
On the Applicationn of Preconditioning Operators for Nonlinear Elliptic Problems
Upload
independentCategory
view
1download
0
Original Contribution
NITROXYL AFFORDS THIOL-SENSITIVE MYOCARDIAL PROTECTIVEEFFECTS AKIN TO EARLY PRECONDITIONING
PASQUALE PAGLIARO,* DANIELE MANCARDI,* RAFFAELLA RASTALDO,* CLAUDIA PENNA,*DONATELLA GATTULLO,* K ATRINA M. MIRANDA,† MARTIN FEELISCH,‡ DAVID A. WINK,† DAVID A. KASS,§ and
NAZARENO PAOLOCCI§
*Dipartimento di Scienze Cliniche e Biologiche, Universita` degli Studi di Torino, Orbassano, Italy;†Radiation Biology Branch,National Cancer Institute, National Institutes of Health, Bethesda, MD, USA;‡Department of Molecular and Cellular Physiology,Louisiana State University Health Sciences Center, Shreveport, LA, USA; and§Division of Cardiology, Department of Medicine,
The Johns Hopkins Medical Institutions, Baltimore, MD, USA
(Received 10 May 2002;Revised 16 August 2002;Accepted 20 September 2002)
Abstract—Nitric oxide (NO) donors mimic the early phase of ischemic preconditioning (IPC). The effects of nitroxyl(HNO/NO�), the one-electron reduction product of NO, on ischemia/reperfusion (I/R) injury are unknown. Here weinvestigated whether HNO/NO�, produced by decomposition of Angeli’s salt (AS; Na2N2O3), has a cardioprotectiveeffect in isolated perfused rat hearts. Effects were examined after intracoronary perfusion (19 min) of either AS (1�M),the NO donor diethylamine/NO (DEA/NO, 0.5�M), vehicle (100 nM NaOH) or buffer, followed by global ischemia(30 min) and reperfusion (30 min or 120 min in a subset of hearts). IPC was induced by three cycles of 3 min ischemiafollowed by 10 min reperfusion prior to I/R. The extent of I/R injury under each intervention was assessed by changesin myocardial contractility as well as lactate dehydrogenase (LDH) release and infarct size. Postischemic contractility,as indexed by developed pressure anddP/dtmax, was similarly improved with IPC and pre-exposure to AS, as opposedto control or DEA/NO-treated hearts. Infarct size and LDH release were also significantly reduced in IPC and ASgroups, whereas DEA/NO was less effective in limiting necrosis. Co-infusion in the triggering phase of AS and thenitroxyl scavenger,N-acetyl-L-cysteine (4 mM) completely reversed the beneficial effects of AS, both at 30 and 120 minreperfusion. Our data show that HNO/NO� affords myocardial protection to a degree similar to IPC and greater thanNO, suggesting that reactive nitrogen oxide species are not only necessary but also sufficient to trigger myocardialprotection against reperfusion through species-dependent, pro-oxidative, and/or nitrosative stress-related mechanisms.© 2002 Elsevier Science Inc.
Keywords—Ischemia/reperfusion, Preconditioning, Myocardial necrosis, Nitroxyl, Angeli’s salt, Nitric oxide, Dieth-ylamine/NO complex,N-acetyl-L-cysteine, Free radicals
INTRODUCTION
Brief episodes of coronary occlusion diminish theextent of myocardial injury during subsequent infarc-tion [1– 4]. This phenomenon, known as ischemic pre-conditioning (IPC), affords both early (1–3 h) andsustained (3– 4 d; late phase) myocardial protectionagainst reperfusion injury [2–7]. Late IPC has beenattributed to the generation of multiple signaling mol-
ecules, metabolites, and ligands [3,4], and recent re-ports have shown that endogenous nitric oxide (NO)plays an important role in late IPC [3,8]. Pretreatmentof conscious rabbits with NO donors under normoxic[9] or ischemic [10] conditions mimics the effects oflate IPC. NO has also been suggested to play a role inearly IPC [10 –12], including a “vascular” IPC effect[13]. However, other data have demonstrated that ni-tric oxide synthase (NOS) inhibitors can attenuateischemia/reperfusion (I/R) damage [14 –16] withoutan effect on IPC [17,18]. The different doses of NOdonors applied and the different basal levels of NOendogenously produced may explain these disparities.Alternatively, exogenous NO may trigger early IPC
Address correspondence to: Pasquale Pagliaro MD, Ph.D., Diparti-mento di Scienze Cliniche e Biologiche Universita` di Torino OspedaleS. Luigi, Regione Gonzole10 10043 Orbassano (TO), Italy; Tel:�39(011) 670-8130/7710; Fax: �39 (011) 903-8639; E-Mail:[email protected].
Free Radical Biology & Medicine, Vol. 34, No. 1, pp. 33–43, 2003Copyright © 2002 Elsevier Science Inc.Printed in the USA. All rights reserved
0891-5849/02/$–see front matter
PII S0891-5849(02)01179-6
33

through a free radical-mediated process not shared byendogenous NO [18].
These uncertainties may reflect the in vivo propertiesof NO, which are dictated by the precise redox status ofthe tissue through formation of reactive nitrogen oxidespecies (RNOS) that have disparate biological actions[19]. The involvement of nitroxyl (HNO/NO�), the one-electron reduction product of NO, in preconditioning hasnot been determined. Although the production of nitroxylin vivo is still a matter of debate [20], it is producedduring oxidation of L-arginine by NOS under certainconditions [21–23]. At physiological pH, HNO can begenerated by decomposition of Angeli’s salt (AS,Na2N2O3), which also produces equimolar nitrite, a prin-cipal metabolite of NO biosynthesis [24,25]. In vitro,high concentrations of AS (� 1 mM) are significantlymore cytotoxic than NO [26]. In addition, myocardialinjury in vivo is exacerbated by exposure to AS atreperfusion but attenuated by NO donors [27]. However,AS may provide protection against neuronal damageunder hypoxic conditions by regulating calcium influxthrough the N-methyl-D-aspartate (NMDA) channel[28]. Furthermore, we recently reported that AS inducesa potent redox-dependent, positive inotropic response inconscious healthy dogs [29], an effect that cannot bereproduced by the NO donor diethylamine/NO (DEA/NO).
Based on these prior observations, we sought to in-vestigate the hypothesis that the effect of HNO on postI/R myocardium would differ substantially from that ofcontrol myocardium. We here demonstrate that HNOinduces a significantly higher degree of cardioprotectionthan NO, resembling that of IPC, and that this beneficialeffect is completely abrogated by co-infusion with thenitroxyl scavenger, N-acetyl-L-cysteine (NAC).
MATERIALS AND METHODS
Animals
Male Wistar rats (n � 115; body weight 450–550 g)were housed in identical cages and were allowed accessto tap water and a standard rodent diet ad libitum. Theanimals received humane care in compliance with theGuide for the Care and Use of Laboratory Animals by theU.S. National Research Council and in accordance withItalian law (DL-116, Jan. 27, 1992).
Isolated heart perfusion
Each animal was anesthetized with urethane (1 g/kgi.p.). The chest was opened 10 min after heparin (2500U, i.m.) treatment, and the heart was rapidly excised,placed in ice-cold buffer solution and weighed. Isolatedrat hearts were attached to the perfusion apparatus and
retrogradely perfused with oxygenated Krebs-Henseleitbuffer (127 mM NaCl, 17.7 mM NaHCO3, 5.1 mM KCl,1.5 mM CaCl2, 1.26 mM MgCl2, 11 mM D-glucosesupplemented with 5 �g/ml lidocaine and gassed with95% O2 and 5% CO2 [30,31]). The hearts were instru-mented as previously described [30,31]. A constant flowwas maintained to obtain a typical coronary perfusionpressure of 85–90 mm Hg. A constant proportion of 10%of the flow rate was applied by means of one of twoperfusion pumps (Terumo, Tokyo, Japan) using a 50 mlsyringe connected to the aortic inflow cannula. Drugapplications (see Experimental Protocols) were per-formed by switching from the syringe containing bufferalone to the syringe of the other pump containing the10� drug stock solution. A small hole in the left ven-tricular wall allowed drainage of the thebesian flow, anda polyvinyl-chloride balloon was placed into the leftventricle and connected to an electromanometer for re-cording of left ventricular pressure (LVP). The heartswere electrically paced at 280–300 bpm and kept in atemperature-controlled chamber (37°C). Coronary perfu-sion pressure (CPP) and coronary flow were monitoredwith a second electromanometer and an electromagneticflow-probe, respectively, both placed along the perfusionline. Coronary flow, CPP, and LVP were recorded usinga TEAC R-71 recorder, digitized at 1000 Hz and ana-lyzed off-line with DataQ-Instruments/CODAS soft-ware, which allowed quantification of the maximum rateof increase of LVP during systole (dP/dtmax).
