
Synthesis of the phorboxazoles—potent, architecturally novel ...
Upload
independentCategory
view
3download
0
Cell. Signal. Vol. 11, No. 6, pp. 453–464, 1999 ISSN 0898-6568/99 $–see front matterCopyright 1999 Elsevier Science Inc. PII S0898-6568(99)00017-0
Potent Inhibitory Ligands of the GRB2SH2 Domain from Recombinant Peptide Libraries
Charles P. Hart,* Jennifer E. Martin, Margaret A. Reed, Aftab A. Keval,Matthew J. Pustelnik, Jeffrey P. Northrop, Dinesh V. Patel1 and J. Russell Grove
Affymax Research Institute, Santa Clara, CA 95051, USA
ABSTRACT. We cloned and expressed the SH2 domain of human GRB2 as glutathione S-transferase and mal-tose binding protein fusion proteins. We screened three phagemid-based fd pVIII-protein phage display librariesagainst SH2 domain fusion proteins. Sequence analysis of the peptide extensions yielded a variety of related pep-tides. By examining the ability of the phage clones to bind other SH2 domains, we demonstrated that the phagewere specific for the SH2 domain of GRB2. Based on the sequence motif identified in the “random” libraryscreening experiment, we also built and screened a phage display library based on a Tyr-X-Asn motif (X5-Tyr-X-Asn-X8). To examine the affinity of the phage derived peptides for GRB2, we set up a radioligand competitionbinding assay based on immobilized GRB2 and radiolabelled autophosphorylated EGFR ICD as the radioligand.Results obtained with peptide competitors derived from the phage sequences demonstrated that nonphosphotyr-osine-containing peptides identified with the phage display technology had an affinity for the receptor similar totyrosine-phosphorylated peptides derived from the EGFR natural substrate. Interestingly, when the phage displaypeptides were then phosphorylated on tyrosine, their affinity for GRB2 increased dramatically. We also demon-strated the ability of the peptides to block the binding of the GRB2 SH2 domain to EGFR in a mammalian cell-based binding assay. cell signal 11;6:453–464, 1999. 1999 Elsevier Science Inc.
KEY WORDS. GRB2, SH2, Phage display
INTRODUCTION binding domains such as SH2 (src homology region 2) do-mains, SH3 (src homology region 3) domains, plextrin ho-The cellular signalling networks through which growth fac-mology (PH) domains, and actin-binding domains [4–6].tors and hormones elicit changes in gene expression are keyBoth receptor tyrosine kinase signalling [e.g., EGF andsystems for a better understanding of cell growth, differenti-platelet-derived growth factor (PDGF)] and cytokine recep-ation, and morphogenesis. In addition, the molecules in-tor JAK/STAT signalling (e.g., IFNg and IL2) utilize tyro-volved in cellular signalling are considered targets for thera-sine phosphorylation and the specific recognition of phos-peutic intervention in a number of disease states [1–3]. Thephotyrosine by SH2 domains [7].biochemical and genetic dissection of these pathways and
One particularly well-studied pathway that serves as athe molecular cloning and characterisation of the signalparadigm of receptor tyrosine kinase-based signalling is cen-transduction proteins has demonstrated a modular structuretered on the Ras GTPase protein and is involved with trans-for many of the proteins, where a small number of con-duction of epidermal growth factor signals [8]. Briefly, di-served sequence motifs are assembled into chimeric modu-meric EGF dimerizes the cell surface EGF receptor (EGFR)lar proteins [4–6]. These modules include catalytic domainsresulting in cross phosphorylation of specific tyrosines inwith kinase, phosphatase, and phospholipase activities, andthe intracellular portion of the receptor by the tyrosine ki-nase domain present there. These phosphotyrosines then
*Author to whom all correspondence should be addressed. Charles P.serve as binding sites for SH2 domains, for example theHart, 3410 Central Expressway, Santa Clara, CA 95051-0703. Tel: 11-
408-522-5741; Fax: 11-408-481-0522; E-mail: charles [email protected] SH2 domain in the adapter protein GRB2 [9, 10]. The asso-1Present address: Versicor Inc., Fremont, CA. ciated SH3 domains in GRB2 mediate the recruitment toAbbreviations: GRB2–Growth factor receptor bound protein 2; SH2–
the cell periphery of the Ras guanine nucleotide exchangeSrc homology region 2; SH3–Src homology region 3; EGF(R)–epidermalgrowth factor (receptor); ICD–intracellular domain; PH–plextrin homol- factor Sos which can then activate Ras [11, 12]. Activatedogy; PDGF–platelet-derived growth factor; JAK–Janus kinase; STAT– Ras then stimulates a cascade of kinases, resulting in the nu-signal transducer and activator of transcription; IFNg–gamma interferon;
clear transport of phosphorylated MAP kinase which phos-IL2–interleukin 2; MAP kinase–mitogen-activated protein kinase; IRS-1–insulin receptor substrate–1; PI 3-kinase–phosphatidylinositol 3–kinase; phorylates and activates nuclear transcription factors. ThisGST–glutathione S-transferase; MBP–maltose binding protein; PLCg– pathway is conserved in nature, being present in a very sim-phospholipase C-gamma; IPTG–isopropylthiogalactoside.
Received 30 October 1998; and accepted 8 December 1998. ilar form in humans, Drosophila, and Caenorhabditis [9].

454 C. P. Hart et al.
In addition to its role in EGF and PDGF signalling, the rich rare binding phage through biological amplificationand re-screening suggested an approach complementary toGRB2 adapter protein has also been shown to be involved
in a variety of other cellular signalling pathways. GRB2 is that of the synthetic phosphopeptide diversity techniquesinvolved in insulin receptor signalling where it binds the described above.insulin receptor substrate-1 (IRS-1) [13, 14]. GRB2 is also Peptide ligands could be subsequently analogued and modi-involved in both B-cell and T-cell signalling. GRB2 associ- fied (through a peptidomimetic approach) to confer enhancedates with SHC upon B-cell receptor activation [15] and be- pharmaceutic properties of stability and availability (reviewedcomes associated with the product of the c-abl proto-onco- in [42]). Peptides can also be modified to achieve intracellu-gene upon T-cell receptor cross-linking [16]. GRB2 also lar accessibility (reviewed in [43]). Alternatively, peptidesbecomes associated with a novel 36-kDa protein upon T-cell can be used for structural studies of the “bound state” withreceptor activation [17–19]. In addition, GRB2 serves as a par- their complexed targets to facilitate a structure-based ratio-adigm for other adapter-type SH2 containing proteins that nal design approach to ligand optimization.have a similar overall structural organization such as Nck We cloned and expressed the SH2 domain of humanand Crk that are involved in T-cell signalling [4, 20]. GRB2 and screened phage display libraries. Sequence anal-
The initial identification of specific binding sites for a ysis of enriched clones yielded a variety of related peptides.particular SH2 domain came from examining the primary se- Based on the sequence motif identified in the “random” li-quences around the phosphotyrosine residues in target pro- brary screening experiment, we also built and screened a phageteins bound by the SH2-containing PI-3-kinase (reviewed in display library based on the Tyr-X-Asn motif (X5-Tyr-X-Asn-[21]). Synthetic phosphopeptides based on these sequences X8). We set up a radioligand competition binding assay to ex-were shown to inhibit SH2/receptor binding [22]. Subse- amine the affinity of the phage-derived peptides for GRB2.quent molecular genetic studies extended these findings by Non-phosphotyrosine-containing peptides derived from themutating individual tyrosine residues in receptor intracellu- phage sequences had affinities for the receptor similar to tyro-lar domains and examining the effect on SH2 binding and sine-phosphorylated peptides derived from the EGFR naturalcellular signalling (see [23]). More recently, Songyang, substrate. When the phage display peptides were then phos-Cantley, and co-workers introduced the use of “libraries” of phorylated on tyrosine their affinity for GRB2 increased dra-phosphotyrosine containing peptides and chromatographi- matically. We also demonstrated the ability of the peptides tocally identified peptide families which bound to particular block the binding of the GRB2 SH2 domain to EGFR in aSH2 domains [24, 25] (reviewed in [26]). In addition to mammalian cell-based binding assay.identifying a consensus-type sequence for most of the SH2 During the preparation of this article, two studies em-domains examined, these studies allowed the grouping of ploying phage display for the identification of GRB2 bind-SH2 domains into subfamilies characterized by similar phos- ing peptides were published. Oligino et al. [44] screened aphopeptide-binding motifs. A major implication of these library of random 9mers flanked by invariant cysteines andstudies was the possibility of identifying candidate inter- recovered a single phage clone specific for GRB2. Similar toacting proteins for a given SH2 domain based on the pres- the results described here, subsequent phosphorylation ofence of a similar tyrosine-containing motif within the pro- the tyrosine increased the affinity of the peptide for GRB2.tein [24, 25]. Coupling the sequence characteristics of the Binding was dependent on a cyclic form of the peptide.bound phosphopeptides with the molecular modelling of Gram et al. [45] specifically sought phosphorylated peptidesthe structure of the SH2 domains (based on the known by pretreating a phage library with Src family kinases. Thethree-dimensional structure of the src SH2 domain, [27]) al- selected clones yielded a consensus sequence related to thatlowed the prediction of specific molecular contacts between described here. In contrast, our results are based on the se-the phosphopeptides and cognate SH2 domains [24]. Both lection of many non-phosphorylated peptides from multiplecrystallographic and spectroscopic analyses of the three- libraries, which allow a more thorough analysis of peptide–dimensional structures of SH2 domains in both the “free” SH2 domain interactions.and “phosphopeptide bound” forms have been determined Peptide ligands for SH2 domains could serve as startingand have allowed a detailed look at the nature of the spe- points for a combinatorial chemistry approach or a medici-cific molecular recognition [27–31]. nal chemistry-based peptidomimetic approach for the de-
