
Liquid overlaying improves somatic embryogenesis in Catharanthus roseus
Upload
independentCategory
view
4download
0
Neurobiology of Disease 36 (2009) 413–420
Contents lists available at ScienceDirect
Neurobiology of Disease
j ourna l homepage: www.e lsev ie r.com/ locate /ynbd i
Y-27632 improves rotarod performance and reduces huntingtin levels in R6/2 mice
Mei Li a,b, Yong Huang c, Aye Aye K. Ma a,b, Emil Lin c, Marc I. Diamond a,b,⁎a Department of Neurology, University of California, San Francisco, CA 94143, USAb Department of Cellular and Molecular Pharmacology, University of California, San Francisco, CA 94143, USAc Department of Pharmaceutical Chemistry, University of California, San Francisco, CA 94143, USA
⁎ Corresponding author. GH-S572B, 600 16th Street,USA. Fax: +1 415 514 4112.
E-mail address: [email protected] (M.I. DiamoAvailable online on ScienceDirect (www.scienced
0969-9961/$ – see front matter © 2009 Published by Edoi:10.1016/j.nbd.2009.06.011
a b s t r a c t
a r t i c l e i n f oArticle history:Received 1 May 2009Revised 24 June 2009Accepted 28 June 2009Available online 8 July 2009
Huntington disease (HD) is a devastating, untreatable, dominantly inherited neurodegenerative disease. It iscaused by an expanded CAG codon repeat that leads to an elongated polyglutamine tract in the N-terminus ofthe huntingtin (Htt) protein. Few mechanism-based therapeutic leads have been developed. Y-27632, aninhibitor of the Rho-associated kinase ROCK, reduces Htt aggregation in cultured cells and Htt-inducedneurodegeneration in Drosophila, but its effect in mice is unknown. We determined that Y-27632 isbioavailable in brain, with a half-life of 60–90 min. We then initiated a trial in R6/2 mice, which express Httexon 1, administering 100 mg/kg/day of Y-27632 in drinking water. We did not observe a significant effect onbrain weight, inclusion number or size, striatal medium spiny neuron number, clasping behavior, or lifespan.However, Y-27632 treatment improved rotarod performance significantly, and also reduced soluble brain Httlevels. The ROCK signaling pathway thus remains a promising therapeutic target for HD, and more potentinhibitors may prove useful.
© 2009 Published by Elsevier Inc.
Introduction
Huntington disease (HD) is an untreatable, devastating autosomaldominant neurodegenerative disease characterized by psychiatricdisturbance, motor impairment, and dementia. CAG codon expansionin the huntingtin gene past 37 repeats leads to an expanded,pathogenic polyglutamine tract within the huntingtin (Htt) protein(1993). This expansion destabilizes Htt, leading to misfolding,aggregation, inclusion formation, and toxicity (Shao and Diamond,2007). Emerging evidence suggests that small oligomeric aggregatesmay constitute the pathologic species in polyglutamine diseases (Li etal., 2007; Takahashi et al., 2007a; Takahashi et al., 2007b; Pattison andRobbins, 2008). Multiple studies have identified aggregation inhibi-tors, and several of these compounds have been tested in mousemodels with modest effects on motor performance (Apostol et al.,2003; Chopra et al., 2007; Kazantsev et al., 2002; Tanaka et al., 2004;Zhang et al., 2005).
We previously established a cellular polyglutamine proteinaggregation assay based on fluorescence resonance energy transfer(FRET), which was used to identify Y-27632, an inhibitor of the Rho-associated kinase ROCK. Y-27632 reduced Htt aggregation ∼15% incells, and inhibited photoreceptor degeneration in a Drosophilamodelof HD based on neuronal expression of Htt exon 1 (Pollitt et al., 2003).
San Francisco, CA 94143-2280,
nd).irect.com).
lsevier Inc.
We have observed that both ROCK and a related kinase PRK-2 areinhibited by Y-27632 to reduce polyglutamine aggregation, and thesekinases each contribute to the effects of Y-27632 in cells (Shao et al.,2008a). In studies of the downstreammolecular mechanism bywhichY-27632 reduces intracellular aggregation, we have identified profilin,an Htt interacting protein (Goehler et al., 2004), as a novel target ofROCK (Shao et al., 2008b). Profilin potently inhibits Htt aggregation incells (Shao et al., 2008b), and a Drosophila homologue has beenobserved to suppress polyglutamine-mediated toxicity (Burnett et al.,2008). Profilin is phosphorylated at Ser-137 by ROCK, which blocks itsability to directly bind the Htt peptide. Thus, ROCK inhibitionpromotes dephosphorylation at Ser-137 and enables profilin's protec-tive effects (Shao et al., 2008b). There is no evidence that PRK-2directly phosphorylates profilin. Importantly, knockdown of profilincompletely blocks the anti-aggregation effects of Y-27632 in cells,suggesting that ROCK signaling through profilin is a key signalingpathway in the regulation of aggregation (Shao et al., 2008b). Givenour emerging knowledge about its molecular mechanisms in cells, andits effects in Drosophila, there is a strong rationale to test Y-27632 in amouse model of HD.
R6/2 mice express the Htt exon 1 peptide from the huntingtinpromoter (Mangiarini et al., 1996), and have been widely used inpreclinical trials for HD (Chen et al., 2000; Ferrante et al., 2002; Hocklyet al., 2003a; Ferrante et al., 2003; Fox et al., 2004; Ferrante et al.,2004; Chopra et al., 2007; Bates and Hockly, 2003). Thesemice exhibita progressive neurologic phenotype that leads to early death,generally by 12–16 weeks. This is associated with early andprogressive accumulation of the Htt peptide in intracellular inclusions

414 M. Li et al. / Neurobiology of Disease 36 (2009) 413–420
(Davies et al., 1997; Scherzinger et al., 1997). We used this model totest the efficacy of orally administered Y-27632.
