
The synthesis of novel spirocyclic heterocycles as potential ...
261
University of Wollongong Thesis Collections University of Wollongong Thesis Collection University of Wollongong Year The synthesis of novel spirocyclic heterocycles as potential cancer therapeutics targeting the cell-cycle Sarah Rebecca Yong University of Wollongong Yong, Sarah Rebecca, The synthesis of novel spirocyclic heterocycles as potential cancer therapeutics targeting the cell-cycle, PhD thesis, Department of Chemistry, University of Wollongong, 2006. http://ro.uow.edu.au/theses/680 This paper is posted at Research Online. http://ro.uow.edu.au/theses/680
-
Upload
khangminh22 -
Category
Documents
-
view
3 -
download
0
Transcript of The synthesis of novel spirocyclic heterocycles as potential ...

University of Wollongong Thesis Collections
University of Wollongong Thesis Collection
University of Wollongong Year
The synthesis of novel spirocyclic
heterocycles as potential cancer
therapeutics targeting the cell-cycle
Sarah Rebecca YongUniversity of Wollongong
Yong, Sarah Rebecca, The synthesis of novel spirocyclic heterocycles as potential cancertherapeutics targeting the cell-cycle, PhD thesis, Department of Chemistry, University ofWollongong, 2006. http://ro.uow.edu.au/theses/680
This paper is posted at Research Online.
http://ro.uow.edu.au/theses/680


The Synthesis of Novel Spirocyclic Heterocycles
as Potential Cancer Therapeutics
Targeting the Cell-cycle.
A thesis submitted in (partial) fulfillment
of the requirements for the award of the degree
DOCTOR OF PHILOSOPHY
from
UNIVERSITY OF WOLLONGONG
By
Sarah Rebecca Yong, B. Med. Chem. (Hons)
Supervisors: Prof. Stephen G. Pyne and Dr. Alison T. Ung
University of Wollongong
Department of Chemistry
Wollongong, Australia
November 2006

THESIS CERTIFICATION
I, Sarah R. Yong, hereby declare that all material in this thesis, submitted in partial
fulfillment of the requirements for the award of Doctor of Philosophy, in the
Department of Chemistry, University of Wollongong, is wholly my own work unless
otherwise referenced or acknowledged. This document has not been submitted for
qualifications at any other academic institution
Sarah Rebecca Yong
Date:

PUBLICATIONS ARISING FROM THIS THESIS
Yong, Sarah R.; Williams, Morwenna C.; Pyne, Stephen G.; Ung, Alison T.; Skelton,
Brian W.; White, Allan H.; Turner, Peter. Synthesis of 2-azaspiro[4.4]nonan-1-ones
via phosphine-catalyzed [3+2]-cycloadditions. Tetrahedron, 2005, 61(34), 8120-8129.
Yong, Sarah R.; Ung, Alison T.; Pyne, Stephen G.; Skelton, Brian W.; White, Allan H.
Syntheses of spiro[cyclopropane-1,3’oxindole]-2-carboxylic acid and
cyclopropa[c]quinoline-7b-carboxylic acid and their derivatives. Tetrahedron, 2007,
63, 1191-1199.
Yong, Sarah R.; Ung, Alison T.; Pyne, Stephen G.; Skelton, Brian W.; White, Allan H.
Synthesis of novel 3’-spirocyclic-oxindole derivatives and assessment of their
cytostatic activities. Tetrahedron, 2007, 63, 5579-5586.

Table of Contents i
TABLE OF CONTENTS
Page No.
LIST OF FIGURES, SCHEMES AND TABLES ...................................................... iv
ABBREVIATIONS .........................................................................................................x
ACKNOWLEDGEMENTS........................................................................................ xiv
ABSTRACT ...................................................................................................................xv
CHAPTER 1: INTRODUCTION ..................................................................................1
1.1 Introduction to Cancer Therapeutics ...........................................................................2
1.1.1 The Cell-cycle .......................................................................................................2
1.2 Molecular Targets in the Cell-cycle ............................................................................3
1.2.1 Cell-cycle dysregulation, a hallmark of cancer .....................................................3
1.2.2 Cyclin-Dependent Kinases (CDKs) ......................................................................4
1.2.2 MDM2-p53..........................................................................................................13
1.3 Naturally Occurring Spirocyclic Oxindoles ..............................................................22
1.4 Aims of this study .....................................................................................................30
CHAPTER 2: SYNTHESIS OF 2-AZASPIRO[4.4]NONAN-1-ONES AND
SPIRO[CYCLOPENTANE-1,1`-[1H]ISOINDOL]-3`(2`H)ONES USING THE
PHOSPHINE-CATALYSED [3+2]-CYCLOADDITION REACTION...................31
2.1 Synthesis of 2-azaspiro[4.4]nonan-1-ones ................................................................34
2.1.1 Synthesis of 2-methylene γ-lactams 57 or 58 .....................................................34
2.1.2 [3+2]-cycloaddition reactions using the 2-methylene γ-lactam 57 .....................35
2.1.2 [3+2]-cycloaddition using the 2-methylene γ-lactam 58 ....................................40
2.2 Synthesis of spiro[cyclopentane-1,1`-[1H]isoindol]-3`(2`H)-ones ...........................41
2.2.1 [3+2]-cycloadditions using acrylate 59 ...............................................................41
2.2.2 Asymmetric [3+2]-cycloaddition reaction ..........................................................44
2.3 Synthesis of spirocyclic derivatives .......................................................................48
2.3.1 Pathway A: Increasing structural diversity of existing structures .......................48
2.3.2 Pathway B: [3+2]-cycloaddition with structural diversity embedded in the
starting ylide............................................................................................................50
2.3.3 Curtius Rearrangement........................................................................................51
2.3.4 Hydrogenation.....................................................................................................53
CHAPTER 3: SYNTHESIS OF SPIROCYCLOPROPANE INDOLINONES.......55
3.1 Synthesis of Spirocyclopropane Indolinone 129.......................................................63

Table of Contents ii
3.1.1 Cyclopropanation of the acrylate 59 using EDSA ..............................................63
3.1.2 Reductive cyclization of 117...............................................................................64
3.1.3 Derivatisation of compounds ..............................................................................67
3.1.4 Synthesis of 129 using the amide-sulfonium salt 132 .........................................68
3.1.5 Cyclopropanation of the acrylate 59 using 132...................................................69
3.2 Cyclopropanations using α-methylene indolinones ..................................................70
3.2.1 Synthesis of α-methylene indolinones 108a and 108b........................................74
3.2.1 Cyclopropanation reaction of the α-methylene indolinones 108a and 108b.......75
CHAPTER 4: SYNTHESIS OF SPIRO[INDOLE-3,5`-ISOXAZOLIDIN]-2(1H)-
ONES AND SPIRO[INDOLE-3,6`-[1,3]OXAZINANE]-2,2`(1H)-DIONES USING
THE [1,3]-DIPOLAR CYCLOADDITION REACTION..........................................79
4.1 Introduction ...............................................................................................................81
4.1.1 Regioselectivity and Stereoselectivity of the [1,3]-DC reaction .........................82
4.2 Synthesis of Nitrones ................................................................................................94
4.2.1 Synthesis of acyclic nitrones 142a and 142b ......................................................95
4.2.2 Synthesis of the cyclic nitrone 143 .....................................................................95
4.3 [1,3]-Dipolar cycloaddition reactions using 59 .........................................................96
4.3.1 [1,3]-DC reactions using acyclic nitrones 142a and 142b ..................................96
4.3.2 [1,3]-Dipolar cycloadditions using the cyclic nitrone 143 ................................106
4.3.3 Derivatisation of the cycloadducts 170 and 171 ...............................................108
4.3.4 Derivatisation of the cycloadducts 172 and 173 ...............................................110
CHAPTER 5: TOWARDS THE SYNTHESIS OF A POTENT PURINE CDK
INHIBITOR.................................................................................................................112
5.1 Towards the Synthesis of a Potent Purine CDK Inhibitor.......................................113
5.1.1 Alternative Synthesis using DoM Chemistry....................................................113
5.1.2 Revised Former Synthesis .................................................................................121
5.1.3 Conclusions .......................................................................................................125
CHAPTER 6: BIOLOGICAL TESTING .................................................................126
RESULTS AND DISCUSSION..................................................................................126
6.1 Introduction .............................................................................................................127
6.2 Cytostatic Cellular Studies ......................................................................................127
6.2.1 Prelimary Cytostaticity Studies of 85a and 87 ..................................................127
6.2.2 Cytostaticity Screening against H460, MCF-7 and SF-268 ..............................129
6.3 Protein Inhibition Studies........................................................................................133

Table of Contents iii
6.3.1 CDK5 and gSK-3 ..............................................................................................133
6.3.2 CDK2 ................................................................................................................133
6.3.3 MDM2-p53........................................................................................................133
6.4 Conclusion and Future Directions...........................................................................135
CHAPTER 7: CONCLUSIONS AND FUTURE DIRECTIONS ...........................136
CHAPTER 8: EXPERIMENTAL .............................................................................137
8.1 General Synthetic Procedures .................................................................................138
8.2 Experimental for Chapter 2 .....................................................................................140
8.3 Experimental for Chapter 3 .....................................................................................172
8.4 Experimental for Chapter 4 .....................................................................................183
8.5 Experimental for Chapter 5 .....................................................................................195
CHAPTER 9: REFERENCES ...................................................................................200
Appendix 1: X-ray Crystal Structures ......................................................................230
Appendix 2: X-ray Crystal Data ................................................................................233
Appendix 3: Biological Testing Procedures ..............................................................237
A3.1 Cytostatic Cellular testing ....................................................................................237
A3.1.1 Prelimary Cytostaticity Studies of 85a and 87 ...............................................237
A3.1.2 Cytostaticity Screening against H460, MCF-7 and SF-268 ...........................239
A3.2 Protein Inhibition Studies.....................................................................................239
A3.2.1 CDK2 Assay...................................................................................................239
A3.2.2 MDM2-p53 interaction using ELISA.............................................................240

List of Figures, Schemes & Tables iv
LIST OF FIGURES, SCHEMES AND TABLES
Page No.
Figure 1.1 The different phases of the eukaryotic cell-cycle (G1-, S-, G2- and M-phases),
is regulated by a complex interplay of cellular pathways. ........................................3
Figure 1.2 The various different CDK/cyclin complexes and their functions. ................5
Figure 1.3 Crystal structure of the cyclin A/CDK2 complex. Cyclin A is coloured in
magenta, CDK2 in cyan; ATP is shown as a ball and stick representation. Portions
of CDK2 that undergo large conformational changes upon cyclin-binding are
highlighted: the PSTAIRE (single-letter amino acids) helix in red and the T-loop in
yellow.27 ....................................................................................................................7
Figure 1.4 The cyclin-induced conformational changes that occur within the CDK unit,
more specifically its PSTAIRE helix and T-loop, upon cyclin binding.13,27 The
ribbon structures of CDK from the complex, coloured in cyan and in monomeric
form, coloured in grey are superimposed upon one another. The inset highlights the
change in position of Glu51 (E51) located on the PSTAIRE helix. The space-filled
structures highlight the differences in substrate accessibility to ATP and CAK
accessibility to Thr160 in the monomeric (middle) and cyclin-bound (bottom) CDK
structures. ..................................................................................................................8
Figure 1.5 Schematic representation of the binding mode of some CDK inhibitors
elucidated by X-ray crystal structural analysis. Yellow represents the ATP-binding
pocket, green the hydrophobic region and red and blue represents hydrogen bond
acceptor and donor interactions, respectively. a) ATP, b) staurosporine,62 c)
dechlorinated flavopiridol,38 d) purvalanol B (2) e) NU6027 (5) f) indirubin-5-
sulfate (12), g) hymenialdisine (16) and h) diarylurea derivative.39 .......................11
Figure 1.6 The essential p53 response to cellular stress.70 .............................................13
Figure 1.7 The regulation of p53 by MDM2.70 ..............................................................14
Figure 1.8 The X-ray crystal structure of the complex of MDM2 and segment of p53.94
.................................................................................................................................15
Figure 1.9 A sectional viewof the X-ray crystal structure, revealing the MDM2 binding
site bound to a) WT-p53 and b) 8-mer peptide.97 ...................................................16
Figure 1.10 Inhibition values for the chalcone derivative 29 and its against various
human breast cancer cell lines and the normal breast cell line, MCF-10A and
MCF-12A.108 ...........................................................................................................18

List of Figures, Schemes & Tables v
Figure 1.11 Overlay of the benzodiazepinedione 23a (yellow) with a 9-mer peptide,
with critical amino acids, Phe19, Trp23 and Leu26 highlighted in green.101..............18
Figure 1.12 A) X-ray crystal structure of the Nutlin-2 (26a) (yellow) bound to MDM2
(red), B) Nutlin-2 (topaz) overlayed with the three critical residues (green) of the
p53 peptide.67 ..........................................................................................................20
Figure 1.13 A) X-ray crystal structure of the MDM2-p53, highlighting critical residues
of p53 (magenta) in this binding interaction (Phe19, Trp23, Leu26 and Leu22) B) and
C) Predicted binding mode of spirooxindole (IC50 = 13 nM) (white) using the
GOLD program. Hydrogen bonds (yellow dashed line). C) Overlay of model
binding of spirooxindole with X-ray crystal structure. ...........................................21
Figure 2.1 The observed cross-peaks and the calculated dihedral angles (φ) of 62 using
Spartan ‘04 (AM1)………………………………………………………………..37
Figure 2.2 Single crystal X-ray crystallographic analysis of 63. ...................................38
Figure 2.3 Enlargements of the A) gCOSY B) gHMBC and C) gHSQC spectra of 71
showing the cross-peaks concerning the protons H-5`β (δ 3.62), H-2`β (δ 3.52), H-
2`α (δ 3.21) and H-2`α (δ 2.99)................................................................................43
Figure 2.4 Single crystal X-ray crystallographic analysis of (S)-76. .............................46
Figure 2.5 Single crystal X-ray crystallographic analysis of 102 (left) and 103 (right).53
Figure 2.6 The important NOE peak between Ha and Ho observed in 104 (left) and
absent in 105 (right) and the calculated measured distance using Spartan ‘04
(AM1)......................................................................................................................54
Figure 3.1 Drawing showing the NOE correlations (inset) and single crystal X-ray
crystallographic structure of 117…………………………………………………64
Figure 3.2 Single crystal X-ray crystallographic analysis of 118 (left) and 119 (right).65
Figure 3.3 Single crystal X-ray crystallographic analysis of 133. .................................70
Figure 3.4 Single crystal X-ray crystallographic analysis of 108a (left) and 108b (right).
.................................................................................................................................75
Figure 3.5 Single crystal X-ray crystallographic analysis of 139a (left) and 139b (right).
.................................................................................................................................77
Figure 4.1 Single crystal X-ray crystallographic analysis of 170a…………………...101
Figure 4.2 The NOE correlations (*) of 170a and 171a displayed on their Spartan
models (Spartan ‘04 (AM1)). ................................................................................102

List of Figures, Schemes & Tables vi
Figure 4.3 The NOE correlations (*) of 170b and 171b displayed on their Spartan
models (Spartan ‘04 (AM1)). ................................................................................104
Figure 4.4 The NOE correlations (*) of 172 and 173 displayed on their Spartan models
(Spartan ‘04 (AM1)...............................................................................................106
Figure 5.1 Single crystal X-ray crystallographic analysis of 187a…………………...123
Figure 5.2 1H NMR spectra of 187a in A) CDCl3 and B) CD3OD. .............................124
Figure 6.1 Inhibition of Mpro murine myeloid cells by drugs: A) 1, B) 85a and C) 87
determined by MTT cell proliferation assay. Results represent means ± standard error of
the mean of 8 replicate cultures……………………………………………………….128
Figure 6.2 Inhibition of HL60 human myeloid cells by drugs: A) 1, B) 85a and C) 87
determined by MTT cell proliferation assay. Results represent means ± standard error of
the mean of 8 replicate cultures……………………………………………………….128
Figure 6.3 Inhibition of B16 murine melanoma cells by drugs: A) 1, B) 85a and C) 87
determined by SRB cell proliferation assay. Results represent means ± standard error of
the mean of 6 replicate cultures……………………………………………………….128
Figure 6.4 Comparison between cell-cycle profiles of B16 melanoma cells treated with
various selective CDK2 inhibitors. A) 1% DMSO as control B) 35µM of 1 C) 100µM of
85a D) 100µM of 87. The percentage of cells in each cell-cycle phase (G1, S and G2/M)
are given………………………………………………………………………………128
Figure 6.5 Viable cell counts of B16 cells taken 48 h after treatment with various
CDK2 inhibitors compared to DMSO control. Results represent means ± standard error
of the mean of cultures performed in triplicate……………………………………….128
Figure 6.6 GI50 curves in duplicate for 174a (A and B) and 172 (C and D) against
MCF-7………………………………………………………………………………...132
Figure 6.7 The protein inhibition results for the synthesised compounds against varying
protein targets, CDK5 and gSK-3 (yellow), CDK2 (topaz) and MDM2-p53 (red)…..134
Scheme 1.1......................................................................................................................30
Scheme 2.1…...………………………………………………………………………...34
Scheme 2.2a ....................................................................................................................35
Scheme 2.3 (Compounds 62 and 63 are racemic)...........................................................35
Scheme 2.4......................................................................................................................39
Scheme 2.5212..................................................................................................................39
Scheme 2.6......................................................................................................................39
0.01
Pe
rcen
t G
row
th
-20
0
20
40
60
80
100

List of Figures, Schemes & Tables vii
Scheme 2.7......................................................................................................................40
Scheme 2.8a ....................................................................................................................41
Scheme 2.9a ....................................................................................................................42
Scheme 2.10 (Compounds 71 and 72 are racemic).........................................................42
Scheme 2.11....................................................................................................................45
Scheme 2.12a ..................................................................................................................47
Scheme 2.13a ..................................................................................................................49
Scheme 2.14a ..................................................................................................................50
Scheme 2.15a (all compounds are racemic)....................................................................51
Scheme 2.16a (all compounds are racemic)....................................................................51
Scheme 2.17a (all compounds are racemic) ....................................................................52
Scheme 2.18....................................................................................................................53
Scheme 2.19....................................................................................................................54
Scheme 3.1...…………………………………………………………………………...56
Scheme 3.2......................................................................................................................57
Scheme 3.3244..................................................................................................................58
Scheme 3.4243..................................................................................................................58
Scheme 3.5242..................................................................................................................59
Scheme 3.6 (all intermediates are racemic) ....................................................................61
Scheme 3.7a (Compound 117 is racemic).......................................................................63
Scheme 3.8a (all compounds are racemic)......................................................................64
Scheme 3.9......................................................................................................................65
Scheme 3.10251................................................................................................................67
Scheme 3.11a (all compounds are racemic)....................................................................68
Scheme 3.12a ..................................................................................................................69
Scheme 3.13a ..................................................................................................................69
Scheme 3.14 (all compounds and intermediates are racemic) ........................................73
Scheme 3.15a 257 .............................................................................................................74
Scheme 3.16a (all compounds are racemic)....................................................................76
Scheme 3.17....................................................................................................................78
Scheme 4.1…...…………………...……………………………………………………80
Scheme 4.2......................................................................................................................81
Scheme 4.3......................................................................................................................82
Scheme 4.4......................................................................................................................83

List of Figures, Schemes & Tables viii
Scheme 4.5......................................................................................................................84
Scheme 4.6299..................................................................................................................91
Scheme 4.7185..................................................................................................................92
Scheme 4.8a 106................................................................................................................93
Scheme 4.9a 301................................................................................................................94
Scheme 4.10....................................................................................................................95
Scheme 4.11....................................................................................................................95
Scheme 4.12a 312..............................................................................................................96
Scheme 4.13311................................................................................................................96
Scheme 4.14..................................................................................................................100
Scheme 4.15..................................................................................................................100
Scheme 4.16a (all compounds are racemic)..................................................................105
Scheme 4.17a (all compounds are racemic)..................................................................107
Scheme 4.18a (all compounds are racemic)..................................................................110
Scheme 4.19a (all compounds are racemic) ..................................................................111
Scheme 5.1a (Yields given are those previously attained by a PhD student at
UNT)316…………………..……………………………………………………...114
Scheme 5.2....................................................................................................................115
Scheme 5.3330................................................................................................................116
Scheme 5.4a ..................................................................................................................117
Scheme 5.5....................................................................................................................118
Scheme 5.6....................................................................................................................119
Scheme 5.7a ..................................................................................................................120
Scheme 5.8....................................................................................................................120
Scheme 5.9a 326..............................................................................................................121
Scheme 5.10..................................................................................................................122
Scheme 5.11a ................................................................................................................122
Scheme 5.12a ................................................................................................................123
Scheme 5.13a ................................................................................................................125
Table 1.1 The occurrence of mutations affecting cell-cycle control in particular cancer
types.4,7,8…………………………………………………………………………...4
Table 1.4 Naturally occurring spirocyclic oxindoles......................................................23
Table 1.5 Cytotoxicity against A341 human epidermoid carcinoma cells.168 ................27
Table 1.6 Cytotoxicity against MDA-MD-468 and MCF-7 breast cancer cell lines.177.28

List of Figures, Schemes & Tables ix
Table 1.7 Inhibition of MDM2-p53 interaction.106.........................................................29
Table 2.1 Summary of rotations and yields for Scheme 2.12……...………………….46
Table 2.2 Summary of rotations and yields for Scheme 2.14 ........................................49
Table 3.1244…………………………………………………………………………….60
Table 3.2221 .....................................................................................................................71
Table 4.1 (all products are racemates)…………………………………………………85
Table 4.2293 .....................................................................................................................86
Table 4.3 .........................................................................................................................88
Table 4.4 .........................................................................................................................90
Table 4.5 .........................................................................................................................98
Table 4.6 .......................................................................................................................102
Table 4.7 .......................................................................................................................104
Table 4.8 .......................................................................................................................107
Table 4.9a (all compounds are racemic) .......................................................................108
Table 5.1...……………………………………………………………………………115
Table 5.2335,336 ..............................................................................................................119
Table 5.3 1H NMR chemical shifts (δ) for 187a in different NMR solvents ...............124
Table 6.1a……...……………………………………………………………………...131
Table 6.2a......................................................................................................................132

Abbreviations x
ABBREVIATIONS
[α]D Specific rotation
ABq AB-quartet (NMR)
AcOH Acetic acid
ArC Aromatic carbon
ArCH Aromatic methine
ATP Adenosine triphosphate
B16 Murine melanoma cell line
Bn Benzyl
Boc tert-Butoxycarbonyl
b.p. Boiling point
bs Broad singlet (NMR)
BTEAC Benzyl triethylammonium chloride
Bu3P Tributylphosphine
CAK CDK-activating kinase
Calcd Calculated
Cat. Catalyst
C6D6 Deuterated Benzene
CDCl3 Deuterated chloroform
CDK Cyclin-dependent kinase
CD3OD Deuterated methanol
CHCl3 Chloroform
CI+ve Chemical ionization (positive ion mode)
CNS Central nervous system
COSY Correlation spectroscopy
CR Curtius rearrangement
d Day
d Doublet (NMR)
δ Chemical shift (NMR)
DBU 1,8-diazobicyclo[5.4.0]undec-7-ene
[1,3]-DC [1,3]-Dipolar cycloaddition
DCM Dichloromethane

Abbreviations xi
dd Doublet of doublets (NMR)
ddd Doublet of doublet of doublets (NMR)
ddt Doublet of doublet of triplets (NMR)
de Diastereomeric excess
DEPT Distortionless enhancement by polarization transfer
DFT Density functional theory
dm Doublet of multiplets
DMAP 4-Dimethylaminopyridine
DMEM Dulbecco’s modified Eagle’s medium
DMF N,N-Dimethylformamide
DMG Directed metalation group
DMSO Dimethylsulfoxide
D2O Deuterated water
DoM Directed ortho metalation
DPPA Diphenylphosphoryl azide
dq Doublet of quartets (NMR)
dr Diastereomer regioselectivity
dt Doublet of triplets (NMR)
ε Dielectric constant
EC50 Effective concentration; concentration having 50% of desired response
compared to control
EDCI 1-[3-(Dimethylamino)propyl]-3-ethylcarbodiimidem hydrochloride
EDSA Ethyl (dimethylsulfuranylidene) acetate
EDTA Ethylenediamine tetraacetic acid
EI Electron impact
ELISA Enzyme-linked immunosorbant assay
eq. Molar equivalents
ESI+ve Electrospray ionisation (positive)
ESI-ve Electrospray ionisation (negative)
Et2O Diethylether
EtOAc Ethyl acetate
EtOH Ethanol
FCS Fetal calf serum
FMO Frontier molecular orbital

Abbreviations xii
GI50 Concentration for 50% of growth inhibition
gSK-3 Glycogen synthase kinase-3
H460 Human non-small cell lung cell line
HDM2 Human double minute 2
HL60 Human leukemic myeloid cell line
HMBC Heteronuclear multiple bond correlation
HOBT 1-Hydroxybenzotriazole
HRMS High resolution mass spectrometry
HSAB Hard and soft acids and bases principle
HSQC Heteronuclear single quantum correlation
Hz Hertz
i ipso
IC50 Concentration for 50% inhibition
IR Infrared spectroscopy
J Coupling constant (NMR)
Ki Inhibition constant
LDA Lithium diisopropylamide
LiHMDS Lithium hexamethyldisilazane
lit. Literature
m Multiplet (NMR)
m-CBA meta-Chlorobenzoic acid
MCF-7 Human breast cell line
m-CPBA meta-Chloroperoxybenzoic acid
MDM2 Murine double minute 2
MeCN Acetonitrile
MIC Minimum inhibitory concentration
min Minutes
MOM Methoxymethoxy
m.p. Melting point
Mpro Murine myeloid cell line
MS Mass spectrometry
n-BuLi n-Butyl lithium
NCI National cancer institute
NMR Nuclear magnetic resonance

Abbreviations xiii
NNTRI Non-nucleoside reverse transcriptase inhibitor
NOESY Nuclear Overhauser enhancement spectroscopy
NPM/B23 Nucleophosmin/B23
od Overlapping doublets (NMR)
φ Dihedral angle
PBS Phosphate buffered saline
Pd/C Palladium on activated carbon
Ph Phenyl
PMB p-Methoxybenzyl
ppm Parts per million (NMR)
PS Petroleum spirit (b.p. 40-60°C)
PTLC Preparative thin layer chromatography
q Quartet (NMR)
QSAR Quantitative structure-activity relationships
Rb Retinoblastoma tumor suppressor protein
Rf Retardation Factor
RT Room temperature
s Singlet (NMR)
SAR Structure activity relationship
sat. Saturated
s-BuLi sec-Butyl lithium
SDS Sodium dodecylsulfate
SF-268 Human central nervous system cell line
SRB Sulphorhodamine B
t Triplet (NMR)
TCA Trichloroacetic acid
TFA Trifluoroacetic acid
THF Tetrahydrofuran
THP Tetrahydropyran
TLC Thin layer chromatography
TMEDA N,N,N`,N`-tetramethylethylenediamine
TMS Tetramethylsilane
tt Triplet of triplets (NMR)
WT Wild-type

Acknowledgements xiv
ACKNOWLEDGEMENTS
The deepest and sincerest of gratitude must be expressed to my supervisor, Professor
Stephen Pyne, for the countless sacrificing of time, wisdom, patience, guidance and
knowledge for the benefit of his students.
To Dr. Alison Ung for her supervision and the loving care and concern you’ve always
shown towards me, in particularly during my PhD.
To Prof. Roger Griffin and Prof. Bernard Golding for the generous care, hospitality
and supervision I received under your care when I was a long way from home.
To the many people that make up the Pyne lab; the Thai Association, Morwenna,
Minyan and Uta, Arife, Andrew, Joe Hartley, Steve Taylor, Leena, and Nicole, for
making my days of my PhD a joy and worthwhile.
To the many technician staff at UOW, especially to Wilford Lie, for always selflessly
giving me your time, wisdom, guidance and NMR expertise and to Dr. John Korth for
helping me in times of distress, whether it be an MS problem or simply opening up a
can of salmon for me.
Lastly to my dear family and friends, thanks for your constant and faithful prayers,
support, love, and friendship during the ups and downs of my PhD. I definitely could
not have endured these last couple of years without you.
Khorpkun ka! Xie xie ni! Terima kasih banyak! Cam òn! Thanks mates! Luv ya lots!

Abstract xv
ABSTRACT
The spirocyclic ring structure is a feature of a number of naturally-occurring and
synthetic products that possess interesting biologically activities. This thesis describes
our efforts towards synthesising various spirocyclic heterocycles as potential cancer
therapeutics targeting the cell-cycle. Three different spirocyclic scaffolds, A-C, were
accessed through a variety of cycloaddition reactions and several of these were
investigated for their cytostaticity and protein-inhibition properties. To gain further
insights into the development of novel CDK2 inhibitors, part of this research was
conducted at the Anti-Cancer Drug Design Initiative (ADDI) laboratory, University of
Newcastle upon the Tyne, UK.
This thesis is divided into three primary synthetic chemistry chapters, based upon
discussion surrounding the main chemistries used to derive the various spirocyclic
oxindole scaffolds of the types A-C. Chapter 2 describes the use of the phosphine-
catalysed [3+2]-cycloaddition reaction to synthesise racemic and enantio-enriched
versions of spirocycles of type A. Chapter 3 describes the use of the cyclopropanation
reaction to synthesise spirocycles of type B. Lastly, Chapter 4 describes the
employment of the [1,3]-dipolar cycloaddition reaction of nitrones to the synthesis of
spirocycles of type C. A discussion on the CDK2 research, performed in the UK,
towards a new synthetic strategy employing directed ortho metalation chemistry is then
provided in Chapter 5. The cellular cytostaticity screening against the cancer cell lines,
H460, MCF-7 and SF-268, and protein inhibition studies against the cell-cycle proteins
CDK2, CDK5, gSK-3 and MDM2, for a range of the spirocycles synthesised in
Chapters 2-4, is then given and discussed in Chapter 6. Final conclusions and future
work are drawn together in Chapter 7. All synthetic methods and physical and
spectroscopic data are provided for all compounds in Chapter 8. Lastly, an appendix
including all X-ray crystal structures and their crystallographic data, and also a section
on the biological testing procedures is provided.

Chapter 1: Introduction 1
CHAPTER 1: INTRODUCTION
Mùa hoa sữa.
“The season of the alstonia flower”
Artist : Bá Việt.
Altonisine

Chapter 1: Introduction 2
1.1 Introduction to Cancer Therapeutics
1.1.1 The Cell-cycle
Cells traverse a series of co-ordinated events collectively known as the cell-cycle
(Figure 1.1A). The cell-cycle is generally regarded as being composed of two primary
phases: the interphase, comprised of subphases G1-, S-, and G2-, and the short mitotic
phase (M-phase).1 DNA replication occurs during the S-phase (S = synthesis) and cell
division (mitosis), resulting in the formation of two daughter cells, occurs in the
following M-phase. Two gap phases, G1- and G2- separate the S- and M- phases, to
provide time for growth and to ensure the fidelity and completion of these critical
events.
The control of the cell-cycle, through its various stages and more importantly its ability
to effectively respond to external and internal cellular stressors is governed by a
complex, highly-ordered web of protein-protein interactions, transcriptional activation,
phosphorylating and dephosphorylating events, and degradation processes (Figure
1.1B).2 As part of this control, several important checkpoints exist to safeguard against
and respond to aberrant processes.2,3 Some important checkpoints are the restriction
point, during the G1-phase; the DNA damage checkpoint, during the S-phase; the DNA
replication checkpoint, during the G2-phase; and the spindle-assembly checkpoint,
during the M-phase.2,3 The restriction point during the G1-phase signifies two important
events: the irreversible commitment to another cell-cycle and the independence of the
cell from extracellular growth promoting signals. Cells can also reversibly exit the G1-
phase to enter the dormant G0-phase, in which they remain metabolically active but are
not embarking upon cell division, or irreversibly exit the cycle such as during apoptosis,
which is programmed cell death, or terminal differentiation.1

Chapter 1: Introduction 3
Figure 1.1 The different phases of the eukaryotic cell-cycle (G1-, S-, G2- and M-phases), is regulated by a complex interplay of cellular pathways.
1.2 Molecular Targets in the Cell-cycle
1.2.1 Cell-cycle dysregulation, a hallmark of cancer
A prominent feature of many cancer types is the lack of cell-cycle control and the
inability to respond correctly to cellular abnormalities.2,4 Indeed many cancer types
correlate to aberrant regulatory pathways caused by genetic mutations, overexpressions
and deletions (Table 1.1).5 Therefore, a promising strategy towards the development of
novel anticancer agents is the development of inhibitors to re-establish cell-cycle
control.2,5,6 By targeting these altered pathways, that are essential for cancer survival
and growth, it is hoped that host toxicity would be minimized and also the possibility of
potentiating current conventional treatments through restoring a normal cell-cycle could
be realized.4 To this end, much research has been given throughout the literature
directed towards a variety of cellular targets, with a number of significant successes.
Paclitaxel (Taxol®) which is clinically used for the treatment of breast and ovarian
cancer, causes G2/M-phase arrest through stabilising microtubules and inhibiting their
depolymerization back to tubulin.5 Because of their relevance to this thesis, two out of
the array of cellular protein targets currently being pursued, the cyclin-dependent
kinases (CDKs) and the MDM2-p53 interaction, will be discussed now in more detail.
A B

Chapter 1: Introduction 4
Table 1.1 The occurrence of mutations affecting cell-cycle control in particular cancer types.4,7,8
Cancer Type Mutation
90% of non-small cell lung cancers Lack of functional Rb
50% of breast cancer Overexpression of cyclin D1
55% of gliomas and mesotriolomas 38% of pancreatic cancer
Deletion or mutation of p16
50% of all human cancers Mutation of p53
70% of breast cancer 90% of colorectal adenocarcinomas
Overexpression of bcl-2
20-30% of sarcomas Overexpression of MDM2
1.2.2 Cyclin-Dependent Kinases (CDKs)
Cyclin-dependent kinases (CDKs) are essential enzymes involved in the control of cell-
cycle progression and cellular proliferation.9 They impart control through their
regulation of host proteins via their active phosphorylation of critical serine and
threonine residues.10 The transient activation of these kinases, at specific cell-cycle
stages, ensures the correct timing and ordering of events required for cell-cycle
progression.11 CDK activity is largely regulated by the binding of their specific
activating cyclin12 or their CDK inhibitory proteins (CKIs).13,14 In addition to these
binding-interactions, other necessary post-translational modifications such as
phosphorylation and dephosphorylation events further contribute to the complex
regulation of CDK activity. Due to their importance in cell-cycle control, inhibitors of
CDKs are anticipated to possess therapeutic utility against a wide array of proliferative
diseases, especially cancer.6,15,16 Indeed many cancers have shown mutations that affect
the regulation of CDK activity.4,10
Currently, there are thirteen different CDKs and sixteen different cyclins that have been
described, which can form in excess of fifteen different active kinase complexes
(Figure 1.2).16,17 The main CDKs involved with the cell-cycle are CDK1 (aka in yeast
CDC2), CDK2, CDK4, CDK6 and CDK7.16,17 The substrates modulated by the activity
of CDKs include, tumor suppressor proteins (e.g., retinoblastoma (Rb) and p53),
transcription factors (e.g., E2F-DP1 and RNA pol II), replication factors (e.g., DNA pol
α and replication protein A), and organizational factors that influence cellular and
chromatin structures (e.g., nucleophosmin/B23 (NPM/B23), histone H1, lamin A, and

Chapter 1: Introduction 5
MAP4).6 Some interesting non cell-cycle roles of CDKs are neurite outgrowth and
neurone migration involving CDK5,18,19 and HIV-Tat-dependent transcription involving
CDK9,20-22 having implications in Alzheimer’s disease and HIV infection, respectively
(Figure 1.2).
Figure 1.2 The various different CDK/cyclin complexes and their functions.
The different phases of cell development are regulated by the transient activity of
specific cyclin/CDK complexes. G1-phase is controlled by D-type cyclins (D1, D2, and
D3), whose expression is initiated by growth factors, which primarily bind to CDKs 4
and 6. The major substrate for these G1-complexes is the retinoblastoma tumor
suppressor protein (Rb). In its hypophosphorylated form, Rb binds to the E2F/DP1
transcription factor and acts as a transcription suppressor. Upon phosphorylation by G1-

Chapter 1: Introduction 6
complexes, Rb dissociates from E2F/DP1, leading to the activation of S-phase genes
that encode for proteins including cyclin E, dihydrofolate reductase (DHFR),
thymidylate synthetase (TS), E2F and Rb.9,23 In late G1-phase, due to the previous
phosphorylation event of Rb, cyclin E is expressed and this activates CDK2. Cyclin
E/CDK2 further phosphorylates Rb resulting in a positive feedback loop that drives
cells through the restriction point, as independence from initial growth factors is now
attained, and into S-phase.9,23 Some additional cyclin E/CDK2 substrates involved with
replication processes, such as DNA and centrosome duplication, are DNA pol α,
replication protein A, histone H1, and nucleophosmin/B23.17 Exiting of the S-phase is
promoted by cyclin A/CDK2. This cyclin change incurs an alteration in substrate
specificity, from Rb to E2F/DP1. The phosphorylation of E2F/DP1 leads to the
inactivation of its transcription promoter activity. Failure to turn off E2F/DP1 in the S-
phase results in apoptosis. CDK1 in association with cyclins A and B complete the
cycle.23,24
These CDKs, which are “switched on” by their association with cyclins are also
“switched off” by the binding of CDK inhibitory proteins (CKIs). Similar to cyclins,
their expression and destruction is regulated, though by converse signals. Most
antiproliferative signals result in the induction of CKIs.13 Antiproliferative signals such
as senescence, the presence of extracellular anti-mitogenic factors like TGFβ, and the
induction of p53 at the DNA damage checkpoint, have been shown to induce the
expression of the respective CKIs, p16INK4a, p15INK4b and p21Cip1,WAF-1.13,17 There are
two major families of CKIs, the INK4 and the Cip/Kip family of CKIs. The CDK4 and
CDK6 monomeric units, or their complexes with cyclin D, are specifically inhibited by
the INK4 family of CKIs: p15INK4b, p16INK4a, p18INK4c, and p19INK4d.25 Whilst, the
Cip/Kip family of inhibitors, p21Cip1,WAF-1, p27Kip1 and p57Kip2, bind to all G1- and S-
phase CDK complexes and are important in p53- and TGFβ-mediated cell-cycle
arrest.14 Cip/Kip inhibitors function as assembly factors for CDK4 and CDK6
complexes but inhibit both CDK2 complexes and cyclin B/CDK1 through blocking
ATP-binding.9,14 Insights into how these inhibitors work have revealed that the CDK4
inhibitor p16INK4a can displace its assembly factor p27Kip1 and hence make it available to
inhibit cyclin E/CDK2 and cause a double G1-phase block.9
A host of X-ray crystallographic structures have been attained for CDKs. The majority
of X-ray crystal structures attained are in regard to CDK2. These include the apoenzyme

Chapter 1: Introduction 7
CDK2,26 CDK2-Mg2+ATP complex,26 CDK2 with an array of synthetic inhibitors,
cyclinA-bound-CDK2 complex,27 and the phosphorylated cyclinA-bound-CDK2
complex with p27Kip1.14 In addition, the crystal structures for CDK6 with p16INK4a,
p19INK4d,25,28 virus-encoded cyclin29 and a synthetic flavonol inhibitor30 and CDK5 with
a indirubin inhibitor have also been obtained.31 High homology is seen among CDK1-7
(40-75%) with a conserved catalytic core of approximately 300 amino acids common to
all eukaryotic protein kinases.32 CDKs possess the same fold and tertiary structure as
many other protein kinases e.g. the cyclic-AMP-dependent protein kinase (PKA).26
CDK4 shares the most similarity to CDK6 (70% homology) and only 45% homology
with CDK2.33 CDK6 and CDK2 share 50% homology. Crystallographic studies have
revealed that the monomeric CDK consists of two lobes, an amino-terminal lobe (~ 90
residues), which is β-sheet rich [containing 5 antiparallel β strands (β1-β5), and a single
large α helix (α1)] and a larger carboxy-terminal lobe (~ 200 residues), which is mainly
α-helical, [containing a pseudo-4-helical bundle (α2, 3, 4, 6), a small β-ribbon (β6-β8),
and two additional helices (α5-7)].26 A deep cleft featured between these two lobes is
the site of ATP-binding and catalysis.26
Figure 1.3 Crystal structure of the cyclin A/CDK2 complex. Cyclin A is coloured in magenta, CDK2 in cyan; ATP is shown as a ball and stick representation. Portions of CDK2 that undergo large conformational changes upon cyclin-binding are highlighted: the PSTAIRE (single-letter amino acids) helix in red and the T-loop in yellow.27
The kinase activity of CDKs is mainly dependent upon the binding of its corresponding
cyclin and the activating phosphorylation of a conserved threonine residue (Thr160) by
the CDK-activating kinase (CAK). The mode in which cyclins activate CDKs has been
elucidated through crystallographic studies comparing the structural changes produced
upon cyclin A binding to CDK2. Upon cyclin binding the important residue Glu51 (E51),

Chapter 1: Introduction 8
is brought into the active site and combined with Lys33 and Asp145, forms the necessary
catalytic triad conserved in all eukaryotic kinases.26,27 This catalytic triad co-ordinates
and orientates the phosphate groups of ATP and a magnesium ion into their optimal
active configuration. Also a flexible loop region on the C-lobe termed the “T-loop”
moves away from the active site, upon cyclin binding, to provide substrate access to
ATP and to expose the activating phosphorylation site located on this loop, Thr160.26,27
In addition to these conformational changes that have been discussed, other changes
such as the movement of the N- and C-lobes of the CDK unit and the exposure of
residues for inactivating phosphorylations by the dual-specificity Wee1 and Myt1
kinases have also been described.25
Figure 1.4 The cyclin-induced conformational changes that occur within the CDK unit, more specifically its PSTAIRE helix and T-loop, upon cyclin binding.13,27 The ribbon structures of CDK from the complex, coloured in cyan and in monomeric form, coloured in grey are superimposed upon one another. The inset highlights the change in position of Glu51 (E51) located on the PSTAIRE helix. The space-filled structures highlight the differences in substrate accessibility to ATP and CAK accessibility to Thr160 in the monomeric (middle) and cyclin-bound (bottom) CDK structures.

Chapter 1: Introduction 9
The development of CDK inhibitors, as a strategy to restore cell-cycle control and to
develop potential cancer therapeutics, has been widely researched and had some notable
successes.
Previously, Chen et al.34 showed that selective killing of cancer cells over normal cells
could be achieved through using a peptide inhibitor of CDK2. The peptides synthesised
were peptidomimetics of either an E2F-derived peptide (PVKRRLCL) or a consensus
peptide (PVKRRLFG) based on the cyclin/CDK2 binding motifs of CKIs p21 and p27.
Their hypothesis was that by inhibiting cyclin A/CDK2 they could achieve selective
killing of cells in which E2F already was deregulated by virtue of Rb inactivation. Their
reasoning was based on the fact that E2F activity needs to be inactivated by cyclin
A/CDK2 phosphorylation or else apoptosis is induced. By inhibiting cyclin A/CDK2,
this altered pathway, unique to the transformed cells, is made lethal.
However, the majority of the research throughout the literature has been devoted to the
design and development of ATP-competitive ligands for the highly conserved CDK
active site. First assumed as an unprofitable mode of inhibition, due to the difficulty, if
not impossibility, in producing selectivity within the wide number of protein kinases (ca.
2000) and hence avoiding toxicity, this view was challenged by a report in 1994. This
report described an exceptionally potent inhibitor of epidermal growth factor receptor
(EGFR) tyrosine kinase, which was ATP-competitive yet highly specific relative to
other receptor tyrosine kinases.35 Since then, a wealth of selective ATP-competitive
ligands has been produced, including CDK selective ligands. Within this subset,
specificity has even been demonstrated between different cyclin/CDK complexes. It is
therefore a realistic and feasible approach to inhibit cellular proliferation through the
design of ATP-competitive ligands. Indeed the number of these CDK antagonists
continues to steadily grow.
Initially, the development of CDK inhibitors was primarily devoted to CDK4 and
CDK6, but due to a few discoveries questioning their importance for tumor genesis and
the selective killing of transformed cells through targeting CDK2, described above, by
Chen et al.34 emphasis was shifted to targeting CDK2 complexes. Indeed the bulk of the
literature and the X-ray crystal structures attained involve the CDK2 enzyme. However,
recently, some surprising results have brought into question the validity of targeting
CDK2 for cancer therapeutics. In small-interfering (si) RNA experiments, depletion of
CDK2 failed to exhibit any cytostaticity on osteosarcomas and Rb-negative cervical

Chapter 1: Introduction 10
cancers.36 Though previously, a cyclin A knockout was found to be embryonically
lethal in mice, unlike the same scenario with cyclin D, recent results conflict with this
finding, showing that embryonic fibroblasts lacking CDK2 proliferate normally and
become immortal in culture.37 Furthermore a knockout CDK2 in mice (Cdk2-/-) gave
rise to viable (up to two years survival), albeit sterile, offspring.37 These results suggest
that possibly a more therapeutically useful strategy would be the development of non-
selective CDK inhibitors rather than ones selective for a particular CDK. Presently a
number of CDK inhibitors are undergoing clinical trials: flavopiridol38 (L868275) (13),
and 7-hydroxystaurosporine39 (UCN-01) (18) exhibiting general non-selective CDK
activity, CDK2-selective inhibitors: roscovitine40 (CYC-202) (1) and BMS-38703241
(17) and CDK4-selective inhibitor, PD033299142,43 (7). However, to date, no small
molecule inhibitor targeting the CDK function is in clinical use.
A diverse range of structures feature within the selection of ATP-competitive inhibitors
of CDK.44 These vary with regard to their specificity against the broader milieu of
kinases and between CDK types. The initial type of inhibitors were purine-based with
notable potent CDK1 and CDK2 selective inhibitors such as roscovitine 1,40 and one of
the most potent purine analogues, purvalanol B 2,45 which was discovered through a
rational design approach. Roscovitine 1, has additionally shown some promising in vitro
and in vivo antitumor properties with an average cytoxicity IC50 of 15.2 µM against a
panel of 19 human tumor cell lines and exhibiting in this screening a selectivity for
rapidly proliferating cells over non-proliferating cells and apoptosis induction. Also, 1
has been shown to reduce tumor growth (45-62%) in Lovo human colorectal tumor and
a human uterine xenograft MESSA-DXS.46 Some other purine-related heterocyclic
scaffolds exhibited in the CDK inhibitors include pyrimidines 5,47 pyridopyrimidines 6
and 7,42,48 diaminopyrimidine 8,49 quinazolines 9,50,51 and oxindoles 10.52 Oxindole 10
was additionally found to reduce alopecia in rats by 33-50%.53 Indirubin-type structures
11 and 12, have also been developed, targeting inhibition at CDK2, CDK5 and gSK-
3.54-56 Some structurally different ligands include flavopiridol 13 from the Indian plant
Dysoxylum binectariferum and its more potent mimic, the benzylidene-benzofuran-3-
one 14;57 butyrolactone 15 from Aspergillus species;58 the alkaloid hymenialdisine 16 a
constituent from several marine sponges;59 BMS-387032 17 which has also displayed
potent cellular cytotoxicity against the A2780 human ovarian cancer line (IC50 = 95 nM)
and a superior efficacy profile than flavopiridol in in vivo tumor and xenograft
models;41 staurosporine and its related analog UCN-01 18, both isolated from

Chapter 1: Introduction 11
Streptomyces species;39 alsterpaullone 19,60 and tricyclic structures like the
indenopyrazoles 20.61 Flavopiridol 13 and roscovitine 1 have additionally showed
interesting inhibitory activity against HIV-1.20-22 Some of the inhibitory activities for
compounds 1-20 are displayed in Table 1.2. It can be seen that a range of structures are
tolerated by the ATP-binding pocket of CDK2. However, some common features within
the CDK2 ligands include they are generally planar, low-molecular weight (< 600 Da),
hydrophobic heterocycles. Essential binding-interactions can also be drawn (Figure 1.5),
with hydrophobic interactions and hydrogen bond interactions, more specifically with
the backbone carbonyl (HBA) and amino side chain (HBD) of Leu83 and with the
backbone carbonyl group of Glu81. Selectivity towards particular CDKs can be achieved
by additional interactions with amino acids outside the ATP-binding pocket, such as
Gln131, Asp145, Lys33 and Asp86.
Figure 1.5 Schematic representation of the binding mode of some CDK inhibitors elucidated by X-ray crystal structural analysis. Yellow represents the ATP-binding pocket, green the hydrophobic region and red and blue represents hydrogen bond acceptor and donor interactions, respectively. a) ATP, b) staurosporine,62 c) dechlorinated flavopiridol,38 d) purvalanol B (2) e) NU6027 (5) f) indirubin-5-sulfate (12), g) hymenialdisine (16) and h) diarylurea derivative.39

dbev
Text Box
See print copy for table 1.2

Chapter 1: Introduction 13
1.2.2 MDM2-p53
The tumor suppressor protein p53 plays an essential role in the regulation of the cellular
response to stress through a complex interplay of proteins known as the p53 pathway, to
induce such events as apoptosis, cell-cycle arrest, DNA-repair pathways, differentiation
and senescence (Figure 1.6).66 p53 is the most frequently altered protein in human
cancer, highlighting its importance in normal cell-cycle regulation.67,68 In approximately
50% of all human cancers, its function has been made void due to deletions or
mutations occurring in its DNA-binding domain.68 Through DNA-binding and
transcriptional activation of multiple-target genes, p53 solicits the suppression of
oncogenesis. The influence that the loss of p53 has is manifested in p53-deficient mice,
where an increased tumorogenesis and greatly reduced survival rate was incurred.69
Figure 1.6 The essential p53 response to cellular stress.70
Regulation of p53 has been described at the level of transcription, translation,
conformational change, and various covalent and non-covalent modifications.71
However one of the key mechanisms of p53 regulation is through control of its stability.
Integral to this is its autoregulatory feedback inhibitor, MDM2 (or its human analogue,
HDM2). MDM2 inhibits p53 activity in multiple ways (Figure 1.7).72 First, MDM2
binds to the N-terminus of p53, overlapping with the transcriptional activation domain
of p53.73,74 Secondly, MDM2 also contains a RING finger domain and hence can
function as an E3 ubiquitin ligase for p53 and target its ubiquitin-dependent
proteasomal degradation.75,76 Finally, MDM2 also plays a role in regulating the

Chapter 1: Introduction 14
subcellular localization of p53 through nuclear export.77,78 In addition to its role in p53
regulation, MDM2 has functions that are independent of p53.79
Figure 1.7 The regulation of p53 by MDM2.70
Indeed the interplay of MDM2 and p53 was manifested by the discovery that MDM2
deficiency causes early embryonic lethality in mice and furthermore that this event can
be rescued by the simultaneous deletion of the TP53 gene.69,80 MDM2 overexpression
has also been shown to block p53-mediated cell-cycle arrest and apoptosis.81 These
results combined indicate that unrestrained p53 activity blocks normal growth and
development and highlights the essential role of MDM2 in p53 regulation. Furthermore,
amplification of MDM2 has been observed in more than forty different types of
malignancies, including solid tumors, sarcomas and leukemias, that retain wild-type p53
(WT-p53).82-84 In soft tissue sarcomas, the highest levels of MDM2 overexpression is
observed (20-30%).7,8 More importantly, overexpression of MDM2 may be related to
increased metastases, resistance to anticancer drugs and the pathnogenicity of
HIV.83,85,86
MDM2 is therefore a promising novel target for cancer therapeutics, with inhibitors
potentially re-establishing p53 function and hence its tumor suppressor effects in WT-
p53 tumors.70,87-89 Though gene therapy has been exploited to this end,88,90 the rest of
this discussion will be concerned about the development of small molecule inhibitors of
MDM2-p53.

Chapter 1: Introduction 15
The inhibition of protein-protein interactions have been long considered difficult for
therapeutic intervention by small molecules.91-93 This is mainly due to the fact that their
interacting surfaces are usually too large and flat for effective disruption by drug-like
components. Structural insights into the MDM2-p53 interaction were revealed by the X-
ray crystal structure of a conserved segment from the transactivation domain of p53
(residues 15-29) bound to MDM2 and through genetic and biochemical studies.94,95 The
crystal structure revealed a relatively deep cavity on the surface of the MDM2 protein
(Figure 1.8). More importantly, three conserved amino acid residues (Phe19, Trp23, and
Leu26) residing on an α-helix of p53, were revealed to project deeply within this
hydrophobic cavity and found to play a critical role in the binding interaction through
amino acid substitution studies (Figure 1.8). Within this complex, there are no salt-
bridges, and only three intermolecular hydrogen bonds, with the stability of the complex
therefore, primarily being due to hydrophobic interactions (70% of the atoms at the
interface are non-polar).70 These advantageous features of a well-defined pocket, and
structural elucidation of the key binding interaction, made the MDM2-p53 complex an
attractive target for protein inhibition and revived research for inhibitors of protein-
protein-interactions.
Figure 1.8 The X-ray crystal structure of the complex of MDM2 and segment of p53.94
Initial efforts and successes in designing inhibitors, were focused on the development of
peptidomimetics of the binding domains of p53 (Table 1.3, entries 1-6). Initial phage
studies, revealed a potent 12-mer natural peptide derivative of p53 that was found to
bind to MDM2 with a 29 times greater efficacy (IC50 = 0.3 µM) than the natural WT-
p53 peptide (Table 1.3, entry 2 c.f. entry 1).96 Decreasing the peptide to the 8-mer
(Table 1.3, entry 3) gave comparable binding to that of the WT-p53. The incorporation

Chapter 1: Introduction 16
of non-natural amino acids such as α-amino isobutyric acid (Aib) and 1-
aminocyclopropanecarboxylic acid (Ac3c) were found to increase binding-affinity by
improving structural organisation of the amino acids for a closer fit (Table 1.3, entries 4
and 5). The increased functionality of natural amino acids with the use of
phosphonomethylphenylalanine (Pmp) for Tyr22 and 6-chlorotryptophan (6ClTrp) for
Trp23 was found to greatly increase potency, by exploiting new binding interactions and
structural space (Table 1.3, entries 5 and 6). The former replacement resulted in an
additional salt bridge with MDM2 and yielded an IC50 of 0.3 µM, whilst the latter was
found to occupy an additional small hydrophobic pocket to attain the most potent
peptide inhibitor of MDM2-p53 to date, with an IC50 of 5 nM. Recently this 8-mer was
co-crystallised with MDM2 and the structure solved by X-ray diffraction (Figure 1.9).97
The X-ray crystal structure indeed revealed a greater complementarity of this 8-mer
with the MDM2 pocket compared with the WT-p53 peptide.
Figure 1.9 A sectional viewof the X-ray crystal structure, revealing the MDM2 binding site bound to a) WT-p53 and b) 8-mer peptide.97
More recently, other potent natural-derived and non-natural synthetic peptides have
come to light. Appella et. al.98 have synthesised a series of non-natural peptides with
their most active analogue inhibiting HDM2-p53 with an IC50 of 6.6 ± 0.7 µM (Table
1.3, entry 7). Through the use of protein-grafting, Schepartz et. al.99 were able to
synthesise a 40-mer natural peptide (Table 1.3, entry 8) which bound to HDM2 with an
IC50 of 1.6 ± 0.2 µM. The fungal metabolite, chlorofusin (21) was also found to be an
MDM2-p53 inhibitor through ELISA studies, with an IC50 of 4.6 µM (Table 1.3, entry
9).53

dbev
Text Box
See print copy for figure 1.3

Chapter 1: Introduction 18
However, the design of non-peptide, small molecule, MDM2-p53 inhibitors has been an
area with little success until quite recently. The first low molecular weight inhibitor
reported of the MDM2-p53 interaction were the chalcones (e.g. 22) (Table 1.3, entry
10), found to bind to the p53-transactivation domain using ELISA, with an IC50 of 117
µM. Recently, more potent chalcone boronic acid derivatives (e.g. 29) were synthesised
in an effort to form a stronger salt-bridge with Lys51 of MDM2 (Figure 1.10). These
derivatives exhibited low micromolar activity against various breast cancer cell lines
and had significant selectivity over normal breast cell lines.108
O
O
B
OH
I
OH
29
Cell Line MDA MB-435 MDA MB-231 MCF-7 MCF-10A MCF-12A IC50 18 µM 11 µM 9.5 µM 38 µM 100 µM
Figure 1.10 Inhibition values for the chalcone derivative 29 and its against various human breast cancer cell lines and the normal breast cell line, MCF-10A and MCF-12A.108
Using a high-throughput direct binding assay technique, named ThermoFluor, a lead
1,4-benzodiazepine-2,5-dione was found to bind to HDM2.101,109,110 Optimization
studies yielded an analogue (23a) with a potency of 0.42 µM (Table 1.3, entry 11a) that
was also co-crystallised with the HDM2 protein (Figure 1.11). This revealed that the
iodophenyl group of 23a was able to occupy a third hydrophobic pocket. Recently,
further optimization studies has lead to the more potent benzodiazepinedione 23b with
an IC50 of 0.25 µM (Table 1.3, entry 11b).102
Figure 1.11 Overlay of the benzodiazepinedione 23a (yellow) with a 9-mer peptide, with critical amino acids, Phe19, Trp23 and Leu26 highlighted in green.101

Chapter 1: Introduction 19
Screening of chemical libraries have yielded some potent and novel molecules. The first
discovery was a small molecule termed “RITA” (24) (Table 1.3, entry 12), which binds
interestingly not to MDM2 but to p53 and causes its increased accumulation in tumor
cells.103 This analogue was found to prevent the HDM2-p53 interaction in vitro and in
vivo in various cell lines and tumors possessing WT-p53, preventing the
ubiquitinylation of p53, transcriptional activation of p53-target genes and apoptosis.
Development of a QSAR model based on MDM2-p53 peptide inhibitors, was used to
screen an NCI database, yielding the sulfonamide 25 (Table 1.3, entry 13) which
showed a dose-dependent inhibition of MDM2 with an IC50 of 31.8 µM.104
By far the most exciting discovery using chemical library screening was the
identification of three cis-imidazoline derivatives 26 (Table 1.3, entry 14) that could
inhibit MDM2-p53 binding.67 These compounds termed “Nutlins” exhibited the first
significant nanomolar activity (90-300 nM) (Table 1.3, entry 14). Chirality was found
to be an important aspect to activity with approximately a 150- to 200-fold difference in
affinity found between enantiomers. The X-ray crystal structure of one of these
analogues, Nutlin-2 (26a) (Table 1.3, entry 14a), was also attained (Figure 1.12) and
verified that the Nutlins were able to occupy the MDM2-binding site and mimic the
three critical residues necessary for binding of p53 binding.67 Furthermore, the different
binding orientation of the Tyr100 residue of MDM2 when binding the Nutlins compared
to the WT-p53, revealed a certain degree of flexibility within the MDM2 pocket and
additional structural space that could be possibly exploited in the future for novel
binding interactions.111 Nutlin-3 (26b) (Table 1.3, entry 14b) was found to be the most
active with an IC50 of 90 nM. In addition, Nutlins were found to exhibit selectivity for
tumors possessing WT-p53 and to activate the p53 pathway. Finally, Nutlins were able
to be orally administered to nude mice bearing human tumor xenografts. The growth of
the osterosarcoma SJSA-1 tumor, which bears an amplified mdm2 gene, was reduced by
90% after a 20 day treatment with racemic Nutlin-3 (26b).105 Purification of the active
enantiomer of 26b, increased the potency of the compound 2-fold and showed over
100% tumor growth inhibition in the SJSA-1 tumor model and also a prostate xenograft
model, LNCaP.67

Chapter 1: Introduction 20
Figure 1.12 A) X-ray crystal structure of the Nutlin-2 (26a) (yellow) bound to MDM2 (red), B) Nutlin-2 (topaz) overlayed with the three critical residues (green) of the p53 peptide.67
By far, the most potent, cell-permeable, non-peptide, small molecule inhibitors to date
are the spirooxindoles 27 (Table 1.3, entry 15).106,112 These derivatives, discovered by a
structure-based, de novo design, possess structural features similar to that of the natural
product, spirotryprostatin B (30). These spirooxindoles boast in low nanomolar activity
against MDM2 and HDM2 proteins and an effective cell-growth inhibition in WT-p53
cancer cells over ones where p53 is deleted. They also showed minimal toxicity to
normal cells. Optimisation studies yielded the most potent derivative 27 (R = F) (IC50 =
3 ± 1.5 nM), with a 12-fold increased potency over Nutlin-3 (26b) and 3 orders of
magnitude greater binding affinity for MDM2 than the natural substrate, p53. A 3-fold
potency was seen in the cell-growth inhibition of LNCaP cells with WT-p53 and a dose-
dependent increase in levels of p53 was observed. The X-ray structure of analogue 27
(R = H, X = O) bound to MDM2 was attained (Figure 1.13). In addition to mimicking
the three critical amino acids of p53, it exploited another over-looked critical residue,
Leu22. In addition the oxygen atom of its morpholino functional group was in close
enough proximity to form a H-bond interaction with the charged amino group of Lys90
residing on the MDM2 protein.
Also recently, the structurally similar isoindolines (e.g. 28) (Table 1.3, entry 16) were
shown to exhibit low micromolar MDM2-p53 inhibitory binding activity, with an IC50
of 15.8 ± 0.8 µM.107

Chapter 1: Introduction 21
NH
HN X
NH
O
N
Cl
Cl
O
HN N
NO
O
O
H
Spirotryprostatin B
R
27 (R = H, X = O) 30
Figure 1.13 A) X-ray crystal structure of the MDM2-p53, highlighting critical residues of p53 (magenta) in this binding interaction (Phe19, Trp23, Leu26 and Leu22) B) and C) Predicted binding mode of spirooxindole (IC50 = 13 nM) (white) using the GOLD program. Hydrogen bonds (yellow dashed line). C) Overlay of model binding of spirooxindole with X-ray crystal structure.
Another field which is emerging is the development of inhibitors of the ubiquitin ligase
activity of HDM2. Small molecule inhibitors such as 31 have shown inhibition of this
type (IC50 of ~ 20 µM).113
HN
N N
NO2
O
O
31
In conclusion, the MDM2-p53 interaction has proven to be a successful target for
developing novel cancer therapeutics, with potent peptide and small molecule non-
peptide inhibitors able to be developed with nanomolar activity. Additionally, it has
been shown that inhibiting this interaction can increase levels of p53 and in some cases
induce apoptosis and reduce tumor growth in WT-p53 cancer cell lines. Preliminary
work has also revealed that MDM2-p53 inhibitors are more toxic for tumor cells than
for normal cells.114
A C B
a

Chapter 1: Introduction 22
1.3 Naturally Occurring Spirocyclic Oxindoles
A range of naturally occurring biologically active compounds possessing the
spiro[pyrrolidine-3,3`-oxindole] ring system have been isolated from plant, marine and
fungal sources (Table 1.4). In general, these oxindole alkaloids possess the common
basic framework derived from tryptamine, and are characterized by their unique spiro
fusion to a pyrrolidine ring at the 3-position of the oxindole core.
The simplest of all oxindole spirocycles to be found in Nature is (+)-coerulescine (32)
isolated from the blue canary grass, Phalaris coerulescens (Table 1.4).115 Various types
of the Phalaris species have been associated with livestock toxicity.116,117 Related
structures are (-)-horsfiline (33) isolated from a small Malaysian-indigenous tree,
Horsfieldia superba (Myristicaceae), elacomine (34) isolated from the roots of the shrub,
Elaegnus commutata and salacin (35) isolated from the plant, Uncaria salaccensis
(Table 1.4). Though many synthetic studies have been undertaken for these simple
analogues, especially for 33, no biological activity studies have been reported.118 The
only insight into the biological activity is that (±)-32 displays a moderate local
anaesthetic effect.119 Rhychnophylline (36) isolated from Uncaria sinensis has been
shown to have neural protective effects from dopamine-induced apoptosis in NT2
cells,120 and from glutamate-induced neuronal death in cultured rat cerebellar granule
cells.121 Pteropodine (37) and its C-3-epimer, isopteropodine are heteroyohimbane-type
oxindole alkaloids isolated from the plant Uncaria tomentosa, a Peruvian medicinal
plant known as “cat’s claw”. Traditionally, this plant has numerous medicinal
applications, including the treatment of arthritis, asthma, digestive and inflammation
disorders.122 A neuropharmacological profile showed that the total alkaloid content of U.
tomentosa produced an ameliorative effect on the central cholinergic system in mice.123
Indeed, 37 and isopteropodine have been further shown to positively modulate the
function of rat muscarinic M1 and 5-HT2 receptors expressed in xenopus oocytes.124
Cellular studies have also revealed that 37 has a cytotoxic effect and induces apoptosis
in human lymphoblastic leukaemia T-cells (CCRF-CEM-C7H2).125

Chapter 1: Introduction 23
Table 1.4 Naturally occurring spirocyclic oxindoles
Natural product: structure, name and source of origin. Activity Total
Synthesis
NH
NMe
O
32
(-)-Coerulescine from blue canary grass, Phalaris coeruscelens.
115
*Source has reported livestock toxicity.116,117 *Racemate has moderate local anaesthetic activity.119
Racemate126-129
NH
NM e
MeO
O
33
(-)-Horsfiline from Malaysian plant, Horsfieldia superba.
118
Not tested
Racemate118,126-
128,130-134 Asymmetric135-
137
NH
NH
O
HO 34
(±)-Elacomine from the roots of the shrub, Elaegnus commutata.
138
Not tested Racemate138-140
NH
N
O
CHOCOEt
35
(±)-Salacin from Uncaria salaccensis.141
Not tested Racemate141
HN N
O
Et
HCO2M e
MeO
36
Rhychnophylline from Uncaria sinensis.142-144
Protective neural effects120,121
Racemate145-147
37
Pteropodine isolated from a number of Uncaria species including U. bernaysii, U.
ferrea, U. pteropoda and U. tomentosa.122
*Positive effect at rat muscarinic (M1) and 5-HT receptors.124 *Cytostaticity in human lymphoblastic leukaemia T-cells and induced apoptotis.125 No cytotoxicity or genotoxicity in mouse model.
Racemate148

Chapter 1: Introduction 24
Natural product: structure, name and source of origin. Activity
Total
Synthesis
NH
O
N
HO
OO
NHMe
R
Citranadin A (38a) Citrinadin B (38b) from
marine fungi, Penicillium citrinum.149,150
Citrinadin B shows modest cytotoxicity against murine leukemia L1210 cells (IC50, 10 µg/ mL).150
Not reported
NH
O
C
H
H
NH
O
H
H
Me
O
39
Alstonisine from Alstonia muelleriana Domin151 and Alstonia angustifolia Wall.152
Not tested Asymmetric153
HN
N
HO
O H H
MeN NHH
40
Strychnofoline from the leaves of Strychnos usambarensis.
154
Antimitotic activity against cultures of mouse melanoma and Ehrlich tumor cells.155
Racemate156
41
Vinblastine from the periwinkle Catharanthus roseus.
157
Clinical cancer therapeutic against a range of cancer types, causing G2/M-phase arrest by preventing microtubule formation.157,158
Racemate159,160
38a: R = H 38b: R =
O
O
N

Chapter 1: Introduction 25
Natural product: name, structure, and source of origin. Activity Total
Synthesis
NMe
N
OO
MeO
OMe 42
Aspidophytine from the leaves of the plant, Haplophyton cimicidum.161
Insecticide162 Racemate162
N
O
NH
O
OMe
H
H
43
Gelsedine from South-east Asian plant Gelsemium elegans Benth.
(Loganiaceae).163,164
*Analgesic, anti-inflammatory, and anti-tumor activities.165-167 *Cytototoxicity against A431 human epidermoid carcinoma cells.168
Racemate169,170 Asymmetric171,172
HN N
N
MeO
O
O
OH
H
44
Spirotryprostatin A from the fermented broth of Aspergillus fumigatus.
173
*G2/M-phase arrest in tsFT210 cells (MIC = 197.5 µM).173
Racemate174-177 Asymmetric178,179
HN N
NO
O
OH
30
Spirotryprostatin B from the fermented broth of Aspergillus fumigatus.
173,180
*G2/M-phase arrest in tsFT210 cells (MIC = 14.0 µM).173 *Growth inhibition of human chronic myelogenous leukemia K562 cells (35 µg/ mL) and human promyelocytic leukemia HL-60 cells (10 µg/ mL).
Racemate175,176,181
,182 Asymmetric183-189

Chapter 1: Introduction 26
Citrinadin A and B (38a,b) are pentacyclic spiroindolinone alkaloids isolated from the
cultured broth of the marine fungi, Penicillium citrinum, separated from the red alga,
Actinotrichia fragilis, and collected at Hedo Cape, Okinawa Island (Table 1.4).149,150
Although several spiroindolinone alkaloids such as brevianamides,190,191
paraherquamides,192,193 and sclerotamide,194 have been isolated from the fungi of the
genuses Penicillium or Aspergillus, Citrinadin A and B belong to a novel class of
alkaloids with its pentacyclic skeleton and epoxy isoprene unit. Citrinadin B (38b)
shows modest cytotoxicity against murine leukaemia L1210 cells (IC50 = 10 µg/ mL).150
Alstonisine (39) is a spirocyclic oxindole alkaloid isolated from Alstonia muelleriana
Domin151 and Alstonia angustifolia Wall.152
Strychnofoline (40) is the most potent analogue of a class of natural alkaloids isolated
from the leaves of Strychnos usambarensis,154 which displayed antimitotic activity
against cultures of mouse melanoma and Ehrlich tumor cells (Table 1.4).155 The first
total synthesis of 40 was reported in 2002.156
The vinca alkaloids, though not spirocyclic in nature, in which vinblastine (41) is a
representative, are a subset of drugs derived from the periwinkle plant, Catharanthus
roseus. Traditionally used for the treatment of diabetes, their current use is as clinical
cancer therapeutics.157,158 The vinca alkaloids have been shown to cause cell-cycle
G2/M-phase arrest and induce apoptosis, through the prevention of microtubule
formation.157,158 Without proper microtubule formation, chromosomes cannot be aligned
and cell division was impossible. The alkaloid aspidophytine (42) is isolated from the
leaves of the plant Haplophyton cimicidum and is used traditionally for its insecticidal
properties (Table 1.4).162
Gelsedine-type indole alkaloids are isolated from the “toxic” plant, Gelsemium elegans
Benth. (Loganiaceae), which is widely distributed in South-East Asia and used in
traditional Asian medicine as a remedy for certain kinds of skin ulcers and
dermatitis.168,195-198 Analgesic, anti-inflammatory, and anti-tumor activities of these
alkaloids have also been described.165-167 The cytototoxicity of selected Gelsemium
alkaloids against A431 human epidermoid carcinoma cells revealed some exhibited
more than 4-fold lower activity than that of cisplatin (Table 1.5).168

Chapter 1: Introduction 27
Table 1.5 Cytotoxicity against A341 human epidermoid carcinoma cells.168
N
O
NH
O
OMeRN
O
N
O
OMe
R1
R2
H
H
H
H
45 46
Compound EC50 (µµµµM) Gelsenicine 45 (R1 = H, R2 = H) 37
45 (R1 = OAc, R2 = H) 0.25
45 (R1 = OAc, R2 = OH) 36
45 (R1 = OH, R2 = OH) 1.3
Gelsedine 46 (R = H) 0.35
Gelsemicine 46 (R = OMe) 0.75
Cisplatina 3.5 a Positive control
Spirotryprostatins A (44) and B (30) were isolated from the fermentation broth of
Aspergillus fumigatus BM939 and were shown to cause G2/M phase cell-cycle arrest in
tsFT210 cells at MICs of 197.5 µM and 14.0 µM, respectively (Table 1.4).173
Spirotryprostatins A and B, were isolated from 400 litres of fermentation broth in
quantities of 1 mg and 11 mg, respectively. Spirotryprostatin B (30) has also been
shown to inhibit growth of human chronic myelogenous leukemia K562 cells and
human promyelocytic leukemia HL-60 cells with MICs of 35 and 10 µg/ mL,
respectively.184
Related structures, tryprostatins A and B, 47 and 48, respectively, were also isolated
from the fermented broth of Aspergillus fumigatus BM939 and caused G2/M phase cell-
cycle arrest at higher MICs of 125 µM and 62.5 µM, respectively.173,180 Tryprostatin A
(47) has also been shown to be a specific and novel inhibitor of microtubule
assembly199,200 and to reverse protein-mediated drug-resistance in breast cancer.201 A
diastereomer of tryprostatin B (48) has also been shown to exhibit cytostaticity at a
concentration of 100 µM against the human cancer cell lines, MCF-7 (breast), H520
(lung) and PC-3 (prostate), greater than that that of the known cancer therapeutic,
etoposide.202

Chapter 1: Introduction 28
NH
NH
NO
O
H
H
R NH
NH
NO
O
H
H
H
47; Tryprostatin A (R = OMe)
48; Tryprostatin B (R = H)
49
In the pursuit of trying to understand the reason behind the difference in activities
between spirotryprostatins A (44) and B (30) and tryprostatins A (47) and B (48), the
synthesis towards the demethoxy analogue of spirotryprostatin A was successfully
achieved by Danishfeshky et al.177 This analogue however exhibited similar biological
activity to the natural product (Table 1.6, entry 2). Surprisingly, simpler starting
analogues, 50 and 51, were extremely potent cell-growth inhibitors of the breast cancer
cell line, MDA-MB-468, being up to 3 to 4 orders of magnitude more potent than
spirotryprostatin A (Table 1.6, entry 6). The simplicity of analogue 51 is undeniable,
with only three steps required for its synthesis. However, to date, their mode of
inhibition has not been elucidated.
Table 1.6 Cytotoxicity against MDA-MD-468 and MCF-7 breast cancer cell lines.177
HN N
N
R
O
O
OH
H
HN N
N
OBn
O
O
O
H
H
HN N Boc
OBn
R
OCO2Me
3 33
44 50 51
IC50 (µµµµM) Entry Compound
MDA MB-468 MCF7 1 (3S)-(+)-44 (R = OMe) 110 >> 300 2 (3S)-44 (R = H) 100 >> 300 3 (3R)-(-)-44 (R = OMe) 85 >> 300
4 (3R)-50 0.025 100
5 (3S)-50 0.02 80
6 51 0.02 40

Chapter 1: Introduction 29
As discussed previously, novel spirocycles by Wang et al.106,112 have shown to be potent
nanomolar MDM2-p53 inhibitors (Table 1.7). Derivatives 27 (R = H, X = O) and
(5`R)-27 (R = F, X = O) were also shown to inhibit LNCaP prostate cancer cells with
WT-p53 with an IC50 of 800 nM and 280 nM, respectively, over cancer cells with
deleted p53 and over normal cells. The most potent analogue, (5`R)-27, showed an
inhibition of four orders of magnitude greater selectivity for the MDM2 protein over
Bcl-2/Bcl-xL proteins.
Table 1.7 Inhibition of MDM2-p53 interaction.106
( R)
5` NH
HN X
NH
O
N
Cl
Cl
O
R
NH
N
NH
O
Cl
Cl
O
R
52 27
Compound Ki ±±±± SD (nM) 52 (R = H) 86 ± 20 52 (R = 2``-F) 38 ± 15 52 (R = 4``-F) 170 ± 70
52 (R = 5``-F) 44 ± 9
27 (R = H, X = O) 13 ± 4
(5`R)-27 R = F, X = O 3 ± 1.5
(5`S)-27 R = F, X = O 4000 ± 500
27 R = F, X = CH2 39 ± 5

Chapter 1: Introduction 30
1.4 Aims of this study
The aims of my project were to synthesise a range of novel spirocyclic oxindoles (C-E)
and related structures for cytotoxicity analysis against cancer cell lines, in particular
MCF-7 breast cancer cell line, and protein inhibition studies against cell-cycle proteins,
CDK and MDM2-p53. These different spirocyclic scaffolds were envisioned to be
accessed through various cycloadditions reactions from alkenes of type A or B (Scheme
1.1). The 2-azaspiro[4.4]nonan-1-ones and spiro[cyclopentante-1,1`-[1H]isoindol]-
3`(2`H)-ones (C) were proposed to be accessed through a phosphine-catalysed [3+2]-
cycloaddition reaction with A or B, respectively. The spiro[cyclopropane-1`,3-
[3H]indol]-2`(1H)-ones (D) were envisioned to be accessed through cyclopropanation
reactions involving sulfur ylides. Lastly, the spiro[indole-3,5`-isoxazolidin]-2(1H)-ones
(E) were planned to be accessed through the [1,3]-dipolar cycloaddition reactions of B
with nitrones. An additional aim of this study was to conduct a six month project,
related to the design and synthesis of novel CDK2 small molecule inhibitors, in
collaboration with the a cancer research laboratory, Anti-cancer Drug Design Initiative
(ADDI), based in the University of Newcastle upon Tyne, UK.
Scheme 1.1
NH
O
CPhosphine-catalysed[3+2]-cycloaddition
CyclopropanationSulfur ylide
R
R

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 31
CHAPTER 2: SYNTHESIS OF 2-
AZASPIRO[4.4]NONAN-1-ONES AND
SPIRO[CYCLOPENTANE-1,1`-
[1H]ISOINDOL]-3`(2`H)ONES USING THE
PHOSPHINE-CATALYSED [3+2]-
CYCLOADDITION REACTION
Spirotryprostatin B isolated from Aspergillus fumigatus exhibits a G2/M phase arrest on
tsFT210 cells with an MIC of 14.0 µM.173

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 32
The phosphine-catalysed cycloaddition reaction of ethyl buta-2,3-dienoate or ethyl 2-
butynoate with electron-deficient alkenes has been established as a useful method for
preparing substituted cyclopentenes203-213 both in racemic and enantio-enriched forms
(General Scheme and Scheme 2.1).214 However, only a few examples of preparing
spiro-heterocyclic derivatives using this method have been reported.207,211-213 The
[3+2]-cycloaddition proceeds through the initial formation of an ylide (54) in situ, via
the nucleophilic attack of the phosphine (R3P) followed by proton transfer from the
allylic, γ-position to the α-position to give a resonance stabilized allylic anion. The
ylide 54 then undergoes a [3+2]-cycloaddition reaction with the electron deficient olefin
(CH2=CH-E) to form the cyclic intermediates 55a and 55b which are in equilibrium
with 56a and 56b, respectively. The reaction yields the two regioisomers, A or B and
the phosphine catalyst is regenerated by an elimination mechanism. Therefore we
envisaged that using either the 2-methylene γ-lactams 57210 or 58215 or the acrylate 59,
racemic and enantio-enriched versions of the 2-azaspiro[4.4]nonan-1-one and
spiro[cyclopentane-1,1`-[1H]isoindol]-3`(2`H)-one ring systems could be accessed
(Scheme 2.1).216 During the initial phase of our study, Lu et al.210 reported the
triphenylphosphine-catalysed cycloaddition reaction of 57 and 53a.

