
Structural studies on enzymes from Salmonella typhimurium ...
234
Structural studies on enzymes from Salmonella typhimurium involved in propionate metabolism: biodegradative threonine deaminase, propionate kinase and 2-methylisocitrate lyase A thesis submitted for the Degree of Doctor of Philosophy in the Faculty of Science by Dhirendra Kumar Simanshu Molecular Biophysics Unit INDIAN INSTITUTE OF SCIENCE Bangalore - 560 012 INDIA September 2006
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of Structural studies on enzymes from Salmonella typhimurium ...

Structural studies on enzymes from Salmonella typhimurium involved in propionate metabolism:
biodegradative threonine deaminase, propionate kinase and 2-methylisocitrate lyase
A thesis submitted for the Degree of
Doctor of Philosophy in the Faculty of Science
by
Dhirendra Kumar Simanshu
Molecular Biophysics Unit
INDIAN INSTITUTE OF SCIENCE Bangalore - 560 012
INDIA September 2006

Dedicated
to “my parents”

Declaration
I hereby declare that the research work reported in the thesis entitled “Structural
studies on enzymes from Salmonella typhimurium involved in propionate metabolism:
biodegradative threonine deaminase, propionate kinase and 2-methylisocitrate lyase” is
entirely original and was carried out by me under the supervision of Prof. M. R. N.
Murthy at the Molecular Biophysics Unit, Indian Institute of Science, Bangalore, India.
I further declare that the scientific contents of this thesis have not been the basis
for award of any degree, diploma, fellowship, associateship or any other similar title of
any University or Institution.
Date:
Dhirendra Kumar Simanshu
SR No. 111501461
Molecular Biophysics Unit
Indian Institute of Science
Bangalore - 560 012, India

Certificate
This is to certify that the work described in the thesis entitled “Structural studies
on enzymes from Salmonella typhimurium involved in propionate metabolism:
biodegradative threonine deaminase, propionate kinase and 2-methylisocitrate lyase” is
the result of investigations carried out by Dhirendra Kumar Simanshu at the Molecular
Biophysics Unit, Indian Institute of Science, Bangalore, India under my supervision, and
the results presented in this thesis have not previously formed the basis for the award of
any other diploma, degree or fellowship.
Date
Prof. M. R. N. Murthy
Molecular Biophysics Unit
Indian Institute of Science
Bangalore - 560 012, India

Acknowledgements
I am indeed fortunate to have worked with Prof. M. R. N. Murthy who has been a great teacher and mentor. He has given me a great research experience and education and I have thoroughly enjoyed working with him. The freedom of thought and action extended by him was immense. His encouragement and help made me feel confident to overcome every problem I encountered. I take this opportunity to express my deep sense of gratitude to him for his concern, guidance, encouragement and constant support during my five years of stay.
Most of the work reported in this thesis has been done in collaboration with Prof. H. S. Savithri. It was wonderful to be associated with her. I am grateful to her for her keen interest in my work, her constructive criticisms and timely suggestions during the scientific discussions. My heartfelt thanks to both Prof. Murthy and Savithri for their parental care and for making me feel at home.
My sincere thanks to Prof. Vijayan and Prof. Suguna for teaching basic crystallography and for their constant encouragement and interest in my progress. Dr. Gopal deserves special thanks for helping us to adopt various new advancements in our research work and for his constant enquiries about my work. I am greatly indebted to Prof. N. Appaji Rao for his valuable suggestions during the course of study. I would like to acknowledge Prof. Balaram, Prof. Manju Bansal, Prof. Saraswathi Vishweshwara, Prof. Srinivasan and Prof. Ramkumar for the wonderful courses offered by them. I would like to sincerely thank Dr. Rajiv Bhat, JNU, for introducing me to the subject of biophysics.
I fall short of words to express my heartfelt gratitude to all my teachers who have guided me during my school and college days. It was their belief that has kept me going. I would like to thank all the scientists who have provided us freely a number of excellent programs for solving and analyzing crystal structures. My heartfelt thanks to the whole CCP4 team for coming and giving some wonderful lectures and training during the CCP4 workshop.
My sincere thanks to Mr. Babu and Mr. James for excellent maintenance of X-ray lab round the clock. I owe a debt of gratitude to Ms. Gayathri, Dr. Lokanath, Dr. Kunishima and Prof. Tsukihara for their help in synchrotron data collection at SPring-8, Hyogo, Japan.
My sincere thanks are due to Dr. Sathesh and Dr. Parthasarathy for teaching me tips and tricks of molecular biology and crystallographic techniques. I gratefully acknowledge my seniors Dr. Isai, Dr. Sangita, Dr. Jeyaprakash and Dr. Saikrishnan for their help during my initial days. My heartfelt thanks to Sougata and Kausik for guiding me during my interview days in MBU.
I thank Anupama, Divya, Nandashree, Sagar and Bharath (JNC) for trying their best to crystallize some of the difficult proteins. I thank Sagar for helping me with the cloning of biodegradative threonine deaminase. I thank Subhash and Rajavel for their help in gel filtration experiments. I acknowledge the help of Swarna during the structure analysis of 2-methylisocitrate lyase. I thank Parimal for his help in analyzing the kinetics data. I thank Poornima for her help in carrying out radioactive experiments. My wholehearted thanks are due to all my past and present lab mates Partha, Isai, Sangita, Rajaram, Gayathri, Somesh, Anju, Sagar, Bharath, Krishna, Vijayalakshmi, Prasad, Archana, Giri, Chandrani, Rajaganapathy, Prasuna, Jothi, Moumita, Anupama, Nandashree, Divya, Venkatesh, Raghavan and Bharath (JNC). I acknowledge late Prof. Naidu for being a part of our group and for his advice on scientific and non-scientific matters. I thank Inbaraj (Raju) and Papanna for their lab assistance. Our lab has been a great and lively place to work and working in the lab has always been fun. I shall cherish lovely and ‘fun’tastic moments I have had with them. Rajaram has been helpful especially during course work and apart from being batch-mates,