Materials and methods
Y-27632 was a generous gift from Mitsubishi Pharma, Japan.
AntibodiesMouse anti-human huntingtin N-terminal antibody clone EM48,
mouse anti-expanded polyglutamine antibody clone 1C2, mouse anti-human and mouse full length huntingtin antibody MAB2166 andrabbit anti-dopamine and cAMP regulated phosphoprotein of 32 kD(DARPP32) were purchased from Chemicon International Inc.; mouseanti-glial fibrillary acidic protein (GFAP) was kindly shared by ZhaolinHua, M.D., Ph.D. and Robert Edwards, M.D. at UCSF. Rabbit anti-phospho-profilin 1 at serine 137 was developed by our laboratory(Shao et al., 2008b).
Y-27632 detection in vivo
Frozen brain specimenswere pulverized, and then homogenized inH2O. Acetonitrile was added to precipitate protein with chloroquine(100 ng/ml) as an internal standard (IS), and Y-27632 concentrationin the supernatantwasmeasured byHPLC-tandemmass spectrometry(LC-MS-MS). To create reference standards, 14 known concentrationsof Y-27632 (1.7–1700 ng/ml) were split into vehicle-treated mouseblood and brain specimens and carried through the extraction/purification procedure. The LC/MS/MS system consisted of ShimadzuLC-10 AD pumps, a Waters intelligent Sample Processor 717 Plusautosampler, and a Micromass Quattro LC Ultima triple quadrupoletandem mass spectrometer. The mass spectrometer was set for elec-trospray ionization in positive ion mode. Y-27632 and IS were de-tected with multiple reaction monitoring (MRM) at 248.3N93.3 m/zand 320.9N247.6 m/z, respectively. The sample cone voltage andcollision energy for both Y-27632 and IS were set at 30 eV and 25 eV,respectively. Y-27632 and IS were separated by silica column(50 mm×4.6 mm) with mobile phase composed of 90% acetonitrile,0.06% TFA and 2.5 mM ammonium formate. The flow rate was 1.8 ml/min and 1/10 was split into the mass detector.
Mouse husbandry
R6/2 mice in the B6/CBA background were backcrossed withC57B/6 females (Jackson Laboratory, Bar Harbor, ME) for more thanseven generations. Genotyping and CAG repeat numbers weredetected from tail DNA by PCR. Mice were maintained in a barrieranimal facility at the University of California San Francisco, housed fiveper cage with food and water available ad libitum, and kept on a 12 hlight/dark cycle. All studies and procedures were done under theapproval of the Institutional Animal Care and Use Committees inaccordance with the National Institutes of Health Guide for the Careand Use of Laboratory Animals.
Therapeutic trials
For analysis of rotarod performance, 16 female R6/2 animals wereused. These consisted of 7 littermate pairs, and two additionalanimals. Littermates were evenly divided between the two groups.For the analysis of Y-27632 on lifespan, 17 female R6/2 animals wereused. These consisted of 8 littermates evenly divided betweentreatment and control groups, and an additional animal was studiedin the treatment group. For lifespan studies, animals were followed inaccordance with IACUC approval until they lost N30% of body weight,or exhibited a moribund appearance (based on poor movement orgrooming) at which point they were euthanized, or were found to
have spontaneously died, which frequently occurred without the firsttwo criteria being met.
Rotarod testing
We performed rotarod evaluations using a rotarod apparatus(Columbus Instruments. Columbus, OH) as described previously (Li etal., 2007). We carried out four rotarod tests per session, allowing atleast 15 min of rest for the mice between each test. The rotarodaccelerated from 4 to 40 rpm over 15 s and was maintained at 40 rpmthereafter. The lowest value from all four tests was discarded. Theremaining three values were averaged, and the data were analyzed bythe student's t-test, assuming equal variance between the two groups.
Histology and cell counts
At age 13.5 weeks, mice were sacrificed and brains were cut insagittal midline sections. The left side of the brain was freshly frozenby ethanol-dry ice for biochemical studies and right side of the brainwas embedded in OCT compound and freshly frozen by ethanol-dryice for histological studies. 12 μm serial coronal sections cut throughthe whole striatum were collected and fixed with 4% paraformalde-hyde. Every 8th slice was immunostained with DARPP32 and GFAPantibodies and DAPI. Adjacent slices were stained with hematoxylin/eosin. Cells in the striatumwere counted using a previously publishedmethod (Chopra et al., 2007), with minor modifications. 36–60 serialadjacent images were taken from one large image of a whole sliceusing a Nikon 6 dimensional microscope (Nikon) at the Nikon ImagingCenter at UCSF (http://nic.ucsf.edu/6D.html). A 10× objective andautomated program NIS-Elements Advanced Research 3.0 (http://www.nis-elements.com/) were used. DARPP32 positive cells, GFAPpositive cells or DAPI positive cells were counted by analyzing thelarge images with Image J (NIH, http://rsbweb.nih.gov/ij/). The areaof the striatum at the level of crossing of the anterior commissure wasalso measured by Image J. Statistical analysis was performed using astudent's t-test.