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 33
General Scheme
54b
Me
O
X
X
OPR3
53E
R3P
54
X
OR3P
COX COX
E
E
H+ transfer
X
OPR3
X
OPR3
E
COX COX
E
E
R3P R3P
+
+
H+ transfer
COX COX
E
E
R3P R3P
R3P
+
+
54a
regioisomer A regio isomer B
55b55a
56b56a
54c
X
OPR3

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 34
Scheme 2.1
SO
O
N
54
Me
O
X X
OBu3PBu3P
CO2Me
NO 2
R
N
O
Boc
N
O
R1
COX
Zn/H+
59
53
57; R = H58; R = CO2Et
b ; X =
a; X = OEt
c; X = N(PM B)Ph
2.1 Synthesis of 2-azaspiro[4.4]nonan-1-ones
2.1.1 Synthesis of 2-methylene γγγγ-lactams 57 or 58
The 2-methylene γ-lactams 57 and 58 were prepared using a method obtained from the
literature.215 The N-Boc protected lactams 60 and 61, were treated with LiHMDS (1.2-
1.5 eq.) in THF at -78 °C for 45 min and the resulting enolate was treated with
Eschenmoser’s salt (N,N-dimethylmethylene ammonium iodide). Methylation of the
resulting dimethylaminomethyl derivative formed the corresponding ammonium salt,
which collapsed to the desired 2-methylene γ-lactam upon base treatment as shown in
Scheme 2.2.

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 35
Scheme 2.2a
N
O
BocR
57; R = H (45%)
58; R = CO2Et (64%)
N
O
BocRN
O
BocR
N
O
BocRN
O
BocR
N I
N
CH3I
N
H
B
a)
d)
60; R = H
61; R = CO2Et
b)
c)
a Reagents and conditions: (a) LiHMDS (1.2-1.5 eq.), THF, -78 °C, 45 min 67% b)
Eschenmoser’s salt, N2, -78 °C�RT, 18 c) MeI (8 eq.), anhydrous DCM, 18 h d) sat. NaHCO3.
Initial model reactions to optimise yields were performed on 60. The conditions that
yielded an optimised yield for 57 of 45% were as follows. In the initial deprotonation
step it was found that the use of 1.2-1.5 equivalents of fresh lithium
hexamethyldisilazane was more effective than the use of potassium
hexamethyldisilazane or lithium diisiopropyl amide. The quality of the light and
moisture-sensitive Eschenmoser’s salt was also found to be critical, with decomposition
ascertained by brown discolouration. It was found that the desired alkene 57 could be
furnished upon washing of the ammonium salt intermediate with a saturated solution of
sodium bicarbonate. Using similar conditions, 61 was converted to 58 in 64% overall
yield.
2.1.2 [3+2]-cycloaddition reactions using the 2-methylene γγγγ-lactam 57
Scheme 2.3 (Compounds 62 and 63 are racemic)

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 36
The reaction of 57 with ethyl 2-butynoate (2 eq.) and TBP (1 eq.) in benzene solution at
RT for 15 h gave a mixture (ca. 80 : 20) of two racemic regio-isomeric cycloadducts, 62
and 63, that were isolated in yields of 51% and 21%, respectively, after column
chromatography (Scheme 2.3). We found that the use of a stoichiometric amount to
TBP was required to obtain good conversion to 62 and 63. The structures of 62 and 63
were confirmed by extensive 2D NMR experiments.
From molecular modelling studies (Spartan ’04 (AM1)) on 62 the dihedral angles (φ)
for C-1, H-6α and C-1, H-9α were found to be close to 90°, indicative of a very small
(zero) 3-bond coupling 1H-13C coupling. Whereas the dihedral angles calculated for C-
1, H-6β and C-1, H-9β were calculated to be ~25°, indicative of a larger 3-bond 1H-13C
coupling (Figure 2.1). The modelling calculations concurred well with the strong cross-
peaks observed in the gHMBC in 62 between C-1 (δ 175) with H-6β (δ 3.04) and H-9β
(δ 2.71). Indeed, these signals at δ 3.04 and δ 2.71 were confirmed from gHSQC
experiments to be protons on different carbons. The gCOSY and TOCSY experiments
provided further differentiation of these protons, through H-9β (δ 2.71) exhibiting a
stronger, three-bond proton-proton coupling to the olefinic proton compared to the
weaker, four-bond proton-proton coupling of H-6β (δ 3.04). These experiments allowed
the unequivocal assignments of the four allylic protons as H-6β (δ 3.04), H-9β (δ 2.71),
H-6α (δ 2.13), and H-9α (δ 1.73). In the gHMBC spectrum of 63, the lactam carbonyl
carbon at δ 177 showed cross-peaks with the methylene protons on C-3, C-4 and C-9
but not to those on C-8.

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 37
δδδδ 3.04 (dq, J 16.5, 2.5 Hz) H-6666ββββ
δδδδ 2.13 (d, J 16.5 Hz) H-6αααα
H-9ββββ δδδδ 2.71 (dq, J 18.5, 2.5 Hz)
H-9αααα δδδδ 1.73 (d, J 18.0 Hz)
gHMBC cross-peaks
φφφφ (C-1, H-6ββββ) = 24.76o
φφφφ (C-1, H-6αααα) = -97.42o
φφφφ (C-1, H-9ββββ) = -27.18o
φφφφ (C-1,H-9999αααα) = 95.71o
Figure 2.1 The observed cross-peaks and the calculated dihedral angles (φ) of 62 using Spartan ‘04 (AM1).
In 62 and 63, the C-9 protons exhibited a slightly larger geminal coupling value ~18 Hz
compared to the protons of C-6 (~16 Hz). Furthermore, from our NMR analyses of
derivatives of 62 it was found that the relative chemical shifts of these allylic protons,
and their corresponding carbons, and their corresponding geminal coupling values
remained relatively the same and could be thus used for their identification. C6D6 was
N
Boc
EtO2C
HβHβ
Hα Hα
O5
3
96

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 38
found to be the better solvent as opposed to CDCl3 for the NMR analysis of 62 with the
clear separation of the critical protons discussed above. This was in contrast with 63.
For 63, a large chemical shift difference was observed in CDCl3 between the α and β
protons on carbons C-3, C-4 and C-9 (∆δ ~ 0.3-0.5). The structure of 63 was established
by single crystal X-ray structural analysis (Figure 2.2). This structure confirmed the
one determined by NMR analysis. The spectroscopic data of these compounds agreed
well with that reported by Lu et al.213 who reported a combined yield for 62 and 63 (d. r.
= 62 : 38) of only 33% when the more hindered and less nucleophilic catalyst,
triphenylphosphine (0.1 eq.), was employed.
Figure 2.2 Single crystal X-ray crystallographic analysis of 63.
Based on steric considerations alone, the regiochemical outcome of this reaction can be
rationalised as occurring via the transition state A (R = H, X = OEt) which would be
expected to be favoured over the more sterically demanding transition state B (R = H, X
= OEt, Scheme 2.4).212,213 An attempt was made to favour the transition state B over
transition state A, as shown previously from our laboratories, on a different alkene,
using the chiral alkyne, N-(2-butynoyl)-(4S)-benzyloxazolidinone 53d (Scheme 2.5).212
However the reaction of 57 and 53d yielded no desired compound 64 or 65 (Scheme
2.6).212

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 39
Scheme 2.4
PBu3
NBocO
X
O
A
Bu3P
NBocO
X
O
B
regioisomer A
regioisomer B
R
R
Scheme 2.5212
N
NOBn
Bn
O Me
X
ON
NOR2
R1
ON
NOR2
R1
O
COX
COX
reg ioisomer A regioisomer B
+ +
N
OBn
O
53a X = CO2Et
53d X =
Using 53a Combined yield of 81%
Ratio A : B = >98:<2
Using 53d Combined yield of 61%
Ratio A : B (de) = 11 (0%): 98 (>98%)
Bu3P
PhH, RT
Scheme 2.6
Bu P COX
COX

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 40
2.1.2 [3+2]-cycloaddition using the 2-methylene γγγγ-lactam 58
Under similar conditions the chiral 2-methylene γ-lactam 58 reacted with the ylide 54a
(X = OEt) to produce a mixture of the three cycloadducts 66, 67 and 68 in a ratio of 63 :
17 : 20, respectively, from 1H NMR analysis of the crude reaction mixture (Scheme 2.7).
Diastereomerically pure samples of 66 (28% yield) and 68 (13% yield) could be
obtained after extensive chromatographic purifications, however a pure sample of 67
could not be obtained due to difficulties in separating 67 from 66 and 68. Although the
absolute stereochemistries of 66 and 68 could not be unequivocally proven from
NOESY NMR experiments we assume that the major cycloadduct 66 arises from attack
of the ylide onto the face of the 2-methylene group of 58 that is anti to the ethyl ester
substituent (via transition state A, R = CO2Et, X = OEt, Scheme 2.4).
Scheme 2.7
(S)N
O
Boc
Me CO2Et
Bu3P
( S)N
(S) O
BocEtO 2C
CO2Et
(S)N
(R) O
BocEtO2C
CO2Et
PhH, RT
( S)N
(S) O
BocEtO 2C
CO 2Et
+
58
53a
+
66 (28%) 67
regioisomer A reg ioisomer B
68 (13%)
EtO2C

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 41
2.2 Synthesis of spiro[cyclopentane-1,1`-[1H]isoindol]-3`(2`H)-
ones
2.2.1 [3+2]-cycloadditions using acrylate 59
It was initially envisaged that through a similar reaction to that of the synthesis of 57
and 58, the methylene indolinone 70 (R = H) could be synthesised from the N-Boc
protected oxindole 69 using the chemistry employed in Scheme 2.2 for the synthesis of
57 and 58 (Scheme 2.8 (Pathway A)). However, this method proved unsuccessful, and
is consistent with the known instability of 70 (R = H) reported in the literature.217
Though a convenient synthesis of 70 (R = H) from indole-3-acetic acid has been
developed,218-220 the use of 70 (R = H) is limited.221 Therefore, an alternative pathway
was devised, Pathway B, which utilised the methyl 2-(2-nitrophenyl)acrylate 59 as the
cycloaddition substrate instead of 70 (R = H) to give the adduct 71, which could then be
readily cyclised to afford the indole spirocyclopentene 72 (R = H) (Scheme 2.8).
Scheme 2.8a
N
O
Boc
R
N
O
Boc
N
O
R
E
E
70 7269
Pathway A
Pathway B
[3+2]cycloaddition
reductivecyclisation
a)-d)
a
Reagents and conditions: For the formation of 70 (R = H): (a) LiHMDS (1.2-1.5 eq.), THF, -78 °C, 45 min, 67% (b) Eschenmoser’s salt, N2, -78 °C�RT, 18 c) MeI (8 eq.), anhydrous DCM, 18 h (d) sat. NaHCO3.

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 42
Acrylate 59127 was prepared initially according to the literature method of Selvakumar.
The malonate 73 was prepared via a nucleophilic substitution of 2-fluoronitrobenzene
by dimethyl malonate under basic conditions. Though several attempts were made, the
best yield using the literature conditions was only 46%. A revised synthesis, offered by
the authors, employed DMF instead of THF and potassium carbonate as the base instead
of sodium hydride.222 This revised method yielded the malonate 73 in 93% yield.
Treatment of malonate 73 with aqueous formaldehyde in the presence of potassium
carbonate afforded the acrylate 59 in 82% yield. Acrylate 59 most likely arises via a
decarboxylation-elimination mechanism (Scheme 2.9).
Scheme 2.9a
CO2Me
NO2
CO2Me
NO2
F
NO2
CO2Me
59 (82%)
a) or b)
73
c)
NO2
MeO 2C
O
O
OH
-OH
a Reagents and conditions: (a) NaH (2.2 eq.), anhydrous THF, (CH2(CO2Me)2) (2 eq.), 0
°C�60 °C, 18 h (46%); (b) (CH2(CO2Me)2) (1.2 eq.), anhydrous DMF, K2CO3 (2 eq.), 0 °C�60 °C, 18 h (93%); (c) formalin, K2CO3 (1.5 eq.), 60 °C, 2 h.
Scheme 2.10 (Compounds 71 and 72 are racemic)
CO2M e
NO2
Me CO2Et
PhH, RT
CO2Me
NO 2
CO2Et
NH
O
CO 2Et
53a
Bu3P (0.2 equiv.)
59 71 72
Zn/ HCl
(93%) (98%)
Treatment of 59 with ethyl 2-butynoate 53a and TBP (0.2 eq.) gave the racemic
cycloadduct 71 as a single regioisomer in 93% yield (Scheme 2.10). The regiochemistry
of 71 was confirmed in a similar manner to that of 62. In the gHMBC spectrum of 71
the carbonyl carbon of the methyl ester at δ 174.0 exhibited cross-peaks to the protons
at δ 3.62 and δ 3.52 (Figure 2.3 B)). Only protons that have a dihedral angle (φ) of 0° to
the ester carbonyl should exhibit cross-peaks (i.e. H-2`β and H-5`β, and not H-2`α and
H-5`α in which the dihedral angle is close to 90°). Indeed, these signals at δ 3.62 and δ
3.52 were seen from gHSQC experiments to be from protons on different carbons
(Figure 2.3 C)). The gCOSY experiments provided further differentiation of these
protons, through δ 3.62 (H-5`β) and 2.99 (H-5`α) exhibiting a stronger three-bond

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 43
proton-proton coupling to the olefinic proton (CH-4`) compared to the significantly
weaker four-bond proton-proton coupling of δ 3.52 (H-2`β) and δ 3.21 (H-2`α) (Figure
2.3 A)). These experiments allowed the unequivocal assignments of the four allylic
protons.
Figure 2.3 Enlargements of the A) gCOSY B) gHMBC and C) gHSQC spectra of 71 showing the cross-peaks concerning the protons H-5`β (δ 3.62), H-2`β (δ 3.52), H-2`α (δ 3.21) and H-2`α (δ 2.99).
Furthermore, during the synthesis of chiral versions of 71 the structure of the crystalline
intermediate (S)-76 was determined by X-ray crystallography (Figure 2.4). (S)-76 was
converted to its methyl ester analogue of 71, (S)-78, which gave similar NMR values
(except for the ester values) to the ethyl ester 71, which further confirmed the
regiochemistry of 71. Based on steric considerations alone, the regiochemical outcome
of this reaction can be rationalised as occurring via the transition state A (X = OEt)
which would be expected to be favoured over the more sterically demanding transition
state B (X = OEt) (Scheme 2.4). Several reductive cyclization methods were tested to
A) B) CO2Me CO2Et C) OCH2 OCH3 CH-5` CH-2`
OCH2 {
NO2
CO2Me
O
OEt
2
11̀
3`
Hαααα
Hαααα
Hββββ
Hββββ
71
CH-4`
H-5`α
H-2`α
H-2`β
H-5`β
OCH3

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 44
form the oxindole moiety, including using hot ethanol and sodium sulfide,223 catalytic
hydrogenation with Pd/C and Cu(acac)2 and sodium borohydride in ethanol.224
However, the best method was found to be exposure to zinc and aqueous HCl, at
reflux,225 which gave the spiro[cyclopentane-1,1'-[1H]isoindol]-3'(2'H)-one derivative
72 in 98% yield (Scheme 2.10). The indole moiety of 72 was confirmed by the
appearance of the lactam NH (δ 9.15 (bs, 1H)). The aromatic proton ortho to the nitro
group when cyclised moved upfield (δ 7.93 to δ 6.93). In addition, upon cyclisation a
loss of the signal corresponding to the methyl group (δ 3.64 (s, 3H)) and a downfield
shift in this carbonyl (δ 174.0 to δ 183.2) was observed. The NMR data collected for the
methyl ester (S)-80 were similar to those of the ethyl ester 72 (except for the ester
values).
2.2.2 Asymmetric [3+2]-cycloaddition reaction
Previously, Zhang et al.226 had showed that chiral phosphines can be utilised to produce
homochiral cycloaddition products in high enantioselectivity. However, the synthesis of
these chiral phosphines required a multistep sequence and was not attempted. In our
efforts to prepare enantio-enriched versions of 72, the corresponding cycloadditions
reactions of 59 with the chiral alkyne 53b, derived from Oppolzer’s (1S)-camphor
sultam,227 were examined. The chiral alkyne 53b, has been previously used by our
research group for examining the asymmetric [3+2]-cycloaddition reaction with 5-
methylenehydantoins.212 The chiral alkyne 53b, was prepared via a one-pot method,
originally developed by Evans et al. (Scheme 2.11).228 This method involves the initial
formation of the lithium salt of the camphorsultam (74) using n-buthyl lithium at -78 °C
in THF. This salt was then added to in situ generated 2-butynoyl pivaloyl anhydride,
prepared from the reaction of a mixture of pivaloyl chloride, 2-butynoic acid and
triethylamine in THF, to give a mixture of the desired product 53b, and an unwanted
pivaloyl derivative, 75.

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 45
Scheme 2.11
O
OHO
O
O
t-BuCOCl,NEt3, THF
SO
O
NH
SO
O
N
n-BuLi, THF
-78 oC-0 oC
Li
SO
O
NS
OO
N
O O+
53b
74
75
Lith ium salt
The tributylphosphine-catalysed reaction between 59 and 53b produced a 3.3 : 1
mixture of the diasteromeric cycloadducts (S)-76 and (R)-77 from which pure samples
of (S)-76 and (R)-77 could be obtained after column chromatography, along with a
mixture of (S)-76 and (R)-77 in a combined yield of 66% (Scheme 2.12). The
regioselectivity can be explained again on steric grounds in which transition state A
would be favoured over B (Scheme 2.4). The absolute (1`S)-configuration of the
cyclopentane ring of (S)-76 ([α]D26 –22.0 (c 0.3, CHCl3)) was established by a single
crystal X-ray structural analysis (Figure 2.4) which then allowed assignment of the
(1`R)-configuration to this ring of the minor diastereomer (R)-77 ([α]D24 +19.0 (c 0.6,
CHCl3)). Several methods, including acid catalysis with either para-toluenesulfonic
acid or concentrated HCl were used in efforts to remove the chiral auxiliary, however
with no success. The chiral auxiliary was finally removed by methanolysis of (S)-76
and (R)-77 in the presence of samarium(III) triflate229 to give the methyl esters, (S)-78
and (R)-79 in yields of 67% and 68 %, respectively. The structures of (S)-78 and (R)-79
were confirmed by the loss of signals corresponding to the chiral auxiliary in the NMR
and MS data. Indeed the NMR data collected for (S)-78 and (R)-79 agreed well with
structurally similar 71. Reductive cyclization of (S)-78 and (R)-79 by treatment with
zinc/aqueous HCl gave the tricyclic lactams, (S)-80 and (R)-81, respectively, that
displayed almost identical NMR spectroscopic data (apart from the ester signals) to
racemic 72 (Scheme 2.12 and Table 2.1).

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 46
Figure 2.4 Single crystal X-ray crystallographic analysis of (S)-76.
Table 2.1 Summary of rotations and yields for Scheme 2.12
Cpd No. Major (S) Cpd No. Minor (R)
(S)-78
(S)-80
67%, [α]D24 = -42.5 (c 0.1, CHCl3)
69%, [α]D24 = -40.8 (c 1.2, CHCl3)
(R)-79
(R)-81
68%, [α]D26 = +50 (c 0.7, CHCl3)
56%, [α]D23 = +57.4 (c 1.0, CHCl3)

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 47
Scheme 2.12a
CO2Me
NO 2
Me COXC
PhH, RT, 18 h
CO 2Me
NO2
COXc
CO2Me
NO 2
COXc
CO2M e
NO2
CO2Me
CO2M e
NO2
CO2Me
NH
O
CO2Me
NH
O
CO2Me
53b
+
Bu3P (0.1 equiv)
59
(S)-76[X-ray]
(R)-77
+
(S)-78 (R)-79
(S)-80 (R)-81
a) a)
b) b)
a
Reagents and conditions: (a) Sm(OTf)3 (1 eq.), MeOH, 50 °C, 18 h, 67% ((S)-78), 68% ((R)-79); (b) Zn dust (24 eq.), 8.9M HCl, MeOH/H2O, reflux, 2 h, 69% ((S)-80), 56% ((R)-81).

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 48
2.3 Synthesis of spirocyclic derivatives
2.3.1 Pathway A: Increasing structural diversity of existing structures
The spiro-cyclic compounds 62, 63 and 72 contain two functional groups, namely the
lactam nitrogen and the ester carbonyl, which can be further derivatised to provide
compounds with increased structural diversity for biological testing. For example, the
N-Boc protecting group in racemic 62 and 63 was readily removed upon exposure to
trifluoroacetic acid (TFA) to give compounds 82 and 88, respectively (Scheme 2.13)
This reaction resulted in the loss of the signals corresponding to the N-Boc protecting
group as observed by MS and NMR spectroscopy and, in the case of 82, the appearance
of a signal (δ 7.50) corresponding to the free lactam nitrogen in the 1H NMR spectrum.
The nitrogen atom of 82 and 88 was readily N-benzylated with benzyl bromide under
basic conditions to give the resulting compounds 83 and 89, respectively. These
compounds were converted to their N-aryl amide derivatives 85a,b and 91, respectively,
through amide bond formation between their respective carboxylic acids, 84 and 90,
obtained through an initial base hydrolysis of their esters, and aniline and 4-
dimethylaminoaniline (Scheme 2.13). Amide 87 was likewise obtained from the
coupling reaction of aniline and the carboxylic acid 86 obtained from base catalysed
hydrolysis of ester 82.
Using related chemistry, ester 72 and chiral esters (S)-80 and (R)-81 were converted to
the N-aryl amides 93a,b and enantiomerically pure (S)-93b and (R)-93b via their
respective racemic carboxylic acid 92 and their enantiomerically pure carboxylic acids
(S)-92 and (R)-92 (Scheme 2.14 and Table 2.2). These two pairs of enantiomeric
products, (S)-92 and (R)-92, and (S)-93b and (R)-93b, gave opposite, though not
exactly equal, optical rotations (Table 2.2). Differences in their magnitude were due to
purification difficulties. The products from all the reactions described in Scheme 2.13
and Scheme 2.14 were confirmed by extensive 2D NMR experiments and mass
spectrometry at both high and low resolution. A more detailed description of how these
structures were assigned and characterised, using the formation of 85a from 82 as an
example, will now be described. The appearance of the N-benzyl methylene protons as
the characteristic ABquartet (J ~ 14 Hz) at δ 4.46 and the presence of aromatic protons
in the 1H NMR spectrum confirmed the N-benzylated product 83. 13C NMR analysis
also confirmed the N-benzyl substituent with the appearance of aromatic carbons and a

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 49
methylene group at δ 47.1. Upon base hydrolysis of 83 product 84 showed a loss of the
signals corresponding to the ethyl group in both the 1H and 13C NMR spectra and the
appearance of a broad hydroxyl peak (δH 9.16). In addition a slight downfield shift was
observed for both carbamate carbonyl (δ 164.6 to 168.8) and olefin proton in both the 1H NMR (δ 6.69 to 6.81) and the 13C NMR (δ 141.0 to 143.5) spectra. The N-aryl amide
derivative 85a was confirmed by the presence of extra aromatic protons, a change in the
chemical shift of methylene protons CH2-6 and CH2-9, an upfield shift in the olefinic
proton in both the 1H (δ 6.81 to δ 6.50) and 13C (δ 143.5 to 135.0) NMR spectra and
also in the carbonyl of concern (δ 168.8 to δ 162.7). Furthermore, compounds 82, 85a,b,
86, 87, 90 and 91 all gave single crystals for X-ray structural analysis (see Appendix 1).
Scheme 2.13a
N
O
Boc
CO 2Et
N
O
Boc
CO2Et
NH
O
CO 2Et
NH
OCO 2Et
NR
O
COX
NR
OCOX
62
63
82*
88*
a)
83, R = Bn, X = OEt
84, R = Bn, X = OH85*, R = Bn, X = NHAr86, R = H, X = OH87*, R = H, X = NHPh
b)
a) b)
a; Ar = Ph, b; Ar = 4-M e2NC6H4-
c
d
89, R = Bn, X = OEt
90*, R = Bn, X = OH91*, R = Bn, X = NHPh
c
d
* Structure confirmed by X-ray a Reagents and conditions: (a) TFA, DCM, 2.5 h, 91% (82), 86% (88); (b) NaH (1.3 eq.), Bu4NI (0.1 eq.), BnBr (1.5 eq.), anhydrous THF, RT, 1-5 h, 74% (83), 47% (89); (c) K2CO3 (2 eq.), MeOH/H2O, high pressure tube, 60 °C, 1 d, 93% (84), 53% (86), 80% (90); (d) Aniline or 4-N,N-dimethylaniline (1.2 eq.), HOBT (1 eq.), EDCI (1 eq.), anhydrous MeCN, 0 °C�60 °C, 1-2 d, 54% (85a), 64% (85b), 91% (87), 91% (91).
Table 2.2 Summary of rotations and yields for Scheme 2.14
Cpd No. Major (S) Cpd No. Minor (R)
(S)-92 (S)-93b
55%, [α]D24 = -42.2 (c 1.7, CHCl3)
48%, [α]D24 = -47.4 (c 1.2, CHCl3)
(R)-92 (R)-93b
84%, [α]D24 = +38.9 (c 0.89, CHCl3)
32%, [α]D25 = +56.7 (c 0.8, CHCl3)

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 50
Scheme 2.14a
NH
O
CO 2R
NH
O
CONHAr
NH
O
CO 2H
(r ac)-72 R = Et
(S)-80 R = Me
(R)-81
a)
(rac)-92 (94%)
(S)-92 (55%)
(R)-92 (84%)
b)
(rac)-93a (52%)
(rac)-93b (44%)
(S)-93b (48%)
(R)-93b (32%)
a; Ar = Ph, b ; Ar = 4-Me2NC6H4-
a Reagents and conditions: (a) K2CO3 (2 eq.), MeOH/H2O, high pressure tube, 60 °C, 5 h, 94% ((rac)-92), 55% ((S)-92), 84% ((R)-92); (b) Aniline or 4-N,N-dimethylaniline (1.7 eq.), HOBT (1 eq.), EDCI (1 eq.), MeCN, 0 °C�RT, 15 h, 52% ((rac)-93a), 44% ((rac)-93b), 48% ((S)-93b), 32% ((R)-93b).
2.3.2 Pathway B: [3+2]-cycloaddition with structural diversity
embedded in the starting ylide
To explore a more direct method to these N-aryl amide derivatives, the phosphine-
catalysed [3+2]-cycloaddition reactions of the 2-methylene γ-lactam 57 and acrylate 59
with the ylide 54 (X = NHPh), that was generated in situ from the reaction of N-phenyl
2-butynamide 94, was examined (Scheme 2.15). These reactions were unsuccessful,
presumably due to internal quenching of the ylide B (X = NHPh) by the relatively acidic
secondary amide NH. In accordance with this hypothesis was the fact that the
corresponding N-PMB protected ylide 54c (X = N(PMB)Ph), generated in situ from the
tertiary amide 53c, gave the racemic cycloadducts 95 and 96, in yields of 14% and 55%,
respectively (Scheme 2.15 and Scheme 2.16). These reactions, while producing poor to
modest yields, were completely regioselective, presumably due to the increased steric
bulk of the ylide 54c which would further destabilize transition state B over transition
state A (Scheme 2.4). Treatment of the cycloaddition product 95 with TFA, gave N-
phenyl amide 87 (Scheme 2.15) that was identical to the compound 87 prepared
according to Scheme 2.13. Similarly, reductive cyclization of 96 followed by
deprotection of the product 97 with TFA gave (rac)-93a (Scheme 2.16) that was
identical to the (rac)-93a prepared according to Scheme 2.14. To the best of our
knowledge the phosphine-catalysed [3+2]-cycloaddition reactions of alkenes and 2-
butynamides has not been previously reported. This more direct route, Pathway B,

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 51
however proved to be a lower yielding synthesis, with overall yields of 8% and 4%
from 57 and 59, respectively, compared to using Pathway A that had overall yields of
32% and 44%, respectively. This was the result of the lower yielding major phosphine-
catalysed cycloaddition reaction with the bulkier ylide 54c.
Scheme 2.15a (all compounds are racemic)
N
O
Boc
Me CONRPhBu3P
N
O
Boc
CON(PMB)Ph
PhH, RTNH
O
CONHPh
57
94, R = H53c, R = PMB
+
95 (14%) 87
a)
a Reagents and conditions: (a) anisole (10 eq.), TFA (125 eq.), DCM, 15 h, 57%.
Scheme 2.16a (all compounds are racemic)
CO 2Me
NO2
Me CON(PM B)Ph
PhH, RT
CO2Me
NO 2
CON(PMB)Ph
NH
O
CONHPh
NH
O
CON(PMB)Ph
53c
Bu3P (0.1 equiv.)
59 96 (55%)
97
a)
(r ac)-93a
b)
a Reagents and conditions: (a) activated Zn dust (3.4 eq.), acetic acid, 1.5 h, 16%; (b) anisole (10 eq.), TFA (125 eq.), DCM, 15 h, 52%.
2.3.3 Curtius Rearrangement
Initially it was anticipated, that the synthesis of analogues with more hydrogen-bonding
substituents, such as the urea derivatives (99), could be accessed by employing the
Curtius rearrangement (CR). The CR proceeds through converting the racemic
carboxylic acid 92 to its corresponding acyl azide (100) by treatment with
diphenylphosphoryl azide (DPPA),230,231 which upon heating with aromatic amines
should afford the urea analogues, 99 (Scheme 2.17). This reaction however did not
yield any of the desired compound, however upon aqueous workup, the novel
spirocyclic ketone, 103 was afforded. Repeating the CR conditions and employing acid
hydrolysis conditions resulted in a mixture of the spiro-cyclic ketones, 102 and 103 in a

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 52
ca 1 : 1 mixture (Scheme 2.17). These compounds were readily separated by column
chromatography and were isolated in yields of 54% and 35%, respectively. The 1H
NMR spectrum of 103 showed two distinct NH resonances (δH (C6D6) 7.96 (bs) and
4.84 (bs)), and a deshielded aromatic proton (δH (C6D6) 8.64 (d, J 8.0 Hz) consistent
with the presence of the N-aminocarbonyl group with internal H-bonding to the lactam
carbonyl group. The structures of 102 and 103 were confirmed by single crystal X-ray
structural analysis (Figure 2.5). The unexpected product 103 could arise from self-
condensation of the intermediate vinyl isocyanate 98 to give the carbamate derivative
100. Acid hydrolysis of 100 then gives, via 101, the spiro-cyclic ketones 102 and 103
(Scheme 2.17). However, no attempts were made to isolate or characterize the
intermediates involved.
Scheme 2.17a (all compounds are racemic)
NH
O
CO2H
O
NH
O
HN N
O
O
OCN
O
OCN
H
O
NH
O
NCO
H O
a)
(rac)-92 98
98
100
NH
O
HN
99
O
HN
Ar
ArNH2
∆
a Reagents and conditions: a) DPPA (2 eq.), NEt3 (2 eq.), anhydrous toluene, 85 °C�reflux, 30 mins b) 8.9 M HCl, reflux (1 h)� RT (15 h).

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 53
Figure 2.5 Single crystal X-ray crystallographic analysis of 102 (left) and 103 (right).
2.3.4 Hydrogenation
To increase structural flexibility of the spirocyclic system and see what effect this had
on biological activity, the cyclopentene ring was hydrogenated to its cyclopentane
structure. Catalytic hydrogenation of the alkene moiety of racemic ester 72 gave a 1.8 :
1 mixture of the diastereomers 104 and 105, respectively that were readily separated by
column chromatography (Scheme 2.18). Using related chemistry, the alkene moiety of
racemic amide 93a gave a mixture of the diastereomers 106 and 107, which were
obtained in yields of 78% and 17%, respectively after purification by column
chromatography (Scheme 2.19). The relative stereochemistry of 104 and 106 to their
diastereomers 105 and 107 was determined by 1D NOE experiments that showed a
significant enhancement of the signal for the methine proton Ha upon radiation of the
aromatic proton Hortho (Ho) and vice versa. The measured distances between Ha and Ho
for 104 and 106, was calculated by Spartan ’04 (AM1) to be 4.316 Å and 3.829 Å,
respectively (Figure 2.6). This was in contrast to the longer distances measured
between Ha and Ho in 105 and 107 of 5.028 Å and 5.109 Å, respectively (Figure 2.6).
Indeed NOEs are normally only observed between protons less than 5 Å apart.
Scheme 2.18

Chapter 2: Phosphine-catalysed [3+2]-Cycloaddition Reaction 54
Scheme 2.19
NH
O
CONHPh
NH
O
CONHPhHa
Ho
NH
O
CONHPhH
Pd/C
H2
EtOAc
106 (78%)
* NOE
107 (17%)
+
*
(r ac)-93a
NOEsignalobserved
4.32 Å
5.03 Å
No NOEobserved
Figure 2.6 The important NOE peak between Ha and Ho observed in 104 (left) and absent in 105 (right) and the calculated measured distance using Spartan ‘04 (AM1).
In conclusion, the phosphine-catalysed (3+2) cycloaddition has been shown to be a
good method to access 2-azaspiro[4.4]nonan-1-one and spiro[cyclopentane-1,1`-
[1H]isoindol]-3`(2`H)-one ring systems. The relative stereochemistry of key compounds
has also been unequivocally determined by single crystal X-ray structural analysis.
Compounds 72, 85a,b, 87, 91, (S)-92, (rac)-93a, (rac)-93b and (S)-93b, 102-107 were
submitted for cytotoxicity studies and protein inhibition studies and these results are
discussed in Chapter 6.

Chapter 3: Synthesis of Spirocyclopropane Indolinones 55
CHAPTER 3: SYNTHESIS OF
SPIROCYCLOPROPANE INDOLINONES
The Madagascan rosy periwinkle (Catharanthus roseus) is the source of various vinca
alkaloids, like vinblastine, which is used clinically as a chemotherapeutic for leukemia,
Hodgkin's disease, and breast and lung cancer.
NM e
N
O
E t
H O H
C O 2 M e
M eO
M e O
N H
N
E tH O
C O 2 M e
H
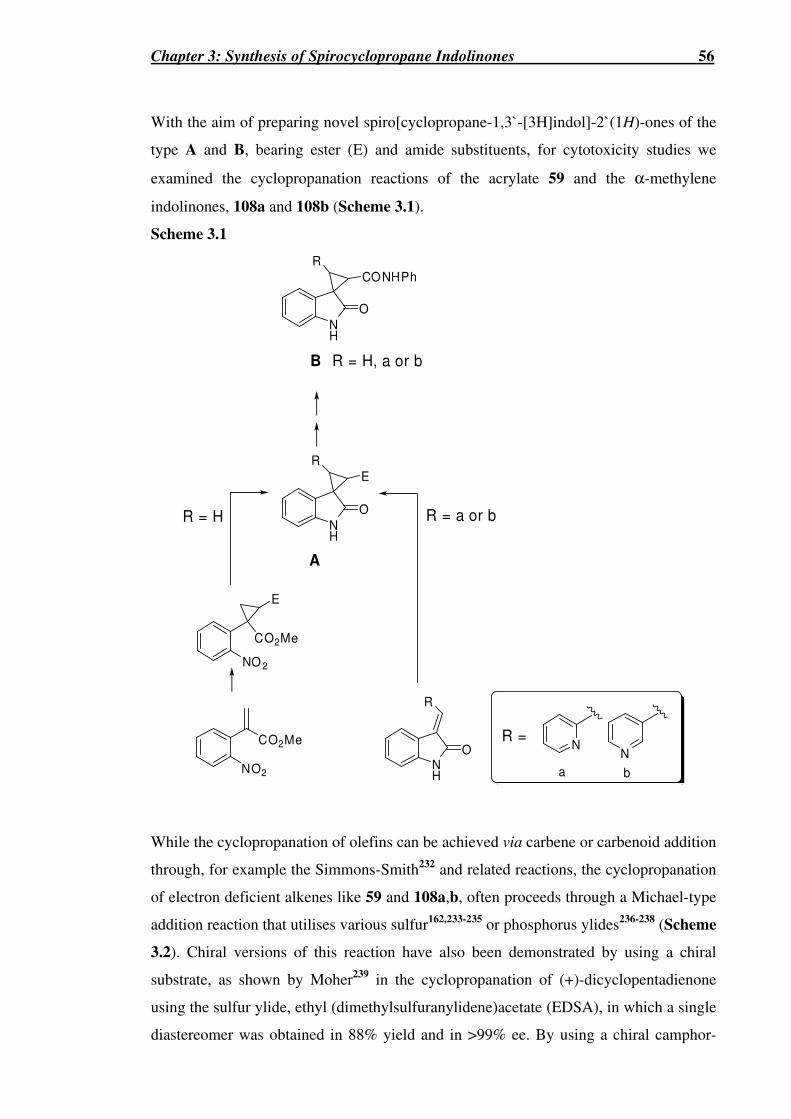
Chapter 3: Synthesis of Spirocyclopropane Indolinones 56
With the aim of preparing novel spiro[cyclopropane-1,3`-[3H]indol]-2`(1H)-ones of the
type A and B, bearing ester (E) and amide substituents, for cytotoxicity studies we
examined the cyclopropanation reactions of the acrylate 59 and the α-methylene
indolinones, 108a and 108b (Scheme 3.1).
Scheme 3.1
CO2Me
NO2
NO 2
CO2Me
E
E
NH
O
NH
O
NH
O
R
R
R
CONHPh
NN
R =
a b
R = H R = a or b
A
R = H, a or bB
While the cyclopropanation of olefins can be achieved via carbene or carbenoid addition
through, for example the Simmons-Smith232 and related reactions, the cyclopropanation
of electron deficient alkenes like 59 and 108a,b, often proceeds through a Michael-type
addition reaction that utilises various sulfur162,233-235 or phosphorus ylides236-238 (Scheme
3.2). Chiral versions of this reaction have also been demonstrated by using a chiral
substrate, as shown by Moher239 in the cyclopropanation of (+)-dicyclopentadienone
using the sulfur ylide, ethyl (dimethylsulfuranylidene)acetate (EDSA), in which a single
diastereomer was obtained in 88% yield and in >99% ee. By using a chiral camphor-

Chapter 3: Synthesis of Spirocyclopropane Indolinones 57
derived sulfur ylide, Tang et al.240,241 showed that high daistereoselectivities with
excellent enantiomeric excesses could be achieved in the cyclopropanation reactions of
pro-chiral electron-deficient alkenes.
The use of the sulfur ylide, EDSA, 110 (R = Et), for cyclopropanation reactions has
been widely established.242,243 This sulfur ylide has the ability to react with α,β-
unsaturated esters, ketones, aldehydes and nitriles to readily afford the corresponding
cyclopropanated products as mixtures of diastereomers depending on the substrate and
reaction conditions.243 Previously, the moisture sensitive ylide EDSA was prepared
beforehand243 and used in subsequent cyclopropanations, however, it has been shown by
Pedregal et al.244 that EDSA can be generated in situ by treatment of ethyl
dimethylsulfonium acetate bromide 109 (R = Et) with DBU (Scheme 3.2). This method
yields greater stereoselectivity than that shown previously by Payne,243 as seen in the
case of 2-cyclohexen-1-one which gave exclusively the exo product in comparison to
the 2 : 1 ratio of exo : endo products obtained using Payne’s original method (Scheme
3.3).243,244
Scheme 3.2
CO2R
SMe2 Br DBUCO2R
DBUMe2S
109 W
R1 R2
R3
R3
R2
W
R1
SMe2 SMe2 R3
R2
**
*

Chapter 3: Synthesis of Spirocyclopropane Indolinones 58
Scheme 3.3244
OH
H
CO2Et
H
H
CO2Et+CO2Et
SMe2Br
DBU4 d
de = 100%(55%)
(0%)
endoexo
The mechanism of this cyclopropanation reaction proceeds by first generation of the
ylide 110 under basic conditions from the sulfonium salt 109 (Scheme 3.2).242 The ylide
then attacks the olefin via a Michael-type addition reaction to form the betaine
intermediate A (Scheme 3.2).242 Ring closure, promoted by the elimination of dimethyl
sulfide, affords the cyclopropanated derivative. This reaction has the possibility of
yielding a number of products through the formation of three contiguous stereogenic
centres (Scheme 3.2). These stereoisomers can be grouped into trans and cis isomers
based on the two electron-withdrawing groups (W and CO2R).242 The stereoselectivity
of this cyclopropanation reaction has been shown to be dependant upon the reaction
conditions, in particular the nature of the solvent.242,244 In the case of the acyclic α,β-
unsaturated aldehyde methacrolein, a mixture of trans- and cis-
cyclopropanecarboxyaldehydes were formed (Scheme 3.4), the ratio of these isomers
was found to be solvent dependent.243
Scheme 3.4243
CO2Et
SMe2 Me
CHOMe
CHOOHC
Me
CO2EtCO2Et
++
These stereoisomers are the result of the equilibrium which exists between the betaine
intermediates A and B (Scheme 3.5).242 While the trans product always predominates, it
has been observed that with decreasing solvent polarity and dielectric constant (ε), the
stereoselectivity for the trans product increases. Electrostatic interactions should favour
the initial formation of the eclipsed betaines A and B. Their subsequent collapse into
their respective cis- and trans- cyclopropane products proceeds via their respective anti-
conformers, C and D. Solvents possessing a lower ε should be less capable of shielding
the proposed internal ion-pair attraction and therefore retarding the formation of C and

Chapter 3: Synthesis of Spirocyclopropane Indolinones 59
D and promoting a greater equilibrium between A and B.242 This would result in an
enhancement of formation of the more stable trans-product, in which the two polarised
carbonyl groups are trans to each other.242 Conversely, in solvents possessing a higher ε,
the rate of cyclopropane formation may become more competitive with the equilibrium
between A and B, resulting in greater proportions of the cis-product.242
Scheme 3.5242
R1
Me2S
CH
RO2C
Me2S
C
HH
R1
CO2R
H
RO 2C
Me2S
C
HH
R1
H
RO2C
cis
tr ans
A
B
C
D
H
H
R1
CHO
+
110
R1
CO2R
H
H
R1
SMe2
CO2R
Me2S
CH
RO 2C
HH
R1
HH
O
O
O
O
O
O
H
H
H
H
H
H
Pedregal et al.244 similarly showed the effect solvent polarity had on diasteroselectivity
in their study of the cyclopropanation reactions of various acyclic enones (Table 3.1).
Overall, it was shown that using the less polar solvent toluene (ε = 2.4) yielded only the
major trans isomer, 111 (Table 3.1, entries 1, 2, 4, 6 and 8). Whereas, the use of the the
more polar solvent chloroform (ε = 4.8) yielded a mixture of the major trans isomer,
111 and the cis isomer, 112 and varying low amounts (~10%) of the minor trans isomer,
113 (Table 3.1, entries 3, 5, 7 and 9). It was also shown by Pedregal et al. that the
minor trans isomer 113 was the product of epimerization under these basic reaction
conditions of the cis product 112 and not the major trans product 111.244 Indeed, when
112 (R1 = R2 = CH3) was subjected to treatment with NaOEt (1 eq.) in EtOH for 4 h, it
was completely transformed into 113. The four possible racemic anti-betaine

Chapter 3: Synthesis of Spirocyclopropane Indolinones 60
intermediates (A-D) involved in this reaction are shown in Scheme 3.6. Upon closer
observation of these four possible intermediates, two are disfavoured (B and C) due to
the unfavourable steric interaction that can occur when the COR1 and R2 groups are cis
to one another. Indeed the major products of Pedregal et al.’s study, 111 and 112, whose
relative stereochemistry was confirmed by NOE studies, are the (1,2)-trans-(1,3)-cis-
product and the (1,2)-cis-(1,3)-trans-product, respectively. The latter product is less
favoured of these two products because of an unfavourable dipole-dipole interaction
that occurs between the ester group (E) and the ketone enolate anion in intermediate A
(Scheme 3.6). The minor trans product 113, corresponds to the less favoured (1,2)-
trans-(1,3)-trans-product (Scheme 3.6). It should be noted to the reader, that
compounds 111-113, and intermediates A-D, are racemic. Furthermore, the
intermediates A and C as drawn, give rise to the enantiomers of 112 and 113,
respectively.
Table 3.1244
R1
O
R2 R2 R2 R2
CO2Et CO2EtCO2Et
O
R1
O
R1
O
R1+ +
(1,2)-t rans-(1,3)-cis (1,2)-cis-(1,3)-tr ans (1,2)-t rans-(1,3)-tr ans
111 112 113
12
3
a)
Entry Substrate Solvent Time Isolated Yield (%)
1 R1 = R2 = CH3 Toluenea 96 h 50 (111)
2 R1 = R2 = CH3 Toluene 48 h 89 (111) (de 80%)
3 R1 = R2 = CH3 CHCl3 18 h 63 (111); 32 (112)
4 R1 = CH3, R2 = C5H11 Toluenea 48 h 84 (111)
5 R1 = CH3, R2 = C5H11 CHCl3 48 h 60 (111); 31 (112)
6 R1 = Ph(CH2)2, R2 = CH3 Toluene 48 h 90 (111)
7 R1 = Ph(CH2)2, R2 = CH3 CHCl3 48 h 67 (111); 26 (112)
8 R1 = Ph(CH2)2, R2 = C5H11 Toluene 48 h 85 (111)
9 R1 = Ph(CH2)2, R2 = C5H11 CHCl3 48 h 55 (111); 27 (112)
a 1.0 eq. of sulfonium salt.

Chapter 3: Synthesis of Spirocyclopropane Indolinones 61
Scheme 3.6 (all intermediates are racemic)
Me2S
C
R2 H
H
E
Me2S
C
R2 H
H
E
(1,2)-tr ans-(1,3)-cis-product**
R1
HO
R1H
O
Me2S
C
H R2
H
E R1H
O
Me2S
C
H R2
H
ER1
HO
(1,2)-t rans-(1,3)-tr ans-product**
(1,2)-cis-(1,3)-tr ans-product**
(1,2)-cis-(1,3)-cis-product**
H
E
E
E
E
H
R2H
H
R1OC R2
R1OC
R2H
H H
R1OC R2
R1OC H
*
*
dipolar repulsion
dipolar repulsion
1
2 3
1
2 3
1
2 3
1
2 3
A
B
C
D [MAJOR PRODUCT]

Chapter 3: Synthesis of Spirocyclopropane Indolinones 62
During the course of this project He et al.245 reported the synthesis and potent antiviral
activity of some ester and amide derivatives of 5-bromo-3-spirocyclopropyloxindole-2`-
carboxylic acid 114 (R = OH, X = H, Y = Br). These derivatives were found to be
potent HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs) on both wild-
type (WT) and drug-resistant mutant viruses.245 The ethyl ester of compound 114 (R =
OEt, X = H, Y = Br) was prepared from the 3-carboethoxymethylene derivative of 5-
bromoisatin (116, Y = Br) by treatment with diazomethane to generate the
cyclopropanated product, 114 (R = OEt, X = H, Y = Br) via its diazo intermediate. This
compound was found to be a potent NNRTI showing an EC50 of a 66 nM. Interestingly,
its 2`-epimer was found to be totally inactive. In addition, removal of the 5-bromo
substituent or its 5-fluoro analogue led to a significantly reduced antiviral activity.245
Similar reactions to He’s have been utilized earlier to prepare the ethyl ester 114 (R =
OEt, X = H, Y = H)246,247 and 2`-substituted-3-spirocyclopropyloxindole-2`-carboxylic
esters.246 The 3`-phenyl-3-spirocyclopropyloxindole-2-carboxylic ester 115 and its
related 3`,3`-diphenyl-3-spirocyclopropyloxindole-2-carboxylic ester have also been
prepared from 116 (Y = H) using phenyldiazomethane246 and diphenyldiazomethane.247
In the same year, Shanmugam et al.248 reported the synthesis of N-alkyl derivatives of
114 (R = O-alkyl, X = H, Y = H or Br) employing a reductive cyclization reaction to
prepare the cyclopropane rings as a diastereomeric mixture of 2`-epimers (dr ~ 1 : 2).
Indeed prior to the work disclosed here, no methods were available to prepare 3`-aryl-3-
spirocyclopropyloxindole-2`-carboxylic esters, including the desired 3`-(2- and 3-
pyridyl)-substituted analogues of 114 that did not employ potentially hazardous diazo
compounds.249
COR
X
Y Y
CO2Et2`3`
1`

Chapter 3: Synthesis of Spirocyclopropane Indolinones 63
3.1 Synthesis of Spirocyclopropane Indolinone 129
3.1.1 Cyclopropanation of the acrylate 59 using EDSA
Scheme 3.7a (Compound 117 is racemic)
CO2Me
NO2NO2
CO2Me
CO2Et
59 117 (80%)
DBU, anhydrous toluene,
109 (R = Et) (2 equiv), 20 h
The cyclopropanation reaction of 59 with EDSA generated in situ from 109 (R = Et) (2
eq.) with DBU (1.5 eq.) in anhydrous toluene for 20 h at RT yielded solely the trans-
product 117 in excellent yield (80%). The use of toluene, with its characteristic low ε of
2.4 may promote the equilibrium between the betaine intermediates (c.f. A and B in
Scheme 3.5), thus leading to the favoured product 117, with the polarised ester groups
(CO2Me and CO2Et) in a trans relationship (Scheme 3.5). The formation of 117 was
confirmed by the loss of olefinic NMR signals (δH 6.54 and δH 5.89 and δC 127.4 and δC
120.1) and the appearance of NMR signals indicative of the cyclopropane ring at δH
3.02 (1H) and ~ δH 2.00 (2H) and δC 36.1, δC 29.7 and δC 22.1. The trans
stereochemistry was consistent with NOESY experiments with observed NOE
correlations between the aromatic methine Ho and the methylene protons, CH2-3` (~ δ
2.00), and the absence of an NOE between the aromatic methine Ho and the
cyclopropane methine proton, CH-2` (δ 3.00) (Figure 3.1 (inset)). From molecular
modelling studies using Spartan ‘04 (AM1) the distances between each of the two
methylene protons CH2-3` and Ho in 117, were found to be 2.255 Å and 3.524 Å
compared with the distance between Ho and the methine proton CH-2` of 4.705 Å. The
latter distance was considered too large for an observable NOE. A NOE correlation
would have been expected between Ho and CH-2` in the cis-isomer of 117 where the
inter proton-proton distance would be 3.924 Å. The structure of 117 however, was
unequivocally established by single crystal X-ray structural analysis (Figure 3.1).

Chapter 3: Synthesis of Spirocyclopropane Indolinones 64
Figure 3.1 Drawing showing the NOE correlations (inset) and single crystal X-ray crystallographic structure of 117.
3.1.2 Reductive cyclization of 117
Scheme 3.8a (all compounds are racemic)
NH
O
MeO2C
NO2
CO 2Me
CO2EtCO 2Et
NH
O
117 118 119
+
a) or b)
1H NMR ratio of 118 : 119a): 12 : 1
b): 4 : 1
a Reagents and conditions: (a) Zn dust (40 eq.), EtOH, HCl, reflux, 3 h, 70% (118), 5% (119); (b) Pd/C, H2, 2 d, 61% (118), 18% (119).
The reductive cyclization of 117 by the previous method using zinc and HCl with
heating under refluxing conditions, led to the formation of two products, 118 and 119,
based upon which ester the resulting nucleophilic amine attacked (Scheme 3.8). In the
previous cyclisation reactions of the spirocyclopentenes (S)-78, (R)-79 and 71, only the
desired indolinone product was obtained and none of its alternative and less likely
seven-membered ring lactam, was formed (Scheme 2.10 and Scheme 2.12). However,
for 117 the ability to cyclise to form a favoured, stable six-membered ring lactam
proved itself to be a strong driving force, with a high selectivity observed of 12 : 1 in
favour of the formation of quinoline 118 over the more strained five-membered lactam
NO2
CO2Me
1
2
1`
2`
HH H
OEt
O
Ho
**
*NOE

Chapter 3: Synthesis of Spirocyclopropane Indolinones 65
ring structure, the indolinone 119. Extensive purification of this mixture furnished 118
and 119 in 70% and 5% yield, respectively. In contrast, milder reducing conditions via
catalytic hydrogenation using Pd/C and H2 led to a less selective reaction, giving a 4 : 1
mixture in favour of the quinoline product 118. Extensive purification of this mixture
furnished 118 and 119 in 61% and 18% yield, respectively. Previously, Selvakumar et
al.250 showed the same regioselectivity for the quinoline product 121 when 120 was
cyclised using similar catalytic hydrogenation conditions (Scheme 3.9). The higher
selectivity found in the cyclisation of 117 using zinc, may be due to Zn2+ activation of
the less hindered ester carbonyl by co-ordination, leading to more of the quinoline
product 118.
Scheme 3.9
NO2
CO2Me
CO 2Et
NH
NH
O
O
CO2Me CO2Et
+
1H NMR ratio of 121 : 122 (4 : 1)
Combined yield (73%)
Pd/C, H2, THF
120 121 122
The structures of 118 and 119 were confirmed from their 1D NMR analysis. Mass
spectrometry at both high and low resolution confirmed their molecular formulae and
revealed fragmentation concurring with the loss of either the methyl ester group for 118
or an ethyl ester group for 119. The presence of their specific alkyl ester group in the 1D
NMR spectra was a key factor in confirming the assignment of products. The structures
of 118 and 119 were also unequivocally established by single crystal X-ray structural
analysis (Figure 3.2).
Figure 3.2 Single crystal X-ray crystallographic analysis of 118 (left) and 119 (right).

Chapter 3: Synthesis of Spirocyclopropane Indolinones 66
From 1H NMR studies, the aromatic proton ArH-4 was more downfield in 118 (δ 7.72)
than 119 (δ 7.34) consistent with the closer proximity of the ester group to ArH-4 in 118
over 119 (Figure 3.2). For 118, the individual cyclopropane methylene protons
displayed a large chemical shift difference of 1.4 ppm (δ 2.43 and δ 1.03). Indeed from
molecular modelling and from the X-ray crystal structure of 118 (Figure 3.2), these
protons are in very different chemical and magnetic environments. The more downfield
methylene proton (δ 2.43) was assumed to be that cis (H-1β) to the ester carbonyl group.
In contrast to this, the individual methylene cyclopropane signals for 119 showed a
smaller chemical shift difference of 0.14 ppm (δ 2.17 and δ 2.03), revealing a more
similar chemical environment as in the cyclopropane structure of 119 (Figure 3.2). The
cyclopropane methine proton (H-1a) of 118 appeared as a ddd (δ 2.58) and slightly
upfield from the analogous proton (H-2`) in 119 which appeared as a dt (δ 2.72). The 1H
NMR spectral data of 119 at 300 MHz was similar to that reported in 1978 by
Bennett246 for the same compound at 60 MHz. Indeed, the spectral data for the similar
N-methyl derivatives of 119 synthesised by Shanmugam et al.,248 also confirmed the
stereochemistry assigned. The N-methyl derivative of 119 with the same (1`R*, 2`R*)
configuration displayed similar 1H NMR values for its cyclopropane protons, δ 2.70
(dd, J 8.7, 7.5 Hz, 1H), 2.13 (dd, J 7.5, 4.5 Hz, 1H), and 2.01 (dd, J 8.7, 4.5 Hz, 1H) as
those of (1`R*, 2`R*)-119, δ 2.72 (dd, J 8.5, 7.5 Hz, 1H), 2.17 (dd, J 7.5, 4.5 Hz, 1H),
and 2.03 (dd, J 8.5, 4.5 Hz, 1H). This was in contrast to the 1H NMR values for their
other diastereomer with the (1`R*, 2`S*) configuration, δ 2.57 (dd, J 8.4, 8.1 Hz, 1H),
2.31 (dd, J 8.1, 4.8 Hz, 1H), and 2.17 (dd, J 8.4, 4.8 Hz, 1H).248
He et al.251 has also recently synthesised similar but isomeric quinoline cyclopropanes
to 118 from 123 as novel NNRTIs (Scheme 3.10). Quinolines 125 displayed potent
anti-HIV activity (EC50 = 1 nM) against wildtype (WT) and some HIV-1 mutant strains,
with activity comparable to or even greater than that of the known NNRTIs, efavirenz
(Sustiva®) (EC50 = 1 nM) and nevirapine (Viramune®) (EC50 = 50 nM).251

Chapter 3: Synthesis of Spirocyclopropane Indolinones 67
Scheme 3.10251
R1 = alkyl, aryl
R2 = OR, NHR, R
Most potent derivative
R1 = butyl, R2 = CO2allyl
Anti-HIV activity of EC50 = 1nM
against WT, and HIV-1 resistant
mutants, F227L and L1001.
123
Cl
N O
CO2Et
R1
PMB
Cl
NH
O
COR2
R1
Cl
N O
CO2Et
R1
PMB
125124
a) (CH3)2SOCH3I,
NaH, DMSO,
0-90 oC
3.1.3 Derivatisation of compounds 118 and 119
The ester groups of 118 and 119 were subsequently both converted to their respective
amides 128 and 129, via their corresponding carboxylic acids 126 and 127 (Scheme
3.11), using the same chemistry to that shown previously for the 2-azaspiro[4.4]nonan-
1-one and spiro[cyclopentane-1,1`-[1H]isoindol]-3`(2`H)-one ring systems (Section
2.3.1). Confirmation of the successful formation of the acids 126 and 127, was shown
by a relative increase in their polarities, shown by a lower Rf from TLC analysis on
silica gel, and the loss of NMR signals corresponding to their respective alkyl ester
groups. In addition, the structure of 126 was established by single crystal X-ray
structural analysis (see Appendix 1). Confirmation of the successful formation of the
amides 128 and 129, was established by a relative decrease in polarity, shown by a
higher Rf from TLC analysis on silica gel, the appearance of an additional amide NH
signal and the appearance of aromatic protons in their respective 1H NMR spectra. In
addition, for 128, a downfield shift was observed for C-7b (δ 30.8 to δ 33.3) and an
upfield shift for the carbonyl amide (δ 173.3 to δ 166.7). A similar upfield shift was also
observed for the carbonyl amide of 129 (δ 178.7 to δ 167.1). Mass spectrometry at both
high and low resolution also confirmed the structures of 126-129.
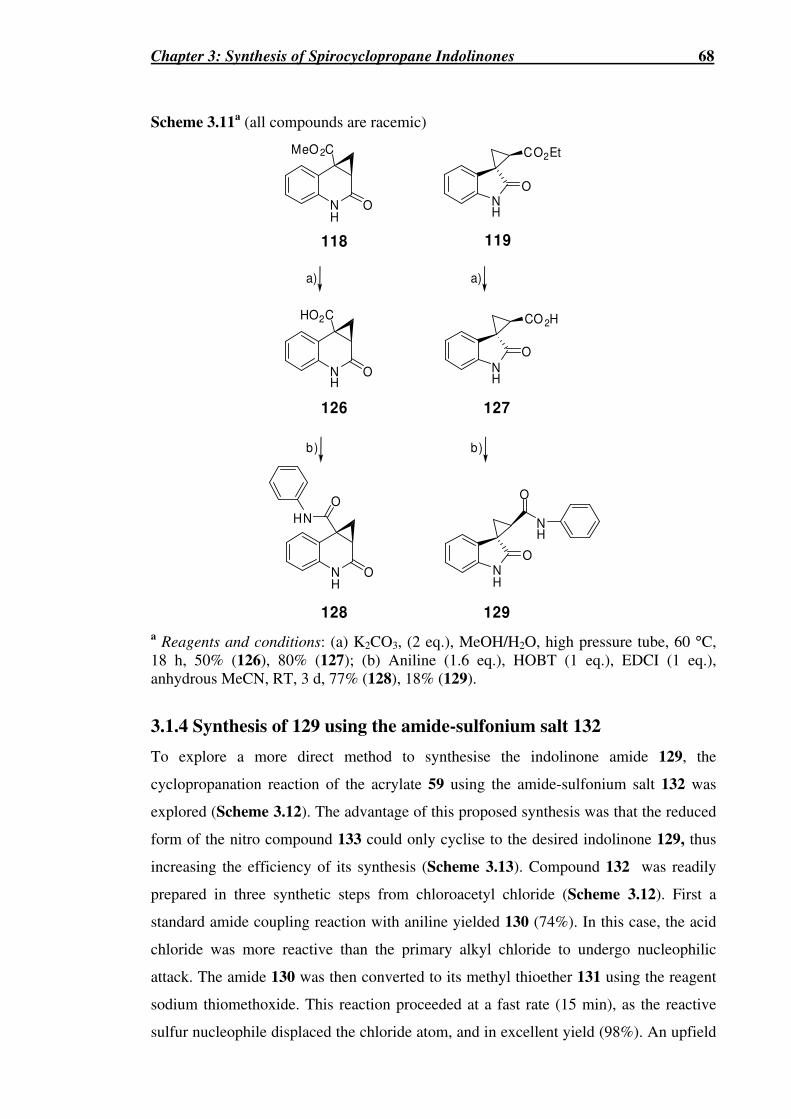
Chapter 3: Synthesis of Spirocyclopropane Indolinones 68
Scheme 3.11a (all compounds are racemic)
NH
O
NH
O
MeO 2C
NH
O
HO2C
CO2Et
NH
O
CO 2H
NH
O
NH
O
NH
O
118 119
128 129
126 127
HN
O
a)
b)
a)
b)
a Reagents and conditions: (a) K2CO3, (2 eq.), MeOH/H2O, high pressure tube, 60 °C, 18 h, 50% (126), 80% (127); (b) Aniline (1.6 eq.), HOBT (1 eq.), EDCI (1 eq.), anhydrous MeCN, RT, 3 d, 77% (128), 18% (129).
3.1.4 Synthesis of 129 using the amide-sulfonium salt 132
To explore a more direct method to synthesise the indolinone amide 129, the
cyclopropanation reaction of the acrylate 59 using the amide-sulfonium salt 132 was
explored (Scheme 3.12). The advantage of this proposed synthesis was that the reduced
form of the nitro compound 133 could only cyclise to the desired indolinone 129, thus
increasing the efficiency of its synthesis (Scheme 3.13). Compound 132 was readily
prepared in three synthetic steps from chloroacetyl chloride (Scheme 3.12). First a
standard amide coupling reaction with aniline yielded 130 (74%). In this case, the acid
chloride was more reactive than the primary alkyl chloride to undergo nucleophilic
attack. The amide 130 was then converted to its methyl thioether 131 using the reagent
sodium thiomethoxide. This reaction proceeded at a fast rate (15 min), as the reactive
sulfur nucleophile displaced the chloride atom, and in excellent yield (98%). An upfield

Chapter 3: Synthesis of Spirocyclopropane Indolinones 69
shift of the methylene protons in both the 1H (δ 4.17 to δ 3.28) and 13C NMR spectra (δ
42.8 to δ 38.7) of 131 and the appearance of the signals corresponding to the methyl
group (δH 2.13, δC 16.0) were observed. The thioether 131 was subsequently methylated
using iodomethane to yield the sulfonium salt 132, as an off-white solid that
precipitated from the reaction mixture. The yield was relatively low (52%), possibly due
to the poor quality of the iodomethane, however the starting material 131 was easily
recovered. Although far less common that their ester-sulfonium analogues, amide
sulfonium salts like 132 have been previously used for the cyclopropanation reactions
of electron deficient alkenes, however normally as their tertiary amide derivatives.252,253
Amide sulfonium salts have also been utilised for epoxidation reactions254 and for the
synthesis of aziridinyl carboxamides.255
Scheme 3.12a
Cl
ClCl
O
HN
O
HN
O
HN
Oa) b) c)MeS Me2S
I
132131130 a Reagents and conditions: (a) Aniline (1.1 eq.), pyridine (1.5 eq.), anhydrous DCM, 0 °C�RT, 1 h, 74%; (b) MeSNa (1.1 eq.), anhydrous MeOH, RT, 15 min, 98%; (c) MeI (10 eq.), anhydrous DCM, RT, 2 d, 52%.
3.1.5 Cyclopropanation of the acrylate 59 using 132
Scheme 3.13a
CO2Me CO 2Me
O
NHa)
NO
NH
O
b)
a Reagents and conditions: (a) 132 (1.5 eq.), DBU (1.1 eq.), anhydrous DCM, RT, 2 d, 39% (b) Fe (8 eq.), AcOH, EtOH, sonication, 2 h, 60%.
The cyclopropanation reaction of the acrylate 59 and the ylide generated in situ from the
amide-sulfonium salt 132 (1.5 eq.) with DBU (1.1 eq.) in anhydrous DCM, at RT for 2
d, yielded solely the trans product 133 in an unoptimised yield of 39%. The formation
of 133 was confirmed by 1D NMR analysis, with the loss of olefinic signals (δH 6.54
and δH 5.89 and δC 127.4 and δC 120.1) and the appearance of signals indicative of a

Chapter 3: Synthesis of Spirocyclopropane Indolinones 70
cyclopropane ring (δH 3.01, δH 2.28 and δH 1.98, and δC 35.9, δC 32.8 and δC 21.0). The
structure of 133 was unequivocally established by single crystal X-ray structural
analysis (Figure 3.3).
Figure 3.3 Single crystal X-ray crystallographic analysis of 133.
The reductive cyclisation of the cycloadduct 133 to provide 129, however proved
difficult. This may have been due to the steric bulk of the amide group. Several methods
were attempted, including catalytic hydrogenation over Pd/C or PdCl2. However these
methods only afforded recovered starting material. Previously, it was discovered that
the reduction of nitro-substituted aromatics can proceed using iron with acetic acid
under sonication.256 This method utilises ultrasonic energy waves to accelerate the
reaction. This method successfully yielded a product with the same 1H NMR spectrum
as 129, synthesised previously, in an unoptimised yield of 60%. This more direct route
(Scheme 3.13), provided a higher yielding synthesis of 129 with an overall yield from
59 of 23% in comparison to the the overall yield from 59 of 2.1% for the longer
synthesis that employed EDSA (Scheme 3.7).
3.2 Cyclopropanations using α-methylene indolinones
To increase the structural diversity around the cyclopropane ring, the cyclopropanation
reaction involving the α-methylene indolinones 108a and 108b, and EDSA was
investigated in order to prepare 2`-substituted 2-pyridyl and 3-pyridyl analogues of 119.
Earlier work by Croce et al.221 reported the synthesis of related 2-aryl substituted
cyclopropanes to these targets (Scheme 3.1) using 3-arylmethylene-indolin-2-ones (134)
or 3-methylene-indolin-2-one and the ylide dimethylsulfonium phenacylide (135)

Chapter 3: Synthesis of Spirocyclopropane Indolinones 71
(Table 3.2). Using either the (E)- or the (Z)-arylmethylene-indol-2-ones 134, yielded
the same result, with only two of the possible four racemic stereoisomers produced
(Table 3.2). Structural confirmation of 136 and 137 was performed through
photoisomerization of the minor isomer 137 to 136, and by 1H NMR studies. The
slightly larger 3J2`,3` values of the minor isomer 137 relative to the major isomer 136,
confirmed its cis stereochemistry and inferred the trans stereochemistry of 136 (Table
3.2).221
Table 3.2221
N
R2
O
R1
SMe2
O
Ph
NO
R1
NO
R1
PhOC PhOCR2 R2
+
134
135
136 137
THF, DMSOor MeCNRT, 48 h
1`
2` 3`
(1`,2`)-t rans-(1`,3`)-cis (1`,2`)-t rans-(1`,3`)-tr ans
δδδδ Substrate
Cpd No. H-2`a H-3`
3J2`,3` (Hz) Ratiob 136 : 137
(Overall Yield)
136 4.37 4.13 8.5 R1 = CH3CO,
R2 = H 137 3.80 4.02 9.5
2 : 1
(75%)
136 4.32 4.09 8.5 R1 = CH3CO,
R2 = OCH3 137 3.75 3.98 9.5
1.8 : 1
(85%)
136 4.36 4.15 8.5 R1 = CH3CO,
R2 = NO2 137 3.78 4.09 9.5
1.5 : 1
(94%)
136 4.35 4.15 8.0 R1 = CH3,
R2 = H 137 3.82 4.03 9.0
2.5 : 1
(60%)
136 4.29 4.04 8.0 R1 = CH3,
R2 = OCH3 137 3.75 3.95 9.0
3 : 1
(70%)
136 4.32 4.10 8.0 R1 = CH3,
R2 = NO2 137 3.87 4.04 9.0
3.5 : 1
(75%) a This proton is deshielded by the benzoyl group in 136. b Ratio determined by 1H NMR analysis of the crude reaction mixture.

