
Huntingtin inclusion bodies are iron-dependent centers of oxidative events
14
Huntingtin inclusion bodies are iron-dependent centers of oxidative events Wance J. J. Firdaus 1 , Andreas Wyttenbach 2 , Paola Giuliano 3 , Carole Kretz-Remy 1 , R. William Currie 1,4 and Andre ´ -Patrick Arrigo 1 1 Laboratoire Stress Oxydant, Chaperons et Apoptose, Universite ´ Claude Bernard Lyon-1, Villeurbanne, France 2 Southampton Neuroscience Group, School of Biological Sciences, University of Southampton, UK 3 Department of Clinical Immunology, National Cancer Institute, G. Pascale Foundation, Naples, Italy 4 Department of Anatomy and Neurobiology, Dalhousie University, Halifax, NS, Canada Huntington’s disease (HD) is a progressive neuro- degenerative disorder that induces neuronal selective loss and formation of intraneuronal protein aggregates in the striatum and cortex. It is caused by abnormally expanded polyglutamine polyQ tracts (larger than 36 glutamine repeats) in exon1 (Ex1) of huntingtin (htt), an iron-regulated neuronal protein implicated in vesicle trafficking [1–6]. In human and transgenic mice, HD correlates with the appearance of intraneuronal, intranuclear and perinuclear aggregates ⁄ inclusions containing the protease-resistant mutated N-terminal htt fragment [2,7–10]. In spite of the fact that inclusion bodies formed by mutated N-terminal htt fragment often correlate with toxicity [11], polyQ proteins can also be toxic even in the absence of detectable forma- tion of aggregates ⁄ inclusion bodies [3,12–15]. Inclusion bodies have also been reported to correlate with cell survival [16]. Moreover, it has been proposed that inclusion body formation is probably a cell-mediated process to concentrate and promote the clearance of Keywords Hsps; Huntingtin; inclusion bodies; iron; ROS Correspondence A.-P. Arrigo, Laboratoire Stress Oxydant, Chaperons et Apoptose, CNRS UMR 5534, Centre de Ge ´ne ´ tique Mole ´ culaire et Cellulaire, Universite ´ Claude Bernard Lyon-1, 43 Blvd du 11 Novembre, 69622 Villeurbanne Cedex, France Fax: +33 0 472432685 Tel: +33 0 472448595 E-mail: [email protected] (Received 31 August 2006, accepted 11 October 2006) doi:10.1111/j.1742-4658.2006.05537.x Recently, we reported that the transient expression of huntingtin exon1 polypeptide containing polyglutamine tracts of various sizes (httEx1-polyQ) in cell models of Huntington disease generated an oxidative stress whose intensity was CAG repeat expansion-dependent. Here, we have analyzed the intracellular localization of the oxidative events generated by the httEx1-polyQ polypeptides. Analysis of live COS-7 cells as well as neuronal SK-N-SH and PC12 cells incubated with hydroethidine or dichlorofluoresc- ein diacetate revealed oxidation of these probes at the level of the inclusion bodies formed by httEx1-polyQ polypeptides. The intensity and frequency of the oxidative events among the inclusions were CAG repeat expansion- dependent. Electron microscopic analysis of cell sections revealed the pres- ence of oxidation-dependent morphologic alterations in the vicinity of httEx1-polyQ inclusion bodies. Moreover, a high level of oxidized proteins was recovered in partially purified inclusions. We also report that the iron chelator deferroxamine altered the structure, localization and oxidative potential of httEx1-polyQ inclusion bodies. Hence, despite the fact that the formation of inclusion bodies may represent a defense reaction of the cell to eliminate httEx1 mutant polypeptide, this phenomenon appears inherent to the generation of iron-dependent oxidative events that can be deleterious to the cell. Abbreviations DCFH-DA, dichlorofluorescein diacetate; 2,4-DNBH, 2,4-dinitrophenylhydrazine; EB, ethidium bromide; Ex1, exon1; FACS, fluorescence- activated cell sorting; FITC, fluorescein isothiocyanate; HA, hemagglutinin; HD, Huntington’s disease; HE, hydroethidine; Hsp, heat shock or stress protein; htt, huntingtin; NAC, N-acetyl-L-cysteine; ROS, reactive oxygen species. 5428 FEBS Journal 273 (2006) 5428–5441 ª 2006 The Authors Journal compilation ª 2006 FEBS
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Huntingtin inclusion bodies are iron-dependent centers of oxidative events

Huntingtin inclusion bodies are iron-dependent centersof oxidative eventsWance J. J. Firdaus1, Andreas Wyttenbach2, Paola Giuliano3, Carole Kretz-Remy1,R. William Currie1,4 and Andre-Patrick Arrigo1
1 Laboratoire Stress Oxydant, Chaperons et Apoptose, Universite Claude Bernard Lyon-1, Villeurbanne, France
2 Southampton Neuroscience Group, School of Biological Sciences, University of Southampton, UK
3 Department of Clinical Immunology, National Cancer Institute, G. Pascale Foundation, Naples, Italy
4 Department of Anatomy and Neurobiology, Dalhousie University, Halifax, NS, Canada
Huntington’s disease (HD) is a progressive neuro-
degenerative disorder that induces neuronal selective
loss and formation of intraneuronal protein aggregates
in the striatum and cortex. It is caused by abnormally
expanded polyglutamine polyQ tracts (larger than 36
glutamine repeats) in exon1 (Ex1) of huntingtin (htt),
an iron-regulated neuronal protein implicated in vesicle
trafficking [1–6]. In human and transgenic mice, HD
correlates with the appearance of intraneuronal,
intranuclear and perinuclear aggregates ⁄ inclusions
containing the protease-resistant mutated N-terminal
htt fragment [2,7–10]. In spite of the fact that inclusion
bodies formed by mutated N-terminal htt fragment
often correlate with toxicity [11], polyQ proteins can
also be toxic even in the absence of detectable forma-
tion of aggregates ⁄ inclusion bodies [3,12–15]. Inclusion
bodies have also been reported to correlate with cell
survival [16]. Moreover, it has been proposed that
inclusion body formation is probably a cell-mediated
process to concentrate and promote the clearance of
Keywords
Hsps; Huntingtin; inclusion bodies; iron;
ROS
Correspondence
A.-P. Arrigo, Laboratoire Stress Oxydant,
Chaperons et Apoptose, CNRS UMR 5534,
Centre de Genetique Moleculaire et
Cellulaire, Universite Claude Bernard Lyon-1,
43 Blvd du 11 Novembre, 69622
Villeurbanne Cedex, France
Fax: +33 0 472432685
Tel: +33 0 472448595
E-mail: [email protected]
(Received 31 August 2006, accepted
11 October 2006)
doi:10.1111/j.1742-4658.2006.05537.x
Recently, we reported that the transient expression of huntingtin exon1
polypeptide containing polyglutamine tracts of various sizes (httEx1-polyQ)
in cell models of Huntington disease generated an oxidative stress whose
intensity was CAG repeat expansion-dependent. Here, we have analyzed
the intracellular localization of the oxidative events generated by the
httEx1-polyQ polypeptides. Analysis of live COS-7 cells as well as neuronal
SK-N-SH and PC12 cells incubated with hydroethidine or dichlorofluoresc-
ein diacetate revealed oxidation of these probes at the level of the inclusion
bodies formed by httEx1-polyQ polypeptides. The intensity and frequency
of the oxidative events among the inclusions were CAG repeat expansion-
dependent. Electron microscopic analysis of cell sections revealed the pres-
ence of oxidation-dependent morphologic alterations in the vicinity of
httEx1-polyQ inclusion bodies. Moreover, a high level of oxidized proteins
was recovered in partially purified inclusions. We also report that the iron
chelator deferroxamine altered the structure, localization and oxidative
potential of httEx1-polyQ inclusion bodies. Hence, despite the fact that the
formation of inclusion bodies may represent a defense reaction of the
cell to eliminate httEx1 mutant polypeptide, this phenomenon appears
inherent to the generation of iron-dependent oxidative events that can be
deleterious to the cell.
Abbreviations
DCFH-DA, dichlorofluorescein diacetate; 2,4-DNBH, 2,4-dinitrophenylhydrazine; EB, ethidium bromide; Ex1, exon1; FACS, fluorescence-
activated cell sorting; FITC, fluorescein isothiocyanate; HA, hemagglutinin; HD, Huntington’s disease; HE, hydroethidine; Hsp, heat shock or
stress protein; htt, huntingtin; NAC, N-acetyl-L-cysteine; ROS, reactive oxygen species.
5428 FEBS Journal 273 (2006) 5428–5441 ª 2006 The Authors Journal compilation ª 2006 FEBS

an undesirable and protease-resistant mutant protein
by activating autophagy [17,18]. Therefore, toxicity
could originate from defects in the cell-mediated pro-
cess leading to inclusion body formation.
In HD transgenic mice and in brains of HD
patients, heat shock or stress proteins (Hsps) are
expressed in affected neurons, where they associate
with polyQ aggregates [19–21]. Analysis performed in
cell models have revealed that Hsps, such as Hsp70
and Hsp40 (Hdj-1), interfere with polyQ toxicity by
decreasing the efficiency of aggregate and inclusion
body formation [11,22–24]. In spite of being less
effective than Hsp70 ⁄Hdj-1 in suppressing polyQ
aggregation, Hsp27 protects neuronal cells against
polyQ-expanded httEx1-mediated oxidative stress [11]
and neuronal apoptotic cell death [25,26].
