
The Roles of Ecdysoneless in Endoplasmic Reticulum Stress ...
191
University of Nebraska Medical Center University of Nebraska Medical Center DigitalCommons@UNMC DigitalCommons@UNMC Theses & Dissertations Graduate Studies Summer 8-18-2017 The Roles of Ecdysoneless in Endoplasmic Reticulum Stress and The Roles of Ecdysoneless in Endoplasmic Reticulum Stress and Oncogenesis Oncogenesis Appolinaire A. Olou University of Nebraska Medical Center Follow this and additional works at: https://digitalcommons.unmc.edu/etd Part of the Other Medicine and Health Sciences Commons Recommended Citation Recommended Citation Olou, Appolinaire A., "The Roles of Ecdysoneless in Endoplasmic Reticulum Stress and Oncogenesis" (2017). Theses & Dissertations. 219. https://digitalcommons.unmc.edu/etd/219 This Dissertation is brought to you for free and open access by the Graduate Studies at DigitalCommons@UNMC. It has been accepted for inclusion in Theses & Dissertations by an authorized administrator of DigitalCommons@UNMC. For more information, please contact [email protected].
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of The Roles of Ecdysoneless in Endoplasmic Reticulum Stress ...

University of Nebraska Medical Center University of Nebraska Medical Center
DigitalCommons@UNMC DigitalCommons@UNMC
Theses & Dissertations Graduate Studies
Summer 8-18-2017
The Roles of Ecdysoneless in Endoplasmic Reticulum Stress and The Roles of Ecdysoneless in Endoplasmic Reticulum Stress and
Oncogenesis Oncogenesis
Appolinaire A. Olou University of Nebraska Medical Center
Follow this and additional works at: https://digitalcommons.unmc.edu/etd
Part of the Other Medicine and Health Sciences Commons
Recommended Citation Recommended Citation Olou, Appolinaire A., "The Roles of Ecdysoneless in Endoplasmic Reticulum Stress and Oncogenesis" (2017). Theses & Dissertations. 219. https://digitalcommons.unmc.edu/etd/219
This Dissertation is brought to you for free and open access by the Graduate Studies at DigitalCommons@UNMC. It has been accepted for inclusion in Theses & Dissertations by an authorized administrator of DigitalCommons@UNMC. For more information, please contact [email protected].

UNIVERSITY OF NEBRASKA, MEDICAL CENTER
Role of Mammalian Ecdysoneless protein in Endoplasmic Reticulum Stress and Oncogenesis
A DISSERTATION
SUBMITTED TO THE GRADUATE SCHOOL
IN PARTIAL FULFILLMENT OF THE REQUIREMENTS
for the degree of
Doctor of Philosophy
By
Appolinaire A. Olou
Omaha, Nebraska
August 2017
Committee Members
Dr. Band Vimla (PI)
Dr. Datta Kaustubh Dr. Bhakat Kishor
Dr. Band Hamid Dr. Lu Runqing*
* Dr. Runqing Lu passed away un-expectedly on March 29, 2016

1
TABLE OF CONTENTS Page Number
Abstract 4
Acknowledgement 6
List of Figures 7
Abbreviation 9
Chapter One: Introduction 14
1.1 Overview of the Endoplasmic Reticulum (ER) and its functions in the cells 15
1.2 Physiological and pathological conditions that perturb ER homeostasis and
induce stress 16
1.3 Upstream signaling pathways of the ER stress response 17
1.4The ER stress in Cancer: a paradox 19
1.5 The Ecdysoneless (ECD) protein, its oncogenic and stress-related potentials 21
References 24
Chapter Two: Mammalian ECD protein functions as a novel negative regulator of
the Unfolded Protein Response 29
2.1 Overview 30

2
2.2 Introduction 33
2.3 Materials and Methods 38
2.4 Results 42
2.4.1 Effect of ER stress induction by Thapsigargin, Tunicamycin, Brefeldin A on
ECD protein levels in MCF-7, MDA-468, and MCF10A 42
2.4.2 Physiological stress by glucose deprivation led to a decrease in ECD protein
levels 45
2.4.3 Effects of other forms of stress on ECD protein levels 48
2.4.4 ER stress by Thapsigargin and glucose deprivation led to increase in ECD
mRNA levels 52
2.4.5 The decrease in the levels of ECD protein upon ER stress induction is PERK-
eIF2α dependent 56
2.4.6 ECD depletion or overexpression distinctly regulates the UPR upon ER stress
induction 66
2.4.7 ECD overexpression protects cells from ER stress-induced cell death 80
2.4.8 ECD co-localizes and associates with PERK, ATF6, IRE1α and GRP78 84
2.4.9 ECD association with GRP78 and PERK upon ER stress induction 91
2.4.10 ECD does not modulate enzymatic activity of PERK in vitro 94

3
2.4.11 ECD is a positive regulator of chaperones, predominantly, GRP78 protein
levels 96
2.4.12 ECD does not affect GRP78 transcription or mRNA stability 101
2.4.13 ECD enhances GRP78 protein stability and this effect is required for ECD to
attenuate ER stress signaling 107
2.5 Discussion 114
References 118
Chapter Three: Summary and Conclusion 125
Chapter Four: Future Directions 132
Appendix 137

4
ABSTRACT
Tumor cells are well known to exhibit ER stress as a result of their altered environment
characterized by redox and calcium imbalance, deregulation of protein synthesis to meet
their oncogenic demands, and decreased vascularization associated with nutrient limitation
and hypoxia, all of which are conducive of ER stress. Accordingly, markers of ER stress
signaling response are up-regulated in various cancers. The outcome of ER stress signaling
response varies from survival and adaption to apoptosis. Although pro-apoptotic ER stress
signaling molecules are up-regulated in cancer, the mechanisms by which cancer cells seem
refractory to, or evade, their apoptotic signaling are not fully understood. In this study, we
investigate roles of the mammalian Ecdysoneless (ECD) in ER stress and oncogenesis.
Certain tumors overexpress ECD protein suggesting possible roles in oncogenesis. In fact,
both normal and cancer cells require ECD for their growth and undergo cell cycle arrest
when the ECD level is deficient. ECD associates with various stress response proteins such
as p53, well known for genotoxic stress response such as DNA damage, and TXNIP, an
oxidative stress protein recently implicated in ER stress response. Furthermore, ECD is a
critical component of chaperone-like complexes such as the R2TP complex whose
members have ER stress-related roles. In this study, we provide evidence that ECD is
involved in ER stress.
Stress induction by multiple stimuli, including Thapsigargin, Tunicamycin, glucose
starvation, H2O2, NOX4 overexpression, led to reduced ECD protein levels, but ECD
mRNA increased, in a PERK-eIF2α dependent manner. Although a phospho-protein, ECD
is not a target for a phosphorylation-mediated degradation by PERK but is rather targeted
for a translation block via the PERK-eIF2α axis. To assess the functional connection

5
between ECD and ER stress signaling pathways, we used cells in which ECD could be
inducibly depleted or overexpressed. Depletion of ECD enhanced PERK signaling and
apoptosis upon ER stress induction while overexpression of ECD produced the opposite
effect by inhibiting PERK signaling and increasing cell survival. IRE1α signaling was
slightly affected by these changes in the cellular level of ECD, as indicated by the slight
increase or decrease of its downstream target, spliced XBP-1, upon ER stress induction in
the presence or absence of ECD; on the other hand, the ATF6 pathway was minimally
affected.
Based on these findings, we examined the possible mechanism by which ECD regulates
ER stress signaling, particularly the PERK pathway. We found that ECD co-localized and
associated with all the ER stress sensors PERK, IRE1α, ATF6 and GRP78. However, ECD
does not modulate the enzymatic activity of PERK toward its substrate eIF2α. Rather, ECD
enhanced chaperones’ levels, predominantly, GRP78 upon ER stress induction. Finally,
disruption of this chaperone-enhancing effect of ECD abrogated the attenuating effect of
ECD on PERK and impaired the pro-survival effect of overexpressed ECD upon ER stress
induction. Taken together, ECD regulates the levels of chaperones, predominantly, GRP78
to enhance the folding capacity of the stressed ER and provide survival advantage in a
stress condition.

6
ACKNOWLEDGMENTS
Dr. Vimla Band is the principal investigator (PI) in the ECD project, which this study was
a part of. She is one of the very few researchers working on ECD. Obviously, this project
could not have gone anywhere without her and I would like to thank her for her active roles
in the study. She did all she could to ensure completion of this project, including countless
meetings and discussions. I would also like to thank my committee members for their
suggestions along the way.
I thank the Band lab members for being part of the journey too. Particularly, I thank Dr.
Mir Riyaz who embraced me from day one as his own student and answered every question
I asked him, including technical ones.

7
LIST OF FIGURES
Figure 2.1 Effect of ER stress induction in MCF7, MDA-468 and MCF-10A
Figure 2.2: Physiological stress by glucose starvation reduces ECD protein levels: (A-B)
Figure 2.3: Effects of other forms of stress (oxidative, reductive and osmotic) on ERCD
protein level
Figure 2.4: ER stress induction by Thapsigargin and glucose starvation led to an increase
in ECD mRNA
Figure 2.5: The decrease in the levels of ECD protein upon ER stress induction is PERK
dependent
Figure 2.6: ECD mRNA levels failed to increase in PERK KO MEFs upon ER stress
Figure 2.7: PERK does not phosphorylate ECD
Figure 2.8: The decrease in ECD protein levels is abrogated in eIF2α phospho-deficient
MEFs
Figure 2.9: Increased UPR in ECD-null cells upon Thapsigargin
Figure 2.10: ECD-null cells exhibited increased UPR and cell death upon Thapsigargin
treatment
Figure 2.11: ECD knock down in Panc-1 cells upon glucose starvation led to increased
UPR and cell death

8
Figure 2.12: Decreased p-PERK and CHOP mRNA induction in ECD inducible MEFs
upon Thapsigargin
Figure 2.13: Decreased UPR signaling in ECD inducible MEFs correlated with in
chaperones induction upon Thapsigargin and Tunicamycin treatment
Figure 2.14: Decreased cleaved caspase 3 and p-SAPK/JNK levels in ECD
overexpressing MEF upon Thapsigargin
Figure 2.15: Increased colony formation in ECD over-expressing MEFs upon
Thapsigargin
Figure 2.15: ECD co-localizes with PERK and GRP78
Figure 2.16: ECD associates with PERK, GRP78, IRE1α and ATF6
Figure 2.17: Defining the domain (s) on ECD required for interaction
Figure 2.18: ECD association with PERK and GRP78 upon ER stress induction
Figure 2.19: ECD does not modulate enzymatic activity of PERK toward eIF2α
Figure 2.20: ECD deletion or knock down led to a reduced induced levels of GRP78.
Figure 2.21: ECD overexpression led to a robust increase in chaperone levels,
predominantly, GRP78
Figure 2.22: Effect of ECD on GRP78 mRNA induction
Figure 2.23: Effect of ECD on GRP78 mRNA stability

9
Figure 2.24: ECD mediated GRP78 protein stability is required for ECD ER stress
function
ABBREVIATIONS
ASK1: Apoptosis-Signaling Kinase 1
ATF2: Activation of Transcription Factor 2
ATF4: Activation of Transcription Factor 4
ATF6; Activation of Transcription Factor 6
ATP: Adenosine Triphosphate
Bcl-2:B-cell Lymphoma 2
BFA: Brefeldin A
CDK2: Cyclin Dependent Kinase 2
CHOP: CCAAT enhancer binding protein Homologous Protein
CK2: Casein Kinase 2
DNA: Deoxyribonucleic Acid

10
Dox: Doxycycline
DTT: Dithiothreitol
ECD: Ecdysoneless
EDEM: ER Degradation Enhancing α-mannosidase-like protein
eIF2α: Eukaryotic Initiation of translation Factor 2
ER: Endoplasmic Reticulum
ERAD: ER Associated Degradation
ERo1a: ER Oxidoreductin- 1 alpha
G1/S: G1 phase to S phase transition
G2/M: G2 phase to M phase
GADD34: Growth Arrest and DNA Damage inducible 34
GADD53: Growth Arrest and DNA Damage inducible 153
GCN2: General Control Non-derepressible 2
GCR2: Glycolysis Regulation 2
GFP: Green Fluorescent Protein
GRP78: Glucose-Regulated Protein 78
GRP94: Glucose-Regulated Protien 94

11
GTP: Guanine Triphosphate
H2O2: Hydrogen Peroxide
hECD: Human Ecdysoneless
HRI: Heme-Regulated Inhibitor
hSGT1: Human Suppressor of GCR two 1
IRE1α: Inositol-Requiring Enzyme 1 alpha
IRES: Internal Ribosomal Entry Site
JNK: c-Jun N-terminal Kinase
KO: Knockout
MEF: Mouse Embryonic Fibroblast
Met: Methionine
mRNA: Messenger RNA
NOX4: NADPH Oxidase 4
O2: Oxygen
P53: Protein 53
p58 IPK: Protein 58 Inhibitor of Protein Kinase
PCR: Polymerase Chain Reaction

12
PDI: Protein Disulfide Isomerase
p-eIF2α: phospho-eIF2α
PERK:PKR-like ER Kinase
PKR: Protein Kinase RNA-dependent
PIH1D1: Protein Interacting with Hsp90 1contining Domain 1
PP1: Protein Phosphatase 1
p-PERK: phospho-PERK
PTM: Post Translation Modification
qRT-PCR: quantitative Real-Time PCR
R2TP: RUVB1/RUVB2/Tah1/PiH comtaining complex
RedOx: Oxidation and Reduction
RIDD: Regulated IRE1 Dependent Decay
RNA: Ribonucleic Acid
ROS: Reactive Oxygen Species
rtTA: Reverse Tetracycline controlled Trans-activator
RUVBL1: RuvB-like AAA ATPase 1
S. Cerevisiae: Saccharomyces cerevisiae

13
SAPK: Stress Activated Protein Kinases
SDS-PAGE: Sodium Dodecylsulfate-polyacrylamide gel electrophoresis
siRNA: Small- Interfering RNA
SP1/2: Site ½ Proteinase
TRAF2: TNF Receptor Associated Factor 2
tRNA: transfer RNA
TXNIP: Thioredoxin-Interacting Protein
UPR: Unfolded Protein Response
WT: Wild-Type
XBP-1: X-box Binding Protien 1

14
CHAPTER ONE: INTRODUCTION

15
1.1 Overview of the Endoplasmic Reticulum (ER) and its functions in the cells
The Unfolded Protein Response (UPR), a term used interchangeably with
Endoplasmic Reticulum (ER) stress response, is a reference to stress in the ER which is a
membrane-bound sub-cellular organelle interconnecting other organelles (1), including
the Golgi apparatus, the lysosome, the mitochondria and the nucleus. It is the largest
organelle and the site for many central cellular functions such as synthesis, folding and
maturation of secreted and membrane proteins, biogenesis of cholesterol, and calcium
storage. Different classes of proteins mediate these functions of the ER; key among them
are proteins involved in translation. In eukaryotic cells, translation occurs directly in the
ER, following assembly and translocation of the ribosomal complex to the translocation
channel. This complex is composed of the eukaryotic initiation factor 2 alpha (eIF2a) in a
ternary complex with bound GTP and the methionine initiator tRNA (Met-tRNAi-eIF2a-
GTP) (2). As the newly synthesized proteins enter the ER, they undergo post-translational
modifications (PTM) in an oxidative environment. Examples of common PTM are
glycosylation, disulfide bond formation and phosphorylation. Proper PTM precedes
proper folding of proteins into their three-dimensional structure with the assistance of ER
resident chaperones such as the Glucose-Regulated Protein 78/ Immunoglobin Binding
Protein (GRP78/ Bip), GRP94, calreticulin, calnexin and protein disulfide isomerases or
PDI (3). The PDIs, in particular, are oxygen-requiring ER oxidoreductase 1 (Ero1)
enzymes which generate disulfide bonds de novo and transfer them to their thioredoxin
motif containing the thiol/disulfide oxidoreductase site (4). Lastly, the ER chaperones are
calcium binding proteins and function calcium-dependently, justifying why the ER is also

16
the main calcium storage in the cell, with ATP-dependent calcium pumps which influx
calcium from the cytosol to the ER. The ER also participates in intra and extracellular
calcium signaling (5).
1.2 Physiological and pathological conditions that perturb ER homeostasis and
induce stress
Physiological conditions that induce stress in the ER, such as glucose deficiency,
are cell type specific. Glycosylation, a critical PTM, is disrupted when cells are glucose-
deprived, as a result of a decreased level of glycosyl group in the cells (6). Moreover,
glucose deficiency also decreases ATP level in the cells, impairing crucial functions of
ATP-dependent calcium pumps in the cells (6). High glucose also perturbs proper ER
function in secretory cells, such as pancreatic beta cells, due to the ensuing increased
production of insulin that must be processed through the ER. Other physiological
perturbants that induce ER stress are calcium deprivation, which impairs the function of
key ER chaperones, free fatty acids and cytokines. In pancreatic beta cells, the best model
for studying ER stress, ER stress can be physiologically induced by mutant insulin (6).
Thus, pathological conditions that cause increased blood glucose levels, such as diabetes,
induce ER stress (7). Viral infection is another type of pathological ER stress inducer
because of the increased synthesis of viral proteins that must be processed in the host ER.
Ischemia, the loss of blood supply to body organs, is also known to induce ER stress (8).
Finally, in order to support proper protein folding, the ER environment is highly

17
oxidative and is sensitive to change in the reduction-oxidation (RedOx) state of the cell
(9).
1.3 Upstream signaling pathways of the ER stress response
ER stress is a condition in cells characterized by protein load in the ER exceeding
the folding capacity of the ER. It can be caused by any of the physiological or
pathological conditions mentioned above. Additionally, pharmaceutical agents such as
Thapsigargin, Tunicamycin, Brefeldin A, Dithiothreitol and hydrogen peroxide also
induce ER stress. Perturbation of the ER function elicits a response termed the unfolded
protein response (UPR). The UPR is an adaptive pro-survival response in its initial phase
but shifts to a pro-apoptotic response when the stress becomes overwhelming (10-12).
Three canonical pathways mediate these two phases of the UPR of which PERK (PKR-
like ER Kinase), IRE1α (Inositol Requiring Enzyme 1 alpha) and ATF6 (Activation of
Transcription Factor 6) are the upstream players.
PERK is an ER transmembrane kinase with luminal, transmembrane and
cytoplasmic domains. The cytoplasmic c-terminal kinase domain of PERK is activated by
homo-dimerization of the protein followed by auto-phosphorylation (10,13,14). The
active PERK kinase phosphorylates the eukaryotic initiation factor 2 alpha (eIF2a) (15),
altering its conformation and leading to a gradual disassembly of the ternary Met-tRNAi-
eIF2a-GTP complex, a critical part of the active translation complex. The resulting
inhibition of the translation machinery causes a decrease in the load of protein entering
the ER lumen (15) as well as cell cycle block, allowing cells time to solve the stress in
the ER. Although global cap-dependent translation is blocked, a cap-independent
translation of specific ER stress response genes proceeds. One such cap-independently

18
translated protein is Activation of Transcription Factor 4 (ATF4) (15), a transcription
factor of the same family as ATF6. ATF4 translocates into the nucleus to activate a
transcription enhancer gene, the CCAAT-enhancer-binding protein homologous protein
(CHOP), which sensitizes cells to ER stress-induced cell death by multiple mechanisms
including a down-regulation of the pro-survival Bcl-2 (B-Cell Lymphoma 2) protein
family while increasing levels of members of the death receptor protein family.
Moreover, CHOP is known to down-regulate glutathione levels, the cell’s anti-oxidant
defense protein, leading to a surge in the levels of reactive oxygen species (ROS), a
subsequent release of cytochrome c into the cytoplasm and activation of the caspase
cascade, culminating in cell death (15-19). Concurrently, CHOP activates another
protein, GADD34 which, together with the type 1 protein phosphatase (PP1), targets
eIF2α for de-phosphorylation and resumption of protein synthesis (20-24). Alternatively,
this recovery from the stress-induced translation block is also achieved in cells by cellular
PERK inhibitor p58 IPK (25,26).
IRE1 exists in two isoforms, IRE1α and IRE1β. IRE1α, the widely studied
isoform, has both kinase and RNAse function. Like PERK, it is activated by dimerization
and auto-phosphorylation. With the RNAse activity, it is known to splice a 26 nucleotide
intron from the X-box binding protein 1 (XBP-1) (16,27-30) to generate an active
transcription factor, which translocates to the nucleus to activate ER response genes, one
of which is the ER degradation-enhancing protein (EDEM) (16,31). In severe ER stress,
EDEM activates the massive degradation of misfolded and unfolded proteins, termed ER-
associated degradation (ERAD) (32,33), a process involving ER chaperone-mediated
retro-translocation of proteins to the cytoplasm for proteasomal degradation. The RNAse

19
function of IRE1α is also associated with a degradation of ER-associated mRNA in a
process termed RIDD or regulated IRE1α-dependent decay (34,35) to further inhibit
protein translation into the stressed ER. With the kinase function, IRE1α is known to
recruit TRAF2 (TNF-receptor activating factor 2) to activate the apoptosis-signaling
kinase 1 (ASK1) and the Jun N-terminal Kinase (JNK), leading to caspase cascade and
cell death (11,36).
ATF6, like IRE1, exists in two isoforms, ATF6α, the most studied isoform, and
ATF6β. Upon ER stress signaling, ATF6α translocates to the Golgi apparatus where it is
proteolytically cleaved by the Golgi Site 1 & 2 proteases (SP 1&2) (37,38) to generate an
active transcription factor which translocates into the nucleus to activate ER stress target
genes. ATF6α activates expression of chaperones upon ER stress, including GRP78,
GRp94 (39).
1.4 The ER stress in Cancer: a paradox
The ER stress is the basis of diseases collectively referred to as ER stress diseases,
including heart diseases, neurodegenerative diseases, diabetes and cancer (40). With regard
to cancer, the ER stress response is known to promote survival mechanisms in cancer,
hence driving oncogenesis. The tumor microenvironment is indeed a very hostile
environment, characterized by Redox imbalance (41,42)], deregulated protein synthesis
(43), and decreased vascularization associated with hypoxia and hypoglycemia (44), all of
which are conducive to ER stress. Cancer cells also generate a higher level of hydrogen
peroxide, a precursor of reactive oxygen species (ROS) which also perturbs ER

20
homeostasis. As such, markers of the ER stress response are up-regulated in various
cancers [22], suggestive of fully activated and operational ER stress signaling to drive
tumor progression. For instance, X-Box binding Protein 1(XBP1) promotes survival in
triple negative breast cancer by enhancing the hypoxic response pathway (45) while CHOP
drives hepatocellular oncogenesis by promoting oncogenic cell growth (46). In the same
vein, the chaperone GRP78 promotes tumor survival, metastasis, angiogenesis and
resistance to a wide variety of therapies (47,48).
Controversially, components of the ER stress response are also implicated in
negative regulatory control of tumor growth. In this context, activated PERK has been
known to induce proliferation block via multiple mechanisms including blocks in DNA
synthesis and protein translation (49-53). Moreover, expression of PERK leads to tissue
atrophy in Drosophila melanogaster, a phenotype that was corrected by co-expressing
GADD34, the phosphatase that represses PERK signaling (54). Although there are
conflicting reports as to the role of PERK in tumor progression, the PERK pathway has a
strong potential to reduce the growth of tumor cells (55). Besides PERK, a second
possibility exists for the ER stress signaling to regulate tissue growth through ATF4
which has also been shown to negatively control neurogenesis in the developing mouse
brain (56). ATF4 levels change during cell cycle progression and the protein undergoes a
phosphorylation-dependent degradation required for cell cycle progression (56). This
negative regulatory role of ATF4 on cell growth was observed in another study in which
overexpression of ATF4 inhibited cell proliferation and mammary gland development
(57).

21
The last and third level of negative regulation of cell growth by components of the
ER stress response involves CHOP, also known as Growth Arrest and DNA-Damage
inducible (GADD153). Induction of CHOP has been reported in in vitro differentiation
experiments of 3T3-L1 cells to adipocytes, a process requiring growth arrest (58),
suggesting a connection of CHOP to growth arrest as indicated by the very nomenclature
of the protein (Growth Arrest and DNA-Damage inducible). Moreover, in tumors of
adipose tissue, a chromosomal translocation has been found to cause a fusion protein
between CHOP and a novel RNA binding protein, TLS (translocated in liposarcoma)
(59-61). The TLS-CHOP fusion protein was oncogenic (62). This oncogenic gain of
function of the mutant form of CHOP, ie TLS-CHOP, further implies an anti-oncogenic
or growth suppressive role of the wild type CHOP. In fact, microinjection of CHOP into
cells inhibited BrdU incorporation and caused proliferative block, a defect reversed by
the TLS-CHOP variant mentioned above (62). Altogether, these studies provide strong
evidence that the ER stress signaling has the capacity to both negatively and positively
affect tumor growth. However, paradoxically, although the ER stress signaling is
activated in various cancers, cells in tumors seem refractory to its growth inhibitory
signaling and only proliferate, even indefinitely. This seeming tolerance of tumor cells to
the stress-induced growth inhibitory signaling hints to the existence of mechanisms to
desensitize and protect tumor cells against the growth inhibitory effect of the ER stress
signaling.
1.5 The Ecdysoneless (ECD) protein, its oncogenic and stress-related potentials

22
ECD was first identified in Drosophila as the mutation that caused a deficiency in
the steroid hormone, Ecdysone, a hormone that plays roles in normal embryogenesis,
metamorphosis and reproduction (63), all of which require cell growth and proliferation.
As a result, these ECD mutants understandably exhibited developmental defects. The
mammalian ECD was discovered from a screen of genes that could rescue the growth
defects in S. Cerevisiae mutant for the Growth Control Regulatory gene 2, GCR2
(64).This was the first indication that the mammalian ECD plays a role in growth. Studies
from my mentor’s lab provided the most direct evidence that ECD does indeed play a key
role in positively regulating cell growth and development. The lack of ECD in cells
caused cell cycle to be arrested in G1 phase, a defect that was rescued by exogenously
expressed ECD (65). The requirement of ECD for cell growth may justify its over-
expression in various cancers and its association with poor prognosis and short patient
survival (66,67). In pancreatic cancer, knock down of ECD reduced proliferation and
tumorigenicity with cells arrested at G1-S phase (66). While ECD overexpression alone
did not accelerate cell proliferation, its overexpression together with Ras enhanced cell
proliferation in epithelial cells did (68). However, ECD overexpression in fibroblasts
cells induced p53 dependent cellular senescence, by p53 stabilization and positively
regulatingp53(69), implying a link to p53. Moreover, ECD interacts with p53 to stabilize
it and positively regulates its transcriptional activity (69), raising the possibility that ECD
may have a connection to p53 pathways. In addition to its tumor suppressive roles, p53 is
also related to stress such as genotoxic stress and oxidative stress. ECD may be part of
protein complexes that modulate p53 stress functions such as the oxidative stress
regulator TXNIP. ECD interacts with TXNIP and together, ECD-TXNIP increases p53

23
activity (70). Similar to ECD, TXNIP interacts with p53 to increase its stability and
function under oxidative stress (71). Significantly, a recent study found an ER-stress-
related role for TXNIP in its interaction with PDI (Protein Disulfide Isomerase) to
increase their enzymatic activity during ER stress thereby reducing stress (72).
Furthermore, TXNIP is implicated in ER stress response as a novel component of the
stress response signaling to mediate the inflammation-related axis of the stress-induced
apoptosis (73). Also, TXNIP may also regulate the physiological function of the ER
because it is part of the cellular redoxisome, comprising Thioredoxin, which regulates the
redox state of the cell including the redox environment in the ER. The PDIs mentioned
above are redox-sensitive ER oxidoreductases with Thioredoxin motifs and changes in
the ER oxidative environment impair their functions (4).
Lastly, we and others have recently shown that ECD associates with R2TP
complex (74,75). The R2TP complex is involved in the assembly and remodeling of large
protein-protein or protein-RNA complexes, such as RNA polymerase, small nucleolar
ribonucleoproteins (snoRNPs), and phosphatidylinositol 3-kinase-related kinases
(PIKKs) (76). Our recent study showed that the N-terminal region of ECD associates
with the RUVBL1 component of the R2TP complex and both the phosphorylation and
interaction of ECD with RUVBL1 are required for ECD’s role to promote cell cycle
progression (75). The R2TP has an ER stress-related role through its RUVBL2
component (77). Notably, RUVBL2, or TIP49b has been shown to inhibit the activity of
ATF2 (77), a downstream target of PERK signaling, suggesting a role of the R2TP
complex component in ER stress. Overall, the multiple lines of evidence presented above

24
suggest some connection of ECD to ER stress. In my thesis, I investigated in detail
whether ECD regulates ER stress.
REFERENCES
1. Friedman, J. R., and Voeltz, G. K. (2011) The ER in 3D: a multifunctional dynamic membrane network. Trends Cell Biol. 21, 709-717
2. Lopez-Lastra, M., Rivas, A., and Barria, M. I. (2005) Protein synthesis in eukaryotes: the growing biological relevance of cap-independent translation initiation. Biol. Res. 38, 121-146
3. Verfaillie, T., Garg, A. D., and Agostinis, P. (2013) Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett. 332, 249-264
4. Sevier, C. S., and Kaiser, C. A. (2008) Ero1 and redox homeostasis in the endoplasmic reticulum. Biochim. Biophys. Acta 1783, 549-556
5. Berridge, M. J., Lipp, P., and Bootman, M. D. (2000) The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1, 11-21
6. Oslowski, C. M., and Urano, F. (2011) Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 490, 71-92
7. Back, S. H., and Kaufman, R. J. (2012) Endoplasmic reticulum stress and type 2 diabetes. Annu. Rev. Biochem. 81, 767-793
8. Nakka, V. P., Gusain, A., and Raghubir, R. (2010) Endoplasmic reticulum stress plays critical role in brain damage after cerebral ischemia/reperfusion in rats. Neurotox. Res. 17, 189-202
9. Bhandary, B., Marahatta, A., Kim, H. R., and Chae, H. J. (2012) An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. International journal of molecular sciences 14, 434-456
10. Szegezdi, E., Logue, S. E., Gorman, A. M., and Samali, A. (2006) Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO reports 7, 880-885
11. Tabas, I., and Ron, D. (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 13, 184-190
12. Hetz, C., Chevet, E., and Oakes, S. A. (2015) Proteostasis control by the unfolded protein response. Nat. Cell Biol. 17, 829-838
13. Lai, E., Teodoro, T., and Volchuk, A. (2007) Endoplasmic reticulum stress: signaling the unfolded protein response. Physiology 22, 193-201
14. Ma, K., Vattem, K. M., and Wek, R. C. (2002) Dimerization and release of molecular chaperone inhibition facilitate activation of eukaryotic initiation factor-2 kinase in response to endoplasmic reticulum stress. J. Biol. Chem. 277, 18728-18735
15. Harding, H. P., Novoa, I., Zhang, Y., Zeng, H., Wek, R., Schapira, M., and Ron, D. (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099-1108

25
16. Boelens, J., Lust, S., Offner, F., Bracke, M. E., and Vanhoecke, B. W. (2007) Review. The endoplasmic reticulum: a target for new anticancer drugs. In Vivo 21, 215-226
17. Ma, Y., Brewer, J. W., Diehl, J. A., and Hendershot, L. M. (2002) Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J. Mol. Biol. 318, 1351-1365
18. Zinszner, H., Kuroda, M., Wang, X., Batchvarova, N., Lightfoot, R. T., Remotti, H., Stevens, J. L., and Ron, D. (1998) CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 12, 982-995
19. Malhotra, J. D., Miao, H., Zhang, K., Wolfson, A., Pennathur, S., Pipe, S. W., and Kaufman, R. J. (2008) Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. U. S. A. 105, 18525-18530
20. Marciniak, S. J., Yun, C. Y., Oyadomari, S., Novoa, I., Zhang, Y., Jungreis, R., Nagata, K., Harding, H. P., and Ron, D. (2004) CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18, 3066-3077
21. Ma, Y., and Hendershot, L. M. (2003) Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J. Biol. Chem. 278, 34864-34873
22. Novoa, I., Zeng, H., Harding, H. P., and Ron, D. (2001) Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol. 153, 1011-1022
23. Connor, J. H., Weiser, D. C., Li, S., Hallenbeck, J. M., and Shenolikar, S. (2001) Growth arrest and DNA damage-inducible protein GADD34 assembles a novel signaling complex containing protein phosphatase 1 and inhibitor 1. Mol. Cell. Biol. 21, 6841-6850
24. Novoa, I., Zhang, Y., Zeng, H., Jungreis, R., Harding, H. P., and Ron, D. (2003) Stress-induced gene expression requires programmed recovery from translational repression. EMBO J. 22, 1180-1187
25. van Huizen, R., Martindale, J. L., Gorospe, M., and Holbrook, N. J. (2003) P58IPK, a novel endoplasmic reticulum stress-inducible protein and potential negative regulator of eIF2alpha signaling. J. Biol. Chem. 278, 15558-15564
26. Yan, W., Frank, C. L., Korth, M. J., Sopher, B. L., Novoa, I., Ron, D., and Katze, M. G. (2002) Control of PERK eIF2alpha kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proc. Natl. Acad. Sci. U. S. A. 99, 15920-15925
27. Ron, D., and Walter, P. (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519-529
28. Prischi, F., Nowak, P. R., Carrara, M., and Ali, M. M. (2014) Phosphoregulation of Ire1 RNase splicing activity. Nature communications 5, 3554
29. Lisbona, F., Rojas-Rivera, D., Thielen, P., Zamorano, S., Todd, D., Martinon, F., Glavic, A., Kress, C., Lin, J. H., Walter, P., Reed, J. C., Glimcher, L. H., and Hetz, C. (2009) BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1alpha. Mol. Cell 33, 679-691
30. Calfon, M., Zeng, H., Urano, F., Till, J. H., Hubbard, S. R., Harding, H. P., Clark, S. G., and Ron, D. (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92-96
31. Lee, A. H., Iwakoshi, N. N., and Glimcher, L. H. (2003) XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23, 7448-7459
32. Wang, T., and Hebert, D. N. (2003) EDEM an ER quality control receptor. Nat. Struct. Biol. 10, 319-321
33. Oda, Y., Hosokawa, N., Wada, I., and Nagata, K. (2003) EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science 299, 1394-1397

26
34. Hollien, J., and Weissman, J. S. (2006) Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313, 104-107
35. Hollien, J., Lin, J. H., Li, H., Stevens, N., Walter, P., and Weissman, J. S. (2009) Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 186, 323-331
36. Urano, F., Wang, X., Bertolotti, A., Zhang, Y., Chung, P., Harding, H. P., and Ron, D. (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664-666
37. Ye, J., Rawson, R. B., Komuro, R., Chen, X., Dave, U. P., Prywes, R., Brown, M. S., and Goldstein, J. L. (2000) ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 6, 1355-1364
38. Chen, X., Shen, J., and Prywes, R. (2002) The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J. Biol. Chem. 277, 13045-13052
39. Baumeister, P., Luo, S., Skarnes, W. C., Sui, G., Seto, E., Shi, Y., and Lee, A. S. (2005) Endoplasmic reticulum stress induction of the Grp78/BiP promoter: activating mechanisms mediated by YY1 and its interactive chromatin modifiers. Mol. Cell. Biol. 25, 4529-4540
40. Schonthal, A. H. (2012) Endoplasmic reticulum stress: its role in disease and novel prospects for therapy. Scientifica 2012, 857516
41. Szatrowski, T. P., and Nathan, C. F. (1991) Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 51, 794-798
42. Jorgenson, T. C., Zhong, W., and Oberley, T. D. (2013) Redox imbalance and biochemical changes in cancer. Cancer Res. 73, 6118-6123
43. Grzmil, M., and Hemmings, B. A. (2012) Translation regulation as a therapeutic target in cancer. Cancer Res. 72, 3891-3900
44. Masson, N., and Ratcliffe, P. J. (2014) Hypoxia signaling pathways in cancer metabolism: the importance of co-selecting interconnected physiological pathways. Cancer & metabolism 2, 3
45. Chen, X., Iliopoulos, D., Zhang, Q., Tang, Q., Greenblatt, M. B., Hatziapostolou, M., Lim, E., Tam, W. L., Ni, M., Chen, Y., Mai, J., Shen, H., Hu, D. Z., Adoro, S., Hu, B., Song, M., Tan, C., Landis, M. D., Ferrari, M., Shin, S. J., Brown, M., Chang, J. C., Liu, X. S., and Glimcher, L. H. (2014) XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature 508, 103-107
46. DeZwaan-McCabe, D., Riordan, J. D., Arensdorf, A. M., Icardi, M. S., Dupuy, A. J., and Rutkowski, D. T. (2013) The stress-regulated transcription factor CHOP promotes hepatic inflammatory gene expression, fibrosis, and oncogenesis. PLoS genetics 9, e1003937
47. Lee, A. S. (2007) GRP78 induction in cancer: therapeutic and prognostic implications. Cancer Res. 67, 3496-3499
48. Dong, D., Ni, M., Li, J., Xiong, S., Ye, W., Virrey, J. J., Mao, C., Ye, R., Wang, M., Pen, L., Dubeau, L., Groshen, S., Hofman, F. M., and Lee, A. S. (2008) Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 68, 498-505
49. Brewer, J. W., Hendershot, L. M., Sherr, C. J., and Diehl, J. A. (1999) Mammalian unfolded protein response inhibits cyclin D1 translation and cell-cycle progression. Proc. Natl. Acad. Sci. U. S. A. 96, 8505-8510
50. Brewer, J. W., and Diehl, J. A. (2000) PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc. Natl. Acad. Sci. U. S. A. 97, 12625-12630

27
51. Hamanaka, R. B., Bennett, B. S., Cullinan, S. B., and Diehl, J. A. (2005) PERK and GCN2 contribute to eIF2alpha phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway. Mol. Biol. Cell 16, 5493-5501
52. Cabrera, E., Hernandez-Perez, S., Koundrioukoff, S., Debatisse, M., Kim, D., Smolka, M. B., Freire, R., and Gillespie, D. A. (2017) PERK inhibits DNA replication during the Unfolded Protein Response via Claspin and Chk1. Oncogene 36, 678-686
53. Bourougaa, K., Naski, N., Boularan, C., Mlynarczyk, C., Candeias, M. M., Marullo, S., and Fahraeus, R. (2010) Endoplasmic reticulum stress induces G2 cell-cycle arrest via mRNA translation of the p53 isoform p53/47. Mol. Cell 38, 78-88
54. Malzer, E., Daly, M. L., Moloney, A., Sendall, T. J., Thomas, S. E., Ryder, E., Ryoo, H. D., Crowther, D. C., Lomas, D. A., and Marciniak, S. J. (2010) Impaired tissue growth is mediated by checkpoint kinase 1 (CHK1) in the integrated stress response. J. Cell Sci. 123, 2892-2900
55. Sequeira, S. J., Ranganathan, A. C., Adam, A. P., Iglesias, B. V., Farias, E. F., and Aguirre-Ghiso, J. A. (2007) Inhibition of proliferation by PERK regulates mammary acinar morphogenesis and tumor formation. PLoS One 2, e615
56. Frank, C. L., Ge, X., Xie, Z., Zhou, Y., and Tsai, L. H. (2010) Control of activating transcription factor 4 (ATF4) persistence by multisite phosphorylation impacts cell cycle progression and neurogenesis. J. Biol. Chem. 285, 33324-33337
57. Bagheri-Yarmand, R., Vadlamudi, R. K., and Kumar, R. (2003) Activating transcription factor 4 overexpression inhibits proliferation and differentiation of mammary epithelium resulting in impaired lactation and accelerated involution. J. Biol. Chem. 278, 17421-17429
58. Ron, D., and Habener, J. F. (1992) CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev. 6, 439-453
59. Aman, P., Ron, D., Mandahl, N., Fioretos, T., Heim, S., Arheden, K., Willen, H., Rydholm, A., and Mitelman, F. (1992) Rearrangement of the transcription factor gene CHOP in myxoid liposarcomas with t(12;16)(q13;p11). Genes Chromosomes Cancer 5, 278-285
60. Crozat, A., Aman, P., Mandahl, N., and Ron, D. (1993) Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 363, 640-644
61. Rabbitts, T. H., Forster, A., Larson, R., and Nathan, P. (1993) Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t(12;16) in malignant liposarcoma. Nat. Genet. 4, 175-180
62. Barone, M. V., Crozat, A., Tabaee, A., Philipson, L., and Ron, D. (1994) CHOP (GADD153) and its oncogenic variant, TLS-CHOP, have opposing effects on the induction of G1/S arrest. Genes Dev. 8, 453-464
63. Garen, A., Kauvar, L., and Lepesant, J. A. (1977) Roles of ecdysone in Drosophila development. Proc. Natl. Acad. Sci. U. S. A. 74, 5099-5103
64. Sato, T., Jigami, Y., Suzuki, T., and Uemura, H. (1999) A human gene, hSGT1, can substitute for GCR2, which encodes a general regulatory factor of glycolytic gene expression in Saccharomyces cerevisiae. Mol. Gen. Genet. 260, 535-540
65. Kim, J. H., Gurumurthy, C. B., Naramura, M., Zhang, Y., Dudley, A. T., Doglio, L., Band, H., and Band, V. (2009) Role of mammalian Ecdysoneless in cell cycle regulation. J. Biol. Chem. 284, 26402-26410
66. Dey, P., Rachagani, S., Chakraborty, S., Singh, P. K., Zhao, X., Gurumurthy, C. B., Anderson, J. M., Lele, S., Hollingsworth, M. A., Band, V., and Batra, S. K. (2012) Overexpression of ecdysoneless in pancreatic cancer and its role in oncogenesis by regulating glycolysis. Clin. Cancer Res. 18, 6188-6198

28
67. Zhao, X., Mirza, S., Alshareeda, A., Zhang, Y., Gurumurthy, C. B., Bele, A., Kim, J. H., Mohibi, S., Goswami, M., Lele, S. M., West, W., Qiu, F., Ellis, I. O., Rakha, E. A., Green, A. R., Band, H., and Band, V. (2012) Overexpression of a novel cell cycle regulator ecdysoneless in breast cancer: a marker of poor prognosis in HER2/neu-overexpressing breast cancer patients. Breast Cancer Res. Treat. 134, 171-180
68. Bele, A., Mirza, S., Zhang, Y., Ahmad Mir, R., Lin, S., Kim, J. H., Gurumurthy, C. B., West, W., Qiu, F., Band, H., and Band, V. (2015) The cell cycle regulator ecdysoneless cooperates with H-Ras to promote oncogenic transformation of human mammary epithelial cells. Cell cycle 14, 990-1000
69. Zhang, Y., Chen, J., Gurumurthy, C. B., Kim, J., Bhat, I., Gao, Q., Dimri, G., Lee, S. W., Band, H., and Band, V. (2006) The human orthologue of Drosophila ecdysoneless protein interacts with p53 and regulates its function. Cancer Res. 66, 7167-7175
70. Suh, H. W., Yun, S., Song, H., Jung, H., Park, Y. J., Kim, T. D., Yoon, S. R., and Choi, I. (2013) TXNIP interacts with hEcd to increase p53 stability and activity. Biochem. Biophys. Res. Commun. 438, 264-269
71. Jung, H., Kim, M. J., Kim, D. O., Kim, W. S., Yoon, S. J., Park, Y. J., Yoon, S. R., Kim, T. D., Suh, H. W., Yun, S., Min, J. K., Lee, H. G., Lee, Y. H., Na, H. J., Lee, D. C., Kim, H. C., and Choi, I. (2013) TXNIP maintains the hematopoietic cell pool by switching the function of p53 under oxidative stress. Cell metabolism 18, 75-85
72. Lee, S., Min Kim, S., Dotimas, J., Li, L., Feener, E. P., Baldus, S., Myers, R. B., Chutkow, W. A., Patwari, P., Yoshioka, J., and Lee, R. T. (2014) Thioredoxin-interacting protein regulates protein disulfide isomerases and endoplasmic reticulum stress. EMBO Mol. Med. 6, 732-743
73. Oslowski, C. M., Hara, T., O'Sullivan-Murphy, B., Kanekura, K., Lu, S., Hara, M., Ishigaki, S., Zhu, L. J., Hayashi, E., Hui, S. T., Greiner, D., Kaufman, R. J., Bortell, R., and Urano, F. (2012) Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell metabolism 16, 265-273
74. Horejsi, Z., Stach, L., Flower, T. G., Joshi, D., Flynn, H., Skehel, J. M., O'Reilly, N. J., Ogrodowicz, R. W., Smerdon, S. J., and Boulton, S. J. (2014) Phosphorylation-dependent PIH1D1 interactions define substrate specificity of the R2TP cochaperone complex. Cell reports 7, 19-26
75. Mir, R. A., Bele, A., Mirza, S., Srivastava, S., Olou, A. A., Ammons, S. A., Kim, J. H., Gurumurthy, C. B., Qiu, F., Band, H., and Band, V. (2015) A Novel Interaction of Ecdysoneless (ECD) Protein with R2TP Complex Component RUVBL1 Is Required for the Functional Role of ECD in Cell Cycle Progression. Mol. Cell. Biol. 36, 886-899
76. Horejsi, Z., Takai, H., Adelman, C. A., Collis, S. J., Flynn, H., Maslen, S., Skehel, J. M., de Lange, T., and Boulton, S. J. (2010) CK2 phospho-dependent binding of R2TP complex to TEL2 is essential for mTOR and SMG1 stability. Mol. Cell 39, 839-850
77. Cho, S. G., Bhoumik, A., Broday, L., Ivanov, V., Rosenstein, B., and Ronai, Z. (2001) TIP49b, a regulator of activating transcription factor 2 response to stress and DNA damage. Mol. Cell. Biol. 21, 8398-8413

29
CHAPTER TWO: Mammalian ECD protein functions as a novel negative regulator
of the Unfolded Protein Response

30
2.1 Overview
The mammalian Ecdysoneless (ECD) is a highly-conserved ortholog of the Drosophila
ECD gene product whose mutations impair the synthesis of Ecdysone and produce cell-
autonomous survival defects but mechanisms by which ECD functions are largely
unknown. This study presents evidence that ECD regulates endoplasmic reticulum (ER)
stress response. Stress induction by multiple stimuli, Thapsigargin, Tunicamycin, glucose
starvation, H2O2, NOX4 overexpression, DTT, and sorbitol, led to reduced ECD protein in
levels in multiple cell lines (MCF7, MDA-468, MCF10A, Panc-1, 76NTERT, MDA231-
NOX4). However, contrary to the decrease in ECD protein levels, ECD mRNA increased
upon Thapsigargin treatment suggesting that the decrease in ECD protein is not at
transcriptional level. To elucidate the ER stress signaling pathway regulating ECD protein
levels, we induced ER stress in MEF cells in which the three ER stress pathways, PERK,
IRE1α and ATF6, had been individually abrogated to produce single knock out (KO)
MEFs. In IRE1α KO and ATF6 KO MEFs, the decrease in ECD protein levels was
comparable with the control WT MEFs upon ER stress induction. However, in PERK KO
MREFs, the levels of ECD protein did not change upon ER stress induction as compared
to WT PERK. Furthermore, in PERK KO MEFs, ECD mRNA levels failed to increase,
suggesting that the decrease in ECD protein and the increase in ECD mRNA may be tied
to PERK activity. Since ECD is a phospho-protein and certain proteins undergo a
phosphorylation-dependent degradation, we first tested the hypothesis that the decrease in
ECD protein levels is PERK phosphorylation mediated. To assess that possibility, we
performed an in vitro kinase assay to examine the potential that ECD could be a substrate
for phosphorylation by PERK, a kinase. However, we did not find that ECD was
phosphorylated by PERK. The second hypothesis for the decrease in ECD protein levels

31
was that ECD protein translation was blocked by PERK phosphorylation of eIF2α. To test
that hypothesis, we induced ER stress in phospho-deficient eIF2α MEFs and found that the
decrease in ECD levels upon ER stress induction was abrogated just as in PERK KO
MEFS. The results suggested that ECD protein levels are reduced via the PERK-eIF2α axis
of the ER stress signaling and that ECD may be functionally linked to that PERK-eIF2α
axis. To examine that possibility, we resorted to cells in which ECD could be inducibly
depleted or overexpressed. Depletion of ECD increased the levels of p-PERK, p-eIF2α,
and these effects were enhanced upon ER stress induction by Thapsigargin treatment or
glucose deprivation along with an increase in CHOP mRNA, and cleaved caspase 3. A
slight increase in the levels of spliced XBP-1, the downstream target of IRE1α, was also
observed but cleaved ATF6 did not significantly change. These effects correlated with a
decrease in the colony formation of the ECD-null cells. Reciprocally, overexpression of
ECD led to a marked decrease in p-PERK and its downstream targets, p-eIF2α, ATF4 and
CHOP mRNA levels. Spliced XBP1 levels were slightly decreased while ATF6 did not
change significantly. These effects correlated with a decrease in caspase 3 cleavage and an
increase in the colony forming ability of the ECD overexpressing cells. Taken together,
these results suggested that ECD functions as a repressor of stress signaling, particularly
as a negative regulator of PERK signaling and that ECD and PERK are linked through a
negative feedback mechanism whereby ECD inhibits PERK while, when activated, PERK
inhibits ECD protein by blocking its translation.
Based on the findings presented above, we sought to investigate the possible mechanisms
by which ECD regulates ER stress signaling, particularly the PERK pathway. First, we
examined the localization of ECD relative to the upstream mediators of the ER stress

32
signaling and found that ECD localized and associated with PERK, ATF6, IRE1α and
GRP78. To assess whether ECD directly regulates PERK signaling, we performed in vitro
kinase assay of PERK with eIF2α in the presence of an increasing concentration of the
ECD protein. However, ECD did not modulate enzymatic activity of PERK toward eIF2α.
Since induction of chaperones during ER stress signaling is well known as a homeostatic
mechanism aimed at restoring the ER function by clearing the stress stimulus, we next
examined the effect of ECD on levels of chaperones, particularly GRP78 which is the major
regulator of PERK signaling. We found that the levels of GRP78 (GRP94 and PDI to a
lesser extent) were dependent on ECD and that depletion or overexpression of ECD
decreased or increased GRP78 protein levels, respectively, upon ER stress induction.
However, GRP78 mRNA was not affected, suggesting a post-translational effect. To
determine whether a change in the levels of ECD affects GRP78 protein stability, we
deleted ECD followed by cycloheximide treatment and found that the decrease in GRP78
protein level was faster in ECD-null cells. Reciprocally, in ECD overexpressing cells, the
decrease in the levels of GRP78 protein was slower upon cycloheximide treatment. Finally,
to determine whether GRP78 was required for ECD effects on PERK signaling, we
knocked down GRP78 and found that even in the presence of overexpressed ECD, a knock
down of GRP78 abrogated the attenuating effect of ECD on PERK, correlating with a
decrease in colony formation of the ECD overexpressing cells. Taken together, these
results argue that ECD enhances the levels of chaperones, predominantly, GRP78 to
enhance the folding capacity of the stressed ER and reduce stress.

