
Structure–activity relationships for thiol reactivity and rat or human hepatocyte toxicity induced...
13
Copyright © 2007 John Wiley & Sons, Ltd. JOURNAL OF APPLIED TOXICOLOGY J. Appl. Toxicol. 2008; 28: 608–620 Published online 2 November 2007 in Wiley InterScience (www.interscience.wiley.com) DOI: 10.1002/jat.1312 Structure–activity relationships for thiol reactivity and rat or human hepatocyte toxicity induced by substituted p-benzoquinone compounds Katie Chan, 1, * Neil Jensen 2 and Peter J. O’Brien 1 1 University of Toronto, Faculty of Pharmacy, Toronto, ON, Canada M5S 2S2 2 Celsis-In Vitro Technologies, 1450 South Rolling Road, Baltimore, MD 21227, USA Received 3 August 2007; Revised 7 September 2007; Accepted 13 September 2007 ABSTRACT: Covalent binding of toxic chemicals to cellular targets is a molecular interaction that initiates a wide array of adverse biological effects. The creation of a covalent bond can be cited as a key initiating step along many toxi- city pathways which must be predicted in order to predict the potential of a chemical to cause specific harmful effects. Currently, quantitative structure-activity relationship (QSAR) models are being improved by focusing on endpoints such as simple electrophile reactivity for covalent interactions rather than on commonly used complex toxicity endpoints. The cytotoxicity and electrophilic reactivity of 10 p-substituted benzoquinone derivatives, which are well known electrophilic alkylating agents, were investigated under the premise that QSAR toxicity models can be improved when the molecular triggering event is considered. Hepatocyte toxicity was determined by incubation of individual compounds with freshly isolated rat or cryopreserved human hepatocyte suspensions. The potential for chemical reactivity between a chemical and cellular target was measured by determining non-enzymic reactivity with glutathione, representing thiol nucleophiles. The decline in free thiol moieties was measured to characterize the electrophile reactivity. It was found that the degree of rat hepatotoxicity induced by benzoquinones correlated with the rate at which they non-enzymically react with glutathione and to various global and atomic electronic frontier orbital parameters which described electrophilicity. Human hepatocytes showed similar results but the statistical significance was much lower. The QSAR expressions suggest that covalent binding reactivity serves as a good correlate to hepatotoxicity and could improve QSAR modeling for potential toxicity risks. Copyright © 2007 John Wiley & Sons, Ltd. KEY WORDS: hepatocytes; QSAR; covalent binding; glutathione; cytotoxicity; benzoquinones; cryopreserved human hepatocytes; chemical reactivity * Correspondence to: Katie Chan, University of Toronto, Faculty of Phar- macy, Toronto, ON, Canada M5S 2S2. E-mail: [email protected] for biological activity. Modeling of adverse outcomes in the past has been a challenge because chemicals from all different structural classes cause similar biological effects, while also having multiple pathways that lead to these same events. A new approach has been initiated to improve the strategy behind QSAR modeling. This in- volves a shift from modeling complex toxicity endpoints, such as organ toxicity, skin irritation/sensitization, mutagenicity and carcinogenicity, to a central event that is common to these various toxicological events. Protein covalent binding, which is the adduction of reactive metabolites to critical proteins, is considered to be a key initiating step that can be used to predict the potential for a chemical to cause specific harmful effects (Schultz et al., 2006). For example, liver toxicity induced by many xenobiotics can be attributed to their electrophilic metabolites reacting with critical cellular nucleophiles (proteins and DNA), forming an irreversible covalent bond that can lead to adverse events such as cell death or hypersensitivity (Liebler and Guengerich, 2005). QSAR modeling therefore has been shifted to focus on the event of covalent binding and the limited chemical reactivities Introduction Quinones are a ubiquitous class of compounds which are common constituents in plants and biologically relevant molecules (Bolton et al., 2000). Quinone moieties are also present in many drugs and can be formed as metabolites from a variety of xenobiotics (Verma, 2006). They repre- sent structural features which have the potential to create a variety of hazardous effects in vivo, as they are highly reactive electrophiles and can bind covalently to critical proteins and DNA causing toxicity (Rossi et al., 1986; Tapper et al., 2000; van Ommen et al., 1988). Quantitative Structure-Activity Relationship (QSAR) modeling is a tool used in toxicity assessments that attempts to correlate biological activity with structural features of molecules. This is based on the general premise that molecular properties that are characteristic of all active compounds must in some way be essential
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Structure–activity relationships for thiol reactivity and rat or human hepatocyte toxicity induced...

608 K. CHAN ET AL.
Copyright © 2007 John Wiley & Sons, Ltd. J. Appl. Toxicol. 2008; 28: 608–620
DOI: 10.1002/jat
JOURNAL OF APPLIED TOXICOLOGYJ. Appl. Toxicol. 2008; 28: 608–620Published online 2 November 2007 in Wiley InterScience(www.interscience.wiley.com) DOI: 10.1002/jat.1312
Structure–activity relationships for thiol reactivity andrat or human hepatocyte toxicity induced by substitutedp-benzoquinone compounds
Katie Chan,1,* Neil Jensen2 and Peter J. O’Brien1
1 University of Toronto, Faculty of Pharmacy, Toronto, ON, Canada M5S 2S22 Celsis-In Vitro Technologies, 1450 South Rolling Road, Baltimore, MD 21227, USA
Received 3 August 2007; Revised 7 September 2007; Accepted 13 September 2007
ABSTRACT: Covalent binding of toxic chemicals to cellular targets is a molecular interaction that initiates a wide
array of adverse biological effects. The creation of a covalent bond can be cited as a key initiating step along many toxi-
city pathways which must be predicted in order to predict the potential of a chemical to cause specific harmful effects.
Currently, quantitative structure-activity relationship (QSAR) models are being improved by focusing on endpoints such
as simple electrophile reactivity for covalent interactions rather than on commonly used complex toxicity endpoints. The
cytotoxicity and electrophilic reactivity of 10 p-substituted benzoquinone derivatives, which are well known electrophilic
alkylating agents, were investigated under the premise that QSAR toxicity models can be improved when the molecular
triggering event is considered. Hepatocyte toxicity was determined by incubation of individual compounds with freshly
isolated rat or cryopreserved human hepatocyte suspensions. The potential for chemical reactivity between a chemical and
cellular target was measured by determining non-enzymic reactivity with glutathione, representing thiol nucleophiles.
The decline in free thiol moieties was measured to characterize the electrophile reactivity. It was found that the degree
of rat hepatotoxicity induced by benzoquinones correlated with the rate at which they non-enzymically react with
glutathione and to various global and atomic electronic frontier orbital parameters which described electrophilicity.
Human hepatocytes showed similar results but the statistical significance was much lower. The QSAR expressions
suggest that covalent binding reactivity serves as a good correlate to hepatotoxicity and could improve QSAR modeling
for potential toxicity risks. Copyright © 2007 John Wiley & Sons, Ltd.
KEY WORDS: hepatocytes; QSAR; covalent binding; glutathione; cytotoxicity; benzoquinones; cryopreserved human hepatocytes;
chemical reactivity
* Correspondence to: Katie Chan, University of Toronto, Faculty of Phar-
macy, Toronto, ON, Canada M5S 2S2.
E-mail: [email protected]
for biological activity. Modeling of adverse outcomes in
the past has been a challenge because chemicals from
all different structural classes cause similar biological
effects, while also having multiple pathways that lead
to these same events. A new approach has been initiated
to improve the strategy behind QSAR modeling. This in-
volves a shift from modeling complex toxicity endpoints,
such as organ toxicity, skin irritation/sensitization,
mutagenicity and carcinogenicity, to a central event that
is common to these various toxicological events. Protein
covalent binding, which is the adduction of reactive
metabolites to critical proteins, is considered to be a key
initiating step that can be used to predict the potential
for a chemical to cause specific harmful effects (Schultz
et al., 2006). For example, liver toxicity induced by many
xenobiotics can be attributed to their electrophilic
metabolites reacting with critical cellular nucleophiles
(proteins and DNA), forming an irreversible covalent
bond that can lead to adverse events such as cell death or
hypersensitivity (Liebler and Guengerich, 2005). QSAR
modeling therefore has been shifted to focus on the event
of covalent binding and the limited chemical reactivities
Introduction
Quinones are a ubiquitous class of compounds which are
common constituents in plants and biologically relevant
molecules (Bolton et al., 2000). Quinone moieties are also
present in many drugs and can be formed as metabolites
from a variety of xenobiotics (Verma, 2006). They repre-
sent structural features which have the potential to create
a variety of hazardous effects in vivo, as they are highly
reactive electrophiles and can bind covalently to critical
proteins and DNA causing toxicity (Rossi et al., 1986;
Tapper et al., 2000; van Ommen et al., 1988).