Neuronal degeneration, including that in Alzheimer’s,
Parkinson’s and Huntington’s diseases, are charac-
terized by oxidative events such as mitochondrial
complex I and IV deficiency, and elevated level of
nitric oxide and reactive oxygen species (ROS) [11,27–
32]. In the case of HD, oxidative modifications are
proportional in intensity with the number of CAG
repeats in the htt-polyQ polypeptide [11,33–36]. In
transgenic HD mice as well as in cellular models of
HD, the expression of mutant httEx1-polyQ polypep-
tide has been reported to generate mitochondrial defi-
ciency as well as elevated levels of nitric oxide, ROS
and protein oxidation [11,33–36] that directly contrib-
ute to cell death [11]. Proteomic analysis aimed at
detecting protein carbonyl residues has revealed that
specific proteins are indeed oxidized in the HD mouse
brain due to httEx1 expression [37]. This recent obser-
vation confirms that oxidative modifications are
observed not only in cellular HD models but also
in vivo in the brains of HD mice. Mitochondrial dys-
function has been proposed to be responsible for the
oxidative events associated with HD, as expression of
proteins containing glutamine repeats usually corre-
lates with mitochondrial depolarization [38–40] and
impaired clearance of oxidized proteins [27]. However,
a comparative analysis of the protection generated by
Hsps with that provided by the antioxidant agent
N-acetyl-l-cysteine (NAC) reveals that, in the cascade
of events responsible for the oxidative stress mediated
by httEx1-polyQ expression, the formation of inclusion
bodies is a phenomenon that is likely to be upstream
of the defects in mitochondria [36].
Here, we have analyzed the intracellular localization
of the oxidative events generated by httEx1-polyQ
expression. Using fluorescent probes and different live
cell types, we observed a high level of oxidative events
at the level of the inclusion bodies formed by HttEx1-
polyQ. These NAC- and Hsp-sensitive oxidative events
were responsible for cellular damage in the vicinity of
the inclusion bodies. Analysis performed in the pres-
ence of the iron chelator deferroxamine revealed that
the oxidation, localization and structural organization
of the inclusion bodies formed by mutant htt were
iron-dependent. Hence, in spite of the fact that inclu-
sion body formation could be a defense reaction of the
cell to eliminate an aberrant polypeptide, our results
suggest that this process is not foolproof, as iron-
dependent oxidative events are often generated that
alter neighboring cellular morphology, including that
of organelles such as mitochondria.
Results
In COS-7 cells as well as in neuronal SK-N-SH
and PC12 cells, HttEx1-polyQ inclusion bodies
are centers of oxidative events
We recently showed [11,36] that the expression of
httEx1-polyQ polypeptide results in the oxidation of
probes [dichlorofluorescein diacetate (DCFH-DA)] and
hydroethidine (HE)] aimed at detecting intracellular
oxidative events. These probes freely penetrate inside
living cells and become fluorescent upon their oxida-
tion by ROS. This study, performed in different types
of cell (i.e COS-7 cells), showed that an abnormal oxi-
dation process, proportional to the number of CAG
repeats, is concomitant with the presence of mutant
httEx1-polyQ polypeptide when it forms inclusion bod-
ies [36]. Illustrations of the inclusion bodies formed by
mutant httEx1 polypeptides fused to enhanced green
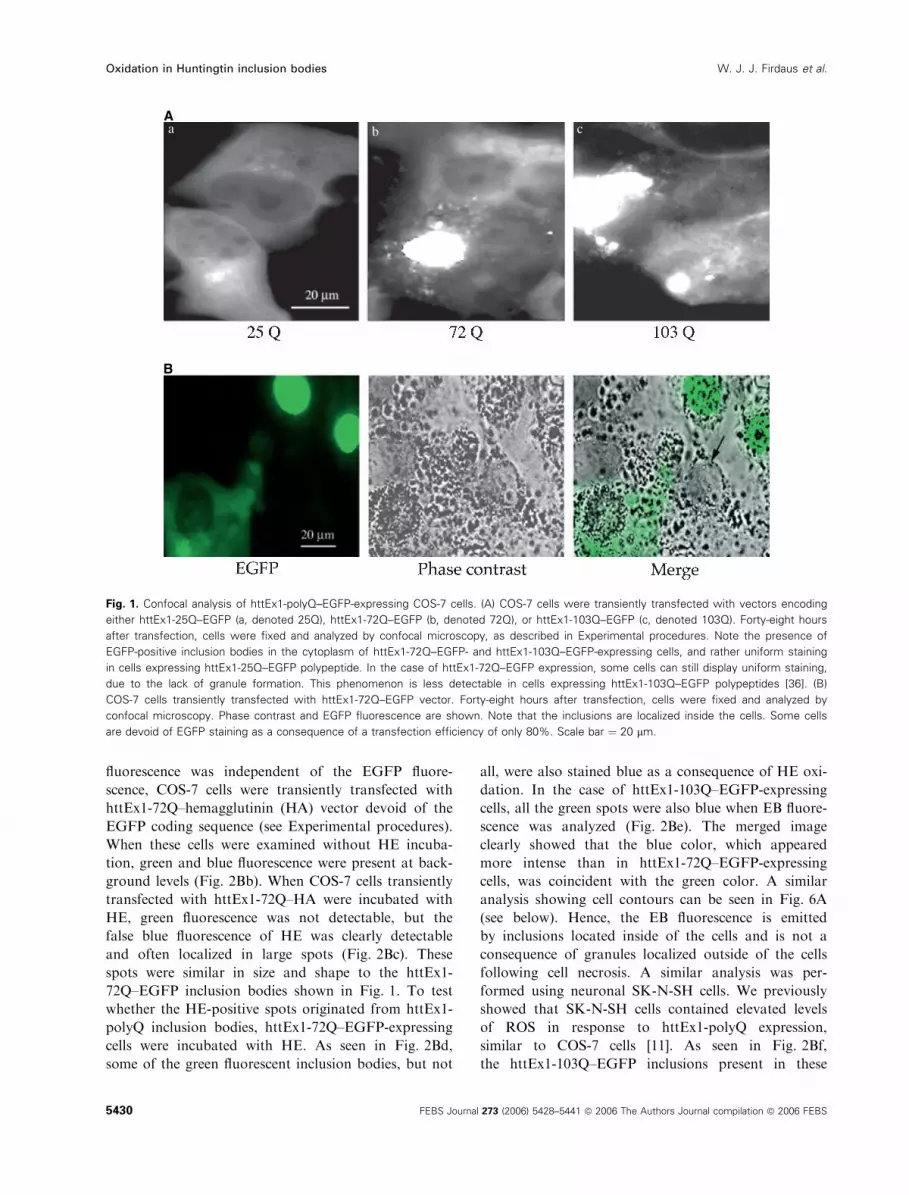
fluorescent protein (EGFP) are presented in Fig. 1.
To further increase our knowledge of the oxidation
process generated by httEx1-polyQ–EGFP expression,
we analyzed where this phenomenon was taking place
inside the cell. This was assessed by confocal micro-
scopic analysis of live COS-7 cells incubated for 1 h
with HE (see Experimental procedures). This probe
was used because, once it is oxidized to ethidium bro-
mide (EB), its fluorescent wavelength (590 nm) differs
from that of EGFP (maximum 510 nm). The red EB
fluorescence was recorded in false blue color. As seen
in Fig. 2A, COS-7 cells exposed for 1 h to 10 lm
myxothiazol, an inhibitor of ubiquinol oxidation, dis-
played cytoplasmic blue spots after HE staining. This
staining, which corresponded to high levels of ROS
in mitochondria [41], was not observed in cells not
treated with myxothiazol. COS-7 cells expressing
httEx1-25Q–EGFP were characterized by diffuse green
cytoplasmic EGFP staining and did not display blue
fluorescence (Fig. 2Ba). To confirm that the EB
W. J. J. Firdaus et al. Oxidation in Huntingtin inclusion bodies
FEBS Journal 273 (2006) 5428–5441 ª 2006 The Authors Journal compilation ª 2006 FEBS 5429
fluorescence was independent of the EGFP fluore-
scence, COS-7 cells were transiently transfected with
httEx1-72Q–hemagglutinin (HA) vector devoid of the
EGFP coding sequence (see Experimental procedures).
