
“The bone marrow-CNS connection: Implications in the pathogenesis of diabetic retinopathy.”
14
Bone marrow-CNS connections: Implications in the pathogenesis of diabetic retinopathy Jane Yellowlees Douglas a, 1 , Ashay D. Bhatwadekar b,1 , Sergio Li Calzi b,1 , Lynn C. Shaw b,1 , Debra Carnegie b, 1 , Sergio Caballero b, 1 , Quihong Li c,1 , Alan W. Stitt e,1 , Mohan K. Raizada d, 1 , Maria B. Grant b, * , 1 a Clinical and Translational Science Institute, College of Medicine, University of Florida, Gainesville, FL 32610, USA b Department of Pharmacology and Therapeutics, College of Medicine, University of Florida, PO Box 100267, 1600 SWArcher Road, Gainesville, FL 32610-0267, USA c Department of Ophthalmology, College of Medicine, University of Florida, Gainesville, FL 32610, USA d Department of Physiology, College of Medicine, University of Florida, Gainesville, FL 32610, USA e Centre for Vision and Vascular Science, Queen’s University Belfast, Belfast BT12 6BA, UK article info Article history: Available online 15 May 2012 Keywords: Diabetic retinopathy Inflammation Bone marrow Central nervous system Therapeutic approaches to diabetic retinopathy Neuroinflammation abstract Diabetic retinopathy is the fourth most common cause of blindness in adults. Current therapies, including anti-VEGF therapy, have partial efficacy in arresting the progression of proliferative diabetic retinopathy and diabetic macular edema. This review provides an overview of a novel, innovative approach to viewing diabetic retinopathy as the result of an inflammatory cycle that affects the bone marrow (BM) and the central and sympathetic nervous systems. Diabetes associated inflammation may be the result of BM neuropathy which skews haematopoiesis towards generation of increased inflam- matory cells but also reduces production of endothelial progenitor cells responsible for maintaining healthy endothelial function and renewal. The resulting systemic inflammation further impacts the hypothalamus, promoting insulin resistance and diabetes, and initiates an inflammatory cascade that adversely impacts both macrovascular and microvascular complications, including diabetic retinopathy (DR). This review examines the idea of using anti-inflammatory agents that cross not only the blood eretinal barrier to enter the retina but also have the capability to target the central nervous system and cross the bloodebrain barrier to reduce neuroinflammation. This neuroinflammation in key sympathetic centers serves to not only perpetuate BM pathology but promote insulin resistance which is characteristic of type 2 diabetic patients (T2D) but is also seen in T1D. A case series of morbidly obese T2D patients with retinopathy and neuropathy treated with minocycline, a well-tolerated antibiotic that crosses both the blooderetina and bloodebrain barrier is presented. Our results indicates that mino- cycine shows promise for improving visual acuity, reducing pain from peripheral neuropathy, promoting weight loss and improving blood pressure control and we postulate that these observed beneficial effects are due to a reduction of chronic inflammation. Ó 2012 Published by Elsevier Ltd. Contents 1. Introduction ...................................................................................................................... 482 2. Mechanism implicated in the pathophysiology of retinopathy ........................................ .................................. 482 Abbreviations: AGE, advanced glycation end products; VEGF, vascular endothelial growth factors; BBB, bloodebrain barrier; BM, bone marrow; BMDC, bone marrow- derived cell; CRP, C-reactive protein; CNS, central nervous system; COX-2, cyclooxygenase-2; DR, diabetic retinopathy; DIOinduced obesity, diet-; EPCs, endothelial progenitor cells; HSCs, haematopoietic stem cells; HFD, high-fat diet; IKK, IkB kinase; IL-1, interleukin-1; IL-6, interleukin-6; IL-10, interleukin-10; MCP-1 or CCL2, monocyte chemoattractant protein-1; EDN1, Endothelin 1; NFkB, Nuclear factorkB; PRV, pseudo rabies virus; SDF-1, stromal derived factor-1; TNF-a, tumor necrosis factor alpha; T1D, Type 1 diabetes; T2D, Type 2 diabetes. * Corresponding author. Tel.: þ1 352 846 0978; fax: þ1 352 392 9696. E-mail address: grantma@ufl.edu (M.B. Grant). 1 Percentage of work contributed by each author in the production of the manuscript is as follows: All authors contributed equally to this manuscript. Douglas 10%, Bhatwadekar 10%, Li Calzi 10%, Shaw 10%, Carnegie 10%, Caballero 10%, Li 10%, Stitt 10%, Raizada 10%, Grant 10%. Contents lists available at SciVerse ScienceDirect Progress in Retinal and Eye Research journal homepage: www.elsevier.com/locate/prer 1350-9462/$ e see front matter Ó 2012 Published by Elsevier Ltd. doi:10.1016/j.preteyeres.2012.04.005 Progress in Retinal and Eye Research 31 (2012) 481e494
Transcript of “The bone marrow-CNS connection: Implications in the pathogenesis of diabetic retinopathy.”

at SciVerse ScienceDirect
Progress in Retinal and Eye Research 31 (2012) 481e494
Contents lists available
Progress in Retinal and Eye Research
journal homepage: www.elsevier .com/locate/prer
Bone marrow-CNS connections: Implications in the pathogenesis of diabeticretinopathy
Jane Yellowlees Douglas a,1, Ashay D. Bhatwadekar b,1, Sergio Li Calzi b,1, Lynn C. Shaw b,1,Debra Carnegie b,1, Sergio Caballero b,1, Quihong Li c,1, Alan W. Stitt e,1, Mohan K. Raizada d,1,Maria B. Grant b,*,1
aClinical and Translational Science Institute, College of Medicine, University of Florida, Gainesville, FL 32610, USAbDepartment of Pharmacology and Therapeutics, College of Medicine, University of Florida, PO Box 100267, 1600 SW Archer Road, Gainesville, FL 32610-0267, USAcDepartment of Ophthalmology, College of Medicine, University of Florida, Gainesville, FL 32610, USAdDepartment of Physiology, College of Medicine, University of Florida, Gainesville, FL 32610, USAeCentre for Vision and Vascular Science, Queen’s University Belfast, Belfast BT12 6BA, UK
a r t i c l e i n f o
Article history:Available online 15 May 2012
Keywords:Diabetic retinopathyInflammationBone marrowCentral nervous systemTherapeutic approaches to diabeticretinopathyNeuroinflammation
Abbreviations: AGE, advanced glycation end prodderived cell; CRP, C-reactive protein; CNS, centralprogenitor cells; HSCs, haematopoietic stem cells; HFDchemoattractant protein-1; EDN1, Endothelin 1; NFkBType 1 diabetes; T2D, Type 2 diabetes.* Corresponding author. Tel.: þ1 352 846 0978; fax
E-mail address: [email protected] (M.B. Grant).1 Percentage of work contributed by each author
Bhatwadekar 10%, Li Calzi 10%, Shaw 10%, Carnegie 1
1350-9462/$ e see front matter � 2012 Published bydoi:10.1016/j.preteyeres.2012.04.005
a b s t r a c t
Diabetic retinopathy is the fourth most common cause of blindness in adults. Current therapies,including anti-VEGF therapy, have partial efficacy in arresting the progression of proliferative diabeticretinopathy and diabetic macular edema. This review provides an overview of a novel, innovativeapproach to viewing diabetic retinopathy as the result of an inflammatory cycle that affects the bonemarrow (BM) and the central and sympathetic nervous systems. Diabetes associated inflammation maybe the result of BM neuropathy which skews haematopoiesis towards generation of increased inflam-matory cells but also reduces production of endothelial progenitor cells responsible for maintaininghealthy endothelial function and renewal. The resulting systemic inflammation further impacts thehypothalamus, promoting insulin resistance and diabetes, and initiates an inflammatory cascade thatadversely impacts both macrovascular and microvascular complications, including diabetic retinopathy(DR). This review examines the idea of using anti-inflammatory agents that cross not only the blooderetinal barrier to enter the retina but also have the capability to target the central nervous systemand cross the bloodebrain barrier to reduce neuroinflammation. This neuroinflammation in keysympathetic centers serves to not only perpetuate BM pathology but promote insulin resistance which ischaracteristic of type 2 diabetic patients (T2D) but is also seen in T1D. A case series of morbidly obeseT2D patients with retinopathy and neuropathy treated with minocycline, a well-tolerated antibiotic thatcrosses both the blooderetina and bloodebrain barrier is presented. Our results indicates that mino-cycine shows promise for improving visual acuity, reducing pain from peripheral neuropathy, promotingweight loss and improving blood pressure control and we postulate that these observed beneficial effectsare due to a reduction of chronic inflammation.
� 2012 Published by Elsevier Ltd.
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4822. Mechanism implicated in the pathophysiology of retinopathy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 482
ucts; VEGF, vascular endothelial growth factors; BBB, bloodebrain barrier; BM, bone marrow; BMDC, bone marrow-nervous system; COX-2, cyclooxygenase-2; DR, diabetic retinopathy; DIOinduced obesity, diet-; EPCs, endothelial, high-fat diet; IKK, IkB kinase; IL-1, interleukin-1; IL-6, interleukin-6; IL-10, interleukin-10; MCP-1 or CCL2, monocyte, Nuclear factor kB; PRV, pseudo rabies virus; SDF-1, stromal derived factor-1; TNF-a, tumor necrosis factor alpha; T1D,
: þ1 352 392 9696.
in the production of the manuscript is as follows: All authors contributed equally to this manuscript. Douglas 10%,0%, Caballero 10%, Li 10%, Stitt 10%, Raizada 10%, Grant 10%.
Elsevier Ltd.

J. Yellowlees Douglas et al. / Progress in Retinal and Eye Research 31 (2012) 481e494482
3. Microvascular disease and bone marrow . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4843.1. Bone marrow, vasculature endothelium and its repair . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4843.2. Bone marrow and nervous system . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4853.3. Bone marrow neuropathy and DR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 485
4. Chronic inflammation and its impacts on DR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4854.1. Diet-induced obesity, inflammation and DR pathogenesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4864.2. Hypothalamic inflammation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4884.3. Endocrine effects of hypothalamic inflammation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4884.4. Insulin resistance in diabetes and DR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 488
5. Interventions addressing DR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4885.1. Interventions addressing inflammation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4885.2. Corticosteroids . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4885.3. Aldose reductase inhibitors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4895.4. Aspirin and salicylates . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4895.5. Minocycline . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 489
5.5.1. Efficacy in humans . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4895.5.2. Case series: minocycline improved visual acuity and neuropathic pain in diabetic subjects . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 489
6. Implications of anti-inflammatory strategies . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4906.1. Conclusions and future directions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 490Contributions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 491Acknowledgments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 491References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 491
1. Introduction
Among Americans of working age, diabetic retinopathy (DR) isthe leading cause of blindness (National Eye Institute, 2004) andranks as the fourth most common source of vision loss in olderadults (Diabetes Group, 2000).Within the last decade, an estimated937,000 Americans aged 40 and over have been classified as blind,with another 2.4 million suffering from low vision (Congdon et al.,2004). In 2004, retinopathy affected 3.4 percent of the US pop-ulation or over 4 million individuals, with 0.75 percent or nearly900,000 people, suffering from vision-threatening retinopathy(Kempen et al., 2004). Moreover, the number of individuals in theUS with blindness or low vision is estimated to nearly double by2020, due largely to the aging population. However, some specificgroups face far higher risks of DR. A 2004 study estimated thatnearly half of all American Hispanics suffered from some form ofDR, with Hispanics of Mexican ancestry at particular risk (Varmaet al., 2004). Diabetes is continuing to rise at an alarming rateworldwide as developing countries adopt the Western lifestyle anddiet characterized by low physical activity and ingestion of foodsrich in fat, refined carbohydrates and animal protein. China is noexception; the Handan Eye Study indicates in China 21.1 millionadults (30 years or older) had diabetes in 2009, and of those adults,nearly 43% had developed DR and more than half of the DR caseshad progressed to vision-threatening status (Wang et al., 2009).
