
Topological switching between an α−β parallel protein and a remarkably helical molten globule
6
Topological Switching between an r- Parallel Protein and a Remarkably Helical Molten Globule Sanne M. Nabuurs, Adrie H. Westphal, Marije aan den Toorn, Simon Lindhoud, and Carlo P. M. van Mierlo* Laboratory of Biochemistry, Wageningen UniVersity, Dreijenlaan 3, 6703 HA Wageningen, The Netherlands Received February 24, 2009; E-mail: [email protected] Abstract: Partially folded protein species transiently exist during folding of most proteins. Often these species are molten globules, which may be on- or off-pathway to native protein. Molten globules have a substantial amount of secondary structure but lack virtually all the tertiary side-chain packing characteristic of natively folded proteins. These ensembles of interconverting conformers are prone to aggregation and potentially play a role in numerous devastating pathologies, and thus attract considerable attention. The molten globule that is observed during folding of apoflavodoxin from Azotobacter vinelandii is off-pathway, as it has to unfold before native protein can be formed. Here we report that this species can be trapped under nativelike conditions by substituting amino acid residue F44 by Y44, allowing spectroscopic characterization of its conformation. Whereas native apoflavodoxin contains a parallel -sheet surrounded by R-helices (i.e., the flavodoxin-like or R- parallel topology), it is shown that the molten globule has a totally different topology: it is helical and contains no -sheet. The presence of this remarkably nonnative species shows that single polypeptide sequences can code for distinct folds that swap upon changing conditions. Topological switching between unrelated protein structures is likely a general phenomenon in the protein structure universe. Introduction Four decades ago, Levinthal 1 predicted that folding of proteins to their native states involves folding pathways on which folding intermediates reside. Today, the considerable advances made in experimental methods have shown that hardly any protein exhibits true two-state folding behavior. 2 Ultrarapid mixing experiments led to the detection of short-lived kinetic intermedi- ates, 3 and techniques such as Förster resonance energy transfer (FRET), fluorescence correlation spectroscopy (FCS), and NMR revealed partially folded states that are difficult to detect. 4,5 Occasionally proteins can be changed by mutagenesis to create a polypeptide that forms a relatively stable folding intermediate. 6-8 In general, experimental data on folding intermediates argue for near-native topology of these species, which have incom- pletely folded or partially misfolded structural elements. 8-10 Most folding proteins encounter folding energy landscapes 11 that are rough. As a result, partially folded intermediates, which may be on- or off-pathway to the native state, are populated. When the intermediate is on-pathway, as is observed for the majority of proteins studied to date, it has nativelike topology and is productive for folding. In contrast, when the intermediate is off-pathway it is trapped in such a manner that the native state cannot be reached without substantial reorganizational events. 2 Several kinetic studies have revealed involvement of off-pathway intermediates during protein folding. 12-18 Structural characterization of folding intermediates is chal- lenging due to the transient nature of their existence. In addition, if an intermediate is observed at equilibrium, it is in most cases sparsely populated relative to the corresponding native and unfolded states. However, characterization of folding intermedi- ates is crucial for the understanding of protein folding. Kinetic intermediates that appear early during folding have been shown to resemble the relatively stable molten globule 19-22 intermedi- ates formed by several proteins under mildly denaturing (1) Levinthal, C. J. Chim. Phys. 1968, 65, 44–45. (2) Jahn, T. R.; Radford, S. E. Arch. Biochem. Biophys. 2008, 469, 100– 117. (3) Roder, H.; Maki, K.; Cheng, H. Chem. ReV. 2006, 106, 1836–1861. (4) Schuler, B. ChemPhysChem 2005, 6, 1206–1220. (5) Kay, L. E. J. Magn. Reson. 2005, 173, 193–207. (6) Schwarzinger, S.; Mohana-Borges, R.; Kroon, G. J.; Dyson, H. J.; Wright, P. E. Protein Sci. 2008, 17, 313–321. (7) Whittaker, S. B.-M.; Spence, G. R.; Grossmann, J. G.; Radford, S. E.; Moore, G. R. J. Mol. Biol. 2007, 366, 1001–1015. (8) Religa, T. L.; Markson, J. S.; Mayor, U.; Freund, S. M.; Fersht, A. R. Nature 2005, 437, 1053–1056. (9) Mayor, U.; Guydosh, N. R.; Johnson, C. M.; Grossmann, J. G.; Sato, S.; Jas, G. S.; Freund, S. M.; Alonso, D. O.; Daggett, V.; Fersht, A. R. Nature 2003, 421, 863–867. (10) Nishimura, C.; Dyson, H. J.; Wright, P. E. J. Mol. Biol. 2006, 355, 139–156. (11) Bryngelson, J. D.; Onuchic, J. N.; Socci, N. D.; Wolynes, P. G. Proteins: Struct., Funct., Genet. 1995, 21, 167–195. (12) Fernandez-Recio, J.; Genzor, C. G.; Sancho, J. Biochemistry 2001, 40, 15234–15245. (13) Bollen, Y. J.; Sanche ´z, I. E.; van Mierlo, C. P. Biochemistry 2004, 43, 10475–10489. (14) Wu, Y.; Vadrevu, R.; Kathuria, S.; Yang, X.; Matthews, C. R. J. Mol. Biol. 2007, 366, 1624–1638. (15) Butler, J. S.; Loh, S. N. J. Mol. Biol. 2005, 350, 906–918. (16) Kathuria, S. V.; Day, I. J.; Wallace, L. A.; Matthews, C. R. J. Mol. Biol. 2008, 382, 467–484. (17) Otzen, D. E.; Giehm, L.; Baptista, R. P.; Kristensen, S. R.; Melo, E. P.; Pedersen, S. Biochim. Biophys. Acta 2007, 1774, 323–333. (18) Lorenz, T.; Reinstein, J. J. Mol. Biol. 2008, 381, 443–455. Published on Web 05/20/2009 10.1021/ja9014309 CCC: $40.75 2009 American Chemical Society 8290 9 J. AM. CHEM. SOC. 2009, 131, 8290–8295
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Topological switching between an α−β parallel protein and a remarkably helical molten globule

Topological Switching between an r-� Parallel Protein and aRemarkably Helical Molten Globule
Sanne M. Nabuurs, Adrie H. Westphal, Marije aan den Toorn, Simon Lindhoud, andCarlo P. M. van Mierlo*
Laboratory of Biochemistry, Wageningen UniVersity, Dreijenlaan 3,6703 HA Wageningen, The Netherlands
Received February 24, 2009; E-mail: [email protected]
Abstract: Partially folded protein species transiently exist during folding of most proteins. Often these speciesare molten globules, which may be on- or off-pathway to native protein. Molten globules have a substantialamount of secondary structure but lack virtually all the tertiary side-chain packing characteristic of nativelyfolded proteins. These ensembles of interconverting conformers are prone to aggregation and potentiallyplay a role in numerous devastating pathologies, and thus attract considerable attention. The molten globulethat is observed during folding of apoflavodoxin from Azotobacter vinelandii is off-pathway, as it has tounfold before native protein can be formed. Here we report that this species can be trapped under nativelikeconditions by substituting amino acid residue F44 by Y44, allowing spectroscopic characterization of itsconformation. Whereas native apoflavodoxin contains a parallel �-sheet surrounded by R-helices (i.e., theflavodoxin-like or R-� parallel topology), it is shown that the molten globule has a totally different topology:it is helical and contains no �-sheet. The presence of this remarkably nonnative species shows that singlepolypeptide sequences can code for distinct folds that swap upon changing conditions. Topological switchingbetween unrelated protein structures is likely a general phenomenon in the protein structure universe.
