
Mutated CYLD affects the functional state of dendritic cells
10
Mutated cylindromatosis gene affects the functional state of dendritic cells Matthias Bros 1 , Nadine Dexheimer 1 , Verena Besche 1 , Joumana Masri 2 , Stefanie Trojandt 1 , Nadine Ho ¨velmeyer 2 , Sonja Reissig 2 , Ramin Massoumi 3 , Stephan Grabbe 1 , Ari Waisman 2 and Angelika B. Reske-Kunz 1 1 Clinical Research Unit Allergology, Department of Dermatology, University Medical Center of the Johannes Gutenberg-University, Mainz, Germany 2 Institute for Molecular Medicine, University Medical Center of the Johannes Gutenberg- University, Mainz, Germany 3 Department of Laboratory Medicine, Lund University, Malmo ¨ University Hospital, Malmo ¨, Sweden Cylindromatosis gene (CYLD) is a ubiquitously expressed deubiquitinating enzyme, which interacts with members of the NF-jB signaling pathway and attenuates NF-jB and JNK signaling. Here, we report that DC derived from transgenic mice, which solely express a naturally occurring CYLD isoform (CYLD ex7/8 ), display a higher content of nuclear RelB and express elevated levels of NF-jB family members as well as of known NF-jB-target genes comprising costimulatory molecules and pro-inflammatory cytokines, as compared with WT DC. Accordingly, unstimulated CYLD ex7/8 DC exhibited a significantly higher primary allogenic T-cell stimulatory capacity than WT DC and exerted no tolerogenic activity. Transduction of unstimulated CYLD ex7/8 DC with relB-specific shRNA reduced their T-cell stimulatory capacity. Treatment with the synthetic glucocorticoid dexamethasone known to inhibit NF-jB and AP-1 activity reverted the pro-immunogenic phenotype and function of CYLD ex7/8 DC and re-established their tolerogenic function. DC derived from CYLD knockout mice showed no functional alterations compared with WT DC. Therefore, although complete loss of CYLD may be compensated for by other endogenous NF-jB inhibitors, CYLD ex7/8 acts in a dominant negative manner. Our findings raise the question of whether genetic defects associated with increased NF-jB activity may result in distur- bed maintenance of peripheral tolerance. Key words: Autoimmunity . Costimulation . DC . Tolerance . Transgenic/knockout mice Introduction Under steady-state conditions, DC reside as sentinel cells in most tissues and sample their micro-environment for antigens. In an immature or semi-mature state, DC exert tolerogenic function and interact with thymus-derived natural as well as induced adaptive Treg to maintain peripheral tolerance [1]. Contact with pathogen-derived components and pro-inflammatory cytokines released by pathogen-sensing cells results in stimulation of DC mediated by activation of NF-kB transcription factors [2]. These transcription factors induce a transition of the DC phenotype characterized by strong upregulation of costimulatory receptors and pro-inflammatory cytokines [3]. In their acquired mature state, DC constitute the most potent APC of the immune system and may transiently override the function of Treg [4, 5] to ensure the activation of T-effector cells. Correspondence: Dr. Matthias Bros e-mail: [email protected] & 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu DOI 10.1002/eji.200939285 Eur. J. Immunol. 2010. 40: 2848–2857 Matthias Bros et al. 2848
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Mutated CYLD affects the functional state of dendritic cells

Mutated cylindromatosis gene affects the functionalstate of dendritic cells
Matthias Bros1, Nadine Dexheimer1, Verena Besche1, Joumana Masri2,
Stefanie Trojandt1, Nadine Hovelmeyer2, Sonja Reissig2,
Ramin Massoumi3, Stephan Grabbe1, Ari Waisman2 and
Angelika B. Reske-Kunz1
1 Clinical Research Unit Allergology, Department of Dermatology, University Medical Center of
the Johannes Gutenberg-University, Mainz, Germany2 Institute for Molecular Medicine, University Medical Center of the Johannes Gutenberg-
University, Mainz, Germany3 Department of Laboratory Medicine, Lund University, Malmo University Hospital, Malmo,
Sweden
Cylindromatosis gene (CYLD) is a ubiquitously expressed deubiquitinating enzyme, which
interacts with members of the NF-jB signaling pathway and attenuates NF-jB and JNK
signaling. Here, we report that DC derived from transgenic mice, which solely express a
naturally occurring CYLD isoform (CYLDex7/8), display a higher content of nuclear RelB and
express elevated levels of NF-jB family members as well as of known NF-jB-target genes
comprising costimulatory molecules and pro-inflammatory cytokines, as compared with
WT DC. Accordingly, unstimulated CYLDex7/8 DC exhibited a significantly higher primary
allogenic T-cell stimulatory capacity than WT DC and exerted no tolerogenic activity.
Transduction of unstimulated CYLDex7/8 DC with relB-specific shRNA reduced their T-cell
stimulatory capacity. Treatment with the synthetic glucocorticoid dexamethasone known
to inhibit NF-jB and AP-1 activity reverted the pro-immunogenic phenotype and function
of CYLDex7/8 DC and re-established their tolerogenic function. DC derived from CYLD
knockout mice showed no functional alterations compared with WT DC. Therefore,
although complete loss of CYLD may be compensated for by other endogenous NF-jB
inhibitors, CYLDex7/8 acts in a dominant negative manner. Our findings raise the question
of whether genetic defects associated with increased NF-jB activity may result in distur-
bed maintenance of peripheral tolerance.
Key words: Autoimmunity . Costimulation . DC . Tolerance . Transgenic/knockout mice
Introduction
Under steady-state conditions, DC reside as sentinel cells in most
tissues and sample their micro-environment for antigens. In an
immature or semi-mature state, DC exert tolerogenic function
and interact with thymus-derived natural as well as induced
adaptive Treg to maintain peripheral tolerance [1]. Contact with
pathogen-derived components and pro-inflammatory cytokines
released by pathogen-sensing cells results in stimulation of DC
mediated by activation of NF-kB transcription factors [2]. These
transcription factors induce a transition of the DC phenotype
characterized by strong upregulation of costimulatory receptors
and pro-inflammatory cytokines [3]. In their acquired mature
state, DC constitute the most potent APC of the immune system
and may transiently override the function of Treg [4, 5] to ensure
the activation of T-effector cells.Correspondence: Dr. Matthias Brose-mail: [email protected]
& 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
DOI 10.1002/eji.200939285 Eur. J. Immunol. 2010. 40: 2848–2857Matthias Bros et al.2848

NF-kB activity is under tight control of endogenous inhibitors
[6] and mutation-associated deregulation of NF-kB activity
results in functional defects of the immune system, immunode-
ficiencies in case of impaired NF-kB activity, and autoimmune
diseases characterized by chronic inflammation due to persistent
NF-kB activity [7]. In addition, hyperactivity of NF-kB is
frequently apparent in cancer cells and may be associated with
their increased resistance toward apoptotic signals and their
enhanced proliferation.
