] See, e.g., R. O. Jones and R. Gunnarsson, Rev. Mod.Phys. 61, 689 (1989); M. C. Payne, M. P. Teter, D. C.Allan, T. A. Arias, and J. D. Joannopoulos, Rev. Mod.Phys. 64, 1045 (1992), and references therein.
[14] D. Chen, M. J. Winokur, M. A. Masse, and F. E. Karasz,Phys. Rev. B 41, 6759 (1990).
[15] C.Y. Yang, F. Hide, M. A. Dıaz-Garcıa, A. J. Heeger, andY. Cao, Polymer 39, 2299 (1998).
[16] D. Chen, M. J. Winokur, M. A. Masse, and F. E. Karaz,Polymer 33, 3116 (1992).
[17] E. M. Conwell, J. Perlstein, and S. Shaik, Phys. Rev. B 54,R2308 (1996).
[18] J. C. Slater and G. F. Koster, Phys. Rev. 94, 1498(1954).
[19] P. Gomes da Costa, R. G. Dandrea, and E. M. Conwell,Phys. Rev. B 47, 1800 (1993).
[20] S. Baroni, A. Dal Corso, S. de Gironcoli, andP. Giannozzi, http://www.pwscf.org (2001).
[21] J.-W. van der Horst, P. A. Bobbert, M. A. J. Michels,G. Brocks, and P. J. Kelly, Phys. Rev. Lett. 83, 4413(1999).
[22] M. Rohlfing, M. L. Tiago, and S. G. Louie, Synth. Met.116, 101 (2001).
[23] A. S. Davydov, Theory of Molecular Excitons (McGraw-Hill, New York, 1962).
[24] A. Calzolari, R. Di Felice, E. Molinari, and A. Garbesi,Appl. Phys. Lett. 80, 3331 (2002).
[25] J. L. Bredas, D. Beljonne, J. Cornil, J. P. Calbert,Z. Shunai, and R. Silbey, Synth. Met. 125, 107(2002).
[26] A. Ruini, M. J. Caldas, G. Bussi, and E. Molinari, Phys.Rev. Lett. 88, 206403 (2002).
P H Y S I C A L R E V I E W L E T T E R S week ending28 FEBRUARY 2003VOLUME 90, NUMBER 8
086401-4 086401-4

Ab initio study of transport parameters in polymer crystals
Andrea Ferretti,* Alice Ruini, Giovanni Bussi, and Elisa MolinariINFM National Center on NanoStructures and BioSystems at Surfaces(S3) and Dipartimento di Fisica,
Universitadi Modena e Reggio Emilia, Via Campi 213/A, 41100 Modena, Italy
Marilia J. CaldasInstituto de Fı´sica, Universidade de Sa˜o Paulo, Cidade Universita´ria, 05508-900 Sa˜o Paulo, Brazil and INFM-S3, Italy
~Received 29 July 2003; revised manuscript received 21 January 2004; published 28 May 2004!
Transfer integrals~TI’s! are essential parameters in the calculation of electron transport both in coherent andincoherent regimes. We show that TI’s for polymer crystals can be obtained from first principles, starting fromplane-wave density-functional calculations of the electronic structure in the local-density approximation, andpropose methods at different levels of approximation. We demonstrate that special choices of single-chainstates can be used very effectively as building blocks for the crystal electronic structure, thus allowing a deeperinsight into the transport properties of molecular crystals. We apply this approach to polymer systems of greatinterest to molecular electronics, such as poly-para-phenylene-vinylene and polythiophene in different crystalpacking morphologies, and show that it offers a very powerful tool to understand and design the impact ofintermolecular interactions on conduction of organic crystals.
DOI: 10.1103/PhysRevB.69.205205 PACS number~s!: 71.20.Rv, 71.70.2d
I. INTRODUCTION
Organic materials based onp-conjugated oligomers andpolymers constitute the active elements in new-generationplastic electronic and optoelectronic devices, such as field-effect transistors, light-emitting diodes, and photovoltaic andsolar cells.1,2 These developments stimulate further investi-gation of the transport properties ofp-conjugated systems,since device performance strongly depends on the efficiencyof charge transport processes. Full understanding of the mi-croscopic mechanisms controlling device operation is diffi-cult to achieve, mainly due to the complex structure of poly-mer films, which are characterized by strong covalentintramolecular bonds and weak van der Waals interactionsbetween adjacent chains. Each chain has, on its own, char-acteristic transport properties along the long chain axis~in-trachain transport!, as confirmed by the high electrical an-isotropy seen for stretched material.3 However, it has beenshown that the film morphology strongly influences thetransport properties, indicating that intramolecular interac-tions are not sufficient to explain experiments.4 We stressthat packing morphologies for same-family polymers can in-deed vary significantly. Taking as an example poly-para-phenylene vinylene~PPV!, the unsubstituted polymer~thatis, with no side chains attached to either the phenylene orvinylene groups! is known to crystallize in a herringbonestructure5 @Fig. 1~a!# typical of several polymeric and oligo-meric crystals; however, due to solubility and other growthproblems it is often useful to functionalize the main chainwith aliphatic or ether side chains, and a displacedp-stack(pS) configuration@Fig. 1~b!# is then observed such as in,for example, poly@28-methoxy, 5-~2-ethyl-hexyloxy!# phe-nylene vinylene~MEH-PPV!.6 While long-chained samplesare mostly disordered, with embedded crystalline regions,oligomeric films can be very organized and, due to the preva-lence of gaps between the relatively short chains, interchaintransport is expected to dominate.
In this context,ab initio approaches based on the density-functional theory~DFT! can provide a valuable input to thefield, as they can take both the atomistic nature and the crys-talline environment fully into account and allow thereby anaccurate investigation of the relevant transport parameters.8
At very low temperatures, the transport mechanisms in well-ordered materials can be described in terms of bandlike mo-
FIG. 1. Representation of three-dimensional arrangements forPPV @(2C6H42C2H22)n# chains. The carbon atoms are repre-sented as circles, and the bonding to hydrogen atoms is also indi-cated by sticks.~a! Unsubstituted herringbone PPV: the unit cell ismonoclinic with two inequivalent chains~carbon atoms representedas solid and open circles, respectively! in the basis; from Ref. 7,
a58.07 Å (x), b56.05 Å, (y), c56.54 Å (z), bc5123°, and set-ting anglefs552°. ~b! Displacedp stack: base-centered ortho-rhombic Bravais lattice with one chain per unit cell, whose dimen-
sions area515.00 Å (x), b57.12 Å (y), and c56.65 Å (z).
Stacking distance alongy is 3.56 Å according to Ref. 6.
PHYSICAL REVIEW B 69, 205205 ~2004!
0163-1829/2004/69~20!/205205~10!/$22.50 ©2004 The American Physical Society69 205205-1

tion, while with increasing temperature the transport pro-cesses may involve sequential hopping between adjacentchains. From a microscopic point of view, both cases areproperly described by means of the interchain transfer inte-grals that express the ‘‘ease of transfer’’ of a charge betweentwo interacting chains, and can be inserted, e.g., in aButtiker-Landauer9–11 expression for conductance12,13 in thecoherent regime, or in the Marcus14 formulation for electrontransfer in the hopping regime. We will present here threealternative schemes for theab initio calculation of interchaintransfer integrals. We apply the formalism to different poly-mer crystals, based on PPV and polythiophene~PT!; thisallows us to discuss the results for the interchain transferintegrals as a function of the nature and arrangement of theinteracting units.
We first present the different theoretical methodologies inSec. II; we present and discuss our results for the prototypecrystals in Sec. III, and conclude.
II. THEORY
Ab initio DFT calculations based on plane-wave~PW!expansion of the wave functions constitute an important toolto understand the electronic properties of polymercrystals;15–17 it is sometimes helpful however to probe ‘‘lo-calized’’ parameters such as interchain hopping, which areformally linked to a tight-binding, localized-basis expansionof the wave function. The two expansion bases are, ofcourse, completely equivalent if taken to convergency; thechoice is one of conveniency only, and it is thus useful to beable to transit from one description to the other. We hereextract tight-binding parameters from ourab initio PW band-structure calculations for typical polymer crystals.
The tight binding~TB! formalism is useful to write theHamiltonian of a complex system starting from subunits thatwe can identify as building blocks; it is usually associatedwith combinations of atomic orbitals, and is also very usefulin its simplest form of one orbital per site~e.g., for disor-deredp-conjugated systems!.18 That scope, however, can besignificantly expanded: the important issue is to single outfrom the electronic structure of the complex system the rel-evant states to be used in a‘‘smart site definition.’’ Three-dimensional~3D! polymer crystals are made of 1D infinitechains arranged in a 2D lattice: it turns out that for manycases of interest we can identifychain statesthat can be usedas building blocks for the 3D system. As an example, we seein Fig. 2 the probability distribution for relevant states of anisolated PPV chain, and for the corresponding crystal statesin the pS structure. In this section we present a specificformulation of the TB scheme, in a Slater-Koster spirit,19
which we apply to the case of polymer crystals in order toobtain interchain transfer integrals~TI’s!. We also evaluateTI’s and chain states overlap directly from the full Blochstructure obtained with the first-principles calculation, wecan thus compare values obtained with the direct and indirectmethods, and allowing for deeper insight in the transportproperties of the systems as a whole.
The polymer crystal structure is generated by arrangingthe individual infinite chains in a 2D lattice, described by
vectors $P%, whose unit cell may containq inequivalentchains located at$t%. We write the eigenfunction for the 1Disolated chain problem, corresponding to thei th inequivalentchain in thesth lattice site as
^r ufmk,Ps
i &5fmki ~r2ti2Ps!, ~1!
wherem is the band index and the scalark points are definedfor the 1D Brillouin zone~BZ!. To expand the crystal eigen-vectorsuc lk& in terms of the isolated chain states we use thefollowing ansatz:8
uc lk&5(s
eik•Ps(m,i
Cmilk ufmkz ,Ps
i &. ~2!
The Bloch sum overPs in Eq. ~2! ensures thek symmetryof the crystal states. The important feature of Eq.~2! is thatthe onlyk state of the isolated chain structure contributing tothe k expansion is the one corresponding to the componentof k along the chain directionkz5k• z; indeed, the crystalstate with k-symmetry k[(kx ,ky ,kz) is orthogonal to allstates differing in one of these quantum numbers~in particu-lar, kz , which is defined for chain states!.
Using Eq.~2! we obtain the master equation
(n, j
Cn jl (
seik•PsEmi,n j
kz ~Ps!
5e l~k!(n, j
Cn jl (
seik•Psami,n j
kz ~Ps! ; m,i ,k, ~3!
where we have defined transfer integralsEmi,n jkz (Ps) and
overlap integralsami,n jkz (Ps) as follows:
FIG. 2. Comparison of HOMO~highest occupied molecular or-bital! and LUMO ~lowest unoccupied molecular orbital! states of~a!PPV ‘‘trimer,’’ ~b! PPV isolated chain, and~c! PPV pS crystal.Calculations have been done using HF-AM1 and ZINDO for~a!and PW-DFT for~b! and ~c!.
FERRETTI, RUINI, BUSSI, MOLINARI, AND CALDAS PHYSICAL REVIEW B69, 205205 ~2004!
205205-2

Emi,n jkz ~Ps!5^fmkz,0
i uHufnkz ,Ps
j &, ~4!
ami,n jkz ~Ps!5^fmkz,0
i ufnkz ,Ps
j &, ~5!
H standing for the crystal Hamiltonian; it will also be usefulto define the sums
Emi,n j~k!5(s
eik•PsEmi,n jkz ~Ps!, ~6!
ami,n j~k!5(s
eik•Psami,n jkz ~Ps!. ~7!
A. Inverse tight-binding scheme
In a standard TB scheme the eigenvaluese l(k) are thesolutions of the master equation and can be obtained oncethe TI’s and overlaps are known. Our final goal here is thecalculation of the TI’s starting from the full band structureobtained by means ofab initio DFT calculation, which wedenote as aninverse tight-bindingapproach. This schemepartly corresponds to the Slater-Koster framework,19 whereTI’s are obtained by interpolating on a finite set ofk pointsfor which eigenvalues are known~either from experimentaldata or from a previous calculation through some othermethod!, in order to obtain the full band structure in a fol-lowing step within a standard TB spirit.
In the following, we describe the application of the in-verse TB scheme to the polymer crystals shown in Figs. 1and 3. These specific crystal structures allow us to investi-gate both the case of one@see Fig. 1~b!# and two@see Figs.1~a! and 3# polymer chains in the unit cell. We will exploit animportant simplification for such systems, namely, that therelevant states—lowest conduction band and top of valenceband—for the crystalline structures can be derived just fromthe corresponding bands of the isolated chains. We will labelthese states as LUMO~lowest unoccupied molecular orbital!and HOMO ~highest occupied molecular orbital! bands, inanalogy to quantum chemistry terms. We assume zero over-lap between states in different chains@analogous to the ne-glect of differential/diatomic overlap~NDO! approximationof quantum chemistry methods#,
ami,n jkz ~Ps!5dmnd i j dPs,0
, ~8!
with implications that we will discuss later on.
1. One chain per unit cell
Here we consider the case of one chain per unit cell (q51), as for thep-stacked PPV crystal, and one level perchain. Restricting ourselves to the HOMO~LUMO! genera-tion, the resulting master equation reads
(s
eik•PsEm1,m1kz ~Ps!5em~k!, ~9!
where we remark that the indexm is related to both crystaland isolated-chain bands; that is, the correspondence be-tween states is taken on a one-to-one basis@the HOMO
~LUMO! band in the chain generates the HOMO~LUMO!band in the crystal#. Equation~9! can be easily inverted byFourier transform~FT!, leading to an explicit expression forTI’s:
Em1,m1kz ~Ps!5
1
Nk(
ke2 ik•Psem~k!. ~10!
2. Two chains per unit cell
In this section we will deal with the case of two sites~chains! in the cell (q52) and one state per site (p51); werewrite the master equation~3! as
(n, j
Cn jl Emi,n j~k!5e l~k!(
n, jCn j
l ami,n j~k!. ~11!
In the NDO approximation Eq.~11! can be read as the eigen-value problem forE, with eigenvaluese l(k). We now obtaintwo crystal bands arising from the two inequivalent sites perunit cell: the indexl runs over these two bands which werefer ase6(k). Using the approximationE115E22, whichwill be discussed in the next paragraph, and the hermiticityof E we write
e6~k!5E116uE12u ~12!
and obtain the final relations involving TI’s:
FIG. 3. Representation of three-dimensional arrangements forPT @(2C4H2S2)n# chains. The carbon atoms are represented assmall circles, sulphur atoms by large circles, and the bonding tohydrogen atoms is also indicated by sticks. Both structures have the
same orthorombic unit cell with~Refs. 20,21! a56.03 Å (x), b
57.85 Å (y), c57.81 Å (z), setting anglefs566° ~not to scale:ydirection has been arbitrarily stretched to make the sketch morereadable!. There are two inequivalent chains~carbon atoms repre-sented as solid and open circles, respectively! in the basis, the dif-ference between PT1~a! and PT2~b! being a shift, by half a unit
cell vector alongz, of the second chain relative to the first.
AB INITIO STUDY OF TRANSPORT PARAMETERS IN . . . PHYSICAL REVIEW B69, 205205 ~2004!
205205-3

(s
eik•PsEm1,m1kz ~Ps!5
1
2@e1~k!1e2~k!#, ~13!
U(s
eik•PsEm1,m2kz ~Ps!U51
2@e1~k!2e2~k!#. ~14!
3. Herringbone crystals
Now we consider the particular case where there exists aspatial inversionJ and an improper rotationS bringing oneof the inequivalent chains in the other, as is the case for allherringbone~HB! crystals. In the general case the conditionE115E22 does not hold, so in principle it is not possible touse Eq.~12!. However, it can be shown that this condition isvalid to a good approximation, and this allows us to use Eqs.~13! and~14! for actual calculations. Equation~13!, referringto TI’s between equivalent chains, can be easily inverted bymeans of FT as before, but the modulus operator in equation~14! precludes a direct FT and requires a further approxima-tion regarding TI’s between inequivalent chains. We willtherefore neglect all TI’s but those related to nearest neigh-bors~NN’s! of the reference chain@those shown in the lowerpanel of Fig. 1~a!#. In the HB packing we have four NN’swhose TI’s are found to be all equal and real due to symme-try properties~see the Appendix!. It is now possible to sumup all the remaining terms in the left-hand side of Eq.~14!and we arrive at the final expression for the TI between in-equivalent chains:
uEm1,m2kz ~0!u5 1
8 @e1~k!2e2~k!#, ~15!
wherek5(0,0,kz).
B. Direct computation
A state-of-the-art PW-DFT calculation provides bandstructures and Bloch eigenstates, and allows one to extractTI’s through direct computation. This approach is more de-manding than inverse TB, because of wave-function han-dling; we apply it here to prove the validity of the approxi-mations adopted in the inverse TB approach. Introducing thespectral resolution for the crystal Hamiltonian into the TIdefinition, Eq.~4!, we obtain
Emi,n jkz ~Ps!5(
lk^fmkz,0
i uc lk&e l~k!^c lkufnkz ,Ps
j &, ~16!
where onlyk points whosez component equalskz ~thus de-fining a plane in the Brillouin zone! give a nonvanishingcontribution to the sum. We next define the projection inte-grals
dmilk 5^fmkz,0
i uc lk&, ~17!
and we arrive at
Emi,n jkz ~Ps!5(
lke l~k!eik•Psdmi
lk dn jlk* . ~18!
Thus it is possible to perform a direct calculation of TI’s byevaluating the projection of crystal eigenstates onto thosecalculated for an isolated chain in the crystalline referencecell (Ps50). We have numerically implemented this proce-dure starting from a plane-wave representation for the crystaleigenstates, combined with a reciprocal lattice integration forscalar products.
We note that the overlap matricesa are not directly used,however the existence of nonzero overlaps implies that re-sults are dependent on the zero of the energy scale chosen forthe spectral resolution of the Hamiltonian. In this case thephysically sound quantities are the matrix elements ofva2H rather than TI’s. However, because we are interested ina small energy window nearby Fermi energy, setting therethe origin of the energies makes all the relevant informationto be recast in the TI’s only and their physical meaning to beunambiguous.
C. Quasidirect computation
We introduce this last approach in order to analyze indepth the effect of the NDO approximation. We can obtainoverlaps between isolated chain states in an exact fashion bymeans of the projection integrals defined in the last section.Once overlaps are computed, in principle it could be possibleto eliminate the NDO approximation from the inverse TBcalculation in the case of one chain per unit cell, but this isstill not straightforward in the case of HB crystals. We can,however, assess the impact of the approximations used~NDO versus NN! in that case, by performing a calculationwhere the only approximation included is the NDO.
Once we obtained the projectionsdmilk and recasted the
master equation as
(n j
Emi,n j~k!Cn jlk5e l~k!dmi
lk ~19!
we could obtain the TI’s giving as input the expansion coef-ficientsCn j
lk ; now, the coefficients can be independently ob-tained from a knowledge of the projections and the overlaps
(n j
ami,n j~k!Cn jlk5dmi
lk . ~20!
Alternatively, we can substitute here the NDO approxima-tion ami,n j
NDO in which case we obtain approximateCn jlk,NDO to
be used for the calculation of TI’s:
(n j
Emi,n jNDO ~k!Cn j
lkNDO5e l~k!dmilk , ~21!
where NDO is the only approximation fully incorporated.
III. RESULTS
We have applied the methods described above to investi-gate the transport properties of poly~para-phenylene vi-nylene! and polythiophene. Both PPV- and PT-based materi-als offer the possibility to investigate the impact ofcrystalline aggregation on electronic and transport properties,
FERRETTI, RUINI, BUSSI, MOLINARI, AND CALDAS PHYSICAL REVIEW B69, 205205 ~2004!
205205-4

because they can be deposited and grown in more than onecrystal structure. While for PPV we investigate HB andpSstructures, in the case of PT we can analyze different ar-rangements of the inequivalent chains in the HB cell. Unsub-stituted oligothiophenes, such asa-nT with n ranging from 4to 16, are experimentally very well studied22 and it is knownthat forn54,6 two possible polymorph HB packings can beobtained depending on the deposition temperature.21 Follow-ing Ref. 20, we construct two different polythiophene struc-tures, PT1 and PT2, having the same symmetry properties ofhigh and low temperaturea-4T respectively: to a good ap-proximation, these ideal polymer systems can give reliableinformation about the intermolecular coupling for real oligo-meric films. The details of PT1 and PT2 packings are shownin Fig. 3; the difference between them is a translation of oneinequivalent chain relative to the other by half a lattice vec-tor along the chain direction, with the effect that the sulphurlone pairs on neighbor chains are closer to~PT1! or fartherapart from~PT2! each other.
A. Band structures
The first step for the computation of TI’s is to obtain theab initio electronic structure for the systems of interest, forwhich we use DFT-LDA calculations with 45 Ry cutoff en-ergy in the plane-wave basis set, norm-conserving pseudopo-tentials, and periodic boundary conditions. We perform thesecalculations through the PWSCF package.23 In Figs. 4 and 5we report band structures and BZ details for the PPV and PTsystems, respectively, including the isolated chains. Disper-sion along the chain direction dominates the bandwidth, but
a non-negligible dispersion is also found for directions or-thogonal to the chains, a first indication of interchain inter-actions. Comparing results forpS versus HB in the case ofPPV @Figs. 4~b! and 4~c!# we note the band doubling presentin the latter, due to the existence of two inequivalent chainsin the unit cell. The same doubling occurs also in the bandstructures of PT1 and PT2@Figs. 5~b! and 5~c!# but, due tothe higher point symmetry of the thiophene crystals, bandsare degenerate except for some low-symmetry lines~as, e.g.,X-G-Y). The splitting between the doublet states is anotherwell-known sign of interaction between inequivalent chains.
In the case of HB-PPV, the HOMO and LUMO states~valence-band top and conduction-band minimum, respec-tively! are aligned in a direct-gap configuration at the samekpoint (B) in the BZ @Fig. 4~c!#, while for pS-PPV theHOMO is located atA and the LUMO atB @Fig. 4~b!#. BothPT1 and PT2 have LUMO states at theX point, while theHOMO is found atG for PT1 and atX for PT2 @Figs. 5~b!and 5~c!#; we note, however, that for PT2 the highest valenceband atG is also quasidegenerate with the HOMO state.
It is interesting at this point to compare results, at leastqualitatively, between PW and TB. We will here perform theextreme comparison between fullab initio DFT-PW calcula-tions for infinite systems, and a semiempirical HF TB calcu-lation for a finite~oligomeric! chain, performed through themost used techniques of the NDO family, AM1 for geom-etries and ZINDO for electronic charge densities andenergies.26,27 These techniques have been carefully param-etrized for isolated small molecules, however, it has beenshown that they can also be applied to ring-structured, rela-tively long oligomers.28 We show in Fig. 2 the probabilitydensities for the relevant one-electron states of PPV systems:
FIG. 4. Brillouin zone~upper panels! and band structures~lowerpanels! for ~a! PPV single chain,~b! pS-PPV, and~c! HB-PPV.Chain direction is parallel toG-Z everywhere, and relevantk pointsare indicated. Wave-vector units are not in scale from graph tograph. Energies in eV. A description of the HOMO-band behavior inthe yz plane can be found in Ref. 8.
FIG. 5. Brillouin zone~upper panels! and band structures~lowerpanels! for ~a! PT single chain,~b! PT1, and~c! PT2. Chain direc-tion is parallel toG-Z everywhere, and relevantk points are indi-cated. Wave-vector units are not in scale from graph to graph. En-ergies in eV. Further details about band structures for these systemscan be found in Refs. 20, and 24, 25.
AB INITIO STUDY OF TRANSPORT PARAMETERS IN . . . PHYSICAL REVIEW B69, 205205 ~2004!
205205-5
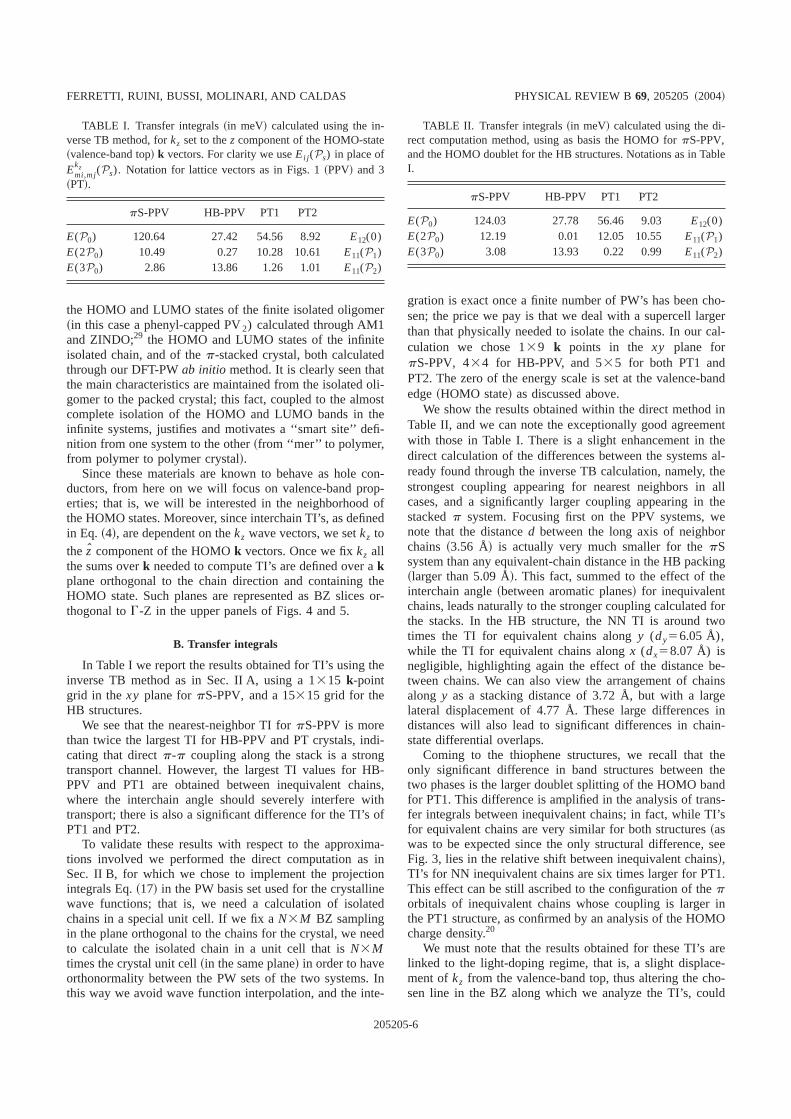
the HOMO and LUMO states of the finite isolated oligomer~in this case a phenyl-capped PV2) calculated through AM1and ZINDO;29 the HOMO and LUMO states of the infiniteisolated chain, and of thep-stacked crystal, both calculatedthrough our DFT-PWab initio method. It is clearly seen thatthe main characteristics are maintained from the isolated oli-gomer to the packed crystal; this fact, coupled to the almostcomplete isolation of the HOMO and LUMO bands in theinfinite systems, justifies and motivates a ‘‘smart site’’ defi-nition from one system to the other~from ‘‘mer’’ to polymer,from polymer to polymer crystal!.
Since these materials are known to behave as hole con-ductors, from here on we will focus on valence-band prop-erties; that is, we will be interested in the neighborhood ofthe HOMO states. Moreover, since interchain TI’s, as definedin Eq. ~4!, are dependent on thekz wave vectors, we setkz tothe z component of the HOMOk vectors. Once we fixkz allthe sums overk needed to compute TI’s are defined over akplane orthogonal to the chain direction and containing theHOMO state. Such planes are represented as BZ slices or-thogonal toG-Z in the upper panels of Figs. 4 and 5.
B. Transfer integrals
In Table I we report the results obtained for TI’s using theinverse TB method as in Sec. II A, using a 1315 k-pointgrid in thexy plane forpS-PPV, and a 15315 grid for theHB structures.
We see that the nearest-neighbor TI forpS-PPV is morethan twice the largest TI for HB-PPV and PT crystals, indi-cating that directp-p coupling along the stack is a strongtransport channel. However, the largest TI values for HB-PPV and PT1 are obtained between inequivalent chains,where the interchain angle should severely interfere withtransport; there is also a significant difference for the TI’s ofPT1 and PT2.
To validate these results with respect to the approxima-tions involved we performed the direct computation as inSec. II B, for which we chose to implement the projectionintegrals Eq.~17! in the PW basis set used for the crystallinewave functions; that is, we need a calculation of isolatedchains in a special unit cell. If we fix aN3M BZ samplingin the plane orthogonal to the chains for the crystal, we needto calculate the isolated chain in a unit cell that isN3Mtimes the crystal unit cell~in the same plane! in order to haveorthonormality between the PW sets of the two systems. Inthis way we avoid wave function interpolation, and the inte-
gration is exact once a finite number of PW’s has been cho-sen; the price we pay is that we deal with a supercell largerthan that physically needed to isolate the chains. In our cal-culation we chose 139 k points in the xy plane forpS-PPV, 434 for HB-PPV, and 535 for both PT1 andPT2. The zero of the energy scale is set at the valence-bandedge~HOMO state! as discussed above.
We show the results obtained within the direct method inTable II, and we can note the exceptionally good agreementwith those in Table I. There is a slight enhancement in thedirect calculation of the differences between the systems al-ready found through the inverse TB calculation, namely, thestrongest coupling appearing for nearest neighbors in allcases, and a significantly larger coupling appearing in thestackedp system. Focusing first on the PPV systems, wenote that the distanced between the long axis of neighborchains ~3.56 Å! is actually very much smaller for thepSsystem than any equivalent-chain distance in the HB packing~larger than 5.09 Å!. This fact, summed to the effect of theinterchain angle~between aromatic planes! for inequivalentchains, leads naturally to the stronger coupling calculated forthe stacks. In the HB structure, the NN TI is around twotimes the TI for equivalent chains alongy (dy56.05 Å),while the TI for equivalent chains alongx (dx58.07 Å) isnegligible, highlighting again the effect of the distance be-tween chains. We can also view the arrangement of chainsalong y as a stacking distance of 3.72 Å, but with a largelateral displacement of 4.77 Å. These large differences indistances will also lead to significant differences in chain-state differential overlaps.
Coming to the thiophene structures, we recall that theonly significant difference in band structures between thetwo phases is the larger doublet splitting of the HOMO bandfor PT1. This difference is amplified in the analysis of trans-fer integrals between inequivalent chains; in fact, while TI’sfor equivalent chains are very similar for both structures~aswas to be expected since the only structural difference, seeFig. 3, lies in the relative shift between inequivalent chains!,TI’s for NN inequivalent chains are six times larger for PT1.This effect can be still ascribed to the configuration of theporbitals of inequivalent chains whose coupling is larger inthe PT1 structure, as confirmed by an analysis of the HOMOcharge density.20
We must note that the results obtained for these TI’s arelinked to the light-doping regime, that is, a slight displace-ment ofkz from the valence-band top, thus altering the cho-sen line in the BZ along which we analyze the TI’s, could
TABLE I. Transfer integrals~in meV! calculated using the in-verse TB method, forkz set to thez component of the HOMO-state~valence-band top! k vectors. For clarity we useEi j (Ps) in place ofEmi,m j
kz (Ps). Notation for lattice vectors as in Figs. 1~PPV! and 3~PT!.
pS-PPV HB-PPV PT1 PT2
E(P0) 120.64 27.42 54.56 8.92 E12(0)E(2P0) 10.49 0.27 10.28 10.61 E11(P1)E(3P0) 2.86 13.86 1.26 1.01 E11(P2)
TABLE II. Transfer integrals~in meV! calculated using the di-rect computation method, using as basis the HOMO forpS-PPV,and the HOMO doublet for the HB structures. Notations as in TableI.
pS-PPV HB-PPV PT1 PT2
E(P0) 124.03 27.78 56.46 9.03 E12(0)E(2P0) 12.19 0.01 12.05 10.55 E11(P1)E(3P0) 3.08 13.93 0.22 0.99 E11(P2)
FERRETTI, RUINI, BUSSI, MOLINARI, AND CALDAS PHYSICAL REVIEW B69, 205205 ~2004!
205205-6