When these cells were examined without HE incuba-
tion, green and blue fluorescence were present at back-
ground levels (Fig. 2Bb). When COS-7 cells transiently
transfected with httEx1-72Q–HA were incubated with
HE, green fluorescence was not detectable, but the
false blue fluorescence of HE was clearly detectable
and often localized in large spots (Fig. 2Bc). These
spots were similar in size and shape to the httEx1-
72Q–EGFP inclusion bodies shown in Fig. 1. To test
whether the HE-positive spots originated from httEx1-
polyQ inclusion bodies, httEx1-72Q–EGFP-expressing
cells were incubated with HE. As seen in Fig. 2Bd,
some of the green fluorescent inclusion bodies, but not
all, were also stained blue as a consequence of HE oxi-
dation. In the case of httEx1-103Q–EGFP-expressing
cells, all the green spots were also blue when EB fluore-
scence was analyzed (Fig. 2Be). The merged image
clearly showed that the blue color, which appeared
more intense than in httEx1-72Q–EGFP-expressing
cells, was coincident with the green color. A similar
analysis showing cell contours can be seen in Fig. 6A
(see below). Hence, the EB fluorescence is emitted
by inclusions located inside of the cells and is not a
consequence of granules localized outside of the cells
following cell necrosis. A similar analysis was per-
formed using neuronal SK-N-SH cells. We previously
showed that SK-N-SH cells contained elevated levels
of ROS in response to httEx1-polyQ expression,
similar to COS-7 cells [11]. As seen in Fig. 2Bf,
the httEx1-103Q–EGFP inclusions present in these
A
B
a b c
Fig. 1. Confocal analysis of httEx1-polyQ–EGFP-expressing COS-7 cells. (A) COS-7 cells were transiently transfected with vectors encoding
either httEx1-25Q–EGFP (a, denoted 25Q), httEx1-72Q–EGFP (b, denoted 72Q), or httEx1-103Q–EGFP (c, denoted 103Q). Forty-eight hours
after transfection, cells were fixed and analyzed by confocal microscopy, as described in Experimental procedures. Note the presence of
EGFP-positive inclusion bodies in the cytoplasm of httEx1-72Q–EGFP- and httEx1-103Q–EGFP-expressing cells, and rather uniform staining
in cells expressing httEx1-25Q–EGFP polypeptide. In the case of httEx1-72Q–EGFP expression, some cells can still display uniform staining,
due to the lack of granule formation. This phenomenon is less detectable in cells expressing httEx1-103Q–EGFP polypeptides [36]. (B)
COS-7 cells transiently transfected with httEx1-72Q–EGFP vector. Forty-eight hours after transfection, cells were fixed and analyzed by
confocal microscopy. Phase contrast and EGFP fluorescence are shown. Note that the inclusions are localized inside the cells. Some cells
are devoid of EGFP staining as a consequence of a transfection efficiency of only 80%. Scale bar ¼ 20 lm.
Oxidation in Huntingtin inclusion bodies W. J. J. Firdaus et al.
5430 FEBS Journal 273 (2006) 5428–5441 ª 2006 The Authors Journal compilation ª 2006 FEBS

neuronal cells were also centers of HE oxidation. A
similar observation was made in neuronal PC12 cells
containing nuclear inclusion bodies formed by httEx1-
43Q–EGFP (Fig. 2Bg). These cells were reported by
Giuliano et al. [34] to contain elevated levels of ROS.
It is interesting to note in Fig. 2Bg (see also Fig. 2Bd)
that not all the EGFP-expressing granules displayed
HE-dependent blue staining. These observations sug-
gest that no spillover of EGFP fluorescence is recov-
ered at the level of the blue HE signal, and that in
cells expressing httEx1-72Q and httEx1-103Q, HE oxi-
dation does not take place throughout the cell and ⁄ormitochondria, but is preferentially localized to the
inclusion bodies. Control experiments were therefore
performed to exclude the possibility that HE could
somehow stick to the inclusion bodies unspecifically.
To address this problem, we tested whether DCFH-
DA oxidation also gave a similar signal in COS-7 cells
expressing HA-tagged httEx1-72Q. Figure 2C shows
that DCFH-DA oxidation was concentrated in spots
of similar size to the inclusion bodies (about 10 lm in
diameter). In addition, smaller stained spots were
clearly visible; these may represent oxidative events
that take place in mitochondria, a phenomenon that
was detectable with HE. No fluorescence was detected
in cells not exposed to DCFH-DA. Taken together,
these observations indicate that HE and DCFH-DA
oxidation are concentrated at the level of htt inclusion
bodies, hence suggesting that these structures are
centers of oxidative events.
HE oxidation at the level of httEx1-polyQ
inclusion bodies is decreased by antioxidants
and Hsp overexpression but not by proteasome
inhibitors
As shown in Fig. 2, confocal analysis of httEx1-72Q–
EGFP-expressing live COS-7 cells incubated with HE
revealed a predominant oxidation of this probe (blue
false color) at the level of the inclusion bodies formed
by this protein (fluorescent green). The presence of an
ongoing oxidation process at the level of httEx1-72Q–
EGFP inclusion bodies was further suggested by the
strong decrease in blue staining (HE oxidation) of the
inclusions in cells treated for 12 h with 2 mm NAC
(Fig. 3A) or 5 mm glutathione ethyl ester (not shown)
before being analyzed. In contrast, these antioxi-
dants did not alter EGFP staining and the size of
A
B
C
COS (590 nm)
COS 25Q EGFP
COS 72Q (-EGFP,-HE)
COS 72Q (-EGFP, +HE)
COS 72Q EGFP (+HE)
COS 103Q EGFP (+HE)
SK-N-SH103Q EGFP (+HE)
PC12 43Q EGFP (+HE)
COS 72Q (-EGFP, -HE+ DCFH-DA)
Fig. 2. Huntingtin inclusion bodies are centers of oxidative events.
(A) COS-7 cells were either exposed or not exposed to 10 lM
myxothiazol for 1 h before being incubated for 1 h with 10 lM HE
and analyzed as living cells by confocal microscopy. HE fluores-
cence (blue false color, 590 nm wavength) was recorded. (B) COS-7
cells were transiently transfected with vectors encoding either
httEx1-25Q–EGFP (COS 25Q EGFP, a), httEx1-72Q–HA without
EGFP [COS 72Q (– EGFP), b, c], httEx1-72Q–EGFP (COS 72Q
EGFP, d) or httEx1-103Q–EGFP (COS 103Q EGFP, e). Forty-eight
hours after transfection, cells were incubated for 1 h with 10 lM
HE [except for COS 72Q (– EGFP, – HE), c] before being analyzed
as live cells by confocal microscopy. EGFP (green, 505–530 nm
wavelength) and HE (blue false color, 552–638 nm wavength)
fluorescence as well as the merged result were recorded. SK-N-SH
neuronal cells transfected with httEx1-103Q–EGFP (SK-N-SH 103Q
EGFP, f) and httEx1-43Q-transfected Tet-Off PC12 neuronal cells
(PC12 43Q EGFP, g) were also analyzed. Note the presence of sev-
eral inclusion bodies adjacent to and in the nuclei of PC12 cells.
(C) COS-7 cells, transiently transfected with the EGFP devoid
httEx1-72Q–HA vector [COS 72Q (– EGFP, – HE, + DCFH-DA)],
were incubated for 20 min in NaCl ⁄ Pi containing 5 lgÆlL)1 DCFH-
DA, and fluorescence was recorded at 510 nm. Scale bar ¼ 20 lm.
W. J. J. Firdaus et al. Oxidation in Huntingtin inclusion bodies
FEBS Journal 273 (2006) 5428–5441 ª 2006 The Authors Journal compilation ª 2006 FEBS 5431

httEx1-72Q–EGFP inclusions [36]. Proteasome inhibi-
tors have been reported to induce intracellular protein
aggregation and to stimulate oxidative protein modifi-
cations such as increased carbonyl formation [42]. We
therefore analyzed whether proteasome inhibitors
could modulate inclusion body oxidation and ⁄or fur-
ther increase ROS production in httEx1-polyQ-expres-
sing cells. HttEx1-72Q–EGFP-expressing COS-7 cells
were therefore exposed or not exposed for 1 h to
10 lm of the inhibitors lactacystin or MG132 prior to
analysis of HE intracellular oxidation by confocal
microscopy (as shown in Fig. 2). Treatment for only
1 h was used to avoid an increase in the size and num-
ber of the inclusion bodies as described in Wyttenbach
et al. (2002) [11]. It can be seen in Fig. 3B that in the
presence of proteasome inhibitors, HE oxidation still
occurred preferentially at the level of the inclusion
bodies. Analysis of the blue color in the merged images
suggested that HE oxidation was even more intense in
treated than in untreated cells. We then analyzed the
level of HE oxidation in httEx1-Q72–EGFP inclusions
of cells overexpressing either Hsp70 ⁄Hdj-1 or Hsp27.
Figure 3C shows that in both cases, HE oxidation was
less intense when compared to cells expressing httEx1-
Q72–EGFP without these Hsps. This phenomenon was
correlated, particularly in the case of Hsp70 ⁄Hdj-1-
overexpressing cells, with reduced size of the inclusion
bodies.
NAC treatment decreases the intracellular
morphologic alterations that occur in the vicinity
of httEx1-polyQ inclusion bodies
The morphology of httEx1-polyQ inclusion bodies was
analyzed by electron microscopy. This was assessed by
analyzing COS-7 cells that were transfected with either
httEx1-Q72–EGFP- or httEx1-Q103–EGFP-encoding
vectors. Two days after transfection, COS-7 cells
were processed for electron microscopy as described
in Experimental procedures. Cells that expressed
HttEx1-72Q–EGFP (Fig. 4A) or HttEx1-103Q–EGFP
(Fig. 4C) polypeptides contained fibrilous inclusion
bodies surrounded by mitochondria and organelles that
showed pathologic morphologies. Moreover, an empty
halo was often detected around the inclusions, partic-
ularly in cells transfected with httEx1-Q103–EGFP-
encoding vector. High magnification of the electron
micrograph presented in Fig. 4C (see Fig. 4E) suggests
that this halo represents a zone where the cell structures
have been destroyed. Indeed, we have recorded several
pictures of dying cells where a large cell domain around
the inclusion bodies is empty and has probably been
destroyed (Fig. 4G). To test whether the halo, or empty
domain, is a consequence of the oxidative stress that
occurs at the surface of inclusion bodies, a similar ana-
lysis was performed, with the difference that COS-7
cells were treated with 2 mm NAC for 12 h before
being processed for electron microscopy. In the pres-
ence of this antioxidant, cells still contained fibrillar
inclusion bodies, but the cellular morphology surround-
ing the inclusions was less altered. Particularly, no
empty halo or empty space could be detected (Fig. 4B,D
and the higher magnification presented in Fig. 4F).