Chapter 3: Synthesis of Spirocyclopropane Indolinones 72
The four possible racemic anti-betaine intermediates (A-D) involved in these reactions
are shown in Scheme 3.14. Of these four anti-betaine intermediates, C would be
expected to be favoured energetically in terms of minimizing unfavourable steric and
dipole-dipole interactions. Indeed the major product 136 would be expected to arise via
this intermediate. Betaine intermediate A would be the least favoured due to adverse
steric interactions between the substituted phenyl ring (Ar) and the aromatic ring of the
oxindole moiety in addition to the adverse dipole-dipole interaction between the ketone
(W) and the oxindole carbonyl groups. The minor product, 137 is the (1`,2`)-trans-
(1`,3`)-trans-product formed via the the anti-betaine intermediate D. The alternative
product that would likewise exhibit a 3J2`,3 cis coupling value, is the (1`,2`)-cis-(1`,3`)-
cis-product that would result from the anti-betaine intermediate B, would be
disfavoured due to the adverse dipole-dipole interaction between the ketone and
oxindole carbonyl groups. It therefore seems that this adverse dipole-dipole interaction
in intermediate B, is more unfavourable than the adverse steric interaction of the aryl
rings in intermediate D.
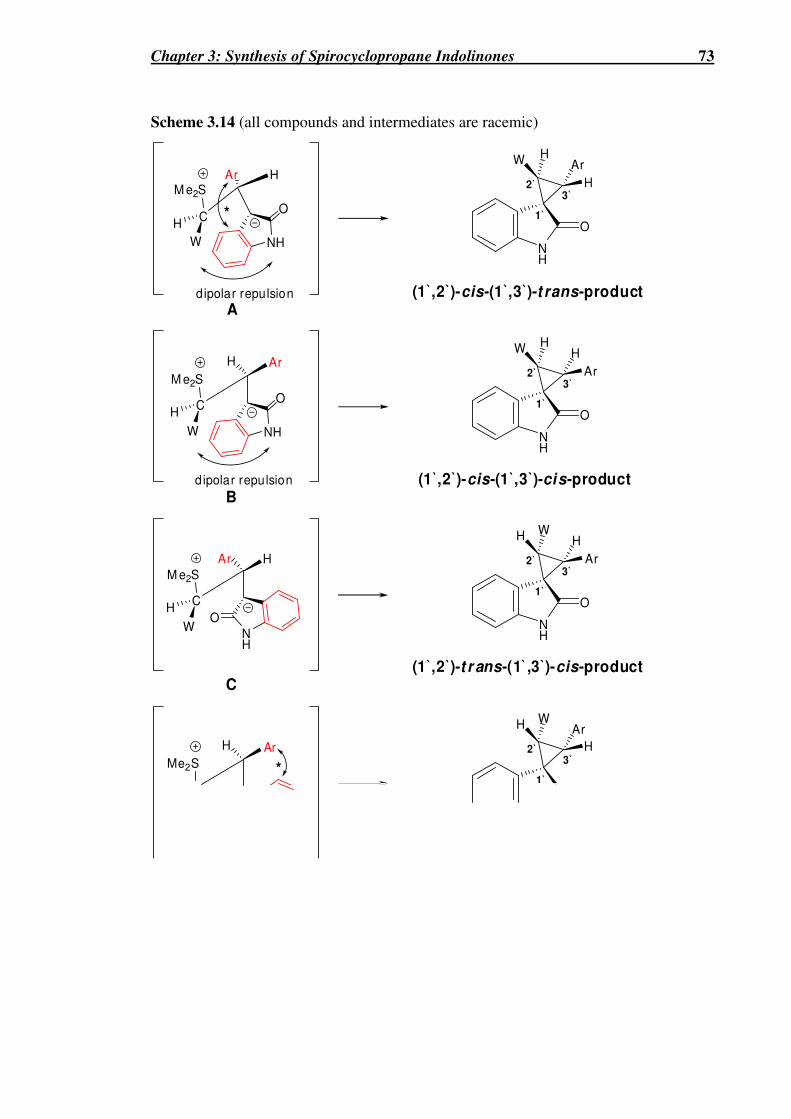
Chapter 3: Synthesis of Spirocyclopropane Indolinones 73
Scheme 3.14 (all compounds and intermediates are racemic)
M e2S
C
Ar H
H
WNH
W H
H
Ar
O
NH
W H
Ar
H
O
M e2S
C
Ar H
H
W
(1`,2`)-t r ans-(1`,3`)-cis-product
NH
H W
Ar
H
O
NH
O
NH
O
M e2S
C
H Ar
H
W NH
O
Me2S
C
H Ar
H W
H
Ar
(1`,2`)-cis-(1`,3`)-cis-product
(1`,2`)-cis-(1`,3`)-t rans-product
*
*
dipolar repulsion
dipolar repulsion
1`
2`3`
1`
2`3`
1`
2`3`
1`
2`3`
A
B
C
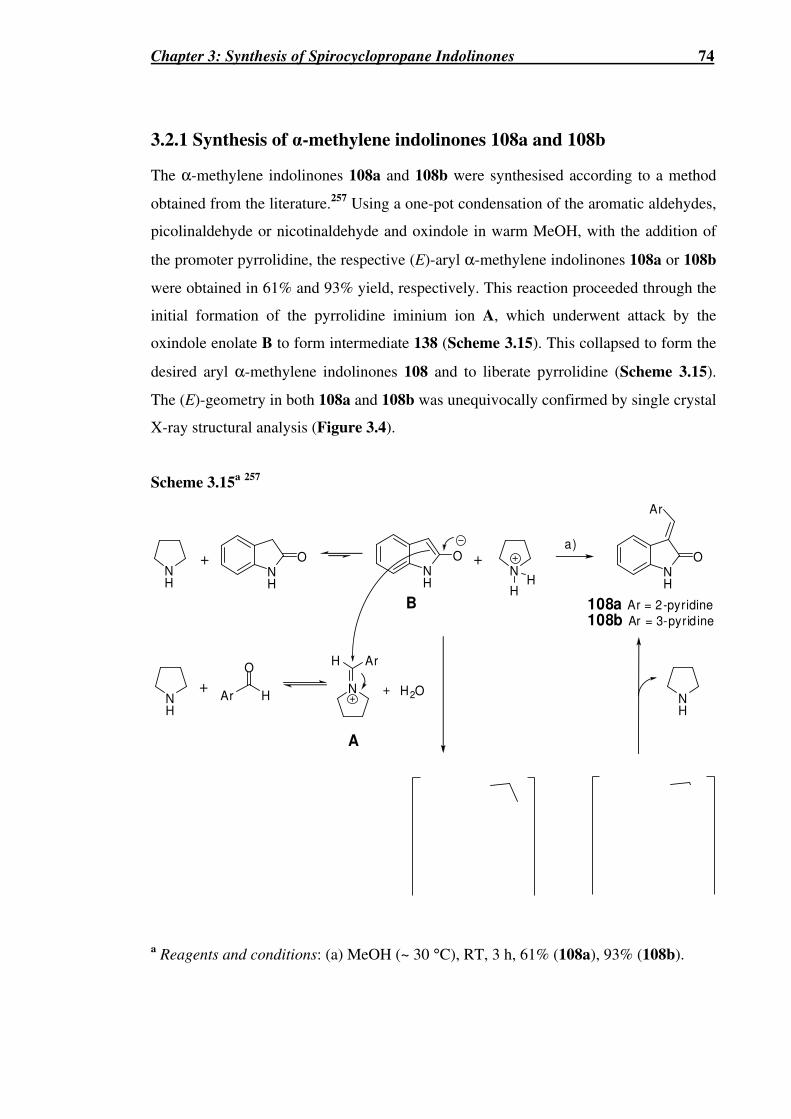
Chapter 3: Synthesis of Spirocyclopropane Indolinones 74
3.2.1 Synthesis of α-methylene indolinones 108a and 108b
The α-methylene indolinones 108a and 108b were synthesised according to a method
obtained from the literature.257 Using a one-pot condensation of the aromatic aldehydes,
picolinaldehyde or nicotinaldehyde and oxindole in warm MeOH, with the addition of
the promoter pyrrolidine, the respective (E)-aryl α-methylene indolinones 108a or 108b
were obtained in 61% and 93% yield, respectively. This reaction proceeded through the
initial formation of the pyrrolidine iminium ion A, which underwent attack by the
oxindole enolate B to form intermediate 138 (Scheme 3.15). This collapsed to form the
desired aryl α-methylene indolinones 108 and to liberate pyrrolidine (Scheme 3.15).
The (E)-geometry in both 108a and 108b was unequivocally confirmed by single crystal
X-ray structural analysis (Figure 3.4).
Scheme 3.15a 257
NH
O
NH
Ar
O
H+ N
H
NH
Ar
O
NH
a)
NH
O
Ar
A
B
NH
+N
+ H2O
HH
+
108a Ar = 2-pyridine
108b Ar = 3-pyrid ine
a Reagents and conditions: (a) MeOH (~ 30 °C), RT, 3 h, 61% (108a), 93% (108b).

Chapter 3: Synthesis of Spirocyclopropane Indolinones 75
Figure 3.4 Single crystal X-ray crystallographic analysis of 108a (left) and 108b (right).
3.2.1 Cyclopropanation reaction of the α-methylene indolinones 108a
and 108b
The cyclopropanation reactions of either 108a or 108b with EDSA generated in situ
from 109 (R = Et) (1.6 eq.) and DBU (1 eq.) in anhydrous acetonitrile for 24 h at RT
yielded a mixture of diastereomeric cyclopropanes (Scheme 3.16). For the
cyclopropanation using 108a, 1H NMR analysis of the crude reaction mixture revealed a
5.6 : 1.8 : 1 mixture of the distereomers, 139a, 140a and 141a, respectively. In contrast,
the cyclopropanation reaction using 108b proved to be a more selective reaction giving
a 43 : 7 : 1 mixture of the diastereomers, 139b, 140b and 141b, respectively. The
purification and separation of these cyclopropanation products by column
chromatography proved difficult. Only the isomers 139a and 141a could be isolated in
diastereomerically pure form, in 27% and 12% yields, respectively from the
cyclopropanation reaction using 108a. The remaining chromatographic fractions
consisted of various mixtures of all three isomers 139a-141a. The major trans-isomer
139b was isolated in diastereomerically pure form in 61% yield from the
cyclopropanation reaction using 108b. The other diastereomers 140b and 141b could
only be obtained as inseparable mixtures.

Chapter 3: Synthesis of Spirocyclopropane Indolinones 76
Scheme 3.16a (all compounds are racemic)
1`
2` 3`
(1`,2`)-tr ans-(1`,3`)-cis (1`,2`)-tr ans-(1`,3`)-t rans
NN
NH
Ar
O
Ar
EtO2C
NH
O
Ar =
a b
ArEtO2C
NH
O
ArEtO 2C
NH
O+ +
108a108b
139 140 141
1H NMR ratio and (yields) of:
139a : 140a : 141a (5.6 : 1.8 : 1) (27%, NA,12%)*
139b : 140b : 141b (43 : 7 : 1) (61%, NA, NA)*
a)
(1`,2`)-cis-(1`,3`)-tr ans
a Reagents and conditions: (a) DBU (1 eq.), anhydrous MeCN, 109 (R = Et) (1.6 eq.), RT, 24 h. *Yields refer to diastereomerical pure compounds.
The structures of 139a and 139b were unequivocally established by single crystal X-ray
structural analysis, although the ester side-chain of 139a is disordered over two sites
(Figure 3.5). The assignment of the relative stereochemistries of the diastereomeric
products produced in Scheme 3.16 was based mainly upon the coupling constants
observed for the cyclopropane methines, CH-3` and CH-2`. The chemical shifts and
coupling constants for the major isomers from both reactions (139a and 139b), and the
corresponding minor isomers according to prevalence ((140a and 140b) and (141a and
141b)) were almost identical, indicative of their same relative configurations. For
simplicity only diastereomic products from the cyclopropanation reaction employing
108a will be described in detail. The methine cyclopropane 1H NMR resonances
appeared as doublets for all diastereomers, with those in the major isomer 139a being
more downfield (δ 3.93 and 3.46). While those for the isomer 141a appeared very close
together, almost like an ABq (δ 3.86 and 3.82) and those for the isomer 140a appeared
more upfield (δ 3.57 and 3.08). The cyclopropane vicinal coupling constants for two of
the products, 139a and 141a, were found to be the same, 3J2`,3` ~ 8 Hz. This was in
contrast to isomer 140a, which had a vicinal coupling constant of 3J2`,3` ~ 10 Hz. Since
in cyclopropanes, cis-vicinal coupling constants (3J2`,3` 6-12 Hz) are usually larger than
trans–vicinal coupling constants (3J2`,3` 4-8 Hz)258 the major diastereomers, 139a and
139b, and the least prominent diastereomers 141a and 141b, were assigned as the 2`,3`-
trans -isomers while, 140a and 140b, were assigned as the 2`,3`-cis-isomers. While the
similar 3`-phenyl derivative 115 was synthesised earlier,246 the relative stereochemistry

Chapter 3: Synthesis of Spirocyclopropane Indolinones 77
at the CH-3` position was not defined in this compound. A comparison of the 1H NMR
data of 115 (δ 3.73, d, J 8 Hz and δ 3.20, d, J 8 Hz) with that of 139a,b suggests that
they have the same relative stereochemistries.
Figure 3.5 Single crystal X-ray crystallographic analysis of 139a (left) and 139b (right).
The four possible racemic anti-betaine intermediates (A-D) involved in these reactions
are shown in Scheme 3.14 (W = CO2Et). The crystal structure attained for the major
products from both reactions accords with the (1`,2`)-trans-(1`,3`)-cis-product. Similar
to the result of Croce et al.221 the observed major products (139a,b) would be expected
to arise via the same anti-betaine intermediate C since the unfavourable steric and
dipole-dipole interactions are minimized. Betaine intermediate A would be the least
favourable due to adverse steric interactions between the 2- or 3-pyridyl subsituent (Ar)
and the aromatic ring of the oxindole moiety in addition to the adverse dipole-dipole
interaction between the ester (W) and oxindole carbonyl groups. We speculate that the
2`,3`-cis-isomer 140a,b has the relative stereochemistry of that shown for (1`,2`)-trans-
(1`,3`)-trans-product that results from the the anti-betaine intermediate D as opposed to
the alternative (1`,2`)-cis-(1`,3`)-cis-product that results from the anti-betaine
intermediate B. The minor product, 141a,b because of the similar 3J2`,3` value to the
trans-product 139a,b should accord with the (1`,2`)-cis-(1`,3`)-trans-product.
Compounds 141a,b may arise from epimerisation of the isomer 140a,b under the basic
reaction conditions as shown previously by Pedregal et al.244 (Table 3.1). Unfortunately
diastereomerically pure samples of 140a,b, could not be obtained to examine this
possibility or to perform meaningful NOESY NMR experiments. The lower
diastereoselectivity observed in the cyclopropanation of 108a compared to 108b, may

Chapter 3: Synthesis of Spirocyclopropane Indolinones 78
be as a result of a more unfavourable dipole-dipole interaction in betaine C between the
oxindole carbonyl group and the pyridine nitrogen atom, in the 2-pyridyl series (C, X =
N, Y =CH) compared to the 3-pyridyl series (C, X = CH, Y = N) (Scheme 3.17). Such
an interaction would destabilize C relative to other reactive anti-betaine conformations
(for example, one in which oxindole carbonyl group is anti to the pyridine ring) leading
to an erosion of product diastereoselectivity by increased formation of the cis-isomer
140a,b.
Scheme 3.17
140a (X = N, Y = CH)140b (X = CH, Y = N)
Me2S
C
H
HE N
H
H EH
O1`
2`3`
C
Y
X
NH
O
XY
In conclusion, the cyclopropanation reaction utilising ester- and amide-stabilized sulfur
ylides has proven to be a successful method to prepare spirocyclopropane-1`,3-indoles,
including novel 3`-(2- and 3-pyridyl)-substituted analogues 139-141 and the novel
cyclopropa[c]quinoline-7b-carboxylates. The relative stereochemistry of key
compounds has also been unequivocally determined by single crystal X-ray structural
analysis. Compounds 108a,b, 139a,b and 141a, were successfully prepared in
diastereomerically pure form and submitted for cytostaticity studies and protein
inhibition studies and the results of these are discussed in Chapter 6.

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 79
CHAPTER 4: SYNTHESIS OF
SPIRO[INDOLE-3,5`-ISOXAZOLIDIN]-2(1H)-
ONES AND SPIRO[INDOLE-3,6`-
[1,3]OXAZINANE]-2,2`(1H)-DIONES USING
THE [1,3]-DIPOLAR CYCLOADDITION
REACTION
Perennial grasses of the genus Phalaris, have been shown to result in poisoning
episodes of livestock, characterized by acute or chronic central nervous signs or by
sudden death. Coerulescine and horsfiline have also been isolated from Phalaris
coerulescens.259,260

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 80
This chapter describes the synthesis of spiro[indole-3,5`-isoxazolidin]-2(1H)-ones and
spiro[indole-3,6`-[1,3]oxazinane]-2,2`(1H)-diones of the type B and C, respectively
through employing [1,3]-dipolar cycloaddition reactions ([1,3]-DC) of the two acyclic
nitrones, 142a and 142b, and the cyclic nitrone, 143 to the acrylate 59 to give
cycloadducts of the type A (Scheme 4.1). These cycloadducts were then converted to
compounds of the type B and C through reduction and cyclisation reactions.
Scheme 4.1
CO2Me
NO 2 NO2
CO 2Me
NH
O
59
O
N
RR`
O
NR`
R
NH
O
OH
NHR`R
NH
O
O
N
R`R
O
[1,3]-dipolarcycloaddition Reduction
Reduction
XCOXCyclisation
R`N
O
RH
or
A B
142a (R = Ph, R` = Ph)
142b (R = Ph, R` = Me)
143
N
O
C

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 81
4.1 Introduction
Through the [1,3]-DC reactions of nitrones with dipolarophiles (A=B, Scheme 4.2) a
wide variety of synthetic and natural biologically active compounds261,262 have been
accessed, including nucleosides, γ-amino alcohols, peptides, amino acids, alkaloids
(quinolizidines, indolizidines and pyrrolizidines), sugars and β-lactams (Scheme 4.2).263
Scheme 4.2
142
R
NO
R`
A B
A B
ON NHR`R
AB
OH
R`
R+
NHR`R
CO2MeR1
OH
ON
R`
R
MeO 2C R1
NR
O
R`
OH
R1
γ-amino alcohols
β -lactam
H
A B =
MeO2C
R`
Cycloaddition reactions have been widely explored as important carbon-carbon bond-
forming reactions. In particular, the [1,3]-DC reaction of nitrones and nitrile oxides with
alkenes is amongst the most widely studied reaction, forming one carbon-carbon bond
and one carbon-oxygen bond. In 1960, Huisgen proposed the now widely accepted
concept of the [1,3]-DC reaction which proceeds as a concerted (but not simultaneous)
process (Scheme 4.3).264,265 This theory rejected Firestone’s proposed reaction
mechanism which proceeded via a diradical intermediate, on the basis of product
stereospecificity (Scheme 4.3).266-268 However, ironically Huisgen further showed the
first example of a two-step cycloaddition, using a thiocarbonyl ylide as the 1,3-
dipole.269 The most common [1,3]-DC reaction of nitrones utilises an alkene
dipolarophile to form isoxazolidines. However, an array of multiply-bonded systems
such as alkynes, allenes,270 isocyanates, nitriles, thiocarbonyls, phosphoranes, sulfenes
and sulfinyl groups have also been demonstrated.265,271 In the case of the addition to
alkenes, the isoxazolidine formed can possess up to three new chiral centres. Therefore,
a combination of regioisomeric and diastereomeric products can result. Finally,
asymmetric versions of the [1,3]-DC reaction have been displayed through either using

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 82
chiral substrates,137,272 or chiral nitrones,273-277 or by precomplexation of the
dipolarophile with various chiral Lewis acids.278-280
Scheme 4.3
ab
cd e
d e
cb
a
e d
cb
a+ +
1,3-dipole dipolarophile regiomeric products
d e
cb
a
diradical
Huisgen's Mechanism
Firestone's Mechanism
4.1.1 Regioselectivity and Stereoselectivity of the [1,3]-DC reaction
4.1.1.1 Regioselectivity of the [1,3]-DC reaction
Regioselectivity and stereoselectivity of the [1,3]-DC reaction using various substituted
alkenes has been heavily theorised and researched. Though recently other theories such
as Density Functional Theory-Hard and Soft Acids and Bases principle (DFT-
HSAB),281 have been developed, the Frontiers Molecular Orbital (FMO) theory
developed by Fukui282,283 contributes important insights into the majority of [1,3]-DC
reactions. Previously, cycloadditions have been categorised by Sustmann284 into three
different types (Scheme 4.4). Type I classifies cycloadditions whose dominant
interaction occurs between the Highest Occupied Molecular Orbital (HOMO) of the
dipole and the Lowest Unoccupied Molecular Orbital (LUMO) of the dipolarophile.
The majority of Diels-Alder reactions fall into this classification (Scheme 4.4). Type III
classifies the opposite situation, where the dominant interaction is the LUMOdipole and
HOMOdipolarophile, e.g. alkene ozonisation (Scheme 4.4). The third classification, Type II,
exists when the similarity between the LUMO and HOMO energies of both dipole and
dipolarophile is such that it implies that both Type I and Type III interactions are
significant in determining the reactivity and regiochemistry (Scheme 4.4).265

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 83
Scheme 4.4
E
LUMO
HOMOType I
Dipolarophile
Dipole
Type II Type III
DipolarophileDipole
Dipolarophile
Dipole
The [1,3]-DC reaction of nitrones is believed to correspond to this Type II classification.
It has been observed with various mono-substituted alkenes, that up to four products
may result. These products can be classified according to their regiochemistry, as either
the 4- or 5- regioisomer, and their stereochemistry, as either endo or exo, in reference to
the position of the withdrawing group of the dipolarophile relative to the dipolar linkage
during the transition state. With most dipolarophiles, the 5-regioisomeric isoxazolidine
has been found to be favoured, in high selectivity. However, with increasing electron
affinity of the dipolarophile and decreasing ionisation potential of the nitrone, the
formation of the 4-regioisomeric isoxazolidine has been shown to increase (Scheme
4.5). This can be explained by the FMO theory.265 In electron-rich dipolarophiles, such
as in the case when W = Ph, the dominant interaction is that of the LUMOdipole and
HOMOdipolarophile, that is it resembles a Type III process and the 5-regioisomer will result.
In electron-poor dipolarophiles, such as in the case when W = NO2, the dominant
interaction is that of the HOMOdipole and LUMOdipolarophile, that is it resembles a Type I
process and leads to the 4-regioisomer. Theoretically, a point must exist in which there
is a switch from the LUMOdipole-HOMOdipolarophile control (ie. Type III) to the
HOMOdipole-LUMOdipolarophile (ie. Type I) as one increases the electron-withdrawing
power of the substituents on the dipolarophile. This point has been shown to be
approached with methyl acrylate (W = CO2Me), which yields a regioisomeric mixture
of cycloadducts in the case of nitrone 142b (Scheme 4.5).285

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 84
Scheme 4.5
ON
ON
R`
H
O2N
NO2 WR`
R
W
5-regioisomer4-regioisomer
R H
142a (R = Ph, R` = Me)142b (R = Ph, R` = Ph)
H N
R
O
R`
Nitrone W Ratio (4-isomer : 5-isomer)a Ref.
142b Ph 0 : 100 286,287
142b CO2Me 30 : 70 285
142a CO2Me 0 : 100 288
142a NO2 100 : 0 286,287 a Ratio determined by 1H NMR analysis.
E
LUMO
HOMO
DipolarophileDipole Dipolarophile
Dipole
O 2N
O 2N
NO N
O
Ph
NO
NO Ph
4.1.1.2 Endo versus exo stereoselectivity of the [1,3]-DC reaction
The factors determining the stereoselectivity of the [1,3]-DC reaction are complex.
They include secondary orbital interactions,289 nitrone E to Z-isomerization,290-292 and
the structure of the substrate (Table 4.1).265,293 In the classic case, stereochemistry is
governed by secondary orbital interactions analogous to those directing the endo/exo
approach in the Diels-Alder reaction.265 When secondary orbital interactions are
negligible, the major product has been observed to be the exo-adduct, 145. However,
when secondary orbital interactions can occur between the nitrogen atom of the nitrone

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 85
and the vicinal sp2-hybridised substituent of the alkene, the transition state is stabilized
and the endo-adduct, 144 predominates (Table 4.1). One such example of this, is the
[1,3]-DC reaction between the acyclic nitrone 142b and dimethyl maleate where
favourable secondary orbital interactions in the endo-transition state, leads to the
stereoselectivity of 90 : 10 of the endo- and exo- isomers 144 and 145, respectively
(Table 4.1, entry 1).289 However, the [1,3]-DC reactions between acyclic nitrones and
dipolarophiles such as methyl acrylate,294 α-methylstyrene (Table 4.1, entry 2),295 and
styrene (Table 4.1, entry 3)296 exhibit poor selectivity. One possible reason why
stereoselectivity is often hard to predict in the case of acyclic nitrones, is at high
temperatures they can undergo E to Z-isomerization. Most cyclic nitrones on the other
hand, are locked into the more reactive (E)-configuration.297 In addition to these effects,
diastereofacial selectivity (or enantioselectivity if the substrates are both achiral) must
also be considered.265
Table 4.1 (all products are racemates)
H
N
O
R
5-endo
R2
R1
R`
R2
N
O
R1
R`R
5-exo
R2
N
O
R1
R`R
H
N
O
R
R1
R`
N
R
R`
H
R2
R1LUMO
HOMOO
N
R
R`
H
LUMO
HOMOO
R3
R3
R3
R3
144
R3
R3
Entry Nitrone R1 R2 R3 144 : 145a Ref.
1 142a CO2Me H CO2Me 90 : 10 289
2 142a Ph Me H 55 : 45 295
3 142a Ph H H 33 : 67 296 a Ratios determined by 1H NMR analysis.

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 86
Furthermore, Mélot et al.,293 in their study of the [1,3]-DC reactions between α-
methylenelactams, 146 and acyclic nitrones 142, observed a diastereomeric mixture of
only the 5-regioisomers, 147 and 148 in all cases. The endo-adduct, 147 was favoured
in all [1,3]-DC reactions, with high or exclusive selectivity. The reactions using α-
benzoyl nitrone 142c were completely stereoselective for 147 for all α-methylene
lactams. This was due to the fact that the exo-product would be disfavoured due to an
adverse dipole-dipole interaction, in the transition state, between the carbonyl of 142c
(R = PhCO) and the lactam carbonyl.
Table 4.2293
H
N
O
R
5-endoR`
NO
R`R
5-exo
N
R
R`
H
LUMO
HOMO O
N
R
R`
H
LUMO
HOMOO
Bonding interaction
Favourable secondary orbital interactionKey:
147
148
Unfavourable dipole interaction
N O
R1
n( )
H R
NR` O
+
to luene∆
N O
n( )
R1
NO
R1
n( )
H
N
O
R
R`
N
O
( )n
R1
N
O
R1
NR1
O
n( )
n( )
146 142
NO
R`R
NO
R1
n( )
Nitrone 146 Time (h) Temp (°°°°C) Yield 147 : 148
142ca R1 = p-MeOPh, n = 0 0.25 110 92%b 100 : 0
142a R1 = p-MeOPh, n = 0 240 55c 62%b >90 : <10
142b R1 = p-MeOPh, n = 0 0.5 110 83%b >95 : <5
142b R1 = p-MePh, n = 0 1 110 78%b >95 : <5
142ca R1 = CH3, n = 1 0.5 80 72%b 100 : 0
142b R1 = CH3, n = 1 96 50 67% 85 : 15
142ca R1 = CH3, n = 2 0.5 80 80%b 100 : 0
142b R1 = CH3, n = 2 96 50 73% 80 : 20 a 142c (R = PhCO, R` = Ph), bYield of pure 147, cReaction at 110 °C resulted in by-products.

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 87
Ali et al.297 has examined the stereoselectivity of the [1,3]-DC reaction in cyclic nitrone
systems employing 2,3,4,5-tetrahydropyridine 1-oxide 149 and monosubstituted or 1,1-
disubstituted alkenes (Table 4.3). The [1,3]-DC reaction of 149 and propene, attained
exclusively the 2-exo cycloadduct 150 (Table 4.3, entry 1), since secondary orbital
interactions are negligible. Stereoselectivity for the 2-exo-product over the 2-endo-
product was observed in the case of ethyl vinyl ether, allyl alcohol, or styrene (Table
4.3, entries 2-4). The substrates, methyl acrylate, acrylonitrile and acrolein (Table 4.3,
entries 5-7) however all afforded a mixture of all four possible adducts, 150, 151, 152
and 153. Interestingly acrolein showed a complete reversal of regioselectivity with the
mixture of diastereomers revealed as 3 : 5 : 24 : 68, respectively (Table 4.3, entry 7).
The [1,3]-DC reaction with various 1,1-disubstituted alkenes with 149 yielded a
diastereomeric mixture of the 2-regioisomers, 150 and 151 (Table 4.3, entries 8-14)
with selectivity for the 2-endo-product, 151.

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 88
Table 4.3
2-endo
R1
N
O
R2
2-ex oNO
R1
R2
R2
N
O
R1
H H
3-endo N
OR2
R1H
150
152 153
151
3-exo N
OR1
R2H
N
O
R1 R2+
149
NO
R2
R1
NO
R2
R1
NO
R1
R2
Entry R1 R2 Temp (°°°°C)
Time (h)
Solvent 150 : 151 : 152 : 153a
(Combined Yield %)
1 Me H 110 4 Toluene 100 : 0 : 0: 0 (53%)
2 OEt H 40 12 EtOH 93 : 7 : 0: 0 (67%)
3 CH2OH H 80 5 Toluene 83 : 17 : 0: 0 (84%)
4 Ph H 110 5 Toluene 78 : 22 : 0: 0 (92%)
5 CO2Me H 0 0.2 DCM 69 : 15 : 10: 6 (96%)
6 CN H 25 0.5 DCM 61 : 20 : 13: 6 (92%)
7 CHO H 25 0.2 DCM 3 : 5 : 24 : 68 (96%)
Entry R1 R2 Temp (°°°°C)
Time (h)
Solvent 150 : 151a (Combined Yield %)
8 CHO Me 25 0.4 DCM 0 : 100 (94%)
9 CO2Me Me 25 1.5 DCM 4 : 96 (86%)
10 CH2OH Me 95 5 Toluene 15 : 85 (77%)
11 CH2OCOMe Me 95 2 Toluene 17 : 83 (66%)
12 CH2OSiButMe2 Me 95 1.5 Toluene 30 : 70 (55%)
13 CH2OTHP Me 95 1.5 Toluene 33 : 67 (58%)
14 Ph Me 95 1 Toluene 42 : 58 (71%) a Ratio determined by 1H NMR analysis

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 89
During the course of this work, Parmar et al.298 reported the synthesis of the related
isoxazolidine spirocyclic oxindoles 154 and 155 to those required in this project using
the [1,3]-DC reaction of 3-methylene-indolinone 116 (Y = H) with various C-
substituted phenyl-N-phenylnitrones 142d (Table 4.4). Utilising microwave radiation,
with a domestic 850 W microwave oven, and solvent-free conditions, yielded a mixture
of both the 4- and 5-regioisomers, 154 and 155, respectively. The 5-regioisomer, 155
(Pathway II) was favoured in all reactions, with the highest regioselectivity observed of
1 : 4.88 based upon product yields (Table 4.4, entry 7). Parmar et al.298 also noted that
only one diastereomer was seen for both regioisomers, 154 and 155, with the relative
stereochemistry confirmed by single crystal X-ray crystallographic analysis. The
stereochemistry elucidated for 154 and 155, indicated that they were formed via an exo-
like and endo-like transition state, respectively, with respect to the ethyl ester group of
116 (Table 4.4). These selectivities show a maximising of the favourable secondary
orbital interactions that can occur between the nitrogen atom of the nitrone and the
oxindole carbonyl or ethyl ester group of 116 (Y = H), and a minimising of the
unfavourable steric interactions that can occur between the aromatic rings.

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 90
Table 4.4
NH
O
NOEtO2C
R2
R1
HN
CO2EtO
NH
O
O
NEtO2C
R1
R2
Pathway I
Pathway II
4`-exo-regioisomer [X-ray]
5`-endo-regioisomer [X-ray]
A
B
H
N
O
R1
R2
154
155
H
N
O
R1
R2
NH
O
EtO2C
ON
H
R1
R2
+µW
116 (Y = H) 142d
Y
NHEtO2C
O
Entry R1 R2 Time (min)a
154 (Yield %)
155 (Yield %)
Ratio (154 : 155)b
1 Cl H 4.0 22.7 33.8 1 : 1.49
2 Br H 4.0 12.1 38.5 1 : 3.18
3 NO2 H 5.0 12.8 27.1 1 : 2.12
4 F H 4.0 10.0 43.7 1 : 4.37
5 CH3 H 4.0 15.6 39.2 1 : 2.51
6 H Cl 5.0 21.4 33.0 1 : 1.54
7 H Br 4.0 9.1 44.4 1 : 4.88
8 H NO2 4.0 12.1 42.1 1 : 3.48
9 H F 5.0 11.7 23.2 1 : 1.98
10 H CH3 4.0 15.8 36.1 1 : 2.28 a Microwave irradiation was performed at 1 min pulses with 10 second intervals. b Ratio based on product yields.

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 91
Mélot et al.299 recently reported the synthesis of spiro[1H-isoindol-1,5`(4`H)-
isoxazole]-7(6H)-ones employing the [1,3]-DC reaction between isoindolinones (156)
and acyclic nitrones (142) under microwave irradiation conditions. These reactions
yielded a mixture of the two 5`-regioisomeric cycloadducts (157 and 158).
Unfortunately, Mélot et al.299 just notes that when R1 = p-NCC6H4 the diastereomeric
excess (de) was 35-60%, without defining which diastereomer was favoured.
Scheme 4.6299
156
N
O
R1
H R
NR` O
+µW
N
O
R1
NO
+
R`R
H
N
O
R1
NO
R`H
R
142 157 158
During our study, synthetic investigations towards natural spirocycles and their
derivatives that employed a chiral, one-pot [1,3]-DC reaction, were reported. This
reaction first utilised by Williams et al.,185 employed a chiral azomethine ylide derived
from 5,6-diphenylmorpholin-2-one (159),300 and various aldehydes (RCHO) and the 3-
methylene-indolinone, 116 (Y = H) (Scheme 4.7). These [1,3]-DC reactions yielded a
mixture of three products, 160, 161 and 162, based on the E/Z-formation of the ylide,
the α/β-approach with respect to the phenyl rings of 159 and the endo/exo approach
with respect to the ethyl ester group. Cycloadduct 160 was found to result from the (E)-
α-exo-transition state, 161 from the (E)-α-endo-transition state and 162 from the (Z)-α-
exo-transition state. Selectivity for cycloadduct 160 over 162 was observed for all
aldehydes tested, due to the bulkiness of the aldehyde substituent (R) favouring the
formation of the E-ylide. High exo-selectivity was also observed, revealing adverse
steric effects with the endo-approach. This reaction set up four contiguous stereogenic
centres and constructed the entire prenylated tryptrophyl moiety of spirotryprostatin B,
in a single step. Removal of the chiral auxiliary was achieved by catalytic
hydrogenation using PdCl2 and then esterification of the resulting carboxylic acid with
TMSCHN2.

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 92
Scheme 4.7185
NH
EtO2C
O
NH
O
Ph
PhO
159
RCHOmol. sieves.Toluene
NH
O
N
NH
N
O Ph
Ph
R
EtO2C
HO
O
NH
N
O Ph
Ph
R
EtO2C
HO
O
O
O
R H
EtO 2C
PhPh
+ +
116 (Y = H)
160 161 162
Y
(E)-α-exo (Z )-α-exo(E)-α-endo
(E )-α-exo
NH
EtO2C
O
N O
PhPh
O
R
Entry Aldehyde Temp 160 (Yield %)
161 (Yield %)
162 (Yield %)
160 : 162
1 R = Ha Reflux 28 11 0 >20 : 1
2 R = BzOCH2 Reflux 44 14 0 >20 : 1
3 R = BzOCH2 60 °C 54 8 0 >20 : 1
4 i-Pr Reflux 43 11 5 8.6 : 1
5 i-Pr 60 °C 74 6 Trace >20 : 1
6 i-Bu Reflux 84 1 0 >20 : 1
7 i-Bu 60 °C 86 0 0 >20 : 1
8 Me2(OMe)CCH2b Reflux 29 0 0 >20 : 1
9 Me2(OMe)CCH2b 60 °C 82 1 0 >20 : 1
10 p-MeOPhc Reflux 60 0 0 >20 : 1 a 9% of another regioisomer was also isolated. b A trisubstituted olefin resulting from elimination of the tertiary alcohol was also isolated c Required prolonged heated (>24 h) to obtain the reported yield, whereas most reaction times were 2-8 h.

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 93
More recently, Wang et al.106 reported a similar synthetic strategy employing instead
163 (R1 = Cl, R2 = aryl), to yield only the (Z)-α-exo-cycloadduct, 162 (Scheme 4.8).
The lactone of 162 was condensed with various amines to form amides, including 164.
Finally, the chiral auxiliary was removed using Pb(OAc)4 to yield 27. Spirocycles 27
were found to be a novel class of potent, non-peptide, small molecule MDM2-p53
inhibitors, with the most potent derivative 27 (R = ButCH2, R1 = Cl, R3 = F and X = O)
displaying a Ki of 3 ± 1.5 nM.106
Scheme 4.8a 106
NH
R2
O
R1NH
N
O Ph
Ph
R
R2
HO
O
R1
163 162 164 27
R2 = Cl
R3
NH
NOH
Ph Ph
RR2
H
O
O
R1
HN
N
X
NH
NH
RR2
H
O
O
R1
HN
N
X
a) b) c)
a Reagents and conditions: (a) 4 Å molecular sieves, 159, toluene, 70 °C; b) 1-(2-aminoethyl)-piperidine, THF; (c) Pb(OAc)4, DCM:MeOH (1 : 1), 0 °C.

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 94
In addition, Schreiber et al.301 highlighted the synthetic utility of this reaction for
combinatorial synthesis, through utilising macrobead-supported aldehydes, either
enantiomer of the chiral auxiliary 159, and isatin-derived dipolarophiles bearing an
allylic ester group (165) (Scheme 4.9).
Scheme 4.9a 301
165159
NH
O
HN
N
OPh
Ph
HO
O
a)Si
i -Pr i -Pr
R CHO
R1
OO
+ +
Sii -Pr i -Pr
RO
O
R1
NH
O
Ph
PhO
a Reagents and conditions: (a) Mg(ClO4)2, pyridine, HC(OMe)3, toluene, RT.
4.2 Synthesis of Nitrones
Generally nitrones are easily prepared through using several well-established methods
in the literature (Scheme 4.10). Nitrones can be obtained through the condensation of
carbonyl compounds with N-monosubstituted hydroxylamines (Scheme 4.10, equation
(1))302-306 and the oxidation of N,N-disubstituted hydroxylamines (Scheme 4.10,
equation (2)).303,307-309 However, unless commercially available, the preparation of these
starting hydroxylamines is quite tedious. Oximes can also be alkylated to afford nitrones
(Scheme 4.10, equation (3)). The tungstate-catalysed oxidation of secondary amines
with hydrogen peroxide is also a well-established method affording acyclic and cyclic
nitrones (Scheme 4.10, equation 4)).310 Recently the use and the generation of a
catalytic amount of Davis reagent 166 in situ by the oxidant m-CPBA to yield nitrones
has been displayed (Scheme 4.10, equation (5)).311

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 95
Scheme 4.10
(1) R1R2C=O + R3NHOH
(3) R1R2C=NOH + R3X
(2) R1R2CHNR3OH
R1R2CHNHR3 + H2O2 (4)
R1R2CHNHR3 (5)
R2 N
R1
O
R3
H2O
HX
[O]
Na2WO4
S N
OO
O166
4.2.1 Synthesis of acyclic nitrones 142a and 142b
To prepare acyclic nitrones 142a and 142b the standard condensation of benzaldehyde
with N-methyl or N-phenyl hydroxylamine was employed. These nitrones were obtained
in excellent yields and were crystallised easily from solution (Scheme 4.11).
Scheme 4.11
O
H
R`
NH
HO R`N
OR`
NOH
HO
R`N
O
OH
H
142a (R` = Me) (99.7%)
142b (R` = Ph) (90%)
+ H2O
4.2.2 Synthesis of the cyclic nitrone 143
To synthesise the cyclic pyrrolidine nitrone, 143 two methods were employed (Scheme
4.12). The first was the common tungstate-catalysed oxidation of pyrrolidine.310,312 The
tungstate reaction proceeds through the N-oxidation of the secondary amine (A),
pyrrolidine in this case, via its corresponding N,N-disubstituted hydroxylamine (B) by
peroxytungstate (w-OOH where w = WO3-, WO6
-).312 B is further oxidised to form the
zwitterion C, which dehydrates to yield the desired nitrone. Peroxytungstate is initially
formed from sodium tungstate and hydrogen peroxide. The dimerisation and
trimerisation of 143 upon attempted isolation, is well known.265 To prevent this event,
the crude reaction mixture was used directly in the subsequent [1,3]-DC reaction,
however no desired product was seen. Nitrone 143 was found to be readily purified by
column chromatography using 10% EtOH:CHCl3 as eluent. However, only the “wet-
yield” for 143 could be ascertained, as 21% and it was immediately diluted upon
isolation with toluene to prevent dimerisation.

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 96
Scheme 4.12a 312
w OH + H2O 2 w OOH + H2O
w OOH W
O
O OO
OH
HN
w OOHw OH+
w OH+
+ H2O
A
B
B
143C
NH
N
O
N
OOH
N
OH
N
OOH
N
OH
a w = WO3
-, WO6-
In an attempt to generate 143 in greater yield, the Davis reagent was used.311,313
Stappers et al.311 in an attempt to develop a safe and scalable amine to nitrone oxidation
method successfully employed a catalytic amount of the Davis reagent precursor, 169 to
generate the Davis reagent 166 in situ through the oxidant m-CPBA (Scheme 4.13). The
Davis reagent 166 was subsequently reduced upon oxidising amine 167 to nitrone 168.
However, using this method, in this work, resulted in none of the desired nitrone, 143.
Scheme 4.13311
NH NO
167 168
4.3 [1,3]-Dipolar cycloaddition reactions using 59
4.3.1 [1,3]-DC reactions using acyclic nitrones 142a and 142b
The [1,3]-DC reaction of 59 with acyclic nitrones, 142a or 142b was studied as part of
this project and the results are tabulated in Table 4.5. Studies to optimise the reaction

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 97
conditions were first performed using 142a. Initial experiments utilised anhydrous
DCM as the solvent, with heating at 60 °C in a sealed tube. This reaction did not
proceed to completion even after 7 days (Table 4.5, entry 1). When the reaction was left
for a shorter period, of 4 days, a 1H NMR analysis of the crude reaction mixture
revealed a mixture of 170a, 171a and 59, in a ratio of 1.0 : 0.57 : 0.14, respectively
(Table 4.5, entry 2). Separation of these three compounds however, proved difficult,
due to 171a and the starting acrylate 59 having very similar Rf values. However, 170a
and 171a could be isolated in pure form, in yields of 26% and 20%, respectively. To
increase the yield and efficiency of the reaction, microwave irradiation was employed.
Initial experiments employed anhydrous toluene, as the solvent. After 10 min at 150 °C,
the reaction was shown to be incomplete, and so an additional 30 min of microwave
radiation at 150 °C was given. 1H NMR analysis of the crude reaction mixture at both
reaction times revealed the same ratio of 170a : 171a : 59 as 1.0 : 1.0 : 0.77,
respectively (Table 4.5, entry 3). With the hope of increasing the yield, the solvent was
abandoned, a higher amount of nitrone was employed (1.2 eq.) and progressively longer
reaction times were used (30 min, 1 h and 2.5 h) (Table 4.5, entry 4). It was seen that
the reaction would not go to completion and upon longer reaction periods, degradation
of products was observed (Table 4.5, entry 4). Thirty minutes was found to be the
optimal time for maximising conversion and minimizing decomposition, which yielded
a mixture of 170a, 171a and 59 in a ratio of 1.0 : 1.5 : 1.3, respectively (Table 4.5,
entry 5). These compounds were isolated, after purification by column chromatography,
in yields of 15%, 30% and 29%, respectively. The cycloaddition of 142b with 59 using
these optimised conditions of 30 min of microwave irradiation at 150 °C yielded a
mixture of 170a, 171a and 59 in a ratio of 3.0 : 1.0 : 0.8, respectively (Table 4.5, entry
7). Purification of the diastereomeric products again proved difficult, but by using a
chromatotron®, pure samples of 170b, 171b, and 59 were isolated in respective yields
of, 58%, 6% and 11%. However, fractions consisting of a mixture of 171b and 59 were
also obtained. The poor yields and the apparent disparity between the 1H NMR ratios
and the corresponding isolated yields for all these [1,3]-DC reactions were due to
purification difficulties, with poor resolution often encountered with the isomeric
products and starting alkene 59.
To understand why complete consumption of the alkene was never observed, pure 171a
was subjected to microwave irradiation, in the absence of solvent, for 30 min at 150 °C

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 98
(Table 4.5, entry 6). This experiment yielded a surprising result, with a similar mixture
of products being seen, with 170a, 171a and 59 being formed in a ratio of 1.0 : 1.1 : 1.5,
respectively. It was assumed that the nitrone 142a was also formed, but perhaps
dimerized or decomposed under the thermal conditions. The same experiment was
conducted on 170b. This resulted in the appearance of alkene 59, however it was
difficult to determine the ratio of 170b and 171b in the mixture due to poor resolution
of the 1H NMR spectrum. These experiments revealed that these [1,3]-DC reactions
were actually reversible and hence why a complete reaction was never observed.
Cycloreversion is a feature of [1,3]-DC reactions that has received little comment
throughout the literature.265 Due to these cycloreversion results, the cycloaddition
products, 170a : 171a must be formed under thermodynamically controlled rather than
kineticically controlled conditions.
Table 4.5
CO2Me
NO2
N
H Ph
OR`
+
5`-exo- 5`-endo-
+
59 142a142b
R` = Me R` = Ph 170 171
NO
CO2Me
NO2
R`
NO
CO2Me
NO2
R`
Reaction Conditions Results
Entry 59 : 142a Solvent ∆∆∆∆ Method Time (Temp) 170a : 171a : 59a (Yields)b 1 1 : 1.45 anhydrous
DCM sealed tube
7 d (60 °C) 1.0 : 0.75 : 0.5
2 1 : 1.1 anhydrous DCM
sealed tube
4 d (60 °C) 1.0 : 0.57 : 0.14 (26%, 20%, e)
3 1 : 1 anhydrous toluene
µW 10 min, 30 mind (150 °C)
1.0 : 1.0 : 0.77 (15%, 29%, 29%)
4 1 : 1.2 no solvent µW 2.5 h (150 °C) 1.0 : 1.0 : 1.3d,e 5 1 : 1.2 no solvent µW 30 min (150 °C) 1.0 : 1.5 : 1.3
(15%, 30%, 29%)
6 171a no solvent µW 30 min (150 °C) 1.0 : 1.1 : 1.5e
Entry 59 : 142b Solvent ∆∆∆∆ Method Time (Temp) 170b : 171b : 59a (Yields) 7 1 : 1.3 no solvent µW 30 min (150 °C) 3.0 : 1.0 : 0.8
(58%, 6%f, 11%f) a Ratio determined by 1H NMR analysis of crude reaction mixture. b Yields after purification by column chromatography. c Additional heating was required for completion. d Degradation of products was observed. e Yield not recorded. f A mixture of 170b, 171b and 59 was also isolated.

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 99
Only the 5-regioisomer and not the 4-regioisomer was observed in all [1,3]-DC
reactions using the acyclic nitrones 142a or 142b. The regiochemistry and relative
stereochemistry of 170a, 171a and 170b, 171b were confirmed through extensive 1D
and 2D NMR experiments. The cycloadducts 170a,b and 171a,b were all assigned as
the 5-regioisomer because the isoxazolidinone protons nearly all appeared as a dd (the
exception was 170a, whose signals were very broad). In the 4-regioisomer the
stereogenic methine proton, geminal to the phenyl substituent would be expected to be a
singlet in the 1H NMR spectrum (Scheme 4.15). The signals for the methylene protons
in the products observed were also more upfield (δ 3-4.5 ppm) than those expected for
the 4-regioisomer, (δ 5-6 ppm) which would be deshielded due to the electronegative
oxygen atom (c.f. Parmar et al.298 and Mélot et al.293). Indeed the J values of 171a, 170b,
and 171b all indicated an ABX system, with both geminal (~ J 12.7-13.5 Hz) and
vicinal couplings (6-9.6 Hz) that would only occur in the 5-regioisomer. For example,
170b had the following 1H NMR (CDCl3, 300 MHz) data: δ 4.83 (dd, J 9.5, 7.5 Hz, 1H,
CH-3`); 4.12 (dd, J 13.5, 7.5 Hz, 1H, CHβCHα-4`); 2.54 (dd, J 13.3, 9.3 Hz, 1H,
CHαCHβ-4`). Furthermore, the spirocarbon for all diastereomers was quite downfield
(δC 82.8-85.6 ppm) in comparison to the typical chemical shift for the 4-regioisomer
(~δC 67 ppm), indicating it was adjacent to an electronegative oxygen.298 Previously
Parmar et al.298 in their similar studies using a m- or p-substituted versions of 142b and
3-methylene-indolinones, did observe both regioisomers, with selectivity for the 5-
regioisomer. These experimental observations were in agreement with our calculated
HOMO and LUMO energies of dipolarophiles 59, 116 (Y = H) and nitrone 142b using
Spartan ‘04 (AM1) (Scheme 4.14). The dominant interaction in both reactions, was
predicted to be that between the LUMOdipolarophile and HOMOdipole leading to the 5-
regioisomer. However for Parmar et al’s case.,298 the HOMOdipolarophile energy was found
to be higher for 116 (Y = H) (-8.51 eV) than for 59 (-9.39 eV), resulting in a smaller
difference between the two possible HOMO and LUMO interactions and is therefore
consistent with the poorer selectivity observed for 116 (Y = H).

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 100
Scheme 4.14
E (eV)
LUMO
HOMO
142b 59 142b 116 (Y = H)
∆ = 9.35∆ = 9.35 ∆ = 10.15∆ = 11.03
-9.39
1.641.14
1.64
-8.51-8.21
1.14
-8.21
Dipole Dipolarophile Dipole Dipolarophile
Scheme 4.15
Pathway I
Pathway II
4-regioisomer
5-regioisomer
H
N
O
R ON
NO2
CO2Me
R
CO2Me
NO2
H
N
O
CO2MeNO2
RN
O
NO 2
CO2Me
R

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 101
The structure of 170a however, was unequivocally established by single crystal X-ray
structural analysis and confirmed the NOE studies done on this compound (Figure 4.1).
Several 1D NOESY experiments were performed to determine the relative
stereochemistry of the stereogenic carbons, C-3` and C-5` of these cycloadducts (Figure
4.2). It must be noted that all diastereomers are racemates. For the diastereomer 170a,
irradiating the aromatic proton ArHo resulted in an enhancement in the signals (hence
indicating an NOE), for CH-3` (δ 3.54) and for one of the protons of the methylene
CH2-4` (δ 2.62 and not δ 3.88). In contrast, irradiating ArHo of the other diastereomer
171a caused an enhancement for only one of the methylene CH2-4` protons (δ 2.39 and
not 3.92) and not CH-3` (δ 3.97). The above studies indicate that the more downfield
signals of CH2-4` in 170a (δ 3.88) and 171a (δ 3.92), corresponded to the proton syn to
the methyl ester, which were clearly deshielded by this ester group. These NOE
correlations were consistent with molecular modelling studies using Spartan ‘04 (AM1)
(Figure 4.2). In 170a, the distance between ArHo and CH-3` was calculated to be 3.036
Å. This was in contrast to 171a, whose calculated distance of 4.514 Å corresponded to
the absence or only a weak NOE correlation. A stronger NOE was predicted to be seen,
in both 170a and 171a, between ArHo and CHα-4` (2.048 Å and 2.014 Å, respectively)
compared to CHβ-4` (3.626 Å and 3.509 Å, respectively). In addition, a stronger NOE
was predicted to occur between CH-3` and the proton on C-4` which is syn to it, over
that which is anti. In 170a, CHα-3` shows a shorter calculated distance to CHα-4` (2.470
Å) than CHβ-4` (3.055 Å). Similarly, 171a, CHβ-3` has a shorter calculated distance to
CHβ-4` (2.281 Å) than CHα-4` (2.689 Å).
Figure 4.1 Single crystal X-ray crystallographic analysis of 170a

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 102
*
*CHα-4`*
*CHα-4`
CH-3`
CH-3`
Ho
Ho
3`
4`
170a 171a
NO
CO 2Me
NO2
Me
Ho Hα
Hβ
3`
4`
NO
CO 2Me
NO2
Me
Ho Hα
Hβ
Figure 4.2 The NOE correlations (*) of 170a and 171a displayed on their Spartan models (Spartan ‘04 (AM1)).
It should be noted that some of the important inter-proton distances measured from the
X-ray crystal structure of 170a differed significantly from that calculated in the gas
phase (Table 4.6). In particular, the distances between ArHo and CH2-4` and CH-3` in
the solid state were significantly longer because of the differences in the orientation of
the C-5`-phenyl ring in the solid-state and gas-phase structures (Figure 4.1 c.f. Figure
4.2). These differences most likely arise due to crystal packing effects in the solid state.
Table 4.6
Measured inter-proton distance from the X-ray crystal of 170a
Calculated inter-proton distance from the AM1 model of 170a
ArHo-CHα-3` 3.543 Å 3.036 Å
ArHo-CHα-4` 3.570 Å 2.048 Å
ArHo-CHβ-4` 4.425 Å 3.626 Å
CHα-3`-CHα-4` 2.266 Å 2.470 Å
CHα-3`-CHβ-4` 2.798 Å 3.055 Å

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 103
Similarly, in the [1,3]-DC reaction using 142b the relative stereochemistry of 170b and
171b was confirmed through extensive 1D and 2D NOESY experiments. Unfortunately
in 171b the signals of ArHo overlapped with those for the ArH-ortho protons of the
other phenyl ring. In the 2D NOESY spectrum of 171b, there was a cross-peak between
the CH-3` proton and one of the methylene protons of CH2-4` (δ 3.93 and not 2.89) with
the overlapping aromatic multiplet. In contrast, in the 1D NOESY of 170b irradiating
the more upfield proton of CH2-4` (δ 2.54) showed important NOE enhancements to
ArHo and CH-3`. Because previously it has been shown that the more downfield proton
of CH2-4` corresponds to that which is syn to the methyl ester, the corresponding endo-
and exo-relative configurations were assigned to 171b and 170b, respectively (Figure
4.3 and Scheme 4.16). Interestingly, the relative chemical shift of the stereogenic CH-3`,
was found to change with the different deshielding effects of the N-substitutent, when
comparing both exo- (170a,b) and endo-products (171a,b). For, in the case using 142a,
CH-3` was more downfield in the endo-product (171a) (δ 3.97 c.f. δ 3.54 for 170a),
however for the [1,3]-DC reaction using 142b, CH-3` was more downfield in the exo-
product (170b) (δ 4.83 c.f. δ 4.37 for 171b). These NOE correlations were again
confirmed by molecular modelling studies using Spartan ‘04 (AM1) (Figure 4.3). In
170b, the distance between ArHo and CH-3` was calculated to be 2.913 Å. This was in
contrast to 171b, in which the calculated inter-proton distance of 4.407 Å corresponded
to the absence or a weak NOE correlation. A stronger NOE was predicted to be seen, in
both 170b and 171b, between ArHo with CHα-4` (2.076 Å and 2.081 Å, respectively)
over CHβ-4` (3.648 Å and 3.637 Å, respectively). In addition a stronger NOE was
predicted to be observed between CH-3` and the proton syn to CH-3` on C-4` over that
which is anti. In 170b, CHα-3` displayed a shorter calculated distance to CHα-4` (2.412
Å) than CHβ-4` (3.034 Å). Similarly, 171b, CHβ-3` had a shorter calculated distance to
CHβ-4` (2.255 Å) than CH-4α (2.731 Å).

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 104
*
*CHα-4`
*
*CHα-4`
CH-3`CH-3`
Ho Ho
3`
4`
170b 171b
NO
CO 2Me
NO2
Ph
Ho Hα
Hβ
3`
4`
NO
CO 2Me
NO2
Ph
Ho Hα
Hβ
Figure 4.3 The NOE correlations (*) of 170b and 171b displayed on their Spartan models (Spartan ‘04 (AM1)).
While the cycloadducts 170a,b and 171a,b are formed under reversible,
thermodynamically (equilibrium) controlled conditions and not under kinetically
controlled conditions, the cycloadducts 170a,b and 171a,b correspond to the exo- and
endo-products, respectively (Scheme 4.16). The difference in the heats of formation
calculated using Spartan ’04 (AM1) (Table 4.7) suggests that, in the gas phase, the
endo-products 171a and 171b have a higher heat of formation over their respective exo-
products 170a and 170b by 5.16 kcal/mol and -0.45 kcal/mol, respectively. A higher
heat of formation indicates less stability. These calculated heats of formation however
are contrary to the experimental results obtained, when a comparison is made between
the relative diastereoselectivities for the nitrones 142a and 142b. The experimental
results suggest a larger difference in the heats of formation for 170b and 171b (Table
4.5, entries 1, 2 and 7), while the calculated values suggest a higher diastereoselectivity
should be observed between 170a and 171a (Table 4.7).
Table 4.7
Compound Heats of formation (kcal/mol) Difference
170a -733 264.3
171a -733 259.2 5.16 kcal/mol 170b -852 143.3
171b -852 142.9 -0.45 kcal/mol

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 105
Scheme 4.16a (all compounds are racemic)
H
N
O
171
CO 2MeNO2
R NO
NO2
CO2Me
R
170
N
O
NO2
CO2Me
RH
N
O
MeO2C
R
O2N
5`-endo
5`-exo

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 106
4.3.2 [1,3]-Dipolar cycloadditions using the cyclic nitrone 143
The [1,3]-DC reaction of 59 with the cyclic nitrone 143 was conducted using a minimal
amount of anhydrous toluene as solvent with microwave irradiation at 150 °C for 30
min. 1H NMR analysis of the crude reaction mixture revealed a mixture of 172, 173 and
59 in a ratio of 2.7 : 1 : 1.8, respectively. Extensive purification of this mixture gave
pure samples of 172 and 173 in 40% and 6% yield, respectively, and an unresolved
mixture of 173 and 59. The discrepancy between the 1H NMR ratios and yields was due
to the difficulty in separation of this unresolved mixture upon purification. The relative
stereochemistry of 172 and 173 was confirmed by extensive 1D and 2D NMR
experiments. Analogous to the NMR spectrum of 170a,b and 171a,b the more
downfield proton of CH2-3` in adducts 172 and 173 was assumed to be the one syn to
the methyl ester group. Hence the relative intensities of the NOEs from the protons syn
or anti to CH-3a` were used to establish the relative configurations of 172 and 173
(Figure 4.4). For 173, a stronger NOE was seen between CH-3a` and the more
downfield proton of CH-3` (δ 3.82). In contrast, for 172, analysis in C6D6 (for resolution
of peaks) showed an NOE between CH-3a` and the more upfield proton of CH-3` (δ
2.38). In addition, NOE correlations were observed between the ArHo and CHα-3` for
both 172 and 173.
CHα-3`
δ 2.38
*
*
CHα-3`
δ 2.07*
*
CH-3a`δ 3.25-3.13
CH-3a`δ 4.02-3.96
CHβ-3`
δ 3.82
CHβ-3`
δ 3.58
Ho Ho
Figure 4.4 The NOE correlations (*) of 172 and 173 displayed on their Spartan models (Spartan ‘04 (AM1).

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 107
Scheme 4.17a (all compounds are racemic)
NO
NO
59
+
143
CO2Me
NO2
a)
1H NMR ratio: (2.7:1)
(40%) (6%)172 173
CO2MeNO2
MeO2C
O2N
NO
CO2Me
NO2
172
173
2-endo
2-exo
H
NO
CO2Me
NO2
H
NO
CO2Me
NO2
NO
CO2Me
NO2
H H
A cycloreversion study was attempted on 172 similar to that performed for the products
of the [1,3]-DC reaction involving the acyclic nitrones 142a and 142b. However due to
the small amount of cycloadduct 172 used, the result of this study was inconclusive
whether cycloreversion had taken place. The regiochemical and stereochemical outcome
of this [1,3]-DC reaction agreed with the results of Ali et al.297 discussed earlier (Table
4.3) with the 2-endo product being favoured over the 2-exo product and none of the 3-
regioisomers being seen. The observed low stereoselectivity of 2.7 : 1 was consistent
with that expected when both groups, the aromatic ring and the carbonyl ester, are able
to make favourable secondary orbital interactions in the transition state with the cyclic
nitrone. The difference in the heats of formation calculated using Spartan ’04 (AM1)
(Table 4.8) accords with the selectivity observed, with a higher energy of formation for
173 compared to 172, in the gas phase, by 11.56 kcal/mol, that is the endo-product 172
is thermodynamically more stable.
Table 4.8
Compound Heats of formation (kcal/mol) Difference 172 -638 034.9
173 -638 023.3 -11.56 kcal/mol

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 108
4.3.3 Derivatisation of the cycloadducts 170 and 171
Efforts were then made towards preparing the tricyclic targets B and C (Scheme 4.1) by
first reductive cyclization of the aryl nitro moiety of the cycloadducts 170a,b and 171a
(Table 4.9). Cycloadduct 171b was not derivitised further due to its limited amount.
First, catalytic hydrogenation was utilised in an attempt to yield indolinones B (Method
a)). For 170a and 171a the indolinones 174a and 176a, respectively were successfully
prepared using this method (Table 4.9, entry 1 and 3). However, it was noted that
prolonged reaction times led to further hydrogenolysis of the isoxazolidine ring in these
products to yield the over-reduced products, 175a and 177a, affecting the yields of these
spirocycles. For 170b however, this method only yielded the over-reduced product 175b
(Table 4.9, entry 5). The use of activated zinc and glacial acetic acid with sonication as
an alternative method to 174a and 176a, (Method b)). However this method yielded
only the over-reduced products, 175a,b and 177a (Table 4.9, entries 2, 4 and 6).
Table 4.9a (all compounds are racemic)
N
O
NO2
CO 2Me
R
H
170a (R = Me)
170b (R = Ph)
HN
HO
R
H
O
NO
N
Me
H
O
N
O
NO2
CO 2Me
MeH
171a
HN
HO
N
MeH
O
NO
N
Me H
O
a) b)a) b)
Entry Starting Material Cyclisation Method Results (Yields)
1 170a a) 174a (24%)b 2 170a b) 175a (58%) 3 171a a) 176a (54%)b 4 171a b) 177a (94%) 5 170b a) 175b (55%)b 6 170b b) 175b (90%)
a Reagents and conditions: (a) Pd/C, H2, EtOAc; (b) Activated Zn dust (10 eq.), glacial AcOH, sonication, 1 h. b Prolonged exposure led to over-reduction to occur.

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 109
The structures of these reduction products were confirmed by extensive 1D and 2D
NMR experiments and from MS data. To distinguish between the over-reduced amino-
alcohol products, 175a,b and 177a, and the spirocyclic products, 174a and 176a, mass
spectrometry was utilised with a higher molecular ion by two mass units, indicating the
amino-alcohol products. In addition, an upfield shift in the the aromatic protons was
observed in all products indicating indolinone formation. The chemical shift of the N-
methyl carbon moved upfield in the over-reduced product 175a relative to 170a (δ 43.0
to 28.3) but remained relatively the same in the over-reduced product 177a (δ 43.9)
indicating that in the latter, the isoxazolidine ring remained intact.
The amino alcohols, 175a,b and 177a, were then converted to the tricyclic derivatives
by treatment with triphosgene under basic conditions in anhydrous THF to form the
desired oxazinanone products, 178a,b and 179a, respectively (Scheme 4.18). The
formation of the oxazinanone ring was confirmed by the presence of another carbonyl
group (~ δC 150) and a downfield shift in the stereogenic proton CH-4`. In addition,
mass spectrometry at both high and low resolution confirmed the formation of the
desired products. Furthermore, similar NOE correlations to those seen in 170a and 171a
were observed, with the relative NOE intensities between the stereogenic proton, CH-4`
and each of the protons of the methylene CH2-5` of 178a and 179a, revealing their
relative configurations. As previously ascertained the more downfield signal of the
methylene CH2-5` corresponded to the proton syn to the oxindole carbonyl. In 178a, a
stronger NOE was seen between CH-4` and CHα-5` and conversely for 179a, a stronger
NOE was seen between CH-4` and CHβ-5`. Compounds 177a, 178a,b and 179a were
sent for X-ray crystallography, however analysis of these compounds unfortunately did
not return before thesis submission.

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 110
Scheme 4.18a (all compounds are racemic)
a)
(61%)175a (R = Me)175b (R = Ph)
178a (R = Me)
HN
HO
NH
Me
H
O
N
O
NH
R
H
O
O
(51%)179a
N
O
NH
Me
H
O
O
177a
HN
HO
NH
MeH
Oa)
(68%)178b (R = Ph)
4`6`
4`6`
a Reagents and conditions: (a) (Cl3CO)2CO, anhydrous THF, anhydrous NEt3, 2-7 d.
4.3.4 Derivatisation of the cycloadducts 172 and 173
The cycloadducts 172 and 173 were also treated under the hydrogenation conditions.
Initially the major diastereomer 172 was used. A variety of reductive cyclization
methods were employed, including the previous two methods (Methods a) and b)),
however these reactions were unsuccessful and furthermore starting material could not
be recovered. By this stage, only the minor diastereomer 173 was available in sufficient
quantities to attempt the catalytic hydrogenation using PdCl2/H2. This reaction
successfully yielded a product (180) with a m/z of 232 from LRMS analysis and a
molecular formula of C13H16N2O2 from HRMS analysis, which corresponded to the
over-reduced product. However, NMR analysis of 180 was impossible, due to the
broadening of all peaks, possibly due to traces of palladium in the sample. Crude 180
was thence subjected to treatment with triphosgene, which furnished the
hexahydropyrrolooxazinane 181 (Scheme 4.19). The formation of 181 was confirmed
with an observed downfield shift in the spirocarbon (δC 85.5 to 90.6) and the appearance
of an extra carbonyl resonance (δC 151.6). Mass spectrometry at both high and low
resolution also confirmed the structure of compound 181.

Chapter 4: [1,3]-Dipolar Cycloaddition Reaction 111
Scheme 4.19a (all compounds are racemic)
b)a)
(25% over 2 steps)
181
NO2
N
O
CO2Me
173 180
H
NH
NH
HO
H
NH
N
O H
O
O O
a Reagents and conditions: (a) anhydrous MeOH, PdCl2 (0.2 eq.), H2, 3 h; (b) (Cl3CO)2CO, anhydrous THF, anhydrous NEt3, 2 d.
In conclusion, the [1,3]-dipolar cycloaddition reaction utilising two acyclic and one
cyclic nitrone has proven to be a successful method to synthesise spiro[indole-3,5`-
isoxazolidin]-2(1H)-ones and spiro[indole-3,6`-[1,3]oxazinane]-2,2`(1H)-diones.
Compounds 170a,b, 172, 175a,b, 174, 178a,b, 179a and 181 were all submitted for
cytostaticity studies and protein inhibition studies and the results of these are discussed
in Chapter 6.

Chapter 5: Towards the Synthesis of Potent Purine CDK Inhibitors 112
CHAPTER 5: TOWARDS THE SYNTHESIS OF
A POTENT PURINE CDK INHIBITOR
Citranadin B is isolated from the marine fungi, Penicillium citrinum and shows modest
cytoxicity against murine leukemia L1210 cells.149,150
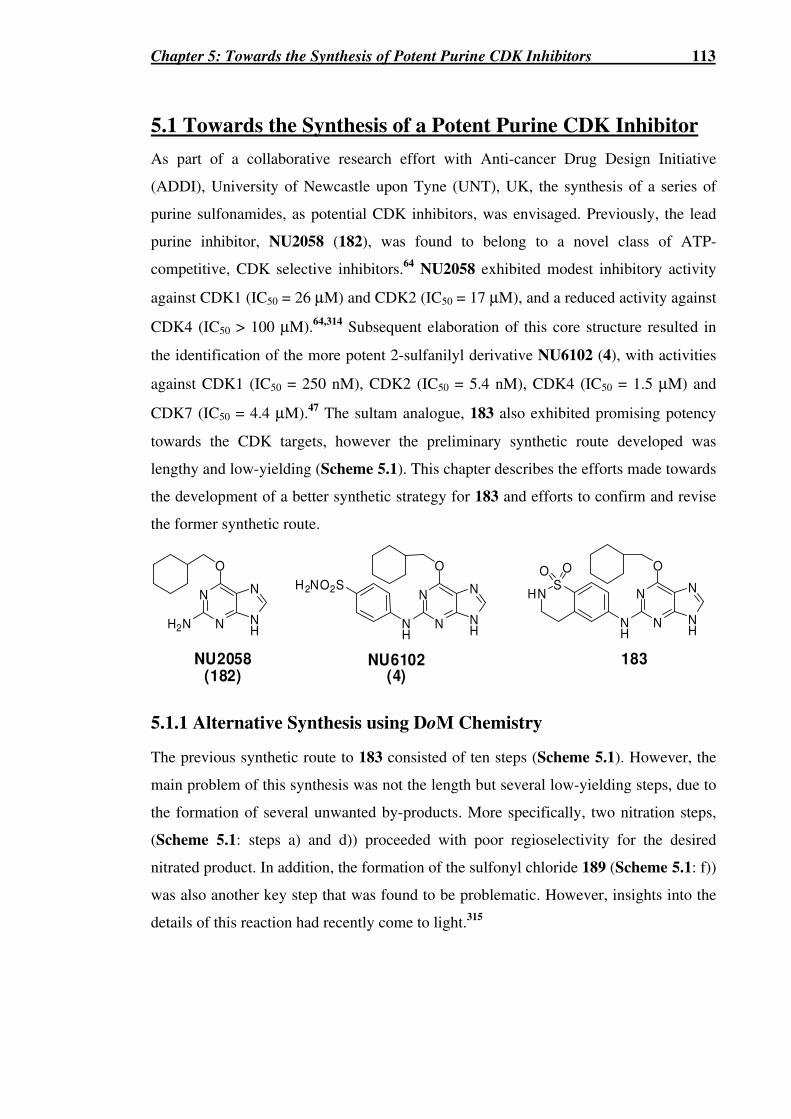
Chapter 5: Towards the Synthesis of Potent Purine CDK Inhibitors 113
5.1 Towards the Synthesis of a Potent Purine CDK Inhibitor
As part of a collaborative research effort with Anti-cancer Drug Design Initiative
(ADDI), University of Newcastle upon Tyne (UNT), UK, the synthesis of a series of
purine sulfonamides, as potential CDK inhibitors, was envisaged. Previously, the lead
purine inhibitor, NU2058 (182), was found to belong to a novel class of ATP-
competitive, CDK selective inhibitors.64 NU2058 exhibited modest inhibitory activity
against CDK1 (IC50 = 26 µM) and CDK2 (IC50 = 17 µM), and a reduced activity against
CDK4 (IC50 > 100 µM).64,314 Subsequent elaboration of this core structure resulted in
the identification of the more potent 2-sulfanilyl derivative NU6102 (4), with activities
against CDK1 (IC50 = 250 nM), CDK2 (IC50 = 5.4 nM), CDK4 (IC50 = 1.5 µM) and
CDK7 (IC50 = 4.4 µM).47 The sultam analogue, 183 also exhibited promising potency
towards the CDK targets, however the preliminary synthetic route developed was
lengthy and low-yielding (Scheme 5.1). This chapter describes the efforts made towards
the development of a better synthetic strategy for 183 and efforts to confirm and revise
the former synthetic route.
N
N NH
N
O
H2N
N
N NH
N
O
NH
NU2058(182)
NU6102(4)
H2NO2SN
N NH
N
O
NH
SHN
O O
183
5.1.1 Alternative Synthesis using DoM Chemistry
The previous synthetic route to 183 consisted of ten steps (Scheme 5.1). However, the
main problem of this synthesis was not the length but several low-yielding steps, due to
the formation of several unwanted by-products. More specifically, two nitration steps,
(Scheme 5.1: steps a) and d)) proceeded with poor regioselectivity for the desired
nitrated product. In addition, the formation of the sulfonyl chloride 189 (Scheme 5.1: f))
was also another key step that was found to be problematic. However, insights into the
details of this reaction had recently come to light.315

Chapter 5: Towards the Synthesis of Potent Purine CDK Inhibitors 114
Scheme 5.1a (Yields given are those previously attained by a PhD student at UNT)316
O 2N
NHS
O O
SO 2Cl
O2N Cl
Cl Cl
NO2
Cl
NH2.HCl
Cl
NHAc
Cl
NHAc
O2N Cl
NH3
O2N
Cl
SO2NH2
O 2N Cl H2N
NHS
O O
NH
HNS
OO
N
N NH
N
O
a) b) c)
e)d) f)
g) h) i) j)
183(16%)
184a(25%)
185(94%)
186(74%)
187a(76%)
188(95%)
189(42%)
190 191(29%)
192(52%)
a
Reagents and conditions: (a) H2SO4, HNO3, 5-10 oC; (b) 10% Pd/C, H2, MeOH, HCl; (c) Ac2O, AcOH; (d) HNO3 (fuming), AcOH, 0 °C; (e) EtOH, HCl, reflux; (f) 1. C6H5CF3, AcOH (aq), NaNO2, HCl, 2. SO2, Cu(I)Cl, AcOH; (g) NH3 (aq); (h) NaOH (aq); (i) NH4CO3, Pd/C, MeOH; (j) TFA, TFE, 70 oC, 6-cyclohexylmethoxy-2-fluoro-9H-purine.
Although other nitration methods have reported increased selectivity for the less-
favoured nitrated product, such as the use of nitronium tetrafluoroborate in
tetramethylenesulfone, the reported selectivities for alkylbenzenes were still poor and
inadequate (~ 1 : 1 (ortho : para)).317,318 The directed ortho metalation (DoM) of
aromatic systems has been utilised widely due to its characteristic high regio-controlled
addition of electrophiles to aromatic systems. The DoM reaction was first discovered by
both Gilmann and Bebb319 and Wittig and Fuhrman,320 with the ortho deprotonation of
anisole using n-BuLi. The DoM reaction consists of the initial deprotonation at the site
ortho to the heteroatom-containing directed metalation group (DMG) by a strong base
(193) (Scheme 5.2).321 The DMG co-ordinates the base, (194) usually an alkyl lithium
reagent, resulting in the ortho-lithiated species (195). Upon subsequent treatment with
various electrophiles, an array of 1,2-disubstituted products (196) can be attained.

Chapter 5: Towards the Synthesis of Potent Purine CDK Inhibitors 115
Scheme 5.2
DMG DMG
Li
DMG
E
(RLi)n E+DMG
H
(RLi)nor
(RLi)nLmn n193 194 195 196
The synthetic utility of this reaction has been manifested by the wide selection of
DMGs (more than 40) that can be utilised by this reaction.321 For a successful ortho-
deprotonation, the DMG has to be one that provides a good co-ordination site for the
alkyl-lithium base, whilst still being a poor electrophilic site for attack by this strong
base. Some DMGs include various carbonyl functional groups (CONR1R2, CO2H,
OCONR2), protected amines, imines, phosphates, sulfonamides, ethers, oxazolines, and
MOM ethers.321 Co-operative metalation effects with multi-substituted aromatics have
also been studied with surprising high regioselectivities being observed (Table 5.1). It
has been ascertained through experimental results that, generally, when the DMG is
CONEt2, N-Boc or OCONEt2 that in concert with groups like O-alkyl, halogens, and
imines, the C-2 substituted product is favoured in high regioselectivity. However,
combined with the NMe2 group, the C-6 substituted product is favoured in high
regioselectivity.
Table 5.1
DMG
X
a) s-BuLi, TM EDA,
THF, -78 oC
b) E+
DMG
X
E
DMG
X
E
+
197 198
2
6
2
6
199
DMG X E+ 198 : 199 (Yields) Ref.
OMe D2O, TMSCl ~ 95 : 5 (90%) 322
Cl MeOD 95 : 5 (90%) 322 OMOMa ICH2CH2I 100 : 0 (35%) 323
CONEt2
NMe2 PhCHO 5 : 95 (NA) 324
OMeb (MeS)2 95 : 5 (82%) 325 N-CO2tBu
OMea,c I(CH2)3Cl 95 : 5 (26%) 326
OMe CO2 67 : 33 (83%) 321
Cl MeI 95 : 5 (83%) 327
OCONEt2
NMe2 TMSCl 0 : 100 (93%) 328 a t-BuLi, Et2O, hexane, -78 °C; b n-BuLi, THF, 0 °C; c -20 °C;

Chapter 5: Towards the Synthesis of Potent Purine CDK Inhibitors 116
In addition, the scope of the DoM reaction has been highlighted by the various aromatic
systems that can be utilised, including naphthalenes, pyridines, thiophenes, furans,
triazolopyridines and quinines.321 The DoM process, usually demands the employment
of powerful alkyllithium bases in organic solvents. Due to their aggregation in
hydrocarbon solvents, the addition of basic solvents (ethers, amines, phosphines) and
bidentate ligands, such as N,N,N`,N`-tetramethylethylenediamine (TMEDA), have been
utilised. These effectively break down these alkyl lithium aggregates, to form
monomers and dimers in solution and hence significantly increase their basicity. The
sec-BuLi⋅TMEDA combination has been found to be the most potent metalating
agent.329
Previously Snieckus et al.330 showed the utility of the N-cumyl protected sulfonamide
functional group as a DMG and its subsequent effective removal by TFA (Scheme 5.3).
It has also been previously shown that the nitro functionality is not tolerated by the
DoM chemistry. However, amines protected by their silicon-based “stabase” group, can
tolerate DoM conditions.331,332
With these aspects in mind, a new synthetic strategy towards the synthesis of 183,
which incorporates the DoM chemistry was envisioned (Scheme 5.4). This synthetic
strategy had a number of advantages over the previous synthetic route. First, the
strategy consisted of only six steps to the target compound, 183. Secondly, the use of
the DoM chemistry should ensure the greater selectivity for the desired product. Lastly,
it possessed greater synthetic flexibility, due to the various electrophiles one can employ
in the DoM reaction, providing access to a wide number of possible derivatives of 183
for further structure activity relationship (SAR) studies.
Scheme 5.3330
O2

Chapter 5: Towards the Synthesis of Potent Purine CDK Inhibitors 117
Scheme 5.4a
203
SO 2Cl
O2N
S
O 2N
O
O
NHSO 2NHR1
H2N
SO2NHR1
R2N
c)
E+ =
SO 2NHR1
R2N
R1 =
R2 =
Si
SiBrBr
BrCl
X H2N
NHS
O O
192207206
205204
a) b)
d) e)
Si
Si
a b
a Reagents and conditions: (a) cumylamine (1 eq.), aqueous 10% NaOH, -40 °C�RT, 2
h 64%; (b) NH4CHO (7 eq.), anhydrous MeOH, 10% Pd/C, 78 h, 97%; (c) n-BuLi (3.1 eq.), anhydrous THF, 208, -78 °C; (d) i) s-BuLi (3.1 eq.), TMEDA, anhydrous THF, -78 °C, ii) E+ (1.2 eq.); (e) TFA.
The first step in the synthesis was the reaction of p-nitrobenzenesulfonyl chloride (203)
with cumylamine. Two methods were employed. The first used anhydrous triethylamine
and anhydrous conditions and furnished 204 in 56% yield. The second employed a
solution of 10% NaOH as base and furnished 204 in 64% yield. Signals in both the
aromatic and methyl region, corresponding to the presence of the cumylamino group
were seen in the 1H and 13C NMR spectra. Compound 204 was then reduced using
catalytic hydrogenation employing Pd/C and ammonium formate as the hydrogen
source, to afford 205 in 97% yield. An upfield shift of the ortho and meta protons were
seen in the 1H NMR spectrum of 205, consistant with reduction of the electron-
withdrawing nitro group. Mass spectrometry at both low and high resolution also
confirmed the formation of 205.