lead to regions with different band curvatures~as seen inRef. 8 for pS-PPV), and could hence modify the TI valuesat higher doping.
C. Approximation analysis
At this point it is interesting to understand which are theapproximations used in the inverse TB calculations thatmostly contribute to the differences in results with respect tothe direct computation results, namely, the smart site choice,the NDO approximation, and the NN limitation for inequiva-lent chains~HB packing!.
In order to check the smart site choice, we can perform ananalysis of the projection of the actual crystal eigenstates forthe HOMO band along the linek• z5kz , ucHOMO,k&, withucHOMO,k
TB & generated from the Bloch sum of single-chainstatesufHOMO,kz ,Ps
i &. This is done for eachk used in the
calculations, and we show in Table III the minimum andmaximum projection norms for each system. In the worstcase, which occurs forpS-PPV, the norm is already quiteclose to unity, so we can see that our ansatz is fully justified.
The second strong approximation we wish to check is theneglect of differential overlap@NDO, Eq. ~8!# used for in-verse TB calculations. This is the only other approximationinvolved in the calculation of TI’s forpS-PPV ~one chainper cell!, while the further approximations of the Appendix Aare needed for the treatment of inequivalent-chain TI’s in thecase of two chains per cell.
To gauge the effect of NDO on these systems we show inTable IV the TI’s calculated through the quasidirect compu-tation method described in Sec. II C, with the smart sitechoice of bands. As expected,pS-PPV TI’s coincide numeri-cally with inverse TB results. Proceeding to HB systems, wesee that results obtained within NDO quasidirect computa-tion reproduce very well the inverse TB results, and is also ingood accord with directly computed TI’s. We note that theNN approximation is not included here. The agree-
ment between the three sets of results shows that there is nocancellation of errors eventually coming from the NDO andNN approximations.
D. Discussion
The structural characteristics of the polymer system arereflected on the transport properties through the TI’s. How-ever, direct comparison of model theoretical results for inter-chain TI’s with experiments probing carrier mobility in realsamples is not straightforward, due to additional effects notusually accounted for, such as structural disorder, defects,electron-phonon interaction~i.e., polarons!, and temperature.We here model the polymer system as an ideal crystal, whichallows us to take into account the long-range, asymptoticeffects of packing, as well as to use a fullab initio calcula-tion method. Other approximate models have been used toextract TI’s for similar systems, the most prominent beingthe construction of molecular aggregates following experi-mental packing structures observed for crystals.30–32 In thatcase, only semiempirical HF techniques have been used ascalculation methods, mainly due to the difficulty of treatinglarge aggregates throughab initio methods. We remark that,although proved to be extremely reliable for the systems forwhich they were specifically parametrized, the HF tech-niques used are not guaranteed to describe nonbonded mo-lecular aggregates; the tests33 for electron transfer, for ex-ample, quoted in Refs. 30 and 32, actually apply to on-chaintransfer across molecular bridges in donor-acceptor mol-ecules, that is, a single-bonded structure. Moreover, the TIvalues are taken from the relevant~e.g., HOMO! splittingsobtained in the aggregate, but extrapolated to the ideal crys-tal bandwidth assuming nearest-neighbor-only interactions;that is, the ideal crystal model is invoked at the end, and inan extremely limiting approximation. As we have shown, theNN approximation is not valida prori, in particular forp-stacked systems. Even for HB systems our results differqualitatively from the results of aggregate/NN models: tak-ing as an example the thiophene systems, the authors of Ref.32 find values of 60 and 82 meV, respectively, for the LT andHT packings ofT6 oligomers, to be compared with our val-ues of;10 and 50 meV, indicating a much more significantdifference in interchain interaction effects.
We next address the issue of polaronic effects on thetransport properties of anisotropic systems, which has beendiscussed over the years34–37 within the framework of Hol-stein’s model for molecular crystals.38 Regarding the isolatedchains, we could expect stabilization of large polarons~thatis, a carrier would be dressed by lattice distortions on a scaleof several lattice parameters or monomer lengths! if the TIalong the chainEi exceeds a value related to the energeticsof elastic displacements, namely
Ei@a2
K, ~22!
wherea refers to the short-range electron-lattice coupling,taken to be linear in nuclear displacements, andK is an ef-fective elastic constant representative of the material. Largepolarons are not expected however for 3D systems, and for
TABLE III. Calculated square norms for projection ofab initiocrystal eigenstates over the subspace generated by the ‘‘smart site’’choice of single-chain states. We report the minimum~first line! andmaximum~second line! values of the norms along the line in the BZ~see text!.
Projections pS-PPV HB-PPV PT1 PT2
Min 0.96460 0.98161 0.99044 0.99100Max 0.99602 0.99117 0.99757 0.99718
TABLE IV. Transfer integrals~in meV! calculated using quasi-direct computation method, with the NDO approximation. Nota-tions as in Table I.
pS-PPV HB-PPV PT1 PT2
E(P0) 120.63 27.54 54.40 9.03 E12(0)E(2P0) 10.50 0.27 10.31 10.50 E11(P1)E(3P0) 2.86 14.01 1.57 1.32 E11(P2)
AB INITIO STUDY OF TRANSPORT PARAMETERS IN . . . PHYSICAL REVIEW B69, 205205 ~2004!
205205-7

anisotropic systems such as polymer crystals Emin34 pro-posed a criterium for the stability of large polarons based onthe the ratio of intrachain to interchainE' TI’s:
Ei
E'
.12R2, R'4Ei
a2/K, ~23!
whereR is the ~adimensional! radius of the 1D polaron. Wedo not here calculate the elastic parameters, however, we canextract some information from the experimentally detected39
polaron radii for PPV and PT,R;2 and 5, respectively. Wefocused our attention on interchain TI’s, however, it is pos-sible to recast our definitions for intrachain interactions byconsidering as is usual each polymer chain as built from‘‘meric’’ orbitals ~as shown in Fig. 2, the chain HOMO bandcan be seen as derived from the mer states!. In this way weare able to estimate the nearest-neighbor intrachain TI in thesame inverse TB formulation, and compare them to the in-terchain TI’s. The dispersion along the chains can be esti-mated from the isolated chain,Ei5550 meV for PPV and;950 meV for PT. The so-obtained intrachain-interchain TIratios start from 4.5 forpS-PPV, increase to;20 for PT1and HB-PPV, and reach;100 for PT2, which are alwaysoutside the condition for polaron stability in Eq.~23!, as isthe case also fortrans-polyacetylene~TPA!. The unquestion-able existence of polarons in real samples was elegantly jus-tified by Mizes and Conwell,35 that obtain stabilization dueto disorder-induced finite-size effects~e.g., conjugationbreaks, and kinks!. In this spirit, polaron kinetics for TPA inquasi-1D aggregates has been recently investigated,36,37 andour values for TI’s can be valuable to similar studies in PPVand PT.
Recognizing the simplifications introduced in our model,we can still compare our results to existing experimentaldata. Many experimental studies clearly point to the impor-tant role of intermolecular interactions and structural orderfor the transport efficiency of organic samples. In particular,alkyl-substituted polythiophenes can also show solid-stateassembly withp-stacking between thiophene rings40–42 andthe corresponding regioregular poly~3-alkylthiophene!~P3AT! can be grown with very high ordering and self-orienting properties. The possibility of achieving very highmobilities with P3AT-based field-effect transistors wasdemonstrated,43–45suggesting that strongp-p interchain in-teractions enhance charge transport. This is qualitatively con-sistent with our results forp-stacked PPV, where we find amuch higher value for TI’s than any HB phase; moreover,our quantitative estimate for the TI’s are generally consistentwith mobilities of the order of few tenths of square centime-ter per volt second, as obtained for highly ordered samples ofP3AT.45
IV. SUMMARY AND CONCLUSIONS
In this paper we present a first-principles study of conju-gated polymer systems, focusing on different possibilities forthe calculation of transfer integrals between polymericchains. We formulated a tight-binding scheme for polymercrystals, identifying isolated-chain states as building blocks
for the electronic structure. This is a useful ansatz as it al-lows for deeper insight in the transport mechanisms of thesecomplex systems, where there is a strong interplay betweenconfinement~in the individual 1D chains! and delocalization~in the 3D structure!. We also present a study of the neglectof differential/diatomic overlap~NDO! approximation forthese chain states.
We apply the suggested methods to polypara-phenylene-vinylene and polythiophene, in different crystalline symme-tries: p-stacks~PPV! and herringbone~PPV and PT!. Forthese polymers, there is a clear separation between thesingle-chain HOMO band and the lower valence bands in theenergy range of interest. We have found that this energyseparation exists in the crystal if it also exists in the corre-sponding single-chain states or groups of states, as is the casehere.
Turning to specific results for the studied polymer crys-tals, we find that ap-stacked chain configuration, as inpS-PPV, leads to TI at least twice larger than any othercoupling in the analyzed structures. The strong coupling canin part be ascribed to the closer packing between neighborchains achieved in that symmetry: this result is in contrast tothe usual assumption that, inp-stacked films, chains are iso-lated due to lateral separation between stacks. In the case ofHB symmetry we always find that interaction betweennearest-neighbor inequivalent chains is the dominant contri-bution, despite the large interchain angles. These resultsshould help choosing growth and deposition techniques toobtain desired film properties.
ACKNOWLEDGMENTS
This work was supported in part by the RTN EU Contract‘‘EXCITING’’ ~Contract No. HPRN-CT-2002-00317! and byMIUR ~Italy! through FIRB ‘‘NOMADE;’’ M.J.C. acknowl-edges support from FAPESP, CNPq, and MCT~Brazil!.
APPENDIX: SYMMETRY PROPERTIES OF HB CRYSTALS
In HB crystals the symmetry operations are the spatialinversion J and the translation-rotationS taking one of theinequivalent chains in the other:
@ J,H#50, @S,H#50. ~A1!
Another property is that also isolated chains have the inver-sion symmetry, and that the inversion center for the crystalcoincides with that for the isolated chain. We put the inver-sion center of one of the equivalent chains, labeled byi51, in the origin of the reference frame so that@see Fig.1~a!#
Jufmkz ,Pa
1 &5ufm2kz ,2Pa
1 &, ~A2!
Jufmkz ,Pa
2 &5ufm2kz ,2Pa2P12P2
2 &. ~A3!
We can associateS to $R,t%, whereR is a point-group rota-tion andt a translation vector:
FERRETTI, RUINI, BUSSI, MOLINARI, AND CALDAS PHYSICAL REVIEW B69, 205205 ~2004!
205205-8

^r uSuf&5f~R21r2t!. ~A4!
For these HB crystals,S can be associated toR5s1, thereflection relative to theyz plane~orthogonal toP1), and thefractional translationt can be chosen to be either1
2 (P11P2) or 1
2 (P11P21c) ~wherec is the lattice vector alongchains! depending on the particular symmetry details.
We first consider the former case in whichc is not in-volved in t; thus, for the genericPa5n1P11n2P2 we canwrite
Sufmkz ,n1P11n2P2
1 &5ufmkz ,2n1P11n2P2
2 &, ~A5!
Sufmkz ,n1P11n2P2
2 &5ufmkz ,2n1P11(n211)P2
1 &. ~A6!
We need the relations between the four nearest neighborsinequivalent TI’s,
^fmkz,01 uHufmkz ,2P12P2
2 &5Em1,m2kz ~2P12P2!, ~A7!
^fmkz,01 uHufmkz ,2P1
2 &5Em1,m2kz ~2P1!, ~A8!
^fmkz,01 uHufmkz ,2P2
2 &5Em1,m2kz ~2P2!, ~A9!
^fmkz,01 uHufmkz,0
2 &5Em1,m2kz ~0!. ~A10!
In the following, we will drop the band indexm to shortenthe notation. Using all the symmetry operations definedabove it is possible to obtain the desired relations:
E1,2kz ~2P1!5^fkz,0
1 uS†HSufkz ,2P1
2 &5^fkz,02 uHufkz ,P11P2
1 &
5^fkz ,P11P2
1 uHufkz,02 &* 5E
1,2kz* ~2P12P2!,
~A11!
E1,2kz ~0!5^fkz,0
1 uS†HSufkz,02 &5^fkz,0
2 uHufkz ,1P2
1 &
5^fkz ,P2
1 uHufkz,02 &* 5E
1,2kz* ~2P2!, ~A12!
E1,2kz ~0!5^fkz,0
1 uJ†HJufkz,02 &5^f2kz,0
1 uHuf2kz ,2P12P2
2 &
5^fkz,01 uHufkz ,2P12P2
2 &* 5E1,2kz* ~2P12P2!.
~A13!
In summary,
E1,2kz ~0!5E1,2
kz* ~2P12P2!, ~A14!
E1,2kz ~0!5E1,2
kz* ~2P2!, ~A15!
E1,2kz ~0!5E1,2
kz ~2P1!. ~A16!
In the case of the fractional translationt5 12 (P11P21c),
Eq. ~A6! must be modified to
Sufmkz ,n1P11n2P 2
2 &5e2 ikzcufmkz ,2n1P11(n211)P2
1 &~A17!
becauseS2 is equivalent to a lattice translation with an inte-ger c component, while Eq.~A5! remains unchanged. Thisleads to a modified version of the previous results:
E1,2kz ~0!5E
1,2kz* ~2P12P2!, ~A18!
E1,2kz ~0!5e2 ikzcE
1,2kz* ~2P2!, ~A19!
E1,2kz ~0!5eikzcE1,2
kz ~2P1!. ~A20!
Returning to our systems, HB-PPV admits just one sym-metry operation likeS ~a reflection with respect to theyzplane! with a fractional translation of the first type, andtherefore Eqs.~A14!–~A16! are valid. Moreover, it is pos-sible to prove that NN inequivalent chain TI’s are all real andequal by considering inversion and time-reversal symmetrytogether with the NN approximation. From these results Eq.~15! is immediately proved.
Because of their orthorombic unit cell, PT1 and PT2 havea higher-symmetry group with two different operations of thesame type ofS ~one related to a reflection with respect to theyz and the other to thexz plane! where the fractional trans-lation is of the first type for one operation and of the secondtype for the other one. Using Eqs.~A14!–~A16! and Eqs.~A18!–~A20!, one can show that TI’s are again all equal.Also in this case, it is possible to demonstrate that Eq.~15!holds.
*Email address: [email protected]. Burroughes, D.D.C. Bradley, A.R. Brown, R.N. Marks, K.
Mackay, R.H. Friend, P.L. Burns, and A.B. Holmes, Nature~London! 347, 539 ~1990!.
2R. Friend, R. Gymer, A. Holmes, J. Burroughes, R. Marks, C.Taliani, D. Bradley, D.D. Santos, J. Bre´das, and M. Lo¨gdlung,Nature~London! 397, 121 ~1999!.
3M. Ahlskog, M. Reghu, A.J. Heeger, T. Noguchi, and T. Ohnishi,Phys. Rev. B53, 15 529~1996!.
4T.Q. Nguyen, R.C. Kwong, M.E. Thompson, and B.J. Schwartz,
Appl. Phys. Lett.76, 2454~2000!.5D. Chen, M.J. Winokur, M.A. Masse, and F.E. Karaz, Polymer
33, 3116~1992!.6C.Y. Yang, F. Hide, M.A. Dı´az-Garcı´a, A.J. Heeger, and Y. Cao,
Polymer39, 2299~1998!.7D. Chen, M.J. Winokur, M.A. Masse, and F.E. Karasz, Phys. Rev.
B 41, 6759~1990!.8A. Ferretti, A. Ruini, E. Molinari, and M.J. Caldas, Phys. Rev.
Lett. 90, 086401~2003!.9R. Landauer, Philos. Mag.21, 863 ~1970!.
AB INITIO STUDY OF TRANSPORT PARAMETERS IN . . . PHYSICAL REVIEW B69, 205205 ~2004!
205205-9

10M. Buttiker, Y. Imry, R. Landauer, and S. Pinhas, Phys. Rev. B31, 6207~1985!.
11M. Buttiker, Phys. Rev. Lett.57, 1761~1986!.12P.A. Schulz, D.S. Galva˜o, and M.J. Caldas, Phys. Rev. B44, 6073
~1991!.13R. Hey, F. Gagel, M. Schreiber, and K. Maschke, Phys. Rev. B55,
4231 ~1997!.14R.A. Marcus and N. Sutin, Biochim. Biophys. Acta811, 265
~1985!.15C. Ambrosch-Draxl, J.A. Majewski, P. Vogl, and G. Leising,
Phys. Rev. B51, 9668~1995!.16M. Rohlfing and S.G. Louie, Phys. Rev. Lett.82, 1959~1999!.17A. Ruini, M.J. Caldas, G. Bussi, and E. Molinari, Phys. Rev. Lett.
88, 206403~2002!.18D.S. Galvao, D.A. dos Santos, B. Laks, C.P. de Melo, and M.J.
Caldas, Phys. Rev. Lett.63, 786 ~1989!.19J.C. Slater and G.F. Koster, Phys. Rev.94, 1498~1954!.20P. Puschnig and C. Ambrosch-Draxl, Synth. Met.119, 245~2001!.21T. Siegrist, C. Kloc, R.A. Laudise, H.E. Katz, and R.C. Haddon,
Adv. Mater.10, 379 ~1998!.22See, e.g., Ref. 20 and references therein.23See S. Baroni, A. Dal Corso, S. de Gironcoli, and P. Giannozzi,
http://www.pwscf.org24G. Bussi, A. Ruini, E. Molinari, M.J. Caldas, P. Puschnig, and C.
Ambrosch-Draxl, Appl. Phys. Lett.80, 4118~2002!.25A. Ruini, G. Bussi, A. Ferretti, M.J. Caldas, and E. Molinari,
Synth. Met.139, 755 ~2003!.26M.J.S. Dewar, E.G. Zoebish, E.F. Healy, and J.J.P. Stewart, J. Am.
Chem. Soc.107, 3902~1985!.27J. Ridley and M. Zerner, Theor. Chim. Acta32, 111 ~1973!.
28L.Y.A. Davila and M.J. Caldas, J. Comput. Chem.23, 1135~2002!.
29M.A. Santos and L. Y. A. Da´vila ~unpublished!.30J. Cornil, J.P. Calbert, D. Beljonne, R. Silbey, and J.L. Bre´das,
Synth. Met.119, 1 ~2001!.31J.L. Bredas, J.P. Calbert, D.A. da Silva Filho, and J. Cornil,
PNAS 99, 5804~2002!.32J.L. Bredas, D. Beljonne, J. Cornil, J.P. Calbert, Z. Shunai, and R.
Silbey, Synth. Met.125, 107 ~2002!.33M.D. Newton, Int. J. Quantum Chem.77, 255 ~2000!.34D. Emin, Phys. Rev. B33, 3973~1986!.35H.A. Mizes and E.M. Conwell, Phys. Rev. Lett.70, 1505~1993!.36Å. Johansson and S. Stafstro¨m, Phys. Rev. Lett.86, 3602~2001!.37Å. Johansson and S. Stafstro¨m, Phys. Rev. B66, 085208~2002!.38T. Holstein, Ann. Phys.8, 325 ~1959!.39K. Marumoto, N. Takeuchi, and S. Kuroda, Chem. Phys. Lett.
382, 541 ~2003!.40R.D. McCullogh, S. Tristram-Nagle, S.P. Williams, R.D. Lowe,
and M. Jayaraman, J. Am. Chem. Soc.115, 4910~1993!.41T.A. Chen, X. Wu, and R.D. Rieke, J. Am. Chem. Soc.117, 233
~1995!.42T.J. Prosa, M.J. Winokur, J. Moulton, P. Smith, and A.J. Heeger,
J. Am. Chem. Soc.51, 159 ~1995!.43H. Sirringhaus, N. Tessler, and R.H. Friend, Science280, 1741
~1998!.44H. Sirringhaus, P.J. Brown, R.H. Friend, M.M. Nielsen, K. Bech-
gaard, B.M.W. Langeveld-Voss, A.J.H. Spiering, R.A.J. Janssen,E.W. Meijer, and P. Herwig, Nature~London! 401, 685 ~1999!.
45G. Wang, J. Swensen, D. Moses, and A.J. Heeger, J. Appl. Phys.93, 6137~2003!.
FERRETTI, RUINI, BUSSI, MOLINARI, AND CALDAS PHYSICAL REVIEW B69, 205205 ~2004!
205205-10

Synthetic Metals 139 (2003) 755–757
Charge transport and radiative recombination in polythiophenecrystals: a first-principles study
A. Ruinia,b,∗, G. Bussia,b, A. Ferrettia,b, M.J. Caldasa,c, E. Molinaria,ba INFM National Center on nanoStructures and bioSystems at Surfaces (S3), Modena, Italy
b Dipartimento di Fisica, Università di Modena e Reggio Emilia, v. Campi 213/A, Modena 41100, Italyc Instituto de Física, Universidade de São Paulo, São Paulo, Brazil
Abstract
We investigate two phases of polythiophene crystals by means of first-principles calculations, focusing on the effect of the differentstructure on charge transport parameters and luminescence quantum yield. The resulting microscopic interpretation highlights the impactof solid-state interchain coupling on both transport and emissive properties of semiconducting polymer crystals.© 2003 Elsevier B.V. All rights reserved.
Keywords: Density-functional theory; Optical absorption; Charge transport; Polythiophene
Thiophene-based materials are very attractive organicsemiconductors due to their high chemical stability and un-common versatility[1]; in fact, oligothiophene films weresuccessfully exploited as active materials in field-effecttransistors[2], light-emitting diodes[3], photodiodes andplastic solar cells[4]. However, a full understanding ofthe microscopic mechanisms controlling device operationis still lacking, mainly due to the complex structure ofpolymer crystals: each chain has, on its own, characteristictransport (along the long chain axis) and optical properties,that may be affected in the film since the chains are weaklybound through van der Waals forces. Owing to the finiteconjugation length in real samples, charge transport prop-erties are strongly influenced by the interchain coupling.Light emission, on the other hand, depends on the existenceof a dipole-allowed transition from the lowest excited stateto the (singlet) ground state, which can be helped or ham-pered by the solid-state interaction between chains. In thispaper, we present a theoretical study of intrinsic transportparameters and luminescence efficiency in polythiophenecrystals, and discuss the relation between the particularchain arrangement in the solid state and optoelectronicperformance.
Structural analysis of unsubstituted oligothiophene crys-tals reveals a typical herring-bone chain arrangement, anddiffraction experiments[5] distinguished two different
∗ Corresponding author. Tel.:+39-059-2055300; fax:+39-059-367488.E-mail address: [email protected] (A. Ruini).
polymorphs depending on the source temperature (high,HT or low, LT) used during crystal growth. We adoptedthese structural data to build two different polymer crystals[6] having essentially the same chain arrangement of theHT and LT phases, which will be denoted PT1 and PT2in the following. In both cases the unit cell contains twochains in herring-bone geometry, and the only differenceconsists in a translation of the second chain relative tothe first by half a unit vector along the chain direction, asshown in the top panel ofFig. 1. This different environmentleads to a larger interchain charge density for PT1 than forPT2.
Single-particle energiesε and wavefunctionsψ were cal-culated from first-principles within the density-functionaltheory and the local density approximation[7], usingplane-waves basis sets and ab initio pseudopotentials. Theelectronic band structure for both crystals[6,8] is shown inFig. 1 for the energy gap region. We note that dispersionalong the chain direction (ΓZ and YD) follows closely thatof the isolated chain, and that the single-chain states are dou-bled due to the presence of two non-equivalent chains in theunit cell. There is noticeable splitting of the doublets alongdirections perpendicular to the chains, which also show anon-negligible dispersion, particularly for PT1. These aresignatures of interchain coupling. It is also important to re-mark that the topmost pair of valence and the lowest pair ofconduction bands originate, respectively, from the highestoccupied band (HOMO) and the lowest unoccupied band(LUMO) of the individual thiophene chains, with virtuallyno mixture from other states.
0379-6779/$ – see front matter © 2003 Elsevier B.V. All rights reserved.doi:10.1016/S0379-6779(03)00319-9

756 A. Ruini et al. / Synthetic Metals 139 (2003) 755–757
Fig. 1. Electronic band structure for PT1 (right) and PT2 (left); the insetsshow the crystalline packings and the Brillouin Zone.
We can investigate further the transport parameters forthese systems; in the case of polymer crystals we haveshown[9] that transfer integrals[10,11], reflecting the wholeband, are more sensitive to interchain geometry than effec-tive masses. Assuming the HOMO bandsφHkz (r −τ i) of theindividual chains (i = 1,2) to generate the valence-band-topdoublet, in a Slater–Koster[12] spirit, we use inverse dis-crete Fourier transforms of the band structures to obtain theintegralsEHkz
ij for coupling between the chains, across therelevant cell vectorsP in the directions orthogonal to thechains:
EHkzij (P ) =
∫d3r φ∗
Hkz (r − τ i)HφHkz (r − P − τj ) (1)
for thek-vectors corresponding to the crystal valence-bandtop. We compare results for PT1 and PT2 inTable 1, fornearest-neighbor chains (all the other integrals are muchsmaller): while values are generally very similar in the twostructures, we find a significant difference associated to thecharge transfer between the nearest-neighbor inequivalentchains, approximately five times larger in PT1. Our studydemonstrates quantitatively that PT1 is expected to showbetter transport properties than PT2, and that the favouredtransport path involves the symmetry-inequivalent chains.
Electroluminescence takes place via the radiative recom-bination of injected electron–hole pairs, which might oth-erwise decay through a nonradiative decay channel. Whensuch pairs are produced on isolated thiophene chains by, e.g.photoexcitation, excitons undergo fast radiative decay, withhigh quantum efficiency. The emissive properties of polymer
Table 1Transfer integrals for holes (HOMO states) in PT1 and PT2 (firstthree columns):E1,2, between nearest-neighbour inequivalent chains, andE(a)(E(b)), between equivalent chains alongx(y), (seeFig. 1)
EHkz12 (P ) E
Hkz11 (P x) E
Hkz11 (P y) ∆H ∆L ∆
PT1 54.6 10.3 1.3 430 210 60PT2 8.9 10.6 1.0 60 210 10
Last three columns report the HOMO (LUMO) band-splitting∆H (∆L ),and the exciton Davydov splitting∆. All energies in meV.
crystals are however known to depend strongly on symme-try and on interchain coupling: due to the doubling of theband levels, each excitonic level splits in the crystal in twoDavydov components[13], the lowest one being opticallyinactive (dark). It is crucial to establish the value of this en-ergy splitting (Davydov splitting,∆), compared the thermalenergykBT : In fact, if ∆ kBT then the two lowest states(active and dark) are practically degenerate and the photo-luminescence process maintains the same efficiency as forthe isolated molecule; otherwise excitations decay into thelowest available state (dark), which leads to non-radiativeemission. Also from the experimental side, an overall con-sensus about the value of∆ is still not achieved[14].
A proper theoretical description of optical excitationsmust go beyond the mean-field study based on the single-particle band structure concept, and requires the solution ofa many-body problem. To this end, we have implementedan ab initiodensity-matrix scheme[15,16], that providesthe exciton states by direct diagonalization of the two-bodySchrödinger equation:
(εeµ − εhν )Aµν +∑µ′ν′
Kµν′,νµ′Aµ′ν′ = ExAµν, (2)
whereK is the electron–hole interaction kernel, includingthe screened direct Coulomb interaction and the unscreenedexchange interaction; this is equivalent to the Bethe–Salpether equation within the Green’s function scheme[17].The solution ofEq. (2)provides the excitonic eigenenergiesEx and eigenstatesA.
Our calculations yield the excitonic spectra of PT1 andPT2, and a real-space analysis of all lowest excitonic states[8] reveals that the lowest active exciton isdirect, that is,confined to one chain (DE), as is also its (inactive) Davy-dov partner; interestingly, we find alsocharge-transferexcitons (CTE) with electron and hole on different neigh-boring chains, that are expected to be inactive due to thelow probability for radiative recombination. These CTE arestill below the single-particle gap, and always very close inenergy to the active DE, in particular for the case of PT1.
The analysis of the excitonic states can also provide infor-mation on the single-particle states that are mainly involvedin the four lowest excitations, as represented schematicallyin Fig. 2: the optically active states are basically producedfrom transitions between the topmost valence and secondconduction band, and between the second-top valence andfirst conduction band. We now consider in greater detailthe two lowest direct Davydov components: for both PT1and PT2 the dark exciton is lower in energy than the ac-tive one, in agreement with the observed quenching of lu-minescence in the solid state with respect to solutions. InTable 1we report our results for∆ in both crystal structures,compared to the single-particle band splittings for HOMOand LUMO states:∆H is larger in the first structure, dueto the stronger interchain coupling, and accordingly∆ isfound to be higher for PT1than for PT2; however, no gen-eral rule can be deduced to quantitatively extract Davydov

A. Ruini et al. / Synthetic Metals 139 (2003) 755–757 757
Fig. 2. Schematic representation of the contribution of different band-pairsto each excitonic transition. In the first (second) row results for thefirst four excited states of PT1 (PT2) structure are shown. Each boxcontains a schematic view of the two upper valence band and the twolowest conduction bands. The thickness of each vertical line representsthe contribution of that particular band-pair to the exciton (see text fordetails). The thick box indicates the optically active state (n = 3 for PT1,n = 2 for PT2).
splittings directly from single-particle eigenvalues. It is in-teresting to observe that∆ is abovekBT at room tempera-ture for PT1, and below for PT2; this indicates the presenceof an efficient non-radiative decay path in PT1, stronglyquenching the photoluminescence efficiency, while the lu-minescence properties of PT2 are preserved. On the otherhand, a large∆ is suitable for photoconductivity, as the de-cay of the exciton to an optically inactive state, where thecarrier-pair will stay trapped for a longer time, allows foreasier separation of electrons and holes giving rise to theelectrical signal. Our findings point therefore to PT1 as asuitable system to be exploited in photoconduction-baseddevices, also in view of its remarkable interchain transportproperties.
In conclusion, our study confirms that chain-packing ex-erts a considerable effect both on transport parameters andon the radiative properties of the excited states, and cantherefore be exploited to design materials with optimal per-formances.
Acknowledgements
RTN EU Contract “Exciting” No. HPRN-CT-2002-00317,CNR and MAE (Italy), FAPESP and CNPq (Brazil) aregratefully acknowledged.
References
[1] G. Gigli, et al., Appl. Phys. Lett. 72 (1998) 1013.[2] H. Sirringhaus, N. Tessler, R.H. Friend, Science 280 (1998) 1741.[3] R.N. Marks, F. Biscarini, R. Zamboni, C. Taliani, Europhys. Lett.
32 (1995) 523.[4] M. Granstrom, K. Petritsch, A.C. Arias, R.H. Friend, Synth. Met.
102 (1999) 957;J. Ackermann, C. Videlot, A. El Kassmi, Thin Solid Films 403(2002) 157.
[5] T. Siegrist, et al., Adv. Mater. 10 (1998) 379.[6] P. Puschnig, C. Ambrosch-Draxl, Synth. Met. 119 (2001) 245.[7] W. Kohn, L.J. Sham, Phys. Rev. 140 (1995) A1133;
S. Baroni, A. Dal Corso, S. de Gironcoli, P. Giannozzi, (2001),http://www.pwscf.org.
[8] G. Bussi, et al., Appl. Phys. Lett. 80 (2002) 4118;A. Ruini, et al., in: Radiation-Matter Intraction in Confined Systems,Soc. Italiana di Fisica, Bologna, 2002, p. 155.
[9] A. Ferretti, A. Ruini, M.J. Caldas, E. Molinari, Phys. Rev. Lett. 90,086401 (2003).
[10] R.A. Marcus, N. Sutin, Biochim. Biophys. Acta 811 (1985) 265;R. Landauer, Philos. Mag. 21 (1970) 863.
[11] J.L. Brédas, J.P. Calbert, D.A. da Silva Filho, J. Cornil, PNAS 99(2002) 5804.
[12] J.C. Slater, G.F. Koster, Phys. Rev. 94 (1954) 1498.[13] A.S. Davydov, Theory of Molecular Excitons, McGraw-Hill, New
York, 1962.[14] M. Muccini, et al., Phys. Rev. B 62 (2000) 6296;
W. Gebauer, et al., Chem. Phys. 227 (1998) 33;S. Möller, et al., Phys. Rev. B 61 (2000) 15749;F. Kouki, et al., J. Chem. Phys. 113 (2000) 385.
[15] U. Hohenester, Phys. Rev. B 64 (2001) 205305.[16] A. Ruini, M.J. Caldas, G. Bussi, E. Molinari, Phys. Rev. Lett. 88
(2002) 206403.[17] M. Rohlfing, S.G. Louie, Phys. Rev. Lett. 81 (1999) 2312;
M. Rohlfing, S.G. Louie, Phys. Rev. B 62 (2000) 4927.