The tumor suppressor gene cylindromatosis gene (CYLD) was
identified as being mutated in many cases of familial cylin-
dromatosis [8]. Patients suffer from benign skin appendages,
which originate from hair follicle cells on the scalp and facial skin
[9]. However, CYLD is expressed rather ubiquitously [8] and in
different types of tumors CYLD was frequently found to be
downregulated [10], which indicates a general importance of
CYLD as a tumor suppressor.
CYLD is a deubiquitinating enzyme, which interacts with the
signaling components TRAF2, TRAF6, TRAF7 (TRAF, TNF recep-
tor-associated factor) and NF-kB essential modulator (NEMO)
[11–14]. Ubiquitination of these proteins triggers assembly of the
IkB kinase complex, which results in NF-kB activation, and
accordingly CYLD was shown to inhibit canonical NF-kB activation
via IkB kinase complex. Moreover, CYLD controls nuclear entry of
the proto-oncogene Bcl3 [15], which forms heterodimers with
NF-kB family members to exert transcriptional control [16].
Recently, a transgenic mouse strain (CYLDex7/8) engineered to
solely express a naturally occurring CYLD isoform devoid of NEMO
and TRAF2 interaction domains but retaining the ability to interact
with Bcl3 was shown by us to harbor an expanded population of
mature B cells displaying higher activity of NF-kB and enhanced
nuclear accumulation of Bcl3 [17].
Here, we show that unstimulated BM-derived DC (BMDC)
isolated from CYLDex7/8 mice showed an increased content of
nuclear RelB which was associated with higher expression of DC-
activation markers, and induced stronger allogenic T-cell prolif-
eration than WT BMDC. Accordingly, CYLDex7/8 BMDC were
unable to induce the differentiation of naıve T cells to Treg in
vitro. Transduction of CYLDex7/8 BMDC with relB-specific shRNA
reduced their T-cell stimulatory capacity. Treatment of CYLDex7/8
BMDC with the synthetic glucocorticoid dexamethasone (DEX)
previously shown to inhibit NF-kB activity, reverted the hyper-
activity of CYLDex7/8 BMDC and re-established their tolerogenic
function.
Results
Murine BMDC express two isoforms of CYLD
It was recently shown that murine B cells expressed a CYLD
isoform lacking exons 7 and 8 (termed CYLDex7/8) [17] that
encode TRAF/NEMO interaction domains (Fig. 1A). Transgenic
mice (C57BL/6 background) engineered to express only the short
isoform CYLDex7/8 showed a strong expansion of the mature
B-cell population due to constitutive NF-kB activation. As shown
in Fig. 1B, BMDC of C57BL/6 origin expressed mRNA of the two
CYLD isoforms both at immature state and following stimulation
with LPS. As expected, BMDC derived from transgenic CYLDex7/8
mice expressed only the shorter CYLD isoform.
CYLDex7=8 BMDC display altered function
Compared with WT BMDC, unstimulated CYLDko BMDC displayed
lower MHCII expression, but similar surface levels of the
costimulatory molecules CD40, CD80 and CD86 (Fig. 1C and D).
On the other hand, CYLDex7/8 BMDC expressed comparable levels
of MHCII and CD80, but moderately higher levels of CD40 and
CD86 as shown by the MFI. Upon stimulation with LPS, the
fraction of cells expressing MHCII at high level was enhanced in all
three groups (Fig. 1C), and the average MHCII expression level
was elevated (Fig. 1D). The expression of the costimulatory
molecules monitored was enhanced in response to stimulation as
well. Interestingly, stimulated CYLDex7/8 BMDC displayed lower
CD40 expression than WT BMDC, but higher CD80 expression.
In MLR, both WT and CYLDko BMDC showed comparable
allogeneic T-cell stimulatory capacity under basal conditions,
which was significantly increased upon LPS stimulation (Fig. 1E).
This finding suggests that the absence of NF-kB inhibitory func-
tion of CYLD (CYLDko) may be compensated for by other endo-
genous NF-kB inhibitors [6]. On the contrary, unstimulated
CYLDex7/8 BMDC were almost as potent as stimulated WT BMDC
in inducing T-cell proliferation (Fig. 1F), suggesting that sole
expression of truncated CYLD lacking TRAF and NEMO interac-
tion domains, but retaining catalytic activity, counteracts
compensatory mechanisms. However, the presence of a WT CYLD
allele (CYLDwt/ex7/8) was sufficient to counteract the excessive
stimulatory potency observed in unstimulated CYLDex7/8 BMDC
(Fig. 1G).
CYLDex7=8 BMDC display a pro-immunogenic geneexpression signature
To analyze more thoroughly the molecular base for the stronger
APC activity of CYLDex7/8 BMDC as compared with WT BMDC at
unstimulated state, we monitored the expression of a panel of
well-known genes whose expression state compared with
unstimulated WT BMDC, unstimulated CYLDex7/8 BMDC showed
significantly higher mRNA expression of NF-kB family member
c-Rel as well as enhanced expression of relB (Fig. 2A), both of
which mediate upregulation of costimulatory molecules and pro-
inflammatory cytokines [18, 19]. Concomitantly, unstimulated
CYLDex7/8 BMDC displayed higher mRNA expression of CD40,
OX40L and SLAM, (Fig. 2B), but significantly lower expression of
mRNA encoding for LIGHT and the coinhibitory molecules B7-H3
and B7-DC (Fig. 2C). All the pro-inflammatory cytokine mRNA
species analyzed showed higher expression in CYLDex7/8 BMDC
than in WT BMDC at basal state (Fig. 2D). Interestingly, mRNA
Eur. J. Immunol. 2010. 40: 2848–2857 Immunomodulation 2849
& 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
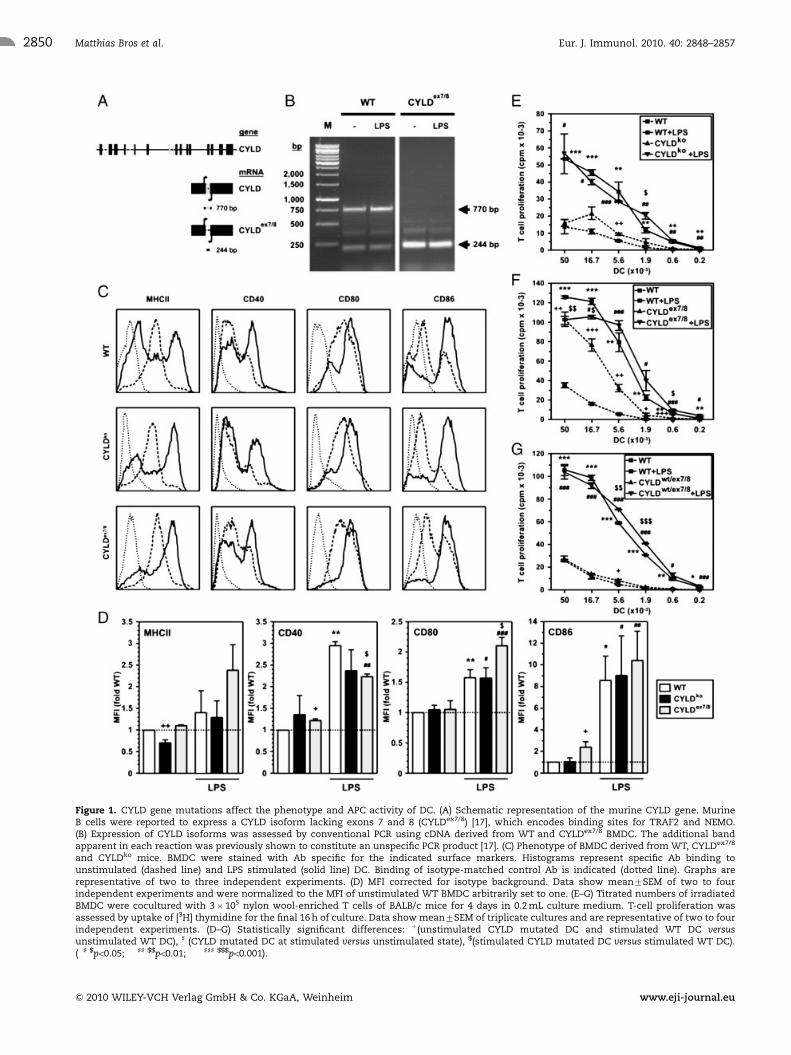
