
Many roads lead to primary autosomal recessive microcephaly
21
Many roads lead to primary autosomal recessive microcephaly Angela M. Kaindl a,b,c,d,1, *, Sandrine Passemard a,b,e,f,1 , Pavan Kumar g , Nadine Kraemer c,d , Lina Issa c,d , Angelika Zwirner c,d , Benedicte Gerard e , Alain Verloes e , Shyamala Mani g,h , Pierre Gressens a,b,f,i a Inserm, U676, Paris, France b Universite ´ Paris 7, Faculte ´ de Me ´decine Denis Diderot, IFR02 and IFR25, Paris, France c Department of Pediatric Neurology, Charite ´ – Universita ¨tsmedizin Berlin, Campus Virchow-Klinikum, Berlin, Germany d Institute for Neuroanatomy and Cell Biology, Charite ´ – Universita ¨tsmedizin Berlin, Campus Mitte, Berlin, Germany e APHP, Department of Clinical Genetics, Hoˆpital Robert Debre ´, Paris, France f APHP, Department of Pediatric Neurology, Hoˆpital Robert Debre ´, Paris, France g National Brain Research Centre, Manesar, India h CNS, IISc, Bangalore, India i Institute for Reproductive and Developmental Biology, Imperial College, Hammersmith Campus, London, United Kingdom Progress in Neurobiology 90 (2010) 363–383 ARTICLE INFO Article history: Received 13 July 2009 Received in revised form 27 October 2009 Accepted 11 November 2009 Keywords: Microcephaly MCPH Centrosome Spindle Cell cycle Mental retardation ABSTRACT Autosomal recessive primary microcephaly (MCPH), historically referred to as Microcephalia vera, is a genetically and clinically heterogeneous disease. Patients with MCPH typically exhibit congenital microcephaly as well as mental retardation, but usually no further neurological findings or malformations. Their microcephaly with grossly preserved macroscopic organization of the brain is a consequence of a reduced brain volume, which is evident particularly within the cerebral cortex and thus results to a large part from a reduction of grey matter. Some patients with MCPH further provide evidence of neuronal heterotopias, polymicrogyria or cortical dysplasia suggesting an associated neuronal migration defect. Genetic causes of MCPH subtypes 1–7 include mutations in genes encoding microcephalin, cyclin-dependent kinase 5 regulatory associated protein 2 (CDK5RAP2), abnormal spindle-like, microcephaly associated protein (ASPM), centromeric protein J (CENPJ), and SCL/TAL1- interrupting locus (STIL) as well as linkage to the two loci 19q13.1–13.2 and 15q15–q21. Here, we provide a timely overview of current knowledge on mechanisms leading to microcephaly in humans with MCPH and abnormalities in cell division/cell survival in corresponding animal models. Understanding the pathomechanisms leading to MCPH is of high importance not only for our understanding of physiologic brain development (particularly of cortex formation), but also for that of trends in mammalian evolution with a massive increase in size of the cerebral cortex in primates, of microcephalies of other etiologies including environmentally induced microcephalies, and of cancer formation. ß 2009 Elsevier Ltd. All rights reserved. Abbreviations: 53BP1, tumor-suppressor protein p53 binding protein 1; APC, anaphase-promoting complex; ASPM, abnormal spindle-like, microcephaly associated protein; ATM, ataxia telangiectasia mutated; ATR, ataxia telangiectasia and RAD3-related protein; BACH1, BRCA1-associated carboxyl-terminal helicase; BRCT, carboxyl-terminal domain of the breast cancer gene 1 BRCA1; BRIT1, BRCT-Repeat Inhibitor of hTert expression; BUBR1, budding uninhibited by benzimidazoles 1 homolog beta; C48, CDK5 activator binding protein C48; CASK, calcium/calmodulin-dependent serine protein kinase gene; CDC24A, cell division cycle 25A; CDK5, cyclin-dependent kinase 5; CDK5R1, cyclin-dependent kinase 5 regulatory subunit 1; CDK5RAP2, cyclin-dependent kinase 5 regulatory associated protein 2; CENPJ, centromeric protein J; CEP215, centrosome associated protein 215; CH, calponin homology domain; CHK1/2, checkpoint kinase 1/2; CMD, calmodulin; cnn, centrosomin; CNS, central nervous system; CPAP, centrosomal P4.1-associated protein; E, embryonal day; EB1, plus-end binding protein EB1; GLI1, glioma-associated oncogene homolog; gTuRC, gamma-tubulin ring complex; Hnf3b, hepatocyte nuclear factor 3-beta, forehead box A2; LAP, LAG-3-associated protein; Lefty2, left–right determination factor 2; LIP1, LYST-interacting protein 1; MAD2, mitotic arrest-deficient 2; MCPH, autosomal recessive primary microcephaly; MCPH1, microcephalin; MDC1, mediator of DNA damage checkpoint protein 1; MOT, microtubule-organizing center; NBS1, Nijmegen breakage syndrome protein 1; NE, neuroepithelium; OFC, occipito-frontal head circumference; PCC, premature chromosome condensation; PCM, pericentriolar matrix; PCNT, pericentrin; Pitx2, paired-like homeodomain transcription factor 2; PLK1, polo-like kinase 1; RELN, reelin; Sas-4, spindle assembly abnormal 4; SD, standard deviation; Shh, sonic hedgehog; shRNA, short hairpin RNA; STIL, SCL/TAL1-interrupting locus; SUFU, Suppressor of Fused; Tbr1, T-box brain 1; TNFalpha, tumor necrosis factor-alpha; zyg-1, ZYGote defective, embryonic lethal. * Corresponding author at: Department of Pediatric Neurology, Charite ´ – University Medicine Berlin, Campus Virchow Klinikum, 13353 Berlin, Germany. Tel.: +49 030 450 566 112; fax: +49 030 450 566 920. E-mail address: [email protected] (A.M. Kaindl). 1 These authors contributed equally to this work. Contents lists available at ScienceDirect Progress in Neurobiology journal homepage: www.elsevier.com/locate/pneurobio 0301-0082/$ – see front matter ß 2009 Elsevier Ltd. All rights reserved. doi:10.1016/j.pneurobio.2009.11.002
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Many roads lead to primary autosomal recessive microcephaly

Progress in Neurobiology 90 (2010) 363–383
Many roads lead to primary autosomal recessive microcephaly
Angela M. Kaindl a,b,c,d,1,*, Sandrine Passemard a,b,e,f,1, Pavan Kumar g, Nadine Kraemer c,d, Lina Issa c,d,Angelika Zwirner c,d, Benedicte Gerard e, Alain Verloes e, Shyamala Mani g,h, Pierre Gressens a,b,f,i
a Inserm, U676, Paris, Franceb Universite Paris 7, Faculte de Medecine Denis Diderot, IFR02 and IFR25, Paris, Francec Department of Pediatric Neurology, Charite – Universitatsmedizin Berlin, Campus Virchow-Klinikum, Berlin, Germanyd Institute for Neuroanatomy and Cell Biology, Charite – Universitatsmedizin Berlin, Campus Mitte, Berlin, Germanye APHP, Department of Clinical Genetics, Hopital Robert Debre, Paris, Francef APHP, Department of Pediatric Neurology, Hopital Robert Debre, Paris, Franceg National Brain Research Centre, Manesar, Indiah CNS, IISc, Bangalore, Indiai Institute for Reproductive and Developmental Biology, Imperial College, Hammersmith Campus, London, United Kingdom
A R T I C L E I N F O
Article history:
Received 13 July 2009
Received in revised form 27 October 2009
Accepted 11 November 2009
Keywords:
Microcephaly
MCPH
Centrosome
Spindle
Cell cycle
Mental retardation
A B S T R A C T
Autosomal recessive primary microcephaly (MCPH), historically referred to as Microcephalia vera, is a
genetically and clinically heterogeneous disease. Patients with MCPH typically exhibit congenital
microcephaly as well as mental retardation, but usually no further neurological findings or
malformations. Their microcephaly with grossly preserved macroscopic organization of the brain is a
consequence of a reduced brain volume, which is evident particularly within the cerebral cortex and thus
results to a large part from a reduction of grey matter. Some patients with MCPH further provide evidence
of neuronal heterotopias, polymicrogyria or cortical dysplasia suggesting an associated neuronal
migration defect. Genetic causes of MCPH subtypes 1–7 include mutations in genes encoding
microcephalin, cyclin-dependent kinase 5 regulatory associated protein 2 (CDK5RAP2), abnormal
spindle-like, microcephaly associated protein (ASPM), centromeric protein J (CENPJ), and SCL/TAL1-
interrupting locus (STIL) as well as linkage to the two loci 19q13.1–13.2 and 15q15–q21. Here, we provide
a timely overview of current knowledge on mechanisms leading to microcephaly in humans with MCPH
and abnormalities in cell division/cell survival in corresponding animal models. Understanding the
pathomechanisms leading to MCPH is of high importance not only for our understanding of physiologic
brain development (particularly of cortex formation), but also for that of trends in mammalian evolution
with a massive increase in size of the cerebral cortex in primates, of microcephalies of other etiologies
including environmentally induced microcephalies, and of cancer formation.
� 2009 Elsevier Ltd. All rights reserved.
Contents lists available at ScienceDirect
Progress in Neurobiology
journa l homepage: www.e lsev ier .com/ locate /pneurobio
Abbreviations: 53BP1, tumor-suppressor protein p53 binding protein 1; APC, anaphase-promoting complex; ASPM, abnormal spindle-like, microcephaly associated protein;
ATM, ataxia telangiectasia mutated; ATR, ataxia telangiectasia and RAD3-related protein; BACH1, BRCA1-associated carboxyl-terminal helicase; BRCT, carboxyl-terminal
domain of the breast cancer gene 1 BRCA1; BRIT1, BRCT-Repeat Inhibitor of hTert expression; BUBR1, budding uninhibited by benzimidazoles 1 homolog beta; C48, CDK5
activator binding protein C48; CASK, calcium/calmodulin-dependent serine protein kinase gene; CDC24A, cell division cycle 25A; CDK5, cyclin-dependent kinase 5; CDK5R1,
cyclin-dependent kinase 5 regulatory subunit 1; CDK5RAP2, cyclin-dependent kinase 5 regulatory associated protein 2; CENPJ, centromeric protein J; CEP215, centrosome
associated protein 215; CH, calponin homology domain; CHK1/2, checkpoint kinase 1/2; CMD, calmodulin; cnn, centrosomin; CNS, central nervous system; CPAP,
centrosomal P4.1-associated protein; E, embryonal day; EB1, plus-end binding protein EB1; GLI1, glioma-associated oncogene homolog; gTuRC, gamma-tubulin ring
complex; Hnf3b, hepatocyte nuclear factor 3-beta, forehead box A2; LAP, LAG-3-associated protein; Lefty2, left–right determination factor 2; LIP1, LYST-interacting protein 1;
MAD2, mitotic arrest-deficient 2; MCPH, autosomal recessive primary microcephaly; MCPH1, microcephalin; MDC1, mediator of DNA damage checkpoint protein 1; MOT,
microtubule-organizing center; NBS1, Nijmegen breakage syndrome protein 1; NE, neuroepithelium; OFC, occipito-frontal head circumference; PCC, premature chromosome
condensation; PCM, pericentriolar matrix; PCNT, pericentrin; Pitx2, paired-like homeodomain transcription factor 2; PLK1, polo-like kinase 1; RELN, reelin; Sas-4, spindle
assembly abnormal 4; SD, standard deviation; Shh, sonic hedgehog; shRNA, short hairpin RNA; STIL, SCL/TAL1-interrupting locus; SUFU, Suppressor of Fused; Tbr1, T-box
brain 1; TNFalpha, tumor necrosis factor-alpha; zyg-1, ZYGote defective, embryonic lethal.
* Corresponding author at: Department of Pediatric Neurology, Charite – University Medicine Berlin, Campus Virchow Klinikum, 13353 Berlin, Germany.
Tel.: +49 030 450 566 112; fax: +49 030 450 566 920.
E-mail address: [email protected] (A.M. Kaindl).1 These authors contributed equally to this work.
0301-0082/$ – see front matter � 2009 Elsevier Ltd. All rights reserved.
doi:10.1016/j.pneurobio.2009.11.002

A.M. Kaindl et al. / Progress in Neurobiology 90 (2010) 363–383364
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 364
2. Phenotype of patients with MCPH . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 364
3. Genotype of patients with MCPH . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 364
4. Pathomechanisms underlying MCPH . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 365
5. Microcephalin (syn. BRIT1; MCPH1) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 365
6. Cyclin-dependent kinase 5, regulatory associated protein 2 (syn. CDK5RAP2, C48, CEP215, KIAA1633, MCPH3) . . . . . . . . . . . . . . . . . . . . . 373
7. Abnormal spindle-like, microcephaly-associated (ASPM, MCPH5) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 376
8. Centromeric protein J (syn. CENPJ, CPAP, LAP, LIP1, MCPH6) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 378
9. SCL/TAL1-interrupting locus (syn. STIL, SIL, MCPH7) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 378
10. MCPH and cerebellum . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 379
11. The role of MCPH proteins in the evolution of the cerebral cortex. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 379
12. Other proteins that show an overlap with MCPH pathomechanisms and phenotype . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 380
13. Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 380
Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 381
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 381
1. Introduction
Microcephaly is defined as a small cranium with a significantlyreduced occipito-frontal head circumference (OFC) of more thantwo standard deviations (SD) below the mean for age, sex, andethnicity (severe microcephaly OFC < �3 SD). Microcephaly can beacquired (caused by environmental factors) or hereditary in originand can become apparent congenitally (primary microcephaly) orpostnatally (secondary microcephaly). The incidence of micro-cephaly at birth, as evaluated in birth defect registers world-wide,varies from 1.3 to 150 per 100,000 live-births, depending on thepopulation and the applied SD threshold to define microcephaly(source: International Clearinghouse for Birth Defects Surveillanceand Research, 2006 report; http://www.icbdsr.org/). Primary, non-syndromal microcephaly has an incidence of 1:30,000 to 1:250,000live-births (Van Den Bosch, 1959).
Microcephalia vera (‘true microcephaly’) is a loosely defined,historical term referring to children with isolated, non-syndromalcongenital microcephaly. This term was coined without consider-ation of etiology or neuropathology, and it is still applied, however,in a narrower sense, to designate patients with non-syndromalautosomal recessive microcephaly without lissencephaly orpachygyria. Syndromic microcephalies as well as secondary, oftenenvironmentally induced microcephalies which can be caused byunderlying processes such as high rates of pathologic apoptosis,proliferation and patterning abnormalities, migration defects,disturbance of extracellular matrix integrity and defects ofsynaptogenesis will not be discussed in this review (please referto Francis et al., 2006; Katyal and McKinnon, 2007; Abuelo, 2007;Woods, 2004).
2. Phenotype of patients with MCPH
Autosomal recessive primary microcephaly (MCPH, for Micro-Cephaly Primary Hereditary) is a rare, genetically heterogeneousdisease reported in about 100 families world-wide. Historically,primary microcephaly was defined by an OFC < �3 at birth, areduced brain volume, mental retardation (IQ between 30 and 70–80) and no further neurological findings except for mild seizures(Woods et al., 2005; Passemard, 2009) (Fig. 1). However, it isbecoming clear that the true phenotype spectrum of patients withMCPH gene mutations is wider than indicated by previouspublications which for the most part provide no detailedphenotype information. In individual patients, the OFC is stillin the normal range (around �2 SD) at birth followed by adevelopment of a microcephaly within the first year of life(Passemard, 2009). As recently noted, MCPH may already be
evident by the 24th week of gestation through ultrasound and/orMRI analysis (Tunca et al., 2006). Also, we were able todemonstrate recently that neurological features can indeed occurin patients with MCPH due to ASPM gene mutations (Passemard,2009). These include speech delay, hyperactivity and attentiondeficit, aggressiveness, focal or generalized seizures, delay ofdevelopmental milestones and pyramidal signs (Passemard, 2009).Hyperactivity and attention deficit appeared to be major childhoodproblems in all patients of our cohort and possibly causedimpairments in performance. Moreover, abnormal height andweight was detected in some patients (Passemard, 2009; Trimbornet al., 2004).
Imaging studies reveal typically brains of ‘normal architecture’but of reduced size (Woods et al., 2005). The latter is particularlyevident in the cerebral cortex, which shows a simplified cerebralcortex structure, and there is also a slightly reduced white mattervolume (Fig. 2). Individual patients with MCPH provide evidenceof periventricular neuronal heterotopias (Woods et al., 2005;Trimborn et al., 2004) suggesting neuronal migration defects.Moreover, we have detected infra-tentorial anomalies (brainstemor cerebellar hypoplasia), dysmorphic and/or enlarged lateralventricles and corpus callosum agenesia as well as focal micro-polygyria and/or dysplasia (Passemard, 2009). Future studies willneed to address in what way white matter disease also contributesto brain size reduction in MCPH patients. Descriptions of thehistological findings in MCPH patients, indicating a significantlyreduced brain volume with an almost preserved convolutionpattern, are rare and were performed at a time when a geneticdiagnosis was not possible (Bamatter and Rabinowicz, 1969;Robain and Lyon, 1972). An overview of MCPH features, genemutations and the reported phenotype by MCPH gene mutationare given in Tables 1–3, respectively.
