
Increased hepatic steatosis and insulin resistance in mice lacking hepatic androgen receptor
Upload
khangminh22Category
view
0download
0

TARGETING OF MACROPHAGES AND HEPATIC STELLATE CELLS FOR THE TREATMENT OF LIVER DISEASES
Dhadhang Wahyu Kurniawan

TARGETING OF MACROPHAGES AND HEPATIC STELLATE CELLS FOR THE TREATMENT OF LIVER DISEASES
DISSERTATION
to obtain the degree of doctor at the University of Twente,
on the authority of the Rector Magnificus, Prof. Dr. Ir. A. Veldkamp,
on account of the decision of the graduation committee, to be publicly defended
on Wednesday the 11th of May 2022 at 16.45 hours
by
DHADHANG WAHYU KURNIAWAN
Born on the 21st of March 1979
in Lamongan, Indonesia

This thesis has been approved by:
Promotors: Prof. Dr. Jai Prakash, University of Twente
Prof. Dr. Gert Storm, University of Twente
Co-promotor: Dr. Ruchi Bansal, University of Twente
Cover design: Arfin Deri Listiandi Printed by: Ipskampp printing Lay-out: Dhadhang Wahyu Kurniawan ISBN: 978-90-365-5344-5 DOI: 10.3990/1.9789036553445
© 2022 Dhadhang Wahyu Kurniawan, The Netherlands. All rights reserved. No parts of this thesis may be reproduced, stored in a retrieval system or transmitted in any form or by any means without permission of the author. Alle rechten voorbehouden. Niets uit deze uitgave mag worden vermenigvuldigd, in enige vorm of op enige wijze, zonder voorafgaande schriftelijke toestemming van de auteur.

Graduation Committee
Chairman: Prof. Dr. J.L. Herek University of Twente, The Netherlands
Supervisors: Prof. Dr. Gert Storm University of Twente, The Netherlands
Prof. Dr. Jai Prakash University of Twente, The Netherlands
Co-supervisor: Dr. Ruchi Bansal University of Twente, The Netherlands
Committee Members: Prof. Dr. H.B.J. Karperien University of Twente, The Netherlands
Prof. Dr. A. Kocer University of Twente, The Netherlands
Prof. Dr. K. Poelstra University of Groningen, The Netherlands
Prof. Dr. P. Olinga University of Groningen, The Netherlands
Prof. Dr. R. Weiskirchen RWTH Aachen University, Germany

The work in this thesis have been performed at the Department of Advanced Organ Bioengineering and Therapeutics (AOT), formerly known as Biomaterials Science and Technology (BST), University of Twente, The Netherlands.
The research in this thesis was financially supported by the Indonesia Endowment Fund for Education (Lembaga Pengelola Dana Pendidikan, LPDP Indonesia, grant no. 20151022024584) and the Netherlands Organization for Health Research and Development (ZonMW, NWO)-funded VENI innovation grant 916.151.94 (awarded to Dr. Ruchi Bansal).
The printing of this thesis was financially supported by the Department of Advanced Organ Bioengineering and Therapeutics, Faculty of Science and Technology, University of Twente, The Netherlands.
Paranymphs:
Iqbal Yulizar Mukti
Ahmed Mohamed Ramadan Hamza Mostafa

Dedicated to my wife, my sons, my parents, my family, friends, and colleagues

TABLE OF CONTENTS
CHAPTER 1 9 General Introduction, Aim and Outline
CHAPTER 2 23 Role of Spleen Tyrosine Kinase in Liver Diseases
CHAPTER 3 49 Therapeutic Inhibition of Spleen Tyrosine Kinase in Inflammatory Macrophages using PLGA nanoparticles for the Treatment of Non-alcoholic Steatohepatitis
CHAPTER 4 81 Src Kinase as a Potential Therapeutic Target in Non-alcoholic and Alcoholic Steatohepatitis
CHAPTER 5 123 Fibroblast growth factor 2 (FGF2) conjugated superparamagnetic iron oxide nanoparticles (SPIONs) ameliorate hepatic stellate cells activation in vitro and acute liver injury in mice
CHAPTER 6 157 Summary
APPENDIX 164
A: Nederlandse Samenvatting
B: Ringkasan Bahasa Indonesia
C: List of Publications
D: Acknowledgements
E: About the Author

9
Chapter 1
General Introduction, Aim and Outline

10
1.1 Introduction
The liver is a vital organ in our body that plays a key role in metabolism and blood
detoxification. Disruption or failure of these functions can result in drastic and lethal
consequences. In general, liver damage can be categorized into different stages, the diagnosis,
treatability, and eventual patient survival is dependent on the stage. The first stage of liver
disease is known as hepatitis (liver inflammation). If this condition is diagnosed early and
treated successfully, the inflammation may vanish. But, if the inflammation continues over a
longer period or upon repeated injury, it can lead to the scarring of the liver (fibrosis) that
eventually leads to chronic liver diseases such as cirrhosis (end-stage liver disease) and
hepatocellular carcinoma (HCC, primary liver cancer) [1, 2] (Figure 1). Liver fibrosis represents
the final pathological pathway irrespective of the etiology.
Figure 1. The illustration of pathogenesis of liver diseases, including the etiologies and the types of liver damage. The etiologies of liver diseases are unhealthy lifestyle, overconsumption of alcohol, and viral hepatitis. Created with www.biorender.com. The etiologies of liver diseases are non-alcoholic fatty liver disease (NAFLD, due to metabolic
disorders and unhealthy lifestyle), alcohol-associated liver disease (ALD, due to excessive
intake of alcohol), viral hepatitis (HBV/HCV, due to hepatitis B/C viral infections), drug-induced
liver injury (DILI, due to drug abuse), primary biliary cholangitis (PBC) or primary sclerosing
cholangitis (PSC) (due to auto-immune disorders) and hemochromatosis or Wilson’s disease

11
(due to genetic factors) etc [3-5]. Among other liver diseases, ALD and NAFLD are the most
common etiologies of chronic liver disease worldwide [6].
Over the past decades, hepatologists have made significant progress in disease understanding,
monitoring and disease management. However, two main challenges remain unresolved, i.e.,
(i) noninvasive, precise and early diagnosis (or disease staging), and (ii) effective treatment of
chronic liver diseases [7]. Currently, the only available treatment for the end-stage liver
diseases is liver transplantation; however, the feasibility of transplantation is limited due to a
lack of sufficient donor organs, and risks and complications associated with liver
transplantation, including transplant rejection, bleeding, infections and long-term use of
immunosuppressants [8]. Other treatments that mainly focus on removing the underlying
cause (e.g., a reduction of alcohol consumption in ALD or healthy diet, exercise and weight
loss in NAFLD that can slow the progression or even reverse early stage fibrosis [9]) are,
however, insufficient for chronic liver disease. Therefore, there is a need to develop effective
and safe therapies (preferably integrated with proper diagnosis, theranostics) for the
treatment of liver diseases.
Below we briefly elaborate on different (etiological) liver diseases:
Liver fibrosis
In chronic liver diseases, fibrosis is the result of an initial wound-healing response of the liver
to a repeated injury, which is associated with the secretion of inflammatory cytokines and
chemokines (produced by inflammatory immune cells, particularly macrophages), and an
excessive deposition of extracellular matrix (ECM) (produced by activated hepatic stellate
cells, HSCs or myofibroblasts). In the normal liver, ECM is a highly dynamic matrix with a
precisely regulated balance between synthesis and degradation. If the hepatic injury persists,
this regulation and ultimately liver regeneration fails and hepatocytes are substituted with
abundant ECM, including fibrillar collagen [10-12].
HSCs or myofibroblasts are the key source of excess ECM including collagen types I and III as
well as other proteins expressed in pathological fibrous tissues [11]. In particular, the
activation and transdifferentiation of quiescent HSCs into an activated myofibroblast-like
phenotype is the key pathogenic event in liver fibrosis characterized by an increased
expression of α-smooth muscle actin (α-SMA), a characteristic HSC activation marker, as well

12
as a large variety of proteins forming the connective tissue [13, 14]. The activation of HSCs is
mainly triggered by a multitude of profibrogenic and promitogenic mediators which are
released from injured liver cells (hepatocytes and sinusoidal endothelial cells) and from
(infiltrating) immune cells. Among these factors, transforming growth factor-β (TGF-β) and
platelet-derived growth factor (PDGF) are the key profibrogenic mediators [15]. Activation of
HSCs includes discrete phenotypic transformation characterized by the loss of retinoids,
increased proliferation, contractility and migration, and aberrant ECM secretion and
decreased matrix degradation, amongst other factors [14, 16].
Chronic liver diseases
Chronic liver disease (CLD) or end-stage liver disease, resulting from different etiologies as
mentioned above, is the leading cause of mortality worldwide [17]. CLD encompasses a large
number of conditions that have different etiologies and exist on a continuum between
hepatitis and cirrhosis [18, 19]. Liver cirrhosis is the result of a chronic inflammation and
fibrosis that can eventually lead to distortion of liver architecture, loss of liver function and,
as a result, end-stage liver failure [20-23]. The major complications associated with cirrhosis
includes ascites, spontaneous bacterial peritonitis, hepatic encephalopathy, portal
hypertension, variceal bleeding, and hepatorenal syndrome [24].
Non-alcoholic fatty liver disease (NAFLD)
As mentioned above, NAFLD represents one of the most common causes for chronic liver
disease worldwide and is one of the leading causes of liver-related mortality [25, 26]. NAFLD
is a disorder characterized by excessive accumulation of fat (in the form of triglycerides) in
hepatocytes (>5% fat content in the liver) [27, 28], and includes a spectrum of diseases ranging
from simple steatosis, steatohepatitis, to fibrosis and irreversible cirrhosis that may further
progress to HCC [29].
NAFLD is a continuously growing problem around the globe often accompanied with
increasing number of metabolic disorders including obesity, dyslipidemia, hypertension, and
type 2 diabetes mellitus (T2DM) [30-32]. Around 40-45 million adults in the United States are
diagnosed with NAFLD, which is directly associated with the high prevalence of obesity [26]
and steatosis present in 70% of T2DM patients [33]. In most NAFLD patients, the initial
symptoms start with lipid accumulation (steatosis), which is influenced by obesity and insulin

13
resistance (diabetes). Progression to steatohepatitis and fibrosis depends on other factors
such as free fatty acids (FFAs), inflammation, oxidative stress, and mitochondrial dysfunction
in a complex interplay with genetic predisposition [29, 34-36].
Non-alcoholic steatohepatitis (NASH) is considered to be the progressive/severe form of
NAFLD [27, 37] and is a leading cause of chronic liver disease worldwide [38, 39]. It is
associated with liver cancer even in the absence of overt liver cirrhosis. Studies suggest that
the incidence of NASH-associated HCC will increase over time and will become one of the
major etiology (together with alcoholic liver disease (ALD)) for HCC in the future [38, 40].
The pathogenesis of NASH and its progression to fibrosis is very complex and involves multiple
mechanisms (multiple hit hypothesis) [37, 41]. Excessive FFAs in the liver are the major driving
forces for steatosis. Steatosis is associated with oxidative stress and endoplasmic reticulum
stress that leads to hepatocyte injury and apoptosis. Prolonged and unrepaired cell injuries
promote inflammation involving Kupffer cells and infiltrating immune cells, and stimulate
fibrogenesis driven by HSCs [42-44].
Alcoholic liver disease (ALD)
As in NAFLD, ALD also varies from simple steatosis to alcohol-associated steatohepatitis (ASH)
that leads to fibrosis with or without cirrhosis and eventually HCC may develop [45, 46]. A
pivotal component in the progression of ALD is the direct toxicity of the first metabolite of
alcohol degradation i.e., acetaldehyde [47]. The universal therapy of ALD patients is prolonged
alcohol abstinence, regardless of the disease stage. However, most patients are diagnosed at
advanced stages of the disease, which leads to higher rates of complications and mortality
[48]. As the only established therapies include nutritional support and corticosteroids, it is
urgent to develop effective therapies based on the understanding of the pathophysiology of
ALD [49, 50].
1.2 Aim of the thesis
Hepatic macrophages and HSCs are main pathogenic drivers that are involved in the initiation
and development of liver inflammation, fibrosis, cirrhosis and hepatocarcinogenesis.
Consequently, hepatic macrophages and HSCs are the promising targets for therapeutics for
the treatment of liver diseases [51]. This thesis aims to identify novel approaches and potential

14
therapeutics to attenuate the inflammation and progression of liver fibrosis by
(mechanistic/molecular) targeting hepatic macrophages and HSCs.
1.3 Outline of the thesis
In this thesis, we developed different approaches to modulate macrophages and HSCs for the
treatment of liver diseases.
Macrophages are important cells of the innate immune system that have crucial roles in many
inflammatory diseases, either acute or chronic. Hepatic macrophages comprise of Kupffer cells
and infiltrating (bone marrow-derived) monocytes/macrophages [52]. During liver injury,
Kupffer cells rapidly respond to the injury by producing cytokines and chemokines, including
interleukin-1β (IL-1β), tumor necrosis factor α (TNF-α), C-C motif chemokine ligand 2 (CCL2),
and C-C motif chemokine ligand 5 (CCL5), resulting in the recruitment of other immune cells,
such as monocytes [53]. In response to exogenous or endogenous danger signals (e.g.
bacterial products or necrotic cell debris) through pattern recognition receptors (PRRs) in the
hepatic microenvironment, macrophages can be polarized into multiple phenotypes,
particularly M1 (classical) or M2 (alternative) activation states as known traditionally, for
simplistic presentation [54]. In general, M1 macrophages are considered as the primary
mediator of inflammation (pro-inflammatory) and M2 macrophages as the primary mediator
of wound healing during pathological fibrosis (anti-inflammatory) [55]. Macrophages
therefore exert a dual role i.e., causing liver inflammation and/or fibrosis resolution.
HSCs are multifunctional non-parenchymal cells located in the space of Disse between
sinusoidal endothelial cells and hepatocytes. In the normal/healthy liver, HSCs remain in a
non-proliferative and quiescent phenotype, and store vitamin A lipid droplets. As mentioned
above, upon liver injury, HSCs get activated and transdifferentiate into proliferative,
contractile, inflammatory, chemotactic, and ECM producing activated HSCs or myofibroblasts
[56, 57]. Activated HSCs produce tremendous amount of aberrant ECM that leads to the scar
tissue formation referred to as liver fibrosis that further progresses to CLD [58, 59].
As a first step in this thesis, to identify molecular targets in macrophages, we performed gene
profiler array in M0, M1- and M2-polarized mouse RAW264.7 macrophages. Two major kinase
families that are involved in the intracellular signaling pathways in innate cells are the Src-

15
family kinases and the SYK-ZAP70 kinases. We therefore assessed the implication of the SYK
and Src kinases in liver diseases.
SYK (spleen tyrosine kinase) is a cytoplasmic tyrosine kinase of 72 KDa that belongs to
ZAP70/SYK family of the non-receptor protein tyrosine kinases (PTKs). SYK was identified and
known to be expressed in hematopoietic cells, including macrophages, and has shown to be
involved in the downstream signaling events that drive inflammatory pathways of both the
innate and adaptive immune system. While most studies focused on hematopoietic cells, SYK
has also been shown to be expressed in non-hematopoietic cells including fibroblasts and
hepatocytes. In chapter 2, we present a review dealing with the SYK pathway in liver diseases.
This review summarizes the current understanding of SYK and its therapeutic implication in
liver diseases.
Understanding the role of SYK in liver and other diseases, we studied the implication of the
SYK pathway in NASH and investigated Poly (lactic-co-glycolic acid) (PLGA)-nanoparticles
based delivery of a SYK pathway inhibitor for the treatment of NASH. In chapter 3, we first
analyzed the SYK expression in livers of NASH and alcoholic hepatitis (AH) patients. We then
examined the expression and activation of the SYK signaling pathway in inflammatory
macrophages. We, thereafter, using the small-molecule SYK inhibitor R406, studied the
therapeutic implication of SYK inhibition in inflammatory macrophages. For the efficient
delivery of the SYK inhibitor R406 in vivo, we formulated R406 in PLGA nanoparticles and
investigated their therapeutic efficacy in vitro in M1-differentiated RAW macrophages and
murine bone marrow derived macrophages, and in vivo in the MCD diet-induced NASH mouse
model (Figure 2). With the promising results obtained during this study, we dwelled into the
other tyrosine kinases i.e., Src family of protein tyrosine kinases (SFKs), particularly Src being
the poorly investigated SFKs, that are the largest family of cytoplasmic tyrosine kinases
expressed in innate immune cells.
Src is a non-receptor tyrosine kinase that belongs to the Src family of protein tyrosine kinases
(SFKs) that includes nine members: Src, Yes, Fyn, Fgr, Lck, Hck, Blk, Lyn and Yrk. While Src, Fyn
and Yes are ubiquitously expressed, others (except Yrk, exclusively expressed in chickens) are
restricted to hematopoietic cells. SFKs regulate many fundamental cellular processes including

16
cell growth, differentiation, migration, survival, and are also identified as proto-oncogenes
involved in the cancer progression.
Figure 2. Graphical abstract depicting the hypothesized therapeutic role of R406-containing PLGA nanoparticles with improved pharmacokinetics of R406 in a NASH mouse model (Kurniawan et. al., Journal of Controlled Release 288 (2018) 227–238 [60]).
Moreover, SFKs play an essential role in the recruitment and activation of monocytes,
macrophages, neutrophils, and other immune cells. Considering the multicellular functions
and regulation of multiple signaling pathways by Src, we hypothesized Src may be a master
regulator involved in the pathophysiology of NAFLD and ALD (Figure 3). Therefore, in chapter
4, we investigated the role of Src kinase in NASH and ASH. We first assessed the expression
and activation of Src in human liver diseases from different etiologies i.e., NASH, alcoholic
hepatitis (AH), hepatitis C virus (HCV)-cirrhosis and biliary atresia (BA); and in respective
disease mouse models that mimic pathophysiological features of human NASH (methionine
choline deficient MCD-diet induced), ASH (chronic Lieber De Carli ethanol EtOH-diet induced
model), and carbon tetrachloride (CCl4)- and bile duct ligation (BDL)-induced liver fibrosis.
Thereafter, we assessed the functional role of Src kinase using a selective Src inhibitor KX2-
391. Functional inhibition of Src was studied in vitro in cell culture, precise-cut liver slices
(PCLS) and 3D human liver spheroids, and in vivo in MCD-diet-induced NASH and EtOH-diet-

17
induced ASH mouse models. We further examined mechanistic aspects involved in Src-
mediated effects.
Figure 3. Graphical abstract depicting the role of Src kinase in liver steatosis, inflammation, and fibrosis. The figure shows that Src kinase activates macrophages and HSCs via FAK/PI3K/AKT pathways, and is involved in fatty acid biosynthesis. KX2-391, a selective Src kinase inhibitor inhibited the key processes involved in alcoholic and non-alcoholic fatty liver diseases (Kurniawan et. al., Clinical and Translational Discovery, 2022 (in press)).
In the last chapter (chapter 5), we focused on targeting HSCs, key cell pathogenic cells involved
in fibrogenesis, with the aim to develop a theranostic approach with combined therapy and
diagnosis for personalized disease management. Fibroblast growth factors (FGFs) have been
shown to regulate HSCs differentiation and liver fibrosis. FGF-FGFR signaling pathways have
been shown to regulate liver homeostasis by regulating metabolism, promoting hepatocyte
proliferation and detoxification, and liver regeneration after partial hepatectomy. Specifically,
FGF2 has been shown to regulate HSCs function and has been investigated previously in liver
fibrosis. However, contradictory results have been reported. In this chapter, we investigated
the role of FGF2 and FGF2-SPIONs (FGF2 conjugated to the surface of superparamagnetic iron
oxide nanoparticles) as a theranostic approach with the aim to achieve an effective therapy
with simultaneous diagnosis of the stage of advanced fibrosis. To accomplish our aim, we first
analyzed the expression of FGFR in human liver cirrhosis and TGFβ-activated human HSCs (LX

18
cells) in vitro. We then investigated the effects of human recombinant FGF2 (low-molecular
weight) on TGFβ-activated human HSCs (LX2 cells) in vitro. In order to improve the stability
and systemic half-life of FGF2, we conjugated FGF2 to dextran- and PEG-coated SPIONs (Figure
4). Subsequently, we examined the therapeutic effects of FGF2-SPIONs versus free FGF2 on
TGFβ-activated human HSCs in vitro and in an acute CCl4 induced mouse model in vivo.
Figure 4. Schematic representation of FGF2 conjugation to SPIONs using carbodiimide chemistry (Kurniawan et. al., Journal of Controlled Release 328 (2020) 640–652 [61]).
Finally, in chapter 6, we summarize and discuss the findings presented in this thesis.
1.4 References
[1] A.L. Foundation, Progression of Liver Disease, American Liver Foundation Handout, 2016. [2] J. Das, Liver disease pathophysiology, Clinical Pharmacist, 3 (2011) 140-144. [3] J. Wiegand, T. Berg, The etiology, diagnosis and prevention of liver cirrhosis: part 1 of a
series on liver cirrhosis, Dtsch Arztebl Int, 110 (2013) 85-91. [4] W.M. Lee, Etiologies of acute liver failure, Semin Liver Dis, 28 (2008) 142-152. [5] J.C. Nault, Pathogenesis of hepatocellular carcinoma according to aetiology, Best Pract Res
Clin Gastroenterol, 28 (2014) 937-947. [6] N. Toshikuni, M. Tsutsumi, T. Arisawa, Clinical differences between alcoholic liver disease
and nonalcoholic fatty liver disease, World J Gastroenterol, 20 (2014) 8393-8406. [7] P. Uhl, G. Fricker, U. Haberkorn, W. Mier, Current status in the therapy of liver diseases,
Int J Mol Sci, 15 (2014) 7500-7512. [8] D.-B. Moon, S.-G. Lee, Liver transplantation, Gut and Liver, 3 (2009) 145-165. [9] N. Pydyn, K. Miekus, J. Jura, J. Kotlinowski, New therapeutic strategies in nonalcoholic fatty
liver disease: a focus on promising drugs for nonalcoholic steatohepatitis, Pharmacol Rep, 72 (2020) 1-12.
[10] R. Bataller, D.A. Brenner, Liver fibrosis, Journal of Clinical Investigation, 115 (2005) 209-218.
[11] K. Bottcher, M. Pinzani, Pathophysiology of liver fibrosis and the methodological barriers to the development of anti-fibrogenic agents, Adv Drug Deliv Rev, 121 (2017) 3-8.
[12] R. Xu, Z. Zhang, F.S. Wang, Liver fibrosis: mechanisms of immune-mediated liver injury, Cell Mol Immunol, 9 (2012) 296-301.
[13] R. Weiskirchen, F. Tacke, Liver fibrosis: which mechanisms matter, Clinical Liver Disease 8(2016) 94-99.
[14] T. Higashi, S.L. Friedman, Y. Hoshida, Hepatic stellate cells as key target in liver fibrosis, Adv Drug Deliv Rev, 121 (2017) 27-42.

19
[15] A.M. Gressner, R. Weiskirchen, Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-b as major players and therapeutic targets, Journal of Cellular and Molecular Medicine, 10 (2006) 76-99.
[16] J.T. Li, Z.X. Liao, J. Ping, D. Xu, H. Wang, Molecular mechanism of hepatic stellate cell activation and antifibrotic therapeutic strategies, J Gastroenterol, 43 (2008) 419-428.
[17] V.W. Setiawan, D.O. Stram, J. Porcel, S.C. Lu, L. Le Marchand, M. Noureddin, Prevalence of chronic liver disease and cirrhosis by underlying cause in understudied ethnic groups: The multiethnic cohort, Hepatology, 64 (2016) 1969-1977.
[18] T.R.R. III, A.M. Bhatti, Preventive strategies in chronic liver disease: Part II. cirrhosis, American Family Physician 64 (2001) 1735-1740.
[19] R.S. Rahimi, D.C. Rockey, Complications and outcomes in chronic liver disease, Curr Opin Gastroenterol, 27 (2011) 204-209.
[20] R.A. Mohammad, Complications of chronic liver disease, 2010, pp. 91-108. [21] A. Tayyeb, F. Azam, R. Nisar, R. Nawaz, U. Qaisar, G. Ali, Regenerative Medicine in Liver
Cirrhosis: Promises and Pitfalls, Liver Cirrhosis - Update and Current Challenges2017. [22] S.A. Almani, A.S. Memon, A.I. Memon, M. Iqbal Shah, M.Q. Rahpoto, R. Solangi, Cirrhosis
of liver: etiological factors, complications and prognosis, JLUMHS, (2008) 61-66. [23] X. Verhelst, A. Geerts, H.V. Vlierberghe, Cirrhosis: reviewing the literature and future
perspectives, European Medical Journal, (2016) 111-117. [24] J.J. Heidelbaugh, M. Sherbondy, Cirrhosis and chronic liver failure: part II. complications
and treatment, American Family Physician 74 (2006). [25] B. Li, C. Zhang, Y.T. Zhan, Nonalcoholic Fatty Liver Disease Cirrhosis: A Review of Its
Epidemiology, Risk Factors, Clinical Presentation, Diagnosis, Management, and Prognosis, Can J Gastroenterol Hepatol, 2018 (2018) 2784537.
[26] M. Lazo, R. Hernaez, M.S. Eberhardt, S. Bonekamp, I. Kamel, E. Guallar, A. Koteish, F.L. Brancati, J.M. Clark, Prevalence of nonalcoholic fatty liver disease in the United States: the Third National Health and Nutrition Examination Survey, 1988-1994, Am J Epidemiol, 178 (2013) 38-45.
[27] E.M. Brunt, V.W. Wong, V. Nobili, C.P. Day, S. Sookoian, J.J. Maher, E. Bugianesi, C.B. Sirlin, B.A. Neuschwander-Tetri, M.E. Rinella, Nonalcoholic fatty liver disease, Nat Rev Dis Primers, 1 (2015) 15080.
[28] N. Katsiki, D.P. Mikhailidis, C.S. Mantzoros, Non-alcoholic fatty liver disease and dyslipidemia: An update, Metabolism, 65 (2016) 1109-1123.
[29] J.K. Dowman, J.W. Tomlinson, P.N. Newsome, Pathogenesis of non-alcoholic fatty liver disease, QJM, 103 (2010) 71-83.
[30] M.E. Rinella, Nonalcoholic fatty liver disease: a systematic review, JAMA, 313 (2015) 2263-2273.
[31] Y. Fazel, A.B. Koenig, M. Sayiner, Z.D. Goodman, Z.M. Younossi, Epidemiology and natural history of non-alcoholic fatty liver disease, Metabolism, 65 (2016) 1017-1025.
[32] S.A. Polyzos, C.S. Mantzoros, Nonalcoholic fatty future disease, Metabolism, 65 (2016) 1007-1016.
[33] G. Targher, L. Bertolini, R. Padovani, S. Rodella, R. Tessari, L. Zenari, C. Day, G. Arcaro, Prevalence of nonalcoholic fatty liver disease and its association with cardiovascular disease among type 2 diabetic patients, Diabetes Care, 30 (2007) 1212-1218.
[34] Y.L. Fang, H. Chen, C.L. Wang, L. Liang, Pathogenesis of non-alcoholic fatty liver disease in children and adolescence: From "two hit theory" to "multiple hit model", World J Gastroenterol, 24 (2018) 2974-2983.

20
[35] J. Yu, S. Marsh, J. Hu, W. Feng, C. Wu, The Pathogenesis of Nonalcoholic Fatty Liver Disease: Interplay between Diet, Gut Microbiota, and Genetic Background, Gastroenterol Res Pract, 2016 (2016) 2862173.
[36] E. Buzzetti, M. Pinzani, E.A. Tsochatzis, The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD), Metabolism, 65 (2016) 1038-1048.
[37] M.V. Machado, A.M. Diehl, Pathogenesis of Nonalcoholic Steatohepatitis, Gastroenterology, 150 (2016) 1769-1777.
[38] A. Canbay, J.P. Sowa, W.K. Syn, J. Treckmann, NASH Cirrhosis - the New Burden in Liver Transplantation: How Should It Be Managed?, Visc Med, 32 (2016) 234-238.
[39] Z.M. Younossi, The epidemiology of nonalcoholic steatohepatitis, Clinical Liver Disease, 11 (2018) 92-94.
[40] M. Arrese, A.E. Feldstein, Nash-Related Cirrhosis: An Occult Liver Disease Burden, Hepatol Commun, 1 (2017) 84-86.
[41] N. Magee, A. Zou, Y. Zhang, Pathogenesis of Nonalcoholic Steatohepatitis: Interactions between Liver Parenchymal and Nonparenchymal Cells, Biomed Res Int, 2016 (2016) 5170402.
[42] W. Liu, R.D. Baker, T. Bhatia, L. Zhu, S.S. Baker, Pathogenesis of nonalcoholic steatohepatitis, Cell Mol Life Sci, 73 (2016) 1969-1987.
[43] K.H. Kim, M.S. Lee, Pathogenesis of Nonalcoholic Steatohepatitis and Hormone-Based Therapeutic Approaches, Front Endocrinol (Lausanne), 9 (2018) 485.
[44] A. Caligiuri, A. Gentilini, F. Marra, Molecular Pathogenesis of NASH, Int J Mol Sci, 17 (2016).
[45] R.S. O'Shea, S. Dasarathy, A.J. McCullough, Alcoholic liver disease, Am J Gastroenterol, 105 (2010) 14-32; quiz 33.
[46] W. Dunn, V.H. Shah, Pathogenesis of Alcoholic Liver Disease, Clin Liver Dis, 20 (2016) 445-456.
[47] F. Stickel, C. Datz, J. Hampe, R. Bataller, Pathophysiology and Management of Alcoholic Liver Disease: Update 2016, Gut Liver, 11 (2017) 173-188.
[48] M.O. Farooq, R. Bataller, Pathogenesis and Management of Alcoholic Liver Disease, Dig Dis, 34 (2016) 347-355.
[49] B. Gao, R. Bataller, Alcoholic liver disease: pathogenesis and new therapeutic targets, Gastroenterology, 141 (2011) 1572-1585.
[50] L.Z. Kong, N. Chandimali, Y.H. Han, D.H. Lee, J.S. Kim, S.U. Kim, T.D. Kim, D.K. Jeong, H.N. Sun, D.S. Lee, T. Kwon, Pathogenesis, Early Diagnosis, and Therapeutic Management of Alcoholic Liver Disease, Int J Mol Sci, 20 (2019).
[51] F. Tacke, Targeting hepatic macrophages to treat liver diseases, J Hepatol, 66 (2017) 1300-1312.
[52] C. Ju, F. Tacke, Hepatic macrophages in homeostasis and liver diseases: from pathogenesis to novel therapeutic strategies, Cell Mol Immunol, 13 (2016) 316-327.
[53] Y.-y. Ma, M.-q. Yang, Z.-g. He, Q. Wei, J.-y. Li, The Biological Function of Kupffer Cells in Liver Disease, Biology of Myelomonocytic Cells2017.
[54] A. Sica, P. Invernizzi, A. Mantovani, Macrophage plasticity and polarization in liver homeostasis and pathology, Hepatology, 59 (2014) 2034-2042.
[55] P.J. Wermuth, S.A. Jimenez, The significance of macrophage polarization subtypes for animal models of tissue fibrosis and human fibrotic diseases, Clin Transl Med, 4 (2015) 2.
[56] C. Yin, K.J. Evason, K. Asahina, D.Y. Stainier, Hepatic stellate cells in liver development, regeneration, and cancer, J Clin Invest, 123 (2013) 1902-1910.

21
[57] T. Tsuchida, S.L. Friedman, Mechanisms of hepatic stellate cell activation, Nat Rev Gastroenterol Hepatol, 14 (2017) 397-411.
[58] R.K. Moreira, Hepatic stellate cells and liver fibrosis, Arch Pathol Lab Med, 131 (2007) 1728-1734.
[59] C. Hoffmann, N.E.H. Djerir, A. Danckaert, J. Fernandes, P. Roux, C. Charrueau, A.M. Lachages, F. Charlotte, I. Brocheriou, K. Clement, J. Aron-Wisnewsky, F. Foufelle, V. Ratziu, B. Hainque, D. Bonnefont-Rousselot, P. Bigey, V. Escriou, Hepatic stellate cell hypertrophy is associated with metabolic liver fibrosis, Sci Rep, 10 (2020) 3850.
[60] D.W. Kurniawan, A.K. Jajoriya, G. Dhawan, D. Mishra, J. Argemi, R. Bataller, G. Storm, D.P. Mishra, J. Prakash, R. Bansal, Therapeutic inhibition of spleen tyrosine kinase in inflammatory macrophages using PLGA nanoparticles for the treatment of non-alcoholic steatohepatitis, J Control Release, 288 (2018) 227-238.
[61] D.W. Kurniawan, R. Booijink, L. Pater, I. Wols, A. Vrynas, G. Storm, J. Prakash, R. Bansal, Fibroblast growth factor 2 conjugated superparamagnetic iron oxide nanoparticles (FGF2-SPIONs) ameliorate hepatic stellate cells activation in vitro and acute liver injury in vivo, J Control Release, 328 (2020) 640-652.

22

23
Chapter 2
Role of Spleen Tyrosine Kinase in Liver Diseases
Dhadhang Wahyu Kurniawan, Gert Storm, Jai Prakash, Ruchi Bansal
Published in: World J Gastroenterol. 2020 Mar 14;26(10):1005-1019. doi: 10.3748/wjg. v26.i10.1005.

24
Abstract
Spleen tyrosine kinase (SYK), a non-receptor tyrosine kinase, is expressed in most
hematopoietic cells and non-hematopoietic cells and play a crucial role in both immune and
non-immune biological responses. SYK mediate diverse cellular responses via an immune-
receptor tyrosine-based activation motifs (ITAMs)-dependent signalling pathways, ITAMs-
independent and ITAMs-semi-dependent signalling pathways. In liver, SYK expression has
been observed in parenchymal (hepatocytes) and non-parenchymal cells (hepatic stellate cells
and Kupffer cells) and found to be positively correlated with the disease severity. The
implication of SYK pathway has been reported in different liver diseases including liver fibrosis,
viral hepatitis, alcoholic liver disease, non-alcoholic steatohepatitis, and hepatocellular
carcinoma. Antagonism of SYK pathway using kinase inhibitors have shown to attenuate the
progression of liver diseases thereby suggesting SYK as a highly promising therapeutic target.
This review summarizes the current understanding of SYK and its therapeutic implication in
liver diseases.

25
2.1 Introduction
Spleen tyrosine kinase (SYK) is a cytoplasmic non-receptor protein tyrosine kinase (PTK) that
consists of two SYK homology 2 domains (SH2) and a C-terminal tyrosine kinase domain. These
domains are linked by two linker regions: interdomain A between the two SH2 domains and
interdomain B between the C-terminal SH2 domain and the kinase domain (Figure 1). SYK is a
member of the Zeta-chain-associated protein kinase 70/SYK family of the PTKs, with the
estimated molecular weight of 70 kDa [1, 2]. SYK is highly expressed in hematopoietic cells
including mast cells, neutrophils, macrophages, platelets, B cells and immature T cells, and is
important in signal transduction in these cells [2, 3]. In Immune cells, SYK mainly functions via
interaction of its tandem SH2 domains with immunoreceptor tyrosine-based activation motifs
(ITAMs). In mast cells, SYK mediates downstream signaling via high-affinity IgE receptors, FcεRI
and in neutrophils, macrophages, monocytes and platelets downstream signalling is mediated
via high affinity Igγ receptors, FcγR [4-6]. SYK plays a key role in signaling downstream of the
B and T cell receptors, hence also play a crucial role in early lymphocyte development [4, 7-
10]. Upon activation, SYK modulates downstream signaling events that drive inflammatory
pathways of both the innate and adaptive immune systems [11]. Besides ITAM-dependent
signalling pathway, SYK also mediates ITAM-independent signaling via integrins and C-type
lectins. For instance, SYK induces β2 integrin-mediated respiratory burst, spreading, and site-
directed migration of neutrophils towards inflammatory lesions [12].
Figure 1. Structure of spleen tyrosine kinase. Spleen tyrosine kinase contains tandem pair of spleen tyrosine kinase homology 2 which connected by interdomain A and separated by interdomain B from the catalytic (kinase) domain. SYK: Spleen tyrosine kinase; SH2: Spleen tyrosine kinase homology 2; ITAM: Immune-receptor tyrosine-based activation motifs.
The multifactorial role of SYK in the immune system has attracted attention in the past years.
SYK is recognized as a potential target for the treatment of inflammatory diseases such as
rheumatoid arthritis, asthma, allergic rhinitis, renal disorders, liver fibrosis and autoimmune

26
diseases [3, 7, 13-23]. In particular, the prevention of activation of cells via immune complexes
or antigen-triggered Fc receptor signaling and prevention of B cell receptor-mediated events
are believed to have increasing therapeutic potential of SYK [24, 25].
Besides hematopoietic cells, SYK has also been shown to be expressed in non-hematopoietic
cells including fibroblasts, epithelial cells, hepatocytes, neuronal cells, and vascular
endothelial cells [7, 26]. Here, SYK has shown to be involved in signalling steps leading to
mitogen activated protein (MAP) kinase activation by G-protein-coupled receptors in
hepatocytes [26, 27]. Besides being implicated in hepatocytes, SYK is also expressed in hepatic
macrophages, hepatic stellate cells (HSCs) and hepatic sinusoidal endothelial cells in liver [28].
However, studies investigating SYK signaling pathway in liver diseases are still limited, hence
this review highlights and discusses the opportunities and challenges of SYK as a potential
target for the treatment of liver diseases.
2.2 Spleen Tyrosine Kinase signaling mechanisms
Immunoreceptor signaling through SYK requires the SYK kinase activity as well as both SH2
domains [29]. The SYK kinase domain is inactive in the resting state of the protein but can be
activated by interaction of both SH2 domains to dual phosphorylated ITAMs [30].
Phosphorylation of tyrosine residues within the linker regions (interdomain A or B) also results
in kinase activation even in the absence of phosphorylated ITAM binding [29, 30]. Binding of
the SH2 domains of SYK to phosphorylated ITAMs is a critical step in SYK activation and
downstream signaling [31]. SYK itself can catalyze the autophosphorylation of its linker
tyrosine’s, leading to sustained SYK activation after a transient ITAM phosphorylation. In
addition, SYK itself can phosphorylate ITAMs, suggesting the existence of a positive-feedback
loop during initial ITAM-mediated SYK activation [32]. Tsang et al. [33] showed that SYK can
be fully triggered by phosphorylation or binding of its SH2 domains to the dual-
phosphorylated immune-receptor tyrosine based activity motif (ppITAM) (Figure 2) [33, 34].
Recently, Slomiany and Slomiany demonstrated LPS-induced SYK activation through protein
kinase Cδ (PKCδ)-mediates SYK phosphorylation on serine residues that is required for its
recruitment to the membrane-anchored TLR4, followed by SYK subsequent activation through
tyrosine phosphorylation. Hence, the intermediate phase of PKCδ-mediated SYK
phosphorylation on serine residues affects the inflammatory response [35]. The activated SYK

27
binds to a number of downstream signaling effectors and amplifies the inflammatory signal
propagation by affecting transcription factors activation and their assembly to transcriptional
complexes involved in expression of the proinflammatory genes [36].
Figure 2. Basis of spleen tyrosine kinase activation. In the resting state, spleen tyrosine kinase is auto-inhibited, because of the binding of interdomain A and interdomain B to the kinase domain. This auto-inhibited conformation can be activated by binding of the two spleen tyrosine kinase homology 2 domains to dually phosphorylated immune-receptor tyrosine-based activation motifs or by phosphorylation of linker tyrosine’s in interdomain A or B. SH2: Spleen tyrosine kinase homology 2; ITAM: Immune-receptor tyrosine-based activation motifs.
2.3 Spleen Tyrosine Kinase in liver fibrosis
Liver fibrosis, triggered by hepatitis B/C viral infection (viral hepatitis), alcohol abuse (alcoholic
liver disease) or non-alcoholic steatohepatitis (NASH) etc., is characterized by an excessive
deposition of extracellular matrix (ECM) proteins [37], leading to tissue scarring that further
progresses to end-stage liver cirrhosis and hepatocellular carcinoma [38].
Liver fibrosis poses a major health problem accounting for more than 1 million people deaths
every year worldwide [39]. Moreover, there is no therapeutic treatment available to date [40].
The central player that produces ECM resulting in liver fibrosis is HSCs [41]. HSCs are normally
localized in the peri-sinusoidal area, termed as space of Disse, as quiescent cells in healthy
liver and functions as retinoid storage cells [42]. Owing to hepatic injury, quiescent HSCs

28
phenotypically transdifferentiate into activated, contractile, highly proliferative, and ECM-
producing myofibroblasts [43].
SYK has been documented to play a critical role in the activation of HSCs and its upregulation
is evidenced in hepatic fibrosis/cirrhosis in hepatitis B and C patients, alcoholic hepatitis as
well as in NASH patients [28, 44]. Upregulated SYK further aggravate fibrosis by augmenting
trans-communication between hepatocytes and HSCs [28]. Blockage of SYK pathway using SYK
inhibitors abrogated HSCs activation, thereby ameliorated liver fibrosis and HCC development
in vivo in animal models [28]. SYK has also shown to mediate its function via expression of
transcription factors associated with HSCs activation (cAMP response element-binding
protein, CBP; myeloblastosis proto-oncogene, MYB and myelocytomatosis proto-oncogene,
MYC) and proliferation (MYC and cyclin D1, CCND1) [28]. Furthermore, two isoforms of SYK
i.e. the full-length SYK(L) and an alternatively spliced SYK(S) have been suggested whereby
SYK(L) but not SYK(S) found to play a major role in liver fibrosis while SYK(S) has been
associated with increased tumorigenicity, HCC invasiveness and metastases [28].
Interestingly, the crosstalk between SYK and Wnt (portamanteau of int and wg, wingless-
related integration site) signaling pathways also mediates activation of HSCs and accumulation
of immune cells at the site of fibrosis [28]. Wnt signaling has been shown to be upregulated in
activated HSCs and blockade of canonical Wnt pathway by adenoviral mediated transduction
of Wnt antagonist (Dickkopf-1) or via selective inhibitors reinstates quiescent phase of HSCs
in cultured cells [45, 46]. In-depth investigation at a genetic level revealed overexpression of
certain transcriptional factors (MYB, CBP and MYC) which plays a vital role in the activation of
HSCs [47, 48]. Notably, both the canonical Wnt pathway and SYK has shown to regulate the
expression of MYC and CBP [23, 49] highlighting SYK-Wnt crosstalk during liver fibrogenesis.
SYK has also shown to promote expression of several target genes including Wnt in activated
macrophages in a similar manner as in HSCs and this potential crosstalk between SYK and
other signaling pathways warrants further investigation. Dissection of the trans-
communication between signaling pathways is of great importance in order to highlight
prominent therapeutic targets to hinder inflammation and fibrogenesis. SYK is the major
signaling pathway and is also shown to be expressed in recruited macrophages, besides HSCs,
in the hepatic fibrosis [20, 28]. Selective blocking of SYK or its deletion in macrophages has
been correlated with the diminished activation of macrophages, which is indicated by a

29
reduction in the expression of Fc gamma receptors, monocyte chemoattractant protein 1
(MCP-1), tumor necrosis factor α (TNF-α) and interleukin 6 (IL-6) [5]. In summary, activation
of HSCs under the influence of SYK signaling leads to the secretion of soluble factors in the
form of cytokines and chemokines. These factors not only facilitate the recruitment of
macrophages (and other immune cells) but also arbitrates their activation to further worsen
the site of fibrosis.
2.4 Spleen Tyrosine Kinase in viral hepatitis
In the recent study, SYK expression was found to be highly induced in the liver tissues of HBV
and HCV infected patients. Furthermore, markedly increased expression of SYK was observed
in HCV-infected hepatocytes which in turn promoted reciprocal higher SYK expression in HSCs
thereby inducing HSCs activation and disease development [28, 50]. Furthermore, the
preliminary study analyzing gene expression profiles in Egyptian HCC patients associated with
HCV, showed that SYK is one of the most up-regulated genes out of 180 of genes were up
regulated [51].
HCV is also associated with B lymphocyte proliferative disorders, as evidenced by the binding
of HCV to B-cell surface receptor CD81 [52]. CD81 (cluster of differentiation 81, also known as
TAPA1), is identified as a target of an antibody that controlled B-cell proliferation. Engagement
of CD81 with HCV [53, 54], leads to ezrin and radixin phosphorylation through SYK activation
[55, 56]. Ezrin and radixin are members of the ERM (ezrin, radixin, moesin) family of actin-
binding proteins [56]. Hence, ezrin-moesin-radixin proteins and SYK are important therapeutic
host targets for the development HCV treatment [57].
SYK is also an important regulator and therapeutic target against HCV infection in hepatocytes
[55]. SYK expression has been observed near the plasma membrane of hepatocytes in HCV-
infected patients [57, 58]. HCV non-structural protein 5A (NS5A) has been shown to physically
and directly interact with SYK thereby promoting the malignant transformation of HCV-
infected hepatocytes [58]. These studies suggests that the strategies blocking SYK activation
before HCV-CD81 interaction, and/or modulating HCV post-entry and trafficking within target
cells involving SYK, F-actin, stable microtubules and EMR proteins provide novel opportunities
for the development of anti-HCV therapies [55].