Figure 1. CYLD gene mutations affect the phenotype and APC activity of DC. (A) Schematic representation of the murine CYLD gene. MurineB cells were reported to express a CYLD isoform lacking exons 7 and 8 (CYLDex7/8) [17], which encodes binding sites for TRAF2 and NEMO.(B) Expression of CYLD isoforms was assessed by conventional PCR using cDNA derived from WT and CYLDex7/8 BMDC. The additional bandapparent in each reaction was previously shown to constitute an unspecific PCR product [17]. (C) Phenotype of BMDC derived from WT, CYLDex7/8
and CYLDko mice. BMDC were stained with Ab specific for the indicated surface markers. Histograms represent specific Ab binding tounstimulated (dashed line) and LPS stimulated (solid line) DC. Binding of isotype-matched control Ab is indicated (dotted line). Graphs arerepresentative of two to three independent experiments. (D) MFI corrected for isotype background. Data show mean7SEM of two to fourindependent experiments and were normalized to the MFI of unstimulated WT BMDC arbitrarily set to one. (E–G) Titrated numbers of irradiatedBMDC were cocultured with 3� 105 nylon wool-enriched T cells of BALB/c mice for 4 days in 0.2 mL culture medium. T-cell proliferation wasassessed by uptake of [3H] thymidine for the final 16 h of culture. Data show mean7SEM of triplicate cultures and are representative of two to fourindependent experiments. (D–G) Statistically significant differences: 1(unstimulated CYLD mutated DC and stimulated WT DC versusunstimulated WT DC), ] (CYLD mutated DC at stimulated versus unstimulated state), $(stimulated CYLD mutated DC versus stimulated WT DC).(�] $po0.05; ��]] $$po0.01; ���]]] $$$po0.001).
Eur. J. Immunol. 2010. 40: 2848–2857Matthias Bros et al.2850
& 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

encoding the DC maturation marker Fascin (Fscn) was signifi-
cantly increased in CYLDex7/8 BMDC as well (Fig. 2E). Upon
stimulation with LPS, CYLDex7/8 BMDC exhibited higher mRNA
expression of the NF-kB family members p52 and relB compared
with stimulated WT BMDC. Further, the mRNA levels of IL-1b
and TGF-b1 and of the DC maturation marker Fascin were
increased, but decreased mRNA expression in case of LIGHT and
the coinhibitory receptors B7-H3 and B7-DC was seen.
NF-kB transcription factors are complexed in the cytoplasm
and exert regulatory activity upon translocation into the nucleus.
Figure 2. CYLDex7/8 DC exhibit a pro-immunogenic gene expression signature and enhanced accumulation of RelB in the nucleus. BMDC werederived from WT and CYLDex7/8 mice and aliquots were stimulated with LPS. The relative mRNA expression level of genes encoding (A) NF-kBfamily members, (B, C) costimulatory/coinhibitory receptors, (D) cytokines and (E) other molecules was assessed by quantitative real-time PCR.Data indicate relative differences in mRNA expression of LPS-stimulated WT and of CYLDex7/8 BMDC at unstimulated and stimulated statecompared with unstimulated WT BMDC (dashed line). Data show mean7SEM of three to five independent experiments performed in duplicate.�po0.05, statistically significant differences versus unstimulated WT DC. (F) Protein extracts were probed for RelB and c-Rel protein content in thecytoplasmic and nuclear compartments. b-Actin detection served as loading control. Data are representative of two (c-Rel) and three (RelB)experiments. (G–I) BMDC of WT and CYLDex7/8 genotype were transduced on days 5 and 6 of culture with lentiviral particles harboring either emptyvector as control (Ctrl) or relB shRNA (sh relB). On day 8, BMDC were harvested and subjected to experiments. (G) Protein extracts of transducedBMDC populations were probed for RelB protein content. b-Tubulin detection served as loading control. Data presented are representative of twoindependent experiments. (H) Titrated numbers of irradiated BMDC of either population were cocultured with 3� 105 nylon wool-enriched T cellsof BALB/c mice for 4 days as described (see legend of Fig. 1E–G). Data show mean7SEM of triplicate cultures and are representative of threeindependent experiments. (I) Quantification of relative allogeneic T-cell proliferation induced by 1.9� 104 differentially transduced BMDC derivedfrom WT and CYLDex7/8 mice cocultured with 3� 105 BALB/c T cells. T-cell proliferation induced by unstimulated WT BMDC transduced withcontrol vector (Ctrl) was arbitrarily set to one. Data show mean7SEM of three independent experiments performed in triplicate. Statisticallysignificant differences: �(versus WT DC transduced with Ctrl vector), 1(CYLDex7/8 DC transduced with relB shRNA versus Ctrl vector), ](CYLDex7/8 DCversus WT DC, each transduced with relB shRNA). �]1po0.05; ��11po0.01; ���po0.001).
Eur. J. Immunol. 2010. 40: 2848–2857 Immunomodulation 2851
& 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

The activity of the NF-kB transcription factor family members
relB and c-Rel correlates with the activation state of DC [20]. Due
to our finding of higher relB and c-Rel mRNA levels in CYLDex7/8
BMDC than in WT BMDC, we assessed their expression on the
protein level by Western blot analysis. WT and CYLDex7/8 BMDC
harbored comparable amounts of RelB in the cytoplasm,
whereas the amounts of cytoplasmic c-Rel were very low
in both cell types (Fig. 2F). Stimulation with LPS induced
enhanced expression of both NF-kB proteins. Most interestingly,
in the nuclear compartment unstimulated CYLDex7/8 BMDC
displayed a higher RelB level than WT BMDC, whereas c-Rel was
apparent at a low level in either BMDC population. In LPS-
stimulated BMDC, a general increase in nuclear RelB and c-Rel
was noted.