2.2 The case of organic-metallic hybrid interfaces 35
2.2 The case of organic-metallic hybrid interfaces
In the main path to the integration of molecular systems into electronic de-vices the need of wiring molecules gained fundamental importance. There-fore large interest arose around the physics and chemistry of organo-metallicinterfaces formed by the adsorption of molecules onto metal electrodes [1].A second important class of findings demonstrated [2] that when molecularobjects adsorb on metallic or semiconducting surfaces they are sometimesable to self-organize and form ordered monolayers (self assembled mono-layers, SAM’s). This property has been further investigated in view of thepossible use of well ordered SAM’s as active layers in electronic devices. Thekey importance of these results largely contributed to further fuel the topicof hybrid interfaces.
Addressing the above issues, we applied ab initio surface science tech-niques to the case of mercapto-benzoxazole (MBO) molecule [3] adsorbedon Copper (100) surface, in conjunction with an experimental gropu in ourlaboratory [4]. This molecule belongs to the family of hetero-aromatic thiols.We first study the most stable configuration of the adsorbed SAM by meansof DFT total energy calculations. We focused on two different coverages anddetermine the most stable adsorption sites and molecular geometries. Ourfindings clearly indicate a chemisorption description of the interface withthe formation of strong molecule-surface bonds.
Then we proceed to analyze the electronic structure of the most sta-ble configurations. The latter study has been directly compared with highresolution angle-resolved photoemission electron-spectroscopy (HR-ARPES)data [4]. This combined experimental-theoretical analysis allows for a deepinsight in the electronic properties of the interface. One important issuehere is the alignment of molecule and metal electronic level after the in-terface is formed. Our results are presented in the first paper [5] of thissection. They are consistent with a bonding-antibonding picture [6] of theS-Cu hybrid states localized at the edge of the d Copper range. The forma-tion of molecule-metal hybrid states is detected. Their energy localizationin the antibonding S-Cu peak near Fermi energy is particularly interestingfor transport. In fact these states are mostly localized near the Sulfur atomsin the very interface region, thus connecting the metal environment to themolecular one. The specific discussion and comparison with experimentalresults can be found in the paper [5]. In the last work [7] more weight isgiven to the details of experimental techniques and related findings.
Bibliography
[1] C. Joachim, J. K. Gimzewski, and A. Aviram, Electronic using hybrid-molecular and mon-molecular devices, Nature 408, 541–548 (Nov. 2000).

36 Electronic structure from a transport point of view
[2] A. Ulman, Formation and structure of self-assembled monolayers,Chem. Rev. 96(4), 1533–1554 (June 1996).
[3] G. Contini, V. D. Castro, S. Stranges, R. Richter, and M. Alagia, Gas-phase photoemission study of 2-mercaptobenzoxazole, J. Phys. Chem. A104, 9675–9680 (Aug. 2000).
[4] C. Mariani, F. Allegretti, V. Corradini, G. Contini, V. D. Castro, C. Bal-dacchini, and M. G. Betti, Electronic band states of long-range orderedaromatic thione molecules assembled on Cu(100), Phys. Rev. B 66(11),115407 (Sept. 2002).
[5] A. Ferretti and R. Di Felice, Electron delocalization at the hybridaromatic-thiol/Cu(100) interface, Phys. Rev. B 70(11), 115412 (Sept.2004).
[6] B. Hammer and J. K. Norskov, Theory of Adsorption ad Surface Reac-tions, in Chemisorption and Reactivity of Supported Clusters and ThinFilms, edited by R. M. Lambert and G. Pacchioni, pages 285–351, KluwerAcademic Publishers, 1997.
[7] R. Di Felice, A. Ferretti, C. Mariani, M. Betti, C. Baldacchini, and V. DiCastro, Surface-science approach to the study of mercaptobenzoxazoleon Cu(111), Surf. Sci. 566–568, 579–584 (June 2004).

Electron delocalization at the hybrid aromatic-thiol/Cu„100… interface
Andrea Ferretti and Rosa Di FeliceINFM National Center on nanoStructures and bioSystems at SurfacessS3d and Dipartimento di Fisica,
Università di Modena e Reggio Emilia, Via Campi 213/A, 41100 Modena, Italy(Received 29 March 2004; revised manuscript received 26 May 2004; published 16 September 2004)
We present an in-depth investigation of the structural and electronic properties of aps232d mercaptoben-zoxazole(MBO) monolayer on the Cus100d surface by means of repeated supercell density functional theorysimulations. Our results show that the formation of the interface, with the molecular S headgroups lying atfour-fold coordination sites on the metal substrate, is a strongly exothermic reaction that brings an energy gainof 1.4 eV/molecule with respect to the free surface and gas-phase thione molecules. The electronic structure ofthe most stable atomic configuration is characterized by bonding and antibonding hybrid metal-moleculeelectron states obtained from coupling between thep orbitals of the S atoms and thed orbitals of the Cu atoms.A detailed assignment of the experimental photoemission peaks, which were revealed by recent measurements,can be traced on the basis of our computational findings. In addition, we are able to show that the depositionof MBO on Cus100d results in chemisorption rather than physisorption and to fix the relative position of themetal Fermi level with respect to the molecular levels of the highest occupied and lowest unoccupied molecu-lar orbitals.
DOI: 10.1103/PhysRevB.70.115412 PACS number(s): 73.20.At, 68.43.Bc, 68.43.Fg
I. INTRODUCTION
Thiol and mercapto functional groups ubiquitously foundin several modular[e.g., HS-sCH2dn-CH3, HS-sC6H4dn-H,with variablen giving the modular chains] organic moleculesare widely exploited as anchoring handles to form hybridorganic-inorganic interfaces for nanotechnologyapplications.1–7Whereas alkanethiols are mostly used for thispurpose,8–10aromatic thiols may represent an attractive alter-native to enhance the electronic coupling between the metaland molecules, because of charge conjugation in the molecu-lar rings.11 In fact, one may expect the carrier mobility to befacilitated through aromatic poly-rings with respect to alkylicchains(once electrons are injected into the molecular com-ponent of the hybrid system through the contact junction).Despite this potential improvement, the use of thiols as “al-ligator clips”12–14 for capturing macromolecules onto metalsubstrates15,16 has been limited thus far to alkylic chains,whereas the study of aromatic thiols has been focused onshort benzene-dithiols between two metal pads or inbreakjunctions.17,18
Few theoretical investigations of benzene-thiol on a goldsurface or benzene-dithiol between two gold surfaces eluci-dated the nature of the coupled metal-molecule orbitals19–21
at atomically resolved interfaces. However, to our knowl-edge, most of the experimental work in the characterizationof the junctions between aromatic thiols and metal supportshas been devoted to conductivity measurements andimaging17,18 rather than to an understanding of the funda-mental nature of the interface electron states, with few ex-ceptions based on electronic structure studies.22,23 Recently,a clear-cut surface science approach has been applied byMariani and co-workers to the investigation of 232 mercap-tobenzoxazole(MBO) overlayers on the Cus100d surface asa study case, to characterize long-range ordered structuresobtained in ultrahigh vacuum(UHV).24,25 A key finding of
their experiments was the identification of six photoemissionpeaks attributed to the presence of the molecules on the in-organic support and the measurement of in-plane band dis-persions for some of them. Despite the accuracy of theiranalysis, the nature and degree of hybridization between themolecule and metal around the edge of the Cu bands re-mained somehow puzzling. In particular, a photoemissionpeak with a binding energy of approximately 2.2 eV wasinterpreted as a purely molecular feature deriving from thehighest occupied molecular orbital(HOMO) of MBO local-ized on the aromaticp bonds. This assignment deviates fromthe simple description of the metal-molecule coupling interms of the Newns-Anderson model,7,26,27 according towhich the molecular HOMO couples through the sulfurporbitals to the metald orbitals, giving rise to a bonding peakbelow the d bands and an antibonding peak above thedbands. The presence of a native MBO HOMO(Ref. 24) wellabove the bonding energy range, fully localized on the aro-matic tail of the molecule withoutp-S hybridization andsubstantially unperturbed upon adsorption, is not explainedwithin this framework. A closer examination of the micro-scopic nature of the electron states and the band structure ofmercaptobenzoxazole, in both the gas-phase and adsorbedconfigurations, is required in order to explain this mismatch.
To clarify the open controversial issues, we have per-formed first-principles periodic supercell plane-wavepseudopotential calculations of the MBO/Cus100d interfacein the 1- and 0.5-monolayer(ML ) regimes. We optimized thesystem starting from several initial conditions and found themost stable configurations for both coverages. For the highcoverage which was the object of the experimental studies,24
we computed the ground-state electronic structure: throughthe analysis of the atom-projected density of states(PDOS)and of the wave function distributions we were able to iden-tify the exact character of the photoemission peaks. Our re-sults are in good agreement with the experimental findings
PHYSICAL REVIEW B 70, 115412(2004)
1098-0121/2004/70(11)/115412(7)/$22.50 ©2004 The American Physical Society70 115412-1

and give us more insight into the nature of the hybrid orbitalsand their delocalization at the molecule-metal junction. Thepartial deviation from the Newns-Anderson bonding-antibonding picture is due to the very nature of the aromaticmolecular orbitals.
II. COMPUTATIONAL METHOD
Our simulations were performed using periodic supercellplane-wave pseudopotential density functional theory(DFT),as implemented in thePWSCFcode.28 The PW91 exchange-correlation functional was adopted.29 The ion cores in theelectron-ion interaction were described by non-norm-conserving pseudopotentials30 for all the species except sul-fur, for which a norm-conserving pseudopotential wasused.31 All the pseudopotentials were consistently generatedemploying in the atomic calculations the same PW91exchange-correlation parametrization used later in the sur-face simulations. The plane-wave expansion of the electronicwave functions was truncated at a kinetic energy cutoff of25 Ry. The above technical specifications were proven suf-ficiently accurate for the study of other thiolated metalsurfaces.7,32 For a detailed discussion of the relative perfor-mance of the PW91 and other gradient-corrected DFT func-tionals(such as PBE) in the computation of adsorption ener-gies for thiol monolayers, the reader is referred to thespecialized literature.33 We note here that our conclusions(regarding the choice of the most stable structure and thenature of the mixed orbitals) would not be compromised by adifferent choice of functional.
The physical systems were modeled by repeated super-cells containing five(100) planes of copper, one MBO mol-ecule adsorbed at one of the two surfaces in the slab, and10 Å of vacuum included to inhibit spurious interactions be-tween neighboring replicas. As232d cell with 4 Cu atomsper plane was chosen to describe the 1-ML-coverage regime,whereas as2Î232Î2d cell with 8 Cu atoms per plane wasselected for the 0.5-ML-coverage regime. The simulations ofthe real systems consisted of structural optimizations tosample local minima of the total energy surface which cor-respond to equilibrium structures. All the atoms were relaxeduntil the atomic forces vanished within a required precisionof 0.03 eV/Å. Brillouin zone(BZ) sums were computed bysampling the two-dimensional(2D) BZ with a uniform 838 mesh of Monkhorst-Packk points.34 The surface energyof the clean Cus100d surface was converged with respect tothe slab thickness and BZ sampling, before dealing with thegeometries of the MBO overlayers.
In Fig. 1(a) [1(b)], we report the structure formula of mer-captobenzoxazole in the thiol(thione) conformation. It is ap-conjugated molecule with a planar structure. Two differenttautomers exist in nature:(i) in the thiol form, the S head-group is saturated by a H atom, as shown in Fig. 1(a), N=C7 is a double bond, and S-C7 is a single bond;(ii ) in thethione form, the S headgroup is unsaturated and the H atomis attached instead to the N atom in the pentagonal ring, asshown in Fig. 1(b), S=C7 is a double bond, and N-C7 is asingle bond. In the remainder of our paper, we present thecomputed interface formation energies with respect to the
thione-like MBO, which is found experimentally to be themost stable isomer:35 therefore, it is expected to be the gas-phase precursor for the overlayer attainment. Indeed, our cal-culations also predict that thione-MBO is more energeticallyfavorable than thiol-MBO by 0.4 eV/molecules.9.2 kcal/mold. Note that the two conformations are meaningless whenthe molecular overlayer is considered on the metal surface,because the H atom whose position discriminates betweenthem is lost upon contact with the substrate and the mol-ecules are adsorbed in the radical form shown in Fig. 1(c).
Figure 1(d) shows a top-view scheme of the simulationsupercell, with identification of the two lattice sites availablefor molecular adsorption, namely the two-fold(bridge) andthe four-fold(fcc) ones. Figure 1(e) shows a sideview of therelaxeds232d MBO monolayer on Cus100d, which we dis-cuss in detail in the next section.
FIG. 1. Simulation setup for MBO/Cus100d. The inset showsthe definition of the azimuthal angleu. n is the surface normal.(a),(b),(c) Structure formula of mercaptobenzoxazole(MBO) in thethiol, thione, radical forms, respectively.(d) Scheme of a 232 por-tion of a Cus100d surface(top view). Solid dots(open circles) in-dicate Cu atoms in the outermost(below-surface) layer. The shadedcircles in the low left corner represent the 131 periodicity of thesurface with cell parameter of 2.57 Å. The small black dots markthe two-fold and four-fold adsorption sites for sulfur.(e) Relaxedstructure for aps232d MBO monolayer on Cus100d, with the mol-ecules at four-fold sites: a 333 cell is shown. The total thickness ofthe supercell is about 26 Å and includes a 10-Å large vacuum re-gion. Black, white, and gray spheres represent Cu, S, and MBOatoms, respectively.
A. FERRETTI AND R. DI FELICE PHYSICAL REVIEW B70, 115412(2004)
115412-2

III. RESULTS AND DISCUSSION
A. Structure and energetics
We first performed a structural investigation to determinethe most favorable adsorption location and 2D lateral pack-ing, testing both two-fold and four-fold sites of the squareCus100d lattice and starting from different molecular orien-tations with respect to the surface normal and to the diagonalof the square unit cell. More specifically, we considered awide range of values for the azimuthal angleu defined inFig. 1 (e.g., the inclination of the S-C bond with respect tothe surface normal) and for the polar anglef defined in Fig.2 (e.g., the rotation of the whole MBO plane around theS-C bond). The results of our simulations indicate that forthe 1-ML coverage the two-fold bridge site is not even ametastable state for molecular adsorption: This is so regard-less of the orientation of the aromatic plane. We sampledinitial configurations with the S headgroup at the two-foldsite and the molecular tailgroup oriented either parallel to thesurface normal and along the cell diagonalsu=f=0°d or atseveral different polar and azimuthal angles: the moleculealways migrates towards the four-fold site. We thus concludethat in the 1-ML regime the S atom is coordinated with four
neighboring Cu atoms: The molecular axis is normal to thesurfacesu=0° ±5°d and the molecular plane is oriented veryclose to thes131d-cell diagonalsf=0° ±7°d. This configu-ration is shown in side view in Fig. 1(e) and in top view inFig. 2(a): it accomplishes as232d reconstruction containingone MBO radical in eachs232d cell of the square Cus100dlattice, and we propose it as the atomic model explaining theexperimental low-energy electron diffraction(LEED)findings.24 The relaxed S-Cu distance is 2.42 Å, typical ofthe thiolate S-metal bond. The stacking distance betweenneighboring aromatic planes is 3.6 Å[d1 in Fig. 2(a)]. Theadjacent planes are laterally shifted so as to minimize repul-sive interactions, whereas eclipsed planes are separated by adistance of 7.2 Å[d2 in Fig. 2(a)]. Although we are awarethat dispersion interactions between stacked planes are notwell reproduced in DFT simulations, we believe that suchinterplanar couplings are minimized in the described con-figuration with noneclipsed nearest neighbors. Hence, theformation energy is reliable to an accuracy of about1 kcal/mol. Since we suspected that the reorientation of themolecule perpendicular to the surface plane could bestrongly driven by the high lateral packing of the monolayer,which does not leave enough space for the conjugated planesto adjust parallel(or close to parallel) to the surface, weinvestigated the lower-coverage regime of 0.5 ML, with adensity of one MBO molecule in everys2Î232Î2d super-cell. This low-density simulation was also aimed at under-standing whether the S-Cu coordination number leading tothe highest energy gain(four-fold vs two-fold) could dependon molecular packing, as suggested by others for the simplemethanethiol.32,33,36A top view of the most favorable relaxedgeometry is shown in Fig. 2(b). Even in this case we foundthat the molecule does not adsorb at the two-fold site anddoes not like to lie at a high angle with the surface normal.Therefore, we conclude that at least in the 0.5–1-ML rangethe molecular assembly(including adsorption site and orien-tation) is determined by the local S-Cu bonding rather thanby the tailgroups, by maximizing the S coordination. Inter-estingly, the S headgroup of mercaptobenzoxazole interact-ing with a Cus100d surface behaves very differently from theS headgroups of methanethiols1 MLd (Ref. 32) and cysteines0.5 MLd (Refs. 7 and 27) interacting with a Aus111d sur-face. In fact, the former tends to assume the highest four-foldcoordination, whereas the latter prefer a lower two-fold co-ordination with the metal atoms.
We computed the formation energy of the most energeti-cally favorable interface configuration at 1-ML coverage ac-cording to the reaction
2MBOthione+ 2Cus100d → 2MBO/Cus100d + H2, s1d
assuming that molecular hydrogen is obtained as a reactionproduct. This is the most reasonable hypothesis on the basisof bond counting arguments and the important fact thatstrong chemical interactions occur between the S atom andthe metal. For this coupling to occur easily, S needs to be inthe radical form when it comes into contact with thesubstrate.7,27,32This is possible only if the S-C7 bond in Fig.1(a) is single(at variance with the thione molecular precur-
FIG. 2. Top-view images for the most stable configurations ofthe MBO/Cus100d interface in the 1-ML(a) and 0.5-ML (b) re-gimes. Only the MBO molecules and the Cu atoms in the outermostsurface plane are shown. Black spheres represent Cu atoms, grayspheres represent the MBO atoms; the S atoms are represented aswhite spheres(in the figure, S atoms are shadowed by the MBOaromatic planes, and only their bonds with Cu are visible as black-and-white sticks). The solid (dashed) square indicates the 232(2Î232Î2) cell. Lattice directions are indicated by arrows.p is theline projection of the aromatic plane onto the(100) lattice plane.The inset shows the definition of the polar anglef. (a) A 333replica of the 232 unit supercell is shown;d1=3.6 Å indicates thedistance between nearest-neighbor aromatic planes which are onlypartially overlapped;d2=7.2 Å indicates the distance betweensecond-neighbor aromatic planes which are eclipsed.(b) A 232replica of the 2Î232Î2 unit supercell is shown.
ELECTRON DELOCALIZATION AT THE HYBRID… PHYSICAL REVIEW B 70, 115412(2004)
115412-3
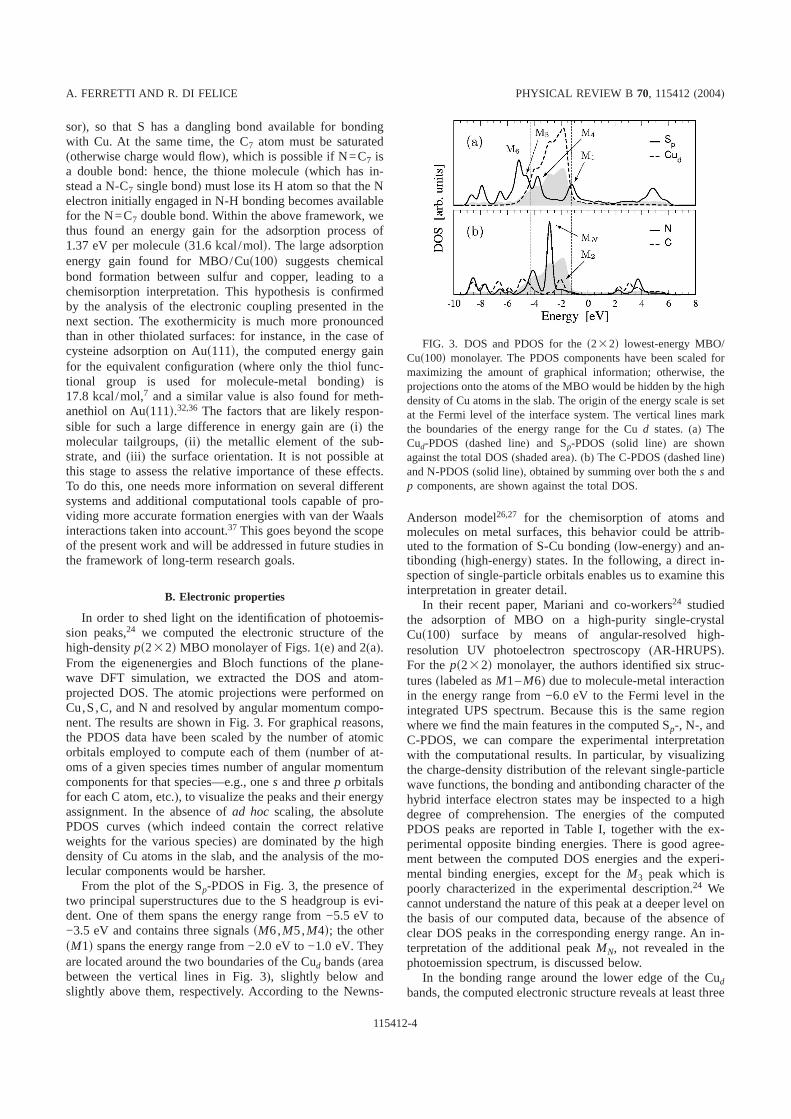
sor), so that S has a dangling bond available for bondingwith Cu. At the same time, the C7 atom must be saturated(otherwise charge would flow), which is possible if N=C7 isa double bond: hence, the thione molecule(which has in-stead a N-C7 single bond) must lose its H atom so that the Nelectron initially engaged in N-H bonding becomes availablefor the N=C7 double bond. Within the above framework, wethus found an energy gain for the adsorption process of1.37 eV per molecules31.6 kcal/mold. The large adsorptionenergy gain found for MBO/Cus100d suggests chemicalbond formation between sulfur and copper, leading to achemisorption interpretation. This hypothesis is confirmedby the analysis of the electronic coupling presented in thenext section. The exothermicity is much more pronouncedthan in other thiolated surfaces: for instance, in the case ofcysteine adsorption on Aus111d, the computed energy gainfor the equivalent configuration(where only the thiol func-tional group is used for molecule-metal bonding) is17.8 kcal/mol,7 and a similar value is also found for meth-anethiol on Aus111d.32,36 The factors that are likely respon-sible for such a large difference in energy gain are(i) themolecular tailgroups,(ii ) the metallic element of the sub-strate, and(iii ) the surface orientation. It is not possible atthis stage to assess the relative importance of these effects.To do this, one needs more information on several differentsystems and additional computational tools capable of pro-viding more accurate formation energies with van der Waalsinteractions taken into account.37 This goes beyond the scopeof the present work and will be addressed in future studies inthe framework of long-term research goals.
B. Electronic properties
In order to shed light on the identification of photoemis-sion peaks,24 we computed the electronic structure of thehigh-densityps232d MBO monolayer of Figs. 1(e) and 2(a).From the eigenenergies and Bloch functions of the plane-wave DFT simulation, we extracted the DOS and atom-projected DOS. The atomic projections were performed onCu,S,C, and N andresolved by angular momentum compo-nent. The results are shown in Fig. 3. For graphical reasons,the PDOS data have been scaled by the number of atomicorbitals employed to compute each of them(number of at-oms of a given species times number of angular momentumcomponents for that species—e.g., ones and threep orbitalsfor each C atom, etc.), to visualize the peaks and their energyassignment. In the absence ofad hocscaling, the absolutePDOS curves(which indeed contain the correct relativeweights for the various species) are dominated by the highdensity of Cu atoms in the slab, and the analysis of the mo-lecular components would be harsher.
From the plot of the Sp-PDOS in Fig. 3, the presence oftwo principal superstructures due to the S headgroup is evi-dent. One of them spans the energy range from −5.5 eV to−3.5 eV and contains three signalssM6,M5,M4d; the othersM1d spans the energy range from −2.0 eV to −1.0 eV. Theyare located around the two boundaries of the Cud bands(areabetween the vertical lines in Fig. 3), slightly below andslightly above them, respectively. According to the Newns-
Anderson model26,27 for the chemisorption of atoms andmolecules on metal surfaces, this behavior could be attrib-uted to the formation of S-Cu bonding(low-energy) and an-tibonding (high-energy) states. In the following, a direct in-spection of single-particle orbitals enables us to examine thisinterpretation in greater detail.
In their recent paper, Mariani and co-workers24 studiedthe adsorption of MBO on a high-purity single-crystalCus100d surface by means of angular-resolved high-resolution UV photoelectron spectroscopy(AR-HRUPS).For theps232d monolayer, the authors identified six struc-tures(labeled asM1–M6) due to molecule-metal interactionin the energy range from −6.0 eV to the Fermi level in theintegrated UPS spectrum. Because this is the same regionwhere we find the main features in the computed Sp-, N-, andC-PDOS, we can compare the experimental interpretationwith the computational results. In particular, by visualizingthe charge-density distribution of the relevant single-particlewave functions, the bonding and antibonding character of thehybrid interface electron states may be inspected to a highdegree of comprehension. The energies of the computedPDOS peaks are reported in Table I, together with the ex-perimental opposite binding energies. There is good agree-ment between the computed DOS energies and the experi-mental binding energies, except for theM3 peak which ispoorly characterized in the experimental description.24 Wecannot understand the nature of this peak at a deeper level onthe basis of our computed data, because of the absence ofclear DOS peaks in the corresponding energy range. An in-terpretation of the additional peakMN, not revealed in thephotoemission spectrum, is discussed below.
In the bonding range around the lower edge of the Cudbands, the computed electronic structure reveals at least three
FIG. 3. DOS and PDOS for thes232d lowest-energy MBO/Cus100d monolayer. The PDOS components have been scaled formaximizing the amount of graphical information; otherwise, theprojections onto the atoms of the MBO would be hidden by the highdensity of Cu atoms in the slab. The origin of the energy scale is setat the Fermi level of the interface system. The vertical lines markthe boundaries of the energy range for the Cud states.(a) TheCud-PDOS (dashed line) and Sp-PDOS (solid line) are shownagainst the total DOS(shaded area). (b) The C-PDOS(dashed line)and N-PDOS(solid line), obtained by summing over both thes andp components, are shown against the total DOS.
A. FERRETTI AND R. DI FELICE PHYSICAL REVIEW B70, 115412(2004)
115412-4

