
6-Membered P-Heterocycles: Ring-Condensed 1,3,2-Diheterophosphorinane 2-Chalcogenides
47
6-membered P-heterocycles: Ring-condensed 1,3,2- diheterophosphorinane 2-chalcogenides Éva Frank* and János Wölfling Department of Organic Chemistry, University of Szeged, Hungary *Address correspondence to this author at the Department of Organic Chemistry, University of Szeged, H-6722 Szeged, Dóm tér 8., Hungary; Tel: +36-62-544275; E-mail: [email protected] szeged.hu 1
Transcript of 6-Membered P-Heterocycles: Ring-Condensed 1,3,2-Diheterophosphorinane 2-Chalcogenides

6-membered P-heterocycles: Ring-condensed 1,3,2-
diheterophosphorinane 2-chalcogenides
Éva Frank* and János Wölfling
Department of Organic Chemistry, University of Szeged, Hungary
*Address correspondence to this author at the Department of
Organic Chemistry, University of Szeged, H-6722 Szeged, Dóm
tér 8., Hungary; Tel: +36-62-544275; E-mail: [email protected]
szeged.hu
1

Keywords: 1,3,2-diheterophosphorinanes, stereostructures,
conformational preferences, 1H, 13C, 31P NMR studies
Abstract
Literature publications (up to the end of 2006) relating to
the synthesis, biological significance and conformational
behaviour of carbocycle- and heterocycle-condensed six-
membered tetracoordinate P(V) 1,3,2-diheterophosphorinanes are
reviewed. The replacement of carbon atoms of cyclohexane by P
and O and/or N to form 1,3,2-dioxa-, 1,3,2-oxaza- or 1,3,2-
diazaphosphorinanes introduces lone pair electrons instead of
hydrogens and changes the bond angles and bond lengths, and
can thereby lead to significantly different stereostructural
preferences of the hetero ring as compared with those of
cyclohexane. Although monocyclic 1,3,2-diheterophosphorinanes
often exist in a chair conformation in both the solid and
solution states, the existence of conformations other than a
chair can predominate in some cases for steric and
stereoelectronic reasons associated with the presence of bulky
2

groups on the P-containing ring or in polycyclic rigid
systems.
After a brief account on the most important pharmacological
and stereochemical features, involving the configuration
assignment and conformation determination of P-
diheterophosphorinanes, the present review mainly focuses on
the synthesis and stereostructural studies of carbo- and
heterocycle-condensed derivatives where the fused ring may
exert considerable effects on the conformational behaviour of
the P-hetero ring.
The bibliography includes 76 references.
1. INTRODUCTION
Monocyclic and condensed six-membered P-heterocycles have been
subjected to intense research during the past few decades [1].
3

Among them, particularly the tetracoordinate P(V) 1,3,2-
dioxaphosphorinanes and 1,3,2-oxazaphosphorinanes have
attracted considerable attention from both stereochemical and
pharmacological aspects (Figure 1) [2-7]. The main aims of
these studies were: (i) to clarify the mode of action of
second-messenger cyclic nucleotides (e.g. cAMP and cGMP), which
contain a 1,3,2-dioxaphosphorinane moiety and play important
roles in hormone action and cell communication [5, 8]; (ii) to
acquire more information on the biotransformations of the
clinically widely used antitumour agent cyclophosphamide and
its analogues, which contain a 1,3,2-oxazaphosphorinane ring,
in order to develop compounds with improved action and to
reveal certain structure-activity relationships [2, 9]; (iii)
to synthesise novel, potentially active 1,3,2-
diheterophosphorinane derivatives [10-13]; and (iv) to
investigate the unique conformational behaviour of differently
substituted six-membered P-hetero rings [1-7]. In contrast to
the 1,3,2-dioxa- and 1,3,2-oxazaphosphorinanes, the
corresponding 1,3,2-diaza analogues have been less
comprehensively studied, although their synthetic importance
increased when 1,3,2-diazaphosphinane phosphoramides proved to
4

be effective auxiliaries for the promotion of stereoselective
carbon-carbon or carbon-hydrogen bond-forming reactions [14].
Figure 1.
The recognition of a chiral bioactive compound via the active
site of a specific enzyme(s) responsible for its
biotransformation depends on the absolute configuration of one
or more asymmetric centres in the molecule, and the
conformation can also be determinative. Accordingly, the
stereochemistry is important for the bioactivities of chiral
heterophosphorinanes. Diheterophosphorinane rings display
stereostructural properties quite different from those of
cyclohexane as a consequence of the electron lone pairs on the
ring heteroatoms and the changed bond angles and lengths. In
general, both in solution and in the solid state, 1,3,2-
dioxaphosphorinanes adopt a chair conformation that is slightly
flattened at the P end, because of the relatively long PO
5

bonds. The interconversion between the two alternative chair
forms, however, is normally rapid in solution [4-7], unless
there is a conformational bias towards the population of one
or other conformer by very bulky groups on the P-ring and in
rigid polycycles. In these latter cases, one of the chair
conformers or an unusual intermediate twist [15], half-chair [16]
or boat [17] form becomes predominant and these systems can be
regarded as anancomeric [7]. The 1,3,2-oxazaphosphorinanes may
have an enhanced propensity towards twist conformations as
compared with the 1,3,2-dioxa series, although the ring N
substituent can have a significant modulating effect [18]. The
conformational preference of these heterocycles is mainly
influenced by the steric and electronic properties of the
substituents on the P, the size of the group on the ring N
atom and the P configuration [19-24]. In contrast to the
conformationally diverse 1,3,2-oxazaphosphorinane analogues,
the corresponding 1,3,2-N,N,P heterocycles are characterized
by chair or twist-chair conformations [25, 26]. Assignment of the
P configuration and detection of the predominant conformation
of the 1,3,2-diheterophosphorinanes in solution necessitate
different NMR techniques [4-7], but X-ray crystallography [27,
6

