
Effects of partial suppression of parkin on huntingtin mutant R6/1 mice
Upload
independentCategory
view
0download
0
www.elsevier.com/locate/ynbdi
Neurobiology of Disease 29 (2008) 22–29Haloperidol protects striatal neurons from dysfunction inducedby mutated huntingtin in vivo
Delphine Charvin,a,b Emmanuel Roze,a,c Valérie Perrin,b Carole Deyts,a Sandrine Betuing,a
Christiane Pagès,a Etienne Régulier,b,1 Ruth Luthi-Carter,b Emmanuel Brouillet,d
Nicole Déglon,d and Jocelyne Cabochea,⁎
aSignalisation Neuronale et Régulations Géniques, Centre National de la Recherche Scientifique,Université Pierre et Marie Curie, UMR 7102, 9 quai Saint Bernard, 75005 Paris, FrancebÉcole Polytechnique Fédérale de Lausanne (EPFL), Lausanne, SwitzerlandcDepartment of Neurology, Saint-Antoine Hospital, AP-HP, Paris, FrancedCEA, Institute of Biomedical Imaging and MIRCen, Orsay, France
Received 20 April 2007; revised 16 July 2007; accepted 23 July 2007Available online 19 August 2007
Huntington’s disease (HD) results from an abnormal polyglutamineextension in the N-terminal region of the huntingtin protein. Thismutation causes preferential degeneration of striatal projection neurons.We previously demonstrated, in vitro, that dopaminergic D2 receptorstimulation acted synergistically with mutated huntingtin (expHtt) toincrease aggregate formation and striatal death. In the present work, weextend these observations to an in vivo system based on lentiviral-mediated expression of expHtt in the rat striatum. The early and chronictreatment with the D2 antagonist haloperidol decanoate protects striatalneurons from expHtt-induced dysfunction, as analyzed by DARPP-32and NeuN stainings. Haloperidol treatment also reduces aggregatesformation, an effect that ismaintained over time. These findings indicatethat D2 receptors activation contributes to the deleterious effects ofexpHtt on striatal function andmay represent an interesting early targetto alter the subsequent course of neuropathology in HD.© 2007 Elsevier Inc. All rights reserved.
Keywords: Dopamine; D2 receptor; Lentiviral infection; Huntington’sdisease; PolyQ huntingtin; NeuN; DARPP-32; Striatal dysfunction
Introduction
Huntington’s disease (HD) is an autosomal dominant neurode-generative disorder resulting in chorea, cognitive impairment andbehavioral disturbances. Clinical symptoms generally appearbetween 35 and 42 years old and progress over a 10- to 20-yearcourse leading to death (Quinn and Schrag, 1998). HD is caused by
⁎ Corresponding author. Fax: +33 1 44 27 26 69.E-mail address: [email protected] (J. Caboche).
1 Present address: Novartis, Target and Lead Discovery Unit, Basel,Switzerland.
Available online on ScienceDirect (www.sciencedirect.com).
0969-9961/$ - see front matter © 2007 Elsevier Inc. All rights reserved.doi:10.1016/j.nbd.2007.07.028
a polyglutamine expansion in the huntingtin (htt) protein (Group,1993) and is characterized by two neuropathological hallmarks:formation of intraneuronal aggregates containing N-terminalfragments of mutated huntingtin (expHtt) and a progressive andpreferential degeneration of striatal neurons (DiFiglia et al., 1997;Reiner et al., 1988). This preferential vulnerability is important inHD pathophysiology since both the evolution and severity ofsymptoms are directly correlated to the rate of striatal neurode-generation (Aylward et al., 2000; Vonsattel et al., 1985). Severalneuro-anatomical and experimental observations propose that thephysiological DA release, that is particularly abundant in thestriatum (Weiner et al., 1991), may be a factor of vulnerability forstriatal neurons. First, HD neuropathology progresses in thestriatum according to the same dorsoventral gradient as local DAconcentration (Cass, 1997; Vonsattel and DiFiglia, 1998; Vonsattelet al., 1985). Furthermore, in animal or cell models where DAconcentration is artificially increased, a spontaneous striatal deathis observed (Bowyer et al., 1996; Luo et al., 1998; Ricaurte et al.,1982). Dopamine transporter knock-out (DAT−/−) mice displayboth spontaneous striatal death and behavioral alterations thatresemble HD (Cyr et al., 2003). When mated to knock-in HD micethese DAT−/− mice show exacerbation of HD pathophysiology andacceleration of expHtt aggregate formation (Cyr et al., 2006).Moreover, in a pharmacological HD rat model generated bysystemic administration of 3-nitropropionic acid (3-NP), removalof the nigrostriatal dopaminergic input protects the striatum fromneurodegeneration (Reynolds et al., 1998).
We recently made important extensions of these observationsand demonstrated, in vitro, that DA accelerates two neuropatho-logical hallmarks induced by expHtt: aggregate formation andstriatal neuron death (Charvin et al., 2005). Thus, in expHtt-expressing striatal neurons, DA potentiated both activation of thepro-apoptotic JNK/c-Jun pathway and aggregate formation, two

23D. Charvin et al. / Neurobiology of Disease 29 (2008) 22–29
DA-mediated effects that occurred early in the process of expHttneurotoxicity. These effects were due to ROS production and D2receptor stimulation, respectively.
Based on our in vitro observations, we hypothesized that anearly treatment with a D2 antagonist might be, at least in part,neuroprotective in HD. To test this, we used a lentivirus-based HDrat model that reproduces, within the striatum, the mainneuropathological features observed in patients: neuritic andintranuclear inclusions, neuronal dysfunction and death (Colinet al., 2005; de Almeida et al., 2002; Pardo et al., 2006; Perrinet al., 2007; Regulier et al., 2003). We show that early treatmentwith haloperidol decanoate significantly reduces both neuronaldysfunction and aggregate formation in the HD rat striatum. Theseresults indicate that early pharmacological blockade of D2receptors attenuates the pathogenic effects of expHtt on striatalfunction and may represent an interesting early target to alter thesubsequent course of neuropathology in HD.