Phage display has been used to identify peptidic ligands velopment of a therapeutic agent. In addition, the determi-for a variety of molecular targets (reviewed in [32]). These nation of the structural basis for the high-binding affinity ofstudies have identified epitopes and mimotopes of antibod- these peptides for their SH2 substrate could contribute toies [33], peptide antagonists and agonists of cytokine recep- the rational design of a therapeutic agent.tor extracellular domains [34, 35], binding sites for SH3 do-mains [36–38]; and substrates for proteases [39, 40]. We
MATERIALS AND METHODSpostulated that phage display could identify novel, non-SH2 Domain Cloning and Expressionphosphotyrosine-containing SH2 ligands, since Escherichia
coli are thought not to contain tyrosine kinases (see [41]). SH2 domains were expressed in bacteria as fusion proteinswith glutathione S-transferase (GST) and maltose bindingIn addition, the ability to screen very large libraries (e.g.,
.1011 different peptides) and the ability selectively to en- protein (MBP). Expression vectors were derived from pGEX-

Inhibitory Ligands of GRB2 SH2 455
3X (Pharmacio, Piscataway, NJ, USA) and pMAL-c2 (New of 12 variable amino acids with 2 3 109 clones, and one of8 variable amino acids flanked by an invariant Gly-Gly-CysEngland Biolabs, Beverly, MA, USA). The polymerase chain
reaction (PCR) was used to clone the SH2 domain regions at the amino terminus and an invariant Cys-Gly-Gly car-boxy to the variable region (2 3 109 clones). These librariesfrom human cDNA (Clonetech, Palo Alto, CA, USA). Ap-were amplified under induction conditions (0.2% arabi-proximately 15 amino acids amino terminal to the SH2 do-nose/0.1% glucose) that resulted in approximately 10% ofmain and six amino acids carboxy terminal were also includedthe surface pVIII as recombinants (~300 of ~3000 mole-in the constructs. For the human GRB2 SH2 domain aminocules per phage) (see [51]).acids Asp45 to Leu164 [9] were isolated with the PCR primers
Construction of specific peptide-displaying phage clones59-GGCGGATCCAAGACGGCTTCATTCCCAAGAA-and a custom SH2 domain phage library utilized similarCTAC-39 and 59-GCCGAATTCAAGAGGGCCTGGAC-cloning vectors to those used for the primary random librar-GTATGTCGGCTGC-39. The amplified DNA was digestedies [52]. The two specific phage clones that were con-with the restriction endonucleases BamH1 and EcoR1 andstructed presented a 13-amino acid peptide from the ICD ofligated into the GST and MBP expression vectors. Thethe human EGFR centered on the tyrosine at 1068 (Phe1062-DNA sequence of the resulting recombinants was deter-Pro1074) or a derivative replacing the Tyr1068 with a Phe. Bothmined with the dideoxy chain termination technique [46]were tethered to pVIII with a (Gly4Ser)3 linker. Theseusing both flanking and internal sequencing primers. Theclones were constructed by annealing two partially overlap-SH2 domains of the human SHC, human PLCg, andping oligonucleotides, extending the overlapping regionchicken v-Src were similarly cloned (Gln365 to Leu475 ofwith the four dNTPs and Sequenase V2.0 (USB, LakeSHC [47]; Arg652 to Lys763 of PLCg [48]; and Arg145 to Lys249
Placid, NY, USA) digesting with restriction endonucleasesof v-Src [49]). For fusion protein production the E. colispecific for the termini (BstXI and BsiHKAI), and cloningstrain TG2 was transformed with expression vector DNA,into the vector p8V5. The custom GRB2 SH2 domain li-grown in LB media with ampicillin (100 mg/ml) to logbrary had the structure X5-Tyr-X-Asn-X8-(Gly4Ser)2 withphase (OD600 ~ 0.5), and induced with 0.4 mM isopropyl-the variable amino acids (X) encoded by the NNK codon.thiogalactoside (IPTG) for 3 h. The cells were harvested byThis library was constructed by annealing a single-strandedcentrifugation, resuspended in lysis buffer (10 mM phos-oligonucleotide with two short primers to generate double-phate pH 7.5, 30 mM NaCl, 0.25% Tween 20, 10 mMstranded termini for ligation into the pSV2 vector digestedb-mercaptoethanol, 10 mM EGTA, 10 mM EDTA), andwith BstXI. The gapped duplex recombinants were used tolysed with three cycles of freezing and thawing followed bytransform E. coli via electroporation. This library containedsonication. The cell extract was centrifuged and the fusion2 3 108 different recombinants.protein-containing soluble fraction was loaded onto the ap-
For library screening, the GRB2 SH2 fusion protein waspropriate affinity chromatography resin column (glutathi-immobilized in microtiter wells (Dynatech Immulon 4; Dy-one agarose or amylose agarose). After washing off unboundnatech, Chantilly, VA, USA) by direct coating with 100 mlproteins, the fusion proteins were eluted with glutathioneof a 25 mg/ml (w/v) solution of the fusion protein in phos-(3 mM) or maltose (10 mM). Fusion protein-containingphate buffered saline (PBS) for 1 h at 378C. The wells werefractions were pooled and dialysed overnight in PBS. Thethen blocked by the addition of a solution of 1% (w/v) bo-purified proteins were characterized by polyacrylamide gelvine serum albumin (BSA) in PBS for 1 h at 378C. For eachelectrophoresis (PAGE) and radioligand binding (see below).library screened, approximately 1 3 1012 phage particlesFor mammalian cell expression of human GRB2, a full-(~100–500 library equivalents) in 600 ml PBS with 0.1%length cDNA was isolated by the PCR using appropriateBSA and 0.05% GST or MBP were added to six GRB2 SH2primers and human Jurkat T-cell cDNA and ligated into ancoated wells and incubated for 2 h at 48C. Unbound phageSRa vector [50]. An amino terminal peptide-encoding oli-was removed by washing the wells with cold PBS/0.05%gonucleotide specifying a FLAG epitope (DYKDDDDK)Tween 20 at 48C followed by a 30-min incubation in coldwas included in the recombinant. Log-phase COS cellsPBS/Tween, and then repeating the wash cycles. Bound(grown in Dulbecco’s modified Eagle’s medium containingphage was eluted from the wells with 100 ml of an acidic so-10% foetal bovine serum) were transfected by electropora-lution of glycine/HCl (100 mM/pH 2.2/0.1% BSA) incu-tion (5 mg DNA per 1 3 107 cells) and cultured overnight.bated for 10 min at 258C and then repeatedly pipetted. Thephage suspension was immediately neutralised with Tris
Phage Library Screening base. The eluted phage were amplified by infecting logphase E. coli (strain ARI 292) and inoculating 10 ml of LBThree phage libraries were employed in the primary library
screening. All were phagemid-based, araB promoter pVIII media containing 0.1% glucose and 100 mg/ml ampicillin.Upon reaching the log phase of growth (OD600 ~ 0.5), thedisplay libraries (see [51]). The variable peptide portion of
each was encoded by the NNK codon motif (N 5 A, C, G, culture was superinfected with helper phage (VCS ~ 13)conferring kanamycin resistance at a multiplicity of infec-T; K 5 G, T) and tethered to the pVIII protein by a
(Gly4Ser)3 peptide linker. The three libraries screened [52] tion of 10:1. Kanamycin (10 mg/ml) and arabinose (0.2%)were added, and the culture was grown overnight with vig-included one of 10 variable amino acids at the amino termi-
nus containing approximately 6 3 109 different clones; one orous shaking at 378C. Phage were harvested by removing

456 C. P. Hart et al.
the bacteria by centrifugation and precipitating the phage- min. Removal of excess [g-32P]ATP was effected with Stra-tagene’s NucTrap Probe Purification Column (Stratagene,containing supernatant with polyethylene glycol (PEG)
and NaCl (to 3.3% and 0.33 M, respectively). After 1 h on La Jolla, CA, USA). The column was hydrated with 70 mlTBS/0.1% BSA/0.05% Tween 20, and the labelling reac-ice, the phage precipitate was recovered by centrifugation.