28] and ab initio calculations [29-33] are also important tools
for stereostructural analysis.
2. SYNTHETIC AND STEREOCHEMICAL ASPECTS OF
TETRACOORDINATE P(V) 1,3,2-DIHETEROPHOSPHORINANES
The synthesis and conformational behaviour of monocyclic and
some bicyclic diheterophosphorinanes (especially the 1,3,2-
dioxa and 1,3,2-oxaza derivatives) substituted at different
positions of the six-membered P-containing ring have been
thoroughly reviewed, by Bentrude and others [3-7]. Besides 2-oxo
derivatives, 2-thio- [34] and 2-seleno analogues [35] have
been reported that possess not only interesting conformational
features, but also variable pharmacological activities.
Accordingly, the in vitro and in vivo antitumour properties of
several cyclophosphamide-like molecules were evaluated in
search for 1,3,2-oxazaphosphorinanes highly cytotoxic to L1210
lymphoid leukaemia [36-38] or which act as tumour-targeted
prodrugs [12]. A number of 1,3,2-dioxaphosphorinanes have been
reported to exhibit diverse bioactivities and have different
synthetic applications, e.g. a potent calcium antagonist [39],
7

microsomal triglyceride-transfer protein inhibitors [40],
cytochrome P450 3A-activated prodrugs [41], efficient resolving
agents for the resolution of amines and amino acids [42] and
chiral auxilaries for diastereoselective condensation
reactions [43]. The most exhaustive studies, however, have
been focused on the conformational analyses of diastereomeric
1,3,2-diheterophosphorinanes. A general method for the
preparation of diheterophosphorinanes is the phosphorylation
of differently functionalized, originally chiral 1,3-diols,
1,3-aminoalcohols or 1,3-diamines with P-containing reagents.
During these ring-closures, two diastereomers differing in P
configuration can be obtained because of the stereogenic P
atom and they are usually separable. When both isomers are
available, different NMR methods can be used for the
assignment of the P configuration, and thus for the
determination of the P=O bond orientation. The following
indicators may be suitable for this purpose: (i) 31P NMR
chemical shifts [4, 7, 44, 45]; (ii) 1H chemical shifts due to
the 1,3-diaxial interactions; (iii) changes in 13C chemical
shifts arising from the shielding effect of oxygen; (iv) NOE
effects between the P substituent and ring protons; and (v) 1JP,N
8

coupling constants [46, 47]. 31P NMR spectroscopy has proved
very helpful for distinguishing between the related P-epimers.
In general, the isomer with the phosphoryl group axial absorbs
downfield of the isomer with the phosphoryl group equatorial.
Anomalous 31P chemical shifts have been reported for isomeric
phosphorinane structures [3, 4, 48], but have been left
unexplained or have been interpreted in terms of a chair to chair
[49] or a chair to twist-boat equilibration [24]. Hence, other
corroborative NMR parameters (such as characteristic 1H and 13C
NMR shifts) are needed in these cases for identification of
the diastereomers. Most electronegative substituents (e.g. Cl or
OR) prefer the axial position on P, with the important
exceptions of amino or substituted amino groups, which tend to
assume an equatorial location. The axial preference is
generally ascribed to the anomeric effect, i.e. stabilization of
n-* interactions between the lone pair electrons on the ring
heteroatom and the axial * P-X orbital [5], in combination
with reduced 1,3-syn-diaxial repulsions. Alkyl and aryl groups
usually prefer the equatorial position, the preference
increasing with the bulk of the substituent. The single
diastereomers display conformational flexibility in solution,
9

and the axial or equatorial propensity of a certain P
substituent can shift the conformational equilibrium, to some
extent, towards the conformer in which its orientation is
preferred. The specific conformational behaviour of these P-
containing rings has been handled in terms of a chair-
alternative boat or a chair-twist equilibrium, and the coupling
constants 3JH,H and 3JH,P have proved to be useful indicators of
possible dynamic processes [5-7]. A typical feature of 2-oxo-
1,3,2-diheterophosphorinanes, which exist predominantly in one
conformation in solution, is the combination of large 3JH,P
values for the protons equatorial to and hence antiperiplanar
to the P, and small 3JH,P values for the axial protons synclinal
to the P.
3. SYNTHESIS AND CONFORMATIONAL STUDY OF RING-CONDENSED
1,3,2-DIHETEROPHOSPHORINANES
3.1. 1,3,2-Diheterophosphorinanes condensed to a five-membered
carbo- or heterocyclic ring
In order to clarify the mode of action of natural second
messenger cyclic nucleoside monophosphates (1: cAMP and 2:
cGMP), the conformational behaviour of different analogous P-
10

derivatized dioxaphosphorinanes has been intensively studied
by means of NMR and crystallographic methods, which have
revealed that the six-membered cyclic phosphates, phosphonates
and phosphoramidates fused trans to a ribofuranose ring can
exist as equilibrium mixtures of chair and twist forms [3-7]
(Scheme 1). The interactions of natural cyclic nucleosides
with the active sites of the enzyme and therefore their
biological activities have been suggested to occur through the
pseudo-axial phosphoryl oxygen of the twist conformer.
Scheme 1
The same chair-twist equilibrium was observed for differently P-
substituted cyclopentane-fused model compounds, revealing that
the P configuration and the nature of the P substituent can
affect this conformational equilibrium, similarly as in the
cyclic nucleotides, decreasing or increasing the contribution
of one of the possible conformers [50]. Thus, the cis isomers (X
11

cis to H1) 3a8a and the trans isomers (X trans to H1) 9b and 10b
adopt exclusively the chair conformation, due to the axial
preferences of the OMe, OPh and Cl substituents in 3a8a and to
the equatorial tendency of NMe2 in 9b and 10b (Scheme 2). When
the OMe, OPh or Cl group (X) is situated equatorially in 3b8b,
its axial propensity drives the equilibrium toward the twist
conformer, where it can take up a pseudo-axial position. The
opposite is true for the axial NMe2 in 9a and 10a. Moreover,
the chair-twist equilibrium has been shown to be solvent-
sensitive.
Scheme 2
However, 2-oxo-1,3,2-dioxaphosphorinanes with cis-annelated to a
xylofuranose or cyclopentane ring, can adopt other
conformational states [6, 51-53] due to the less strained
12