Li et al. [21] have shown that inhibition of RelB activity by
RNA interference in BMDC diminished their T-cell stimulatory
capacity. Based on our finding of a higher expression of relB both
on the mRNA and on the protein level in unstimulated CYLDex7/8
BMDC, we asked for the relevance of this NF-kB member for the
observed T-cell hyperstimulatory capacity of these cells. Inhibi-
tion of relB by transduction of unstimulated BMDC with specific
shRNA attenuated RelB expression in BMDC of both genotypes
(Fig. 2G), and resulted in impaired T-cell stimulatory capacity of
CYLDex7/8 BMDC (Fig. 2H and I).
CYLDex7=8 BMDC treated with DEX exert tolerogenicfunction
CYLD was shown to limit the activity of various AP-1 and NF-kB
transcription factors besides relB [6, 22]. Therefore, we tested,
whether glucocorticoids (GC), which were shown to broadly
inhibit NF-kB and AP-1 activity and are frequently prescribed in
the treatment of autoimmune diseases and severe allergic
diseases [23], were able to diminish the T-cell hyperstimulatory
capacity of CYLDex7/8 BMDC more efficiently than relB shRNA.
GC activates the normally retained GC receptor, which exerts its
anti-inflammatory effects by direct inhibition of NF-kB and AP-1
transcription factors through protein–protein interaction. In
addition, activated GC receptor binds to GC-response elements
and diminishes the expression of genes encoding pro-inflamma-
tory products.
We treated DC progenitors from day 3 of culture onward with
the synthetic GC DEX. After treatment with DEX, with or without
LPS stimulation, BMDC of WT or CYLDex7/8 origin expressed
MHCII as well as the costimulatory molecules CD40, CD80 and
CD86 at significantly reduced levels as compared with BMDC
cultivated in the absence of DEX (Fig. 3A and B).
Compared with unstimulated WT BMDC, in many cases DEX-
treated unstimulated BMDC of either genotype displayed lowered
expression of mRNA encoding for different NF-kB family
members (Fig. 3C), costimulatory (Fig. 3D) and coinhibitory
(Fig. 3E) molecules, pro-inflammatory cytokines (Fig. 3F) as well
as Fscn and CCR7 (Fig. 3G). However, DEX treatment induced
mRNA expression of LIGHT in BMDC of either genotype and of
B7-H2 in CYLDex7/8 BMDC (Fig. 3E). DEX treatment, in general,
prevented stimulation-dependent increases of NF-kB family
members as well as of costimulatory molecules and diminished
LPS-mediated upregulation of pro-inflammatory cytokine mRNA
species, most evident in case of IL-1b and IL12p40 mRNA
(compare with the data in Fig. 2). In addition, expression of Fscn
and CCR7 was strongly inhibited.
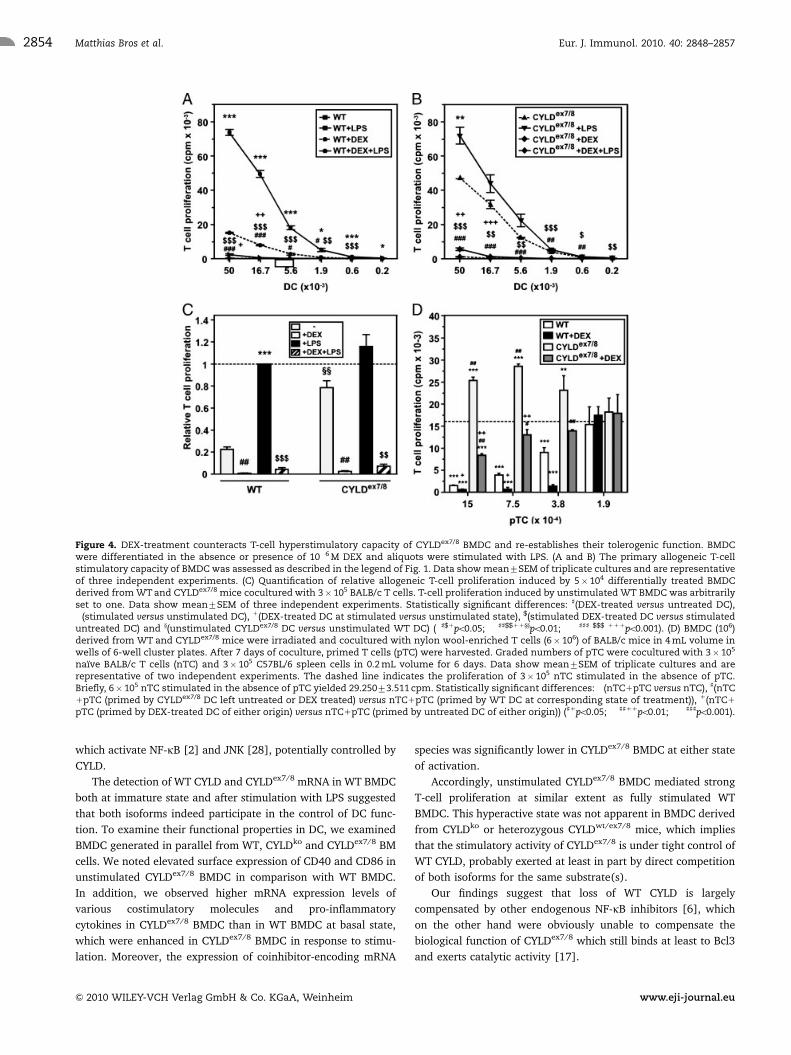
In accordance with their pro-tolerogenic phenotype, both WT
and CYLDex7/8 DEX-DC did not mediate efficient allogeneic T-cell
stimulation (Fig. 4A and B). Their T-cell stimulatory capacity was
lower than the moderate stimulatory potential exerted by unsti-
mulated WT BMDC (Fig. 4C; n 5 3). The same effect was noted
for CYLDko BMDC (data not shown).
Unstimulated WT BMDC were shown to exert tolerogenic
activity [1, 4]. Accordingly, T cells primed by unstimulated
WT BMDC inhibited proliferation of naıve T cells. On the
contrary, unstimulated CYLDex7/8 BMDC, which displayed a
strongly enhanced T-cell stimulatory capacity, did not exert
tolerogenic function (Fig. 4D). In accordance with our finding of
a drastically reduced allogeneic T-cell stimulatory potential of
DEX-DC, T cells primed by CYLDex7/8 DEX-DC exerted suppres-
sive activity on cocultured naıve T cells, although at lower
potency than T cells primed by unstimulated WT BMDC or WT
DEX-DC.
Discussion
Although the tumor suppressor gene CYLD was initially identified
as a candidate gene mutated in patients suffering from familial
cylindromatosis [8], the finding that its expression is strongly
diminished in tumors of distinct origin like colon and hepatocel-
lular carcinomas [10] suggested a more general role of CYLD.
Since the aa sequences of human and murine CYLD are highly
conserved (around 95% homology), several groups have gener-
ated CYLD knockout mouse strains in order to elucidate its role
in vivo.