3. Genotype of patients with MCPH
Genetic causes of MCPH subtypes 1–7 include mutations ingenes encoding microcephalin (MCPH1; Jackson et al., 2002, 1998;OMIM #251200), cyclin-dependent kinase 5 regulatory associatedprotein 2 CDK5RAP2 (MCPH3; Bond et al., 2005; Moynihan et al.,2000; OMIM #604804), abnormal spindle-like, microcephalyassociated ASPM (MCPH5; Pattison et al., 2000; Shen et al.,2005; OMIM #608716), centromeric protein J CENPJ (MCPH6;Bond et al., 2005; Leal et al., 2003; OMIM #608393), SCL/TAL1-interrupting locus STIL (MCPH7; Kumar et al., 2009; OMIM#612703) as well as linkage to the two loci 19q13.1–13.2 (MCPH2;Roberts, 1999); OMIM%604317) and 15q15–q21 (MCPH4; Jamie-son et al., 1999); OMIM%604321) (Kaindl, 2008); see Table 2 and
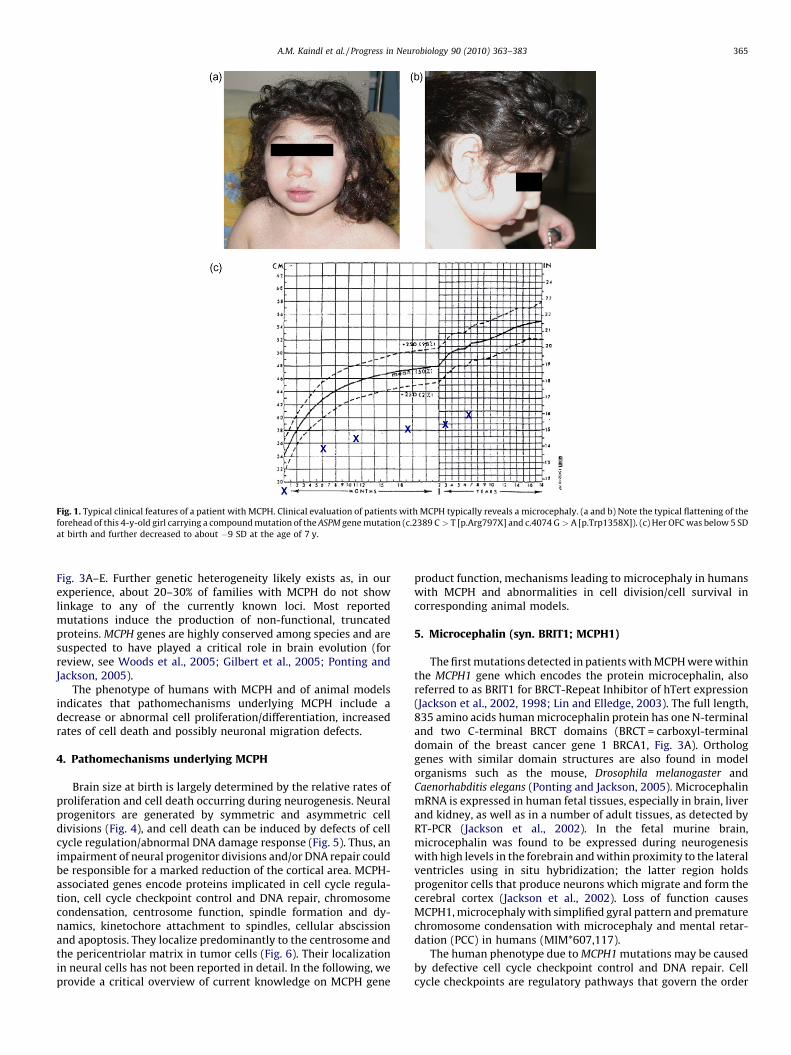
Fig. 1. Typical clinical features of a patient with MCPH. Clinical evaluation of patients with MCPH typically reveals a microcephaly. (a and b) Note the typical flattening of the
forehead of this 4-y-old girl carrying a compound mutation of the ASPM gene mutation (c.2389 C > T [p.Arg797X] and c.4074 G > A [p.Trp1358X]). (c) Her OFC was below 5 SD
at birth and further decreased to about �9 SD at the age of 7 y.
A.M. Kaindl et al. / Progress in Neurobiology 90 (2010) 363–383 365
Fig. 3A–E. Further genetic heterogeneity likely exists as, in ourexperience, about 20–30% of families with MCPH do not showlinkage to any of the currently known loci. Most reportedmutations induce the production of non-functional, truncatedproteins. MCPH genes are highly conserved among species and aresuspected to have played a critical role in brain evolution (forreview, see Woods et al., 2005; Gilbert et al., 2005; Ponting andJackson, 2005).
The phenotype of humans with MCPH and of animal modelsindicates that pathomechanisms underlying MCPH include adecrease or abnormal cell proliferation/differentiation, increasedrates of cell death and possibly neuronal migration defects.
4. Pathomechanisms underlying MCPH
Brain size at birth is largely determined by the relative rates ofproliferation and cell death occurring during neurogenesis. Neuralprogenitors are generated by symmetric and asymmetric celldivisions (Fig. 4), and cell death can be induced by defects of cellcycle regulation/abnormal DNA damage response (Fig. 5). Thus, animpairment of neural progenitor divisions and/or DNA repair couldbe responsible for a marked reduction of the cortical area. MCPH-associated genes encode proteins implicated in cell cycle regula-tion, cell cycle checkpoint control and DNA repair, chromosomecondensation, centrosome function, spindle formation and dy-namics, kinetochore attachment to spindles, cellular abscissionand apoptosis. They localize predominantly to the centrosome andthe pericentriolar matrix in tumor cells (Fig. 6). Their localizationin neural cells has not been reported in detail. In the following, weprovide a critical overview of current knowledge on MCPH gene
product function, mechanisms leading to microcephaly in humanswith MCPH and abnormalities in cell division/cell survival incorresponding animal models.
5. Microcephalin (syn. BRIT1; MCPH1)
The first mutations detected in patients with MCPH were withinthe MCPH1 gene which encodes the protein microcephalin, alsoreferred to as BRIT1 for BRCT-Repeat Inhibitor of hTert expression(Jackson et al., 2002, 1998; Lin and Elledge, 2003). The full length,835 amino acids human microcephalin protein has one N-terminaland two C-terminal BRCT domains (BRCT = carboxyl-terminaldomain of the breast cancer gene 1 BRCA1, Fig. 3A). Orthologgenes with similar domain structures are also found in modelorganisms such as the mouse, Drosophila melanogaster andCaenorhabditis elegans (Ponting and Jackson, 2005). MicrocephalinmRNA is expressed in human fetal tissues, especially in brain, liverand kidney, as well as in a number of adult tissues, as detected byRT-PCR (Jackson et al., 2002). In the fetal murine brain,microcephalin was found to be expressed during neurogenesiswith high levels in the forebrain and within proximity to the lateralventricles using in situ hybridization; the latter region holdsprogenitor cells that produce neurons which migrate and form thecerebral cortex (Jackson et al., 2002). Loss of function causesMCPH1, microcephaly with simplified gyral pattern and prematurechromosome condensation with microcephaly and mental retar-dation (PCC) in humans (MIM*607,117).
The human phenotype due to MCPH1 mutations may be causedby defective cell cycle checkpoint control and DNA repair. Cellcycle checkpoints are regulatory pathways that govern the order

Fig. 2. Radiological features of patients with MCPH. Cranial MRI of the patient depicted in Fig. 1 with an ASPM gene mutation (A) compared to an age matched control (B). Axial
(1) and coronal (2) T2-weighted images show the typical reduction of brain volume, especially of cerebral cortex in a patient with MCPH (A) in comparison to an age-matched
control (B). Note the typical appearance of a simplified gyral pattern with scarce gyri in the patient. Here, the number of gyri is reduced and the sulci are shallow compared to
the control person. Sagittal (3) T1-weighted images illustrate the typical MRI features of patients with ASPM mutations in comparison to controls. Note the poorly developed
frontal lobes in the patient and the agenesis of the rostrum of the corpus callosum (white arrow).
A.M. Kaindl et al. / Progress in Neurobiology 90 (2010) 363–383366
and timing of cell cycle transitions to ensure completion of onecellular event prior to commencement of another one. Thesecheckpoint control mechanisms are linked to those of DNA damagerepair as, in response to DNA damage, the cell cycle needs to bedelayed until the damage is repaired to restore the integrity of theorganism or, if repair is not possible, arrested with a subsequentinduction of cell death. The BRCT domains occurring in MCPH1 areevolutionarily conserved phosphor-peptide binding amino acidtandem repeat domains involved in cell cycle control (Bork et al.,1997; Huyton et al., 2000; Yu et al., 2003). The BRCT domain wasfirst identified within the tumor-suppressor gene product BRCA1(Yu et al., 2003); there it interacts with phosphorylated BRCA1-associated carboxyl-terminal helicase (BACH1) in a cell cycleregulated manner, and this interaction is required for DNAdamage-induced checkpoint control during the transition fromG2 phase to M phase of the cell cycle (Yu et al., 2003). Genomicmutations of BRCA1 as well as reduced expression can be found infamilial breast cancer and other tumor specimen (Yu et al., 2003;Rai et al., 2006; Hagemann et al., 2008).
Microcephalin is induced by DNA damage and forms nuclearfoci minutes after irradiation-induced DNA damage in tumor cells(Rai et al., 2006). The protein is recruited to double-strand breaksvia interaction of its C-terminal BRCT domains with phosphory-lated H2AX, a histone variant that marks sites of damaged DNA(Wood et al., 2007). This then allows for recruitment of othermediator proteins that help promote the damage signal to CHK1and CHK2, which can phosphorylate various substrates tomodulate the cell cycle, DNA repair and apoptosis (Wood et al.,2007). Microcephalin specifically co-localizes to and activates DNAdamage response proteins, such as mediator of DNA damagecheckpoint protein 1 (MDC1), tumor-suppressor protein p53binding protein 1 (53BP1), Nijmegen breakage syndrome protein1 (NBS1) and phosphorylated ATM (ataxia telangiectasia, mutated)(Rai et al., 2006). Mutated MCPH1-transcript detected in thebreast cancer specimen was unable to complement the defectiveactivation of DNA damage response proteins, in contrast to fulllength MCPH1 (Rai et al., 2006). MCPH1 interacts with condensin IIin the homologous repair of DNA damage (Wood, 2008). Moreover,

Table 1Overview of MCPH features.
Main features � Microcephaly (�2 SD) at birth, further
‘relative’ reduction of OFC in the first years of life
� Reduction of cerebral cortex volume
� Simplification of gyral pattern
� Mild to severe mental retardation
(normal IQ possible)
Inconsistent features � Neurological/neuropsychological
- Delay of motor milestones
- Pyramidal signs
- Speech delay
- Hyperactivity and attention deficit
- Aggressiveness
- Sleep disorder
- Seizures (tonic-clonic, focal, generalized)
� Further intracranial malformations
- Agenesis of the corpus callosum
- Focal dysplasia
- Focal microgyria
- Perisylvanian polymicrogyria
- Dysmorphic or enlarged lateral ventricles
- Large pituitary gland
- Reduction of white matter
- Infra-tentorial anomalies (brainstem/
cerebellar hypoplasia)
� Endocrinology
- Short stature
- Early puberty
� Extracranial malformations
- Syndactyly
- Renal agenesia
- Multicystic kidney
� Other
- Premature chromosome condensation
(e.g. in MCPH1)
- So far no increased cancer risk noted
A.M. Kaindl et al. / Progress in Neurobiology 90 (2010) 363–383 367
MCPH1 gene mutations result in premature onset of condensin II-mediated chromosome condensation (Trimborn et al., 2006).Further evidence that MCPH1 may be involved in checkpointcontrol comes from experiments using RNA interference. Suchan approach lead to a severe impairment of the intra-S phase andG2/M DNA damage checkpoint in U2OS cells (human osteosarcomacell line) with a loss of checkpoint control allowing cells to proceedinto mitosis despite exposure to considerable doses of ionizingirradiation (IR), an increased sensibility to DNA damage via IR and adecrease of BRCA1 and the checkpoint kinase 1 (CHK1) (Xu et al.,2004; Lin et al., 2005). The latter are key regulators in the control ofintra-S and G2/M checkpoints. MCPH1 also acts downstream fromCHK1 in regulating the stability of cell division cycle 25A (Cdc25A),a critical regulator of cell cycle progression, and consequentlypreventing premature entry into mitosis (Alderton et al., 2006).Microcephalin may thus affect DNA damage checkpoints by bothdirectly transmitting DNA damage signals and controlling theexpression levels of other checkpoint regulators. It has recentlybeen shown that Microcephalin promotes the expression of CHK1
Table 2Overview of MCPH genes.
MCPH Protein Gene
MCPH1 Microcephalin MCPH
MCPH2 –
MCPH3 Cyclin-dependent kinase 5 regulatory associated protein 2 CDK5
MCPH4 –
MCPH5 Abnormal spindle-like, microcephaly associated ASPM
MCPH6 Centromeric protein J CENP
MCPH7 SCL/TAL1 interrupting locus STIL
and BRCA1 through the interaction with the transcription factorE2F1 and enhancement of their transcriptional activities on thepromoters of both genes as well as other E2F targeted genesinvolved in DNA repair and apoptosis such as RAD51, DDB2,TOPBP1, p73 and caspases (Yang et al., 2008). In humanlymphoblastoid cell lines with MCPH1 truncating mutations, lossof MCPH1 function induces defective G2/M checkpoint arrest,nuclear fragmentation after DNA damage and supernumerarymitotic centrosomes (Alderton et al., 2006).
Several findings underline the role of MCPH1 as a regulator ofchromosome condensation. Mutations in the MCPH1 gene have notonly been reported in patients with MCPH but also in those withpremature chromosome condensation (PCC) syndrome, character-ized by microcephaly, short stature and misregulated chromosomecondensation (Trimborn et al., 2004). Intriguingly, the cellularphenotype of patients with PCC or MCPH1 includes high numbersof prophase-like cells due to premature chromosome condensationin early G2-phase and delayed decondensation in post-mitosis.This cellular phenotype can be reproduced through MCPH1-siRNAmediated reduction of the mRNA (Trimborn et al., 2004, 2006).MCPH1 inhibits condensin II, a protein important for chromosomearchitecture, potentially via CDK-cyclin A (Trimborn et al., 2006).MCPH1 appears to also influence centrosome function as itlocalizes to the centrosome throughout the cell cycle in U2OScells (Zhong et al., 2006) and also in other model organisms (Brunket al., 2007; Rickmyre et al., 2007; Jeffers et al., 2008). The N-terminal BRCT1 has been reported to be required for centrosomallocalization in irradiated cells (Jeffers et al., 2008). Knock-down ofMicrocephalin in U2OS cells via siRNA induced centrosomeanomalies (possibly a secondary effect), spindle misalignment aswell as compacted and lagging chromosomes with defectivespindle checkpoint activation and delayed cytokinesis by nega-tively regulating Aurora and polo-like kinase 1 (PLK1) (Rai et al.,2008). Spindle positioning is critical to ensure the unequalsegregation of polarity factors and generate daughter cells withdifferent sizes or fates during asymmetric cell division. Treatmentof MCPH1 patient lymphoblastoid cell lines with nocodazoleresults in supernumerary centrosomes (Alderton et al., 2006).Mcph1 appears to be essential for the coordination of mitosis in theDrosophila embryo as, in mcph1 mutant flies, mitotic entry isslowed from the very first mitosis with prolonged prophase andmetaphase stages frequent premature separation as well asdetachment of centrosomes (Brunk et al., 2007; Table 4). As aconsequence, centrosome and nuclear cycles become uncoordi-nated, resulting in premature chromosome condensation, mitoticentry with unreplicated DNA, genomic instability and arrestedembryonic development (Brunk et al., 2007; Rickmyre et al.,2007). Brains of adult male mcph1�/� flies are reportedly ofnormal size but have defects in mushroom body structure,suggesting an evolutionarily conserved role for MCPH1 inbrain development (Rickmyre et al., 2007). Microcephaly occurringin MCPH1-deficiency may thus also be due to a defectivecentrosomal function of MCPH1 influencing the number ofproliferative, symmetric cell divisions of neuronal stem cellsduring neurogenesis.