33
2.2 INTRODUCTION
The endoplasmic reticulum (ER) is a central subcellular organelle with essential roles in
the synthesis, folding and maturation of secreted and membrane proteins, biogenesis of
cholesterol, calcium homeostasis and regulation of survival and apoptosis pathways (1-10).
Aberrations in these ER functions are sensed by well-conserved ER transmembrane
sensors, Inositol-Requiring Enzyme 1 alpha (IRE1α), PKR-like ER kinase (PERK) and
Activating Transcription Factor 6 (ATF6), that activate homeostatic signaling pathways
collectively referred to as the unfolded protein response (UPR) (11,12). These ER stress
sensors exhibit a dynamic and reversible interaction with the ER chaperone GRP78 (13).
In un-stressed cells, GRP78 is bound to luminal domains of UPR sensors, which maintains
them in an inactive state (14). During ER stress, the increased load of unfolded proteins
competes for GRP78 binding, leading to activation of UPR sensors (15).
As an important chaperone, the level of GRP78 is also increased during ER stress
signaling, mainly via the ATF6 pathway (16). The induction of GRP78 and other
chaperones such as GRP94 (17-19) and PDI (20,21) is a homeostatic mechanism to
restore normal functions in the ER and protect cells from stress-induced cell death. Part
of GRP78’s cyto-protective role involves its regulation of the ER stress sensors during
stress signaling to prevent their hyper-activation and ensuing cell death. To achieve that
effect, GRP78 is known to reversibly bind the ER stress sensors, when induced, and
inhibit their signaling (13). Consequently, overexpression of GRP78 has been shown to
inhibit PERK signaling, in particular (13). Furthermore, GRP78 has been shown to
physically interact with caspases (22) and prevent their cleavage (23).

34
The GRP78 protein is known to contain the canonical KDEL ER retention signal
(24,25) which allows it to be retro-transported from the Golgi to the ER during its
journey through the secretory pathway. Accordingly, GRP78 was traditionally thought of
as an ER-resident protein. However, recent evidence points to a ubiquitous nature of the
protein in cells. GRP78 has been found in the cytoplasm where it interacts with
cytoplasmic proteins such as caspace-7 and clusterin protein (22,26); there is also
evidence of GRP78 presence in the nucleus and mitochondria (27). This ubiquitous
nature of GRP78 in cells suggests that the ER functions of GRP78 may not necessarily
require its ER- localization, especially as studies have identified a cytoplasmic, non-
KDEL containing variant of the GRP78 protein which still has ER stress-related
functions via cytoplasmic regulators of the ER stress signaling (28).
The PERK pathway has emerged as a key pathway in cellular UPR response as well
as homeostasis under unstressed conditions and in disease states (29-34). Release of
GRP78 from PERK leads to its dimerization, auto-phosphorylation and activation
(15,35,36). A major PERK substrate is the eukaryotic translation initiation factor 2 alpha
(eIF2α) whose phosphorylation by PERK inactivates it leading to a block in general cap-
dependent protein translation and consequent decrease in the protein load entering the ER
(37); concurrently, PERK signaling selectively enhances the cap-independent translation
of specific mRNAs, such as Activating Transcription Factor 4 (ATF4) (37). ATF4 induces
the expression of CCAAT/enhancer-binding protein-homologous protein (CHOP) which
promotes apoptosis in response to stress (37-45). PERK-induced phosphorylation of eIF2α
also inhibits cell cycle progression by reducing the levels of cyclins and hence cyclin-
dependent kinase 2 (CDK2) activity (46-48). Termination of PERK signaling, together

35
with de-phosphorylation of eIF2α, is required to re-initiate protein synthesis and resume
the cell cycle. Thus, PERK-mediated UPR leads to a coordinated program of cellular
protection and mitigation of stress. However, PERK also contributes to an alternate
outcome through CHOP-mediated activation of a cellular death pathway to eliminate
severely damaged cells (37-45). Mechanisms to fine-tune the outcomes of UPR pathways
are important to mitigate the negative consequences of ER stress. This is of particular
importance under conditions where cells experience ER stress as part of their physiological
responses, such as antibody-secreting plasma cells or insulin-secreting pancreatic islet cells
(49). We have identified ECD as a negative regulator of the PERK-mediated UPR.
The ECD gene was first identified based on genetic mutations in Drosophila that lead to
reduced production of the developmentally-regulated steroid hormone Ecdysone, which is
synthesized in the ER, hence the designation of Ecdysoneless for such fly mutants (50).
The mammalian ECD gene was cloned based on the rescue of growth defects in S.
cerevisiae mutant in the growth control regulatory gene 2 (GCR2), a glycolysis regulatory
gene (51). Thus, ECD was thought to be involved in mammalian glycolysis gene
expression and initially named hSGT1 (human suppressor of gcr2)(51). My lab identified
the same gene in a screen for interacting partners of human papilloma virus E6 oncoprotein
and found it to interact with p53 and transactivate p53-regulated genes (52,53).
To elucidate the functional role(s) of mammalian ECD, my lab generated ECD-null mice
and demonstrated that homozygous deletion of ECD was early embryonic lethal while ex
vivo Cre-mediated deletion of ECD in ECDfl/fl mouse embryonic fibroblasts (MEFs) led to
proliferative block and a significant decrease in cell survival (54,55). ECD was found to
be essential for E2F target gene expression by facilitating the dissociation of the

36
retinoblastoma RB protein from E2F and promoting the G1-to-S phase cell cycle
progression (54). As a consequence, ECD-null MEFs showed a decrease in the levels of
cyclins A, B1, E and D1, reduction in CDK2 kinase activity and were arrested in G1 phase
of the cell cycle (54). Interestingly, E2F family proteins such as the E2F1 have been
implicated in UPR-mediated cell death (56). In addition to its promotion of the G1/S phase,
ECD also promotes G2/M phase of the cell cycle and its knockdown induced not just a
G2/M arrest but also apoptosis (57). Induction of UPR not only induces apoptosis but also
halts cell cycle progression in G1 and G2 phases (46-48,58) and these effects are mediated
by PERK (46,47), suggesting a potential connection between ECD and the PERK arm of
the UPR.
More recently, we uncovered another mechanism by which ECD regulates cellular
proliferation, through its interaction with RUVBL1 and PIH1D1 components of the
prefoldin co-chaperone R2TP (55) which is involved in the assembly or remodeling of a
number of protein and protein-RNA complexes to regulate many physiological processes
(59-65). Recently, it was reported that knock down of the RUVBL1 component of the
R2TP complex induced cell cycle block and ER stress (66). Furthermore, ECD has been
shown to associate with the stress response protein Thioredoxin-Interacting Protein
(TXNIP) (67) which is known to bind to the ER chaperone PDI (protein disulfide bond
isomerase) to increase its enzymatic activity to relieve ER stress (20). Lastly, TXNIP was
recently shown to be a novel component of the UPR and is regulated by the PERK pathway
(68). Based on these multiple lines of suggestive evidence pointing to a potential role of
ECD in the regulation of ER stress, we investigated ECD and demonstrated that it
modulates ER stress signaling, particularly the PERK arm. Induction of ER stress, using

37
both chemical ER stress inducers (Thapsigargin and Tunicamycin) and physiological ER
stress inducers (glucose starvation), led to a reduced level of the ECD protein, and this
effect was not seen in PERK KO or phospho-deficient eIF2α MEFs. Notably, ECD mRNA
levels were increased, suggesting impaired ECD translation as a mechanism for reduced
protein levels. ECD depletion increased the levels of p-PERK and its downstream targets,
p-eIF2α and CHOP mRNA while ECD overexpression markedly decreased their levels
upon ER stress induction. Spliced XBP-1, the downstream target of IRE1α and cleaved
AFT6 were slightly affected. Significantly, ECD overexpression and depletion distinctly
affected the survival outcome of cells upon ER stress. Additionally, we found that the
levels of GRP78 (GRP94 and PDI to a lesser extent) were dependent on ECD and that
depletion or overexpression of ECD decreased or increased GRP78 protein levels,
respectively, upon ER stress induction. However, GRP78 mRNA was not affected,
suggesting a post-translational effect. Deletion of ECD followed by cycloheximide
treatment showed that the decrease in GRP78 protein level was faster in ECD-null cells.
Reciprocally, in ECD overexpressing cells, the decrease in the levels of GRP78 protein
was slower upon cycloheximide treatment. Finally, to determine whether GRP78 was
required for ECD effects on PERK signaling, we knocked down GRP78 and found that
even in the presence of overexpressed ECD, a knock down of GRP78 abrogated the
attenuating effect of ECD on PERK, correlating with a decrease in the colony formation of
the ECD overexpressing cells. Taken together, these results argue that ECD enhances the
levels of chaperones, predominantly, GRP78 to enhance the folding capacity of the stressed
ER and reduce stress.

38
2.3 MATERIALS AND METHODS
Reagents and antibodies
Thapsigargin, (Cat# 12758) and Tunicamycin (Cat# 12819) and Brefeldin A (cat# 9972)
were from Cell Signaling and dissolved in DMSO. Dithiothreitol (cat# 9776), Sorbitol
(cat# 1876) and Hydrogen Peroxide (cat# H1009) were from Sigma. A monoclonal
antibody against ECD, generated in our laboratory, has been described previously (69).
Antibodies against p-PERK (cat# 3179), ATF4 (cat# 11815), p-eIF2α (cat# 9721), eIF2α
(cat# 9722), Caspase 3 (cat# 9662), s-XBP1 (cat# 12782), IRE1α (cat# 3294), GRP78 (cat#
3177), p-SAPK/JNK (cat# 9251) and total PERK (cat# 3192) were from Cell signaling.
ATF6 antibody was purchased from Enzo Life (cat# ALX-804-381-C100).
Cell lines and culture conditions
ECD inducible MEFs were generated from the lab. 13.5 days old embryos of mice of the
genotype Tet(O)-Flag-hECD-IRES-eGFP were used. rtTA was introduced retrovirally to
generate Tet(O)-Flag-hECD-IRES-eGFP; rtTA. ECD fl/fl MEFs were maintained in
DMEM medium supplemented with 10% fetal bovine serum and treated with adenoviruses
coding for GFP (adeno-GFP, control) or Cre (adeno-Cre) as described previously (54,55).
The culture condition for the ECD inducible MEFs was same as the ECD fl/fl MEFs. Panc-

39
1 cells have been previously described (70). For ECD overexpression studies, control
MEFs or ECD over-expressing MEFs were cultured with 1μg/ml of Doxycycline (Cat #
231286, BD Biosciences) to induce expression of the ECD transgene. MEFs with a non-
phosphorylatable eIF2α were obtained from Dr. Thomas Rutkowski’s lab, Carver College
of Medicine, Iowa and has been described elsewhere (71). PERK knockout (KO) MEFs
were from ATCC (CRL-2976) while IRE1α KO and ATF6 KO were gifts from Dr. Urano’s
lab, Washington University St. Louis and Dr. Hendershot’s lab, St. Jude Children Research
Hospital, Tennessee, respectively. Immortal mammary epithelial cell lines MCF-10A,
76NTERT were from the lab and was cultured in DFCI-1 medium (55). Cancer cells line
MCF7 and MDA-468 were cultured in alpha media. NOX4 expressing MDA-231 were
obtained from Dr. Teoh-Fitzgerald’s lab (UNMC, Biochemistry).
Western blotting
For western blotting, cell lysates were prepared in RIPA buffer (Cat# 89901, Thermo
Scientific) supplemented with protease inhibitors (Roche, Cat# 11836145001). Lysates
were resolved on SDS-PAGE gel, transferred on to nylon membrane (IPVH00010,
Millipore), and subjected to ECL-based Western blotting as described (54,55).
Colony Formation Assay- For clonogenic assay, 1,000 cells were plated in triplicate and
allowed to attach to the plate (about 8 h) followed by treatment with Thapsigargin for 24h.
10 days later, colony formation was assessed as previously described (54,55).

40
In vitro kinase assay-A total of 500 ng of purified recombinant ECD proteins or its
mutants was incubated with 0.2 mM ATP, 1 µCi of [32P]ATP (Perkin-Elmer), and 0.2 µl
(10 U) of human recombinant PERK or CK2 (NEB, Beverly, MA) at 30°C for 30 min.
The reaction was stopped by adding SDS-PAGE sample buffer. The 32P-labeled proteins
were detected by autoradiography after SDS-PAGE and then transfer to polyvinylidene
difluoride (PVDF) membranes (Millipore).
Induction of ER stress by glucose starvation- Panc-1 cells were maintained in DMEM
supplemented with 10% fetal bovine serum and then switched to glucose-free DMEM
medium (cat# 11966-025, life technologies) for indicated times.
Statistical analysis- for assessment of statistical significance, a Student t test was used. P
values of < 0.05 were considered statistically significant
RNA isolation and real-time quantitative PCR (qRT-PCR)
Total RNA was isolated using the TRizol reagent (Invitrogen cat# 15596018). 1 µg of
RNA was reverse- transcribed using SuperScript TMII reverse transcriptase (Invitrogen
cat# 18064014). The qPCR was performed with primer sets indicated in the table
belowbelow:

41
Gene Forward primers Reverse primer
Mouse
CHOP
CTGCCTTTCACCTTGGAGAC CGTTTCCTGGGGATGAGATA
Huma
n
CHOP
CATTGCCTTTCTCCTTCGGG CCAGAGAAGCAGGGTCAAGA
Mouse
ECD
CCGGTCTGGCACAAACTTCTGC
TG
AGGGTCGAAGCATCCCTCCATC
GA
Huma
n
ECD
ACTTTGAAACACACGAACCTGG
CG
TGATGCAGGTGTGTGCTAGTTC
CT
Mouse
GRP7
8
AGTGGTGGCCACTAATGGAG CAATCCTTGCTTGATGCTGA

42
2.4 Results
2.4.1 Effect of ER stress induction by Thapsigargin, Tunicamycin, Brefeldin A on
ECD protein levels in MCF-7, MDA-468 and MCF-10A
To begin to explore the potential link of ECD to stress pathways, we assessed the effects
of various chemical ER stress inducers on ECD protein levels. For this purpose, cancer
cell lines MCF7 and MDA-468 and normal mammary epithelial cell line MCF-10A were
treated with Thapsigargin, Tunicamycin and BFA, and ECD protein levels were analyzed
by western blot. Treatment with either chemical increased the levels of eIF2α
phosphorylation, a marker of ER stress (Figure 2.1A-C). ECD protein levels did not
change much in MCF7 and MDA-468 cells with either chemical but the levels decrease
more dramatically in MCF-10A (Figure 2.1 A-C)

43
UT Tg Tun BFA
ECD
p-eIF2α
β-actin
MCF7 A
UT Tg Tun BFA
MDA-468
ECD
p-eIF2α
β-actin
B
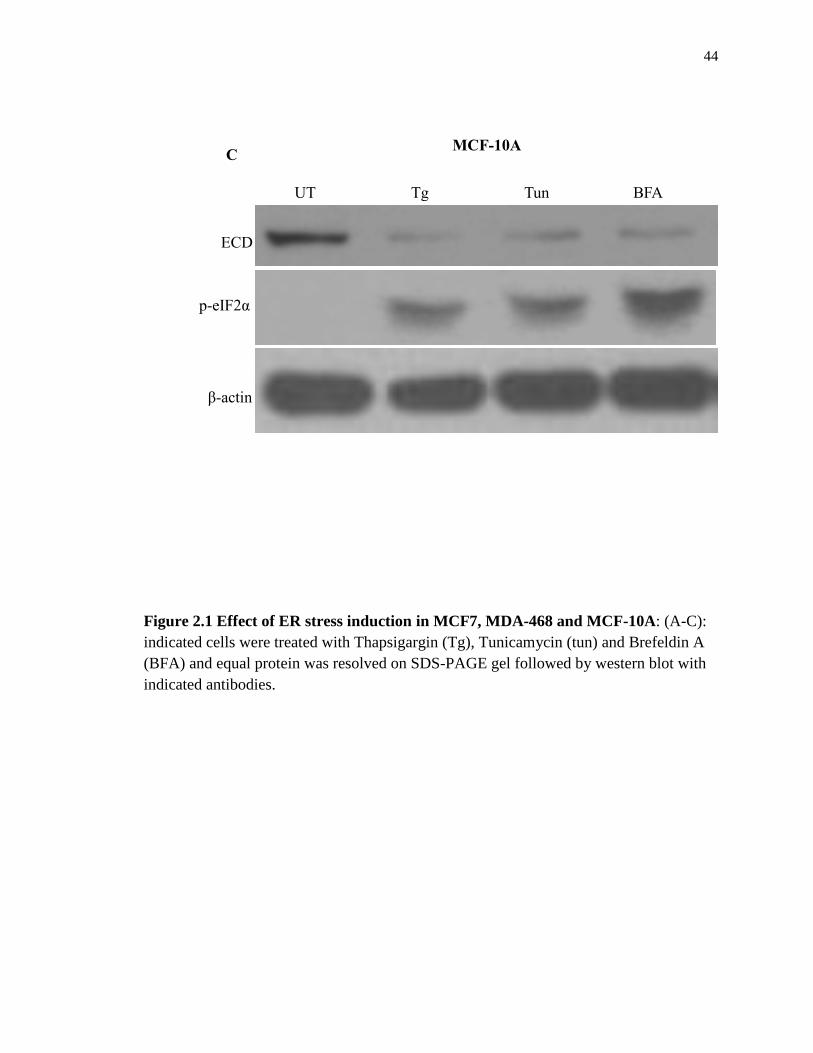
44
Figure 2.1 Effect of ER stress induction in MCF7, MDA-468 and MCF-10A: (A-C):
indicated cells were treated with Thapsigargin (Tg), Tunicamycin (tun) and Brefeldin A
(BFA) and equal protein was resolved on SDS-PAGE gel followed by western blot with
indicated antibodies.
UT Tg Tun BFA
MCF-10A
ECD
p-eIF2α
β-actin
C

45
2.4.2 Physiological stress by glucose deprivation led to a decrease in ECD protein
levels
Given the effect of chemical ER stress inducers on ECD protein levels presented above,
we next assessed whether physiological stress such as glucose starvation would have
similar effects. For this purpose, we used a human pancreatic carcinoma cell line (Panc-
1) and mouse beta cells (MIN-6) cells both of which are known to exhibit ER stress upon
glucose starvation (21). These cells were switched from their normal complete DMEM
medium to glucose-free medium for various time points and ECD protein levels were
assessed by western blot. This glucose-free culture condition increased the levels of the
glucose-regulated protein 78 (GRP78), PERK phosphorylation (p-PERK) and p-eIF2α as
expected (21). Significantly, similar to chemical ER stress, glucose starvation-induced
ER stress also exhibited reduced levels of the ECD protein (Figure 2.2 A-B)

46
Time, h: 0 12 24 48 72
Glucose starvation
ECD
β-actin
p-eIF2α
Panc-1
GRP78
A

47
Figure 2.2: Physiological stress by glucose starvation reduces ECD protein levels:
(A-B) Human pancreatic carcinoma and mouse beta cell lines were cultured in glucose-
free medium for indicated time points. Equal protein was resolved on SDS-PAGE gel
followed by western blot with indicated antibodies.
Time, h: 0 6 1 2 24 48
Glucose starvation
ECD
β-actin
p-eIF2α
p-PERK
MIN6 cells B

48
2.4.3 Effects of other forms of stress on ECD protein levels
The effect of both chemically-induced and physiologically-induced stress on the levels
of ECD protein prompted us to further examine the effects of other stresses (oxidative,
osmotic, and reducing) on ECD protein levels. For that purpose, we first treated
MCF10A, 76NTERT and MCF7 cells with H2O2, a commonly used oxidative stress
inducer (72). Significantly, treatment with H2O2, also led to a decrease in ECD protein
levels (Figure 2.3 A). To further assess these effects, we used another type of cells
(MDA-231) which are under a physiologically-induced oxidative stress. These cells
express the NOX4 system, which generates ROS and oxidative stress (73,74) as
diagrammed in Figure 2.3 B. Again, here too, even in the absence of any exogenously-
derived stress, ECD protein levels are lower in the NOX4-expressing cells as compared
to the control cells. Reductive and osmotic stresses, by DTT and sorbitol treatment,
respectively, also led to a decrease in the ECD protein level (Figure 2.3 C-D).

49
ECD
H2O
2 (μM) UT 50 100 300
MCF10A
β-actin
H2O
2 (μM) - + - +
ECD
β-actin
76NTert MCF7
A

50
NADPH NADP+
+ e-
+ H +
O2
O2
- Oxidative stress
ER Stress
The NOX4 system and Oxidative stress:
H2O
2
B
MDA231
ECD
β-actin

51
Figure 2.3: Effects of other forms of stress (oxidative, reductive and osmotic) on
ERCD protein level: (A) MCF-10A, 76NTERT and MCF7 cell lines were treated with
indicated doses of H2O2, (B) MDA-231 expressing NOX4 and their control cells were
lysed, (C) MCF10A and MCF7 cell lines were treated with DTT, (D) T98G cells were
treated with sorbitol. Equal protein was resolved on SDS-PAGE gel followed by western
blot with indicated antibodies.
DTT - + - +
ECD
β-actin
MCF10A MCF7 C
Sorbitol: - +
β-actin
ECD
p-p38
T98G D

52
2.4.4 ER stress induction by Thapsigargin and glucose deprivation led to increase in
ECD mRNA levels
Given that all forms of stress led to a decrease in ECD protein levels, we assessed
whether this decrease in ECD protein levels was due to reduced ECD mRNA levels. We
treated MCF10A cells with Thapsigargin and measured ECD mRNA levels using qRT-
PCR. Induction of CHOP mRNA was used as a control (Figure 2.4 A). Notably, ECD
mRNA levels were not only not reduced but in fact showed an increase (Figure 2.4 B),
suggesting that the reduction in the ECD protein level was not at the transcriptional level.
Likewise, physiological stress by glucose starvation inPanc-1 cells also increased ECD
mRNA levels (Figure 2.4 D) with GRP78 mRNA induction used as control (Figure 2.4
C).

53
0
10
20
30
40
50
CHOP mRNA
Tg: 0h 8h 12h 24h
Rel
ativ
e m
RN
A
A
0
1
2
3
4
5
6
7
8
9
Tg: 0h 8h 12h 24h
Rel
ativ
e m
RN
A
*
*
ECD mRNA B

54
0
10
20
30
40
50
60
GRP78 nRNAR
elat
ive
mR
NA
C
Glucose starvation: 0h 12h 24h 48h 72h
Rel
ativ
e m
RN
A
0
0.5
1
1.5
2
2.5
3
3.5
4
ECD mRNA
Glucose starvation: 0h 12h 24h 48h 72h
D

55
Figure 2.4: ER stress induction by Thapsigargin and glucose starvation led to an
increase in ECD mRNA. MCF-10A cells were treated with Thapsigargin (A-B) and
Panc-1 cells were cultured in glucose-free medium (C-D), then total RNA was isolated
followed by qRT-PCR using primers against CHOP, GRP78, and ECD(Mean +/- SD and
p-values are shown from 3 independent experiments * p<0.05). CHOP and GRP78
mRNA induction served as control for Thapsigargin-induced and glucose starvation-
induced ER stress, respectively.

56
2.4.5 The decrease in the levels of ECD protein upon ER stress induction is PERK-
eIF2α dependent
Given the results above that various stresses led to a decrease in ECD protein level but
that ECD mRNA increased, we sought to determine the mechanism by which ECD
protein and mRNA levels inversely correlate. All stresses mentioned above are
interconnected. For instance, Oxido-reductive stress induces ER stress and vice versa, as
summarized in the diagram in Figure 2.3B. ER stress activates three canonical signaling
pathways summarized in Figure 1.1. To determine which pathway controls ECD protein
and mRNA levels, we induced ER stress in cells in which the upstream mediators of the
pathways (PERK, IREα and ATF6) have been genetically abrogated. For that purpose,
we used ATF6 KO, IRE1α KO and PERK KO MEFs in which the pathways have been
individually knocked out (KO). The absence of the protein was confirmed by western
blot with antibody against the protein that was knocked out (Figure 2.5A-C). In ATF6
KO and IRE1 KO MEFs, the decrease in ECD protein levels was comparable to that in
the control wild-type (Figure 2.5A-B). However, in PERK KO MEFs, the decrease in
ECD protein levels was abrogated (Figure 2.5C), suggesting that the decrease in ECD
protein levels is PERK dependent.

57
Tg: - + - +
WT KO
ATF6
ECD
β-actin
A
IRE1α
ECD
IRE1
β-actin
WT KO
Tg - + - +
B
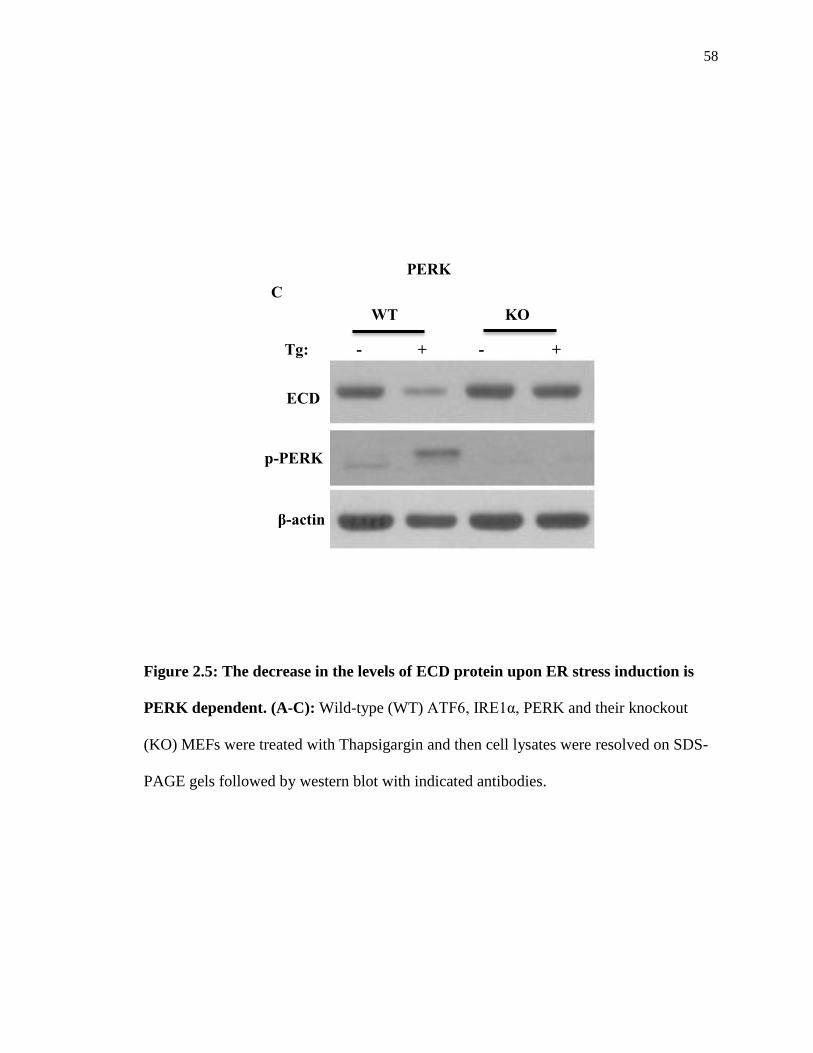
58
Figure 2.5: The decrease in the levels of ECD protein upon ER stress induction is
PERK dependent. (A-C): Wild-type (WT) ATF6, IRE1α, PERK and their knockout
(KO) MEFs were treated with Thapsigargin and then cell lysates were resolved on SDS-
PAGE gels followed by western blot with indicated antibodies.
Tg: - + - +
WT KO
ECD
β-actin
p-PERK
PERK
C

59
As we previously observed that ECD mRNA increased upon ER stress induction but the
protein levels decreased, here we sought to assess whether ECD mRNA changes in
PERK WT vs. KO MEFs. Again, we treated PERK WT and KO MEFs with
Thapsigargin and measured the change in ECD mRNA by qPCR. The identity of the
PERK KO MEFs in this experiment was determined by analyzing the product of the PCR
product obtained with primers against the PERK KO domain. As shown in Figure 2.6A,
there was no PCR product in the PERK KO samples, confirming the identity of the cells.
To further confirm that, we used primers against CHOP mRNA in the qPCR. As
expected, while CHOP mRNA was induced in the PERK WT cells, the induction was
very minimal in the PERK KO cells (Figure 2.6B), since CHOP is downstream of PERK
(44). Significantly, while ECD mRNA increased in PERK WT MEFs, it did not increase
much in PERK KO MEFs (Figure 2.6C). This result that ECD mRNA failed to increase
when the protein levels didn’t change, suggests that ECD mRNA only increases when the
protein levels are down, suggesting a feedback mechanism.

60
β-actin
KO domain
A
0
5
10
15
20
25
30
35
40CHOP mRNA
PERK WT
PERK KO
UT 7h 14h
Rel
ativ
e m
RN
A
B
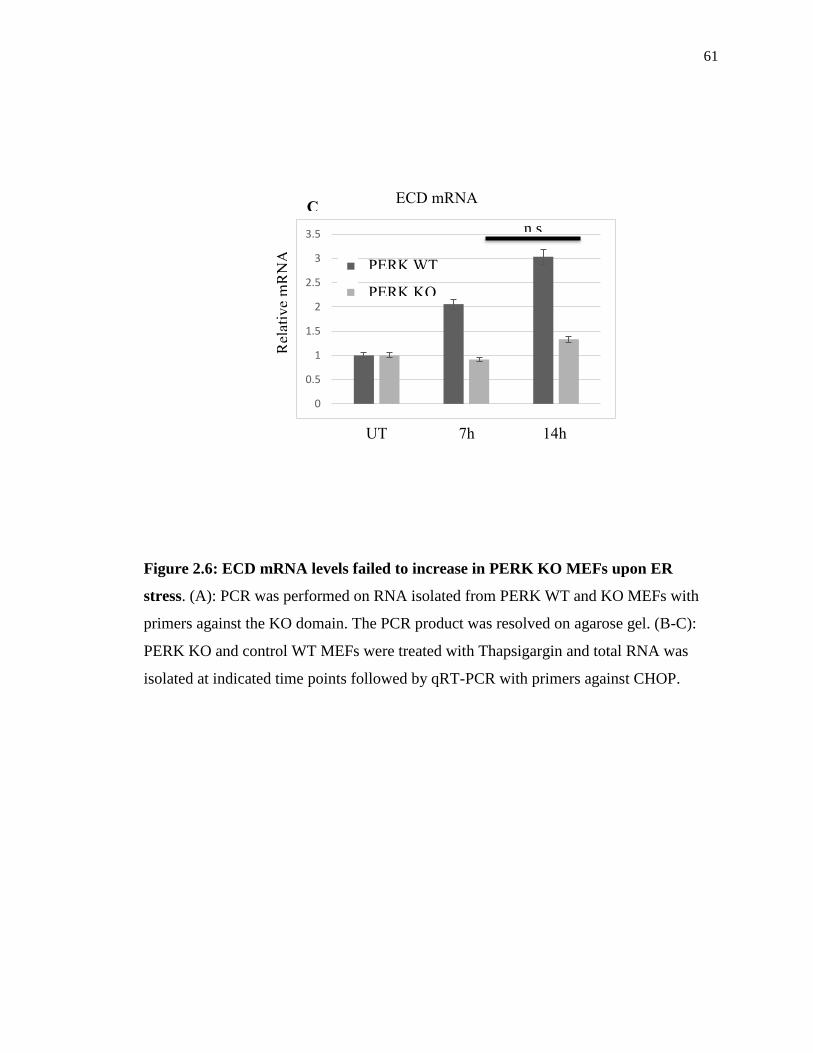
61
Figure 2.6: ECD mRNA levels failed to increase in PERK KO MEFs upon ER
stress. (A): PCR was performed on RNA isolated from PERK WT and KO MEFs with
primers against the KO domain. The PCR product was resolved on agarose gel. (B-C):
PERK KO and control WT MEFs were treated with Thapsigargin and total RNA was
isolated at indicated time points followed by qRT-PCR with primers against CHOP.
0
0.5
1
1.5
2
2.5
3
3.5
PERK WT
PERK KO R
elat
ive
mR
NA
UT 7h 14h
ECD mRNA
n.s
C

62
To explain the PERK-dependent decrease in ECD protein levels, we proposed two
hypotheses:
1) PERK phosphorylates ECD leading to ECD degradation since (i) PERK is a
kinase, (ii) ECD is a phospho-protein as a substrate for CK2 phosphorylation and
(iii) certain proteins are known to undergo a phosphorylation-dependent
degradation (75).
2) PERK phosphorylates eIF2α to inhibit ECD protein translation since PERK is
known to inhibit translation during ER stress signaling by phosphorylating eIF2α
(37).
To investigate the first hypothesis that ECD undergoes a PERK phosphorylation-
dependent degradation, we first assessed whether ECD could be a substrate for PERK
phosphorylation by performing an in vitro kinase assay of ECD by PERK. EIF2α was
used as the positive control for PERK phosphorylation since eIF2α is a substrate for
PERK (37); and CK2 was used as a positive control for phosphorylation of ECD since
CK2 is known to phosphorylate ECD (55). As expected, eIF2α and ECD were
phosphorylated in vitro by PERK and CK2, respectively (Figure 2.7, lanes 1-2 and 3,
respectively). However, a phosphorylation of ECD by PERK was not observed (Figure
2.7, lane 4-5).

63
Figure 2.7: PERK does not phosphorylate ECD: In vitro kinase assay of recombinant
human ECD prepared from two different sources (bacterial and insect cell derived) was
performed using human recombinant GST fused PERK. Proteins were separated by SDS-
PAGE, transferred to PVDF membrane, and phosphate labeling was detected by
autoradiography. Phosphorylation of eIF2α by PERK was used as the positive control for
PERK mediated phosphorylation while phosphorylation of ECD by CK2 was used as the
positive control for ECD phosphorylation.
p-ECD
p-eIF2α
p-PERK
+ + + PERK
+ - - eIF2α
- + - Bacterial ECD
- - + Baculoviral ECD
WT ECD: + -
3S/A ECD: - +
CK2: + +
Lane: 1 2 3 4 5
+ + + + + 32P

64
To investigate the second possibility that ECD translation is blocked by PERK
phosphorylation of eIF2α, we resorted to eIF2α MEFs. Since PERK activation and
subsequent phosphorylation of eIF2α mediates translational block in response to ER
stress (37), we utilized MEFs from mice that carry a serine 51 to alanine mutation in
eIF2α which prevents its phosphorylation by activated PERK and makes the cells
resistant to PERK-mediated translation block upon ER stress induction (71). As expected,
the levels of p-eIF2α increased in WT MEFs but not in the phospho-deficient eIF2α
mutant MEFs when ER stress was induced with Thapsigargin or Tunicamycin (Figure
2.8, compare lanes 1-3 with lanes 4-6). Importantly, while ECD protein levels decreased
in WT MEFs treated with ER stress inducers (Fig. 1J, compare lane 1 to lanes 2-3), the
levels of ECD protein did not change in identically-treated mutant eIF2α MEFs (Figure
2.8, compare lane 4 to lanes 5-6). Together, these results support a conclusion that ECD
protein levels are downregulated upon ER stress in a PERK-eIF2α dependent manner,
and that ER stress upregulates ECD mRNA levels, suggesting a regulatory link between
ECD and the PERK pathway of ER stress.

65
Figure 2.8: The decrease in ECD protein levels is abrogated in eIF2α phospho-
deficient MEFs. WT eIF2α MEFs or mutant eIF2α phospho-deficient MEFs were treated
with Thapsigargin (Tg, 50 nM) or Tunicamycin (Tun, 50 ng/ml) for 14 h and then cell
lysates were analyzed by western blotting with indicated antibodies.
ECD
p-eIF2α
β-actin
- Tg Tun - Tg Tun
Wild-type Phospho-deficient
eIF2α
Lane: 1 2 3 4 5 6

66
2.4.6 ECD depletion or over-expression distinctly regulate ER stress signaling upon
ER stress induction
The abrogation of the decrease and increase in ECD protein and mRNA levels,
respectively, upon disruption of PERK pathway (Figure 2.5-2.8) suggested that ECD may
be functionally linked to UPR through PERK pathway. Therefore, we assessed the
activation of the PERK pathway in the absence or presence of UPR inducers in cells in
which ECD could be inducibly deleted. For this purpose, we utilized ECDfl/fl MEFs which
have been previously established and shown to exhibit ECD gene deletion and loss of ECD
protein expression upon infection with adenovirus expressing Cre recombinase (54).
Previously, it was observed that induction of ECD deletion in these ECDfl/fl MEFs, aside
from inducing cell cycle arrest, led to a decrease in cell survival even in the absence of any
stress (54,55).
Notably, Cre-meditated deletion of ECD in the ECDfl/fl MEFs led to an increase in the
levels of phospho-PERK (p-PERK) and p-eIF2α, when compared to control adeno-GFP
infected MEFs (Figure 2.9 compare lane 1 with 3), and these effects were further enhanced
by treatment with Thapsigargin (Tg), the inhibitor of the Sarco/Endoplasmic Reticulum
Calcium ATPase (Figure 2.9, compare lane 2 with 4). Spliced XBP1, the downstream target
of IRE1α signaling also showed some increase in ECD null cells upon Thapsigargin
treatment while ATF6 did not show a significant change (Figure 2.9).
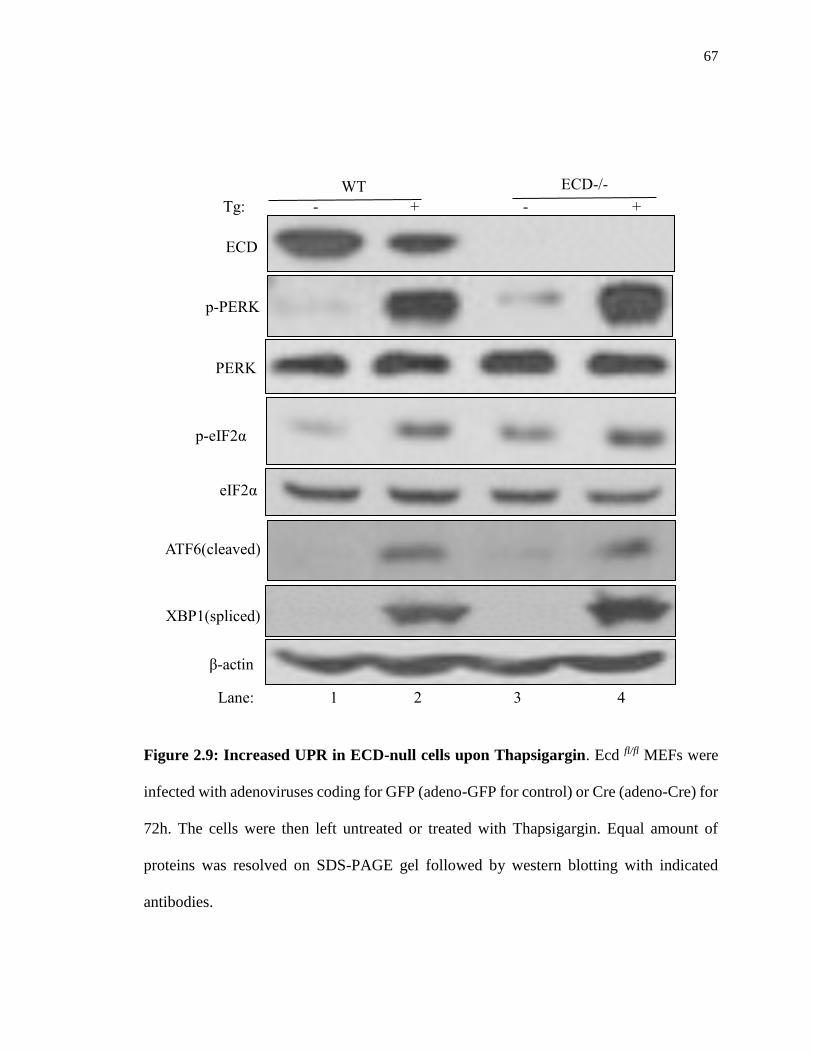
67
Figure 2.9: Increased UPR in ECD-null cells upon Thapsigargin. Ecd fl/fl MEFs were
infected with adenoviruses coding for GFP (adeno-GFP for control) or Cre (adeno-Cre) for
72h. The cells were then left untreated or treated with Thapsigargin. Equal amount of
proteins was resolved on SDS-PAGE gel followed by western blotting with indicated
antibodies.
Tg: - + - +
Lane: 1 2 3 4
ECD
p-PERK
PERK
p-eIF2α
eIF2α
ATF6(cleaved)
XBP1(spliced)
β-actin
WT ECD-/-

68
Consistent with the increased p-PERK levels upon ECD deletion, Thapsigargin treated
ECD-depleted MEFs also exhibited increased expression of CHOP mRNA (Figure 2.10A),
a downstream effector of the PERK pathway whose target genes promote cell death (37-
41,43,44). The increased CHOP mRNA correlated with an increased cleaved caspases 3 in
ECD null cells (Figure 2.10B, compare lane 2 with 4) and a significant reduction in cell
survival, as measured by colony-forming assays (Figure 2.10C).

