
In Situ Evidence of Neoplastic Cell Phagocytosis by Macrophages in Papillary Thyroid Cancer 1
Upload
independentCategory
view
0download
0
Activation of Hepatic Stellate Cells After Phagocytosis ofLymphocytes: A Novel Pathway of Fibrogenesis
Nidal Muhanna, Sarit Doron1, Ori Wald1, Amjad Horani1, Ahmed Eid2, Orit Pappo3, Scott L.Friedman4, and Rifaat Safadi51Liver and Gastroenterology Units; Division of Medicine, Hadassah Hospital, Jerusalem, Israel2Division of Surgery, Hadassah Hospital, Jerusalem, Israel3Division of Pathology Department, Hadassah Hospital, Jerusalem, Israel4Division of Liver Diseases, New York, NY5Mount Sinai School of Medicine, New York, NY
AbstractIncreased CD8-T lymphocytes and reduced natural killer (NK) cells contribute to hepatic fibrosis.We have characterized pathways regulating the interactions of human hepatic stellate cells (HSCs)with specific lymphocyte0020subsets in vivo and in vitro. Fluorescence-activated cell sorting (FACS)was used to characterize human peripheral blood lymphocytes (PBLs) and intrahepatic lymphocytes(IHLs) obtained from healthy controls and from patients with either hepatitis B virus (HBV) orhepatitis C virus (HCV) with advanced fibrosis. Liver sections were analyzed byimmunohistochemistry and confocal microscopy. To investigate in vitro interactions, PBLs fromhealthy controls or patients with HCV cirrhosis were co-cultured with an immortalized human HSCline (LX2 cells) or with primary HSCs. Significant alterations in lymphocyte distribution wereidentified in IHLs but not PBLs. The hepatic CD4/CD8 ratio and NK cells were significantly reducedin HBV/ HCV patients. Expression of alpha-smooth muscle actin and infiltration of CD4, CD8, andNK cells were readily apparent in liver sections from patients with cirrhosis but not in healthycontrols. Lymphocytes from each subset were in proximity to HSCs primarily within the periportalregions, and some were directly attached or engulfed. In culture, HSC activation was stimulated byHCV-derived CD8-subsets but attenuated by NK cells. Confocal microscopy identified lymphocytephagocytosis within HSCs that was completely prevented by blocking intracellular adhesionmolecule 1 (ICAM-1) and integrin molecules, or by irradiation of HSCs. LX2 knockdown of eitherCdc42 or Rac1 [members of the Rho-guanosine triphosphatase (GTPase) family] prevented bothphagocytosis and the activation of HSC by HCV-derived lymphocytes.
Conclusion—The CD4/CD8 ratio and NK cells are significantly decreased in livers with advancedhuman fibrosis. Moreover, disease-associated but not healthy lymphocytes are engulfed by culturedHSCs, which is mediated by the Rac1 and Cdc42 pathways. Ingestion of lymphocytes by HSCs inhepatic fibrosis is a novel and potentially important pathway regulating the impact of lymphocyteson the course of hepatic fibrosis. (HEPATOLOGY 2008;48:963–977.)
Hepatic fibrosis associated with inflammatory cell infiltration is a prominent feature ofpersistent infection by hepatitis B virus (HBV) and hepatitis C virus (HCV). The hepatic stellate
Copyright © 2008 by the American Association for the Study of Liver Diseases.Address reprint requests to: Rifaat Safadi, MD, Liver and Gastroenterology Units, Division of Medicine Hadassah University Hospitaland Hebrew University-Hadassah Medical School, POB 12000, IL-91120 Jerusalem, Israel. [email protected]; fax: 972-2-6420338.Potential conflict of interest: Nothing to report.
NIH Public AccessAuthor ManuscriptHepatology. Author manuscript; available in PMC 2010 June 3.
Published in final edited form as:Hepatology. 2008 September ; 48(3): 963–977. doi:10.1002/hep.22413.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

cell (HSC) has assumed a central role in this response after its activation by inflammatorycytokines and mediators.1–5 The cell-mediated immune response after viral hepatitis reflectsthe activity of CD4+ helper T and CD8+ cytotoxic T lymphocytes. CD4+ T cells are activatedthrough the interaction of major histocompatibility complex (MHC)-II with antigen-presentingcells, Kupffer cells, dendritic cells, and macrophages. CD8+ T cells are MHC-I restricted andare a major mechanism of cytotoxic clearance of infected cells.6 CD4+ T cells secrete cytokinessuch as tumor necrosis factor alpha, interferon gamma (IFN-γ), and interleukin 2. Thesecytokines are responsible for the activation of macrophages, Kupffer cells, and natural killer(NK) cells, leading to phagocytosis and nonspecific lysis of infected cells. Viral clearanceduring HCV infection is made possible through vigorous HCV-specific CD4+ and CD8+ Tcell responses.7 It is thought that CD4+ T cell activation and priming are required for CD8+T cells’ ability to achieve viral clearance through cytotoxic effects.8,9 These processes canalso lead to activation of HSCs in response to injury, which in the absence of viral clearanceis followed by matrix deposition, fibrosis, and eventually cirrhosis.10,11 HCV-specific CD4activity is correlated with histological fibrosis and portal tract inflammation.12–14
A role of HSCs in the inflammatory response to viral infection has been established based ontheir capacity to present antigen and modulate lymphocyte behavior.15,16 Althoughlymphocytes directly interact with HSCs by adhesion,14 the pathways mediating interaction oflymphocytes and HSCs are not well understood. Moreover, HSC are phagocytic based on theirinternalization of latex particles, bacteria16 as well as apoptotic bodies.17 In murine models,fibrosis is mainly mediated by direct activation of HSCs by CD8 lymphocytes,18 and adoptivelytransferred CD8 cells are fibrogenic in naïve mice. In addition, NK cells display anti-fibroticactivity by killing activated HSCs that have lost the self-recognition marker, MHC class I.19–21 In this study, we have investigated the morphological and functional interactions betweenhuman HSCs and lymphocyte subsets. Our results indicate that HSCs not only physicallyinteract with lymphocytes but may contribute to lymphocyte clearance by cellular ingestion.
Patients and MethodsStudy Population
Blood and liver samples of patients referred for liver biopsy because of chronic HBV and HCVwere obtained with informed consent after the approval of the Hadassah Hospital EthicsCommittee. All HCV patients were positive for serum HCV antibodies (Abbot) and HCV RNA(tested by HCV amplicor, Roche). Similarly, HBV patients were positive for serum hepatitisB surface antigen (Abbot) and HBV-DNA (HBV Monitor, Roche). Only patients withadvanced but compensated fibrosis were included in this cohort. Advanced fibrosis wasclinically established by the presence of splenomegaly, thrombocytopenia, and irregular liverechotexture, and confirmed by liver biopsy demonstrating Metavir F3 or F4,22 as assessed bya single pathologist. No patient had evidence of HBV/HCV, HBV/human immunodeficiencyvirus or HCV/human immunodeficiency virus co-infection. Control liver biopsy specimensfrom HBV/HCV-negative patients were obtained during hepatobiliary surgery for eitherresection of hemangioma or benign tumors, in which histologically normal liver tissuesurrounding the resected lesion was used.
Lymphocyte IsolationHuman peripheral blood lymphocytes (PBLs) were collected in heparinized tubes from patientsand healthy volunteers after the guidelines approved by the Hadassah Hospital EthicsCommittee. Mononuclear cells were isolated by centrifugation over Ficoll-Hypaque(Pharmacia) according to Boyum.23 After three washes in saline, cells were resuspended inmedium. For freezing, Roswell Park Memorial Institute 1640 medium supplemented with 50%heat-inactivated fetal bovine serum (FBS; Gibco) and 20% dimethyl sulfoxide was used, and
Muhanna et al. Page 2
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

cells were stored at −70°C. The culture medium for PBLs was Roswell Park Memorial Institute1640 with 10% FBS. Human NK, CD4, and CD8 cells were isolated from PBLs using amagnetic cell sorting kit (Miltenyi Biotec) according to manufacturer’s instructions. Forintrahepatic lymphocytes (IHL),24 liver tissue obtained from needle biopsies or surgery waswashed twice in 10 mL phosphate-buffered saline (PBS) to remove remaining PBLs. Tissuewas cut into 1-mm pieces and incubated in 2 mL Roswell Park Memorial Institute 1640 (Gibco,Great Britain) for 15 minutes with collagenase type IV (Worthington, Lakewood, NJ) anddeoxyribonuclease I (Worthington) at 37°C. After incubation, the tissue was run through a 1-mL pipette and then filtered through a cell strainer fluorescence-activated cell sorting (FACS)tube (Becton Dickinson, Mountain View, CA). Cells were washed and centrifuged at 92 g for5 minutes at 4°C. The pellet was resuspended in FACS buffer for flow cytometry analysis. Thecell yield was between 100,000 to 200,000 cells per biopsy.
In Vitro Analysis of Human Lymphocyte Interactions with HSCsThe human hepatic stellate cell line LX2 was used to examine interactions of lymphocytes withHSC in culture. This cell line has been extensively validated in a number of studies that establishits relevance to primary HSCs.25 For validation of results, primary human HSCs were used.
Primary Human HSC Isolation and CultureHSCs were isolated as previously described.26 Isolated HSCs were cultured in M199containing 10% FBS, changed 24 hours after plating and every 3 to 4 days thereafter. Whenthe cultures reached confluence, they were trypsinized (0.05% trypsin/0.53 mMethylenediaminetetra-acetic acid) and passaged at a ratio of 1:3. Subsequent passages wereperformed every 7 to 10 days.
Activation of HSCs by PBLsHealthy or cirrhotic HCV-derived PBLs (106 cells) were isolated from eight donors in eachgroup. Isolated PBLs were then co-cultured individually with LX2 cells or primary isolatedHSCs in 18-mm dishes 1% FBS (Atlantic Biologicals) medium. Triplicate cultures wereperformed from each PBL donor. HSCs (105 cells) were previously cultured 3 days (up tosubcomplete confluence) before PBL constitution. Either 106 mixed PBL cells or separatesubsets (CD4, CD8, or NK cell) were used in the co-culture. After 24 hours of co-cultureactivation of HSC medium with lymphocytes was removed, and cells were washed, harvestedby cell scraping, and analyzed for alpha0smooth muscle actin (α-SMA) Western blotting andmessenger RNA (mRNA) expression.
PhagocytosisConfocal imaging was used to investigate phagocytosis of lymphocytes by HSCs using mixedPBLs either derived from HCV-infected patients with cirrhosis or from healthy volunteers.HSC/PBL co-cultures were incubated for 0, 6, 12, 24, and 72 hours. Circular glass slides(Marienfeld, Laboratory Glassware, Germany) placed in the bottom of 12-well flasks (NuncBrand Products, Denmark) were used in each co-culture. Expressions of CD4, CD8, NK, andα-SMA markers were analyzed at each time point by immunocytochemistry. Thirty-six imagesfrom different fields were acquired from each co-culture dish.
Phagocytosis was further assessed using confocal microscopy of 1-µm sections. In addition,PBLs from three different donors were preincubated with 3,3′-dioctadecy-loxacarbocyanineperchlorate (DiOC; Sigma). DiOC, a cyanine dye, was dissolved in dimethylsulfoxide (2 mg/mL; Sigma) as described previously to stain intracellular lipids green.27 After overnightincubation with agitation, DiOC-stained cells were washed several times and then culturedwith HSCs.