A
B
C
Fig. 3. Analysis of factors that could modulate oxidation of HE at
the level of htt inclusion bodies.(A) COS-7 cells were transiently
transfected with vector encoding httEx1-72Q–EGFP. Thirty-six
hours after transfection, cells were either kept untreated (NT) or
treated for 12 h with 2 mM NAC before being processed to analyze
HE oxidation at the level of htt inclusion bodies (as described in the
legend of Fig. 2 and in Experimental procedures). (B) Same as (A),
but in this case, 47 h after transfection, cells were treated or not
treated for 1 h with 10 lM lactacystin or MG132 before being ana-
lyzed. (C) Cotransfection of the cells with vectors encoding either
Hsp70 ⁄ Hdj-1 or Hsp27 was also performed. Note the reduced size
of the inclusions. Confocal microscopy of live cells is presented.
EGFP (green) and HE (blue false color) fluorescence as well as the
merged result were recorded. Scale bar ¼ 20 lm.
Oxidation in Huntingtin inclusion bodies W. J. J. Firdaus et al.
5432 FEBS Journal 273 (2006) 5428–5441 ª 2006 The Authors Journal compilation ª 2006 FEBS

These cellular alterations close to inclusions may induce
a necrotic type of death (Fig. 4G) or, if they are less
intense, apoptotic death characterized by nuclear dis-
ruption and apoptotic body formation (Fig. 4H).
Partially purified inclusion bodies are enriched
in oxidized proteins
To further analyze the oxidative stress that occurs at
the level of httEx1-polyQ inclusions, experiments were
performed to obtain and analyze cell extracts enriched
in these structures. We did not plan to obtain pure
inclusion bodies, as we were interested in analyzing the
oxidation (using an oxyblot approach) of the cellular
material surrounding these structures. As inclusion
body purification necessitated a large number of cells,
we used stably transfected PC12 43Q–EGFP Tet-Off
cells, which, upon stimulation in doxycycline hydro-
chloride-devoid medium, allow the nuclear accumu-
lation of httEx1-43Q–EGFP polypeptide. Like the
cytoplamic inclusions observed in COS-7 cells, these
nuclear granules are also centers of oxidative events
(Fig. 2Bg). HttEx1-43Q–EGFP inclusion bodies were
partially purified as described in Experimental proce-
dures. Fluorescent and phase contrast microscope ana-
lysis findings are presented in Fig. 5A. The partially
purified material shows the presence of EGFP granules
surrounded by cellular material. Analysis of the pro-
tein content revealed that the partially purified mater-
ial is enriched in several polypeptides, particularly in
the 20 kDa range (Fig. 5B). Immunoblot analysis
using anti-EGFP serum also revealed that the purified
A72Q 72Q+NAC
103Q 103+NAC
5 µm
5 µm
5 µm
gr
chap
gr
1 µm
B G
H
DC
E F
Fig. 4. Electron microscopic analysis of htt inclusion bodies in transfected COS-7 cells. COS-7 cells transiently expressing httEx1-72Q–EGFP
(A, B) or httEx1-103Q–EGFP (C, D, E, F) were either kept untreated (A, C, E) or treated (B, D, F) with 2 mM NAC. The NAC treatment started
36 h after transfection and lasted for 12 h. At the end of the NAC treatment, i.e. 48 h after transfection, cells were processed for electron
microscopy analysis as described in Experimental procedures. High-magnification micrographs are shown to illustrate the cellular morphology
in the vicinity of httEx1-polyQ–EGFP inclusion bodies (E, F). Arrowheads in (C) and (D) denote the regions of the inclusion that were enlarged
in the (E) and (F) magnification panels. Note, in (A), (C) and (E), the altered cellular morphology in the vicinity of httEx1-polyQ–EGFP inclusion
bodies. In NAC-treated cells (B, D, F), the halo surrounding the inclusions is no longer detectable and the cellular morphology is almost nor-
mal. In (G) and (H), images of dying httEx1-72Q–EGFP-expressing COS-7 cells are presented. G, necrotic morphology with enhanced cellular
destruction in the vicinity of the inclusion; H, apoptotic death; ap, apoptotic body; ch, chromatin; gr, htt granule or aggregate. Scale bars are
indicated in the figure.
W. J. J. Firdaus et al. Oxidation in Huntingtin inclusion bodies
FEBS Journal 273 (2006) 5428–5441 ª 2006 The Authors Journal compilation ª 2006 FEBS 5433

material was enriched (about 2.5-fold, see Experimen-
tal procedures) in httEx1-43Q–EGFP polypeptide
(about 45 kDa). Immunoblot detection of protein car-
bonyl residues using 2,4-dinitrophenylhydrazine (2,4-
DNPH)-treated extracts was performed as described in
Experimental procedures. Quantification analysis of
the blot revealed a three-fold overall increase in the
level of oxidized proteins in the partially purified
material. Moreover, it was also possible to detect oxid-
ized polypeptides in the enriched fraction that were
undetectable in total cell extracts. Note that no major
oxidized polypeptide had an SDS gel migration similar
to that of httEx1-polyQ, suggesting that the oxidation
process occurs mainly at the level of the cellular pro-
teins surrounding or trapped in the inclusions rather
than at the level of httEx1 polypeptide itself.
The structural organization and localization of
httEx1-polyQ inclusion bodies, as well as the
oxidative stress associated with these structures,
are iron-dependent phenomena
A paradoxical role is believed to be played by transition
metals such as copper and iron in the pathology of
neurodegenerative diseases [43,44]. For example, htt is
an iron-regulated protein [4], and the formation of ROS
is detected in in vitro assays in which aggregated a-syn-uclein [45], b-amyloid [46,47] or prion protein fragment
[48] are incubated in the presence of redox-active
transition metals. We therefore tested the effect of
deferroxamine, an iron-chelating agent, on the oxidative
stress generated by inclusion bodies in COS-7 cells tran-
siently transfected with httEx1-103Q–EGFP vector and
in PC12 43Q–EGFP-Tet-Off cells grown in doxycycline
hydrochloride-devoid medium. Twenty-four hours after
transfection, COS-7 cells were treated with 5 mm defer-
roxamine for 24 h before being analyzed. As seen in
Fig. 6A, these cells displayed a drastic decrease in the
size of most inclusion bodies formed by httEx1-103Q,
suggesting that, in COS-7 cells, iron plays a key role in
the formation and ⁄or structural integrity of httEx1-
polyQ inclusion bodies. Moreover, most of the deferrox-
amine-generated small inclusions were unable to oxidize
HE. In some cells, both the original large inclusions
stained with HE and the small inclusions, usually less
prone to oxidation, coexisted (Fig. 6Ab, arrows). The
antioxidant effect generated by deferroxamine was
further quantified by analyzing the fluorescence emitted
by oxidized HE by fluorescence-activated cell sorting
(FACS) analysis (see Experimental procedures). As seen
A Ba
b
Fig. 5. Analysis of the oxidative protein status of partially purified httEx1-43Q–EGFP nuclear inclusions isolated from stably transfected PC12
neuronal cells. httEx1-43Q–EGFP nuclear inclusions were partially purified from httEx1-43Q–EGFP-transfected Tet-Off PC12 cells grown for
6 days in culture medium devoid of doxycycline hydrochloride to induce the accumulation of httEx1-43Q–EGFP nuclear inclusions as des-
cribed in Experimental procedures. (A) Detection of EGFP-positive inclusions: (a) starting material; (b) enriched nuclear fraction. Merged ima-
ges of EGFP fluorescence (arrows) and phase contrast analysis are presented. Arrows point to EGFP-positive inclusions. Bar: 10 lm (a) and
5 lm (b). (B) SDS ⁄ PAGE of proteins. Quantitatively equivalent amounts of proteins present in total cell lysate (Tot) and the enriched nuclear
fraction (NF) were analyzed. Coomassie blue-stained gels, red Ponceau staining of nitrocellulose membrane after blotting, immunoblot detec-
tion of EGFP and oxyblot analysis (see Experimental procedures) are presented. The immunoblots were probed with anti-EGFP serum and
the oxyblot was probed with anti-2,4-DNPH serum. Visualization was with enhanced chemiluminescence (ECL), as described in Experimental
procedures. The samples obtained from the derivation-control solution (negative controls of oxyblots; see Experimental procedures) were
devoid of any signals and are not presented in the figure. A, actin; H, histones.
Oxidation in Huntingtin inclusion bodies W. J. J. Firdaus et al.
5434 FEBS Journal 273 (2006) 5428–5441 ª 2006 The Authors Journal compilation ª 2006 FEBS

in Fig. 6B, in COS cells expressing httEx1-103Q–EGFP,
oxidized HE fluorescence was more intense (about 2.5-
fold) than in httEx1-25Q–EGFP-expressing cells. The
effect was not observed if httEx1-103Q–EGFP-expres-
sing cells were exposed to deferroxamine (24 h, 5 mm)
prior to analysis. In httEx1-25Q–EGFP-expressing cells,
deferroxamine had no significant effect (not shown).
Hence, HE oxidation in httEx1-103Q–EGFP-expressing
COS-7 cells appears to be iron-dependent.
Similar analysis performed in PC12 43Q–EGFP Tet-
Off cells revealed that deferroxamine interfered with
the nuclear localization of the inclusion bodies formed
by httEx1-43Q–EGFP and strongly reduced their abil-
ity to oxidize HE (Fig. 7). However, no significant
changes in the size of the 43Q–EGFP inclusions were
detectable, probably because the inclusions formed by
httEx1-43Q–EGFP are rather small compared to those
formed by httEx1-103Q in COS-7 cells. It is also inter-
esting to note that some HE-negative inclusions were
recovered outside of PC-12 cells.