Quantitative Structure-Activity Relationship (QSAR)
modeling is a tool used in toxicity assessments that
attempts to correlate biological activity with structural
features of molecules. This is based on the general
premise that molecular properties that are characteristic of
all active compounds must in some way be essential

STRUCTURE-ACTIVITY RELATIONSHIPS FOR THIOL REACTIVITY 609
Copyright © 2007 John Wiley & Sons, Ltd. J. Appl. Toxicol. 2008; 28: 608–620
DOI: 10.1002/jat
Table 1. Structure of p-benzoquinone derivatives
Substituents at position: C′p-Benzoquinone Abbreviation 2 3 5 6
1 chloroanil -Cl4 Cl Cl Cl Cl
2 2,5-dichloro-benzoquinone -2,5-Cl2 Cl H Cl H
3 2-bromo-benzoquinone -2Br Br H H H
4 2-tert butyl-benzoquinone -2(CH3)4 (CH3)4 H H H
5 2-methyl-benzoquinone -2CH3 CH3 H H H
6 p-benzoquinone -none H H H H
7 2,6-dimethyl-benzoquinone -2,6-(CH3)2 CH3 H H CH3
8 2,6-dimethoxy-benzoquinone -2,6-(OCH3)2 OCH3 H H OCH3
9 2,3,5-trimethyl-benzoquinone -2,3,5-(CH3)3 CH3 CH3 CH3 H
10 2,3,5,6-tetramethyl-benzoquinone -2,3,5,6-(CH3)4 CH3 CH3 CH3 CH3
that drive this event (Aptula et al., 2005; Aptula and
Roberts, 2006). A method for modeling the reactivity of
electrophiles has been developed using glutathione (GSH)
as a surrogate for soft nucleophiles. Schultz et al. pro-
posed that this model will simulate the relative rates at
which a reactive chemical is likely to bind protein targets
and provide a measure of the inherent risk of reactivity
(Schultz et al., 2005). Using this method in combination
with QSAR modeling (to identify structural indicators of
electrophilic reactivity), is proposed as an alternative high
throughput approach to determining the potential toxicity
of chemicals.
This study investigated the cytotoxicity and electrophi-
lic reactivity of ten p-benzoquinone derivatives (Table 1).
Cytotoxicity was determined in both rat and human
hepatocytes. Electrophilic reactivity was measured using a
non-enzymic thiol assay based on GSH. Structure-activity
relationship models were derived to determine the physico-
chemical parameters that underlie p-benzoquinone alkylat-
ing activities and toxicity. The goal was to determine the
key structural features which impact reactivity and whether
intrinsic reactivity and potential for covalent binding can
serve as a risk indicator for hepatotoxicity. This may play a
significant role in improving future toxicological evaluation.
Materials and Methods
Chemicals
All chemicals were of the highest commercial grade avail-
able. Chloranil, 2,5-dichlorobenzoquinone, 2-tert-butyl
benzoquinone, 2-methylbenzoquinone, benzoquinone,
2,6-dimethylbenzoquinone, 2,6-dimethoxybenzoquinone,
2,3,5,6-tetramethylbenzoquinone, trypan blue, 3-[4,5,-
dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide
(MTT) and 5,5′-dithio-bis-(2-nitrobenzoic acid) (DTNB)
were purchased from Sigma Aldrich Co. Canada Ltd
(Oakville, ON, Canada).
2,3,5-(CH3)3-benzoquinone and 2-bromobenzoquinone
were prepared as described by Rossi et al. (1986). Briefly,
their respective quinols (5% w/v) were oxidized with
Ag2O (5 eq) in dry diethyl ether until complete, followed
by crystallization of the quinone from ether onto fresh
dry Ag2O under a slow stream of helium gas. Quinones
were then sublimed three times to yield brilliant yellow
crystals.
Hepatocytes
Male Sprague-Dawley rats weighing 275–300 g (Charles
River Canada Laboratories, Montreal, QC, Canada) were
housed in ventilated plastic cages over PWI 8-16 hard-
wood bedding. There were 12 air changes per hour, 12 h
light photoperiod (lights on at 0800 h) and an environ-
mental temperature of 21–23 °C with a 50–60% relative
humidity. The animals were fed with a normal standard
chow diet and tap water ad libitum. Fresh hepatocytes
were isolated from rats by collagenase perfusion of the
liver according to the method described by Moldeus and
coworkers (Moldeus et al., 1978). Cells (106 cells ml−1)
(10 ml) were suspended in Krebs-Henseleit buffer (pH 7.4)
containing 12.5 mM HEPES in continually rotating 50 ml

610 K. CHAN ET AL.
Copyright © 2007 John Wiley & Sons, Ltd. J. Appl. Toxicol. 2008; 28: 608–620
DOI: 10.1002/jat
round-bottomed flasks and maintained under an atmo-
sphere of 95% O2 and 5% CO2 in a water bath of 37 °C
throughout the entire experiment. The hepatocytes used
were at least 85% viable immediately after isolation. Fresh
rat hepatocytes were prepared before each experiment.
Cryopreserved human male hepatocytes were obtained
from Celsis-In Vitro Technologies. Briefly, cryopreserved
cells were thawed at 37 °C, and suspended in pre-warmed
(37 °C) InVitroGRO™ CP medium (Celsis-In Vitro Tech-
nologies, Baltimore, MD). The cell suspension was mixed
gently, centrifuged at 50 g for 5 min at 4 °C, and the
supernatant discarded. The cells were then resuspended in
InVitroGRO™ HI medium (Celsis-In Vitro Technologies,
Baltimore, MD). Hepatocytes (0.5 × 106 viable cells ml−1)
(0.2 ml) were suspended in 48-well plates, under 37 °C
and 5% CO2, on a rocking apparatus. Limited to avail-
ability, human hepatocytes used in these experiments
were from one donor (lot. ZAG, see www.celsis.com for
details). The viability of human hepatocytes post-thaw
was 77% ± 2% (viability before cryopreservation, 85%).
An advantage of using one donor in these experiments
was that it avoided variation between cell batches from
different humans. Such variation occurs to a lesser extent
in laboratory cross-bred rats. Both freshly isolated rat and
cryopreserved male hepatocytes were treated with various
doses of p-benzoquinone compounds and incubated for
up to 3 h.
Cell Viability
The viability of freshly isolated hepatocytes was deter-
mined by the trypan blue (0.4% w/v) exclusion test
(Chan et al., 2007). The viability of cryopreserved
hepatocytes was assessed by adding 0.02 ml of a solution
of 3-[4,5,-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium
bromide (MTT, 5 mg ml−1 in H2O) to each well. The
plates were returned to the incubator for another 3 h.
After the incubation 0.04 N acidified isopropanol (0.2 ml)
was added per well to dissolve the MTT formazan. The
absorbance of MTT formazan was measured at 572 nm
and 690 nm. The corrected absorbance was determined
by subtracting the 690 nm value from the 572 nm value
as a background correction. Cytotoxicity was defined
by LC50, the concentration of a chemical (μM) added
to hepatocytes which caused 50% of cells to be dead at
2 h. The results were expressed as the mean ± SEM from
three separate experiments.
Non-enzymatic Glutathione Reactivity (ThiolReactivity)
The non-enzymatic reaction of various electrophiles
with glutathione (GSH) was assayed using DTNB (5,5′-dithio-bis-(2-nitrobenzoic acid)). Stock solutions of GSH
were made fresh in 0.1 M potassium phosphate buffer
containing 1 mM EDTA to prevent oxidation. Stock solu-
tions of electrophile compounds were prepared in DMSO
at 100× their desired concentrations. All reactions took place
at 25 °C in 0.1 M potassium phosphate buffer (pH 8.0).
The final volume of the reaction mixture was 10 ml, to
which 1 ml of GSH stock solution was added with 0.1 ml
of electrophile stock solution. After a given reaction time
ranging from 10 to 180 min, 0.1 ml of a DTNB stock
solution (0.01 g ml−1) dissolved in 1% sodium citrate,
was added to the reaction. The absorbance at 412 nm was
read immediately with a Pharmacia Biotech Ultraspec
1000 spectrophotometer. The optical density of the
chromophore corresponds to the free thiol concentrations
of the solution, i.e. unreacted GSH. To determine the
reactivity of compounds with GSH, pseudo first-order
kinetics was assumed for the decrease of GSH concentra-
tion, with electrophile added in excess. From the loss
of GSH concentration over time, the rate of reactivity
of electrophiles toward GSH, kGSH, was calculated using
the following equations derived by Freidig et al. (1999).
kGSH,obs = (ln CGSH,0 − ln CGSH,t)/t (1)
kGSH = kGSH,obs/CEl,0 (2)
where, CGSH,0 is the initial concentration of GSH, CGSH,t is
the final concentration of GSH, t is the reaction incu-
bation time, and CEl,0 is the electrophile concentration.
All reaction rates were measured with a control (with
GSH) and a blank (without GSH). No decrease of GSH
was found in control incubations which were spiked with
DMSO only. Negative controls (run without GSH) were
performed to make sure test compounds did not interfere
with absorbance of DTNB-conjugates. The reaction rates
were expressed as the mean ± SEM from three separate
experiments.
Quantitative-Structure Activity Relationships(QSAR) and Calculation of PhysicochemicalParameters
Molecular structures of all p-benzoquinone derivatives
were prepared using CAChe molecular modeling soft-
ware (Version 6.1.12 for Windows 2000/XP, (2000–2003)
Fujitsu Ltd). The physico-chemical parameters used
were restricted to those hypothesized to be associated
with the general cytotoxic and reactivity mechanism
for p-benzoquinones and calculated using the CAChe
MOPAC quantum mechanics application. ELUMO (energy
of the lowest unoccupied molecular orbital), EHOMO
(energy of the highest molecular orbital), dipole moment
(μ), and electron density properties (nucleophilic frontier
density (NufD) and LUMO) for each carbon atom on
the aromatic ring, were calculated using the MOPAC PM5
STRUCTURE-ACTIVITY RELATIONSHIPS FOR THIOL REACTIVITY 611
Copyright © 2007 John Wiley & Sons, Ltd. J. Appl. Toxicol. 2008; 28: 608–620
DOI: 10.1002/jat
semi-empirical Hamiltonian algorithm. Log P and mole-
cular refractivity (MR) were also calculated by CAChe,
using an atom typing scheme of Ghose and Crippen
(Ghose et al., 1988). SPARC On Line Calculator v3.1,
(Sparc OnLine Calculator. http://ibmlc2.chem.uga.edu/
sparc [Jan 2007]) was used to calculate one electron
reduction potentials (E1/2) of the molecules. Statistical
calculations of correlations between physiochemical
parameters and biochemical data were carried out by
multiple linear regression analysis using Sigma Stat
(V2.03, 1992–1997 SPSS Inc.).
