
A redox active site containing murrel cytosolic thioredoxin: Analysis of immunological properties

JCP-09-0092.R1(21852)ORIGINAL ARTICLE 1
J o u r n a l o fJ o u r n a l o f
CellularPhysiologyCellularPhysiology
Thioredoxin Interacting Protein(TXNIP) Induces InflammationThrough Chromatin Modificationin Retinal Capillary EndothelialCells Under Diabetic ConditionsQ1
LORENA PERRONE,1 TAKHELLAMBAM S. DEVI,1 KEN-ICHI HOSOYA,2
TETSUYA TERASAKI,3 AND LALIT P. SINGH1,4*1Department of Anatomy/Cell Biology, Wayne State University School of Medicine, Detroit, Michigan2Department of Pharmaceutics, University of Toyama, Toyama, Japan3Department of Molecular Biopharmacy and Genetics, Tohoku University, Aoba, Japan4Department of Ophthalmology, Wayne State University School of Medicine, Detroit, Michigan
Chronic hyperglycemia and activation of receptor for advanced glycation end products (RAGE) are known risk factors for microvasculardisease development in diabetic retinopathy. Thioredoxin-interacting protein (TXNIP), an endogenous inhibitor of antioxidantthioredoxin (TRX), plays a causative role in diabetes and its vascular complications. Herein we investigate whether HG and RAGE induceinflammation in rat retinal endothelial cells (EC) under diabetic conditions in culture through TXNIP activation and whether epigeneticmechanisms play a role in inflammatory gene expression. We show that RAGE activation by its ligand S100B or HG treatment of retinal ECinduces the expression of TXNIP and inflammatory genes such as Cox2, VEGF-A, and ICAM1. TXNIP silencing by siRNA impedes RAGEand HG effects while stable over-expression of a cDNA for human TXNIP in EC elevates inflammation. p38 MAPK-NF-kB signalingpathway and histone H3 lysine (K) nine modifications are involved in TXNIP-induced inflammation. Chromatin immunoprecipitation(ChIP) assays reveal that TXNIP over-expression in EC abolishes H3K9 tri-methylation, a marker for gene inactivation, and increasesH3K9 acetylation, an indicator of gene induction, at proximal Cox2 promoter bearing the NF-kB-binding site. These findings haveimportant implications toward understanding the molecular mechanisms of ocular inflammation and endothelial dysfunction in diabeticretinopathy.
J. Cell. Physiol. 9999: 1–12, 2009. � 2009 Wiley-Liss, Inc.
Additional Supporting Information may be found in the onlineversion of this article.
Contract grant sponsor: AmericanQ2 Diabetes Association CareerDevelopment Award;Contract grant number: 7-03-CD-14.Contract grant sponsor: Mid-West Eye Bank, Michigan.Contract grant sponsor: Marie Curie International Reintegration;Contract grant number: 224892.
Lorena Perrone’s present address is IPBS, CNRS UMR 5089, 205Route de Narbonne, 31 077 Toulouse, France.
*Correspondence to: Lalit P. Singh, Department of Anatomy/CellBiology, Wayne State University School of Medicine, 540 EastCanfield Avenue, #8332, Detroit, MI 48201.E-mail: [email protected]
Received 2 March 2009; Accepted 12 May 2009
Published online in Wiley InterScience(www.interscience.wiley.com.), 00 Month 2009.DOI: 10.1002/jcp.21852
Diabetes leads to various microvascular complications, such asnephropathy, neuropathy, and retinopathy, that are believed toshare common underlying mechanisms. Different metabolicpathways have been considered involved, including enhancedpolyol metabolism, activation of PKC, increased oxidativestress, enhanced hexosamine biosynthesis pathway (HBP), andenhanced expression of the receptor of advanced glycation endproducts (RAGE) and its ligands (Aronson, 2008). However,the molecular causes responsible for the development ofdiabetic complications remain a major challenge so far. Recentstudies suggest that environmental components (i.e., nutrition,exercise, stress) participate in the development of type 1 and2 diabetes and its (micro)vascular complications by inducingaberrant gene expression (Liu et al., 2008). Environmentalfactors affect gene expression through epigenetic changes ofthe genome, such as DNA methylation and histonemodifications (Liu et al., 2008). However, these studies are stillat an early stage and the identification of the environmentalfactors and intracellular mechanisms responsible for theepigenetic changes in diabetes will be important. Sustainedhyperglycemia (HG) and increased RAGE ligands areconsidered environmental factors involved in the developmentof diabetic complications.
RAGE is a plasma membrane receptor of theimmunoglobulin superfamily (Bierhaus et al., 2005) and itsexpression is increased in response to biological stress, such asmechanical vascular injury, inflammation, diabetes, andneurodegenerative diseases. RAGE ligands up-regulate RAGEexpression itself, further enhancing local cellular response(Schmidt et al., 2000). RAGE ligands include AGEs, S100/
� 2 0 0 9 W I L E Y - L I S S , I N C .
calgranulins, amphoterin (HMGB1), and b-amyloid peptides(Bierhaus et al., 2005). It is important to note thatenvironmental factors such as diet lead to AGEs formation andRAGE activation (Goldin et al., 2006). S100/calgranulins andHMGB1 are also RAGE ligands whose expressions are stronglyincreased in diabetic retinopathy (Pachydaki et al., 2006).
Endothelium dysfunction promotes the clinicalcomplications of diabetes because of increased vascular

2 P E R R O N E E T A L .
permeability and thrombogenicity. RAGE induces endotheliumdysfunction in vivo by promoting oxidative stress and vascularhyper-permeability. Blockade of RAGE function by solubleRAGE (sRAGE) inhibits endothelial barrier dysfunction(Wautier et al., 1996). Over-expression of both RAGE and itsligands in the vitreous cavity of patients affected by diabeticproliferative retinal disease leads to amplified RAGE-inducedpro-inflammatory response (Pachydaki et al., 2006).Attenuation of RAGE axis by administration of sRAGE reducesthe development of vascular lesions in vivo (Barile et al., 2005).
The duration and severity of hyperglycemia play a keyfunction in the development of diabetic retinopathy.Hyperglycemia induces biochemical alterations in vascularendothelial cells (EC) leading to cellular dysfunction (Khan andChakrabarti, 2007). Interestingly, the involvement of highglucose (HG)-induced epigenetic mechanisms has been alreadyhypothesized (Khan and Chakrabarti, 2007). Because of therelevance of hyperglycemia and RAGE activation in diabeticretinopathy, it will be important to gain insight into themolecular mechanisms downstream of HG and RAGEactivation and investigate whether they lead to epigeneticmodification and aberrant gene expression.
Hyperglycemia induces the expression ofthioredoxin-interacting protein (TXNIP), which is theendogenous inhibitor of reactive oxygen species (ROS)-scavenging protein thioredoxin (TRX) (Schulze et al., 2004;Chung et al., 2006; Turturro et al., 2007). TXNIP is a critical linkin glucose toxicity and beta-cell apoptosis in diabetic animals(Chen et al., 2008a), and its role in oxidative stress and aorticEC inflammation has been shown previously (Schulze et al.,2004; Chung et al., 2006; Turturro et al., 2007). Mostinterestingly, recent studies reveal that TXNIP plays a keyfunction in diabetes development. Indeed, mice defective forTXNIP are resistant to streptozotocin (STZ)-induced diabetes(Chen et al., 2008b). However, the role of TXNIP in retinalmicrovascular inflammation and dysfunction in diabetes as wellas its influence on retinal EC in vitro has not been investigatedso far.
The aim of the present study was to investigate as to whetherHG and RAGE induce inflammatory gene expression via TXNIPexpression and epigenetic modifications at gene promoters.Therefore, to understand the contribution of TXNIP inendothelial dysfunction in diabetic retinopathy, we firstinvestigated whether TXNIP plays a role in inflammation andchromatin remodeling in retinal EC under in vitro diabeticconditions, and whether RAGE signal involves in TXNIP mRNAexpression and inflammatory gene induction. S100/calgranulin-type proteins are known to increase in diabetic retinopathy(Pachydaki et al., 2006) and they are known ligands for RAGE inaddition to AGEs. Since TXNIP plays a critical role inSTZ-induced diabetes in animal models (Chen et al., 2008Q3),we hypothesized that TXNIP mediates RAGE and HG-inducedinflammation in retinal capillary EC in culture. In this study, wedemonstrate that TXNIP mediates both RAGE- and HG-induced over-expression of inflammatory Cox2, VEGF-A, andRAGE itself. We also observed that TXNIP causes epigeneticchromatin remodeling at inflammatory gene promoter inretinal capillary EC. Pro-inflammatory p38 MAPK and NF-kBsignals are involved in histone H3 chromatin remodeling andinflammatory gene expression. Thus, TXNIP up-regulation indiabetes may play a role in epigenetic chromatin remodeling andsustained inflammation leading to microvascular complicationsincluding diabetic retinopathy.
Materials and MethodsMaterials
Antibodies: anti-TXNIP (MBL Biotechnology, Beaver, MA);anti-actin and anti-p65 NF-kB subunit (Cell Signaling Solutions,
JOURNAL OF CELLULAR PHYSIOLOGY
Lake Placid, NY); anti-acetylated K9 histone H3 and anti-trimethylated histone H3 in K9 (Abcam, Cambridge, MA);anti-RAGE blocking antibody (R&D System, Minneapolis, MN)(Perrone et al., 2008). The enhanced chemiluminescence (ECL)system was from Amersham (Arlington Heights, IL). DMEM and F-12 nutrient mixture (Ham’s) were from GIBCO (Grand Island,NY). Bovine brain S100B protein was purchased from Calbiochem(EMD, San Diego, CA). (E)-2-Fluoro-40-methoxystilbene (NF-kBinhibitor IV), SN50 Cell-Permeable Inhibitor Peptide, p38 MAPKinhibitor SB202190, and PP1 (pyrazolo pyrimidine-type inhibitor 1)were also from Calbiochem (San Diego, CA).
Cell culture
The temperature-sensitive rat TR-iBRB2 were grown as previouslydescribed (Hosoya and Tomi, 2005; Nagase et al., 2006). TR-iBRB2(denotes transgenic-inner blood–retinal barrier EC line 2)differentiates at 378C, but becomes conditionally immortalized at338C. These cells maintain spindle fiber shapes, multicellularnodules, express EC-specific markers, exhibit bipolar morphologylike the primary retinal EC, and maintain other endothelialproperties (Hosoya and Tomi, 2005). TR-iBRB2 cells weremaintained at 338C in DMEM and F-12 nutrient mixture (Ham’s)(4:1 ratio) containing 5.0 mM D-glucose, 10% FCS, 100mg/mlstreptomycin, 100 U/ml penicillin, and 2 mM glutamine. At70–80% confluence, they were differentiated for 2 days at 378C andincubated in the starvation medium (0.2% serum in DMEM mediumwith 100mg/ml streptomycin, 100 U/ml penicillin, and 2 mMglutamine) the desired concentrations of glucose (5 mM, LG or25 mM, HG) or S100B (20mg/ml), or glucosamine (GlcN,1.5 mMþ LG) for different time periods. Cells were serum starved16 h before short time stimulation (from 10 min to 2 h) with HG,GlcN, S100B. For long-term stimulation, cells were serum starved16 h before addition HG, GlcN, or S100B and further continued for24 h. Azaserine (an inhibitor of HBP, 0.5mM) (Sigma, St Louis, MO)was introduced 1 h before adding glucose and maintainedthroughout the period of incubation. Primary human retinal ECwere obtained from ScienCell (San Diego, CA), and employed insome experiments. Human retinal EC were grown in mediumsupplied by the company and according to their instructions andwere maintained at 378C in a humidified incubator of 5% CO2.
Diabetes induction of rats
Diabetes of adult male Sprague–Dawley rats (275–300 g) wasinduced by injection of a single dose of STZ (65 mg/kg i.v., Sigma)dissolved in 0.01 M citrate buffer, pH 4.5, as described previously(Jiang et al., 2006; Singh et al., 2007). Normal rats received a similarvolume of vehicle alone (n¼ 6–8 for diabetic and normal rats). Therats were treated in accordance with the principles of NIHGuidelines for the Care and Use of Laboratory Animals andapproved by the Institutional Animal Investigation Committee. At 4or 8 weeks of diabetes, which achieved a blood-glucose level400–450 mg/dl compare to �117 mg/dl in control rats, they weresacrificed with an overdose of pentobarbital. The retina wereremoved, frozen immediately in liquid N2, and stored at �708Cuntil used. Cellular extracts were prepared in RIPA buffercontaining protease inhibitors by homogenization with a Teflonpiston. Cell debris was removed by centrifugation and the proteinamount in the supernatant was measured by the Coomassie-basedmethod. The levels of TXNIP and TRX were examined on tissueextracts by Western blotting.
Generation of stably transfectant TR-iBRB2 cell lines
The CMV-human TXNIP and CMV-LacZ in pcDNA3.1 plasmidwere used to generate stably over-expressing TXNIP andcorresponding control LacZ cell lines as previously described(Cheng et al., 2008Q4). Several individual clones were selected fromTXNIP and LacZ expressing transfectants. A control LacZ andhTXNIP-expressing TR-iBRB2 cell lines were selected for further