A B C
FEDDEF
NT
EGFP HE Merge
Fig. 7. Iron as a key modulator of the localization and oxidation of
htt inclusion bodies present in the nucleus of PC12 neuronal
cells. httEx1-43Q-transfected Tet-Off PC12 cells were grown for
6 days in culture medium devoid of doxycycline hydrochloride to
induce the accumulation of httEx1-43Q–EGFP nuclear inclusions.
Cells were either kept untreated (NT) or treated for 24 h with
5 mM deferroxamine (DEF) before being processed to analyze
HE oxidation at the level of EGFP-positive htt nuclear inclusion
bodies (as described in Experimental procedures). Note the intra-
cellular redistribution of the inclusions in the cytoplasm or outside
of the cells (arrow) and their decreased ability to oxidize HE.
Scale bar ¼ 20 lm.
A B
Fig. 6. Iron chelation alters the structure and oxidation of htt inclusion bodies present in the cytoplasm of COS-7 cells. (A) Effect of iron on
the shape and oxidation of htt inclusions. COS-7 cells were transiently transfected with httEx1-103Q–EGFP-encoding vector. Twenty-four
hours after transfection, cells were either kept untreated or treated for 24 h with 5 mM deferroxamine before being processed for confocal
microscopy analysis of HE oxidation at the level of EGFP-positive htt cytoplasmic inclusion bodies (as described in the legend of Fig. 2 and
in Experimental procedures). (a) Untreated cells. Two examples of deferroxamine-treated cells are presented in (b) and (c). Note the
decreased size of most inclusions and their weak ability to oxidize HE. In (b), the disruption of the inclusion is not complete: the arrows point
to inclusions that are still large and positive for HE oxidation. In (c), only small granules are detected. In (a), (b) and (c), the EGFP background
was artificially increased to allow better detection of the cell contours. The images shown are representative of three independent analyses.
Bar ¼ 20 lm. (B) ROS produced by htt inclusion bodies are iron-dependent. COS-7 cells were transiently transfected with vectors encoding
either httEx1-25Q–EGFP (25Q) or httEx1-103Q–EGFP (103Q). Twenty-four hours after transfection, cells were either kept untreated or trea-
ted for 24 h with 5 mM deferroxamine (DEF) before being processed for ROS analysis using HE as a probe and fluorescence detection by
FACS cytometry (see Experimental procedures). A representative experiment is presented.
W. J. J. Firdaus et al. Oxidation in Huntingtin inclusion bodies
FEBS Journal 273 (2006) 5428–5441 ª 2006 The Authors Journal compilation ª 2006 FEBS 5435

Discussion
In transiently transfected mammalian cells, the forma-
tion of cytoplasmic perinuclear inclusion bodies in
response to httEx1-polyQ expression correlates, in a
CAG repeat expansion-dependent manner, with
increased levels of intracellular ROS [11]. A similar
observation was made in yeast cells expressing mutant
httEx1 with 103 CAG repeats [35]. This oxidative
stress, which appears to be linked to the presence of
perinuclear inclusion bodies [36], alters the cellular
environment, particularly mitochondria [49] concentra-
ted in the vicinity of the inclusion bodies [50,51]. Here
we have analyzed the intracellular localization of the
oxidative events generated in response to httEx1-polyQ
expression. Using confocal microscopic analysis of live
cells incubated with HE, we observed that this probe
was oxidized at the level of httEx1 inclusion bodies.
In httEx1-72Q–EGFP cells, the intensity and size of
oxidized HE fluorescent spots were often less than
those of spots showing EGFP fluorescence, but the
intensity of fluorescence was more intense than, and
the size closely matched, those of EGFP fluorescent
spots in httEx1-103Q–EGFP-expressing cells. Similar
HE oxidation has been observed in nuclear httEx1
bodies of neuronal SK-N-SH and PC12 cells, suggest-
ing that the oxidative events induced by this neuronal
protein can occur in the cytoplasm or nucleus and are
not cell type-specific. It is of interest that in httEx1-
72Q–EGFP-expressing cells, some inclusion bodies
appeared to be devoid of oxidative events. In contrast,
inclusion bodies formed by httEx1-103Q polypeptide
were all positive. Is the oxidation of HE a consequence
of a high level of ROS in the vicinity of the inclusion
bodies? Experiments performed in the presence of anti-
oxidants, such as NAC, suggest that this is indeed the
case, as in cells exposed to NAC, a drastic loss of
oxidized HE fluorescence was noticed, confirming that
the phenomenon recorded is representative of oxidative
events at the level of inclusion bodies. We observed
that Hsp70 ⁄Hdj-1 or Hsp27 overexpression decreased
the overall cellular intensity of HE oxidation. This
phenomenon may be a consequence of the decreased
size of httEx1-polyQ bodies and ⁄or of the antioxidant
chaperone activity of some of the Hsps, particularly
Hsp27, which maintains glutathione in its reduced
form and decreases intracellular levels of oxidized pro-
teins [52–54] and redox-sensitive Fe(II) [55,56]. In cells
exposed to proteasome inhibitors, which are known to
stimulate oxidative protein modifications [42], a higher
level of HE oxidation at the level of htt inclusion bod-
ies was noted (this study), as well as an increased level
of oxidized proteins [36].
To further investigate the oxidative events taking
place at the level of htt inclusion bodies, we performed
electron microscope analysis. We show here that the
cell structure is destroyed in the vicinity of the inclu-
sion bodies, a phenomenon that was CAG repeat
expansion-dependent and did not develop in cells
exposed to antioxidants. Furthermore, analysis of par-
tially purified inclusion bodies, which still contained
cellular material tags, revealed the high level of oxid-
ized proteins associated or copurifying with the inclu-
sions. These observations confirm that oxidative events
are taking place at the level of htt inclusion bodies and
that these events could be deleterious to the cell.
How can inclusion bodies formed by httEx1-polyQ
generate oxidative events? No easy answer can be given
to this question; however, several recent observations
point to a role played by redox-active transition metals,
such as iron and copper, which are important factors in
the pathology of neurodegenerative diseases [43,44].
For example, the brain regions that are most affected in
these diseases also often contain deposits of iron and
copper [43,44,57–59]. Membrane-associated oxidative
stress, which is a metal-catalyzed oxidative disruption
of membrane protein and lipid signaling, occurs in the
pathogenesis of Alzheimer’s, disease, Parkinson’s dis-
ease and Huntington’s diseases [60]. Moreover, purified
aggregated a-synuclein and b-amyloid, which do not
spontaneously form free radicals [61], can do so if they
are in contact with redox-active Fe(II) [45,47]. Finally,
htt is an iron-regulated polypeptide [4]. This prompted
us to analyze the role played by the iron chelator defer-
roxamine in the oxidative events associated with inclu-
sion bodies. We observed that chelatable iron played a
key role in the formation and ⁄or structural integrity
and localization of httEx1-polyQ inclusion bodies.
Chelatable iron was also necessary to promote the oxi-
dative events associated with these aggregated structu-
res. These observations are reminiscent of an earlier
observation [47] suggesting that the formation of aggre-
gated a-synuclein and b-amyloid is a metal-dependent
mechanism that releases ROS as side products of the
process. ROS produced during aggregate formation are
subsequently converted to hydroxyl radicals by an
Fe(II)-catalyzed Fenton reaction. Our results support
the hypothesis that the formation of htt inclusion bodies
is also an iron-dependent process that generates ROS.
What is the role of the oxidative events initiated by
httEx1-polyQ inclusion bodies? Do they lead to the
oxidation of mutant htt and therefore enhance its de-
gradation? This is probably not the case, as oxyblot
analysis of httEx1-polyQ-expressing cells or partially
purified inclusions revealed no specific oxidation of
httEx1-polyQ polypeptides but only enhanced oxida-
Oxidation in Huntingtin inclusion bodies W. J. J. Firdaus et al.
5436 FEBS Journal 273 (2006) 5428–5441 ª 2006 The Authors Journal compilation ª 2006 FEBS

tion of cellular proteins (our results and [36]). Hence,
our results are probably not suggestive of an ‘oxido-
some type’ of mechanism that involves the specific
oxidation of aggregated httEx1-polyQ proteins.
The provocative idea has been proposed that htt
inclusion bodies are in fact beneficial to the cell, by
promoting the clearance of mutant protein by activa-
ting autophagy through the inhibition of mammalian
target for rapamycin (mTOR) [18] and by neutralizing
the toxicity of dispersed filamentous htt aggregates,
such as proteasome inhibition [62–64]. The fact that
htt inclusion bodies can be centers of iron-dependent
oxidative events may therefore represent a problem
that the cell has to deal with to promote the formation
and clearance of these bodies. Inclusion clearance
could therefore be balanced between the rate of htt
aggregation and the oxidative stress that is generated
by this phenomenon. Both phenomena depend on the
intracellular level of iron that can be chelated by defer-
roxamine. However, the level of iron required in each
case is not known, and the levels may be different,
allowing only a few possibilities for the cell to promote
inclusion formation without generating oxidative side-
effects. Further studies will be required to test whether
inclusions (formed by httEx1-43Q or httEx1-72Q) that
are devoid of oxidative events are better able to
undergo an autophagy process than those that are
oxidized, and whether antioxidants or iron chelators
that abolish httEx1-polyQ toxicity could stimulate the
autophagy process.