The phage pellet was resuspended in PBS and any residual tion was loaded onto the column and pushed through withanother 70 ml of TBS/0.1% BSA/0.05% Tween 20 into di-bacteria were killed by heat treatment (708C for 15 min).lution buffer (TBS/0.1% BSA).The amplified phage pool was then added to SH2 domain-
For competition binding assays the SH2 domain was im-coated wells and incubated, washed, eluted, and amplifiedmobilized with the GRB2 SH2 domain fusion protein di-as above. Four cycles of affinity enrichment were carriedluted to allow a maximum of 10% of the radioligand to bindout. Phage titers (of primary and amplified libraries, elutedto the wells, to avoid depletion of the competing ligandphage, and amplified phage) were determined by injecting[54]. White 96-well plates (Packard, Meriden, CT, USA)100 ml of a 10-fold-concentrated log-phase culture of E. coliwere coated with GST-GRB2 SH2 at 125 ng/well in 100 ml(K91) with 10 ml of the PBS-diluted phage and plating onfor 1 h at 378C. Plates were washed with 200 ml/well of coldLB agar plates containing 100 mg/ml ampicillin. The num-TBS three times and then blocked with 180 ml/well of TBS/ber of resulting colonies was used to determine transforming1% BSA for 1 h at 378C. Wells were washed again with 200units (TU) per ml. To amplify individual phage clones forml of cold TBS three times. Nonspecific background was de-SH2 binding analysis and DNA sequencing, single coloniestermined by co-incubating probe with a vast excess of awere isolated from the titering plates and grown to log phasehigh-affinity peptide for the GRB2 SH2 domain (at 100in LB/ampicillin. The culture was then superinfected withtimes the concentration required to inhibit 50% of the ra-the helper phage as above, and after addition of arabinosedioligand binding, IC50). Serial dilutions of test peptidesand kanamycin, grown overnight with vigorous shaking.and ~60,000 cpm of radioligand were added to the wells.The phage stock was precipitated as above with PEG/NaClThe assay buffer was TBS containing 0.1% BSA. Assays in-and resuspended in PBS. DNA sequencing of recombinantvolving peptides with free sulfhydryls included 5 mM dithi-phage inserts was also by the dideoxy chain terminationothreitol (DTT). After the binding reached equilibriumtechnique using double-stranded phage DNA and appro-(1 h at 258C), the wells were washed with cold TBS, 200priate primers.ml scintillation fluid was added (MicroScint 20, Packard),and the plates were counted (TopCount, Packard) to deter-
SH2 Domain Binding Assays mine the IC50. Each of the test peptide dilution points wasdone in triplicate. The IC50 value was determined with aTo assess the SH2 domain-binding properties of individual
phage clones, a phage ELISA [53] was used. The SH2 do- non-linear least squares regression to a four-parameter logis-tic equation [55].main was immobilized and blocked as described above (ex-
cept using only 0.25 mg of fusion protein per well). Approx- The GRB2 SH2/EGFR immunoprecipitation inhibitionassay used the GRB2 expressing COS cells 48 h after trans-imately 2 3 109 phage particles in PBS/0.1% BSA were
added and incubated for 2 h at 48C, and the wells were fection with the FLAG epitope-tagged full-length GRB2mammalian expression vector plasmid and 24 h after remov-washed with cold PBS. The primary antibody, a polyclonal
rabbit antiphage antiserum [34] diluted 1:5000, was added, ing serum from the growth medium. Cells were suspended inPBS, epidermal growth factor (EGF, R&D Systems, Minne-incubated for 1 h at 48C, and the wells were again washed
with cold PBS. The secondary antibody, an alkaline phos- apolis, MN, USA) was added (final concentration of 100 ng/ml), and after 3 min at 378C, a whole cell extract (WCE) wasphatase conjugated goat anti-rabbit IgG (Tago, Camarillo,
CA, USA) diluted 1:2000 was then added, incubated for 1 produced by lysis with ice-cold lysis buffer (30 mM HEPESpH 7.4, 100 mM NaCl, 1 mM EGTA, 1% Triton X-100,h at 48C, and washed. The alkaline phosphatase substrate
p-nitrophenyl phosphate in diethanolamine buffer (Biorad, 0.1% BSA, 1 mM Na3VO4, 10 mM NaF, 20 mg/ml leupep-tin, 8 m/ml aprotinin) followed by spinning out insolubleHercules, CA, USA) was added, and the colorimetric reac-
tion was read as an O.D. at 405 nm. For peptide competi- debris at 100,000 3 g for 10 min. Test peptide (1 mM) wasthen added to aliquots of WCE and incubated for 20 min attion phage ELISAs, synthetic peptides were added to the
wells with the phage. These peptides were prepared by stan- 48C. The anti-FLAG antibody M2 (Kodak, Rochester, NY,USA) was added to the extract (at 1 mg/ml) followed by 10dard solid-phase methodologies (SynPep Corp., Dublin,
CA, USA). Verification of peptide cyclization was per- ml of protein A-Sepharose beads (Sigma, St. Louis, MO,USA). After 1 h at 48C the beads were washed three timesformed by mass spectrometry.
The radioligand competition binding assay used as a in ice-cold buffer (PBS, 0.1% Tween 20, 1 mM Na3VO4).Sepharose beads were pelleted by centrifugation and theprobe a baculovirus produced intracellular domain of the
human EGFR (EGFR-ICD; a gift of B. Gay, Novartis, Basel, pellet extracted with PAGE sample buffer (60 mM Tris pH6.7, 3% SDS, 5 mM b-mercaptoethanol, 0.05% bromophe-Switzerland) autophosphorylated in the presence of [g-
32P]ATP. The labelling was carried out in 20 mM Tris-HCl nol blue, 8% glycerol). For Western blot analysis of the im-munoprecipitated proteins, the samples were heated to 1008CpH 7.4, 10 mM MgCl2, 10 mM MnCl2, 0.5 mM ATP, 1 mCi/
well [g-32P]ATP, and 0.86 nmol/well EGFR-ICD in 70 ml. for 4 min, separated electrophoretically on a denaturing 7%polyacrylamide gel, and transferred to a nitrocellulose mem-The labelling was performed at room temperature for 30

Inhibitory Ligands of GRB2 SH2 457
FIGURE 1. PCR cloning and bacterial expression of GRB2 SH2 domain. Left panel shows the agarose gel electrophoresis of the PCR.Left lane, molecular weight markers; middle lane, product of a reaction lacking the cDNA target (primers only); right lane, amplifica-tion product obtained with the human spleen cDNA as target. The size of the product is indicated. Middle panel is an SDS-PAGE imageof the total protein of the E. coli harbouring the GST-GRB2 SH2 expression plasmid. Left lane, molecular weight markers; middle lane,proteins from cells before induction; right lane, proteins from cells after 3 h of induction with IPTG. Arrow indicates induced fusionprotein. Right panel is an SDS-PAGE image of the induced protein products after glutathione agarose affinity purification of GST orthe GST-GRB2 SH2 fusion protein. The size of the GST-GRB2 SH2 fusion protein is indicated.
brane (Schleicher & Schuell, Keene, NH, USA). The filter agarose for MBP, glutathione-agarose for GST). Fusionwas blocked in 5% non-fat dry milk (NFDM) in PBS con- proteins directly immobilized on plastic microtiter wellstaining 0.05% Tween 20 (PBST) for 60 min at room tem- demonstrated wild-type affinity for radiolabelled phospho-perature (RT), incubated with an anti-EGFR monoclonal peptide and phosphoprotein probes (see below).antibody (466, at 2 mg/ml in PBST containing 0.5%NFDM) for 60 min at RT and washed in PBST for 15 min.
