
Synthesis of the phorboxazoles—potent, architecturally novel ...
33
REVIEW ARTICLE Synthesis of the phorboxazoles—potent, architecturally novel marine natural products Zachary Shultz 1 and James W Leahy 1,2,3 The phorboxazoles have attracted the attention of synthetic chemists around the globe due to their potent biological activity and novel structure. A review of the recent synthetic efforts as well as new phorboxazole analogs is discussed. The Journal of Antibiotics (2016) 69, 220–252; doi:10.1038/ja.2016.8; published online 9 March 2016 INTRODUCTION In 1995, Searle and Molinski reported the isolation of a pair of novel natural products from the Indian Ocean marine sponge Phorbas sp. collected off the Muiron Islands that demonstrated potent antifungal and cytostatic activity. 1 The subsequent structural elucidation of these two macrolides, 2–4 phorboxazoles A (1, Figure 1) and B (2), has inspired considerable attention and synthetic creativity over the succeeding two decades that has resulted in 10 total syntheses as well as a number of additional synthetic approaches, the discovery of new members of the phorboxazole family, the creation of biological probes and the generation of analogs to uncover hints about their bioactivity. Some of the highlights of these efforts are reviewed here. More recently, Capon et al. 5 isolated both 1 and 2 from a Raspailia sp. sponge collected along the Australian Northern Rottnest Shelf while searching for nematocidal agents. This second isolation from such a taxonomically distinct source suggests a potential microbial biosynthetic origin or at least contribution that has yet to be uncovered, leaving synthesis as the primary source for further consideration of the natural products. It is not surprising that the distinctive structural features of the phorboxazoles, which includes the presence of four unique pyran rings and two oxazoles, attracted the interest of a number of synthetic groups. In fact, the first synthetic approach to the molecule was reported before the final structure elucidation was published! 6 Seitz and Hoffmann have previously examined some of the earliest syntheses of the phorboxazoles, 7,8 and therefore we will primarily focus on the efforts that were not previously highlighted. With a target as structurally diverse as the phorboxazoles, there is an array of potential approaches to be considered. However, the need to form a macrolide left a limited number of options for the cyclization. The most commonly exploited approach was to close the ring via an olefination of the α,β-unsaturated macrolactone—a strategy that has proven to be quite effective in this regard. Notably, the Evans total synthesis 9 and the Paterson synthetic approach 10 utilized a Yamaguchi macrolactonization as an alternative. The other common disconnec- tion was about the C16–C18 oxazole or the C19–C20 olefin to join the two macrolide fragments. As the two natural products differ only about the stereochemical disposition of the hydroxyl substituent at the C13 position, the phorboxazoles serve as an excellent vehicle for the study of the various approaches to solve the synthetic challenges posed. In this review, we will first evaluate the completed syntheses of 1 and 2, respectively, followed by the approaches of other groups in this regard and the efforts reported to date on the discovery of other members of the phorboxazole family, biological probes and additional analogs. DISCUSSION Total syntheses Earliest syntheses. As the earlier reviews covered the first generation syntheses of 1 by Forsyth and Smith as well as the syntheses of 1 by Pattenden and Williams and the synthesis of 2 by Evans, we will only point out a few of the salient highlights from these seminal works. Forsyth, who was the first to complete the synthesis of either natural product (Figure 2), 11 established the Michael cyclization to the pentasubstituted pyran and developed a biomimetic route to the oxazoles. He observed the same lack of diastereoselectivity in the formation of the C38 center observed by Molinski and Leahy, 2 ascertained the viability of the hetero-Diels–Alder approach to the pyrans and established the feasibility of the Still–Gennari macrocycli- zation. Before 2006, Evans had achieved the only synthesis of 2, 9 which featured a macrolactonization strategy of an acetylene to enable a selective reduction to the Z C2–C3 olefin (Figure 3). In addition, Evans’ lithiation of the oxazole methyl, optimization of the C19–C20 Wittig and use of β-ketoesters enabled many of the efforts that followed. Smith’s pioneering use of the Petasis–Ferrier rearrangement serves as a hallmark of his synthesis of 1 (Figure 4), 12 and he implemented a Stille protocol for introduction of the exocyclic oxazole, allowing it to serve as a linchpin for the two complex 1 Department of Chemistry, University of South Florida, Tampa, FL, USA; 2 Department of Molecular Medicine, Morsani College of Medicine, University of South Florida, Tampa, FL, USA and 3 Florida Center of Excellence for Drug Discovery and Innovation, University of South Florida, Tampa, FL, USA Correspondence: Professor JW Leahy, Department of Chemistry, University of South Florida, 4202 E. Fowler Avenue, CHE 205, Tampa, FL 33620, USA. E-mail: [email protected] Dedicated to Professor Amos B Smith, III - an outstanding chemist, mentor and friend. Received 11 December 2015; revised 14 January 2016; accepted 19 January 2016; published online 9 March 2016 The Journal of Antibiotics (2016) 69, 220–252 & 2016 Japan Antibiotics Research Association All rights reserved 0021-8820/16 www.nature.com/ja
-
Upload
khangminh22 -
Category
Documents
-
view
3 -
download
0
Transcript of Synthesis of the phorboxazoles—potent, architecturally novel ...

REVIEW ARTICLE
Synthesis of the phorboxazoles—potent, architecturallynovel marine natural products
Zachary Shultz1 and James W Leahy1,2,3
The phorboxazoles have attracted the attention of synthetic chemists around the globe due to their potent biological activity and
novel structure. A review of the recent synthetic efforts as well as new phorboxazole analogs is discussed.
The Journal of Antibiotics (2016) 69, 220–252; doi:10.1038/ja.2016.8; published online 9 March 2016
INTRODUCTION
In 1995, Searle and Molinski reported the isolation of a pair of novelnatural products from the Indian Ocean marine sponge Phorbas sp.collected off the Muiron Islands that demonstrated potent antifungaland cytostatic activity.1 The subsequent structural elucidation of thesetwo macrolides,2–4 phorboxazoles A (1, Figure 1) and B (2), hasinspired considerable attention and synthetic creativity over thesucceeding two decades that has resulted in 10 total syntheses as wellas a number of additional synthetic approaches, the discovery of newmembers of the phorboxazole family, the creation of biological probesand the generation of analogs to uncover hints about their bioactivity.Some of the highlights of these efforts are reviewed here.More recently, Capon et al.5 isolated both 1 and 2 from a Raspailia
sp. sponge collected along the Australian Northern Rottnest Shelfwhile searching for nematocidal agents. This second isolation fromsuch a taxonomically distinct source suggests a potential microbialbiosynthetic origin or at least contribution that has yet to beuncovered, leaving synthesis as the primary source for furtherconsideration of the natural products.It is not surprising that the distinctive structural features of the
phorboxazoles, which includes the presence of four unique pyran ringsand two oxazoles, attracted the interest of a number of syntheticgroups. In fact, the first synthetic approach to the molecule wasreported before the final structure elucidation was published!6 Seitzand Hoffmann have previously examined some of the earliestsyntheses of the phorboxazoles,7,8 and therefore we will primarilyfocus on the efforts that were not previously highlighted.With a target as structurally diverse as the phorboxazoles, there is an
array of potential approaches to be considered. However, the need toform a macrolide left a limited number of options for the cyclization.The most commonly exploited approach was to close the ring via anolefination of the α,β-unsaturated macrolactone—a strategy that hasproven to be quite effective in this regard. Notably, the Evans totalsynthesis9 and the Paterson synthetic approach10 utilized a Yamaguchi
macrolactonization as an alternative. The other common disconnec-tion was about the C16–C18 oxazole or the C19–C20 olefin to join thetwo macrolide fragments. As the two natural products differ onlyabout the stereochemical disposition of the hydroxyl substituent at theC13 position, the phorboxazoles serve as an excellent vehicle for thestudy of the various approaches to solve the synthetic challengesposed. In this review, we will first evaluate the completed syntheses of1 and 2, respectively, followed by the approaches of other groups inthis regard and the efforts reported to date on the discovery of othermembers of the phorboxazole family, biological probes and additionalanalogs.
DISCUSSION
Total synthesesEarliest syntheses. As the earlier reviews covered the first generationsyntheses of 1 by Forsyth and Smith as well as the syntheses of 1 byPattenden and Williams and the synthesis of 2 by Evans, we will onlypoint out a few of the salient highlights from these seminal works.Forsyth, who was the first to complete the synthesis of either naturalproduct (Figure 2),11 established the Michael cyclization to thepentasubstituted pyran and developed a biomimetic route to theoxazoles. He observed the same lack of diastereoselectivity in theformation of the C38 center observed by Molinski and Leahy,2
ascertained the viability of the hetero-Diels–Alder approach to thepyrans and established the feasibility of the Still–Gennari macrocycli-zation. Before 2006, Evans had achieved the only synthesis of 2,9
which featured a macrolactonization strategy of an acetylene to enablea selective reduction to the Z C2–C3 olefin (Figure 3). In addition,Evans’ lithiation of the oxazole methyl, optimization of the C19–C20Wittig and use of β-ketoesters enabled many of the efforts thatfollowed. Smith’s pioneering use of the Petasis–Ferrier rearrangementserves as a hallmark of his synthesis of 1 (Figure 4),12 and heimplemented a Stille protocol for introduction of the exocyclicoxazole, allowing it to serve as a linchpin for the two complex
1Department of Chemistry, University of South Florida, Tampa, FL, USA; 2Department of Molecular Medicine, Morsani College of Medicine, University of South Florida, Tampa, FL,USA and 3Florida Center of Excellence for Drug Discovery and Innovation, University of South Florida, Tampa, FL, USACorrespondence: Professor JW Leahy, Department of Chemistry, University of South Florida, 4202 E. Fowler Avenue, CHE 205, Tampa, FL 33620, USA.E-mail: [email protected] to Professor Amos B Smith, III - an outstanding chemist, mentor and friend.Received 11 December 2015; revised 14 January 2016; accepted 19 January 2016; published online 9 March 2016
The Journal of Antibiotics (2016) 69, 220–252& 2016 Japan Antibiotics Research Association All rights reserved 0021-8820/16
www.nature.com/ja