Chromosome References
1 8p23 Jackson et al. (2002, 1998)
19q13.1–q13.2 Roberts (1999)
RAP2 9q33.3 Bond et al. (2005), Moynihan et al. (2000)
15q15–q21 Jamieson et al. (1999)
1q31 Pattison et al. (2000), Shen et al. (2005)
J 13q12.2 Bond et al. (2005), Leal et al. (2003)
1p32 Kumar et al. (2009)
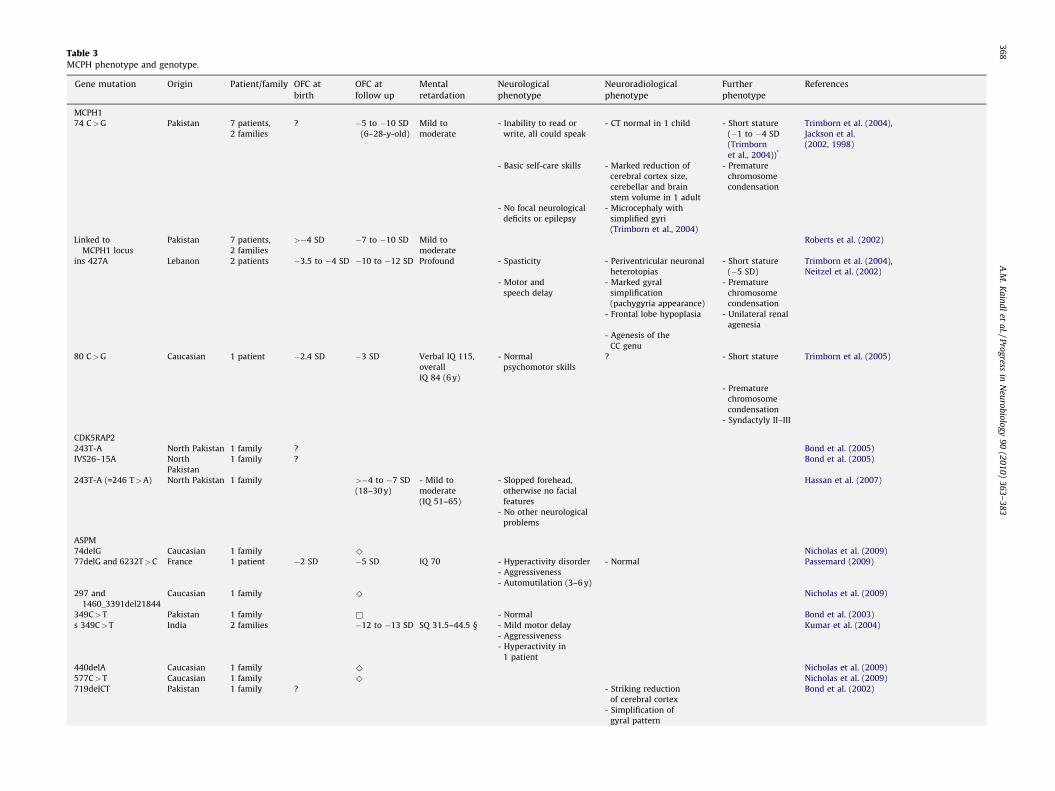
Table 3MCPH phenotype and genotype.
Gene mutation Origin Patient/family OFC at
birth
OFC at
follow up
Mental
retardation
Neurological
phenotype
Neuroradiological
phenotype
Further
phenotype
References
MCPH1
74 C>G Pakistan 7 patients,
2 families
? �5 to �10 SD
(6–28-y-old)
Mild to
moderate
- Inability to read or
write, all could speak
- CT normal in 1 child - Short stature
(�1 to �4 SD
(Trimborn
et al., 2004))*
Trimborn et al. (2004),
Jackson et al.
(2002, 1998)
- Basic self-care skills - Marked reduction of
cerebral cortex size,
cerebellar and brain
stem volume in 1 adult
- Premature
chromosome
condensation
- No focal neurological
deficits or epilepsy
- Microcephaly with
simplified gyri
(Trimborn et al., 2004)
Linked to
MCPH1 locus
Pakistan 7 patients,
2 families
>�4 SD �7 to �10 SD Mild to
moderate
Roberts et al. (2002)
ins 427A Lebanon 2 patients �3.5 to �4 SD �10 to �12 SD Profound - Spasticity - Periventricular neuronal
heterotopias
- Short stature
(�5 SD)
Trimborn et al. (2004),
Neitzel et al. (2002)
- Motor and
speech delay
- Marked gyral
simplification
(pachygyria appearance)
- Premature
chromosome
condensation
- Frontal lobe hypoplasia - Unilateral renal
agenesia
- Agenesis of the
CC genu
80 C>G Caucasian 1 patient �2.4 SD �3 SD Verbal IQ 115,
overall
IQ 84 (6 y)
- Normal
psychomotor skills
? - Short stature Trimborn et al. (2005)
- Premature
chromosome
condensation
- Syndactyly II–III
CDK5RAP2
243T-A North Pakistan 1 family ? Bond et al. (2005)
IVS26–15A North 1 family ? Bond et al. (2005)
Pakistan
243T-A (=246 T>A) North Pakistan 1 family >�4 to �7 SD
(18–30 y)
- Mild to
moderate
(IQ 51–65)
- Slopped forehead,
otherwise no facial
features
Hassan et al. (2007)
- No other neurological
problems
ASPM
74delG Caucasian 1 family ^ Nicholas et al. (2009)
77delG and 6232T>C France 1 patient �2 SD �5 SD IQ 70 - Hyperactivity disorder - Normal Passemard (2009)
- Aggressiveness
- Automutilation (3–6 y)
297 and
1460_3391del21844
Caucasian 1 family ^ Nicholas et al. (2009)
349C>T Pakistan 1 family & - Normal Bond et al. (2003)
s 349C>T India 2 families �12 to �13 SD SQ 31.5–44.5 § - Mild motor delay Kumar et al. (2004)
- Aggressiveness
- Hyperactivity in
1 patient
440delA Caucasian 1 family ^ Nicholas et al. (2009)
577C>T Caucasian 1 family ^ Nicholas et al. (2009)
719delCT Pakistan 1 family ? - Striking reduction
of cerebral cortex
Bond et al. (2002)
- Simplification of
gyral pattern
A.M
.K
ain
dl
eta
l./Pro
gress
inN
euro
bio
log
y9
0(2
01
0)
36
3–
38
33
68

1002delA Pakistan 1 family Muhammad et al. (2009)
1152delAG Caucasian 1 family ^ Nicholas et al. (2009)
1258delTCTCAAG Pakistan 2 families ? & - Normal Bond et al. (2002, 2003)
1260delTCAAGTC Pakistan 1 family # - Moderate - Normal Gul et al. (2006)
1630delATCTT and
full length ASPM
gene del
France 1 patient �3.5 SD �7 SD IQ 30 - Mild motor and
speech delay
- Simplification of
frontal and occipital
gyral pattern
- Early puberty Passemard (2009)
- Hyperactivity
disorder (3–12 y)
- Enlarged occipital
horns of the LV
1366G>T Turkey 1 family ^ Nicholas et al. (2009)
1406delATCCTAAAA Caucasian 1 family ^ Nicholas et al. (2009)
1590delA Caucasian 1 family ^ Nicholas et al. (2009)
1727delAG Yemen 1 family �10 to �11 SD - Severe - Normal Bond et al. (2003)
1959delCAAA Saudi Arabia 1 family & - Normal Bond et al. (2003),
Nicholas et al. (2009)
Caucasian 1 family
1990C>T Pakistan 1 family & - Normal Bond et al. (2003)
2389C>T and
4074G>A
Morocco 1 patient �5 SD �9 SD IQ 42 - Motor and speech
delay
- Agenesis of CC rostrum Passemard (2009)
- Hyperactivity
disorder
- Enlarged LV
- Coarse gyri
- Simplified frontal and
occipital gyral pattern
- Large pituitary
2389C>T and
6686delTTAAA
Lebanon/
Algeria
1 patient �6 SD �7 SD IQ 42 - Normal motor
development
- Agenesis of CC rostrum Passemard (2009)
- Hyperactivity disorder - Enlarged LV occipital horns
- Pyramidal signs - Simplified gyral pattern
- Sleep disturbance - Posterior focal parietal
dysplasia
2761-25A>G Caucasian 1 family ^ Nicholas et al. (2009)
IVS9 and 5G>T
(2936 and 5G>T)
Pakistan 1 family & - Normal Bond et al. (2003)
2938C>T Pakistan 1 family Muhammad et al. (2009)
2967G>A Caucasian 1 family ^ Nicholas et al. (2009)
3055C>T Caucasian 2 families ^ Nicholas et al. (2009)
3055C>T and
7894C>T
Pakistan� 1 family Muhammad et al. (2009)
3082GA Pakistan 1 family & - Normal Bond et al. (2003)
3188T>G Pakistan 1 family ^ Nicholas et al. (2009)
3477delCGCTA Pakistan 1 family Muhammad et al. (2009)
3527C>G Jordani 1 family & - Normal Bond et al. (2003)
3663delG Pakistan 2 families �11 SD - Normal Bond et al. (2003)
3710C>G Caucasian 1 family ^ Nicholas et al. (2009)
3741 and 1G>A Caucasian 1 family ^ Nicholas et al. (2009)
3796G>T Africa 1 family ^ Nicholas et al. (2009)
3811C>T Germany 2 families & - Normal Bond et al. (2003)
Asia 1 family Nicholas et al. (2009)
Reunion
island
1 patient �5 SD �8 SD - Mild motor delay.
Hyperactivity disorder
- Agenesis of CC splenium Passemard (2009)
- Sleep disturbance
3945delAG and
9191delGAAA
Germany 1 patient �4 SD �7 SD IQ 104 - Normal motor
development
- Simplified frontal
gyral pattern
- Short stature
(�2 SD at birth,
�3 SD at 4 y)
Passemard (2009)
- Enlarged LV occipital
horns
- Dysmorphic frontal
ventricles
A.M
.K
ain
dl
eta
l./Pro
gress
inN
euro
bio
log
y9
0(2
01
0)
36
3–
38
33
69
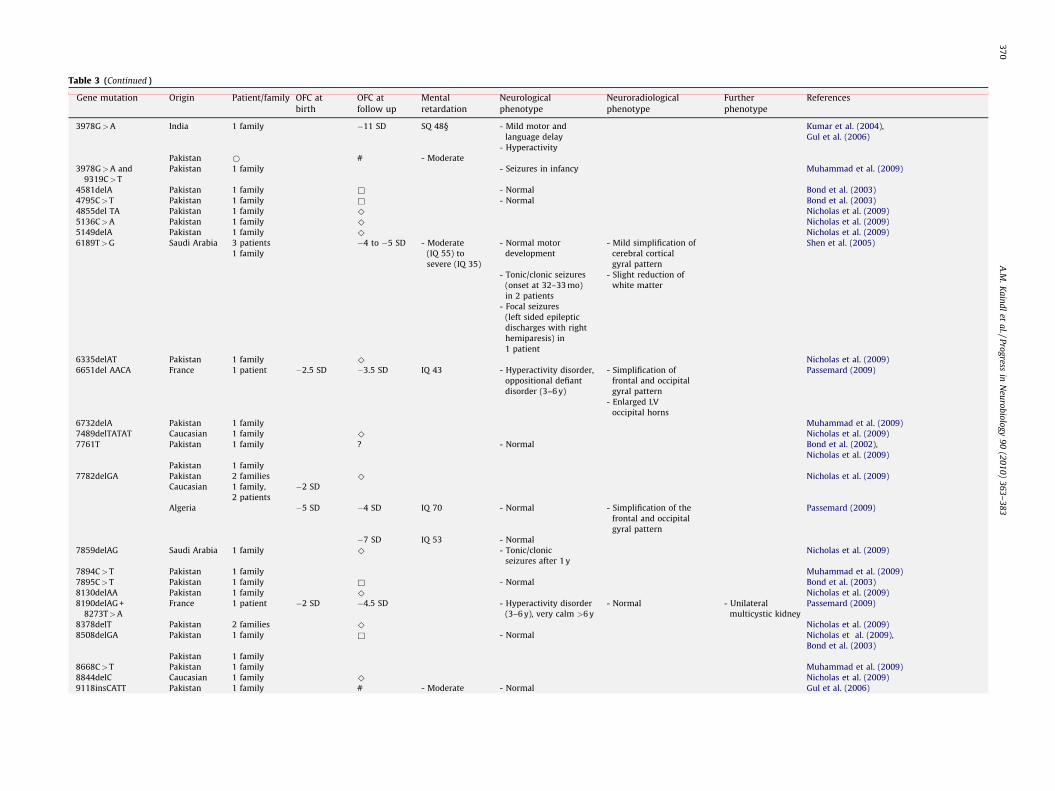
Table 3 (Continued )
Gene mutation Origin Patient/family OFC at
birth
OFC at
follow up
Mental
retardation
Neurological
phenotype
Neuroradiological
phenotype
Further
phenotype
References
3978G>A India 1 family �11 SD SQ 48§ - Mild motor and
language delay
Kumar et al. (2004),
Gul et al. (2006)
- Hyperactivity
Pakistan * # - Moderate
3978G>A and
9319C>T
Pakistan 1 family - Seizures in infancy Muhammad et al. (2009)
4581delA Pakistan 1 family & - Normal Bond et al. (2003)
4795C>T Pakistan 1 family & - Normal Bond et al. (2003)
4855del TA Pakistan 1 family ^ Nicholas et al. (2009)
5136C>A Pakistan 1 family ^ Nicholas et al. (2009)
5149delA Pakistan 1 family ^ Nicholas et al. (2009)
6189T>G Saudi Arabia 3 patients
1 family
�4 to �5 SD - Moderate
(IQ 55) to
severe (IQ 35)
- Normal motor
development
- Mild simplification of
cerebral cortical
gyral pattern
Shen et al. (2005)
- Tonic/clonic seizures
(onset at 32–33 mo)
in 2 patients
- Slight reduction of
white matter
- Focal seizures
(left sided epileptic
discharges with right
hemiparesis) in
1 patient
6335delAT Pakistan 1 family ^ Nicholas et al. (2009)
6651del AACA France 1 patient �2.5 SD �3.5 SD IQ 43 - Hyperactivity disorder,
oppositional defiant
disorder (3–6 y)
- Simplification of
frontal and occipital
gyral pattern
Passemard (2009)
- Enlarged LV
occipital horns
6732delA Pakistan 1 family Muhammad et al. (2009)
7489delTATAT Caucasian 1 family ^ Nicholas et al. (2009)
7761T Pakistan 1 family ? - Normal Bond et al. (2002),
Nicholas et al. (2009)
Pakistan 1 family
7782delGA Pakistan 2 families ^ Nicholas et al. (2009)
Caucasian 1 family,
2 patients
�2 SD
Algeria �5 SD �4 SD IQ 70 - Normal - Simplification of the
frontal and occipital
gyral pattern
Passemard (2009)
�7 SD IQ 53 - Normal
7859delAG Saudi Arabia 1 family ^ - Tonic/clonic
seizures after 1 y
Nicholas et al. (2009)
7894C>T Pakistan 1 family Muhammad et al. (2009)
7895C>T Pakistan 1 family & - Normal Bond et al. (2003)
8130delAA Pakistan 1 family ^ Nicholas et al. (2009)
8190delAG +
8273T>A
France 1 patient �2 SD �4.5 SD - Hyperactivity disorder
(3–6 y), very calm >6 y
- Normal - Unilateral
multicystic kidney
Passemard (2009)
8378delT Pakistan 2 families ^ Nicholas et al. (2009)
8508delGA Pakistan 1 family & - Normal Nicholas et al. (2009),
Bond et al. (2003)
Pakistan 1 family
8668C>T Pakistan 1 family Muhammad et al. (2009)
8844delC Caucasian 1 family ^ Nicholas et al. (2009)
9118insCATT Pakistan 1 family # - Moderate - Normal Gul et al. (2006)
A.M
.K
ain
dl
eta
l./Pro
gress
inN
euro
bio
log
y9
0(2
01
0)
36
3–
38
33
70
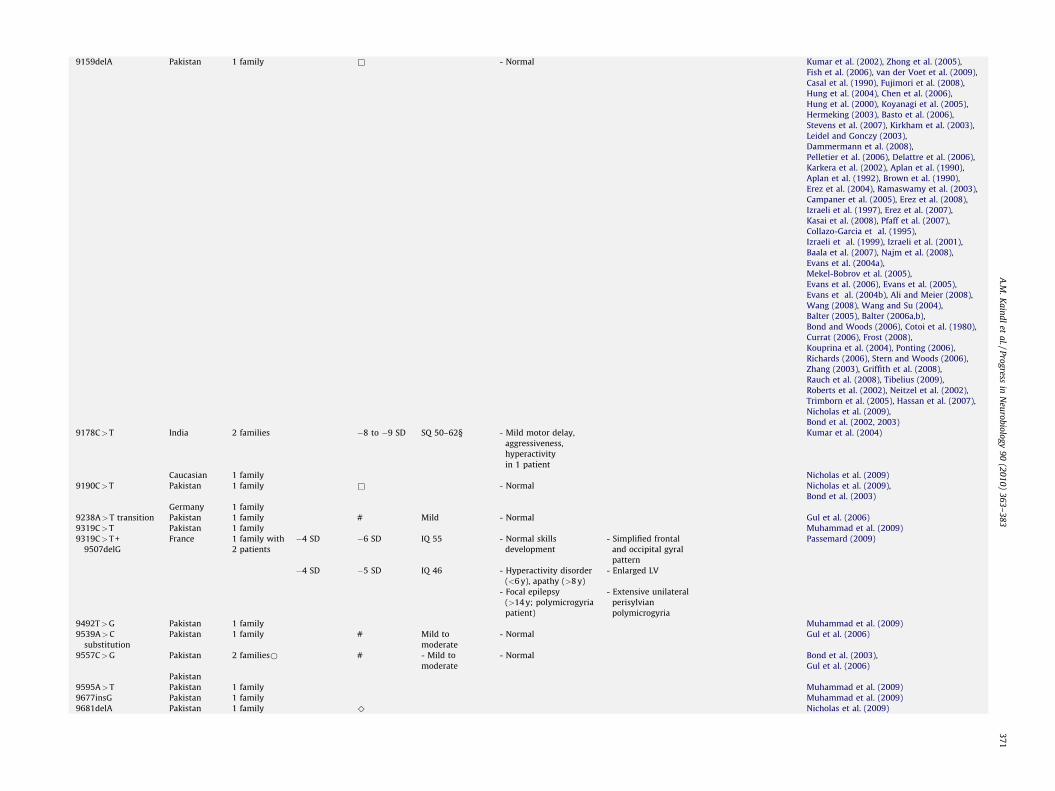
9159delA Pakistan 1 family & - Normal Kumar et al. (2002), Zhong et al. (2005),
Fish et al. (2006), van der Voet et al. (2009),
Casal et al. (1990), Fujimori et al. (2008),
Hung et al. (2004), Chen et al. (2006),
Hung et al. (2000), Koyanagi et al. (2005),
Hermeking (2003), Basto et al. (2006),
Stevens et al. (2007), Kirkham et al. (2003),
Leidel and Gonczy (2003),
Dammermann et al. (2008),
Pelletier et al. (2006), Delattre et al. (2006),
