
Superoxide dismutase-1 and other proteins in inclusions from transgenic amyotrophic lateral...
11
*Department of Medical Biosciences, Clinical Chemistry, Umea ˚ University, Umea ˚, Sweden Department of Pathology, Umea ˚ University, Umea ˚, Sweden àDepartment of Forest Genetics and Plant Physiology, Swedish University of Agricultural Sciences, Umea ˚, Sweden §Department of Pharmacology and Clinical Neuroscience, Umea ˚ University, Umea ˚, Sweden Amyotrophic lateral sclerosis (ALS) is a devastating neuro- degenerative disease characterized by loss of motor neurons in the motor cortex, brainstem, and spinal cord. This results in progressive muscular atrophy, and the patients usually succumb to respiratory failure within a few years. About 10% of ALS cases appear in families (Haverkamp et al. 1995), and in some of these the disease is linked to mutations in the gene of the ubiquitously expressed antioxidant enzyme CuZn-superoxide dismutase (SOD1) (Rosen et al. 1993). Overall, about 6% of all cases with ALS show SOD1 mutations, and more than 140 such mutations have been identified [(Andersen et al. 2003); http://alsod.iop.kcl.ac.uk/ Als/Summary/summary.aspx]. The mutations confer a cyto- toxic gain of function of unknown character to the enzyme (Gurney et al. 1994; Andersen et al. 1995). There is now considerable evidence to suggest that the demise of motor neurons is non-cell autonomous and dependent on noxious effects of mutant SOD1s in several cell types in the motor areas (Boillee et al. 2006; Nagai et al. 2007; Henkel et al. 2009). A hallmark of ALS caused by mutant SOD1s, both in patients and transgenic models, is inclusions that are immunoreactive for SOD1 (Bruijn et al. 1997; Johnston et al. 2000; Jonsson et al. 2004, 2006b). In the animal models, inclusions appear concomitantly with the onset of symptoms and become markedly more abundant in the terminal phase of the disease (Johnston et al. 2000; Wang et al. 2002b; Jonsson et al. 2004, 2006a). They are present in Received December 21, 2009; revised manuscript received March 13, 2010; accepted April 12, 2010. Address correspondence and reprint requests to Stefan L. Marklund, Department of Medical Biosciences, Clinical Chemistry, Umea University, SE-901 85 Umea ˚, Sweden. E-mail: [email protected] Abbreviations used: ALS, amyotrophic lateral sclerosis; ER, endo- plasmic reticulum; hSOD1, human SOD1; LC-ESI, Liquid chromato- graphy-electrospray ionization; MALDI-TOF, Matrix-assisted laser desorption ionization time of flight; MS, Mass spectrometry; NP-40, Nonidet P-40; SOD1, superoxide dismutase-1. Abstract Mutant superoxide dismutase-1 (SOD1) causes amyotrophic lateral sclerosis (ALS) through a cytotoxic mechanism of un- known nature. A hallmark in ALS patients and transgenic mouse models carrying human SOD1 (hSOD1) mutations are hSOD1-immunoreactive inclusions in spinal cord ventral horns. The hSOD1 inclusions may block essential cellular functions or cause toxicity through sequestering of other proteins. Inclusions from four different transgenic mouse models were examined after density gradient ultracentrifuga- tion. The inclusions are complex structures with heteroge- neous densities and are disrupted by detergents. The aggregated hSOD1 was mainly composed of subunits that lacked the native stabilizing intra-subunit disulfide bond. A proportion of subunits formed hSOD1 oligomers or was bound to other proteins through disulfide bonds. Dense inclusions could be isolated and the protein composition was analyzed using proteomic techniques. Mutant hSOD1 accounted for half of the protein. Ten other proteins were identified. Two were cytoplasmic chaperones, four were cytoskeletal pro- teins, and 4 were proteins that normally reside in the endo- plasmic reticulum (ER). The presence of ER proteins in inclusions containing the primarily cytosolic hSOD1 further supports the notion that ER stress is involved in ALS. Keywords: amyotrophic lateral sclerosis, endoplasmic retic- ulum, inclusion, proteomics, superoxide dismutase-1, trans- genic mice. J. Neurochem. (2010) 114, 408–418. JOURNAL OF NEUROCHEMISTRY | 2010 | 114 | 408–418 doi: 10.1111/j.1471-4159.2010.06753.x 408 Journal Compilation Ó 2010 International Society for Neurochemistry, J. Neurochem. (2010) 114, 408–418 Ó 2010 The Authors
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Superoxide dismutase-1 and other proteins in inclusions from transgenic amyotrophic lateral...

*Department of Medical Biosciences, Clinical Chemistry, Umea University, Umea, Sweden
�Department of Pathology, Umea University, Umea, Sweden
�Department of Forest Genetics and Plant Physiology, Swedish University of Agricultural Sciences, Umea, Sweden
§Department of Pharmacology and Clinical Neuroscience, Umea University, Umea, Sweden
Amyotrophic lateral sclerosis (ALS) is a devastating neuro-degenerative disease characterized by loss of motor neuronsin the motor cortex, brainstem, and spinal cord. This resultsin progressive muscular atrophy, and the patients usuallysuccumb to respiratory failure within a few years. About10% of ALS cases appear in families (Haverkamp et al.1995), and in some of these the disease is linked to mutationsin the gene of the ubiquitously expressed antioxidant enzymeCuZn-superoxide dismutase (SOD1) (Rosen et al. 1993).
Overall, about 6% of all cases with ALS show SOD1mutations, and more than 140 such mutations have beenidentified [(Andersen et al. 2003); http://alsod.iop.kcl.ac.uk/Als/Summary/summary.aspx]. The mutations confer a cyto-toxic gain of function of unknown character to the enzyme(Gurney et al. 1994; Andersen et al. 1995). There is nowconsiderable evidence to suggest that the demise of motorneurons is non-cell autonomous and dependent on noxiouseffects of mutant SOD1s in several cell types in themotor areas(Boillee et al. 2006; Nagai et al. 2007; Henkel et al. 2009).
A hallmark of ALS caused by mutant SOD1s, both inpatients and transgenic models, is inclusions that areimmunoreactive for SOD1 (Bruijn et al. 1997; Johnstonet al. 2000; Jonsson et al. 2004, 2006b). In the animalmodels, inclusions appear concomitantly with the onset ofsymptoms and become markedly more abundant in theterminal phase of the disease (Johnston et al. 2000; Wanget al. 2002b; Jonsson et al. 2004, 2006a). They are present in
Received December 21, 2009; revised manuscript received March 13,2010; accepted April 12, 2010.Address correspondence and reprint requests to Stefan L. Marklund,
Department of Medical Biosciences, Clinical Chemistry, UmeaUniversity, SE-901 85 Umea, Sweden.E-mail: [email protected] used: ALS, amyotrophic lateral sclerosis; ER, endo-
plasmic reticulum; hSOD1, human SOD1; LC-ESI, Liquid chromato-graphy-electrospray ionization; MALDI-TOF, Matrix-assisted laserdesorption ionization time of flight; MS, Mass spectrometry; NP-40,Nonidet P-40; SOD1, superoxide dismutase-1.
Abstract
Mutant superoxide dismutase-1 (SOD1) causes amyotrophic
lateral sclerosis (ALS) through a cytotoxic mechanism of un-
known nature. A hallmark in ALS patients and transgenic
mouse models carrying human SOD1 (hSOD1) mutations are
hSOD1-immunoreactive inclusions in spinal cord ventral
horns. The hSOD1 inclusions may block essential cellular
functions or cause toxicity through sequestering of other
proteins. Inclusions from four different transgenic mouse
models were examined after density gradient ultracentrifuga-
tion. The inclusions are complex structures with heteroge-
neous densities and are disrupted by detergents. The
aggregated hSOD1 was mainly composed of subunits that
lacked the native stabilizing intra-subunit disulfide bond. A
proportion of subunits formed hSOD1 oligomers or was bound
to other proteins through disulfide bonds. Dense inclusions
could be isolated and the protein composition was analyzed
using proteomic techniques. Mutant hSOD1 accounted for
half of the protein. Ten other proteins were identified. Two
were cytoplasmic chaperones, four were cytoskeletal pro-
teins, and 4 were proteins that normally reside in the endo-
plasmic reticulum (ER). The presence of ER proteins in
inclusions containing the primarily cytosolic hSOD1 further
supports the notion that ER stress is involved in ALS.
Keywords: amyotrophic lateral sclerosis, endoplasmic retic-
ulum, inclusion, proteomics, superoxide dismutase-1, trans-
genic mice.