Muhanna et al. Page 3
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

To assess the existence of ligand/receptor-mediated phagocytosis, confocal assessment wasrepeated in different modifications of HSC/PBL co-cultures. These included preincubation ofHSCs with blocking antibodies at a final concentration of 100 µg/mL for 30 minutes and thencultured with HCV-derived PBLs. HSCs were preincubated with either mouse anti-humanclass I (HLA-ABC Antigen, DakoCytomation), class II (HLA-DP, DQ, DR Antigen,DakoCytomation), intercellular adhesion molecule (ICAM-1) (CD54, ICAM-1,DakoCytomation), the type I membrane glycoprotein adhesion molecule “IntegrinalphaV” (polyclonal Intigren, CHEMICON International), MIC-A or MIC-B (Bio-Legend,6D4), or control antibodies. For T-cell receptor blocking, mouse anti-human T-cell receptorantibodies were incubated with lymphocytes before co-culture under similar conditions. Inseparate experiments, either HSCs or PBL cells were irradiated to obtain apoptosis,16,28 beforeco-culture to distinguish whether HSCs are responsible for phagocytosis of lymphocytes. HSCswere washed twice with PBS, trypsinized, and irradiated with 6000 rads, a dose shown to inhibitproliferation without affecting cell viability or membrane protein expression.15,16Experiments with irradiated nonproliferating PBLs were performed using 3000 rads.15,16
Fluorescent-Activated Cell Sorting Analysis (FACS)Briefly, isolated lymphocytes were adjusted to 106/mL in staining buffer (in saline containing1% bovine albumin; Biological Industries, Israel). Fifty microliters cell suspension wasincubated with antibody on ice for 30 minutes, washed with staining buffer, and fixed with 2%paraformaldehyde. Fc receptors were blocked by incubation with 1% human plasma for 15minutes on ice. Blocked lymphocytes were then mixed with either fluorescein isothiocyanate(FITC), phycoerythrin (PE), allophycocyanin, or peridinin-chlorophyll-α-protein conjugatedanti-CD4, anti-CD8, anti-CD16, and anti-CD45 antibodies, respectively (IQ Products,Groningen, Netherlands; antibodies were diluted 1:40) or isotype controls for 20 minutes onice, and were washed with FACS buffer. CD45 was used as a global marker for leukocytes.Intracellular staining of lymphocytes with anti-IFN-γ and transforming growth factor beta(TGF-β) antibodies (FITC and PE conjugated, respectively) was performed according to themanufacturer’s protocol (BD Bio-sciences). For apoptosis measurements of lymphocytesinside the HSCs, propidium iodide (PI) staining of fragmented DNA and phosphatidylserinestaining by annexin V conjugated to FITC (R&D Systems, Minneapolis, MN) were usedaccording to the manufacturer’s instruction. Lymphocyte apoptosis was therefore defined asCD45+ , annexin-V(+) but PI(−). FACS data were acquired using a FACS Caliber FlowCytometer (Becton Dickinson); the data were analyzed using the CellQuest 3.3 software. Forall analyses, lymphocyte gating was performed using the forward scatter versus side scatterplot.
ImmunofluorescenceLiver biopsies or cell culture slides were incubated overnight at room temperature with isotonicPBS, 10% sucrose, and 4% formaldehyde solution. Then they were frozen at −80°C for storage,and 7-µm-thick frozen sections were prepared using Cryostat (Leica CM 3000). Cells werepermeabilized using the 0.2% Triton (Sigma). To block nonspecific background staining, 1%bovine serum albumin was used for 20 minutes. After PBS washing, slides were incubatedwith primary antibodies for 45 minutes at room temperature in the dark. Primary antibodiesused in liver biopsies were human FITC-anti-CD4 (at a dilution of 1:100), anti-CD8 (at adilution of 1:70), and anti-CD16 (at a dilution of 1:100) markers (IQ Products, Groningen,Netherlands). Primary antibodies used in cell culture slides were human FITC-anti-CD4, PE-anti-CD8, PE-anti-CD16, FITC, or PE-anti-CD45 (at a dilution of 1:50), FITC-anti-Rac1, andFITC-anti-Cdc42 (at a dilution of 1:50, Santa Cruz Biotechnology, INC.) markers. Foridentification of HSCs: α–SMA (DAKO, cat# M0851) primary antibody (at a dilution of 1:150)conjugated to Cy-5 (at a dilution of 1:40) as the secondary antibody (Jackson Immunoresearch)were used. The Cy-5 secondary antibody was then added, preceded and followed by three PBS
Muhanna et al. Page 4
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

washes. To preserve staining, sections were stacked and covered with Fluoromount-G(Southern Biotechnology Associates). Sections were then stored at 4°C while awaitinganalysis.29,30
Confocal Microscopy and Image CaptureA Zeiss LSM 410 Confocal laser scanning system (Zeiss, Germany) attached to a ZeissAxiovert 135 M microscope was used. The fluorescence images were collected by employingplan-apochromat Zeiss, 40 × 1.4 lens. The system was equipped with an argon laser (488 nmexcitation line) for green fluorescence and two helium-neon lasers (543 nm and 633 nm lines)for red fluorescence. Triple-labeled specimens were excited with three lasers and monitoredsimultaneously using triple detectors and filter-block combinations. The excitation powers andemission filters were tuned to keep the overlap from each channel at a minimum. In eachexperiment, laser intensity, background level, contrast, aperture, and electronic zoom size werecollected at the same level. Fifty images were collected from each specimen and converted totiff format and processed using Zeiss LSM Image Browser software. Image processing wasperformed using Adobe Photoshop software (Adobe Systems UK, Uxbridge, and Middlesex,UK) and ImagePro Plus programs (Media Cybernatics, USA).31,32
α-SMA ImmunoblotImmunoblot analysis of α-SMA in cultured HSC protein extracts was performed as previouslydescribed.18,19,21
Real-Time Polymerase Chain Reaction AnalysisWashed LX2 cells were harvested by a scraper for RNA extraction and complementary DNAtranscription as previously described.18,19 The complementary DNA product was used for real-time polymerase chain reaction as previously described.18,19 β-Actin served as internal controlsand H2O served as a negative control. Primers used for the β-actin were as follows:
Forward: GAT GAG ATT GGC ATG GCT TT
Reverse: AGA GAA GTG GGG TGG CTT TT
For SMA:
Forward: TCC TCC CTG GAG AAG AGC TAC
Reverse: TAT AGG TGG TTT CGT GGA TGC
HSC Transfection by Small Interfering RNASmall interfering RNA (siRNA) were diluted in 100:l in medium without serum to aconcentration minimum of 5 nM and maximum of 25 nM. Three microliters HiPerFecttransfection reagent (Qiagene) was added to the diluted siRNA and mixed well. The mixturewas incubated at room temperature for 10 minutes to allow the formation of transfectioncomplexes. The complex was then added to 105 cells seeded the day before on 24-well plates.The efficiency of transfection was performed using positive and negative siRNA silencingcontrols provided in Qiagen siRNA human/mouse starter kit. siRNA transfection was validatedby immunofluorescence and the gene silencing confirmed by real-time polymerase chainreaction.
siRNA Silencing of Rac1/Cdc42LX2 cells or primary HSCs were transfected either with commercial Rac1 or Cdc42 siRNAsusing RNA interference human/mouse starter kit (HiPerFect Transfection Reagent, QIAGEN)according to manufacturer instructions: complementary DNA sequences of human Cdc42 and
Muhanna et al. Page 5
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Rac1: 5′-GUG UCG GCA UCAUAC UAAAdTdT-3′ (Cdc42#1) and 5′-CAG CAA UGC AGACAA UUA AdTdT-3′ (Cdc42#4); 5′-GGU UGG UAU UAU CAG GAA AdTdT-3′ (Rac1#1)and 5′-GAC AUA ACA UUG UAC UGU AdTdT-3′ (Rac1#3). siRNA transfected HSC werethen co-cultured with cirrhotic HCV-derived lymphocytes. For controls (available in the samekit), HSCs were transfected with control-siRNA before co-culture. Nonsilencing controlsiRNA (Alexa Fluor 488 Labeled, QIAGEN) were used for negative control. As a positivecontrol, the specific positive siRNA (MAPK1) was transfected into HSCs but without co-culture with lymphocytes. Six triplicates of co-culture from six different patients were used ineach of the first two groups. Activation of HSCs was assessed after 24 hours of co-culture.Medium containing free lymphocytes was removed, and HSCs were harvested by scraping forα–SMA Western blotting assessment.
Statistical MethodsResults are illustrated as mean ± standard deviation. For statistically significant differences;paired and unpaired Student t test and analysis of variance were used.
ResultsAltered Distribution of Lymphocyte Subsets in Chronic HCV and HBV
Accumulating data implicate several different lymphocyte subsets in hepatic inflammation,but their relative abundance and importance are uncertain.33–36 To clarify this issue, thecomposition of PBL and IHL from six control cases was compared with 25 HCV and sevenHBV patients with advanced fibrosis (Table 1). Significant differences were mainly confinedto IHL and not PBL (Fig. 1). Intrahepatic CD4 cells were 34% ± 7.5% of total lymphocytes inhealthy controls but only 22.6% ± 6.6% in HCV patients (P = 0.004) and 22% ± 5.3% amongHBV patients (P = 0.04). Differences in CD8 cells between healthy controls and patients witheither HBV or HCV were not significant (Fig. 1, upper panel). However, the CD4/CD8 ratiowas significantly decreased from 0.88% ± 0.17% in healthy donors to 0.53% ± 0.12% (P =0.007) in HCV and 0.44 ± 0.12 (P = 0.02) in HBV cases. As previously reported,18,19,21
significant NK alterations in the current study were confined to IHLs (Fig. 1). Specifically,intrahepatic NK cells were significantly decreased (P = 0.04) from 24.9% ± 2.8% of healthylymphocytes to 17.7% ± 5% and 19.3% ± 4.1% in HCV-infected and HBV-infectedindividuals, respectively.
HSCs and Lymphocytes Interact Directly In VivoLymphocytes were assessed in situ in liver sections from HCV patients with cirrhosis usingconfocal microscopy. Scattered, weakly positive α-SMA–positive cells were apparent incontrol livers (Fig. 2A) but became prominent in fibrotic livers (Fig. 2B–D). Immunostainingfor lymphocyte subsets was negative in normal livers, suggesting that lymphocytes do notadhere to liver parenchyma in the absence of injury (Fig. 2A). In contrast, fibrotic livers werepositively stained for CD4, CD8, and NK (CD16) cells (Fig. 2B–D, respectively).Lymphocytes were located only in direct proximity to the α-SMA–positive cells along fibroticsepta and not elsewhere. (The classic green color of FITC fluorescence was remapped to bluefor easier viewing.) Therefore, FITC-conjugated subsets with the classic blue color becamepink when merged with the red Cy-5 α–SMA (Fig. 2B–D). Similar results were also obtainedin HBV cirrhotic livers (data not shown).
Immune Cell Interactions Contribute to FibrosisAlterations of human lymphocyte subsets associated with hepatic fibrosis were similar to thoseseen in rodent fibrosis models.18–21 To evaluate the functional impact of interactions oflymphocytes with HSCs, a co-culture system was employed, combining mixed PBLs, isolated
Muhanna et al. Page 6
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

CD4, CD8, or NK cells with LX2 cells for 48 hours, comparing the effects of cells from normaldonors with those from donors with HCV.
