
Mice lacking asparaginyl endopeptidase develop disorders resembling hemophagocytic syndrome
Upload
independentCategory
view
5download
0
Increased Hepatic Steatosis and Insulin Resistance inMice Lacking Hepatic Androgen Receptor
Hung-Yun Lin,1* I-Chen Yu,1* Ruey-Shen Wang,1,2 Yei-Tsung Chen,1 Ning-Chun Liu,1 Saleh Altuwaijri,1
Cheng-Lung Hsu,1,3 Wen-Lung Ma,1 Jenny Jokinen,1 Janet D. Sparks,1 Shuyuan Yeh,1 and Chawnshang Chang1
Early studies demonstrated that whole-body androgen receptor (AR)–knockout mice withhypogonadism exhibit insulin resistance. However, details about the mechanisms underly-ing how androgen/AR signaling regulates insulin sensitivity in individual organs remainunclear. We therefore generated hepatic AR-knockout (H-AR�/y) mice and found that maleH-AR�/y mice, but not female H-AR�/� mice, fed a high-fat diet developed hepatic steatosisand insulin resistance, and aging male H-AR�/y mice fed chow exhibited moderate hepaticsteatosis. We hypothesized that increased hepatic steatosis in obese male H-AR�/y miceresulted from decreased fatty acid �-oxidation, increased de novo lipid synthesis arising fromdecreased PPAR�, increased sterol regulatory element binding protein 1c, and associatedchanges in target gene expression. Reduced insulin sensitivity in fat-fed H-AR�/y mice wasassociated with decreased phosphoinositide-3 kinase activity and increased phosphenol-pyruvate carboxykinase expression and correlated with increased protein-tyrosine phospha-tase 1B expression. Conclusion: Together, our results suggest that hepatic AR may play a vitalrole in preventing the development of insulin resistance and hepatic steatosis. AR agoniststhat specifically target hepatic AR might be developed to provide a better strategy fortreatment of metabolic syndrome in men. (HEPATOLOGY 2008;47:1924-1935.)
Nonalcoholic fatty liver disease (NAFLD) is acommon hepatic disorder with abnormal liverfunction, hyperlipidemia, and steatohepatitis
that can progress to cirrhosis.1 The estimated prevalenceof NAFLD in the United States is approximately 20% butis as high as 75% in obese and type 2 diabetic patients.2,3
Studies have suggested that ectopic triglyceride (TG) ac-
cumulation in the liver is a key element in the pathogen-esis of insulin resistance,4 with increased endogenousglucose production in both lean and obese subjects.5,6
Insulin resistance is an important component of type 2diabetes and metabolic syndrome. Feeding rodents ahigh-fat diet (HFD) leads to hepatic steatosis, with im-paired insulin signaling and hepatic insulin resistance.7,8
Together, these results strongly suggest that hepatic lipidaccumulation may be directly responsible for the subse-quent development of hepatic insulin resistance and in-creased endogenous glucose production, key elements inthe pathogenesis of type 2 diabetes. They also suggest thatreducing hepatic lipid accumulation might be an effectivetherapeutic strategy for both NAFLD and type 2 diabetes.
Epidemiological evidence suggests that sex differencesexist in type 2 diabetes, with a higher prevalence in menthan in women,9 possibly because of differences in insulinsensitivity and regional deposition of body fat.10 Testos-terone effects are mediated by the androgen receptor(AR).11,12 The total and general AR-knockout mousemodel (T-AR�/y) demonstrates progressively reduced in-sulin sensitivity and impaired glucose tolerance at ad-vanced ages, similar to that observed in people withtestosterone deficiency. The loss of the AR contributes toincreased triglyceride content in skeletal muscle and liverin these animals, and serum leptin concentrations are el-
Abbreviations: AR, androgen receptor; AR�/y, wild-type mice; H-AR�/y, hepaticAR-knockout mice; HFD, high-fat diet; NAFLD, nonalcoholic fatty liver disease;PEPCK, phosphenolpyruvate carboxykinase; PTP1B, protein tyrosine phosphatase1B; SREBP-1c, sterol regulatory element binding protein 1c; TG, triglyceride.
From the 1George Whipple Lab for Cancer Research, Departments of Pathology,Urology, and the Cancer Center, University of Rochester Medical Center, Rochester,NY; the 2Department of Gynecology and Obstetrics, Taipei Medical University,Taipei, Taiwan; and 3Division of Hematology-Oncology, Department of InternalMedicine, Chang Gung Memorial Hospital, Chang Gung University College ofMedicine, Taoyuan, Taiwan.
Received September 13, 2007; accepted January 22, 2008.Supported by National Institutes of Health grant DK073414 and George
Whipple Professor Endowment.*These authors contributed equally to this study.Address reprint requests to: Chawnshang Chang, Ph.D., 601 Elmwood Avenue,
Box 626, Rochester, NY 14642. E-mail: [email protected]; fax: (585)756-4133.
Copyright © 2008 by the American Association for the Study of Liver Diseases.Published online in Wiley InterScience (www.interscience.wiley.com).DOI 10.1002/hep.22252Potential conflict of interest: Nothing to report.Supplementary material for this article can be found on the HEPATOLOGY Web
site (http://interscience.wiley.com/jpages/0270-9139/suppmat/index.html).
1924

evated.13 One mechanism by which testosterone via ARmight facilitate insulin sensitivity is by regulating the ex-pression of insulin-dependent proteins, and dose-depen-dent effects of testosterone on insulin receptorsubstrate–1 (IRS-1) and glucose transporter–4(GLUT-4) have been documented in cell models.14 An-other potential mechanism involves AR-dependentchanges in mitochondrial function, and decreased tran-scription of oxidative phosphorylation genes has been ob-served in skeletal muscle of insulin-resistant subjects.Decreased mitochondrial function leads to decreasedlipid oxidation, ectopic TG accumulation, and, ulti-mately, to insulin resistance.15 In male subjects, testoster-one level has been shown to correlate positively withmitochondrial capacity, as assessed by maximal aerobiccapacity and expression of oxidative phosphorylationgenes.16
Another potential mechanism involves protein ty-rosine phosphatase 1B (PTP1B), a member of the proteintyrosine phosphatase family. PTP1B is a negative regula-tor of insulin action,17 attenuating insulin signalingthrough dephosphorylation of IR and IRS-1.18 PTP1B-knockout mice have increased insulin sensitivity and obe-sity resistance.19,20 Recently, it was shown that androgenwithdrawal in LNCaP (human prostate cancer cell line)cells increased expression of PTP1B with a correspondingincrease in its tyrosine phosphatase activity.21
To determine whether AR is required and important inglucose and lipid homeostasis in the liver, we used a Cre-loxP strategy to specifically delete AR in the liver withoutimpaired development of genital organs and subsequenthypogonadism. Our results demonstrate that male he-patic AR-knockout (H-AR�/y) mice have increased he-patic steatosis and insulin resistance on HFD feeding,with decreased �-oxidation and increased PTP1B expres-sion. Results suggest that the AR plays an important rolein hepatic glucose and lipid homeostasis.
Materials and Methods
Animals. All animal procedures were approved by theanimal care and use committee of the University of Roch-ester School of Medicine, in accordance with NationalInstitutes of Health guidelines. Construction of targetingvectors and generation of the chimera founder mice havebeen described previously.13,22 The floxAR mice werebred into a C57BL/6 background. Albumin is specificallyexpressed in the liver, and its promoter was used to driveCre (Alb-Cre; C57BL/6; The Jackson Laboratory) anddelete floxed AR fragments specifically in the liver. TheH-AR�/y mice were genotyped by PCR, as described pre-viously.13 Animals were housed in pathogen-free facilities,
maintained on a 12-hour light/12-hour dark schedule(light on at 6:00 AM), and had access to standard labora-tory chow (5010; PMI Lab Diet) and water ad libitum.