T X N I P I S R E Q U I R E D F O R R A G E - I N D U C E D I N F L A M M A T I O N 3
experiments between 4 and 8 passages. To knock down TXNIPexpression, we performed four independent transfections withfour different plasmids carrying a double stranded oligonucleotidetargeted to different regions of the rat TXNIP mRNA (Super ArrayBioscience Corporation, Frederick, MD). As control, a plasmidencoding a scramble RNA was transfected. The clone that stronglysilenced TXNIP expression was identified by Q-RTPCR andWestern blot analysis.
Real-time quantitative PCR
Gene expression was analyzed by real-time quantitative PCR(Q-RTPCR) using the BioRad Chromo 4 detection system andSYBR Green PCR Master Mix from Applied Biosystems (FosterCity, CA). Primers were designed using Primer Express v 2.0(Applied Biosystems) and synthesized by Invitrogen(Carlsbad, CA).
Primer sequences of genes for the Q-RTPCR are:
(1) Rat TXNIP
JOURNAL OF CELLU
Forward: 50-CTGAAGTTACCCGAGTCAAAGC-30
Reverse: 50-CTCACCTGTAGCCTGGTCTTCT-3
(2) Rat COX 2 Forward: 50-TACCCGGACTGGATTCTACG-30Reverse: 50-AAGTTGGTGGGCTGTCAATC-30
(3) Mouse actin
Forward: 50-ATTATCACCAACTGGGATGACATGG-30Reverse: 50-CCAGCAGATTCCATACCAATGAAAG-30
(4) Rat RAGE
Forward: 50-ACAGAAACCGGTGATGAAGG-30Reverse: 50-CTCTCCTCGAGTC TGGGTTG-30
(5) Rat VEGF
Forward: 50-AGAAAGCCCAATGAAGTGGTG-30Reverse: 50-ACTCCAGGGCTTCATCATTG-30
(6) Rat ICAM1
Forward: 50-CCCCACCTACATACATTCCTAC-30Reverse: 50-ACATTTTCTCCCAGGCATTC-30
(7) Rat iNOS
Forward: 50-ATGGTTCCTGGGCGTGTTGGAATA-30Reverse: 50-TGTAAAGCTGTGGCCCTGACAGTA-30
Total RNA was isolated using TRIzol reagent (Invitrogen).cDNA was synthesized from 1mg of RNA using iScript cDNAsynthesis Kit (BioRad, Hercules, CA) in 20ml according tomanufacturer’s instructions. The Q-RTPCR reaction mixturecontained 1� SYBR Green PCR Master Mix, 400 nM forward andreverse primers, and 2ml of cDNA in a final volume of 25ml. ThePCR cycling was programmed as 958C for 15 s, 558C for 35 s, and728C for 30 s for 40 cycles followed by the construction of a meltingcurve through increasing the temperature from 55 to 958C at aramp rate of 2% for 20 min. The Q-RTPCR samples were evaluatedusing a single predominant peak as a quality control. Ct values wereused to calculate the relative expression level of messenger RNAand they were normalized to actin, as previously described (Chenget al., 2006). As negative controls, the same reaction wasperformed on RNA samples without the reverse transcriptasereaction, and no PCR products were detected.
Chromatin Immunoprecipitation (ChIP)
We used an EZ-ZymeTM Enzymatic Chromatin Prep Kit fromUpstate (Millipore Corp., Bilerica, MA) according to theirinstructions. Cells were fixed in 1% paraformaldehyde for 10 min atroom temperature. Chromatin was precleaned with proteinA/G-sepharose saturated with BSA and salmon sperm DNA.Antibodies to acetylated H3K9 and phosphorylated in Ser10,trimethylated H3K9, and NF-kB were used at 1:100 dilutions. ChIPwas carried out in RIPA buffer (10 mM Tris/Cl, pH 8; 1% TX-100;0.1% DOC; 0.1% SDS, 140 mM NaCl; 1 mM PMSF) at 48C overnightwith continuous rotation. Saturated protein A/G-sepharose wasthen added to the immune complexes and incubated for 1 h at 48Cwith shaking. The immunoprecipitates were washed three timeswith RIPA buffer and once with LiCl buffer (0.25 M LiCl,0.5% NP-40, 0.5% Na deoxycholate, 1 mM EDTA,10 mM Tris/Cl, pH 8). Beads were resuspended in 0.1 ml TE buffer(10 mM Tris/Cl, pH 8; 1 mM EDTA) and treated 30 min with RNase50mg/ml at 378C. Reversion of the cross-linking was carried out byadding Proteinase K 500mg/ml and SDS 0.5% at a finalconcentration for 3 h at 568C and further at 658C overnight. DNAsamples were extracted with phenol/chloroform and ethanol
LAR PHYSIOLOGY
precipitated. The promoter region for Cox2 and iNOS (containingthe NF-kB site) were analyzed by quantitative andsemi-quantitative PCR using the following primers:
1.
Cox2 promF: 50-CAG CAG CCC TCT CAT TTC ATT-30Cox2 promR: 50-TCT TTG AGG TCT CGG GTT TCC-30
2.
iNOS promF: 50-ATGGTTCCTGGGCGTGTTGGAATA-30 ; iNOS promR: 50-TGTAAAGC TGTGGCCCTGACAGTA-30The same promoter regions were also analyzed by semi-quantitative PCR using hot start gold Taq polymerase (AppliedBiosystems). Input DNA amplification was used as a control forDNA loading of the immunoprecipitates. To distinguish thepromoter DNA analysis by real-time PCR from the mRNA analysisby reverse transcriptase cDNA synthesis and real-time PCR, wenamed promoter DNA PCR as quantitative for real-time PCR andsemi-quantitative for Thermal Cycler PCR followed by agarose gelanalysis.
SDS–PAGE and Western blotting
SDS–PAGE, Western blot analysis of proteins, andimmunohistology were performed as described previously (Jianget al., 2006; Vincent et al., 2007).
Intracellular reactive oxygen species (ROS) measurement
The formation of intracellular ROS in cells was detected aspreviously described (Singh et al., 2007).
Immunofluorescence analysis of NF-kB subcellular localization
TR-iBRB2 cells were growth on coverslips coated with collagenand serum starved for 48 h in LG conditions before stimulationwith S100B (20mg/ml) or the vehicle (LG medium without serum)for 2 h. After stimulation, cells were washed twice with PBS andfixed 20 min with 4% paraformaldehyde in PBS. Cells were treatedfor 5 min with PBS containing 0.15% TX-100, blocked with PBS0.05% TX-100 and 1% bovine albumin for 30 min. Cells wereincubated at room temperature for 1 h with rabbit anti-NF-kBantibody (Cell Signaling Solutions). Texas-Red-conjugated anti-rabbit IgG were used for staining (Molecular Probes). Specimenswere mounted in PRO LONG GOLD mounting mediumcontaining DAPI. We analyzed these experiments by an ApoTomeFluoresence Microscope (ZeissQ5).
Statistical analysis
Comparison between two sets of experiments was analyzed byStudent’s t-test while one-way ANOVA was used to determinedifferences among means in multiple sets of experiments followedby Bonferroni post hoc test. In both cases, a preset P-value of<0.05was considered statistically significant.
ResultsRAGE and high glucose induce TXNIP expression inretinal endothelial cells
We have previously demonstrated that high glucose (HG)induces TXNIP expression in glomerular mesangial cells(Cheng et al., 2006) and increases in the diabetic rat kidney(Singh et al., 2007). TXNIP expression is also increased inSTZ-induced diabetic rat retina while TRX expression isunvaried (Supplementary Data Fig. S1C). The fact that TXNIPexpression is induced significantly in the retina of diabetic rats,and by high ambient glucose in culture in both rat and primaryhuman retinal EC (Supplementary Data Fig. S1A,B) suggest thatTXNIP may contribute to endothelial inflammation and oculardisease development in diabetes. RAGE is known to play animportant role in endothelial inflammation and long-termdisease progression in diabetic retinopathy. Therefore, we