69
0
5
10
15
20
25
30
35
40
0h 7h 12h
WT ECD-/-
Rel
ativ
e m
RN
A
Tg
CHOP mRNA
*
*
A
Tg: - + - +
ECD
Caspase 3
Cleaved-Caspase 3
β-actin
WT ECD-/-
Lane: 1 2 3 4
B

70
Figure 2.10: ECD-null cells exhibited increased ER stress and cell death upon
Thapsigargin treatment. (A): After adenoviruses infection as described in Figure 2.9,
cells were treated with Thapsigargin. Total RNA was isolated followed by qRT-PCR with
Tg: - +
WT
ECD-/-
0
0.2
0.4
0.6
0.8
1
1.2
WT ECD-/-
UT Tg
Rel
ativ
e su
rviv
al
*
**
C

71
CHOP primers; Mean +/- SD and p-values from 3 independent experiments are indicated
* p<0.05. (B): After Adenoviruses infection, the cells were then left untreated or treated
with Thapsigargin. Equal amount of proteins was resolved on SDS-PAGE gel followed by
western blotting with indicated antibodies. (C): After adenoviruses infection as described
in (A), equal number (1,000) of wild-type (WT) or ECD-/- cells were plated in triplicate
and treated with Thapsigargin for 24h. 10 days later, surviving colonies were assessed after
crystal blue (0.5% in 25% Methanol) staining. The color retained after the wash was
dissolved in 10% acetic acid and the absorbance was read at 590 nm. The graph below
represents the relative absorbance; Mean +/- SD and p-values from 4 independent
experiments are indicated * p<0.05; ** p≤0.002

72
To further assess these effects, we used physiological stress in Panc-1 cells in which ECD
was knocked down followed by glucose starvation. Again, here too, a siRNA knock down
of ECD plus glucose starvation led to an enhanced eIF2α phosphorylation, the downstream
target of PERK, and increased cleaved caspase 3 as compared to control cells (Figure 2.11,
compare lanes 1-5 with lanes 6-10), suggesting more cell death in ECD-knocked down
cells upon glucose starvation. Moreover, spliced XBP-1 levels, the downstream target of
IRE1α, were increased in ECD siRNA-treated cells as compared to the control cells
whereas ATF6 levels were comparable (Figure 2.11, compare lanes 1-5 with lanes 6-10).
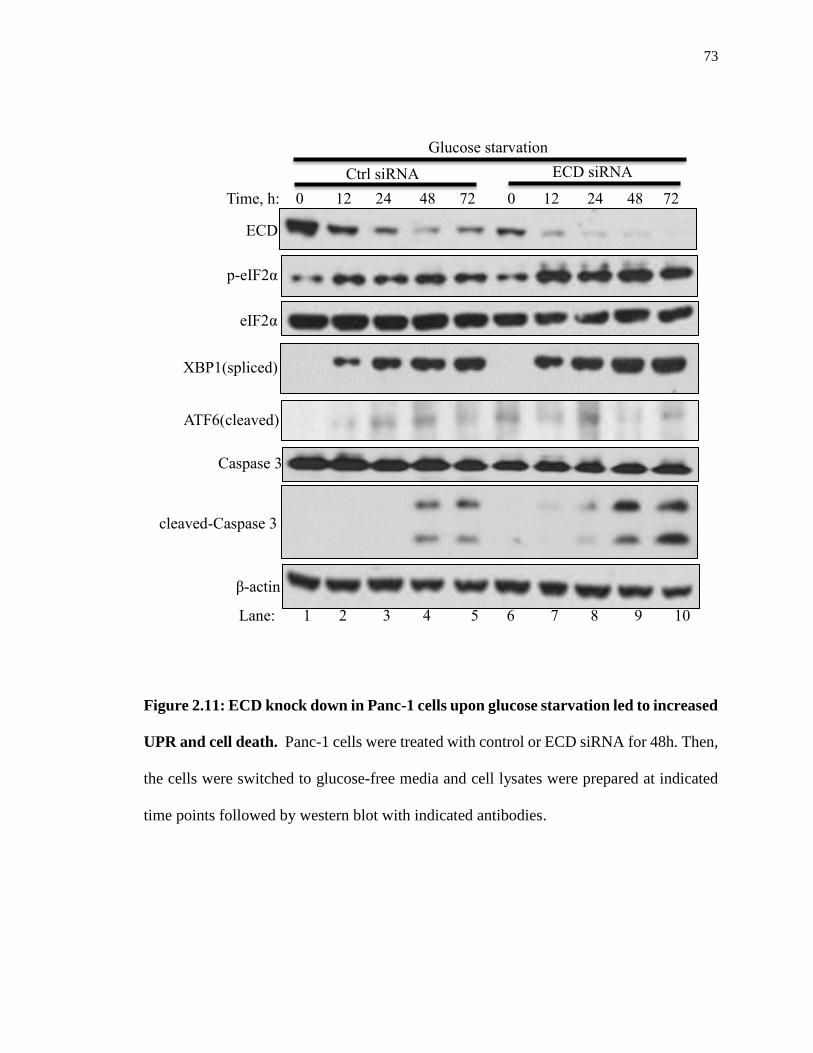
73
Figure 2.11: ECD knock down in Panc-1 cells upon glucose starvation led to increased
UPR and cell death. Panc-1 cells were treated with control or ECD siRNA for 48h. Then,
the cells were switched to glucose-free media and cell lysates were prepared at indicated
time points followed by western blot with indicated antibodies.
cleaved-Caspase 3
Time, h: 0 12 24 48 72 0 12 24 48 72
p-eIF2α
ECD
β-actin
XBP1(spliced)
eIF2α
ECD siRNA Ctrl siRNA
ATF6(cleaved)
Caspase 3
Lane: 1 2 3 4 5 6 7 8 9 10
Glucose starvation

74
Given the effects of ECD depletion on the UPR presented above, we used a reciprocal
approach to overexpress ECD and then examined its impact on the UPR, in particular the
PERK pathway, upon ER stress induction. To this end, and because of ongoing difficulties
overexpressing ECD in cancer cell lines, we resorted to MEFs generated in the lab from
mice that carry a Doxycycline (Dox)-inducible ECD transgene ((Tet(O)-Flag-hECD-IRES-
eGFP; rtTA)) or a control transgenic mouse without rtTA ((Tet(O)-Flag- hECD-
IRESeGFP)). These MEFs were treated with Dox to induce ECD overexpression, followed
by treatment with Thapsigargin. As expected, control MEFs exhibited an increase in p-
PERK and CHOP mRNA; however, the MEFs with Dox-induced ECD overexpression
exhibited a substantially reduced level of p-PERK and CHOP mRNA upon Thapsigargin
treatment as compared to control MEFs (Figure 2.12A-B).

75
Figure 2.12: Decreased p-PERK and CHOP mRNA induction in ECD inducible
MEFs upon Thapsigargin. (A): ECD inducible MEFs [(Tet(O)-Flag- hECD-
IRESeGFP;rtTA)] or control MEFs [(Tet(O)-Flag- hECD-IRESeGFP)] were treated with
Tg : - + - +
Dox: + + + +
p-PERK
ECD
β-actin
Lane: 1 2 3 4
A
0
10
20
30
40
50
60
Rel
ativ
e m
RN
A
Tet(O)-Flag- hECD
-IRES eGFP
Tet(O)-Flag- hECD
-IRES eGFP; rtTA
CHOP mRNA
Tg: - +
*
B

76
Dox for 48 h followed by treatment with Thapsigargin. Cell lysates were prepared at
indicated time points and equal amount of proteins was resolved on SDS-PAGE gel
followed by western blot with indicated antibodies. (B): Following ECD induction and
Thapsigargin treatment as described above, CHOP mRNA levels were assessed using
qRT-PCR.

77
Likewise, when the ECD inducible MEF cell lines were treated with Thapsigargin for
various time points (3, 6, 12 or 24h), ECD overexpressing MEFs exhibited lower levels of
p-PERK compared to those in control MEFs (Figure 2.13A, compare lanes 1-5 with lanes
6-10). A corresponding reduction in the levels of ATF4, a downstream target of PERK
signaling, was seen in ECD overexpressing MEFs compared to their control MEFs (Figure
2.13A, compare lanes 3-5 with lanes 8-10); s-XBP1 and ATF6 also were slightly reduced
in ECD over-expressing MEFs (Figure 2.13A, compare lanes 1-5 with lanes 6-10). Similar
to Thapsigargin, induction of ER stress with Tunicamycin, which induces ER stress by
inhibiting glycosylation of proteins in the ER, was also associated with lower levels of p-
PERK and p-eIF2α in ECD overexpressing MEFs as compared to control MEFs (Figure
2.13B, compare lanes 2-5 with lanes 7-10). Taken together, these results strongly support
a conclusion that ECD negatively regulates PERK signaling.

78
p-PERK
PERK
ATF4
ATF6 (cleaved)
XBP1(spliced)
Tg, h: 0 3 6 12 24 0 3 6 12 24
Tet(O)-Flag- hECD
-IRES eGFP; rtTA
Tet(O)-Flag- hECD
-IRES eGFP
β-actin
Lane: 1 2 3 4 5 6 7 8 9 10
ECD
A

79
Figure 2.13: Decreased UPR signaling in ECD inducible MEFs correlated with in
chaperones induction upon Thapsigargin treatment. (A-B): ECD inducible MEFs
[(Tet(O)-Flag- hECD-IRESeGFP;rtTA)] or control MEFs [(Tet(O)-Flag- hECD-
IRESeGFP)] were treated with Dox for 48 h followed by treatment with Thapsigargin. Cell
lysates were prepared at indicated time points and equal amount of proteins was resolved
on SDS-PAGE gel followed by western blot with indicated antibodies
Tun, h: 0 3 6 12 24 0 3 6 12 24
Tet(O)-Flag- hECD
-IRES eGFP; rtTA Tet(O)-Flag- hECD
-IRES eGFP
ECD
p-PERK
PERK
p-eIF2α
eIF2α
β-actin
Lane: 1 2 3 4 5 6 7 8 9 10
B

80
2.4.7 ECD overexpression protects cells from ER stress-induced cell death
ER stress response is initially aimed at the survival of cells (76); however severe ER stress
shifts the response from a pro-survival to a pro-apoptotic response (36,44) through PERK-
mediated induction of CHOP expression, a transcription factor that enhances the
expression of pro-apoptotic pathway genes (37-41,43,44,77). Several studies found that
inhibition of the PERK pathway protected cells against stress-induced cell death (78-81).
Given our observations that ECD functions as a modulator of PERK signaling, we assessed
if the level of ECD determines a differential survival vs. apoptotic cell fate upon ER stress
induction. For that purpose, we treated control MEFs or Dox-inducible ECD
overexpressing MEFs with Thapsigargin and then assessed the level of apoptosis induction
by examining caspase 3 cleavage (82). As anticipated, a Thapsigargin dose-dependent
increase in cleaved caspase 3 levels was observed in control MEFs, whereas the levels of
cleaved caspase 3 were markedly lower in ECD-overexpressing MEFs (Figure 2.14).
Moreover, given the role of stress-activated protein kinases SAPK/JNK in mediating ER
stress-induced cell death, as a downstream target of IRE1α (44,83,84), we further assessed
the levels of phospho-SAPK/JNK. Notably, while the p-SAPK/JNK level increased in
response to Thapsigargin in control MEFs, no changes were observed in ECD-
overexpressing MEFs (Figure 2.14, compare lane 1-3 to 4-6).

81
Figure 2.14: Decreased cleaved caspase 3 and p-SAPK/JNK levels in ECD
overexpressing MEF upon Thapsigargin. ECD was induced as described in (Fig. 3 and
4) followed by Thapsigargin treatment for 24 h. Equal amount of proteins was resolved on
SDS-PAGE gel followed by western blot with indicated antibodies
Tg (nM): - 50 100 - 50 100
ECD
Caspase 3
(total)
cleaved-Caspase 3
Lane: 1 2 3 4 5 6
Dox: + + + + + +
Tet(O)-Flag- hECD
-IRES eGFP; rtTA
Tet(O)-Flag- hECD
-IRES eGFP
p-SAPK/JNK
β-actin

82
To further assess the pro-survival effects of ECD against apoptotic cell fate upon ER stress
induction, we assessed the abilities of control vs. ECD-overexpressing MEFs to form
colonies after their exposure to ER stress. For this purpose, equal numbers of control or
ECD overexpressing MEFs were treated with Thapsigargin for 24h, and the cells were
maintained in Thapsigargin-free medium for 10 days followed by crystal violet staining
and counting of surviving colonies. Notably, more colonies were observed in ECD
overexpressing MEFs as compared to control MEFs (Figure 2.15), further supporting the
conclusion that ECD provides survival advantage upon ER stress.

83
Figure 2.15: Increased colony formation in ECD over-expressing MEFs upon
Thapsigargin. After ECD induction, control and ECD inducible MEFs were trypsinized
and equal number of cells (1,000) were plated in triplicates. 8 h later, the cells were
treated with Thapsigargin for 24h. 10 days later, surviving colonies were assessed by
Tg: - 50nM 100nM Dox: + + +
Tet(O)-Flag- hECD
-IRES eGFP; rtTA
Tet(O)-Flag- hECD
-IRES eGFP
0
0.2
0.4
0.6
0.8
1
1.2
Rel
ativ
e su
rviv
al
Tet(O)-Flag- hECD
-IRES eGFP
Tet(O)-Flag- hECD
-IRES eGFP; rtTA
**
Tg: - 50 nM 100 nM

84
Crystal blue staining (0.5% in 25% Methanol). The color retained after the wash was
dissolved in 10% Acetic acid and the absorbance was read at 590 nm. The graph below
represents the relative absorbance; Mean +/- SD and p-values from 4 independent
experiments are indicated ** p≤0.002.
----------------------------------
2.4.8 ECD co-localizes and associates with PERK, ATF6, IRE1α and GRP78
Given the roles of ECD in modulating ER stress as presented above, we sought to define
the possible mechanism (s). Previously, ECD has been reported to localize, predominantly,
in the cytoplasm with a fast nuclear export, allowing it to shuttle through the nucleus (85).
Given the roles of ECD in attenuating ER stress signaling, particularly PERK, we first
asked whether ECD could also be localizing to the ER or co-localizes with the upstream
mediators of the ER stress signaling, especially as previous works from the lab showed a
peri-nuclear localization for ECD, which is the classic definition of an ER localization.
Therefore, we performed a proximity ligation assay (PLA) to assess the localization of
ECD relative to PERK and GRP78. Known ECD interaction with PIH1D1 component of
the R2TP served as the positive control (55,59). Indeed, ECD and PIH1D1 formed distinct
foci detectable by PLA (Figure 2.15B, red dots). Significantly, PLA signals were also
observed for ECD and GRP78 or ECD and PERK (Figure 2.15 C-D). Negative controls
(IgG) did not show any PLA signal (Figure 2.15 E).
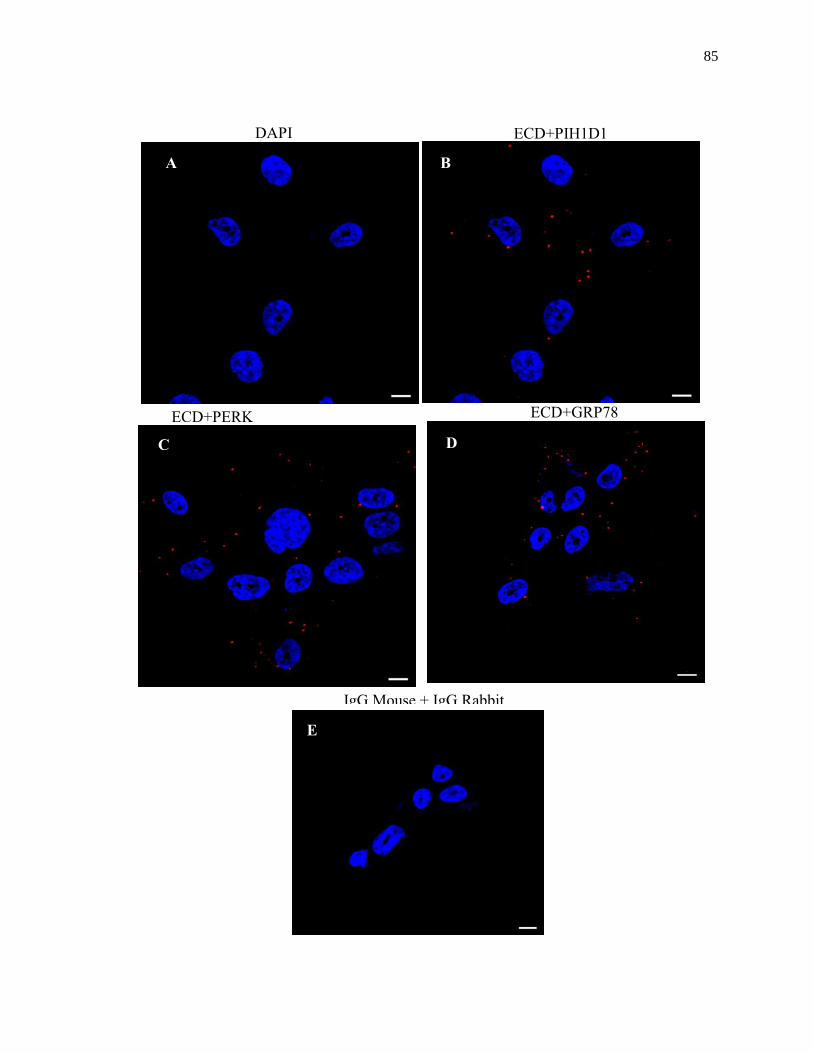
85
F
H
ECD+GRP78 ECD+PERK
ECD+PIH1D1 DAPI
A B
C D
IgG Mouse + IgG Rabbit
E

86
Figure 2.15: ECD co-localizes with PERK and GRP78. MCF-10A cells were fixed with
3% PFA and stained with indicated antibodies (all antibodies were generated in rabbit
except anti-ECD mouse antibody) followed by species-specific secondary antibodies
linked to complementary DNA probes to allow fluorescent probe-based detection of the
PCR amplification product as distinct foci. Incubation with Proximity Ligation Assay plus
and minus probes, followed by ligation and amplification, was carried out according to the
manufacturer protocol. Red dots indicate interaction. ECD and PIH1D1 served as the
positive control.

87
The localization of ECD close to the ER stress signaling mediators prompted us to assess
their association. For that purpose, we carried out immunoprecipitation (IP) of ECD from
lysates from MCF-10A cell followed by western blotting for PERK, GRP78, IRE1α, and
ATF6 antibodies to assess their association. IP of ECD co-immunoprecipitated PIH1D1
(Figure 2.16) as expected based on known interaction of ECD with PIH1D1 (55,59).
Importantly, PERK, GRP78, IRE1α and ATF6 also co-immunoprecipitated with ECD
(Figure 2.16).

88
Figure 2.16: ECD associates with PERK, GRP78, IRE1α and ATF6. Lysates from
MCF-10A cells were subjected to IP with anti-ECD antibody overnight followed by
Western blotting with indicated antibodies. ECD and PIH1D1 served as the positive
controls.
GRP78
PIH1D1
IRE1α
PERK
ECD
ATF6

89
To further asses these results, we carried out a GST pull down experiment by incubating
bacterially purified GST-fused full length ECD, C-terminal and N-terminal deletion
mutants ECD with cell lysate containing overexpressed GRP78, IRE1α, PERK and ATF6
followed by western blot with the respective antibodies; this GST pull down experiment
will help define the domain(s) on ECD required for their association. Known interaction of
full length GST-ECD with RUVBL1/Pontin was used as a positive control (55). As
expected, full length GST-ECD pulled down RUVBL1/Pontin (Figure 2.17). Significantly,
full length ECD interacts with GRP78, IRE1α and ATF6. N-terminal and C-terminal
deletions of ECD abrogated interaction with GRP78 and ATF6 (Figure 2.17) while only
the C-terminal deletion of ECD abrogated interaction with IRE1α (Figure 2.17), suggesting
that both the N and C termini are required for interaction with GRP78 and ATF6 but only
the C-terminus is required for interaction with IRE1α. GST-ECD did not pull down PERK.

90
Figure 2.17: Defining the domain (s) on ECD required for interaction. 3µg of
bacterially purified GST fused ECD full length (FL) and its mutants were incubated with
lysates from 293T cells overexpressing GRP78, IRE1α and ATF6 for 3h at 4°. The GST
beads were washed and resolved on SDS-PAGE gel followed by western blot with
indicated antibodies. Pontin and ECD interaction served as a positive control. Below the
blot is the ponceau.
GST-ECD
GRP78
Pontin
IRE1a
ATF6
N-ter C-ter FL
1 644
1 438
150 644
GST
GST-ECD (1-644)
GST-ECD (1-438) GST-ECD (150-644)

91
2.4.9 ECD associates with GRP78 and PERK upon ER stress induction
We further examined the ER stress-related roles of ECD by assessing its association with
PERK and GRP78 upon ER stress induction. For that purpose, we carried out
immunoprecipitation (IP) of ECD from lysates from the MCF-10A cell, treated or left
untreated with Thapsigargin, followed by western blotting for PERK and GRP78
antibodies to assess their association. IP of ECD co-immunoprecipitated PIH1D1 (Figure
2.18A), as expected based on known interaction of ECD with PIH1D1 (55,59).
Importantly, PERK and GRP78 also co-immunoprecipitated with ECD (Figure 2.18A). A
reverse IP of GRP78 also co-immunoprecipitated ECD, with PERK used as a positive
control (Figure 2.18B). Taken together, these results suggest that ECD associates with
PERK and GRP78.
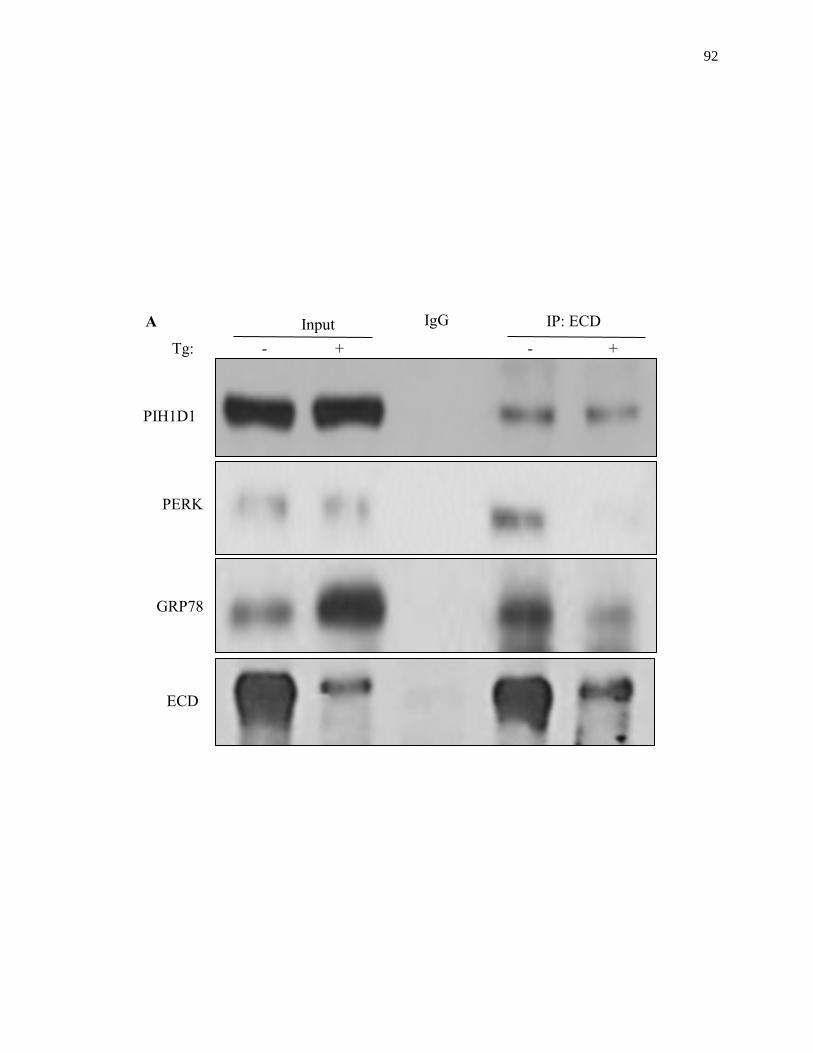
92
ECD
Tg: - + - +
Input IP: ECD IgG
PERK
GRP78
PIH1D1
A

93
Figure 2.18: ECD association with PERK and GRP78 upon ER stress induction.
Lysates from MCF-10A cells, treated or left untreated with Thapsigargin, were subjected
to IP with anti-ECD antibody (A) or anti-GRP78 (B) followed by Western blotting with
indicated antibodies. ECD and PIH1D1(A) or GRP78 and PERK (B) served as positive
controls.
Tg: - + - +
Input IgG
ECD
GRP78
PERK
IP: GRP78 B

94
2.4.10 ECD does not modulate enzymatic activity of PERK in vitro
Given that ECD associates with PERK and modulates its signaling, we hypothesized that
ECD could be affecting PERK signaling either directly or indirectly. To test the direct
hypothesis, we assessed the possibility that ECD affects PERK enzymatic activity. We
performed an in vitro kinase assay of PERK with its substrate eIF2α in the presence of an
increasing concentration of pure ECD protein. This experiment showed that eIF2α
phosphorylation did not change in the presence of an increasing concentration of ECD
(Figure 2.19), suggesting that ECD does not directly modulate PERK enzymatic activity.

95
Figure 2.19: ECD does not modulate enzymatic activity of PERK toward eIF2α. In
vitro kinase assay of recombinant human GST fused PERK with its substrate eIF2α in the
presence of increasing concentration of pure ECD protein. Proteins were separated by
SDS-PAGE, transferred to PVDF membrane, and phosphate labeling was detected by
autoradiography.
p-PERK
PERK + + + + + + +
eIF2α - + + + + + +
ECD (ng) - - 50 100 200 400 800
32P + + + + + + +
p-eIF2α

96
2.4.11 ECD positively regulates levels of chaperones, predominantly, GRP78 protein
The results above that ECD does not modulate the enzymatic activity of PERK ruled out
the possibility of a direct effect on PERK signaling. Therefore, we examined the next
possibility of an indirect mechanism whereby ECD acts through another protein to
modulate PERK. GRP78 is a repressor of the three arms of the ER stress signaling,
including PERK, of which it is known to bind to the luminal domain to repress its
activation in unstressed cells (13,14). The dissociation of GRP78 from PERK upon ER
stress is key to activation of PERK signaling, but it is also a reversible process, as GRP78
is also known to bind back to PERK to inhibit its signaling when the stress is relieved.
Accordingly, we asked whether ECD could be cooperating with GRP78 to modulate
PERK signaling especially since ECD and GRP78 associate (Figure 2.16-2.18). To
explore that possibility, we first examined the changes in the levels of GRP78 in the
presence and absence of ECD upon ER stress induction. We first exposed the wild-type
MEFs or MEFs with ECD deletion induced by adeno-Cre to Thapsigargin, and then
assessed the levels of GRP78 protein, at various time points, by western blotting. The
levels of GRP78, over time, were reduced in ECD-null MEFs compared to GRP78 levels
in control MEFs (Figure 2.20A, compare lanes 3-5 with 8-10). Likewise, using
physiological stress, a knock down of ECD in Panc-1 cells led to a decrease in GRP78
levels upon glucose starvation (Figure 2.20B, compare lanes 1-5 with lanes 6-10).

97
Tg, h: 0 2 4 7 11 0 2 4 7 11
ECD
GRP78
β-actin
WT ECD-/-
Lane: 1 2 3 4 5 6 7 8 9 10
MEF A

98
Figure 2.20: ECD deletion or knock down led to a reduced induced levels of GRP78.
(A): Ecd fl/fl MEFs were treated with adenovirus as previously described and then the
cells were treated with Thapsigargin (50 nM); (B): ECD was knocked down by siRNA
(20 nM) in Panc-1 cells followed by exposure to glucose-free media. Cell lysates were
collected at indicated time points followed by western blot with indicated antibodies.
ECD siRNA Ctrl siRNA
Time, h: 0 12 24 48 72 0 12 24 48 72
GRP78
β-actin
Lane: 1 2 3 4 5 6 7 8 9 10
ECD
Glucose starvation
Panc-1
B

99
To further examine these effects, we used a reciprocal approach by overexpressing ECD
and assessing GRP78 protein levels upon ER stress induction. In the presence of
overexpressed ECD, upon induction of ER stress by Thapsigargin treatment, the levels of
GRP78 were robustly elevated (Figure 2.21A). GRP94 and PDI, two other chaperones
with ER stress-related functions, were also increased although not as robustly as GRP78
(Figure 2.21A). Similar results were obtained with Tunicamycin treatment, another ER
stress inducing chemical (Figure 2.21B).

100
Figure 2.21: ECD overexpression led to a robust increase in chaperone levels,
predominantly, GRP78. ECD inducible MEFs [(Tet(O)-Flag- hECD-IRESeGFP;rtTA)]
or control MEFs [(Tet(O)-Flag- hECD-IRESeGFP)] were treated with Dox for 48 h
followed by treatment with Thapsigargin (50 nM, A) or Tunicamycin (50 ng/ml, B). Cell
lysates were prepared at indicated time points and equal amount of proteins was resolved
on SDS-PAGE gel followed by western blot with indicated antibodies.
Tun, h: 0 3 6 12 24 0 3 6 12 24
Tet(O)-Flag- hECD
-IRES eGFP; rtTA Tet(O)-Flag- hECD
-IRES eGFP
ECD
β-actin
GRP78
Lane: 1 2 3 4 5 6 7 8 9 10
B
GRP78
Tg, h: 0 3 6 12 24 0 3 6 12 24
GRP94
Tet(O)-Flag- hECD
-IRES eGFP; rtTA
Tet(O)-Flag- hECD
-IRES eGFP
β-actin
Lane: 1 2 3 4 5 6 7 8 9 10
ECD
PDI
A

101
2.4.12 ECD does not affect GRP78 transcription or mRNA stability
Based on the distinct effect of ECD depletion or overexpression on GRP78 protein levels,
we next examined GRP78 mRNA induction in the presence or absence of ECD. We
carried out qPCR analyses of mRNA isolated from WT or ECD-null MEFs treated with
Thapsigargin. As expected, GRP78 mRNA levels increased in WT MEFs upon
Thapsigargin treatment (Figure 2.22A); however, the levels of GRP78 mRNA induction
in ECD-null MEFs was comparable to that in control MEFs (Figure 2.22A). Similarly,
GRP78 mRNA levels in control vs. ECD overexpressing MEFs treated with Thapsigargin
were comparable (Figure 2.22B). Likewise, we examined the post-transcriptional effect
of ECD on GRP78 mRNA stability by treating cells with Actinomycin D, to inhibit
mRNA synthesis, and collected mRNA at various time points followed by qPCR with
primers against GRP78 mRNA. The rate of decrease of GRP78 mRNA over time is
comparable between ECD depleted or overexpressed cells and their respective controls
(Figure 2.23A-B), suggesting that ECD does not affect GRP78 mRNA stability. Taken
together, these results suggest that ECD does not affect GRP78 mRNA transcription or
post-transcriptionally.
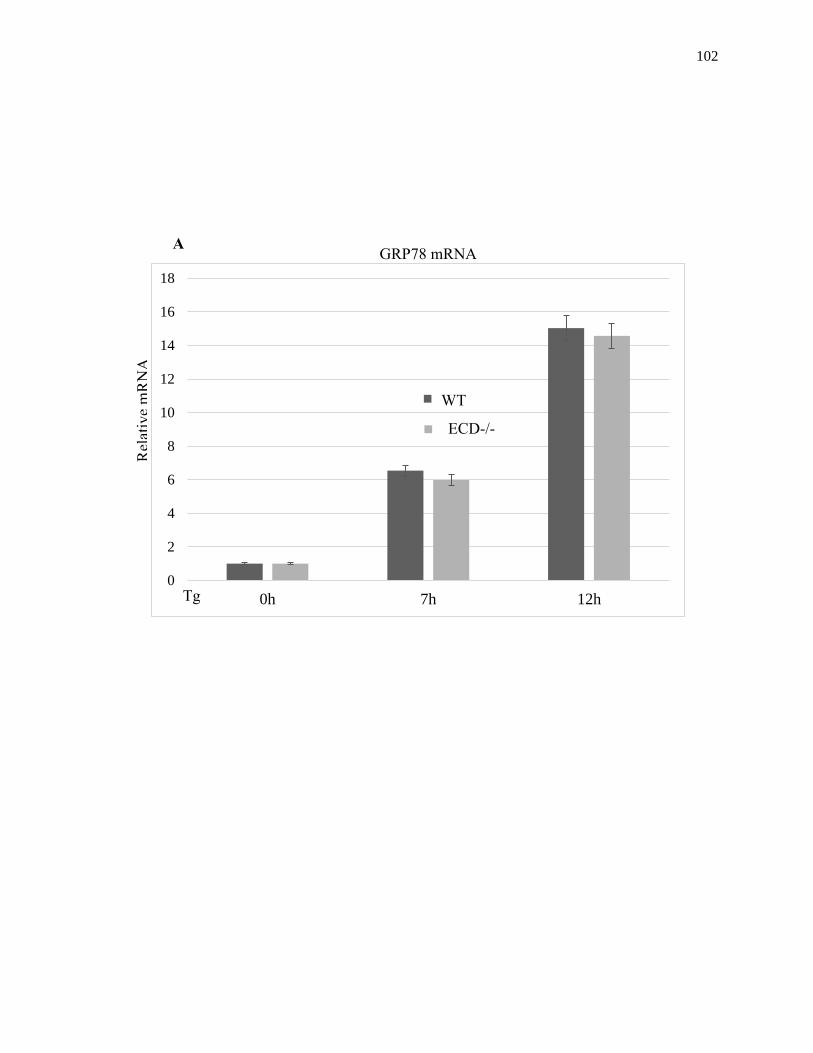
102
0
2
4
6
8
10
12
14
16
18
0h 7h 12h
Rel
ativ
e m
RN
A
GRP78 mRNA
Tg
WT
ECD-/-
A

103
Figure 2.22: Effect of ECD on GRP78 mRNA induction. Following ECD deletion by
Adeno-CRE infection (A) or ECD overexpression (B) and Thapsigargin treatment, the
levels of GRP78 mRNA were assessed in WT (control) vs. ECD-/- (Cre-adenovirus
treated) or control vs. ECD overexpressing MEFs using qRT-PCR.
0
5
10
15
20
25
0h 7h 12h
GRP78 mRNA
Tet(O)-Flag-hECD-IRES eGFP Tet(O)-Flag-hECD-IRES eGFP; rttTA
Rel
ativ
e m
RN
A
Tg
B

104
0
0.2
0.4
0.6
0.8
1
1.2
GRP78 mRNA stability
WT ECD-/- Linear (WT) Linear (ECD-/-)
0
0.2
0.4
0.6
0.8
1
1.2
0h 30 min 1h 2h 4h 8h
GRP78 mRNA stability
WT ECD-/-
Rel
ativ
e m
RN
A
Rel
ativ
e m
RN
A
Act D: 0h 30 min 1h 2h 4h 8h
A

105
0
0.2
0.4
0.6
0.8
1
1.2
0h 30 min 1h 2h 4h 8h
GRP78 mRNA stability
CTRL ECD O.E
0
0.2
0.4
0.6
0.8
1
1.2
GRP78 mRNA stability
CTRL ECD O.E Linear (CTRL) Linear (ECD O.E)Act D: 0h 30 min 1h 2h 4h 8h
Rel
ativ
e m
RN
A
Rel
ativ
e m
RN
A
B

106
Figure 2.23: Effect of ECD on GRP78 mRNA stability. Following ECD deletion by
Adeno-CRE infection (A) or ECD overexpression (B) and Thapsigargin treatment, the
cells were treated with Actinomycin D and mRNA was collected at indicated time points.
The levels of GRP78 mRNA were assessed in WT (control) vs. ECD-/- (Cre-adenovirus
treated) or control vs. ECD overexpressing MEFs using qRT-PCR.

107
2.4.13 ECD enhances GRP78 protein stability and this effect is required for ECD to
attenuate ER stress signaling
The results above showing that ECD does not regulate GRP78 protein at mRNA level
raised a strong possibility that ECD might instead regulate GRP78 at protein level
especially since the two protein associated. To determine whether GRP78 may be less
stable in ECD null cells or more stable in ECD overexpressing cells, we first knocked
down ECD in Panc-1 cells, which have high levels of GRP78, followed by inhibition of
protein synthesis by cycloheximide treatment over various time points and GRP78
protein levels were analyzed by western blot. Significantly, the time dependent decrease
in GRP78 protein levels following cycloheximide treatment was faster in ECD-knocked
down cells compared to their control cells (Figure 2.24A). Next, to examine the
possibility that ECD overexpression is associated with increased GRP78 protein stability,
control or ECD overexpressing cells were treated with Thapsigargin to induce GRP78
protein, followed by cycloheximide (CHX) treatment over various time points. Notably,
the time-dependent decrease in GRP78 levels was slower in ECD overexpressing MEFs
as compared to control MEFs (Figure 2.24B). Finally, to determine whether the increase
in GRP78 protein stability upon ECD overexpression was required for the attenuation of
PERK activation seen upon ER stress induction, we depleted GRP78 using siRNA in
both control and ECD overexpressing MEFs followed by Thapsigargin treatment.
Notably, even in the presence of overexpressed ECD, the attenuating effect of ECD
overexpression on PERK phosphorylation upon Thapsigargin treatment was abrogated by
GRP78 knockdown (Figure 2.24C; compare lane 5-6 with 7-8). Furthermore, this knock
down of GRP78 reduced the survival effect of ECD overexpression by reducing the

108
colonies’ ability to form the ECD overexpressing cells (Figure 2.24D, compare lane 2
and 4). Taken together, these results support the conclusion that ECD positively regulates
GRP78 protein level upon ER stress induction to attenuate PERK activation and provide
survival advantage.

109
GRP78
CHX, h: 0 12 24 36 48 0 12 24 36 48
Ctrl siRNA ECD siRNA
ECD
β-actin
Lane: 1 2 3 4 5 6 7 8 9 10
-0.2
0
0.2
0.4
0.6
0.8
1
1.2
Ctrl siRNA
ECD siRNA
CHX, h: 0 12 24 36 48
T1/2
> 36h
T1/2
~24h
Panc-1 A

110
β-actin
Tg: + + + + + + + + + +
CHX, h: - 2 6 12 15 - 2 6 12 15
GRP78
ECD
Lane: 1 2 3 4 5 6 7 8 9 10
Tet(O)-Flag- hECD
-IRES eGFP; rtTA Tet(O)-Flag- hECD
-IRES eGFP
0
0.2
0.4
0.6
0.8
1
1.2
Tet(O)-Flag-hECD-IRES eGFP
Tet(O)-Flag-hECD-IRES eGFP; rtTA
Rel
ativ
e G
RP
78
CHX, h: - 2 6 12 15
B

111
Tg - + - + - + - +
Scrambled + + - - + + - -
GRP78 SiRNA - - + + - - + +
p-PERK
ECD
GRP78
β-actin
Lane: 1 2 3 4 5 6 7 8
PERK
Tet(O)-Flag- hECD
-IRES eGFP; rtTA Tet(O)-Flag- hECD
-IRES eGFP
C
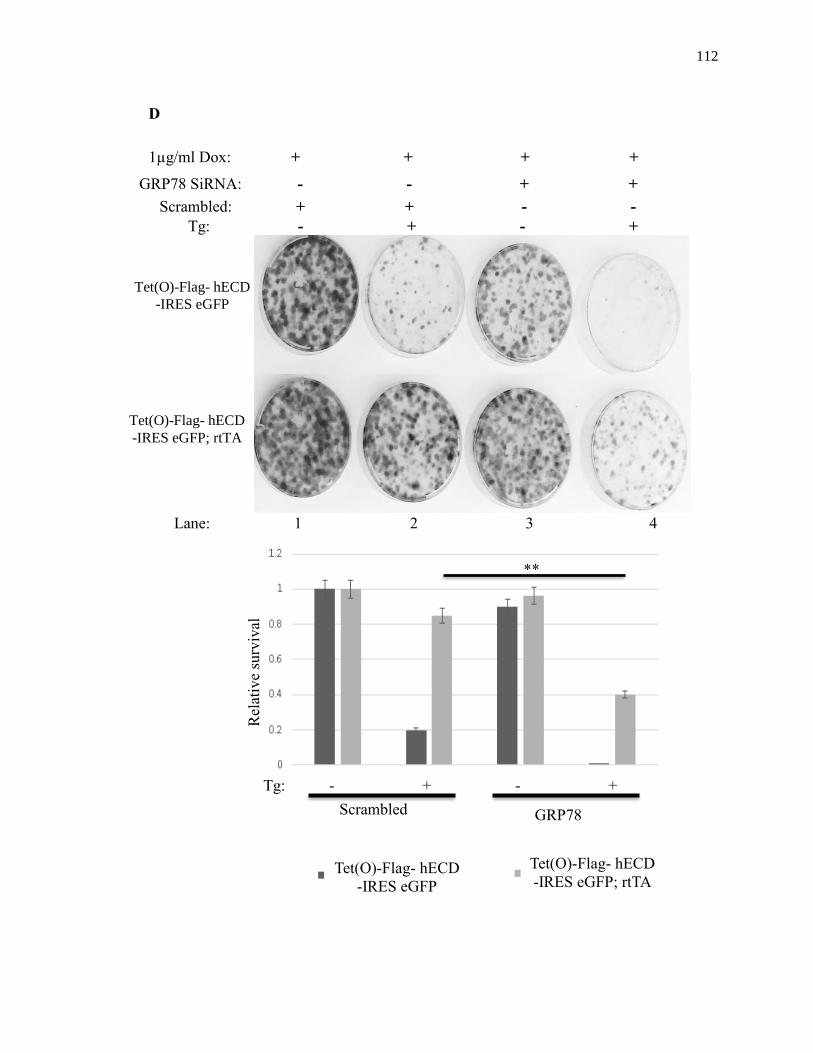
112
Tet(O)-Flag- hECD -IRES eGFP; rtTA
Tet(O)-Flag- hECD -IRES eGFP
Tg: - + - +
Lane: 1 2 3 4
GRP78 SiRNA: - - + +
Scrambled: + + - -
Tg: - + - +
Scrambled
Tet(O)-Flag- hECD
-IRES eGFP
Tet(O)-Flag- hECD
-IRES eGFP; rtTA
GRP78
SiRNA
Rel
ativ
e su
rviv
al
1µg/ml Dox: + + + +
D
**

113
Figure 2.24: ECD mediated GRP78 protein stability is required for ECD ER stress
function. (A): ECD was knocked down in Panc-1 cells followed by cycloheximide
treatment (25 uM). Cell lysates were prepared at indicated time points followed by
western blot with indicated antibodies. (B): ECD inducible MEFs and their control MEFs
were treated with Dox, as described previously, followed by treatment with Thapsigargin
and then Cycloheximide treatment (25 uM) for indicated time points. Cell lysates were
prepared followed by western blotting with indicated antibodies. (C): ECD
overexpressing and their control MEFs were treated with GRP78 SiRNA (30 nM) or
control siRNA (scrambled). 24 h later, the cells were treated with Dox for 48 h to induce
ECD overexpression followed by Thapsigargin treatment (50 nM). Equal amount of
proteins was resolved on SDS-PAGE gel followed by western blotting with indicated
antibodies. (D): After ECD induction, control and ECD inducible MEFs were trypsinized
and equal number of cells (1,000) were plated in triplicates. 8 h later, the cells were
treated with Thapsigargin for 24h. 10 days later, surviving colonies were assessed by
crystal blue staining (0.5% in 25% Methanol). The color retained after the wash was
dissolved in 10% Acetic acid and the absorbance was read at 590 nm. The graph below
represents the relative absorbance; Mean +/- SD and p-values from 3 independent
experiments are indicated ** p≤0.002

114
2.5 DISCUSSION
The ER-localized stress response pathway referred to as the UPR is a well-conserved
response to a number of cellular stresses such as unfolding of proteins in the ER. The UPR
elicits a spectrum of downstream responses whose outcomes range from restoration of
homeostasis to cellular apoptosis if the stress is extreme and prolonged (36,44). The UPR
thus represents a double-edged sword and must be intricately regulated to prevent
inappropriate cellular outcomes. Mechanisms that help modulate the magnitude and type
of the UPR in response to physiological or pathological stress stimuli are not fully
understood. In this study, we provide evidence that ECD protein is a repressor of stress
response.
Several lines of circumstantial evidence discussed in the Introduction section
suggested a potential involvement of ECD in UPR but direct support for such a role has
been lacking. To explore a connection of ECD to stress response pathways, we first
assessed the effects of multiple stress stimuli on ECD protein levels. Induction of stress
both chemically (Thapsigargin, Tunicamycin, BFA, DTT, sorbitol, and H2O2) and
physiologically (glucose starvation and NOX4 expression) in multiple cell lines led to
downregulation of ECD protein levels (Figure 2.1-2.3). The concomitant increase in ECD
mRNA levels (Figure 2.4) suggests that the protein was transcriptionally induced. By using
AFT6 KO, IRE1α and PERK kinase knockout (KO), we found that PERK is the upstream
mediator of the decrease in ECD protein levels and that in PERK KO MEFs, ECD protein
and mRNA levels did not change upon ER stress induction (Figure 2.5-2.6). Since ECD is
not a substrate for PERK phosphorylation (Figure 2.7) but ECD protein levels did not

115
change when eIF2α phosphorylation, the downstream effect of PERK activation, was
disrupted (Figure 2.8), the results strongly suggest that ECD protein levels are regulated
via the PERK-eIF2α axis upon ER stress induction. Since that axis controls translation
upon stress induction, then the results further suggest that ECD protein undergoes a PERK-
eIF2α-dependent translation block upon ER stress. The decrease in ECD protein levels in
other forms of stress, including oxido-reductive and osmotic stresses, further argues that
induction of stress in general leads to an eIF2α-dependent decrease in ECD protein levels.
In fact, all forms of cellular stress, including viral infection and nutrient limitation in
addition to those mentioned above, converge to a common node, eIF2α. Moreover, they
are interconnected. For instance, viral infection induces ER stress (86) due to the increase
viral protein synthesis. Oxidative stress and nutrient limitation also induce ER stress (87-
89).
With regard to ER stress, these findings linked ECD to the PERK-eIF2α pathway.
A functional connection of ECD to the PERK arm of the UPR is supported by the distinct
modulation of the PERK-mediated responses elicited by perturbations of the cellular levels
of ECD. Depletion of ECD sensitized cells to PERK signaling in response to Thapsigargin
treatment, with increase in p-PERK and p-eIF2α levels, as well as an increase in the
downstream effector of PERK, the transcription factor CHOP, altogether led to increased
caspase 3 cleavage and reduced survival of these ECD depleted MEFs (Figure 2.10).
Similarly, physiological stress by glucose deprivation in a human pancreatic carcinoma cell
line (Panc-1) upon ECD knock down resulted in increased ER stress with enhanced p-
eIF2α, spliced XBP-1 and cell death (Figure 2.11). Reciprocally, upregulation of ECD
reduced PERK signaling upon induction of ER stress (Figure 2.12). As activation of the

116
PERK pathway promotes cell death in response to ER stress, primarily through CHOP-
dependent expression of pro-apoptotic genes (37-45) and abrogation of PERK signaling
protects cells against stress-induced cell death (78-80), our results supported the likelihood
that ECD functions to modulate PERK pathway activity and promote cell survival during
ER stress. Indeed, assessment of cellular survival in response to ER stress showed that
reduction in ECD levels impaired cell survival to ER stress (Figure 2.10) while
overexpression of ECD promoted cell survival (Figure 2.14).
Collectively, these results support the conclusion that ECD and PERK are linked
through a negative feedback mechanism whereby ECD exerts an inhibitory effect on PERK
pathway signaling and activated PERK in turn reduces ECD protein levels via eIF2α-
dependent translational block. Consistent with a reciprocal negative feedback relation
between ECD and PERK, activated PERK negatively regulates cell growth (46-48);
conversely, ECD positively regulates cell growth (54) and is overexpressed in human
breast and pancreatic cancer specimens, correlating with poor prognostic markers and
shorter survival (69,90). While the physiological benefit of an increased ECD mRNA upon
ER stress is not yet understood, a speculation is that this effect may reflect a feedback
response to the decrease in ECD protein levels whereby cells may try to compensate for
the loss of ECD but ECD translation is blocked. Although ECD has been reported to play
roles in pre-mRNA splicing in drosophila (91), it is unlikely that ECD alters its own mRNA
splicing upon ER stress because ECD protein levels decrease upon ER stress.
Modulation of ECD levels using knockdown or overexpression demonstrated that
ECD is a positive regulator of GRP78 levels (Figure 2.20 and 2.21). While slight increases
in GRP94 and PDI were observed, the effect on GRP78 was more dramatic. However, this

117
effect may be post-translational since ECD did not have a transcriptional or post-
transcriptional effect on GRP87 mRNA. The slight increase in GRP94 and PDI, both of
which have ER stress-related functions, further suggests that ECD may affect chaperones’
levels, in general, when overexpressed upon ER stress induction.
As increased expression of GRP78 and other chaperones is known to promote the
clearing of ER stress-causing unfolded protein load and reduce the activation of UPR
sensors (13-15,20), this increase in chaperones levels in the presence of ECD upon ER
stress induction argue that ECD may be repressing stress by elevating chaperone levels.
Indeed, a knock down of GRP78 abrogated the attenuating effect of ECD on PERK
signaling even in the presence of overexpressed ECD (Figure 2.24C). Notably, GRP78 is
required for cell survival not only in response to ER stress (23,92-94) but also in other
stressful and hostile conditions such as glucose deficiency encountered in tumor
microenvironments (95-100). Given that the PERK arm of the UPR is also activated in
cancer (77,101,102) and both ECD and GRP78 are overexpressed in cancer (69,90,95-
100), we suggest that ECD overexpression may play a similar role to mitigate the negative
consequences of elevated PERK signaling found in cancer. Given the mechanistic link
between ECD and GRP78 in inhibiting the PERK pathway to promote cell survival, it will
be of great interest to explore if GRP78 and ECD are co-overexpressed in tumors that use
the UPR to promote tumor cell survival and hence may be suitable targets for UPR-directed
therapeutic agents.