Histological Analysis. Tissues were fixed in 4% (g/v)para-formaldehyde and embedded in paraffin, and sec-tions were with hematoxylin/eosin. Images were acquiredusing an E800 microscope (Nikon) and a SPOT camera(Diagnostic Instruments) and were analyzed using Sig-maScan Pro software (version 5.0; SPSS).
Analytical Procedures. Fasting blood samples weretaken from mice 14 hours after withdrawal of food.Blood samples designated as random-fed state weredrawn 6 hours after introducing food into the cages ofmice that had been subjected to a preceding 14-hourfast. Blood glucose concentration was measured using aglucometer (One Touch Ultra; Lifescan). Insulin andleptin levels were determined in duplicate 5-�L serumsamples, using a mouse insulin and leptin enzyme-linked immunosorbent assay kit (Crystal Chem) ac-cording to the manufacturer’s instruction. Serumadiponectin level was determined in duplicate in20-�L serum samples using an enzyme-linked immu-nosorbent assay kit (eBioscience). For the glucose tol-erance test (GTT), after a 14-hour fast, mice were givenan oral bolus of D-glucose (2 g/kg body weight), andblood glucose concentration was measured in samplestaken after 0, 30, 60, 90, and 120 minutes. Insulintolerance tests (ITTs) were performed on 6-hour-fast-ing mice by intraperitoneal injection of 1 unit/kg bodyweight human insulin (Sigma Aldrich). Blood glucoseconcentrations were determined 0, 60, and 120 min-utes after insulin administration. The triglyceride levelin sera from fasting animals was determined using aGPO-Trinder assay (Sigma Aldrich). The serum-freefatty acid level in fasting animals was measured using aNEFA-Kit-U (Wako Pure Chemical). For determina-tion of tissue triglyceride content, 50-100 mg of tissuewas homogenized on ice in pH 7.3 extraction buffer(20 mmol/L Tris, 1 mmol/L �-mercaptoethanol, 1mmol/L EDTA). After centrifugation, the glycerolcontent of the supernatants was determined using aGPO-Trinder assay (Sigma Aldrich) according to themanufacturer’s instructions.
PI3k Activity by ELISA. PI3k activity was deter-mined using PI3k ELISA (Echelon Biosciences Inc.).This kit measures PI3k activity as a conversion ofPI(4,5)P2 into PI(3,4,5)P3. We used this kit in conjunc-tion with anti-p85 PI3k antibody (Upstate Biotechnol-ogy). Briefly, cells were washed 3 times with ice-coldbuffer A and then lysed in buffer A containing 1% NP40and 1 mM phenylmethylsulfonyl fluoride. Lysates wereincubated on ice for 20 to 30 minutes and then cleared at
HEPATOLOGY, Vol. 47, No. 6, 2008 LIN, YU, ET AL. 1925

16,000g for 20 minutes. PI3k was immunoprecipitatedby polyclonal anti-p85 PI3k antibody at 4°C for 1 hour.Binding of the coimmunoprecipitated p85/p110 PI3kcomplexes to protein A–agarose beads was carried out at4°C for 1 hour, and the beads then were washed 3 timeswith buffer A containing 1% NP40, 3 times with bufferB, and twice with buffer C, as suggested by the manufac-turer. The kinase reaction was carried out for 1 hour atroom temperature in 4 mM MgCl2, 20 mM Tris-HCl(pH 7.4), and 20 mM NaCl supplemented with 25 �MATP.
Mouse Primary Hepatocytes. Mouse hepatocyteswere obtained by nonrecirculating collagenase perfusionthrough the portal vein of male wild-type (AR�/y) or age-matched H-AR�/y mice. This standard procedure wascarried out as described previously.23
Fatty Acid Oxidation. Isolated hepatocytes were in-cubated for 1 hour in scintillation vials in Krebs bufferwith 2% BSA, with isotope-labeled 1 mM palmitic acid.Radiolabeled CO2 was collected in center wells withWhatman filter paper and 300 �L of 1M methylbenze-thonium hydroxide in methanol. At the end of the incu-bation, 300 �L of 5M H2SO4 was added to volatilize theremaining CO2, and the solution was incubated for an-other 30 minutes. The center wells were then placed inother scintillation vials, and 8 mL of aqueous scintillantwas added and counted on a � counter.
Real-Time Quantitative PCR. Total RNA was pre-pared from cells or tissues with Trizol (Initrogen) accord-ing to the manufacture’s instructions. We used a real-time
PCR method to quantify the mRNA as described previ-ously.13
Statistical Analysis. Data are means � SEM. Dif-ferences between 2 groups were assessed by the unpaired2-tailed Student’s t test. Data involving more than 2groups were assessed by 1-way ANOVA plus the Student-Newman-Keuls method (SigmaStat, SyStat).
Results
Generation of Hepatic ARKO Mice. Using a Cre-loxP conditional knockout strategy, we mated female het-erozygous floxed AR mice with male Alb-Cre mice togenerate H-AR�/y and wild type AR (AR�/y; Alb Cre�)littermates. We detected Cre and floxed AR DNA frag-ments in tail genomic DNA of weaned 21-day-oldH-AR�/y mice (Fig. 1A). Various tissues including epi-didymis, heart, kidney, muscle, spleen, testis, and adiposewere harvested from 8-week-old H-AR�/y and AR�/y
mice, and only liver tissue showed deletion of AR exon 2with an 180-bp product from RT-PCR using primers forexons 1 and 3 (Fig. 1B). This finding suggests thatH-AR�/y mice have selective disruption of AR specificallyin the liver.
Male H-AR�/y Mice But Not Female H-AR�/� MiceWere More Susceptible to Diet-Induced Obesity. Pre-vious findings demonstrated that T-AR�/y mice develophepatic steatosis and insulin resistance at an advanced age.To determine if hepatic AR plays major roles in the de-velopment of obesity and obesity-associated hepatic ste-
Fig. 1. Generation of mice with conditional knockout of AR in hepatocytes (H-AR�/y). (A) Identification and confirmation of H-AR�/y mice. GenomicDNA was isolated from tail snips and used as a template for PCR with primers “select” and “2-3” or Cre. The detailed method and primer sequenceswere described previously.13 The expression of floxed AR and Alb-Cre in the tail genomic DNA of H-AR�/y mice was confirmed by PCR. (B) RT-PCRof various tissues harvested from AR�/y and H-AR�/y mice. Only the mRNA from the livers of the H-AR�/y mice showed a knockout band (ko) anda wt AR band when primers exons 1 and 3 were used; other tissues had only a wt band. Data are representative images from the two experimentalgroups (n � 5).