These statistics are congruent with studies in the United Statesand Europe, indicating that some form of retinopathy affects 40percent of the adult populationwith diabetes (Kempen et al., 2004)or 138 million people worldwide of the estimated 346 millionalready diagnosed with T2D alone (WHO, 2011). Despite recentadvances in drug therapeutics and lifestyle management, theprevalence of T2D is estimated to nearly double by 2030 (WHO,2011), and, with it, the prevalence of DR.
Pharmacotherapy using anti-vascular endothelial growth factor(VEGF) has shown some efficacy in addressing DR in patients withproliferative diabetic retinopathy or macular edema. In trials per-taining to PDR, treatment with bevacizumab has resulted indecreasing leakage in neovascular lesions. Mirshahi et al. con-ducted a large clinical studies in individuals with bilateral PDR withhigh-risk characteristics (Mirshahi et al., 2008). Eyes receivedscatter laser treatment according to the ETDRS protocol and one eye
also received 1.25 mg bevacizumab injection while the other eyereceived sham injection. Ninety percent of the intervention eyeshad complete regression of neovascularization versus 25% in thesham group; however, after initial beneficial response, therewas nodifference in the two groups after 4 months. Clinical trials of bev-acizumab for treatment of PDR show increased risk of tractionalretinal detachment (TRD) following a rapid retinal neovascularregression (Arevalo et al., 2008; Moradian et al., 2008). Moreover,anti-VEGF therapy does little to address the underlying causes thatcontribute to the occlusion and degeneration of retinal capillaries(Kern, 2007).
Recent research has identified chronic inflammation resultingfrom diabetes as a potential cause of damage to the retina via pro-inflammatory proteins, including cyclooxygenase-2 (COX-2),interleukin-1 (IL-1), and tumor necrosis factor-alpha (TNF-a)(Joussen et al., 2004; Kern, 2007). Still other studies have impli-cated changes to bone marrow-derived cells (BMDC), cells partic-ularly adversely influenced by hyperglycaemia and unable tofunction to promote revascularisation and remodelling. Moreover,hyperglycaemia contributes to the production of pro-inflammatorycells (Loomans et al., 2009). Furthermore, studies have consistentlyestablished causal links between diet-induced obesity (DIO), a pro-inflammatory response and hypothalamic inflammation (Choiet al., 2010; De Souza et al., 2005; Thaler et al.). This reviewfocuses on the links between the bone marrow, the central nervoussystem (CNS), diabetes and DR and provides an innovative, mech-anistic persective to the pathogenesis of DR, particularly in light ofthe promising results from our case series showing the efficacy ofan anti-inflammatory therapy in addressing aspects of diabetes. Bysuggesting a novel approach to treating T2D and DR targeting theCNS, this strategy improved visual acuity and symptoms associatedwith painful diabetic neuropathy. It may be determined that use ofminocycline may serve to treat all vascular complications.
2. Mechanism implicated in the pathophysiology ofretinopathy
Epidemiological and prospective data have revealed that thestressors of the diabetic vasculature persist beyond the point whengood glycaemic control had been achieved (Diabetes_Group, 2000)and is referred to as ‘metabolic memory’. The first suggestion of this

Fig. 1. Acellular capillaries-Trypsin digested image of diabetic rat retina showingacellular capillary (red arrow) which is the result of loss of both pericytes and endo-thelial cells. Acellular capillaries are the pathologic hallmark of diabetic retinopathyand their quantification is a useful way to assess the extent of retinopathy.
J. Yellowlees Douglas et al. / Progress in Retinal and Eye Research 31 (2012) 481e494 483
phenomenon came from a study by Engerman and Kern in the1980s. Alloxan-induced diabetic dogs were divided into threegroups according to glycaemic control: poor control for five years,good control for five years, and poor control for two-and-a-halfyears followed by good control for two-and-a-half years. Retinop-athy developed in the latter group, despite subsequent sustainedgood glycaemic control having been achieved (Engerman andKern, 1987).
More recently, studies by Kowluru and coworkers have shownthat hyperglycaemia-induced elevations in inflammatory media-tors in retinal microvascular cells resist reversal after re-institutionof normal glucose conditions (Kowluru et al., 2010).
The standard parameter of glycaemic control, hemoglobin A1Cis an advanced glycation endproduct (AGE). This particular cross-link formation of AGEs serves as an excellent tool to evaluateglucose control. Similar chemical changes in extracellular matrices(ECM) lead to pathological thickening of vascular basementmembranes, including those of the retinal capillaries (Bailey, 2001;Gardiner et al., 2003). Accumulation of AGEs has many deleteriouseffects on basement membrane physiology and cellular function(Boyd-White and Williams, 1996; Monnier et al., 1986; Wells-Knecht et al., 1995). For example, retinal vascular endothelialcells and pericytes are co-dependent on complex, two-waycommunication via paracrine growth factors secreted into theirshared basement to ensure continued survival of both cell types.AGE formation on this basement membrane alters retinal vascularcell function and compromises cell survival by cross-linking orotherwise inactivating vital heparin-binding growth factors andintegrin-recognition domains (Paul and Bailey, 1999).
Accelerated death of endothelial cells, pericytes and microvas-cular smoothmuscle cells is known to occur in diabetic retinopathy(Mizutani et al., 1996; Stitt et al., 1994). This is believed due to theAGE-modification of basement membrane removing key survivalfactors from the matrix (Stitt et al., 2004). The accumulation ofAGEs and associated cross-links on the arginine and lysine residuesof ECM is a recognized phenomenon during diabetes (Fu et al.,1994).
Significantly, this hyperglycaemic damage targets endothelialcells as well as pericytes. The death of both cell types is a hallmark ofdiabetic retinopathy and leads to the formation of naked basementmembrane tubes. These non-perfused, degenerated tubes/capil-laries can be recognized histologically as ‘acellular capillaries’ indiabetic patients, dogs, rats and mice (Engerman and Kern, 1995;Joussen et al., 2004; Kern and Engerman, 1996a,b; Mizutani et al.,1996; Tang et al., 2003), Fig. 1. In turn, acellular capillaries result inlesions that produce irreversible retinal ischemia through theirinability to support blood flow. This vessel destruction leading toretinal ischemia and increased expression of angiogenic growthfactors ultimately triggers retinal neovascularization.While all thesehistological features are well established, the mechanisms under-lying diabetes-induced retinal cell injury and death are incompletelyunderstood. Degeneration of retinal capillaries is recognized to playan important role in the pathogenesis of DR (Davis et al., 1997), withmany investigators believing that the resulting retinal ischemiastimulates pre-retinal neovascularization. Acute ischemia/reperfu-sion of the retina also has been recognized to cause formation ofthese acellular capillaries, even in non-diabetic rats andmice (Zhenget al., 2005).
Endothelial progenitor cells (EPCs) play a role in maintenance ofthe normal vasculature (Fig. 2) and defects in EPCs may contributeto capillary degeneration. In Fig. 2, a trypsin digested rat retina wasstained with CD133, CD14 and lectin. EPCs are present in thevasculature in the diabetic retina and typically appear at thebifurcations of the retinal capillaries and are seen as clusters as wellas single cells.
The collective evidence indicates that the loss of retinal micro-vascular cells, a critical early step in diabetic retinopathy, may notonly be due to increased cell death but also to altered repairmechanisms. In the retina, a defect in EPCs could prevent repara-tion of endothelial injury early on, leading to acellular capillarylesions and retinal ischemia.
As we show in Fig. 3, CD34þ cells are dysfunctional in diabeticsand have inability to home to areas of injury to orchestrate therepair process. In Fig. 3, a model of ischemia reperfusion (I/R) injurythat demonstrates a key feature of diabetic retinopathy, acellularcapillaries, was used to examine the function of CD34þ cells. Mouseeyes were subjected to I/R injury and CD34þ were injected into thevitreous after injury. For this study we isolated CD34þ cells fromdiabetic patients with vascular complications or age- and gender-matched controls. Immunofluorescence studies show that controlcells but not diabetic cells home to areas of injury.
This defect in migration is due in part to a diabetes-associatedreduction in phosphorylation and intracellular redistribution ofvasodilator-stimulated phosphoprotein (VASP), a critical actinmotor protein required for cell migration that also controls vaso-dilation and platelet aggregation (Li Calzi et al., 2008). The effect ofthe chemokine stromal derived factor-1(SDF-1) on VASP redistri-bution is markedly reduced in diabetics (Fig. 4) whereas VASPredistributes to filopodia-like structures following SDF-1 treat-ment in controls. Thus, SDF-1 promotes cytoskeletal changesthrough site- and cell type-specific VASP phosphorylation inhealthy CD34þ cell but in diabetes, blunted responses to this factorand other growth factors may lead to reduced vascular repair andtissue perfusion.
Endothelial injury, acellular capillaries and subsequent retinalischemia result in the compensatory release of chemokines andgrowth factors such as VEGF, SDF-1, andmonocyte chemoattractantprotein-1 (MCP-1 or CCL2) by the ischemia injured retina. Studieshave already revealed correlations between the vitreous levels ofSDF-1, MCP-1 and VEGF and the degree of macular edema and

Fig. 2. EPC staining in rat retina. (A) Representative trypsin digested rat retina stained with CD133 i) CD14 (ii) and lectin (iii) showing presence of EPCs (white arrow) in the retinalvasculature of a diabetic rat. These cells appeared at the bifurcations of the retinal capillaries (B) with occasional clusters (white arrow) (C) or a single cell (D).
J. Yellowlees Douglas et al. / Progress in Retinal and Eye Research 31 (2012) 481e494484
retinopathy in patients with DR (Butler et al., 2005). However,many additional factors have been shown to be increased in thevitreous of diabetics including IGF-1 (Grant et al., 1986); IL1B, IL6,IL8, CCL2, and EDN1 (Zhou et al., 2012) and these factors likelyimpact also DR outcomes.
Restoration of EPC function would result in repair of areas ofcapillary injury (acellular capillaries). This would prevent thedevelopment of ischemia and the subsequent ‘compensatory’retinal expression of chemokines and growth factors that accu-mulate in the vitreous. Central to understanding the role of EPCs invascular repair is an understanding of how BMDC become endo-thelial cells.
3. Microvascular disease and bone marrow
In animal studies EPC-enriched haematopoietic stem cells(HSCs), injected into the eyes of mice, promoted revascularisation,selectively targeting retinal astrocytes, cells that serve as a templatefor both developmental and injury-associated retinal angiogenesis
(Otani et al., 2002). Furthermore, BMDC contribute to vascularisa-tion, providing reparative functions throughout the body, fromtissue perfusion during wound healing to promoting endotheliali-sation of the ischaemic retina (Grant et al., 2002).
3.1. Bone marrow, vasculature endothelium and its repair
In the embryonic blood island, a central HSC is surrounded byhaematopoietic and endothelial precursors (Flamme and Risau,1992). This physical association between endothelial precursorsand HSCs, as well as the realisation that these cells share antigenicdeterminants such as Flk-1/KDR/VEGFR2, Tie-2, and CD34 (Flammeand Risau, 1992; Weiss and Orkin, 1996), suggested that these cellsare derived from a common precursor, the haemangioblast. SinceHSCs isolated from the peripheral circulation are able to providesustained haematopoietic function (Brugger et al., 1995), it wasreasonable that circulating cells may also function as EPCs.
In fact, Asahara et al. demonstrated that cells isolated on thebasis of the expression of the haematopoietic stem cell marker

Fig. 3. Human peripheral blood CD34þ cells from diabetic patients are dysfunctional invascular homing and association and cannot repair damaged retinal vessels. Mouseeyes subjected to ischemia/reperfusion insult underwent injection (into the vitreous)with CD34þ cells isolated from long-term diabetic patients or age- and gender-matched non-diabetic controls. Two days after injection of the cells, the mouse eyeswere harvested and examined by immunofluorescent laser scanning confocalmicroscopy. (A) CD34þ cells (identified by green fluorescence) from diabetic patientsare found near vessels (visualized by red fluorescence), albeit in a random distribution(Zheng et al., 2007). (B) Most cells from non-diabetic control patients are associatedwith vessels. The yellow color is the result of co-localized fluorescence and indicatesthe CD34þ cells are associating and possibly some of the cells actually incorporatinginto vessel walls (Caballero et al., 2007). Images are compressed z-series (approxi-mately 30 mm total thickness). Scale bar ¼ 50 mm.