118
REFERENCES
1. Voeltz, G. K., Rolls, M. M., and Rapoport, T. A. (2002) Structural organization of the
endoplasmic reticulum. EMBO reports 3, 944-950
2. Reid, D. W., and Nicchitta, C. V. (2015) Diversity and selectivity in mRNA translation
on the endoplasmic reticulum. Nature reviews. Molecular cell biology 16, 221-231
3. Jan, C. H., Williams, C. C., and Weissman, J. S. (2014) Principles of ER cotranslational
translocation revealed by proximity-specific ribosome profiling. Science 346, 1257521
4. Ron, D., and Walter, P. (2007) Signal integration in the endoplasmic reticulum unfolded
protein response. Nat. Rev. Mol. Cell Biol. 8, 519-529
5. Ikonen, E. (2008) Cellular cholesterol trafficking and compartmentalization. Nature
reviews. Molecular cell biology 9, 125-138
6. Ron, D., and Hampton, R. Y. (2004) Membrane biogenesis and the unfolded protein
response. The Journal of cell biology 167, 23-25
7. Fagone, P., and Jackowski, S. (2009) Membrane phospholipid synthesis and endoplasmic
reticulum function. Journal of lipid research 50 Suppl, S311-316
8. Kim, I., Xu, W., and Reed, J. C. (2008) Cell death and endoplasmic reticulum stress:
disease relevance and therapeutic opportunities. Nature reviews. Drug discovery 7, 1013-
1030
9. Berridge, M. J., Bootman, M. D., and Roderick, H. L. (2003) Calcium signalling:
dynamics, homeostasis and remodelling. Nature reviews. Molecular cell biology 4, 517-
529
10. Pinton, P., Giorgi, C., Siviero, R., Zecchini, E., and Rizzuto, R. (2008) Calcium and
apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 27,
6407-6418
11. Wu, J., and Kaufman, R. J. (2006) From acute ER stress to physiological roles of the
Unfolded Protein Response. Cell death and differentiation 13, 374-384
12. Bravo-Sagua, R., Rodriguez, A. E., Kuzmicic, J., Gutierrez, T., Lopez-Crisosto, C.,
Quiroga, C., Diaz-Elizondo, J., Chiong, M., Gillette, T. G., Rothermel, B. A., and
Lavandero, S. (2013) Cell death and survival through the endoplasmic reticulum-
mitochondrial axis. Current molecular medicine 13, 317-329
13. Bertolotti, A., Zhang, Y., Hendershot, L. M., Harding, H. P., and Ron, D. (2000)
Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response.
Nat. Cell Biol. 2, 326-332
14. Gardner, B. M., Pincus, D., Gotthardt, K., Gallagher, C. M., and Walter, P. (2013)
Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring
Harbor perspectives in biology 5, a013169
15. Lai, E., Teodoro, T., and Volchuk, A. (2007) Endoplasmic reticulum stress: signaling the
unfolded protein response. Physiology 22, 193-201
16. Baumeister, P., Luo, S., Skarnes, W. C., Sui, G., Seto, E., Shi, Y., and Lee, A. S. (2005)
Endoplasmic reticulum stress induction of the Grp78/BiP promoter: activating
mechanisms mediated by YY1 and its interactive chromatin modifiers. Mol. Cell. Biol.
25, 4529-4540
17. Lee, A. S. (1992) Mammalian stress response: induction of the glucose-regulated protein
family. Curr. Opin. Cell Biol. 4, 267-273
18. Marzec, M., Eletto, D., and Argon, Y. (2012) GRP94: An HSP90-like protein specialized
for protein folding and quality control in the endoplasmic reticulum. Biochim. Biophys.
Acta 1823, 774-787

119
19. Eletto, D., Dersh, D., and Argon, Y. (2010) GRP94 in ER quality control and stress
responses. Semin. Cell Dev. Biol. 21, 479-485
20. Lee, S., Min Kim, S., Dotimas, J., Li, L., Feener, E. P., Baldus, S., Myers, R. B.,
Chutkow, W. A., Patwari, P., Yoshioka, J., and Lee, R. T. (2014) Thioredoxin-interacting
protein regulates protein disulfide isomerases and endoplasmic reticulum stress. EMBO
Mol. Med. 6, 732-743
21. Oslowski, C. M., and Urano, F. (2011) Measuring ER stress and the unfolded protein
response using mammalian tissue culture system. Methods Enzymol. 490, 71-92
22. Reddy, R. K., Mao, C., Baumeister, P., Austin, R. C., Kaufman, R. J., and Lee, A. S.
(2003) Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis
induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7
activation. J. Biol. Chem. 278, 20915-20924
23. Rao, R. V., Peel, A., Logvinova, A., del Rio, G., Hermel, E., Yokota, T., Goldsmith, P.
C., Ellerby, L. M., Ellerby, H. M., and Bredesen, D. E. (2002) Coupling endoplasmic
reticulum stress to the cell death program: role of the ER chaperone GRP78. FEBS Lett.
514, 122-128
24. Meunier, L., Usherwood, Y. K., Chung, K. T., and Hendershot, L. M. (2002) A subset of
chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum
to bind nascent proteins. Mol. Biol. Cell 13, 4456-4469
25. Munro, S., and Pelham, H. R. (1986) An Hsp70-like protein in the ER: identity with the
78 kd glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell
46, 291-300
26. Li, N., Zoubeidi, A., Beraldi, E., and Gleave, M. E. (2013) GRP78 regulates clusterin
stability, retrotranslocation and mitochondrial localization under ER stress in prostate
cancer. Oncogene 32, 1933-1942
27. Ni, M., Zhang, Y., and Lee, A. S. (2011) Beyond the endoplasmic reticulum: atypical
GRP78 in cell viability, signalling and therapeutic targeting. Biochem. J. 434, 181-188
28. Ni, M., Zhou, H., Wey, S., Baumeister, P., and Lee, A. S. (2009) Regulation of PERK
signaling and leukemic cell survival by a novel cytosolic isoform of the UPR regulator
GRP78/BiP. PLoS One 4, e6868
29. Teske, B. F., Wek, S. A., Bunpo, P., Cundiff, J. K., McClintick, J. N., Anthony, T. G.,
and Wek, R. C. (2011) The eIF2 kinase PERK and the integrated stress response facilitate
activation of ATF6 during endoplasmic reticulum stress. Molecular biology of the cell 22,
4390-4405
30. DuRose, J. B., Tam, A. B., and Niwa, M. (2006) Intrinsic capacities of molecular sensors
of the unfolded protein response to sense alternate forms of endoplasmic reticulum stress.
Molecular biology of the cell 17, 3095-3107
31. Wang, S., and Kaufman, R. J. (2012) The impact of the unfolded protein response on
human disease. The Journal of cell biology 197, 857-867
32. Harding, H. P., Zhang, Y., Bertolotti, A., Zeng, H., and Ron, D. (2000) Perk is essential
for translational regulation and cell survival during the unfolded protein response.
Molecular cell 5, 897-904
33. Liu, Z., Lv, Y., Zhao, N., Guan, G., and Wang, J. (2015) Protein kinase R-like ER kinase
and its role in endoplasmic reticulum stress-decided cell fate. Cell death & disease 6,
e1822
34. Scheuner, D., Vander Mierde, D., Song, B., Flamez, D., Creemers, J. W., Tsukamoto, K.,
Ribick, M., Schuit, F. C., and Kaufman, R. J. (2005) Control of mRNA translation
preserves endoplasmic reticulum function in beta cells and maintains glucose
homeostasis. Nature medicine 11, 757-764

120
35. Ma, K., Vattem, K. M., and Wek, R. C. (2002) Dimerization and release of molecular
chaperone inhibition facilitate activation of eukaryotic initiation factor-2 kinase in
response to endoplasmic reticulum stress. J. Biol. Chem. 277, 18728-18735
36. Szegezdi, E., Logue, S. E., Gorman, A. M., and Samali, A. (2006) Mediators of
endoplasmic reticulum stress-induced apoptosis. EMBO reports 7, 880-885
37. Harding, H. P., Novoa, I., Zhang, Y., Zeng, H., Wek, R., Schapira, M., and Ron, D.
(2000) Regulated translation initiation controls stress-induced gene expression in
mammalian cells. Mol. Cell 6, 1099-1108
38. Ma, Y., Brewer, J. W., Diehl, J. A., and Hendershot, L. M. (2002) Two distinct stress
signaling pathways converge upon the CHOP promoter during the mammalian unfolded
protein response. J. Mol. Biol. 318, 1351-1365
39. Zinszner, H., Kuroda, M., Wang, X., Batchvarova, N., Lightfoot, R. T., Remotti, H.,
Stevens, J. L., and Ron, D. (1998) CHOP is implicated in programmed cell death in
response to impaired function of the endoplasmic reticulum. Genes Dev. 12, 982-995
40. Oyadomari, S., Koizumi, A., Takeda, K., Gotoh, T., Akira, S., Araki, E., and Mori, M.
(2002) Targeted disruption of the Chop gene delays endoplasmic reticulum stress-
mediated diabetes. The Journal of clinical investigation 109, 525-532
41. Song, B., Scheuner, D., Ron, D., Pennathur, S., and Kaufman, R. J. (2008) Chop deletion
reduces oxidative stress, improves beta cell function, and promotes cell survival in
multiple mouse models of diabetes. The Journal of clinical investigation 118, 3378-3389
42. Malhotra, J. D., Miao, H., Zhang, K., Wolfson, A., Pennathur, S., Pipe, S. W., and
Kaufman, R. J. (2008) Antioxidants reduce endoplasmic reticulum stress and improve
protein secretion. Proc. Natl. Acad. Sci. U. S. A. 105, 18525-18530
43. Thorp, E., Li, G., Seimon, T. A., Kuriakose, G., Ron, D., and Tabas, I. (2009) Reduced
apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe-/- and Ldlr-/-
mice lacking CHOP. Cell metabolism 9, 474-481
44. Tabas, I., and Ron, D. (2011) Integrating the mechanisms of apoptosis induced by
endoplasmic reticulum stress. Nat. Cell Biol. 13, 184-190
45. Pennuto, M., Tinelli, E., Malaguti, M., Del Carro, U., D'Antonio, M., Ron, D., Quattrini,
A., Feltri, M. L., and Wrabetz, L. (2008) Ablation of the UPR-mediator CHOP restores
motor function and reduces demyelination in Charcot-Marie-Tooth 1B mice. Neuron 57,
393-405
46. Brewer, J. W., Hendershot, L. M., Sherr, C. J., and Diehl, J. A. (1999) Mammalian
unfolded protein response inhibits cyclin D1 translation and cell-cycle progression. Proc.
Natl. Acad. Sci. U. S. A. 96, 8505-8510
47. Brewer, J. W., and Diehl, J. A. (2000) PERK mediates cell-cycle exit during the
mammalian unfolded protein response. Proc. Natl. Acad. Sci. U. S. A. 97, 12625-12630
48. Hamanaka, R. B., Bennett, B. S., Cullinan, S. B., and Diehl, J. A. (2005) PERK and
GCN2 contribute to eIF2alpha phosphorylation and cell cycle arrest after activation of the
unfolded protein response pathway. Mol. Biol. Cell 16, 5493-5501
49. Nutt, S. L., Hodgkin, P. D., Tarlinton, D. M., and Corcoran, L. M. (2015) The generation
of antibody-secreting plasma cells. Nature reviews. Immunology 15, 160-171
50. Garen, A., Kauvar, L., and Lepesant, J. A. (1977) Roles of ecdysone in Drosophila
development. Proc. Natl. Acad. Sci. U. S. A. 74, 5099-5103
51. Sato, T., Jigami, Y., Suzuki, T., and Uemura, H. (1999) A human gene, hSGT1, can
substitute for GCR2, which encodes a general regulatory factor of glycolytic gene
expression in Saccharomyces cerevisiae. Mol. Gen. Genet. 260, 535-540
52. Gao, Q., Srinivasan, S., Boyer, S. N., Wazer, D. E., and Band, V. (1999) The E6
oncoproteins of high-risk papillomaviruses bind to a novel putative GAP protein, E6TP1,
and target it for degradation. Molecular and cellular biology 19, 733-744

121
53. Zhang, Y., Chen, J., Gurumurthy, C. B., Kim, J., Bhat, I., Gao, Q., Dimri, G., Lee, S. W.,
Band, H., and Band, V. (2006) The human orthologue of Drosophila ecdysoneless protein
interacts with p53 and regulates its function. Cancer Res. 66, 7167-7175
54. Kim, J. H., Gurumurthy, C. B., Naramura, M., Zhang, Y., Dudley, A. T., Doglio, L.,
Band, H., and Band, V. (2009) Role of mammalian Ecdysoneless in cell cycle regulation.
J. Biol. Chem. 284, 26402-26410
55. Mir, R. A., Bele, A., Mirza, S., Srivastava, S., Olou, A. A., Ammons, S. A., Kim, J. H.,
Gurumurthy, C. B., Qiu, F., Band, H., and Band, V. (2015) A Novel Interaction of
Ecdysoneless (ECD) Protein with R2TP Complex Component RUVBL1 Is Required for
the Functional Role of ECD in Cell Cycle Progression. Mol. Cell. Biol. 36, 886-899
56. Pagliarini, V., Giglio, P., Bernardoni, P., De Zio, D., Fimia, G. M., Piacentini, M., and
Corazzari, M. (2015) Downregulation of E2F1 during ER stress is required to induce
apoptosis. Journal of cell science 128, 1166-1179
57. Xu, S. H., Huang, J. Z., Chen, M., Zeng, M., Zou, F. Y., Chen, D., and Yan, G. R. (2015)
Amplification of ACK1 promotes gastric tumorigenesis via ECDdependent p53
ubiquitination degradation. Oncotarget
58. Bourougaa, K., Naski, N., Boularan, C., Mlynarczyk, C., Candeias, M. M., Marullo, S.,
and Fahraeus, R. (2010) Endoplasmic reticulum stress induces G2 cell-cycle arrest via
mRNA translation of the p53 isoform p53/47. Mol. Cell 38, 78-88
59. Horejsi, Z., Stach, L., Flower, T. G., Joshi, D., Flynn, H., Skehel, J. M., O'Reilly, N. J.,
Ogrodowicz, R. W., Smerdon, S. J., and Boulton, S. J. (2014) Phosphorylation-dependent
PIH1D1 interactions define substrate specificity of the R2TP cochaperone complex. Cell
reports 7, 19-26
60. Ahn, S., Kim, J., and Hwang, J. (2013) CK2-mediated TEL2 phosphorylation augments
nonsense-mediated mRNA decay (NMD) by increase of SMG1 stability. Biochimica et
biophysica acta 1829, 1047-1055
61. Boulon, S., Marmier-Gourrier, N., Pradet-Balade, B., Wurth, L., Verheggen, C., Jady, B.
E., Rothe, B., Pescia, C., Robert, M. C., Kiss, T., Bardoni, B., Krol, A., Branlant, C.,
Allmang, C., Bertrand, E., and Charpentier, B. (2008) The Hsp90 chaperone controls the
biogenesis of L7Ae RNPs through conserved machinery. The Journal of cell biology 180,
579-595
62. Boulon, S., Pradet-Balade, B., Verheggen, C., Molle, D., Boireau, S., Georgieva, M.,
Azzag, K., Robert, M. C., Ahmad, Y., Neel, H., Lamond, A. I., and Bertrand, E. (2010)
HSP90 and its R2TP/Prefoldin-like cochaperone are involved in the cytoplasmic
assembly of RNA polymerase II. Molecular cell 39, 912-924
63. Horejsi, Z., Takai, H., Adelman, C. A., Collis, S. J., Flynn, H., Maslen, S., Skehel, J. M.,
de Lange, T., and Boulton, S. J. (2010) CK2 phospho-dependent binding of R2TP
complex to TEL2 is essential for mTOR and SMG1 stability. Mol. Cell 39, 839-850
64. Kim, S. G., Hoffman, G. R., Poulogiannis, G., Buel, G. R., Jang, Y. J., Lee, K. W., Kim,
B. Y., Erikson, R. L., Cantley, L. C., Choo, A. Y., and Blenis, J. (2013) Metabolic stress
controls mTORC1 lysosomal localization and dimerization by regulating the TTT-
RUVBL1/2 complex. Molecular cell 49, 172-185
65. Zhao, R., Kakihara, Y., Gribun, A., Huen, J., Yang, G., Khanna, M., Costanzo, M., Brost,
R. L., Boone, C., Hughes, T. R., Yip, C. M., and Houry, W. A. (2008) Molecular
chaperone Hsp90 stabilizes Pih1/Nop17 to maintain R2TP complex activity that regulates
snoRNA accumulation. The Journal of cell biology 180, 563-578
66. Yuan, X. S., Wang, Z. T., Hu, Y. J., Bao, F. C., Yuan, P., Zhang, C., Cao, J. L., Lv, W.,
and Hu, J. (2016) Downregulation of RUVBL1 inhibits proliferation of lung
adenocarcinoma cells by G1/S phase cell cycle arrest via multiple mechanisms. Tumour
Biol.

122
67. Suh, H. W., Yun, S., Song, H., Jung, H., Park, Y. J., Kim, T. D., Yoon, S. R., and Choi, I.
(2013) TXNIP interacts with hEcd to increase p53 stability and activity. Biochem.
Biophys. Res. Commun. 438, 264-269
68. Oslowski, C. M., Hara, T., O'Sullivan-Murphy, B., Kanekura, K., Lu, S., Hara, M.,
Ishigaki, S., Zhu, L. J., Hayashi, E., Hui, S. T., Greiner, D., Kaufman, R. J., Bortell, R.,
and Urano, F. (2012) Thioredoxin-interacting protein mediates ER stress-induced beta
cell death through initiation of the inflammasome. Cell metabolism 16, 265-273
69. Zhao, X., Mirza, S., Alshareeda, A., Zhang, Y., Gurumurthy, C. B., Bele, A., Kim, J. H.,
Mohibi, S., Goswami, M., Lele, S. M., West, W., Qiu, F., Ellis, I. O., Rakha, E. A.,
Green, A. R., Band, H., and Band, V. (2012) Overexpression of a novel cell cycle
regulator ecdysoneless in breast cancer: a marker of poor prognosis in HER2/neu-
overexpressing breast cancer patients. Breast Cancer Res. Treat. 134, 171-180
70. Singh, P. K., Wen, Y., Swanson, B. J., Shanmugam, K., Kazlauskas, A., Cerny, R. L.,
Gendler, S. J., and Hollingsworth, M. A. (2007) Platelet-derived growth factor receptor
beta-mediated phosphorylation of MUC1 enhances invasiveness in pancreatic
adenocarcinoma cells. Cancer Res. 67, 5201-5210
71. Scheuner, D., Song, B., McEwen, E., Liu, C., Laybutt, R., Gillespie, P., Saunders, T.,
Bonner-Weir, S., and Kaufman, R. J. (2001) Translational control is required for the
unfolded protein response and in vivo glucose homeostasis. Molecular cell 7, 1165-1176
72. Giorgio, M., Trinei, M., Migliaccio, E., and Pelicci, P. G. (2007) Hydrogen peroxide: a
metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol.
8, 722-728
73. Kuroda, J., Ago, T., Matsushima, S., Zhai, P., Schneider, M. D., and Sadoshima, J.
(2010) NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart.
Proc. Natl. Acad. Sci. U. S. A. 107, 15565-15570
74. Hubackova, S., Kucerova, A., Michlits, G., Kyjacova, L., Reinis, M., Korolov, O.,
Bartek, J., and Hodny, Z. (2016) IFNgamma induces oxidative stress, DNA damage and
tumor cell senescence via TGFbeta/SMAD signaling-dependent induction of Nox4 and
suppression of ANT2. Oncogene 35, 1236-1249
75. Yada, M., Hatakeyama, S., Kamura, T., Nishiyama, M., Tsunematsu, R., Imaki, H.,
Ishida, N., Okumura, F., Nakayama, K., and Nakayama, K. I. (2004) Phosphorylation-
dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 23,
2116-2125
76. Hetz, C., Chevet, E., and Oakes, S. A. (2015) Proteostasis control by the unfolded protein
response. Nat. Cell Biol. 17, 829-838
77. Bu, Y., and Diehl, J. A. (2016) PERK Integrates Oncogenic Signaling and Cell Survival
During Cancer Development. Journal of cellular physiology 231, 2088-2096
78. Ding, X., Ma, M., Teng, J., Shao, F., Wu, E., and Wang, X. (2016) Numb Protects
Human Renal Tubular Epithelial Cells From Bovine Serum Albumin-Induced Apoptosis
Through Antagonizing CHOP/PERK Pathway. Journal of cellular biochemistry 117,
163-171
79. Zhu, X., Zelmer, A., Kapfhammer, J. P., and Wellmann, S. (2016) Cold-inducible RBM3
inhibits PERK phosphorylation through cooperation with NF90 to protect cells from
endoplasmic reticulum stress. FASEB journal : official publication of the Federation of
American Societies for Experimental Biology 30, 624-634
80. Sanson, M., Auge, N., Vindis, C., Muller, C., Bando, Y., Thiers, J. C., Marachet, M. A.,
Zarkovic, K., Sawa, Y., Salvayre, R., and Negre-Salvayre, A. (2009) Oxidized low-
density lipoproteins trigger endoplasmic reticulum stress in vascular cells: prevention by
oxygen-regulated protein 150 expression. Circulation research 104, 328-336
81. Oommen, D., and Prise, K. M. (2013) Down-regulation of PERK enhances resistance to
ionizing radiation. Biochemical and biophysical research communications 441, 31-35

123
82. Shiraishi, H., Okamoto, H., Yoshimura, A., and Yoshida, H. (2006) ER stress-induced
apoptosis and caspase-12 activation occurs downstream of mitochondrial apoptosis
involving Apaf-1. Journal of cell science 119, 3958-3966
83. Urano, F., Wang, X., Bertolotti, A., Zhang, Y., Chung, P., Harding, H. P., and Ron, D.
(2000) Coupling of stress in the ER to activation of JNK protein kinases by
transmembrane protein kinase IRE1. Science 287, 664-666
84. Chen, L., Xu, S., Liu, L., Wen, X., Xu, Y., Chen, J., and Teng, J. (2014) Cab45S inhibits
the ER stress-induced IRE1-JNK pathway and apoptosis via GRP78/BiP. Cell Death Dis.
5, e1219
85. Kim, J. H., Gurumurthy, C. B., Band, H., and Band, V. (2010) Biochemical
characterization of human Ecdysoneless reveals a role in transcriptional regulation. Biol.
Chem. 391, 9-19
86. Dash, S., Chava, S., Aydin, Y., Chandra, P. K., Ferraris, P., Chen, W., Balart, L. A., Wu,
T., and Garry, R. F. (2016) Hepatitis C Virus Infection Induces Autophagy as a
Prosurvival Mechanism to Alleviate Hepatic ER-Stress Response. Viruses 8
87. Cao, S. S., and Kaufman, R. J. (2014) Endoplasmic reticulum stress and oxidative stress
in cell fate decision and human disease. Antioxidants & redox signaling 21, 396-413
88. Bhandary, B., Marahatta, A., Kim, H. R., and Chae, H. J. (2012) An involvement of
oxidative stress in endoplasmic reticulum stress and its associated diseases. International
journal of molecular sciences 14, 434-456
89. Bunpo, P., Dudley, A., Cundiff, J. K., Cavener, D. R., Wek, R. C., and Anthony, T. G.
(2009) GCN2 protein kinase is required to activate amino acid deprivation responses in
mice treated with the anti-cancer agent L-asparaginase. J. Biol. Chem. 284, 32742-32749
90. Dey, P., Rachagani, S., Chakraborty, S., Singh, P. K., Zhao, X., Gurumurthy, C. B.,
Anderson, J. M., Lele, S., Hollingsworth, M. A., Band, V., and Batra, S. K. (2012)
Overexpression of ecdysoneless in pancreatic cancer and its role in oncogenesis by
regulating glycolysis. Clin. Cancer Res. 18, 6188-6198
91. Claudius, A. K., Romani, P., Lamkemeyer, T., Jindra, M., and Uhlirova, M. (2014)
Unexpected role of the steroid-deficiency protein ecdysoneless in pre-mRNA splicing.
PLoS genetics 10, e1004287
92. Visioli, F., Wang, Y., Alam, G. N., Ning, Y., Rados, P. V., Nor, J. E., and Polverini, P. J.
(2014) Glucose-regulated protein 78 (Grp78) confers chemoresistance to tumor
endothelial cells under acidic stress. PloS one 9, e101053
93. Fu, Y., Li, J., and Lee, A. S. (2007) GRP78/BiP inhibits endoplasmic reticulum BIK and
protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer
Res. 67, 3734-3740
94. Wang, M., Ye, R., Barron, E., Baumeister, P., Mao, C., Luo, S., Fu, Y., Luo, B., Dubeau,
L., Hinton, D. R., and Lee, A. S. (2010) Essential role of the unfolded protein response
regulator GRP78/BiP in protection from neuronal apoptosis. Cell Death Differ. 17, 488-
498
95. Lee, H. K., Xiang, C., Cazacu, S., Finniss, S., Kazimirsky, G., Lemke, N., Lehman, N. L.,
Rempel, S. A., Mikkelsen, T., and Brodie, C. (2008) GRP78 is overexpressed in
glioblastomas and regulates glioma cell growth and apoptosis. Neuro-oncology 10, 236-
243
96. Chang, Y. J., Chen, W. Y., Huang, C. Y., Liu, H. H., and Wei, P. L. (2015) Glucose-
regulated protein 78 (GRP78) regulates colon cancer metastasis through EMT biomarkers
and the NRF-2/HO-1 pathway. Tumour biology : the journal of the International Society
for Oncodevelopmental Biology and Medicine 36, 1859-1869
97. Xing, X., Li, Y., Liu, H., Wang, L., and Sun, L. (2011) Glucose regulated protein 78
(GRP78) is overexpressed in colorectal carcinoma and regulates colorectal carcinoma cell
growth and apoptosis. Acta histochemica 113, 777-782

124
98. Daneshmand, S., Quek, M. L., Lin, E., Lee, C., Cote, R. J., Hawes, D., Cai, J., Groshen,
S., Lieskovsky, G., Skinner, D. G., Lee, A. S., and Pinski, J. (2007) Glucose-regulated
protein GRP78 is up-regulated in prostate cancer and correlates with recurrence and
survival. Human pathology 38, 1547-1552
99. Xing, X., Lai, M., Wang, Y., Xu, E., and Huang, Q. (2006) Overexpression of glucose-
regulated protein 78 in colon cancer. Clinica chimica acta; international journal of
clinical chemistry 364, 308-315
100. Fernandez, P. M., Tabbara, S. O., Jacobs, L. K., Manning, F. C., Tsangaris, T. N.,
Schwartz, A. M., Kennedy, K. A., and Patierno, S. R. (2000) Overexpression of the
glucose-regulated stress gene GRP78 in malignant but not benign human breast lesions.
Breast cancer research and treatment 59, 15-26
101. Wang, M., and Kaufman, R. J. (2014) The impact of the endoplasmic reticulum protein-
folding environment on cancer development. Nature reviews. Cancer 14, 581-597
102. Nagelkerke, A., Bussink, J., Mujcic, H., Wouters, B. G., Lehmann, S., Sweep, F. C., and
Span, P. N. (2013) Hypoxia stimulates migration of breast cancer cells via the
PERK/ATF4/LAMP3-arm of the unfolded protein response. Breast Cancer Res. 15, R2

125
CHAPTER 3: Summary and Conclusion

126
ECD protein has been known for many years as an important cell growth regulatory
protein. The overexpression of ECD in cancer, including breast, prostate and pancreatic
cancers, strongly suggests oncogenic roles for ECD. In this study, we presented evidence
for an involvement of ECD in ER stress in cancer. Normal or cancer cell lines with low
levels of ECD exhibited increased ER stress and cell death upon ER stress induction.
Conversely, ECD overexpressing cells exhibited a decreased ER stress and cell death upon
ER stress. Together, these distinct effects of ECD knock down or overexpression on cells
argue that ECD may be functioning as a buffering protein against stress in cells and as a
regulator of sensitivity of cells to ER stress. The increase in ECD mRNA upon ER stress
indicates a transcriptional induction of ECD and further suggests an ER stress-related role.
The functional connection of ECD to the PERK-eIF2α arm of the ER stress signaling
further argues in favor of the pro-survival roles for ECD in stress conditions, since that axis
mediates cell death during ER stress signaling via CHOP induction (1-8). ECD inhibits
PERK-eIF2α signaling while PERK-eIF2α inhibits ECD protein translation, thus
establishing a negative feedback loop between ECD and the PERK arm of the ER stress
signaling.
Mechanistically, ECD may be exerting its effects by multiple ways. Particularly,
the ECD-GRP78 axis is a novel pathway for regulating ER stress. ECD enhances the level
of GRP78, a key chaperone and inhibitor of PERK signaling. As increased expression of
GRP78 and other chaperones is known to promote the clearing of ER stress-causing
unfolded protein load and reduce the activation of UPR sensors (9-12), this increase in
chaperones’ levels in the presence of ECD upon ER stress induction argues that ECD may
be attenuating stress by elevating chaperone levels and enhancing the folding capacity in

127
the stressed ER. Indeed, a disruption of the chaperone-enhancing effect of ECD abrogated
the attenuating effect of ECD on PERK signaling even in the presence of overexpressed
ECD. Notably, GRP78 is required for cell survival not only in response to ER stress (13-
16) but also in other stressful and hostile conditions such as glucose deficiency encountered
in tumor microenvironments (17-22). Given that the PERK arm of the UPR is also activated
in cancer (8,23,24) and both ECD and GRP78 are overexpressed in cancer (17-22,25,26),
we suggest that ECD overexpression may play a similar role to mitigate the negative
consequences of elevated PERK signaling found in cancer. Given the mechanistic link
between ECD and GRP78 in inhibiting the PERK pathway to promote cell survival, it
would be of great interest to explore if GRP78 and ECD are co-overexpressed in tumors
that use the UPR to promote tumor cell survival and hence may be suitable targets for
therapeutic agents directed against ER stress in cancer.
Conclusion:
• Stress induction by multiple stimuli, Thapsigargin, Tunicamycin, glucose
starvation, H2O2, NOX4 overexpression, DTT, and sorbitol, led to a reduced ECD
protein in multiple cell lines (MCF7, MDA-468, MCF10A, Panc-1, 76NTERT,
MDA231-NOX4).
• ECD mRNA increased upon Thapsigargin treatment.
• In PERK KO MREFs, the levels of ECD protein did not change upon ER stress
induction as compared to WT PERK.

128
• In PERK KO MEFs, ECD mRNA levels failed to increase as compared to WT
PERK.
• PERK does not phosphorylate ECD.
• The decrease in ECD protein levels is via the PERK-eIF2α axis of the ER stress
signaling.
• Loss of ECD in cells increased the levels of p-PERK, p-eIF2α, and these effects
were enhanced upon ER stress induction by Thapsigargin treatment or glucose
deprivation along with an increased in CHOP mRNA, and cleaved caspase 3.
• Loss of ECD slightly increased the levels of spliced XBP-1, the downstream target
of IRE1α but cleaved ATF6 did not significantly change.
• Loos of ECD correlated with a decrease in the colony formation of ECD-null cells
upon ER stress induction.
• Overexpression of ECD led to a marked decrease in p-PERK and its downstream
targets, p-eIF2α, ATF4 and CHOP mRNA levels upon ER stress induction
• Overexpression of ECD upon ER stress induction slightly increased the levels of
spliced XBP1 but ATF6 did not change significantly.
• Overexpression of ECD upon ER stress induction correlated with a decrease in
caspase 3 cleavage and an increase in the colony formation of ECD overexpressing
cells
• ECD localized and associated with PERK, ATF6, IRE1α and GRP78.
• ECD did not modulate enzymatic activity of PERK toward eIF2α.
• Depletion of ECD upon ER stress induction was associated with decreased GRP78
protein levels.

129
• Overexpression of ECD upon ER stress induction increased the levels of GRP78
protein.
• GRP78 mRNA was not affected by deletion or overexpression of ECD upon ER
stress induction.
• Knock down of ECD followed by cycloheximide treatment was associated with a
faster decrease in GRP78 protein level as compared to control cells with intact
cellular ECD levels.
• ECD overexpression plus cycloheximide treatment correlated with a slower
decrease in GRP78 protein levels.
• A knock down of GRP78 abrogated the attenuating effect of ECD on PERK, even
in the presence of overexpressed ECD. This effect further correlated with a decrease
in the colony formation of ECD overexpressing cells.

130
REFERENCES
1. Harding, H. P., Novoa, I., Zhang, Y., Zeng, H., Wek, R., Schapira, M., and Ron, D. (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099-1108
2. Ma, Y., Brewer, J. W., Diehl, J. A., and Hendershot, L. M. (2002) Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J. Mol. Biol. 318, 1351-1365
3. Oyadomari, S., Koizumi, A., Takeda, K., Gotoh, T., Akira, S., Araki, E., and Mori, M. (2002) Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. The Journal of clinical investigation 109, 525-532
4. Song, B., Scheuner, D., Ron, D., Pennathur, S., and Kaufman, R. J. (2008) Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. The Journal of clinical investigation 118, 3378-3389
5. Tabas, I., and Ron, D. (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 13, 184-190
6. Thorp, E., Li, G., Seimon, T. A., Kuriakose, G., Ron, D., and Tabas, I. (2009) Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe-/- and Ldlr-/- mice lacking CHOP. Cell metabolism 9, 474-481
7. Zinszner, H., Kuroda, M., Wang, X., Batchvarova, N., Lightfoot, R. T., Remotti, H., Stevens, J. L., and Ron, D. (1998) CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 12, 982-995
8. Bu, Y., and Diehl, J. A. (2016) PERK Integrates Oncogenic Signaling and Cell Survival During Cancer Development. Journal of cellular physiology 231, 2088-2096
9. Bertolotti, A., Zhang, Y., Hendershot, L. M., Harding, H. P., and Ron, D. (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2, 326-332
10. Gardner, B. M., Pincus, D., Gotthardt, K., Gallagher, C. M., and Walter, P. (2013) Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harbor perspectives in biology 5, a013169
11. Lai, E., Teodoro, T., and Volchuk, A. (2007) Endoplasmic reticulum stress: signaling the unfolded protein response. Physiology 22, 193-201
12. Lee, S., Min Kim, S., Dotimas, J., Li, L., Feener, E. P., Baldus, S., Myers, R. B., Chutkow, W. A., Patwari, P., Yoshioka, J., and Lee, R. T. (2014) Thioredoxin-interacting protein regulates protein disulfide isomerases and endoplasmic reticulum stress. EMBO Mol. Med. 6, 732-743
13. Rao, R. V., Peel, A., Logvinova, A., del Rio, G., Hermel, E., Yokota, T., Goldsmith, P. C., Ellerby, L. M., Ellerby, H. M., and Bredesen, D. E. (2002) Coupling endoplasmic reticulum stress to the cell death program: role of the ER chaperone GRP78. FEBS Lett. 514, 122-128
14. Visioli, F., Wang, Y., Alam, G. N., Ning, Y., Rados, P. V., Nor, J. E., and Polverini, P. J. (2014) Glucose-regulated protein 78 (Grp78) confers chemoresistance to tumor endothelial cells under acidic stress. PloS one 9, e101053
15. Fu, Y., Li, J., and Lee, A. S. (2007) GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 67, 3734-3740
16. Wang, M., Ye, R., Barron, E., Baumeister, P., Mao, C., Luo, S., Fu, Y., Luo, B., Dubeau, L., Hinton, D. R., and Lee, A. S. (2010) Essential role of the unfolded protein response regulator GRP78/BiP in protection from neuronal apoptosis. Cell Death Differ. 17, 488-498

131
17. Lee, H. K., Xiang, C., Cazacu, S., Finniss, S., Kazimirsky, G., Lemke, N., Lehman, N. L., Rempel, S. A., Mikkelsen, T., and Brodie, C. (2008) GRP78 is overexpressed in glioblastomas and regulates glioma cell growth and apoptosis. Neuro-oncology 10, 236-243
18. Chang, Y. J., Chen, W. Y., Huang, C. Y., Liu, H. H., and Wei, P. L. (2015) Glucose-regulated protein 78 (GRP78) regulates colon cancer metastasis through EMT biomarkers and the NRF-2/HO-1 pathway. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine 36, 1859-1869
19. Xing, X., Li, Y., Liu, H., Wang, L., and Sun, L. (2011) Glucose regulated protein 78 (GRP78) is overexpressed in colorectal carcinoma and regulates colorectal carcinoma cell growth and apoptosis. Acta histochemica 113, 777-782
20. Daneshmand, S., Quek, M. L., Lin, E., Lee, C., Cote, R. J., Hawes, D., Cai, J., Groshen, S., Lieskovsky, G., Skinner, D. G., Lee, A. S., and Pinski, J. (2007) Glucose-regulated protein GRP78 is up-regulated in prostate cancer and correlates with recurrence and survival. Human pathology 38, 1547-1552
21. Xing, X., Lai, M., Wang, Y., Xu, E., and Huang, Q. (2006) Overexpression of glucose-regulated protein 78 in colon cancer. Clinica chimica acta; international journal of clinical chemistry 364, 308-315
22. Fernandez, P. M., Tabbara, S. O., Jacobs, L. K., Manning, F. C., Tsangaris, T. N., Schwartz, A. M., Kennedy, K. A., and Patierno, S. R. (2000) Overexpression of the glucose-regulated stress gene GRP78 in malignant but not benign human breast lesions. Breast cancer research and treatment 59, 15-26
23. Wang, M., and Kaufman, R. J. (2014) The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nature reviews. Cancer 14, 581-597
24. Nagelkerke, A., Bussink, J., Mujcic, H., Wouters, B. G., Lehmann, S., Sweep, F. C., and Span, P. N. (2013) Hypoxia stimulates migration of breast cancer cells via the PERK/ATF4/LAMP3-arm of the unfolded protein response. Breast Cancer Res. 15, R2
25. Dey, P., Rachagani, S., Chakraborty, S., Singh, P. K., Zhao, X., Gurumurthy, C. B., Anderson, J. M., Lele, S., Hollingsworth, M. A., Band, V., and Batra, S. K. (2012) Overexpression of ecdysoneless in pancreatic cancer and its role in oncogenesis by regulating glycolysis. Clin. Cancer Res. 18, 6188-6198
26. Zhao, X., Mirza, S., Alshareeda, A., Zhang, Y., Gurumurthy, C. B., Bele, A., Kim, J. H., Mohibi, S., Goswami, M., Lele, S. M., West, W., Qiu, F., Ellis, I. O., Rakha, E. A., Green, A. R., Band, H., and Band, V. (2012) Overexpression of a novel cell cycle regulator ecdysoneless in breast cancer: a marker of poor prognosis in HER2/neu-overexpressing breast cancer patients. Breast Cancer Res. Treat. 134, 171-180

132
CHAPTER 4: Future Directions

133
• Characterize ER stress elements (ERSE) in ECD gene promoter
The increase in ECD mRNA upon ER stress induction suggests existence of
ERSE in ECD gene promoter. A follow-up study to characterize these ERSE
would be interesting.
• Assess potential roles of ECD in autophagy through GRP78
In addition to further examining the ECD-GRP78 connection in tumor context, it
would also be interesting to assess whether ECD pro-survival effect upon ER stress
is via GRP78-mediated autophagy. The rationale is that ECD enhanced GRP78
while GRP78 has been implicated in autophagy. Although a clear mechanism was
not presented, it was reported in numerous studies that GRP78 was required for
autophagy induction and that inhibition of GRP78 inhibited autophagy induction
(1-3).
• Assess potential ECD-R2TP connection in regulating ER stress
The R2TPcomplex, composed of RUVBL1, RUVBL2, PIH1D1 and RPAP3 is a
multi-protein complex which regulates the assembly and stability of many
complexes (4). A study identified a role for RUVBL2 in the regulation of ATF2 in
response to stress through direct interaction with ATF2 (5). ECD is a component
of the R2TP complex (6,7). ECD interacts with the RUVBL1 component of the

134
R2TP complex, an interaction required for ECD roles in cell growth (33). It is
reported that knock of RUVBL1 affected cellular level of GRP78 an induced ER
stress (8). Therefore, it would be curious to examine the potential ECD-R2TP
connection in ER stress regulation, especially as ECD overexpression correlated
with enhanced of both GRP78 and RUVBL1 upon ER stress induction (Figure 4.1
below) .
Figure 4.1: ECD overexpression correlates with enhancement with RUVBL1 and
GRP78. ECD overexpressing and their control MEFs were treated with Tunicamycin and
protein lysate was prepared at indicated time points followed by western blot with indicated
antibodies.
ECD
Tun: - 3h 6h 12h 24h - 3h 6h 12h 24h
Tet(O)-Flag- hECD -IRES eGFP; rtTA
Tet(O)-Flag- hECD -IRES eGFP
GRP78
RUVBL1
β-actin

135
• Assess the requirement of ECD in chaperone (GRP78, GRP94)-mediated ER
calcium regulation. The ER is well known as a site for calcium storage in cells.
ECD regulates the levels of the critical chaperone proteins (GRP78 and GRP94)
that mediate that function of the ER (9,10).
• Assess the roles of ECD in protein translation through GRP78. GRP78 is
known to be involved in protein translation not only by binding nascent protein in
the ER but importantly by forming the translocation channel in complex with Sec61
(11,12).
• Explore in vivo studies of the ECD-GRP78 axis in regulating oncogenesis-
induced ER stress in mouse models. Works in the lab indicated potential
oncogenic roles of ECD, as its overexpression induced tumors. Tumor initiation
and progression are complex processes characterized by ER stress as a result of the
increased bioenergetics demands in the tumor. Overcoming this early ER stress is
paramount for tumor progression. It would be interesting to assess whether the
ECD-mediated upregulation in the levels of GRP78 is required for tumor initiation
and progression.