1926 LIN, YU, ET AL. HEPATOLOGY, June 2008
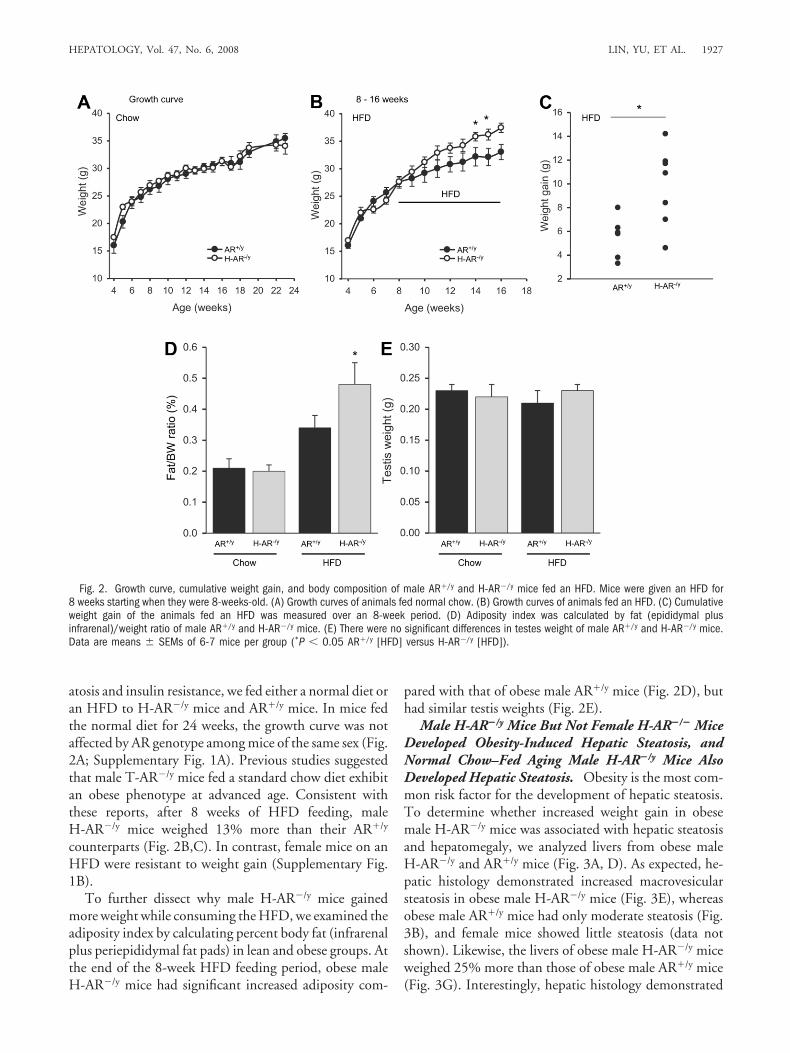
atosis and insulin resistance, we fed either a normal diet oran HFD to H-AR�/y mice and AR�/y mice. In mice fedthe normal diet for 24 weeks, the growth curve was notaffected by AR genotype among mice of the same sex (Fig.2A; Supplementary Fig. 1A). Previous studies suggestedthat male T-AR�/y mice fed a standard chow diet exhibitan obese phenotype at advanced age. Consistent withthese reports, after 8 weeks of HFD feeding, maleH-AR�/y mice weighed 13% more than their AR�/y
counterparts (Fig. 2B,C). In contrast, female mice on anHFD were resistant to weight gain (Supplementary Fig.1B).
To further dissect why male H-AR�/y mice gainedmore weight while consuming the HFD, we examined theadiposity index by calculating percent body fat (infrarenalplus periepididymal fat pads) in lean and obese groups. Atthe end of the 8-week HFD feeding period, obese maleH-AR�/y mice had significant increased adiposity com-
pared with that of obese male AR�/y mice (Fig. 2D), buthad similar testis weights (Fig. 2E).
Male H-AR�/y Mice But Not Female H-AR�/� MiceDeveloped Obesity-Induced Hepatic Steatosis, andNormal Chow–Fed Aging Male H-AR�/y Mice AlsoDeveloped Hepatic Steatosis. Obesity is the most com-mon risk factor for the development of hepatic steatosis.To determine whether increased weight gain in obesemale H-AR�/y mice was associated with hepatic steatosisand hepatomegaly, we analyzed livers from obese maleH-AR�/y and AR�/y mice (Fig. 3A, D). As expected, he-patic histology demonstrated increased macrovesicularsteatosis in obese male H-AR�/y mice (Fig. 3E), whereasobese male AR�/y mice had only moderate steatosis (Fig.3B), and female mice showed little steatosis (data notshown). Likewise, the livers of obese male H-AR�/y miceweighed 25% more than those of obese male AR�/y mice(Fig. 3G). Interestingly, hepatic histology demonstrated
Fig. 2. Growth curve, cumulative weight gain, and body composition of male AR�/y and H-AR�/y mice fed an HFD. Mice were given an HFD for8 weeks starting when they were 8-weeks-old. (A) Growth curves of animals fed normal chow. (B) Growth curves of animals fed an HFD. (C) Cumulativeweight gain of the animals fed an HFD was measured over an 8-week period. (D) Adiposity index was calculated by fat (epididymal plusinfrarenal)/weight ratio of male AR�/y and H-AR�/y mice. (E) There were no significant differences in testes weight of male AR�/y and H-AR�/y mice.Data are means � SEMs of 6-7 mice per group (*P � 0.05 AR�/y [HFD] versus H-AR�/y [HFD]).
HEPATOLOGY, Vol. 47, No. 6, 2008 LIN, YU, ET AL. 1927
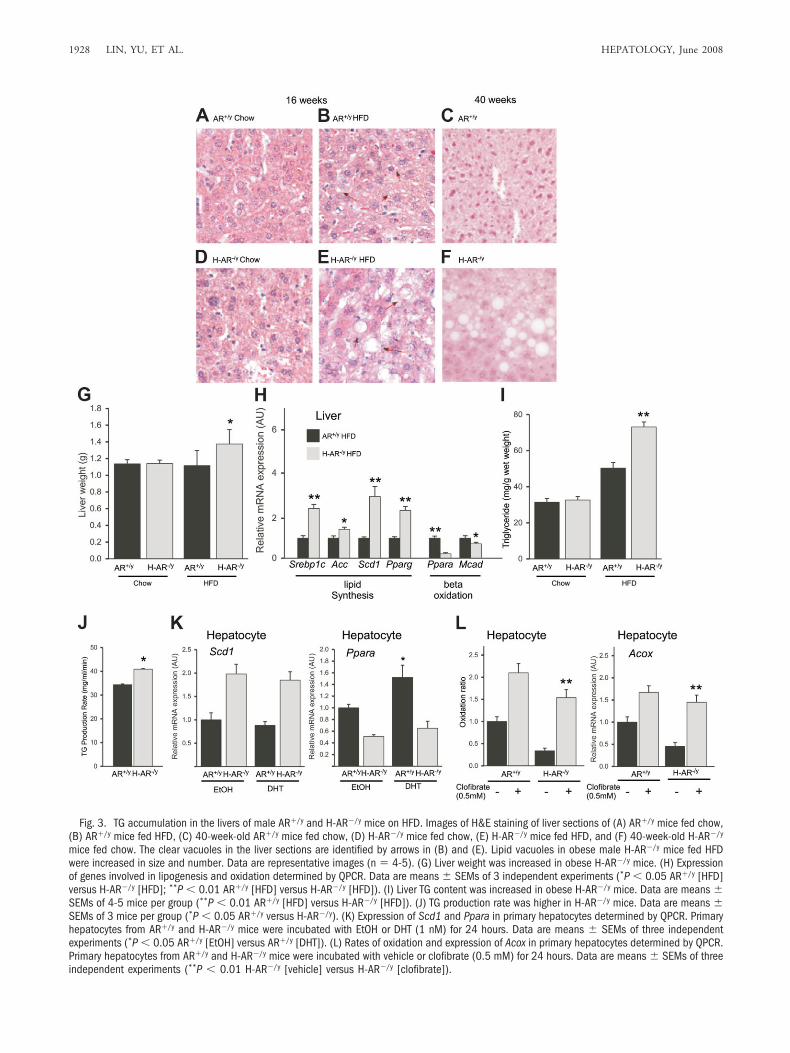
Fig. 3. TG accumulation in the livers of male AR�/y and H-AR�/y mice on HFD. Images of H&E staining of liver sections of (A) AR�/y mice fed chow,(B) AR�/y mice fed HFD, (C) 40-week-old AR�/y mice fed chow, (D) H-AR�/y mice fed chow, (E) H-AR�/y mice fed HFD, and (F) 40-week-old H-AR�/y
mice fed chow. The clear vacuoles in the liver sections are identified by arrows in (B) and (E). Lipid vacuoles in obese male H-AR�/y mice fed HFDwere increased in size and number. Data are representative images (n � 4-5). (G) Liver weight was increased in obese H-AR�/y mice. (H) Expressionof genes involved in lipogenesis and oxidation determined by QPCR. Data are means � SEMs of 3 independent experiments (*P � 0.05 AR�/y [HFD]versus H-AR�/y [HFD]; **P � 0.01 AR�/y [HFD] versus H-AR�/y [HFD]). (I) Liver TG content was increased in obese H-AR�/y mice. Data are means �SEMs of 4-5 mice per group (**P � 0.01 AR�/y [HFD] versus H-AR�/y [HFD]). (J) TG production rate was higher in H-AR�/y mice. Data are means �SEMs of 3 mice per group (*P � 0.05 AR�/y versus H-AR�/y). (K) Expression of Scd1 and Ppara in primary hepatocytes determined by QPCR. Primaryhepatocytes from AR�/y and H-AR�/y mice were incubated with EtOH or DHT (1 nM) for 24 hours. Data are means � SEMs of three independentexperiments (*P � 0.05 AR�/y [EtOH] versus AR�/y [DHT]). (L) Rates of oxidation and expression of Acox in primary hepatocytes determined by QPCR.Primary hepatocytes from AR�/y and H-AR�/y mice were incubated with vehicle or clofibrate (0.5 mM) for 24 hours. Data are means � SEMs of threeindependent experiments (**P � 0.01 H-AR�/y [vehicle] versus H-AR�/y [clofibrate]).