Immunohistochemistry
Brain, pancreas and muscle embedded in OCT compound were cuton a cryostat (Leica 1850). 12 μm sagittal brain sections cut from themidline, and coronal sections cut from half of the brain were air-driedfor 20 min. Dried sections were fixed with 4% paraformaldehyde for10 min at 4 °C, washed with 0.05% Tween-20 in phosphate-bufferedsaline, and then incubated with 5% goat serum and 0.1% Triton X-100in phosphate-buffered saline for 30 min at room temperaturefollowed by overnight primary antibody incubation at 4 °C (mouseanti-huntingtin N-terminal antibody EM48 (1:400) and rabbit anti-phospho profilin 1 at serine137(1:1000; n=12 each in Y-27632 andvehicle-treated groups), or rabbit anti-DARPP32(1:500) and mouseanti-GFAP(1:250; n=6 each in Y-27632 and vehicle-treated groups)),and washing with 0.05% Tween-20 in phosphate-buffered saline. Goatanti-mouse IgG conjugated with Alexa 546 and goat anti-rabbit IgGconjugated with Alexa 488 (1:4000; Molecular Probes) were appliedto the sections for 2 h at room temperature in the dark. The sectionswere then washed for 1 h and counterstained with 4′6′ diamidino-2-phenylindole (DAPI) (50 ng/ml) for 30 s, washed, and mounted withProLong Gold anti-fade reagent (Molecular Probes). Fluorescenceimages were collected with a Nikon spectral laser confocal microscopeC1s1 with excitation wavelength at 405 nm, 488 nm and 561 nm anddetection at 435–450 nm, 515–530 nm, and 605–675 nm.
Immunoprecipitation
50 mg of brain powder was dounce-homogenized with 25 strokeson ice in 500 μl RIPA buffer and centrifuged 15,000×g for 30 min at

Fig.1. Evaluation of Y-27632 clearance in serum and brain.Wild-typemicewere given an intraperitoneal dose of 10mg/kg Y-27632. Two animals were sacrificed at the indicated timeintervals, and serum (A) and brain tissue (B) was harvested for mass spectrometry to determine compound levels. Error bars represent the range. Absolute concentrations weredetermined by using control tissue extracts spiked with known amounts of compound.
415M. Li et al. / Neurobiology of Disease 36 (2009) 413–420
4 °C. 2 μl of 1C2 antibody (Chemicon) was covalently conjugated to a20 μl bead volume of anti-mouse IgG beads (Sigma) by DSS (Pierce) aspreviously described (Li et al., 2007). Beads were washed thoroughlywith RIPA buffer. 200 μl supernatant extracted with the above RIPAbuffer was added with 1C2-mouse IgG beads overnight at 4°Cfollowed by at least four washes in RIPA buffer. Samples were boiledfor 5 min in 1% SDS sample buffer prior to SDS-PAGE andWestern blotanalyses. Blots were then probed with 1C2 (1:2500 dilution), washedwith TBS and 0.5% Tween-20, and then incubated with horseradishperoxidase-conjugated mouse True Blot (1:5000 dilution,eBioscience) that does not recognize denatured rabbit IgG heavy orlight chain. Horseradish peroxidase signal was detected by ECL-Plus(Amersham Biosciences).
Quantitative PCR
RNA was prepared from whole brain by using the RNeasy minikit (Qiagen) according to the manufacturer's protocol. Real time PCRwas performed as described previously (Wang et al., 2006), exceptthe internal control was actin instead of Rpl19. R6/2 transgeneprimers were: TCA GGT TCT GCT TTT ACC TGC (forward) and CTTCAT CAG CTT TTC CAG GG (reverse). Relative Htt exon 1 transcriptwas measured by [sample(Ct Htt exon1−Ct actin)−minimumsample(Ct Htt exon1−Ct actin)]2.
Results
Y-27632 bioavailability
We first determined whether Y-27632 would penetrate the bloodbrain barrier. We administered 10 mg/kg as a single intraperitonealdose to a group of 14 mice and sacrificed two of them at 15 min,30 min, 1.5 h, 6 h, 8 h, 12 h and 24 h after injection. We then measuredY-27632 serum and brain concentrations by HPLC-tandem massspectrometry. Peak serum concentration was 3–3.5 μM and brainconcentrationwas 0.3–0.65 μM 15min after injection (Fig. 1). Half-lifein each tissue was ∼60–90 min and the brain/serum ratio was ∼1/6
Table 1Concentrations in serum and brain after oral administration of Y-27632.
Sample no. Serum conc. (nM) Brain conc. (nM) Serum/brain ratio
1 59 6.9 8.72 91 17.6 5.23 298 47.5 6.34 393 20.2 19.55 51 10.3 4.9
Five animals were administered 50 mg/kg/day of Y-27632 orally for one week. Animalswere then sacrificed, and serum and brain levels were determined by liquidchromatography–mass spectrometry.
(Fig. 1). These studies indicated that at this dose Y-27632 readilypenetrates the blood brain barrier, although with a fairly short half-life. Peak CNS concentrationswere slightly less than desired to achieveoptimal ROCK inhibition. Next, we tested for oral availability, firstconfirming the stability of Y-27632 in drinking water (Supp. Fig. 1).We treated a cohort of 5 wild-type mice with 50 mg/kg/day Y-27632in drinking water for 7 days and determined concentrations in serumand brain (Table 1). Finally, we used dose escalation to determine thehighest amount tolerated.We administered Y-27632 in drinkingwaterto a cohort of 5 of wild-type mice beginning at 75 mg/kg/day andgradually increasing by 25 mg/kg/day every 3–4 days. The micetolerated up to 250 mg/kg/day, however we were concerned aboutusing 250 mg/kg/day for an extended period due to limitingcompound and possible toxicity. Thus, we decided to use 100 mg/kg/day administered orally for an efficacy trial.