J. Yellowlees Douglas et al. / Progress in Retinal and Eye Research 31 (2012) 481e494 485
CD34 not only express CD31, Flk-1, and E-selectin after 7 days inculture, but also express endothelial NOS, produce nitric oxide, andhome to areas of angiogenesis in vivo (Asahara et al., 1997). Theirresults were confirmed by Shi et al. (1998) with CD34þ cells thatwere 95% pure. Other investigators have demonstrated that thenumber of circulating CD34þ cells and potentially their ability todirect endothelial repair is increased by treatment with HMG CoAreductase inhibitors (Dimmeler et al., 2001) as well as witherythropoietin (Bahlmann et al., 2004). In addition, injection ofhealthy non-diabetic CD34þ cells into diabetic mice acceleratesrevascularisation of ischaemic limbs (Schatteman et al., 2000) andwound healing (Sivan-Loukianova et al., 2003). Only a subset ofCD34þ cells, those expressing VEGFR-2 and CD133 (Peichev et al.,2000), likely represent circulating cells capable of differentiatinginto endothelial cells. CD34þ cells, as well as the resultant coloniesand/or cells that express endothelial markers as a result of culturingperipheral mononuclear cells under the appropriate conditions, aretypically identified and called EPCs.
Interestingly, HMG CoA reductase inhibitors (Dimmeler et al.,2001; Llevadot et al., 2001) and erythropoietin (Bahlmann et al.,2004) not only increase the number of CD34þ in circulation but
also have been shown to enhance EPC chemotactic activity andenhance their ability to form capillary tubes on matrigel.
3.2. Bone marrow and nervous system
Communication between the bone marrow, CNS, and peripheralnervous system is critical in order to affect rapid immune responsesand to sustain vascular health. Trans-synaptically connectedneurons can be traced using pseudo rabies virus (PRV) expressinggreen fluorescent protein and when PRV is injected into bonemarrow of mice, green fluorescent protein (gfp)þ neurons can bedetected in the brain (Card et al.). The sympathetic nervous system(SNS) regulates the release of BM cells into the circulation(Mendez-Ferrer et al., 2008).
3.3. Bone marrow neuropathy and DR
In models of both T2D and T1D, diabetes is associated witha reduced number of EPCs in the circulation. Moreover, the circa-dian rhythmicity of release of EPCs from the BM is altered, and thedevelopment of BM neuropathy is responsible for these events(Busik et al., 2009). BM neuropathy precedes the development ofDR (Busik et al., 2009), thus connecting DR for the first time toperipheral nerve dysfunction in the BM. These concepts representa paradigm shift in the pathogenesis of DR. BM neuropathy iscentral to the loss of protective vascular progenitors.
Previous studies showed a decrease in tyrosine hydroxylase(THþ) nerves in diabetic rats, suggesting that diabetes is associatedwith loss of adrenergic neurotransmitters, secondary to peripheralneuropathy affecting the BM (Katayama et al., 2006). Based onexperiments with chemically (6-OH-DA) sympathectomized adultmice and the a1-adrenergic antagonist prazosin, Maestroni et al.established that regulated production of monocytes was under aninhibitory noradrenergic tone (Maestroni et al., 1992), suggestingthat loss of this noradrenergic influence, as is seen in rodents withdiabetic peripheral neuropathy, would result in the generation ofmore monocytes (Rameshwar and Gascon, 1992), which is exactlywhat we observe in diabetes.
HSCs reside in two basic places in the marrow: 1) adherent tostromal cells at the endosteal bone surface (primitive HSCs such asLTR-HSCs) or 2) at the sinusoids where they are adherent to endo-thelial cells (more mature HSC). BM stromal cells are derived frommesenchymal stem cells and have a ‘fibroblast-like phenotype(Sorrell et al., 2009). Neurotransmitters andneuropeptides stimulateBM stromal cells to produce cytokines/factors that regulate HSC self-renewal, proliferation and differentiation (Takahashi et al., 2003).
Acting on BM stromal cells, neuropeptides released by sensoryneurons regulate anti-inflammatory cytokine production by thesecells, including interleukin-10 (IL-10). Thus, the loss of key neuro-peptides would result in loss of IL-10, contributing to a pro-inflammatory BM environment in diabetes (Sakagami et al., 1993).Taken together, these findings suggest that loss of neurotransmittersand neuropeptides promotes changes in the BM microenvironmentthat leads to an increased numbers of monocytes and a phenotypicswitch of these cells to a more pathological phenotype.
4. Chronic inflammation and its impacts on DR
Metabolic and physiologic abnormalities in the retina stemmingfrom diabetes suggest inflammation plays a direct role in thedevelopment of DR (Kern, 2007). We observe an increase ininflammatory cells throughout the retina of the diabetic asdemonstrated by CD45þ immunohistochemistry of retinal crosssections compared to control mice (Fig. 5). One key component ofthe inflammatory process involves the attraction and adhesion of

Fig. 4. VASP redistribution is impaired in diabetic EPCs. Immunofluorescence images of EPCs from normal (A, B) and diabetic (C, D) donors left untreated (A, C) or treatedwith 100 nM SDF-1 for 4 h (B, D). Normal EPCs show a dramatic VASP redistribution following treatment (A, B) while EPCs from a diabetic donor do not show any response toSDF-1 (C, D).
J. Yellowlees Douglas et al. / Progress in Retinal and Eye Research 31 (2012) 481e494486
leukocytes to the vascular wall. This process, leukostasis, isparticularly increased in the retinas of diabetic mice (Vincent andMohr, 2007), while in rats leukostasis is associated with retinalendothelial cell injury and death (Joussen et al., 2001). In Fig. 6, weshow flat mounts of trypsin digested retinas from diabetic andcontrol mice. The diabetic vasculature shows the presence of CD133cells with positive staining for the monocyte marker CD14. The
Fig. 5. Diabetic retina contains higher numbers of CD45þcells. Immunohistochemicalanalysis of CD45þ cells in retinal cross sections from control (A) and mice with onemonth of STZ-induced diabetes was performed. (C) Quantitation of the numberof CD45þ cells/section is shown and demonstrates increased number in diabeticcompared to controls.
choice of these phenotypic markers was based on CD133 being theearliest stem cell marker expressed by circulating EPCs (Quiriciet al., 2001) while it has been previously reported that CD14 mayserve as therapeutic alternative for diabetic patients withcompromised CD34 function (Awad et al., 2006). Also CD34 is oftenexpressed by capillary endothelium (Fina et al., 1990) and latedifferentiating EPCs (Thill et al., 2008; Yoder et al., 2007), whichwould make it difficult to separate circulating EPCs from capillaryendothelium. Thus the use of CD133 as a selection marker for EPCsenabled us to easily identify this distinct population of EPCs in theretinal vasculature.
Both T1D and T2D represent forms of chronic inflammation,triggered by metabolic surplus that engages the same signallingpathways present in classic inflammation. Hotamisligil (2006)distinguishes this form of inflammation from the classic responseto injury or immune system challenge with the term ‘meta-flammation’ for metabolically-triggered inflammation. This chronicmetaflammation is characterized by abnormal cytokine production,as well as the activation of a network of inflammatory signallingpathways, in particular the IKK/NFkB pathway that, in non-diabetics,provides protection against insulin resistance (Ye, 2008). Moreover,diabetes is associated with an altered inflammatory response(Chang et al., 1996; Graves et al., 2005; Hotamisligil et al., 1993; Huet al., 2004; Lalla et al., 2007; Liu et al., 2006; Naguib et al., 2004;Pickup, 2004; Pradhan et al., 2001; Yan et al., 2009). Diabeticsdemonstrate increased serum levels of inflammatory markers,including C-reactive protein (CRP) and Interleukin-6 (IL-6), as wellas pro-inflammatory cytokines such as TNF-a (Shoelson et al., 2006).
4.1. Diet-induced obesity, inflammation and DR pathogenesis
The first definitive link between obesity, T2D and chronicinflammationemerged fromstudies revealing theover-expression of

Fig. 6. Immature progenitors are present in the diabetic retina. (A) Trypsin digested diabetic retinal vasculature (i) shows presence of CD133þ cells in diabetic vasculature (ii) withpositive staining for CD14 marker (iii). Surprizingly, vehicle control rat retina (iv) did not show CD133þ cells (v) in 60% of the trypsin-digests although monocytic CD14þ expressionwere detected (vi). N ¼ 6 each vehicle control or diabetic treatment. (B) Individual count of number of CD133þ and CD14þ cells showing two fold increase in CD133þ cells(p < 0.001).
J. Yellowlees Douglas et al. / Progress in Retinal and Eye Research 31 (2012) 481e494 487
TNF-a in the adipose tissue of obese mice (Hotamisligil et al., 1993).A pro-inflammatory cytokine, TNF-a, is similarly over-expressed inboth the adipose and muscle tissue of obese humans (Hotamisligilet al., 1995; Kern and Engerman, 1995). Strikingly, Hotamisligilet al. (1993) and Feinstein et al. (1993) both induced insulinresistance via TNF-a in rats in vivo and in vitro (Kanety et al., 1995).Moreover, after initiation of a high-fat diet (HFD), the endotheliumexpresses cell adhesion molecules to bind leukocytes. In contrast,circulating leukocytes in healthy, non-obese patients do notadhere to the endothelium (Blake and Ridker, 2002). Over the pastdecade, studies have consistently documented the impact of anHFD and of diet-induced obesity (DIO) on the expression of pro-inflammatory cytokines and, perhaps more importantly, oninflammatory responsive proteins in the hypothalamus (De Souzaet al., 2005; Posey et al., 2009).
Central to the pathogenesis of DR is increased oxidative stressand subsequent activation of inflammatory pathways includingTNF-a, IL-1a and NFk-b activation (Kowluru and Chan, 2007).Inflammatory cytokines increase iNOS and the resulting patho-logically high levels of nitric oxide (NO) diffuse to adjacent tissues
where they can adversely impact retinal tissues (Caldwell et al.,2005). Receptors for cytokines are found on multiple cell types inthe retina including endothelial cells, neuronal cells, Müller cells,microglia, and astrocytes. Both circulating cytokines, entering theretina via disruption of retinal vasculature, and cytokines releasedby cells within the retina (neurons, microglia, and astrocytes)contribute to the inflammatory process. While the role of braininflammation in T2D is well established, T1D is also associated withincreased expression of pro-inflammatory factors, Interleukin 1beta (IL-1b), Interleukin 2 (IL-2), IL-6, TNF-a, receptor for advancedglycation end products (RAGE), and NFkB, compared to agematched control brains (Gebicke-Haerter et al., 2001; Sima et al.,2009). In addition, TNF-a and IL-1b can induce COX-2 activity inperivascular macrophages of the blood brain barrier (BBB) andgenerate prostaglandin E2, which enters the brain and stimulatesPVN neurons regulating ACTH release and sympathetic drive(Beishuizen and Thijs, 2003). Increased expression of JAM-1induces leukocytes accumulation in the microvasculature whichleads to enhancement of cell transmigration across tight junctionsand increased production of cytokines (Persidsky et al., 2006).

J. Yellowlees Douglas et al. / Progress in Retinal and Eye Research 31 (2012) 481e494488
4.2. Hypothalamic inflammation
In DIO, the earliest markers of hypothalamic inflammation arepresent a mere eight weeks after beginning an HFD (Calegari et al.)and, in rodent models, within 24 h of HFD feeding (Thaler et al.).This inflammation is marked by activation, recruitment andproliferation of microglia, immune cells of the brain that actsimilarly to macrophages (Sofroniew, 2009). This hypothalamicinflammation results in disruption of pancreatic b-cell function anda dysfunctional increase in insulin secretion.
Neuroinflammation also plays a role in T1D. Vargus et al induceddiabetes using streptozotocin (STZ) and showed increased expres-sions of Ang II, ICAM-1, LFA-1 and CD8 positive cells in diversezones of cerebrum and cerebellum of diabetic rats (week 8).Treatment of diabetic animals with losartan or enalapril reducedthe expression of those molecules. These authors concluded thatthe presence of Ang II-mediated brain inflammatory events indiabetes is probably mediated by AT1 receptors. These results areconsistent with our observations that activated microglia arepresent in increased numbers in the paraventricular nucleus (PVN)of T1D mice compared to controls [Grant MB, unpublished results].