1928 LIN, YU, ET AL. HEPATOLOGY, June 2008

that aging male H-AR�/y mice fed normal chow also de-veloped moderate hepatic steatosis at 40 weeks of age (Fig.3F).
As early studies suggested that adipokines such as lep-tin and adiponectin are able to reverse hepatic steatosis inob/ob mice and lipodystrophic mice, we therefore assayedleptin and adiponectin levels in male H-AR�/y mice withhepatic steatosis. The refed serum leptin level was signif-icantly higher in obese male H-AR�/y mice, along with areduced fasting serum adiponectin level in lean maleH-AR�/y mice (Table 1). However, increased serum lep-tin was not sufficient to reduce hepatic steatosis and insu-lin resistance in obese male H-AR�/y mice (Table 1).Together, these results suggest that obese male H-AR�/y
mice had abnormal adipokine profiles and systemic leptinresistance that might contribute to the development ofobesity-induced hepatic steatosis.
Obese Male H-AR�/y Mice Developed Hepatic Ste-atosis Via Reduced Lipid Oxidation and Increased DeNovo Lipid Synthesis. We first observed reduced fattyacid oxidation in isolated primary hepatocytes fromH-AR�/y mice (Fig. 3L). Mechanistic dissection foundthat PPAR� and sterol regulatory element binding pro-tein 1c (SREBP-1c), 2 key genes involved in the modula-tion of fatty acid oxidation, were altered in the livers ofobese male H-AR�/y mice (Fig. 3H). Increased SREBP-1cexpression is consistent with the up-regulation of acetylCoA carboxylase (ACC), the first committed step in fattyacid synthesis. With increased malonyl CoA levels leadingto inhibition of carnitine palmitoyl transferase 1 (CPT-1), it then results in reduced mitochondrial fatty oxida-tion via reduction of free fatty acid transport from thecytosol to mitochondria. Reduced expression of PPAR�in H-AR�/y hepatocytes consistent with decreased malo-nyl CoA decaboxylase (MCAD) (Fig. 3H) could also con-tribute to increased malonyl CoA levels.
Increased De Novo Fatty Acid Synthesis. We notedincreased ectopic TG accumulation in the livers of obesemale H-AR�/y mice (Fig. 3I) and increased exportation of
VLDL-TG from the livers of obese male H-AR�/y mice(Fig. 3J). SREBP-1c, a key gene in the regulation of fattyacid synthesis, and its major target gene, stearoyl-CoAdesaturase 1 (SCD1), were both increased in H-AR�/y
hepatocytes (Fig. 3H). These results suggest increased denovo fatty acid synthesis may be partially responsible forectopic TG accumulation in liver.
Confirmation of Involvement of PPAR� in Devel-opment of Hepatic Steatosis in Obese Male H-AR�/y
Mice. Both decreased PPAR� and increased SREBP-1cmight play key roles in the development of hepatic steato-sis in obese male H-AR�/y mice. To support the involve-ment of AR in PPAR�, we further confirmed these resultsin primary hepatocytes. The addition of DHT to primaryhepatocytes increased PPAR� expression only in cellsfrom AR�/y mice (Fig. 3K), indicating that lack of ARsprevented androgen-induced PPAR�. However, the ad-dition of the PPAR� agonist clofibrate resulted in therestoration of fatty acid oxidation and expression ofACOX in isolated H-AR�/y hepatocytes (Fig. 3L), indi-cating that the PPAR� ligand was still able to activatefatty acid oxidation in an androgen-independent fashion.
Male H-AR�/y Mice But Not Female H-AR�/� MiceDeveloped Obesity-Induced Hepatic Insulin Resis-tance. Hepatic insulin resistance in obese mice and hu-mans is strongly associated with hepatic steatosis andhepatomegaly. Increased fat mass in male mice fed anHFD would be expected to influence systemic metabolicparameters, including insulin sensitivity. We measuredfasting blood glucose and serum insulin levels and foundno differences between lean male H-AR�/y and leanAR�/y mice (Fig. 4A-C). Similarly, we found no differ-ences in glucose level in female mice of either AR geno-type (Supplementary Fig. 2). However, HFD-fed maleH-AR�/y mice had increased fasting, refed blood glucose,and serum insulin levels compared with those in maleAR�/y counterparts (Fig. 4A-C). The calculated ho-meostasis model of insulin resistance (HOMA-IR) valuesof obese male H-AR�/y mice were 1.2-fold higher than
Table 1. Metabolic Parameters During Fasting in AR�/y and H-AR–/y Mice Fed Chow or HFD at 16 Weeks Old
Group
AR�/y H-AR–/y
Chow Diet High-Fat Diet Chow Diet High-Fat Diet
Triglyceride (mg/dL) 20.1 � 2.6 27.3 � 3.3 30.7 � 2.4 35.2 � 2.6*NEFA (mEq/L) 0.86 � 0.07 0.76 � 0.11 0.96 � 0.08 0.71 � 0.13Cholesterol (mg/dL) 113.2 � 3.4 142.6 � 5.1 105.7 � 8.3 137.3 � 10.6Leptin (ng/mL)† 2.3 � 0.2 9.6 � 2.1 3.2 � 0.1 15.1 � 3.4‡Adiponectin (�g/mL) 2.13 � 0.09 nd 1.42 � 0.19§ ndIGF-1 (ng/mL) 342.9 � 33.6 nd 156.7 � 18.6 ndGH (ng/mL) 62 � 8.1 nd 240 � 36.4 ndTestosterone (ng/mL) 3.34 � 0.59 nd 3.49 � 0.91 nd
Fasting refers to an overnight fast. Data are mean � SEM of 6-7 mice per group. *P � 0.05 AR�/y (HFD) versus H-AR-/y (HFD). †Leptin was assayed in fed animals.‡P � 0.01 AR�/y (HFD) versus H-AR-/y (HFD). §P � 0.05 AR�/y (chow) versus H-AR-/y (chow). P � 0.01 AR�/y (chow) versus H-AR-/y (chow). nd, not determined.