Results
Hepatocyte Cytotoxicity
Concentrations of a series of ten p-benzoquinone con-
geners required to cause a 50% decrease in hepatocyte
cell viability (LC50 μM) in 2 h were determined and are
given in Table 2. The viability of control (untreated)
hepatocytes remained constant for both rat and human
hepatocytes during the 2 h incubation and up to as long
as 3 h incubation. At longer times the viability for both
cells in suspension began to decline. The cytotoxic effec-
tiveness of p-benzoquinone congeners towards rat hepato-
cytes ranged from a LC50 of 18 μM to 800 μM, resulting
in a 44-fold difference between the most and least cyto-
toxic p-benzoquinone. Chloranil was the most cytotoxic
benzoquinone and 2,3,5-6-tetramethyl benzoquinone, was
the least toxic. In human hepatocytes, the cytotoxic effec-
tiveness ranged from 80 μM to 1600 μM, resulting in a 20-
fold difference between the most and least cytotoxic
p-benzoquinone. The most cytotoxic p-benzoquinone in
human hepatocytes was 2,6-dimethylbenzoquinone, while
halobenzoquinones were the most cytotoxic towards rat
hepatocytes. The order of cytotoxicity towards rat hepa-
tocytes was: 2,3,5,6-Cl4 > 2,5-Cl2 > 2Br > 2-tbutyl > 2CH3
> BQ > 2,6-(CH3)2 > 2,6-(OCH3)2 > 2,3,5-(CH3)3 > 2,3,5,6-
(CH3)4. The order of cytotoxicity in human hepatocytes,
differed from what was seen with rat hepatocytes, and
was: 2,6-(CH3)2 > 2,6-(OCH3)2 > 2CH3 > 2,5-Cl2 > BQ >2-tbutyl > -Cl4 > 2,3,5-(CH3)3 > 2Br > 2,3,5,6-(CH3)4.
Quantitative Structure Activity Relationships(QSAR) and Cytotoxicity
Rat Hepatocytes
Multiple linear regressions (MLR) analysis was used to
derive structure activity relationships using the cytotoxi-
city data in Table 2 and the physico-chemical descriptors
calculated in Table 3. The derived QSAR models are
Table 2. p-Benzoquinone hepatocyte cytotoxicity and non-enzymic glutathione reactivity
Rat hepatocyte Human hepatocyte Non-enzymictoxicity toxicity glutathione reactivity
p-Benzoquinone LC50 (2 h) μM log LC50 LC50 (2 h) μM log LC50 kGSH [M−1 min−1] log kGSH
1 chloroanil 18 ± 2 1.26 200 ± 20 2.30 4116 ± 440 3.61
2 2,5-dichloro-benzoquinone 23 ± 2 1.36 140 ± 15 2.15 6052 ± 600 3.78
3 2-bromo-benzoquinone 31 ± 5 1.49 500 ± 45 2.70 1435 ± 180 3.16
4 2-tert butyl-benzoquinone 32 ± 3 1.51 160 ± 20 2.20 846 ± 80 2.93
5 2-methyl-benzoquinone 46 ± 5 1.66 117 ± 13 2.07 2423 ± 225 3.38
6 p-benzoquinone 57 ± 6 1.76 143 ± 15 2.16 1712 ± 148 3.23
7 2,6-dimethyl-benzoquinone 80.5 ± 7 1.91 83 ± 10 1.92 1601 ± 177 3.20
8 2,6-dimethoxy-benzoquinone 81.5 ± 8 1.91 100 ± 10 2.00 696 ± 70 2.84
9 2,3,5-trimethyl-benzoquinone 300 ± 35 2.48 233 ± 25 2.37 376 ± 45 2.58
10 2,3,5,6-tetramethyl-benzoquinone 800 ± 77 2.90 1600 ± 100 3.20 113 ± 11 2.05
Table 3. Calculated physico-chemical parameters for p-benzoquinones
ELUMO EHOMO E1/2 Dipole Molecularp-Benzoquinone (eV) (eV) log P (eV) moment (μ) refractivity (MR)
1 chloroanil −2.832 −9.841 2.527 −0.394 0.004 47.00
2 2,5-dichloro-benzoquinone −2.624 −10.289 1.491 −0.23 0.004 37.39
3 2-bromo-benzoquinone −2.46 −10.65 1.247 −0.169 0.682 35.40
4 2-tert butyl-benzoquinone −2.124 −10.702 2.082 −1.177 1.079 46.44
5 2-methyl-benzoquinone −2.204 −10.787 0.922 −1.046 0.749 32.82
6 p-benzoquinone −2.286 −10.921 0.455 −0.984 0.006 27.78
7 2,6-dimethyl-benzoquinone −2.126 −10.727 1.389 −1.099 1.185 37.86
8 2,6-dimethoxy-benzoquinone −2.122 −10.233 −0.05 −0.873 0.322 40.70
9 2,3,5-trimethyl-benzoquinone −2.049 −10.278 1.86 −1.143 0.671 42.90
10 2,3,5,6-tetramethyl-benzoquinone −1.976 −10.253 2.324 −1.182 0.001 47.94
612 K. CHAN ET AL.
Copyright © 2007 John Wiley & Sons, Ltd. J. Appl. Toxicol. 2008; 28: 608–620
DOI: 10.1002/jat
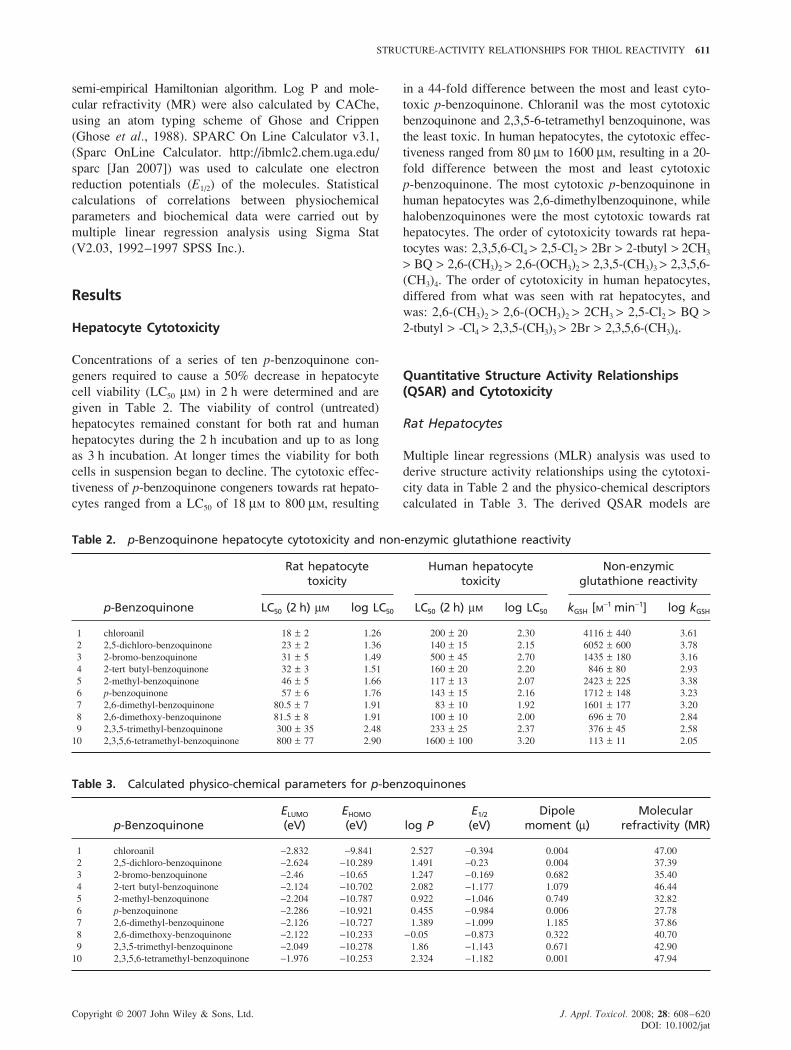
Figure 1. Plot of log LC50 cytotoxicity values for substituted p-benzoquinones in rat hepatocytes against their ELUMO
values. The line represents Eqn. (1) log LC50 = 24.54 + 17.7 ELUMO + 3.36 (ELUMO)2, n = 9, r2 = 0.90, s = 0.04, F = 25.9,P = 0.002, (�) correlated data; (�) outlier
reported in Table 5. In QSAR studies chemical congeners
contain common templates, which in this study is a rigid
aromatic ring with para substituted carbonyl groups
(Table 1). These carbonyl groups are important as they
identify this benzoquinone group into a specific reaction
mechanism mode. p-Benzoquinones to a greater or lesser
extent act as Michael acceptor electrophiles depending
on the substituents present. Since quinones are known
reactive Michael acceptors, they were categorized under
this electrophilic reaction mechanism domain. Structure-
activity trends were evaluated within this mechanism
domain as well as by structural similarity. The variation
between each derivative is limited to the substituents
present on the 2,3,5 and 6 carbons of the aromatic ring
template (Table 1). Rat hepatocyte cytotoxicity for this
group of p-benzoquinones correlated best to the energy of
the lowest unoccupied molecular orbital (ELUMO) (Fig. 1).