J. Neurochem. (2010) 114, 408–418.
JOURNAL OF NEUROCHEMISTRY | 2010 | 114 | 408–418 doi: 10.1111/j.1471-4159.2010.06753.x
408 Journal Compilation � 2010 International Society for Neurochemistry, J. Neurochem. (2010) 114, 408–418� 2010 The Authors

motor neurons, astrocytes, and the neuropil in affected areasof the CNS (Bruijn et al. 1997; Watanabe et al. 2001;Jonsson et al. 2004), but they have also been observed insome peripheral tissues (Jonsson et al. 2008). The SOD1inclusions might conceivably cause cytotoxicity by blockingessential cellular functions and/or by entrapment of otherproteins which might become inactivated, depleted orerroneously activated. Alternatively they are merely markersof failing protein quality control and clearance in terminallyinjured cells. Studies of these cellular entities have so farbeen limited to histochemical techniques. To shed furtherlight on these issues, we report the isolation and character-ization of inclusions from four transgenic mouse models. Thebasic separation was by density gradient ultracentrifugationusing a protocol developed for separation of organelles(Bergemalm et al. 2006).
Material and methods
Transgenic miceThe G127insTGGG (G127X) human SOD1 (hSOD1) and D90A
mouse models were developed in our laboratory and backcrossed
with C57/BL6BomTac mice for more than 25 generations (Jonsson
et al. 2004, 2006b). The strain expressing G85R mutant hSOD1 was
obtained from Dr D. W. Cleveland and was similarly backcrossed
for > 15 generations (Bruijn et al. 1997). The strain expressing the
G93A mutant (Gurney et al. 1994) was obtained from the Jackson
Laboratory (Bar Harbor, ME, USA) and was backcrossed with C57/
BL6BomTac for > 20 generations. Mice were regarded as terminal
when they were no longer able to reach for food. This happened at
216, 447, 370, and 135 days of age for G127X, D90A, G85R and
G93A mice respectively. As controls, 200 days old C57/BL6Bom-
Tac (Taconic Europe, Bomholt, Denmark) or non-transgenic
littermates from the breeding program were used.
Homogenization of tissue and separation of inclusionsMice were killed by intraperitoneal injection of pentobarbital. The
thorax was cut open and the animal perfused through the left
ventricle with 20 mL 0.15 M NaCl. The spinal cord was dissected
by cutting the vertebral column at the base of the scull and just
above the hip bone. A syringe was inserted at the lower opening and
the spinal cord was flushed out with 0.15 M NaCl. The spinal cord
was then gently homogenized in a Dounce glass-pestle homogenizer
with 12 + 2 strokes using loose-fit and tight-fit pestles respectively,
at 4�C in 25 volumes of mitochondrial buffer [0.21 M mannitol,
0.11 M sucrose, 10 mM K HEPES (pH 7.2), 1 mM EGTA, and the
antiproteolytic cocktail Complete� without EDTA (Roche Diag-
nostics, Basel, Switzerland)]. The homogenate was passed through
a 5-lm nylon mesh by centrifugation at 20 g at 4�C for 10 min and
was then separated by density-gradient ultracentrifugation as
described previously (Bergemalm et al. 2006). Briefly, the filtrates
were supplied with iodixanol (OptiPrep; Axis-Shield PoC AS, Oslo,
Norway) to a final concentration of 8%. The filtrates (1–1.5 mL)
were loaded on top of continuous 9-mL 12–38% iodixanol gradients
in mitochondrial buffer and centrifuged at 100 000 g for 90 min,
at 4�C in a swinging-bucket rotor (45 Ti; Beckman-Coulter,
Fullerton, CA, USA). The tubes were punctured with a cannula and
tapped from the bottom in 500-lL portions and, if not analyzed
immediately, were stored at )80�C. The localization of organelles
was determined by marker protein assays as previously
described (Bergemalm et al. 2006). For further information, see
Appendix S1.
Filter trapping of hSOD1 aggregates and inclusionsFor trapping of hSOD1 aggregates, a dot blot apparatus with 0.15-
lm cellulose acetate filters (Schleicher & Schuell, Dassel,
Germany) was used, essentially as described by Wang et al.(Wang et al. 2002a). In the gradients from the G93A and D90A
models, substantial amounts of hSOD1 are present inside mito-
chondria (Bergemalm et al. 2006). To prevent trapping of such
hSOD1, organelles were disrupted by addition of 0.5% Nonidet
P-40 (NP-40) to the fractions. Following washing with the
mitochondrial buffer, the dots were punched out and extracted in
sodium dodecyl sulfate–polyacrylamide gel electrophoresis sample
buffer.
For capture of intact inclusions present in pooled dense fractions
(fractions 1–4) from terminal G85R and G127X mice, the procedure
was carried out without addition of detergent. The punched-out dots
were extracted in DeStreak 2-D rehydration buffer (GE Healthcare,
Uppsala, Sweden).
Immunocapture and protease susceptibility of inclusionsA dense fraction (fraction 5) from G85R and G127X mice was
incubated for 1 h at 22�C with three different anti-hSOD1 antibodies
(to residues 24–39, 100–115, and 131–153) immobilized on CNBr-
Sepharose gel beads. The beads were washed thrice and the final
pellet was incubated in sample buffer for immunoblotting.
For analysis of proteinase susceptibility, fraction 5 was incubated
with proteinase K (50 or 10 lg/mL) at 23�C for 30 min with or
without the addition of 0.5% NP-40. The reaction was stopped with
40 mM phenylmethylsulphonylflouride.
Reduced and non-reduced immunoblots, in-gel reduction, andquantificationThe immunoblots were generally carried out using a human-specific
antibody raised against a peptide corresponding to amino acids
24–39 of the human SOD1 sequence (Jonsson et al. 2004). Thesodium dodecyl sulfate–polyacrylamide gel electrophoresis step
was run both with and without (non-reduced) the reductant
mercaptoethanol in the sample buffer. In the cases non-reduced
immunoblots were run, 40 mM iodoacetamide was added to the
‘mitochondrial buffer’ used for homogenization to alkylate cystein
thiols. Human SOD1 subunits lacking the intrasubunit C57–C146
disulfide bond show much higher immunoreactivity in the blots
than subunits with the bond intact (Zetterstrom et al. 2007). Toachieve more equal reactivity for all hSOD1 species (subunits with
and without the C57–C146 bond, or disulfide-linked to form
hSOD1 polymers or complexes with other proteins), after electro-
phoresis non-reducing gels were immersed for 10 min at 23�C in
transfer buffer containing 2% mercaptoethanol prior to electroblot-
ting onto polyvinylidene difluoride membranes (in-gel reduction).
The chemiluminescence of the blots was recorded in a ChemiDoc
apparatus and analyzed with Quantity One software (Bio-Rad
Laboratories, Hercules, CA, USA).
� 2010 The AuthorsJournal Compilation � 2010 International Society for Neurochemistry, J. Neurochem. (2010) 114, 408–418
SOD1 inclusions in ALS transgenic mice | 409

Two-dimensional electrophoresis and protein identificationFiltered-trapped inclusions (fraction 1–4 pools) from pools of 2–3
mice were run on 11-cm 3-11NL IPG strips (GE Healthcare).
Analytic 2-D gels were stained with silver stain and preparative with
Coomassie Brilliant Blue G-250. Transgene-specific proteins were
identified manually on the 2-D gels and the proteins identified by
Matrix-assisted laser desorption ionization time of flight (MALDI-
TOF) Mass spectrometry (MS) as described previously (Bergemalm
et al. 2009). For further details, see Appendix S1.
Liquid chromatography-electrospray ionization (LC-ESI) MS/MSand data analysisFilter-trapped inclusions (a pool of fractions 1–4) from control (C57/
BL6BomTac), G85R and G127X mice were incubated at 55�C for
60 min in 0.1 M Tris, pH. 9.0, containing 10 mM dithiothreitol
and 6 M guanidine hydrochloride. Iodoacetamide was then added
to a final concentration of 55 mM, followed by incubation at 37�Cfor 30 min in the dark. The reactionmixture (without cellulose acetate
membranes) was filtered through Microcon YM-10 filters (Millipore,
Billerica, MA, USA) and retained material was washed twice with
0.2 M NH4HCO3. Trypsin was added at an enzyme-to-substrate ratio
of 1 : 50 and digestion was carried out overnight at 37�C on the top of
the Microcon YM-10 filter in the presence of 0.05 M NH4HCO3
[Correction added after online publication: 26/05/10: 7�C changed to
37�C]. The resulting peptides were collected by centrifugation,
lyophilized, and resuspended in 0.1% formic acid. Peptide identifi-
cationwas performed as previously described (Srivastava et al. 2009).For further details, see Appendix S1.