Chapter 5: Towards the Synthesis of Potent Purine CDK Inhibitors 118
To synthesise the stabase adduct 206a several methods were employed, however both
methods failed to yield any of desired product. First, a standard procedure was
attempted (Scheme 5.5).331,333 This method employed first n-BuLi to deprotonate the
substituted aniline and then addition of 1,2-bis(chlorodimethylsilyl)ethane (208).
However the formation of 206a was not observed. An excess of n-BuLi (3.1 eq.) was
employed to further deprotonate the more acidic sulfonamide NH group. The
deprotonated sulfonamide moiety in 205 may have been responsible to the lack of
stabase formation, since this anionic group would make deprotonation of the less acidic
aniline amino group more difficult.
Scheme 5.5
Si
Si Cl
Cl
208
205
NHS
O
O
H2Nn-BuLi (3.1 equiv)
dry THF
-78 oC
N
NHS
O
O
Si
Si
206a
Secondly, a method outlined by Guggenheim334 was also attempted, which boasted of
being an easier procedure for preparing large amounts of the desired stabase adducts,
especially for the more acidic aniline-type systems (Scheme 5.6). This method involved
the initial preparation of silizane 209 through using dimethylamine or diethylamine.
Compound 209 was purified by distillation and this moisture-sensitive liquid was then
heated in equimolar amounts with the various anilines to yield the stabase adduct
quantitatively (Scheme 5.6). The catalytic use of zinc iodide (0.5 mol %) was also
found to accelerate this reaction. This reaction however failed to yield any of the desired
stabase adduct 206a. Possible reasons for the failure of this reaction were, first,
purification of 209 proved to be difficult and secondly, all anilines used in the literature
examples, were liquids and therefore these reactions were performed neat. Compound
205 however, was not a liquid and was found to possess a high melting point (194.5 °C),
and though this reaction was attempted neat and at higher temperatures (180 °C), no
product was observed. In addition, in both attempts of synthesising 206a it must be
noted that these stabase adducts are renown for their acid-lability, and decomposition of
the desired product may have occurred upon purification by column chromatography.

Chapter 5: Towards the Synthesis of Potent Purine CDK Inhibitors 119
Scheme 5.6
N
NHS
O
O
Si
Si
Si
Si Cl
Cl Si
Si NR2
NR2
R = Me or Et
HNR2,dry THF ZnI2, 140 oC
208 206a209
205
In response to the adverse lability of the stabase group, more acid-stable “stabase-like”
protecting groups have also been synthesised. These included the benzostabase
(BSB),335,336 (211a) and 1,1,3,3-tetraethyl-1,3-disilaisoindolines (TEDI),337 (211b)
protecting groups. Similar to the stabase adducts these protecting groups are amenable
to DoM conditions. BSB and TEDI, however, have been shown to have 36 and 75 times,
respectively, greater stability towards acidic conditions and hence column
chromatography, than their corresponding stabase derivatives.335,337 The synthesis of
211a,b proceeds through dehydrogenative silylation employing various transition metal
catalysts (Table 5.2).335,336 Indeed this dehydrogenative silylation method can be
employed for the former stabase adducts to give a cleaner result.335
Table 5.2335,336
211a; R = Me211b; R = Et210
R `N
Si
Si
RR
R R
Si
R
H
R
Si
R
H
R
R`NH2+ + H2
R` Catalyst (mol %) Reaction Conditions Yielda (%)
Ph (Ph3P)2RhCl (0.4) No solvent, 80 °C, 4 h 72b (95%)
(Ph3P)2RhCl (0.2) Toluene, 120 °C, 25 h 77b p-Br-C6H4 CsF (90) HMPA, 120 °C, 3.5 h 92b (100%)
m-Br-C6H4 CsF (30) HMPA, 120 °C, 4 h 71b
10% Pd/C PhH, 50 °C, 48 h 87c (95%)
CsF (0.75) HMPA, 120 °C, 16 h (95%)
CsF (0.75) DMF, 100 °C, 30 h (95%)
PhCH2
TBAF (0.5) No solvent, 80 °C, 96 h (95%)
PhCH2CH(NH2)CO2Et PdCl2 (0.02) Toluene, reflux, 36 h 82c a Crude yield estimated by 1H NMR analysis. b Isolated by recrystallisation. c Isolated by distillation.

Chapter 5: Towards the Synthesis of Potent Purine CDK Inhibitors 120
The synthesis of corresponding BSB derivative 206b was attempted using Wilkinson’s
catalyst ((Ph3P)2RhCl) however none of the desired 206b was observed (Scheme 5.7).
Though the reaction was not successful, finding a suitable solvent to dissolve the amine,
205 was probably the major determining factor in its failure. In addition, further
investigations to optomise the reaction conditions, such as the use of other transition
metal catalysts, solvent, temperature and time, may lead to the success of this reaction.
Scheme 5.7a
206b205
H2N
NHS
O
O
N
NHS
O
O
Si
Si
a)
a
Reagents and conditions: (a) 210 (1.3 eq.), anhydrous toluene (1 mL), (Ph3P)3RhCl (0.1 mol %), 80 ºC, N2, 15 h.
An attempt of the DoM reaction using 204 was also attempted, in accordance with the
method of Snieckus et al.330 (Scheme 5.8). However the reaction yielded none of the
desired product, 212 and only starting material was recovered.
Scheme 5.8
204
O2N
NHS
O
O
O2N
NHS
O
Oa) TMEDA, s-BuLi (2.1 equiv)
THF, -78 oC, 2 h
b) 1-bromo-2-chloroethaneCl
212
Reed et al.326 showed the ability to form various quinolines (213) and naphthyridines by
using the DoM conditions on aromatic systems with an N-Boc group as a DMG and
dihalogenated alkanes as the electrophiles (Scheme 5.9). By analogy, the suitably
amino-protected sulfonamide 206c may undergo DoM and then cyclization to furnish
the desired cyclic sulfonamide 214 (Scheme 5.9). Possible amino-protecting groups for
206c could be the N,N-dibenzyl group (R = R` = Bn) or the N-Boc-protecting group (R
= Boc, R` =H). However due to time constraints these variations could not be tested.

Chapter 5: Towards the Synthesis of Potent Purine CDK Inhibitors 121
Scheme 5.9a 326
213
NHa)
Boc
R
Cl
N Boc
R
N
Boc
R
SO2NHR1
N
206c (R1 = CH(CH3)2Ph)
SO2N
N
R1b)
Cl
O2S
N
214
R
R `
R
R`
R
R`
NH
a
Reagents and conditions: (a) i) t-BuLi (2.5 eq.), THF; ii) I(CH2)2Cl; iii) reflux; (b) s-BuLi, THF ii) Br(CH2)2Cl
5.1.2 Revised Former Synthesis
To confirm the products and to improve yields, the former synthetic strategy outlined in
Scheme 5.1 was also attempted. The first step in this synthesis was the nitration of 2-
phenylethyl chloride. When considering the regiochemical outcome, the resonance
structures reveal that the 2-chloroethyl group is an ortho, para-directing group (Scheme
5.10). Standard nitrating conditions using sulfuric acid and nitric acid were used. 1H
NMR analysis of the crude reaction mixture revealed a regioisomeric mixture of 184a
and 184b in a ratio of 1 : 2.2, respectively.316 Purification of these regioisomers by
column chromatography, using the 5% Et2O:PS gave pure samples of 184a and 184b, in
26% and 57% yields, respectively. Confirmation of the regiochemistry of both 184a and
184b was confirmed by 1D NMR analysis. The aromatic region of 184a revealed a
system typical of an ortho-disubstituted aromatic ring (i.e. d, t, t, and d). Likewise 184b
showed the characteristic pair of doublets, typical of a para-disubstituted aromatic ring.
The meta-disubstituted product, with its characteristic substitution pattern (s, d, t, and d),
was not observed.

Chapter 5: Towards the Synthesis of Potent Purine CDK Inhibitors 122
Scheme 5.10
Cl Cl
NO2
HNO 3, H2SO4,
0-10 oC, RT, 2h
184a (26%)
Cl
+O2N
184b (57%) 184c (0%)
Cl
+
NO2
Cl
NO2
Cl
O2N
Cl
NO2
Cl
NO2
Cl
O2N
Cl
NO2
Cl
NO2
Cl
O2N
Cl
NO2
Major resonancecontributor
Major resonancecontributor
The nitro-compound 184a was reduced by catalytic hydrogenation under acidic
conditions to yield 185 in 99% yield (Scheme 5.11). The formation of 185 as its salt
form was necessary to prevent intramolecular cyclisation to form the unwanted
quinoline product. The protection of the amine with the acetate group under standard
conditions, proceeded smoothly to yield 186 in a 95% yield (Scheme 5.11).
Scheme 5.11a
Cl
NO2
Cl
NH2.HCl
Cl
NHAca) b)
184a 185 (99%) 186 (95%) a
Reagents and conditions: (a) anhydrous MeOH, conc. HCl, 10% Pd/C, -78 °C� RT, H2, 18 h (b) Ac2O, AcOH, RT, 5 h.
To nitrate 186, stronger nitrating conditions were necessary (Scheme 5.12). Previously,
the molar ratio of fuming nitric acid to glacial acetic acid for a successful nitration was
found to be 2.2 : 1, respectively.316 Attempts to vary this ratio or scale up the reaction
were performed, however with limiting success at improving the yield of the desired
adduct 187a. The predicted regiochemical outcome is unclear, as both substituents have
ortho, para-directing effects, however, the effects of the N-acetamide should dominate
and possible steric effects should favour the desired para-substituted product, 187a. 1H

Chapter 5: Towards the Synthesis of Potent Purine CDK Inhibitors 123
NMR analysis of the crude reaction mixture using the former reaction conditions,
revealed a mixture of 187a and 187b in a ratio of 1 : 1. Previously the solvent system
used to separate 187a and 187b by column chromatography was 0-100% EtOAc:PS. It
was found however that the crude mixture was difficult to dissolve in pure EtOAc and
hence purification by column chromatography was made impossible. Therefore
recrystallisation was utilised. Recrystallisation using MeOH, yielded 187a as fine
feathery white needles, and recrystallisation from EtOAc or DCM, yielded 187b as a
cream powder. The structure of 187a was unequivocally established by single crystal X-
ray structural analysis (Figure 5.1). 1H NMR analysis of 187a was initially performed
in CDCl3. The corresponding NMR spectrum previously reported of 187a was found to
be very different, however the NMR solvent had not been disclosed (Figure 5.2 and
Table 5.3).316 In particular, the aromatic region showed all aromatic protons
overlapping as a broad doublet. Upon conducting the NMR experiment in CD3OD these
protons were found to separate, revealing the characteristic aromatic pattern for a 1, 2,
4-trisubstituted system of d, dd and d, with J values indicative of ortho (9.0 Hz) and
meta coupling (2.7 Hz). The 1H NMR data now correlated well with that previously
reported (Table 5.3). Likewise 187b showed the characteristic aromatic pattern (dd, t
and dd) and corresponding J values indicative of a 1, 2, 6-trisubstituted system.
Scheme 5.12a
Cl
NHAc
186
Cl
NHAc
O2N
a)
187a (26%)
Cl
NHAc
187bb
+
NO2
Cl
NHAc
187c (0%)
O2N
+
1H NMR ratio of 187a : 187b : 187c (1 : 1 : 0) a
Reagents and conditions: (a) glacial AcOH, fuming HNO3, -5 °C-0 °C�RT. b Yield not recorded.
Figure 5.1 Single crystal X-ray crystallographic analysis of 187a

Chapter 5: Towards the Synthesis of Potent Purine CDK Inhibitors 124
Figure 5.2 1H NMR spectra of 187a in A) CDCl3 and B) CD3OD.
Table 5.3 1H NMR chemical shifts (δδδδ) for 187a in different NMR solvents
Previous Values for 187aa 316 187a (CD3OD, 300 MHz) 8.22 (d, J 2.7 Hz, 1H, ArCH-3); 8.12 (dd, J 9.0, 2.7 Hz, 1H, ArCH-5); 7.78 (d, J 9.0 Hz, 1H, ArCH-6); 3.80 (t, J 7.5 Hz, 2H, CH2CH2Cl); 3.20 (t, J 7.5 Hz, 2H, CH2CH2Cl); 2.23 (s, 3H, CH3)
187a (CDCl3, 300 MHz)
9.70 (s, 1H, NH); 8.21 (d, J 2.7 Hz, 1H, ArCH-3); 8.12 (dd, J 9.0, 2.7 Hz, 1H, ArCH-5); 7.93 (d, J 8.9 Hz, 1H, ArCH-6); 3.89 (t, J 6.9 Hz, 2H, CH2CH2Cl); 3.23 (t, J 7.0 Hz, 2H, CH2CH2Cl).
8.16 (d, J 12.0 Hz, 3H, ArH); 7.40 (bs, 1H, NH); 3.61 (t, J 6.0 Hz, 2H, CH2CH2Cl); 2.90 (t, J 6.0 Hz, 2H, CH2CH2Cl); 2.00 (s, 3H, CH3).
a Deuterated solvent used was not reported.316
Deprotection of the amino group using acidic conditions yielded 188. Purification at this
point was found to be problematic so continuation of the synthesis without purification
9.0 8.0 7.0 6.0 5.0 4.0 3.0 2.0 1.0
8.0 7.0
8.0 7.0 6.0 5.0 4.0 3.0 2.0
8.2 8.1 8.0 7.9 7.8 7.7
A)
B)

Chapter 5: Towards the Synthesis of Potent Purine CDK Inhibitors 125
was necessary. Insights into the diazotization and subsequent sulfonylation had been
described by Hoffman.315 In this report details necessary for the success of the reaction
were recorded, such as the use of Cu(I)Cl and not Cu(II)Cl and the critical maintainance
of the temperature between -10 to -5 ºC to prevent formation of dark red adducts. The
reaction was performed in exact accordance to the procedure outlined by Hoffman, and
though dark red adducts were not observed in the reaction mixture, none of the desired
sulfonyl chloride 189 was observed and only 188 was recovered (Scheme 5.13).
Scheme 5.13a
SO 2Cl
O2N ClCl
NHAc
O2N Cl
NH3
O2N
Cla) b)
187a 188 189 a
Reagents and conditions: (a) i) Solution A: conc. HCl, glacial AcOH (dry-ice EtOH bath (-72 ºC), NaNO2 in H2O -5 ºC. -10 ºC to -5 ºC, 45 min ii) Solution B: SO2, glacial AcOH, Cu (I) Cl, RT � -5 °C, b) Solution A to Solution B, <30 °C.
5.1.3 Conclusions
Synthesis towards the sultam 183 using a new strategy employing DoM and the former
strategy were made. Though completion was not realised, important insights were made
in both approaches. It was highlighted in our new efforts, the problematic use of stabase
analogues, and possibly revision of this approach with the future use of the N,N-
dibenzyl or N-Boc analogues or further optimization studies for the BSB-protection.
Confirmation and revision of the former synthetic approach was also made, in particular
the unequivocal determination by single crystal X-ray structural analysis of key nitro-
compound, 187a and its cleaner purification by recrystalisation was developed.

Chapter 6: Biological Testing Results and Discussion 126
CHAPTER 6: BIOLOGICAL TESTING
RESULTS AND DISCUSSION
A number of spirooxindole alkaloids, in which pteropodine is an example, have been
isolated from the Peruvian medicinal plant, Uncaria tomentosa, known as “cat’s claw”.
Pteropodine has been shown to exhibit cytostaticity against human lymphoblastic
leukaemia T-cells and induce apoptosis.125

Chapter 6: Biological Testing Results and Discussion 127
6.1 Introduction
To ascertain the potential utility of synthesised spirocyclic compounds (described in
Chapters 2-4) as potential cancer therapeutics and in particular as cell-cycle inhibitors,
cytostaticity studies against the cancer cell lines Mpro (murine myeloid), B16 (murine
melanoma), HL60 (human myeloid), H460 (human non small cell lung), MCF-7 (human
breast) and SF-268 (human CNS), as well as protein inhibition studies against key cell-
cycle proteins MDM2, CDK2, CDK5 and gSK-3 were performed. The importance of both
investigations is undeniable, with the former giving insights concerning the drug’s
cytostatic activity, phase inhibition and pharmacokinetic properties, and the latter studies
elucidating the possible mode of action of the drugs.
6.2 Cytostatic Cellular Studies
6.2.1 Prelimary Cytostaticity Studies of 85a and 87
Prelimary cytostaticity studies carried out by Dr. Grant McArthur, Peter MacCallum
Cancer Institute, Melbourne, involved the assaying of synthesised 2-azaspiro[4.4]nonan-
1-ones, 85a and 87 against the three cancer cell lines, Mpro, B16 and HL60.338 The
cytostatic activities of 85a and 87, were assessed against the known CDK2 inhibitor,
roscovitine (1). Roscovitine is a selective CDK inhibitor recording an IC50 activity of 0.70
µM, >100 µM and 0.45 µM against CDKs 2, 4, and 1, respectively. In these cell line
assays, 87 exhibited no detectable inhibitory activity against the three cell lines tested. In
contrast, 85a displayed an inhibitory activity against B16 cells, with an IC50 of 77 µM,
approximately 3-fold reduced activity than that of 1 (IC50 = 28 µM) (Figure 6.3). Further
assessment revealed that the inhibitory activity of 85a was due to a mild to moderate G1-
phase block (Figure 6.4).
PhHNOC PhHNOC

dbev
Text Box
See print copy for figure 6.1, 6.2 & 6.3

Chapter 6: Biological Testing Results and Discussion 129
6.2.2 Cytostaticity Screening against H460, MCF-7 and SF-268
To assess the cytostaticity of synthesised compounds and their utility as potential cancer
therapeutics, cytostaticity screening against the cell lines, H460, MCF-7 and SF-268
were performed by Dr. Carleen Cullinane at the Andrew Durant Drug Testing Facility,
Research Division of the Peter MacCallum Cancer Centre, Melbourne. These cell lines
were chosen because they are common cell lines used in screening by the NCI. Also
structurally similar spirocyclic oxindoles have shown activity against breast cancer cell
lines in the literature.177 SF-268 was included to reveal the toxicity of the drug to the
CNS.
6.2.2.1 Cytostaticity Screening of 2-Azaspiro[4.4]nonan-1-ones
The results of the cytostaticity studies of the 2-azaspiro[4.4]nonan-1-ones, 85a, 85b, 87
and 91, are displayed in Table 6.1. These compounds showed poor to modest activities.
The most active of this series was 85b (Table 6.1, entry 4) which inhibited the cell
growth of H460 cells by only 74% at a concentration of 25 µM.
6.2.2.2 Cytostaticity Screening of Spiro[cyclopentane-1,1`-[1H]isoindol]-3`(2`H)-
ones
The results of the cytostaticity studies of the spiro[cyclopentane-1,1`-[1H]isoindol]-
3`(2`H)-ones, 72, 92, 93b, 102-107, are displayed in Table 6.1. The most active of this
series were the oxindoles with an amide bond to 4-(N,N-dimethyl)-aniline (Table 6.1,
entries 8 and 16). To ascertain if one enantiomer was more active than the other, the
enantiomerically pure versions of 93b was tested against its racemic mixture (Table 6.1,
entries 8, 16-18). It was found that neither the (S)- nor (R)-enantiomer was markedly
responsible for the activity of the racemic mixture. In fact the racemic mixture was
more active than the enantiomerically pure samples. This suggests that a mixture is
necessary for activity, and that a potentiation or other type of synergistic effect might be
involved.
6.2.2.3 Cytostaticity Screening of Spiro[indole-3,5`-isoxazolidin]-2(1H)-ones and
spiro[indole-3,6`-[1,3]oxazinane]-2,2`(1H)-diones
The results of the cytostaticity studies of the spiro[indole-3,5`-isoxazolidin]-2(1H)-ones,
170a, 172, 174a and 175b, and spiro[indole-3,6`-[1,3]oxazinane]-2,2`(1H)-diones,
178a,b, and 179a, are displayed in Table 6.2. These compounds proved to be the most
active series, with notable activity in six of the seven compounds tested (Table 6.2,
entries 1, 3-7). It was found that cytostaticity was moderately dependent on the relative

Chapter 6: Biological Testing Results and Discussion 130
chirality at the 4`-stereocentre for the diastereomeric oxazinan-2-ones, 178a and 179a
(Table 6.2, entries 1 and 2), with a reduction in activity observed in the (4`S*, 6`R*)
diastereomer, 179a over the more active (4`R*, 6`R*) diastereomer, 178a. Compounds
178b and 170a, which shared the same relative configuration to that of 178a, displayed
similar cytostaticity activities to 178a (Table 6.2, entries 3 and 4). Interestingly,
compounds 170a and 174a had similar activities, even though the latter had an
uncyclised oxindole ring (Table 6.2, entries 4 and 6). The bicycle 175b (Table 6.2,
entry 5) showed similar activity to the tricycle 178b (Table 6.2, entry 3) suggesting that
activity may not be dependent upon the intact isoxazolidine ring. However, compounds
174a and 172 with their smaller isoxazolidine ring showed a very low level of cell
growth at 25 µM (Table 6.2, entries 6 and 7). Indeed 174a was our most active
compound with a GI50 against MCF-7 cells of 2.6 ± 0.1 µM. The
hexahydropyrroloisoxazole ring was also found to be advantageous for activity with 172
(entry 7) showing an activity comparable to our most active compound 174a.
Compound 172 had a GI50 against MCF-7 cells of 4.1 ± 0.2 µM. The GI50s for
compounds 174a and 172 were extrapolated from duplicate studies of growth inhibition
over a drug concentration of 0-25 µM (Figure 6.6). It was observed that 174a had a
much sharper GI curve (Figure 6.6, A) and B)) than 172 (Figure 6.6, C) and D)), which
may suggest that it has a narrow therapeutic index, that is there is a fineline between no
activity and high toxicity. Furthermore at higher doses of 174a, cytotoxicity as opposed
to cytostaticity was clearly demonstrated as fewer cells remained at the end of the assay
than when at the beginning.
6.2.2.4 Cytostaticity Screening of Spiro[cyclopropane-1`,3-[3H]indol]-2`(1H)-ones
The results of the cytostaticity studies and the structures of spiro[cyclopropane-1`,3-
[3H]indol]-2`(1H)-ones, 139a, 139b and 141a, are displayed in Table 6.2.
Unfortunately none of these compounds showed any cytostaticity against the selected
cancer cell lines (Table 6.2, entries 8-10). Interestingly, the indirubin-like precursors,
108a and 108b showed notable cytostaticity (Table 6.2, entries 11 and 12). The 3-
pyridine structure 108b was shown to be slightly more active than the 2-pyridine 108a.
The GI50 of 108b against H460 cells was found to be 9.5 ± 3.5 µM. Structurally similar
indirubins have been shown to be CDK2 inhibitors.52,55,56,339,340

dbev
Text Box
See print copy for table 6.1

dbev
Text Box
See print copy for table 6.2

Chapter 6: Biological Testing Results and Discussion 133
6.3 Protein Inhibition Studies
To assess the utility of synthesised compounds as potential cancer therapeutics and to
determine their mode of action, protein inhibition studies were performed against CDK2,
by Dr. Yuzhu Cheng and Lan-Zhen Wang, Northern Institute for Cancer Research
(NICR), Medical School, University of Newcastle upon Tyne (UNT), UK; CDK5 and
gSK-3, by Laurent Meijer’s laboratory, USA; and MDM2-p53, by Dr. Ian Hardcastle’s
laboratory, NICR, Medical School, UNT. These proteins were chosen because they are
important cell-cycle regulators and their inhibition has resulted in cytostaticity of
different cancers. Furthermore, structurally similar spirocyclic oxindoles have been
shown to inhibit the cell-cycle protein MDM2-p53.106,112 The array of compounds tested
for protein inhibition are displayed in a venn diagram (Figure 6.7).
6.3.1 CDK5 and gSK-3
Only five compounds, 85a,b, 87, 91 and (rac)-93 were tested against CDK5 and gSK-3
(glycogen synthase kinase-3) (Figure 6.7 (yellow)). Unfortunately, none of these
compounds were active against these two targets, with the IC50 of all compounds found
to be greater than 10 µM.
6.3.2 CDK2
Seventeen compounds, 72, 85a,b, 87, 91, (rac)-93a,b, 102-107, 108a,b, and 139a,b
were tested against CDK2 (Figure 6.7 (topaz)). The most active CDK2 inhibitor was
found to be compound 85b with 25% inhibition of the protein at a concentration of 100
µM. Unfortunately, most compounds tested against this target were inactive.
6.3.3 MDM2-p53
Thirteen compounds, 85b, (rac)-93a, 108b, 170a,b, 172, 174a, 175b, 177a, 178a,b,
179a and 181, were tested against MDM2-p53 (Figure 6.7 (red)). The most active
compounds against this receptor were 108b and 181, showing moderate inhibition
against this target with an IC50 of 45.9 ± 2.0 µM and 55.7 ± 3.5 µM, respectively.
Interestingly, the most active cytostatic compounds (174a and 172, Table 6.2, entries 6
and 7) were found to exhibit no activity at this target. Unfortunately due to the limited
amounts of 181, it was not submitted for cytostaticity studies.

dbev
Text Box
See print copy for figure 6.7

Chapter 6: Biological Testing Results and Discussion 135
6.4 Conclusion and Future Directions
From the cytostatic and the protein inhibition studies, some conclusions can be
ascertained. First, the spiro[indole-3,5`-isoxazolidin]-2(1H)-one and spiro[indole-3,6`-
[1,3]oxazinane]-2,2`(1H)-dione series proved to be the most active and promising series
over the spiro[cyclopentane-1,1`-[1H]isoindol]-3`(2`H)-ones and 2-azaspiro[4.4]nonan-
1-ones and then the spiro[cyclopropane-1`,3-[3H]indol]-2`(1H)-ones. A possible mode
of action was not elucidated, though the more flexible MDM2-p53 binding pocket is a
possibility. Generally, throughout the literature CDK2 and CDK5 inhibitors tend to be
planar heteroaromatics suggesting that the observed inactivity of our spirocyclic
compounds was possibly due to unfavourable steric interactions within the CDK active
site.39 Future biological studies should be directed at elucidating the possible mode of
action through screening against other cell-cycle protein targets, such as gSK-3 and bcl-
2, and other protein kinases, and to investigate the phase-inhibition of the spiro[indole-
3,5`-isoxazolidin]-2(1H)-one and spiro[indole-3,6`-[1,3]oxazinane]-2,2`(1H)-dione
series. Also an assessment of the anti-HIV activity of the spiro[cyclopropane-1`,3-
[3H]indol]-2`(1H)-ones may prove interesting, due to recent literature revealing similar
structures being potent HIV-1 non-nucleoside reverse transcriptase inhibitors.245,251

Chapter 7: Conclusions & Future Directions 136
CHAPTER 7: CONCLUSIONS AND FUTURE
DIRECTIONS The main aim of this project was to synthesise several interesting spirocyclic scaffolds
as potential novel cancer therapeutics targeting the cell-cycle. The three different
spirocyclic scaffolds (A-C) were successfully synthesised using the phosphine-
catalysed [3+2]-cycloaddition reaction, the cyclopropanation reaction utilising sulfur
ylides and the [1,3]-dipolar cycloaddition reaction, respectively. Enantio-enriched
versions of some of these spirocycles were also successfully synthesised utilising the
Oppolzer’s chiral auxiliary. Efforts were also made towards improving the synthetic
strategy towards these spirocycles by using novel amide ylides. In the case of the
spirocyclopropanes this methodology yielded greater amounts of the desired
spirocyclopropane 129 however for the spirocyclopentanes, 87 and (rac)-93a the
bulkier amide ylide resulted in lower yields, although the regioselectivities of these
reactions were enhanced.
NH
ONH
HN
O
O
A
NH
O
B C
RR
R
R = functional group
These spirocyclic derivatives were indeed shown to possess interesting cytostatic
activity, with notable low micromolar growth inhibition activity being observed for
spiro[indole-3,5`-isoxazolidin]-2(1H)-one 172 (GI50 = 4.1 ± 0.2 µM) and
hexahydropyrrolo[1,2-b]isoxazole-2`-carboxylate 174a (GI50 = 2.6 ± 0.1 µM) against
the breast cancer cell line MCF-7. Though initial studies for spirocyclopentanes 85a and
87 revealed a mild to moderate G1-phase block further elucidation of the mode of action
for the range of spirocycles synthesised, by protein inhibition studies, were inconclusive.
However these studies revealed that these spirocycles were not CDK2 inhibitors and
had the potential of acting as MDM2-p53 inhibitors with 181 displaying an IC50 of 55.7
± 3.5 µM against this target. Further biological assessment would be to demonstrate
cell-cycle phase-inhibition and protein-inhibition studies against other possible target
receptors. Furthermore, CDK2 research performed in the UK, with efforts towards a
new synthetic strategy for a potent CDK2 inhibitor and the revision of the former
synthetic route were successfully made.

Chapter 8: Experimental 137
CHAPTER 8: EXPERIMENTAL
Gelsemium elegans Benth. (Loganiaceae), is a toxic plant, widely distributed in
Southeast Asia and used in traditional Chinese medicine as a remedy for certain kinds
of skin ulcers and dermatitis. The gelsemium alkaloids possess potent cytotoxicity
against A431 epidermoid carcinoma cells.168

Chapter 8: General Synthetic Procedures 138
8.1 General Synthetic Procedures
All reagents used were of commercial grade and, unless specified, used as received.
THF was distilled over Na/benzophenone before use while DCM was distilled over
CaH2 before use. Anhydrous MeOH, MeCN and DMF were purchased from Aldrich in
sure-sealed bottles and used as supplied. Anhydrous benzene was dried over sodium. In
procedures involving petroleum spirit, only b.p. range 40-60 oC was used.
Microwave assisted reactions were perfomed using either the Milestone Microwave
Laboratory Systems, Microwave Solvent Extraction Lab Station connected to a Lab
Terminal 800 Controller operating at a maximum of 1000 Watts or using a CEM
EXPLORER 24-position Microwave connected to a Discover operating system, running
at a maximum of 300 Watts. Melting points were measured using a Gallenkamp melting
point apparatus and all measurements are reported in degrees celcius (oC). For Chapter 5,
melting points were determined on a Stuart Scientific SMP3 apparatus. Infrared spectra
(IR) were recorded neat on an Excalibur series BioRad Spectrophotometer. Ultraviolet
(UV) spectra were recorded in MeOH or EtOH on a U-2001 Hitachi Spectrophotometer.
Optical rotations were measured at the specified temperature using a 10 mm cell in a
JASCO DIP-370 Digital Polarimeter at a known concentration (g/100 mL) in CHCl3.
TLCs were preformed using Merck aluminium sheets precoated with Kieselgel 60 F254
(25 mm) as the adsorbent. These plates were also used for the separation of compounds
by PTLC. Purification of compounds using a chromatotron® were performed using
glass plates precoated with Kieselgel 60 PF254 (1 mm or 4 mm) silica gel and using a
Harrisson Research chromatotron® (Model 7924T). Both TLC and chromatotron®
plates were visualized with UV light at 254 and 365 nm. Purification by flash column
chromatography was conducted under medium pressure using Kieselgel 60 (0.040-
0.063 mm) silica gel.
Mass spectra were obtained using electron impact (EI), chemical ionization (CI) or
electrospray ionization (ESI) using either, a Shimadzu QP-5000 (LRMS: EI and CI), or
a VG Autospec (HRMS: EI and CI) or a Micromass QTofII (LR and HRMS: ESI)
spectrometer. The m/z readings were recorded and the intensity of the peak stated, in
brackets, as a percentage of the base peak.
NMR experiments were performed on a Varian Mercury 300 spectrometer in all
chapters except Chapter 5 where a Bruker Avance 300 spectrometer was used, both at
300 MHz (1H NMR) or 75.5 MHz (13C NMR), unless otherwise stated. CDCl3 was used

Chapter 8: General Synthetic Procedures 139
as the NMR solvent unless otherwise stated. All spectra were referenced to CDCl3 (1H
NMR δ 7.26 ppm and 13C NMR δ 77.00 ppm), C6D6 (1H NMR δ 7.16 ppm and 13C
NMR δ 128.40 ppm), CD3OD (1H NMR δ 3.31 ppm and 13C NMR δ 49.00 ppm) or
DMSO (1H NMR δ 2.50 ppm and 13C NMR δ 39.51 ppm).
NMR signals were assigned with the following nomenclature; singlet (s), doublet (d),
triplet (t), doublet of doublets (dd), doublet of triplets (dt), doublet of doublet of doublet
of doublets (dddd), doublet of quartets (dq) and AB quartet (ABq) or multiplet (m). All
chemical shifts (δ) are reported in parts per million (ppm) and coupling constants are
calculated in Hertz (Hz). 1H NMR assignments were achieved with the aid of gCOSY,
TOCSY, gHSQC and in some cases NOESY experiments. 13C NMR assignments were
based upon DEPT, gHSQC and gHMBC experiments.
X-ray crystallography was performed by Prof. A. H. White and Dr. B. W. Skelton at the
Department of Chemistry, University of Western Australia. Molecular modelling was
performed using PC Spartan ‘04 (AM1).
All solvents were removed under reduced pressure employing a Büchi rotary evaporator
and compounds were dried over anhydrous magnesium sulfate, unless otherwise stated.
All compounds are named according to Chemical Abstracts (CA) index nomenclature.
All compounds were determined to be of >95% purity by 1H NMR and TLC analysis.

Chapter 8: Experimental for Chapter 2 140
8.2 Experimental for Chapter 2
(3aS,6R,7aR)-1-But-2-ynoyl-8,8-dimethylhexahydro-3a,6-methano-2,1-
benzisothiazole 2,2-dioxide (53b) and (3aS,6R,7aR)-1-(2,2-Dimethylpropanoyl)-8,8-
dimethylhexahydro-3a,6-methano-2,1-benzisothiazole 2,2-dioxide (75)227,341
To a solution of 2-butynoic acid (784 mg, 9.15 mmol) in anhydrous THF (50 mL) at -78 oC (dry ice-acetone bath) under a N2 atmosphere was added in succession
trimethylacetyl chloride (1.24 mL, 10 mmol) and NEt3 (1.4 mL, 10 mmol). The reaction
mixture was allowed to stir at -78 oC for 15 min, then at 0 oC for 45 min and then
recooled to -78 oC. In a second flask containing a solution of n-butyllithium (6.3 mL of
1.5 M in hexane, 10 mmol) under a N2 atmosphere was added dropwise a solution of 74
(1.965 g, 9.15 mmol) in anhydrous THF (20 mL) and at -78 oC. This mixture was stirred
for 15 min and then cooled to -78 oC. With the whole system at -78 oC, the contents of
the second flask were transferred to the first flask via a cannula. The reaction mixture
was then slowly warmed to RT and stirred for 18 h. The crude mixture was washed with
KHSO4 solution (28 mL, 2 M) and extracted with EtOAc. The combined organic
extracts were washed with H2O and brine, and then dried, filtered and the solvent was
removed in vacuo. A 1H NMR analysis of the crude reaction mixture crude revealed a
1.6 : 1 of products 53b : 75. The products were purified by column chromatography
using 20% EtOAc:PS as eluent to yield the major product 53b as a white powder (1.40
g, 5.0 mmol, 55%) and the minor product 75 as white crystals (1.17 g, 3.9 mmol, 43%).
53b: Rf = 0.49 in 20% EtOAc:PS, m.p. 170-176 oC (lit.227 m.p.
170-172 oC), [α]D24 = -72.4 (c 0.5, CHCl3), (lit.
227 [α]D25 = -
108.4 (c 2, CHCl3)). MS (EI) m/z 281 (12%) [M.+]; HRMS
(CI+ve) Calcd for C14H20NO3S [MH+] 282.1164. Found:
282.1170. 1H NMR δ 3.87 (dd, J 7.6, 5.1 Hz, 1H, CH-7a);
3.46 (ABq, J 13.5 Hz, 2H, CH2-3); 2.22 (d, J 17.0 Hz, 1H, CH-7β); 2.11-2.03 (m, 1H,
CH-7α); 2.08 (s, 3H, CH-4`); 1.93-1.89 (m, 3H, CH-4β, CH-5β, CH-6); 1.41-1.35 (m,
2H, CH-4α, CH-5α); 1.18 (s, 3H, CH3-9); 0.98 (s, 3H, CH3-10). 13C NMR δ 150.0 (C-1`);
90.9 (C≡CCH3); 73.8 (C≡CCH3); 64.9 (CH-7a); 53.0 (CH2-3); 48.3 (C-3a); 47.8 (C-8);
44.8 (CH-6); 38.3 (CH2-7); 32.8 (CH2-4); 26.4 (CH2-5); 20.9 (CH3-9); 19.9 (CH3-10);
4.32 (CH3-4`). NMR data collected for 53b agreed with that reported in the literature.227
O 2 S N
H
O
C H 33 a
6
9
7
11 `

Chapter 8: Experimental for Chapter 2 141
75: Rf = 0.93 in 20% EtOAc:PS, m.p. 112-114 oC (lit.341 m.p. 124-
126 oC), [α]D23 = -29.4 (c 1.1, CHCl3) (lit.
342 [α]D25 = -55.5 (c 1.18,
CHCl3). MS (ESI+ve) m/z 299 (11%) [M+]. HRMS (CI+ve) Calcd
for C15H26NO3S [MH+] 300.1633. Found: 300.1644. 1H NMR δ
4.02 (dd, J 7.8 Hz, 4.5 Hz, 1H, CH-7a); 3.45 (ABq, J 13.8 Hz, 2H,
CH2-3); 2.01 (dd, J 13.5, 7.8 Hz, 1H, CH-7β); 1.90-1.83 (m, 3H,
CH-4β, CH-5β, CH-6); 1.81-1.73 (m, 1H, CH-7α); 1.49-1.42 (m, 1H, CH-4α); 1.34 (s,
9H, C(CH3)3); 1.26 (d, J 3.6 Hz, 1H, CH-5α); 1.15 (s, 3H, CH3-9); 0.95 (s, 3H, CH3-10). 13C NMR δ 179.5 (C-1`); 67.3 (CH-7a); 53.8 (CH2-3); 48.1 (C-3a); 47.8 (C-8); 44.2
(CH-6); 42.4 (C(CH3)3); 38.9 (CH2-7); 32.7 (CH2-4); 27.3 (C(CH3)3); 26.7 (CH2-5);
20.5 (CH3-9); 19.9 (CH3-10). NMR data collected for 75 agreed with that reported in
the literature.341
N-(4-Methoxybenzyl)-N-phenylbut-2-ynamide (53c)
The title compound was prepared from N-(4-
Methoxybenzyl)-N-phenylamine (117 mg, 0.6 mmol) and 2-
butynoic acid (50.7 mg, 0.6 mmol) using a similar method
to that described below for the synthesis of 85a. However
the reaction mixture was left stirring for 18 h. The crude
product was purified by gradient column chromatography
using 10-50% EtOAc:PS as the eluent to yield 53c as a brown oil (296.5 mg, 1.06 mmol,
66%, Rf = 0.62 in 50% EtOAc:PS). MS (ESI+ve) m/z 280.2 (99%) [MH+]; HRMS (EI)
Calcd for C18H17NO2 [M.+] 279.1259. Found: 279.1280. 1H NMR δ 7.31-7.29 (m, 2H,
ArCH-m); 7.11 (d, J 8.7 Hz, 2H, ArCH-o`); 7.07-7.04 (m, 1H, ArCH-p); 7.06 (d, J 7.8
Hz, 2H, ArCH-o); 6.78 (d, J 8.7 Hz, 2H, ArCH-m`); 4.86 (s, 2H, NCH2); 3.77 (s, 3H,
OCH3); 1.70 (s, 3H, CCH3). 13C NMR δ 158.9 (ArC-p`); 154.3 (CONH); 141.6 (ArC-i);
130.1 (ArCH-o`); 129.0 (ArC-i`); 128.8 (ArCH-m); 128.4 (ArCH-o); 127.8 (ArCH-p);
113.7 (ArCH-m`); 90.3 (C≡CCH3); 74.1 (C≡CCH3); 55.0 (OCH3); 51.5 (NCH2); 3.68
(CCH3).
tert-Butyl 3-methylene-2-oxopyrrolidine-1-carboxylate (57)
To a solution of 60 (2.02 g, 10.9 mmol) in anhydrous THF (32 mL) at -78
°C (dry ice/acetone bath) and under an atmosphere of N2 was added via
N5 1 OB o c
N
OMe
o
p
m
m'
o'
p'
i'
i
O
O2S N
H
O
3a
6
9
7
11̀

Chapter 8: Experimental for Chapter 2 142
syringe lithium hexamethyldisilazane (16 mL of a 1.0 M solution in hexanes, 15.9
mmol) and the solution was left stirring at -78 °C (dry ice/acetone bath) for 45 min. The
solution was transferred via cannula into a flask containing Eschenmoser`s salt (N,N-
dimethylmethylene ammonium iodide) (3.86 g, 20.9 mmol) under N2 and at -78 °C. The
reaction mixture was left to stir under a N2 atmosphere, vigorously and at -78 °C for the
first 15 min, and then at RT for 18 h. The reaction mixture was then quenched with H2O
(2 × 30 mL) and extracted with EtOAc (2 × 50 mL). The organic combined extracts
were washed with brine, dried, and evaporated in vacuo. To a solution of the crude
product in anhydrous DCM (50 mL), was added CH3I (5 mL, 80.3 mmol), and the
mixture was left to stir at RT for 3 d. The reaction mixture was evaporated under
reduced pressure. The oily residue was washed with sat. NaHCO3 solution (2 mL) and
extracted with EtOAc (3 × 10 mL). The organic extracts were dried, filtered and the
solvent was removed in vacuo. The crude product was purified by column
chromatography using 40-50% EtOAc:PS as the eluent to give 57 as a yellow custard
gum (935.1 mg, 4.74 mmol, 45%, Rf = 0.69 in 40% EtOAc:PS, m.p. 56-58 °C, (lit.213
m.p. 73-75 °C)). MS (ESI+ve) m/z 198 (14%) [MH+], 153 (47%) [MH+-(CH3)3], 98
(100%) [MH+-Boc]; HRMS (ESI+ve) Calcd for C10H16NO3 [MH+] 198.1130. Found:
198.1150. 1H NMR δ 6.18 (dt, J 3.0, 0.6 Hz, 1H, CHACHB=); 5.48 (dt, J 3.0, 0.6 Hz, 1H,
CHBCHA=); 3.73 (t, J 7.5 Hz, 2H, NCH2); 2.75 (tt, J 7.5, 3.0 Hz, 2H, CH2-4); 1.55 (s,
9H, C(CH3)3). 13C NMR δ 166.5 (C-2); 150.9 (NCO2); 139.3 (C-3); 119.9 (CH2=); 83.3
(C(CH3)3); 43.3 (NCH2); 28.4 (C(CH3)3); 23.5 (CH2-4). NMR data collected for 57
agreed well with those found in the literature.213
1-tert-Butyl 2-ethyl (2S)-4-methylene-5-oxopyrrolidine-1,2-dicarboxylate (58)
The title compound was prepared from 61 (722.7 mg, 2.8 mmol)
using a similar method to that described above for the synthesis of
57. The crude product was purified by column chromatography
using 25-50% EtOAc:PS as eluent to afford 58 as a clear oil (125.8
mg, 0.47 mmol, 21%, Rf = 0.42 in 25% EtOAc:PS, (lit. values215: Rf = 0.38 in 2:1
Hexane:EtOAc, [α]D25= -13.9 (c 1.5, CHCl3)). MS (CI+ve) m/z 269 (3%) [MH+];
HRMS Calcd for C13H20NO5 [MH+] 270.1341. Found: 270.1352. 1H NMR δ 6.24 (t, J
2.7 Hz, 1H, CHACHB=); 5.52 (t, J 2.4 Hz, 1H, CHBCHA=); 4.61 (dd, J 10.2, 3.3 Hz, 1H,
CH-2); 4.23 (q, J 7.2 Hz, 2H, CH2CH3); 3.08 (ddt, J 17.4, 9.9, 3.0 Hz, 1H, CHACHB-3);
21 N 5 OE t O 2 C B o c

Chapter 8: Experimental for Chapter 2 143
2.71 (ddt, J 17.4, 3.3, 2.4 Hz, 1H, CHBCHA-3); 1.52 (s, 9H, C(CH3)3); 1.28 (t, J 7.0 Hz,
3H, CH3CH2). 13C NMR δ 171.2 (CO2Et); 165.7(C-5); 150.1 (NCO2); 136.8 (C-4);
121.1 (CH2=); 84.0 (C(CH3)3); 61.9 (CH2CH3); 56.0 (CH-2); 28.1 (C(CH3)3); 28.0
(CH2-3); 14.3 (CH3CH2). NMR data collected for 58 agreed well with those found in
the literature.215
Methyl 2-(2-nitrophenyl)acrylate (59)127
To a solution of the malonate 73 (701 mg, 2.8 mmol) in formalin
(34-38% wt, 3 mL, which had previously been thoroughly shaken)
was added a solution of K2CO3 (580 mg, 4.2 mmol) in H2O (2 mL).
The resultant dark brown solution was then heated to 60 °C for 2 h
(a reflux condenser was attached to the flask) and then cooled to RT. The reaction
mixture was then quenched with H2O and extracted with Et2O. The combined extracts
were subsequently washed with brine, dried and the solvent was removed in vacuo. The
crude product was purified by column chromatography using 15-30% EtOAc:PS as
eluent to yield 59 as a yellow oil (471 mg, 2.27 mmol, 82%, Rf = 0.57 in 20%
EtOAc:PS). MS (CI+ve) m/z 208 (96%) [MH+], 176 (100%) [M+-OMe]; HRMS (CI+ve)
Calcd for C10H10NO4 [MH+] 208.0610. Found: 208.0604. 1H NMR δ 8.11 (dd, J 8.1, 1.2
Hz, 1H, ArCH-3); 7.66 (dt, J 7.5, 1.4 Hz, 1H, ArCH-5); 7.54 (dt, J 7.9, 1.5 Hz, 1H,
ArCH-4); 7.40 (dd, J 7.5, 1.8 Hz, 1H, ArCH-6); 6.54 (d, J 0.9 Hz, 1H, CHACHB=); 5.89
(d, J 0.9 Hz, 1H, CHBCHA=); 3.72 (s, 3H, OCH3). 13C NMR δ 165.2 (CO2Me); 147.7
(ArC-2); 133.6 (ArCH-5); 132.8 (ArC-1); 132.0 (ArCH-6); 129.3 (ArCH-4); 127.4
(CH2); 124.5 (ArCH-3); 120.1 (C=); 52.2 (CH3).
tert-Butyl 2-oxopyrrolidine-1-carboxylate (60)
To a solution of 2-pyrrolidinone (4.40 g, 51.6 mmol) in anhydrous
MeCN (100 mL) was added di-(tert-butoxylcarbonate) (12.87 g, 59.0
mmol) and a catalytic amount of DMAP (a few crystals). The
reaction mixture was then heated at reflux under a N2 atmosphere for
2 h. The reaction mixture was then allowed to cool to RT and then the solvent was
removed under reduced pressure. Compound 60 was isolated through purification by
column chromatography, using 60% EtOAc:PS as the eluent, to afford 60 as a brown oil
(8.77 g, 47.4 mmol, 92%, Rf = 0.6 in 60% EtOAc:PS). MS (ESI+ve) m/z 186.1 (5%)
NO2
CO2Me
61
2
NO 1 OO

Chapter 8: Experimental for Chapter 2 144
[MH+]; HRMS (ESI+ve) Calcd for C9H16NO3 [MH+] 186.1130. Found: 186.1136. 1H
NMR δ 3.75 (t, J 7.2 Hz, 2H, NCH2); 2.51 (t, J 8.1 Hz, 2H, CH2-3); 2.00 (q, J 7.5 Hz,
2H, CH2-4); 1.53 (s, 9H, C(CH3)3). 13C NMR δ 174.4 (C-2); 150.3 (NCO2); 83.0
(C(CH3)3); 46.8 (NCH2); 33.3 (CH2-3); 28.4 (C(CH3)3); 17.8 (CH2-4). NMR data
collected for 60 agreed well with those found in the literature.113
1-tert-Butyl 2-ethyl (2S)-5-oxopyrrolidine-1,2-dicarboxylate (61)
The title compound was prepared from (S)-(+)-ethyl 2-
pyrrolidone-5-carboxylate (684.7 mg, 4.35 mmol) using a
similar method to that described for the synthesis of 60.
Compound 61 was isolated, through purification by column chromatography using 50%
EtOAc:PS as the eluent, as a yellow oil (722.7 mg, 2.8 mmol, 64%, Rf = 0.51 in 50%
EtOAc:PS). MS (CI+ve) m/z 258 (34%) [MH+]; HRMS (CI+ve) Calcd for C12H20NO5
[MH+] 258.1341. Found: 258.1349. 1H NMR δ 4.60 (dd, J 9.4, 2.8 Hz, 1H, CH-2); 4.24
(q, J 7.2 Hz, 2H, CH2CH3); 2.70-2.25 (m, 3H, CH2-4 and CHACHB-3); 2.08-1.98 (m,
1H, CHBCHA-3); 1.50 (s, 9H, (CH3)3); 1.30 (t, J 7.0 Hz, 3H, CH2CH3). 13C NMR δ
173.4 (C-5); 171.3 (CO2Et); 149.2 (NCO2); 83.5 (C(CH3)3); 61.8 (CH2CH3); 59.1 (CH-
2); 31.3 (CH2-4); 28.1 (C(CH3)3); 21.7 (CH2-3); 14.4 (CH3CH2).
2-tert-Butyl 7-ethyl (5R*)-1-oxo-2-azaspiro[4.4]non-7-ene-2,7-dicarboxylate (62)
and 2-tert-butyl 6-ethyl (5S*)-1-oxo-2-azaspiro[4.4]non-6-ene-2,6-dicarboxylate (63)
To a solution of 57 (200.7 mg, 1.02 mmol) in
anhydrous benzene (3 mL) was added ethyl 2-
butynoate (0.13 mL, 1.12 mmol) and
tributylphosphine (0.25 mL, 1.01 mmol). The
reaction mixture was allowed to stir at RT for 18
h, under an atmosphere of N2. The solvent was
then evaporated in vacuo. A 1H NMR analysis of crude mixture revealed an 82 : 18
mixture of the two regioisomers, 62 and 63, respectively. Compounds 62 and 63 were
purified first by column chromatography using 10-30% EtOAc:PS as the eluent and
further using PTLC (30% EtOAc:PS) to give pure samples of 62 and 63.
62: (Yellow oil, 161.7 mg, 0.52 mmol, 51%, Rf = 0.78 in 30% EtOAc:PS) MS (ESI+ve)
m/z 310.0 (32%) [MH+], 332.1 (60%) [M++Na+], 348 (15%) [M++K+], 254.1 (100%)
2
1N
5OEtO2C
OO
N
B o c
E t O 2 C
H ββββH ββββ
H αααα H αααα
N
B o c
E t O 2 C96
5
3 1
96
5
3 1O O
6 2 6 3

Chapter 8: Experimental for Chapter 2 145
[MH+-C(CH3)3], 210.1 (85%) [MH+-Boc]; HRMS (CI+ve) Calcd for C16H24NO5 [MH+]
310.1654. Found: 310.1664. 1H NMR (C6D6, 500 MHz) δ 6.37 (t, J 2.0 Hz, 1H, CH=);
3.98 (dd, J 14.0, 7.5 Hz, 2H, CH2CH3); 3.20 (ddd, J 13.0, 6.0, 6.0 Hz, 2H, NCH2); 3.04
(dq, J 16.5, 2.5 Hz, 1H, CH-6β); 2.71 (dq, J 18.5, 2.5 Hz, 1H, CH-9β); 2.13 (d, 16.5 Hz,
1H, CH-6α); 1.73 (d, 18.0 Hz, 1H, CH-9α); 1.48 (s, 9H, C(CH3)3); 1.12 (ddd, J 13.0, 6.0,
6.0 Hz, 1H, CHACHB-4); 1.04 (ddd, J 13.0, 6.0, 6.0 Hz, 1H, CHBCHA-4); 0.97 (t, 7.5 Hz,
3H, CH3CH2). 13C NMR (C6D6) δ 175.4 (C-1); 163.7 (CO2Et); 151.0 (NCO2); 139.8
(CH=); 134.3 (C-7); 82.1 (C(CH3)3); 60.1 (CH2CH3); 51.6 (C-5); 43.3 (CH2-9); 42.9
(NCH2); 42.3 (CH2-6); 33.2 (CH2-4); 28.1 (C(CH3)3); 14.3 (CH3CH2). NMR data for 62
agreed well with the literature when performed under literature conditions.213
63: (White crystalline solid, 44.3 mg, 0.14 mmol, 21%, Rf = 0.58 in 30% EtOAc:PS,
m.p. 100-102 °C (lit.213 m.p. 107-110.5 °C). MS (ESI+ve) m/z 310.2 (53%) [MH+],
332.1 (29%) [M++Na+], 348.1 (23%) [M++K+], 254.1 (100%) [MH+-C(CH3)3], 209.8
(95%) [MH+-Boc]; HRMS (ESI+ve) Calcd for C16H24NO5 [MH+] 310.1654. Found:
310.1654. 1H NMR (500 MHz) δ 6.99 (t, J 2.5 Hz, 1H, CH=); 4.17 (ddd, J 14.5, 7.0, 0.5
Hz, 2H, CH2CH3); 3.90 (ddd, 10.5, 9.5, 3.5 Hz, 1H, NCHACHB); 3.63 (ddd, J 10.5, 8.5,
8.5 Hz, 1H, NCHBCHA); 2.69-2.62 (m, 1H, CHACHB-8); 2.58-2.51 (m, 1H, CHBCHA-8);
2.46-2.38 (om, 2H, CHACHB-9 and CHACHB-4); 1.98 (ddd, J 13.0, 8.5, 4.5 Hz,
CHBCHA-9); 1.92 (ddd, J 12.5, 8.5, 4.0 Hz, CHBCHA-4); 1.54 (s, 9H, (C(CH3)3); 1.26
(dt, J 7.5, 2.5 Hz, 3H, CH3CH2). 13C NMR δ 176.8 (C-1); 163.6 (CO2Et); 150.6 (NCO2);
147.5 (CH=); 138.0 (C-6); 83.0 (C(CH3)3); 60.8 (CH2CH3); 59.7 (C-5); 44.2 (NCH2);
37.4 (CH2-9); 31.7 (CH2-8); 29.8 (CH2-4); 28.4 (C(CH3)3); 14.5 (CH3CH2). The
structure of 63 was confirmed by X-ray crystallography (see Appendix 1: X-ray 1:
Compound 63). NMR data collected for 63 agreed well with those found in the
literature.213

Chapter 8: Experimental for Chapter 2 146
2-tert-Butyl 3,7-diethyl (3S,5R)-1-oxo-2-azaspiro[4.4]non-7-ene-2,3,7-tricarboxylate
(66), 2-tert-butyl 3,6-diethyl (3S,5S)-1-oxo-2-azaspiro[4.4]non-6-ene-2,3,6-
tricarboxylate (67) and 2-tert-butyl 3,7-diethyl (3S,5S*)-1-oxo-2-azaspiro[4.4]non-
7-ene-2,3,7-tricarboxylate (68)
To a solution of 58 (125.8 mg, 0.47 mmol) in
anhydrous benzene (3 mL) was added ethyl-
2-butynoate (0.06 mL, 1.12 mmol) and Bu3P
(0.25 mL, 1.01 mmol). The reaction was
allowed to stir at RT for 15 h, under an
atmosphere of N2. The solvent was
evaporated in vacuo and 1H NMR analysis
showed a mixture of the two diastereomers, 66 and 68, and one regioisomer 67 (66 : 67
: 68 = 63 : 17 : 20). Compounds 66 and 68 were purified by column chromatography
using 20-90% EtOAc:PS as eluent and further by PTLC (30% EtOAc:PS). Compound
67 was unable to be isolated as a pure sample.
66: (Yellow oil, 50.5 mg, 0.13 mmol, 28%, Rf = 0.57 in 30% EtOAc:PS, [α]D22= -17.5
(c 0.5, CHCl3). MS (ESI+ve) m/z 382.2 (5%) [MH+]; HRMS (ESI+ve) Calcd for
C19H28NO7 [MH+] 382.1866. Found: 382.1892. 1H NMR (500 MHz) δ 6.57 (s, 1H,
CH=); 4.54 (dd, J 9.5, 4.5 Hz, 1H, CH-5); 4.21 (dq, J 6.5, 1.5 Hz, 2H, NCHCO2CH2);
4.16 (q, J 6.7 Hz, 2H, CCO2CH2); 3.15 (dd, J 16.5, 2 Hz, 1H, CH-6β); 3.04 (dd, J 18.5,
1.5 Hz, 1H, CH-9β); 2.55 (d, J 16.5 Hz, 1H, CH-6α); 2.39 (d, J 17.5 Hz, 1H, CH-9α);
2.55-2.33 (m, 1H, CHACHB-4); 2.09 (dd, J 13.0, 4.0 Hz, 1H, CHBCHA-4); 1.49 (s, 9H,
C(CH3)3); 1.29-1.23 (m, 6H, CH2CH3). 13C NMR δ 176.9 (C-2); 171.5 (NCHCO2Et);
164.3 (CCO2Et); 149.6 (NCO2) 139.8 (CH=); 134.4 (C-7); 84.0 (C(CH3)3); 61.9
(NCHCO2CH2); 60.7 (CCO2CH2); 56.7 (CH-5); 51.0 (C-3); 45.2 (CH2-9); 43.5 (CH2-6);
37.9 (CH2-4); 28.1 (C(CH3)3); 14.4 (CH3CH2); 14.3 (CH3CH2).
68: (Yellow oil, 24 mg, 63 µmol, 13%, Rf = 0.36 in 30% EtOAc:PS, [α]D23= -1.88 (c
0.014, CHCl3). MS (ESI+ve) m/z 382.2 (5%) [MH+]; HRMS (ESI+ve) Calcd for
C19H28NO7 [MH+] 382.1866. Found: 382.1914. 1H NMR (500 MHz) δ 6.65 (s, 1H,
CH=); 4.57 (dd, J 9.5, 3.5 Hz, 1H, CH-5); 4.29-4.21 (m, 2H, NCHCO2CH2); 4.18 (q, J
7.5 Hz, 2H, CCO2CH2); 3.17-3.12 (m, 1H, CH-6β); 3.12-3.08 (m, 1H, CH-9β); 2.51 (d, J
16.5 Hz, 1H, CH-6α); 2.45 (d, J 18.5 Hz, 1H, CH-9α); 2.33 (dd, J 13.5, 9.5 Hz, 1H,
CHACHB-4); 2.20 (dd, J 13.5, 4.0 Hz, 1H, CHBCHA-4); 1.51 (s, 9H, C(CH3)3); 1.31 (t, J
N
Boc
EtO2C
O
EtO2C N
Boc
EtO2C
O
EtO2C
96
5
3 1
96
5
3 1
(5R)-66(5S)-68
67

Chapter 8: Experimental for Chapter 2 147
7.0 Hz, 3H, CCO2CH2CH3); 1.27 (t, J 7.0 Hz, NCHCO2CH2CH3). 13C NMR δ 177.0 (C-
2); 171.1 (NCHCO2Et); 164.3 (CCO2Et); 149.4 (NCO2); 139.9 (C-8); 133.8 (C-7); 83.8
(C(CH3)3); 61.8 (NCHCO2CH2); 60.4 (CCO2CH2); 56.4 (CH-5); 50.7 (C-3); 44.8 (CH2-
9); 43.6 (CH2-6); 37.8 (CH2-4); 27.8 (C(CH3)3); 14.2 (CH3CH2); 14.1 (CH3CH2).
1-(tert-Butoxycarbonyl)-oxindole (69)
The title compound was prepared from oxindole (1.09 g, 8.2 mmol)
using a similar method to that described above for the synthesis of
60. The crude product was purified by column chromatography
using 10% EtOAc:PS as eluent to yield 69 as a creamy orange crystalline solid (840.3
mg, 3.6 mmol, 44%, m.p. 50-54 °C (lit.343 m.p. 67 °C), Rf = 0.42 in 10% EtOAc:PS).
MS (ESI+ve) m/z 234 (23%) [MH+]; HRMS (ESI+ve) Calcd for C13H16NO3 [MH+]
234.1130. Found: 234.1138. 1H NMR δ 7.75 (d, J 8.1 Hz, 1H, ArCH-7); 7.29-7.19 (m,
2H, ArCH-4 and ArCH-6); 7.10 (t, J 7.5 Hz, 1H, ArCH-5); 3.61 (s, 2H, CH2CO); 1.64
(s, 9H, C(CH3)3). 13C NMR δ 173.1 (C-2); 149.3 (NCO2); 141.1 (ArC-7a); 128.2
(ArCH); 124.4 (ArCH); 123.4 (ArC-3a); 115.2 (ArCH-7); 84.5 (C(CH3)3); 36.8
(CH2CO); 28.4 (C(CH3)3).
(1`S*)-3`-Ethyl 1`-methyl 1`-(2-nitrophenyl)cyclopent-3`-ene-1`,3`-dicarboxylate
(71)
To a solution of the alkene 59 (1.013 g, 4.9 mmol) and ethyl-2-
butynoate (0.63 mL, 5.4 mmol) in anhydrous benzene (35 mL)
was slowly added Bu3P (0.24 mL, 9.8 × 10-4 mol). The reaction
mixture was left to stir at RT for 6 h. Upon evaporation in vacuo
of the volatiles the crude product was purified by column chromatography using 20-
50% EtOAc:PS as eluent to yield a peach coloured oil (1.45 g, 4.5 mmol, 93%, Rf =
0.81 in 50% EtOAc:PS). MS (CI+ve) m/z 320 (100%) [MH+], 288 (41%) [M+-OMe],
260 (13%) [M+-CO2Me], 246 (22%) [M+-(CO2Me, CH3)], 206 (68%), 188 (21%) [M+-
(CO2Me, CO2Et)]; HRMS (CI+ve) Calcd for C16H18NO6 [MH+] 320.1134. Found:
320.1132. 1H NMR δ 7.93 (dd, J 8.1, 1.5Hz, 1H, ArCH-3); 7.58 (dt, J 7.8, 1.8 Hz, 1H,
ArCH-5); 7.42 (dt, J 7.6, 1.5 Hz, 1H, ArCH-4); 7.40 (d, 7.8 Hz, 1H, ArCH-6); 6.74 (t, J
1.8 Hz, 1H, CH=); 4.20 (q, J 6.9 Hz, 2H, CH2CH3); 3.64 (s, 3H, CO2CH3); 3.62 (dq, J
19.2, 2.7 Hz, 1H, CH-5`β); 3.52 (dq, J 17.4, 2.5 Hz, 1H, CH-2`β); 3.21 (dm, J 17.1 Hz,
N
Boc
O
3a
7a7
5
NO2
CO2Me
O
OEt
2
11`
3`
Hαααα
Hαααα
Hββββ
Hββββ

Chapter 8: Experimental for Chapter 2 148
1H, CH-2`α); 2.99 (dt, J 19.2, 2.4 Hz, 1H, CH-5`α); 1.29 (t, J 6.9 Hz, 3H, CH3CH2). 13C
NMR δ 174.0 (CO2Me); 164.0 (CO2Et); 148.1 (ArC-2); 140.1 (CH=); 138.1 (ArC-1);
133.8 (C-3`); 133.2 (ArCH-5); 128.3 (ArCH-6); 128.0 (ArCH-4); 125.3 (ArCH-3); 60.6
(CH2CH3); 55.6 (C-1`); 52.4 (CO2CH3); 45.8 (CH2-5`); 44.2 (CH2-2`); 14.2 (CH2CH3).
Ethyl (1`S*)-2-oxo-1,2-dihydrospiro[cyclopent-3`-ene-1`,3-indole]-3`-carboxylate
(72)
To a solution of 71 (29.6 mg, 9.3 × 10-5 mol) in EtOH (0.7 mL)
and H2O (0.18 mL) was added activated Zn dust (96 mg, 1.5
mmol) and 8.9 M HCl (0.14 mL).225 The reaction was heated at
reflux (100 °C) for 2 h. Another aliquot of activated Zn dust
(96 mg, 1.5 mmol) was added and the reaction was left at reflux for an additional 4 h.
The mixture was then cooled and filtered through a bed of celite and diluted with H2O.
The filtrate was then extracted with EtOAc and the organic extracts were combined and
dried to yield 71 as a creamy brown oil (23.4 mg, 9.1 × 10-5 mol, 98%, Rf = 0.5 in 50%
EtOAc:PS), which was pure upon 1H NMR analysis and did not require further
purification. MS (CI+ve) m/z 258 (100%) [MH+], 212 (12%) [MH+-OEt], 184 (12%)
[MH+-CO2Et]; HRMS (EI) Calcd for C15H15NO3 [M.+] 257.1052. Found: 257.1048. 1H
NMR (500 MHz) δ 9.15 (bs, 1H, NH); 7.21 (d, J 7.5 Hz, 1H, ArCH-4); 7.20 (t, J 8.0 Hz,
1H, ArCH-6); 7.01 (t, J 7.7 Hz, 1H, ArCH-5); 6.93 (d, J 8.0 Hz, 1H, ArCH-7); 6.86 (bs,
1H, CH=); 4.23 (q, J 7.0 Hz, 2H, CH2CH3); 3.27 (dd, J 16.5, 2.5 Hz, 1H, CH-2`β); 3.19
(dd, J 18.7, 2.3 Hz, 1H, CH-5`β); 2.90 (d, J 16.5 Hz, 1H, CH-2`α); 2.80 (d, J 18.5 Hz,
1H, CH-5`α); 1.31 (t, J 7.3 Hz, 3H, CH3CH2). 13C NMR δ 183.2 (C-2); 164.2 (CO2Et);
140.6 (CH=); 139.7 (ArC-7a); 136.6 (ArC-3a); 134.8 (C-3`); 128.1 (ArCH-6); 123.0
(ArCH-5); 122.1 (ArCH-4); 109.9 (ArCH-7); 60.5 (CH2CH3); 52.5 (C-3); 44.9 (CH2-5`);
43.4 (CH2-2`); 14.2 (CH3CH2).
Dimethyl (2-nitrophenyl)malonate (73)
The title compound was prepared using two methods. Method 1:127
To a suspension of NaH (0.75 g, 15.6 mmol, 50% dispersion in
paraffin wax) in anhydrous THF (120 mL) at 0 °C (ice bath) was
added dimethyl malonate (1.62 mL, 14.2 mmol) dropwise over a
period of 20 min followed by the introduction of a solution of 2-fluoronitrobenzene
O
OEt
NH
O
Hββββ
Hββββ
Hαααα
Hαααα
33a
7a
5
7
1`
3`
N O 2C O 2 M e2

Chapter 8: Experimental for Chapter 2 149
(0.75 mL, 7.1 mmol) in anhydrous THF (50 mL) via cannula under N2. The mixture was
warmed to RT, then heated to 60 °C and left to stir for 18 h. The solution turned a dark
orange colour. The mixture was cooled to 0 °C before quenching with sat. NH4Cl
solution. The resulting light yellow solution was then extracted with EtOAc (4 × 100
mL). The combined extracts were washed successively with H2O and brine and then
dried and the solvent was removed in vacuo. The crude product was purified by column
chromatography, elution with 10-20% EtOAc:PS yielded 73 as a yellow oil (837.5 mg,
3.3 mmol, 46.5%). Method 2:222 To a solution of dimethyl malonate (1.31 mL, 12.7
mmol) in anhydrous DMF (30 mL) at 0 °C (ice bath) under Ar was added anhydrous
K2CO3 (3.3 g, 21 mmol). The mixture was allowed to stir for 15 min at 0 °C before the
addition of 2-fluoronitrobenzene (1.12 mL, 10.5 mmol). The mixture was allowed to
warm to RT, then heated at 60 °C for 18 h. The reaction mixture was then cooled to RT
before the addition of water and extraction with EtOAc (4 × 30 mL). The combined
extracts were washed multiple times with H2O and brine before drying and the solvent
was removed in vacuo. The crude product was purified by column chromatography
using 10-20% EtOAc:PS as eluent to afford 73 as a light semi-crystalline yellow oil
(3.00 g, 12 mmol, 93%, Rf = 0.3 in 20% EtOAc:PS). MS (CI+ve) m/z 254 (100%)
[MH+], 178 (20%) [MH+-(NO2, OMe)], 133 (29%); HRMS (CI+ve) Calcd for
C11H12NO6 [MH+] = 254.0665. Found: 254.0661. 1H NMR δ 8.08 (dd, J 8.1, 1.5 Hz, 1H,
ArCH-3); 7.67 (dt, J 7.5, 1.5 Hz, 1H, ArCH-5); 7.56 (dt, J 7.5, 1.8 Hz, 1H, ArCH-4);
7.51 (dd, J 7.8, 1.5 Hz, 1H, ArCH-6); 5.33 (s, 1H, CH); 3.81 (s, 6H, ((CO2CH3)2)). 13C
NMR 167.1 (CO2Me); 148.2 (ArC-2); 133.2 (ArCH-5); 131.0 (ArCH-6); 129.0 (ArCH-
4); 127.5 (ArC-1); 124.7 (ArCH-3); 53.8 (CH); 52.5 (CH3). The NMR data collected for
73 corresponded to the NMR data given by the author.222
(3aS,6R,7aR)-8,8-Dimethylhexahydro-3a,6-methano-2,1-benzisothiazole 2,2-
dioxide (74)
To a solution of (3aS,6R)-8,8-Dimethyl-4,5,6,7-tetrahydro-3a,6-methano-
2,1-benzisothiazole 2,2-dioxide (2.08 g, 9.76 mmol) in H2O (3.2 mL) and
MeOH (9.5 mL) at 5 oC was added, in portions, NaBH4 (2.08 g, 9.76 mmol).
The reaction was then allowed to stir for 19 h at RT. The MeOH was then removed in
vacuo and the residue was diluted with DCM (~10 mL). The mixture was then washed
with aqueous 51% H2SO4 (13 mL) and extracted with DCM. The combined organic
O 2 S N H3 a 6 71

Chapter 8: Experimental for Chapter 2 150
extracts were washed with brine and subsequently dried. Evaporation and
recrystallisation from EtOH yielded 74 as white crystals (1.26 g, 5.87 mmol, 61%, Rf =
0.25 in 20% EtOAc:PS, m.p. 172-176 oC (lit.344 m.p. 190-191 oC), [α]D23 = -20.4 (c 1.0,
CHCl3), (lit.344 [α]D20 = -31.3 (c 5, CHCl3))). MS (ESI+ve) m/z 216 (100%) [MH+];
HRMS (EI) Calcd for C10H17NO2S [M.+] 215.0980. Found: 215.0971. 1H NMR (500
MHz) δ 4.14 (bs, 1H, NH); 3.43 (m, 1H, CH-7a); 3.12 (ABq, J 14.8 Hz, 2H, CH2-3);
2.01-1.95 (m, 1H, CH-7β); 1.94-1.83 (m, 4H, CH-4β, CH-5β, CH-6, CH-7α); 1.46 (t, J
9.0 Hz, 1H, CH-4α); 1.34-1.28 (m, 1H, CH-5α); 1.13 (s, 3H, CH3-9); 0.94 (s, 3H, CH3-
10). 13C NMR (125 MHz) δ 62.8 (CH-7a); 55.0 (C-3a); 50.3 (CH2-3); 47.4 (C-8); 44.7
(CH-6); 36.0 (CH2-7); 31.8 (CH2-4); 26.8 (CH2-5); 20.5 (CH3-9); 20.4 (CH3-10). NMR
data collected for 74 agreed with that reported in the literature.344
Methyl (3aS,6R,7aR,1`S)-3`-[(8,8-dimethylhexahydro-3a,6-methano-2,1-
benzisothiazole 2,2-dioxide)carbonyl]-1`-(2-nitrophenyl)cyclopent-3`-ene-1`-
carboxylate ((S)-76) and Methyl (3aS,6R,7aR,1`R)-3`-[(8,8-dimethylhexahydro-
3a,6-methano-2,1-benzisothiazole 2,2-dioxide)carbonyl]-1`-(2-
nitrophenyl)cyclopent-3`-ene-1`-carboxylate ((R)-77)
To a solution of 59 (147 mg, 0.71 mmol) and 53b (198 mg, 0.71 mmol) in anhydrous
benzene (1.5 mL) under a N2 atmosphere was added Bu3P (0.02 mL, 71 µmol). The
reaction was stirred at RT for 18 h and was then concentrated. The diastereomeric
products were obtained in a ratio of 3.3 : 1 ((S)-76 : (R)-77) from 1H NMR analysis of
the crude reaction mixture. The crude mixture was purified by column chromatography
using 15% EtOAc:PS as eluent, yielding pure diastereomeric products (S)-76 (140.6 mg,
0.29 mmol, 13%), as colourless crystals, and (R)-77 (154.5 mg, 0.32 mmol, 15%) as
colourless crystals and a mixture (399.8 mg, 0.82 mmol, 38%) containing both
diastereomeric products in the ratio of 4.3:1 ((S)-76 : (R)-77). Further purification by
PTLC (20% EtOAc:PS) yielded more of the pure diastereomeric products.