Experimental procedures
Cell culture, reagents, DNA vectors and
transfection
African green monkey kidney (COS-7) and neuronal SK-
N-SH and Tet-Off PC12 cells were cultured, seeded in
DMEM (Gibco, InVitrogen, Paisley, UK) supplemented
with 10% heat-inactivated neonatal bovine serum. The
medium included penicillin and streptomycin (50 UÆmL)1)
(Gibco, InVitrogen), and 1 lgÆmL)1 Fungizone (Gibco,
InVitrogen). The growing medium of PC12 cells was
further supplemented with 10 lgÆmL)1 of doxycycline
hydrochloride (Gibco, InVitrogen). The cells were main-
tained at 37 �C in a 5% CO2 atmosphere with 95%
humidity. EGFP-encoding DNA control vector (called
pCineo-EGFP) as well as the same vector containing
httEx1 with 25, 72 or 103 glutamine repeats fused to EGFP
(called httEx1-25Q–EGFP, httEx1-72Q–EGFP and httEx1-
103Q–EGFP) have already been described [11]. We have
also used vectors encoding Ex1 of htt fused to HA that
contain 25 or 72 glutamine repeats (called httEx1-25Q–HA
and httEx1-72Q–HA) [11]. Control vector (pCineo) and the
same vector bearing Hsp27 (pCIneohsp27) have already
been characterized [65], as well as vectors encoding human
Hdj-1(Hsp40), Hdj-2, and Hsp70 [11]. For transfection
experiments, exponentially growing COS-7 and SK-N-SH
cells were plated in 60 mm dishes (5 · 105 cells per dish) 1
day before transfection. Each transfection experiment was
performed with 3 lg of DNA encoding httEx1-polyQ or
the various chaperones (Hsp70 ⁄Hdj-1 and Hsp27) using
lipofectamine (Gibco, InVitrogen) according to the manu-
facturer’s instructions. In the case of transfection with mul-
tiple vectors, we used identical amounts of each vector
(1–2 lg of DNA to a total of 4 lg). Forty-eight hours aftertransfection, the cell medium was removed and replaced
with DMEM supplemented with 10% fetal bovine serum.
PC12 cells expressing httEx1-Q43–EGFP polypeptide under
the control of a Tet-Off promoter have already been
described [34]. Induction was performed by growing PC12
Tet-Off cells for 6 days in culture medium devoid of doxy-
cycline hydrochloride (Sigma–Aldrich, St-Quentin-Fallavier,
France). Deferroxamine, NAC, lactacystin and MG132
were obtained from Sigma–Aldrich.
Determination of intracellular ROS levels
Forty-eight hours after transfection, COS-7 cells were pla-
ted in triplicate at a concentration of 5 · 104 cells per well
(96-well culture plates) and allowed to grow for 24 h at
37 �C. Cells were then washed three times in NaCl ⁄Pi
(devoid of calcium and magnesium) before being incubated
for 10 min in NaCl ⁄Pi containing 40 lgÆmL)1 of HE. Flow
cytometric analysis was performed using a FACScalibur
cytometer (Becton Dickinson, Mountain View, CA) using a
488 nm excitation wavelength. The emission filter was
610 nm bandpassed for EB fluorescence (FL2-H).
In vivo intracellular EGFP and ROS localization
by confocal microscopy
Transiently transfected COS-7 and neuronal SK-N-SH cells
were grown on coverslips in 60 mm dishes, as were stably
transfected PC-12 cells. Forty-eight hours after transfection,
cells were rinsed once in DMEM supplemented with 10%
fetal bovine serum. Control of oxidative stress in mitochon-
dria was assessed by treating cells for 1 h with 10 lLÆmL)1
of an inhibitor of ubiquinol oxidation, myxothiazol
(Sigma–Aldrich). Stock solutions of 1 mm HE were prepared
in dimethylsulfoxide on the day of the experiments and were
kept in the dark. HE at a concentration of 10 lm was added,
and the cells were incubated for 1 h at 37 �C. We also used
DCFH-DA to detect ROS in live cells transfected with
HA-tagged httEx1-72Q. DCFH-DA at a concentration of
5 lm was added, and the cells were incubated for 20 min at
37 �C. After incubation with HE or DCFH-DA probes, cells
W. J. J. Firdaus et al. Oxidation in Huntingtin inclusion bodies
FEBS Journal 273 (2006) 5428–5441 ª 2006 The Authors Journal compilation ª 2006 FEBS 5437

were washed once with NaCl ⁄Pi devoid of calcium and mag-
nesium before being observed alive (in NaCl ⁄Pi devoid of
calcium and magnesium) using a confocal inverted micro-
scope equipped with 40· oil immersion objective lens (Zeiss
LSM 510 Meta, Axiovert 200X, Jena, Germany) in k mode.
This configuration was used to achieve separation of the
different fluorescences. The microscope was able to scan
the whole visible spectrum (detection every 10 nm). Using
this configuration, the spectrum of each fluorochrome was
determined, and these spectra were used to separate, in the
final image, the fluorescence of each signal by spectral de-
convolution. The fluorochromes were excited at 488 nm,
and the emission wavelength was 590 nm for oxidized HE
and 510 nm for EGFP or oxidized DCFH-DA. For con-
venient visualization of the phenomenon, a false color
(blue) was used to monitor the red fluorescence generated
by oxidized HE (EB). Groups of cells as well as single cells
were randomly selected from the microscope field and ana-
lyzed for the fluorescence of either HE (blue), EGFP
(green), DCFH-DA (green) or the superimposed merged
images. In the merged images, a blue color is indicative of
intense oxidation of HE. Image processing was performed
using lsm 510 meta software (Zeiss, Jena, Germany) and
photoshop 7.0 (Adobe Inc., Mountainview, CA, USA).
Partial purification of httEx1-43Q–EGFP nuclear
inclusions from PC12 cells
HttEx1-43Q–EGFP nuclear inclusions were partially puri-
fied according to the method of Mitsui et al. [66], with some
modifications. PC12 43Q–EGFP Tet-Off cells (5 · 107) were
plated in 10 100 mm tissue culture dishes (5 · 106 cells per
dish) and cultured for 6 days at 37 �C under 5% CO2 in
DMEM containing 10% fetal bovine serum, 50 UÆmL)1
penicillin and streptomycin (Gibco, InVitrogen) and
1 lgÆmL)1 Fungizone (Gibco, InVitrogen). During the incu-
bation, the medium was devoid of doxycycline hydrochlo-
ride to allow expression of httEx1-43Q–EGFP polypeptide.
Cells were collected in an ice-cooled homogenization glass
potter with 3 mL of lysis buffer composed of NaCl ⁄Pi con-
taining 10 mm MgCl2, 1500 U of DNase I (Sigma–Aldrich)
and 3 U of RNase A (Sigma–Aldrich). Homogenization
was performed with a Potter–Elvehjem-type homogenizer
with 30 up–down strokes on ice. Lysates were transferred in
eppendorf tubes and centrifuged at 1500 g for 30 min with
an Eppendorf centrifuge (Hamburg, Germany), rotor type
10 · 1.5 mL ⁄ 2 mL aerosol tight motor with lid. The pres-
ence of inclusion bodies in the pellets was detected using a
fluorescence inverted microscope equipped with a 40· oil
immersion objective lens (Zeiss Axiovert 200M; Zeiss, Jena,
Germany) using a fluorescein isothiocyanate filter. The pel-
lets were then washed several times in NaCl ⁄Pi containing
4% sarcosyl. The partially purified inclusion bodies were
then stored at ) 80 �C prior to gel electrophoresis and
immunoblot analysis.
Electron microscopy
Forty-eight hours after transfection, COS-7 cells grown in
60 mm diameter dishes (TPP, Zurich, Switzerland) were
fixed for 30 min at 4 �C in a buffer containing 2% glutaral-
dehyde in 0.1 m sodium cacodylate ⁄HCl buffer (pH 7.4).
Cells were then rinsed three times (overnight) at 4 �C in
0.1 m sodium cacodylate ⁄HCl buffer (pH 7.4) containing
0.2 m sucrose before being postfixed for 30 min at 4 �C in a
buffer composed of 1% osmium tetroxide and 0.15 m
sodium cacodylate ⁄HCl adjusted to pH 7.4. Cells were then
dehydrated with graded ethanol, scraped and pelleted in
70% ethanol, and embedded in Epon as a cell pellet. After
polymerization at 60 �C for 3 days, ultrathin sections
(60–80 nm) were cut using an RMC MTX ultramicrotome
(Ventana, France), collected on 200 mesh copper grids,
stained with uranyl acetate and lead citrate, and observed
with a JEOL 1200 CX transmission electron microscope
(Tokyo, Japan). Images were recorded with a Megaview II
numeric camera (New York, NY, USA), and analysis
software (Eloise, Roissy, France) was used to analyze the
images.
Immunoblot analysis and protein carbonyl
residue determination (oxyblot)
Protein concentration was determined in aliquots using the
Bradford protein assay. Total protein samples were separ-
ated in 12% SDS ⁄PAGE before being analyzed in immu-
noblots probed with anti-EGFP (1 : 1000) (Molecular
Probes ⁄ Interchim, Montlucon, France) and peroxidase-
labeled secondary antibody (1 : 1000) (Tebu, Le Perray en
Yvelines, France). Protein bands were visualized with the
ECLTM system (GE Healthcare, Chalfont St Giles, UK)
and autoradiographs were recorded onto X-Omat LS films
(Eastman Kodak Co., Rochester, NY). Immunoblot detec-
tion of carbonyl residues was done as previously described
[67] using the S7150 OxyblotTM Protein Oxidation Detec-
tion Kit from Chemicon International (Temecula, CA).
