
The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated...
6
article 160 nature genetics • volume 29 • october 2001 The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis Yi Yang 1 , Afif Hentati 1 , Han-Xiang Deng 1 , Omar Dabbagh 2 , Toru Sasaki 1 , Makito Hirano 1 , Wu-Yen Hung 1 , Karim Ouahchi 1 , Jianhua Yan 1 , Anser C. Azim 1 , Natalie Cole 1 , Generoso Gascon 3 , Ayesha Yagmour 4 , Mongi Ben-Hamida 5 , Margaret Pericak-Vance 6 , Fayçal Hentati 5 & Teepu Siddique 1,7,8 Amyotrophic lateral sclerosis (ALS) and primary lateral sclerosis (PLS) are neurodegenerative conditions that affect large motor neurons of the central nervous system. We have identified a familial juvenile PLS (JPLS) locus overlap- ping the previously identified ALS2 locus on chromosome 2q33. We report two deletion mutations in a new gene that are found both in individuals with ALS2 and those with JPLS, indicating that these conditions have a common genetic origin. The predicted sequence of the protein (alsin) may indicate a mechanism for motor-neuron degener- ation, as it may include several cell-signaling motifs with known functions, including three associated with gua- nine-nucleotide exchange factors for GTPases (GEFs). 1 Department of Neurology, Northwestern University Medical School, Chicago, Illinois, USA. 2 King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia. 3 Division of Pediatric Neurology, Brown University, Providence, Rhode Island, USA. 4 King Fahad Military Hospital, Jeddah, Saudi Arabia. 5 Institute of Neurology, Tunis, Tunisia. 6 Medical Genetics, Duke University Medical Center, Durham, North Carolina, USA. 7 Department of Cell and Molecular Biology, Northwestern University Medical School, Chicago, Illinois, USA. 8 Northwestern University Institute of Neuroscience, Chicago, Illinois, USA. Correspondence should be addressed to T.S. (e-mail: [email protected]). Introduction Amyotrophic lateral sclerosis and primary lateral sclerosis (PLS) are closely related but clinically distinct neurodegenerative disor- ders 1–3 . Following the first description of these disorders, there was a period of diagnostic confusion, but the distinction between ALS and PLS has since become clear and diagnostic criteria have been established 1,2,4,5 . Both ALS and PLS are progressive paralytic disorders that result from dysfunction of the motor systems comprising the upper motor neurons (UMN) of the motor cortex and lower motor neurons (LMN) of the brainstem and the spinal cord. In PLS, the degeneration is confined to the UMN 1,6 , whereas in ALS, both sets of neurons are affected 5 . ALS is a more severe dis- ease, and its phenotype is influenced by the relative ratio of UMN to LMN involvement. The PLS phenotype 7–13 shows a slowly progressive spastic paresis involving all four extremities and mus- cles of facial expression, speech, swallowing, and sometimes eye movements and sphincter control. The diagnosis of PLS is essen- tially one of exclusion, as the UMN pathways can be involved in other disorders such as spondylitic myelopathy, vitamin B 12 defi- ciency, human T-lymphotropic virus 1 (HTLV-1) infection, ‘pure’ spastic paraparesis, multiple sclerosis, tumors, strokes or syringo- or bulbomyelia. The exact prevalence of PLS is not known 1 , but it is rarer than ALS and can be differentiated from it by the absence of denervation or reinnervation upon electromyo- graphic or muscle-biopsy examination 5 . PLS is insidious and may, at first, affect only the bulbar inner- vated muscles or cause spasticity in the lower extremities. It may be initially diagnosed as pseudobulbar palsy, spastic paraparesis, multiple sclerosis, early ALS 13 or other conditions. As symptoms of PLS evolve over time, early diagnostic confusion is common. The progression of symptoms over three to five years is therefore an essential consideration in the diagnosis of PLS 1,4 . It usually takes several years before the appearance of essential characteris- tics such as bulbospinal spasticity and continued absence of LMN involvement. Consistent with the anatomic pathology, positron emission tomography (PET) scanning and magnetic resonance imaging (MRI) show decreased 18-flurodeoxyglucose uptake and atrophy of the precentral gyrus, respectively 1 . Magnetic cortical stimulation shows prolonged central-motor conduction times 1,12,14 . Although PLS is a sporadic disorder of adult middle age 1 , it has been described in children (juvenile PLS, JPLS), both in iso- lated 15 as well as in familial circumstances 12,16 . To establish the diagnosis of JPLS, additional childhood-onset disorders such as the leukodystrophies, biotin-responsive encephalopathies and juvenile Leigh-like syndromes and other neurometabolic entities must be ruled out 12 . Because of the rigor of the diagnostic criteria and the rarity of familial disease, PLS has not been the subject of any intensive genetic investigation until this report, and no locus related to it has been mapped. The genetics of ALS is relatively better studied. In 10% of ALS cases, the disease is transmitted as a recessive or dominant trait. Four gene loci have been mapped to chromosomes 21q21, 2q33, 15q15–q21 and 9q34 (refs. 17–24). ALS linked to 21q is caused by mutations in SOD1 (refs. 5,6) that lead to a dominant gain of toxic function. The genes for other ALS loci have not been identified. We first linked the recessive form of juvenile ALS type 3 (ALS2, © 2001 Nature Publishing Group http://genetics.nature.com © 2001 Nature Publishing Group http://genetics.nature.com
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated...

article
160 nature genetics • volume 29 • october 2001
The gene encoding alsin, a protein withthree guanine-nucleotide exchange factordomains, is mutated in a form of recessiveamyotrophic lateral sclerosisYi Yang1, Afif Hentati1, Han-Xiang Deng1, Omar Dabbagh2, Toru Sasaki1, Makito Hirano1, Wu-Yen Hung1,Karim Ouahchi1, Jianhua Yan1, Anser C. Azim1, Natalie Cole1, Generoso Gascon3, Ayesha Yagmour4, MongiBen-Hamida5, Margaret Pericak-Vance6, Fayçal Hentati5 & Teepu Siddique1,7,8
Amyotrophic lateral sclerosis (ALS) and primary lateral sclerosis (PLS) are neurodegenerative conditions that affect
large motor neurons of the central nervous system. We have identified a familial juvenile PLS (JPLS) locus overlap-
ping the previously identified ALS2 locus on chromosome 2q33. We report two deletion mutations in a new gene
that are found both in individuals with ALS2 and those with JPLS, indicating that these conditions have a common
genetic origin. The predicted sequence of the protein (alsin) may indicate a mechanism for motor-neuron degener-
ation, as it may include several cell-signaling motifs with known functions, including three associated with gua-
nine-nucleotide exchange factors for GTPases (GEFs).
1Department of Neurology, Northwestern University Medical School, Chicago, Illinois, USA. 2King Faisal Specialist Hospital and Research Center, Riyadh,Saudi Arabia. 3Division of Pediatric Neurology, Brown University, Providence, Rhode Island, USA. 4King Fahad Military Hospital, Jeddah, Saudi Arabia.5Institute of Neurology, Tunis, Tunisia. 6Medical Genetics, Duke University Medical Center, Durham, North Carolina, USA. 7Department of Cell andMolecular Biology, Northwestern University Medical School, Chicago, Illinois, USA. 8Northwestern University Institute of Neuroscience, Chicago, Illinois,USA. Correspondence should be addressed to T.S. (e-mail: [email protected]).