Introduction
Four decades ago, Levinthal1 predicted that folding of proteinsto their native states involves folding pathways on which foldingintermediates reside. Today, the considerable advances madein experimental methods have shown that hardly any proteinexhibits true two-state folding behavior.2 Ultrarapid mixingexperiments led to the detection of short-lived kinetic intermedi-ates,3 and techniques such as Förster resonance energy transfer(FRET), fluorescence correlation spectroscopy (FCS), and NMRrevealed partially folded states that are difficult to detect.4,5
Occasionally proteins can be changed by mutagenesis to createa polypeptide that forms a relatively stable folding intermediate.6-8
In general, experimental data on folding intermediates arguefor near-native topology of these species, which have incom-pletely folded or partially misfolded structural elements.8-10
Most folding proteins encounter folding energy landscapes11
that are rough. As a result, partially folded intermediates, whichmay be on- or off-pathway to the native state, are populated.When the intermediate is on-pathway, as is observed for themajority of proteins studied to date, it has nativelike topologyand is productive for folding. In contrast, when the intermediateis off-pathway it is trapped in such a manner that the nativestate cannot be reached without substantial reorganizationalevents.2 Several kinetic studies have revealed involvement ofoff-pathway intermediates during protein folding.12-18
Structural characterization of folding intermediates is chal-lenging due to the transient nature of their existence. In addition,if an intermediate is observed at equilibrium, it is in most casessparsely populated relative to the corresponding native andunfolded states. However, characterization of folding intermedi-ates is crucial for the understanding of protein folding. Kineticintermediates that appear early during folding have been shownto resemble the relatively stable molten globule19-22 intermedi-ates formed by several proteins under mildly denaturing(1) Levinthal, C. J. Chim. Phys. 1968, 65, 44–45.
(2) Jahn, T. R.; Radford, S. E. Arch. Biochem. Biophys. 2008, 469, 100–117.
(3) Roder, H.; Maki, K.; Cheng, H. Chem. ReV. 2006, 106, 1836–1861.(4) Schuler, B. ChemPhysChem 2005, 6, 1206–1220.(5) Kay, L. E. J. Magn. Reson. 2005, 173, 193–207.(6) Schwarzinger, S.; Mohana-Borges, R.; Kroon, G. J.; Dyson, H. J.;
Wright, P. E. Protein Sci. 2008, 17, 313–321.(7) Whittaker, S. B.-M.; Spence, G. R.; Grossmann, J. G.; Radford, S. E.;
Moore, G. R. J. Mol. Biol. 2007, 366, 1001–1015.(8) Religa, T. L.; Markson, J. S.; Mayor, U.; Freund, S. M.; Fersht, A. R.
Nature 2005, 437, 1053–1056.(9) Mayor, U.; Guydosh, N. R.; Johnson, C. M.; Grossmann, J. G.; Sato,
S.; Jas, G. S.; Freund, S. M.; Alonso, D. O.; Daggett, V.; Fersht, A. R.Nature 2003, 421, 863–867.
(10) Nishimura, C.; Dyson, H. J.; Wright, P. E. J. Mol. Biol. 2006, 355,139–156.
(11) Bryngelson, J. D.; Onuchic, J. N.; Socci, N. D.; Wolynes, P. G.Proteins: Struct., Funct., Genet. 1995, 21, 167–195.
(12) Fernandez-Recio, J.; Genzor, C. G.; Sancho, J. Biochemistry 2001,40, 15234–15245.
(13) Bollen, Y. J.; Sanchez, I. E.; van Mierlo, C. P. Biochemistry 2004,43, 10475–10489.
(14) Wu, Y.; Vadrevu, R.; Kathuria, S.; Yang, X.; Matthews, C. R. J. Mol.Biol. 2007, 366, 1624–1638.
(15) Butler, J. S.; Loh, S. N. J. Mol. Biol. 2005, 350, 906–918.(16) Kathuria, S. V.; Day, I. J.; Wallace, L. A.; Matthews, C. R. J. Mol.
Biol. 2008, 382, 467–484.(17) Otzen, D. E.; Giehm, L.; Baptista, R. P.; Kristensen, S. R.; Melo,
E. P.; Pedersen, S. Biochim. Biophys. Acta 2007, 1774, 323–333.(18) Lorenz, T.; Reinstein, J. J. Mol. Biol. 2008, 381, 443–455.