In brief, 48 h after transfection, cells were lysed in SDS
(6% final concentration) in the presence of 50 mm
dithiothreitol. Ten microliters of each sample lysate was
transferred into each of two eppendorf tubes and treated
for 15 min with either 10 lL of the 1· 2,4-DNPH solution
or 10 lL of the derivation-control solution (negative con-
trol). After incubation, 7.5 lL of neutralization solution
was added to both tubes. Proteins were then analyzed by
gel electrophoresis, and immunoblotting was performed
using anti-2,4-DNPH according to the manufacturer’s
instructions. Immune complexes were detected by
chemiluminescence using the ECLTMsystem (Amersham-
Biosciences). Autoradiographs were recorded onto X-Omat
LS films (Eastman Kodak Co.). Quantification of the blots
was performed using nih image version 1.62 software
(NIH, Bethesda, MD).
Oxidation in Huntingtin inclusion bodies W. J. J. Firdaus et al.
5438 FEBS Journal 273 (2006) 5428–5441 ª 2006 The Authors Journal compilation ª 2006 FEBS

Acknowledgements
We wish to thank Dominique Guillet for excellent tech-
nical assistance, Dr Simone Peyrol at CeCiL (Centre
Commun de Microscope Electronique et d’Imagerie
Laennec, Faculte de Medicine RTH Laennec, UCB-
Lyon1, France) for assistance with electron microscopy,
and Dr Beatrice Burdin (Centre Technologique des
Microstructures, UCBL-Lyon1, France) for assistance
with confocal microscopy. This work was supported by
the Association pour la recherche sur le cancer (grant
no. 4602) and the Region Rhone-Alpes (Thematique
Cancer) (to APA). Wance Firdaus was a postgraduate
scholarship holder from CNOUS (Centre National des
Oeuvres Universitaires et Scolaires), Paris. Dr Andreas
Wyttenbach thanks the HighQ Foundation and the
Medical Research Council (MRC) for financial support.
William Currie was a Visiting-Professor from Dalhousie
University, NS, Canada, and held a CIHR ⁄CNRS
International Scientific Exchange Scholarship from the
Canadian Institutes of Health Research and the Centre
National de la Recherche Scientifique, France.
References
1 Ambrose CM, Duyao MP, Barnes G, Bates GP, Lin
CS, Srinidhi J, Baxendale S, Hummerich H, Lehrach H,
Altherr M et al. (1994) Structure and expression of the
Huntington’s disease gene: evidence against simple inac-
tivation due to an expanded CAG repeat. Somat Cell
Mol Genet 20, 27–38.
2 Ross CA (1997) Intranuclear neuronal inclusions: a
common pathogenic mechanism for glutamine-repeat
neurodegenerative diseases? Neuron 19, 1147–1150.
3 Perutz MF (1999) Glutamine repeats and neurodegener-
ative diseases: molecular aspects. Trends Biochem Sci
24, 58–63.
4 Hilditch-Maguire P, Trettel F, Passani LA, Auerbach A,
Persichetti F & MacDonald ME (2000) Huntingtin: an
iron-regulated protein essential for normal nuclear and
perinuclear organelles. HumMol Genet 9, 2789–2797.
5 Kegel KB, Kim M, Sapp E, McIntyre C, Castano JG,
Aronin N & DiFiglia M (2000) Huntingtin expression
stimulates endosomal-lysosomal activity, endosome
tubulation, and autophagy. J Neurosci 20, 7268–7278.
6 Sherman MY & Goldberg AL (2001) Cellular defenses
against unfolded proteins: a cell biologist thinks about
neurodegenerative diseases. Neuron 29, 15–32.
7 Davies SW, Turmaine M, Cozens BA, DiFiglia M,
Sharp AH, Ross CA, Scherzinger E, Wanker EE, Man-
giarini L & Bates GP (1997) Formation of neuronal
intranuclear inclusions underlies the neurological dys-
function in mice transgenic for the HD mutation. Cell
90, 537–548.
8 Petersen A, Mani K & Brundin P (1999) Recent
advances on the pathogenesis of Huntington’s disease.
Exp Neurol 157, 1–18.
9 Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald
M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Hous-
man DE & Thompson LM (2000) The Huntington’s dis-
ease protein interacts with p53 and CREB-binding
protein and represses transcription. Proc Natl Acad Sci
USA 97, 6763–6768.
10 Bates G (2003) Huntingtin aggregation and toxicity in
Huntington’s disease. Lancet 361, 1642–1644.
11 Wyttenbach A, Sauvageot O, Carmichael J, Diaz-
Latoud C, Arrigo A-P & Rubinsztein DC (2002) Heat
shock protein 27 prevents cellular polyglutamine toxicity
and suppresses the increase of reactive oxygen species
caused by huntingtin. Hum Mol Genet 11, 1137–1151.
12 Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM,
Clark HB, Zoghbi HY & Orr HT (1998) Ataxin-1 nuclear
localization and aggregation: role in polyglutamine-
induced disease in SCA1 transgenic mice. Cell 95, 41–53.
13 Saudou F, Finkbeiner S, Devys D & Greenberg ME
(1998) Huntingtin acts in the nucleus to induce apopto-
sis but death does not correlate with the formation of
intranuclear inclusions. Cell 95, 55–66.
14 Carmichael J, Chatellier J, Woolfson A, Milstein C,
Fersht AR & Rubinsztein DC (2000) Bacterial and yeast
chaperones reduce both aggregate formation and cell
death in mammalian cell models of Huntington’s dis-
ease. Proc Natl Acad Sci USA 97, 9701–9705.
15 Marsh JL, Walker H, Theisen H, Zhu YZ, Fielder T,
Purcell J & Thompson LM (2000) Expanded polygluta-
mine peptides alone are intrinsically cytotoxic and cause
neurodegeneration in Drosophila. Hum Mol Genet 9,
13–25.
16 Arrasate M, Mitra S, Schweitzer ES, Segal MR & Fink-
beiner S (2004) Inclusion body formation reduces levels
of mutant huntingtin and the risk of neuronal death.
Nature 431, 805–810.
17 Kopito RR (2000) Aggresomes, inclusion bodies and
protein aggregation. Trends Cell Biol 10, 524–530.
18 Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S,
Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane
CJ et al. (2004) Inhibition of mTOR induces autophagy
and reduces toxicity of polyglutamine expansions in fly
and mouse models of Huntington disease. Nat Genet 36,
585–595.
19 Jana NR, Zemskov EA, Wang G & Nukina N (2001)
Altered proteasomal function due to the expression of
polyglutamine-expanded truncated N-terminal hunting-
tin induces apoptosis by caspase activation through
mitochondrial cytochrome c release. Hum Mol Genet 10,
1049–1059.
20 Sakahira H, Breuer P, Hayer-Hartl MK & Hartl FU
(2002) Molecular chaperones as modulators of poly-
W. J. J. Firdaus et al. Oxidation in Huntingtin inclusion bodies
FEBS Journal 273 (2006) 5428–5441 ª 2006 The Authors Journal compilation ª 2006 FEBS 5439

glutamine protein aggregation and toxicity. Proc Natl
Acad Sci USA 99, 16412–16418.
21 Wyttenbach A (2004) Role of heat shock proteins dur-
ing polyglutamine neurodegeneration: mechanisms and
hypothesis. J Mol Neurosci 23, 69–96.
22 Muchowski PJ, Schaffar G, Sittler A, Wanker EE,
Hayer-Hartl MK & Hartl FU (2000) Hsp70 and hsp40
chaperones can inhibit self-assembly of polyglutamine
proteins into amyloid-like fibrils. Proc Natl Acad Sci
USA 97, 7841–7846.
23 Wyttenbach A, Carmichael J, Swartz J, Furlong RA,
Narain Y, Rankin J & Rubinsztein DC (2000) Effects
of heat shock, heat shock protein 40 (HDJ-2), and pro-
teasome inhibition on protein aggregation in cellular
models of Huntington’s disease. Proc Natl Acad Sci
USA 97, 2898–2903.
24 Muchowski PJ (2002) Protein misfolding, amyloid for-
mation, and neurodegeneration: a critical role for
molecular chaperones? Neuron 35, 9–12.
25 Mehlen P, Coronas V, Ljubic-Thibal V, Ducasse C,
Granger L, Jourdan F & Arrigo A-P (1999) Small stress
protein Hsp27 accumulation during dopamine-mediated
differentiation of rat olfactory neurons counteracts
apoptosis. Cell Death Differ 6, 227–233.
26 Wagstaff MJ, Collaco-Moraes Y, Smith J, de Belleroche
JS, Coffin RS & Latchman DS (1999) Protection of
neuronal cells from apoptosis by Hsp27 delivered with a
herpes simplex virus-based vector. J Biol Chem 274,
5061–5069.
27 Halliwell B & Jenner P (1998) Impaired clearance of
oxidised proteins in neurodegenerative diseases. Lancet
351, 1510–1520.
28 Pettmann B & Henderson CE (1998) Neuronal cell
death. Neuron 20, 633–647.
29 Facchinetti F, Dawson VL & Dawson TM (1998) Free
radicals as mediators of neuronal injury. Cell Mol
Neurobiol 18, 667–682.
30 Browne SE, Ferrante RJ & Beal MF (1999) Oxidative
stress in Huntington’s disease. Brain Pathol 9, 147–
163.
31 Bogdanov MB, Andreassen OA, Dedeoglu A, Ferrante
RJ & Beal MF (2001) Increased oxidative damage to
DNA in a transgenic mouse model of Huntington’s dis-
ease. J Neurochem 79, 1246–1249.