fragments. Pattenden was the first to use a carbohydrate startingmaterial (D-xylose) in his synthesis of 1,13 and his utilization of theJulia olefination for the introduction of the polyene side chain wasadopted by several subsequent synthetic approaches (Figure 5). Inaddition to using a Horner–Wadsworth–Emmons approach toC19–C20 and C27–C28, Williams was able to take advantage of his
in situ asymmetric transmetallation/allylation protocol in his synthesisof 1 (Figure 6).14
Recent synthesesSecond generation synthesis of 1 by Amos B Smith, III. In the yearssince those syntheses were completed, five additional total syntheses
N
O
O
R1R2
OO
O
ON
O
OMeO
HO
BrMeO
OH
2
5
9
11
1519
22
26
2831
33
37
40
46
R2
OO
O
N
O
O
N
OO
OH
O
BrO
HO
13
16
19
22
25 2
3
28
33
40
R1
Phorboxazole A (1, R1 = H, R2 = OH)Phorboxazole B (2, R1 = OH, R2 = H)
O
Figure 1 Structures of phorboxazoles A and B.
N
O
O
OO
O
O
HO
H3NCl
OTBDPS
OMeO
MeO
BrMeO
OH
OH
O
OTES
O
HO
H3NCl
OH
O
H2N
HO
O
O
OTBDPS
PMBO
OMe
I
HO OMe
O
OMe
OMeO
H
O
HO
OTBDPS
OO
PMBO
BrTESO
OTES
OH
MeO
O
N
O
Boc
1
Figure 2 Retrosynthetic analysis of Craig Fosyth’s first-generation total synthesis of phorboxazole A.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
221
The Journal of Antibiotics
I
Br
MeO
O
N
O
O
TESO
MeO
TESO OTIPS
H
O
N
O
O
O
Bu3PMsO-
OTIPS
PhHN
O
O
OTIPS
OTMS
O
O
OMeBnO
H
OHOH O
TESO
H
O
NHPh
O
ON
O
Bn
N
O
N
O N
O
TIPSO
O
O
OBn
OO
2
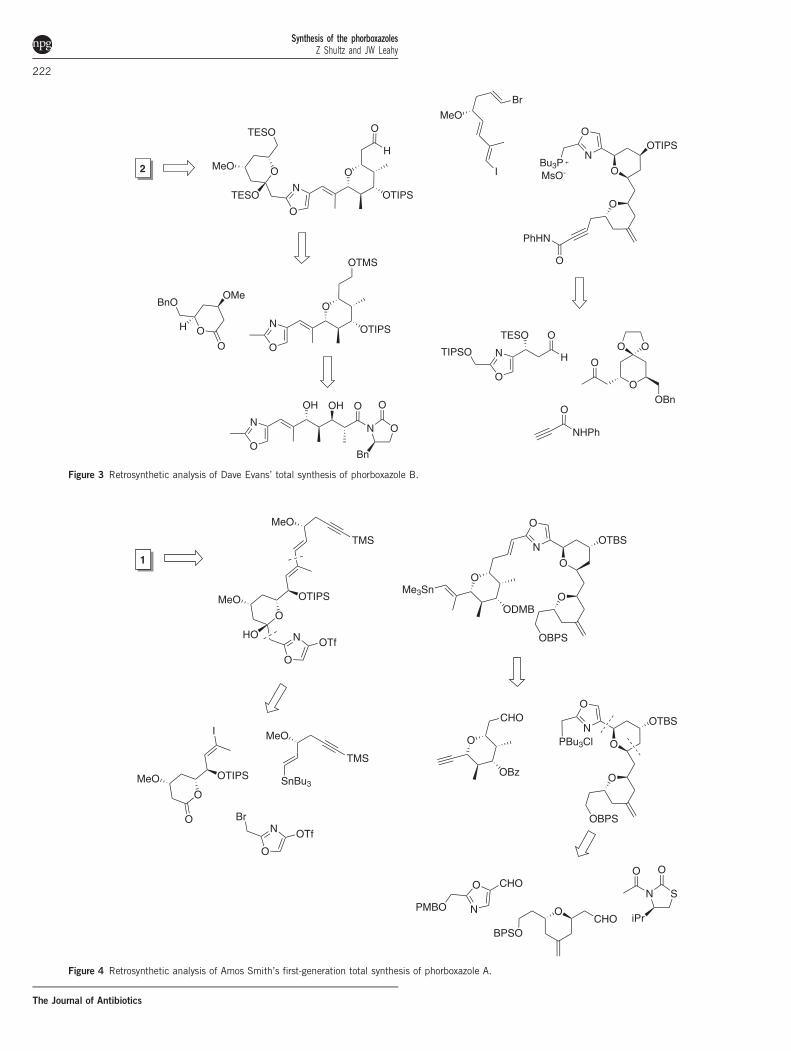
Figure 3 Retrosynthetic analysis of Dave Evans’ total synthesis of phorboxazole B.
N
O
O
MeO
HO
MeO
OTIPS
OTf
TMSN
O
O
OODMB
OMe3Sn
OBPS
OTBS
O
MeO OTIPS
I
N
O
OTfO Br
MeO
TMS
SnBu3OBz
O
CHON
O
O
O
OBPS
OTBS
PBu3Cl
OCHO
BPSO
N
O
S
O
iPrN
O
PMBO
CHO
1
Figure 4 Retrosynthetic analysis of Amos Smith’s first-generation total synthesis of phorboxazole A.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
222
The Journal of Antibiotics
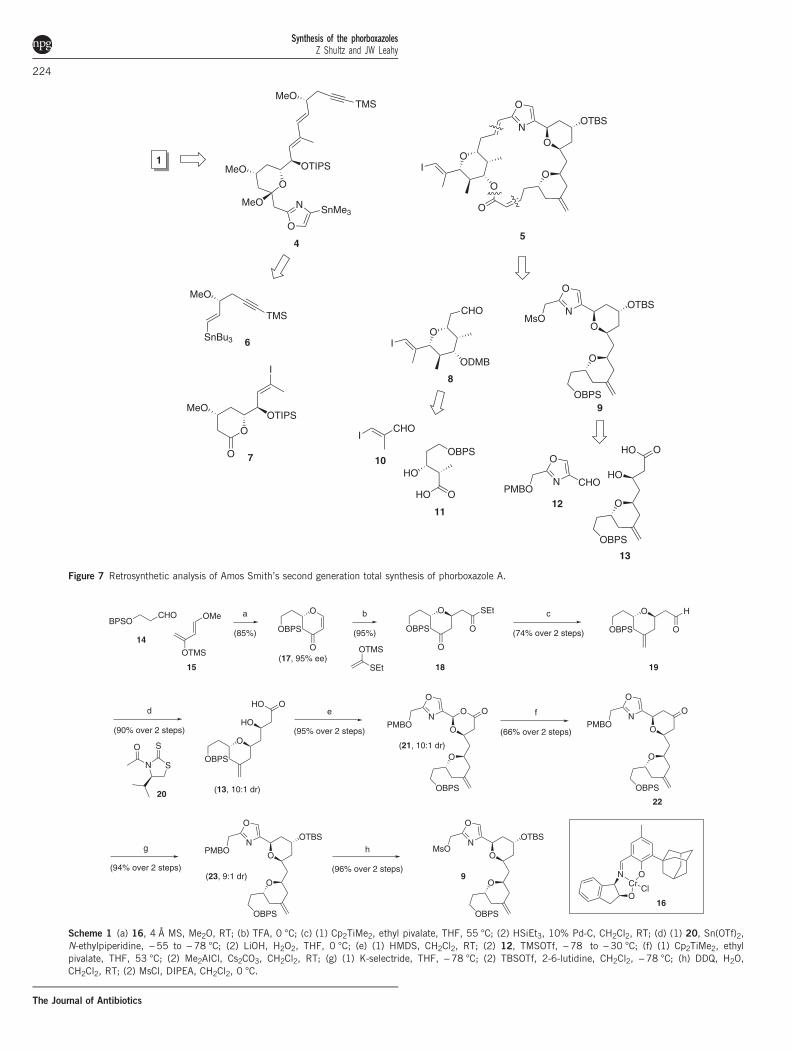
have been reported. The first of these was a second generationapproach to 1 by Smith,15 who has shown an admirable commitmentto developing scalable syntheses of scarce natural products in order tofacilitate their biological investigation.16,17 His retrosynthesis, which isa bit more convergent than his first generation approach but usesmany of the same disconnections, is shown in Figure 7. Asymmetrichetero-Diels–Alder cyclization of 14 with Danishefsky’s diene (15)
gave 17 in 95% ee using Jacobsen’s catalyst (16) on a 470-g scale(Scheme 1). Michael addition of an acetate provided excellent transselectivity, which allowed for the completion of the initial pyran 18.An acetate aldol set the stage for the formation of acetal 21, whichthen underwent the Petasis–Ferrier rearrangement to bis-pyran 22.Once the ketone was reduced to the axial alcohol, the C3–C19fragment (9) was completed as expected. Smith also used 14 as his
O
MeO
BrMeO
OH OPMP
OHO
N
O
O
O
MsOOTIPS
TBSOO
HO
N
OP(O)(OMe)2
NO O
O
OPMB
OTIPS
I
O O
HOO
PMBO OH
HOCO2Me
S
N
SO
O
BrMeO
OPMB
O
MeO OH
MeO OMe
3
OHOH
OHC
OHOH
1
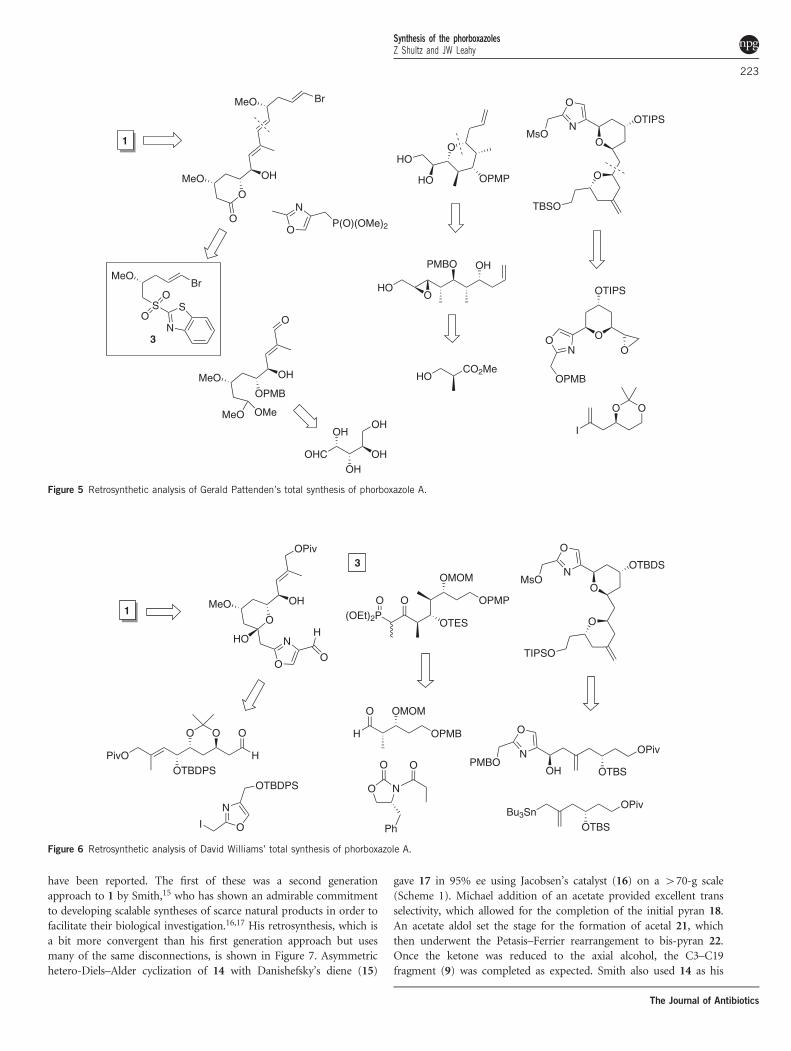
Figure 5 Retrosynthetic analysis of Gerald Pattenden’s total synthesis of phorboxazole A.
N
O
O
MeO
HO
OH
OPiv
H
O
(OEt)2PO O
OMOM
OPMP
OTES
N
O
O
O
MsOOTBDS
TIPSO
N
O
PMBOOH OTBS
OPiv
Bu3SnOPiv
OTBS
PivOOTBDPS
O O
H
O
N
O
OTBDPS
I
H
O
OPMB
OMOM
O N
O O
Ph
3
1
Figure 6 Retrosynthetic analysis of David Williams’ total synthesis of phorboxazole A.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
223
The Journal of Antibiotics
N
O
O
OO
O
O
OTBS
I
N
O
O
MeO
MeO
MeO
OTIPS
SnMe3
TMS
CHO
ODMB
OI
HO
HO
O
OBPS
O
N
O
PMBOCHO
N
O
O
O
OTBSMsO
OBPS
OBPS
HO O
HO
CHOI
TMS
MeO
SnBu3
O
MeOOTIPS
O
I
54
6
7
8
9
10
1112
13
1
Figure 7 Retrosynthetic analysis of Amos Smith’s second generation total synthesis of phorboxazole A.
OMe
OTMS
O
O
OBPS
OTMS
SEt
O
O
OBPS
SEt
O
O
OBPS
H
O
N S
SOO
OBPS
HO O
HO
BPSOCHO a
(85%)
(17, 95% ee)
b
(95%)
c
(74% over 2 steps)
d
(90% over 2 steps)
(13, 10:1 dr)
e
(95% over 2 steps)
N
O
O
O
O
PMBO
OBPS
O
(21, 10:1 dr)
f
(66% over 2 steps)
N
O
O
O
PMBO
OBPS
O
g
(94% over 2 steps)
N
O
O
O
PMBO
OBPS
OTBS
h
(96% over 2 steps)
N
O
O
O
OTBSMsO
OBPS
(23, 9:1 dr)
14
15 18 19
2022
9 N O
O
CrCl
16
Scheme 1 (a) 16, 4 Å MS, Me2O, RT; (b) TFA, 0 °C; (c) (1) Cp2TiMe2, ethyl pivalate, THF, 55 °C; (2) HSiEt3, 10% Pd-C, CH2Cl2, RT; (d) (1) 20, Sn(OTf)2,N-ethylpiperidine, −55 to −78 °C; (2) LiOH, H2O2, THF, 0 °C; (e) (1) HMDS, CH2Cl2, RT; (2) 12, TMSOTf, −78 to −30 °C; (f) (1) Cp2TiMe2, ethylpivalate, THF, 53 °C; (2) Me2AICl, Cs2CO3, CH2Cl2, RT; (g) (1) K-selectride, THF, −78 °C; (2) TBSOTf, 2-6-lutidine, CH2Cl2, −78 °C; (h) DDQ, H2O,CH2Cl2, RT; (2) MsCl, DIPEA, CH2Cl2, 0 °C.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
224
The Journal of Antibiotics
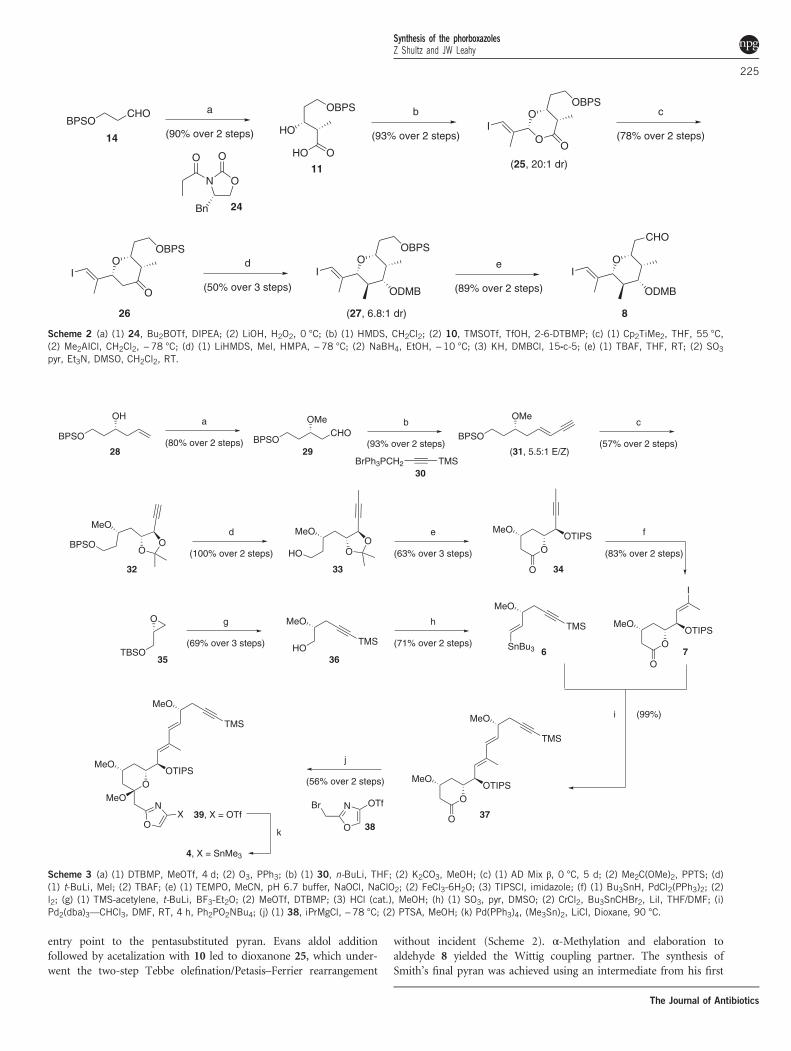
entry point to the pentasubstituted pyran. Evans aldol additionfollowed by acetalization with 10 led to dioxanone 25, which under-went the two-step Tebbe olefination/Petasis–Ferrier rearrangement
without incident (Scheme 2). α-Methylation and elaboration toaldehyde 8 yielded the Wittig coupling partner. The synthesis ofSmith’s final pyran was achieved using an intermediate from his first
BPSOCHO a
(90% over 2 steps)
ON
OO
Bn
OBPS
HO O
HO
b
(93% over 2 steps)
O
OI
O
OBPS
(25, 20:1 dr)
c
(78% over 2 steps)
OI
O
OBPSd
(50% over 3 steps)
OI
ODMB
OBPS
(27, 6.8:1 dr)
e
(89% over 2 steps)
OI
ODMB
CHO
14
24
11
26 8
Scheme 2 (a) (1) 24, Bu2BOTf, DIPEA; (2) LiOH, H2O2, 0 °C; (b) (1) HMDS, CH2Cl2; (2) 10, TMSOTf, TfOH, 2-6-DTBMP; (c) (1) Cp2TiMe2, THF, 55 °C,(2) Me2AICl, CH2Cl2, −78 °C; (d) (1) LiHMDS, Mel, HMPA, −78 °C; (2) NaBH4, EtOH, −10 °C; (3) KH, DMBCl, 15-c-5; (e) (1) TBAF, THF, RT; (2) SO3pyr, Et3N, DMSO, CH2Cl2, RT.
BPSO
OH
BPSOCHO
OMea
(80% over 2 steps)BPSO
OMeb
(93% over 2 steps)
c
(57% over 2 steps)
MeO
BPSOO
Od
(100% over 2 steps)
MeO
HO OO
O
MeOOTIPS
O
e
(63% over 3 steps)
f
(83% over 2 steps)
O
MeOOTIPS
O
I
TBSO
O
HOTMS
MeOg
(69% over 3 steps)
h
(71% over 2 steps)
TMS
MeO
SnBu3
O
MeOOTIPS
O
TMS
MeO
BrPh3PCH2 TMS(31, 5.5:1 E/Z)
i (99%)
O
MeOOTIPS
TMS
MeO
MeO
O
NX
j
(56% over 2 steps)
39, X = OTf
4, X = SnMe3
k
28 29
30
32 33 34
763635
37N
O
Br OTf
38
Scheme 3 (a) (1) DTBMP, MeOTf, 4 d; (2) O3, PPh3; (b) (1) 30, n-BuLi, THF; (2) K2CO3, MeOH; (c) (1) AD Mix β, 0 °C, 5 d; (2) Me2C(OMe)2, PPTS; (d)(1) t-BuLi, Mel; (2) TBAF; (e) (1) TEMPO, MeCN, pH 6.7 buffer, NaOCl, NaClO2; (2) FeCl3-6H2O; (3) TIPSCl, imidazole; (f) (1) Bu3SnH, PdCl2(PPh3)2; (2)l2; (g) (1) TMS-acetylene, t-BuLi, BF3-Et2O; (2) MeOTf, DTBMP; (3) HCl (cat.), MeOH; (h) (1) SO3, pyr, DMSO; (2) CrCl2, Bu3SnCHBr2, Lil, THF/DMF; (i)Pd2(dba)3—CHCl3, DMF, RT, 4 h, Ph2PO2NBu4; (j) (1) 38, iPrMgCl, −78 °C; (2) PTSA, MeOH; (k) Pd(PPh3)4, (Me3Sn)2, LiCl, Dioxane, 90 °C.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
225
The Journal of Antibiotics