Y-27632 improves rotarod function
R6/2 mice are very well characterized, and determinants ofexperimental variability include littermate status, gender, weight,and CAG repeat length (Hockly et al., 2002; Hockly et al., 2003b). Toreduce variationwe only used female mice, controlling for initial bodyweight and littermate status, and controlling for CAG repeat numberby PCR (all animals contained 110 to 120 CAG repeats within the Httexon 1 transgene). For the rotarod trial, 16 identically aged R6/2females, including 7 littermate pairs, were divided into two groups. 8R6/2 females received 100 mg/kg/day Y-27632 in drinking water. Thesecond group received vehicle control (water). We administered Y-27632 beginning at age 4 weeks. After an initial drop in the weight ofthe treated animals, we observed no significant differences betweenthe two groups over the course of the study (Fig. 2A). No significantchanges were observed in clasping responses, a pathological limbreflex observed in these animals (Fig. 2B). 3 weeks after beginningtreatment we initiated rotarod testing once per week, as soon as it waspossible (rotarod testing in very young animals is not feasible). Anaccelerating rotarod (Columbus Instruments) was set to increasegradually from 4–40 rpm over 15 s, and maintained at 40 rpmthereafter. Four trials were carried out at each time point, and theshortest trial was discarded; the remaining three trials were averaged.Both groups exhibited a decline in performance over time. At age 7.5,8.5 and 9.5 weeks, there were no significant differences betweentreatment and control groups. At age 10.5 weeks there was asignificant improvement in performance in the treated vs. controlanimals: 55.8±9.0 s vs. 24.4±16.2 s, (pb0.001, t-test); at 12.5 weeks33.8±16.0 vs. 13.7±12.1 s, (pb0.05, t-test) (Fig. 2C). A directcomparison of the rotarod performance of littermates at 12.5 weeksillustrates a relatively consistent benefit of the compound (Fig. 2D).An individual comparison of performance for each animal indicatedan improvement in median function in the treated animals after

Fig. 2. Analysis of Y-27632 on behavior in R6/2mice. Cohorts of female R6/2micewere randomized according to littermate status, weight, and CAG repeat length into two groups of 8animals. Y-27632 at 100 mg/kg/day was administered by putting the compound in drinking water, controlling for daily intake to ensure accurate dosing. Dosing began at age4 weeks. Rotarod testing began at age 7.5 weeks. (A) Y-27632 treatment had no significant effect on weight. (B) Y-27632 treatment had no significant effect on development of theclasping reflex. (C) Y-27632 treatment significantly improved rotarod performance beginning at age 10.5 weeks. ⁎pb0.05; ⁎⁎pb0.01 (t-test). (D) Comparison of littermates testedshows a consistent improvement in rotarod performance in the treated animals at 12.5 weeks. (E) Plots of individual animal's performance at each time point indicate animprovement in median performance on the rotarod in the treated group.
416 M. Li et al. / Neurobiology of Disease 36 (2009) 413–420
10 weeks (Fig. 2E), ruling out a large effect on a small number ofanimals. At 13.5 weeks, three animals in the treatment group died dueto handling-induced seizures, but no animals in the control group.
Y-27632 has no effect on lifespan
Because we observed handling-induced seizures, especially in thetreated animals, wewere concerned that despite its benefit on rotarodperformance that Y-27632 might somehow lower the seizure thresh-old. Thus, we decided to test its effects on lifespan in the absence ofthe extensive handling required for rotarod testing. We repeated thetrial with an additional 17 female R6/2 littermate mice, sorted by thesame criteria used previously, with 9 animals in the Y-27632treatment group and 8 animals in control group. Y-27632 was givenfrom age 4 weeks. The median lifespan of Y-27632 treated animalswas 126 days and control animals was 120 days, which matched thelifespan of other cohorts of R6/2 mice maintained in our colony(Supp. Fig. 2). There was no significant effect of treatment on lifespan(Fig. 3A), and no effect on body weight in this group (Fig. 3B).
Y-27632 does not change medium spiny cell density in the striatum
Striatal medium spiny neuron loss is a principal feature ofHuntington disease pathology. Thus, we tested whether Y-27632treatment would change the medium spiny neuron density in R6/2mice. We sacrificed 6 pairs of treated and control mice at 13.5 weeks.Brains were weighed and cut in a sagittal midline section. Half of thebrain was embedded in OCT compound, and frozen in ethanol-dry icefor pathological studies; the other half was snap-frozen for biochem-ical studies. The brains embedded in OCT compound were cut on acoronal plane, with 12 μmserial sections cut from the anterior throughthe posterior striatum. Every 8th section was collected and doublestained with DARPP32 (dopamine and cAMP regulated phosphopro-tein of 32 kD), a marker for medium spiny neurons, and GFAP (glialfibrillary acidic protein), a marker for astrocytes. Adjacent imageswere collected by an automated fluorescence microscope (Nikon 6D).The images were then recombined to encompass a large image of theentire slice section. By analyzing the data using Image J (n=6 samplesper condition), we found that Y-27632 treatment had no statisticallysignificant effect on the area of the striatum, medium spiny neuron