Results
Characterization of the hSOD1-containing inclusionsInclusions immunoreactive for hSOD1 are found in motorneurons, glial cells, and the neuropil of both ALS patientsand transgenic model mice that carry mutant hSOD1s(Figure S1). To preserve the integrity of the inclusions, weused a protocol developed for organelle separation by densitygradient ultracentrifugation (Bergemalm et al. 2006). Thehomogenization is gentle, no sonication is used, and there isno pelleting by centrifugation. Detergents were avoided, asthey disrupt the inclusions (see below) and might also releaseproteins bound to hSOD1 through hydrophobic interactions.The typical density gradient and the localization of markersfor organelles is shown in Fig. 1a. Both pre-symptomatic andterminal G93A, D90A, G85R, and G127X transgenic micewere examined. The G127X mice have very low levels of themutant hSOD1, and virtually no mutant protein entered thegradient when pre-symptomatic mice were examined(Fig. 1b), as shown previously (Bergemalm et al. 2006). Incontrast, a significant proportion of the hSOD1 in spinal cordhomogenates from pre-symptomatic G93A and D90A miceentered the gradient (Fig. 1c, Figure S2). These mice havevery high levels of mutant hSOD1 (Jonsson et al. 2006b).
The two identical subunits in hSOD1 contain four cysteins(C6, C57, C111 and C146), two of which (C57 and C146)
form a stabilizing intrasubunit disulfide bond. With G93Amice, virtually all of the hSOD1 that entered the gradientcarried the C57–C146 disulfide bond, as determined by non-reducing immunoblots (Fig. 1c, upper and middle panels).The distribution of this disulfide-oxidized hSOD1 closelyfollowed the distribution of the mitochondrial marker, and itprobably represents the hSOD1 previously shown to artifi-cially overload mitochondria in these models (Fig. 1c)(Bergemalm et al. 2006). There was no evidence for thepresence of significant amounts of hSOD1 aggregates in pre-symptomatic G93A mice, as examined by filter-trap analysisin the presence of NP-40 (Fig. 1c, lower panel).
In terminally ill mice from all the transgenic models, muchmore mutant hSOD1 entered the gradient (Figs 1d, 2 and3a–f) and hSOD1 was now captured by filter-trapping,showing that the protein was aggregated.
The density of proteins is around 1.37 (Schachman 1959).Simple protein aggregates should thus accumulate at thebottom of the ultracentrifugation tube (c.f. gradient inFig. 1a). In the present transgenic models, the inclusionswere distributed all over the gradient and there was almost nohSOD1 in fraction 1 (i.e. at the bottom) (Figs 1d, 2a and3a–f). One possible explanation could be that the inclusions/aggregates are small and therefore have not yet reached theirequilibrium positions in the density gradient after 90 min at100 000 g. Prolongation of the centrifugation to 360 min,however, did not significantly alter the position of theinclusions (Fig. 2a). In contrast, addition of the detergentNP-40 to the spinal cord homogenate prior to the centrifu-gation caused a major shift of hSOD1 towards the bottom ofthe tube as expected for protein aggregates (Fig. 2b). Weconclude from this that the inclusions are not simple proteinaggregates but more complex structures.
Next, attempts were made to capture hSOD1 from denseinclusions derived from G127X and G85R mice (fraction 5,Fig. 1d) using anti-hSOD1 peptide antibodies immobilizedon Sepharose. We used three different antibodies to increasethe likelihood of capturing corresponding reactive epitopesexposed in the aggregates. In the absence of detergent,virtually no hSOD1 was captured (Fig. 2c). With the additionof 1% NP-40, the capture of hSOD1 increased markedly.This indicates that the aggregates of hSOD1 in inclusionsmay be covered by a coating of other proteins or othercellular components. Exposure to proteinase K, however,resulted in near complete degradation of the hSOD1 both inthe presence and absence of NP-40 (Fig. 2d).
The nature of hSOD1 in inclusionsStructural disulfide bonds are rare in proteins in the stronglyreducing cytosol. The C57–C146 intrasubunit disulfide bondis a potential weak spot in hSOD1, and significant fractionslacking the linkage have been found previously in transgenicmodels (Jonsson et al. 2006a; Zetterstrom et al. 2007). The
Journal Compilation � 2010 International Society for Neurochemistry, J. Neurochem. (2010) 114, 408–418� 2010 The Authors
410 | D. Bergemalm et al.

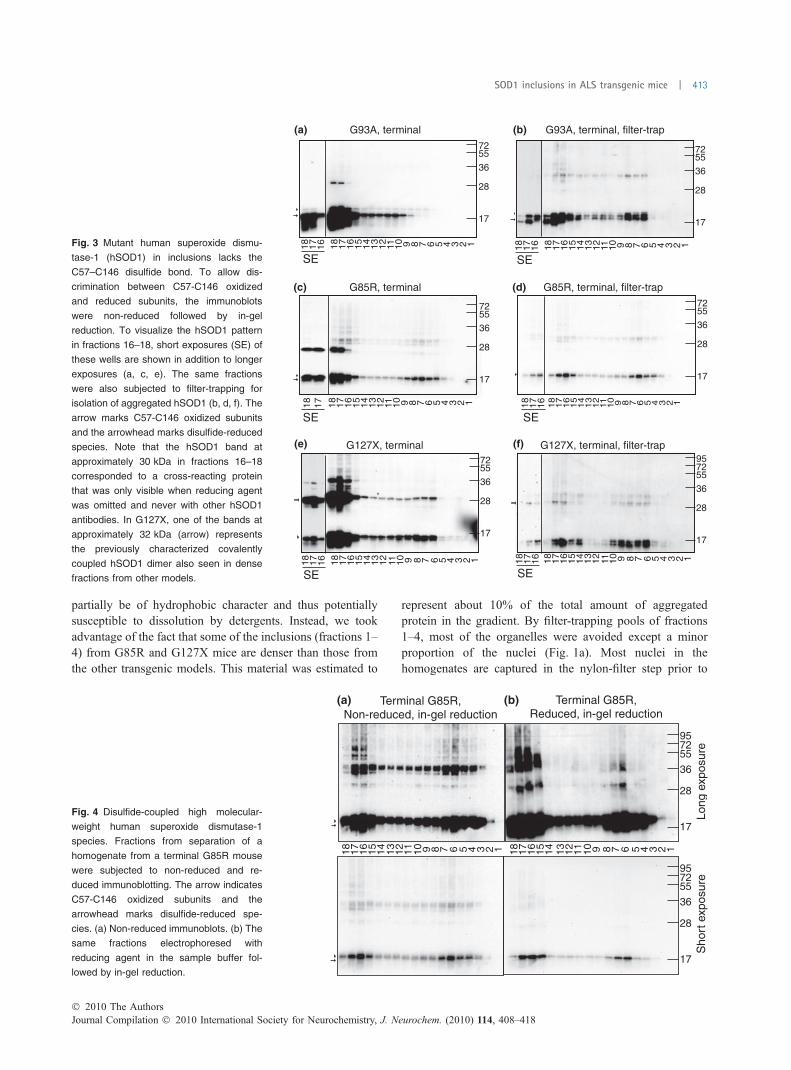
resulting loss of stability leads to increased sampling ofdisordered conformations and such hSOD1s should have anincreased propensity to aggregate (Zetterstrom et al. 2007;Chattopadhyay et al. 2008; Furukawa et al. 2008; Karchet al. 2009). We therefore studied the status of the disulfidebond in hSOD1 from our separations. Fig. 3a shows a non-reduced, in-gel reduced immunoblot of ultracentrifugationfractions from a terminal G93A mouse. Most of the hSOD1remained in the top fractions before the density gradient. Thelargest proportion of hSOD1 in these fractions, as well as inthe minor portion entering the gradient, carries the nativeC57–C146 disulfide bond. Human SOD1 lacking the bond ismuch less abundant, and has previously been estimated toaccount for 8% of the total in this model (Jonsson et al.2006a). Even in terminally ill G93A mice, aggregated
hSOD1 represents only a small subfraction (< 10%) of thetotal hSOD1 in spinal cords (Jonsson et al. 2006a; Zetter-strom et al. 2007). To specifically study aggregated hSOD1,the fractions were filter-trapped in a dot-blot apparatus. Thedetergent NP-40 was added to the fractions to solubilizeorganelles and prevent capture of any soluble hSOD1 inthem. From the terminal G93A mice, all the aggregatedhSOD1 that had entered the gradient was found to lack theC57–C146 disulfide bond (Fig. 3b). Some hSOD1 with thebond oxidized was found in fractions that remained on top ofthe gradient (fractions 17–18). These fractions contained theabundant sticky lipid materials present in CNS homogenatesthat were not completely solubilized by the weak NP-40detergent. A band with mobility intermediate between that ofreduced hSOD1 and C57–C146 disulfide-oxidized hSOD1
Presymptomatic G93A
0
10
20
30
40
50
0
10
20
30
40
50
Tot
al a
mou
nt (
%)
Tot
al a
mou
nt (
%)
Tot
al a
mou
nt (
%)
Tot
al a
mou
nt (
%)
Fraction
hSOD1
Presymptomatic G127X
18 16 14 12 10 8 6 4 20
5
10
15
20
25
0
5
10
15
20
25
Fraction
G127X G85R
Terminal G127X and G85R
Non
-red
uced
IBG
93A
Filt
er-t
rap
IBG
93A