4 P E R R O N E E T A L .
treated retinal EC (TR-iBRB2) with 20mg/ml bovinebrain-derived S100B protein for 2 h to stimulate RAGE aspreviously described (Hofmann et al., 1999; Vincent et al., 2007;Perrone et al., 2008). We also treated TR-iBRB2 cells for 2 hwith 25 mM glucose (HG) and compared the results withRAGE. Initially, we tested S100B concentration both at 10mg/ml as previously described to stimulate EC (Harja et al., 2008)and S100B 20mg/ml described in this study. We found thatalthough both concentrations of S100B are effective in inducingTXNIP expression, S100B at 20mg/ml produces higheractivation without affecting cell viability (data not shown).Therefore, we utilized 20mg/ml S100B for all our experiments.Furthermore, we observed that TXNIP expression is enhancedby S100B, HG, or GlcN within 30 min of incubation and reachesthe maximal level at 2 h. There was no further increase inTXNIP expression either at 6 or 24 h (data not shown), hencewe performed further experiments after 2 h treatment. Wealso observed that 20 mM mannitol in LG does not induceTXNIP expression suggesting that the effect of high glucoseobserved here is not due to increased osmolarity (data notshown).
As shown in Figure 1A, S100B induces TXNIP mRNAexpression at the same extent of HG when compared with LG(P< 0.05 for both HG and S100B vs. LG). A RAGE-specificblocking antibody (Perrone et al., 2008) completely abolishesS100B-induced TXNIP expression, demonstrating that RAGE is
Fig. 1. RAGEinducesTXNIPexpression.A:TXNIPmRNAexpressionwaTXNIP expression was analyzed in control cells expressing a scramble seinterference(siTXNIPcells).Bothcell linesweretreatedwithLG(5 mMgluthe average of three independent experiments carried out in triplicate (n Ueach treatment, P < 0.05 for both. B: TR-iBRB2 cells were treated for 2 h wTXNIP mRNA expression was analyzed by Q-RTPCR. Azaserine compleconditions and the decrement is statistically significant (P < 0.01 HG vs. H(P < 0.01vs.LG); (C)Westernblotting analysisofTXNIPexpression in siScrisrepresentativeofthree independentexperiments,n U 3).Cellsweretreatof the Western blot analysis shown in (C). MSignificant increase versus LG
JOURNAL OF CELLULAR PHYSIOLOGY
responsible for TXNIP expression in response to S100Btreatment (Fig. 1A). Knock down of TXNIP by RNAinterference (siTXNIP cells) results in a decrement of TXNIPmRNA expression in low glucose (LG) conditions and totallyinhibits TXNIP expression following either S100B or HGstimulation (Fig. 1A). We have previously shown that the HBP isresponsible for TXNIP expression in renal mesangial cells(Cheng et al., 2006). Here again, we show that HG inducesTXNIP expression in retinal EC through the HBP. This isevident by the observation that azaserine, an inhibitor of theHBP, completely abolishes HG-induced TXNIP expression(Fig. 1B). Furthermore, stimulation for 2 h with GlcN, whichincreases HBP, induces TXNIP expression (Fig. 1B). Westernblot analysis shows that 2 h of stimulation with either HG orS100B leads to increased TXNIP protein levels (�3-folds,P< 0.5 for both S100B and HG vs. LG), while knock down ofTXNIP in siTXNIP-expressing cells results in a significantreduction of TXNIP both in LG and HG or S100B conditions(Fig. 1C).
TXNIP is required for RAGE-induced VEGF-A, ICAM1,Cox2, and RAGE itself expression
RAGE ligands induce the expression of VEGF-A, ICAM1, Cox2,and RAGE itself in various cellular systems (Lu et al., 1998;Bierhaus et al., 2005; Bianchi et al., 2007; Kaji et al., 2007). We
sanalyzedbyQ-RTPCRusingactinasacontrolgene fornormalization.quence (siScramble cells) and in cells depleted of TXNIP by RNAcose),HG(25 mMglucose),andS100B(20mg/ml).Thedatashowedare9). MSignificant increase versus LG; #significant decrease by TXNIP forith HG, with or without azaserine (0.5mM), with 1.5 mM glucosaminetely blocks the HG-induced TXNIIP expression to the level of LGG R Azaserine). GlcN also increases TXNIP expression significantlyamble (control) andsiTXNIPsilencingTR-iBRB2cells (thedatashownedwithLG,HG,andS100Basdescribedabovein(A).D:Quantification; #significant decrease by TXNIP silencing, P < 0.05 for both.

T X N I P I S R E Q U I R E D F O R R A G E - I N D U C E D I N F L A M M A T I O N 5
concentrated in this study predominantly on RAGE activationof TXNIP and inflammation because this is the firstdemonstration that RAGE is involved in TXNIP activation. Weverified that stimulation for 2 h with either HG or S100Binduces a significant increment of RAGE, VEGF-A, ICAM1, andCox2 mRNA expression in retinal EC (Fig. 2A–D, statisticalanalysis in Supplementary Data Fig. S1). RAGE is responsiblefor S100B-induced effects since a RAGE-specific blockingantibody inhibits S100B-induced pro-inflammatory geneexpression (Fig. 2A–D, data analysis in S1). It has beenpreviously shown that short-term stimulation by HG does notinitiate a signaling cascade through RAGE (Esposito et al.,2001). We further investigated whether TXNIP is required forRAGE- and HG-induced expression of these pro-inflammatorygenes. Knock down of TXNIP in siTXNIP cells totally abolishesHG and S100B-induced RAGE, VEGF-A, ICAM1, and Cox2expressions (Fig. 2A–D, data analysis in S1) in retinal EC. StableTXNIP over-expression induces VEGF-A mRNA expressionlower than the other genes but significant when compared tocontrol cells. We observed that Cox2 is the highestinduced gene by TXNIP in this study and we focus on it.Conversely, we observed that RAGE induces iNOS activationin retinal EC, however, it is independent of TXNIP signaling,because TXNIP silencing does not block S100B-mediated iNOSexpression, whereas anti-RAGE blocking antibodies do (Fig. 3),although the precise mechanism is yet to be determined.Furthermore, after 2 h treatment with high glucose, iNOSexpression was not changed (data not shown) demonstratingthat at short time stimulation HG and RAGE pathways areseparated.
Fig. 2. TXNIP is required for RAGE-induced RAGE, VEGF-A, ICAM1, andgenes was analyzed by Q-RTPCR in siScramble (control) and siTXNIP (depfornormalization.Bothcell linesweretreatedwithLG,HG,S100B,andS100three independent experiments carried out in triplicate (n U 9). MSignificatreatment; P values for both were <0.05.
JOURNAL OF CELLULAR PHYSIOLOGY
TXNIP induction of pro-inflammatory gene expressioninvolves p38 MAPK and NF-kB
Previously, TXNIP was shown to cause inflammation in aorticvessel endothelium (Reddy et al., 2002; Schulze et al., 2004;Chung et al., 2006; Turturro et al., 2007). To further investigatethe function of TXNIP in pro-inflammatory gene expression,we produced a TR-iBRB2 cell line over-expressing the humanTXNIP (hTXNIP) stably. As control, TR-iBRB2 cells were stablytransfected with a plasmid encoding LacZ protein. hTXNIP cellsover-express TXNIP protein (�4-folds) compared to LacZcontrol cells (Fig. 4A). Over-expression of TXNIP in TR-iBRB2cells itself leads to a slight but significant increment of ROSaccumulation in LG conditions similar to HG treatment of LacZcontrol cells (P< 0.05 vs. LG), and HG addition does notproduce further ROS accumulation in hTXNIP cells (Fig. 4B).TXNIP over-expression itself results in a significant incrementof RAGE, ICAM1, and Cox2 mRNA expression in LGconditions compared to LacZ control cells (Fig. 4C, statisticalanalysis in Supplementary Data Fig. S2). TXNIPover-expression induces VEGF-A mRNA expression marginallybut to a significant extent compared to control cells(Fig. 4C, statistical analysis in Supplementary Data Fig. S2).Among the genes analyzed, Cox2 showed the highest degree ofstimulation by TXNIP over-expression (17�4.3-folds,P¼ 2.33E-07 vs. Lac control). To investigate the extent towhich ROS accumulation plays a role in TXNIP-inducedpro-inflammatory gene expression, we treated hTXNIP cellsfor 24 h with the anti-oxidant a-lipoic acid (150mM, LA).Addition of LA totally inhibits RAGE, ICAM1, and VEGF-A
Cox2 expression. A–D: Messenger RNA expression for inflammatoryleted of TXNIP by RNA interference) cells using actin as a control geneBplusanti-RAGEblockingantibody.Thedatashowedaretheresultsofnt increase versus LG; ^significant decrease in siTXNIP cells for each
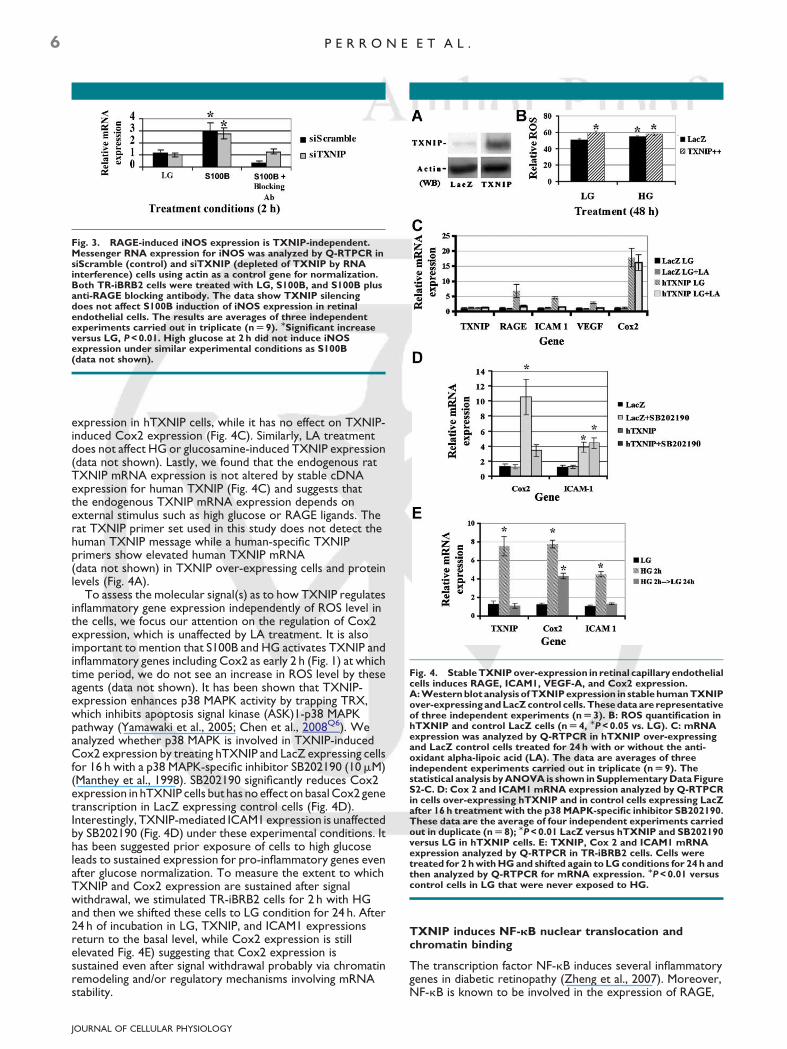
Fig. 4. Stable TXNIP over-expression in retinal capillary endothelialcells induces RAGE, ICAM1, VEGF-A, and Cox2 expression.A: Western blot analysis of TXNIP expression in stable human TXNIPover-expressing and LacZ control cells. These data are representativeof three independent experiments (n U 3). B: ROS quantification inhTXNIP and control LacZ cells (n U 4, MP < 0.05 vs. LG). C: mRNAexpression was analyzed by Q-RTPCR in hTXNIP over-expressingand LacZ control cells treated for 24 h with or without the anti-oxidant alpha-lipoic acid (LA). The data are averages of threeindependent experiments carried out in triplicate (n U 9). Thestatistical analysis by ANOVA is shown in Supplementary Data FigureS2-C. D: Cox 2 and ICAM1 mRNA expression analyzed by Q-RTPCRin cells over-expressing hTXNIP and in control cells expressing LacZafter 16 h treatment with the p38 MAPK-specific inhibitor SB202190.These data are the average of four independent experiments carriedout in duplicate (n U 8); MP < 0.01 LacZ versus hTXNIP and SB202190versus LG in hTXNIP cells. E: TXNIP, Cox 2 and ICAM1 mRNAexpression analyzed by Q-RTPCR in TR-iBRB2 cells. Cells weretreated for 2 h with HG and shifted again to LG conditions for 24 h andthen analyzed by Q-RTPCR for mRNA expression. MP < 0.01 versuscontrol cells in LG that were never exposed to HG.
Fig. 3. RAGE-induced iNOS expression is TXNIP-independent.Messenger RNA expression for iNOS was analyzed by Q-RTPCR insiScramble (control) and siTXNIP (depleted of TXNIP by RNAinterference) cells using actin as a control gene for normalization.Both TR-iBRB2 cells were treated with LG, S100B, and S100B plusanti-RAGE blocking antibody. The data show TXNIP silencingdoes not affect S100B induction of iNOS expression in retinalendothelial cells. The results are averages of three independentexperiments carried out in triplicate (n U 9). MSignificant increaseversus LG, P < 0.01. High glucose at 2 h did not induce iNOSexpression under similar experimental conditions as S100B(data not shown).
6 P E R R O N E E T A L .
expression in hTXNIP cells, while it has no effect on TXNIP-induced Cox2 expression (Fig. 4C). Similarly, LA treatmentdoes not affect HG or glucosamine-induced TXNIP expression(data not shown). Lastly, we found that the endogenous ratTXNIP mRNA expression is not altered by stable cDNAexpression for human TXNIP (Fig. 4C) and suggests thatthe endogenous TXNIP mRNA expression depends onexternal stimulus such as high glucose or RAGE ligands. Therat TXNIP primer set used in this study does not detect thehuman TXNIP message while a human-specific TXNIPprimers show elevated human TXNIP mRNA(data not shown) in TXNIP over-expressing cells and proteinlevels (Fig. 4A).
To assess the molecular signal(s) as to how TXNIP regulatesinflammatory gene expression independently of ROS level inthe cells, we focus our attention on the regulation of Cox2expression, which is unaffected by LA treatment. It is alsoimportant to mention that S100B and HG activates TXNIP andinflammatory genes including Cox2 as early 2 h (Fig. 1) at whichtime period, we do not see an increase in ROS level by theseagents (data not shown). It has been shown that TXNIP-expression enhances p38 MAPK activity by trapping TRX,which inhibits apoptosis signal kinase (ASK)1-p38 MAPKpathway (Yamawaki et al., 2005; Chen et al., 2008Q6). Weanalyzed whether p38 MAPK is involved in TXNIP-inducedCox2 expression by treating hTXNIP and LacZ expressing cellsfor 16 h with a p38 MAPK-specific inhibitor SB202190 (10mM)(Manthey et al., 1998). SB202190 significantly reduces Cox2expression in hTXNIP cells but has no effect on basal Cox2 genetranscription in LacZ expressing control cells (Fig. 4D).Interestingly, TXNIP-mediated ICAM1 expression is unaffectedby SB202190 (Fig. 4D) under these experimental conditions. Ithas been suggested prior exposure of cells to high glucoseleads to sustained expression for pro-inflammatory genes evenafter glucose normalization. To measure the extent to whichTXNIP and Cox2 expression are sustained after signalwithdrawal, we stimulated TR-iBRB2 cells for 2 h with HGand then we shifted these cells to LG condition for 24 h. After24 h of incubation in LG, TXNIP, and ICAM1 expressionsreturn to the basal level, while Cox2 expression is stillelevated Fig. 4E) suggesting that Cox2 expression issustained even after signal withdrawal probably via chromatinremodeling and/or regulatory mechanisms involving mRNAstability.
JOURNAL OF CELLULAR PHYSIOLOGY
TXNIP induces NF-kB nuclear translocation andchromatin binding
The transcription factor NF-kB induces several inflammatorygenes in diabetic retinopathy (Zheng et al., 2007). Moreover,NF-kB is known to be involved in the expression of RAGE,