Materials and methods
Animals
Adult female Wistar rats weighing between 180 and 200 g wereused. The animals were housed in a controlled temperature room thatwas maintained on a 12 h light/dark cycle. Food and water wereavailable ad libitum. The experiments were performed in accordancewith the European Community Council directive 86/609/EEC forcare and use of laboratory animals.
Injection of the lentiviruses
Concentrated viral stocks were thawed and resuspended byrepeated pipetting. Htt171-19Q- and htt171-82Q-expressinglentiviral vectors (de Almeida et al., 2002) were stereotaxicallyinjected into the right or the left striatum, respectively, of ketamine(75 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.) anesthetized animalsusing a 10 μl Hamilton syringe (Hamilton, Reno, NV). The animalsreceived 800 ng p24 (5 μl) of lentiviral vectors in each side at thefollowing coordinates: 0.5 mm rostral to bregma, 3 mm lateral tomidline and 5 mm ventral from the skull surface. The viruses wereinjected at 0.2 μl/min by means of an automatic injector (StoeltingCo.). The needle was then left in place for an additional 5 min andgently withdrawn. The skin was closed using wound chips autoclips(Phymep, Paris, France).
Treatment with haloperidol decanoate
Haloperidol decanoate (GensiaSicor pharmaceuticals) (50 mg/ml;30 mg/kg) was injected intramuscularly every 3 weeks (firstinjection the day after viral infection) and equal volumes of thevehicle (sesame oil, Sigma France) were injected intramuscularly tosham rats. Haloperidol decanoate-treated rats were sacrificed 2 or 8weeks, respectively, after the beginning of the treatment (n=7 foreach time point). Two groups of vehicle-treated rats were treated inparallel in the same conditions (n=7 for each time point).
Histological processing
Tissue preparationTwo or eight weeks after lentiviral injection, the animals were
anesthetized with a sodium pentobarbital overdose, transcardially
perfused, post-fixed in 4% paraformaldehyde, 10% picric acid for24 h and finally cryoprotected in 15% sucrose and 0.1 Mphosphate buffer for 48 h. The brains were frozen in isopentane at−25 °C and 25 μm coronal sections were cut on a slidingmicrotome cryostat (Leitz/Wetslar; Leica Microsystems, Nussloch,Germany). Slices throughout the entire striatum were collected andstored in 24-well trays as free-floating sections in TB containing30% ethylene glycol and 30% glycerol. The trays were stored at−20 °C until immunohistochemical processing.
Primary antibodiesThe following primary antibodies were used: the mouse anti-
human huntingtin monoclonal EM48 antibody (MAB5374,Chemicon), a mouse monoclonal antibody recognizing thedopamine- and cAMP-regulated phosphoprotein of 32 kDa(DARPP-32; a generous gift from Dr. P. Greengard, RockefellerUniversity, New York, NY) and the mouse monoclonal anti-body recognizing the neuronal-specific nuclear protein (NeuN;VMA377B, Abcys).
Immunohistochemical procedureFor light microscopy, sections were first incubated for 5 min in
a PBS solution containing 10% methanol, 0.9% H2O2, next in0.2% Triton. Free-floating sections were then incubated at RT for1 h in 0.1 M PBS containing 10% NGS (Vector Laboratories)followed by a reaction with the DARPP-32 antibody (1:100,000, o/n, 4 °C) or with the NeuN antibody (1:400, o/n, 4 °C) diluted in a5% NGS/PBS solution. Sections were incubated with an anti-mouse biotinylated secondary antibody (1:200, Vector Labora-tories) diluted in 1% NGS/PBS for 2 h at RT, and bound antibodywas visualized by using the Vectastain ABC kit (PK-4000, VectorLaboratories), with 3,3′-diaminobenzidine tetrahydrochloride(DAB metal concentrate; Pierce) as substrate. The sections weremounted, dehydrated by passing twice through ethanol and xyleneand coverslipped with Eukit.
The nuclear localization of huntingtin was investigated onsections double-stained for htt (EM48 Ab) and Hoechst. Httimmunohistochemistry was done as previously described.
Cell counting and lesion measurements
The number of huntingtin aggregates was determined bycounting EM48-positive objects. The striatal dysfunction wasevaluated by measuring the DARPP-32- and NeuN-depletedstriatal volume. The number of cells in the expHtt-infectedstriatum was determined by counting Hoechst-positive objectsand was expressed as a percentage of the number of cells countedin the contralateral normal htt-infected striatum. In all cases animage analysis software (Image Pro Plus 4.5.0.19, MediaCybernetics) was used. The aggregates were counted at amagnification of 10× on 25 μm thick sections separated by150 μm throughout the entire striatum. The total number ofaggregates per brain was obtained by multiplying the counts by 6.The area of the striatum showing a loss of DARPP-32 or of NeuNstaining was measured for each animal with an operator-independent process. The volume was then estimated with thefollowing formula: volume=d(a1+a2+a3…), where d is distancebetween serial sections (150 μm), and a1, a2, a3… are DARPP-32-or NeuN-depleted areas for individual serial sections. Determina-tion of the areas showing depletion in DARPP-32 or NeuNimmunoreactivity was performed using a 1.6× objective.

24 D. Charvin et al. / Neurobiology of Disease 29 (2008) 22–29
Data analyses
Data are expressed as mean±sem and were evaluated by aStudent’s t test. The significance level was set at P≤0.05. APearson analysis was performed to determine the coefficient ofcorrelation (R2) between the volume of DARPP32 lesion and thenumber of aggregates.