arrangement of the fused rings [54, 55]. The P-epimeric pairs
of 5(R) and 5(S)-substituted P-OPh cyclic phosphates 1417 with
a cis-fused 1,2-O-isopropylidene--D-xylofuranose moiety, were
synthesised in two steps from 11 by applying a one-pot
hydrolysis–oxidation and Grignard reagent addition protocol
and subsequent phosphorylation of the corresponding 1,3-diol
precursors 12ac and 13ac [51] (Scheme 3). The existence of
certain conformers, or rather their dynamic equilibrium in
solution, was found to depend on the P configuration, and not
only the chair-twist, but also the chair-boat equilibrium was
observed in solution [50]. The coupling constants 3JH,H and 3JH,P
indicated the chair-twist equilibrium for the derivatives ac of
14 and 16 in solution, while in the solid state the chair
conformation was demonstrated to be more favourable by an X-
ray crystallographic study of 14a. In contrast, the strong
pseudo-axial-seeking force of the P-OPh group in 15and17 led
to a chair-boat equilibration in solution, with the exception of
15b, for which a chair-twist equilibrium was deduced. An upfield
shift of the axially oriented H1 signal of the furanose ring,
generated by the anisotropic shielding effect of the aromatic
ring, was noted as a result of the proximity of the H1 and the
13

pseudo-axial OPh group in the boat conformation of 15 and 17
[51, 52]. The contribution of the boat conformer was shown to
increase when the temperature was lowered, and this conformer
was confirmed by an X-ray analysis of 17b in the solid state.
Scheme 3
Despite the unfavourable equatorial orientation of the OPh
substituent in the chair conformer of 15, this conformation
becomes predominant when substituent R is a hydroxymethyl
group [53] (Scheme 3, 15d). The free OH function in 15d can
stabilize the chair form by intramolecular H-bonding with the
14

phosphoryl oxygen atom; as indicated by NMR and computational
calculations, this interaction can overcompensate the axial
preference of the OPh group.
Besides the previously-mentioned T, C1 and B1 forms
additional chair (C2) and distorted boat (B2) conformations have
been suggested for cyclic phosphates cis-fused to a cyclopentane
ring [56] (Scheme 4). However, on the basis of 1H NMR
measurements the existence of C2 was excluded for both P
epimers of 2-oxo- and 2-thio-1,3,2-dioxaphosphorinane
derivatives 1825, its non-occurrence being explained by the
strong steric interaction between the cis-fused phosphate and
cyclopentane rings. Accordingly, the cis phosphates (Y cis to H1)
18a23a and the trans isomers (X trans to H1) 24b and 25b were
demonstrated to populate almost exclusively the same C1
conformation as observed for 3’,5’-xylo-cAMP analogues [55]
with OMe, OPh or Cl in the preferred axial orientation in
18a23a, and with equatorial NMe2 in 24b and 25b. At the same
time, 18b23b, 24a and 25a were reported to exist as C1 B2
[55], rather than T C1 or C1 B1 equilibrium mixtures.
15

Scheme 4
However, conformation B2 can become predominant for the
epimeric pairs of D-ring-fused 1,3,2-dioxaphosphorinanes and
also for their 1,3,2-oxazaphosphorinane analogues, where the
five-membered ring D is part of a more rigid estrane
framework. In this regard, we recently reported the syntheses
and stereochemical analyses of dioxa- (2729) and
oxazaphosphorino[16,17]estrone derivatives (30a, 4650) [31,
32, 57] obtained from estrone precursors 26 [58, 59] and 27
[60] by direct ring-closures with different P reagents or via
phosphorylation of the reduced imines 4145 of 27 with
benzaldehydes 3135 (Scheme 5).
16

Scheme 5
The related epimers a and b of 2729 and 46-50 were separated
by column chromatography and their P configurations were
assigned by means of 31P, 1H and 13C NMR measurements. Analyses
of the coupling constants 3JH,H and 3JH,P and ab initio calculations
for the isomers of 2730 and 4750, supported by single-
crystal X-ray analysis for 27a, revealed that the P-
containing rings of the P-Ph and P-OPh-substituted epimers of
17

27, 28, 4650 and the only isolated isomer 30a, all exist in
a distorted boat conformation B2, both in solution and in the
solid state, regardless of the P configuration and the
substituent preferences (Scheme 6). The P substituent is
situated pseudo-axially in the a series (Y cis to H17), and
pseudo-equatorially in the b series (Y trans to H17) of
compounds. An approximately 0.6 ppm upfield shift of the H17
signal generated by the anisotropic shielding effect of the
P-Ph ring in 27a, 30a and 46a50a also supported conformation
B2. Nevertheless, the coupling pattern of 29a and the results
of stereochemical calculations suggested a T C2 equilibrium
mixture strongly shifted towards C2, flattened at the
phosphorus end, in view of the strong equatorial preference
of the N(CH2CH2Cl)2 group. The stereostructures were
demonstrated to be appreciably influenced by the rigidity of
the molecular structure, and the ordinary chair conformation
did not predominate in most cases. The angular methyl group
at C-13 may have a further steric modifying effect on the
preferred conformations.
18

Scheme 6
Besides the above-mentioned steroidal compounds 4650
containing a five-membered ring-condensed 1,3,2-
oxazaphosphorinane moiety, a further noteworthy example (54)
of this structural building block [61] can be found in the
literature with the N atom in a bridgehead position (Scheme
7). The bidentate P-ligand 54 was synthesised from
phosphinylisoxazolidine 51 by the reductive ring-opening and
subsequent ring-closure of 52 with
bis(diethylamino)phenylphosphine, followed by oxidation of the
intermediate phosphinyl-oxazaphosphorinane 53 to afford 54 as
a single diastereomer. Single-crystal X-ray analysis of 54
demonstrated that the oxazaphosphorinane ring adopted a twist
19

conformation with the P(O)Ph2 and P-Ph substituents in a cis
relationship.
Scheme 7
3.2. 1,3,2-Diheterophosphorinanes condensed to a six-membered
carbo- or heterocyclic ring
Bicyclic systems involving a dioxa- and an N-unsubstituted
oxazaphosphorinane moiety trans-fused to a cyclohexane ring were
thoroughly studied by Gorenstein et al. [23, 24, 62, 63] (Scheme
8). It was pointed out that a flexible chair-twist-boat
interconversion can occur in solution to a certain extent,
depending on the P configuration and the P-substituent
preferences. Thus, the P-OAr dioxaphosphorinane isomers 55a58a
and oxazaphosphorinane 60a adopt almost exclusively a chair
conformation in solution, with the OAr substituent in its
20