18 17 16 15 14 13 12 11 10 9 8 7 6 5 1 2 3 418 17 16
SE
17
28
36
5572
18 16 14 12 10 8 6 4 2
18 16 14 12 10 8 6 4 2 18 16 14 12 10 8 6 4 2
0
10
20
30
40
50
0
10
20
30
40
50
Fraction
hSOD1 (Oxidized) hSOD1 (Reduced) Mitochondria
0
10
20
30
40
50
1.06
1.08
1.10
1.12
1.14
1.16
1.18
1.20
1.22
Fraction
Nuclei Mitochondria Cytosol Density
Den
sity
(g/
mL)
Control mouse(a) (b)
(c) (d)
Fig. 1 Density-gradient separations. Homogenates of spinal cords
from different mutant human superoxide dismutase-1 (hSOD1)
transgenic mouse models were subjected to density-gradient sepa-
rations. Mutant hSOD1 was detected by immunoblotting (IB). For
detection of mutant hSOD1 aggregates, fractions were filter-trapped
in a dot-blot apparatus. (a) Distribution of nuclei, cytosol, and mito-
chondria in the density gradient located by marker proteins (hnRNP,
LD, SDH, and respectively). (b) Distribution of hSOD1 in a pre-
symptomatic (100 days) low hSOD1-level model (G127X). (c) Dis-
tribution of C57-C146 disulfide-reduced and oxidized mutant hSOD1
in a high hSOD1-level model (G93A) at a pre-symptomatic stage
(70 days) (upper panel). The middle panel shows the non-reduced
western blot. Arrow marks oxidized and arrowhead marks C57–C146
disulfide-reduced species. SE = short exposure of the blot. Note that
the weak band at approximately 30 kDa in fractions 17–18 was a
cross-reacting protein, only visible when reducing agent was omitted
and never with other anti-hSOD1 antibodies. Distribution of aggre-
gated hSOD1 as revealed by filter-trapping (lower panel). (d) Distri-
bution of hSOD1 in terminal low hSOD1-level models (G85R,
G127X).
� 2010 The AuthorsJournal Compilation � 2010 International Society for Neurochemistry, J. Neurochem. (2010) 114, 408–418
SOD1 inclusions in ALS transgenic mice | 411

also appeared. This hSOD1 apparently carries a non-nativedisulfide bond and has previously been observed in solubleform in the G93A model (Zetterstrom et al. 2007).
The G85R and G127X mice contain ‡ 20-fold less hSOD1in the spinal cord than G93Amice (Jonsson et al. 2006a). Theaggregates in terminally ill mice of these models accountedfor about 50% of the total hSOD1, and the patterns of totalhSOD1 and filter-trapped hSOD1 were much more similar(Fig. 3c and d). In G127X hSOD1, there is no native disulfidebond owing to the C-terminal truncation, and only traces ofhSOD1 carrying the C57–C146 bond were seen in the G85Rmodel (Fig. 3c). Accordingly, the filter-trapped hSOD1 isdisulfide-reduced in these mice (Fig. 3d and f). In the G85Raggregates, we did not detect any band corresponding to theputative non-natively disulfide-coupled hSOD1 subunits ofthe G93A model (Fig. 3d). A band with increased mobility,which disappeared in common reduced immunoblots was,however, seen in non-reduced in-gel reduced fractions fromG127X mice (Fig. 3f). This species with higher mobility isprobably a subunit with a non-native disulfide bond.
From terminal mice belonging to all models, there werealso hSOD1 bands of lower mobility than SOD1 subunits(Fig. 4). In reduced immunoblots of hSOD1 aggregates,most of the protein appeared at the position of disulfide-reduced subunits (Fig. 4, right panels). The low-mobilitybands thus mainly correspond to hSOD1 that is disulfide-linked to form hSOD1 oligomers and complexes with otherproteins. We estimate that such disulfide-coupled hSOD1represents £ 30% of the aggregated hSOD1.
In immunoblots for detection of G127X hSOD1, a highmolecular weight band is present at roughly the position onewould estimate for a dimer (33 kDa) (Fig. 5a, arrow). This isa common theme among mutant hSOD1 variants (Jonssonet al. 2006a) and has recently been reported to represent acovalently coupled hSOD1 dimer in G127X mice (Berge-malm et al. 2009). On 2-D immunoblots (which are reducingin the first dimension) of spinal cords from terminally illG127X mice, in addition to monomers and dimers, spotswith higher molecular weights were present (Fig. 5b, arrow-heads). These represent ubiquinated hSOD1 (Jonsson et al.2004). We extended this finding by running 2-D westernblots of dense inclusions (fractions 1–4) from G85R mice;we also examined fraction 18 in the top of the gradient fordetection of hSOD1 and ubiquitin (Fig. 5c). In the inclu-sions, trains of faint spots coinciding with hSOD1 andubiquitin were seen, in addition to hSOD1 dimers. No otherubiquitinated proteins were detected. In fraction 18, noubiquinated hSOD1 was detected but there was weakimmunostaining of some other ubiquitinated proteins.
Other proteins in inclusionsFor this study, detergents were avoided as they disrupt theinclusions (Fig. 2b and c). Furthermore, the associationbetween hSOD1 aggregates and other proteins might
0
10
20
30
40
50(a)
(b)
(c)
(d)
0
10
20
30
40
50
Fraction
90 min
360 min 90 min 5x
360 min 5x
Effect of centrifugation time
18 16 14 12 10 8 6 4 2
hS
OD
1 (%
of
tota
l am
ou
nt)
18 16 14 12 10 8 6 4 20
10
20
30
40
50
0
10
20
30
40
50
hS
OD
1 (%
of
tota
l am
ou
nt)
Fraction
No det.
+NP40 No det. 5x
+NP40 5x
Effect of detergent
G85R
G127X
Fr. 5
Filtrat
e
Ab. be
ads
Fr. 5
Filtrat
e
Ab. be
ads
+NP-40
ProtK 0 50 10 50 10 NP-40 – – – + +
G85R
Fig. 2 Characterization of human superoxide dismutase-1 (hSOD1)
inclusions. (a) Homogenate of a spinal cord from a terminal G127X
mutant hSOD1 transgenic mouse was subjected to density-gradient
ultracentrifugation, for 90 min or 360 min. All fractions were subjected
to immunoblotting for hSOD1. To better visualize the hSOD1 pattern in
the gradient, the concentrations were multiplied by 5 (5·). (b) To study
the effect of detergent on hSOD1 inclusions, a G127X spinal cord
homogenate was split in two and 0.5% Nonidet P-40 (NP-40) was
added to one of the samples prior to density-gradient separation. After
density-gradient separation as before, all fractions were subjected to
immunoblotting for hSOD1. To better visualize the hSOD1 pattern in
the gradient, the concentrations were multiplied by 5 (5·). (c) Dense
inclusions (fraction 5) from terminal G127X and G85R mice were
incubated with anti-hSOD1 antibodies immobilized on Sepharose
beads with or without the addition of the non-ionic detergent NP-40.
The beads were captured by filtration on a 5-lm nylon filter and then
incubated in sample buffer for immunoblotting. (d) Dense inclusions
(fraction 5) were subjected to treatment with two different concentra-
tions of proteinase K (50 or 10 lg/mL), with or without the addition of
non-ionic detergent (0.5% NP-40). Remaining hSOD1 was analyzed
by immunoblotting.