Published on Web 05/20/2009
10.1021/ja9014309 CCC: $40.75 2009 American Chemical Society8290 9 J. AM. CHEM. SOC. 2009, 131, 8290–8295

conditions.19 This resemblance has been demonstrated forR-lactalbumin,23 apomyoglobin,24 RNase H,25 T4 lysozyme,26
and Im727 and suggests that these molten globules can beconsidered as models of transient intermediates.2 Due to exposedhydrophobic groups, molten globules are prone to aggregation.Particularly, a decrease of the folding rate due to the presenceof an off-pathway molten globule, which is kinetically trappedand partially folded, increases the likelihood of protein aggrega-tion. This aggregation phenomenon can have detrimental effectson organisms.28 Understanding the formation and conformationof molten globules offers potential insights into factors respon-sible for protein misfolding, aggregation, and, potentially, fornumerous devastating pathologies.29
Here, we report the trapping under nativelike conditions ofthe molten globule folding intermediate of a 179-residueflavodoxin from Azotobacter Vinelandii, enabling characteriza-tion of its conformation. Flavodoxins are monomeric proteinsinvolved in electron transport and contain a noncovalently boundflavin mononucleotide (FMN) cofactor. These proteins consistof a single structural domain and adopt the flavodoxin-like orR-� parallel topology, which is widely prevalent in nature. Bothdenaturant-induced equilibrium and kinetic (un)folding offlavodoxin and apoflavodoxin (i.e., flavodoxin without FMN)have been characterized by use of guanidine hydrochloride(GuHCl) as denaturant.13,30-34 The folding data show thatapoflavodoxin autonomously folds to its native state, which isstructurally identical to flavodoxin with the exception of residuesin the flavin-binding region of the apoprotein. These residueshave considerable dynamics in apoflavodoxin.35,36 The last stepin flavodoxin folding is binding of the cofactor to nativeapoflavodoxin.30
Kinetic folding of apoflavodoxin involves an energy landscapewith two intermediates and is described by Ioff S unfoldedapoflavodoxinS IonS native apoflavodoxin.13 Intermediate Ion
lies on the productive route from unfolded to native protein, ishighly unstable and is therefore not observed during denaturant-induced equilibrium unfolding. Approximately 90% of foldingmolecules fold via off-pathway intermediate Ioff, which is arelatively stable species that needs to unfold to produce nativeprotein and thus acts as a trap.13 The formation of an off-pathway species is typical for proteins with a flavodoxin-liketopology.32 An off-pathway intermediate is experimentallyobserved for all other R-� parallel proteins of which the kinetic
folding has been investigated: apoflavodoxin from Anabaena,12
CheY,16 cutinase,17 and UMP/CMP kinase.18
Equilibrium unfolding of apoflavodoxin is described by thethree-state model Ioff S unfolded apoflavodoxin S nativeapoflavodoxin (see Figure S1 in Supporting Information).13
Unfolded apoflavodoxin contains several transiently structuredregions that dock, causing formation of Ioff.
37 The intermediatefolds in a noncooperative manner38 and populates significantlyin the concentration range of 1-3 M GuHCl. Its maximalpopulation is at 1.76 M GuHCl with a mole fraction of 0.63(Figure S1d in Supporting Information). The off-pathway speciesis molten globule-like: it is compact, its three tryptophans aresolvent-exposed, and it has severely broadened NMR resonancesdue to exchange between different conformers on the micro- tomillisecond time scale.13,33,39 Although the conformation of thismolten globule is currently unknown and cannot be determinedby using NMR spectroscopy, intermediate Ioff probably containshelices.13 Elevated protein concentrations39 and molecularcrowding40 cause severe aggregation of this species.
Anfinsen’s thermodynamic hypothesis41 states that eachamino acid sequence codes for a protein that folds to the statethat has the lowest free energy. This state is called the nativestate and is characterized by a unique tertiary fold. In this studyit is shown that the equilibrium between native and moltenglobule apoflavodoxin can be shifted, in the absence ofdenaturant, from one monomeric species to the other. Fullpopulation of the molten globule folding state of apoflavodoxinis possible through covalent introduction of just a single extraoxygen atom in the protein, achieved by substituting F44 byY44. This substitution leads to significant destabilization ofnative Y44-apoflavodoxin compared to WT-apoflavodoxin.Upon a mild change in conditions, that is, lowering saltconcentration, virtually all protein molecules exist as moltenglobule, as it has become the lowest energy species. Nowcharacterization of an off-pathway folding intermediate ispossible in the absence of denaturant. We show that it has adrastically different topology compared to native protein: it ishelical and lacks the parallel �-sheet of native apoflavodoxin.Our observations show that interconversion between twounrelated protein folds can be dictated by a single amino acidsequence.
Experimental Section
Proteins. The single cysteine at position 69 in A. Vinelandii(strain ATCC 478) flavodoxin II was substituted by an alanine toavoid covalent dimerization of apoflavodoxin. This protein variantis largely similar to wild-type flavodoxin33,42 and is referred to asWT-flavodoxin. Subsequently, in addition, the phenylalanine atposition 44 was substituted by a tyrosine via site-directed mutagen-esis. The latter protein variant is referred to as Y44-flavodoxin.Both flavodoxin variants were obtained from transformed Escheri-chia coli cells and purified as described.33
To obtain native apoprotein, holoprotein was denatured in 6 MGuHCl. Subsequently, both FMN and denaturant were removed
(19) Arai, M.; Kuwajima, K. AdV. Protein Chem. 2000, 53, 209–282.(20) Ohgushi, M.; Wada, A. FEBS Lett. 1983, 164, 21–24.(21) Redfield, C. Methods 2004, 34, 121–132.(22) Ptitsyn, O. B. AdV. Protein Chem. 1995, 47, 83–229.(23) Arai, M.; Kuwajima, K. Fold. Des. 1996, 1, 275–287.(24) Jennings, P. A.; Wright, P. E. Science 1993, 262, 892–896.(25) Raschke, T. M.; Marqusee, S. Nat. Struct. Biol. 1997, 4, 298–304.(26) Kato, H.; Feng, H.; Bai, Y. J. Mol. Biol. 2007, 365, 870–880.(27) Spence, G. R.; Capaldi, A. P.; Radford, S. E. J. Mol. Biol. 2004, 341,
215–226.(28) Chiti, F.; Dobson, C. M. Annu. ReV. Biochem. 2006, 75, 333–366.(29) Dobson, C. M. Nature 2003, 426, 884–890.(30) Bollen, Y. J.; Nabuurs, S. M.; van Berkel, W. J.; van Mierlo, C. P.
J. Biol. Chem. 2005, 280, 7836–7844.(31) Bollen, Y. J.; Kamphuis, M. B.; van Mierlo, C. P. Proc. Natl. Acad.
Sci. U.S.A. 2006, 103, 4095–4100.(32) Bollen, Y. J.; van Mierlo, C. P. Biophys. Chem. 2005, 114, 181–189.(33) van Mierlo, C. P.; van Dongen, W. M.; Vergeldt, F.; van Berkel, W. J.;
Steensma, E. Protein Sci. 1998, 7, 2331–2344.(34) Visser, N. V.; Westphal, A. H.; van Hoek, A.; van Mierlo, C. P.; Visser,
A. J.; van Amerongen, H. Biophys. J. 2008, 95, 2462–2469.(35) Steensma, E.; Nijman, M. J.; Bollen, Y. J.; de Jager, P. A.; van den
Berg, W. A.; van Dongen, W. M.; van Mierlo, C. P. Protein Sci. 1998,7, 306–317.