sharp peaks(M6,M5,M4 in Table I) in the Sp-PDOS[Fig.3(a), solid line] whose computed energies are −5.4,−4.6, and−3.8 eV. This is consistent with the concept that bondingwith the metal would occur through the molecular sulfurhook. In the same energy window(−5.5 to −3.5 eV), lesspronounced maxima are present also in the C- and N-PDOScurves: the overlap of peaks in the different PDOS compo-nents suggests that the bonding interface electron states arenot completely localized on sulfur as far as the molecularside of the MBO/Cu junction is concerned, but derive frommolecular orbitals which are hybrids between the headgroupand the aromatic rings(see Fig. 4). Examples of such bond-ing states contributing to theM6 peak are shown in Figs.5(a) and 5(b). In Fig. 5(a), the isosurface is maximum on theS atom, but also has somep-like character on the C atoms ofthe aromatic rings in the tailgroup; this orbital clearly stemsfrom one of the two degenerate HOMO’s of thione-MBOshown in Figs. 4(a) and 4(b)—namely, that withp character[Fig. 4(b)]. In Fig. 5(b) the conjugated contribution is stron-ger; this orbital derives from rehybridization of the twohighest-energy occupied orbitals of thiol-MBO in Figs. 4(e)and 4(f) or, alternatively, from the twop-like highest-energyoccupied orbitals of thione-MBO in Figs. 4(b) and 4(c), ex-cluding thes-like HOMO of Fig. 4(a). The latter is insteadresponsible for interface coupled orbitals of the kind shownin Fig. 5(c). This complex situation can be explained by thefact that, when a bond is formed with the metal, the HOMOtypical of the stable molecular phase is shifted to lower en-ergies and hybridizes with other molecular orbitals[Figs.4(d) and 4(h)] at this lower-energy range. Note that in thiol-MBO, where sulfur is saturated by hydrogen, thep-likeHOMO [Fig. 4(e)] is not degenerate and has density contri-butions on both the headgroup and tailgroup: This conditionis more similar but not completely equivalent to the interfaceenvironment, where the S atom becomes saturated by bond-ing with the metal atoms. Hence, the interface electron statesare reminiscent of the fact that the molecule impinges on thesurface with bothp and s sulfur p orbitals available, butrehybridize to account for metal-molecule coupling, thus re-sembling more closely the thiol than the thione molecularorbitals. As a final comment on the bonding energy region,we discuss the identification ofM4, which was left uncertainby the experiment.24 An early investigation attributed it tosulfur-copper coupling,38 whereas a more recent study25 re-lated it to a molecule-molecule interaction on the basis of itspresence also in the condensed MBO phase. Our results sup-
port the former interpretation, because it lies within thebonding energy window and is found to be a maximum inthe Sp-PDOS.
In the antibonding range around the upper edge of the Cudbands we recognize a peakM2 in the C-PDOS at −2.1 eV[Fig. 3(b), dashed line] and another peakM1 in the Sp-PDOSat −1.3 eV[Fig. 3(a), solid line]. Two electron states contrib-uting to M1 are shown as isosurface plots in Figs. 5(g) and5(h): They are clearly antibonding orbitals deriving from thetwo degenerate thione HOMO’s(p-like and s-like on sul-fur). Thus, our findings clearly confirm the assignment ofpeak M1 in the photoemission spectra by Mariani andco-workers24 as the antibonding fingerprint of the metal-MBO coupling. The slight energy shift that we find withrespect to the experimental binding energy is most likely tobe attributed to the lack of quasiparticle corrections in theDFT ground-state energy levels,39 which have been demon-strated to be important for bulk copper40 and Cu surfaces41,42
and are therefore expected to affect the interface states thathave a strong Cu contribution. Nonetheless, it is generallyaccepted43 that for systems such as the one we are investi-gating and for occupied states with a large molecular local-
TABLE I. The main peaks in the computed PDOS curves(lowerrow) are compared to the binding energies of features due tomolecule-surface interaction(upper row) from the integrated pho-toemission measurements of Ref. 24. All energies are in eV. Theexperimental binding energies are related to the absolute energieswith respect to the Fermi level(reported in this table) by a changeof sign.
M6 M5 M4 M3 MN M2 M1
Expt. −5.35 −4.75 −3.80 −3.40 — −2.23 −1.50
Theory −5.4 −4.6 −3.8 — −2.9 −2.1 −1.3
FIG. 4. Charge density plots of selected molecular orbitals forthe thione(a),(b),(c),(d) and thiol(e),(f),(g),(h) tautomers of MBO.(a),(b),(c) are the highest occupied levels for the thione,(e),(f),(g)for the thiol. (d) and (h) are not immediately below(c) and (g),respectively, but are shown because representative ofs-likeS-aromatic hybridization. The energies are in eV, relative to theFermi level of the adsorbate system, therefore comparable to thoseof Table I and of Fig. 5. Since the vacuum level is arbitrary in ourcalculation, to fix the energy scale we aligned the lowest-lying levelfrom the isolated thione, isolated thiol, and interface calculations. Infact this level pertains to a pure molecular orbital, identical in thethione and thiol forms and unaffected by adsorption. We use theorbitals of the free thione molecule to compare with the adsorbedphase(radical-MBO), because thione-MBO is the experimental gas-phase precursor, thus most representative to illustrate the inducedchanges. The highest occupied orbitals of the radical form are asthose in(a),(b),(c), because the S headgroup is similarly unsatur-ated. The occurrence of an unsaturated N in the adsorbed radical isrepresented by an orbital similar to that in panel(g) characteristic ofthiol-MBO.
ELECTRON DELOCALIZATION AT THE HYBRID… PHYSICAL REVIEW B 70, 115412(2004)
115412-5

ization, the DFT eigenvalues undergo smallk-point-dependent energy shifts. The assignment of theM2 phote-mission peak as due to the unaltered HOMO of MBO(Ref.24) was the most controversial with respect to the generalbonding-antibonding framework verified for thiolated metalsurfaces,27 as mentioned in the Introduction. By inspectingthe isosurface plots of computed electron states in the corre-sponding energy range, we are able to recognize the molecu-lar features that determine this peak. Two such plots areshown in Figs. 5(e) and 5(f). The former contains thes-likeS character of the thione HOMO, antibonding with copper,and hybridized with a lower-energy thione molecular orbital(at −1.72 eV, not shown in Fig. 4) that gives thep-likearomatic+C7 shape of Fig. 5(e). The latter fully resemblesthe aromatic component of the thiol HOMO but lacks S con-tribution. None of them is unambiguously equal to theHOMO of either MBO tautomers. Therefore, we concludethat even theM2 photoemission structure is due to the com-plex rehybridization of the molecule when it comes into con-tact with the inorganic substrate and contributes to the anti-bonding coupling, although shifted to energies lower thanthose of the main antibonding peakM1. However, we wishto point out that our interpretation does not contradict butrefines the experimental understanding forM2 and, in par-ticular, it supports the identification of a predominant aro-matic character.
At energies intermediate between the bonding and the an-tibonding range, from the N-PDOS[Fig. 3(b), solid line] anew sharp feature at −2.9 eV appears: it suggests weakmetal-molecule hybridization mainly localized on nitrogen inthe aromatic five-ring structure. One electron state contribut-ing to this peak is shown in the isosurface plot in Fig. 5(d).
This feature, which is due to the HOMO-2 of thiol-MBO[Fig. 4(g)], is probably hidden in the AR-HRUPS data be-cause it coincides with a strong Cu feature. The existence ofa sharp N-related peak below the S-Cu antibonding peak,with weak metal coupling, is comparable to a similar thi-olated interface studied recently—e.g., the cysteine/Aus111dwith cysteine binding through only the thiol functionalgroup.7,27
In Table II the Löwdin atomic charges onC,N,O, and Satoms are reported for the free molecule in both thione andthiol conformations, and for the radical MBO adsorbed onCus100d in the most favorable 232 reconstruction that wediscussed above. It is evident that important changes amongthe various phases occur only for the S atom, which loses0.55 electrons in passing from the gas-phase thione precursorto the adsorbed configuration at high coverage. However, adeeper inspection of the copper charges shows that only 0.22electrons are transferred to the Cu atoms of the two outer-most surface layers, whereas the remaining 0.33 electronsremain in the interface region.
Finally, the computed electronic structure allows us to fixthe energy shift between the molecular HOMO and the metalFermi level. We find that after rehybridization the Fermilevel lies 1.3 eV above the antibonding peak due to theHOMO and is much closer to the occupied than to the unoc-cupied states. This finding, which becomes even more evi-dent by taking into account the underestimation of DFT en-ergy gaps, is in good agreement with what occurs in othersimilar systems.7,19
IV. SUMMARY
We presented a DFT-based characterization of the struc-tural and electronic properties of an MBO monolayer on theCus100d surface. For this system, chemisorption is attainedvia a large energy gain of 1.4 eV/molecule. In addition to the
FIG. 5. Charge density plots of bonding and antibonding states,computed for the stable 232 MBO monolayer on Cus100d. The Cusubstrate lays in the lower part of each panel. The energies are ineV, relative to the Fermi level. The isosurface levels are arbitrary.(a), (b), and(c) are related to the low-energy bonding structures,(d)to the nitrogen localized structure at −2.9 eV,(e) and (f) to theantibondingshoulder M2, and(g) and (h) to the main antibondingpeakM1.
TABLE II. Atomic charges for the MBO molecule in the freeand adsorbed phases, computed after the Löwdin scheme. The ad-sorbed molecule is considered in the 232 monolayer structure.Although the absolute Löwdin charges must be taken with care,they can be used to estimate the variation of the charge state atdifferent atoms when a molecule undergoes a transition from thegas-phase to an adsorbed configuration.
Atom MBOthione MBOthiol MBO/Cus100d
S −0.15 +0.09 +0.40
N −0.26 −0.29 −0.24
O −0.20 −0.22 −0.18
Cs7d +0.23 +0.27 +0.26
Cs6d +0.12 +0.13 +0.13
Cs5d −0.17 −0.15 −0.14
Cs4d −0.14 −0.15 −0.15
Cs3d −0.15 −0.14 −0.14
Cs2d −0.15 −0.16 −0.15
Cs1d +0.23 +0.21 +0.21
A. FERRETTI AND R. DI FELICE PHYSICAL REVIEW B70, 115412(2004)
115412-6

usual bonding and antibonding orbitals arising from S-metalcoupling, the C-projected density of states reveals the pres-ence of a strongM2 peak mostly localized on the aromatictailgroup. M2 develops into a shoulder of the S-projectedantibonding peak, because of the specific coupling betweenthe S headgroup and the aromatic tailgroup in this molecule.In addition, a sharp feature due to N, which is not evident inphotoemission spectra, is found between the bonding and theantibonding peaks. We remark the excellent agreement withphotoemission data and the power of our computational ap-proach to complement experimental results and to affordmuch insight into the correct interpretation of the interfaceelectronic structure.
Finally, the metal Fermi level lies very close to the occu-pied molecular orbitals, and the molecule donates 0.55 elec-trons to the interface environment in the adsorption process.
ACKNOWLEDGMENTS
Elisa Molinari is acknowledged for inspiring this workand for a critical reading of the manuscript. We are gratefulto Carlo Mariani and his collaborators for insightful discus-sions. The work was supported by INFM through PRA-SINPROT and through the Parallel Computing Initiative forthe allocation of computing time at CINECA(Bologna), bythe EC through Contract No. IST-2000-28024, and by MIURthrough FIRB “NOMADE.”
1Y. Xia, J. A. Rogers, K. E. Paul, and G. M. Whitesides, Chem.Rev. (Washington, D.C.) 99, 1823(1999).
2R. M. Zimmermann and E. C. Cox, Nucleic Acids Res.22, 492(1994).
3A. Alessandrini, M. Gerunda, G. W. Canters, M. P. Verbeet, andP. Facci, Chem. Phys. Lett.376, 625 (2003).
4X. D. Cui, A. Primak, X. Zarate, J. Tomfohr, O. F. Sankey, A. L.Moore, T. A. Moore, D. Gust, G. Harris, and S. M. Lindsay,Science294, 571 (2001).
5C. Joachim, J. K. Gimzewski, and A. Aviram, Nature(London)408, 541 (2000).
6J. M. Tour, Acc. Chem. Res.33, 791 (2000).7R. Di Felice and A. Selloni, J. Chem. Phys.120, 4906(2004).8A. Ulman, Chem. Rev.(Washington, D.C.) 96, 1533(1996).9F. Schreiber, Prog. Surf. Sci.65, 151 (2000).
10R. M. Metzger, Chem. Rev.(Washington, D.C.) 103, 3803(2003).
11A. Ferretti, A. Ruini, E. Molinari, and M. J. Caldas, Phys. Rev.Lett. 90, 086401(2003).
12L. Jones and J. M. Tour, Polym. Prepr.(Am. Chem. Soc. Div.Polym. Chem.) 36, 233 (1995).
13J. M. Seminario, C. E. De La Cruz, and P. A. Derosa, J. Am.Chem. Soc.123, 5616(2001).
14M. L. Chabinyc, X. Chen, R. E. Holmlin, H. Jacobs, H. Skulason,C. D. Frisbie, V. Mujica, M. A. Ratner, M. A. Rampi, and G. M.Whitesides, J. Am. Chem. Soc.124, 11 730(2002).
15D. P. Allison, L. A. Bottomley, T. Thundat, G. M. Brown, R. P.Woychik, J. J. Schrick, K. B. Jacobson, and R. J. Warmack,Proc. Natl. Acad. Sci. U.S.A.89, 10 129(1992).
16E. Braun, Y. Eichen, U. Sivan, and G. Ben-Yoseph, Nature(London) 391, 775 (1988).
17M. A. Reed, C. Zhou, C. J. Muller, T. P. Burgin, and J. M. Tour,Science278, 252 (1997).
18J. Reichert, R. Ochs, D. Beckmann, H. B. Weber, M. Mayor, andH. v. Löhneysen, Phys. Rev. Lett.88, 176804(2002).
19Y. Xue, S. Datta, and M. Ratner, J. Chem. Phys.115, 4292(2001).
20S. Piccinin, A. Selloni, S. Scandolo, R. Car, and G. Scoles, J.Chem. Phys.119, 6729(2003).
21J. Taylor, M. Brandbyge, and K. Stokbro, Phys. Rev. Lett.89,138301(2002).
22P. A. Agron, T. A. Carlson, W. B. Dress, and G. L. Nyberg, J.
Electron Spectrosc. Relat. Phenom.42, 313 (1987).23W. Shen, G. L. Nyberg, and J. Liesegang, Surf. Sci.298, 143
(1993).24C. Mariani, F. Allegretti, V. Corradini, G. Contini, V. D. Castro,
C. Baldacchini, and M. G. Betti, Phys. Rev. B66, 115407(2002).
25V. D. Castro, F. Allegretti, C. Baldacchini, M. G. Betti, G. Con-tini, V. Corradini, F. Lamastra, and C. Mariani, Surf. Sci.507–511, 7 (2002).
26B. Hammer and J. K. Norskov, inChemisorption and Reactivityof Supported Clusters and Thin Films, edited by R. M. Lambertand G. Pacchioni(Kluwer Academic, Dordrecht, 1997), pp.285–351.
27R. Di Felice, A. Selloni, and E. Molinari, J. Phys. Chem. B107,1151 (2003).
28S. Baroni, A. Dal Corso, S. de Gironcoli, and P. Giannozzi, 2001,http://www.pwscf.org
29J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R.Pederson, D. J. Singh, and C. Fiolhais, Phys. Rev. B46, 6671(1992).
30D. Vanderbilt, Phys. Rev. B41, 7892(1990).31N. Troullier and J. L. Martins, Phys. Rev. B43, 1993(1991).32M. C. Vargas, P. Giannozzi, A. Selloni, and G. Scoles, J. Phys.
Chem. B 105, 9509(2001).33J. Gottschalck and B. Hammer, J. Chem. Phys.116, 784 (2002).34H. J. Monkhorst and J. D. Pack, Phys. Rev. B13, 5188(1976).35G. Contini, V. D. Castro, S. Stranges, R. Richter, and M. Alagia,
J. Phys. Chem. A104, 9675(2000).36H. Grönbeck, A. Curioni, and W. Andreoni, J. Am. Chem. Soc.
122, 3839(2000).37A. J. Misquitta, B. Jeziorski, and K. Szalewicz, Phys. Rev. Lett.
91, 033201(2003).38D. T. Ling, J. N. Miller, D. L. Weissman, P. Pianetta, P. M. Stefan,
I. Lindau, and W. E. Spicer, Surf. Sci.124, 175 (1983).39L. Hedin and S. Lundqvist, Solid State Phys.23, 1 (1969).40A. Marini, G. Onida, and R. Del Sole, Phys. Rev. Lett.88,
016403(2002).41C. Baldacchini, L. Chiodo, F. Allegretti, C. Mariani, M. G. Betti,
P. Monachesi, and R. Del Sole, Phys. Rev. B68, 195109(2003).42C. Baldacchini, L. Chiodo, F. Allegretti, C. Mariani, M. G. Betti,
P. Monachesi, and R. Del Sole, Phys. Rev. B69, 129901(2004).43M. Rohlfing and S. G. Louie, Phys. Rev. B62, 4927(2000).
ELECTRON DELOCALIZATION AT THE HYBRID… PHYSICAL REVIEW B 70, 115412(2004)
115412-7

Surface-science approach to the studyof mercaptobenzoxazole on Cu(1 0 0)
R. Di Felice a,*, A. Ferretti a,b, C. Mariani a,c, M.G. Betti c,C. Baldacchini c, V. Di Castro d
a INFM Center for nanoStructures and bioSystems at Surfaces (S3),
c/o Dipartimento di Fisica Via Campi 213A, 41100 Modena, Italyb Dipartimento di Fisica, Universita di Modena e Reggio Emilia, 41100 Modena, Italyc INFM and Dipartimento di Fisica, Universita di Roma ‘‘La Sapienza’’, Roma, Italy
d Dipartimento di Chimica and INFM, Universita di Roma ‘‘La Sapienza’’, Roma, Italy
Available online 9 June 2004
Abstract
We present a combined approach, based on photoemission experiments and DFT calculations, to the study of the
adsorption of aromatic organic molecules on metal substrates. Our main purpose was to characterize the interface
electronic properties and to search for electron states delocalized between the substrate and the molecular overlayer. We
demonstrate how the interplay between theory and experiment, within a surface-science framework, is a powerful tool
to gain insight into these issues. In particular, the computational results allow us to give a microscopic characterization,
in terms of relative molecule–substrate coupling, of the photoemission peaks.
2004 Elsevier B.V. All rights reserved.
Keywords: Density functional calculations; Angle resolved photoemission; Chemisorption; Aromatics; Low index single crystal
surfaces
1. Introduction
Organic molecules with a sulfur head-group are
attracting considerable interest because of theirwide use in nanotechnology-related fields, like
surface patterning and functionalization [1] and
molecular electronics [2]. Among the several
interesting applications, the strong affinity of sul-
fur to different metals can be exploited to form
contacts, to link other species to a supporting
metallic surface, or to form well-ordered self-
assembled monolayers (SAMs) [3]. Among the
huge variety of SAMs, particular attention hasbeen devoted to layers of alkanethiols and dialkyl
disulfides on Au(1 1 1) [4,5], to investigate the
adsorption site of the S head-group [4] and the
chemical reactions involved in the adsorption
process (dissociation of S–S bonds, sulfur–sub-
strate interactions). An interesting alternative is
represented by aromatic thiolate layers whose
molecular tail contains aromatic p-rings.Despite the growing interest in such systems,
still open questions remain about the intimate
*Corresponding author. Tel.: +39-059-2055320; fax: +39-
059-374794.
E-mail address: [email protected] (R. Di Felice).
0039-6028/$ - see front matter 2004 Elsevier B.V. All rights reserved.
doi:10.1016/j.susc.2004.05.111
Surface Science 566–568 (2004) 579–584
www.elsevier.com/locate/susc

molecule-to-substrate interface properties, the
evolution of the molecular levels (formation of
extended band states, bandwidth), the conduction
mechanisms at the molecular scale, and the
microscopic control of the transport properties.
Moreover, it is not clear how and to which extentall such issues depend on the specific molecular
tail-group (aromatic versus alkylic) and its possi-
ble functionalization (addition of a reactive group
or insertion of a hetero-atom in the aromatic
rings).
In this article, we show the results of a combined
experimental and theoretical investigation of the
electronic properties at an organic–inorganicinterface selected as a relevant study case, i.e. the
adsorption of the heterocyclic aromatic mercapto-
benzoxazole (MBO, chemical formula C7H5NOS)
on Cu(1 0 0). We discuss the experimental photo-
emission spectra in light of the computed band-
structure and electron states. This comparative
analysis allows us to interpret the chemisorption
mechanism in terms of S–Cu bonding and anti-bonding hybridization, similarly to what happens
for isolated sulfur atoms [6] and for non-aromatic
molecules [5] on close-packed metal surfaces, and
to identify the molecular orbitals from which the
photoemission peaks take origin, attaining a more
refined and complete statement of the previous
interpretation [7].
2. Experimental and theoretical methods
The experiments were performed in a UHV
chamber containing an angular resolved high
resolution ultraviolet photoelectron (ARUPS)
spectroscopy apparatus, low-energy electron dif-
fraction (LEED), and facilities for sample prepa-ration [7]. Photoelectron spectra were excited with
a high intensity He discharge lamp (HeIa and
HeIIa photons, hm ¼ 21:218 and 40.814 eV,
respectively). Angular-integrated spectra were
taken with an integration angle of about ±6 withrespect to the normal emission direction. The en-
ergy resolution, determined on the Cu Fermi level
(EF), was 16 meV. Calibration of the binding en-ergy (BE) scale was carried out in the adsorbed
systems, using the Cu Fermi edge at 0 eV.
The Cu(1 0 0) single crystal substrate was
repeatedly cleaned by a series of sputtering–
annealing cycles to remove surface contaminants,
and its quality was then tested by Auger spec-
troscopy and LEED. MBO was supplied by Al-
drich Chemical Company Inc. in the form ofpowder, with 98.4% purity. The main impuri-
ties were removed before measurements by re-
peated sublimation cycles. MBO depositions
were carried out at vapor pressure around 106 Pa.
The theoretical investigation consisted of first-
principle atomistic calculations using the state-
of-the-art plane-wave periodic DFT machinery
within the repeated supercell approach [8]. Gra-dient corrections were included in the exchange-
correlation functional in the PW91 formulation
[9]. The positions of all the atoms in the super-
cells were relaxed in the potential energy deter-
mined by the full quantum mechanical electronic
structure, until the forces vanished within a
precision of 0.03 eV/A. The electron–ion inter-
action in the DFT total energy functional wasdescribed by non-norm-conserving pseudopoten-
tials [10] for all the species except S, which
was instead represented by a norm-conserving
pseudopotential [11]. The electron wavefunctions
were expanded in a plane-wave basis-set up to a
kinetic energy cutoff of 25 Ry. The thiolated
surfaces were simulated with supercells having a
2 · 2 periodicity, containing a slab of five Culayers with four atoms each, one MBO molecule
adsorbed at one surface in the slab, and a vac-
uum thickness of 12 A to avoid spurious inter-
actions between neighboring replicas: the chosen
configurations correspond to the system at satu-
ration coverage. Ten special k points in the
irreducible wedge were included in Brillouin zone
(BZ) sums. The computational details weretested on isolated MBO molecules and Cu(1 0 0)
free surfaces, and on similar thiolated systems
[4,5].
In the experimental results the energies are re-
ported as binding energies and therefore they are
positive. In the computational results the energies
are relative to EF and therefore they are negative.
The calculated energies are related to the BE’s byjust a change of sign (except for the computational
error).
580 R. Di Felice et al. / Surface Science 566–568 (2004) 579–584
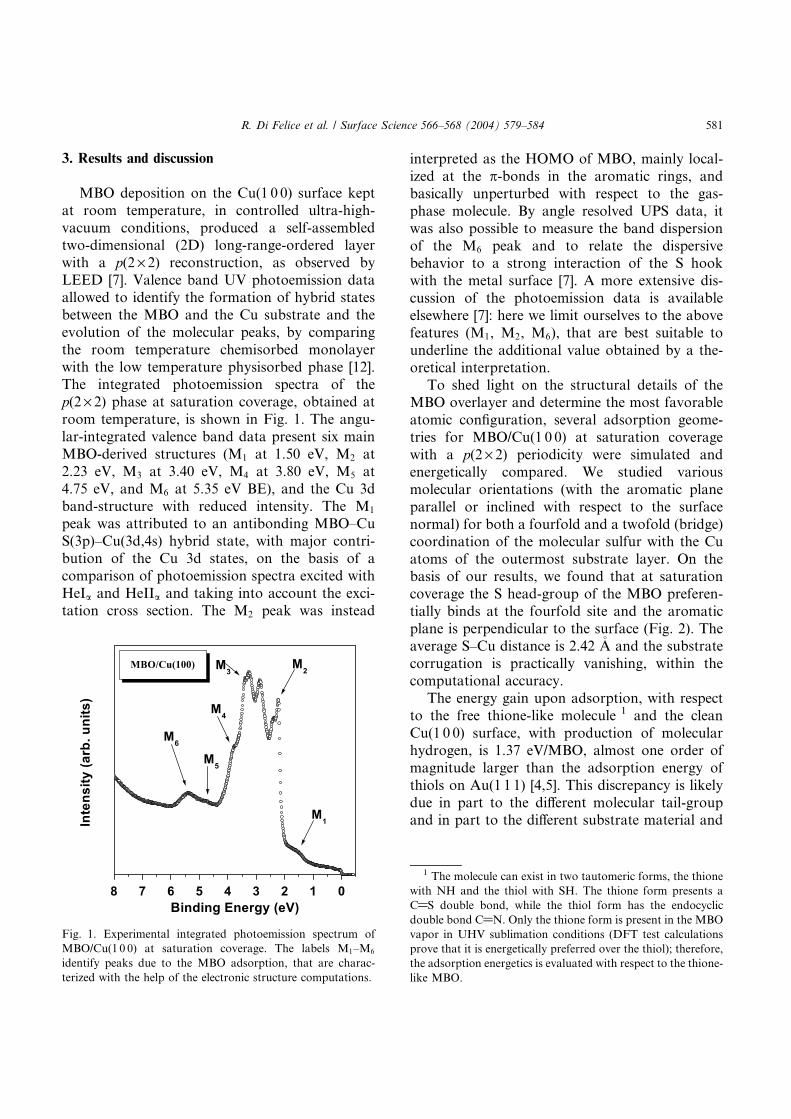
3. Results and discussion
MBO deposition on the Cu(1 0 0) surface kept
at room temperature, in controlled ultra-high-
vacuum conditions, produced a self-assembledtwo-dimensional (2D) long-range-ordered layer
with a p(2 · 2) reconstruction, as observed by
LEED [7]. Valence band UV photoemission data
allowed to identify the formation of hybrid states
between the MBO and the Cu substrate and the
evolution of the molecular peaks, by comparing
the room temperature chemisorbed monolayer
with the low temperature physisorbed phase [12].The integrated photoemission spectra of the
p(2 · 2) phase at saturation coverage, obtained at
room temperature, is shown in Fig. 1. The angu-
lar-integrated valence band data present six main
MBO-derived structures (M1 at 1.50 eV, M2 at
2.23 eV, M3 at 3.40 eV, M4 at 3.80 eV, M5 at
4.75 eV, and M6 at 5.35 eV BE), and the Cu 3d
band-structure with reduced intensity. The M1
peak was attributed to an antibonding MBO–Cu
S(3p)–Cu(3d,4s) hybrid state, with major contri-
bution of the Cu 3d states, on the basis of a
comparison of photoemission spectra excited with
HeIa and HeIIa and taking into account the exci-
tation cross section. The M2 peak was instead
interpreted as the HOMO of MBO, mainly local-
ized at the p-bonds in the aromatic rings, and
basically unperturbed with respect to the gas-
phase molecule. By angle resolved UPS data, it
was also possible to measure the band dispersion
of the M6 peak and to relate the dispersivebehavior to a strong interaction of the S hook
with the metal surface [7]. A more extensive dis-
cussion of the photoemission data is available
elsewhere [7]: here we limit ourselves to the above
features (M1, M2, M6), that are best suitable to
underline the additional value obtained by a the-
oretical interpretation.
To shed light on the structural details of theMBO overlayer and determine the most favorable
atomic configuration, several adsorption geome-
tries for MBO/Cu(1 0 0) at saturation coverage
with a p(2 · 2) periodicity were simulated and
energetically compared. We studied various
molecular orientations (with the aromatic plane
parallel or inclined with respect to the surface
normal) for both a fourfold and a twofold (bridge)coordination of the molecular sulfur with the Cu
atoms of the outermost substrate layer. On the
basis of our results, we found that at saturation
coverage the S head-group of the MBO preferen-
tially binds at the fourfold site and the aromatic
plane is perpendicular to the surface (Fig. 2). The
average S–Cu distance is 2.42 A and the substrate
corrugation is practically vanishing, within thecomputational accuracy.
The energy gain upon adsorption, with respect
to the free thione-like molecule 1 and the clean
Cu(1 0 0) surface, with production of molecular
hydrogen, is 1.37 eV/MBO, almost one order of
magnitude larger than the adsorption energy of
thiols on Au(1 1 1) [4,5]. This discrepancy is likely
due in part to the different molecular tail-groupand in part to the different substrate material and
Fig. 1. Experimental integrated photoemission spectrum of
MBO/Cu(1 0 0) at saturation coverage. The labels M1–M6
identify peaks due to the MBO adsorption, that are charac-
terized with the help of the electronic structure computations.
1 The molecule can exist in two tautomeric forms, the thione
with NH and the thiol with SH. The thione form presents a
C@S double bond, while the thiol form has the endocyclic
double bond C@N. Only the thione form is present in the MBO
vapor in UHV sublimation conditions (DFT test calculations
prove that it is energetically preferred over the thiol); therefore,
the adsorption energetics is evaluated with respect to the thione-
like MBO.
R. Di Felice et al. / Surface Science 566–568 (2004) 579–584 581