Journal Compilation � 2010 International Society for Neurochemistry, J. Neurochem. (2010) 114, 408–418� 2010 The Authors
412 | D. Bergemalm et al.
partially be of hydrophobic character and thus potentiallysusceptible to dissolution by detergents. Instead, we tookadvantage of the fact that some of the inclusions (fractions 1–4) from G85R and G127X mice are denser than those fromthe other transgenic models. This material was estimated to
represent about 10% of the total amount of aggregatedprotein in the gradient. By filter-trapping pools of fractions1–4, most of the organelles were avoided except a minorproportion of the nuclei (Fig. 1a). Most nuclei in thehomogenates are captured in the nylon-filter step prior to
G93A, terminal
18 17 16 15 14 13 12 11 10 9 8 7 6 5 1 2 3 418 17 16
SE
18 17 16 15 14 13 12 11 10 9 8 7 6 5 1 2 3 418 17 16
SE
SE
18
17
16
15
14
13
12
11 10
9 8 7 6 5 1 2 3 418 17 16
SE
G93A, terminal, filter-trap
G85R, terminal G85R, terminal, filter-trap
18 17 16 15 14 13 12 11 10 9 8 7 6 5 1 2 3 418 17
18 17 16 15 14 13 12 11 10 9 8 7 6 5 1 2 3 418 17 16
SE
18 17 16 15 14 13 12 11 10 9 8 7 6 5 1 2 3 418 17 16
SE
G127X, terminal(e)
(c)
(a)
(f)
(d)
(b)
G127X, terminal, filter-trap
17
28
36
557295
17
28
36
5572
17
28
36
5572
17
28
36
5572
17
28
36
5572
17
28
36
5572
Fig. 3 Mutant human superoxide dismu-
tase-1 (hSOD1) in inclusions lacks the
C57–C146 disulfide bond. To allow dis-
crimination between C57-C146 oxidized
and reduced subunits, the immunoblots
were non-reduced followed by in-gel
reduction. To visualize the hSOD1 pattern
in fractions 16–18, short exposures (SE) of
these wells are shown in addition to longer
exposures (a, c, e). The same fractions
were also subjected to filter-trapping for
isolation of aggregated hSOD1 (b, d, f). The
arrow marks C57-C146 oxidized subunits
and the arrowhead marks disulfide-reduced
species. Note that the hSOD1 band at
approximately 30 kDa in fractions 16–18
corresponded to a cross-reacting protein
that was only visible when reducing agent
was omitted and never with other hSOD1
antibodies. In G127X, one of the bands at
approximately 32 kDa (arrow) represents
the previously characterized covalently
coupled hSOD1 dimer also seen in dense
fractions from other models.
17
28
36
557295
Terminal G85R,(a) (b)Non-reduced, in-gel reduction
Terminal G85R,Reduced, in-gel reduction
18 17 16 15 14 13 12 11 10 9 8 7 6 5 1 2 3 4 18 17 16 15 14 13 12 11 10 9 8 7 6 5 1 2 3 4
Long
exp
osur
e
17
28
36
557295
Sho
rt e
xpos
ure
Fig. 4 Disulfide-coupled high molecular-
weight human superoxide dismutase-1
species. Fractions from separation of a
homogenate from a terminal G85R mouse
were subjected to non-reduced and re-
duced immunoblotting. The arrow indicates
C57-C146 oxidized subunits and the
arrowhead marks disulfide-reduced spe-
cies. (a) Non-reduced immunoblots. (b) The
same fractions electrophoresed with
reducing agent in the sample buffer fol-
lowed by in-gel reduction.
� 2010 The AuthorsJournal Compilation � 2010 International Society for Neurochemistry, J. Neurochem. (2010) 114, 408–418
SOD1 inclusions in ALS transgenic mice | 413

the ultracentrifugation (Bergemalm et al. 2006). In fractions5 and upwards there was too much contamination ofmitochondria to allow delineation of proteins present in theSOD1-containing inclusions.
The captured material was then solubilized and subjected to2-D electrophoresis, and representative resulting spot patternsare shown in Fig. 6a–c. Four replicate preparations of 2-3
spinal cords from terminally ill G85R and G127X mice andC57BL/6JBomTac controls were analyzed. Some 20 spots,whichwere only seen in both transgenic strains and never in thecontrols, were picked for identification by MALDI-TOFanalysis. The major train of spots, which represented about50% of all protein, was found to be hSOD1 subunits. Ten otherproteins were identified (Table 1, Fig. 6d). Some prominent
Homog
enat
e
Frac
tion
4 Fractions 1–4
G127X
UbiquitinG85R hSOD1
(a)
(c)
(b)
Fra
ctio
n1–
4F
ract
ion
18
17
28
36
557295
130
17
28
36
557295
130
17
28
36
557295
130
17
28
36
557295
130
17
28
36
Fig. 5 Ubiquitination of human superoxide
dismutase-1 (hSOD1) in inclusions.
Reducing 2-D immunoblots of hSOD1 re-
vealed a series of spots of high molecular
weight. To explore the nature of these
entities, dense and light fractions (fractions
1–4 pooled and fraction 18) were analyzed
further. (a) The G127X SOD-1 covalent di-
mer (arrow) previously characterized. (b)
From 2-dimensional immunoblotting with
anti-hSOD1 on pooled fractions 1–4 of ter-
minal homogenates, a ladder of spots with
decreasing mobility was seen (arrow-
heads). Arrow indicates the covalent
G127X dimer. (c) To determine whether
these represented ubiquitinated hSOD1,
pooled fractions 1–4 and a fraction con-
taining mostly soluble protein (fraction 18)
from a terminal G85R mouse, were sub-
jected to 2-D immunoblotting with anti-
hSOD1 and anti-ubiquitin antibodies (arrow
marks suspected dimer).
Control
(a) (b) (c)
G85R G127X15
2025
3750
75
(d) (e)
P63017P20029
P23927
G85R SOD1
P08551P09103P09103 P46660
P20152
G85R SOD1+ Ubiq.
G85R SOD1+ Ubiq.
P08113
P14211
15
20
25
37
50
75
SOD1-dimer
Vimentin
HSC70
B6 G85R G127X
Fig. 6 2-D analysis of inclusions and vali-
dation of proteins. Filter-trapped pooled
fractions 1–4 of terminal G85R, G127X, and
control mice were subjected to 2-dimen-
sional electrophoresis. (a–c) Silver-stained
gels for analysis. (d) For identification of
proteins, spots from Coomassie-stained
gels were subjected to MALDI-TOF MS.
Identifiers are in Swissprot formatting as in
Table 1. (e) Pools of fractions 1–4 from
G85R, G127X, and C57/BL6BomTac con-
trol mice were subjected to immunoblotting
with anti-vimentin and anti-HSC70 anti-
bodies.
Journal Compilation � 2010 International Society for Neurochemistry, J. Neurochem. (2010) 114, 408–418� 2010 The Authors
414 | D. Bergemalm et al.

spots that were present in material from both controls andtransgenic mice were also analyzed and found to be cytoskel-etal and nuclear proteins (not shown). Similar preparationsfrom three transgenic and two controlmicewere also subjectedto LC-MS/MS analysis. We found one additional transgene-specific protein (tubulin-beta) and most MALDI-TOF identi-fications were replicated (Table 1).
To validate our findings with a different method, we run1-dimensional immunoblots for HSC70 and vimentin. Thestaining was only seen in extracts of pooled dense fractions(fractions 1–4) from transgenic mice (Fig. 6e). The interme-diate filament protein vimentin is primarily expressed in glialand mesenchymal cells, but is also expressed in adult motorneurons and found in inclusions from symptomatic G93Amice (Perrin et al. 2005).
Discussion
Here we report the first isolation and partial characterizationof hSOD1-containing inclusions from spinal cords of ALSmodel mice. The inclusions appear to be relatively complexdetergent-susceptible structures that equilibrate in the densitygradient at lower densities than simple protein aggregates.The cause of the reduced density is not quite clear; theprotease-susceptibility suggest that this is not explained byencapsulation by a continuous membrane. The presence ofseveral proteins from the endoplasmic reticulum (ER)suggests that ER membranes may possibly be constituentsof the inclusions (see further below).
Similar separations were recently carried out on homogen-ates of spinal ventral horns from an ALS patient carrying theG127X SOD1 mutation (Jonsson et al. 2008). It is notewor-thy that the density patterns of the inclusions were very similarto those found here for spinal cords from terminal G127Xmice. This suggests that the inclusions in the transgenicmouse models are of the same nature as those in ALS patients.