Results
Haloperidol decanoate treatment protects striatal neurons from thedysfunction induced by expHtt expression
To analyze the in vivo effect of a D2 antagonist treatment on HDneuropathology, we chose a rat model of HD based on lentiviral-mediated expression of expHtt in the striatum (de Almeida et al.,2002). In this model, lentiviral vectors (LV) overexpressingN-terminal fragments of expHtt (171 amino acids) with a patho-logical stretch of 82 glutamines (LV-htt-171-82Q) are injectedunilaterally into the rat striatum and progressively producecharacteristic features observed in HD patients, including neuronalinclusions and progressive striatal neuron dysfunction. Inclusionsfirst appear 1week after LV-htt-171-82Q injection and striatal neurondysfunction is observed from the fourth week after infection. By
Fig. 1. Haloperidol decanoate treatment decreases striatal neurons dysfunction,evaluated by the loss of DARPP-32 immunoreactivity at 8 weeks. While overexpr(left panels), a drastic loss of DARPP-32 immunoreactive neurons was observed in tpanel). Note that haloperidol decanoate treatment (Hal) strongly reduces this loss ofmagnification showing the DARPP-32 depleted region in the striata presented in squdepleted region in the htt171-82Q-expressing striata of vehicle- (Veh) or haloperidoHal: n=7. Statistical test: unpaired Student's t test; ∗Pb0.05. Values are expresse
contrast, overexpression of the normal form of the same htt fragment(htt-171-19Q) is devoid of striatal toxicity, and this treatment servesas a control. Thus, LV-htt-171-82Q was injected into the rightstriatum and htt-171-19Q into the left. To evaluate the therapeuticeffect of early D2 receptor blockade in HD, we administered a depotformulation of haloperidol, haloperidol decanoate, which allows alimited number of injections and a relatively stable D2 receptorsoccupancy over time (Nyberg et al., 1997). Haloperidol decanoate(one intramuscular, i.m., injection, 30 mg/kg, every 3 weeks) orvehicle (sesame oil, same protocol and volume) was injected in LV-htt171-19/82Q rats before the appearance of neuropathologicalhallmarks starting 1 day after lentiviral injection. The impact ofhaloperidol decanoate versus vehicle treatment was examined onstriatal dysfunction and inclusion formation.
We investigated the effect of haloperidol decanoate treatment onneuronal dysfunction, measured by the loss of DARPP-32 andNeuNimmunoreactivities at 8 weeks. DARPP-32 is expressed in 96% ofthe striatal medium-sized spiny neurons and is down-regulated inHD transgenic mice and in LV-based HD rat models (Bibb et al.,2000; Canals et al., 2004; de Almeida et al., 2002; Luthi-Carter et al.,2000). All striata overexpressing expHtt presented a clear loss ofDARPP-32 immunoreactivity (Figs. 1A, B). In contrast, no loss ofDARPP-32 staining was detected in striata overexpressing thenormal form of htt (Fig. 1A, left panels). Wemeasured the volume of
as revealed by DARPP-32 staining. (A) Striatal neurons dysfunction wasession of the normal form of htt (19Q) had no effect on DARPP-32 staininghe LV-htt171-82Q-infected striatum (82Q) of vehicle animals (Veh) (right upDARPP-32 immunoreactivity (right low panel). Scale bar, 500 μm. (B) Highares in A. Scale bar, 100 μm. (C) Quantification of the volume of DARPP-32l decanoate-treated (Hal) animals, 8 weeks after LV injection. In C, Veh: n=7;d as means±sem.

25D. Charvin et al. / Neurobiology of Disease 29 (2008) 22–29
the DARPP-32 depleted region in LV-htt171-82Q-expressingstriatum of rat treated either with haloperidol decanoate or vehicle.In control rats, chronic haloperidol treatment did not modifyDARPP-32 immunostaining. Of interest, the volume of the DARPP-32-depleted region was importantly reduced in rats treated withhaloperidol when compared to animals treated with vehicle(haloperidol, 1.4±0.2 mm3; vehicle, 2.0±0.2 mm3; Pb0.05) (Fig.1C). Thus, haloperidol decanoate treatment produced a 30%reduction in the volume of expHtt-related loss of DARPP-32.Similar results were obtained with the neuronal marker NeuN (Figs.2A, B). In rats treated with vehicle, LV-htt171-82Q infectionproduced a NeuN-depleted region of 1.8±0.2 mm3. This NeuN-depleted region was significantly reduced in rats treated withhaloperidol decanoate (1.1±0.2 mm3, P≤0.01) (Fig. 2B). Thus,haloperidol decanoate treatment reduced by 38% the extent ofexpHtt-induced loss of NeuN.
Fig. 2. Haloperidol decanoate treatment decreases striatal neurons dysfunc-tion, as revealed by NeuN staining. (A) Striatal neurons dysfunction wasevaluated by the loss of NeuN immunoreactivity at 8 weeks. As for DARPP-32 staining (Fig. 1), overexpression of the normal form of htt (19Q) had noeffect on NeuN staining (left panels) whereas expHtt (82Q) induced a drasticloss of NeuN immunoreactive neurons of vehicle animals (Veh) (right uppanel). Note that haloperidol decanoate treatment (Hal) strongly reduces thisloss of NeuN immunoreactivity (right low panel). (B) Quantification of thevolume of NeuN-depleted region in the htt171-82Q-expressing striata ofvehicle- (Veh) or haloperidol decanoate-treated (Hal) animals, 8 weeks afterLV injection. In B, Veh: n=7; Hal: n=7. Statistical test: unpaired Student'st test; ∗∗Pb0.01. Values are expressed as means±sem.