30
2.5 Spleen Tyrosine Kinase in alcoholic liver disease (ALD)
The pathogenesis of ALD is multifactorial involving many complex processes including ethanol-
mediated liver injury, inflammation in response to the injury, and intestinal permeability and
microbiome changes [59-61] as shown in Figure 3. Alcohol and its metabolites generate
reactive oxygen species (ROS) and induce hepatocyte injury through mitochondrial damage
and endoplasmic reticulum (ER) stress [62-64]. Damaged hepatocytes release pro-
inflammatory cytokines and chemokines resulting in recruitment and activation of immune
cells. Central cell types involved in ALD progression are macrophages that have an important
role in inducing liver inflammation [65] by stimulating infiltration of immune cells (mainly
monocytes) and activation of Kupffer cells (KCs, resident macrophages) [59]. The early
communication of hepatocyte damage is mediated by KCs through damage-associated
molecular patterns (DAMPs) released by dying hepatocytes or pathogen-associated molecular
patterns (PAMPs) including lipopolysaccharides (LPS) via pattern recognition receptors (PRRs)
such as TLRs (Toll-like receptors), and NF-κB (nuclear factor kappa-light-chain-enhancer of
activated B cells) signaling and inflammasome activation etc. In ALD, resident and recruited
macrophages in the liver are activated by TLR4 (Toll-like receptor 4) signaling pathway
regulated by bacterial endotoxin (LPS) that is elevated in the portal and systemic circulation
owing to increased intestinal permeability after excessive alcohol intake [66, 67]. However,
there are also other mechanisms that regulate macrophage activation, such as hepatocyte
injury and lipid accumulation, histone acetylation in ethanol-exposed macrophages and
complement system [68]. SYK also plays an important role in TLR4 signaling, and SYK
phosphorylation in neutrophils and monocytes has been correlated with pro-inflammatory
cytokine secretion including TNF-α and MCP-1 [69]. Interestingly, SYK phosphorylation has
been shown to be regulated by LPS/TLR adaptor molecules MyD88/IRAKM (IL-1R-associated
kinase M)-mincle axis linking LPS-induced hepatocyte cell death with inflammation during ALD
disease pathogenesis. Zhou et al., has shown that damaged hepatocytes release endogenous
Mincle ligand spliceosome-associated protein 130 (SAP130) as a danger signal that
synergistically with LPS drives inflammation including inflammasome activation during ALD
[70].
Several studies have documented the increased SYK expression and phosphorylation in the
livers of alcoholic hepatitis (AH) patients [44]. Interestingly, increased SYK phosphorylation

31
was observed in ballooned hepatocytes with Mallory-Denk Bodies, co-localized with
ubiquitinated proteins in the cytoplasm suggesting the critical role of SYK in hepatocytes
during ER stress [71, 72]. SYK regulates hepatic cell death via TRAF family member-associated
NF-Ƙβ activator (TANK)-binding kinase 1/interferon (IFN) regulatory factor 3 (TBK1/IRF3)
signaling [20]. SYK has also been reported to play an important role in lipid accumulation, and
treatment with SYK inhibitor prevented progressive steatosis by suppressing lipid biogenesis
and increasing lipid metabolism in both in vitro cell culture and in vivo in ALD mouse models
exhibiting moderate ASH and chronic alcohol drinking [20]. SYKY525/526 phosphorylation
indicates SYK activation and is a prerequisite for its downstream modulatory function [73]. In
addition, total SYK and activated pSYKY525/526 expression was found to be significantly
increased in the circulating blood monocytes, and PBMCs in AH/cirrhosis patients [20]. Since
SYK is closely involved in the pathogenesis of ALD, SYK inhibition could prevent and/or
attenuate alcohol-induced liver inflammation, cell death, steatosis and subsequently fibrosis
in various phases of ALD [20, 73].
2.6 Spleen Tyrosine Kinase in Non-alcoholic Steatohepatitis (NASH)
NASH is characterized by increasing accumulation of so-called toxic lipids in hepatocytes, that
can develop into cirrhosis and primary liver cancer [74]. NASH is the more severe and clinically
significant form of NAFLD (non-alcoholic fatty liver disease) [75], characterized by hepatic cell
injury, steatosis together with inflammation, resulting into fibrosis signified by deposition of
extracellular matrix mainly composed of collagen/fibrin fibrils [76]. The progression of NASH
is associated with a progressive build-up of danger signals particularly PRRs including TLRs,
and nucleotide oligomerization domain (NOD)-like receptors (NLRs) [77] that engage multiple
receptors during immune response [78].
As also mentioned earlier, the interaction of LPS with TLR4 plays a major role in linking innate
immunity with inflammatory response and the activation of KCs [77, 79, 80]. Activated KCs
produce inflammatory cytokines and chemokines such as IL-1β, IL-6, iNOS, FcγR1, and CCL2
that contribute to the recruitment of circulating monocytes and macrophages into the
inflammed liver during NASH development mostly similar to ASH [81]. Activated KCs also
secrete TNF superfamily ligands such as TNF-α and TNF-related apoptosis-inducing ligand
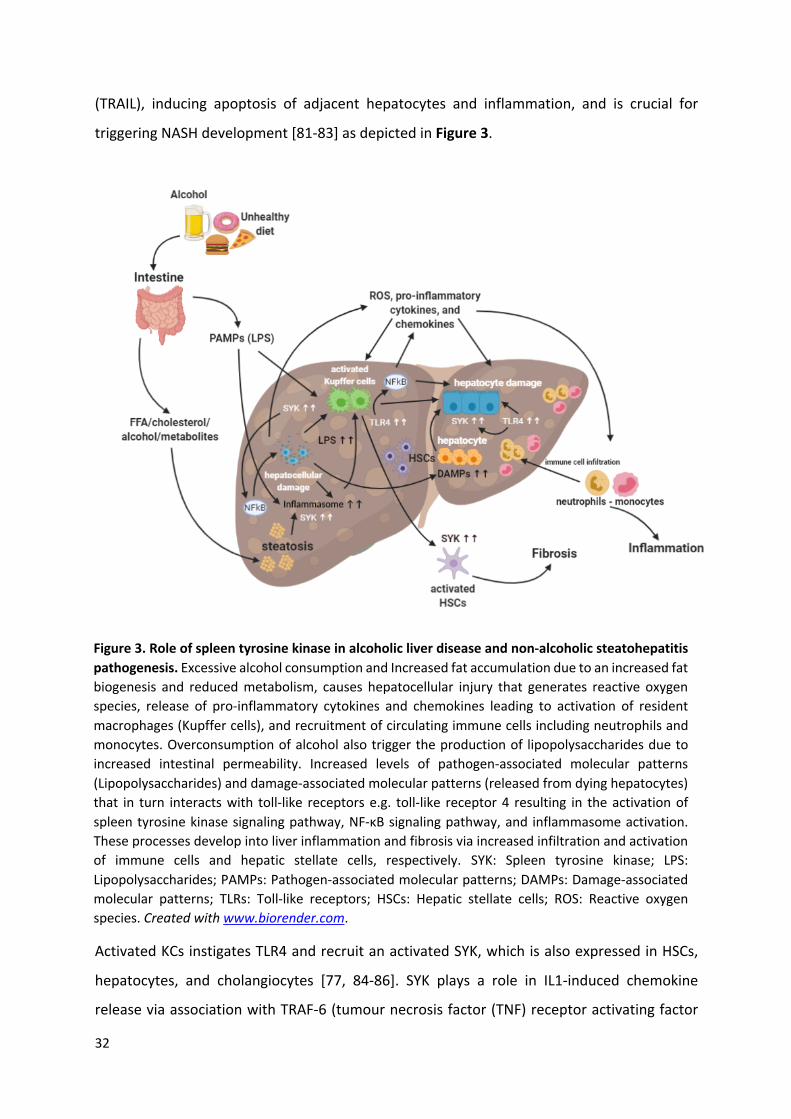
32
(TRAIL), inducing apoptosis of adjacent hepatocytes and inflammation, and is crucial for
triggering NASH development [81-83] as depicted in Figure 3.
Activated KCs instigates TLR4 and recruit an activated SYK, which is also expressed in HSCs,
hepatocytes, and cholangiocytes [77, 84-86]. SYK plays a role in IL1-induced chemokine
release via association with TRAF-6 (tumour necrosis factor (TNF) receptor activating factor
Figure 3. Role of spleen tyrosine kinase in alcoholic liver disease and non-alcoholic steatohepatitis pathogenesis. Excessive alcohol consumption and Increased fat accumulation due to an increased fat biogenesis and reduced metabolism, causes hepatocellular injury that generates reactive oxygen species, release of pro-inflammatory cytokines and chemokines leading to activation of resident macrophages (Kupffer cells), and recruitment of circulating immune cells including neutrophils and monocytes. Overconsumption of alcohol also trigger the production of lipopolysaccharides due to increased intestinal permeability. Increased levels of pathogen-associated molecular patterns (Lipopolysaccharides) and damage-associated molecular patterns (released from dying hepatocytes) that in turn interacts with toll-like receptors e.g. toll-like receptor 4 resulting in the activation of spleen tyrosine kinase signaling pathway, NF-κB signaling pathway, and inflammasome activation. These processes develop into liver inflammation and fibrosis via increased infiltration and activation of immune cells and hepatic stellate cells, respectively. SYK: Spleen tyrosine kinase; LPS: Lipopolysaccharides; PAMPs: Pathogen-associated molecular patterns; DAMPs: Damage-associated molecular patterns; TLRs: Toll-like receptors; HSCs: Hepatic stellate cells; ROS: Reactive oxygen species. Created with www.biorender.com.

33
6), which is a shared molecule in multiple signaling pathways and is recruited through
interactions of adaptor MyD88 and IRAK-1 (interleukin-1 (IL1) receptor-associated kinase 1)
with TLR4 [87-89]. Likewise, TLR4 transduces signals via the BCR (B-cell receptor) leading to
activation of SYK, which is important for B-cell survival, proliferation [90], and BCR-mediated
immune response [5]. Lipid peroxidation products, derived from phospholipid oxidation are
one of the sources of neo-antigens that are able to promote an adaptive immune response in
NASH [91]. The involvement of T and B cells in the progression of NASH automatically implicate
role of SYK in this process.
Recently, we have shown the positive correlation of SYK expression with the increasing NAS
score (NAFLD activity score) in livers from NASH patients as compared to normal livers [44].
As aforementioned, the role of SYK in NASH is not only via PRR pathways, but also through
NLR pathways. The role of several NLRs have been crucial in the formation of inflammasomes
and the nomenclature of inflammasomes is hence based on the NLR [92]. SYK is required for
NLRP3 (NLR protein 3) inflammasome activation [93], that forms an IL-1β-processing
inflammasome complex. Inflammasome activation has been shown to be associated with the
late stages of NASH, and not in early steatosis in mice [94]. Inflammasome activation can be
induced by free fatty acids (FFAs) and these FFAs can also induce apoptosis and the release of
danger signals in hepatocytes [94, 95]. Consequently, pharmacological inhibition of NLRP3
inflammasome in vivo has been demonstrated to reduce liver inflammation, hepatocyte
injury, and liver fibrosis in NASH [44, 96].
2.7 Spleen Tyrosine Kinase in Hepatocellular Carcinoma (HCC)
Hepatocyte apoptosis and compensatory proliferation are the key drivers for HCC
development, and SYK has been suggested to play a key role in HCC progression. in HCC,
intestinal microbiota and TLR4 link inflammation and carcinogenesis in the chronically injured
liver, and SYK regulate this link mediated via LPS-TLR4 interaction [97]. The intimate
correlation between SYK methylation and loss-of-expression, together with the role of SYK
methylation in gene silencing, indicates that epigenetic inactivation of SYK contributes to the
progression of HCC [98] signifying SYK methylation and loss of SYK expression as predictors of
poor overall survival in patients with HCC. Furthermore, methylation of SYK promoter was
found to be inversely regulated in HCC cells. Restoring SYK expression in SYK-silenced HCC cell

34
lines decreased hepatocellular growth, cell migration and invasion but increased cell adhesion
[99, 100].
On the other hand, checkpoint kinase 1 (CHK1) was found to be overexpressed and correlated
with poor survival of HCC patients. CHK1 phosphorylate tumor suppressor SYK isoform, SYK(L)
at Ser295 and inducing its proteasomal degradation. However, non-phosphorylated mutant
form of SYK(L) has been shown to suppress proliferation, colony formation, migration, and
tumor growth in HCC lines. Therefore, a strong inverse correlation between the expression
levels of CHK1 and SYK(L) was observed in patients with HCC [101]. Interestingly, Hong et al.
showed that another SYK isoform, SYK(S) promotes tumor growth, downregulate apoptosis,
enhances metastasis and counteract the opposing effects of SYK(L) [102]. These studies
suggest that SYK(L) downregulation or SYK(S) upregulation are the strong predictors of poor
clinical outcome in patients with HCC.
2.8 Small Molecules SYK inhibitors
Over the past decade, SYK signaling pathway has been recognized as a promising target for
the therapeutic intervention in different disease including autoimmune and inflammatory
disorders, fibrotic diseases and tumor. However, specificity and selectivity remain the major
concern for the development of drugs targeting ubiquitously expressed kinases. Hence,
debate about the specificity of SYK inhibitors has been a major point of discussion and has still
not reached an appropriate conclusion since the first SYK inhibitors entered into medicinal
chemistry optimization [25, 103, 104]. Over the past few years, several SYK inhibitors have
been designed while many are still in development, and the molecular structures of some of
these SYK inhibitors are depicted in Figure 4. Several SYK inhibitors are been evaluated in
preclinical and clinical studies in different diseases [103, 105], as highlighted in Table 1.
Table 1. Summary of pre-clinical and clinical studies using SYK inhibitors
Compound Medical Condition Description/Effect Ref. Fostamatinib (R788)
Ulcerative colitis Suppression of TNFα, T cells and neutrophils [106] Rheumatoid arthritis Reduced inflammation and tissue damage,
suppressed clinical arthritis, pannus formation and synovitis.
[107, 108]
Chronic lymphocytic leukemia and non-Hodgkin lymphoma
Disruption of BCR signaling inhibiting the proliferation and survival of malignant B cells.
[109], [110]

35
Ischemia-reperfusion induced intestinal and lung damage
Impaired release of pro-inflammatory and coagulation mediators, reduced neutrophils, macrophages and platelet accumulations
[111]
Glomerulonephritis Reduced proteinuria, glomerular macrophage and CD8 cells, MCP-1 and IL-1β, and renal injury
[112]
Entospletinib (GS-9973)
Chronic lymphocytic leukemia (CLL)
Decreased inflammation and disruption of chemokine/cytokine circuits (BCR signaling).
[113-115]
Diffuse large B-cell lymphoma (DLBCL)
Disruption of BCR signaling inhibiting the proliferation and survival of malignant B cells.
[116]
Cherubisme (craniofacial disorder)
Ameliorates inflammation and bone destruction in the mouse model of cherubism
[117]
Cerdulatinib (PRT062070)
Diffuse large B-cell lymphoma (DLBCL)
Disruption of BCR signalling inhibiting the proliferation and survival of malignant B cells
[118, 119]
TAK-659
Epstein-Barr virus-associated lymphoma
Inhibited tumour development and metastases [120]
Chronic lymphocytic leukemia (CLL)
Decreased tumour survival, myeloid cell proliferation and metastasis.
[121]
R406 (tamatinib)
Immunocomplexes mediated inflammation
Inhibits several critical modes of the inflammatory cascade
[122]
Human platelets Inhibition of activation of CLEC-2 (C-type lectin 2, platelet receptor), and platelet activation
[123]
Chronic lymphocytic leukemia (CLL)
Inhibition of constitutive and BCR-induced SYK activation, abrogation of CLL cell survival, migration, and paracrine signalling.
[124]
Leukemia Reduced tyrosine phosphorylation and c-Myc expression, blockade of tumorigenic cells proliferation transformed by oncogenes
[125]
Megakaryocytic leukemia
induced apoptosis, reduced cell proliferation and blockade of STAT5 signalling
[126]
Glomerulonephritis Downregulated MCP-1 production from mesangial cells and macrophages
[112]
Piceatannol
Oral squamous cell carcinoma (OSCC)
Inhibited tumour cell proliferation, induced of apoptosis, attenuated VEGF and MMP9 expression, and decreased metastases.
[127]
Some of the above mentioned SYK inhibitors have been explored in liver diseases and are
presented in Table 2. R406 has been shown to reduce SYK expression and phosphorylation in
macrophages, and other hepatic cells and has been shown to ameliorate non-alcoholic and
alcoholic steatohepatitis by inhibiting steatosis, inflammation and fibrosis suggesting multi-
faceted effects of this highly selective SYK inhibitor [20, 44]. GS-9973 is a new emerging,
selective and potent inhibitor of SYK that was evaluated in activated HSCs and showed anti-
fibrotic effects in rodent liver fibrosis models [28].

36
Very recently, two new inhibitors PRT062607 and Piceatannol have been investigated in
myeloid cells to reveal their protective effect against liver fibrosis and hepatocarcinogenesis
in vivo. Both inhibitors selectively blocked SYK phosphorylation, significantly reduced the
infiltration of inflammatory cells and HSCs trans-differentiation, and inhibited malignant
transformation in fibrotic livers [128].
Despite the encouraging results with SYK inhibitors, some issues remain unsolved (e.g. their
long-term safety has not yet been demonstrated). Moreover, due to the ubiquitous expression
of SYK in different cells, concerns have been raised about the possibility of side-effects owing
to the overall inhibition of the multiple cellular functions [2, 127]. A major challenge therefore
is how to inhibit pathological processes without disrupting physiological cell functions [129].
Nanotechnology is an interesting and promising alternative to improve the efficacy and
therapeutic effect of the SYK inhibitor e.g. using poly lactic-co-glycolic acid (PLGA)
nanoparticles, we have demonstrated improved therapeutic effectivity of R406 in MCD-diet
induced NASH [44]. In this study, we have shown that R406, encapsulated in PLGA
Fostamatinib (R788)
Cerdulatinib (PRT062070) TAK-659
Tamatinib (R406)
Entospletinib (GS-9973)
Piceatannol
Figure 4. Molecular structure of several SYK inhibitors. R406, GS-9973, PRT062070, and Piceatannol have been studied in liver diseases, while R788 and TAK-659 are being investigated in other diseases.

37
nanoparticles, reduced expression of SYK in macrophages in vitro, and attenuated steatosis,
inflammation, and fibrosis in vivo in NASH mouse model [44].
Table 2. SYK inhibitors implicated in liver diseases
2.9 Conclusion
In this review, we have highlighted the implication of SYK signaling pathways in different
diseases, more importantly in liver diseases. SYK plays a multifaceted role in liver diseases such
as liver fibrosis, alcoholic liver disease, non-alcoholic steatohepatitis, viral hepatitis, and
hepatocellular carcinoma. Furthermore, several SYK-related mechanisms have been
understood in the past decade which led to the development of numerous small-molecule
inhibitors that have been and are currently evaluated in vitro, in vivo in different animal
models and in clinical trials in patients for different indications. These inhibitors have shown
highly potent effects in the tested models and therefore is a promising therapeutic target that
should be explored further. To improve the therapeutic efficacy and clinical use of SYK
inhibitors with improved safety profile and reduce the side effects, nanotechnology
approaches, such as polymeric nanoparticles, liposomal-mediated delivery, or micelles, and
finally organ (tumor)-targeted drug delivery could be explored.
2.10 Acknowledgment
Inhibitor Mechanism of action Therapeutic effect Ref.
R406 Blocking of Fc receptor signalling pathway, NF-κB signalling pathway and inflammasome activation.
Reduced SYK expression and phosphorylation resulting in attenuated liver steatosis, inflammation and fibrosis in ASH and NASH murine models.
[20, 44]
GS-9973 Decreased expression of HSCs activation (CBP, MYB, MYC) and HSCs proliferation factors (MYC and CCND1).
Inhibition of HSCs proliferation and HSC activation resulting in amelioration of fibrosis and hepatocarcinogenesis.
[28]
PRT062607 and Piceatannol
Increased intra-tumoral p16, p53 and decreased expression of Bcl-xL and SMAD4. Decreased expression of genes regulating angiogenesis, apoptosis, cell cycle regulation and cellular senescence. Down-regulation of mTOR, IL-8 signalling and oxidative phosphorylation.
Reduced HSCs differentiation and infiltration of inflammatory cells including T cells, B cells and myeloid cells, reduced oncogenic progression. Marked attenuation of toxin-induced liver fibrosis, associated hepatocellular injury, intra-hepatic inflammation and hepatocarcinogenesis.
[128]

38
D.W.K has been supported by a PhD scholarship received from the Endowment Fund for the
Education Republic of Indonesia (Lembaga Pengelola Dana Pendidikan/LPDP, RI).
2.11 References
[1] C.A. Lowell, Src-family and Syk kinases in activating and inhibitory pathways in innate immune cells: signaling cross talk, Cold Spring Harb Perspect Biol, 3 (2011) 1-16.
[2] M. Riccaboni, I. Bianchi, P. Petrillo, Spleen tyrosine kinases: biology, therapeutic targets and drugs, Drug Discov Today, 15 (2010) 517-530.
[3] O.N. Pamuk, G.C. Tsokos, Spleen tyrosine kinase inhibition in the treatment of autoimmune, allergic, and autoinflammatory diseases, Arthritis Research & Therapy, 12 (2010) 1-11.
[4] A. Mocsai, J. Ruland, V.L. Tybulewicz, The SYK tyrosine kinase: a crucial player in diverse biological functions, Nat Rev Immunol, 10 (2010) 387-402.
[5] E. Schweighoffer, J. Nys, L. Vanes, N. Smithers, V.L.J. Tybulewicz, TLR4 signals in B lymphocytes are transduced via the B cell antigen receptor and SYK, J Exp Med, 214 (2017) 1269-1280.
[6] M. Turner, E. Schweighoffer, F. Colucci, J.P.D. Santo, V.L. Tybulewicz, Tyrosine kinase SYK: essential functions for immunoreceptor signalling, Immunologi Today, 21 (2000) 148-154.
[7] J.M. Bradshaw, The Src, Syk, and Tec family kinases: distinct types of molecular switches, Cell Signal, 22 (2010) 1175-1184.
[8] R.J. Cornall, A.M. Cheng, T. Pawson, C.C. Goodnow, Role of Syk in B-cell development and antigen-receptor signaling, PNAS, 97 (2000) 1713-1718.
[9] B. Keller, I. Stumpf, V. Strohmeier, S. Usadel, E. Verhoeyen, H. Eibel, K. Warnatz, High SYK Expression Drives Constitutive Activation of CD21(low) B Cells, J Immunol, 198 (2017) 4285-4292.
[10] A.R. Burton, L.J. Pallett, L.E. McCoy, K. Suveizdyte, O.E. Amin, L. Swadling, E. Alberts, B.R. Davidson, P.T. Kennedy, U.S. Gill, C. Mauri, P.A. Blair, N. Pelletier, M.K. Maini, Circulating and intrahepatic antiviral B cells are defective in hepatitis B, J Clin Invest, 128 (2018) 4588-4603.
[11] D. Frommhold, I. Mannigel, J. Schymeinsky, A. Mocsai, J. Poeschl, B. Walzog, M. Sperandio, Spleen tyrosine kinase Syk is critical for sustained leukocyte adhesion during inflammation in vivo, BMC Immunol, 8 (2007) 31.
[12] A. Mocsai, M. Zhou, F. Meng, V.L. Tybulewicz, C.A. Lowell, Syk is required for integrin signaling in neutrophils, Immunity, 16 (2002) 547-558.
[13] O.N. Pamuk, P.H. Lapchak, P. Rani, P. Pine, J.J. Dalle Lucca, G.C. Tsokos, Spleen tyrosine kinase inhibition prevents tissue damage after ischemia-reperfusion, Am J Physiol Gastrointest Liver Physiol, 299 (2010) G391-399.
[14] R.L. Geahlen, Getting Syk: spleen tyrosine kinase as a therapeutic target, Trends Pharmacol Sci, 35 (2014) 414-422.
[15] S.P. McAdoo, J. Reynolds, G. Bhangal, J. Smith, J.P. McDaid, A. Tanna, W.D. Jackson, E.S. Masuda, H.T. Cook, C.D. Pusey, F.W. Tam, Spleen tyrosine kinase inhibition attenuates autoantibody production and reverses experimental autoimmune GN, J Am Soc Nephrol, 25 (2014) 2291-2302.
[16] H. Patterson, R. Nibbs, I. McInnes, S. Siebert, Protein kinase inhibitors in the treatment of inflammatory and autoimmune diseases, Clin Exp Immunol, 176 (2014) 1-10.

39
[17] M. Kaur, M. Singh, O. Silakari, Inhibitors of switch kinase 'spleen tyrosine kinase' in inflammation and immune-mediated disorders: a review, Eur J Med Chem, 67 (2013) 434-446.
[18] T.K. Ma, S.P. McAdoo, F.W. Tam, Spleen Tyrosine Kinase: A Crucial Player and Potential Therapeutic Target in Renal Disease, Nephron, 133 (2016) 261-269.
[19] K.H. Chen, H.H. Hsu, H.Y. Yang, Y.C. Tian, Y.C. Ko, C.W. Yang, C.C. Hung, Inhibition of spleen tyrosine kinase (syk) suppresses renal fibrosis through anti-inflammatory effects and down regulation of the MAPK-p38 pathway, Int J Biochem Cell Biol, 74 (2016) 135-144.
[20] T.N. Bukong, A. Iracheta-Vellve, B. Saha, A. Ambade, A. Satishchandran, B. Gyongyosi, P. Lowe, D. Catalano, K. Kodys, G. Szabo, Inhibition of spleen tyrosine kinase activation ameliorates inflammation, cell death, and steatosis in alcoholic liver disease, Hepatology, 64 (2016) 1057-1071.
[21] S. McAdoo, F.W.K. Tam, Role of the Spleen Tyrosine Kinase Pathway in Driving Inflammation in IgA Nephropathy, Semin Nephrol, 38 (2018) 496-503.
[22] P.S. Michael, W.L. Christine, S. Andreas, C. Chung-Wai, Syk: A Novel Target for Treatment of Inflammation in Lung Disease, Inflammation & Allergy - Drug Targets (Discontinued), 8 (2009) 87-95.
[23] G. Coffey, F. DeGuzman, M. Inagaki, Y. Pak, S.M. Delaney, D. Ives, A. Betz, Z.J. Jia, A. Pandey, D. Baker, S.J. Hollenbach, D.R. Phillips, U. Sinha, Specific Inhibition of Spleen Tyrosine Kinase Suppresses Leukocyte Immune Function and Inflammation in Animal Models of Rheumatoid Arthritis, Journal of Pharmacology and Experimental Therapeutics, 340 (2012) 350-359.
[24] Y. Koyama, D.A. Brenner, Liver inflammation and fibrosis, The Journal of clinical investigation, 127 (2017) 55-64.
[25] R. Singh, E.S. Masuda, D.G. Payan, Discovery and development of spleen tyrosine kinase (SYK) inhibitors, J Med Chem, 55 (2012) 3614-3643.
[26] S. Yanagi, R. Inatome, T. Takano, H. Yamamura, Syk expression and novel function in a wide variety of tissues, Biochem Biophys Res Commun, 288 (2001) 495-498.
[27] S. Tsuchida, S. Yanagi, R. Inatome, J. Ding, P. Hermann, T. Tsujimura, N. Matsui, H. Yamamura, Purification of a 72-kDa protein-tyrosine kinase from rat liver and its identification as Syk: Involvement of Syk in signaling events of hepatocytes, J. Biochem, 127 (2000) 321-327.
[28] C. Qu, D. Zheng, S. Li, Y. Liu, A. Lidofsky, J.A. Holmes, J. Chen, L. He, L. Wei, Y. Liao, H. Yuan, Q. Jin, Z. Lin, Q. Hu, Y. Jiang, M. Tu, X. Chen, W. Li, W. Lin, B.C. Fuchs, R.T. Chung, J. Hong, Tyrosine kinase SYK is a potential therapeutic target for liver fibrosis, Hepatology, (2018).
[29] L.M. Keshvara, C. Isaacson, M.L. Harrison, R.L. Geahlen, Syk activation and dissociation from the B-cell antigen receptor is mediated by phosphorylation of tyrosine 130, The Journal of Biological Chemistry, 272 (1997) 10377-10381.
[30] Y. Zhang, H. Oh, R.A. Burton, J.W. Burgner, R.L. Geahlen, C.B. Post, Tyr130 phosphorylation triggers Syk release from antigen receptor by long-distance conformational uncoupling, PNAS 105 (2008) 11760-11765.
[31] R.A. Grucza, K. Fu¨tterer, A.C. Chan, G. Waksman, Thermodynamic study of the binding of the tandem-SH2 domain of the Syk kinase to a dually phosphorylated ITAM peptide: evidence for two conformers, Biochemistry, 38 (1999) 5024-5033.
[32] V.r. Rolli, M. Gallwitz, T. Wossning, Alexandra Flemming, W.W.A. Schamel, C.Z. rn, M. Reth, Amplification of B cell antigen receptor signaling by a Syk/ITAM positive feedback loop, Molecular Cell, 10 (2002) 1057-1069.

40
[33] Y. Kulathu, E. Hobeika, G. Turchinovich, M. Reth, The kinase Syk as an adaptor controlling sustained calcium signalling and B-cell development, EMBO J, 27 (2008) 1333-1344.
[34] E. Tsang, A.M. Giannetti, D. Shaw, M. Dinh, J.K. Tse, S. Gandhi, H. Ho, S. Wang, E. Papp, J.M. Bradshaw, Molecular mechanism of the Syk activation switch, J Biol Chem, 283 (2008) 32650-32659.
[35] B.L. Slomiany, A. Slomiany, Helicobacter pylori LPS-induced gastric mucosal spleen tyrosine kinase (Syk) recruitment to TLR4 and activation occurs with the involvement of protein kinase Cdelta, Inflammopharmacology, 26 (2018) 805-815.
[36] B.L. Slomiany, A. Slomiany, Syk: a new target for attenuation of Helicobacter pylori-induced gastric mucosal inflammatory responses, Inflammopharmacology, 27 (2019) 203-211.
[37] R. Bataller, D.A. Brenner, Liver fibrosis, Journal of Clinical Investigation, 115 (2005) 209-218.
[38] M. Parola, M. Pinzani, Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues, Mol Aspects Med, 65 (2019) 37-55.
[39] S.K. Asrani, H. Devarbhavi, J. Eaton, P.S. Kamath, Burden of liver diseases in the world, J Hepatol, 70 (2019) 151-171.
[40] D. Schuppan, M. Ashfaq-Khan, A.T. Yang, Y.O. Kim, Liver fibrosis: Direct antifibrotic agents and targeted therapies, Matrix Biol, 68-69 (2018) 435-451.
[41] R.L. Gieseck Iii, M.S. Wilson, T.A. Wynn, Type 2 immunity in tissue repair and fibrosis, Nature Reviews Immunology, 18 (2017) 62.
[42] T. Tsuchida, S.L. Friedman, Mechanisms of hepatic stellate cell activation, Nat Rev Gastroenterol Hepatol, 14 (2017) 397-411.
[43] K. Nishikawa, Y. Osawa, K. Kimura, Wnt/beta-Catenin Signaling as a Potential Target for the Treatment of Liver Cirrhosis Using Antifibrotic Drugs, Int J Mol Sci, 19 (2018).
[44] D.W. Kurniawan, A.K. Jajoriya, G. Dhawan, D. Mishra, J. Argemi, R. Bataller, G. Storm, D.P. Mishra, J. Prakash, R. Bansal, Therapeutic inhibition of spleen tyrosine kinase in inflammatory macrophages using PLGA nanoparticles for the treatment of non-alcoholic steatohepatitis, J Control Release, 288 (2018) 227-238.
[45] J.H. Cheng, H. She, Y.P. Han, J. Wang, S. Xiong, K. Asahina, H. Tsukamoto, Wnt antagonism inhibits hepatic stellate cell activation and liver fibrosis, Am J Physiol Gastrointest Liver Physiol, 294 (2008) G39-49.
[46] B.O. Akcora, G. Storm, R. Bansal, Inhibition of canonical WNT signaling pathway by beta-catenin/CBP inhibitor ICG-001 ameliorates liver fibrosis in vivo through suppression of stromal CXCL12, Biochim Biophys Acta Mol Basis Dis, 1864 (2018) 804-818.
[47] X. Wang, X. Tang, X. Gong, E. Albanis, S.L. Friedman, Z. Mao, Regulation of hepatic stellate cell activation and growth by transcription factor myocyte enhancer factor 2, Gastroenterology, 127 (2004) 1174-1188.
[48] J. Mann, D.A. Mann, Transcriptional regulation of hepatic stellate cells, Adv Drug Deliv Rev, 61 (2009) 497-512.
[49] Y. Tokunaga, Y. Osawa, T. Ohtsuki, Y. Hayashi, K. Yamaji, D. Yamane, M. Hara, K. Munekata, K. Tsukiyama-Kohara, T. Hishima, S. Kojima, K. Kimura, M. Kohara, Selective inhibitor of Wnt/beta-catenin/CBP signaling ameliorates hepatitis C virus-induced liver fibrosis in mouse model, Sci Rep, 7 (2017) 325.
[50] B. Aouar, D. Kovarova, S. Letard, A. Font-Haro, J. Florentin, J. Weber, D. Durantel, L. Chaperot, J. Plumas, K. Trejbalova, J. Hejnar, J.A. Nunes, D. Olive, P. Dubreuil, I. Hirsch, R.

41
Stranska, Dual Role of the Tyrosine Kinase Syk in Regulation of Toll-Like Receptor Signaling in Plasmacytoid Dendritic Cells, PLoS One, 11 (2016) e0156063.
[51] A.R. Zekri, M.M. Hafez, A.A. Bahnassy, Z.K. Hassan, T. Mansour, M.M. Kamal, H.M. Khaled, Genetic profile of Egyptian hepatocellular-carcinoma associated with hepatitis C virus Genotype 4 by 15 K cDNA microarray: preliminary study, BMC Res Notes, 1 (2008) 106.
[52] P. Pileri, Y. Uematsu, S. Campagnoli, G. Galli, F. Falugi, R. Petracca, A.J. Weiner, M. Houghton, D. Rosa, G. Grandi, S. Abrignani, Binding of hepatitis C virus to CD81, Science, 282 (1998) 938-941.
[53] M. Brazzoli, A. Bianchi, S. Filippini, A. Weiner, Q. Zhu, M. Pizza, S. Crotta, CD81 is a central regulator of cellular events required for hepatitis C virus infection of human hepatocytes, J Virol, 82 (2008) 8316-8329.
[54] J. Zhang, G. Randall, A. Higginbottom, P. Monk, C.M. Rice, J.A. McKeating, CD81 is required for hepatitis C virus glycoprotein-mediated viral infection, J Virol, 78 (2004) 1448-1455.
[55] T.N. Bukong, K. Kodys, G. Szabo, Human ezrin-moesin-radixin proteins modulate hepatitis C virus infection, Hepatology, 58 (2013) 1569-1579.
[56] G.P. Coffey, R. Rajapaksa, R. Liu, O. Sharpe, C.C. Kuo, S.W. Krauss, Y. Sagi, R.E. Davis, L.M. Staudt, J.P. Sharman, W.H. Robinson, S. Levy, Engagement of CD81 induces ezrin tyrosine phosphorylation and its cellular redistribution with filamentous actin, J Cell Sci, 122 (2009) 3137-3144.
[57] T.N. Bukong, K. Kodys, G. Szabo, A Novel Human Radixin Peptide Inhibits Hepatitis C Virus Infection at the Level of Cell Entry, Int J Pept Res Ther, 20 (2014) 269-276.
[58] S. Inubushi, M. Nagano-Fujii, K. Kitayama, M. Tanaka, C. An, H. Yokozaki, H. Yamamura, H. Nuriya, M. Kohara, K. Sada, H. Hotta, Hepatitis C virus NS5A protein interacts with and negatively regulates the non-receptor protein tyrosine kinase Syk, J Gen Virol, 89 (2008) 1231-1242.
[59] W. Dunn, V.H. Shah, Pathogenesis of Alcoholic Liver Disease, Clin Liver Dis, 20 (2016) 445-456.
[60] B. Gao, R. Bataller, Alcoholic liver disease: pathogenesis and new therapeutic targets, Gastroenterology, 141 (2011) 1572-1585.
[61] G. Szabo, P. Mandrekar, A recent perspective on alcohol, immunity, and host defense, Alcohol Clin Exp Res, 33 (2009) 220-232.
[62] F. Stickel, C. Datz, J. Hampe, R. Bataller, Pathophysiology and Management of Alcoholic Liver Disease: Update 2016, Gut Liver, 11 (2017) 173-188.
[63] J. Petrasek, A. Iracheta-Vellve, T. Csak, A. Satishchandran, K. Kodys, E.A. Kurt-Jones, K.A. Fitzgerald, G. Szabo, STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease, Proc Natl Acad Sci U S A, 110 (2013) 16544-16549.
[64] C.S. Lieber, Alcoholic fatty liver: its pathogenesis and mechanism of progression to inflammation and fibrosis, Alcohol, 34 (2004) 9-19.
[65] J. Guo, S.L. Friedman, Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis, Fibrogenesis & Tissue Repair, 3 (2010) 1-19.
[66] G. Szabo, Gut-liver axis in alcoholic liver disease, Gastroenterology, 148 (2015) 30-36. [67] Y.S. Roh, E. Seki, Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis
and carcinogenesis, J Gastroenterol Hepatol, 28 Suppl 1 (2013) 38-42. [68] J. Petrasek, P. Mandrekar, G. Szabo, Toll-like receptors in the pathogenesis of alcoholic
liver disease, Gastroenterol Res Pract, 2010 (2010).

42
[69] Y.I. Miller, S.H. Choi, P. Wiesner, Y.S. Bae, The SYK side of TLR4: signalling mechanisms in response to LPS and minimally oxidized LDL, Br J Pharmacol, 167 (2012) 990-999.
[70] H. Zhou, M. Yu, J. Zhao, B.N. Martin, S. Roychowdhury, M.R. McMullen, E. Wang, P.L. Fox, S. Yamasaki, L.E. Nagy, X. Li, IRAKM-Mincle axis links cell death to inflammation: Pathophysiological implications for chronic alcoholic liver disease, Hepatology, 64 (2016) 1978-1993.
[71] N. Afifiyan, B. Tillman, B.A. French, M. Masouminia, S. Samadzadeh, S.W. French, Over expression of proteins that alter the intracellular signaling pathways in the cytoplasm of the liver cells forming Mallory-Denk bodies, Exp Mol Pathol, 102 (2017) 106-114.
[72] N. Afifiyan, B. Tillman, B.A. French, O. Sweeny, M. Masouminia, S. Samadzadeh, S.W. French, The role of Tec kinase signaling pathways in the development of Mallory Denk Bodies in balloon cells in alcoholic hepatitis, Exp Mol Pathol, 103 (2017) 191-199.
[73] T.N. Bukong, A. Iracheta-Vellve, B. Gyongyosi, A. Ambade, D. Catalano, K. Kodys, G. Szabo, Therapeutic Benefits of Spleen Tyrosine Kinase Inhibitor Administration on Binge Drinking-Induced Alcoholic Liver Injury, Steatosis, and Inflammation in Mice, Alcoholism: Clinical and Experimental Research, 40 (2016) 1524-1530.
[74] M.V. Machado, A.M. Diehl, Pathogenesis of Nonalcoholic Steatohepatitis, Gastroenterology, 150 (2016) 1769-1777.
[75] J.K. Dowman, J.W. Tomlinson, P.N. Newsome, Pathogenesis of non-alcoholic fatty liver disease, QJM, 103 (2010) 71-83.
[76] Y.L. Fang, H. Chen, C.L. Wang, L. Liang, Pathogenesis of non-alcoholic fatty liver disease in children and adolescence: From "two hit theory" to "multiple hit model", World J Gastroenterol, 24 (2018) 2974-2983.
[77] V. Bieghs, C. Trautwein, Innate immune signaling and gut-liver interactions in non-alcoholic fatty liver disease, Hepatobiliary Surg Nutr, 3 (2014) 377-385.
[78] M. Ganz, T.N. Bukong, T. Csak, B. Saha, J.K. Park, A. Ambade, K. Kodys, G. Szabo, Progression of non-alcoholic steatosis to steatohepatitis and fibrosis parallels cumulative accumulation of danger signals that promote inflammation and liver tumors in a high fat-cholesterol-sugar diet model in mice, J Transl Med, 13 (2015) 193.
[79] S. Kiziltas, Toll-like receptors in pathophysiology of liver diseases, World J Hepatol, 8 (2016) 1354-1369.
[80] B. Nath, I. Levin, T. Csak, J. Petrasek, C. Mueller, K. Kodys, D. Catalano, P. Mandrekar, G. Szabo, Hepatocyte-specific hypoxia-inducible factor-1alpha is a determinant of lipid accumulation and liver injury in alcohol-induced steatosis in mice, Hepatology, 53 (2011) 1526-1537.
[81] A. Alisi, G. Carpino, F.L. Oliveira, N. Panera, V. Nobili, E. Gaudio, The Role of Tissue Macrophage-Mediated Inflammation on NAFLD Pathogenesis and Its Clinical Implications, Mediators Inflamm, 2017 (2017) 8162421.
[82] C.P. Day, From fat to inflammation, Gastroenterology, 130 (2006) 207-210. [83] G. Carpino, V. Nobili, A. Renzi, C. De Stefanis, L. Stronati, A. Franchitto, A. Alisi, P. Onori,
R. De Vito, G. Alpini, E. Gaudio, Macrophage Activation in Pediatric Nonalcoholic Fatty Liver Disease (NAFLD) Correlates with Hepatic Progenitor Cell Response via Wnt3a Pathway, PLoS One, 11 (2016) e0157246.
[84] A. Caligiuri, A. Gentilini, F. Marra, Molecular Pathogenesis of NASH, Int J Mol Sci, 17 (2016).

43
[85] J. Yu, S. Marsh, J. Hu, W. Feng, C. Wu, The Pathogenesis of Nonalcoholic Fatty Liver Disease: Interplay between Diet, Gut Microbiota, and Genetic Background, Gastroenterol Res Pract, 2016 (2016) 2862173.
[86] Y. Mao, F. Yu, J. Wang, C. Guo, X. Fan, Autophagy: a new target for nonalcoholic fatty liver disease therapy, Hepat Med, 8 (2016) 27-37.
[87] S. Chattopadhyay, G.C. Sen, Tyrosine phosphorylation in Toll-like receptor signaling, Cytokine Growth Factor Rev, 25 (2014) 533-541.
[88] E. Seki, D.A. Brenner, Toll-like receptors and adaptor molecules in liver disease: update, Hepatology, 48 (2008) 322-335.
[89] A. Chaudhary, T.M. Fresquez, M.J. Naranjo, Tyrosine kinase Syk associates with toll-like receptor 4 and regulates signaling in human monocytic cells, Immunol Cell Biol, 85 (2007) 249-256.
[90] R. Flynn, J.L. Allen, L. Luznik, K.P. MacDonald, K. Paz, K.A. Alexander, A. Vulic, J. Du, A. Panoskaltsis-Mortari, P.A. Taylor, J.C. Poe, J.S. Serody, W.J. Murphy, G.R. Hill, I. Maillard, J. Koreth, C.S. Cutler, R.J. Soiffer, J.H. Antin, J. Ritz, N.J. Chao, R.A. Clynes, S. Sarantopoulos, B.R. Blazar, Targeting Syk-activated B cells in murine and human chronic graft-versus-host disease, Blood, 125 (2015) 4085-4094.
[91] N. Magee, A. Zou, Y. Zhang, Pathogenesis of Nonalcoholic Steatohepatitis: Interactions between Liver Parenchymal and Nonparenchymal Cells, Biomed Res Int, 2016 (2016) 5170402.
[92] G. Szabo, T. Csak, Inflammasomes in liver diseases, J Hepatol, 57 (2012) 642-654. [93] A.F. Malik, R. Hoque, X. Ouyang, A. Ghani, E. Hong, K. Khan, L.B. Moore, G. Ng, F. Munro,
R.A. Flavell, Y. Shi, T.R. Kyriakides, W.Z. Mehal, Inflammasome components Asc and caspase-1 mediate biomaterial-induced inflammation and foreign body response, Proc Natl Acad Sci U S A, 108 (2011) 20095-20100.
[94] T. Csak, M. Ganz, J. Pespisa, K. Kodys, A. Dolganiuc, G. Szabo, Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells, Hepatology, 54 (2011) 133-144.
[95] W.Z. Mehal, The inflammasome in liver injury and non-alcoholic fatty liver disease, Dig Dis, 32 (2014) 507-515.
[96] A.R. Mridha, A. Wree, A.A.B. Robertson, M.M. Yeh, C.D. Johnson, D.M. Van Rooyen, F. Haczeyni, N.C. Teoh, C. Savard, G.N. Ioannou, S.L. Masters, K. Schroder, M.A. Cooper, A.E. Feldstein, G.C. Farrell, NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice, J Hepatol, 66 (2017) 1037-1046.
[97] I.J. Song, Y.M. Yang, S. Inokuchi-Shimizu, Y.S. Roh, L. Yang, E. Seki, The contribution of toll-like receptor signaling to the development of liver fibrosis and cancer in hepatocyte-specific TAK1-deleted mice, Int J Cancer, 142 (2018) 81-91.
[98] Y. Yuan, J. Wang, J. Li, L. Wang, M. Li, Z. Yang, C. Zhang, J.L. Dai, Frequent epigenetic inactivation of spleen tyrosine kinase gene in human hepatocellular carcinoma, Clin Cancer Res, 12 (2006) 6687-6695.
[99] S.H. Shin, K.H. Lee, B.H. Kim, S. Lee, H.S. Lee, J.J. Jang, G.H. Kang, Downregulation of spleen tyrosine kinase in hepatocellular carcinoma by promoter CpG island hypermethylation and its potential role in carcinogenesis, Lab Invest, 94 (2014) 1396-1405.
[100] C. Carone, A. Olivani, R. Dalla Valle, R. Manuguerra, E.M. Silini, T. Trenti, G. Missale, E. Cariani, Immune Gene Expression Profile in Hepatocellular Carcinoma and Surrounding Tissue Predicts Time to Tumor Recurrence, Liver Cancer, 7 (2018) 277-294.

44
[101] J. Hong, K. Hu, Y. Yuan, Y. Sang, Q. Bu, G. Chen, L. Yang, B. Li, P. Huang, D. Chen, Y. Liang, R. Zhang, J. Pan, Y.X. Zeng, T. Kang, CHK1 targets spleen tyrosine kinase (L) for proteolysis in hepatocellular carcinoma, J Clin Invest, 122 (2012) 2165-2175.
[102] J. Hong, Y. Yuan, J. Wang, Y. Liao, R. Zou, C. Zhu, B. Li, Y. Liang, P. Huang, Z. Wang, W. Lin, Y. Zeng, J.L. Dai, R.T. Chung, Expression of variant isoforms of the tyrosine kinase SYK determines the prognosis of hepatocellular carcinoma, Cancer Res, 74 (2014) 1845-1856.
[103] D. Liu, A. Mamorska-Dyga, Syk inhibitors in clinical development for hematological malignancies, J Hematol Oncol, 10 (2017) 145.
[104] G. Thoma, S. Veenstra, R. Strang, J. Blanz, E. Vangrevelinghe, J. Berghausen, C.C. Lee, H.G. Zerwes, Orally bioavailable Syk inhibitors with activity in a rat PK/PD model, Bioorg Med Chem Lett, 25 (2015) 4642-4647.
[105] M.C. Lucas, N. Bhagirath, E. Chiao, D.M. Goldstein, J.C. Hermann, P.Y. Hsu, S. Kirchner, J.J. Kennedy-Smith, A. Kuglstatter, C. Lukacs, J. Menke, L. Niu, F. Padilla, Y. Peng, L. Polonchuk, A. Railkar, M. Slade, M. Soth, D. Xu, P. Yadava, C. Yee, M. Zhou, C. Liao, Using ovality to predict nonmutagenic, orally efficacious pyridazine amides as cell specific spleen tyrosine kinase inhibitors, J Med Chem, 57 (2014) 2683-2691.
[106] G. Can, S. Ayvaz, H. Can, S. Demirtas, H. Aksit, B. Yilmaz, U. Korkmaz, M. Kurt, T. Karaca, The Syk Inhibitor Fostamatinib Decreases the Severity of Colonic Mucosal Damage in a Rodent Model of Colitis, J Crohns Colitis, 9 (2015) 907-917.
[107] M.E. Weinblatt, M.C. Genovese, M. Ho, S. Hollis, K. Rosiak-Jedrychowicz, A. Kavanaugh, D.S. Millson, G. Leon, M. Wang, D.v.d. Heijde, Effects of fostamatinib, an oral spleen tyrosine kinase inhibitor, in rheumatoid arthritis patients with an inadequate response to methotrexate, Arthritis & Rheumatology, 66 (2014) 3255-3264.
[108] J.A. Gomez-Puerta, A. Mocsai, Tyrosine kinase inhibitors for the treatment of rheumatoid arthritis, Current Topics in Medicinal Chemistry, 13 (2013) 760-773.
[109] M. Suljagic, P.G. Longo, S. Bennardo, E. Perlas, G. Leone, L. Laurenti, D.G. Efremov, The Syk inhibitor fostamatinib disodium (R788) inhibits tumor growth in the Emu- TCL1 transgenic mouse model of CLL by blocking antigen-dependent B-cell receptor signaling, Blood, 116 (2010) 4894-4905.
[110] J.W. Friedberg, J. Sharman, J. Sweetenham, P.B. Johnston, J.M. Vose, A. Lacasce, J. Schaefer-Cutillo, S. De Vos, R. Sinha, J.P. Leonard, L.D. Cripe, S.A. Gregory, M.P. Sterba, A.M. Lowe, R. Levy, M.A. Shipp, Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia, Blood, 115 (2010) 2578-2585.
[111] P.H. Lapchak, L. Kannan, P. Rani, O.N. Pamuk, A. Ioannou, J.J. Dalle Lucca, P. Pine, G.C. Tsokos, Inhibition of Syk activity by R788 in platelets prevents remote lung tissue damage after mesenteric ischemia-reperfusion injury, Am J Physiol Gastrointest Liver Physiol, 302 (2012) G1416-1422.
[112] J. Smith, J.P. McDaid, G. Bhangal, R. Chawanasuntorapoj, E.S. Masuda, H.T. Cook, C.D. Pusey, F.W. Tam, A spleen tyrosine kinase inhibitor reduces the severity of established glomerulonephritis, J Am Soc Nephrol, 21 (2010) 231-236.
[113] R.T. Burke, S. Meadows, M.M. Loriaux, K.S. Currie, S.A. Mitchell, P. Maciejewski, A.S. Clarke, J.A. Dipaolo, B.J. Druker, B.J. Lannutti, S.E. Spurgeon, A potential therapeutic strategy for chronic lymphocytic leukemia by combining idelalisib and GS-9973, a novel spleen tyrosine kinase (Syk) inhibitor, Oncotarget, 5 (2013) 908-915.