Experimental compounds
The HNO donor Angeli’s salt (sodium trioxodinitrate,Na2N2O3) was a generous gift of Dr. Jon M. Fukuto(University of California, Los Angeles, CA, USA) andthe NO donor DEA/NO [NaEt2NN(O)NO] was a gener-ous gift of Dr. Joseph Saavedra (National Cancer Insti-tute, Frederick, MD, USA). Stock solutions (100 mM in10 mM NaOH) of these compounds were diluted inbuffer immediately prior to use. All other chemicals werepurchased from Sigma (St. Louis, MO, USA).
Experimental protocols
Each heart was allowed to stabilize for 30 min atwhich time baseline parameters were recorded. Typi-cally, coronary flow (9 � 2 ml/min/g wet weight) wasadjusted to obtain the desired CPP (85–90 mm Hg)within the first 10 min and kept constant thereon. Afterthis stabilization period, hearts were randomly assignedto one of the treatment groups described below (n � 12in each group) and then subjected to 30 min of global,no-flow ischemia followed by 30 min of reperfusion(I/R) (see Fig. 1A). Pacing was discontinued at the be-
34 P. PAGLIARO et al.
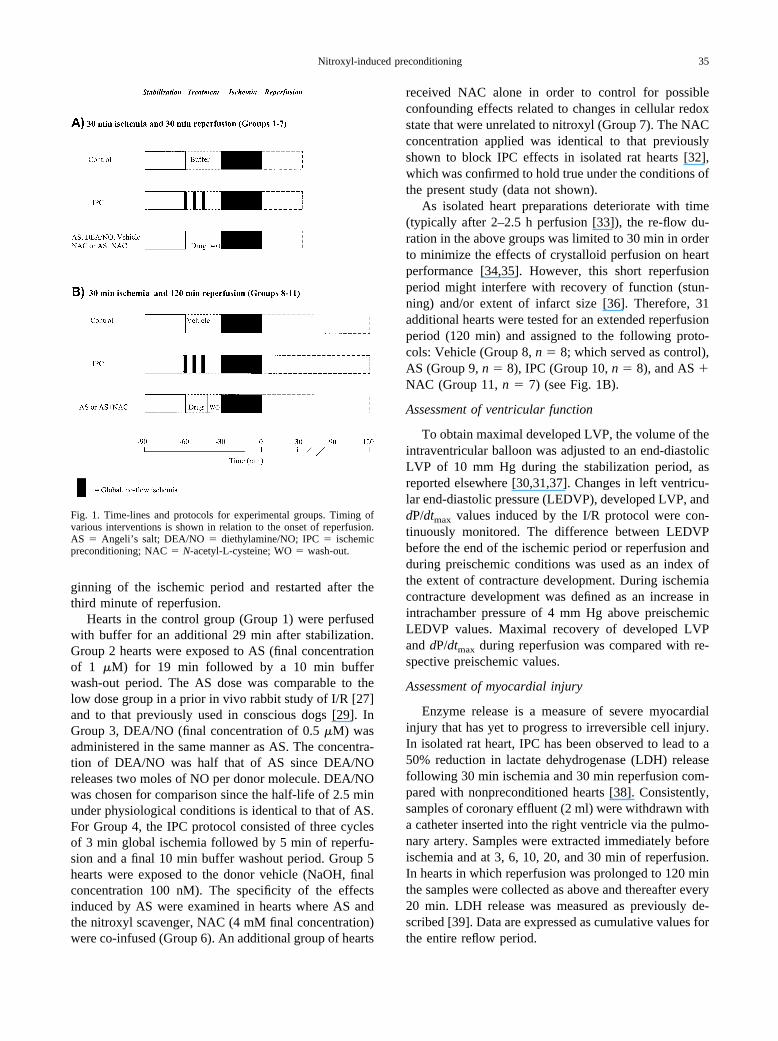
ginning of the ischemic period and restarted after thethird minute of reperfusion.
Hearts in the control group (Group 1) were perfusedwith buffer for an additional 29 min after stabilization.Group 2 hearts were exposed to AS (final concentrationof 1 �M) for 19 min followed by a 10 min bufferwash-out period. The AS dose was comparable to thelow dose group in a prior in vivo rabbit study of I/R [27]and to that previously used in conscious dogs [29]. InGroup 3, DEA/NO (final concentration of 0.5 �M) wasadministered in the same manner as AS. The concentra-tion of DEA/NO was half that of AS since DEA/NOreleases two moles of NO per donor molecule. DEA/NOwas chosen for comparison since the half-life of 2.5 minunder physiological conditions is identical to that of AS.For Group 4, the IPC protocol consisted of three cyclesof 3 min global ischemia followed by 5 min of reperfu-sion and a final 10 min buffer washout period. Group 5hearts were exposed to the donor vehicle (NaOH, finalconcentration 100 nM). The specificity of the effectsinduced by AS were examined in hearts where AS andthe nitroxyl scavenger, NAC (4 mM final concentration)were co-infused (Group 6). An additional group of hearts
received NAC alone in order to control for possibleconfounding effects related to changes in cellular redoxstate that were unrelated to nitroxyl (Group 7). The NACconcentration applied was identical to that previouslyshown to block IPC effects in isolated rat hearts [32],which was confirmed to hold true under the conditions ofthe present study (data not shown).
As isolated heart preparations deteriorate with time(typically after 2–2.5 h perfusion [33]), the re-flow du-ration in the above groups was limited to 30 min in orderto minimize the effects of crystalloid perfusion on heartperformance [34,35]. However, this short reperfusionperiod might interfere with recovery of function (stun-ning) and/or extent of infarct size [36]. Therefore, 31additional hearts were tested for an extended reperfusionperiod (120 min) and assigned to the following proto-cols: Vehicle (Group 8, n � 8; which served as control),AS (Group 9, n � 8), IPC (Group 10, n � 8), and AS �NAC (Group 11, n � 7) (see Fig. 1B).
Assessment of ventricular function
To obtain maximal developed LVP, the volume of theintraventricular balloon was adjusted to an end-diastolicLVP of 10 mm Hg during the stabilization period, asreported elsewhere [30,31,37]. Changes in left ventricu-lar end-diastolic pressure (LEDVP), developed LVP, anddP/dtmax values induced by the I/R protocol were con-tinuously monitored. The difference between LEDVPbefore the end of the ischemic period or reperfusion andduring preischemic conditions was used as an index ofthe extent of contracture development. During ischemiacontracture development was defined as an increase inintrachamber pressure of 4 mm Hg above preischemicLEDVP values. Maximal recovery of developed LVPand dP/dtmax during reperfusion was compared with re-spective preischemic values.
Assessment of myocardial injury
Enzyme release is a measure of severe myocardialinjury that has yet to progress to irreversible cell injury.In isolated rat heart, IPC has been observed to lead to a50% reduction in lactate dehydrogenase (LDH) releasefollowing 30 min ischemia and 30 min reperfusion com-pared with nonpreconditioned hearts [38]. Consistently,samples of coronary effluent (2 ml) were withdrawn witha catheter inserted into the right ventricle via the pulmo-nary artery. Samples were extracted immediately beforeischemia and at 3, 6, 10, 20, and 30 min of reperfusion.In hearts in which reperfusion was prolonged to 120 minthe samples were collected as above and thereafter every20 min. LDH release was measured as previously de-scribed [39]. Data are expressed as cumulative values forthe entire reflow period.