4.3. Endocrine effects of hypothalamic inflammation
Various hypothalamic nuclei regulate satiety and metabolic andfluid homeostasis (Masuo et al.). Thus, hypothalamic inflammationcauses resistance to both leptin (Enriori et al., 2007; Ozcan et al.,2009) and insulin (De Souza et al., 2005; Posey et al., 2009),resulting in further increases in adipose tissue and insulin resis-tance (Choi et al., 2010).
4.4. Insulin resistance in diabetes and DR
A typical Western diet can thus cause a cycle of increasinghypothalamic inflammation, which leads to insulin (and leptin)resistance, leading to weight gain (Velloso et al., 2008). Evenindependent of obesity, hypothalamic inflammation can impairinsulin release from b cells, impair peripheral insulin action, andpotentiate hypertension (Kang et al., 2009; Purkayastha et al.),which is pertinent to T1D as well as T2D.
In T2D, patients enter into a vicious cycle where their insulinresistance increases adiposity, which, in turn, also spurs morehypothalamic inflammation that increases insulin resistance. Evenif patients initiate low-fat, moderate carbohydrate diets, theirinsulin resistance can make losing weight difficult and lessen theodds of patients remaining compliant with a restricted intake offats and carbohydrates (Fletcher et al., 2006). Decreased sensitivityto insulin and resulting obesity can ultimately lead to T2D, whichleads to still higher levels of hypothalamic inflammation andinsulin resistance (Shoelson et al., 2006). However, patients wholost massive amounts of body mass following bariatric surgeryshowed lowered amounts of hypothalamic inflammation afterweight loss, as detected via fMRI (Ochner et al.), as well asdecreased concentrations of IL-6 (van de Sande-Lee et al., 2011).
Recently, there has been great excitement regarding the almostimmediate improvement in blood glucose (remission of T2D) inpatients undergoing Roux-en-Y gastric bypass. Pournaras et al.showed that exclusion of glucose passage through the proximalsmall bowel results in enhanced insulin and gut hormoneresponses in patients after gastric bypass (Pournaras et al.). Thusthis strategy to restore metabolic control will likely favorablyimpact CNS inflammation in T2D.
Obviously, significant weight loss, resulting in decreasedadiposity and improved glucose control, provides the ideal meansfor treating T2D. Unfortunately, some obese and morbidly obese
patients with T2D remain poor candidates for procedures like theRoux-en-Y gastric bypass used in van de Sande-Lee et al. due topotentially high rates of complications or their status as high riskcases for anaesthesia (van de Sande-Lee et al., 2011).
5. Interventions addressing DR
Hyperglycaemia is at the center of DR pathogenesis as large-scale T1D and T2D clinical trials have been conducted world-wide and have demonstrated that early intensive glycaemiccontrol can reduce the incidence and progression of micro andmacrovascular complications (American Diabetes Association,1998; Diabetes Group, 2000). The benefits of tight control havebeen documented in Type 1 in the Diabetes Control and Compli-cation Trial (DCCT) study (DCCT, 1993) and the United KingdomProspective Diabetes Study (UKPDS) (UKPDS, 1998) demonstratedthat controlling blood glucose levels reduced the risk of diabeticretinopathy and nephropathy in T2D.
The beneficial effects of intensive glycaemic control achieved byearly intervention can persist for several years, a phenomenon thatfirst described as ‘metabolic memory’ by the Diabetes Control andComplications Trial and Epidemiology of Diabetes Interventionsand Complications (DCCT/EDIC) collaboration (Group, 2003).
While prevention of hyperglycaemia is the cornerstone ofprevention of DR, once DR occurs, the impact of tight controlremains important. As mentioned above, tight glucose controlequally benefits patients with T1D or T2D while the strategies toachieve this in T1D includes insulin pumps and continuous glucosesensors, the judicious use of multiple agents from the armamen-tarium of drugs available make this equally possible in T2D. The keyrole of diet and exercise is equally effective and important for bothT1D and T2D. However, tight control is not possible in all patients,thus supplemental strategies are needed to prevent progression ofthis disease and to treat it. Several therapeutic approaches showpromise in addressing DR by decreasing systemic inflammation,with the full range best covered in Kern’s 2007 review (Kern, 2007).However, given this article’s focus on the links between BM, theCNS and inflammation, this review will here focus only on thoseinterventions that touch on the BM and CNS inflammation.
5.1. Interventions addressing inflammation
Four pharmacologic interventions have displayed some efficacyin reducing inflammatory response in patients, thus improving DRin human and animal studies. Of these four, the first, intravitrealcorticosteroids, treat DR locally, in a manner similar to current anti-VEGF therapy. The second, aldose reductase inhibitors, havedemonstrated inconsistent outcomes across animal and humanstudies. The third, aspirin and salicylates, shows some efficacy,albeit with side effects. Minocycline, an antibiotic with anti-inflammtory properties, is currently proving successful in animalstudies (Yune et al., 2007) and, most recently, in our case series ofobese, diabetic patients with DR.
5.2. Corticosteroids
Known to exert profound anti-inflammatory effects on a varietyof conditions, corticosteroids have proven effective as a localizedtherapy for inflammation in DR. Intravitreal injections showpromise in reversing or preventing vascular permeability in DR(Dubey, 2006; Felinski and Antonetti, 2005). Currently, glucocor-ticoids prove particularly useful in preserving the BBB in thetreatment of brain tumors and show similar effects on retinalvasculature, making them a promising treatment option for aspectsof DR management.

J. Yellowlees Douglas et al. / Progress in Retinal and Eye Research 31 (2012) 481e494 489
5.3. Aldose reductase inhibitors
The polyol pathway enzyme, aldose reductase, inhibits expres-sion of ICAM-1, COX-2, and leukostasis via inhibition of pro-inflammatory NF-KB activity (Ramana et al., 2004). As a result,aldose reductase inhibitors have been extensively studied for theirinhibition of aldose reductase under hyperglycemic conditions(Kern, 2007). However, this class of drugs has shown only mixedresults, at best, in inhibiting DR in animal models (Kojima et al.,1985) and, moreover, has proven unsuccessful in treating diabeticpatients (Arauz-Pacheco et al., 1992; Group, 1990). Currently,further studies remain to establish the possibility that aldosereductase inhibitors provide beneficial effects due to their anti-inflammatory actions (Kern, 2007).
5.4. Aspirin and salicylates
Researchers beginning in the late nineteenth century discoveredhigh doses of salicylates dramatically lowered the glucose levels ofdiabetic patients (Ebstein, 1876), an effect confirmed a quarter-century later in the impact of salicylates on reducing sugarsecreted in the urine of diabetics (Williamson, 1901). The effec-tiveness of salicylates in treating T2Dwas rediscovered accidentallyin the 1950s when a physician administered a high dose of aspirinto treat arthritis associated with rheumatic fever and discovered hispatient no longer required daily insulin injections (Reid et al., 1957).When the patient’s joint symptoms resolved, the aspirin was dis-continued, and a subsequent glucose tolerance test showedelevated blood glucose. Intrigued, Reid and his colleagues admin-istered aspirin to a small series of diabetics, whose blood glucosereturned to normal, non-diabetic levels in every patient (Reid et al.,1957). Unfortunately, follow-up studies focused on the mechanismof salicylates in reducing insulin secretionwith inconclusive results(Shoelson et al., 2006).
Only recently have studies begun to focus on the efficacy ofsalicylates in reducing inflammation associated with the develop-ment of obesity and T2D. Aspirin, at a typical dose of 650 mg,inhibits NF-KB (Kopp and Ghosh, 1994), as well as leukocyteadhesion (Pierce et al., 1996). Hundal et al. demonstrated that thesame high dose of aspirin fomented substantial reductions infasting and postprandial glucose (Hundal et al., 2002). Moreover,aspirin, initiated with onset of induced diabetes in dogs overa 5-year trial, significantly inhibited retinal aneurysm and theformation of acellular capillaries but proved less successful indiminishing pericyte ghosts and the frequency of micoraneurysms.Notably, over the course of this trial, aspirin was administered atonly 20 mg/kg of body weight, a level significantly lower than the650 mg that proved efficacious in earlier human studies but judgedto represent the maximum tolerable chronic dose to avoid severeside effects (Kern and Engerman, 2001). However, the therapeuticbenefits of aspirin in reducing or preventing DR in T2D patients arecurrently offset by the effects of the requisite high dosage, includinggastrointestinal irritation and unacceptably high risks of bleeding(Hundal et al., 2002).
5.5. Minocycline
A second generation, chemically modified tetracycline, mino-cycline is commonly prescribed for its antimicrobial action but alsodisplays significant anti-inflammatory effects (Amin et al., 1996;Yrjanheikki et al., 1999). Moreover, minocycline shows promise indecreasing the microglial activation in the hypothalamus (Lai andTodd, 2006) that accompanies the inflammatory cycle connectingthe BMDC and CNS to insulin resistance and T2D (Rothwell et al.,1999). Animal studies of the efficacy of minocycline in treating
DR in mice show that long-term administration of the drugsuccessfully prevented retinal capillary degeneration in diabeticmice (Vincent and Mohr, 2007). Minocycline has been successfullyused to treat painful diabetic neuropathy (Dray, 2008). Moreover,minocycline’s ability to decrease hypothalamic inflammation couldprovide a particularly effective strategy in treating DR by improvingpatients’ blood glucose control.
5.5.1. Efficacy in humansThis potential efficacy was recently tested by our laboratory
through a case series of patientswith obesity, insulin resistance, T2D,and DR. As an anti-inflammatory agent, minocycline freely crossesthe BBB, is neuro-protective and inhibits microglial activation in thebrain (Zhu et al., 2002). Minocycline also produces beneficial effectsin acute and chronic brain disorders, including stroke, traumaticbrain injury and neurodegenerative diseases (Kreuter et al., 2004;Sanchez et al., 2001; Tikka and Koistinaho, 2001).
5.5.2. Case series: minocycline improved visual acuity andneuropathic pain in diabetic subjects
Nine T2D patients with either mild to moderate DR came fromthree different settings: three patients from a diabetes and weightmanagement clinic, three from a weight management clinic at theUniversity of Florida’s College of Medicine, and three from clinicianreferrals at the University of Florida’s Shands Hospital. Notably, allnine morbidly obese patients began the study with minocyclineprescribed on a compassionate basis, secondary to their inadequateresponse to weight management programs and the use of multipleoral hypoglycaemic and anti-hypertensive agents. The short dura-tion of the study limited the interpretation of retinal changes;however all patients had mild to moderate non-proliferative dia-betic retinopathy at onset and also experienced blurred vision andreduced visual acuity with episodes of hyperglycaemia. At the timeof minocycline initiation, all nine patients were also already takingatorvastatin (Lipitor�) or other HMG CoA reductase inhibitors. Allpatients had a diagnosis of Type 2 diabetes for a minimum of tenyears but not more than 21 years. Patients also had body massindices (BMI) between 37 and 60 kg/m2. An adult with a BMIbetween 25 and 29 kg/m2 is classified as overweight, with obeseadults having a BMI greater than 30 kg/m2, and morbidly obeseadults as having a BMI greater than 40 kg/m2. Consistent with linksestablished by epidemiology studies, all patients also had hyper-tension. All nine patients also experienced symptoms consistentwith peripheral neuropathy, confirmed on physical examination asdecreased sensation to light touch or pin prick, although nopatients underwent nerve conduction studies to document degreeof neuropathy. Although the four patients with the highest initialBMIs had been previously diagnosed with sleep apnea, all ninepatients complained of sleep disturbances. Hypertension wasmanaged with either beta-blockers, centrally acting alpha-agonists,ACE inhibitors or angiotensin receptor blockers, with a medianblood pressure of 168 � 16/93 � 10 mmHg.