32 Halliwell B (2001) Role of free radicals in the neuro-
degenerative diseases: therapeutic implications for anti-
oxidant treatment. Drugs Aging 18, 685–716.
33 Tabrizi SJ, Workman J, Hart PE, Mangiarini L, Mahal
A, Bates G, Cooper JM & Schapira AH (2000) Mito-
chondrial dysfunction and free radical damage in the
Huntington R6 ⁄ 2 transgenic mouse. Ann Neurol 47,
80–86.
34 Giuliano P, De Cristofaro T, Affaitati A, Pizzulo GM,
Feliciello A, Criscuolo C, De Michele G, Filla A, Avv-
edimento EV & Varrone S (2003) DNA damage induced
by polyglutamine-expanded proteins. Hum Mol Genet
12, 2301–2309.
35 Sokolov S, Pozniakovsky A, Bocharova N, Knorre D &
Severin F (2006) Expression of an expanded polygluta-
mine domain in yeast causes death with apoptotic
markers. Biochim Biophys Acta 12, 12–19.
36 Firdaus WJ, Wyttenbach A, Diaz-Latoud C, Currie
RW & Arrigo A-P (2006) Analysis of oxidative events
induced by expanded polyglutamine huntingtin exon 1
that are differentially restored by expression of heat
shock proteins or treatment with an antioxidant. FEBS
J 273, 3076–3093.
37 Perluigi M, Poon HF, Maragon W, Pierce WM, Klein
JB, Calabrese V, Cini C, De Marco C & Butterfield DA
(2005) Proteomic analysis of protein expression and oxi-
dative modification in R6 ⁄ 2 transgenic mice ) a model
of Huntington’s disease. Mol Cell Proteomics 20, 20–30.
38 Sawa A (2001) Mechanisms for neuronal cell death and
dysfunction in Huntington’s disease: pathological cross-
talk between the nucleus and the mitochondria? J Mol
Med 79, 375–381.
39 Panov AV, Gutekunst CA, Leavitt BR, Hayden MR,
Burke JR, Strittmatter WJ & Greenamyre JT (2002)
Early mitochondrial calcium defects in Huntington’s
disease are a direct effect of polyglutamines. Nat Neuro-
sci 5, 731–736.
40 Panov AV, Burke JR, Strittmatter WJ & Greenamyre
JT (2003) In vitro effects of polyglutamine tracts on
Ca2+-dependent depolarization of rat and human
mitochondria: relevance to Huntington’s disease. Arch
Biochem Biophys 410, 1–6.
41 Sun J & Trumpower BL (2003) Superoxide anion gen-
eration by the cytochrome bc1 complex. Arch Biochem
Biophys 419, 198–206.
42 Demasi M & Davies KJ (2003) Proteasome inhibitors
induce intracellular protein aggregation and cell death
by an oxygen-dependent mechanism. FEBS Lett 542,
89–94.
43 Rottkamp CA, Nunomura A, Hirai K, Sayre LM, Perry
G & Smith MA (2000) Will antioxidants fulfill their
expectations for the treatment of Alzheimer disease?
Mech Ageing Dev 116, 169–179.
44 Rottkamp CA, Nunomura A, Raina AK, Sayre LM,
Perry G & Smith MA (2000) Oxidative stress, antioxi-
dants, and Alzheimer disease. Alzheimer Dis Assoc
Disord 14, S62–S66.
45 Turnbull S, Tabner BJ, El-Agnaf OM, Moore S, Davies
Y & Allsop D (2001) alpha-Synuclein implicated in Par-
kinson’s disease catalyses the formation of hydrogen
peroxide in vitro. Free Radic Biol Med 30, 1163–1170.
46 Tabner BJ, Turnbull S, El-Agnaf O & Allsop D (2001)
Production of reactive oxygen species from aggregating
proteins implicated in Alzheimer’s disease, Parkinson’s
disease and other neurodegenerative diseases. Curr Top
Med Chem 1, 507–517.
Oxidation in Huntingtin inclusion bodies W. J. J. Firdaus et al.
5440 FEBS Journal 273 (2006) 5428–5441 ª 2006 The Authors Journal compilation ª 2006 FEBS

47 Tabner BJ, Turnbull S, El-Agnaf OM & Allsop D
(2002) Formation of hydrogen peroxide and hydroxyl
radicals from A (beta) and alpha-synuclein as a possible
mechanism of cell death in Alzheimer’s disease and Par-
kinson’s disease. Free Radic Biol Med 32, 1076–1083.
48 Turnbull S, Tabner BJ, Brown DR & Allsop D (2003)
Copper-dependent generation of hydrogen peroxide
from the toxic prion protein fragment PrP106–126.
Neurosci Lett 336, 159–162.
49 Beal MF (1998) Mitochondrial dysfunction in neurode-
generative diseases. Biochim Biophys Acta 1366, 211–223.
50 Muchowski PJ, Ning K, D’Souza-Schorey C & Fields S
(2002) Requirement of an intact microtubule cytoskele-
ton for aggregation and inclusion body formation by a
mutant huntingtin fragment. Proc Natl Acad Sci USA
99, 727–732.
51 Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder
G, Lehrach H & Wanker EE (2001) Accumulation of
mutant huntingtin fragments in aggresome-like inclusion
bodies as a result of insufficient protein degradation.
Mol Biol Cell 12, 1393–1407.
52 Preville X, Salvemini F, Giraud S, Chaufour S, Paul
C, Stepien G, Ursini MV & Arrigo A-P (1999) Mam-
malian small stress proteins protect against oxidative
stress through their ability to increase glucose-6-phos-
phate dehydrogenase activity and by maintaining opti-
mal cellular detoxifying machinery. Exp Cell Res 247,
61–78.
53 Arrigo A-P (2001) Hsp27: novel regulator of intracellu-
lar redox state. IUBMB Life 52, 303–307.
54 Arrigo A-P, Firdaus WJ, Mellier G, Moulin M, Paul C,
Diaz-Latoud C & Kretz-Remy C (2005) Cytotoxic
effects induced by oxidative stress in cultured mamma-
lian cells and protection provided by Hsp27 expression.
Methods 35, 126–138.
55 Arrigo AP, Virot S, Chaufour S, Firdaus W, Kretz-
Remy C & Diaz-Latoud C (2005) Hsp27 consolidates
intracellular redox homeostasis by upholding glu-
tathione in its reduced form and by decreasing iron
intracellular levels. Antioxid Redox Signal 7, 414–422.
56 Chen H, Zheng C, Zhang Y, Chang YZ, Qian ZM &
Shen X (2006) Heat shock protein 27 downregulates the
transferrin receptor 1-mediated iron uptake. Int J
Biochem Cell Biol 38, 1402–1416.
57 Sayre LM, Perry G, Atwood CS & Smith MA (2000)
The role of metals in neurodegenerative diseases. Cell
Mol Biol 46, 731–741.
58 Sayre LM, Smith MA & Perry G (2001) Chemistry and
biochemistry of oxidative stress in neurodegenerative
disease. Curr Med Chem 8, 721–738.
59 Fernandes Leite J (2001) Huntington s disease: a bimo-
lecular vision. Rev Neurol 32, 762–767.
60 Mattson MP (2004) Metal-catalyzed disruption of mem-
brane protein and lipid signaling in the pathogenesis of
neurodegenerative disorders. Ann NY Acad Sci 1012,
37–50.
61 Turnbull S, Tabner BJ, El-Agnaf OM, Twyman LJ &
Allsop D (2001) New evidence that the Alzheimer beta-
amyloid peptide does not spontaneously form free radi-
cals: an ESR study using a series of spin-traps. Free
Radic Biol Med 30, 1154–1162.
62 Holmberg CI, Staniszewski KE, Mensah KN, Matou-
schek A & Morimoto RI (2004) Inefficient degradation
of truncated polyglutamine proteins by the proteasome.
EMBO J 23, 4307–4318.
63 Venkatraman P, Wetzel R, Tanaka M, Nukina N &
Goldberg AL (2004) Eukaryotic proteasomes cannot
digest polyglutamine sequences and release them during
degradation of polyglutamine-containing proteins. Mol
Cell 14, 95–104.
64 Diaz-Hernandez M, Valera AG, Moran MA, Gomez-
Ramos P, Alvarez-Castelao B, Castano JG, Hernandez F
& Lucas JJ (2006) Inhibition of 26S proteasome activity
by huntingtin filaments but not inclusion bodies isolated
from mouse and human brain. J Neurochem 19, 19–25.
65 Paul C, Manero F, Gonin S, Kretz-Remy C, Virot S &
Arrigo A-P (2002) Hsp27 as a negative regulator of
cytochrome C release. Mol Cell Biol 22, 816–834.
66 Mitsui K, Nakayama H, Akagi T, Nekooki M, Ohtawa
K, Takio K, Hashikawa T & Nukina N (2002) Purifica-
tion of polyglutamine aggregates and identification of
elongation factor-1alpha and heat shock protein 84 as
aggregate- interacting proteins. J Neurosci 22, 9267–9277.
67 Levine RL, Williams JA, Stadtman ER & Shacter E
(1994) Carbonyl assays for determination of oxidatively
modified proteins. Methods Enzymol 233, 346–357.
W. J. J. Firdaus et al. Oxidation in Huntingtin inclusion bodies
FEBS Journal 273 (2006) 5428–5441 ª 2006 The Authors Journal compilation ª 2006 FEBS 5441




