Figure 3A shows α-SMA (upper lane) and β-Actin (second lane) expression as assessed bywestern blotting from cultured protein extracts of these co-cultures. Both CD8 and to a lesserextent CD4 cells (middle panel), as well as mixed HCV lymphocytes (upper panel) from HCV-infected patients could activate HSCs, as manifested by increased α-SMA expression in theLX2 cells. In contrast, NK cells had minimal effects (middle panel). Moreover, neither mixednor isolated subsets from healthy controls had any effect on LX2 HSCs (Fig. 3A, lower panel).Expression of α–SMA mRNA (Fig. 3B) corresponded to western blot results. Specifically, α-SMA mRNA expression was increased twofold ± 0.8-fold in mixed HCV-derived lymphocytesco-cultured with LX2 cells (Fig. 3B, upper panel), compared with LX2 cells cultured alone.The expression in mixed cultures with CD4 cells was 1.6 ± 0.9, CD8 was 3 ± 0.4, and in NKcells 0.9 ± 0.7. Significant differences were seen when CD8-LX2 co-cultures were comparedwith co-cultures containing mixed (P = 0.04), CD4 (P = 0.01), and NK (P = 0.004) subsets.Primary isolated HSC revealed the same pattern seen with LX2 cells (Fig. 3B, lower panel).Similar to results in mouse fibrosis models,18–20 NK cells attenuated HSC activation comparedwith mixed lymphocytes (P = 0.03), reinforcing the relevance of these earlier mouse studiesto human disease. Intracellular cytokine analysis of cultured lymphocytes was studied by FACSusing healthy or HCV-derived PBL (Fig. 3C). The findings further support the effect of eachsubset on HSCs: compared with healthy PBLs, HCV-derived NK cells showed a significantdecrease of TGF-β secretion from 2.3% ± 1.2% to 1.5% ± 0.8% of NK cells (P = 0.05) and asignificant increase (P = 0.03) of IFN-γ secretion from 0.2% ± 0.1% to 0.5% ± 0.4% of NKcells (Fig. 3C, left upper and lower panels). Decreased TGF-β and increased IFN-γ secretionby NK cells are consistent with an antifibrotic effect. HCV-derived CD8 cells, however,showed a significant increase of TGF-β secretion from 9.2% ± 3.6% to 46.1% ± 19.7% (P <0.0001) and a significant decrease (P = 0.009) of IFN-γ secretion from 5% ± 5.3% to 0.6% ±0.5% of CD8 cells (Fig. 3C, middle upper and lower panels). Increased TGF-β and decreasedIFN-γ secretion from CD8 cells are compatible with their profibrotic effect. TGF-β was alsoincreased from 122 ± 15.2 (P = 0.04) in the case of CD4 subsets; however, IFN-γ secretionwas similar in both healthy and HCV-derived CD4 lymphocytes (Fig. 3C, left upper and leftlower panels), indicating a milder profibrogenic effect as compared with CD8 cells.
Very similar results to Fig. 3A–C were also obtained using primary HSCs instead of LX2 cells(data not shown).
Direct Contact/Attachment Is Followed by Phagocytosis of Lymphocytes by HSCsWe previously demonstrated direct contact of HCV-derived NK cells with cultured humanHSCs.19 To extend these findings, LX2 cells were co-cultured with mixed PBLs from HCV-in-fected donors with cirrhosis and healthy donors for 0, 6, 12, 24, and 72 hours (as detailed).LX2 cells were stained red by Cy-5 antibody conjugated to anti-α–SMA (Fig. 4, red arrows).Figure 4 demonstrates representative images of interactions (for 6 hours’ co culture) of thethree different lymphocyte subsets (white arrows) in proximity to HSCs (red arrows). This isincluding FITC-conjugated CD4 (Fig. 4A, blue), PE-CD8 (Fig. 4B, green), as well as PE-CD16(NK) HCV-derived cells (Fig. 4C, green). These interactions were not present when LX2 cellswere cultured with healthy lymphocytes (Fig. 4D). Thus, direct interaction between PBL andLX2 cells could contribute to the functional effects of lymphocytes from HCV patients onHSCs. Similar to findings in LX2 cells, HCV-derived lymphocyte subsets (Fig. 5A–C) but nothealthy PBLs (Fig. 5D) were also engulfed within primary HSCs.
Based on the direct proximity of lymphocytes to HSC at early time points (Fig. 2, Fig. 4, andFig. 5), we monitored the HSC/PBL co-culture longer than 6 hours. Phagocytosis of CD45cells (as a pan leukocyte marker, conjugated to FITC in the case of co-culture with LX2 cells
Muhanna et al. Page 7
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

and to PE in the case of primary isolated HSC) by HSCs was found at 12, 24, and 72 hours ofco-culture. HSCs (Fig. 6A–D; LX2 in Fig. 6A–B; as well as primary HSCs in Fig. 6C–D) werestained red by the Cy-5 conjugated to α–SMA (red arrows), and lymphocytes were stained blue(white arrows), phagocytosis was associated with α-SMA–positive encapsulation aroundingested lymphocytes (Fig. 6B and D, green arrows). Furthermore, serial 1-µm section byconfocal images demonstrated HSCs within the same image plane because both appear anddisappear simultaneously, confirming the presence of phagocytosed lymphocytes (whitearrows) within HSCs (red arrows; selective sections are presented in Fig. 7A–D with LX2 cellsand Fig. 7G–J with primary isolated HSCs). To further demonstrate phagocytosis and notphysical overlay of the two cell types, HCV-derived lymphocytes were preincubated withDiOC to stain intracellular lipid content, washed, and then cultured with HSCs. Fig. 7E–F withLX2 cells and Fig. 7K–M with primary isolated HSCs show lymphocytes inside HSCs withdiffusion of lymphocyte-derived DiOC within the cytoplasm of HSCs.
The apoptosis of lymphocytes was then evaluated using HCV-derived PBLs in culture for 48hours either alone or with HSC (Fig. 8). In the case of co-culture with LX2 cells, apoptosiswas evaluated only in the engulfed PBLs after washing the floating cells. Apoptotic PBLs (Fig.8A), defined as CD45+ , Annexin+ , and PI(−), were significantly increased from 19% ± 4.5%in the case of PBL culture alone to 25.7% ± 1.7% of total CD45+ cells in the case of engulfedlymphocytes (P = 0.002). Interestingly, the dead lymphocytes (Fig. 8b), defined as CD45+ ,Annexin+, and PI(+), were also increased even more significantly from 7.7% ± 0.8% to 26.2%± 2.2%, respectively (P < 0.0001). The data suggest a rapid killing of lymphocytes inside HSCs.Apoptosis of lymphocytes co-cultured with primary HSCs showed the same pattern (Fig. 8C–D).
We next explored whether phagocytosis of lymphocytes by HSCs was regulated by specificligand–receptor interactions, focusing on several families of molecules. Phagocytic receptorsare very diverse and include at least one member of each prototypical adhesion receptor family(integrin, cadherin, and so forth).37 Figure 9 demonstrates selected examples from eachcondition in which LX2 cells were stained red by the Cy-5 conjugated to α-SMA (red arrows),and lymphocytes were stained with PE-CD45 cells (white arrows). Compared with thephagocytosis of lymphocytes seen in untreated coculture (Fig. 9A), phagocytosis was notaffected when HSCs were preblocked by specific antibodies to either MHC class I and classII molecules (Fig. 9B–C) or natural-killer group 2, member D (NKG2D) receptors for MHCclass I chain-related gene A (MIC-A) and MHC class I chain-related gene B (MIC-B) ligands(data not shown). Similarly, phagocytosis was not affected when lymphocytes were preblockedby specific antibodies to T-cell receptor (Fig. 9F). In contrast, phagocytosis was blocked bypreincubation with antibodies to either ICAM-1 or integrin alphaV before co culture (Fig. 9D–E). To confirm that LX2 cells are responsible for phagocytosis of lymphocytes rather thanpenetration of LX2 cells by lymphocytes, either lymphocytes (Fig. 9G) or LX2 cells (Fig. 9H)were irradiated to attenuate apoptosis in only one of the two cell types before co-culture. Onlywhen HSCs (but not lymphocytes) were irradiated was phagocytosis completely blocked. Thisfinding is consistent with results in which radiation has attenuated allogeneic T-lymphocyteproliferation when co-cultured with HSCs.16 Primary HSC followed similar responses as LX2cells (Fig. 10).
The blocking of ICAM-1 or integrin alphaV before co-culture with HCV-derived PBLs (Fig.9D–E) was accompanied by a decrease of HSC activation (Fig. 11). Specifically, expressionof α–SMA assessed by western blotting was decreased after ICAM-1 or integrin alphaVblocking in the presence of equal β-actin (Fig. 11, lower panel). Expression of α–SMA mRNA(Fig. 11, upper panel) corresponded to the western blot results. Specifically, α-SMA mRNAexpression was increased 2.8 ± 0.05-fold in mixed HCV-derived lymphocytes co-cultured withLX2 cells, compared with LX2 cells cultured alone. The expression significantly decreased to
Muhanna et al. Page 8
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

1.9 ± 0.09 (P = 0.005) after ICAM-1 blocking, and to 1.6 ± 0.02 (P = 0.005) in integrin alphaVblocking. Significant differences were also seen when ICAM-1 blocking was compared withintegrin alphaV blocking (P = 0.02). Primary HSCs responded similarly to LX2 cells afterICAM-1 or integrin alphaV blocking (data not shown).
Rac1 & Cdc42 Are Involved in the Phagocytosis of Lymphocytes by HSCAfter the recognition that intercellular adhesion molecules contribute to the phagocyticresponse of HSCs, we sought to identify the underlying intracellular signaling pathways.Signaling cascades regulating cellular adhesion parallel those activated during phagocytosis.37 In particular, Rho-family guanosine triphosphatases (GTPases) are activated downstreamof adhesion receptors and control the cytoskeletal changes that underlie adhesive events.38
Accordingly, we examined the potential roles of Cdc42 and Rac1 (as part of the Rho-familyGTPases) by characterizing these two molecules with specific antibodies in the co-culturesystem (Fig. 12). Overlay of both molecules together with that of Cy-5 red α–SMA stainingthe HSC was apparent (Fig. 12), suggesting that Cdc42 and Rac1 recruitment contribute toHSC–lymphocyte adhesion and phagocytosis.
To uncover the functional importance of Rac 1 and Cdc42 to the adhesion and phagocytosisof lymphocytes by HSCs, we employed selective knockdown using specific siRNAs,38
confirmed by using confocal microscopy of fluorescent control siRNA (data not shown). Figure13 documents Cdc42 down-regulation by specific siRNAs as assessed by western blotting(lower panel), which was associated with attenuated phagocytosis as demonstrated by confocalmicroscopy (data not shown because they are similar to Fig. 9D–E, H) associated with reducedHSC activation as assessed by α-SMA expression (upper panel). Results of the Rac1 siRNAsilencing were identical to those using Cdc42 silencing (data not shown). Cdc42 and Rac 1were also involved in the phagocytosis of PBLs by primary HSC (data not shown).
DiscussionDuring viral infections, cytotoxic CD8+ T cells are activated by specific peptides, and theirresponse is enhanced by specific helper CD4+ T cells. Attack of activated T cells on HCV-infected hepatocytes may explain, at least in part, histological damage.12,39–44 Our study hasevaluated T-cell subset distribution in PBLs and intrahepatic T cells from patients with HBV-related and HCV-related liver fibrosis. Significant differences were confined to IHLs and notPBLs, suggesting that lymphocytes may be sequestered at the site of infection and thatassessment of PBL may not be representative of local interactions in liver.45,46 Indeed,alterations of the intrahepatic cytokine network may be the basis for T-cell alterations in liverinjury.47
As others have previously reported in inflammatory liver injury,14,48, we have found a decreasein the percentage of CD4 T cells in the fibrotic livers compared with the peripheral blood,leading to a significantly reduced CD4/ CD8 ratio, even though there is no significant changein CD8 cell abundance.