HEPATOLOGY, Vol. 47, No. 6, 2008 LIN, YU, ET AL. 1929
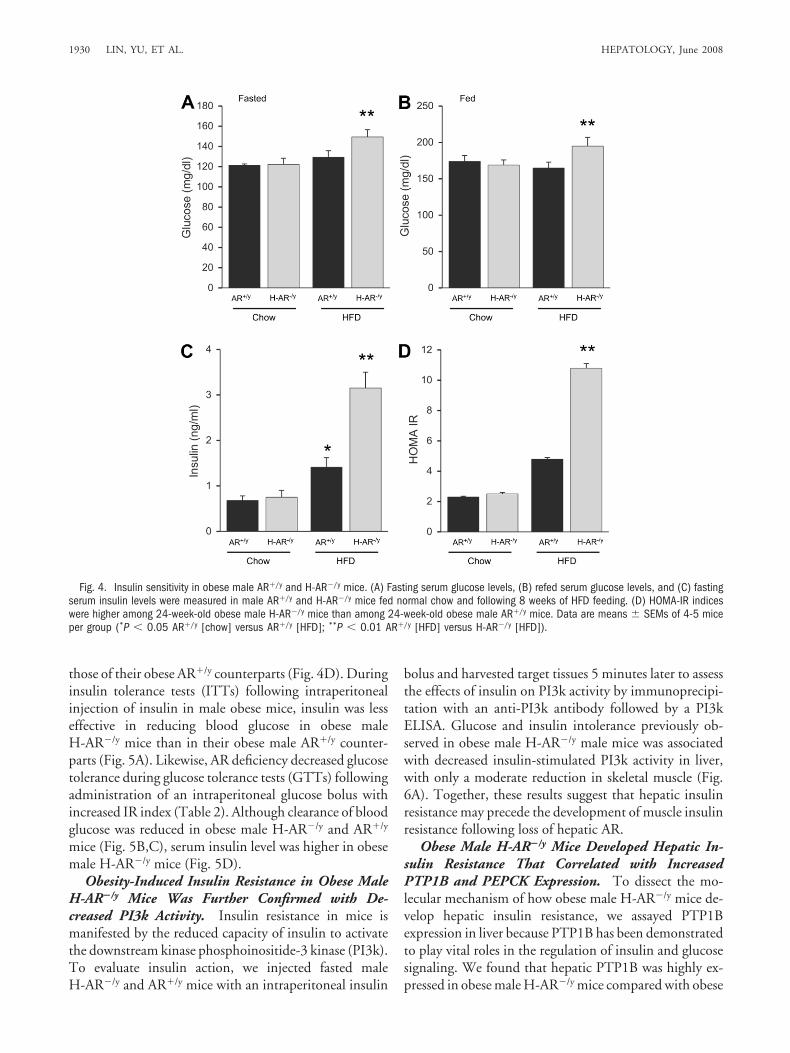
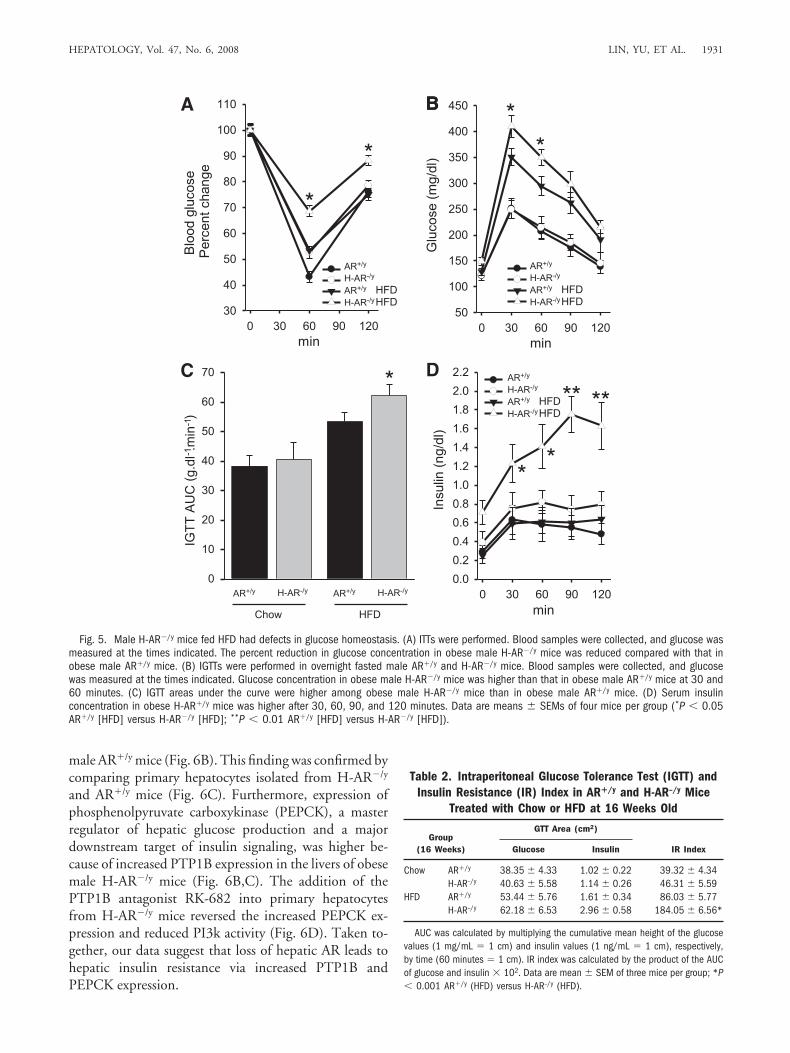
those of their obese AR�/y counterparts (Fig. 4D). Duringinsulin tolerance tests (ITTs) following intraperitonealinjection of insulin in male obese mice, insulin was lesseffective in reducing blood glucose in obese maleH-AR�/y mice than in their obese male AR�/y counter-parts (Fig. 5A). Likewise, AR deficiency decreased glucosetolerance during glucose tolerance tests (GTTs) followingadministration of an intraperitoneal glucose bolus withincreased IR index (Table 2). Although clearance of bloodglucose was reduced in obese male H-AR�/y and AR�/y
mice (Fig. 5B,C), serum insulin level was higher in obesemale H-AR�/y mice (Fig. 5D).
Obesity-Induced Insulin Resistance in Obese MaleH-AR�/y Mice Was Further Confirmed with De-creased PI3k Activity. Insulin resistance in mice ismanifested by the reduced capacity of insulin to activatethe downstream kinase phosphoinositide-3 kinase (PI3k).To evaluate insulin action, we injected fasted maleH-AR�/y and AR�/y mice with an intraperitoneal insulin
bolus and harvested target tissues 5 minutes later to assessthe effects of insulin on PI3k activity by immunoprecipi-tation with an anti-PI3k antibody followed by a PI3kELISA. Glucose and insulin intolerance previously ob-served in obese male H-AR�/y male mice was associatedwith decreased insulin-stimulated PI3k activity in liver,with only a moderate reduction in skeletal muscle (Fig.6A). Together, these results suggest that hepatic insulinresistance may precede the development of muscle insulinresistance following loss of hepatic AR.
Obese Male H-AR�/y Mice Developed Hepatic In-sulin Resistance That Correlated with IncreasedPTP1B and PEPCK Expression. To dissect the mo-lecular mechanism of how obese male H-AR�/y mice de-velop hepatic insulin resistance, we assayed PTP1Bexpression in liver because PTP1B has been demonstratedto play vital roles in the regulation of insulin and glucosesignaling. We found that hepatic PTP1B was highly ex-pressed in obese male H-AR�/y mice compared with obese
Fig. 4. Insulin sensitivity in obese male AR�/y and H-AR�/y mice. (A) Fasting serum glucose levels, (B) refed serum glucose levels, and (C) fastingserum insulin levels were measured in male AR�/y and H-AR�/y mice fed normal chow and following 8 weeks of HFD feeding. (D) HOMA-IR indiceswere higher among 24-week-old obese male H-AR�/y mice than among 24-week-old obese male AR�/y mice. Data are means � SEMs of 4-5 miceper group (*P � 0.05 AR�/y [chow] versus AR�/y [HFD]; **P � 0.01 AR�/y [HFD] versus H-AR�/y [HFD]).
1930 LIN, YU, ET AL. HEPATOLOGY, June 2008
male AR�/y mice (Fig. 6B). This finding was confirmed bycomparing primary hepatocytes isolated from H-AR�/y
and AR�/y mice (Fig. 6C). Furthermore, expression ofphosphenolpyruvate carboxykinase (PEPCK), a masterregulator of hepatic glucose production and a majordownstream target of insulin signaling, was higher be-cause of increased PTP1B expression in the livers of obesemale H-AR�/y mice (Fig. 6B,C). The addition of thePTP1B antagonist RK-682 into primary hepatocytesfrom H-AR�/y mice reversed the increased PEPCK ex-pression and reduced PI3k activity (Fig. 6D). Taken to-gether, our data suggest that loss of hepatic AR leads tohepatic insulin resistance via increased PTP1B andPEPCK expression.
