
Potent Inhibitory Ligands of the GRB2 SH2 Domain from Recombinant Peptide Libraries
Upload
independentCategory
view
1download
0
The isoform-speci®c stretch of hSos1 de®nes a new Grb2-binding domain
Natasha Zarich1, Jose Luis Oliva1, Rocõ o Jorge1, Eugenio Santos2,3 and Jose M Rojas*,1
1Unidad de BiologõÂa Celular, Centro Nacional de BiologõÂa Fundamental, Instituto de Salud Carlos III, 28220 Majadahonda,Madrid, Spain; 2Laboratory of Cellular and Molecular Biology, National Cancer Institute, National Institutes of Health, Bethesda,Maryland, MD 20892, USA
hSos1 isoform II, de®ned by the presence of a 15 aminoacid stretch in its carboxy-terminal region, exhibitshigher Grb2 a�nity than hSos1 isoform I. In this study,we investigated the cause for this di�erence and observedthat, in addition to the four currently accepted Grb2-binding motifs, a number of additional, putative SH3-minimal binding sites (SH3-MBS) could be identi®ed.The isoform II-speci®c 15 amino acid stretch containedone of them. Indeed, we demonstrated by site-directedmutagenesis that these SH3-MBS were responsible forthe Grb2 interaction, and we found that C-terminalfragments of the two hSos1 isoforms (lacking the fourcannonical Grb2-binding motifs, but containing the SH3-minimal binding sites) were able to bind Grb2, with theisoform II fragment showing higher Grb2 a�nity thanthe corresponding isoform I fragment. Furthermore, weprovide evidence that C-terminal truncated mutants ofeither hSos1 isoform, containing only the SH3-minimalbinding sites, were able to originate in vivo stablecomplexes with Grb2. Although, Grb2-binding remainshigher in both full-length isoforms, compared to the C-terminal truncated mutants, these mutants were also ableto activate Ras, supporting a potential role of this C-terminal region as negative modulator of Sos1 activity.These ®ndings document the existence of a new,functional, SH3-minimal binding site located in thespeci®c stretch of hSos1 isoform II which may beresponsible for the increased Grb2 a�nity of this isoformin comparison to isoform I, and for the physiologicalproperties di�erences between both isoforms. Moreover,these SH3-minimal binding sites may be su�cient toattain stable and functional hSos1-Grb2 complexes.Oncogene (2000) 19, 5872 ± 5883.
Keywords: hSos1; isoforms; Grb2; SH3 domains
Introduction
The Ras guanine nucleotide exchange factor Sos isinvolved in the coupling of growth factor receptors toRas-dependent mitogenic signaling pathways (Boguskiand McCormick, 1993; Downward, 1994; Feig, 1994;Quilliam et al., 1995; Schlessinger, 1993). A proposedmechanism suggests that recruitment of Sos to theplasma membrane via formation of a complex withGrb2 adapter proteins is responsible for activation of themature, membrane-bound Ras proteins (Egan and
Weinberg, 1993; McCormick, 1993; Pawson and Schles-singer, 1993). In this model, both the cytosolic andmembrane-bound Sos forms are thought to exhibitsimilar nucleotide exchange activity, and no change ofthis exchange activity is supposed to occur as aconsequence of relocation inside the cell. In support ofthis notion, constitutive or conditional membranetargeting of these exchange factors has been shown topotentiate Ras activation in transfected cells (Aronheimet al., 1994; Holsinger et al., 1995; Quilliam et al., 1994).However, some reports suggest that, irrespective ofsubcellular location, the intrinsic guanine nucleotideexchange activity of Sos may be di�erent before andafter stimulation of surface tyrosine kinase receptors (Liet al., 1993a, 1995, 1996; Rojas et al., 1999). Sos consistsof several de®ned domains, each involving a distinctfunction. For example, Ras activation is mediated by acentral domain of Sos that is highly conserved amongdi�erent Ras guanine nucleotide exchange factors(Boguski and McCormick, 1993) and its structure hasbeen determined in complex with Ras (Boriack-Sjodin etal., 1998). The SH3 domains of Grb2 bind to speci®cproline-rich sequences located in the C-terminal regionof Sos (Chardin et al., 1993; Li et al., 1993b; Rozakis-Adcock et al., 1993), that adopt a left-handed polypro-line type II helix conformation (Feng et al., 1994; Limand Richards, 1994; Yu et al., 1994), and some reportssuggest that the C-terminal portion of Sos may exertnegative regulation over the activity of the whole Sos1protein (Byrne et al., 1996; Corbalan-Garcia et al., 1998;Karlovich et al., 1995; Kim et al., 1998; McCollam et al.,1995; Wang et al., 1995). The proline-rich, C-terminalregion of Sos contains a number of phosphorylationsites for MAPK and p90 RSK-2 (Cherniack et al., 1994;Corbalan-Garcia et al., 1996; Douville and Downward,1997; Rozakis-Adcock et al., 1995), and this phosphor-ylation may play a negative feedback role on the Raspathway. Finally, the N-terminal region of Sos containsregions of homology to Dbl (DH) and pleckstrin (PH)domains implicated in phospholipid binding and Rac1activation (Nimnual et al., 1998) and their structureshave also been determined (Koshiba et al., 1997; Soissonet al., 1998).
Previously, we identi®ed two distinct human Sos1isoforms (hSos1 Isf I and hSos1 Isf II) with di�erentGrb2 binding a�nity (Rojas et al., 1996). Theseisoforms di�er only by the presence in hSos1 Isf II ofa 15 amino acid stretch located close the ®rst proline-rich motif required for Grb2 binding. Some humantissues express only one isoform (fetal brain, and adultskeletal muscle, liver, lung and pancreas) whereasothers express di�erent proportions of both in fetaland adult stages. In vitro binding assays and yeast two-hybrid analysis showed that hSos1 Isf II exhibits higherGrb2 a�nity than hSos1 Isf I (Rojas et al., 1996).
Oncogene (2000) 19, 5872 ± 5883ã 2000 Macmillan Publishers Ltd All rights reserved 0950 ± 9232/00 $15.00
www.nature.com/onc
*Correspondence: JM Rojas3Current address: IBM-CC, CSIC-USAL. Campus Unamuno,Universidad de Salamanca, 37007 Salamanca, SpainReceived 27 December 1999; revised 18 September 2000; accepted22 September 2000

Additionally, hSos1 Isf II is signi®cantly more e�ectivethan hSos1 Isf I to induce transforming phenotype ofNIH3T3 cells when transfected alone or in conjunctionwith normal H-Ras (Rojas et al., 1999). Furthermore,direct Ras guanine nucleotide exchange activity assaysin cellular lysates showed that hSos1 Isf II transfec-tants consistently exhibited higher activity than hSos1Isf I transfectants under unstimulated conditions(Rojas et al., 1999).
We undertook the present study to explore theexistence of a new SH3-minimal binding site into the15 amino stretch speci®c of hSos1 Isf II, and toexamine further its implication in the higher Grb2binding a�nity of this isoform that hSos1 Isf I. Thus,we analysed in vitro and in vivo association of Grb2with either hSos1 isoform C-terminal truncatedmutants, containing only the SH3-minimal bindingsites, and their functionality in the physiologicalactivation of Ras.
Results
The C-terminal isoform-specific stretch of hSos1 containsa new putative SH3-minimal binding site
The C-terminal region of hSos1 contains four proline-rich Grb2-Binding Motifs (Grb2-BM) responsible forthe interaction with Grb2 (PCCPPR) (Chardin et al.,
1993; 1995; Cussac et al., 1994, 1999; Simon andSchreiber, 1995). As shown in Figure 1a, in addition ofthese Grb2-BM, other putative domains containing theSH3-Minimal Binding Site (SH3-MBS) (CPXCP)(Sudol, 1998) could be identi®ed in the C-terminalregion of hSos1. The human Sos1 isoforms (hSos1 Isf Iand hSos1 Isf II) di�er only by the presence in one ofthem (hSos1 Isf II) of a 15 amino acid stretch locatedin the C-terminal of hSos1 close to the ®rst Grb2-BM(Figure 1a) (Rojas et al., 1996). Furthermore, hSos1 IsfII exhibits much higher Grb2 binding a�nity thanhSos1 Isf I (Rojas et al., 1996). Since the 15 amino acidstretch speci®c of hSos1 Isf II contains one putativeSH3-MBS (LPHGP) (Figure 1a), we asked whetherthis additional domain could be responsible for thedi�erent Grb2 a�nity with either hSos1 isoform.