Outliers were determined as data points that had residuals
exceeding three sigma (Hall et al., 2003; Huuskonen,
2001; Lipnick, 1991). Removal of 2-tert butyl benzoquin-
one as an outlier resulted in the derived parabolic equation:
log LC50 = 24.54 + 17.7 ELUMO + 3.36 (ELUMO)2;
n = 9, r2 = 0.90, P = 0.002 (1)
ELUMO serves as a global reactivity parameter which
measures the readiness of a molecule to accept electrons,
i.e. electrophilicity. The derived parabolic line of best fit
suggests that at first cytotoxicity increases as the values
of ELUMO become more negative, but then at a specific
point cytotoxicity turns around and decreases. An ELUMO
value of −2.63 eV is where the inversion point occurs,
which theoretically is the most cytotoxic p-benzoquinone.
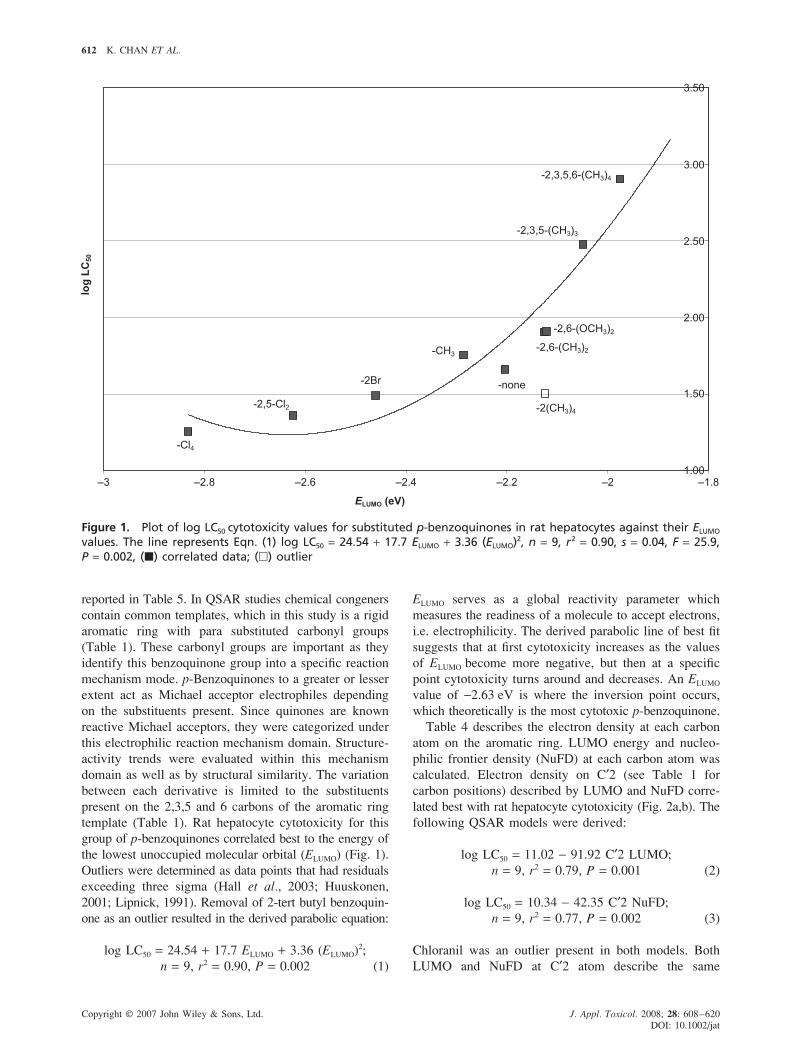
Table 4 describes the electron density at each carbon
atom on the aromatic ring. LUMO energy and nucleo-
philic frontier density (NuFD) at each carbon atom was
calculated. Electron density on C′2 (see Table 1 for
carbon positions) described by LUMO and NuFD corre-
lated best with rat hepatocyte cytotoxicity (Fig. 2a,b). The
following QSAR models were derived:
log LC50 = 11.02 − 91.92 C′2 LUMO;
n = 9, r2 = 0.79, P = 0.001 (2)
log LC50 = 10.34 − 42.35 C′2 NuFD;
n = 9, r2 = 0.77, P = 0.002 (3)
Chloranil was an outlier present in both models. Both
LUMO and NuFD at C′2 atom describe the same
STRUCTURE-ACTIVITY RELATIONSHIPS FOR THIOL REACTIVITY 613
Copyright © 2007 John Wiley & Sons, Ltd. J. Appl. Toxicol. 2008; 28: 608–620
DOI: 10.1002/jat
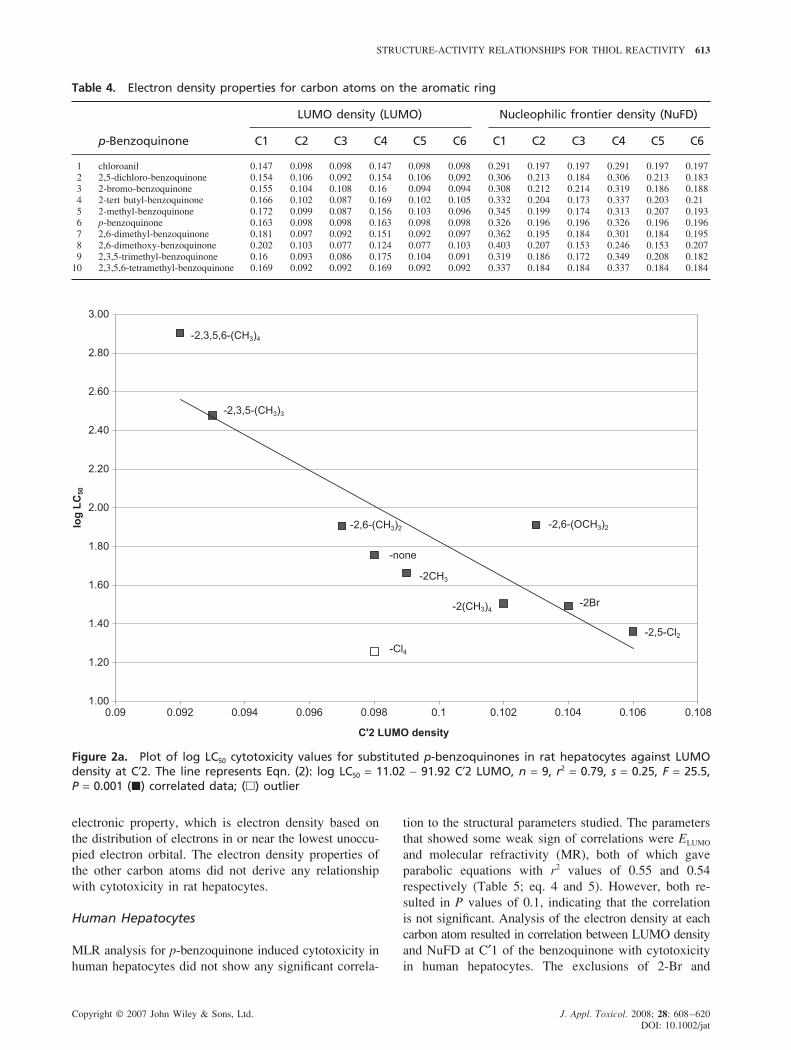
Table 4. Electron density properties for carbon atoms on the aromatic ring
LUMO density (LUMO) Nucleophilic frontier density (NuFD)
p-Benzoquinone C1 C2 C3 C4 C5 C6 C1 C2 C3 C4 C5 C6
1 chloroanil 0.147 0.098 0.098 0.147 0.098 0.098 0.291 0.197 0.197 0.291 0.197 0.197
2 2,5-dichloro-benzoquinone 0.154 0.106 0.092 0.154 0.106 0.092 0.306 0.213 0.184 0.306 0.213 0.183
3 2-bromo-benzoquinone 0.155 0.104 0.108 0.16 0.094 0.094 0.308 0.212 0.214 0.319 0.186 0.1884 2-tert butyl-benzoquinone 0.166 0.102 0.087 0.169 0.102 0.105 0.332 0.204 0.173 0.337 0.203 0.21
5 2-methyl-benzoquinone 0.172 0.099 0.087 0.156 0.103 0.096 0.345 0.199 0.174 0.313 0.207 0.193
6 p-benzoquinone 0.163 0.098 0.098 0.163 0.098 0.098 0.326 0.196 0.196 0.326 0.196 0.1967 2,6-dimethyl-benzoquinone 0.181 0.097 0.092 0.151 0.092 0.097 0.362 0.195 0.184 0.301 0.184 0.195
8 2,6-dimethoxy-benzoquinone 0.202 0.103 0.077 0.124 0.077 0.103 0.403 0.207 0.153 0.246 0.153 0.207
9 2,3,5-trimethyl-benzoquinone 0.16 0.093 0.086 0.175 0.104 0.091 0.319 0.186 0.172 0.349 0.208 0.18210 2,3,5,6-tetramethyl-benzoquinone 0.169 0.092 0.092 0.169 0.092 0.092 0.337 0.184 0.184 0.337 0.184 0.184
electronic property, which is electron density based on
the distribution of electrons in or near the lowest unoccu-
pied electron orbital. The electron density properties of
the other carbon atoms did not derive any relationship
with cytotoxicity in rat hepatocytes.