IntroductionAmyotrophic lateral sclerosis and primary lateral sclerosis (PLS)are closely related but clinically distinct neurodegenerative disor-ders1–3. Following the first description of these disorders, therewas a period of diagnostic confusion, but the distinction betweenALS and PLS has since become clear and diagnostic criteria havebeen established1,2,4,5.
Both ALS and PLS are progressive paralytic disorders thatresult from dysfunction of the motor systems comprising theupper motor neurons (UMN) of the motor cortex and lowermotor neurons (LMN) of the brainstem and the spinal cord. InPLS, the degeneration is confined to the UMN1,6, whereas inALS, both sets of neurons are affected5. ALS is a more severe dis-ease, and its phenotype is influenced by the relative ratio of UMNto LMN involvement. The PLS phenotype7–13 shows a slowlyprogressive spastic paresis involving all four extremities and mus-cles of facial expression, speech, swallowing, and sometimes eyemovements and sphincter control. The diagnosis of PLS is essen-tially one of exclusion, as the UMN pathways can be involved inother disorders such as spondylitic myelopathy, vitamin B12 defi-ciency, human T-lymphotropic virus 1 (HTLV-1) infection,‘pure’ spastic paraparesis, multiple sclerosis, tumors, strokes orsyringo- or bulbomyelia. The exact prevalence of PLS is notknown1, but it is rarer than ALS and can be differentiated from itby the absence of denervation or reinnervation upon electromyo-graphic or muscle-biopsy examination5.
PLS is insidious and may, at first, affect only the bulbar inner-vated muscles or cause spasticity in the lower extremities. It maybe initially diagnosed as pseudobulbar palsy, spastic paraparesis,
multiple sclerosis, early ALS13 or other conditions. As symptomsof PLS evolve over time, early diagnostic confusion is common.The progression of symptoms over three to five years is thereforean essential consideration in the diagnosis of PLS1,4. It usuallytakes several years before the appearance of essential characteris-tics such as bulbospinal spasticity and continued absence of LMNinvolvement. Consistent with the anatomic pathology, positronemission tomography (PET) scanning and magnetic resonanceimaging (MRI) show decreased 18-flurodeoxyglucose uptake andatrophy of the precentral gyrus, respectively1. Magnetic corticalstimulation shows prolonged central-motor conductiontimes1,12,14.
Although PLS is a sporadic disorder of adult middle age1, it hasbeen described in children (juvenile PLS, JPLS), both in iso-lated15 as well as in familial circumstances12,16. To establish thediagnosis of JPLS, additional childhood-onset disorders such asthe leukodystrophies, biotin-responsive encephalopathies andjuvenile Leigh-like syndromes and other neurometabolic entitiesmust be ruled out12. Because of the rigor of the diagnostic criteriaand the rarity of familial disease, PLS has not been the subject ofany intensive genetic investigation until this report, and no locusrelated to it has been mapped.
The genetics of ALS is relatively better studied. In 10% of ALScases, the disease is transmitted as a recessive or dominant trait.Four gene loci have been mapped to chromosomes 21q21, 2q33,15q15–q21 and 9q34 (refs. 17–24). ALS linked to 21q is caused bymutations in SOD1 (refs. 5,6) that lead to a dominant gain of toxicfunction. The genes for other ALS loci have not been identified.We first linked the recessive form of juvenile ALS type 3 (ALS2,
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://g
enet
ics.
nat
ure
.co
m© 2001 Nature Publishing Group http://genetics.nature.com
article
nature genetics • volume 29 • october 2001 161
MIM#205100; ref. 23) tochromosome 2q33–35(ref. 18), and subse-quently also linked juve-nile-onset PLS (JPLS) to2q33. We now report thata differentially spliced gene encoding short and long forms of aprotein, which we named alsin, is mutated in both ALS2 and JPLS.
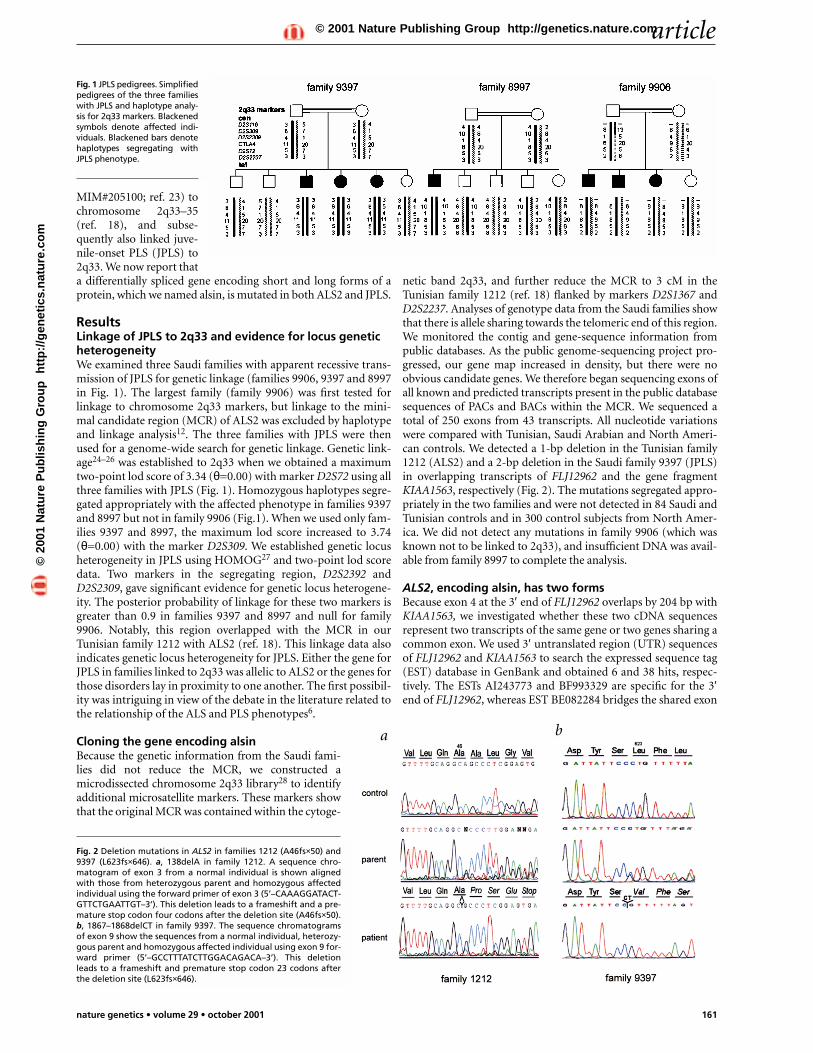
ResultsLinkage of JPLS to 2q33 and evidence for locus geneticheterogeneityWe examined three Saudi families with apparent recessive trans-mission of JPLS for genetic linkage (families 9906, 9397 and 8997in Fig. 1). The largest family (family 9906) was first tested forlinkage to chromosome 2q33 markers, but linkage to the mini-mal candidate region (MCR) of ALS2 was excluded by haplotypeand linkage analysis12. The three families with JPLS were thenused for a genome-wide search for genetic linkage. Genetic link-age24–26 was established to 2q33 when we obtained a maximumtwo-point lod score of 3.34 (θ=0.00) with marker D2S72 using allthree families with JPLS (Fig. 1). Homozygous haplotypes segre-gated appropriately with the affected phenotype in families 9397and 8997 but not in family 9906 (Fig.1). When we used only fam-ilies 9397 and 8997, the maximum lod score increased to 3.74(θ=0.00) with the marker D2S309. We established genetic locusheterogeneity in JPLS using HOMOG27 and two-point lod scoredata. Two markers in the segregating region, D2S2392 andD2S2309, gave significant evidence for genetic locus heterogene-ity. The posterior probability of linkage for these two markers isgreater than 0.9 in families 9397 and 8997 and null for family9906. Notably, this region overlapped with the MCR in ourTunisian family 1212 with ALS2 (ref. 18). This linkage data alsoindicates genetic locus heterogeneity for JPLS. Either the gene forJPLS in families linked to 2q33 was allelic to ALS2 or the genes forthose disorders lay in proximity to one another. The first possibil-ity was intriguing in view of the debate in the literature related tothe relationship of the ALS and PLS phenotypes6.