Despite several in vitro studies which congruently showed that
CYLD inhibits NF-kB [11–13] and JNK [22] activity by interfering
with signaling molecules TRAF2, TRAF6 and NEMO as well as
Bcl3 [15], the various CYLDko mouse strains showed marked
phenotypical differences [24–26]. Recently, we identified a
naturally occurring CYLD isoform, which lacks exons 7 and 8
(CYLDex7/8) that encode aa 394–557 which encompass the
binding sites for TRAF2 and NEMO. Notably, transgenic mice
engineered to solely express CYLDex7/8 showed very similar B-cell
defects as noted for the CYLDko mouse strain described by Jin
et al. [27].
Taken together, CYLD seems to be critically involved in the
differentiation of T and B lymphocytes and controls their state of
activation. In light of this concept, we asked whether CYLD
function may influence APC function as well. We focused on DC
because they are known to exert tolerogenic function in imma-
ture or semi-mature states [1], and to acquire potent T-cell
stimulatory function upon activation by pathogen-derived signals
Eur. J. Immunol. 2010. 40: 2848–2857Matthias Bros et al.2852
& 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
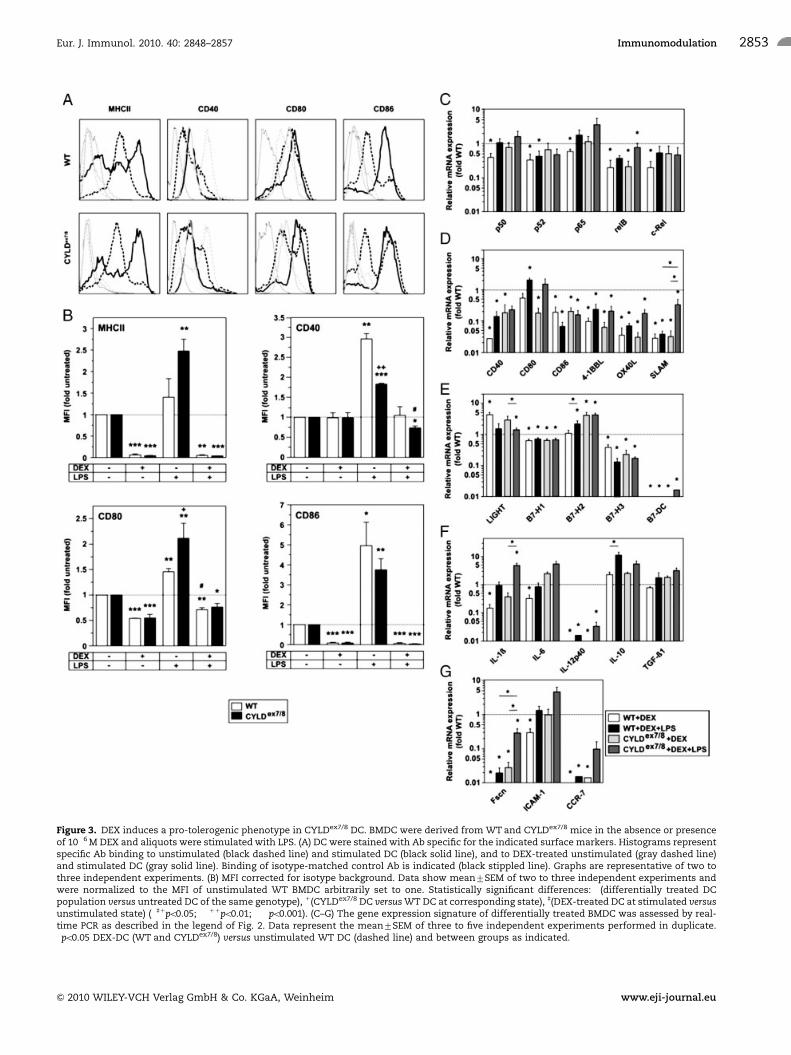
Figure 3. DEX induces a pro-tolerogenic phenotype in CYLDex7/8 DC. BMDC were derived from WT and CYLDex7/8 mice in the absence or presenceof 10�6 M DEX and aliquots were stimulated with LPS. (A) DC were stained with Ab specific for the indicated surface markers. Histograms representspecific Ab binding to unstimulated (black dashed line) and stimulated DC (black solid line), and to DEX-treated unstimulated (gray dashed line)and stimulated DC (gray solid line). Binding of isotype-matched control Ab is indicated (black stippled line). Graphs are representative of two tothree independent experiments. (B) MFI corrected for isotype background. Data show mean7SEM of two to three independent experiments andwere normalized to the MFI of unstimulated WT BMDC arbitrarily set to one. Statistically significant differences: �(differentially treated DCpopulation versus untreated DC of the same genotype), 1(CYLDex7/8 DC versus WT DC at corresponding state), ](DEX-treated DC at stimulated versusunstimulated state) (�]1po0.05; ��11po0.01; ���po0.001). (C–G) The gene expression signature of differentially treated BMDC was assessed by real-time PCR as described in the legend of Fig. 2. Data represent the mean7SEM of three to five independent experiments performed in duplicate.�po0.05 DEX-DC (WT and CYLDex7/8) versus unstimulated WT DC (dashed line) and between groups as indicated.
Eur. J. Immunol. 2010. 40: 2848–2857 Immunomodulation 2853
& 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
which activate NF-kB [2] and JNK [28], potentially controlled by
CYLD.
The detection of WT CYLD and CYLDex7/8 mRNA in WT BMDC
both at immature state and after stimulation with LPS suggested
that both isoforms indeed participate in the control of DC func-
tion. To examine their functional properties in DC, we examined
BMDC generated in parallel from WT, CYLDko and CYLDex7/8 BM
cells. We noted elevated surface expression of CD40 and CD86 in
unstimulated CYLDex7/8 BMDC in comparison with WT BMDC.
In addition, we observed higher mRNA expression levels of
various costimulatory molecules and pro-inflammatory
cytokines in CYLDex7/8 BMDC than in WT BMDC at basal state,
which were enhanced in CYLDex7/8 BMDC in response to stimu-
lation. Moreover, the expression of coinhibitor-encoding mRNA
species was significantly lower in CYLDex7/8 BMDC at either state
of activation.
Accordingly, unstimulated CYLDex7/8 BMDC mediated strong
T-cell proliferation at similar extent as fully stimulated WT
BMDC. This hyperactive state was not apparent in BMDC derived
from CYLDko or heterozygous CYLDwt/ex7/8 mice, which implies
that the stimulatory activity of CYLDex7/8 is under tight control of
WT CYLD, probably exerted at least in part by direct competition
of both isoforms for the same substrate(s).
Our findings suggest that loss of WT CYLD is largely
compensated by other endogenous NF-kB inhibitors [6], which
on the other hand were obviously unable to compensate the
biological function of CYLDex7/8 which still binds at least to Bcl3
and exerts catalytic activity [17].