preferred axial orientation. The strong equatorial-seeking
force of the bis(2-chloroethyl)amino group in 59a, however,
strongly shifts the conformational equilibrium towards the twist-
boat form. The opposite is true for the b series of compounds;
while the NMR measurements supported the existence of the chair
conformer for 59b with an equatorial P-N(CH2CH2Cl)2 substituent,
compounds 55b58b and 60b, containing a P-OAr group, prefer to
adopt the twist-boat form due to the substituent preferences.
Depopulation of the chair conformation was found to occur to the
largest degree for 58b and 60b. Similar results were obtained
for the epimeric pairs a and b of the tetrahydropyran-fused 2-
chloro-1,3,2-dioxaphosphorinane-2-thione derivative 61 [64]
and the bicyclic cyclophosphamide analogue 62 [65]. The
coupling constants indicated that 61a and 62b are
predominantly in the chair, while 61b and 62a are in the twist-
chair conformation, with an axial or pseudo-axial Cl in 61 and
an equatorial or pseudo-equatorial bis(2-chloroethyl)amino
substituent in 62. However, single-crystal X-ray analyses
often reveal stereostructures in which the P-substituent is
situated in a disfavoured position [62, 65]. This can be
21

attributed to the small energy difference between the possible
conformers.
Scheme 8
In order to study the effect of the hetero ring N substituent
on the conformations of 2-[bis(2-chloroethyl)amino]-1,3,2-
oxazaphosphorinane 2-oxide derivatives not only trans- but also
cis-fused to cyclohexane, the N-H (69, 72) [63, 67], N-Me (70,
73) [29, 67] and N-Bn (71, 74) analogues [67] were synthesised
from the corresponding aminoalcohols (6365 and 6668) by
22

treatment with bis(2-chloroethyl)phosphoramidic dichloride in
THF (Scheme 9).
Scheme 9
As concerns the characteristic coupling constants 3JH,H and 3JH,P
of the trans-annelated derivatives 6971, the a series of
compounds (H8a cis to NR2) can presumably exist in an equilibrium
mixture of chair, twist-boat and two kinds of boat conformations
(Scheme 10). While a ca 50/50 mixture of chair/boat-1
conformation was assumed by Gorenstein et al. for 69a on the basis
of 1H and 13C measurements [63], a further detailed NMR study by
Pihlaja et al. also involving 15N NMR spectroscopy [46], on the
23

conformational behaviour of 69a and its N-substituted
derivatives 70a and 71a suggested the existence of the chair-
twist-boat equilibrium with only minor amounts of the two
alternative boat forms [46, 67]. However, conformational
optimization at the MP2 level additionally indicated the
population of the boat-1 conformer in 70a [29]. Moreover, the
nature of the N-3 substituent can affect the equilibration to
varying extents: the contribution of the twist-boat conformer
seems to decrease in the sequence methyl > benzyl > hydrogen
instead of benzyl > methyl > hydrogen, which would be feasible
considering the fact that the amount of the twist-boat
conformation depends on the size of the N substituent. The
anomalous sequence was explained by the unfavourable
interactions between the Ph group and other moieties of the
molecule in the twist form. The twist-boat conformation may be less
preferable for 71a because the benzyl group makes the electron
lone pair of the ring N less available for the nN → *
interaction with the axial P=O bond. The characteristic
couplings of the trans-fused isomers in the b series indicated
the chair conformation for 69b71b with an equatorial NR2
substituent [63, 67]. The geminal coupling between H4ax and H4eq
24

(ranging from -11.0 to -11.2) implies the equatorial
orientation of the ring N substituent in 69b71b, while in the
a epimers of 6971 the more negative values demonstrate that R1
displays some tendency to be located axially in the sequence
69a > 71a > 70a. Geometry optimizations utilizing the B3LYP
DFT method and subsequent vibrational analyses also confirmed
that the axial position of the N-3 substituent is less stable
for 69b and 70b and more favourable for 69a and 70a [29].
Scheme 10
The hetero ring of the cis-fused derivatives 7274 can exist in
two different chair conformations (O-in and O-out forms), this
situation being complicated by the possibility of boat and twist-
boat conformations for the a epimers (Scheme 11) [67]. On NMR
analysis of the a series of compounds, the O-in/O-out
25

equilibrium was found to shift towards the O-out conformation
to some extent, in the sequence methyl > benzyl > hydrogen;
the driving force was for the NR2 group to be located
equatorially, though a minor contribution of the twist-1
conformer was also suggested, especially for 74a. In contrast,
the b isomers of 7274 existed largely in the O-in chair
conformation, with NR2 in its preferred orientation.
Scheme 11
Since the substituents of the endocyclic N may exert
considerable effects on the conformation of the
diheterophosphorinane ring, cyclohexane-fused derivatives
containing the N next to the ring annelation position were
26

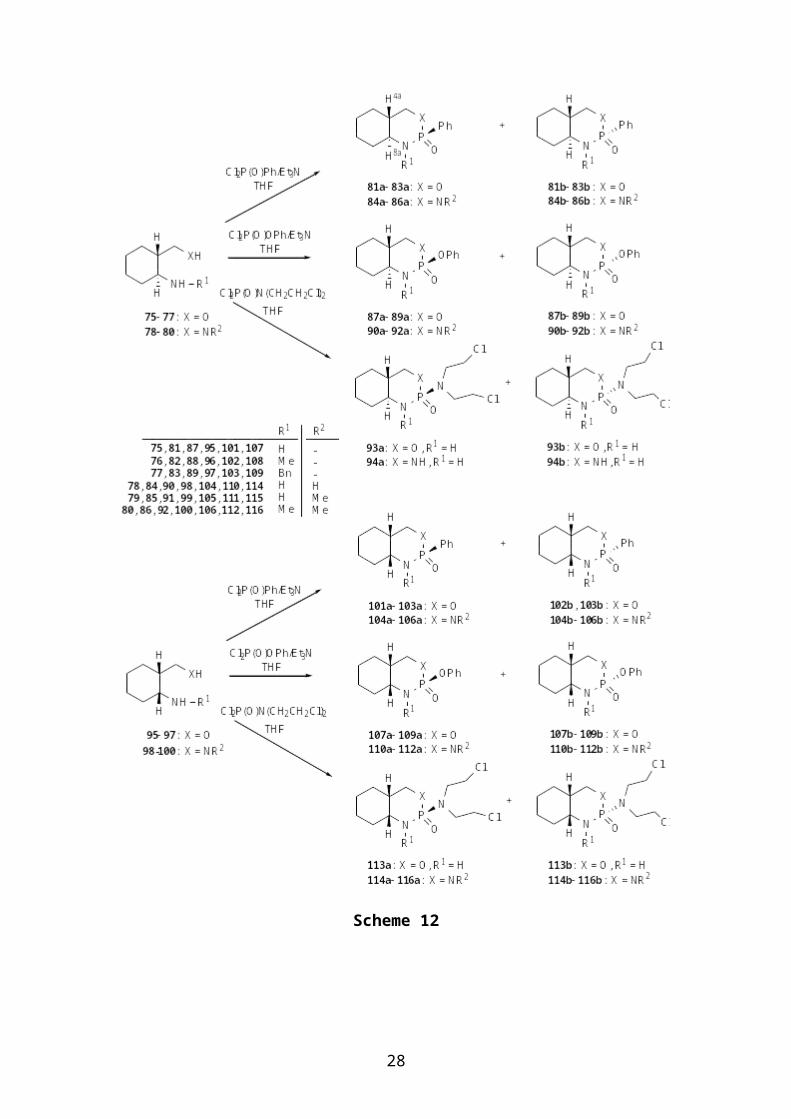
also examined. Thus, differently P- and N-substituted
oxazaphosphorinanes (8183, 8789, 93, 101103, 107109, 113)
[33, 68] and their diazaphosphorinane analogues (8486, 9092,
94, 104106, 110112, 114116) [14] trans- and cis-fused to
cyclohexane were synthesised from the corresponding
aminoalcohols 7577 and 9597 or diamines 7880 and 98100 with
three different P(V) reagents (Scheme 12). The yields of the
products and the epimeric ratios are listed in Table 1.
27
Scheme 12
28