Phage Library ScreeningAnti-EGFR monoclonal antibody 466 was raised againstthe extracellular domain of the human EGFR. The filter For initial naive library screening three phagemid-basedwas incubated with secondary antibody (horseradish peroxi- pVIII-protein display libraries [52] were screened againstdase-conjugated goat anti-mouse polyclonal IgG from the GRB2 SH2 domain. One of these consisted of 10 ran-Pierce (Rockford, IL, USA) at 1:10000 in PBST/0.5% dom residues (encoded by the NNK motif, see MaterialsNFDM) for 60 min at RT and washed with PBST for 15 and Methods) linked to the pVIII protein by a linker con-min. The filter was then developed with a chemilumines- sisting of three tandem copies of the sequence (Gly4Ser).cence kit from Amersham (Piscataway, NJ, USA) and ex- This library comprised approximately 6 3 109 different re-posed to film for 5–20 s. combinants. The second library used was similar, but displayed
12 random residues and had 2 3 109 different members, whilethe third library included two invariant cysteines thatRESULTS
SH2 Domain Cloning and Expression flanked an 8-amino acid random region. The size of this li-brary was 2 3 109. All three of these phage libraries wereSH2 domains were expressed as chimeric fusion proteinsbuilt using an arabinose operon inducible promoter that al-with maltose binding protein (MBP) and glutathionelowed the titrating of relative expression of the foreign pep-S-transferase (GST). Figure 1 shows an example of SH2 do-tide on the phage surface [51]. The phage libraries used weremain cloning, expression, and purification. The polymeraseamplified under nutritional growth conditions (0.2% arabi-chain reaction (PCR) was used to isolate the SH2 domainnose, 0.1% glucose) that resulted in approximately 10% ofcoding region from human cDNA. The PCR primer oligo-the surface pVIII proteins as recombinants (~300/3000).nucleotides were designed to allow the in-frame fusion of
For each library, a total of four enrichment cycles of affin-the SH2 domain to the carboxy terminus of the fusion part-ity selection and amplification were performed. After eachners. The specific boundaries chosen for the SH2 domainround of affinity enrichment, individual phage clones werecloning junctions (see Materials and Methods) were similarisolated and amplified in liquid culture. A phage-basedto those in previous studies utilizing GST-SH2 fusion pro-ELISA [53] was employed to assess the binding specificityteins (for example, see [24]). DNA sequencing was used toof the individual phage clones. Both MBP and GST fusionsconfirm the sequence of the engineered fusion proteins.were used as affinity selection reagents in separate experi-E. coli bearing the expression vectors were grown to log-ments. Our initial library screening experiment (data notphase and fusion protein expression was induced by the ad-shown) resulted in the isolation of phage that bound specifi-dition of isopropyl b-d-thiogalactopyranoside (IPTG). Thecally to the fusion partner proteins (MBP and GST), andfusion proteins were soluble in a cell lysate and were affinity
purified on the appropriate chromatography resin (amylose- not to the SH2 domain itself. To enhance the possibility of
458 C. P. Hart et al.
isolating SH2 domain-specific phage, we “subtracted” thefusion partner-specific phage by co-incubating the phagewith soluble GST or MBP during the affinity selection. Se-quential cycles of affinity enrichment were characterized byan increasing percentage of the input phage being recoveredby the acid elution (data not shown).
SH2 Domain Binding by Individual Phage Clones
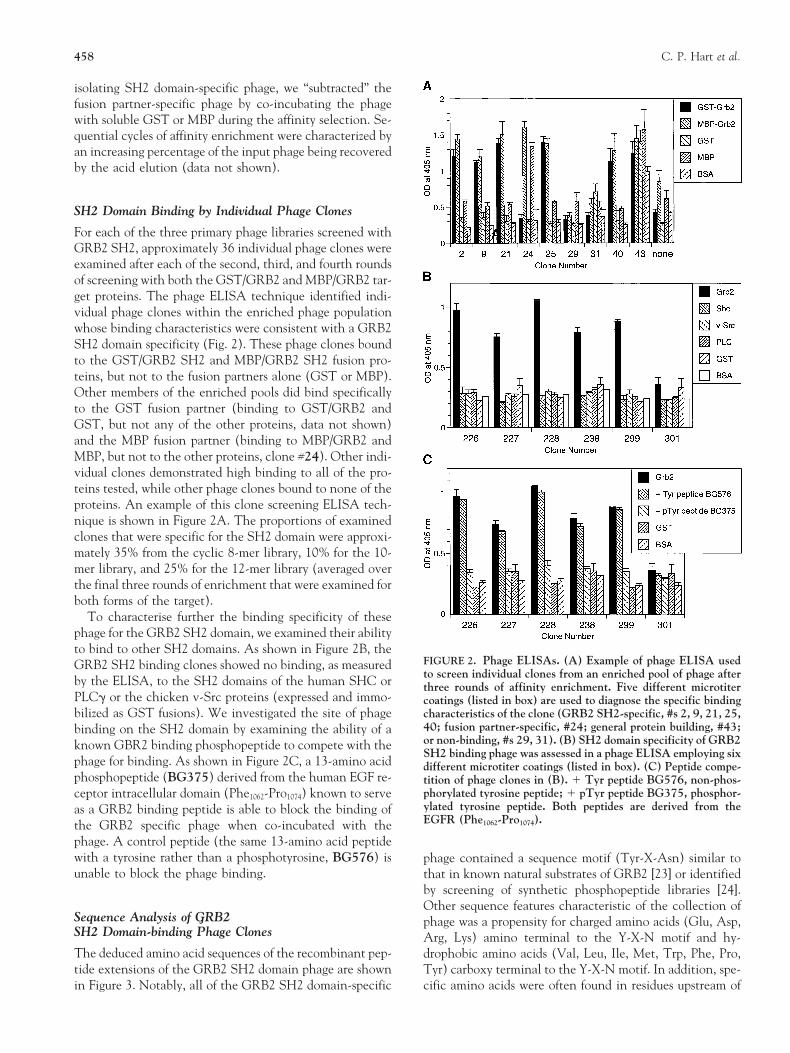
For each of the three primary phage libraries screened withGRB2 SH2, approximately 36 individual phage clones wereexamined after each of the second, third, and fourth roundsof screening with both the GST/GRB2 and MBP/GRB2 tar-get proteins. The phage ELISA technique identified indi-vidual phage clones within the enriched phage populationwhose binding characteristics were consistent with a GRB2SH2 domain specificity (Fig. 2). These phage clones boundto the GST/GRB2 SH2 and MBP/GRB2 SH2 fusion pro-teins, but not to the fusion partners alone (GST or MBP).Other members of the enriched pools did bind specificallyto the GST fusion partner (binding to GST/GRB2 andGST, but not any of the other proteins, data not shown)and the MBP fusion partner (binding to MBP/GRB2 andMBP, but not to the other proteins, clone #24). Other indi-vidual clones demonstrated high binding to all of the pro-teins tested, while other phage clones bound to none of theproteins. An example of this clone screening ELISA tech-nique is shown in Figure 2A. The proportions of examinedclones that were specific for the SH2 domain were approxi-mately 35% from the cyclic 8-mer library, 10% for the 10-mer library, and 25% for the 12-mer library (averaged overthe final three rounds of enrichment that were examined forboth forms of the target).
To characterise further the binding specificity of thesephage for the GRB2 SH2 domain, we examined their abilityto bind to other SH2 domains. As shown in Figure 2B, the
FIGURE 2. Phage ELISAs. (A) Example of phage ELISA usedGRB2 SH2 binding clones showed no binding, as measuredto screen individual clones from an enriched pool of phage afterby the ELISA, to the SH2 domains of the human SHC or three rounds of affinity enrichment. Five different microtiter
PLCg or the chicken v-Src proteins (expressed and immo- coatings (listed in box) are used to diagnose the specific bindingbilized as GST fusions). We investigated the site of phage characteristics of the clone (GRB2 SH2-specific, #s 2, 9, 21, 25,
40; fusion partner-specific, #24; general protein building, #43;binding on the SH2 domain by examining the ability of aor non-binding, #s 29, 31). (B) SH2 domain specificity of GRB2known GBR2 binding phosphopeptide to compete with theSH2 binding phage was assessed in a phage ELISA employing six
phage for binding. As shown in Figure 2C, a 13-amino acid different microtiter coatings (listed in box). (C) Peptide compe-phosphopeptide (BG375) derived from the human EGF re- tition of phage clones in (B). 1 Tyr peptide BG576, non-phos-
phorylated tyrosine peptide; 1 pTyr peptide BG375, phosphor-ceptor intracellular domain (Phe1062-Pro1074) known to serveylated tyrosine peptide. Both peptides are derived from theas a GRB2 binding peptide is able to block the binding ofEGFR (Phe1062-Pro1074).the GRB2 specific phage when co-incubated with the
phage. A control peptide (the same 13-amino acid peptidewith a tyrosine rather than a phosphotyrosine, BG576) is phage contained a sequence motif (Tyr-X-Asn) similar tounable to block the phage binding. that in known natural substrates of GRB2 [23] or identified
by screening of synthetic phosphopeptide libraries [24].Other sequence features characteristic of the collection of
Sequence Analysis of GRB2 phage was a propensity for charged amino acids (Glu, Asp,SH2 Domain-binding Phage Clones Arg, Lys) amino terminal to the Y-X-N motif and hy-The deduced amino acid sequences of the recombinant pep- drophobic amino acids (Val, Leu, Ile, Met, Trp, Phe, Pro,tide extensions of the GRB2 SH2 domain phage are shown Tyr) carboxy terminal to the Y-X-N motif. In addition, spe-
cific amino acids were often found in residues upstream ofin Figure 3. Notably, all of the GRB2 SH2 domain-specific

Inhibitory Ligands of GRB2 SH2 459
estingly, the 10-mer library yielded no cysteine-containingclones. Note that a single phage clone was identified (227)that contained two residues between the tyrosine and aspar-agine (rather than the canonical single residue).