(36) Steensma, E.; van Mierlo, C. P. J. Mol. Biol. 1998, 282, 653–666.
(37) Nabuurs, S. M.; Westphal, A. H.; van Mierlo, C. P. M. J. Am. Chem.Soc. 2008, 130, 16914–16920.
(38) Nabuurs, S. M.; Westphal, A. H.; van Mierlo, C. P. M. J. Am. Chem.Soc. 2009, 131, 2739–2746.
(39) van Mierlo, C. P.; van den Oever, J. M.; Steensma, E. Protein Sci.2000, 9, 145–157.
(40) Engel, R.; Westphal, A. H.; Huberts, D. H.; Nabuurs, S. M.; Lindhoud,S.; Visser, A. J.; van Mierlo, C. P. J. Biol. Chem. 2008, 283, 27383–27394.
(41) Anfinsen, C. B. Science 1973, 181, 223–230.(42) Steensma, E.; Heering, H. A.; Hagen, W. R.; Van Mierlo, C. P. Eur.
J. Biochem. 1996, 235, 167–172.
J. AM. CHEM. SOC. 9 VOL. 131, NO. 23, 2009 8291
Topological Switching between Unrelated Structures A R T I C L E S

via gel filtration on a Superdex 75 column, which is loaded with100 mM potassium pyrophosphate (KPPi), pH 6.0. During thisremoval step, the protein folds to native apoprotein.
KPPi was the buffer during all experiments, as the use of thisbuffer gave rise to high-quality NMR spectra of A. Vinelandiiflavodoxin (i.e., sharp NMR resonances were obtained).
Fluorescence. Steady-state fluorescence measurements of de-naturant-induced equilibrium unfolding were done at 25 °C on aCary Eclipse fluorometer (Varian). Excitation was at 280 nm andemission was measured at 340 nm. Protein concentration was 4µM in 100 mM KPPi at pH 6.0.
Fluorescence emission spectra were obtained at 25 °C. Excitationwas at 280 nm. Protein concentration was 4 µM. The buffer usedwas 100 mM KPPi, at pH 6.0, or a 10-fold dilution of this buffer.
Thermal protein unfolding was followed by fluorescence emis-sion. Temperature was increased in a 1.5 mL stirred quartz cuvette(path length 0.4 cm) from 12 to 75 °C at a rate of 1 °C/min.Excitation was at 280 nm and emission was recorded at 340 nm.Protein was in 100 mM KPPi, pH 6.0, and protein concentrationranged between 2.5 - 3.0 µM.
In all experiments, excitation and emission slits were set to awidth of 5 nm.
Far-UV CD. CD measurements were acquired by use of a JascoJ715 spectropolarimeter.
Denaturant-induced equilibrium unfolding was followed at 25°C in a 2-mm quartz cuvette by measuring ellipticities for a periodof 3 min at 222 and 255 nm, and the obtained signals weresubsequently averaged. The averaged ellipticity at 255 nm wassubtracted from the averaged ellipticity at 222 nm. Proteinconcentration was 4 µM in 100 mM KPPi at pH 6.0.
CD spectra of protein in 1-mm quartz cuvettes were obtainedby averaging 20 scans and corrected by subtracting spectra ofcorresponding blank solutions. Protein concentration ranged be-tween 1 and 2 µM. Buffer used was 100 mM KPPi, at pH 6.0, ora 10-fold dilution of this buffer.
Thermal unfolding of Y44-apoflavodoxin was followed at 210nm. Temperature was increased from 10 to 75 °C at a rate of 1°C/min in a 1.5 mL stirred quartz cuvette (path length 0.4 cm).Protein concentration was 3 µM in 100 mM KPPi at pH 6.0.
Fluorescence Anisotropy. Anisotropy was measured at 25 °Con a Fluorolog 3.2.2 fluorometer (Horiba Jobin Yvon Ltd.).Excitation was at 300 nm with a 1 nm slit, and emission was at345 nm with a 14 nm slit. For each protein sample, fivemeasurements were done at each setting and the data weresubsequently averaged. In the case of denaturant-induced equilib-rium unfolding of Y44-apoflavodoxin, protein concentration was4 µM in 100 mM KPPi, pH 6.0. In the experiments in which theKPPi concentration was decreased by diluting the buffer solution,protein concentration ranged between 2.7 and 3.0 µM.
NMR. Gradient-enhanced 1H-15N heteronuclear single quantumcoherence (HSQC) spectra were recorded on a Bruker AMX 500MHz instrument. Sample temperature was 25 °C.
Data Analysis. a. Denaturant-Induced Equilibrium Unfold-ing. A three-state model for apoflavodoxin equilibrium unfoldingwas globally fitted to fluorescence emission intensity data at 340nm, CD data at 222 nm, and fluorescence anisotropy data at 345nm (see Supporting Information).
b. Thermal-Induced Equilibrium Unfolding. A two-statemodel of unfolding, in which the change in free energy for thermal-induced protein unfolding is described by the modified Gibbs-Helmholtz equation, was fitted to individual thermal unfoldingcurves (see Supporting Information).
c. Dissociation Constant of the Y44-Apoflavodoxin-FMNcomplex. The dissociation constant was determined via quenchingof FMN fluorescence upon binding of cofactor to apoprotein30 (seeSupporting Information).
Results and Discussion
Design of Y44-Apoflavodoxin. Residue F44 is a highlyconserved amino acid residue in flavodoxins.43 It is part of themajor hydrophobic core of flavodoxin, containing residues thatare in a rigid three-dimensional geometry in native protein(Figure 1).44 Consequently, substitution of F44 by the slightlylarger and more hydrophilic residue tyrosine is expected todecrease the stability of the native state of apoflavodoxin againstunfolding. It potentially avoids significant population of thenative state of Y44-apoflavodoxin under conditions that favorfolding. This subtle residue substitution will have much lesseffect on the stability of the molten globule intermediate of Y44-apoflavodoxin than it has on the stability of native Y44-apoflavodoxin, as this intermediate with exposed hydrophobicgroups is highly dynamic.13,33,39 Unfolded apoflavodoxin isexpected to remain largely unaltered upon substituting F44 byY44, as unfolded molecules are more dynamic than moltenglobule protein molecules.37
FMN Is Tightly Bound to Y44-Flavodoxin. The dissociationconstant of the Y44-apoflavodoxin-FMN complex is deter-mined by titrating a solution containing Y44-apoflavodoxin toa solution containing FMN (see Supporting Information). Uponcofactor binding to apoflavodoxin, FMN fluorescence quenches.The corresponding FMN binding curve is shown in Figure S2in Supporting Information, and the fitted dissociation constantKD of the Y44-apoflavodoxin-FMN complex is (4.6 ( 0.1) ×10-10 M. This value differs only slightly from the one thatcharacterizes the WT-apoflavodoxin-FMN complex [i.e., KD
is (3.4 ( 0.6) × 10-10 M30]. Consequently, substitution of F44by Y44 hardly affects the capacity of apoflavodoxin to bindFMN tightly.