Fig. 1. Time-lines and protocols for experimental groups. Timing ofvarious interventions is shown in relation to the onset of reperfusion.AS � Angeli’s salt; DEA/NO � diethylamine/NO; IPC � ischemicpreconditioning; NAC � N-acetyl-L-cysteine; WO � wash-out.
35Nitroxyl-induced preconditioning

Infarct areas were assessed in a blinded fashion tocorroborate the data relative to myocardial injury asdetermined by LDH release. At the end of the experi-ment, i.e., directly after reperfusion, each heart was rap-idly removed from the perfusion apparatus, and the leftventricle (LV) was dissected into 2–3 mm circumferen-tial slices. Following 15 min of incubation at 37°C in0.1% solution of nitro blue tetrazolium in phosphatebuffer [27], unstained necrotic tissue was carefully sep-arated from stained viable tissue by an independent ob-server who was not aware of the nature of the interven-tion. The weights of the necrotic and nonnecrotic tissueswere then determined, and the necrotic mass was ex-pressed as a percentage of total left ventricular mass.
Statistical analysis
All values are presented as means � SEM. All datawere subjected to ANOVA followed by the Bonferronicorrection for post hoc t-tests. Significance was acceptedat a p level of � .05.
RESULTS
Cardiac weight (1.34 � 0.01 g; n � 115) and thecardiac to body weight ratio (0.003 � 0.00003; n � 115)were equivalent among the eleven treatment groups.
Hearts subjected to 30 min ischemia and 30 minreperfusion (Groups 1–7)
Preischemic function. Table 1 displays baseline cardiacfunction after stabilization and prior to ischemia ob-served in these seven groups. There were no significantdifferences among groups.
Contracture development during ischemia. The develop-ment of contracture occurred after 15 � 2 min of globalischemia in control hearts and after 15 � 1 min in thevehicle groups. This effect was somewhat delayed(17–20 min; p � NS vs. controls) in all other groups. Atthe end of the ischemic period, LVEDP was 24–28 mmHg in control, vehicle, NAC-, and NAC � AS-treatedhearts with no statistically significant differences amongthese groups. In contrast, AS, DEA/NO, and IPC treat-ments lowered LVEDP values to 16–19 mm Hg (p � NSbetween these groups; p � .05 for all compared tocontrol, vehicles, and NAC � AS treatments), as shownin Fig. 2A.
Contracture development during reperfusion. LVEDPfurther increased at reperfusion. The maximal incrementduring reperfusion was significantly lower in AS- andDEA/NO-treated hearts when compared to control, ve-hicle, NAC, and NAC � AS groups. AS was even moreeffective than DEA/NO in preventing LVEDP increase.
Tab
le1.
Bas
elin
eC
ardi
acFu
nctio
nA
fter
Stab
iliza
tion
and
Prio
rto
Isch
emia
inE
xper
imen
tal
Gro
ups
1to
7
End
ofst
abili
zatio
npe
riod
Post
trea
tmen
t(p
reis
chem
ic)
Con
trol
AS
DE
A/N
OIP
CV
ehic
leN
AC
NA
C�
AS
Con
trol
AS
DE
A/N
OIP
CV
ehic
leN
AC
NA
C�
AS
CPP
(mm
Hg)
85�
589
�4
91�
488
�9
92�
588
�3
84�
487
�5
88�
690
�4
93�
894
�7
88�
482
�4
LV
ED
P(m
mH
g)8
�0.
69
�0.
78
�0.
711
�0.
59
�0.
78
�0.
810
�0.
99
�0.
610
�0.
89
�0.
711
�1.
610
�0.
810
�1.
710
�0.
9D
evel
oped
LV
P(m
mH
g)89
�7
99�
698
�5
85�
688
�8
89�
796
�7
88�
898
�7
99�
683
�6
88�
886
�6
93�
3
dP/d
t max
(mm
Hg/
s)27
94�
212
3145
�15
530
47�
120
2692
�66
2661
�10
127
60�
216
3011
�19
526
49�
151
3223
�14
729
76�
9326
34�
6727
01�
103
2481
�24
527
14�
187
CPP
�co
rona
rype
rfus
ion
pres
sure
;LV
ED
P�
left
vent
ricu
lar
end-
dias
tolic
pres
sure
;LV
P�
left
vent
ricu
lar
pres
sure
;dP/
dt m
ax�
max
imum
rate
ofin
crea
seof
LV
Pdu
ring
syst
ole;
AS
�A
ngel
i’s
salt;
DE
A/N
O�
diet
hyla
min
e/N
O(N
Odo
nor)
;IP
C�
isch
emic
prec
ondi
tioni
ng;
NA
C�
N-a
cety
l-L
-cys
tein
e.
36 P. PAGLIARO et al.
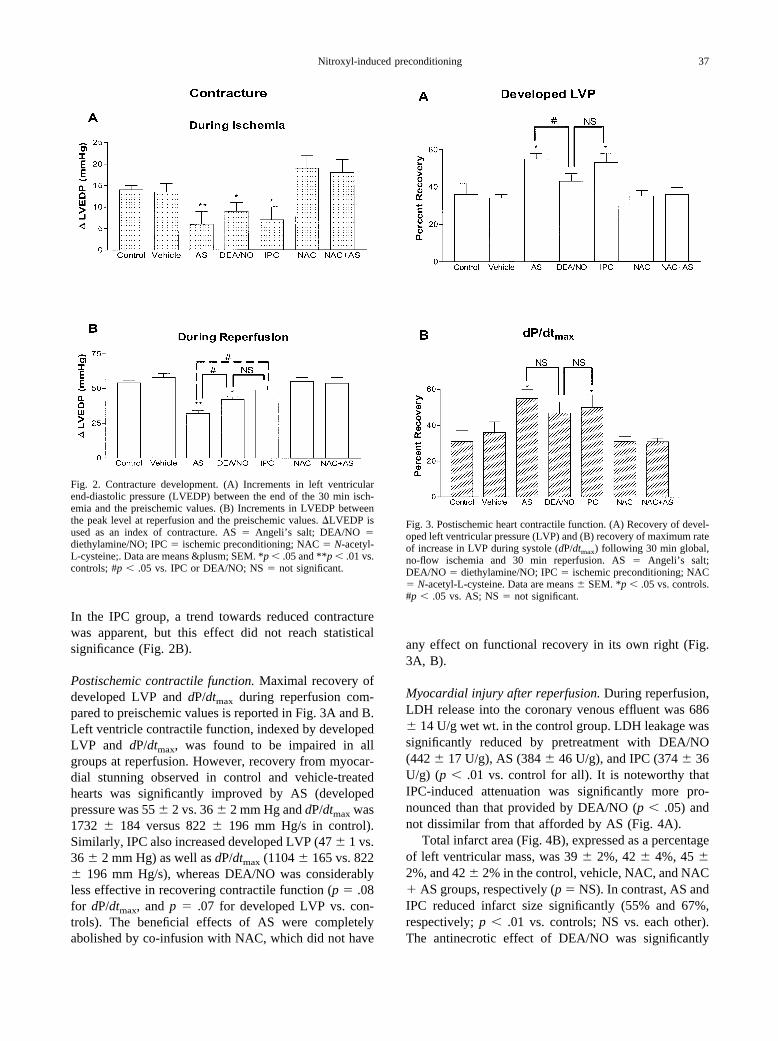
In the IPC group, a trend towards reduced contracturewas apparent, but this effect did not reach statisticalsignificance (Fig. 2B).
Postischemic contractile function. Maximal recovery ofdeveloped LVP and dP/dtmax during reperfusion com-pared to preischemic values is reported in Fig. 3A and B.Left ventricle contractile function, indexed by developedLVP and dP/dtmax, was found to be impaired in allgroups at reperfusion. However, recovery from myocar-dial stunning observed in control and vehicle-treatedhearts was significantly improved by AS (developedpressure was 55 � 2 vs. 36 � 2 mm Hg and dP/dtmax was1732 � 184 versus 822 � 196 mm Hg/s in control).Similarly, IPC also increased developed LVP (47 � 1 vs.36 � 2 mm Hg) as well as dP/dtmax (1104 � 165 vs. 822� 196 mm Hg/s), whereas DEA/NO was considerablyless effective in recovering contractile function (p � .08for dP/dtmax, and p � .07 for developed LVP vs. con-trols). The beneficial effects of AS were completelyabolished by co-infusion with NAC, which did not have
any effect on functional recovery in its own right (Fig.3A, B).