NK cells are cytotoxic cells of the innate immune system with inhibitory receptors recognizingclass I MHC, and a variety of activating receptors including NKG2D.49 We and others havereported an anti-fibrotic effect of NK cells in vivo and in vitro.19,20,50 in which NK cellsbecome stimulated in liver injury as the expression of inhibitory killing immunoglobulinreceptors decrease, compared with the activation receptors. HSC activation, conversely, leadsto reduced expression of class I molecules, leading NK cells to recognize HSCs as ‘non-self’that provokes HSC killing.19 NK cells (CD16+) significantly decrease in fibrotic infectedpatients (Fig. 1), thereby reducing an important anti-fibrotic subset. In support of thispossibility, Morishima et al.51 reported a decreased frequency of NK cells, similar to earlier
Muhanna et al. Page 9
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

studies in blood52–54 and liver.55,56 NK cell function is reduced in patients with primary biliarycirrhosis57 and in HCV-infected individuals.58–60 Our current findings further support an anti-fibrotic role of NK cells, in part through direct interaction with HSC in vivo and in vitro (Fig.4–Fig. 7) as well as through altered cytokine expression that reduces their activation (Fig. 3C).Moreover, NK cell numbers are reduced in virally infected livers, thereby attenuating animportant anti-fibrotic pathway (Fig. 1). Direct NK–HSC adhesion is accompanied by evidenceof increased apoptosis of activated HSCs and liver NK cells in situ.19
Although lymphocyte infiltration is a prominent feature of viral hepatitis, their functionalimpact on HSC activation has not been well established in human disease, whereas animalmodels have suggested a direct fibrogenic activity.18,19,21 In this study, evidence of fibrogenicstimulation associated with a direct physical interaction and an enhanced fibrogenic cytokineprofile is suggested by the co-culture results, wherein CD8+ T-cell subsets harbored the greatestfibrogenic activity, and NK cells decreased the activation of HSC (Fig. 3). The findingsignificantly expands earlier observations implicating CD8+ T cells in biliary fibrosisassociated with murine graft-versus-host disease34 and in alcoholic cirrhosis.35
Phagocytosis of lymphocytes by HSCs is consistent with recent reports documenting thephagocytic capacity of HSCs towards macromolecules.16,61,62 Moreover, because engulfmentof hepatocyte apoptotic bodies is fibrogenic,17 the same may hold true for phagocytosis oflymphocytes by HSCs. The capacity to internalize extracellular material has also beendescribed in other fibrogenic cell types (i.e., mesangial cells) as well as in hepaticnonprofessional antigen presenting cells such as sinusoidal endothelial cells.62,63 Our resultsare consistent with the findings of a recent study showing that activated HSCs can phagocytoseapoptotic bodies.17 HSCs can phagocytose both apoptotic and non-apoptotic lymphocytes (Fig.8), and increased apoptosis and death of lymphocytes inside HSC may be an inactivationmechanism of lymphocytes. The loss of lymphocyte engulfment by irradiated HSC implicatesHSC phagocytosis in this process. The translocation of cytoplasmic lymphocyte contents intothe HSC cytoplasm demonstrated by DiOC diffusion into the HSC cytoplasm providesadditional further evidence and explains the different effects of each lymphocyte subset onHSC responses. Phagocytosis of activated lymphocytes by the HSCs was also found in vitroin liver injury because of graft-versus-host disease after bone marrow transplantation in theabsence of viral infection (data not shown). Therefore, activated but not infected lymphocytesare responsible for in vitro HSC activation. We therefore suggest that phagocytosis oflymphocytes in the human in vivo setting is an additional inflammatory mechanism to activateHSC in addition to the recognized paracrine inflammatory pathways. The engulfment ofapoptotic cells by phagocytes was reported to prevent the release of potentially toxic orimmunogenic intracellular contents from the dying cells.65 Similar results have been foundwith fibroblasts and epithelial cells.63 Therefore, the phagocytosis of lymphocytes inside HSCsprovokes the concentration of immunogenic intracellular contents within the cells. In this case,the prominent engulfed lymphocyte subsets will dictate the behavior of HSCs. We reportedpreviously that apoptosis of LX2 cells occurs when they are co-cultured with NK cells,19 andhere we demonstrate that those NK cells are IFN-γ secretors, and their phagocytosis leads toinactivation of LX2 cells (Fig. 3). Conversely, LX2 proliferation and activation are increasedin the case of engulfed TGF-β–enriched CD8 subsets (Fig. 3). Because healthy lymphocytesdo not undergo phagocytosis, their co-culture with LX2 cells did not alter the expression ofSMA (Fig. 3A, lower panel).
We further explored the potential pathways specifically mediating phagocytosis. Phagocytosiswas initiated among different HSC surface adhesion ligands/ receptors that are involved withlymphocyte interaction, including CD11c, and ICAM-1.65 In contrast, neither MHC class Iand II, nor NK cell receptor–ligand interactions are important for HSCs to display antigen
Muhanna et al. Page 10
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

presentation properties,16 whereas both class II and CD11c are essential for antigenpresentation.66
Recent studies implicate cytoskeletal rearrangements mediated by Rho GTP-binding proteins.67 Rho proteins or their downstream effectors can be recruited to the adhesion structure, forexample, Rac1 and Cdc42, and co-localize at cell–cell contacts.68 Indeed, Rac1 and Cdc42silencing blocked phagocytosis and prevented HSC activation. Rac1/Cdc42 is not the onlypathway involved in phagocytosis, and other mechanisms could be also involved.Phosphatidylserine and phosphatidylserine receptor interactions have been suggested as analternative mechanism in HSCs. Phosphatidylserine on apoptotic cells promotes their uptakeand induces antiinflammatory responses in phagocytes, including TGF-β release.69 Interactionbetween phosphatidylserine and the phosphatidylserine receptor inhibits immune responses invivo.70 This interaction is relevant to the LX2 cells, because they express the phosphatidylserinereceptor.61
In summary, our results imply that human lymphocytes may mediate not only liver injury butalso the fibrogenic response through direct, regulated interactions with hepatic stellate cells.The findings further expand a growing body of evidence that defines specific lymphocyteinteractions in hepatic fibrogenesis.
Abbreviations
α-SMA alpha-smooth muscle actin
DiOC dioctadecyloxacarbocyanine perchlorate
FACS fluorescence-activated cell sorting
FBS fetal bovine serum
FITC fluorescein isothiocyanate
GTPase guanosine triphosphatase
HBV hepatitis B virus
HCV hepatitis C virus
HSC hepatic stellate cell
ICAM-1 intracellular adhesion molecule 1
IFN-γ interferon gamma
IHL intrahepatic lymphocyte
MHC major histocompatibility complex
mRNA messenger RNA
NK natural killer
PBL peripheral blood lymphocyte
PBS phosphate-buffered saline
PE phycoerythrin
PI propidium iodide
siRNA small interfering RNA
Muhanna et al. Page 11
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

AcknowledgmentsSupported by grants from NIH (DK 56621) and US-Israel Binational Scientific Foundation Awards (grant No.8033603, 8033605).
References1. Wake K. Cell-cell organization and functions of ’sinusoids’ in liver microcirculation system. J Electron
Microsc (Tokyo) 1999;48:89–98. [PubMed: 10356785]2. Gressner A, Bachem M. Cellular sources of noncollagenous matrix proteins: role of fatstoring cells in
fibrogenesis. Semin Liver Dis 1990;10:30–46. [PubMed: 2186487]3. Pinzani M, Gesualdo L, Sabbah G, Abboud H. Effects of platelet-derived growth factor and other
polypeptide mitogens on DNA synthesis and growth of cultured rat liver fatstoring cells. J Clin Invest1989;84:1786–1793. [PubMed: 2592560]
4. Matsuoka M, Tsukamoto H. Stimulation of hepatic lipocyte collagen production by Kupffer cell-derived transforming growth factor beta: implication for a pathogenetic role in alcoholic liverfibrogenesis. Hepatology 1990;11:599–605. [PubMed: 2328954]
5. Reiter Z. Interferon: a major regulator of natural killer cell-mediated cytotoxicity. J Interferon Res1993;13:247–257. [PubMed: 7693829]
6. Cramp ME, Rossol S, Chokshi S, Carucci P, Williams R, Naoumov NV. Hepatitis C virus-specific T-cell reactivity during interferon and ribavirin treatment in chronic hepatitis C. Gastroenterology2000;118:346–355. [PubMed: 10648463]
7. Thimme R, Oldach D, Chang K, Steiger C, Ray SC, Chisari FV. Determinants of viral clearance andpersistence during acute hepatitis C virus infection. J Exp Med 2001;194:1395–1406. [PubMed:11714747]
8. Grun ̈er NH, Gerlach TJ, Jung M, Diepolder HM, Schirren CA, Schraut WW, et al. Association ofhepatitis C virus-specific CD8+ T cells with viral clearance in acute hepatitis C. J Infect Dis2000;181:1528–1536. [PubMed: 10823750]
9. Koziel M. Cytokines in viral hepatitis. Semin Liver Dis 1999;19:157–169. [PubMed: 10422198]10. Brown PMJ, Neuman MG. Immunopathogenesis of hepatitis C viral infection: H1/TH2 responses
and the role of cytokines. Clin Biochem 2001;34:167–171. [PubMed: 11408013]11. Iredale JP. Hepatic stellate cell behavior during resolution of liver injury. Semin Liver Dis
2001;21:427–435. [PubMed: 11586470]12. Nelson DR, Marousis CG, Davis GL, Rice CM, Wong J, Houghton M, et al. The role of hepatitis C
virus-specific cytotoxic T lymphocytes in chronic hepatitis C. J Immunol 1997;1:1473–1481. 158.[PubMed: 9013994]
13. Napoli J, Bishop G, McGuinness P, Painter D, McCaughan G. Progressive liver injury in chronichepatitis C infection correlates with increased intrahepatic expression of Th1-associated cytokines.Hepatology 1996;24:759–765. [PubMed: 8855173]
14. Roger PM, Chaillou S, Breittmayer JP, Dahman M, St Paul MC, Chevallier P, et al. Intrahepatic CD4T-cell apoptosis is related to METAVIR score in patients with chronic hepatitis C virus. Scand JImmunol 2005;62:168–175. [PubMed: 16101824]
15. Winau F, Hegasy G, Weiskirchen R, Weber S, Cassan C, Sieling PA, et al. Ito cells are liver-residentantigen-presenting cells for activating T cell responses. Immunity 2007;26:117–129. [PubMed:17239632]
16. Vinas O, Bataller R, Sancho-Bru P, Gines P, Berenguer C, Enrich C, et al. Human hepatic stellatecells show features of antigen-presenting cells and stimulate lymphocyte proliferation. Hepatology2003;38:919–929. [PubMed: 14512879]
17. Zhan SS, Jiang JX, Wu J, Halsted C, Friedman SL, Zern MA, et al. Phagocytosis of apoptotic bodiesby hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo.Hepatology 2006;43:435–443. [PubMed: 16496318]
18. Safadi R, Ohta M, Alvarez CE, Fiel I, Bansal M, Mehal W, et al. Immune stimulation of hepaticfibrogenesis by CD8 lymphocytes and its attenuation by transgenic interleukin 10 from hepatocytes.Gastroenterology 2004;127:870–882. [PubMed: 15362042]
Muhanna et al. Page 12
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

19. Melhem A, Muhanna N, Bishara A, Alvarez CE, Ilan Y, Bishara T, et al. Anti-fibrotic activity of NKcells in experimental liver injury through killing of activated HSC. J Hepatol 2006;45:60–71.[PubMed: 16515819]
20. Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z, Gao B. Natural killer cells ameliorate liver fibrosisby killing activated stellate cells in NKG2D–dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology 2006;130:435–452. [PubMed: 16472598]
21. Muhanna N, Horani A, Doron S, Safadi R. Lymphocyte-hepatic stellate cell proximity suggests adirect interaction. Clin Exp Immunol 2007;148:338–347. [PubMed: 17437422]
22. Bedossa P, Poynard T. An algorithm for the grading of activity in chronic hepatitis C: the METAVIRCooperative Study Group. Hepatology 1996;24:289–293. [PubMed: 8690394]