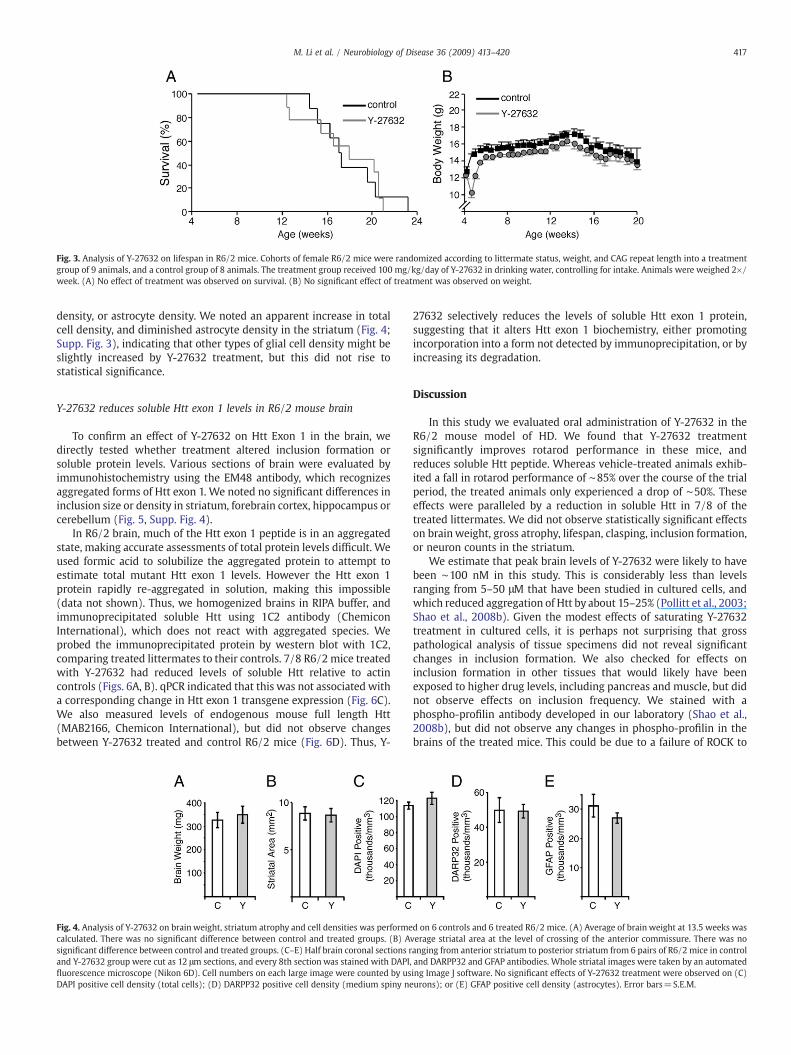
Fig. 3. Analysis of Y-27632 on lifespan in R6/2 mice. Cohorts of female R6/2 mice were randomized according to littermate status, weight, and CAG repeat length into a treatmentgroup of 9 animals, and a control group of 8 animals. The treatment group received 100 mg/kg/day of Y-27632 in drinking water, controlling for intake. Animals were weighed 2×/week. (A) No effect of treatment was observed on survival. (B) No significant effect of treatment was observed on weight.
417M. Li et al. / Neurobiology of Disease 36 (2009) 413–420
density, or astrocyte density. We noted an apparent increase in totalcell density, and diminished astrocyte density in the striatum (Fig. 4;Supp. Fig. 3), indicating that other types of glial cell density might beslightly increased by Y-27632 treatment, but this did not rise tostatistical significance.
Y-27632 reduces soluble Htt exon 1 levels in R6/2 mouse brain
To confirm an effect of Y-27632 on Htt Exon 1 in the brain, wedirectly tested whether treatment altered inclusion formation orsoluble protein levels. Various sections of brain were evaluated byimmunohistochemistry using the EM48 antibody, which recognizesaggregated forms of Htt exon 1. We noted no significant differences ininclusion size or density in striatum, forebrain cortex, hippocampus orcerebellum (Fig. 5, Supp. Fig. 4).
In R6/2 brain, much of the Htt exon 1 peptide is in an aggregatedstate, making accurate assessments of total protein levels difficult. Weused formic acid to solubilize the aggregated protein to attempt toestimate total mutant Htt exon 1 levels. However the Htt exon 1protein rapidly re-aggregated in solution, making this impossible(data not shown). Thus, we homogenized brains in RIPA buffer, andimmunoprecipitated soluble Htt using 1C2 antibody (ChemiconInternational), which does not react with aggregated species. Weprobed the immunoprecipitated protein by western blot with 1C2,comparing treated littermates to their controls. 7/8 R6/2 mice treatedwith Y-27632 had reduced levels of soluble Htt relative to actincontrols (Figs. 6A, B). qPCR indicated that this was not associated witha corresponding change in Htt exon 1 transgene expression (Fig. 6C).We also measured levels of endogenous mouse full length Htt(MAB2166, Chemicon International), but did not observe changesbetween Y-27632 treated and control R6/2 mice (Fig. 6D). Thus, Y-
Fig. 4. Analysis of Y-27632 on brain weight, striatum atrophy and cell densities was performecalculated. There was no significant difference between control and treated groups. (B) Asignificant difference between control and treated groups. (C–E) Half brain coronal sections rand Y-27632 group were cut as 12 μm sections, and every 8th section was stained with DAPI,fluorescence microscope (Nikon 6D). Cell numbers on each large image were counted by usDAPI positive cell density (total cells); (D) DARPP32 positive cell density (medium spiny ne
27632 selectively reduces the levels of soluble Htt exon 1 protein,suggesting that it alters Htt exon 1 biochemistry, either promotingincorporation into a form not detected by immunoprecipitation, or byincreasing its degradation.
Discussion
In this study we evaluated oral administration of Y-27632 in theR6/2 mouse model of HD. We found that Y-27632 treatmentsignificantly improves rotarod performance in these mice, andreduces soluble Htt peptide. Whereas vehicle-treated animals exhib-ited a fall in rotarod performance of ∼85% over the course of the trialperiod, the treated animals only experienced a drop of ∼50%. Theseeffects were paralleled by a reduction in soluble Htt in 7/8 of thetreated littermates. We did not observe statistically significant effectson brainweight, gross atrophy, lifespan, clasping, inclusion formation,or neuron counts in the striatum.
We estimate that peak brain levels of Y-27632 were likely to havebeen ∼100 nM in this study. This is considerably less than levelsranging from 5–50 μM that have been studied in cultured cells, andwhich reduced aggregation of Htt by about 15–25% (Pollitt et al., 2003;Shao et al., 2008b). Given the modest effects of saturating Y-27632treatment in cultured cells, it is perhaps not surprising that grosspathological analysis of tissue specimens did not reveal significantchanges in inclusion formation. We also checked for effects oninclusion formation in other tissues that would likely have beenexposed to higher drug levels, including pancreas and muscle, but didnot observe effects on inclusion frequency. We stained with aphospho-profilin antibody developed in our laboratory (Shao et al.,2008b), but did not observe any changes in phospho-profilin in thebrains of the treated mice. This could be due to a failure of ROCK to
d on 6 controls and 6 treated R6/2 mice. (A) Average of brain weight at 13.5 weeks wasverage striatal area at the level of crossing of the anterior commissure. There was noanging from anterior striatum to posterior striatum from 6 pairs of R6/2 mice in controland DARPP32 and GFAP antibodies. Whole striatal images were taken by an automateding Image J software. No significant effects of Y-27632 treatment were observed on (C)urons); or (E) GFAP positive cell density (astrocytes). Error bars=S.E.M.