Cloning the gene encoding alsinBecause the genetic information from the Saudi fami-lies did not reduce the MCR, we constructed amicrodissected chromosome 2q33 library28 to identifyadditional microsatellite markers. These markers showthat the original MCR was contained within the cytoge-
netic band 2q33, and further reduce the MCR to 3 cM in theTunisian family 1212 (ref. 18) flanked by markers D2S1367 andD2S2237. Analyses of genotype data from the Saudi families showthat there is allele sharing towards the telomeric end of this region.We monitored the contig and gene-sequence information frompublic databases. As the public genome-sequencing project pro-gressed, our gene map increased in density, but there were noobvious candidate genes. We therefore began sequencing exons ofall known and predicted transcripts present in the public databasesequences of PACs and BACs within the MCR. We sequenced atotal of 250 exons from 43 transcripts. All nucleotide variationswere compared with Tunisian, Saudi Arabian and North Ameri-can controls. We detected a 1-bp deletion in the Tunisian family1212 (ALS2) and a 2-bp deletion in the Saudi family 9397 (JPLS)in overlapping transcripts of FLJ12962 and the gene fragmentKIAA1563, respectively (Fig. 2). The mutations segregated appro-priately in the two families and were not detected in 84 Saudi andTunisian controls and in 300 control subjects from North Amer-ica. We did not detect any mutations in family 9906 (which wasknown not to be linked to 2q33), and insufficient DNA was avail-able from family 8997 to complete the analysis.
ALS2, encoding alsin, has two formsBecause exon 4 at the 3′ end of FLJ12962 overlaps by 204 bp withKIAA1563, we investigated whether these two cDNA sequencesrepresent two transcripts of the same gene or two genes sharing acommon exon. We used 3′ untranslated region (UTR) sequencesof FLJ12962 and KIAA1563 to search the expressed sequence tag(EST) database in GenBank and obtained 6 and 38 hits, respec-tively. The ESTs AI243773 and BF993329 are specific for the 3′end of FLJ12962, whereas EST BE082284 bridges the shared exon
Fig. 1 JPLS pedigrees. Simplifiedpedigrees of the three familieswith JPLS and haplotype analy-sis for 2q33 markers. Blackenedsymbols denote affected indi-viduals. Blackened bars denotehaplotypes segregating withJPLS phenotype.
Fig. 2 Deletion mutations in ALS2 in families 1212 (A46fs×50) and9397 (L623fs×646). a, 138delA in family 1212. A sequence chro-matogram of exon 3 from a normal individual is shown alignedwith those from heterozygous parent and homozygous affectedindividual using the forward primer of exon 3 (5’–CAAAGGATACT-GTTCTGAATTGT–3’). This deletion leads to a frameshift and a pre-mature stop codon four codons after the deletion site (A46fs×50).b, 1867–1868delCT in family 9397. The sequence chromatogramsof exon 9 show the sequences from a normal individual, heterozy-gous parent and homozygous affected individual using exon 9 for-ward primer (5’–GCCTTTATCTTGGACAGACA–3’). This deletionleads to a frameshift and premature stop codon 23 codons afterthe deletion site (L623fs×646).
a b
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://g
enet
ics.
nat
ure
.co
m© 2001 Nature Publishing Group http://genetics.nature.com

article
162 nature genetics • volume 29 • october 2001
and the 5′ exon of KIAA1563 (Fig. 3). We successfully amplifiedand sequenced the reverse transcriptase–polymerase chain reac-tion (RT–PCR) product of FLJ12962 and the putative long formobtained from lymphoblasts of an individual with ALS2 fromfamily 1212, indicating that the two transcripts are alternateshort and long forms of the same gene (now defined as ALS2).Sequences of both RT–PCR products carry the expected 1-bpdeletion in the shared exon 3. There may be decreased RT–PCRproducts in the long form for the affected person and carrier par-ent compared with the control, but the RT–PCR is not quantita-tive (Fig. 3, middle panel). We did not have transformedlymphoblastoid cell lines or tissue from families with JPLS (9397and 8997) to examine mRNA expression of the gene. TheFLJ12962 transcript is 2,657 bp long, with an open reading frame(ORF) of 1,191 bp encoding a 396–amino acid protein. Align-ment of mRNA shows four exons. The assembled long form con-taining KIAA1563 and most of FLJ12962 thus has a full-lengthsequence of 6,394 bp, with an ORF of 4,974 bp that encodes 1,657amino acids. An mRNA alignment shows distribution of 34exons in a genomic region of 83 kb. Both deletions (A46fs×50)and (L623fs×647) are thus expected to encode markedly smallerproteins with additional amino acids at their carboxy termini: a49–amino acid protein in the family with ALS2 and a 645–aminoacid protein in the family with JPLS. Both the short and longforms of alsin can be detected by northern blots as bands ofapproximately 2.3 kb and 6 kb, respectively (data not shown).RT-PCR amplifies both forms from cDNA derived from multiplehuman tissues with a profile similar to that of KIAA1563 (Fig. 3).There is evidence from the EST database of at least three addi-tional alternatively spliced forms of the gene. We also detected
nine single-nucleotide polymorphisms (SNPs), including threepredicted to effect an amino acid change, in ALS2 (Table 1).