A BB
C D
Fig. 5. Male H-AR�/y mice fed HFD had defects in glucose homeostasis. (A) ITTs were performed. Blood samples were collected, and glucose wasmeasured at the times indicated. The percent reduction in glucose concentration in obese male H-AR�/y mice was reduced compared with that inobese male AR�/y mice. (B) IGTTs were performed in overnight fasted male AR�/y and H-AR�/y mice. Blood samples were collected, and glucosewas measured at the times indicated. Glucose concentration in obese male H-AR�/y mice was higher than that in obese male AR�/y mice at 30 and60 minutes. (C) IGTT areas under the curve were higher among obese male H-AR�/y mice than in obese male AR�/y mice. (D) Serum insulinconcentration in obese H-AR�/y mice was higher after 30, 60, 90, and 120 minutes. Data are means � SEMs of four mice per group (*P � 0.05AR�/y [HFD] versus H-AR�/y [HFD]; **P � 0.01 AR�/y [HFD] versus H-AR�/y [HFD]).
Table 2. Intraperitoneal Glucose Tolerance Test (IGTT) andInsulin Resistance (IR) Index in AR�/y and H-AR–/y Mice
Treated with Chow or HFD at 16 Weeks Old
Group(16 Weeks)
GTT Area (cm2)
IR IndexGlucose Insulin
Chow AR�/y 38.35 � 4.33 1.02 � 0.22 39.32 � 4.34H-AR–/y 40.63 � 5.58 1.14 � 0.26 46.31 � 5.59
HFD AR�/y 53.44 � 5.76 1.61 � 0.34 86.03 � 5.77H-AR–/y 62.18 � 6.53 2.96 � 0.58 184.05 � 6.56*
AUC was calculated by multiplying the cumulative mean height of the glucosevalues (1 mg/mL � 1 cm) and insulin values (1 ng/mL � 1 cm), respectively,by time (60 minutes � 1 cm). IR index was calculated by the product of the AUCof glucose and insulin � 102. Data are mean � SEM of three mice per group; *P� 0.001 AR�/y (HFD) versus H-AR–/y (HFD).
HEPATOLOGY, Vol. 47, No. 6, 2008 LIN, YU, ET AL. 1931
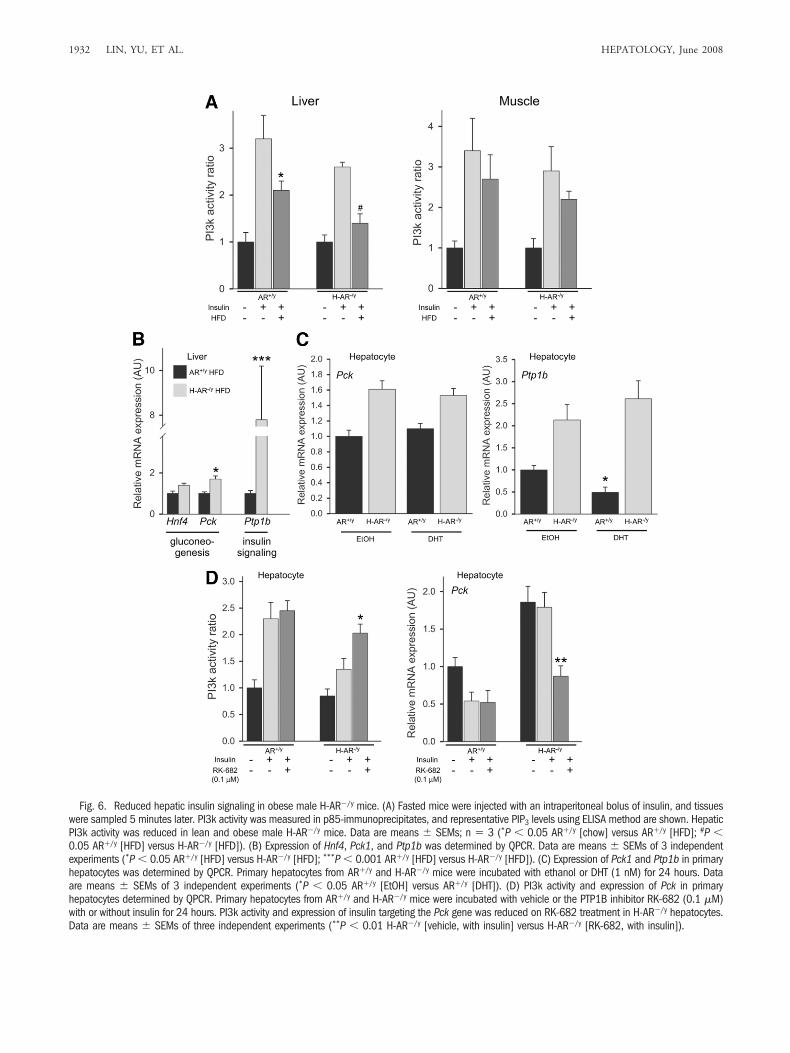
Fig. 6. Reduced hepatic insulin signaling in obese male H-AR�/y mice. (A) Fasted mice were injected with an intraperitoneal bolus of insulin, and tissueswere sampled 5 minutes later. PI3k activity was measured in p85-immunoprecipitates, and representative PIP3 levels using ELISA method are shown. HepaticPI3k activity was reduced in lean and obese male H-AR�/y mice. Data are means � SEMs; n � 3 (*P � 0.05 AR�/y [chow] versus AR�/y [HFD]; #P �0.05 AR�/y [HFD] versus H-AR�/y [HFD]). (B) Expression of Hnf4, Pck1, and Ptp1b was determined by QPCR. Data are means � SEMs of 3 independentexperiments (*P � 0.05 AR�/y [HFD] versus H-AR�/y [HFD]; ***P � 0.001 AR�/y [HFD] versus H-AR�/y [HFD]). (C) Expression of Pck1 and Ptp1b in primaryhepatocytes was determined by QPCR. Primary hepatocytes from AR�/y and H-AR�/y mice were incubated with ethanol or DHT (1 nM) for 24 hours. Dataare means � SEMs of 3 independent experiments (*P � 0.05 AR�/y [EtOH] versus AR�/y [DHT]). (D) PI3k activity and expression of Pck in primaryhepatocytes determined by QPCR. Primary hepatocytes from AR�/y and H-AR�/y mice were incubated with vehicle or the PTP1B inhibitor RK-682 (0.1 �M)with or without insulin for 24 hours. PI3k activity and expression of insulin targeting the Pck gene was reduced on RK-682 treatment in H-AR�/y hepatocytes.Data are means � SEMs of three independent experiments (**P � 0.01 H-AR�/y [vehicle, with insulin] versus H-AR�/y [RK-682, with insulin]).
1932 LIN, YU, ET AL. HEPATOLOGY, June 2008

DiscussionAndrogen–AR signaling plays a pivotal role in repro-
ductive organ development and prostate cancer. The roleof the AR in metabolic disorders has not been previouslyappreciated. Although hepatic insulin resistance and ste-atosis are associated, a causal relationship between ectopicfat accumulation and insulin sensitivity has not beenclearly established. We present evidence from the presentstudy that supports the importance of the hepatic AR inthe regulation of lipid and glucose homeostasis and dem-onstrates that hepatic deficiency of ARs significantly re-duces glucose tolerance and insulin sensitivity andincreases the accumulation of lipid in livers derived fromobese male H-AR�/y mice.