Karkera et al. (2002), Aplan et al. (1990),
Aplan et al. (1992), Brown et al. (1990),
Erez et al. (2004), Ramaswamy et al. (2003),
Campaner et al. (2005), Erez et al. (2008),
Izraeli et al. (1997), Erez et al. (2007),
Kasai et al. (2008), Pfaff et al. (2007),
Collazo-Garcia et al. (1995),
Izraeli et al. (1999), Izraeli et al. (2001),
Baala et al. (2007), Najm et al. (2008),
Evans et al. (2004a),
Mekel-Bobrov et al. (2005),
Evans et al. (2006), Evans et al. (2005),
Evans et al. (2004b), Ali and Meier (2008),
Wang (2008), Wang and Su (2004),
Balter (2005), Balter (2006a,b),
Bond and Woods (2006), Cotoi et al. (1980),
Currat (2006), Frost (2008),
Kouprina et al. (2004), Ponting (2006),
Richards (2006), Stern and Woods (2006),
Zhang (2003), Griffith et al. (2008),
Rauch et al. (2008), Tibelius (2009),
Roberts et al. (2002), Neitzel et al. (2002),
Trimborn et al. (2005), Hassan et al. (2007),
Nicholas et al. (2009),
Bond et al. (2002, 2003)
9178C>T India 2 families �8 to �9 SD SQ 50–62§ - Mild motor delay,
aggressiveness,
hyperactivity
in 1 patient
Kumar et al. (2004)
Caucasian 1 family Nicholas et al. (2009)
9190C>T Pakistan 1 family & - Normal Nicholas et al. (2009),
Bond et al. (2003)
Germany 1 family
9238A>T transition Pakistan 1 family # Mild - Normal Gul et al. (2006)
9319C>T Pakistan 1 family Muhammad et al. (2009)
9319C>T +
9507delG
France 1 family with
2 patients
�4 SD �6 SD IQ 55 - Normal skills
development
- Simplified frontal
and occipital gyral
pattern
Passemard (2009)
�4 SD �5 SD IQ 46 - Hyperactivity disorder
(<6 y), apathy (>8 y)
- Enlarged LV
- Focal epilepsy
(>14 y; polymicrogyria
patient)
- Extensive unilateral
perisylvian
polymicrogyria
9492T>G Pakistan 1 family Muhammad et al. (2009)
9539A>C
substitution
Pakistan 1 family # Mild to
moderate
- Normal Gul et al. (2006)
9557C>G Pakistan 2 families* # - Mild to
moderate
- Normal Bond et al. (2003),
Gul et al. (2006)
Pakistan
9595A>T Pakistan 1 family Muhammad et al. (2009)
9677insG Pakistan 1 family Muhammad et al. (2009)
9681delA Pakistan 1 family ^ Nicholas et al. (2009)
A.M
.K
ain
dl
eta
l./Pro
gress
inN
euro
bio
log
y9
0(2
01
0)
36
3–
38
33
71

Table 3 (Continued )
Gene mutation Origin Patient/family OFC at
birth
OFC at
follow up
Mental
retardation
Neurological
phenotype
Neuroradiological
phenotype
Further
phenotype
References
9686delTTAAA Lebanon 1 family,
4 patients
�7 to �8 SD - Profound - Motor and speech
delay
Passemard (2009)
- Generalized
tonic/clonic seizures
(onset 18 mo and 8 y)
9697C>T Pakistan� Muhammad et al. (2009)
9730C>T Pakistan ^ Nicholas et al. (2009)
9754delA Yemen 1 family �10 to �11 SD - Severe - Normal Bond et al. (2003)
9789T>A Pakistan 1 family ^ Nicholas et al. (2009)
IVS25 + 1G>T
(9984 + 1G>T)
Pakistan 1 family �7 SD - Normal Bond et al. (2003)
IVS25 + 1G>T
(9984 + 1G>T)/
3663delG
Pakistan 1 patient �9 SD - Profound Bond et al. (2003)
10,059C>A Pakistan 1 family ^ Nicholas et al. (2009)
Translocation
(1;4)(q31;p15.3)
? 1 patient Pichon et al. (2004)
large deletion
21.844b (loss of
exons2 to 13)
1 patient ^ Nicholas et al. (2009)
2389C>T and
7781delAG
Algeria 1 family,
3 patients
�4.5 to �6 SD Mild to severe Speech delay Hypoplasia of the
frontal lobes, moderate
posterior parietal atrophy,
anterior orientation of the
insula, thin CC, gyral
simplification
Saadi (2009)
Unable to write,
read and count
at adult age
CENPJ
17delC North 2 families ? ? Bond et al. (2005)
Pakistan
3704A-T Brazil 1 family ? ? Bond et al. (2005)
c.3243–
3246delTCAG
Pakistan 1 family <�3 to 5 SD
(8–13 y)
2 with moderate
mental retardation
(IQ 45–50)
- Unable to read or
write, could not speak
simple phrases and
did not have basic
self-care skills
- No malformations - Normal standard
lymphocyte
karyotype
Gul et al. (2006)
1 severe mental
retardation
(IQ 30–35)
- No other neurological
problems
- EEG normal
STIL
3655 delG India 3 families <�5 to 13 SD
(5–23 y)
- Mild to severe - Delayed developmental
milestones, speech delay
Kumar et al. (2009)
3715C>T - ‘‘No history of any
neurological deficits or
any malformations’’
IVS16 + 1G>A
All mutations are homozygous unless noted. (&) �5 to �11 SD for all patients from Bond et al. (2003); (#) �4 to �9 SD for all patients from Gul et al. (2006); (*) 5 families for both 3978G>A and 9557C>G from Gul; (§) Social
Quotient; (^) OFC<�3 SD for all patients from Nicholas et al. (2009); (�) heterozygous mutation carriers with small heads (OFC �2 to �3.5 SD).* Patients (n = 2) with primary microcephaly due to MCPH1 mutations with PPC syndrome (microcephaly, short stature and Premature Chromosome Condensation): for the first one, height was about �1 SD, for the second one,
between�4 and �5 SD. In PCC syndrome, Trimborn et collaborators noticed that gyral simplification was more marked (resembling pachygyri) than in MCPH1 related patients without PPC phenotype and periventricular neuronal
heterotopias were also present.
A.M
.K
ain
dl
eta
l./Pro
gress
inN
euro
bio
log
y9
0(2
01
0)
36
3–
38
33
72

Fig. 3. Position of mutations within the MCPH genes MCPH1, CDK5RAP2, ASPM, CENPJ and STIL. The coding and non-coding regions of the following MCPH genes are drawn to scale,
and the exons are depicted as boxes (reference sequences according to NCBI genome viewer build 35.1 and Ensembl release 54): (A) Microcephalin, MCPH1; (B) CDK5RAP2, cyclin-
dependent kinase 5 regulatory associated protein 2; (C) ASPM, Abnormal spindle-like, microcephaly associated; (D) CENPJ, Centromeric protein J; (E) SIL, SCL/TAL1 interrupting
locus. The localization and type of published mutations are given as well as the number of times a specific mutation has been reported. For cDNA sequence, the reference sequence.
KnownandpredictedMCPHproteindomainsarepresented,andthefull lengthproteinsaregivenwhenseveralisoformsexist.ForCDK5RAP2,theexactpositionsofthetworeported
structuralmaintenanceofchromosome (SMC)domains(Evans etal.,2006)withintheprotein(SMCAA137–499,SMC_NAA793–1040;accessionno.CAI16963)aswell asthatofthe
interaction site with CDK5R1 (AA1726–1768) are not clear and can only be predicted. For the CENPJ gene, Tang and coworkers reported a PN2-3 domain (AA311–422), which
containsa microtubuledestabilizing motif,and a microtubule bindingdomain(AA423–607) (Cormier etal.,2009).For theSTIL gene, a PIN1-bindingdomainhas beenreported tobe
localized at AA567–704 (Campaner et al., 2005), but its location is given as AA583–759 in Genecard and AA584–779 in Uniprot. This site is not cited in the NCBI database.
A.M. Kaindl et al. / Progress in Neurobiology 90 (2010) 363–383 373
6. Cyclin-dependent kinase 5, regulatory associated protein 2(syn. CDK5RAP2, C48, CEP215, KIAA1633, MCPH3)
The human cyclin-dependent kinase 5, regulatory associatedprotein 2 gene encodes CDK5RAP2, also referred to as centrosome
associated protein 215 (CEP215) or CDK5 activator binding proteinC48 (C48). Four isoforms have been reported so far. The human fulllength, 1893 amino acid CDK5RAP2 protein contains an N-terminalinteraction site with the gamma-tubulin ring complex (gTuRC),a C-terminal interaction site with CDK5 regulatory subunit 1
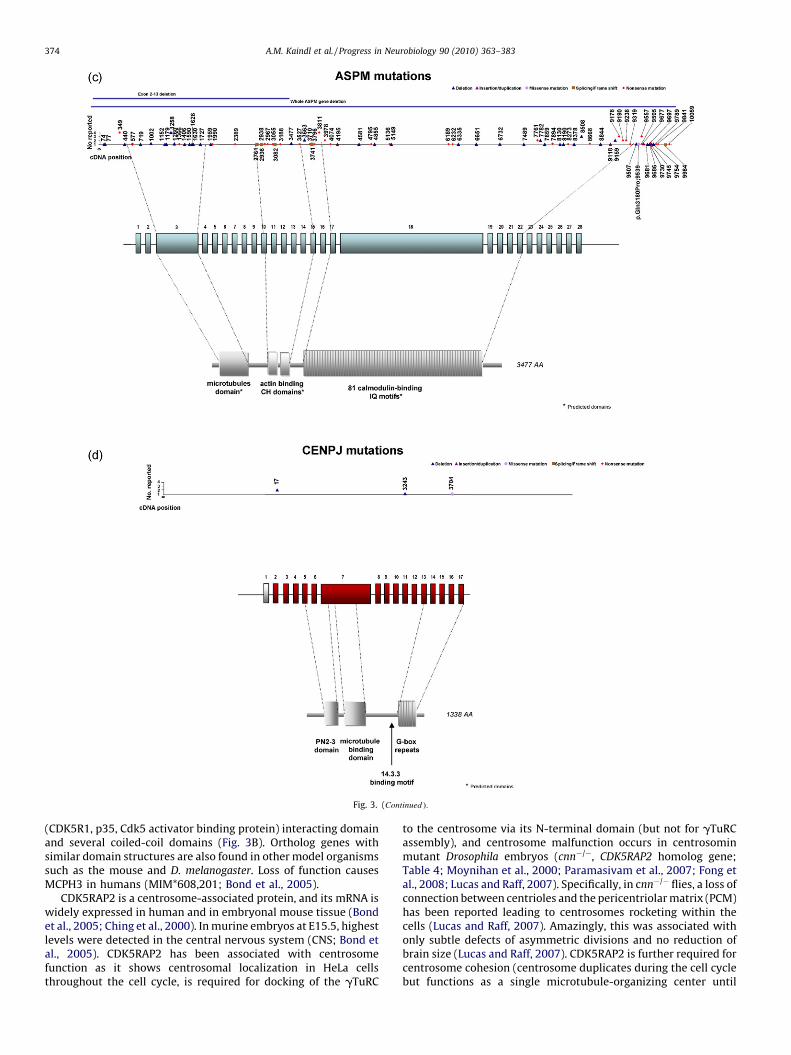
Fig. 3. (Continued ).
A.M. Kaindl et al. / Progress in Neurobiology 90 (2010) 363–383374
(CDK5R1, p35, Cdk5 activator binding protein) interacting domainand several coiled-coil domains (Fig. 3B). Ortholog genes withsimilar domain structures are also found in other model organismssuch as the mouse and D. melanogaster. Loss of function causesMCPH3 in humans (MIM*608,201; Bond et al., 2005).
CDK5RAP2 is a centrosome-associated protein, and its mRNA iswidely expressed in human and in embryonal mouse tissue (Bondet al., 2005; Ching et al., 2000). In murine embryos at E15.5, highestlevels were detected in the central nervous system (CNS; Bond etal., 2005). CDK5RAP2 has been associated with centrosomefunction as it shows centrosomal localization in HeLa cellsthroughout the cell cycle, is required for docking of the gTuRC
to the centrosome via its N-terminal domain (but not for gTuRCassembly), and centrosome malfunction occurs in centrosominmutant Drosophila embryos (cnn�/�, CDK5RAP2 homolog gene;Table 4; Moynihan et al., 2000; Paramasivam et al., 2007; Fong etal., 2008; Lucas and Raff, 2007). Specifically, in cnn�/� flies, a loss ofconnection between centrioles and the pericentriolar matrix (PCM)has been reported leading to centrosomes rocketing within thecells (Lucas and Raff, 2007). Amazingly, this was associated withonly subtle defects of asymmetric divisions and no reduction ofbrain size (Lucas and Raff, 2007). CDK5RAP2 is further required forcentrosome cohesion (centrosome duplicates during the cell cyclebut functions as a single microtubule-organizing center until
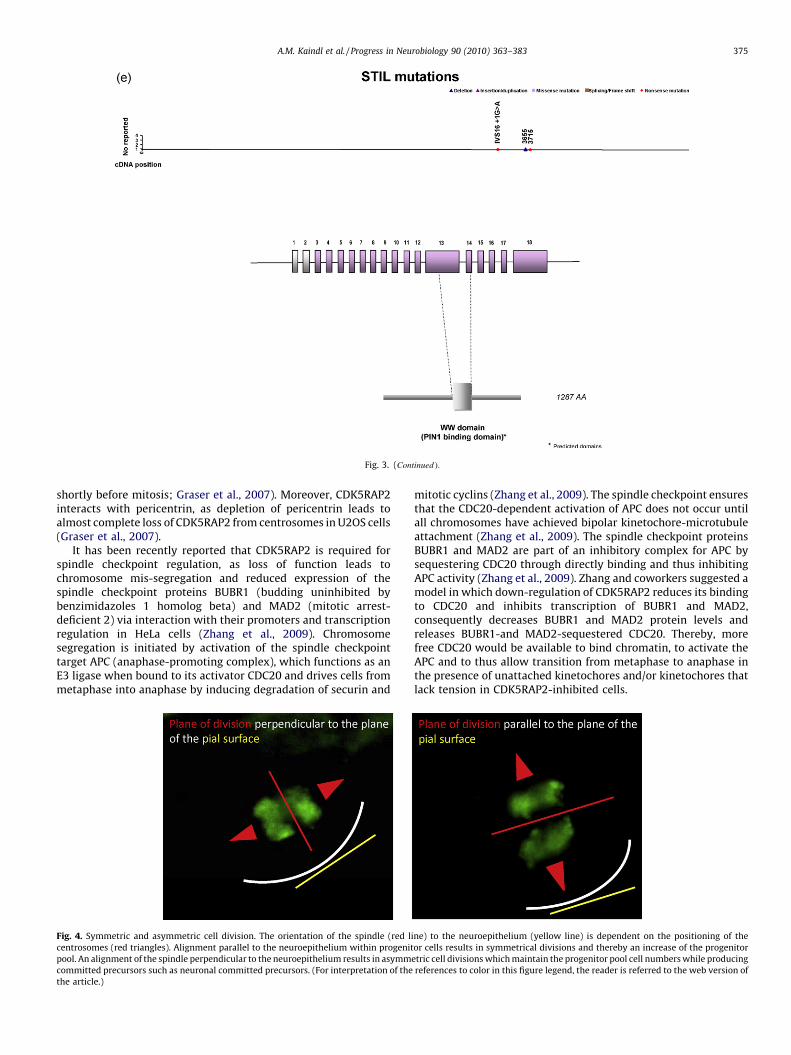
Fig. 3. (Continued ).