Substitution of F44 by Y44 Does Not Alter the Conforma-tion of (Apo)flavodoxin. Tight FMN binding occurs primarilythrough a very specific combination and geometry of hydrogen
(43) Larkin, M. A.; Blackshields, G.; Brown, N. P.; Chenna, R.; McGet-tigan, P. A.; McWilliam, H.; Valentin, F.; Wallace, I. M.; Wilm, A.;Lopez, R.; Thompson, J. D.; Gibson, T. J.; Higgins, D. G. Bioinfor-matics 2007, 23, 2947–2948.
(44) Alagaratnam, S.; van Pouderoyen, G.; Pijning, T.; Dijkstra, B. W.;Cavazzini, D.; Rossi, G. L.; Van Dongen, W. M.; van Mierlo, C. P.;van Berkel, W. J.; Canters, G. W. Protein Sci. 2005, 14, 2284–2295.
Figure 1. Residue F44 resides in a hydrophobic pocket of WT-flavodoxin.(a) Cartoon representation of WT-flavodoxin in which F44 (red) and FMN(yellow) are shown in stick representation (PDB ID 1YOB). (b) Residueswithin 5 Å distance of F44 (red) are highlighted.
8292 J. AM. CHEM. SOC. 9 VOL. 131, NO. 23, 2009
A R T I C L E S Nabuurs et al.
bonds and aromatic interactions with apoflavodoxin.45 Conse-quently, the observation that Y44-apoflavodoxin binds FMNalmost as firmly as WT-apoflavodoxin implies that the three-dimensional structures of Y44- and WT-flavodoxin must besimilar. Indeed, secondary structure content of both proteins isidentical, as revealed by far-UV CD spectroscopy (Figure 2a).In addition, the 1H-15N HSQC spectra of both holoproteinsare alike (Figure 2b). Only a few, relatively small chemicalshift changes due to changing the chemical identity of theside chain of residue 44 are observed. As HSQC spectra arefingerprints of protein conformations, the similarity in cross-peak positions of the backbone amides of Y44- and WT-flavodoxin implies that the corresponding three-dimensionalstructures are nearly indistinguishable.
Just as observed for flavodoxin, it is expected that introductionof only a single extra oxygen atom into the protein, achievedby substituting F44 by Y44, does not affect the three-dimensional structure of native apoflavodoxin. This structurecontains three tryptophans that have specific relative positionsand orientations in the hydrophobic core of the molecule.44
Indeed, in 100 mM KPPi, pH 6.0, at 25 °C, both protein variantsgive rise to almost identical fluorescence emission spectra(wavelengths of emission maxima are identical, i.e., 330 nm,and corresponding fluorescence emission intensities differ onlyslightly). Most importantly, under this experimental circum-stance, native WT-apoflavodoxin has a strikingly low fluores-cence anisotropy of 0.035 ( 0.00313 and anisotropy of Y44-apoflavodoxin is comparably low (i.e., anisotropy is 0.038 (0.002). The low anisotropy of native WT-apoflavodoxin is dueto rapid unidirectional FRET from W167 to W128, as thesetryptophans are only 7 Å apart.34 Consequently, the conforma-tions of native WT- and native Y44-apoflavodoxin must benearly identical.
Although the substitution of F44 by Y44 does not alter theconformation of native apoflavodoxin, it is expected that thissubstitution decreases the stability of native protein againstunfolding, as is shown below.
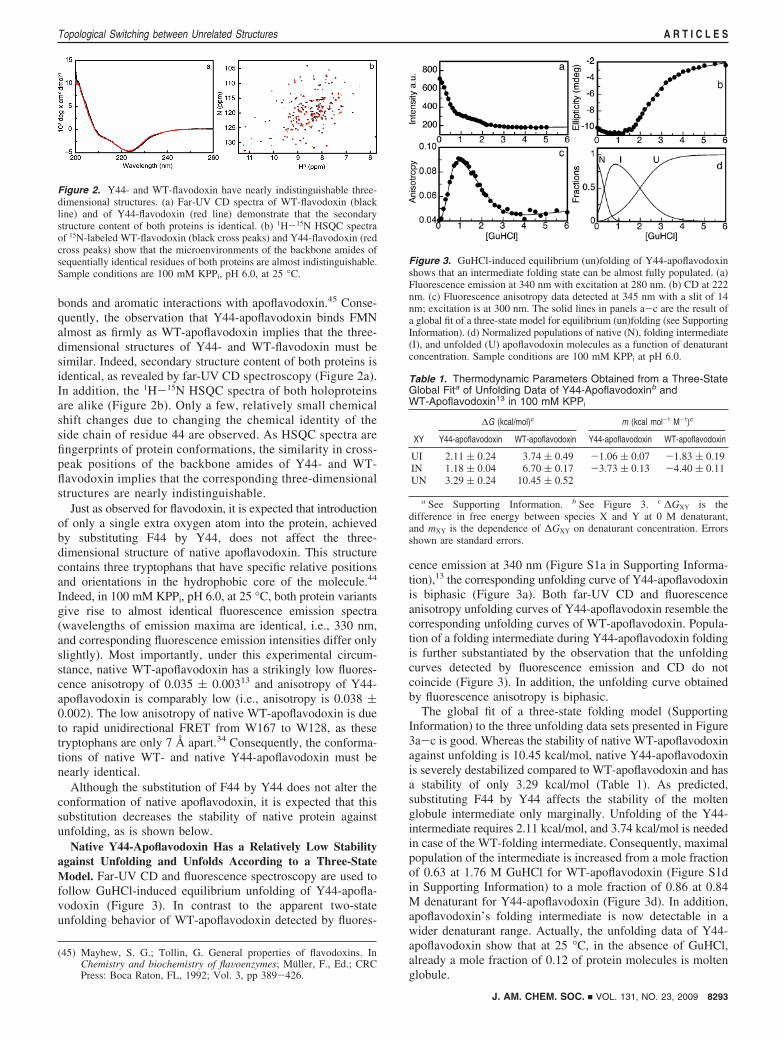
Native Y44-Apoflavodoxin Has a Relatively Low Stabilityagainst Unfolding and Unfolds According to a Three-StateModel. Far-UV CD and fluorescence spectroscopy are used tofollow GuHCl-induced equilibrium unfolding of Y44-apofla-vodoxin (Figure 3). In contrast to the apparent two-stateunfolding behavior of WT-apoflavodoxin detected by fluores-
cence emission at 340 nm (Figure S1a in Supporting Informa-tion),13 the corresponding unfolding curve of Y44-apoflavodoxinis biphasic (Figure 3a). Both far-UV CD and fluorescenceanisotropy unfolding curves of Y44-apoflavodoxin resemble thecorresponding unfolding curves of WT-apoflavodoxin. Popula-tion of a folding intermediate during Y44-apoflavodoxin foldingis further substantiated by the observation that the unfoldingcurves detected by fluorescence emission and CD do notcoincide (Figure 3). In addition, the unfolding curve obtainedby fluorescence anisotropy is biphasic.