generation synthesis (which could also be prepared from aldehyde 14,meaning all of the fragments emanate from the same startingmaterial), and features an asymmetric Sharpless dihydroxylation toset the C37 and C38 stereocenters, a Stille coupling of 6 and 7 toinstall the diene and the introduction of the linchpin oxazole (Scheme3). With all of the intermediates in hand, Wittig coupling of 8 and 9proceeded with outstanding selectivity (Scheme 4). Macrocyclizationwas achieved using the Still–Gennari conditions to give the Z-olefinwith 2.5:1 selectivity. The two fragments were united via a Stillecoupling, and the peripheral vinyl bromide was installed followed byglobal deprotection to give 1.
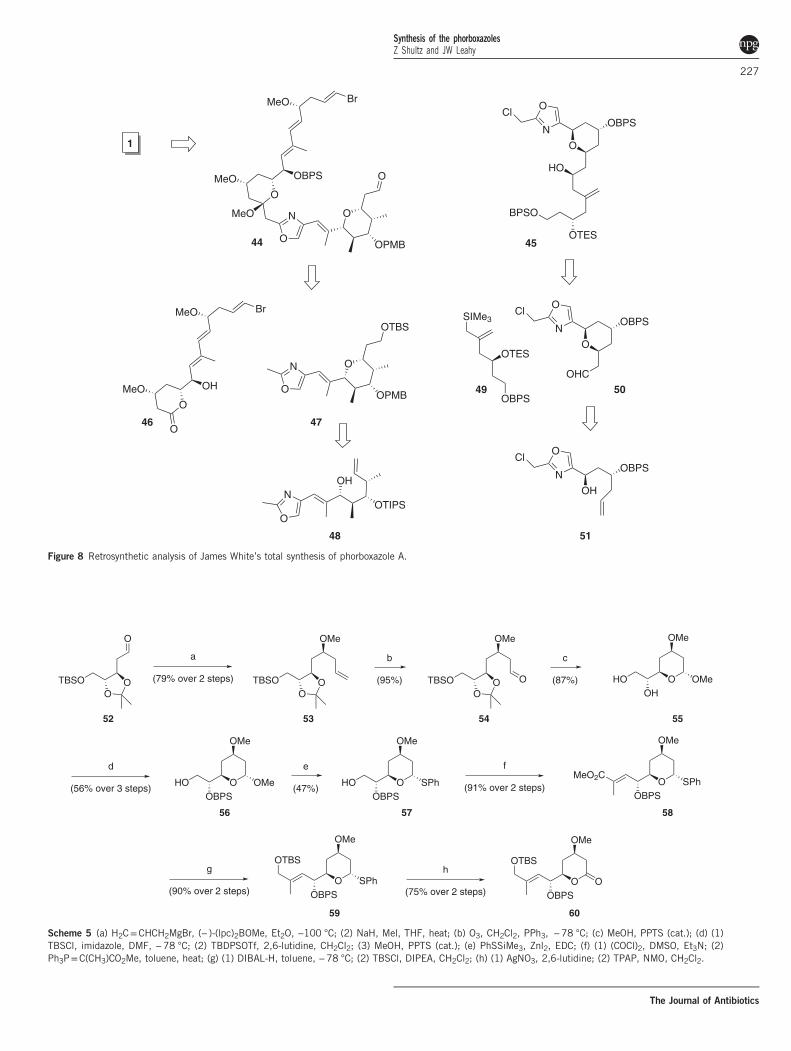
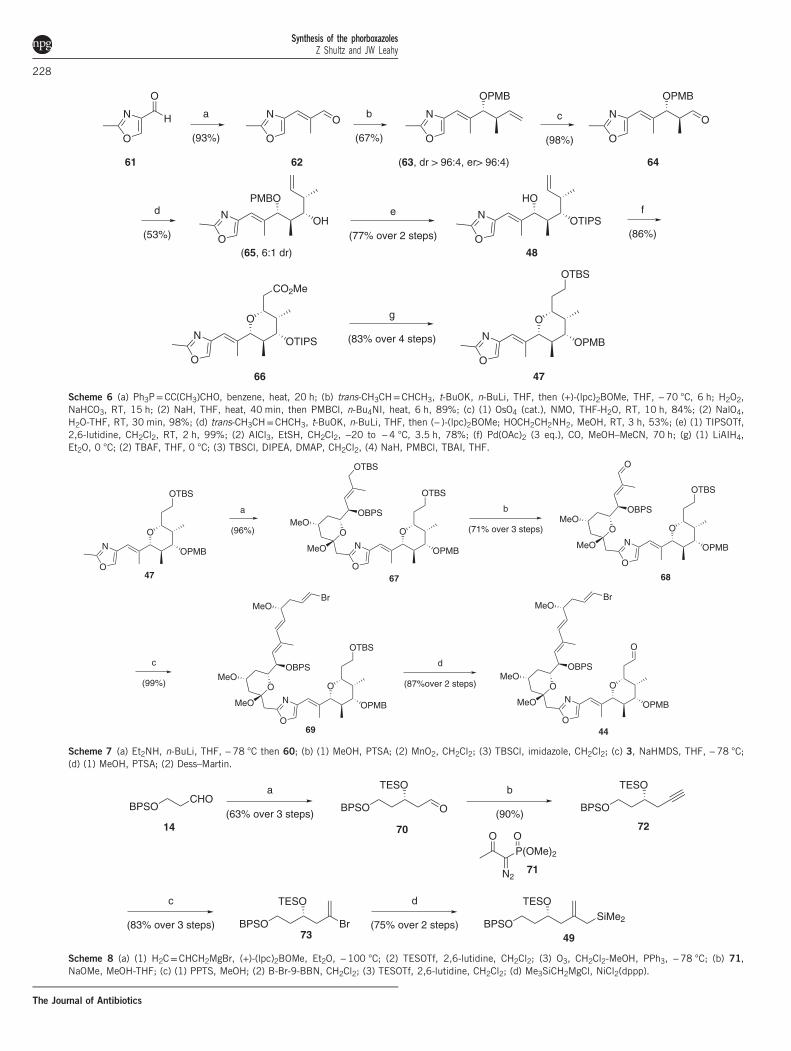
Total synthesis of 1 by James White. In their synthetic approach to 1,the White group installed the elaborated polyene before introducingthe bis-pyran portion of the macrolide (Figure 8).18,19 Asymmetricallylation of tartrate-derived 52 leads to 53, which was oxidized andacetalized (Scheme 5). Protection of the secondary alcohol allows forthe Wittig homologation to 58, which facilitated the generation oflactone 60. Asymmetric crotylation of 62 installed the C25 and C26stereocenters with 92% ee (Scheme 6), which was further elaboratedvia a second asymmetric crotylation with good diastereocontrol. Apalladium-mediated alkoxycarbonylation then created the pentasub-stituted pyran 66, which was converted to 47 in anticipation ofcoupling to 46, which was achieved via deprotonation of the oxazolemethyl (Scheme 7). Formation of enal 68 and Julia olefinationprovided the C20–C46 fragment. It should be noted that the vinylbromide had to be installed after the introduction of the oxazole, as itpromoted elimination to the undesired terminal alkyne.White prepared the bis pyran from the asymmetric allylation of 14
(Scheme 8). Seyferth–Gilbert homologation to the alkyne precededbromoboration and extension to allylsilane 49. Asymmetric allylationof 74 proceeded with outstanding enantiocontrol (Scheme 9), whichwas extended to 77 via a second asymmetric allylboration. The cispyran was again formed via a carbonylative process, which was thencoupled with 49 to complete the C3–C19 fragments. The undesired
diastereomer at C9 was able to be converted to 45 using an oxidation/reduction protocol. The final assembly of 1 was achieved via the Wittigunion of the fragments, which selectively gave the trans product(Scheme 10). The trans-pyran was then closed and the macrolideformed via intramolecular Horner–Wadsworth–Emmons olefinationwith 4:1 selectivity. Global deprotection yielded 1.
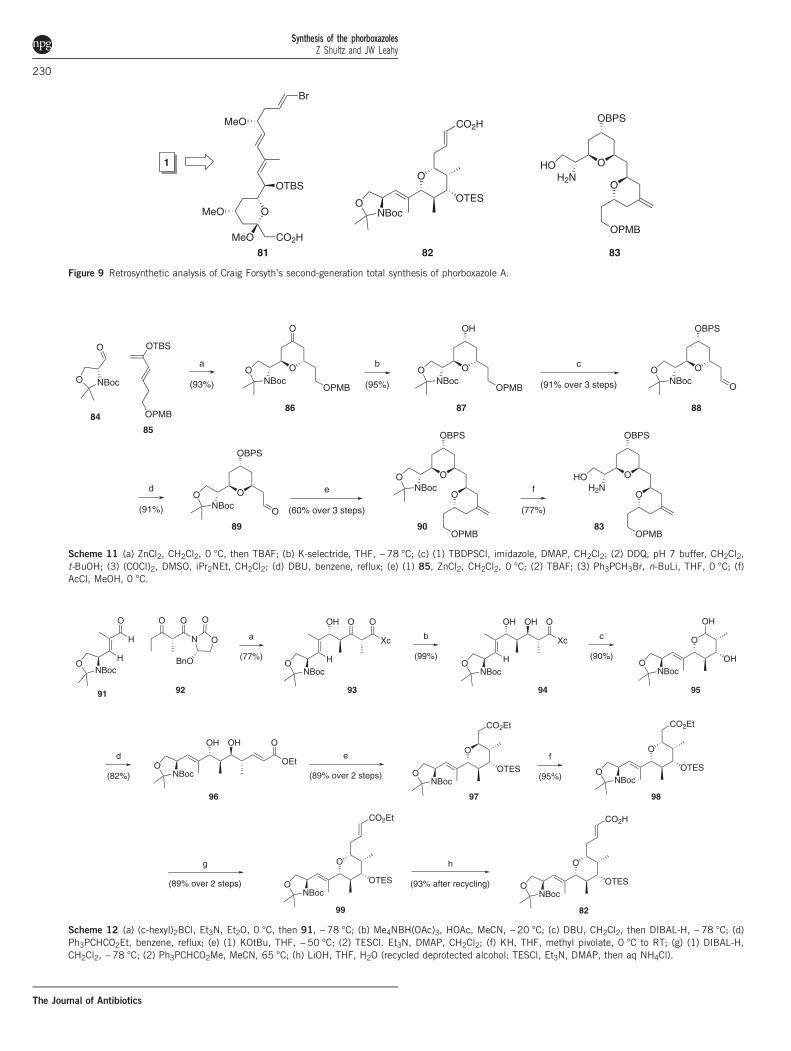
Second generation syntheses of 1 by Craig Forsyth. Forsyth has alsorecently disclosed an extensive study that culminated in a second-generation synthesis of 1, expanding on his de novo oxazole approach(Figure 9).20,21 Hetero–Diels–Alder cyclization with Garner’s aldehyde(84) gave an excellent yield of exclusively 86, which is initially thetrans-pyran (Scheme 11). Manta has also reported the synthesis of 86using this methodology. Upon elaboration to aldehyde 88, this couldbe equilibrated to the desired cis product (89). A second hetero-Diels–Alder cyclization with diene 85 then provided bis-pyran 90.β-Ketoimide aldol addition to 91 and directed reduction gave thestereotetrad of the pentasubstituted pyran, which was extended toα,β-unsaturated ester 96 and closed via the Michael protocol(Scheme 12) to render primarily the trans-pyran. Equilibration via aretro-Michael/Michael process then afforded the desired stereochem-istry, and the ester was extended and saponified to 82.Forsyth’s approach to the exoxyclic pyran ultimately emanated from
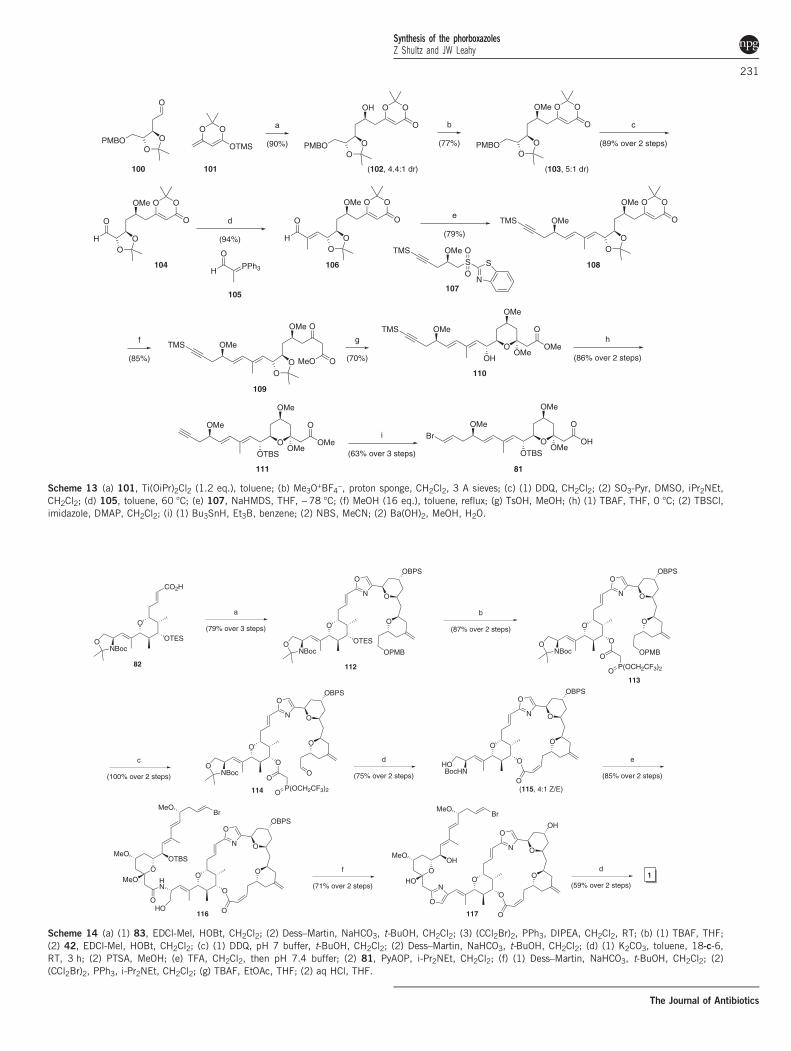
D-xylose derived 100 (Scheme 13). Silylketene acetal addition gave44:1 selectivity for 102, which was converted to the aldehyde andextended to 106. Julia addition of the terminus gave 108, which couldbe ketalized and advanced to vinyl bromide 81.Forsyth explored an impressive array of approaches to optimize the
final formation of 1, including a ring closing metathesis thatdramatically improved the C2–C3 Z:E selectivity and a tandem denovo oxazole formation of both rings. The most direct synthesis isshown in Scheme 14. Ultimately, his second-generation synthesisachieves 1 in an overall yield of 6% with a longest linear sequence of19 steps from 84.
N
O
O
O
OTBSMsO
OBPS
a
(96% over 2 steps)
N
O
O
O
OTBS
OBPSODMB
O
I
N
O
O
O
OTBS
OOH
O
I
HO
OP(OCH2CF3)2
O
d (88% over 2 steps)
N
O
O
OO
O
O
OTBS
I
(40, 20:1 E/Z)
b
(81% over 2 steps)
N
O
O
OO
O
ON
O
O
MeO
MeO
MeO
OTIPS
TMS
OTBSe
(68%)
9 41
42c,
5
43
f
(33% over 4 steps)1
Scheme 4 (a) (1) PBu3, DMF, RT; (2) 8, DBU, DMF, RT; (b) (1) KOH, 18-c-6, THF, 4% H2O; (2) Dess–Martin, NaHCO3, CH2Cl2, 0 °C; (3) DDQ, pH 7buffer, CH2Cl2; (c) (1) EDCl-Mel, HOBT, 42, CH2Cl2; (d) K2CO3, 18-c-6, toluene, RT; (e) Pd2(dba)3, CHCl3, 4, AsPh3, DIPEA, Ph2PO2NBu4, DMF, RT; (f)(1) AgNO3, NBS, acetone; (2) PdCl2(PPh3)2, HSnBu3, THF (6:1, ext:int); (3) NBS, MeCN, 0 °C; (4) 6% HCl, THF, RT.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
226
The Journal of Antibiotics
a
(79% over 2 steps)
O
TBSOO
O
OMe
TBSOO
O
b
(95%)
OMe
TBSOO
O O
c
(87%) O
OMe
OMeOH
HO
d
(56% over 3 steps)O
OMe
OMeOBPS
HO
e
(47%)O
OMe
SPhOBPS
HO
f
(91% over 2 steps)O
OMe
SPhOBPS
MeO2C
g
(90% over 2 steps)O
OMe
SPhOBPS
OTBSh
(75% over 2 steps)O
OMe
OOBPS
OTBS
52 53 54 55
56 57 58
59 60
Scheme 5 (a) H2C=CHCH2MgBr, (− )-(lpc)2BOMe, Et2O, −100 °C; (2) NaH, Mel, THF, heat; (b) O3, CH2Cl2, PPh3, −78 °C; (c) MeOH, PPTS (cat.); (d) (1)TBSCl, imidazole, DMF, −78 °C; (2) TBDPSOTf, 2,6-lutidine, CH2Cl2; (3) MeOH, PPTS (cat.); (e) PhSSiMe3, Znl2, EDC; (f) (1) (COCl)2, DMSO, Et3N; (2)Ph3P=C(CH3)CO2Me, toluene, heat; (g) (1) DIBAL-H, toluene, −78 °C; (2) TBSCl, DIPEA, CH2Cl2; (h) (1) AgNO3, 2,6-lutidine; (2) TPAP, NMO, CH2Cl2.
OPMB
ON
O
OTBS N
O
O
OHC
Cl
O
O
MeO
BrMeO
OH
O
OPMB
ON
O
O
MeO
MeO
BrMeO
OBPS
N
O
O
HO
ClOBPS
OTES
BPSO
OBPSSIMe3
OBPS
OTES
OTIPS
OHN
O
N
O
OH
ClOBPS
44 45
46 47
48
49 50
51
1
Figure 8 Retrosynthetic analysis of James White’s total synthesis of phorboxazole A.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
227
The Journal of Antibiotics
N
O
H
O
N
O
O N
O
OPMB
N
O
O
OPMB
N
O
PMBO
OH
N
O
O
OTIPS
CO2Me
a
(93%)
b
(67%)
c
(98%)
d
(53%)
e
(77% over 2 steps)
(63, dr > 96:4, er> 96:4)
(65, 6:1 dr)
f
(86%)
N
O
HO
OTIPS
g
(83% over 4 steps) N
O
O
OPMB
OTBS
61 62 64
48
66 47
Scheme 6 (a) Ph3P=CC(CH3)CHO, benzene, heat, 20 h; (b) trans-CH3CH=CHCH3, t-BuOK, n-BuLi, THF, then (+)-(lpc)2BOMe, THF, −70 °C, 6 h; H2O2,NaHCO3, RT, 15 h; (2) NaH, THF, heat, 40 min, then PMBCl, n-Bu4NI, heat, 6 h, 89%; (c) (1) OsO4 (cat.), NMO, THF-H2O, RT, 10 h, 84%; (2) NalO4,H2O-THF, RT, 30 min, 98%; (d) trans-CH3CH=CHCH3, t-BuOK, n-BuLi, THF, then (− )-(lpc)2BOMe; HOCH2CH2NH2, MeOH, RT, 3 h, 53%; (e) (1) TIPSOTf,2,6-lutidine, CH2Cl2, RT, 2 h, 99%; (2) AICl3, EtSH, CH2Cl2, −20 to −4 °C, 3.5 h, 78%; (f) Pd(OAc)2 (3 eq.), CO, MeOH–MeCN, 70 h; (g) (1) LiAIH4,Et2O, 0 °C; (2) TBAF, THF, 0 °C; (3) TBSCl, DIPEA, DMAP, CH2Cl2, (4) NaH, PMBCl, TBAI, THF.
N
O
O
OPMB
OTBS
a
(96%)
N
O
O
OPMB
OTBS
O
MeO
MeOOBPS
OTBS
b
(71% over 3 steps)
c
(99%)
N
O
O
OPMB
OTBS
O
MeO
MeOOBPS
O
N
O
O
OPMB
OTBS
O
MeO
MeOOBPS
BrMeO
d
(87%over 2 steps)
N
O
O
OPMB
O
O
MeO
MeOOBPS
BrMeO
47 67 68
69 44
Scheme 7 (a) Et2NH, n-BuLi, THF, −78 °C then 60; (b) (1) MeOH, PTSA; (2) MnO2, CH2Cl2; (3) TBSCl, imidazole, CH2Cl2; (c) 3, NaHMDS, THF, −78 °C;(d) (1) MeOH, PTSA; (2) Dess–Martin.
a
(63% over 3 steps)BPSO O
TESO b
(90%)
OP(OMe)2
O
N2
BPSO
TESO
c
(83% over 3 steps) BPSO
TESO
Br
d
(75% over 2 steps) BPSO
TESOSiMe2
BPSOCHO
14 70
71
72
73 49
Scheme 8 (a) (1) H2C=CHCH2MgBr, (+)-(lpc)2BOMe, Et2O, −100 °C; (2) TESOTf, 2,6-lutidine, CH2Cl2; (3) O3, CH2Cl2-MeOH, PPh3, −78 °C; (b) 71,NaOMe, MeOH-THF; (c) (1) PPTS, MeOH; (2) B-Br-9-BBN, CH2Cl2; (3) TESOTf, 2,6-lutidine, CH2Cl2; (d) Me3SiCH2MgCl, NiCl2(dppp).
Synthesis of the phorboxazolesZ Shultz and JW Leahy
228
The Journal of Antibiotics

Total synthesis of 2 by Guo-Qiang Lin and Wei-Shan Zhou. Two totalsyntheses of 2 have also been achieved in the last decade. The first ofthese came from the labs of Lin and Zhou (Figure 10).22 Asymmetricallylation and Wacker oxidation of the protected product gave an 87%
ee of ketone 126 (Scheme 15). Aldol addition and oxidation led todiketone 128, which was closed to pyranone 129. Hydrogenationsimultaneously set the C13 and C15 stereocenters, leading to aldehyde122. Aldol addition of 123 (derived as the opposite antipode to 126,
N
O
ClO
H a
(84%)
N
O
ClOH
N
O
ClOTBS
Ob
(69% over 2 steps)(75, er > 20:1)
N
O
ClOH
OBPSc
(56% over 3 steps)(77, dr > 12:1)
d
(77% over 2 steps)
N
O
ClO
OBPS
CHO
f
(55-65%)
N
O
ClO
OBPS
OH
BPSO
OTES(45, 1:1 mixture at C9)
74 76
50
Scheme 9 (a) (+)-(lpc)2BOMe, CH2=CHCH2MgBr, Et2O, −100 °C; (b) (1) TBSOTf, 2,6-lutidine, CH2Cl2, RT; (2) OsO4(cat.), NalO4, H2O-THF; (c) (1)(+)-(lpc)2BOMe, CH2=CHCH2MgBr, Et2O, −100 °C; (2) TBDPSOTf, 2,6-lutidine, CH2Cl2, RT; (3) HCl (3N), THF–H2O; (d) (1) PdCl2(MeCN)2, (10 mol%), CO(1 atm), MeOH, p-benzoquinone; (2) DIBAL-H, CH2Cl2, −78 °C; (f) 49, SnCl4, CH2Cl2, −78 °C (inverted unwanted diastereomer by [O]/[H] process).
N
O
ClO
OBPS
OH
BPSO
OTES
a
(85%)
N
O
O
OBPS
OH
BPSO
OTESN
O
O
OPMB
O
MeO
MeOOBPS
BrMeO
N
O
O
OBPS
ON
O
O
OPMB
O
MeO
MeOOBPS
BrMeO
OBPS
c
(53% over 4 steps)
N
O
O
OBPS
ON
O
O
O
O
MeO
MeOOBPS
BrMeO
OP(OMe)2O
O
d (15% over 3 steps)(4:1 Z/E)
45
b
(83% over 3 steps)
78
79 80
1
Scheme 10 (a) Bu3P, DMF, DBU, then 44; (b) (1) MsCl, Et3N; (2) MeOH, PPTS, CH2Cl2; (3) Et3N, MeCN; (c) (1) DDQ, CH2Cl2;(2) (MeO)2P(O)CH2CO2H,DCC, Ch2Cl2; (3) NH4F (excess), MeOH, 50°C; (4) Dess–Martin, CH2Cl2; (d) K2CO3, 18-c-6, toluene, −78°C to RT; (2) TBAF, THF; HCl (6%), THF.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
229
The Journal of Antibiotics
OTBS
OMeO
CO2HMeO
Br
MeO
O
OTESNBoc
O
CO2H
O
OBPS
HOH2N
O
OPMB
81 82 83
1
Figure 9 Retrosynthetic analysis of Craig Forsyth’s second-generation total synthesis of phorboxazole A.
OPMB
OTBS
O
O
OPMB
ONBoc
a
(93%)
b
(95%)
O
OH
OPMB
ONBoc
c
(91% over 3 steps)
O
OBPS
O
ONBoc
d
(91%)
O
OBPS
O
ONBoc
e
(60% over 3 steps)
O
OBPS
ONBoc
O
OPMB
O
OBPS
HOH2N
O
OPMB
f
(77%)
O
O NBoc
8485
86 87 88
89 90 83
Scheme 11 (a) ZnCl2, CH2Cl2, 0 °C, then TBAF; (b) K-selectride, THF, −78 °C; (c) (1) TBDPSCl, imidazole, DMAP, CH2Cl2; (2) DDQ, pH 7 buffer, CH2Cl2,t-BuOH; (3) (COCl)2, DMSO, iPr2NEt, CH2Cl2; (d) DBU, benzene, reflux; (e) (1) 85, ZnCl2, CH2Cl2, 0 °C; (2) TBAF; (3) Ph3PCH3Br, n-BuLi, THF, 0 °C; (f)AcCl, MeOH, 0 °C.
O
N O
O
BnO
O
NBocO
H
O
H a
(77%)
OOOH
ONBoc
H
Xc
OOHOH
ONBoc
H
Xcb
(99%)
c
(90%)
O
OHNBoc
O
OH
d
(82%)
OH
NBocO
OH O
OEte
(89% over 2 steps)
O
CO2Et
OTESNBoc
O
f
(95%)
O
CO2Et
OTESNBoc
O
g
(89% over 2 steps)
O
OTESNBoc
O
CO2Et
O
OTESNBoc
O
CO2H
h
(93% after recycling)
91 92 93 94 95
96 97 98
99 82
Scheme 12 (a) (c-hexyl)2BCl, Et3N, Et2O, 0 °C, then 91, −78 °C; (b) Me4NBH(OAc)3, HOAc, MeCN, −20 °C; (c) DBU, CH2Cl2, then DIBAL-H, −78 °C; (d)Ph3PCHCO2Et, benzene, reflux; (e) (1) KOtBu, THF, −50 °C; (2) TESCl. Et3N, DMAP, CH2Cl2; (f) KH, THF, methyl pivolate, 0 °C to RT; (g) (1) DIBAL-H,CH2Cl2, −78 °C; (2) Ph3PCHCO2Me, MeCN, 65 °C; (h) LiOH, THF, H2O (recycled deprotected alcohol: TESCl, Et3N, DMAP, then aq NH4Cl).
Synthesis of the phorboxazolesZ Shultz and JW Leahy
230
The Journal of Antibiotics
O
OOPMBO
O O
OTMS
a
(90%)
OH
OOPMBO
O
O
O
(102, 4.4:1 dr)
b
(77%)
OMe
OOPMBO
O
O
O
c
(89% over 2 steps)
OMe
OO
O
O
O
H
O d
(94%)
OMe
OO
O
O
O
H
O
PPh3H
O
e
(79%)
TMS OMeS
N
S
O
O
OMe
OO
O
O
O
TMS OMe
f
(85%)
OMe
OO
TMS OMe
MeO O
O
g
(70%)
TMS OMe
OHO
OMe
OMe
O
OMe
h
(86% over 2 steps)
OMe
OTBSO
OMe
OMe
O
OMe
i
(63% over 3 steps)
OMe
OTBSO
OMe
OH
O
OMeBr
100 101 (103, 5:1 dr)
104
105
106
107
108
109
110
111 81
Scheme 13 (a) 101, Ti(OiPr)2Cl2 (1.2 eq.), toluene; (b) Me3O+BF4−, proton sponge, CH2Cl2, 3 A sieves; (c) (1) DDQ, CH2Cl2; (2) SO3-Pyr, DMSO, iPr2NEt,CH2Cl2; (d) 105, toluene, 60 °C; (e) 107, NaHMDS, THF, −78 °C; (f) MeOH (16 eq.), toluene, reflux; (g) TsOH, MeOH; (h) (1) TBAF, THF, 0 °C; (2) TBSCl,imidazole, DMAP, CH2Cl2; (i) (1) Bu3SnH, Et3B, benzene; (2) NBS, MeCN; (2) Ba(OH)2, MeOH, H2O.
O
OTESNBoc
O
CO2H
a
(79% over 3 steps) O
OTESNBoc
O
O
OBPS
N
O
OPMB
O
O
ONBoc
O
O
OBPS
N
O
OPMB
O
b
(87% over 2 steps)
P(OCH2CF3)2
O
O
c
(100% over 2 steps)
O
ONBoc
O
O
OBPS
N
O
O
O
P(OCH2CF3)2
O
O
O
OBocHN
HO
O
OBPS
N
O
O
O(115, 4:1 Z/E)
d
(75% over 2 steps)
e
(85% over 2 steps)
O
OHN
O
OBPS
N
O
O
OHOO
OMeO
BrMeO
OTBSMeO
f
(71% over 2 steps)O
O
O
OH
O
O
OHO
MeOOH
MeO
N
O
O
N
Br
d
(59% over 2 steps)
82 112
113
114
116 117
1
Scheme 14 (a) (1) 83, EDCl-Mel, HOBt, CH2Cl2; (2) Dess–Martin, NaHCO3, t-BuOH, CH2Cl2; (3) (CCl2Br)2, PPh3, DIPEA, CH2Cl2, RT; (b) (1) TBAF, THF;(2) 42, EDCl-Mel, HOBt, CH2Cl2; (c) (1) DDQ, pH 7 buffer, t-BuOH, CH2Cl2; (2) Dess–Martin, NaHCO3, t-BuOH, CH2Cl2; (d) (1) K2CO3, toluene, 18-c-6,RT, 3 h; (2) PTSA, MeOH; (e) TFA, CH2Cl2, then pH 7.4 buffer; (2) 81, PyAOP, i-Pr2NEt, CH2Cl2; (f) (1) Dess–Martin, NaHCO3, t-BuOH, CH2Cl2; (2)(CCl2Br)2, PPh3, i-Pr2NEt, CH2Cl2; (g) TBAF, EtOAc, THF; (2) aq HCl, THF.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
231
The Journal of Antibiotics