T X N I P I S R E Q U I R E D F O R R A G E - I N D U C E D I N F L A M M A T I O N 7
ICAM1, VEGF-A, and Cox2 in diabetes (Shanmugam et al.,2003; Bierhaus et al., 2005; Nagai et al., 2007). Therefore, weinvestigated whether NF-kB is required for the TXNIP-inducedpro-inflammatory gene expression in retinal EC usingpharmacological inhibitors of NF-kB. Stable hTXNIPover-expressing cells were incubated for 16 h with varyingconcentrations of an NF-kB inhibitor, (E)-2-Fluoro-40-methoxystilbene (Heynekamp et al., 2006) (0.5mM and 15mM)in LG condition. (E)-2-Fluoro-40-methoxystilbene is a derivativeof resveratrol that has been isolated for his increased capabilityto inhibit NF-kB-mediated gene expression compared toresveratrol (Heynekamp et al., 2006). Resveratrol affectsNF-kB-mediated gene expression by down-regulating NF-kBexpression (Sun et al., 2006). At the concentrations used, bothinhibitors significantly blocks NF-kB DNA binding activity in anELISA assay (Figure S3). We observed a dose–responseinhibition of RAGE, VEGF-A, Cox2, and ICAM1 expression byincreasing concentrations of the inhibitor (Fig. 5A). A statisticalanalysis shows significant inhibition for all genes (P< 0.0005 vs.respective control), which is shown in Supplementary Data Fig.S3. A cell permeable NF-kB inhibitory peptide (50mg/ml),which inhibits NF-kB nuclear translocation (Lin et al., 1995) alsoreduces RAGE, VEGF-A, Cox2, and ICAM1 expression inhTXNIP cells significantly (Fig. 5A, data analysis in S3). Theseresults indicate that NF-kB is a mediator of TXNIP-inducedinflammatory gene expression in retinal EC.
Next, we demonstrate that TXNIP expression is requiredfor RAGE-induced NF-kB nuclear translocation.Immunofluorescence analysis of control cells expressing thescramble siRNA (siScramble) shows nuclear translocation ofNF-kB following 2 h of stimulation with S100B, while there is noevident NF-kB nuclear localization in siScramble cells in LGconditions (Fig. 5B). Knock down of TXNIP in siTXNIP cellscompletely abolishes NF-kB nuclear translocation following 2 hof S100B stimulation. Over-expression of hTXNIP leads to NF-kB nuclear localization in LG conditions (Fig. 5B). There is noevident nuclear localization of NF-kB in LacZ control cells in LGconditions (not shown). We further ask whether NF-kBaccumulation in the nucleus leads to chromatin binding andTXNIP plays a role in NF-kB-mediated Cox2 expressionfollowing S100B stimulation. We employed ChIP assay usingNF-kB antibodies and followed by semi-quantitative (Fig. 5C) orquantitative PCR (Fig. 5D) to measure NF-kB accessibility tochromatin at Cox2 promoter region bearing the NF-kBconsensus motif. In siScramble (control) cells, NF-kB binds toCox2 promoter following 2 h stimulation with S100B(Fig. 5C,D). On the other hand, knock down of TXNIPcompletely abolishes NF-kB binding to Cox2 promoterfollowing S100B stimulation in siTXNIP cells (Fig. 5C,D). Tofurther demonstrate the role of TXNIP in inducing NF-kBbinding to Cox-2 promoter, we also analyzed by ChIP theassociation of NF-kB to Cox-2 promoter following GlcNstimulation (quantitative PCR in Supplementary Data Fig. S2A,and semi-quantitative PCR analysis in Supplementary Data Fig.S2B). In siScramble cells, NF-kB binds to Cox2 promoterfollowing 2 h stimulation with GlcN, while the binding is absentin LG controls (Supplementary Data Fig. S2A,B). Knock down ofTXNIP completely abolishes NF-kB binding to Cox2 promoterby GlcN in siTXNIP cells (Supplementary Data Fig. S2A,B).These results suggest that TXNIP increases transcription factoraccessibility to gene promoters and induces inflammatory geneexpression for Cox2.
RAGE and TXNIP induce histone H3 modifications onCox2 promoter via Np38 MAPK activation
As mentioned above, TXNIP increases chromatin accessibilityof transcription factor NF-kB at Cox2 gene promoter andHG-induced Cox2 expression is still elevated after shifting the
JOURNAL OF CELLULAR PHYSIOLOGY
cells to LG for 24 h. Therefore, we hypothesize that TXNIP maycause histone remodeling at Cox2 promoter. p38MAPK-dependent phosphorylation of histone H3 inducesCox2 expression in enterocytes (Karrasch et al., 2006).Phosphorylation and acetylation of histone H3 are indicative ofinduced gene transcription (Hamon et al., 2007), while histoneH3 tri-methylation is a marker of gene silencing (Barski et al.,2007). We investigated whether TXNIP over-expression isnecessary and sufficient to induce histone H3 modifications atCox2 promoter by ChIP analysis followed by semi-quantitativeand quantitative PCR (Fig. 6A,B, respectively, and theirstatistical analysis is presented in Supplementary Data Fig. S4).To assess as to whether TXNIP induces epigenetic remodelingand gene expression, we used an antibody capable ofrecognizing acetylated histone H3 at K9 unambiguous markerof induced transcriptional activation as previously reported(Hamon et al., 2007). H3 K9 acetylation is initiated byphosphorylation at H3 S10, therefore, the acetylated antibodyis known as K9 acetylated S10 phosphorylated antibody.Antibodies that recognize either acetylated K9 orphosphorylated S10 alone histone H3 have poor accessibility tochromatin in ChIP assays, therefore, we use the K9 acetylatedantibody that also recognize phosphorylated S10. In siScramblecells, S100B induces H3 K9 acetylation in Cox2 promoter onNF-kB site (�8.13-folds vs. LG, P¼ 5.21E-08), while histone H3is tri-methylated at residue K9 in LG condition (Fig. 6A,B). UponsiRNA silencing of TXNIP in siTXNIP cells, histone H3 is tri-methylated at K9 both in LG and S100B treatment (Fig. 6A,B)and no H3 K9 acetylation is observed. Conversely, over-expression of TXNIP in hTXNIP cells alone is capable ofinducing H3K9 acetylation and abolishes H3K9 tri-methylationin Cox2 promoter bearing NF-kB consensus site (Fig. 6A,B). Toexamine further if p38 MAPK is involved in TXNIP-mediatedhistone H3 modifications, we treated hTXNIP cells for 30 minwith SB202190 (Manthey et al., 1998). The results show thattri-methylation of H3 at K9 is increased (Fig. 6A,B; last lane) andreduces K9 acetylation of H3, which correlate with theinhibition of Cox2 expression by SB202190 (Fig. 4D). Theseresults demonstrate that TXNIP is involved in H3K9remodeling at Cox2 promoter, which involves the p38 MAPKsignaling pathway.
To verify further that the observed epigenetic modificationson Cox-2 promoter are specifically induced by TXNIP, and notby cellular manipulations such as conditional immortalizationand gene transfection, we analyzed histone H3 tri-methylationand acetylation status of K9 residue on iNOS promoter bearingthe NF-kB site, because iNOS expression is independent ofTXNIP in S100B stimulation (Fig. 3). As shown in Figure 7, insiScramble control cells, H3 K9 acetylation is minimal at iNOSpromoter in LG conditions and is induced twofold by treatmentwith S100B. K9 tri-methylation is higher under LG conditionand is strongly reduced following treatment with S100B (Fig. 7).In siTXNIP cells, histone H3 is tri-methylated at K9 in LG, but isagain demethylated following S100B treatment (lanes 4 and 5),which is similar to siScramble controls. Furthermore, over-expression of TXNIP itself does not produce any significantepigenetic remodeling of H3 K9 in iNOS promoter whencompared with the siScramble cell (lane 2 vs. lane 6). Indeed,histone H3 K9 is still tri-methylated in cells over-expressinghTXNIP and K3 K9 acetylation is suppressed. Thus, this lack ofH3 K9 acetylation on iNOS promoter in hTXNIP cells parallelsthe absence of iNOS expression in these cells. Furthermore,SB202190 treatment of the siTXNIP cells does not produce asignificant reduction in histone H3 K9 tri-methylation (Fig. 7 lastlane). Perhaps, H3K9 still exists as di-or mono-methylatedforms, which prevent K9 acetylation and iNOS expression.Future studies will clarify this question. Thus, TXNIPover-expression nor TXNIP knock down do not affectepigenetic modification iNOS promoter by S100B indicating