Figure 4. DEX-treatment counteracts T-cell hyperstimulatory capacity of CYLDex7/8 BMDC and re-establishes their tolerogenic function. BMDCwere differentiated in the absence or presence of 10�6 M DEX and aliquots were stimulated with LPS. (A and B) The primary allogeneic T-cellstimulatory capacity of BMDC was assessed as described in the legend of Fig. 1. Data show mean7SEM of triplicate cultures and are representativeof three independent experiments. (C) Quantification of relative allogeneic T-cell proliferation induced by 5� 104 differentially treated BMDCderived from WT and CYLDex7/8 mice cocultured with 3� 105 BALB/c T cells. T-cell proliferation induced by unstimulated WT BMDC was arbitrarilyset to one. Data show mean7SEM of three independent experiments. Statistically significant differences: ](DEX-treated versus untreated DC),�(stimulated versus unstimulated DC), 1(DEX-treated DC at stimulated versus unstimulated state), $(stimulated DEX-treated DC versus stimulateduntreated DC) and y(unstimulated CYLDex7/8 DC versus unstimulated WT DC) (�]$1po0.05; ��]]$$11yypo0.01; ���]]] $$$ 111po0.001). (D) BMDC (106)derived from WT and CYLDex7/8 mice were irradiated and cocultured with nylon wool-enriched T cells (6� 106) of BALB/c mice in 4 mL volume inwells of 6-well cluster plates. After 7 days of coculture, primed T cells (pTC) were harvested. Graded numbers of pTC were cocultured with 3� 105
naıve BALB/c T cells (nTC) and 3� 105 C57BL/6 spleen cells in 0.2 mL volume for 6 days. Data show mean7SEM of triplicate cultures and arerepresentative of two independent experiments. The dashed line indicates the proliferation of 3� 105 nTC stimulated in the absence of pTC.Briefly, 6� 105 nTC stimulated in the absence of pTC yielded 29.25073.511 cpm. Statistically significant differences: �(nTC1pTC versus nTC), ](nTC1pTC (primed by CYLDex7/8 DC left untreated or DEX treated) versus nTC1pTC (primed by WT DC at corresponding state of treatment)), 1(nTC1
pTC (primed by DEX-treated DC of either origin) versus nTC1pTC (primed by untreated DC of either origin)) (]1po0.05; ��]]11po0.01; ���]]]po0.001).
Eur. J. Immunol. 2010. 40: 2848–2857Matthias Bros et al.2854
& 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

Interestingly, unstimulated CYLDex7/8 BMDC showed higher
transcript expression levels of NF-kB family members relB and
c-Rel than WT BMDC, and displayed a higher nuclear RelB
content. Similarly, CYLDex7/8 B cells were shown by us to express
higher amounts of RelB protein than WT B cells, but the relB
mRNA level remained unaltered in these B cells, suggesting post-
transcriptional regulation [17]. In line with our finding of higher
nuclear RelB levels in CYLDex7/8 than in WT BMDC at basal state,
we noted elevated expression of costimulatory molecules and
pro-inflammatory cytokines in unstimulated CYLDex7/8 BMDC on
the transcriptional and protein level as compared with WT
BMDC. Accordingly, in contrast to unstimulated WT BMDC,
unstimulated CYLDex7/8 BMDC induced hyperproliferation of
interacting allogeneic T cells and lacked suppressive activity. In
accordance with the prominent nuclear localization of RelB in
CYLDex7/8 BMDC, but not WT BMDC at unstimulated state, we
achieved partial inhibition of the T-cell hyperstimulatory activity
of CYLDex7/8 BMDC by their transduction with lentivirus encod-
ing relB-specific shRNA. Our results suggest that in CYLDex7/8
BMDC other factors besides RelB may contribute to the pro-
immunogenic phenotype and T-cell hyperstimulatory activity of
CYLDex7/8 BMDC. This notion is in line with the reports showing
that CYLD limits the activity of various NF-kB and AP-1 tran-
scription factors [6, 22]. A more extensive inhibition of NF-kB
and AP-1 family members is achieved by GC [23]. Therefore, in
an attempt to re-establish tolerogenic function of CYLDex7/8
BMDC, we have employed the synthetic GC DEX. We have
recently shown that DEX-treated DC acquired a tolerogenic
phenotype and induced Treg [29]. Cotreatment of BMDC of
either genotype with DEX diminished expression of NF-kB family
members, costimulatory molecules and pro-inflammatory cyto-
kines. In accordance with their DEX-mediated pro-tolerogenic
phenotype, DEX-treated BMDC of either origin displayed low APC
activity, and CYLDex7/8 BMDC regained the ability to induce Treg.
In an analogous manner, Brummelkamp et al. [12] have shown
that hyperactivation of NF-kB induced by CYLD gene silencing
can be compensated by applying the NF-kB inhibitor aspirin.
Our study indicates that germline mutations affecting NF-kB
activity may determine the functional state of DC, if not compen-
sated by other endogenous control mechanisms. In clinical settings,
it may be possible to overcome these defects by generating DC
ex vivo, educated by applying suitable drugs to prevent DC hyper-
activity [30] (as shown here) or to induce strong DC activation,
followed by reinfusion of antigen-loaded DC into the patient.
Materials and methods
Mice
Female C57BL/6 and BALB/c mice as well as female CYLD
mutant mice (knockout and ex7/8) on C57BL/6 background [15,
17] were bred and maintained in the Central Animal Facilities of
the University of Mainz under specific pathogen-free conditions
on a standard diet. The ‘‘Principles of Laboratory Animal Care’’
(NIH publication no. 85-23, revised 1985) were followed.
Cells
BMDC were generated from BM progenitors of mice as first
described by Scheicher et al. [31] with some modifications [32]
using BMDC medium (IMDM with 10% FBS; (PAA, Colbe,
Germany), 2 mM L-glutamine (Biochrom AG, Berlin, Germany),
100 U/mL penicillin, 100mg/mL streptomycin (Gibco, Paisly, UK)),
supplemented with 5% of GM-CSF containing cell culture super-
natant [33] (a kind gift from Dr. B. Stockinger, MRC National
Institute for Medical Research, Mill Hill, London). Where indicated
DEX (Sigma-Aldrich, Deisenhofen, Germany) was added on day 3
of culture (10�6 M). On days 7 or 8 of culture, part of the
nonadherent cells was harvested (485% CD11c1 cells), reseeded
on 100-mm2 Cellstar tissue-culture dishes (Greiner) and stimu-
lated with LPS (1mg/mL; Calbiochem, Schwalbach, Germany) for
24 h. GM-CSF and DEX were replenished in the course of change of
culture media on days 3 and 6, and were added to cell cultures
stimulated with LPS. HEK 293T cells used for production of
lentiviral particles were kept as described previously [34].
Flow cytometry
BMDC (5� 105) were washed in staining buffer (PBS/2% FBS).
To block Fc receptor-mediated staining, cells were incubated with
rat anti-mouse CD16/CD32 (2.4.G2) (Dianova, Hamburg,
Germany) for 15 min on ice. Afterward, cells were incubated
with FITC-conjugated rat mAb recognizing MHC class II I-A/I-E
(2G9), CD80 (1G10), CD86 (GL1) (BD Pharmingen, San Diego,
CA, USA) and CD40 (1C10) (Southern Biotechnology, Birming-
ham, AL, USA) for 20 min on ice. Appropriate isotype controls
were used. Flow cytometric analysis was performed using a
FACScan flow cytometer (BD Biosciences) equipped with
CellQuest Software.