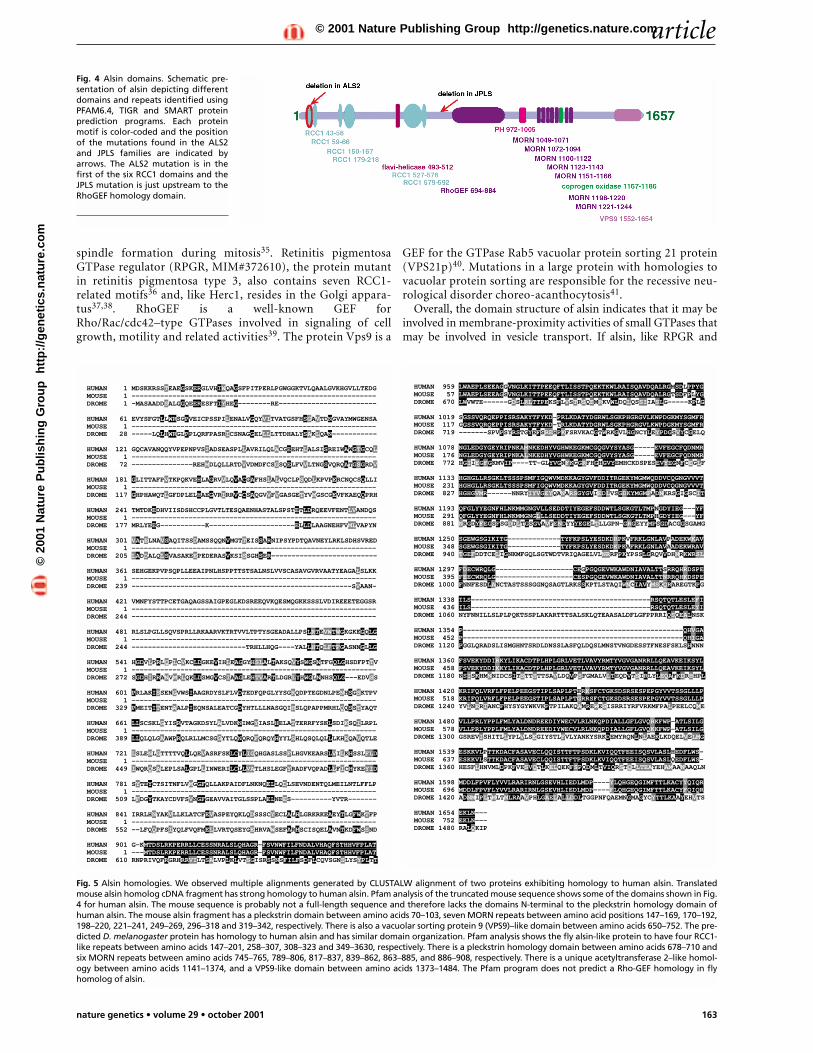
Long form of ALS2 has three cell-signaling motifsAnalyses using PFAM 6.4, TIGR and SMART programs29–31 ofthe predicted protein alsin show three distinct domains: regula-tor of chromatin condensation–like domain (RCC1-like domain,or RLD), guanine-nucleotide exchange factor for Rho protein(RhoGEF) and vacuolar protein sorting 9 (VPS9), all of whichare guanine-nucleotide exchange factors (GEFs) that activatemembers of the Ras super family of GTPases (Fig. 4). A pleckstrinhomology domain was also identified. Pleckstrin homologydomains target their host proteins to membranes by binding tophophoinositides, and are involved in cell signaling andcytoskeletal rearrangement, among other things32. We alsoobserved seven membrane occupation and recognition nexus(MORN) repeats. MORN repeats are present in phosphatidyli-nositol signaling proteins33. We identified two proteins withdomain organization similar to alsin in Mus musculus andDrosophila melanogaster by BLAST analyses. The protein pre-dicted to encode transcript CG7158 of D. melanogaster has theRCC1, pleckstrin homology, MORN and VPS9 domains butlacks the RhoGEF motif. The mouse protein fragment for tran-script has pleckstrin homology and VPS9 domains (Fig. 5). Themouse alsin ortholog is probably incomplete and lacks theamino-terminal domains.
DiscussionThe presence of several well-studied cell signaling domains inalsin may provide clues to its function and its role in thedegeneration of motor neurons. Besides RCC1, RLDs occur inproteins of several conserved genes, including the HERC(domain homologous to the E6-associated protein C terminusand RCC1 domain protein) family of proteins34, which alsoincludes HECT, an E-3 ubiquitin ligase motif. Although thefunction of the RLD in Herc is not known, RCC1 itself (alsoknown as RanGEF) promotes exchange of GDP for GTP forthe GTPase Ran. The protein RCC1 is involved in nuclearimport and export during interphase of the cell cycle and in
Fig. 3 Expression of ALS2. a, Schema of long and short transcripts of ALS2. ALS2mRNAs are fully or partially covered by known sequences (gray boxes) ofFLJ12962 for the short form and KIAA1563 for the long form. Open boxdenotes non-coding region and closed box denotes coding region. ESTs(AI243773 and BF993329) support the presence of a short form, while EST(BE082284) bridges exon 4–5, corresponding to the long form. RT–PCR usingoligo-dT–derived cDNA from control lymphoblasts (shaded boxes) shows evi-dence of expression of both forms as full-length mRNAs. ∆, deletion site foundin patients in the present study. b, ALS2 mRNA in lymphoblasts. Both forms ofALS2 mRNA containing the entire coding sequences were amplified by RT–PCRfrom a patient (Pt) and a carrier (Ca) in ALS2 family 1212 as well as from a con-trol (C). M, molecular size marker; NC, negative control (no cDNA template).We confirmed a one-base deletion in exon 3 in both forms of ALS2 mRNA bysequencing of RT-PCR fragments from the ALS2 patient. c, Tissue distribution ofALS2 mRNA. We carried out RT–PCR using specific primers for short and longforms. Both short and long forms are ubiquitously expressed in various tissues.M, 100-bp ladder (500–200 bp is shown); SC, spinal cord; He, heart; Br, brain; Pl,placenta; Lu, lung; Li, liver; SM, skeletal muscle; Ki, kidney; Pa, pancreas; andNC, negative control.
Table 1 • Polymorphic nucleotide changes detected in ALS2
1 2 3 4 5 6 7 8 9
exon/intron intron 2 exon 4 exon 4 exon 6 exon 13 exon 25 exon 26 exon 27 intron 29
nucleotidea change IVS2+7 305A→G 1102G→A 1632T→A 2466G→A 3885G→A 4015C→T 4217G→A IVS29-71T→C C→T
amino acid change H102R V368M R1406KaThe nucleotide changes follow previously described rules for nomenclature50.
a
b
c
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://g
enet
ics.
nat
ure
.co
m© 2001 Nature Publishing Group http://genetics.nature.com
article
nature genetics • volume 29 • october 2001 163
spindle formation during mitosis35. Retinitis pigmentosaGTPase regulator (RPGR, MIM#372610), the protein mutantin retinitis pigmentosa type 3, also contains seven RCC1-related motifs36 and, like Herc1, resides in the Golgi appara-tus37,38. RhoGEF is a well-known GEF forRho/Rac/cdc42–type GTPases involved in signaling of cellgrowth, motility and related activities39. The protein Vps9 is a
GEF for the GTPase Rab5 vacuolar protein sorting 21 protein(VPS21p)40. Mutations in a large protein with homologies tovacuolar protein sorting are responsible for the recessive neu-rological disorder choreo-acanthocytosis41.
Overall, the domain structure of alsin indicates that it may beinvolved in membrane-proximity activities of small GTPases thatmay be involved in vesicle transport. If alsin, like RPGR and
Fig. 4 Alsin domains. Schematic pre-sentation of alsin depicting differentdomains and repeats identified usingPFAM6.4, TIGR and SMART proteinprediction programs. Each proteinmotif is color-coded and the positionof the mutations found in the ALS2and JPLS families are indicated byarrows. The ALS2 mutation is in thefirst of the six RCC1 domains and theJPLS mutation is just upstream to theRhoGEF homology domain.