Human Hepatocytes
MLR analysis for p-benzoquinone induced cytotoxicity in
human hepatocytes did not show any significant correla-
tion to the structural parameters studied. The parameters
that showed some weak sign of correlations were ELUMO
and molecular refractivity (MR), both of which gave
parabolic equations with r2 values of 0.55 and 0.54
respectively (Table 5; eq. 4 and 5). However, both re-
sulted in P values of 0.1, indicating that the correlation
is not significant. Analysis of the electron density at each
carbon atom resulted in correlation between LUMO density
and NuFD at C′1 of the benzoquinone with cytotoxicity
in human hepatocytes. The exclusions of 2-Br and
Figure 2a. Plot of log LC50 cytotoxicity values for substituted p-benzoquinones in rat hepatocytes against LUMOdensity at C′2. The line represents Eqn. (2): log LC50 = 11.02 − 91.92 C′2 LUMO, n = 9, r2 = 0.79, s = 0.25, F = 25.5,P = 0.001 (�) correlated data; (�) outlier
614 K. CHAN ET AL.
Copyright © 2007 John Wiley & Sons, Ltd. J. Appl. Toxicol. 2008; 28: 608–620
DOI: 10.1002/jat
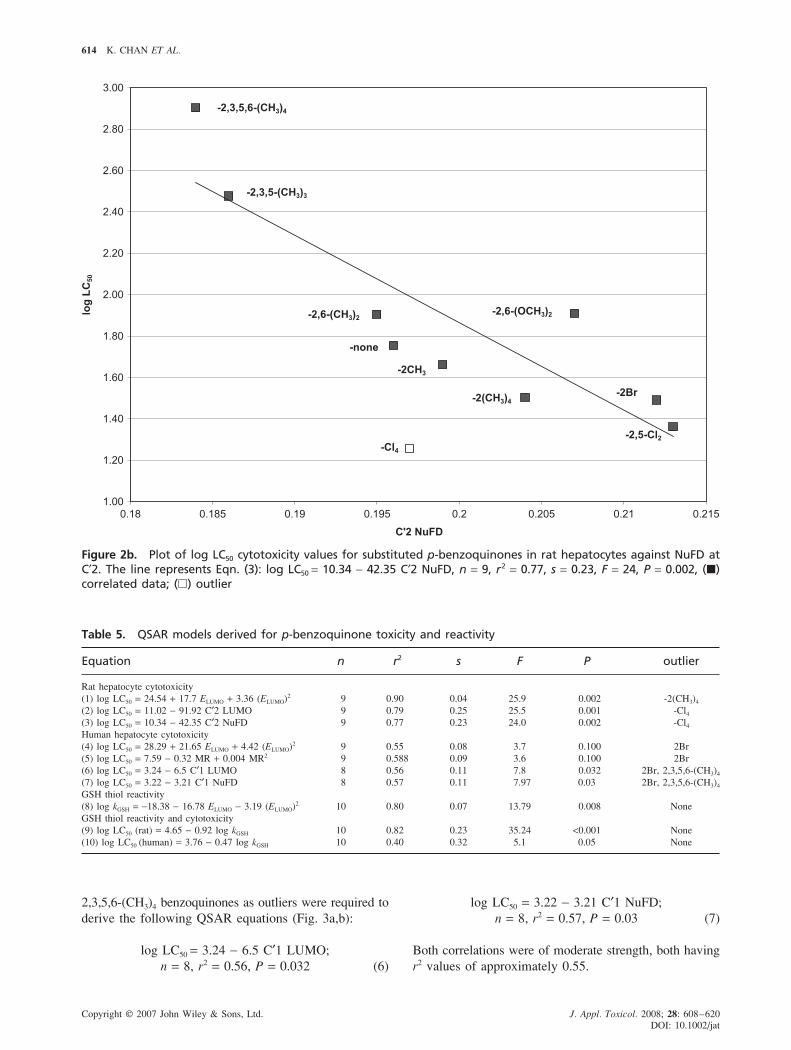
log LC50 = 3.22 − 3.21 C′1 NuFD;
n = 8, r2 = 0.57, P = 0.03 (7)
Both correlations were of moderate strength, both having
r2 values of approximately 0.55.
2,3,5,6-(CH3)4 benzoquinones as outliers were required to
derive the following QSAR equations (Fig. 3a,b):
log LC50 = 3.24 − 6.5 C′1 LUMO;
n = 8, r2 = 0.56, P = 0.032 (6)
Figure 2b. Plot of log LC50 cytotoxicity values for substituted p-benzoquinones in rat hepatocytes against NuFD atC′2. The line represents Eqn. (3): log LC50 = 10.34 − 42.35 C′2 NuFD, n = 9, r2 = 0.77, s = 0.23, F = 24, P = 0.002, (�)correlated data; (�) outlier
Table 5. QSAR models derived for p-benzoquinone toxicity and reactivity
Equation n r2 s F P outlier
Rat hepatocyte cytotoxicity
(1) log LC50 = 24.54 + 17.7 ELUMO + 3.36 (ELUMO)2 9 0.90 0.04 25.9 0.002 -2(CH3)4
(2) log LC50 = 11.02 − 91.92 C′2 LUMO 9 0.79 0.25 25.5 0.001 -Cl4
(3) log LC50 = 10.34 − 42.35 C′2 NuFD 9 0.77 0.23 24.0 0.002 -Cl4
Human hepatocyte cytotoxicity
(4) log LC50 = 28.29 + 21.65 ELUMO + 4.42 (ELUMO)2 9 0.55 0.08 3.7 0.100 2Br
(5) log LC50 = 7.59 − 0.32 MR + 0.004 MR2 9 0.588 0.09 3.6 0.100 2Br
(6) log LC50 = 3.24 − 6.5 C′1 LUMO 8 0.56 0.11 7.8 0.032 2Br, 2,3,5,6-(CH3)4
(7) log LC50 = 3.22 − 3.21 C′1 NuFD 8 0.57 0.11 7.97 0.03 2Br, 2,3,5,6-(CH3)4
GSH thiol reactivity
(8) log kGSH = −18.38 − 16.78 ELUMO − 3.19 (ELUMO)2 10 0.80 0.07 13.79 0.008 None
GSH thiol reactivity and cytotoxicity
(9) log LC50 (rat) = 4.65 − 0.92 log kGSH 10 0.82 0.23 35.24 <0.001 None
(10) log LC50 (human) = 3.76 − 0.47 log kGSH 10 0.40 0.32 5.1 0.05 None
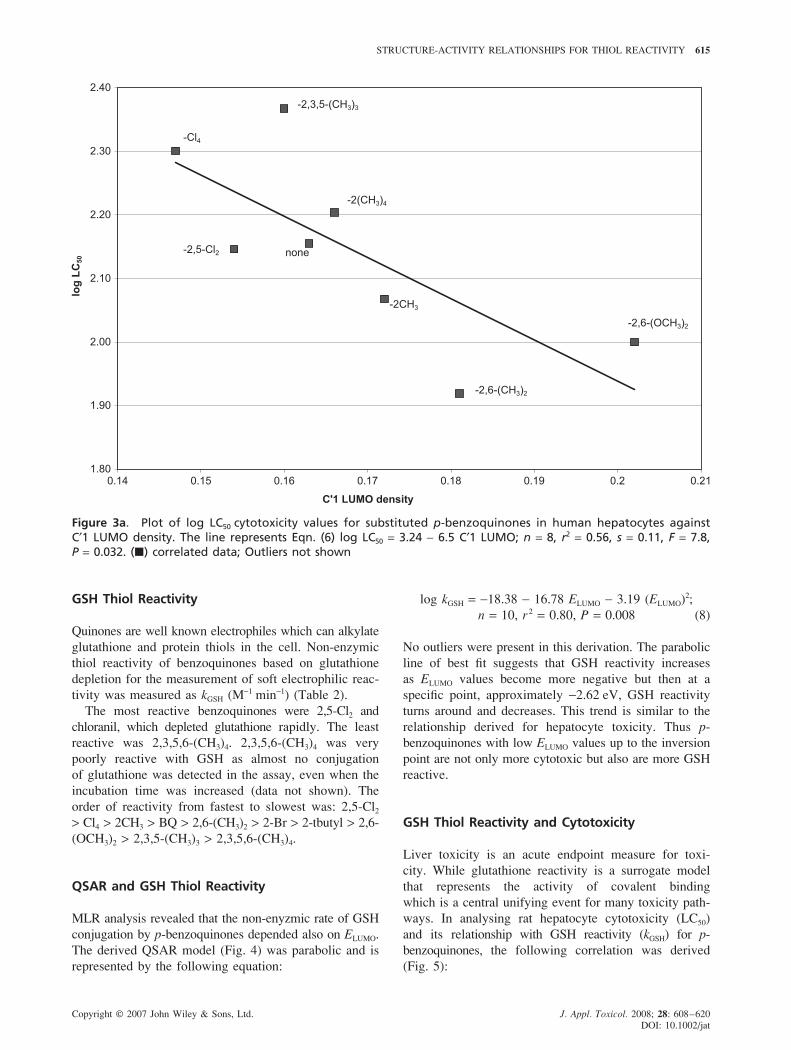
STRUCTURE-ACTIVITY RELATIONSHIPS FOR THIOL REACTIVITY 615
Copyright © 2007 John Wiley & Sons, Ltd. J. Appl. Toxicol. 2008; 28: 608–620
DOI: 10.1002/jat
Figure 3a. Plot of log LC50 cytotoxicity values for substituted p-benzoquinones in human hepatocytes againstC′1 LUMO density. The line represents Eqn. (6) log LC50 = 3.24 − 6.5 C′1 LUMO; n = 8, r2 = 0.56, s = 0.11, F = 7.8,P = 0.032. (�) correlated data; Outliers not shown
GSH Thiol Reactivity
Quinones are well known electrophiles which can alkylate
glutathione and protein thiols in the cell. Non-enzymic
thiol reactivity of benzoquinones based on glutathione
depletion for the measurement of soft electrophilic reac-
tivity was measured as kGSH (M−1 min−1) (Table 2).