Myocardial injury after reperfusion. During reperfusion,LDH release into the coronary venous effluent was 686� 14 U/g wet wt. in the control group. LDH leakage wassignificantly reduced by pretreatment with DEA/NO(442 � 17 U/g), AS (384 � 46 U/g), and IPC (374 � 36U/g) (p � .01 vs. control for all). It is noteworthy thatIPC-induced attenuation was significantly more pro-nounced than that provided by DEA/NO (p � .05) andnot dissimilar from that afforded by AS (Fig. 4A).
Total infarct area (Fig. 4B), expressed as a percentageof left ventricular mass, was 39 � 2%, 42 � 4%, 45 �2%, and 42 � 2% in the control, vehicle, NAC, and NAC� AS groups, respectively (p � NS). In contrast, AS andIPC reduced infarct size significantly (55% and 67%,respectively; p � .01 vs. controls; NS vs. each other).The antinecrotic effect of DEA/NO was significantly
Fig. 2. Contracture development. (A) Increments in left ventricularend-diastolic pressure (LVEDP) between the end of the 30 min isch-emia and the preischemic values. (B) Increments in LVEDP betweenthe peak level at reperfusion and the preischemic values. �LVEDP isused as an index of contracture. AS � Angeli’s salt; DEA/NO �diethylamine/NO; IPC � ischemic preconditioning; NAC � N-acetyl-L-cysteine;. Data are means &plusm; SEM. *p � .05 and **p � .01 vs.controls; #p � .05 vs. IPC or DEA/NO; NS � not significant.
Fig. 3. Postischemic heart contractile function. (A) Recovery of devel-oped left ventricular pressure (LVP) and (B) recovery of maximum rateof increase in LVP during systole (dP/dtmax) following 30 min global,no-flow ischemia and 30 min reperfusion. AS � Angeli’s salt;DEA/NO � diethylamine/NO; IPC � ischemic preconditioning; NAC� N-acetyl-L-cysteine. Data are means � SEM. *p � .05 vs. controls.#p � .05 vs. AS; NS � not significant.
37Nitroxyl-induced preconditioning
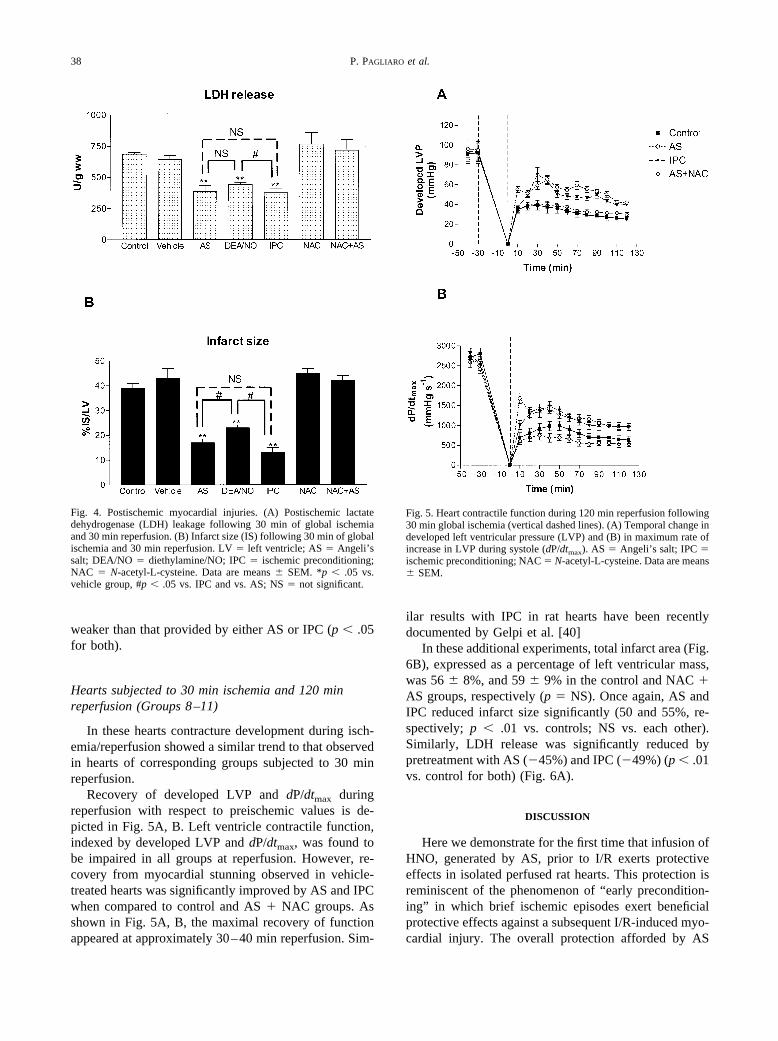
weaker than that provided by either AS or IPC (p � .05for both).
Hearts subjected to 30 min ischemia and 120 minreperfusion (Groups 8–11)
In these hearts contracture development during isch-emia/reperfusion showed a similar trend to that observedin hearts of corresponding groups subjected to 30 minreperfusion.
Recovery of developed LVP and dP/dtmax duringreperfusion with respect to preischemic values is de-picted in Fig. 5A, B. Left ventricle contractile function,indexed by developed LVP and dP/dtmax, was found tobe impaired in all groups at reperfusion. However, re-covery from myocardial stunning observed in vehicle-treated hearts was significantly improved by AS and IPCwhen compared to control and AS � NAC groups. Asshown in Fig. 5A, B, the maximal recovery of functionappeared at approximately 30–40 min reperfusion. Sim-
ilar results with IPC in rat hearts have been recentlydocumented by Gelpi et al. [40]
In these additional experiments, total infarct area (Fig.6B), expressed as a percentage of left ventricular mass,was 56 � 8%, and 59 � 9% in the control and NAC �AS groups, respectively (p � NS). Once again, AS andIPC reduced infarct size significantly (50 and 55%, re-spectively; p � .01 vs. controls; NS vs. each other).Similarly, LDH release was significantly reduced bypretreatment with AS (�45%) and IPC (�49%) (p � .01vs. control for both) (Fig. 6A).
DISCUSSION
Here we demonstrate for the first time that infusion ofHNO, generated by AS, prior to I/R exerts protectiveeffects in isolated perfused rat hearts. This protection isreminiscent of the phenomenon of “early precondition-ing” in which brief ischemic episodes exert beneficialprotective effects against a subsequent I/R-induced myo-cardial injury. The overall protection afforded by AS
Fig. 4. Postischemic myocardial injuries. (A) Postischemic lactatedehydrogenase (LDH) leakage following 30 min of global ischemiaand 30 min reperfusion. (B) Infarct size (IS) following 30 min of globalischemia and 30 min reperfusion. LV � left ventricle; AS � Angeli’ssalt; DEA/NO � diethylamine/NO; IPC � ischemic preconditioning;NAC � N-acetyl-L-cysteine. Data are means � SEM. *p � .05 vs.vehicle group, #p � .05 vs. IPC and vs. AS; NS � not significant.
Fig. 5. Heart contractile function during 120 min reperfusion following30 min global ischemia (vertical dashed lines). (A) Temporal change indeveloped left ventricular pressure (LVP) and (B) in maximum rate ofincrease in LVP during systole (dP/dtmax). AS � Angeli’s salt; IPC �ischemic preconditioning; NAC � N-acetyl-L-cysteine. Data are means� SEM.