A.M. Kaindl et al. / Progress in Neurobiology 90 (2010) 363–383 375
shortly before mitosis; Graser et al., 2007). Moreover, CDK5RAP2interacts with pericentrin, as depletion of pericentrin leads toalmost complete loss of CDK5RAP2 from centrosomes in U2OS cells(Graser et al., 2007).
It has been recently reported that CDK5RAP2 is required forspindle checkpoint regulation, as loss of function leads tochromosome mis-segregation and reduced expression of thespindle checkpoint proteins BUBR1 (budding uninhibited bybenzimidazoles 1 homolog beta) and MAD2 (mitotic arrest-deficient 2) via interaction with their promoters and transcriptionregulation in HeLa cells (Zhang et al., 2009). Chromosomesegregation is initiated by activation of the spindle checkpointtarget APC (anaphase-promoting complex), which functions as anE3 ligase when bound to its activator CDC20 and drives cells frommetaphase into anaphase by inducing degradation of securin and
Fig. 4. Symmetric and asymmetric cell division. The orientation of the spindle (red li
centrosomes (red triangles). Alignment parallel to the neuroepithelium within progenit
pool. An alignment of the spindle perpendicular to the neuroepithelium results in asymm
committed precursors such as neuronal committed precursors. (For interpretation of the
the article.)
mitotic cyclins (Zhang et al., 2009). The spindle checkpoint ensuresthat the CDC20-dependent activation of APC does not occur untilall chromosomes have achieved bipolar kinetochore-microtubuleattachment (Zhang et al., 2009). The spindle checkpoint proteinsBUBR1 and MAD2 are part of an inhibitory complex for APC bysequestering CDC20 through directly binding and thus inhibitingAPC activity (Zhang et al., 2009). Zhang and coworkers suggested amodel in which down-regulation of CDK5RAP2 reduces its bindingto CDC20 and inhibits transcription of BUBR1 and MAD2,consequently decreases BUBR1 and MAD2 protein levels andreleases BUBR1-and MAD2-sequestered CDC20. Thereby, morefree CDC20 would be available to bind chromatin, to activate theAPC and to thus allow transition from metaphase to anaphase inthe presence of unattached kinetochores and/or kinetochores thatlack tension in CDK5RAP2-inhibited cells.
ne) to the neuroepithelium (yellow line) is dependent on the positioning of the
or cells results in symmetrical divisions and thereby an increase of the progenitor
etric cell divisions which maintain the progenitor pool cell numbers while producing
references to color in this figure legend, the reader is referred to the web version of
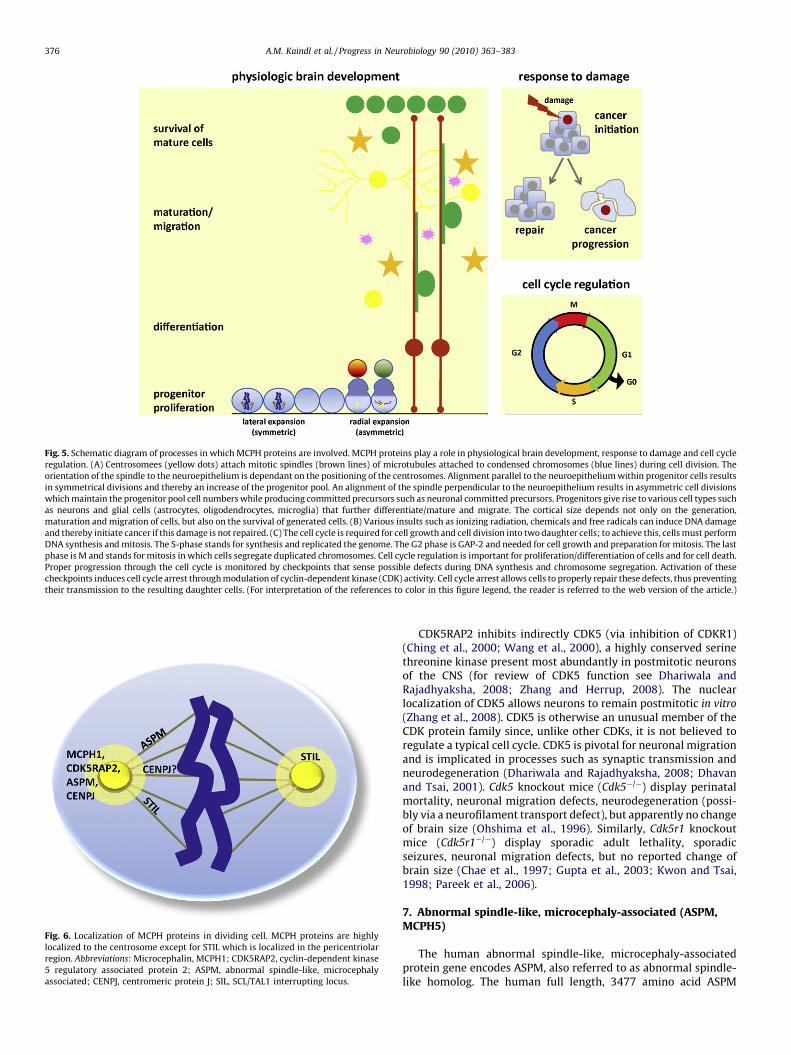
Fig. 5. Schematic diagram of processes in which MCPH proteins are involved. MCPH proteins play a role in physiological brain development, response to damage and cell cycle
regulation. (A) Centrosomees (yellow dots) attach mitotic spindles (brown lines) of microtubules attached to condensed chromosomes (blue lines) during cell division. The
orientation of the spindle to the neuroepithelium is dependant on the positioning of the centrosomes. Alignment parallel to the neuroepithelium within progenitor cells results
in symmetrical divisions and thereby an increase of the progenitor pool. An alignment of the spindle perpendicular to the neuroepithelium results in asymmetric cell divisions
which maintain the progenitor pool cell numbers while producing committed precursors such as neuronal committed precursors. Progenitors give rise to various cell types such
as neurons and glial cells (astrocytes, oligodendrocytes, microglia) that further differentiate/mature and migrate. The cortical size depends not only on the generation,
maturation and migration of cells, but also on the survival of generated cells. (B) Various insults such as ionizing radiation, chemicals and free radicals can induce DNA damage
and thereby initiate cancer if this damage is not repaired. (C) The cell cycle is required for cell growth and cell division into two daughter cells; to achieve this, cells must perform
DNA synthesis and mitosis. The S-phase stands for synthesis and replicated the genome. The G2 phase is GAP-2 and needed for cell growth and preparation for mitosis. The last
phase is M and stands for mitosis in which cells segregate duplicated chromosomes. Cell cycle regulation is important for proliferation/differentiation of cells and for cell death.
Proper progression through the cell cycle is monitored by checkpoints that sense possible defects during DNA synthesis and chromosome segregation. Activation of these
checkpoints induces cell cycle arrest through modulation of cyclin-dependent kinase (CDK) activity. Cell cycle arrest allows cells to properly repair these defects, thus preventing
their transmission to the resulting daughter cells. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
Fig. 6. Localization of MCPH proteins in dividing cell. MCPH proteins are highly
localized to the centrosome except for STIL which is localized in the pericentriolar
region. Abbreviations: Microcephalin, MCPH1; CDK5RAP2, cyclin-dependent kinase
5 regulatory associated protein 2; ASPM, abnormal spindle-like, microcephaly
associated; CENPJ, centromeric protein J; SIL, SCL/TAL1 interrupting locus.
A.M. Kaindl et al. / Progress in Neurobiology 90 (2010) 363–383376
CDK5RAP2 inhibits indirectly CDK5 (via inhibition of CDKR1)(Ching et al., 2000; Wang et al., 2000), a highly conserved serinethreonine kinase present most abundantly in postmitotic neuronsof the CNS (for review of CDK5 function see Dhariwala andRajadhyaksha, 2008; Zhang and Herrup, 2008). The nuclearlocalization of CDK5 allows neurons to remain postmitotic in vitro
(Zhang et al., 2008). CDK5 is otherwise an unusual member of theCDK protein family since, unlike other CDKs, it is not believed toregulate a typical cell cycle. CDK5 is pivotal for neuronal migrationand is implicated in processes such as synaptic transmission andneurodegeneration (Dhariwala and Rajadhyaksha, 2008; Dhavanand Tsai, 2001). Cdk5 knockout mice (Cdk5�/�) display perinatalmortality, neuronal migration defects, neurodegeneration (possi-bly via a neurofilament transport defect), but apparently no changeof brain size (Ohshima et al., 1996). Similarly, Cdk5r1 knockoutmice (Cdk5r1�/�) display sporadic adult lethality, sporadicseizures, neuronal migration defects, but no reported change ofbrain size (Chae et al., 1997; Gupta et al., 2003; Kwon and Tsai,1998; Pareek et al., 2006).
7. Abnormal spindle-like, microcephaly-associated (ASPM,MCPH5)
The human abnormal spindle-like, microcephaly-associatedprotein gene encodes ASPM, also referred to as abnormal spindle-like homolog. The human full length, 3477 amino acid ASPM

Table 4MCPH animal models overview.
MCPH (human gene) Animal (gene) Animal model phenotype References
MCPH1 (MCPH1) mcph1�/� - Uncoordinated centrosome and nuclear cycles Brunk et al. (2007), Rickmyre et al. (2007)
- Slowed mitotic entry, premature chromosome
condensation, mitotic entry with unreplicated DNA,
genomic instability, premature separation and
centrosome detachment
- Chk2-mediated mitotic arrest
- Adult brains are reportedly of normal size but have
defects in mushroom body structure
MCPH3 (CDK5RAP2) cnn�/� - Centrosome dysfunction (centrosome ‘rocketing’ within
the cells due to loss of connection between
centrosomes and pericentriolar matrix)
Lucas and Raff (2007)
- Only subtle defects of asymmetric divisions
- Brains are reportedly of normal size
MCPH5 (ASPM) asp�/� - Mitotic arrest in flies Casal et al. (1990)
- Centrosome do not function correctly as
microtubule-organizing center
- Mitotic spindle positioning defect
- Cell cleavage plane positioning defect during asymmetric
cell division
- Spermatogenesis defect
MCPH6 (CENPJ) dsas4�/� - Centrioles are lost during embryonic development Basto et al. (2006), Stevens et al. (2007)
- Slow mitotic spindle assembly
- Abnormal asymmetric divisions of larval neuroblasts
- Mutant flies develop into morphologically normal adults
without cilia or flagella and die shortly after birth
MCPH7 (STIL) - Embryonal lethal (E10.5) Pfaff et al. (2007), Izraeli et al. (1999)
Sil�/� - Developmental abnormalities appear around E7.5–8.5
- Reduced size, limited developmental progress, midline
neural tube defects, abnormal left–right development
- Increased rates of apoptosis
- Decreased rates of proliferation
- Abnormal expression of lefty2, nodal, Pitx2, Patched,
Gli1, Shh, Hnf3b
- Embyryonal lethal
Sil�/� - Disorganized mitotic spindles
- Lack one or both centrosomes
- Increased rates of mitosis
cnn, Centrosomin; dsas, drosophila spindle assembly abnormal. ( ) Drosophila; ( ) mouse; ( ) zebra fish.
A.M. Kaindl et al. / Progress in Neurobiology 90 (2010) 363–383 377
protein contains two calponin homology (CH) domains and severalputative calmodulin-binding IQ motifs (Fig. 3C). Orthologgenes with similar domain structures are also found in othermodel organisms. Loss of function causes MCPH5 in humans(MIM*605,481; Bond et al., 2002; Kumar et al., 2002).
ASPM plays a role in mitotic spindle function includingorientation of cleavage plane. The spindle apparatus dictates theplane of cell cleavage, which is critical in the choice betweensymmetric or asymmetric division. Spindle positioning is con-trolled by an evolutionarily conserved pathway. ASPM localizes tothe centrosome in the interphase and to mitotic spindle poles, fromprophase through telophase, in murine embryonic neuroepithelial(NE) cells and primary stem cells as well as progenitor cells of themammalian brain (Zhong et al., 2005; Fish et al., 2006). ASPMmaintains symmetrical cell divisions and is down-regulated withthe switch from proliferative to neurogenic divisions (Fish et al.,2006). Knock-down of Aspm in telencephalic NE cells by RNAinterference induced a lack of Aspm protein at mitotic spindlepoles and an alteration of the cleavage plane (orientation lessfrequently perpendicular to the ventricular surface of theneuroepithelium) (Fish et al., 2006). The authors reported thatthe latter alteration in the cleavage plane orientation increased theprobability that the cells underwent asymmetric division andconcomitantly decreased the number of neuroepithelial progenitorcells since a large proportion of neuroepithelial cell progeny wasfound in the neuronal layer. Recently, it has been reported that
ASPM promotes meiotic spindle organization/positioning viaaccumulation of LIN5 at meiotic and mitotic spindle poles (vander Voet et al., 2009); spindle rotation during maternal meiosisrequires LIN5, ASPM, calmodulin CMD1, and dynein. The authorsargue that the LIN5 complex LIN5/ASPM/CMD� at spindle poles isimportant for dynein function and thus cleavage plane orientationduring mitotic cell divisions (van der Voet et al., 2009). ASPM,moreover, localizes at midbodies and has a possible role in cellularabscission (Paramasivam et al., 2007).
The study of mutant Drosophila (asp�/�) revealed that ASPMmight not only be important for mitotic spindle positioning andfor correct positioning of cell cleavage plane during asymmetriccell division, but also for centrosome function as a microtubule-organizing center ((MTOC) Table 4). Asp gene mutations causemitotic arrest in flies, and Drosophila males display defectsduring spermatogenesis: (i) variable size of nuclei and neben-kerns of early spermatids, which are also multinucleate insteadof having single and uniformly sized nuclei; (ii) elongatingspermatids in which abnormal-sized mitochondrial derivativeselongate alongside more than one axoneme; (iii) failures in theindividualization process, where abnormal spermatids remainsyncytial, and seem to be eliminated during the coiling stage(Casal et al., 1990).
ASPM is down-regulated post ionizing radiation (Fujimori et al.,2008), and Aspm down-regulation decreases BRCA1 (Zhong et al.,2005). ASPM mRNA is over-expressed in transformed human cell

A.M. Kaindl et al. / Progress in Neurobiology 90 (2010) 363–383378
lines and tumors, and its increased expression is positivelyassociated with proliferation of glioblastoma cells (Hagemannet al., 2008).
8. Centromeric protein J (syn. CENPJ, CPAP, LAP, LIP1, MCPH6)
The human centromeric protein J gene encodes CENPJ, alsoreferred to as centrosomal P4.1-associated protein (CPAP), LAG-3-associated protein (LAP) or LYST-interacting protein 1 (LIP1). Thehuman full length, 1338 amino acid CENPJ protein contains a 112-amino acid long microtubule destabilizing motif PN2-3 (Hung etal., 2004) and two C-terminal 14-3-3 binding sites (Chen et al.,2006) (Fig. 3D). Ortholog genes with similar domain structures arealso found in other model organisms. Loss of function causesMCPH6 in humans (MIM*609,279).
CENPJ plays a role in centrosome and spindle function. Theprotein shows centrosomal localization throughout the cell cyclein a microtubule-independent way and is associated to thegamma-tubulin complex (Hung et al., 2000). It has been suggestedthat CENPJ regulated microtubule dynamics at the centrosome,and such a precise regulation of microtubule assembly anddisassembly at kinetochores and centrosomes is thought to beimportant for the maintenance of the spindle structure andchromosome segregation during mitosis (Hung et al., 2004).Expression of a ‘destabilizing domain’ (residues 311–422) was ableto destabilize microtubule nucleation in HeLa cells (Hung et al.,2004). This domain directly recognized the plus end of micro-tubules and inhibited microtubule nucleation from the centro-some, and it also bound to tubulin dimers, resulting in thedestabilization of microtubules (Hung et al., 2004). Over-expres-sion of this CENPJ domain inhibited cell proliferation and inducedapoptosis in HeLa cells after G2/M arrest (Hung et al., 2004).
CENPJ interacts with the N-terminal domain of the p65 subunit(RelA) of NF-kappaB, a transcription factor important for variouscellular events such as inflammation, immune response, prolifera-tion and apoptosis, in 293T (immortalized human embryonickidney HEK) and MCF-7 (human breast adenocarcinoma) cell lines(Koyanagi et al., 2005). It has been suggested that CENPJ functionsas a co-activator of NF-kappaB-mediated transcription (likely viap300/CREB-binding protein) as (i) CENPJ over-expression in theabove named cell lines enhanced NF-kappaB-dependent transcrip-tion induced by tumor necrosis factor-alpha (TNFalpha), (ii) CENPJdown-regulation via siRNA resulted in decreased activation of NF-kappaB by TNFalpha, and (iii) CENPJ interacts with the co-activatorp300/CREB-binding protein (Koyanagi et al., 2005). CENPJ deple-tion via siRNA further lead to an increase of multiple spindle poles,mitosis arrest and apoptosis. Direct interaction between CENPJ and14-3-3 in a cell cycle-dependant manner (significant reductionwhen cells enter mitosis) has been demonstrated in293T andMCF-7 cells (Chen et al., 2006). While the most common mode of14-3-3 function is the sequestration of its target proteins into acompartment such as the cytoplasm to modify their functions, adisruption of the 14-3-3 binding site did not interfere with CENPJtargeting the centrosome (Chen et al., 2006). Since CENPJ has beenreported to translocate to the nucleus after extracellular stimula-tion for example with TNFalpha, it needs to be further analyzedwhether an association with 14-3-3 leads to such a translocation(Chen et al., 2006). Further, the association between CENPJ and 14-3-3 may be important for cell cycle control since 14-3-3 proteinsplay a role in cell cycle control (Chen et al., 2006; Hermeking,2003).