The most reactive benzoquinones were 2,5-Cl2 and
chloranil, which depleted glutathione rapidly. The least
reactive was 2,3,5,6-(CH3)4. 2,3,5,6-(CH3)4 was very
poorly reactive with GSH as almost no conjugation
of glutathione was detected in the assay, even when the
incubation time was increased (data not shown). The
order of reactivity from fastest to slowest was: 2,5-Cl2
> Cl4 > 2CH3 > BQ > 2,6-(CH3)2 > 2-Br > 2-tbutyl > 2,6-
(OCH3)2 > 2,3,5-(CH3)3 > 2,3,5,6-(CH3)4.
QSAR and GSH Thiol Reactivity
MLR analysis revealed that the non-enyzmic rate of GSH
conjugation by p-benzoquinones depended also on ELUMO.
The derived QSAR model (Fig. 4) was parabolic and is
represented by the following equation:
log kGSH = −18.38 − 16.78 ELUMO − 3.19 (ELUMO)2;
n = 10, r2 = 0.80, P = 0.008 (8)
No outliers were present in this derivation. The parabolic
line of best fit suggests that GSH reactivity increases
as ELUMO values become more negative but then at a
specific point, approximately −2.62 eV, GSH reactivity
turns around and decreases. This trend is similar to the
relationship derived for hepatocyte toxicity. Thus p-
benzoquinones with low ELUMO values up to the inversion
point are not only more cytotoxic but also are more GSH
reactive.
GSH Thiol Reactivity and Cytotoxicity
Liver toxicity is an acute endpoint measure for toxi-
city. While glutathione reactivity is a surrogate model
that represents the activity of covalent binding
which is a central unifying event for many toxicity path-
ways. In analysing rat hepatocyte cytotoxicity (LC50)
and its relationship with GSH reactivity (kGSH) for p-
benzoquinones, the following correlation was derived
(Fig. 5):

616 K. CHAN ET AL.
Copyright © 2007 John Wiley & Sons, Ltd. J. Appl. Toxicol. 2008; 28: 608–620
DOI: 10.1002/jat
Figure 3b. Plot of log LC50 cytotoxicity values for substituted p-benzoquinones in human hepatocytes against C′1NuFD. The line represents Eqn. (7) log LC50 = 3.22 − 3.21 C′1 NuFD; n = 8, r2 = 0.57, s = 0.11, F = 7.97, P = 0.03. (�)correlated data; Outliers not shown
Log LC50 = 4.65 − 0.92 log kGSH;
n = 10, r2 = 0.82 and P < 0.001 (9)
No outliers were apparent in this model. This indicates
that there is a strong and significant correlation between
the reactivity of benzoquinones with nucleophiles and rat
hepatocyte toxicity. Comparison of GSH reactivity and
cytotoxicity in human hepatocytes resulted in a much
weaker relationship compared with that found in the rat
model:
log LC50 = 3.76 − 0.47 log kGSH;
n = 10, r2 = 0.40, P = 0.05 (10)
Discussion
The hepatotoxic potential and electrophilic reactivity
of ten substituted p-benzoquinones were investigated
here. QSAR modeling was also explored to assess the
physico-chemical parameters which underlie quinone
hepatotoxicity in relation to their electrophilic reactivity
with glutathione.
In rat hepatocytes the most cytotoxic benzoquinone
was chloranil, a tetra-chloro substituted benzoquinone,
while the least cytotoxic was the tetra methylated
benzoquinone, 2,3,5,6-(CH3)4. In human hepatocytes, 2,6-
dimethyl benzoquinone was the most toxic, and similar
to rat hepatocytes duroquinone was the least toxic. To
understand the reason for the higher toxicity of the
dimethyl benzoquinone in human hepatocytes versus
chloranil in rat hepatocytes, a more detailed investigation
would be needed to compare the effects of metabolizing
and detoxifying enzymes in the hepatocytes of these two
species.
Frontier molecular orbital energies have been shown to
play a major role in governing many chemical reactions.
They are responsible for the formation of charge transfer
complexes involved in chemical reactions and have
become commonly used quantum chemical descriptors
for molecular interactions. The energy (E) of the lowest
unoccupied molecular orbital (LUMO) serves as a global
reactivity parameter which measures the energy added
when an electron is placed in the LUMO to produce
a negative ion (electron affinity). ELUMO essentially char-
acterizes the susceptibility of the molecule toward attack
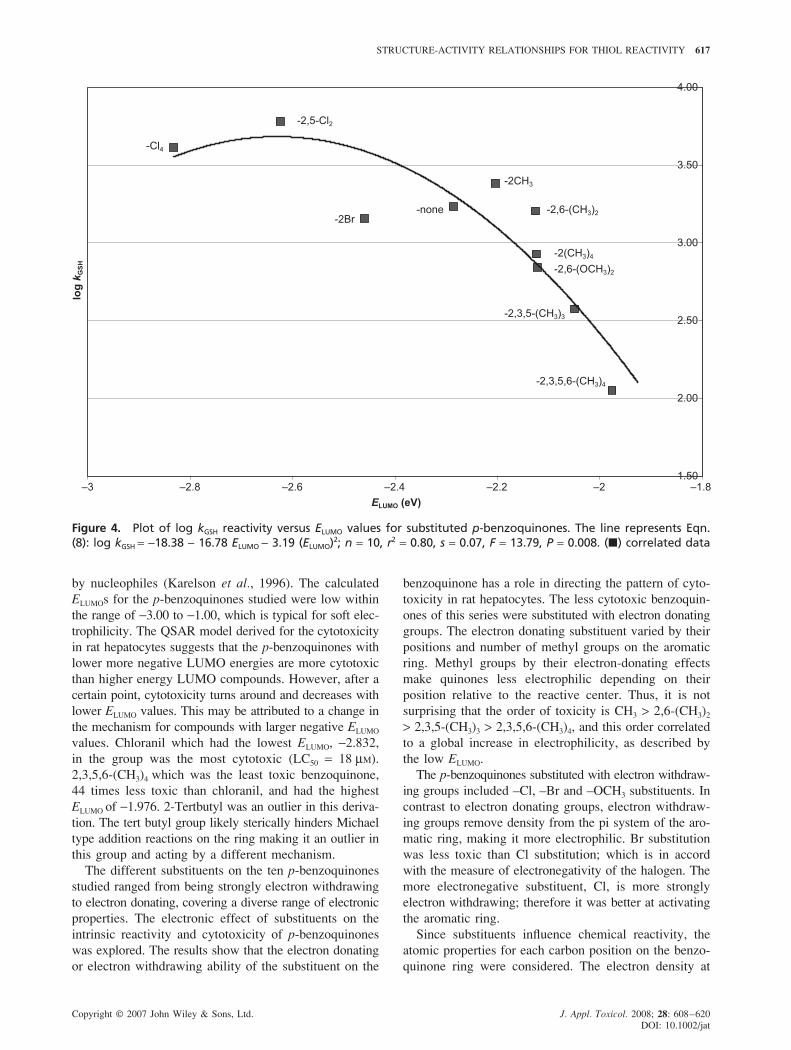
STRUCTURE-ACTIVITY RELATIONSHIPS FOR THIOL REACTIVITY 617
Copyright © 2007 John Wiley & Sons, Ltd. J. Appl. Toxicol. 2008; 28: 608–620
DOI: 10.1002/jat
Figure 4. Plot of log kGSH reactivity versus ELUMO values for substituted p-benzoquinones. The line represents Eqn.(8): log kGSH = −18.38 − 16.78 ELUMO − 3.19 (ELUMO)2; n = 10, r2 = 0.80, s = 0.07, F = 13.79, P = 0.008. (�) correlated data
by nucleophiles (Karelson et al., 1996). The calculated
ELUMOs for the p-benzoquinones studied were low within
the range of −3.00 to −1.00, which is typical for soft elec-
trophilicity. The QSAR model derived for the cytotoxicity
in rat hepatocytes suggests that the p-benzoquinones with
lower more negative LUMO energies are more cytotoxic
than higher energy LUMO compounds. However, after a
certain point, cytotoxicity turns around and decreases with
lower ELUMO values. This may be attributed to a change in
the mechanism for compounds with larger negative ELUMO
values. Chloranil which had the lowest ELUMO, −2.832,
in the group was the most cytotoxic (LC50 = 18 μM).
2,3,5,6-(CH3)4 which was the least toxic benzoquinone,
44 times less toxic than chloranil, and had the highest
ELUMO of −1.976. 2-Tertbutyl was an outlier in this deriva-
tion. The tert butyl group likely sterically hinders Michael
type addition reactions on the ring making it an outlier in
this group and acting by a different mechanism.
The different substituents on the ten p-benzoquinones
studied ranged from being strongly electron withdrawing
to electron donating, covering a diverse range of electronic
properties. The electronic effect of substituents on the
intrinsic reactivity and cytotoxicity of p-benzoquinones
was explored. The results show that the electron donating
or electron withdrawing ability of the substituent on the
benzoquinone has a role in directing the pattern of cyto-
toxicity in rat hepatocytes. The less cytotoxic benzoquin-
ones of this series were substituted with electron donating
groups. The electron donating substituent varied by their
positions and number of methyl groups on the aromatic
ring. Methyl groups by their electron-donating effects
make quinones less electrophilic depending on their
position relative to the reactive center. Thus, it is not
surprising that the order of toxicity is CH3 > 2,6-(CH3)2
> 2,3,5-(CH3)3 > 2,3,5,6-(CH3)4, and this order correlated
to a global increase in electrophilicity, as described by
the low ELUMO.