Acknowledgements
together we have seen many “ups and downs” and I thank him for all his help and cooperation. My heartfelt thanks to Anju and Gayathri for their concern, moral support and warm friendship extended to me. Gayathri stands out as one of the most intelligent, gracious and humble individuals I have ever known. I have benefited immensely from the numerous scientific discussions I have had with her during the course of investigation. I gratefully acknowledge her for proofreading most of my manuscripts and the present thesis. I express my sincere thanks to her for friendly advice and help during my difficult times. I shall cherish my association with her.
I extend my heartfelt thanks to all the past and present members of Prof. Savithri’s lab Sathesh, Poornima, Anindya, Uma, Bhavani, Farida, Soumya, Lokesh, Smita, Chhavi, Subash, Sathiya, Purnima, Govind, Saraswati, Vinitha, Ramachandra and Rajani for their friendships and for the help with their lab facilities. I treasure the warm friendship I share with Poornima.
All the members of X-ray group and D’Cryst have been very cooperative. I have benefited immensely from the scientific discussions on various topics of structural biology in D’Cryst. I thank all the lab members of Prof. Vijayan’s group, Prof. Suguna’s group and Dr. Gopal’s group for their friendship and for the help with their lab facilities. I thank Rajan and Bhaskar for excellent maintenance of the computing facility for the X-ray group.
I take this opportunity to thank all my batchmates Ashima, Ashima (DIC), Chanakya, Fredrick, Gowri, Gyanendra, Padmashri, Prajapati, Rajaram, Sabareesh, Sathyapriya, Siddharth, Shailendra, Swarna and Vikas for the fun times we had together. I thank all the MBUites for their friendly gestures, refreshing smiles and for their cooperation. I also enjoyed working with Ramya, Shailendra and Swarna in organizing Nu biophysical society (NuBS) events. I thank Shilpi for her efforts in keeping all the centrifuges in working condition. I would like to thank my “C mess” gang Bharath, Anju, Vijayalakshmi and Vetri for their great companionship during last two years.
I have been blessed with many friends who have extended their unconditional love and support during my good and bad times. I would like to acknowledge all my B. Sc and M. Sc. friends for their help and encouragement. I have always cherished the company of my school and college friends. I thank Birendra, Durganand, Jamal, Manjeet, Mustkim and Rakesh for being wonderful friends.
I have thoroughly enjoyed playing cricket for MBU during my five years of stay in the campus. Memory of winning BICS cup continuously for four years is something I will cherish. I take this opportunity to thank all my MBU teammates and my friends in BC, MCBL, MRDG and various engineering departments. I also acknowledge Gymkhana for providing excellent facility.
My thanks are due to Supercomputer Education Research Centre and their supporting staff for providing excellent computing and graphics facility. I thank Mr. Raju for helping with network facility and Mr. Govindaraju for maintenance of spectroscopic instruments. I would like to thank Mr. Prakash (proteomics facility) for helping me with the mass spectroscopic studies. I thank MBU office staff Mrs. Radha, Mr. Ravindran and Mr. Shivshankar especially for making sure that I got my fellowship on time. The financial assistance by the Council for Scientific and Industrial Research, Government of India and Indian Institute of Science is gratefully acknowledged. I acknowledge Department of Science and Technology and Department of Biotechnology, Government of India, for supporting the X-ray facility at MBU.
I express my heartfelt thanks to brothers, sisters and brothers/sisters-in-law for their love, support and encouragement.
Words are not enough to thank my parents for educating me with aspects from both arts and sciences, for their sacrifice, unconditional love, trust, constant support and encouragement to pursue my interests. I dedicate this thesis to them.
Dhirendra Kumar Simanshu
vi