Therefore, we carried out pull-down assays toevaluate the Grb2 binding a�nity of di�erent hSos1 C-terminal fragments. We expressed as glutathione S-transferase (GST)-fusion proteins (Guan and Dixon,1991) either full-length C-terminal region of bothisoforms (PRI, residues 1066 ± 1318 and PRII residues1066 ± 1333, see Figure 1), or without the four Grb2-BM(CMRI, residues 1021 ± 1130; and CMRII, residues1021 ± 1145, see Figure 1) (Chardin et al., 1993; Cussacet al., 1994; Li et al., 1993b). Cytoplasmic extracts fromhuman T24 bladder carcinoma cells were incubated withpuri®ed GST-PRI, GST-PRII, GST-CMRI and GST-CMRII proteins coupled to glutathione sepharose
Figure 1 Binding of cellular Grb2 to GST-CMRII and GST-CMRI fusion proteins in cell-free extracts. (a) C-terminal region ofhSos1 showing the four Grb2 binding motifs (Grb2-BM), the minimal SH3 binding sites (SH3-MBS) and the 15 amino acid stretch(underlined) speci®c of hSos1-Isf II. The arrow highlighted the last amino acid of CMRI and CMRII proteins. (b) T-24 cell-freeextracts were incubated with 10 mg of GST, GST-CMRI, GST-CMRII, GST-PRI and GST-PRII proteins coupled to glutathione-sepharose beads. After washing, cellular proteins bound to the beads were run in SDS alongside the corresponding cell extract andimmunoblotted against monoclonal Grb2 antibody. The top of the panel shows the scheme of domains of hSos1 and the C-terminalfragments of this protein used in the in vitro binding assays. DH, Dbl-homology. PH, Pleckstrin homology. REM, Ras exchangermotif. CDC25-H, CDC25 homology. PR, Proline-rich domain. Position of the 15 amino acid insert de®ning Isf II is indicated
Oncogene
SH3 binding site in the specific stretch of hSos1 IsfIIN Zarich et al
5873

beads, and the proteins bound to the beads analysed byimmunoblotting with antibodies to Grb2 (Figure 1b),the same results were obtained with human M426®broblasts (data not shown). Whereas puri®ed GSTalone did not bind any Grb2, similar amounts of Grb2were bound to GST-PRI and PRII, and we also detectedthat GST-CMRI and CMRII bind Grb2. Controlimmunoblots with anti-GST antibodies showed similarsignals in each case (data not shown). Normalizing thesignal of Grb2 to that of GST in the immunoblotsshowed that GST-CMR II (containing the 15 amino acidstretch) proteins could bind up to threefold higheramounts of Grb2 than GST-CMRI and twofold lowerthan GST-PRII/PRI.
Yeast two hybrid analysis of interaction between Grb2 andC-terminal regions of hSos1 (Isf I and Isf II) without thefour cannonical Grb2-binding motifs
We con®rmed the di�erential Grb2 binding a�nity ofCMRI and CMRII by evaluating their functionalinteraction with Grb2 in a yeast two hybrid system(Mendelsohn and Brent, 1994) (Figure 2). A yeast hoststrain (EGY48), harboring both LacZ and Leureporter genes under control of LexA, was cotrans-formed with a plasmid carrying Grb2 fused to the B42activation domain and another plasmid containingeither the hSos1 C-terminal truncated fragmentdescribed (CMRI or CMRII) or PRII (as positivecontrol) fused to the LexA DNA-binding domain. Thefunctional interaction between Grb2 and each con-struct was assessed as a function of detectable b-galactosidase activity and growth on medium withoutleucine (LEU+ phenotype). Controls harboring theDHPH domain of hSos1 together with Grb2, or Grb2or either hSos1 C-terminal fragment alone, did notelicit any b-galactosidase activity, (Figure 2a) and didnot grow well on leu7 plates (Figure 2b). Furthermore,transformants harboring both Grb2 and PRII yieldedhigh levels of enzymatic activity, easily detected onyeast colonies (Figure 2a) and displayed fast and highgrown on medium without leucine. The interactionlevel between Grb2 and CMRII displayed by this two-hybrid system was in accordance with the pull-downresults, since b-galactosidase activity was clearlydetectable, though somewhat weaker that Grb2+PRIIcolonies (Figure 2a), and showed LEU+ phenotype(Figure 2b). In sharp contrast, transformants carryingGrb2 and CMRI elicit an almost undetectable b-galactosidase activity and did not grow well on leu7
plates (Figure 2). We used an on/o� model to test thespeci®city of these results. Thus, b-galactosidaseactivity and LEU+ phenotype was detected only whenthe yeast colonies were patched on galactose-ra�noseselected medium (on state: expression of B42-Grb2)and was undetectable on glucose selected medium (o�state: not expression of B42-Grb2) (Mendelsohn andBrent, 1994). Again, these results con®rmed thatCMRII is able to bind Grb2 and with higher a�nitythan CMRI.
The putative SH3-minimal binding sites of CMRI andCMRII are responsible for the interaction with Grb2
Because our results suggested that the C-terminalregion of either hSos1 isoform without the four
cannonical Grb2-BM were able to bind Grb2, weasked if the putative SH3-MBS located in theseregions (one in CMRI and two in CMRII) could beresponsible for the binding with Grb2. We mutated allprolines of these putative SH3-MBS to alanines andwe obtained three di�erent GST constructs (Figure 3):GST-CMRI AA (with the two prolines of the uniqueputative SH3-MBS mutated to alanines), GST-CMRIIAA (with the two prolines of the 15 amino acidstretch mutated to alanines) and GST-CMRII AA-AA(containing either putative SH3-MBS mutated theprolines to alanines). Again, cytoplasmic extracts fromhuman T24 bladder carcinoma cells were incubatedwith puri®ed GST, GST-CMRII, GST-CMRI, GST-CMRII AA, GST-CMRII AA-AA and GST-CMRIAA proteins coupled to glutathione sepharose beadsand the proteins bound to the beads analysed byimmunoblotting with antibodies to Grb2 (Figure 3),same results were obtained with human M426®broblasts (data not shown). Whereas puri®ed GSTalone did not bind any Grb2, we detected that GST-CMRII and GST-CMRI bind Grb2 (although withhigher a�nity for CMRII). However, we observed adrastic reduction of the Grb2 amount associated withGST-CMRII AA (consistently with the mutations intothe 15 amino acid stretch) in comparison with thewild type GST-CMRII. As expected the mutantsGST-CMRI AA (mutated in its unique putativeSH3-MBS) and GST-CMRII AA-AA (mutated ineither putative SH3-MBS) did not bind Grb2. Controlimmunoblots with anti-GST antibodies showed similarsignals in each case (Figure 3). Taken together, allthese results demonstrated that the described putativeSH3-MBS were responsible of the Grb2 binding actingas real binding-sites and supported that the isoform-speci®c stretch of hSos1 de®nes a new Grb2 binding-domain.