Fig. 5 Alsin homologies. We observed multiple alignments generated by CLUSTALW alignment of two proteins exhibiting homology to human alsin. Translatedmouse alsin homolog cDNA fragment has strong homology to human alsin. Pfam analysis of the truncated mouse sequence shows some of the domains shown in Fig.4 for human alsin. The mouse sequence is probably not a full-length sequence and therefore lacks the domains N-terminal to the pleckstrin homology domain ofhuman alsin. The mouse alsin fragment has a pleckstrin domain between amino acids 70–103, seven MORN repeats between amino acid positions 147–169, 170–192,198–220, 221–241, 249–269, 296–318 and 319–342, respectively. There is also a vacuolar sorting protein 9 (VPS9)–like domain between amino acids 650–752. The pre-dicted D. melanogaster protein has homology to human alsin and has similar domain organization. Pfam analysis shows the fly alsin-like protein to have four RCC1-like repeats between amino acids 147–201, 258–307, 308–323 and 349–3630, respectively. There is a pleckstrin homology domain between amino acids 678–710 andsix MORN repeats between amino acids 745–765, 789–806, 817–837, 839–862, 863–885, and 886–908, respectively. There is a unique acetyltransferase 2–like homol-ogy between amino acids 1141–1374, and a VPS9-like domain between amino acids 1373–1484. The Pfam program does not predict a Rho-GEF homology in flyhomolog of alsin.
HUMAN 1 MDSKKRSSTEAEGSKERGLVHIWQAGSFPITPERLPGWGGKTVLQAALGVKHGVLLTEDGMOUSE 1 ------------------------------------------------------------DROME 1 -MASAADDSALGGQEERESFTIYHEG--------RE------------------------
HUMAN 61 EVYSFGTLLWRSGPVEICPSSPILENALVGQYVITVATGSFHSGAVTDNGVAYMWGENSAMOUSE 1 ------------------------------------------------------------DROME 28 -----LQLHWRGLPPLQRFPASRICSNAGGELLLLTTDHALYSAKLQAN-----------
HUMAN 121 GQCAVANQQYVPEPNPVSIADSEASPLLAVRILQLACGEEHTLALSISREIWAWGTGCQLMOUSE 1 ------------------------------------------------------------DROME 72 ---------------REHMDLQLLRTDVVDMDFCSGSQELFVVLTNGSVQRQATGSGRDV
HUMAN 181 GLITTAFPVTKPQKVEHLAGRVVLQVACGAFHSLALVQCLPSQDLKPVPERCNQCSQLLIMOUSE 1 ------------------------------------------------------------DROME 117 GHPHAWQTLGFDPLELHAEGVRIRRVCCSAQGVVFVGASGETYVMGSCGEVFKAEQQPRH
HUMAN 241 TMTDKEDHVIISDSHCCPLGVTLTESQAENHASTALSPSTETLDRQEEVFENTLVANDQSMOUSE 1 ------------------------------------------------------------DROME 177 MRLYEEG-----------K---------------------ELLDLAAGNEHFVMLVAPYN
HUMAN 301 VATELNAVSAQITSSDAMSSQQNVMGTTEISSARNIPSYPDTQAVNEYLRKLSDHSVREDMOUSE 1 ------------------------------------------------------------DROME 205 LADDALQLSVASAKEEPEDERASVKSISSGHSER--------------------------
HUMAN 361 SEHGEKPVPSQPLLEEAIPNLHSPPTTSTSALNSLVVSCASAVGVRVAATYEAGALSLKKMOUSE 1 ------------------------------------------------------------DROME 239 ------------------------------------------------------SVAAN-
HUMAN 421 VMNFYSTTPCETGAQAGSSAIGPEGLKDSREEQVKQESMQGKKSSSLVDIREEETEGGSRMOUSE 1 ------------------------------------------------------------DROME 244 ------------------------------------------------------------
HUMAN 481 RLSLPGLLSQVSPRLLRKAARVKTRTVVLTPTYSGEADALLPSLRTEVWTWGKGKEGQLGMOUSE 1 ------------------------------------------------------------DROME 244 ----------------------------TRHLLHQG----YALLHTQLFTFGASNNGLLG
HUMAN 541 HGDVLPRLQPLCVKCLDGKEVIHLEAGGYHSLALTAKSQVYSWGSNTFGQLGHSDFPTTVMOUSE 1 ------------------------------------------------------------DROME 272 SGDHIRRANVMRLQKLDSMGVCSIAAGLEHTVARTLDGRLYHWGLNNHSQLG---EDVSS
HUMAN 601 PRLAKISSENGVWSIAAGRDYSLFLVDTEDFQPGLYYSGRQDPTEGDNLPENHSGSKTPVMOUSE 1 ------------------------------------------------------------DROME 329 PMEITITENTAALPIEQNSALEATCGDYHTLLLNASGQIHSLQPAPPMRHLQQSSTYAQT
HUMAN 661 LLSCSKLGYISRVTAGKDSYLALVDKNIMGYIASLHELATTERRFYSKLSDIKSQILRPLMOUSE 1 ------------------------------------------------------------DROME 389 LLQLQLGAAWPRQLRLLMCSGGYTLQNQRQFQRQYHYYLSHLQSQLQLLLKHRQAVQTLE
HUMAN 721 LSLENLGTTTTVQLLQEVASRFSKLCYLIGQHGASLSSFLHGVKEARSLVILKHSSLFLDMOUSE 1 ------------------------------------------------------------DROME 449 IWQRQSALEPLSALGPLLINWERILCLLVATLHSLEGFYRADFVQPADLLFICHYKEYID
HUMAN 781 SYTEYCTSITNFLVMGGFQLLAKPAIDFLNKNQELLQDLSEVNDENTQLMEILNTLFFLPMOUSE 1 ------------------------------------------------------------DROME 509 LFDGYTKAYCDVFSVNGFGEAVVAITGLSSPLAELNEES----------YVTR-------
HUMAN 841 IRRLHNYAKVLLKLATCFEVASPEYQKLQDSSSCYECLALHLGRKRKEAEYTLGFWKTFPMOUSE 1 ------------------------------------------------------------DROME 552 --LFQQPFSIYQLFVQFMELLVRTQSEYGEHRVAWSEFARHSCISQELAVNTKDFWSSND
HUMAN 901 G-KMTDSLRKPERRLLCESSNRALSLQHAGR-FSVNWFILFNDALVHAQFSTHHVFPLATMOUSE 1 ---MTDSLRKPERRLLCESSNRALSLQHAGR-FSVNWFILFNDALVHAQFSTHHVFPLATDROME 610 RNPRIVQFRGRHRRVILTSALVPLKLVTSGISRSSNSFILFSDFLCQVSGNSLYSYPLTT
HUMAN 959 LWAEPLSEEAGGVNGLKITTPEEQFTLISSTPQEKTKWLRAISQAVDQALRGMSDLPPYGMOUSE 57 LWAEPLSEEAGSVNGLKITTPEEQFTLISSTPQEKTKWLRAISQAVDQALRGTSDFPLYGDROME 670 LWVWTE------GDSLRLTTPEKSFLVSTRSQEMRKVWLDQLQSSIIASLG-----KPLG