Based on these two different neuronal markers, our data clearlyshow that haloperidol decanoate treatment attenuates expHtt-related striatal neuron dysfunction.
Haloperidol decanoate treatment decreases expHtt aggregateformation
The appearance of intraneuronal inclusions, a hallmark of thepathology, was assessed in our model system. Inclusions can bedetected by immunofluorescence with the EM48 antibody, whichspecifically recognizes the aggregated form of htt (Fig. 3A). Asexpected, none of the striata infected with LV-htt171-19Q wasstained with the EM48 antibody (not shown). In contrast, LV-mediated expression of the htt171-82Q protein induced theformation of expHtt aggregates as early as 2 weeks after viralinjection. Inclusions accumulated and their number was furtherincreased 8 weeks after infection (Figs. 3A, B). For each expHtt-expressing animal, the total number of aggregates in the striatumwas quantified. D2 receptor blockade by haloperidol significantlydecreased expHtt aggregate formation in striatal neurons (Fig. 3A),what is consistent with our previous results obtained in vitro(Charvin et al., 2005). Haloperidol decanoate treatment decreasedthe number of expHtt-containing aggregates at 2 weeks by 42%(vehicle- versus haloperidol decanoate-treated rats, P=0.05). Adecrease in expHtt aggregates (−28%) was still found 8 weeks afterLV injection (vehicle- versus haloperidol decanoate-treated rats,P=0.001) (Fig. 3B). To assess whether the decreased number ofaggregates was attributable to a decreased number of cells, wequantified the percentage of cells in the expHtt-infected striatacompared to the contralateral normal htt-infected side. Thisquantification showed no difference between both groups of animals(haloperidol, 101±2%; vehicle, 103±3%; PN0.2) (Fig. 3C),revealing that the decreased number of aggregates does not reflecta decreased number of cells under haloperidol decanoate treatment.
Thus, altogether, these data demonstrate that chronic blockadeof D2 receptors by haloperidol decanoate produced a significantdecrease in aggregate formation over time.
Haloperidol decanoate treatment preferentially decreases somaticand neuritic expHtt aggregates formation
It was previously shown that, in htt171-82Q-infected ratstriatum, most of the inclusions are present in the nuclei of striatalneurons, with some aggregates observed in neuritic compartments(de Almeida et al., 2002). To distinguish these two types ofinclusions, a double staining of aggregates and nuclei wasperformed with the EM48 antibody and Hoechst, respectively.This double staining confirmed that aggregates can take the shapeof a dense staining in large spherical nuclear inclusions, or ofsmaller-sized spheres dotting the extra-nuclear compartments (i.e.neurites and soma) (Fig. 4A). Based on these characteristics, wequantified separately nuclear and extra-nuclear inclusions in LV-htt171-82Q-expressing striata of rats treated with haloperidoldecanoate (or vehicle). Interestingly, haloperidol decanoate treat-ment affected extra-nuclear inclusion formation preferentially,decreasing their number by 59% (extra-nuclear aggregates ofvehicle-treated rats versus extra-nuclear aggregates of haloperidoldecanoate-treated rats, P=0.018), while it had no or little effect onnuclear inclusion formation (nuclear inclusions of vehicle-treatedrats versus nuclear inclusions of haloperidol decanoate-treated rats,P=0.196) (Fig. 4B).
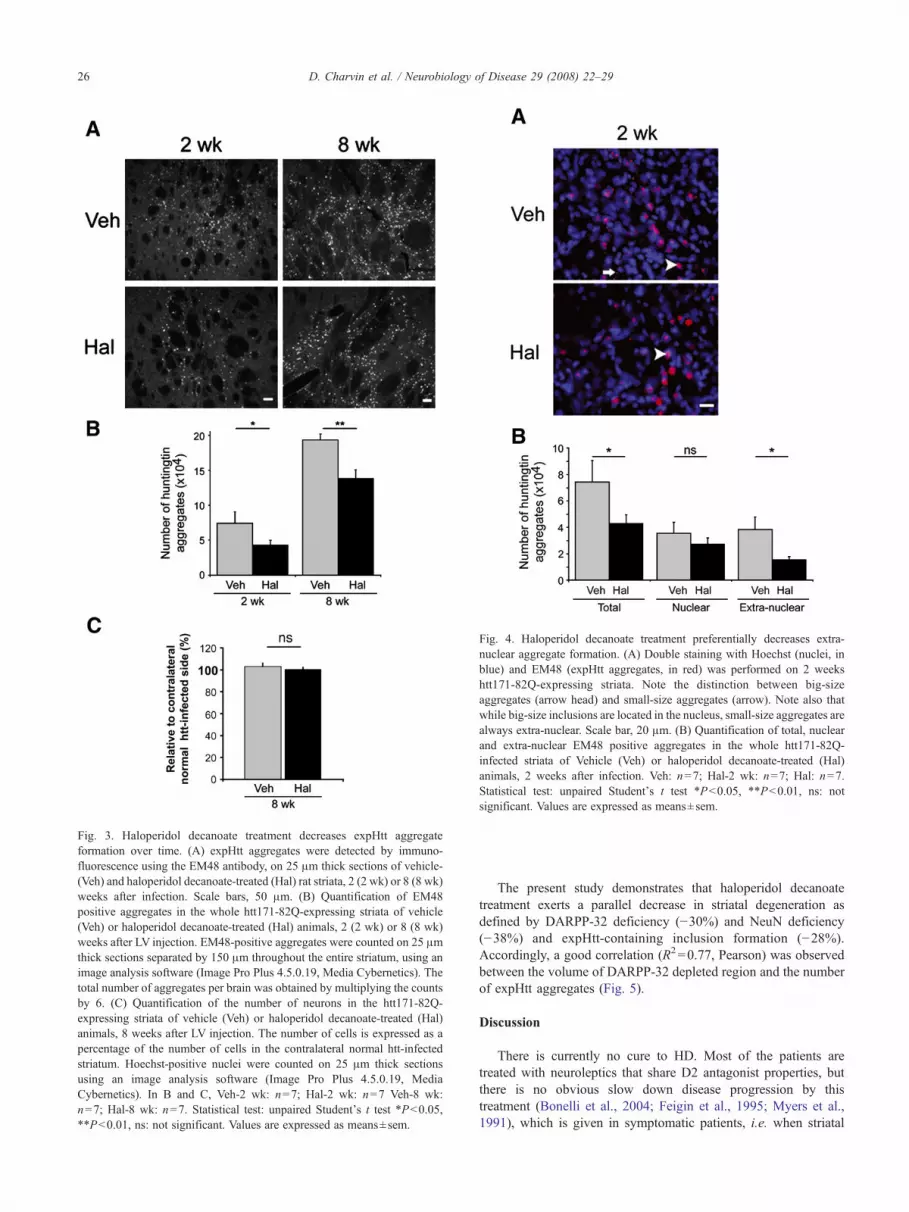