The hSos1 isoforms lacking the four cannonicalGrb2-binding motifs are able to originate in vivostable complexes with Grb2
To extrapolate the results to an in vivo situation, wecarried out truncations in the C-terminal of eitherhSos1 isoform. To this end, we obtained hSos1-CMRIand hSos1-CMRII constructs consisting of full-lengthhSos1 lacking the four Grb2-BM. Transient transfec-tions of 293T cells with the epitope-tagged (HA)constructs of these truncated mutants or the wild typeisoforms of hSos1, showed the overexpression tosimilar levels of all these proteins and with thepredicted sizes (Figure 4). Cellular lysates and anti-Grb2 immunoprecipitates of those transfectants werefurther analysed by immunoblotting with anti-HAantibodies (Figure 4). We consistently detected higheramounts of coimmunoprecipitated HA-hSos1 Isf IIthan HA-hSos1 Isf I. Interestingly, the immunoblotanalyses with anti-HA demonstrated that both HA-hSos1-CMRI and HA-hSos1-CMRII were in vivoassociated with Grb2. Furthermore, the amounts ofcoimmunoprecipitated HA-hSos1-CMRII were higherthan HA-hSos1-CMRI (and lower than hSos1 Isf I/IsfII) (Figure 4), in agreement with the higher capacity tobind Grb2 by the CMR II than CMR I fragmentsdescribed in the above assays. The speci®city of thisinteraction was demonstrated by using the plasmid
SH3 binding site in the specific stretch of hSos1 Isf IIN Zarich et al
5874
Oncogene

pCEFL-KZ-HA-NDP (coding for the N-terminal halfof hSos1). When cell extracts of 293T cells over-expressing HA-NDP were incubated in parallel with
anti-Grb2 antibody, we did not detect HA-NDPprotein associated to the anti-Grb2 immunoprecipitates(Figure 4). Similar results were obtained in Cos 1 cells
Figure 2 Interaction between Grb2 and hSos1 C-terminal fragments in a yeast two-hybrid system. A B42 activation domain fusionplasmid coding for B42-Grb2 (construct in pJG4-5) was cotransformed into yeast strain EGY48 (ura3, his3, trp1, LexAop-leu2)with the plasmid pSH18-34 (LacZ reporter with LexA binding sites) and with LexA DNA binding domain fusion plasmids(constructs in pEG202) coding respectively for the LexA fused to the same CMRI, CMRII and PRII (positive control) fragmentstested in Figure 1a and for the DHPH region of hSos1 (negative control). As others negative controls, plasmid pJG4-5 wascotransfected with pEG202-CMRI (CMRI+Vector), pEG202-CMRII (CMRII+Vector), pEG202-PRII (PRII+Vector) or pEG202alone (Vector+Vector), and plasmid pEG202 was cotransformed with pJG4-5-Grb2 (Vector+Grb2). Four independent coloniesfrom each cotransformation were analysed for b-galactosidase activity in color X-Gal plates assay (a) and for LEU+ phenotype bypatching these colonies in medium lacking leucine (b). The speci®city of the interactions was tested by dependence to galactose-ra�nose (a and b)
Oncogene
SH3 binding site in the specific stretch of hSos1 IsfIIN Zarich et al
5875

(data not shown) and all these data indicate that thetruncated mutants hSos1-CMRI and hSos1 CMRIImay originate in vivo stable complexes with Grb2.
C-terminal truncated mutants of the hSos1 isoforms(lacking the four cannonical Grb2-binding motifs) arefunctional in vivo and physiologically active
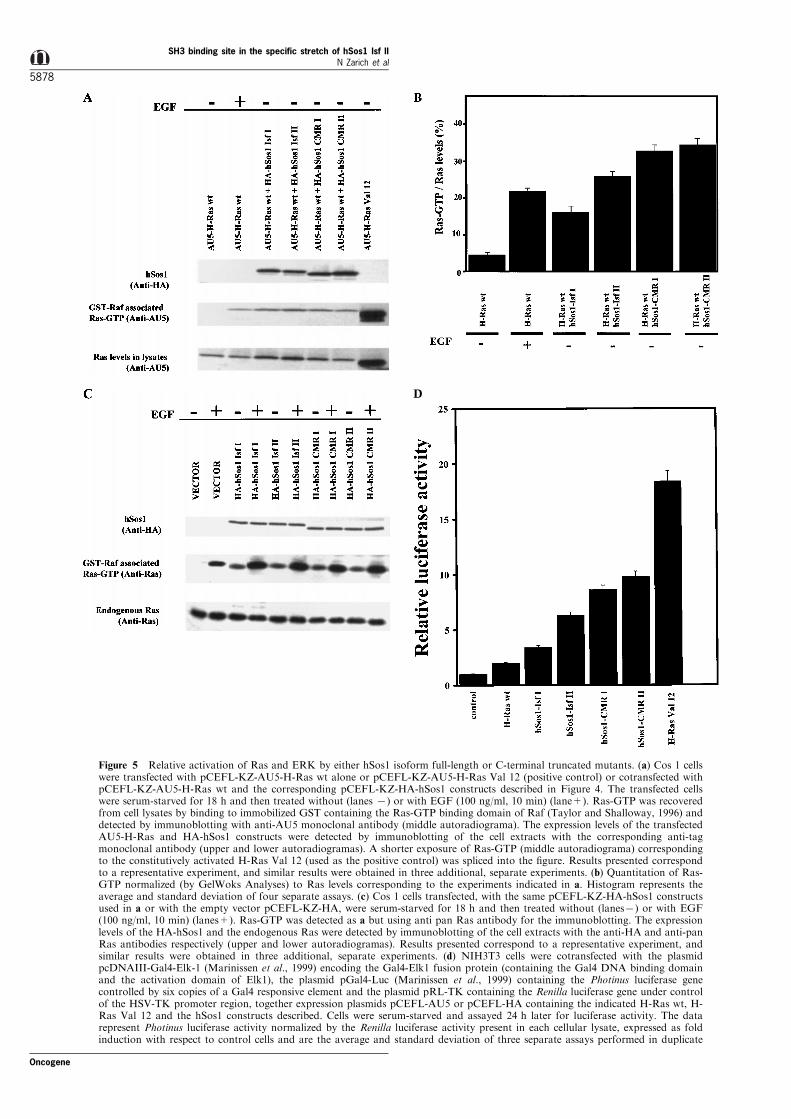
To go further into whether the hSos1 C-terminaltruncated mutants were functionally active to catalysethe exchange of bound GDP for GTP on Ras, weinvestigate whether the overexpression of the abovemutants was per se su�cient to induce Ras activation.Thus, we decided to measure the level of Ras-GTP intransient transfectants overexpressing either hSos1construct. We used a non-radioactive Ras-GTPdetection assay by incubating the cell lysates withsepharose-GST-RBD (Ras Binding Domain of Raf-1)beads and immunoblotting with speci®c antibodies(Taylor and Shalloway, 1996). Figure 5a showsrepresentative results comparing the levels of Ras-GTP in transient cotransfected Cos 1 cells, expressingAU5-H-Ras wild type and HA-tagged constructs ofeither hSos1 isoform (full-length or truncated), underbasal conditions. As controls, Cos 1 cells harboringAU5-H-Ras wild type alone (basal conditions or EGFstimulated) or AU5-H-Ras Val 12 were analysed inparallel. Analysis of GTP-bound Ras in cell lysatesexpressing either full-length or truncated mutants ofhSos1 showed (in all cases) triggering of Rasactivation, compared with the obtained in cellsharboring AU5-H-Ras wild type alone under basal
conditions (Figure 5a). The amounts of AU5-H-Ras-GTP ± normalized to AU5-H-Ras levels (Figure 5b) ±in the cells harboring HA-hSos1 Isf I were slightlylower than those obtained by EGF stimulation in cellstransfected with AU5-H-Ras wild type alone. Inaccordance with previously published data (Rojas etal., 1999), the basal levels of AU5-H-Ras-GTP (Figure5b) in cotransfectants overexpressing HA-hSos1 Isf IIwere higher than those measured in Isf I-expressingcells, but lower than those produced in cells harboringAU5-H-Ras Val 12 (Figure 5a). Interestingly, in thesame unstimulated conditions, both truncated mutantsinduced Ras-GTP levels twofold higher than full-lengthhSos1 Isf I and slightly higher than hSos1 Isf II (Figure5b).
A more adequate model to investigate the physiolo-gical behavior of the hSos1 truncated mutants is theanalysis of endogenous Ras activation induced by thestimulation of receptor protein tyrosine kinases, suchas the epidermal growth factor receptor (EGFR).Therefore, Cos 1 cells transiently transfected with thehSos1 constructs or vector alone were serum-starvedfor 18 h and then stimulated with EGF (Figure 5c),and the endogenous Ras-GTP was detected by theprevious non-radioactive method (Taylor and Shallo-way, 1996). Unlike the results under unstimulatedconditions (Figure 5a,b), cells overexpressing hSos1(full-lengths or truncated mutants) showed a similarincrease of Ras-GTP upon EGF stimulation (Figure 5cand normalizing data not shown). Taken together,these results indicate that both hSos1-CMRI andhSos1-CMRII may activate Ras as the same level
Figure 3 The SH3-Minimal Binding Sites of CMRI and CMRII are responsible for the interaction with Grb2. T-24 cell-freeextracts were incubated with 10 mg of GST, GST-CMRII, GST-CMRI, and the SH3-MBS mutants: GST-CMRII AA, GST-CMRIIAA-AA and GST-CMRI AA, proteins coupled to glutathione-sepharose beads. After washing, cellular proteins bound to the beadswere run in SDS alongside the corresponding cell extract and immunoblotted against monoclonal Grb2 antibody. As control, the®lter was stripped and re-blotted against polyclonal GST antibody. The top shows the scheme of the C-terminal fragments mutantsof hSos1 used in the in vitro binding assays
SH3 binding site in the specific stretch of hSos1 Isf IIN Zarich et al
5876
Oncogene

and physiological pattern as hSos1 full-length proteinsupon EGF stimulation.