Patients were started on 100 mg twice daily of minocycline forten months. Patients also received counseling on continuing thehigh protein, moderate fat and low carbohydrate dietdwithcarbohydrate intake limited to 30 g or less at each mealdwhichthey had been on at the time of the initiation of minocycline.Every month, patients’ weight loss and blood pressure was recor-ded. Blood glucose control, established via HbA1c levels, wasmeasured every three months during the study’s ten months.
All nine patients achieved consistent weight loss of an averageof 37.9� 4.0 kg over a ten-month period, decreasing their BMI by anaverage of 30 � 7%. Patients not in either a diabetes or weightmanagement clinic achieved the same weight loss as patientsreceiving more aggressive counseling on diet, exercise, and
J. Yellowlees Douglas et al. / Progress in Retinal and Eye Research 31 (2012) 481e494490
behavioural modifications. Patients’ weight loss was also accom-panied by improvement in HbA1c, with a reduction of an average of22 � 12%. All patients reported improved visual acuity during thestudy and improvement of neuropathic pain of their extremities.
No patients reported adverse effects from minocycline. Onthe contrary, patients on minocycline reported mild appetitesuppression and earlier satiety. Following their weight loss, allpatients noted improved quality of sleep. Patients also reportedimproved mood and increased ability to exercise, due in part totheir substantial weight loss but also to minocycline’s apparenteffects in decreasing neuromuscular pain resulting frominflammation (McMahon and Wells, 2004; Zhang et al., 2009).These same effects, coupled with weight loss, resulted in patients’subjective improvement of pain symptoms associated withperipheral neuropathy.
6. Implications of anti-inflammatory strategies
All patients uniformly reported improved visual acuity. Theseresults are likely due to improved blood glucose and blood pressurecontrol but may also be due to decreased retinal and CNS inflam-mation. The mechanism(s) by which minocycline produces thesedramatic beneficial outcomes remains speculative at the presenttime. However, based on existing evidence, we believe that itseffects may, in part, be mediated by its anti-inflammatory actions
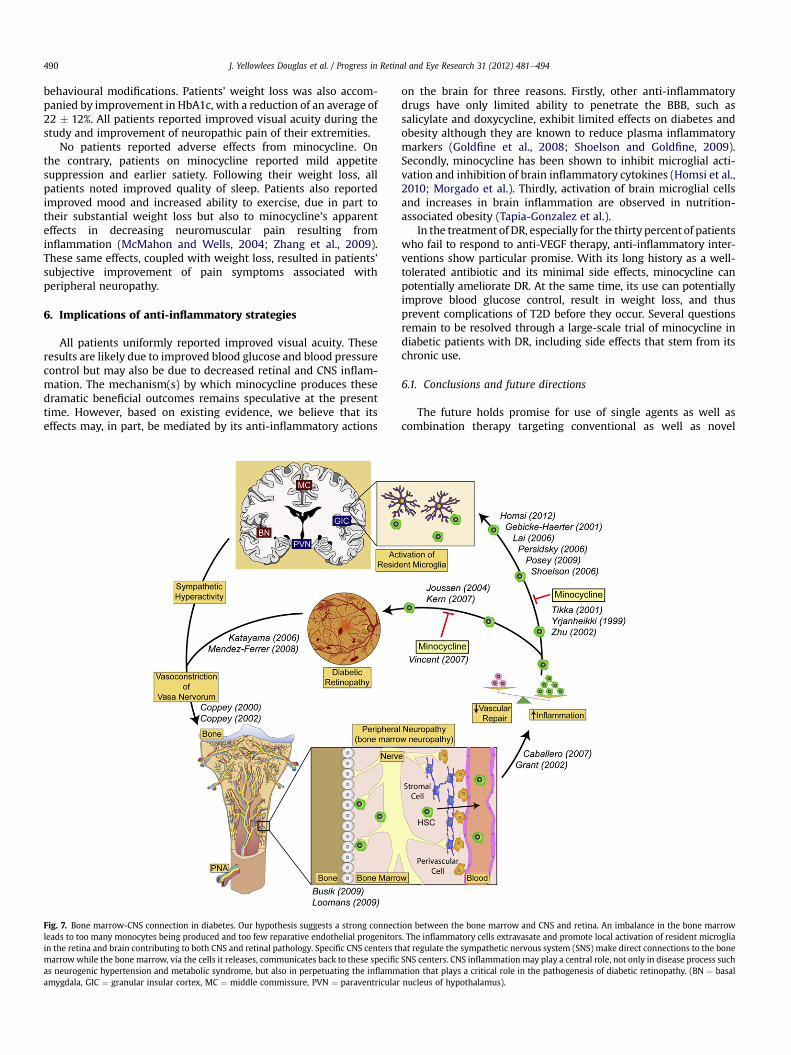
Fig. 7. Bone marrow-CNS connection in diabetes. Our hypothesis suggests a strong connectleads to too many monocytes being produced and too few reparative endothelial progenitorin the retina and brain contributing to both CNS and retinal pathology. Specific CNS centers thmarrow while the bone marrow, via the cells it releases, communicates back to these specificas neurogenic hypertension and metabolic syndrome, but also in perpetuating the inflammamygdala, GIC ¼ granular insular cortex, MC ¼ middle commissure, PVN ¼ paraventricular
on the brain for three reasons. Firstly, other anti-inflammatorydrugs have only limited ability to penetrate the BBB, such assalicylate and doxycycline, exhibit limited effects on diabetes andobesity although they are known to reduce plasma inflammatorymarkers (Goldfine et al., 2008; Shoelson and Goldfine, 2009).Secondly, minocycline has been shown to inhibit microglial acti-vation and inhibition of brain inflammatory cytokines (Homsi et al.,2010; Morgado et al.). Thirdly, activation of brain microglial cellsand increases in brain inflammation are observed in nutrition-associated obesity (Tapia-Gonzalez et al.).
In the treatment of DR, especially for the thirty percent of patientswho fail to respond to anti-VEGF therapy, anti-inflammatory inter-ventions show particular promise. With its long history as a well-tolerated antibiotic and its minimal side effects, minocycline canpotentially ameliorate DR. At the same time, its use can potentiallyimprove blood glucose control, result in weight loss, and thusprevent complications of T2D before they occur. Several questionsremain to be resolved through a large-scale trial of minocycline indiabetic patients with DR, including side effects that stem from itschronic use.
6.1. Conclusions and future directions
The future holds promise for use of single agents as well ascombination therapy targeting conventional as well as novel
ion between the bone marrow and CNS and retina. An imbalance in the bone marrows. The inflammatory cells extravasate and promote local activation of resident microgliaat regulate the sympathetic nervous system (SNS) make direct connections to the boneSNS centers. CNS inflammation may play a central role, not only in disease process suchation that plays a critical role in the pathogenesis of diabetic retinopathy. (BN ¼ basalnucleus of hypothalamus).

J. Yellowlees Douglas et al. / Progress in Retinal and Eye Research 31 (2012) 481e494 491
signalling pathways (Afzal et al., 2007; Kramerov et al., 2008).Viewing DR as an expression of neuroinflammation, allows the useof agents such as minocycline to simultaneous address micro andmacrovascular complications. Complications from diabetes includenot only vascular complications but also Alzheimer’s disease, whichis correlated with diabetes (Arvanitakis et al., 2004a,b; Luchsingeret al., 2001). In a recent Dutch study, T2D doubled the risk ofpatients for developing dementia, with patients receiving insulinhaving a four-fold risk, compared to non-diabetics (Ott andKivipelto, 1999).
This novel approach to treating DR via neuroinflammationcounters the shortcomings of existing treatments. While evidenceshows that anti-inflammatory agents, such as tetracycline, canprove efficacious in treating DR by reducing inflammation locally,tetracycline’s inability to cross the BBB inhibits its ability toreduce neuroinflammation. Given the low cost of minocyclineand its potential to replace substantial pharmacologic regimens,including insulin sensitisers, anti-hyperglycaemic agents, anti-hypertensive agents, and anti-VEGF therapies, this approachto treating DR via central inflammation deserves furtherinvestigation.
In conclusion, conceptually we are proposing that BM neurop-athy is central to the inciting events of DR. The concepts proposedrepresent a paradigm shift in the pathogenesis of DR (Fig. 7). TheBM is a rich source of reparative cells for the retina (Grant et al.,2002; Otani et al., 2002). However, these key cellular componentsbecome dysfunctional in diabetes. In combination with BMneuropathy that promotes the generation of an excess of mono-cytes, the loss of reparative cells with the simultaneous increase indetrimental inflammatory cells, such as monocytes, promote thedevelopment of endothelial dysfunction.
The communication between the BM, CNS and peripheralnervous system is critical to affect rapid immune responses andto sustain vascular health. The development of BM neuropathysets up systemic inflammation by shifting haematopoiesistowards the generation of inflammatory cells. Importantly, BMneuropathy precedes the development of DR, thus connecting DRfor the first time to peripheral nerve dysfunction in the BM. Thealtered bone marrow population can, in turn, perpetuate not onlyretina, but also CNS inflammation. Inflammation in these keyregions of the SNS that regulate the BM perpetuate thedysfunction. Thus, strategies that lead to specific reduction ofmonocyte subpopulations may represent an optimal strategy forDR management, as well as management of generalized inflam-mation implicated in both microvascular and macrovasculardisease in diabetes.
Contributions
MKR and MBG conceived the case series. ADB, AS and QLgenerated data for figures. ADB edited the manuscript. MBGprovided follow-up and outcomes. LCS modelled the data. DCprovided dietary counseling. SC edited the manuscript and gener-ated data for figure. SLC edited the manuscript and generated datafor figure. YD and MBG contributed to the writing of the paper.
None of the authors have any competing interests.No ethical or Institutional Review Board (IRB) approval was
required for this study.All authors have completed the Unified Competing Interest form
at http://www.icmje.org/coi_disclosure.pdf (available on requestfrom the corresponding author) and declare: no support from anyorganisation for the submitted work; no financial relationshipswith any organisations that might have an interest in the submittedwork in the previous three years, no other relationships or activitiesthat could appear to have influenced the submitted work.
Acknowledgments
NIH/NCRR Clinical and Translational Science Award to theUniversity of Florida UL1 RR029890; EY 007739, EY012601, DK096221 to MBG and HL110170 to MBG and MKR.
References
Afzal, A., Shaw, L.C., Ljubimov, A.V., Boulton, M.E., Segal, M.S., Grant, M.B., 2007.Retinal and choroidal microangiopathies: therapeutic opportunities. Microvasc.Res. 74, 131e144.
Amin, A.R., Attur, M.G., Thakker, G.D., Patel, P.D., Vyas, P.R., Patel, R.N., Patel, I.R.,Abramson, S.B., 1996. A novel mechanism of action of tetracyclines: effects onnitric oxide synthases. Proc. Natl. Acad. Sci. U.S.A. 93, 14014e14019.
Arauz-Pacheco, C., Ramirez, L.C., Pruneda, L., Sanborn, G.E., Rosenstock, J., Raskin, P.,1992. The effect of the aldose reductase inhibitor, ponalrestat, on the progres-sion of diabetic retinopathy. J. Diabetes Complications 6, 131e137.
Arevalo, J.F., Fromow-Guerra, J., Sanchez, J.G., Maia, M., Berrocal, M.H., Wu, L.,Saravia, M.J., Costa, R.A., 2008. Primary intravitreal bevacizumab for subfovealchoroidal neovascularization in age-related macular degeneration: results ofthe Pan-American Collaborative Retina Study Group at 12 months follow-up.Retina 28, 1387e1394.
Arvanitakis, Z., Wilson, R.S., Bienias, J.L., Evans, D.A., Bennett, D.A., 2004a. Diabetesmellitus and risk of Alzheimer disease and decline in cognitive function. Arch.Neurol. 61, 661e666.
Arvanitakis, Z., Wilson, R.S., Schneider, J.A., Bienias, J.L., Evans, D.A., Bennett, D.A.,2004b. Diabetes mellitus and progression of rigidity and gait disturbance inolder persons. Neurology 63, 996e1001.
Asahara, T., Murohara, T., Sullivan, A., Silver, M., van der Zee, R., Li, T.,Witzenbichler, B., Schatteman, G., Isner, J.M., 1997. Isolation of putativeprogenitor endothelial cells for angiogenesis. Science 275, 964e967.