Table 1. Yields and epimeric ratios of cyclohexane-condensedoxaza- and diazaphosphorinanes [33, 14]
Comp. Y R1 Yield(%)[b]
Ratio(a:b)
[c]
Comp.
Y R1 R2 Yield
(%)[b]
Ratio(a:b)
[c]
81 Ph H 47 [d] 84 Ph H H 29 67:3382b Ph Me 24 [e] 85 Ph H Me 31 36:6483 Ph Bn 33 55:45 86 Ph Me Me 25 55:4587 OPh H 62 48:52 90 OPh H H 47 50:5088 OPh Me 60 50:50 91 OPh H Me 65 49:5189 OPh Bn 58 41:59 92 OPh Me Me 44 50:5093 Mu[a] H 60 45:55 104 Ph H H 11 [d]
101a Ph H 22 [e] 105 Ph H Me 38 68:32102 Ph Me 32 18:82 106 Ph Me Me 22 13:87103 Ph Bn 53 56:44 110 OPh H H 44 52:48107 OPh H 48 73:27 111 OPh H Me 62 55:45108 OPh Me 44 49:51 112 OPh Me Me 67 50:50109 OPh Bn 41 49:51 114 Mu[a] H H 55 62:38113 Mu[a] H 65 50:50 115 Mu[a] H Me 32 43:57
116 Mu[a] Me Me 43 50:50[a]Mu = N(CH2CH2Cl)2[b]Sum of the pure isolated isomers a and b[c]Determined from the 1H NMR spectra of the crude products[d]Not given[e]Single epimerConformational analyses by NMR methods and DFT calculations
revealed that the hetero rings of the trans-fused
oxazaphosphorinanes 8183 and 93, together with the similar
diazaphosphorinanes 8486 and 94, predominantly adopt a chair
conformation, regardless of the P configuration, although in
29

93b and 94b a small proportion of non-chair conformations can be
predicted due to the equatorial preference of the N(CH2CH2Cl)2
group. A similar chair form was assumed for 87b89b and 90b92b,
whereas the conformational equilibrium was shifted towards skew
conformers such as B and T in 87a89a and 90a92a, since the
strong axial preference of the OPh substituent depopulates the
unfavourable chair conformation (Scheme 13). In the cis-fused
compounds 101106, 107112 and 113116, the hetero ring N can be
either axial (N-in) or equatorial (N-out) with the expected chair
conformation of the fused cyclohexane moiety. The axial or
equatorial preference of the P substituents can shift the N-
in/N-out equilibrium in a direction such that the substituents
can occupy their preferred positions in the chair conformation,
but both chair forms can depopulate towards more preferable skew
conformations when the P configuration is unfavourable (Scheme
13). The fragmentations of compounds 81and114 have been
thoroughly studied by means of electron ionization MS [69,
70].
30