45
[114] J. Sharman, M. Hawkins, K. Kolibaba, M. Boxer, L. Klein, M. Wu, J. Hu, S. Abella, C. Yasenchak, An open-label phase 2 trial of entospletinib (GS-9973), a selective spleen tyrosine kinase inhibitor, in chronic lymphocytic leukemia, Blood, 125 (2015) 2336-2343.
[115] J. Sharman, J. Di Paolo, Targeting B-cell receptor signaling kinases in chronic lymphocytic leukemia: the promise of entospletinib, Ther Adv Hematol, 7 (2016) 157-170.
[116] J.M. Burke, A. Shustov, J. Essell, D. Patel-Donnelly, J. Yang, R. Chen, W. Ye, W. Shi, S. Assouline, J. Sharman, An Open-label, Phase II Trial of Entospletinib (GS-9973), a Selective Spleen Tyrosine Kinase Inhibitor, in Diffuse Large B-cell Lymphoma, Clin Lymphoma Myeloma Leuk, 18 (2018) e327-e331.
[117] T. Yoshimoto, T. Hayashi, T. Kondo, M. Kittaka, E.J. Reichenberger, Y. Ueki, Second-Generation SYK Inhibitor Entospletinib Ameliorates Fully Established Inflammation and Bone Destruction in the Cherubism Mouse Model, J Bone Miner Res, 33 (2018) 1513-1519.
[118] G. Coffey, A. Betz, F. DeGuzman, Y. Pak, M. Inagaki, D.C. Baker, S.J. Hollenbach, A. Pandey, U. Sinha, The novel kinase inhibitor PRT062070 (Cerdulatinib) demonstrates efficacy in models of autoimmunity and B-cell cancer, J Pharmacol Exp Ther, 351 (2014) 538-548.
[119] J. Ma, W. Xing, G. Coffey, K. Dresser, K. Lu, A. Guo, G. Raca, A. Pandey, P. Conley, H. Yu, Y.L. Wang, Cerdulatinib, a novel dual SYK-JAK kinase inhibitor, has broad anti-tumor activity in both ABC and GCB types of diffuse large B cell lymphoma, Oncotarget, 6 (2015) 43881-43896.
[120] O. Cen, K. Kannan, J. Huck Sappal, J. Yu, M. Zhang, M. Arikan, A. Ucur, D. Ustek, Y. Cen, L. Gordon, R. Longnecker, Spleen Tyrosine Kinase Inhibitor TAK-659 Prevents Splenomegaly and Tumor Development in a Murine Model of Epstein-Barr Virus-Associated Lymphoma, mSphere, 3 (2018).
[121] N. Purroy, J. Carabia, P. Abrisqueta, L. Egia, M. Aguilo, C. Carpio, C. Palacio, M. Crespo, F. Bosch, Inhibition of BCR signaling using the Syk inhibitor TAK-659 prevents stroma-mediated signaling in chronic lymphocytic leukemia cells, Oncotarget, 8 (2017) 742-756.
[122] S. Braselmann, V. Taylor, H. Zhao, S. Wang, C. Sylvain, M. Baluom, K. Qu, E. Herlaar, A. Lau, C. Young, B.R. Wong, S. Lovell, T. Sun, G. Park, A. Argade, S. Jurcevic, P. Pine, R. Singh, E.B. Grossbard, D.G. Payan, E.S. Masuda, R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation, J Pharmacol Exp Ther, 319 (2006) 998-1008.
[123] J.C. Spalton, J. Mori, A.Y. Pollitt, C.E. Hughes, J.A. Eble, S.P. Watson, The novel Syk inhibitor R406 reveals mechanistic differences in the initiation of GPVI and CLEC-2 signaling in platelets, J Thromb Haemost, 7 (2009) 1192-1199.
[124] M.P. Quiroga, K. Balakrishnan, A.V. Kurtova, M. Sivina, M.J. Keating, W.G. Wierda, V. Gandhi, J.A. Burger, B-cell antigen receptor signaling enhances chronic lymphocytic leukemia cell migration and survival: specific targeting with a novel spleen tyrosine kinase inhibitor, R406, Blood, 114 (2009) 1029-1037.
[125] T. Wossning, S. Herzog, F. Kohler, S. Meixlsperger, Y. Kulathu, G. Mittler, A. Abe, U. Fuchs, A. Borkhardt, H. Jumaa, Deregulated Syk inhibits differentiation and induces growth factor-independent proliferation of pre-B cells, J Exp Med, 203 (2006) 2829-2840.
[126] C. Sprissler, D. Belenki, H. Maurer, K. Aumann, D. Pfeifer, C. Klein, T.A. Muller, S. Kissel, J. Hulsdunker, J. Alexandrovski, T. Brummer, H. Jumaa, J. Duyster, C. Dierks, Depletion of STAT5 blocks TEL-SYK-induced APMF-type leukemia with myelofibrosis and myelodysplasia in mice, Blood Cancer J, 4 (2014) e240.

46
[127] M. Baluom, E.B. Grossbard, T. Mant, D.T. Lau, Pharmacokinetics of fostamatinib, a spleen tyrosine kinase (SYK) inhibitor, in healthy human subjects following single and multiple oral dosing in three phase I studies, Br J Clin Pharmacol, 76 (2013) 78-88.
[128] A. Torres-Hernandez, W. Wang, Y. Nikiforov, K. Tejada, L. Torres, A. Kalabin, Y. Wu, M.I.U. Haq, M.Y. Khan, Z. Zhao, W. Su, J. Camargo, M. Hundeyin, B. Diskin, S. Adam, J.A.K. Rossi, E. Kurz, B. Aykut, S.A.A. Shadaloey, J. Leinwand, G. Miller, Targeting SYK signaling in myeloid cells protects against liver fibrosis and hepatocarcinogenesis, Oncogene, (2019).
[129] V.C. Kyttaris, G.C. Tsokos, Syk kinase as a treatment target for therapy in autoimmune diseases, Clin Immunol, 124 (2007) 235-237.
47
2.12 Supplementary Information
Figure S1. Crystal structure of spleen tyrosine kinase complexed with R406 (Protein Data Bank code 3FQS).

48

49
Chapter 3
Therapeutic Inhibition of Spleen Tyrosine Kinase in Inflammatory
Macrophages using PLGA Nanoparticles for the Treatment of Non-
alcoholic Steatohepatitis
Dhadhang Wahyu Kurniawan, Arun Kumar Jajoriya, Garima Dhawan, Divya Mishra, Josepmaria Argemi, Ramon Bataller, Gert Storm, Durga Prasad Mishra,
Jai Prakash, Ruchi Bansal
Published in: J Control Release. 2018 Oct 28; 288:227-238. doi: 10.1016/j.jconrel.2018.09.004.

50
Abstract
Non-alcoholic steatohepatitis (NASH) is the leading cause of cirrhosis worldwide and the most
rapidly growing indication for liver transplantation. Macrophages are the important cellular
component in the inflammatory milieu in NASH. Inflammatory and pro-fibrotic mediators
produced by macrophages causes significant tissue injury in many inflammatory diseases.
Therefore, inhibition of the inflammatory macrophages would be a promising approach to
attenuate NASH. In this study, we studied the implication of SYK pathway in NASH, and
investigated PLGA nanoparticles-based delivery of SYK pathway inhibitor as an effective and
promising therapeutic approach for the treatment of NASH. We found positive correlation
between SYK expression with the pathogenesis of NASH and alcoholic hepatitis in patients.
Importantly, SYK expression was significantly induced in M1-differentiated inflammatory
macrophages. To inhibit SYK pathway specifically, we used a small-molecule inhibitor R406
that blocks Fc-receptor signalling pathway and reduces immune complex-mediated
inflammation. R406 dose-dependently inhibited nitric-oxide release and M1-specific markers
in M1-differentiated macrophages. Thereafter, we synthesized PLGA nanoparticles to deliver
R406 to increase the drug pharmacokinetics for the efficient treatment of NASH. We
investigated the therapeutic efficacy of R406-PLGA in-vitro in differentiated macrophages, and
in-vivo in Methionine-Choline-deficient (MCD)-diet induced NASH mouse model. R406-PLGA
inhibited M1-specific differentiation markers in RAW and bone-marrow-derived
macrophages. In-vivo, R406 and more strongly R406-PLGA ameliorated fibrosis, inflammation
and steatosis in mice. R406 and more significantly R406-PLGA reduced ALT, AST and total
cholesterol serum levels. These results suggest that delivery of SYK inhibitor using PLGA
nanoparticles can be a potential therapeutic approach for the treatment of Non-alcoholic
steatohepatitis.

51
3.1 Introduction
Cirrhosis and hepatocellular carcinoma (HCC) are the most common liver-related causes of
morbidity associated with NASH (Non-alcoholic steatohepatitis) [1]. In the United States,
NASH has already become the second-leading etiology of liver disease requiring liver
transplantation in adults [2-4]. Advanced fibrosis or cirrhosis has been identified as the most
important histological feature associated with NASH [5]. Liver fibrosis is the condition
characterized by excessive accumulation of abnormal extracellular matrix (ECM) proteins,
which results from chronic non-resolving inflammation [6, 7]. The inflammation triggers the
tissue destruction which further leads to scar tissue formation. Cell death and inflammation
represent two characteristic but intricately linked features of the chronic liver disease that
promote the development of fibrosis [6, 7].
Hepatic injury initiates a cascade of a fibrogenic process initiated by inflammatory and
fibrogenic signals. The activation process is initiated by the release of growth factors,
profibrogenic cytokines and chemokines by the injured hepatocytes, and inflammatory cells
particularly macrophages and other non-parenchymal cells [6-8]. Hepatic macrophages arise
from circulating bone-marrow-derived monocytes, which are recruited to the injured liver or
from proliferating resident macrophages (Kupffer cells). Resident macrophages have shown
to play a significant role in initiaton of the inflammatory responses during tissue injury, while
infiltrating monocyte-derived macrophages leads to chronic liver inflammation and
fibrogenesis [7]. During enhanced recruitment owing to liver damage and environmental cues,
infiltrating monocytes undergo differentiation into two broad subsets of macrophages that
are categorized as classically-activated (M1) or alternatively-activated (M2). The initial
inflammatory response is predominantly mediated by classically-activated (M1-
differentiated) macrophages (activated by Th1 cytokines e.g. IFN-γ and LPS). In contrast, the
resolution phase of inflammation is driven by alternatively-activated (M2-differentiated)
macrophages, stimulated by Th2 cytokines (IL-4 or IL-13) [7].
We have earlier demonstrated crucial role of signaling pathways in liver fibrogenesis [9-11].
Another major signaling pathway involved in the pathogenesis of liver disease is Spleen
tyrosine kinase (SYK) signaling pathway [12-15]. SYK is a cytoplasmic tyrosine kinase of 72 KDa
that belongs to ZAP70/SYK family of the non-receptor protein tyrosine kinases (PTKs) [16]. SYK

52
was identified and known to be expressed in hematopoietic cells, including macrophages, and
has shown to be important in the downstream signalling events that drive inflammatory
pathways of both the innate and adaptive immune systems [16]. While most studies focused
on hematopoietic cells, SYK has also been shown to be expressed in non-hematopoietic cells
including fibroblasts and hepatocytes [15, 17]. A recent study has reported the up-regulation
of SYK in hepatocytes and activated HSCs during liver fibrosis that promotes HSCs activation
and proliferation via crosstalk between cytokines secreted by hepatocytes and HSCs [15].
Many studies have also demonstrated the role of SYK in various inflammatory and fibrotic
diseases, such as Rheumatoid Arthritis [18, 19], allergic asthma/rhinitis [20, 21], renal
interstitial fibrosis [22] and that SYK inhibition could be a therapeutic strategy for the
treatment of inflammation- and fibrosis-related diseases [15, 16, 18, 20, 22]. SYK functions via
an immuno-receptor tyrosine-based activation motif (ITAM) or Toll-like receptors (TLRs) which
are activated by pathogen-associated molecular patterns (PAMP) [16, 23]. Selective SYK
inhibition or deletion in macrophages has shown to reduce macrophage activation as shown
by reduced production of FcγR (Fc gamma receptor), CCL2 (or MCP1, macrophage chemotactic
protein 1), IL-6 and TNF-α (Tumor necrosis factor alpha) [16, 23]. Therapeutic inhibition of SYK
has shown to ameliorate inflammation and steatosis in Alcoholic liver diseases [12, 13], and
suppressed liver fibrosis via inhibition of HSC activation in animal models [15]. However,
implication and inhibition of SYK pathway in NASH and specifically in M1 inflammatory
macrophages has not been investigated yet.
Macrophages and especially the liver-resident Kupffer cells are in the focus of nanomedicine
due to their highly efficient and non-specific uptake of many nanomaterials as well as due to
their critical pathogenic functions during inflammation and fibrogenesis [24]. One of the
nanoparticles is Poly(lactic-co-glycolic acid) (PLGA)-based nanoparticles that are widely used
as drug delivery carriers in various biomedical applications such as vaccination, cancer,
inflammation, and other diseases [25, 26]. PLGA is a FDA-approved biodegradable polymer
with favorable mechanical properties, biodegradation kinetics, drug compatibility and
biocompatibility since PLGA is biologically hydrolyzed to metabolite monomers, lactic acid and
glycolic acid [25]. These two monomers are endogenously metabolized by the body via the
Krebs cycle therefore no systemic toxicity is associated with the utilization of PLGA for drug

53
delivery. PLGA-nanoparticles are internalized in cells partly through fluid-phase pinocytosis
and through Clathrin-mediated endocytosis [27].
In this study, we have analyzed the SYK expression in NASH and alcoholic hepatitis (AH)
patients. We have further examined the expression and activation of SYK signaling pathway in
inflammatory macrophages. We thereafter, using small-molecule SYK inhibitor R406, studied
the implication of therapeutic SYK inhibition in inflammatory macrophages. For the efficient
delivery of SYK inhibitor R406 in vivo, we synthesized R406-encapsulated PLGA nanoparticles
and investigated their therapeutic efficacy in vitro in M1-differentiated RAW macrophages
and murine bone marrow derived macrophages, and in vivo in MCD diet-induced NASH mouse
model.
3.2 Materials and methods
3.2.1 Cell lines
Murine RAW 264.7 macrophages were obtained from the American Type Culture Collection
(ATCC, Manassas, VA, USA). RAW macrophages were cultured in Roswell Park Memorial
Institute (RPMI) 1640 medium (Lonza, Verviers, Belgium) supplemented with 2 mM L-
glutamine (Sigma, St. Louis, MO), 10 % fetal bovine serum (FBS, Lonza) and antibiotics (50
U/mL Penicillin and 50 µg/mL streptomycin, Sigma).
3.2.2 Bone marrow derived macrophages
Bone marrow-derived macrophage (BMDM) cultures were freshly isolated from C57BL/6 mice
as described previously elsewhere [28]. Briefly, femurs and tibias were flushed with
Dulbecco's modified Eagle's medium (DMEM) (Life Technologies, Carlsbad, CA, USA) with 10
% FBS (Lonza) to collect bone marrow. Cells were then triturated 3-5 times through an 18-
gauge needle and centrifuged at 1200 rpm for 5 minutes. After removing supernatant, RBCs
were lysed with the lysis buffer and the remaining cells were washed in DMEM + 10 % FBS and
plated at 1 × 106 cells/ml in DMEM supplemented with 1% penicillin/streptomycin, 1 % HEPES,
0.001% β-mercaptoethanol, 10 % FBS, and 20 % supernatant from mouse 3T3 fibroblasts
(obtained from ATCC and cultured in cultured in (DMEM) medium supplemented with 2 mM
L-glutamine (Sigma), 10 % FBS (Lonza) and antibiotics (Sigma)). The fibroblasts conditioned
medium is required to promote differentiation of bone marrow cells into macrophages (7 –

54
10 days). Media was changed on days 2, 4, and 6 and cells were re-plated on day 7 for
performing experiments.
3.2.3 PLGA nanoparticles preparation
Poly (D,L-lactide-co-glycolide) (PLGA) nanoparticles were prepared by the emulsification
solvent evaporation method as described earlier [29]. Briefly, the internal phase containing
100 µL ethyl acetate containing 2.5 mg PLGA (Poly (D,L-lactide-co-glycolide, with a co-
monomer ratio of 50 : 50 (lactic/glycolic acid), Mw = 17,000, Corbion Purac, Gorinchem, The
Netherlands) and 40 μg R406 (Mw = 628.63, Selleckchem, Boston, NY) was emulsified into an
external aqueous phase of 100 µL of 2 % Poly (vinyl-alcohol) (PVA, Sigma) (w/v), drop by drop
under constant vortexing at maximum speed. The formed o/w microemulsion was
subsequently sonicated for 2 min at 5% power output. The emulsion was transferred into 40
mL of 0.3 % PVA (w/v) under magnetic stirring, and stirred overnight at room temperature to
evaporate organic solvent ethyl acetate and to solidify the emulsified nanodroplets. The
formed nanoparticles were isolated by centrifugation for 60 min at 16,000 rpm (rotor SS-34,
Sorvall RC-5C Plus, Kendro Lab, USA) and the supernatant was discarded. The resulting
nanoparticles were characterized for their average size and size distribution by dynamic light
scattering (DLS) and zeta potential measurements using a Nano ZS Zetasizer (Malvern
Instruments Ltd., Malvern, UK). R406-PLGA nanoparticles were characterized using Scanning
electron microscope (JEOL JSM-IT 100, Akishima, Japan) with a secondary electron detector
and an acceleration voltage of 3–5 kV. The nanoparticle dispersions were freeze-dried before-
hand and mounted on conductive carbon tape.
3.2.3.1 Determination of R406 encapsulation
R406 drug encapsulation (% DE) was done by developed HPLC method using UPLC
(Thermoscientific™ Dionex™ Ultimate™ 3000 system). Briefly, 50 µL samples R406-PLGA in
suspension were centrifuged using MicroCL 17 centrifuge (Thermoscientific) for 5 min at
13,300 rpm. Filtrates were discarded and the pellet was dissolved in DMSO and measured with
UPLC at λmax 261 nm (Figure S1A). Different drug concentrations were used, and measured
with UPLC and plotted as a standard curve (Figure S1B) for determination of drug
concentration.

55
3.2.3.2 In vitro drug release and uptake by RAW macrophage
To perform the drug release study of R406 and R406-PLGA, we plated the RAW cells (5 x 105
cells/well) in 12 well plates and cultured overnight. Then we added R406 and R406-PLGA in
the cells. Furthermore, 100 µL of samples were collected and replaced by an equal volume of
the medium at time intervals (0, 6, 12, 24, and 48 h). The amount of drug released into the
medium was calculated according to a standard curve of R406 which is measured with UPLC.
Each experiment was conducted as three independent experiment (n = 3). We also performed
the drug release study of R406 and R406-PLGA without RAW cells, i.e. in the complete medium
for RAW cells. The drug and the nanoparticles with the medium incubated in a shaking
incubator at 37°C. The rest of the procedure was same as drug release study with RAW cells.
The drug uptake was measured as % total drug (at different time points) - % total drug left in
the medium (at different time points). The graphs were ploted as % cumulative drug release
(to depict drug release) and % total drug uptake (to depict drug uptake by RAW macrophages).
3.2.4 In vitro efficacy studies in RAW cell macrophages
For differentiation of RAW macrophages, cells were plated (1 x 106 cells/well) in 12 well plates
and cultured overnight. The cells were then incubated with LPS (100 ng/mL, Sigma) and IFNγ
(10 ng/mL, Peprotech, Rocky Hill, NJ) for M1 differentiation, and IL-4 (10 ng/mL, Peprotech)
and IL-13 (10 ng/mL, Peprotech) for M2 differentiation for 24 h. To study the effect of SYK
inhibitor; RAW cells were incubated with medium alone, 0.5, 1.0 and 5.0 μM of SYK inhibitor
R406 (Selleckchem, Boston, NY) together with M1 differentiation stimulus for 24 h. For the
effect of SYK inhibitor R406 loaded nanoparticles (R406-PLGA), M1-differentiated
macrophages were incubated either with 5 μM R406, R406-PLGA (equivalent dose, 5 μM) or
PLGA (empty nanoparticles, equimolar concentration as R406-PLGA). Cells were then lysed
either with protein lysis buffer for western blot analysis or RNA lysis buffer for quantitative
real-time PCR analysis. All the efficacy studies were performed at least 3 times independently.
3.2.5 In vitro efficacy studies in freshly isolated murine bone marrow derived macrophages
Bone marrow derived macrophages were plated at a density of 1 × 106/mL and differentiated
into M1 macrophages using LPS (100 ng/mL, sigma) and IFNγ (10 ng/mL, Peprotech), or M2
macrophages using IL-4 (10 ng/mL, Peprotech) and IL-13 (10 ng/mL, Peprotech). For efficacy
studies, cells were incubated with medium alone, SYK-loaded nanoparticles (R406-PLGA),

56
empty nanoparticles (PLGA) and free drug (R406) together with M1 stimulus. After 24 h of
incubation, cells were lysed for RNA isolation and real-time PCR analysis. Three independent
experiments were performed.
3.2.6 Nitric oxide (NO) release bioassay
The effect of R406 on M1 macrophages was assessed by measuring the inhibition of nitric
oxide (NO) release as described earlier [30], a stable NO metabolite produced by M1
macrophages. RAW cells (2x105 cells/200 μL/well) were seeded in 96-well plates, were
cultured overnight and incubated with IFNγ (10 ng/mL, Peprotech) and LPS (100 ng/mL, Sigma)
together with different concentrations of SYK inhibitor R406 (0, 0.5, 1.0, and 5.0 μM). After 24
hrs, 100 μL culture supernatant was added to 100 μL of Griess reagent (1% sulfanilamide
(Sigma), 0.1% naphthyl ethylenediamine dihydrochloride (Sigma); 3% phosphoric acid (Sigma))
and the absorbance at 540 nm was measured with a microplate reader. NO release assays
were performed in triplicates in three independent experiments.
3.2.7 Cell viability assay
To assess the effects on cell viability, cells were plated in 96 well plates, cultured overnight
and incubated with different concentrations of R406 (0.1, 0.5, 1.0 and 5.0 uM) together with
M1 stimulus (100 ng/mL LPS and 10 ng/mL IFNγ) for 24 h. To study the effects of R406-PLGA
on cell viability, cells were incubated with R406-PLGA (equivalent dose, 5 μM), PLGA (empty
nanoparticles, equimolar concentration as R406-PLGA) or free drug R406 (5 μM) together with
M1 stimulus for 48 h. Cell viability assays were performed using Alamar Blue reagent
(Invitrogen, Carlsbad, CA, USA) as per manufacturer's instructions. The results are represented
as % cell viability normalized to untreated control cells (at 100%). All measurements were
performed in triplicates in three independent experiments.
3.2.8 Methionine and choline-deficient (MCD) diet model
Animal studies were conducted in strict accordance with the guidelines and ethical regulations
for the Care and Use of Laboratory Animals, CSIR-Central Drug Research Institute, India. The
protocols were approved by the Institutional Animal Ethics Committee at the CSIR-Central
Drug Research Institute, India. 8-week old male C57BL/6 mice (Jackson Laboratories, Bar
Harbor, USA) were fed on MCD diet (C1070, Altromin GmbH, Lage, Germany) for 4 weeks while
the controls received regular chow diet (n=4) for 4 weeks. In this study, we used male mice

57
since it has been shown that male mice develop more severe fatty liver disease as compared
to female mice. Furthermore, male mice were shown to be more vulnerable to hepatic lipid
accumulation while on MCD diet as compared to female mice. This study also suggested the
reduction of fatty liver disease development in female mice due to the sex hormones e.g.
estrogen [31]. MCD mice were treated for two weeks intravenously (three times per week)
with vehicle (PBS, n=5), R406 (10 mg/kg, n=5), R406-PLGA (equivalent dose of 10 mg/kg, n=5)
or PLGA (empty nanoparticles, equimolar concentrations as R406-PLGA, n=5) while continuing
MCD diet (Figure S2) At the end of the experiment, mice were euthanised and the liver tissues
and blood samples were collected for analysis. Alanine aminotransferase (ALT), aspartate
aminotransferase (AST), total plasma cholesterol levels, and plasma triglycerides levels were
measured by standard automated laboratory methods.
3.2.9 RNA extraction, reverse transcription, quantitative real-time PCR
Total RNA from cells and liver tissues was isolated using GenElute Total RNA Miniprep Kit
(Sigma) and SV total RNA isolation system (Promega Corporation, Madison, WI, USA),
respectively, according to manufacturer’s instructions. The RNA concentration was
quantitated by a UV spectrophotometer (NanoDrop Technologies, Wilmington, DE). Total RNA
(1 μg) was reverse-transcribed using iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). All the
primers were purchased from Sigma-Genoysis (Haverhill, UK). Real-time PCR was performed
using 2x SensiMix SYBR and Fluorescein Kit (Bioline, QT615-05, Luckenwalde, Germany), 20 ng
cDNA and pre-tested gene-specific primer sets. The cycling conditions for the BioRad CFX384
Real-Time PCR detection system were 95°C for 10 min, 40 cycles of 95°C/15 sec, 72°C/15 sec,
and 58°C/15 sec. Finally, cycle threshold (Ct) values were normalized to reference gene GAPDH
and fold changes in expression were calculated using the 2-ΔΔCt method. All primers were
purchased from Sigma-Genosys (Haverhill, UK). The primer sequences are given in Table S1.
3.2.10 Western blot analysis
Cells were lysed using 1x Cell Lysis Buffer containing 1x DTT reducing agent (Cell Signaling
Technology, Leiden, the Netherlands) and homogenized using ultra-sonication on ice. The
samples were boiled and subjected to SDS-PAGE with 10% Tris-glycine gels (Life Technologies)
followed by protein transfer onto PVDF membrane. The membranes were developed
according to the standard protocols using primary and secondary antibodies (as mentioned in

58
Table S2). The bands were developed using ECL detection reagent (Perkin Elmer Inc., Walthan,
MA) and photographed using FluorChem M Imaging System (ProteinSimple, Alpha Innotech,
San Leandro CA). Intensity of individual bands were quantified using NIH ImageJ densitometry
software (NIH, Bethesda, MD), and expressed as % relative to β-actin or SYK.
3.2.11 Histological stainings
3.2.11.1 Immunostainings
Liver tissues were harvested and transferred to Tissue-Tek OCT embedding medium (Sakura
Finetek, Torrance, CA) and snap-frozen in 2-methyl butane chilled in a dry ice. Cryosections (6
µm) were cut using a Leica CM 3050 cryostat (Leica Microsystems, Nussloch, Germany). The
sections were air-dried and fixed with acetone for 20 min. Tissue sections were rehydrated
with PBS and incubated with the primary antibody for overnight at 4°C. Sections then were
incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h at room
temperature. Then incubated with HRP-conjugated tertiary antibody for 1 h at room
temperature. Afterwards, peroxidase activity was developed using AEC (3-amino-9-ethyl
carbazole) substrate kit (Life Technologies, Carlsbad, CA) for 20 min and nuclei were
counterstained with hematoxylin (Fluka Chemie, Buchs, Switzerland). Endogenous peroxidase
activity was blocked by 3% H2O2 prepared in methanol. The sections were mounted with
Aquatex mounting medium (Merck, Darmstadt, Germany) and were scanned using
Hamamatsu NanoZoomer Digital slide scanner 2.0HT (Hamamatsu Photonics, Bridgewater,
NJ). The details about the antibodies used for immunostainings are provided in Table S2.
3.2.11.2 Oil-Red-O staining
Oil-Red-O stock solution was prepared by dissolving 0.3 g Oil-Red-O (Sigma) in 100 mL
isopropanol. Sections were fixed in 4% formalin for 20 min and were then stained in Oil-Red-
O solution as per manufacturer’s instructions. Briefly, formalin-fixed sections were rinsed with
60% isopropanol followed by staining with freshly prepared Oil Red O working solution for 15
min. Thereafter, sections were rinsed with 60% isopropanol and nuclei were counterstained
with hematoxylin (Fluka Chemie). Finally, sections were rinsed with tap water and were
mounted with aquatex mounting medium (Merck).

59
3.2.11.3 Hematoxylin and Eosin staining
Sections were fixed with 4% formalin for 20 min and then rinsed with distilled water. The
sections were incubated with hematoxylin for 15 min followed by washings with tap water.
Thereafter, sections were incubated with eosin solution for 1.5 min followed by wash in 96%
ethanol, dehydration with ethanol and were mounted with VectaMount mounting medium
(Vector Laboratories, Burlingame, CA).
3.2.11.4 Quantitative histological analyses
For quantitation, stained sections were scanned at high resolution using Hamamatsu
NanoZoomer Digital slide scanner 2.0HT (Hamamatsu Photonic). High resolution scans were
viewed using NanoZoomer Digital Pathology (NDP2.0) viewer software (Hamamatsu
Photonic). About 20 images (100 x) of each entire section (from NDP) were imported into
ImageJ software and were analyzed quantitatively at a fixed threshold.
3.2.12 SYK gene expression in the human liver tissues
SYK mRNA expression was assessed in the publicly available transcriptome datasets of liver
tissue from patients with NASH (GSE48452) [32] obtained from the National Center for
Biotechnology Information Gene Expression Omnibus database (GEO). Patients were
categorized into 4 groups as per biopsy-based pathological investigation: control group
(n=23); NAFLD activity score NAS 1-2 (n=13); NAS 3-4 (n=7) and NAS 5-6 (n=15).
High-throughput transcriptome profiling was performed using Illumina HiSeq2000 platform
(San Diego, CA) with liver samples provided by the InTeam Consortium (Human Biorepository
Core, University of Pittsburgh, PA) as described recently [18]. SYK mRNA expression was
evaluated in patients with different phenotypes: 11 patients with non-severe alcoholic
hepatitis (with Maddrey’s discriminant function, MDF ≤32); AH responders (with severe
alcoholic hepatitis (MDF>32) patients responsive to steroids, n=9) AH non-responders (AH
severe alcoholic hepatitis (MDF>32) patients non-responsive to steroids therapy, n=9) and 10
patients with explants who underwent early liver transplantation for severe alcoholic
hepatitis. Controls (n=10) were non-diseased livers obtained from biopsied single nodules,
whose histology was without significant alterations.

60
3.2.13 Statistical analyses
All the data are presented as a mean ± standard error of the mean (SEM). The graphs and
statistical analyses were performed using GraphPad Prism version 5.02 (GraphPad Prism
Software, Inc., La Jolla, CA). Comparison to control group were analyzed using unpaired
students’ t-test while multiple comparisons between different groups were performed by one-
way analysis of variance (ANOVA) with Bonferroni post-hoc test. The differences were
considered significant at p < 0.05.
3.3 Results and Discussion
3.3.1 Upregulation of SYK expression in NASH and AH patients, and inflammatory M1 macrophages
Previous studies have documented that the functional inhibition of SYK pathway attenuates
the severity of inflammatory diseases and fibrotic diseases [12, 13, 15, 16]. In this study, we
investigated the association of SYK expression with pathogenesis of NASH (non-alcoholic
steatohepatitis) and AH (Alcoholic hepatitis). Using transcriptome analysis, we found a highly
significant induction of SYK expression that correlated with the increasing NAS score (NAFLD
activity score) as compared to normal livers (GEO accession number: GSE48452) (Figure 1A).
In AH patients, we found a significant correlation between AH severity and SYK expression
(Figure 1B). Likewise, previous studies have shown correlation of SYK and SYK pathway
activation in liver fibrosis and alcoholic liver disease (ALD) patients as compared to healthy
controls [13, 15]. Since macrophages are the key pathogenic cells in NASH and inflammatory
liver diseases [33], we analyzed the SYK expression in differentiated murine RAW
macrophages i.e. LPS- and IFNγ-induced M1-differentiated inflammatory macrophages, and
IL-4- and IL-13-induced M2-differentiated restorative macrophages. We first confirmed the
differentiation of macrophages using M1 and M2-specific markers and found that M1-
differentiated macrophages significantly expressed M1 markers i.e. iNOS (inducible nitric
oxide synthase), IL-1β (Interleukin-1-beta), CCL2 (or MCP1, macrophage chemotactic protein-
1) and IL-6 (Interleukin-6) while M2-differentiated macrophages expressed M2 markers i.e.
MRC1 (mannose receptor complex 1) and ARG (Arginase I) (Figure 1C). We then analyzed SYK
expression in these differently differentiated macrophages. We observed significant
upregulation of SYK mRNA and protein expression in LPS- and IFNγ-differentiated M1
inflammatory macrophages as compared to undifferentiated macrophages, and IL-4 and IL-

61
13-mediated M2-differentiated restorative macrophages (Figure 1D,E). These results suggest
SYK as an appropriate molecular target in macrophages and inflammatory liver diseases
especially NASH.
Figure 1. Upregulation of SYK expression in NASH and AH patients, and differentiated macrophages. (A) Normalized SYK mRNA expression in the liver tissues from NASH patients: Control (n=23); NAFLD activity score (NAS) 1-2 (n=13); NAS 3-4 (n=7); NAS 5-6 (n=15). (B) Normalized SYK mRNA expression in the alcoholic hepatitis (AH) patients: controls (non-diseased livers, n=10); AH non-severe (n=11 patients); AH responders (AH patients responsive to steroids, n=9) AH non-responders (AH patients non-responsive to steroids therapy, n=9); AH explants (AH severe patients with early liver trannsplantation, n=11). (C) Gene expression of macrophage differentiation markers: iNOS, IL-1β, CCL2, IL-6 (M1 macrophages); MRC1 and ARG1 (M2 macrophages). (D) SYK gene expression in the undifferentiated macrophage (control), M1 and M2 macrophages. (E) SYK protein expression as compared to β-actin and the respective quantitative analysis. Data are presented as mean + SEM. *p<0.05, **p<0.01 and ***p<0.001 denotes significance.

62
3.3.2 SYK inhibitor R406 inhibited LPS- and IFNγ-induced activation of murine RAW macrophages
Since SYK expression was upregulated in M1-differentiated inflammatory macrophages, we
explored the activation of this receptor in macrophages. We used R406 which is a potent SYK
inhibitor (Figure 2A) to inhibit SYK signaling pathway, that has been used earlier [13]. We first
examined the phosphorylation of SYK and observed that increased SYK expression in M1
macrophages was accompanied with increased phosphorylation of SYK (Figure 2B,C). We then
tested the effect of SYK inhibitor, R406 on SYK phosphorylation and functional activation i.e.
nitric oxide (NO) release in M1-differentiated macrophages. We found that R406
concentration dependently reduced SYK phosphorylation i.e. SYK signalling pathway (Figure
2B,C) and LPS- and IFNγ-induced NO release (Figure 2D). In addition, we performed viability
studies and found no toxicity with R406 (Figure S3).
Figure 2. Inhibition of SYK phosphorylation and nitric oxide release by SYK inhibitor R406 in RAW macrophages. (A) Structure of R406, a SYK inhibitor. (B) Protein expression and (C) quantitative densitometry analysis of phosphorylated SYK (pSYK) as compared to SYK in RAW 264.7 macrophages after incubation with medium alone, M2 stimulus (10 ng/mL IL-4 and 10 ng/mL IL-13) and M1 stimulus (100 ng/mL LPS + 10 ng/mL IFNγ) without and with R406 (0, 0.5, 1, and 5 µM). (D) Nitric oxide (NO) release as measured in the culture supernatant of RAW 264.7 cells incubated with medium alone (M0 or control) or M1 stimulus (100 ng/mL LPS + 10 ng/mL IFNγ) with R406 (0, 0.5, 1, and 5 µM). ##p<0.01 denotes significance versus control M0 undifferentiated macrophages. *p < 0.05, **p < 0.01, and ***p < 0.001 denotes significance versus M1-differentiated macrophages.

63
3.3.3 SYK inhibitor R406 inhibited gene expression of inflammatory markers in mouse RAW macrophages
We further examined the efficacy of R406 on gene expression of inflammatory markers in M1-
differentiated macrophages. We observed that R406 dose-dependently inhibited LPS- and
IFNγ-induced gene expression of M1 markers i.e. IL1β, Fc gamma receptor 1 (FcγR1), iNOS,
CCL2 (or MCP1), IL-6 and CCR2 (C-C motif chemokine receptor 2) (Figure 3A-F). These results
are consistent with previous studies in experimental animal models [13, 34, 35]. Altogether,
these results suggest that SYK inhibitor R406 treatment can potentially inhibit inflammatory
macrophages and hence R406 has high potency for the treatment of inflammation-associated
diseases. Inflammation in the liver protects this organ from infection and injury, but excessive
inflammation may lead to extensive loss of hepatocytes, ischemia-reperfusion injury,
metabolic alterations, and eventually hepatic damage. Therefore, inhibition of inflammation
or inflammatory macrophages via SYK pathway inhibition might be promising approach for
the treatment of inflammatory liver diseases.
Figure 3. Inhibition of M1-specific differentiation and inflammatory markers by R406 in RAW macrophages. Gene expression of M1 markers IL-1β (A); FcγR1 (B); iNOS (C); CCL2 (D); IL-6 (E); and CCR2 (F) in RAW 264.7 cells after incubation with medium alone (M0) or M1 stimulus with R406 (0, 0.5, 1, and 5 µM). expression values for the respective genes in untreated M1 macrophages were set

64
at 1.0 to calculate the relative gene expression. Data are presented as mean + SEM. #p<0.05, ##p<0.01 denotes significance versus control M0 macrophages. *p<0.05, **p<0.01 and ***p<0.001 denotes significance versus M1-differentiated macrophages.
3.3.4 Preparation and characterization of R406-PLGA nanoparticles
Since small-molecule tyrosine kinase inhibitors have been shown to have poor
pharmacokinetics and bioavailability [36], we prepared R406-encapsulated PLGA
nanoparticles (R406-PLGA, Figure 4A) to improve the pharmacokinetics and thereof in vivo
therapeutic efficacy of R406. R406-PLGA nanoparticles (R406-PLGA) were analyzed for the
morphology, size and zeta potential (Figure 4B,C). R406-PLGA has Z-average 160 nm, with
almost comparable size to the control empty PLGA nanoparticles (PLGA) (Figure 4C). The zeta
potential was towards negative as shown in Figure 4C. We also measured the drug
encapsulation of R406 in the PLGA nanoparticles using HPLC (Figure S1A,B).
Figure 4. Characterization of R406-PLGA nanoparticles. (A) Representative image of R406-PLGA nanoparticles as characterized by scanning electron microscope (SEM); (B) Schematic presentation depicting the drug (R406) encapsulated in the PLGA nanoparticles; (C) The average particle size, polydispersity index (PDI), and zeta potential (ZP) of R406-PLGA NPs (or R406-PLGA) as compared to empty-NPs (PLGA) (n=3); (D) Physical stability (size stability) of R406-PLGA NPs for 46 days; (E) Protein expression and (F) quantitative densitometry analysis of pSYK compared to SYK on RAW 264.7 cells after incubation with medium alone (control), M2 stimulus (10 ng/mL IL-4 and 10 ng/mL IL-13), M1 stimulus (100 ng/mL LPS + 10 ng/mL IFNγ) with or without R406 (free drug), R406-PLGA or empty-NPs (PLGA). Data are presented as mean + SEM. *p < 0.05 and **p < 0.01 denotes significance.
The drug encapsulation of R406 in the PLGA nanoparticles was found to be 37,49%. We further
examined the physical stability of the R406-PLGA nanoparticles and observed that the particle

65
size remained relatively stable at around 160 nm size after 46 days. These results suggest the
successful synthesis of R406-PLGA nanoparticles with favorable particle size and stability for
appropriate delivery of SYK inhibitor.
3.3.5 R406-PLGA nanoparticles inhibited the SYK signalling pathway and gene expression of inflammatory markers in RAW macrophages
Following successful synthesis of R406-PLGA nanoparticles, we examined the R406 efficacy
after encapsulation in PLGA nanoparticles. We first tested R406-PLGA nanoparticles in RAW
macrophages for inhibition of SYK pathway and we found that both R406 and R406-PLGA
nanoparticles significantly inhibited the SYK signalling pathway (SYK phosphorylation) as
shown in Figure 4E,F. While empty PLGA nanoparticles alone didn’t show any effect in SYK
phosphorylation (Figure 4E,F). We further investigated the therapeutic effects of R406-PLGA
nanoparticles in M1-differentiated inflammatory macrophages. Interestingly, both R406 and
R406-PLGA nanoparticles significantly inhibited the gene expression of M1-specific markers
i.e. iNOS, FcγR1, IL-6, IL-1β, CCL2 (or MCP1) and IL-1α (Figure 5A-F).
However, we observed R406 was more effective than R406-PLGA nanoparticles which is
probably due to slow drug release kinetics of PLGA nanoparticles [25]. Generally, higher
content of polyglycolide leads to quicker rates of degradation with an exception of 50:50 ratio
of PLA/PGA which is used in this study exhibits the fastest degradation. As per modeling
studies, PLA/PGA 50:50 demonstrated release kinetics of about 2-3 weeks to achieve 100%
drug release [25]. Furthermore, studies have found, that the type of drug also plays a critical
role in determining the release kinetics [37]. We performed drug release studies using R406-
PLGA nanoparticles and found that % cumulative release of R406 from PLGA nanoparticles
increases gradually after incubation for 48 h as compared to the free drug (Figure S4A).
Furthermore, we performed drug uptake studies and observed that R406 (in free form) was
significantly taken up more in RAW macrophages (about 54% at 48 h) as compared to R406
(from PLGA nanoparticles) (about 45% at 48 h) as can be seen in Figure S4B. These data
supports the reduced in vitro therapeutic efficacy of R406-PLGA as compared to free R406,
however further suggest slow and gradual release of R406 which will be desired in vivo.
Essentially, these results suggest that R406 retained its therapeutic effects following
encapsulation in PLGA nanoparticles. We also performed the cell viability studies with R406-
PLGA nanoparticles and observed no effect on cell viability as shown in Figure S5.

66
3.3.6 R406-PLGA nanoparticles inhibited the SYK signalling pathway and gene expression of inflammatory markers in murine bone marrow derived macrophages
In general, immortalized cells are not considered normal as they divide indefinitely and
express unique gene patterns that are not found in cell type in vivo and hence they might not
have the relevant attributes of normal cells. Thus, it is important to validate the characteristics
and results on primary cells that have not been passaged many times. To mimic the in vivo
more closely, we performed studies on primary murine bone marrow derived macrophages
(BMDMs).
We first investigated the expression of SYK in BMDMs and observed significant upregulation
of SYK expression in M1-differentiated BMDMs as compared to undifferentiated and M2-
differentiated BMDMs (Figure 6A). Thereafter, we examined the effect of R406-PLGA
nanoparticles on the gene expression of inflammatory markers in M1-differentaited
inflammatory BMDMs. We found that both R406 and R406-PLGA nanoparticles induced highly
significant down-regulation in the expression levels of major inflammatory markers i.e. CCL2,
Figure 5. Inhibition of inflammatory markers or M1 differentiation markers by R406 and R406-PLGA in RAW macrophages. Gene expression of inflammatory or M1-differentiation macrophages markers in RAW 264.7 cells: (A) iNOS; (B) FcγR1; (C) IL-6; (D) IL-1β; (E) CCL2; (F) IL-1α after incubation with medium alone (M0 undifferentiated macrophages) or M1 stimulus (100 ng/mL LPS + 10 ng/mL IFNγ) with or without R406 (free drug), R406-PLGA or empty-NPs (PLGA). Data are presented as mean ± SEM. #p<0.05, ##p<0.01 denotes significance versus control M0 macrophages. Data are presented as mean + SEM. *p<0.05, **p<0.01, and ***p<0.001 denotes significance versus M1-differentiated macrophages.

67
IL-1α and IL-6 as shown in Figure 6B-D. These results further confirmed our previous results
in RAW macrophages and suggest that R406 retained its therapeutic efficacy after
encapsulation in PLGA nanoparticles. Notably, we did not observe any effects from empty
PLGA nanoparticles in RAW macrophages and in BMDMs (Figure 5A-F, 6B-D).
Figure 6. Inhibition of inflammatory markers or M1 differentiation markers by R406 and R406-PLGA in murine bone marrow derived macrophages. (A) Normalized SYK expression in undifferentiated M0, M1-differentiated and M2-differentiated macrophages. Gene expression of inflammatory markers: (B) CCL2; (C) IL-1α; and (D) IL-6 after incubation with medium alone (undifferentiated M0 macrophages) or M1 stimulus (100 ng/mL LPS + 10 ng/mL IFNγ) with or without R406 (free drug), R406-PLGA or empty-NPs (PLGA). Data are presented as mean + SEM. #p<0.05, ##p<0.01 denotes significance versus control M0 macrophages. *p<0.05, **p<0.01 and ***p<0.001 denotes significance versus M1-differentiated macrophages.
3.3.7 R406-PLGA nanoparticles ameliorated liver inflammation, fibrosis and steatosis in MCD diet induced NASH mouse model
Finally, we investigated the therapeutic efficacy of R406-PLGA nanoparticles in MCD-diet
induced NASH mouse model. We first examined the expression of SYK expression in mice with
MCD-diet induced NASH and found that SYK expression was highly significantly induced in
mice with NASH as compared to healthy controls (Figure 7B). With this observation, we
surmised that SYK play a major role in MCD-diet induced liver steatosis. We then examined

68
the therapeutic efficacy of R406 and R406-PLGA nanoparticles in MCD-diet induced NASH. We
followed the clinical treatment regimen, where we induced NASH for 4 weeks and treated
mice for last 2 weeks with in total 4 intravenous administrations while continuing with MCD
diet (Figure S2).
Figure 7. R406-PLGA ameliorates inflammation, steatosis, and fibrosis in methionine and choline-deficient (MCD) diet induced NASH mouse model. (A) Representative images of liver tissue from control group (chow-fed mice, n = 4), MCD group (MCD diet-fed mice, n = 5), MCD + R406 group (MCD diet-fed mice treated with free drug R406, n = 5), MCD + R406-PLGA group (MCD diet-fed mice treated

69
with R406-PLGA nanoparticles, n = 5), and MCD + PLGA group (MCD diet-fed mice treated with empty PLGA nanoparticles, n = 5). (B) Normalized SYK expression in liver tissue from MCD diet NASH mice (n = 5) compared to normal mice (n = 5). **p < 0.01 denotes significance. (C) Representative images of liver section stained with hematoxylin and eosin (H&E) staining, oil-red-O staining, and collagen I, F4/80 and MHC II as performed on healthy controls and MCD diet mice treated with respective treatments. (D) Quantitative image analysis of liver sections from healthy controls and MCD diet mice treated with respective treatments stained with oil-red-O, collagen I, F4/80 and MHC II. Data are presented as mean + SEM. #p < 0.05 denotes significance versus healthy controls; *p < 0.05, **p < 0.01, ***p < 0.001 denotes significance versus MCD diet mice.
Morphological examination of liver tissues at the end of the study showed the visible pale and
gross appearance suggesting the hepatocellular damage in MCD-diet induced NASH mice as
compared to normal healthy control group (Figure 7A). Treatment with R406 and R406-PLGA
showed considerably better appearance of livers in contrast to MCD-diet induced NASH livers
and PLGA-treated NASH livers (Figure 7A). Interestingly, we found that SYK inhibitor R406 and
more significantly R406-PLGA nanoparticles ameliorated (a) hepatic inflammation as
examined by H&E, F4/80 (macrophage marker) and MHC-II staining’s; (b) hepatic fibrosis as
confirmed by collagen I (major extracellular matrix protein) expression; and (c) steatosis as
examined using oil-red-o staining (Figure 7C,D).
We further examined plasma liver transaminases i.e. ALT and AST levels, and found that R406
and more efficiently R406-PLGA inhibited MCD-diet induced Ast and ALT levels (Figure 8A,B).
We also analyzed intrahepatic expression of SYK pathway downstream signaling molecule
CARD9 (caspase recruitment domain-containing protein 9) [38, 39] and found highly
significant inhibition of CARD9 by R406-PLGA nanoparticles as compared to R406 (Figure 8C).
In in vitro studies, we observed that both R406 and R406-PLGA nanoparticles decreased CCL2
(MCP1) expression in RAW macrophages and BMDMs, which was further implicated in the in
vivo studies. Reduced intrahepatic CCL2 expression by R406 and R406-PLGA nanoparticles
inhibits the macrophage chemotaxis/migration to liver therefore further inhibiting ongoing
liver inflammation (Figure 7C,D). Previous studies have demonstrated the crucial role of CCL2
in liver inflammation and shown that pharmacological inhibition of CCL2 ameliorated liver
inflammation by inhibition of macrophage infiltration [28]. We also analyzed gene expression
of F4/80 and crucial inflammatory markers i.e. CCL2, TNFα, iNOS, and intracellular cell
adhesion molecule 1 (ICAM1), and found that MCD-diet induced the macrophage influx (as
shown by F4/80 expression) and inflammatory markers which were inhibited by R406 and
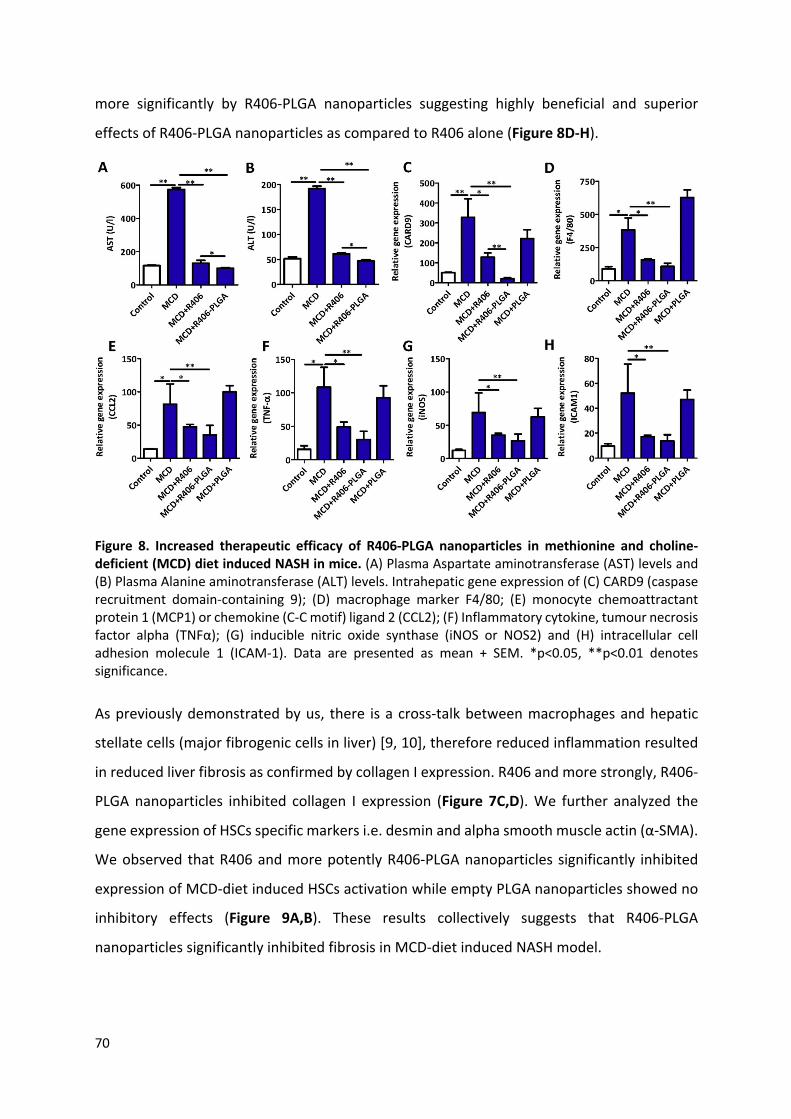
70
more significantly by R406-PLGA nanoparticles suggesting highly beneficial and superior
effects of R406-PLGA nanoparticles as compared to R406 alone (Figure 8D-H).
Figure 8. Increased therapeutic efficacy of R406-PLGA nanoparticles in methionine and choline-deficient (MCD) diet induced NASH in mice. (A) Plasma Aspartate aminotransferase (AST) levels and (B) Plasma Alanine aminotransferase (ALT) levels. Intrahepatic gene expression of (C) CARD9 (caspase recruitment domain-containing 9); (D) macrophage marker F4/80; (E) monocyte chemoattractant protein 1 (MCP1) or chemokine (C-C motif) ligand 2 (CCL2); (F) Inflammatory cytokine, tumour necrosis factor alpha (TNFα); (G) inducible nitric oxide synthase (iNOS or NOS2) and (H) intracellular cell adhesion molecule 1 (ICAM-1). Data are presented as mean + SEM. *p<0.05, **p<0.01 denotes significance.
As previously demonstrated by us, there is a cross-talk between macrophages and hepatic
stellate cells (major fibrogenic cells in liver) [9, 10], therefore reduced inflammation resulted
in reduced liver fibrosis as confirmed by collagen I expression. R406 and more strongly, R406-
PLGA nanoparticles inhibited collagen I expression (Figure 7C,D). We further analyzed the
gene expression of HSCs specific markers i.e. desmin and alpha smooth muscle actin (α-SMA).
We observed that R406 and more potently R406-PLGA nanoparticles significantly inhibited
expression of MCD-diet induced HSCs activation while empty PLGA nanoparticles showed no
inhibitory effects (Figure 9A,B). These results collectively suggests that R406-PLGA
nanoparticles significantly inhibited fibrosis in MCD-diet induced NASH model.