Fig. 4. Haloperidol decanoate treatment preferentially decreases extra-nuclear aggregate formation. (A) Double staining with Hoechst (nuclei, inblue) and EM48 (expHtt aggregates, in red) was performed on 2 weekshtt171-82Q-expressing striata. Note the distinction between big-sizeaggregates (arrow head) and small-size aggregates (arrow). Note also thatwhile big-size inclusions are located in the nucleus, small-size aggregates arealways extra-nuclear. Scale bar, 20 μm. (B) Quantification of total, nuclearand extra-nuclear EM48 positive aggregates in the whole htt171-82Q-infected striata of Vehicle (Veh) or haloperidol decanoate-treated (Hal)animals, 2 weeks after infection. Veh: n=7; Hal-2 wk: n=7; Hal: n=7.Statistical test: unpaired Student's t test ∗Pb0.05, ∗∗Pb0.01, ns: notsignificant. Values are expressed as means±sem.
Fig. 3. Haloperidol decanoate treatment decreases expHtt aggregateformation over time. (A) expHtt aggregates were detected by immuno-fluorescence using the EM48 antibody, on 25 μm thick sections of vehicle-(Veh) and haloperidol decanoate-treated (Hal) rat striata, 2 (2 wk) or 8 (8 wk)weeks after infection. Scale bars, 50 μm. (B) Quantification of EM48positive aggregates in the whole htt171-82Q-expressing striata of vehicle(Veh) or haloperidol decanoate-treated (Hal) animals, 2 (2 wk) or 8 (8 wk)weeks after LV injection. EM48-positive aggregates were counted on 25 μmthick sections separated by 150 μm throughout the entire striatum, using animage analysis software (Image Pro Plus 4.5.0.19, Media Cybernetics). Thetotal number of aggregates per brain was obtained by multiplying the countsby 6. (C) Quantification of the number of neurons in the htt171-82Q-expressing striata of vehicle (Veh) or haloperidol decanoate-treated (Hal)animals, 8 weeks after LV injection. The number of cells is expressed as apercentage of the number of cells in the contralateral normal htt-infectedstriatum. Hoechst-positive nuclei were counted on 25 μm thick sectionsusing an image analysis software (Image Pro Plus 4.5.0.19, MediaCybernetics). In B and C, Veh-2 wk: n=7; Hal-2 wk: n=7 Veh-8 wk:n=7; Hal-8 wk: n=7. Statistical test: unpaired Student's t test ∗Pb0.05,∗∗Pb0.01, ns: not significant. Values are expressed as means±sem.
26 D. Charvin et al. / Neurobiology of Disease 29 (2008) 22–29
The present study demonstrates that haloperidol decanoatetreatment exerts a parallel decrease in striatal degeneration asdefined by DARPP-32 deficiency (−30%) and NeuN deficiency(−38%) and expHtt-containing inclusion formation (−28%).Accordingly, a good correlation (R2=0.77, Pearson) was observedbetween the volume of DARPP-32 depleted region and the numberof expHtt aggregates (Fig. 5).
Discussion
There is currently no cure to HD. Most of the patients aretreated with neuroleptics that share D2 antagonist properties, butthere is no obvious slow down disease progression by thistreatment (Bonelli et al., 2004; Feigin et al., 1995; Myers et al.,1991), which is given in symptomatic patients, i.e. when striatal

Fig. 5. Haloperidol decanoate treatment exerts a parallel effect betweenneuronal dysfunction and expHtt aggregate formation. (A) DARPP-32 andaggregates immunostainings on adjacent LV-htt171-82Q-infected striatalsections from a vehicle- (Veh; upper panels) and haloperidol decanoate-treated (Hal, lower panels) animal, 8 weeks after LV infection. Note that thehaloperidol decanoate-treated rat presents a smaller DARPP-32 depletedregion and less aggregates than the vehicle-treated rats. Scale bar forDARPP-32 stained sections, 200 μm. Scale bar for aggregates stainedsections, 20 μm. (B) Correlation between the volume of DARPP-32 depletedregion and the number of expHtt aggregates for each animal. Blackdiamonds: haloperidol decanoate-treated animals; gray diamonds: vehicle-treated rats. Statistical test: Pearson coefficient of correlation: R2=0.77.