Scheme 16) gave a nearly 8:1 diastereomeric ratio of 132. Nystedolefination and cyclization resulted in bis-pyran 135, which was thenprepped for the Wittig coupling. Sequential asymmetric crotylation ofglyceraldehyde 136 using Roush’s tartrate-derived methodology pro-duced 140 (Scheme 17), which was closed to the cis pyran using anintramolecular oxymercuration cyclization with good stereocontrol.Once the resultant iodide was transformed into a protected silyl ether,removal of the acetonide and periodate cleavage yielded ketone 144,which was adorned with the oxazole to complete the C20–C32 piece.The exocyclic pyran was generated from the Sharpless asymmetric
dihydroxylation of enoate 147 (Scheme 18). Upon extension to theallylic acetate, the primary TBS ether was converted to the corre-sponding aldehyde and subjected to a titanium silylketene acetaladdition with 4:1 selectivity at C35. Deprotection allowed selectiveδ-lactone cyclization, which led to 154. Methyl deprotonation andketalization was used to prepare 155 (Scheme 19), which underwentJulia olefination with 3 to C20–C46 fragment 118. Although its use inthe synthesis of the phorboxazoles was well established by Pattenden,
Lin and Zhou prepared 3 via dihydroxylation of bis-allyl ether 157 andWipf’s hydrozirconation protocol (Scheme 20). The synthesis of 2 wascompleted via Wittig coupling of the C19-C20 olefin and Horner–Wadsworth–Emmons macrocyclization (Scheme 21).
Total synthesis of 2 by Steven Burke. The Burke synthesis of 2(Figure 11) is a beautiful illustration of the use of two-directionalsynthesis.23 Readily available diol 171 was ozonolyzed and convertedto achiral tetraol 170 (Scheme 22). Palladium-catalyzed desymmetri-zation with Trost’s DPPBA ligand gave the bis-pyran 174 in 98% ee.Selective asymmetric dihydroxylation of the equatorial vinyl groupcompleted the desymmetrization, which was able to be oxidized tocarboxylate 177 and cyclized under Mitsunobu conditions to lactone178, thus differentiating the two secondary alcohols. Upon reductiveopening of the lactone, the exocyclic alkene was installed using theTebbe reagent to generate 180. Removal of the acetonide and selectiveprimary protection allowed for the introduction of nitrogen withdiphenylphosphoryl azide. Burke was the first to utilize an asymmetric
ON
O
O
MeO
MeO
BrMeO
OTIPS
N
O
O
O
BPSO
MsO
O
OPMB
O
OMe
OTIPSOTBSO
ON
OOPMB
OBPS
OTBS
N
OTBSO
O
OTBS
H
O OPMB
OBPS
O
118
119
120
121
122
123
3
2
Figure 10 Retrosynthetic analysis of Gua-Lin Qiang and Wei-Shan Zhou’s total synthesis of phorboxazole B.
O OPMB OPMB
OTBS
N
O
H
O
TBSO
OPMB
OTBSO
OPMB
OTBSO
N
O
OH
TBSO
N
OTBSO
O
O
OPMB HON
OTBSO OH
H
OPMB
a
(68% over 2 steps)
b
(89%)
d
(87% over 2 steps)
e
(65%)
(125, 87% ee)
c
(90% over 2 steps)
f
(82% over 3 steps)
N
OTBSO
O
OTBS
H
O
124 126
127
128
129 130 122
Scheme 15 (a) (1) (− )-lpc2BAIIyl, Et2O, −78 °C; (2) TBSCl, imidazole, DMF; (b) PdCl2 (cat.), CuCl, O2, DMF/H2O, RT, 6 h; (c) (1) LDA, 127, THF, −78 °C;(2) Dess–Martin, CH2Cl2, RT; (d) (1) HF in CH3CN, RT, 12 h; (2) TBSCl, imidazole, DMF, RT; (e) H2, 10% Pd/C, EtOAc (saturated with 0.1N HCl), 8 h (f)(1) TBSCl, imidazole, DMF; (2) DDQ, CH2Cl2, H2O; (3) Dess–Martin, CH2Cl2, RT.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
232
The Journal of Antibiotics

O OBPS OBPS
OPMBa
(64% over 2 steps)
b
(86%)OBPS
OPMBOc
(68%)
O
OTBSO
N
OTBS
O
BPSO
O
OTBSO
N
OTBSHO
OOPMB
BPSO
(132, 7.8:1 dr)
O
OTBSO
N
OTBS
BzO
OPMB
BPSO
d
(66% over 2 steps)
O
OTBSO
N
OTBS
MsO
OH
BPSO
e
(67% over 3 steps)
f
(83%)
g
(73% over 2 steps)
O
OTBSO
N
OMs
O
BPSO
(131, 87% ee)
OZn Zn
Zn
Br Br
Nysted Reagent
14 123
133 134 135
119
Scheme 16 (a) (1) (+)-lpc2BAIIyl, Et2O, −78 °C; (2) PMBOC(=NH)CCl3, cyclohexane/CH2Cl2, BF3OEt2, 0 °C; (b) PdCl2 (cat.), CuCl, O2, DMF/H2O, RT, 6 h;(c) (1) LiHMDS, TMSCl, THF, −78 °C; (2) 122, TiCl4, −78 °C; (d) (1) BzCl, pyr, RT; (2) Nysted reagent, TiCl4, RT, 30 min; (e) (1) DIBAL-H, CH2Cl2, −78 °C; (2) MsCl, Et3N, CH2Cl2, 0 °C; (3) DDQ, CH2Cl2/H2O; (f) Et3N, MeCN, reflux; (g) (1) NH4F, MeOH, 50 °C; (2) DIPEA, MsCl.
OO
H
OO
O
OAc
OO
OAc
O OO
OH
OH
BO
O
R1
R3R2
R4
(-) 137: R1=R4=CO2iPr, R2=R3=H(+) 137: R1=R4=H, R2=R3=CO2iPr
OO
O
I
OH
b
(85%)
c
(70%)
a
(62% over 2 steps)
d
(86%)
(141, 5:1 dr)
OO
O
CN
OPMB
e
(64% over 2 steps) OO
O
OPMB
f
(72% over 2 steps)
OH
g
(53% over 4 steps)
O
OPMB
OBPS
O
h
(78% BORSM)
O
OPMB
OTBDPS
N
ON
O
PO(OMe)2
136 138 139 140
142 143
144145
121
Scheme 17 (a) (1) (− )-137, 4 Å MS, toluene, −78 °C, 7 h; (2) Ac2O, Et3N, DMAP (cat.), CH2Cl2, RT, overnight; (b) O3, MeOH, −78 °C; then PPh3, RT; (c)(+)-137, 4 Å MS, toluene, −78 °C, 6 h; (d) Hg(OAc)2, toluene, 0 °C, l2, 30 °C; (e) (1) PMBOC(=NH)CCl3, BF3OEt2, 0 °C; (2) NaCN, DMSO, 70 °C; (f) (1)DIBAL-H, CH2Cl2, 0 °C, then 1N HCl; (2) NaBH4, MeOH; (g) (1) TBDPSCl, Et3N, CH2Cl2; (2) HIO4, EtOAc; (3) MeLi, THF, −78 °C; (4) Dess–Martin,CH2Cl2, RT; (h) 145, LDA, THF, −78 °C.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
233
The Journal of Antibiotics
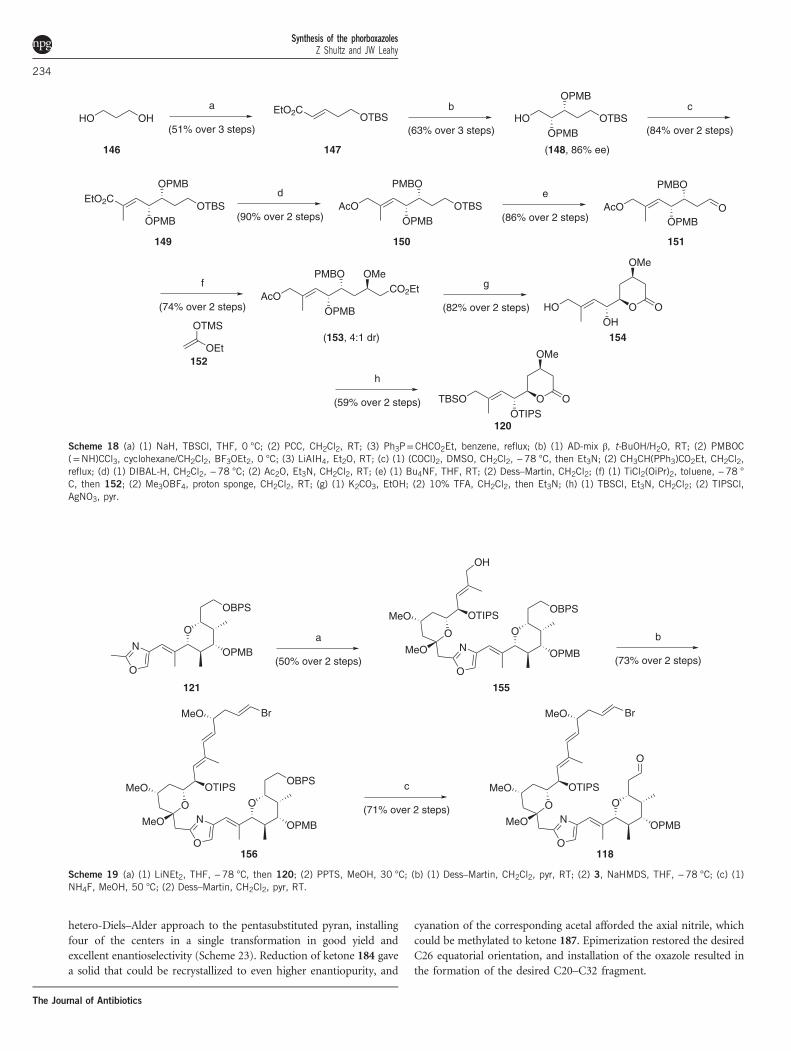
hetero-Diels–Alder approach to the pentasubstituted pyran, installingfour of the centers in a single transformation in good yield andexcellent enantioselectivity (Scheme 23). Reduction of ketone 184 gavea solid that could be recrystallized to even higher enantiopurity, and
cyanation of the corresponding acetal afforded the axial nitrile, whichcould be methylated to ketone 187. Epimerization restored the desiredC26 equatorial orientation, and installation of the oxazole resulted inthe formation of the desired C20–C32 fragment.
OHHOa
(51% over 3 steps)OTBS
EtO2C b
(63% over 3 steps)OTBSHO
OPMB
OPMB
(148, 86% ee)
c
(84% over 2 steps)
OTBSOPMB
OPMBEtO2C
OTBSOPMB
PMBOd
(90% over 2 steps)AcO O
OPMB
PMBOe
(86% over 2 steps)AcO
OPMB
PMBOf
(74% over 2 steps)AcO
OMeCO2Et
(153, 4:1 dr)OTMS
OEt
g
(82% over 2 steps) HOOH
O
OMe
O
h
(59% over 2 steps) TBSOOTIPS
O
OMe
O
146 147
149 150 151
154
120
152
Scheme 18 (a) (1) NaH, TBSCl, THF, 0 °C; (2) PCC, CH2Cl2, RT; (3) Ph3P=CHCO2Et, benzene, reflux; (b) (1) AD-mix β, t-BuOH/H2O, RT; (2) PMBOC(=NH)CCl3, cyclohexane/CH2Cl2, BF3OEt2, 0 °C; (3) LiAIH4, Et2O, RT; (c) (1) (COCl)2, DMSO, CH2Cl2, −78 °C, then Et3N; (2) CH3CH(PPh3)CO2Et, CH2Cl2,reflux; (d) (1) DIBAL-H, CH2Cl2, −78 °C; (2) Ac2O, Et3N, CH2Cl2, RT; (e) (1) Bu4NF, THF, RT; (2) Dess–Martin, CH2Cl2; (f) (1) TiCl2(OiPr)2, toluene, −78 °C, then 152; (2) Me3OBF4, proton sponge, CH2Cl2, RT; (g) (1) K2CO3, EtOH; (2) 10% TFA, CH2Cl2, then Et3N; (h) (1) TBSCl, Et3N, CH2Cl2; (2) TIPSCl,AgNO3, pyr.
O
OPMB
OBPS
N
O
a
(50% over 2 steps)
O
OPMB
OBPS
N
O
O
MeO OTIPS
OH
MeOb
(73% over 2 steps)
O
OPMB
OBPS
N
O
O
MeO OTIPS
MeO
BrMeO
c
(71% over 2 steps)O
OPMBN
O
O
MeO OTIPS
MeO
BrMeO
O
121 155
156 118
Scheme 19 (a) (1) LiNEt2, THF, −78 °C, then 120; (2) PPTS, MeOH, 30 °C; (b) (1) Dess–Martin, CH2Cl2, pyr, RT; (2) 3, NaHMDS, THF, −78 °C; (c) (1)NH4F, MeOH, 50 °C; (2) Dess–Martin, CH2Cl2, pyr, RT.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
234
The Journal of Antibiotics

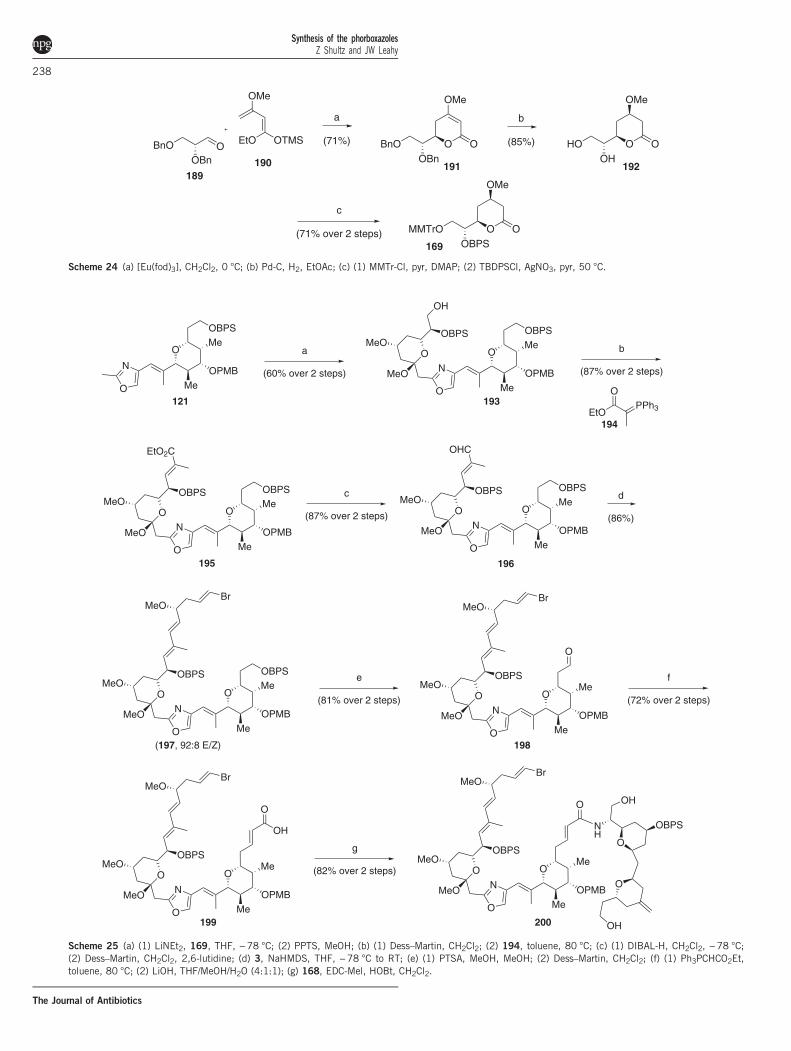
The exocyclic pyran was also prepared using a hetero-Diels–Aldercyclization which, under europium catalysis, returned 191 as a singlediastereomer (Scheme 24). Hydrogenation set the C35 stereocenterand liberated the diol, which could be manipulated to selectivelyprotected 169. Lithiation of 121 and addition of 169 formed the C32–C33 bond (Scheme 25), and the peripheral triene moiety was realized
via the established Wittig/Julia methodology. Selective deprotection ofthe primary alcohol allowed the Wittig extension to α,β-unsaturatedacid 199, which was coupled to amine 168 to provide the oxazoleprecursor. Wipf cyclodehydration achieved the oxazole formation, andthe macrolide was closed via the Horner–Wadsworth–Emmonsolefination (Scheme 26). Desilylation completed the synthesis of 2.
TMSOH
OMeO
O
a, b
O
O
O
O
c
O
OOH
OH TMS
TMS
d
N
SHS
TMS
SS
NOMe
SS
NOMe
BrO
O
f, g
157 158 159
160
161
162 3
e
Scheme 20 (a) AD-mix-α, t-BuOH/H2O, RT, 87% ee, 66%; (b) (1) HBr, AcOH; (2) K2CO3, MeOH, 88%; (c) (1) TMSC2H, BuLi, BF3OEt2, THF, −78 °C; (2)Me3OBF4, proton sponge, CH2Cl2, RT, 70% for two steps; (d) CAN, CH3CN/H2O, RT, 91%; (e) 161, PPh3, DEAD, THF, 0 °C, 81%; (f) TBAF, THF, RT,99%; (g) (1) Cp2ZrHCl, THF; then NBS; (2) (NH4)6Mo7O24·4H2O, 30% H2O2, EtOH, 56% for two steps.
O
OTBSO
N
OMs
O
TBDPSO
a
(50% over 2 steps) O
OPMB
N
O
O
MeOOTIPS
MeO
BrMeO
O
OTBSO
N
O
OH
O
OH
N
O
O
MeOOTIPS
MeO
BrMeO
O
OTBSO
N
O
O
b
(73% over 2 steps)
c
(57% over 2 steps) O
O
N
O
O
MeOOTIPS
MeO
BrMeO
O
OTBSO
N
O
O
d(51% over 2 steps)
(165, 5:1 Z/E)
119 163
164
2
Scheme 21 (a) (1) Bu3P, DMF, then 118, DBU, RT; (2) NH4F, MeOH, 50 °C; (b) (1) Dess–Martin, pyr, CH2Cl2, RT; (2) DDQ, CH2Cl2, pH 7 buffer; (c) (1)(EtO)2P(O)CH2CO2H, DCC, CH2Cl2; (2) K2CO3, 18-c-6, toluene, −20 °C; (d) TBAF, THF, RT; (2) 6% HCl, THF.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
235
The Journal of Antibiotics