71
Figure 9. Increased therapeutic efficacy of R406-PLGA nanoparticles in methionine and choline deficiency (MCD) diet induced NASH in mice. Intrahepatic gene expression of (A) Desmin and (B) α-SMA; (C) Total serum cholesterol levels expressed in mg/dL; (D) Total serum triglyceride levels in mg/dL; intrahepatic gene expression of (E) uncoupling protein 1 (UCP1); (F) PR domain containing 16 (PDRM16); (G) Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) and (H) c/EBPβ (CCAAT/enhancing binding protein β). Data are presented as mean + SEM. *p < 0.05, **p < 0.01 denotes significance.
Finally, we analyzed the effect of R406-PLGA on hepatic steatosis. Oil-red-o staining showed
significant induction in hepatic steatosis in MCD-induced NASH which was inhibited by R406
and more strongly by R406-PLGA nanoparticles (Figure 7C,D). We first examined total plasma
cholesterol and triglyceride levels, and found significant upregulation of these parameters in
NASH mice as compared to healthy controls. Interestingly, MCD-diet-induced cholesterol and
triglyceride levels were highly significantly inhibited by R406 and, more efficiently, by R406-
PLGA nanoparticles (Figure 9C,D). Furthermore, we analyzed expression of steatosis related
markers: UCP1 (uncoupling protein 1), PDRM16 (PR-domain containing 16), PGC1α
(Peroxisome proliferator-activator receptor gamma coactivator 1 alpha) and c/EBPβ
(CCAAT/enhancing binding protein β). UCP1, PDRM16 and PGC1α has been shown to induce
lipid oxidation and hence are involved in PGC1α/PDRM16/UCP1-mediated lipid energy
dissipation [39]. c/EBPβ (CCAAT/enhancing binding protein β), has shown to have a key role in
steatosis and the pathophysiology of MCD diet-mediated model of NASH through its effect on

72
lipid synthesis, inflammation, and ER stress [40]. We found that R406-PLGA nanoparticles
alone induced the expression of PGC1α, PDRM16, UCP1 and reduced MCD-diet induced
C/EBPβ expression suggesting increased lipid oxidation, reduced lipid synthesis and reduced
steatosis (Figure 9E-H). Altogether, these results suggest that R406 and more significantly
R406-PLGA nanoparticles significantly ameliorated fibrosis, inflammation, and steatosis in
NASH mouse model. Importantly, PLGA-mediated delivery significantly increased the in vivo
therapeutic efficacy of R406 and therefore potentially reduce the frequency of drug
administration.
3.4 Conclusion
In conclusion, we have demonstrated schematically a novel role of SYK signaling pathway in
modulating liver inflammation, fibrosis and steatosis attributed to non-alcoholic
steatohepatitis. We have also shown the role of SYK pathway in inflammatory M1
macrophages. Furthermore, this study highlights SYK inhibitor R406 as a potential therapeutic
for NASH. Using PLGA nanoparticles, we have demonstrated the efficient intrahepatic delivery
of SYK inhibitor for increased therapeutic efficacy as shown by decreased inflammation,
hepatic injury, fibrosis, and steatosis by suppressing SREBP1-mediated lipogenesis and
increased PGC1α/PDRM16/UCP1-mediated lipid energy dissipation. To our best knowledge,
this is the first study highlighting the increased therapeutic effects of SYK inhibitor using PLGA-
mediated delivery in NASH. This promising strategy can potentially be applicable for the
treatment of other inflammatory diseases and should be further explored for the systematic
evaluation for the clinical applications.
3.5 Acknowledgements
This study was funded by Endowment Fund for the Education Republic of Indonesia (Lembaga
Pengelola Dana Pendidikan/LPDP RI). This project was partially supported by the Netherlands
Organization for Health Research and Development (ZonMW, NWO)-funded VENI innovation
grant 916.151.94 (to R.B.). Authors thank Hetty ten Hoopen for her technical assistance.
3.6 References
[1] L. Pimpin, H. Cortez-Pinto, F. Negro, E. Corbould, J.V. Lazarus, L. Webber, N. Sheron, Burden of liver disease in Europe: epidemiology and analysis of risk factors to identify prevention policies, J Hepatol, (2018).

73
[2] S.M. Francque, D. van der Graaff, W.J. Kwanten, Non-alcoholic fatty liver disease and cardiovascular risk: Pathophysiological mechanisms and implications, J Hepatol, 65 (2016) 425-443.
[3] N. Katsiki, D.P. Mikhailidis, C.S. Mantzoros, Non-alcoholic fatty liver disease and dyslipidemia: An update, Metabolism, 65 (2016) 1109-1123.
[4] S.A. Polyzos, C.S. Mantzoros, Nonalcoholic fatty future disease, Metabolism, 65 (2016) 1007-1016.
[5] J. Wattacheril, D. Issa, A. Sanyal, Nonalcoholic Steatohepatitis (NASH) and Hepatic Fibrosis: Emerging Therapies, Annu Rev Pharmacol Toxicol, 58 (2018) 649-662.
[6] R. Bataller, D.A. Brenner, Liver fibrosis, Journal of Clinical Investigation, 115 (2005) 209-218.
[7] E. Seki, R.F. Schwabe, Hepatic inflammation and fibrosis: functional links and key pathways, Hepatology, 61 (2015) 1066-1079.
[8] R. Bansal, B. Nagorniewicz, J. Prakash, Clinical Advancements in the Targeted Therapies against Liver Fibrosis, Mediators Inflamm, 2016 (2016) 7629724.
[9] R. Bansal, J. van Baarlen, G. Storm, J. Prakash, The interplay of the Notch signaling in hepatic stellate cells and macrophages determines the fate of liver fibrogenesis, Sci Rep, 5 (2015) 18272.
[10] B. Ozturk Akcora, G. Storm, J. Prakash, R. Bansal, Tyrosine kinase inhibitor BIBF1120 ameliorates inflammation, angiogenesis and fibrosis in CCl4-induced liver fibrogenesis mouse model, Sci Rep, 7 (2017) 44545.
[11] B.O. Akcora, G. Storm, R. Bansal, Inhibition of canonical WNT signaling pathway by beta-catenin/CBP inhibitor ICG-001 ameliorates liver fibrosis in vivo through suppression of stromal CXCL12, Biochim Biophys Acta, 1864 (2018) 804-818.
[12] T.N. Bukong, A. Iracheta-Vellve, B. Gyongyosi, A. Ambade, D. Catalano, K. Kodys, G. Szabo, Therapeutic Benefits of Spleen Tyrosine Kinase Inhibitor Administration on Binge Drinking-Induced Alcoholic Liver Injury, Steatosis, and Inflammation in Mice, Alcohol Clin Exp Res, 40 (2016) 1524-1530.
[13] T.N. Bukong, A. Iracheta-Vellve, B. Saha, A. Ambade, A. Satishchandran, B. Gyongyosi, P. Lowe, D. Catalano, K. Kodys, G. Szabo, Inhibition of spleen tyrosine kinase activation ameliorates inflammation, cell death, and steatosis in alcoholic liver disease, Hepatology, 64 (2016) 1057-1071.
[14] S.H. Shin, K.H. Lee, B.H. Kim, S. Lee, H.S. Lee, J.J. Jang, G.H. Kang, Downregulation of spleen tyrosine kinase in hepatocellular carcinoma by promoter CpG island hypermethylation and its potential role in carcinogenesis, Lab Invest, 94 (2014) 1396-1405.
[15] C. Qu, D. Zheng, S. Li, Y. Liu, A. Lidofsky, J.A. Holmes, J. Chen, L. He, L. Wei, Y. Liao, H. Yuan, Q. Jin, Z. Lin, Q. Hu, Y. Jiang, M. Tu, X. Chen, W. Li, W. Lin, B.C. Fuchs, R.T. Chung, J. Hong, Tyrosine kinase SYK is a potential therapeutic target for liver fibrosis, Hepatology, (2018).
[16] A. Mocsai, J. Ruland, V.L. Tybulewicz, The SYK tyrosine kinase: a crucial player in diverse biological functions, Nat Rev Immunol, 10 (2010) 387-402.
[17] T.N. Bukong, K. Kodys, G. Szabo, Human ezrin-moesin-radixin proteins modulate hepatitis C virus infection, Hepatology, 58 (2013) 1569-1579.

74
[18] T. Oton, L. Silva-Fernandez, J.L. Andreu, Spleen tyrosine kinase (Syk) inhibitor for rheumatoid arthritis, N Engl J Med, 364 (2011) 83; author reply 84.
[19] H. Okamoto, A. Kobayashi, Spleen tyrosine kinase (Syk) inhibitor for rheumatoid arthritis, N Engl J Med, 364 (2011) 83-84; author reply 84.
[20] E.S. Masuda, J. Schmitz, Syk inhibitors as treatment for allergic rhinitis, Pulm Pharmacol Ther, 21 (2008) 461-467.
[21] M. Ulanova, F. Duta, L. Puttagunta, A.D. Schreiber, A.D. Befus, Spleen tyrosine kinase (Syk) as a novel target for allergic asthma and rhinitis, Expert Opin Ther Targets, 9 (2005) 901-921.
[22] K.H. Chen, H.H. Hsu, H.Y. Yang, Y.C. Tian, Y.C. Ko, C.W. Yang, C.C. Hung, Inhibition of spleen tyrosine kinase (syk) suppresses renal fibrosis through anti-inflammatory effects and down regulation of the MAPK-p38 pathway, Int J Biochem Cell Biol, 74 (2016) 135-144.
[23] Y. Kulathu, G. Grothe, M. Reth, Autoinhibition and adapter function of Syk, Immunol Rev, 232 (2009) 286-299.
[24] M. Bartneck, K.T. Warzecha, F. Tacke, Therapeutic targeting of liver inflammation and fibrosis by nanomedicine, Hepatobiliary Surg Nutr, 3 (2014) 364-376.
[25] H.K. Makadia, S.J. Siegel, Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier, Polymers (Basel), 3 (2011) 1377-1397.
[26] M. Mir, N. Ahmed, A.U. Rehman, Recent applications of PLGA based nanostructures in drug delivery, Colloids Surf B Biointerfaces, 159 (2017) 217-231.
[27] G. Sahay, D.Y. Alakhova, A.V. Kabanov, Endocytosis of nanomedicines, J Control Release, 145 (2010) 182-195.
[28] C. Baeck, X. Wei, M. Bartneck, V. Fech, F. Heymann, N. Gassler, K. Hittatiya, D. Eulberg, T. Luedde, C. Trautwein, F. Tacke, Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice, Hepatology, 59 (2014) 1060-1072.
[29] D.L. Priwitaningrum, J.G. Blonde, A. Sridhar, J. van Baarlen, W.E. Hennink, G. Storm, S. Le Gac, J. Prakash, Tumor stroma-containing 3D spheroid arrays: A tool to study nanoparticle penetration, J Control Release, 244 (2016) 257-268.
[30] R. Bansal, J. Prakash, E. Post, L. Beljaars, D. Schuppan, K. Poelstra, Novel engineered targeted interferon-gamma blocks hepatic fibrogenesis in mice, Hepatology, 54 (2011) 586-596.
[31] Y.H. Lee, S.H. Kim, S.N. Kim, H.J. Kwon, J.D. Kim, J.Y. Oh, Y.S. Jung, Sex-specific metabolic interactions between liver and adipose tissue in MCD diet-induced non-alcoholic fatty liver disease, Oncotarget 7 (2016) 46959-46971.
[32] M. Ahrens, O. Ammerpohl, W. von Schonfels, J. Kolarova, S. Bens, T. Itzel, A. Teufel, A. Herrmann, M. Brosch, H. Hinrichsen, W. Erhart, J. Egberts, B. Sipos, S. Schreiber, R. Hasler, F. Stickel, T. Becker, M. Krawczak, C. Rocken, R. Siebert, C. Schafmayer, J. Hampe, DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery, Cell Metab, 18 (2013) 296-302.
[33] F. Tacke, Targeting hepatic macrophages to treat liver diseases, J Hepatol, 66 (2017) 1300-1312.

75
[34] Y.I. Miller, S.H. Choi, P. Wiesner, Y.S. Bae, The SYK side of TLR4: signalling mechanisms in response to LPS and minimally oxidized LDL, Br J Pharmacol, 167 (2012) 990-999.
[35] O. Gross, H. Poeck, M. Bscheider, C. Dostert, N. Hannesschlager, S. Endres, G. Hartmann, A. Tardivel, E. Schweighoffer, V. Tybulewicz, A. Mocsai, J. Tschopp, J. Ruland, Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence, Nature, 459 (2009) 433-436.
[36] M. Herbrink, B. Nuijen, J.H. Schellens, J.H. Beijnen, Variability in bioavailability of small molecular tyrosine kinase inhibitors, Cancer Treat Rev, 41 (2015) 412-422.
[37] S.J. Siegel, J.B. Kahn, K. Metzger, K.I. Winey, K. Werner, N. Dan, Effect of drug type on the degradation rate of PLGA matrices, Eur J Pharm Biopharm, 64 (2006) 287-293.
[38] R.A. Drummond, S. Saijo, Y. Iwakura, G.D. Brown, The role of Syk/CARD9 coupled C-type lectins in antifungal immunity, Eur J Immunol, 41 (2011) 276-281.
[39] Z. Zhang, L. He, S. Hu, Y. Wang, Q. Lai, P. Yang, Q. Yu, S. Zhang, F. Xiong, S. Simsekyilmaz, Q. Ning, J. Li, D. Zhang, H. Zhang, X. Xiang, Z. Zhou, H. Sun, C.Y. Wang, AAL exacerbates pro-inflammatory response in macrophages by regulating Mincle/Syk/Card9 signaling along with the Nlrp3 inflammasome assembly, Am J Transl Res, 7 (2015) 1812-1825.
[40] S.M. Rahman, J.M. Schroeder-Gloecker, R.C. Janssen, H. Jiang, I. Qadri, K.N. Maclean, J.E. Friedman, CCAAT/enhancing binding protein beta deletion in mice attenuate inflammation, endoplasmic reticulum stress, and lipid accumulation in diet-induced nonalcoholic steatohepatitis, Hepatology 45 (2007) 1108-1117.

76
3.7 Supplementary Information
Table S1. Sequence of the mouse primers used for quantitative real-time PCR
Table S2. Antibodies used for the immunohistochemistry and western blotting
Gene
Forward primer sequence (5’-3’) Reverse primer sequence (5’-3’) Accession no. iNOS GGTGAAGGGACTGAGCTGTT GCTACTCCGTGGAGTGAACAA NM_010927.4 IL-1β GCCAAGACAGGTCGCTCAGGG CCCCCACACGTTGACAGCTAGG NM_008361.3 CCL2 GTGCTGACCCCAAGAAGGAA GTGCTGAAGACCTTAGGGCA NM_011333.3 IL-6 TGATGCTGGTGACAACCACGGC TAAGCCTCCGACTTGTGAAGTGTA NM_031168.1 MRC1 TGGATGGATGGGAGCAAAGT GCTGCTGTTATGTCTCTGGC NM_008625.2 ARG1 GTGAAGAACCCACGGTCTGT CTGGTTGTCAGGGGAGTGTT NM_007482.3 GAPDH ACAGTCCATGCCATCACTGC GATCCACGACGGACACATTG NM_008084.2
SYK TCCATTGCAGCCATTCCCAT GTCCTGGAGTCACAGAAGGC NM_011518.2
FcγR1 GTGCTCTCATTGGGTGGTGA TGTAAGCCGTGCCTTCCAAT NM_010186.5
CCR2 AGGAGCCATACCTGTAAATGC TGTGGTGAATCCAATGCCCT NM_009915.2
IL-1α CGCTTGAGTCGGCAAAGAAA CTGATACTGTCACCCGGCTC NM_010554.4
CARD9 GGCTCTGAACAAGGAGCATCT TCCTGCGGTAGTTCTCGAAG NM_001037747.1
UCP1 GGATGGTGAACCCGACAACT GCTGAAACTCCGGCTGAGAA NM_009463.3
PDRM16 TGTGCGAAGGTGTCCAAACT TCTCTCCTCCTCCTTGAGCC NM_027504.3
PGC1α ACTCTCAGTAAGGGGCTGGTT GACGCCAGTCAAGCTTTTTCA NM_008904.2
SREBP1 CTGGTGAGTGGAGGGACCAT GACCGGTAGCGCTTCTCAAT NM_011480.4
Primary Antibody Source Immunostaining Western blotting
Polyclonal rabbit anti-SYK Cell Signalling 1:400
Monoclonal mouse anti-β-actin Sigma 1:5000
Polyclonal rabbit anti-pSYK Cell Signal Tech. 1:400
Polyclonal goat anti-collagen I Southern Biotech 1:100
Rat anti mouse F4/80 BioRad 1:100
Rat monoclonal MHC II Santa Cruz 1:100
Secondary Antibody Source
Polyclonal goat anti-rabbit IgG DAKO 1:100 1:1000
Polyclonal rabbit anti-mouse IgG DAKO 1:100 1:1000
Polyclonal goat anti-mouse IgG DAKO 1:100 1:1000
Polyclonal rabbit anti-goat IgG DAKO 1:100 1:1000
Polyclonal goat anti-rat IgG Southern Biotech 1:100 1:1000

77
Figure S1. Determination of % encapsulation of R406 in PLGA nanoparticles using HPLC. (A) Chromatogram of R406 in R406-PLGA nanoparticles. (B) Calibration curve for R406.
Figure S2. Schematic representation of the experimental design and treatment regimen in MCD-diet induced NASH mouse model.

78
Figure S3. Effect of R406 on RAW cell viability. Viability of RAW 264.7 macrophages after treatment with different concentrations of R406 in the presence of M1 stimulus (100 ng/mL LPS + 10 ng/mL IFNγ). Bars represent mean + SEM from n=3 independent experiments performed in triplicates.
Cell viability
M0 0 0.5 1 50
20
40
60
80
100M0M1
R406 (µM)
% ce
ll vi
abili
ty

79
Figure S4. In vitro drug release and uptake by RAW macrophages. (A) % cumulative release rate of R406 from R406-PLGA nanoparticles in medium (containing 10% FBS) as analyzed at different time intervals (0, 6, 12, 24, and 48 h). (B) % drug uptake by RAW macrophages as measured at different time intervals (0, 6, 12, 24, and 48 h).

80
Figure S5. Effect of R406-PLGA nanoparticles on the RAW cell viability. Viability of RAW 264.7 macrophages after treatment with R406, R406-PLGA nanoparticles and empty PLGA nanoparticles in the presence of M1 stimulus (100 ng/mL LPS + 10 ng/mL IFNγ). Bars represent mean + SEM from n=3 independent experiments performed in triplicates.
Control
MediumR406
R406-PLGA
PLGA
0
20
40
60
80
100M0M1
% C
ell v
iabi
lity

81
Chapter 4
Src Kinase as a Potential Therapeutic Target in Non-alcoholic and
Alcoholic Steatohepatitis
Dhadhang Wahyu Kurniawan, Richell Booijink, Arun Kumar Jajoriya, Garima Dhawan, Divya Mishra, Dorenda Oosterhuis, Josepmaria Argemi, Gert Storm, Peter Olinga, Ramon Bataller,
Sujit Komar Mohanty, Durga Prasad Mishra, Jai Prakash, Ruchi Bansal
Published in: Clinical and Translational Discovery vol. 2 issue 1 March 2022, DOI: 10.1002/ctd2.18

82
Abstract
Non-alcoholic steatohepatitis (NASH) and alcohol-associated steatohepatitis (ASH) are the
major cause of liver-related mortality with limited therapeutic options available. In this study,
we investigated the role of Src tyrosine kinase in the pathogenesis of (N)ASH. We examined
Src kinase expression in livers from patients with NASH, ASH, cirrhosis and biliary atresia, and
from preclinical mouse models: methionine choline deficient (MCD)-diet-induced NASH,
Lieber-DeCarli ethanol (EtOH)-diet-induced ASH, carbon tetrachloride (CCl4)- and bile duct
ligation (BDL)-induced liver fibrosis. Functional studies, using Src kinase inhibitor KX2-391,
were performed in NASH and ASH mouse models, and in relevant in vitro models.
Transcriptomic analysis revealed increased Src kinase expression in human and mouse
diseased livers that positively correlated with disease progression. Src kinase inhibition
ameliorated lipid biosynthesis (steatosis); monocytes/macrophages infiltration and
inflammatory cytokines expression (inflammation); and hepatic stellate cells (HSCs) activation
and collagen expression (fibrosis) in MCD-diet and EtOH-diet liver disease mouse models. In
vitro Src inhibition attenuated FFA-induced hepatocytic lipid accumulation; LPS/IFNγ-induced
nitric-oxide release, and expression of several inflammatory signatures in macrophages and
TGFβ-induced HSCs activation, contractility and collagen expression. Moreover, Src inhibition
diminished gene expression of inflammatory markers in LPS/IFNγ-stimulated BMDMs and LPS-
stimulated PCLS, and reduced FA accumulation, macrophage activation and collagen
deposition in 3D human liver spheroids. Mechanistic studies further revealed that Src kinase
mediated the effects through FAK/PI3K/AKT pathway. Our results demonstrate a multicellular
and functional role of Src kinase highlighting Src kinase as a promising therapeutic target in
(non)alcoholic steatohepatitis.

83
4.1 Introduction
Non-alcoholic steatohepatitis (NASH) and alcohol-associated steatohepatitis (ASH) are the
major cause of cirrhosis and hepatocellular carcinoma (HCC) associated morbidity and
mortality worldwide [1-3]. Currently, NASH and ASH are the common indication for liver
transplantation [4, 5]. Both non-alcoholic fatty liver diseases (NAFLD) and alcoholic liver
diseases (ALD) encompass a spectrum of pathologies ranging from simple steatosis to fibrosis,
cirrhosis and HCC [6, 7]. Although NASH and ASH share common pathogenesis and many
histopathological features, they differ in etiology. NASH is characterized by an excessive fat
accumulation in the liver while ASH is associated with high alcohol consumption (>20-30g/day)
[8]. Recent studies have deciphered the involvement of diverse signaling pathways that drive
the pathophysiology of NAFLD and ALD [9, 10]. Though significant progress has been made
towards (N)ASH disease understanding at the cellular and molecular level, currently there is
no clinically approved therapy available. Considering the alarming prevalence and global
burden, cutting-edge focused research is needed to discover novel therapeutic targets for the
development of effective treatment approaches.
When defining the pathogenic drivers of (N)ASH, it has been conceptualized that both
parenchymal cells (e.g. hepatocytes) and non-parenchymal cells (e.g. macrophages and
hepatic stellate cells, HSCs) are severely affected [6, 7]. Hepatocyte injury and steatosis are
triggered by toxic alcoholic metabolites during ALD, and fatty acid and cholesterol biosynthesis
with dysregulated lipid metabolism and transport during NAFLD. This hepatocyte injury is
accompanied by endoplasmic reticulum stress and initiates chronic inflammation through
reactive oxygen species and release of pro-inflammatory cytokines and chemokines [6, 7].
Furthermore, ASH and NASH are associated with altered intestinal permeability resulting in
increased intrahepatic levels of endotoxins further contributing to inflammation [11-13]. Due
to the ongoing inflammation, inflammatory mediators are released by the immune cells
mainly hepatic macrophages (resident Kupffer cells and infiltrating monocytes-derived
macrophages), leading to the trans-differentiation of quiescent HSCs into proliferative,
contractile and activated HSCs (myofibroblast phenotype) that contribute to fibrosis and
cirrhosis, and genomic instability predisposes to HCC [14].

84
Src is a non-receptor tyrosine kinase that belongs to the Src family of protein tyrosine kinases
(SFKs) that includes nine members: Src, Yes, Fyn, Fgr, Lck, Hck, Blk, Lyn and Yrk. While Src, Fyn
and Yes are ubiquitously expressed, others (except Yrk, exclusively expressed in chickens) are
restricted to hematopoietic cells [15, 16]. SFKs regulate many fundamental cellular processes
including cell growth, differentiation, migration, survival, and are also identified as proto-
oncogenes involved in the cancer progression [15, 16]. Moreover, SFKs play an essential role
in the recruitment and activation of monocytes, macrophages, neutrophils and other immune
cells [17]. Besides, Src and its family members have been implicated in several intracellular
signaling pathways in macrophages mediated by integrins and toll-like receptors [17, 18]. SFKs
play an important role in many pathologies including lung fibrosis, renal fibrosis, liver fibrosis
and systemic sclerosis [19-22]. These studies have reported TGFβ-induced activation of SFKs
mainly Src and Fyn, and the blockade of Src and Fyn is associated with reduced ECM
production and inhibited tissue fibrosis [20-22]. Particularly in context to liver fibrosis, Fyn
inhibitor saracatinib was shown to attenuate liver fibrosis via HSCs inhibition mediated via
focal adhesion kinase (FAK)/neural Wiskott-Aldrich syndrome protein (N-WASP) signaling [19].
Using the same inhibitor (saracatinib), Seo et al., recently demonstrated that Src inhibition
attenuated liver fibrosis, by preventing HSCs activation and suppressing hepatocytic
connective tissue growth factor (CTGF) expression, associated with SMAD3 downregulation
[23]. TGFβ1/Src axis is also involved in hepatocyte de-differentiation regulated by hepatocyte
nuclear factor 4 alpha (HNF4A) in alcoholic hepatitis [24]. SFKs interacts closely with Syk-ZAP70
family kinases with Src-family being ‘upstream’ hence regulating Syk-ZAP70 family, and the
FAK/proline-rich tyrosine kinase 2 (pyk2) involved in integrin signaling [25]. Several studies,
including ours, have reported the implication of Syk in liver diseases [26-28]. Considering the
multicellular functions and regulation of multiple signaling pathways by Src, we hypothesized
Src may be a master regulator involved in the pathophysiology of ALD and NAFLD.
In this study, we investigated the role of Src kinase in NASH and ASH. We first assessed the
expression and activation of Src in human liver diseases from different etiologies i.e. NASH,
alcoholic steatohepatitis (ASH), Hepatitis C virus (HCV)-cirrhosis and biliary atresia (BA); and
in respective disease mouse models that mimic pathophysiological features of human NASH
(methionine choline deficient MCD-diet induced [29]), ASH (chronic Lieber-DeCarli ethanol
EtOH-diet induced model [30]), and carbon tetrachloride (CCl4)- and bile duct ligation (BDL)-

85
induced liver fibrosis. Thereafter, we assessed the functional role of Src kinase using a
selective inhibitor KX2-391. Functional inhibition of Src kinase pathway was studied in vitro in
cell culture, PCLS and 3D human liver spheroids, and in vivo in MCD-diet-induced NASH and
EtOH-diet-induced ASH mouse models. We further examined the mechanisms involved in Src
kinase-mediated effects. To our best knowledge, this is the first study demonstrating the role
of Src kinase in (N)ASH.
4.2 Materials and methods
4.2.1 Gene expression analysis of the human liver tissues
Src kinase mRNA expression was assessed using the publicly available transcriptome datasets
of liver tissues from patients with non-alcoholic steatohepatitis (GSE48452) [31], Hepatitis C
virus (HCV)-related cirrhosis (GSE6764) [32] and biliary atresia (GSE15235) [33] obtained from
the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus database
(GEO) and analyzed using GEO2R online database. NASH patients were categorized into 4
groups as per biopsy-based pathological investigation: control group (n=36); NAFLD activity
score (NAS) 1-2 (n=15); NAS 3-4 (n=7); and NAS 5-6 (n=15); HCV-cirrhosis patients were
categorized into 2 groups: control group (n=10) and cirrhosis group (n=9); biliary atresia
patients were divided into 3 groups: control group (n=4); biliary atresia with liver inflammation
(n=17) and biliary atresia with fibrosis (n=26).
High-throughput transcriptome profiling was performed using Illumina HiSeq2000 platform
(San Diego, CA) with liver samples provided by the InTeam Consortium (Human Biorepository
Core, University of Pittsburgh, PA) and deposited in the Database of Genotypes and
Phenotypes of the National Institutes of Health (dbGaP Study Accession: phs001807.v1.p1)
[24]. Src kinase mRNA expression was evaluated in patients with different phenotypes: 11
patients with non-severe alcoholic hepatitis (with Maddrey’s discriminant function, MDF ≤32);
ASH responders (with severe alcoholic steatohepatitis (MDF>32) patients responsive to
steroids, n=9) ASH non-responders (severe alcoholic steatohepatitis (MDF>32) patients non-
responsive to steroids therapy, n=9) and 10 patients with explants who underwent early liver
transplantation for severe alcoholic steatohepatitis. Controls (n=10) were non-diseased livers
obtained from biopsied single nodules, without significant alterations as per histopathological
analysis.

86
4.2.2 Human liver specimens
Human liver specimens were obtained from the autopsy of patients with NASH (n=5)
anonymously provided by the Laboratory Pathology Netherlands (LabPON). Normal liver
tissue (n=4) was collected from patients receiving hepatic resections for non-tumoral diseases,
including hepatic adenoma and focal nodular hyperplasia. Upon Institutional Review Board
approval and after written informed consent from patients, the tissue specimens were
collected. The use of human tissues was approved by the Local Medical Ethics Committee, and
the experimental protocols were performed in accordance with institutional guidelines and
regulations.
4.2.3 CCl4-induced chronic liver fibrosis mouse model
These animal experiments were performed in strict accordance with the guidelines and
regulations for the Care and Use of Laboratory Animals, Utrecht University, The Netherlands.
The protocols were approved by the Institutional Animal Ethics Committee of the University
of Twente, The Netherlands. All the animals received ad libitum normal chow diet and normal
water, and were housed with 12h-light/12h-dark cycle. 8-week old male C57BL/6 mice (Janvier
Labs, Le Genest, St. Isle, France) were treated with multiple intraperitoneal injections (twice
per week) of olive oil (Sigma, St. Louis, MO, USA) or CCl4 (Sigma, 1mL/kg, diluted in olive oil in
1 CCl4:5 olive oil) for 8 weeks (n=9 per group). At the end of the experiment, all mice were
sacrificed and livers were harvested for the subsequent analysis.
4.2.4 Bile-duct ligation mouse model
These animal experiments were performed in strict accordance with the guidelines and
regulations for the Care and Use of Laboratory Animals, Utrecht University, The Netherlands.
The protocols were approved by the Institutional Animal Ethics Committee of the University
of Twente, The Netherlands. All the animals received ad libitum normal chow diet and normal
water, and were housed with 12h-light/12h-dark cycle. 8-week old male BALB/c mice (Janvier
Labs) were anesthetized with Isoflurane (2%). After midline laparotomy, the bile duct was
ligated with 6-0 silk suture. The abdomen was then washed with sterile saline and closed in
two layers with 4-0 vicryl suture. Mice were sacrificed after 48 h (n=3) and 28 days (n=3)
postoperatively and livers were harvested for the subsequent analysis. Control (sham-
operated) mice (n=6) were sacrificed and livers were harvested for the subsequent analysis.

87
4.2.5 Methionine and choline-deficient (MCD) diet model
These animal experiments were performed in strict accordance to the guidelines and ethical
regulations for the Care and Use of Laboratory Animals, CSIR-Central Drug Research Institute
(CDRI), India. The experimental procedures conducted in this study have been approved by
the Institutional Animal Ethics Committee at the CSIR-Central Drug Research Institute, India.
MCD model was performed as reported previously [27]. 8-week old male C57BL/6 mice
(Jackson Laboratories, Bar Harbor, USA) were fed a MCD diet (C1070, Altromin GmbH, Lage,
Germany) for 4 weeks while the controls received normal chow diet (n=8). MCD diet fed mice
were treated for two weeks intravenously (three times per week) with vehicle (PBS, phosphate
buffered saline, n=8) or KX2-391 (10 mg/kg, n=5) while continuing MCD diet feeding. At the
end of the experiment, mice were euthanized, the liver tissues and blood were collected for
the subsequent analysis [27]. Alanine aminotransferase (ALT), aspartate aminotransferase
(AST), total plasma cholesterol, and total plasma triglycerides were measured using standard
automated laboratory methods.
4.2.6 Lieber-DeCarli chronic ethanol feeding model
These animal experiments were performed in strict accordance with the guidelines and
regulations for the Care and Use of Laboratory Animals, Utrecht University, The Netherlands.
The protocols were approved by the Institutional Animal Ethics Committee of the University
of Twente, The Netherlands. 10-12-week old female C57BL/6 mice were fed a Lieber-DeCarli
diet (F1258SP, Bio-Serv, Frenchtown, NJ, USA) with 3% ethanol (Thermo Fisher Scientific,
Rockford, IL, USA) for 8 weeks while control mice (n=5) were pair-fed with isocaloric control
liquid diet (F1259SP, Bio-Serv) [30]. 3% ethanol-containing Lieber-DeCarli diet fed mice (EtOH
mice) were treated for three weeks intravenously (twice per week) with vehicle (PBS, n=6) or
KX2-391 (10 mg/kg, n=5) whilst continuing alcoholic diet. Mice were euthanized, blood and
liver tissues were collected for analysis at the end of the experiment. Alanine
aminotransferase (ALT), aspartate aminotransferase (AST), total plasma cholesterol and total
plasma triglycerides were measured using standard automated laboratory methods.
4.2.7 Cell Lines
Human hepatic stellate cells (LX2 cells), provided by Prof. Scott Friedman (Mount Sinai
Hospital, New York, NY, USA), were cultured in DMEM-Glutamax medium (Invitrogen,

88
Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS, Lonza, Verviers, Belgium),
and antibiotics (50 U/mL Penicillin and 50µg/mL streptomycin, Sigma). Human hepatocytes
(HepG2) (ATCC, Manassas, VA, USA) and mouse NIH3T3 fibroblasts (ATCC) were cultured in
Dulbecco's modified Eagle's (DMEM) medium (Lonza) supplemented with 2 mM L-glutamine
(Sigma), 10% FBS (Lonza) and antibiotics [50U/mL Penicillin (Sigma) and 50 µg/mL
streptomycin (Sigma)]. Mouse RAW264.7 macrophages (ATCC) and human THP1 monocytes
(ATCC) were cultured in FBS, L-glutamine and antibiotics supplemented Roswell Park
Memorial Institute (RPMI) 1640 medium (Lonza). Human umbilical vein endothelial cells
(HUVECs) were purchased from Lonza and cultured in endothelial basal medium (EBM; Lonza)
supplemented with supplements (hydrocortisone, bovine brain extract, epidermal growth
factor, gentamycin sulphate, amphotericin-B) and 10% fetal bovine serum (FBS, Lonza).
4.2.8 Bone marrow derived macrophages
Bone marrow-derived macrophages (BMDMs) were freshly isolated from C57BL/6 mice as
described previously [27, 34]. Briefly, femurs and tibias were flushed with Dulbecco's modified
Eagle's medium (DMEM) (Life Technologies, Carlsbad, CA, USA) with 10% FBS (Lonza) to collect
bone marrow. Cells were then triturated 3–5 times through an 18-gauge needle and
centrifuged at 1200 rpm for 5 min. After removing supernatant, RBCs were lysed with the lysis
buffer and the remaining cells were washed in DMEM + 10% FBS and plated at 1×106 cells/mL
in DMEM supplemented with 1% penicillin/streptomycin, 1% HEPES, 0.001% β-
mercaptoethanol, 10% FBS, and 20% supernatant from mouse 3T3 fibroblasts [obtained from
ATCC and cultured in cultured in (DMEM) medium supplemented with 2 mM L-glutamine
(Sigma), 10% FBS (Lonza) and antibiotics (Sigma)]. The fibroblasts conditioned medium is
required to promote differentiation of bone marrow cells into macrophages (7–10 days).
Media was changed on days 2, 4 and 6, and cells were re-plated on day 7 for performing
experiments.
4.2.9 Effects of Src inhibitor KX2-391 on human LX2 cells
KX2-391, a highly selective and potent Src kinase inhibitor used in this study has been
purchased from Selleckchem (Houston, TX, USA). Cells were seeded in 24 well plates (5x104
cells/well) or 12 well plates (1x105 cells/well) and cultured overnight. To study the effects, cells
were starved overnight with serum-free medium and incubated with starvation medium
alone, 5 ng/mL of human recombinant TGFβ1 (Roche, Mannheim, Germany) with and without

89
50µM KX2-391 for 24h. Cells (24-well plates) were then fixed with chilled acetone : methanol
(1:1), dried and stained for different markers (collagen-I antibodies are summarized in
supplementary table 1). In addition, cells (12 well plates) were lysed with RNA lysis buffer to
perform quantitative real-time PCR analyses. Experiments were performed as three
independent experiments.
4.2.10 3D collagen-I gel contraction assay
3D collagen-I gel contraction assay has been performed as per standardized protocol. Briefly,
collagen suspension (5.0 mL) containing 3.0 mL Collagen G1 (5 mg/mL, Matrix biosciences,
Mörlenbach, Germany), 0.5 mL 10x M199 medium, 85µl 1N NaOH (Sigma) and sterile water
was prepared, and mixed with 1.0 mL (2×106) LX2 cells. Collagen gel-cells suspension (0.6
mL/well) was plated in a 24-well culture plates and allowed to polymerize for 1h at 37°C.
Polymerized gel was then incubated with 1 mL of serum-free medium with or without TGFβ
(5 ng/mL) together with 50µM KX2-391 followed by detachment of the gels from the culture
wells. Photographs were made with a digital camera at 24 h. The size of the gels was digitally
measured and normalized with their respective well size in each image. Gel contraction
experiments were performed in duplicates as three independent experiments.
4.2.11 Effects of Src inhibitor KX2-391 on Nitric Oxide (NO) release
The effect of KX2-391 on pro-inflammatory macrophages was assessed by measuring the
inhibition in nitrite (NO2) release. Nitrite was measured as a surrogate marker of NO
production. RAW264.7 cells (2 × 105 cells/200μl/well) were seeded in 96-well plates, cultured
with IFNγ (10 ng/mL) and LPS (100 ng/mL) together with different concentrations of Src
inhibitor KX2-391 (1, 5 and 50 μM). After 24 h, 100 μL culture supernatant was added to 100
μL of Griess reagent (1% sulfanilamide; 0.1% naphthylethylendiamine dihydrochloride; 3%
phosphoric acid) and absorbance at 540 nm was measured using microplate reader.
4.2.12 Effects of Src inhibitor KX2-391 on RAW264.7 and THP1 macrophages
Macrophages were plated in 12 well plates (1x105 cells/well) and cultured overnight [with 50
ng/mL phorbol-12-myristate-13-acetate (PMA) for THP1 macrophages for differentiation] at
37°C/5% CO2. To assess the effects, cells were incubated with medium alone, M2 stimulus (10
ng/mL IL-4 and IL-13) or M1 stimulus (10 ng/mL of IFNγ and 10 ng/mL LPS) with and/or without
KX2-391 (1, 5 and 50 μM) for 24h. Cells were lysed with RNA lysis buffer to perform

90
quantitative real-time PCR analyses or with protein lysis buffer for western blot analyses. All
the experiments were performed as three independent experiments.
4.2.13 In vitro efficacy studies of Src inhibitor KX2-391 in freshly isolated bone marrow derived macrophages
Bone marrow derived macrophages were plated at a density of 1 x 106 cells/mL and
differentiated into M1 macrophages using LPS (100 ng/mL, Sigma) and IFNγ (10 ng/mL,
Peprotech) or M2 macrophages using IL-4 (10 ng/mL, Peprotech) and IL-13 (10 ng/mL,
Peprotech). For efficacy studies, the cells were incubated with medium alone, 1, 10, and 50
µM of KX2-391 together with M1 stimulus. Incubated for 24 h, then cells were lysed for RNA
isolation and quantitative real-time PCR. At least 3 independent experiments were performed
for these efficacy studies.
4.2.14 Efficacy studies on murine precision-cut liver slices (PCLS)
Murine precision-cut liver slices (PCLS) were prepared as described previously [35]. We used
murine liver cores were obtained using a 5-mm biopsy-punch. Subsequently, the slices were
made using a Krumdieck tissue slicer (Alabama Research and Development, USA), was filled
with ice-cold Krebs-Henseleit buffer (KHB) supplemented with 25 mM D-glucose (Merck,
Darmstadt, Germany), 25 mM NaHCO3 (Merck), 10 mM HEPES (MP Biomedicals, Aurora, OH),
saturated with carbogen (95% O2/5% CO2) and adjusted to pH 7.4. A wet weight of PCLS
approximately 3 mg, have an estimated thickness of 300-400 µm. To avoid rapid loss of
viability after slicing, PCLS were directly transferred to ice-cold University of Wisconsin organ
preservation solution (UW-solution). After slicing, PCLS were cultured in 12-well plates in
William’s medium E + GlutaMAX (Gibco, New York, USA) supplemented with 14 mM Glucose
(Merck, Darmstadt, Germany) and 50 µg/mL gentamycin (Gibco). The slices were incubated
for 24 h at 37°C in an 80% O2/5% CO2 atmosphere. The culture plates were horizontally
shaken at 90 rpm (amplitude 2cm). For these experiments, PCLS were incubated with medium
alone, M1 stimulus (100 ng/mL LPS + 10 ng/mL IFNγ) with and without KX2-391 (25 µM). The
viability of the slices was assessed by measuring the adenosine triphosphate (ATP) content
using the ATP bioluminescence kit (Roche Diagnostics, Mannheim, Germany). Determined ATP
values (pmol) were normalized to the total amount of protein (µg) estimated by the Lowry
method (Bio-rad RC DC Protein Assay, Bio-Rad, Veenendaal, The Netherlands). Values
displayed are relative values compared to the related controls.

91
4.2.15 In vitro lipid biogenesis in human hepatocytes (HepG2 cells)
HepG2 cells were treated with palmitate (0.3 mM) in complete cell culture medium with or
without 50 μM KX2-391 for 48 h. At the end of the experiment, cells were washed twice with
PBS followed by fixation and staining using with oil-red-O staining kit (Sigma) as per
manufacturer’s instructions.
4.2.16 In vitro Human 3D spheroid NASH model
To prepare the human 3D spheroids, human hepatocytes HepG2 (80%), human monocytes
THP1 (10%), human hepatic stellate cells LX2 (5%) and human endothelial cells HUVECs (5%)
were mixed using complete medium containing 10% FBS as described elsewhere [36].
Spheroids containing 1,200 total cells were plated in ultra-low adhesion U bottomed plates
(ULA) (1,200 cells/100 μL) followed by addition of 100 μL of medium. After 7 days of culturing,
3D spheroids were incubated with 10 μg/mL LPS with and without KX2-391 (50 μM). After 7
days of culturing, images of the 3D spheroids were captured using light microscope and
spheroids were embedded in Tissue-Tek optimum-cutting temperature (O.C.T) embedding
medium (Thermo fisher Scientific), fixed and snap-frozen in 2-methyl butane (Sigma) chilled
on a dry ice. The spheroids were cryo-sectioned into 4 μm cryosections and stained as
described below.
4.2.17 RNA extraction, reverse transcription and quantitative real time PCR
Total RNA from cells and liver tissues was isolated using GenElute Total RNA Miniprep Kit
(Sigma) and SV total RNA isolation system (Promega Corporation, Fitchburg, WI, USA)
respectively according to manufacturer’s instructions. The RNA concentration was
quantitated by a UV spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA).
Total RNA (500 ng – 1 μg) was reverse-transcribed using iScript cDNA Synthesis Kit (Bio-Rad,
Hercules, CA, USA). All the primers were purchased from Sigma-Genosys (Haverhill, UK). Real-
time PCR was performed using 2x SensiMix SYBR and Fluorescein Kit (Bioline, QT615-05,
Luckenwalde, Germany), 20 ng cDNA and pre-tested gene-specific primer sets (listed in Table
S1 and S2). The cycling conditions for the BioRad CFX384 Real-Time PCR detection system
were 95°C for 10 min, 40 cycles of 95°C/15 sec, 58°C/15 sec and 72°C/15 sec. Finally, cycle
threshold (Ct) values were normalized to reference gene GAPDH and fold changes in
expression were calculated using the 2-ΔΔCt method.