27D. Charvin et al. / Neurobiology of Disease 29 (2008) 22–29
degeneration is already in an advanced stage (Reiner et al., 1988;Rosas et al., 2003). Our in vivo study shows that treatment withthe D2 antagonist haloperidol decanoate beginning at an earlystage significantly reduces both expHtt-induced neuronal dys-function and aggregate formation in the striatum of rats infectedwith a lentiviral vector overexpressing N-terminal fragment ofexpHtt. Based on striatal neuron dysfunction and expHttaggregate formation, the present data provide the first in vivoevidence that D2 receptor blockade can slow down striataldysfunction in HD.
Previous reports demonstrated that striatal neuron dysfunctionis more important in HD pathophysiology than neurodegeneration.Brain imaging studies indicated that neuronal alterations arepresent before the appearance of clinical symptoms (Andrewset al., 1999; Antonini et al., 1996; Aylward et al., 1994; Aylwardet al., 1996; Aylward et al., 2000; Aylward et al., 2004; Graftonet al., 1990; Mazziotta et al., 1987). Decrease in DARPP-32expression can be observed in early symptomatic transgenic micethat do not present any obvious cell loss (Bibb et al., 2000; Luthi-
Carter et al., 2000; Menalled et al., 2000; van Dellen et al., 2000).We used DARPP-32 and NeuN labelings as markers of neuronaldysfunction as previously reported (Colin et al., 2005; de Almeidaet al., 2002; Pardo et al., 2006; Perrin et al., 2007; Regulier et al.,2003) and found a significant protection against striatal neuronsdysfunction with the haloperidol treatment. While the role of D2receptors in expHtt-induced striatal vulnerability that we pre-viously described in vitro (Charvin et al., 2005) only implicated D2receptors expressed by these neurons, in vivo the scenario isdifferent since D2 receptors are also present in cortico-striatalterminals. Stimulation of these pre-synaptic D2 receptors isassociated with reduced glutamate release (Centonze et al., 2004)and increased excitotoxicity should be expected upon chronichaloperidol treatment. Instead, long-term treatment with haloper-idol does not modify glutamate concentration within the striatum(Bustillo et al., 2006). Our data extend these observations andclearly show a neuroprotective effect of chronic haloperidoltreatment in an in vivo HD model.
We found a good correlation between the volume of NeuN andDARPP-32 depleted region and the number of expHtt aggregates,with a preferential decrease of extra-nuclear aggregate formation,i.e. somatic and neuritic aggregates. There is still a strong debateabout the role of aggregates in neurotoxicity. Aggregates of mutatedhuntingtin are observed in HD post-mortem patients’ brains as wellas in different animal models of HD, where they typically precedeonset of symptoms (Davies et al., 1997; Gutekunst et al., 1999). InHD patients, expHtt inclusions are found in affected areas of thebrain, such as cortex and striatum, but not in unaffected areas, suchas cerebellum, and they are almost exclusively found in neurons, andnot in unaffected glial cells (Hoffner et al., 2005). Furthermore,formation of neuritic aggregates has been shown to be highlycorrelated to the clinical course of the disease (Gutekunst et al.,1999; Li et al., 1999). Thus, aggregate formation was firstconsidered as a pathological hallmark of the disease. Experimentsfrom animal models of HD remain, however, puzzling. For examplethe transgenic R6/2 mouse model, where a large number ofinclusions are found in several regions, shows no striatal lesionbut a reduction of striatal volume at the time when the mice die(between 10 and 13 weeks old) (Mangiarini et al., 1996). In a mousemodel of HD that expresses an N-terminal human fragment of Httwith an expanded polyglutamine repeat (120) under the control ofthe endogenous human promoter (shortstop), frequent and wide-spread nuclear htt inclusions occur early, with however, no clinicalevidence of neuronal dysfunction (Slow et al., 2005). While sometreatments that improve motor performance and delay theneuropathological sequelae in HD mouse models do not affectaggregates formation (Ferrante et al., 2003; Gardian et al., 2005;Hockly et al., 2003; Andreassen et al., 2001), other treatments,including trehalose, that reduce striatal atrophy and degeneration,improve motor symptoms and increase survival and producedecreased aggregates (Dedeoglu et al., 2003; Keene et al., 2002;Nguyen et al., 2005; Sanchez et al., 2003; Tanaka et al., 2004). Fromin vitro data, Arrasate et al. (2004) provided evidence about aneuroprotective role of aggregates, at least in early stages, sincestriatal neurons that contained aggregates had a decreasedcumulative risk of death when compared to diffused expHtt. Bycontrast, Li et al. (1999) clearly established that extra-nuclearaggregates were toxic for striatal cells in culture, and neuriticaggregates were associated with axonal degeneration as an earlypathological event. This was confirmed in a Drosophila model ofHD (Lee et al., 2004). Thus, whether the potential benefit of haldol

28 D. Charvin et al. / Neurobiology of Disease 29 (2008) 22–29
decanoate treatment on striatal dysfunction is associated with thedecrease in neuritic aggregates remains to be determined.
The current treatment of HD patients with neuroleptics that shareD2 receptor antagonist properties, such as haloperidol, is given afterthe onset of symptoms. Brain imaging studies of HD patients clearlyindicate that striatal dysfunctions occur before the appearance ofclinical symptoms (Andrews et al., 1999; Antonini et al., 1996;Aylward et al., 2004; Grafton et al., 1990; Mazziotta et al., 1987).Thus neuroleptic treatments may not be effective because thetemporal opportunity for neuronal rescue has passed. The presentstudy indicates that the blockade of the D2 receptor signaling, whendone early, may delay the onset of HD neuropathology, as analyzedherein at a molecular level, and thereby represents an interestingtherapeutic option, isolated or in association with other potentialneuroprotective therapy. As patients with chronic neurolepticstreatment may experience numerous side effects, further elucidationof intracellular events mediated by D2 receptors in expHtt-inducedstriatal toxicity could help to develop better tolerated therapeuticstrategies, an issue under investigation in our laboratory.