HUMAN 1019 SGSSVQRQEPPISRSAKYTFYKD-PRLKDATYDGRWLSGKPHGRGVLKWPDGKMYSGMFRMOUSE 117 GGSSVQRQEPPISRSAKYTFYKD-TRLKDATYDGRWLSGKPHGRGVLKWPDGKMYSGMFRDROME 719 -------SPVPSYRSTGYEFSREHPKFSRVKACGTWRKGVLHGNCYLEYPDGSVYCGELQ
HUMAN 1078 NGLEDGYGEYRIPNKAMNKEDHYVGHWKEGKMCGQGVYSYASG-----EVFEGCFQDNMRMOUSE 176 NGLEDGYGEYRIPNKALNKEDHYVGHWKEGKMCGQGVYSYASG-----EVFEGCFQDNMRDROME 772 HGIIEGFGKMVIP----TT-GLYVGNFKGGRFHGHGVYEMHCKDSPESEVYEGNFCEGLF
HUMAN 1133 HGHGLLRSGKLTSSSPSMFIGQWVMDKKAGYGVFDDITRGEKYMGMWQDDVCQGNGVVVTMOUSE 231 HGHGLLRSGKLTSSSPSMFIGQWVMDKKAGYGVFDDITRGEKYMGMWQDDVCQGNGVVVTDROME 827 HGHGVMR------NNRYIYVGEYQANARSGYGVIEDLVSGDKYMGMFADNKRSGIGSCIT
HUMAN 1193 QFGLYYEGNFHLNKMMGNGVLLSEDDTIYEGEFSDDWTLSGKGTLTMPNGDYIEG---YFMOUSE 291 QFGLYYEGNFHLNKMMGNGLLLSEDDTIYEGEFSDDWTLSGKGTLTMPHGDYIEG---YFDROME 881 NRGDYFEGSFSGDDLTGSGVAVFENDYYYEGELTLLGPN-GRGEYYMPSGDACGGSGAMG
HUMAN 1250 SGEWGSGIKITG-------------TYFKPSLYESDKDRPKVFRKLGNLAVPADEKWKAVMOUSE 348 SGEWGSGIKITG-------------TYFKPSLYESDKDKPKAFRKLGNLAVAADEKWRAVDROME 940 TGEFDDTCELIGNKMFGQLSGTWDTVRIQAGELVLNRRFPKYPSSLGRQVVDHNRKWRSL
HUMAN 1297 FDECWRQLG-------------------CEGPGQGEVWKAWDNIAVALTTSRRQHRDSPEMOUSE 395 FEECWRQLG-------------------CESPGQGEVWKAWDNIAVALTTNRRQHKDSPEDROME 1000 FNNFESDLANCTASTSSSGGNQSAGTLRKSSKPTLSTAQIWNCIAVYMSKQRAREGTKPG
HUMAN 1338 ILS--------------------------------------------RSQTQTLESLEFIMOUSE 436 ILS--------------------------------------------RSQTQTLESLEYIDROME 1060 NYFNNILLSLPLPQKTSSPLAKARTTTSALSKLQTEAASALDFLGFPPRRIQSQEALNSK
HUMAN 1354 P------------------------------------------------------QHVGAMOUSE 452 P------------------------------------------------------QHIGADROME 1120 PGGLQRADSLISMGHNTSRDLDNSSLASFQLDQSLMNSTVNGDESSTFNESFSKLSHNNN
HUMAN 1360 FSVEKYDDIRKYLIKACDTPLHPLGRLVETLVAVYRMTYVGVGANRRLLQEAVKEIKSYLMOUSE 458 FSVEKYDDIKKYLIKACDTPLHPLGRLVETLVAVYRMTYVGVGANRRLLQEAVKEIKSYLDROME 1180 NSISKHMNNIDCSITSTTSTTSAVLDQVPSFGMALVLTEQDVTSIRLYLEQAFKDRHHPL
HUMAN 1420 KRIFQLVRFLFPELPEEGSTIPLSAPLPTERKSFCTGKSDSRSESPEPGYVVTSSGLLLPMOUSE 518 KRIFQLVRFLFPELPEEGSTIPLSAPLPTGRRSFCTGKSDSRSESPEPGYVVTSSGLLLPDROME 1240 YVLNERIANCFHYSYGYWKVKPTPILAKQAMREWESISRRIYRFVRKMFPALPEELCQME
HUMAN 1480 VLLPRLYPPLFMLYALDNDREEDIYWECVLRLNKQPDIALLGFLGVQRKFWP-ATLSILGMOUSE 578 VLLPRLYPPLFMLYALDNDREEDIYWECVLRLNKQPDIALLGFLGVQKKFWP-ATLSILGDROME 1300 GSREVISHITLLYPLVLSEGIYSTLFVLYANKYSRKDEMYRQNLNLAEKLKDQELVELMG
HUMAN 1539 ESKKVLPTTKDACFASAVECLQQISTTFTPSDKLKVIQQTFEEISQSVLASLHEDFLWS-MOUSE 637 ESKKVLSTTKDACFASAVECLQQISTTFTPSDKLKVIQQTFEEISQSVLASLQEDFLWS-DROME 1360 HESFLHNVMLDPKFVESVQTLKELQEKFSPQDMLTVIQRSTQLLTEAYEHAMAANAAQLN
HUMAN 1598 MDDLFPVFLYVVLRARIRNLGSEVHLIEDLMDP----YLQHGEQGIMFTTLKACYYQIQRMOUSE 696 MDDLFPVFLYVVLRARIRNLGSEVHLIEDLMDP----FLQHGEQGIMFTTLKACYFQIQRDROME 1420 ADNMIPLTMLTMLRAAVPHLGAELALLDDLTGGPNFQAEMNGMAGYCYTTLKAAYEHVTS
HUMAN 1654 EKLN---MOUSE 752 EKLN---DROME 1480 RALQKIP
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://g
enet
ics.
nat
ure
.co
m© 2001 Nature Publishing Group http://genetics.nature.com

article
164 nature genetics • volume 29 • october 2001
HERc1, is also resident in the Golgi apparatus, its loss in ALS2may provide a clue to the disruption of the Golgi apparatusobserved in ALS42.
We have identified the gene whose mutation causes ALS2 andshow that it has a common genetic origin with JPLS. The reces-sively inherited deletion mutations suggest that the motor neurondegeneration in these disorders is a loss of function. Loss-of-func-tion paradigms provide an opportunity for direct examination ofthe molecular consequences of the loss in model systems. Such areductive approach is inherently difficult to apply in ALS causedby dominant mutations in SOD1 (refs. 43–46), as the mechanismof disease for ALS is the gain of an unknown function. The dele-tion mutation in JPLS occurs in an exon (exon 9) present only inthe long form of ALS2, while the deletion mutation in ALS2occurs in an exon (exon 3) shared by both the long and shortforms, upstream of the JPLS mutation. The mRNA of both formsof ALS2 from the lymphoblasts of an individual with ALS2 is eas-ily amplified by RT–PCR (Fig. 3, middle panel), but the stabilityof the mutant RNA and proteins, commonly observed to beunstable in truncation mutations47–49, remains to be determined.We hypothesize that the intact short form of alsin and/or thelonger mutant (645–amino acid) protein may retain partial func-tion and spare the motor neurons of the spinal cord and brainstem, thus exhibiting the JPLS and not the ALS2 phenotype. Thepresence of RCC1-like domains in both RPGR and alsin suggestthat GEFs and/or GTPases may be a common link in neuronaldegeneration of both sensory (RPGR) and motor (ALS) neurons,and should be investigated in other forms of neurodegeneration.
MethodsJPLS family acquisition. Individuals with JPLS in all three of the families withJPLS had childhood-onset, severe bulbar spasticity and extremity spasticitywith preserved cognition and sensation. Central-motor conduction timeswere severely delayed or unrecordable12. In contrast to ALS2 (ref. 23 and fam-ily 1212 in ref. 18), we did not detect changes in denervation and reinnerva-tion on electromyographic examination in the families with JPLS.