92
4.2.18 Immunohistochemistry
Liver tissues were harvested and transferred to Tissue-Tek optimum-cutting temperature
(O.C.T.) embedding medium (Sakura Finetek, Torrance, CA, USA), and snap-frozen in 2-methyl
butane chilled on a dry ice. Cryosections (4 μm) were cut using a Leica CM 3050 cryostat (Leica
Microsystems, Nussloch, Germany). The sections were air-dried and fixed with acetone for 10
min. Cells or tissue sections were rehydrated with PBS and incubated with the primary
antibody (refer to Table S3) for 1 h at room temperature. Cells or sections were then incubated
with horseradish peroxidase (HRP)-conjugated/fluorescent secondary antibody for 1 h at
room temperature. Then incubated with HRP-conjugated/fluorescent tertiary antibody for 1
h at room temperature. Thereafter, peroxidase activity (where HRP-conjugated antibodies
were used) was developed using AEC (3-amino-9-ethyl carbazole) substrate kit (Life
Technologies, Carlsbad, CA, USA) for 20 min and nuclei were counterstained with hematoxylin
(Sigma). In case of fluorescent stainings, nuclei were stained with DAPI (4ʹ,6-diamidino-2-
phenylindole). For liver sections, endogenous peroxidase activity was blocked by 3% H2O2
prepared in methanol. Cells or sections were mounted with Aquatex mounting medium
(Merck, Darmstadt, Germany) or anti-fade DAPI-containing mounting medium (Merck). The
staining was visualized and the images were captured using Nikon eclipse E600 microscope
(Nikon, Tokyo, Japan).
4.2.18.1 Oil-Red-O staining
Oil-Red-O stock solution was prepared by dissolving 0.3 g Oil-Red-O (Sigma) in 10 mL
isopropanol. The sections were fixed in 4% formalin for 20 min and were then stained in Oil-
Red-O solution as per manufacturer’s instructions. Briefly, formalin-fixed sections were rinsed
with 60% isopropanol followed by staining with freshly prepared Oil-Red-O working solution
for 15 min. Thereafter, the sections were rinsed with 60% isopropanol and nuclei were
counterstained with hematoxylin (Fluka Chemie). Eventually, the sections were rinsed with
tap water and were mounted with aquatex mounting medium (Merck).
4.2.18.2 Hematoxylin and Eosin staining
The sections were fixed with 4% formalin for 20 min and then rinsed with distilled water. The
sections were incubated with hematoxylin for 15 min followed by washing with tap water.
Thereafter, the sections were incubated with eosin solution for 1.5 min followed by wash in

93
96% ethanol, dehydration with ethanol and were mounted with VectaMount mounting
medium (Vector Laboratories, Burlingame, CA).
4.2.18.3 Quantitative histological analysis
For quantitation, sections were scanned using Hamamatsu NanoZoomer Digital slide scanner
2.0HT (Hamamatsu Photonics, Bridgewater, NJ, USA) for quantitative histological analysis.
High resolution scans were viewed using NanoZoomer Digital Pathology (NDP2.0) viewer
software (Hamamatsu Photonics). About 20 images (100x) of each entire section (from NDP)
were imported into ImageJ and were analyzed quantitatively at a fixed threshold. All the
primary antibodies used in this study have been pre-tested for specificity. The staining’s
performed in the study included the negative control (without primary antibody and isotype
control antibodies) to confirm the specificity of the staining and showed no non-specific
staining.
4.2.19 Western blot analysis
Cells were lysed using standard western blot lysis buffer (Life Technologies) and the prepared
cell lysates were stored at -80°C until use. The samples were boiled and subjected to SDS-
PAGE with 10% Tris-glycine gels (Life Technologies) followed by protein transfer onto PVDF
membrane. The membranes were developed according to the standard protocols using
primary and secondary antibodies as mentioned in Table S3. The bands were visualized using
ECL detection reagent (Perkin Elmer Inc., Waltham, MA) and photographed using FluorChem
M Imaging System (ProteinSimple, Alpha Innotech, San Leandro CA). Intensity of individual
bands was quantified using ImageJ densitometry software, and expressed in relative %.
4.2.20 Statistical analyses
All the graphs and heat maps were made using GraphPad Prism version 8.4.1 (GraphPad Prism,
La Jolla, CA, USA). All the results are expressed as the mean + standard error of the mean
(SEM). Statistical analyses were performed using GraphPad Prism version 9.1.2 (GraphPad
Prism, La Jolla, CA, USA). Multiple comparisons between different groups were calculated
using the one-way analysis of variance (ANOVA) with the Bonferroni post hoc test. Statistical
differences between two groups were calculated using a two-tailed unpaired t test.
Differences were considered significant when #p < 0.05, ##p < 0.01, ###p < 0.001 or *p < 0.05,
**p < 0.01, ***p < 0.001.

94
4.3 Results
4.3.1 Upregulation of Src expression is associated with liver disease severity
We first analyzed Src gene expression in different etiological liver diseases using the publicly
available database. In NASH patients, we found a highly significant induction of Src gene
expression that correlated with the increasing NAS (NAFLD activity score) compared to healthy
control livers (Figure 1A). Likewise in ASH patients, we found a significant correlation between
ASH severity with increasing Src gene expression from ASH non-severe, ASH responders
(responsive to steroids), ASH non-responders (non-responsive to steroids) and ASH explants
(with liver transplantation) (Figure 1A). Moreover, Src gene expression was upregulated in
liver tissues obtained from patients with other etiologies i.e. HCV-induced cirrhosis, and
infants with BA-induced liver inflammation and fibrosis (Figure 1A). Consistent with the
human data, Src gene expression was significantly increased in MCD diet-induced NASH,
Lieber-DeCarli EtOH diet-induced ASH, CCl4- and BDL-induced liver fibrosis mouse models
(Figure 1B).
We further examined the protein expression of Src in human and mouse livers by
immunostainings. We found that Src expression was increased in human NASH livers
compared to normal livers (Figure 1C). Furthermore, Src expression was higher in the livers
from MCD-induced NASH, Lieber-DeCarli EtOH-diet induced ASH and CCl4-induced fibrosis
mouse model compared to control livers (Figure 1D). Moreover, we observed an increased
nuclear localization of Src suggesting active Src kinase in the diseased livers as indicated by
arrows (Figure 1C-D). Histological analysis revealed significant liver damage in human NASH
liver tissues, and MCD-diet and EtOH-diet induced mouse models (hematoxylin and eosin;
H&E), while CCl4-induced liver fibrosis was confirmed using collagen I immunostainings when
compared to control livers (Figure 1C-D).

95
Figure 1. Upregulation of Src expression in human and mouse liver tissues. (A) Intrahepatic Src gene expression (from publicly available datasets) in NASH patients characterized by NAFLD activity score (NAS): NAS1-2 (n=15), NAS3-4 (n=7), NAS5-6 (n=15) compared to healthy controls (n=36); ASH patients with a spectrum of ALD: non-severe ASH (n=11), ASH responders (responsive to steroids, n=9), ASH non-responders (non-responsive to steroids, n=9) and ASH explants (with liver explants, n=10) compared to control livers (n=10); HCV-cirrhotic patients (n=9) compared to healthy controls (n=10); Biliary atresia (BA) patients with BA inflammation (n=17); BA fibrosis (n=26) compared to healthy controls (n=4). (B) Relative Src gene expression in the MCD-diet-fed (n=8) compared to control chow-diet fed mice (n=8); EtOH-diet-fed (n=11) compared to isocaloric liquid diet-fed mice (n=11); CCl4 (n=9) compared to control (olive oil) mice (n=9); and BDL 48hrs (n=3) and 28days (n=3) post-ligation compared to control (sham-operated) mice (n=6); (C) Representative images (scale=100μm) of Src-

96
and H&E-stained liver tissue sections from human NASH (n=5) compared to healthy controls (n=4). (D) Representative images (scale=100μm) of Src-, H&E- and collagen-I-stained liver sections from MCD-diet-fed NASH (n=8), EtOH-diet-fed ASH (n=8) and CCl4-induced liver fibrosis (n=8) mouse models compared to respective controls (n=8). Inserts are magnified images with arrows depicting Src nuclear staining. Mean + SEM, two-tailed Student’s t-test or one-way ANOVA with Bonferroni post-hoc test for comparisons between 2 or more than 2 groups respectively, *p<0.05, **p<0.01, ***p<0.001.
4.3.2 Src kinase inhibition ameliorated liver steatosis, inflammation and fibrosis in MCD-diet induced NASH mouse model
To understand the implication of Src kinase pathway in NASH, we tested the highly selective
and potent Src kinase inhibitor KX2-391 that binds specifically to the substrate binding site of
Src kinase. We investigated the therapeutic efficacy of KX2-391 in MCD-diet-induced NASH in
mice as per the regimen depicted in Figure 2A. Macroscopic visualization of liver tissues
demonstrated the visible pale and gross appearance of the liver suggesting the liver damage
in MCD-diet-fed NASH mice compared to control (chow-diet-fed) mice (Figure 2B). Treatment
with KX2-391 showed a noticeably improved appearance of livers compared to untreated
NASH livers (Figure 2B).
Steatosis, inflammation, and fibrosis are the hallmarks of NASH. We assessed the gene
expression of several NASH-related genes i.e. steatosis-related genes: lipogenesis-related
genes i.e. stearoyl CoA desaturase-1 (SCD1) that catalyzes the biosynthesis of
monounsaturated fatty acids [37], and diacylglycerol acyl transferase 2 (DGAT2) that catalyzes
the first step in triglyceride (TG) synthesis [38], endoplasmic reticulum (ER) stress and
unfolded protein response (ER/UPR) induced genes i.e. activating transcription factor 6 (ATF6)
and X-box-binding protein 1 (XBP1) [39], and fatty acid biosynthesis regulating protein i.e.
sterol regulatory element-binding protein 1c (SREBP1c) [40]; inflammation-related genes:
tumor necrosis factor alpha (TNFα), interleukin-1-beta (IL-1β), inducible nitric-oxide synthase
(iNOS), C-C motif chemokine ligand 2 (CCL2), interleukin-6 (IL-6); and fibrosis-related genes:
HSCs markers i.e. Desmin and alpha-smooth muscle actin (α-SMA). These genes were found
to be significantly upregulated in MCD-diet fed mice compared to controls, and were reduced
following treatment with KX2-391 (Figure 2C and Figure S1).
Intriguingly, KX2-391 significantly ameliorated liver damage as confirmed by H&E staining.
KX2-391 inhibited liver steatosis, inflammation, and fibrosis as assessed by Oil-red-O, F4/80,
and collagen-I (immuno)stainings respectively (Figure 2D-E).

97

98
Figure 2. Src inhibition ameliorated steatosis, inflammation and fibrosis in MCD-diet induced NASH in mice. (A) Schematic showing NASH induction and KX2-391 treatment regimen. (B) Representative macroscopic images of livers from control (chow) (n=8), MCD (n=8), and MCD+KX2-391 (n=5) group. (C) Heatmap showing relative intrahepatic gene expression (normalized with 18s rRNA) for steatosis (SCD1, DGAT2, XBP1, SREBP1 and ATF6); inflammation (TNFα, IL-1β, iNOS, CCL2 and IL-6) and fibrosis (Desmin and α-SMA) markers in different groups (Refer to Figure S1). *p<0.05, **p<0.01 denotes significance versus control group and #p<0.05, ##p<0.01 denotes significance versus MCD group. (D) Representative images (scale=100μm) and (E) quantitative analysis of liver sections stained with H&E, oil-red-O, F4/80 and collagen-I, and total liver collagen content (Hydroxyproline). (E) Plasma AST, ALT, triglycerides, and cholesterol levels. Mean + SEM, one-way ANOVA with Bonferroni post-hoc test, *p<0.05, **p<0.01, ***p<0.001.
Moreover, we found that KX2-391 significantly inhibited total collagen as assessed by
hydroxyproline assay, and AST, ALT, total triglycerides, and total cholesterol plasma levels
(Figure 2E-F). These results suggest Src kinase inhibition as a prospective approach to treat
NASH.
4.3.3 Src kinase inhibition attenuated liver steatosis, inflammation and fibrosis in EtOH-diet induced ASH mouse model
We further investigated the therapeutic efficacy of KX2-391 in chronic ethanol feeding in mice
as described elsewhere [30] and depicted in Figure 3A. Macroscopic visualization of liver
tissues at the end of the experiment morphologically showed mild pale appearance suggesting
the hepatocellular damage in EtOH-diet fed mice compared to isocaloric-diet fed control mice
(Figure 3B). Appearance of liver tissue after treatment with KX2-391 was improved when
compared to livers derived from EtOH-diet fed mice (Figure 3B).
We also assessed the gene expression of steatosis-related markers: SCD1, DGAT2, ATF6 and
XBP1; inflammation-related markers: IL-1β, iNOS, TNFα, Arg1, and IL-6; and fibrosis-related
markers: Collagen I, Desmin and α-SMA. KX2-391 reduced the EtOH-induced expression of
SCD1, DGAT2, ATF6 and XBP1 significantly (Figure 3C and Figure S2). Furthermore,
inflammation markers (IL-1β, iNOS, TNFα and IL-6) upregulated during EtOH-induced ASH
were inhibited by KX2-391 treatment (Figure 3C, Figure S2). Interestingly, KX2-391
upregulated the expression of Arginase-I (Arg1), as known as anti-inflammatory/restorative
macrophage marker (Figure 3C, Figure S2). We then analyzed the expression of fibrosis
markers in the liver tissues and observed that KX2-391 significantly reduced collagen I, desmin
and α-SMA expression as shown in Figure 3C and Figure S2.

99

100
Figure 3. Src inhibition attenuated steatosis, inflammation and fibrosis in EtOH-diet induced ASH in mice. (A) Schematic showing ASH induction and KX2-391 treatment regimen. (B) Representative macroscopic images of livers from control (n=6), EtOH (n=6), and EtOH+KX2-391 (n=6) group. (C) Heatmap showing relative intrahepatic gene expression (normalized with 18s rRNA) for steatosis (SCD1, DGAT2, ATF6 and XBP1); inflammatory (IL-1β, iNOS, TNFα, IL-6 and Arg1) and fibrosis (Collagen I, Desmin and α-SMA) markers in different groups (Refer to Figure S2). *p<0.05, **p<0.01 denotes significance versus control group and #p<0.05, ##p<0.01 denotes significance versus EtOH group. (D) Representative images (scale=100μm) and (E) quantitative analysis of liver sections stained with H&E, oil-red-O, F4/80 and collagen-I, and total liver collagen content (Hydroxyproline). (F) Plasma AST, ALT, triglycerides, and cholesterol levels. Mean + SEM, one-way ANOVA with Bonferroni post-hoc test, *p<0.05, **p<0.01, ***p<0.001.
We further observed increased liver damage as evidenced by H&E staining’s, steatosis (lipid
accumulation) as confirmed by Oil-Red-O staining’s, inflammation as assessed by F4/80
immuno-staining, and fibrosis as analyzed by collagen I immunostaining and total
hydroxyproline content in the EtOH-diet fed ASH mouse model (Figure 3D-E). KX2-391
treatment significantly inhibited liver steatosis, hepatic macrophage infiltration and
inflammation, and fibrogenesis (Figure 3D-E). We then analyzed plasma AST, ALT, triglycerides
and cholesterol levels and found that these parameters were upregulated in EtOH-fed mice
and strongly reduced following KX2-391 treatment (Figure 3F). These findings suggest that Src
inhibition ameliorates steatosis, inflammation and fibrosis associated with ASH.
4.3.4 Inhibition of Src diminished hepatocytic lipid accumulation and HSCs activation
To dissect the cellular effects of Src kinase activation in the liver during NASH and ALD, we
examined Src expression in different (non-activated) human cells i.e. THP1 (monocytes),
HUVECs (endothelial cells), HepG2 (hepatocytes) and LX2 cells (HSCs), and found increased Src
gene expression in hepatocytes and HSCs compared to THP1 and HUVECs (Figure 4A). To
confirm these in vitro results, we performed co-immunostainings of Src with hepatocyte
marker (EpCAM), macrophage marker (CD68) and fibrosis marker (Collagen I) in human NASH
livers. Indeed, we observed colocalization (indicated with arrows) of Src with these markers
further confirming the expression of Src in different liver cells (Figure S3). We then
investigated the implication of Src in hepatocytes and HSCs. We treated HepG2 cells with
palmitate (free fatty acid, FFA) to mimic lipid biogenesis and lipid storage. In accordance to
the in vivo data, Src inhibition in hepatocytes reduced hepatocytic lipid accumulation, as
assessed by oil-red-o staining, suggesting that Src activation directly regulates lipid
accumulation/metabolism in hepatocytes (Figure S4). In LX2 cells, we found that TGFβ-

101
induced collagen (gene and protein) expression and contractility was significantly attenuated
following treatment with KX2-391 (Figure 4B-E) corroborating with the anti-fibrotic effects of
KX2-391 observed in the mouse models.
Figure 4. Src inhibition decreased TGFβ-induced HSCs activation, contraction and collagen expression via FAK/PI3K/AKT pathway. (A) Relative Src gene expression (normalized with 18s rRNA) in THP1 monocytes, HUVECs, HepG2 and HSCs (n=3). (B) Collagen-I-stained control and TGFβ-activated LX2 cells ± 50μM KX2-391 (n=3) (C) Relative collagen-I and -III gene expression (normalized with 18s rRNA) in control and TGFβ-activated LX2 cells ± 50μM KX2-391 (n=3). (D) Quantitative analysis and (E) representative images showing contractility of control and TGFβ-activated LX2 cells ± 50μM KX2-391 (n=3). (F) Representative image showing western-blot analysis for phosphorylated Src, FAK, PI3K and AKT normalized with respective non-phosphorylated proteins in control and TGFβ-activated LX2 cells ± KX2-391 (n=3) (Refer to Figure S5). (G) % relative cell viability using Alamar blue assay on control and TGFβ-activated LX2 cells ± 50μM KX2-391 (n=3). Mean + SEM, one-way ANOVA with Bonferroni post-hoc test, *p<0.05, **p<0.01, ns=non-significant.

102
4.3.5 Src mediates HSCs activation via FAK/PI3K/AKT pathway
We performed western blot analysis to understand the underlying mechanism of anti-fibrotic
effects mediated by Src inhibitor in HSCs. We observed induction in the Src phosphorylation
upon TGFβ activation in HSCs which was inhibited by KX2-391 treatment (Figure 4F and Figure
S5). We further observed an increased phosphorylation of focal adhesion kinase (FAK),
phosphatidylinositol 3-kinase (PI3K) and serine/threonine kinase (AKT) after treatment with
TGFβ in HSCs. FAK/PI3K/AKT pathway plays an important role in the proliferation and
activation of HSCs, and fibrosis progression [41]. Furthermore, previously it has been
suggested that Src regulates FAK/PI3K/AKT pathway [42] as further confirmed in this study.
Here, we observed that Src inhibitor inhibited phosphorylation and hence activation of
FAK/PI3K/AKT (Figure 4F and Figure S5) suggesting Src-mediated regulation of FAK/PI3K/AKT
pathway in HSCs and liver fibrogenesis. Importantly, we observed about 15% inhibition in cell
viability reflected in the metabolic activity as assessed by Alamar blue assay (Figure 4G). The
inhibition in cell viability (or metabolic activity) might be attributed to inhibition of
FAK/PI3K/AKT pathway resulting in reduced HSCs proliferation and activation.
4.3.6 Src inhibition decreased expression of inflammatory markers in LPS- and IFNγ-stimulated pro-inflammatory macrophages and in precision-cut liver slices
Several studies have documented a significant increase in intrahepatic monocyte/macrophage
infiltration, phenotypic differentiation and activation in vitro, in vivo in experimental models,
and patients with (N)ASH [43]. To determine the implication of Src in macrophages, we first
assessed Src expression and Src pathway activation in non-activated, pro-inflammatory
(stimulated with LPS/IFNγ) and pro-resolving (stimulated with IL-4/IL-13) macrophages. In
RAW264.7 macrophages, we found significant induction of Src (gene and protein) expression
in LPS+IFNγ-stimulated pro-inflammatory macrophages compared to non-stimulated and
IL4+IL13-stimulated macrophages (Figure 5A-B). We also found an increased pSrc expression
in LPS+IFNγ-stimulated macrophages (Figure 5B). We found dose-dependent inhibition in cell
viability with 15% inhibition using highest concentration of KX2-391 (50µM) reflecting the
effect of KX2-391 on RAW264.7 macrophages metabolic activity following Src inhibition
(Figure 5C).
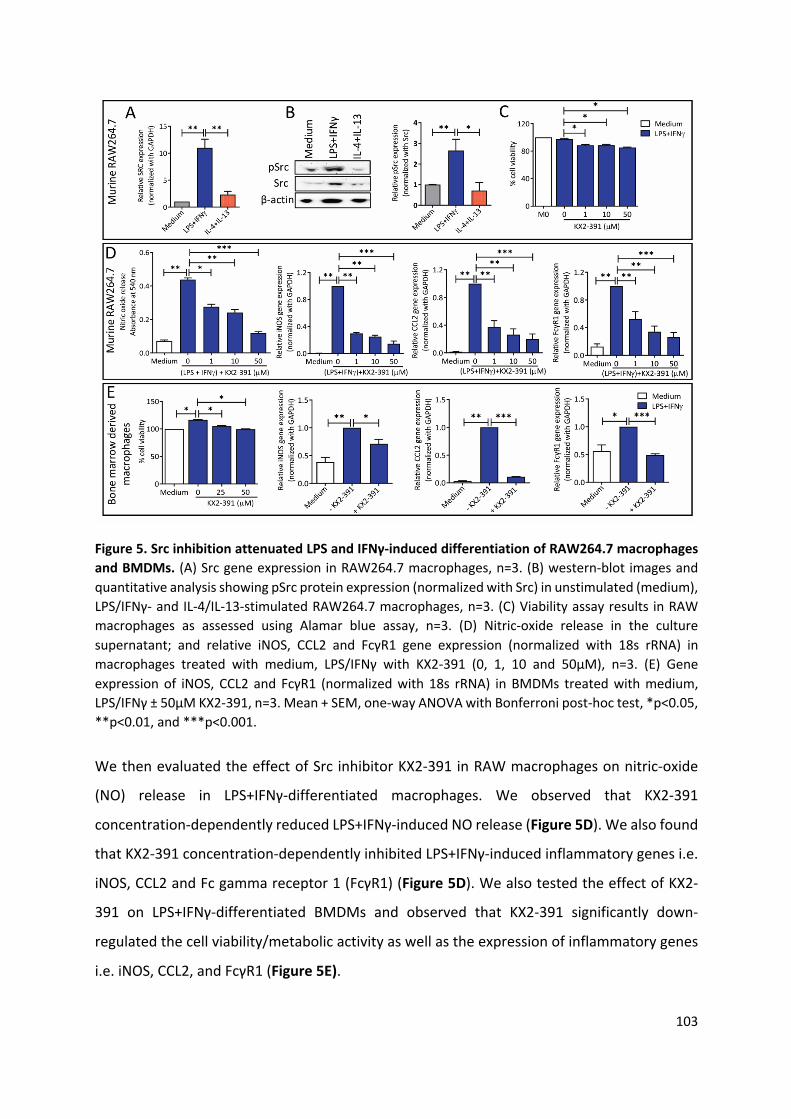
103
Figure 5. Src inhibition attenuated LPS and IFNγ-induced differentiation of RAW264.7 macrophages and BMDMs. (A) Src gene expression in RAW264.7 macrophages, n=3. (B) western-blot images and quantitative analysis showing pSrc protein expression (normalized with Src) in unstimulated (medium), LPS/IFNγ- and IL-4/IL-13-stimulated RAW264.7 macrophages, n=3. (C) Viability assay results in RAW macrophages as assessed using Alamar blue assay, n=3. (D) Nitric-oxide release in the culture supernatant; and relative iNOS, CCL2 and FcγR1 gene expression (normalized with 18s rRNA) in macrophages treated with medium, LPS/IFNγ with KX2-391 (0, 1, 10 and 50μM), n=3. (E) Gene expression of iNOS, CCL2 and FcγR1 (normalized with 18s rRNA) in BMDMs treated with medium, LPS/IFNγ ± 50μM KX2-391, n=3. Mean + SEM, one-way ANOVA with Bonferroni post-hoc test, *p<0.05, **p<0.01, and ***p<0.001.
We then evaluated the effect of Src inhibitor KX2-391 in RAW macrophages on nitric-oxide
(NO) release in LPS+IFNγ-differentiated macrophages. We observed that KX2-391
concentration-dependently reduced LPS+IFNγ-induced NO release (Figure 5D). We also found
that KX2-391 concentration-dependently inhibited LPS+IFNγ-induced inflammatory genes i.e.
iNOS, CCL2 and Fc gamma receptor 1 (FcγR1) (Figure 5D). We also tested the effect of KX2-
391 on LPS+IFNγ-differentiated BMDMs and observed that KX2-391 significantly down-
regulated the cell viability/metabolic activity as well as the expression of inflammatory genes
i.e. iNOS, CCL2, and FcγR1 (Figure 5E).

104
Finally, we analyzed the effect of KX2-391 in PCLS (prepared as shown schematically in Figure
6A and described elsewhere [35]) and human THP1 monocytes incubated with LPS+IFNγ.
Results revealed upregulation of Src expression in LPS+IFNγ-stimulated PCLS (as observed in
LPS+IFNγ treated BMDMs and THP1 macrophages) as compared to unstimulated and IL-4+IL-
13-stimulated PCLS (Figure S6 and Figure 6B). We treated PCLS with KX2-391 and examined
the effect of KX2-391 on PCLS viability (assessed using ATP assays, normalized with total
protein). In accordance with the in vitro results, we observed 15% inhibition (non-significant)
of cell viability/metabolic activity as compared to LPS+IFNγ PCLS (without KX-291), which
could be attributed to reduced cell proliferation and activation (Figure 6C).
Figure 6. KX2-391 inhibited LPS and IFNγ-induced inflammation in human THP1 macrophages, and precision-cut liver slices (PCLS). (A) Schematic showing PLCS preparation (refer to the methods section for details). (B) Src gene expression in PCLS, n=3. (C) Viability (ATP) assay normalized with total protein as assessed in PCLS, n=3 (3 liver slices per experiment). (D) Gene expression of iNOS and CCL2 (normalized with 18s rRNA) in PCLS treated with medium, LPS/IFNγ ± 25μM KX2-391, n=3 (3 liver slices per experiment). (E) Gene expression of iNOS, CCL2 and IL-6 (normalized with 18s rRNA) in PMA-

105
treated THP1 macrophages treated with medium alone, LPS/IFNγ ± 50μM KX2-391, n=3. Mean + SEM, one-way ANOVA with Bonferroni post-hoc test, *p<0.05, **p<0.01, and ***p<0.001.
Upon treatment with KX2-391, We further observed that KX2-391 significant inhibited
LPS+IFNγ-induced gene expression of inflammatory markers in PCLS (iNOS and CCL2) (Figure
6D) and THP1 macrophages (iNOS, CCL2 and IL-6) (Figure 6E). These results are consistent with
our findings in the mouse models.
4.3.7 Pharmacological Src inhibition attenuated pro-inflammatory macrophage activation via FAK/PI3K/AKT pathway
Finally, we performed western-blot analysis of unstimulated, LPS+IFNγ-stimulated, IL-4+IL-13-
stimulated, and LPS+IFNγ-stimulated and treated with KX2-391, to understand the underlying
mechanism of Src in macrophages. We observed stronger induction in Src protein expression
and phosphorylation upon LPS+IFNγ stimulation which was diminished by KX2-391 treatment
(Figure 7A and Figure S7). We further observed increased phosphorylation of FAK, PI3K and
AKT following LPS+IFNγ stimulation. Studies have reported the involvement of FAK/PI3K/AKT
pathway in the survival and activation of macrophages, and inflammatory responses [44].
Figure 7. Src inhibition attenuated LPS+IFNγ stimulated pro-inflammatory macrophages via FAK/PI3K/AKT pathway. (A) Representative image showing western-blot analysis for phosphorylated Src, FAK, PI3K and AKT normalized with respective non-phosphorylated proteins, in unstimulated, IL-4/IL-13- and LPS/IFNγ-stimulated ± KX2-391, n=3. Refer to Figure S7. (B) Schematic depicting the mechanism-of-action of Src-mediated effects.

106
In this study, we have observed that KX2-391 inhibited phosphorylation and activation of
FAK/PI3K/AKT in LPS+IFNγ-stimulated macrophages (Figure 7A and Figure S7) suggesting the
Src-mediated regulation of FAK/PI3K/AKT pathway in inflammatory macrophages. The
proposed mechanism of Src-mediated effects is schematically presented in Figure 7B.
Finally, we investigated the effect of KX2-391 in in vitro 3D-human spheroids model whereby
3D-spheroids were formed from human hepatocytes (HepG2), human hepatic stellate cells
(LX2 cells), human monocytes (THP1) and human endothelial cells (HUVEC) as schematically
presented in Figure S8A. These spheroids or liver microtissues display characteristic mild
steatohepatitis phenotype when incubated with LPS (endotoxin) as shown in Figure S8B. In
this study, we used LPS is a pleiotropic inflammatory stimulus as it is proposed to reach the
liver via hepatic portal vein after release from Gastrointestinal tract contributing to disease
progression. We treated 7-day spheroids with LPS, and incubated with KX2-391 for 7 days, and
found that treatment with LPS increased collagen-I expression and macrophage activation
marker, MHC-II (major histocompatibility complex class II) expression as well as steatosis
assessed by oil-red-o staining, and all these markers were reduced following Src inhibition
(Figure S8B). However, it is important to stress that LPS only induces pathways associated with
inflammatory signaling therefore NASH-relevant cues such as fructose, free fatty acids,
cholesterol and TGFβ in combination with LPS could be supplemented to model different
aspects of clinical NASH [45].
4.4 Discussion and conclusions
In this study, we report Src kinase as a promising therapeutic target in non-alcoholic and
alcoholic steatohepatitis. We demonstrated that Src was significantly expressed in NASH, ASH,
BA and HCV-cirrhotic patients suggesting that Src kinase is a common pathway involved in
liver pathophysiology irrespective of etiology. Furthermore, Src was upregulated in
hepatocytes, HSCs and inflammatory macrophages. Using a selective Src inhibitor KX2-391,
we have demonstrated amelioration of steatosis, inflammation and fibrogenesis in vitro and
in vivo in NASH and ASH mouse models. We further investigated the mechanism-of-action of
Src and found that Src regulates inflammation and fibrosis via a FAK/PI3K/AKT pathway.
Src family kinases (SFKs), comprising of nine members, have been earlier shown to be involved
in myofibroblasts activation in fibrotic diseases [19-22], and in macrophage activation in

107
inflammatory diseases [17, 18]. However, SFKs and especially Src kinase, one of the SFKs, has
not been explored yet in liver diseases especially in (N)ASH. We observed an increased Src
expression in different liver pathologies in patients and in mouse models. We also evaluated
Src inhibition by KX2-391 in vivo in NASH and ASH mouse models. MCD- and EtOH-diet induced
NASH and ASH respectively in mouse models and showed characteristic accumulation of fat
in hepatocytes (hepatic steatosis), intra-hepatic inflammation, and fibrosis as reported
previously [29, 30]. Triglycerides, FFAs and/or ketones have been hypothesized to be a key
initiating factor in NASH [46] and alcoholic fatty liver pathogenesis. Increased dietary FFA
intake and impaired FFA oxidation causes an increase in the flux of FFA within hepatocytes.
Once the threshold of lipid storage is exceeded, leads to production of toxic lipid metabolites.
The toxic lipid and alcoholic metabolites promote the overproduction of reactive oxygen
species (ROS), which cause liver damage [6, 7]. In this study, we have demonstrated in vitro
that KX2-391 rescued FFA accumulation in HepG2 cells when treated with palmitate. Likewise,
we showed in vivo that KX2-391 ameliorated steatosis in the liver induced by MCD and EtOH
diet.
We further found significant upregulation of SCD1, DGAT2, XBP1 and ATF6 gene expression in
liver tissues from NASH and ASH mouse models. SCD1 and XBP1 is required for de novo FA
synthesis in the liver [37, 47] and KX2-391 inhibited FA synthesis by reducing the SCD1 and
XBP1 expression significantly. Furthermore, de novo lipogenesis is enhanced by DGAT2 and
ATF6 overexpression [38, 48], based on our results, KX2-391 could prevent the overexpression
of DGAT2 and ATF6 and hence lipogenesis. SREBP1 is also shown to be upregulated which
plays a key role in hepatic transcriptional regulation of lipogenic enzymes [49] and
triglycerides in the liver [50]. In this study, we also observed significant downregulation in
SREBP1 expression after treatment with KX2-391.
Inflammation plays an important role in NASH and ASH pathogenesis [6, 7]. In this study, we
found that Src was highly upregulated in LPS/IFNγ-stimulated macrophages and Src inhibition
strongly inhibited LPS/IFNγ-induced NO release and inflammatory markers. Previous studies
demonstrated that pharmacological inhibition of CCL2 ameliorated liver inflammation by
inhibition of macrophage infiltration [34]. In this study, we showed that KX2-391 inhibited
CCL2 expression hence macrophage infiltration in vivo. Notably, we observed increased Src
kinase expression and phosphorylation in pro-inflammatory macrophages, a phenomenon

108
that is not specific to (N)ASH alone but also relevant for several inflammatory diseases
suggesting the implication of Src Kinase beyond (N)ASH.
Fibrosis is an end-stage disease in ASH and NASH patients, we therefore also analyzed the
expression of Src in HSCs and found increased expression of Src, which upon inhibition
attenuated TGFβ-induced collagen deposition and HSCs contractility. Consistent with in vitro
findings, KX2-391 inhibited fibrotic parameters in vivo in MCD- and EtOH-diet mouse models
and 3D human spheroids. Notably, KX2-391 was found to be non-toxic as assessed by the
viability studies in vitro and in PCLS, and in vivo as confirmed by the body weight of the animals
before and after treatment. Notably, Src modulates several signaling pathways, hence it is
possible that Src inhibitor treatment might lead to off-target effects and increasing risk for
infections due to immuno-suppression. Such potential adverse effects should be considered
during evaluation of Src inhibitors. Targeting approaches including organ-/cell-specific
delivery of potential Src inhibitors may be explored to improve the therapeutic efficacy while
reducing adverse effects.
Based on the results, we have clearly demonstrated a novel role of Src kinase in NASH and
ASH. We have also demonstrated the role of Src pathway in inflammatory macrophages, HSCs,
and hepatocytes, and further unraveled the mechanism-of-action of Src kinase. However
detailed mechanistic studies especially in in vivo disease models and human liver tissues are
required to validate our findings. To our best knowledge, this is the first comprehensive study
exploring the role of Src kinase and Src kinase inhibitor KX2-391 in NASH and ASH, and suggests
Src tyrosine Kinase as a potential therapeutic target in non-alcoholic and alcoholic
steatohepatitis. More studies using Src kinase inhibitor in other (N)ASH animal models, also
considering the lack of metabolic syndrome in MCD diet model, are warranted to validate
these findings. Since Src kinase is expressed on different cell types including hepatocytes,
inflammatory macrophages and HSCs, it is relevant that KX2-391 would be effective at
different stages of disease progression. Notwithstanding, given the ubiquitous expression of
Src kinase and as it regulates several signaling pathways, it is possible that (long-term) Src
treatment might potentially lead to adverse off-target effects including immunosuppression
with an increased risk for infections. Such potential adverse effects should be kept in mind
while investigating the therapeutic potential of Src inhibition in (N)ASH. To circumvent the

109
adverse effects, organ/cell-specific delivery of a Src inhibitor could be explored to improve the
short half-life and therapeutic efficacy.
4.5 Acknowledgements
Authors thank Busra Ozturk for the technical assistance during the animal experiments.
Authors also thank Ana Ortiz-Perez and Alexandros Vassilios Gabriel for their help in 3D human
spheroid study. This project was supported by the Indonesian Endowment Fund for Education
from the Republic of Indonesia (Lembaga Pengelola Dana Pendidikan/LPDP to DWK) and
Netherlands Organization for Health Research and Development, ZonMW, NWO, VENI
innovation grant 916.151.94 (to RB), and the University of Twente.
4.6 References
[1] L. Pimpin, H. Cortez-Pinto, F. Negro, E. Corbould, J.V. Lazarus, L. Webber, N. Sheron, E.H.S. Committee, Burden of liver disease in Europe: Epidemiology and analysis of risk factors to identify prevention policies, J Hepatol, (2018).
[2] M. Arrese, A.E. Feldstein, Nash-Related Cirrhosis: An Occult Liver Disease Burden, Hepatol Commun, 1 (2017) 84-86.
[3] S. Mitra, A. De, A. Chowdhury, Epidemiology of non-alcoholic and alcoholic fatty liver diseases, Transl Gastroenterol Hepatol, 5 (2020) 16.
[4] J.L. Mohler, E.S. Antonarakis, A.J. Armstrong, A.V. D'Amico, B.J. Davis, T. Dorff, J.A. Eastham, C.A. Enke, T.A. Farrington, C.S. Higano, E.M. Horwitz, M. Hurwitz, J.E. Ippolito, C.J. Kane, M.R. Kuettel, J.M. Lang, J. McKenney, G. Netto, D.F. Penson, E.R. Plimack, J.M. Pow-Sang, T.J. Pugh, S. Richey, M. Roach, S. Rosenfeld, E. Schaeffer, A. Shabsigh, E.J. Small, D.E. Spratt, S. Srinivas, J. Tward, D.A. Shead, D.A. Freedman-Cass, Prostate Cancer, Version 2.2019, NCCN Clinical Practice Guidelines in Oncology, J Natl Compr Canc Netw, 17 (2019) 479-505.
[5] D. Haldar, B. Kern, J. Hodson, M.J. Armstrong, R. Adam, G. Berlakovich, J. Fritz, B. Feurstein, W. Popp, V. Karam, P. Muiesan, J. O'Grady, N. Jamieson, S.J. Wigmore, J. Pirenne, S.A. Malek-Hosseini, E. Hidalgo, Y. Tokat, A. Paul, J. Pratschke, M. Bartels, P. Trunecka, U. Settmacher, M. Pinzani, C. Duvoux, P.N. Newsome, S. Schneeberger, L. European, A. Intestine Transplant, Outcomes of liver transplantation for non-alcoholic steatohepatitis: A European Liver Transplant Registry study, J Hepatol, 71 (2019) 313-322.
[6] W. Dunn, V.H. Shah, Pathogenesis of Alcoholic Liver Disease, Clin Liver Dis, 20 (2016) 445-456.
[7] S.M. Francque, D. van der Graaff, W.J. Kwanten, Non-alcoholic fatty liver disease and cardiovascular risk: Pathophysiological mechanisms and implications, J Hepatol, 65 (2016) 425-443.
[8] F. Scaglioni, S. Ciccia, M. Marino, G. Bedogni, S. Bellentani, ASH and NASH, Dig Dis, 29 (2011) 202-210.
[9] M.A. Konerman, J.C. Jones, S.A. Harrison, Pharmacotherapy for NASH: Current and emerging, J Hepatol, 68 (2018) 362-375.

110
[10] B. Gao, R. Bataller, Alcoholic liver disease: pathogenesis and new therapeutic targets, Gastroenterology, 141 (2011) 1572-1585.
[11] A.A. Kolodziejczyk, D. Zheng, O. Shibolet, E. Elinav, The role of the microbiome in NAFLD and NASH, EMBO Mol Med, 11 (2019).
[12] K. Ray, NAFLD. Leaky guts: intestinal permeability and NASH, Nat Rev Gastroenterol Hepatol, 12 (2015) 123.
[13] G. Szabo, Gut-liver axis in alcoholic liver disease, Gastroenterology, 148 (2015) 30-36. [14] N. Goossens, Y. Hoshida, Is Hepatocellular Cancer the Same Disease in Alcoholic and
Nonalcoholic Fatty Liver Diseases?, Gastroenterology, 150 (2016) 1710-1717. [15] R. Roskoski, Jr., Src protein-tyrosine kinase structure and regulation, Biochem Biophys Res
Commun, 324 (2004) 1155-1164. [16] S.J. Parsons, J.T. Parsons, Src family kinases, key regulators of signal transduction,
Oncogene, 23 (2004) 7906-7909. [17] C.A. Lowell, Src-family kinases: rheostats of immune cell signaling, Mol Immunol, 41
(2004) 631-643. [18] C.L. Abram, C.A. Lowell, The diverse functions of Src family kinases in macrophages, Front
Biosci, 13 (2008) 4426-4450. [19] G. Du, J. Wang, T. Zhang, Q. Ding, X. Jia, X. Zhao, J. Dong, X. Yang, S. Lu, C. Zhang, Z. Liu, Z.
Zeng, R. Safadi, R. Qi, X. Zhao, Z. Hong, Y. Lu, Targeting Src family kinase member Fyn by Saracatinib attenuated liver fibrosis in vitro and in vivo, Cell Death Dis, 11 (2020) 118.
[20] M. Hu, P. Che, X. Han, G.Q. Cai, G. Liu, V. Antony, T. Luckhardt, G.P. Siegal, Y. Zhou, R.M. Liu, L.P. Desai, P.J. O'Reilly, V.J. Thannickal, Q. Ding, Therapeutic targeting of SRC kinase in myofibroblast differentiation and pulmonary fibrosis, J Pharmacol Exp Ther, 351 (2014) 87-95.
[21] H.Y. Seo, J.H. Jeon, Y.A. Jung, G.S. Jung, E.J. Lee, Y.K. Choi, K.G. Park, M.S. Choe, B.K. Jang, M.K. Kim, I.K. Lee, Fyn deficiency attenuates renal fibrosis by inhibition of phospho-STAT3, Kidney Int, 90 (2016) 1285-1297.
[22] Y. Yan, L. Ma, X. Zhou, M. Ponnusamy, J. Tang, M.A. Zhuang, E. Tolbert, G. Bayliss, J. Bai, S. Zhuang, Src inhibition blocks renal interstitial fibroblast activation and ameliorates renal fibrosis, Kidney Int, 89 (2016) 68-81.
[23] H.Y. Seo, S.H. Lee, J.H. Lee, Y.N. Kang, J.S. Hwang, K.G. Park, M.K. Kim, B.K. Jang, Src Inhibition Attenuates Liver Fibrosis by Preventing Hepatic Stellate Cell Activation and Decreasing Connetive Tissue Growth Factor, Cells, 9 (2020).
[24] J. Argemi, M.U. Latasa, S.R. Atkinson, I.O. Blokhin, V. Massey, J.P. Gue, J. Cabezas, J.J. Lozano, D. Van Booven, A. Bell, S. Cao, L.A. Vernetti, J.P. Arab, M. Ventura-Cots, L.R. Edmunds, C. Fondevilla, P. Stärkel, L. Dubuquoy, A. Louvet, G. Odena, J.L. Gomez, T. Aragon, J. Altamirano, J. Caballeria, M.J. Jurczak, D.L. Taylor, C. Berasain, C. Wahlestedt, S.P. Monga, M.Y. Morgan, P. Sancho-Bru, P. Mathurin, S. Furuya, C. Lackner, I. Rusyn, V.H. Shah, M.R. Thursz, J. Mann, M.A. Avila, R. Bataller, Defective HNF4alpha-dependent gene expression as a driver of hepatocellular failure in alcoholic hepatitis, Nature Communications, 10 (2019) 3126.
[25] C.A. Lowell, Src-family and Syk kinases in activating and inhibitory pathways in innate immune cells: signaling cross talk, Cold Spring Harb Perspect Biol, 3 (2011).
[26] T.N. Bukong, A. Iracheta-Vellve, B. Saha, A. Ambade, A. Satishchandran, B. Gyongyosi, P. Lowe, D. Catalano, K. Kodys, G. Szabo, Inhibition of spleen tyrosine kinase activation ameliorates inflammation, cell death, and steatosis in alcoholic liver disease, Hepatology, 64 (2016) 1057-1071.

111
[27] D.W. Kurniawan, A.K. Jajoriya, G. Dhawan, D. Mishra, J. Argemi, R. Bataller, G. Storm, D.P. Mishra, J. Prakash, R. Bansal, Therapeutic inhibition of spleen tyrosine kinase in inflammatory macrophages using PLGA nanoparticles for the treatment of non-alcoholic steatohepatitis, J Control Release, 288 (2018) 227-238.
[28] C. Qu, D. Zheng, S. Li, Y. Liu, A. Lidofsky, J.A. Holmes, J. Chen, L. He, L. Wei, Y. Liao, H. Yuan, Q. Jin, Z. Lin, Q. Hu, Y. Jiang, M. Tu, X. Chen, W. Li, W. Lin, B.C. Fuchs, R.T. Chung, J. Hong, Tyrosine kinase SYK is a potential therapeutic target for liver fibrosis, Hepatology, 68 (2018) 1125-1139.
[29] L. Hebbard, J. George, Animal models of nonalcoholic fatty liver disease, Nat Rev Gastroenterol Hepatol, 8 (2011) 35-44.
[30] A. Bertola, S. Mathews, S.H. Ki, H. Wang, B. Gao, Mouse model of chronic and binge ethanol feeding (the NIAAA model), Nat Protoc, 8 (2013) 627-637.
[31] M. Ahrens, O. Ammerpohl, W. von Schonfels, J. Kolarova, S. Bens, T. Itzel, A. Teufel, A. Herrmann, M. Brosch, H. Hinrichsen, W. Erhart, J. Egberts, B. Sipos, S. Schreiber, R. Hasler, F. Stickel, T. Becker, M. Krawczak, C. Rocken, R. Siebert, C. Schafmayer, J. Hampe, DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery, Cell Metab, 18 (2013) 296-302.
[32] E. Wurmbach, Y.B. Chen, G. Khitrov, W. Zhang, S. Roayaie, M. Schwartz, I. Fiel, S. Thung, V. Mazzaferro, J. Bruix, E. Bottinger, S. Friedman, S. Waxman, J.M. Llovet, Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma, Hepatology, 45 (2007) 938-947.
[33] K. Moyer, V. Kaimal, C. Pacheco, R. Mourya, H. Xu, P. Shivakumar, R. Chakraborty, M. Rao, J.C. Magee, K. Bove, B.J. Aronow, A.G. Jegga, J.A. Bezerra, Staging of biliary atresia at diagnosis by molecular profiling of the liver, Genome Med, 2 (2010) 33.
[34] C. Baeck, X. Wei, M. Bartneck, V. Fech, F. Heymann, N. Gassler, K. Hittatiya, D. Eulberg, T. Luedde, C. Trautwein, F. Tacke, Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice, Hepatology, 59 (2014) 1060-1072.
[35] I.A. de Graaf, P. Olinga, M.H. de Jager, M.T. Merema, R. de Kanter, E.G. van de Kerkhof, G.M. Groothuis, Preparation and incubation of precision-cut liver and intestinal slices for application in drug metabolism and toxicity studies, Nat Protoc, 5 (2010) 1540-1551.
[36] M. Kozyra, I. Johansson, Å. Nordling, S. Ullah, V.M. Lauschke, M. Ingelman-Sundberg, Human hepatic 3D spheroids as a model for steatosis and insulin resistance, Scientific Reports, 8 (2018) 14297.
[37] D. Bhattacharya, B. Basta, J.M. Mato, A. Craig, D. Fernandez-Ramos, F. Lopitz-Otsoa, D. Tsvirkun, L. Hayardeny, V. Chandar, R.E. Schwartz, A. Villanueva, S.L. Friedman, Aramchol downregulates stearoyl CoA-desaturase 1 in hepatic stellate cells to attenuate cellular fibrogenesis, JHEP Rep, 3 (2021) 100237.
[38] D.G. McLaren, S. Han, B.A. Murphy, L. Wilsie, S.J. Stout, H. Zhou, T.P. Roddy, J.N. Gorski, D.E. Metzger, M.K. Shin, D.F. Reilly, H.H. Zhou, M. Tadin-Strapps, S.R. Bartz, A.M. Cumiskey, T.H. Graham, D.M. Shen, K.O. Akinsanya, S.F. Previs, J.E. Imbriglio, S. Pinto, DGAT2 Inhibition Alters Aspects of Triglyceride Metabolism in Rodents but Not in Non-human Primates, Cell Metab, 27 (2018) 1236-1248 e1236.
[39] S. Lee, S. Kim, S. Hwang, N.J. Cherrington, D.Y. Ryu, Dysregulated expression of proteins associated with ER stress, autophagy and apoptosis in tissues from nonalcoholic fatty liver disease, Oncotarget, 8 (2017) 63370-63381.