The densities of the inclusions were very heterogeneous,covering those of all organelles. This should make it virtually
impossible to separate organelles in terminal spinal cordsfrom the inclusions using differential centrifugation. Thisdifficulty is the basis of the (in our view) misconception thatthe hSOD1 aggregates are mainly associated with mitochon-dria (Liu et al. 2004; Vijayvergiya et al. 2005; Vande Veldeet al. 2008). It may also lead to erroneous conclusionsregarding the properties of hSOD1 in mitochondria. Thepresent results do not formally exclude the possibility thatsome aggregated hSOD1 actually is associated with thisorganelle, as a proportion of the inclusions cofractionate withthe mitochondrial marker. There was, however, no evidencein any of the cases for peaks in aggregated hSOD1 at thelocation of the mitochondrial marker peak (fractions 9–10);troughs or comparatively low levels were found instead. InD90A and G93A mice, the hSOD1 in the cytosol on the topof the gradient is partially disulfide-reduced (Fig. 1, middleand lower panels, Fig. 3a, Figure S2), and in these models 8–12% of the total hSOD1 has been estimated to lack the nativedisulfide bond (Jonsson et al. 2006a). In contrast, the hSOD1that co-fractionates with the mitochondrial marker appears tobe completely C57–C146 disulfide-oxidized (Fig. 1c, upperand middle panels, Fig. 3a, Figure S2). Reduced subunitsentering the intermembrane space are apparently efficientlyoxidized by copper-chaperone for SOD1 (CCS) and thedisulfide-oxidizing machinery present in this compartment(Reddehase et al. 2009). As oxidized subunits cannot leavethe intermembrane space, this is a likely explanation for theartificial hSOD1 overloading of mitochondria in the D90Aand G93A models (Bergemalm et al. 2006). The mitochon-drial hSOD1 should have a low propensity to aggregate, ashSOD1 aggregates appear to be derived mainly from reducedsubunits (see discussion below).
Human SOD1 in inclusionsWe have previously shown that in the present transgenicmurine ALS models, spinal cords are enriched with solublehydrophobic disulfide-reduced hSOD1 throughout life (Jons-son et al. 2006a; Zetterstrom et al. 2007). We found here that
Table 1 Transgene-specific proteins iden-
tified. Fractions 1–4 from terminal G127X,
G85R and C57/BL6BomTac control mice
were pooled and inclusions filter-trapped.
After solubilisation in rehydration buffer the
inclusions were separated on 2-D gels and
transgene-specific spots picked and identi-
fied using Matrix-assisted laser desorption
ionization time of flight (MALDI-TOF). Sim-
ilar preparations (filter-trapped inclusions)
from both control and transgenic mice were
also subjected to Liquid chromatography-
electrospray ionization mass spectrometry
(LC-ESI MS)
Protein name
Accession no.
(Swissprot) Compartment 2-D gels LC-ESI MS/MS
Alpha-crystallin B chain P23927 Cytoplasm X X
HSC70 P63017 Cytoplasm X X
Vimentin P20152 Cytoplasm X X
Tubulin beta-5 chain P99024 Cytoplasm – X
Neurofilament light polypeptide P08551 Cytoplasm X X
Alpha-internexin P46660 Cytoplasm X X
GRP78 P20029 ER X –
Endoplasmin (HSP90B1/GRP94) P08113 ER X –
Calreticulin (ERP60) P14211 ER X X
Protein-disulfide isomerase P09103 ER X –
ER, endoplasmic reticulum; LC-ESI, Liquid chromatography-electrospray ionization; MS, Mass
spectrometry.
� 2010 The AuthorsJournal Compilation � 2010 International Society for Neurochemistry, J. Neurochem. (2010) 114, 408–418
SOD1 inclusions in ALS transgenic mice | 415

the inclusions contained such hSOD1 in aggregated form.There was also some disulfide-coupled hSOD1 of highermolecular weight and minute amounts of ubiquitinatedsubunits. Human SOD1 has already been shown to beubiquitinated in transgenic models (Jonsson et al. 2004; Bassoet al. 2006), and herewe found that all such species are trappedin inclusions and that this modification is restricted to hSOD1.Similar to our findings, Karch et al. recently reported that thehSOD1 in detergent-resistant aggregates from transgenic miceis mainly composed of subunits lacking the native C57–C146disulfide bond (Karch et al. 2009). Thus, absence of the C57–C146 disulfide bond appears to be the fundamental prerequisitefor aggregation of hSOD1 in vivo. This conclusion is supportedby studies on hSOD1 aggregation in vitro (Chattopadhyayet al. 2008; Furukawa et al. 2008) and on various cysteinemutants of hSOD1 expressed in cultured cells (Karch et al.2009). Several studies both in vitro (Banci et al. 2007; OztugDurer et al. 2009) and in cultured cells (Niwa et al. 2007;Karch and Borchelt 2008) have reported that C6 and C111 canengage in formation of multimers and aggregates. In thepresent study and in the previous study on detergent-resistantaggregates in mice (Karch et al. 2009), the amounts ofdisulfide-coupled species were substantial but they wereprobably formed secondarily to aggregation of large amountsof hSOD1 with reduced cysteines.
Other proteins in inclusionsHuman SOD1 was found to account for about 50% of theprotein. Around 20 other protein spots were found, whichwere not detected in fractions from control mice. Out of tenthat could be identified, two were cytoplasmic chaperones,four were cytoskeletal proteins, and four were proteins thatnormally reside in the ER.
The second most abundant protein in inclusions was thesmall heat-shock protein ab-crystallin. It has previously beenfound in detergent-insoluble fractions containing mutanthSOD1 (Wang et al. 2003) and can prevent aggregation ofmutant hSOD1 in vitro (Wang et al. 2005). HSC70 has beenfound to co-immunoprecipitate with mutant hSOD1 (Shinderet al. 2001; Choi et al. 2004) and therapeutic interventionaimed at increasing the amount of heat-shock proteins hasbeen shown to increase the lifespan of an ALS transgenicmouse model (Kieran et al. 2004).
Out of the four proteins detected that normally reside inthe ER, one – calreticulin – has not been implicated in ALSbefore. Protein-disulfide isomerase and GRP78 have beenfound by immunohistochemistry in hSOD1-positive inclu-sions in transgenic mice (Tobisawa et al. 2003; Wate et al.2005; Atkin et al. 2006) and also in inclusions from sporadicALS patients (Atkin et al. 2008). Endoplasmin has beenidentified in detergent-insoluble fractions of G93A mice(Basso et al. 2009). Each of these four proteins is involved inrecognition of misfolded proteins and targeting as part of aprocess called ER-associated degradation. It is notable that
along with the cytoplasmic chaperones HSC70 and ab-crystallin, in total six key players in protein quality controlcan be found in the terminal inclusions.
In a previous study, aggregates in spinal cords fromtransgenic mice were isolated by two different approaches: aspellets collected by centrifugation following a harsh deter-gent extraction procedure and by antibody capture (Shawet al. 2008). The major aggregated protein was found to bemutant hSOD1. The only other protein found with reasonableconsistency was vimentin. In another study, symptomaticmice expressing YFP-G85R hSOD1 were compared withmice expressing YFP-wild type hSOD1 (Wang et al. 2009).Homogenates of spinal cords were washed several times withdetergent, and final pellets after centrifugation were analyzedusing a modified LC-MS protocol. Vimentin was again theonly reasonably abundant mutant-enriched protein, whichcoincided with the present findings. Glial fibrillary acidicprotein (GFAP), neurofilament-M and -H, and peripherinwere also found at higher levels in YFP-G85R hSOD1 mice.In a recent similar study, detergent-resistant pellets fromspinal cords from symptomatic G93A mice and wt-hSOD1mice were compared. 42 proteins were more abundant in theG93A mutant mice, and among them, similar to our findings,vimentin, ab-crystallin, protein-disulfide isomerase and en-doplasmin were found (Basso et al. 2009). Whereas mutantSOD1 was the major component in our present study, suchmaterial was a minor component in both studies ondetergent-resistant pellets (Basso et al. 2009; Wang et al.2009). This suggests that the pellets contain aggregatedmaterial from compromised spinal cords that is not present inSOD1 inclusions. Detergent-induced disruption of inclusionsmight also have caused loss of some proteins coaggregatedwith hSOD1. Analysis of inclusions and detergent-resistantpellets thus yields different and complementary informationon protein perturbations in spinal cords of ALS model mice.
Conclusions
The inclusions analyzed in these experiments have only beenfound in terminal transgenic mice. The obvious question iswhether this late entity is of any major importance for thepathogenic process of ALS. The protein content of theinclusions probably reflects interactions that are establishedbetween individual soluble proteins, and these have beenshown to be highly regulated (Rajan et al. 2001). As theamounts of proteins apart from hSOD1 are minute, it appearsunlikely that a major depletion of the proteins results fromentrapment in the inclusions. This study was, however, madeon the tissue level and the picture might be different inindividual cells. Given that SOD1 is primarily a cytosolicprotein, it is remarkable that 4 of the proteins identifiednormally reside in the ER. SOD1 has recently been shown tobe present in the ER and to be secreted by neural cells throughan ER-dependent mechanism (Turner et al. 2005; Kikuchi
Journal Compilation � 2010 International Society for Neurochemistry, J. Neurochem. (2010) 114, 408–418� 2010 The Authors
416 | D. Bergemalm et al.