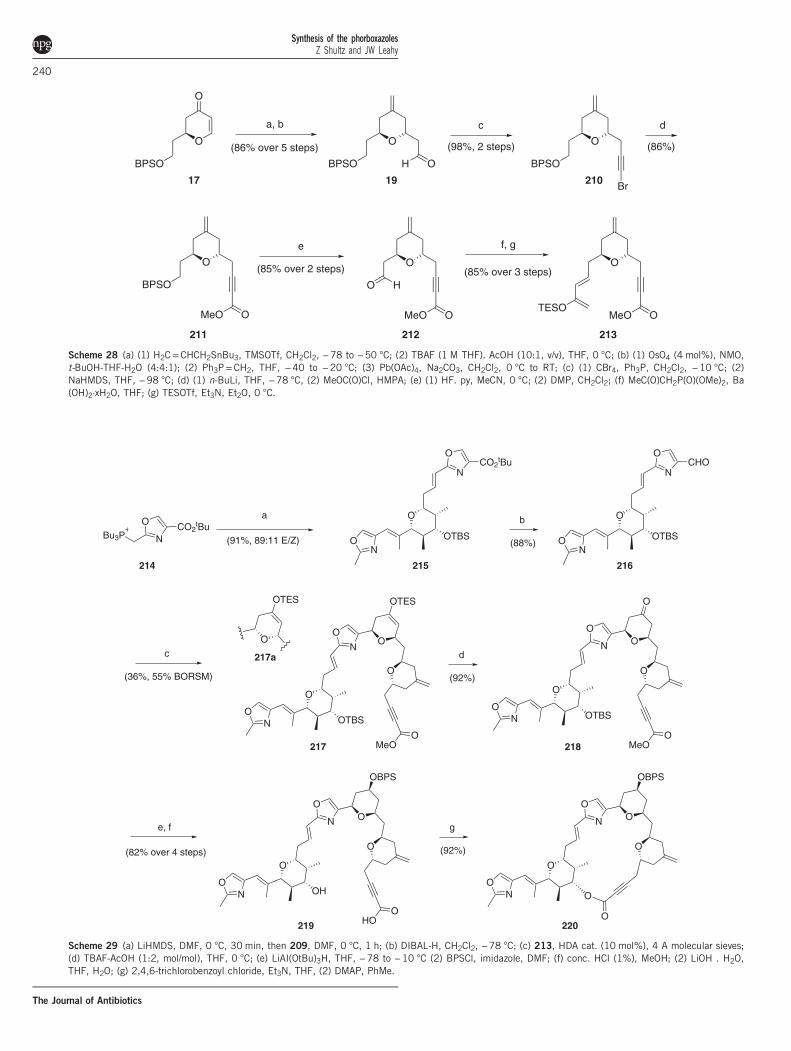
Additional synthetic approaches to the phorboxazolesPhorboxazole macrolide. Among the reported synthetic approachesto the phorboxazoles, only the Paterson group has published acompleted macrolide.10 The pentasubstituted pyran was assembledusing the anti-aldol addition of chiral ketone 204 to aldehyde 62,intramolecular Tishchenko reduction of the ketone and eventuallyMichael cyclization to the pyran (Scheme 27). The C5–C9 pyran wasprepared via the Michael allylation of enone 17 to set the requisitetrans-pyran with excellent diastereoselectivity (94% de) using allyltributylstannane (Scheme 28), and the subsequent aldehyde wasconverted into the corresponding acetylenic ester 211. Finally, theC9 side chain was deprotected and converted into siloxydiene 213 toset the stage for a second hetero-Diels–Alder cyclization. Following theWittig coupling of 214 to 209, this was achieved to provide a nearly1:1 mixture of diastereomeric pyrans 217 and 217a that provedresistant to improved diastereocontrol (Scheme 29). Reductionof ketone 218 followed by generation of the seco acid allowed forthe Yamaguchi macrolactonization developed by Evans and synthesisof 220.
The Hoffmann group recognized that the 8-oxabicyclo[3.2.1]octanering system mapped well onto the phorboxazole pyrans, and exploitedthat in their approach.24 Reduction of meso ketone 221 withL-Selectride and protection set the stage for the asymmetric hydro-boration of the olefin with diisopinocampheylborane to give 223 in96% ee (Scheme 30).25 Oxidation to the ketone and subsequentBaeyer–Villiger ring expansion provided 225, which underwentmethanolysis to provide pyran acetal 226. Allylation proceeded withexcellent trans selectivity (499% de), which set the stage for theelaboration to aldehyde 230. The C11 stereocenter was introduced viaasymmetric allylation, which allowed for the remaining elaboration ofC3–C13 fragment 233.Hoffmann’s approach to the pentasubstituted pyran started from
the racemic dimethylated bicycle 234 (Scheme 31).26 DIBAL reductionof the ketone and benzylation gave 235, which underwent asymmetrichydroboration to a mixture of 236 and 237. Both components ofthis mixture proved useful, as 236 was transformed into theC1–C7 portion of discodermolide while 237 was carried ontoward the phorboxazoles.27 Utilizing their PCC/Baeyer–Villiger
NH
O
OOPMB
O
ON
O
O
MeO
HO
BrMeO
OH OBPS
O O
O
OPMB
ON
O
O
MeO
HO
BrMeO
OH
TESO
O
O
OTES
OBPSH2N
O
O
MeO
OH
OMMTr OBPS
OPMB
ON
O
OAc
OH OH OH OH
OAc
166
167
168
169 121
170
3
2
Figure 11 Retrosynthetic analysis of Steve Burke’s total synthesis of phorboxazole B.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
236
The Journal of Antibiotics

HO OHa
(42% over 2 steps)
OH OH OH OH b
(72%)
AcO OAc
OH OH OH OH
c
(75%)
O O
OHOH(174, 98% ee)
d
(68% over 2 steps)O
O
OAc
OAc
HO
HO e
(83% over 2 steps)O
O
OAc
OAc
O
O
HO
f
(91%)
O
O
OAc
OAc
O
O
OOH
g
(75% over 3 steps)
O
O
OBPS
O
O
OO
OAc OAc
h
(90% over 2 steps)
O
O
OH
OBPS
O
O
TESO
i
(72% over 2 steps)
O
O
OBPS
O
O
TESO
j
(70% over 3 steps)
O
O
OBPS
TESO
HO
TESO
O
O
OBPS
TESO
N3
TESO
k
(88%)
O
O
OBPS
TESO
H2N
TESO
l
(76%)
170171 172
173
175 176
177178 179
180 181 182 168
Scheme 22 (a) (1) O3, DMS, −78 °C to RT; (2) In, allyl bromide, H2O, RT, 24 h; (b) 173, Grubbs' second-generation, CH2Cl2:DMF (20:1), RT, 30 min; (c)Pd2dba-CHCl3 (2 mol%), (R,R),DPPBA (6 mol%), Et3N, CH2Cl2, 0 °C; (d) (1) Ac2O, DMAP, pyr, RT, 1 h; (2) AD-mix β, 0 °C, 3 h, (96% BORSM); (e) (1)2,2-DMP, PPTS (cat.), 1,2-DCE, 40 °C, 30 min; (2) disiamylborane, RT, 1.5 h, NaBO3, H2O, RT, 1.5 h; (f) RuCl3 (3 mol%), NalO4, 0 °C, 5 h; (g) (1) NaOH,MeOH, then Dowex CCR-3; (2) DIAD, PPh3, THF, 1 h; (3) TBDPSCl, Et3N, RT (h) (1) LiBH4, THF, 0 °C; (2) TESCl, 2,6-lutidine, CH2Cl2; (i) (1) Dess–Martin,CH2Cl2, RT, 4 h; (2) Cp2TiMe2, THF/toluene, 80 °C; (j) 1) PTSA, MeOH, 40 °C; (2) TESCl, imidazole, −78 °C; (k) DEAD, PPh3, N3-PO(OMe)2; (l) PPh3,H2O, THF, reflux.
O
OPMBMe Me
MeO
OBPS
O
OPMB
Me Me
NC
OBPS
O
OPMBMe Me
OBPS
O
Me
O
OPMBMe Me
OBPS
O
Me
MeO
MeOTES
Me
O
OBPS
O
O
Me Me
MeO
OBPSb
(81% over 2 steps)
c
(82%)
a
(77%)
d
(65%, 99% BORSM)
e
(77%)
f
(68%, 78% BORSM)
O
OPMB
Me
Me
OBPS
N
O
183
14
184 185
186 187
188 121
Scheme 23 (a) 16 (2 mol%), RT, 14 h, then HF/pyr/MeOH, −20 °C, 15 min; (b) (1) NaBH4, EtOH, −10 °C, 15 min (98.5% ee after recrystallization); (2)PMB-Cl, KH, RT, 2 h; (c) 0.5 eq. THM-OTf, TMS-CN, MeCN, 0 °C 2 h; (d) AIMe3 5% Ni(acac)2, 0 °C, 42 h; (e) KHMDS, LiCl, PhCH3, 55 °C 2 h, 91%; (f)145, LDA, THF, −78 °C to RT.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
237
The Journal of Antibiotics
OTMSEtO
OMe
BnOOBn
O O
OMe
OBnOOBn
c
(71% over 2 steps)
a
(71%)
b
(85%) O
OMe
OHOOH
O
OMe
OMMTrOOBPS169
189190 191 192
Scheme 24 (a) [Eu(fod)3], CH2Cl2, 0 °C; (b) Pd-C, H2, EtOAc; (c) (1) MMTr-Cl, pyr, DMAP; (2) TBDPSCl, AgNO3, pyr, 50 °C.
d
(86%)
O
OPMB
Me
Me
OBPS
N
O
O
OPMB
Me
Me
OBPS
N
O
O
MeO
MeOOBPS
OH
a
(60% over 2 steps)
O
OPMB
Me
MeOBPS
N
O
O
MeO
MeOOBPS
b
(87% over 2 steps)
EtO2C
c
(87% over 2 steps)O
OPMB
Me
Me
OBPS
N
O
O
MeO
MeOOBPS
OHC
O
OPMB
Me
Me
OBPS
N
O
O
MeO
MeOOBPS
BrMeO
(197, 92:8 E/Z)
e
(81% over 2 steps)O
OPMB
Me
Me
N
O
O
MeO
MeOOBPS
BrMeO
O
f
(72% over 2 steps)
O
OPMB
Me
Me
N
O
O
MeO
MeOOBPS
BrMeO
O
OH
g
(82% over 2 steps) O
OPMB
Me
Me
N
O
O
MeO
MeOOBPS
BrMeO
O
O
O
OBPS
OH
NH
OH
EtO
OPPh3
121 193
194
195 196
198
199 200
Scheme 25 (a) (1) LiNEt2, 169, THF, −78 °C; (2) PPTS, MeOH; (b) (1) Dess–Martin, CH2Cl2; (2) 194, toluene, 80 °C; (c) (1) DIBAL-H, CH2Cl2, −78 °C;(2) Dess–Martin, CH2Cl2, 2,6-lutidine; (d) 3, NaHMDS, THF, −78 °C to RT; (e) (1) PTSA, MeOH, MeOH; (2) Dess–Martin, CH2Cl2; (f) (1) Ph3PCHCO2Et,toluene, 80 °C; (2) LiOH, THF/MeOH/H2O (4:1:1); (g) 168, EDC-Mel, HOBt, CH2Cl2.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
238
The Journal of Antibiotics

protocol, Hoffmann synthesized 238, which was elaborated toacrylonitrile 241 and subjected to rhodium catalyzed oxazole cycliza-tion. Finally, LAH reduction simultaneously reduced the oxazole β-methoxy substituent and the ester to provide the C15–C26 fragment.Yadav initiated his synthetic approach to the macrolide portion
from vinyl furan 244, readily available from D-glucose (Scheme 32),28
which provided the C13 and C15 stereocenters of phorboxazole A.29 Itis notable that D-galactose would correspond to the phorboxazole B
stereochemistry. Upon opening the furan, the olefin was extended viaa hydroboration/oxidation/Wittig sequence, and Michael addition ofthe secondary alcohol of 249 formed cis-pyran 250. Asymmetricallylation followed by acylation with acryloyl chloride gave 252, whichwas closed with first-generation Grubbs catalyst to lactone 253.Reduction to the ethyl acetal and Lewis acid mediated allylationyielded expected trans-pyran 255, which represents the C2–C16portion of phorboxazole A.
ON
H
O O
OPMB a
f
ON
OH O
OPMB ON
O OTBS
OHb, c
hO
N
O OTBS
O
iPr
O
iPr
d, e O
OMe
ON
OH OTBS
OHO
N
O
OTBS
H
O
(94%, > % ds) (65% over 3 steps)
(91%)(86%) (81%, 80% ds)
62 204 205 206
207 208 209
Scheme 27 (a) (c-Hex)2BCl, Et3N, Et2O, 0 °C, 1 h; 62, −78 to −20 °C, 18 h; H2O2, MeOH, pH 7 buffer; (b) Sml2 (30 mol%), iPrCHO, THF, −20 °C; (c)(1) TBSCl, Imidazole, DMAP, DMF, 80 °C, 50 h; (2) DDQ, 20:1 CH2Cl2/H2O, 20 °C; (d) (COCl)2, DMSO, −78 °C, 10 min, −78°C, 1 h; Et3N, −78 to −40 °C, 40 min; (e) LiCl, DBU, (MeO)2P(O)CH2CO2Me, 4:1 CH3CN/CH2Cl2, 20 °C, 2.5 h; (f) DIBAL, CH2Cl2, −78 °C, 5 h; (h) SO3-pyr, Et3N, 2:1 CH2Cl2/DMSO,0 °C, 2 h.
O
OPMB
Me
Me
N
O
O
MeO
MeOOBPS
BrMeO
O
O
O
OBPS
OH
NH
OH
a
(71% over 2 steps)
O
OPMB
Me
Me
N
O
O
MeO
MeOOBPS
BrMeO
O
O
O
OBPS
N
O
b
(71% over 2 steps)O
O
Me
Me
N
O
O
MeO
MeOOBPS
BrMeO
O
O
O
OBPS
N
OO
P(OMe)2O
c
(93%)
(203, 3:1 Z/E)
O
O
N
O
O
MeOOBPS
MeO
BrMeO
O
OBPSO
N
O
O
200 201
202
d(61% over 2 steps)
2
Scheme 26 (a) (1) Dess–Martin, CH2Cl2; (2) 2,6-DBMP, PPh3, (CCl2Br)2, 0 °C, then DBU, MeCN, RT; (b) (1) DDQ, CH2Cl2, pH 7 buffer; (2) DIC, (MeO)2P(O)CH2CO2H, CH2Cl2; (c) K2CO3, 18-c-6, toluene, −40 °C to RT; (d) (1) TBAF, THF; (2) 0.72 N HCl, THF RT, 60 h.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
239
The Journal of Antibiotics
O
O
BPSO
O
BPSO
a, b
H O
O
BPSO
c
Br
d
O
BPSO
MeO O
O
O
MeO O
H
e
O
MeO O
f, g
TESO
(86% over 5 steps) (98%, 2 steps) (86%)
(85% over 2 steps) (85% over 3 steps)
17 19 210
211 212 213
Scheme 28 (a) (1) H2C=CHCH2SnBu3, TMSOTf, CH2Cl2, −78 to −50 °C; (2) TBAF (1 M THF). AcOH (10:1, v/v), THF, 0 °C; (b) (1) OsO4 (4 mol%), NMO,t-BuOH-THF-H2O (4:4:1); (2) Ph3P=CH2, THF, −40 to −20 °C; (3) Pb(OAc)4, Na2CO3, CH2Cl2, 0 °C to RT; (c) (1) CBr4, Ph3P, CH2Cl2, −10 °C; (2)NaHMDS, THF, −98 °C; (d) (1) n-BuLi, THF, −78 °C, (2) MeOC(O)Cl, HMPA; (e) (1) HF. py, MeCN, 0 °C; (2) DMP, CH2Cl2; (f) MeC(O)CH2P(O)(OMe)2, Ba(OH)2·xH2O, THF; (g) TESOTf, Et3N, Et2O, 0 °C.
ON
O
OTBS
N
OCHO
b
cO
O
OMeO
OTES
O
NO
OTES
O
O
OMeO
O
O
N
ON
O
OTBS
e, fO
O
OHO
OBPS
O
N
ON
O
OH
O
O
OBPS
O
N
ON
O
O
O
(88%)
(36%, 55% BORSM)
(82% over 4 steps)
N
OBu3P
CO2tBu
a
ON
O
OTBS
N
OCO2
tBu
(91%, 89:11 E/Z)
214 215
ON
O
OTBS
d
(92%)
g
(92%)
216
217 218
219 220
217a
Scheme 29 (a) LiHMDS, DMF, 0 °C, 30 min, then 209, DMF, 0 °C, 1 h; (b) DIBAL-H, CH2Cl2, −78 °C; (c) 213, HDA cat. (10 mol%), 4 A molecular sieves;(d) TBAF-AcOH (1:2, mol/mol), THF, 0 °C; (e) LiAI(OtBu)3H, THF, −78 to −10 °C (2) BPSCl, imidazole, DMF; (f) conc. HCl (1%), MeOH; (2) LiOH . H2O,THF, H2O; (g) 2,4,6-trichlorobenzoyl chloride, Et3N, THF, (2) DMAP, PhMe.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
240
The Journal of Antibiotics