112
[40] T. Nagaya, N. Tanaka, T. Suzuki, K. Sano, A. Horiuchi, M. Komatsu, T. Nakajima, T. Nishizawa, S. Joshita, T. Umemura, T. Ichijo, A. Matsumoto, K. Yoshizawa, J. Nakayama, E. Tanaka, T. Aoyama, Down-regulation of SREBP-1c is associated with the development of burned-out NASH, J Hepatol, 53 (2010) 724-731.
[41] S. Reif, A. Lang, J.N. Lindquist, Y. Yata, E. Gabele, A. Scanga, D.A. Brenner, R.A. Rippe, The role of focal adhesion kinase-phosphatidylinositol 3-kinase-akt signaling in hepatic stellate cell proliferation and type I collagen expression, J Biol Chem, 278 (2003) 8083-8090.
[42] V. Thamilselvan, D.H. Craig, M.D. Basson, FAK association with multiple signal proteins mediates pressure-induced colon cancer cell adhesion via a Src-dependent PI3K/Akt pathway, FASEB J, 21 (2007) 1730-1741.
[43] F. Tacke, Targeting hepatic macrophages to treat liver diseases, J Hepatol, 66 (2017) 1300-1312.
[44] X. Sun, X. Wang, T. Chen, T. Li, K. Cao, A. Lu, Y. Chen, D. Sun, J. Luo, J. Fan, W. Young, Y. Ren, Myelin activates FAK/Akt/NF-kappaB pathways and provokes CR3-dependent inflammatory response in murine system, PLoS One, 5 (2010) e9380.
[45] T. Kostrzewski, S. Snow, A.L. Battle, S. Peel, Z. Ahmad, J. Basak, M. Surakala, A. Bornot, J. Lindgren, M. Ryaboshapkina, M. Clausen, D. Linden, C. Maass, L.M. Young, A. Corrigan, L. Ewart, D. Hughes, Modelling human liver fibrosis in the context of non-alcoholic steatohepatitis using a microphysiological system, Commun Biol, 4 (2021) 1080.
[46] N. Katsiki, D.P. Mikhailidis, C.S. Mantzoros, Non-alcoholic fatty liver disease and dyslipidemia: An update, Metabolism, 65 (2016) 1109-1123.
[47] L.H. Glimcher, A.H. Lee, From sugar to fat: How the transcription factor XBP1 regulates hepatic lipogenesis, Ann N Y Acad Sci, 1173 Suppl 1 (2009) E2-9.
[48] D.L. Howarth, C. Lindtner, A.M. Vacaru, R. Sachidanandam, O. Tsedensodnom, T. Vasilkova, C. Buettner, K.C. Sadler, Activating transcription factor 6 is necessary and sufficient for alcoholic fatty liver disease in zebrafish, PLoS Genet, 10 (2014) e1004335.
[49] J.C. Chow, P.R. Ling, Z. Qu, L. Laviola, A. Ciccarone, B.R. Bistrian, R.J. Smith, Growth hormone stimulates tyrosine phosphorylation of JAK2 and STAT5, but not insulin receptor substrate-1 or SHC proteins in liver and skeletal muscle of normal rats in vivo, Endocrinology, 137 (1996) 2880-2886.
[50] N. Yahagi, H. Shimano, A.H. Hasty, T. Matsuzaka, T. Ide, T. Yoshikawa, M. Amemiya-Kudo, S. Tomita, H. Okazaki, Y. Tamura, Y. Iizuka, K. Ohashi, J. Osuga, K. Harada, T. Gotoda, R. Nagai, S. Ishibashi, N. Yamada, Absence of sterol regulatory element-binding protein-1 (SREBP-1) ameliorates fatty livers but not obesity or insulin resistance in Lep(ob)/Lep(ob) mice, J Biol Chem, 277 (2002) 19353-19357.

113
4.7 Supplementary Information
Table S1. Sequence of the mouse primers used for quantitative real-time PCR
Table S2. Sequence of the human primers used for quantitative real-time PCR
Table S3. Antibodies used for the immunohistochemistry and western blotting
Gene
Forward primer sequence (5’-3’) Reverse primer sequence (5’-3’) Accession no. 18s rRNA AACTTTCGATGGTAGTCGCCGT TCCTTGGATGTGGTAGCCGTTT 6YAN_2
ARG1 GTGAAGAACCCACGGTCTGT CTGGTTGTCAGGGGAGTGTT NM_007482.3
ATF6 TGGGAATGGAAGCCTAAAGAGG TGGGAGGACAGAGAAACAAGC NM_001081304.1 CCL2 GTGCTGACCCCAAGAAGGAA GTGCTGAAGACCTTAGGGCA NM_011333.3
Col1a1 TGACTGGAAGAGCGGAGAGT ATCCATCGGTCATGCTCTCT NM_007742.3
Desmin ATGCAGCCACTCTAGCTCGT CTCATACTGAGCCCGGATGT NM_010043.1
DGAT2 AGTTTCCTGGCATAAGGCCC ACATCAGGTACTCGCGAAGC NM_026384
F4/80 TGCATCTAGCAATGGACAGC GCCTTCTGGATCCATTTGAA NM_010130.4
FcγR1 GTGCTCTCATTGGGTGGTGA TGTAAGCCGTGCCTTCCAAT NM_010186.5 GAPDH ACAGTCCATGCCATCACTGC GATCCACGACGGACACATTG NM_008084.2 IL-1β GCCAAGACAGGTCGCTCAGGG CCCCCACACGTTGACAGCTAGG NM_008361.3 IL-6 TGATGCTGGTGACAACCACGGC TAAGCCTCCGACTTGTGAAGTGTA NM_031168.1
iNOS GGTGAAGGGACTGAGCTGTT GCTACTCCGTGGAGTGAACAA NM_010927.4 SCD1 GGAGACGGGAGTCACAAGAG TGCATCATTAACACCCCGAT NM_009127
Src GTGAGGTTCTGGACCAGGTG CTGGTACTGTGGCTCAGTGG NM_009271.3
SREBP1 CTGGTGAGTGGAGGGACCAT GACCGGTAGCGCTTCTCAAT NM_011480.4
TNF-α AGGCTGCCCCGACTACGTGC CAGCGCTGAGTTGGTCCCCC NM_013693.2
XBP1 AAGGGGAGTGGAGTAAGGCT TGCTGCAGAGGTGCACATAG NM_013842.3
α-SMA ACTACTGCCGAGCGTGAGAT CCAATGAAAGATGGCTGGAA NM_007392.2
Gene
Forward primer sequence (5’-3’) Reverse primer sequence (5’-3’) Accession no. Col1a1 GTACTGGATTGACCCCAACC CGCCATACTCGAACTGGAAT NM_000088.3 col3a1 AAGAAGGCCCTGAAGCTGAT GTGTTTCGTGCAACCATCCT NM_000090 Src CAACACAGAGGGAGACTGGT CACGTAGTTGCTGGGGATGT NM_198291.3 18s rRNA TGAGGTGGAACGTGTGATCA CCTCTATGGGCCCGAATCTT NM_022551.2
Primary Antibody Source Immunostaining Western blotting Polyclonal rabbit anti-Src Cell Signaling 1:100 1:500
Polyclonal rabbit anti-pSRC Cell Signaling 1:500
Polyclonal rabbit anti-pFAK (Tyr397) Cell Signaling 1:500
Polyclonal rabbit anti-FAK Cell Signaling 1:500
Polyclonal rabbit anti-pAKT (Ser473) Cell Signaling 1:500
Polyclonal rabbit anti-AKT Cell Signaling 1:500

114
Polyclonal rabbit pPI3K p85 (Tyr458)/p55 (Tyr199)
Cell Signaling 1:500
Polyclonal rabbit anti-PI3K P85 Cell Signaling 1:500
Polyclonal goat anti-collagen I Southern Biotech 1:100
Monoclonal rat anti-F4/80 BioRad 1:100
Monoclonal mouse anti-β-actin Sigma 1:5000
Monoclonal mouse anti-EpCAM HansaBiomed 1:100
Monoclonal mouse anti-CD68 eBioScience 1:100
Secondary Antibody Source
Polyclonal goat anti-rabbit IgG DAKO 1:100 1:1000
Polyclonal rabbit anti-mouse IgG DAKO 1:100 1:1000
Polyclonal goat anti-mouse IgG DAKO 1:100 1:1000
Polyclonal rabbit anti-goat IgG DAKO 1:100 1:1000
Polyclonal goat anti-rat IgG Southern Biotech 1:100 1:1000
Donkey anti-goat Alexa 488 ThermoScientific 1:100
Donkey anti-rabbit Alexa 594 ThermoScientific 1:100

115
Figure S1. Src kinase inhibitor, KX2-391 inhibits the expression of steatosis, inflammation and fibrosis related genes in MCD diet-induced NASH in mice. Relative gene expression (normalized with 18s rRNA and relative to control mice) for (A) steatosis markers (SCD1, DGAT2, XBP1, SREBP1c and ATF6); (B) inflammation markers (TNFα, IL-1β, IL-6, iNOS and CCL2); and (C) fibrosis-HSCs markers (Desmin and α-SMA) as analyzed in the liver tissues from control chow-diet fed healthy mice (n = 8), MCD-diet fed mice (n = 8), MCD-diet fed mice treated with 10mg/kg KX2-391 (n = 5). Graphs represent mean + SEM, statistical differences were calculated using one-way ANOVA with the Bonferroni post hoc test. *p < 0.05, **p < 0.01 denotes significance.

116
Figure S2. Src kinase inhibitor, KX2-391 inhibits the expression of steatosis, inflammation and fibrosis and related genes in EtOH diet-induced ASH in mice. Relative gene expression (normalized with 18s rRNA and relative to control mice) for (A) steatosis markers (SCD1, DGAT2, XBP1 and ATF6); (B) inflammation markers (IL-1β, iNOS, TNFα, IL-6) (C) Arg1; and (D) fibrosis markers (Col1a1, Desmin and α-SMA) as analyzed in the liver tissues from isocaloric diet-fed healthy mice (n = 5), EtOH-diet fed mice (n = 6), EtOH-diet fed mice treated with 10mg/kg KX2-391 (n = 5). Graphs represent mean + SEM, statistical differences were calculated using one-way ANOVA with the Bonferroni post hoc test. *p < 0.05, **p < 0.01 denotes significance.

117
Figure S3. Colocalization of Src kinase with hepatocytes, macrophages and HSCs. Representative images (scale=100μm) of EpCAM (hepatocyte marker), CD68 (pan-macrophage marker) and collagen I (ECM, fibrosis-HSCs-related marker) (in green, first column) and Src (in red, second column) stained liver sections from human NASH livers. Blue staining (third column) indicates DAPI-nuclear staining. Arrows shown in merge images (fourth column) depict colocalization.

118
Figure S4. Inhibition of palmitate-induced lipid deposition in HepG2 cells. Oil-red-O staining and respective quantitative analysis showing inhibition of Palmitate-induced lipid deposition in HepG2 cells after 24h of treatment with 50µM KX2-391. Data presented as mean + SEM, statistical differences were calculated using two-tailed unpaired t test. **p < 0.01 denotes significance.
Palmitate Palmitate+KX2-391
Palmitate Palmitate+KX2-3910
20
40
60**
% o
il-re
d-O
stai
ning
(pos
itive
are
a pe
r fie
ld)

119
Figure S5. Src mediates HSCs activation via FAK/PI3K/AKT pathway. Quantitative western blot analysis (refer to Figure 4F) for phosphorylated Src, FAK, PI3K and AKT normalized with the respective non-phosphorylated Src, FAK, PI3K and AKT respectively as examined in control and TGFβ-activated HSCs (LX2 cells) with and without 24h of treatment with 50µM Src kinase inhibitor, KX2-391, n = 3. Graphs represent mean + SEM, statistical differences were calculated using one-way ANOVA with the Bonferroni post hoc test, *p < 0.05, **p < 0.01 denotes significance.
Control TGFb TGFb+KX2-3910.0
0.5
1.0
1.5
Rela
tive
pFAK
exp
ress
ion
(nor
mal
ized
with
FAK
)
✱ ✱
Control TGFb TGFb+KX2-3910.0
0.5
1.0
1.5
Rela
tive
pAkt
exp
ress
ion
(nor
mal
ized
with
Akt
)
✱ ✱✱
Control TGFb TGFb+KX2-3910.0
0.5
1.0
1.5
Rela
tive
pPI3
K ex
pres
sion
(nor
mal
ized
with
PI3
K)
✱ ✱
Control TGFb TGFb+KX2-3910.0
0.5
1.0
1.5Re
lativ
e pS
rc e
xpre
ssio
n(n
orm
alize
d w
ith S
rc)
✱ ✱

120
Figure S6. Src gene expression in LPS and IFNγ-stimulated BMDMs and human THP1 macrophages. Gene expression of Src (normalized with GAPDH) in unstimulated, LPS/IFNγ- and IL-4/IL-13-stimulated macrophages, n=3. Graphs represent mean + SEM, statistical differences were calculated using one-way ANOVA with Bonferroni post-hoc test, **p < 0.01 denotes significance.
Medium
LPS+
IFNg
IL-4+IL-
130
5
10
15 ✱✱ ✱✱Re
lativ
e Sr
c exp
ress
ion
(nor
mal
ized
with
GAP
DH)
BMDMs
Medium
LPS+
IFNg
IL-4+IL-
130.0
0.5
1.0
1.5
2.0
2.5✱✱ ✱✱
Rela
tive
Src e
xpre
ssio
n(n
orm
alize
d w
ith G
APDH
)
Human THP1

121
Figure S7. Pharmacological Src inhibition attenuated pro-inflammatory macrophage activation via FAK/PI3K/AKT pathway. Quantitative western blot analysis (refer to Figure 7A) for phosphorylated Src, FAK, PI3K and AKT normalized with the respective non-phosphorylated Src, FAK, PI3K and AKT respectively as examined in unstimulated (medium), LPS+IFNγ stimulated, IL-4+IL-13 stimulated and LPS+IFNγ stimulated treated with 50µM KX2-391 macrophages (refer to Figure 6A) and quantitatively analyzed as presented here, n = 3. Graphs represent mean + SEM, statistical differences were calculated using one-way ANOVA with the Bonferroni post hoc test, *p < 0.05, **p < 0.01 denotes significance.

122
Figure S8. Therapeutic efficacy of KX2-391 in 3D human spheroids model. (A) Spheroid assay schematic illustration: The cell-mixture containing HepG2, LX2, HUVECS and THP1 was plated in a 96-well ULA plates at 1,200 cells/well of density and grown for 7 days; At day 7, spheroids were treated with or without LPS and WP1066; After 7 days of treatment, the spheroids were retrieved, snap frozen and sectioned. The corresponding cryosections were used for immunostaining. (B) First panel of images depicts 3D spheroids stained with fibrosis marker, collagen I (in red); second panel depicts 3D spheroids stained with HSC activation marker, α-SMA (in green); third panel of images presents 3D spheroids stained with macrophage activation marker, MHC-II (in red); and fourth panel of images depicts 3D spheroids stained with M2 macrophage marker, mannose receptor (in green). Blue color shows DAPI nuclear staining.
A
B Control LPS + KX2-391 LPS
MH
CII
Colla
gen
I O
il-Re
d-O

123
Chapter 5
Fibroblast growth factor 2 (FGF2) conjugated superparamagnetic
iron oxide nanoparticles (SPIONs) ameliorate hepatic stellate cells
activation in vitro and acute liver injury in mice
Dhadhang Wahyu Kurniawan, Richell Booijink, Lena Pater, Irene Wols, Aggelos Vrynas, Gert Storm, Jai Prakash, Ruchi Bansal
Published in: J Control Release. 2020 Dec 10; 328:640-652. doi: 10.1016/j.jconrel.2020.09.041.

124
Abstract
Liver diseases are the growing health problem with no clinically approved therapy available.
Activated hepatic stellate cells (HSCs) are the key driver cells responsible for extracellular
matrix deposition, the hallmark of liver fibrosis. Fibroblast growth factor 2 (FGF2) has shown
to possess anti-fibrotic effects in fibrotic diseases including liver fibrosis, and promote tissue
regeneration. Among the fibroblast growth factor receptors (FGFRs), FGF2 interact primarily
with FGFR1, highly overexpressed on activated HSCs and inhibit HSCs activation. However,
FGF2 poses several limitations including short systemic half-life and stability owing to
enzymatic degradation. The aim of this study is to improve the stability and half-life of FGF2
thereby improving the therapeutic efficiency of FGF2 for the treatment of liver fibrosis. We
found that FGFR1-3 mRNA levels were overexpressed in cirrhotic human livers, while FGFR1c,
2c, 3c, 4 and FGF2 mRNA levels were overexpressed in TGFβ-activated HSCs (LX2 cells), and
FGFR1 protein expression were highly increased in TGFβ-activated HSCs. Treatment with FGF2
inhibited TGFβ-induced HSCs activation, migration, and contraction in vitro. FGF2 was
conjugated to superparamagnetic iron-oxide nanoparticles (SPIONs) using carbodiimide
chemistry, and the resulting FGF2-SPIONs was confirmed by dynamic light scattering (DLS),
zeta potential, dot-blot analysis, and Prussian Blue iron-staining. In vitro, treatment with
FGF2-SPIONs evidenced increased therapeutic effects (attenuated TGFβ-induced HSCs
activation, migration, and contraction) of FGF2 in TGFβ-activated HSCs and ameliorated early
liver fibrogenesis in vivo in acute carbon tetrachloride (CCl4)-induced liver injury mouse
model. In contrast, free FGF2 showed no significant effects in vivo. Altogether, this study
presents a promising therapeutic approach using FGF2-SPIONs for the treatment of liver
fibrosis.

125
5.1 Introduction
Liver diseases caused by viral infections (mainly hepatitis B and C viruses), metabolic
disorders, alcohol or drug abuse and autoimmune disorders affecting millions of people,
represents a major health problem with high morbidity and mortality [1]. Acute liver injury is
a transient, often reversible, wound healing response however persistent chronic injury to
the liver results in progressive accumulation of extracellular matrix (ECM) components
eventually resulting in liver cirrhosis, end-stage liver failure and hepatocellular carcinoma [2,
3]. Due to the lack of an effective therapy, liver diseases poses a major clinical impact with an
increasing number of patients requiring liver transplantation [1, 4, 5].
Hepatic stellate cells (HSCs) play a key role in the progression of liver fibrosis, regardless of an
underlying cause [3, 6, 7]. Upon injury, hepatocytes undergo apoptosis or necrosis and release
pro-inflammatory and pro-fibrogenic mediators that stimulates recruitment and activation of
inflammatory cells resulting in chronic liver inflammation. The resident and infiltrated
immune cells, in turn, secrete pro-inflammatory and pro-fibrogenic factors that activates
quiescent HSCs [2, 3]. Quiescent HSCs transdifferentiate into myofibroblast-like cells and
become highly proliferative, migratory, and contractile cells producing excessive amounts of
ECM components that accumulates in liver parenchyma, disrupts liver architecture and forms
the characteristic scar tissue [3, 6, 7].
Fibroblasts growth factors (FGFs) have been shown to regulate HSCs differentiation and liver
fibrosis. There are seven subfamilies of FGFs: FGF1 subfamily (FGF1, FGF2); FGF4 subfamily
(FGF4, FGF5, FGF6); FGF8 subfamily (FGF8, FGF17, FGF18); FGF9 subfamily (FGF9, FGF16,
FGF20); FGF10 subfamily (FGF3, FGF7, FGF10, FGF22); FGF11 subfamily (FGF11, FGF12, FGF13,
FGF14) and FGF19 subfamily (FGF15, FGF19, FGF21, FGF23) [8, 9]. These subfamilies of FGFs
are tissue specific and have different binding affinity with FGF receptors (FGFRs). There are
four isoforms of FGFRs: FGFR1, FGFR2, FGFR3, and FGFR4. FGFRs have different splice variants
and display tissue specific expression [8, 9]. FGF-FGFR signaling is critical in several
developmental processes including cellular proliferation, migration, differentiation,
morphogenesis and organogenesis [10]. Furthermore, FGF-FGFR signaling pathway regulate
liver homeostasis by regulating metabolism of lipids, cholesterol and bile acids, promotes

126
hepatocyte proliferation and detoxification, and facilitating liver regeneration after partial
hepatectomy [11].
Among other FGFs, FGF2 (also known as basic FGF) play a crucial role in numerous cellular
processes including organ development, wound healing and tissue regeneration [12]. FGF2
has been shown to regulate HSCs function and has been investigated extensively in liver
fibrosis however showed contradictory results [9, 13]. Some studies have demonstrated the
pro-fibrotic effects of FGF2, while numerous studies have demonstrated the anti-fibrotic
effects of FGF2 in vitro and in vivo in preclinical animal models [13]. Using FGF1-/-FGF2-/-
mouse models, Yu et al., demonstrated that acute carbon-tetrachloride (CCl4) administration
in FGF1-/-FGF2-/- mice resulted in elevated serum alanine aminotransferase (ALT) levels, HSCs
activation marked by increased intra-hepatic expression of alpha smooth muscle actin (α-
SMA) and desmin, accompanied with activation and migration of HSCs to the site-of-injury,
while chronic CCl4 administration led to decreased collagen expression and fibrosis [14]. Pan
et al., has demonstrated that 18 KDa low- (FGF2lmw) and 21 or 22 KDa high-molecular weight
(FGF2hmw) forms have distinct role in liver fibrogenesis, and that the exogenous FGF2lmw
treatment attenuated HSCs activation and fibrosis [15]. The presence of different FGF2
isoforms with distinct roles might explain the conflicting results reported on the effects of
FGF2. Besides hepatic fibrosis, FGF2 treatment has also shown to attenuate bleomycin-
induced pulmonary fibrosis [16] and ischemia reperfusion induced renal injury [17].
With respect to myofibroblast (activated HSCs in liver) activation and differentiation,
evidences have revealed that FGF2 promotes myofibroblast apoptosis in vivo, antagonizes
activation and transforming growth factor beta (TGFβ) signaling, antagonizes contractile
phenotypes and myofibroblast differentiation of non-fibroblasts progenitors, suppresses pro-
fibrogenic gene expression, and promotes regenerative healing [13]. Mapping studies and
crystal structures of FGF-FGFR complexes have revealed that FGF2 binds to different FGFR
splice variants with varying affinity [18-20]. In human liver myofibroblasts, FGF2 has been
shown to interact with FGFR1 which is highly overexpressed on myofibroblasts [21]. Although
the underlying mechanism through which FGF2 affects liver fibrosis is not entirely
understood, different FGF2-regulated signalling pathways have been proposed in several
diseases and cell types including Janus kinase (JAK), signal transducer and activator of
transcription (STAT), extracellular signal regulated kinase (ERK), mitogen-activated protein

127
kinase (MAPK), c-jun N-terminal kinase (JNK) and serine/threonine kinase AKT (also known as
protein kinase B, PKB) pathways [13]. Particularly, it has been demonstrated that selective
inhibitor of phosphatidylinositol 3-kinase (PI3K) and mitogen-activated protein kinase kinase
(MAPKK or MEK) abolished the protective effects of FGF2 suggesting involvement of PI3K/AKT
and MEK/ERK signaling pathways in FGF2-mediated effects [17].
While the attractiveness of using FGF2 as therapeutic is evident, there are several limitations
including a short systemic half-life after intravenous administration (due to small size) and
poor stability (owing to susceptibility to degradation by proteases) [22]. Conjugation of FGF2
to superparamagnetic iron-oxide nanoparticles (SPIONs) could be a promising alternative for
eliminating the drawbacks hampering future clinical application. SPIONs possess tailored
surface chemistry, low cytotoxicity and unique magnetic properties [23, 24]. Recent
developments of polymer (dextran/PEG)-coated SPIONs has shown tremendous
improvements in biocompatibility and blood circulation. We have previously demonstrated
an increased magnetic resonance imaging (MRI) contrast and therapeutic efficacy using
Relaxin-SPIONs in liver fibrosis [25].
In this study, we have first analyzed the expression of FGFR in human liver cirrhosis and TGFβ-
activated human HSCs (LX cells) in vitro. We then investigated the therapeutic effects of
human recombinant FGF2 (low-molecular weight) on TGFβ-activated human HSCs (LX cells)
in vitro. In order to improve the stability and systemic half-life of FGF2 thereby therapeutic
efficacy, we conjugated FGF2 to dextran- and PEG-coated SPIONs. Subsequently, we
examined the therapeutic effect of FGF2-SPIONs versus free FGF2 on TGFβ-activated human
HSCs in vitro and in an acute CCl4 induced mouse model in vivo.
5.2 Materials and methods
5.2.1 FGFR gene expression analysis in the liver tissues from healthy and cirrhosis patients
Publicly available transcriptome datasets of liver tissues (GSE6764) from normal healthy
individuals (n=10) and cirrhotic patients (n=12) [26] were analysed using the Gene Expression
Omnibus (GEO) database of the National Centre for Biotechnology Information (NCBI) to
assess FGF receptor (FGFR) gene expression i.e. FGFR1, FGFR2, FGFR3 and FGFR4 in normal
and cirrhotic human livers.

128
5.2.2 Cell lines
Human hepatic stellate cells (LX2 cells), an immortalized human derived cell line, was
provided by Prof. Scott Friedman (Mount Sinai Hospital, New York, NY, USA). LX2 cells were
cultured in Dulbecco’s Modified Eagle’s Medium (DMEM)-Glutamax (Invitrogen, Carlsbad, CA,
USA) supplemented with 10% fetal bovine serum (FBS, Lonza, Verviers, Belgium), 50 U/mL
penicillin (Sigma, St. Louis, MO, USA) and 50 µg/mL streptomycin (Sigma).
5.2.3 Conjugation of FGF2 to SPIONs
Human FGF2 (Peprotech, Rocky Hill, NJ, USA) was conjugated to dextran-coated PEG-COOH
functionalized super-paramagnetic iron-oxide nanoparticles, SPIONs (micromod
Partikeltechnologie, GmbH, Rostock, Germany) using carbodiimide chemistry as described
previously [25, 27] and depicted in Figure 5A. Briefly, 100 µL of SPIONs (5 mg/mL) were
activated with 35 µmol NHS (N-hydroxysuccinimide, Sigma) and 10 µmol EDC (1-ethyl-3-(3-
dimethylaminopropyl)-carbodiimide HCl, Sigma) prepared in 125 µL of MES ([2(-N-
morpholino) ethanesulfonic acid, Sigma) buffer (pH 6.3). After 45 min of reaction at RT with
gentle shaking, SPIONs were washed thrice with PBS and purified using 30 kDa AmiconTM Ultra
Centrifugal Filters (Merck Millipore, Darmstadt, Germany) by centrifugation at 5000 rpm.
Afterwards, 15 µg of FGF2 (0.06 nmol in 15 µL) was added to the activated SPIONs and left to
react for overnight at 4°C with gentle shaking. Samples were then purified and unconjugated
COOH groups (on SPIONs) were reacted with 10 µg of glycine (Sigma) for 30 min at RT.
Eventually, FGF2-coated SPIONs (FGF2-SPIONs) were purified, resuspended in 100 µL PBS and
stored at 4°C.
5.2.4 Characterization of FGF2-SPIONs
5.2.4.1 Size and zeta potential measurements
The size of SPIONs and FGF2-SPIONs was measured using dynamic light scattering (DLS) with
Zetasizer Nano (Malvern Instruments, UK). Briefly, 5 µL of SPIONs or FGF2-SPIONs were
diluted in 1 mL PBS and measured in 1mL disposable polystyrene cuvettes. For zeta potential
measurements, 5 µL of SPIONs or FGF2-SPIONs were diluted in 1 mL KCl (10mM) and
measured in folded capillary cells DTS1060 (Malvern Instruments).

129
5.2.4.2 Dot blot analysis
Conjugation efficiency of FGF2 on SPIONs was determined by Dot blot analysis as reported
previously [25, 27]. Briefly, FGF2, SPIONs and FGF2-SPIONs were serially diluted in TBS buffer
(Thermo Scientific, Rockford, IL, USA). 2 µL of the samples were spotted on the nitrocellulose
membrane and allowed to dry for 10 min. The membrane was blocked for 1 h with 5%
blotting-grade blocker (BioRad, Hercules, CA, USA) prepared in TBS buffer Tween®-20 (TBST)
(Thermo Scientific). Afterwards, the membrane was incubated with rabbit anti-FGF2
monoclonal antibody (1:1000; Cell Signaling Technology, Massachusetts, MA, USA) for 1 h.
Membrane was washed three times in TBST and incubated for 1 h with secondary rabbit anti-
rat HRP-conjugated polyclonal antibody (1:1000, Dako, Glostrup, Denmark) followed by
tertiary goat-anti rabbit anti-rat HRP-conjugated polyclonal antibody (1:1000, Dako).
Afterwards, the blot was developed using PierceTM ECL Plus Western Blotting substrate
(Thermo Scientific) and was imaged using FluorChem Imaging System (ProteinSimple, Alpha
Innotech, San Leandro, CA, USA). The intensity of the dots were quantified using NIH ImageJ
software (NIH, Bethesda, MD) and the conjugation efficiency was calculated using the
standard curves prepared from known concentrations of FGF2 dots.
5.2.4.3 Prussian blue iron staining
To estimate the recovery of SPIONs during the conjugation process, Prussian Blue staining kit
(Sigma) was performed on the dot blots as per the manufacturer’s instructions and described
previously [25, 27]. Briefly, FGF2, SPIONs and FGF2-SPIONs were serially diluted in TBS buffer
(Thermo Scientific). 2 µL of the samples were spotted on the nitrocellulose membrane and
allowed to dry for 10 min. Iron was detected using Prussian Blue staining kit (Sigma)
containing potassium ferrocyanide and hydrochloric acid in 1:1 ratio. Images were captured
using a normal digital camera. The intensity of the dots were quantified using NIH ImageJ
software (NIH, Bethesda, MD) and the amount of iron staining (representative of SPIONs) was
calculated using the standard curves prepared from known amounts of SPIONs dots.
5.2.5 Cell binding and uptake experiments
Cell binding and uptake studies were performed as per the standardized protocols reported
previously [25]. Briefly, LX2 cells were seeded at 1 x 104 cells/well and cultured overnight.
Cells were then serum-starved for overnight and then incubated with 5 ng/mL TGFβ (Roche,

130
Mannheim, Germany) for 24 h. TGFβ-activated LX2 cells were then incubated with SPIONs or
FGF2-SPIONs at RT for 2 h (binding study) or at 37°C for 4 h (uptake study) in serum-free
medium supplemented with 0.5 % BSA (bovine serum albumin). After incubation, cells were
washed thrice with Dulbecco’s phosphate buffer saline (DPBS, Lonza) and were fixed with 4%
formalin and stained using Prussian Blue iron staining as per manufacturer’s instructions.
Images were captured using Nikon E400 microscope (Nikon, Tokyo, Japan), cellular uptake
was assessed by counting iron-positive cells/per field and has been depicted as % uptake.
5.2.6 Immunofluorescent staining
Cells were seeded in 24-well plates (3 x 104 cells/well) and cultured overnight. Cells were then
serum-starved for overnight and incubated with either starvation medium alone, different
concentrations of FGF2 (50 ng/mL, 100 ng/mL or 250 ng/mL) or 250 ng/mL FGF2, SPIONs, or
FGF2-SPIONs, and 5 ng/mL TGFβ for 24 h. Afterwards the cells were washed with 1x PBS, fixed
with ice-cold acetone and methanol (1:1 ratio) for 30 min at -20 °C followed by drying for 30
min at RT and rehydration with 1x PBS. The staining was performed using rabbit anti-FGFR1
(1:100) or goat anti-collagen I (1:100). Briefly, cells were incubated with the respective
primary antibodies (refer to Table S1) followed by incubation with Alexa 488-conjugated
secondary antibodies (Life Technologies, Gaithersburg, MD, USA). Cells were then mounted
with DAPI-containing mounting medium (Sigma). The staining was visualized, the images were
captured using fluorescent microscopy (Evos microscope, Tokyo, Japan), analysed using NIH
ImageJ software and presented as relative expression versus TGFβ-treated LX2 cells.
5.2.7 Cell viability studies
Cells were seeded in 96-well plates (5000 cells/well) and cultured overnight. Cells were
serum-starved for overnight and incubated with starvation medium alone, different
concentrations of FGF2 (50 ng/mL, 100 ng/mL or 250 ng/mL) or 250 ng/mL FGF2, SPIONs, or
FGF2-SPIONs, and 5 ng/mL TGFβ for 24 h. Cells were then incubated with Alamar blue reagent
(Invitrogen), incubated for 4 h and fluorescent signal was measured using a VIKTORTM plate
reader (Perkin Elmer, Waltham, MA).
5.2.8 Western blot analysis
Cells were seeded in 12-well plates (8 x 104 cells/well) and cultured overnight. Cells were
serum-starved for overnight and incubated with starvation medium alone, different

131
concentrations of FGF2 (50 ng/mL, 100 ng/mL or 250 ng/mL) or 250 ng/mL FGF2, SPIONs, or
FGF2-SPIONs, and 5 ng/mL TGFβ for 24 h. Cells were lysed using 1x lysis buffer prepared from
3x blue loading buffer and 30x reducing agent (1.25 M dithiothreitol, DTT) (Cell Signaling
Technology, Massachusetts, MA, USA) as per manufacturer’s instructions. The prepared
samples were loaded on 10 % Tris-glycine gels (Life Technologies) followed by transfer to the
PVDF membrane (Roche). The membranes were developed according to the standard
protocols using primary and secondary antibodies (refer to Table S1). The bands were
visualized using PierceTM ECL Plus Western Blotting substrate (Thermo Scientific) and
photographed using FluorChem Imaging System. Intensity of individual bands was quantified
using NIH ImageJ software, normalized with respective β-actin bands and presented as
relative expression versus TGFβ-treated LX2 cells.
5.2.9 Quantitative real time PCR
Cells were seeded in 12-well plates (8 x 104 cells/well) and cultured overnight. Cells were then
serum-starved for overnight and incubated with starvation medium alone, different
concentrations of FGF2 (50 ng/mL, 100 ng/mL or 250 ng/mL) or 250 ng/mL FGF2, SPIONs, or
FGF2-SPIONs, and 5 ng/mL TGFβ for 24 h. Cells were then lysed using RNA lysis buffer. Total
RNA from cells was isolated using the GenElute Total RNA Miniprep Kit (Sigma) according to
the manufacturer’s instructions. The RNA concentration was quantified using NanoDrop® ND-
1000 Spectrophotometer (Thermo Scientific, Waltham, USA). Total RNA (1 µg) was reverse
transcribed using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). Real-time PCR
was performed using 20 ng of cDNA, pre-tested gene-specific primer sets (listed in Tables S2)
and the 2x SensiMix SYBR and Fluorescein Kit (Bioline GmbH, QT615-05, Luckenwalde,
Germany) according to the manufacturer’s instructions. Finally, cycle threshold (Ct) values
were normalized to the reference gene 18s rRNA, and relative expression were calculated
using the 2-ΔΔCt method versus TGFβ-treated LX2 cells.
5.2.10 3D collagen gel cell contraction assay
A collagen suspension (5 mL) containing 3 mL of collagen G1 (5 mg/mL, Matrix biosciences,
Morlenbach, Germany), 0.5 mL of 10x M199 medium (Sigma), 85 µL of 1 N NaOH (Sigma) and
sterile water was mixed with 1 mL (2 x 106) of LX2 cells. The gel and cell suspension (0.6
mL/well) was plated in a 24-well culture plate and was allowed to polymerize. Polymerized

132
gels were incubated with 1 mL of serum-starved medium with or without human recombinant
TGFβ (5 ng/mL) together with different concentrations of FGF2 (50 ng/mL, 100 ng/mL or 250
ng/mL), or 250 ng/mL of FGF2, FGF2-SPIONs, and SPIONs (equimolar concentration), followed
by detachment of the gels. The size of the gels were digitally measured, normalized with their
respective well size in each image and presented as relative gel contraction versus TGFβ-
treated LX2 cells.
5.2.11 Cell migration assay
Cells were plated in 12-well culture plates (1 x 105 cells/well), cultured overnight and serum-
starved for 24 h. A standardized scratch was made using a 200 µL pipette tip fixed in a custom-
designed holder. Afterwards, cells were washed twice and incubated with starvation medium
(control), or with 5 ng/mL TGFβ prepared with or without different concentrations of FGF2
(50 ng/mL, 100 ng/mL or 250 ng/mL), or 250 ng/mL FGF2, SPIONs, or FGF2-SPIONs.
Microscopic images were taken at 0 and 24 h to measure the size of the scratch. Photographs
were analyzed using NIH ImageJ software, normalized with 0 h time point and presented as
relative wound healing versus TGFβ-treated LX2 cells.
5.2.12 CCl4-induced acute liver injury mouse model
All the animal experiments were carried out strictly according to the ethical guidelines for the
Care and Use of Laboratory Animals (Utrecht University, The Netherlands). Male Balb/c mice
(8-10 weeks old) were given single intraperitoneal injection of 1.0 mL/kg carbon tetrachloride
(CCl4, Sigma). CCl4-treated mice were treated with two intravenous administrations of PBS
(n=5), FGF2 (250 ng/dose, n=5) or FGF2-SPIONs (250 ng/dose, n=6) at day 3 and day 5. Healthy
controls (n=6) received olive oil alone. All the animals were euthanized at day 6. Liver tissues
and blood samples were retrieved for further analyses. All the animals were weighed before
sacrificing, and the respective organs were weighed directly after sacrificing. Alanine
aminotransferase (ALT) activity was determined in the plasma samples using a colorimetric
ALT activity assay kit (Sigma MAK052) according to the manufacturer’s instructions.
5.2.13 Histological immunostainings
Collected liver tissues were transferred to Tissue-Tek OCT embedding medium (Sakura
Finetek, Torrance, CA, USA) and snap-frozen in 2-methyl butane in a dry ice. Cryosections (5
µm) were cut using a Leica CM 3050 cryostat (Leica Microsystems, Nussloch, Germany). The

133
cryosections were air-dried and fixed with acetone for 20 minutes. Tissue sections were
rehydrated with PBS and incubated with the primary antibody (Collagen I or F4/80) overnight
at 4°C (refer to Table S1). This was followed by incubation with horseradish peroxidase (HRP)-
conjugated secondary antibody for 1 h at RT. Next, the samples were incubated with HRP-
conjugated tertiary antibody for 1 h at RT. Thereafter, peroxidase activity was developed
using the AEC (3-amino-9-ethyl carbazole) substrate kit (Life Technologies) for 20 minute and
nuclei were counterstained with hematoxylin (Fluka Chemie, Buchs, Switzerland).
Endogenous peroxidase activity was blocked by 3% H2O2 prepared in methanol. The sections
were mounted with Aquatex mounting medium (Merck) and were scanned using Hamamatsu
NanoZoomer Digital slide scanner 2.0HT (Hamamatsu Photonics). For quantitation, the high
resolution scans were viewed using NanoZoomer Digital Pathology (NDP2.0) viewer software
(Hamamatsu Photonics). About 20 images (100x) of each stained liver tissue section (from
NDP) were imported into ImageJ software and were analyzed quantitatively at a fixed
threshold.
5.2.13.1 Prussian blue staining combined with collagen I immunostaining
Cryosections were dried and fixed with 4% formaldehyde (Sigma) for 30 min. The sections
were washed thrice with 1x PBS and incubated with primary collagen I antibody for 1 h at RT,
followed by incubation with 0.3% hydrogen peroxide for 30 min. Next, the sections were
incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h and
HRP-conjugated tertiary antibody for 1 h. Sections were then stained with AEC for 20 min as
per manufacturer’s instructions. Thereafter, the sections were washed with Milli Q water and
incubated with freshly prepared Prussian blue solution (Sigma). The sections were incubated
with Prussian blue mix solution for 30 min, washed in deionized water followed by mounting
with aquatex mounting medium, and imaged using NanoZoomer.
5.2.14 Graphs and statistical analyses
All graphs were made using GraphPad Prism version 8.4.1 (GraphPad Prism, La Jolla, CA, USA).
The results are expressed as the mean ± standard error of the mean (SEM). Statistical analyses
were performed using GraphPad Prism version 8.4.1 (GraphPad Prism, La Jolla, CA, USA).
Multiple comparisons between different groups were calculated using the one-way analysis
of variance (ANOVA) with the Bonferroni post hoc test. Statistical differences between two

134
groups were calculated using a two-tailed unpaired t test. Differences were considered
significant when #p < 0.05, ##p < 0.01, ###p < 0.001 or *p < 0.05, **p < 0.01, ***p < 0.001.
5.3 Results
5.3.1 Upregulation of FGF receptors (FGFRs) mRNA expression in cirrhosis patients
We first examined the mRNA expression levels of fibroblast growth factor receptors (FGFR)
and fibroblast growth factor 2 (FGF2) in human cirrhotic liver tissues using the publicly
available human microarray datasets GSE6764 from NCBI GEO database [26]. Patients were
categorized into two groups: normal and cirrhosis representing normal and cirrhotic livers
respectively. The available corresponding microarray datasets were analyzed using an online
database GEO2R. There are four known isoforms of FGFRs: FGFR1, FGFR2, FGFR3, FGFR4, and
all FGFRs are shown to be expressed in the liver [28]. Transcriptomic data analysis revealed a
significant increase in the mRNA expression levels of FGFR1 (p<0.05), FGFR2 (p<0.001) and
FGFR3 (p<0.05) in the cirrhotic livers compared to the normal livers (p<0.05) (Figure 1A).
However, no significant difference in FGFR4 mRNA expression (p=0.1037) was observed in
cirrhotic versus normal livers. Plasma levels of basic FGF (FGF2) were found to be significantly
elevated with the progression of liver disease (chronic hepatitis > liver cirrhosis >
hepatocellular carcinoma (HCC) [29]. However, no significant difference in FGF2 mRNA
expression, in the analyzed dataset GSE6764, was observed in the cirrhotic human livers as
compared to the normal healthy livers (Figure 1A).
5.3.2 Upregulation of FGFRs expression in vitro in human HSCs (LX2 cells)
FGF receptors have shown to be expressed by hepatocytes and HSCs, and are involved in
epithelium-mesenchymal paracrine signaling thereby regulating organogenesis [8]. During
liver injury, different FGFs produced by HSCs bind and activate the FGFRs on hepatocytes, and
hepatocyte-derived FGFs bind and activate FGFRs on HSCs thereby regulating hepatic
fibrogenesis [9]. Here, we have examined the expression levels of different FGF receptors and
FGF2 in control and human HSCs activated by TGFβ since TGFβ is the major fibrogenic cytokine
responsible for the activation and trans-differentiation of quiescent HSCs in the liver. We
activated LX2 cells (a human HSC cell line) with 5 ng/mL of recombinant human TGFβ for 24 h
and examined the mRNA expression levels of FGFRs and FGF2 in TGFβ-activated LX2 cells
versus control LX2 cells. We observed that mRNA expression of the FGFRs isoforms: FGFR1c

135
(p<0.05), FGFR2c (p<0.05), FGFR3c (p<0.05) and FGFR4 (p<0.05) was significantly upregulated
in TGFβ-activated LX2 cells compared to control cells as shown in Figure 1B. No significant
differences in the mRNA expression for FGFR1b, FGFR2b and FGFR3b was observed in
activated human HSCs [30]. Moreover, we found a significant increase in FGF2 mRNA levels
(Figure 1B), which is in accord with the recent study whereby induced FGF2 mRNA was
observed in activated humas HSCs [30]. Since FGF2 primarily interacts with FGFR1,
overexpressed by human myofibroblasts [21], we assessed the protein expression of FGFR1
using western blot analysis and immunofluorescence staining in TGFβ-activated HSCs versus
control cells. We found that FGFR1 was highly upregulated in the TGFβ-induced LX2 cells as
compared to control cells as shown in Figure 1C,D.
Figure 1. Expression of fibroblast growth factor receptors (FGFR) and FGF2 in human cirrhotic livers and TGFβ-activated human hepatic stellate cells (HSCs, LX2 cells). (A) Normalized FGFR1, FGFR2, FGFR3, FGFR4, and FGF2 mRNA expression levels from publicly available human microarray datasets (GSE6764). Normal livers (n = 10) and cirrhosis livers (n = 12). (B) Relative mRNA expression (normalized with 18 s rRNA) of FGFR1c, FGFR2c, FGFR3c, FGFR4, and FGF2 in control and TGFβ-activated LX2 cells, n = 6. (C) Representative image and quantitative analysis of western blot showing expression of FGFR1 and β-actin, performed on control and TGFβ-activated LX2 cells, n = 6. (D) Representative immunofluorescent images showing expression of FGFR1 (green) in control and TGFβ-activated HSCs (LX2 cells). Blue staining represents DAPI-nuclear staining, n = 3. Graphs represent

136
mean + SEM; statistical differences were calculated using two-tailed unpaired t-test, *P < 0.05, ***P < 0.001.
5.3.3 FGF2 inhibited collagen and α-SMA expression in TGFβ-activated human HSCs (LX2 cells) via pAKT signaling pathway
FGFs are categorized into seven subfamilies: FGF1, 4, 8, 9, 10, 11 and 19 as mentioned before
[8, 9]. Among other subfamilies, FGF1 subfamily, FGF1 and FGF2 have been investigated for
their effects on HSCs and liver fibrogenesis [9]. In particular, FGF2 has been shown to regulate
HSCs activation [9].
Figure 2. Effects of FGF2 on collagen I and α-SMA expression on TGFβ-activated LX2 cells. (A) Representative immunofluorescent images and (B) quantitative analysis of collagen I (green colour) stained control and TGFβ-activated LX2 cells treated with medium alone or FGF2 (50, 100 and 250 ng/mL), n=7. Blue staining represents DAPI-nuclear staining. (C) Relative gene expression analysis (normalized with 18 s rRNA) for α-SMA and collagen I as examined in control and TGFβ-activated LX2

137
cells treated with medium alone or FGF2 in different concentration (50, 100 and 250 ng/mL), n = 4. Graphs represent mean + SEM, statistical differences were calculated using one-way ANOVA with the Bonferroni post hoc test, ##P < 0.01, ###P < 0.001 represent significance versus control cells; *P < 0.05, **P < 0.01, ***P < 0.001 represent significance versus TGFβ-treated cells.
In this study, we investigated the effects of FGF2 on TGFβ-activated HSCs. Upon incubation
with TGFβ, we observed a significant upregulation of major extracellular matrix (ECM)
protein, collagen I and mRNA expression of major HSCs activation markers, alpha-smooth
muscle actin (α-SMA), collagen I and TGFβ, in TGFβ-activated LX2 compared to control LX2
cells. After treatment with increasing concentrations of FGF2, dose-dependent inhibition in
collagen I protein expression and mRNA expression of α-SMA, collagen I and TGFβ was found
(Figure 2A-C and Figure S1).
Moreover, FGF2-mediated dose-dependent inhibition of TGFβ-induced collagen I and α-SMA
protein expression which was confirmed by western blot analysis (Figure 3). We also
investigated the possible mechanism of action underlying FGF2-mediated inhibitory effects
on TGFβ-activated HSCs. Previously, it has been shown that FGF2 regulates proliferation,
migration, and invasion of cancer cells through phosphatidylinositol 3-kinase (PI3K)/AKT
signaling pathway [31]. In an ischemia-reperfusion induced renal injury model, it has been
demonstrated that selective inhibition of phosphatidylinositol 3-kinase (PI3K) [and mitogen-
activated protein kinase kinase (MAPKK or MEK)] abolished the protective effects of FGF2
confirming an involvement of PI3K/AKT (and MEK/ERK signaling pathways) in FGF2-mediated
effects [17]. Moreover, activation of PI3K/AKT signaling pathway, regulated by TGFβ, has been
shown to stimulate collagen synthesis by fibroblasts in different fibrotic diseases e.g. by HSCs
in hepatic fibrogenesis [32]. In this study, we examined the expression of p-AKT in control LX2
cells and TGFβ-activated LX2 cells treated without and with increasing concentrations of
FGF2. We observed that TGFβ led to an increased expression of pAKT in LX2 cells which was
dose-dependently inhibited with increasing concentrations of FGF2 (Figure 3).