The p-benzoquinones substituted with electron withdraw-
ing groups included –Cl, –Br and –OCH3 substituents. In
contrast to electron donating groups, electron withdraw-
ing groups remove density from the pi system of the aro-
matic ring, making it more electrophilic. Br substitution
was less toxic than Cl substitution; which is in accord
with the measure of electronegativity of the halogen. The
more electronegative substituent, Cl, is more strongly
electron withdrawing; therefore it was better at activating
the aromatic ring.
Since substituents influence chemical reactivity, the
atomic properties for each carbon position on the benzo-
quinone ring were considered. The electron density at
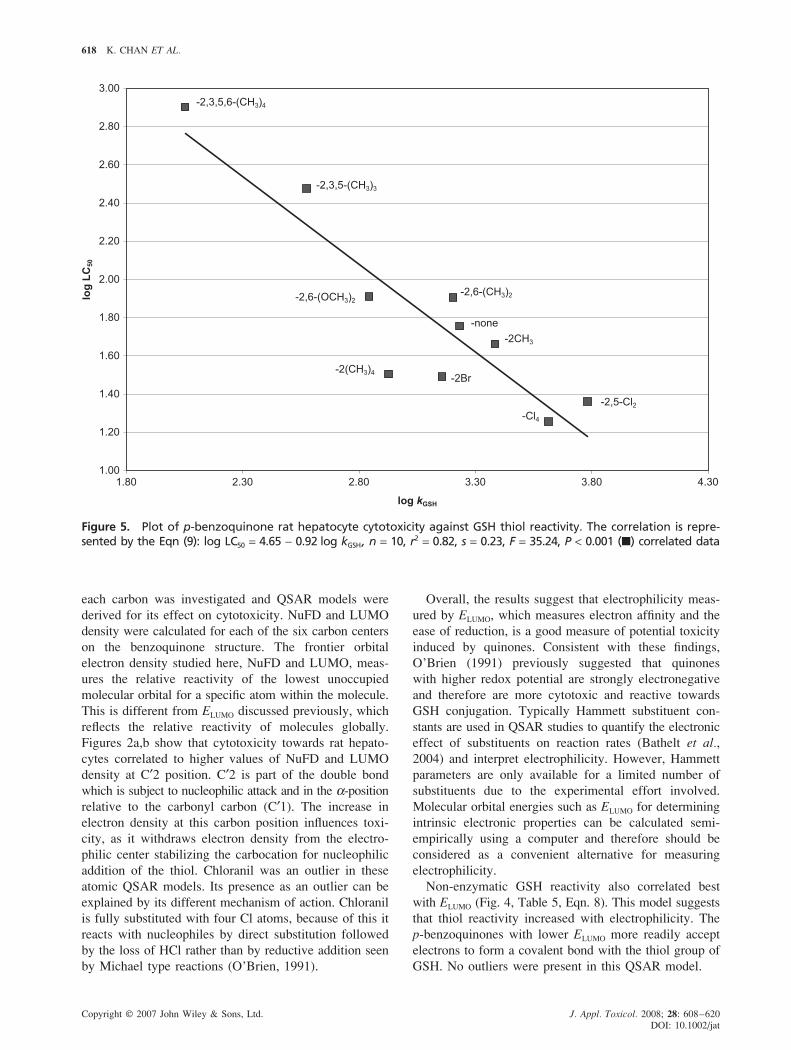
618 K. CHAN ET AL.
Copyright © 2007 John Wiley & Sons, Ltd. J. Appl. Toxicol. 2008; 28: 608–620
DOI: 10.1002/jat
Figure 5. Plot of p-benzoquinone rat hepatocyte cytotoxicity against GSH thiol reactivity. The correlation is repre-sented by the Eqn (9): log LC50 = 4.65 − 0.92 log kGSH, n = 10, r2 = 0.82, s = 0.23, F = 35.24, P < 0.001 (�) correlated data
each carbon was investigated and QSAR models were
derived for its effect on cytotoxicity. NuFD and LUMO
density were calculated for each of the six carbon centers
on the benzoquinone structure. The frontier orbital
electron density studied here, NuFD and LUMO, meas-
ures the relative reactivity of the lowest unoccupied
molecular orbital for a specific atom within the molecule.
This is different from ELUMO discussed previously, which
reflects the relative reactivity of molecules globally.
Figures 2a,b show that cytotoxicity towards rat hepato-
cytes correlated to higher values of NuFD and LUMO
density at C′2 position. C′2 is part of the double bond
which is subject to nucleophilic attack and in the α-position
relative to the carbonyl carbon (C′1). The increase in
electron density at this carbon position influences toxi-
city, as it withdraws electron density from the electro-
philic center stabilizing the carbocation for nucleophilic
addition of the thiol. Chloranil was an outlier in these
atomic QSAR models. Its presence as an outlier can be
explained by its different mechanism of action. Chloranil
is fully substituted with four Cl atoms, because of this it
reacts with nucleophiles by direct substitution followed
by the loss of HCl rather than by reductive addition seen
by Michael type reactions (O’Brien, 1991).
Overall, the results suggest that electrophilicity meas-
ured by ELUMO, which measures electron affinity and the
ease of reduction, is a good measure of potential toxicity
induced by quinones. Consistent with these findings,
O’Brien (1991) previously suggested that quinones
with higher redox potential are strongly electronegative
and therefore are more cytotoxic and reactive towards
GSH conjugation. Typically Hammett substituent con-
stants are used in QSAR studies to quantify the electronic
effect of substituents on reaction rates (Bathelt et al.,
2004) and interpret electrophilicity. However, Hammett
parameters are only available for a limited number of
substituents due to the experimental effort involved.
Molecular orbital energies such as ELUMO for determining
intrinsic electronic properties can be calculated semi-
empirically using a computer and therefore should be
considered as a convenient alternative for measuring
electrophilicity.
Non-enzymatic GSH reactivity also correlated best
with ELUMO (Fig. 4, Table 5, Eqn. 8). This model suggests
that thiol reactivity increased with electrophilicity. The
p-benzoquinones with lower ELUMO more readily accept
electrons to form a covalent bond with the thiol group of
GSH. No outliers were present in this QSAR model.

STRUCTURE-ACTIVITY RELATIONSHIPS FOR THIOL REACTIVITY 619
Copyright © 2007 John Wiley & Sons, Ltd. J. Appl. Toxicol. 2008; 28: 608–620
DOI: 10.1002/jat
Quinones react with GSH via the 1,4-Michael addition
mechanism, producing GSH hydroquinone conjugates
in vitro (O’Brien, 1991). Substitution with electron donat-
ing substituents like, –CH3, and –OCH3, decreased the
rate of GSH addition. While those with electron with-
drawing substituents, –Br or –Cl, increased reactivity.
To confirm that measuring GSH reactivity can reflect the
mechanistic reactivity of quinones to cause cytotoxicity
by alkylation, a relationship between GSH reactivity
and rat hepatotoxicity was assessed. A strong and signifi-
cant correlation between LC50 and kGSH (Fig. 5, Table 5,
Eqn. 9) was derived. This verified that GSH reactivity is
a good indicator of p-benzoquinone cytotoxicity in rat
hepatocytes and both are dependent on the electrophilicity
of the p-benzoquinone.
Similar to this study, Siraki et al. (2004) showed that
electron affinity (EA) was the best parameter for describing
p-benzoquinone induced cytotoxicity and GSH depletion
in rat hepatocytes. EA is essentially the positive value of
ELUMO. Siraki suggested that EA parameterizes for one
electron reduction pathways that leads to ROS formation,
presumably from redox cycling and also it may partially
parameterize alkylation pathways, however, another variable
may be required. In this study, the results confirm that
ELUMO parameterizes for an alkylation pathway.
In human hepatocytes cytotoxicity was moderately
correlated to ELUMO and molecular refractivity (MR;
describes the overall molecular bulk based on size and
polarizability), however, the correlation failed at the
statistical validation stage, P = 0.1. Cytotoxicity also
moderately correlated to electron density described by
NuFD and LUMO at C′1. C′1 is part of the carbonyl
moiety, which has electron withdrawing ability, thus
lowering the electron density in the ring increasing the
likelihood for nucleophilic conjugation of thiol groups.
The relationships derived in Eqns 6 and 7, suggests that
the higher the energy of the unoccupied orbital localized
on the double bond of this carbon, the more toxic the
compound. Increases in the energy of the lowest unoccu-
pied orbital increases the electron density on this carbon.
This affects the electrostatic and polarization component
of the molecule at the electrophilic center, increasing its
susceptibly to thiol addition (Osman et al., 1988). No
robust relationships were found in any of these models.
The structure-activity conclusion that can be drawn as yet
is that sterics in addition to electrophilicity has an impor-
tant role in the cytotoxic mechanism of p-benzoquinones
towards human hepatocytes. Non-enzymic GSH reactiv-
ity and human hepatocyte toxicity also had a relatively
weak relationship in comparison with what was found for
rat hepatocytes. Unsuccessfully, no relationship was con-
firmed between human and rat hepatocyte cytotoxicity.