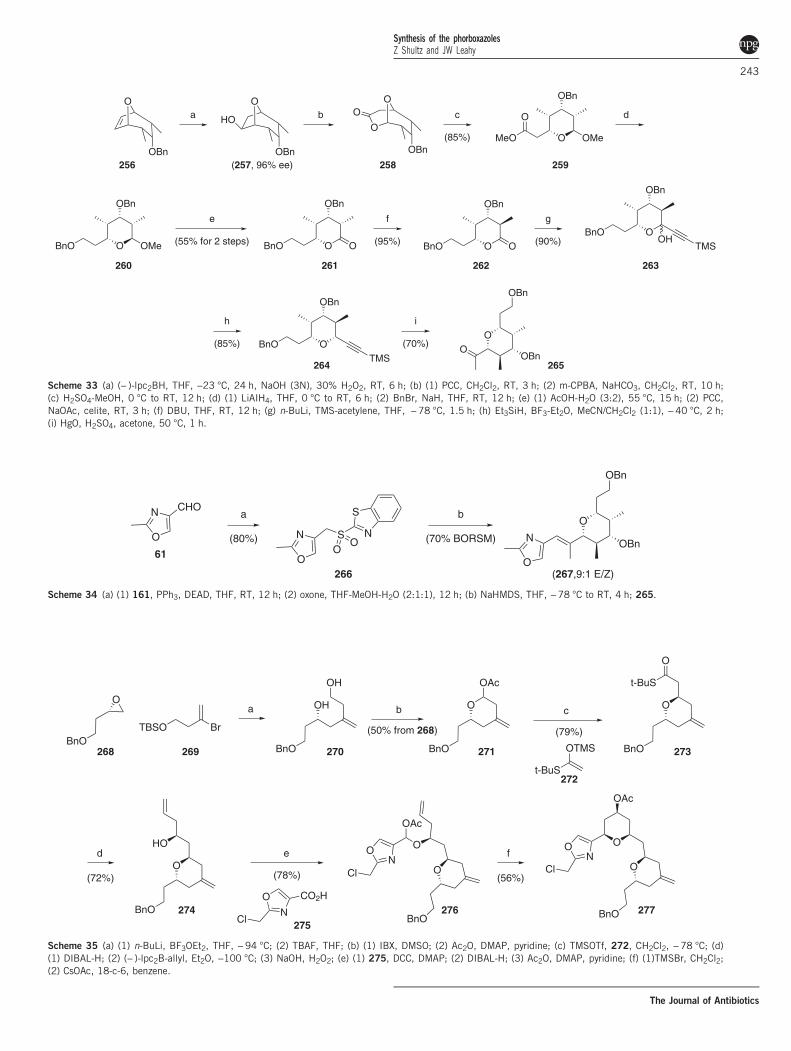
Yadav also prepared the pentasubstituted pyran from the8-oxabicyclo[3.2.1]octane ring system, starting from the asymmetrichydroboration of achiral dimethylated derivative 256 (Scheme 33).30
Ring opening of Baeyer–Villiger lactone 258 revealed pyran 259 which,
upon protection of the C20 Wittig precursor as a benzyl ether, wasoxidized to lactone 262. This allowed for the epimerization of the C25methyl using DBU with excellent selectivity and conversion. The C27side chain was then introduced via acetylenation, and hemiketal
O
O
O
HO OPMB
O
O OPMB
O
OPMBOO O
OPMB
MeOCO2Me
O
OH
CO2Me
O
OPMB
b ca d
e f
(85%)
(223, 96% ee)
(92%) (96%)
(91%) (85%)
(227, 96% de)
g
(97%) O
OTIPS
CO2Me
h
(88%) O
OTIPS
CH2OTBS
i
(100%) O
OTIPS
CH2OTBSCHO
221 222 224
225 226 228
229 230
j
(72%) O
OTIPS
CH2OTBS
k
(93%)
OH
(231, 92% de)
O
OTIPS
CH2OTBS
OTBS l
(98%)O
OTIPS
CH2OTBS
OTBSO
232 233
Scheme 30 (a) (1)24; (2) NaH, PMBCl, (n-Bu)4Nl, THF, reflux, 6 h; (b) (+)-lpc2BH, THF, −10 °C, 1 week; (c) PCC, DCM, RT, 5 h; (d) m-CPBA DCM, RT,overnight; (e) MeOH, conc. H2SO4 (cat.), RT, overnight; (f) allyltrimethylsilane, TMSOTf, MeCN, −20 °C to RT, 1 h; (g) TIPSCl, imidazole, DMF, RT, 16 h;(h) (1) DIBAL-H, −20 °C, THF, 2 h; (2) TBSCl, imidazole, DMF, RT, 16 h; (i) O3, DCM, −78 °C then PPh3 (j) (− )B-allyl-β-diisopinocampheyl-borane,toluene, −78 °C, 5 h then H2O2, NaOH; (k) TBSOTf, 2,6-lutidine, DCM, 0 °C, 30 min; (l) O3, DCM, −78 °C then PPh3.
O
O
Oa
(85%) OBn
b
(93%)
O
OBnHO
c
(82%)
O
O OBn
O
(237, > 95% ee)
d
(84%)
OMeO
OBn
CO2Me e
(71%)
OMeO
OBn
CHO f
(85%)
OMeO
OBn
CN
(241, 9:1 E/Z)
g
(61%)
OMeO
OBn
N
O
CO2Me
OMe h
(49%)
OMeO
OBn
N
O
CH2OH
(±) 234 (±) 235
239 240
242 243
O
OBnHO
236 238
Scheme 31 (a) (1) DIBAL-H, THF, −78 °C, 11 h; (2) NaH, THF, BnBr, reflux, 16 h; (b) (− )-(lpc)2BH, THF, −10 °C, 14 d, then NaOH, H2O2, 3 h; (c) (1)PCC/SiO2, DCM, RT, 15 h; (2) m-CPBA, NaHCO3, DCM, RT, 15 h; (d) H2SO4 (cat.), MeOH, 16 h; (e) (1) DIBAL-H, THF, 0 °C, 4 h; (2) PCC, DCM, RT, 15 h;(f) Ph3PCHCN, toluene, LiCl, RT, 20 h; (g) N2C(CO2Me)2, Rh2(OAc)4, CHCl3, 15 h, reflux; (h) LiAIH4, THF, −78 °C, 3 h.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
241
The Journal of Antibiotics

reduction gave the elaborated pyran as a single isomer. Oxymercura-tion then gave ketone 265. The oxazole was introduced via a modifiedJulia olefination of 265 with sulfone 266 (Scheme 34), giving C20–C32fragment 267 as a 9:1 mixture of olefin isomers.The synthesis of the bis pyran was also achieved by the Rychnovsky
group using their expertise with the Prins reaction.31 Epoxide 268 wasopened with the lithium anion of 269 to give diol 270, which wassmoothly closed to the pyran and extended to trans thioester 273(Scheme 35). The C11 stereocenter was formed via reduction to thealdehyde and asymmetric allylation. Acylation of the C11 alcohol withoxazole 275 and conversion to the α-acetoxyether generated the Prinsprecursor, which proceeded as expected to the cis-pyran, representingthe C3–C19 portion of phorboxazole B. Rychnovsky showed that thePrins cyclization could also be achieved to install the C11 stereocenter,but the yields were diminished presumably due to the proximity of theoxazole nitrogen.The Prins reaction was also used by Rychnovsky to make the
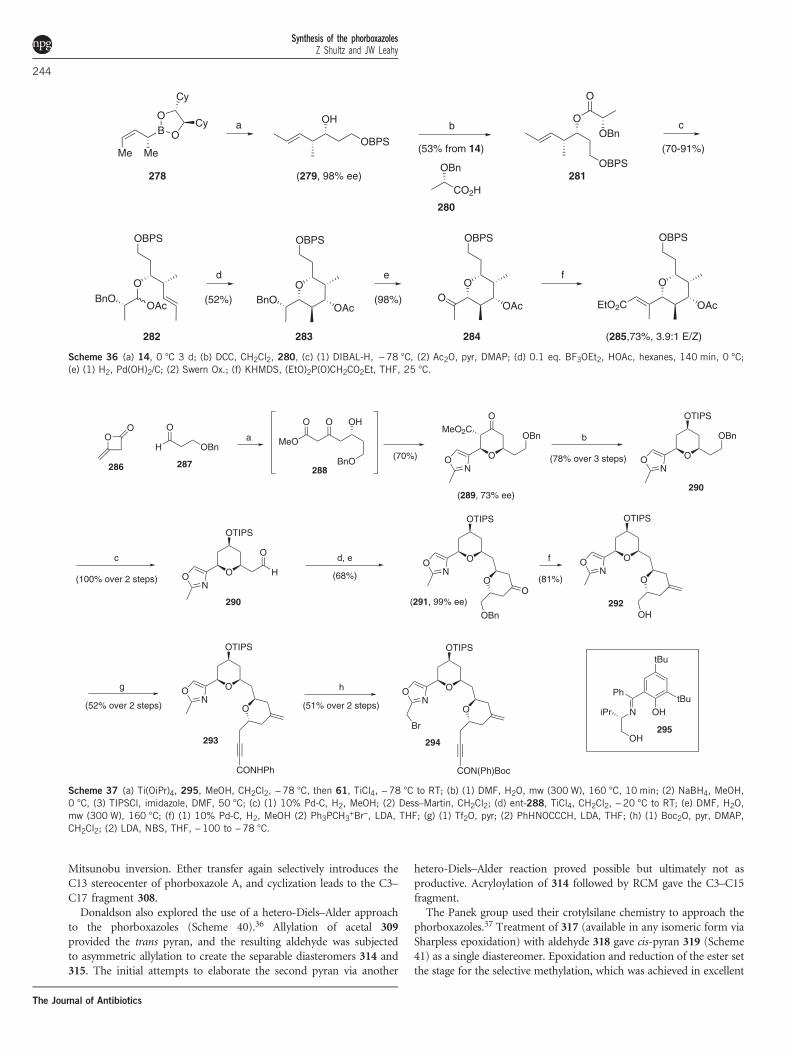
pentasubstituted pyran as well.32 Asymmetric crotylation of aldehyde14 with 278 gave 279 in 98% ee (Scheme 36), setting the C22stereocenter. Acylation with protected lactic acid yielded 281, whichwas transformed into Prins precursor 282 in standard fashion. Thiscyclization proceeded as expected with catalytic BF3 etherate to set theremaining stereocenters of the pyran. Elaboration to ketone 284 wasfollowed by Horner Wadsworth Emmons olefination to afford theC20–C29 fragment as a 3.9:1 mixture of olefin isomers.The Clarke group also used the macrolide pyrans as a showcase for
their work on the asymmetric Maitland–Japp reaction. Toward thisend, diketene was able to be coupled to aldehyde 287 (setting the C11stereocenter) and oxazole aldehyde 61 in a one-pot transformation
using Ti(IV) and chiral ligand 295 to prepare pyranone 289 as amixture of C15 epimers in 73% ee (Scheme 37).33 The trans isomercould be converted to the desired cis isomer with Yb(OTf)3.Decarboxylation, reduction and protection thus gave pyran 290, whichwas converted to the aldehyde and subjected to a second Maitland–Japp reaction using Chan’s diene and Evans’ pybox ligand, whichestablished the trans pyran ring with excellent stereocontrol andcomplete enantiomeric enrichment. Once the exocyclic olefin at C7was introduced, the primary alcohol was liberated and activated fordisplacement with an acetylenic anion. Radical bromination of theoxazole methyl finally gave C1–C19 fragment 294 functionalized forfurther elaboration.Clarke’s initial efforts to utilize the Maitland–Japp reaction to
generate the pentasubstituted pyran resulted in a C23-epimericproduct, so he ultimately initiated his synthesis of the remainder ofthe macrolide fragment using the anti-aldol addition of 296 to 62(Scheme 38).34 Reductive removal of the chiral auxiliary followedsilylation of the C26 alcohol, which allowed for outstanding Felkin–Ahn control of the subsequent crotyl addition. Acrylate 300 was addedvia a metathesis reaction, which set the stage for the Michaelcyclization (13:1 selectivity) to the C20–C32 fragment.Taylor was able to demonstrate that his electrophile-induced ether
transfer methodology allowed access to the bis-pyran portion of 1(Scheme 39).35 Ether transfer of the BOM-protected analog of 28 withICl conferred the C5 stereocenter with exquisite control (20:1), andthe subsequent sulfone (303) could be cyclized and allylated to thetrans pyran nearly quantitatively. Following elaboration to aldehyde305, allylation gave a 1:1 mixture of C11 alcohols that could beseparated and completely converted into the desired 306 via
OOH
OH
OH
OH
HO
D-Glucose
aO
O
O b
(67% for 2 steps)
OH
OH OH
c
(73% for 2 steps)O
OBn O
OOBn O
HOd
(80%)
e
(72% for 2 steps)
OOBn O
EtO
O
f
(91%)HO
OBnHO
OEt
O
O OEt
O
MOMO
OBn
g
(74% for 2 steps) OMOMO
OBn
OH
h
(67% for 2 steps)
i
(93%) OMOMO
OBn
O O
j
(94%) OMOMO
OBn
O O
k
(86%) OMOMO
OBn
O OEt
l
(85%)
OMOMO
OBn
O
11
244 245 246
249248247
251252
255254253
(250, 20:1cis/trans)
Scheme 32 (a)27; (b) (1) 50% AcOH, H2SO4 (cat.), 2 h; (2) NaBH4, MeOH, 0 °C to RT, 1 h; (c) (1) PTSA (cat.), acetone, RT, 3 h; (2) NaH, BnBr, THF,reflux, 8 h; (d) BH3-DMS, NaOH, H2O2, THF, 8 h; (e) (1) IBX, THF, DMSO, RT, 2 h; (2) Ph3P=CHCO2Et, benzene, RT, 12 h; (f) CSA, MeOH, RT, 2 h; (g)(1) NaH, THF, −78 °C, 4 h; (2) iPr2NEt, MOMCl, CH2Cl2, 0 °C, 6 h. (h) (1) DIBAL-H, CH2Cl2, −78 °C, 1 h; (2) (+)-allyl diisopinocampheylborane, Et3N,H2O2, 1 h; (i) iPr2NEt, acryoyl chloride, CH2Cl2, 0 °C to RT, 4 h; (j) Grubb's 1st gen., Ti(iOPr)4, CH2Cl2, 60 °C, 6 h; (k) DIBAL-H, CH2Cl2, −78 °C, 30 minthen CSA, EtOH; (l) allyltrimethyl silane. TMSOTf, CH2Cl2, 0 °C, 1 h.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
242
The Journal of Antibiotics
OBn
Oa
OBn
O
HOO
OBn
O
b O c
(85%) O
OBn
OMeMeO
O
O
OBn
OMeBnO
d
e
(55% for 2 steps) O
OBn
OBnO
f
(95%) O
OBn
OBnO
g
(90%)O
OBn
BnOOH
TMS
h
(85%) O
OBn
BnOTMS
i
(70%)O
OBn
OBn
O
(257, 96% ee)256 258 259
260 261 262 263
264 265
Scheme 33 (a) (− )-lpc2BH, THF, −23 °C, 24 h, NaOH (3N), 30% H2O2, RT, 6 h; (b) (1) PCC, CH2Cl2, RT, 3 h; (2) m-CPBA, NaHCO3, CH2Cl2, RT, 10 h;(c) H2SO4-MeOH, 0 °C to RT, 12 h; (d) (1) LiAIH4, THF, 0 °C to RT, 6 h; (2) BnBr, NaH, THF, RT, 12 h; (e) (1) AcOH-H2O (3:2), 55 °C, 15 h; (2) PCC,NaOAc, celite, RT, 3 h; (f) DBU, THF, RT, 12 h; (g) n-BuLi, TMS-acetylene, THF, −78 °C, 1.5 h; (h) Et3SiH, BF3-Et2O, MeCN/CH2Cl2 (1:1), −40 °C, 2 h;(i) HgO, H2SO4, acetone, 50 °C, 1 h.
N
O
CHOa
(80%) N
O
S N
S
OO
b
(70% BORSM)
O
OBn
OBn
N
O(267,9:1 E/Z)
61
266
Scheme 34 (a) (1) 161, PPh3, DEAD, THF, RT, 12 h; (2) oxone, THF-MeOH-H2O (2:1:1), 12 h; (b) NaHMDS, THF, −78 °C to RT, 4 h; 265.
BnO
TBSO Br
OH
OH
BnO
Oa b
BnO
O
OAc
c
t-BuS
OTMS
(50% from 268) (79%)
d
BnO
O
HOe
N
O CO2H
Cl BnO
O
O
OAc
ON
Cl
f
BnO
O
t-BuS
O
BnO
O
OAc
OON
Cl(72%) (78%) (56%)
268 269 270 271
272
273
274 276 277
275
Scheme 35 (a) (1) n-BuLi, BF3OEt2, THF, −94 °C; (2) TBAF, THF; (b) (1) IBX, DMSO; (2) Ac2O, DMAP, pyridine; (c) TMSOTf, 272, CH2Cl2, −78 °C; (d)(1) DIBAL-H; (2) (− )-lpc2B-allyl, Et2O, −100 °C; (3) NaOH, H2O2; (e) (1) 275, DCC, DMAP; (2) DIBAL-H; (3) Ac2O, DMAP, pyridine; (f) (1)TMSBr, CH2Cl2;(2) CsOAc, 18-c-6, benzene.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
243
The Journal of Antibiotics
Mitsunobu inversion. Ether transfer again selectively introduces theC13 stereocenter of phorboxazole A, and cyclization leads to the C3–C17 fragment 308.Donaldson also explored the use of a hetero-Diels–Alder approach
to the phorboxazoles (Scheme 40).36 Allylation of acetal 309provided the trans pyran, and the resulting aldehyde was subjectedto asymmetric allylation to create the separable diasteromers 314 and315. The initial attempts to elaborate the second pyran via another
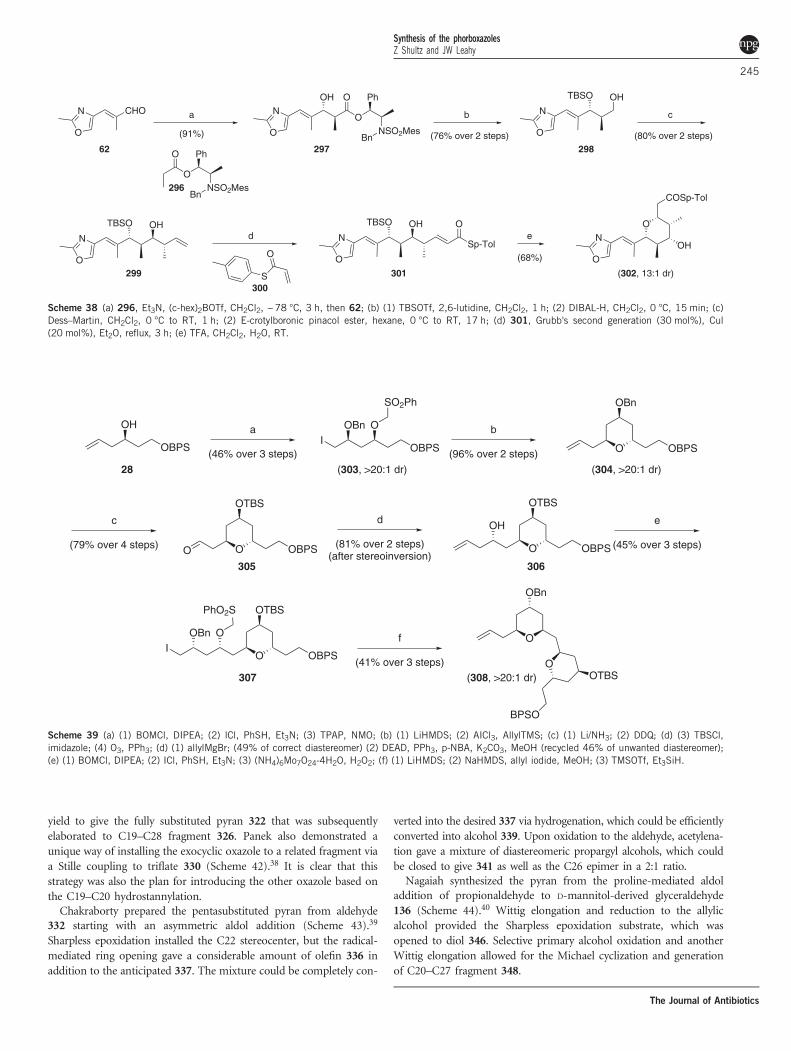
hetero-Diels–Alder reaction proved possible but ultimately not asproductive. Acryloylation of 314 followed by RCM gave the C3–C15fragment.The Panek group used their crotylsilane chemistry to approach the
phorboxazoles.37 Treatment of 317 (available in any isomeric form viaSharpless epoxidation) with aldehyde 318 gave cis-pyran 319 (Scheme41) as a single diastereomer. Epoxidation and reduction of the ester setthe stage for the selective methylation, which was achieved in excellent
OB O
MeMe
Cy
Cy c
OBPS
OHb
(279, 98% ee)CO2H
OBn
O
O
OBPS
OBn
O
OAcBnO
a
O
OAc
OBPS
BnO
d
OBPS
eO
OAc
OBPS
O
fO
OAc
OBPS
EtO2C
(70-91%)
(52%) (98%)
(285,73%, 3.9:1 E/Z)
(53% from 14)
278
280
281
282 283 284
Scheme 36 (a) 14, 0 °C 3 d; (b) DCC, CH2Cl2, 280, (c) (1) DIBAL-H, −78 °C, (2) Ac2O, pyr, DMAP; (d) 0.1 eq. BF3OEt2, HOAc, hexanes, 140 min, 0 °C;(e) (1) H2, Pd(OH)2/C; (2) Swern Ox.; (f) KHMDS, (EtO)2P(O)CH2CO2Et, THF, 25 °C.
OO
H
O
OBn
tBu
tBuOHN
Ph
OH
iPr
MeO
O O OH
BnO
a
OON
OBn
OMeO2C
(289, 73% ee)
b
288
OON
OBn
OTIPS
(78% over 3 steps)
OON
O
OTIPS
H
c
(100% over 2 steps)
d, e OON
OTIPS
O
OBn
O
f OON
OTIPS
O
OH
g OON
OTIPS
O
CONHPh
h OON
OTIPS
O
CON(Ph)Boc
(52% over 2 steps) (51% over 2 steps)
Br
(291, 99% ee)
(68%) (81%)
286 287
290
290 292
293 294295
(70%)
Scheme 37 (a) Ti(OiPr)4, 295, MeOH, CH2Cl2, −78 °C, then 61, TiCl4, −78 °C to RT; (b) (1) DMF, H2O, mw (300 W), 160 °C, 10 min; (2) NaBH4, MeOH,0 °C, (3) TIPSCl, imidazole, DMF, 50 °C; (c) (1) 10% Pd-C, H2, MeOH; (2) Dess–Martin, CH2Cl2; (d) ent-288, TiCl4, CH2Cl2, −20 °C to RT; (e) DMF, H2O,mw (300 W), 160 °C; (f) (1) 10% Pd-C, H2, MeOH (2) Ph3PCH3
+Br−, LDA, THF; (g) (1) Tf2O, pyr; (2) PhHNOCCCH, LDA, THF; (h) (1) Boc2O, pyr, DMAP,CH2Cl2; (2) LDA, NBS, THF, −100 to −78 °C.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
244
The Journal of Antibiotics
yield to give the fully substituted pyran 322 that was subsequentlyelaborated to C19–C28 fragment 326. Panek also demonstrated aunique way of installing the exocyclic oxazole to a related fragment viaa Stille coupling to triflate 330 (Scheme 42).38 It is clear that thisstrategy was also the plan for introducing the other oxazole based onthe C19–C20 hydrostannylation.Chakraborty prepared the pentasubstituted pyran from aldehyde
332 starting with an asymmetric aldol addition (Scheme 43).39
Sharpless epoxidation installed the C22 stereocenter, but the radical-mediated ring opening gave a considerable amount of olefin 336 inaddition to the anticipated 337. The mixture could be completely con-
verted into the desired 337 via hydrogenation, which could be efficientlyconverted into alcohol 339. Upon oxidation to the aldehyde, acetylena-tion gave a mixture of diastereomeric propargyl alcohols, which couldbe closed to give 341 as well as the C26 epimer in a 2:1 ratio.Nagaiah synthesized the pyran from the proline-mediated aldol
addition of propionaldehyde to D-mannitol-derived glyceraldehyde136 (Scheme 44).40 Wittig elongation and reduction to the allylicalcohol provided the Sharpless epoxidation substrate, which wasopened to diol 346. Selective primary alcohol oxidation and anotherWittig elongation allowed for the Michael cyclization and generationof C20–C27 fragment 348.
Ph
O
O
O
N CHO a
O
N
OH
O
O Ph
NSO2MesBn
NSO2MesBn
(91%)
b
O
N
TBSO OH
(76% over 2 steps)
c
(80% over 2 steps)
O
N
TBSO OHd
S
O (68%)O
N
TBSO OH O
Sp-Tole
O
N
O
OH
COSp-Tol
(302, 13:1 dr)
62
296
297 298
299
300
301
Scheme 38 (a) 296, Et3N, (c-hex)2BOTf, CH2Cl2, −78 °C, 3 h, then 62; (b) (1) TBSOTf, 2,6-lutidine, CH2Cl2, 1 h; (2) DIBAL-H, CH2Cl2, 0 °C, 15 min; (c)Dess–Martin, CH2Cl2, 0 °C to RT, 1 h; (2) E-crotylboronic pinacol ester, hexane, 0 °C to RT, 17 h; (d) 301, Grubb's second generation (30 mol%), Cul(20 mol%), Et2O, reflux, 3 h; (e) TFA, CH2Cl2, H2O, RT.
OBPS
OH a
(46% over 3 steps) OBPS
OI
OBn
SO2Ph
b
(96% over 2 steps)
(303, >20:1 dr)
O
OBn
OBPS
c
(79% over 4 steps) O
OTBS
O OBPS
d
(81% over 2 steps)(after stereoinversion)
O
OTBS
OBPS
OH e
(45% over 3 steps)
O
OTBS
OBPS
OI
OBn
PhO2S
f
(41% over 3 steps)
O
OOTBS
BPSO
OBn
28 (304, >20:1 dr)
305 306
307 (308, >20:1 dr)
Scheme 39 (a) (1) BOMCl, DIPEA; (2) lCl, PhSH, Et3N; (3) TPAP, NMO; (b) (1) LiHMDS; (2) AICl3, AllylTMS; (c) (1) Li/NH3; (2) DDQ; (d) (3) TBSCl,imidazole; (4) O3, PPh3; (d) (1) allylMgBr; (49% of correct diastereomer) (2) DEAD, PPh3, p-NBA, K2CO3, MeOH (recycled 46% of unwanted diastereomer);(e) (1) BOMCl, DIPEA; (2) ICl, PhSH, Et3N; (3) (NH4)6Mo7O24-4H2O, H2O2; (f) (1) LiHMDS; (2) NaHMDS, allyl iodide, MeOH; (3) TMSOTf, Et3SiH.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
245
The Journal of Antibiotics