The mechanisms leading to the development of he-patic steatosis are likely to be complex. Previous studieshave shown that genes involved in lipogenesis are elevatedin livers derived from ob/ob mice,24 and the transcrip-tional factor SREBP-1c was shown to contribute to theincreased rate of lipogenesis in livers of these mice.24,25
Hepatic expression of fatty acid synthase is increased inlivers of ob/ob mice, and the fatty acid synthase gene istranscriptionally regulated by SREBP-1c.26 Indeed,SREBP-1c expression was markedly increased in the liversof obese male H-AR�/y mice; they showed significantlyincreased hepatic steatosis on HFD feeding. Aging maleH-AR�/y mice spontaneously developed hepatic steatosiseven when consuming normal chow. Our study demon-strates that under short-term HFD feeding conditions,hepatic steatosis was substantially increased in maleH-AR�/y mice and was associated with increasing rates oflipogenesis, a contributor to TG accumulation. In addi-tion to affecting de novo lipogenesis, hepatic knockout ofthe AR also affected �-oxidation. Lipogenesis and �-oxi-dation are inversely related because malonyl-CoA pro-duced by ACC, the first committed enzyme in fatty acidsynthesis, appears to regulate fatty acid oxidation by allo-steric inhibition of CPT-1, the rate-limiting enzyme of�-oxidation.27 In rodents with NAFLD, suppression ofACC with antisense oligonucleotide significantly reduceshepatic malonyl-CoA level, lowers hepatic lipids, and im-proves insulin sensitivity.28 Consistent with previousfindings, ACC content was higher in livers of obese maleH-AR�/y mice, which likely led to reduced activity ofCPT-1. Another transcriptional factor, PPAR�, governsthe expression of major enzymes of the �-oxidation path-way, as well as CPT-1 expression.29 PPAR� expressionwas reduced in the livers of obese male H-AR�/y mice.However, the lower expression of PPAR� in human liverthan in rodent liver,30 as well the dominant-negativesplice variant of PPAR� in human liver,31 suggests a more
modest role of PPAR� in humans. Therefore, the coordi-nated modulation of gene expression in lipogenesis andoxidation was influenced in livers derived from obesemales lacking ARs.
Interestingly, we found that the sex of the animals ap-pears to influence the effectiveness of AR deletion in in-creasing body weight and hepatic fat accumulation. Notonly did male H-AR�/y mice weigh more and have morehepatic steatosis with short-term HFD feeding, but a sim-ilar effect also was observed in male H-AR�/y mice fedstandard chow at advanced age. Female H-AR�/� micefed an HFD for 8 weeks, however, did not gain moreweight than standard chow–fed female AR�/� mice, sug-gesting that in females, deletion of hepatic ARs does notinfluence obesity induced by HFD and that female miceare resistant to diet-induced obesity. In comparison, onlymale H-AR�/y mice fed an HFD for 8 weeks, rather thanstandard chow for 8 weeks, accumulated more bodyweight, and had increased hepatic steatosis and insulinresistance compared with their similarly fed male AR�/y
counterparts. If these sexually dimorphic differences inthe responses to changes in hepatic androgen–AR signal-ing were to hold true in humans, different outcomes inmales and females with hyperandrogenic or hypoandro-genic conditions might be expected. Indeed, hyperan-drogenic conditions, such as polycystic ovariansyndrome, have been strongly associated with reducedglucose tolerance and insulin sensitivity in women,32,33
whereas hypoandrogenism has been linked with insulinresistance16,34 and adiposity16,35 in men.
PTP1B is a well-characterized protein phosphatase andhas been shown to function as a negative regulator ofinsulin signaling and the leptin-signaling pathways and isa mediator of resistance to both hormones.36,37 Leptinimproves insulin sensitivity by direct action on hepato-cytes.38 Markedly elevated hepatic PTP1B expression im-pairs the ability of leptin to reduce serum glucose levels byinhibition of hepatic PEPCK activity.39 Hepatic PTP1Band serum leptin levels were increased in obese maleH-AR�/y mice, whereas hepatic leptin signaling was im-paired. Leptin resistance and consequently up-regulatedleptin secretion are believed to play a pivotal role in obe-sity. Testosterone supplementation in hypogonadal menreduces leptin secretion by fat cells through an AR-medi-ated pathway,40 breaking the circuit of leptin resistanceand obesity.41 The liver has long been implicated in theregulation of food intake and body weight, but the signal-ing molecules and pathways involved have not been fullyestablished. One hypothesis is that an increase in glucoseutilization can produce a satiety signal.42 Hepatic sensorsdetecting changes in energy expenditure trigger a signal inthe vagal afferents acting centrally to inhibit food intake.42
HEPATOLOGY, Vol. 47, No. 6, 2008 LIN, YU, ET AL. 1933

The lipolytic effect of leptin in the liver, depleting TG con-tent and increasing �-oxidation,43,44 might provide an alter-nate satiety signal that could also be transmitted throughvagal afferents.42 A recent study demonstrated induction ofleptin receptor expression in the liver by leptin and by fooddeprivation, in association with a large increase in a shortform of the leptin receptor that might bind and reduce thebioavailability of leptin to satiety centers.43
In H-AR�/y mice with loss of AR specifically in theliver, feeding an HFD also induced insulin resistance aswas observed in T-AR�/y mice. Reduced PPAR� expres-sion and consequent reduced lipid oxidation with ectopictriglyceride accumulation in the liver were also observedin H-AR�/y mice fed an HFD. Reduced PPAR� expres-sion in AR�/y mice and H-AR�/y mice because of loss ofthe T-AR and increased PPAR� expression in AR�/y
hepatocytes with DHT treatment indicate AR directlyregulates PPAR� expression. Reduced PI3k activity onlyin liver not in skeletal muscle in H-AR�/y mice furtherproved that increased hepatic PTP1B levels reduce phos-phorylation on tyrosine residues and consequently re-duces PI3k activity specifically in the liver. The H-AR�/y
mice model also suggests a plausible novel model of he-patic steatosis that might be a key element in hepaticinsulin resistance.
In summary, our results have demonstrated that HFDfeeding leading to hepatic steatosis and insulin resistanceis associated with AR deficiency in obese male H-AR�/y
mice. We found increased expression of PTP1B in theliver through an androgen-dependent mechanism thatcurtails the action of insulin and leptin. Our findingsimplicate the hepatic AR as a positive factor in preventingthe development of hepatic steatosis and insulin resis-tance. Strategies aimed at increasing AR activity specifi-cally in the liver through tissue-selective AR modulatorscould therefore improve both hepatic insulin and leptinsensitivity and improve both lipid and glucose homeosta-sis. Further understanding of the role of the AR in thedevelopment of insulin and leptin resistance in other tis-sues, such as brain and skeletal muscle, may provide ad-ditional insights into the pathophysiology of metabolicdiseases.
References1. Marchesini G, Brizi M, Morselli-Labate AM, Bianchi G, Bugianesi E,
McCullough AJ, et al. Association of nonalcoholic fatty liver disease withinsulin resistance. Am J Med 1999;107:450-455.
2. Angulo P. Nonalcoholic fatty liver disease. N Engl J Med 2002;346:1221-1231.
3. Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD,Cohen JC, et al. Prevalence of hepatic steatosis in an urban population
in the United States: impact of ethnicity. HEPATOLOGY 2004;40:1387-1395.
4. Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M, etal. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome.Diabetes 2001;50:1844-1850.
5. Kim HJ, Kim HJ, Lee KE, Kim DJ, Kim SK, Ahn CW, et al. Metabolicsignificance of nonalcoholic fatty liver disease in nonobese, nondiabeticadults. Arch Intern Med 2004;164:2169-2175.
6. Seppala-Lindroos A, Vehkavaara S, Hakkinen AM, Goto T, WesterbackaJ, Sovijarvi A, et al. Fat accumulation in the liver is associated with defectsin insulin suppression of glucose production and serum free fatty acidsindependent of obesity in normal men. J Clin Endocrinol Metab 2002;87:3023-3028.
7. Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, et al. Mechanismof hepatic insulin resistance in non-alcoholic fatty liver disease. J BiolChem 2004;279:32345-32353.
8. Wang J, Obici S, Morgan K, Barzilai N, Feng Z, Rossetti L. Overfeedingrapidly induces leptin and insulin resistance. Diabetes 2001;50:2786-2791.
9. Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabe-tes: estimates for the year 2000 and projections for 2030. Diabetes Care2004;27:1047-1053.
10. Gale EA, Gillespie KM. Diabetes and gender. Diabetologia 2001;44:3-15.11. Chang CS, Kokontis J, Liao ST. Molecular cloning of human and rat
complementary DNA encoding androgen receptors. Science 1988;240:324-326.