38 P. PAGLIARO et al.
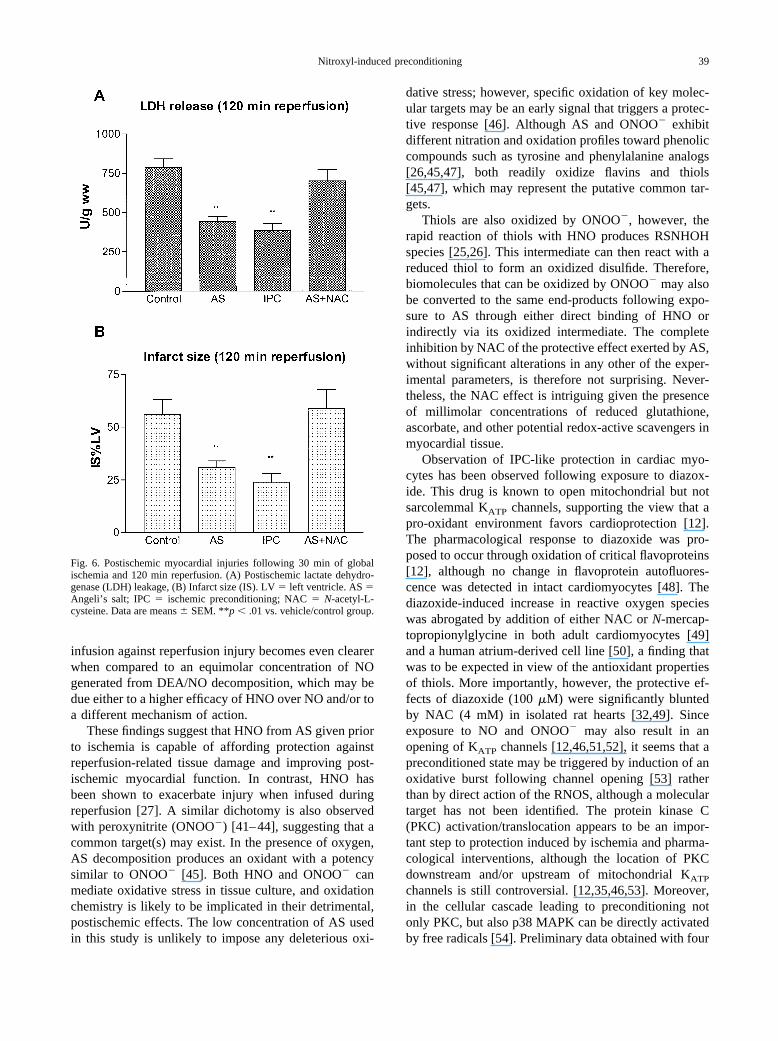
infusion against reperfusion injury becomes even clearerwhen compared to an equimolar concentration of NOgenerated from DEA/NO decomposition, which may bedue either to a higher efficacy of HNO over NO and/or toa different mechanism of action.
These findings suggest that HNO from AS given priorto ischemia is capable of affording protection againstreperfusion-related tissue damage and improving post-ischemic myocardial function. In contrast, HNO hasbeen shown to exacerbate injury when infused duringreperfusion [27]. A similar dichotomy is also observedwith peroxynitrite (ONOO�) [41–44], suggesting that acommon target(s) may exist. In the presence of oxygen,AS decomposition produces an oxidant with a potencysimilar to ONOO� [45]. Both HNO and ONOO� canmediate oxidative stress in tissue culture, and oxidationchemistry is likely to be implicated in their detrimental,postischemic effects. The low concentration of AS usedin this study is unlikely to impose any deleterious oxi-
dative stress; however, specific oxidation of key molec-ular targets may be an early signal that triggers a protec-tive response [46]. Although AS and ONOO� exhibitdifferent nitration and oxidation profiles toward phenoliccompounds such as tyrosine and phenylalanine analogs[26,45,47], both readily oxidize flavins and thiols[45,47], which may represent the putative common tar-gets.
Thiols are also oxidized by ONOO�, however, therapid reaction of thiols with HNO produces RSNHOHspecies [25,26]. This intermediate can then react with areduced thiol to form an oxidized disulfide. Therefore,biomolecules that can be oxidized by ONOO� may alsobe converted to the same end-products following expo-sure to AS through either direct binding of HNO orindirectly via its oxidized intermediate. The completeinhibition by NAC of the protective effect exerted by AS,without significant alterations in any other of the exper-imental parameters, is therefore not surprising. Never-theless, the NAC effect is intriguing given the presenceof millimolar concentrations of reduced glutathione,ascorbate, and other potential redox-active scavengers inmyocardial tissue.
Observation of IPC-like protection in cardiac myo-cytes has been observed following exposure to diazox-ide. This drug is known to open mitochondrial but notsarcolemmal KATP channels, supporting the view that apro-oxidant environment favors cardioprotection [12].The pharmacological response to diazoxide was pro-posed to occur through oxidation of critical flavoproteins[12], although no change in flavoprotein autofluores-cence was detected in intact cardiomyocytes [48]. Thediazoxide-induced increase in reactive oxygen specieswas abrogated by addition of either NAC or N-mercap-topropionylglycine in both adult cardiomyocytes [49]and a human atrium-derived cell line [50], a finding thatwas to be expected in view of the antioxidant propertiesof thiols. More importantly, however, the protective ef-fects of diazoxide (100 �M) were significantly bluntedby NAC (4 mM) in isolated rat hearts [32,49]. Sinceexposure to NO and ONOO� may also result in anopening of KATP channels [12,46,51,52], it seems that apreconditioned state may be triggered by induction of anoxidative burst following channel opening [53] ratherthan by direct action of the RNOS, although a moleculartarget has not been identified. The protein kinase C(PKC) activation/translocation appears to be an impor-tant step to protection induced by ischemia and pharma-cological interventions, although the location of PKCdownstream and/or upstream of mitochondrial KATP
channels is still controversial. [12,35,46,53]. Moreover,in the cellular cascade leading to preconditioning notonly PKC, but also p38 MAPK can be directly activatedby free radicals [54]. Preliminary data obtained with four
Fig. 6. Postischemic myocardial injuries following 30 min of globalischemia and 120 min reperfusion. (A) Postischemic lactate dehydro-genase (LDH) leakage, (B) Infarct size (IS). LV � left ventricle. AS �Angeli’s salt; IPC � ischemic preconditioning; NAC � N-acetyl-L-cysteine. Data are means � SEM. **p � .01 vs. vehicle/control group.
39Nitroxyl-induced preconditioning

hearts show that the PKC inhibitor GF109203X (0.5 �M;Calbiochem, San Diego, CA, USA; IC50 for PKC inhi-bition � 0.01 �M [55]), co-infused with AS is unable toprevent AS-induced protection. This finding is consistentwith the concept that during the triggering phase ofprotection, the pathways between the membrane recep-tors, free radicals from one hand, and kinases and KATP
channel from the other appear to contain redundancy[54]. Studies are currently underway to examine whetherHNO also promotes KATP channel opening and, if so,whether these effects are involved in the signaling cas-cade leading to protection [46].
Calcitonin gene-related peptide (CGRP) has been re-cently shown to precondition the rat heart [11,56], andwe demonstrated that AS-induced positive inotropic ac-tion can be abrogated by blocking CGRP receptors [29].Accordingly, a preliminary set of experiments (n � 4)assessed the impact of the selective CGRP receptor an-tagonist CGRP-(8–37) (0.1 �M; Sigma) on myocardialprotection evoked by AS. As expected, CGRP-(8–37)prevented the beneficial action of nitroxyl on both func-tional recovery and infarct size. More extensive studiesare currently underway to evaluate whether AS precon-ditioning action is associated with the release of CGRP,as previously described with ischemic- and nitroglycer-in-induced preconditioning [11,56].