Scheme 13
31

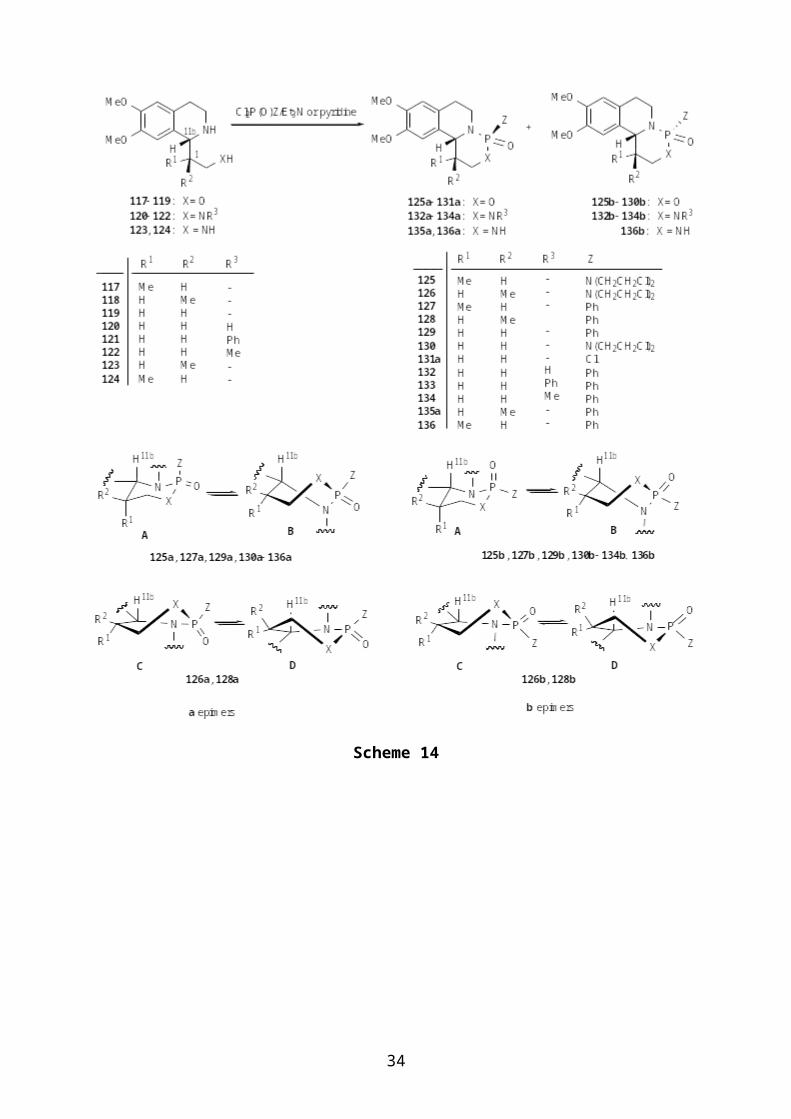
Synthetic and conformational studies of
tetrahydroisoquinoline-condensed oxazaphosphorinanes 125131
[71, 72] and diazaphosphorinanes 132136 [26], which are part
of a more rigid system containing their N atom in a bridgehead
position, were carried out by Fülöp et al. (Scheme 14). The
diastereomeric ratios (Table 2) and the conformational
distributions in these tricyclic systems were strongly
influenced by the substituents (R1, R2 and R3) of the starting
aminoalcohols 117119 and diamines 120-124. The fused hetero
rings in compounds 125, 127, 129 and 130136 proved to exist in
an ordinary chair (A)-twist (C) equilibrium, although one or
other of the conformers (A or B) can predominate, depending on
the P configuration and P-substituent preferences. Thus, the
equilibrium is shifted completely towards the chair conformation
A in the oxazaphosphorinanes 125b, 130b and 127b, 129b, due to
the equatorial tendency of the N(CH2CH2Cl)2 and Ph groups.
However, unusual distorted conformational states (C D) with
a planar bridgehead N were reported for both epimers of 126
and 128, with a predominance of conformer C for 126a, 126b and
128b and of conformer D for 128a, where the steric interaction
between the aromatic moiety of the connecting isoquinoline and
32

the Me-1 substituent (R2) can force the hetero ring to adopt
distorted twist conformations. Minor amounts of C and D
besides A and B were also suggested for the N-substituted
diazaphosphorinanes 133b and 134a. The existence of
conformations A and C in the solid state was also confirmed by
X-ray analysis of 125b and 126a. The bis(2-chloroethyl)amino-
substituted benzologues of 1,3,2-diazaphosphorino
isoquinolines (132-136) were found to possess anticancer
activity [73].
33
Scheme 14
34

Table 2. Yields and epimeric ratios of tetrahydroisoquinoline-condensed oxaza- and diazaphosphorinanes
Comp.
R1 R2 Z Yield (%)[b]
Ratio (a:b)[c]
Ref.
125 Me H N(CH2CH2Cl)2 35 50:50 [73]126 H Me N(CH2CH2Cl)2 32 50:50 [73]127 Me H Ph 51 50:50 [73]128 H Me Ph 35 50:50 [73]129 H H Ph 26 50:50 [72]130 H H N(CH2CH2Cl)2 34 50:50 [72]131a[a]
H H Cl 27 - [72]
Comp.
R1 R2 R3 Z Yield (%)[b]
Ratio (a:b)[d]
Ref.
132 H H H Ph 49 56:44 [26]133 H H Ph Ph 40 29:71 [26]134 H H Me Ph 40 27:73 [26]135a[a]
H Me H Ph 44 - [26]
136 Me H H Ph 25 57:43 [26][a]Single epimer[b]Sum of the pure isolated isomers a and b[c]Determined from the 31P NMR spectra of the crude products[d]Determined from the 1H NMR spectra of the crude products
A few benzene-fused dioxa-, oxaza- and diazaphosphorinanes are
also mentioned in the literature, but from pharmacological
35

rather than stereochemical aspects, though the condensed
aromatic moiety further restricts the conformational mobility
of the hetero ring. Nitrobenzocyclophosphamide analogues 137-
140 were synthesised and their antiproliferative activities
were examined on cell cultures (Scheme 15) [12, 74]. Compounds
137 and 139 with benzylic oxygen in the phosphorinane ring para
to the nitro group exhibited modestly enhanced cytotoxicity in
E. coli nitroreductase and could be suitable “lead” compounds in
the development of special gene-directed enzyme prodrugs. The
antibacterial activities of benzologues 141149 were evaluated
and found effective against Gram-positive and Gram- negative
bacteria at different concentrations [75]. The hybrid
derivatives 150 or 151, involving thiazolidine (150) or
benzothiazine (151) and a benzodiazaphosphorinanone moiety in
the same molecule can be valuable model compounds for research
into new pharmaceuticals [76].
36

Scheme 15
4. SUMMARY
37

The synthetic routes and stereostructural features of
carbo- and heterocycle-condensed tetracoordinate P(V)
diheterophosphorinanes have been discussed in detail.
The extreme sensitivity of the conformational behaviour of
the P ring to substitution in both mono- and poly-cyclic
systems makes these derivatives excellent model compounds for
investigation of the steric and electronic effects of
functional groups in different positions on the hetero ring.
Interest has recently been shifted towards fused
cycloalkane- and heterocycle-condensed derivatives, and new
questions have been raised concerning the bioactivities and
stereostructural properties of these compounds.
The results published so far are already of great value,
but further studies on different structures will be motivated
by the intention to develop new pharmacologically active
agents and to reveal additional important findings on the
stereochemistry of these ring systems.
ACKNOWLEDGEMENTS
Thanks are due to the Hungarian Scientific Research Fund (OTKA
grant TO49366) for financial support.
38