23. Boyum M. Isolation of mononuclear cells and granulocytes from human blood. Scand J Clin LabInvest 1968;97:81–85.
24. Wald O, Pappo O, Safadi R, Dagan-Berger M, Beider K, Wald H, et al. Involvement of the CXCL12/CXCR4 pathway in the advanced liver disease that is associated with hepatitis C virus or hepatitis Bvirus. Eur J Immunol 2004;34:1164–1174. [PubMed: 15048728]
25. Taimr P, Higuchi H, Kocova E, Rippe RA, Friedman S, Gores GJ. Activated human stellate cellsexpress the TRAIL receptor-2/Death receptor-5 and undergo TRAIL-mediated apoptosis.Hepatology 2003;37:87–95. [PubMed: 12500193]
26. Friedman SL, Rockey DC, McGuire RF, Maher JJ, Boyles JK, Yamasaki G. Isolated hepatic lipocytesand Kupffer cells from normal human liver: morphological and functional characteristics in primaryculture. Hepatology 1992;15:234–243. [PubMed: 1735526]
27. Hoppner M, Luhm J, Schlenke P, Koritke P, Frohn C. A flowcytometry based cytotoxicity assayusing stained effector cells in combination with native target cells. J Immunol Methods 2002;15:157–163. 267. [PubMed: 12165437]
28. Qamar A, Sheikh SZ, Masud A, Jhandier MN, Inayat IB, Hakim W, et al. In vitro and in vivo protectionof stellate cells from apoptosis by leptin. Dig Dis Sci 2006;51:1697–1705. [PubMed: 16957995]
29. Forbes SJ, Russo FP, Rey V, Burra P, Rugge M, Wright NA, et al. A significant proportion ofmyofibroblasts are of bone marrow origin in human liver fibrosis. Gastroenterology 2004;126:955–963. [PubMed: 15057733]
30. Sham RL, Packman CH, Abboud CN, Lichtman MA. Signal transduction and the regulation of actinconformation during myeloid maturation: studies in HL60 cells. Blood 1991;77:363–370. [PubMed:1985701]
31. Brelje TC, Wessendorf MW, Sorenson RL. Multicolor laser scanning focal microscopy: practicalapplication and limitations. Methods Cell Biol 2002;70:165–244. [PubMed: 12512325]
32. Tarshis M, Yavlovich A, Katzenell A, Ginsburg I, Rottem S. Intracellular location and survival ofMycoplasma penetrans within HeLa cells. Curr Microbiol 2004;49:136–140. [PubMed: 15297920]
33. Inada S, Suzuki K, Kimura T, Hayashi A, Narita T, Yui R, et al. Concentric fibrosis and cellularinfiltration around bile ducts induced by graft-versus-host reaction in mice: a role of CD8+ cells.Autoimmunity 1995;22:163–171. [PubMed: 8734570]
34. Laso F, Iglesias-Osma C, Ciudad J, Lopez A, Pastor I, Orfao A. Chronic alcoholism is associatedwith an imbalanced production of Th-1/Th-2 cytokines by peripheral blood T cells. Alcohol Clin ExpRes 1999;23:1306–1311. [PubMed: 10470972]
35. Laso F, Iglesias-Osma C, Ciudad J, Lopez A, Pastor I, Torres E, et al. Alcoholic liver cirrhosis isassociated with a decreased expression of the CD28 costimulatory molecule, a lower ability of Tcells to bind exogenous IL-2, and increased soluble CD8 levels. Cytometry 2000;42:290–295.[PubMed: 11025487]
36. Lombardo L, Capaldi A, Poccardi G, Vineis P. Peripheral blood CD3 and CD4 T- lymphocytereduction correlates with severity of liver cirrhosis. Int J Clin Lab Res 1995;25:153–156. [PubMed:8562979]
37. Cougoule C, Wiedemann A, Lim J, Caron E. Phagocytosis, an alternative model system for the studyof cell adhesion. Semin Cell Dev Biol 2004;15:679–689. [PubMed: 15561587]
38. Endo Y, Even-Ram S, Pankov R, Matsumoto K, Yamada KM. Inhibition of Rho GTPases by RNAInterference. Methods Enzymol 2006;406:345–361. [PubMed: 16472669]
Muhanna et al. Page 13
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

39. Valiante NM, D’Andrea A, Crotta S, Lechner F, Klenerman P, Nuti S, et al. Life, activation and deathof intrahepatic lymphocytes in chronic hepatitis C. Immunol Rev 2000;174:77–89. [PubMed:10807508]
40. Mehal WZ, Azzaroli F, Crispe IN. Immunology of the healthy liver: old questions and new insights.Gastroenterology 2001;120:250–260. [PubMed: 11208734]
41. Lechner F, Wong DK, Dunbar PR, Chapman R, Chung RT, Dohren-wend P, et al. Analysis ofsuccessful immune responses in persons infected with hepatitis C virus. J Exp Med 2000;191:1499–1512. [PubMed: 10790425]
42. Cooper S, Erickson AL, Adams EJ, Kansopon J, Weiner AJ, Chien DY, et al. Analysis of a successfulimmune response against hepatitis C virus. Immunity 1999;10:439–449. [PubMed: 10229187]
43. Takaki A, Wiese M, Maertens G, Depla E, Seifert U, Liebetrau A, et al. Cellular immune responsespersist and humoral responses decrease two decades after recovery from a single-source outbreak ofhepatitis C. Nat Med 2000;6:578–582. [PubMed: 10802716]
44. Gerlach JT, Diepolder HM, Jung MC, Gruener NH, Schraut WW, Za-choval R, et al. Recurrence ofhepatitis C virus after loss of virus-specific CD4 (+) T-cell response in acute hepatitis C.Gastroenterology 1999;117:933–941. [PubMed: 10500077]
45. Bertoletti A, D’Elios MM, Boni C, De Carli M, Zignego AL, Durazzo M, et al. Different cytokineprofiles of intraphepatic T cells in chronic hepatitis B and hepatitis C virus infections.Gastroenterology 1997;112:193–199. [PubMed: 8978359]
46. Ferrari C, Urbani S, Penna A, Cavalli A, Valli A, Lamonaca V, et al. Immunopathogenesis of hepatitisC virus infection. J Hepatol 1999;31:31–38. [PubMed: 10622557]
47. Yao ZQ, Nguyen DT, Hiotellis AI, Hahn YS. Hepatitis C virus core protein inhibits human Tlymphocyte responses by a complement-dependent regulatory pathway. J Immunol 2001;167:5264–5272. [PubMed: 11673541]
48. Tran A, Yang G, Doglio A, Ticchioni M, Laffont C, Durant J, et al. Phenotyping of intrahepatic andperipheral blood lymphocytes in patients with chronic hepatitis C. Dig Dis Sci 1997;42:2495–2500.[PubMed: 9440626]
49. Lanier LL. NK cell recognition. Ann Rev Immunol 2005;23:225–274. [PubMed: 15771571]50. Nattermann J, Feldmann G, Ahlenstiel G, Langhans B, Sauerbruch T, Spengler U. Surface expression
and cytolytic function of NK cell receptors is altered in chronic hepatitis C. Gut 2006;55:869–877.[PubMed: 16322112]
51. Morishima C, Paschal DM, Wang CC, Yoshihara CS, Wood BL, Yeo AE, et al. Decreased NK cellfrequency in chronic hepatitis C does not affect ex vivo cytolytic killing. Hepatology 2006;43:573–580. [PubMed: 16496327]
52. Lin AW, Gonzalez SA, Cunningham-Rundles S, Dorante G, Marshall S, Tignor A, et al. CD56(+dim) and CD56(+ bright) cell activation and apoptosis in hepatitis C virus infection. Clin ExpImmunol 2004;137:408–416. [PubMed: 15270860]
53. Meier UC, Owen RE, Taylor E, Worth A, Naoumov N, Willberg C, et al. Shared alterations in NKcell frequency, phenotype, and function in chronic human immunodeficiency virus and hepatitis Cvirus infections. J Virol 2005;79:12365–12374. [PubMed: 16160163]
54. Par G, Rukavina D, Podack ER, Horanyi M, Szekeres-Bartho J, Hegedus G, et al. Decrease in CD3-negative-CD8dim(+) and Vdelta2/Vgamma9 TcR+ peripheral blood lymphocyte counts, lowperforin expression and the impairment of natural killer cell activity is associated with chronichepatitis C virus infection. J Hepatol 2002;37:514–522. [PubMed: 12217606]
55. Deignan T, Curry MP, Doherty DG, Golden-Mason L, Volkov Y, Norris S, et al. Decrease in hepaticCD56(+) T cells and V alpha 24(+) natural killer T cells in chronic hepatitis C viral infection. JHepatol 2002;37:101–108. [PubMed: 12076868]
56. Kawarabayashi N, Seki S, Hatsuse K, Ohkawa T, Koike Y, Aihara T, et al. Decrease of CD56(+)Tcells and natural killer cells in cirrhotic livers with hepatitis C may be involved in their susceptibilityto hepatocellular carcinoma. Hepatology 2000;32:962–969. [PubMed: 11050046]
57. Matheson DS, Green BJ, Minuk GY. Natural killer-cell activity and the response to interferons alpha,beta, and gamma in patients with primary biliary cirrhosis. J Allergy Clin Immunol 1989;84:214–218. [PubMed: 2503552]
Muhanna et al. Page 14
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

58. Corado J, Toro F, Rivera H, Bianco NE, Deibis L, De Sanctis JB. Impairment of natural killer (NK)cytotoxic activity in hepatitis C virus (HCV) infection. Clin Exp Immunol 1997;109:451–457.[PubMed: 9328121]
59. Crotta S, Stilla A, Wack A, D’Andrea A, Nuti S, D’Oro U, et al. Inhibition of natural killer cellsthrough engagement of CD81 by the major hepatitis C virus envelope protein. J Exp Med2002;195:35–41. [PubMed: 11781363]
60. Tseng CT, Klimpel GR. Binding of the hepatitis C virus envelope protein E2 to CD81 inhibits naturalkiller cell functions. J Exp Med 2002;195:43–49. [PubMed: 11781364]
61. Canbay A, Taimr P, Torok N, Higuchi H, Friedman S, Gores GJ. Apoptotic body engulfment by ahuman stellate cell line is profibrogenic. Lab Invest 2003;83:655–663. [PubMed: 12746475]
62. Kosugi I, Muro H, Shirasawa H, Ito I. Endocytosis of soluble IgG immune complex and its transportto lysosomes in hepatic sinusoidal endothelial cells. J Hepatol 1992;16:106–114. [PubMed: 1484143]
63. Hughes J, Liu Y, Van Damme J, Savill J. Human glomerular mesangial cell phagocytosis of apoptoticneutrophils. J Immunol 1997;158:4389–4397. [PubMed: 9127003]
64. Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature 2000;407:784–788.[PubMed: 11048729]
65. Diamond MS, Garcia-Aguilar J, Bickford JK, Corbi AL, Springer TA. The I domain is a majorrecognition site on the leukocyte integrin Mac-1 (CD11b/ CD18) for four distinct adhesion ligands.J Cell Biol 1993;120:1031–1043. [PubMed: 7679388]
66. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature 1998;19:245–252.392. [PubMed: 9521319]
67. Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature 2002;420:629–635. [PubMed:12478284]
68. Fukata M, Kaibuchi K. Rho-family GTPases in cadherin-mediated cell-cell adhesion. Nat Rev MolCell Biol 2001;2:887–897. [PubMed: 11733768]
69. Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cellspromotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest 2002;109:41–50.[PubMed: 11781349]
70. Hoffmann PR, Kench JA, Vondracek A, Kruk E, Daleke DL, Jordan M, et al. Interaction betweenphosphatidylserine and the phosphatidylserine receptor inhibits immune responses in vivo. JImmunol 2005;174:1393–1404. [PubMed: 15661897]
Muhanna et al. Page 15
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Fig. 1.Lymphocyte alterations in fibrotic patients. FACS analysis of isolated intrahepatic andperipheral blood lymphocytes from healthy donors (black bars), HCV (open bars), and HBV(gray bars) fibrotic patients are illustrated (see Patients and Methods). Significant differenceswere confined to intrahepatic lymphocytes, in which CD4 decreased, leading to an increase ofCD4/CD8 ratio (Fig. 1); NK cells decreased in both fibrotic groups.