American Diabetes Association, 1998. Implications of the United Kingdomprospective diabetes study. Diabetes Care 21, 2180e2184.
Awad, O., Dedkov, E.I., Jiao, C., Bloomer, S., Tomanek, R.J., Schatteman, G.C., 2006.Differential healing activities of CD34þ and CD14þ endothelial cell progenitors.Arterioscler. Thromb. Vasc. Biol. 26, 758e764.
Bahlmann, F.H., De Groot, K., Spandau, J.M., Landry, A.L., Hertel, B., Duckert, T.,Boehm, S.M., Menne, J., Haller, H., Fliser, D., 2004. Erythropoietin regulatesendothelial progenitor cells. Blood 103, 921e926.
Bailey, A.J., 2001. Molecular mechanisms of ageing in connective tissues. Mech.Ageing Dev. 122, 735e755.
Beishuizen, A., Thijs, L.G., 2003. Endotoxin and the hypothalamo-pituitary-adrenal(HPA) axis. J. Endotoxin Res. 9, 3e24.
Blake, G.J., Ridker, P.M., 2002. Tumour necrosis factor-alpha, inflammatorybiomarkers, and atherogenesis. Eur. Heart J. 23, 345e347.
Boyd-White, J., Williams Jr., J.C., 1996. Effect of cross-linking on matrix permeability.A model for AGE-modified basement membranes. Diabetes 45, 348e353.
Brugger, W., Heimfeld, S., Berenson, R.J., Mertelsmann, R., Kanz, L., 1995. Reconsti-tution of hematopoiesis after high-dose chemotherapy by autologous progen-itor cells generated ex vivo. N. Engl. J. Med. 333, 283e287.
Busik, J.V., Tikhonenko, M., Bhatwadekar, A., Opreanu, M., Yakubova, N., Caballero, S.,Player, D., Nakagawa, T., Afzal, A., Kielczewski, J., Sochacki, A., Hasty, S., Li Calzi, S.,Kim, S., Duclas, S.K., Segal, M.S., Guberski, D.L., Esselman, W.J., Boulton, M.E.,Grant, M.B., 2009. Diabetic retinopathy is associated with bone marrowneuropathy and a depressed peripheral clock. J. Exp. Med. 206, 2897e2906.
Butler, J.M., Guthrie, S.M., Koc, M., Afzal, A., Caballero, S., Brooks, H.L., Mames, R.N.,Segal, M.S., Grant, M.B., Scott, E.W., 2005. SDF-1 is both necessary and sufficientto promote proliferative retinopathy. J. Clin. Invest. 115, 86e93.
Caldwell, R.B., Bartoli, M., Behzadian, M.A., El-Remessy, A.E., Al-Shabrawey, M.,Platt, D.H., Liou, G.I., Caldwell, R.W., 2005. Vascular endothelial growth factorand diabetic retinopathy: role of oxidative stress. Curr. Drug Targets 6, 511e524.
Caballero, S., Sengupta, N., Afzal, A., Chang, K.H., Li Calzi, S., Guberski, D.L., Kern, T.S.,Grant, M.B., 2007. Ischemic vascular damage can be repaired by healthy, but notdiabetic, endothelial progenitor cells. Diabetes 56, 960e967.
Calegari, V.C., Torsoni, A.S., Vanzela, E.C., Araujo, E.P., Morari, J., Zoppi, C.C., Sbragia,L., Boschero, A.C., Velloso, L.A. Inflammation of the hypothalamus leads todefective pancreatic islet function. J. Biol. Chem. 286, 12870e12880.
Card, J.P., Kobiler, O., Ludmir, E.B., Desai, V., Sved, A.F., Enquist, L.W. A dual infectionpseudorabies virus conditional reporter approach to identify projections tocollateralized neurons in complex neural circuits. PLoS One 6, e21141.
Chang, K.M., Lehrhaupt, N., Lin, L.M., Feng, J., Wu-Wang, C.Y., Wang, S.L., 1996.Epidermal growth factor in gingival crevicular fluid and its binding capacity ininflamed and non-inflamed human gingiva. Arch. Oral Biol. 41, 719e724.
Choi, S.J., Kim, F., Schwartz, M.W., Wisse, B.E., 2010. Cultured hypothalamic neuronsare resistant to inflammation and insulin resistance induced by saturated fattyacids. Am. J. Physiol. Endocrinol. Metab. 298, E1122eE1130.
Congdon,N., O’Colmain, B., Klaver, C.C., Klein, R.,Munoz, B., Friedman,D.S., Kempen, J.,Taylor,H.R.,Mitchell, P., 2004. Causes andprevalenceof visual impairment amongadults in the United States. Arch. Ophthalmol. 122, 477e485.
Davis, T.M., Stratton, I.M., Fox, C.J., Holman, R.R., Turner, R.C., 1997. U.K. ProspectiveDiabetes Study 22. Effect of age at diagnosis on diabetic tissue damage duringthe first 6 years of NIDDM. Diabetes Care 20, 1435e1441.

J. Yellowlees Douglas et al. / Progress in Retinal and Eye Research 31 (2012) 481e494492
DCCT, 1993. The effect of intensive treatment of diabetes on the development andprogression of long-term complications in insulin-dependent diabetes mellitus.The Diabetes Control and Complications Trial Research Group. N. Engl. J. Med.329, 977e986.
De Souza, C.T., Araujo, E.P., Bordin, S., Ashimine, R., Zollner, R.L., Boschero, A.C.,Saad, M.J., Velloso, L.A., 2005. Consumption of a fat-rich diet activates a proin-flammatory response and induces insulin resistance in the hypothalamus.Endocrinology 146, 4192e4199.
Diabetes Group, 2000. Retinopathy and nephropathy in patients with type 1 dia-betes four years after a trial of intensive therapy. The Diabetes Control andComplications Trial/Epidemiology of Diabetes Interventions and ComplicationsResearch Group. N. Engl. J. Med. 342, 381e389.
Dimmeler, S., Aicher, A., Vasa, M., Mildner-Rihm, C., Adler, K., Tiemann, M.,Rutten, H., Fichtlscherer, S., Martin, H., Zeiher, A.M., 2001. HMG-CoA reductaseinhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Aktpathway. J. Clin. Invest. 108, 391e397.
Dray, A., 2008. Neuropathic pain: emerging treatments. Br. J. Anaesth. 101, 48e58.Dubey, A.K., 2006. Intravitreal injection of triamcinolone acetonide for diabetic
macular edema: principles and practice. Indian J. Ophthalmol. 54, 290 (authorreply 290e291).
Ebstein, W., 1876. Zur therapie des diabetes mellitus, insbesondere uber die ane-wendeng des salicylsuren natron bei demselben. Klin. Wochschr. 13, 337e340.
Engerman, R.L., Kern, T.S., 1987. Progression of incipient diabetic retinopathy duringgood glycemic control. Diabetes 36, 808e812.
Engerman, R.L., Kern, T.S., 1995. Retinopathy in animal models of diabetes. DiabetesMetab. Rev. 11, 109e120.
Enriori, P.J., Evans, A.E., Sinnayah, P., Jobst, E.E., Tonelli-Lemos, L., Billes, S.K.,Glavas, M.M., Grayson, B.E., Perello, M., Nillni, E.A., Grove, K.L., Cowley, M.A.,2007. Diet-induced obesity causes severe but reversible leptin resistance inarcuate melanocortin neurons. Cell Metab. 5, 181e194.
Feinstein, R., Kanety, H., Papa, M.Z., Lunenfeld, B., Karasik, A., 1993. Tumor necrosisfactor-alpha suppresses insulin-induced tyrosine phosphorylation of insulinreceptor and its substrates. J. Biol. Chem. 268, 26055e26058.
Felinski, E.A., Antonetti, D.A., 2005. Glucocorticoid regulation of endothelial celltight junction gene expression: novel treatments for diabetic retinopathy. Curr.Eye Res. 30, 949e957.
Fina, L., Molgaard, H.V., Robertson, D., Bradley, N.J., Monaghan, P., Delia, D.,Sutherland, D.R., Baker, M.A., Greaves, M.F., 1990. Expression of the CD34 genein vascular endothelial cells. Blood 75, 2417e2426.
Flamme, I., Risau, W., 1992. Induction of vasculogenesis and hematopoiesis in vitro.Development 116, 435e439.
Fletcher, B.R., Calhoun, M.E., Rapp, P.R., Shapiro, M.L., 2006. Fornix lesions decouplethe induction of hippocampal arc transcription from behavior but not plasticity.J. Neurosci. 26, 1507e1515.
Fu, M.X., Wells-Knecht, K.J., Blackledge, J.A., Lyons, T.J., Thorpe, S.R., Baynes, J.W.,1994. Glycation, glycoxidation, and cross-linking of collagen by glucose.Kinetics, mechanisms, and inhibition of late stages of the Maillard reaction.Diabetes 43, 676e683.
Gardiner, T.A., Anderson, H.R., Stitt, A.W., 2003. Inhibition of advanced glycationend-products protects against retinal capillary basement membrane expansionduring long-term diabetes. J. Pathol. 201, 328e333.
Gebicke-Haerter, P.J., Spleiss, O., Ren, L.Q., Li, H., Dichmann, S., Norgauer, J.,Boddeke, H.W., 2001. Microglial chemokines and chemokine receptors. Prog.Brain Res. 132, 525e532.
Goldfine, A.B., Silver, R., Aldhahi, W., Cai, D., Tatro, E., Lee, J., Shoelson, S.E., 2008. Useof salsalate to target inflammation in the treatment of insulin resistance andtype 2 diabetes. Clin. Transl. Sci. 1, 36e43.
Grant, M.B., May, W.S., Caballero, S., Brown, G.A., Guthrie, S.M., Mames, R.N.,Byrne, B.J., Vaught, T., Spoerri, P.E., Peck, A.B., Scott, E.W., 2002. Adult hema-topoietic stem cells provide functional hemangioblast activity during retinalneovascularization. Nat. Med. 8, 607e612.
Grant, M.B., Schmetz, I., Russell, B., Harwood Jr., H.J., Silverstein, J., Merimee, T.J.,1986. Changes in insulin-like growth factors I and II and their binding proteinafter a single intramuscular injection of growth hormone. J. Clin. Endocrinol.Metabol. 63, 981e984.
Graves, D.T., Naguib, G., Lu, H., Leone, C., Hsue, H., Krall, E., 2005. Inflammation ismore persistent in type 1 diabetic mice. J. Dent. Res. 84, 324e328.
Group, S.R., 1990. A randomized trial of sorbinil, an aldose reductase inhibitor, indiabetic retinopathy. Sorbinil Retinopathy Trial Research Group. Arch. Oph-thalmol. 108, 1234e1244.
Group W.T.f.t.D.C.a.C.T.E.o.D.I.a.C.R, 2003. Sustained effect of intensive treatment oftype 1 diabetes mellitus on development and progression of diabeticnephropathy: the Epidemiology of Diabetes Interventions and Complications(EDIC) study. JAMA 290, 2159e2167.
Homsi, S., Piaggio, T., Croci, N., Noble, F., Plotkine, M., Marchand-Leroux, C., Jafarian-Tehrani, M., 2010. Blockade of acute microglial activation by minocyclinepromotes neuroprotection and reduces locomotor hyperactivity after closed headinjury in mice: a twelve-week follow-up study. J. Neurotrauma 27, 911e921.
Hotamisligil,G.S., 2006. Inflammationandmetabolic disorders.Nature444, 860e867.Hotamisligil, G.S., Arner, P., Caro, J.F., Atkinson, R.L., Spiegelman, B.M., 1995.
Increased adipose tissue expression of tumor necrosis factor-alpha in humanobesity and insulin resistance. J. Clin. Invest. 95, 2409e2415.
Hotamisligil, G.S., Shargill, N.S., Spiegelman, B.M., 1993. Adipose expression oftumor necrosis factor-alpha: direct role in obesity-linked insulin resistance.Science 259, 87e91.
Hu, F., Lu, R., Huang, B., Liang, M., 2004. Free radical scavenging activity of extractsprepared from fresh leaves of selected Chinese medicinal plants. Fitoterapia 75,14e23.