REFERENCES
[1] Keglevich G. Curr. Org. Chem. 2006, 10, 93-111.
[2] Calvin, M. In Clinical Pharmacology of Anti-Neoplastic Drugs, Pinedo,
H. M., Ed.; Elsevier: Amsterdam, 1978, 245, and references
cited therein.
[3] Maryanoff, B. E.; Hutchins, R. O.; Maryanoff, C. A. Top.
Stereochem. 1979, 11, 187, and references cited therein.
[4] Gallagher, M. J. In Phosphorus-31 NMR Spectroscopy in
Stereochemical Analysis, Verkade, J. G., Quin, L. D., Eds; VCH:
Deerfield Beach, FL, 1987, Chapter 9, and references cited
therein.
[5] Bentrude, W. G. In Phosphorus-31 NMR Spectral Properties in
Compound Characterization and Structural Analysis, Quin, L. D., Verkade,
39

J. G., Eds; VCH: New York, NY, 1994, Chapter 4, and
references cited therein.
[6] Bentrude, W. G. In Methods in Stereochemical Analysis; Juaristi,
E., Ed.; VCH: New York, NY, 1995, Chapter 7, and references
cited therein.
[7] Bentrude, W. G., Setzer, W. N. In Phosphorus-31 NMR
Spectroscopy in Stereochemical Analysis; Verkade, J. G., Quin, L. D.,
Eds.; VCH: Deerfield Beach, FL, 1987, Chapter 11, and
references cited therein.
[8] Zhou, J., Chen, R. J. Chem. Soc., Perkin Trans 1, 1998, 2917.
[9] Wardle, N. J., Bligh, S. W. A., Hudson, H. R. Curr. Org.
Chem. 2005, 9, 1803.
[10] Sulsky, R., Robl, J. A., Biller, S. A., Harrity, T. W.,
Wetterau, J., Connolly, F., Jolibois, K., Kunselman, L. Bioorg.
Med. Chem. Lett. 2004, 14, 5067.
[11] Erion, M. D., Reddy, K. R., Boyer, S. H., Matelich, M. C.,
Gomez-Galeno, J., Lemus, R. H., Ugarkar, B. G., Colby, T.
J., Schanzer, J., van Poelje, P. D. J. Am. Chem. Soc. 2004,
126, 5154.
[12] Jiang, Y., Han, J., Yu, C., Vass, S. O., Searle, P. F.,
Browne, P., Knox, R. J., Hu, L. J. Med. Chem. 2006, 49, 4333.
40

[13] Misiura, K., Kinas, R. W., Kuśnierczyk, H., Radzikowski,
C., Stec, W. J. Anti-Cancer Drugs 2001, 12, 453.
[14] Zalán, Z., Kivelä, H., Lázár, L., Fülöp, F., Pihlaja, K.
Eur. J. Org. Chem. 2006, 2145.
[15] Kinas, R., Stec, W. J., Krüger, C. Phosphorus Sulfur 1978, 4,
295.
[16] Polozov, A. M., Litvinov, I. A., Kataeva, O. N., Stolov,
A. A., Yarkova, E. G., Khotinen, A. V., Klimovitskii, E. N. J.
Mol. Struct. 1995, 356, 125.
[17] Day, R., O., Bentrude, W. G., Yee, K. C., Setzer, W. N.,
Deiters, J. A., Holmes, R.R. J. Am. Chem. Soc. 1984, 106, 103.
[18] Maryanoff, B. E., McPhail, A. T., Hutchins, R. O. J. Am.
Chem. Soc. 1981, 103, 4432.
[19] Bentrude, W. G., Day, R. O., Holmes, J. M., Quin Szakál,
G., Setzer, W. N., Sopchik, A. E., Holmes, R. R. J. Am. Chem.
Soc. 1984, 106, 106.
[20] Holmes, R. R., Day, R. O., Setzer, W. N., Sopchik, A. E.,
Bentrude, W. G. J. Am. Chem. Soc. 1984, 106, 2353.
[21] Bajwa G. S., Chandrasekaran, S., Hargis, J. H., Sopchik,
A. E., Blatter, D., Bentrude, W. G. J. Am. Chem. Soc. 1982,
104, 6385.
41

[22] Setzer, W. N., Sopchik, A. E., Bentrude, W. G. J. Am. Chem.
Soc. 1985, 107, 2083.
[23] Gorenstein, D. G., Rowell, R., Findlay, J. J. Am. Chem. Soc.
1980, 102, 5077.
[24] Gorenstein, D. G., Rowell, R. J. Am. Chem. Soc. 1979, 101,
4925.
[25] Gholivand, K., Pourayoubi, M., Farshadian, S., Molani, S.,
Shariatinia, Z. Anal. Sci. 2005, 21, x55.
[26] Zalán Z., Martinek, T. A., Lázár, L., Fülöp, F. Tetrahedron
2003, 59, 9117.
[27] Ślepokura, K., Lis, T. Acta Cryst. 2004, C60, o315.
[28] Wan, S-G., Liu, C., Yu, Y., Yang, X-Y. Acta Cryst. 2005, E61,
o2117.
[29] Tähtinen, P., Bagno, A., Koch, A., Pihlaja, K. Eur. J. Org.
Chem. 2004, 4921.
[30] Frank, É., Schäfer, B., Mucsi, Z., Keglevich, G.
Heteroatom Chem. (in press)
[31] Frank, É., Körtvélyesi, T., Czugler, M., Mucsi, Z.,
Keglevich, G. Steroids 2007, 72, 437-445.
[32] Frank, É., Kazi, B., Mucsi, Z., Ludányi, K., Keglevich, G.
Steroids 2007, 72, 446-458.
42

[33] Kivelä, H., Zalán, Z., Tähtinen, P., Sillanpää, R., Fülöp,
F., Pihlaja, K. Eur. J. Org. Chem. 2005, 1189.
[34] see e.g.: Dros, A. C., Zijlstra, W. J., Van Duijnen, P.
Th., Spek, A. L., Kooijman, H., Kellogg, R. M. Tetrahedron
1998, 54, 7787.
[35] see e.g.: Bartczak, T. J., Galdecki, Z., Wolf, W. M.,
Lesiak, K., Stec, W. J. Acta Cryst. 1986, C42, 244.
[36] Borch, R. F., Canute, G. W. J. Med. Chem. 1991, 34, 3044.
[37] Misiura, K., Kinas, R. W., Stec, W. J., Kusnierczyk, H.,
Radzikowski, C., Sonoda, A. J. Med. Chem. 1988, 31, 226.
[38] Ludeman, S. M., Boyd, V. L., Regan, J. B., Gallo, K. A.,
Zon, G., Ishii, K. J. Med. Chem. 1986, 29, 716.
[39] Sakoda, R., Kamikawaji, Y., Seto, K. Chem. Pharm. Bull. (Tokyo)
1992, 40, 2370.
[40] Sulsky, R., Robl, J. A., Biller, S. A., Harrity, T. W.,
Wetterau, J., Connolly, F., Jolibois, K., Kunselman, L. Bioorg.
Med. Chem. Lett. 2004, 14, 5067.
[41] Erion, M. D., Reddy, K. R., Boyer, S. H., Matelich, M. C.,
Gomez-Galeno, J., Lemus, R. H., Ugarkar, B. G., Colby, T.
J., Schanzer, J., van Poelje, P. D. J. Am. Chem. Soc. 2004,
126, 5154.
43