Fig. 5. TXNIP induces NF-kB nuclear translocation and chromatin binding. A: NF-kB inhibitors block inflammatory gene expression incellsover-expressinghTXNIP.Thesedataaretheaverageofthreeindependentexperimentscarriedoutintriplicate(n U 9).ThestatisticalanalysisbyANOVA test is shown in Supplementary Data FigureS2-E. Theconcentrationsused for NF-kBinhibitors in this studysignificantlyblocks NF-kBconsensus DNAbinding activity inELISA assay (Figure S3).B:TR-iBRB2cells were grownon coverslipscoated withcollagen andserumstarved for48 h in LG conditions before stimulation with S100B (20mg/ml) or the vehicle (LG alone in serum starved medium) for 2 h. NF-kBsubcellular localization was investigated by immunofluorescence analysis using an anti-NF-kB antibody as described in Materials and MethodsSection. In the figure, the localization of NF-kB in the cytoplasm (red) and nucleus (blue) under different conditions are shown: in siScramble cells(control cells expressing a scramble silencing RNA) treated with either LG or S100B for 2 h; in siTXNIP cells (knock down of TXNIP by RNAinterference) stimulated with S100B for 2 h; in hTXNIP cells (cells over-expressing hTXNIP) in LG conditions. In siScramble cells, NF-kB islocalized in the cytoplasm while treatment with S100B induces nuclear NF-kB localization as evident from the merge with the nuclear staining.There is no nuclear localization of NF-kB in LacZ control cells in LG condition (not shown). These data are representative of three independentexperiments. C,D: In vivo binding of NF-kB p65 subunit with Cox2 promoter chromatin was determined by ChIP assay. ChIP analysis ofNF-kBbindingtoCox2promotercontaining theNF-kBconsensussitewasperformedinsiScramble (control) andsiTXNIP(depletedofTXNIPbyRNA interference) cells. C: Semi-quantitative PCR analysis (these data are representative of four independent experiments) and (D) quantitativePCR analysis show that S100B induces NF-kB association with Cox2 promoter in siScramble (control) cells, which is abolished upon TXNIPsilencing in retinal endothelial cells. These data are the average of three independent experiments carried out in duplicate, n U 6; P < 0.01 S100Bversus LG.
JOURNAL OF CELLULAR PHYSIOLOGY
8 P E R R O N E E T A L .

Fig. 6. TXNIP is required for chromatin remodeling of Cox2 promoter. ChIP analysis of histone H3 modifications (acetylation in K9 versustri-methylation in K9) in siScramble (control), siTXNIP (depleted of TXNIP by RNA interference), and hTXNIP over-expressing cells in thepresence or absence of p38 MAPK inhibition by SB202190. A: Semi-quantitative PCR analysis of ChIP DNA (these data are representative of fourindependent experiments). B: Quantititave PCR analysis (these data are the average of three independent experiments carried out in duplicate,n U 6.ThestatisticalanalysisofthedataisshowninSupplementaryDataFig.S4).MrepresentsMockDNAprecipitationwithnon-specificIgG.Inputis the total DNA used in ChIP.
T X N I P I S R E Q U I R E D F O R R A G E - I N D U C E D I N F L A M M A T I O N 9
that the epigenetic modifications induced by TXNIP on Cox2promoter are specific.
Discussion
In the present study we demonstrate for the first time thatTXNIP is activated by RAGE and high glucose in causinginflammation in retinal EC and provide a novel function ofTXNIP in producing the pathogenic effects in diabetes. It hasbeen previously shown that in short-term stimulation by HG
Fig. 7. Chromatin remodeling of iNOS promoter by S100B isTXNIP-independent. ChIP analysis by quantitative PCR of histone H3modifications at K9, acetylation versus tri-methylation in iNOSpromoter bearing NF-kB-binding site, in siScramble (control),siTXNIP (depleted of TXNIP by RNA interference), and hTXNIPover-expressing cells. H3K9 tri-methylation is higher in controlsiScramble cells, which is reduced significantly by S100B treatment(P < 0.01, n U 3; lane 2 vs. 3, gray bar) while H3K9 acetylation isincreased (black bar). The effect of S100B on H3K9 modification wasnot difference between siScramble and siTXNIP-expressing cellssuggesting that TXNIP is not involved in iNOS chromatinremodeling. Similarly, TXNIP over-expression does not interferewith S100B’s effect on H3 K9 acetylation or methylation.Interestingly, S100B-mediated H3 K9 tri-methylation was also notreduced by p38 MAPK inhibitor, SB202190. Mock DNA precipitationwith non-specific IgG was used as a background for non-specific DNAbinding in ChIP assay. These data are the average of threeindependent experiments carried out in duplicate, n U 6.
JOURNAL OF CELLULAR PHYSIOLOGY
does not initiate a signaling cascade through RAGE (Espositoet al., 2001). Thus, we can conclude that both HG and RAGEactivate TXNIP expression and the two pathways are separatedat this point. We also found that high glucose effect on TXNIPexpression is mediated by its metabolic flux through the HBP inretinal EC as was previously shown for renal mesangial cells byour laboratory (Cheng et al., 2006; Singh et al., 2007). Our newfindings are: (i) TXNIP is indispensable for HG- andRAGE-induced inflammatory gene expression; (ii) TXNIP isnecessary for RAGE-mediated epigenetic remodeling ofhistone H3 at K9 at cox2 promoter; (iii) TXNIPover-expression in retinal EC itself causes inflammatory geneexpression; and (iv) p38 MAPK and transcription factor NF-kBare involved in TXNIP-mediated inflammatory geneexpression.
TXNIP is an early response gene to high ambient glucose andin diabetes, and TXNIP itself is able to induce RAGE expression;therefore, hyperglycemia-induced TXNIP expression willenhance RAGE expression leading to feed back TXNIP-expression loop, which could produce a long-lasting chromatinremodeling and sustained inflammatory gene expression inretinal capillary EC in diabetes. It has been shown that RAGEtriggering induces the expression of adhesion molecules andpro-inflammatory factors (Yan et al., 2004) leading to retinalblood vessel leakage in diabetic retinopathy. For the first time,we demonstrate here that TXNIP expression is increased in thediabetic retina and in retinal capillary EC by high glucose andRAGE, and that TXNIP mediates RAGE-induced inflammatoryCox2, VEGF-A, ICAM1, and RAGE expression itself in retinalEC in culture. It was proposed that the RAGE may participate inglycemic ‘‘memory’’ formation leading to the development andprogression of vascular and microvascular complications ofdiabetes (Yan et al., 2004). We propose that hyperglycemia- andRAGE-induced TXNIP expression is essential for a vicious cycleof diabetic cell injury and endothelial inflammation becauseepigenetic chromatin modifications are mitotically inheritableand they can potentiate long-term sustained inflammatory geneexpression and ocular disease development. Indeed, aproposed model for HG- and RAGE-mediated TXNIPexpression and inflammatory gene expression is depicted inFigure 8. Although TXNIP can increase ROS in retinal EC, it mayalso induce inflammation before detectable ROS generationand cellular oxidative stress occur. The present study wasfocused on RAGE activation by S100B and early events inTXNIP-mediated inflammatory gene expression, which is