Hundal, R.S., Petersen, K.F., Mayerson, A.B., Randhawa, P.S., Inzucchi, S.,Shoelson, S.E., Shulman, G.I., 2002. Mechanism by which high-dose aspirinimproves glucose metabolism in type 2 diabetes. J. Clin. Invest. 109, 1321e1326.
National Eye Institute, 2004. Statement on the prevalence of diabetic retinopathyand age-related macular degeneration among Hispanic/Latino Americans (pressrelease).
Joussen, A.M., Murata, T., Tsujikawa, A., Kirchhof, B., Bursell, S.E., Adamis, A.P., 2001.Leukocyte-mediated endothelial cell injury and death in the diabetic retina.Am. J. Pathol. 158, 147e152.
Joussen, A.M., Poulaki, V., Le, M.L., Koizumi, K., Esser, C., Janicki, H., Schraermeyer, U.,Kociok, N., Fauser, S., Kirchhof, B., Kern, T.S., Adamis, A.P., 2004. A central role forinflammation in the pathogenesis of diabetic retinopathy. FASEB J. 18,1450e1452.
Kanety, H., Feinstein, R., Papa, M.Z., Hemi, R., Karasik, A., 1995. Tumor necrosis factoralpha-induced phosphorylation of insulin receptor substrate-1 (IRS-1). Possiblemechanism for suppression of insulin-stimulated tyrosine phosphorylation ofIRS-1. J. Biol. Chem. 270, 23780e23784.
Kang, H.W., Kim, D., Kim, H.J., Kim, C.H., Kim, Y.S., Park, M.J., Kim, J.S., Cho, S.H.,Sung, M.W., Jung, H.C., Lee, H.S., Song, I.S., 2009. Visceral obesity and insulinresistance as risk factors for colorectal adenoma: a cross-sectional, case-controlstudy. Am. J. Gastroenterol. 105, 178e187.
Katayama, Y., Battista, M., Kao, W.M., Hidalgo, A., Peired, A.J., Thomas, S.A.,Frenette, P.S., 2006. Signals from the sympathetic nervous system regulatehematopoietic stem cell egress from bone marrow. Cell 124, 407e421.
Kempen, J.H., O’Colmain, B.J., Leske, M.C., Haffner, S.M., Klein, R., Moss, S.E.,Taylor, H.R., Hamman, R.F., 2004. The prevalence of diabetic retinopathy amongadults in the United States. Arch. Ophthalmol. 122, 552e563.
Kern, T.S., 2007. Contributions of inflammatory processes to the development of theearly stages of diabetic retinopathy. Exp. Diabetes Res. 2007, 95103.
Kern, T.S., Engerman, R.L., 1995. Vascular lesions in diabetes are distributed non-uniformly within the retina. Exp. Eye Res. 60, 545e549.
Kern, T.S., Engerman, R.L., 1996a. Capillary lesions develop in retina rather thancerebral cortex in diabetes and experimental galactosemia. Arch. Ophthalmol.114, 306e310.
Kern, T.S., Engerman, R.L., 1996b. A mouse model of diabetic retinopathy. Arch.Ophthalmol. 114, 986e990.
Kern, T.S., Engerman, R.L., 2001. Pharmacological inhibition of diabetic retinopathy:aminoguanidine and aspirin. Diabetes 50, 1636e1642.
Kojima, K., Matsubara, H., Harada, T., Mizuno, K., Suzuki, M., Hotta, N., Kakuta, H.,Sakamoto, N., 1985. Effects of aldose reductase inhibitor on retinal micro-angiopathy in streptozotocin-diabetic rats. Jpn. J. Ophthalmol. 29, 99e109.
Kopp, E., Ghosh, S., 1994. Inhibition of NF-kappa B by sodium salicylate and aspirin.Science 265, 956e959.
Kowluru, R.A., Chan, P.S., 2007. Oxidative stress and diabetic retinopathy. Exp.Diabetes Res. 2007, 43603.
Kowluru, R.A., Zhong, Q., Kanwar, M., 2010. Metabolic memory and diabetic reti-nopathy: role of inflammatory mediators in retinal pericytes. Exp. Eye Res. 90,617e623.
Kramerov, A.A., Saghizadeh, M., Caballero, S., Shaw, L.C., Li Calzi, S., Bretner, M.,Montenarh, M., Pinna, L.A., Grant, M.B., Ljubimov, A.V., 2008. Inhibition ofprotein kinase CK2 suppresses angiogenesis and hematopoietic stem cellrecruitment to retinal neovascularization sites. Mol. Cell Biochem. 316,177e186.
Kreuter, M., Langer, C., Kerkhoff, C., Reddanna, P., Kania, A.L., Maddika, S.,Chlichlia, K., Bui, T.N., Los, M., 2004. Stroke, myocardial infarction, acute andchronic inflammatory diseases: caspases and other apoptotic molecules astargets for drug development. Arch. Immunol. Ther. Exp. (Warsz) 52, 141e155.
Lai, A.Y., Todd, K.G., 2006. Hypoxia-activated microglial mediators of neuronalsurvival are differentially regulated by tetracyclines. Glia 53, 809e816.
Lalla, E., Kaplan, S., Yang, J., Roth, G.A., Papapanou, P.N., Greenberg, S., 2007. Effectsof periodontal therapy on serum C-reactive protein, sE-selectin, and tumornecrosis factor-alpha secretion by peripheral blood-derived macrophages indiabetes. A pilot study. J. Periodontal Res. 42, 274e282.
Li Calzi, S., Purich, D.L., Chang, K.H., Afzal, A., Nakagawa, T., Busik, J.V., Agarwal, A.,Segal, M.S., Grant, M.B., 2008. Carbon monoxide and nitric oxide mediatecytoskeletal reorganization in microvascular cells via vasodilator-stimulatedphosphoprotein phosphorylation: evidence for blunted responsiveness in dia-betes. Diabetes 57, 2488e2494.
Liu, R., Bal, H.S., Desta, T., Krothapalli, N., Alyassi, M., Luan, Q., Graves, D.T., 2006.Diabetes enhances periodontal bone loss through enhanced resorption anddiminished bone formation. J. Dent. Res. 85, 510e514.
Llevadot, J., Murasawa, S., Kureishi, Y., Uchida, S., Masuda, H., Kawamoto, A., Walsh, K.,Isner, J.M., Asahara, T., 2001. HMG-CoA reductase inhibitor mobilizes bone mar-rowederived endothelial progenitor cells. J. Clin. Invest. 108, 399e405.
Loomans, C.J., van Haperen, R., Duijs, J.M., Verseyden, C., de Crom, R., Leenen, P.J.,Drexhage, H.A., de Boer, H.C., de Koning, E.J., Rabelink, T.J., Staal, F.J.,van Zonneveld, A.J., 2009. Differentiation of bone marrow-derived endothelialprogenitor cells is shifted into a proinflammatory phenotype by hyperglycemia.Mol. Med. 15, 152e159.
Luchsinger, J.A., Tang, M.X., Stern, Y., Shea, S., Mayeux, R., 2001. Diabetes mellitusand risk of Alzheimer’s disease and dementia with stroke in a multiethniccohort. Am. J. Epidemiol. 154, 635e641.

J. Yellowlees Douglas et al. / Progress in Retinal and Eye Research 31 (2012) 481e494 493
Maestroni,G.J., Conti, A., Pedrinis, E.,1992. Effectof adrenergic agentsonhematopoiesisafter syngeneic bone marrow transplantation in mice. Blood 80, 1178e1182.
Masuo, K., Lambert, G.W., Esler, M.D., Rakugi, H., Ogihara, T., Schlaich, M.P. The roleof sympathetic nervous activity in renal injury and end-stage renal disease.Hypertens. Res. 33, 521e528.
McMahon, J.M., Wells, D.J., 2004. Electroporation for gene transfer to skeletalmuscles: current status. BioDrugs 18, 155e165.
Mendez-Ferrer, S., Lucas, D., Battista, M., Frenette, P.S., 2008. Haematopoietic stemcell release is regulated by circadian oscillations. Nature 452, 442e447.
Mirshahi, A., Roohipoor, R., Lashay, A., Mohammadi, S.F., Abdoallahi, A., Faghihi, H.,2008. Bevacizumab-augmented retinal laser photocoagulation in proliferativediabetic retinopathy: a randomized double-masked clinical trial. Eur. J. Oph-thalmol. 18, 263e269.
Mizutani, M., Kern, T.S., Lorenzi, M., 1996. Accelerated death of retinal microvascularcells in human and experimental diabetic retinopathy. J. Clin. Invest. 97,2883e2890.
Monnier, V.M., Vishwanath, V., Frank, K.E., Elmets, C.A., Dauchot, P., Kohn, R.R., 1986.Relation between complications of type I diabetes mellitus and collagen-linkedfluorescence. N. Engl. J. Med. 314, 403e408.
Moradian, S., Ahmadieh, H., Malihi, M., Soheilian, M., Dehghan, M.H., Azarmina, M.,2008. Intravitreal bevacizumab in active progressive proliferative diabeticretinopathy. Graefes Arch. Clin. Exp. Ophthalmol. 246, 1699e1705.
Morgado, C., Pereira-Terra, P., Cruz, C.D., Tavares, I. Minocycline completely reversesmechanical hyperalgesia in diabetic rats through microglia-induced changes inthe expression of the potassium chloride co-transporter 2 (KCC2) at the spinalcord. Diabetes Obes. Metab. 13, 150e159.
Naguib, G., Al-Mashat, H., Desta, T., Graves, D.T., 2004. Diabetes prolongs theinflammatory response to a bacterial stimulus through cytokine dysregulation.J. Invest. Dermatol. 123, 87e92.
Ochner, C.N., Kwok, Y., Conceicao, E., Pantazatos, S.P., Puma, L.M., Carnell, S.,Teixeira, J., Hirsch, J., Geliebter, A. Selective reduction in neural responses tohigh calorie foods following gastric bypass surgery. Ann. Surg. 253, 502e507.
Otani, A., Kinder, K., Ewalt, K., Otero, F.J., Schimmel, P., Friedlander, M., 2002. Bonemarrow-derived stem cells target retinal astrocytes and can promote or inhibitretinal angiogenesis. Nat. Med. 8, 1004e1010.
Ott, E.A., Kivipelto, J., 1999. Influence of chromium tripicolinate on growth andglucose metabolism in yearling horses. J. Anim. Sci. 77, 3022e3030.
Ozcan, L., Ergin, A.S., Lu, A., Chung, J., Sarkar, S., Nie, D., Myers Jr., M.G., Ozcan, U.,2009. Endoplasmic reticulum stress plays a central role in development ofleptin resistance. Cell Metab. 9, 35e51.
Paul, R.G., Bailey, A.J., 1999. The effect of advanced glycation end-product formationupon cell-matrix interactions. Int. J. Biochem. Cell Biol. 31, 653e660.
Peichev, M., Naiyer, A.J., Pereira, D., Zhu, Z., Lane, W.J., Williams, M., Oz, M.C.,Hicklin, D.J., Witte, L., Moore, M.A., Rafii, S., 2000. Expression of VEGFR-2 andAC133 by circulating human CD34(þ) cells identifies a population of functionalendothelial precursors. Blood 95, 952e958.
Persidsky, Y., Ramirez, S.H., Haorah, J., Kanmogne, G.D., 2006. Bloodebrain barrier:structural components and function under physiologic and pathologic condi-tions. J. Neuroimmune Pharmacol. 1, 223e236.
Pickup, J.C., 2004. Inflammation and activated innate immunity in the pathogenesisof type 2 diabetes. Diabetes Care 27, 813e823.
Pierce, J.W., Read, M.A., Ding, H., Luscinskas, F.W., Collins, T., 1996. Salicylatesinhibit I kappa B-alpha phosphorylation, endothelial-leukocyte adhesionmolecule expression, and neutrophil transmigration. J. Immunol. 156,3961e3969.
Posey, K.A., Clegg, D.J., Printz, R.L., Byun, J., Morton, G.J., Vivekanandan-Giri, A.,Pennathur, S., Baskin, D.G., Heinecke, J.W., Woods, S.C., Schwartz, M.W.,Niswender, K.D., 2009. Hypothalamic proinflammatory lipid accumulation,inflammation, and insulin resistance in rats fed a high-fat diet. Am. J. Physiol.Endocrinol. Metab. 296, E1003eE1012.