Acknowledgments
This work was supported by Centre National de la RechercheScientifique (CNRS) and Université Pierre et Marie Curie for JC,by CEA for ND and by Ecole Polytechnique Fédérale de Lausanne(EPFL) for RL-C. Delphine Charvin was supported by fellowshipsof Ministère de la Recherche et de la Technologie and Fondationpour la Recherche Médicale (FRM). Valérie Perrin was supportedby a fellowship of Association Française contre les Myopathies(AFM). The authors thank Philippe Colin and Noelle Dufour fortheir excellent technical assistance.
References
Andreassen, O.A., et al., 2001. Dichloroacetate exerts therapeutic effects intransgenic mouse models of Huntington’s disease. Ann. Neurol. 50,112–117.
Andrews, T.C., et al., 1999. Huntington’s disease progression. PET andclinical observations. Brain 122 (Pt 12), 2353–2363.
Antonini, A., et al., 1996. Striatal glucose metabolism and dopamine D2receptor binding in asymptomatic gene carriers and patients withHuntington’s disease. Brain 119 (Pt 6), 2085–2095.
Arrasate, M., et al., 2004. Inclusion body formation reduces levels of mutanthuntingtin and the risk of neuronal death. Nature 431, 805–810.
Aylward, E.H., et al., 1994. Reduced basal ganglia volume associated withthe gene for Huntington’s disease in asymptomatic at-risk persons.Neurology 44, 823–828.
Aylward, E.H., et al., 1996. Basal ganglia volume and proximity to onset inpresymptomatic Huntington disease. Arch. Neurol. 53, 1293–1296.
Aylward, E.H., et al., 2000. Rate of caudate atrophy in presymptomaticand symptomatic stages of Huntington’s disease. Mov. Disord. 15,552–560.
Aylward, E.H., et al., 2004. Onset and rate of striatal atrophy in preclinicalHuntington disease. Neurology 63, 66–72.
Bibb, J.A., et al., 2000. Severe deficiencies in dopamine signaling inpresymptomatic Huntington’s disease mice. Proc. Natl. Acad. Sci.U. S. A. 97, 6809–6814.
Bonelli, R.M., et al., 2004. Huntington’s disease: present treatmentsand future therapeutic modalities. Int. Clin. Psychopharmacol. 19,51–62.
Bowyer, J.F., et al., 1996. Parenterally administered 3-nitropropionic acidand amphetamine can combine to produce damage to terminals and cellbodies in the striatum. Brain Res. 712, 221–229.
Bustillo, J., et al., 2006. Long-term treatment of rats with haloperidol: lack ofan effect on brain N-acetyl aspartate levels. Neuropsychopharmacology31, 751–756.
Canals, J.M., et al., 2004. Brain-derived neurotrophic factor regulates theonset and severity of motor dysfunction associated with enkephali-nergic neuronal degeneration in Huntington’s disease. J. Neurosci.24, 7727–7739.
Cass, W.A., 1997. Decreases in evoked overflow of dopamine in rat striatumafter neurotoxic doses of methamphetamine. J. Pharmacol. Exp. Ther.280, 105–113.
Centonze, D., et al., 2004. Differential contribution of dopamine D2S andD2L receptors in the modulation of glutamate and GABA transmissionin the striatum. Neuroscience 129, 157–166.
Charvin, D., et al., 2005. Unraveling a role for dopamine in Huntington’sdisease: the dual role of reactive oxygen species and D2 receptorstimulation. Proc. Natl. Acad. Sci. U. S. A. 102, 12218–12223.
Colin, E., et al., 2005. Akt is altered in an animal model of Huntington’sdisease and in patients. Eur. J. Neurosci. 21, 1478–1488.
Cyr, M., et al., 2003. Sustained elevation of extracellular dopamine causesmotor dysfunction and selective degeneration of striatal GABAergicneurons. Proc. Natl. Acad. Sci. U. S. A. 100, 11035–11040.
Cyr, M., et al., 2006. Dopamine enhances motor and neuropathologicalconsequences of polyglutamine expanded huntingtin. FASEB J. 20,2541–2543.
Davies, S.W., et al., 1997. Formation of neuronal intranuclear inclusionsunderlies the neurological dysfunction in mice transgenic for the HDmutation. Cell 90, 537–548.
de Almeida, L.P., et al., 2002. Lentiviral-mediated delivery of mutanthuntingtin in the striatum of rats induces a selective neuropathologymodulated by polyglutamine repeat size, huntingtin expression levels,and protein length. J. Neurosci. 22, 3473–3483.
Dedeoglu, A., et al., 2003. Creatine therapy provides neuroprotection afteronset of clinical symptoms in Huntington’s disease transgenic mice. J.Neurochem. 85, 1359–1367.
DiFiglia, M., et al., 1997. Aggregation of huntingtin in neuronalintranuclear inclusions and dystrophic neurites in brain. Science 277,1990–1993.
Feigin, A., et al., 1995. Functional decline in Huntington’s disease. Mov.Disord. 10, 211–214.
Ferrante, R.J., et al., 2003. Histone deacetylase inhibition by sodiumbutyrate chemoterapy ameliorates the neurodegenerative phenotype inHuntington’s Disease mice. J. Neurosci. 23, 9418–9427.
Gardian, G., et al., 2005. Neuroprotective effects of phenylbutyrate in theN171-82Q transgenic mouse model of Huntington’s disease. J. Biol.Chem. 280, 556–563.
Grafton, S.T., et al., 1990. A comparison of neurological, metabolic,structural, and genetic evaluations in persons at risk for Huntington’sdisease. Ann. Neurol. 28, 614–621.