Chapter 8: Experimental for Chapter 2 151
(S)-76: Rf = 0.53 in 30% EtOAc:PS, m.p. 196-200 oC, [α]D26 = -
22.0 (c 0.3, CHCl3). MS (EI) m/z 488 (5%) [M.+]; HRMS (CI+ve)
Calcd for C24H29N2O7S [MH+] 489.1695. Found: 489.1690. 1H
NMR (500 MHz) δ 7.94 (d, J 7.5 Hz, 1H, ArCH-3); 7.56 (bs, 2H,
ArCH-5, ArCH-6); 7.41 (bs, 1H, ArCH-4); 6.74 (bs, 1H, CH=);
4.07 (t, J 5.5 Hz, 1H, CH-7a); 3.74 (d, J 19.5 Hz, 1H, CH-5`β);
3.66 (s, 3H, CO2CH3); 3.64 (d, J 19.0 Hz, 1H, CH-2`β); 3.45 (ABq,
J 13.5 Hz, 2H, CH2-3); 3.19 (d, J 19.0 Hz, 1H, CH-2`α); 3.06 (d, J
19.5 Hz, 1H, CH-5`α); 2.09-1.99 (m, 2H, H-7α, CH-7β ); 1.97-1.91 (m, 3H, CH-4β, CH-
5β, CH-6); 1.44-1.37 (m, 2H, CH-4α, CH-5α); 1.24 (s, 3H, CH3-9); 1.00 (s, 3H, CH3-10). 13C NMR (125 MHz) δ 174.8 (CO2CH3); 171.3 (=CCO); 148.5 (ArC-2); 141.6 (CH=);
138.4 (ArC-1) 134.7 (C-3`); 133.4 (ArCH-5); 129.2 (ArCH-6); 128.0 (ArCH-4); 125.1
(ArCH-3); 65.6 (CH-7a); 54.6 (C-1`); 53.7 (CH2-3); 52.4 (CO2CH3); 48.1 (C-3a); 47.7
(C-8); 47.0 (CH2-5`); 45.5 (CH2-2`); 45.2 (CH-6); 38.3 (CH2-7); 33.3 (CH2-4); 26.5
(CH2-5); 21.3 (CH3-9); 19.9 (CH3-10). The structure of (S)-76 was confirmed by X-ray
crystallography (see Appendix 1: X-ray 13: Compound (S)-76)
(R)-77: Rf = 0.43 in 30% EtOAc:PS, m.p. 198-202 oC, [α]D24 =
+19.0 (c 0.6, CHCl3). MS (EI) m/z 488 (2.6%) [M.+]; HRMS
(CI+ve) Calcd for C24H29N2O7S [MH+] 489.1695. Found:
489.1713. 1H NMR (500 MHz) δ 7.95 (d, J 8.0 Hz, 1H, ArCH-3);
7.60 (t, J 7.5 Hz, 1H, ArCH-5); 7.54(d, J 8.0 Hz, 1H, ArCH-6);
7.43 (t, J 7.5 Hz, 1H, ArCH-4); 6.66 (s, 1H, CH=); 4.08-4.05 (m,
1H, CH-7a); 3.80 (d, J 19.0 Hz, 1H, CH-5`α); 3.68 (s, 3H,
CO2CH3); 3.50 (d, J 13.5 Hz, 1H, CH-3α); 3.46-3.42 (m, 3H,
CH-2`α, CH-2`β, CH-3β); 2.97 (d, J 19.0 Hz, 1H, CH-5`β); 2.06-2.00 (m, 2H, CH-7α,
CH-7β); 1.98-1.90 (m, 3H, CH-4β, CH-5β, CH-6); 1.46-1.40 (m, 2H, CH-4α, CH-5α);
1.23 (s, 3H, CH3-9); 1.00 (s, 3H, CH3-10). 13C NMR (125 MHz) δ 173.9 (CO2CH3);
165.7 (=CCO); 148.2 (ArC-2); 139.9 (CH=); 138.1 (ArC-1); 134.7 (C-3`); 133.3
(ArCH-5); 128.6 (ArCH-6); 128.0 (ArCH-4); 125.4 (ArCH-3); 65.5 (CH-7a); 55.9 (C-
1`); 53.6 (CH2-3); 52.5 (CO2CH3); 48.1 (C-3a); 47.7 (C-8); 46.3 (CH2-5`); 44.8 (CH2-
2`); 45.2 (CH-6); 38.4 (CH2-7); 33.2 (CH2-4); 26.5 (CH2-5); 21.3 (CH3-9); 19.9 (CH3-
10).
NO2
CO2Me
O
Hαααα
Hαααα
Hββββ
Hββββ
O2S N
H
2
11`
3`
3a
6
9
7
1
NO 2
CO2Me
O
H αααα
Hαααα
H ββββ
H ββββ
O2S N
H
2
11`
3`
3a
6
9
7
1

Chapter 8: Experimental for Chapter 2 152
Dimethyl (1`S)-1`-(2-nitrophenyl)cyclopent-3`-ene-1`,3`-dicarboxylate ((S)-78)
To a solution of (S)-76 (199 mg, 0.4 mmol) in anhydrous
MeOH (10 mL) was added Sm(OTf)3 (258 mg, 0.43 mmol).
The reaction was heated at 50 oC for 15 h. The mixture was
cooled and the solvent was removed in vacuo. The residue was
then diluted with DCM and washed with brine and sat. NaHCO3
solution, dried and the solvent was removed in vacuo. The crude mixture was purified
by column chromatography using 8:11:1 (DCM:PS:EtOAc) as eluent to afford (S)-78 as
a peach oil (82.4 mg, 0.27 mmol, 67%) and the the recovered chiral auxiliary as white
crystals.
(S)-78: Rf = 0.42 in 20% EtOAc:PS, [α]D24 = -42.5 (c 0.1, CHCl3), MS (ESI+ve) m/z
306 (13%) [MH+]; HRMS (ESI+ve) Calcd for C15H16NO6 [MH+] 306.0978. Found:
306.0966. 1H NMR δ 7.95 (dd, J 7.8, 1.5 Hz, 1H, ArCH-3); 7.59 (dt, J 7.5, 1.5 Hz, 1H,
ArCH-5); 7.44 (dt, J 7.5, 1.5 Hz, 1H, ArCH-4); 7.42 (dd, J 7.8, 1.5 Hz, 1H, ArCH-6);
6.78-6.75 (m, 1H, CH=); 3.77 (s, 3H, =CCO2CH3); 3.67 (s, 3H, PhCCO2CH3); 3.62 (dq,
J 19.5, 2.7 Hz, 1H,CH-5`β); 3.51 (dq, J 17.4, 2.4 Hz, 1H, CH-2`β); 3.22 (dt, J 17.1, 1.5
Hz, 1H, CH-2`α); 2.98 (ddt, J 19.1, 2.4, 0.9 Hz, 1H, CH-5`α). 13C NMR (125 MHz) δ
174.0 (PhCCO2Me); 164.4 (=CCO2Me); 148.2 (ArC-2); 140.4 (CH=); 138.0 (ArC-1);
133.6 (C-3`); 133.2 (ArCH-5); 128.3 (ArCH-6); 128.1 (ArCH-4); 125.3 (ArCH-3); 55.7
(C-1`); 52.4 (PhCCO2CH3); 51.7 (=CCO2CH3); 45.8 (CH2-5`); 44.2 (CH2-2`). NMR
data collected for (S)-78 agreed well with the spectroscopic data (apart from the ester
signals) to racemic 71.
Dimethyl (1`R)-1`-(2-nitrophenyl)cyclopent-3`-ene-1`,3`-dicarboxylate ((R)-79)
The title compound was prepared from a method similar to that
described above for the synthesis of (S)-78 using (R)-77 (81.9
mg, 0.17 mmol). Purification by column chromatography using
solvent system 8:11:1 (DCM:PS:EtOAc) as eluent yielded (R)-79
as a brown oil (34.7 mg, 0.1 mmol, 68%) and recovered chiral
auxiliary as white crystals. (R)-79: Rf = 0.24 in 20% EtOAc:PS, [α]D26 = +50.0 (c 0.7,
CHCl3). MS (ESI+ve) m/z 306 (40%) [MH+]; HRMS (ESI+ve) Calcd for C15H16NO6
[MH+] 306.0978. Found: 306.0985. NMR data collected for (R)-79 was identical to that
of its enantiomer (S)-78.
NO2
CO2Me
O
OMe
2
11`
3`
Hαααα
Hαααα
Hββββ
Hββββ
N O 2C O 2 M e21 1 ` H αH β H β

Chapter 8: Experimental for Chapter 2 153
Methyl (1`S)-2-oxo-1,2-dihydrospiro[cyclopent-3`-ene-1`,3-indole]-3`-carboxylate
((S)-80)
To a solution of (S)-78 (21.7 mg, 0.07 mmol) in MeOH (0.5
mL) and H2O (0.17 mL) was added activated Zn dust (112 mg,
1.7 mmol) and 8.9 M HCl (0.1 mL). The reaction was heated at
reflux for 2 h. The mixture was then filtered through a bed of
celite and the precipitate was washed with H2O and MeOH. The filtrate was then
concentrated in vacuo. The crude product was purified by column chromatography
using 30% EtOAc:PS as eluent and further purified by PTLC (30% EtOAc:PS) to yield
(S)-80 as a yellow oil (11.9 mg, 49 µmol, 69%, Rf = 0.23 in 30% EtOAc:PS, [α]D24 = -
40.8 (c 1.2, CHCl3). MS (EI) m/z 243 (11%) [M.+]; HRMS (ESI+ve) Calcd for
C14H14NO3 [MH+] 244.0974. Found: 244.0966. 1H NMR δ 8.73 (bs, 1H, NH); 7.208 (dd,
J 7.5, 1.2 Hz, 1H, ArCH-4); 7.206 (dt, J 7.8, 1.2 Hz, 1H, ArCH-6); 7.01 (dt, J 7.8, 0.9
Hz, 1H, ArCH-5); 6.92 (d, J 7.8 Hz, 1H, ArCH-7); 6.88-6.84 (m, 1H, CH-4`); 3.79 (s,
3H, OMe); 3.26 (ddd, J 16.5, 5.1, 2.7 Hz, 1H, CH-2`β); 3.20 (ddd, J 18.4, 5.1, 2.7 Hz,
1H, CH-5`β); 2.90 (dm, J 16.5 Hz, 1H, CH-2`α); 2.80 (dm, J 18.9 Hz, 1H, CH-5`α). 13C
NMR δ 182.9 (C-2); 164.6 (CO2CH3); 140.9 (CH=); 139.7 (ArC-7a); 136.5 (ArC-3a);
134.5 (C-3`); 128.1 (ArCH-6); 123.0 (ArCH-5); 122.2 (ArCH-4); 109.8 (ArCH-7); 52.5
(C-3); 51.7 (CH3); 45.0 (CH2-5`); 43.4 (CH2-2`). NMR data collected for (S)-80 agreed
well with the spectroscopic data (apart from the ester signals) to racemic 72.
Methyl (1`R)-2-oxo-1,2-dihydrospiro[cyclopent-3`-ene-1`,3-indole]-3`-carboxylate
((R)-81)
The title compound was prepared using a similar method to
that described above for the synthesis of (S)-80 using (R)-79
(14.6 mg, 48 µmol). (R)-81 was obtained as a peach oil (6.5
mg, 27 µmol, 56%, Rf = 0.52 in 30% EtOAc:PS, [α]D23 =
+57.4 (c 1.0, CHCl3),. MS (EI) m/z 243 (50%) [M.+]; HRMS (ESI+ve) Calcd for
C14H14NO3 [MH+] 244.0974. Found: 244.0963. NMR data collected for (R)-81 was
identical to that of its enantiomer.
O
OMe
NH
O
Hββββ
Hββββ
Hαααα
Hαααα
33a
7a
5
7
1`
3`
O
O M e
NH
O
H ββ ββ
H ββ ββ
H αα αα
H αα αα
33 a
7 a
5
7
1 `
3 `

Chapter 8: Experimental for Chapter 2 154
Ethyl (5R*)-1-oxo-2-azaspiro[4.4]non-7-ene-7-carboxylate (82)
To a solution of 62 (852.4 mg, 2.76 mmol) in anhydrous DCM (2.5
mL) was added TFA (2.5 mL). The solution was left to stir for 2.5 h
under an atmosphere of N2. The solvent was removed in vacuo, and
the oily residue was then treated with sat. NaHCO3 solution (2 × 10
mL) and extracted with DCM (2 × 20 mL). The organic combined
extracts were dried, and evaporated in vacuo to yield compound 82 as brown needle-like
crystals (524.2 mg, 2.5 mmol, 91%, Rf = 0.26 in 70% EtOAc:PS, m.p. 72-76°C). MS
(CI+ve) m/z 210 (100%) [MH+]; HRMS (CI+ve) Calcd for C11H16NO3 [MH+] 210.1130.
Found: 210.1132. 1H NMR δ 7.50 (bs, 1H, NH); 6.69 (t, J 2.7 Hz, 1H, CH=); 4.19 (q, J
7.2 Hz, 2H, CH2CH3); 3.35 (t, J 7.1 Hz, 2H, NCH2); 3.02 (od, J 16.5, 2H, CH-6β and
CH-9β); 2.59 (d, J 15.9 Hz, 1H, CH-6α); 2.46 (d, J 18.9 Hz, 1H, CH-9α); 2.16-2.14 (m,
2H, CH2-4); 1.32 (t, J 7.0 Hz, 3H, CH2CH3). 13C NMR δ 182.3 (C-1); 164.6 (CO2Et);
140.9 (CH=); 134.5 (C-7); 60.4 (CH2CH3); 49.4 (C-5); 43.5 (CH2-9); 42.0 (CH2-6); 39.5
(NCH2); 37.8 (CH2-4); 14.4 (CH2CH3). The structure of 82 was confirmed by X-ray
crystallography (see Appendix 1: X-ray 2: Compound 82).
Ethyl (5R*)-2-benzyl-1-oxo-2-azaspiro[4.4]non-7-ene-7-carboxylate (83)
To a stirred solution of 82 (255.7 mg, 1.22 mmol) in anhydrous THF
(15 mL), under an atmosphere of N2, was added in quick succession,
NaH (76 mg, 1.6 mmol, 50% dispersion in paraffin wax),
tetrabutylammonium iodide (45 mg, 0.12 mmol) and benzyl bromide
(0.22 mL, 1.85 mmol). The reaction mixture was left stirring for 1 h
under a N2 atmosphere. The reaction mixture was then quenched with H2O (50 mL) and
extracted with DCM (3 × 40 mL). The combined organic extracts were then dried,
filtered and evaporated in vacuo. The crude product was purified by column
chromatography using 40-60% EtOAc:PS as the eluent to give 83 as a brown oil (271.5
mg, 0.91 mmol, 74%, Rf = 0.56 in 50% EtOAc:PS). MS (CI+ve) m/z 300 (8%) [MH+];
HRMS (EI) Calcd for C18H21NO3 [M.+] 299.1521. Found: 299.1508. 1H NMR δ 7.34-
7.21 (m, 5H, ArCH); 6.69 (s, 1H, CH=); 4.46 (ABq, J 14.5 Hz, 2H, NCH2Ph); 4.19 (dq,
J 6.9, 2.4 Hz, 2H, CH2CH3); 3.21-3.16 (m, 2H, CH2-3); 3.05 (od, J 16.2 Hz, 2H, CH-6β
and CH-9β); 2.56 (d, J 15.3 Hz, 1H, CH-6α); 2.43 (d, J 18.9 Hz, 1H, CH-9α); 2.04-1.91
(m, 2H, CH2-4); 1.28 (dt, J 7.2, 2.4 Hz, 3H, CH2CH3). 13C NMR δ 178.0 (C-1); 164.6
NH
EtO2C
HββββHββββ
Hαααα Hαααα96
5
3 1O
N
EtO2C
HββββHββββ
Hαααα Hαααα96
5
3 1O
Bn

Chapter 8: Experimental for Chapter 2 155
(CO2Et); 141.0 (CH=); 136.6 (C-7); 134.4 (ArC-i); 128.9 (ArCH-m); 128.2 (ArCH-o);
127.8 (ArCH-p); 60.5 (CH2CH3); 50.3 (C-5); 47.1 (NCH2Ph); 43.81 (CH2-9); 43.78
(CH2-3); 42.2 (CH2-6); 35.5 (CH2-4); 14.5 (CH2CH3).
(5R*)-2-Benzyl-1-oxo-2-azaspiro[4.4]non-7-ene-7-carboxylic acid (84)
To a solution of 83 (271.5 mg, 0.91 mmol) in MeOH (2 mL),
contained within a sealed tube, was added a solution of K2CO3 (251
mg, 1.82 mmol) in water (2.5 mL). The tube was sealed and the
mixture was left stirring at 40 °C for 4 d, another equivalent of
K2CO3 was added and temperature was raised to 60 °C for 1 d. The
solvent was removed in vacuo and H2O (15 mL) was added to the oily residue. The
solution was then washed with Et2O (2 × 25 mL). The aqueous fraction was acidified
(pH ~ 1) with 10% HCl and extracted with EtOAc (3 × 25 mL). The organic combined
extracts were combined, dried and evaporated in vacuo to yield a white solid (229.7 mg,
0.85 mmol, 93 %, Rf = 0.06 in 50% EtOAc:PS). MS (CI+ve) m/z 272 (100%) [MH+];
HRMS (CI+ve) Calcd for C16H17NO3 [M+] 271.1208. Found: 271.1123. 1H NMR δ 9.16
(bs, 1H, OH); 7.35-7.24 (m, 3H, ArCH); 7.22 (d, J 6.3 Hz, 2H, ArCH-o); 6.81 (s, 1H,
CH=); 4.49 (ABq, J 14.7 Hz, 2H, NCH2Ph); 3.23-3.17 (m, 2H, CH2-3), 3.11 (ddd, J
18.3, 4.9, 2.2 Hz, 1H, CH-9β), 3.05 (ddd, J 16.5, 4.9, 2.2, 1H, CH-6β); 2.56 (d, J 17.1
Hz, 1H, CH-6α); 2.45 (d, J 18.6 Hz, 1H, CH-9α); 2.08-1.92 (m, 2H, CH2-4). 13C NMR δ
178.1 (C-1); 168.8 (CO2H); 143.5 (CH=); 136.4 (C-7); 133.9 (ArC-i); 128.9 (ArCH-m);
128.2 (ArCH-o); 127.8 (ArCH-p); 50.7 (C-5); 47.4 (NCH2Ph); 44.1 (CH2-9); 44.0
(CH2-3); 42.0 (CH2-6); 35.6 (CH2-4).
N
HO2C
HββββHββββ
Hαααα Hαααα96
5
3 1O
Bn

Chapter 8: Experimental for Chapter 2 156
(5R*)-2-Benzyl-N-phenyl-1-oxo-2-azaspiro[4.4]non-7-ene-7-carboxamide (85a)
To a solution of 84 (52.2 mg, 0.21 mmol) and HOBT (26 mg, 0.2
mmol) in anhydrous MeCN (2 mL) at 0 °C (ice-bath) was added
aniline (0.02 mL, 0.25 mmol). The solution was stirred for 10 min at
0 °C before the addition of EDCI (38.2 mg, 0.2 mmol) and left to stir
at RT for 18 h and then at 60 °C for 2 h. The solvent was then
removed, and the residue diluted with DCM and washed
successively with H2O and brine and then dried and evaporated in
vacuo. Purification of the crude product was achieved through
column chromatography using 70% EtOAc:PS as the eluent to yield
85a as white crystals (36.2 mg, 0.11 mmol, 54%, Rf = 0.32 in 60%
EtOAc:PS, m.p. 148-150°C). MS (CI+ve) m/z 347 (80%) [MH+], 256 (8%) [MH+-Bn],
188 (28%), 113 (26%), 97 (58%), 85 (100%); HRMS (CI+ve) Calcd for C22H22N2O2
[M+] 346.1681. Found: 346.1632. 1H NMR (500 MHz) δ 7.55 (d, J 8.0 Hz, 2H, ArCH-
o`); 7.34-7.29 (m, 5H, ArCH); 7.23 (d, J 7.5 Hz, 2H, ArCH-m`); 7.11 (t, J 7.3 Hz, 1H,
ArCH-p`); 6.50 (s, 1H, CH=); 4.49 (ABq, J 14.5 Hz, 2H, NCH2); 3.21 (q, J 7.0 Hz, 2H,
CH2-3); 3.15 (d, J 16.0 Hz, 1H, CH-6β); 3.08 (d, J 18.0 Hz, 1H, CH-9β); 2.68 (d, J 15.0
Hz, 1H, CH-6α); 2.49 (d, 18.0 Hz, 1H, CH-9α); 2.11-2.00 (m, 2H, CH2-4). 13C NMR
(125 MHz) δ 177.6 (C-1); 162.7 (CONHPh); 137.8 (ArC-i`); 137.7 (C-7); 136.2 (ArC-
i); 135.0 (CH=); 128.8 (ArCH); 128.6 (ArCH-m); 127.9(ArCH); 127.5 (ArCH); 124.2
(ArCH-p); 119.9 (ArCH-o); 50.3 (C-5); 47.1 (NCH2); 43.7 (CH2-3 and CH2-9); 42.4
(CH2-6); 35.3 (CH2-4). The structure of 85a was confirmed by X-ray crystallography
(see Appendix 1: X-ray 5: Compound 85a).
io
m
p
i`o`
m`
p`
N
Hββββ
Hββββ
Hαααα
Hαααα96
53
1 O
NHO

Chapter 8: Experimental for Chapter 2 157
(5R*)-2-Benzyl-N-[4-(dimethylamino)phenyl]-1-oxo-2-azaspiro[4.4]non-7-ene-7-
carboxamide (85b)
A solution of N,N-dimethylaminoaniline (26.8 mg, 0.2 mmol) in
anhydrous MeCN (2 mL) at 0 °C (ice-bath) was added via cannula to
a mixture of 84 (48.5 mg, 0.18 mmol) and HOBT (26.6 mg, 0.2
mmol). The solution was stirred at 0 °C for 10 min before the
addition of EDCI (35.4 mg, 0.18 mmol) and the mixture was allowed
to stir under N2 at RT for 18 h. The solvent was then removed, and
the residue diluted with DCM and washed successively with H2O
and brine and then dried and evaporated in vacuo. The crude
compound was purified by column chromatography using 70%
EtOAc:PS as the eluent to yield 85b as brown crystals (44.4 mg,
0.11 mmol, 64%, Rf = 0.30 in 70% EtOAc:PS, m.p. 168-170 °C). MS (CI+ve) m/z 390
(100%) [MH+]; HRMS (CI+ve) Calcd for C24H28N3O2 [MH+] 390.2181. Found:
390.2170. 1H NMR (500 MHz) δ 7.39 (d, J 8.5 Hz, 2H, ArCH-o`); 7.34 (t, J 7.3 Hz, 2H,
ArCH-m); 7.29 (t, J 7.0 Hz, 2H, ArCH-p); 7.23 (d, J 7.0 Hz, 2H, ArCH-o); 6.70 (d, J
9.0 Hz, 2H, ArCH-m`); 6.47 (bs, 1H, CH=); 4.49 (ABq, J 15.0 Hz, 2H, NCH2Ph); 3.20
(q, J 6.3 Hz, 2H, CH2-3); 3.14 (dd, J 15.5, 2.5 Hz, 1H, CH-6β); 3.08 (dd, J 18.0, 2.5 Hz,
1H, CH-9β); 2.92 (bs, 6H, N(CH3)2); 2.66 (d, J 15.5 Hz, 1H, CH-6α); 2.47 (d, J 17.5 Hz,
1H, CH-9α); 2.10-1.99 (m, 2H, CH2-4). 13C NMR δ 178.1 (C-1); 148.3 (ArCH-p`);
138.2 (C-7); 136.6 (ArC-i); 134.8 (CH=); 129.0 (ArCH-m); 128.3 (ArCH-o); 127.9
(ArCH-p); 127.7 (ArC-i`); 122.0 (ArCH-o`); 113.2 (ArCH-m`); 50.7 (C-5); 47.3
(NCH2Ph); 43.9 (CH2-3); 43.8 (CH2-9); 42.6 (CH2-6); 41.1 (N(CH3)2); 35.5 (CH2-4).
The structure of 85b was confirmed by X-ray crystallography (see Appendix 1: X-ray
7: Compound 85b)
io
m
p
i`o`
m`
p`
N
Hββββ
Hββββ
Hαααα
Hαααα96
53
1 O
NHO
N

Chapter 8: Experimental for Chapter 2 158
(5R*)-1-Oxo-2-azaspiro[4.4]non-7-ene-7-carboxylic acid (86)
The title compound was prepared from 82 (90.5 mg, 0.43 mmol) using
a similar method to that described above for the synthesis of 84.
Compound 86 was obtained as brown crystals (41.6 mg, 0.23 mmol,
53%, Rf = 0.03 in EtOAc, m.p. 168 °C). MS (CI+ve) m/z 182 (100%)
[MH+], 164 (10%) [MH+-H2O], 149 (8%) [MH+-CO2H]; HRMS (CI+ve) Calcd for
C9H12NO3 [MH+] 182.0817. Found: 182.0818. 1H NMR δ 6.79 (bs, 1H, NH); 6.68 (s,
1H, CH=); 3.76-3.68 (m, 2H, NCH2); 3.03 (od, J 14.4 Hz, 2H, CH-6β and CH-9β); 2.59
(d, J 15.0 Hz, 1H, CH-6α); 2.47 (d, J 18.0 Hz, 1H, CH-9α); 2.21-2.04 (m, 2H, CH2-4). 13C NMR δ 181.7 (C-1); 165.0 (CO2H); 141.2 (CH=); 134.1 (C-7); 49.4 (C-5); 43.7
(CH2-9); 42.1 (CH2-6); 39.5 (NCH2); 37.9 (CH2-4).
(5R*)-1-Oxo-N-phenyl -2-azaspiro[4.4]non-7-ene-7-carboxamide (87)
The title compound was prepared by two methods. Method 1:
The title compound was prepared from 86 (38 mg, 0.21 mmol)
and aniline (0.02 mL, 0.22 mmol) using a similar method to that
described above for the synthesis of 85a. Compound 87 was
obtained as a semi-crystalline yellow oil which crystallised upon
standing, after purification by column chromatography using 5%
MeOH:EtOAc as the eluent (48.9 mg, 1.91 × 10-4 mol, 91%, Rf =
0.23 in 5% MeOH:EtOAc, m.p. 148-150 °C). Method 2: To a
solution of spiroamide 95 (75.7 mg, 0.16 mmol) was added anisole (0.18 mL, 1.65
mmol) and TFA (1.5 mL). The reaction was left to stir for 15 h. The volatiles were then
removed and residue dissolved in CHCl3 (10 mL) and poured slowly onto sat. Na2CO3
solution. The mixture was repeatedly extracted with CHCl3, yielded a yellow oil which
crystallised upon standing (23.1 mg, 90 µmol, 57%). MS (CI+ve) m/z 257 (32%)
[MH+], 153 (100%), 101 (91%); HRMS (CI+ve) Calcd for C15H16N2O2 [M+] 256.1212.
Found: 256.1227. 1H NMR δ 7.55 (d, J 9.0 Hz, 2H, ArCH-o); 7.35-7.26 (m, 2H, ArCH-
m); 7.10 (t, J 7.3 Hz, 1H, ArCH-p); 6.50 (s, 1H, CH=); 6.15 (bs, 1H, CONHPh); 3.37 (t,
J 6.6 Hz, 2H, NCH2); 3.14 (dd, J 15.7, 2.5 Hz, 1H, CH-6β); 3.05 (dd, J 18.1, 2.5 Hz, 1H,
CH-9β); 2.71 (d, J 16.2 Hz, 1H, CH-6α); 2.52 (d, J 18.3 Hz, 1H, CH-9α); 2.26-2.08 (m,
2H, CH2-4). 13C NMR δ 183.0 (C-1); 166.0 (CO2NHPh); 137.7 (C-7); 134.9 (CH=);
130.3 (ArC-i); 128.8 (ArCH-m); 124.2 (ArCH-p); 119.8 (ArCH-o); 49.1 (C-5); 43.3
NH
HO2C
HββββHββββ
Hαααα Hαααα96
5
3 1O
io
p
m
NH
HββββHββββ
Hαααα Hαααα96
5
3 1O
NH
O

Chapter 8: Experimental for Chapter 2 159
(CH2-9); 42.1 (CH2-6); 39.2 (NCH2); 37.4 (CH2-4). The structure of 87 was confirmed
by X-ray crystallography (see Appendix 1: X-ray 8: Compound 87)
Ethyl (5S*)-1-oxo-2-azaspiro[4.4]non-6-ene-6-carboxylate (88)
The title compound was prepared using a similar method to that
described above for 82, using 63 (337.8 mg, 1.09 mmol) to yield
compound 88 as white needle-like crystals (197.0 mg, 0.94 mmol,
86%, Rf = 0.13 in 70% EtOAc:PS, m.p. 102-104 °C). MS (CI+ve)
m/z 210 (53%) [MH+]; 149 (39%) [MH+-O2Et]; 89 (100%); HRMS (CI+ve) Calcd for
C11H16NO3 [MH+] 210.1130. Found: 210.1133. 1H NMR δ 6.99 (t, J 2.7 Hz, 1H, CH=);
4.18 (q, J 7.2 Hz, 2H, CH2CH3); 3.50 (ddt, J 9.3, 3.6, 0.9 Hz, 1H, NCHACHB); 3.35 (dt,
J 9.3, 7.8 Hz, 1H, NCHBCHA); 2.68 (ddd, J 9.3, 4.5, 2.7 Hz, 1H, CHACHB-8); 2.62
(ddd, J 9.3, 4.2, 2.7 Hz, 1H, CHBCHA-8); 2.59-2.46 (m, 1H, CHACHB-4); 2.44-2.37 (m,
1H, CHACHB-9); 2.09-2.01 (m, 1H, CHBCHA-4); 1.99 (ddd, 12.6, 8.1, 4.5 Hz, 1H,
CHBCHA-9); 1.28 (ddt, J 7.2, 7.2, 3.0 Hz, 3H, CH2CH3). 13C NMR (125 MHz) δ 180.9
(C-1); 163.7 (CO2Et); 147.0 (CH=); 137.8 (C-6); 60.1 (CH2CH3); 55.8 (C-5); 39.7
(NCH2); 36.4 (CH2-9); 32.9 (CH2-4); 31.2 (CH2-8); 14.0 (CH2CH3). The structure of 88
was confirmed by X-ray crystallography (see Appendix 1: X-ray 3: Compound 88).
Ethyl (5S*)-2-benzyl-1-oxo-2-azaspiro[4.4]non-6-ene-6-carboxylate (89)
The title compound was prepared using a similar method to that
described above for the preparation of 83 using 88 (129 mg, 0.62
mmol). The crude product was purified by column chromatography
using 50% EtOAc:PS as the eluent to give 89 as a yellow oil (87.3
mg, 0.3 mmol, 47%, Rf = 0.42 in 50% EtOAc:PS). MS (CI+ve) m/z 300 (8%) [MH+];
HRMS (EI) Calcd for C18H21NO3 [M.+] 299.1521. Found: 299.1508. 1H NMR δ 7.36-
7.23 (m, 5H, ArCH); 6.99 (t, J 2.5 Hz, 1H, CH=); 4.50 (ABq, J 17.4 Hz, 2H, NCH2Ph);
4.23-4.08 (m, 2H, CH2CH3); 3.35 (dt, J 9.4, 3.9 Hz, 1H, CHACHB-3); 3.21 (ddd, J 9.3,
8.4, 6.9 Hz, 1H, CHBCHA-3); 2.73-2.61 (m, 1H, CHACHB-8); 2.55 (ddd, J 8.7, 6.3, 2.4
Hz, 1H, CHBCHA-8); 2.46-2.33 (om, 2H, CHACHB-9 and CHACHB-4); 2.00-1.89 (om,
2H, CHBCHA-9 and CHBCHA-4); 1.29-1.22 (m, 3H, CH2CH3). 13C NMR δ 176.9 (C-1);
163.8 (CO2Et); 147.0 (CH=); 138.3 (C-7); 136.6 (ArC-i); 128.5 (ArCH-m); 128.0
N
EtO2C5
3 1
96
O
Bn
NH
EtO2C5
3 1
96
O

Chapter 8: Experimental for Chapter 2 160
(ArCH-o); 127.3 (ArCH-p); 60.2 (CH2CH3); 56.7 (C-5); 47.0 (NCH2Ph); 44.2 (CH2-3);
36.9 (CH2-9); 31.2 (CH2-8); 30.5 (CH2-4); 14.1 (CH2CH3).
(5S*)-2-Benzyl-1-oxo-2-azaspiro[4.4]non-6-ene-6-carboxylic acid (90)
The title compound was prepared from 89 (87.3 mg, 0.3 mmol) as
described for the synthesis of 84 to yield 90 as white needle-like
crystals (63.3 mg, 0.23 mmol, 80%) which were further purified by
recrystallisation from 1% MeOH:EtOAc (22.2 mg, 82 µmol, 28%,
m.p. 200 °C). MS (CI+ve) m/z 272 (100%) [MH+]; HRMS (CI+ve) Calcd for
C16H17NO3 [M+] 271.1208. Found: 271.1199. 1H NMR (CD3OD, 500 MHz) δ 7.34-7.25
(m, 5H, ArCH); 6.99 (t, J 2.5 Hz, 1H, CH=); 4.48 (ABq, J 14.7 Hz, 2H, NCH2Ph); 3.39-
3.25 (m, 2H, CH2-3); 2.59 (dt, J 7.3, 2.5 Hz, 1H, CH2-8); 2.37 (ddd, J 13.0, 9.0, 8.0 Hz,
1H, CHACHB-4); 2.28 (ddd, J 12.5, 8.75, 8.5 Hz, 1H, CHACHB-9); 2.08-2.00 (m, 2H,
CHBCHA-9 and CHBCHA-4). 13C NMR (CD3OD, 125 MHz) δ 180.0 (C-1); 167.7
(CO2H); 148.6 (CH=); 137.6 (C-7); 139.9 (ArC-i); 129.7 (ArCH-m); 128.9 (ArCH-o);
128.6 (ArCH-p); 58.8 (C-5); 47.8 (NCH2Ph); 45.6 (CH2-3); 37.3 (CH2-9); 32.1 (CH2-8);
31.4 (CH2-4). The structure of 90 was confirmed by X-ray crystallography (see
Appendix 1: X-ray 4: Compound 90)
(5S*)-2-Benzyl-N-phenyl-1-oxo-2-azaspiro[4.4]non-6-ene-6-carboxamide (91)
The title compound was prepared from 90 (22.2 mg,
82 µmol) as described above for the synthesis of
85a however the reaction mixture was left stirring at
60 °C for 4 d under N2. The crude product was
purified by column chromatography using 60-100%
EtOAc:PS to yield 91 as white crystals (27.4 mg, 79 µmol, 97%, Rf = 0.18 in 60%
EtOAC:PS, m.p. 164-165 °C). MS (CI+ve) m/z 347 (57%) [MH+], 346 (26%) [M+], 225
(17%) [M+-CONHPh], 149 (45%) [M+-(CONHPh, Ph)]; HRMS (CI+ve) Calcd for
C22H22N2O2 [M+] 346.1681. Found: 346.1671. 1H NMR δ 8.38 (bs, 1H, NH); 7.52 (d, J
7.5 Hz, 2H, ArCH-o`); 7.29-7.24 (m, 7H, ArCH); 7.06 (t, J 7.35 Hz, 1H, ArCH-p); 6.68
(bs, 1H, CH=); 4.53 (ABq, J 14.7 Hz, 2H, NCH2Ph); 3.39 (dt, J 9.3, 3 Hz, 1H,
CHACHB-3); 3.24 (dd, J 17.4, 7.8 Hz, 1H, CHBCHA-3); 2.62-2.54 (m, 2H, CH2-8); 2.49
(dt, J 12, 8.4 Hz, 1H, CHACHB-4); 2.34 (ddd, 12.6, 9, 8.1 Hz, 1H, CHACHB-9); 2.01-
N
HO2C5
3 1
96
O
Bn
i
i '
o
p
m
o 'm '
p '
N
5
3 1
96
O
HN
O

Chapter 8: Experimental for Chapter 2 161
1.93 (om, 2H, CHBCHA-9 and CHBCHA-4). 13C NMR δ 177.9 (C-1); 163.6 (CONHPh);
142.4 (C-7); 139.9 (CH=); 138.3 (ArC-i`); 136.5 (ArC-i); 129.0 (ArCH); 128.9 (ArCH);
128.1 (ArCH); 127.7 (ArCH-p); 124.2 (ArCH-p`); 120.2 (ArCH-o`); 58.8 (C-5); 47.4
(NCH2Ph); 44.7 (CH2-3); 36.4 (CH2-9); 31.3 (CH2-8); 30.7 (CH2-4). The structure of 91
was confirmed by X-ray Crystallography (see Appendix 1: X-ray 6: Compound 91).
(1`S*)-2-Oxo-1,2-dihydrospiro[cyclopent-3`-ene-1`,3-indole]-3`-carboxylic acid
((rac)-92)
The title compound was prepared from 72 using a similar
method to that described for the synthesis of 84. However the
mixture was left stirring at 60 °C for 5 h and no further
additions of K2CO3 were needed. After extraction, the
compound required no further purification yielding a creamy brown solid (29.9 mg, 1.3
× 10-4 mol, 94%, Rf = 0.38 in EtOAc, m.p. 108-110 °C). MS (EI) m/z 229 (12%) [M.+],
211 (6%) [M+-H2O], 183 (14%) [M+-CO2H]; HRMS (EI) Calcd for C13H11NO3 [M.+]
229.0739. Found: 229.0744. 1H NMR δ 9.13 (bs, 1H, NH); 7.23 (d, J 7.8 Hz, 1H,
ArCH-4); 7.22 (t, J 7.6 Hz, 1H, ArCH-6); 7.03 (t, J 7.6 Hz, 1H, ArCH-5); 6.98 (s, 1H,
CH=); 6.94 (d, J 7.5 Hz, 1H, ArCH-7); 3.28 (dd, J 16.5, 2.7 Hz, 1H, CH-2`β); 3.22 (dd,
J 16.5, 2.1 Hz, 1H, CH-5`β); 2.91 (d, J 16.5 Hz, 1H, CH-2`α); 2.83 (d, J 18.9 Hz, CH-
5`α). 13C NMR δ 183.6 (C-2); 168.5 (CO2H); 143.1 (CH=); 139.5 (ArC-7a); 136.4
(ArC-3a); 134.4 (C-3`); 128.2 (ArCH-6); 123.3 (ArCH-5); 122.1 (ArCH-4); 110.1
(ArCH-7); 52.7 (C-3); 45.0 (CH2-5`); 43.0 (CH2-2`).
(1`S)-2-Oxo-1,2-dihydrospiro[cyclopent-3`-ene-1`,3-indole]-3`-carboxylic acid ((S)-
92)
The title compound was prepared from (S)-80 (31.9 mg, 0.13
mmol) using a similar method to that described for the
synthesis of 84. However no further additions of K2CO3 were
needed. After extraction, the compound required no further
purification yielding (S)-92 (16.5 mg, 72 µmol, 55%) as a clear oil. [α]D24 = -42.4 (c 1.7,
CHCl3), MS (EI) m/z 229 (71 %) [M.+]; HRMS (ESI+ve) Calcd for C13H12NO3 [MH+]
230.0817. Found: 230.0826. NMR data collected for (S)-92 was identical to that of its
racemate (rac)-92.
O
OH
NH
O
Hββββ
Hββββ
Hαααα
Hαααα
33a
7a
5
7
1`
3`
NH O H βα33 a7 a5 7 1 `

Chapter 8: Experimental for Chapter 2 162
(1`R)-2-Oxo-1,2-dihydrospiro[cyclopent-3`-ene-1`,3-indole]-3`-carboxylic acid ((R)-
92)
The title compound was prepared using a similar method to that
described above for the synthesis of (S)-92 using (R)-81 (10.2
mg, 42 µmol). (R)-92 was obtained as a white solid (8.1 mg, 35
µmol, 84%, [α]D24 = +38.9 (c 0.89, CHCl3),. MS (EI) m/z 243
(50%) [M.+]; HRMS (ESI+ve) Calcd for C14H14NO3 [MH+] 244.0974. Found: 244.0963.
NMR data collected for (R)-92 was identical to that of (S)-92 and its racemate (rac)-92.
(1`S*)-2-Oxo-N-phenyl-1,2-dihydrospiro[cyclopent-3`-ene-1`,3-indole]-3`-
carboxamide ((rac)-93a)
The title compound was prepared using two methods.
Method 1: The title compound was prepared from
(rac)-92 (29.3 mg, 1.28 × 10-4 mol) and aniline (0.02
mL, 2.2 × 10-4 mol) using a similar method to that
described for the above synthesis of 85a, however the reaction mixture was allowed to
stir under N2 for 15 h. The crude compound was extracted with DCM, and washed with
H2O and brine. The combined extracts were dried and solvent was evaporated in vacuo
to yield white crystals (35.8 mg, 1.2 × 10-4 mol, 92%, Rf = 0.13 in 40% EtOAc:PS, m.p.
184 °C). Method 2: To a solution of amide 97 (12.4 mg, 2.9 × 10-5 mol) in anhydrous
DCM (5 mL) was added sequentially anisole (0.03 mL, 0.3 mmol) and TFA (0.28 mL,
3.6 mmol). The reaction was left stirring for 15 h. The volatiles were then removed and
the residue dissolved in CHCl3 and poured slowly onto sat. Na2CO3 solution. The crude
mixture was repeatedly extracted with CHCl3. The solvent was evaporated in vacuo to
yield white crystals (4.6 mg, 1.5 × 10-5 mol, 52%). MS (EI) m/z 304 (8%) [M.+], 184
[M+-CONHPh], 159 [M+-CONHPhC=CH]; HRMS (EI) Calcd for C19H16N2O2 [M.+]
304.1212. Found: 304.1207. 1H NMR δ 8.87 (bs, 1H, NH); 7.78 (bs, 1H, CONHPh);
7.58 (d, J 8.1 Hz, 2H, ArCH-o); 7.32 (t, J 7.8 Hz, 2H, ArCH-m), 7.24 (d, J 7.8 Hz, 1H,
ArCH-4); 7.20 (t, J 7.2 Hz, 1H, ArCH-6); 7.10 (t, J 7.5 Hz, 1H, ArCH-p); 7.01 (t, J 7.6
Hz, 1H, ArCH-5); 6.91 (d, J 7.8 Hz, 1H, ArCH-7); 6.68 (s, 1H, CH=); 3.32 (dd, J 16.0,
2.1 Hz, 1H, CH-2`β); 3.18 (d, J 18.3, 2.1 Hz, 1H, CH-5`β); 3.00 (d, J 16.2 Hz, 1H, CH-
2`α); 2.82 (d, J 18.0 Hz, 1H, CH-5`α). 13C NMR δ 182.9 (C-2); 162.5 (CONH); 139.7
(ArC-7a); 138.2 (C-3`); 137.6 (ArC-i); 136.1 (ArC-3a); 135.6 (CH=); 129.0 (ArCH-m);
O
OH
NH
O
Hββββ
Hββββ
Hαααα
Hαααα
33a
7a
5
7
1`
3`
O
NH
NH
O
Hββββ
Hββββ
Hαααα
Hαααα
33a
7a
5
7
1`
3`
o m
p

Chapter 8: Experimental for Chapter 2 163
128.2 (ArCH-6); 124.4 (ArCH-p); 123.2 (ArCH-5); 122.3 (ArCH-4); 120.1 (ArCH-o);
109.9 (ArCH-7); 52.5 (ArCH-3); 44.8 (CH2-5`); 43.5 (CH2-2`). The structure of (rac)-
93a was confirmed by X-ray crystallography (see Appendix 1: X-ray 12: Compound
(rac)-93a)
(1`S*)-2-Oxo-N-[4-(dimethylamino)phenyl]-1,2-dihydrospiro[cyclopent-3`-ene-
1`,3-indole]-3`-carboxamide ((rac)-93b)
The title compound was prepared from (rac)-92 (23.8 mg, 0.1
mmol) and N,N-dimethylaminoaniline (24.1 mg, 0.2 mmol) using
a similar method to that described for the above synthesis of 85b.
The crude compound was purified by column chromatography
using 50-70% EtOAc:PS as eluent to yield a black powder (15.9
mg, 45µmol, 44%, Rf = 0.73 in 80% EtOAc:PS). MS (EI) m/z
347 (3%) [M.+], 167 (35%), 149 (100%); HRMS (EI) Calcd for
C21H21N3O2 [M.+] 347.1634. Found: 347.1633. 1H NMR δ 8.35
(bs, 1H, NH-1); 7.40 (d, J 9.3 Hz, 2H, ArCH-o); 7.40 (bs, 1H, NHPhNMe2); 7.26 (d, J
7.2 Hz, 1H, ArCH-4); 7.20 (dt, J 7.5, 1.5 Hz, 1H, ArCH-6); 7.02 (dt, J 7.5, 0.9 Hz, 1H,
ArCH-5); 6.90 (d, J 7.5 Hz, 1H, ArCH-7); 6.69 (d, J 9.0 Hz, 2H, ArCH-m); 6.64 (bs, 1H,
CH=); 3.33 (dd, J 16.0, 2.2 Hz, 1H, CH-2`β); 3.19 (dq, J 18.0, 2.4 Hz, 1H, CH-5`β);
2.98 (d, J 16.0 Hz, 1H, CH-2`α); 2.91 (bs, 6H, N(CH3)2); 2.82 (d, J 17.4 Hz, 1H, CH-
5`α). 13C NMR (125 MHz) δ 182.6 (C-2); 162.1 (=CCO); 148.1 (ArC-p); 139.6 (ArC-
7a); 138.3 (C-3`); 136.4 (ArC-3a); 135.0 (CH=); 128.2 (ArCH-6); 127.3 (ArC-i); 123.1
(ArCH-5); 122.4 (ArCH-4); 121.9 (ArCH-o); 112.9 (ArCH-m); 109.8 (ArCH-7); 52.6
(C-3); 44.9 (CH2-5`); 43.6 (CH2-2`); 40.8 (N(CH3)2).
NH
NH
O
Hββββ
Hββββ
Hαααα
Hαααα
33a
7a
5
7
1`
3`
N
O
o
mp
i

Chapter 8: Experimental for Chapter 2 164
(1`S)-2-Oxo-N-[4-(dimethylamino)phenyl]-1,2-dihydrospiro[cyclopent-3`-ene-1`,3-
indole]-3`-carboxamide ((S)-93b)
The title compound was prepared from (S)-92 (16.5 mg, 72
µmol) and N,N-dimethylaminoaniline hydrochloride (30 mg,
0.14 mmol) using a similar method to that described for the
above synthesis of 85b, however diisopropylamine (0.02 mL)
was added to the reaction mixture and it was allowed to stir
under N2 at RT for 18 h. The crude compound was purified by
column chromatography using 50-80% EtOAc:PS to yield (S)-
as a brown oil (11.9 mg, 3.4 µmol, 48%, Rf = 50% in 80%
EtOAc:PS, [α]D24 = -47.4 (c 1.2, CHCl3). The 1H NMR spectral data of (S)-93b
matched with that of its racemate (rac)-93b.
(1`R)-2-Oxo-N-[4-(dimethylamino)phenyl]-1,2-dihydrospiro[cyclopent-3`-ene-1`,3-
indole]-3`-carboxamide ((R)-93b)
The title compound was prepared using a similar method to
that described above for the synthesis of (S)-93b using (R)-92
(16.3 mg, 71 µmol). (R)-93b was obtained as a dark brown oil
(7.8 mg, 22 µmol, 32%, [α]D25 = +56.7 (c 0.8, CHCl3),. The 1H
NMR spectrum of (R)-93b was identical to that of (S)-93b and
its racemate (rac)-93b.
But-2-ynoic acid phenylamide (94)
The title compound was prepared from aniline (0.15 mL,
1.6 mmol) and 2-butynoic acid (123 mg, 1.46 mmol) using
a similar method to that described for the synthesis of 85a.
However the reaction mixture was left stirring for 2 d. The
crude product was purified by gradient column chromatography using 20-50%
EtOAc:PS as the eluent to yield 94 as peach coloured crystals (170.8 mg, 1.07 mmol,
67%, Rf = 0.65 in 40% EtOAc:PS, m.p. 90 °C). MS (CI+ve) m/z 160 (100%) [MH+];
HRMS (CI+ve) Calcd for C10H9NO [M+] 159.0684. Found: 159.0675. 1H NMR δ 7.59
(bs, 1H, NH); 7.51 (d, J 7.8 Hz, 2H, ArCH-o); 7.32 (t, J 7.8 Hz, 2H, ArCH-m); 7.12 (t, J
Op m o i
NH
NH
O
Hββββ
Hββββ
Hαααα
Hαααα
33a
7a
5
7
1`
3`
N
O
o
mp
i
NH
NH
O
Hββββ
Hββββ
Hαααα
Hαααα
33a
7a
5
7
1`
3`
N
O
o
mp
i

Chapter 8: Experimental for Chapter 2 165
7.5 Hz, 1H, ArCH-p); 1.98 (s, 3H, CH3). 13C NMR δ 151.1 (CO); 137.3 (ArC-i); 129.0
(ArCH-m); 124.7 (ArCH-p); 119.8 (ArCH-o); 84.5 (C≡CCH3); 75.3 (C≡CCH3); 3.71
(CH3). The structure of 94 was confirmed by X-ray crystallography (see Appendix 1:
X-ray 9: Compound 94)
tert-Butyl (5R*)-7-{[4-methoxybenzyl(phenyl)amino]carbonyl]}-1-oxo-2-
azaspiro[4.4]non-7-ene-2-carboxylate (95)
The title compound was prepared using a similar method
described above for the synthesis of 62 and 63 using 57 (190
mg, 0.97 mmol) and 53c (296.5 mg, 1.06 mmol) instead of
ethyl 2-butynoate. The crude mixture was purified by gradient
column chromatography using 10-90% EtOAc:PS as eluent to
yield 95 as a brown oil (64.3 mg, 0.13 mmol, 14%, Rf = 0.59 in
50% EtOAc:PS). MS (ESI+ve) m/z 499 (12%) [MH++Na+],
477.3 (12%) [MH+], 377.3 (100%) [MH+-(Boc, CH3)]; HRMS
(ESI+ve) Calcd for C28H33N2O5 [MH+] 477.2389. Found: 477.2412. 1H NMR (500
MHz) δ 7.33-7.22 (m, 3H, ArCH-m and ArCH-p); 7.13 (d, J 7.0 Hz, 2H, ArCH-o`);
6.97 (dd, J 8.5, 2H, ArCH-o); 6.78 (d, J 9.0 Hz, 2H, ArCH-m`); 5.56 (s, 1H, CH=); 4.89
(s, 2H, NCH2); 3.77 (s, 3H, OCH3); 3.58-3.50 (m, 2H, CH2-3); 2.74 (d, J 18.0 Hz, 1H,
CH-9β); 2.73 (d, J 14.7 Hz, 1H, CH-6β); 2.26 (d, J 14.4 Hz, 1H, CH-6α); 2.18 (d, J 18.3
Hz, 1H, CH-9α); 1.73-1.71 (m, 2H, CH2-4); 1.52 (s, 9H, C(CH3)3). 13C NMR δ 177.0
(C-1); 166.2 (CONPhPMB); 158.7 (ArC-p`); 150.2 (NCO2); 142.5 (ArC-i); 136.5 (C-7);
135.1 (CH=); 129.9 (ArCH-o`); 129.3 (ArC-i`); 129.1 (ArCH-m); 127.9 (ArCH-o);
127.5 (ArCH-p); 113.6 (ArCH-m`); 82.8 (C(CH3)3; 55.1 (OCH3); 52.9 (NCH2); 51.5 (C-
5); 43.8 (CH2-9); 43.1 (CH2-6); 43.0 (CH2-3); 33.2 (CH2-4); 27.9 C(CH3)3.
Methyl (1`S*)-3`-{[4-Methoxybenzyl(phenyl)amino]carbonyl}-1`-(2-
nitrophenyl)cyclopent-3`-ene-1`-carboxylate (96)
To a solution of the alkene 59 (26 mg, 1.2 × 10-4 mol)
and amide 53c (39.1 mg, 1.4 × 10-4 mol) in anhydrous
benzene (3 mL) was slowly added Bu3P (0.02 mL, 80
µmol). The reaction was left to stir for 2 d. Upon
evaporation in vacuo of the volatiles the resulting crude
i'
p'
o'
m'
io
p
m
N
HββββHββββ
Hαααα Hαααα96
5
3 1O
N
O
OMe
Boc
N O 2C O 2 M e O M eH ββ αα12 1 ` o 'm 'p 'i '

Chapter 8: Experimental for Chapter 2 166
product was purified by column chromatography using 30-50% EtOAc:PS as eluent to
yield a yellow oil (33.4 mg, 6.9 × 10-5 mol, 55%, Rf = 0.65 in 40% EtOAc:PS). MS
(CI+ve) m/z 487 (16%) [MH+], 455 (18%) [MH+-OMe], 425 (17%) [MH+-(OMe)2];
HRMS (ESI+ve) Calcd for C28H27N2O6 [MH+] 487.1869. Found: 487.1871. 1H NMR
(C6D6, 500 MHz) δ 7.52 (d, J 8.0 Hz, 1H, ArCH-3); 7.25 (d, J 8.5 Hz, 2H, ArCH-o`);
7.10 (d, J 8.5 Hz, 1H, ArCH-6); 6.90 (t, J 7.5 Hz, 1H, ArCH-5); 6.88-6.86 (m, 3H,
ArCH-m and ArCH-p); 6.74-6.72 (m, 4H, ArCH-m` and ArCH-o); 6.67 (t, J 7.7 Hz, 1H,
ArCH-4); 5.44 (s, 1H, CH=); 4.94 (ABq, J 14.5 Hz, 2H, NCH2); 3.54 (dd, J 17.0, 2.0
Hz, 1H, CH-2`β); 3.32 (dd, J 18.5 Hz, 1H, CH-5`β); 3.28 (s, 6H, CO2CH3 and OCH3);
3.18 (d, J 17.5 Hz, 1H, CH-2`α); 2.45 (d, J 18.5 Hz, 1H, CH-5`α). 13C NMR (C6D6, 125
MHz) δ 173.8 (CO2Me); 165.7 (CON); 159.5 (ArC-p`); 148.8 (ArC-2); 143.2 (ArC-i);
138.7 (ArC-1); 137.1 (C-3`); 134.7 (CH=); 132.7 (ArCH-5); 130.6 (ArCH-o`); 130.1
(ArC-i`); 129.2 (ArCH-m); 128.7 (ArCH-6); 128.3 (ArCH-o); 127.5 (ArCH-4); 127.2
(ArCH-p); 125.1 (ArCH-3); 114.1(ArCH-m`); 55.4 (C-1`); 54.6 (CO2CH3); 53.1
(NCH2); 51.9 (OCH3); 47.0 (CH2-2`); 45.9 (CH2-5`).
(1`S*)-N-(4-Methoxybenzyl)-2-oxo-N-phenyl-1,2-dihydrospiro[cyclopent-3-ene-
1`,3-indole]-3-carboxamide (97)
To a stirred solution of 96 (87.8 mg, 0.18 mmol) in acetic
acid (15 mL) was added activated Zn dust345 (40 mg,
6.12 × 10-4 mol). Stirring was continued for 1.5 h. The
solution was then filtered through a bed of celite and the
filtrate washed with sat. Na2CO3 solution and then
extracted with EtOAc. The extracts were combined and dried to yield 97 as a brown oil
(12.4 mg, 29.2 µmol, 16%, Rf = 0.63 in 70% EtOAc:PS), which was pure upon 1H
NMR analysis and did not require further purification. MS (CI+ve) m/z 425 (37%)
[MH+]; HRMS (EI) Calcd for C27H24N2O3 [M.+] 424.1787. Found: 424.1786. 1H NMR
(C6D6, 500 MHz) δ 7.99 (bs, 1H, NH); 7.26 (d, J 8.5 Hz, 2H, ArCH-o`); 6.96 (t, J 7.5
Hz, 2H, ArCH-m); 6.94 (t, J 6.5 Hz, 1H, ArCH-6); 6.90-6.88 (m, 1H, ArCH-p); 6.88 (d,
J 7.0 Hz, 1H, ArCH-4); 6.81 (d, J 7.5 Hz, 2H, ArCH-o); 6.77 (t, J 7.8 Hz, 1H, ArCH-5);
6.74 (d, J 8.5 Hz, 2H, ArCH-m`); 6.43 (d, J 8.0 Hz, 1H, ArCH-7); 5.69 (s, 1H, CH=);
4.95 (ABq, J 14.0 Hz, 2H, NCH2); 3.29 (s, 3H, OCH3); 3.23 (dd, J 16.0, 2.5 Hz, 1H,
CH-2`β); 2.88 (dd, J 18.0, 2.5 Hz, 1H, CH-5`β); 2.71 (d, J 16.5 Hz, 1H, CH-2`α); 2.18 (d,
O
N
OMe
NH
O
3a4
77a
1`
3`
3
o m
pHββββ
Hββββ
Hαααα
Hαααα
o'
m'
p'
i'
i

Chapter 8: Experimental for Chapter 2 167
J 17.5 Hz, 1H, CH-5`α). 13C NMR (C6D6,125 MHz) δ 182.0 (C-2); 165.9 (CON); 159.5
(ArC-p`); 143.4 (ArC-i); 140.4 (ArC-7a); 138.3 (C-3`); 137.1 (ArC-3a); 135.3 (CH=);
130.6 (ArCH-o`); 130.3 (ArC-i`); 129.3 (ArCH-m); 128.5 (ArCH-o); 128.3 (ArCH-6);
127.2 (ArCH-p); 122.6 (ArCH-5); 122.3 (ArCH-4); 114.1 (ArCH-m`); 109.6 (ArCH-7);
54.6 (CH3); 53.1 (NCH2); 52.5 (C-3); 46.0 (CH2-2`); 44.8 (CH2-5).
(1`S*)-3H-Spiro[cyclopentane-1`,3-indole]-2,3`(1H)-dione (102) and (1`S*)-2,3`-
Dioxospiro[cyclopentane-1`,3-indole]-1(2H)-carboxamide (103).
A solution of the racemic acid (rac)-92 (55.6 mg, 0.24 mmol),
diphenylphosphoryl azide (DPPA) (0.11 mL, 0.48 mmol) and NEt3
(0.07 mL, 0.5 mmol) in anhydrous toluene (3 mL) was heated at 85
°C for 5 h and then heated at reflux for 30 min. Then 8.9 M HCl
(0.05 mL) was cautiously added. The mixture was then heated at
reflux for another 1 h, then cooled to RT and stirred for 15 h. The
solvent was removed in vacuo. 1H NMR analysis of the crude reaction mixture revealed
a 1 : 1 mixture of products 102 and 103, respectively. The crude mixture was purified
by column chromatography using 30-50% EtOAc:PS as eluent and then further purified
by column chromatography using 2:1:1 (DCM:PS:EtOAc) to give pure samples of 102
and 103.
102: A semicrystalline yellow oil which crystallized upon standing (26.5 mg, 0.13
mmol, 54%, Rf = 0.28 in 50% EtOAc:PS). (MS (EI) m/z 201 (67%) [M.+], 145 (100%)
[M+-(CH2)2CO], 117 (77%); HRMS (EI) Calcd for C12H12NO2 [MH+] 202.0868. Found:
202.0874. 1H NMR (C6D6, 500 MHz) δ 8.81 (bs, 1H, NH); 6.96 (t, J 7.8 Hz, 1H, ArCH-
6); 6.79 (t, J 7.8 Hz, 1H, ArCH-5); 6.66 (d, J 7.5 Hz, 1H, ArCH-4); 6.55 (d, J 7.5 Hz,
1H, ArCH-7); 2.62-2.53 (m, 1H, CH-4`β); 2.51 (d, J 17.5 Hz, 1H, CH-2`β); 2.15-2.06
(m, 1H, CH-4`α); 2.05 (d, J 18.0 Hz, 1H, CH-2`α); 2.06-2.01 (m, 1H, CH-5`β); 1.60 (dt,
J 13.0, 8.5 Hz, 1H, CH-5`α). 13C NMR (C6D6, 125 MHz) δ 214.0 (C-3`); 182.7 (C-2);
141.0 (ArC-7a); 133.4 (ArC-3a); 128.4 (ArCH-6); 122.7 (ArCH-5); 122.5 (ArCH-4);
110.3 (ArCH-7); 51.1 (C-3); 46.7 (CH2-2`); 36.5 (CH2-4`); 33.4 (CH2-5`) The structure
of 102 was confirmed by X-ray crystallography (see Appendix 1: X-ray 10:
Compound 102).
103: White crystals (21.1 mg, 86 µmol, 36%, Rf = 0.73 in 50% EtOAc:PS, m.p. 139-
143°C). MS (EI) m/z 244 (2%) [M.+], 201 (36%) [M+-CONH2], 167 (39%), 149 (100%);
N
O
Hββββ
Hββββ
Hαααα
Hαααα
HααααHββββ O
R
33a
7a
5
7
1`
3`
102: R = H103: R = CONH2

Chapter 8: Experimental for Chapter 2 168
HRMS (EI) Calcd for C13H12N2O3 [M.+], 244.0848. Found: 244.0823. 1H NMR (C6D6,
500 MHz) δ 8.64 (d, J 8.0 Hz, 1H, ArCH-7); 7.96 (bs, 1H, NHAHB); 7.08 (t, J 8.0 Hz,
1H, ArCH-6); 6.85 (t, J 7.5 Hz, 1H, ArCH-5); 6.55 (d, J 7.5 Hz, 1H, ArCH-4); 4.84 (bs,
1H, NHBHA); 2.37 (ddd, J 18.0, 9.0, 9.0 Hz, 1H, CH-4`β); 2.22 (d, J 18.5 Hz, 1H, CH-
2`β); 2.01 (ddd, J 18.5, 9.0, 6.0 Hz, 1H, CH-4`α); 1.87 (d, J 18.5 Hz, 1H, CH-2`α); 1.74-
1.68 (m, 1H, CH-5`β); 1.43-1.37 (m, 1H, CH-5`α). 13C NMR (C6D6, 125 MHz) δ 212.5
(C-3`); 182.0 (C-2); 152.1 (CONH2); 139.9 (ArC-7a); 131.5 (ArC-3a); 128.8 (ArCH-6);
125.0 (ArCH-5); 121.6 (ArCH-4); 117.0 (ArCH-7); 51.3 (C-3); 47.0 (CH2-2`); 36.1
(CH2-4`); 34.0 (CH2-5`). The structure of 103 was confirmed by X-ray crystallography
(see Appendix 1: X-ray 11: Compound 103).
Ethyl (1`R*)-2-oxo-1,2-dihydrospiro[cyclopentane-1`,3-indole]-3`-carboxylate (104)
and Ethyl (1`S*)-2-oxo-1,2-dihydrospiro[cyclopentane-1`,3-indole]-3`-carboxylate
(105)
To a solution of spiroalkene 72 (34.9 mg, 0.14 mmol) in EtOAc
(2.2 mL) under N2 was added 10 wt. % palladium on activated
carbon (Pd/C) (9.4 mg). The system was then flushed with H2 and
was left to stir under a H2 atmosphere (balloon) for 18 h. The crude
reaction mixture was filtered through a bed of celite and washed
multiple times with EtOAc. The combined organic extracts were
evaporated in vacuo. 1H NMR analysis of the crude reaction
mixture revealed a 1 : 1.8 mixture of 104 and 105, respectively. The crude product was
purified by column chromatography using 20-30% EtOAc:PS as the eluent and then
further by PTLC in 30% EtOAc:PS to give pure samples of 104 and 105.
104: A yellow oil (6.9 mg, 0.26 µmol, 20%, Rf = 0.38 in 30% EtOAc:PS). MS (EI) m/z
259 (64%) [M.+], 260 (12%) [MH+]; HRMS (EI) Calcd for C15H17NO3 [M.+] 259.1208.
Found: 259.1220. 1H NMR (C6D6, 500 MHz) δ 8.14 (bs, 1H, NH); 7.22 (d, 1H, J 7.5 Hz,
ArCH-4); 6.96 (dt, J 7.5, 1.0 Hz, 1H, ArCH-6); 6.86 (dt, J 7.5, 1.0 Hz, 1H, ArCH-5);
6.48 (d, J 8.0 Hz, 1H, ArCH-7); 3.99 (q, J 7.0 Hz, 2H, CH2CH3); 3.38 (quintet, J 8.0 Hz,
1H, CH-3`α); 2.43 (dd, J 13.5, 8.5 Hz, 1H, CH-2`β); 2.38-2.33 (m, 1H, CH-4`β); 2.31
(dd, J 14.0, 8.0 Hz, 1H, CH-2`α); 2.25-2.19 (m, 1H, CH-4`α); 2.10 (dt, J 13.0, 7.5 Hz,
1H, CH-5`β); 1.87 (dt, J 12.5, 7.5 Hz, 1H, CH-5`α); 0.96 (t, J 7.0 Hz, 3H, CH3CH2). 13C
NMR (C6D6, 125 MHz) δ 183.6 (C-2); 175.2 (CO2Et); 140.9 (ArC-7a); 135.6 (ArC-3a);
Hββββ
Hββββ
Hαααα
Hαααα
R1
NH
O
R2
33a
7a
5
7
1`
3`
104: R1= H
R2= CO2Et
105: R1= CO2Et
R2= H

Chapter 8: Experimental for Chapter 2 169
123.4 (ArCH-4); 127.7 (ArCH-6); 122.7 (ArCH-5); 109.5 (ArCH-7); 60.3 (CH2CH3);
54.3 (C-3); 44.5 (CH-3`); 40.6 (CH2-2`); 38.1 (CH2-5`); 30.8 (CH2-4`); 14.2 (CH3CH2).
105: A creamy white oil, 20.1 mg, 0.78 µmol, 57%, Rf = 0.28 in 30% EtOAc:PS). MS
(EI) m/z 259 (72%) [M.+], 260 (12%) [MH+]; HRMS (EI) Calcd for C15H17NO3 [M.+]
259.1208. Found: 259.1219. 1H NMR (500 MHz) δ 8.91 (bs, 1H, NH); 7.20 (t, J 7.7 Hz,
1H, ArCH-6); 7.18 (d, J 7.0 Hz, 1H, ArCH-4); 7.02 (t, J 7.7 Hz, 1H, ArCH-5); 6.93 (d,
J 7.5 Hz, 1H, ArCH-7); 4.18 (q, J 7.2 Hz, 2H, CH2CH3); 3.25 (ABq, J 8.0 Hz, 1H, CH-
3`β); 2.51 (dd, J 13.0, 10.0 Hz, 1H, CH-2`β); 2.40-2.28 (m, 3H, CH2-4` and CH-5`β);
2.14 (dd, J 13.0, 8.0 Hz, 1H, CH-2`α); 1.95-1.84 (m, 1H, CH-5`α); 1.28 (t, J 7.3 Hz, 3H,
CH3CH2). 13C NMR (125 MHz) δ 183.1 (C-2); 174.4 (CO2Et); 140.1 (ArC-7a); 136.1
(ArC-3a); 127.7 (ArCH-6); 122.53 (ArCH-4); 122.49 (ArCH-5); 109.8 (ArCH-7); 60.6
(CH2CH3); 54.3 (C-3); 44.8 (CH-3`); 40.8 (CH2-2`); 37.3 (CH2-5`); 29.6 (CH2-4`); 14.2
(CH3CH2).
(1`R*)-2-Oxo-N-phenyl-1,2-dihydrospiro[cyclopentane-1`,3-indole]-3`-
carboxamide (106) and (1`S*)-2-Oxo-N-phenyl-1,2-dihydrospiro[cyclopentane-
1`,3-indole]-3`-carboxamide (107)
The title compounds were prepared using a similar method to that
described above for the synthesis of 104 and 105 using 93.
However, additional Pd/C (3 mg) and EtOAc (1 mL) was needed
after 18 h. The system was again flushed with H2 gas and left
stirring under a H2 atmosphere (balloon) for 4 h. The crude
reaction mixture was filtered through a bed of celite and washed
multiple times with EtOAc. The combined organic extracts were
evaporated in vacuo. 1H NMR analysis of the crude reaction mixture revealed a 1.5 : 1
mixture of products 106 and 107, respectively. The crude product was purified by
column chromatography using 40-70% EtOAc:PS as the eluent to give pure samples of
106 and 107.
106: (9.5 mg, 31 µmol, 78%). MS (EI) m/z 306 (100%) [M.+], 307 (10%) [MH+], 213
(68%) [M+-NHPh], 186 (89%) [M+-CONHPh]; HRMS (EI) Calcd for C19H18N2O2 [M.+]
306.1368. Found: 306.1374. 1H NMR (C6D6, 500 MHz) δ 7.69 (d, J 8.0 Hz, 2H, ArCH-
o); 7.33 (d, J 7.5 Hz, 1H, ArCH-4); 7.24-7.17 (m, 2H, ArCH-m); 7.02-6.97 (m, 1H,
ArCH-6); 6.94 (t, J 7.5 Hz, 1H, ArCH-p); 6.88 (t, J 7.5 Hz, 1H, ArCH-5); 6.41 (d, J 7.5
Hββββ
Hββββ
Hαααα
Hαααα
R1
NH
O
R2
33a
7a
5
7
1`3`
106: R1= H
R2= CONHPh
107: R1= CONHPh
R2= H

Chapter 8: Experimental for Chapter 2 170
Hz, 1H, ArCH-7); 3.01 (quintet, J 8.0 Hz, 1H, CH-3`α); 2.44 (dd, J 13.5, 9.0 Hz, CH-
2`β); 2.31-2.22 (om, 3H, CH-2`α and CH2-4`); 2.10 (dt, J 13.0, 7.5 Hz, 1H, CH-5`β);
1.98 (ddd, J 13.0, 11.0, 8.5 Hz, 1H, CH-5`α). 13C NMR (C6D6, 500 MHz) δ 183.2 (C-2);
172.7 (CONHPh); 140.4 (ArC-7a); 139.0 (ArC-7a); 135.5 (ArC-3a); 129.0 (ArCH-m);
128.3 (ArCH-6); 123.9 (ArCH-p); 123.8 (ArCH-4); 123.0 (ArCH-5); 119.8 (ArCH-o);
109.2 (ArCH-7); 54.1 (C-3); 47.1 (CH-3`); 41.2 (CH2-2`); 38.2 (CH2-5`); 31.4(CH2-4`).
107: (2.1 mg, 6.9 µmol, 17%). MS (EI) m/z 306 (31%) [M.+], 307 (8%) [MH+], 214
(68%) [M+-NHPh], 186 (91%) [M+-CONHPh]; HRMS (EI) Calcd for C19H18N2O2 [M.+]
306.1368. Found: 306.1383. 1H NMR (500 MHz) δ 9.44 (bs, 1H, NHPh); 7.68 (d, J 7.5
Hz, 2H, ArCH-o); 7.53 (bs, 1H, NH-1); 7.32 (t, J 7.5 Hz, 2H, ArCH-m); 7.24 (t, J 7.5
Hz, 1H, ArCH-6); 7.23 (d, J 7.5 Hz, 1H, ArCH-4); 7.10 (t, J 7.5 Hz, 1H, ArCH-5); 7.08
(t, J 7.5 Hz, 1H, ArCH-p); 6.91 (d, J 7.5 Hz, 1H, ArCH-7); 3.41-3.34 (m, 1H, CH-3`β);
2.61-2.53 (m, 1H, CH-4`β); 2.51-2.42 (m, 3H, CH2-2` and CH-4`α); 2.36-2.31 (m, 1H,
CH-5`β), 2.05 (ddd, J 13.5, 10.2, 7.1 Hz, 1H, CH-5`α). 13C NMR δ 184.7 (C-2); 174.1
(CONHPh); 139.7 (ArC-7a); 139.0 (ArC-i); 134.3 (ArC-3a); 128.9 (ArCH-m); 128.1
(ArCH-6); 123.7 (ArCH-p); 123.3 (ArCH-5); 122.6 (ArCH-4); 119.5 (ArCH-o); 109.8
(ArCH-7); 54.9 (C-3); 48.8(CH-3`); 40.0 (CH2-2`); 39.6 (CH2-5`); 31.7 (CH2-4`).
N-(4-Methoxybenzyl)-N-phenylamine346
A mixture of p-anisaldehyde (0.61 mL, 5 mmol), aniline (0.46 mL, 5
mmol) and montmorillonite K10 clay (0.1 g) contained in a microwave
reaction vessel was placed in a microwave oven and irradiated for 15
min at 120 °C with an additional 2 min preceeding and 30 min post-
irradiation for cooling down. To the resultant crude product was added
DCM (10 mL), and K2CO3 (~ 30 g). This mixture was then filtered
through a column of celite under vacuum and the filtrate was then evaporated in vacuo
to yield the imine which required no further purifcation. To a solution of the resulting
imine (1.17 g, 5.5 mmol) in MeOH (80 mL) was added NaBH4 (166 mg, 4.4 mmol).
The reaction mixture was allowed to stir for 3 h, whereupon the solvent was removed in
vacuo. The resulting residue was dissolved in EtOAc (25 mL), and the solution was
then washed with water (2 × 30 mL), dried and the solvent evaporated in vacuo. The
crude product was then purified by gradient column chromatography using 0-80%
EtOAc:PS as the eluent to yield N-(4-Methoxybenzyl)-N-phenylamine as a yellow oil
H N
O M e
o
p
m
m '
o '
p '
i '
i