Exocyclic fragments. In addition to completing the macrolide, Pater-son also completed the entire exocyclic portion of thephorboxazoles.10 Mukaiyama aldol addition of 101 to 349 usingEvans’ pybox catalyst installed the C37 stereocenter in 495% ee, andacid-catalyzed lactonization gave β-ketoester 351 (Scheme 45). Hydro-genation of the corresponding vinylogous carbonate simultaneously
installed the C35 center and liberated the primary alcohol, which wasoxidized to aldehyde 354. Meanwhile, the triene was formed fromasymmetric acetate aldol addition to aldehyde 357 (Scheme 46) in astraightforward manner. The Nozaki–Kishi addition of 362 to 354proceeded to give the fully elaborated C33–C46 fragment, though thereaction proceeded in disappointing yield and selectivity. The mixture
O
O
BPSO
a
(69%)
O
O
BPSO
OMe
b
(92%)
O
OH
BPSO
OMe
H H H
d
(73%)
O
OAc
BPSO
OMe
Hc
(92%)
O
OAc
BPSO
H
H
e
(89%)
O
OAc
BPSO
H
OHCH
O
OAc
BPSO
H
Hf
HO
O
OAc
BPSO
H
HHO
(42%) (41%)
(±) 17 (±) 309 (±) 311
(±) 312 (±) 313 315314
(±) 310
g
h
O
OAc
BPSO
H
HOO
O
OAc
BPSO
H
HOO
(52%)
(73%)
316317
Scheme 40 (a) Hg(OAc)2, MeOH, NaBH3CN, THF; (b) LiAI(OtBu)3, THF; (c) Ac2O, NaOAc, (d) TMS-allyl, TMSOTf, CH3CN; (e) O3, CH2Cl2, −78 °C, PPh3;(f) (− )-(lpc)2B-allyl, toluene, −78 °C, salt-free then NaBO3, toluene, H2O; (g) H2C=CHCOCl, Et3N, DMAP; (h) Grubb's cat. (0.2 eq.), Ti(iPrO)4.
CO2Me
OTES
SiMe2Ph O
TIPS
MeO2C
a
(60-70%)
O
TIPS
MeO2CO
b
(85%, dr = 13:1)
c
(92%)
O
TIPS
OH
OH
O
TIPS
OOH
OO
OHTIPS
Me-
O
TIPS
OH
OTf
eO
TIPS
OH
f
(80% for 3 steps)
TMS
d
(>90%)
O
TIPS
OH
TMS
g
(86%, dr = 8:1)
O
TIPSOTMS
h
(70% for 3 steps)
SnBu3
H
O
TIPS
317
318
319 320 321
322323324325326
Scheme 41 (a) TMSOTf (0.5 eq.), CH2Cl2, 20 °C; (b) m-CPBA, CCl4, RT; (c) LiAIH4, THF, 0 °C; (d) CH3MgBr/Cul, THF, 0 °C; (e) Tf2O, pyr, CH2Cl2, −15 °C; (f) (1) TMSOTf, 2,6-lutidine; (2) LDA, THF, TMS-acetylene, HMPA, −10 °C; (g) (1) Dess–Martin, CH2Cl2, RT; (2) LiAIH4, THF, 0 °C; (h) (1) TMSOTf, 2,6-lutidine; (2) NBS, AgNO3; (3)Bu3SnH, PdCl2(PPh3)2.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
246
The Journal of Antibiotics

O
NO
Phb
(90%)
PhOTf
N
O
OOTBS
TBSO
a
(70%)
OOTBS
TBSO
c
(60%)
OOTBS
TBSO
N
OPh
Me3Sn
327 328
329 330
331
Scheme 42 (a) (Me3Sn)2CuCNLi2, THF, −78 °C; then Mel, DMPU, −78 °C to RT; (b) Tf2O, Et3N, THF, −78 °C; (c) Pd2(dba)3-CHCl3 (6 mol%), P(t-Bu)3(12 mol%), LiCl, NMP, 60 °C.
BnOCHO
N O
S
Bn
O
a
(53% over 3 steps)
BnO
OH
OBPSBnO
OH
OBPSb
(81%)
O
(335, 83% dr)
BnO
OH
OBPSc
OH
d
(65% over 2 steps)
BnO
OH
OBPS
OH
e
(56% over 2 steps) OH
OBPS
OO
f
(58% over 3 steps)
OTBSOO
g
(76% over 2 steps)
TMS
OH
g
(76% over 2 steps)
O
OTBS
OH
TMS
OTBS
OH
OO
O
OTBS
OH
TMS(37%) (21%)
332 333 334 336
337 338 339
340 341 342
Scheme 43 (a) (1) 333, TiCl4, DIPEA, CH2Cl2, −78 to 0 °C, 0.5 h then 332, −78 to 0 °C, 1 h; (2) NaBH4, EtOH, 0 °C, 15 min; (3) TBDPSCl, Et3N,DMAP, CH2Cl2, 0 °C to RT, 4 h; (b) Ti(Oi-Pr)4, (− )-DIPT, TBHP, CH2Cl2, 4 Å MS, −20 °C, 3 h; (c) Cp2TiCl2, Zn, ZnCl2, THF, −20 °C to RT, 12 h; (d) H2,10% Pd-C, CH3COONH4, MeOH, RT, 0.5 h; (e) (1) H2, Pd-C, MeOH, RT, 24 h; (2) 2,2-dimethoxypropane, CSA, CH2Cl2, 0 °C, 1 h; (f) (1) TBAF, THF, 0 °Cto RT, 4 h; (2) TBS-OTf, 2,6-lutidine, CH2Cl2, 0 °C, 0.5 h; (3) HF-pyr, THF, 0 °C to RT, 18 h; (g) SO3-pyr, Et3N, DMSO:CH2Cl2 (2:1.6), 0 °C, 0.5 h; (2)TMS-acetylene, n-BuLi, THF, −78 °C to RT, 1 h, then aldehyde, 0 °C, 15 min; (g) (1) MsCl, DMAP, pyridine, 0 °C, 0.5 h; (2) CSA, CH2Cl2, 0 °C to RT, 3 h.
O
HOO
a
OH
OO
H
O
(343, 90:10 syn/anti)
b
(76% over 2 steps)
OH
OO
COOEtOTBS
OO
OHc
(82% over 2 steps)
d
(53% over 2 steps)
TBSO
OO
OH
OH
e
(70% over 2 steps)
TBSO
OO
OH
OEt
O
f
O
CO2Et
OHOO
136 344 345
346 347 348
Scheme 44 (a) L-proline, EtCHO, DMF, 4 °C, 36 h; (b) Ph3P=CHCOOEt, benzene, 80 °C, 12 h; (c) (1) TBSOTf, 2-6,lutidine, CH2Cl2, 0 °C, 1 h; (2) DIBAL-H, CH2Cl2, −78 °C to RT, 4 h; (d) (1) (− )-DIPT, Ti(iPrO)4, TBHP, CH2Cl2, −20 °C, 10 h; (2) MeLi, Cul, Et2O, −20 °C, 6 h; (e) (1) TEMPO, BAIB, CH2Cl2,RT, 1 h; (2) Ph3P=CHCOOEt, benzene, 80 °C, 12 h; (f) TBAF, THF.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
247
The Journal of Antibiotics

BnOO
H a, bO O
O
OHBnO(87% over 2 steps)
(350, ee >= 95%)
c
(99%) OBnO
O
O
d
(99%)
OBnO
O
OMe
e
(84%) OHO
O
OMe
f
(88%) OO
O
OMe
H
349 351
352 353 354
Scheme 45 (a) 101, [Cu((R,R)-Ph-pybox)]-(SbF6)2 (8 mol%), CH2Cl2, −98 °C to −78 °C; (b) PPTs, MeOH; (c) K2CO3, MeOH; (d) (MeO)2SO2, K2CO3,acetone; (e) H2, Pd-C, EtOAc; (f) Swern oxidation.
HO I a, b
(82% over 2 steps)IN
MeOO
IH
Oc
(95%)
I
OH
NS
O
d
(96%)
NS
O
O
O(359, R/S > 97:3)
I
OMeO
NMeOe
(72%)I
OMeOf
(94%)H I
OMeg, h
(99% over 2 steps)(7.5:1 E/Z)
Br
355 356 357
358
360 361 362
i
(29%)
OMeBr
O O
OMe
OH
(363, 1:3.4 dr)
j
(58%)
OMeBr
O O
OMe
O
364
Scheme 46 (a) MnO2, CH2Cl2; (b) MeON(Me)C(O)CH2,P(O)(OEt)2, LiCl, DBU, MeCN-CH2Cl2 (3:1); (c) DIBAL-H, THF, −78 °C; (d) (1) 358, Sn(OTf), N-ethylpiperidine, CH2Cl2, −40 °C, (2) 357, −98 to −78 °C; (e) (1) MeNHOMe.HCl, ImH, CH2Cl2; (2) Mel, Ag2O, Et2O, reflux; (f) DIBAL-H, THF, −78 °C; (g)nBu3SnCHI2, CrCl2, DMF; (h) NBS, MeCN, 0 °C; (i) NiCl2-CrCl2 (10:1), tBuC5H4N, THF; 354; (j) TPAP, 4 Å molecular sieves, CH2Cl2.
OTHPO OMPM
a
(71% for 3 steps)
OMe
OMPMHO
b
(86% for 2 steps)
OMe
OMPMCl
c
(78%)
OH
OH d
(86% for 2 steps)
OMe
OHO
O
OMe
OMPMCl
Cl
e
(79% for 2 steps)
OMeO
O
Cl
OH
OEt
O f
(80%)
OMeO
O
Cl
O
OEt
O g
(50%) O OEt
O
OMeHCl
OMe
OH
h
(63% for 3 steps) O OEt
O
OMeHHO
OMe
OMOM
i
(quant) O OEt
O
OMeHH
OMe
OMOM
O
(369, 94% de)
OMeBr S
O O
S
N j
(70%) O OEt
O
OMeH
OMe
OMOM
OMeBr
(377, 1:1 E/Z)
365 366 367 368
370 371
372 373 374 375
376
Scheme 47 (a) (1) n-BuLi, BF3Et2O, −78 °C; (2) NaH, Mel, THF; (3) PTSA, MeOH; (b) (1) LiAIH4, THF, reflux, 2 h; (2) PPh3, CCl4, reflux, 12 h; (c) AD-mixβ. 0 °C, 48 h; (d) (1) 2,2-DMP, PTSA, acetone; (2) DDQ, CH2Cl2-H2O (7:3); (e) (1) IBX; (2) LiHMDS; CH3COOEt, −78 °C; (f) PDC; (g) PPTS, MeOH, 36 h;(h) (1) MOMCl, DIPEA; (2) HCOONa, Nal, DMF, 80 °C, 3 d; (3) NaBH4, MeOH, 0 °C; (i) Dess–Martin Ox.; (j) 375, NaHMDS, THF, −78 °C.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
248
The Journal of Antibiotics