Muhanna et al. Page 16
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
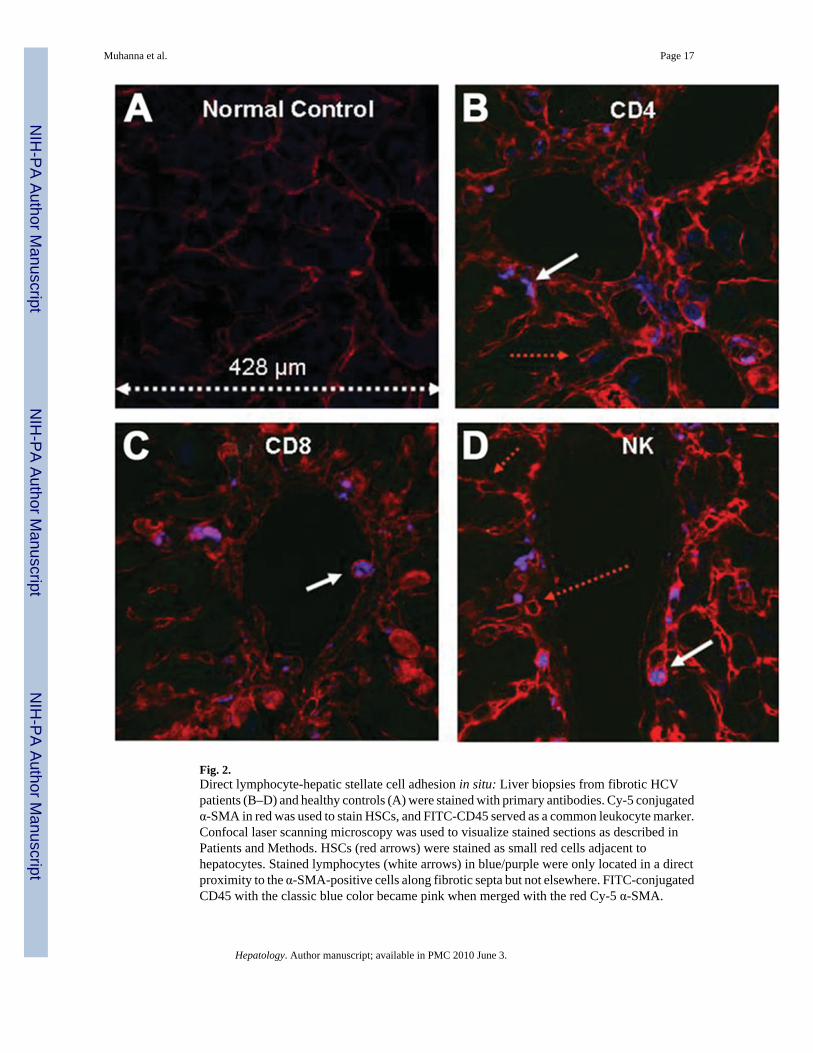
Fig. 2.Direct lymphocyte-hepatic stellate cell adhesion in situ: Liver biopsies from fibrotic HCVpatients (B–D) and healthy controls (A) were stained with primary antibodies. Cy-5 conjugatedα-SMA in red was used to stain HSCs, and FITC-CD45 served as a common leukocyte marker.Confocal laser scanning microscopy was used to visualize stained sections as described inPatients and Methods. HSCs (red arrows) were stained as small red cells adjacent tohepatocytes. Stained lymphocytes (white arrows) in blue/purple were only located in a directproximity to the α-SMA-positive cells along fibrotic septa but not elsewhere. FITC-conjugatedCD45 with the classic blue color became pink when merged with the red Cy-5 α-SMA.
Muhanna et al. Page 17
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Fig. 3.The profibrogenic and antifibrogenic effect of lymphocyte subsets in culture: LX2 cells wereco-cultured 24 hours with lymphocytes from either HCV patients or healthy controls, either asmixed or isolated subsets. (A) Results from two individuals of each group. Protein extractswere evaluated for α-SMA (upper lanes) as a marker for HSC activation and compared withβ-actin (second lanes). The upper panel demonstrates the activation of LX2 cells after co-culture with mixed HCV-derived PBLs. The middle panel indicates that HCV CD8 cells, andto a lesser extent CD4 cells as well as mixed HCV lymphocytes, activate HSCs, as manifestedby a more intense expression of α-SMA protein as assessed by western blot. NK cells barelyactivate HSCs. Healthy lymphocytes, in the lower part of the figure, fail to activate HSCs.
Muhanna et al. Page 18
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Results from mRNA analysis for expression of α-SMA (B) correspond to results from westernblot (LX2 in the upper panel and primary isolated HSCs in the lower one). Experiments wererepeated three times with nearly identical results. Moreover, although this figure derived fromtwo HCV and two healthy donors, results are reproducible when the experiment is repeatedwith lymphocytes from other cases (data not shown). Intracellular cytokine analysis of culturedlymphocytes was studied by FACS using healthy or HCV-derived PBLs (C). Results showdecreased TGF-β and increased IFN-γ secretion by NK cells, decreased TGF-β from CD4 andCD8 cells with increased IFN-γ secretion only in the CD8 subsets.
Muhanna et al. Page 19
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
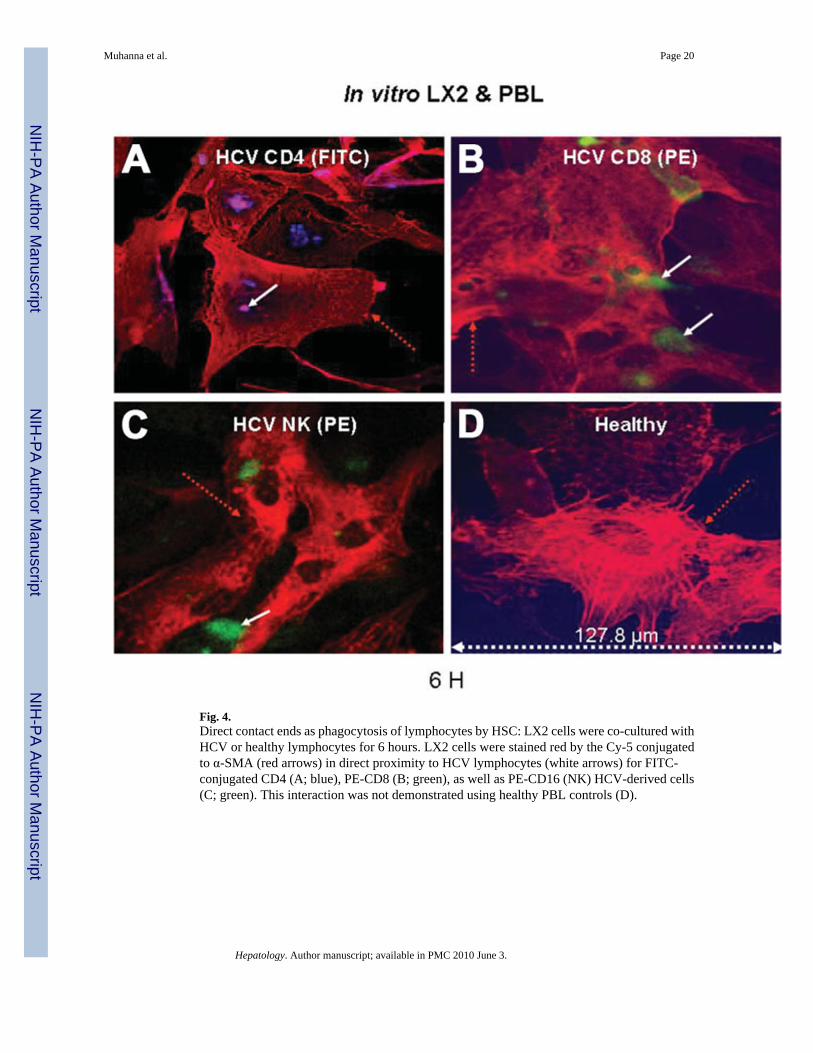
Fig. 4.Direct contact ends as phagocytosis of lymphocytes by HSC: LX2 cells were co-cultured withHCV or healthy lymphocytes for 6 hours. LX2 cells were stained red by the Cy-5 conjugatedto α-SMA (red arrows) in direct proximity to HCV lymphocytes (white arrows) for FITC-conjugated CD4 (A; blue), PE-CD8 (B; green), as well as PE-CD16 (NK) HCV-derived cells(C; green). This interaction was not demonstrated using healthy PBL controls (D).
Muhanna et al. Page 20
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
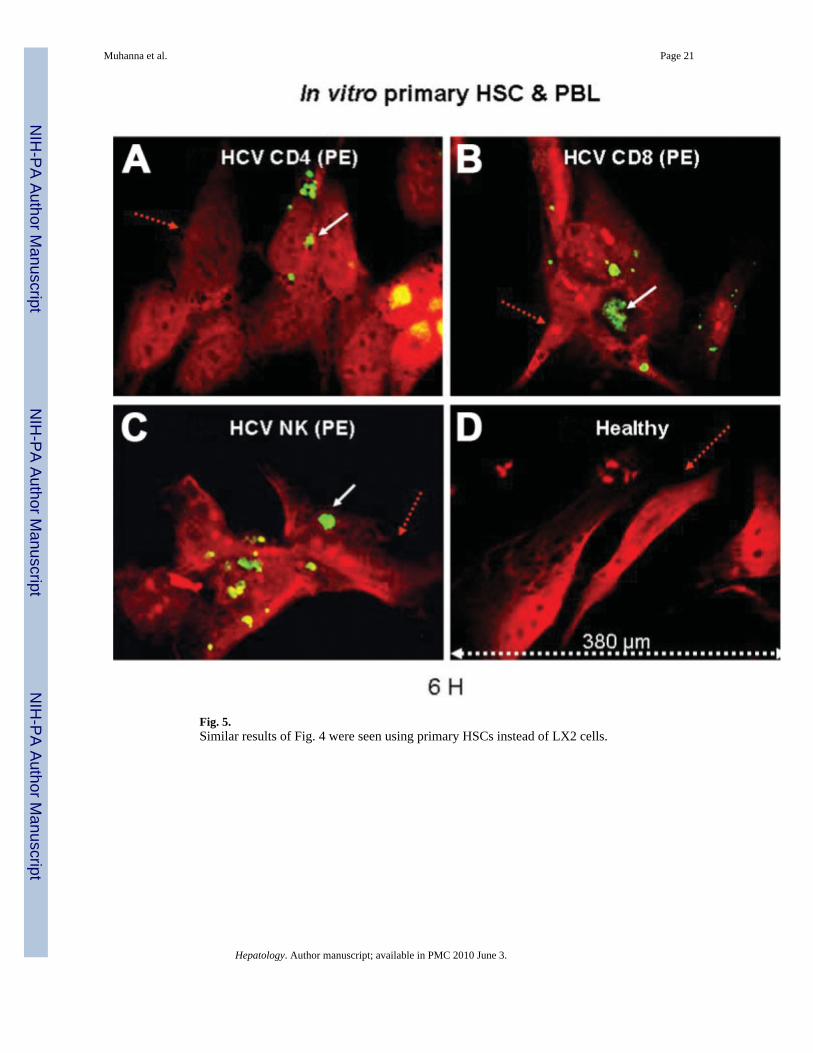
Fig. 5.Similar results of Fig. 4 were seen using primary HSCs instead of LX2 cells.