Global ischemia typically results in multiple smallareas of necrosis with a heterogeneous distribution ofviable tissue in all groups. This fact, together with theoccurrence of myocardial stunning, may provide an ex-planation for the high reductions in inotropy in allgroups. Temporal changes in left ventricular functionduring reperfusion over 2 h show that maximal recoverycan be appreciated at 30–40 min reperfusion (Fig. 5).Whether the subsequent progressive reduction in devel-oped LVP is due to increasing infarct size, accentuationof stunning and/or preparate deterioration cannot be eas-ily determined. Limiting analysis to the first 30 min ofreperfusion (i.e., when preparate deterioration is limited)suggests that the improvement in postischemic myocar-dial function in the three groups in which infarct size isreduced (i.e., AS-, IPC-, and DEA/NO-pretreated hearts)is mainly due to infarct size limitation (Figs. 3 and 4)rather than to a reduction of stunning of the viablemyocardium. Yet, recovery of function was statisticallysignificant vs. controls only in the AS- and IPC-pre-treated hearts. This suggests that these two treatmentsreduced not only infarct size but also the degree to whichthe surviving myocardium had been stunned. Since freeradicals markedly contribute to stunning of reperfusedmyocardium [36,40], it may be speculated that IPC andAS pretreatment induce moderate oxidative stresses thatlead to alteration of kinases activity, KATP channel acti-vation, or changes in sulfhydryl group redox state, which
could be involved in the antistunning effect followingprolonged ischemia [57,58]. An alternative explanationof the antistunning effect may derive from the recentstudy of Gelpi et al. [40] in which it was suggested thatimprovement of recovery of function in ischemic pre-conditioned rat heart may be attributed to reduced for-mation of free radicals via a xanthine oxidase (XO)pathway due to the reduced release of purine by precon-ditioned heart. In fact, in XO-deficient rabbit hearts,recovery of function was not a function of ischemicpreconditioning despite marked reduction in infarct sizein preconditioned hearts [40]. Most notably Gelpi et al.[40] suggested that prevention of contracture might be amore robust indicator of protection than contractility.Therefore, the beneficial effects exerted by AS on bothmyocardial contractility and relaxation (i.e., reduction incontracture development) at reperfusion is an intriguingresult that deserves further investigation, as it may indi-cate an improvement of myofilament responsiveness toCa2� [58].
As in previous in vivo studies [27,29], the presentinvestigation was purposely designed to discriminate theactions of HNO and NO, bearing in mind that AS re-leases both HNO and nitrite, a principal oxidation prod-uct of NO. Since DEA/NO and AS similarly affectvascular tone (approximately �15% in perfusion pres-sure) and inotropy (approximately �10% in dP/dtmax)during drug infusion (data not shown) and since theseeffects disappear during the following wash out, changesin tissue perfusion and contractility are unlikely to ac-count for the different impact of the two agents onI/R-induced myocardial injury. This may reflect the or-thogonal profiles of NO and HNO biochemistry andpharmacology observed to date. NO is a relatively inertmolecule whose myocardial actions, including its pre-conditioning effect, are well known and appreciated [59].In contrast, HNO has a more diverse reactivity[19,26,45,47,60], and its functional properties, with theexception of vasodilatation [29,61,62], are still largelyunknown and, perhaps, quite dissimilar from those at-tributed to NO [27,29]. For instance, regulation ofNMDA function by HNO has been shown to differ fromthat of NO [28]. Moreover, unlike the NO donor S-nitrosoglutathione, administration of AS (3 �mol/kg) atreperfusion markedly enhanced neutrophil-derived my-eloperoxidase (MPO) activity in the in vivo reperfusedmyocardium. This increase correlated with plasma crea-tine kinase (CK) release, offering an explanation for thedetrimental effects of AS on blood-reperfused myocar-dium [27]. However, it should be noted that the AS (1�mol/kg) given at reperfusion was without effect onMPO and CK activities and on I/R-mediated tissue ne-crosis. A similar dose, comparable to that used in thepresent study, induced a redox-dependent positive ino-
40 P. PAGLIARO et al.

tropic response in conscious dogs [29]. It is intriguing tospeculate that there may be a concentration window inwhich HNO may serve as a pro-oxidant triggering stim-ulus [63] during the preconditioning phase to affordprotection against subsequent ischemia/reperfusion-in-duced myocardial injury.
In conclusion, HNO generated by Angeli’s salt in-duces early IPC-like effects in isolated rat hearts result-ing in reduced LDH release and myocardial necrosis andimproved cardiac performance at reperfusion. To a lesserextent, these effects were mimicked by the NO donorDEA/NO. Whether HNO may be produced endog-enously remains to be elucidated. There is increasingevidence for formation of HNO by nitric oxide synthases[21–23] and the present data, combined with our pre-vious findings [27,29], lend further support to the as-sumption that this redox sibling of NO is involved inmodulation of cardiac function under both normal (pre-ischemic) and pathological (postischemic) conditions.Finally, the enhanced, thiol-dependent protection in-duced by HNO compared to equimolar NO suggests amechanism involving either an oxidative alteration or theinvolvement of critical thiols or metals [60,64].
Acknowledgements — We thank Prof. Fabio Di Lisa and Prof. Gi-useppe Poli for helpful advice and suggestions. The authors are alsograteful to Dr. Myron L. Weisfeldt for critically reviewing the manu-script and to Drs. Jon M. Fukuto and Joseph E. Saavedra for providingus with AS and DEA/NO. This study was supported by Compagnia diSan Paolo, University of Turin and MIUR [(Cofin), PP] and by aBeginning Grant-in-Aid (0265435U) by the American Heart Associa-tion (NP).
REFERENCES
[1] Murry, C. E.; Jennings, R. B.; Reimer, K. A. Preconditioning withischemia: a delay of lethal cell injury in ischemic myocardium.Circulation 74:1124–1136; 1986.
[2] Downey, J. M.; Cohen, M. V. Signal transduction in ischemicpreconditioning. Adv. Exp. Med. Biol. 430:39–55; 1987.
[3] Bolli, R. The late phase of preconditioning. Circ. Res. 87:972–983; 2000.
[4] Pagliaro, P.; Gattullo, D.; Rastaldo, R.; Losano, G. Ischemicpreconditioning: from the first to the second window of protec-tion. Life Sci. 69:1–15; 2001.
[5] Marber, M. S.; Latchman, D. S.; Walker, J. M.; Yellon, D. M.Cardiac stress protein elevation 24 hours after brief ischemia orheat stress is associated with resistance to myocardial infarction.Circulation 88:1264–1272; 1993.
[6] Tang, X. L.; Qiu, Y.; Park, S. W.; Sun, J. Z.; Kalya, A.; Bolli, R.Time course of late preconditioning against myocardial stunningin conscious pigs. Circ. Res. 79:424–434; 1996.
[7] Baxter, G. F.; Goma, F. M.; Yellon, D. M. Characterisation of theinfarct-limiting effect of delayed preconditioning: timecourse anddose-dependency studies in rabbit myocardium. Basic Res. Car-diol. 92:159–167; 1997.
[8] Bolli, R.; Bhatti, Z. A.; Tang, X. L.; Qiu, Y.; Zhang, Q.; Guo, Y.;Jadoon, A. K. Evidence that late preconditioning against myocar-dial stunning in conscious rabbits is triggered by the generation ofnitric oxide. Circ. Res. 81:42–52; 1997.
[9] Takano, H.; Tang, X. L.; Qiu, Y.; Guo, Y.; French, B. A.; Bolli,R. Nitric oxide donors induce late preconditioning against myo-
cardial stunning and infarction in conscious rabbits via an anti-oxidant-sensitive mechanism. Circ. Res. 83:73–84; 1998.
[10] Hill, M.; Takano, H.; Tang, X. L.; Kodani, E.; Shirk, G.; Bolli, R.Nitroglycerin induces late preconditioning against myocardialinfarction in conscious rabbits despite development of nitratetolerance. Circulation 104:694–699; 2001.
[11] Hu, C.-P.; Li, Y.-J.; Deng, H.-W. The cardioprotective effects ofnitroglycerin-induced preconditioning are mediated by calcitoningene-related peptide. Eur. J. Pharmacol. 369:189–194; 1999.
[12] Sasaki, N.; Sato, T.; Ohler, A.; O’Rourke, B.; Marban, E. Acti-vation of mitochondrial ATP-dependent potassium channels bynitric oxide. Circulation 101:439–445; 2000.
[13] Gattullo, D.; Linden, R. J.; Losano, G.; Pagliaro, P.; Westerhof,N. Ischaemic preconditioning changes the pattern of coronaryreactive hyperaemia in the goat: role of adenosine and nitricoxide. Cardiovasc. Res. 42:57–64; 1999.