The global fit of a three-state folding model (SupportingInformation) to the three unfolding data sets presented in Figure3a-c is good. Whereas the stability of native WT-apoflavodoxinagainst unfolding is 10.45 kcal/mol, native Y44-apoflavodoxinis severely destabilized compared to WT-apoflavodoxin and hasa stability of only 3.29 kcal/mol (Table 1). As predicted,substituting F44 by Y44 affects the stability of the moltenglobule intermediate only marginally. Unfolding of the Y44-intermediate requires 2.11 kcal/mol, and 3.74 kcal/mol is neededin case of the WT-folding intermediate. Consequently, maximalpopulation of the intermediate is increased from a mole fractionof 0.63 at 1.76 M GuHCl for WT-apoflavodoxin (Figure S1din Supporting Information) to a mole fraction of 0.86 at 0.84M denaturant for Y44-apoflavodoxin (Figure 3d). In addition,apoflavodoxin’s folding intermediate is now detectable in awider denaturant range. Actually, the unfolding data of Y44-apoflavodoxin show that at 25 °C, in the absence of GuHCl,already a mole fraction of 0.12 of protein molecules is moltenglobule.
(45) Mayhew, S. G.; Tollin, G. General properties of flavodoxins. InChemistry and biochemistry of flaVoenzymes; Muller, F., Ed.; CRCPress: Boca Raton, FL, 1992; Vol. 3, pp 389-426.
Figure 2. Y44- and WT-flavodoxin have nearly indistinguishable three-dimensional structures. (a) Far-UV CD spectra of WT-flavodoxin (blackline) and of Y44-flavodoxin (red line) demonstrate that the secondarystructure content of both proteins is identical. (b) 1H-15N HSQC spectraof 15N-labeled WT-flavodoxin (black cross peaks) and Y44-flavodoxin (redcross peaks) show that the microenvironments of the backbone amides ofsequentially identical residues of both proteins are almost indistinguishable.Sample conditions are 100 mM KPPi, pH 6.0, at 25 °C.
Figure 3. GuHCl-induced equilibrium (un)folding of Y44-apoflavodoxinshows that an intermediate folding state can be almost fully populated. (a)Fluorescence emission at 340 nm with excitation at 280 nm. (b) CD at 222nm. (c) Fluorescence anisotropy data detected at 345 nm with a slit of 14nm; excitation is at 300 nm. The solid lines in panels a-c are the result ofa global fit of a three-state model for equilibrium (un)folding (see SupportingInformation). (d) Normalized populations of native (N), folding intermediate(I), and unfolded (U) apoflavodoxin molecules as a function of denaturantconcentration. Sample conditions are 100 mM KPPi at pH 6.0.
Table 1. Thermodynamic Parameters Obtained from a Three-StateGlobal Fita of Unfolding Data of Y44-Apoflavodoxinb andWT-Apoflavodoxin13 in 100 mM KPPi
∆G (kcal/mol)c m (kcal mol-1 M-1)c
XY Y44-apoflavodoxin WT-apoflavodoxin Y44-apoflavodoxin WT-apoflavodoxin
UI 2.11 ( 0.24 3.74 ( 0.49 -1.06 ( 0.07 -1.83 ( 0.19IN 1.18 ( 0.04 6.70 ( 0.17 -3.73 ( 0.13 -4.40 ( 0.11UN 3.29 ( 0.24 10.45 ( 0.52
a See Supporting Information. b See Figure 3. c ∆GXY is thedifference in free energy between species X and Y at 0 M denaturant,and mXY is the dependence of ∆GXY on denaturant concentration. Errorsshown are standard errors.