Preface
I formally joined Prof. M. R. N. Murthy’s laboratory at the Molecular Biophysics
Unit, Indian institute of Science, on 1st August 2001. During that time, the interest in the
laboratory was mainly focused on structural studies on a number of capsid mutants of two
plant viruses, sesbania mosaic virus and physalis mottle virus, to gain an insight into the
virus structure and its assembly. Besides these two projects, there were a few other
collaborative projects running in the lab at that time such as NIa protease from pepper
vein banding virus and diaminopropionate ammonia lyase from Escherichia coli with
Prof. H. S. Savithri, triosephosphate isomerase from Plasmodium falciparum with Prof.
P. Balaram and Prof. H. Balaram and a DNA binding protein (TP2) with Prof. M. R. S.
Rao. During my first semester, along with my course work, I was assigned to make an
attempt to purify and crystallize recombinant NIa protease and TP2 protein. I started with
NIa protease which could be purified using one step Ni-NTA affinity column
chromatography. Although the expression and protein yield were reasonably good,
protein precipitated with in a couple of hours after purification. Attempts were made to
prevent the precipitation of the purified enzyme and towards this end we were successful
to some extent. However, during crystallization trials most of the crystallization drops
precipitated completely even at low protein concentration. TP2 protein was purified using
three-step chromatographic techniques by one of the project assistant in Prof. M. R. S.
Rao’s laboratory. Because of low expression level and three step purification protocol,
protein yield was not good enough for complete crystallization screening. Hits obtained
from our initial screening could not be confirmed because of low protein yield as well as
batch to batch variation. My attempts to crystallize these two proteins remained
unsuccessful but in due course I had learnt a great deal about the tips and tricks of
expression, purification and mainly crystallization. To overcome the problems faced with
these two proteins, we decided to make some changes in the gene construct and try
different expression systems.
By this time (beginning of 2002), I had finished my first semester and a major
part of the course work, so we decided to start a new project focusing on some of the
unknown enzymes from a metabolic pathway. Dr. Parthasarathy, who had finished his
Ph. D. from the lab, helped me in literature work and in finding targets for structural