Muhanna et al. Page 21
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
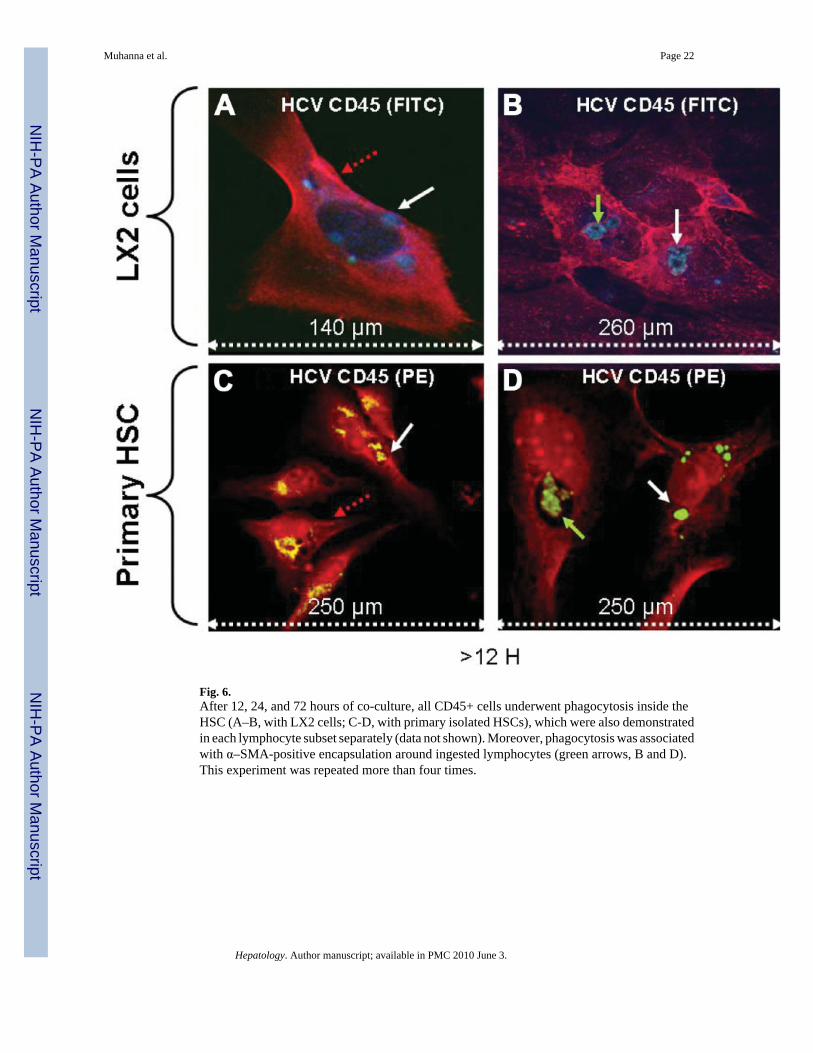
Fig. 6.After 12, 24, and 72 hours of co-culture, all CD45+ cells underwent phagocytosis inside theHSC (A–B, with LX2 cells; C-D, with primary isolated HSCs), which were also demonstratedin each lymphocyte subset separately (data not shown). Moreover, phagocytosis was associatedwith α–SMA-positive encapsulation around ingested lymphocytes (green arrows, B and D).This experiment was repeated more than four times.
Muhanna et al. Page 22
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Fig. 7.Confocal microscopy documents phagocytosis: Serial 1-µm section images showed HSC andCD45+ cells appear and disappear at the same confocal channel, confirming phagocytosis(selective sections are presented in A–D with LX2 cells and in G-J with primary isolated HSCs).HCV-derived lymphocytes were preincubated with DiOC, washed, and then cultured with HSCcells. (E–F) LX2 cells and (K and L) primary isolated HSCs demonstrate lymphocytes insidethe HSCs, with release of their DiOC into the HSC cytoplasm. The same was also demonstratedfor each lymphocyte subset individually (data not shown). The experiment was repeated fourtimes.
Muhanna et al. Page 23
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
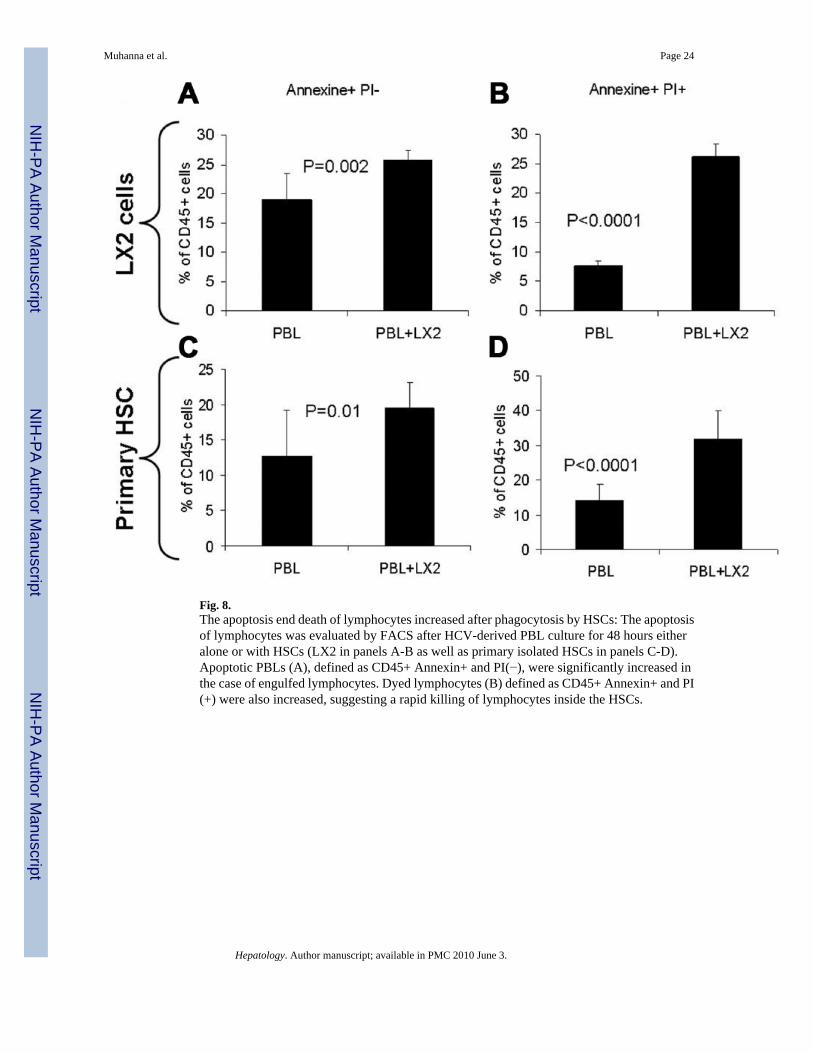
Fig. 8.The apoptosis end death of lymphocytes increased after phagocytosis by HSCs: The apoptosisof lymphocytes was evaluated by FACS after HCV-derived PBL culture for 48 hours eitheralone or with HSCs (LX2 in panels A-B as well as primary isolated HSCs in panels C-D).Apoptotic PBLs (A), defined as CD45+ Annexin+ and PI(−), were significantly increased inthe case of engulfed lymphocytes. Dyed lymphocytes (B) defined as CD45+ Annexin+ and PI(+) were also increased, suggesting a rapid killing of lymphocytes inside the HSCs.
Muhanna et al. Page 24
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Fig. 9.Phagocytosis is mediated by a ligand/receptor adhesion. Compared with the nonmanipulatedco-culture and unstimulated phagocytosis (A), prevention of phagocytosis was achieved whenthe HSC-related ICAM1 and integrin alphaV were blocked before co-culture as described inPatients and Methods (D and E, respectively). Blocking of HSC-related class I as well as classII before co-culture with lymphocytes did not affect phagocytosis (B and C, respectively).Blocking of lymphocyte-related T9cell receptor before co-culture with lymphocytes did notaffect phagocytosis (F). Only when HSC (H) but not lymphocytes (G) were irradiated wasphagocytosis blocked. (I) Absence of phagocytosis when HSC were co-cultured with healthylymphocytes.
Muhanna et al. Page 25
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
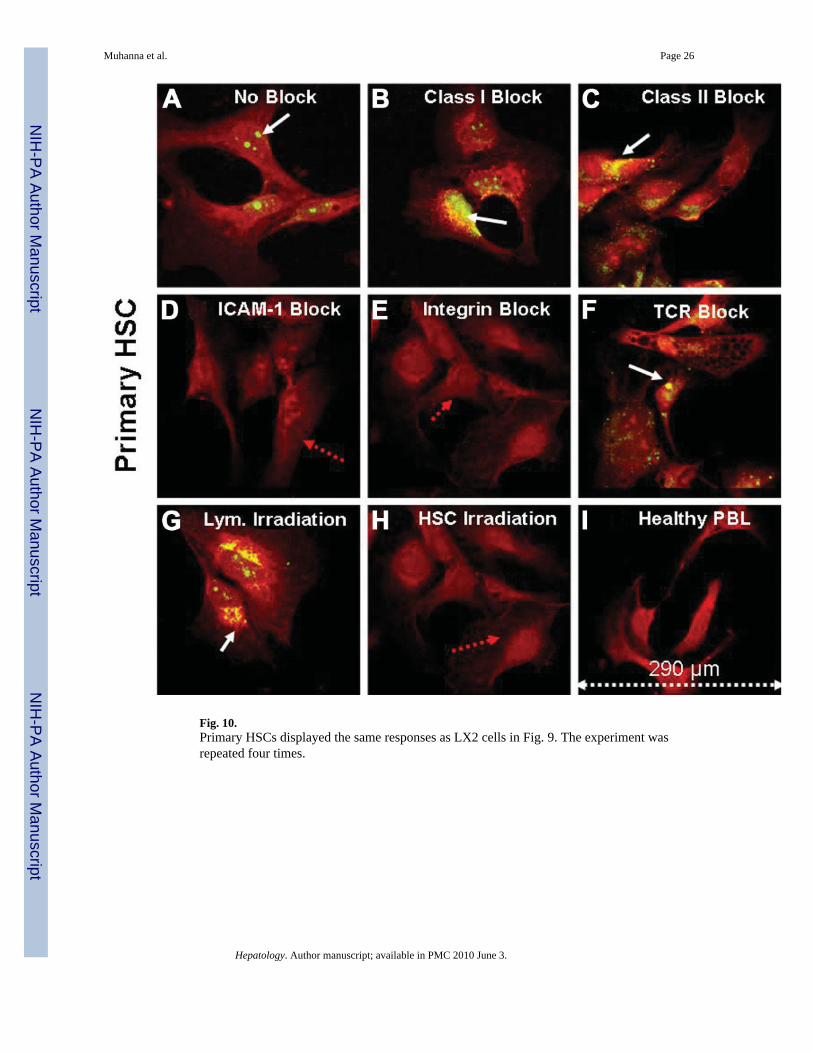
Fig. 10.Primary HSCs displayed the same responses as LX2 cells in Fig. 9. The experiment wasrepeated four times.
Muhanna et al. Page 26
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
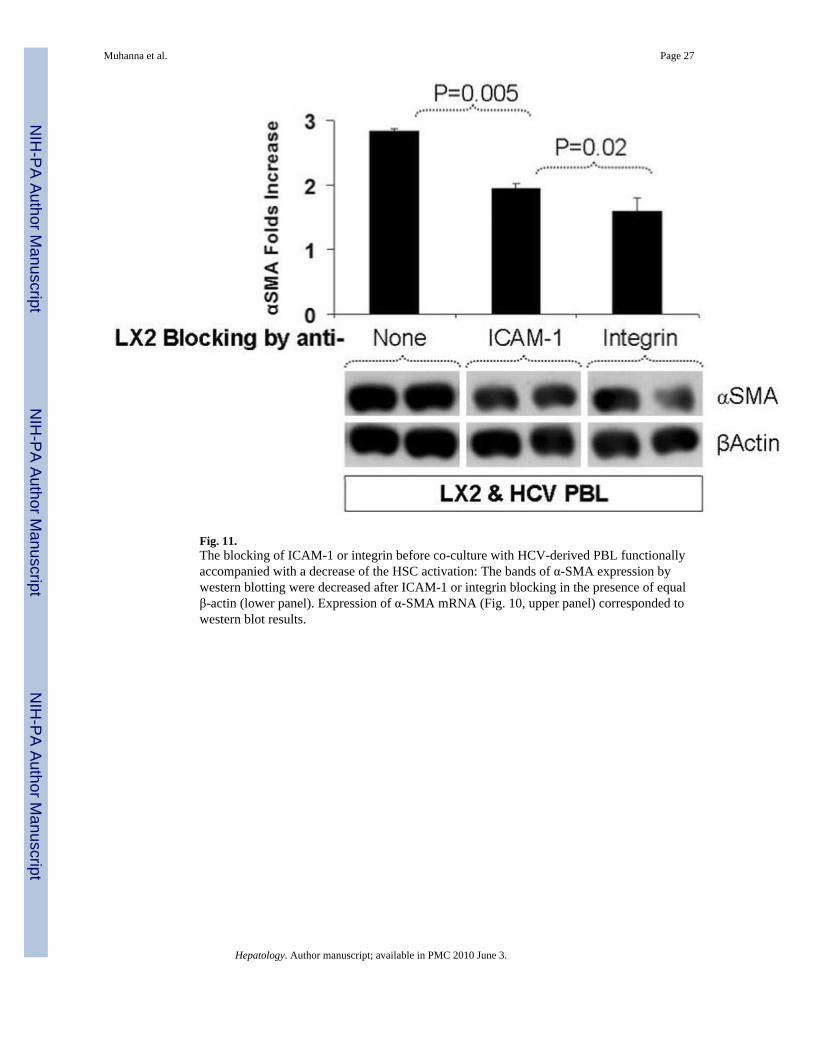
Fig. 11.The blocking of ICAM-1 or integrin before co-culture with HCV-derived PBL functionallyaccompanied with a decrease of the HSC activation: The bands of α-SMA expression bywestern blotting were decreased after ICAM-1 or integrin blocking in the presence of equalβ-actin (lower panel). Expression of α-SMA mRNA (Fig. 10, upper panel) corresponded towestern blot results.