[42] Hulst, R., Zijlstra, R. W. J., De Vries, N. K., Feringa,
B. L. Tetrahedron: Asymmetry 1994, 5, 1701.
[43] Weener, J-W., Versleijen, J. P. G., Meetsma, A., Ten
Hoeve, W., Van Leusen, A. M. Eur. J. Org. Chem. 1998, 1511.
[44] Gorenstein, D. G. In Phosphorus-31 NMR: Principles and Applications,
Gorenstein, D. G., Ed.; Academic Press: New York, NY, 1984,
1-53, and references cited therein.
[45] Gorenstein, D. G. In Progress in Nuclear Magnetic Resonance
Spectroscopy, Emsley, J. W., Feeney, J., Sutcliffe, L. H.,
Eds.; Pergamon Press: Oxford, 1983, Chapter 1, and
references cited therein.
[46] Viljanen, T., Klika, K. D., Fülöp, F., Pihlaja, K., J. Chem.
Soc., Perkin Trans. 2, 1998, 1479.
[47] Modro, A. M., Modro, T. A., Bernatowitcz, P., Stefaniak,
L. Magn. Reson. Chem. 1997, 35, 774.
[48] Zhou, J., Chen, R. J. Chem. Soc., Perkin Trans 1, 1998, 2917.
[49] Bentrude, W. G., Tan, H-W., Yee, K. C. J. Am. Chem. Soc.
1975, 97, 573.
[50] Hermans, R. J. M., Buck, H. M. J. Org. Chem. 1987, 52, 5150.
[51] Sartillo-Piscil, F., Cruz, S., Sánchez, M., Höpfl, H., de
Parodi, C. A., Quintero, L. Tetrahedron 2003, 59, 4077.
44

[52] Sartillo-Piscil, F., Sánchez, M., Cruz-Gregorio, S.,
Quintero, L. Tetrahedron 2004, 60, 3001.
[53] Cruz-Gregorio, S., Sánchez, M., Clara-Rosa, A., Bèrnes,
S., Quintero, L., Sartillo- Piscil F. J. Org. Chem. 2005, 70,
7107.
[54] Miljkovic, D. A., Vukojevic, N. S., Hadzic, P. A., Minic,
D. J., Hughes, N. A., Hill, M. N. S., Harangi, J. J. Chem. Soc.,
Perkin Trans. 2, 1990, 1093.
[55] Neeser, J-R., Tronchet, J. M. J., Charollais, E. J. Can. J.
Chem. 1983, 61, 1387.
[56] Hermans, R. J. M., Buck, H. M. J. Org. Chem. 1988, 53, 2077.
[57] Frank, É., Kazi, B., Ludányi, K., Keglevich, G. Tetrahedron
Lett. 2006, 47, 1105.
[58] Schneider, G., Vass, A., Vincze, I., Sohár, P. Liebigs Ann.
Chem. 1988, 267.
[59] Wölfling, J., Schneider, G., Péter, A. J. Chromatogr. A,
1999, 852, 433.
[60] Hajnal, A., Wölfling, J., Schneider, G. Collect. Czech. Chem.
Commun. 1998, 63, 1613.
[61] Pietrusiewicz, K. M., Salamonczyk, I., Wieczorek, W.,
Brandi, A., Cicchi, S., Goti, A. Tetrahedron 1991, 47, 9083.
45

[62] Day, R. O., Gorenstein, D. G., Holmes, R. R. Inorg. Chem.
1983, 22, 2192.
[63] Yang, J-C., Shah, D. O., Rao, N. U. M., Freeman, W. A.,
Sosnovsky, G., Gorenstein, D. G. Tetrahedron 1988, 44, 6305.
[64] Bouchu, D., Dreux, J. Tetrahedron Lett. 1980, 21, 2513.
[65] Lilo, B., Moreau, M., Bouchu, D. Tetrahedron Lett. 1990, 31,
887.
[66] Van Nuffel, P., Lenstra, A. T. H., Geise, H. J. Bull. Soc.
Chim. Belg. 1982, 91, 43.
[67] Viljanen, T., Tähtinen, P., Pihlaja, K., Fülöp, F. J. Org.
Chem. 1998, 63, 618.
[68] Goodridge, R. J., Hambley, T. W., Ridley, D. D. Aust. J.
Chem. 1986, 39, 591.
[69] Juhász, M., Martiskainen, O., Zalán, Z., Fülöp, F.,
Pihlaja, K. Rapid Commun. Mass. Spectrom. 2006, 20, 433.
[70] Martikainen, O., Juhász, M., Zalán, Z., Fülöp, F.,
Pihlaja, K. Rapid Commun. Mass. Spectrom. 2006, 20, 1621.
[71] Fülöp, F., Forró E., Martinek, T., Günther, G., Sillanpää,
R. J. Mol. Struct. 2000, 554, 119.
[72] Martinek, T., Forró E., Günther, G., Sillanpää, R., Fülöp,
F. J. Org. Chem. 2000, 65, 316.
46

[73] Bull, J. E. O., Naidu, M. S. R., Phosphorus, Sulfur, Silicon
2000, 162, 231.
[74] Li, Z., Han, J., Jiang, Y., Browne, P., Knox, R. J., Hu,
L. Bioorg. Med. Chem. 2003, 11, 4171.
[75] Prasad, G. S., Babu, B. H., Reddy, K. R., Haranath, P. R.,
Reddy, C. S. ARKIVOC 2006 (xiii) 165.
[76] Gröger, H., Wilken, J., Martens, J., Neda, I., Schmutzler,
R. Heteroatom Chem. 1998, 9, 679.
47




