A consequence of the Ras-GTP state is theactivation of the Raf-MEK-MAPK pathway (Jonesonand Bar-Sagi, 1997). After threonine and tyrosinephosphorylation, the MAPK (ERK1 and ERK2) aretranslocated to the cell nucleus and they activate (byphosphorylation) several transcription factors (Elk1,Ets1-2, etc.). The mutated hSos1 proteins were studiedfor their ability to stimulate MAPK by using a reporterassay in NIH3T3 cells cotransfected with the abovehSos1 constructs or H-Ras (wild type or Val 12),together with a chimerical Gal4-Elk1 transcriptionfactor (containing the DNA binding domain of Gal4and the activation domain of Elk1) and the reporterplasmid TATA-Gal4-Luc. Figure 5d shows the resultsobtained in a set of transient transfection experimentswhere we measured the induction of luciferase activityin starved conditions. Again, H-Ras Val 12 wassigni®cantly potent to activate Gal4-Elk1, and thetranscriptional activation induced by hSos1 (full-lengthor truncated) was, in all cases, higher than thecorresponding H-Ras wild type alone and more potentwith either hSos1 truncated mutants than the full-length isoforms. These results con®rm that both hSos1-CMRI and hSos1-CMRII may activate the ERKpathway like hSos1 Isf I and hSos1 Isf II full-lengthproteins do, in accordance with previous Ras-GTPresults.
Biological characterization of NIH3T3 cells transfectedwith the hSos1 isoforms C-terminal truncated mutants
To ascertain possible biological di�erences between thewild type isoforms and the C-terminal truncatedmutants of hSos1, we tested the ability of mammalianexpression vectors driving expression of either isoformto induce formation of transformed foci after transfec-tion into NIH3T3 cells. The di�erence in transformingpotency between the isoforms I and II and C-terminaltruncated mutants was manifested in experimentswhere the hSos1 constructs were cotransfected withnormal ras genes. To this end, NIH3T3 cells werecotransfected with the above hSos1 constructs and H-Ras wild type, and the transformed foci (Figure 6)were detected as described previously (Rojas et al.,1996). The overexpression of H-Ras wild type aloneproduced weak, but reproducible, transforming activitythat was enhanced several fold when hSos1 constructs(full-length or truncated mutants) were included in thecotransfection experiments (Figure 6). The same hSos1constructs, when they were overexpressed alone,induced a similar number of foci than H-Ras wildtype alone (data not shown). As shown in Figure 6,hSos1-Isf I constructs produced only a modest increasein focus formation over the number of foci producedby H-Ras wild type alone. In contrast, cotransfectionof hSos1-Isf II with H-Ras wild type resultedconsistently in a more than threefold increase over
Figure 4 Analysis of Grb2-hSos1 complexes in vivo from di�erent hSos1 transfectants. 293T cells were transiently transfected with(2 mg/plate) pCEFL-KZ-HA-hSos1-Isf I (coding full-length HA-hSos1-Isf I), pCEFL-KZ-HA-hSos1-Isf II (coding full-length HA-hSos1-Isf II), pCEFL-KZ-HA-hSos1-CMRI and pCEFL-KZ-HA-hSos1-CMRII (coding either hSos1 isoform C-terminal truncatedmutants) and as control pCEFL-KZ-HA-NDP (coding the N-terminal half of hSos1). Cell extracts expressing similar levels of HA-hSos1-Isf I, Ha-hSos1-Isf II, HA-hSos1-CMRI, HA-hSos1-CMRII, and HA-NDP were incubated with anti-Grb2 polyclonalantibodies. The anti-Grb2 immunoprecipitates or whole cell extracts were then analysed by immunoblotting using anti-HAmonoclonal antibody as described in Materials and methods. The autoradiograma shown correspond to a representative experimentthat was repeated three times. The top shows the di�erent HA-hSos1 constructs used in this assay. DH, Dbl-homology. PH,Pleckstrin homology. REM, Ras exchanger motif. CDC25-H, CDC25 homology. PR, proline-rich domain. Position of the 15 aminoacid insert de®ning Isf II is indicated
Oncogene
SH3 binding site in the specific stretch of hSos1 IsfIIN Zarich et al
5877
D
Figure 5 Relative activation of Ras and ERK by either hSos1 isoform full-length or C-terminal truncated mutants. (a) Cos 1 cellswere transfected with pCEFL-KZ-AU5-H-Ras wt alone or pCEFL-KZ-AU5-H-Ras Val 12 (positive control) or cotransfected withpCEFL-KZ-AU5-H-Ras wt and the corresponding pCEFL-KZ-HA-hSos1 constructs described in Figure 4. The transfected cellswere serum-starved for 18 h and then treated without (lanes 7) or with EGF (100 ng/ml, 10 min) (lane+). Ras-GTP was recoveredfrom cell lysates by binding to immobilized GST containing the Ras-GTP binding domain of Raf (Taylor and Shalloway, 1996) anddetected by immunoblotting with anti-AU5 monoclonal antibody (middle autoradiograma). The expression levels of the transfectedAU5-H-Ras and HA-hSos1 constructs were detected by immunoblotting of the cell extracts with the corresponding anti-tagmonoclonal antibody (upper and lower autoradiogramas). A shorter exposure of Ras-GTP (middle autoradiograma) correspondingto the constitutively activated H-Ras Val 12 (used as the positive control) was spliced into the ®gure. Results presented correspondto a representative experiment, and similar results were obtained in three additional, separate experiments. (b) Quantitation of Ras-GTP normalized (by GelWoks Analyses) to Ras levels corresponding to the experiments indicated in a. Histogram represents theaverage and standard deviation of four separate assays. (c) Cos 1 cells transfected, with the same pCEFL-KZ-HA-hSos1 constructsused in a or with the empty vector pCEFL-KZ-HA, were serum-starved for 18 h and then treated without (lanes7) or with EGF(100 ng/ml, 10 min) (lanes+). Ras-GTP was detected as a but using anti pan Ras antibody for the immunoblotting. The expressionlevels of the HA-hSos1 and the endogenous Ras were detected by immunoblotting of the cell extracts with the anti-HA and anti-panRas antibodies respectively (upper and lower autoradiogramas). Results presented correspond to a representative experiment, andsimilar results were obtained in three additional, separate experiments. (d) NIH3T3 cells were cotransfected with the plasmidpcDNAIII-Gal4-Elk-1 (Marinissen et al., 1999) encoding the Gal4-Elk1 fusion protein (containing the Gal4 DNA binding domainand the activation domain of Elk1), the plasmid pGal4-Luc (Marinissen et al., 1999) containing the Photinus luciferase genecontrolled by six copies of a Gal4 responsive element and the plasmid pRL-TK containing the Renilla luciferase gene under controlof the HSV-TK promoter region, together expression plasmids pCEFL-AU5 or pCEFL-HA containing the indicated H-Ras wt, H-Ras Val 12 and the hSos1 constructs described. Cells were serum-starved and assayed 24 h later for luciferase activity. The datarepresent Photinus luciferase activity normalized by the Renilla luciferase activity present in each cellular lysate, expressed as foldinduction with respect to control cells and are the average and standard deviation of three separate assays performed in duplicate
SH3 binding site in the specific stretch of hSos1 Isf IIN Zarich et al
5878
Oncogene

the number of transformed foci produced by normalH-Ras alone (Figure 6). Furthermore, either hSos1-CMR I or hSos1-CMR II cotransfected with H-Raswild type were more active on the induction of focusformation than either hSos1 full-length isoformcotransfected with H-Ras wild type (Figure 6), andthis di�erence of foci was consistent with the aboveresults of Ras and MAPK activation and with thepublished observations about the negative-regulatorye�ect of the carboxyl terminal region of Sos (Corbalan-Garcia et al., 1998; Kim et al., 1998; McCollam et al.,1995; Rojas et al., 1999; Wang et al., 1995). Theseresults support that either hSos1-CMRI or hSos1-CMRII may display the same biological e�ect than thefull-length hSos1 Isf I and hSos1 Isf II to synergizewith H-Ras wild type on the induction of celltransforming phenotype.