Muhanna et al. Page 27
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
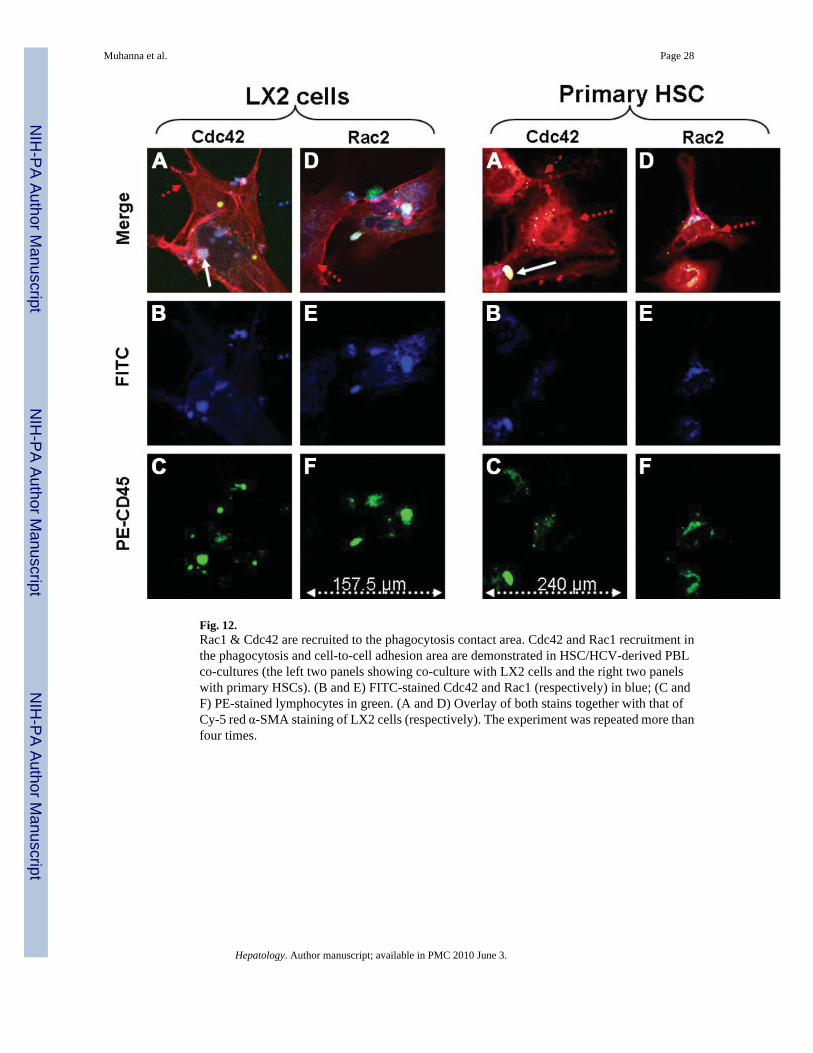
Fig. 12.Rac1 & Cdc42 are recruited to the phagocytosis contact area. Cdc42 and Rac1 recruitment inthe phagocytosis and cell-to-cell adhesion area are demonstrated in HSC/HCV-derived PBLco-cultures (the left two panels showing co-culture with LX2 cells and the right two panelswith primary HSCs). (B and E) FITC-stained Cdc42 and Rac1 (respectively) in blue; (C andF) PE-stained lymphocytes in green. (A and D) Overlay of both stains together with that ofCy-5 red α-SMA staining of LX2 cells (respectively). The experiment was repeated more thanfour times.
Muhanna et al. Page 28
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
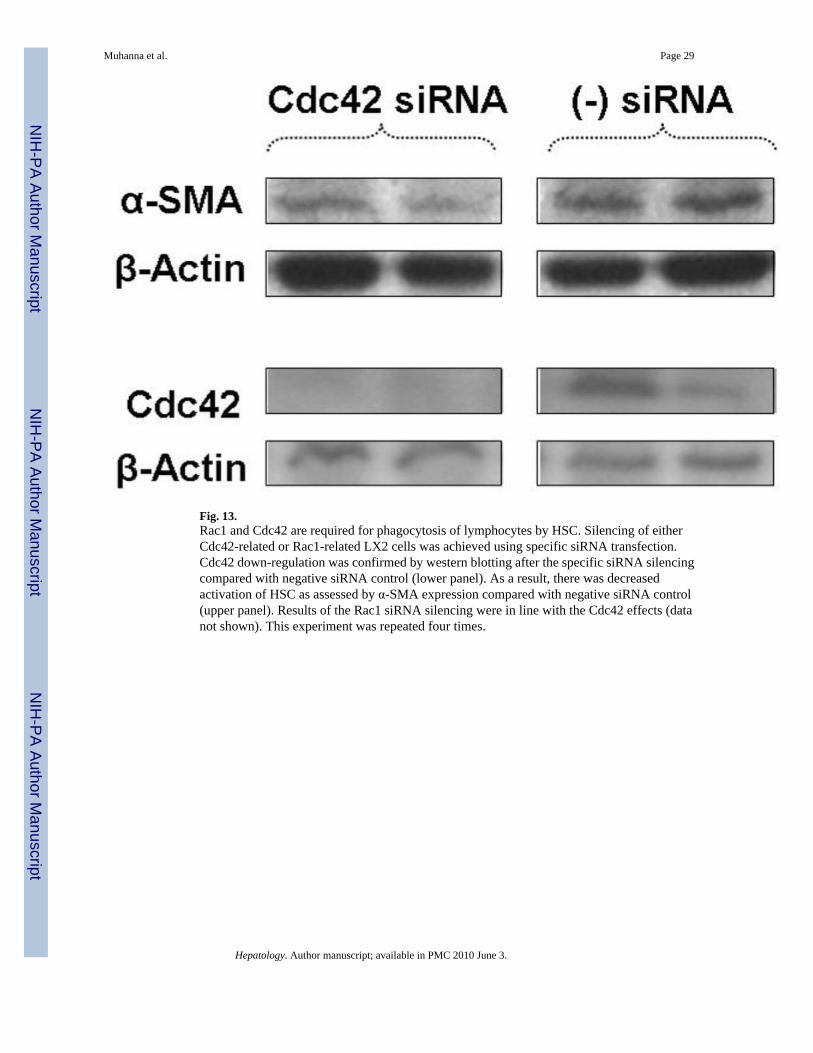
Fig. 13.Rac1 and Cdc42 are required for phagocytosis of lymphocytes by HSC. Silencing of eitherCdc42-related or Rac1-related LX2 cells was achieved using specific siRNA transfection.Cdc42 down-regulation was confirmed by western blotting after the specific siRNA silencingcompared with negative siRNA control (lower panel). As a result, there was decreasedactivation of HSC as assessed by α-SMA expression compared with negative siRNA control(upper panel). Results of the Rac1 siRNA silencing were in line with the Cdc42 effects (datanot shown). This experiment was repeated four times.
Muhanna et al. Page 29
Hepatology. Author manuscript; available in PMC 2010 June 3.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
Muhanna et al. Page 30
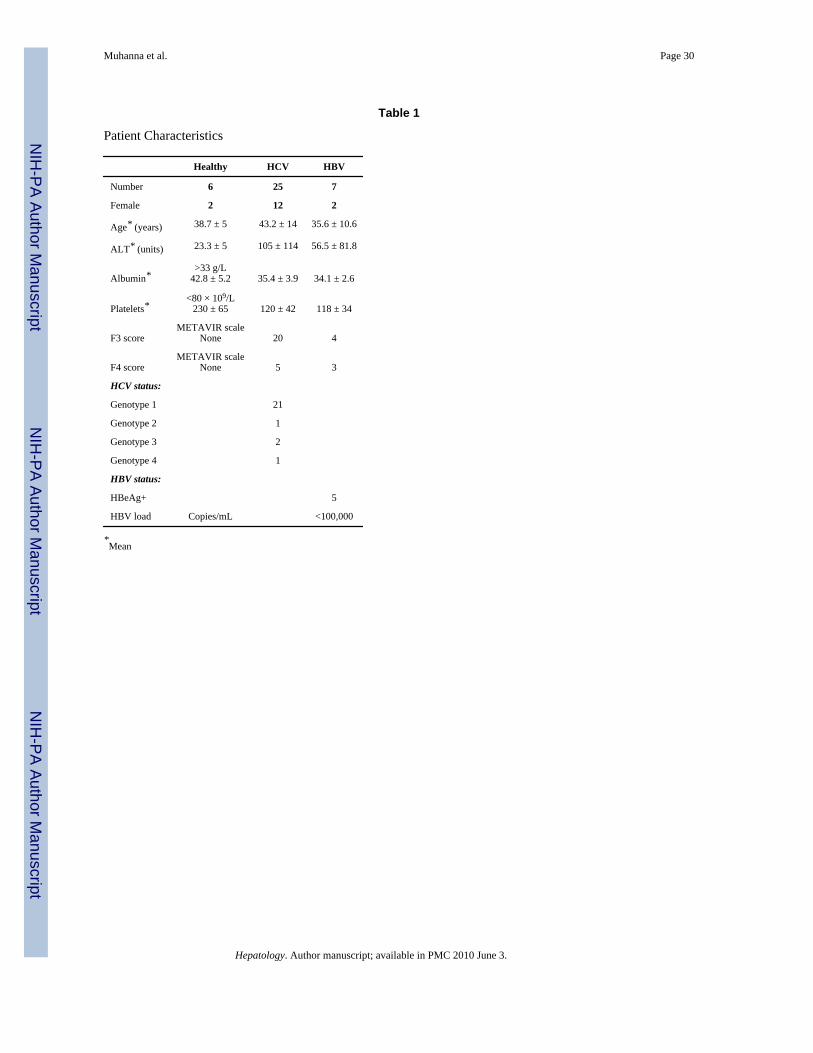
Table 1
Patient Characteristics
Healthy HCV HBV
Number 6 25 7
Female 2 12 2
Age* (years) 38.7 ± 5 43.2 ± 14 35.6 ± 10.6
ALT* (units) 23.3 ± 5 105 ± 114 56.5 ± 81.8
Albumin*>33 g/L
42.8 ± 5.2 35.4 ± 3.9 34.1 ± 2.6
Platelets*<80 × 109/L
230 ± 65 120 ± 42 118 ± 34
F3 scoreMETAVIR scale
None 20 4
F4 scoreMETAVIR scale
None 5 3
HCV status:
Genotype 1 21
Genotype 2 1
Genotype 3 2
Genotype 4 1
HBV status:
HBeAg+ 5
HBV load Copies/mL <100,000
*Mean
Hepatology. Author manuscript; available in PMC 2010 June 3.
Copyright © 2022 FDOKUMEN




