Pournaras, D.J., Aasheim, E.T., Bueter, M., Ahmed, A.R., Welbourn, R., Olbers, T., LeRoux, C.W. Effect of bypassing proximal gut on gut hormones involved withglycemic control and weight loss. Surg. Obes. Relat. Dis.
Pradhan, A.D., Manson, J.E., Rifai, N., Buring, J.E., Ridker, P.M., 2001. C-reactiveprotein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA286, 327e334.
Purkayastha, S., Zhang, H., Zhang, G., Ahmed, Z., Wang, Y., Cai, D. Neural dysregu-lation of peripheral insulin action and blood pressure by brain endoplasmicreticulum stress. Proc. Natl. Acad. Sci. U.S.A. 108, 2939e2944.
Quirici, N., Soligo, D., Caneva, L., Servida, F., Bossolasco, P., Deliliers, G.L., 2001.Differentiation and expansion of endothelial cells from human bone marrowCD133(þ) cells. Br. J. Haematol. 115, 186e194.
Ramana, K.V., Friedrich, B., Srivastava, S., Bhatnagar, A., Srivastava, S.K., 2004.Activation of nuclear factor-kappaB by hyperglycemia in vascular smoothmuscle cells is regulated by aldose reductase. Diabetes 53, 2910e2920.
Rameshwar, P., Gascon, P., 1992. Release of interleukin-1 and interleukin-6 fromhuman monocytes by antithymocyte globulin: requirement for de novosynthesis. Blood 80, 2531e2538.
Reid, J., Macdougall, A.I., Andrews, M.M., 1957. Aspirin and diabetes mellitus. Br.Med. J. 2, 1071e1074.
Rothwell, P.M., Gutnikov, S.A., McKinney, P.A., Schober, E., Ionescu-Tirgoviste, C.,Neu, A., 1999. Seasonality of birth in children with diabetes in Europe: multi-centre cohort study. European Diabetes Study Group. BMJ 319, 887e888.
Sakagami, Y., Girasole, G., Yu, X.P., Boswell, H.S., Manolagas, S.C., 1993. Stimulationof interleukin-6 production by either calcitonin gene-related peptide or para-thyroid hormone in two phenotypically distinct bone marrow-derived murinestromal cell lines. J. Bone Miner. Res. 8, 811e816.
Sanchez, M., Rene, O.B.A., Ona, V., Li, H., Mingwei, M.D., Friedlander, M., 2001.Minocycline reduces traumatic brain injury-mediated caspase-1 activation,tissue damage, and neurological dysfunction. Neurosurgery 48, 1399e1401.
Schatteman, G.C., Hanlon, H.D., Jiao, C., Dodds, S.G., Christy, B.A., 2000. Blood-derived angioblasts accelerate blood-flow restoration in diabetic mice. J. Clin.Invest. 106, 571e578.
Shi, Q., Rafii, S., Wu, M.H., Wijelath, E.S., Yu, C., Ishida, A., Fujita, Y., Kothari, S.,Mohle, R., Sauvage, L.R., Moore, M.A., Storb, R.F., Hammond, W.P., 1998. Evidencefor circulating bone marrow-derived endothelial cells. Blood 92, 362e367.
Shoelson, S.E., Goldfine, A.B., 2009. Getting away from glucose: fanning the flamesof obesity-induced inflammation. Nat. Med. 15, 373e374.
Shoelson, S.E., Lee, J., Goldfine, A.B., 2006. Inflammation and insulin resistance.J. Clin. Invest. 116, 1793e1801.
Sima, A.A., Zhang, W., Muzik, O., Kreipke, C.W., Rafols, J.A., Hoffman, W.H., 2009.Sequential abnormalities in type 1 diabetic encephalopathy and the effects of C-Peptide. Rev. Diabet. Stud. 6, 211e222.
Sivan-Loukianova, E., Awad, O.A., Stepanovic, V., Bickenbach, J., Schatteman, G.C.,2003. CD34þ blood cells accelerate vascularization and healing of diabeticmouse skin wounds. J. Vasc. Res. 40, 368e377.
Sofroniew, M.V., 2009. Molecular dissection of reactive astrogliosis and glial scarformation. Trends Neurosci. 32, 638e647.
Sorrell, J.M., Baber, M.A., Caplan, A.I., 2009. Influence of adult mesenchymal stemcells on in vitro vascular formation. Tissue Eng. Part A 15, 1751e1761.
Stitt, A.W., Anderson, H.R., Gardiner, T.A., Archer, D.B., 1994. Diabetic retinopathy:quantitative variation in capillary basement membrane thickening in arterial orvenous environments. Br. J. Ophthalmol. 78, 133e137.
Stitt, A.W., Frizzell, N., Thorpe, S.R., 2004. Advanced glycation and advanced lip-oxidation: possible role in initiation and progression of diabetic retinopathy.Curr. Pharm. Des. 10, 3349e3360.
Takahashi, K., Mitsui, K., Yamanaka, S., 2003. Role of ERas in promoting tumour-likeproperties in mouse embryonic stem cells. Nature 423, 541e545.
Tang, J., Mohr, S., Du, Y.D., Kern, T.S., 2003. Non-uniform distribution of lesions andbiochemical abnormalities within the retina of diabetic humans. Curr. Eye Res.27, 7e13.
Tapia-Gonzalez, S., Garcia-Segura, L.M., Tena-Sempere, M., Frago, L.M., Castellano,J.M., Fuente-Martin, E., Garcia-Caceres, C., Argente, J., Chowen, J.A. Activation ofmicroglia in specific hypothalamic nuclei and the cerebellum of adult ratsexposed to neonatal overnutrition. J. Neuroendocrinol. 23, 365e370.
Thaler, J.P., Yi, C.X., Schur, E.A., Guyenet, S.J., Hwang, B.H., Dietrich, M.O., Zhao, X.,Sarruf, D.A., Izgur, V., Maravilla, K.R., Nguyen, H.T., Fischer, J.D., Matsen, M.E.,Wisse, B.E., Morton, G.J., Horvath, T.L., Baskin, D.G., Tschop, M.H., Schwartz,M.W. Obesity is associated with hypothalamic injury in rodents and humans. J.Clin. Invest. 122, 153e162.
Thill, M., Strunnikova, N.V., Berna, M.J., Gordiyenko, N., Schmid, K., Cousins, S.W.,Thompson, D.J., Csaky, K.G., 2008. Late outgrowth endothelial progenitor cells inpatients with age-related macular degeneration. Invest. Ophthalmol. Vis. Sci.49, 2696e2708.
Tikka, T.M., Koistinaho, J.E., 2001. Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J. Immunol. 166,7527e7533.
UKPDS, 1998. Intensive blood-glucose control with sulphonylureas or insulincompared with conventional treatment and risk of complications in patientswith type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS)Group. Lancet 352, 837e853.
van de Sande-Lee, S., Pereira, F.R., Cintra, D.E., Fernandes, P.T., Cardoso, A.R.,Garlipp, C.R., Chaim, E.A., Pareja, J.C., Geloneze, B., Li, L.M., Cendes, F.,Velloso, L.A., 2011. Partial reversibility of hypothalamic dysfunction and changesin brain activity after body mass reduction in obese subjects. Diabetes 60,1699e1704.
Varma, R., Paz, S.H., Azen, S.P., Klein, R., Globe, D., Torres, M., Shufelt, C., Preston-Martin, S., 2004. The Los Angeles Latino Eye Study: design, methods, andbaseline data. Ophthalmology 111, 1121e1131.
Velloso, L.A., Araujo, E.P., de Souza, C.T., 2008. Diet-induced inflammation of thehypothalamus in obesity. Neuroimmunomodulation 15, 189e193.
Vincent, J.A., Mohr, S., 2007. Inhibition of caspase-1/interleukin-1beta signalingprevents degeneration of retinal capillaries in diabetes and galactosemia. Dia-betes 56, 224e230.
Wang, F.H., Liang, Y.B., Zhang, F., Wang, J.J., Wei, W.B., Tao, Q.S., Sun, L.P.,Friedman, D.S., Wang, N.L., Wong, T.Y., 2009. Prevalence of diabetic retinopathyin rural China: the Handan Eye Study. Ophthalmology 116, 461e467.
Weiss, M.J., Orkin, S.H., 1996. In vitro differentiation of murine embryonic stemcells. New approaches to old problems. J. Clin. Invest. 97, 591e595.
Wells-Knecht, K.J., Zyzak, D.V., Litchfield, J.E., Thorpe, S.R., Baynes, J.W., 1995.Mechanism of autoxidative glycosylation: identification of glyoxal and arabi-nose as intermediates in the autoxidative modification of proteins by glucose.Biochemistry 34, 3702e3709.
WHO, 2011. WHO Diabetes Fact Sheet N312. Diabetes.Williamson, R.T., 1901. On the treatment of glycosuria and diabetes mellitus with
sodium salicylate. Br. Med. J. 1, 760e762.

J. Yellowlees Douglas et al. / Progress in Retinal and Eye Research 31 (2012) 481e494494
Yan, S.F., Ramasamy, R., Schmidt, A.M., 2009. Receptor for AGE (RAGE) and itsligands-cast into leading roles in diabetes and the inflammatory response.J. Mol. Med. (Berl.) 87, 235e247.
Ye, J., 2008. Regulation of PPARgamma function by TNF-alpha. Biochem. Biophys.Res. Commun. 374, 405e408.
Yoder, M.C., Mead, L.E., Prater, D., Krier, T.R., Mroueh, K.N., Li, F., Krasich, R.,Temm, C.J., Prchal, J.T., Ingram, D.A., 2007. Redefining endothelial progenitorcells via clonal analysis and hematopoietic stem/progenitor cell principals.Blood 109, 1801e1809.
Yrjanheikki, J., Tikka, T., Keinanen, R., Goldsteins, G., Chan, P.H., Koistinaho, J., 1999.A tetracycline derivative, minocycline, reduces inflammation and protectsagainst focal cerebral ischemia with a wide therapeutic window. Proc. Natl.Acad. Sci. U.S.A. 96, 13496e13500.
Yune, T.Y., Lee, J.Y., Jung, G.Y., Kim, S.J., Jiang, M.H., Kim, Y.C., Oh, Y.J., Markelonis, G.J.,Oh, T.H., 2007. Minocycline alleviates death of oligodendrocytes by inhibitingpro-nerve growth factor production in microglia after spinal cord injury.J. Neurosci. 27, 7751e7761.
Zhang, Z.Y., Zhang, Z., Fauser, U., Schluesener, H.J., 2009. Improved outcome of EAN,an animal model of GBS, through amelioration of peripheral and centralinflammation by minocycline. J. Cell Mol. Med. 13, 341e351.
Zheng, L., Gong, B., Hatala, D.A., Kern, T.S., 2007. Retinal ischemia and reperfusioncauses capillary degeneration: similarities to diabetes. Invest. Ophthalmol. Vis.Sci. 48, 361e367.
Zheng, M.H., Chen, J., Kirilak, Y., Willers, C., Xu, J., Wood, D., 2005. Porcine smallintestine submucosa (SIS) is not an acellular collagenous matrix and containsporcine DNA: possible implications in human implantation. J. Biomed. Mater.Res. B Appl. Biomater. 73, 61e67.
Zhou, J., Wang, S., Xia, X., 2012. Role of intravitreal inflammatory cytokines andangiogenic factors in proliferative diabetic retinopathy. Curr. Eye Res. 37,416e420.
Zhu, S., Stavrovskaya, I.G., Drozda, M., Kim, B.Y.S., Ona, V., Li, M., Sarang, S., Liu, A.S.,Hartley, D.M., Wuk, D.C., Gullans, S., Ferrante, R.J., Przedborskikq, S., Kristal, B.S.,Friedlander, M., 2002. Minocycline inhibits cytochrome c release and delaysprogression of amyotrophic lateral sclerosis in mice. Nature 417, 74e78.




