The SH3-MBS of the 15 amino acid stretch is responsiblefor the physiological property differences betweenhSos1 isoforms
Previously, we found that hSos1 Isf II is more e�ectivethan hSos1 Isf I to induce activation of the Ras-Raf-MAPK pathway (Rojas et al., 1996, 1999). In thiscontext, a critical question is whether the SH3-MBSinto the stretch speci®c of isoform II, which increasethe a�nity to Grb2, is also in vivo responsible for thedi�erent functionality of hSos1 Isf II versus hSos1 IsfI. To explore this possibility, we mutated the twoprolines of the 15 amino acid stretch to alanines in thefull-length hSos1 Isf II, obtaining the mutant hSos1 IsfII AA. We compared the capacity of hSos1-Isf I, hSos1Isf II and hSos1 Isf II AA to activate MAPK using aGal4-Elk1/TATA-Gal4-Luc reporter assay. Figure 7shows the results obtained in a set of transienttransfection experiments, where we measured theinduction of luciferase activity in starved conditions.Again, hSos1 Isf II was more potent than hSos1 Isf I in
inducing MAPK activation, whereas the mutant hSos1Isf II AA showed the same level of luciferase activity asisoform I. These results provide a functional evidenceof the importance of the speci®c isoform II SH3-MBSin the context of the full-length hSos1 Isf II molecule,and suggest that this motif is responsible for thephysiological property di�erences between hSos1 iso-forms.
Discussion
We previously identi®ed the existence of two distincthuman Sos1 isoforms (designated hSos1 Isf I andhSos1 Isf II) di�ering only by the presence in Isf II of a15 amino acid stretch located close the ®rst proline-richmotif required for Grb2 binding. In vitro bindingassays using GST-fusion proteins and yeast two-hybridanalysis showed that hSos1-Isf II exhibits signi®cantlyhigher Grb2 a�nity than hSos1 Isf I (Rojas et al.,1996). In addition, we found that hSos1 Isf II issigni®cantly more e�ective than hSos1 Isf I to inducemalignant transformation of rodent ®broblasts whentransfected alone or in conjunction with H-Ras wildtype, and direct Ras guanine nucleotide exchangeactivity assays in cellular lysates showed that hSos1Isf II transfectants consistently exhibited about three-fold higher activity than hSos1 Isf I transfectants underbasal, unstimulated conditions (Rojas et al., 1999).
The increased Grb2 binding a�nity of hSos1 Isf IIas compared to hSos1 Isf I was also consistent withcomputer predictions of the secondary structure ofhSos1 protein, caused by the presence of the 15 aminoacid stretch in its C-terminal proline-rich region (Rojas
Figure 6 Focus formation assay in NIH3T3 cells cotransfectedwith H-Ras wt and hSos1 constructs. NIH3T3 cells werecotransfected with 1 mg of pCEFL-KZ-AU5-H-Ras wt and 1 mgof each pCEFL-KZ-HA-hSos1 constructs described in Figure 4.After 14 days the dishes were stained with Giemsa to score thetransformed foci. All plasmids DNAs produced similar numbersof marker-selectable colonies. The data are averages and standarddeviation of three independent assays performed in duplicate
Figure 7 Role of the SH3-MBS speci®c of hSos1 Isf II in thedi�erent capacity to activate ERK pathway by either hSos1isoform. NIH3T3 cells were cotransfected with the plasmidspcDNAIII-Gal4-Elk-1, pGal4-Luc and pRL-TK (Figure 5d),together with expression vector pCEFL-HA empty or containingthe full-length hSos1 Isf I, hSos1 Isf II or hSos1 Isf II AA (twoprolines of speci®c SH3-MBS mutated for alanines), respectively.Cells were serum-starved and assayed 24 h later for luciferaseactivity. The data represent Photinus luciferase activity normalizedby the Renilla luciferase activity present in each cellular lysate,expressed as fold induction with respect to control cells and arethe average and standard deviation of three separate assaysperformed in duplicate
Oncogene
SH3 binding site in the specific stretch of hSos1 IsfIIN Zarich et al
5879

et al., 1996). According to those predictions the 15amino acid stretch could cause a very marked increasein overall surface accessibility of the neighboring ®rstGrb2-binding motif (Grb2-BM) (PPVPPR) requiredfor optimal Grb2 SH3 binding (Chardin et al., 1993,1995; Cussac et al., 1994, 1999; Li et al., 1993b; Simonand Schreiber, 1995). In conclusion, our ®rst hypoth-esis was than an increased accessibility to solvent, dueto the 15 amino acid stretch, of this proline-rich regioncould contribute to the higher Grb2 binding abilitydisplayed by hSos1 Isf II (Rojas et al., 1996).
However, another possibility could be argued as anexplanation of the Grb2 binding di�erence, namely, anew SH3 binding site into the 15 amino acid stretch.The SH3 domains bind proline-rich motifs that adopt aleft-handed polyproline type II helix conformation(Feng et al., 1994; Lim and Richards, 1994; Yu etal., 1994), and the C-terminal region of hSos1 containsfour proline-rich Grb2-Binding Motifs (Grb2-BM),which have the core sequence: PCCPPR (Chardin etal., 1993, 1995; Cussac et al., 1994, 1999; Simon andSchreiber, 1995). Independently of these motifs, thegeneral consensus sequence for SH3 domain ligands isCPXCP (Mayer and Eck, 1995; Ren et al., 1993;Sudol, 1998) that we denoted as SH3-Minimal BindingSite (SH3-MBS). Eight SH3-MBS are located in the C-terminal of hSos1 (positions 1021 ± 1333) (Figure 1):two of them are positioned downstream of the Grb2-BS, four of them are included in the Grb2-BS, and theother two are located upstream of the four Grb2-BS.Speci®cally, one of these last two SH3-MBS is commonto both hSos1 isoforms (LPRFP: positions 1024 ± 1028)and the other one is speci®c for hSos1 Isf II (LPHGP:®ve last residues of the 15 amino acid stretch).
Here, we decided to analyse whether the SH3-MBSlocated into isoform II stretch could be relevantregarding the more e�ective binding of Grb2 to hSos1Isf II than hSos1 Isf I. The results obtained by in vitro(pull-down assays) and yeast two hybrid experimentsdemonstrate that the proline-rich region of hSos1 Isf IIwithout the four Grb2-BM is able to bind Grb2. Thispeptide contains the two upstream SH3-MBS denotedabove. The same peptide of hSos1 Isf I (containingonly one SH3-MBS) binds slightly Grb2 by pull-downassays, and it did not show positive results in yeast twohybrid experiments. In relation with this, some reportshave indicated that the C-terminal region of hSos1 Isf Icontaining only the ®rst two Grb2-BM is negative tobind Grb2 by yeast two hybrid approach (Chardin etal., 1993; Rojas et al., 1996), unlike the same region ofhSos1 Isf II (Rojas et al., 1996). Given that the uniquedi�erence among these proline-rich peptides is the 15amino acid stretch, the above and previous dataprovide support for the hypothesis about that thenew SH3-MBS contained into the 15 amino acidstretch could be responsible for the higher Grb2binding a�nity of hSos1 Isf II than hSos1 Isf I. Inaccordance with this, the results obtained by Grb2pull-down assays with di�erent mutants, originated bysite-directed mutagenesis in which the proline of SH3-MBS were substituted for alanine, demonstrated thatthese putative SH3-MBS are real domains responsiblefor the Grb2 binding and that the isoform-speci®cstretch of hSos1 de®nes a new Grb2 binding-domain.