Fig. 5. Analysis of Y-27632 on inclusion formation and phospho-profilin levels. Representative histopathology is shown. Various brain regions were stained with EM48 antibody(red), phospho-profilin antibody (green), and DAPI (blue). EM48 recognizes diffuse and aggregated forms of Htt exon 1. Phospho-profilin was distributed in the cytoplasm offorebrain cortical neurons, hippocampal CA3 neurons and cerebellar Purkinje neurons but not in striatal neurons. No effect of Y-27632 treatment was observed on cellular atrophy,phospho-profilin level or distribution, inclusion size or frequency. Scale bars are shown.
418 M. Li et al. / Neurobiology of Disease 36 (2009) 413–420
phosphorylate profilin in brain, but ismore likely due to sub-saturatingdoses of Y-27632. Whether a more potent ROCK inhibitor, or higherbrain concentrations, would producemore apparent effects on profilinphosphorylation or inclusion formation awaits further study.
Y-27632 treatment selectively reduced brain Htt exon 1 levels,without affecting Htt exon 1 gene expression or full length mouse Htt.This suggests that the compound exerts some effect on the protein'smetabolism in vivo, and is in keeping with a recent report whichindicates an enhancing effect of Y-27632 treatment on Htt degradationvia proteasome and autophagy pathways (Bauer et al., 2009). Ourprior work suggests that Htt exon 1 is not cleared via the proteasome(Chandra et al., 2008), but autophagy may play an important role(Ravikumar and Rubinsztein, 2006; Ravikumar et al., 2004). As wehavementioned, it is very difficult to quantify precisely total Htt levelsin vivo due to its rapid aggregation. However, our analysis of Htt Exon1 transgene expression via qPCR and endogenous Htt levels effectivelyrules out a primary effect of Y-27632 on the Htt promoter, andsuggests either that the total protein level is reduced, or that solubleprotein is more rapidly cleared. In cultured cells we have not observeda consistent effect of Y-27632 treatment on Htt exon 1 levels, but this
may relate to the fact that it has been relatively over-expressed viatransient transfection. As we learn more about mechanisms thatgovern Htt exon 1 solubility vs. aggregation, it will be interesting todeterminewhether these conformational states have a direct effect onprotein degradation.
Multiple parameters likely govern lifespan of R6/2 mice. Theseinclude peripheral effects such as weight loss and muscle atrophy,and central effects such as seizures. Consistent with this, two studiesbesides our own have noted a dissociation between lifespan andimproved performance in behavior assays (Chopra et al., 2007; Chouet al., 2005). In this study, we did not observe significant weight lossin either group of animals, even at later points in life, or profounddefects in glucose metabolism (data not shown), suggesting thatmuscle wasting or systemic illness did not play a major role. After therotarod trials, we observed fatal handling-induced seizures in 3/8animals treated with Y-27632, but in none of the controls. Thus, it ispossible that despite a benefit in motor function, drug treatment mayhave lowered the seizure threshold. However, one report indicatesthat Y-27632 may reduce seizures in a mouse model (Inan andBuyukafsar, 2008).
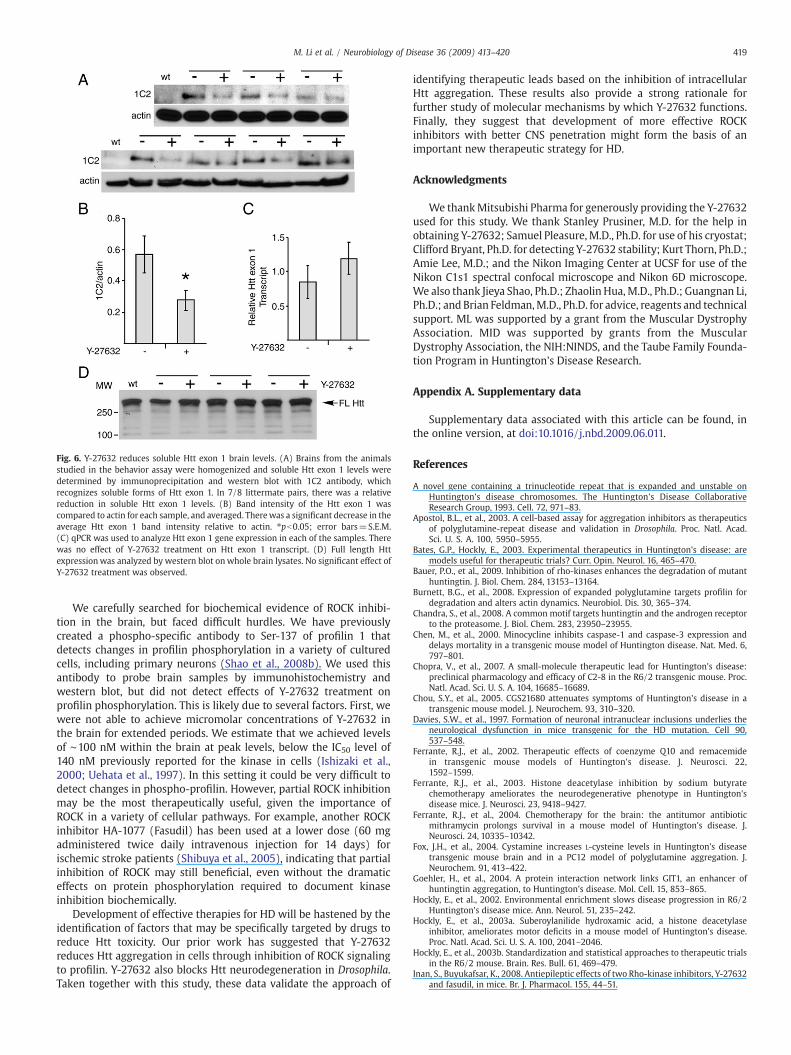
Fig. 6. Y-27632 reduces soluble Htt exon 1 brain levels. (A) Brains from the animalsstudied in the behavior assay were homogenized and soluble Htt exon 1 levels weredetermined by immunoprecipitation and western blot with 1C2 antibody, whichrecognizes soluble forms of Htt exon 1. In 7/8 littermate pairs, there was a relativereduction in soluble Htt exon 1 levels. (B) Band intensity of the Htt exon 1 wascompared to actin for each sample, and averaged. Therewas a significant decrease in theaverage Htt exon 1 band intensity relative to actin. ⁎pb0.05; error bars=S.E.M.(C) qPCR was used to analyze Htt exon 1 gene expression in each of the samples. Therewas no effect of Y-27632 treatment on Htt exon 1 transcript. (D) Full length Httexpressionwas analyzed by western blot on whole brain lysates. No significant effect ofY-27632 treatment was observed.