136
REFERENCES
1. Zhang, X. Y., Zhang, T. T., Song, D. D., Zhou, J., Han, R., Qin, Z. H., and Sheng, R.
(2015) Endoplasmic reticulum chaperone GRP78 is involved in autophagy activation induced by ischemic preconditioning in neural cells. Molecular brain 8, 20
2. Cook, K. L., Shajahan, A. N., Warri, A., Jin, L., Hilakivi-Clarke, L. A., and Clarke, R. (2012) Glucose-regulated protein 78 controls cross-talk between apoptosis and autophagy to determine antiestrogen responsiveness. Cancer Res. 72, 3337-3349
3. Li, J., Ni, M., Lee, B., Barron, E., Hinton, D. R., and Lee, A. S. (2008) The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ. 15, 1460-1471
4. Horejsi, Z., Takai, H., Adelman, C. A., Collis, S. J., Flynn, H., Maslen, S., Skehel, J. M., de Lange, T., and Boulton, S. J. (2010) CK2 phospho-dependent binding of R2TP complex to TEL2 is essential for mTOR and SMG1 stability. Mol. Cell 39, 839-850
5. Cho, S. G., Bhoumik, A., Broday, L., Ivanov, V., Rosenstein, B., and Ronai, Z. (2001) TIP49b, a regulator of activating transcription factor 2 response to stress and DNA damage. Mol. Cell. Biol. 21, 8398-8413
6. Horejsi, Z., Stach, L., Flower, T. G., Joshi, D., Flynn, H., Skehel, J. M., O'Reilly, N. J., Ogrodowicz, R. W., Smerdon, S. J., and Boulton, S. J. (2014) Phosphorylation-dependent PIH1D1 interactions define substrate specificity of the R2TP cochaperone complex. Cell reports 7, 19-26
7. Mir, R. A., Bele, A., Mirza, S., Srivastava, S., Olou, A. A., Ammons, S. A., Kim, J. H., Gurumurthy, C. B., Qiu, F., Band, H., and Band, V. (2015) A Novel Interaction of Ecdysoneless (ECD) Protein with R2TP Complex Component RUVBL1 Is Required for the Functional Role of ECD in Cell Cycle Progression. Mol. Cell. Biol. 36, 886-899
8. Yuan, X. S., Wang, Z. T., Hu, Y. J., Bao, F. C., Yuan, P., Zhang, C., Cao, J. L., Lv, W., and Hu, J. (2016) Downregulation of RUVBL1 inhibits proliferation of lung adenocarcinoma cells by G1/S phase cell cycle arrest via multiple mechanisms. Tumour Biol.
9. Lamb, H. K., Mee, C., Xu, W., Liu, L., Blond, S., Cooper, A., Charles, I. G., and Hawkins, A. R. (2006) The affinity of a major Ca2+ binding site on GRP78 is differentially enhanced by ADP and ATP. J. Biol. Chem. 281, 8796-8805
10. Schauble, N., Lang, S., Jung, M., Cappel, S., Schorr, S., Ulucan, O., Linxweiler, J., Dudek, J., Blum, R., Helms, V., Paton, A. W., Paton, J. C., Cavalie, A., and Zimmermann, R. (2012) BiP-mediated closing of the Sec61 channel limits Ca2+ leakage from the ER. EMBO J. 31, 3282-3296
11. Alder, N. N., Shen, Y., Brodsky, J. L., Hendershot, L. M., and Johnson, A. E. (2005) The molecular mechanisms underlying BiP-mediated gating of the Sec61 translocon of the endoplasmic reticulum. J. Cell Biol. 168, 389-399
12. Lang, S., Benedix, J., Fedeles, S. V., Schorr, S., Schirra, C., Schauble, N., Jalal, C., Greiner, M., Hassdenteufel, S., Tatzelt, J., Kreutzer, B., Edelmann, L., Krause, E., Rettig, J., Somlo, S., Zimmermann, R., and Dudek, J. (2012) Different effects of Sec61alpha, Sec62 and Sec63 depletion on transport of polypeptides into the endoplasmic reticulum of mammalian cells. J. Cell Sci. 125, 1958-1969

137
Chapter 5: Appendix
1. Appolinaire A. Olou, Aniruddha Sarkar, Aditya Bele, C.B. Gurumurthy, Riyaz A. Mir, Shalis A. Ammons, Sameer Mirza, Irfana Saleem, Fumihiko Urano, Hamid Band and Vimla Band. Mammalian ECD protein is a novel negative regulator of the PERK arm of unfolded protein response. 2017. Mol. Cell. Biol.
2. Riyaz A. Mir, Aditya Bele, Sameer Mirza, Shashank Srivastava, Appolinaire A. Olou, Shalis A. Ammons, Jun Hyun Kim, Channabasavaiah B. Gurumurthy, Fang Qiu, Hamid Band, Vimla Band. A Novel Interaction of Ecdysoneless (ECD) Protein with R2TP Complex Component RUVBL1 Is Required for the Functional Role of ECD in Cell Cycle Progression. 2015. Mol. Cell. Biol.

Fig. 1:
B
Tun: 0h 12h 24h
ECD
p-eIF2α
β-actin
MCF10A
Tg, h: 0h 12h 24h
A
β-actin
p-eIF2α
ECD
MCF10A Time, h: 0 12 24 48 72
Glucose starvation
ECD
β-actin
p-eIF2α
C Panc-1
0
10
20
30
40
50CHOP mRNA
Tg: 0h 8h 12h 24h
Rel
ativ
e m
RN
A
D
G
Tg: - + - +
WT KO
ECD
β-actin
p-PERK
PERK
Lane: 1 2 3 4
ECD
p-eIF2α
β-actin
- Tg Tun - Tg Tun
Wild-type Phospho-deficient eIF2α J
Lane: 1 2 3 4 5 6
0
0.5
1
1.5
2
2.5
3
3.5
PERK WT
PERK KO
Rel
ativ
e m
RN
A
UT 7h 14h
ECD mRNA
I
*
0
5
10
15
20
25
30
35
40CHOP mRNA
PERK WT
PERK KO
UT 7h 14h
Rel
ativ
e m
RN
A
H
0
1
2
3
4
5
6
7
8
9
Tg: 0h 8h 12h 24h R
elat
ive
mR
NA
E
*
*
ECD mRNA
Rel
ativ
e m
RN
A
F ECD mRNA
Glucose starvation: 0h 12h 24h 48h 72h
0
0.5
1
1.5
2
2.5
3
3.5
4
MCB Accepted Manuscript Posted Online 26 June 2017Mol. Cell. Biol. doi:10.1128/MCB.00030-17Copyright © 2017 Olou et al.This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International license.
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

Fig. 2:
C
A
PERK GRP78 DAPI MERGE
B
ECD
ECD
PERK
GRP78
DAPI
DAPI
MERGE
MERGE
Merged DAPI Ms IgG Ms IgG
DAPI Merged Rb IgG Rb IgG
D
E
WCL
Soluble
Fraction Microsomal
Fraction
GRP78
GAPDH
ECD
Ex
per
imen
t 1
Ex
per
imen
t 2
Ex
per
imen
t 3
SERCA2
RCAS1
PERK
Ex
per
imen
t 1
Ex
per
imen
t 2
Ex
per
imen
t 3
Ex
per
imen
t 1
Ex
per
imen
t 2
Ex
per
imen
t 3
F
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

Fig. 2: Continued
DAPI PLA signal
ECD
+
PIH1D1
MERGED
ECD
+
GRP78
ECD
+
PERK
IgG
G
H
I
J
Tg: - + - +
Input IgG
ECD
GRP78
PERK
ECD
Tg: - + - +
Input IP: ECD IgG
PERK
GRP78
PIH1D1
K
IP: GRP78
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from
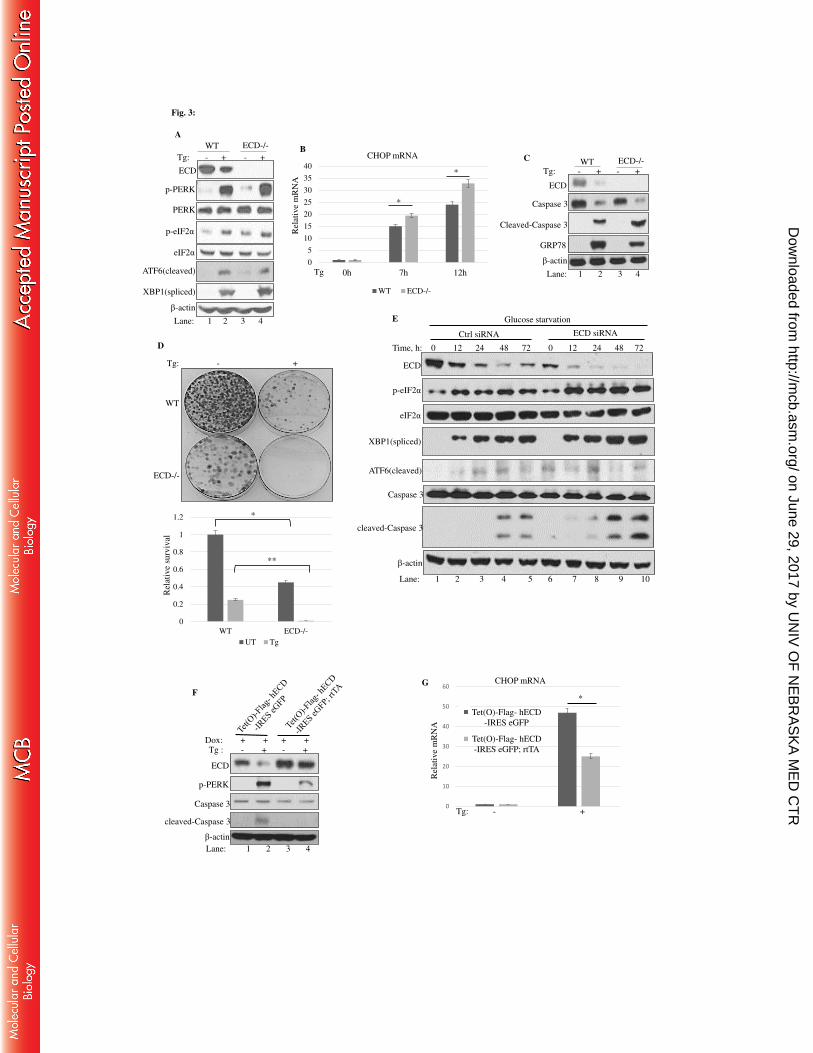
Fig. 3:
Tg: - + - +
Lane: 1 2 3 4
ECD
p-PERK
PERK
p-eIF2α
eIF2α
ATF6(cleaved)
XBP1(spliced)
β-actin
WT ECD-/-
A
Tg: - + - +
ECD
Caspase 3
Cleaved-Caspase 3
β-actin
C
GRP78
WT ECD-/-
Lane: 1 2 3 4
D
Tg: - +
WT
ECD-/-
0
0.2
0.4
0.6
0.8
1
1.2
WT ECD-/-
UT Tg
Rel
ativ
e su
rviv
al
*
**
0
5
10
15
20
25
30
35
40
0h 7h 12h
WT ECD-/-
B
Rel
ativ
e m
RN
A
Tg
CHOP mRNA
*
*
cleaved-Caspase 3
Time, h: 0 12 24 48 72 0 12 24 48 72
p-eIF2α
ECD
β-actin
XBP1(spliced)
eIF2α
ECD siRNA Ctrl siRNA
ATF6(cleaved)
Caspase 3
E
Lane: 1 2 3 4 5 6 7 8 9 10
Glucose starvation
F
Caspase 3
cleaved-Caspase 3
Tg : - + - + Dox: + + + +
p-PERK
ECD
β-actin
Lane: 1 2 3 4
0
10
20
30
40
50
60
Rel
ativ
e m
RN
A
Tet(O)-Flag- hECD
-IRES eGFP
Tet(O)-Flag- hECD
-IRES eGFP; rtTA
CHOP mRNA
Tg: - +
*
G
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from
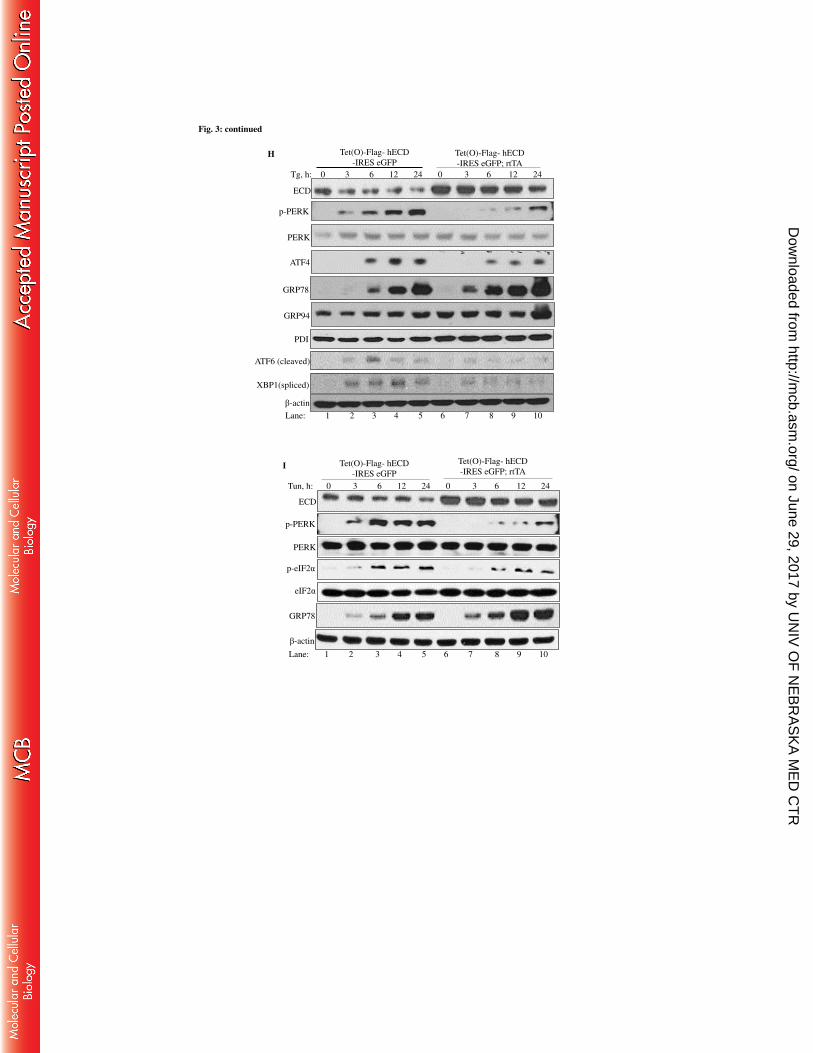
Fig. 3: continued
p-PERK
PERK
ATF4
GRP78
ATF6 (cleaved)
XBP1(spliced)
Tg, h: 0 3 6 12 24 0 3 6 12 24
GRP94
Tet(O)-Flag- hECD
-IRES eGFP; rtTA
Tet(O)-Flag- hECD
-IRES eGFP
β-actin
H
Lane: 1 2 3 4 5 6 7 8 9 10
ECD
PDI
Tun, h: 0 3 6 12 24 0 3 6 12 24
Tet(O)-Flag- hECD
-IRES eGFP; rtTA
Tet(O)-Flag- hECD
-IRES eGFP
ECD
p-PERK
PERK
p-eIF2α
eIF2α
β-actin
GRP78
I
Lane: 1 2 3 4 5 6 7 8 9 10
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from
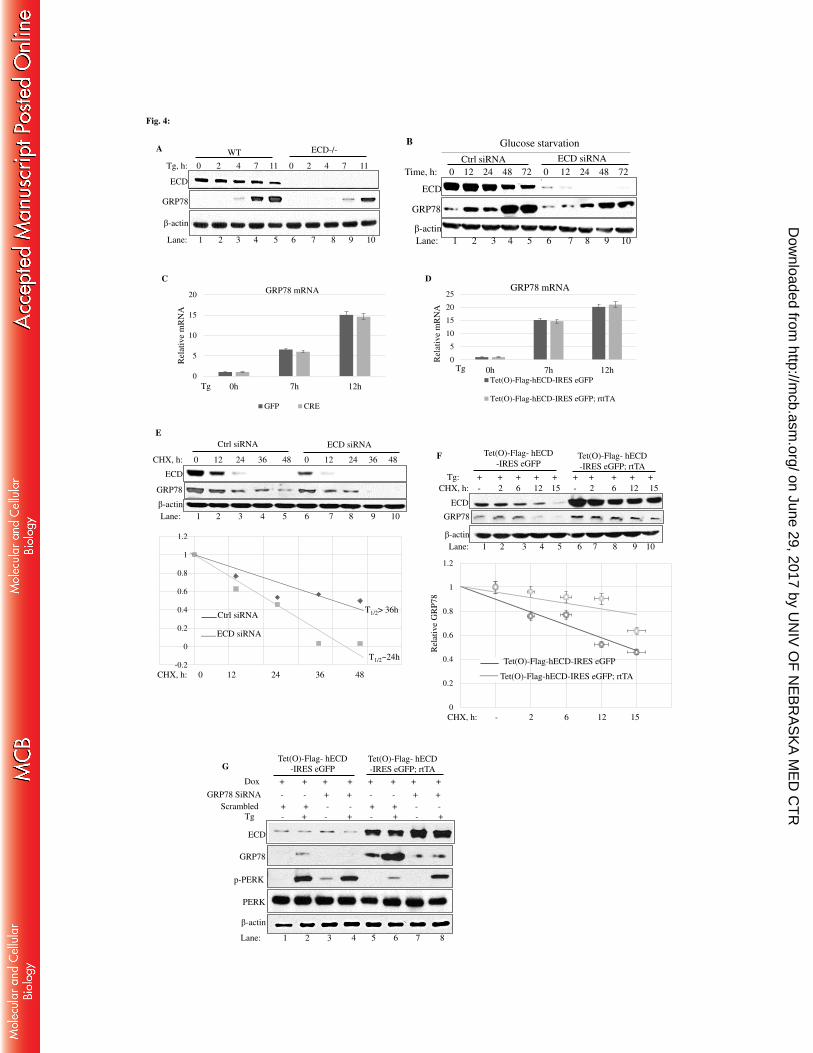
Tg, h: 0 2 4 7 11 0 2 4 7 11
ECD
GRP78
β-actin
WT ECD-/-
Lane: 1 2 3 4 5 6 7 8 9 10
A
Fig. 4:
ECD siRNA Ctrl siRNA
Time, h: 0 12 24 48 72 0 12 24 48 72
GRP78
β-actin
Lane: 1 2 3 4 5 6 7 8 9 10
ECD
B Glucose starvation
0
5
10
15
20
0h 7h 12h
GFP CRE
Rel
ativ
e m
RN
A
GRP78 mRNA
Tg
C
0
5
10
15
20
25
0h 7h 12h
GRP78 mRNA
Tet(O)-Flag-hECD-IRES eGFP
Tet(O)-Flag-hECD-IRES eGFP; rttTA
D
Rel
ativ
e m
RN
A
Tg
GRP78
CHX, h: 0 12 24 36 48 0 12 24 36 48
Ctrl siRNA ECD siRNA
ECD
β-actin
E
Lane: 1 2 3 4 5 6 7 8 9 10
-0.2
0
0.2
0.4
0.6
0.8
1
1.2
Ctrl siRNA
ECD siRNA
CHX, h: 0 12 24 36 48
T1/2> 36h
T1/2~24h
β-actin
Tg: + + + + + + + + + +
CHX, h: - 2 6 12 15 - 2 6 12 15
GRP78
ECD
F
Lane: 1 2 3 4 5 6 7 8 9 10
Tet(O)-Flag- hECD
-IRES eGFP; rtTA
Tet(O)-Flag- hECD
-IRES eGFP
0
0.2
0.4
0.6
0.8
1
1.2
Tet(O)-Flag-hECD-IRES eGFP
Tet(O)-Flag-hECD-IRES eGFP; rtTA
Rel
ativ
e G
RP
78
CHX, h: - 2 6 12 15
Tg - + - + - + - +
Scrambled + + - - + + - -
GRP78 SiRNA - - + + - - + +
p-PERK
Dox + + + + + + + +
ECD
GRP78
β-actin
G
Lane: 1 2 3 4 5 6 7 8
PERK
Tet(O)-Flag- hECD
-IRES eGFP; rtTA
Tet(O)-Flag- hECD
-IRES eGFP
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

Fig. 5:
Tg (nM): - 50 100 - 50 100
ECD
β-actin
Caspase 3 (total)
cleaved-Caspase 3
Lane: 1 2 3 4 5 6
Dox: + + + + + +
A Tet(O)-Flag- hECD
-IRES eGFP; rtTA
Tet(O)-Flag- hECD
-IRES eGFP
Tg: - 50nM 100nM
C Dox: + + +
Tet(O)-Flag- hECD
-IRES eGFP; rtTA
Tet(O)-Flag- hECD
-IRES eGFP
0
10
20
30
40
50
60
70
80
0h 50nM 100nM
CHOP mRNA
Rel
ativ
e m
RN
A
Tg:
B
Tet(O)-Flag- hECD
-IRES eGFP
Tet(O)-Flag- hECD
-IRES eGFP; rtTA
*
0
0.2
0.4
0.6
0.8
1
1.2
Rel
ativ
e su
rviv
al
Tet(O)-Flag- hECD
-IRES eGFP
Tet(O)-Flag- hECD
-IRES eGFP; rtTA
**
Tg: - 50 nM 100 nM
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

1
Mammalian ECD protein is a novel negative regulator of the PERK arm of unfolded -1
protein response 2
3
Appolinaire A. Olou1, Aniruddha Sarkar1, Aditya Bele1, C.B. Gurumurthy1, Riyaz A. Mir1, 4
Shalis A. Ammons1, Sameer Mirza1, Irfana Saleem2, Fumihiko Urano6,7, Hamid Band1;3-5 and 5
Vimla Band1,5# 6
7
Departments of 1Genetics, Cell Biology and Anatomy, 2Biochemistry and Molecular Biology 8
3Pathology & Microbiology, College of Medicine; ,4Eppley Institute for Research in Cancer and 9
Allied Diseases; and 5Fred & Pamela Buffett Cancer Center, University of Nebraska Medical 10
Center, 985805 Nebraska Medical Center, Omaha, NE 68198, 6Division of Endocrinology, 11
Metabolism and Lipid Research, and 7Department of Pathology, Washington University School 12
of Medicine, St. Louis, Missouri. 13
14
Running Title: ECD modulates PERK pathway 15
# Corresponding author: Email: [email protected]; Phone: (402)-559-8565; Fax: (402)-559-7328 16
Keywords: ECD, ER, PERK, UPR, Cell survival, GRP78 17
Word count: 18
Material and Method: 765 19
Introduction, Results and Discussion: 4368 20
21
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

2
22
ABSTRACT 23
The mammalian Ecdysoneless (ECD) is a highly-conserved ortholog of the Drosophila Ecd gene 24
product whose mutations impair the synthesis of Ecdysone and produce cell-autonomous 25
survival defects but mechanisms by which ECD functions are largely unknown. Here, we present 26
evidence that ECD regulates endoplasmic reticulum (ER) stress response. ER stress induction led 27
to a reduced ECD protein but this effect was not seen in PERK KO or phospho-deficient eIF2α 28
MEFs; moreover, ECD mRNA levels were increased, suggesting impaired ECD translation as 29
the mechanism for reduced protein levels. ECD co-localizes and co-immunoprecipitates with 30
PERK and GRP78. ECD depletion increased the levels of p-PERK, p-eIF2α, and these effects 31
were enhanced upon ER stress induction. Reciprocally, overexpression of ECD led to a marked 32
decrease in p-PERK, p-eIF2α and ATF4 levels, but a robust increase in GRP78 protein levels. 33
However, GRP78 mRNA levels were unchanged, suggesting a post-transcriptional event. 34
Knockdown of GRP78 reversed the attenuating effect of ECD over-expression on PERK 35
signaling. Significantly, overexpression of ECD provided a survival advantage to cells upon ER 36
stress induction. Taken together, we demonstrate that ECD promotes survival upon ER stress by 37
increasing GRP78 protein levels to enhance the adaptive folding protein in the ER to attenuate 38
PERK signaling. 39
40
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

3
INTRODUCTION 41
The endoplasmic reticulum (ER) is a central subcellular organelle with essential roles in the 42
synthesis, folding and maturation of secreted and membrane proteins, biogenesis of cholesterol, 43
calcium homeostasis and regulation of survival and apoptosis pathways (1-10). Aberrations in 44
these ER functions are sensed by well-conserved ER transmembrane sensors, Inositol-Requiring 45
Enzyme 1 alpha (IRE1α), PKR-like ER kinase (PERK) and Activating Transcription Factor 6 46
(ATF6), that activate homeostatic signaling pathways collectively referred to as the unfolded 47
protein response (UPR) (11,12). These ER stress sensors exhibit a dynamic and reversible 48
interaction with the ER chaperone GRP78 (13). In un-stressed cells, GRP78 is bound to luminal 49
domains of UPR sensors, which maintains them in an inactive state (14). During ER stress, the 50
increased load of unfolded proteins competes for GRP78 binding, leading to activation of UPR 51
sensors (15) which, through intermediate signaling, evoke overlapping as well as pathway-52
specific responses to restore ER homeostasis and promote cell survival or alternatively eliminate 53
stressed cells through apoptosis if homeostasis cannot be restored. 54
The PERK pathway has emerged as a key pathway in cellular UPR response as well as 55
homeostasis under unstressed conditions and in disease states (16-21). Release of GRP78 from 56
PERK leads to its dimerization, auto-phosphorylation and activation (15,22,23). A major PERK 57
substrate is the eukaryotic translation initiation factor 2 alpha (eIF2α) whose phosphorylation by 58
PERK inactivates it leading to a block in general cap-dependent protein translation and 59
consequent decrease in the protein load entering the ER (24); concurrently, PERK signaling 60
selectively enhances the cap-independent translation of specific mRNAs, such as Activating 61
Transcription Factor 4 (ATF4) (24). ATF4 induces the expression of CCAAT/enhancer-binding 62
protein-homologous protein (CHOP) which promotes apoptosis in response to stress (24-32). 63
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

4
PERK-induced phosphorylation of eIF2α also inhibits cell cycle progression by reducing the 64
levels of cyclins and hence cyclin-dependent kinase 2 (CDK2) activity (33-35). Termination of 65
PERK signaling, together with de-phosphorylation of eIF2α, is required to re-initiate protein 66
synthesis and resume cell cycle. Thus, PERK-mediated UPR leads to a coordinated program of 67
cellular protection and mitigation of stress. However, PERK also contributes to an alternate 68
outcome through CHOP-mediated activation of a cellular death pathway to eliminate severely 69
damaged cells (24-32). Mechanisms to fine-tune the outcomes of UPR pathways are important to 70
mitigate the negative consequences of ER stress. This is of particular importance under 71
conditions where cells experience ER stress as part of their physiological responses, such as 72
antibody-secreting plasma cells or insulin-secreting pancreatic islet cells (36). We have identified 73
ECD as a negative regulator of the PERK-mediated UPR. 74
The Ecd gene was first identified based on genetic mutations in Drosophila that lead to reduced 75
production of the developmentally-regulated steroid hormone Ecdysone, which is synthesized in 76
the ER, hence the designation of ecdysoneless for such fly mutants (37). The mammalian Ecd 77
gene was cloned based on the rescue of growth defects in S. cerevisiae mutant in the growth 78
control regulatory gene 2 (GCR2), a glycolysis regulatory gene (38). Thus, ECD was thought to 79
be involved in mammalian glycolysis gene expression and initially named hSGT1 (human 80
suppressor of gcr2)(38). We identified the same gene in a screen for interacting partners of 81
human papilloma virus E6 oncoprotein and found it to interact with p53 and transactivate p53-82
regulated genes (39,40). 83
To elucidate the functional role(s) of mammalian ECD, we generated Ecd-null mice and 84
demonstrated that homozygous deletion of Ecd was early embryonic lethal while ex vivo Cre-85
mediated deletion of Ecd in Ecdfl/fl mouse embryonic fibroblasts (MEFs) led to proliferative 86
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

5
block and a significant decrease in cell survival (41,42). ECD was found to be essential for E2F 87
target gene expression by facilitating the dissociation of the retinoblastoma RB protein from 88
E2F and promoting the G1-to-S phase cell cycle progression (41). As a consequence, Ecd-null 89
MEFs showed a decrease in the levels of cyclins A, B1, E and D1, reduction in CDK2 kinase 90
activity and were arrested in G1 phase of the cell cycle (41). Interestingly, E2F family proteins 91
such as the E2F1 have been implicated in UPR-mediated cell death (43). In addition to its 92
promotion of the G1/S phase, ECD also promotes G2/M phase of the cell cycle and its 93
knockdown induced not just a G2/M arrest but also apoptosis (44). Induction of UPR not only 94
induces apoptosis but also halts cell cycle progression in G1 and G2 phases (33-35,45) and these 95
effects are mediated by PERK (33,34), suggesting a potential connection between ECD and the 96
PERK arm of the UPR. 97
More recently, we uncovered another mechanism by which ECD regulates cellular proliferation, 98
involving its interaction with RUVBL1 and PIH1D1 components of the prefoldin co-chaperone 99
R2TP (42) which is involved in the assembly or remodeling of a number of protein and protein-100
RNA complexes to regulate many physiological processes (46-52). Recently, it was reported that 101
knock down of the RUVBL1 component of the R2TP complex induced cell cycle block and ER 102
stress (53). Furthermore, ECD has been shown to associate with the stress response protein 103
Thioredoxin-Interacting Protein (TXNIP) (54) which is known to bind to the ER chaperone PDI 104
(protein disulfide bond isomerase) to increase its enzymatic activity to relieve ER stress (55). 105
Lastly, TXNIP was recently shown to be a novel component of the UPR and is regulated by the 106
PERK pathway (56). Thus, multiple lines of suggestive evidence pointed to a potential role of 107
ECD in the regulation of ER stress. In this study, we demonstrate that induction of ER stress, 108
using both chemical ER stress inducers (Thapsigargin and Tunicamycin) and physiological ER 109
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

6
stress inducers (glucose starvation), led to a reduced ECD protein, and this effect was not seen in 110
PERK KO or phospho-deficient eIF2α MEFs. Notably, ECD mRNA levels were increased, 111
suggesting impaired ECD translation as a mechanism for reduced protein levels. Moreover, ECD 112
localizes and co-immunoprecipitates with the unfolded protein response mediators PERK and 113
GRP78. While ECD depletion increased the levels of p-PERK and its downstream targets, p-114
eIF2α and ATF4, ECD overexpression markedly decreased their levels upon ER stress induction, 115
whereas induction of GRP78 protein robustly increased, suggesting an enhanced adaptive folding 116
capacity of the ER. Knockdown of GRP78 reversed the attenuation of PERK signaling seen upon 117
ECD overexpression. Significantly, ECD over-expression and depletion distinctly affected the 118
survival outcome of cells upon ER stress. 119
120
MATERIALS AND METHODS 121
Reagents and antibodies- Thapsigargin, (Cat# 12758) and Tunicamycin (Cat# 12819) were 122
from Cell Signaling, dissolved in DMSO and used at indicated concentrations. SiRNA against 123
GRP78 was from Santa Cruz (Sc-35522) and Cycloheximide was purchased from Sigma (cat# 124
7698). A monoclonal antibody against ECD, generated in our laboratory, has been described 125
previously (57). Antibodies against p-PERK (cat# 3179), ATF4 (cat# 11815), p-eIF2α (cat# 126
9721), eIF2α (cat# 9722), GRP78 (cat# 3177), Caspase 3 (cat# 9662), GRP94 (cat# 2104), s-127
XBP1 (cat# 12782), RCAS1 (cat# 6960), PDI (cat# 3501) and total PERK (cat# 3192) were from 128
Cell signaling. ATF6 and SERCA2 antibodies were purchased from Enzo Life (cat# ALX-804-129
381-C100) and abcam (ab2861), respectively. PERK and GRP78 used for immunofluorescence 130
and proximity ligation assay (PLA) were from Santa Cruz (cat# sc-9477) and abcam (ab21685), 131
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

7
respectively. PIH1D1 was from Santa Cruz (18Y9 sc-101000). GRP78 for IP/WB was purchased 132
from abcam (cat#21685). 133
134
Establishment of MEFs with doxycycline-inducible ECD over-expression from Ecd transgenic 135
mice- MEFs were generated from 13.5 days old embryos of mice of the genotype Tet(O)-Flag-136
hEcd-IRES-eGFP used as control. rtTA was introduced retrovirally to generate Tet(O)-Flag-137
hEcd-IRES-eGFP; rtTA (the derivation of the transgenic mice will be described separately). 138
139
Cell lines and culture conditions- Ecd fl/fl MEFs were maintained in DMEM medium 140
supplemented with 10% fetal bovine serum and treated with adenoviruses coding for GFP 141
(adeno-GFP, control) or Cre (adeno-Cre) as described previously (41,42). Panc-1 cells have been 142
previously described (58). For ECD overexpression studies, control MEFs or ECD over-143
expressing MEFs were cultured with 1μg/ml of Doxycycline (Cat # 231286, BD Biosciences) to 144
induce expression of the Ecd transgene. MEFs with a non-phosphorylatable eIF2α were obtained 145
from Dr. Thomas Rutkowski’s lab, Carver College of Medicine, Iowa and has been described 146
elsewhere (59). PERK knockout (KO) MEFs were from ATCC (CRL-2976). Immortal human 147
mammary epithelial cell line MCF-10A was cultured in DFCI-1 medium (42). 148
149
Cellular Fractionation and Immunoprecipitation- Subcellular fractionation of MCF-10A cells 150
into soluble (GAPDH was used as a marker(42,60)) and microsomal fractions (PERK and 151
SERCA2 were used as a marker) was carried out according to the manufacturer protocol (Sigma 152
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

8
Aldrich, Cat# ER0100)(61). For immunoprecipitation (I.P), cells were washed in Phosphate 153
Buffered Saline (PBS) and lysed in CHAPS buffer [(0.3% CHAPS, 20 mM Tris-HCl (pH 7.4), 154
120 mM NaCl, 10% glycerol, 5 mM EDTA supplemented with protease and phosphatase 155
inhibitor (Roche, Cat# 04906837001 and 11836145001)]. 156
157
Western blotting- For western blotting, cell lysates were prepared in RIPA buffer (Cat# 89901, 158
Thermo Scientific) supplemented with protease inhibitors (Roche, Cat# 11836145001). Lysates 159
were resolved on SDS-PAGE gel, transferred on to nylon membrane (IPVH00010, Millipore), 160
and subjected to ECL-based Western blotting as described (41,42). 161
162
Immunofluorescence analysis- Immortal MEFs were cultured on coverslips, fixed for 30 min in 163
3% Paraformaldehyde (PFA) and permeabilized in PBS (containing 0.5 % Triton X-100 and 164
10% goat serum) for 20 min at room temperature, followed by blocking in PBS (containing 10% 165
goat serum) for 1 hour at room temperature. The cells were then incubated with primary 166
antibodies in blocking buffer overnight at 4°C followed by 1-hour incubation with their 167
corresponding secondary antibodies in blocking buffer at room temperature. Cells were then 168
washed in PBS (containing 0.1% Tween-20) and then mounted for confocal with the super-169
resolution structure-illuminated microscopy (SIM). Confocal images were obtained using the 170
Carl Zeiss LSM 510 microscope and 3D-SIM images were collected with a Zeiss ELYRA PS.1 171
illumination system (Carl Zeiss). 172
173
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

9
Proximity Ligation Assay (PLA)- PLA was previously described (62). Briefly, MCF-10A cells 174
were fixed with 3% PFA, stained with the indicated antibodies and incubated with Proximity 175
Ligation Assay plus and minus probes followed by ligation and amplification reaction according 176
to the manufacturer’s protocol (DuoLink Sigma). 177
178
Colony Formation Assay- For clonogenic assay, 1,000 cells were plated in triplicate and allowed 179
to attach to the plate (about 8 h) followed by treatment with Thapsigargin for 24h. 10 days later, 180
colony formation was assessed as previously described (41,42). 181
182
RNA isolation and real-time quantitative PCR (qRT-PCR)- Total RNA was isolated using the 183
TRizol reagent (Invitrogen cat# 15596018). 1 µg of RNA was reverse- transcribed using 184
SuperScript TMII reverse transcriptase (Invitrogen cat# 18064014). The qPCR was performed 185
with primer sets indicated in the table: 186
Primers used for the Real-time quantitative PCR
Genes Forward primer Reverse primer
mouse CHOP CTGCCTTTCACCTTGGAGAC CGTTTCCTGGGGATGAGATA
human CHOP CATTGCCTTTCTCCTTCGGG CCAGAGAAGCAGGGTCAAGA
mouse GRP78 AGTGGTGGCCACTAATGGAG CAATCCTTGCTTGATGCTGA
mouse Ecd CCGGTCTGGCACAAACTTCTGCTG AGGGTCGAAGCATCCCTCCATCGA
human Ecd ACTTTGAAACACACGAACCTGGCG TGATGCAGGTGTGTGCTAGTTCCT
187
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

10
188
Induction of ER stress by glucose starvation- Panc-1 cells were maintained in DMEM 189
supplemented with 10% fetal bovine serum and then switched to glucose-free DMEM medium 190
(cat# 11966-025, life technologies) for indicated times. 191
192
Statistical analysis- for assessment of statistical significance, a Student t test was used. P values 193
of < 0.05 were considered statistically significant. 194
195 196
RESULTS 197
Induction of ER stress leads to reduced ECD protein expression in a PERK-eIF2α-dependent 198
manner- To begin to explore the potential link of ECD to ER stress, we asked if the levels of 199
ECD protein are affected by induction of ER stress. For this purpose, immortal human mammary 200
epithelial cell line MCF-10A was treated with Thapsigargin or Tunicamycin, two commonly 201
used chemical ER stress inducers (63), and ECD protein levels were analyzed by western 202
blotting. Treatment with both ER stress inducers increased the level of p-eIF2α as expected (63) 203
(Fig. 1A&B). Notably, a concomitant time-dependent decrease in ECD protein levels was 204
observed upon treatment with either Thapsigargin (Fig. 1A) or Tunicamycin (Fig. 1B). 205
Given the effect of chemical ER stress inducers on ECD protein levels, we next assessed whether 206
physiological stress such as glucose starvation would have similar effects. For this purpose, we 207
used human pancreatic carcinoma cell line (Panc-1) which is known to exhibit ER stress upon 208
glucose starvation (63). Significantly, similar to chemical ER stress, glucose starvation-induced 209
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

11
ER stress also exhibited a reduced levels of ECD protein(Fig. 1C). To assess whether the 210
decrease in ECD protein levels was due to reduced ECD mRNA levels, we measured ECD 211
mRNA levels using qRT-PCR. Induction of CHOP mRNA was used as a control (Fig. 1D). 212
Notably, ECD mRNA levels were not only not reduced but in fact showed an increase (Fig. 1E), 213
suggesting that the reduction in ECD protein level was not at the transcriptional level; likewise, 214
physiological stress by glucose starvation also increased ECD mRNA levels (Fig. 1F). Since 215
PERK activation and subsequent phosphorylation of eIF2α mediates translational block in 216
response to ER stress (24), we used MEFs in which this pathway is genetically abrogated. First, 217
we treated Wild-Type (WT) MEFs or MEFs from PERK kinase domain knockout (PERK-KO) 218
mice (64) with Thapsigargin and analyzed ECD protein levels by western blotting. The expected 219
lack of PERK pathway activation in PERK-KO MEFs was confirmed by lack of induction of 220
PERK phosphorylation in PERK-KO MEFs compared to WT MEFs (Fig.1G). Significantly, 221
while a decrease in ECD protein levels was observed in WT MEFs treated with Thapsigargin, the 222
levels of ECD protein were unchanged in PERK-KO MEFs (Fig. 1G). Then, ECD mRNA were 223
assessed in PERK KO and control MEFs to determine whether the levels are altered upon 224
Thapsigargin treatment. Again, CHOP mRNA induction was used as a positive control (Fig. 1H). 225
As expected, in PERK KO MEFs, CHOP mRNA is very minimally induced upon Thapsigargin 226
treatment as compared to control cells (Fig. 1H), since CHOP is downstream of PERK (24-32). 227
Notably, while ECD mRNA increased in control WT MEFs, in PERK KO MEFs, induction of 228
ECD mRNA is low as compared to control WT PERK MEFs (Fig. 1I). 229
To further examine the role of the PERK pathway in the downregulation of ECD protein 230
expression, we utilized MEFs from mice that carry a serine 51 to alanine mutation in eIF2α 231
which prevents its phosphorylation by activated PERK and makes the cells resistant to PERK-232
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

12
mediated translation block upon ER stress induction (59). As expected, the levels of p-eIF2α 233
increased in WT MEFs but not in the phospho-deficient eIF2α mutant MEFs when ER stress was 234
induced with Thapsigargin or Tunicamycin (Fig. 1J, compare lanes 1-3 with lanes 4-6). 235
Importantly, while ECD protein levels decreased in WT MEFs treated with ER stress inducers 236
(Fig. 1J, compare lane 1 to lanes 2-3), the levels of ECD protein did not change in identically-237
treated mutant eIF2α MEFs (Fig. 1J compare lane 4 to lanes 5-6). Together, these results support 238
a conclusion that ECD protein levels are downregulated upon ER stress in a PERK-eIF2α 239
dependent manner, and that ER stress upregulates ECD mRNA levels, suggesting a regulatory 240
link between ECD and the PERK pathway of ER stress. 241
242
ECD co-localizes and associates with PERK and GRP78- To further explore a potential link 243
between ECD and the ER stress pathway, we assessed the localization of ECD. We first 244
performed immunofluorescence using Super-resolution Structured Illumination Microscopy 245
(SIM), a technique that offers a much higher resolution than conventional confocal microscopy 246
(65,66). Co-localization of PERK and GRP78 served as a positive control since PERK and 247
GRP78 are known interacting partners (13). Indeed, we observed that endogenous PERK and 248
GRP78 were co-localized in a punctate distribution (Fig. 2A). Significantly, ECD also co-249
localized with PERK and GRP78 (Fig. 2B & C), although not as extensive as GRP78-PERK co-250
localization. Negative controls rabbit and mouse IgGs showed no staining (Fig. 2 D & E). Next, 251
we performed sub-cellular fractionation of MCF-10A cells to biochemically assess the ER 252
localization of ECD. This analysis showed expected presence of ECD in the soluble fraction 253
(40,42). Importantly, ECD was prominently present in the ER-containing microsomal fraction 254
(Fig. 2F). Given the co-localization of ECD with PERK and GRP78 (Fig. 2A-C), we assessed if 255
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

13
ECD associates with PERK and/or GRP78. First, we used PLA to assess the proximity of ECD 256
relative to GRP78 and PERK in MCF-10A cells. Known ECD interaction with 257
PIH1D1component of the R2TP served as positive control (42,46). Indeed, ECD and PIH1D1 258
formed distinct foci detectable by PLA (Fig. 2G, red dots). Significantly, PLA signals were also 259
observed for ECD and GRP78 or ECD and PERK (Fig. 2H-I). Finally, we carried out 260
immunoprecipitation (IP) of ECD from lysates from MCF-10A cell, treated or left untreated with 261
Thapsigargin, followed by western blotting for PERK and GRP78 antibodies to assess their 262
association. IP of ECD co-immunoprecipitated PIH1D1 (Fig. 2K), as expected based on known 263
interaction of ECD with PIH1D1 (42,46). Importantly, PERK and GRP78 also co-264
immunoprecipitated with ECD (Fig. 2K, left panel). A reverse IP of GRP78 also co-265
immunoprecipitated ECD, with PERK used as a positive control (Fig. 2K right panel). Taken 266
together, these results suggest that ECD associates with PERK and GRP78. 267
268
ECD regulates the PERK arm of the UPR- The association of ECD with PERK and GRP78 269
(Fig. 2) and abrogation of the decrease in ECD levels in PERK KO cells upon ER stress (Fig.1) 270
suggested that ECD may be functionally linked to UPR through PERK pathway. Therefore, we 271
assessed the activation of the PERK pathway in the absence or presence of UPR inducers in cells 272
in which ECD could be inducibly deleted. For this purpose, we utilized Ecdfl/fl MEFs which have 273
been previously established and shown to exhibit Ecd gene deletion and loss of ECD protein 274
expression upon infection with adenovirus expressing Cre recombinase (41). Previously, we 275
have observed that induction of Ecd deletion in these Ecdfl/fl MEFs, aside from inducing cell 276
cycle arrest, led to a decrease in cell survival even in the absence of any stress (41,42). 277
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