In vivo assays showed that either hSos1 isoformwithout the four Grb2-BM were able to originate
stable complexes with Grb2 (although with morea�nity in the case of isoform II), and may activatethe Ras-MAPK pathway. Speci®cally, this Ras andMAPK induced activation were higher, under basalconditions, and at same level and following samefunctional pattern upon EGF stimulation, that than ofhSos1 full-length proteins (Egan and Weinberg, 1993;McCormick, 1993; Pawson and Schlessinger, 1993).Obviously, we cannot discard that other domains ofhSos1 (di�erent of the C-terminal region) could beresponsible for the stability of association with Grb2(although it is very improbable), but the results of thisreport argue against the idea that the four Grb2-BMare strictly necessary for Grb2-hSos1 complexing andphysiological activity (upon tyrosine kinase receptorstimulation) of hSos1 proteins (Chardin et al., 1993,1995; Li et al., 1993b; Rozakis-Adcock et al., 1992,1993). Moreover, the mutation of the prolines withinthe SH3-MBS speci®c of isoform II reduces thefunctionality of this protein to the levels of hSos1 IsfI in the Ras-Raf-MAPK activation, suggesting a roleof that motif in the physiological property di�erencesbetween both isoforms. Furthermore, cotransfectionexperiments showed that either hSos1 isoform, withoutthe four Grb2-BM, caused a higher increase in thenumber of foci produced by cotransfected normal H-ras genes, than cotransfection of either hSos1 full-length isoform with normal H-ras genes, in accordancewith the results of Ras and MAPK activation underunstimulated conditions. These data are consistent withother reports suggesting that the C-terminal portion ofSos1 may exert negative regulation over the activity ofthe whole Sos1 protein (Aronheim et al., 1994; Byrneet al., 1996; Corbalan-Garcia et al., 1998; Karlovich etal., 1995; Kim et al., 1998; McCollam et al., 1995;Quilliam et al., 1995; Rojas et al., 1999; Wang et al.,1995). The stoichiometry of the Grb2-Sos complex isnot clearly de®ned and, though the relation 1 : 1 ispossible, the existence in the C-terminal region ofhSos1 of four Grb2-BM and four independent SH3-MBS suggests that several molecules of Grb2 may beassociated with one molecule of hSos1. Our hypothesisis that the role of Grb2 is not only to recruit Sos to theplasma membrane, upon tyrosine kinase receptorstimulation, but also to control the nucleotideexchange activity of Sos. In agreement with this, theabsence of the four Grb2-BM diminished the Grb2binding capacity of hSos but increased the synergistice�ect with normal H-Ras on the induction oftransforming foci. In that regard, it is interesting tonote that microinjection with bacterially expressedpeptides fragments of the C-terminal region of hSos1Isf II induce Xenopus oocyte maturation (by activationof the Ras pathway), suggesting that they do so byinteracting with some of the intracellular target(s) ofendogenous Sos in the oocyte (Rojas et al., 1999).Furthermore, the proline-rich, C-terminal region of Soscontains a number of phosphorylation sites for MAPKand p90 RSK-2 (Cherniack et al., 1994; Corbalan-Garcia et al., 1996; Douville and Downward, 1997;Rozakis-Adcock et al., 1995), and this phosphorylationmay play a negative feedback role on the Ras pathway.Obviously, several of these phosphorylation sites areabsent in the C-terminal truncated mutants.
The existence of two functionally di�erent hSos1isoforms, with di�erential expression patterns and
SH3 binding site in the specific stretch of hSos1 Isf IIN Zarich et al
5880
Oncogene

distribution in adult and fetal tissues (Rojas et al.,1996), suggests ®nely modulated mechanisms control-ling Ras activation in di�erent tissues or at di�erentstages of development. As an alternative scenario, it ispossible that hSos1 isoforms with di�erent Grb2binding capacity could also regulate signal transduction¯uxes by disrupting the association of Grb2 with otherproteins. In this respect, Grb2 SH3 domains has beenshown to interact with a plethora of unrelatedmolecules besides Sos, including Rap-(C3G) exchangefactors (Gotoh et al., 1995; Guerrero et al., 1998),regulators of the Rac/Rho family (Vav) (Ye andBaltimore, 1994), cytoskeletal proteins such as dynaminand synapsin I (McPherson et al., 1994; Pawson, 1995)as well as protein tyrosine phosphatases (den Hertog etal., 1994). Since these proteins bind to the Grb2 SH3domains, the presence of two hSos1 isoforms withdi�erent Grb2 binding capacity could di�erentiallymodulate the separate pools of intracellular complexescontaining Grb2. Furthermore, it has been recentlyidenti®ed that the endocytic protein intersectin com-petes with the SH3 domains of Grb2 in binding to Sos1(Tong et al., 2000). Current studies exploring themechanism(s) whereby the C-terminal region of hSos1exerts a negative regulation on the activity of thisprotein, including the functional role of Grb2, as wellas the modulation of other Grb2 complexes areexpected to help de®ne the physiological responsesmediated by the two hSos1 isoforms.
Materials and methods
Cell lines
NIH3T3 cells were maintained in Dulbecco's modi®edEagle's medium (DMEM: Life Technologies, Inc.) supple-mented with 10% calf serum. Cos 1 and the human 293T(kidney keratinocyte), M426 (lung embryonic ®broblasts) andT24 (bladder carcinoma) cell lines were maintained inDMEM (Life Technologies, Inc.) supplemented with 10%fetal bovine serum.
DNA constructs
C-terminal coding regions of hSos1 Isf I (PRI, residues1066 ± 1318) from pSP72-hSos1 Isf I and hSos1 Isf II (PRII,residues 1066 ± 1333) from pGEM-hSos1 Isf II, weresubcloned between sites BamHI and SmaI of vector pGEX-3X (Pharmacia) as BamHI±ClaI (blunt-ended) and BamHI±SmaI fragments, respectively. The C-terminal truncatedmutants of hSos1 Isf I (CMRI, residues 1021 ± 1130) andhSos1 Isf II (CMRII, residues 1021 ± 1045) were PCR-ampli®ed using as primers oligonucleotides corresponding topositions 3061 ± 3390 of hSos1-Isf I and 3061 ± 3435 of hSos1-Isf II and providing restriction sites EcoRI and XhoI at the 5'and 3' ends, respectively. The sequences of the oligonucleo-tides utilized will be made available upon request. Theampli®ed products were then subcloned into pGEX-4T-1(Pharmacia). The GST-CMR I and CMR II mutants inresidues 1025 and 1028 (both CMR), and 1127, 1130 (onlyCMR II) were generated using speci®c primers in whichproline was substituted for alanine, using the quickchangesite-directed mutagenesis Kit (Stratagene). The sequence ofall PCR-generated constructs was veri®ed by direct sequen-cing using Sequenase T7 (Amersham Corp.). The epitope-tagged hSos1 Isf I (pCEFL-KZ-HA-hSos1 Isf I) and hSos1Isf II (pCEFL-KZ-HA-hSos1 Isf II) were generated byinserting the full-length coding region of hSos1 (isoform I
and II respectively) into the pCEFL-KZ-HA expressionvector. To obtain the truncated mutants hSos1 CMR I andhSos1 CMR II, the 3'-region of hSos1 Isf I (position 3200 ±3390) and the 3'-region of hSos1 Isf II (positions 3200 ± 3435)were PCR-ampli®ed providing restriction sites BamHI at 5'end and SacI and NotI at 3' end. The sequences of theoligonucleotides utilized will be made available upon request.The ampli®ed products were subcloned between sites BamHIand SacI of plasmids pSP72-hSos1 Isf I and pGEM3-hSos1Isf II respectively. The epitope-tagged truncated mutantshSos1 CMRI (pCEFL-KZ-HA-hSos1 CMRI) and hSos1-CMRII (pCEFL-KZ-HA-hSos1 CMRII) were generated byinserting the coding region of the above hSos1 truncatedmutants into the pCEFL-KZ-HA expression vector. Simi-larly, the plasmid pCEFL-KZ-HA-NDP (coding the N-terminal half of hSos1) was obtained by subcloning the 5'-region of hSos1 (positions 1 ± 1790) in the above pCEFL-KZ-HA vector. The double mutant hSos1 Isf II AA (pCEFL-KZ-HA-hSos1 Isf II AA) was obtained by subcloning betweenthe two BglII sites of HA-hSos1 Isf II a PCR productgenerated by using speci®c primers of hSos1 Isf II in whichreverse oligonucleotide the prolines 1127 and 1130 weresubstituted for alanines (like that CMRII AA). The plasmidspCEFL-KZ-AU5-H-RAS wt, pCEFL-KZ-AU5-H-RAS Val12, pGal4-Luc and pcDNAIII-Gal4-Elk-1 were the kind giftof SJ Gutkind (Zohar et al., 1998).