Group, T.H.s.D.C.R., 1993. A novel gene containing a trinucleotide repeatthat is expanded and unstable on Huntington’s disease chromosomes.Cell 72, 971–983.
Gutekunst, C.A., et al., 1999. Nuclear and neuropil aggregates inHuntington’s disease: relationship to neuropathology. J. Neurosci. 19,2522–2534.
Hockly, E., et al., 2003. Suberoylanilide hydroxamic acid, a histonedeacetylase inhibitor, ameliorates motor deficits in a mouse model ofHuntington’s disease. Proc. Natl. Acad. Sci. U. S. A. 100, 2041–2046.
Hoffner, G., et al., 2005. Purification of neuronal inclusions of patients withHuntington’s disease reveals a broad range of N-terminal fragments ofexpanded huntingtin and insoluble polymers. J. Neurochem. 95,125–136.
Keene, C.D., et al., 2002. Tauroursodeoxycholic acid, a bile acid, isneuroprotective in a transgenic animal model of Huntington’s disease.Proc. Natl. Acad. Sci. U. S. A. 99, 10671–10676.
Lee, W.C., et al., 2004. Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila modelof Huntington’s disease. Proc. Natl. Acad. Sci. U. S. A. 101, 3224–3229.

29D. Charvin et al. / Neurobiology of Disease 29 (2008) 22–29
Li, H., et al., 1999. Ultrastructural localization and progressive formation ofneuropil aggregates in Huntington’s disease transgenic mice. Hum. Mol.Genet. 8, 1227–1236.
Luo, Y., et al., 1998. Dopamine induces apoptosis through an oxidation-involved SAPK/JNK activation pathway. J. Biol. Chem. 273, 3756–3764.
Luthi-Carter, R., et al., 2000. Decreased expression of striatal signalinggenes in a mouse model of Huntington’s disease. Hum. Mol. Genet. 9,1259–1271.
Mangiarini, L., et al., 1996. Exon 1 of the HD gene with an expanded CAGrepeat is sufficient to cause a progressive neurological phenotype intransgenic mice. Cell 87, 493–506.
Mazziotta, J.C., et al., 1987. Reduced cerebral glucose metabolism inasymptomatic subjects at risk for Huntington’s disease. N. Engl. J. Med.316, 357–362.
Menalled, L., et al., 2000. Decrease in striatal enkephalin mRNA in mousemodels of Huntington’s disease. Exp. Neurol. 162, 328–342.
Myers, R.H., et al., 1991. Factors associated with slow progression inHuntington’s disease. Arch. Neurol. 48, 800–804.
Nguyen, T., et al., 2005. Clioquinol down-regulates mutant huntingtinexpression in vitro and mitigates pathology in a Huntington’s diseasemouse model. Proc. Natl. Acad. Sci. U. S. A. 102, 11840–11845.
Nyberg, S., et al., 1997. Delayed normalization of central D2 dopaminereceptor availability after discontinuation of haloperidol decanoate.Preliminary findings. Arch. Gen. Psychiatry 54, 953–958.
Pardo, R., et al., 2006. Inhibition of calcineurin by FK506 protects againstpolyglutamine-huntingtin toxicity through an increase of huntingtinphosphorylation at S421. J. Neurosci. 26, 1635–1645.
Perrin, V., et al., 2007. Neuroprotection by Hsp104 and Hsp27 in lentiviral-based rat models of Huntington’s disease. Mol. Ther. 15 (5), 903–911.
Quinn, N., Schrag, A., 1998. Huntington’s disease and other choreas. J.Neurol. 245, 709–716.
Regulier, E., et al., 2003. Early and reversible neuropathology induced bytetracycline-regulated lentiviral overexpression of mutant huntingtin inrat striatum. Hum. Mol. Genet. 12, 2827–2836.
Reiner, A., et al., 1988. Differential loss of striatal projection neurons inHuntington disease. Proc. Natl. Acad. Sci. U. S. A. 85, 5733–5737.
Reynolds, D.S., et al., 1998. Dopamine modulates the susceptibility ofstriatal neurons to 3-nitropropionic acid in the rat model of Huntington’sdisease. J. Neurosci. 18, 10116–10127.
Ricaurte, G.A., et al., 1982. Dopamine nerve terminal degenerationproduced by high doses of methylamphetamine in the rat brain. BrainRes. 235, 93–103.
Rosas, H.D., et al., 2003. Evidence for more widespread cerebral pathologyin early HD: an MRI-based morphometric analysis. Neurology 60,1615–1620.
Sanchez, I., et al., 2003. Pivotal role of oligomerization in expandedpolyglutamine neurodegenerative disorders. Nature 421, 373–379.
Slow, E.J., et al., 2005. Absence of behavioral abnormalities andneurodegeneration in vivo despite widespread neuronal huntingtininclusions. Proc. Natl. Acad. Sci. U. S. A. 102, 11402–11407.
Tanaka, M., et al., 2004. Trehalose alleviates polyglutamine-mediatedpathology in a mouse model of Huntington disease. Nat. Med. 10,148–154.
van Dellen, A., et al., 2000. N-Acetylaspartate and DARPP-32 levelsdecrease in the corpus striatum of Huntington’s disease mice.NeuroReport 11, 3751–3757.
Vonsattel, J.P., DiFiglia, M., 1998. Huntington disease. J. Neuropathol. Exp.Neurol. 57, 369–384.
Vonsattel, J.P., et al., 1985. Neuropathological classification of Huntington’sdisease. J. Neuropathol. Exp. Neurol. 44, 559–577.
Weiner, D.M., et al., 1991. D1 and D2 dopamine receptor mRNA in ratbrain. Proc. Natl. Acad. Sci. U. S. A. 88, 1859–1863.
Copyright © 2022 FDOKUMEN




