Preface
studies. Finally, we decided to target enzymes involved in the propionate metabolism.
The pathways for propionate metabolism in Escherichia coli as well as Salmonella
typhimurium were just established and there were no structural information available for
most of the enzymes involved in these pathways. Since, propionate metabolic pathways
were well described in the case of Salmonella typhimurium, we decided to use this as the
model organism. We first started with the enzymes present in the propionate catabolic
pathway “2-methylcitrate pathway”, which converts propionate into pyruvate and
succinate. 2-methylcitrate pathway resembles the well-studied glyoxylate and TCA cycle.
Most of the enzymes involved in 2-methylcitrate pathway were not characterized
biochemically as well as structurally. First, we cloned all the four enzymes PrpB, PrpC,
PrpD and PrpE present in the prpBCDE operon along with PrpR, a transcription factor,
with the help of Dr. P.S. Satheshkumar from Prof. H. S. Savithri’s laboratory. Since these
five proteins were cloned with either N- or C-terminal hexa-histidine tag, they could be
purified easily using one-step Ni-NTA affinity column chromatography. PrpB, PrpC and
PrpD had good expression levels but with PrpE and PrpR, more than 50% of the
expressed protein went into insoluble fraction, probably due to the presence of membrane
spanning domains in these two enzymes. Around this time, crystallization report for the
PrpD from Salmonella was published by Ivan Rayment’s group, so after that we focused
only on the remaining four proteins leaving out PrpD. Our initial attempts to crystallize
these proteins became successful in case of PrpB, 2-methylisocitrate lyase. We collected
a complete diffraction data to a resolution of 2.5 Å which was later on extended to a
resolution of 2.1 Å using another crystal. Repeated crystallization trials with PrpC also
gave small protein crystals but they were not easy to reproduce and size and diffraction
quality always remained a problem. Using one good crystal obtained for PrpC, data to a
resolution of 3.5 Å could be collected. Unfortunately, during data collection due to failure
of the cryo-system, a complete dataset could not be collected. Further attempts to
crystallize this protein made by Nandashree, one of my colleagues in the lab at that time,
was also without much success. Attempts to purify and crystallize PrpE and PrpR were
made by me as well as one of my colleagues, Anupama. In this case, besides
crystallization, low expression and precipitation of the protein after purification were
major problems.
viii

Preface
Our attempt to phase the PrpB data using the closest search model
(phosphoenolpyruvate mutase) by molecular replacement technique was unsuccessful,
probably because of low sequence identity between them (24%). Further attempts were
made to obtain heavy atom derivatives of PrpB crystal. We could obtain a mercury
derivative using PCMBS. However, an electron density map based on this single
derivative was not interpretable. Around this time, the structure of 2-methylisocitrate
lyase (PrpB) from E. coli was published by Grimm et. al. The structure of Salmonella
PrpB could easily be determined using the E. coli PrpB enzyme as the starting model. We
also solved the structure of PrpB in complex with pyruvate and Mg2+. Our attempts to
crystallize PrpB with other ligands were not successful. Using the structures of PrpB and
its complex with pyruvate and Mg2+, we carried out comparative studies with the well-
studied structural and functional homologue, isocitrate lyase. These studies provided the
plausible rationale for different substrate specificities of these two enzymes. Due to
unavailability of PrpB substrate commercially and the extensive biochemical and
mutational studies carried out by two different groups made us turn our attention to other
enzymes in this metabolic pathway. Since our repeated attempts to obtain good
diffraction quality crystals of PrpC, PrpE and PrpR continued to be unsuccessful, we
decided to target other enzymes involved in propionate metabolism.
We looked into the literature for the metabolic pathways by which propionate is
synthesized in the Salmonella typhimurium and finally decided to target enzymes present
in the metabolic pathway which converts L-threonine to propionate. Formation of
propionate from L-threonine is the most direct route in many organisms. During February
2003, we initiated these studies with the last enzyme of this pathway, propionate kinase
(TdcD), and within a couple of months we could obtain a well-diffracting crystal in
complex with ADP and with a non-hydrolysable ATP analog, AMPPNP. TdcD structure
was solved by molecular replacement using acetate kinase as a search model. Propionate
kinase, like acetate kinase, contains a fold with the topology βββαβαβα, identical with
that of glycerol kinase, hexokinase, heat shock cognate 70 (Hsc70) and actin, the
superfamily of phosphotransferases. Examination of the active site pocket in propionate
kinase revealed a plausible structural rationale for the greater specificity of the enzyme
towards propionate than acetate.
ix