orientation: however, the present status of our
simulations does not allow us to discriminate be-
tween the two effects. When the simulations were
started with the S head-group occupying a twofoldlocation, we observed a migration of the molecule
towards the highest-coordination site. Preliminary
calculations indicate that the same distinct pref-
erence for the highest S–Cu coordination (four-
fold) is also characteristic of the MBO/Cu(1 0 0)
system at lower coverages, although in this case
the orientation of the molecule may change.
After obtaining and describing the energeticallyfavorable p(2 · 2) high-coverage geometry, we
computed its electronic structure, the total density
of states (DOS), the angular-component-resolved
DOS projected onto atoms (S, C, N, O) of the
MBO, and the single-particle wavefunctions. We
compared such quantities with those of the gas-
phase molecule in its thione and thiol configura-
tions. This analysis allows us to identify thepresence of bonding and antibonding S–Cu hybrid
states [5], and to give a more detailed interpreta-
tion of the peaks identified in the photoemission
spectra. GW corrections calculated for bulk cop-
per [13] indicate that a downward shift by 0.6 eV
should be applied to the computed DFT eigen-
values to obtain a reliable positioning of the Cu d
edge with respect to the Fermi level: in our analysis
below, we take into account this shift (which is not
uniform throughout the relevant energy range) todiscuss the positions of the molecular features in
the DOS with respect to the upper limit of the
substrate d bands, in the frame of the Newns–
Anderson chemisorption model [6].
In Fig. 3, we show the computed DOS (total and
Sp-projected) and the isosurface plots of an anti-
bonding and a bonding electron states that we
identify as contributions to the M2 (because of thestrong component on the molecular aromatic tail)
and M6 peaks in the experimental data. The cor-
respondence between the computed orbitals and
the experimental peaks is assigned on the basis of
the orbital energy; for instance, the computed en-
ergy of the electron state shown in Fig. 3b is )2.3eV, which compares well with the peak at 2.2 eV
binding energy in the photoemission spectrum ofFig. 1. Its strong tail-group component is consis-
tent with the experimental dependence of the M2
intensity on the photon energy. Moreover, we can
identify this state as originating from the HOMO
of the gas-phase MBO in the thiol form (see Fig.
3d–f). 2 With respect to the previous attribution of
the M2 peak as a purely molecular signal because
of the strong tail localization, we can actuallypoint out also an antibonding character between S
and Cu, thus making the picture quite similar
to other systems discussed previously in the litera-
ture [5].
Similar states of antibonding character, in
which however the charge distribution has only
Sp–Cud behavior with no contribution on the
MBO tail-group, are found to contribute to theantibonding peak of Fig. 3a, centered at )1.4 eV;
these are related to the peak M1 of the photo-
emission experiment. To summarize the molecular
features above the Cu d edge, we found that the
M1 antibonding peak has a low-energy tail around
Fig. 2. Left: top view of the 2· 2 supercell periodically repli-
cated to show the 2D molecular arrangement. Only the up-
permost Cu layer is shown. To minimize the p-stack repulsion,
the MBO aromatic plane lies slightly off-tilted with respect to
the diagonal of the 2· 2 cell. Right: 3D view of the relaxed
lowest-energy geometry. All the atoms in the simulation cell are
shown. The fourfold coordination of the S atoms with the Cu
atoms is evident in both panels. The purple spheres represent
Cu. Yellow, green, red, blue, and white spheres represent S, C,
O, N, and H, respectively. (For interpretation of the references
in colour in this figure legend, the reader is referred to the web
version of this article.)
2 Of the two gas-phase highest occupied molecular orbitals
(Fig. 3d and e), that in Fig. 3d with both S and aromatic charge
distribution is responsible for the M2 peak, and that in Fig. 3e
is responsible for the M1 peak, not shown here.
582 R. Di Felice et al. / Surface Science 566–568 (2004) 579–584
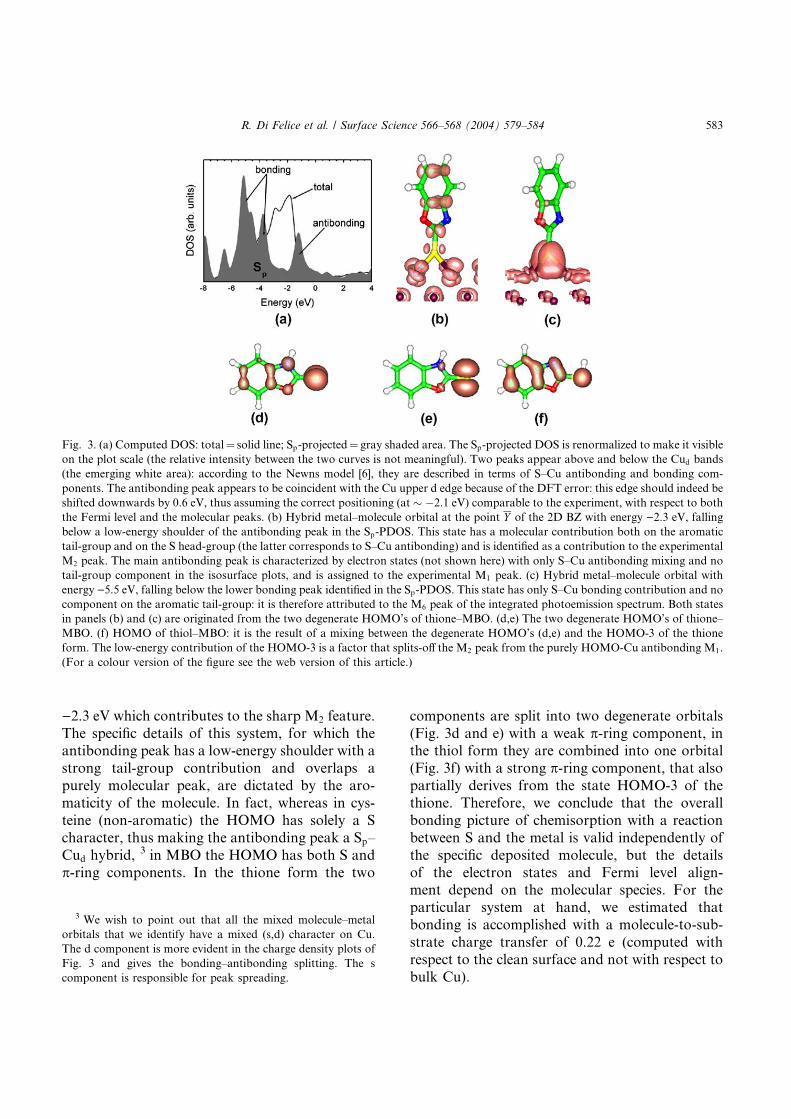
)2.3 eV which contributes to the sharp M2 feature.
The specific details of this system, for which theantibonding peak has a low-energy shoulder with a
strong tail-group contribution and overlaps a
purely molecular peak, are dictated by the aro-
maticity of the molecule. In fact, whereas in cys-
teine (non-aromatic) the HOMO has solely a S
character, thus making the antibonding peak a Sp–
Cud hybrid,3 in MBO the HOMO has both S and
p-ring components. In the thione form the two
components are split into two degenerate orbitals
(Fig. 3d and e) with a weak p-ring component, inthe thiol form they are combined into one orbital
(Fig. 3f) with a strong p-ring component, that also
partially derives from the state HOMO-3 of the
thione. Therefore, we conclude that the overall
bonding picture of chemisorption with a reaction
between S and the metal is valid independently of
the specific deposited molecule, but the details
of the electron states and Fermi level align-ment depend on the molecular species. For the
particular system at hand, we estimated that
bonding is accomplished with a molecule-to-sub-
strate charge transfer of 0.22 e (computed with
respect to the clean surface and not with respect to
bulk Cu).
3 We wish to point out that all the mixed molecule–metal
orbitals that we identify have a mixed (s,d) character on Cu.
The d component is more evident in the charge density plots of
Fig. 3 and gives the bonding–antibonding splitting. The s
component is responsible for peak spreading.
Fig. 3. (a) Computed DOS: total¼ solid line; Sp-projected¼ gray shaded area. The Sp-projected DOS is renormalized to make it visible
on the plot scale (the relative intensity between the two curves is not meaningful). Two peaks appear above and below the Cud bands
(the emerging white area): according to the Newns model [6], they are described in terms of S–Cu antibonding and bonding com-
ponents. The antibonding peak appears to be coincident with the Cu upper d edge because of the DFT error: this edge should indeed be
shifted downwards by 0.6 eV, thus assuming the correct positioning (at 2:1 eV) comparable to the experiment, with respect to both
the Fermi level and the molecular peaks. (b) Hybrid metal–molecule orbital at the point Y of the 2D BZ with energy )2.3 eV, falling
below a low-energy shoulder of the antibonding peak in the Sp-PDOS. This state has a molecular contribution both on the aromatic
tail-group and on the S head-group (the latter corresponds to S–Cu antibonding) and is identified as a contribution to the experimental
M2 peak. The main antibonding peak is characterized by electron states (not shown here) with only S–Cu antibonding mixing and no
tail-group component in the isosurface plots, and is assigned to the experimental M1 peak. (c) Hybrid metal–molecule orbital with
energy )5.5 eV, falling below the lower bonding peak identified in the Sp-PDOS. This state has only S–Cu bonding contribution and no
component on the aromatic tail-group: it is therefore attributed to the M6 peak of the integrated photoemission spectrum. Both states
in panels (b) and (c) are originated from the two degenerate HOMO’s of thione–MBO. (d,e) The two degenerate HOMO’s of thione–
MBO. (f) HOMO of thiol–MBO: it is the result of a mixing between the degenerate HOMO’s (d,e) and the HOMO-3 of the thione
form. The low-energy contribution of the HOMO-3 is a factor that splits-off the M2 peak from the purely HOMO-Cu antibonding M1.
(For a colour version of the figure see the web version of this article.)
R. Di Felice et al. / Surface Science 566–568 (2004) 579–584 583

4. Conclusions
We presented and discussed the results of a
combined experimental-computational investiga-
tion of an aromatic thiolate monolayer on theCu(1 0 0) surface. The complex analysis resulted in
the detailed identification of the experimental pho-
toemission peaks in terms of molecular orbitals and
molecule–metal mixing. We found that the selected
system satisfies a general model of atomic chemi-
sorption on metal surfaces [6], which was recently
extended to organic molecule adsorption [5]. How-
ever, specific details that may affect the electricalbehavior of the system and its reaction to other
impacting species for organic heterostructures, de-
pend on the choice of the molecules employed to
attain the first interface organic monolayer.
Acknowledgements
This work was partially funded by INFM
through PRA SINPROT, by the EC through
contract IST-2000-28024 ‘‘SAMBA’’, and by
MIUR through FIRB ‘‘NOMADE’’. Computing
time at CINECA was provided by INFM through
the Commitee for Parallel Computation.
References
[1] A. Ulman, Chem. Rev. 96 (1996) 1533;
Y. Xia, J.A. Rogers, K.A. Paul, G.M. Whitesides, Chem.
Rev. 99 (1999) 1823.
[2] C. Joachim, J.K. Gimzewski, A. Aviram, Nature 408
(2000) 541.
[3] F. Schreiber, Prog. Surf. Sci. 65 (2000) 151.
[4] M.C. Vargas, P. Giannozzi, A. Selloni, G. Scoles, J. Phys.
Chem. B 105 (2001) 9509.
[5] R. Di Felice, A. Selloni, E. Molinari, J. Phys. Chem. B 107
(2003) 1151.
[6] B. Hammer, J.K. Nørskov, Theory of adorption and
surface reactions, in: R.M. Lambert, G. Pacchioni (Eds.),
Chemisorption and Reactivity of Supported Clusters
and Thin Films, Kluwer Academic, The Netherlands, 1997.
[7] C. Mariani, F. Allegretti, V. Corradini, G. Contini, V. Di
Castro, C. Baldacchini, M.G. Betti, Phys. Rev. B 66 (2002)
115407.
[8] We employed the computer code PWSCF by S. Baroni, A.
Dal Corso, S. de Gironcoli, P. Giannozzi, available at
http://www.pwscf.org.
[9] J.P. Perdew, J.A. Chevary, S.H. Vosko, K.A. Jackson,
D.J. Singh, C. Fiolhais, Phys. Rev. B 46 (1992) 6671.
[10] D. Vanderbilt, Phys. Rev. B 41 (1990) 7892.
[11] N. Troullier, J.L. Martins, Phys. Rev. B 46 (1992) 1754.
[12] V. Di Castro, F. Allegretti, C. Baldacchini, M.G. Betti, G.
Contini, V. Corradini, F. Lamastra, C. Mariani, Surf. Sci.
507–510 (2002) 7.
[13] A. Marini, G. Onida, R. Del Sole, Phys. Rev. Lett. 88
(2002) 016403.
584 R. Di Felice et al. / Surface Science 566–568 (2004) 579–584


Chapter 3
Transport in a devicegeometry with the inclusionof correlation
The study of nano-junctions largely increased its importance as a possibleway to overcome the intrinsic limits of current electronics. One of the openquestions in the field is the formulation of a theoretical framework ableto describe transport properties. A detailed formalism for transport existsdue to the studies of mesoscale physics [1]. At the same time, ab initiomethods were demonstrated to be successful in describing the electronicproperties of nanoscale systems. The matching of DFT electronic structurecalculations and the out-of-equilibrium statistics and boundary conditionsproper of transport is still a challenging and controversial topic [2,3,4,5]. Inorder to work around this difficulty new basic formulation of the transportproblem are appearing [6, 7, 8, 9]. In the same spirit, recent experimentalfindings [10,11,12] highlight the need for a description able to treat electron-electron interactions in transport.
In the present work we address the issue of including electron correlationin the calculation of transport properties. In the first paper of this Chap-ter [13] (still unpublished) we first derive a formalism suitable to express thecurrent in a Landauer-like form valid in the general interacting case. Weimplement the method using the matrix Green’s functions formalism andthe maximally localized Wannier functions as done in the WanT code [14].We apply the above formulation to the case of a conductor exploiting shortrange correlation, which we treat by means of the 3BS method [15,16]. Forthis system we compute the equilibrium transmittance explicitly analyzingthe proper effects arising from the interactions. Our results demonstratethat the inclusion of correlation is particularly effective in quenching thetransmittance. We address this finding to the presence of finite lifetimesfor quasi-particle (QP) levels in the conductor. The second work presented

52 Transport in a device geometry with the inclusion of correlation
in this Chapter, also still unpublished, reports a detailed derivation of theformalism and further analysis of the previous application, including a com-parison with the standard LDA+U approach [17]. It is shown that LDA+Uresults are consistent with those obtained neglecting finite QP lifetimes inour formalism which are not able to fully describe the transmittance quench-ing due to correlation.
Bibliography
[1] H. Haug and A.-P. Jauho, Quantum kinetics in transport and optics ofsemiconductors, Springer Berlin, 1996.
[2] J. Taylor, H. Guo, and J. Wang, Ab initio modeling of quantum trans-port properties of molecular electronic devices, Phys. Rev. B 63(24),245407–245419 (June 2001).
[3] M. Brandbyge, J.-L. Mozos, P. Ordejon, J. Taylor, and K. Stok-bro, Density-functional method for nonequilibrium electron transport,Phys. Rev. B 65, 165401 (Mar. 2002).
[4] Y. Xue, S. Datta, and M. A.Ratner, First-principles based matrixGreen’s function approach to molecular electronic devices: general for-malism, Chem. Phys. 281(2–3), 151–170 (2002).
[5] F. Evers, F. Weigend, and M. Koentopp, Conductance of molecularwires and transport calculations based on density-functional theory,Phys. Rev. B 69(23), 235411 (2004).
[6] D. S. Kosov, Kohn–Sham equations for nanowires with direct current,J. Chem. Phys. 119(1), 1–5 (2003).
[7] N. T. Maitra, I. Souza, and K. Burke, Current-density functional theoryof the response of solids, Phys. Rev. B 68(4), 045109 (2003).
[8] P. Delaney and J. C. Greer, Correlated Electron Transport in MolecularElectronics, Phys. Rev. Lett. 93(3), 036805 (July 2004).
[9] R. Gebauer and R. Car, Current in open quantum systems, Cond-Mat, 0410105v1 (Oct. 2004).
[10] W. Liang, M. P. Shores, M. Bockrath, J. R. Long, and H. Park, Kondoresonance in a single-molecule transistor, Nature 417, 725–729 (June2002).
[11] J. Park, A. N. Pasupathy, J. I. Goldsmith, C. Chang, Y. Yaish, J. R.P. M. Rinkoski, J. P. Sethna, H. D. Abrunna, P. L. McEuen, and D. C.Ralph, Coulomb blockade andthe Kondo effect in single-atom transis-tors, Nature 417, 722–725 (June 2002).

53
[12] P. Jarillo-Herrero, S. Sapmaz, C. Dekker, L. P. Kouwenhoven, andH. S. J. van der Zant, Electron-hole symmetry in a semiconductingcarbon nanotube quantum dot, Nature 429, 389–392 (May 2004).
[13] A. Ferretti, A. Calzolari, R. Di Felice, F. Manghi, M. J. Caldas,M. Buongiorno Nardelli, and E. Molinari, First principle theory ofcorrelated transport through nano-junctions, Cond-Mat , 0409222v1(Sept. 2004).
[14] A. Calzolari, C. Cavazzoni, N. Marzari, and M. Buongiorno Nardelli,2004, http://www.wannier-transport.org.
[15] C. Calandra and F. Manghi, Three-body scattering theory of correlatedhole and electron states, Phys. Rev. B 50(4), 2061–2073 (July 1994).
[16] F. Manghi, V. Bellini, and C. Arcangeli, On-site correlation in valenceand core states of ferromagnetic nickel, Phys. Rev. B 56(12), 7149–7161(Sept. 1997).
[17] M. Cococcioni and S. de Gironcoli, A linear response approach tothe calculation of the effective interaction parameters in the LDA+Umethod, Cond-Mat , 0405160v1 (May 2004).

First principle theory of correlated transport through nano-junctions
A. Ferretti,1 A. Calzolari,1 R. Di Felice,1 F. Manghi,1 M. J. Caldas,2 M. Buongiorno Nardelli,3, 4 and E. Molinari11INFM National Center on nanoStructures and bioSystems at Surfaces (S3) and Dipartimento di Fisica,
Universita di Modena e Reggio Emilia, 41100 Modena, Italy2Instituto de Fısica, Universidade de Sao Paulo, 05508-900 Sao Paulo, Brazil
3Department of Physics, North Carolina State University, Raleigh, NC 27695, USA4CCS-CSM, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
(Dated: December 10, 2004)
We report the inclusion of electron-electron correlation in the calculation of transport propertieswithin an ab initio scheme. A key step is the reformulation of Landauer’s approach in terms ofan effective transmittance for the interacting electron system. We apply this framework to analyzethe effect of short range interactions on Pt atomic wires and discuss the coherent and incoherentcorrection to the mean-field approach.
One of the most pressing problems in nanotechnologyis the need for recasting all the know-how about meso-scopic transport physics into the fully quantum mechan-ical limit appropriate for atomic scale phenomena. Inthe case of electronic and transport properties of atomicand molecular conductors, we must address at the micro-scopic level both the chemical complexity of the conduc-tor and the complexity of the interactions between thedifferent components of an extended open system.
The combination of Green’s function methods with aDensity Functional Theory (DFT) description of elec-tronic states has become a standard approach to studycharge transport at the nanoscale [1–5]. However, caremust be taken in comparing the computed transportcharacteristics to experiments [6]. Indeed, some impor-tant features — such as electron correlations, dissipa-tion, decoherence and temperature effects — are at themoment partly or fully neglected. These deficiencies be-come more and more crucial when the dimensions of atleast one part of the system reach a confinement sit-uation where for instance electron-electron (e-e) inter-actions may become dominant. Recent observations ofKondo effect and Coulomb blockade in molecules con-nected to external electrodes [7, 8], and in nanotubeswith magnetic impurities [9] or size confinement [10], in-dicate that correlations do play an important role in themechanism of charge transport in nano-devices. Whereasmany efforts [6, 11–14] have been directed to study otheraspects of the transport problem, a standard approach toinclude correlation effects does not yet exist. One mayexpect that some of the difficulties in the interpretationof transport experiments on simple atomic chains andindividual molecules could be ascribed to the neglect ormistreatment of correlations.
In this Letter we develop a new method for the abinitio computation of quantum transport in the strongcorrelation regime, and then apply it to specifically ad-dress the effect of e-e interactions on electronic transportthrough atomic-scale conductors. Following closely Meirand Wingreen [15], we derive a Landauer-like expressionfor the current through a correlated conductor between
uncorrelated leads. Short range e-e interactions in theconductor are described through the Three-Body Scat-tering (3BS) formalism [16, 17]. The method is imple-mented through use of “maximally-localized” Wannierfunctions [5, 18] (MLWF). A particularly well-adaptedsystem for gauging the relevance of e-e correlations isa late-transition-metal break-junction: in such a config-uration, e-e effects are negligible in the bulk but, as aconsequence of dimensionality, may acquire relevance inthe junction region. We apply our method to simulatea model Platinum break-junction of varying length. Ourfindings show a large suppression of the transmittancethat we ascribe to the inclusion of e-e elastic decoher-ence in the simulation. Moreover, the strong conduc-tance reduction, also for small wire lengths, suggests thatcorrelation cannot be neglected when studying transportproperties of systems with localized electrons.
The system is modelled as three different regions, theleft (L) and right (R) electrodes and a central conductor(C). We express our operators in a localized basis set [19]which allows us to write the Hamiltonian and the Green’sfunctions of the whole system as 3 × 3 block matricesdefined on the basis in the L, R, and C regions [Hxy,Gxy(ω) where x, y = L,C,R].
The Hamiltonian reads:
H =∑
ll′∈LorR
Hll′c†l cl′ + Hint +
∑l∈LorR
i∈C
[Hli c†l di + h.c.
],
(1)where cl and c†l (di and d†i ) are the one-electron annihila-tion and creation operators in the leads (conductor). Inthe above expression, the first term describes the L andR leads, Hint the conductor, the last term the coupling ofthe conductor with the L and R leads. We stress that theleads and the coupling Hamiltonian are non-interactingand all the e-e interaction is restricted to the conductor.
From the continuity equation for the steady-state cur-rent in the system and using the Keldysh non-equilibriumGreen’s function formalism [20], the following expression

for the current [15] is derived:
I =2ei
h
∫dω Tr
[Σ<
L − Σ<R
]AC + [ΓL − ΓR]G<
C
. (2)
Here AC = i[GrC − Ga
C ] is the spectral function andGr,a,<,>
C are the (retarded, advanced, lesser, greater)Green’s functions in the conductor. The trace should betaken on the conductor. The interaction between conduc-tor and leads is described through the lead self-energies(SE’s), defined as Σr,a,<,>
x (ω) = HCx Gr,a,<,>x (ω)HxC ,
where x=L,R. Finally, the ΓL,R terms in Eq. (2) are de-fined as twice the imaginary part of the retarded lead-SE’s, i.e. ΓL,R = i
[Σr
L,R − ΣaL,R
]. Note also that
Σ<L,R = ifL,R ΓL,R and Σ>
L,R = −i(1− fL,R) ΓL,R, wherefL and fR are the Fermi occupation functions for the leftand right leads.
While in the non-interacting case the above expressionbrings to the usual Landauer formula [15], in the presenceof interaction between electrons this is no longer trueand further assumptions are needed. We here adopt theansatz proposed by Ng [21] which relates Σ<
C (Σ>C) to
Σr,aC , thus defining the statistics of energy levels, in the
general out-of-equilibrium interacting case. The startingpoint is the assumption [22]:
Σ<,>C (ω) = Σ<,>
0 C (ω) Λ(ω) , (3)
where Σ<,>0 C = Σ<,>
L + Σ<,>R refer to the non-interacting
case and include only the coupling with the leads, whilethe full Σ<,>
C include also e-e interactions. Λ(ω), alsocalled A in other formulations [22], is determined by theidentity Σ>
C − Σ<C = Σr
C − ΣaC leading to:
Λ(ω) = [Σr0 C(ω)− Σa
0 C(ω)]−1 [ΣrC(ω)− Σa
C(ω)] . (4)
Here the interacting SE’s take the form:
Σr,aC (ω) = Σr,a
L (ω) + Σr,aR (ω) + Σr,a
corr(ω) (5)
where Σr,acorr account for the e-e interactions (while Σr,a
0 C
just drop this last term). Relations (3-5) are the keyto relate Eq. (2) to a Landauer-like formula. In fact,following Eq. (6) of Ref. [22] and using G<
C = GrCΣ<
CGaC ,
it is possible to derive G<C = iGr
C [fLΓL + fRΓR] ΛGaC
and therefore GrC −Ga
C = −iGrC [ΓL + ΓR] ΛGa
C . Thesesteps lead to the final expression for the current:
I =e
~
∫dω
2π[fL − fR] Tr ΓL Gr
C ΓR Λ GaC . (6)
We remark that the e-e correlation plays a twofold role:it renormalizes the Green’s functions, which should nowbe calculated for the interacting system, and also mod-ifies the expression for the current, introducing the cor-rective factor Λ(ω). The quantity traced in Eq. (6), evenif not a true transmittance across the scattering regiondue to the breakdown of Landauer’s theory, still plays
the same role as regards transport. For this reason werefer to it as an effective transmittance and compare itto the transmittance of the non-interacting case. In par-ticular, since the imaginary part of the e-e self-energyexactly vanishes at the Fermi energy (EF ), the correc-tion Λ(ω = EF ) becomes the identity operator and theeffective transmittance computed at EF is proportionalto the conductance at zero temperature, provided thatthe SE’s are calculated for the interacting system.
An accurate evaluation of the effective transmittanceneeds to be based on: (i) a good description of the non-interacting system, and (ii) the calculation of the e-ecorrelated SE to include many-body effects arising fromthe interaction in the conductor. The first problem issolved by exploiting a recent methodology for the ab ini-tio calculation of the transmittance in the coherent trans-port regime [5]. The ground state of the mean-field sys-tem is described within the Local Density Approximationto DFT, using norm-conserving pseudopotentials and aplane-wave basis set [23]. The geometry of the L-C-Rnano-junction are as in Ref. [1]. To obtain a consistentdescription of the system in a real-space localized basisset, the MLWF’s are computed both for the leads andfor the conductor [24]. The details of this transformationand its application to electronic transport are describedelsewhere [5, 18]. We remark that the basis change fromBloch eigenvectors to MLWF’s preserves orthonormal-ity and completeness in the original Hilbert subspace,thereby avoiding typical problems arising very often forother localized basis functions. The same features al-low us to use a minimal basis set for computing trans-port properties, while employing the system independentplane-wave basis set for the DFT calculation.
To discuss the inclusion of e-e correlations we first needto define the interaction hamiltonian in Eq. (1). We focuson the short range e-e interaction for two main reasons.On one hand, this regime is characterized by strong de-viations from the non-interacting behavior, for instancein terms of quasiparticle lifetimes (which include elasticdecoherence in the transport formalism). On the otherhand, it allows us to adopt an Anderson-like form [25]of the interaction which is suitable for the localization inthe conductor region only, as required by our approach.
In this work the e-e self-energy Σrcorr is computed using
a non-perturbative approach based on an effective Ander-son hamiltonian, whose U Coulomb integrals could be ei-ther calculated ab initio [26] or used as adjustable inputparameters. It is solved by means of a configuration in-teraction scheme with up to three bodies (3BS) added tothe non-interacting Fermi sea: two (one) electron(s) andone (two) hole(s) for conduction (valence) states. Thismethod has been successfully applied to describe photoe-mission experiments on strongly correlated systems [17].The 3BS self-energy is formally given [17] as a sum overprojectors onto atomic states (those with non-negligibleU integrals) and thus can be properly localized in the
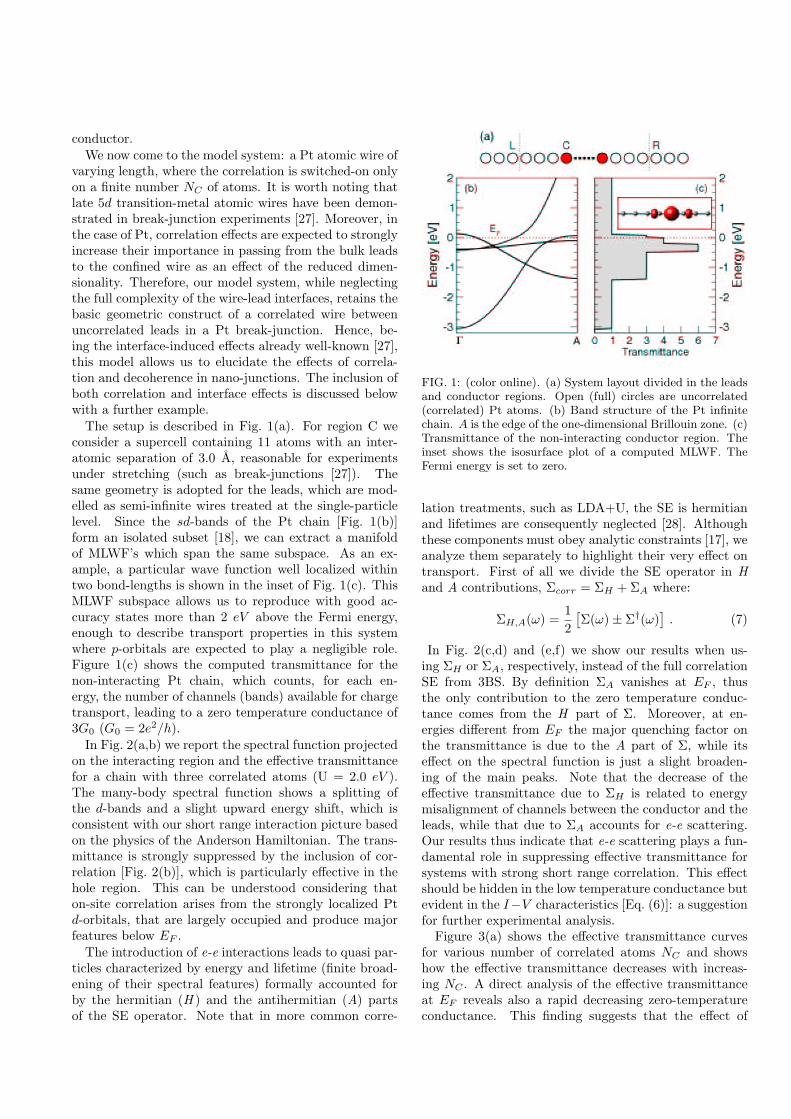
conductor.We now come to the model system: a Pt atomic wire of
varying length, where the correlation is switched-on onlyon a finite number NC of atoms. It is worth noting thatlate 5d transition-metal atomic wires have been demon-strated in break-junction experiments [27]. Moreover, inthe case of Pt, correlation effects are expected to stronglyincrease their importance in passing from the bulk leadsto the confined wire as an effect of the reduced dimen-sionality. Therefore, our model system, while neglectingthe full complexity of the wire-lead interfaces, retains thebasic geometric construct of a correlated wire betweenuncorrelated leads in a Pt break-junction. Hence, be-ing the interface-induced effects already well-known [27],this model allows us to elucidate the effects of correla-tion and decoherence in nano-junctions. The inclusion ofboth correlation and interface effects is discussed belowwith a further example.
The setup is described in Fig. 1(a). For region C weconsider a supercell containing 11 atoms with an inter-atomic separation of 3.0 A, reasonable for experimentsunder stretching (such as break-junctions [27]). Thesame geometry is adopted for the leads, which are mod-elled as semi-infinite wires treated at the single-particlelevel. Since the sd-bands of the Pt chain [Fig. 1(b)]form an isolated subset [18], we can extract a manifoldof MLWF’s which span the same subspace. As an ex-ample, a particular wave function well localized withintwo bond-lengths is shown in the inset of Fig. 1(c). ThisMLWF subspace allows us to reproduce with good ac-curacy states more than 2 eV above the Fermi energy,enough to describe transport properties in this systemwhere p-orbitals are expected to play a negligible role.Figure 1(c) shows the computed transmittance for thenon-interacting Pt chain, which counts, for each en-ergy, the number of channels (bands) available for chargetransport, leading to a zero temperature conductance of3G0 (G0 = 2e2/h).
In Fig. 2(a,b) we report the spectral function projectedon the interacting region and the effective transmittancefor a chain with three correlated atoms (U = 2.0 eV ).The many-body spectral function shows a splitting ofthe d-bands and a slight upward energy shift, which isconsistent with our short range interaction picture basedon the physics of the Anderson Hamiltonian. The trans-mittance is strongly suppressed by the inclusion of cor-relation [Fig. 2(b)], which is particularly effective in thehole region. This can be understood considering thaton-site correlation arises from the strongly localized Ptd-orbitals, that are largely occupied and produce majorfeatures below EF .
The introduction of e-e interactions leads to quasi par-ticles characterized by energy and lifetime (finite broad-ening of their spectral features) formally accounted forby the hermitian (H ) and the antihermitian (A) partsof the SE operator. Note that in more common corre-
FIG. 1: (color online). (a) System layout divided in the leadsand conductor regions. Open (full) circles are uncorrelated(correlated) Pt atoms. (b) Band structure of the Pt infinitechain. A is the edge of the one-dimensional Brillouin zone. (c)Transmittance of the non-interacting conductor region. Theinset shows the isosurface plot of a computed MLWF. TheFermi energy is set to zero.
lation treatments, such as LDA+U, the SE is hermitianand lifetimes are consequently neglected [28]. Althoughthese components must obey analytic constraints [17], weanalyze them separately to highlight their very effect ontransport. First of all we divide the SE operator in Hand A contributions, Σcorr = ΣH + ΣA where:
ΣH,A(ω) =12
[Σ(ω)± Σ†(ω)
]. (7)
In Fig. 2(c,d) and (e,f) we show our results when us-ing ΣH or ΣA, respectively, instead of the full correlationSE from 3BS. By definition ΣA vanishes at EF , thusthe only contribution to the zero temperature conduc-tance comes from the H part of Σ. Moreover, at en-ergies different from EF the major quenching factor onthe transmittance is due to the A part of Σ, while itseffect on the spectral function is just a slight broaden-ing of the main peaks. Note that the decrease of theeffective transmittance due to ΣH is related to energymisalignment of channels between the conductor and theleads, while that due to ΣA accounts for e-e scattering.Our results thus indicate that e-e scattering plays a fun-damental role in suppressing effective transmittance forsystems with strong short range correlation. This effectshould be hidden in the low temperature conductance butevident in the I−V characteristics [Eq. (6)]: a suggestionfor further experimental analysis.
Figure 3(a) shows the effective transmittance curvesfor various number of correlated atoms NC and showshow the effective transmittance decreases with increas-ing NC . A direct analysis of the effective transmittanceat EF reveals also a rapid decreasing zero-temperatureconductance. This finding suggests that the effect of
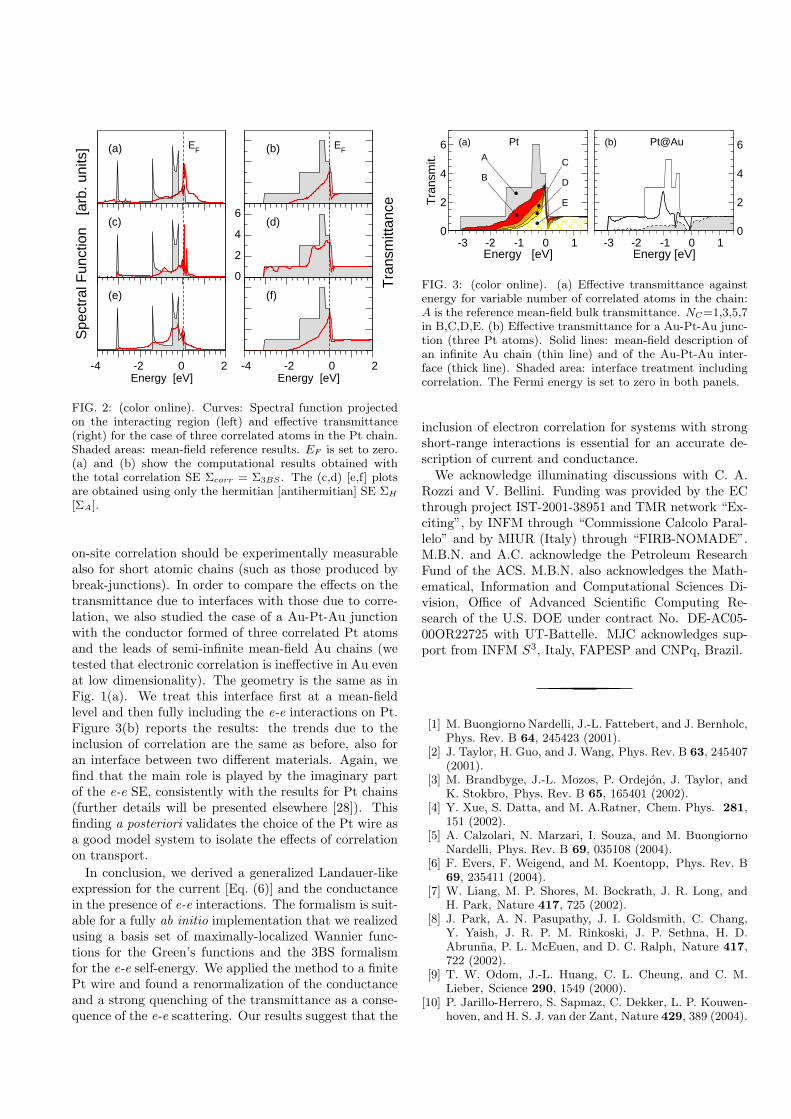
0
2
4
6
-4 -2 0 2Energy [eV]
-4 -2 0 2Energy [eV]
(a) (b)
(c) (d)
(e) (f)
Spe
ctra
l Fun
ctio
n [
arb.
uni
ts]
Tra
nsm
ittan
ce
EF EF
FIG. 2: (color online). Curves: Spectral function projectedon the interacting region (left) and effective transmittance(right) for the case of three correlated atoms in the Pt chain.Shaded areas: mean-field reference results. EF is set to zero.(a) and (b) show the computational results obtained withthe total correlation SE Σcorr = Σ3BS . The (c,d) [e,f] plotsare obtained using only the hermitian [antihermitian] SE ΣH
[ΣA].
on-site correlation should be experimentally measurablealso for short atomic chains (such as those produced bybreak-junctions). In order to compare the effects on thetransmittance due to interfaces with those due to corre-lation, we also studied the case of a Au-Pt-Au junctionwith the conductor formed of three correlated Pt atomsand the leads of semi-infinite mean-field Au chains (wetested that electronic correlation is ineffective in Au evenat low dimensionality). The geometry is the same as inFig. 1(a). We treat this interface first at a mean-fieldlevel and then fully including the e-e interactions on Pt.Figure 3(b) reports the results: the trends due to theinclusion of correlation are the same as before, also foran interface between two different materials. Again, wefind that the main role is played by the imaginary partof the e-e SE, consistently with the results for Pt chains(further details will be presented elsewhere [28]). Thisfinding a posteriori validates the choice of the Pt wire asa good model system to isolate the effects of correlationon transport.
In conclusion, we derived a generalized Landauer-likeexpression for the current [Eq. (6)] and the conductancein the presence of e-e interactions. The formalism is suit-able for a fully ab initio implementation that we realizedusing a basis set of maximally-localized Wannier func-tions for the Green’s functions and the 3BS formalismfor the e-e self-energy. We applied the method to a finitePt wire and found a renormalization of the conductanceand a strong quenching of the transmittance as a conse-quence of the e-e scattering. Our results suggest that the
-3 -2 -1 0 1Energy [eV]
0
2
4
6
Tra
nsm
it.
-3 -2 -1 0 1Energy [eV]
0
2
4
6A
B
C
E
D
(a) (b) Pt@AuPt
FIG. 3: (color online). (a) Effective transmittance againstenergy for variable number of correlated atoms in the chain:A is the reference mean-field bulk transmittance. NC=1,3,5,7in B,C,D,E. (b) Effective transmittance for a Au-Pt-Au junc-tion (three Pt atoms). Solid lines: mean-field description ofan infinite Au chain (thin line) and of the Au-Pt-Au inter-face (thick line). Shaded area: interface treatment includingcorrelation. The Fermi energy is set to zero in both panels.
inclusion of electron correlation for systems with strongshort-range interactions is essential for an accurate de-scription of current and conductance.
We acknowledge illuminating discussions with C. A.Rozzi and V. Bellini. Funding was provided by the ECthrough project IST-2001-38951 and TMR network “Ex-citing”, by INFM through “Commissione Calcolo Paral-lelo” and by MIUR (Italy) through “FIRB-NOMADE”.M.B.N. and A.C. acknowledge the Petroleum ResearchFund of the ACS. M.B.N. also acknowledges the Math-ematical, Information and Computational Sciences Di-vision, Office of Advanced Scientific Computing Re-search of the U.S. DOE under contract No. DE-AC05-00OR22725 with UT-Battelle. MJC acknowledges sup-port from INFM S3, Italy, FAPESP and CNPq, Brazil.
[1] M. Buongiorno Nardelli, J.-L. Fattebert, and J. Bernholc,Phys. Rev. B 64, 245423 (2001).
[2] J. Taylor, H. Guo, and J. Wang, Phys. Rev. B 63, 245407(2001).
[3] M. Brandbyge, J.-L. Mozos, P. Ordejon, J. Taylor, andK. Stokbro, Phys. Rev. B 65, 165401 (2002).
[4] Y. Xue, S. Datta, and M. A.Ratner, Chem. Phys. 281,151 (2002).
[5] A. Calzolari, N. Marzari, I. Souza, and M. BuongiornoNardelli, Phys. Rev. B 69, 035108 (2004).
[6] F. Evers, F. Weigend, and M. Koentopp, Phys. Rev. B69, 235411 (2004).
[7] W. Liang, M. P. Shores, M. Bockrath, J. R. Long, andH. Park, Nature 417, 725 (2002).
[8] J. Park, A. N. Pasupathy, J. I. Goldsmith, C. Chang,Y. Yaish, J. R. P. M. Rinkoski, J. P. Sethna, H. D.Abrunna, P. L. McEuen, and D. C. Ralph, Nature 417,722 (2002).
[9] T. W. Odom, J.-L. Huang, C. L. Cheung, and C. M.Lieber, Science 290, 1549 (2000).
[10] P. Jarillo-Herrero, S. Sapmaz, C. Dekker, L. P. Kouwen-hoven, and H. S. J. van der Zant, Nature 429, 389 (2004).