Chapter 8: Experimental for Chapter 2 171
(1.042 g, 4.9 mmol, 98%, Rf = 0.73 in 10% EtOAc:PS). MS (CI+ve) m/z 214 (22%)
[MH+]; HRMS (CI+ve) Calcd for C14H16NO [MH+] 214.1232. Found: 214.1222. 1H
NMR δ 7.29 (d, J 8.5Hz, 2H, ArCH-o`); 7.17 (t, J 7.8 Hz, 2H, ArCH-m); 6.87 (d, J 8.7
Hz, 2H, ArCH-o); 6.71 (t, J 7.4 Hz, 1H, ArCH-p); 6.63 (d, J 8.4 Hz, 2H, ArCH-m`);
4.25 (s, 2H, CH2NH); 3.93 (bs, 1H, NH); 3.80 (s, 3H, OCH3). 13C NMR δ 158.7 (ArC-
p`); 145.8 (ArC-i); 129.1 (ArCH-m); 128.7 (ArCH-o`); 117.4 (ArCH-p); 113.9 (ArCH-
m`); 112.7 (ArCH-o); 55.3 (OCH3); 47.8 (CH2N).
(3aS,6R)-8,8-Dimethyl-4,5,6,7-tetrahydro-3a,6-methano-2,1-benzisothiazole 2,2-
dioxide
To a solution of concentrated NH3 (113 mL) cooled to 0 oC (ice-bath) was
added dropwise by syringe, a solution of (+)-(S)-camphorsulfonyl chloride
(7.70 g, 30.8 mmol) dissolved in 1,4-dioxane (19 mL). The reaction mixture
was stirred at 0 oC for 2 h and then heated at reflux at 90 oC for 4 h. Upon cooling the
reaction mixture, off-white crystals were formed, which were collected and dried to give
(3aS,6R)-8,8-Dimethyl-4,5,6,7-tetrahydro-3a,6-methano-2,1-benzisothiazole 2,2-
dioxide (4.34 g, 20.4 mmol, 66%, Rf = 0.10 in 20% EtOAc:PS, m.p. 217-220 oC (lit.344
m.p. 230 oC), [α]D23 = -17.2 (c 1.09, CHCl3), (lit.
344 [α]D20 = -32.2 (c 2, CHCl3))). MS
(EI) m/z 213 (1.3%) [M.+]; HRMS (EI) Calcd for C10H15NO2S [M.+] 213.0824. Found:
213.0820. 1H NMR δ 3.07 (ABq, J 13.2 Hz, 2H, CH2-3); 2.77 (ddd, J 19.4 Hz, 1H, CH-
7β); 2.38 (d, J 19.2 Hz, 1H, CH-7α); 2.26 (t, J 4.2 Hz, 1H, CH-6); 2.09-2.07 (m, 1H,
CH-5β); 2.05-2.04 (m, 1H, CH-4β); 1.78 (t, J 9.0 Hz, 1H, CH-5α); 1.50-1.44 (m, 1H,
CH-4α), 1.08 (s, 3H, (CH3-10); 0.87 (s, 3H, (CH3-9). 13C NMR δ 195.4 (C-7a); 64.4 (C-
3a); 49.4 (CH2-3); 47.9 (C-8); 44.6 (CH-6); 35.8 (CH2-7); 28.3 (CH2-5); 26.6 (CH2-4);
19.4 (CH3-10); 18.9 (CH3-9). NMR data collected for (3aS,6R)-8,8-Dimethyl-4,5,6,7-
tetrahydro-3a,6-methano-2,1-benzisothiazole 2,2-dioxide agreed with that reported in
the literature.344
O2S N
3a
6
9
H
7
1

Chapter 8: Experimental for Chapter 3 172
8.3 Experimental for Chapter 3 (3E)-(Pyridin-2-ylmethylene)-1,3-dihydro-2H-indol-2-one (108a)
To a solution of oxindole (391.1 mg, 2.9 mmol) and picolinaldehyde
(0.3 mL, 3.1 mmol) in MeOH (1 mL) at ~30 °C was added
pyrrolidine (0.22 mL, 2.6 mmol). The mixture was left stirring at RT
for 3 h. The crystals were filtered off and the supernatant purified by
column chromatography using 30-50% EtOAc:PS as eluent to yield
108a as orange crystals (400.2 mg, 1.8 mmol, 61%, Rf = 0.37 in 30%
EtOAc:PS, m.p. 192-198 °C (lit.43 m.p. 205-206 °C)). MS (EI) m/z 222 (49%) [M.+],
221 (72%) [M+-1], 194 (94%); 166 (15%), 144 (52%) [M+-C5H4N]; HRMS (EI) Calcd
for C14H10N2O [M+-1] 221.0715. Found: 221.0720. 1H NMR (500 MHz) δ 9.00 (d, J 8.0
Hz, 1H, ArCH-4); 8.87 (d, J 4.0 Hz, 1H, ArCH-3`); 8.57 (bs, 1H, NH); 7.79 (dt, J 8.0,
0.5 Hz, 1H, ArCH-5`); 7.72 (s, 1H, CH=); 7.62 (d, J 8.0 Hz, 1H, ArCH-6`); 7.32 (t, J
7.0 Hz, 1H, ArCH-4`); 7.28 (t, J 7.5 Hz, 1H, ArCH-6); 7.05 (t, J 7.7 Hz, 1H, ArCH-5);
6.90 (d, J 7.5 Hz, 1H, ArCH-7). 13C NMR (125 MHz) δ 170.8 (C-2); 153.8 (C-1`);
149.6 (ArCH-3`); 142.2 (ArC-7a); 136.6 (ArCH-5`); 134.8 (CH=); 130.6 (ArCH-6);
129.5 (C=); 128.2 (ArCH-4); 127.9 (ArCH-6`); 123.7 (ArCH-4`); 122.3 (ArCH-5);
122.1 (ArC-3a); 109.7 (ArCH-7). The structure of 108a was confirmed by X-ray
crystallography (see Appendix 1: X-ray 19: Compound 108a). The literature did not
assign the stereochemistry or give any spectral data for 108a.257
(3E)-Pyridiny-3-ylmethylene-1,3-dihydroindol-2-one (108b)
To a solution of oxindole (528 mg, 4.0 mmol) and nicotinaldehyde
(0.4 mL, 4.2 mmol) in MeOH (6.3 mL) at ~30 °C was added
pyrrolidine (0.3 mL, 3.6 mmol). The mixture was left stirring at RT
for 3 h. The solvent was then evaporated in vacuo and purified by
column chromatography using 50% EtOAc:PS to yield needle-like
orange crystals (816.3 mg, 3.7 mmol, 93%, Rf = 0.3 in 60%
EtOAc:PS, m.p. 190-192 °C, (lit.257 m.p. 193.5-196 °C)). MS (EI) m/z 222 (94%) [M.+],
194 (32%), 166 (20%), 144 (39%) [M+-C5H4N]; HRMS (EI) Calcd for C14H10N2O [M.+]
222.0793. Found: 222.0796. 1H NMR δ 8.93 (s, 1H, ArCH-2`); 8.75 (bs, 1H, NH); 8.68
(d, J 4.2 Hz, 1H, ArCH-4`); 7.96 (dt, J 6.3, 1.5 Hz, 1H, ArCH-6`); 7.75 (s, 1H, CH=);
7.51 (d, J 7.8 Hz, 1H, ArCH-4); 7.43 (dd, J 8.1, 1.5 Hz, 1H, ArCH-5`); 7.25 (dt, J 7.5,
NH
O
N
3 a
7 a
4
7
2
1 `
2 `
NH
O
N
3a
7a
4
7
2
1`
3`

Chapter 8: Experimental for Chapter 3 173
1.3 Hz, 1H, ArCH-6); 6.91 (d, J 7.8 Hz, 1H, ArCH-7), 6.88 (dt, J 7.5, 0.7 Hz, 1H,
ArCH-5). 13C NMR δ 169.5 (C-2); 150.2 (ArCH-4`); 149.9 (ArCH-2`); 141.9 (ArC-7a);
136.3 (ArCH-6`); 132.8 (CH=); 130.8 (C-1`); 130.6 (ArCH-6); 129.5 (C=); 123.5
(ArCH-5`); 122.9 (ArCH-4); 122.1 (ArCH-5); 121.2 (ArC-3a); 110.5 (ArCH-7). The
structure of 108b was confirmed by X-ray crystallography (see Appendix 1: X-ray 20:
Compound 108b). The literature did not assign the stereochemistry or give any spectral
data for 108b.257
(1`R*, 2`R*) 2`-Ethyl 1`-methyl 1`-(2-nitrophenyl)cyclopropane-1`,2`-dicarboxylate
(117)
A solution of ethyl dimethylsulfonium acetate bromide (1.56 g, 6.8
mmol) and 1,8-diazobicyclo[5.4.0]undec-7-ene (DBU) (0.76 mL,
5.1 mmol) in anhydrous toluene (30 mL) was stirred under a N2
atmosphere at RT for 30 min. A solution of 59 (709.7 mg, 3.4
mmol) in anhydrous toluene (10 mL) was added and stirring was continued for 20 h.
The reaction mixture was washed with 10% HCl solution (2 × 40 mL) and the aqueous
washings extracted with EtOAc (3 × 100 mL). The organic combined extracts were
dried, filtered and evaporated under reduced pressure. The crude mixture was purified
by column chromatography, elution with 10-25% EtOAc:PS yielded 117 as a light-
yellow semi-crystalline oil which crystallised upon standing (799.7 mg, 2.7 mmol, 80%,
Rf = 0.5 in 20% EtOAc:PS) and recovered starting 59 (41.8 mg, 2 × 10-4 mol, 6%). MS
(ESI+ve) m/z 294 (100%) [MH+], 248 (43%) [M+-OEt]; HRMS (ESI+ve) Calcd for
C14H16NO6 [MH+] 294.0978. Found: 294.0981. 1H NMR δ 8.06 (bd, J 6.6 Hz, 1H,
ArCH-3); 7.62 (bd, J 7.5 Hz, 1H, ArCH); 7.51-7.46 (m, 2H, ArCH and ArCH-4); 3.92
(q, J 7.0 Hz, 2H, CH2CH3); 3.64 (s, 3H, OCH3); 3.02 (bs, 1H, CH-2`); 2.02-1.99 (m, 2H,
CH2-3`); 1.08 (t, J 7.0 Hz, 3H, CH3CH2). 13C NMR δ 171.3 (CO2Me); 169.5 (CO2Et);
149.2 (ArC-1); 133.5 (ArCH-5 and ArCH-6); 131.0 (ArC-2); 128.9 (ArCH-4); 124.8
(ArCH-3); 61.2 (CH2CH3); 53.0 (OCH3); 36.1 (C-1`); 29.7 (CH-2`); 22.1 (CH2-3`); 13.8
(CH2CH3). The structure of 117 was confirmed by X-ray crystallography (see
Appendix 1: X-ray 14: Compound 117).
NO2
CO2Me
O
OEt1
2
1`
2̀

Chapter 8: Experimental for Chapter 3 174
Methyl (1aR*,7bR*)-2-oxo-1,1a,2,3-tetrahydro-7bH-cyclopropa[c]quinoline-7b-
carboxylate (118) and Ethyl (1`R*, 2`R*)-2-oxo-1,2-dihydrospiro[cyclopropane-
1`,3-indole]-2`-carboxylate (119)
The title compounds were prepared using two methods. Method 1: To a solution of 117
(493.8 mg, 1.68 mmol) in EtOH : H2O (12.8 mL : 3.2 mL) was added activated Zn dust
(2.627 g, 40 mmol) and 8.9 M HCl (2.54 mL). The mixture was stirred and heated at
reflux for 3 h. The mixture was filtered through a bed of celite and washed with EtOH. 1H NMR analysis of the crude reaction mixture revealed a mixture of 118 and 119, in a
ratio of 12 : 1, respectively. The crude product was purified by column chromatography
using 20-50% EtOAc:PS as eluent and then further purified using DCM:PS:EtOAc (2 :
2 : 1) as eluent to yield 118, as white crystals (253.6 mg, 1.17 mmol, 70%) and 119, as
white crystals (19.3 mg, 8.3 × 10-5 mol, 5%. Method 2: To a solution of 117 (167.6 mg,
5.7 × 10-4 mol) in EtOAc (8.6 mL) was added 10% Pd/C (33 mg). The system was
flushed with H2 gas and left to stir under a H2 atmosphere (balloon) for 2 d. 1H NMR
analysis of the crude reaction mixture revealed a mixture of 118 and 119, in a ratio of 4 :
1, respectively. The crude product was purified by column chromatography and then by
PTLC using 30% EtOAc:PS as eluent to yield 118, as white crystals (76.1 mg, 3.5 × 10-
4 mol, 61%, Rf = 0.15 in 30% EtOAc:PS, m.p.166-170 °C) and 119, as white crystals
(23.9 mg, 0.1 mmol, 18%, Rf = 0.3 in 30% EtOAc:PS, m.p. 136-138 °C (lit.245 m.p. 154-
156 °C).
118: MS (EI) m/z 217 (55%) [M.+], 202 (58%) [M+-Me], 158 (53%)
[M+-CO2Me]; HRMS (EI) Calcd for C12H11NO3 [M.+] 217.0739.
Found: 217.0735. 1H NMR δ 8.75 (bs, 1H, NH); 7.72 (dd, J 8.1,
1.2 Hz, 1H, ArCH-4); 7.20 (dt, J 7.8, 1.5 Hz, 1H, ArCH-6); 7.07
(dt, J 7.5, 1.5 Hz, 1H, ArCH-5); 6.81 (dd, J 8.1, 1.0 Hz, 1H, ArCH-7); 3.80 (s, 3H, CH3)
2.58 (ddd, J, 10.5, 5.1, 1.3 Hz, 1H, CH-1a); 2.43 (dd, J 4.2, 10.5 Hz, 1H, CHAHB-1);
1.03 (dd, J 5.7, 4.8 Hz, 1H, CHBHA-1). 13C NMR δ 170.4 (CO2Me); 167.2 (C-2); 134.3
(ArC-7a); 129.8 (ArCH-4); 127.8 (ArCH-6); 123.0 (ArCH-5); 119.2 (ArC-3a); 115.7
(ArCH-7); 52.7 (CH3); 29.9 (C-7b); 28.6 (CH-1a); 17.9 (CH2-1). The structure of 118
was confirmed by X-ray crystallography (see Appendix 1: X-ray 15: Compound 118).
NH
O
MeO2C
H
4
7b7a
3a
2
1
61a

Chapter 8: Experimental for Chapter 3 175
119: MS (EI) m/z 231 (68%) [M.+], 186 (32%) [M+-OEt]; HRMS
(EI) Calcd for C13H13NO3 [M.+] 231.0895. Found: 231.0896. 1H
NMR (500 MHz) δ 9.26 (bs, 1H, NH); 7.34 (d, J 7.5 Hz, 1H,
ArCH-4); 7.22 (dt, J 7.5, 1.5 Hz, 1H, ArCH-6); 7.00 (dt, J 8.0,
1.0 Hz, 1H, ArCH-5); 6.98 (d, J 7.5 Hz, 1H, ArCH-7); 4.08-4.21 (m, 2H, CH2CH3);
2.72 (dd, J 8.5, 7.5 Hz, 1H, CH-2`); 2.17 (dd, J 7.5, 4.5 Hz, 1H, CHACHB-3`); 2.03 (dd,
J 8.5, 4.5 Hz, 1H, CHBCHA-3`); 1.21 (t, J 7.3 Hz, 3H, CH3). 13C NMR (125 MHz) δ
177.2 (C-2); 168.6 (CO2Et); 141.5 (ArC-7a); 127.7 (ArCH-6); 126.2 (ArC-3a); 122.9
(ArCH-4): 122.2 (ArCH-5); 110.0 (ArCH-7); 61.3 (CH2CH3); 34.0 (C-3); 32.9 (CH-2`);
20.8 (CH2-3`); 14.1 (CH3). The structure of 119 was confirmed by X-ray
crystallography (see Appendix 1: X-ray 16: Compound 119). NMR data collected for
119 agreed well with those found in the literature.245
(1aR*, 7bR*)-2-Oxo-1,1a,2,3-tetrahydro-7bH-cyclopropa[c]quinoline-7b-
carboxylic acid (126)
To a solution of 118 (91.5 mg, 0.4 mmol) in MeOH (1.5 mL)
contained within a sealed tube was added a solution of K2CO3 (109
mg, 0.8 mmol) in H2O (1 mL). The tube was sealed and the
mixture was left stirring at 60 °C for 18 h. The solvent was removed by evaporation in
vacuo and the residue was dissolved in H2O (15 mL) and washed with Et2O. The
aqueous solution was then acidified to a ~ pH 1 with 10% HCl and extracted with Et2O.
The combined extracts were dried to yield 126 as a white powder (40.3 mg, 2.0 × 10-4
mol, 50%, Rf = 0 in 30% EtOAc:PS, m.p. 152-156°C). MS (EI) m/z 203 (35%) [M.+],
159 (24%), 130 (30%), 111 (32%), 97 (45%), 71 (60%), 57 (97%), 43 (87%); HRMS
(EI) Calcd for C11H9NO3 [M.+] 203.0582. Found: 203.0580. 1H NMR (CD3OD) δ 7.77
(dd, J 7.8, 1.5 Hz, 1H, ArCH-4); 7.17 (dt, J 7.5, 1.2 Hz, 1H, ArCH-6); 7.03 (dt, J 7.5,
1.2 Hz, 1H, ArCH-5); 6.87 (dd, J 8.1, 0.9 Hz, 1H, ArCH-7); 2.46 (dd, J 10.5, 5.4 Hz,
1H, CH-1a); 2.36 (dd, J 10.5, 3.9 Hz, 1H, CHACHB-1); 0.92 (dd, J 5.8, 4.0 Hz, 1H,
CHBCHA-1). 13C NMR (CD3OD, 125 MHz) δ 173.3 (CO2H); 169.6 (C-2); 136.2 (ArC-
7a); 131.0 (ArCH-4); 128.6 (ArCH-6); 123.7 (ArCH-5); 121.1 (ArC-3a); 116.8 (ArCH-
7); 30.8 (C-7b); 29.3 (CH-1a); 18.3 (CH2-1). The structure of 126 was confirmed by X-
ray crystallography (see Appendix 1: X-ray 17: Compound 126).
NH
O
HOOC
4
7b7a
3a
2
1
61a
H
CO2Et
NH
O
3a
7a
4
7
3
3`
2`

Chapter 8: Experimental for Chapter 3 176
(1`R*, 2`R*)-2-Oxo-1,2-dihydrospiro[cyclopropane-1`,3-indole]-2`-carboxylic acid
(127)
The title compound was prepared using a similar method to that
described above for the synthesis of 126 except starting with 119
(38.2 mg, 0.16 mmol) to yield 127 as white crystals (25.4 mg, 0.13
mmol, 80 %, Rf = 0 in 30% EtOAc:PS, m.p. 142-145 °C). MS (EI) m/z 203 (29%) [M.+];
HRMS (EI) Calcd for C11H9NO3 [M.+] 203.0582. Found: 203.0579. 1H NMR (CD3OD)
δ 7.27 (dd, J 7.5, 1.0 Hz, 1H, ArCH-4); 7.21 (dt, J 7.5, 1.3 Hz, 1H, ArCH-6); 6.96 (d, J
7.5 Hz, 1H, ArCH-7); 6.95 (dt, J 7.5, 1.2 Hz, 1H, ArCH-5); 2.48 (dd, J 8.5, 7.3 Hz, 1H,
CH-2`); 2.00 (dd, J 7.2, 4.3 Hz, 1H, CHACHB-3`); 1.82 (dd, J 8.4, 4.5 Hz, 1H,
CHBCHA-3`). 13C NMR (CD3OD, 125 MHz) δ 178.7 (CO2H); 171.2 (C-2); 143.5 (ArC-
7a); 128.8 (ArCH-6); 127.6 (ArC-3a); 123.6 (ArCH-4); 123.0 (ArCH-5); 111.0 (ArCH-
7); 34.7 (C-3); 33.9 (CH-2`); 21.0 (CH2-3`).
(1aR*, 7bR*)-2-Oxo-N-phenyl-1,1a,2,3-tetrahydro-7bH-cyclopropa[c]quinoline-7b-
carboxamide (128)
To a solution of 126 (52.4 mg, 2.6 × 10-4 mol) and HOBT (34.9 mg,
2.6 × 10-4 mol) in anhydrous MeCN (3 mL) at 0 °C (ice-bath) was
added aniline (0.04 mL, 4.1 × 10-4 mol). The solution was stirred
for 10 min at 0 °C before the addition of EDCI (49.5 mg, 2.6 × 10-4
mol) and left to stir at RT for 2 h, then at 50 °C for 18 h, then again
at RT for 3 d. The solvent was then removed, and the residue was diluted with DCM
and washed successively with 10% HCl, H2O and brine. The organic combined extracts
were then collected, dried and evaporated in vacuo. Purification of the crude product
was achieved through column chromatography using 10% MeOH:CHCl3 as the eluent
to yield 128 as an amber coloured oil (56 mg, 2.0 × 10-4 mol, 77 %, Rf = 0.28 in 50%
EtOAc:PS). MS (EI) m/z 278 (63%) [M.+], 279 (15%) [MH+], 263 (26%), 206 (12%),
186 (27%), 158 (47%), 130 (94%); HRMS (EI) Calcd for C17H14N2O2 [M.+] 278.1055.
Found: 278.1051. 1H NMR (500 MHz) δ 9.57 (bs, 1H, NH-3); 8.23 (bs, 1H, NHPh);
7.56-7.54 (m, 3H, ArCH-7 and ArCH-o); 7.33 (t, J 8.0 Hz, 2H, ArCH-m); 7.16 (t, J 7.0
Hz, 1H, ArCH-5); 7.13 (t, J 7.3 Hz, 1H, ArCH-p); 7.06 (t, J 7.7 Hz, 1H, ArCH-6); 6.90
(d, J 8.0 Hz, 1H, ArCH-4); 2.58 (dd, J 10.7, 4.7 Hz, 1H, (CHACHB-1); 2.18 (dd, J 10.5,
5.7 Hz, 1H, CH-1a); 0.89 (t, J 5.7 Hz, 1H, CHBCHA-1). 13C NMR (125 MHz) δ 168.8
NH
O4
7b7a
3a
2
1
61a
H
O
HN
i
m
o
p
CO2H
NH
O
3a
7a
4
7
3
3`
2`

Chapter 8: Experimental for Chapter 3 177
(C-2); 166.7 (CONHPh); 137.7 (ArC-i); 135.3 (ArC-3a); 129.1 (ArCH-m); 128.4
(ArCH-7); 128.3 (ArCH-5); 124.6 (ArCH-p); 123.7 (ArCH-6); 120.0 (ArC-7a); 119.6
(ArCH-o); 116.6 (ArCH-4); 33.3 (C-7b); 27.6 (CH-1a); 15.1 (CH2-1).
(1`R*, 2`R*)-2`-Oxo-N-phenyl-1`,2`-dihydrospiro[cyclopropane-1,3`-indole]-2-
carboxamide (129)
The title compound was prepared using two methods.
Method 1. The title compound was prepared using a
similar method to that described above for the
synthesis of 128 except starting with 127 (25.9 mg, 1.3 × 10-4 mol). The crude product
after acidic workup was purified initially by column chromatography using 10%
MeOH:CHCl3 as eluent and then further purified on a chromatotron® (0-1%
MeOH:CHCl3) to yield 129 as a beige powder (6.4 mg, 2.3 × 10-5 mol, 18%, Rf = 0.23
in 50% EtOAc:PS). MS (EI) m/z 278 (94%) [M.+], 279 (10%) [MH+], 263 (31%), 206
(13%), 186 (34%), 158 (64%); HRMS (EI) Calcd for C17H14N2O2 [M.+] 278.1055.
Found: 278.1051. Method 2. To a solution of 133 (15.7 mg, 4.6 × 10-5 mol) in a
mixture of H2O (1 mL), AcOH (2 mL) and EtOH (2 mL), contained within a sealed tube,
was added Fe (20 mg, 3.6 × 10-4 mol). The mixture was subjected to sonication for 2 h.
The mixture was diluted with DCM (100 mL) and washed successively with sat. K2CO3
solution (20 mL) and H2O (100 mL). The solution was dried, filtered and the solvent
was removed in vacuo. The crude product was purified by chromatotron® using 40-
100% EtOAc:PS and then MeOH to yield 129, as light brown solid (7.8 mg, 2.8 × 10-5
mol, 60 %) and recovered 133 (0.4 mg, 1.2 × 10-6 mol, 2%). MS (EI) m/z 278 (34%)
[M.+], 235 (15%), 223 (10%), 185 (47%) [M+-NHPh], 157 (30%) [M+-CONHPh], 146
(42%), 130 (96%), 103 (30%); HRMS (EI) Calcd for C17H14N2O2 [M.+] 278.1055.
Found: 278.1049. 1H NMR (500 MHz, CH3OD) δ 7.47 (d, J 7.5 Hz, 2H, ArCH-o); 7.30
(d, J 8.0 Hz, 1H, ArCH-4); 7.25 (t, J 8.3 Hz, 2H, ArCH-m); 7.18 (dt, J 7.0, 1.0 Hz, 1H,
ArCH-6); 7.04 (t, J 7.8 Hz, 1H, ArCH-p); 6.95 (d, J 8.0 Hz, 1H, ArCH-7); 6.92 (t, J 8.0
Hz, 1H, ArCH-5); 2.81 (dd, J 8.5, 7.5 Hz, 1H, CH-2`); 2.25 (dd, J 7.3, 4.3 Hz, 1H,
CHACHB-3`); 1.91 (dd, J 8.7, 4.3 Hz, 1H, CHBCHA-3`). 13C NMR (125 MHz, CH3OD)
δ 179.0 (C-2); 167.1 (CONH); 143.5 (ArC-7a); 139.7 (ArC-i); 129.7 (ArCH-m); 128.6
(ArCH-6); 128.0 (ArC-3a); 125.2 (ArCH-p); 123.5 (ArCH-5); 123.0 (ArCH-4); 121.2
(ArCH-o); 110.9 (ArCH-7); 36.2 (CH-2`); 35.0 (C-3); 20.1 (CH2-3`).
CONH
NH
O
3a
7a
4
7
3
3`
2` i
o m
p

Chapter 8: Experimental for Chapter 3 178
2-Chloro-N-phenylacetamide (130)
To a solution of pyridine (0.46 mL, 5.6 mmol) and aniline
(0.38 mL, 4.1 mmol) in anhydrous DCM (100 mL) at 0 °C
was added chloroacetyl chloride (0.3 mL, 3.8 mmol). The
mixture was allowed to warm to RT and left stirring at RT for 1 h. The crude mixture
was washed with citric acid, then sat. K2CO3 solution, and dried and the solvent was
removed in vacuo to yield white crystals (796.5 mg, 4.7 mmol) which were
recrystallised from EtOAc:PS, to yield off-white crystalline plates (465.5 mg, 2.8 mmol,
74%, Rf = 0.61 in 30% EtOAc:PS, m.p. 132-136 °C (lit.347 m.p. 132-134 °C (from
MeOH))). MS (EI) m/z 169 (70%), 171 (36%) [M.+], 120 (54%) [M+-CH2Cl]; HRMS
(EI) Calcd for C8H8NO35Cl [M.+] 169.0294. Found: 169.0295. 1H NMR δ 8.30 (bs, 1H,
NH); 7.54 (dd, J 7.5, 1.2 Hz, 2H, ArCH-o); 7.35 (dt, J 7.8, 1.9 Hz, 2H, ArCH-m); 7.16
(tt, J 7.2, 1.2 Hz, 1H, ArCH-p); 4.17 (s, 2H, CH2). 13C NMR δ 163.9 (CO); 136.6 (ArC-
i); 129.0 (ArCH-m); 125.2 (ArCH-p); 120.1 (ArCH-o); 42.8 (CH2). NMR data for 130
have not been reported in literature.
2-(Methylsulfanyl)-N-phenylacetamide (131)348
To a solution of 130 (257 mg, 1.52 mmol) in anhydrous
MeOH (35 mL) was added sodium thiomethoxide (95%,
123.7 mg, 1.68 mmol) The reaction was stirred at RT for
15 min. The solvent was removed in vacuo in a fume
cupboard. The residue was diluted with DCM and washed with sat. K2CO3 solution and
dried to yield a cream solid (270.1 mg, 1.5 mmol, 98%, Rf = 0.52 in 30% EtOAc:PS).
MS (EI) m/z 181 (49%) [M.+], 135 (45%) [MH+-SMe]; HRMS (EI) Calcd for
C9H11NOS [M.+] 181.0561. Found: 181.0562. 1H NMR δ 8.85 (bs, 1H, NH); 7.54 (d, J
7.5 Hz, 2H, ArCH-o); 7.29 (t, J 7.9 Hz, 2H, ArCH-m); 7.09 (t, J 7.4 Hz, 1H, ArCH-p);
3.28 (s, 2H, CH2); 2.14 (s, 3H, CH3). 13C NMR δ 167.0 (CO); 137.4 (ArC-i); 128.7
(ArCH-m); 124.3 (ArCH-p); 119.7 (ArCH-o); 38.7 (CH2); 16.0 (CH3). 1H NMR data
collected for 131 agreed well with those found in the literature.349
Cl
HN
O
i
o m
p
HN
OMeS
i
o m
p

Chapter 8: Experimental for Chapter 3 179
2-Anilino-2-oxoethyl-(dimethyl)sulfonium iodide (132)
To a solution of 131 (270.1 mg, 1.5 mmol) in anhydrous
DCM (2mL) was added MeI (0.94 mL, 15 mmol). The flask
was sealed and the superseal tightly wrapped with parafilm
and left stirring for 2 d at RT. The reaction was found to be
incomplete by TLC analysis and so further MeI (0.94 mL, 15 mmol) was added and the
mixture left stirring at RT for 2 d. The mixture was diluted with DCM and 132 was
filtered off as an off-white solid (253.3 mg, 7.84 × 10-4 mol, 52%, Rf = 0 in 30%
EtOAc:PS) and the starting material 131 was recovered from evaporation of the filtrate
(105.1 mg, 5.8 × 10-4 mol, 39%). MS (ESI+ve) m/z 196 (100%) [M+-I], ESI(-ve) m/z
127 (100%) [I-]. The NMR data for this salt proved impossible to assign due to peak
broadening.
Methyl (1`R*, 2`R*)-2`-(anilinocarbonyl)-1`-(2-
nitrophenyl)cyclopropanecarboxylate (133)
To a solution of 132 (214.3 mg, 6.6 × 10-5 mol) in
anhydrous DCM (3 mL) was added DBU (0.07 mL, 4.7
× 10-4 mol) and the solution was stirred under a N2 atmosphere at RT for 30 min. A
solution of 59 (91.5 mg, 4.4 × 10-4 mol) in anhydrous DCM (2 mL) was then added and
stirring continued for 2 d. The reaction mixture was diluted with DCM and the solution
was washed with 1 M HCl solution (2 × 40 mL). The aqueous layers were back-
extracted with DCM (2 × 50 mL). The combined extracts were dried and evaporated
under reduced pressure to yield a brown yellow solid. The crude product was purified
by column chromatography using 20-50% EtOAc:PS as eluent to yield 133 as a white
solid (59 mg, 1.7 × 10-4 mol, 39%, Rf = 0.4 in 30% EtOAc:PS, m.p. 226-228°C),
recovered 59 (51.5 mg, 2.4 × 10-5 mol, 56%) and 131 (60 mg, 3.0 × 10-4 mol). MS (EI)
m/z 340 (40%) [M.+], 294 (90%) [M+-NO2]; HRMS (EI) Calcd for C18H16N2O5 [M.+]
340.1058. Found: 340.1055. 1H NMR δ 8.04 (dd, J 8.1, 1.3 Hz, 1H, ArCH-3); 7.65-7.54
(m, 2H, ArCH-5 and ArCH-6); 7.45 (dt, J 7.5, 1.6 Hz, 1H, ArCH-4); 7.24-7.19 (m, 4H,
ArCH-o and ArCH-m); 7.07-7.02 (m, 1H, ArCH-p); 3.65 (s, 3H, CH3); 3.01 (bs, 1H,
CH-2`); 2.28 (bs, 1H, CHACHB-3`); 1.98 (dd, J 8.4, 4.8 Hz, 1H, CHBCHA-3`). 13C NMR
δ 171.9 (CO2Me); 165.3 (CONH); 149.0 (ArC-1); 137.4 (ArC-i); 133.4 (ArCH-5 and
ArCH-6); 130.8 (ArC-2); 129.0 (ArCH-4); 128.8 (ArCH-o); 125.2 (ArCH-3); 124.5
HN
OMe2S
i
o m
p
NO2
CO2Me
O
NH1
2
1`
2` i
o m
p

Chapter 8: Experimental for Chapter 3 180
(ArCH-p); 120.0 (ArCH-m); 53.0 (OCH3); 35.9 (C-1`); 32.8 (CH-2`); 21.0 (CH2-3`).
The structure of 133 was confirmed by X-ray crystallography (see Appendix 1: X-ray
18: Compound 133).
Ethyl (1`R*, 2`R*, 3`R*)-2-oxo-3`-pyridin-2-yl-1,2-dihydrospiro[cyclopropane-1`,3-
indole]-2`-carboxylate (139a), Ethyl (1`R*, 2`R*, 3`S*)-2-oxo-3`-pyridin-2-yl-1,2-
dihydrospiro[cyclopropane-1`,3-indole]-2`-carboxylate (140a) and Ethyl (1`R*,
2`S*, 3`S*)-2-oxo-3`-pyridin-2-yl-1,2-dihydrospiro[cyclopropane-1`,3-indole]-2`-
carboxylate (141a)
EtO 2C
NH
O
N3a
7a
4
7
3`2` 1``
3``
5``
2
EtO2C
NH
O
N3a
7a
4
7
3`2` 1``
3``
5``
2
EtO2C
NH
O
N3a
7a
4
7
3`2` 1``
3``
5``
2
139a 140a 141a
To a solution of ethyl dimethylsulfoniumacetate bromide (173.6 mg, 7.6 × 10-4 mol) in
anhydrous MeCN (3.7 mL) was added DBU (0.07 mL, 4.7 × 10-4 mol) and the solution
was stirred under a N2 atmosphere at RT for 30 min. A solution of 108a (108.8 mg, 4.7
× 10-4 mol) in anhydrous MeCN (1 mL) was added and stirring was maintained for 24 h.
The reaction mixture was diluted with EtOAc and washed with 10% HCl solution (2 ×
40 mL). The organic extracts were dried and evaporated under reduced pressure to yield
a crude product with the appearance of peach coloured crystals (155.6 mg, 5.0 × 10-4
mol). 1H NMR analysis of the crude reaction mixture revealed a 5.6: 1.8 : 1 mixture of
products 139a : 140a : 141a, respectively. The crude mixture was purified by column
chromatography using 30-50% EtOAc:PS as eluent to yield 139a as clear needle-like
crystals (41.3 mg, 1.3 × 10-4 mol, 27%, Rf = 0.24 in 50% EtOAc:PS, m.p. 178-180 °C)
and 141a as a cream oil (17.4 mg, 5.6 × 10-5 mol, 12%, Rf = 0.31 in 50% EtOAc:PS).
Compound 140a was unable to be isolated as a pure sample but was identified as the cis
isomer, and had 1H NMR resonances at δ 3.57 (d, J 9.9 Hz, 1H, CH-2`) and 3.08 (d, J
10.2 Hz, 1H, CH-3`).
139a: MS (EI) m/z 308 (26%) [M.+], 309 (6%) [MH+], 262 (46%), 235 (94%), 217
(31%), 205 (44%); HRMS (EI) Calcd for C18H16N2O3 [M.+] 308.1161. Found: 308.1165.
1H NMR (500 MHz) δ 8.88 (bs, 1H, NH); 8.52 (d, J 4.0 Hz, 1H, ArCH-3``); 7.62 (dt, J

Chapter 8: Experimental for Chapter 3 181
7.5, 1.5 Hz, 1H, ArCH-5``); 7.45 (d, J 7.5 Hz, 1H, ArCH-4); 7.33 (d, J 7.5 Hz, 1H,
ArCH-6``); 7.18-7.14 (m, 2H, ArCH-4`` and ArCH-6); 6.99 (t, J 7.7 Hz, 1H, ArCH-5);
6.74 (d, J 7.5 Hz, 1H, ArCH-7); 4.26-4.14 (m, 2H, CH2); 3.93 (d, J 8.0 Hz, 1H, CH-2`);
3.46 (d, J 8.0 Hz, 1H, CH-3`); 1.25 (t, J 7.0 Hz, 3H, CH3). 13C NMR (125 MHz) δ 173.8
(C-2); 168.1 (CO2Et); 153.1 (ArC-1``); 149.1 (ArCH-3``); 141.4 (ArC-7a); 136.1
(ArCH-5``); 127.7 (ArCH-6); 126.1 (ArC-3a); 123.9 (ArCH-6``); 122.8 (ArCH-4);
122.4 (ArCH-4``); 122.1 (ArCH-5); 109.8 (ArCH-7); 61.5 (CH2); 40.7 (CH-2`); 39.5
(C-3); 36.8 (CH-3`); 14.1 (CH3). The structure of 139a was confirmed by X-ray
crystallography (see Appendix 1: X-ray 21: Compound 139a).
141a: 1H NMR δ 8.60 (bs, 1H, NH); 8.55 (dm, J 4.8 Hz, 1H, ArCH-3``); 7.51 (dt, J 7.5,
1.9 Hz, 1H, ArCH-5``); 7.19 (t, J 4.0 Hz, 1H, ArCH-6``); 7.11 (ddd, J 7.5, 4.8, 1.2 Hz,
1H, ArCH-4``); 7.03 (dt, J 7.5, 1.5 Hz, 1H, ArCH-6); 6.80 (d, J 7.8 Hz, 1H, ArCH-7);
6.75 (d, J 7.5 Hz, 1H, ArCH-4); 6.68 (dt, J 7.2, 1.0 Hz, 1H, ArCH-5); 4.21-4.13 (m, 2H,
CH2); 3.86 (d, J 8.1 Hz, 1H, CH-2`); 3.82 (d, J 8.1 Hz, 1H, CH-3`); 1.21 (t, J 7.0 Hz,
3H, CH3). 13C NMR δ 174.7 (C-2); 167.2 (CO2Et); 152.9 (ArC-1``); 148.8 (ArCH-3``);
141.3 (ArC-7a); 136.5 (ArCH-5``); 127.5 (ArCH-6); 126.1 (ArC-3a); 125.6 (ArCH-6``);
122.6 (ArCH-4``); 122.5 (ArCH-4); 121.8 (ArCH-5); 109.7 (ArCH-7); 61.4 (CH2); 40.2
(CH-2`); 40.1 (C-3); 35.7 (CH-3`); 14.2 (CH3).
Ethyl (1`R*, 2`R*, 3`R*)-2`-oxo-3-pyridin-3-yl-1`,2`-dihydrospiro[cyclopropane-
1,3`-indole]-2-carboxylate (139b), Ethyl (1`R*, 2`R*, 3`S*)-2`-oxo-3-pyridin-3-yl-
1`,2`-dihydrospiro[cyclopropane-1,3`-indole]-2-carboxylate (140b) and Ethyl (1`R*,
2`S*, 3`S*)-2`-oxo-3-pyridin-3-yl-1`,2`-dihydrospiro[cyclopropane-1,3`-indole]-2-
carboxylate (141b)
5`` 5`` 5``
The title compounds were prepared using a similar method to that described above for
the synthesis of 139a, 140a and 141a, except starting with 108b (104.4 mg, 4.7 × 10-4
mol). After extraction the crude product had the appearance of a peach coloured solid
(101.7 mg, 3.3 × 10-4 mol, 70%). 1H NMR analysis of the crude reaction mixture

Chapter 8: Experimental for Chapter 3 182
revealed a 43 : 7 : 1 mixture of products 139b : 140b : 141b, respectively. The crude
product was purified by column chromatography using 40-60% EtOAc:PS as eluent to
yield 139b as cream crystals (88.2 mg, 2.9 × 10-4 mol, 61%, Rf = 0.38 in 10%
MeOH:CHCl3, m.p. 208-210 °C). Compounds 140b and 141b were unable to be
isolated as pure samples but were identified as the cis- and trans- isomers, respectively.
140b: 1H NMR δ 3.45 (dd, J 9.6, 0.6 Hz, 1H, CH-2`) and 3.05 (d, J 9.6 Hz, 1H, CH-3`).
141b: 1H NMR δ 3.65 (d, J 5.7 Hz, 1H, CH-2`) and 3.63 (d, J 5.1 Hz, 1H, CH-3`).
139b: MS (EI) m/z 308 (46%) [M.+], 262 (57%) [M+-OEt], 235 (96%) [M+-CO2Et];
HRMS (EI) Calcd for C18H16N2O3 [M.+] 308.1161. Found: 308.1158. 1H NMR (500
MHz) δ 9.40 (bs, 1H, NH); 8.57 (bs, 1H, ArCH-2``); 8.49 (bs, 1H, ArCH-4``); 7.66 (d, J
8.5 Hz, 1H, ArCH-6``); 7.46 (d, J 7.5 Hz, 1H, ArCH-4); 7.22 (t, J 7.7 Hz, 2H, ArCH-6
and ArCH-5``); 7.03 (t, J 7.5 Hz, 1H, ArCH-5); 6.82 (d, J 7.7 Hz, 1H, ArCH-7); 4.27-
4.15 (m, 2H, CH2); 3.73 (d, J 8.0 Hz, 1H, CH-2`); 3.34 (d, J 8.0 Hz, 1H, CH-3`); 1.26 (t,
J 7.7 Hz, 3H, CH3). 13C NMR (125 MHz) δ 173.7 (C-2); 167.9 (CO2Et); 150.4 (ArCH-
2``); 148.4 (ArCH-4``); 141.5 (ArC-7a); 136.6 (ArCH-6``); 128.8 (ArC-1``); 127.9
(ArCH-6); 125.9 (ArC-3a); 122.8 (ArCH-5``); 122.6 (ArCH-4); 122.1 (ArCH-5); 110.0
(ArCH-7); 61.7 (CH2CH3); 39.5 (C-3); 37.0 (CH-2`); 36.6 (CH-3`); 14.1 (CH3CH2).
The structure of 139b was confirmed by X-ray crystallography (see Appendix 1: X-ray
22: Compound 139b).

Chapter 8: Experimental for Chapter 4 183
8.4 Experimental for Chapter 4
C-Phenyl-N-methylnitrone (142a)
A solution of N-methylhydroxyamine-hydrochloric salt (1.902 g, 23
mmol) in anhydrous DCM (23 mL) was added to benzaldehyde (1.84
mL, 18 mmol). Solid NaHCO3 (4.7 g, 56 mmol), was added and
reaction mixture was heated at 80 °C for 18 h. The mixture was then
cooled, filtered and the solids were washed with DCM (10 mL). The filtrate was
removed in vacuo to yield yellow crystalline plates (2.423 g, 17.9 mmol, 99.7%, Rf =
0.42 in 10% MeOH:CHCl3) m.p. 70-72 °C (lit.350 m.p. 84-86 °C). MS (EI) m/z 135
(62%) [M.+], 134 (95%) [M+-1]. 1H NMR (500 MHz) δ 8.21-8.19 (m, 2H, ArCH-o);
7.38-7.35 (m, 4H, ArCH); 3.77 (s, 3H, CH3). 13C NMR (125 MHz) δ 130.0 (ArC-i);
129.7 (ArCH); 127.8 (ArCH), 127.7 (ArCH), 53.7 (CH3).
C-Phenyl-N-phenylnitrone (142b)
The title compound was prepared previously by Javad Safaei
(University of Wollongong) using the following method and
donated for my use. A solution of N-phenyl hydroxylamine (19.8 g,
0.18 mol) and benzaldehyde (18.3 g, 0.18 mol) in a minimal
amount of EtOH was allowed to stand at RT for 18 h. The
crystalline precipitate was filtered and recrystallised from benzene to afford 142b as off-
white fine needle-like crystals (32 g, 0.16 mol, 90%, Rf = 0.7 in 10% MeOH:CHCl3,
m.p. 114 °C (lit m.p.351 114-115 °C). 1H NMR δ 8.42-8.38 (m, 2H, ArCH-o); 7.92 (s,
1H, CH=); 7.79-7.76 (m, 2H, ArCH); 7.50-7.45 (m, 6H, ArCH). 13C NMR δ 149.1
(ArC-i); 134.6 (CH=); 130.9 (ArC-i); 130.6 (ArCH); 129.9 (ArCH); 129.1 (ArCH);
129.0 (ArCH-o); 128.6 (ArCH); 121.7 (ArCH).
3,4-Dihydro-2H-pyrrole 1-oxide (143)
The preparation of cyclic nitrone 143 was performed using the following
method. As 143 is known to dimerise readily, only a crude yield was recorded.
In a 3-necked round bottom flask fitted with a thermometer was added
Na2WO4·2H2O (0.264 g, 8.0 × 10-4 mol) and then the flask was flushed with N2. To this
was added H2O (4 mL) and pyrrolidine (1.67 mL, 20 mmol). The mixture was then
NO
NO Me
NO

Chapter 8: Experimental for Chapter 4 184
cooled to -5 °C (ice-salt bath) before the addition of 30% aqueous H2O2 solution (4.5
mL), over a period of 30 min maintaining the temperature below 20 °C. Upon
completion of addition, the cooling bath was removed and the mixture was stirred at RT
for 3 h. The mixture was then cooled again before the addition of NaHSO3 (300 mg),
with testing against starch-iodide paper to ensure removal of excess H2O2. NaCl (2.5 g)
was then added and the mixture was extracted with DCM (4 × 100 mL). The organic
extracts were combined, dried and the solvent then was removed in vacuo. The crude
mixture was purified by column chromatography using 10% EtOH: CHCl3 as the eluent
to yield 143 as a brown oil (0.366 g, 4.3 mmol, 21%). The pyrrolidine was immediately
diluted with anhydrous toluene (2 mL) and the solution flushed with N2 and used
immediately in subsequent cycloaddition reactions. Rf = 0.24 in 10% EtOH:CHCl3. 1H
NMR δ 6.90-6.89 (m, 1H, CH-2); 4.01-3.95 (m, 2H, CH2-5); 2.77-2.71 (m, 2H, CH2-3);
2.31-2.21 (m, 2H, CH2-4).
Methyl (3`R*, 5`R*)-2`-methyl-5`-(2-nitrophenyl)-3`-phenylisoxazolidine-5`-
carboxylate (170a) and Methyl (3`S*, 5`R*)-2`-methyl-5`-(2-nitrophenyl)-3`-
phenylisoxazolidine-5`-carboxylate (171a)
The title compounds, 170a and 171a were prepared using two methods. Method 1: To a
solution of 59 (110 mg, 5.3 × 10-4 mol) in anhydrous DCM (1 mL), contained within a
sealed tube was added nitrone 142a (85.5 mg, 6.3 × 10-4 mol). The tube was sealed and
the mixture was left stirring at 60 °C for 4 d. 1H NMR analysis of the crude reaction
mixture revealed the ratio of 170a : 171a : 59 was 1.0 : 0.57 : 0.14. The mixture was
purified by column chromatography using 30% EtOAc:PS as eluent to yield 170a as a
yellow oil (47.2 mg, 1.3 × 10-4 mol, 26%) and 171a as a yellow oil (36.8 mg, 1.1 × 10-4
mol, 20%) and a mixture of 59 and 171a. Method 2: A mixture of 59 (581.4 mg, 2.8
mmol) and nitrone 142a (379 mg, 2.8 mmol) was placed in a sealed glass microwave
reaction vessel. The mixture was subjected to microwave-assisted heating at 150 °C for
30 min. 1H NMR analysis of the crude reaction mixture revealed the ratio of 170a :
171a : 59 was 1.0 : 1.5 : 1.3. The mixture was purified by column chromatography
using 20% DCM:PS 1% MeOH as eluent to yield 170a as off-white clear crystals
(149.7 mg, 4.4 ×10-4 mol, 15%, Rf = 0.37 in 10% EtOAc:PS, m.p. 124-126 °C) and a
mixture of 171a and 59. The mixture was further purified by column chromatography

Chapter 8: Experimental for Chapter 4 185
using 20% EtOAc:PS as eluent to yield 171a as a yellow oil (285 mg, 8.3 × 10-4 mol,
30%, Rf = 0.16 in 10% EtOAc:PS) and recovered 59 (168 mg, 8.0 × 10-4 mol, 29%).
170a: MS (EI) m/z 342 (19%) [M.+], 296 (9%), 220 (11%),
134 (88%); 118 (5%), 104 (89%); HRMS (EI) Calcd for
C18H18N2O5 [M.+] 342.1216. Found: 342.1217. 1H NMR
(500 MHz) δ 8.22 (d, J 7.0 Hz, 1H, ArCH-6); 8.14 (d, J 7.5
Hz, 1H, ArCH-3); 7.74 (t, J 7.5 Hz, 1H, ArCH-5 ); 7.53 (t, J
7.7 Hz, 1H, ArCH-4); 7.49 (d, J 7.5 Hz, 2H, ArCH-o); 7.36 (t, J 7.3 Hz, 2H, ArCH-m);
7.32 (t, J 7.3 Hz, 1H, ArCH-p); 3.88 (bt, J 11.7 Hz, 1H, CHβCHα-4`); 3.75 (s, 3H,
CO2CH3); 3.54 (bs, 1H, CHα-3`); 2.73 (s, 3H, NCH3); 2.62 (dd, J 13.5, 7.0 Hz, 1H,
CHαCHβ-4`). 13C NMR (125 MHz) δ 169.6 (CO2); 146.3 (ArC-2); 137.3 (ArC-1); 136.9
(ArC-i); 133.8 (ArCH-5); 128.75 (ArCH-4); 128.72 (ArCH-m); 128.4 (ArCH-o); 128.2
(ArCH-p); 127.7 (ArCH-6); 125.3 (ArCH-3); 82.8 (C-5`); 73.4 (CH-3`); 53.0 (CO2CH3);
49.9 (CH2-4`); 43.0 (NCH3). The structure of 170a was confirmed by X-ray
crystallography (see Appendix 1: X-ray 23: Compound 170a)
171a: MS (EI) m/z 342 (13%) [M.+], 296 (4%), 220 (9%), 134
(89%); 118 (28%), 104 (72%); HRMS (EI) Calcd for
C18H18N2O5 [M.+] 342.1216. Found: 342.1220. 1H NMR (500
MHz) δ 8.29 (dd, J 8.0, 1.5 Hz, 1H, ArCH-6); 8.14 (dd, J 8.0,
1.5 Hz, 1H, ArCH-3); 7.77 (dt, J 8.0, 1.5 Hz, 1H, ArCH-5);
7.51 (dt, J 8.0, 1.5 Hz, 1H, ArCH-4); 7.30-7.23 (m, 5H,
ArCH-o, ArCH-m and ArCH-p); 3.97 (t, J 9.0, 7.5 Hz, 1H, CHβ-3`); 3.92 (dd, J 12.7,
6.5 Hz, 1H, CHβCHα-4`); 3.73 (s, 3H, CO2CH3); 2.72 (s, 3H, NCH3); 2.39 (dd, J 13.0,
9.5 Hz, 1H, CHαCHβ-4`). 13C NMR (125 MHz) δ 169.0 (CO2); 146.3 (ArC-2); 139.2
(ArC-1); 137.1 (ArC-i); 134.3 (ArCH-5); 128.7 (ArCH-o); 128.5 (ArCH); 128.3
(ArCH); 128.2 (ArCH); 127.7 (ArCH-m); 125.2 (ArCH-3); 85.5 (C-5`); 74.1 (CH-3`);
53.0 (CO2CH3); 51.8 (CH2-4`); 43.0 (NCH3).
1
2
5`3`
om
p
iN
O
CO2Me
Hββββ
Hαααα
HααααMe
NO2
1
2
5`3`
o
mp
iN
O
CO2Me
Hββββ
Hαααα
Me
NO2
Hββββ

Chapter 8: Experimental for Chapter 4 186
Methyl (3`R*, 5`R*)-5`-(2-nitrophenyl)-2`,3`-diphenylisoxazolidine-5`-carboxylate
(170b) and Methyl (3`S*, 5`R*)-5`-(2-nitrophenyl)-2`,3`-diphenylisoxazolidine-5`-
carboxylate (171b)
A mixture of 59 (133.7 mg, 6.5 × 10-4 mol) and nitrone 142b (164.7 mg, 8.4 × 10-4 mol)
was placed in a sealed glass microwave reaction vessel. The mixture was subjected to
microwave-assisted heating at 150 °C for 30 min. 1H NMR analysis of the crude
reaction mixture revealed the ratio of 170b : 171b : 59 was 3 : 1 : 0.8. The mixture was
purified by column chromatography using 0-10% EtOAc:PS as eluent to yield 170b as a
bright yellow oil (151.3 mg, 3.7 ×10-4 mol, 58%, Rf = 0.72 in 10% EtOAc:PS) and a
mixture of 171b and 59. The mixture was further purified using a chromatotron® (0-
2.5% EtOAc:PS) to yield 171b as a yellow oil (14.9 mg, 3.7 × 10-5 mol, 6%, Rf = 0.31
in 10% EtOAc:PS) and recovered 59 (15.3 mg, 7.4 × 10-5 mol, 11%) and a mixture of
171b and 59 (43.6 mg).
170b: MS (EI) m/z 404 (58%) [M.+], 345 (2%) [M+-CO2Me],
296 (7%), 220 (17%), 194 (21%), 180 (32%), 134 (26%), 104
(91%); HRMS (EI) Calcd for C23H20N2O5 [M.+] 404.1372.
Found: 404.1357. 1H NMR (500 MHz) δ 8.37 (dd, J 8.3, 1.3 Hz,
1H, ArCH-6); 8.14 (dd, J 7.7, 1.0 Hz, 1H, ArCH-3); 7.76 (dt, J
8.3, 1.3 Hz, 1H, ArCH-5); 7.52 (dt, 8.5, 1.5 Hz, 1H, ArCH-4);
7.31 (d, J 8.0 Hz, 2H, ArCH-o); 7.27 (t, J 7.0 Hz, 2H, ArCH-m); 7.24 (t, J 7.3 Hz, 1H,
ArCH-p); 7.21 (t, J 8.0 Hz, 2H, ArCH-m`); 7.05 (d, J 8.0 Hz, 2H, ArCH-o`); 7.01 (t, J
7.3 Hz, 1H, ArCH-p`); 4.83 (dd, J 9.5, 7.5 Hz, 1H, CHα-3`); 4.12 (dd, J 13.5, 7.5 Hz,
1H, CHβCHα-4`); 3.67 (s, 3H, CH3); 2.54 (dd, J 13.3, 9.3 Hz, 1H, CHαCHβ-4`). 13C
NMR (125 MHz) δ 168.6 (CO2Me); 149.3 (ArC-i`); 146.5 (ArC-2); 139.3 (ArC-i);
137.9 (ArC-1); 134.3 (ArCH-5); 128.6 (ArCH-m`); 128.84 (ArCH-m`); 128.82 (ArCH-
4); 128.1 (ArCH-6); 127.9 (ArCH-p); 127.0 (ArCH-o); 125.2 (ArCH-3); 123.8 (ArCH-
p`); 117.7 (ArCH-o`); 85.6 (C-5`); 71.0 (CH-3`); 52.9 (CH3); 51.9 (CH2-4`).
171b: MS (EI) m/z 404 (52%) [M.+], 345 (2%) [M+-CO2Me],
296 (10%), 220 (18%), 194 (22%), 180 (39%), 134 (26%), 104
(92%). HRMS (EI) Calcd for C23H20N2O5 [M.+] 404.1372.
Found: 404.1358. 1H NMR δ 8.15 (dd, J 8.1, 1.2 Hz, 1H,
ArCH-3); 8.12 (dd, J 7.8, 1.2 Hz, 1H, ArCH-6); 7.68 (dt, J 7.8,
1.2 Hz, 1H, ArCH-5); 7.53 (dt, J 8.1, 1.5 Hz, 1H, ArCH-4);
1 2 5 ` 3 `NO C O 2 M eH ββH ααN O 2 H ββ
1
2
5`3`
om
p
iN
O
CO2Me
Hββββ
Hαααα
Hαααα
NO2
i`o`
m`
p`

Chapter 8: Experimental for Chapter 4 187
7.51 (d, J 6.9 Hz, 2H, ArCH-o); 7.36 (t, J 7.1 Hz, 2H, ArCH-m); 7.32 (t, J 6.9 Hz, 1H,
ArCH-p); 7.20 (t, J 6.9 Hz, 2H, ArCH-m`); 7.02 (d, J 6.9 Hz, 2H, ArCH-o`); 7.02-6.98
(m, 1H, ArCH-p`); 4.37 (dd, J 9.3, 7.7 Hz, 1H, CHβ-3`); 3.93 (dd, J 13.2, 9.6 Hz, 1H,
CHβCHα-4`); 3.78 (s, 3H, CH3); 2.89 (dd, J 13.5, 7.5 Hz, 1H, CHαCHβ-4`). 13C NMR
(125 MHz) δ 169.1 (CO2Me); 148.5 (ArC-i`); 146.5 (ArC-2); 138.8 (ArC-i); 136.9
(ArC-1); 133.8 (ArCH-5); 128.95 (ArCH-4); 128.89 (ArCH-m); 128.5 (ArCH-m`);
128.0 (ArCH-p); 128.1 (ArCH-6); 127.6 (ArCH-o); 125.4 (ArCH-3); 123.5 (ArCH-p`);
118.0 (ArCH-o`); 84.2 (C-5`); 69.0 (CH-3`); 53.1 (CH3); 50.9 (CH2-4`).
Methyl (2`R*, 3a`S*)-2`-(2-nitrophenyl)hexahydropyrrolo[1,2-b]isoxazole-2`-
carboxylate (172) and Methyl (2`R*, 3a`R*)-2`-(2-
nitrophenyl)hexahydropyrrolo[1,2-b]isoxazole-2`-carboxylate (173)
To 59 (87.7 mg, 4.2 × 10-4 mol) in a sealed glass microwave reaction vessel was added a
solution of nitrone 143 (72 mg, 8.5 × 10-4 mol) in anhydrous toluene (0.4 mL). The
mixture was subjected to microwave-assisted heating at 150 °C for 30 min. 1H NMR
analysis of the crude reaction mixture revealed the ratio of 172 : 173 : 59 was 2.7 : 1 :
1.8. The crude mixture was purified by column chromatography using 20-100%
EtOAc:PS as eluent to yield 172 as a light-yellow crystalline solid (51.2 mg, 1.7 × 10-4
mol, 40%, Rf = 0.54 in 30% EtOAc:PS) and 173 as a light yellow semicrystalline oil
(7.3 mg, 2.5 × 10-5 mol, 6%, Rf = 0.22 in 30% EtOAc:PS) and recovered 59 (35.6 mg,
1.7 × 10-4 mol, 40%).
172: MS (EI) m/z 292 (33%) [M.+], 257 (34%), 244 (52%), 233
(85%), 104 (96%); HRMS (EI) Calcd for C14H16N2O5 [M.+],
292.1059. Found: 292.1051. 1H NMR δ 8.20 (dd, J 8.1, 1.8 Hz, 1H,
ArCH-6); 8.13 (dd, J 8.4, 1.5 Hz, ArCH-3); 7.71 (dt, J 7.2, 1.2 Hz,
1H, ArCH-5); 7.47 (dt, J 7.2, 1.5 Hz, 1H, ArCH-4); 3.66 (s, 3H, OCH3); 3.63-3.55 (m,
2H, CHACHB-6` and CHα-3a`); 3.50 (dd, J 13.2, 3.6 Hz, 1H, CHβCHα-3`); 3.05 (dt, J
13.5, 8.1 Hz, 1H, CHBCHA-6`); 2.58 (dd, J 13.2, 7.6 Hz, 1H, CHαCHβ-3`); 2.21-2.01 (m,
2H, CHACHB-4` and CHACHB-5`); 1.99-1.89 (m, 1H, CHBCHA-4`); 1.84-1.74 (m, 1H,
CHBCHA-5`). 13C NMR δ 168.9 (CO2); 146.1 (ArC-2); 139.2 (ArC-1); 134.2 (ArCH-5);
128.5 (ArCH-4); 128.4 (ArCH-6); 125.3 (ArCH-3); 87.5 (C-2`); 66.7 (CHα-3a`); 56.8
(CH2-6`); 53.0 (OCH3); 47.9 (CH2-3`); 29.9 (CH2-4`); 23.7 (CH2-5`). 1H NMR (C6D6) δ
8.47 (dd, J 7.8, 1.5 Hz, 1H, ArCH-6); 7.86 (dd, J 8.1, 1.5 Hz, ArCH-3); 7.13 (dt, J 7.3,
N O 2
N
O
C O 2 M e
1
2
3 a `4 `
6 `
2 `
H ααααH αααα
H ββββ

Chapter 8: Experimental for Chapter 4 188
1.5 Hz, 1H, ArCH-5); 6.75 (dt, J 7.2, 1.5 Hz, 1H, ArCH-4); 3.58 (dd, J 13.3, 3.4 Hz, 1H,
CHβCHα-3`); 3.54-3.48 (m, 1H, CHACHB-6`); 3.26 (s, 3H, CO2CH3); 3.13-3.25 (m, 1H,
CHα-3a`); 2.77 (dt, J 13.5, 7.9 Hz, 1H, CHBCHA-6`); 2.38 (dd, J 13.2, 7.5 Hz, 1H,
CHαCHβ-3`); 1.96-2.09 (m, 2H, CHACHB-4` and CHACHB-5`); 1.48-1.59 (m, 1H,
CHBCHA-4`); 1.35-1.45 (m, 1H, CHBCHA-5`). 13C NMR (C6D6) 169.1 (CO2); 147.0
(ArC-2); 140.1 (ArC-1); 133.7 (ArCH-5); 129.4 (ArCH-6); 128.0 (ArCH-4); 125.1
(ArCH-3); 87.9 (C-2`); 67.0 (CH-3a`); 57.0 (CH2-6`); 52.5 (OCH3); 48.5 (CH2-3`); 30.2
(CH2-4`); 24.1 (CH2-5`).
173: MS (EI) m/z 292 (12%) [M.+], 257 (18%), 244 (25%), 233
(39%), 104 (49%); HRMS (ESI+ve) Calcd for C14H17N2O5 [MH+]
293.1137. Found: 293.1130. 1H NMR (500 MHz) δ 8.06 (dd, J 8.3,
1.3 Hz, 1H, ArCH-3); 8.01 (dd, J 8.0, 1.0 Hz, 1H, ArCH-6), 7.67 (dt,
J 7.7 Hz, 1.3 Hz, 1H, ArCH-5); 7.50 (dt, J 8.5, 1.5 Hz, 1H, ArCH-4); 4.02-3.96 (m, 1H,
CHβ-3a`); 3.82 (dd, J 13.0, 8.0 Hz, 1H, CHβCHα-3`); 3.71 (s, 3H, OCH3); 3.54 (ddd, J
13.8, 7.5, 3.5 Hz, 1H, CHACHB-6`); 3.11 (dt, J 13.8, 8.0 Hz, 1H, CHBCHA-6`); 2.07 (dd,
J 13.0, 4.0 Hz, 1H, CHαCHβ-3`); 2.11-2.02 (m, 1H, CHACHB-5`); 1.94 (dt, J 13.0, 8.0
Hz, 1H, CHACHB-4`); 1.84-1.76 (m, 1H, CHBCHA-5`); 1.46 (ddt, J 13.0, 9.0, 4.5 Hz,
1H, CHBCHA-4`). 13C NMR δ 169.7 (CO2); 146.6 (ArC-2); 136.9 (ArC-1); 133.8
(ArCH-5); 128.7 (ArCH-4); 126.9 (ArCH-6); 125.1 (ArCH-3); 85.5 (C-2`); 66.9 (CHβ-
3a`); 56.9 (CH2-6`); 52.9 (OCH3); 46.0 (CH2-3`); 31.0 (CH2-4`); 24.1 (CH2-5`).
2`-Methyl-(3`S*, 5`R*)-3`-phenylspiro[indole-3,5`-isoxazolidin]-2(1H)-one (174a)
To a solution of 170a (54 mg, 1.6 × 10-4 mol) in EtOAc (1
mL) under an atmosphere of N2 was added 10% Pd/C (9
mg). The vessel was then flushed with H2 and left stirring
under an atmosphere of H2 (balloon) for 18 h. The crude
mixture was filtered through a bed of celite, washed with
EtOAc (3 × 50 mL) and the filtrate was evaporated in vacuo. The crude product was
purified by column chromatography using 30-50% EtOAc:PS as eluent to yield 174 as a
yellow oil (10.6 mg, 3.8 × 10-5 mol, 24%, Rf = 0.18 in 30% EtOAc:PS). MS (EI) m/z
280 (10%) [M.+], 263 (15%), 145 (37%) [M+-C6H5CHNCH3O], 134 (92%), 117 (42%);
HRMS (EI) Calcd for C17H16N2O2 [M.+] 280.1212. Found: 280.1206. 1H NMR (500
MHz) δ 8.79 (bs, 1H, NH); 7.57(d, J 7.0 Hz, 2H, ArCH-o); 7.44 (d, J 7.5 Hz, 1H,
5 `
3 `
o m
piN
O
H ββββ
H αααα
H ααααM e
NH
3 a
7 a
5
7
O2
1
2
3a`4̀
6`
2`
NO2
N
O
CO2Me
Hββββ
Hββββ
Hαααα

Chapter 8: Experimental for Chapter 4 189
ArCH-4); 7.39 (t, J 7.5 Hz, 2H, ArCH-m); 7.34(d, J 7.5 Hz, 1H, ArCH-p); 7.28 (t, J 7.7
Hz, 1H, ArCH-6); 7.10 (t, J 7.5 Hz, 1H, ArCH-5); 6.94 (d, J 8.0 Hz, 1H, ArCH-7); 3.85
(bs, 1H, CHα-3`); 3.01 (t, J 11.5 Hz, 1H, CHβCHα-4`); 2.77 (dd, J 12.7, 6.5 Hz, 1H,
CHαCHβ-4`); 2.71 (s, 3H, NCH3). 13C NMR (125 MHz) δ 179.4 (C-2); 141.1 (ArC-7a);
137.0 (ArC-i); 130.5 (ArC-3a); 130.1 (ArCH-6); 128.8 (ArCH-m); 128.5 (ArCH-o);
128.4 (ArCH-p); 124.3 (ArCH-4); 123.1 (ArCH-5); 110.5 (ArCH-7); 80.5 (C-3); 74.5
(CH-3`); 49.3 (CH2-4`); 43.7 (NCH3).
(2R*, 3R*)-3-Hydroxy-3-[2-(methylamino)-2-phenylethyl]-1,3-dihydro-2H-indol-2-
one (175a)
To a solution of 170a (78.4 mg, 2.3 × 10-4 mol) in glacial
AcOH (9.2 mL) was added activated Zn dust (150 g, 2.3
mmol). The mixture was sonicated for 1 h. The crude
mixture was then filtered through a bed of celite and
washed with EtOAc. The filtrate was washed with sat. Na2CO3 solution and then H2O,
then dried, filtered and evaporated in vacuo. The crude was purified by column
chromatography using 10-30% EtOAc:PS as eluent to yield 175a as a yellow oil (37.6
mg, 1.3 × 10-4 mol, 58%, Rf = 0.16 in 30% EtOAc:PS). MS (EI) m/z 282 (12%) [M.+],
206 (6%), 146 (13%), 134 (21%), 120 (91%), 104 (12%); HRMS (EI) Calcd for
C17H18N2O2 [M.+] 282.1368. Found: 282.1365. 1H NMR (500 MHz) δ 7.32 (t, J 7.3 Hz,
2H, ArCH-m); 7.30-7.26 (m, 1H, ArCH-p); 7.15-7.11 (m, 3H, ArCH-4 and ArCH-o);
7.08 (t, J 7.5 Hz, 1H ArCH-6); 6.71 (t, J 7.3 Hz, 1H ArCH-5); 6.67 (d, J 7.5 Hz, 1H
ArCH-7); 4.70 (dd, J 7.5, 6.0 Hz, 1H, CH); 2.98 (dd, J 14.0, 8.0 Hz, 1H, CHACHB);
2.76 (s, 3H, NHCH3); 2.48 (dd, J 14.2, 5.7 Hz, 1H, CHBCHA). 13C NMR (125 MHz) δ
175.7 (C-2); 145.4 (ArC-7a); 139.7 (ArC-i); 129.2 (ArCH-6); 128.9 (ArCH-m); 128.1
(ArCH–p); 126.8 (ArCH-o); 126.7 (ArC-3a and ArCH-4); 118.6 (ArCH-5);
118.4(ArCH-7); 79.4 (C-3); 61.8 (CH); 42.4 (CH2); 28.8 (NHCH3).
o m
p
iMeHN
HO
H
NH
3a
7a
5
7
O2

Chapter 8: Experimental for Chapter 4 190
(2R*, 3S*)-3-(2-Anilino-2-phenylethyl)-3-hydroxy-1,3-dihydro-2H-indol-2-one
(175b)
The title compound was prepared using two methods.
Method 1: The title compound was prepared from 170b
(61 mg, 1.5 × 10-4 mol) using a similar method to that
described for the preparation for 174a. The crude product
was purified by column chromatography using 20-40%
EtOAc:PS as eluent to yield 175b as a yellow oil (28.6 mg, 8.3 × 10-5 mol, 55%, Rf =
0.66 in 40% EtOAc:PS). Method 2: The title compound was prepared from 170b (72.7
mg, 1.8 × 10-4 mol) using a similar method to that described for the preparation for 175a.
After following the same workup procedure, the crude mixture was purified by column
chromatography using 10-20% EtOAc:PS as eluent to yield 175b as a cream solid (55.5
mg, 1.6 × 10-4 mol, 90%) and purified further by recrystallision to yield 175b as a cream
solid (29.8 mg, 8.7 × 10-5 mol, 48%). MS (EI) m/z 344 (15%) [M.+], 148 (11%), 196
(46%), 120 (43%), 182 (92%), 104 (16%). HRMS (EI) Calcd for C22H20N2O2 [M.+]
344.1525. Found: 344.1505. 1H NMR (500 MHz) δ 7.36 (d, J 8.5 Hz, 2H, ArCH-o`);
7.27-7.21 (m, 6H, ArCH-m`, ArCH-o, and ArCH-m); 7.20-7.15 (m, 2H, ArCH-p and
ArCH-6); 7.09 (t, J 7.5 Hz, 1H, ArCH-p`) 6.94 (d, J 7.5 Hz, 1H, ArCH-4); 6.77 (d, J 7.5
Hz, 1H, ArCH-7); 6.71 (t, J 7.5 Hz, 1H, ArCH-5); 4.97 (dd, J 9.7, 5.7 Hz, 1H, CHα);
4.66 (bs, 1H, NH); 3.34 (dd, J 13.0, 6.0 Hz, 1H, CHβCHα); 2.42 (dd, J 12.7, 9.7 Hz, 1H,
CHαCHβ). 13C NMR (125 MHz) δ 175.3 (C-2); 146.0 (ArC-7a); 139.1 (ArC-i); 136.8
(ArC-i`); 129.5 (ArCH-6); 128.8 (ArCH-m`); 128.7 (ArCH-m); 128.0 (ArCH-p); 127.0
(ArCH-o); 125.9 (ArCH-4); 125.8 (ArCH-p`); 124.4 (ArC-3a); 123.4 (ArCH-o`);
118.3(ArCH-5); 118.0(ArCH-7); 79.8 (C-3); 60.1 (CHPh); 43.5 (CH2).
2`-Methyl-(3`R*, 5`R*)-3`-phenylspiro[indole-3,5`-isoxazolidin]-2(1H)-one (176a)
The title compound was prepared from 171a (78 mg, 2.3 × 10-4
mol) using a similar method to that described above for the
preparation of 174a. However, the reaction was left for only 2 h.
The crude product was purified by column chromatography
using 30-50% EtOAc:PS to yield 176a as a yellow oil (34.8 mg,
1.2 × 10-4 mol, 54%, Rf = 0.16 in 30% EtOAc:PS). MS (EI) m/z
280 (13%) [M.+], 263 (22%), 145 (58%), 134 (94%), 117 (63%);
NO H ββH ααH ββNH3 a7 a5 7 O25 ` 3 `
2
o m
pii`o`
m`
p`
3a
7a
5
7
HNHO
H
NH
O

Chapter 8: Experimental for Chapter 4 191
HRMS (ESI+ve) Calcd for C17H17N2O2 [MH+] 281.1290. Found: 281.1293. 1H NMR
(500 MHz) δ 8.05 (bs, 1H, NH); 7.53 (d, J 7.5 Hz, 1H, ArCH-4); 7.50 (d, J 7.5 Hz, 2H,
ArCH-o); 7.38 (d, J 7.5 Hz, 2H, ArCH-m); 7.33 (t, J 7.3 Hz, 1H, ArCH-p); 7.25 (t, J 7.5
Hz, 1H, ArCH-6); 7.08 (t, J 7.3 Hz, 1H, ArCH-5); 6.85 (d, J 7.5 Hz, 1H, ArCH-7); 4.23
(bm, 1H, CHβ-3`); 3.01 (dd, J 13.0, 6.0 Hz, 1H, CHβCHα-4`); 2.79 (s, 3H, NCH3); 2.74-
2.70 (m, 1H, CHαCHβ-4`). 13C NMR δ 178.2 (C-2); 140.7 (ArC-7a); 137.7 (ArC-i);
130.7 (ArC-3a); 129.7 (ArCH-6); 128.8 (ArCH-m); 128.1 (ArCH-p); 127.6 (ArCH-o);
124.7 (ArCH-4); 123.3 (ArCH-5); 110.3 (ArCH-7); 81.8 (C-3); 72.9 (CH-3`); 49.7
(CH2-4`); 43.9 (NCH3).
(2R*, 3S*)-3-hydroxy-3-[2-(methylamino)-2-phenylethyl]-1,3-dihydro-2H-indol-2-
one (177a)
The title compound was prepared from 171a (49 mg, 1.43 ×
10-4 mol) using a similar method to that described above for
the synthesis of 175a. Compound 177a was obtained as a
yellow oil, which required no further purification (38 mg,
1.35 × 10-4 mol, 94%, Rf = 0.45 in 30% EtOAc:PS). MS (EI)
m/z 282 (31%) [M.+], 146 (18%), 134 (26%), 120 (89%), 104 (13%); HRMS (EI) Calcd
for C17H18N2O2 [M.+], 282.1368. Found: 282.1366. 1H NMR δ 7.39-7.34 (m, 3H,
ArCH-m and ArCH-p); 7.26 (dd, J 6.0, 2.1 Hz, 2H, ArCH-o); 7.13 (dt, J 8.1, 1.5 Hz, 1H,
ArCH-6); 6.89 (dd, J 8.4, 1.5 Hz, 1H, ArCH-4); 6.71 (t, 7.2 Hz, 2H, ArCH-5 and
ArCH-7); 4.27 (dd, J 9.0, 5.7 Hz, 1H, CH); 3.15 (dd, J 13.2, 6.0 Hz, 1H, CHACHB);
2.71 (s, 3H, NHCH3); 2.34 (dd, J 13.2, 9.3 Hz, 1H, CHBCHA). 13C NMR δ 176.1 (C-2);
146.0 (ArC-7a); 138.7 (ArC-i); 129.0 (ArCH-m); 128.5 (ArCH-p); 127.4 (ArCH-o);
129.2 (ArCH-6); 125.7 (ArCH-4); 124.8 (ArC-3a); 118.0 (ArCH-5); 117.8 (ArCH-7);
79.5 (C-3); 61.0 (CH); 43.5 (CH2); 28.3 (NHCH3). Crystals of 177a were sent for X-ray
crystallography, however analysis did not return before thesis submission.
3`-Methyl-(4`R*, 6`R*)-4`-phenyl-2`H-spiro[indole-3,6`-[1,3]oxazinane]-2,2`(1H)-
dione (178a)
To a solution of 175a (68.2 mg, 2.4 × 10-4 mol) in
anhydrous THF (2 mL) was added triphosgene (21.5 mg,
7.2 × 10-5 mol) and anhydrous NEt3 (0.07 mL, 4.8 × 10-4
3 a7 a5 7 2O NH O6 ` H ββH αα
3a
7a
5
7
2
o
mp
iHN
HO
Me
NH
O
H