Preface
One of the datasets of TdcD obtained in the presence of ATP showed extra
continuous density beyond the γ-phosphate. Careful examination of this extra electron
density finally allowed us to build diadenosine tetraphosphate (Ap4A) into the active site
pocket, which fitted the density very well. Since the data was collected at a synchrotron
source to a resolution of 1.98 Å, we could identify the ligand in the active site pocket
solely on the basis of difference Fourier map. Later on, co-crystallization trials of TdcD
with commercially available Ap4A confirmed its binding to the enzyme. These studies
suggested the presence of a novel Ap4A synthetic activity in TdcD, which is further being
examined by biochemical experiments using mass-spectrometry as well as thin-layer
chromatography experiments.
By the end of 2004, we shifted our focus to the first enzyme involved in the
anaerobic degradation of L-threonine to propionate, a biodegradative threonine
deaminase (TdcB). Sagar Chittori, who had joined the lab as an integrated Ph. D student,
helped me in cloning this enzyme. My attempt to crystallize this protein became finally
successful and datasets in three different crystal forms were collected. Dataset for TdcB
in complex with CMP was collected during a synchrotron trip to SPring8, Japan by my
colleague P. Gayathri and Prof. Murthy. TdcB structure was solved by molecular
replacement using the N-terminal domain of biosynthetic threonine deaminase as a search
model. Structure of TdcB in the native form and in complex with CMP helped us to
understand several unanswered questions related to ligand mediated oligomerization and
enzyme activation observed in this enzyme.
The structural studies carried out on these three enzymes have provided structural
as well as functional insights into the catalytic process and revealed many unique features
of these metabolic enzymes. All these have been possible mainly due to proper guidance
and encouragement from Prof. Murthy and Prof. Savithri. Prof. Murthy’s teaching as well
as discussions during the course of investigation has helped me in a great deal to learn
and understand crystallography. Collaboration with Prof. Savithri kept me close to
biochemistry and molecular biology, the background with which I entered the world of
structural biology. The freedom to choose the project and carry forward some of my own
ideas has given me enough confidence to enjoy doing research in future.
x

CONTENTS Abbreviations . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
Abstract . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3
1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . 8 1.1 Salmonella . . . . . . . . . . . . . . . . . . . . . . . . . . 9
1.1.1 General characteristics . . . . . . . . . . . . . . . . . . 9 1.1.2 Classification . . . . . . . . . . . . . . . . . . . . . . 10 1.1.3 Salmonella enterica serovar Typhimurium . . . . . . . . . . . 10
1.2 Propionate metabolism . . . . . . . . . . . . . . . . . . . . . 12 1.3 Anaerobic degradation of L-threonine to propionate . . . . . . . . 13 1.3.1 tdc operon . . . . . . . . . . . . . . . . . . . . . . . 14 1.3.2 Regulation of the tdc operon . . . . . . . . . . . . . . . . 16 1.3.3 Enzymes encoded by the tdc operon and phosphotransacetylase . . 17 1.3.3.1 TdcB: Biodegradative threonine deaminase . . . . . . . 18 1.3.3.2 TdcC: Threonine/Serine transport protein. . . . . . . . 20 1.3.3.3 TdcD: Propionate kinase . . . . . . . . . . . . . . 21 1.3.3.4 TdcE: 2-ketobutyrate formate lyase . . . . . . . . . . 22 1.3.3.5 TdcF: A protein of unknown function . . . . . . . . . 23 1.3.3.6 TdcG: L-serine deaminase . . . . . . . . . . . . . 24 1.3.3.7 Pta: Phosphotransacetylase . . . . . . . . . . . . . 25 1.4 Propionate catabolism: 2-methylcitrate pathway . . . . . . . . . . 26 1.4.1 prp operon and 2-methylcitrate pathway in S. typhimurium . . . . 28 1.4.2 prp operon and 2-methylcitrate pathway in E. coli . . . . . . . . 31 1.4.3 Enzymes encoded by the prp operon and AcnA/AcnB . . . . . . 32 1.4.3.1 PrpR: A member of σ54 family of transcriptional activators . . 33 1.4.3.2 PrpE: Propionyl-CoA synthetase . . . . . . . . . . . 34 1.4.3.3 PrpC: 2-methylcitrate synthase . . . . . . . . . . . . 35 1.4.3.4 PrpD: 2-methylcitrate dehydratase . . . . . . . . . . 36 1.4.3.5 AcnA/AcnB: Aconitase A or Aconitase B . . . . . . . . 37 1.4.3.6 PrpB: 2-methylisocitrate lyase . . . . . . . . . . . . 39 1.4.4 Presence of 2-methylcitrate pathway among prokaryotes . . . . . 41 1.5 Objectives of the present study . . . . . . . . . . . . . . . . . 42 2 Materials and methods . . . . . . . . . . . . . . . . . . . . . . 44 2.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . 45 2.2 Materials . . . . . . . . . . . . . . . . . . . . . . . . . . . 45 2.2.1 Chemicals used in the study . . . . . . . . . . . . . . . . 45 2.2.2 Plasmids used in the study . . . . . . . . . . . . . . . . 45 2.2.3 Bacterial strains used in the study . . . . . . . . . . . . . . 47