Fig. 8. Schematic representation of RAGE- and HG-inducedinflammation through TXNIP expression in retinal endothelial cells.S100B stimulation induces RAGE-dependent TXNIP expression andROS production. HG induces TXNIP expression via the HBP.Inhibition of NF-kB abolishes TXNIP-mediated expression of Cox2,RAGE, ICAM1, and VEGF-A mRNA expression. Anti-oxidanta-lipoicacid treatment of retinal capillary endothelial cells revert TXNIP-induced RAGE, ICAM1, and VEGF mRNA expression. On the otherhand, TXNIP-mediated Cox2 expression is insensitive toa-lipoic acidtreatment. While a role for ROS generation is accepted in long-termmicrovascular complications of diabetes, TXNIP is also capable ofinducing inflammatory gene expression early without causing ROSgeneration. p38 MAPK inhibitor SB202190 blocks TXNIP-mediatedhistone H3 modification at Cox2 promoter and gene expression. Insummary, TXNIP mediates RAGE- and HG-induced inflammation bychromatin remodeling and enhanced NF-kB binding to genepromoters, for example, of Cox2.
10 P E R R O N E E T A L .
independent of ROS generation. We cannot exclude, howeverthat, at later stages elevated oxidative/nitrosative stresses willstrength the hyperglycemia- and diabetes-induced pro-inflammatory signals such as p38 MAPK-NF-kB axis and causeocular disease development and progression. Herein, wedemonstrate that high glucose, RAGE, and diabetes-inducedTXNIP over-expression in the retinal EC could play a triggeringfactor in early retinal inflammation and endothelial dysfunctionin diabetes.
Previous studies showed that RAGE triggering generatesnuclear translocation of NF-kB and target gene expression (Yanet al., 2004). Moreover, it is well documented that TXNIPexpression leads to NF-kB activation (Aitken et al., 2004). Ourdata further reinforce the hypothesis that the expression ofRAGE, VEGF-A, ICAM1, and Cox2 is NF-kB dependent anddemonstrate further that TXNIP is required for NF-kB nucleartranslocation in retinal EC. Indeed, we show that stable TXNIPexpression itself is sufficient to cause RAGE, VEGF-A, ICAM1,and Cox2 expressions and NF-kB nuclear translocation. Weand others demonstrated that RAGE activation can also induceROS production via activation of NADPH oxidase (Wendtet al., 2006; Vincent et al., 2007). However, as mentioned abovein the present study, we did not focus our attention on RAGE orTXNIP-mediated ROS production since we observed thatTXNIP can induce early inflammatory gene expression and notall TXNIP-mediated gene expressions are mediated by ROS.Instead, we present a novel aspect of TXNIP function ininflammatory gene expression that might lead to epigeneticchromatin remodeling, memory formation, long-term aberrantgene expression in diabetic retinopathy. We demonstrate thatRAGE, VEGF-A, and ICAM1 expression can be activated byboth ROS and NF-kB-dependent pathways in retinal ECover-expressing TXNIP stably, which showed higher ROSlevels as previously demonstrated (Wendt et al., 2006; Vincent
JOURNAL OF CELLULAR PHYSIOLOGY
et al., 2007). On the other hand, we also show that Cox2 isinsensitive to anti-oxidant treatment by a-lipoic acid.
Here for the first time, we also present data to demonstratethat TXNIP is responsible for epigenetic histone H3 K9modification and regulation of Cox2 expression in retinalcapillary EC. Environmental factors, such as diet, chronichyperglycemia, stress, and inflammatory cytokines and growthfactors, can influence epigenetic histone and DNA remodelingand aberrant gene expression under various pathologicalconditions (Liu et al., 2008). Modifications in the nuclear histoneacetylation/methylation balance results in enhancedtranscription of specific pro-inflammatory genes. A previousstudy demonstrated that p38 MAPK-dependentphosphorylation of histone H3 induces Cox2 expression inenterocytes (Karrasch et al., 2006). Activation of p38 MAPKleads to histone H3 phosphorylation, probably via activation ofdownstream nuclear mitogen- and stress-activated proteinkinase (MSK) 1, inducing modification of chromatin structureand increased DNA binding of NF-kB (Marwick et al., 2004;Dyson et al., 2005). Tri-methylation of histone H3 at K9correlates with gene silencing (Barski et al., 2007), whilephosphorylation at residue Serine 10 (S10) in a p38MAPK-dependent manner and acetylation of histone H3 K9correlate with gene transcription (Hamon et al., 2007).Activated NF-kB recruits transcriptional co-factors such asp300/CBP and pCAF that bear histone acetylase activity andthey may participate in TXNIP-induced H3 K9 acetylation(Gerritsen et al., 1997; Rahman Q7et al., 2004; Deng et al., 2008;Villeneuve et al., 2008).
It has been previously shown that epigenetic modificationscontrol NF-kB-dependent expression of the pro-inflammatoryfactor TNFa (El Gazzar et al., 2007). Tri-methylation of histoneH3 K9 leads to TNFa silencing and reduces the binding ofNF-kB to its proximal promoter (El Gazzar et al., 2007). Herewe show that in the absence of TXNIP expression by siRNAsilencing, H3 K9 is tri-methylated on Cox2 promoter andinhibits NF-kB binding. On the contrary, TXNIP over-expression induces phosphorylation, acetylation, anddemethylation of histone H3, resulting in TXNIP-dependentNF-kB association with the Cox2 promoter. TXNIP-dependent p38 MAPK activation is also documented(Yamawaki et al., 2005; Chen et al., 2008c). TXNIP-dependentepigenetic modifications may result in sustained Cox2expression since lowering glucose level to normal after an initialexposure of retinal EC to high glucose does not reduce Cox2expression to the basal level, a process known as epigeneticmemory formation. We observed that Cox2 expression is stillelevated in LG condition up to 24 h when the cells werepreviously treated with HG for 2 h only (Fig. 4E). A recent studydemonstrated that S100B-mediated RAGE activation increasesCox2 mRNA stability (Shanmugam et al., 2008) and we cannotexclude this phenomenon in retinal EC as well. However, wefocus our attention on TXNIP-mediated epigeneticmodifications leading to increased Cox2 transcription in retinalEC. In agreement, it has been shown that Cox2 induction byS100B is transcriptionally regulated (Shanmugam et al., 2003). Inthe present study, we demonstrate a role for TXNIP inchromatin remodeling at histone H3 K9 in Cox2 promoter andgene expression since a site-specific alternative K9 tri-methylation and acetylation is observed on Cox2 promoterupon TXNIP silencing or over-expression, respectively, but noton iNOS promoter. Indeed, we show that RAGE-induced iNOSexpression is TXNIP-independent. Thus, the epigeneticmodifications at H3 K9 observed in this study are to a certaindegree a specific effect of TXNIP-dependent induced on Cox-2promoter remodeling. Furthermore, we have not examinedchromatin remodeling patterns for H3 K9 at other genesdescribed in this study such as RAGE, VEGF-A, and ICAM1 andthey will be investigated in further studies. In addition, there are