14
Notably, Cre-meditated deletion of Ecd in the Ecdfl/fl MEFs led to an increase in the levels of 278
phospho-PERK (p-PERK) and p-eIF2α, when compared to control adeno-GFP infected MEFs 279
(Fig. 3A, compare lane 1 with 3), and these effects were further enhanced by treatment with 280
Thapsigargin (Tg), the inhibitor of the Sarco/Endoplasmic Reticulum Calcium ATPase (Fig. 3A, 281
compare lane 2 with 4). Spliced XBP1 also showed some increase in ECD null cells upon 282
Thapsigargin treatment while ATF6 did not show a significant change (Fig. 3A). Consistent with 283
the increased p-PERK levels upon ECD deletion, Thapsigargin treated ECD-depleted MEFs also 284
exhibited increased expression of CHOP mRNA (Fig. 3B), a downstream effector of the PERK 285
pathway whose target genes promote cell death (24-28,30,31). The increased CHOP mRNA 286
correlated with an increased cleaved caspases 3 in ECD null cells (Fig. 3C, compare lane 2 with 287
4) and a significant reduction in cell survival, as measured by colony-forming assays (Fig. 3D). 288
To further assess these effects, we used physiological stress in Panc-1 cells (Fig. 3E) in which 289
ECD was knocked down followed by glucose starvation. Again, here too, a siRNA knock down 290
of ECD plus glucose starvation led to an enhanced eIF2α phosphorylation, the downstream target 291
of PERK, and increased cleaved caspase 3 as compared to control cells (Fig. 3E, compare lanes 292
1-5 with lanes 6-10), suggesting more cell death in ECD-knocked down cells upon glucose 293
starvation. 294
In a reciprocal approach, we overexpressed ECD and then examined its impact on the UPR, in 295
particular the PERK pathway, upon ER stress induction. To this end, we generated MEFs from 296
mice that carry a Doxycycline (Dox)-inducible Ecd transgene ((Tet(O)-Flag-hECD-IRES-eGFP; 297
rtTA)) or a control transgenic mouse without rtTA ((Tet(O)-Flag- hECD-IRESeGFP)). These 298
MEFs were treated with Dox to induce ECD overexpression, followed by treatment with 299
Thapsigargin. As expected, control MEFs exhibited an increase in p-PERK and CHOP mRNA; 300
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

15
however, the MEFs with Dox-induced ECD overexpression exhibited a substantially reduced 301
level of p-PERK and CHOP mRNA upon Thapsigargin treatment as compared to control MEFs 302
(Fig. 3F & G). Likewise, when the MEF cell lines were treated with Thapsigargin for various 303
time points (3, 6, 12 or 24h), ECD over-expressing MEFs exhibited lower levels of p-PERK 304
compared to those in control MEFs (Fig. 3H, compare lanes 1-5 with lanes 6-10). A 305
corresponding reduction in the levels of ATF4, a downstream target of PERK signaling, was 306
seen in ECD over-expressing MEFs compared to their control MEFs (Fig. 3H, compare lanes 3-5 307
with lanes 8-10); s-XBP1 and ATF6 also were slightly reduced in ECD over-expressing MEFs 308
(Fig. 3H, compare lanes 1-5 with lanes 6-10). Importantly, ECD overexpression was associated 309
with a substantially higher level of GRP78 following Thapsigargin treatment as compared to 310
control MEFs (Fig. 3H compare lanes 2-5 to lanes 7-10); GRP94 protein and PDI, two other 311
chaperones, were also slightly increased in ECD over-expressing MEFs but not as dramatically 312
increased as GRP78 upon Thapsigargin treatment (Fig. 3H compare lanes 1-5 to lanes 6-10). 313
Similar to Thapsigargin, induction of ER stress with Tunicamycin, which induces ER stress by 314
inhibiting glycosylation of proteins in the ER, was also associated with lower levels of p-PERK 315
and p-eIF2α in ECD overexpressing MEFs as compared to control MEFs (Fig. 3I compare lanes 316
2-5 with lanes 7-10). Furthermore, higher levels of GRP78 protein was also observed in ECD 317
overexpressing MEFs as compared to control MEFs upon Tunicamycin treatment (Fig. 3I 318
compare lanes 2-5 with lanes 7-10). Taken together, these results strongly support a conclusion 319
that ECD negatively regulates PERK signaling while enhancing the adaptive capacity of the cells 320
by increasing levels of chaperones, predominantly, GRP78 protein in response to ER stress. 321
The increased induction of GRP78 is required for ECD to attenuate PERK signaling- 322
Induction of GRP78 expression during ER stress is a homeostatic mechanism to reduce the 323
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

16
unfolded protein load in the ER, thereby reducing the activation of UPR sensors. As a major 324
regulator of PERK, overexpression of GRP78 has been shown to reduce PERK signaling upon 325
ER stress induction (13). Given the enhanced induction of GRP78, together with attenuation of 326
PERK signaling in ECD overexpressing MEFs exposed to ER stress (Fig. 3H-I), we examined 327
the possibility that the increased GRP78 levels upon ECD overexpression may be 328
mechanistically linked to the reduction of PERK signaling. To investigate this possibility, we 329
first examined changes in the levels of GRP78 in ECD deleted MEFs where we observed 330
increased PERK phosphorylation (Fig. 3A). To assess if ECD was required for optimal GRP78 331
protein levels upon ER stress, we exposed the wild-type MEFs or MEFs with Ecd deletion 332
induced by adeno-Cre (as in Fig. 3A-D) to Thapsigargin, and then assessed the levels of GRP78 333
protein, at various time points, by western blotting. The levels of GRP78, over time, were 334
reduced in ECD-null MEFs compared to GRP78 levels in control MEFs (Fig. 4A, compare lanes 335
3-5 with 8-10). Likewise, using physiological stress, a knock down of ECD in Panc-1 cells led to 336
a decrease in GRP78 levels upon glucose starvation (Fig. 4B, compare lanes 1-5 with lanes 6-337
10). To examine whether the reduced induction of GRP78 in ECD-null MEFs was a result of 338
reduced GRP78 mRNA expression, we carried out qPCR analyses of mRNA isolated from WT 339
or Ecd-null MEFs treated with Thapsigargin. As expected, GRP78 mRNA levels increased in 340
WT MEFs upon Thapsigargin treatment (Fig. 4C); however, the levels of GRP78 mRNA 341
induction in Ecd-null MEFs was comparable to that in control MEFs (Fig. 4C). Similarly, 342
GRP78 mRNA levels in control vs. ECD over-expressing MEFs treated with Thapsigargin were 343
comparable (Fig. 4D). To determine whether GRP78 may be less stable in ECD null cells, we 344
first knocked down ECD in Panc-1 cells, which have high level of GRP78, followed by 345
inhibition of protein synthesis by cycloheximide treatment over various time points and GRP78 346
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

17
protein levels were analyzed by western blot. Significantly, the time dependent decrease in 347
GRP78 protein levels following cycloheximide treatment was faster in ECD-knocked down cells 348
compared to their control cells (Fig. 4E). Next, to examine the possibility that ECD 349
overexpression is associated with increased GRP78 protein stability, control or ECD over-350
expressing cells were treated with Thapsigargin to induce GRP78 protein, followed by 351
cycloheximide (CHX) treatment over various time points. Notably, the time-dependent decrease 352
in GRP78 levels was slower in ECD over-expressing MEFs as compared to control MEFs (Fig. 353
4F). Finally, to determine whether the increase in GRP78 protein levels upon ECD 354
overexpression was required for the attenuation of PERK activation seen upon ER stress 355
induction, we depleted GRP78 using siRNA in both control and ECD overexpressing MEFs 356
followed by Thapsigargin treatment. Notably, the attenuating effect of ECD overexpression on 357
PERK phosphorylation upon Thapsigargin treatment was abrogated by GRP78 knockdown (Fig. 358
4G; compare lane 5-6 with 7-8). Taken together, these results support the conclusion that ECD 359
positively regulates GRP78 protein level upon ER stress induction to attenuate PERK activation. 360
361
ECD overexpression protects cells from ER stress-induced cell death- ER stress response is 362
initially aimed at the survival of cells (67); however severe ER stress shifts the response from a 363
pro-survival to a pro-apoptotic response (23,31) through PERK-mediated induction of CHOP 364
expression, a transcription factor that enhances the expression of pro-apoptotic pathway genes 365
(24-28,30,31,68). Several studies found that inhibition/attenuation of the PERK pathway 366
protected cells against stress-induced cell death (69-72). Given our observations that ECD 367
functions as a modulator of PERK signaling, we assessed if the levels of ECD determine a 368
differential survival vs. apoptotic cell fate upon ER stress induction. For that purpose, we treated 369
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

18
control MEFs or Dox-inducible ECD overexpressing MEFs with Thapsigargin and then assessed 370
the level of apoptosis induction by examining caspase 3 cleavage (73). As anticipated, a 371
Thapsigargin dose-dependent increase in cleaved caspase 3 levels was observed in control 372
MEFs, whereas the levels of cleaved caspase 3 were markedly lower in ECD-overexpressing 373
MEFs (Fig. 5A). Real-time qPCR analyses demonstrated that Thapsigargin-induced expression 374
of CHOP, a PERK-regulated mediator of cell death (24-28,30-32), was lower in ECD 375
overexpressing MEFs as compared to control MEFs (Fig. 5B). To further assess the pro-survival 376
effects of ECD against apoptotic cell fate upon ER stress induction, we assessed the abilities of 377
control vs. ECD-overexpressing MEFs to form colonies after their exposure to ER stress. For this 378
purpose, equal numbers of control or ECD overexpressing MEFs were treated with Thapsigargin 379
for 24h, and the cells were maintained in Thapsigargin-free medium for 10 days followed by 380
crystal violet staining and counting of surviving colonies. Notably, more colonies were observed 381
in ECD overexpressing MEFs as compared to control MEFs (Fig. 5C), further supporting the 382
conclusion that ECD provides survival advantage upon ER stress. 383
384
DISCUSSION 385
The ER-localized stress response pathway referred to as the UPR is a well-conserved 386
response to a number of cellular stresses such as unfolding of proteins in the ER. The UPR elicits 387
a spectrum of downstream responses whose outcomes range from restoration of homeostasis to 388
cellular apoptosis if the stress is extreme and prolonged (23,31). The UPR thus represents a 389
double-edged sword and must be intricately regulated to prevent inappropriate cellular outcomes. 390
Mechanisms that help modulate the magnitude and type of the UPR in response to physiological 391
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

19
or pathological stress stimuli are not fully understood. In this study, we provide evidence that 392
ECD protein is a negative regulator of the PERK arm of the UPR through GRP78. 393
Several lines of circumstantial evidence discussed in the Introduction section suggested a 394
potential involvement of ECD in UPR but direct support for such a role has been lacking. We 395
established that induction of ER stress, using both chemical ER stress inducers (Thapsigargin 396
and Tunicamycin) and physiological ER stressors (glucose starvation), leads to downregulation 397
of ECD protein levels while the mRNA levels were elevated (Fig. 1A-F). By using PERK kinase 398
knockout (KO) or phosphorylation-deficient eIF2α MEFs (Fig. 1G-J), we established that ECD 399
was linked to the PERK pathway of the UPR. The association of ECD with PERK and GRP78 400
(Fig. 2) further linked ECD to the PERK arm of the UPR. A functional connection of ECD to the 401
PERK arm of the UPR is supported by the distinct modulation of the PERK-mediated responses 402
elicited by perturbations of the cellular levels of ECD. Depletion of ECD sensitized cells to 403
PERK signaling in response to ER stress, with increase in p-PERK and p-eIF2α levels, as well as 404
an increase in the downstream effector of PERK, the transcription factor CHOP, leading to a 405
reduced survival of these ECD depleted MEFs (Fig. 3A-E). Reciprocally, upregulation of ECD 406
reduced PERK signaling upon induction of ER stress (Fig. 3F-I). As activation of the PERK 407
pathway promotes cell death in response to ER stress, primarily through CHOP-dependent 408
expression of pro-apoptotic genes (24-32) and abrogation of PERK signaling protects cells 409
against stress-induced cell death (69-71), our results supported the likelihood that ECD functions 410
to modulate PERK pathway activity and promote cell survival during ER stress. Indeed, 411
assessment of cellular survival in response to ER stress showed that reduction in ECD levels 412
impaired cell survival to ER stress (Fig. 3C-E) while overexpression of ECD promoted cell 413
survival (Fig. 5). Collectively, these results support the conclusion that ECD and PERK are 414
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

20
linked through a negative feedback mechanism whereby ECD exerts an inhibitory effect on 415
PERK pathway signaling whereas activated PERK in turn reduces ECD protein levels via eIF2α-416
dependent translational block. Consistent with a reciprocal negative feedback relation between 417
ECD and PERK, activated PERK negatively regulates cell growth (33-35); conversely, ECD 418
positively regulates cell growth (41) and is overexpressed in human breast and pancreatic cancer 419
specimens, correlating with poor prognostic markers and shorter survival (57,74). While the 420
physiological benefit of an increased ECD mRNA upon ER stress is not yet understood, a 421
speculation is that this effect may reflect a feedback response to the decrease in ECD protein 422
levels to compensate for the loss of ECD but ECD translation is blocked. Although ECD has 423
been reported to play roles in pre-mRNA splicing in drosophila (75), it is unlikely that ECD 424
alters its own mRNA splicing upon ER stress because ECD protein levels decrease upon ER 425
stress. 426
Modulation of ECD levels using knockdown or overexpression demonstrated that ECD is 427
a positive regulator of GRP78 levels (Fig. 3H-I; Fig 4.). While slight increases in GRP94 and 428
PDI were observed, the effect on GRP78 was more dramatic. As increased expression of GRP78 429
and other chaperones is known to promote the clearing of ER stress-causing unfolded protein 430
load and reduce the activation of UPR sensors (13-15,55), we surmised that upregulation of 431
GRP78 and other chaperones (Fig. 3H) may represent one potential mechanism by which ECD 432
negatively regulates the PERK pathway activation and relieves ER stress. Indeed, knockdown of 433
GRP78 eliminated the ability of overexpressed ECD to attenuate PERK pathway signaling (Fig. 434
4G). Notably, GRP78 is required for cell survival not only in response to ER stress (76-79) but 435
also in other stressful and hostile conditions such as glucose deficiency encountered in tumor 436
microenvironment (80-85). Given that the PERK arm of the UPR is also activated in cancer 437
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

21
(68,86,87) and both ECD and GRP78 are overexpressed in cancer (57,74,80-85), we suggest that 438
ECD overexpression may play a similar role to mitigate the negative consequences of elevated 439
PERK signaling found in cancer. Given the mechanistic link between ECD and GRP78 in 440
inhibiting the PERK pathway to promote cell survival, it will be of great interest to explore if 441
GRP78 and ECD are co-overexpressed in tumors that use the UPR to promote tumor cell 442
survival and hence may be suitable targets for UPR-directed therapeutic agents. 443
444
Acknowledgements: We thank the members of the Band laboratory for helpful discussions. The 445
WT and mutant eIF2α MEFs were a gift from Dr. Thomas Rutkowski’s lab, Carver College of 446
Medicine, Iowa. 447
Funding--This work was supported by the NIH grants CA116552, CA87986 and CA105489 to 448
HB, and CA96844 and CA144027 to VB; Department of Defense grants W81WH-11-1-0167 to 449
HB, W81XWH-07-1-0351, W81XWH-11-1-0171 and W81XWH-14-1-0567 to VB. We 450
acknowledge the support to the UNMC Confocal, Flow Cytometry and other Core facilities from 451
the NCI Cancer Center Support Grant (P30CA036727) to Fred & Pamela Buffett Cancer Center 452
and the Nebraska Research Initiative. SM was a postdoctoral fellow of the Susan G. Komen 453
Foundation. AO was supported by supplement to CA144027 NCI grant. 454
455
456
Figure Legends: 457
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

22
Figure 1. Induction of ER stress leads to reduced ECD protein expression in a PERK-458
eIF2α-dependent manner. (A-C): MCF-10A cells were treated with Thapsigargin (50 nM, A) 459
or Tunicamycin (50 ng/ml, B); Panc-1 (C) were cultured in glucose-free medium and then cell 460
lysates were prepared at the indicated time points. Equal amount of proteins was resolved on 461
SDS-PAGE followed by western blotting with indicated antibodies. Increase in the levels of p-462
eIF2α served as marker for induction of ER stress. (D-F): MCF-10A cells were treated with 463
Thapsigargin (D-E) and Panc-1 cells were cultured in glucose-free medium (F), then total RNA 464
was isolated followed by qRT-PCR using CHOP primers and ECD primer (Mean +/- SD and p-465
values are shown from 3 independent experiments * p<0.05). CHOP mRNA induction served as 466
control for Thapsigargin-induced ER stress. (G): Wild-type (WT) PERK and PERK kinase 467
domain knockout (PERK-KO) MEFs were treated with Thapsigargin (50 nM) for 14 h and then 468
cell lysates were resolved on SDS-PAGE gels followed by western blot with indicated 469
antibodies. (H-I): PERK KO and control WT MEFs were treated with Thapsigargin and total 470
RNA was isolated at indicated time points followed by qRT-PCR with primers against CHOP 471
(Fig. H) or ECD (Fig. I). (J): WT eIF2α MEFs or mutant eIF2α phospho-deficient MEFs were 472
treated with Thapsigargin (Tg, 50 nM) or Tunicamycin (Tun, 50 ng/ml) for 14 h and then cell 473
lysates were analyzed by western blotting with indicated antibodies. 474
475
Figure 2: ECD co-localizes and associates with PERK and GRP78. (A-E): Immortal MEFs 476
were fixed in 3% paraformaldehyde (PFA) and stained with indicated antibodies and mounted 477
for analyses with structure-illuminated microscopy (SIM). PERK and GRP78 co-localization 478
served as positive control. DAPI was used to stain the nucleus. (F): MCF-10A cells were 479
fractionated into soluble and microsomal fractions. The purity of the fractions was assessed by 480
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

23
GAPDH (a marker for the soluble/cytoplasmic fraction), PERK and SERCA (both markers for 481
microsomal fraction) and RCAS1 (a Golgi-predominant protein) (88). (G-J): MCF-10A cells 482
were fixed with 3% PFA and stained with indicated antibodies (all antibodies were generated in 483
rabbit except anti-ECD mouse antibody) followed by species-specific secondary antibodies 484
linked to complementary DNA probes to allow fluorescent probe-based detection of the PCR 485
amplification product as distinct foci. Incubation with Proximity Ligation Assay plus and minus 486
probes, followed by ligation and amplification, was carried out according to the manufacturer 487
protocol. Red dots indicate interaction. ECD and PIH1D1 served as positive control. (K): Lysates 488
from MCF-10A cells, treated or left un-treated with Thapsigargin, were subjected to IP with anti-489
ECD antibody (left panel) or anti-GRP78 (right panel) followed by Western blotting with 490
indicated antibodies. ECD and PIH1D1(left panel) or GRP78 and PERK (right panel) served as 491
positive controls. 492
493
Figure 3. ECD regulates the PERK pathway of the UPR. (A & C): Ecd fl/fl MEFs were 494
infected with adenoviruses coding for GFP (adeno-GFP for control) or Cre (adeno-Cre) for 72h. 495
The cells were then left un-treated or treated with Thapsigargin (50 nM). Equal amount of 496
proteins was resolved on SDS-PAGE gel followed by western blotting with indicated antibodies. 497
(B): After adenoviruses infection as described in (A), cells were treated with Thapsigargin. Total 498
RNA was isolated followed by qRT-PCR with CHOP primers; Mean +/- SD and p-values from 3 499
independent experiments are indicated * p<0.05. (D): After adenoviruses infection as described 500
in (A), equal number (1,000) of wild-type (WT) or ECD-/- cells were plated in triplicate and 501
treated with Thapsigargin for 24h. 10 days later, surviving colonies were assessed after crystal 502
blue (0.5% in 25% Methanol) staining. The color retained after the wash was dissolved in 10% 503
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

24
acetic acid and the absorbance was read at 590 nm. The graph below represents the relative 504
absorbance; Mean +/- SD and p-values from 4 independent experiments are indicated * p<0.05; 505
** p≤0.002. (E): Panc-1 cells were treated with control or ECD siRNA for 48h. Then, the cells 506
were switched to glucose-free media and cell lysates were prepared at indicated time points 507
followed by western blot with indicated antibodies. (F; H-I): ECD inducible MEFs [(Tet(O)-508
Flag- hECD-IRESeGFP;rtTA)] or control MEFs [(Tet(O)-Flag- hECD-IRESeGFP)] were treated 509
with Dox for 48 h followed by treatment with Thapsigargin (50 nM) or Tunicamycin (50 ng/ml). 510
Cell lysates were prepared at indicated time points and equal amount of proteins was resolved on 511
SDS-PAGE gel followed by western blot with indicated antibodies. (G): Following ECD 512
induction and Thapsigargin treatment as described above, CHOP mRNA levels were assessed 513
using qRT-PCR. 514
515
Figure 4. The increased induction of GRP78 expression is required for ECD to 516
downregulate PERK signaling. (A): Ecd fl/fl MEFs were treated with adenovirus as described in 517
(Fig. 3A-C) and then the cells were treated with Thapsigargin (50 nM). Cell lysates were 518
prepared at the indicated time points. Equal amount of proteins was resolved on SDS-PAGE gel 519
followed by western blotting with indicated antibodies. (B): ECD was knocked down by siRNA 520
(20 nM) in Panc-1 cells followed by exposure to glucose-free media and cell lysates were 521
collected at indicated times points followed by western blot with indicated antibodies. (C-D): 522
Following ECD deletion (C) or ECD overexpression (D) and Thapsigargin treatment as 523
described above, the levels of GRP78 mRNA were assessed in WT (control) vs. ECD-/- (Cre-524
adenovirus treated) or control vs. ECD over-expressing MEFs using qRT-PCR. (E): ECD was 525
knocked down in Panc-1 cells followed by cycloheximide treatment (25 uM). Cell lysates were 526
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

25
prepared at indicated time points followed by western blot with indicated antibodies. (F): ECD 527
inducible MEFs and their control MEFs were treated with Dox, as described previously, 528
followed by treatment with Thapsigargin and then Cycloheximide treatment (25 uM) for 529
indicated time points. Cell lysates were prepared followed by western blotting with indicated 530
antibodies. (G): ECD overexpressing and their control MEFs were treated with GRP78 SiRNA 531
(30 nM) or control siRNA (scrambled). 24 h later, the cells were treated with Dox for 48 h to 532
induce ECD overexpression followed by Thapsigargin treatment (50 nM). Equal amount of 533
proteins was resolved on SDS-PAGE gel followed by western blotting with indicated antibodies. 534
535
Figure 5. ECD overexpression provides survival advantage. (A): ECD was induced as 536
described in (Fig. 3 and 4) followed by Thapsigargin treatment for 24 h. Equal amount of 537
proteins was resolved on SDS-PAGE gel followed by western blot with indicated antibodies. 538
(B): Following ECD induction with Dox and Thapsigargin treatment as described above, total 539
RNA was isolated and CHOP mRNA was assessed by qPCR; Mean +/- SD and p-values from 3 540
independent experiments are indicated * p<0.05. (C): After ECD induction, control and ECD 541
inducible MEFs were trypsinized and equal number of cells (1,000) were plated in triplicates. 8 h 542
later, the cells were treated with Thapsigargin for 24h. 10 days later, surviving colonies were 543
assessed by Crystal blue staining (0.5% in 25% Methanol). The color retained after the wash was 544
dissolved in 10% Acetic acid and the absorbance was read at 590 nm. The graph below 545
represents the relative absorbance; Mean +/- SD and p-values from 4 independent experiments 546
are indicated ** p≤0.002. 547
548
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

26
549
550 551 552 553
REFERENCES 554 555
1. Voeltz, G. K., Rolls, M. M., and Rapoport, T. A. (2002) Structural organization of the endoplasmic 556 reticulum. EMBO reports 3, 944-950 557
2. Reid, D. W., and Nicchitta, C. V. (2015) Diversity and selectivity in mRNA translation on the 558 endoplasmic reticulum. Nature reviews. Molecular cell biology 16, 221-231 559
3. Jan, C. H., Williams, C. C., and Weissman, J. S. (2014) Principles of ER cotranslational 560 translocation revealed by proximity-specific ribosome profiling. Science 346, 1257521 561
4. Ron, D., and Walter, P. (2007) Signal integration in the endoplasmic reticulum unfolded protein 562 response. Nature reviews. Molecular cell biology 8, 519-529 563
5. Ikonen, E. (2008) Cellular cholesterol trafficking and compartmentalization. Nature reviews. 564 Molecular cell biology 9, 125-138 565
6. Ron, D., and Hampton, R. Y. (2004) Membrane biogenesis and the unfolded protein response. 566 The Journal of cell biology 167, 23-25 567
7. Fagone, P., and Jackowski, S. (2009) Membrane phospholipid synthesis and endoplasmic 568 reticulum function. Journal of lipid research 50 Suppl, S311-316 569
8. Kim, I., Xu, W., and Reed, J. C. (2008) Cell death and endoplasmic reticulum stress: disease 570 relevance and therapeutic opportunities. Nature reviews. Drug discovery 7, 1013-1030 571
9. Berridge, M. J., Bootman, M. D., and Roderick, H. L. (2003) Calcium signalling: dynamics, 572 homeostasis and remodelling. Nature reviews. Molecular cell biology 4, 517-529 573
10. Pinton, P., Giorgi, C., Siviero, R., Zecchini, E., and Rizzuto, R. (2008) Calcium and apoptosis: ER-574 mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 27, 6407-6418 575
11. Wu, J., and Kaufman, R. J. (2006) From acute ER stress to physiological roles of the Unfolded 576 Protein Response. Cell death and differentiation 13, 374-384 577
12. Bravo-Sagua, R., Rodriguez, A. E., Kuzmicic, J., Gutierrez, T., Lopez-Crisosto, C., Quiroga, C., Diaz-578 Elizondo, J., Chiong, M., Gillette, T. G., Rothermel, B. A., and Lavandero, S. (2013) Cell death and 579 survival through the endoplasmic reticulum-mitochondrial axis. Current molecular medicine 13, 580 317-329 581
13. Bertolotti, A., Zhang, Y., Hendershot, L. M., Harding, H. P., and Ron, D. (2000) Dynamic 582 interaction of BiP and ER stress transducers in the unfolded-protein response. Nature cell 583 biology 2, 326-332 584
14. Gardner, B. M., Pincus, D., Gotthardt, K., Gallagher, C. M., and Walter, P. (2013) Endoplasmic 585 reticulum stress sensing in the unfolded protein response. Cold Spring Harbor perspectives in 586 biology 5, a013169 587
15. Lai, E., Teodoro, T., and Volchuk, A. (2007) Endoplasmic reticulum stress: signaling the unfolded 588 protein response. Physiology 22, 193-201 589
16. Teske, B. F., Wek, S. A., Bunpo, P., Cundiff, J. K., McClintick, J. N., Anthony, T. G., and Wek, R. C. 590 (2011) The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 591 during endoplasmic reticulum stress. Molecular biology of the cell 22, 4390-4405 592
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

27
17. DuRose, J. B., Tam, A. B., and Niwa, M. (2006) Intrinsic capacities of molecular sensors of the 593 unfolded protein response to sense alternate forms of endoplasmic reticulum stress. Molecular 594 biology of the cell 17, 3095-3107 595
18. Wang, S., and Kaufman, R. J. (2012) The impact of the unfolded protein response on human 596 disease. The Journal of cell biology 197, 857-867 597
19. Harding, H. P., Zhang, Y., Bertolotti, A., Zeng, H., and Ron, D. (2000) Perk is essential for 598 translational regulation and cell survival during the unfolded protein response. Molecular cell 5, 599 897-904 600
20. Liu, Z., Lv, Y., Zhao, N., Guan, G., and Wang, J. (2015) Protein kinase R-like ER kinase and its role 601 in endoplasmic reticulum stress-decided cell fate. Cell death & disease 6, e1822 602
21. Scheuner, D., Vander Mierde, D., Song, B., Flamez, D., Creemers, J. W., Tsukamoto, K., Ribick, M., 603 Schuit, F. C., and Kaufman, R. J. (2005) Control of mRNA translation preserves endoplasmic 604 reticulum function in beta cells and maintains glucose homeostasis. Nature medicine 11, 757-605 764 606
22. Ma, K., Vattem, K. M., and Wek, R. C. (2002) Dimerization and release of molecular chaperone 607 inhibition facilitate activation of eukaryotic initiation factor-2 kinase in response to endoplasmic 608 reticulum stress. The Journal of biological chemistry 277, 18728-18735 609
23. Szegezdi, E., Logue, S. E., Gorman, A. M., and Samali, A. (2006) Mediators of endoplasmic 610 reticulum stress-induced apoptosis. EMBO reports 7, 880-885 611
24. Harding, H. P., Novoa, I., Zhang, Y., Zeng, H., Wek, R., Schapira, M., and Ron, D. (2000) Regulated 612 translation initiation controls stress-induced gene expression in mammalian cells. Molecular cell 613 6, 1099-1108 614
25. Ma, Y., Brewer, J. W., Diehl, J. A., and Hendershot, L. M. (2002) Two distinct stress signaling 615 pathways converge upon the CHOP promoter during the mammalian unfolded protein response. 616 Journal of molecular biology 318, 1351-1365 617
26. Zinszner, H., Kuroda, M., Wang, X., Batchvarova, N., Lightfoot, R. T., Remotti, H., Stevens, J. L., 618 and Ron, D. (1998) CHOP is implicated in programmed cell death in response to impaired 619 function of the endoplasmic reticulum. Genes & development 12, 982-995 620
27. Oyadomari, S., Koizumi, A., Takeda, K., Gotoh, T., Akira, S., Araki, E., and Mori, M. (2002) 621 Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. 622 The Journal of clinical investigation 109, 525-532 623
28. Song, B., Scheuner, D., Ron, D., Pennathur, S., and Kaufman, R. J. (2008) Chop deletion reduces 624 oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse 625 models of diabetes. The Journal of clinical investigation 118, 3378-3389 626
29. Malhotra, J. D., Miao, H., Zhang, K., Wolfson, A., Pennathur, S., Pipe, S. W., and Kaufman, R. J. 627 (2008) Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. 628 Proceedings of the National Academy of Sciences of the United States of America 105, 18525-629 18530 630
30. Thorp, E., Li, G., Seimon, T. A., Kuriakose, G., Ron, D., and Tabas, I. (2009) Reduced apoptosis and 631 plaque necrosis in advanced atherosclerotic lesions of Apoe-/- and Ldlr-/- mice lacking CHOP. 632 Cell metabolism 9, 474-481 633
31. Tabas, I., and Ron, D. (2011) Integrating the mechanisms of apoptosis induced by endoplasmic 634 reticulum stress. Nature cell biology 13, 184-190 635
32. Pennuto, M., Tinelli, E., Malaguti, M., Del Carro, U., D'Antonio, M., Ron, D., Quattrini, A., Feltri, 636 M. L., and Wrabetz, L. (2008) Ablation of the UPR-mediator CHOP restores motor function and 637 reduces demyelination in Charcot-Marie-Tooth 1B mice. Neuron 57, 393-405 638
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

28
33. Brewer, J. W., Hendershot, L. M., Sherr, C. J., and Diehl, J. A. (1999) Mammalian unfolded 639 protein response inhibits cyclin D1 translation and cell-cycle progression. Proceedings of the 640 National Academy of Sciences of the United States of America 96, 8505-8510 641
34. Brewer, J. W., and Diehl, J. A. (2000) PERK mediates cell-cycle exit during the mammalian 642 unfolded protein response. Proceedings of the National Academy of Sciences of the United 643 States of America 97, 12625-12630 644
35. Hamanaka, R. B., Bennett, B. S., Cullinan, S. B., and Diehl, J. A. (2005) PERK and GCN2 contribute 645 to eIF2alpha phosphorylation and cell cycle arrest after activation of the unfolded protein 646 response pathway. Molecular biology of the cell 16, 5493-5501 647
36. Nutt, S. L., Hodgkin, P. D., Tarlinton, D. M., and Corcoran, L. M. (2015) The generation of 648 antibody-secreting plasma cells. Nature reviews. Immunology 15, 160-171 649
37. Garen, A., Kauvar, L., and Lepesant, J. A. (1977) Roles of ecdysone in Drosophila development. 650 Proceedings of the National Academy of Sciences of the United States of America 74, 5099-5103 651
38. Sato, T., Jigami, Y., Suzuki, T., and Uemura, H. (1999) A human gene, hSGT1, can substitute for 652 GCR2, which encodes a general regulatory factor of glycolytic gene expression in Saccharomyces 653 cerevisiae. Molecular & general genetics : MGG 260, 535-540 654
39. Gao, Q., Srinivasan, S., Boyer, S. N., Wazer, D. E., and Band, V. (1999) The E6 oncoproteins of 655 high-risk papillomaviruses bind to a novel putative GAP protein, E6TP1, and target it for 656 degradation. Molecular and cellular biology 19, 733-744 657
40. Zhang, Y., Chen, J., Gurumurthy, C. B., Kim, J., Bhat, I., Gao, Q., Dimri, G., Lee, S. W., Band, H., 658 and Band, V. (2006) The human orthologue of Drosophila ecdysoneless protein interacts with 659 p53 and regulates its function. Cancer research 66, 7167-7175 660
41. Kim, J. H., Gurumurthy, C. B., Naramura, M., Zhang, Y., Dudley, A. T., Doglio, L., Band, H., and 661 Band, V. (2009) Role of mammalian Ecdysoneless in cell cycle regulation. The Journal of 662 biological chemistry 284, 26402-26410 663
42. Mir, R. A., Bele, A., Mirza, S., Srivastava, S., Olou, A. A., Ammons, S. A., Kim, J. H., Gurumurthy, C. 664 B., Qiu, F., Band, H., and Band, V. (2015) A Novel Interaction of Ecdysoneless (ECD) Protein with 665 R2TP Complex Component RUVBL1 Is Required for the Functional Role of ECD in Cell Cycle 666 Progression. Molecular and cellular biology 36, 886-899 667
43. Pagliarini, V., Giglio, P., Bernardoni, P., De Zio, D., Fimia, G. M., Piacentini, M., and Corazzari, M. 668 (2015) Downregulation of E2F1 during ER stress is required to induce apoptosis. Journal of cell 669 science 128, 1166-1179 670
44. Xu, S. H., Huang, J. Z., Chen, M., Zeng, M., Zou, F. Y., Chen, D., and Yan, G. R. (2015) Amplification 671 of ACK1 promotes gastric tumorigenesis via ECDdependent p53 ubiquitination degradation. 672 Oncotarget 673
45. Bourougaa, K., Naski, N., Boularan, C., Mlynarczyk, C., Candeias, M. M., Marullo, S., and 674 Fahraeus, R. (2010) Endoplasmic reticulum stress induces G2 cell-cycle arrest via mRNA 675 translation of the p53 isoform p53/47. Molecular cell 38, 78-88 676
46. Horejsi, Z., Stach, L., Flower, T. G., Joshi, D., Flynn, H., Skehel, J. M., O'Reilly, N. J., Ogrodowicz, R. 677 W., Smerdon, S. J., and Boulton, S. J. (2014) Phosphorylation-dependent PIH1D1 interactions 678 define substrate specificity of the R2TP cochaperone complex. Cell reports 7, 19-26 679
47. Ahn, S., Kim, J., and Hwang, J. (2013) CK2-mediated TEL2 phosphorylation augments nonsense-680 mediated mRNA decay (NMD) by increase of SMG1 stability. Biochimica et biophysica acta 1829, 681 1047-1055 682
48. Boulon, S., Marmier-Gourrier, N., Pradet-Balade, B., Wurth, L., Verheggen, C., Jady, B. E., Rothe, 683 B., Pescia, C., Robert, M. C., Kiss, T., Bardoni, B., Krol, A., Branlant, C., Allmang, C., Bertrand, E., 684 and Charpentier, B. (2008) The Hsp90 chaperone controls the biogenesis of L7Ae RNPs through 685 conserved machinery. The Journal of cell biology 180, 579-595 686
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

29
49. Boulon, S., Pradet-Balade, B., Verheggen, C., Molle, D., Boireau, S., Georgieva, M., Azzag, K., 687 Robert, M. C., Ahmad, Y., Neel, H., Lamond, A. I., and Bertrand, E. (2010) HSP90 and its 688 R2TP/Prefoldin-like cochaperone are involved in the cytoplasmic assembly of RNA polymerase II. 689 Molecular cell 39, 912-924 690
50. Horejsi, Z., Takai, H., Adelman, C. A., Collis, S. J., Flynn, H., Maslen, S., Skehel, J. M., de Lange, T., 691 and Boulton, S. J. (2010) CK2 phospho-dependent binding of R2TP complex to TEL2 is essential 692 for mTOR and SMG1 stability. Molecular cell 39, 839-850 693
51. Kim, S. G., Hoffman, G. R., Poulogiannis, G., Buel, G. R., Jang, Y. J., Lee, K. W., Kim, B. Y., Erikson, 694 R. L., Cantley, L. C., Choo, A. Y., and Blenis, J. (2013) Metabolic stress controls mTORC1 lysosomal 695 localization and dimerization by regulating the TTT-RUVBL1/2 complex. Molecular cell 49, 172-696 185 697
52. Zhao, R., Kakihara, Y., Gribun, A., Huen, J., Yang, G., Khanna, M., Costanzo, M., Brost, R. L., 698 Boone, C., Hughes, T. R., Yip, C. M., and Houry, W. A. (2008) Molecular chaperone Hsp90 699 stabilizes Pih1/Nop17 to maintain R2TP complex activity that regulates snoRNA accumulation. 700 The Journal of cell biology 180, 563-578 701
53. Yuan, X. S., Wang, Z. T., Hu, Y. J., Bao, F. C., Yuan, P., Zhang, C., Cao, J. L., Lv, W., and Hu, J. (2016) 702 Downregulation of RUVBL1 inhibits proliferation of lung adenocarcinoma cells by G1/S phase 703 cell cycle arrest via multiple mechanisms. Tumour biology : the journal of the International 704 Society for Oncodevelopmental Biology and Medicine 705
54. Suh, H. W., Yun, S., Song, H., Jung, H., Park, Y. J., Kim, T. D., Yoon, S. R., and Choi, I. (2013) TXNIP 706 interacts with hEcd to increase p53 stability and activity. Biochemical and biophysical research 707 communications 438, 264-269 708
55. Lee, S., Min Kim, S., Dotimas, J., Li, L., Feener, E. P., Baldus, S., Myers, R. B., Chutkow, W. A., 709 Patwari, P., Yoshioka, J., and Lee, R. T. (2014) Thioredoxin-interacting protein regulates protein 710 disulfide isomerases and endoplasmic reticulum stress. EMBO Mol. Med. 6, 732-743 711
56. Oslowski, C. M., Hara, T., O'Sullivan-Murphy, B., Kanekura, K., Lu, S., Hara, M., Ishigaki, S., Zhu, L. 712 J., Hayashi, E., Hui, S. T., Greiner, D., Kaufman, R. J., Bortell, R., and Urano, F. (2012) Thioredoxin-713 interacting protein mediates ER stress-induced beta cell death through initiation of the 714 inflammasome. Cell metabolism 16, 265-273 715
57. Zhao, X., Mirza, S., Alshareeda, A., Zhang, Y., Gurumurthy, C. B., Bele, A., Kim, J. H., Mohibi, S., 716 Goswami, M., Lele, S. M., West, W., Qiu, F., Ellis, I. O., Rakha, E. A., Green, A. R., Band, H., and 717 Band, V. (2012) Overexpression of a novel cell cycle regulator ecdysoneless in breast cancer: a 718 marker of poor prognosis in HER2/neu-overexpressing breast cancer patients. Breast cancer 719 research and treatment 134, 171-180 720
58. Singh, P. K., Wen, Y., Swanson, B. J., Shanmugam, K., Kazlauskas, A., Cerny, R. L., Gendler, S. J., 721 and Hollingsworth, M. A. (2007) Platelet-derived growth factor receptor beta-mediated 722 phosphorylation of MUC1 enhances invasiveness in pancreatic adenocarcinoma cells. Cancer 723 Res. 67, 5201-5210 724
59. Scheuner, D., Song, B., McEwen, E., Liu, C., Laybutt, R., Gillespie, P., Saunders, T., Bonner-Weir, 725 S., and Kaufman, R. J. (2001) Translational control is required for the unfolded protein response 726 and in vivo glucose homeostasis. Molecular cell 7, 1165-1176 727
60. Taha, M. S., Nouri, K., Milroy, L. G., Moll, J. M., Herrmann, C., Brunsveld, L., Piekorz, R. P., and 728 Ahmadian, M. R. (2014) Subcellular fractionation and localization studies reveal a direct 729 interaction of the fragile X mental retardation protein (FMRP) with nucleolin. PloS one 9, e91465 730
61. Dallner, G. (1974) Isolation of rough and smooth microsomes--general. Methods in enzymology 731 31, 191-201 732
62. Mohibi, S., Srivastava, S., Wang-France, J., Mirza, S., Zhao, X., Band, H., and Band, V. (2015) 733 Alteration/Deficiency in Activation 3 (ADA3) Protein, a Cell Cycle Regulator, Associates with the 734
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

30
Centromere through CENP-B and Regulates Chromosome Segregation. The Journal of biological 735 chemistry 290, 28299-28310 736
63. Oslowski, C. M., and Urano, F. (2011) Measuring ER stress and the unfolded protein response 737 using mammalian tissue culture system. Methods Enzymol. 490, 71-92 738
64. Harding, H. P., Zeng, H., Zhang, Y., Jungries, R., Chung, P., Plesken, H., Sabatini, D. D., and Ron, D. 739 (2001) Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for 740 translational control in secretory cell survival. Molecular cell 7, 1153-1163 741
65. Huang, B., Bates, M., and Zhuang, X. (2009) Super-resolution fluorescence microscopy. Annual 742 review of biochemistry 78, 993-1016 743
66. Gustafsson, M. G., Shao, L., Carlton, P. M., Wang, C. J., Golubovskaya, I. N., Cande, W. Z., Agard, 744 D. A., and Sedat, J. W. (2008) Three-dimensional resolution doubling in wide-field fluorescence 745 microscopy by structured illumination. Biophysical journal 94, 4957-4970 746
67. Hetz, C., Chevet, E., and Oakes, S. A. (2015) Proteostasis control by the unfolded protein 747 response. Nature cell biology 17, 829-838 748
68. Bu, Y., and Diehl, J. A. (2016) PERK Integrates Oncogenic Signaling and Cell Survival During 749 Cancer Development. Journal of cellular physiology 231, 2088-2096 750
69. Ding, X., Ma, M., Teng, J., Shao, F., Wu, E., and Wang, X. (2016) Numb Protects Human Renal 751 Tubular Epithelial Cells From Bovine Serum Albumin-Induced Apoptosis Through Antagonizing 752 CHOP/PERK Pathway. Journal of cellular biochemistry 117, 163-171 753
70. Zhu, X., Zelmer, A., Kapfhammer, J. P., and Wellmann, S. (2016) Cold-inducible RBM3 inhibits 754 PERK phosphorylation through cooperation with NF90 to protect cells from endoplasmic 755 reticulum stress. FASEB journal : official publication of the Federation of American Societies for 756 Experimental Biology 30, 624-634 757
71. Sanson, M., Auge, N., Vindis, C., Muller, C., Bando, Y., Thiers, J. C., Marachet, M. A., Zarkovic, K., 758 Sawa, Y., Salvayre, R., and Negre-Salvayre, A. (2009) Oxidized low-density lipoproteins trigger 759 endoplasmic reticulum stress in vascular cells: prevention by oxygen-regulated protein 150 760 expression. Circulation research 104, 328-336 761
72. Oommen, D., and Prise, K. M. (2013) Down-regulation of PERK enhances resistance to ionizing 762 radiation. Biochemical and biophysical research communications 441, 31-35 763
73. Shiraishi, H., Okamoto, H., Yoshimura, A., and Yoshida, H. (2006) ER stress-induced apoptosis 764 and caspase-12 activation occurs downstream of mitochondrial apoptosis involving Apaf-1. 765 Journal of cell science 119, 3958-3966 766
74. Dey, P., Rachagani, S., Chakraborty, S., Singh, P. K., Zhao, X., Gurumurthy, C. B., Anderson, J. M., 767 Lele, S., Hollingsworth, M. A., Band, V., and Batra, S. K. (2012) Overexpression of ecdysoneless in 768 pancreatic cancer and its role in oncogenesis by regulating glycolysis. Clinical cancer research : 769 an official journal of the American Association for Cancer Research 18, 6188-6198 770
75. Claudius, A. K., Romani, P., Lamkemeyer, T., Jindra, M., and Uhlirova, M. (2014) Unexpected role 771 of the steroid-deficiency protein ecdysoneless in pre-mRNA splicing. PLoS genetics 10, e1004287 772
76. Rao, R. V., Peel, A., Logvinova, A., del Rio, G., Hermel, E., Yokota, T., Goldsmith, P. C., Ellerby, L. 773 M., Ellerby, H. M., and Bredesen, D. E. (2002) Coupling endoplasmic reticulum stress to the cell 774 death program: role of the ER chaperone GRP78. FEBS Lett. 514, 122-128 775
77. Visioli, F., Wang, Y., Alam, G. N., Ning, Y., Rados, P. V., Nor, J. E., and Polverini, P. J. (2014) 776 Glucose-regulated protein 78 (Grp78) confers chemoresistance to tumor endothelial cells under 777 acidic stress. PloS one 9, e101053 778
78. Fu, Y., Li, J., and Lee, A. S. (2007) GRP78/BiP inhibits endoplasmic reticulum BIK and protects 779 human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 67, 3734-780 3740 781
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