The differences in strength of the human models com-
pared with the rat may be due partly to differences in the
preparation of the hepatocytes. Thawing of cryopreserved
hepatocytes itself can result in a 10–20% loss in viabil-
ity and yield. This loss is minimal and considered accept-
able because of the relatively high viability of the re-
maining cells (Li et al., 1999) and is therefore more or
less on a par with fresh hepatocytes. Sohlenius-Sternbeck
and Schmidt (2005), demonstrated that cryopreserved
hepatocytes from rat and humans had much lower levels
of intracellular glutathione than freshly prepared rat
hepatocytes (approx 18-fold less) and suggests that the
cryopreservation procedure decreased the hepatocyte
glutathione conjugating capacity (Sohlenius-Sternbeck
and Schmidt, 2005). This may explain the less robust
viability derived for human hepatocytes and the poor
correlation to non-enzymic GSH reactivity. Nevertheless,
the derived models although statistically poorer for human
hepatocytes, point in the direction of electrophilic reactiv-
ity as an important indicator for potential liver toxicity.
In this study correlation of p-benzoquinone induced rat
and human hepatocyte cytotoxicity with other parameters
such as log P, EHOMO, redox potential, dipole moment and
molecular refractivity were not significant compared with
ELUMO or the atomic frontier electron density descriptors
on C′2 and C′1. Normally in toxicity QSAR modeling,
increases in hydrophobic character results in increased
toxic potency. However, in this study hepatocyte
cytotoxicity for all ten analogs did not correlate to log P.
Generally log P is related to biomembrane permeation
of the toxicant. However, in addition to uptake, log P in
mammalian cells has been related to intracellular meta-
bolism which includes binding, induction and inhibition
of phase I and II metabolizing enzymes (Chan et al.,
2007). The insignificance of log P in this study suggests
that membrane permeation and interaction with quinone
metabolizing enzymes, particularly, membrane bound
reductases are not rate limiting steps in the toxicity
mechanism, and that electrophilic reactivity has a more
significant role.
There is an increasing need to be able to predict toxic
potency for risk assessment and regulatory purposes for
both environmental and human endpoints (Schultz et al.,
2007). It is also necessary in drug development to reduce
the number of unnecessary toxicities that are seen in drugs
released on to the market (Guengerich and Macdonald,
2007). Protein covalent binding by reactive drug meta-
bolites is recognized to be a critical event in initiating
organ toxicities such as hepatotoxicity (Guengerich and
Macdonald, 2007). Application of QSAR to this process
has concentrated on describing molecular triggering
events in toxicity such as protein covalent binding, rather
than on typical toxicity endpoints which are complex.
It has been shown that for substituted p-benzoquinones
their rate of soft electrophilic reactivity with GSH can
potentially predict cytotoxicity in rat hepatocytes. As a
first step, the potential for surrogate models of a critical
event in toxicity to improve toxicity-based QSAR
models was shown. QSAR is a tool that should be looked
upon as an aid in chemical risk assessments, and an

620 K. CHAN ET AL.
Copyright © 2007 John Wiley & Sons, Ltd. J. Appl. Toxicol. 2008; 28: 608–620
DOI: 10.1002/jat
alternative non-animal method for predicting toxicity
that is easy and efficient in terms of time and financial
costs (Lessigiarska et al., 2006). However, as advised by
Harder et al. (2003) the potential influence of metabolism
on the occurrence of reactive modes of toxic action
should be realized, and therefore we should be looking
for toxicity indicators that clearly link observable toxi-
city with mechanisms of toxic action. QSARs should not
be looked at as a final decision predictive tool, but rather
as an exploratory tool to provide insight into the func-
tionalities of structure on a particular biological effect
(Harder et al., 2003). This method may help to screen out
potentially toxic compounds during the early drug discov-
ery stages and help access risks for chemical toxins.
References
Aptula AO, Patlewicz G, Roberts DW. 2005. Skin sensitization:reaction mechanistic applicability domains for structure-activity rela-tionships. Chem. Res. Toxicol. 18: 1420–1426.
Aptula AO, Roberts DW. 2006. Mechanistic applicability domainsfor nonanimal-based prediction of toxicological end points: generalprinciples and application to reactive toxicity. Chem. Res. Toxicol.
19: 1097–1105.Bathelt CM, Ridder L, Mulholland AJ, Harvey JN. 2004. Mechanism
and structure-reactivity relationships for aromatic hydroxylation bycytochrome P450. Org. Biomol. Chem. 2: 2998–3005.
Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ. 2000. Roleof quinones in toxicology. Chem. Res. Toxicol. 13: 135 –160.
Chan K, Jensen NS, Silber PM, O’Brien PJ. 2007. Structure-activityrelationships for halobenzene induced cytotoxicity in rat and humanhepatoctyes. Chem. Biol. Interact. 165: 165–174.
Freidig AP, Verhaar HJM, Hermens JL. 1999. Quantitative structure-property relationships for the chemical reactivity of acrylates andmethacrylates. Environ. Toxicol. Chem. 18: 1133–1139.
Ghose A, Pritchett A, Crippen G. 1988. Atomic physicochemicalparameters for three dimensional structure directed quantitativestructure-activity relationships 3. J. Comput. Chem. 9: 80–90.
Guengerich FP, Macdonald JS. 2007. Applying mechanisms ofchemical toxicity to predict drug safety. Chem. Res. Toxicol. 20:344–369.
Hall LM, Hall LH, Kier LB. 2003. Modeling drug albumin bindingaffinity with e-state topological structure representation. J. Chem Inf.
Comput. Sci. 43: 2120–2128.Harder A, Escher BI, Schwarzenbach RP. 2003. Applicability and
limitation of OSARs for the toxicity of electrophilic chemicals.Environ. Sci. Technol. 37: 4955–4961.
Huuskonen J. 2001. QSAR modeling with the electrotopological state:TIBO derivatives. J. Chem Inf. Comput. Sci. 41: 425–429.
Karelson M, Lobanov VS, Katritzky AR. 1996. Quantum-chemicaldescriptors in QSAR/QSPR studies. Chem. Rev. 96: 1027–1044.
Lessigiarska I, Worth AP, Netzeva TI, Dearden JC, Cronin MT. 2006.Quantitative structure-activity-activity and quantitative structure-activity investigations of human and rodent toxicity. Chemosphere
65: 1878–1887.Li AP, Gorycki PD, Hengstler JG, Kedderis GL, Koebe HG,
Rahmani R, de Sousas G, Silva JM, Skett P. 1999. Present statusof the application of cryopreserved hepatocytes in the evaluation ofxenobiotics: consensus of an international expert panel. Chem. Biol.
Interact. 121: 117–123.Liebler DC, Guengerich FP. 2005. Elucidating mechanisms of
drug-induced toxicity. Nat. Rev. Drug Discov. 4: 410–420.Lipnick RL. 1991. Outliers: their origin and use in the classification
of molecular mechanisms of toxicity. Sci. Total Environ. 109–110:131–153.
Moldeus P, Hogberg J, Orrenius S. 1978. Isolation and use of livercells. Methods Enzymol. 52: 60–71.
O’Brien PJ. 1991. Molecular mechanisms of quinone cytotoxicity.Chem. Biol. Interact. 80: 1–41.
Osman R, Namboodiri K, Weinstein H, Rabinowitz JR. 1988.Reactivities of acrylic acid and methacrylic acid in a nucleophilicaddition model of their biological activity. J. Am. Chem. Soc. 110:1701–1707.
Rossi L, Moore GA, Orrenius S, O’Brien PJ. 1986. Quinone toxicity inhepatocytes without oxidative stress. Arch. Biochem. Biophys. 251:25–35.
Schultz TW, Carlson RE, Cronin MT, Hermens JL, Johnson R, O’BrienPJ, Roberts DW, Siraki A, Wallace KB, Veith GD. 2006. A conceptualframework for predicting the toxicity of reactive chemicals: modelingsoft electrophilicity. SAR QSAR. Environ. Res. 17: 413–428.
Schultz TW, Ralston KE, Roberts DW, Veith GD, Aptula AO. 2007.Structure-activity relationships for abiotic thiol reactivity and aquatictoxicity of halo-substituted carbonyl compounds. SAR QSAR.
Environ. Res. 18: 21–29.Schultz TW, Yarbrough JW, Johnson EL. 2005. Structure-activity
relationships for reactivity of carbonyl-containing compounds withglutathione. SAR QSAR. Environ. Res. 16: 313–322.
Siraki AG, Chan TS, O’Brien PJ. 2004. Application of quantitativestructure-toxicity relationships for the comparison of the cytotoxicityof 14 p-benzoquinone congeners in primary cultured rat hepatocytesversus PC12 cells. Toxicol. Sci. 81: 148–159.
Sohlenius-Sternbeck AK, Schmidt S. 2005. Impaired glutathione-conjugating capacity by cryopreserved human and rat hepatocytes.Xenobiotica 35: 727–736.
Tapper MA, Sheedy BR, Hammermeister DE, Schmieder PK. 2000.Depletion of cellular protein thiols as an indicator of arylation inisolated trout hepatocytes exposed to 1,4-benzoquinone. Toxicol. Sci.
55: 327–334.van Ommen B, Voncken JW, Muller F, van Bladeren PJ. 1988. The
oxidation of tetrachloro-1,4-hydroquinone by microsomes andpurified cytochrome P-450b. Implications for covalent binding toprotein and involvement of reactive oxygen species. Chem. Biol.
Interact. 65: 247–259.Verma RP. 2006. Anti-cancer activities of 1,4-naphthoquinones: a
QSAR study. Anticancer Agents Med. Chem. 6: 489–499.




