138
Figure 3. FGF2 mediates the effects on TGFβ-activated LX2 cells via pAKT signaling pathway. Representative images and quantitative analysis of western blot depicting bands for collagen I, pAkt, Akt, α-SMA and β-actin, examined in control and TGFβ-activated LX2 cells treated with medium alone or increasing concentrations of FGF2 (50, 100 and 250 ng/mL), n = 3. Graphs represent mean + SEM, statistical differences were calculated using one-way ANOVA with the Bonferroni post hoc test, ##P < 0.01, ###P < 0.001 represent significance versus control cells; *P < 0.05, **P < 0.01, ***P < 0.001 represent significance versus TGFβ-treated cells.
5.3.4 FGF2 inhibited TGFβ-induced migration and collagen gel contractility of human HSCs (LX2 cells)
Upon TGFβ activation, HSCs become highly migratory and contractile cells that drives the
fibrotic alterations associated with chronic liver disease [3, 6]. Therefore, we also assessed
the effects of FGF2 on TGFβ-induced HSCs migration and contraction. We performed scratch
assay to assess HSCs migration and observed that TGFβ potentiated migration of HSCs which
was significantly and dose-dependently inhibited by an increasing concentrations of FGF2
(Figure 4A,C). We also examined the contraction using a 3D collagen gel contraction assay
and found that FGF2 dose-dependently decreased TGFβ-induced HSCs contraction (Figure

139
4B,D). Finally, we analysed the effect of FGF2 on cell viability and observed no significant
differences on cell viability with tested FGF2 concentrations (Figure 4E).
Figure 4. Efficacy of FGF2 on TGFβ-induced migration and contractility of TGFβ-activated LX2 cells. (A) Representative images (at 0 and 24 h) and (C) quantitative analysis (after 24 h) of migration by control and TGFβ-induced LX2 cells treated with medium alone or FGF2 (50, 100 and 250 ng/mL). (B) Representative images (after 72 h) and (D) quantitative analysis of 3D collagen gel contraction by control and TGFβ-activated LX2 cells treated with medium alone or FGF2 (50, 100 and 250 ng/mL). (E) % cell viability of control cells and TGFβ-activated LX2 cells treated with medium alone or FGF2 (50, 100 and 250 ng/mL). Graphs represent mean + SEM, statistical differences were calculated using one-way ANOVA with the Bonferroni post hoc test, ##P < 0.01, ###P < 0.001 represent significance versus control cells; **P < 0.01, ***P < 0.001 represent significance versus TGFβ-treated cells.

140
5.3.5 Characterization of FGF2-SPIONs
After confirming the pharmacological effects of FGF2 in vitro on TGFβ-activated HSCs, we
conjugated FGF2 protein to SPIONs. Biologics such as peptides and proteins have very short
half-life and are highly prone to enzymatic degradation and therefore are rapidly eliminated
from the body or degraded reducing their therapeutic efficacy [33]. Several strategies
including nanocarriers-mediated peptide/protein drugs delivery have been evolved over
years to protect them from degradation and to prolong their systemic half-life [34]. In this
study, we used SPIONs to improve the half-life of FGF2 and to target FGF2 to the livers. We
conjugated FGF2 to dextran-coated PEG-COOH functionalized SPIONs using carbodiimide
reaction chemistry as illustrated in a schematic picture in Figure 5A.
We conjugated the cysteine part of the FGF2 to the functionalized SPIONs and left the FGF2
receptor-binding domain abide to interact with FGFR1 on the HSCs surface. The conjugation
of FGF2 to SPIONs was confirmed by changes in size and zeta potential, and by dot-blot
analysis. Dynamic light scattering (DLS) measurements showed a slight increase in the mean
hydrodynamic size of SPIONs after conjugation with FGF2 (95 nm FGF2-SPIONs versus-80 nm
SPIONs) (Figure 5B). We also observed an increase in the negative surface charge (zeta
potential) of the particles after FGF2 conjugation (mean value of about -10 mV FGF2-SPIONs
versus -4 mV SPIONs) (Figure 5B). The successful conjugation of FGF2 to SPIONs was
confirmed by a dot blot analysis using anti-FGF2 antibody (Figure 5C). Based on the
quantitative analysis of FGF2 and FGF2-SPIONs, about 82% of FGF2 was estimated to be
conjugated to the SPIONs as derived from FGF2 standard curves presented in Figure S2.
Prussian blue iron-staining and the respective quantitative analysis confirmed that about 88%
SPIONs were recovered after conjugation due to the nanoparticle loss during the purification
steps as determined by SPIONs standard curves presented in Figure S2.
Quantitative analysis of dot blot FGF2 staining and iron staining (Figure S2) suggested 93%
conjugation of FGF2 to SPIONs indicating 4-5 FGF2 molecules per SPION. Finally, we
characterized the binding and uptake of FGF2-SPIONs in the TGFβ-activated LX2 cells. The
result demonstrated an increase in binding and uptake of FGF2-SPIONs as compared to the
unconjugated SPIONs (Figure 5D).

141
Subsequently, we analyzed the effect of FGF2-SPIONs and SPIONs on cell viability and
observed no significant differences on cell viability with tested concentrations (up to 250
ng/mL) (Figure 5E). FGF2-SPIONs were examined for long-term stability at 4°C using DLS
measurement (size) and dot blot (FGF2 conjugation/release) analysis. The results showed that
FGF2-SPIONs retained their size and FGF2 conjugation after 4 weeks of storage at 4°C.
Figure 5. Characterization and HSCs-specific binding/uptake of FGF2-SPIONs. (A) Schematic representation of FGF2 conjugation to SPION using carbodiimide chemistry. (B) Schematic of FGF2-SPIONs and table showing the hydrodynamic size, polydispersity index (PDI) and zeta potential of SPIONs and FGF2-SPIONs from n = 3 independent conjugations. (C) Dot blot image displaying FGF2 conjugation on SPIONs performed by FGF2 staining and SPIONs detection by Prussian Blue iron staining. FGF2-SPIONs, FGF2 and SPIONs were serially diluted and spotted on the membrane, thus the different dots in the figure represents the serial dilution of the respective samples. (D) Representative microscopic images and quantitative image analysis showing binding and uptake of FGF2-SPIONs versus SPIONs in TGFβ-activated LX2 cells, n = 8. (E) % cell viability of control LX2 cells and TGFβ-

142
activated LX2 cells with and without SPIONs, FGF2, FGF2-SPIONs treatment, n=8. Graphs represent mean + SEM; statistical differences were calculated using two-tailed unpaired t test, ***P < 0.001.
5.3.6 FGF2-SPIONs inhibited expression fibrosis markers, migration, and contractility of human HSCs (LX2) in vitro
Following FGF2 conjugation to SPIONs, we examined whether FGF2 retains its
pharmacological activity after chemical conjugation. To assess the effects of FGF2-SPIONs, we
used 250 ng/mL FGF2 based on our previous dose-dependent studies (Figure. 2,3,4). We
investigated the effects of FGF2-SPIONs on collagen I and α-SMA protein expression, and
collagen I, α-SMA and TGFβ1 mRNA expression on TGFβ-activated LX2 cells and found that
FGF2-SPIONs reduced both TGFβ-induced α-SMA and collagen I expression, and TGFβ1 mRNA
expression (Figure 6A-D and Figure S3).
Figure 6. Effects of FGF2-SPIONs on TGFβ-activated LX2 cells. (A) Representative immunofluorescent images (n = 4) and quantitative analysis of collagen I stained control and TGFβ-activated LX2 cells treated with medium alone, FGF2, FGF2-SPIONs or SPIONs. (B) Gene expression analysis for α-SMA and collagen I (normalized with 18 s rRNA) in control and TGFβ-activated LX2 cells treated with

143
medium alone, FGF2, FGF2-SPIONs or SPIONs, n = 6. (C) Representative images and (D) quantitative analysis of western blot depicting bands for collagen I, pAkt, Akt, α-SMA and β-actin, examined in control and TGFβ-activated LX2 cells treated with medium alone, FGF2, FGF2-SPIONs or SPIONs, n = 3. Graphs represent mean + SEM, statistical differences were calculated using one-way ANOVA with the Bonferroni post hoc test, #p<0.05, ##P < 0.01, ###P < 0.001 represent significance versus control cells; *P < 0.05, **P < 0.01, ***P < 0.001 represent significance versus TGFβ-treated cells.
Collaborating with our previous results, FGF2-SPIONs also inhibited the protein expression of
pAkt in TGFβ-activated LX2 cells (Figure 6C,D). We further examined the effect of FGF2-
SPIONs on TGFβ-induced HSCs migration and contraction and observed that FGF2-SPIONs
attenuated TGFβ-induced HSCs migration (Figure 7A) and contraction (Figure 7B). The results
clearly demonstrated that conjugation of FGF2 to SPIONs retained the pharmacological
effects of FGF2 and showed similar or slightly improved HSCs inhibitory effects as compared
to free unconjugated FGF2. Of note, SPIONs alone did not show significant inhibition of HSCs
as also reported previously [25].
Figure 7. Effects of FGF2-SPIONs on TGFβ-induced migration and contractility of human HSCs (LX2). (A) Representative images (at 0 and 24 h) and quantitative analysis (after 24 h) of migration by control and TGFβ-induced LX2 cells treated with medium alone, FGF2, FGF2-SPIONs or SPIONs. (B) Representative images (after 72 h) and quantitative analysis of 3D collagen gel contraction containing control and TGFβ-activated LX2 cells treated with medium alone, FGF2, FGF2-SPIONs or SPIONs, n = 6. Graphs represent mean + SEM, statistical differences were calculated using one-way ANOVA with the Bonferroni post hoc test. #P < 0.05, ##P < 0.05 versus control LX2 cells; *P < 0.05, **P < 0.01, ***P < 0.001 versus TGFβ-treated LX2 cells.

144
5.3.7 FGF2-SPIONs ameliorated fibrosis and inflammation in CCl4-induced acute liver injury mouse model
FGF2-SPIONs were subsequently evaluated for their therapeutic effects in the acute CCl4-
induced liver injury mouse model. Single CCl4 administration resulted in an increased intra-
hepatic collagen I (major ECM marker indicative of HSCs activation and ECM production, and
fibrogenesis) and increased F4/80 (macrophage marker indicative of macrophage-driven liver
inflammation) expression in CCl4-treated mice when compared with olive oil treated control
mice. FGF2-SPIONs (and not free FGF2) significantly attenuated collagen I and F4/80 protein
expression when compared with CCl4 mice or CCl4 mice treated with free FGF2 (P < 0.05) as
shown in Figure 8A,B.
Furthermore, % liver weight (normalized with respective body weight) was found to be
increased in CCl4 mice and CCl4 mice treated with free FGF2. Notably CCl4 mice treated with
FGF2-SPIONs showed significantly lowered % liver weights when compared with CCl4 mice (P
< 0.001), or CCl4 mice treated with free FGF2 (P < 0.01) (Figure 8C). We further examined the
localization of FGF2-SPIONs in the fibrotic livers. Following Prussian blue iron staining of
collagen-I stained liver sections, we observed localization of FGF2-SPIONs (stained with
Prussian blue) in the fibrotic regions stained with collagen-I secreted by activated HSCs
confirming HSCs-specific localization of FGF2-SPIONs in the fibrotic livers (Figure S4). This
observation aligns and supports our previous findings where we have shown an increased,
possibly HSCs-specific, uptake of RLX-SPIONs (relaxin-conjugated SPIONs) in the fibrotic livers
when compared to unconjugated SPIONs using MRI [25].
Finally, we examined plasma ALT levels and observed highly significant downregulation in ALT
levels following treatment with FGF2-SPIONs when compared with CCl4 mice (P < 0.001) or
CCl4 mice treated with free FGF2 (P < 0.05) (Figure 8D). Based on the previous in vitro results
and our previous studies [25, 27, 35] demonstrating that multiple administrations of SPIONs
didn’t show any beneficial therapeutic effects, SPIONs were not tested in vivo in the acute
CCl4-induced liver injury mouse model.
5.4 Discussion
In this study, we demonstrated the improved therapeutic efficacy of FGF2 after conjugation
with SPIONs, as a promising approach for the treatment of liver fibrosis. We confirmed an

145
upregulation of intrahepatic mRNA expression of FGFRs in cirrhotic patients and TGFβ-
activated HSCs. We further observed an increased protein expression of FGFR1, a major FGF2-
binding receptor in TGFβ-activated HSCs. Human recombinant FGF2 attenuated TGFβ-
induced HSCs activation, collagen I production, migration and contraction mediated via the
pAKT pathway. FGF2-conjugated SPIONs showed improved inhibition of TGFβ-activated HSCs
in vitro, and ameliorated inflammation and collagen I production in vitro in the acute CCl4-
induced liver injury mouse model.
Acute liver injury is a transient wound healing response characterized by liver inflammation
(increased infiltration and activation of macrophages) and fibrogenesis (enhanced activation
of quiescent HSCs). When the injury persists, liver undergous progressive scarring and
degeneration in a process known as fibrosis. HSCs are the main pathogenic cells responsible
for the production of abnormal fibrillar collagens in liver fibrosis. Therefore, substantial
efforts have been made to target HSCs for the treatment of liver fibrosis which showed
promising results [3-5, 7]. Upon hepatocellular injury, HSCs are activated in the presence of
growth factors, importantly TGFβ that results in activation, proliferation, migration,
contraction and collagen production by HSCs. Activation of HSCs are regulated by several
growth factors and signaling pathways [3, 4, 6]. Among others, FGFs have also been shown to
regulate HSCs [13], in particular FGF2 that has been shown to possess both pro- and anti-
fibrotic effects [9, 13]. In the present study, we found that exogenous low-molecular weight
FGF2 attenuated HSCs activation, migration, contraction and collagen expression. Our results
are in complete agreement with a study where high- and low-molecular weight FGF2 isoforms
were compared and demonstrated that low-molecular weight FGF2, as also used in this study,
potently ameliorated HSCs activation [15].
FGF2 can bind to different FGFR receptors with varying affinity, however it mainly interacts
with FGFR1 that has shown to be overexpressed on HSCs [21]. In this study, we also assessed
the expression levels of different types of FGFRs in human cirrhosis and in TGFβ-activated
human HSCs. Interestingly we found that almost all of the different FGFRs (except FGFR4)
were upregulated in human cirrhotic livers. FGFR1c, FGFR2c, FGFR3c and FGFR4 mRNA levels
were upregulated in TGFβ-activated human HSCs, and FGFR1 protein was highly expressed
on TGFβ-activated human HSCs.

146
Figure 8. Effects of FGF2-SPIONs in CCl4-induced acute liver injury mouse model. (A) Representative images and (B) quantitative analysis of collagen I (ECM marker) and F4/80 (macrophage marker) stained liver sections from control group (n = 6), CCl4 group (CCl4 mice treated with PBS, n = 5), CCl4 + FGF2 group (CCl4 mice treated with free FGF2, n = 5), and CCl4 + FGF2-SPIONs group (CCl4 mice treated with FGF2-SPIONs, n = 6). (C) % liver weights (normalized with respective body weights) from control group (n = 6), CCl4 group (CCl4 mice treated with PBS, n = 5), CCl4 + FGF2 group (CCl4 mice treated with free FGF2, n = 5), and CCl4 + FGF2-SPIONs group (CCl4 mice treated with FGF2-SPIONs, n = 6). (D) Alanine aminotransferase (ALT) levels (U/I) as analyzed in the plasma from different treatment groups. (B-D): Each symbol represents individual mice. Graphs represent mean + SEM, statistical differences were calculated using one-way ANOVA with the Bonferroni post hoc test. *P < 0.05, **P < 0.01, ***P < 0.001.
Although several mechanisms have been proposed with regard to FGF2-FGFR1 [13, 15, 36], in
this study we found an involvement of PI3K/AKT signaling pathway since pAKT expression was
significantly dose-dependently inhibited by an increasing concentrations of FGF2. These
results corroborates with the previous reports where selective inhibition of the PI3K/AKT
signaling pathway was shown to abrogate the protective functions of FGF2 [13, 17, 31, 32].
Our results confirmed the anti-fibrotic effects of FGF2. However, FGF2, like other biologics,
has several limitations such as short systemic half-life and susceptibility to enzymatic
degradation thereby reduced stability [22]. rFGF2 when administered with heparin showed

147
improved systemic circulation of FGF2 with slower clearance of heparin/rFGF-2 complexes
[22]. Nanotechnological advancements in the last decades have improved the
pharmacokinetics and targeted delivery of biologics e.g. using nanocarriers [33, 37]. SPIONs
provide several other advantages such as small size and dextran-PEG surface coating for
evading the mononuclear phagocytic system (MPS) and a large surface area allowing surface
conjugation and detection using MRI [23, 37, 38].
FGF2 has been shown to possess anti-fibrotic effects as also reported by others [13, 14, 15,
21, 36]. In this study, by coupling SPIONs to the SPIONs, we aimed to enhance the systemic
half-life and stability, and improve the targeting of FGF2 to the liver. Following successful
synthesis of FGF2-SPIONs, we confirmed the biological activity of FGF2, and found that FGF2-
SPIONs showed enhanced inhibition of HSCs activation, migration, contraction and collagen
production, most likely, due to the increased FGF2 stability. Subsequently, we investigated
the effects of FGF2 and FGF2-SPIONs in acute CCl4-induced liver injury mouse model. We
found that two doses of 250ng/dose/mouse FGF2-SPIONs attenuated fibrosis and
inflammation in early liver fibrogenesis model as confirmed by collagen I (ECM) expression
and F4/80 (macrophage) expression respectively. FGF2, in unconjugated free didn’t show the
significant inhibition in fibrogenesis in vivo at the tested doses. These results suggests that
SPIONs improved the half-life and possibly liver accumulation and HSCs-specific, and stability
of FGF2 resulting in improved biological efficacy of FGF2 in vivo. Since FGF2 is also known to
be involved in liver homeostasis, tissue repair and regeneration [11, 39, 40], it is also possible
that FGF2-SPIONs positively improve hepatocyte proliferation and hence promote liver
regeneration. To our best knowledge, this is the first study that explore the delivery of FGF2
to the diseased liver. However, this study has been performed in acute liver injury (earlier
liver fibrosis) model that does not correspond to the clinical situation. Patients normally
presents to the clinic when liver damage progresses to cirrhosis associated with liver
dysfunction. Nevertheless, this study provides the first proof-of-concept results highlighting
that FGF2-SPIONs can be a promising therapeutic approach, and therefore should to be
further explored in an advanced models of liver cirrhosis.
In conclusion, this study demonstrates that the SPIONs-mediated delivery of FGF2 potentiates
the therapeutic efficacy of FGF2 in vitro and in vivo thereby suggesting FGF2-SPIONs as a
potential therapeutic approach for the treatment of liver fibrosis. Moreover, SPIONs also

148
provides a possibility for MRI-based diagnosis therefore may also provide a personalized
theranostic approach with combined therapy and diagnosis for personalized disease
management. Altogether, this study presents a novel approach for the delivery of FGF2 as an
effective treatment of liver fibrosis.
5.5 Acknowledgements
This study was supported by the Indonesian Endowment Fund for Education from the
Republic of Indonesia (Lembaga Pengelola Dana Pendidikan/LPDP) and the University of
Twente.
5.6 References
[1] S.K. Asrani, H. Devarbhavi, J. Eaton, P.S. Kamath, Burden of liver diseases in the world, J Hepatol, 70 (2019) 151-171.
[2] R. Bataller, D.A. Brenner, Liver fibrosis, Journal of Clinical Investigation, 115 (2005) 209-218.
[3] S.L. Friedman, Mechanisms of hepatic fibrogenesis, Gastroenterology, 134 (2008) 1655-1669.
[4] R. Bansal, B. Nagorniewicz, J. Prakash, Clinical Advancements in the Targeted Therapies against Liver Fibrosis, Mediators Inflamm, 2016 (2016) 7629724.
[5] D. Schuppan, Y.O. Kim, Evolving therapies for liver fibrosis, J Clin Invest, 123 (2013) 1887-1901.
[6] S.L. Friedman, Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver, Physiol Rev, 88 (2008) 125-172.
[7] S. Yazdani, R. Bansal, J. Prakash, Drug targeting to myofibroblasts: Implications for fibrosis and cancer, Adv Drug Deliv Rev, 121 (2017) 101-116.
[8] D.M. Ornitz, N. Itoh, The Fibroblast Growth Factor signaling pathway, Wiley Interdiscip Rev Dev Biol, 4 (2015) 215-266.
[9] J.D. Schumacher, G.L. Guo, Regulation of Hepatic Stellate Cells and Fibrogenesis by Fibroblast Growth Factors, Biomed Res Int, 2016 (2016) 8323747.
[10] C.M. Teven, E.M. Farina, J. Rivas, R.R. Reid, Fibroblast growth factor (FGF) signaling in development and skeletal diseases, Genes Dis, 1 (2014) 199-213.
[11] S.M. Tsai, D.W. Liu, W.P. Wang, Fibroblast growth factor (Fgf) signaling pathway regulates liver homeostasis in zebrafish, Transgenic Res, 22 (2013) 301-314.
[12] A. Bikfalvi, S. Klein, G. Pintucci, D.B. Rifkin, Biological roles of fibroblast growth factor-2, Endocr Rev, 18 (1997) 26-45.
[13] D.M. Dolivo, S.A. Larson, T. Dominko, Fibroblast Growth Factor 2 as an Antifibrotic: Antagonism of Myofibroblast Differentiation and Suppression of Pro-Fibrotic Gene Expression, Cytokine Growth Factor Rev, 38 (2017) 49-58.
[14] C. Yu, F. Wang, C. Jin, X. Huang, D.L. Miller, C. Basilico, W.L. McKeehan, Role of fibroblast growth factor type 1 and 2 in carbon tetrachloride-induced hepatic injury and fibrogenesis, Am J Pathol, 163 (2003) 1653-1662.

149
[15] R.L. Pan, L.X. Xiang, P. Wang, X.Y. Liu, L. Nie, W. Huang, J.Z. Shao, Low-molecular-weight fibroblast growth factor 2 attenuates hepatic fibrosis by epigenetic down-regulation of Delta-like1, Hepatology, 61 (2015) 1708-1720.
[16] H.Y. Koo, L.M. El-Baz, S. House, S.N. Cilvik, S.J. Dorry, N.M. Shoukry, M.L. Salem, H.S. Hafez, N.O. Dulin, D.M. Ornitz, R.D. Guzy, Fibroblast growth factor 2 decreases bleomycin-induced pulmonary fibrosis and inhibits fibroblast collagen production and myofibroblast differentiation, J Pathol, 246 (2018) 54-66.
[17] X. Tan, Q. Tao, G. Li, L. Xiang, X. Zheng, T. Zhang, C. Wu, D. Li, Fibroblast Growth Factor 2 Attenuates Renal Ischemia-Reperfusion Injury via Inhibition of Endoplasmic Reticulum Stress, Front Cell Dev Biol, 8 (2020) 147.
[18] A.N. Plotnikov, J. Schlessinger, S.R. Hubbard, M. Mohammadi, Structural basis for FGF receptor dimerization and activation, Cell, 98 (1999) 641-650.
[19] A. Chellaiah, W. Yuan, M. Chellaiah, D.M. Ornitz, Mapping ligand binding domains in chimeric fibroblast growth factor receptor molecules. Multiple regions determine ligand binding specificity, J Biol Chem, 274 (1999) 34785-34794.
[20] A.N. Plotnikov, S.R. Hubbard, J. Schlessinger, M. Mohammadi, Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity, Cell, 101 (2000) 413-424.
[21] J. Rosenbaum, S. Blazejewski, A.-M. Preaux, A. Mallat, D. Dhumeaux, P. Mavier, Fibroblast growth factor 2 and transforming growth factor b1 interactions in human liver myofibroblast, Gastroenterology 109 (1995) 1986-1996.
[22] M.A. Bush, E. Samara, M.J. Whitehouse, C. Yoshizawa, D.L. Novicki, M. Pike, R.J. Laham, M. Simons, N.A. Chronos, Pharmacokinetics and pharmacodynamics of recombinant FGF-2 in a phase I trial in coronary artery disease, J Clin Pharmacol, 41 (2001) 378-385.
[23] J.E. Rosen, L. Chan, D.B. Shieh, F.X. Gu, Iron oxide nanoparticles for targeted cancer imaging and diagnostics, Nanomedicine, 8 (2012) 275-290.
[24] C. Tassa, S.Y. Shaw, R. Weissleder, Dextran-coated iron oxide nanoparticles: a versatile platform for targeted molecular imaging, molecular diagnostics, and therapy, Acc Chem Res, 44 (2011) 842-852.
[25] B. Nagorniewicz, D.F. Mardhian, R. Booijink, G. Storm, J. Prakash, R. Bansal, Engineered Relaxin as theranostic nanomedicine to diagnose and ameliorate liver cirrhosis, Nanomedicine, 17 (2019) 106-118.
[26] E. Wurmbach, Y.B. Chen, G. Khitrov, W. Zhang, S. Roayaie, M. Schwartz, I. Fiel, S. Thung, V. Mazzaferro, J. Bruix, E. Bottinger, S. Friedman, S. Waxman, J.M. Llovet, Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma, Hepatology, 45 (2007) 938-947.
[27] D.F. Mardhian, A. Vrynas, G. Storm, R. Bansal, J. Prakash, FGF2 engineered SPIONs attenuate tumor stroma and potentiate the effect of chemotherapy in 3D heterospheroidal model of pancreatic tumor, Nanotheranostics, 4 (2020) 26-39.
[28] S.E. Hughes, Differential expression of the fibroblast growth factor receptor (FGFR) multigene family in normal human adult tissues, J Histochem Cytochem, 45 (1997) 1005-1019.
[29] K. Jim-No, M. Tanimizu, I. Hyodo, F.Kurimoto, T. Yamashita, Plasma level of basic fibroblast growth factor increases with progression of chronic liver disease, J. Gastroenterol. 32 (1997)119-121.

150
[30] T. Seitz, K. Freese, P. Dietrich, W.E. Thasler, A. Bosserhoff, C. Hellerbrand, Fibroblast growth factor 9 is expressed by activated hepatic stellate cells and promotes progression of hepatocellular carcinoma, Sci. Rep. 10 (2020) 4546.
[31] H. Shi, J. Xu, R. Zhao, H. Wu, L. Gu, Y. Chen, FGF2 regulates proliferation, migration, and invasion of ECA109 cells through PI3K/Akt signalling pathway in vitro, Cell Biol Int, 40 (2016) 524-533.
[32] M.K. Son, Y.L. Ryu, K.H. Jung, H. Lee, H.S. Lee, H.H. Yan, H.J. Park, J.K. Ryu, J.K. Suh, S. Hong, S.S. Hong, HS-173, a novel PI3K inhibitor, attenuates the activation of hepatic stellate cells in liver fibrosis, Sci Rep, 3 (2013) 3470.
[33] B.J. Bruno, G.D. Miller, C.S. Lim, Basics and recent advances in peptide and protein drug delivery, Ther Deliv, 4 (2013) 1443-1467.
[34] D.S. Pisal, M.P. Kosloski, S.V. Balu-Iyer, Delivery of therapeutic proteins, J Pharm Sci, 99 (2010) 2557-2575.
[35] D.F. Mardhian, G. Storm, R. Bansal, J. Prakash, Nano-targeted relaxin impairs fibrosis and tumor growth in pancreatic cancer and improves the efficacy of gemcitabine in vivo, J Control Release, 290 (2018) 1-10.
[36] M. Sato-Matsubara, T. Matsubara, A. Daikoku, Y. Okina, L. Longato, K. Rombouts, L.T.T. Thuy, J. Adachi, T. Tomonaga, K. Ikeda, K. Yoshizato, M. Pinzani, N. Kawada, Fibroblast growth factor 2 (FGF2) regulates cytoglobin expression and activation of human hepatic stellate cells via JNK signaling, J Biol Chem, 292 (2017) 18961-18972.
[37] M. Arruebo, R. Fernández-Pacheco, M.R. Ibarra, J. Santamaría, Magnetic nanoparticles for drug delivery, Nano Today, 2 (2007) 22-32.
[38] R. Tietze, J. Zaloga, H. Unterweger, S. Lyer, R.P. Friedrich, C. Janko, M. Pottler, S. Durr, C. Alexiou, Magnetic nanoparticle-based drug delivery for cancer therapy, Biochem Biophys Res Commun, 468 (2015) 463-470.
[39] L. Maddaluno, C. Urwyler, S. Werner, Fibroblast growth factors: key players in regeneration and tissue repair, Development, 144 (2017) 4047-4060.
[40] Y.R. Yun, J.E. Won, E. Jeon, S. Lee, W. Kang, H. Jo, J.H. Jang, U.S. Shin, H.W. Kim, Fibroblast growth factors: biology, function, and application for tissue regeneration, J Tissue Eng, 2010 (2010) 218142.

151
5.7 Supplementary Information
Table S1. Antibodies used for the immunofluorescence staining and western blotting
Table S2: Sequence of the human primers used for quantitative real-time PCR
Primary Antibody Source Immunostaining Western blotting
Polyclonal rabbit anti-FGF2 Cell Signalling 1:1000
Monoclonal mouse anti-β-actin Sigma 1:5000
Polyclonal rabbit anti-α-SMA Sigma 1:500
Polyclonal goat anti-collagen I Southern Biotech 1:100 1:600
Rat anti-mouse F4/80 BioRad 1:100
Polyclonal rabbit anti-FGFR1 Cell Signaling 1:100 1:1000
Secondary Antibody Source
HRP-conjugated goat anti-rabbit IgG DAKO 1:100 1:2000
HRP-conjugated rabbit anti-mouse IgG DAKO 1:100 1:2000
HRP-conjugated goat anti-mouse IgG DAKO 1:100 1:2000
HRP-conjugated rabbit anti-goat IgG DAKO 1:100 1:2000
Alexa 488 conjugated donkey anti-rabbit IgG Thermo Fisher Scientific 1:200
Alexa 488 conjugated donkey anti-goat IgG Thermo Fisher Scientific 1:200
Gene
Forward primer sequence (5’-3’) Reverse primer sequence (5’-3’) Accession no. FGFR1c GGACTCTCCCATCACTCTGCAT
GGCCCCTGTGCAATAGATGA
M34186.1 FGFR2c GCCAAGCCTGAGTCCTTTCT
ACGCAGAAGAGTGGTCCTTG
NM_001144919
FGFR3c GACGTACACGCTGGACGTGCTGGA
AGCACCACCAGCCACGCAGAGTGA
NM_000142 FGFR4 AGTTCTGCCTACAGGACACG ACAGGAGTCCCACCGTGTAT NG_012067.1
FGF2 GGCTTCTTCCTGCGCATCCA GCTCTTAGCAGACATTGGAAGA NM_002006 α-SMA CCCCATCTATGAGGGCTATG
CAGTGGCCATCTCATTTTCA
NM_001613.2 Col-1α1 GTACTGGATTGACCCCAACC CGCCATACTCGAACTGGAAT NM_000088.3
TGFβ1 GCGTGCTAATGGTGGAAACC GAGCAACACGGGTTCAGGTA NM_000660.4 18 s rRNA TGAGGTGGAACGTGTGATCA CCTCTATGGGCCCGAATCTT NM_022551.2
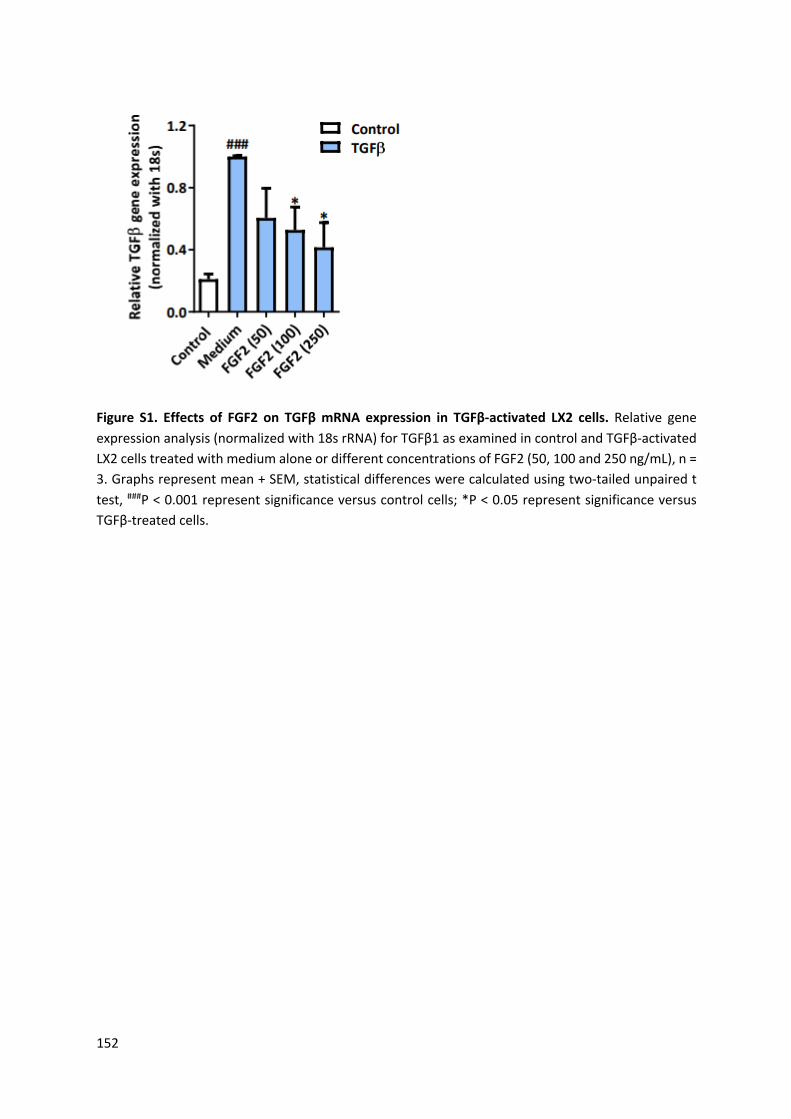
152
Figure S1. Effects of FGF2 on TGFβ mRNA expression in TGFβ-activated LX2 cells. Relative gene expression analysis (normalized with 18s rRNA) for TGFβ1 as examined in control and TGFβ-activated LX2 cells treated with medium alone or different concentrations of FGF2 (50, 100 and 250 ng/mL), n = 3. Graphs represent mean + SEM, statistical differences were calculated using two-tailed unpaired t test, ###P < 0.001 represent significance versus control cells; *P < 0.05 represent significance versus TGFβ-treated cells.
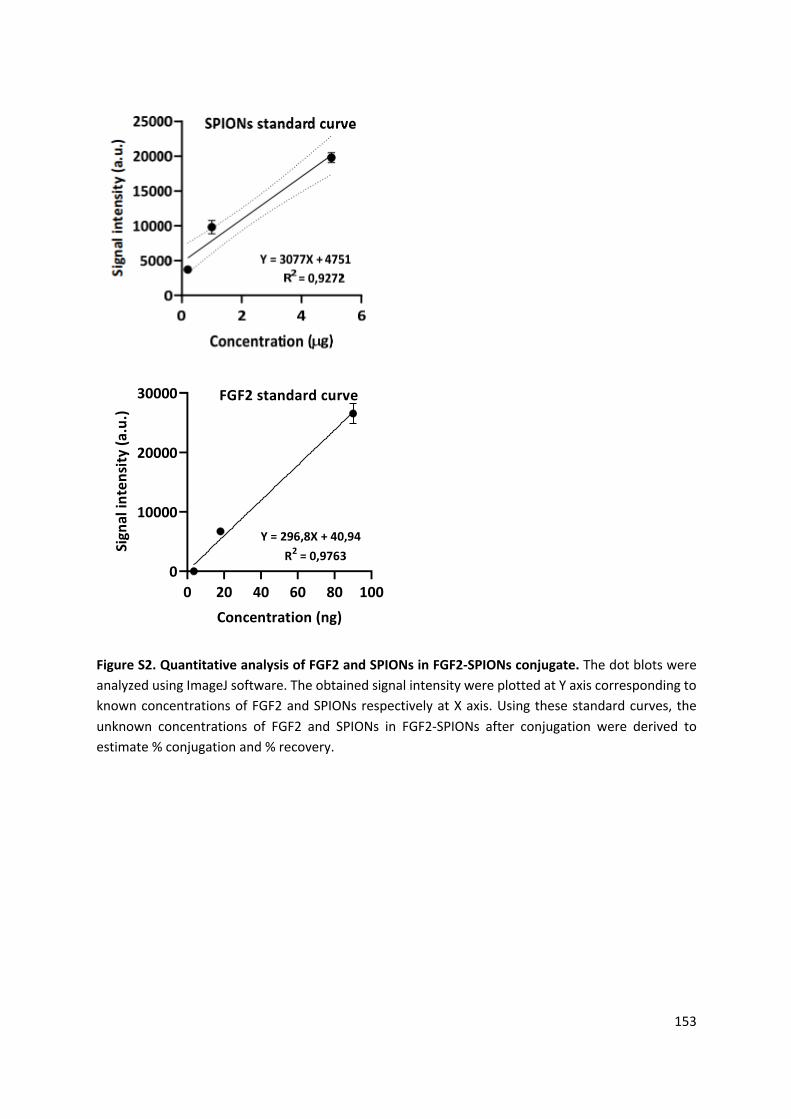
153
Figure S2. Quantitative analysis of FGF2 and SPIONs in FGF2-SPIONs conjugate. The dot blots were analyzed using ImageJ software. The obtained signal intensity were plotted at Y axis corresponding to known concentrations of FGF2 and SPIONs respectively at X axis. Using these standard curves, the unknown concentrations of FGF2 and SPIONs in FGF2-SPIONs after conjugation were derived to estimate % conjugation and % recovery.
0 20 40 60 80 1000
10000
20000
30000 FGF2 standard curve
Concentration (ng)
Sign
al in
tens
ity (a
.u.)
Y = 296,8X + 40,94R2 = 0,9763

154
Figure S3. Effects of FGF2 on TGFβ mRNA expression in TGFβ-activated LX2 cells. Relative gene expression analysis (normalized with 18s rRNA) for TGFβ1 as examined in control and TGFβ-activated LX2 cells treated with medium alone or different concentrations of FGF2 (50, 100 and 250 ng/mL), n = 3. Graphs represent mean + SEM, statistical differences were calculated using two-tailed unpaired t test, ###P < 0.001 represent significance versus control cells; *P < 0.05 represent significance versus TGFβ-treated cells.

155
Figure S4. HSCs-specific localization of FG2-SPIONs in liver in vivo. Representative microscopic image showing HSCs-specific localization of FGF2-SPIONs (in blue color, indicated by arrows) in the fibrotic regions stained with collagen I immunostaining (in red color). The collagen I immunostaining together with Prussian blue iron staining was performed on cryosections prepared from liver tissues obtained from fibrotic mice treated with FGF2-SPIONs.

156

157
Chapter 6
Summary

158
Summary
Liver fibrosis and its progression to liver cirrhosis and hepatocellular carcinoma (HCC) is a
growing health problem affecting millions of people worldwide. This growing global health
burden is attributed to hepatitis viral infections (viral hepatitis), metabolic disorders such as
obesity and diabetes (non-alcoholic fatty liver disease, NAFLD) and alcohol abuse (alcohol-
associated liver disease, ALD), among others [1]. Upon persistent liver injury, chronic
inflammation ensues that develops into fibrosis, that further develops into cirrhosis (end-
stage liver failure) and/or hepatocellular carcinoma (HCC).
Following hepatocellular damage, liver inflammation is triggered by the resident immune cells
followed by activation and infiltration of immune cells, mainly monocytes/macrophages, that
lead to chronic liver inflammation. Liver inflammation instigates proliferation, activation and
trans-differentiation of quiescent hepatic stellate cells (HSCs) to myofibroblasts that secretes
excessive amounts of extracellular matrix proteins, mainly collagen, resulting in tissue stiffness
and distortion of liver architecture referred to liver fibrosis that further develops into liver
cirrhosis (liver dysfunction) and cancer development [2, 3].
Despite the increasing prevalence of such liver diseases, unfortunately no clinically approved
drugs are available for their treatment. Removal of the underlying cause e.g. by alcohol
abstinence, lifestyle modifications i.e. healthy diet and exercise (weight loss), and regular
monitoring of liver function is recommended for patients with acute/early liver disease (liver
fibrosis). Liver transplantation is the only option available in case of end-stage liver failure
(liver cirrhosis). Due to the lack of sufficient liver donors, lifelong immunosuppressive drugs,
high costs and other associated problems, liver transplantation is used for only a minority of
patients. Therefore, there is an urgent need to develop a drug-based treatment for fatty liver
and fibrotic liver diseases.
Macrophages and hepatic stellate cells (HSCs) are known to play a crucial role in the
development of liver diseases, both during acute and chronic liver injury [4, 5]. Therefore, in
this thesis, we developed and investigated novel therapeutic approaches targeting
macrophages and HSCs. As summarized in chapter 1, we investigated the utility of novel
compounds and nanoparticle-based approaches to achieve this goal.

159
In an initial preliminary study, we used a profiler array tool, applied to murine RAW 264.7 cells.
Unstimulated M0 macrophages, lipopolysaccharide (LPS)/interferon gamma (IFNγ)-stimulated
pro-inflammatory M1 macrophages and interleukin (IL)-4/IL-13-stimulated pro-resolving M2
macrophages were analyzed for differential gene expression. In the profiler array, we found
that spleen tyrosine kinase (SYK) was highly upregulated in M1 macrophages, among other
genes that were upregulated in M1 macrophages (17 in total). This phenomenon is also
highlighted in our literature review (chapter 2), in which we describe and discuss the role of
the SYK signaling pathway in liver diseases [6]. This review points out that SYK is highly
expressed in liver diseases, and that SYK expression and activation of its signaling pathway
correlates with the disease severity in different etiological liver diseases including viral
hepatitis (B and C), NASH, ALD, liver cirrhosis and HCC [7-9] [10]. SYK is known to be widely
expressed in hepatocytes, hepatic macrophages and activated HSCs [11]. We further provide
an overview of different SYK inhibitors that have/can be explored for the treatment of liver
diseases. We have also highlighted the limitations of using SYK inhibitors e.g., poor
pharmacokinetics (small molecule inhibitors) and adverse effects due to involvement of SYK
in normal cellular functions, and future applications of targeting SYK inhibitors using
nanoparticles.
In chapter 3, motivated with the results obtained from the profiler array, we explored the
effects of a small molecule SYK inhibitor, R406 [12] (without and with nanoparticles) in vitro
and in vivo [8]. As shown previously by others [10, 11], we found a positive correlation of SYK
expression with the pathogenesis of NASH and alcoholic hepatitis in patients. But not shown
previously, we observed that SYK expression was particularly induced in M1-differentiated
pro-inflammatory macrophages. Following this observation, we assessed the implication of
the SYK pathway in macrophages and found that inhibition of the SYK pathway using R406
resulted in the inhibition of inflammation markers i.e., nitric oxide (NO) release, and enhanced
gene expression of IL-1β, FcγR1, iNOS, CCL2, IL-6, and CCR2, in a dose-dependent manner.
Based on the significant inhibition of the SYK pathway by R406 in M1 (pro-inflammatory)
macrophages and to overcome the limitations as mentioned above, we encapsulated this drug
into poly lactic-co-glycolic acid (PLGA) nanoparticles. PLGA was chosen considering its
biodegradability, biocompatibility as well as the fact that it is a FDA approved polymer [13].
After extensive characterization of the R406-PLGA nanoparticles, we examined the efficacy of

160
R406-PLGA nanoparticles in in vitro cultured RAW macrophages and BMDMs polarized
towards the M1 phenotype. We found that both R406-PLGA nanoparticles as well as R406
itself could significantly inhibit the gene expression of several inflammatory markers in vitro
[14]. In a subsequent in vivo study, using the MCD-diet NASH mouse model, we found that
R406-PLGA nanoparticles significantly ameliorated liver inflammation, fibrosis, and steatosis
as compared to free R406 [14]. We concluded that R406-PLGA nanoparticles could be a
promising approach for the treatment of NASH. However, further studies in mouse models i.e.
western diet models and/or the Stelic Animal Model (STAM) model, that more closely mimic
the clinical NASH situation, are needed.
Since SYK pathway is closely associated with SRC kinase pathway [15], we then explored the
involvement and role of SRC kinase pathway in liver diseases in chapter 4. We confirmed that
the Src gene was highly upregulated in human liver tissues obtained from patients with NASH,
alcoholic hepatitis, cirrhosis, and biliary atresia. We also observed that the Src gene was highly
upregulated in RAW 264.7 macrophages, murine BMDMs, and human MDMs (marrow-
derived macrophages), when polarized to their respective M1 phenotype. For a further
investigation of the role of the Src kinase pathway and potential therapeutic benefit of
inhibition of this pathway, we used the selective small molecule Src kinase inhibitor, KX2-391
[16].
KX2-391 inhibited the phosphorylation of Src, SYK and significantly reduced the NO release in
RAW macrophages. Moreover, gene expression analysis in RAW macrophages and BMDMs
evidenced that Src pathway inhibition mediated by KX2-391 attenuated the expression of
different inflammatory markers including iNOS, CCL2, and FcγR1. Additionally, in a precision-
cut liver slices (PCLS) model [17], KX2-391 decreased the gene expression of Src, iNOS and
CCL2 without affecting the cell/tissue viability. Moreover, KX2-391 attenuated hepatocytic
lipid accumulation; TGFβ-induced HSCs activation, contractility and collagen expression.
Next, we investigated the relevance of the Src pathway in NASH and ASH using a MCD-diet
NASH mouse model and a Lieber-De Carli ALD mouse model [18], respectively. The results
indicated that KX2-391 attenuated inflammation, fibrosis, and steatosis in both NASH and ASH
mouse models. Mechanistic studies further revealed that Src mediated the effects through
the FAK/PI3K/AKT pathway.