Chapter 8: Experimental for Chapter 4 192
mol). The mixture was stirred under N2 for 7 d. The reaction mixture was diluted with
EtOAc and the solution was washed successively with H2O, sat. NaHCO3 solution and
brine and then dried and evaporated under reduced pressure. The crude product was
purified by column chromatography using 30-100% EtOAc:PS to yield 178a as a white
crystalline solid (45.9 mg, 1.5 × 10-4 mol, 61%, Rf = 0.25 in 50% EtOAc:PS, m.p. 200-
204 °C). MS (EI) m/z 308 (67%) [M.+], 309 (15%) [MH+], 251 (91%) [M+-NMeCO],
206 (84%), 146 (94%) [M+-CH2C6H5CHNCH3CO], 130 (80%) [M+-
CH2C6H5CHNCH3CO2], 118 (38%), 102 (43%); HRMS (EI) Calcd for C18H16N2O3
[M.+] 308.1161. Found: 308.1161. 1H NMR (500 MHz) δ 9.02 (bs, 1H, NH); 7.45 (t, J
7.3 Hz, 2H, ArCH-m); 7.39 (t, J 7.5 Hz, 1H, ArCH-p); 7.29-7.25 (m, 3H, ArCH-o and
ArCH-6); 7.09-7.05 (m, 2H, ArCH-4 and ArCH-5); 6.89 (d, J 8.5 Hz, 1H, ArCH-7);
4.88 (dd, J 7.5, 7.0 Hz, 1H, CHα-4`); 3.09 (dd, J 15.0, 7.0 Hz, 1H, CHβCHα-5`); 2.76 (s,
3H, NCH3); 2.39 (dd, J 14.5, 7.5 Hz, 1H, CHαCHβ-5`). 13C NMR (125 MHz) δ 169.9
(C-2); 150.8 (C-2`); 138.8 (ArC-i); 135.6 (ArC-7a); 130.0 (ArCH-6); 129.3 (ArCH-m);
128.7 (ArCH-p); 126.7 (ArCH-o); 124.4 (ArCH-4); 123.6 (ArCH-5); 118.4 (ArC-3a);
115.0 (ArCH-7); 86.1 (C-3); 60.8 (CH-4`); 43.6 (CH2-5`); 29.0 (NCH3). Crystals of
178a were sent for X-ray crystallography, however analysis did not return before thesis
submission.
3`-Phenyl-(4`R*, 6`R*)-4`-phenyl-2`H-spiro[indole-3,6`-[1,3]oxazinane]-2,2`(1H)-
dione (178b)
The title compound was prepared from 175b (20.9 mg, 6.1
× 10-5 mol) using a similar method to that described above
for the synthesis of 178a. However, the reaction was left
for only 2 d. The crude product was then purified by
column chromatography using 30-100% EtOAc:PS as
eluent to yield 178b as a white crystalline solid (15.4 mg,
4.2 × 10-5 mol, 68%, Rf = 0.73 in 10% MeOH:CHCl3, m.p. 256-258 °C). MS (EI) m/z
370 (21%) [M.+], 251 (65%) [M+-PhNCO], 206 (46%), 180 (30%), 146 (89%), 130
(53%), 103 (32%); HRMS (EI) Calcd for C23H18N2O3 [M.+] 370.1317. Found: 370.1319.
1H NMR (d6-DMSO, 500 MHz) δ 10.4 (s, 1H, NH); 7.50 (d, J 7.5 Hz, 1H, ArCH-4);
7.43 (d, J 8.0 Hz, 2H, ArCH-o`); 7.39 (d, J 7.5 Hz, 2H, ArCH-o); 7.33 (t, J 7.0 Hz, 1H,
ArCH-6); 7.29 (t, J 8.0 Hz, 2H, ArCH-m); 7.26 (t, J 8.0 Hz, 2H, ArCH-m`); 7.20 (t, J
2
o m
pi
N
O
H αααα
NH
i `
o `
m `p `
3 a
7 a
5
7
6 `
2 `4 `
O
O
H ββββ
H αααα

Chapter 8: Experimental for Chapter 4 193
7.3 Hz, 1H, ArCH-p); 7.09 (t, J 7.5 Hz, 1H, ArCH-5); 7.08 (t, J 7.5 Hz, 1H, ArCH-p`);
6.91 (d, J 7.5 Hz, 1H, ArCH-7); 5.83 (dd, J 7.5, 7.0 Hz, 1H, CHα-4`); 3.37-3.30 (m, 1H,
CHαCHβ-5`); 2.40 (dd, J 14.3, 7.3 Hz, 1H, CHαCHβ-5`). 13C NMR (dDMSO, 125 MHz)
δ 169.5 (C-2); 149.1 (C-2`); 140.2 (ArC-i); 136.6 (ArC-i`); 135.9 (ArC-7a); 130.0
(ArCH-6); 128.8 (ArCH-m); 128.6 (ArCH-m`); 127.9 (ArCH-p); 127.0 (ArCH-o); 125.9
(ArCH-p`); 123.8 (ArCH-4); 123.6 (ArCH-o`); 122.9 (ArCH-5); 119.7 (ArC-3a); 114.4
(ArCH-7); 84.2 (C-3); 58.8 (CH-4`); 42.5 (CH2-5`). Crystals of 178b were sent for X-
ray crystallography, however analysis did not return before thesis submission.
3`-Methyl-(4`S*, 6`R*)-4`-phenyl-2`H-spiro[indole-3,6`-[1,3]oxazinane]-2,2`(1H)-
dione (179a)
The title compound was prepared from 177a (41.8 mg, 1.5 ×
10-4 mol) using a similar method to that described above for
the synthesis of 178a. However, the reaction was left for only
2 d. The crude product was purified by column
chromatography using 30-100% EtOAc:PS as eluent to yield
179a as a white crystalline solid (23.5 mg, 7.6 × 10-5 mol, 51%, Rf = 0.39 in 50%
EtOAc:P, m.p. 244-248 °C). MS (EI) m/z 308 (43%) [M.+], 251 (72%), 206 (68%), 146
(94%), 130 (65%); HRMS (EI) Calcd for C18H16N2O3 [M.+] 308.1161. Found: 308.1166.
1H NMR (500 MHz, (CD3)2CO) δ 9.27 (bs, 1H, NH); 7.49-7.48 (m, 4H, ArCH-o and
ArCH-m); 7.43-7.40 (m, 1H, ArCH-p); 7.35 (d, J 7.5 Hz, 1H, ArCH-4); 7.32 (dt, J 7.8,
1.2 Hz, 1H, ArCH-6); 7.07 (dt, J 7.5, 1.0 Hz, 1H, ArCH-5); 7.03 (d, J 8.0 Hz, 1H,
ArCH-7); 4.96 (dd, J 7.5, 7.0 Hz, 1H, CHβ-4`); 3.27 (dd, J 14.5, 7.5 Hz, 1H, CHβCHα-
5`); 2.64 (s, 3H, NCH3); 2.47 (dd, J 15.0, 7.5 Hz, 1H, CHαCHβ-5`). 13C NMR
((CD3)2CO) δ 171.0 (C-2); 150.2 (C-2`); 141.0 (ArC-i); 137.4 (ArC-7a); 130.6 (ArCH-
6); 129.9 (ArCH-m), 129.3 (ArCH-p); 128.1 (ArCH-o); 124.5 (ArCH-4); 123.6 (ArCH-
5); 121.7 (ArC-3a); 115.1 (ArCH-7); 85.0 (C-3); 61.4 (CH-4`); 43.9 (CH2-5`); 28.6
(NCH3). Crystals of 179a were sent for X-ray crystallography, however analysis did not
return before thesis submission.
3a
7a
5
7
2
o
m
p
iMeN
O
NH
O
Hββββ
O
6`
2`4`
Hββββ
Hαααα

Chapter 8: Experimental for Chapter 4 194
(3R*, 3R*)-3-Hydroxy-3-(pyrrolidin-2-ylmethyl)indol-2-one (180) and (3R*,
4a`R*)-2`H-spiro[indoline-3,3`-pyrrolo[1,2-c][1,3`]oxazine]-1`,2(1H)-dione (181)
To a solution of 173 (41.3 mg, 1.4 × 10-4 mol) in anhydrous MeOH (2 mL) under an
atmosphere of N2 was added PdCl2 (5.2 mg, 2.8 × 10-5 mol) and the vessel flushed with
H2 and left stirring for 3 h under a H2 atmosphere (balloon). The crude mixture was then
filtered through a bed of celite and the solid was washed with MeOH (10 mL). The
solvent was evaporated in vacuo. The crude product was purified by column
chromatography using 50-100% EtOAc:PS as eluent, to yield material (180) that was
impossible to analyse by NMR analysis due to the broadening of all peaks, perhaps due
to traces of palladium. To a solution of this material (28 mg, 1.2 × 10-4 mol) in
anhydrous THF (1 mL) was added triphosgene (10.7 mg, 3.6 × 10-5 mol) and anhydrous
NEt3 (0.03 mL, 2.4 × 10-4 mol). The mixture was stirred under N2 for 2 d. The crude
was then washed with H2O and extracted with EtOAc. The organic extracts were then
successively washed with sat. NaHCO3 solution and brine, dried and evaporated under
reduced pressure. The crude product was purified by
chromatotron® (0-4% MeOH:CHCl3) to yield 181 as a brown
semicrystalline oil (9.2 mg, 3.6 × 10-5 mol, 25% over 2 steps).
180: MS (EI) m/z 232 (10%) [M.+], 214 (6%) [M+-H2O], 149
(22%), 120 (34%), 86 (37%), 70 (77%), 43 (96%); HRMS (EI)
Calcd for C13H16N2O2 [M.+] 232.1211. Found: 232.1206.
181: MS (EI) m/z 258 (70%) [M.+], 259 (19%) [MH+], 214
(32%), 186 (24%), 174 (94%), 146 (94%), 133 (50%), 117
(35%), 104 (29%). HRMS (EI) Calcd for C14H14N2O3 [M.+]
258.1004. Found: 258.0997. 1H NMR δ 7.34-7.29 (m, 2H,
ArCH-6 and ArCH-4); 7.10 (dt, J 7.5, 0.9 Hz, 1H, ArCH-5);
6.91 (dd, J 8.1, 1.2 Hz, 1H, ArCH-7); 4.11-4.01 (m, 1H, CHβ-4a`); 3.63-3.52 (m, 1H,
CHACHB-7`); 3.20-3.12 (m, 1H, CHBCHA-7`); 3.00 (dd, J 13.3, 6.1 Hz, 1H, CHβCHα-
4`); 2.36 (dd, J 13.2, 7.8.0 Hz, 1H CHαCHβ-4`); 2.26-2.09 (m, 3H, CHACHB-5` and
CH2-6`); 1.53-1.47 (m, 1H, CHBCHA-5`). 13C NMR δ 171.5 (C-2); 151.6 (C-1`); 136.6
(ArC-7a); 121.5 (ArC-3a); 90.6 (C-3`); 131.2 (ArCH-6); 124.3 (ArCH-4); 124.7
(ArCH-5); 115.6 (ArCH-7); 59.5 (CHβ-4a`); 42.5 (CH2-7`); 43.1 (CH2-4`); 33.5 (CH2-
5`); 27.1 (CH2-6`).
NH
N
OH ββββ
O
O
3
3 a5
7 7 a
3 `
4 a `5`
7 `
H ββββH αααα
2
NH
HN
HO H
O3
3a5
77a

Chapter 8: Experimental for Chapter 5 195
8.5 Experimental for Chapter 5
1-(2-Chloroethyl)-2-nitrobenzene (184a) and 1-(2-chloroethyl)-4-nitrobenzene
(184b) Warning: the nitration part of this experiment was performed behind a
safety screen.
To HNO3 (68% in H2O, 7.25 mL, 110 mmol) previously cooled to < 10 ºC (ice-salt bath)
was added conc. H2SO4 (97% in H2O, 8.5 mL, 153 mmol) dropwise. Maintaining the
temperature between 0-10 ºC, the nitrating mixture was then added dropwise to 2-
phenylethyl chloride (9.35 mL, 70.4 mmol). The temperature was allowed to warm to
RT and then left to stir for 2 h before carefully pouring the mixture into ice-water (~
400 mL). The aqueous phase was then extracted with DCM (3 × 100 mL) and the
combined organic extracts were washed with H2O (~100 mL) followed by sat. NaHCO3
solution (~100 mL) and H2O (~100 mL). The organic extracts were subsequently dried
(Na2SO4) and concentrated in vacuo to yield a yellow oil. 1H NMR analysis of the crude
reaction revealed a 1 : 2.2 mixture of isomers 184a and 184b, respectively. The crude
reaction mixture was purified by column chromatography using 5% Et2O:PS as eluent,
to yield 184a as a yellow oil (3.40 g, 18 mmol, 26%, Rf = 0.4 in 5% Et2O:PS) and 184b
as pale yellow crystals (7.45 g, 40 mmol, 57%, Rf = 0.27 in 5% Et2O:PS).
184a: UV λmax 226, 255.5 nm (EtOH); IR (film) 2959, 2932, 2861,
1609, 1521, 1439, 1343, 1200, 1161, 951, 855, 785, 722, 699, 660
cm-1; MS (EI) m/z 185 (36%) [M.+]. HRMS (EI) Calcd for
C8H8NO2Cl [M.+] 185.0243. Found: 185.0239. 1H NMR δ 7.99 (d, J 9.0 Hz, 1H, ArCH-
3); 7.58 (t, J 7.5 Hz, ArCH-5); 7.44 (t, J 6.0 Hz, 1H, ArCH-4); 7.43 (d, J 9.0 Hz, 1H,
ArCH-6); 3.84 (t, J 7.5 Hz, 2H, CH2CH2Cl); 3.36 (t, J 7.5 Hz, 2H, CH2CH2Cl). 13C
NMR δ 133.6 (ArC-2); 133.5 (ArC-1); 133.3 (ArCH-3); 128.6 (ArCH-5 and ArCH-6),
125.5 (ArCH-4); 44.2 (CH2CH2Cl); 36.7 (CH2CH2Cl). HRMS (EI) m/z 185.0243 [M.+],
calc. 185.0244; MS (EI) m/z 185 (8%) [M.+], 168 (100%), 140 (9%), 136 (30%), 92
(88%).
184b: UV λmax 214, 269 nm (EtOH); IR (powdered solid) 3111,
3083, 2955, 2848, 1597, 1506, 1453, 1339, 1255, 1101, 1032,
854, 812, 750, 719, 683 cm-1; MS (EI) m/z 185 (40%) [M.+], 187 (16%) [M++2], 167
(20%), 149 (59%), 136 (94%). HRMS (EI) Calcd for C8H8NO2Cl [M.+], 185.0243.
Found: 185.0243. 1H NMR δ 8.18 (d, J 9.0 Hz, 2H, ArCH-3 and ArCH-5); 7.40 (d, J
NO2
Cl1
2
C l1

Chapter 8: Experimental for Chapter 5 196
9.0 Hz, 2H, ArCH-2 and ArCH-6); 3.78 (t, J 6.0 Hz, 2H, CH2CH2Cl); 3.18 (t, J 6.0 Hz,
2H, CH2CH2Cl). 13C NMR δ 145.5 (ArC-1 and ArC-4); 129.7 (ArCH-2 and ArCH-6);
123.7 (ArCH-3 and ArCH-5); 43.7 (CH2CH2Cl); 38.6 (CH2CH2Cl).
2-(2-Chloroethyl)aniline hydrochloride (185)
To a solution of 184a (2.571 g) in anhydrous MeOH (40.2 mL) and
conc. HCl (37%) (2.41 mL) at –78 ºC (dry ice-acetone bath) was
added 10% Pd/C (200.8 mg). The mixture was allowed to warm to RT before the
introduction of H2 and was left to stir under an atmosphere of H2 (balloon) for 18 h. The
reaction mixture was then filtered through a bed of celite and washed with MeOH (3 ×
100 mL). The solvent was removed in vacuo to furnish 185 as a pink solid (2.632 g,
13.7 mmol, 99%, Rf = 0.76 in 30% EtOAc:PS, m.p. 173-175 ºC). UV λmax 235, 288.5
nm (EtOH); IR (powdered solid) 2920 (NH), 2851 (NH), 2776 (NH), 1588, 1564, 1481,
1420, 1358, 1294, 1269, 1188, 1155, 1124, 1083, 1042, 995, 905, 814, 792, 757, 646
cm-1; 1H NMR δ 7.46-7.41 (m, 2H, ArCH); 7.38-7.30 (m, 2H, ArCH); 3.92 (t, J 7.5 Hz,
2H, CH2CH2Cl); 3.68 (bs, 2H, NH2); 3.16 (t, J 7.5 Hz, CH2CH2Cl). 13C NMR δ 131.8
(ArC); 131.6 (ArC); 128.4 (ArCH); 127.8 (ArCH); 123.7 (ArCH); 43.9 (CH2CH2Cl);
33.4 (CH2CH2Cl).
N-[2-(2-Chloroethyl)phenyl]acetamide (186)
To a solution of 185 (3.648g, 19 mmol) in acetic anhydride (109.4
mL) was added glacial acetic acid (61 mL). This mixture was left to
stir at RT for 5 h, and then poured into ice-water (300 mL). The aqueous phase was then
extracted with DCM (3 × 100 mL). The combined organic layers were then washed with
5% aqueous HCl (100 mL) followed by sat. NaHCO3 solution (100 mL), dried and
concentrated in vacuo to give 186 as an off-white solid (3.565 g, 18 mmol, 95%, Rf =
0.3 in 50% EtOAc:PS, m.p. 119-121 ºC). IR (powdered solid) 3278 (NHCO), 2958 (C-
H), 1645, 1586, 1528, 1437, 1371, 1290, 1153, 1096, 1042, 1015, 972, 903, 858, 748,
708, 652, 602 cm-1; 1H NMR δ 7.57 (d, J 9.0 Hz, 1H, ArCH-6); 7.38 (bs, 1H, NH);
7.31-7.19 (m, 3H, ArCH); 3.76 (t, J 7.5 Hz, CH2CH2Cl); 3.06 (t, J 7.5 Hz, CH2CH2Cl);
2.21 (s, 3H, CH3). 13C NMR δ 168.9 (CO); 135.8 (ArC-1); 132.1 (ArC-2); 130.4
(ArCH); 128.2 (ArCH); 126.8 (ArCH); 126.3 (ArCH); 45.6 (CH2CH2Cl); 34.9
(CH2CH2Cl); 24.5 (CH3).
NHAc
Cl
1
2
1
2
NH2 .HCl
Cl

Chapter 8: Experimental for Chapter 5 197
N-[2-(2-Chloroethyl)-4-nitrophenyl]acetamide (187a) and N-[2-(2-Chloroethyl)-6-
nitrophenyl]acetamide (187b)
Warning: the nitration part of this experiment was performed behind a safety
screen. To a solution of 186 (505 mg, 2.55 mmol) in glacial acetic acid (1.1 mL, 19
mmol), initially stirred at RT until completely dissolved then placed in an ice-salt bath
was added fuming HNO3 (>90% in H2O, 2.0 mL, 42.5 mmol) very slowly dropwise,
whilst maintaining the temperature at 0 ºC (alternating between ice-salt bath and dry
ice-EtOAc bath). The resulting mixture was allowed to warm to RT and was left to stir
for 15 h before careful pouring into ice-water (100 mL). The aqueous phase was
extracted with DCM (3 × 100 mL), dried (Na2SO4) and concentrated in vacuo to give a
cream powder (456 mg, 1.56 mmol). A 1H NMR analysis of the crude product revealed
a 1 : 1 mixture of isomers 187a and 187b, respectively. This mixture was purified by
recrystallisation from MeOH to yield 187a as fine feathery white needle-like crystals
(163.2 mg, 6.73 × 10-4 mol, 26%, m.p. 190 ºC, Rf = 0.27 in 50% EtOAc:PS) and further
purified by column chromatography using 50-100% EtOAc:PS as eluent.
187a: UV λmax 298.0 nm (MeOH); IR (powdered solid) 3276
(NH), 1667, 1581, 1348, 1013, 830, 697, 669 cm-1; MS (EI)
m/z 242 (10%) [M.+], 207 (75%), 164 (13%), 151 (94%), 134
(18%), 117 (37%), 105 (16%). HRMS (EI) Calcd for C10H11N2O3Cl [M.+] 242.0458.
Found: 244.0451. Calcd for C10H11N2O337Cl [M++2] 244.0429. Found: 244.0429. 1H
NMR (CD3OD) δ 8.22 (d, J 2.7 Hz, 1H, ArCH-3); 8.12 (dd, J 9.0, 2.7 Hz, 1H, ArCH-5);
7.78 (d, J 9.0 Hz, 1H, ArCH-6); 3.80 (t, J 7.5 Hz, 2H, CH2CH2Cl); 3.20 (t, J 7.5 Hz, 2H,
CH2CH2Cl); 2.23 (s, 3H, CH3). 1H NMR δ 2.00 (s, 3H, CH3), 2.90 (t, J = 6 Hz, 2H,
CH2CH2Cl), 3.61 (t, J = 6 Hz, 2H, CH2CH2Cl), 7.40 (bs, 1H, NH); 8.16 (d, J = 12 Hz,
3H, ArH); 13C NMR δ 168.2 (NHCOCH3); 144.6 (ArC-4); 141.9 (ArC-1); 129.9 (ArC-
2), 125.3 (ArCH-3); 123.8 (ArCH-6); 123.5 (ArCH-5); 44.3 (CH2CH2Cl); 34.4
(CH2CH2Cl); 24.4 (CH3). The structure of 187a was confirmed by X-ray
crystallography (see Appendix 1: X-ray 24: Compound 187a)
187b: cream powder/ crystal (Rf = 0.48 in 50% EtOAc:PS, m.p.
171 ºC). UV λmax 205 nm (MeOH); IR (powdered solid) 3279
(NH), 1663, 1583, 1505, 1443, 1347, 1271, 885, 806, 784, 735,
680 cm-1; 1H NMR δ 8.22 (bs, 1H, NH); 7.93 (dd, J 9.0, 3.0 Hz, ArCH-6); 7.61 (dd, J
9.0, 3.0 Hz, 1H, ArCH-3); 7.41 (t, J 9.0 Hz, 1H, ArCH-4); 3.83 (dd, J 15.0, 9.0 Hz, 2H,
NHAc
Cl2
1
4O2N
C l24

Chapter 8: Experimental for Chapter 5 198
CH2CH2Cl); 3.14 (dd, J 13.5, 7.5 Hz, 2H, CH2CH2Cl); 2.25 (s, 1H, CH3). 13C NMR δ
169.0 (NHCOCH3); 139.0 (ArC-6); 135.1 (ArCH-3); 130.0 (ArC-2); 127.1 (ArCH-4);
123.9 (ArCH-5); 119.5 (ArC-1); 43.5 (CH2CH2Cl); 35.0 (CH2CH2Cl); 23.6 (CH3).
2-(2-Chloroethyl)-4-nitroaniline hydrochloride (188)
To a solution of EtOH : HCl (2 : 1), (99 mL) was added 187a
(320.9 mg, 1.3 mmol) and the resulting solution was stirred and
heated at reflux (110 ºC) for 15 h. The EtOH was removed in vacuo and TLC performed
to reveal only a base line spot, Rf = 0 in 50% EtOAc:PS), the solvents were then slowly
evaporated to yield 188 as a yellow/brown crystal.
188: 1H NMR δ 8.00 (dd, J 9.0, 1.5 Hz, 1H, ArCH-5); 7.96-7.95 (m, 1H, ArCH-3); 6.49
(d, J 9.0 Hz, 1H, ArCH-6); 5.91 (bs, 2H, NH2); 3.78 (t, J 9.0 Hz, 2H, CH2CH2Cl); 3.15
(t, J 9.0 Hz, 2H, CH2CH2Cl). 13C NMR δ 157.0 (ArC-1); 140.0 (ArC-4); 129.7 (ArC-2);
125.9 (ArCH-3); 121.0 (ArCH-5); 107.3 (ArCH-6); 47.3 (CH2CH2Cl); 28.5
(CH2CH2Cl).
N-(1-Methyl-1-phenylethyl)-4-nitrobenzenesulfonamide (204)
The title compound was prepared using two methods.
Method 1: To a solution of p-nitrobenzenesulfonyl
chloride (51.2 mg, 2.3 × 10-4 mol) in anhydrous EtOH
(1 mL) was added in succession, cumylamine (0.04
mL, 2.8 × 10-4 mol) and anhydrous NEt3 (0.05 mL,
2.8 × 10-4 mol). The mixture was left stirring at RT for 15 h and under an atmosphere of
N2. The mixture was then diluted with DCM and washed successively with H2O and 5%
HCl solution. The organic extracts were dried (Na2SO4) and the solvent was removed in
vacuo. The crude product was purified by column chromatography using 30%
EtOAc:PS as eluent to yield 204 as cream crystals (41.7 mg, 1.3 × 10-4 mol, 56%, Rf =
0.51 in 30% EtOAc:PS, m.p. 138.8 ºC). Method 2: To a solution of cumylamine (0.03
mL, 2.2 × 10-4 mol) in aqueous 10% NaOH (5 mL) was added p-nitrobenzenesulfonyl
chloride (50 mg, 2.2 × 10-4 mol) at a rate so that the reaction temperature did not exceed
40 °C (placed in an ice-bath). After addition was complete, the mixture was allowed to
stir at RT for 2 h. The mixture was then diluted with DCM and washed successively
with H2O and 5% HCl solution. The organic extracts were dried (Na2SO4) to yield 204
1
4O2N
S
O
ONH
i`
o`m`
p`
NH2
ClO2N
.HCl1
24

Chapter 8: Experimental for Chapter 5 199
as cream crystals (45.4 mg, 1.4 × 10-4 mol, 64%). UV λmax 206, 269 nm (EtOH); IR
(powdered solid) 3319, 1521, 1312, 1144, 981, 849, 773, 705 cm-1; 1H NMR δ 8.04 (d,
J 9.0 Hz, 2H, ArCH-3 and ArCH-5); 7.60 (d, J 9.0 Hz, 2H, ArCH-2 and ArCH-6); 7.13
(d, J 9.0 Hz, 2H, ArCH-m`); 7.06 (t, J 6.0 Hz, 1H, ArCH-p`); 7.04 (d, J 9.0 Hz, 2H,
ArCH-o`); 5.23 (bs, 1H, NH), 1.63 (s, 6H, (CH3)2). 13C NMR (125 MHz) δ 149.4 (ArC-
4), 147.9 (ArC-1), 143.3 (ArC-i`), 128.2 (ArCH-2 and ArCH-6), 128.1 (ArCH-m`),
127.5 (ArCH-p`), 125.8 (ArCH-o`), 123.7 (ArCH-3 and ArCH-5), 58.9 (C(CH3)2), 30.0
(C(CH3)2).
4-Amino-N-(1-methyl-1-phenylethyl)benzenesulfonamide (205)
To a stirred suspension of 204 (766 mg, 2.08 mmol) in
anhydrous MeOH (26 mL) was added 10% Pd/C (115
mg) and ammonium formate (689.5 mg, 10.9 mmol).
The reaction was left stirring at RT for 18 h and under
an atmosphere of N2. Upon finding the reaction to be
incomplete additional ammonium formate (222.5 mg, 3.5 mmol) and 10% Pd/C (76.6
mg) was added. The reaction was then left to stir at RT for 2.5 d. The mixture was
filtered through a bed of celite and the precipitate washed with MeOH. The solvent
from the filtrate was removed in vacuo and the residue taken up in EtOAc and washed
successively with H2O and brine. The washings were back extracted with EtOAc. The
extracts were dried (Na2SO4) and the solvent removed in vacuo. The crude product was
purified via recrystallisation from EtOH and also by column chromatography using 5%
MeOH:DCM as eluent to yield greyish-pink, needle-like crystals (556.6 mg, 2.02 mmol,
97%, Rf = 0.16 in 30% EtOAc:PS, m.p. 194.5 ºC). UV λmax 210, 266 nm (EtOH); IR
(powdered solid) 3443, 3364, 3267, 1593, 1499, 1428, 1286, 1133, 1091, 1029, 1001,
918, 824, 762, 676 cm-1; MS (EI) m/z 290 (19%) [M.+], 291 (4%) [MH+], 275 (65%),
172 (14%), 156 (95%), 118 (44%), 108 (35%). HRMS (EI) Calcd for C15H18N2O2S [M.+]
290.1089. Found: 290.1089. 1H NMR δ 7.47 (d, J 6.0 Hz, 2H, ArCH-2 and ArCH-6);
7.33 (dd, J 6.0 Hz, 2H, ArCH-m`); 7.24 (t, J 7.5 Hz, 1H, ArCH-p`); 7.22 (d, J 6.0 Hz,
2H, ArCH-o`); 6.56 (d, J 9.0 Hz, 2H, ArCH-3 and ArCH-5); 4.71 (bs, 1H, NH); 4.04
(bs, 2H, NH2); 1.58 (s, 6H, (CH3)2). 13C NMR δ 168.0 (ArC-4); 146.2 (ArC-i`); 132.0
(ArC-1); 129.2 (ArCH-m`); 128.3 (ArCH-2 and ArCH-6); 127.0 (ArCH-p`); 125.5
(ArCH-o`); 113.9 (ArCH-3 and ArCH-5); 58.7 (C(CH3)2); 29.8 (C(CH3)2).
1
4H2N
S
O
ONH
i`
o`m`
p`

Chapter 9: References 200
CHAPTER 9: REFERENCES
1. Alberts, B.; Bray, D.; Lewis, J.; Raff, M.; Roberts, K.; Watson, J. D., Molecular
Biology of the Cell. 3rd ed.; Garland Publishing: New York, 1994.
2. Hartwell, L. H.; Kastan, M. B., Cell cycle control and cancer. Science 1994, 266,
1821-1828.
3. Hartwell, L. H.; Weinert, T. A., Checkpoints: controls that ensure the order of
cell cycle events. Science 1989, 246, 629-634.
4. Hall, M.; Peters, G., Genetic alterations of cyclins, cyclin-dependent kinases,
and CDK inhibitors in human cancer. Adv. Cancer Res. 1996, 68, 67-108.
5. Brooks, G.; La Thangue, N. B., The cell cycle and drug discovery: the promise
and the hope. Drug Discovery Today 1999, 4, 455-464.
6. Webster, K. R., Therapeutic Potential of Targeting the Cell Cycle. Chem. Res.
Toxicol. 2000, 13, 940-943.
7. Dworakowska, D.; Jassem, E.; Jassem, J.; Peters, B.; Dziadziuszko, R.; Zylicz,
M.; Jakobkiewicz-Banecka, J.; Kobierska-Gulida, G.; Szymanowska, A.; Skokowski, J.;
Roessner, A.; Schneider-Stock, R., MDM2 gene amplification: a new independent
factor of adverse prognosis in non-small cell lung cancer (NSCLC). Lung Cancer 2004,
43, 285-295.
8. Momand, J.; Jung, D.; Wilczynski, S.; Niland, J., The MDM2 gene
amplification database. Nucleic Acids Res. 1998, 26, 3453-3459.
9. Toogood, P. L., Cyclin-dependent kinase inhibitors for treating cancer. Med. Res.
Rev. 2001, 21, 487-498.
10. Sielecki, T. M.; Boylan, J. F.; Benfield, P. A.; Trainor, G. L., Cyclin-dependent
kinase inhibitors: useful targets in cell cycle regulation. J. Med. Chem. 2000, 43, 1-18.
11. Harper, J. W.; Adams, P. D., Cyclin-Dependent Kinases. Chem. Rev. 2001, 101,
2511-2526.
12. Connell-Crowley, L.; Solomon, M. J.; Wei, N.; Harper, J. W., Phosphorylation
independent activation of human cyclin-dependent kinase 2 by cyclin A in vitro. Mol.
Biol. Cell 1993, 4, 79-92.
13. Pavletich, N. P., Mechanisms of Cyclin-dependent Kinase Regulation:
Structures of CDKs, their Cyclin Activators, and Cip and INK4 Inhibitors. J. Mol. Biol.
1999, 287, 821-828.

Chapter 9: References 201
14. Russo, A. A.; Jeffrey, P. D.; Patten, A. K.; Massague, J.; Pavletich, N. P.,
Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin
A-CDK2 complex. Nature 1996, 382, 325-331.
15. Senderowicz, A. M.; Sausville, E. A., Preclinical and clinical development of
cyclin-dependent kinase modulators. J. Natl. Cancer Inst. 2000, 92, 376-387.
16. Knockaert, M.; Greengard, P.; Meijer, L., Pharmacological inhibitors of cyclin-
dependent kinases. Trends Pharmacol. Sci. 2002, 23, 417-425.
17. Fischer, P. M.; Lane, D. P., Inhibitors of cyclin-dependent kinases as anti-cancer
therapeutics. Curr. Med. Chem. 2000, 7, 1213-1245.
18. Maccioni, R. B.; Otth, C.; Concha, I. I.; Munoz, J. P., The protein kinase Cdk5:
structural aspects, roles in neurogenesis and involvement in Alzheimer's pathology. Eur.
J. Biochem. 2001, 268, 1518-1527.
19. Smith, D. S.; Tsai, L.-H., Cdk5 behind the wheel: a role in trafficking and
transport? Trends Cell Biol. 2002, 12, 28-36.
20. Wang, D.; De la Fuente, C.; Deng, L.; Wang, L.; Zilberman, I.; Eadie, C.;
Healey, M.; Stein, D.; Denny, T.; Harrison, L. E.; Meijer, L.; Kashanchi, F., Inhibition
of human immunodeficiency virus type 1 transcription by chemical cyclin-dependent
kinase inhibitors. J. Virol. 2001, 75, 7266-7279.
21. Chao, S.-H.; Fujinaga, K.; Marion, J. E.; Taube, R.; Sausville, E. A.;
Senderowicz, A. M.; Peterlin, B. M.; Price, D. H., Flavopiridol inhibits P-TEFb and
blocks HIV-1 replication. J. Biol. Chem. 2000, 275, 28345-28348.
22. Chao, S.-H.; Price, D. H., Flavopiridol inactivates P-TEFb and blocks most
RNA polymerase II transcription in vivo. J. Biol. Chem. 2001, 276, 31793-31799.
23. Sherr, C. J., Cancer cell cycles. Science 1996, 274, 1672-1677.
24. Morisaki, H.; Ando, A.; Nagata, Y.; Pereira-Smith, O.; Smith, J. R.; Ikeda, K.;
Nakanishi, M., Complex Mechanisms Underlying Impaired Activation of Cdk4 and
CDK2 in Replicative Senescence: Roles of p16, p21, and Cyclin D1. Exp. Cell Res.
1999, 253, 503-510.
25. Russo, A. A.; Tong, L.; Lee, J.-O.; Jeffrey, P. D.; Pavletich, N. P., Structural
basis for inhibition of the cyclin-dependent kinase CDK6 by the tumor suppressor
p16INK4a. Nature 1998, 395, 237-243.
26. De Bondt, H. L.; Rosenblatt, J.; Jancarik, J.; Jones, H. D.; Morgan, D. O.; Kim,
S. H., Crystal structure of cyclin-dependent kinase 2. Nature 1993, 363, 595-602.

Chapter 9: References 202
27. Jeffrey, P. D.; Russo, A. A.; Polyak, K.; Gibbs, E.; Hurwitz, J.; Massague, J.;
Pavletich, N. P., Mechanism of CDK activation revealed by the structure of a cyclin A-
CDK2 complex. Nature 1995, 376, 313-320.
28. Brotherton, D. H.; Dhanaraj, V.; Wick, S.; Brizuela, L.; Domaille, P. J.;
Volyanik, E.; Xu, X.; Parisini, E.; Smith, B. O.; Archer, S. J.; Serrano, M.; Brenner, S.
L.; Blundell, T. L.; Laue, E. D., Crystal structure of the complex of the cyclin D-
dependent kinase CDK6 bound to the cell-cycle inhibitor p19INK4d. Nature 1998, 396,
390.
29. Schulze-Gahmen, U.; Kim, S. H., Crystallization of a complex between human
CDK6 and a virus-encoded cyclin is critically dependent on the addition of small
charged organic molecules. Acta Crystallogr. D. Biol. Crystallogr. 2001, D57, 1287-
1289.
30. Lu, H.; Chang, D. J.; Baratte, B.; Meijer, L.; Schulze-Gahmen, U., Crystal
Structure of a Human Cyclin-Dependent Kinase 6 Complex with a Flavonol Inhibitor,
Fisetin. J. Med. Chem. 2005, 48, 737-743.
31. Polychronopoulos, P.; Magiatis, P.; Skaltsounis, A.-L.; Myrianthopoulos, V.;
Mikros, E.; Tarricone, A.; Musacchio, A.; Roe, S. M.; Pearl, L.; Leost, M.; Greengard,
P.; Meijer, L., Structural Basis for the Synthesis of Indirubins as Potent and Selective
Inhibitors of Glycogen Synthase Kinase-3 and Cyclin-Dependent Kinases. J. Med.
Chem. 2004, 47, 935-946.
32. Taylor, S. S.; Radzio-Andzelm, E., Three protein kinase structures define a
common motif. Structure 1994, 2, 345-355.
33. Honma, T.; Hayashi, K.; Aoyama, T.; Hashimoto, N.; Machida, T.; Fukasawa,
K.; Iwama, T.; Ikeura, C.; Ikuta, M.; Suzuki-Takahashi, I.; Iwasawa, Y.; Hayama, T.;
Nishimura, S.; Morishima, H., Structure-Based Generation of a New Class of Potent
Cdk4 Inhibitors: New de Novo Design Strategy and Library Design. J. Med. Chem.
2001, 44, 4615-4627.
34. Chen, Y.-N. P.; Sharma, S. K.; Ramsey, T. M.; Jiang, L.; Martin, M. S.; Baker,
K.; Adams, P. D.; Bair, K. W.; Kaelin, W. G., Jr., Selective killing of transformed cells
by cyclin/cyclin-dependent kinase 2 antagonists. Proc. Natl. Acad. Sci. U. S. A. 1999,
96, 4325-4329.
35. Fry, D. W.; Kraker, A. J.; McMichael, A.; Ambroso, L. A.; Nelson, J. M.;
Leopold, W. R.; Connors, R. W.; Bridges, A. J., A specific inhibitor of the epidermal
growth factor receptor tyrosine kinase. Science 1994, 265, 1093-1095.

Chapter 9: References 203
36. Tetsu, O.; McCormick, F., Proliferation of cancer cells despite CDK2 inhibition.
Cancer Cell 2003, 3, 233-245.
37. Ortega, S.; Prieto, I.; Odajima, J.; Martin, A.; Dubus, P.; Sotillo, R.; Barbero, J.
L.; Malumbres, M.; Barbacid, M., Cyclin-dependent kinase 2 is essential for meiosis but
not for mitotic cell division in mice. Nat. Genet. 2003, 35, 25-31.
38. de Azevedo, W. F., Jr.; Mueller-Dieckmann, H.-J.; Schulze-Gahmen, U.;
Worland, P. J.; Sausville, E.; Kim, S.-H., Structural basis for specificity and potency of
a flavonoid inhibitor of human CDK2, a cell cycle kinase. Proc. Natl. Acad. Sci. U. S. A.
1996, 93, 2735-2740.
39. Huwe, A.; Mazitschek, R.; Giannis, A., Small molecules as inhibitors of cyclin-
dependent kinases. Angew. Chem., Int. Ed. 2003, 42, 2122-2138.
40. De Azevedo, W. F.; Leclerc, S.; Meijer, L.; Havlicek, L.; Strnad, M.; Kim, S. H.,
Inhibition of cyclin-dependent kinases by purine analogs. Crystal structure of human
CDK2 complexed with roscovitine. Eur. J. Biochem. 1997, 243, 518-526.
41. Misra, R. N.; Xiao, H.-y.; Kim, K. S.; Lu, S.; Han, W.-C.; Barbosa, S. A.; Hunt,
J. T.; Rawlins, D. B.; Shan, W.; Ahmed, S. Z.; Qian, L.; Chen, B.-C.; Zhao, R.; Bednarz,
M. S.; Kellar, K. A.; Mulheron, J. G.; Batorsky, R.; Roongta, U.; Kamath, A.; Marathe,
P.; Ranadive, S. A.; Sack, J. S.; Tokarski, J. S.; Pavletich, N. P.; Lee, F. Y. F.; Webster,
K. R.; Kimball, S. D., N-(cycloalkylamino)acyl-2-aminothiazole inhibitors of cyclin-
dependent kinase 2. N-[5-[[[5-(1,1-dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-
4- piperidinecarboxamide (BMS-387032), a highly efficacious and selective antitumor
agent. J. Med. Chem. 2004, 47, 1719-1728.
42. Toogood, P. L.; Harvey, P. J.; Repine, J. T.; Sheehan, D. J.; VanderWel, S. N.;
Zhou, H.; Keller, P. R.; McNamara, D. J.; Sherry, D.; Zhu, T.; Brodfuehrer, J.; Choi, C.;
Barvian, M. R.; Fry, D. W., Discovery of a Potent and Selective Inhibitor of Cyclin-
Dependent Kinase 4/6. J. Med. Chem. 2005, 48, 2388-2406.
43. Fry, D. W.; Harvey, P. J.; Keller, P. R.; Elliott, W. L.; Meade, M.; Trachet, E.;
Albassam, M.; Zheng, X.; Leopold, W. R.; Pryer, N. K.; Toogood, P. L., Specific
inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor
activity in human tumor xenografts. Mol. Cancer Ther. 2004, 3, 1427-1438.
44. Garrett, M. D.; Fattaey, A., CDK inhibition and cancer therapy. Curr. Opin.
Genet. Dev. 1999, 9, 104-111.
45. Gray, N. S.; Wodicka, L.; Thunnissen, A.-M. W. H.; Norman, T. C.; Kwon, S.;
Espinoza, F. H.; Morgan, D. O.; Barnes, G.; LeClerc, S.; Meijer, L.; Kim, S.-H.;

Chapter 9: References 204
Lockhart, D. J.; Schultz, P. G., Exploiting chemical libraries, structure, and genomics in
the search for kinase inhibitors. Science 1998, 281, 533-538.
46. McClue, S. J.; Blake, D.; Clarke, R.; Cowan, A.; Cummings, L.; Fischer, P. M.;
MacKenzie, M.; Melville, J.; Stewart, K.; Wang, S.; Zhelev, N.; Zheleva, D.; Lane, D.
P., In vitro and in vivo antitumor properties of the cyclin dependent kinase inhibitor
CYC202 (R-roscovitine). Int. J. Cancer 2002, 102, 463-468.
47. Davies, T. G.; Bentley, J.; Arris, C. E.; Boyle, F. T.; Curtin, N. J.; Endicott, J. A.;
Gibson, A. E.; Golding, B. T.; Griffin, R. J.; Hardcastle, I. R.; Jewsbury, P.; Johnson, L.
N.; Mesquiche, V.; Newell, D. R.; Noble, M. E. M.; Tucker, J. A.; Wang, L.; Whitfield,
H. J., Structure-based design of a potent purine-based cyclin-dependent kinase inhibitor.
Nature Struct. Biol. 2002, 9, 745-749.
48. Barvian, M.; Boschelli, D.; Cossrow, J.; Dobrusin, E.; Fattaey, A.; Fritsch, A.;
Fry, D.; Harvey, P.; Keller, P.; Garrett, M.; La, F.; Leopold, W.; McNamara, D.; Quin,
M.; Trumpp-Kallmeyer, S.; Toogood, P.; Wu, Z.; Zhang, E., Pyrido[2,3-d]pyrimidin-7-
one Inhibitors of Cyclin-Dependent Kinases. J. Med. Chem. 2000, 43, 4606-4616.
49. Chu, X.-J.; DePinto, W.; Bartkovitz, D.; So, S.-S.; Vu, B. T.; Packman, K.;
Lukacs, C.; Ding, Q.; Jiang, N.; Wang, K.; Goelzer, P.; Yin, X.; Smith, M. A.; Higgins,
B. X.; Chen, Y.; Xiang, Q.; Moliterni, J.; Kaplan, G.; Graves, B.; Lovey, A.; Fotouhi,
N., Discovery of [4-Amino-2-(1-methanesulfonylpiperidin-4-ylamino)pyrimidin-5-
yl](2,3-difluoro-6- methoxyphenyl)methanone (R547), A Potent and Selective Cyclin-
Dependent Kinase Inhibitor with Significant in vivo Antitumor Activity. J. Med. Chem.
2006, 49, 6549-6560.
50. Shewchuk, L.; Hassell, A.; Wisely, B.; Rocque, W.; Holmes, W.; Veal, J.;
Kuyper, L. F., Binding mode of the 4-anilinoquinazoline class of protein kinase
inhibitor: X-ray crystallographic studies of 4-anilinoquinazolines bound to cyclin-
dependent kinase 2 and p38 kinase. J. Med. Chem. 2000, 43, 133-138.
51. Sielecki, T. M.; Johnson, T. L.; Liu, J.; Muckelbauer, J. K.; Grafstrom, R. H.;
Cox, S.; Boylan, J.; Burton, C. R.; Chen, H.; Smallwood, A.; Chang, C. H.; Boisclair,
M.; Benfield, P. A.; Trainor, G. L.; Seitz, S. P., Quinazolines as cyclin dependent kinase
inhibitors. Bioorg. Med. Chem. Lett. 2001, 11, 1157-1160.
52. Bramson, H. N.; Corona, J.; Davis, S. T.; Dickerson, S. H.; Edelstein, M.; Frye,
S. V.; Gampe, R. T., Jr.; Harris, P. A.; Hassell, A.; Holmes, W. D.; Hunter, R. N.;
Lackey, K. E.; Lovejoy, B.; Luzzio, M. J.; Montana, V.; Rocque, W. J.; Rusnak, D.;
Shewchuk, L.; Veal, J. M.; Walker, D. H.; Kuyper, L. F., Oxindole-Based Inhibitors of

Chapter 9: References 205
Cyclin-Dependent Kinase 2 (CDK2): Design, Synthesis, Enzymatic Activities, and X-
ray Crystallographic Analysis. J. Med. Chem. 2001, 44, 4339-4358.
53. Davis, S. T.; Benson, B. G.; Bramson, H. N.; Chapman, D. E.; Dickerson, S. H.;
Dold, K. M.; Eberwein, D. J.; Edelstein, M.; Frye, S. V.; Gampe, R. T., Jr.; Griffin, R. J.;
Harris, P. A.; Hassell, A. M.; Holmes, W. D.; Hunter, R. N.; Knick, V. B.; Lackey, K.;
Lovejoy, B.; Luzzio, M. J.; Murray, D.; Parker, P.; Rocque, W. J.; Shewchuk, L.; Veal,
J. M.; Walker, D. H.; Kuyper, L. F., Prevention of chemotherapy-induced alopecia in
rats by CDK inhibitors. Science (Washington, D. C.) 2001, 291, 134-137.
54. Hoessel, R.; Leclerc, S.; Endicott, J. A.; Nobel, M. E. M.; Lawrie, A.; Tunnah,
P.; Leost, M.; Damiens, E.; Marie, D.; Marko, D.; Niederberger, E.; Tang, W.;
Eisenbrand, G.; Meijer, L., Indirubin, the active constituent of a Chinese antileukemia
medicine, inhibits cyclin-dependent kinases. Nature Cell Biol. 1999, 1, 60-67.
55. Lane, M. E.; Yu, B.; Rice, A.; Lipson, K. E.; Liang, C.; Sun, L.; Tang, C.;
McMahon, G.; Pestell, R. G.; Wadler, S., A novel cdk2-selective inhibitor, SU9516,
induces apoptosis in colon carcinoma cells. Cancer Res. 2001, 61, 6170-6177.
56. Li, X.; Huang, P.; Cui, J. J.; Zhang, J.; Tang, C., Novel pyrrolyllactone and
pyrrolyllactam indolinones as potent cyclin-dependent kinase 2 inhibitors. Bioorg. Med.
Chem. Lett. 2003, 13, 1939-1942.
57. Schoepfer, J.; Fretz, H.; Chaudhuri, B.; Muller, L.; Seeber, E.; Meijer, L.;
Lozach, O.; Vangrevelinghe, E.; Furet, P., Structure-Based Design and Synthesis of 2-
Benzylidene-benzofuran-3-ones as Flavopiridol Mimics. J. Med. Chem. 2002, 45, 1741-
1747.
58. Kitagawa, M.; Okabe, T.; Ogino, H.; Matsumoto, H.; Suzuki-Takahashi, I.;
Kokubo, T.; Higashi, H.; Saitoh, S.; Taya, Y.; et al., Butyrolactone I, a selective
inhibitor of cdk2 and cdc2 kinase. Oncogene 1993, 8, 2425-2432.
59. Havlicek, L.; Hanus, J.; Vesely, J.; Leclerc, S.; Meijer, L.; Shaw, G.; Strnad, M.,
Cytokinin-Derived Cyclin-Dependent Kinase Inhibitors: Synthesis and cdc2 Inhibitory
Activity of Olomoucine and Related Compounds. J. Med. Chem. 1997, 40, 408-412.
60. Gussio, R.; Zaharevitz, D. W.; McGrath, C. F.; Pattabiraman, N.; Kellogg, G. E.;
Schultz, C.; Link, A.; Kunick, C.; Leost, M.; Meijer, L.; Sausville, E. A., Structure-
based design modifications of the paullone molecular scaffold for cyclin-dependent
kinase inhibition. Anticancer. Drug Des. 2000, 15, 53-66.

Chapter 9: References 206
61. Nugiel, D. A.; Etzkorn, A.-M.; Vidwans, A.; Benfield, P. A.; Boisclair, M.;
Burton, C. R.; Cox, S.; Czerniak, P. M.; Doleniak, D.; Seitz, S. P., Indenopyrazoles as
Novel Cyclin Dependent Kinase (CDK) Inhibitors. J. Med. Chem. 2001, 44, 1334-1336.
62. Lawrie, A. M.; Noble, M. E. M.; Tunnah, P.; Brown, N. R.; Johnson, L. N.;
Endicott, J. A., Protein kinase inhibition by staurosporine revealed in details of the
molecular interaction with CDK2. Nat. Struct. Biol. 1997, 4, 796-801.
63. Dreyer, M. K.; Borcherding, D. R.; Dumont, J. A.; Peet, N. P.; Tsay, J. T.;
Wright, P. S.; Bitonti, A. J.; Shen, J.; Kim, S.-H., Crystal structure of human cyclin-
dependent kinase 2 in complex with the adenine-derived inhibitor H717. J. Med. Chem.
2001, 44, 524-530.
64. Arris, C. E.; Boyle, F. T.; Calvert, A. H.; Curtin, N. J.; Endicott, J. A.; Garman,
E. F.; Gibson, A. E.; Golding, B. T.; Grant, S.; Griffin, R. J.; Jewsbury, P.; Johnson, L.
N.; Lawrie, A. M.; Newell, D. R.; Noble, M. E. M.; Sausville, E. A.; Schultz, R.; Yu,
W., Identification of Novel Purine and Pyrimidine Cyclin-Dependent Kinase Inhibitors
with Distinct Molecular Interactions and Tumor Cell Growth Inhibition Profiles. J. Med.
Chem. 2000, 43, 2797-2804.
65. Davis, S. T.; Benson, B. G.; Bramson, H. N.; Chapman, D. E.; Dickerson, S. H.;
Dold, K. M.; Eberwein, D. J.; Edelstein, M.; Frye, S. V.; Gampe, R. T., Jr.; Griffin, R. J.;
Harris, P. A.; Hassell, A. M.; Holmes, W. D.; Hunter, R. N.; Knick, V. B.; Lackey, K.;
Lovejoy, B.; Luzzio, M. J.; Murray, D.; Parker, P.; Rocque, W. J.; Shewchuk, L.; Veal,
J. M.; Walker, D. H.; Kuyper, L. F., Prevention of chemotherapy-induced alopecia in
rats by CDK inhibitors. Science 2001, 291, 134-137.
66. Vousden, K. H.; Lu, X., Live or let die: the cell's response to p53. Nat. Rev.
Cancer 2002, 2, 594-604.
67. Vassilev, L. T., p53 Activation by Small Molecules: Application in Oncology. J.
Med. Chem. 2005, 48, 4491-4499.
68. Hollstein, M.; Rice, K.; Greenblatt, M. S.; Soussi, T.; Fuchs, R.; Sorlie, T.;
Hovig, E.; Smith-Sorensen, B.; Montesano, R.; Harris, C. C., Database of p53 gene
somatic mutations in human tumors and cell lines. Nucleic Acids Res. 1994, 22, 3551-5.
69. Jones, S. N.; Roe, A. E.; Donehower, L. A.; Bradley, A., Rescue of embryonic
lethality in Mdm2-deficient mice by absence of p53. Nature 1995, 378, 206-208.
70. Chene, P., Inhibiting the p53-MDM2 interaction: An important target for cancer
therapy. Nat. Rev. Cancer 2003, 3, 102-109.

Chapter 9: References 207
71. Ashcroft, M.; Vousden, K. H., Regulation of p53 stability. Oncogene 1999, 18,
7637-7643.
72. Balint, E.; Vousden, K. H., Activation and activities of the p53 tumor suppressor
protein. Br. J. Cancer 2001, 85, 1813-1823.
73. Momand, J.; Zambetti, G. P.; Olson, D. C.; George, D.; Levine, A. J., The mdm-
2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated
transactivation. Cell 1992, 69, 1237-1245.
74. Wadgaonkar, R.; Collins, T., Murine double minute (MDM2) blocks p53-
coactivator interaction, a new mechanism for inhibition of p53-dependent gene
expression. J. Biol. Chem. 1999, 274, 13760-13767.
75. Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M., Mdm2 promotes the rapid
degradation of p53. Nature 1997, 387, 296-299.
76. Kubbutat, M. H. G.; Jones, S. N.; Vousden, K. H., Regulation of p53 stability by
Mdm2. Nature 1997, 387, 299-303.
77. Boyd, S. D.; Tsai, K. Y.; Jacks, T., An intact HDM2 RING-finger domain is
required for nuclear exclusion of p53. Nature Cell Biol. 2000, 2, 563-568.
78. Geyer, R. K.; Yu, Z. K.; Maki, C. G., The MDM2 RING-finger domain is
required to promote p53 nuclear export. Nature Cell Biol. 2000, 2, 569-573.
79. Zhang, Z.; Zhang, R., p53-Independent activities of MDM2 and their relevance
to cancer therapy. Curr. Cancer Drug Targets 2005, 5, 9-20.
80. de Oca Luna, R. M.; Wagner, D. S.; Lozano, G., Rescue of early embryonic
lethality in mdm2-deficient mice by deletion of p53. Nature 1995, 378, 203-206.
81. Chen, J.; Wu, X.; Lin, J.; Levine, A. J., mdm-2 inhibits the G1 arrest and
apoptosis functions of the p53 tumor suppressor protein. Mol. Cell. Biol. 1996, 16,
2445-2452.
82. Muthusamy, V.; Hobbs, C.; Nogueira, C.; Cordon-Cardo, C.; McKee, P. H.;
Chin, L.; Bosenberg, M. W., Amplification of CDK4 and MDM2 in malignant
melanoma. Genes Chromosomes Cancer 2006, 45, 447-454.
83. Rayburn, E.; Zhang, R.; He, J.; Wang, H., MDM2 and human malignancies:
expression, clinical pathology, prognostic markers, and implications for chemotherapy.
Curr. Cancer Drug Targets 2005, 5, 27-41.
84. Bueso-Ramos, C. E.; Yang, Y.; deLeon, E.; McCown, P.; Stass, S. A.; Albitar,
M., The human MDM-2 oncogene is overexpressed in leukemias. Blood 1993, 82,
2617-2623.

Chapter 9: References 208
85. Sun, Y.; Huang, P. L.; Li, J. J.; Huang, Y. Q.; Zhang, L.; Huang, P. L.; Lee-
Huang, S., Anti-HIV Agent MAP30 Modulates the Expression Profile of Viral and
Cellular Genes for Proliferation and Apoptosis in AIDS-Related Lymphoma Cells
Infected with Kaposi's Sarcoma-Associated Virus. Biochem. Biophys. Res. Commun.
2001, 287, 983-994.
86. Lee-Huang, S.; Zhang, L.; Huang, P. L.; Chang, Y.-T.; Huang, P. L., Anti-HIV
activity of olive leaf extract (OLE) and modulation of host cell gene expression by HIV-
1 infection and OLE treatment. Biochem. Biophys. Res. Commun. 2003, 307, 1029-1037.
87. Fesik, S. W., Promoting apoptosis as a strategy for cancer drug discovery. Nat.
Rev. Cancer 2005, 5, 876-885.
88. Lane, D. P.; Lain, S., Therapeutic exploitation of the p53 pathway. Trends in
Mol. Med. 2002, 8, S38-S42.
89. Zhang, R.; Wang, H., MDM2 oncogene as a novel target for human cancer
therapy. Curr. Pharm. Des. 2000, 6, 393-416.
90. Wiman, K. G., Strategies for therapeutic targeting of the p53 pathway in cancer.
Cell Death Differ. 2006, 13, 921-926.
91. Chene, P., Inhibition of the p53-MDM2 Interaction: Targeting a Protein-Protein
Interface. Mol. Cancer Res. 2004, 2, 20-28.
92. Toogood, P. L., Inhibition of Protein-Protein Association by Small Molecules:
Approaches and Progress. J. Med. Chem. 2002, 45, 1543-1558.
93. Arkin, M. R.; Wells, J. A., Small-molecule inhibitors of protein-protein
interactions: progressing towards the dream. Nat. Rev. Drug Disc. 2004, 3, 301-317.
94. Kussie, P. H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A. J.;
Pavletich, N. P., Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor
transactivation domain. Science 1996, 274, 948-953.
95. Lin, J.; Chen, J.; Elenbaas, B.; Levine, A. J., Several hydrophobic amino acids in
the p53 amino-terminal domain are required for transcriptional activation, binding to
mdm-2 and the adenovirus 5 E1B 55-kDa protein. Genes Dev. 1994, 8, 1235-1246.
96. Garcia-Echeverria, C.; Chene, P.; Blommers, M. J. J.; Furet, P., Discovery of
Potent Antagonists of the Interaction between Human Double Minute 2 and Tumor
Suppressor p53. J. Med. Chem. 2000, 43, 3205-3208.
97. Sakurai, K.; Schubert, C.; Kahne, D., Crystallographic Analysis of an 8-mer p53
Peptide Analogue Complexed with MDM2. J. Am. Chem. Soc. 2006, 128, 11000-11001.

Chapter 9: References 209
98. Hara, T.; Durell, S. R.; Myers, M. C.; Appella, D. H., Probing the Structural
Requirements of Peptoids That Inhibit HDM2-p53 Interactions. J. Am. Chem. Soc. 2006,
128, 1995-2004.
99. Kritzer, J. A.; Zutshi, R.; Cheah, M.; Ran, F. A.; Webman, R.; Wongjirad, T. M.;
Schepartz, A., Miniature protein inhibitors of the p53-hDM2 interaction. ChemBioChem
2006, 7, 29-31.
100. Stoll, R.; Renner, C.; Hansen, S.; Palme, S.; Klein, C.; Belling, A.; Zeslawski,
W.; Kamionka, M.; Rehm, T.; Muehlhahn, P.; Schumacher, R.; Hesse, F.; Kaluza, B.;
Voelter, W.; Engh, R. A.; Holak, T. A., Chalcone Derivatives Antagonize Interactions
between the Human Oncoprotein MDM2 and p53. Biochemistry 2001, 40, 336-344.
101. Grasberger, B. L.; Lu, T.; Schubert, C.; Parks, D. J.; Carver, T. E.; Koblish, H.
K.; Cummings, M. D.; LaFrance, L. V.; Milkiewicz, K. L.; Calvo, R. R.; Maguire, D.;
Lattanze, J.; Franks, C. F.; Zhao, S.; Ramachandren, K.; Bylebyl, G. R.; Zhang, M.;
Manthey, C. L.; Petrella, E. C.; Pantoliano, M. W.; Deckman, I. C.; Spurlino, J. C.;
Maroney, A. C.; Tomczuk, B. E.; Molloy, C. J.; Bone, R. F., Discovery and Cocrystal
Structure of Benzodiazepinedione HDM2 Antagonists That Activate p53 in Cells. J.
Med. Chem. 2005, 48, 909-912.
102. Leonard, K.; Marugan, J. J.; Raboisson, P.; Calvo, R.; Gushue, J. M.; Koblish, H.
K.; Lattanze, J.; Zhao, S.; Cummings, M. D.; Player, M. R.; Maroney, A. C.; Lu, T.,
Novel 1,4-benzodiazepine-2,5-diones as Hdm2 antagonists with improved cellular
activity. Bioorg. Med. Chem. Lett. 2006, 16, 3463-3468.
103. Issaeva, N.; Bozko, P.; Enge, M.; Protopopova, M.; Verhoef, L. G. G. C.;
Masucci, M.; Pramanik, A.; Selivanova, G., Small molecule RITA binds to p53, blocks
p53-HDM-2 interaction and activates p53 function in tumors. Nature Med. 2004, 10,
1321-1328.
104. Galatin, P. S.; Abraham, D. J., A Nonpeptidic Sulfonamide Inhibits the p53-
mdm2 Interaction and Activates p53-Dependent Transcription in mdm2-Overexpressing
Cells. J. Med. Chem. 2004, 47, 4163-4165.
105. Vassilev, L. T.; Vu, B. T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.;
Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; Fotouhi, N.; Liu, E. A., In vivo
Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science 2004,
303, 844-848.
106. Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Wang, G.; Qiu, S.; Shangary, S.; Gao,
W.; Qin, D.; Stuckey, J.; Krajewski, K.; Roller, P. P.; Wang, S., Structure-Based Design

Chapter 9: References 210
of Spiro-oxindoles as Potent, Specific Small-Molecule Inhibitors of the MDM2-p53
Interaction. J. Med. Chem. 2006, 49, 3432-3435.
107. Hardcastle, I. R.; Ahmed, S. U.; Atkins, H.; Calvert, A. H.; Curtin, N. J.; Farnie,
G.; Golding, B. T.; Griffin, R. J.; Guyenne, S.; Hutton, C.; Kallblad, P.; Kemp, S. J.;
Kitching, M. S.; Newell, D. R.; Norbedo, S.; Northen, J. S.; Reid, R. J.; Saravanan, K.;
Willems, H. M. G.; Lunec, J., Isoindolinone-based inhibitors of the MDM2-p53 protein-
protein interaction. Bioorg. Med. Chem. Lett. 2005, 15, 1515-1520.
108. Kumar, S. K.; Hager, E.; Pettit, C.; Gurulingappa, H.; Davidson, N. E.; Khan, S.
R., Design, Synthesis, and Evaluation of Novel Boronic-Chalcone Derivatives as
Antitumor Agents. J. Med. Chem. 2003, 46, 2813-2815.
109. Parks, D. J.; LaFrance, L. V.; Calvo, R. R.; Milkiewicz, K. L.; Gupta, V.;
Lattanze, J.; Ramachandren, K.; Carver, T. E.; Petrella, E. C.; Cummings, M. D.;
Maguire, D.; Grasberger, B. L.; Lu, T., 1,4-Benzodiazepine-2,5-diones as small
molecule antagonists of the HDM2-p53 interaction: discovery and SAR. Bioorg. Med.
Chem. Lett. 2005, 15, 765-770.
110. Raboisson, P.; Marugan, J. J.; Schubert, C.; Koblish, H. K.; Lu, T.; Zhao, S.;
Player, M. R.; Maroney, A. C.; Reed, R. L.; Huebert, N. D.; Lattanze, J.; Parks, D. J.;
Cummings, M. D., Structure-based design, synthesis, and biological evaluation of novel
1,4-diazepines as HDM2 antagonists. Bioorg. Med. Chem. Lett. 2005, 15, 1857-1861.
111. Buolamwini, J. K.; Addo, J.; Kamath, S.; Patil, S.; Mason, D.; Ores, M., Small
molecule antagonists of the MDM2 oncoprotein as anticancer agents. Curr. Cancer
Drug Targets 2005, 5, 57-68.
112. Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Qiu, S.; Ding, Y.; Gao, W.; Stuckey,
J.; Krajewski, K.; Roller, P. P.; Tomita, Y.; Parrish, D. A.; Deschamps, J. R.; Wang, S.,
Structure-Based Design of Potent Non-Peptide MDM2 Inhibitors. J. Am. Chem. Soc.
2005, 127, 10130-10131.
113. Yasuda, N.; Hsiao, Y.; Jensen, M. S.; Rivera, N. R.; Yang, C.; Wells, K. M.;
Yau, J.; Palucki, M.; Tan, L.; Dormer, P. G.; Volante, R. P.; Hughes, D. L.; Reider, P. J.,
An Efficient Synthesis of an αvβ3 Antagonist. J. Org. Chem. 2004, 69, 1959-1966.
114. Chene, P.; Fuchs, J.; Carena, I.; Furet, P.; Garcia Echeverria, C., Study of the
cytotoxic effect of a peptidic inhibitor of the p53-hdm2 interaction in tumor cells. FEBS
Lett. 2002, 529, 293-297.

Chapter 9: References 211
115. Anderton, N.; Cockrum, P. A.; Colegate, S. M.; Edgar, J. A.; Flower, K.; Vit, I.;
Willing, R. I., Oxindoles from Phalaris coerulescens. Phytochemistry 1998, 48, 437-
439.
116. Colegate, S. M.; Anderton, N.; Edgar, J.; Bourke, C. A.; Oram, R. N., Suspected
blue canary grass (Phalaris coerulescens) poisoning of horses. Aust. Vet. J. 1999, 77,
537-538.
117. Cheeke, P. R., Natural Toxicants in Feeds, Forages and Poisonous Plants. 2nd
ed.; Interstate Publishers Inc.: Danville, p 275.
118. Jossang, A.; Jossang, P.; Hadi, H. A.; Sevenet, T.; Bodo, B., Horsfiline, an
oxindole alkaloid from Horsfieldia superba. J. Org. Chem. 1991, 56, 6527-6530.
119. Kornet, M. J.; Thio, A. P., Oxindole-3-spiropyrrolidines and -piperidines.
Synthesis and local anesthetic activity. J. Med. Chem. 1976, 19, 892-898.
120. Shi, J.-S.; Kenneth, H. G., Effect of rhynchophylline on apoptosis induced by
dopamine in NT2 cells. Acta Pharmacol. Sinica 2002, 23, 445-449.
121. Shimada, Y.; Goto, H.; Itoh, T.; Sakakibara, I.; Kubo, M.; Sasaki, H.; Terasawa,
K., Evaluation of the protective effects of alkaloids isolated from the hooks and stems
of Uncaria sinensis on glutamate-induced neuronal death in cultured cerebellar granule
cells from rats. J. Pharm. Pharmacol. 1999, 51, 715-722.
122. Laus, G.; Brossner, D.; Keplinger, K., Alkaloids of Peruvian Uncaria tomentosa.
Phytochemistry 1997, 45, 855-860.
123. Abdel-Fattah, M. A. F.; Matsumoto, K.; Tabata, K.; Takayama, H.; Kitajima, M.;
Aimi, N.; Watanabe, H., Effects of Uncaria tomentosa total alkaloids and its
components on experimental amnesia in mice: elucidation using the passive avoidance
test. J. Pharm. Pharmacol. 2000, 52, 1553-1561.
124. Kang, T.-H.; Murakami, Y.; Matsumoto, K.; Takayama, H.; Kitajima, M.; Aimi,
N.; Watanabe, H., Rhynchophylline and isorhynchophylline inhibit NMDA receptors
expressed in Xenopus oocytes. Eur. J. Pharmacol. 2002, 455, 27-34.
125. Bacher, N.; Tiefenthaler, M.; Sturm, S.; Stuppner, H.; Ausserlechner, M. J.;
Kofler, R.; Konwalinka, G., Oxindole alkaloids from Uncaria tomentosa induce
apoptosis in proliferating, G0/G1-arrested and bcl-2-expressing acute lymphoblastic
leukaemia cells. Br. J. Haematol. 2006, 132, 615-622.
126. Chang, M.-Y.; Pai, C.-L.; Kung, Y.-H., Synthesis of (+/-)-coerulescine and a
formal synthesis of (+-)-horsfiline. Tetrahedron Lett. 2005, 46, 8463-8465.

Chapter 9: References 212
127. Selvakumar, N.; Azhagan, A. M.; Srinivas, D.; Krishna, G. G., A direct
synthesis of 2-arylpropenoic acid esters having nitro groups in the aromatic ring: a short
synthesis of (+/-)-coerulescine and (+/-)-horsfiline. Tetrahedron Lett. 2002, 43, 9175-
9178.
128. Lizos, D. E.; Murphy, J. A., Concise synthesis of (+/-)-horsfiline and (+/-)-
coerulescine by tandem cyclisation of iodoaryl alkenyl azides. Org. Biomol. Chem.
2003, 1, 117-122.
129. Somei, M.; Noguchi, K.; Yamagami, R.; Kawada, Y.; Yamada, K.; Yamada, F.,
Chemistry of indoles. 95. Preparation and a novel rearrangement reaction of 1,2,3,4-
tetrahydro-9-hydroxy-β-carboline, and their applications for the total synthesis of (+/-)-
coerulescine. Heterocycles 2000, 53, 7-10.
130. Marti, C. Novel approach to spiro-pyrrolidine-oxindoles and its application to
the synthesis of (+/-)-horsfiline and (-)-spirotryprostatin B. 2003.
131. Bascop, S.-I.; Sapi, J.; Laronze, J.-Y.; Levy, J., The synthesis of the oxindole
alkaloid: (+/-)-horsfiline. Heterocycles 1994, 38, 725-732.
132. Fischer, C.; Meyers, C.; Carreira, E. M., Efficient synthesis of (+/-)-horsfiline
through the MgI2-catalyzed ring-expansion reaction of a spiro[cyclopropane-1,3-indol]-
2'-one. Helv. Chim. Acta 2000, 83, 1175-1181.
133. Jones, K.; Wilkinson, J., A total synthesis of horsfiline via aryl radical
cyclization. J. Chem. Soc., Chem. Commun. 1992, 1767-1769.
134. Murphy, J. A.; Tripoli, R.; Khan, T. A.; Mali, U. W., Novel Phosphorus Radical-
Based Routes to Horsfiline. Org. Lett. 2005, 7, 3287-3289.
135. Lakshmaiah, G.; Kawabata, T.; Shang, M.; Fuji, K., Total Synthesis of (-)-
Horsfiline via Asymmetric Nitroolefination. J. Org. Chem. 1999, 64, 1699-1704.
136. Pellegrini, C.; Straessler, C.; Weber, M.; Borschberg, H.-J., Synthesis of the
oxindole alkaloid (-)-horsfiline. Tetrahedron: Asymmetry 1994, 5, 1979-1992.
137. Palmisano, G.; Annunziata, R.; Papeo, G.; Sisti, M., Oxindole alkaloids. A novel
non-biomimetic entry to (-)-horsfiline. Tetrahedron: Asymmetry 1996, 7, 1-4.
138. Pellegrini, C.; Weber, M.; Borschberg, H.-J., Total synthesis of (+)-elacomine
and (-)-isoelacomine, two hitherto unnamed oxindole alkaloids from Elaeagnus
commutata. Helv. Chim. Acta 1996, 79, 151-168.
139. Miyake, F. Y.; Yakushijin, K.; Horne, D. A., Preparation and synthetic
applications of 2-halotryptophan methyl esters: Synthesis of spirotryprostatin B. Angew.
Chem., Int. Ed. 2004, 43, 5357-5360.

Chapter 9: References 213
140. Miyake, F. Y.; Yakushijin, K.; Horne, D. A., Preparation and Synthetic
Applications of 2-Halotryptamines: Synthesis of Elacomine and Isoelacomine. Org. Lett.
2004, 6, 711-713.
141. Ponglux, D.; Wongseripipatana, S.; Aimi, N.; Nishimura, M.; Ishikawa, M.;
Sada, H.; Haginiwa, J.; Sakai, S., Structure and synthesis of two new types of oxindole
alkaloids from Uncaria salaccensis. Chem. Pharm. Bull. (Tokyo). 1990, 38, 573-575.
142. Shi, J.-S.; Yu, J.-X.; Chen, X.-P.; Xu, R.-X., Pharmacological actions of uncaria
alkaloids, rhynchophylline and isorhynchophylline. Acta Pharmacol. Sinica 2003, 24,
97-101.
143. Zhang, J.; Yang, C.; Wu, D., Chemical constituents of Uncaria rhynchophylla
(III). Zhongcaoyao 1999, 30, 12-14.
144. Nozoye, T., Uncaria alkaloid. XX. Structure of rhynchophylline. 5. Structure of
rhynchophylline and isorhynchophylline. Chem. Pharm. Bull. (Tokyo). 1958, 6, 309-312.
145. Ban, Y.; Oishi, T., Synthesis of 3-spirooxindole derivatives. III. Stereospecific
syntheses of rac-N-methylrhynchophyllane for the stereochemistry of rhynchophylline
and isorhynchophylline. Chem. Pharm. Bull. (Tokyo). 1963, 11, 451-460.
146. Ban, Y.; Seto, M.; Oishi, T., Stereoselective total synthesis of (+/-)-
rhynchophylline and (+/-)-isorhynchophylline. Tetrahedron Lett. 1972, 2113-2116.
147. Ban, Y.; Seto, M.; Oishi, T., Synthesis of 3-spirooxindole derivatives. VII. Total
synthesis of alkaloids (+/-)-rhynchophylline and (+/-)-isorhynchophylline. Chem.
Pharm. Bull. (Tokyo). 1975, 23, 2605-2613.
148. Martin, S. F.; Mortimore, M., New methods for the synthesis of oxindole
alkaloids. Total syntheses of isopteropodine and pteropodine. Tetrahedron Lett. 1990,
31, 4557-4560.
149. Tsuda, M.; Kasai, Y.; Komatsu, K.; Sone, T.; Tanaka, M.; Mikami, Y.;
Kobayashi, J., Citrinadin A, a novel pentacyclic alkaloid from marine-derived fungus
Penicillium citrinum. Org. Lett. 2004, 6, 3087-3089.
150. Mugishima, T.; Tsuda, M.; Kasai, Y.; Ishiyama, H.; Fukushi, E.; Kawabata, J.;
Watanabe, M.; Akao, K.; Kobayashi, J. i., Absolute Stereochemistry of Citrinadins A
and B from Marine-Derived Fungus. J. Org. Chem. 2005, 70, 9430-9435.
151. Elderfield, R. C.; Gilman, R. E., Alstonia alkaloids. XI. Alkaloids of Alstonia
muelleriana. Phytochemistry 1972, 11, 339-343.