Bacterial expression of GST fusion proteins
The plasmid pGEX-RBD containing the Raf Ras-bindingdomain (amino acids 1 ± 149) fused to glutathione S-transferase (GST) was kindly provided by SJ Taylor and DShalloway (Taylor and Shalloway, 1996). A bacterial cultureof Escherichia coli BL21 (DE3) harboring that plasmid, andgrowing in 500 ml of Luria-Bertani (LB) medium, wasinduced at an optical density at 600 nm of 0.5 for 3 h at378 with 1 mM isopropyl-1-thio-b-D-galactopyranoside(IPTG, Calbiochem). The cells were collected by centrifuga-tion at 3000 g for 30 min and the GST-RBD puri®cation wasby following the previously described method (Taylor andShalloway, 1996). Similarly, the BL21(DE3) strain ofEscherichia coli was also transformed with the vectorpGEX-4T-1 encoding the fusion protein GST-PRI or GST-PRII, -CMRI or GST-CMRII. The transformed bacteriawere grown in 500 ml of LB medium until the optical densityat 600 nm was 0.5 at which time IPTG (1 mM) was added for3 h at 378C. The cells were collected by centrifugation at3000 g for 30 min and the protocol of protein puri®cationwas as previously published (Rojas et al., 1996).
Transfections
Transient transfections in Cos 1 and 293T cells wereperformed with the Lipofectamine reagent (Life Technolo-gies, Inc.). Cos 1 cells to be serum starved received DMEMcontaining 0.5% fetal bovine serum 24 h after transfectionand were then incubated for another 24 h. All assays weredone 48 h after transfection. NIH3T3 cells were transfected(transient or stable) by the calcium phosphate precipitationtechnique. Morphologically transformed foci were scoredafter 2 ± 3 weeks in culture (Rojas et al., 1996). Transfectedcells were also selected in medium supplemented, asappropriate, with Geneticin 750 mg/ml (Life Technologies,Inc.).
Grb2 binding assays
Cell extracts from human T-24 and M426 cell lines wereprepared by lysis in Nonidet P-40 bu�er (Cussac et al., 1994;Li et al., 1993b). The lysates were cleared twice bycentrifugation at 12 000 g for 15 min to obtain the nucleusfree supernatants. The GST, GST-CMRI, GST-CMRII,
Oncogene
SH3 binding site in the specific stretch of hSos1 IsfIIN Zarich et al
5881

GST-PRI and GST-PRII proteins, 10 mg, coupled toglutathione-sepharose beads were added to 1 mg of cellextracts and incubated for 120 min at 48C following theprevious described methodology (Rojas et al., 1996). Thepull-down proteins were resolved by SDS-polyacrylamide gelelectrophoresis (PAGE), transferred to nitrocellulose mem-branes (Schliehcer & Schuell) and probed with monoclonalanti-Grb2 (Transduction Laboratories) or with rabbit anti-GST polyclonal serum. Immunocomplexes were visualized byenhanced chemiluminescence detection (ECL, AmershamCorp.) using goat anti-mouse or goat anti-rabbit IgGscoupled to horseradish peroxidase as secondary antibody(Amersham Corp.).
Raf Ras-binding domain pull-down
Transfected cells, some stimulated for 10 min with EpidermalGrowth Factor (EGF, Calbiochem), were lysed in cold lysisbu�er containing 25 mM HEPES pH 7.5, 1% Triton X-100,150 mM NaCl, 10 mM MgCl2, 1 mM sodium orthovanadate(Na3VO4), 25 mM NaF, 1 mM phenylmethylsulfonyl ¯uoride(PMSF), 10 mg/ml of leupeptin, aprotinin, pepstatin A andtrypsin inhibitor. Nucleus-free supernatants were incubatedwith GST-RBD on glutathione-sepharose beads at 48C for1 h. The beads were then collected by centrifugation andwashed three times with ice-cold PBS, 0.1% Triton X-100,10 mM MgCl2. Ras proteins were separated by SDS ±PAGEand visualized by immunoblotting and ECL (Amersham) onnitrocellulose ®lters (Schleicher & Schuell) with anti-AU5(Berkeley Antibody Company) or pan-Ras monoclonalantibodies (Transduction Laboratories).
Immunoprecipitation and antibodies
Transfected cells, some stimulated as described, were lysed incold lysis bu�er containing 50 mM HEPES pH 7.5, 150 mM
NaCl, 5 mM EDTA, 5 mM EGTA, 1% Triton X-100, 10%glycerol with protease and phosphatase inhibitors (1 mM
PMSF, 10 mg/ml of leupeptin, aprotinin, pepstatin A andtrypsin inhibitor, 10 mM benzamidine, 50 mM NaF and2 mM sodium orthovanadate). Nucleus free supernatantswere incubated with the appropriate antibody at 48C for2 h. Protein G-Sepharose Plus (Pharmacia) was then added,and the incubation was carried out for an additional 1 h.Immune complexes were collected by centrifugation, washed®ve times with lysis bu�er before being resolved by SDS±PAGE, and visualized by immunoblotting and ECL. Grb2polyclonal antibody was from Santa Cruz Biotechnologyand anti-HA monoclonal antibody was from BerkeleyAntibody Company.
Yeast two-hybrid system binding assays
PCR derived fragments encoding for the appropriate CMRI,CMRII and PRII coding regions were subcloned intoplasmid pEG202, expressing the LexA DNA binding domain.The full-length coding sequence for Grb2 was subcloned intoplasmid pJG45, encoding the B42 activation domain.Saccharomyces cerevisiae EGY48 strain (MATa, his3, trp1,ura3-52, leu2: : pLEU2-LexAop6) was used as a host.Cotransformation of two hybrid vectors into yeast wereperformed as described (Rojas et al., 1996, 1999). Protein ±protein interaction in the two-hybrid system was monitoredon plates by Lac Z assay in the presence of 5-bromo-4-chloro-3-indoyl-b-D-galactopyranoside (X-Gal, BoehringerMannheim) and by growing on medium without leucine.All these assays were comparatively performed in galactose-ra�nose (induction of Grb2-B42 expression) and glucose(repression of Grb2-B42 expression) conditions (Mendelsohnand Brent, 1994). EGY48 strain and plasmids pEG202 andpJG45 were the kind gift of R Brent (Mendelsohn and Brent,1994).
Reporter gene analysis
NIH3T3 cells were transfected with 0.6 mg of hSos1 or Rasconstructs, together with 16 ng of pcDNAIII-Gal4-Elk-1,0.1 mg of pRL-TK (a plasmid expressing the enzyme Renillaluciferase) and 0.3 mg of the reporter plasmid (pGal4-Luc),adjusting the total amount of plasmid DNA with emptyvector. After overnight incubation, the cells were washed andkept for 24 h in DMEM supplemented with 0.5% calf serum.Cells were then lysed with passive lysis bu�er (Promega).Photinus and Renilla luciferase presents in the nucleus freesupernatants were assayed with the Dual-Luciferase ReporterAssay System (Promega), and light emission was quantitatedwith a Monolight 2010 luminometer as speci®ed by themanufacturer (Analytical Luminescence Laboratory). TheRenilla luciferase activity present in each sample was usedto normalize Photinus luciferase activity for transfectione�ciency.
AcknowledgmentsWe thank Drs R Brent, SJ Taylor, D Shalloway and SJGutkind and for providing yeast strain and DNA con-structs. This work was supported by DGICYT (PM95-0053), FIS (98/1336), CAM (08.1/0003.1/99) and Ratio-pharm grants to JM Rojas. N Zarich, JL Oliva and RJorge were recipients of predoctoral fellowships fromInstituto de Salud Carlos III (N Zarich and R Jorge),FIS-BEFI (JL Oliva) and AECC (R Jorge).
References
Aronheim A, Engelberg L, Al-Alawi N, Schlessinger J andKarin M. (1994). Cell, 78, 949 ± 961.