Western blot analysis
Western blot analysis was carried out as described previously
[17] using the following Ab: anti-RelB (sc-226) and anti-c-Rel
(sc-71) (Santa Cruz, Santa Cruz, CA, USA), as well as anti-b-Actin
(AC-15; Sigma-Aldrich) and b-tubulin (ab21058; Abcam,
Cambridge, UK). Preparation of nuclear extracts was performed
as described previously [35]. Nuclear and cytoplasmic fractions
were prepared according to the standard procedures [36].
Lentiviral transduction
HEK 293T cells were cotransfected with HIV-1-derived packaging
plasmid pCMVDR8.91 and the vesicular stomatitis virus
Eur. J. Immunol. 2010. 40: 2848–2857 Immunomodulation 2855
& 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

glycoprotein G-encoding plasmid pMD2.G, and either relB shRNA
encoding or empty TRIPZ vector (Open Biosystems, Huntsville,
AL, USA) to generate lentiviral particles as described previously
[34]. Unstimulated BMDC were transduced on days 5 and 6 as
described and were used for experiments on day 8.
MLR
BALB/c splenocytes were depleted of erythrocytes by incubation
in lysis buffer (155 mM NH4Cl, 10 mM KHCO3, 100 mM EDTA-
sodium, pH 7.4) for 1 min and were passed over nylon wool
columns to enrich T cells. T cells (3� 105) were cocultured for 4
days on flat-bottom 96-well tissue culture plates with graded
numbers of irradiated (30 Gy) BMDC in 200 mL of BMDC
medium. Cell proliferation was assessed by the uptake of [3H]
thymidine (0.25 mCi/well) for the last 16 h of culture. Cells were
harvested onto glass fiber filters and retained radioactivity was
measured in a liquid scintillation counter (1205 Betaplate, LKB
Wallac, Turku, Finland).
T-cell restimulation and suppression assays
BMDC (106/well) were cocultured on 6-well tissue culture plates
with nylon wool-enriched BALB/c T cells (6� 106/well) in a
volume of 4 mL for 7 days, and prestimulated T cells were
harvested. C57BL/6 splenocytes, depleted of erythrocytes and
g-irradiated (30 Gy), were cocultured with freshly isolated
BALB/c T cells (3�105 each) and graded numbers of prestimu-
lated T cells for 6 days. T-cell proliferation was assessed as
described above.
PCR and real-time PCR analysis
Total RNA was isolated from at least 5� 105 BMDC by using the
RNeasy Plus Mini kit (Qiagen, Hilden, Germany) as recom-
mended. RNA was reverse-transcribed applying a 1:1 mix of
Oligo-dT and random hexamer primers by using iScript (Bio-Rad,
Munich, Germany) as recommended. Expression of CYLDex7/8
and the corresponding WT sequence was detected by PCR using
primers binding to sequences located in neighboring exons as
described previously [17]. Primer sequences have been described
[29] and primers were purchased from Operon (Koln, Germany).
The house-keeping gene ubiquitin C served as internal control.
Real-time PCR reactions included 200 ng of cDNA and SYBR-
Green mastermix (ABgene, Hamburg, Germany) and were
performed and analyzed as described previously [29].
Statistical analysis
Data were analyzed for statistically significant differences by
applying Student’s t-test.
Acknowledgements: This work was supported by the
Deutsche Forschungsgemeinschaft, Sonderforschungsbereich
548 (to M. B., A. W. and A. B. R. -K.) and 432 (to A. W.), and
the Stiftung Rheinland-Pfalz fur Innovation (15212-386261/761)
(to A. B. R. -K.).
Conflict of interest: The authors declare no financial or
commercial conflict of interest.
References
1 Tang, Q. and Bluestone, J. A., The Foxp31 regulatory T cell: a jack of all
trades, master of regulation. Nat. Immunol. 2008. 9: 239–244.
2 Hofer, S., Rescigno, M., Granucci, F., Citterio, S., Francolini, M. and
Ricciardi-Castagnoli, P., Differential activation of NF-kappa B subunits in
dendritic cells in response to Gram-negative bacteria and to lipopoly-
saccharide. Microbes Infect. 2001. 3: 259–265.
3 Adams, S., O’Neill, D. W. and Bhardwaj, N., Recent advances in dendritic
cell biology. J. Clin. Immunol. 2005. 25: 87–98.
4 Fehervari, Z. and Sakaguchi, S., Control of Foxp31 CD251CD41 regulatory
cell activation and function by dendritic cells. Int. Immunol. 2004. 16:
1769–1780.
5 Ahn, J. S., Krishnadas, D. K. and Agrawal, B., Dendritic cells partially
abrogate the regulatory activity of CD41CD251 T cells present in the
human peripheral blood. Int. Immunol. 2007. 19: 227–237.
6 Chen, F., Endogenous inhibitors of nuclear factor-kappaB, an opportunity
for cancer control. Cancer Res. 2004. 64: 8135–8138.
7 Courtois, G. and Gilmore, T. D., Mutations in the NF-kappaB signaling
pathway: implications for human disease. Oncogene 2006. 25: 6831–6843.
8 Bignell, G. R., Warren, W., Seal, S., Takahashi, M., Rapley, E., Barfoot, R.,
Green, H. et al., Identification of the familial cylindromatosis tumour-
suppressor gene. Nat. Genet. 2000. 25: 160–165.
9 Massoumi, R. and Paus, R., Cylindromatosis and the CYLD gene: new
lessons on the molecular principles of epithelial growth control. Bioessays
2007. 29: 1203–1214.
10 Hellerbrand, C., Bumes, E., Bataille, F., Dietmaier, W., Massoumi, R. and
Bosserhoff, A. K., Reduced expression of CYLD in human colon and
hepatocellular carcinomas. Carcinogenesis 2007. 28: 21–27.
11 Trompouki, E., Hatzivassiliou, E., Tsichritzis, T., Farmer, H., Ashworth, A.
and Mosialos, G., CYLD is a deubiquitinating enzyme that negatively
regulates NF-kappaB activation by TNFR family members. Nature 2003.
424: 793–796.
12 Brummelkamp, T. R., Nijman, S. M., Dirac, A. M. and Bernards, R., Loss of
the cylindromatosis tumour suppressor inhibits apoptosis by activating
NF-kappaB. Nature 2003. 424: 797–801.
13 Kovalenko, A., Chable-Bessia, C., Cantarella, G., Israel, A., Wallach, D.
and Courtois, G., The tumour suppressor CYLD negatively regulates NF-
kappaB signalling by deubiquitination. Nature 2003. 424: 801–805.
14 Yoshida, H., Jono, H., Kai, H. and Li, J. D., The tumor suppressor
cylindromatosis (CYLD) acts as a negative regulator for toll-like receptor 2
signaling via negative cross-talk with TRAF6 and TRAF7. J. Biol. Chem.