et al. 2006; Urushitani et al. 2006). Furthermore, the unfoldedprotein response, which follows severe ER-stress, has beenshown to be activated in both transgenic mice and humanswith ALS (Atkin et al. 2006, 2008; Kikuchi et al. 2006), andinterference with ER stress has been found to prolong thelifespan of transgenic mice (Saxena et al. 2009). The presentfindings lend further support to the idea that the ER, and ERstress and unfolded protein response, are involved in ALS.
Acknowledgements
This work was supported by the Swedish Science Council, the
Swedish Brain Fund/Hallsten Fund, the Swedish Medical Society
including the Bjorklund Fund for ALS Research, the Swedish
Association of Persons with Neurological Disabilities, Vasterbotten
County Council, The Kempe Foundations, and Konung Gustaf V’s
and Drottning Victorias Foundation. We thank Eva Bern, Karin
Hjertkvist, Ann-Charlott Nilsson, Ulla-Stina Spetz, and Agneta
Oberg for technical assistance and Dr D.W. Cleveland for supplying
the G85R SOD1 transgenic mouse strain.
Supporting Information
Additional Supporting Information may be found in the onlineversion of this article:
Appendix S1. Supplementary Materials and methods.Figure S1. Human SOD1-immunoreactive inclusions.Figure S2. A spinal cord homogenate from a pre-symptom-
atic D90A (100 days) transgenic mouse was subjected todensity-gradient ultracentrifugation.
As a service to our authors and readers, this journal providessupporting information supplied by the authors. Such materialsare peer-reviewed and may be re-organized for online delivery,but are not copy-edited or typeset. Technical support issuesarising from supporting information (other than missing files)should be addressed to the authors.
References
Andersen P. M., Nilsson P., Ala-Hurula V., Keranen M. L., Tarvainen I.,Haltia T., Nilsson L., Binzer M., Forsgren L. and Marklund S. L.(1995) Amyotrophic lateral sclerosis associated with homozygosityfor an Asp90Ala mutation in CuZn-superoxide dismutase. Nat.Genet. 10, 61–66.
Andersen P. M., Sims K. B., Xin W. W. et al. (2003) Sixteen novelmutations in the Cu/Zn superoxide dismutase gene in amyotrophiclateral sclerosis: a decade of discoveries, defects and disputes.Amyotroph. Lateral Scler. Other Motor Neuron Disord. 4, 1–12.
Atkin J. D., Farg M. A., Turner B. J. et al. (2006) Induction of theunfolded protein response in familial amyotrophic lateral sclerosisand association of protein-disulfide isomerase with superoxidedismutase 1. J. Biol. Chem. 281, 30152–30165.
Atkin J. D., Farg M. A., Walker A. K., McLean C., Tomas D. and HorneM. K. (2008) Endoplasmic reticulum stress and induction of theunfolded protein response in human sporadic amyotrophic lateralsclerosis. Neurobiol. Dis. 30, 400–407.
Banci L., Bertini I., Durazo A., Girotto S., Gralla E. B., Martinelli M.,Valentine J. S., Vieru M. and Whitelegge J. P. (2007) Metal-freesuperoxide dismutase forms soluble oligomers under physiological
conditions: a possible general mechanism for familial ALS. Proc.Natl. Acad. Sci. USA 104, 11263–11267.
Basso M., Massignan T., Samengo G., Cheroni C., De B. S., SalmonaM., Bendotti C. and Bonetto V. (2006) Insoluble mutant SOD1 ispartly oligoubiquitinated in amyotrophic lateral sclerosis mice.J. Biol. Chem. 281, 33325–33335.
Basso M., Samengo G., Nardo G. et al. (2009) Characterization ofdetergent-insoluble proteins in ALS indicates a causal link betweennitrative stress and aggregation in pathogenesis.PLoSONE 4, e8130.
Bergemalm D., Jonsson P. A., Graffmo K. S., Andersen P. M., Brann-strom T., Rehnmark A. and Marklund S. L. (2006) Overloading ofstable and exclusion of unstable human superoxide dismutase-1variants in mitochondria of murine amyotrophic lateral sclerosismodels. J. Neurosci. 26, 4147–4154.
Bergemalm D., Forsberg K., Jonsson P. A., Graffmo K. S., BrannstromT., Andersen P. M., Antti H. and Marklund S. L. (2009) Changes inthe spinal cord proteome of an amyotrophic lateral sclerosis murinemodel determined by differential in-gel electrophoresis. Mol. CellProteomics 8, 1306–1317.
Boillee S., Yamanaka K., Lobsiger C. S., Copeland N. G., Jenkins N. A.,Kassiotis G., Kollias G. and Cleveland D. W. (2006) Onset andprogression in inherited ALS determined by motor neurons andmicroglia. Science 312, 1389–1392.
Bruijn L. I., Becher M. W., Lee M. K. et al. (1997) ALS-linked SOD1mutant G85R mediates damage to astrocytes and promotes rapidlyprogressive disease with SOD1-containing inclusions. Neuron 18,327–338.
Chattopadhyay M., Durazo A., Sohn S. H., Strong C. D., Gralla E. B.,Whitelegge J. P. and Valentine J. S. (2008) Initiation and elonga-tion in fibrillation of ALS-linked superoxide dismutase. Proc. NatlAcad. Sci. USA 105, 18663–18668.
Choi J. S., Cho S., Park S. G., Park B. C. and Lee D. H. (2004) Co-chaperone CHIP associates with mutant Cu/Zn-superoxidedismutase proteins linked to familial amyotrophic lateral sclerosisand promotes their degradation by proteasomes. Biochem. Biophys.Res. Commun. 321, 574–583.
Furukawa Y., Kaneko K., Yamanaka K., O’Halloran T. V. and Nukina N.(2008) Complete loss of post-translational modifications triggersfibrillar aggregation of SOD1 in familial form of ALS. J. Biol.Chem. 283, 24167–24176.
Gurney M. E., Pu H., Chiu A. Y. et al. (1994) Motor neuron degener-ation in mice that express a human Cu,Zn superoxide dismutasemutation. Science 264, 1772–1775.
Haverkamp L. J., Appel V. and Appel S. H. (1995) Natural history ofamyotrophic lateral sclerosis in a database population. Validationof a scoring system and a model for survival prediction. Brain 118,707–719.
Henkel J. S., Beers D. R., Zhao W. and Appel S. H. (2009) Microglia inALS: the good, the bad, and the resting. J. Neuroimmune Phar-macol. 4, 389–398.
Johnston J. A., Dalton M. J., Gurney M. E. and Kopito R. R. (2000)Formation of high molecular weight complexes of mutant Cu,Zn-superoxide dismutase in a mousemodel for familial amyotrophiclateral sclerosis. Proc. Natl Acad. Sci. USA 97, 12571–12576.
Jonsson P. A., Ernhill K., Andersen P. M., Bergemalm D., BrannstromT., Gredal O., Nilsson P. and Marklund S. L. (2004) Minutequantities of misfolded mutant superoxide dismutase-1 causeamyotrophic lateral sclerosis. Brain 127, 73–88.
Jonsson P. A., Graffmo K. S., Andersen P. M., Brannstrom T., LindbergM., Oliveberg M. and Marklund S. L. (2006a) Disulphide-reducedsuperoxide dismutase-1 in CNS of transgenic amyotrophic lateralsclerosis models. Brain 129, 451–464.
Jonsson P. A., Graffmo K. S., Brannstrom T., Nilsson P., Andersen P. M.and Marklund S. L. (2006b) Motor neuron disease in mice
� 2010 The AuthorsJournal Compilation � 2010 International Society for Neurochemistry, J. Neurochem. (2010) 114, 408–418
SOD1 inclusions in ALS transgenic mice | 417

expressing the wild type-like D90A mutant superoxide dismutase-1. J. Neuropathol. Exp. Neurol. 65, 1126–1136.
Jonsson P. A., Bergemalm D., Andersen P. M., Gredal O., Brannstrom T.and Marklund S. L. (2008) Inclusions of amyotrophic lateralsclerosis-linked superoxide dismutase in ventral horns, liver, andkidney. Ann. Neurol. 63, 671–675.