Genotyping. We extracted DNA from blood samples obtained after informedconsent. For the genome scan of JPLS, we carried out automated genotypingusing ABI Linkage Mapping Set Version 2 together with ABI 377 genotypingsoftware. For fine mapping of additional microsatellite polymorphic markersfrom chromosome 2q33 microdissection library, we used YACs and BACs toreduce the ALS2 locus. We carried out PCR reactions using 20 ng of genomicDNA, 200 µM dNTP including radioactively labeled [32P]dGTP, 0.1 U TaqGold polymerase and 1.5 µM MgCl2 in a 10-µl volume, using initial denatur-ing at 95 ºC for 10 min and then 30 cycles of dissociation at 94 ºC, annealing at55 ºC and extension at 72 ºC with 30-s steps. We resolved PCR products byrunning samples on 6% polyacrylamide gels. Two-point lod scores weredetermined using Fastlink2.2 version of the LINKAGE computer programs.We assumed autosomal recessive inheritance with complete penetrance with agene frequency of 0.0001. We genotyped 16 unrelated Saudi Arabian controlindividuals to derive control allele frequencies.
RT–PCR. We carried out two sets of RT–PCR to verify the presence of twotranscripts of ALS2 and their expression profiles in different tissues. For thefirst set of RT–PCR, we used the following primer: the forward primer(5´–GGACCCACTGGGTTGCCAAGCT–3´) is located at the 5′ UTR that isshared by both forms, the reverse primer for the short form is located at the 3′UTR for the short-form transcript (5´–TTCCTTTATCTAGGCTTGCCTGAT–3´) and the reverse primer for the long form is located at the 3′ UTR forthe long-form transcript (5´–GGTAAGGCTCATTCTAACAACCT–3´). Wesynthesized the first-strand cDNA using total RNA isolated from human lym-phoblastoid cells with an oligo-dT primer. For the second RT–PCR, we used aforward primer located in the fourth exon (5´–CAAGCAGCGATGCCATGTCCTCT–3´) for both short and long forms. The reverse primer for the shortform is the same as for the first set, and the reverse primer for the long form islocated in exon 5 (5´–CTGCCTGCCTGAGCTCCAGTTTC–3´). We synthe-sized the first-strand cDNA from poly(A) mRNA isolated from various
human tissues (Clontech). We carried out PCR using the Long and AccuratePCR system (LA PCR, Takara Shuzo) for 40 cycles: at 94 °C for 30 s, at 58 °Cfor 30 s and at 70 °C for 1–6 min, depending on the sizes of the PCR products.
Mutation detection. We used intronic primers to amplify the 33 codingexons of ALS2 by PCR. To avoid missing the intronic mutations that mayaffect the splicing, we designed all the primers at least 20 bp away from thesplicing sites. We divided the large exon 4 into five overlapping fragmentsfor PCR amplification. After 32 cycles of PCR amplification, we purifiedthe PCR products using 1.5% agarose gel and QIAquick gel extraction kit(Qiagen). We sequenced PCR products using the Big DyeDeoxy Termina-tor Cycle Sequencing kit (Perkin Elmer) on ABI 377 automatic sequencerand/or the Beckman Coulter CEQ 2000XL DNA analysis system. Wesequenced both strands with respective primers.
Northern blot analysis. We labeled the exon 1–3 fragment shared by twomajor transcripts of ALS2 using [32P]dGTP, and used the fragment as aprobe for northern hybridization on FirstChoice Northern Blots (Ambion)containing 2 µg of poly(A) RNA per lane from various tissues. We washedthe hybridized membrane with 0.1×SSC at 42 °C and analyzed it withphosphor imager (Storm 860, Molecular Dynamics).
Computer analysis. We carried out computer analysis of the cDNA andgenomic sequences to identify the transcribed sequences and to assemblethe full-length cDNA and genomic DNA sequences(www.ncbi.nlm.nih.gov/, www.ensembl.org/ and http://genome.ucsc.edu/). We used GENSCAN analysis to predict potential exons (http://genes.mit.edu/GENSCAN.html), and used Pfam, SMART and TIGRanalysis to identify the domain and motifs present in alsin (http://pfam.wustl.edu/hmmsearch.shtml, http://embl-heidelberg.de/, www.tigr.org/). We used the KAZUSA program to profile gene expression (www.kazusa.or.jp/en/).
Genbank accession numbers. Human alsin, AF391100, FLJ12962,AK023024, KIAA1563, AB046783; BAC clone RP11-182H9, AC007242;BAC clone RP11-309N8, AC007279; BAC clone RP11-129K3, AC018615;ESTs for human alsin, AI243773, BF993329, BE006110, AU125528,BE082284, AW867853, BE535882, BE003282, AW905587, AW905617;mouse alsin cDNA fragment, AK014320; mouse alsin protein fragment,BAB29271; D. melanogaster alsin, AAF51764.
AcknowledgmentsWe are deeply indebted to the members of the Tunisian and Saudi Arabianfamilies for their participation. This study was funded by grants from theNIH (PO1 NS21442, RO1 NS37912 and RO1 NS40308), the Les Turner ALSFoundation, Grant Healthcare Foundation, the Michael Jordan Foundationand the Ralph and Marian Falk Medical Research Trust. K.O. is a MuscularDystrophy Association funded fellow and W.-Y.H. is a Muriel Heller Fellow.
Received 20 June; accepted 28 August 2001.
1. Pringle, C.E. et al. Primary lateral sclerosis: clinical features, neuropathology anddiagnostic criteria. Brain 115, 495–520 (1992).
2. Charcot, J.M. Sclerose des cordons laeraux de la moelle epinière, chez une femmehystèrique, atteinte de contracture permanente des quatre members. Bull. Soc.Med. Hop. Paris 2, 24–42 (1965).
3. Erb, W.A. Über einen wenig bekannten spinalen symptomenkomplex. Berl. Klin.Wochenschr 12, 357–359 (1975).
4. Stark, F.M. & Moersch, F.P. Primary lateral sclerosis. J. Nerv. Ment. Dis. 102, 332–352(1945).
5. El Escorial World Federation of Neurology criteria for the diagnosis ofamyotrophic lateral sclerosis. J. Neurol. Sci. 124, 96–107 (1994).
6. Hudson, A.J. et al. Clinicopathological features of primary lateral sclerosis aredifferent from amyotrophic lateral sclerosis. Brain Res. Bull. 30, 359–364 (1993).
7. Beal, M.F. & Richardson, E.P. Primary lateral sclerosis: a case report. Arch. Neurol.38, 630–633 (1981).
8. Fisher, C.M. Pure spastic paralysis of corticospinal origin. Can. J. Neuro. Sci. 4,251–258 (1977).
9. Russo, L.S. Jr. Clinical and electrophysiological studies in primary lateral sclerosis.Arch. Neurol. 39, 662–664 (1982).
10. Sotaniemi, K.A. & Myllylla, V.V. Primary lateral sclerosis, a debated entity. ActaNeurol. Scand. 71, 334–336 (1985).
11. Younger, D.S. et al. Primary lateral sclerosis: a clinical diagnosis reemerges. Arch.Neurol. 45, 1304–1307 (1988).
12. Gascon, G.G. et al. Familial childhood primary lateral sclerosis with associatedgaze paresis. Neuorpediatrics 26, 313–319 (1995).
13. Heene, R., Kolander, D. & Knisatschek, H. Die chronisch-progrediente
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://g
enet
ics.
nat
ure
.co
m© 2001 Nature Publishing Group http://genetics.nature.com

article
nature genetics • volume 29 • october 2001 165
spinobulbäre spastik (primare lateralsklerose). Fortschr. Neurol. Psychiat. 64,192–203 (1996).