2005. 280: 41111–41121.
Eur. J. Immunol. 2010. 40: 2848–2857Matthias Bros et al.2856
& 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

15 Massoumi, R., Chmielarska, K., Hennecke, A., Pfeifer, A. and Fassler, R.,
Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-
kappaB signaling. Cell 2006. 125: 665–677.
16 Hayden, M. S., West, A. P. and Ghosh, S., NF-kappaB and the immune
response. Oncogene 2006. 25: 6758–6780.
17 Hovelmeyer, N., Wunderlich, F. T., Massoumi, R., Jakobsen, C. G., Song, J.,
Worns, M. A., Merkwirth, C. et al., Regulation of B cell homeostasis and
activation by the tumor suppressor gene CYLD. J. Exp. Med. 2007. 204:
2615–2627.
18 Boffa, D. J., Feng, B., Sharma, V., Dematteo, R., Miller, G., Suthanthiran,
M., Nunez, R. and Liou, H. C., Selective loss of c-Rel compromises
dendritic cell activation of T lymphocytes. Cell. Immunol. 2003. 222:
105–115.
19 Li, M., Zhang, X., Zheng, X., Lian, D., Zhang, Z. X., Ge, W., Yang, J. et al.,
Immune modulation and tolerance induction by RelB-silenced dendritic
cells through RNA interference. J. Immunol. 2007. 178: 5480–5487.
20 Neumann, M., Fries, H., Scheicher, C., Keikavoussi, P., Kolb-Maurer, A.,
Brocker, E., Serfling, E. and Kampgen, E., Differential expression of Rel/
NF-kappaB and octamer factors is a hallmark of the generation and
maturation of dendritic cells. Blood 2000. 95: 277–285.
21 Li, M., Zhang, X., Zheng, X., Lian, D., Zhang, Z. X., Ge, W., Yang, J. et al.,
Immune modulation and tolerance induction by RelB-silenced dendritic
cells through RNA interference. J. Immunol. 2007. 178: 5480–5487.
22 Reiley, W., Zhang, M. and Sun, S. C., Negative regulation of JNK signaling
by the tumor suppressor CYLD. J. Biol. Chem. 2004. 279: 55161–55167.
23 Smoak, K. A. and Cidlowski, J. A., Mechanisms of glucocorticoid receptor
signaling during inflammation. Mech. Ageing Dev. 2004. 125: 697–706.
24 Reiley, W. W., Zhang, M., Jin, W., Losiewicz, M., Donohue, K. B., Norbury,
C. C. and Sun, S. C., Regulation of T cell development by the
deubiquitinating enzyme CYLD. Nat. Immunol. 2006. 7: 411–417.
25 Reiley, W. W., Jin, W., Lee, A. J., Wright, A., Wu, X., Tewalt, E. F., Leonard,
T. O. et al., Deubiquitinating enzyme CYLD negatively regulates the
ubiquitin-dependent kinase Tak1 and prevents abnormal T cell
responses. J. Exp. Med. 2007. 204: 1475–1485.
26 Zhang, J., Stirling, B., Temmerman, S. T., Ma, C. A., Fuss, I. J., Derry, J. M.
and Jain, A., Impaired regulation of NF-kappaB and increased suscept-
ibility to colitis-associated tumorigenesis in CYLD-deficient mice. J. Clin.
Invest. 2006. 116: 3042–3049.
27 Jin, W., Reiley, W. R., Lee, A. J., Wright, A., Wu, X., Zhang, M. and Sun, S.
C., Deubiquitinating enzyme CYLD regulates the peripheral development
and naive phenotype maintenance of B cells. J. Biol. Chem. 2007. 282:
15884–15893.
28 Nakahara, T., Uchi, H., Urabe, K., Chen, Q., Furue, M. and Moroi, Y., Role
of c-Jun N-terminal kinase on lipolpolysaccharide induced maturation of
human monocyte-derived dendritic cells. Int. Immunol. 2004. 16:
1701–1709.
29 Bros, M., Jahrling, F., Renzing, A., Wiechmann, N., Dang, N. A., Sutter, A.,
Ross, R. et al., A newly established murine immature dendritic cell line
can be differentiated into a mature state, but exerts tolerogenic function
upon maturation in the presence of glucocorticoid. Blood 2007. 109:
3820–3829.
30 Morelli, A. E. and Thomson, A. W., Tolerogenic dendritic cells and the
quest for transplant tolerance. Nat. Rev. Immunol. 2007. 7: 610–621.
31 Scheicher, C., Mehlig, M., Zecher, R. and Reske, K., Dendritic cells from
mouse bone marrow: in vitro differentiation using low doses of
recombinant granulocyte-macrophage colony-stimulating factor. J. Immu-
nol. Methods 1992. 154: 253–264.
32 Lutz, M. B., Kukutsch, N., Ogilvie, A. L., Rossner, S., Koch, F., Romani, N.
and Schuler, G., An advanced culture method for generating large
quantities of highly pure dendritic cells from mouse bone marrow.
J. Immunol. Methods 1999. 223: 77–92.
33 Zal, T., Volkmann, A. and Stockinger, B., Mechanisms of tolerance
induction in major histocompatibility complex class II-restricted T cells
specific for a blood-borne self-antigen. J. Exp. Med. 1994. 180: 2089–2099.
34 Besche, V., Wiechmann, N., Castor, T., Trojandt, S., Hohn, Y., Kunkel, H.,
Grez, M. et al., Dendritic cells lentivirally engineered to overexpress IL-10
inhibit contact hypersensitivity responses, despite their partial activation
induced by transduction-associated physical stress. J. Gene Med. 2010. 12:
231–243.
35 Weih, F., Carrasco, D. and Bravo, R., Constitutive and inducible Rel/NF-
kappa B activities in mouse thymus and spleen. Oncogene 1994. 9:
3289–3297.
36 Schreiber, E., Matthias, P., Muller, M. M. and Schaffner, W., Rapid
detection of octamer binding proteins with ‘mini-extracts’, prepared
from a small number of cells. Nucleic Acids Res. 1989. 17: 6419.
Abbreviations: BMDC: BM-derived DC � CYLD: cylindromatosis gene �DEX: dexamethasone � GC: glucocorticoid � NEMO: NF-kB essential
modulator � nTC: naıve BALB/c T cells � pTC: primed T cells � TRAF:
TNF receptor-associated factor
Full correspondence: Dr. Matthias Bros, Clinical Research Unit
Allergology, Department of Dermatology, University Medical Center of
the Johannes Gutenberg-University, Ob. Zahlbacher Street 63, D-55131
Mainz, Germany
Fax: 14961313933360
e-mail: [email protected]
Received: 27/1/2009
Revised: 15/6/2010
Accepted: 19/7/2010
Article accepted online: 28/7/2010
Eur. J. Immunol. 2010. 40: 2848–2857 Immunomodulation 2857
& 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu




