Sas-4�/� mutant Drosophila (spindle assembly abnormal Sas isCENPJ homolog) start to lose their centrioles during embryonicdevelopment until, by third-instar larval stages, no centrioles orcentrosomes are detectable (Basto et al., 2006; Stevens et al., 2007;Table 4). Mitotic spindle assembly is slow in mutant cells, and
approximately 30% of the asymmetric divisions of larval neuro-blasts are abnormal. Nevertheless, mutant flies develop with nearnormal timing into morphologically normal adults. These flies,however, have no cilia or flagella and die shortly after birth becausetheir sensory neurons lack cilia. Thus, centrioles are essential forthe formation of centrosomes, cilia and flagella, but, remarkably,they are not essential for most aspects of Drosophila development.
In C. elegans, sas-4 is a centriolar protein that controlscentrosome size (centrosomal organization) (Kirkham et al.,2003). Centrosomes consist of a centriole pair surrounded bypericentriolar materia, and it is acknowledged that centrioles arerequired to organize PCM to form a structurally stable organelle.Centriole duplication fails in the absence of sas-4, and partialdepletion of sas-4 results in structurally defective centrioles thatcontain reduced levels of sas-4 and organize proportionally lesspericentriolar material (stunted centrioles, smaller centrosomes(Kirkham et al., 2003)). The role of sas-4 in centrosome duplicationwas further underlined by FRAP (fluorescence recovery afterphotobleaching) studies demonstrating that sas-4 was recruited tothe centrosome once per cell cycle, at the onset of the organelleduplication cycle in the S phase (Leidel and Gonczy, 2003;Dammermann et al., 2008). Centriolar sas-4 remains in dynamicequilibrium with the cytoplasmic pool until late prophase, when itis stably incorporated in a step that requires gamma-tubulin andmicrotubule assembly (Dammermann et al., 2008). It has beendemonstrated in staged one-cell C. elegans embryos that daughtercentriole assembly begins with the formation and elongation of acentral tube followed by the peripheral assembly of nine singletmicrotubules (Pelletier et al., 2006). The five proteins zyg-1(ZYGote defective, embryonic lethal), sas-4, sas-5, sas-6, and spd-2(spindle defective 2) have been identified as being essential forcentriole formation, and they are recruited in a specific order(Pelletier et al., 2006; Delattre et al., 2006): (i) tube formation andelongation is dependent on the sas-5 and sas-6, and the assemblyof singlet microtubules onto the central tube depends on sas-4; (ii)centriole assembly is triggered by an upstream signal mediated byspd-2 and zyg-1. Knowledge on the pathway of daughter centrioleassembly in C. elegans could have general relevance for centrioleassembly in other organisms.
9. SCL/TAL1-interrupting locus (syn. STIL, SIL, MCPH7)
The human SCL/TAL1-interrupting locus gene encodes STIL, alsoreferred to as SCL-interrupting locus (SIL). STIL is an immediateearly gene that encodes a cytosolic protein of 150 kDa whosefunction is not fully understood and which lacks homology to anyknown protein families or motifs. Two isoforms have beenreported. The human full length 1287 amino acid protein containsa putative nuclear localization signal and a C-terminal domainsimilar to that of TGF-beta (Karkera et al., 2002) (Fig. 3E). Orthologgenes with similar domain structures are also found in other modelorganisms such as the mouse and D. melanogaster. Three mutationsof the STIL gene leading to a truncated gene product in five Indianfamilies with MCPH have been reported (Kumar et al., 2009).
STIL has been in the focus of cancer research for decades sincegenomic rearrangements containing the STIL gene have beenassociated with T-cell acute lymphoblastic leukemia (ALL) (Aplanet al., 1990, 1992; Brown et al., 1990). In these tumor cells,deletions of the STIL gene lead to a juxtaposition of 50 element(promoter) to the downstream SCL gene and thus resulted in aninappropriate SCL mRNA expression regulated by the STIL
promoter (Aplan et al., 1992). Moreover, an increased expressionof STIL in multiple cancers that correlated with the expression ofmitotic spindle checkpoint genes and with increased metastaticpotential (Erez et al., 2004; Ramaswamy et al., 2003). Only

A.M. Kaindl et al. / Progress in Neurobiology 90 (2010) 363–383 379
recently, it has been reported that homozygous mutations of theSTIL gene cause MCPH in humans (Kumar et al., 2009).
STIL is expressed throughout the cytosol with increasedexpression in the perinuclear region that likely plays a role inmitotic entry (cell cycle progression G2-M), apoptosis control andcentrosome function. In HeLa and HEK293T cells, endogenous STILwas detected at the poles of the mitotic spindle in the metaphase(where the microtubules coalesce adjacent to the centrosome),while cells in the anaphase did not show this localization andinterphase cells expressed almost no STIL. STIL is phosphorylatedin mitosis following the mitotic spindle checkpoint. Phosphory-lated STIL interacts with PIN1 (peptidyl prolyl isomerase) which inturn regulates a subset of mitotic phospoproteins (Campaner et al.,2005). STIL expression is regulated by the transcription factor E2F,and it is required for E2F induced transition through mitosis (Erezet al., 2008). The protein is expressed in proliferating cells and isdown-regulated when cellular proliferation ceases because ofserum starvation, contact inhibition or induction of terminaldifferentiation (Izraeli et al., 1997). STIL is essential for mitoticentry and survival of cancer cells (Erez et al., 2007). It influencescancer cell proliferation via de-repression of GLI1 (glioma-associated oncogene homolog) from Suppressor of Fused (SUFU)(Kasai et al., 2008). Moreover, knock-down of STIL in cancer cellscaused a mitotic arrest through a severely delayed entrance intomitosis (G2 to M phase transition), decreased the activation of theCDK1 (CDC2)-cyclin B complex and induced apoptosis in a p53-independent manner (Erez et al., 2007).
Data obtained from zebrafish and HeLa cells indicate that STILmay not only play a role in centrosome function/duplication, butalso in organizing the mitotic spindle. Zebrafish Danio rerio
cassiopeia mutants (loss-of-function mutation in zebrafish sil)have an embryonic lethal defect (Pfaff et al., 2007). They showincreased rates of mitosis, as detected by phospho-H3 immunos-taining, but mitotic cells have extremely disorganized mitoticspindles and often lack one or both centrosomes. In accordancewith these findings, knock-down of STIL by short hairpin RNA(shRNA) in HeLa cells resulted in dividing cells with disorganizedmitotic spindles.
Sil mRNA is expressed in many mouse tissues with highestlevels in bone marrow, thymus, spleen, colon and stomach(Collazo-Garcia et al., 1995). Homozygous sil knockout mice(Sil�/�) display developmental abnormalities around embryonalday (E) 7.5–8.5 and die after E10.5 (Izraeli et al., 1999) (Table 4). Inaddition to reduced size and limited developmental progresscompared to wild-type embryos, Sil mutants have prominentmidline neural tube defects including a delay/failure of neural tubeclosure and holoprosencephaly, abnormal left–right development(randomized heart looping direction), abnormal expression ofLefty2 (left–right determination factor 2), Nodal, Pitx2 (paired-likehomeodomain transcription factor 2), Patched, Gli1, Shh (sonichedgehog), and Hnf3b (hepatocyte nuclear factor 3-beta, foreheadbox A2), increased rates of apoptosis and decreased rates ofproliferation. These latter findings, the reduced expression of genesdownstream of sonic hedgehog (Shh), indicate that STIL may bepart of the Shh pathway. Indeed double mutants of STIL andPatched is consistent with the hypothesis that STIL functionsdownstream of Patched in the Shh pathway (Izraeli et al., 2001).
10. MCPH and cerebellum
Developmentally controlled proliferation, differentiation, mi-gration and apoptosis of cells through processes such ascentrosome function, cell cycle control, spindle assembly anddivision plane orientation are not unique for the cerebral cortex.They also occur throughout the development of the cerebellarcortex. Why a loss of MCPH protein function does not lead to a
gross reduction of the cortex in the cerebellum as it does in thecerebrum is unknown and raises questions regarding the differ-ence in cortex development within the brain. So far, little attentionhas been paid to measuring exactly the size of the cerebellar cortexin patients with MCPH. When doing this, we did detectedcerebellar atrophy in individual patients (unpublished data).Moreover, the role of MCPH proteins have not been studied incerebellar development, and it is, thus, not known whether MCPH
genes are expressed in this context.Defects in other, non-MCPH genes also result in primary
autosomal recessive microcephaly. In a large consanguineousMoroccan family with marked prenatal-onset microcephaly,mutations in the T-box transcription factor gene EOMES (homologof Xenopus Eomesodermin) were detected in affected individuals.In addition to primary microcephaly (Baala et al., 2007), theseindividuals also showed corpus callosum agenesis, bilateralpolymicrogyria, ventricular dilatation, disproportionate pons andcerebellar hypoplasia (Baala et al., 2007). Further, mutations in thecalcium/calmodulin-dependent serine protein kinase gene CASK
have been reported in patients with mental retardation, primarymicrocephaly and hypoplasia of the brainstem and cerebellum(Najm et al., 2008). CASK, a member of the membrane associatedguanylate kinase family, is a multi-domain scaffolding protein thatinteracts with the transcription factor Tbr1 (T-box brain 1) andother genes involved in cortical development such as reelin (RELN).It targets to neuronal synapses and regulates trafficking, targetingand signaling of ion channels (Najm et al., 2008). It will beintriguing to decipher the interaction of these proteins with thoseassociated with MCPH.
11. The role of MCPH proteins in the evolution of the cerebralcortex
The rapid increase in brain size over the past 3–4 million years,and the associated increase in cognitive skills is one of the moststriking features of hominid evolution. MCPH genes are obviouscandidates for this strong evolutionary movement. Early studies ofthe ASPM sequence in great apes show that its evolution issignificantly accelerated, especially in the lineage from the lasthuman/chimpanzee ancestor to humans shows an excess of non-synonymous over synonymous substitutions, a characteristic ofpositive selection. On average, ASPM fixed one mutation every300,000–400,000 years since the human lineage diverged fromchimpanzees some 5–6 million years ago (Evans et al., 2004b). Onegenetic variant of ASPM in humans arose about 5800 years ago andhas since reached an allelic frequency of about 30% (Mekel-Bobrovet al., 2005). Similarly, in MCPH1, a recent variation, whichoccurred 37,000 years ago spread in human population morerapidly than what can be explained by simple genetic drift in asmall population, to reach a frequency of about 70% (Evans et al.,2006, 2005). A similar phenomenon has been observed with thetwo other MCPH genes.
The role of ASPM in the evolution of brain size and cognitivefunction is, however, not without controversy. Studies haveaddressed whether the changes in brain size across the clade ofprimates are correlated to the molecular evolution of the ASPMgene. Two large exons 3 and 18 of ASPM for 23 primate specieswere sequenced. These exons were chosen because they accountfor 70% of the ASPM protein and most of the mutations that occurin microcephaly are found in these exons. Results showed thatpositive selection in ASPM is significantly correlated with cerebralcortex size and not with whole brain size or cerebellum size. This isconsistent with the expression report of ASPM which shows thatASPM expression is limited to the cerebral cortex. This studysuggests that different parts of the brain may have differentevolutionary and functional differentiation based on their distinct

A.M. Kaindl et al. / Progress in Neurobiology 90 (2010) 363–383380
genetic basis. This is in contrast with several studies which havetried to correlate ASPM genotype with brain size variation inhumans (Evans et al., 2004a; Ali and Meier, 2008). Similar analysishas also shown that there have been 45 variations in themicrocephalin gene positively selected within the last 25–30million years of evolution from simian ancestors to modernhumans.
Obviously, the central question is which physiological parame-ter is affected by the Darwinian selection process. The functionalrole of the adaptive evolution of the MCPH genes remainsspeculative, and no clear answer has emerged from geneticepidemiology. The motor of positive selection of the new alleleshas been reckoned to be increased cognitive power. A largeassociation study between the two most recent polymorphismsand IQ has been applied to five replicate samples from fiveindependent populations with a total of about 2400 individuals.The authors concluded that intelligence, as measured by these IQtests, was not detectably associated with the presence of the mostrecent allele of either the ASPM or the Microcephalin gene. In anassociation study of OFC and SNPs located in the ASPM and MCPH1
genes, the non-synonymous SNP rs1057090 (V761A) located in theBRCT domain of the MCPH1 gene is significantly associated with anincreased cranial volume in a Chinese population of 867 unrelatedmales. No recent selection signal could be detected regarding thisSNP, leading the authors to suggest that brain volume variation inhuman populations is neutral or under very weak selection inrecent human history. These results (Wang, 2008) remain to beaffirmed. From such an evolutionary perspective, primary micro-cephaly can be viewed as an example of ‘evolutionary retrogres-sion’ (or an atavistic mutation) whereby the brain dimensions ofthe affected individuals revert back to the level resembling that ofvery early hominids (Evans et al., 2004b; Wang and Su, 2004), aprobably simplistic interpretation as hominids had small brains,but also functional ASPM. However, other studies have come to adifferent conclusion.
It has been noted by Bond et al. that the predicted ASPMproteins encode systematically larger numbers of repeated ‘IQ’domains between flies, mice, and humans, with the predominantdifference between Aspm and ASPM being a single large insertioncoding for IQ domains. One of the most notable trends inmammalian evolution is the massive increase in size of thecerebral cortex, especially in primates. The authors suggested that,in view of evolutionary considerations, these findings suggest thatbrain size is controlled in part through modulation of mitoticspindle activity in neuronal progenitor cells. Humans withautosomal recessive primary microcephaly show a small butotherwise grossly normal cerebral cortex associated with mild tomoderate mental retardation. The authors suggested that geneslinked to this condition offer potential insights into the develop-ment and evolution of the cerebral cortex. This hypothesis is highlydiscussed with subsequent studies supporting or opposing it(Ponting and Jackson, 2005; Evans et al., 2004b; Mekel-Bobrovet al., 2005; Ali and Meier, 2008; Balter, 2005, 2006a,b; Bond andWoods, 2006; Cotoi et al., 1980; Currat, 2006; Frost, 2008;Kouprina et al., 2004; Ponting, 2006; Richards, 2006; Stern andWoods, 2006; Zhang, 2003).
Interestingly, the expansion of IQ repeats has been observedin other non-primate mammalians (see http://www.uniprot.org/uniprot/?query=domain:%22IQ+domain*%22), and raises thequestion of the physiological process which really benefits fromthe selection pressure. As those mammals have not seen thedramatic brain expansion observed in primates, we canspeculate that positive selection is exerted on another parameterregulating reproductive fitness: even if ASPM function seems tobe redundant in tissues outside brain in fetuses and postnatally,nothing is known on its role in gametogenesis or early
embryonic development. Positive selection pressure could beexerted on a more fundamental process than brain size—and, tobe provocative, increased brain size in the primate taxon couldperhaps be a ‘‘side effect’’ of a selection mechanism that affectsall mammalians.
12. Other proteins that show an overlap with MCPHpathomechanisms and phenotype
Centrosome, cell cycle and/or DNA repair-linked proteins havebeen associated with microcephaly phenotypes close to that ofMCPH. The most striking example is the centrosomal proteinpericentrin (PCNT). Mutations in the PCNT gene cause a clinicalphenotype continuum from MCPH1-reminicient patients to Seckeltype 4 syndrome (Griffith et al., 2008) and microcephalicosteodysplastic primordial dwarfism type II (MOPDII) patients(Rauch et al., 2008). Microcephaly, short stature and mentalretardation are the common points of these three syndromes.Microcephaly is isolated in MCPH1 and more pronounced for intwo other diseases. Short stature has been reported only inindividual MCPH patients while striking growth retardation(Seckel) or dwarfism (MOPDII) is prominent in the other diseases.In these diseases, the facial appearance with sloping forehead,micrognathia, prominent midface and upward slanting of thepalpebral fissures is characteristic. Tibelius and coworkers recentlyshowed that a lack of MCPH1 or PCNT results in a loss of CHK1 fromcentrosomes, using two approaches (patient-derived cells carryingMCPH1 or PCNT mutations and cells treated with MCPH1 and PCNTspecific siRNAs) (Tibelius, 2009). Immunoprecipitation of GFP-MCPH1 using an anti-GFP antibody leads to co-immunoprecipita-tion of endogenous CHK1, suggesting that MCPH1 and CHK1interact with each other and that MCPH1 targets CHK1 to thecentrosome. PCNT immunostaining was diminished in lympho-blastoid cell lines derived from patients with MCPH1 mutationsand in U2OS cells treated with MCPH1 siRNA. Immunoprecipitationof GFP-MCPH1 leads to co-immunoprecipitation of endogenousPCNT. Based on their data, the authors suggest that MCPH1 andPCNT interact with each other. As a further finding that underlinessuch an interaction between PCNT and CHK1, Tibelius andcoworkers demonstrated that PCNT was immunoprecipitated bothfrom control or MCPH1 patient-derived cell lines and thatimmunoprecipitation of endogenous CHK1 lead to co-immuno-precipitation of endogenous PCNT. The authors concluded that (i)PCNT and CHK1 interact with each other independent from thepresence of MCPH1, (ii) MCPH1 contributes to the centrosomallocalization of PCNT and (iii) both MCPH1 and PCNT are required torecruit CHK1 to the centrosome. Microcephaly is also a commonclinical feature in disorders involving DNA repair abnormalitiessuch as ataxia telangiectasia and RAD3-related protein (ATR) genemutation or defective ATR signaling caused Seckel syndrome type1, Fanconi Anemia or Nijmegen syndrome. Future studies devotedto resolving the exact overlap of such gene products known to beinvolved in DNA repair pathways in patients with syndromeswhich include microcephaly as a clinical sign with thosemechanisms implicated in MCPH are needed.