31
79. Wang, M., Ye, R., Barron, E., Baumeister, P., Mao, C., Luo, S., Fu, Y., Luo, B., Dubeau, L., Hinton, 782 D. R., and Lee, A. S. (2010) Essential role of the unfolded protein response regulator GRP78/BiP 783 in protection from neuronal apoptosis. Cell Death Differ. 17, 488-498 784
80. Lee, H. K., Xiang, C., Cazacu, S., Finniss, S., Kazimirsky, G., Lemke, N., Lehman, N. L., Rempel, S. 785 A., Mikkelsen, T., and Brodie, C. (2008) GRP78 is overexpressed in glioblastomas and regulates 786 glioma cell growth and apoptosis. Neuro-oncology 10, 236-243 787
81. Chang, Y. J., Chen, W. Y., Huang, C. Y., Liu, H. H., and Wei, P. L. (2015) Glucose-regulated protein 788 78 (GRP78) regulates colon cancer metastasis through EMT biomarkers and the NRF-2/HO-1 789 pathway. Tumour biology : the journal of the International Society for Oncodevelopmental 790 Biology and Medicine 36, 1859-1869 791
82. Xing, X., Li, Y., Liu, H., Wang, L., and Sun, L. (2011) Glucose regulated protein 78 (GRP78) is 792 overexpressed in colorectal carcinoma and regulates colorectal carcinoma cell growth and 793 apoptosis. Acta histochemica 113, 777-782 794
83. Daneshmand, S., Quek, M. L., Lin, E., Lee, C., Cote, R. J., Hawes, D., Cai, J., Groshen, S., 795 Lieskovsky, G., Skinner, D. G., Lee, A. S., and Pinski, J. (2007) Glucose-regulated protein GRP78 is 796 up-regulated in prostate cancer and correlates with recurrence and survival. Human pathology 797 38, 1547-1552 798
84. Xing, X., Lai, M., Wang, Y., Xu, E., and Huang, Q. (2006) Overexpression of glucose-regulated 799 protein 78 in colon cancer. Clinica chimica acta; international journal of clinical chemistry 364, 800 308-315 801
85. Fernandez, P. M., Tabbara, S. O., Jacobs, L. K., Manning, F. C., Tsangaris, T. N., Schwartz, A. M., 802 Kennedy, K. A., and Patierno, S. R. (2000) Overexpression of the glucose-regulated stress gene 803 GRP78 in malignant but not benign human breast lesions. Breast cancer research and treatment 804 59, 15-26 805
86. Wang, M., and Kaufman, R. J. (2014) The impact of the endoplasmic reticulum protein-folding 806 environment on cancer development. Nature reviews. Cancer 14, 581-597 807
87. Nagelkerke, A., Bussink, J., Mujcic, H., Wouters, B. G., Lehmann, S., Sweep, F. C., and Span, P. N. 808 (2013) Hypoxia stimulates migration of breast cancer cells via the PERK/ATF4/LAMP3-arm of the 809 unfolded protein response. Breast cancer research : BCR 15, R2 810
88. Engelsberg, A., Hermosilla, R., Karsten, U., Schulein, R., Dorken, B., and Rehm, A. (2003) The 811 Golgi protein RCAS1 controls cell surface expression of tumor-associated O-linked glycan 812 antigens. J. Biol. Chem. 278, 22998-23007 813
814 815
on June 29, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

A Novel Interaction of Ecdysoneless (ECD) Protein with R2TPComplex Component RUVBL1 Is Required for the Functional Role ofECD in Cell Cycle Progression
Riyaz A. Mir,a Aditya Bele,a Sameer Mirza,a Shashank Srivastava,a Appolinaire A. Olou,a Shalis A. Ammons,a Jun Hyun Kim,a*Channabasavaiah B. Gurumurthy,a Fang Qiu,g Hamid Band,a,b,c,d,e,f Vimla Banda,f
Departments of Genetics, Cell Biology, and Anatomy,a Pathology and Microbiology,b Pharmacology and Experimental Therapeutics,c and Biochemistry and MolecularBiology,d College of Medicine, Eppley Institute for Research in Cancer and Allied Diseases,e Fred and Pamela Buffett Cancer Center,f and Department of Biostatistics,College of Public Health,g University of Nebraska Medical Center, Omaha, Nebraska, USA
Ecdysoneless (ECD) is an evolutionarily conserved protein whose germ line deletion is embryonic lethal. Deletion of Ecd in cellscauses cell cycle arrest, which is rescued by exogenous ECD, demonstrating a requirement of ECD for normal mammalian cellcycle progression. However, the exact mechanism by which ECD regulates cell cycle is unknown. Here, we demonstrate that ECDprotein levels and subcellular localization are invariant during cell cycle progression, suggesting a potential role of posttransla-tional modifications or protein-protein interactions. Since phosphorylated ECD was recently shown to interact with the PIH1D1adaptor component of the R2TP cochaperone complex, we examined the requirement of ECD phosphorylation in cell cycle pro-gression. Notably, phosphorylation-deficient ECD mutants that failed to bind to PIH1D1 in vitro fully retained the ability tointeract with the R2TP complex and yet exhibited a reduced ability to rescue Ecd-deficient cells from cell cycle arrest. Biochemi-cal analyses demonstrated an additional phosphorylation-independent interaction of ECD with the RUVBL1 component of theR2TP complex, and this interaction is essential for ECD’s cell cycle progression function. These studies demonstrate that interac-tion of ECD with RUVBL1, and its CK2-mediated phosphorylation, independent of its interaction with PIH1D1, are importantfor its cell cycle regulatory function.
Precisely regulated cell proliferation is essential for embryonicdevelopment as well as homeostasis in adult organs and tis-
sues, whereas uncontrolled cell proliferation is a hallmark of can-cer (1). A more in-depth understanding of the regulatory controlsof cell cycle progression is therefore of great interest.
The Ecd gene was originally inferred from studies of Drosophilamelanogaster ecdysoneless (or ecd) mutants that exhibit defectivedevelopment due to reduced production of the steroid hormoneecdysone (2). Subsequent cloning of drosophila ecd helped iden-tify a cell-autonomous role of ECD protein in cell survival asidefrom its non-cell-autonomous role in ecdysis (molting) (3). How-ever, the molecular basis of how ECD functions remains unknown(3). The human ECD homologue was initially identified in ascreen of human open reading frames that complemented the S.cerevisiae mutants lacking Gcr2 (glycolysis regulation 2) gene, andit rescued the growth defect caused by reduced glycolytic enzymeactivity in Gcr2 mutants. The human gene was initially designatedHSGT1 (human suppressor of Gcr2) and was suggested to func-tion as a coactivator of glycolytic gene transcription (4). However,ECD protein bears no structural homology to Gcr2, and a trueECD orthologue is absent in S. cerevisiae, suggesting that ECDlikely functions by distinct mechanisms.
We identified human ECD in a yeast two-hybrid screen of hu-man mammary epithelial cell cDNA-encoded proteins for novelbinding partners of the human papillomavirus 16 (HPV16) E6oncogene (5). We showed that deletion of Ecd gene in mice causesembryonic lethality, identifying an essential role of ECD duringearly embryonic development (6). Notably, Cre-mediated condi-tional deletion of Ecd in Ecdfl/fl mouse embryonic fibroblasts(MEFs) led to a G1/S cell cycle arrest, and this phenotype wasrescued by the ectopic expression of human ECD (6), indicating
an essential role of ECD in promoting cell cycle progression. Weshowed that ECD can interact with the retinoblastoma (RB) pro-tein and reduces the repression of RB on E2F transcription factors,providing a novel mechanism by which ECD functions as a posi-tive factor of mammalian cell cycle progression (6). Recently, ECDwas shown to play a vital role in pre mRNA splicing by interactingwith the pre-mRNA-processing-splicing factor 8 (PRPF8) (7). Weand others have shown that ECD shuttles between nucleus and thecytoplasm, with a predominantly cytoplasmic steady-state local-ization due to rapid nuclear export (7, 8). Consistent with thesekey cellular roles of ECD, we found that ECD is significantly over-expressed in breast and pancreatic cancers, and its overexpressioncorrelates positively with poor prognostic factors and poor patientsurvival (9, 10).
A pulldown screen using the phospho-peptide-binding do-
Received 15 June 2015 Returned for modification 6 July 2015Accepted 18 December 2015
Accepted manuscript posted online 28 December 2015
Citation Mir RA, Bele A, Mirza S, Srivastava S, Olou AA, Ammons SA, Kim JH,Gurumurthy CB, Qiu F, Band H, Band V. 2016. A novel interaction of Ecdysoneless(ECD) protein with R2TP complex component RUVBL1 is required for thefunctional role of ECD in cell cycle progression. Mol Cell Biol 36:886 – 899.doi:10.1128/MCB.00594-15.
Address correspondence to Vimla Band, [email protected].
* Present address: Jun Hyun Kim, Molecular Biology Program, Memorial Sloan-Kettering Cancer Center, New York, New York, USA.
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00594-15.
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
crossmark
886 mcb.asm.org March 2016 Volume 36 Number 6Molecular and Cellular Biology
on June 23, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

main of PIH1D1, the adaptor component of the evolutionarilyconserved prefoldin-like cochaperone complex R2TP, recentlyidentified ECD as one of the binding partners (11). This interac-tion was shown to require dual phosphorylation of Ser-505 andSer-518 on ECD (11), suggesting that ECD phosphorylation maymediate its interaction with the R2TP complex. To date, this in-teraction has not been demonstrated in the context of endogenousECD nor has a functional role of this interaction been determined.The core R2TP complex is composed of four proteins: PIH1D1,RPAP3, RUVBL1, and RUVBL2 (each with a number of othernames) (12). The R2TP complex is involved in the assembly ofmultisubunit complexes, including the small nucleolar ribo-nucleoproteins, RNA polymerase II, and phosphatidylinositol3-kinase-related kinases and their complexes (13–15). As such, theR2TP complex is involved in a number of essential cellular pro-cesses. The closely related RUVBL1 and RUVBL2 proteins areAAA� (ATPases associated with diverse cellular activities) thatare essential for R2TP function (16). Recent studies have shownthat RUVBL1 (Pontin) plays an important role in cell cycle regu-lation (17, 18). Germ line deletion of Ruvbl1 was shown to be earlyembryonic lethal (18, 19). Depletion of RUVBL1 in AML1-ETOfusion oncogene-expressing leukemic cells was shown to causecell cycle arrest (17) and Cre-mediated deletion of Ruvbl1 inRuvbl1fl/fl cells also led to G1/S cell cycle arrest (18). The apparentsimilarities in the embryonic lethality and cell cycle arrest pheno-types imparted by the loss of ECD or RUVBL1 expression sug-gested the likelihood that the recently described interaction withthe R2TP complex (11) may underlie the functional requirementof ECD in cell cycle progression.
In this study, we extensively analyzed the mechanism of ECD-R2TP interaction and how disabling this interaction by mutationsin ECD affects the latter’s role in cell cycle progression. We dem-onstrate that ECD levels and localization do not vary during cellcycle progression. We show that casein kinase 2 (CK2) phosphorylates ECD in cells at 6 major sites and a mutant ECD (6S/A)disabled for CK2-mediated phosphorylation exhibits reducedability to rescue the cell cycle arrest caused by Ecd gene deletion.Notably, whereas ECD can interact with PIH1D1, loss of this in-teraction by mutating CK2 phosphorylation sites did not impactthe ECD-R2TP association in cells. We identified a novel interac-tion of ECD with RUVBL1, independent of ECD’s interactionwith PIH1D1, which we show to be essential for ECD’s cellcycle progression function. Notably, a phosphomimetic mu-tant (6S/D) of ECD failed to bind PIH1D1 and was incompetent atrescuing the cell cycle arrest caused by Ecd gene deletion, suggest-ing a potential accessory role for PIH1D1-ECD interaction. Takentogether, our results demonstrate that although CK2-mediatedphosphorylation of ECD is important for its role in cell cycle pro-gression, ECD’s interaction with PIH1D1 is dispensable, suggest-ing that the novel RUVBL1-ECD interaction that we identified isparticularly critical for ECD’s function in cell cycle.
MATERIALS AND METHODSReagents. � protein phosphatase (catalog no. P9614) was purchased fromSigma-Aldrich USA, and the treatment was given according to the man-ufacturer’s instructions. 12.5% SuperSep Phos-tag (50 �mol/liter) waspurchased from Wako Laboratory Chemicals (catalog no. 195-16391).Electrophoresis was performed according to the manufacturer’s protocol.PreScission protease was purchased from GE Healthcare Life Sciences.
Cell cultures. HEK-293T, MEFs, and T98G glioblastoma cell lineswere grown in DMEM (Life Technologies, Grand Island, NY) supple-mented with 10% fetal calf serum. Immortal mammary epithelial cell line76NTERT was cultured in DFCI-1 medium, as described previously (20).U2OS cell line was cultured in �-minimum essential medium (�-MEM).The CK2 inhibitor TBB (4,5,6,7-tetrabromo-2-azabenzimidazole) wasdissolved in dimethyl sulfoxide and used at 50 �M.
Plasmid constructs, site-directed mutagenesis, and transfection.Generation of the pMSCV-puro (Clontech)-based expression constructsfor FLAG-ECD, and its truncated versions has been described previously(6, 8). The pMSCV-puro construct expressing ECD with deletion ofamino acids 499 to 527 was generated using a thee-fragment ligation intoBglII and HpaI sites. C-terminal His6-tagged ECD truncations (1 to 567, 1to 534, and 1 to 432) were generated through PCR amplification andcloning into XbaI and SalI sites of pET-28b� vector (Invitrogen), andrecombinant proteins were purified after expression in Escherichia coliBL21(DE3) strain using a nickel affinity column (GE Healthcare). C-ter-minal His6-tagged full-length ECD was generated by cloning ECD codingsequence into SalI and NotI sites of the pFastBac1 vector (Invitrogen),expressed in Sf21 insect cells, and purified by using a nickel affinity col-umn. The PIH1D1-specific and control siRNAs catalog no. Sc-97385;Santa Cruz) were transfected into subconfluent cells using DharmaFECT1transfection reagent (Thermo Scientific). Green fluorescent protein(GFP)-tagged full-length or truncated ECD expression constructs inthe pGEn2 vector were generated by replacing the ST6-Gal1 insertin the ST6-Gal1-pGEn2 construct (ST6GAL1-pXLG-NtermTCMhis-Strep-DEST) (21) with PCR-amplified ECD coding sequences at theEcoRI and HindIII sites by infusion cloning kit (Clontech). The primersequences used for cloning are indicated in Table S2 in the supplementalmaterial. Human PIH1D1 cDNA sequences clone SC321317; Origene)were subcloned into BamHI and XhoI sites of pGEX-6p-1 for expressionas a glutathione S-transferase (GST) fusion protein in E. coli BL21. Arecombinant GST-PERK kinase domain was expressed in BL21(DE3)cells purified as a GST fusion protein.
Point mutants of ECD were generated using a PCR-based commercialkit (GENEART site-directed mutagenesis system; Invitrogen), accordingto the manufacturer’s instructions, cloned into the pET28b� vector forHis-tagged recombinant protein expression, and purified by using anickel affinity column. The PCR primer sequences are listed in Table S2 inthe supplemental material. All constructs were verified by sequencing.
DNA constructs were transfected in HEK-293T cells using theX-tremeGENE transfection reagent (Roche). Retroviral infection was car-ried out as described previously (6).
Flow cytometry for cell cycle analysis and biochemical fraction-ation. 76NTERT cells were plated at 5 � 105 cells per 100-mm dish for 12h, subjected to growth factor deprivation by culturing in growth factor-free DFCI-3 medium for 72 h (20) and released from synchrony usinggrowth factor-containing DFCI-1 medium (20). Half of the cells werefixed for fluorescence-activated cell sorter (FACS) analysis after fixation inchilled 70% ethanol and staining with propidium iodide; the remainingcells were used for Western blotting. G2/M-to-G1 progression in MEFswas similarly assessed using FACS analysis after nocodazole (100 ng/ml)-dependent arrest in the early G2/M phase of cell cycle (22). Nuclear andcytoplasmic fractions were prepared from cells at various times pointsduring cell cycle progression using the NE-PER kit (Thermo Scientific,catalog no. 78833). Ecdflox/flox MEFs were infected with adeno-Cre-GFP orcontrol adeno-GFP to assess the mitotic index. Cells were collected andfixed as described above. The cell pellet was resuspended in 100 �l ofphosphate-buffered saline (PBS) containing 1% bovine serum albumin(BSA) and 0.25 �g of phospho-H3-S10 (catalog no. ab14955; Abcam) andthen incubated for 1 h at room temperature. Cells were washed in 150 �lof PBS, resuspended in Alexa Fluor 647 (catalog no. A212235; Life Tech-nologies)-conjugated goat anti-mouse antibody diluted at a ratio of 1:300in 100 �l of PBS containing 1% BSA, and incubated at room temperaturein the dark for 30 min, followed by FACS analysis. The median fluores-
ECD-RUVBL1 Interaction Regulates the Cell Cycle
March 2016 Volume 36 Number 6 mcb.asm.org 887Molecular and Cellular Biology
on June 23, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

cence intensity (MFI) of GFP-positive cells at 488- and 633-nm wave-lengths was recorded as an indicator of mitosis in the control and Ecd-nullcells.
Immunoblotting and IP. Cells were lysed in radioimmunoprecipita-tion assay (RIPA) buffer (20 mM Tris [pH 7.2], 150 mM NaCl, 1% TritonX-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS]),and the protein concentration was measured using the bicinchoninicacid (BCA) protein assay reagent (Pierce). Immunoblotting was per-formed with primary antibodies against ECD (9), RB (catalog no.554136; Pharmingen), anti-phospho-Ser (05-1000; Millipore), anti-phospho-Thr (AB1607; Millipore), PIH1D1 (sc-101000 or sc-390810;Santa Cruz), RUVBL1 (12300S [Cell Signaling] or SAB4200194 [Sigma])RUVBL2 (ab36569; Abcam), RPAP3 (HPA038311; Sigma), PARP (sc-8007; Santa Cruz), histone H3 (06-755; Millipore), PRPF8 (ab137694;Abcam), �-actin (A5441; Sigma), or �-tubulin (T6199; Sigma), as indi-cated. For immunoprecipitations (IPs), the cells were lysed in NP-40 lysisbuffer (20 mM Tris-HCl [pH 7.5], 200 mM NaCl, 0.5% Nonidet P-40[NP-40], 1 mM NaF, 0.1 mM Na3VO4, and protease inhibitor mixture[Roche Applied Science]) and then immunoprecipitated with 3 �g ofantibodies against ECD or 35 �l of Ezview red anti-FLAG M2 affinity gel(Sigma) for 2 h to overnight at 4°C. The immune complexes were cap-tured with protein A/G-agarose (sc-2003; Santa Cruz Biotechnology). Toelute FLAG-tagged proteins from anti-FLAG beads before analysis, im-mune complexes were incubated with 150 ng of 3� FLAG peptide (Sig-ma)/�l for 15 min at room temperature, and the supernatants were col-lected for SDS-PAGE. For PIH1D1 interaction with ECD, the cell lysateswere prepared in CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate} lysis buffer (0.3% CHAPS, 0.20 mM Tris-HCl [pH7.4], 120 mM NaCl, 10% glycerol, 5 mM EDTA) supplemented with pro-tease and phosphatase inhibitor (Roche). RUVBL1 immunoprecipitationto assess association with ECD was carried out using a monoclonal anti-RUVBL1 antibody (catalog no. SAB4200194-200UL [Sigma]; 2 �g). Immu-noprecipitated RUVBL1 (close to IgG heavy chain) was detected by West-ern blotting with an anti-RUVBL1 antibody (catalog no. 12300s; CellSignaling) that was conjugated to horseradish peroxidase (HRP) using theLightning-Link HRP conjugation kit (Novus Biologicals).
In vitro kinase assay. A total of 500 ng of purified recombinant ECDproteins or its mutants was incubated with 0.2 mM ATP, 1 �Ci of [�-32P]ATP (Perkin-Elmer), and 0.2 �l (10 U) of human recombinant CK2(NEB, Beverly, MA) at 30°C for 30 min or as indicated. The reaction wasstopped by adding SDS-PAGE sample buffer. The 32P-labeled proteinswere detected by autoradiography after SDS-PAGE and then transfer topolyvinylidene difluoride (PVDF) membranes (Millipore). Once the ra-dioactive signals had decayed, the membranes were blotted with anti-p-Ser antibodies. Ten nanograms of recombinant GST-PERK kinasedomain was autophosphorylated in kinase assay buffer (50 mM HEPES[pH 8.0], 10 mM MgCl2, 2.5 mM EGTA) supplemented with 20 �M coldATP (NEB). Next, 0.5 ng was loaded onto SDS-PAGE gels and subjectedto Western blotting with anti-p-Ser and anti-p-Thr antibodies.
32P metabolic labeling and immunoprecipitation. Exponentiallygrowing or serum-deprived T98G cells were washed with phosphate-freeDulbecco modified Eagle medium (DMEM) supplemented with 10% di-alyzed fetal bovine serum and incubated in the same medium for 1 hbefore adding 0.1 mCi of [32P]orthophosphate (NEN) per 10-cm plate.The cells were labeled for 4 h at 37°C (or for 2, 5, or 16 h for cell cycleanalyses), rinsed once in ice-cold PBS, and lysed in ice-cold lysis buffer(250 mM NaCl, 1% NP-40, 20 mM Tris-HCl [pH 7.4], 1 mM EDTA, 2 �gof aprotinin/ml, 2 �g of leupeptin/ml, 1 �g of pepstatin/ml, 2.5 �g ofantipain/ml, 1 �g of chymostatin/ml, 1 mM Na3VO4, 10 mM NaF, 1 mMsodium molybdate, 0.5 mM phenylmethylsulfonyl fluoride). LabeledECD was immunoprecipitated with affinity-purified mouse anti-Ecdmonoclonal antibody or anti-FLAG beads overnight at 4°C, and Protein GPlus/Protein A-agarose beads were added for 1 h. The beads were washedthee times with ice-cold wash buffer (150 mM NaCl, 1% NP-40, 20 mMTris-HCl [pH 7.4], 1 mM EDTA, and protease inhibitors). The immuno-
precipitated proteins were resolved on 7.5% SDS-polyacrylamide gels,transferred to PVDF membranes, and visualized by autoradiography.
In vitro binding assays. GST-or His-tagged protein pulldowns wereperformed as described previously (6). FLAG-tagged wild-type (WT)ECD or its mutants (3S/A and 6S/A) were expressed by transient transfec-tion in 293T cells and lysed in CHAPS lysis buffer as described above.Then, 1,000 �g of lysate protein was incubated with 2 �g of bead-boundpurified GST-PIH1D1 for 2 h at room temperature, followed by fivewashes, and then the bound proteins were detected by Western blottingwith anti-FLAG antibody. Membranes were stained with Ponceau S tovisualize the GST fusion proteins. In vitro tandem affinity purification(TAP) was performed as described previously (23) using purified recom-binant ECD with a C-terminal FLAG tag.
Cell proliferation and colony formation assays. The cell proliferationwas analyzed as described previously (6). Briefly, Ecdflox/flox MEFs wereinfected with adenoviruses encoding GFP-Cre or GFP (control) (Univer-sity of Iowa Gene Transfer Vector Core) and plated at 104 cells/well insix-well plates, followed by counting of cells at the indicated time points.For assessing colony formation ability, infected cells were plated at 5,000or 1,000 per well in six-well plates for 10 days, the colonies were stainedwith crystal violet (0.5% crystal violet in 25% methanol) and solubilized in10% acetic acid, and then the extent of colony formation was measured bydetermining the absorbance at 590 nm.
Statistical analysis. A generalized estimating equation method wasused to assess the differences among cell types accounting for the corre-lated measurement within a sample. Comparisons between the WT andother cell types at a given time were made with a simulation correction(random sampling from a probability distribution). Some results wereanalyzed using a paired two-tailed Student t test. P values of �0.05 wereconsidered statistically significant.
RESULTSECD levels and localization do not change during cell cycle pro-gression. Given the requirement of ECD for cell cycle progres-sion and its direct association with RB (6), we assessed whetherECD levels or localization are altered during cell cycle progres-sion. For this purpose, an immortal mammary epithelial cellline, 76NTERT, was arrested in the G1 cell cycle phase by growthfactor deprivation, and the cells were then allowed to proceedsynchronously though cell cycle phases by culture in regulargrowth factor-containing medium. FACS analyses showed that amajority of growth factor-deprived cells were growth arrested,with 98% cells in the G1 phase, and only 0.75% cells in the S phaseand 1.25% cells in the G2/M phase (Fig. 1A). Western blotting oflysates showed no significant differences in the levels of ECD pro-tein in cells at various times during cell cycle progression (Fig. 1B).Analysis of nuclear and cytoplasmic fractions prepared at varioustimes during cell cycle progression showed that ECD localizes pri-marily in the cytoplasm (Fig. 1C), which is consistent with itsrapid nuclear export, as previously reported (8). Overall, our re-sults indicate that ECD levels and its subcellular localization donot change significantly during cell cycle progression.
ECD is phosphorylated on serine residues, but overall phos-phorylation does not change during cell cycle progression.Given the known roles of phosphorylation in regulating the cellcycle machinery (24), we sought to determine whether ECD is aphosphoprotein and whether its phosphorylation varies with cellcycle progression. For these analyses, T98G cells (a human brainglioblastoma cell line that expresses a wild-type RB) (25) werecultured in low-serum medium for 48 h to induce growth arrestand then allowed to progress though cell cycle by adding serum-containing medium with 32P-labeled sodium orthophosphate.Autoradiography of anti-ECD immunoprecipitates showed that
Mir et al.
888 mcb.asm.org March 2016 Volume 36 Number 6Molecular and Cellular Biology
on June 23, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

ECD is indeed a phosphoprotein; however, the levels of phosphor-ylation were comparable at various time points during cell cycleprogression (Fig. 2A). As a control, RB showed an expected cellcycle-related increase in phosphorylation at the 16- and 20-h timepoints (Fig. 2A). Further analyses using anti-FLAG immunopre-cipitations (IPs) from [32P]orthophosphate-labeled cells express-ing an exogenous FLAG-tagged ECD protein confirmed the phos-phorylation of ECD in cells (Fig. 2B). These analyses demonstratethat ECD is a phosphoprotein; however, the phosphorylation lev-els do not change during cell cycle progression.
It was reported that a peptide sequence derived from ECD wasphosphorylated by casein kinase 2 (CK2) in vitro (11). To assesswhether the phosphorylation of ECD corresponds to phosphoser-ine (p-Ser) or phosphothreonine (p-Thr) residues, anti-FLAGimmunoprecipitates of T98G cells expressing a FLAG-tagged ECDwere blotted with anti-p-Ser or anti-p-Thr antibodies. A recom-binant GST-PERK kinase domain, known to undergo autophos-phorylation on serine and threonine residues during an in vitrokinase reaction (26), was used as a positive control for serine andthreonine phosphorylation. Indeed, both p-Ser and p-Thr signalswere detected by blotting of autophosphorylated GST-PERK ki-nase domain (Fig. 2C). Although no signals were detected withanti-p-Thr antibody blotting of anti-ECD immunoprecipitation,even after long exposures, a specific band was observed with theanti-p-Ser antibody (Fig. 2C). These results suggested that cellularECD is predominantly phosphorylated on serine residues.
CK2-mediated phosphorylation of ECD is important for itscell cycle regulation function. In view of our results presentedabove, and a recent study that used an array of spotted peptides to
identify CK2 phosphorylation of an ECD peptide on Ser-505 andSer-518 (11), we performed a detailed analysis of potential phos-phorylation sites on ECD using the publically available Kinase-Phos 2.0 tool (http://kinasephos2.mbc.nctu.edu.tw/). This analy-sis identified multiple sites on ECD that could be phosphorylatedby various Ser/Thr kinases. Among these, CK2 was predicted topreferentially phosphorylate multiple serine residues, and this wasof obvious interest in view of our results that cellular ECD is pri-marily phosphorylated on Ser residues (Fig. 2C). The potentialCK2 phosphorylation sites near the C terminus, including Ser-505and Ser-518 reported in the peptide array screen (11), were pre-dicted with the highest confidence (Fig. 3A). To directly assesswhether ECD is a CK2 substrate, we performed an in vitro kinaseassay with purified CK2 and recombinant full-length ECD proteinor its C-terminal truncated versions. Phosphorylation was ob-served with full-length ECD (residues 1 to 644) and its fragmentsencompassing residues 1 to 567 or 1 to 534, whereas substantiallyless phosphorylation was observed with the ECD 1-432 fragment(Fig. 3A and B). These results indicated that ECD was indeed asubstrate for CK2 in vitro and that CK2-dependent phosphoryla-tion occurs predominantly within the C-terminal region of ECD.
CK2 is known to phosphorylate its substrates in clusters, withphosphorylation at one site priming the substrate for phosphory-lation at additional sites (27, 28). The C-terminal region containstwo potential Ser clusters: a proximal cluster of S503, S505, andS518 and a distal cluster of S572, S579, and S584 (Fig. 3C). Toassess the contribution of these clusters to CK2-dependent phos-phorylation of ECD, we introduced Ser-to-Ala mutations in theseresidues, individually as well as in combinations (Fig. 3C and D).
FIG 1 ECD localization and expression do not change during cell cycle progression. 76NTERT cells were cell cycle arrested by culturing in growth factor-freeDFCI-3 medium for 72 h and then switched to growth factor-containing DFCI-1 medium to initiate cell cycle progression. (A) Cells were fixed in 70% ethanolat the indicated time points, stained with propidium iodide, and subjected to FACS analysis. (B) Lysates were collected at the indicated time points and subjectedto Western blotting for ECD or �-actin. ImageJ software was used to quantify ECD signals at various time points during cell cycle progression and expressed asrelative to �-actin signals. (C) Nuclear (N) and cytoplasmic (C) fractions were isolated from cells at various time points during cell cycle progression andsubjected to Western blot analysis with anti-ECD antibody. PARP and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) served as positive controls fornuclear and cytoplasmic proteins, respectively. All experiments were carried out in triplicate. H.E., higher exposure.
ECD-RUVBL1 Interaction Regulates the Cell Cycle
March 2016 Volume 36 Number 6 mcb.asm.org 889Molecular and Cellular Biology
on June 23, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from
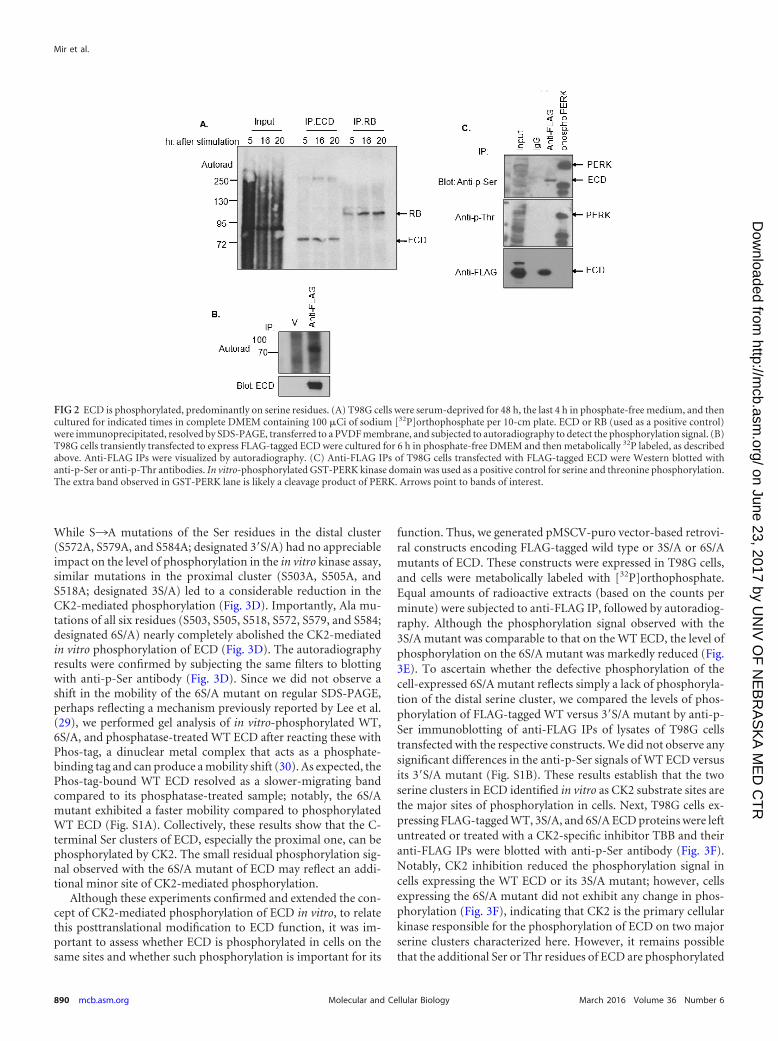
While S¡A mutations of the Ser residues in the distal cluster(S572A, S579A, and S584A; designated 3=S/A) had no appreciableimpact on the level of phosphorylation in the in vitro kinase assay,similar mutations in the proximal cluster (S503A, S505A, andS518A; designated 3S/A) led to a considerable reduction in theCK2-mediated phosphorylation (Fig. 3D). Importantly, Ala mu-tations of all six residues (S503, S505, S518, S572, S579, and S584;designated 6S/A) nearly completely abolished the CK2-mediatedin vitro phosphorylation of ECD (Fig. 3D). The autoradiographyresults were confirmed by subjecting the same filters to blottingwith anti-p-Ser antibody (Fig. 3D). Since we did not observe ashift in the mobility of the 6S/A mutant on regular SDS-PAGE,perhaps reflecting a mechanism previously reported by Lee et al.(29), we performed gel analysis of in vitro-phosphorylated WT,6S/A, and phosphatase-treated WT ECD after reacting these withPhos-tag, a dinuclear metal complex that acts as a phosphate-binding tag and can produce a mobility shift (30). As expected, thePhos-tag-bound WT ECD resolved as a slower-migrating bandcompared to its phosphatase-treated sample; notably, the 6S/Amutant exhibited a faster mobility compared to phosphorylatedWT ECD (Fig. S1A). Collectively, these results show that the C-terminal Ser clusters of ECD, especially the proximal one, can bephosphorylated by CK2. The small residual phosphorylation sig-nal observed with the 6S/A mutant of ECD may reflect an addi-tional minor site of CK2-mediated phosphorylation.
Although these experiments confirmed and extended the con-cept of CK2-mediated phosphorylation of ECD in vitro, to relatethis posttranslational modification to ECD function, it was im-portant to assess whether ECD is phosphorylated in cells on thesame sites and whether such phosphorylation is important for its
function. Thus, we generated pMSCV-puro vector-based retrovi-ral constructs encoding FLAG-tagged wild type or 3S/A or 6S/Amutants of ECD. These constructs were expressed in T98G cells,and cells were metabolically labeled with [32P]orthophosphate.Equal amounts of radioactive extracts (based on the counts perminute) were subjected to anti-FLAG IP, followed by autoradiog-raphy. Although the phosphorylation signal observed with the3S/A mutant was comparable to that on the WT ECD, the level ofphosphorylation on the 6S/A mutant was markedly reduced (Fig.3E). To ascertain whether the defective phosphorylation of thecell-expressed 6S/A mutant reflects simply a lack of phosphoryla-tion of the distal serine cluster, we compared the levels of phos-phorylation of FLAG-tagged WT versus 3=S/A mutant by anti-p-Ser immunoblotting of anti-FLAG IPs of lysates of T98G cellstransfected with the respective constructs. We did not observe anysignificant differences in the anti-p-Ser signals of WT ECD versusits 3=S/A mutant (Fig. S1B). These results establish that the twoserine clusters in ECD identified in vitro as CK2 substrate sites arethe major sites of phosphorylation in cells. Next, T98G cells ex-pressing FLAG-tagged WT, 3S/A, and 6S/A ECD proteins were leftuntreated or treated with a CK2-specific inhibitor TBB and theiranti-FLAG IPs were blotted with anti-p-Ser antibody (Fig. 3F).Notably, CK2 inhibition reduced the phosphorylation signal incells expressing the WT ECD or its 3S/A mutant; however, cellsexpressing the 6S/A mutant did not exhibit any change in phos-phorylation (Fig. 3F), indicating that CK2 is the primary cellularkinase responsible for the phosphorylation of ECD on two majorserine clusters characterized here. However, it remains possiblethat the additional Ser or Thr residues of ECD are phosphorylated
FIG 2 ECD is phosphorylated, predominantly on serine residues. (A) T98G cells were serum-deprived for 48 h, the last 4 h in phosphate-free medium, and thencultured for indicated times in complete DMEM containing 100 �Ci of sodium [32P]orthophosphate per 10-cm plate. ECD or RB (used as a positive control)were immunoprecipitated, resolved by SDS-PAGE, transferred to a PVDF membrane, and subjected to autoradiography to detect the phosphorylation signal. (B)T98G cells transiently transfected to express FLAG-tagged ECD were cultured for 6 h in phosphate-free DMEM and then metabolically 32P labeled, as describedabove. Anti-FLAG IPs were visualized by autoradiography. (C) Anti-FLAG IPs of T98G cells transfected with FLAG-tagged ECD were Western blotted withanti-p-Ser or anti-p-Thr antibodies. In vitro-phosphorylated GST-PERK kinase domain was used as a positive control for serine and threonine phosphorylation.The extra band observed in GST-PERK lane is likely a cleavage product of PERK. Arrows point to bands of interest.
Mir et al.
890 mcb.asm.org March 2016 Volume 36 Number 6Molecular and Cellular Biology
on June 23, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

FIG 3 Phosphorylation of ECD is important for its ability to rescue cell cycle arrest in Ecd-null MEFs. (A) Schematic of ECD protein, its C-terminal deletedconstructs, and CK2 phosphorylation sites predicted by the KinasePhos 0.2 tool (http://kinasephos2.mbc.nctu.edu.tw/). (B) ECD is predominantly phosphor-ylated near its C terminus. In vitro kinase reactions of full-length (aa 1 to 644) ECD and various C-terminal deletion fragments (aa 1 to 567, 1 to 534, and 1 to 432)with human recombinant CK2 were separated by SDS-PAGE, transferred to PVDF membranes, and subjected to autoradiography to detect 32P signals. Thepurity of proteins was assessed by Coomassie brilliant blue staining (CBB). (C) Schematic representation of various point mutants. Black rectangles, WT Serresidues; white rectangles, mutant Ala residues. (D) CK2 phosphorylates ECD at six sites. His-tagged wild-type ECD or its point mutants were purified by nickelaffinity purification and subjected to an in vitro kinase assay, as described above. 32P labeling was detected using autoradiography, and the filters weresubsequently subjected to Western blotting with anti-p-Ser antibody and reprobed with anti-ECD antibody for equal loading. The purity of recombinantproteins was assessed by CBB staining. (E) CK2-dependent phosphorylation of ECD at multiple residues in cultured cells. T98G cells expressing FLAG-taggedECD or its phosphorylation site mutants 3S/A or 6S/A were metabolically labeled with 32P, as described above. FLAG-tagged ECD and its mutants wereimmunoprecipitated and subjected to autoradiography. IP of cells expressing WT ECD subjected to phosphatase treatment is shown. The blot was reprobed
ECD-RUVBL1 Interaction Regulates the Cell Cycle
March 2016 Volume 36 Number 6 mcb.asm.org 891Molecular and Cellular Biology
on June 23, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