14. Brown, W.F., Ebers, G.C., Hudson, A.J., Pringle, C.E. & Veitch, J. Motor evokedresponses in primary lateral sclerosis. Muscle Nerve 15, 626–629 (1992).
15. Grunnet, M.L., Leicher, C., Zimmerman, A., Zalneraitis, E. & Barwick, M. Primarylateral sclerosis in a child. Neurology 39, 1530–1532 (1989).
16. Lerman-Sagie, T., Filliano, J., Smith, D.W. & Korson, M. Infantile onset ofhereditary ascending spastic paralysis with bulbar involvement. J. Child. Neurol.11, 54–57 (1996).
17. Siddique, T. et al. Linkage of a gene causing familial amyotrophic lateral sclerosisto chromosome 21 and evidence of genetic-locus heterogeneity. New Engl. J.Med. 324, 1381–1384 (1991).
18. Hentati, A. et al. Linkage of recessive familial amyotrophic lateral sclerosis tochromosome 2q33–q35. Nature Genet. 7, 425–428 (1994).
19. Hentati, A. et al. Linkage of a commoner form of recessive amyotrophic lateralsclerosis to chromosome 15q15-q22 markers. Neurogenetics 2, 55–60 (1998).
20. Chance, P. F. et al. Linkage of the gene for an autosomal dominant form ofjuvenile amyotrophic lateral sclerosis to chromosome 9q34. Am. J. Hum. Genet.62, 633–640 (1998).
21. Rosen, D. R. et al. Mutations in Cu/Zn superoxide dismutase gene are associatedwith familial amyotrophic lateral sclerosis. Nature 362, 59–62 (1993).
22. Deng, H.-X. et al. Amyotrophic lateral sclerosis and structural defects in Cu,Znsuperoxide dismutase. Science 261, 1047–1051 (1993).
23. Ben Hamida, M., Hentati, F. & Ben Hamida, C. Hereditary motor system diseases(chronic juvenile amyotrophic lateral sclerosis). Brain 113, 347–363 (1990).
24. Cottingham, R.W. Jr, Idury, R.M. & Schaffer, A.A. Faster sequential genetic linkagecomputations. Am. J. Hum. Genet. 53, 252–263 (1993).
25. Lathrop, G.M., Lalouel, J.M., Julier, C. & Ott, J. Strategies for multilocus linkageanalysis in humans. Proc. Natl Acad. Sci. USA 81, 3443–3446 (1984).
26. Schaffer, A.A., Gupta, S.K., Shriram, K. & Cottingham, R.W. Jr. Avoidingrecomputation in linkage analysis. Hum. Hered. 44, 225–237 (1994).
27. Ott, J. Analysis of Human Genetic Linkage revised edition 194–216 (Johns HopkinsUniversity, Baltimore, 1991).
28. Deng, H.X. et al. Chromosome-band-specific painting: chromosome in situsuppression hybridization using PCR products from a microdissected chromosomeband as a probe pool. Hum. Genet. 89, 13–17 (1992).
29. Sonnhammer, E.L.L., Eddy, S.R., Birney, E., Bateman, A. & Durbin, R. Pfam: multiplesequence alignments and HMM-profiles of protein domains. Nucleic Acids Res.26, 320–322 (1998).
30. Delcher, A.L. Alignment of whole genomes. Nucleic Acids Res. 27, 2369–2376(1999).
31. Schultz, J., Milpetz, F., Bork, P. & Ponting, C.P. SMART: identification andannotation of domains from signalling and extracellular protein sequences.Nucleic Acids Res. 27, 229–232 (1999).
32. Lemmon, M.A. and Ferguson, K.M. Signal-dependent membrane targeting by
pleckstrin homology (PH) domains. Biochem. J. 350, Pt 1: 1–18 (2000).33. Takeshima, H. et al. Junctophilins: a novel family of junctional membrane
complex proteins. Mol. Cell. 6, 11–22 (2000).34. Lehman, A. et al. A very large protein with diverse functional motifs is deficient in
rjs (runty, jerky, sterile) mice. Proc. Natl Acad. Sci. USA 95, 9436–9441 (1998).35. Kahana, J.A. & Cleveland, D.W. Some importin news about spindle assembly.
Science 291, 1718–1719 (2001).36. Roepman, R. et al. Positional cloning of the gene for X-linked retinitis pigmentosa
3: homology with the guanine-nucleotide-exchange factor RCC1. Hum. Mol.Genet. 5, 1035–1041 (1996).
37. Yan, D. et al. Biochemical characterization and subcellular localization of themouse retinitis pigmentosa GTPase regulator (mRpgr). J. Biol. Chem. 273,19656–19663 (1998).
38. Rosa, J.L., & Casaroli-Marano, R.P. A giant protein that stimulates guaninenucleotide exchange on ARF1 and Rab proteins forms a cytosolic ternary complexwith clathrin and Hsp70. Oncogene 15, 1–6 (1997).
39. Bar-Sagi, D. & Hall, A. Ras and Rho GTPases: a family reunion. Cell 103, 227–238(2000).
40. Hama, H., Tall, G.G. & Horazdovsky, B.F. Vps9p is a guanine nucleotide exchangefactor involved in vesicle-mediated vacuolar protein transport. J. Biol. Chem. 274,15284–15291 (1999).
41. Rampoldi, L. et al. A conserved sorting-associated protein is mutant in chorea-acanthocytosis. Nature Genet. 28, 119–120 (2001).
42. Mourelatos, Z., Gonatas, N.K., Stieber, A., Gurney, M.E. & Dal Canto, M.C. TheGolgi apparatus of spinal cord motor neurons in transgenic mice expressingmutant Cu,Zn superoxide dismutase becomes fragmented in early, preclinicalstages of the disease. Proc. Natl Acad. Sci. USA 93, 5472–5477 (1996).
43. Gurney, M.E. et al. Motor neuron degeneration in mice that express a humanCu,Zn superoxide dismutase mutation. Science 264, 1172–1175 (1994).
44. Brown, R.H. SOD1 aggregates in ALS: cause, correlate or consequence? NatureMed. 4, 1362–1364 (1998).
45. Cleveland, D.W. From Charcot to SOD1: mechanisms of selective motor neurondeath in ALS. Neuron 24, 515–520 (1999).
46. Deng, H.X. and Siddique, T. Transgenic mouse models and humanneurodegenerative disorders. Arch. Neurol. 57, 1695–1702 (2000).
47. Atawater, J.A., Wisdom, R. & Verma, I.M. Regulated mRNA stability. Annu. Rev.Genet. 24, 519–541 (1990).
48. Shoshani, T. et al. Similar levels of mRNA from the W1282X and the DeltaF508cystic fibrosis alleles, in nasal epithelial cells. J. Clin. Invest. 93, 1502–1507 (1994).
49. Haardt, M., Benharourga, M., Lechardeur, D., Kartner, N. & Lukacs, L. C-terminaltruncations destabilize the cystic fibrosis transmembrane conductance regulatorwithout impairing its biogenesis. J. Biol. Chem. 274, 21873–21877 (1999).
50. Dunnen, J.T. & Antonarakis, S.E. Mutation nomenclature extensions andsuggestions to describe complex mutations: a discussion. Human Mutat. 15, 7–12(2000).
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://g
enet
ics.
nat
ure
.co
m© 2001 Nature Publishing Group http://genetics.nature.com




