Chapter 9: References 214
152. Ghedira, K.; Zeches-Hanrot, M.; Richard, B.; Massiot, G.; Le Men-Olivier, L.;
Sevenet, T.; Goh, S. H., Alkaloids of Alstonia angustifolia. Phytochemistry 1988, 27,
3955-3962.
153. Wearing, X. Z.; Cook, J. M., Enantiospecific, Stereospecific Total Synthesis of
the Oxindole Alkaloid Alstonisine. Org. Lett. 2002, 4, 4237-4240.
154. Marti, C.; Carreira, E. M., Construction of spiro[pyrrolidine-3,3'-oxindoles] -
recent applications to the synthesis of oxindole alkaloids. Eur. J. Org. Chem. 2003,
2209-2219.
155. Bassleer, R.; Depauw-Gillet, M. C.; Massart, B.; Marnette, J. M.; Wiliquet, P.;
Caprasse, M.; Angenot, L., Effects of three alkaloids isolated from Strychnos
usambarensis on cancer cells in culture. Planta Med. 1982, 45, 123-126.
156. Lerchner, A.; Carreira, E. M., First Total Synthesis of (+/-)-Strychnofoline via a
Highly Selective Ring-Expansion Reaction. J. Am. Chem. Soc. 2002, 124, 14826-14827.
157. Noble, R. L., The discovery of the vinca alkaloids - chemotherapeutic agents
against cancer. Biochem. Cell Biol. 1990, 68, 1344-1351.
158. Van Tellingen, O.; Sips, J. H. M.; Beijnen, J. H.; Bult, A.; Nooijen, W. J.,
Pharmacology, bio-analysis and pharmacokinetics of the vinca alkaloids and semi-
synthetic derivatives (review). Anticancer Res. 1992, 12, 1699-1715.
159. Schneider, C., First de novo synthesis of the bisindole alkaloid vinblastine.
Angew. Chem., Int. Ed. 2002, 41, 4217-4219.
160. Hasan, M., Strategies for the production of vinblastine. Adv. Nat. Prod. Chem.,
Proc. Int. Symp. Pak.-US Binatl. Workshop, 5th 1992, 249-256.
161. Yates, P.; MacLachlan, F. N.; Rae, I. D.; Rosenberger, M.; Szabo, A. G.; Willis,
C. R.; Cava, M. P.; Behforouz, M.; Lakshmikantham, M. V.; Zeiger, W., Haplophytine.
Novel type of indole alkaloid. J. Am. Chem. Soc. 1973, 95, 7842-7850.
162. He, F.; Bo, Y.; Altom, J. D.; Corey, E. J., Enantioselective Total Synthesis of
Aspidophytine. J. Am. Chem. Soc. 1999, 121, 6771-6772.
163. Ponglux, D.; Wonseripipatana, S.; Subhadhirasakul, S.; Takayama, H.; Yokota,
M.; Ogata, K.; Phisalaphong, C.; Aimi, N.; Sakai, S., Studies on the indole alkaloids of
Gelsemium elegans (Thailand). Structure elucidation and proposal of a biogenetic route.
Tetrahedron 1988, 44, 5075-5094.
164. Sakai, S.; Takayama, H.; Yokota, M.; Kitajima, M.; Masubuchi, K.; Ogata, K.;
Yamanaka, E.; Aimi, N.; Wongseripipatana, S.; Ponglux, D., Studies on the indole
alkaloids from Gelsemium elegans. Structural elucidation, proposal of biogenetic route,

Chapter 9: References 215
and biomimetic synthesis of koumine and humantenine skeletons. Tennen Yuki
Kagobutsu Toronkai Koen Yoshishu 1987, 29, 224-231.
165. Rujjanawate, C.; Kanjanapothi, D.; Panthong, A., Pharmacological effect and
toxicity of alkaloids from Gelsemium elegans Benth. J. Ethnopharmacol. 2003, 89, 91-
95.
166. Zhang, Z.; Liang, X.; Sun, F.; Lu, Y.; Yang, J.; Xing, Q., Studies on the indole
alkaloids of Gelsemium elegans. Chin. Chem. Lett. 1991, 2, 365-368.
167. Zhang, L.; Lin, J.; Wu, Z., Advances in the study on chemical constituents and
pharmacology of Gelsemium elegans (Gardn. et Champ.) Benth. Zhongyaocai 2003, 26,
451-453.
168. Kitajima, M.; Nakamura, T.; Kogure, N.; Ogawa, M.; Mitsuno, Y.; Ono, K.;
Yano, S.; Aimi, N.; Takayama, H., Isolation of Gelsedine-Type Indole Alkaloids from
Gelsemium elegans and Evaluation of the Cytotoxic Activity of Gelsemium Alkaloids
for A431 Epidermoid Carcinoma Cells. J. Nat. Prod. 2006, 69, 715-718.
169. Takayama, H.; Tominaga, Y.; Kitajima, M.; Aimi, N.; Sakai, S., First Synthesis
of the Novel Gelsemium Alkaloids, Gelselegine, Gelsenicine, and Gelsedine Using a
Biomimetic Approach. J. Org. Chem. 1994, 59, 4381-4385.
170. Van Henegouwen, W. G. B.; Hiemstra, H., Studies toward the Total Synthesis of
the Oxindole Alkaloid Gelsedine: An Efficient Allene-Terminated N-Acyliminium Ion
Cyclization. J. Org. Chem. 1997, 62, 8862-8867.
171. Van Henegouwen, W. G. B.; Fieseler, R. M.; Rutjes, F. P. J. T.; Hiemstra, H.,
First Total Synthesis of ent-Gelsedine via a Novel Iodide-Promoted Allene N-
Acyliminium Ion Cyclization. J. Org. Chem. 2000, 65, 8317-8325.
172. Van Henegouwen, W. G. B.; Fieseler, R. M.; Rutjes, F. P. J. T.; Hiemstra, H.,
Total synthesis of (+)-gelsedine. Angew. Chem., Int. Ed. 1999, 38, 2214-2217.
173. Cui, C.-B.; Kakeya, H.; Osada, H., Novel mammalian cell cycle inhibitors,
spirotryprostatins A and B, produced by Aspergillus fumigatus, which inhibit
mammalian cell cycle at G2/M phase. Tetrahedron 1996, 52, 12651-12666.
174. Edmondson, S. D.; Danishefsky, S. J., The total synthesis of spirotryprostatin A.
Angew. Chem., Int. Ed. 1998, 37, 1138-1140.
175. Lindel, T., Short syntheses of spirotryprostatins. Nachrichten aus der Chemie
2000, 48, 1498-1501.
176. Miyake, F. Y.; Yakushijin, K.; Horne, D. A., A Concise Synthesis of
Spirotryprostatin A. Org. Lett. 2004, 6, 4249-4251.

Chapter 9: References 216
177. Edmondson, S.; Danishefsky, S. J.; Sepp-Lorenzino, L.; Rosen, N., Total
Synthesis of Spirotryprostatin A, Leading to the Discovery of Some Biologically
Promising Analogs. J. Am. Chem. Soc. 1999, 121, 2147-2155.
178. Onishi, T.; Sebahar, P. R.; Williams, R. M., Concise, asymmetric total synthesis
of spirotryprostatin A. Tetrahedron 2004, 60, 9503-9515.
179. Onishi, T.; Sebahar, P. R.; Williams, R. M., Concise, Asymmetric Total
Synthesis of Spirotryprostatin A. Org. Lett. 2003, 5, 3135-3137.
180. Cui, C. B.; Kakeya, H.; Osada, H., Spirotryprostatin B, a novel mammalian cell
cycle inhibitor produced by Aspergillus fumigatus. J. Antibiot. 1996, 49, 832-835.
181. Lindel, T., Short syntheses of the spirotryprostatins. Org. Synth. Highlights V
2003, 360-367.
182. Von Nussbaum, F.; Danishefsky, S. J., A rapid total synthesis of
spirotryprostatin B: proof of its relative and absolute stereochemistry. Angew. Chem.,
Int. Ed. 2000, 39, 2175-2178.
183. Meyers, C.; Carreira, E. M., Total synthesis of (-)-spirotryprostatin B. Angew.
Chem., Int. Ed. 2003, 42, 694-696.
184. Marti, C.; Carreira, E. M., Total Synthesis of (-)-Spirotryprostatin B: Synthesis
and Related Studies. J. Am. Chem. Soc. 2005, 127, 11505-11515.
185. Sebahar, P. R.; Osada, H.; Usui, T.; Williams, R. M., Asymmetric,
stereocontrolled total synthesis of (+) and (-)-spirotryprostatin B via a diastereoselective
azomethine ylide [1,3]-dipolar cycloaddition reaction. Tetrahedron 2002, 58, 6311-
6322.
186. Bagul, T. D.; Lakshmaiah, G.; Kawabata, T.; Fuji, K., Total synthesis of
spirotryprostatin B via asymmetric nitroolefination. Org. Lett. 2002, 4, 249-251.
187. Overman, L. E.; Rosen, M. D., Total synthesis of (-)-spirotryprostatin B and
three stereoisomers. Angew. Chem., Int. Ed. 2000, 39, 4596-4599.
188. Wang, H.; Ganesan, A., A Biomimetic Total Synthesis of (-)-Spirotryprostatin B
and Related Studies. J. Org. Chem. 2000, 65, 4685-4693.
189. Sebahar, P. R.; Williams, R. M., The Asymmetric Total Synthesis of (+)- and (-
)-Spirotryprostatin B. J. Am. Chem. Soc. 2000, 122, 5666-5667.
190. Birch, A. J.; Wright, J. J., Biosynthesis. XLII. Structural elucidation and some
aspects of the biosynthesis of the brevianamides-A and -E. Tetrahedron 1970, 26, 2329-
2344.

Chapter 9: References 217
191. Birch, A. J.; Russell, R. A., Biosynthesis. XLIV. Structural elucidations of
brevianamides-B, -C, -D, and -F. Tetrahedron 1972, 28, 2999-3008.
192. Blanchflower, S. E.; Banks, R. M.; Everett, J. R.; Manger, B. R.; Reading, C.,
New paraherquamide antibiotics with anthelmintic activity. J. Antibiot. 1991, 44, 492-7.
193. Blanchflower, S. E.; Banks, R. M.; Everett, J. R.; Reading, C., Further novel
metabolites of the paraherquamide family. J. Antibiot. 1993, 46, 1355-1363.
194. Whyte, A. C.; Gloer, J. B.; Wicklow, D. T.; Dowd, P. F., Sclerotiamide: a new
member of the paraherquamide class with potent antiinsectan activity from the sclerotia
of Aspergillus sclerotiorum. J. Nat. Prod. 1996, 59, 1093-1095.
195. Xu, Y.-K.; Yang, S.-P.; Liao, S.-G.; Zhang, H.; Lin, L.-P.; Ding, J.; Yue, J.-M.,
Alkaloids from Gelsemium elegans. J. Nat. Prod. 2006, 69, 1347-1350.
196. Kogure, N.; Ishii, N.; Kitajima, M.; Wongseripipatana, S.; Takayama, H., Four
Novel Gelsenicine-Related Oxindole Alkaloids from the Leaves of Gelsemium elegans
Benth. Org. Lett. 2006, 8, 3085-3088.
197. Lin, L. Z.; Yeh, S.; Cordell, G. A.; Ni, C. Z.; Clardy, J., Three oxindole
alkaloids from Gelsemium species. Phytochemistry 1991, 30, 679-683.
198. Lin, L.; Cordell, G. A.; Ni, C.; Clardy, J., Oxindole alkaloids from Gelsemium
elegans. Phytochemistry 1991, 30, 1311-1315.
199. Kondoh, M.; Usui, T.; Mayumi, T.; Osada, H., Effects of tryprostatin derivatives
on microtubule assembly in vitro and in situ. J. Antibiot. 1998, 51, 801-804.
200. Usui, T.; Kondoh, M.; Cui, C.-B.; Mayumi, T.; Osada, H., Tryprostatin A, a
specific and novel inhibitor of microtubule assembly. Biochem. J. 1998, 333, 543-548.
201. Woehlecke, H.; Osada, H.; Herrmann, A.; Lage, H., Reversal of breast cancer
resistance protein-mediated drug resistance by tryprostatin A. Int. J. Cancer 2003, 107,
721-728.
202. Zhao, S.; Smith, K. S.; Deveau, A. M.; Dieckhaus, C. M.; Johnson, M. A.;
Macdonald, T. L.; Cook, J. M., Biological Activity of the Tryprostatins and Their
Diastereomers on Human Carcinoma Cell Lines. J. Med. Chem. 2002, 45, 1559-1562.
203. Zhang, C.; Lu, X., Phosphine-Catalyzed Cycloaddition of 2,3-Butadienoates or
2-Butynoates with Electron-Deficient Olefins. A Novel [3+2] Annulation Approach to
Cyclopentenes. J. Org. Chem. 1995, 60, 2906-2908.
204. Shu, L.-H.; Sun, W.-Q.; Zhang, D.-W.; Wu, S.-H.; Wu, H.-M.; Xu, J.-F.; Lao,
X.-F., Phosphine-catalyzed [3+2] cycloadditions of buta-2,3-dienoates with
[60]fullerene. Chem. Commun. 1997, 79-80.

Chapter 9: References 218
205. Xu, Z.; Lu, X., Phosphine-catalyzed [3+2] cycloaddition reaction of methyl 2,3-
butadienoate and N-tosylimines. A novel approach to nitrogen heterocycles.
Tetrahedron Lett. 1997, 38, 3461-3464.
206. O'Donovan, B. F.; Hitchcock, P. B.; Meidine, M. F.; Kroto, H. W.; Taylor, R.;
Walton, D. R. M., Phosphine-catalyzed cycloaddition of buta-2,3-dienoates and but-2-
ynoates to [60]fullerene. Chem. Commun. 1997, 81-82.
207. Pyne, S. G.; Schafer, K.; Skelton, B. W.; White, A. H., Synthesis of novel
conformationally restricted L-glutamate analogs. Chem. Commun. 1997, 2267-2268.
208. Zhu, G.; Chen, Z.; Jiang, Q.; Xiao, D.; Cao, P.; Zhang, X., Asymmetric [3+2]
Cycloaddition of 2,3-Butadienoates with Electron-Deficient Olefins Catalyzed by Novel
Chiral 2,5-Dialkyl-7-phenyl-7-phosphabicyclo[2.2.1]heptanes. J. Am. Chem. Soc. 1997,
119, 3836-3837.
209. Xu, Z.; Lu, X., A Novel [3+2] Cycloaddition Approach to Nitrogen
Heterocycles via Phosphine-Catalyzed Reactions of 2,3-Butadienoates or 2-Butynoates
and Dimethyl Acetylenedicarboxylate with Imines: A Convenient Synthesis of
Pentabromopseudilin. J. Org. Chem. 1998, 63, 5031-5041.
210. Lu, X.; Zhang, C.; Xu, Z., Reactions of Electron-Deficient Alkynes and Allenes
under Phosphine Catalysis. Acc. Chem. Res. 2001, 34, 535-544.
211. Ung, A. T.; Schafer, K.; Lindsay, K. B.; Pyne, S. G.; Amornraksa, K.; Wouters,
R.; Van der Linden, I.; Biesmans, I.; Lesage, A. S. J.; Skelton, B. W.; White, A. H.,
Synthesis and Biological Activities of Conformationally Restricted Cyclopentenyl-
Glutamate Analogues. J. Org. Chem. 2002, 67, 227-233.
212. Pham, T. Q.; Pyne, S. G.; Skelton, B. W.; White, A. H., Regioselective and
diastereoselective phosphine-catalyzed [3+2] cycloadditions to 5-methylenehydantoins:
reversal of regioselectivity using chiral N-2-butynoyl-(4S)-benzyloxazolidinone.
Tetrahedron Lett. 2002, 43, 5953-5956.
213. Du, Y.; Lu, X.; Yu, Y., Highly Regioselective Construction of Spirocycles via
Phosphine-Catalyzed [3+2]-Cycloaddition. J. Org. Chem. 2002, 67, 8901-8905.
214. Methot, J. L.; Roush, W. R., Nucleophilic phosphine organocatalysis. Adv.
Synthesis Catal. 2004, 346, 1035-1050.
215. Ezquerra, J.; Pedregal, C.; Mico, I.; Najera, C., Efficient synthesis of 4-
methylene-L-glutamic acid and its cyclopropyl analog. Tetrahedron: Asymmetry 1994,
5, 921-926.

Chapter 9: References 219
216. Yong, S. R. Unpublished Honours Thesis; UOW: Wollongong, NSW, Australia,
2003.
217. Grigg, R.; Coulter, T., Sequential 1,3-dipolar cycloaddition-palladium catalyzed
cyclization. A powerful new tactical combination. Tetrahedron Lett. 1991, 32, 1359-
1362.
218. Hinman, R. L.; Bauman, C. P., Reactions of 3-bromooxindoles. The synthesis of
3-methyleneoxindole. J. Org. Chem. 1964, 29, 2431-2437.
219. Grigg, R.; Millington, E. L.; Thornton-Pett, M., Spiro-oxindoles via bimetallic
[Pd(0)/Ag(I)] catalytic intramolecular Heck-1,3-dipolar cycloaddition cascade reactions.
Tetrahedron Lett. 2002, 43, 2605-2608.
220. Rossiter, S., A convenient synthesis of 3-methyleneoxindoles: cytotoxic
metabolites of indole-3-acetic acids. Tetrahedron Lett. 2002, 43, 4671-4673.
221. Chiericato, M.; Dalla Croce, P.; Licandro, E., Heterocycles from ylides. Part 7.
Reactivity of 3-arylmethyleneindolin-2-ones with stabilized sulfur ylides. J. Chem. Soc.,
Perkin Trans. 1 1979, 211-213.
222. Selvakumar, N., Personal communication by author. In January 2004.
223. Vitry, C.; Vasse, J. L.; Dupas, G.; Levacher, V.; Queguiner, G.; Bourguignon, J.,
Stable annelated chiral NADH models with a rigidified amide part in the quinoline
series: synthesis, reactivity and grafting on a Merrifield resin. Tetrahedron 2001, 57,
3087-3098.
224. Murphy, J. A.; Rasheed, F.; Gastaldi, S.; Ravishanker, T.; Lewis, N., Synthesis
of functionalized indolines by radical-polar crossover reactions. J. Chem. Soc., Perkin
Trans. 1 1997, 1549-1558.
225. Blay, G.; Fernandez, I.; Monje, B.; Pedro, J. R., Diastereoselective Michael
addition of (S)-mandelic acid enolate to nitroalkenes. Enantioselective synthesis of α-
hydroxy-α,β-diaryl-γ-lactams. Tetrahedron 2004, 60, 165-170.
226. Longmire, J. M.; Wang, B.; Zhang, X., Highly Enantioselective Ag(I)-Catalyzed
[3+2] Cycloaddition of Azomethine Ylides. J. Am. Chem. Soc. 2002, 124, 13400-13401.
227. Fonquerna, S.; Moyano, A.; Pericas, M. A.; Riera, A., A convenient preparation
of N-(2-alkynoyl) derivatives of chiral oxazolidin-2-ones and bornane-10,2-sultam.
Tetrahedron: Asymmetry 1997, 8, 1685-1691.
228. Evans, D. A.; Ellman, J. A., The total syntheses of the isodityrosine-derived
cyclic tripeptides OF4949-III and K-13. Determination of the absolute configuration of
K-13. J. Am. Chem. Soc. 1989, 111, 1063-1072.

Chapter 9: References 220
229. Evans, D. A.; Scheidt, K. A.; Downey, C. W., Synthesis of (-)-epibatidine. Org.
Lett. 2001, 3, 3009-3012.
230. Gemma, S.; Butini, S.; Fattorusso, C.; Fiorini, I.; Nacci, V.; Bellebaum, K.;
McKissic, D.; Saxena, A.; Campiani, G., A palladium-catalyzed synthetic approach to
new Huperzine A analogues modified at the pyridone ring. Tetrahedron 2002, 59, 87-93.
231. Snowden, R. L.; Linder, S. M.; Wuest, M., A regioselective cyclohexannulation
procedure via dienamine [4+2] cycloaddition. Synthesis of functionalized decalins. Helv.
Chim. Acta 1989, 72, 892-905.
232. Simmons, H. E.; Cairns, T. L.; Vladuchick, S. A.; Hoiness, C. M.,
Cyclopropanes from unsaturated compounds, methylene iodide, and zinc-copper couple.
Org. React. 1973, 20, 1-131.
233. Helquist, P., Comprehensive Organic Synthesis. Pergamon Press: New York,
1991; Vol. 4, p 968.
234. Ferrara, M.; Cordero, F. M.; Goti, A.; Brandi, A.; Estieu, K.; Paugam, R.;
Ollivier, J.; Salaun, J., Intramolecular cycloaddition/rearrangement of
alkylidenecyclopropane nitrones from palladium(0)-catalyzed alkylation of amino acid
derivatives. Eur. J. Org. Chem. 1999, 2725-2739.
235. Krief, A.; Provins, L.; Froldbise, A., Diastereoselective synthesis of dimethyl
cyclopropane-1,1-dicarboxylates from a (γ-alkoxyalkylidene)malonate and sulfur and
phosphorus ylides. Tetrahedron Lett. 1998, 39, 1437-1440.
236. Chinchilla, R.; Najera, C.; Garcia-Granda, S.; Menendez-Velazquez, A.,
Synthesis of (R)- and (S)-2,3-methanovaline from (2S)-N-benzoyl-2-tert-butyl-4-
methylene-1,3-oxazolidin-5-one. Tetrahedron Lett. 1993, 34, 5799-5802.
237. Avery, T. D.; Fallon, G.; Greatrex, B. W.; Pyke, S. M.; Taylor, D. K.; Tiekink, E.
R. T., Mechanistic Investigations on the Reaction between 1,2-Dioxines and Bulky
Stabilized Phosphorus Ylides: An Efficient Route to Closely Related Cyclopropane
Stereoisomers. J. Org. Chem. 2001, 66, 7955-7966.
238. Avery, T. D.; Haselgrove, T. D.; Rathbone, T. J.; Taylor, D. K.; Tiekink, E. R.
T., New synthetic methodology utilizing 1,2-dioxines and stabilized phosphorus ylides:
a highly diastereoselective cyclopropanation reaction. Chem. Commun. 1998, 333-334.
239. Moher, E. D., Novel asymmetric synthesis of a bicyclo[3.1.0]hexane derivative
by an efficient retro-Diels-Alder strategy. Tetrahedron Lett. 1996, 37, 8637-8640.
240. Tang, C. S. F.; Rapoport, H. Reactions of sulfonium ylides with diene esters. J.
Org. Chem. 1973, 38, 2806-2809.

Chapter 9: References 221
241. Ye, S.; Huang, Z.-Z.; Xia, C.-A.; Tang, Y.; Dai, L.-X., A Novel Chiral
Sulfonium Ylide: Highly Enantioselective Synthesis of Vinylcyclopropanes. J. Am.
Chem. Soc. 2002, 124, 2432-2433.
242. Curley, R. W., Jr.; DeLuca, H. F., A study of the stereoselectivity of
cyclopropanation of some α,β-unsaturated aldehydes by ethyl
(dimethylsulfuranylidene)acetate. J. Org. Chem. 1984, 49, 1944-1946.
243. Hoenke, N.; Payne, L. J.; Parsons, P. J.; Hitchcock, P. B., Studies on the base-
catalyzed addition of amines to alkynes and allenes: A novel cascade sequence for the
construction of spirocyclic aminals. Synlett 2006, 654-656.
244. Collado, I.; Dominguez, C.; Ezquerra, J.; Pedregal, C.; Monn, J. A.,
Stereoselective cyclopropanation of enones with ethyl dimethylsulfonium acetate
bromide in the presence of DBU. Tetrahedron Lett. 1997, 38, 2133-2136.
245. Jiang, T.; Kuhen, K. L.; Wolff, K.; Yin, H.; Bieza, K.; Caldwell, J.; Bursulaya,
B.; Wu, T. Y.-H.; He, Y. Bioorg. Med. Chem. Lett. 2006, 49, 3432-3435.
246. Bennett, G. B.; Mason, R. B.; Shapiro, M. J., Reactivity of oxoindole-D3,a-
acrylates toward diazoalkanes: an unusual ring expansion. J. Org. Chem. 1978, 43,
4383-4385.
247. Franke, A., Spirocyclic 2-indolinones by 1,3-dipolar cycloaddition. Justus
Liebigs Ann. Chem. 1978, 717-725.
248. Shanmugam, P.; Vaithiyanathan, V.; Viswambharan, B., Synthesis of
functionalized 3-spirocyclopropane-2-indolones from isomerized Baylis-Hillman
adducts of isatin. Tetrahedron 2006, 62, 4342-4348.
249. Eistert, B.; Kurze, W.; Mueller, G. W., Preparation and reactions of 3-pyridyl-
and 4-pyridyldiazomethanes. Justus Liebigs Ann. Chem. 1970, 732, 1-6.
250. Selvakumar, N.; Yadi Reddy, B.; Sunil Kumar, G.; Iqbal, J., Dimethyl malonate
as a one-carbon source: a novel method of introducing carbon substituents onto
aromatic nitro compounds. Tetrahedron Lett. 2001, 42, 8395-8398.
251. Ellis, D.; Kuhen, K. L.; Anaclerio, B.; Wu, B.; Wolff, K.; Yin, H.; Bursulaya, B.;
Caldwell, J.; Karanewsky, D.; He, Y. Bioorg. Med. Chem. Lett. 2006, 16, 4246-4251.
252. Heras-Lopez, A. M.; Pino-Gonzalez, M. S.; Sarabia-Garcia, F.; Lopez-Herrera,
F. J., Reaction of O-Benzyl- and 4,6-O-Benzylidene-D-Gluco- and D-Galactopyranose
Derivatives with Amide-Stabilized Sulfur Ylides: Stereoselectivity and Reactivity. J.
Org. Chem. 1998, 63, 9630-9634.

Chapter 9: References 222
253. Ma, D.; Jiang, Y., Stereocontrolled cyclopropanation of Garner's aldehyde
derived enones. Tetrahedron: Asymmetry 2000, 11, 3727-3736.
254. Ratts, K. W.; Yao, A. N., Chemistry of resonance-stabilized sulfonium ylides. J.
Org. Chem. 1966, 31, 1689-1693.
255. Zhou, Y.-G.; Li, A.-H.; Hou, X.-L.; Dai, L.-X., The aziridination of N-
tosylimines with amide-stabilized sulfonium ylides: a simple and efficient preparation
of aziridinyl carboxamides. Tetrahedron Lett. 1997, 38, 7225-7228.
256. Gamble, A. B., Unpublished work. In University of Wollongong, 2005.
257. Walker, G. N.; Smith, R. T.; Weaver, B. N., Synthesis of new 3-
(pyridylmethylene)-, 3-(pyridylmethyl)-, 3-(piperidylmethyl)-, and 3-(β-
alkylaminoethyl)-2-indolinones. The reduction of isoindogenides, nitro compounds, and
pyridines in a series of 2-indolinones. J. Med. Chem. 1965, 8, 626-637.
258. Pavia, D. L.; Lampman, G. M.; Kriz, G. S., Introduction to Spectroscopy: A
Guide for Students of Organic Chemistry. Harcourt, Inc.: USA, 2001; p A-13.
259. Anderton, N.; Cockrum, P. A.; Colegate, S. M.; Edgar, J. A.; Flower, K.;
Gardner, D.; Willing, R. I., (-)-Phalarine, a furanobisindole alkaloid from Phalaris
coerulescens. Phytochemistry 1999, 51, 153-157.
260. Anderton, N.; Cockrum, P. A.; Walker, D. W.; Edgar, J. A., Plant-associated
toxins: agricultural, phytochemical and ecological aspects CAB International:
Wallingford, 1994; p 269.
261. Tufariello, J. J., Alkaloids from nitrones. Acc. Chem. Res. 1979, 12, 396-403.
262. Black, J. D., Protein kinase C-mediated regulation of the cell cycle. Frontiers in
Bioscience [Electronic Publication] 2000, 5, D406-D423.
263. Padwa, A.; Pearson, W. H.; Editors, Synthetic Applications of 1,3-Dipolar
Cycloaddition Chemistry Toward Heterocycles and Natural Products. [In: Chem.
Heterocycl. Comp. (Chichester, U. K.), 2002; p 940.
264. Huisgen, R., 1,3-Dipolar cycloaddition - introduction, survey, mechanism. 1,3
[One,Three]-Dipolar Cycloaddit. Chem. 1984, 1, 1-176.
265. Padwa, A.; Editor, 1,3-Dipolar Cycloaddition Chemistry, Vol. 2. 1984; p 704.
266. Garcia, H. H.; Brar, G. A.; Nguyen, D. H. H.; Bjeldanes, L. F.; Firestone, G. L.,
Indole-3-carbinol (I3C) inhibits cyclin-dependent kinase-2 function in human breast
cancer cells by regulating the size distribution, associated cyclin E forms, and
subcellular localization of the CDK2 protein complex. J. Biol. Chem. 2005, 280, 8756-
8764.

Chapter 9: References 223
267. Huisgen, R.; Weinberger, R., Are any nonstereospecific 1,3-dipolar
cycloadditions known? A revision. Tetrahedron Lett. 1985, 26, 5119-5122.
268. Huisgen, R., 1,3-Dipolar cycloadditions. 76. Concerted nature of 1,3-dipolar
cycloadditions and the question of diradical intermediates. J. Org. Chem. 1976, 41, 403-
419.
269. Huisgen, R.; Mloston, G.; Langhals, E., The first two-step 1,3-dipolar
cycloadditions: interception of intermediate. J. Org. Chem. 1986, 51, 4085-4087.
270. Padwa, A.; Kline, D. N.; Koehler, K. F.; Matzinger, M.; Venkatramanan, M. K.,
Cycloaddition of nitrones with allenes. An example of steric control of regiochemistry.
J. Org. Chem. 1987, 52, 3909-3917.
271. Padwa, A.; Editor, 1,3-Dipolar Cycloaddition Chemistry, Vol. 1. 1984; p 817 pp.
272. Hein, J. E.; Hultin, P. G., Recyclable supports for stereoselective 1,3-dipolar
cycloadditions: application of a fluorous oxazolidinone chiral auxiliary. Tetrahedron:
Asymmetry 2005, 16, 2341-2347.
273. Wang, P.-F.; Gao, P.; Xu, P.-F., Synthesis and 1,3-dipolar cycloadditions of two
new chiral geometrically fixed α-alkoxycarbonyl nitrones from a single chiral source.
Synlett 2006, 1095-1099.
274. Stecko, S.; Pasniczek, K.; Jurczak, M.; Urbanczyk-Lipkowska, Z.; Chmielewski,
M., Double asymmetric induction in 1,3-dipolar cycloaddition of five-membered cyclic
nitrones to 2-(5H)-furanones. Tetrahedron: Asymmetry 2006, 17, 68-78.
275. Closa, M.; De March, P.; Figueredo, M.; Font, J., One step preparation and 1,3-
dipolar cycloadditions of (S)-5-hydroxymethyl-1-pyrroline N-oxide. Tetrahedron:
Asymmetry 1997, 8, 1031-1037.
276. Hanessian, S.; Bayrakdarian, M., Asymmetric synthesis of diversely substituted
N-hydroxypyrrolidines using cycloadditions with chiral nitrone enolate/ylides.
Tetrahedron Lett. 2002, 43, 967-971.
277. Belzecki, C.; Panfil, I., Cycloaddition of chiral nitrones. Asymmetric synthesis
of isoxazolidines. J. Org. Chem. 1979, 44, 1212-1218.
278. Suga, H.; Nakajima, T.; Itoh, K.; Kakehi, A., Highly Exo-Selective and
Enantioselective Cycloaddition Reactions of Nitrones Catalyzed by a Chiral
Binaphthyldiimine-Ni(II) Complex. Org. Lett. 2005, 7, 1431-1434.
279. Dugovic, B.; Fisera, L.; Cyranski, M. K.; Hametner, C.; Pronayova, N.; Obranec,
M., Reversal of regioselectivity of nitrone cycloadditions by Lewis acids. Helv. Chim.
Acta 2005, 88, 1432-1443.

Chapter 9: References 224
280. Jensen, K. B.; Roberson, M.; Jorgensen, K. A., Catalytic enantioselective 1,3-
dipolar cycloaddition reactions of cyclic nitrones: A simple approach for the formation
of optically active isoquinoline derivatives. J. Org. Chem. 2000, 65, 9080-9084.
281. Herrera, R.; Nagarajan, A.; Morales, M. A.; Mendez, F.; Jimenez-Vazquez, H.
A.; Zepeda, L. G.; Tamariz, J., Regio- and stereoselectivity of captodative olefins in
1,3-dipolar cycloadditions. A DFT/HSAB theory rationale for the observed
regiochemistry of nitrones. J. Org. Chem. 2001, 66, 1252-1263.
282. Fukui, K., Recognition of stereochemical paths by orbital interaction. Acc. Chem.
Res. 1971, 4, 57-64.
283. Houk, K. N., Frontier molecular orbital theory of cycloaddition reactions. Acc.
Chem. Res. 1975, 8, 361-369.
284. Sustmann, R., Orbital energy control of cycloaddition reactivity. Pure Appl.
Chem. 1974, 40, 569-593.
285. Asrof, A. S.; Senaratne, P. A.; Illig, C. R.; Meckler, H.; Tufariello, J. J., Nitrone
cycloadditions. Regiochemistry. Tetrahedron Lett. 1979, 4167-4170.
286. Houk, K. N.; Bimanand, A.; Mukherjee, D.; Sims, J.; Chang, Y.-M.; Kaufman,
D. C.; Domelsmith, L. N., Nitrone ionization potentials and cycloaddition
regioselectivities. Heterocycles 1977, 7, 293-299.
287. Sims, P. A.; Wong, C. F.; McCammon, J. A., A Computational Model of
Binding Thermodynamics: The Design of Cyclin-dependent Kinase 2 Inhibitors. J. Med.
Chem. 2003, 46, 3314-3325.
288. Huisgen, R.; Seidl, H.; Bruening, I., 1,3-Dipolar cycloadditions. XLIX. Kinetics
and mechanism of nitrone addition to unsaturated compounds. Chem. Ber. 1969, 102,
1102-1116.
289. Gree, R.; Tonnard, F.; Carrie, R., The 1,3-dipolar cycloadditions. XXIII.
Interpretation for the approach of the dipolarophile to the 1,3-dipole. Bull. Soc. Chim.
Fr. 1975, 1325-1330.
290. Burdisso, M.; Gamba, A.; Gandolfi, R.; Toma, L.; Rastelli, A.; Schiatti, E., Syn-
anti isomerism in the 1,3-dipolar cycloaddition to cis-3,4-disubstituted cyclobutenes. 5.
Diastereoselectivity in the reaction with diazoalkanes. J. Org. Chem. 1990, 55, 3311-
3321.
291. Whitham, G. H.; Boyle, L. W.; Peagram, M. J., Relative reactivities of normal
and angle strained cycloolefins in 1,3-dipolar cycloaddition reactions. Rotational
barriers in nitrones. J. Chem. Soc. [Sect] B: Phys. Org. 1971, 1728-1733.

Chapter 9: References 225
292. Padwa, A.; Fisera, L.; Koehler, K. F.; Rodriguez, A.; Wong, G. S. K.,
Regioselectivity associated with the 1,3-dipolar cycloaddition of nitrones with electron-
deficient dipolarophiles. J. Org. Chem. 1984, 49, 276-281.
293. Rigolet, S.; Melot, J. M.; Vebrel, J.; Chiaroni, A.; Riche, C., The reaction of a-
and w-methylene lactams with nitrones. Influence of electronic and geometric factors on
the stereoselectivity of their 1,3-dipolar cycloaddition. Perkin 1 2000, 1095-1103.
294. Joucla, M.; Hamelin, J., 1,3-Dipolar cycloadditions. Part 27. The two modes of
addition of nitrones to methyl acrylate and to trimethyl ethylenetricarboxylate. J. Chem.
Res., Synop. 1978, 276-277.
295. Huisgen, R.; Grashey, R.; Seidl, H.; Hauck, H., 1,3-Dipolar cycloadditions. XLII.
Nitrone additions to other aryl-conjugated ethylenes and vinyl ethers. Chem. Ber. 1968,
101, 2559-2567.
296. Huisgen, R.; Grashey, R.; Hauck, H.; Seidl, H., 1,3-Dipolar cycloadditions. XLI.
Nitrone addition to styrene. Orientation and steric course. Chem. Ber. 1968, 101, 2548-
2558.
297. Ali, S. A.; Wazeer, M. I. M., Regiochemistry and stereochemistry of 1,3-dipolar
cycloaddition of a cyclic nitrone. J. Chem. Soc., Perkin Trans. 1: Org. Bioorg. Chem.
1988, 597-605.
298. Raunak; Kumar, V.; Mukherjee, S.; Poonam; Prasad, A. K.; Olsen, C. E.;
Schaeffer, S. J. C.; Sharma, S. K.; Watterson, A. C.; Errington, W.; Parmar, V. S.,
Microwave-mediated synthesis of spiro-(indoline-isoxazolidines): mechanistic study
and biological activity evaluation. Tetrahedron 2005, 61, 5687-5697.
299. Rigolet, S.; Goncalo, P.; Melot, J. M.; Vebrel, J., The 1,3-dipolar cycloaddition
of 3-methylene-N-substituted isoindolones and nitrones by classical and microwave
techniques: reactivity and stereochemical studies. J. Chem. Res. 1998, 686-687.
300. Williams, R. M.; Zhai, W.; Aldous, D. J.; Aldous, S. C., Asymmetric [1,3]-
dipolar cycloaddition reactions: synthesis of highly substituted proline derivatives. J.
Org. Chem. 1992, 57, 6527-6532.
301. Lo, M. M. C.; Neumann, C. S.; Nagayama, S.; Perlstein, E. O.; Schreiber, S. L.,
A Library of Spirooxindoles Based on a Stereoselective Three-Component Coupling
Reaction. J. Am. Chem. Soc. 2004, 126, 16077-16086.
302. Cope, A. C.; Haven, A. C., Jr., Rearrangement of oxime N-ethers. J. Am. Chem.
Soc. 1950, 72, 4896-4903.

Chapter 9: References 226
303. Utzinger, G. E.; Regenass, F. A., N-Arylnitrones. II. Helv. Chim. Acta 1954, 37,
1892-1901.
304. Wheeler, O. H.; Gore, P. H., Absorption spectra of azo and related compounds.
II. Substituted phenylnitrones. J. Am. Chem. Soc. 1956, 78, 3363-3366.
305. Bonnett, R.; Clark, V. M.; Giddey, A.; Todd, A., Experiments towards the
synthesis of corrins. I. Preparation and reactions of some D1-pyrrolines. A novel proline
synthesis. J. Chem. Soc. 1959, 2087-2093.
306. Robl, J. A.; Hwu, J. R., An efficient method for the generation of N-methyl
nitrones. J. Org. Chem. 1985, 50, 5913-5916.
307. Rupe, H.; Wittwer, R., The condensation products of phenylhydroxylamine with
hydroxymethylene compounds and carbinols. III. Diphenylbromomethane and
phenylhydroxylamine. Helv. Chim. Acta 1922, 5, 217-220.
308. Thesing, J.; Mayer, H., Cyclic nitrones. I. Dimeric 2,3,4,5-tetrahydropyridine N-
oxides. VI. Hydrazine and hydroxylamine derivatives. Chem. Ber. 1956, 89, 2159-2167.
309. Murahasi, S.; Mitsui, H.; Watanabe, T.; Zenki, S., The reaction of N-mono- and
N,N-disubstituted hydroxylamines with palladium catalyst. Tetrahedron Lett. 1983, 24,
1049-1052.
310. Murahashi, S.; Mitsui, H.; Shiota, T.; Tsuda, T.; Watanabe, S., Tungstate-
catalyzed oxidation of secondary amines to nitrones. α-Substitution of secondary
amines via nitrones. J. Org. Chem. 1990, 55, 1736-1744.
311. Stappers, F.; Broeckx, R.; Leurs, S.; Van den Bergh, L.; Agten, J.; Lambrechts,
A.; Van den Heuvel, D.; De Smaele, D., Development of a Safe and Scalable Amine-to-
Nitrone Oxidation: A Key Step in the Synthesis of R107500. Org. Proc. Res. Dev. 2002,
6, 911-914.
312. Murahashi, S.-I.; Imada, Y.; Ohtake, H., Tungstate-Catalyzed Decarboxylative
Oxidation of N-Alkyl-α-amino Acids: An Efficient Method for Regioselective
Synthesis of Nitrones. J. Org. Chem. 1994, 59, 6170-6172.
313. Davis, F. A.; Stringer, O. D., Chemistry of oxaziridines. 2. Improved synthesis
of 2-sulfonyloxaziridines. J. Org. Chem. 1982, 47, 1774-1775.
314. Gibson, A. E.; Arris, C. E.; Bentley, J.; Boyle, F. T.; Curtin, N. J.; Davies, T. G.;
Endicott, J. A.; Golding, B. T.; Grant, S.; Griffin, R. J.; Jewsbury, P.; Johnson, L. N.;
Mesguiche, V.; Newell, D. R.; Noble, M. E. M.; Tucker, J. A.; Whitfield, H. J., Probing
the ATP Ribose-Binding Domain of Cyclin-Dependent Kinases 1 and 2 with O6-
Substituted Guanine Derivatives. J. Med. Chem. 2002, 45, 3381-3393.

Chapter 9: References 227
315. Hoffman, R. V., m-Trifluoromethylbenzenesulfonyl chloride. Org. Synth. 1981,
60, 121-126.
316. Harlow, L. PhD Thesis. UNT, Newcastle upon Tyne, 2004.
317. Olah, G. A.; Kuhn, S. J.; Flood, S. H., Aromatic substitution. VIII. Mechanism
of the nitronium tetrafluoroborate nitration of alkylbenzenes in tetramethylene sulfone
solution. Remarks on certain aspects of electrophilic aromatic substitution. J. Am. Chem.
Soc. 1961, 83, 4571-4580.
318. Kuhn, S. J.; Olah, G. A., Aromatic substitution. VII. Friedel-Crafts type nitration
of aromatics. J. Am. Chem. Soc. 1961, 83, 4564-4571.
319. Gilman, H.; Bebb, R. L., Relative reactivities of organometallic compounds. XX.
Metalation. J. Am. Chem. Soc. 1938, 61, 109-112.
320. Wittig, G.; Fuhrmann, G., Exchange reactions with phenyllithium. V. Behavior
of the halogenated anisoles toward phenyllithium. Berichte der Deutschen Chemischen
Gesellschaft [Abteilung] B: Abhandlungen 1940, 73B, 1197-1218.
321. Sibi, M. P.; Snieckus, V., The directed ortho lithiation of O-aryl carbamates. An
anionic equivalent of the Fries rearrangement. J. Org. Chem. 1983, 48, 1935-1937.
322. Beak, P.; Brown, R. A., The tertiary amide as an effective director of ortho
lithiation. J. Org. Chem. 1982, 47, 34-46.
323. Winkle, M. R.; Ronald, R. C., Regioselective metalation reactions of some
substituted (methoxymethoxy)arenes. J. Org. Chem. 1982, 47, 2101-2108.
324. Reed, J. N. PhD Thesis. University of Waterloo, 1985.
325. Fuhrer, W.; Gschwend, H. W., Ortho functionalization of aromatic amines:
ortho lithiation of N-pivaloylanilines. J. Org. Chem. 1979, 44, 1133-1136.
326. Reed, J. N.; Rotchford, J.; Strickland, D., Synthesis of 1,2,3,4-
tetrahydroquinolines and 1,2,3,4-tetrahydro-1,6-naphthyridines by directed lithiation
reactions. Tetrahedron Lett. 1988, 29, 5725-5728.
327. Chattopadhyah, S.; Snieckus, V., Unpublished results.
328. Skowronska-Ptasinska, M.; Verboom, W.; Reinhoudt, D. N., Effect of different
dialkylamino groups on the regioselectivity of lithiation of O-protected 3-
(dialkylamino)phenols. J. Org. Chem. 1985, 50, 2690-2698.
329. Langer, A. W., Jr., Mechanistic aspects of N-chelated organolithium catalysis.
Advances in Chemistry Series 1974, 130, 1-22.
330. Metallinos, C.; Nerdinger, S.; Snieckus, V., N-Cumyl Benzamide, Sulfonamide,
and Aryl O-Carbamate Directed Metalation Groups. Mild Hydrolytic Lability for Facile

Chapter 9: References 228
Manipulation of Directed Ortho Metalation Derived Aromatics. Org. Lett. 1999, 1,
1183-1186.
331. Djuric, S.; Venit, J.; Magnus, P., Silicon in synthesis: STABASE
[tetramethyldisilylazacyclopentane] adducts. A new primary amine protecting group:
alkylation of ethyl glycinate. Tetrahedron Lett. 1981, 22, 1787-1790.
332. Hoegberg, T., Protection of primary amines as stabase adducts. Acta Pharm.
Suec. 1986, 23, 414-415.
333. Grega, K. C.; Barbachyn, M. R.; Brickner, S. J.; Mizsak, S. A., Regioselective
Metalation of Fluoroanilines. An Application to the Synthesis of Fluorinated
Oxazolidinone Antibacterial Agents. J. Org. Chem. 1995, 60, 5255-61.
334. Guggenheim, T. L., Protection of substituted anilines with 1,1,4,4-tetramethyl-
1,4-bis(N,N-dimethylamino)disilethylene. Tetrahedron Lett. 1984, 25, 1253-1254.
335. Bonar-Law, R. P.; Davis, A. P.; Dorgan, B. J., The benzostabase protecting
group for primary amines: application to aromatic amines. Tetrahedron Lett. 1990, 31,
6721-6724.
336. Bonar-Law, R. P.; Davis, A. P.; Dorgan, B. J.; Reetz, M. T.; Wehrsig, A., The
benzostabase protecting group for primary amines: application to aliphatic amines.
Tetrahedron Lett. 1990, 31, 6725-6728.
337. Davis, A. P.; Gallagher, P. J., 1,1,3,3-Tetraethyl-1,3-disilaisoindolines;
chromatographically stable silicon-based protection for primary amines. Tetrahedron
Lett. 1995, 36, 3269-3272.
338. Yong, S. R. Rational design of novel selective CDK2 inhibitors. University of
Wollongong, Wollongong, 2002.
339. Dermatakis, A.; Luk, K.-C.; DePinto, W., Synthesis of Potent Oxindole CDK2
Inhibitors. Bioorg. Med. Chem. 2003, 11, 1873-1881.
340. Moon, M. J.; Lee, S. K.; Lee, J.-W.; Song, W. K.; Kim, S. W.; Kim, J. I.; Cho,
C.; Choi, S. J.; Kim, Y.-C., Synthesis and structure-activity relationships of novel
indirubin derivatives as potent anti-proliferative agents with CDK2 inhibitory activities.
Bioorg. Med. Chem. 2006, 14, 237-246.
341. Pham, T. Q., Unpublished work.
342. Harding, J. R.; Hughes, R. A.; Kelly, N. M.; Sutherland, A.; Wilis, C. L.,
Syntheses of isotopically labelled L-α-amino acids with an asymmetric center at C-3.
Perkin 1 2000, 3406-3416.

Chapter 9: References 229
343. Rajeswaran, W. G.; Cohen, L. A., Studies on protection of oxindoles.
Tetrahedron 1998, 54, 11375-11380.
344. Capet, M.; David, F.; Bertin, L.; Hardy, J. C., A two-step synthesis of
camphosultam. Synth. Commun. 1995, 25, 3323-3327.
345. Vogel, A. I.; Furniss, B. S.; Hannaford, A. J.; Rogers, V.; Smith, P. W. G.;
Tatchell, A. R., Vogel's Textbook of Practical Organic Chemistry. 1978; p 1280.
346. Varma, R. S.; Dahiya, R., Sodium borohydride on wet clay: solvent-free
reductive amination of carbonyl compounds using microwaves. Tetrahedron 1998, 54,
6293-6298.
347. Jaszay, Z. M.; Petnehazy, I.; Toke, L., Phase-transfer catalyzed synthesis of
amides and esters of carboxylic acids. Synthesis 1989, 745-747.
348. Suzuki, T.; Kouketsu, A.; Matsuura, A.; Kohara, A.; Ninomiya, S.-i.; Kohda, K.;
Miyata, N., Thiol-based SAHA analogues as potent histone deacetylase inhibitors.
Bioorg. Med. Chem. Lett. 2004, 14, 3313-3317.
349. Tamura, Y.; Uenishi, J.; Maeda, H.; Choi, H.-D.; Ishibashi, H., Facile syntheses
of oxindoles and 3-oxo-1,2,3,4-tetrahydroisoquinolines. Synthesis 1981, 534-537.
350. Dicken, C. M.; DeShong, P., Reactions at high pressures. [3+2] Dipolar
cycloaddition of nitrones with vinyl ethers. J. Org. Chem. 1982, 47, 2047-2051.
351. Ciesielski, W.; Kudzin, Z. H.; Tasz, M.; Drabowicz, J., The application of the
trifluoroacetic anhydride-sodium iodide (TFAA-I) system for quantitative determination
of nitrones. Can. J. Chem. 1990, 68, 679-684.
352. Havlicek, L.; Hanus, J.; Vesely, J.; Leclerc, S.; Meijer, L.; Shaw, G.; Strnad, M.,
Cytokinin-derived cyclin-dependent kinase inhibitors: synthesis and cdc2 inhibitory
activity of olomoucine and related compounds. J. Med. Chem. 1997, 40, 408-412.
353. Bottger, V.; Bottger, A.; Howard, S. F.; Picksley, S. M.; Chene, P.; Garcia-
Echeverria, C.; Hochkeppel, H.-K.; Lane, D. P., Identification of novel mdm2 binding
peptides by phage display. Oncogene 1996, 13, 2141-2147.

Appendix 1: X-ray Crystal Structures 230
Appendix 1: X-ray Crystal Structures
X-ray 1: Compound 63
X-ray 2: Compound 82
X-ray 3: Compound 88
X-ray 4: Compound 90
X-ray 5: Compound 85a
X-ray 6: Compound 91
X-ray 7: Compound 85b
X-ray 8: Compound 87
X-ray 9: Compound 94

Appendix 1: X-ray Crystal Structures 231
X-ray 10: Compound 102
X-ray 11: Compound 103 X-ray 12: Compound (rac)-93a
X-ray 13: Compound (S)-76
X-ray 14: Compound 117
X-ray 15: Compound 118
X-ray 16: Compound 119
X-ray 17: Compound 126 X-ray 18: Compound 133

Appendix 1: X-ray Crystal Structures 232
X-ray 19: Compound 108a
X-ray 20: Compound 108b
X-ray 21: Compound 139a
X-ray 22: Compound 139b
X-ray 23: Compound 170a
X-ray 24: Compound 187a

Appendix 2: X-ray Crystal Data 233
Appendix 2: X-ray Crystal Data
Full spheres of CCD area-detector diffractometer data were measured (Bruker AXS
instrument, ω-scans; monochromatic Mo Kα radiation; λ = 0.71073 Å) yielding Nt(otal)
reflections, these merging to N unique after 'empirical'/multiscan 'absorption correction'
(proprietary software) (Rint cited), No (I > 2σ(I)) considered 'observed'. All reflections
were used in the full matrix refinements on F2, refining anisotropic displacement
parameters for the non-hydrogen atoms, hydrogen atoms being included according to a
riding model. Reflection weights were of the form (σ2(F2) + nw F2)-1. Neutral atom
complex scattering factors were employed within the SHELXL 97 and Xtal 3.7 program
systems.
CCDC depositions: 266132 (63), 266133 ((S)-76), 266134 (82), 266135 (85a), 266136
(87), 266137 (88), 266138 (103), 268591 (85b), 268592 (90), 268593 (91), 268594
(102), 622516 (126), 622517 (117), 622518 (118), 622519 (119), 622520 (133), 622521
(108a), 622522 (108b) and 622523 (139b). Crystal/refinement data: 63. C16H23NO5, M
= 309.4. Triclinic, space group 1P (#2), a = 6.085(1), b = 9.534(2), c = 14.682(2) Å, α
= 79.674(3), β = 80.484(3), γ = 77.536(3)°, V = 811.0 Å3. Dc (Z = 2) = 1.267 g cm-3.
µMo = 0.09 mm-1; specimen: 0.27 x 0.20 x 0.12 mm; 'T'min/max = 0.76. 2θmax = 58°; Nt =
7868, N = 3938 (Rint = 0.025), No = 2934; R = 0.047, Rw = 0.052 (nw = 0.4). |∆ρmax| =
0.34(3) e Å-3. (x,y,z,Uiso)H refined. T ca. 153 K. (S)-76. C24H28N2O7S, M = 488.6.
Monoclinic, space group P21 (#4), a = 12.085(5), b = 7.243(1), c = 13.924(2) Å, β =
101.040(3)°, V = 1196 Å3. Dc (Z = 2) = 1.356 g cm-3. µMo = 0.18 mm-1; specimen: 0.28
x 0.18 x 0.14 mm; 'T'min/max = 0.89. 2θmax = 58°; Nt = 11606, N = 3210 (Rint = 0.025),
No = 2241; R = 0.053, Rw = 0.056 (nw = 0.6). |∆ρmax| = 0.28(3) e Å-3. xabs = 0.06(16). T
ca. 300 K. 82. C11H15NO3, M = 209.3. Triclinic, space group , a = 5.597(1), b =
7.910(2), c = 12.861(3) Å, α = 86.683(5), β = 79.353(5), γ = 75.871(5)°, V = 542.6 Å3.
Dc (Z = 2) = 1.281 g cm-3. µMo = 0.09 mm-1; specimen: 0.63 x 0.12 x 0.08 mm;
'T'min/max = 0.78. 2θmax = 58°; Nt = 6707, N = 2861 (Rint = 0.027), No = 2274; R = 0.044,
Rw = 0.051 (nw = 0.3). |∆ρmax| = 0.36(3) e Å-3. T ca. 153 K. 85a. C22H22N2O2, M =

Appendix 2: X-ray Crystal Data 234
346.5. Orthorhombic, space group Pca21 (#29), a = 24.140(16), b = 7.734(5), c =
9.836(7) Å, V = 1836 Å3. Dc (Z = 4) = 1.253 g cm-3. µMo = 0.08 mm-1; specimen: 0.13 x
0.07 x 0.03 mm; 'T'min/max = 0.76. 2θmax = 50°; Nt = 13466, N = 1698 (Rint = 0.11), No =
1284; R = 0.066, Rw = 0.090 (nw = 3). |∆ρmax| = 0.30(6) e Å-3. xabs not refined. T ca.
153 K. 85b. C24H27N3O2, M = 389.5. Triclinic, space group 1P , a = 8.260(3), b =
10.892(4), c = 12.538(4) Å, α = 67.764(6), β = 82.723(6), γ = 81.564(6)°, V = 1030 Å3.
Dc (Z = 2) = 1.256 g cm-3. µMo = 0.08 mm-1; specimen: 0.12 x 0.08 x 0.04 mm;
'T'min/max = 0.88. 2θmax = 50°; Nt = 9351, N = 3586 (Rint = 0.046), No = 2207; R = 0.057,
Rw = 0.072 (nw = 0.2). |∆ρmax| = 0.32(4) e Å-3. T ca. 153 K. 87. C15H10N2O2, M = 256.3.
Monoclinic, space group P21/c (#14), a = 8.204(2), b = 18.128(5)), c = 8.914(2) Å, β =
107.823(4)°, V = 1262 Å3. Dc (Z = 4) = 1.349 g cm-3. µMo = 0.09 mm-1; specimen: 0.12
x 0.08 x 0.08 mm; 'T'min/max = 0.92. 2θmax = 50°; Nt = 10582, N = 2207 (Rint = 0.065),
No = 1747; R = 0.056, Rw = 0.081 (nw = 2). |∆ρmax| = 0.30(5) e Å-3. T ca. 153 K. 88.
C11H15NO3, M = 209.3. Monoclinic, space group P21/c, a = 14.2396(11), b = 6.2065(5),
c = 11.9144(9) Å, β = 102.276(4)°, V = 1029 Å3. Dc (Z = 4) = 1.351 g cm-3. µMo = 0.06
mm-1; specimen: not recorded; 'T'min/max = 1.00. 2θmax = 45°; Nt = 28315, N = 2908
(Rint = 0.97), No = 2306; R = 0.043, Rw = 0.052 (nw = 0.8). |∆ρmax| = 0.49(4) e Å-3.
(x,y,z,Uiso)H refined. T ca. 120 K. 90. C16H17NO3, M = 271.3. Orthorhombic, space
group Pbca (# 61), a = 11.667(7), b = 10.836(6), c = 21.167(12) Å, V = 2676 Å3. Dc (Z
= 8) = 1.347 g cm-3. µMo = 0.09 mm-1; specimen: 0.13 x 0.06 x 0.03 mm; 'T'min/max =
0.00. 2θmax = 50°; Nt = 20960, N = 2394 (Rint = 0.16), No = 975; R = 0.087, Rw = 0.011
(nw = 4.5). |∆ρmax| = 0.36(5) e Å-3. Comment. The carboxylate group was modelled as
disordered over a pair of sites set at equal occupancy after trial refinement; the
associated hydrogen bonding interactions appear to be with the C=O group oxygen of a
neighbouring molecule. 91. C22H22N2O2, M = 346.4. Triclinic, space group , a =
5.121(1), b = 9.606(1), c = 12.379(1) Å, α = 69.657(3), β = 81.925(3), γ = 74.622(3)°, V
= 871.8 Å3. Dc (Z = 2) = 1.320 g cm-3. µMo = 0.09 mm-1; specimen: 0.25 x 0.22 x 0.04
mm; 'T'min/max = 0.89. 2θmax = 65°; Nt = 12099, N = 6128 (Rint = 0.025), No = 4580; R =

Appendix 2: X-ray Crystal Data 235
0.055, Rw = 0.077 (nw = 3.5). |∆ρmax| = 0.53(3) e Å-3. 102. C12H11NO2, M = 201.2.
Triclinic, space group 1P , a = 6.667(4), b = 10.528(6), c = 14.730(8) Å, α = 105.632(9),
β = 98.876(10), γ = 90.305(10)°, V = 983 Å3. Dc (Z = 4) = 1.360 g cm-3. µMo = 0.09
mm-1; specimen: 0.13 x 0.11 x 0.03 mm; 'T'min/max = 0.74. 2θmax = 50°; Nt = 9300, N =
3355 (Rint = 0.25), No = 1645; R = 0.12, Rw = 0.25 (nw = 2.5). |∆ρmax| = 1.2(2) e Å-3.
Comment. The conformations of the two C5 rings differ slightly, the torsions being 'flat'
in the bonds to either side of the CO group respectively. 103. C13H12N2O3, M = 244.3.
Monoclinic, space group P21/n (#14), a = 16.203(3), b = 6.373(3), c = 29.233(2) Å, β =
90.207(7)°, V = 1156 Å3. Dc (Z = 4) = 1.404 g cm-3. µMo = 0.10 mm-1; specimen: 0.50 x
0.10 x 0.04 mm; 'T'min/max = 0.89. 2θmax = 52°; Nt = 7690, N = 2009 (Rint = 0.043), No =
1528; R = 0.059, Rw = 0.096 (nw = 6). |∆ρmax| = 0.62(4) e Å-3. T ca. 153 K. 126.
C11H9NO3, M = 203.2. Monoclinic, space group C2/c )15No.,( 62hC , a = 16.77(2), b =
8.15(1), c = 14.62(2) Å, β = 108.87(2)°, V = 1891 Å3. Dc (Z = 8) = 1.427 g cm-3. µMo =
0.11mm-1; specimen: 0.19 x 0.03 x 0.03 mm; 'T'min/max = 0.74. 2θmax = 50°; Nt = 7254,
N = 1677 (Rint = 0.26), No = 762; R = 0.15, Rw = 0.15. |∆ρmax| = 1.1 e Å-3. T ca. 153 K.
Variata. Weak and limited data would support meaningful refinement of isotropic
displacement parameter forms only for C, N, O. Reflection weights were (σ2(F) + 0.006
F2)-1 (refinement on |F|). 117. C14H15NO6, M = 293.3. Triclinic, space group 1P
2)No.,( 1iC , a = 8.752(4), b = 11.117(4), c = 14.046(6) Å, α = 85.74(3), β = 89.75(3), γ
= 89.20(3)°, V = 1363 Å3. Dc (Z = 4) = 1.429 g cm-3. µMo = 0.11 mm-1; specimen: 0.39
x 0.31 x 0.25 mm (no correction). 2θmax = 71°; Nt = 28823, N = 12033 (Rint = 0.022), No
= 7238; R1 = 0.043, wR2 = 0.12. |∆ρmax| = 0.44 e Å-3. T ca. 100 K. Variata. Reflections
weights were (σ2(F2) + (0.0629P)2)-1 (P = . 118. C12H11NO3, M = 217.2.
Monoclinic, space group P21/c , a = 6.114(2), b = 5.987(2), c = 27.476(7)
Å, β = 95.166(4)°, V = 1002 Å3. Dc (Z = 4) = 1.440 g cm-3. µMo = 0.105 mm-1;
specimen: 0.48 x 0.40 x 0.30 mm; 'T'min/max = 0.85. 2θmax = 56°; Nt = 8868, N = 2407
(Rint = 0.033), No = 2128; R1 = 0.044, wR2 = 0.082 (nw = 1.2). |∆ρmax| = 0.34 e Å-3. T

Appendix 2: X-ray Crystal Data 236
ca. 153 K. 119. C13H13NO3, M = 231.3. Monoclinic, space group P21/c, a = 11.9436(6),
b = 8.4060(10), c = 11.3500(10) Å, β = 95.829(3)°, V = 1134 Å3. Dc (Z = 4) = 1.355 g
cm-3. µMo = 0.097 mm-1; specimen: 0.37 x 0.24 x 0.16 mm; 'T'min/max = 0.99. 2θmax =
52°; Nt = 6250, N = 2290 (Rint = 0.032), No = 1732; R1 = 0.048, wR2 = 0.11 (nw = 3).
|∆ρmax| = 0.36 e Å-3. T ca. 100 K. 133. C18H16N2O5, M = 340.3. Monoclinic, space
group P21/c, a = 8.1790(10), b = 23.616(4), c = 8.8730(10) Å, β = 109.230(10)°, V =
1618 Å3. Dc (Z = 4) = 1.397 g cm-3. µMo = 0.10 mm-1; specimen: 0.17 x 0.07 x 0.008
mm; 'T'min/max = 0.98. 2θmax = 55°; Nt = 14944, N = 3611 (Rint = 0.058), No = 2086; R1
= 0.053, wR2 = 0.12 (nw = 0.067). |∆ρmax| = 0.29 e Å-3. T ca. 100 K. Variata. Reflection
weights were (σ2(F2) + (0.067P)2)-1 (P = )3/)2( 2c
2c FF + . 108a. C14H10N2O, M = 222.3.
Monoclinic, space group P21/n variant);14No.,( 52hC , a = 15.23(2), b = 4.005(4), c =
17.85(2) Å, β = 90.15(2)°, V = 1089 Å3. Dc (Z = 4) = 1.356 g cm-3. µMo = 0.088 mm-1;
specimen: 0.15 x 0.14 x 0.06 mm; 'T'min/max = 0.71. 2θmax = 50°; Nt = 8083, N = 1869
(Rint = 0.10), No = 882; R1 = 0.071, wR2 = 0.22 (nw = 7.3). |∆ρmax| = 0.54 e Å-3. T ca.
298 K. 108b. C14H10N2O, M = 222.3. Monoclinic, space group C2/c, a = 15.819(2), b
=8.722(1), c = 16.153(2) Å, β = 105.780(2)°, V = 2145 Å3. Dc (Z = 8) = 1.377 g cm-3.
µMo = 0.089 mm-1; specimen: 0.45 x 0.12 x 0.09 mm; 'T'min/max = 0.95. 2θmax = 57°; Nt
= 9413, N = 2602 (Rint = 0.23), No = 2117; R1 = 0.041, wR2 = 0.091 (nw = 5.3). |∆ρmax|
= 0.28 e Å-3. T ca. 160 K. 139b. C18H16N2O3, M = 308.3. Orthorhombic, space group
Pna21 )33No.,( 92vC , a = 25.698(1), b = 11.048(3), c = 5.395(7) Å, V = 1532 Å3. Dc (Z
= 4) = 1.337 g cm-3. µMo = 0.092 mm-1; specimen: 0.25 x 0.12 x 0.09 mm; 'T'min/max =
0.87. 2θmax = 59°; Nt = 10953, N = 2071 (Rint = 0.0.57), No = 1722; R1 = 0.060, wR2 =
0.14 (nw = 5.3). |∆ρmax| = 0.44 e Å-3. T ca. 160 K.

Appendix 3: Biological Testing Procedures 237
Appendix 3: Biological Testing Procedures
A3.1 Cytostatic Cellular testing
A3.1.1 Prelimary Cytostaticity Studies of 85a and 87
Preliminary cytostaticity testing for 85a and 87 was performed by Dr. Grant McArthur,
Peter MacCallum Cancer Institute, Melbourne. In general, chemicals used in biological
testing were obtained from Sigma, the CDK drugs from Calbiochem (Roscovitine), 85a
and 87 from Wollongong University, and the cell lines from ATCC (B16 and HL60)
and Dr. S. Collins, Fred Hutchinson Cancer Research Centre, Seattle, Washington
(Mpro). Optical density measurements for all assays were performed on a Molecular
Devices ThermoMax microplate reader. Cell counting was performed using a Sysmex
500 particle counter. Flow cytometry analysis was performed on a FACS Calibur flow
cytometer. The following procedures described herein are accordance with information
relayed by Dr. G. McArthur.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) Assay
A solution of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (5
mg/mL) was prepared in sterile PBS. This solution was filtered, sterilised and stored at -
20 °C and in the absence of light. A solution of lysis buffer was prepared by the
addition of two solutions: 20% w/v SDS to a 37 °C solution of 50% N,N-
dimethylformamide (DMF) in demineralised water. The pH was adjusted to 4.7 through
the addition of 2.5% of 80% acetic acid and 2.5% of 1 N HCl. Mpro and HL60 cells
were plated out in flat bottom 96-well microtiter plates at a concentration of 5000
cells/mL and 10000 cells/mL respectively, in 100 µL of DMEM and 10% FCS (CSL)
and assayed under exponential growth phase. Eight replicates of each set of drug
dilutions were made and incubated overnight. Roscovitine, 85a and 87 were then added
from 10 mM stocks in DMSO. A constant DMSO concentration of either 0.25% or 1%
was maintained at each drug dilution by the addition of DMSO. The plates were
incubated for a further 48 or 72 h before analysis. 20 µL of MTT (5 mg/ml) was added
to each well and then the plates were incubated for 4 h at 37 °C. 100 µL of lysis buffer
was then added and the plates left at RT and in the absence of light overnight. The
optical densities were measured at 550 nm using a microplate reader. A blank
containing media treated with MTT and lysis buffer and a DMSO control were used.

Appendix 3: Biological Testing Procedures 238
Sulphorhodamine B (SRB) Assay
Exponentially growing B16 F1 cells were plated at 1000 cells/well in 96 well microtiter
plates. After 24 h the drugs were added at different concentrations. 72 h after drug
addition, cells were fixed in the wells through the addition of TCA (final concentration
10%) and incubation at 4 °C for 1 h. Plates were then washed in water to remove all
media and fixative, and allowed to dry. 100 µL/well of sulphorhodamine B (SRB) was
added to stain the protein present, and left for 30 min. Plates were again washed in
water, then quickly in 1% acetic acid, and left to dry. The dye was then solubilised by
the addition of 10 mM Tris (100 µL/ well) and allowed to mix on a shaker for 5
minutes. The optical density of each well was measured at 550 nm.
Cell-cycle Analysis
Cell-cycle phase analysis was performed on in vitro samples using the propidium iodide
(PI) stain. B16 murine melanoma cells were incubated in suspension cultures for 24 h
before the addition of the drug diluted in 1% DMSO or with DMSO alone. 48 h after
the addition of the drug, samples were harvested with 0.5 mM EDTA, normalised for
cell number using a Sysmex 500 particle counter and fixed in a solution of fetal bovine
serum (2%) in PBS (1 mL) and 100% ethanol (4 mL). Samples were then centrifuged
and resuspended in a PI buffer which contained 10 µg/mL PI, 250 µg/mL RNase in PBS
and 2% FCS. The samples were then incubated at 37 °C for 30 min. Analysis of the
DNA content was conducted using flow cytometry. The proportion of cells in different
phases of the cell-cycle was determined using ModFit software.
Whole Cell Assays
The first assessment of biological activity was the conducting of standard whole cell
assays. Two standard assays, MTT and SRB, were used to assess the cytostatic and/or
proliferative activity of the three drugs. The MTT assay is a commonly used and well-
established quantitative colorimetric assay for the measurement of cellular proliferation,
viability, and cytostaticity. This assay utilises an indirect staining measurement to assess
the relative proportion of metabolically active cells. The assay is based upon the
cleavage of the yellow tetrazolium salt MTT, to form the water-insoluble, dark blue
formazan crystals. This event can only occur within living cells via the mitochondrial
enzyme succinate-dehydrogenase. The SRB assay is another quantitative colorimetric
assay that in contrast to MTT ascertains the proportion of viable cells by the staining of
cellular protein. The absorbances plotted as a function of concentration of converted dye

Appendix 3: Biological Testing Procedures 239
directly correlates to the number of live cells in the culture. These assays are simple,
rapid, and suitable for large scale analysis through the utilisation of the 96-well
microtiter plates and scanning by a multiwell spectrophotometer.
In general, cultured cell lines can be grouped into two categories, non-adherent and
adherent, based on the way they proliferate. Non-adherent cell lines, for example
myeloid cells, are cultured in suspension whereas adherent cell lines, like melanoma
cells need to form attachments to develop. In these experiments, the MTT assay was
used for the non-adherent cell lines, Mpro and HL60 and the SRB assay was used for
the adherent cell line, B16.
A3.1.2 Cytostaticity Screening against H460, MCF-7 and SF-268
Cytostaticity screening against the cell lines, H460 (human non small cell lung), MCF-7
(human breast) and SF-268 (human CNS) were performed by Dr. C Cullinane at the
Andrew Durant Drug Testing Facility, Research Division of the Peter MacCallum
Cancer Centre, Melbourne. First, 5 mM drug stocks were prepared in DMSO. Cells
were then exposed to 25 µM of each drug for 72 h. The cells were then fixed, stained
with SRB and the percentage cell growth relative to the solvent control determined.
A3.2 Protein Inhibition Studies
A3.2.1 CDK2 Assay
CDK2: Recombinant CDK2/cyclin A3 (C-terminal cyclin A fragment encoding residues
171-432). CDK2 Buffer: 50 mM Tris-HCl pH 7.5, 5 mM MgCl2. Histone H1 (type III-S)
final concentration 0.83 mg/ mL. 12.5 µM ATP in final assay (containing [32P] ATP at
0.01 µCi/ µL). Human CDK2/ cyclin A3 was used in the assays.
The assay involved reacting the purified protein with [32P] ATP and the substrate, so
that 1 pmol of ATP is incorporated for 1 µL of kinase per minute. Histone H1 was used
as the substrate in the CDK2 assays. The assays involved detecting the radiolabelled γ-
phosphate group from ATP to histone. Inhibition studies were performed using an assay
established by Vesely et al.352 Inhibitors were tested in 1% DMSO, as it was assumed
that using greater than 1% DMSO would increasingly disrupt the tertiary structure of
the enzyme. Radiolabelled [32P] ATP was added to the buffered inhibitor, which was
incubated at 30 °C for 10 min. After incubation the assay mixture was spotted onto
phosphocellulose filter paper to bind the protein, allowed to dry for 20-30 seconds and
then transferred to stirring 1% phosphoric acid to dilute the phosphate substrate, thereby
terminating the reaction. The filters were washed a further 5-6 times, and dried, before

Appendix 3: Biological Testing Procedures 240
being placed on a scintillation counter. The amount of [32P] incorporated into histone
was measured by counting the number of disintegrations per minute. This gave the
values of pmol enzyme activity. Control assays were also carried out in 1% DMSO,
allowing the percentage inhibition to be calculated since the control is classed as having
a 100% activity. % Activity remaining = inhibitor pmol / pmol in control. % Inhibition
= 100 - % activity remaining. IC50 values for the inhibitors were determined by plotting
% inhibition values against inhibitor concentration.
A3.2.2 MDM2-p53 interaction using ELISA
96-Well black and white high binding luminometry isoplates (Wallac, Cat N0 140-155)
were coated by overnight incubation at 35 °C with 200 µL per well of 5 µg mL-1
streptavidin (Chemicon International) in coating buffer (0.1 M Na2HPO4.2H2O; 0.1 M
citric acid; pH 5.0). The plates were washed five times in 1X Dissociation Enhanced
Lanthanide Fluorescence ImmunoAssay (DELFIA) buffer (Wallac) and then incubated
for 3 h, RT with saturation buffer (0.3 M D-sorbitol; 50 mM Tris; 150 mM NaCl; 0.1%
BSA; 0.05% sodium azide; pH 7.0) to block non-specific protein binding sites on the
plate. After removal of the buffer from the plates, they were allowed to dry in a sterile
laminar air flow hood at RT before incubation for 1 h at 4 °C with 200 µL per well of
100 µg mL-1 biotinylated IP3 peptide (b-IP3: Ac-Met-Pro-Arg-Phe19-Met-Asp-Tyr-Trp-
Glu-Gly-Leu26-Asn-NH2)353 dissolved in 0.05% DMSO-PBS pH 7.4 buffer. After
washing the wells three times with PBS, the plates were ready to use for MDM2
binding. For initial testing, the compounds and controls were plated out in triplicate into
clear 96 well plates (Nunc) in 10 µL aliquots to give final concentrations of 500 µM,
100 µM and 20 µM in the assay. Control samples consisted of 5% DMSO carrier alone
as a negative control and 100 nM active peptide (AP-B: Ac-Phe19-Met-Aib-Pmp-6-Cl-
Trp-Glu-Ac3-Leu26-NH2) as a positive control peptide antagonist of the MDM2-p53
interaction (IC50 = 5 nM). Compounds and controls aliquoted in 96-well plates were
pre-incubated at 20 oC for 20 min with 190 µL aliquots of optimised concentrations of
in vitro translated MDM2, before transfer of the MDM2-compound mixture to the b-IP3
streptavidin plates, and incubation at 4 oC for 90 min. After washing three times with
PBS to remove unbound MDM2, each well was incubated at 20 oC for 1 hour with a
TBS-Tween (50 mM Tris pH 7.5; 150 mM NaCl; 0.05% Tween 20 nonionic detergent)
buffered solution of primary mouse monoclonal anti-MDM2 antibody (Ab-5,
Calbiochem, used at a 1/200 dilution), then washed three times with TBS-Tween before

Appendix 3: Biological Testing Procedures 241
incubation for 45 min at 20 oC with a goat-anti-mouse horseradish peroxidase (HRP)
conjugated secondary antibody (Dako, used at 1/2000). The unbound secondary
antibody was removed by washing three times with TBS-Tween. The bound HRP
activity was measured by enhanced chemiluminesence (ECLTM, Amersham
Biosciences) using the oxidation of the diacylhydrazide substrate, luminol, to generate a
quantifiable light signal. The luminol substrate together with enhancer was
automatically injected into each well and the relative luminescence units (RLU)
measured over a 30 seccond interval using a Berthold MicroLumat-Plus LB 96V
microplate luminometer. The percentage MDM2 inhibition at a given concentration is
calculated as the (RLU detected in the compound treated sample ÷ RLU of DMSO
controls) x 100. The IC50 was calculated using a plot of % MDM2 inhibition versus
concentration and is the average of three independent experiments.




