[14] Patel, V. C.; Yellon, D. M.; Singh, K. J.; Neidl, G. H.; Woolfson,R. G. Inhibition of nitric oxide limits infarct size in the in siturabbit heart. Biochem. Biophys. Res. Commun. 194:234–238;1993.
[15] Schulz, R.; Wambolt, R. Inhibition of nitric oxide synthesisprotects the isolated working rabbit heart from ischaemia-reper-fusion injury. Cardiovasc. Res. 30:432–439; 1995.
[16] Woolfson, R. G.; Patel, V. C.; Neild, G. H.; Yellon, D. M.Inhibition of nitric oxide synthesis reduces infarct size by anadenosine-dependent mechanism. Circulation 91:1545–1551;1995.
[17] Weselcouch, E. O.; Baird, A. J.; Sleph, P.; Grover, G. J. Inhibitionof nitric oxide synthesis does not affect ischemic preconditioningin isolated perfused rat hearts. Am. J. Physiol. 268:H242–H249;1995.
[18] Nakano, A.; Liu, G. S.; Heusch, G.; Downey, J. M.; Cohen, M. V.Exogenous nitric oxide can trigger a preconditioned state througha free radical mechanism, but endogenous nitric oxide is not atrigger of classical ischemic preconditioning. J. Mol. Cell. Car-diol. 32:1159–1167; 2000.
[19] Miranda, K.; Espey, M. G.; Jourd’heuil, D.; Grisham, M. B.;Fukuto, M.; Feelisch, M.; Wink, D. A. The chemical biology ofnitric oxide. In: Ignarro, L. J., ed. Nitric oxide: biology andpathobiology. San Diego, London: Academic Press; 2000:41–55.
[20] Xia, Y.; Zweier, J. L. Direct measurement of nitric oxide gener-ation from nitric oxide synthase. Proc. Natl. Acad. Sci. USA94:12705–12710; 1997.
[21] Hobbs, A. J.; Fukuto, J. M.; Ignarro, L. J. Formation of free nitricoxide from l-arginine by nitric oxide synthase: direct enhance-ment of generation by superoxide dismutase. Proc. Natl. Acad.Sci. USA 91:10992–10996; 1994.
[22] Pufahl, R. A.; Wishnok, J. S.; Marletta, M. A. Hydrogen peroxidesupported oxidation of NG-hydroxyl-L-arginine by purified nitricoxide synthase. Biochemistry 34:1930–1941; 1995.
[23] Schmidt, H. H. H. W.; Hoffmann, H.; Schindler, U.; Shutenko,Z. S.; Cunningham, D. D.; Feelisch, M. No NO from NO syn-thase. Proc. Natl. Acad. Sci. USA 93:14492–14497; 1996.
[24] Maragos, C. M.; Morley, D.; Wink, D. A.; Dunams, T. M.;Saavedra, J. E.; Hoffman, A.; Bove, A. A.; Isaac, L.; Hrabie, J. A.;Keefer, L. K. Complexes of NO with nucleophiles as agents forthe controlled biological release of nitric oxide. Vasorelaxanteffects. J. Med. Chem. 34:3242–3247; 1991.
[25] Fukuto, J. M.; Cho, J. Y.; Switzer, C. H. The chemical propertiesof nitric oxide and related nitrogen oxides. In: Ignarro, L. J., ed.Nitric oxide: biology and pathobiology. San Diego, London:Academic Press; 2000:23–40.
[26] Wink, D. A.; Feelisch, M.; Fukuto, J.; Chistodoulou, D.;Jourd’heuil, D.; Grisham, M. B.; Vodovotz, Y.; Cook, J. A.;Krishna, M.; DeGraff, W. G.; Kim, S.; Gamson, J.; Mitchell, J. B.The cytotoxicity of nitroxyl: possible implications for the patho-physiological role of NO. Arch. Biochem. Biophys. 351:66–74;1998.
[27] Ma, X. L.; Gao, F.; Liu, G.-L.; Lopez, B. L.; Christopher, T. A.;Fukuto, J. M.; Wink, D. A.; Feelisch, M. Opposite effects of nitric
41Nitroxyl-induced preconditioning

oxide and nitroxyl on postischemic myocardial injury. Proc. Natl.Acad. Sci. USA 96:14617–14622; 1999.
[28] Colton, C. A.; Gbadegesin, M.; Wink, D. A.; Miranda, K. M.;Espey, M. G.; Vicini, S. Nitroxyl anion regulation of the NMDAreceptor. J. Neurochem. 78:1126–1134; 2001.
[29] Paolocci, N.; Saavedra, W. F.; Miranda, K. M.; Martignani, C.;Isoda, T.; Hare, J. M.; Espey, M. G.; Fukuto, J. M.; Feelisch, M.;Wink, D. A.; Kass, D. A. Nitroxyl anion exerts redox-sensitivepositive cardiac inotropy in vivo by calcitonin gene-related pep-tide signaling. Proc. Natl. Acad. Sci. USA 98:10463–10468; 2001.
[30] Rastaldo, R.; Paolocci, N.; Chiribiri, A.; Penna, C.; Gattullo, D.;Pagliaro, P. Cytochrome P-450 metabolite of arachidonic acidmediates bradykinin-induced negative inotropic effect. Am. J.Physiol. Heart Circ. Physiol. 280:H2823–H2832; 2001.
[31] Paolocci, N.; Ekelund, U. E.; Isoda, T.; Ozaki, M.; Vandegaer, K.;Georgakopoulos, D.; Harrison, R. W.; Kass, D. A.; Hare, J. M.cGMP-independent inotropic effects of nitric oxide and peroxyni-trite donors: potential role for nitrosylation. Am. J. Physiol. HeartCirc. Physiol. 279:H1982–H1988; 2000.
[32] Chen, W.; Gabel, S.; Steenbergen, C.; Murphy, E. A redox-basedmechanism for cardioprotection induced by ischemic precondi-tioning in perfused rat heart. Circ. Res. 77:424–429; 1995.
[33] Muller-Strahl, G.; Kottenberg, K.; Zimmer, H. G.; Noack, E.;Kojda, G. Inhibition of nitric oxide synthase augments the posi-tive inotropic effect of nitric oxide donors in the rat heart.J. Physiol. 522:311–320; 2000.
[34] Tani, M.; Suganuma, Y.; Takayama, M.; Hasegawa, H.; Shin-mura, K.; Ebihara, Y.; Tamaki, K. Low concentrations of aden-osine receptor blocker decrease protection by hypoxic precondi-tioning in ischemic rat hearts. J. Mol. Cell. Cardiol. 30:617–626;1998.
[35] Wang, Y.; Takashi, E.; Xu, M.; Ayub, A.; Ashraf, M. Downregu-lation of protein kinase C inhibits activation of mitochondrialK(ATP) channels by diazoxide. Circulation 104:85–90; 2001.
[36] Cohen, M. V.; Yang, X. M.; Downey, J. M. Smaller infarct afterpreconditioning does not predict extent of early functional im-provement of reperfused heart. Am. J. Physiol. 277:H1754–H1761; 1999.
[37] Hare, J. M.; Kim, B.; Flavahan, N. A.; Ricker, K. M.; Peng, X.;Colman, L.; Weiss, R. G.; Kass, D. A. Pertussis toxin-sensitive Gproteins influence nitric oxide synthase III activity and proteinlevels in rat heart. J. Clin. Invest. 101:1424–1431; 1998.
[38] Weselcouch, E. O.; Baird, A. J.; Sleph, P. G.; Dzwonczyk, S.;Murray, H. N.; Grover, G. J. Endogenous catecholamines are notnecessary for ischaemic preconditioning in the isolated perfusedrat heart. Cardiovasc. Res. 29:126–132; 1995.
[39] Bergmeyer, H. U.; Bernt, E., eds. Methods of enzymatic analysis.Weinheim, Germany: Verlag Chemie; 1974.
[40] Gelpi, R. J.; Morales, C.; Choen, M. V.; Downey, J. M. Xanthineoxidase contributes to preconditioning’s preservation of left ven-tricular developed pressure in isolated rat heart: developed pres-sure may not be an appropriate end-point for studies of precon-ditioning. Basic Res. Cardiol. 97:40–46; 2002.