Contents
2.3 Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . 47 2.3.1 Preparation of E. coli competent cells and transformation . . . . 47 2.3.2 Cloning and overexpression . . . . . . . . . . . . . . . . 48 2.3.3 Purification of the recombinant enzyme . . . . . . . . . . . 48 2.3.4 Crystallization . . . . . . . . . . . . . . . . . . . . . 49 2.3.4.1 Hanging drop vapor diffusion method . . . . . . . . . 49 2.3.4.2 Sitting drop vapor diffusion method . . . . . . . . . . 49 2.3.4.3 Microbatch method . . . . . . . . . . . . . . . . 50 2.3.5 Intensity data collection . . . . . . . . . . . . . . . . . . 50 2.3.6 Data processing . . . . . . . . . . . . . . . . . . . . . 51 2.3.7 Structure solution . . . . . . . . . . . . . . . . . . . . 53 2.3.7.1 Multiple isomorphous replacement . . . . . . . . . . 53 2.3.7.1.1 Preparation of heavy atom derivative . . . . . . 54 2.3.7.1.2 Scaling between native and derivative datasets . . 54 2.3.7.1.3 Identifying heavy atom positions by difference Patterson . . . . . . . . . . . . . . . . 54 2.3.7.1.4 Refinement of heavy atom parameters . . . . 55 2.3.7.2 Multiple-wavelength anomalous dispersion . . . . . . 55 2.3.7.3 Molecular replacement . . . . . . . . . . . . . . 56 2.3.7.1 AMoRe . . . . . . . . . . . . . . . . 57 2.3.7.2 MOLREP . . . . . . . . . . . . . . . . 58 2.3.8 Structure refinement . . . . . . . . . . . . . . . . . . . 58 2.3.8.1 CNS . . . . . . . . . . . . . . . . . . . . . 61 2.3.8.1.1 Rigid body refinement . . . . . . . . . . . 61 2.3.8.1.2 B-factor refinement . . . . . . . . . . . . 62 2.3.8.1.3 Simulated annealing . . . . . . . . . . . . 62 2.3.8.1.4 Positional refinement . . . . . . . . . . . 62 2.3.8.1.5 Automatically locating solvent molecules . . . . 63 2.3.8.1.6 Reducing model bias by OMIT maps . . . . . . 63 2.3.8.1.7 Electron density map calculation . . . . . . . 63 2.3.8.2 CCP4 . . . . . . . . . . . . . . . . . . . . . 64 2.3.8.2.1 SCALEPACK2MTZ . . . . . . . . . . . . 64 2.3.8.2.2 TRUNCATE . . . . . . . . . . . . . . . 64 2.3.8.2.3 CAD . . . . . . . . . . . . . . . . . 64 2.3.8.2.4 SCALEIT . . . . . . . . . . . . . . . . 65 2.3.8.2.5 FFT . . . . . . . . . . . . . . . . . . 65 2.3.8.2.6 MLPHARE . . . . . . . . . . . . . . . 65 2.3.8.2.7 REFMAC5 . . . . . . . . . . . . . . . 65 2.3.8.2.8 LIBCHECK . . . . . . . . . . . . . . . 66 2.3.9 Model Building . . . . . . . . . . . . . . . . . . . . . 66 2.3.9.1 COOT . . . . . . . . . . . . . . . . . . . . . 67 2.3.9.2 O . . . . . . . . . . . . . . . . . . . . . . 67 2.3.9.3 ARP/wARP . . . . . . . . . . . . . . . . . . 67
xii