[11] M. Di Ventra and S. T. Pantelides, Phys. Rev. B 61,16207 (2000).
[12] D. S. Kosov, J. Chem. Phys. 119, 1 (2003).[13] N. T. Maitra, I. Souza, and K. Burke, Phys. Rev. B 68,
045109 (2003).[14] P. Delaney and J. C. Greer, Phys. Rev. Lett. 93, 036805
(2004).[15] Y. Meir and N. S. Wingreen, Phys. Rev. Lett. 68, 2512
(1992).[16] C. Calandra and F. Manghi, Phys. Rev. B 50, 2061
(1994).[17] F. Manghi, V. Bellini, and C. Arcangeli, Phys. Rev. B
56, 7149 (1997).[18] N. Marzari and D. Vanderbilt, Phys. Rev. B 56, 12847
(1997).[19] S. Datta, Electronic transport in mesoscopic systems,
Cambridge University Press, 1995.[20] H. Haug and A.-P. Jauho, Quantum kinetics in transport
and optics of semiconductors, Springer Berlin, 1996.[21] T.-K. Ng, Phys. Rev. Lett. 76, 487 (1996).[22] N. Sergueev, Q. feng Sun, H. Guo, B. G. Wang, and
J. Wang, Phys. Rev. B 65, 165303 (2002).[23] S. Baroni, A. Dal Corso, S. de Gironcoli, and P. Gian-
nozzi, 2001, http://www.pwscf.org.[24] A. Calzolari, C. Cavazzoni, N. Marzari, and
M. Buongiorno Nardelli, 2004, WanT code,http://www.wannier-transport.org; see also Ref. [[5]].
[25] G. D. Mahan, Many-particle physics, Plenum, New York,1981.
[26] M. Springer and F. Aryasetiawan, Phys. Rev. B 57, 4364(1998).
[27] N. Agraıt, A. L. Yeyati, and J. M. van Ruitenbeek,Phys. Rep. 377, 81 (2003).
[28] A. Ferretti et al., unpublished.

First principles theoretical description of transport including electron-electroncorrelation
A. Ferretti,1 A. Calzolari,1 R. Di Felice,1 and F. Manghi11INFM National Center on nanoStructures and bioSystems at Surfaces (S3) and Dipartimento di Fisica,
Universita di Modena e Reggio Emilia, 41100 Modena, Italy(Dated: December 28, 2004)
We report about the inclusion of many-body electron interactions in the simulation of transportproperties. We derive a general Landauer-like expression for the current, valid also in the case ofconductors in which the charge carriers undergo generic scattering processes. An important focusis put on the derivation of the theoretical framework, both for the general formalism and for theactual implementation of the method, including the treatment of electronic correlation. We thenshow an example of application and compare the results on the electronic and conduction propertiesobtained with our new scheme to those given by alternative computational frameworks.
PACS numbers:
I. INTRODUCTION
In order to continue to scale down the size of elec-tronic devices, modern science and technology are cur-rently engaged in replacing the usual top-down litho-graphic approach to device fabrication with an alter-native scheme1–11 that would start from ad hoc nano-sized building blocks such as, e.g., carbon nanotubes ormolecules. In this scenario a large effort is being devotedto develop theoretical and computational tools able to de-scribe transport properties of systems obtained connect-ing two (or more) electrodes with a bridging nanoscaleunit. A detailed theory of transport in mesoscopic sys-tems is known since a long time, but the matching ofthose ideas with the new problems arising in nanoscopicobjects is not obvious. One needs to describe at the sametime an open and non-periodic system, as well as de-tails of the bridging conductor at the atomic level (thisis fundamental in the case of molecular conductors, forexample).
Furthermore, the use of standard electronic structureapproaches based on density functional theory (DFT)for calculating transport properties is not straightfor-ward and suffers from additional problems in the de-scription of nanodevices. Even if not rigorously exactDFT is quite well established and it is usually easyto recognize reliable from out-of-scope results. We canroughly separate the problems for DFT in the new fieldof nanoscience in two blocks: The first comprises ques-tions on the applicability of DFT to the transport proper-ties in the out-of-equilibrium situation; few such schemesalready exist,12–14 but several open questions about theinternal consistency of the method appeared in view ofsome claimed (but reproducible) quantitative failures.15The second block of problems concerns the emergence– or even dominance – of other effects, not well de-scribed in standard DFT approaches, in the extremelyconfined systems representative of nanoscale conductors.This is typically the case of electron-phonon coupling orelectron-electron (e-e) interaction. Recent experiments
have in fact demonstrated that some well known meso-scopic effects,16 such as Coulomb blockade,17–20 Kondoeffect19,21,22 or Luttinger-liquid behaviors,2,23,24 occuralso in the case of nanoscale conductors. In view of thesefindings a suitable description of e-e interaction effects inthe treatment of transport is highly desirable: New for-mulations including these effects25–29 are appearing, buta standard approach does not exist yet.
In the present work we tackle the problem of includ-ing e-e interactions in transport simulations within anab initio framework. In order to do that, we first derivea general Landauer-like expression for the current andthus define an effective transmittance across the conduc-tor accounting also for interactions among charge carri-ers and scatterers. We propose a specific implementa-tion and apply our scheme to the case of a finite, shortplatinum atomic chain between leads, modeling a pro-totypical break-junction. We focus on the short-rangee-e correlation regime, and model the junction by turn-ing on the interaction only in the confined conductor, ina finite number of atoms. The calculation requires theuse of maximally localized Wannier functions30,31 for thebasis set, and a matrix Green’s functions (GF’s) frame-work32 in the WanT code.33 Our results demonstratethat both the conductance and the transmittance acrossthe conductor are renormalized by the interactions. Wealso show that a large part of the effect can be ascribed tothe incoherent component of charge motion, arising froma genuine many body treatment of the e-e interaction.We enforce this interpretation by means of a comparisonwith the well established LDA+U scheme.34–36
The paper is organized as follows. In Sec. II we firstgive (II A) a critical survey of the general expression forthe current obtained by Meir and Wingreen,37 then weexplicitly derive (II B) a Landauer-like formula valid alsoin the interacting regime, and finally describe (II C) therelation of this formalism with a scattering picture. InSec. III we give the details of the actual first princi-ples implementation of the method, including (III A) thedescription of the mean-field transport approach usingmaximally localized Wannier functions and (III B) the

2
approach to treat electronic correlation. In Sec. IV wedescribe the system that was selected for a prototype ap-plication. We analyze the results that were obtained withthe new computational method and compare to thosegiven by the LDA+U framework. In Appendix A we givesome more details on the out-of-equilibrium frameworkused throughout the paper.
II. FORMALISM
A. The current through a lead-conductor-leadjunction
Let us consider a system composed of three main re-gions, two leads [that we label left (L) and right (R)] anda central conductor (C) connecting them. By using a lo-calized basis set, it is possible to take advantage of thisgeometry and relate each basis element to a precise regionthus writing the Hamiltonian and other objects of inter-est in block-matrix form, i.e. HXY = Hij, i ∈ X, j ∈ Ywhere X, Y = L,C,R. The L-C-R junction may be de-scribed by the following Hamiltonian:
H =∑
ll′∈LorR
Hll′c†l cl′ + HC +
∑l∈LorR
i∈C
[Hli c†l di + h.c.
].
(1)Here, cl and di (c†l and d†i ) are annihilation (creation)operators in the leads and conductor respectively, whileHC represents a general Hamiltonian including two-bodyinteractions acting in the C-region (written by meansof di and d†i). In this picture the description ofthe leads is cast at the single particle level, while onlythe coupling between the leads and the conductor regionmakes the problem to be fully interacting. Physically,this approximation is justified by the large screening ofthe Coulomb interaction, which takes place in usuallyadopted metallic leads. The assumption breaks down inthe presence of metals with a high degree of charge lo-calization and consequent less effective screening, such assome d- or f -metals and their oxides.
In a seminal work,37 Meir and Wingreen define thecurrent flowing through a lead-conductor-lead junctionas the time derivative of the number operator of one lead,namely NX =
∑l∈X c†l cl where X = L,R. The current
entering the L region can then be written as:
IL = −e〈NL〉 = − ie
~〈[H,NL]〉 , (2)
where the averages given by 〈·〉 are defined on a suitablestatistical non-equilibrium ensemble. Doing the algebraiccalculations for the commutators, the current turns outto be expressed as:
IL =e
~
∫dω
2πTr
G<
CL(ω)HLC −HCLG<LC(ω)
, (3)
where summations have been hidden in the block-matrixmultiplication notation and the trace should be taken on
the conductor basis elements, including spin degrees offreedom. G<
il (t1− t2) = i〈c†l (t1)di(t2)〉 is the lesser Greenfunction and G<
il (ω) is its Fourier transform wrt t1−t2.38
It is interesting to note that, should the interactionterm in the Hamiltonian of Eq. (1) be spread over theentire L-C-R junction, instead of being localized in HC
only, a further term in the current would appear, gener-ated by a two-particle GF:
∆IL =e
~Im
∑ijk∈LCR
l∈L
〈c†i c†jckcl〉 [Vijkl − Vijlk] , (4)
where V is the two-body spread interaction. It is possi-ble to translate this expression in terms of some interac-tion energy expectation values. This indicates that theapproximation of the Hamiltonian as in Eq. (1) meanstreating at the mean-field level the energy fluctuationsof the Coulomb interaction that involve the leads. Inturn, it implies that such fluctuations are neglected asdriving forces for current flow. As mentioned before, thisapproximation at the basis of our treatment, is well justi-fied for the class of physical systems under investigation(e.g. organic nanodevices). It leads also to an impor-tant reduction of the complexity of the whole theory, byavoiding the inclusion of two-particle GF’s.
From Eq. (3) and using the Keldysh non-equilibriumGF techniques38,39 (for the specific details see AppendixA of Ref. [40]) it is possible to write the final expressionfor the current given by Meir and Wingreen:
I =e
2h
∫dω Tr
[fLΓL − fRΓR]AC + i [ΓL − ΓR]G<
C
.
(5)Here AC = i[Gr
C − GaC ] is the spectral function and
Gr,a,<,>C are the (retarded, advanced, lesser, greater)
Green’s functions in the conductor. As in Eq. (3), thetrace should be taken on the conductor basis states (in-cluding spin). The terms ΓL,R represent the couplingmatrices with the L-,R-leads and are defined as:
ΓX = i(ΣrX − Σa
X) (6)Σr,a
X = HCX gr,aX HXC , where X = L, R .
In the latter expression, gr,aX are the retarded and ad-
vanced GF’s of the X-lead, which is considered to bein equilibrium with Fermi occupation function fX(ω).Note also that Σ<
L,R = ifL,RΓL,R and Σ>L,R = −i(1 −
fL,R)ΓL,R.Both terms in Eq. (5) reflect the composition of the
current as a combination of three ingredients: (i) the cou-pling between the conductor and the leads, accounted forby the Γ’s; (ii) the position of energy levels in the con-ductor, given by the spectral function AC(ω); (iii) theoccupations of energy levels, given by fL,R(ω) for theleads and by G<
C(ω) for the conductor. Indeed, whilein the equilibrium case the fluctuation-dissipation theo-rem38 gives G<
C(ω) = ifC(ω)AC(ω), which is by defini-tion a relation between the lesser and the retarded GF’s,

3
in the general out-of-equilibrium case no similar relationsexist and the “occupations” in the conductor should bedetermined by the so called Keldysh equation for G<,to be solved together with the Dyson equation for Gr,38namely:
G<,>C (ω) = Gr
C(ω) Σ<,>C (ω)Ga
C(ω) , (7)Gr,a
C (ω) = Gr,a0,C(ω) + Gr,a
0,C(ω)Σr,aC (ω)Gr,a
C (ω) . (8)
Gr,a0,C are the reference non-interacting (retarded, ad-
vanced) GF’s for the C-region and Σ<,>C (ω), Σr,a
C (ω) arethe self-energies for the interacting system that shouldbe determined by suitable approximations.
B. A generalized Landauer-like formula
If the interaction term in HC of Eq. (1) is neglectedone is left with a mean-field description of the systemand further analysis can be carried out. The mean-fieldquantities are identified here by the subscript 0. An ex-pression for G<,>
0C is available for the (mean-field) out-of-equilibrium regime (details in Appendix A)
G<,>0C = Gr
0C
[Σ<,>
L + Σ<,>R
]Ga
0C , (9)
which is equivalent [cfr Eq. (7)] to the definition of thenon-interacting lesser and greater self-energies as Σ<,>
0,C =Σ<,>
L + Σ<,>R . Subtracting the lesser and greater terms
in Eq. (9) and using the definition Gr −Ga = G> −G<,valid for any Green’s function, one easily derives
Gr0C −Ga
0C = −iGr0C [ΓL + ΓR]Ga
0C . (10)
Inserting Eqs. (9) and (10) into Eq. (5) and taking intoaccount that ΓL Gr
0C ΓR Ga0C = ΓR Gr
0C ΓL Ga0C by virtue
of the particle number conservation in the whole system,one arrives to the so-called Landauer formula,41 as orig-inally expressed by Fisher and Lee:42
I =e
~
∫dω
2π[fL − fR] Tr ΓL Gr
0C ΓR Ga0C . (11)
This equation connects transport quantities such as thecurrent or the zero-temperature conductance to a scat-tering quantity like the transmittance across the scat-tering region, thus allowing for a deeper insight into themechanism of ballistic transport itself. All this formalismand its interpretation break down when any many-bodycoupling among charge carriers is switched on in the con-ductor.
In order to coherently introduce correlation effects intransport calculations we adopted29 an ansatz previouslyproposed in the literature,43,44 to connect retarded andlesser GF’s in the general interacting out-of-equilibriumcase. According to this ansatz, the matrices Σ<,>
C thatappear in Eq. (7) are defined as:
Σ<C(ω) = Σ<
0C(ω)Λ(ω) , (12)Σ>
C(ω) = Σ>0C(ω)Λ(ω) .
Λ(ω) is a suitable dynamical operator to be determinedby imposing the well known identity Σ>−Σ< = Σr−Σa.The above definitions lead to an explicit expression forΛ(ω) which reads:
Λ(ω) = (Σr0C − Σa
0C)−1 (ΣrC − Σa
C) (13)
where Σr,a0C = Σr,a
L + Σr,aR and Σr,a
C = Σr,aL + Σr,a
R + Σr,acorr.
The Σr,acorr term is a contribution to the self-energy op-
erators due specifically to the presence of an electron-electron interaction in the system (more details are inAppendix A). The ansatz is obviously valid in the non-equilibrium mean-field case by definition (just setting Λequal to the identity), but it is also exact in the equi-librium many-body case, since in equilibrium conditionsthe fluctuation-dissipation theorem holds, and the Gr
and G< are no longer independent. An exact relationto connect Σr and Σ< should exist in any equilibriumsituation, even in the presence of many-body couplingsamong charge carriers. The ansatz of Eq. (12) generalizesthe existence of a relation between Gr and G< to the non-equilibrium many-body case, by inspiration from the twoexact limits described above, i.e. non-equilibrium mean-field and equilibrium many-body. It can be viewed as atool partly playing the role of the fluctuation-dissipationtheorem itself.
Once an explicit form of Σ<C(ω) is known in terms of
Σr,aC (ω), one can write down the two following crucial
equations:
G<,>C = Gr
C
[Σ<,>
L + Σ<,>R
]ΛGa
C , (14)Gr
C −GaC = −iGr
C [ΓL + ΓR] ΛGaC . (15)
These are formally analogous to Eqs. (9) and (10) thatallow to obtain the Landauer formula Eq. (11) from theMeir-Wingreen expression for the current, Eq. (5). Thus,they can be used in the same way to write a general“Landauer-like” expression29 which is valid in the generalout-of-equilibrium many-body case:
I =e
~
∫dω
2π[fL − fR] Tr ΓL Gr
C ΓR Λ GaC . (16)
This is one of the central formulae in this work. Here,the inclusion of electronic correlation gives a two-fold ef-fect, namely the presence of the corrective term Λ andthe renormalization of the conductor GF’s, which do nolonger have the subscript 0 as in the non-interacting caseof Eq. (11).
C. Relation to scattering properties
A direct comparison of Eq. (16) with the Landauerformula (which is equal except for the presence of Λ andthe mean-field Green’s functions) gives a scattering inter-pretation also in the many-body case. In fact, the tracein Eq. (11) is identified as the transmittance (across theC-region) in the mean-field case. In analogy, the term

4
Tr ΓL GrC ΓR Λ Ga
C in Eq. (16) plays the same rolein the many-body case and can be defined as an effec-tive transmittance, Teff(ω), for what concerns the rolein transport phenomena. This is an important observa-tion, because it implies that the introduction of e-e in-teractions can be interpreted a posteriori in terms of therenormalization of a well defined quantity, characterizedby a simple and straightforward interpretation.
From Eq. (16) we are also able to obtain an expressionfor the conductance at zero temperature, just derivingthe current wrt the applied voltage (i.e. the differencebetween the Fermi levels of the L and R leads) and settingω = Ef , as in the standard Landauer approach. Theresult reads:
G =e2
hTeff(Ef ) =
e2
hTr ΓL Gr
C ΓR GaC ω=Ef
, (17)
which is exactly the same result obtained in the mean-field case, except that GF’s are renormalized by the e-einteraction. The Λ factor disappears here because it isevaluated at the Fermi level of the whole system (we arein the limit of almost vanishing bias voltage in whichthe equilibrium is re-established), where the imaginarypart of the correlation self-energy Σr
corr −Σacorr vanishes
by definition, thus leading to Σr0,C − Σa
0,C = ΣrC − Σa
C
in Eq. (13), which gives Λ(Ef ) = I. This partly agreeswith other formulations15 that claim the validity of aLandauer formula also in the general interacting case:we agree with Evers and coworkers for what concernsthe conductance, but not for the current because of thepresence of Λ in our approach.
In closing this section, we wish to emphasize that oneof the main advantages of Eq. (16) over its exact counter-part Eq. (5) is the possibility of identifying an effectivetransmittance. This allows us to have a taste of the lin-earized behavior of the current without explicitly facethe out-of-equilibrium problem. The outcome is a use-ful way of separating the effect of introducing correlationand that of handling the mean-field non-equilibrium sit-uation, which is still open and controversial.15
III. IMPLEMENTATION
We numerically implemented the method describedabove in two steps: first we used the recently releasedWanT package33 for the calculation of coherent trans-port properties based on the DFT electronic structureand on the use of maximally localized Wannier functions(MLWF’s) as basis set; then we generalized the above for-mulation to the case in which an interaction is switchedon between charge carriers in the conductor. We de-veloped the new computational framework by mergingthe WanT treatment of the real space GF method witha suitable way of computing the electron-electron SE’s.The SE’s are dealt with here for the case of short rangecorrelations by means of a non perturbative approachbased on solving an effective Anderson Hamiltonian.45
The adopted SE approach is known as Three Body Scat-tering (3BS) method.46,47
A. Coherent transport using maximally localizedWannier Functions
The treatment of the transport problem, in the presentdescription, needs a localized basis set in order to take ad-vantage of the different physical properties of the variousregions (L, C, R) that compose the system. It was re-cently proposed48 that such a basis set could be obtainedby computing the maximally localized Wannier functions(WF’s) through the algorithm given by Marzari and Van-derbilt,30,31 starting from a plane wave electronic struc-ture calculation. Plane-wave implementations of DFTare very popular and successful in the solid state re-search community. The combination of WF’s and GF’stechniques for the transport problem was developed inthe WanT package. The use of WF’s gives highly desir-able features such as the completeness of the basis in achosen subset of eigenvectors of the system and the or-thonormality of the localized basis. Whereas the secondissue is useful from a practical point of view, the firstone is of fundamental importance to solve – in principles– some typical difficulties in the representability (under-and over-completeness) of wavefunctions on other local-ized bases.
The WanT method is based on a DFT computationof the electronic structure of the system. The code isinterfaced with the PWscf49 package which adopts planewaves as basis set and pseudopotentials to describe theions.
Given a periodic system and its Bloch eigenstates |mk〉computed with the PWscf code, the i-th WF at site Rcan be defined as
|iR〉 =1
Nk
∑k∈BZ
e−ik·R∑m
Uim(k) |mk〉 (18)
where the sum for k ∈ BZ runs over the Nk uniform k-points in the Brillouin zone and U(k) is a unitary matrixmixing different bands at the same k. This form ensuresthe orthonormality and the completeness stemming fromthe properties of the Bloch states. The matrix U(k) rep-resents a further set of degrees of freedom which havebeen used to maximize the localization of the resultingWF’s. In practice, a spread functional is defined as
Ω[U ] =∑
i
[〈i0|r2|i0〉 − 〈i0|r|i0〉2
]. (19)
The larger is Ω for a given a set of U(k), the moreextended in space are the WF’s. For our purposes, Ω[U ]must be iteratively minimized with respect to the U(k)matrix to attain the desired maximal localization. For afull description see Refs. [30,31].
In order to solve the transport problem we need tocompute the WF’s for the L,C,R regions, and the mean-

5
field Hamiltonians on such a basis. Three sets of cal-culations are needed for the L and R leads and the Cconductor. DFT calculations are performed by applyingperiodic boundary conditions to a unit supercell. Oneneeds to include in the conductor supercell a part of eachlead that should be large enough to reproduce the behav-ior of the respective bulk at the edges of the cell. Thisis required in order to enable a well-defined matching ofthe three regions, and it is also useful to extract the HCL
and HCR Hamiltonian blocks entering the lead SE’s cal-culation, from the conductor Hamiltonian. The WanTscheme applied with this care contains all the ingredi-ents for the computation of the mean-field transmittancethrough the C-region according to Fisher and Lee as de-scribed in Sec. II C. Further details are in Ref. [48]. Inthe next section we show how this method can be com-plemented by an accurate description of the SE’s to gobeyond the mean-field restriction.
B. Introduction of many-body effects
Electron-electron interactions are directly accountedfor via the inclusion of a related SE operator. It is im-portant to stress here that the large part of the mean-field machinery for transport calculations can be usedalso in the presence of interactions: this property is de-sired for the implementation of a Landauer-like formulaas Eq. (16). This has clearly important consequences inthe actual implementation of the method. Moreover, dueto the fact that we aim at computing equilibrium proper-ties such as the effective transmittance, the e-e retardedSE is easily connected to the lesser one and thus we needto compute just one of them.
The way the SE is calculated depends on the systemunder study and therefore on the specific range of correla-tion. In this work we focus on the case of short range in-teractions used to model conductors with highly localizedorbitals (typical cases in transition metals). We includesome effective on-site Anderson terms and consistentlyavoid double counting eliminating the respective mean-field terms. We then compute the self-energy for theconductor region (which must contain part of the leads,as described above) considering a fictitious periodicity(making the Anderson Hamiltonian an actual HubbardHamiltonian). This is important in order to simulate thenon-finiteness of the open system without treating it ex-plicitly, giving the correct thermodynamic limit and an-alytical properties of the interacting GF’s.50,51 The con-ductor Hamiltonian in the presence of Hubbard-like e-e
interactions reads [see Eq. (1)]:
HC =∑
σ
∑ij∈C
Hij,σ d†i,σdj,σ+
+12
∑σ
∑pq
(Upq − Jpq) np,σnq,σ+
+12
∑σ
∑pq
Upq np,σnq,−σ , (20)
where site and spin indexes are explicitly separated, Upq
and Jpq are the direct and exchange Coulomb integrals,np,σ = d†p,σdp,σ is the density operator for orbital p. In-dexes p and q run over the orbitals defined on sites wherecharge carriers are correlated. The U and J matrices arenot supposed to couple orbitals on different sites.
The solution of the Hamiltonian in Eq. (20) (e.g., interms of GF’s) is not straightforward in the general caseand some further approximations are needed. Here weadopt a scheme46,47 which is non perturbative in the U/tparameter; t is a measure of the mean-field hopping termgiven by Hij,σ in Eq. (20). The non perturbative ap-proach allows us to focus on the regime U ' t, which isforbidden by alternative schemes designed specifically tosolve the two perturbative limits U/t → 0 and t/U → 0.The method is based on a configuration interaction ex-pansion of the states with one particle (an electron or ahole) added to the Fermi sea. This expansion is trun-cated after a certain number Neh of electron-hole pairsis added to the state with N ± 1 electrons. With thechoice to cut the expansion within one added electron-hole pair, the problem can be recast as an effective threebody scattering (3BS) and its solution determines the GFfor the original Hamiltonian. Because the generic Hub-bard Hamiltonian H is periodic in real space, we moveto the basis of Bloch eigenvectors ak mσ of the mean-field problem, defining the electron (+) and hole (−) GFaccording to the Lehmann representation:51
G+k mσ nσ′
(ω) = 〈GS|ak mσ
1z −H
a†k nσ′
|GS〉 (21)
G−k mσ nσ′
(ω) = 〈GS|a†k nσ′
1z −H
ak mσ|GS〉 (22)
where z = ω + E0[N ] + iη+ for electrons and z =ω − E0[N ] − iη+ for holes, E0[N ] being the energy ofthe many-body ground state (GS) with N electrons (in-dicated as |GS〉). In order to calculate the GF, one needsto evaluate the Hamiltonian H in the subspace relatedto N + 1 and N − 1 electron states for G+ and G−
respectively. The expansion of the N − 1 Hamiltonianup to three bodies added to the mean-field ground state(|GS0〉) contains, for each k and σ, the terms:
|s−〉 = ak mσ|GS0〉, (23)
|t−〉 = a†q1 m1σ1aq2 m2σ2
aq3 m3σ3|GS0〉,
where
q1 − q2 − q3 = k, σ1 − σ2 − σ3 = σ (24)