419M. Li et al. / Neurobiology of Disease 36 (2009) 413–420
We carefully searched for biochemical evidence of ROCK inhibi-tion in the brain, but faced difficult hurdles. We have previouslycreated a phospho-specific antibody to Ser-137 of profilin 1 thatdetects changes in profilin phosphorylation in a variety of culturedcells, including primary neurons (Shao et al., 2008b). We used thisantibody to probe brain samples by immunohistochemistry andwestern blot, but did not detect effects of Y-27632 treatment onprofilin phosphorylation. This is likely due to several factors. First, wewere not able to achieve micromolar concentrations of Y-27632 inthe brain for extended periods. We estimate that we achieved levelsof ∼100 nM within the brain at peak levels, below the IC50 level of140 nM previously reported for the kinase in cells (Ishizaki et al.,2000; Uehata et al., 1997). In this setting it could be very difficult todetect changes in phospho-profilin. However, partial ROCK inhibitionmay be the most therapeutically useful, given the importance ofROCK in a variety of cellular pathways. For example, another ROCKinhibitor HA-1077 (Fasudil) has been used at a lower dose (60 mgadministered twice daily intravenous injection for 14 days) forischemic stroke patients (Shibuya et al., 2005), indicating that partialinhibition of ROCK may still beneficial, even without the dramaticeffects on protein phosphorylation required to document kinaseinhibition biochemically.
Development of effective therapies for HD will be hastened by theidentification of factors that may be specifically targeted by drugs toreduce Htt toxicity. Our prior work has suggested that Y-27632reduces Htt aggregation in cells through inhibition of ROCK signalingto profilin. Y-27632 also blocks Htt neurodegeneration in Drosophila.Taken together with this study, these data validate the approach of
identifying therapeutic leads based on the inhibition of intracellularHtt aggregation. These results also provide a strong rationale forfurther study of molecular mechanisms by which Y-27632 functions.Finally, they suggest that development of more effective ROCKinhibitors with better CNS penetration might form the basis of animportant new therapeutic strategy for HD.
Acknowledgments
We thankMitsubishi Pharma for generously providing the Y-27632used for this study. We thank Stanley Prusiner, M.D. for the help inobtaining Y-27632; Samuel Pleasure, M.D., Ph.D. for use of his cryostat;Clifford Bryant, Ph.D. for detecting Y-27632 stability; Kurt Thorn, Ph.D.;Amie Lee, M.D.; and the Nikon Imaging Center at UCSF for use of theNikon C1s1 spectral confocal microscope and Nikon 6D microscope.We also thank Jieya Shao, Ph.D.; Zhaolin Hua, M.D., Ph.D.; Guangnan Li,Ph.D.; and Brian Feldman,M.D., Ph.D. for advice, reagents and technicalsupport. ML was supported by a grant from the Muscular DystrophyAssociation. MID was supported by grants from the MuscularDystrophy Association, the NIH:NINDS, and the Taube Family Founda-tion Program in Huntington's Disease Research.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.nbd.2009.06.011.
References
A novel gene containing a trinucleotide repeat that is expanded and unstable onHuntington's disease chromosomes. The Huntington's Disease CollaborativeResearch Group, 1993. Cell. 72, 971–83.
Apostol, B.L., et al., 2003. A cell-based assay for aggregation inhibitors as therapeuticsof polyglutamine-repeat disease and validation in Drosophila. Proc. Natl. Acad.Sci. U. S. A. 100, 5950–5955.
Bates, G.P., Hockly, E., 2003. Experimental therapeutics in Huntington's disease: aremodels useful for therapeutic trials? Curr. Opin. Neurol. 16, 465–470.
Bauer, P.O., et al., 2009. Inhibition of rho-kinases enhances the degradation of mutanthuntingtin. J. Biol. Chem. 284, 13153–13164.
Burnett, B.G., et al., 2008. Expression of expanded polyglutamine targets profilin fordegradation and alters actin dynamics. Neurobiol. Dis. 30, 365–374.
Chandra, S., et al., 2008. A common motif targets huntingtin and the androgen receptorto the proteasome. J. Biol. Chem. 283, 23950–23955.
Chen, M., et al., 2000. Minocycline inhibits caspase-1 and caspase-3 expression anddelays mortality in a transgenic mouse model of Huntington disease. Nat. Med. 6,797–801.