Boguski M and McCormick F. (1993). Nature, 366, 643 ±654.
Boriack-Sjodin PA, Margarit SM, Bar-Sagi D and KuriyanJ. (1998). Nature, 394, 337 ± 343.
Byrne JL, Paterson HF and Marshall CJ. (1996). Oncogene,13, 2055 ± 2065.
Chardin P, Camonis JH, Gale NW, van Aelst L, SchlessingerJ, Wigler MH and Bar-Sagi D. (1993). Science, 260, 1338 ±1343.
Chardin P, Cussac D, Maignan S and Ducruix A. (1995).FEBS Lett., 369, 47 ± 51.
Cherniack A, Klarlund J and Czech M. (1994). J. Biol.Chem., 269, 4717 ± 4720.
Corbalan-Garcia S, Margarit SM, Galron D, Yang SS andBar-Sagi D. (1998). Mol. Cell Biol., 18, 880 ± 886.
Corbalan-Garcia S, Yang SS, Degenhardt KR and Bar-SagiD. (1996). Mol. Cell Biol., 16, 5674 ± 5682.
Cussac D, Frech M and Chardin P. (1994). EMBO J., 13,4011 ± 4021.
Cussac D, Vidal M, Leprince C, Liu WQ, Cornille F,Tiraboschi G, Roques BP and Garbay C. (1999). FASEBJ., 13, 31 ± 38.
den Hertog J, Tracy S and Hunter T. (1994). EMBO J., 13,3020 ± 3032.
Douville E and Downward J. (1997). Oncogene, 15, 373 ±383.
Downward J. (1994). FEBS Lett., 338, 113 ± 117.Egan S and Weinberg R. (1993). Nature, 365, 781 ± 783.Feig L. (1994). Curr. Op. Cell Biol., 6, 204 ± 211.Feng S, Chen JK, Yu H, Simon JA and Schreiber SL. (1994).
Science, 266, 1241 ± 1247.
SH3 binding site in the specific stretch of hSos1 Isf IIN Zarich et al
5882
Oncogene

Gotoh T, Hattori S, Nakamura S, Kitayama H, Noda M,Takai Y, Kaibuchi K, Matsui H, Hatase O, Takahashi H,Kurata T and Matsuda M. (1995). Mol. Cell. Biol., 15,6746 ± 6753.
Guan KL and Dixon JE. (1991). Anal. Biochem., 192, 262 ±267.
Guerrero C, Fernandez-Medarde A, Rojas JM, Font deMora J, Esteban LM and Santos E. (1998). Oncogene, 16,613 ± 624.
Holsinger LJ, Spencer DM, Austin DJ, Schreiber SL andCrabtree GR. (1995). Proc. Natl. Acad. Sci. USA, 92,9810 ± 9814.
Joneson T and Bar-Sagi D. (1997). J. Mol. Med., 75, 587 ±593.
Karlovich CA, Bon®ni L, McCollam L, Rogge RD, Daga A,Czech MP and Banerjee U. (1995). Science, 268, 576 ± 579.
Kim JH, Shirouzu M, Kataoka T, Bowtell D and YokoyamaS. (1998). Oncogene, 16, 2597 ± 2607.
Koshiba S, Kigawa T, Kim JH, Shirouzu M, Bowtell D andYokoyama S. (1997). J. Mol. Biol., 269, 579 ± 591.
Li BQ, Subleski M, Fusaki N, Yamamoto T, Copeland T,Princler GL, Kung H and Kamata T. (1996). Proc. Natl.Acad. Sci. USA, 93, 1001 ± 1005.
Li BQ, Subleski M, Shalloway D, Kung HF and Kamata T.(1993a). Proc. Natl. Acad. Sci. USA, 90, 8504 ± 8508.
Li BQ, Wang MH, Kung HF, Ronsin C, Breathnach R,Leonard EJ and Kamata T. (1995). Biochem. Biophys. Res.Commun., 216, 110 ± 118.
Li N, Batzer A, Daly R, Yajnik V, Skolnik E, Chardin P,Bar-Sagi D, Margolis B and Schlessinger J. (1993b).Nature, 363, 85 ± 88.
Lim WA and Richards FM. (1994). Nat. Struct. Biol., 1,221 ± 225.
Marinissen MJ, Chiariello M, Pallante M and Gutkind JS.(1999). Mol. Cell. Biol., 19, 4289 ± 4301.
Mayer BJ and Eck MJ. (1995). Curr. Biol., 5, 364 ± 367.McCollam L, Bon®ni L, Karlovich CA, Conway BR, Kozma
LM, Banerjee U and Czech MP. (1995). J. Biol. Chem.,270, 15954 ± 15957.
McCormick F. (1993). Nature, 363, 15 ± 16.McPherson PS, Czernik AJ, Chilcote TJ, Onofri F, Benfenati
F, Greengard P, Schlessinger J and De Camilli P. (1994).Proc. Natl. Acad. Sci. USA, 91, 6486 ± 6490.
Mendelsohn AR and Brent R. (1994). Curr. Opin. Biotech-nol., 5, 482 ± 486.
Nimnual AS, Yatsula BA and Bar-Sagi D. (1998). Science,279, 560 ± 563.
Pawson T. (1995). Nature, 373, 573 ± 580.Pawson T and Schlessinger J. (1993). Curr. Biol., 3, 4345 ±
4442.Quilliam LA, Hu� SY, Rabun KM, Wei W, Park W, Broeck
D and Der CJ. (1994). Proc. Natl. Acad. Sci. USA, 91,8512 ± 8516.
Quilliam LA, Khosravi-Far R, Hu� SY and Der CJ. (1995).Bioessays, 17, 395 ± 404.
Ren R, Mayer BJ, Cicchetti P and Baltimore D. (1993).Science, 259, 1157 ± 1161.
Rojas JM, Coque JJR, Guerrero C, Aroca P, Font de MoraJ, de la Cruz X, Lorenzi MV, Esteban LM and Santos E.(1996). Oncogene, 12, 2291 ± 2300.
Rojas JM, Subleski M, Coque JJ, Guerrero C, Saez R, Li BQ,Lopez E, Zarich N, Aroca P, Kamata T and Santos E.(1999). Oncogene, 18, 1651 ± 1661.
Rozakis-Adcock M, Fernley R, Wade J, Pawson T andBowtell D. (1993). Nature, 363, 83 ± 85.
Rozakis-Adcock M, McGlade J, Mbamalu G, Pelicci G,Daly R, Li W, Batzer A, Thomas S, Brugge J, Pelicci PG,Schlessinger J and Pawson T. (1992). Nature, 360, 689 ±692.
Rozakis-Adcock M, van der Geer P, Mbamalu G andPawson T. (1995). Oncogene, 11, 1417 ± 1426.
Schlessinger J. (1993). Trends Biochem. Sci., 18, 273 ± 276.Simon JA and Schreiber SL. (1995). Chem. Biol., 2, 53 ± 60.Soisson SM, Nimnual AS, Uy M, Bar-Sagi D and Kuriyan J.
(1998). Cell, 95, 259 ± 268.Sudol M. (1998). Oncogene, 17, 1469 ± 1474.Taylor S and Shalloway D. (1996). Current Biology, 6, 1621 ±
1627.Tong X-K, Hussain NK, de Heuvel E, Kurakin A, Abi-
Jaoude E, Quinn CC, Olson MF, Marais R, Baranes D,Kay BK and McPherson PS. (2000). EMBO J., 19, 1263 ±1271.
Wang W, Fisher E, Jia Q, Dunn J, Por®ri E, Downward Jand Egan S. (1995). Nature Gen., 10, 294 ± 300.
Ye ZS and Baltimore D. (1994). Proc. Natl. Acad. Sci. USA,91, 12629 ± 12633.
Yu H, Chen JK, Feng S, Dalgarno DC, Brauer AW andSchreiber SL. (1994). Cell, 76, 933 ± 949.
Zohar M, Teramoto H, Katz BZ, Yamada KM and GutkindJS. (1998). Oncogene, 17, 991 ± 998.
Oncogene
SH3 binding site in the specific stretch of hSos1 IsfIIN Zarich et al
5883
Copyright © 2022 FDOKUMEN




