6
Analogous relations hold for the N +1 expansion. For in-ternal consistency, in the calculation of |GS〉 one shouldinclude zero- and two-configuration states in the config-uration interaction expansion. It was demonstrated else-where47 that this procedure does not lead to a renormal-ization of the ground state: therefore, |GS3BS〉 = |GS0〉.We are able to write the interacting GF as the matrixelement of the propagator operator on the |sp〉 states:
Gp
ss′(ω) = 〈sp| 1
zp −H|s′ p〉 (25)
where |s′ p〉 and |sp〉 differ only in the band index and withp = +,− for the electron and hole case according to theabove definitions. We also note that, because the Hub-bard Hamiltonian commutes with the total z−componentof the spin, |GS〉 can be chosen as an eigenstate of Sz,so that the GF is diagonal in the spin index σ (besidesbeing diagonal in the k-vector index by virtue of latticesymmetry).
By projecting the Hamiltonian on the N − 1 elec-tron states through the closure relation (the label “−”is dropped)
∑s |s〉〈s| +
∑t |t〉〈t| = I, we obtain three
terms:
H1 =∑ss′
|s〉〈s|H|s′〉〈s′| (26)
H3 =∑tt′
|t〉〈t|H|t′〉〈t′| (27)
V =∑st
|s〉〈s|H|t〉〈t|+ h.c. (28)
which give the Hamiltonian as H ' H1+H3+V . Definingthe resolvent of the three body interaction H3 as
F3(z) =1
z −H3, (29)
and following Ref. [47] we are able to write the GF as:
G−ss′(ω) =
[z −H1ss
′ +∑tt′
VstF3tt′Vt′s′
]−1
. (30)
The last term in the rhs of Eq. (30) is an effective self-energy for the hole propagator only: we denote it Σ−.Analogous relations are valid for the N +1 electron case.A detailed discussion of the theoretical framework ofthe methods (including the calculation of the three-bodyresolvent F3) can be found in Ref. [47] and referencestherein.
The calculation of the resolvent operator and then ofthe SE’s Σ± requires in input the projected density ofstates (pDOS’s) related to the localized orbitals in theexpression for the Hubbard Hamiltonian, and directlyextracted from the DFT calculation. The final expres-sion for the hole and electron SE’s according to the threebody formalism are then given as:
Σ±qσ(ω) = |qσ〉Σorb±
qσ (ω) 〈qσ| (31)
where q’s are the localized orbitals where Hubbard termsU and J are switched on. The terms Σorb±
qσ (ω) are theso-called orbital self-energies which are the output of thethree body scattering calculation. We remark that these|qσ〉 orbitals are not the calculated Wannier functionsthat we presented in Sec. III A: here the localized orbitalsenter directly the form of the Hamiltonian in Eq. (20)and thus carry physical information. On the other hand,WF’s are used only as basis set and not to define the op-erators themselves, because of their intrinsic non-uniquedefinition.52 We thus decide to define the p-,q-orbitals inEq. (20) (as well as the q’s in the last relation) as thed- or f -atomic orbitals present in the system. Once theSE is written on this basis we can move to the Blochstates by knowing the atomic projections of the bandeigenvectors and then again to the WF basis by a furthertransformation (which is known once the WF’s have beencalculated).
In order to obtain the time-ordered SE’s (which arethe needed objects to introduce correlation in the trans-port calculation) we must calculate both electron andhole GF’s [from Eq. 30]. As it is common in most approx-imated calculation the correct analytic properties of theGF are not garanteed and they can be recovered by con-sidering only its imaginary part and determining anewthe real part by Kramers-Kronig relations. Another pos-sibility, which is reliable and numerically stable when theoccupations of the localized orbitals are far from half-filling,47,53 is to do first the Kramers-Kroenig transformon the imaginary part of the hole and electron orbitalSE’s, and then use the result as the full retarded self-energy. The latter is our choice in the remainder of thispaper.
IV. RESULTS
Break-junction techniques54 have recently obtainedlarge success in producing ultimate nano-junctions. Therealization of a break-junction is conceptually simple, al-though practically sophisticated: A metal wire is firstetched and then mechanically stretched until it is broken.Due to the high mechanical precision of the procedure,tiny junctions and ultimate atomic contacts have beenattained.54 Some metals, such as gold, platinum and irid-ium, are able to form55–58 one-dimensional (1D) atomicchains of variable length. This fact has been recently54
related to an interplay between localization of d-orbitalsand spin-orbit effects, which is typical of 5d transitionmetals. According to the above description, the system ina break-junction configuration is divided into two physi-cally distinct regions: the massive bulk tips and the 1Dwire in between them, as depicted in Fig. 1(a). Fromthe point of view of short range electronic correlations itis expected that the 1D chain gives rise to larger effectsthan the bulk tips, because of a more prominent U/tratio: we remind the reader that t is a measure of theaverage hopping terms of Eq. (20), hence of the band-
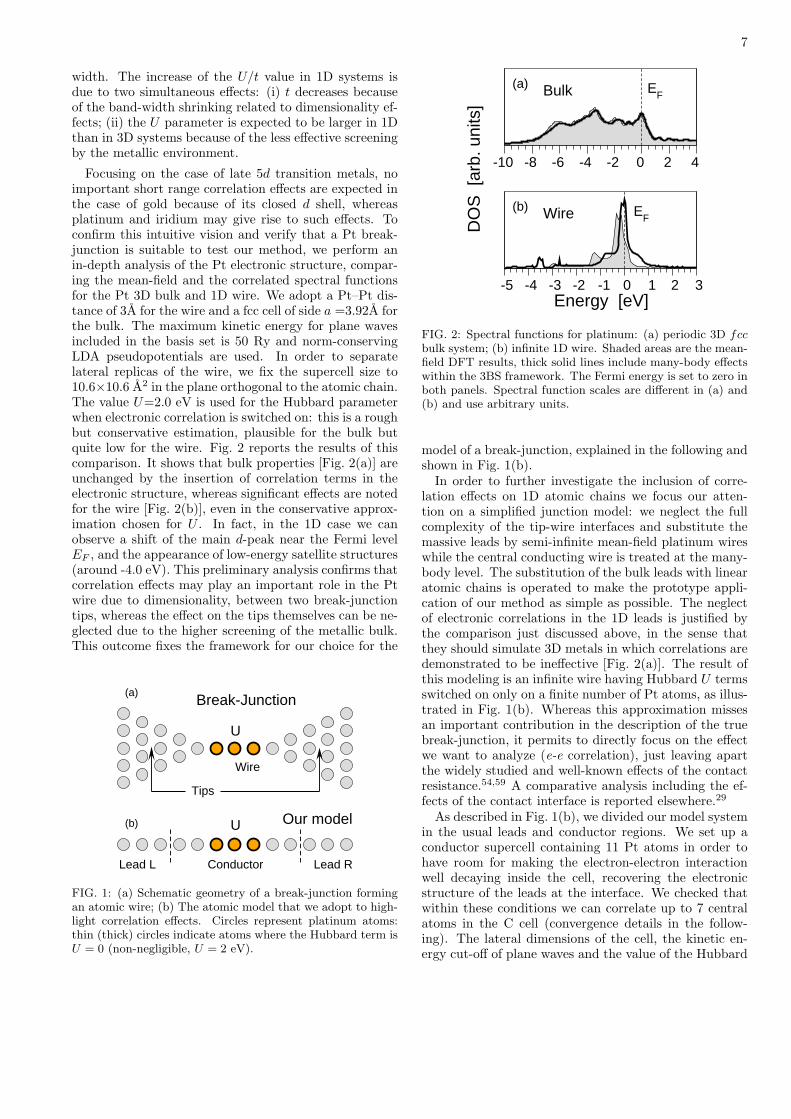
7
width. The increase of the U/t value in 1D systems isdue to two simultaneous effects: (i) t decreases becauseof the band-width shrinking related to dimensionality ef-fects; (ii) the U parameter is expected to be larger in 1Dthan in 3D systems because of the less effective screeningby the metallic environment.
Focusing on the case of late 5d transition metals, noimportant short range correlation effects are expected inthe case of gold because of its closed d shell, whereasplatinum and iridium may give rise to such effects. Toconfirm this intuitive vision and verify that a Pt break-junction is suitable to test our method, we perform anin-depth analysis of the Pt electronic structure, compar-ing the mean-field and the correlated spectral functionsfor the Pt 3D bulk and 1D wire. We adopt a Pt–Pt dis-tance of 3A for the wire and a fcc cell of side a =3.92A forthe bulk. The maximum kinetic energy for plane wavesincluded in the basis set is 50 Ry and norm-conservingLDA pseudopotentials are used. In order to separatelateral replicas of the wire, we fix the supercell size to10.6×10.6 A2 in the plane orthogonal to the atomic chain.The value U=2.0 eV is used for the Hubbard parameterwhen electronic correlation is switched on: this is a roughbut conservative estimation, plausible for the bulk butquite low for the wire. Fig. 2 reports the results of thiscomparison. It shows that bulk properties [Fig. 2(a)] areunchanged by the insertion of correlation terms in theelectronic structure, whereas significant effects are notedfor the wire [Fig. 2(b)], even in the conservative approx-imation chosen for U . In fact, in the 1D case we canobserve a shift of the main d-peak near the Fermi levelEF , and the appearance of low-energy satellite structures(around -4.0 eV). This preliminary analysis confirms thatcorrelation effects may play an important role in the Ptwire due to dimensionality, between two break-junctiontips, whereas the effect on the tips themselves can be ne-glected due to the higher screening of the metallic bulk.This outcome fixes the framework for our choice for the
Wire
Break-Junction
Our model
(a)
(b) U
U
Tips
ConductorLead L Lead R
FIG. 1: (a) Schematic geometry of a break-junction formingan atomic wire; (b) The atomic model that we adopt to high-light correlation effects. Circles represent platinum atoms:thin (thick) circles indicate atoms where the Hubbard term isU = 0 (non-negligible, U = 2 eV).
-5 -4 -3 -2 -1 0 1 2 3Energy [eV]
-10 -8 -6 -4 -2 0 2 4
(a)
(b) Wire
Bulk
EF
DO
S [
arb.
uni
ts]
EF
FIG. 2: Spectral functions for platinum: (a) periodic 3D fccbulk system; (b) infinite 1D wire. Shaded areas are the mean-field DFT results, thick solid lines include many-body effectswithin the 3BS framework. The Fermi energy is set to zero inboth panels. Spectral function scales are different in (a) and(b) and use arbitrary units.
model of a break-junction, explained in the following andshown in Fig. 1(b).
In order to further investigate the inclusion of corre-lation effects on 1D atomic chains we focus our atten-tion on a simplified junction model: we neglect the fullcomplexity of the tip-wire interfaces and substitute themassive leads by semi-infinite mean-field platinum wireswhile the central conducting wire is treated at the many-body level. The substitution of the bulk leads with linearatomic chains is operated to make the prototype appli-cation of our method as simple as possible. The neglectof electronic correlations in the 1D leads is justified bythe comparison just discussed above, in the sense thatthey should simulate 3D metals in which correlations aredemonstrated to be ineffective [Fig. 2(a)]. The result ofthis modeling is an infinite wire having Hubbard U termsswitched on only on a finite number of Pt atoms, as illus-trated in Fig. 1(b). Whereas this approximation missesan important contribution in the description of the truebreak-junction, it permits to directly focus on the effectwe want to analyze (e-e correlation), just leaving apartthe widely studied and well-known effects of the contactresistance.54,59 A comparative analysis including the ef-fects of the contact interface is reported elsewhere.29
As described in Fig. 1(b), we divided our model systemin the usual leads and conductor regions. We set up aconductor supercell containing 11 Pt atoms in order tohave room for making the electron-electron interactionwell decaying inside the cell, recovering the electronicstructure of the leads at the interface. We checked thatwithin these conditions we can correlate up to 7 centralatoms in the C cell (convergence details in the follow-ing). The lateral dimensions of the cell, the kinetic en-ergy cut-off of plane waves and the value of the Hubbard
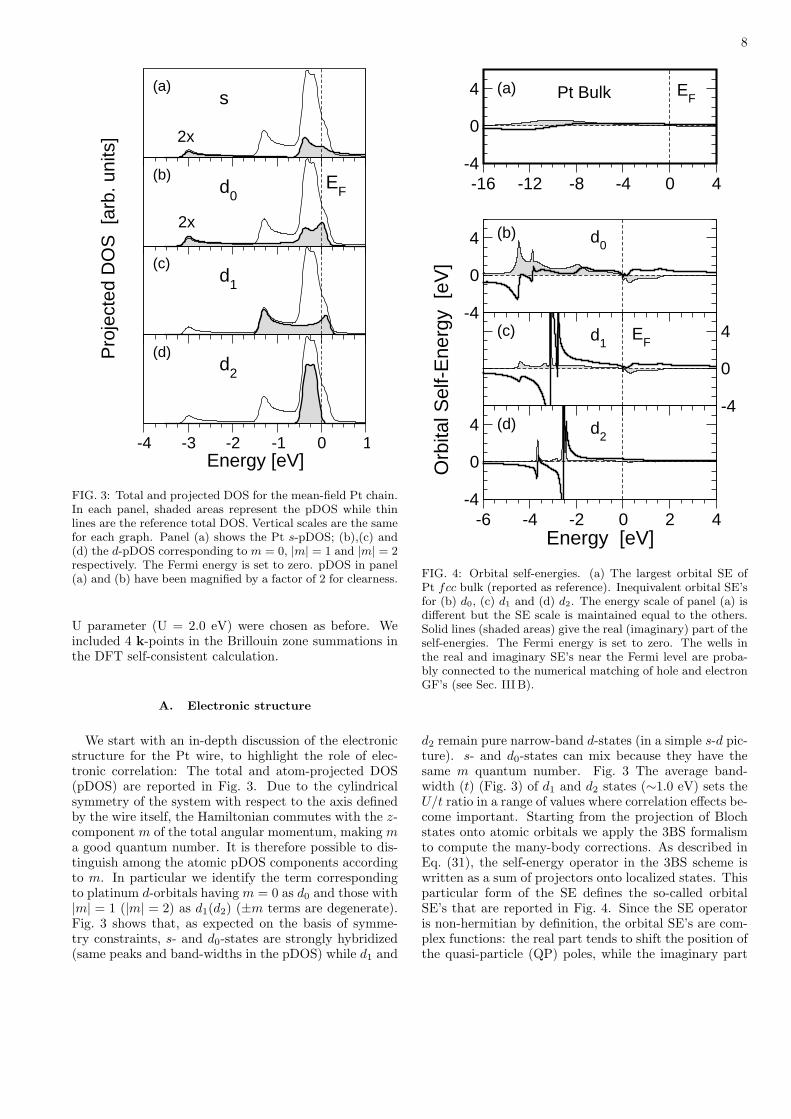
8
-4 -3 -2 -1 0 1Energy [eV]
Pro
ject
ed D
OS
[ar
b. u
nits
]
d2
d1
d0
s(a)
EF(b)
(c)
(d)
2x
2x
FIG. 3: Total and projected DOS for the mean-field Pt chain.In each panel, shaded areas represent the pDOS while thinlines are the reference total DOS. Vertical scales are the samefor each graph. Panel (a) shows the Pt s-pDOS; (b),(c) and(d) the d-pDOS corresponding to m = 0, |m| = 1 and |m| = 2respectively. The Fermi energy is set to zero. pDOS in panel(a) and (b) have been magnified by a factor of 2 for clearness.
U parameter (U = 2.0 eV) were chosen as before. Weincluded 4 k-points in the Brillouin zone summations inthe DFT self-consistent calculation.
A. Electronic structure
We start with an in-depth discussion of the electronicstructure for the Pt wire, to highlight the role of elec-tronic correlation: The total and atom-projected DOS(pDOS) are reported in Fig. 3. Due to the cylindricalsymmetry of the system with respect to the axis definedby the wire itself, the Hamiltonian commutes with the z-component m of the total angular momentum, making ma good quantum number. It is therefore possible to dis-tinguish among the atomic pDOS components accordingto m. In particular we identify the term correspondingto platinum d-orbitals having m = 0 as d0 and those with|m| = 1 (|m| = 2) as d1(d2) (±m terms are degenerate).Fig. 3 shows that, as expected on the basis of symme-try constraints, s- and d0-states are strongly hybridized(same peaks and band-widths in the pDOS) while d1 and
-4
0
4
-4
0
4
-6 -4 -2 0 2 4Energy [eV]
-4
0
4
-16 -12 -8 -4 0 4-4
0
4
EF
EF(a)
(b)
(c)
d0
d2
d1
Orb
ital S
elf-
Ene
rgy
[eV
](d)
Pt Bulk
FIG. 4: Orbital self-energies. (a) The largest orbital SE ofPt fcc bulk (reported as reference). Inequivalent orbital SE’sfor (b) d0, (c) d1 and (d) d2. The energy scale of panel (a) isdifferent but the SE scale is maintained equal to the others.Solid lines (shaded areas) give the real (imaginary) part of theself-energies. The Fermi energy is set to zero. The wells inthe real and imaginary SE’s near the Fermi level are proba-bly connected to the numerical matching of hole and electronGF’s (see Sec. III B).
d2 remain pure narrow-band d-states (in a simple s-d pic-ture). s- and d0-states can mix because they have thesame m quantum number. Fig. 3 The average band-width (t) (Fig. 3) of d1 and d2 states (∼1.0 eV) sets theU/t ratio in a range of values where correlation effects be-come important. Starting from the projection of Blochstates onto atomic orbitals we apply the 3BS formalismto compute the many-body corrections. As described inEq. (31), the self-energy operator in the 3BS scheme iswritten as a sum of projectors onto localized states. Thisparticular form of the SE defines the so-called orbitalSE’s that are reported in Fig. 4. Since the SE operatoris non-hermitian by definition, the orbital SE’s are com-plex functions: the real part tends to shift the position ofthe quasi-particle (QP) poles, while the imaginary part
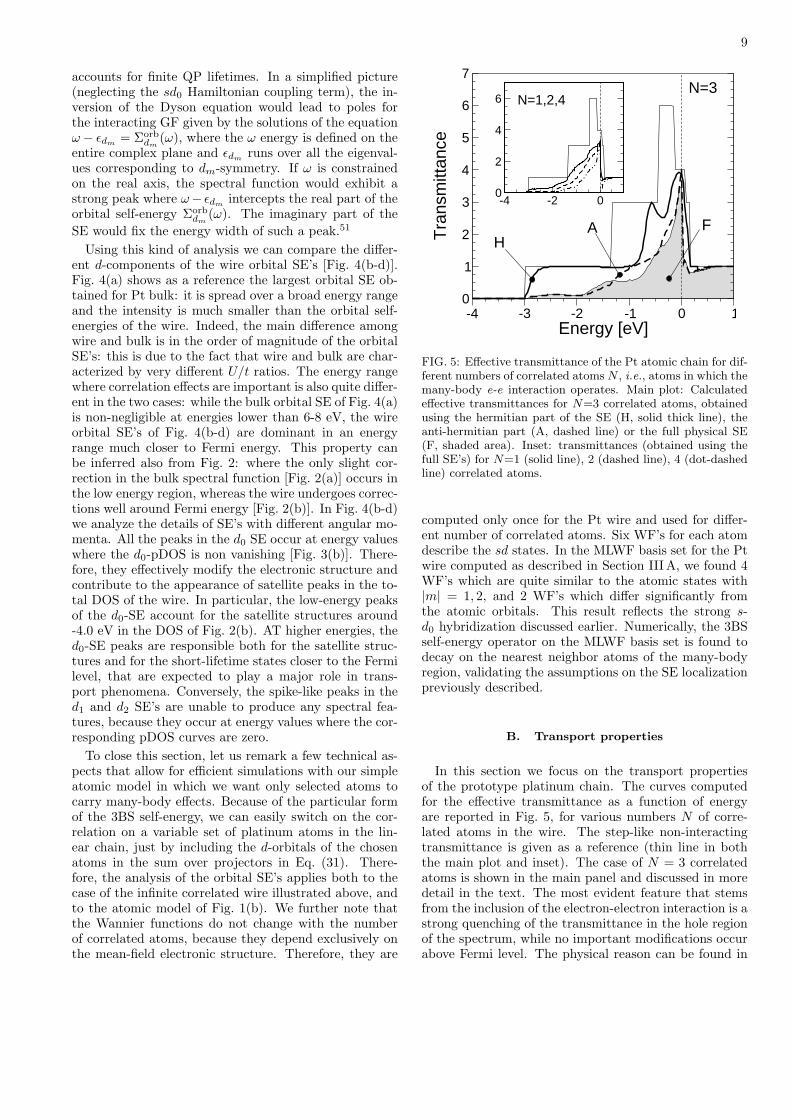
9
accounts for finite QP lifetimes. In a simplified picture(neglecting the sd0 Hamiltonian coupling term), the in-version of the Dyson equation would lead to poles forthe interacting GF given by the solutions of the equationω− εdm = Σorb
dm(ω), where the ω energy is defined on the
entire complex plane and εdm runs over all the eigenval-ues corresponding to dm-symmetry. If ω is constrainedon the real axis, the spectral function would exhibit astrong peak where ω− εdm
intercepts the real part of theorbital self-energy Σorb
dm(ω). The imaginary part of the
SE would fix the energy width of such a peak.51
Using this kind of analysis we can compare the differ-ent d-components of the wire orbital SE’s [Fig. 4(b-d)].Fig. 4(a) shows as a reference the largest orbital SE ob-tained for Pt bulk: it is spread over a broad energy rangeand the intensity is much smaller than the orbital self-energies of the wire. Indeed, the main difference amongwire and bulk is in the order of magnitude of the orbitalSE’s: this is due to the fact that wire and bulk are char-acterized by very different U/t ratios. The energy rangewhere correlation effects are important is also quite differ-ent in the two cases: while the bulk orbital SE of Fig. 4(a)is non-negligible at energies lower than 6-8 eV, the wireorbital SE’s of Fig. 4(b-d) are dominant in an energyrange much closer to Fermi energy. This property canbe inferred also from Fig. 2: where the only slight cor-rection in the bulk spectral function [Fig. 2(a)] occurs inthe low energy region, whereas the wire undergoes correc-tions well around Fermi energy [Fig. 2(b)]. In Fig. 4(b-d)we analyze the details of SE’s with different angular mo-menta. All the peaks in the d0 SE occur at energy valueswhere the d0-pDOS is non vanishing [Fig. 3(b)]. There-fore, they effectively modify the electronic structure andcontribute to the appearance of satellite peaks in the to-tal DOS of the wire. In particular, the low-energy peaksof the d0-SE account for the satellite structures around-4.0 eV in the DOS of Fig. 2(b). AT higher energies, thed0-SE peaks are responsible both for the satellite struc-tures and for the short-lifetime states closer to the Fermilevel, that are expected to play a major role in trans-port phenomena. Conversely, the spike-like peaks in thed1 and d2 SE’s are unable to produce any spectral fea-tures, because they occur at energy values where the cor-responding pDOS curves are zero.
To close this section, let us remark a few technical as-pects that allow for efficient simulations with our simpleatomic model in which we want only selected atoms tocarry many-body effects. Because of the particular formof the 3BS self-energy, we can easily switch on the cor-relation on a variable set of platinum atoms in the lin-ear chain, just by including the d-orbitals of the chosenatoms in the sum over projectors in Eq. (31). There-fore, the analysis of the orbital SE’s applies both to thecase of the infinite correlated wire illustrated above, andto the atomic model of Fig. 1(b). We further note thatthe Wannier functions do not change with the numberof correlated atoms, because they depend exclusively onthe mean-field electronic structure. Therefore, they are
-4 -3 -2 -1 0 1Energy [eV]
0
1
2
3
4
5
6
7
Tra
nsm
ittan
ce
-4 -2 00
2
4
6
H
N=3
A F
N=1,2,4
FIG. 5: Effective transmittance of the Pt atomic chain for dif-ferent numbers of correlated atoms N , i.e., atoms in which themany-body e-e interaction operates. Main plot: Calculatedeffective transmittances for N=3 correlated atoms, obtainedusing the hermitian part of the SE (H, solid thick line), theanti-hermitian part (A, dashed line) or the full physical SE(F, shaded area). Inset: transmittances (obtained using thefull SE’s) for N=1 (solid line), 2 (dashed line), 4 (dot-dashedline) correlated atoms.
computed only once for the Pt wire and used for differ-ent number of correlated atoms. Six WF’s for each atomdescribe the sd states. In the MLWF basis set for the Ptwire computed as described in Section III A, we found 4WF’s which are quite similar to the atomic states with|m| = 1, 2, and 2 WF’s which differ significantly fromthe atomic orbitals. This result reflects the strong s-d0 hybridization discussed earlier. Numerically, the 3BSself-energy operator on the MLWF basis set is found todecay on the nearest neighbor atoms of the many-bodyregion, validating the assumptions on the SE localizationpreviously described.
B. Transport properties
In this section we focus on the transport propertiesof the prototype platinum chain. The curves computedfor the effective transmittance as a function of energyare reported in Fig. 5, for various numbers N of corre-lated atoms in the wire. The step-like non-interactingtransmittance is given as a reference (thin line in boththe main plot and inset). The case of N = 3 correlatedatoms is shown in the main panel and discussed in moredetail in the text. The most evident feature that stemsfrom the inclusion of the electron-electron interaction is astrong quenching of the transmittance in the hole regionof the spectrum, while no important modifications occurabove Fermi level. The physical reason can be found in
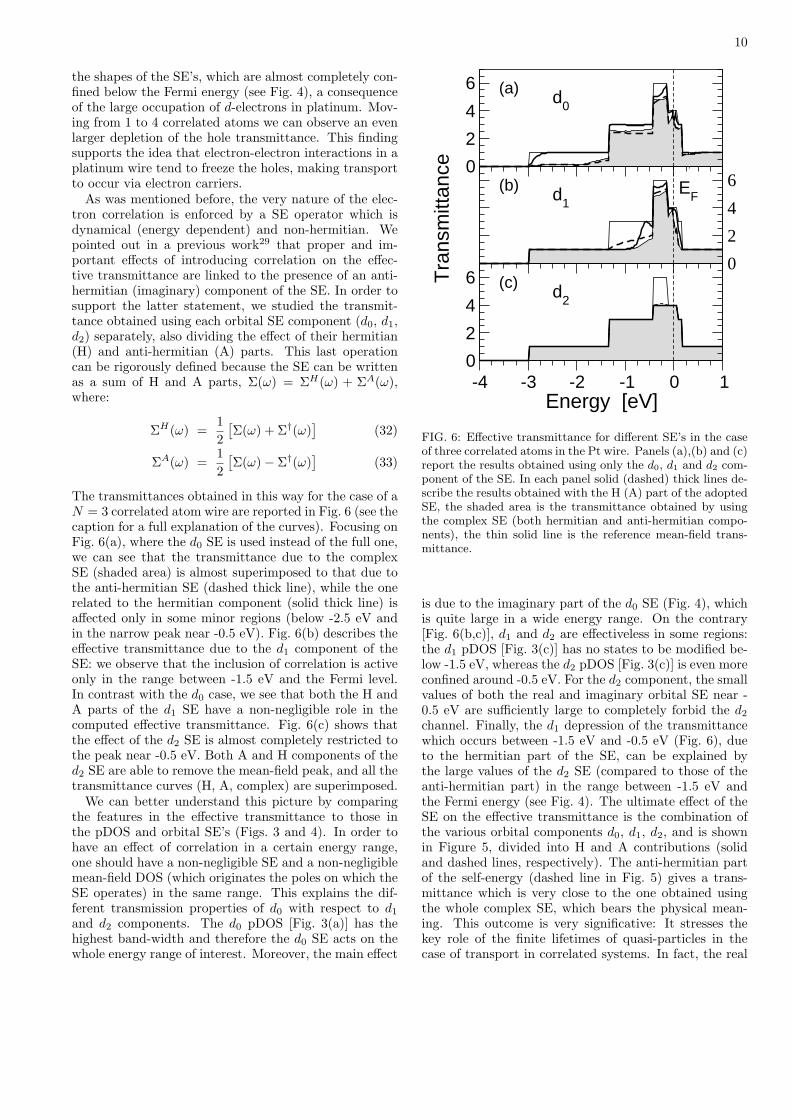
10
the shapes of the SE’s, which are almost completely con-fined below the Fermi energy (see Fig. 4), a consequenceof the large occupation of d-electrons in platinum. Mov-ing from 1 to 4 correlated atoms we can observe an evenlarger depletion of the hole transmittance. This findingsupports the idea that electron-electron interactions in aplatinum wire tend to freeze the holes, making transportto occur via electron carriers.
As was mentioned before, the very nature of the elec-tron correlation is enforced by a SE operator which isdynamical (energy dependent) and non-hermitian. Wepointed out in a previous work29 that proper and im-portant effects of introducing correlation on the effec-tive transmittance are linked to the presence of an anti-hermitian (imaginary) component of the SE. In order tosupport the latter statement, we studied the transmit-tance obtained using each orbital SE component (d0, d1,d2) separately, also dividing the effect of their hermitian(H) and anti-hermitian (A) parts. This last operationcan be rigorously defined because the SE can be writtenas a sum of H and A parts, Σ(ω) = ΣH(ω) + ΣA(ω),where:
ΣH(ω) =12
[Σ(ω) + Σ†(ω)
](32)
ΣA(ω) =12
[Σ(ω)− Σ†(ω)
](33)
The transmittances obtained in this way for the case of aN = 3 correlated atom wire are reported in Fig. 6 (see thecaption for a full explanation of the curves). Focusing onFig. 6(a), where the d0 SE is used instead of the full one,we can see that the transmittance due to the complexSE (shaded area) is almost superimposed to that due tothe anti-hermitian SE (dashed thick line), while the onerelated to the hermitian component (solid thick line) isaffected only in some minor regions (below -2.5 eV andin the narrow peak near -0.5 eV). Fig. 6(b) describes theeffective transmittance due to the d1 component of theSE: we observe that the inclusion of correlation is activeonly in the range between -1.5 eV and the Fermi level.In contrast with the d0 case, we see that both the H andA parts of the d1 SE have a non-negligible role in thecomputed effective transmittance. Fig. 6(c) shows thatthe effect of the d2 SE is almost completely restricted tothe peak near -0.5 eV. Both A and H components of thed2 SE are able to remove the mean-field peak, and all thetransmittance curves (H, A, complex) are superimposed.
We can better understand this picture by comparingthe features in the effective transmittance to those inthe pDOS and orbital SE’s (Figs. 3 and 4). In order tohave an effect of correlation in a certain energy range,one should have a non-negligible SE and a non-negligiblemean-field DOS (which originates the poles on which theSE operates) in the same range. This explains the dif-ferent transmission properties of d0 with respect to d1
and d2 components. The d0 pDOS [Fig. 3(a)] has thehighest band-width and therefore the d0 SE acts on thewhole energy range of interest. Moreover, the main effect
0
2
4
6
0
2
4
6
-4 -3 -2 -1 0 1Energy [eV]
0
2
4
6
EF
(a)
(b)
(c)
d0
d1
d2
Tra
nsm
ittan
ce
FIG. 6: Effective transmittance for different SE’s in the caseof three correlated atoms in the Pt wire. Panels (a),(b) and (c)report the results obtained using only the d0, d1 and d2 com-ponent of the SE. In each panel solid (dashed) thick lines de-scribe the results obtained with the H (A) part of the adoptedSE, the shaded area is the transmittance obtained by usingthe complex SE (both hermitian and anti-hermitian compo-nents), the thin solid line is the reference mean-field trans-mittance.
is due to the imaginary part of the d0 SE (Fig. 4), whichis quite large in a wide energy range. On the contrary[Fig. 6(b,c)], d1 and d2 are effectiveless in some regions:the d1 pDOS [Fig. 3(c)] has no states to be modified be-low -1.5 eV, whereas the d2 pDOS [Fig. 3(c)] is even moreconfined around -0.5 eV. For the d2 component, the smallvalues of both the real and imaginary orbital SE near -0.5 eV are sufficiently large to completely forbid the d2
channel. Finally, the d1 depression of the transmittancewhich occurs between -1.5 eV and -0.5 eV (Fig. 6), dueto the hermitian part of the SE, can be explained bythe large values of the d2 SE (compared to those of theanti-hermitian part) in the range between -1.5 eV andthe Fermi energy (see Fig. 4). The ultimate effect of theSE on the effective transmittance is the combination ofthe various orbital components d0, d1, d2, and is shownin Figure 5, divided into H and A contributions (solidand dashed lines, respectively). The anti-hermitian partof the self-energy (dashed line in Fig. 5) gives a trans-mittance which is very close to the one obtained usingthe whole complex SE, which bears the physical mean-ing. This outcome is very significative: It stresses thekey role of the finite lifetimes of quasi-particles in thecase of transport in correlated systems. In fact, the real
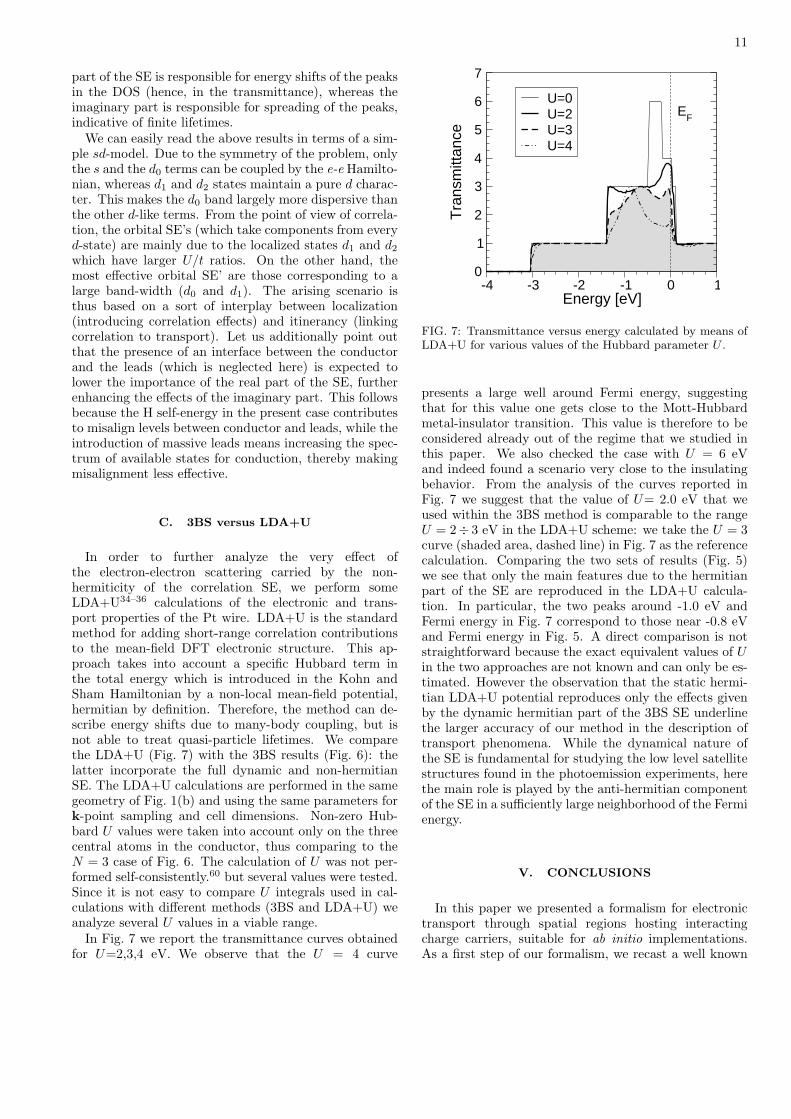
11
part of the SE is responsible for energy shifts of the peaksin the DOS (hence, in the transmittance), whereas theimaginary part is responsible for spreading of the peaks,indicative of finite lifetimes.
We can easily read the above results in terms of a sim-ple sd-model. Due to the symmetry of the problem, onlythe s and the d0 terms can be coupled by the e-e Hamilto-nian, whereas d1 and d2 states maintain a pure d charac-ter. This makes the d0 band largely more dispersive thanthe other d-like terms. From the point of view of correla-tion, the orbital SE’s (which take components from everyd-state) are mainly due to the localized states d1 and d2
which have larger U/t ratios. On the other hand, themost effective orbital SE’ are those corresponding to alarge band-width (d0 and d1). The arising scenario isthus based on a sort of interplay between localization(introducing correlation effects) and itinerancy (linkingcorrelation to transport). Let us additionally point outthat the presence of an interface between the conductorand the leads (which is neglected here) is expected tolower the importance of the real part of the SE, furtherenhancing the effects of the imaginary part. This followsbecause the H self-energy in the present case contributesto misalign levels between conductor and leads, while theintroduction of massive leads means increasing the spec-trum of available states for conduction, thereby makingmisalignment less effective.
C. 3BS versus LDA+U
In order to further analyze the very effect ofthe electron-electron scattering carried by the non-hermiticity of the correlation SE, we perform someLDA+U34–36 calculations of the electronic and trans-port properties of the Pt wire. LDA+U is the standardmethod for adding short-range correlation contributionsto the mean-field DFT electronic structure. This ap-proach takes into account a specific Hubbard term inthe total energy which is introduced in the Kohn andSham Hamiltonian by a non-local mean-field potential,hermitian by definition. Therefore, the method can de-scribe energy shifts due to many-body coupling, but isnot able to treat quasi-particle lifetimes. We comparethe LDA+U (Fig. 7) with the 3BS results (Fig. 6): thelatter incorporate the full dynamic and non-hermitianSE. The LDA+U calculations are performed in the samegeometry of Fig. 1(b) and using the same parameters fork-point sampling and cell dimensions. Non-zero Hub-bard U values were taken into account only on the threecentral atoms in the conductor, thus comparing to theN = 3 case of Fig. 6. The calculation of U was not per-formed self-consistently.60 but several values were tested.Since it is not easy to compare U integrals used in cal-culations with different methods (3BS and LDA+U) weanalyze several U values in a viable range.
In Fig. 7 we report the transmittance curves obtainedfor U=2,3,4 eV. We observe that the U = 4 curve
-4 -3 -2 -1 0 1Energy [eV]
0
1
2
3
4
5
6
7
Tra
nsm
ittan
ce
U=0U=2U=3U=4
EF
FIG. 7: Transmittance versus energy calculated by means ofLDA+U for various values of the Hubbard parameter U .
presents a large well around Fermi energy, suggestingthat for this value one gets close to the Mott-Hubbardmetal-insulator transition. This value is therefore to beconsidered already out of the regime that we studied inthis paper. We also checked the case with U = 6 eVand indeed found a scenario very close to the insulatingbehavior. From the analysis of the curves reported inFig. 7 we suggest that the value of U= 2.0 eV that weused within the 3BS method is comparable to the rangeU = 2÷ 3 eV in the LDA+U scheme: we take the U = 3curve (shaded area, dashed line) in Fig. 7 as the referencecalculation. Comparing the two sets of results (Fig. 5)we see that only the main features due to the hermitianpart of the SE are reproduced in the LDA+U calcula-tion. In particular, the two peaks around -1.0 eV andFermi energy in Fig. 7 correspond to those near -0.8 eVand Fermi energy in Fig. 5. A direct comparison is notstraightforward because the exact equivalent values of Uin the two approaches are not known and can only be es-timated. However the observation that the static hermi-tian LDA+U potential reproduces only the effects givenby the dynamic hermitian part of the 3BS SE underlinethe larger accuracy of our method in the description oftransport phenomena. While the dynamical nature ofthe SE is fundamental for studying the low level satellitestructures found in the photoemission experiments, herethe main role is played by the anti-hermitian componentof the SE in a sufficiently large neighborhood of the Fermienergy.
V. CONCLUSIONS
In this paper we presented a formalism for electronictransport through spatial regions hosting interactingcharge carriers, suitable for ab initio implementations.As a first step of our formalism, we recast a well known