Chopra, V., et al., 2007. A small-molecule therapeutic lead for Huntington's disease:preclinical pharmacology and efficacy of C2-8 in the R6/2 transgenic mouse. Proc.Natl. Acad. Sci. U. S. A. 104, 16685–16689.
Chou, S.Y., et al., 2005. CGS21680 attenuates symptoms of Huntington's disease in atransgenic mouse model. J. Neurochem. 93, 310–320.
Davies, S.W., et al., 1997. Formation of neuronal intranuclear inclusions underlies theneurological dysfunction in mice transgenic for the HD mutation. Cell 90,537–548.
Ferrante, R.J., et al., 2002. Therapeutic effects of coenzyme Q10 and remacemidein transgenic mouse models of Huntington's disease. J. Neurosci. 22,1592–1599.
Ferrante, R.J., et al., 2003. Histone deacetylase inhibition by sodium butyratechemotherapy ameliorates the neurodegenerative phenotype in Huntington'sdisease mice. J. Neurosci. 23, 9418–9427.
Ferrante, R.J., et al., 2004. Chemotherapy for the brain: the antitumor antibioticmithramycin prolongs survival in a mouse model of Huntington's disease. J.Neurosci. 24, 10335–10342.
Fox, J.H., et al., 2004. Cystamine increases L-cysteine levels in Huntington's diseasetransgenic mouse brain and in a PC12 model of polyglutamine aggregation. J.Neurochem. 91, 413–422.
Goehler, H., et al., 2004. A protein interaction network links GIT1, an enhancer ofhuntingtin aggregation, to Huntington's disease. Mol. Cell. 15, 853–865.
Hockly, E., et al., 2002. Environmental enrichment slows disease progression in R6/2Huntington's disease mice. Ann. Neurol. 51, 235–242.
Hockly, E., et al., 2003a. Suberoylanilide hydroxamic acid, a histone deacetylaseinhibitor, ameliorates motor deficits in a mouse model of Huntington's disease.Proc. Natl. Acad. Sci. U. S. A. 100, 2041–2046.
Hockly, E., et al., 2003b. Standardization and statistical approaches to therapeutic trialsin the R6/2 mouse. Brain. Res. Bull. 61, 469–479.
Inan, S., Buyukafsar, K., 2008. Antiepileptic effects of two Rho-kinase inhibitors, Y-27632and fasudil, in mice. Br. J. Pharmacol. 155, 44–51.

420 M. Li et al. / Neurobiology of Disease 36 (2009) 413–420
Ishizaki, T., et al., 2000. Pharmacological properties of Y-27632, a specific inhibitor ofrho-associated kinases. Mol. Pharmacol. 57, 976–983.
Kazantsev, A., et al., 2002. A bivalent Huntingtin binding peptide suppressespolyglutamine aggregation and pathogenesis in Drosophila. Nat. Genet. 30,367–376.
Li, M., et al., 2007. Soluble androgen receptor oligomers underlie pathology in a mousemodel of spinobulbar muscular atrophy. J. Biol. Chem. 282, 3157–3164.
Mangiarini, L., et al., 1996. Exon 1 of the HD gene with an expanded CAG repeat issufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87,493–506.
Pattison, J.S., Robbins, J., 2008. Protein misfolding and cardiac disease: establishingcause and effect. Autophagy 4, 821–823.
Pollitt, S.K., et al., 2003. A rapid cellular FRET assay of polyglutamine aggregationidentifies a novel inhibitor. Neuron 40, 685–694.
Ravikumar, B., Rubinsztein, D.C., 2006. Role of autophagy in the clearance of mutanthuntingtin: a step towards therapy? Mol. Aspects Med. 27, 520–527.
Ravikumar, B., et al., 2004. Inhibition of mTOR induces autophagy and reduces toxicityof polyglutamine expansions in fly and mouse models of Huntington disease. Nat.Genet. 36, 585–595.
Scherzinger, E., et al., 1997. Huntingtin-encoded polyglutamine expansions formamyloid-like protein aggregates in vitro and in vivo. Cell 90, 549–558.
Shao, J., Diamond, M.I., 2007. Polyglutamine diseases: emerging concepts in pathogen-esis and therapy. Hum. Mol. Genet. 16 (Spec No. 2), R115–R123.
Shao, J., et al., 2008a. ROCK and PRK-2 mediate the inhibitory effect of Y-27632 onpolyglutamine aggregation. FEBS Lett. 582, 1637–1642.
Shao, J., et al., 2008b. Phosphorylation of profilin by ROCK1 regulates polyglutamineaggregation. Mol. Cell. Biol. 28, 5196–5208.
Shibuya, M., et al., 2005. Effects of fasudil in acute ischemic stroke: results of aprospective placebo-controlled double-blind trial. J. Neurol. Sci. 238, 31–39.
Takahashi, T., et al., 2007a. Soluble polyglutamine oligomers formed prior to inclusionbody formation are cytotoxic. Hum. Mol. Genet.
Takahashi, Y., et al., 2007b. Detection of polyglutamine protein oligomers in cells byfluorescence correlation spectroscopy. J. Biol. Chem. 282, 24039–24048.
Tanaka, M., et al., 2004. Trehalose alleviates polyglutamine-mediated pathology in amouse model of Huntington disease. Nat. Med. 10, 148–154.
Uehata, M., et al., 1997. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 389, 990–994.
Wang, J.C., et al., 2006. Novel arylpyrazole compounds selectively modulate gluco-corticoid receptor regulatory activity. Genes Dev. 20, 689–699.
Zhang, X., et al., 2005. A potent small molecule inhibits polyglutamine aggregation inHuntington's disease neurons and suppresses neurodegeneration in vivo. Proc.Natl. Acad. Sci. U. S. A. 102, 892–897.
Copyright © 2022 FDOKUMEN




