Tyr-X-Asn Focused Phage Library
We were interested in extending our observations concern-ing the selection of preferred flanking residues and the se-lection of paired cysteines with a larger sample set. Based onthe sequence analysis of the GRB2 SH2 domain-specific pep-tide sequences derived from “random” phage display libraries,we built and screened a custom, focused library around theinvariant Tyr-X-Asn motif identified. A phagemid-basedpVIII-protein display vector was used to construct a 16-merlibrary consisting of five random (NNK-encoded) residuesfollowed by an invariant tyrosine, followed by a randomresidue, an invariant asparagine, and then eight randomresidues (X5-Y-X-N-X8). This peptide was tethered to theamino terminus of pVIII by a (Gly4Ser)3 tether. A library of1 3 109 independent recombinants was constructed. Thenucleotide sequence analysis of 25 randomly selected mem-bers of the unscreened library showed the expected quasi-random distribution of amino acid residues (data notshown). 1011 phage particles (100 library equivalents) werescreened as above, with the exception that only the GST-GRB2 SH2 domain fusion was used as the immobilized tar-get protein (with soluble GST as a co-subtractant). Thescreening enrichment kinetics of this experiment were simi-lar to that observed with the screening of the primary naivelibraries. Sixty individual clones were isolated and amplifiedafter each of rounds three, four, and five of affinity selectionand tested by the phage ELISA technique. Thirty percent
FIGURE 3. Deduced amino acid sequences of GRB2 specific of the clones examined after the third round of enrichmentphage. The amino acid sequences of the randomized variable re- demonstrated specific binding to the GRB2 SH2 domain, asgions of the GRB2 specific phage are listed. Standard single let- did 60% of the round four clones, and 82% of the round fiveter amino acid abbreviations are used. Portions of the “invari-
clones. The deduced amino acid sequences of representativeant” flanking regions are also shown. The numerical names ofmembers of these enriched phage pools are shown in Figure 4.the phage are listed to the left. Phage clones that were indepen-
dently isolated more than once are indicated by multiple desig- Similar to the results obtained with the random peptidenating numbers. The phage at the top of the figure are from the libraries, we identified the presence of preferential amino12 amino acid variable region library, the phage listed in the mid- acids amino terminal, between, and carboxy terminal to thedle of the figure are from the 8 amino acid variable region with
invariant tyrosine and asparagine. Although most of theinvariant flanking cysteines library, and the phage listed at theisolated clones were unique, a few were isolated repeatedly,bottom of the figure are from the 10 amino acid variable region
library. The conserved tyrosines (Y) and asparagines (N) are both within and between rounds of screening. Nearly 75%boxed, and used to align the peptide sequences. Cysteine residues of the clones contained paired cysteines. Interestingly,are circles, amino terminal charged residues are underdotted, and
there was not a rigid requirement for size or location of thecarboxy terminal hydrophobic residues are underlined. Deviationscysteines with respect to the Tyr-X-Asn motif. The propen-in the “invariant” regions are shown (E rather than G in 40, 225;
Y rather than C in 226, 297; D rather than G in 237). sity for the presence of charged amino acids amino terminalto the tyrosine and hydrophobic amino acids carboxy termi-nal to the tyrosine was again apparent.
the tyrosine and residues downstream of the asparagine thatyielded a mixed consensus sequence (E/D-E-R-Y-E-N-V/W-P).
Binding Affinity of the Phage-derived PeptidesCysteine pairs were observed in the two clones derived fromthe random 12-mer library. The 8-mer library with the in- To examine the binding affinities of the phage-derived pep-variant cysteines showed the quickest enrichment kinetics tides, we set up a radioligand binding assay. The GRB2 SH2during the rounds of affinity selection and yielded the high- domain, as a GST fusion, was immobilized by direct coating
of plastic microtiter wells. The intracellular domain (ICD)est percentage of GRB2 SH2 domain-specific clones. Inter-

460 C. P. Hart et al.
no binding was seen to the GST fusion partner alone. Unla-belled autophosphorylated EGFR ICD as a competitor withthe radioligand demonstrated the specificity of the interac-tion (data not shown). The phosphopeptide BG375, de-rived from the ICD of the EGFR, containing Tyr1068, wasalso able to block the binding of the radioligand to the im-mobilized GRB2 SH2 domain, while the de-phospho formof the same peptide (BG576) was inactive in the assay (Ta-ble 1). Co-incubation of the radioligand with an excess ofsoluble GST-GRB2 SH2 “neutralised” the radioligand andblocked its ability to bind to immobilized GRB2 SH2, whilesoluble GST alone had no effect (data not shown). To esti-mate the binding constants of the phage-derived peptides,we optimised the parameters of the binding assay to allowthe accurate quantification of inhibition. We optimised ra-dioligand tracer concentrations and amount of immobilisedtarget molecules so that conditions were non-depleting forthe tracer, and we identified the incubation time to reachequilibrium, as described in Materials and Methods. Figure5 shows an example of a competition binding experimentwhere a range of concentrations of the experimental andcontrol peptides were mixed with the radioligand and incu-bated with the immobilized GST-GRB2 SH2 domain. Theconcentration of peptide that resulted in half-maximalbinding of the radioligand tracer (50% inhibition of totalbinding or IC50) was determined by the use of a four parame-ter logistic equation [55]. The 13-amino acid phosphopep-tide derived from the EGFR ICD (BG375) had an IC50
value of approximately 2 mM, which is similar to publishedreports [23, 56]. Table 1 summarizes data derived from theanalysis of a selected number of the phage-derived peptidesand controls. Note that the nonphosphotyrosine peptidesderived from the phage had affinities in the mM range (e.g.,3.2 mM for UT902) whereas de-phospho forms of naturalpeptide ligands for GRB2 (e.g., BG576) have very low orundetectable binding.