T X N I P I S R E Q U I R E D F O R R A G E - I N D U C E D I N F L A M M A T I O N 11
other H3 lysine residues that are also targeted by methylationand/or acetylation processes at the chromatin level and theirrole in epigenetic regulation of inflammatory gene expressionneeds further studies.
Recently, it has been demonstrated that histone H3tri-methylation is reduced in human vascular smooth musclecells (VSMC) maintained under high glucose in culture and inVSMC derived from a type 2 diabetic model of db/db mouse(Villeneuve et al., 2008). These in vivo data further strength ourhypothesis that environmental factors such as RAGE ligands andhyperglycemia can induce TXNIP and chromatin remodelingleading inflammation and disease development in diabetes. Weare currently testing whether TXNIP mediate epigeneticchromatin remodeling and inflammation in diabeticretinopathy. Thus, our study opens the way to developtherapeutic strategies targeting the epigenetic signalingpathways and chromatin reprogramming to prevent or slowdown the progression of diabetic ocular complications.
Acknowledgments
This study was supported by the American DiabetesAssociation Career Development Award (7-03-CD-14) andResearch to Prevent Blindness to the Department ofOphthalmology, WSU School of Medicine, Detroit, to L.P.Singh. Research grants from Mid-West Eye Bank, Michigan, andJuvenile Diabetes Research Foundation International toDr. Singh, and Marie Curie International Reintegration Grantfrom European Community (number 224892) to L. Perrone arealso acknowledged.
Literature Cited
Aitken C, Hodge J, Nishinaka Y, Vaughan T, Yodoi J, Day C, Morrison N, Nicholson G. 2004.Regulation of human osteoclast differentiation by thioredoxin binding protein-2 andredox-sensitive signaling. J Bone Miner Res 19:2057–2064.
Aronson D. 2008. Hyperglycemia and the pathobiology of diabetic complications. AdvCardiol 45:1–16.
Barile GR, Pachydaki SI, Tari SR, Lee SE, Donmoyer CM, Ma W, Rong LL, Buciarelli LG,Wendt T, Horig H, Hudson BI, Qu W, Weinberg AD, Yan SF, Schmidt AM. 2005. TheRAGE axis in early diabetic retinopathy. Invest Ophthalmol Vis Sci 46:2916–2924.
Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. 2007.High-resolution profiling of histone methylations in the human genome. Cell 129:823–837.
BerkQ8 BC. 2007. Novel approaches to treat oxidative stress and cardiovascular diseases.Trans Am Clin Climatol Assoc 118:209–214.
Bianchi R, Adami C, Giambanco I, Donato R. 2007. S100B binding to RAGE in microgliastimulates COX-2 expression. J Leukoc Biol 81:108–118.
Bierhaus A, Humpert PM, Morcos M, Wendt T, ChavakisT, Arnold B, Stern DM, Nawroth PP.2005. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med83:876–886.
Chen J, Saxena G, Mungrue IN, Lusis AJ, Shalev A. 2008a. Thioredoxin-interacting protein: Acritical link between glucose toxicity and beta-cell apoptosis. Diabetes 57:938–1044.
Chen J, Hui ST, Couto FM, Mungrue IN, Davis DB, Attie AD, Lusis AJ, Davis RA, Shalev A.2008b. Thioredoxin-interacting protein deficiency induces Akt/Bcl-xL signaling andpancreatic beta-cell mass and protects against diabetes. FASEB J 22:3581–3594.
Chen CL, Lin CF, Chang WT, Huang WC, Teng CF, Lin YS. 2008c. Ceramide induces p38MAPK and JNK activation through a mechanism involving a thioredoxin-interactingprotein-mediated pathway. Blood 111:4365–4374.
Cheng DW, Jiang Y, Shalev A, Kowluru R, Crook ED, Singh LP. 2006. An analysis of highglucose and glucosamine-induced gene expression and oxidative stress in renal mesangialcells. Arch Physiol Biochem 112:189–218.
Chung J, Jeon J, Yoon S, Choi I. 2006. Vitamin D3 upregulated protein 1 (VDUP1) is a regulatorfor redox signaling and stress-mediated diseases. J Dermatol 33:662–669.
Deng W-G, Zhu Y, Wu K. 2008. Role of p300 and pCAF in regulating cycloxygenase 2promoter activation by inflammatory mediators. Blood 103:2135–2142.
Dyson MH, Thomson S, Inagaki M, Goto H, Arthur SJ, Nightingale K, Iborra FJ, Mahadevan LC.2005. MAP kinase-mediated phosphorylation of distinct pools of histone H3 at S10 or S28via mitogen- and stress-activated kinase 1/2. J Cell Sci 118:2247–2259.
El Gazzar M, Yoza BK, Hu JY, Cousart SL, McCall CE. 2007. Epigenetic silencing of tumornecrosis factor alpha during endotoxin tolerance. J Biol Chem 282:26857–26864.
Esposito C, Fasoli G, Plati AR, Bellotti N, Conte MM, Cornacchia F, Foschi A, Mazzullo T,Semeraro L, Dal Canton A. 2001. Long-term exposure to high glucose up-regulatesVCAM-induced endothelial cell adhesiveness to PBMC. Kidney Int 59:1842–1849.
Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T. 1997. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci USA 94:2927–2932.
Goldin A, Beckman JA, Schmidt AM, Creager MA. 2006. Advanced glycation end products:Sparking the development of diabetic vascular injury. Circulation 114:597–605.
Hamon MA, Batsche E, Regnault B, Tham TN, Seveau S, Muchardt C, Cossart P. 2007. Histonemodifications induced by a family of bacterial toxins. Proc Natl Acad Sci USA104:13467–13472.
Harja E, Bum DX, Hudson BI, Chang JS, Shen X, Hallam K, Kalea AZ, Lu Y, Rosario RH,Oruganti S, Nikolla Z, Belov D, Lalla E, Ramasamy R, Yan SF, Schmidt AM. 2008. Vascularand inflammatory stresses mediate atherosclerosis via RAGE and its ligands inapoE�/� mice. J Clin Invest 118:183–194.
JOURNAL OF CELLULAR PHYSIOLOGY
Heynekamp J, Weber W, Hunsaker L, Gonzales A, Orlando R, Deck L, Jagt D. 2006.Substituted trans-stilbenes, including analogues of the natural product resveratrol, inhibitthe human tumor necrosis factor alpha-induced activation of transcription factor nuclearfactor kappaB. J Med Chem 49:7182–7189.
Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A,Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D,Schmidt AM. 1999. RAGE mediates a novel proinflammatory axis: A central cell surfacereceptor for S100/calgranulin polypeptides. Cell 97:889–901.
Hosoya K, Tomi M. 2005. Advances in the cell biology of transport via the inner blood-retinalbarrier: Establishment of cell lines and transport functions. Biol Pharm Bull 28:1–8.
Jiang Y, Cheng DW, Levi E, Singh LP. 2006. IGF-1 increases laminin, cyclin D1, and P21Cip1expression in glomerular mesangial cells: An investigation of the intracellular signalingpathway and cell-cycle progression. J Cell Biochem 98:208–220.
Kaji Y, Usui T, Ishida S, Yamashiro K, Moore T, Moore J, Yamamoto Y, Yamamoto H, AdamisAP. 2007. Inhibition of diabetic leukostasis and blood-retinal barrier breakdown with asoluble form of a receptor for advanced glycation end products. Invest Ophthalmol Vis Sci48:858–865.
Karrasch T, Steinbrecher KA, Allard B, Baldwin AS, Jobin C. 2006. Wound-inducedp38MAPK-dependent histone H3 phosphorylation correlates with increased COX-2expression in enterocytes. J Cell Physiol 207:809–815.
Khan ZA, Chakrabarti S. 2007. Cellular signaling and potential new treatment targets indiabetic retinopathy. Exp Diabetes Res 2007:31867.
Lin Y, Yao S, Veach R, Torgerson T, Hawiger J. 1995. Inhibition of nuclear translocation oftranscription factor NF-kappa B by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J Biol Chem 270:14255–14258.
Liu L, Li Y, Tollefsbol TO. 2008. Gene-environment interactions and epigenetic basis ofhuman diseases. Curr Issues Mol Biol 10:25–36.
Lu M, Kuroki M, Amano S, Tolentino M, Keough K, Kim I, Bucala R, Adamis AP. 1998.Advanced glycation end products increase retinal vascular endothelial growth factorexpression. J Clin Invest 101:1219–1224.
Manthey CL, Wang SW, Kinney SD, Yao Z. 1998. SB202190, a selective inhibitor of p38mitogen-activated protein kinase, is a powerful regulator of LPS-induced mRNAs inmonocytes. J Leukoc Biol 64:409–417.
Marwick JA, Kirkham PA, Stevenson CS, Danahay H, Giddings J, Butler K, Donaldson K,Macnee W, Rahman I. 2004. Cigarette smoke alters chromatin remodeling and inducesproinflammatory genes in rat lungs. Am J Respir Cell Mol Biol 31:633–642.
Nagai N, Izumi-Nagai K, Oike Y, Koto T, Satofuka S, Ozawa Y, Yamashiro K, Inoue M,Tsubota K, Umezawa K, Ishida S. 2007. Suppression of diabetes-induced retinalinflammation by blocking the angiotensin II type 1 receptor or its downstream nuclearfactor-kappaB pathway. Invest Ophthalmol Vis Sci 48:4342–4350.
Nagase K, Tomi M, Tachikawa M, Hosoya K. 2006. Functional and molecular characterizationof adenosine transport at the rat inner blood-retinal barrier. Biochim Biophys Acta1758:13–19.
Pachydaki SI, Tari SR, Lee SE, Ma W, Tseng JJ, Sosunov AA, Cataldergirmen G, Scarmeas N,Caspersen C, Chang S, Schiff WM, Schmidt AM, Barile GR. 2006. Upregulation of RAGEand its ligands in proliferative retinal disease. Exp Eye Res 82:807–815.
Perrone L, Peluso G, Melone MA. 2008. RAGE recycles at the plasma membrane in S100Bsecretory vesicles and promotes Schwann cells morphological changes. J Cell Physiol217:60–71.
Reddy M, Thimmalapura P, Lanting L, Nadler J, Fatima S, Natarajan R. 2002. The oxidized lipidand lipoxygenase product 12(S)-hydroxyeicosatetraenoic acid induces hypertrophy andfibronectin transcription in vascular smooth muscle cells via p38 MAPK and cAMPresponse element-binding protein activation. J Biol Chem 277:9920–9928.
Schmidt AM, Yan SD, Yan SF, Stern DM. 2000. The biology of the receptor for advancedglycation end products and its ligands. Biochim Biophys Acta 1498:99–111.
Schulze PC, Yoshioka J, Takahashi T, He Z, King GL, Lee RT. 2004. Hyperglycemia promotesoxidative stress through inhibition of thioredoxin function by thioredoxin-interactingprotein. J Biol Chem 279:30369–30374.
Shanmugam N, Kim YS, Lanting L, Natarajan R. 2003. Regulation of cyclooxygenase-2expression in monocytes by ligation of the receptor for advanced glycation end products.J Biol Chem 278:34834–34844.
Shanmugam N, Reddy MA, Natarajan R. 2008. Distinct roles of heterogenous nuclearribonuclear protein K and microrna-16 in cyclooxygenase-2 RNA stability induced byS100B, a ligand of the receptor for advanced glycation end productsQ9. J Biol Chem10.1074/jbc.M80632220.
Singh LP, Cheng DW, Kowluru R, Levi E, Jiang Y. 2007. Hexosamine induction of oxidativestress, hypertrophy and laminin expression in renal mesangial cells: Effect of the anti-oxidant alpha-lipoic acid. Cell Biochem Funct 25:537–550.
SinghQ10 LP, Cheng DW, Jiang Y, Crook ED. 2008. Transforming growth gactorb1-mediatedmRNA expression and oxidative stress in renal mesangial cells: Comparison with highglucose and hexosamine-induced gene expression profiles. Biochem Ind J 1:68–82.
Sun C, Hu Y, Liu X, Wu T, Wrang Y, He W, Wei W. 2006. Resveratrol downregulates theconstitutional activation of nuclear factor-kappaB in multiple myeloma cells, leading tosuppression of proliferation and invasion, arrest of cell cycle, and induction of apoptosis.Cancer Genet Cytogenet 165:9–19.
Turturro F, Friday E, Welbourne T. 2007. Hyperglycemia regulates thioredoxin-ROS activitythrough induction of thioredoxin-interacting protein (TXNIP) in metastatic breast cancer-derived cells MDA-MB-231. BMC Cancer 7:96.
Villeneuve LM, Reddy MA, Lanting LL, Wang M, Meng L, Natarajan R. 2008. Epigenetic histoneH3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascularsmooth muscle cells in diabetes. Proc Natl Acad Sci USA 105:9047–9052.
Vincent AM, Perrone L, Sullivan KA, Backus C, Sastry AM, Lastoskie C, Feldman EL. 2007.Receptor for advanced glycation end products activation injures primary sensory neuronsvia oxidative stress. Endocrinology 148:548–558.
Wautier JL, Zoukourian C, Chappey O, Wautier MP, Guillausseau PJ, Cao R, Hori O, Stern D,Schmidt AM. 1996. Receptor-mediated endothelial cell dysfunction in diabeticvasculopathy. Soluble receptor for advanced glycation end products blockshyperpermeability in diabetic rats. J Clin Invest 97:238–243.
Wendt T, Harja E, Bucciarelli L, Qu W, Lu Y, Rong LL, Jenkins DG, Stein G, Schmidt AM, YanSF. 2006. RAGE modulates vascular inflammation and atherosclerosis in a murine model oftype 2 diabetes. Atherosclerosis 185:70–77.
Yamawaki H, Pan S, Lee R, Berk B. 2005. Fluid shear stress inhibits vascular inflammation bydecreasing thioredoxin-interacting protein in endothelial cells. J Clin Invest 115:733–738.
Yan SF, Ramasamy R, Bucciarelli LG, Wendt T, Lee LK, Hudson BI, Stern DM, Lalla E, Du YS,Rong LL, Naka Y, Schmidt AM. 2004. RAGE and its ligands: A lasting memory in diabeticcomplications? Diab Vasc Dis Res 1:10–20.
Zheng L, Howell S, Hatala D, Huang K, Kern T. 2007. Salicylate-based anti-inflammatory drugsinhibit the early lesion of diabetic retinopathy. Diabetes 56:337–345.

12 P E R R O N E E T A L .
Q1: Author: The Journal’s copyeditors have taken care to format your authorship according to journal style (First name, Middle Initial,
Surname). In the event a formatting error escaped their inspection, or there was insufficient information to apply journal style, please
take a moment to review all author names and sequences to ensure the accuracy of the authorship in the published article. Please note
that this information will also affect external indexes referencing this paper (e.g., PubMed).
Q2: Author: Please check the grant sponsors.
Q3: Author: Please specify whether it is 2008a, 2008b, or 2008c.
Q4: Author: Please specify whether it is 2008a, 2008b, or 2008c.
Q5: Author: Please provide complete location.
Q6: Author: Please specify whether it is 2008a, 2008b, or 2008c.
Q7: Author: Please add in the reference list.
Q8: Author: Please provide citation in the text.
Q9: Author: Please provide the volume number and page range.
Q10: Author: Please provide citation in the text.
JOURNAL OF CELLULAR PHYSIOLOGY

These proofs have been typeset using figure files transmitted to production when this article was accepted for publication. Please review all figures and note your approval with your submitted proof corrections. You may contact the journal production team at [email protected] if you wish to discuss specific concerns. Because of the high cost of color printing, we can only print figures in color if authors cover the expense. If you have submitted color figures, please indicate your consent to cover the cost on the table listed below by marking the box corresponding to the approved cost on the table. The rate for this journal is $500 USD per printed page of color, regardless on the number of figures appearing on that page. Please note, all color images will be reproduced online in Wiley InterScience at no charge, whether or not you opt for color printing. You will be invoiced for color charges once the article has been published in print.
Failure to return this form with your article proofs may delay the publication of your article.
JOURNAL
JOURNAL OF CELLULAR PHYSIOLOGY MS. NO. NO. COLOR PAGES
MANUSCRIPT TITLE
AUTHOR(S)
No. Color Pages Color Charge No. Color Pages Color Charge No. Color Pages Color Charge
1 $500 5 $2500 9 $4500 2 $1000 6 $3000 10 $5000 3 $1500 7 $3500 11 $5500 4 $2000 8 $4000 12 $6000
***Contact [email protected] for a quote if you have more than 12 pages of color***
Please print my figures color Please print my figures in black and white
Please print the following figures in color and convert these figures to black and white
Approved by
Billing Address
Telephone
Fax