J. AM. CHEM. SOC. 9 VOL. 131, NO. 23, 2009 8293
Topological Switching between Unrelated Structures A R T I C L E S
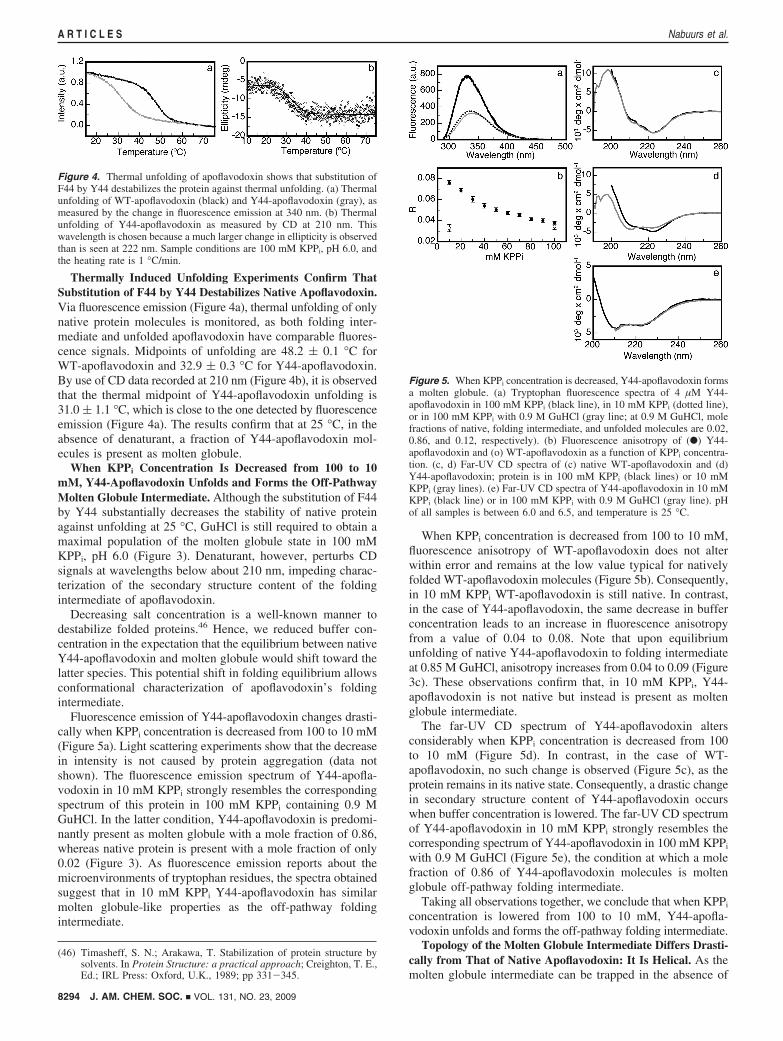
Thermally Induced Unfolding Experiments Confirm ThatSubstitution of F44 by Y44 Destabilizes Native Apoflavodoxin.Via fluorescence emission (Figure 4a), thermal unfolding of onlynative protein molecules is monitored, as both folding inter-mediate and unfolded apoflavodoxin have comparable fluores-cence signals. Midpoints of unfolding are 48.2 ( 0.1 °C forWT-apoflavodoxin and 32.9 ( 0.3 °C for Y44-apoflavodoxin.By use of CD data recorded at 210 nm (Figure 4b), it is observedthat the thermal midpoint of Y44-apoflavodoxin unfolding is31.0 ( 1.1 °C, which is close to the one detected by fluorescenceemission (Figure 4a). The results confirm that at 25 °C, in theabsence of denaturant, a fraction of Y44-apoflavodoxin mol-ecules is present as molten globule.
When KPPi Concentration Is Decreased from 100 to 10mM, Y44-Apoflavodoxin Unfolds and Forms the Off-PathwayMolten Globule Intermediate. Although the substitution of F44by Y44 substantially decreases the stability of native proteinagainst unfolding at 25 °C, GuHCl is still required to obtain amaximal population of the molten globule state in 100 mMKPPi, pH 6.0 (Figure 3). Denaturant, however, perturbs CDsignals at wavelengths below about 210 nm, impeding charac-terization of the secondary structure content of the foldingintermediate of apoflavodoxin.
Decreasing salt concentration is a well-known manner todestabilize folded proteins.46 Hence, we reduced buffer con-centration in the expectation that the equilibrium between nativeY44-apoflavodoxin and molten globule would shift toward thelatter species. This potential shift in folding equilibrium allowsconformational characterization of apoflavodoxin’s foldingintermediate.
Fluorescence emission of Y44-apoflavodoxin changes drasti-cally when KPPi concentration is decreased from 100 to 10 mM(Figure 5a). Light scattering experiments show that the decreasein intensity is not caused by protein aggregation (data notshown). The fluorescence emission spectrum of Y44-apofla-vodoxin in 10 mM KPPi strongly resembles the correspondingspectrum of this protein in 100 mM KPPi containing 0.9 MGuHCl. In the latter condition, Y44-apoflavodoxin is predomi-nantly present as molten globule with a mole fraction of 0.86,whereas native protein is present with a mole fraction of only0.02 (Figure 3). As fluorescence emission reports about themicroenvironments of tryptophan residues, the spectra obtainedsuggest that in 10 mM KPPi Y44-apoflavodoxin has similarmolten globule-like properties as the off-pathway foldingintermediate.
When KPPi concentration is decreased from 100 to 10 mM,fluorescence anisotropy of WT-apoflavodoxin does not alterwithin error and remains at the low value typical for nativelyfolded WT-apoflavodoxin molecules (Figure 5b). Consequently,in 10 mM KPPi WT-apoflavodoxin is still native. In contrast,in the case of Y44-apoflavodoxin, the same decrease in bufferconcentration leads to an increase in fluorescence anisotropyfrom a value of 0.04 to 0.08. Note that upon equilibriumunfolding of native Y44-apoflavodoxin to folding intermediateat 0.85 M GuHCl, anisotropy increases from 0.04 to 0.09 (Figure3c). These observations confirm that, in 10 mM KPPi, Y44-apoflavodoxin is not native but instead is present as moltenglobule intermediate.
The far-UV CD spectrum of Y44-apoflavodoxin altersconsiderably when KPPi concentration is decreased from 100to 10 mM (Figure 5d). In contrast, in the case of WT-apoflavodoxin, no such change is observed (Figure 5c), as theprotein remains in its native state. Consequently, a drastic changein secondary structure content of Y44-apoflavodoxin occurswhen buffer concentration is lowered. The far-UV CD spectrumof Y44-apoflavodoxin in 10 mM KPPi strongly resembles thecorresponding spectrum of Y44-apoflavodoxin in 100 mM KPPi
with 0.9 M GuHCl (Figure 5e), the condition at which a molefraction of 0.86 of Y44-apoflavodoxin molecules is moltenglobule off-pathway folding intermediate.
Taking all observations together, we conclude that when KPPi
concentration is lowered from 100 to 10 mM, Y44-apofla-vodoxin unfolds and forms the off-pathway folding intermediate.
Topology of the Molten Globule Intermediate Differs Drasti-cally from That of Native Apoflavodoxin: It Is Helical. As themolten globule intermediate can be trapped in the absence of
(46) Timasheff, S. N.; Arakawa, T. Stabilization of protein structure bysolvents. In Protein Structure: a practical approach; Creighton, T. E.,Ed.; IRL Press: Oxford, U.K., 1989; pp 331-345.
Figure 4. Thermal unfolding of apoflavodoxin shows that substitution ofF44 by Y44 destabilizes the protein against thermal unfolding. (a) Thermalunfolding of WT-apoflavodoxin (black) and Y44-apoflavodoxin (gray), asmeasured by the change in fluorescence emission at 340 nm. (b) Thermalunfolding of Y44-apoflavodoxin as measured by CD at 210 nm. Thiswavelength is chosen because a much larger change in ellipticity is observedthan is seen at 222 nm. Sample conditions are 100 mM KPPi, pH 6.0, andthe heating rate is 1 °C/min.