12
expression for the current37 in a Landauer-like form usingan ansatz to relate greater and lesser Green’s functionsto advanced and retarded Green’s functions.43 From theLandauer-like formula for the current in a correlated con-ductor, we then defined an effective transmittance. Thecomputation of the latter quantity may unravel the ef-fects of electronic correlation on transport. We numeri-cally implemented the method in the WanT package,33which describes transport by means of the matrix Green’sfunction technique using maximally localized Wannierfunctions as real space basis set. Many-body terms inthe electronic structure were included following the non-perturbative three body scattering formalism.46,47 We fo-cused our analysis on the case of short-range electron-electron interactions (Anderson regime) and applied theformalism to an atomic platinum chain, where only someatoms are considered beyond the mean-field regime, as aschematic simulator of break-junction experiments.
Our results indicate that short-range many-body inter-actions influence the transport properties. The specificnature of the correlation gives rise to quasi-particle spec-tral features which include the renormalization of energylevels and the appearance of finite lifetimes. The latterare found to be the dominant feature in determining theshape of the effective conductance of the correlated con-ductor. Indeed, a direct comparison with LDA+U resultsfor the same system shows that the inclusion of finite life-times, neglected in the LDA+U approach and accountedfor in the 3BS method, is needed to describe the majoreffects of transmittance quenching.
VI. ACKNOWLEDGMENTS
We acknowledge M. Buongiorno Nardelli, M. J. Cal-das and E. Molinari for inspiring the work and thank C.Jacoboni, C. Calandra and G. Bussi for fruitful discus-sions. Funding was provided by the EC through projectIST-2001-38951 and TMR network “Exciting”, by INFMthrough “Commissione Calcolo Parallelo” and by MIUR(Italy) through “FIRB-NOMADE”.
APPENDIX A: KELDYSH FORMALISM INDEVICE CONFIGURATION
For the sake of completeness, we report a derivationof the final form of Eqs. (7,8) presented in this paper,starting from the general non-equilibrium Green’s func-tions (NEGF’s) formalism as proposed by Keldysh. Thisis particularly useful in view of the study of interactingconductors. More detailed descriptions can be found inRefs. [38–40] and references therein. It is important toremark that in the original Keldysh formalism no distinc-tion is made between different parts of the investigatedsystem, whereas in our approach the target system is aL-C-R junction, where it is important to describe therelevant quantities (GF’s and SE’s) as block-matrices in
the L, C, R zones. Hence, this appendix is essentially de-voted to establish a connection between the SE operatorsspread over the whole many-body system (denoted witha ˜ overhead in this appendix), and the same operatorsin a block-matrix form in the conductor region (denotedwith a C subscript).
The starting point is fixed by the coupled equationsinvolving the retarded, advanced and lesser GF’s for thewhole system, that is, in the cases of interest for us, thesum of the L,C,R regions:
Gr = Gr0 + Gr
0 Σr Gr (A1)
G< = (I + GrΣr)G<0 (I + ΣaGa) + GrΣ<Ga (A2)
The G0 GF’s are related to a non-interacting out-of-equilibrium reference state and the self-energies (SE’s)account for the inclusion of the interactions. In our tar-get L-C-R problem all the GF’s and SE’s can be cast ina 3× 3 block-matrix form defined on the L,C,R regions.The purpose of this Appendix is to show a procedure bywhich Eqs (A1,A2) can be modified in order to extractsome expressions for the conductor-diagonal block of theGF’s.
The Dyson equation [Eq. (A1)] can be rewritten in thefinal form of Eq. (8) by introducing the lead self-energiesdefined as
Σr,aX = (HCX + Σr
CX) gr,aX (HXC + Σr
XC) X = L,R(A3)
and adding them to the C-block ΣrC of Σr. This
comes from a direct inversion of the 3 × 3 block-matrixωI−H − Σr. Note that the above definition of the leadSE’s differs from the one given in Eq. (6) because of thepresence of the non-diagonal Σr
CX block elements. Dueto the approximation of considering the leads at a mean-field level, the SE quickly decays inside the C region(eventually making the conductor larger and larger), andtherefore Σr
CX can be neglected, thus justifying Eq. (6).In systems where this approximation does not hold, theexpressions in Eq. (6) should be updated as in Eq. (A3).The final effective self-energy thus reads
ΣrC = Σr
L + ΣrR + Σr
C (A4)
which is the one given in Eq. (13), once set Σrcorr = Σr
C .The sum in Eq. (A4) defines the conductor self-energy ofEq. (8).
To obtain instead Eq. (7) from the general Keldyshformalism, we observe that it is possible to write theequilibrium mean-field lesser GF as
G<0,eq = Gr
0,eq Σ<0,eq Ga
0,eq , (A5)
where Σ<0,eq = 2feq(ω) η+I and η+ is the limit η → 0+.
Therefore, using the same Eq. (7), with the initial statethe equilibrium mean-field system and the final state thenon-equilibrium mean-field system, one obtains
G<0,neq = Gr
0,neq [Σ<0,eq + Σ<
0,neq]Ga0,neq , (A6)

13
where Σ<0,neq accounts for the non-equilibrium properties
of G<0,neq. In the same way, it is possible to add the
full non-equilibrium many-body correction by adding asuitable SE operator Σ<:
G< = Gr [Σ<0,eq + Σ<
0,neq + Σ<]Ga. (A7)
This last expression describes the lesser GF we are inter-ested in for transport calculations.
We make explicit use of the assumption that the Σ<0,neq
and Σ< SE’s can be neglected out of the conductor-diagonal block, or described as constant mean-field termsin the L or R diagonal blocks. This accounts for a possi-ble rigid shift of energy levels. The operation is viable byvirtue of the fact that the system is out-of-equilibriumand interacting only in the conductor region. Within theabove discussed approximations the expression for theC-block of the lesser GF reads:
G<C(ω) = Gr
CL
[Σ<
0,eq L + Σ<0,neq L
]Ga
LC+
GrC
[Σ<
0,eq C + Σ<0,neq C + Σ<
C
]Ga
C+
GrCR
[Σ<
0,eq R + Σ<0,neq R
]Ga
RC (A8)
Using the same techniques as for the retarded, advancedlead SE’s it is possible to derive:
Gr,aLC = gr,a
L HLC Gr,aC (A9)
Gr,aRC = gr,a
R HRC Gr,aC
Gr,aCL = Gr,a
C HCL gr,aL
Gr,aCR = Gr,a
C HCR gr,aR
which finally give:
G<C(ω) = Gr
C
[Σ<
L + Σ<R + Σ<
0,eq C + Σ<0,neq C + Σ<
C
]Ga
C .
(A10)Here the definitions of the lead lesser SE’s is coherentwith the ones given in Eq. (6).
It should be noted that, whereas some other SE’s per-sist, the Σ<
0,eq C term in Eq. (A10) can be neglected be-cause the η+ term is overcome by other leading terms.Moreover, it can be demonstrated15,59 by means of scat-tering considerations that the term Σ<
0,neq C is also negli-gible. These approximations lead to the final expressionfor the lesser self-energy as Σ<
C = Σ<L +Σ<
R +Σ<corr, where
Σ<corr = Σ<
C , as given in Sec. II B after Eq. (13).
1 M. A. Reed, C. Zhou, C. J. Muller, T. P. Burgin, and J. M.Tour, Science 278, 252 (1997).
2 Z. Yao, H. W. C. Postma, L. Balents, and C. Dekker, Na-ture 402, 273 (1999).
3 C. Joachim, J. K. Gimzewski, and A. Aviram, Nature 408,541 (2000).
4 J. M. Tour, Acc. Chem. Res. 33, 791 (2000).5 J. M. Seminario, A. G. Zacarias, and P. A. Derosa,
J. Phys. Chem. A 5, 791 (2001).6 J. G. Park, G. T. Kim, J. H. Park, H. Y. Yu, G. McIntosh,
V. Krstic, S. H. Jhang, B. Kim, S. H. Lee, S. W. Lee,M. Burghard, S. Roth, and Y. W. Park, Thin Solid Films393, 161 (2001).
7 J. Reichert, R. Ochs, D. Beckmann, H. B. Weber,M. Mayor, and H. v. Lohneysen, Phys. Rev. Lett. 88,176804 (2002).
8 C. Loppacher, M. Guggisberg, O. Pfeiffer, E. Meyer,M. Bammerlin, R. Luthi, R. Schlittler, J. K. Gimzewski,H. Tang, and C. Joachim, Phys. Rev. Lett. 90, 066107(2003).
9 R. M. Metzger, Chem. Rev. 103, 3803 (2003).10 G.-H. Kim and T.-S. Kim, Phys. Rev. Lett. 92, 137203
(2004).11 S. Li, Z. Yu, S.-F. Yen, W. C. Tang, and P. J. Burke,
Nano Lett. 4, 753 (2004).12 J. Taylor, H. Guo, and J. Wang, Phys. Rev. B 63, 245407
(2001).13 Y. Xue, S. Datta, and M. A.Ratner, Chem. Phys. 281,
151 (2002).14 M. Brandbyge, J.-L. Mozos, P. Ordejon, J. Taylor, and
K. Stokbro, Phys. Rev. B 65, 165401 (2002).15 F. Evers, F. Weigend, and M. Koentopp, Phys. Rev. B 69,
235411 (2004).16 D. Ferry and S. M. Goodnik, Transport in nanosctructures,
Cambridge University press, 1997.17 S. J. Tans, M. H. Devoret, R. J. A. Groeneveld, and
C. Dekker, Nature 394, 761 (1998).18 W. A. Schoonveld, J. Wildeman, D. Fichou, P. A. Bobbert,
B. J. van Wees, and T. M. Klapwijk, Nature 404, 977(2000).
19 J. Park, A. N. Pasupathy, J. I. Goldsmith, C. Chang,Y. Yaish, J. R. P. M. Rinkoski, J. P. Sethna, H. D.Abrunna, P. L. McEuen, and D. C. Ralph, Nature 417,722 (2002).
20 P. Jarillo-Herrero, S. Sapmaz, C. Dekker, L. P. Kouwen-hoven, and H. S. J. van der Zant, Nature 429, 389 (2004).
21 T. W. Odom, J.-L. Huang, C. L. Cheung, and C. M. Lieber,Science 290, 1549 (2000).
22 W. Liang, M. P. Shores, M. Bockrath, J. R. Long, andH. Park, Nature 417, 725 (2002).
23 M. Bockrath, D. H. Cobden, J. Lu, A. G. Rinzler, R. E.Smalley, L. Balents, and P. L. McEuen, Nature 397, 598(1999).
24 J. N. Crain, A. Kirakosian, K. N. Altmann, C. Bromberger,S. C. Erwin, J. L. McChesney, J.-L. Lin, and F. J. Himpsel,Phys. Rev. Lett. 90, 176805 (2003).
25 D. S. Kosov, J. Chem. Phys. 119, 1 (2003).26 N. T. Maitra, I. Souza, and K. Burke, Phys. Rev. B 68,
045109 (2003).27 P. Delaney and J. C. Greer, Phys. Rev. Lett. 93, 036805
(2004).28 Y.-C. Chen, M. Zwolak, and M. Di Ventra, Nano Lett. 4,
1709 (2004).29 A. Ferretti, A. Calzolari, R. Di Felice, F. Manghi, M. J.

14
Caldas, M. Buongiorno Nardelli, and E. Molinari, Cond-Mat , 0409222v1 (2004).
30 N. Marzari and D. Vanderbilt, Phys. Rev. B 56, 12847(1997).
31 I. Souza, N. Marzari, and D. Vanderbilt, Phys. Rev. B 65,035109 (2002).
32 M. B. Nardelli, Phys. Rev. B 60, 7828 (1999).33 WanT code by Arrigo Calzolari, C. Cavazzoni,
N. Marzari, and M. Buongiorno Nardelli, 2004,http://www.wannier-transport.org. Ref. [48].
34 V. I. Anisimov, J. Zaanen, and O. K. Andersen,Phys. Rev. B 44, 943 (1991).
35 A. I. Liechtenstein, V. I. Anisimov, and J. Zaanen,Phys. Rev. B 52, R5467 (1995).
36 V. I. Anisimov, F. Aryasetiawan, and A. I. Lichtenstein,J. Phys.-Condens. Mat. 9, 767 (1997).
37 Y. Meir and N. S. Wingreen, Phys. Rev. Lett. 68, 2512(1992).
38 H. Haug and A.-P. Jauho, Quantum kinetics in transportand optics of semiconductors, Springer Berlin, 1996.
39 J. Rammer and H. Smith, Rev. Mod. Phys. 58, 323 (1986).40 A.-P. Jauho, N. S. Wingreen, and Y. Meir, Phys. Rev. B
50, 5528 (1994).41 R. Landauer, Philos. Mag. 21, 863 (1970).42 D. S. Fisher and P. A. Lee, Phys. Rev. B 23, R6851 (1981).43 T.-K. Ng, Phys. Rev. Lett. 76, 487 (1996).44 N. Sergueev, Q. feng Sun, H. Guo, B. G. Wang, and
J. Wang, Phys. Rev. B 65, 165303 (2002).45 G. D. Mahan, Many-particle physics, Plenum, New York,
1981.46 C. Calandra and F. Manghi, Phys. Rev. B 50, 2061 (1994).47 F. Manghi, V. Bellini, and C. Arcangeli, Phys. Rev. B 56,
7149 (1997).48 A. Calzolari, N. Marzari, I. Souza, and M. Buongiorno
Nardelli, Phys. Rev. B 69, 035108 (2004).49 S. Baroni, A. Dal Corso, S. de Gironcoli, and P. Giannozzi,
2001, http://www.pwscf.org.50 B. Farid, Ground and low-lying excited states of interact-
ing electron systems; a survey and some critical analyses,in Electron Correlation in the Solid State, edited by N. H.March, pages 103–261, Imperial College Press, 1999.
51 A. L. Fetter and J. D. Walecka, Quantum theory of many-particle systems, McGraw-Hill, 1971.
52 Wannier functions are linked to physical measurable quan-tities such as the polarization which is given as the sum oftheir centers.
53 S. Monastra, F. Manghi, and C. Ambrosch-Draxl,Phys. Rev. B 64, 020507(R) (2001).
54 N. Agraıt, A. L. Yeyati, and J. M. van Ruitenbeek,Phys. Rep. 377, 81 (2003).
55 H. Ohnishi, Y. Kondo, and K. Takayanagi, Nature 395,780 (1998).
56 A. Yanson, G. R. Bollinger, H. van den Brom, N. Agraıt,and J. M. van Ruitenbeek, Nature 395, 783 (1998).
57 V. Rodrigues, T. Fuhrer, and D. Ugarte,Phys. Rev. Lett. 85, 4124 (2000).
58 R. H. M. Smit, C. Untiedt, A. I. Yanson, and J. M. vanRuitenbeek, Phys. Rev. Lett. 87, 266102 (2001).
59 S. Datta, Electronic transport in mesoscopic systems,Cambridge University Press, 1995.
60 M. Cococcioni and S. de Gironcoli, Cond-Mat , 0405160v1(2004).

Conclusions and outlook
I have addressed the first principles study of transport properties in nanosys-tems focusing on three different problems: (i) the analysis of intermolecularcoupling in polymer crystals (Sec. 2.1), (ii) the study of molecular adsorptionon metallic substrates (Sec. 2.2) and (iii) the inclusion of electron-electroncorrelation in the calculation of transport properties (Sec. 3).
In the first line (i), I compared the DFT electronic structures of twopolymer crystals – namely poly para-phenylene-vinylene and poly-thiophene– in different packings. We ad hoc recasted the problem in terms of interchaintransfer integrals, to link electronic properties and transport quantities andimplement the method at two different levels of approximation. We thencompared the results obtained for different systems and find that interchaininteractions can largely tune the electronic properties of interest by a suitablemodification of the crystalline arrangement.
In the second part (ii) of this thesis, I studied the case of self-assembledmonolayers (SAM’s) of hetero-aromatic thiols (Sec. 2.2) on Copper (100)surface at different coverages. First we addressed the structural proper-ties of the SAM’s by means of DFT total energy calculations. Our resultsidentify the adsorption geometries and energetics for the medium and highcoverages, resulting in a chemisorption picture. We then focused on theelectronic properties of the interface in a combined theoretical-experimentalwork, comparing our findings with photoemission spectroscopy data. Thisapproach allows for a detailed identification and characterization of the spec-tral features, indicating a strong hybridization between metal and molecularstates at the interface. The lineup of these states was determined showingthe presence of hybrid states close to Fermi energy and delocalized on the in-terface. We consider this last result particularly interesting for the transportcharacterization of the interface.
Finally I studied (iii) the effects of including electron correlation inthe simulations of transport properties. I re-derived the formalism leadingto the usually adopted Landauer formula, and attained an expressionrigorously accounting for correlation and similar to the Landauer one. Iimplemented this method using the maximally localized Wannier functionsand the matrix Green’s function approach as in the WanT package (SeeSec. 3). We focused on the short range e-e interactions, describing the elec-

74 Conclusions
tron self-energies by the use of the three-body scattering (3BS) formalism(Sec. 3). We then applied the method to the case Platinum chains andcompute the transmittance accounting for the effect of correlation. Ourresults show an important transmission quenching which is addressed to thepresence of finite lifetimes for quasi-particles in the interacting conductor.This finding is further supported by a direct comparison with standardLDA+U calculations.
Further developments of this work are foreseen along two lines: the appli-cation of the above described methods to other classes of systems, and thefurther development of the theoretical and computational tools presentedin this thesis. As to the former issue, we would like to calculate trans-port properties including correlation in other examples of hybrid molecular-devices where an important role is played by open-shell transition-metalatoms. Such systems became prototypes of single-molecule junctions and amany-body description of transport is required. Other applications involvethe introduction different kind of interactions in the calculation of transportproperties. One example of this is the inclusion of long range correlations(e.g. in a GW spirit). Their effects may have an important role in betterdescribing the alignment of molecular and electrode levels in devices, whichis usually described at the DFT level in most calculations. Other develop-ments are those concerning the calculation of short range correlations withinthe 3BS approach. We would like to remove some of the basic approxima-tions of the method and improve its numerical stability in order to increaseits range of applicability.

Acknowledgments
I am warmly grateful to Elisa Molinari, the tutor of my PhD work, for herconstant encouragement and for supporting my activity in a friendly andlively environment. I am deeply in debt with Alice Ruini and Rosa Di Felicefor their supervision and, more importantly, for their kind help and dailypresence. I also wish to express my personal gratitude to Giovanni Bussiand Arrigo Calzolari, whose contribution to the research topics addressedin this Thesis, in terms of discussions and participation, was extremely pre-cious. The continuative supervision of Marilia J. Caldas is acknowledged:she fundamentally contributed in planning and tailoring my Thesis. Thecollaboration and help of Marco Buongiorno Nardelli was of great impor-tance for the last part of my PhD work. I also received important supportby Franca Manghi, through long and illuminating discussions and patientguide.
I want to thank all the friends at the Physics Department. Undoubtedly,they form a dynamic, sometimes chaotic (better fractal), but familiar andenjoyable group of people that I am proud to belong to.
Finally, last but not least, this work is dedicated to my family.


Full list of publications
• A. Ferretti, A. Calzolari, R. Di Felice, F. Manghi, M. J. Caldas,M. Buongiorno Nardelli, and E. Molinari, First principles theoreticaldescription of transport including electron-electron correlation, submit-ted (October 2004).
• A. Ferretti, A. Calzolari, R. Di Felice, F. Manghi, M. J. Caldas,M. Buongiorno Nardelli, and E. Molinari, First principles theory ofcorrelated transport through nano-junctions, submitted (September2004).
• A. Ferretti and R. Di Felice, Electron delocalization at the hy-brid aromatic-thiol/Cu(100) interface, Phys. Rev. B 70(11), 115412(September 2004).
• R. Di Felice, A. Ferretti, C. Mariani, M. Betti, C. Baldacchini, andV. Di Castro, Surface-science approach to the study of mercaptoben-zoxazole on Cu(111), Surf. Sci. 566–568, 579–584 (June 2004).
• A. Ferretti, A. Ruini, G. Bussi, E. Molinari, and M. J. Caldas, Abinitio study of transport parameters in polymer crystals, Phys. Rev. B69(20), 205205 (May 2004).
• A. Ruini, A. Ferretti, G. Bussi, E. Molinari, and M. J. Caldas, Rela-tionship between structural and optoelectronic properties in semicon-ducting polymers, Semicond. Sci. Technol. 19(4), S362–S364 (Mar.2004).
• A. Ruini, G. Bussi, A. Ferretti, M. J. Caldas, and E. Molinari, Chargetransport and radiative recombination in polythiophene crystals: afirst-principles study, Synth. Met. 139, 755–757 (Oct. 2003).
• G. Bussi, A. Ferretti, A. Ruini, M. J. Caldas, and E. Molinari, Opticsand Transport in Conjugated Polymer Crystals: Interchain InteractionEffects, Adv. Solid State Phys. 43, 313– 326 (Sept. 2003).
• A. Ferretti, A. Ruini, E. Molinari, and M. J. Caldas, Electronic Prop-erties of Polymer Crystals: The Effect of Interchain Interactions,Phys. Rev. Lett. 90(8), 086401 (Feb. 2003).

78 Full list of publications
• A. Ruini, J. Caldas, G. Bussi, A. Ferretti, B. M. Silva, G. Goldoni,and E. Molinari, Optical properties of organic materials: from singlemolecules to solid state, in Radiation-Matter Interaction in ConfinedSystems, edited by L. Andreani, G. Benedek, and E. Molinari, 2002.




