13. Conclusion
Brain size is largely determined by the relative rates ofproliferation and cell death, and proteins encoded by MCPH geneshave been shown to play a prominent role in several key steps ofdevelopmental processes that could be responsible for a markedreduction of the cortical area, such as cell cycle progression, DNA/spindle checkpoints as well as centrosome and spindle apparatusintegrity. Some common pathways have been identified for MCPHproteins (e.g. interaction of MCPH1 and STIL with the transcription

A.M. Kaindl et al. / Progress in Neurobiology 90 (2010) 363–383 381
factor E2F). Identifying the pathomechanisms underlying MCPHwill not only increase our knowledge of these rare diseases but alsoenhance our understanding of normal brain development and ofevolution of the brain development. Still, many studies have beenperformed in cancer cells in vitro, and murine models are largelylacking. Murine models will be particularly relevant to determinethe precise roles of MCPH proteins in the control of proliferation ofnormal mammalian neural precursor cells as compared to theirrole in lower organisms or cancer cells. Deciphering the molecularpathways associated with MCPH proteins will also allow todetermine the possible interplays between these different MCPHproteins. Future directions for research in the field could also aimat determining the potential involvement of MCPH proteins andrelated pathways in the pathophysiology of microcephalies ofother etiologies including environmentally induced microcepha-lies.
Conflict of interest
None.
Acknowledgements
The authors are members of the m-icronet consortium(Microcephaly-international collaborative research on etiology).Our research is supported by Inserm, Paris 7, Fondation Lejeune,the German Research Foundation (SFB665), PremUp and theFondation pour la Recherche Medicale et Societe Francaise deNeuropediatrie.
References
Abuelo, D., 2007. Microcephaly syndromes. Semin. Pediatr. Neurol. 14 (September(3)), 118–127.
Alderton, G.K., Galbiati, L., Griffith, E., Surinya, K.H., Neitzel, H., Jackson, A.P., et al.,2006. Regulation of mitotic entry by microcephalin and its overlap with ATRsignalling. Nat. Cell. Biol. 8 (July (7)), 725–733.
Ali, F., Meier, R., 2008. Positive selection in ASPM is correlated with cerebral cortexevolution across primates but not with whole-brain size. Mol. Biol. Evol. 25(November (11)), 2247–2250.
Aplan, P.D., Lombardi, D.P., Ginsberg, A.M., Cossman, J., Bertness, V.L., Kirsch, I.R.,1990. Disruption of the human SCL locus by ‘‘illegitimate’’ V-(D)-J recombinaseactivity. Science 250 (December (4986)), 1426–1429.
Aplan, P.D., Lombardi, D.P., Reaman, G.H., Sather, H.N., Hammond, G.D., Kirsch, I.R.,1992. Involvement of the putative hematopoietic transcription factor SCL in T-cell acute lymphoblastic leukemia. Blood 79 (March (5)), 1327–1333.
Baala, L., Briault, S., Etchevers, H.C., Laumonnier, F., Natiq, A., Amiel, J., et al., 2007.Homozygous silencing of T-box transcription factor EOMES leads to microceph-aly with polymicrogyria and corpus callosum agenesis. Nat. Genet. 39 (April(4)), 454–456.
Balter, M., 2005. Evolution. Are human brains still evolving? Brain genes show signsof selection. Science 309 (September (5741)), 1662–1663.
Balter, M., 2006a. Bruce Lahn profile. Brain man makes waves with claims of recenthuman evolution. Science 314 (December (5807)), 1871–1873.
Balter, M., 2006b. Bruce Lahn profile. Links between brain genes, evolution, andcognition challenged. Science 314 (December (5807)), 1872.
Bamatter, F., Rabinowicz, T., 1969. Study of a familial case of microcephaly andmicrencephaly. Clinical and anatomo-pathologic considerations on a prelimi-nary basis. J. Genet. Hum. 17 (October (3)), 247–274.
Basto, R., Lau, J., Vinogradova, T., Gardiol, A., Woods, C.G., Khodjakov, A., et al., 2006.Flies without centrioles. Cell 125 (June (7)), 1375–1386.
Bond, J., Woods, C.G., 2006. Cytoskeletal genes regulating brain size. Curr. Opin. CellBiol. 18 (February (1)), 95–101.
Bond, J., Roberts, E., Mochida, G.H., Hampshire, D.J., Scott, S., Askham, J.M., et al.,2002. ASPM is a major determinant of cerebral cortical size. Nat. Genet. 32(October (2)), 316–320.
Bond, J., Scott, S., Hampshire, D.J., Springell, K., Corry, P., Abramowicz, M.J., et al.,2003. Protein-truncating mutations in ASPM cause variable reduction in brainsize. Am. J. Hum. Genet. 73 (November (5)), 1170–1177.
Bond, J., Roberts, E., Springell, K., Lizarraga, S.B., Scott, S., Higgins, J., et al., 2005. Acentrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size.Nat. Genet. 37 (April (4)), 353–355.
Bork, P., Hofmann, K., Bucher, P., Neuwald, A.F., Altschul, S.F., Koonin, E.V., 1997. Asuperfamily of conserved domains in DNA damage-responsive cell cycle check-point proteins. FASEB J. 11 (January (1)), 68–76.
Brown, L., Cheng, J.T., Chen, Q., Siciliano, M.J., Crist, W., Buchanan, G., et al., 1990.Site-specific recombination of the tal-1 gene is a common occurrence in humanT cell leukemia. EMBO J. 9 (October (10)), 3343–3351.
Brunk, K., Vernay, B., Griffith, E., Reynolds, N.L., Strutt, D., Ingham, P.W., et al., 2007.Microcephalin coordinates mitosis in the syncytial Drosophila embryo. J. Cell.Sci. 120 (October (Pt 20)), 3578–3588.
Campaner, S., Kaldis, P., Izraeli, S., Kirsch, I.R., 2005. Sil phosphorylation in a Pin1binding domain affects the duration of the spindle checkpoint. Mol. Cell Biol. 25(August (15)), 6660–6672.
Casal, J., Gonzalez, C., Wandosell, F., Avila, J., Ripoll, P., 1990. Abnormal meioticspindles cause a cascade of defects during spermatogenesis in asp males ofDrosophila. Development 108 (February (2)), 251–260.
Chae, T., Kwon, Y.T., Bronson, R., Dikkes, P., Li, E., Tsai, L.H., 1997. Mice lacking p35, aneuronal specific activator of Cdk5, display cortical lamination defects, seizures,and adult lethality. Neuron 18 (January (1)), 29–42.
Chen, C.Y., Olayioye, M.A., Lindeman, G.J., Tang, T.K., 2006. CPAP interacts with 14-3-3 in a cell cycle-dependent manner. Biochem. Biophys. Res. Commun. 342 (April(4)), 1203–1210.
Ching, Y.P., Qi, Z., Wang, J.H., 2000. Cloning of three novel neuronal Cdk5 activatorbinding proteins. Gene 242 (January (1–2)), 285–294.
Collazo-Garcia, N., Scherer, P., Aplan, P.D., 1995. Cloning and characterization of amurine SIL gene. Genomics 30 (December (3)), 506–513.
Cormier, A., Clement, M.J., Knossow, M., Lachkar, S., Savarin, P., Toma, F., et al., 2009.The PN2-3 domain of centrosomal P4.1-associated protein implements a novelmechanism for tubulin sequestration. J. Biol. Chem. 284 (March (11)), 6909–6917.
Cotoi, S., Incze, A., Georgescu, C., Carasca, E., 1980. Pacing below the ventricular ratein terminating ventricular tachycardia and flutter. Am. Heart J. 100 (November(5)), 763–764.
Currat, M., Excoffier, L., Maddison, W., Otto, S.P., Ray, N., Whitlock, M.C., et al.,2006. Comment on ‘‘Ongoing adaptive evolution of ASPM, a brain sizedeterminant in Homo sapiens’’ and ‘‘Microcephalin, a gene regulating brainsize, continues to evolve adaptively in humans’’. Science 313 (July (5784)),172 (author reply).
Dammermann, A., Maddox, P.S., Desai, A., Oegema, K., 2008. SAS-4 is recruited to adynamic structure in newly forming centrioles that is stabilized by the gamma-tubulin-mediated addition of centriolar microtubules. J. Cell Biol. 180 (February(4)), 771–785.
Delattre, M., Canard, C., Gonczy, P., 2006. Sequential protein recruitment in C.elegans centriole formation. Curr. Biol. 16 (September (18)), 1844–1849.
Dhariwala, F.A., Rajadhyaksha, M.S., 2008. An unusual member of the Cdk family:Cdk5. Cell. Mol. Neurobiol. 28 (May (3)), 351–369.
Dhavan, R., Tsai, L.H., 2001. A decade of CDK5. Nat. Rev. Mol. Cell Biol. 2 (October(10)), 749–759.
Erez, A., Perelman, M., Hewitt, S.M., Cojacaru, G., Goldberg, I., Shahar, I., et al., 2004.Sil overexpression in lung cancer characterizes tumors with increased mitoticactivity. Oncogene 23 (July (31)), 5371–5377.
Erez, A., Castiel, A., Trakhtenbrot, L., Perelman, M., Rosenthal, E., Goldstein, I., et al.,2007. The SIL gene is essential for mitotic entry and survival of cancer cells.Cancer Res. 67 (May (9)), 4022–4027.
Erez, A., Chaussepied, M., Castiel, A., Colaizzo-Anas, T., Aplan, P.D., Ginsberg, D.,et al., 2008. The mitotic checkpoint gene, SIL is regulated by E2F1. Int. J. Cancer123 (October (7)), 1721–1725.
Evans, P.D., Anderson, J.R., Vallender, E.J., Gilbert, S.L., Malcom, C.M., Dorus, S., et al.,2004. Adaptive evolution of ASPM, a major determinant of cerebral cortical sizein humans. Hum. Mol. Genet. 13 (March (5)), 489–494.
Evans, P.D., Anderson, J.R., Vallender, E.J., Choi, S.S., Lahn, B.T., 2004. Reconstructingthe evolutionary history of microcephalin, a gene controlling human brain size.Hum. Mol. Genet. 13 (June (11)), 1139–1145.
Evans, P.D., Gilbert, S.L., Mekel-Bobrov, N., Vallender, E.J., Anderson, J.R., Vaez-Azizi, L.M., et al., 2005. Microcephalin, a gene regulating brain size, continuesto evolve adaptively in humans. Science 309 (September (5741)), 1717–1720.
Evans, P.D., Vallender, E.J., Lahn, B.T., 2006. Molecular evolution of the brain sizeregulator genes CDK5RAP2 and CENPJ. Gene 375 (June), 75–79.
Fish, J.L., Kosodo, Y., Enard, W., Paabo, S., Huttner, W.B., 2006. Aspm specificallymaintains symmetric proliferative divisions of neuroepithelial cells. Proc. Natl.Acad. Sci. U.S.A. 103 (July (27)), 10438–10443.
Fong, K.W., Choi, Y.K., Rattner, J.B., Qi, R.Z., 2008. CDK5RAP2 is a pericentriolarprotein that functions in centrosomal attachment of the {gamma}-tubulin ringcomplex. Mol. Biol. Cell 19 (January (1)), 115–125.
Francis, F., Meyer, G., Fallet-Bianco, C., Moreno, S., Kappeler, C., Socorro, A.C., et al.,2006. Human disorders of cortical development: from past to present. Eur JNeurosci. 23 (February (4)), 877–893.
Frost, P., 2008. The spread of alphabetical writing may have favored the latestvariant of the ASPM gene. Med. Hypotheses 70 (1), 17–20.
Fujimori, A., Yaoi, T., Ogi, H., Wang, B., Suetomi, K., Sekine, E., et al., 2008. Ionizingradiation downregulates ASPM, a gene responsible for microcephaly in humans.Biochem. Biophys. Res. Commun. 369 (May (3)), 953–957.
Gilbert, S.L., Dobyns, W.B., Lahn, B.T., 2005. Genetic links between brain develop-ment and brain evolution. Nat. Rev. Genet. 6 (July (7)), 581–590.
Graser, S., Stierhof, Y.D., Nigg, E.A., 2007. Cep68 and Cep215 (Cdk5rap2) are requiredfor centrosome cohesion. J Cell Sci. 120 (December (Pt 24)), 4321–4331.
Griffith, E., Walker, S., Martin, C.A., Vagnarelli, P., Stiff, T., Vernay, B., et al., 2008.Mutations in pericentrin cause Seckel syndrome with defective ATR-dependentDNA damage signaling. Nat. Genet. 40 (February (2)), 232–236.

A.M. Kaindl et al. / Progress in Neurobiology 90 (2010) 363–383382
Gul, A., Hassan, M.J., Hussain, S., Raza, S.I., Chishti, M.S., Ahmad, W., 2006. A noveldeletion mutation in CENPJ gene in a Pakistani family with autosomal recessiveprimary microcephaly. J. Hum. Genet. 51 (9), 760–764.
Gupta, A., Sanada, K., Miyamoto, D.T., Rovelstad, S., Nadarajah, B., Pearlman, A.L.,et al., 2003. Layering defect in p35 deficiency is linked to improper neuronal-glial interaction in radial migration. Nat. Neurosci. 6 (December (12)), 1284–1291.
Hagemann, C., Anacker, J., Gerngras, S., Kuhnel, S., Said, H.M., Patel, R., et al., 2008.Expression analysis of the autosomal recessive primary microcephaly genesMCPH1 (microcephalin) and MCPH5 (ASPM, abnormal spindle-like, microceph-aly associated) in human malignant gliomas. Oncol. Rep. 20 (August (2)), 301–308.
Hassan, M.J., Khurshid, M., Azeem, Z., John, P., Ali, G., Chishti, M.S., et al., 2007.Previously described sequence variant in CDK5RAP2 gene in a Pakistani familywith autosomal recessive primary microcephaly. BMC Med. Genet. 8, 58.
Hermeking, H., 2003. The 14-3-3 cancer connection. Nat. Rev. Cancer. 3 (December(12)), 931–943.
Hung, L.Y., Tang, C.J., Tang, T.K., 2000. Protein 4.1 R-135 interacts with a novelcentrosomal protein (CPAP) which is associated with the gamma-tubulincomplex. Mol. Cell Biol. 20 (October (20)), 7813–7825.
Hung, L.Y., Chen, H.L., Chang, C.W., Li, B.R., Tang, T.K., 2004. Identification of a novelmicrotubule-destabilizing motif in CPAP that binds to tubulin heterodimers andinhibits microtubule assembly. Mol. Biol. Cell 15 (June (6)), 2697–2706.
Huyton, T., Bates, P.A., Zhang, X., Sternberg, M.J., Freemont, P.S., 2000. The BRCA1 C-terminal domain: structure and function. Mutat. Res. 460 (August (3–4)), 319–332.
Izraeli, S., Colaizzo-Anas, T., Bertness, V.L., Mani, K., Aplan, P.D., Kirsch, I.R., 1997.Expression of the SIL gene is correlated with growth induction and cellularproliferation. Cell Growth Differ. 8 (November (11)), 1171–1179.
Izraeli, S., Lowe, L.A., Bertness, V.L., Good, D.J., Dorward, D.W., Kirsch, I.R., et al.,1999. The SIL gene is required for mouse embryonic axial development and left-right specification. Nature 399 (June (6737)), 691–694.
Izraeli, S., Lowe, L.A., Bertness, V.L., Campaner, S., Hahn, H., Kirsch, I.R., et al., 2001.Genetic evidence that Sil is required for the Sonic Hedgehog response pathway.Genesis 31 (October (2)), 72–77.
Jackson, A.P., McHale, D.P., Campbell, D.A., Jafri, H., Rashid, Y., Mannan, J., et al.,1998. Primary autosomal recessive microcephaly (MCPH1) maps to chromo-some 8p22-pter. Am. J. Hum. Genet. 63 (August (2)), 541–546.
Jackson, A.P., Eastwood, H., Bell, S.M., Adu, J., Toomes, C., Carr, I.M., et al., 2002.Identification of microcephalin, a protein implicated in determining the size ofthe human brain. Am. J. Hum. Genet. 71 (July (1)), 136–142.