12. Heinlein CA, Chang C. Androgen receptor (AR) coregulators: an over-view. Endocr Rev 2002;23:175-200.
13. Lin HY, Xu QQ, Yeh SY, Wang RS, Sparks JD, Chang CS. Insulin andleptin resistance with hyperleptinemia in mice lacking androgen receptor.Diabetes 2005;54:1717-1725.
14. Chen X, Li X, Huang HY, Li X, Lin JF. [Effects of testosterone on insulinreceptor substrate-1 and glucose transporter 4 expression in cells sensitiveto insulin]. Zhonghua Yi Xue Za Zhi 2006;86:1474-1477.
15. Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, LeharJ, et al. PGC-1alpha-responsive genes involved in oxidative phosphoryla-tion are coordinately downregulated in human diabetes. Nat Genet 2003;34:267-273.
16. Pitteloud N, Mootha VK, Dwyer AA, Hardin M, Lee H, Eriksson KF, etal. Relationship between testosterone levels, insulin sensitivity, and mito-chondrial function in men. Diabetes Care 2005;28:1636-1642.
17. Zhang ZY. Protein tyrosine phosphatases: prospects for therapeutics. CurrOpin Chem Biol 2001;5:416-423.
18. Ukkola O, Santaniemi M. Protein tyrosine phosphatase 1B: a new targetfor the treatment of obesity and associated co-morbidities. J Intern Med2002;251:467-475.
19. Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, etal. Increased insulin sensitivity and obesity resistance in mice lacking theprotein tyrosine phosphatase-1B gene. Science 1999;283:1544-1548.
20. Tonks NK. PTP1B: from the sidelines to the front lines! FEBS Lett 2003;546:140-148.
21. Wu C, Zhang L, Bourne PA, Reeder JE, di Sant’Agnese PA, Yao JL, et al.Protein tyrosine phosphatase PTP1B is involved in neuroendocrine differ-entiation of prostate cancer. Prostate 2006;66:1125-1135.
22. Yeh S, Tsai MY, Xu Q, Mu XM, Lardy H, Huang KE, et al. Generationand characterization of androgen receptor knockout (ARKO) mice: an invivo model for the study of androgen functions in selective tissues. ProcNatl Acad Sci U S A 2002; 99:13498-13503.
23. Chirieac DV, Davidson NO, Sparks CE, Sparks JD. PI3-kinase activ-ity modulates apo B available for hepatic VLDL production in apobec-1-/- mice. Am J Physiol Gastrointest Liver Physiol 2006;291:G382-G388.
24. Shimomura I, Bashmakov Y, Horton JD. Increased levels of nuclearSREBP-1c associated with fatty livers in two mouse models of diabetesmellitus. J Biol Chem 1999;274:30028-30032.
1934 LIN, YU, ET AL. HEPATOLOGY, June 2008

25. Yahagi N, Shimano H, Hasty AH, Matsuzaka T, Ide T, Yoshikawa T, et al.Absence of sterol regulatory element-binding protein-1 (SREBP-1) ame-liorates fatty livers but not obesity or insulin resistance in Lep(ob)/Lep(ob)mice. J Biol Chem 2002;277:19353-19357.
26. Shimomura I, Hammer RE, Richardson JA, Ikemoto S, Bashmakov Y,Goldstein JL, et al. Insulin resistance and diabetes mellitus in transgenicmice expressing nuclear SREBP-1c in adipose tissue: model for congenitalgeneralized lipodystrophy. Genes Dev 1998;12:3182-3194.
27. McGarry JD, Foster DW. Regulation of hepatic fatty acid oxidation andketone body production. Annu Rev Biochem 1980;49:395-420.
28. Savage DB, Choi CS, Samuel VT, Liu ZX, Zhang D, Wang A, et al.Reversal of diet-induced hepatic steatosis and hepatic insulin resistance byantisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2.J Clin Invest 2006;116:817-824.
29. Brandt JM, Djouadi F, Kelly DP. Fatty acids activate transcription of themuscle carnitine palmitoyltransferase I gene in cardiac myocytes via theperoxisome proliferator-activated receptor alpha. J Biol Chem 1998;273:23786-23792.
30. Palmer CN, Hsu MH, Griffin KJ, Raucy JL, Johnson EF. Peroxisomeproliferator activated receptor-alpha expression in human liver. Mol Phar-macol 1998;53:14-22.
31. Gervois P, Torra IP, Chinetti G, Grotzinger T, Dubois G, Fruchart JC, etal. A truncated human peroxisome proliferator-activated receptor alphasplice variant with dominant negative activity. Mol Endocrinol 1999;13:1535-1549.
32. Kalish GM, Barrett-Connor E, Laughlin GA, Gulanski BI. Association ofendogenous sex hormones and insulin resistance among postmenopausalwomen: results from the Postmenopausal Estrogen/Progestin InterventionTrial. J Clin Endocrinol Metab 2003;88:1646-1652.
33. Gambineri A, Pelusi C, Manicardi E, Vicennati V, Cacciari M, Morselli-Labate AM, et al. Glucose intolerance in a large cohort of mediterraneanwomen with polycystic ovary syndrome: phenotype and associated factors.Diabetes 2004;53:2353-2358.
34. Muller M, Grobbee DE, den T, I, Lamberts SW, van der Schouw YT.Endogenous sex hormones and metabolic syndrome in aging men. J ClinEndocrinol Metab 2005;90:2618-2623.
35. Oh JY, Barrett-Connor E, Wedick NM, Wingard DL. Endogenous sexhormones and the development of type 2 diabetes in older men and wom-en: the Rancho Bernardo study. Diabetes Care 2002;25:55-60.
36. Johnson TO, Ermolieff J, Jirousek MR. Protein tyrosine phosphatase 1Binhibitors for diabetes. Nat Rev Drug Discov 2002;1:696-709.
37. Zhang ZY, Lee SY. PTP1B inhibitors as potential therapeutics in thetreatment of type 2 diabetes and obesity. Expert Opin Investig Drugs2003;12:223-233.
38. Lam NT, Lewis JT, Cheung AT, Luk CT, Tse J, Wang J, et al. Leptinincreases hepatic insulin sensitivity and protein tyrosine phosphatase 1Bexpression. Mol Endocrinol 2004;18:1333-1345.
39. Lam NT, Covey SD, Lewis JT, Oosman S, Webber T, Hsu EC, et al.Leptin resistance following over-expression of protein tyrosine phospha-tase 1B in liver. J Mol Endocrinol 2006;36:163-174.
40. Zitzmann M, Gromoll J, von Eckardstein A, Nieschlag E. The CAG repeatpolymorphism in the androgen receptor gene modulates body fat mass andserum concentrations of leptin and insulin in men. Diabetologia 2003;46:31-39.
41. Mayes JS, Watson GH. Direct effects of sex steroid hormones on adiposetissues and obesity. Obes Rev 2004;5:197-216.
42. Langhans W, Grossmann F, Geary N. Intrameal hepatic-portal infusion ofglucose reduces spontaneous meal size in rats. Physiol Behav 2001;73:499-507.
43. Cohen P, Yang G, Yu X, Soukas AA, Wolfish CS, Friedman JM, et al.Induction of leptin receptor expression in the liver by leptin and fooddeprivation. J Biol Chem 2005;280:10034-10039.
44. Lee Y, Wang MY, Kakuma T, Wang ZW, Babcock E, McCorkle K, et al.Liporegulation in diet-induced obesity. The antisteatotic role of hyperlep-tinemia. J Biol Chem 2001;276:5629-5635.
HEPATOLOGY, Vol. 47, No. 6, 2008 LIN, YU, ET AL. 1935
Copyright © 2022 FDOKUMEN


















![[Hepatic steatosis, visceral fat and metabolic alterations in apparently healthy overweight/obese individuals]](https://static.fdokumen.com/doc/165x107/6324f8237fd2bfd0cb03375f/hepatic-steatosis-visceral-fat-and-metabolic-alterations-in-apparently-healthy.jpg)