BnO
O
CHOO PPh3
N
O
a
(80%, 99:1 dr)
BnO
OO O
N
O
b
(94%)
BnO
OO
N
OOHc
(92%)
BnO
OO
OEt
OOH
d
(65% over 4 steps)
HO
OO
OTBS
OMe OO
OH
OMe
f
(80% over 2 steps)
e
(57% over 2 steps)O
OOTBS
OMe
33
378 379 380 381 382
383 384
Scheme 48 (a) 379, toluene, 65 °C, 36 h, then H8-BINOL (5 mol%), Sm(O-i-Pr)3 (5 mol%), CMHP, THF/toluene 25 °C, 0.7 h; (b) PhSeSePh, NaBH4, EtOH/AcOH, 25 °C, 15 min; (c) EtSLi, EtOH, 25 °C, 40 min; (d) (1) CH3I, Ag2O, MS 3 Å, toluene, 45 °C, 36 h; (2) LiAIH4, EtO2, 25 °C for 30 min then reflux for40 min; (3) TBSCl imidazole, CH2Cl2, 25 °C, 50 min, (4) H2 (5 atm), Pd(OH)2, AcOEt/EtOH, NaHCO3, 25 °C, 18 h, (e) (1) PCC, AcONa, MS 3 Å, CH2Cl2,25 °C, 20 min then (CH3O)2P(O)C(N2)COCH3, K2CO3, CH3OH, 25 °C, 80 min.
OHHO
OH
OH
OH
OHO
O
OO
HOOH
a OO
OO
b
(58% over 3 steps) OBn
c
(62% over 3 steps)
D-Mannitol
OO
OBn
MeOO
O
d
(85%) OBn
H
OO
O
e
OBn
OH
S N
S O
Bn
N
O
S
S
Bn
f
(52% over 2 steps)
OO
OBn
OMeO
EtO
O
(62%)
gO OBn
OHOH
OMe
O
EtO
385 386
387 388
389
390
391 392
Scheme 49 (a)42; (b) (1) NalO4, CH2Cl2, 0 °C, 1 h; (2) NaBH4, MeOH; (3) NaH, THF, BnBr, 0 °C to RT, 12 h; (c) (1) 50% aq. AcOH, RT, overnight; (2)NalO4, CH2Cl2, NaHCO3, 0 °C to RT, 4 h; (3) Ph3P+CH2OMeCl−, t-BuOK, THF, −78 °C to RT, 4 h; (d) (AcO)2Hg; NaBH4, CO2, 0 °C, THF/H2O (1:4); (e)389, TiCl4, EtN(iPr)2, CH2Cl2, 0 to −78 °C, 1 h; (f) (1) Proton Sponge, Me3OBF4, CH2Cl2, 0 °C, 24 h; (2) EtOC(O)CH2COOK, MgCl2, imidazole, THF, RT,12 h; (g) PPTS, MeOH/CH2Cl2, 0 °C to RT, 3 h.
O O
MeO O
OS
O(+)menthyl
p-Tol a
(70%)
O OH
MeO O
SO
p-Tolb
(42-60%)
OH O
MeO O
SO
p-Tol
(396, dr > 98%)
c
(66% over 2 steps)
(S)
OH OH
N O
SO
p-Tol
OMe
(63% over 2 steps)
O O
O
SO
p-Tol (S)
(93%)
O
OH
Me
p-TolOSOMeH
(399, dr > 98%)(S)
(59%)O
OMe
Me
p-TolOSOMeH (49%)
OOHC
OMe
MeOMeH
393 394 395 397
398 400 401
Scheme 50 (a) NaH (2 eq.), t-BuLi (2 eq.), THF, 0 °C; (b) DIBAL-H, THF, −78 °C, 30 min; (c) (1) Me4NHB(OAc)3, HOAc, RT, 2 h; (2) HCl-N(OMe)Me,Me3Al, CH2Cl2, heat, 2 h; (d) (1) (MeO)2C(CH3)2, acetone, CSA, RT, 24 h; (2) MeMgBr, THF, 0 °C to RT, 1 h; (e) PPTS, MeOH, RT, 24 h; (f) NaH, Mel,THF, 0 °C, 10 h; (g) TFAA, 2,6-lutidine, CH3CN, RT, 10 min.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
249
The Journal of Antibiotics
a
(54% over 3 steps)O
OMe
MeOCO2Me
402
O
OMe
MeO
403
N
O
CO2Meb
(78% over 2 steps)
404
c
(79% over 2 steps)
405
HO
OMe
N
O
OH
S
S
TBSO
OMe
N
O
OTBS
H
Od
(71% over 2 steps)
406
TBSO
OMe
N
O
OTBSOH
OH
EtO2C
Scheme 51 (a) (1) IBCF, N-methylmorpholine, L-serine methyl ester, CH2Cl2; (2) Burgess' reagent, THF; (3) CuBr2, HMTA, DBU, CH2Cl2; (b) (1) DIBAL-H,THF; (2) propane-1,3-dithiol, BF3OEt2; (c) (1) TBSOTf, 2,6-lutidine, CH2Cl2; (2) Hg(ClO4)2, CaCO3, MeCN/H2O; (d) (1) E-(EtO)2P(O)CH2CH=CHCO2Et, NaH,CH2Cl2 2) AD-Mix β, t-BuOH/H2O.
N
O
O
X
OO
O
O
R
N
Hemi-phorboxazole A (407, X = CH2, R = OH)408 X = O, R = H
N
O
O
X
OO
O
ON
O
O
MeO
HO
RMeO
OH
409 R =
410 R =
H, X = CH2, R = OH
Cl, X = CH2, R = OH
R
411 R = H, X = O, R = H
NNN
O
OO
O
ON
O
O
MeO
MeO
MeO
OH OH
412
N
O
O
OO
O
ON
O
O
MeO
RO
MeO
OH OH
O
O
X
413 R = H, X = OO
O
S
NHHN
O
414 R = Me, X =
OO
N
Figure 12 Examples of synthetic analogs of the phorboxazoles.
Synthesis of the phorboxazolesZ Shultz and JW Leahy
250
The Journal of Antibiotics

could be oxidized for potential controlled reduction, but those resultshave not been published.The Yadav group has also completed the entire exocyclic
fragment.41 Acetylenic opening of 366 with 365 set the C35 center,which was converted into allyl chloride 368 (Scheme 47). Sharplessdihydroxylation was then used to install the C37 and C38 stereo-centers, and the pyran was elaborated by formation of β-ketoester 372and ketalization to 373. Upon formation of aldehyde 375, Juliaolefination with 376 gave the full C31–C46 fragment as a 1:1 mixtureof olefin isomers.Shibasaki synthesized an intermediate from Smith’s phorboxazole
synthesis to demonstrate the utility of his catalytic asymmetricMichael additions to unsaturated acylpyrroles, installing the C35stereocenter with 499% control (Scheme 48) and efficiently generat-ing alkyne 33.42
Nagaiah was able to demonstrate that D-mannitol, which is theestablished synthetic precursor to glyceraldehyde 136, could also beused for the generation of the exocyclic pyran.40 Periodate cleavage ofdiol 385 allowed access to 386,43 which could be selectively convertedinto aldehyde 388 (Scheme 49). An asymmetric acetate aldol was thenused to install the C35 stereocenter, which was then extended toβ-ketoester 391 and cyclized to the C31–C39 fragment.Another approach to the exocyclic pyran from chiral sulfoxide 395
was reported from the Carreño and Solladié groups (Scheme 50).44
Reduction of this diketone proceeded with excellent diastereocontrolto set C37, which was then used to set C35 via a directed reduction.The terminal ester was converted into the methyl ketone, which couldthen be ketalized to 399. The sulfoxide could then be converted intoaldehyde 401 via the Pummerer reaction.Hoffmann also completed a synthesis of a ring-opened exocyclic
pyran that incorporated the exocyclic oxazole as well (Scheme 51).45
Addition of serine to 402 and aromatization gave 403, which could beconverted into aldehyde 405. Extended homologation via vinylogousHorner–Wadsworth–Emmons reaction and installation of the C37and C38 stereocenters completed the C28–C41 fragment.
Additional phorboxazoles and analogsIn 2009, Molinski reported the isolation of hemi-phorboxazole A(407, Figure 12),46 which contains the full macrolide but only a nitrileinstead of the exocyclic oxazole and hinting at its biosynthetic origin.Smith was able to confirm the structure of 407 via its synthesis froman advanced synthetic intermediate.47 This synthesis provided suffi-cient material to allow for the biological evaluation of 407, whichproved to be essentially inactive compared with 1. Interestingly,synthetic analog 408 showed much greater activity than the naturalproduct.The extensive synthetic efforts detailed above have also allowed for
the generation of phorboxazole analogs and tool compounds useful forunderstanding its mechanism of action. Both the Forsyth and Smithgroups prepared and evaluated alkyne analog 409,48,49 demonstratingthat it was nearly as active as 1 against several cancerous cell lines. Infact, changes to the terminus are reasonably well tolerated and evencould be used to make more active analogs such as vinyl chloride 410.Although modifications such as ketalization at the C33 position do notdestroy activity, the loss of either the macrolide or a significant portionof the exocyclic terminus dramatically reduces activity. Some purelysynthetic analogs such as acetal 411 are quite active, while the activityof others such as triazole 412 (formed through a copper catalyzed click1,3-dipolar cycloaddition) remain unreported.50
Forsyth has prepared some phorboxazole-based biological toolcompounds such as biotinylated analog 413 and fluorescent analog
414.51,52 The latter was shown to be taken up in HeLa cells and toinduce extranuclear cytokeratin intermediate filaments to associatewith cyclin-dependent kinase 4 (Cdk-4), a kinase that has beenexplored as a cancer target.53 The full mechanism of action of thephorboxazoles remains undetermined at this stage.
CONCLUSIONS
The novel structure and biological properties of the phorboxazoleshave clearly attracted the interest of the synthetic community. It hasbeen through these efforts that we have begun to understand some ofthe structural requirements for their impressive biological activity andgained a glimpse of their biochemical target. Given the impressivestrides that have been made in this regard, it is reasonable to think thatthere remain chapters of the phorboxazole story to be written.
CONFLICT OF INTERESTThe authors declare no conflict of interest.
1 Searle, P. A. & Molinski, T. F. Phorboxazoles A and B: potent cytostatic macrolides frommarine sponge Phorbas species. J. Am. Chem. Soc. 117, 8126–8131 (1995)..
2 Searle, P. A., Molinski, T. F., Brzezinski, L. J. & Leahy, J. W. Absolute configuration ofphorboxazoles A and B from the marine sponge Phorbas sp. 1. Macrolide andhemiketal rings. J. Am. Chem. Soc. 118, 9422–9423 (1996).
3 Molinski, T. F. Absolute configuration of phorboxazoles A and B from the marinesponge, Phorbas sp. 2. C43 and complete stereochemistry. Tetrahedron Lett. 37,7879–7880 (1996).
4 Molinski, T. F., Brzezinski, L. J. & Leahy, J. W. Absolute configuration of phorboxazole AC32-C43 analogs by CD exciton-coupling of allylic 2-naphthoate esters. TetrahedronAsymmetry 13, 1013–1016 (2002).
5 Capon, R. J. et al. Esmodil: an acetylcholine mimetic resurfaces in a southernAustralian marine sponge Raspailia (Raspailia) sp. Nat. Prod. Res. 18,305–309 (2004).
6 Lee, C. S. & Forsyth, C. J. Synthesis of the central C18-C30 core of the phorboxazolenatural products. Tetrahedron Lett. 37, 6449–6452 (1996).
7 Seitz, O. Total synthesis of phorboxazole. Nachr. Chem. 49, 1189–1195 (2001).8 Haustedt, L. O., Hartung, I. V. & Hoffmann, H. M. R. The total syntheses of
phorboxazoles–new classics in natural product synthesis. Angew .Chem. Int. Ed. 42,2711–2716 (2003).
9 Evans, D. A. & Fitch, D. M. Asymmetric synthesis of phorboxazole B. Part II: synthesisof the C1-C19 subunit and fragment assembly. Angew. Chem. Int. Ed. 39,2536–2540 (2000).
10 Paterson, I., Steven, A. & Luckhurst, C. A. Phorboxazole B synthetic studies:construction of C(1-32) and C(33-46) subtargets. Org. Biomol. Chem. 2,3026–3038 (2004).
11 Forsyth, C. J., Ahmed, F., Cink, R. D. & Lee, C. S. Total synthesis of phorboxazole A. J.Am. Chem. Soc. 120, 5597–5598 (1998).
12 Smith, A. B. III, Minbiole, K. P., Verhoest, P. R. & Schelhaas, M. Total synthesis of(+)-phorboxazole A exploiting the Petasis-Ferrier rearrangement. J. Am. Chem. Soc.123, 10942–10953 (2001).
13 Pattenden, G. et al. Total synthesis of (+)-phorboxazole A, a potent cytostatic agent fromthe sponge Phorbas sp. Org. Biomol. Chem. 1, 4173–4208 (2003).
14 Williams, D. R. et al. Total synthesis of phorboxazole A. Angew. Chem., Int. Ed. 42,1258–1262 (2003).
15 Smith, A. B. III et al. A second-generation total synthesis of (+)-phorboxazole A. J. Org.Chem. 73, 1192–1200 (2008).
16 Smith, A. B. III et al. Evolution of a Gram-scale synthesis of (+)-discodermolide. J. Am.Chem. Soc. 122, 8654–8664 (2000).
17 Smith, A. B. III, Tomioka, T., Risatti, C. A., Sperry, J. B. & Sfouggatakis, C. Gram-scalesynthesis of (+)-spongistatin 1: development of an improved, scalable synthesis of theF-ring subunit, fragment union, and final elaboration. Org. Lett. 10,4359–4362 (2008).
18 White, J. D., Lee, T. H. & Kuntiyong, P. Total synthesis of phorboxazole A. 2. Assemblyof subunits and completion of the synthesis. Org. Lett. 8, 6043–6046 (2006).
19 White, J. D., Kuntiyong, P. & Lee, T. H. Total synthesis of phorboxazole A. 1.Preparation of four subunits. Org. Lett. 8, 6039–6042 (2006).
20 Wang, B. et al. Total synthesis of phorboxazole A via de Novo oxazole formation: strategyand component assembly. J. Am. Chem. Soc. 133, 1484–1505 (2011).
21 Wang, B. et al. Total synthesis of phorboxazole A via de Novo oxazole formation:convergent total synthesis. J. Am. Chem. Soc. 133, 1506–1516 (2011).
22 Li, D.-R. et al. Total synthesis of phorboxazole B. Chemistry 12, 1185–1204 (2006).23 Lucas, B. S., Gopalsamuthiram, V. & Burke, S. D. Total synthesis of phorboxazole B.
Angew. Chem. Int. Ed. 46, 769–772 (2007).
Synthesis of the phorboxazolesZ Shultz and JW Leahy
251
The Journal of Antibiotics

24 Wolbers, P. & Hoffmann, H. M. R. trans-C-glycosides from 8-oxabicyclo[3.2.1]oct-6-en-3-one - synthesis of the C3-C13 segment of the phorboxazoles A and B. Tetrahedron 55,1905–1914 (1999).
25 Dunkel, R. & Hoffmann, H. M. R. Asymmetric synthesis of polyacetate derived buildingblocks with α-oxyanion functionality. Lewis acid catalyzed opening of 2,9-dioxabicyclo[3.3.1]nonan-3-ones. Tetrahedron 55, 8385–8396 (1999).
26 Wolbers, P., Misske, A. M. & Hoffmann, H. M. R. Synthesis of the enantiopureC15-C26 segment of phorboxazole A and B. Tetrahedron Lett. 40, 4527–4530(1999).
27 Misske, A. M. & Hoffmann, H. M. R. Asymmetric synthesis of seven-carbonsegments of the phorboxazoles and (-)-discodermolide: complementary route fromracemic trans-2,4-dimethyl-8-oxabicyclo[3.2.1]oct-6-en-3-one. Tetrahedron 55,4315–4324 (1999).
28 Ma, P., Martin, V. S., Masamune, S., Sharpless, K. B. & Viti, S. M. Synthesis ofsaccharides and related polyhydroxylated natural products. 2. Simple deoxyalditols. J.Org. Chem. 47, 1378–1380 (1982).
29 Yadav, J. S., Prakash, S. J. & Gangadhar, Y. General strategy towards chiral methylenebis-pyrans: synthesis of the C2-C16 fragment of phorboxazole A. Tetrahedron Asym-metry 16, 2722–2728 (2005).
30 Yadav, J. S., Satyanarayana, M., Srinivasulu, G. & Kunwar, A. C. Astereoselective synthesis of the C20-C32 fragment of the phorboxazoles. Synlett 2007,1577–1580 (2007).
31 Vitale, J. P., Wolckenhauer, S. A., Do, N. M. & Rychnovsky, S. D. Synthesis of the C3-C19 segment of phorboxazole B. Org. Lett. 7, 3255–3258 (2005).
32 Rychnovsky, S. D. & Thomas, C. R. Synthesis of the C22-C26 tetrahydropyransegment of phorboxazole by a stereoselective prins cyclization. Org. Lett. 2,1217–1219 (2000).
33 Clarke, P. A., Santos, S., Mistry, N., Burroughs, L. & Humphries, A. C. The asymmetricMaitland-Japp reaction and its application to the construction of the C1-C19 bis-pyranunit of phorboxazole B. Org. Lett. 13, 624–627 (2011).
34 Clarke, P. A. & Ermanis, K. Synthesis of the C20-C32 tetrahydropyran core of thephorboxazoles and the C22 epimer via a stereodivergent Michael reaction. Org. Lett. 14,5550–5553 (2012).
35 Kartika, R. & Taylor, R. E. Electrophile-Induced ether transfer: stereoselective synthesisof 2,4,6-trisubstituted tetrahydropyrans. Angew. Chem. Int. Ed. 46,6874–6877 (2007).
36 Greer, P. B. & Donaldson, W. A. Phorboxazole synthetic studies: the C3-C15 bis-oxanesegment. Tetrahedron Lett. 41, 3801–3803 (2000).
37 Huang, H. & Panek, J. S. Formal [4+2]-annulation of chiral crotylsilanes:synthesis of the C19-C28 fragment of phorboxazoles. Org. Lett. 3, 1693–1696(2001).
38 Schaus, J. V. & Panek, J. S. Palladium-catalyzed cross-coupling of terminal alkynes with4-trifloyloxazole: studies toward the construction of the C26-C31 subunit ofphorboxazole A. Org. Lett. 2, 469–471 (2000).
39 Chakraborty, T. K., Reddy, V. R. & Reddy, T. J. Synthesis of highly substitutedtetrahydropyrans: preparation of the C20-C28 moiety of phorboxazoles. Tetrahedron 59,8613–8622 (2003).
40 Raju, K. B., Kumar, B. N., Kumar, B. S. & Nagaiah, K. Towards stereoselectivesynthesis of the C(31)-C(39) and C(20)-C(27) fragments of phorboxazole A. Helv. Chim.Acta 98, 386–399 (2015).
41 Yadav, J. S. & Rajaiah, G. A convergent synthesis of the C31-C46 fragment ofphorboxazoles. Synlett 2004, 1743–1746 (2004).
42 Matsunaga, S., Kinoshita, T., Okada, S., Harada, S. & Shibasaki, M. CatalyticAsymmetric 1,4-Addition Reactions Using α,β-Unsaturated N-Acylpyrroles as HighlyReactive Monodentate α,β-Unsaturated Ester Surrogates. J. Am. Chem. Soc. 126,7559–7570 (2004).
43 Wiggins, L. F. The acetone derivatives of hexahydric alcohols; triacetone mannitol andits conversion into d-arabinose. J. Chem. Soc. 13 (1946).
44 Brinkmann, Y., Carreno, M. C., Urbano, A., Colobert, F. & Solladie, G. Asymmetricsynthesis of the tetrahydropyran ring, C32-C38 fragment, of phorboxazoles. Org. Lett. 6,4335–4338 (2004).
45 Wolbers, P. & Hoffmann, H. M. R. Asymmetric synthesis of the enantiopure C(28)-C(41)segment of the phorboxazoles A and B. Synthesis 1999, 797–802 (1999).
46 Dalisay, D. S. & Molinski, T. F. Structure elucidation at the nanomole scale. 2. Hemi-phorboxazole A from Phorbas sp. Org. Lett. 11, 1967–1970 (2009).
47 Smith, A. B. III, Liu, Z., Hogan, A.-M. L., Dalisay, D. S. & Molinski, T. F. Hemi-phorboxazole A: structure confirmation, analogue design and biological evaluation. Org.Lett. 11, 3766–3769 (2009).
48 Uckun, F. M. & Forsyth, C. J. Anticancer activity of synthetic analogues of thephorboxazoles. Bioorg. Med. Chem. Lett. 11, 1181–1183 (2001).
49 Smith, A. B. III et al. Phorboxazole synthetic studies: design, synthesis and biologicalevaluation of phorboxazole A and hemi-phorboxazole A related analogues. Tetrahedron67, 5069–5078 (2011).
50 Ying, L. & Forsyth, C. J. Synthesis of a 16-triazole phorboxazole A analog viaintramolecular triazole formation. Heterocycles 73, 841–855 (2007).
51 Hansen, T. M., Engler, M. M. & Forsyth, C. J. Total synthesis of a biotinylated derivativeof phorboxazole A via sonogashira coupling. Bioorg. Med. Chem. Lett. 13,2127–2130 (2003).
52 Chen, J. et al. Design and total synthesis of a fluorescent phorboxazole A analog forcellular studies. Bioorg. Med. Chem. Lett. 16, 901–904 (2006).
53 Forsyth, C. J., Lu, Y., Chen, J. & La Clair, J. J. Phorboxazole analogues induceassociation of cdk4 with extranuclear cytokeratin intermediate filaments. J. Am. Chem.Soc. 128, 3858–3859 (2006).
Synthesis of the phorboxazolesZ Shultz and JW Leahy
252
The Journal of Antibiotics




