Figure 5. When KPPi concentration is decreased, Y44-apoflavodoxin formsa molten globule. (a) Tryptophan fluorescence spectra of 4 µM Y44-apoflavodoxin in 100 mM KPPi (black line), in 10 mM KPPi (dotted line),or in 100 mM KPPi with 0.9 M GuHCl (gray line; at 0.9 M GuHCl, molefractions of native, folding intermediate, and unfolded molecules are 0.02,0.86, and 0.12, respectively). (b) Fluorescence anisotropy of (b) Y44-apoflavodoxin and (o) WT-apoflavodoxin as a function of KPPi concentra-tion. (c, d) Far-UV CD spectra of (c) native WT-apoflavodoxin and (d)Y44-apoflavodoxin; protein is in 100 mM KPPi (black lines) or 10 mMKPPi (gray lines). (e) Far-UV CD spectra of Y44-apoflavodoxin in 10 mMKPPi (black line) or in 100 mM KPPi with 0.9 M GuHCl (gray line). pHof all samples is between 6.0 and 6.5, and temperature is 25 °C.
8294 J. AM. CHEM. SOC. 9 VOL. 131, NO. 23, 2009
A R T I C L E S Nabuurs et al.

denaturant, its conformational characterization is now possible.Native apoflavodoxin contains a parallel �-sheet that is sur-rounded by five R-helices. It gives rise to a far-UV CD spectrumtypical for flavodoxins; that is, the spectrum has a minimum at222 nm and a shoulder at 210 nm (Figure 6). In contrast, thefar-UV CD spectrum of the Y44-folding intermediate undernativelike conditions is typical for R-helical proteins. Thespectrum has minima at 222 and 210 nm and is similar in shapeto the far-UV CD spectrum of the apomyoglobin molten globuleat pH 4.3, a species that is helical.47 Consequently, the topologyof apoflavodoxin’s molten globule drastically differs from theR-� parallel topology of native apoflavodoxin.
A Single Polypeptide Sequence Can Code for MonomericProtein Folds That Are Largely Different. In contrast to mostfolding intermediates studied to date, the molten globule foldingspecies of apoflavodoxin has a remarkably nonnative character.What causes the off-pathway folding intermediate of apofla-vodoxin to be helical?
Upon protein folding, helices are formed much more rapidlythan sheets, especially when parallel �-sheets are involved. Thisrapid helix formation is due to the highly local character of theinteractions in helices. In contrast, the residues that form thestrands of a parallel �-sheet are separated by many interveningresidues,48,49 as is the case for apoflavodoxin. Unfolded apofla-vodoxin contains four regions with reduced flexibility, of whichtwo transiently form native R-helical structures and one formsnonnative R-helical structure.32 These helices are sufficientlystable to be present about 10% of the time and comprise residuesA41-I52, E104-K118, and T160-A169. Rapid formation of
R-helices and their subsequent nonnative docking throughhydrophobic interactions causes formation of the off-pathwayintermediate during apoflavodoxin folding.32 This docking ofhelices prevents formation of the parallel �-sheet of apofla-vodoxin, and as a result it seems likely that the intermediate ishelical. In the study reported here, conclusive data are presentedthat show that the molten globule intermediate of apoflavodoxinindeed is of helical nature. Most likely, the regions that formhelices in unfolded apoflavodoxin are helical in the off-pathwayintermediate as well.
Further support for the absence of a �-sheet in the off-pathwayfolding intermediate of apoflavodoxin is provided by theobserved differences in cooperativity of folding of nativeapoflavodoxin and molten globule. Native apoflavodoxin isformed highly cooperatively,39 which is inherent to the formationof a parallel �-sheet involving many residues. In contrast, theformation of the off-pathway molten globule of apoflavodoxinis clearly noncooperative.38 Consequently, this species cannotcontain a �-sheet, as is indeed confirmed by the data presentedin this study.
Anfinsen’s thermodynamic hypothesis41 states that eachamino acid sequence codes for a single tertiary fold with varyingamounts of local flexibility. However, monomeric proteins like,for example, prions can assume alternative conformations in amultimeric form.28,29 The study presented here shows that asingle polypeptide sequence can code for monomeric proteinfolds that are largely different under nativelike conditions. Theamino acid sequence of apoflavodoxin codes for the R-�parallel topology of the native state as well as for a helicalprotein species. Upon a mild change of conditions, that is,lowering buffer concentration in the case of Y44-apoflavodoxin,topological switching between both folds occurs and a mono-meric protein species with a distinct fold becomes energeticallymost favorable. Our observations, together with those reportedon lymphotactin,50 show that interconversion between unrelated,monomeric protein structures is likely a common phenomenonin the protein structure universe.
Acknowledgment. The Netherlands Organization for ScientificResearch supported this work.
Supporting Information Available: Figures showing GuHCl-induced equilibrium (un)folding data of WT-apoflavodoxin andquenching of FMN fluorescence upon FMN binding to Y44-apoflavodoxin; and description of procedures used to analyze(a) denaturant-induced equilibrium unfolding data, (b) thermal-induced unfolding data, and (c) dissociation constant of the Y44-apoflavodoxin-FMN complex. This material is available freeof charge via the Internet at http://pubs.acs.org.
JA9014309(47) Hughson, F. M.; Wright, P. E.; Baldwin, R. L. Science 1990, 249,1544–1548.
(48) Bieri, O.; Kiefhaber, T. Biol. Chem. 1999, 380, 923–929.(49) Plaxco, K. W.; Simons, K. T.; Baker, D. J. Mol. Biol. 1998, 277,
985–994.(50) Tuinstra, R. L.; Peterson, F. C.; Kutlesa, S.; Elgin, E. S.; Kron, M. A.;
Volkman, B. F. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 5057–5062.
Figure 6. Conformation of Y44-apoflavodoxin in low salt concentrationdiffers drastically from the conformation of WT-apoflavodoxin in its nativestate. In low salt concentration, native Y44-protein is destabilized and formsa helical molten globule intermediate. Shown are far-UV CD spectra ofnative WT-apoflavodoxin in 100 mM KPPi (black line) and of Y44-apoflavodoxin in 10 mM KPPi (gray line), both at 25 °C. Proteinconcentration is ∼2 µM.
J. AM. CHEM. SOC. 9 VOL. 131, NO. 23, 2009 8295
Topological Switching between Unrelated Structures A R T I C L E S








![A chemically modified [alpha]-amylase with a molten-globule state has entropically driven enhanced thermal stability](https://static.fdokumen.com/doc/165x107/631965ccbc8291e22e0f1555/a-chemically-modified-alpha-amylase-with-a-molten-globule-state-has-entropically.jpg)