C1
REPRINT BILLING DEPARTMENT •• 111 RIVER STREET •• HOBOKEN, NJ 07030 FAX: (201) 748-7670
E-MAIL: [email protected] REPRINT ORDER FORM
Please complete this form even if you are not ordering reprints. This form MUST be returned with your correctedproofs and original manuscript. Your reprints will be shipped approximately 4 weeks after publication. Reprints orderedafter printing will be substantially more expensive.
JOURNAL JOURNAL OF CELLULAR PHYSIOLOGY VOLUME ISSUE
TITLE OF MANUSCRIPT
MS. NO. NO. OF PAGES AUTHOR(S)
No. of Pages 100 Reprints 200 Reprints 300 Reprints 400 Reprints 500 Reprints$ $ $ $ $
1-4 336 501 694 890 10525-8 469 703 987 1251 14779-12 594 923 1234 1565 1850
13-16 714 1156 1527 1901 227317-20 794 1340 1775 2212 264821-24 911 1529 2031 2536 303725-28 1004 1707 2267 2828 338829-32 1108 1894 2515 3135 375533-36 1219 2092 2773 3456 414337-40 1329 2290 3033 3776 4528
**REPRINTS ARE ONLY AVAILABLE IN LOTS OF 100. IF YOU WISH TO ORDER MORE THAN 500 REPRINTS, PLEASE CONTACT OUR REPRINTSDEPARTMENT AT (201) 748-6353 FOR A PRICE QUOTE.
Please send me _____________________
reprints of the above article at $
Please add appropriate State and Local Tax (Tax ExemptNo.____________________)
$
for United States orders only.Please add 5% Postage and Handling $
TOTAL AMOUNT OF ORDER** $**International orders must be paid in currency and drawn on a U.S. bankPlease check one: Check enclosed Bill me Credit CardIf credit card order, charge to: American Express Visa MasterCard
Credit Card No Signature Exp. Date
BILL TO: SHIP TO: (Please, no P.O. Box numbers)Name Name
Institution Institution
Address Address
Purchase Order No. Phone Fax

A. COPYRIGHT
1. The Contributor assigns to Wiley-Blackwell, during the full term of copy-right and any extensions or renewals, all copyright in and to the Contribution,and all rights therein, including but not limited to the right to publish, repub-lish, transmit, sell, distribute and otherwise use the Contribution in whole or inpart in electronic and print editions of the Journal and in derivative worksthroughout the world, in all languages and in all media of expression nowknown or later developed, and to license or permit others to do so.
2. Reproduction, posting, transmission or other distribution or use of the finalContribution in whole or in part in any medium by the Contributor as permit-ted by this Agreement requires a citation to the Journal and an appropriatecredit to Wiley-Blackwell as Publisher, and/or the Society if applicable, suitablein form and content as follows: (Title of Article, Author, Journal Title and Volume/Issue, Copyright © [year], copyright owner as specified in the Journal).Links to the final article on Wiley-Blackwell’s website are encouraged whereappropriate.
B. RETAINED RIGHTS
Notwithstanding the above, the Contributor or, if applicable, the Contributor’sEmployer, retains all proprietary rights other than copyright, such as patentrights, in any process, procedure or article of manufacture described in theContribution.
C. PERMITTED USES BY CONTRIBUTOR
1. Submitted Version. Wiley-Blackwell licenses back the following rights tothe Contributor in the version of the Contribution as originally submitted forpublication:
a. After publication of the final article, the right to self-archive on the Con-tributor’s personal website or in the Contributor’s institution’s/employer’sinstitutional repository or archive. This right extends to both intranets andthe Internet. The Contributor may not update the submission version orreplace it with the published Contribution. The version posted must containa legend as follows: This is the pre-peer reviewed version of the followingarticle: FULL CITE, which has been published in final form at [Link to finalarticle].
b. The right to transmit, print and share copies with colleagues.
2. Accepted Version. Re-use of the accepted and peer-reviewed (but notfinal) version of the Contribution shall be by separate agreement with Wiley-Blackwell. Wiley-Blackwell has agreements with certain funding agencies governing reuse of this version. The details of those relationships, and otherofferings allowing open web use, are set forth at the following website:http://www.wiley.com/go/funderstatement. NIH grantees should check thebox at the bottom of this document.
3. Final Published Version. Wiley-Blackwell hereby licenses back to the Contributor the following rights with respect to the final published version ofthe Contribution:
a. Copies for colleagues. The personal right of the Contributor only to sendor transmit individual copies of the final published version in any format tocolleagues upon their specific request provided no fee is charged, and further-provided that there is no systematic distribution of the Contribu-tion, e.g. posting on a listserve, website or automated delivery.
b. Re-use in other publications. The right to re-use the final Contribution orparts thereof for any publication authored or edited by the Contributor(excluding journal articles) where such re-used material constitutes lessthan half of the total material in such publication. In such case, any modifi-cations should be accurately noted.
c. Teaching duties. The right to include the Contribution in teaching ortraining duties at the Contributor’s institution/place of employment includ-ing in course packs, e-reserves, presentation at professional conferences,in-house training, or distance learning. The Contribution may not be usedin seminars outside of normal teaching obligations (e.g. commercial semi-nars). Electronic posting of the final published version in connection withteaching/training at the Contributor’s institution/place of employment ispermitted subject to the implementation of reasonable access controlmechanisms, such as user name and password. Posting the final publishedversion on the open Internet is not permitted.
d. Oral presentations. The right to make oral presentations based on theContribution.
4. Article Abstracts, Figures, Tables, Data Sets, Artwork and SelectedText (up to 250 words).
a. Contributors may re-use unmodified abstracts for any non-commercialpurpose. For on-line uses of the abstracts, Wiley-Blackwell encourages butdoes not require linking back to the final published versions.
b. Contributors may re-use figures, tables, data sets, artwork, and selectedtext up to 250 words from their Contributions, provided the following conditions are met:
(i) Full and accurate credit must be given to the Contribution.(ii) Modifications to the figures, tables and data must be noted.
Otherwise, no changes may be made.(iii) The reuse may not be made for direct commercial purposes, or for
financial consideration to the Contributor.(iv) Nothing herein shall permit dual publication in violation of journal
ethical practices.
COPYRIGHT TRANSFER AGREEMENT
Date: Contributor name:
Contributor address:
Manuscript number (Editorial office only):
Re: Manuscript entitled
(the “Contribution”)
for publication in (the “Journal”)
published by (“Wiley-Blackwell”).
Dear Contributor(s):Thank you for submitting your Contribution for publication. In order to expedite the editing and publishing process and enable Wiley-Blackwell to disseminate your Contribution to the fullest extent, we need to have this Copyright Transfer Agreement signed and returned as directed in the Journal’sinstructions for authors as soon as possible. If the Contribution is not accepted for publication, or if the Contribution is subsequently rejected, this Agreement shall be null and void. Publication cannot proceed without a signed copy of this Agreement.
CTA-A

D. CONTRIBUTIONS OWNED BY EMPLOYER
1. If the Contribution was written by the Contributor in the course of the Contributor’s employment (as a “work-made-for-hire” in the course ofemployment), the Contribution is owned by the company/employer whichmust sign this Agreement (in addition to the Contributor’s signature) in thespace provided below. In such case, the company/employer hereby assigns toWiley-Blackwell, during the full term of copyright, all copyright in and to theContribution for the full term of copyright throughout the world as specified inparagraph A above.
2. In addition to the rights specified as retained in paragraph B above and therights granted back to the Contributor pursuant to paragraph C above, Wiley-Blackwell hereby grants back, without charge, to such company/employer, itssubsidiaries and divisions, the right to make copies of and distribute the finalpublished Contribution internally in print format or electronically on the Com-pany’s internal network. Copies so used may not be resold or distributed externally.However the company/employer may include information and text from theContribution as part of an information package included with software orother products offered for sale or license or included in patent applications.Posting of the final published Contribution by the institution on a public accesswebsite may only be done with Wiley-Blackwell’s written permission, and paymentof any applicable fee(s). Also, upon payment of Wiley-Blackwell’s reprint fee,the institution may distribute print copies of the published Contribution externally.
E. GOVERNMENT CONTRACTS
In the case of a Contribution prepared under U.S. Government contract orgrant, the U.S. Government may reproduce, without charge, all or portions ofthe Contribution and may authorize others to do so, for official U.S. Govern-
ment purposes only, if the U.S. Government contract or grant so requires. (U.S.Government, U.K. Government, and other government employees: see notesat end)
F. COPYRIGHT NOTICE
The Contributor and the company/employer agree that any and all copies ofthe final published version of the Contribution or any part thereof distributedor posted by them in print or electronic format as permitted herein will includethe notice of copyright as stipulated in the Journal and a full citation to theJournal as published by Wiley-Blackwell.
G. CONTRIBUTOR’S REPRESENTATIONS
The Contributor represents that the Contribution is the Contributor’s originalwork, all individuals identified as Contributors actually contributed to the Con-tribution, and all individuals who contributed are included. If the Contributionwas prepared jointly, the Contributor agrees to inform the co-Contributors ofthe terms of this Agreement and to obtain their signature to this Agreement ortheir written permission to sign on their behalf. The Contribution is submittedonly to this Journal and has not been published before. (If excerpts from copy-righted works owned by third parties are included, the Contributor will obtainwritten permission from the copyright owners for all uses as set forth in Wiley-Blackwell’s permissions form or in the Journal’s Instructions for Contributors,and show credit to the sources in the Contribution.) The Contributor also warrants that the Contribution contains no libelous or unlawful statements,does not infringe upon the rights (including without limitation the copyright,patent or trademark rights) or the privacy of others, or contain material orinstructions that might cause harm or injury.
CHECK ONE BOX:
Contributor-owned work
Contributor’s signature Date
Type or print name and title
Co-contributor’s signature Date
Type or print name and title
Company/Institution-owned work
Company or Institution (Employer-for-Hire) Date
Authorized signature of Employer Date
U.S. Government work Note to U.S. Government EmployeesA contribution prepared by a U.S. federal government employee as part of the employee’s official duties, orwhich is an official U.S. Government publication, is called a “U.S. Government work,” and is in the publicdomain in the United States. In such case, the employee may cross out Paragraph A.1 but must sign (in theContributor’s signature line) and return this Agreement. If the Contribution was not prepared as part of theemployee’s duties or is not an official U.S. Government publication, it is not a U.S. Government work.
U.K. Government work Note to U.K. Government Employees(Crown Copyright) The rights in a Contribution prepared by an employee of a U.K. government department, agency or other
Crown body as part of his/her official duties, or which is an official government publication, belong to theCrown. U.K. government authors should submit a signed declaration form together with this Agreement.The form can be obtained via http://www.opsi.gov.uk/advice/crown-copyright/copyright-guidance/publication-of-articles-written-by-ministers-and-civil-servants.htm
Other Government work Note to Non-U.S., Non-U.K. Government EmployeesIf your status as a government employee legally prevents you from signing this Agreement, please contactthe editorial office.
NIH Grantees Note to NIH GranteesPursuant to NIH mandate, Wiley-Blackwell will post the accepted version of Contributions authored by NIHgrant-holders to PubMed Central upon acceptance. This accepted version will be made publicly available 12 months after publication. For further information, see www.wiley.com/go/nihmandate.
ATTACH ADDITIONAL SIGNATURE
PAGES AS NECESSARY
(made-for-hire in thecourse of employment)
CTA-A
Copyright © 2022 FDOKUMEN




