Karch C. M. and Borchelt D. R. (2008) A limited role for disulfide cross-linking in the aggregation of mutant SOD1 linked to familialamyotrophic lateral sclerosis. J. Biol. Chem. 283, 13528–13537.
Karch C. M., Prudencio M., Winkler D. D., Hart P. J. and Borchelt D. R.(2009) Role of mutant SOD1 disulfide oxidation and aggregationin the pathogenesis of familial ALS. Proc. Natl Acad. Sci. USA106, 7774–7779.
Kieran D., Kalmar B., Dick J. R., Riddoch-Contreras J., Burnstock G.and Greensmith L. (2004) Treatment with arimoclomol, a coin-ducer of heat shock proteins, delays disease progression in ALSmice. Nat. Med. 10, 402–405.
Kikuchi H., Almer G., Yamashita S., Guegan C., Nagai M., Xu Z.,Sosunov A. A., McKhann G. M. and Przedborski S. (2006) Spinalcord endoplasmic reticulum stress associated with a microsomalaccumulation of mutant superoxide dismutase-1 in an ALS model.Proc. Natl Acad. Sci. USA 103, 6025–6030.
Liu J., Lillo C., Jonsson P. A. et al. (2004) Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mito-chondria. Neuron 43, 5–17.
Nagai M., Re D. B., Nagata T., Chalazonitis A., Jessell T. M., WichterleH. and Przedborski S. (2007) Astrocytes expressing ALS-linkedmutated SOD1 release factors selectively toxic to motor neurons.Nat. Neurosci. 10, 615–622.
Niwa J. I., Yamada S. I., Ishigaki S., Sone J., Takahashi M., Katsuno M.,Tanaka F., Doyu M. and Sobue G. (2007) Disulfide bond mediatesaggregation, toxicity and ubiquitylation of familial amyotrophiclateral sclerosis-linked mutant SOD1. J. Biol. Chem. 282, 28087–28095.
Oztug Durer Z. A., Cohlberg J. A., Dinh P. et al. (2009) Loss of metalions, disulfide reduction and mutations related to familial ALSpromote formation of amyloid-like aggregates from superoxidedismutase. PLoS ONE 4, e5004.
Perrin F. E., Boisset G., Docquier M., Schaad O., Descombes P. andKato A. C. (2005) No widespread induction of cell death genesoccurs in pure motoneurons in an amyotrophic lateral sclerosismouse model. Hum. Mol. Genet. 14, 3309–3320.
Rajan R. S., Illing M. E., Bence N. F. and Kopito R. R. (2001) Speci-ficity in intracellular protein aggregation and inclusion body for-mation. Proc. Natl Acad. Sci. USA 98, 13060–13065.
Reddehase S., Grumbt B., Neupert W. and Hell K. (2009) The disulfiderelay system of mitochondria is required for the biogenesis ofmitochondrial Ccs1 and Sod1. J. Mol. Biol. 385, 331–338.
Rosen D. R., Siddique T., Patterson D. et al. (1993) Mutations in Cu/Znsuperoxide-dismutase gene are associated with familial amyo-trophic-lateral-sclerosis. Nature 362, 59–62.
Saxena S., Cabuy E. and Caroni P. (2009) A role for motoneuron sub-type-selective ER stress in disease manifestations of FALS mice.Nat. Neurosci. 12, 627–636.
Schachman H. K. (1959) Ultracentrifugation in biochemistry. AcademicPress, New York.
Shaw B. F., Lelie H. L., Durazo A. et al. (2008) Detergent insolubleaggregates associated with amyotrophic lateral sclerosis in trans-genic mice contain primarily full-length, unmodified superoxidedismutase-1. J. Biol. Chem. 283, 8340–8350.
Shinder G. A., Lacourse M. C., Minotti S. and Durham H. D. (2001)Mutant Cu/Zn-superoxide dismutase proteins have altered solu-bility and interact with heat shock/stress proteins in models ofamyotrophic lateral sclerosis. J. Biol. Chem. 276, 12791–12796.
Srivastava V., Srivastava M. K., Chibani K., Nilsson R., Rouhier N.,Melzer M. and Wingsle G. (2009) Alternative splicing studies ofthe reactive oxygen species gene network in Populus reveal twoisoforms of high-isoelectric-point superoxide dismutase. PlantPhysiol. 149, 1848–1859.
Tobisawa S., Hozumi Y., Arawaka S., Koyama S., Wada M., Nagai M.,Aoki M., Itoyama Y., Goto K. and Kato T. (2003) Mutant SOD1linked to familial amyotrophic lateral sclerosis, but not wild-typeSOD1, induces ER stress in COS7 cells and transgenic mice.Biochem. Biophys. Res. Commun. 303, 496–503.
Turner B. J., Atkin J. D., Farg M. A., Zang d. W., Rembach A., Lopes E.C., Patch J. D., Hill A. F. and Cheema S. S. (2005) Impairedextracellular secretion of mutant superoxide dismutase 1 associateswith neurotoxicity in familial amyotrophic lateral sclerosis.J. Neurosci. 25, 108–117.
Urushitani M., Sik A., Sakurai T., Nukina N., Takahashi R. and Julien J.P. (2006) Chromogranin-mediated secretion of mutant superoxidedismutase proteins linked to amyotrophic lateral sclerosis. Nat.Neurosci. 9, 108–118.
Vande Velde C., Miller T. M., Cashman N. R. and Cleveland D. W.(2008) Selective association of misfolded ALS-linked mutantSOD1 with the cytoplasmic face of mitochondria. Proc. Natl Acad.Sci. USA 105, 4022–4027.
Vijayvergiya C., Beal M. F., Buck J. and Manfredi G. (2005) Mutantsuperoxide dismutase 1 forms aggregates in the brain mitochon-drial matrix of amyotrophic lateral sclerosis mice. J. Neurosci. 25,2463–2470.
Wang J., Xu G. and Borchelt D. R. (2002a) High molecular weightcomplexes of mutant superoxide dismutase 1: age-dependent andtissue-specific accumulation. Neurobiol. Dis. 9, 139–148.
Wang J., Xu G., Gonzales V., Coonfield M., Fromholt D., Copeland N.G., Jenkins N. A. and Borchelt D. R. (2002b) Fibrillar inclusionsand motor neuron degeneration in transgenic mice expressingsuperoxide dismutase 1 with a disrupted copper-binding site.Neurobiol. Dis. 10, 128–138.
Wang J., Slunt H., Gonzales V., Fromholt D., Coonfield M., Copeland N.G., Jenkins N. A. and Borchelt D. R. (2003) Copper-binding-site-null SOD1 causes ALS in transgenic mice: aggregates of non-native SOD1 delineate a common feature. Hum. Mol. Genet. 12,2753–2764.
Wang J., Xu G., Li H., Gonzales V., Fromholt D., Karch C., Copeland N.G., Jenkins N. A. and Borchelt D. R. (2005) Somatodendriticaccumulation of misfolded SOD1-L126Z in motor neurons medi-ates degeneration: alphaB-crystallin modulates aggregation. Hum.Mol. Genet. 14, 2335–2347.
Wang J., Farr G. W., Zeiss C. J. et al. (2009) Progressive aggregationdespite chaperone associations of a mutant SOD1-YFP in trans-genic mice that develop ALS. Proc. Natl Acad. Sci. USA 106,1392–1397.
Watanabe M., Dykes-Hoberg M., Culotta V. C., Price D. L., Wong P. C.and Rothstein J. D. (2001) Histological evidence of proteinaggregation in mutant SOD1 transgenic mice and in amyotrophiclateral sclerosis neural tissues. Neurobiol. Dis. 8, 933–941.
Wate R., Ito H., Zhang J. H., Ohnishi S., Nakano S. and Kusaka H.(2005) Expression of an endoplasmic reticulum-resident chaper-one, glucose-regulated stress protein 78, in the spinal cord of amouse model of amyotrophic lateral sclerosis. Acta Neuropathol.110, 557–562.
Zetterstrom P., Stewart H. G., Bergemalm D., Jonsson P. A., GraffmoK. S., Andersen P. M., Brannstrom T., Oliveberg M. and MarklundS. L. (2007) Soluble misfolded subfractions of mutant superoxidedismutase-1s are enriched in spinal cords throughout life in murineALS models. Proc. Natl Acad. Sci. USA 104, 14157–14162.
Journal Compilation � 2010 International Society for Neurochemistry, J. Neurochem. (2010) 114, 408–418� 2010 The Authors
418 | D. Bergemalm et al.




