161
Accordingly, we proposed that inhibiting the Src kinase pathway using inhibitors such as KX2-
391 represents a potential treatment for liver diseases like NASH and ALD. The Src kinase
inhibitor KX2-391 is also very promising to be studied as a potential therapy for other chronic
diseases including cancer.
In chapter 5, we focused on targeting of HSCs to inhibit fibrosis. Since the fibroblast growth
factor receptor 1 (FGFR1) is highly overexpressed on activated HSCs, we decided to use basic
FGF (FGF2) to inhibit HSCs activation. We observed that FGF2 was able to inhibit TGFβ-induced
HSCs activation (confirmed by reduced collagen-I and α-SMA expression), migration of HSCs
(confirmed by wound healing assays) and contraction of HSCs (confirmed by 3D-gel
contraction assays). These results laid the foundation for our further studies in which we
conjugated FGF2 to superparamagnetic iron oxide nanoparticles (FGF2-SPIONs) to improve
the stability and half-life of FGF2 and thereby to improve the therapeutic efficacy of FGF2 for
the treatment of liver fibrosis. In addition to enhancing therapeutic efficacy, conjugation of
FGF2 with SPIONs is also expected to be applied for diagnostic-related purposes due to its
paramagnetic properties, providing theranostic potential [19]. After performing several in
vitro and in vivo experiments, the results revealed that the potency of FGF2-SPIONs conjugate
is significantly improved as compared to free FGF2. Besides improved therapeutic efficacy, we
also concluded that the conjugation of FGF2 with SPIONs may provide a personalized
theranostic approach with combined therapy and diagnosis for personalized disease
management. However, more studies are required to validate our findings and theranostic
potential of FGF2-SPIONs in more advanced and representative liver fibrosis animal models
[20].
In conclusion, the approaches described in this thesis are promising to be explored further for
the treatment of liver diseases like NASH or ALD-based liver fibrosis. With nanotechnological
approaches such as polymeric nanoparticles and SPIONs, the efficacy of active compounds
with poor pharmacokinetic profile can be significantly increased. In addition to increasing
efficacy, nanoparticulate delivery systems can also reduce the adverse effects of these active
compounds. Since the majority of administered nanoparticles will accumulate and be
sequestered in the liver after intravenous administration into the body, this thesis work
confirms our initial hypothesis that the design of nano-delivery systems can effectively

162
improve drug delivery to the liver and therefore have great potential to ameliorate the
therapy of liver diseases as presented in this thesis.
References
[1] M. Blachier, H. Leleu, M. Peck-Radosavljevic, D.-C. Valla, F. Roudot-Thoraval, The burden of liver disease in Europe: A review of available epidemiological data, in: E.A.f.T.S.o.T.L. (EASL) (Ed.), 2013.
[2] R.S. Rahimi, D.C. Rockey, Complications and outcomes in chronic liver disease, Curr Opin Gastroenterol, 27 (2011) 204-209.
[3] R.A. Mohammad, Complications of chronic liver disease, 2010, pp. 91-108. [4] C. Ju, F. Tacke, Hepatic macrophages in homeostasis and liver diseases: from pathogenesis
to novel therapeutic strategies, Cell Mol Immunol, 13 (2016) 316-327. [5] C. Yin, K.J. Evason, K. Asahina, D.Y. Stainier, Hepatic stellate cells in liver development,
regeneration, and cancer, J Clin Invest, 123 (2013) 1902-1910. [6] D.W. Kurniawan, G. Storm, J. Prakash, R. Bansal, Role of spleen tyrosine kinase in liver
diseases, World J Gastroenterol, 26 (2020) 1005-1019. [7] A.R. Zekri, M.M. Hafez, A.A. Bahnassy, Z.K. Hassan, T. Mansour, M.M. Kamal, H.M. Khaled,
Genetic profile of Egyptian hepatocellular-carcinoma associated with hepatitis C virus Genotype 4 by 15 K cDNA microarray: preliminary study, BMC Res Notes, 1 (2008) 106.
[8] D.W. Kurniawan, A.K. Jajoriya, G. Dhawan, D. Mishra, J. Argemi, R. Bataller, G. Storm, D.P. Mishra, J. Prakash, R. Bansal, Therapeutic inhibition of spleen tyrosine kinase in inflammatory macrophages using PLGA nanoparticles for the treatment of non-alcoholic steatohepatitis, J Control Release, 288 (2018) 227-238.
[9] I.J. Song, Y.M. Yang, S. Inokuchi-Shimizu, Y.S. Roh, L. Yang, E. Seki, The contribution of toll-like receptor signaling to the development of liver fibrosis and cancer in hepatocyte-specific TAK1-deleted mice, Int J Cancer, 142 (2018) 81-91.
[10] T.N. Bukong, A. Iracheta-Vellve, B. Saha, A. Ambade, A. Satishchandran, B. Gyongyosi, P. Lowe, D. Catalano, K. Kodys, G. Szabo, Inhibition of spleen tyrosine kinase activation ameliorates inflammation, cell death, and steatosis in alcoholic liver disease, Hepatology, 64 (2016) 1057-1071.
[11] C. Qu, D. Zheng, S. Li, Y. Liu, A. Lidofsky, J.A. Holmes, J. Chen, L. He, L. Wei, Y. Liao, H. Yuan, Q. Jin, Z. Lin, Q. Hu, Y. Jiang, M. Tu, X. Chen, W. Li, W. Lin, B.C. Fuchs, R.T. Chung, J. Hong, Tyrosine kinase SYK is a potential therapeutic target for liver fibrosis, Hepatology, (2018).
[12] S. Braselmann, V. Taylor, H. Zhao, S. Wang, C. Sylvain, M. Baluom, K. Qu, E. Herlaar, A. Lau, C. Young, B.R. Wong, S. Lovell, T. Sun, G. Park, A. Argade, S. Jurcevic, P. Pine, R. Singh, E.B. Grossbard, D.G. Payan, E.S. Masuda, R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation, J Pharmacol Exp Ther, 319 (2006) 998-1008.
[13] F. Danhier, E. Ansorena, J.M. Silva, R. Coco, A. Le Breton, V. Preat, PLGA-based nanoparticles: an overview of biomedical applications, J Control Release, 161 (2012) 505-522.
[14] H.H. Hansen, M. Feigh, S.S. Veidal, K.T. Rigbolt, N. Vrang, K. Fosgerau, Mouse models of nonalcoholic steatohepatitis in preclinical drug development, Drug Discov Today, 22 (2017) 1707-1718.
[15] C.A. Lowell, Src-family and Syk kinases in activating and inhibitory pathways in innate immune cells: signaling cross talk, Cold Spring Harb Perspect Biol, 3 (2011) 1-16.

163
[16] M. Anbalagan, L. Carrier, S. Glodowski, D. Hangauer, B. Shan, B.G. Rowan, KX-01, a novel Src kinase inhibitor directed toward the peptide substrate site, synergizes with tamoxifen in estrogen receptor alpha positive breast cancer, Breast Cancer Res Treat, 132 (2012) 391-409.
[17] P. Olinga, D. Schuppan, Precision-cut liver slices: a tool to model the liver ex vivo, J Hepatol, 58 (2013) 1252-1253.
[18] A. Bertola, S. Mathews, S.H. Ki, H. Wang, B. Gao, Mouse model of chronic and binge ethanol feeding (the NIAAA model), Nat Protoc, 8 (2013) 627-637.
[19] B. Nagorniewicz, D.F. Mardhian, R. Booijink, G. Storm, J. Prakash, R. Bansal, Engineered Relaxin as theranostic nanomedicine to diagnose and ameliorate liver cirrhosis, Nanomedicine, 17 (2019) 106-118.
[20] D.W. Kurniawan, R. Booijink, L. Pater, I. Wols, A. Vrynas, G. Storm, J. Prakash, R. Bansal, Fibroblast growth factor 2 conjugated superparamagnetic iron oxide nanoparticles (FGF2-SPIONs) ameliorate hepatic stellate cells activation in vitro and acute liver injury in vivo, J Control Release, 328 (2020) 640-652.

164
Appendix A – Nederlandse Samenvatting
Lever fibrose, en zijn gevorderde stadia lever cirrose en hepatocellulair carcinoom (HCC),
vormen een groeiend gezondheidsprobleem voor miljoenen mensen wereldwijd. Deze globaal
groeiende last op de gezondheidszorg is onder andere te wijten aan virale infecties van de
lever (virale hepatitis), metabolische stoornissen zoals obesitas en diabetes (niet-alcoholische
vette lever ziekte, NAFLD) en alcohol misbruik (alcoholische lever ziekte, ALD). Chronische
ontsteking in de lever, dat resulteert in fibrose, ontstaat als gevolg van aanhoudende lever
beschadiging, wat vervolgens verder kan ontwikkelen in lever cirrose (eind stadium lever
falen) en/of hepatocellulair carcinoom (HCC).
Als gevolg van hepatocellulaire schade worden lever eigen immuun cellen geactiveerd,
gevolgd door de activatie en infiltratie van circulerende immuun cellen, voornamelijk
monocyten en macrofagen, wat leidt tot chronische leverontsteking. Leverontsteking zet
daarna aan tot de proliferatie, activatie en trans-differentiatie van rustende hepatische
stellaat cellen (HSCs) naar myofibroblasten, die overmatige hoeveelheden extracellulaire
matrix eiwitten uitscheiden, voornamelijk collageen. Dit resulteert in stijfheid en vervorming
van de lever, gekarakteriseerd als lever fibrose, wat zich verder ontwikkelt in lever cirrose
(lever falen) en kanker.
Helaas zijn er, ondanks de toenemende prevalentie van deze lever ziektes, geen klinisch
goedgekeurde medicijnen beschikbaar voor de behandeling. Voor patiënten met acute lever
ziekte of lever ziekte in een vroeg stadium (lever fibrose), wordt aangeraden de onderliggende
oorzaak van de ziekte aan te pakken, bijvoorbeeld door onthouding van alcohol, of door
veranderingen aan te brengen in levensstijl met een gezond dieet en voldoende beweging, en
door de lever functie te monitoren. Voor patiënten in een laat stadium lever falen (lever
cirrose), is lever transplantatie de enige optie. Echter, lever transplantatie is alleen
beschikbaar voor een minderheid van de patiënten door een gebrek aan voldoende donoren,
levenslange afweeronderdrukkende medicatie, hoge kosten en andere aangesloten
problemen. Daarom is de ontwikkeling van medicijn-gerichte behandeling van vette lever en
fibrotische lever ziektes noodzakelijk.
Macrofagen en hepatische stellaat cellen (HSCs) spelen een cruciale rol in de ontwikkeling van
lever ziektes, zowel bij acute als chronische lever schade. Daarom ontwikkelden en

165
onderzochten we in deze theses nieuwe therapeutische benaderingen gericht op macrofagen
en HSCs. Om dit te bereiken onderzochten we de bruikbaarheid van nieuwe middelen en
technieken gebaseerd op nano-deeltjes, samengevat in hoofdstuk 1.
In een aanvankelijk inleidend onderzoek gebruikten we een profiel array, toegepast op muis
RAW 264.7 cellen. Niet-gestimuleerde M0 macrofagen, lipopolysaccharide (LPS)/interferon
gamma (IFNγ)-gestimuleerde pro-inflammatoire M1 macrofagen en interleukine (IL)-4/IL-13-
gestimuleerde wondgenezing bevorderende M2 macrofagen werden geanalyseerd voor
differentiële gen expressie. In de profiel array vonden we in totaal 17 genen die opgereguleerd
waren in M1 macrofagen, waaronder spleen tyrosine kinase (SYK). Dit wordt ook benadrukt
in onze literatuur recensie (hoofdstuk 2), waarin we de rol van het SYK signaaltransductiepad
in lever ziektes beschrijven en bespreken. Deze literatuurrecensie duidt aan dat SYK een hoge
expressie heeft in lever ziektes, en dat SYK expressie en activatie van zijn
signaaltransductiepad correleert aan de hevigheid van de ziekte in verscheidene etiologische
lever ziektes, waaronder virale hepatitis (B en C), NASH, ALD, lever cirrose en HCC. Het is
bekend dat SYK op grote schaal tot expressie komt in hepatocyten, lever macrofagen en
geactiveerde HSCs. Verder geven we een overzicht van de verschillende SYK inhibitoren die
zijn/kunnen worden onderzocht voor de behandeling van lever ziektes. Ook hebben we de
beperkingen van het gebruik van SYK inhibitoren onderschreven, bijvoorbeeld slechte
farmacokinetiek (kleine molecuul inhibitoren) en negatieve bijwerkingen door betrokkenheid
van SYK in normale cel functies, en verdere applicaties gericht op de combinatie van SYK
inhibitors met nano-deeltjes.
In hoofdstuk 3, gemotiveerd door de resultaten van de profiel array, onderzochten we het
effect van een klein molecuul SYK inhibitor, R406 (met en zonder nano-deeltjes), in vitro en in
vivo. Vergelijkbaar met wat anderen voor ons hebben onderzocht, vonden we een positieve
correlatie tussen SYK expressie en de pathogenese in patiënten met NASH of alcoholische
hepatitis. Echter, wij stelden vast dat SYK expressie voornamelijk opgewekt was in M1-
gedifferentieerde macrofagen, een observatie nog niet eerder beschreven. Na deze
observatie analyseerden we de implicatie van het SYK signaaltransductiepad in macrofagen,
en zagen dat SYK inhibitie met R406 leidde tot een inhibitie van ontstekingsmarkers zoals
stikstofoxide (NO) afgifte, en verhoogde gen expressie van IL-1β, FcγR1, iNOS, CCL2, IL-6 en
CCR2 op een dosisafhankelijke manier.

166
Naar aanleiding van de significante inhibitie van het SYK signaaltransductiepad door R406 in
M1 (pro-inflammatoire) macrofagen, en om voorgenoemde beperkingen te voorkomen,
hebben we dit medicijn ingekapseld in poly lactic-co-glycolic acid (PLGA) nano-deeltjes. PLGA
is gekozen omdat het zowel biodegradeerbaar als biocompatibel is, in combinatie met het feit
dat PLGA een FDA goedgekeurd polymeer is. Na uitgebreide karakterisering van de R406-
PLGA nano-deeltjes, onderzochten we de effectiviteit van de R406-PLGA nano-deeltjes in vitro
in gekweekte RAW macrofagen en BMDMs gepolariseerd richting een M1 fenotype. Hierbij
zagen we dat zowel R406-PLGA nano-deeltjes als R406 zelf de genexpressie van verschillende
ontstekingsmarkers significant kunnen reduceren. In een aaneensluitende in vivo studie,
waarbij we gebruik maakten van het MCD-dieet NASH muis model, vonden we dat R406-PLGA
nano-deeltjes lever ontsteking, fibrose en steatose significant reduceerden, in vergelijking met
vrije R406. We concludeerden dat R406-PLGA nano-deeltjes een veelbelovende strategie
kunnen zijn voor de behandeling van NASH. Echter, additionele studies in muis modellen, die
een betere representatie zijn van klinische NASH ontwikkeling, zoals Westers Dieet modellen
en/of het Stelic Animal Model (STAM model), zijn nodig.
Gezien het SYK signaaltransductiepad en het SRC kinase signaaltransductiepad nauw
verbonden zijn, onderzochten we de betrokkenheid en rol van het SRC kinase
signaaltransductiepad in lever ziektes in hoofdstuk 4. We bevestigden dat het SRC gen een
hoge expressie heeft in menselijk lever weefsel van patiënten met NASH, alcoholische
hepatitis, cirrose en biliaire atresie. We observeerden ook dat het Src gen een hoge expressie
heeft in RAW 264.7 macrofagen, muis BMDMs en menselijke MDMs (macrofagen uit het
beenmerg), wanneer deze gepolariseerd zijn naar hun M1 fenotype. Om de rol en eventuele
therapeutische voordelen van inhibitie van dit SRC kinase signaaltransductiepad verder te
onderzoeken, gebruikten we de selectieve klein molecuul SRC kinase inhibitor KX2-391.
KX2-391 zorgde voor inhibitie van de fosforylering van SRC, SYK en verminderde de NO afgifte
significant in RAW macrofagen. Bovendien, gen expressie analyse in RAW macrofagen en
BMDMs toonde aan dat inhibitie van SRC signaaltransductiepad door KX2-391 de expressie
van ontstekingsmarkers zoals iNOS, CCL2, en FcγR1 verminderde. Ook verminderde KX2-391
lipide accumulatie in hepatocyten; TGFβ-geïnduceerde HSCs activatie, contractiliteit en
collageen expressie.

167
Hierna onderzochten we de relevantie van het SRC signaaltransductiepad in NASH en ASH
door middel van een MCD-dieet NASH muis model, en een Lieber-De Carli ALD muis model,
respectievelijk. De resultaten lieten zien dat KX2-391 zorgde voor een vermindering van
ontsteking, fibrose en steatose in zowel het NASH als het ASH muis model. Verder toonden
mechanistische studies aan dat SRC deze effecten veroorzaakt via het FAK/PI3K/AKT
signaaltransductiepad.
We suggereren dat inhibitie van het SRC kinase signaaltransductiepad met inhibitoren zoals
KX2-391 kan dienen als een potentiële behandeling voor lever ziektes als NASH en ALD. De
SRC kinase inhibitor KX2-391 is ook veelbelovend als potentiële therapie voor andere
chronische ziektes, waaronder kanker.
In hoofdstuk 5 concentreerden we ons op het inhiberen van fibrose, door middel van gerichte
medicatie tegen HSCs. Gezien de hoge expressie van fibroblast growth factor receptor 1
(FGFR1) in actieve HSCs, besloten we basaal FGF (FGF2) te gebruiken voor het remmen van
HSCs activatie. Hierbij zagen we dat FGF2 zorgde voor een remming van TGFβ-geïnduceerde
HSCs activatie (bevestigd door een afname van collageen-1 en α-SMA expressie), migratie van
HSCs (bevestigd door wond heling-assays) en contractie van HSCs (bevestigd door 3D-gel
contractie-assays). Deze resultaten legden de fundering voor onze vervolgonderzoeken,
waarin we de stabiliteit en halveringstijd van FGF2 verbeteren door FGF2 te conjugeren aan
super paramagnetische ijzer oxide nano-deeltjes (FGF2-SPIONs). Hierdoor verbeteren we de
therapeutische efficiëntie van FGF2 voor de behandeling van lever fibrose. Tevens kan de
conjugatie van FGF2 met SPIONs worden gebruikt voor diagnostiek-gerelateerde doeleinden
gezien zijn paramagnetische eigenschappen, potentieel theranostische toepassingen. De
resultaten van verscheidene in vitro en in vivo experimenten toonden aan dat potentie van
het geconjugeerde FGF2-SPION hoger is dan vrije FGF2.
Afgezien van verhoogde therapeutische efficiëntie, concludeerden we dat conjugatie van
FGF2 met SPIONs de potentie heeft voor gepersonaliseerd theranostisch onderzoek, waarbij
therapie en diagnostiek gecombineerd kan worden voor gepersonaliseerd ziektebeheer.
Echter, aanvullend onderzoek is nodig om onze bevindingen te valideren en de theranostische
mogelijkheden van FGF2-SPIONs te analyseren in geavanceerde en representatieve lever
fibrose modellen.

168
In conclusie, deze thesis beschrijft veelbelovende methodes voor de behandeling van lever
ziektes zoals NASH of ALD-gebaseerde lever fibrose. Met behulp van nanotechnologie,
waaronder polymeer nano-deeltjes en SPIONs, wordt de efficiëntie van actieve middelen met
slechte farmacokinetiek significant verbeterd. Naast een toename in efficiëntie, kan een
bezorgsysteem gebaseerd op nano-deeltjes de negatieve bijwerkingen van de actieve
middelen reduceren.
Deze theses bevestigt onze initiële hypothese dat het ontwerpen van bezorgsystemen op
nano-schaal zorgt voor een effectieve verbetering van de medicijn afgifte naar de lever,
aangezien de meerderheid van de nano-deeltjes, na een intraveneuze injectie, accumuleert in
de lever. Daarom, zoals gepresenteerd in deze theses, hebben deze nano-deeltjes veel
potentie als therapeutische behandelingen voor lever ziektes.

169
Appendix B – Ringkasan Bahasa Indonesia
Liver fibrosis yang dapat memburuk menjadi liver sirosis dan hepatocellular carcinoma (HCC)
merupakan salah satu masalah kesehatan utama yang berkembang saat ini yang
mempengaruhi jutaan orang di seluruh dunia. Masalah kesehatan global yang meningkat ini
berhubungan antara lain dengan infeksi virus hepatitis, gangguan metabolisme seperti
obesitas dan diabetes (non-alcoholic fatty liver disease, NAFLD) dan penyalahgunaan alkohol
(alcoholic liver disease, ALD). Pada liver yang luka secara persisten, terjadi inflamasi kronis
yang kemudian dapat berkembang menjadi fibrosis, yang selanjutnya berkembang menjadi
sirosis (gagal liver stadium akhir) dan/atau hepatocellular carcinoma (HCC).
Setelah terjaid kerusakan liver secara seluler, inflamasi liver dipicu oleh sel imun bawaan yang
diikuti oleh aktivasi dan infiltrasi sel imun, terutama monosit/makrofag, yang menyebabkan
inflamasi liver secara kronis. Inflamasi hati memicu terjadinya proliferasi, aktivasi, dan trans-
diferensiasi hepatik stellate cells (HSCs) menjadi miofibroblas yang mensekresi protein matriks
ekstraseluler dalam jumlah berlebihan, terutama kolagen, yang mengakibatkan kekakuan
jaringan dan distorsi arsitektur liver yang disebut liver fibrosis yang selanjutnya berkembang
menjadi liver sirosis (disfungsi liver) dan berkembang memicu terjadinya kanker.
Meskipun prevalensi penyakit liver meningkat sangat tajam, sayangnya belum ada obat yang
telah disetujui secara klinis tersedia untuk pengobatan penyakit-penyakit liver tersebut.
Penghentian penyebab yang memicu terjadinya penyakit liver misalnya dengan berhenti
minum alkohol, modifikasi gaya hidup seperti diet sehat dan olahraga (penurunan berat
badan), dan pemantauan rutin fungsi liver dianjurkan untuk pasien dengan penyakit liver
akut/awal (liver fibrosis). Transplantasi liver adalah satu-satunya pilihan yang tersedia saat ini
dalam kasus gagal liver stadium akhir (liver sirosis). Karena kurangnya donor hati yang cukup,
obat imunosupresif seumur hidup, biaya yang tinggi dan masalah terkait lainnya, transplantasi
liver digunakan hanya untuk sebagian kecil pasien. Oleh karena itu, ada kebutuhan mendesak
untuk mengembangkan pengobatan untuk penyakit liver akibat penumpukan lemak dan
penyakit liver fibrotik.
Makrofag dan hepatic stellate cells (HSCs) diketahui memainkan peranan penting dalam
perkembangan penyakit liver, baik selama cedera liver akut maupun kronis. Oleh karena itu,
di dalam tesis ini, kami mengembangkan dan menginvestigasi pendekatan terapeutik baru

170
yang menargetkan makrofag dan HSCs. Seperti yang dirangkum dalam bab 1 , kami meneliti
kegunaan senyawa baru dan pendekatan terapi berbasis nanopartikel untuk mencapai tujuan
ini.
Pada studi pendahuluan, kami menggunakan profiler-array, yang diaplikasikan pada sel RAW
264,7 murine. Selanjutnya dilakukan analisis ekspresi gen diferensial untuk makrofag M0 yang
tidak distimulasi, makrofag M1 pro-inflamasi yang distimulasi menggunakan lipopolisakarida
(LPS)/interferon gamma (IFNg) dan makrofag M2 anti-inflamasi yang distimulasi dengan
interleukin (IL)-4/IL-13. Hasil profiler-array tersebut, kami menemukan bahwa spleen tyrosine
kinase (SYK) diregulasi sangat tinggi di dalam makrofag M1, terdapat 17 gen yang diregulasi
dalam makrofag M1. Fenomena ini juga dibahas dalam tinjauan literatur kami (bab 2), di mana
kami menggambarkan dan mendiskusikan peran jalur pensinyalan SYK pada penyakit liver.
Review ini menunjukkan bahwa SYK sangat diekspresikan pada penyakit liver, dan bahwa
ekspresi SYK dan aktivasi jalur pensinyalannya berkorelasi dengan tingkat keparahan penyakit
pada berbagai etiologis penyakit liver termasuk hepatitis virus (B dan C), non-alcoholic
steatohepatitis (NASH), ALD, sirosis hati, dan HCC. SYK diketahui diekspresikan secara luas
dalam hepatosit, makrofag hati, dan HSC yang teraktivasi. Kami selanjutnya memberikan
gambaran tentang berbagai inhibitor SYK yang telah/dapat dieksplorasi untuk pengobatan
penyakit liver. Kami juga telah mengkaji keterbatasan penggunaan inhibitor SYK misalnya,
farmakokinetik yang buruk (inhibitor bermolekul kecil) dan efek samping akibat keterlibatan
SYK dalam fungsi seluler normal, dan aplikasi penargetan inhibitor SYK menggunakan
nanopartikel pada masa yang akan datang.
Pada bab 3, merujuk pada hasil yang diperoleh dari profiler-array, kami mengeksplorasi efek
inhibitor SYK bermolekul kecil, R406 (tanpa dan dengan nanopartikel) secara in vitro dan in
vivo. Seperti yang sudah ditunjukkan sebelumnya oleh peneliti lain, kami menemukan korelasi
positif antara ekspresi SYK dengan patogenesis NASH dan hepatitis alkoholik pada pasien.
Tetapi tidak diperlihatkan seperti sebelumnya, kami mengamati bahwa ekspresi SYK secara
khusus diinduksi dalam makrofag pro-inflamasi yang berdiferensiasi M1. Setelah pengamatan
ini, kami meneliti implikasi jalur SYK dalam makrofag dan menemukan bahwa penghambatan
jalur SYK menggunakan R406 mengakibatkan penghambatan marker inflamasi yaitu
pelepasan nitrit oksida (NO), dan peningkatan ekspresi gen IL- 1 , FcgR1, iNOS , CCL2 , IL-6, dan
CCR2, pada dosis yang berkaitan.

171
Berdasarkan penghambatan signifikan jalur SYK oleh R406 dalam makrofag M1 (pro-inflamasi)
dan untuk mengatasi keterbatasan senyawa R406 seperti yang disebutkan di atas, kami
mengenkapsulasi obat ini ke dalam nanopartikel poly lactic-co-glycolic acid (PLGA). PLGA
dipilih karena mempertimbangkan factor biodegradabilitas, biokompatibilitas, dan fakta
bahwa PLGA merupakan polimer yang telah disetujui oleh food and drug administration (FDA).
Setelah melakukan karakterisasi ekstensif terhadap nanopartikel R406-PLGA, kami
mengevaluasi efektivitas nanopartikel R406-PLGA dalam makrofag sel RAW yang dikultur dan
bone marrow derived macrophage (BMDM) secara in vitro yang terpolarisasi menuju fenotipe
M1. Kami menemukan bahwa baik nanopartikel R406-PLGA maupun R406 itu sendiri dapat
secara signifikan menghambat ekspresi beberapa gen penanda inflamasi secara in vitro. Dalam
penelitian in vivo berikutnya, menggunakan model tikus NASH diet methylene-coline
deficiency (MCD), kami mendapati bahwa nanopartikel R406-PLGA secara signifikan
memperbaiki inflamasi hati, fibrosis, dan steatosis jika dibandingkan dengan R406 bebas. Kami
menyimpulkan bahwa nanopartikel R406-PLGA dapat menjadi salah satu pendekatan yang
menjanjikan untuk pengobatan NASH. Namun demikian, penelitian lebih lanjut pada model
tikus yaitu model diet barat dan/atau model Stelic Animal Model (STAM), yang lebih mirip
dengan situasi klinis NASH, diperlukan.
Berkaitan kedekatan jalur SYK dengan jalur SRC kinase, kami kemudian mengeksplorasi
keterlibatan dan peran jalur SRC kinase pada penyakit liver pada bab 4. Kami mengkonfirmasi
bahwa gen Src diregulasi sangat tinggi dalam jaringan hati manusia yang diperoleh dari pasien
dengan kondisi NASH, hepatitis alkoholik, sirosis, dan biliary atresia. Kami juga mengamati
bahwa gen Src diregulasi sangat tinggi dalam sel makrofag RAW 264,7, murine BMDM, dan
marrow-derived macrophage (MDM) manusia, ketika dipolarisasi ke fenotipe M1 masing-
masing. Untuk penelitian lebih lanjut tentang peran jalur SRC kinase dan manfaat potensial
terapeutik dari penghambatan jalur ini, kami menggunakan inhibitor SRC kinase bermolekul
kecil selektif, KX2-391.
KX2-391 menghambat fosforilasi SRC, SYK dan secara signifikan mengurangi pelepasan NO
dalam sel makrofag RAW. Selain itu, analisis ekspresi gen dalam sel makrofag RAW dan BMDM
membuktikan bahwa penghambatan jalur SRC yang dimediasi oleh KX2-391 melemahkan
ekspresi gen penanda inflamasi yang berbeda termasuk iNOS, CCL2, dan FcgR1. Selain itu,
dalam model precise-cut liver slices (PCLS), KX2-391 menurunkan ekspresi gen SRC, iNOS, dan

172
CCL2 tanpa mempengaruhi viabilitas sel/jaringan. Selain itu, KX2-391 melemahkan akumulasi
lipid hepatositik; aktivasi, kontraktilitas, dan ekspresi kolagen yang diinduksi TGFβ pada HSCs.
Selanjutnya, kami menginvestigasi relevansi jalur SRC di dalam NASH dan alcoholic
steatohepatitis (ASH) menggunakan model tikus NASH diet MCD dan model tikus Lieber-De
Carli ALD. Hasilnya menunjukkan bahwa KX2-391 melemahkan inflamasi, fibrosis, dan
steatosis pada model tikus NASH dan ASH. Studi mekanistik lebih lanjut mengungkapkan
bahwa SRC memediasi efek melalui jalur FAK/PI3K/AKT.
Oleh karena itu, kami mengusulkan bahwa menghambat jalur SRC kinase menggunakan
inhibitor seperti KX2-391 memiliki potensi tinggu untuk pengobatan penyakit hati seperti
NASH dan ALD. Inhibitor SRC kinase KX2-391 juga sangat menjanjikan untuk diteliti sebagai
potensial terapi untuk penyakit kronis lainnya termasuk kanker.
Pada bab 5, kami berfokus pada pentargetan HSCs untuk menghambat fibrosis. Terkait
tingginya ekspresi reseptor fibroblast growth factor 1 (FGFR1) pada HSCs yang diaktifkan, kami
memutuskan untuk menggunakan FGF basic (FGF2) untuk menghambat aktivasi HSCs. Kami
mengamati bahwa FGF2 mampu menghambat aktivasi HSCs yang diinduksi TGFβ (dikonfirmasi
dengan pengurangan ekspresi kolagen-I dan α-SMA), migrasi HSCs (dikonfirmasi oleh uji
penyembuhan luka) dan kontraksi HSCs (dikonfirmasi oleh uji kontraksi gel 3D). Hasil ini
menjadi dasar untuk penelitian lanjut kami, di mana kami mengkonjugasikan FGF2 ke
superparamagnetic iron oxide nanoparticles (FGF2-SPIONs) untuk meningkatkan stabilitas dan
waktu paruh FGF2 sehingga meningkatkan kemanjuran terapi FGF2 untuk pengobatan fibrosis
hati. Selain meningkatkan kemanjuran terapeutik, konjugasi FGF2 dengan SPIONs juga
diharapkan diterapkan untuk tujuan terkait diagnostik karena sifat paramagnetiknya yang
dapat memberikan potensi theranostik. Setelah melakukan beberapa percobaan in vitro dan
in vivo, hasilnya menunjukkan bahwa potensi konjugat FGF2-SPIONs meningkat secara
signifikan dibandingkan dengan FGF2 bebas. Selain peningkatan kemanjuran terapi, kami juga
menyimpulkan bahwa konjugasi FGF2 dengan SPIONs dapat memberikan pendekatan
theranostik yang dipersonalisasi dengan gabungan terapi dan diagnosis untuk manajemen
penyakit yang dipersonalisasi. Namun, diperlukan lebih banyak penelitian untuk memvalidasi
temuan kami dan potensi teranostik FGF2-SPIONs pada model hewan fibrosis hati yang lebih
maju dan representatif.

173
Sebagai kesimpulan, pendekatan yang dijelaskan dalam tesis ini menjanjikan untuk
dieksplorasi lebih lanjut untuk pengobatan penyakit hati seperti fibrosis hati berbasis NASH
atau ALD. Dengan pendekatan nanoteknologi seperti nanopartikel polimer dan SPIONs,
kemanjuran senyawa aktif dengan profil farmakokinetik yang buruk dapat ditingkatkan secara
signifikan. Selain meningkatkan efikasi, sistem penghantaran nanopartikel juga dapat
mengurangi efek merugikan dari senyawa aktif tersebut. Karena, sebagian besar nanopartikel
yang diberikan akan terakumulasi dan dilokalisasi di hati setelah pemberian intravena ke
dalam tubuh, karya tesis ini menegaskan hipotesis awal kami bahwa desain sistem
penghantara nano dapat secara efektif meningkatkan penghantaran obat ke hati dan oleh
karena itu memiliki potensi besar untuk memperbaiki terapi penyakit hati seperti yang
disajikan dalam tesis ini.

174
Appendix C
List of Publications (from this thesis)
1. Dhadhang Wahyu Kurniawan, Gert Storm, Jai Prakash, Ruchi Bansal, “Role of spleen tyrosine kinase in liver diseases”, World Journal of Gastroenterology (2020); 26(10): 1005–1019. doi: 10.3748/wjg.v26.i10.1005. (Chapter 2)
2. Dhadhang Wahyu Kurniawan, Arun Kumar Jajoriya, Garima Dhawan, Divya Mishra, Josepmaria Argemi, Ramon Bataller, Gert Storm, Durga Prasad Mishra, Jai Prakash, Ruchi Bansal, “Therapeutic inhibition of spleen tyrosine kinase in inflammatory macrophages using PLGA nanoparticles for the treatment of non-alcoholic steatohepatitis”, Journal of Controlled Release (2018); 288: 227-238. https://doi.org/10.1016/j.jconrel.2018.09.004. (Chapter 3)
3. Dhadhang Wahyu Kurniawan, Richell Booijink, Arun Kumar Jajoriya, Garima Dhawan, Divya Mishra, Dorenda Oosterhuis, Josepmaria Argemi, Gert Storm, Peter Olinga, Ramon Bataller, Sujit Kumar Mohanty, Durga Prasad Mishra, Jai Prakash, Ruchi Bansal, “Src kinase as a potential therapeutic target in non-alcoholic and alcoholic steatohepatitis”, Clinical and Translational Discovery (2022); 2(1): e18. https://doi.org/10.1002/ctd2.18. (Chapter 4)
4. Dhadhang Wahyu Kurniawan, Richell Booijink, Lena Pater, Irene Wols, Aggelos Vrynas, Gert Storm, Jai Prakash, Ruchi Bansal, “Fibroblast growth factor 2 conjugated superparamagnetic iron oxide nanoparticles (FGF2-SPIONs) ameliorate hepatic stellate cells activation in vitro and acute liver injury in vivo”, Journal of Controlled Release (2020); 328: 640-652. https://doi.org/10.1016/j.jconrel.2020.09.041. (Chapter 5)

175
Appendix D – Acknowledgements
Alhamdulillahirobbil'alamin, verily, all praise to Allah Subhanahu wa ta'ala for all the
abundance of His blessings, mercy, grace, and guidance that I accomplished my PhD work at
the University of Twente. Only because of His help I could reach the finish line of my PhD, after
a long and tiring journey as people say about the PhD journey. Now I have to write this section,
one of the simplest parts but not really easy as I should remember pieces of memories that
no one missed being acknowledged.
First of all, I would like to express my big gratitude to Dr. Ruchi Bansal, the person who has
been the most meritorious to my PhD journey. Without you, probably I went back to Indonesia
after doing the qualifier exam, in the failed condition. You are like a fairy godmother to me,
during my PhD work. While I was still in Indonesia and interviewed via skype by you, to be
honest, I had not understood yet what is liver fibrosis, the theme that you proposed to me.
You guided me in the laboratory directly, trained me in skills on how to handle the cell lines,
and how to do research with them, something that I had not been done in Indonesia at all.
You coached me until I was capable to do experiments in molecular biochemistry, molecular
pharmacology, and molecular immunology, independently. After having the results of the
experiments, you taught me how to process and analyze the data to be meaningful numbers
and graphics, good to see, and easy to read. You led me to write 4 manuscripts which were
successfully published in prominent peer-review scientific journals, something that I did not
imagine before I arrived in the Netherland 6 years ago. Your time to supervise me is more than
24 hours per day even on the weekend, you were always available to answer my questions
and received my reports. You educated me about perfection achievement, great scientific
work, wonderful performance, and speed as well as accuracy in taking a decision. Really, what
I achieved at this moment is truly beyond my expectations, I will never forget your direction,
your spirit, and your challenges.
Meeting with Prof. Dr. Gert Storm in Jakarta on 6th of June 2015 changed my life. Although
you did not read my proposal and my CV completely, it doesn’t matter, the most important
you gave me the opportunity to continue to study in the Netherlands. Thank you, Prof, your
generosity brought me to the University of Twente. I always enjoyed our discussions and
talking with you from the first time we met until now. Your style really makes an impression

176
on me and greatly affects my style when interacting with my students. You are truly a great
professor with excellent expertise in academics and a cool communicator. Your reputation is
my inspiration.
Thanks to Prof. Dr. Jai Prakash, as one of my promotors, you always motivate and advised me
to finish my PhD work remarkably. I never forget your statement about writing a paper as well
as I can and publishing it in a highly reputable scientific journal. I have acquired a lot of
scientific mentoring from you.
My gratitude also goes to the members of my graduation committee Prof. Dr. H.B.J.
Karperien, Prof. Dr. A. Kocer, Prof. Dr. K. Poelstra, Prof. Dr. P. Olinga, and Prof. Dr. R.
Weiskirchen for your time, for evaluating my PhD thesis, and taking part in my PhD defense
ceremony.
After being confirmed I was accepted as a PhD student at the University of Twente, the first
Indonesian I contacted was Dwi Lestari. She is my senior in the School of Pharmacy ITB
Bandung. From her, I obtained a lot of information about Enschede. Thank you Mbak Dwi for
your services and your help. You always answered my questions and my requests patiently.
You and your family always gave a warm welcome when I and my family visited your house on
Haaksbergerstraat 234. We miss the nice memories between our families.
Many thanks to Jonas Schnittert, my long-time office mate in ZH253, for the talks, discussions,
and every day jokes. I really remember our early interactions when you were grumpy because
of my mistakes in doing the lab duties. After that, gradually we had good teamwork and we
were more closer to each other, even now though we can’t meet every day physically we keep
in contact via WhatsApp. Thanks for the lesson about the very professional life. Our
relationship for about 2.5 years become an inspirational story to my students and colleagues
in Indonesia.
Another colleague who was in the same office for a long-time period was Marcel Alexander
Heinrich. I often call him "Prof" because of his super talent and ability. As far as I can
remember, during my stay in the BST Department, you were the only master student who has
got 10 for the master's thesis. Your ability to research, excellence in art and creativity make
you a very complete academic figure. I always remember your style after doing something
great. As I predicted, you got cum laude for your PhD work and you truly deserve it. Thanks

177
Marcel, I wish that your style of doing research, data processing, and writing manuscripts can
be transferred to me. Having a comrade like you is my another my wish, I still have another
prediction for you and can’t wait to see that you will receive a Nobel prize in the future.
Another senior figure in the targeted therapeutics group is Praneeth Reddy Kuninty. When
we have a discussion, you always tried to give a solution to the results of my experiments.
Thank Praneeth, discussion with you really enlightened me, your solutions were practical and
applicable. We did not only discuss in the room, but anywhere and anytime, even one very
cool memory with you is a discussion while pedaling a bicycle. I don’t know when we will have
a discussion again while riding a bicycle.
After Jonas graduated, Ahmed Mostafa came as a new PhD student and was sitting at the
desk next to me. Thank you for all the talks and chats about the PhD research as well as the
pharmaceutical worldwide topic. Thanks for your willingness to be my paranymph for my PhD
defense.
I am immeasurably grateful for the support system of the Artificial Organ and Bioengineering
Therapeutics/AOT Department (formerly BST Department). Thank you so much Karin
Hendriks, your service and dedication will not be repaid by me. From Indonesia to Netherlands
and from Netherlands to Indonesia, you have always supported me and was always responsive
to my questions and my requests. Hopefully, God rewards your kindness with multiplied
goodness. Thanks to Hetty ten Hoopen who very patiently and painstakingly supported the
continuity of my research, helped me with the UHPLC analysis, and for all the help during my
PhD project. If you want to visit your uncle’s grave in Indonesia just contact me. Thanks to
Marc Ankone for the very valuable discussion and information regarding our research in the
laboratory, and also for the full of insightful talks. Thank Lydia Bolhuis for organizing, and
distributing lab work very well.
The early weeks of my PhD days were almost always accompanied by Deby and Büsra Ozturk
in the laboratory. Thanks for the accompanying me and supporting me while preforming basic
molecular experiments in the lab.
After you (Büsra) finished your internship, you joined as a research assistant with Ruchi in
2018-2019 period. During this period, we were working together doing in vitro and in vivo

178
experiments. Thank Büsra for the discussion and cooperation, good luck with your life and
your future career.
Thanks to Richell Booijink, the longest partner in the group of liver research supervised by
Ruchi. It’s very pleasing to have a partner like you and work together with you since you were
a Master student. Also big thanks for your help in translating summary of my PhD thesis into
Nederlandse samenvatting. I wish you lots of success with your PhD work!
Thanks to Lena Heinrich (Pater) for the teamwork in the laboratory and for the very helpful,
friendly, and warm attitude. Your picture with Marcel when visiting my baby at my home is
stored neatly in our album.
For my Dutch friend in the department, Thijs Pasman, thank you for the unofficial course
about the Netherlands’ life and culture. Your warmness and jokes will always stay in my deep
memory.
Thanks to Tushar Satav and Tao Lu, two Post-Doctoral colleagues in the targeted therapeutics
research group. Discussion with both of you about working, research, and laboratory is almost
never-ending.
For fellow Dutch colleagues, Albert Jan, a football lover, thank you for the football coaching.
Also for your devour when eating the Indonesian foods. Good luck with your career and your
life!
To Franck Assayag, thank you for offering your apartment in Paris. Staying in your apartment
during the holiday together with my family was very helpful, imagine how many euros I would
have spent if we rented an apartment ourselves.
Thanks to Aysun, Denys, Natalia, Ilaria, Odyl, Iris, Karin Postma, Kasia, Chao, Pia, Aga, Bas,
Duco, Jia Liang, Kim, and Mike for togetherness in the BST Department. Also thanks to Eline,
Eshwari, Alex, Ana, Andreas, and Hilde for togetherness in the liver research group supervised
by Ruchi.
Hero Marhaento and his wife Astuti, our closest neighbor in the Pathmos area, thank you for
warmth, intimacy, kinship, and everything that makes us is like living in Yogyakarta. You also
often to be a lunch partner on the red sofa in the Zuidhorst Building and pray together around
campus. Discussion with you about PhD was always a pleasure even until we’re back in

179
Indonesia still continued. Thank you and an apology to you, if I was often troublesome to you
and your family.
Big thanks to Aulia Akbar for your assistance to drop and pick-up Ghazy goes to school when
I went to Indonesia in November 2017. Our talks on the train provided me with more insights
into life. Good luck with your career after your PhD!
Thanks to Taufiq Arifin, Sunu Widianto, and Ridwan Saptoto for the place of sholat. Instead
of going to Vriehof quite far, sholat in Mushola Taufiq then changed to Musholla Ridwan to be
a good option.
For Kunaifi, Pesi, Nova, Aji, Diah, Khafidz, Habib, Dwi Mandaris, Nico, Jarot, Krishna Aryanto,
Andri, Rizal, Irena, Heksi, Nissa, Abdul Rahman, Wibi, Imam, Yan, Muthia, Rindia, Andani,
Nisa, Yasmin, and all of PhD students from Indonesia who works at the University of Twente,
thank you for everything. Discussions, jokes, and kinship all of you will always chill and I hope
will be a spirit of collaboration to contribute to our beloved homeland, Indonesia Raya.
For the group of the February 2016 intake, including Akbar Aryanto, Aditya Fernando, Rheza,
Bima, Yasir, Aden, Hana, Ratri, Kania, Siti, etc thank you for the teamwork and your warmth.
Still remember at the beginning of our arrival at Enschede we eat together with all of you,
frequently. Especially Mas Akbar, thank you for already willing to accommodate me for a week
before I moved to the new rent house.
Deden Noor Rahman as chief of LPDP UT awardee when my first-time arrival at Enschede,
thank you for subletting your room for a week, while I occupy the housing facilitated by the
University of Twente. Thanks to all of LPDP Enschede awardees for all of your kinship.
To Iqbal Yulizar Mukti, a chief of LPDP UT awardee recently, thanks a lot for your willingness
to be one of my paranymphs in my PhD public defense.
Thanks to Yosia, Adi, Arif, Laksamana, Giri, Wildan, Ega, Ilus, Niswa, Dzulqornaen, Fauzan,
Fajar Eko, and all of PPI Enschede's volunteers who have always been compact and passionate
about guard kinship and the Indonesian atmosphere in Enschede.
Edi Trihartono and Kamia Handayani, a couple of husband and wife along with his family,
became the first Indonesian neighbors for our stay at Oosveenweg 86. Thank a lot for
everything, hopefully, sometime we will meet again warmly in Indonesia.

180
For Kartika Ratna Pratiwi, Erlis Saputra, Agung Budiono (deceased), Hakim, Afif, Didik
Setiawan, Mas Azis, Mbak Ary, Iwan Suharyanto, Karim, Pugo, Adityo, and all members of
Kagama NL, thank you for the kinship. Memories of KMF (kumpul-kumpul, makan-makan,
foto-foto) become sweet memoriest to be always remembered. With Kagama NL members
we are truly feel friendly atmosphere, guyup, rukun, and migunani.
For Amalia Muhaimin and Zulfan Zazuli, thank you for coming to Enschede and helping
prepare menus, food, drink, and everything to celebrate aqiqah of our baby. For Amalia and
Joko Mulyanto, I hope we keep going collaborating to make Unsoed much better.
Meeting with Adhyatmika and Ivan Surya Pradipta for the first time physically in the
Netherlands, I have a familiar feeling directly to both of you. This happens probably because
we have the same background in the pharmacy field. Thank you for your hospitality while I do
research experiments at the University of Groningen.
Dear Muslim colleagues affiliated with IMEA, thanks for the friendship for about 4 years at
Enschede. With IMEA, we still have an atmosphere of Ramadhan, Eid Fitri, and Eid Adha like
in Indonesia, even though we are living in the Netherland. The technical help and spirits of Bu
Sophie, Mbak Enung, Mbak Ely, Mbak Sifa, Mbak Ranny, Mbak Pipit, Mbak Wenny, and other
ladies to my family really make us touched and happy, even until approaching our return to
Indonesia. Big thanks IMEA- ers.
Thanks to Raymon, Ambar, Bu Ria, Pak Bambang, Fajar Sidiq, Monika, Fiki, Charles, Dewi,
and all members of Paguyuban IA ITB the Netherlands. Our togetherness and kinship are very
memorable, although only a few years.
Another community and place in the Netherlands that we will always remember, namely SGB
Utrecht. There we met noble-hearted people, thanks to Pak Gustiandi, Pak Supardi, and Pak
Dian, the trio who is well known as the sovereign of SGB Utrecht. Thank Pak Deden, because
of your network me and family can go for a walk around Europe and meet very kind-hearted
people. Thank Pak Makky for your reception during Fatih doing of UN in Den Haag. Thank Pak
Didin, the owner of Toko Nusantara, go to Den Haag is not complete before enjoying your
meal and drink, of course with the discount. Thank Pak Gunaryadi for your help during my
sons join in the SIDH Den Haag. To my very good senior at ITB, Pak Eko Hardjanto, thank you
very much for your goodness. Even until I’m back already in Indonesia I still bother you often.

181
Regardless, the struggling story in my PhD works at the University of Twente resulted from
the prayers of my mother Ibu Windrati and my mother-in-law Ibu Titiek Ekowati. Your prayers
every time is accepted and ijabah by Allah SWT. Thanks Moms, really, I cannot repay prayers
from you, may Allah reward your prayers with a lot of goodness in the world and the hereafter.
My thanks to my sons Fatih, Ghazy, and Rafid, who became my spirit, inspiration, and soul.
To Fatih, I'm sorry to have made your school move around, hopefully, the story of your
changing school would have added experience and enhanced your fight to reach your goal. To
Ghazy, though your first formal school in the Netherlands, always keep your spirit for go on
and on spirit go to school realize your dreams. To my little one who was born in Enschede, I
hope your childhood memories could motivate you and deliver you reach anything you want.
The last and the ultimate of everything, especially half of my soul dear beloved wife
Yudyawati, thank you have together me in all conditions, joy and sorrow. Besides moan sigh
to Allah SWT, to you my wife I tell all my feelings. Sorry if almost every day I outpour my heart
to you. Without you, maybe I am not able to complete this PhD journey. Your patience, your
attention, your love, and your maturity become my spirit for always being optimistic and
enthusiastic to finish the PhD story. May Allah continue together us and always present
happiness in our family.

182
Appendix E – About the Author
Dhadhang Wahyu Kurniawan was born on March 21st, 1979, in
Lamongan, East Java, Indonesia. He graduated bachelor and
pharmacist (Apotheker) from the School of Pharmacy Institut
Teknologi Bandung (ITB), Indonesia. He got married to his
lovely wife, Yudyawati (Iyut), and has three sons, Fatih, Ghazy,
and Rafid.
From May 2003 until March 2005, he worked as a Production
Supervisor at PT Hexpharm Jaya Laboratories (Kalbe Farma
holding company). In April 2005, he worked as a Formulation (Research and Development)
Senior Supervisor at PT Guardian Pharmatama. Then, from September 2005 until April 2006,
he worked as a lecturer at the Faculty of Pharmacy Universitas Pancasila (UP), Jakarta,
Indonesia. Since April 2006, he is a lecturer and researcher at the Department of Pharmacy
Universitas Jenderal Soedirman (Unsoed) Purwokerto, Indonesia. He became a student affairs
secretary at the Department of Pharmacy Unsoed from 2007 until 2009. In September 2011,
he graduated Master of Pharmacy from the Faculty of Pharmacy Universitas Gadjah Mada
(UGM) Yogyakarta, Indonesia. One year later, he became a general secretary at the
Department of Pharmacy Unsoed from 2012 until 2013. Then, he became the head of the
Department of Pharmacy, Faculty of Health Sciences Unsoed from 2013 until 2015.
At the end of 2015, he decided to resign as the head of the department to continue his study
at the University of Twente, the Netherlands. His PhD study was funded by the Indonesia
Endowment Fund for Education (Lembaga Pengelola Dana Pendidikan/LPDP), Ministry of
Finance Indonesia.
After finishing his PhD, he returned to Unsoed to continue his career in academia.

Copyright © 2022 FDOKUMEN




