Binding Affinity Consequence ofPeptide Phosphorylation and Cyclization
As shown in Table 1, we examined the effect of both phos-phorylating the tyrosine present in the phage-derived pep-tides and cyclizing the dicysteine peptides via disulfidebonds. Tyrosine phosphorylation had a dramatic effect onthe affinity of the phage-derived peptides, increasing theiraffinity from approximately 10-fold to greater than 100-fold
FIGURE 4. Peptide sequences from the Y-X-N focused library. in all of the cases examined. For example, compare UT771Abbreviations and designations are as in Figure 3. Phage listed with UT825 (24 mM vs. 0.24 mM); compare UT902 withat the top of the figure are from the enriched pools of phage
UT903 (3.2 mM vs. 0.4 mM); and compare UT401 witheluted after the third round of affinity enrichment (R3), phageUT402 (62 mM vs. 1 mM).listed in the middle of the figure are from the fourth round of
screening (R4), and phage listed at the bottom of the figure are The disulfide-mediated cyclization of the dicysteine-con-from after the fifth round of screening (R5). taining peptides could also increase the affinity of the phage-
derived peptides. For example, compare UT825 with UT826(0.2 mM vs. 0.04 mM). In other cases examined there was lit-of the human epidermal growth factor receptor (EGFR) wastle, or no, increased potency through cyclization (compareautophosphorylated in vitro in the presence of [g32P]ATP.UT771 with UT403 and compare UT794 with UT823).This radioligand was shown to bind to the immobilized
GRB2 SH2 domain in a specific and saturable manner and In one case we added cysteines to a cysteine-less peptide de-

Inhibitory Ligands of GRB2 SH2 461
TABLE 1. IC50s of synthetic GRB2 inhibitor peptides
Peptide Sequencea IC50b
BG375 FLPVPEpYINQSVP-NH2 2.4 6 1.0 (10)BG576 FLPVPE YINQSVP-NH2 .500 (10)
UT768 Ac-CpYINVPFTC-NH2 0.15 6 0.04 (3)UT793 Ac-CpYINVPFTC-NH2 0.010 6 0.007 (9)UT825 ECpYINVPFTC 0.24 6 0.27 (3)UT826 ECpYINVPFTC 0.035 6 0.025 (5)UT771 EC YINVPFTC 24 6 11 (5)UT403 EC YINVPFTC 26 6 20 (5)
UT795 Ac-CDEVpYVNWSC-NH2 0.15 6 0.08 (4)UT824 Ac-CDEVpYVNWSC-NH2 0.094 6 0.064 (3)UT794 CDEVpYVNWSC-NH2 0.031 (1)UT823 CDEVpYVNWSC-NH2 0.041 (1)
UT822 QERpYENVPG-NH2 2.1 6 1.7 (3)UT901 QER YENVPG-NH2 .1000 (10)
UT903 RERpYENVWY-NH2 0.42 6 0.24 (3)UT902 RER YENVWY-NH2 3.2 6 2.0 (4)UJ259 CRERpYENVWYC-NH2 0.016 6 0.009 (6) FIGURE 6. GRB2 SH2 binding peptides inhibit EGFR/GRB2UJ263 CRER YENVWYC-NH2 4.6 6 1.4 (3) binding in a mammalian cell-derived assay. COS cells transientlyNL260 RECpYENVC-NH2 1.8 6 1.3 (3) transfected with a FLAG epitope-tagged full-length GRB2 pro-NL399 RECpYENVWYC-NH2 0.054 6 0.024 (4) tein were treated with EGF for 3 min and a soluble extract pre-NL400 CRERpYENVC-NH2 0.46 6 0.27 (4) pared. Peptide UT793 (1 mM) was added, followed by anti-
FLAG antibody, protein A Sepharose beads, centrifugation, andUT402 Ac-GCpYWQNVPESCG-NH2 1.0 6 0.3 (3)washing. The immunoprecipitated proteins were separated byUT401 Ac-GC YWQNVPESCG-NH2 62 6 7.1 (3)PAGE, blotted to nitrocellulose, and probed with an anti-EGFRantibody. The first lane are “non-EGF stimulated” cells, the sec-a Single letter abbreviations for amino acids are used.
pY is phoshotyrosine. ond lane is from “non-peptide treated” extracts, and the thirdAc denotes any acetylated amino terminus. lane is from UT793 peptide treated extracts. Arrow points to theNH2 denotes any amidated carboxy terminus. immunoprecipitated EGFR protein band. Molecular weightThe underline signifies a disulfide-mediated cyclization. markers are indicated at left.b Mean IC50 6 standard error in mM. Number of determinations is in
parentheses.
(compare UT903 with UJ259 and NL399, 0.4 mM vs.0.02 mM and 0.05 mM).
rived from the 10mer library and examined the effect ofcyclization (see UJ259, UJ260, NL263, NL399, and
Mammalian Cell-derived in vitro Binding AssayNL400). Whether the cysteines were added flanking thepeptide or in some cases used to substitute for internal resi- We also examined the ability of peptides to inhibit a mam-dues, a significant increase in affinity could be achieved malian cell-based GRB2 SH2 domain phosphoprotein in-
teraction. COS cells were transiently transfected with anexpression vector driving the expression of FLAG-taggedfull-length GRB2 protein. After treatment with EGF, cell-free extracts were produced and the ability of phage-derivedpeptides to inhibit the interaction between the EGFR andthe GRB2 protein was examined by means of an immuno-precipitation assay. An example of one of these experimentsis shown in Figure 6. Peptide UT793 is seen to inhibitEGFR-GRB2 association in this assay, showing a clear cor-relation between the prokaryotic and eukaryotic-basedGRB2 binding assays.
Phosphorylation of the Peptides on the Phage
We wondered about the formal possibility that phage wereFIGURE 5. Measurement of peptide affinities for the GRB2 selected because of some low level of tyrosine phosphoryla-SH2 domain. Competition binding experiments with immobi- tion, even though bacteria do not contain tyrosine kinaseslized GST-GRB2 SH2 domain and radiolabelled EGFR-ICD as
[41]. We generated pVIII recombinant phage bearing theprobe were used to estimate the binding affinities of the phage-13mer that is centered around Tyr1068 in the EGFR (Phe1062-derived peptides. IC50 determinations of selected peptides are
listed in Table 1. Pro1074), with either the natural tyrosine or with the unphos-

462 C. P. Hart et al.
variant asparagine. Using a synthetic peptide library of tet-ramers, where phosphotyrosine was followed by three “ran-dom” residues (18 of the 20 natural amino acids—exceptCys and Trp), Songyang and coworkers [24] enriched forGRB2 SH2-binding peptides by passing the library over anaffinity column and sequencing the “pool” of enriched pep-tides. In addition to the invariant asparagine, they observeda propensity for Tyr, Gln, and Phe (in order of frequencyobserved) in the Tyr 1 3 position. Conversely, Mueller et al.[58], using an immobilised (bead-bound) phosphopeptidelibrary and a method they call “fluorescence-activated beadsorting,” identified Asp and Glu as the most favoured resi-FIGURE 7. EGFR ICD peptide phage ELISA. The ELISA sig-
nal of the affinity enriched phage were compared to phage bear- dues in the Tyr 1 3 position, followed by the uncharged res-ing peptides derived from the intracellular domain of the epider- idues Val, Gln, Tyr, Gly, and Ile. Oligino et al. [44], also us-mal growth factor receptor (EGFR-ICD). The names of the ing a phage display approach, identified a peptide that hadphage are listed at the bottom. 1068F and 1068Y refer to the
Val at the Tyr 1 3 position. Interestingly, at the amino acidEGFR-derived phage, with either a phenylalanine (F) or tyrosineposition between the invariant tyrosine and asparagine, we(Y) in the position equivalent to Tyr1068.isolated a majority of phage with Glu, Similarly, bothMueller et al. [58] and Oligino et al. [44] found Glu, and
phorylatable replacement phenylalanine at the middle posi- Gram et al. [45] found Glu or Met at this position. However,tion. We tested phage preparations of both types, compared Songyang et al. [25] identified uncharged residues at this po-to “hit” phage, in a phage ELISA (see Fig. 7). Both Tyr1068- sition (Gln, Tyr, and Val in order of frequency).derived phage failed to generate the positive signal observed A key distinction between the motifs we identified andwith four phage that had been isolated following the pan- those identified through natural substrate, synthetic pep-ning described above. We also tested whether the phospho- tide, or phosphorylated phage approaches is that all of thetyrosine-specific monoclonal antibody PY20 could detect latter peptides contained an invariant phosphotyrosine,phosphorylated tyrosine either in the ELISA format or from while our selected sequences bore tyrosine. Phosphorylateda blotted gel of the phage proteins. Both of these experi- tyrosines have not been identified in bacteria or bacterio-ments were negative (data not shown). The results support phage (see [41, 59]). Consistent with this was our finding nothe conclusion that phage were selected based on their ex- phosphotyrosine in the positive phage coat proteins blottedpression of unphosphorylated peptides. onto nitrocellulose and probed with an anti-phosphotyro-
sine antibody (data not shown). Our binding analyses of thepeptides (see below) were also consistent with the phage
DISCUSSION not bearing phosphorylated tyrosines. Putative natural sub-strates for the GRB2 SH2 domain (for example, those pres-In the work described here, we employed phage display pep-ent in the EGFR) also demonstrate a strict requirement fortide libraries to identify high-affinity ligands for the humanthe phosphorylated form of the tyrosine for measurableGRB2 SH2 domain. Our goal was to identify SH2 ligandsbinding. Thus, the selection of adjacent and interveningthat did not contain phosphorylated tyrosine residues. We
postulated that SH2-binding peptides could be identified amino acids (with respect to the invariant tyrosine and as-paragine in the phage peptides) may reflect a requirementwhich inhibit SH2/phosphoprotein interactions by: (1)
binding to the phosphotyrosine binding pocket on the SH2 for additional compensatory molecular interactions due tothe absence of phosphate group/SH2 interactions. For ex-domain, interacting with the same residues responsible for
normal binding; (2) blocking the normal binding site by ample, we observed a strong selection for Glu at the Tyr22position, while Gram et al. [45] found no selection for acidicbinding over or around it and sterically blocking ligand
binding; and (3) binding to the SH2 domain and conferring residues when phosphotyrosine was present.Structural analyses of SH2 domain/phosphopeptidesa conformational change which abolishes normal binding.