Jamieson, C.R., Govaerts, C., Abramowicz, M.J., 1999. Primary autosomal recessivemicrocephaly: homozygosity mapping of MCPH4 to chromosome 15. Am. J.Hum. Genet. 65 (November (5)), 1465–1469.
Jeffers, L.J., Coull, B.J., Stack, S.J., Morrison, C.G., 2008. Distinct BRCT domains inMcph1/Brit1 mediate ionizing radiation-induced focus formation and centro-somal localization. Oncogene 27 (January (1)), 139–144.
Kaindl, A.M., Passemard, S., Gressens, P., 2008. Gene table: autosomal recessiveprimary microcephalies (MCPH). Eur. J. Paediatr. Neurol. (September).
Karkera, J.D., Izraeli, S., Roessler, E., Dutra, A., Kirsch, I., Muenke, M., 2002. Thegenomic structure, chromosomal localization, and analysis of SIL as a candidategene for holoprosencephaly. Cytogenet. Genome Res. 97 (1–2), 62–67.
Kasai, K., Inaguma, S., Yoneyama, A., Yoshikawa, K., Ikeda, H., 2008. SCL/TAL1interrupting locus derepresses GLI1 from the negative control of suppressor-of-fused in pancreatic cancer cell. Cancer Res. 68 (October (19)), 7723–7729.
Katyal, S., McKinnon, P.J., 2007. DNA repair deficiency and neurodegeneration. CellCycle 6 (October (19)), 2360–2365.
Kirkham, M., Muller-Reichert, T., Oegema, K., Grill, S., Hyman, A.A., 2003. SAS-4 is aC. elegans centriolar protein that controls centrosome size. Cell 112 (February(4)), 575–587.
Kouprina, N., Pavlicek, A., Mochida, G.H., Solomon, G., Gersch, W., Yoon, Y.H., et al.,2004. Accelerated evolution of the ASPM gene controlling brain size beginsprior to human brain expansion. PLoS Biol. 2 (May (5)), E126.
Koyanagi, M., Hijikata, M., Watashi, K., Masui, O., Shimotohno, K., 2005. Centroso-mal P4.1-associated protein is a new member of transcriptional coactivators fornuclear factor-kappaB. J. Biol. Chem. 280 (April (13)), 12430–12437.
Kumar, A., Markandaya, M., Girimaji, S.C., 2002. Primary microcephaly: microce-phalin and ASPM determine the size of the human brain. J. Biosci. 27 (December(7)), 629–632.
Kumar, A., Blanton, S.H., Babu, M., Markandaya, M., Girimaji, S.C., 2004. Geneticanalysis of primary microcephaly in Indian families: novel ASPM mutations.Clin. Genet. 66 (October (4)), 341–348.
Kumar, A., Girimaji, S.C., Duvvari, M.R., Blanton, S.H., 2009. Mutations in STIL,encoding a pericentriolar and centrosomal protein, cause primary microceph-aly. Am. J. Hum. Genet. 84 (February (2)), 286–290.
Kwon, Y.T., Tsai, L.H., 1998. A novel disruption of cortical development in p35(�/�)mice distinct from reeler. J. Comp. Neurol. 395 (June (4)), 510–522.
Leal, G.F., Roberts, E., Silva, E.O., Costa, S.M., Hampshire, D.J., Woods, C.G., 2003. Anovel locus for autosomal recessive primary microcephaly (MCPH6) maps to13q12.2. J. Med. Genet. 40 (July (7)), 540–542.
Leidel, S., Gonczy, P., 2003. SAS-4 is essential for centrosome duplication in C.elegans and is recruited to daughter centrioles once per cell cycle. Dev. Cell 4(March (3)), 431–439.
Lin, S.Y., Elledge, S.J., 2003. Multiple tumor suppressor pathways negatively regulatetelomerase. Cell 113 (June (7)), 881–889.
Lin, S.Y., Rai, R., Li, K., Xu, Z.X., Elledge, S.J., 2005. BRIT1/MCPH1 is a DNA damageresponsive protein that regulates the Brca1-Chk1 pathway, implicating check-point dysfunction in microcephaly. Proc. Natl. Acad. Sci. U.S.A. 102 (42), 15105–15109.
Lucas, E.P., Raff, J.W., 2007. Maintaining the proper connection between thecentrioles and the pericentriolar matrix requires Drosophila centrosomin. J.Cell Biol. 178 (August (5)), 725–732.
Mekel-Bobrov, N., Gilbert, S.L., Evans, P.D., Vallender, E.J., Anderson, J.R., Hudson,R.R., et al., 2005. Ongoing adaptive evolution of ASPM, a brain size determinantin Homo sapiens. Science 309 (September (5741)), 1720–1722.
Moynihan, L., Jackson, A.P., Roberts, E., Karbani, G., Lewis, I., Corry, P., et al., 2000. Athird novel locus for primary autosomal recessive microcephaly maps tochromosome 9q34. Am. J. Hum. Genet. 66 (February (2)), 724–727.
Muhammad, F., Mahmood Baig, S., Hansen, L., Sajid Hussain, M., Anjum Inayat, I.,Aslam, M., et al., 2009. Compound heterozygous ASPM mutations in PakistaniMCPH families. Am. J. Med. Genet. A 149A (May (5)), 926–930.
Najm, J., Horn, D., Wimplinger, I., Golden, J.A., Chizhikov, V.V., Sudi, J., et al., 2008.Mutations of CASK cause an X-linked brain malformation phenotype withmicrocephaly and hypoplasia of the brainstem and cerebellum. Nat. Genet.40 (September (9)), 1065–1067.
Neitzel, H., Neumann, L.M., Schindler, D., Wirges, A., Tonnies, H., Trimborn, M., et al.,2002. Premature chromosome condensation in humans associated with micro-cephaly and mental retardation: a novel autosomal recessive condition. Am. J.Hum. Genet. 70 (April (4)), 1015–1022.
Nicholas, A.K., Swanson, E.A., Cox, J.J., Karbani, G., Malik, S., Springell, K., et al., 2009.The molecular landscape of ASPM mutations in primary microcephaly. J. Med.Genet. 46 (April (4)), 249–253.
Ohshima, T., Ward, J.M., Huh, C.G., Longenecker, G., Veeranna, Pant, H.C., et al., 1996.Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormalcorticogenesis, neuronal pathology and perinatal death. Proc. Natl. Acad. Sci.U.S.A. 93 (October (20)), 11173–11178.
Paramasivam, M., Chang, Y.J., LoTurco, J.J., 2007. ASPM and citron kinase co-localizeto the midbody ring during cytokinesis. Cell Cycle 6 (July (13)), 1605–1612.
Pareek, T.K., Keller, J., Kesavapany, S., Pant, H.C., Iadarola, M.J., Brady, R.O., et al.,2006. Cyclin-dependent kinase 5 activity regulates pain signaling. Proc. Natl.Acad. Sci. U.S.A. 103 (January (3)), 791–796.
Passemard, S.T.L., Elmaleh, M., Afenjar, A., Alessandri, J.L., Andria, G., Billette deVillemeur, T., Boespflug-Tanguy, O., Burglen, L., Del Gundice, E., Guimiot, F.,Hyon, C., Isidor, B., Megarbane, A., Moog, U., Odent, S., Hernandez, K., Pouvreau,N., Scala, I., Schaer, M., Gressens, P., Gerard, B., Verloes, A., 2009. Expanding theclinical and neuroradiological phenotype of primary microcephaly (MCPH) dueto ASPM mutations. Neurology.
Pattison, L., Crow, Y.J., Deeble, V.J., Jackson, A.P., Jafri, H., Rashid, Y., et al., 2000. Afifth locus for primary autosomal recessive microcephaly maps to chromosome1q31. Am. J. Hum. Genet. 67 (December (6)), 1578–1580.
Pelletier, L., O’Toole, E., Schwager, A., Hyman, A.A., Muller-Reichert, T., 2006.Centriole assembly in Caenorhabditis elegans. Nature 444 (Nov (7119)), 619–623.
Pfaff, K.L., Straub, C.T., Chiang, K., Bear, D.M., Zhou, Y., Zon, L.I., 2007. The zebra fishcassiopeia mutant reveals that SIL is required for mitotic spindle organization.Mol. Cell Biol. 27 (August (16)), 5887–5897.
Pichon, B., Vankerckhove, S., Bourrouillou, G., Duprez, L., Abramowicz, M.J., 2004. Atranslocation breakpoint disrupts the ASPM gene in a patient with primarymicrocephaly. Eur. J. Hum. Genet. 12 (May (5)), 419–421.
Ponting, C.P., 2006. A novel domain suggests a ciliary function for ASPM, a brain sizedetermining gene. Bioinformatics 22 (May (9)), 1031–1035.
Ponting, C., Jackson, A.P., 2005. Evolution of primary microcephaly genes andthe enlargement of primate brains. Curr. Opin. Genet. Dev. 15 (June (3)), 241–248.
Rai, R., Dai, H., Multani, A.S., Li, K., Chin, K., Gray, J., et al., 2006. BRIT1 regulates earlyDNA damage response, chromosomal integrity, and cancer. Cancer Cell 10(August (2)), 145–157.
Rai, R., Phadnis, A., Haralkar, S., Badwe, R.A., Dai, H., Li, K., et al., 2008. Differentialregulation of centrosome integrity by DNA damage response proteins. Cell Cycle7 (July (14)), 2225–2233.
Ramaswamy, S., Ross, K.N., Lander, E.S., Golub, T.R., 2003. A molecular signature ofmetastasis in primary solid tumors. Nat. Genet. 33 (January (1)), 49–54.
Rauch, A., Thiel, C.T., Schindler, D., Wick, U., Crow, Y.J., Ekici, A.B., et al., 2008.Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science319 (February (5864)), 816–819.
Richards, G.D., 2006. Genetic, physiologic and ecogeographic factors contributing tovariation in Homo sapiens: Homo floresiensis reconsidered. J. Evol. Biol. 19(November (6)), 1744–1767.
Rickmyre, J.L., Dasgupta, S., Ooi, D.L., Keel, J., Lee, E., Kirschner, M.W., et al., 2007. TheDrosophila homolog of MCPH1, a human microcephaly gene, is required forgenomic stability in the early embryo. J. Cell. Sci. 120 (October (Pt 20)), 3565–3577.
Robain, O., Lyon, G., 1972. Familial microcephalies due to cerebral malformation.Anatomical and clinical study. Acta Neuropathol. 20 (2), 96–109.
Roberts, E., Jackson, A.P., Carradice, A.C., Deeble, V.J., Mannan, J., Rashid, Y., et al.,1999. The second locus for autosomal recessive primary microcephaly (MCPH2)maps to chromosome 19q13.1-13.2. Eur. J. Hum. Genet. 7 (October–November(7)), 815–820.
Roberts, E., Hampshire, D.J., Pattison, L., Springell, K., Jafri, H., Corry, P., et al., 2002.Autosomal recessive primary microcephaly: an analysis of locus heterogeneityand phenotypic variation. J. Med. Genet. 39 (October (10)), 718–721.

A.M. Kaindl et al. / Progress in Neurobiology 90 (2010) 363–383 383
Saadi, A., Borck, G., Boddaert, N., Chekkour, M.C., Imessaoudene, B., Munnich, A.,et al., 2009. Compound heterozygous ASPM mutations associated with micro-cephaly and simplified cortical gyration in a consanguineous Algerian family.Eur. J. Med. Genet. (March).
Shen, J., Eyaid, W., Mochida, G.H., Al-Moayyad, F., Bodell, A., Woods, C.G., et al.,2005. ASPM mutations identified in patients with primary microcephaly andseizures. J. Med. Genet. 42 (September (9)), 725–729.
Stern, R., Woods, C.G., 2006. Evolutionary genetics: is brain evolution still continu-ing in modern humans? Eur. J. Hum. Genet. 14 (July (7)), 799–800.
Stevens, N.R., Raposo, A.A., Basto, R., St Johnston, D., Raff, J.W., 2007. From stem cellto embryo without centrioles. Curr. Biol. 17 (September (17)), 1498–1503.
Tibelius, A., Marhold, J., Zentgraf, H., Heilig, C.E., Neitzel, H., Ducommun, B., et al.,2009. Microcephalin and pericentrin regulate mitotic entry via centrosome-associated Chk1. J. Cell Biol. (June).
Trimborn, M., Bell, S.M., Felix, C., Rashid, Y., Jafri, H., Griffiths, P.D., et al., 2004.Mutations in microcephalin cause aberrant regulation of chromosome conden-sation. Am. J. Hum. Genet. 75 (August (2)), 261–266.
Trimborn, M., Richter, R., Sternberg, N., Gavvovidis, I., Schindler, D., Jackson, A.P.,et al., 2005. The first missense alteration in the MCPH1 gene causes autosomalrecessive microcephaly with an extremely mild cellular and clinical phenotype.Hum. Mutat. 26 (November (5)), 496.
Trimborn, M., Schindler, D., Neitzel, H., Hirano, T., 2006. Misregulated chromosomecondensation in MCPH1 primary microcephaly is mediated by condensin II. CellCycle 5 (February (3)), 322–326.
Tunca, Y., Vurucu, S., Parma, J., Akin, R., Desir, J., Baser, I., et al., 2006. Prenataldiagnosis of primary microcephaly in two consanguineous families by confron-tation of morphometry with DNA data. Prenat. Diagn. 26 (May (5)), 449–453.
Van Den Bosch, J., 1959. Microcephaly in The Netherlands: a clinical and geneticalstudy. Ann. Hum. Genet. 23 (April (2)), 91–116.
van der Voet, M., Berends, C.W., Perreault, A., Nguyen-Ngoc, T., Gonczy, P., Vidal, M.,et al., 2009. NuMA-related LIN-5. ASPM-1, calmodulin and dynein promotemeiotic spindle rotation independently of cortical LIN-5/GPR/Galpha. Nat. CellBiol. 11 (March (3)), 269–277.
Wang, J.K., Li, Y., Su, B., 2008. A common SNP of MCPH1 is associated with cranialvolume variation in Chinese population. Hum. Mol. Genet. (January).
Wang, Y.Q., Su, B., 2004. Molecular evolution of microcephalin, a gene determininghuman brain size. Hum. Mol. Genet. 13 (June (11)), 1131–1137.
Wang, X., Ching, Y.P., Lam, W.H., Qi, Z., Zhang, M., Wang, J.H., 2000. Identification of acommon protein association region in the neuronal Cdk5 activator. J. Biol.Chem. 275 (October (41)), 31763–31769.
Wood, J.L., Li, K., Liang, Y., Chen, J., 2008. Microcephalin/MCPH1 associates with thecondensin II complex to function in homologous recombination repair. J. Biol.Chem. (August).
Wood, J.L., Singh, N., Mer, G., Chen, J., 2007. MCPH1 functions in an H2AX-dependentbut MDC1-independent pathway in response to DNA damage. J. Biol. Chem. 282(November (48)), 35416–35423.
Woods, C.G., 2004. Human microcephaly. Curr Opin Neurobiol. 14 (February (1)),112–117.
Woods, C.G., Bond, J., Enard, W., 2005. Autosomal recessive primary microcephaly(MCPH): a review of clinical, molecular, and evolutionary findings. Am. J. Hum.Genet. 76 (May (5)), 717–728.
Xu, X., Lee, J., Stern, D.F., 2004. Microcephalin is a DNA damage response proteininvolved in regulation of CHK1 and BRCA1. J. Biol. Chem. 279 (August (33)),34091–34094.
Yang, S.Z., Lin, F.T., Lin, W.C., 2008. MCPH1/BRIT1 cooperates with E2F1 in theactivation of checkpoint. DNA repair and apoptosis. EMBO Rep. 9 (September(9)), 907–915.
Yu, X., Chini, C.C., He, M., Mer, G., Chen, J., 2003. The BRCT domain is a phospho-protein binding domain. Science 302 (October (5645)), 639–642.
Zhang, J., 2003. Evolution of the human ASPM gene, a major determinant of brainsize. Genetics 165 (December (4)), 2063–2070.
Zhang, X., Liu, D., Lv, S., Wang, H., Zhong, X., Liu, B., et al., 2009. CDK5RAP2 isrequired for spindle checkpoint function. Cell Cycle 8 (April (8)).
Zhang, J., Herrup, K., 2008. Cdk5 and the non-catalytic arrest of the neuronal cellcycle. Cell Cycle 7 (November (22)), 3487–3490.
Zhang, J., Cicero, S.A., Wang, L., Romito-Digiacomo, R.R., Yang, Y., Herrup, K.,2008. Nuclear localization of Cdk5 is a key determinant in the postmitoticstate of neurons. Proc. Natl. Acad. Sci. U.S.A. 105 (June (25)), 8772–8777.
Zhong, X., Liu, L., Zhao, A., Pfeifer, G.P., Xu, X., 2005. The abnormal spindle-like,microcephaly-associated (ASPM) gene encodes a centrosomal protein. CellCycle 4 (September (9)), 1227–1229.
Zhong, X., Pfeifer, G.P., Xu, X., 2006. Microcephalin encodes a centrosomal protein.Cell Cycle 5 (February (4)), 457–458.




