[41] Altup, S.; Demiryurek, A. T.; Ak, D.; Tungel, M.; Kanzik, I.Contribution of peroxynitrite to the beneficial effects of precon-ditioning on ischaemia-induced arrhythmias in rat isolated hearts.Eur. J. Pharmacol. 415:239–246; 2001.
[42] Conska, C.; Csont, T.; Onody, A.; Ferdinandy, P. Preconditioningdecreases ischemia/reperfusion-induced peroxynitrite formation.Biochem. Biophys. Res. Commun. 285:1217–1219; 2001.
[43] Altug, S.; Demiryurek, A. T.; Kane, K. A.; Kanzik, I. Evidencefor the involvement of peroxynitrite on ischemic preconditioningin rat isolated hearts. Br. J. Pharmacol. 130:125–131; 2000.
[44] Ma, X. L.; Gao, F.; Lopez, B. L.; Christopher, T. A.; Vinten-Johansen, J. Peroxynitrite, a two-edged sword in post-ischemicmyocardial injury-dichotomy of action in crystalloid- versusblood-perfused hearts. J. Pharmacol. Exp. Ther. 292:912–920;2000.
[45] Miranda, K. M.; Espey, M. G.; Yamada, K.; Krishna, M.; Lud-wick, N.; Kim, S.; Jourd’heuil, D.; Grisham, M. B.; Feelisch, M.;Fukuto, J. M.; Wink, D. A. Unique oxidative mechanisms for the
reactive nitrogen species, nitroxyl anion. J. Biol. Chem. 276:1720–1727; 2001.
[46] Patel, H. H.; Gross, G. J. Diazoxide induced cardiprotecion: whatcomes first, KATP channels or reactive oxygen species? Cardio-vasc. Res. 51:633–636; 2001.
[47] Miranda, K. M.; Yamada, K.-I.; Thomas, D. D.; DeGraff, W.;Espey, M. G.; Krishna, M.; Mitchell, J. B.; Colton, C. A.; Wink,D. A. Further evidence for distinctive reactive intermediates fromnitroxyl and peroxynitrite: effects of buffer conditions on thechemistry of angeli’s salt and synthetic peroxynitrite. Arch. Bio-chem. Biophys. 401:134–144; 2002.
[48] Lawrence, C. L.; Billups, B.; Rodrigo, G. C.; Standen, N. B. TheK(ATP) channel opener diazoxide protects cardiac myocytes duringmetabolic inhibition without causing mitochondrial depolarization orflavoprotein oxidation. Br. J. Pharmacol. 134:535–542; 2001.
[49] Forbes, R. A.; Steenbergen, C.; Murphy, E. Diazoxide-inducedcardioprotection requires signaling through a redox-sensitivemechanism. Circ. Res. 88:802–809; 2001.
[50] Carroll, R.; Gant, V. A.; Yellon, D. M. Mitochondrial KATP channelsprotects a human atrial-derived cell line by a mechanism involvingfree radical generation. Cardiovasc. Res. 51:691–700; 2001.
[51] Ockaili, R.; Emani, V. R.; Okubo, S.; Brown, M.; Krottapalli, K.;Kukreja, R. C. Openeing of mitochondrial KATP channel inducesearly and delayed cardioprotective effect: role of nitric oxide.Am. J. Physiol. 277:H2425–H2434; 1999.
[52] Wei, E. P.; Kontos, H. A.; Beckman, J. S. Mechanisms of cerebralvasodilation by superoxide, hydrogen peroxide and peroxynitrite.Am. J. Physiol. 271:H1262–H1266; 1996.
[53] Pain, T.; Yang, X. M.; Critz, S. D.; Yue, Y.; Nakano, A.; Liu,G. S.; Heusch, G.; Cohen, M. V.; Downey, J. M. Opening ofmitochondrial KATP channels triggers the preconditioned state bygenerating free radicals. Circ. Res. 87:460–466; 2000.
[54] Cohen, M. V.; Yang, X. M.; Liu, G. S.; Heusch, G.; Downey,J. M. Acetylcholine, bradykinin, opioids, and phenylephrine, butnot adenosine, trigger preconditioning by generating free radicalsand opening mitochondrial K(ATP) channels. Circ. Res. 89:273–278; 2001.
[55] Inagaki, K.; Kihara, Y.; Hayashida, W.; Izumi, T.; Iwanaga, Y.;Yoneda, T.; Takeuchi, Y.; Suyama, K.; Muso, E.; Sasayama, S.Anti-ischemic effect of a novel cardioprotective agent, JTV519, ismediated through specific activation of delta-isoform of proteinkinase C in rat ventricular myocardium. Circulation 101:797–804; 2000.
[56] Li, Y. J.; Peng, J. The cardioprotection of calcitonin gene-relatedpeptide-mediated preconditioning. Eur. J. Pharmacol. 442:173–177; 2002.
[57] Tritto, I.; Ambrosio, G. Role of oxidants in the signaling pathwayof preconditioning. Antioxid. Redox Signal. 3:3–10; 2001.
[58] An, J.; Varadarajan, S. G.; Novalija, E.; Stowe, D. F. Ischemicand anesthetic preconditioning reduces cytosolic [Ca2�] and im-proves Ca2� responses in intact hearts. Am. J. Physiol. HeartCirc. Physiol. 281:H1508–H1523; 2001.
[59] Balligand, J.-L.; Feron, O.; Kelly, R. A. Role of nitric oxidein myocardial function. In: Ignarro, L. J., ed. Nitric oxide: bi-ology and pathobiology. San Diego, London: Academic Press;2000:585–632.
[60] Miranda, K. M.; Nims, R. W.; Thomas, D. D.; Espey, M. G.;Citrin, D.; Bartberger, M. D.; Paolocci, N.; Fukuto, J. M.; Feel-isch, M.; Wink, D. A. Comparison of the reactivity of nitric oxideand nitroxyl with heme proteins. A chemical discussion of thedifferential biological effects of these redox related products ofNOS. J. Inorg. Biochem. in press; 2002.
[61] Pino, R. Z.; Feelisch, M. Bioassay discrimination between nitricoxide (NO) and nitroxyl (NO�) using L-cysteine. Biochem. Bio-phys. Res. Commun. 201:54–62; 1994.
[62] Ellis, A.; Li, C. G.; Rand, M. J. Differential actions of L-cysteineon responses to nitric oxide, nitroxyl anions and EDRF in the rataorta. Br. J. Pharmacol. 129:315–322; 2000.
[63] Reif, A.; Zecca, L.; Riederer, P.; Feelisch, M.; Schmidt, H. H.Nitroxyl oxidizes NADPH in a superoxide dismutase inhibitablemanner. Free Radic. Biol. Med. 30:803–808; 2001.
42 P. PAGLIARO et al.

[64] Wong, P. S.-Y.; Hyun, J.; Fukuto, J. M.; Shirota, F. N.; DeMaster,E. G.; Shoeman, D. W.; Nagasawa, H. T. Reaction betweenS-nitrosothiols and thiols: generation of nitroxyl (HNO) and sub-sequent chemistry. Biochemistry 37:5362–5371; 1998.
ABBREVIATIONS
AS—Angeli’s salt (Na2N2O3)CGRP—calcitonin gene-related peptideCK—creatine kinaseCPP—coronary perfusion pressureDEA/NO—diethylamine/NO adductdP/dtmax—maximum rate of increase of LVP during sys-
tole
I/R—ischemia/reperfusionIPC—ischemic preconditioningLDH—lactate dehydrogenaseLEDVP—left end-diastolic ventricular pressureLV—left ventricleLVP—left ventricular pressureMPO—myeloperoxidaseNAC—N-acetyl-L-cysteineNMDA—N-methyl-D-aspartateNOS—nitric oxide synthasePKC—protein kinase CRNOS—reactive nitrogen oxide speciesXO—xanthine oxidase
43Nitroxyl-induced preconditioning
Copyright © 2022 FDOKUMEN




