Interestingly, all of the SH2 domain-binding phage we have utilized both X-ray crystallography and NMR [27, 30,31, 60]; (reviewed in [42]). The peptide binds in an ex-identified bore peptides which resembled the sequences of
the phosphopeptides known to interact with GRB2 (Tyr-X- tended conformation. Most of the contacts are hydrogenbonds formed with the phosphate oxygens. In addition toAsn) [8, 13, 23–25, 44, 45, 57, 58]. However, a detailed
comparison of the peptide sequences we identified with these, there are also interactions between conserved basicamino acids and the aromatic ring of the tyrosine [27]. Rele-those obtained from either peptide library approaches or
with putative natural substrates from target gene products vant to our finding of conserved residues present in thephage peptides out to the Tyr 1 5 position (see Figs. 3 andshowed both similarities and differences. For example, we
observed a preponderance of charged residues amino termi- 4), the NMR study of the PLCgl SH2 domain bound to ahigh-affinity peptide, performed by Pascal et al. [30], identi-nal to the Tyr-X-Asn consensus and a preponderance of
large hydrophobic amino acids carboxy terminal to the in- fied a number of interactions of SH2 amino acids to residues

Inhibitory Ligands of GRB2 SH2 463
at the 14, 15, and the 16 position of the phosphopeptide. pared linear and cyclic forms of peptides containing eitherThis could account for some of the compensatory binding phosphotyrosine, phosphotyrosine analogues, or tyrosine.hypothesized in the phage-derived non-phosphopeptides, as Although binding activity was retained in the cyclic pep-natural substrates diverge after the 13 position and 85% of tides, they did not observe an increase in binding activitythe interactions are at the phosphotyrosine (pTyr), the 11, upon cyclization. Oligino et al. [44], however, observed aand the 13 positions. dramatic loss of binding affinity with a linear analogue of
The selection for phage containing cysteines flanking the their peptide sequence.conserved Tyr-X-Asn motif is consistent with a disulfide- Future work could investigate the effects of these pep-bridged cyclic peptide being a preferred binding form. The tides on cellular signalling. A variety of chemical, physical,periplasm of E. coli, where the phage particles are assembled, and biological approaches to the intracellular delivery ofis an oxidizing environment and favours the formation of di- peptides into living cells have been investigated (reviewedsulfide-bridged cysteines. The presence of disulfide-mediated in [43]). These include encapsulation in liposomes, fusionpeptide loops on phage has been demonstrated experimen- to bacterial toxins and fusion to protein ligands for recep-tally (S. Cwirla and W. Dower, unpublished observation). tor-mediated endocytosis. It may be possible to confer intra-Using a protease substrate between two cysteines and an cellular access to the GRB2 SH2 binding peptides with oneamino terminal antibody epitope, they were able to demon- of these approaches. Hall et al. [65] introduced PLCgSH2strate directly the presence of the loop by proteolytically binding phosphopeptides into cells and were able to inhibitcleaving the amide bond and retaining the epitope. The se- inositol phosphate accumulation and neurite outgrowthlection for dicysteine peptides on screening phage display li- stimulated by FGF. Similarly, Rojas et al. [66] inhibitedbraries has also been reported. An increased potency upon EGF-stimulated Ras activation with a signal sequence-cyclization of the integrin-binding peptides (containing based internalization of a phosphopeptide containing theArg-Gly-Asp) was also seen. Pasqualini et al. [61] screened GRB2 binding site at Tyr1068 of the EGFR.a hexamer pIII library with the integrin a5b1, and one of the The metabolic instability of the phosphate group and the32 different sequences they identified encoded a di-cysteine proteolytic instability of the amide backbone may also limitpeptide. The activity of the di-cysteine peptide was 10-fold the functional characterization or pharmaceutic utility ofgreater than any of the linear peptides they identified and these ligands. A peptidomimetic approach using these pep-tested. Previously, O’Neil et al. [62] built a library of hexa- tides as a starting point or the de novo identification of apeptides on phage where an invariant cysteine was placed non-peptide inhibitor from a combinatorial library may fa-on both sides of the hexamers and screened against the in- cilitate more detailed analyses of GRB2 function in normaltegrin aIIbb3. Peptides derived from selected positive phage and aberrant cellular signalling.showed much higher affinity when tested as oxidized cyclic
We thank B. Dower, S. Cwirla, C. Wagstrom, L. Aldwin, and R.versions, compared to reduced linear versions. Similarly, weBarrett (Affymax) and U. Trinks, B. Gay, G. Caravatti, and A.observed higher binding activities for cyclic versions ofMatter (Novartis) for their contribution of reagents and for critical dis-some of the tested peptides (see Table 1). Note too that the cussions of this work.
phage were selected in the context of a non-phosphorylatedtyrosine, and an increase in affinity through cyclization
Referenceswould a priori need to be manifested in that context. In the1. Brugge J. S. (1993) Science 260, 918–919.cases where cyclization did not increase affinity (Table 1),2. Beattie J. (1996) Cell. Signal. 8, 75–86.it is possible that the constrained conformation was not one3. Saltiel A. R. and Sawyer T. K. (1996) Chem. Biol 3, 887–893.
more favourable for binding. 4. Pawson T. and Gish G. D. (1992) Cell 71, 359–362.Future efforts could investigate alternative approaches to 5. Pawson T. (1995) Nature 373, 573–580.
6. Cohen G. B., Ren R. B. and Baltimore D. (1995) Cell 80,the cyclization of the peptides. These include the use of thi-237–248.oether or alkyl linkages. Note that we found a range of loca-
7. Darnell J. E. Jr., Kerr I. M. and Stark G. R. (1994) Sciencetions and sizes of the loops containing the Tyr-X-Asn bind-264, 1415–1421.
ing motif. Thus, there did not appear to be a strict 8. Van der Geer P., Hunter T. and Lindberg R. A. (1994) Annu.requirement for the locations of the cysteines or the size of Rev. Cell. Biol. 10, 251–337.
9. Lowenstein E. J., Daly R. J., Batzer A. G., Li W., Margolis B.,the resulting loop. Although cysteine peptides have beenLammers R., Ullrich A. and Schlessinger J. (1992) Cell 70,shown to form loops more readily when there is an even431–442.number of residues between the cysteines [63], we observed
10. Matuoka K., Shibata M., Yamakawa A. and Takenawa T.an equivalent number of even and odd numbered residues (1992) Proc. Natl. Acad. Sci. USA 89, 9015–9019.between the cysteines. 11. Buday L. and Downward J. (1993) Cell 73, 611–620.
12. Egan S. E., Giddings B. W., Brooks M. W., Buday L., SizelandThere have been other reports investigating the effects ofA. M. and Weinberg R. A. (1993) Nature 363, 45–51.the cyclization of SH2 domain-binding peptides. Burke et al.
13. Skolnik E. Y., Lee C. H., Batzer A., Vicentini L. M., Zhou M.,[64] created cyclic analogues of hexameric peptides known toDaly R., Myers M. J., Backer J. M., Ullrich A., White M. F.
bind to the p85 subunit of PI 3-kinase by catalyzing amide and Schlessinger J. (1993) EMBO J. 12, 1929–1936.bond formation between the amine and carboxy termini of 14. Tobe K., Matuoka K., Tamemoto H., Ueki K., Kaburagi Y.,
Asai S., Noguchi T., Matsuda M., Tanaka S., Hattori S., Fu-the peptides with diphenyl phosphoryl azide. They com-

464 C. P. Hart et al.
kui Y., Akanuma Y., Yazaki Y., Takenawa T. and Kadowaki 40. Smith M. M., Shi L. and Navre M. (1995) J. Biol. Chem. 270,6440–6449.T. (1993) J. Biol. Chem. 268, 11167–11171.
15. Smit L., Devriessmits A. M. M., Bos J. L. and Borst J. (1994) 41. Beeler J. F., Larochelle W. J., Chedid M., Tronick S. R. andAaronson S. A. (1994) Mol. Cell. Biol. 14, 982–988.J. Biol. Chem. 269, 20209–20212.