by other kinases, depending on the cell type or varying functionalstates.
Next, we assessed whether the phosphorylation of ECD onCK2-dependent serine clusters is relevant to its cell cycle regula-tory function. We have previously demonstrated that introduc-tion of Cre recombinase in Ecdfl/fl MEFs, using adenovirus Cre,causes G1 cell cycle arrest that is largely rescued by introducinghuman ECD (6). We used this approach to compare the extent ofthe rescue of cell cycle arrest induced by endogenous ECD dele-tion upon introducing the wild-type ECD or its phosphodefectivemutants. In initial experiments, we expressed the WT humanECD or its 3S/A or 6S/A mutants in Ecdfl/fl MEFs (Fig. 3G) andthen assessed the ability of the cells to progress through cell cyclewithout or with Cre-induced Ecd deletion. In each case, the ex-pression of exogenous human ECD proteins and the depletion ofendogenous mouse ECD were confirmed by Western blotting(Fig. 3G). As expected, the deletion of Ecd in Ecdfl/fl MEFs arrestedproliferation with no recovery during the entire observation pe-riod (Fig. 3H), and ectopic WT ECD significantly rescued the cellsfrom growth arrest (Fig. 3H; also see Fig. S2A in the supplementalmaterial). Notably, while the 3S/A mutant behaved comparably toWT ECD in rescuing cells from growth arrest, the 6S/A mutantonly exhibited a partial rescue in comparison to that seen with WTECD in repeated experiments (P 0.001) (Fig. 3H) (all P valuesare shown in Table S1 in the supplemental material). Further-more, we examined a mutant in which serine residues 503, 505,and 518 were removed by deletion (499 –527) in the cell cyclerescue experiment and observed that this mutant behaved simi-larly to WT ECD in rescuing the proliferation block (see Fig. S2Aand B in the supplemental material). Next, we generated a phos-phomimetic mutant in which the six serine residues identified tobe phosphorylated were mutated to aspartic acid residues (6S/D).Notably, the phosphomimetic mutant 6S/D was completely defec-tive in cell cycle rescue experiment (Fig. 3I and J). In these exper-iments, we also examined the 3=S/A mutant and observed a partialrescue with this mutant (Fig. 3J). Although the complete lack ofrescue seen with the 6S/D mutant of ECD was surprising, it hasbeen reported that aspartic acid phosphomimics are unsuitablefor biological readouts due to different chemical properties of thetwo residues (31, 32). Taken together, our results underscore theimportance of ECD phosphorylation for cell cycle progression.
Phosphodefective ECD mutants retain their ability to inter-act with PIH1D1 protein, as well as with other components ofthe R2TP complex. In view of the complete lack of any functionalimpact of mutating ECD on S503/505/518 residues, we first reex-amined the previously reported dependence of ECD binding tothe isolated phospho-reader domain of PIH1D1 (11). For thispurpose, GST-PIH1D1 pulldown was carried out with lysates ofHEK-293T cells transiently transfected to express WT ECD or its
3S/A and 6S/A mutants. Confirming previous findings (11), WTECD, but not its 3S/A or 6S/A mutant, was pulled down withGST-PIH1D1 (Fig. 4A). These results suggested that either theR2TP association was unnecessary for ECD function in cell cycleprogression or an alternate mechanism may recruit ECD to theR2TP complex. To distinguish between these possibilities, we firstcarried out anti-ECD IPs of U2OS and MEF cell lysates, followedby anti-PIH1D1 blotting. These analyses confirmed the interac-tion of endogenous ECD and PIH1D1 (Fig. 4B and C). To exam-ine the nature of ECD/PIH1D1 interaction in cells, we carried outanti-PIH1D1 immunoprecipitations from HEK-293T cells ex-pressing untagged (Fig. 4D) or GFP-tagged ECD (Fig. 4E; see alsoFig. S3A in the supplemental material), WT ECD or its phosphor-ylation site mutants (defective in binding to PIH1D1 in the pull-down assay), followed by blotting for ECD (Fig. 4D) or GFP (Fig.4E, see also Fig. S3A in the supplemental material), as well as forthe four R2TP complex components (Fig. 4D and E; see also Fig.S3A in the supplemental material). Since a phosphorylatedDSpDD/E motif, conserved between human and mouse ECD pro-teins (Fig. 4F), was previously found to promote the interaction ofECD with PIH1D1 in vitro (11), we also examined a deletion con-struct (499 –527) of ECD that lacks the DSDD/E motif in ad-dition to the S6/A mutant lacking all CK2-phosphorylatedsites. As expected, PIH1D1 IPs were able to co-IP RPAP3,RUVBL1, or RUVBL2 to a similar extent in all lanes (Fig. 4Dand E). Notably, compared to the levels of endogenous ECD co-IPwith PIH1D1 in vector control lanes, increased amounts of ectop-ically expressed ECD were coimmunoprecipitated from WTECD-transfected cell lysates. Unexpectedly, however, the WT andmutant ECD proteins were coimmunoprecipitated with PIH1D1to comparable levels (Fig. 4D and E; see also Fig. S3A in the sup-plemental material). These results demonstrate that ECD interactswith the R2TP complex in cells but that the phosphorylation-dependent interaction of ECD with PIH1D1 is dispensable for thisassociation. In further support of this conclusion, we carried outanti-ECD and anti-PIH1D1 IPs, treated these IPs with lambdaphosphatase, and then assessed the levels of coimmunoprecipi-tated PIH1D1. Notably, although the phosphatase treatment ro-bustly eliminated the phosphorylation signal on ECD (anti-p-Serblot), no reduction in PIH1D1or ECD coimmunoprecipitationwas seen (see Fig. S3B and C in the supplemental material). Thus,while CK2-phosphorylated ECD can directly interact withPIH1D1, as reported previously (11), this interaction is not re-quired for the association of ECD with the R2TP complex. Next,we examined the ability of 6S/D or 3=S/A mutants of ECD tointeract with PIH1D1. For this purpose, lysates from 293T cellsexpressing FLAG-tagged 3=S/A or 6S/D mutants were used for anin vitro pulldown assay with GST-PIH1D1. As expected, GST-PIH1D1 was able to pull down the 3=S/A mutant but failed to pull
with anti-ECD antibody for equal IP loading. (F) Inhibition of CK2 reduces ECD phosphorylation. T98G cells expressing FLAG-tagged ECD or its phosphor-ylation site mutants 3S/A and 6S/A were treated with CK2 inhibitor TBB (50 �M) for 4 h, subjected to anti-FLAG IP, and Western blotted with anti-p-Ser oranti-FLAG antibodies. The intensity of anti-p-Ser signals was quantified using ImageJ software and normalized relative to FLAG-ECD signals. (G) Phosphory-lation of ECD at six CK2 sites is important for its cell cycle progression function. Ecdflox/flox MEFs stably expressing vector control, wild-type human ECD, or itsindicated mutants were infected with control (ctrl) adeno-GFP or adeno-GFP-Cre (cre) viruses for the indicated times, and lysates were analyzed by anti-ECDand �-tubulin (loading control). Note that human ECD-reconstituted cells express both endogenous mouse (mEcd higher band) and ectopic hECD or itsmutants (hECD, lower band). (H to J) Analysis of hECD or its mutants for rescue of Ecdfl/fl MEFs from cell cycle arrest induced by Cre-mediated Ecd deletion.Ecdfl/fl MEFs expressing vector (V), WT hECD or its 3S/A, 6S/A, 6S/D or 3=S/A mutants were infected with control (ctrl) or Cre adenoviruses, followed by cellcounting at the indicated time points. The Cre/Ctrl cell number ratios at each time point were plotted to assess the level of rescue relative to vector-expressingMEFs. Simulation’s correction was applied to control for multiple testing in the calculation of the mean ratio. The experiment is representative of three repeats.
Mir et al.
892 mcb.asm.org March 2016 Volume 36 Number 6Molecular and Cellular Biology
on June 23, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from
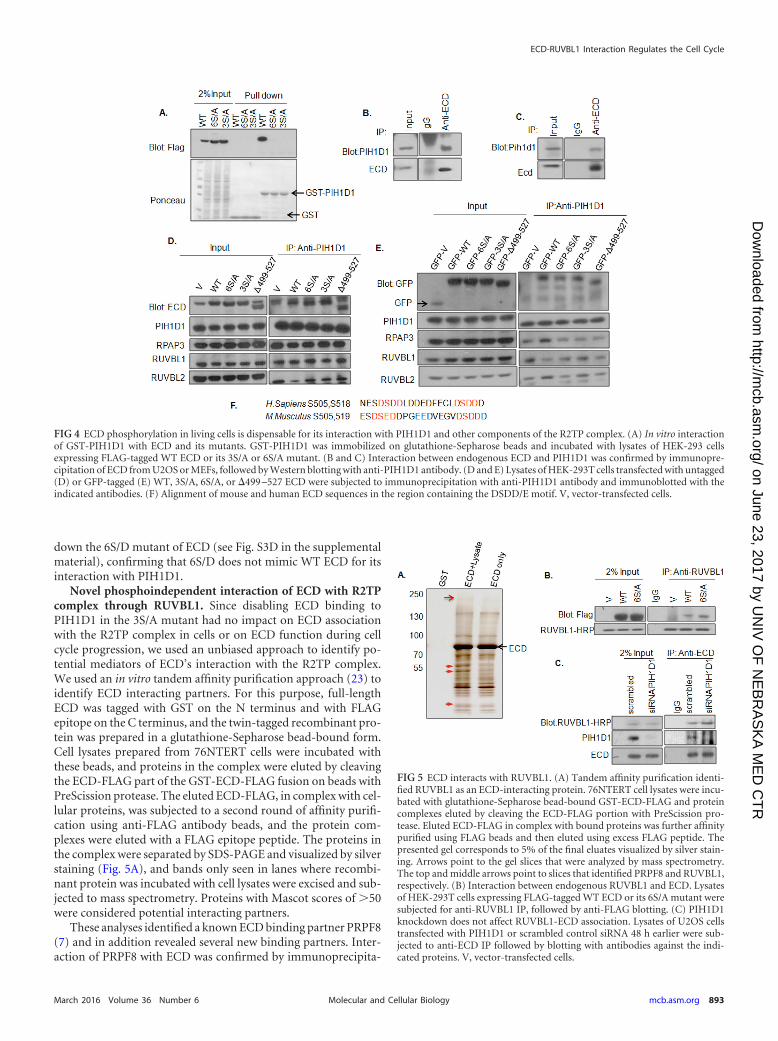
down the 6S/D mutant of ECD (see Fig. S3D in the supplementalmaterial), confirming that 6S/D does not mimic WT ECD for itsinteraction with PIH1D1.
Novel phosphoindependent interaction of ECD with R2TPcomplex through RUVBL1. Since disabling ECD binding toPIH1D1 in the 3S/A mutant had no impact on ECD associationwith the R2TP complex in cells or on ECD function during cellcycle progression, we used an unbiased approach to identify po-tential mediators of ECD’s interaction with the R2TP complex.We used an in vitro tandem affinity purification approach (23) toidentify ECD interacting partners. For this purpose, full-lengthECD was tagged with GST on the N terminus and with FLAGepitope on the C terminus, and the twin-tagged recombinant pro-tein was prepared in a glutathione-Sepharose bead-bound form.Cell lysates prepared from 76NTERT cells were incubated withthese beads, and proteins in the complex were eluted by cleavingthe ECD-FLAG part of the GST-ECD-FLAG fusion on beads withPreScission protease. The eluted ECD-FLAG, in complex with cel-lular proteins, was subjected to a second round of affinity purifi-cation using anti-FLAG antibody beads, and the protein com-plexes were eluted with a FLAG epitope peptide. The proteins inthe complex were separated by SDS-PAGE and visualized by silverstaining (Fig. 5A), and bands only seen in lanes where recombi-nant protein was incubated with cell lysates were excised and sub-jected to mass spectrometry. Proteins with Mascot scores of �50were considered potential interacting partners.
These analyses identified a known ECD binding partner PRPF8(7) and in addition revealed several new binding partners. Inter-action of PRPF8 with ECD was confirmed by immunoprecipita-
FIG 4 ECD phosphorylation in living cells is dispensable for its interaction with PIH1D1 and other components of the R2TP complex. (A) In vitro interactionof GST-PIH1D1 with ECD and its mutants. GST-PIH1D1 was immobilized on glutathione-Sepharose beads and incubated with lysates of HEK-293 cellsexpressing FLAG-tagged WT ECD or its 3S/A or 6S/A mutant. (B and C) Interaction between endogenous ECD and PIH1D1 was confirmed by immunopre-cipitation of ECD from U2OS or MEFs, followed by Western blotting with anti-PIH1D1 antibody. (D and E) Lysates of HEK-293T cells transfected with untagged(D) or GFP-tagged (E) WT, 3S/A, 6S/A, or 499 –527 ECD were subjected to immunoprecipitation with anti-PIH1D1 antibody and immunoblotted with theindicated antibodies. (F) Alignment of mouse and human ECD sequences in the region containing the DSDD/E motif. V, vector-transfected cells.
FIG 5 ECD interacts with RUVBL1. (A) Tandem affinity purification identi-fied RUVBL1 as an ECD-interacting protein. 76NTERT cell lysates were incu-bated with glutathione-Sepharose bead-bound GST-ECD-FLAG and proteincomplexes eluted by cleaving the ECD-FLAG portion with PreScission pro-tease. Eluted ECD-FLAG in complex with bound proteins was further affinitypurified using FLAG beads and then eluted using excess FLAG peptide. Thepresented gel corresponds to 5% of the final eluates visualized by silver stain-ing. Arrows point to the gel slices that were analyzed by mass spectrometry.The top and middle arrows point to slices that identified PRPF8 and RUVBL1,respectively. (B) Interaction between endogenous RUVBL1 and ECD. Lysatesof HEK-293T cells expressing FLAG-tagged WT ECD or its 6S/A mutant weresubjected for anti-RUVBL1 IP, followed by anti-FLAG blotting. (C) PIH1D1knockdown does not affect RUVBL1-ECD association. Lysates of U2OS cellstransfected with PIH1D1 or scrambled control siRNA 48 h earlier were sub-jected to anti-ECD IP followed by blotting with antibodies against the indi-cated proteins. V, vector-transfected cells.
ECD-RUVBL1 Interaction Regulates the Cell Cycle
March 2016 Volume 36 Number 6 mcb.asm.org 893Molecular and Cellular Biology
on June 23, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

tion (see Fig. S3E in the supplemental material). Among the newpartners, RUVBL1 was one of the top candidate proteins with aMascot score of 168. To validate the purification results, we ex-pressed the FLAG-tagged WT ECD or its 6S/A mutant in 293Tcells and performed coimmunoprecipitation experiments with ananti-RUVBL1 antibody. Both the WT and the 6S/A ECD proteinswere coimmunoprecipitated with RUVBL1, suggesting that ECDinteracts with RUVBL1 and that this interaction is independent ofECD phosphorylation (Fig. 5B). To further establish that ECD-RUVBL1 interaction is independent of PIH1D1, we knockeddown the endogenous PIH1D1 with siRNA and then performed aco-IP experiment using an anti-ECD antibody. Notably, we ob-served equal co-IP of RUVBL1 in both control and PIH1D1knockdown cells, confirming that ECD interaction with RUVBL1is PIH1D1 independent (Fig. 5C). Taken together, these resultsdemonstrate that ECD associates with the R2TP complex though anovel interaction with RUVBL1, independent of ECD’s interac-tion with PIH1D1.
Interaction with RUVBL1 is important for the role of ECD incell cycle progression. Germ line deletion of Ecd or Ruvbl1 isembryonic lethal (6, 18), and silencing of either protein in cellsleads to cell cycle arrest (6, 17, 18), suggesting that interaction ofECD with RUVBL1 may play a role in the cell cycle regulationfunction of ECD. To test this hypothesis, we first expressed GFP-tagged ECD or its several C-terminal deletion mutants in HEK-293Tcells and performed co-IP experiments using an anti-RUVBL1 anti-body. Notably, only full-length ECD protein (amino acids [aa] 1 to
644) coimmunoprecipitated with RUVBL1, whereas none of the C-terminal deletions of ECD was able to coimmunoprecipitate withRUVBL1 (see Fig. S4A and B in the supplemental material). Tovalidate these results, we constructed several GST-tagged and His-tagged ECD deletion fragments based on the predicted secondarystructure (by Garnier Robson predictions and PONDR VL-XTsecondary structure prediction) and then examined the direct in-teraction of these ECD mutant proteins with FLAG-RUVBL1 orendogenous RUVBL1 using pulldown with glutathione-Sephar-ose or nickel beads. As shown in (Fig. 6A and B; see also Fig. S4C inthe supplemental material), only the full-length ECD interactswith RUVBL1, whereas all C-terminal or N-terminal deletionsrendered ECD defective in binding to RUVBL1. Next, we com-pared various FLAG-tagged deletion fragments of ECD (residues150 to 438, 438 to 644, 1 to 438, and 150 to 644) (see Fig. S4D in thesupplemental material) with wild-type ECD (residues 1 to 644) fortheir abilities to rescue the growth arrest of Ecdfl/fl MEFs uponadeno-Cre-mediated endogenous Ecd deletion by analyzing cellproliferation by cell counting or colony formation (Fig. 6D). Ineach case, the expression of exogenous human ECD proteins andloss of expression of endogenous mouse Ecd in Cre-expressingcells was confirmed using Western blotting (Fig. 6C; see also Fig.S4D in the supplemental material). As expected, deletion of Ecd inEcdfl/fl MEFs arrested cell proliferation, and ectopic WT humanECD significantly rescued the cells from growth arrest (Fig. 6D).However, none of the deletion mutants was able to rescue the cellproliferation block imposed by endogenous ECD depletion. These
FIG 6 Interaction with RUVBL1 is important for ECD function in cell cycle progression. (A and B) Interaction between FLAG-RUVBL1 and ECD. GST-taggedor His-tagged full-length ECD or its truncated mutants immobilized on glutathione-Sepharose or nickel beads, respectively, were incubated with lysates ofHEK-293T cells expressing FLAG-tagged RUVBL1. (C) Western blotting to show the expression of WT human ECD or its deletion mutants in Ecdfl/fl MEFs withcontrol or Cre adenovirus infection (arrowheads point to the human ECD or mutants). Also note that anti-ECD antibody blot does not detect the C-terminallydeleted ECD 1-438 mutant. (D) Colony formation assay. Ecdfl/fl MEFs expressing full-length WT ECD (aa 1 to 644) or its truncations were infected with ctrl orCre adenoviruses, colonies were stained with crystal violet after 10 days, and the solubilized dye absorbance was read at 590 nm. A histogram shows the relativerescue efficiency of each construct compared to vector control cells. Error bars represent the means � the standard deviations of three independent experiments.A statistical comparison used a Student two-tailed t test. (E) Interaction of FLAG-tagged ECD or its deletion fragments with PIH1D1. Lysates of HEK-293T cellsexpressing the indicated FLAG-tagged ECD fragments were used for GST-PIH1D1 pulldown, followed by anti-FLAG blotting. V, vector-transfected cells.
Mir et al.
894 mcb.asm.org March 2016 Volume 36 Number 6Molecular and Cellular Biology
on June 23, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

results demonstrate that only the full-length ECD, which interactswith RUVBL1, supports cell cycle progression. The lack of rescuewith ECD deletion fragments was not due to lack of their expres-sion (Fig. 6C; see also Fig. S4D in the supplemental material).Notably, two deletion fragments (residues 438 to 644 and 150 to644) that failed to rescue cell cycle arrest still retained their ability tointeract with PIH1D1 (Fig. 6E), further underscoring the conclusionthat interaction of ECD with PIH1D1 is dispensable, while its inter-action with RUVBL1 is indispensable for a role in cell cycle progres-sion. PIH1D1 is known to directly interact with other components ofthe R2TP complex, such as RUVBL1 (33). Reprobing of the samemembrane with antibodies against RUVBL1 and RUVBL2 showedthe expected interaction of PIH1D1 with RUVBL1 or RUVBL2 (seeFig. S4E in the supplemental material).
Our previous studies showed that ECD interacts with RB, afunction important for the ECD role in cell cycle progression (6).Notably, in addition to the expected interaction of full-lengthECD with RB, one mutant (aa 150 to 644) that is defective inrescuing cell cycle arrest (Fig. 6D) was earlier shown to retain itsability to interact with RB (6), suggesting that interaction withRUVBL1 is required for ECD to promote cell cycle progressionwhile interaction with RB in the absence of interaction withRUVBL1 is insufficient for this function. In further support ofthis conclusion cell cycle function competent (3S/A) and deficientmutant (6S/A) of ECD show comparable interaction with RB (seeFig. S4F in the supplemental material).
Ecd deletion leads to reduced mitotic index and delayed mi-totic progression. We have previously reported that the prolifer-ation arrest upon Ecd deletion is not associated with any increasein apoptosis (6). To examine the effect of Ecd deletion on mitosis,
we used adeno-Cre to delete Ecd in Ecdfl/fl MEFs and measured theMFI of phospho-histone H3 (S10) as an indicator of the propor-tion of cells in mitosis using flow cytometry (34). Ecd-deleted cellsshowed a marked decrease in the MFI (45.7) of pH 3 (S10) com-pared to control cells (89.8) (Fig. 7A), indicating that Ecd deletioncells are cell cycle arrested prior to entering mitosis. Low levels ofpH 3 (S10) were further confirmed by Western blotting (Fig. 7B).Next, we assessed the G2/M to G1 progression of MEFs arrested inthe S phase by nocodazole treatment (Fig. 7C). Flow cytometryanalysis revealed a significant impairment in G2/M-to-G1-phasetransition upon Ecd deletion compared to control, in addition to ahigher percentage of Ecd-deleted MEFs in the G1 phase (Fig. 7C toE). Taken together, these results demonstrate a critical role of ECDin both G1-to-S and G2/M-to-G1 transitions. These results areconsistent with the known function of CK2 and RUVBL1 in cellcycle regulation (35–37).
DISCUSSION
Precise regulation of the entry into, progression through, and exitfrom the cell cycle is fundamental to developmental programs andmaintenance of adult tissues in multicellular organisms. Notably,components of the cell cycle machinery and the pathways thatregulate their functions are commonly altered in cancer and otherdiseases (1). Thus, elucidating how the cell cycle machinery iscontrolled is an important area of research in cell and cancer bi-ology.
We have previously shown that ECD, the mammalian ortho-logue of Drosophila ecdysoneless gene, is required for embryonicdevelopment and progression of mammalian cells through theG1-S phase of cell cycle progression (6). Here, we identify a novel
FIG 7 Ecd deletion leads to a mitotic block. (A) Ecdflox/flox MEFs infected with control (blue) or Cre adenoviruses (red) were fixed in 70% ethanol, stained withanti-pH3(S10), and analyzed by flow cytometry. The median fluorescence intensity (MFI), representing the peak channel number on the x axis, is shown. (B)Lysates of control (Ctrl) or Ecd-deleted (Cre) Ecdflox/flox MEFs were blotted with the indicated antibodies. (C) Control (Ctrl) or Ecd deletion (Cre) MEFs weretreated with 100 ng of nocodazole/ml for 20 h, and cells were switched to nocodazole-free medium to initiate cell cycle progression for the indicated time points.The cells were stained with propidium iodide and analyzed by FACS for cell cycle analysis. (D) A graph shows the percentage of cells entering the G1 phase afterrelease from nocodazole treatment at various time points. Data points are means � the standard deviations of results from three independent experiments (*, P �0.05, as determined by two-tailed Student t test). (E) The deletion of Ecd in the experiment shown in panel C was confirmed by Western blotting.
ECD-RUVBL1 Interaction Regulates the Cell Cycle
March 2016 Volume 36 Number 6 mcb.asm.org 895Molecular and Cellular Biology
on June 23, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

mechanism by which ECD functions as an essential element ofmammalian cell cycle progression. Using multiple complemen-tary approaches, we demonstrate a novel interaction of ECD withthe R2TP chaperone complex, mediated by the RUVBL1 compo-nent of R2TP, which we establish is required for ECD to promotecell cycle progression. We also identify a role for the CK2-depen-dent phosphorylation of ECD in cell cycle progression. In contrastto predictions from a previous study (11), this role is independentof the ECD interaction with PIH1D1, the phospho-reader com-ponent of the R2TP complex.
Our findings establish that the phosphorylation of ECD posi-tively regulates its function in promoting the cell cycle progres-sion. Bioinformatics analysis, followed by mass spectroscopy-based phosphoproteomics, identified a number of sites that couldbe phosphorylated by cellular kinases, but we focused on two clus-ters of potential CK2 phosphorylated serine residues since a recentstudy (11) showed that CK2-mediated phosphorylation of twosuch serine residues in the context of a peptide created a bindingsite for the phospho-reader subunit of the R2TP complex. CK2-dependent phosphorylation of site-directed mutants of ECD invitro and in cultured cells identified six serine residues in twospatially separated clusters to be the major CK2 phosphorylationsites on ECD. Notably, however, ECD phosphorylation does notchange during cell cycle progression. This is not entirely surpris-ing since our in vitro analyses, as well as phosphorylation studies incells in the presence of a CK2 inhibitor (Fig. 3D and F), establishthat CK2 is the predominant kinase that phosphorylates ECD;CK2 is considered to be constitutively active and ubiquitous ser-ine/threonine protein kinase (38). Despite its constitutive activity,however, numerous studies point to a role for CK2 in cell prolif-eration and survival (39). Yet, the molecular pathways that medi-ate the function of CK2 in cell proliferation are largely unknown.We suggest that phosphorylation of ECD by CK2 provides onemechanism for CK2’s role in cell proliferation. Although the over-all levels and the subcellular localization of ECD remain invariantduring cell cycle progression (Fig. 1 and 2), it remains possible thatECD phosphorylation at specific sites may vary during cell cycleprogression. As phospho-specific antibodies against specific ser-ine residues on ECD become available, it should be feasible to testthis notion further.
Our findings that ECD is indeed a CK2 substrate in vitro (Fig.3) suggested that CK2-dependent phosphorylation and subse-quent interaction of ECD with the R2TP complex could provide apotential mechanism by which ECD could promote the cell cycletransit. Our co-IP studies in cell cultures demonstrate that ECD infact is in a complex that includes the four core subunits of theR2TP complex (Fig. 4D and E). Remarkably, however, multiplemutant ECD proteins, rendered incapable of directly interactingwith PIH1D1, including mutations of critical serine residues in the3S/A mutant or the 6S/A mutant or deletion of the region incor-porating the major CK2 phosphorylation sites and the acidic mo-tif DSDD that facilitates PIH1D1 interaction (11), fully retainedthe ability to associate with the R2TP complex. Furthermore, theECD-R2TP association was retained in PIH1D1-depleted cells(Fig. 5C). Thus, our results support a PIH1D1-independentmechanism of ECD association with the R2TP complex. Impor-tantly, S¡A mutation of ECD residues that impart PIH1D1 bind-ing (3S/A and 499 –527) had no impact on its ability to functionin cell cycle progression. However, 6S/A and 6S/D were defectivein a cell cycle rescue experiment, underscoring the importance of
ECD phosphorylation for its function that encompasses aminoacids beyond PIH1D1 interaction. Thus, the role of phosphoryla-tion in regulating ECD function during cell cycle appears to beindependent of mediating an interaction with PIH1D1. It remainspossible, however, that phosphorylation-dependent interaction ofECD with PIH1D1, and consequently with the R2TP complex, isrequired for other functions of ECD aside from its role in promot-ing cell cycle progression (6). We have shown that ECD overex-pression in cells leads to p53 stabilization and increased p53-de-pendent target gene expression and to the induction of asenescence phenotype in primary fibroblasts (5). ECD was alsofound to interact with thioredoxin-interacting protein (TXNIP),which was shown to promote p53 stabilization (40). TXNIP has anumber of other functions, including the regulation of glucoseuptake, oxidative stress, and endoplasmic reticulum stress-in-duced apoptosis (41, 42), Thus, ECD phosphorylation and inter-action with PIH1D1 may play a role in regulating these functions.The availability of Ecdfl/fl MEFs in which ECD can be conditionallydeleted, together with the phosphorylation-defective mutants thatwe have characterized here, should allow these notions to be testedin the future.
In view of a novel, PIH1D1-independent mechanism of ECDassociation with the R2TP complex, we sought to answer two keymechanistic questions: first, what are the determinants of ECD-R2TP association, and second, whether this unique mode of in-teraction is functionally relevant in the context of cell cycle pro-gression role of ECD. Unbiased proteomics analysis of cellularproteins that interacted with a recombinant full-length ECD pro-tein, followed by biochemical analyses in cells, demonstrated thatECD interacts with another component of the R2TP complex,RUVBL1 (Fig. 5 and 6). Structure-function studies of ECD usingdeletion fragments demonstrated a strong correlation between thecell cycle progression function of ECD and its ability to interactwith RUVBL1, with only the full-length ECD competent at bothfunctions (Fig. 6A and B; see also Fig. S4B and C in the supple-mental material). Interestingly, the 499 –527 mutant which in-teracts with RUVBL1, but not PIH1D1, was able to rescue the cellcycle arrest caused by Ecd deletion (see Fig. S2A in the supplemen-tal material). Thus, our studies identify a novel interaction of ECDwith RUVBL1 and suggest that this mode of interaction with theR2TP complex is a key to the regulation of cell cycle progression byECD. The delineation of sequences in ECD and RUVBL1 thatmediate their interaction should help directly test whether selec-tive abrogation of this interaction is functionally critical in cellcycle progression, as well as to assess the potential role of ECD inother roles of RUVBL1 within the R2TP complex. Interestingly,mouse ECD and RUVBL1 knockouts are phenotypically similar,since both are embryonic lethal at the blastocyst stage (6, 19).RUVBL1 is essential for cellular proliferation as seen in knockoutcells or upon knockdown of RUVBL1 expression (18). A recentstudy demonstrated that RUVBL1 functions as a critical factor forp300 recruitment to OCT4 target genes (18). It is of interest thatECD also interacts with p300 and promotes its transcriptionalcoactivator function (8). Thus, ECD may function in close coor-dination with RUVBL1.
Given the evidence we present that ECD can physically interactwith two distinct components of the R2TP complex, it is conceiv-able that certain ECD functions require both modes of interac-tion. Recent studies have shown that, aside from the R2TP com-plex, RUVBL1/2 are also parts of other functionally relevant
Mir et al.
896 mcb.asm.org March 2016 Volume 36 Number 6Molecular and Cellular Biology
on June 23, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

complexes, such as chromatin remodeling complexes TIP60,SWR/SRCAP, and INO80 and the Fanconi anemia core complexthat controls DNA interstrand cross-link repair and function, andregulate telomerase biogenesis and mitosis (19, 43–45). Given thePIH1D1-independent interaction of ECD with RUVBL1, the po-tential roles of ECD via these alternative complexes will be of greatfuture interest.
An essential role of ECD in cell cycle progression was estab-lished by our previous observation that ECD is essential for em-bryogenesis and its conditional deletion in MEFs leads to a G1-Scell cycle arrest, together with an inability to initiate an E2F-de-pendent transcriptional program essential for cell cycle progres-sion (6). Notably, we demonstrated that ECD competes with E2Ffor binding to the pocket domain of RB and that the cell cycleprogression defect in Ecd-null MEFs could be overcome by re-moving the RB-mediated suppression of E2F using a pocket-bind-ing oncogene HPV16 E7. Since a key mechanism by which theR2TP complex regulates biochemical processes is by facilitatingprotein complex remodeling, we speculate that the interaction ofECD with the R2TP complex through RUVBL1 may facilitate theECD-RB complex formation and helps dissociate RB from E2Fs,thereby derepressing the E2F-mediated transcription and pro-moting cell cycle progression. Consistent with this speculativemodel, our previous studies showed that binding to RB was notsufficient for the cell cycle progression function of ECD, since weidentified one ECD mutant that was able to interact with RB butwas defective in cell cycle rescue.
Our previous studies demonstrated that ECD is overexpressedin breast and pancreatic cancer patient tissues and that ECDoverexpression correlates with poor prognosis and poor sur-vival in breast cancer patients (9, 10). It is noteworthy thatseveral components of the R2TP/prefoldin complex, includingPIH1D1, RUVBL1, and RUVBL2, are also overexpressed invarious cancers and are predicted to play important roles in on-cogenesis (46, 47). A comprehensive meta-analysis of The CancerGenome Atlas (TCGA) data sets (46) revealed that expression ofmany RUVBL complex genes was significantly higher in breastand colorectal carcinomas compared to their normal tissuecontrols. These investigations suggested a correlation betweenRUVBL complex component overexpression and increasedmTORC1 signaling and metabolic processes necessary for tumorcell growth (46). Another study demonstrated that PIH1D1 isoverexpressed in various breast cancer cell lines, where it plays amajor role in rRNA transcription (48). Our recent studies showeda co-oncogenic role of ECD with Ras when introduced into im-mortal human mammary epithelial cells (49), further suggestingthe potential collaborative role of ECD and the R2TP or otherRUVBL-containing complexes in cell cycle regulation and onco-genesis.
A positive role of ECD in pre-mRNA splicing was reportedrecently based on rescue of splicing defects in the prothoracicglands of Ecd-deficient flies by human ECD and interaction ofECD with a complex containing the spliceosome componentPRP8 (7, 50). Our affinity purification/mass spectrometry analy-ses confirmed the interaction of ECD with PRPF8. The R2TPcomplex regulates mRNA and ribosome biogenesis by facilitatingthe assembly of small nucleolar ribonucleoproteins (snoRNPs),which are known to be involved in spliceosome modification (51,52). Upregulation of R2TP and snoRNP components is thought topromote ribosome synthesis in cancer cells (47). Whether overex-
pressed ECD in tumors may function in concert with R2TP andother RUVBL1-containing complexes to promote oncogenesis re-quires further investigation. Taken together, the findings pre-sented here demonstrate that CK2-mediated phosphorylation andinteraction with RUVBL1 are essential for ECD’s ability to regu-late cell cycle progression.
ACKNOWLEDGMENTS
We thank members of the Band laboratories for their thoughtful discus-sions and suggestions throughout this work, James H. Prestegard (Uni-versity of Georgia) for kindly providing the ST6GAL1-pXLG-NtermTC-MhisStrep-DEST construct, and the UNMC Protein Structure CoreFacility for help with protein purification.
FUNDING INFORMATIONFred & Pamela Buffett Cancer Center Support Grant provided funding toVimla Band under grant number P30CA036727. Susan G. Komen for theCure (postdoctoral fellowship) provided funding to Sameer Mirza undergrant number KG111248. HHS | NIH | National Cancer Institute (NCI)provided funding to Vimla Band under grant numbers CA96844 andCA144027. HHS | NIH | National Cancer Institute (NCI) provided fund-ing to Hamid Band under grant numbers CA87986 and CA99163. DOD |Congressionally Directed Medical Research Programs (CDMRP) pro-vided funding to Vimla Band under grant numbers W81XWH-11-1-0171, W81XWH-07-1-0351, and W81XWH-14-1-0567.
Work in our laboratories is supported by National Institutes of Health(NIH) grants CA96844 and CA144027 to V.B. and CA87986 andCA99163 to H.B., by Department of Defense grants W81XWH-07-1-0351, W81XWH-11-1-0171, and W81XWH-14-1-0567 to V.B., and by aFred & Pamela Buffett Cancer Center Support Grant (P30CA036727).S.M. was supported by Susan G. Komen postdoctoral fellowship grantKG111248. A.B. was supported by DOD BCRP predoctoral fellowshipgrant W8X1XWH-11-0020.
REFERENCES1. Hanahan D, Weinberg RA. 2000. The hallmarks of cancer. Cell 100:57–
70. http://dx.doi.org/10.1016/S0092-8674(00)81683-9.2. Garen A, Kauvar L, Lepesant JA. 1977. Roles of ecdysone in Drosophila
development. Proc Natl Acad Sci U S A 74:5099 –5103. http://dx.doi.org/10.1073/pnas.74.11.5099.
3. Gaziova I, Bonnette PC, Henrich VC, Jindra M. 2004. Cell-autonomousroles of the ecdysoneless gene in Drosophila development and oogenesis.Development 131:2715–2725. http://dx.doi.org/10.1242/dev.01143.
4. Sato T, Jigami Y, Suzuki T, Uemura H. 1999. A human gene, hSGT1, cansubstitute for GCR2, which encodes a general regulatory factor of glyco-lytic gene expression in Saccharomyces cerevisiae. Mol Gen Genet 260:535–540. http://dx.doi.org/10.1007/s004380050926.
5. Zhang Y, Chen J, Gurumurthy CB, Kim J, Bhat I, Gao Q, Dimri G, LeeSW, Band H, Band V. 2006. The human orthologue of Drosophila Ecdy-soneless protein interacts with p53 and regulates its function. Cancer Res66:7167–7175. http://dx.doi.org/10.1158/0008-5472.CAN-06-0722.
6. Kim JH, Gurumurthy CB, Naramura M, Zhang Y, Dudley AT, DoglioL, Band H, Band V. 2009. Role of mammalian Ecdysoneless in cell cycleregulation. J Biol Chem 284:26402–26410. http://dx.doi.org/10.1074/jbc.M109.030551.
7. Claudius AK, Romani P, Lamkemeyer T, Jindra M, Uhlirova M. 2014.Unexpected role of the steroid-deficiency protein Ecdysoneless in pre-mRNAsplicing. PLoS Genet 10:e1004287. http://dx.doi.org/10.1371/journal.pgen.1004287.
8. Kim JH, Gurumurthy CB, Band H, Band V. 2010. Biochemicalcharacterization of human Ecdysoneless reveals a role in transcrip-tional regulation. Biol Chem 391:9 –19. http://dx.doi.org/10.1515/BC.2010.004.
9. Zhao X, Mirza S, Alshareeda A, Zhang Y, Gurumurthy CB, Bele A,Kim JH, Mohibi S, Goswami M, Lele SM, West W, Qiu F, Ellis IO,Rakha EA, Green AR, Band H, Band V. 2012. Overexpression of anovel cell cycle regulator Ecdysoneless in breast cancer: a marker of
ECD-RUVBL1 Interaction Regulates the Cell Cycle
March 2016 Volume 36 Number 6 mcb.asm.org 897Molecular and Cellular Biology
on June 23, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

poor prognosis in HER2/neu-overexpressing breast cancer patients.Breast Cancer Res Treat 134:171–180. http://dx.doi.org/10.1007/s10549-011-1946-8.
10. Dey P, Rachagani S, Chakraborty S, Singh PK, Zhao X, GurumurthyCB, Anderson JM, Lele S, Hollingsworth MA, Band V, Batra SK. 2012.Overexpression of Ecdysoneless in pancreatic cancer and its role in onco-genesis by regulating glycolysis. Clin Cancer Res 18:6188 – 6198. http://dx.doi.org/10.1158/1078-0432.CCR-12-1789.
11. Horejsi Z, Stach L, Flower TG, Joshi D, Flynn H, Skehel JM, O’ReillyNJ, Ogrodowicz RW, Smerdon SJ, Boulton SJ. 2014. Phosphoryla-tion-dependent PIH1D1 interactions define substrate specificity of theR2TP cochaperone complex. Cell Rep 7:19 –26. http://dx.doi.org/10.1016/j.celrep.2014.03.013.
12. Kakihara Y, Houry WA. 2012. The R2TP complex: discovery and func-tions. Biochim Biophys Acta 1823:101–107. http://dx.doi.org/10.1016/j.bbamcr.2011.08.016.
13. Boulon S, Bertrand E, Pradet-Balade B. 2012. HSP90 and the R2TPco-chaperone complex: building multi-protein machineries essential forcell growth and gene expression. RNA Biol 9:148 –154. http://dx.doi.org/10.4161/rna.18494.
14. Horejsi Z, Takai H, Adelman CA, Collis SJ, Flynn H, Maslen S, SkehelJM, de Lange T, Boulton SJ. 2010. CK2 phospho-dependent binding ofR2TP complex to TEL2 is essential for mTOR and SMG1 stability. MolCell 39:839 – 850. http://dx.doi.org/10.1016/j.molcel.2010.08.037.
15. Zhao R, Kakihara Y, Gribun A, Huen J, Yang G, Khanna M, CostanzoM, Brost RL, Boone C, Hughes TR, Yip CM, Houry WA. 2008. Molec-ular chaperone Hsp90 stabilizes Pih1/Nop17 to maintain R2TP complexactivity that regulates snoRNA accumulation. J Cell Biol 180:563–578.http://dx.doi.org/10.1083/jcb.200709061.
16. Matias PM, Baek SH, Bandeiras TM, Dutta A, Houry WA, Llorca O,Rosenbaum J. 2015. The AAA� proteins Pontin and Reptin enter adultage: from understanding their basic biology to the identification of selec-tive inhibitors. Front Mol Biosci 2:17. http://dx.doi.org/10.3389/fmolb.2015.00017.
17. Breig O, Bras S, Martinez Soria N, Osman D, Heidenreich O,Haenlin M, Waltzer L. 2014. Pontin is a critical regulator for AML1-ETO-induced leukemia. Leukemia 28:1271–1279. http://dx.doi.org/10.1038/leu.2013.376.
18. Boo K, Bhin J, Jeon Y, Kim J, Shin HJ, Park JE, Kim K, Kim CR, JangH, Kim IH, Kim VN, Hwang D, Lee H, Baek SH. 2015. Pontin functionsas an essential coactivator for Oct4-dependent lincRNA expression inmouse embryonic stem cells. Nat Commun 6:6810. http://dx.doi.org/10.1038/ncomms7810.
19. Rajendra E, Garaycoechea JI, Patel KJ, Passmore LA. 2014. Abundanceof the Fanconi anaemia core complex is regulated by the RuvBL1 andRuvBL2 AAA� ATPases. Nucleic Acids Res 42:13736 –13748. http://dx.doi.org/10.1093/nar/gku1230.
20. Band V, Sager R. 1989. Distinctive traits of normal and tumor-derivedhuman mammary epithelial cells expressed in a medium that supportslong-term growth of both cell types. Proc Natl Acad Sci U S A 86:1249 –1253. http://dx.doi.org/10.1073/pnas.86.4.1249.
21. Barb AW, Meng L, Gao Z, Johnson RW, Moremen KW, Prestegard JH.2012. NMR characterization of immunoglobulin G Fc glycan motion onenzymatic sialylation. Biochemistry 51:4618 – 4626. http://dx.doi.org/10.1021/bi300319q.
22. Mohibi S, Gurumurthy CB, Nag A, Wang J, Mirza S, Mian Y, Quinn M,Katafiasz B, Eudy J, Pandey S, Guda C, Naramura M, Band H, Band V.2012. Mammalian alteration/deficiency in activation 3 (Ada3) is essentialfor embryonic development and cell cycle progression. J Biol Chem 287:29442–29456. http://dx.doi.org/10.1074/jbc.M112.378901.
23. Kataria R, Xu X, Fusetti F, Keizer-Gunnink I, Jin T, van Haastert PJ,Kortholt A. 2013. Dictyostelium Ric8 is a nonreceptor guanine exchangefactor for heterotrimeric G proteins and is important for development andchemotaxis. Proc Natl Acad Sci U S A 110:6424 – 6429. http://dx.doi.org/10.1073/pnas.1301851110.
24. Fisher D, Krasinska L, Coudreuse D, Novak B. 2012. Phosphorylationnetwork dynamics in the control of cell cycle transitions. J Cell Sci 125:4703– 4711. http://dx.doi.org/10.1242/jcs.106351.
25. Ren S, Rollins BJ. 2004. Cyclin C/cdk3 promotes Rb-dependent G0 exit.Cell 117:239 –251. http://dx.doi.org/10.1016/S0092-8674(04)00300-9.
26. Su Q, Wang S, Gao HQ, Kazemi S, Harding HP, Ron D, Koromilas AE.2008. Modulation of the eukaryotic initiation factor 2 alpha-subunit ki-
nase PERK by tyrosine phosphorylation. J Biol Chem 283:469 – 475. http://dx.doi.org/10.1074/jbc.M704612200.
27. St-Denis N, Gabriel M, Turowec JP, Gloor GB, Li SS, Gingras AC,Litchfield DW. 2015. Systematic investigation of hierarchical phosphor-ylation by protein kinase CK2. J Proteomics 118:49 – 62. http://dx.doi.org/10.1016/j.jprot.2014.10.020.
28. Kappes F, Damoc C, Knippers R, Przybylski M, Pinna LA, Gruss C.2004. Phosphorylation by protein kinase CK2 changes the DNA bindingproperties of the human chromatin protein DEK. Mol Cell Biol 24:6011–6020. http://dx.doi.org/10.1128/MCB.24.13.6011-6020.2004.
29. Lee CR, Park YH, Kim M, Kim YR, Park S, Peterkofsky A, Seok YJ.2013. Phosphorylation-dependent mobility shift of proteins on SDS-PAGE is due to decreased binding of SDS. Bull Korean Chem Soc88:473– 485.
30. Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. 2006. Phos-phate-binding tag, a new tool to visualize phosphorylated proteins. MolCell Proteomics 5:749 –757.
31. de Chiara C, Menon RP, Strom M, Gibson TJ, Pastore A. 2009.Phosphorylation of S776 and 14-3-3 binding modulate ataxin-1 interac-tion with splicing factors. PLoS One 4:e8372. http://dx.doi.org/10.1371/journal.pone.0008372.
32. Menon RP, Soong D, de Chiara C, Holt MR, Anilkumar N, Pastore A.2012. The importance of serine 776 in Ataxin-1 partner selection: a FRETanalysis. Sci Rep 2:919. http://dx.doi.org/10.1038/srep00919.
33. Boulon S, Marmier-Gourrier N, Pradet-Balade B, Wurth L, Verheg-gen C, Jady BE, Rothe B, Pescia C, Robert MC, Kiss T, Bardoni B,Krol A, Branlant C, Allmang C, Bertrand E, Charpentier B. 2008.The Hsp90 chaperone controls the biogenesis of L7Ae RNPs thoughconserved machinery. J Cell Biol 180:579 –595. http://dx.doi.org/10.1083/jcb.200708110.
34. Johansen KM, Johansen J. 2006. Regulation of chromatin structure byhistone H3S10 phosphorylation. Chromosome Res 14:393– 404. http://dx.doi.org/10.1007/s10577-006-1063-4.
35. Pepperkok R, Lorenz P, Ansorge W, Pyerin W. 1994. Casein kinase II isrequired for transition of G0/G1, early G1, and G1/S phases of the cell cycle.J Biol Chem 269:6986 – 6991.
36. Homma MK, Homma Y. 2005. Regulatory role of CK2 during the pro-gression of cell cycle. Mol Cell Biochem 274:47–52. http://dx.doi.org/10.1007/s11010-005-3111-3.
37. Gentili C, Castor D, Kaden S, Lauterbach D, Gysi M, Steigemann P,Gerlich DW, Jiricny J, Ferrari S. 2015. Chromosome missegregationassociated with RUVBL1 deficiency. PLoS One 10:e0133576. http://dx.doi.org/10.1371/journal.pone.0133576.
38. Meggio F, Pinna LA. 2003. One-thousand-and-one substrates of pro-tein kinase CK2? FASEB J 17:349 –368. http://dx.doi.org/10.1096/fj.02-0473rev.
39. Ahmed K, Gerber DA, Cochet C. 2002. Joining the cell survival squad: anemerging role for protein kinase CK2. Trends Cell Biol 12:226 –230. http://dx.doi.org/10.1016/S0962-8924(02)02279-1.
40. Suh HW, Yun S, Song H, Jung H, Park YJ, Kim TD, Yoon SR, Choi I.2013. TXNIP interacts with hEcd to increase p53 stability and activity.Biochem Biophys Res Commun 438:264 –269. http://dx.doi.org/10.1016/j.bbrc.2013.07.036.
41. Shalev A. 2014. Minireview: Thioredoxin-interacting protein: regulationand function in the pancreatic beta-cell. Mol Endocrinol 28:1211–1220.http://dx.doi.org/10.1210/me.2014-1095.
42. Elgort MG, O’Shea JM, Jiang Y, Ayer DE. 2010. Transcriptional andtranslational downregulation of thioredoxin interacting protein is re-quired for metabolic reprogramming during G1. Genes Cancer 1:893–907.http://dx.doi.org/10.1177/1947601910389604.
43. Rosenbaum J, Baek SH, Dutta A, Houry WA, Huber O, Hupp TR,Matias PM. 2013. The emergence of the conserved AAA� ATPases Pon-tin and Reptin on the signaling landscape. Sci Signal 6:mr1. http://dx.doi.org/10.1126/scisignal.2003906.
44. Venteicher AS, Meng Z, Mason PJ, Veenstra TD, Artandi SE. 2008.Identification of ATPases Pontin and Reptin as telomerase componentsessential for holoenzyme assembly. Cell 132:945–957. http://dx.doi.org/10.1016/j.cell.2008.01.019.
45. Magalska A, Schellhaus AK, Moreno-Andres D, Zanini F, Schooley A,Sachdev R, Schwarz H, Madlung J, Antonin W. 2014. RuvB-like ATPasesfunction in chromatin decondensation at the end of mitosis. Dev Cell 31:305–318. http://dx.doi.org/10.1016/j.devcel.2014.09.001.
46. Kim SG, Hoffman GR, Poulogiannis G, Buel GR, Jang YJ, Lee KW, Kim
Mir et al.
898 mcb.asm.org March 2016 Volume 36 Number 6Molecular and Cellular Biology
on June 23, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from

BY, Erikson RL, Cantley LC, Choo AY, Blenis J. 2013. Metabolic stresscontrols mTORC1 lysosomal localization and dimerization by regulatingthe TTT-RUVBL1/2 complex. Mol Cell 49:172–185. http://dx.doi.org/10.1016/j.molcel.2012.10.003.
47. Kakihara Y, Saeki M. 2014. The R2TP chaperone complex: its involve-ment in snoRNP assembly and tumorigenesis. Biomol Concepts 5:513–520. http://dx.doi.org/10.1515/bmc-2014-0028.
48. Kamano Y, Saeki M, Egusa H, Kakihara Y, Houry WA, Yatani H,Kamisaki Y. 2013. PIH1D1 interacts with mTOR complex 1 and enhancesribosome RNA transcription. FEBS Lett 587:3303–3308. http://dx.doi.org/10.1016/j.febslet.2013.09.001.
49. Bele A, Mirza S, Zhang Y, Mir R, Lin S, Kim JH, Gurumurthy CB, WestW, Qiu F, Band H, Band V. 2015. The cell cycle regulator Ecdysonelesscooperates with H-Ras to promote oncogenic transformation of human
mammary epithelial cells. Cell Cycle 14:990 –1000. http://dx.doi.org/10.1080/15384101.2015.1006982.
50. Grainger RJ, Beggs JD. 2005. Prp8 protein: at the heart of the spliceo-some. RNA 11:533–557. http://dx.doi.org/10.1261/rna.2220705.
51. Bizarro J, Charron C, Boulon S, Westman B, Pradet-Balade B, Vander-moere F, Chagot ME, Hallais M, Ahmad Y, Leonhardt H, Lamond A,Manival X, Branlant C, Charpentier B, Verheggen C, Bertrand E. 2014.Proteomic and 3D structure analyses highlight the C/D box snoRNP as-sembly mechanism and its control. J Cell Biol 207:463– 480. http://dx.doi.org/10.1083/jcb.201404160.
52. Bizarro J, Dodre M, Huttin A, Charpentier B, Schlotter F, Branlant C, Ver-heggen C, Massenet S, Bertrand E. 2015. NUFIP and the HSP90/R2TP chaper-one bind the SMN complex and facilitate assembly of U4-specific proteins. Nu-cleic Acids Res 43:8973–8989. http://dx.doi.org/10.1093/nar/gkv809.
ECD-RUVBL1 Interaction Regulates the Cell Cycle
March 2016 Volume 36 Number 6 mcb.asm.org 899Molecular and Cellular Biology
on June 23, 2017 by UN
IV O
F N
EB
RA
SK
A M
ED
CT
Rhttp://m
cb.asm.org/
Dow
nloaded from




