16. Meisner H. and Czech M. P. (1995) J. Biol. Chem. 270, 42. Shoelson S. E. (1997) Curr. Opin. Chem. Biol. 1, 227–234.43. Hawiger J. (1997) Curr. Opin. Immunol. 9, 189–194.25332–25335.
17. Sieh M., Batzer A., Schlessinger J. and Weiss A. (1994) Mol. 44. Oligino L., Lung F.-D. T., Sastry L., Bigelow J., Cao T., Cur-ran M., Burke T. R., Wangs S., Krag D., Roller P. P. and KingCell. Biol. 14, 4435–4442.
18. Motto D. G., Ross S. E., Jackman J. K., Sun Q. M., Olson C. R. (1997) J. Biol. Chem. 272, 29046–29052.45. Gram H., Schmitz R., Zuber J. F. and Baumann G. (1997)A. L., Findell P. R. and Koretzky G. A. (1994) J. Biol. Chem.
269, 21608–21613. Eur. J. Biochem. 246, 633–637.46. Sanger F., Nicklen S. and Coulson A. R. (1977) Proc. Natl.19. Buday L, Khwaja A., Sipeki S., Farago A. and Downward J.
(1996) J. Biol. Chem. 271, 6159–6163. Acad. Sci. USA 74, 5463–5467.47. Pelicci G., Lanfrancome L., Grignani F., McGlade J., Cavallo20. Feller S. M., Ren R. B., Hanafusa H. and Baltimore D. (1994)
Trends. Biochem. Sci. 19, 453–458. F., Forni G., Nicoletti I., Grignani F., Pawson T. and PelicciP. G. (1992) Cell 70, 93–104.21. Cantley L. C., Augur K. R., Carpenter C., Duckworth B., Grazi-
ani A., Kapeller R. and Soltoff S. (1991) Cell 64, 281–302. 48. Burgess W. H., Dionne C. A., Kaplow J., Mudd R., Friesel R.,Zilberstein A., Schlessinger J. and Jaye M. (1990) Mol. Cell.22. Escobedo J. A., Kaplan D. R., Kavanaugh D. R., Turck W. M.
and Williams L. T. (1991) Mol. Cell. Biol. 11, 1125–1132. Biol. 10, 4770–4777.49. Takeya T. and Hanafusa H. (1983) Cell 32, 881–890.23. Batzer A. G., Rotin D., Urena J. M., Skolnik E. Y. and Schles-
singer J. (1994) Mol. Cell. Biol. 14, 5192–5201. 50. Takebe Y., Seiki M., Fujisawa J.-I., Hoy P., Yokota K., AraiK.-I., Yoshida M. and Arai N. (1988) Mol. Cell. Biol. 8, 466–24. Songyang Z., Shoelson S. E., Chaudhuri M., Gish G., Pawson
T., Haser W. G., King F., Roberts T., Ratnofsky S., Lechleider 472.51. Gallop M. A., Barrett R. W., Dower W. J., Fodor S. P. andR. J., Neel B. G., Birge R. B., Fajardo J. E., Chou M. M., Ha-
nafusa H., Schaffhausen B. and Cantley L. C. (1993) Cell 72, Gordon E. M. (1994) J. Med. Chem. 37, 1233–1251.52. Cwirla S. E., Balasubramanian P., Duffin D. J., Wagstrom767–778.
25. Songyang Z., Shoelson S. E., Mcglade J., Olivier P., Pawson C. R., Gates C. M., Singer S. C., Davis A. M., Tansik R. L.,Mattheakis L. C., Boytos C. M., Schatz P. J., Baccanari D. P.,T., Bustelo X. R., Barbacid M., Sabe H., Hanafusa H., Yi T.,
Ren R., Baltimore D., Ratnofsky S., Feldman R. A. and Can- Wrighton N. C., Barrett R. W. and Dower W. J. (1997) Sci-ence 276, 1696–1699.tley L. C. (1994) Mol. Cell. Biol. 14, 2777–2785.
26. Cantley L. C. and Songyang Z. (1994) J. Cell. Sci. 18, 121–126. 53. Barrett R. W., Cwirla S. E., Ackerman M. S., Olson A. M.,Peters E. A. and Dower W. J. (1992) Anal. Biochem. 204,27. Waksman G., Kominos D., Robertson S. C., Pant N., Balti-
more D., Birge R. B., Cowburn D., Hanafusa H., Mayer B. J., 357–364.54. Goldstein A. and Barrett R. W. (1987) Mol. Pharmacol. 31,Overduin M., Resh M. D., Rios C. B., Silverman L. and Kuri-
yan J. (1992) Nature 358, 646–653. 603–609.55. DeLean A., Munson P. J. and Rodbard D. (1978) Amer. J.28. Waksman G., Shoelson S. E., Pant N., Cowburn D. and Kuri-
yan J. (1993) Cell 72, 779–790. Physiol. 235, E97–102.56. Braunwalder A. F., Wennogle L., Gay B., Lipson K. E. and29. Lee C. H., Kominos D., Jacques S., Margolis B., Schlessinger
J., Shoelson S. E. and Kuriyan J. (1994) Structure 2, 423–438. Sills M. A. (1996) J. Biomolecular Screening 1, 23–26.57. Pendergast A. M., Quilliam L. A., Cripe L. D., Bassing C. H.,30. Pascal S. M., Singer A. U., Gish G., Yamazaki T., Shoelson
S. E., Pawson T., Kay L. E. and Formankay J. D. (1994) Cell Dai Z., Li N., Batzer A., Rabun K. M., Der C. J., SchlessingerJ. and Gishizky M. L. (1993) Cell 75, 175–185.77, 461–472.
31. Mikol V., Baumann G., Keller T. H., Manning U. and Zurini 58. Mueller K., Gombert F. O., Manning U., Grossmueller F.,Graff P., Zaegel H., Zuber J. F., Freuler F., Tschopp C. andM. G. M. (1995) J. Mol. Biol. 246, 344–355.
32. Winter J. (1994) Drug Devel. Res. 33, 71–89. Baumann G. (1996) J. Biol. Chem. 271, 16500–16505.59. Schmitz R., Baumann G. and Gram H. (1996) J. Mol. Biol.33. Cwirla S., Peters E. A., Barrett R. W. and Dower W. J. (1990)
Proc. Natl. Acad. Sci. USA 87, 6378–6382. 261, 664–677.60. Booker G. W., Breeze A. L., Downing A. K., Panayotou G.,34. Martens C. L., Cwirla S. E., Lee R. Y. Whitehorn E., Chen
E. Y., Bakker A., Martin E. L., Wagstrom C., Gopalan P., Gout I., Waterfield M. D. and Campbell I. D. (1992) Nature358, 684–687.Smith C. W., Tate E., Koller K. J., Schatz P. J., Dower W. J.
and Barrett R. W. (1995) J. Biol. Chem. 270, 21129–21136. 61. Pasqualini R., Koivunen E. and Ruoslahti E. (1995) J. Cell.Biol. 130, 1189–1196.35. Wrighton N., Balasubramanian P., Barbone F., Kashyap A.,
Farrell F., Jolliffe L., Barrett R. and Dower W. (1997) Nature 62. O’Neil K., Hoess R., Jackson S., Ramachandran N., Mousa S.and DeGrado W. (1992) Proteins 14, 509–515.Biotech. 15, 1261–1265.
36. Rickles R. J., Botfield M. C., Zhou X. M., Henry P. A., Brugge 63. Zhang R. and Snyder G. H. (1989) J. Biol. Chem. 264,18472–18479.J. S. and Zoller M. J. (1995) Proc. Natl. Acad. Sci. USA 92,
10909–10913. 64. Burke T. R. J., Nomizu M., Otaka A., Smyth M. S., RollerP. P., Case R. D., Wolf G. and Shoelson S. E. (1994) Bioch.37. Sparks A. B., Quilliam L. A., Thorn J. M., Der C. J. and Kay
B. K. (1994) J. Biol. Chem. 269, 23853–23856. Biophy. Res. Comm. 201, 1148–1153.65. Hall H., Williams E. J., Moore S. E., Walsh F. S., Prochiantz38. Cheadle C., Ivashchenko Y., South V., Searfoss G. H., French
S., Howk R., Ricca G. A. and Jaye M. (1994) J. Biol. Chem. A. and Doherty P. (1996) Curr. Biol. 6, 580–587.66. Rojas M., Yao S. and Lin Y. (1996) J. Biol. Chem. 271,269, 24034–24039.
39. Matthews D. J. and Wells J. A. (1993) Science 260, 1113–1117. 27456–27461.
Copyright © 2022 FDOKUMEN




