Contents
2.3.10 Structure validation and deposition . . . . . . . . . . . . . 67 2.3.10.1 PROCHECK . . . . . . . . . . . . . . . . . . 68 2.3.10.2 SFCHECK . . . . . . . . . . . . . . . . . . . 68 2.3.10.3 MolProbity . . . . . . . . . . . . . . . . . . 68 2.3.10.4 PDB: ADIT . . . . . . . . . . . . . . . . . . 69 2.3.11 Structure visualization . . . . . . . . . . . . . . . . . . 69 2.3.11.1 PyMOl . . . . . . . . . . . . . . . . . . . . 69 2.3.11.2 MolScript and BobScript . . . . . . . . . . . . . . 69 2.3.11.3 Raster3D . . . . . . . . . . . . . . . . . . 70 2.3.11.4 GRASP . . . . . . . . . . . . . . . . . . . . 70 2.3.11.5 TopDraw . . . . . . . . . . . . . . . . . . 70 2.3.12 Sequence and structure analysis . . . . . . . . . . . . . . 70 2.3.12.1 ProtParam . . . . . . . . . . . . . . . . . . . 70 2.3.12.2 CLUSTAL-W . . . . . . . . . . . . . . . . . . 71 2.3.12.3 ESPript . . . . . . . . . . . . . . . . . . . . 71 2.3.12.4 DSSP . . . . . . . . . . . . . . . . . . . . . 71 2.3.12.5 PROMOTIF . . . . . . . . . . . . . . . . . . 71 2.3.12.6 ALIGN . . . . . . . . . . . . . . . . . . . . 72 2.3.12.7 NACCESS . . . . . . . . . . . . . . . . . . . 72 2.3.12.8 CONTACT . . . . . . . . . . . . . . . . . . 72 2.3.12.9 BAVERAGE . . . . . . . . . . . . . . . . . . 72 2.3.12.10 DynDom . . . . . . . . . . . . . . . . . . . 73 2.3.12.11 VOIDOO . . . . . . . . . . . . . . . . . . . 73 2.3.12.12 DALI . . . . . . . . . . . . . . . . . . . 73 3 Crystal structures of Salmonella typhimurium biodegradative threonine deaminase (TdcB) and its complex with CMP provide structural insights into ligand induced oligomerization and enzyme activation . . . . . . 74 3.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . 75 3.2 Materials and methods . . . . . . . . . . . . . . . . . . . . . 77 3.2.1 Cloning, overexpression and purification . . . . . . . . . . . 77 3.2.2 Activity assay and kinetic studies . . . . . . . . . . . . . . 78 3.2.3 Gel filtration chromatography and glutaraldehyde cross-linking . . 78 3.2.4 Crystallization and data collection . . . . . . . . . . . . . . 78 3.2.5 Structure solution and refinement . . . . . . . . . . . . . 80 3.3 Results and discussion . . . . . . . . . . . . . . . . . . . . . 81 3.3.1 Cloning, overexpression and purification . . . . . . . . . . . 81 3.3.2 Biochemical studies on TdcB . . . . . . . . . . . . . . . . 82 3.3.3 Crystallization and structure solution . . . . . . . . . . . . 84 3.3.4 Model quality . . . . . . . . . . . . . . . . . . . . . . 87 3.3.5 Tertiary structure of TdcB . . . . . . . . . . . . . . . . . 87 3.3.6 Dimeric TdcB . . . . . . . . . . . . . . . . . . . . . . 89
xiii

Contents
3.3.7 Inter-subunit interactions in the dimeric TdcB . . . . . . . . . 91 3.3.8 Tetrameric TdcB in complex with CMP . . . . . . . . . . . . 92 3.3.9 Inter-subunit interactions in the tetrameric TdcB . . . . . . . . 93 3.3.10 Active site pocket . . . . . . . . . . . . . . . . . . . . 98 3.3.11 CMP binding site . . . . . . . . . . . . . . . . . . . . 100 3.3.12 Role of CMP in oligomerization and enzyme activation . . . . . 102 3.3.13 Structural comparison with related enzymes . . . . . . . . . 103 3.3.14 Mechanistic considerations . . . . . . . . . . . . . . . . 105 3.4 Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . 107 4 Crystal structures of ADP- and AMPPNP-bound propionate kinase (TdcD) from Salmonella typhimurium: Comparison with members of acetate and sugar kinase/heat shock cognate 70/actin superfamily . . . . . . . . 108 4.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . 109 4.2 Materials and methods . . . . . . . . . . . . . . . . . . . . . 111
4.2.1 Cloning, overexpression and purification . . . . . . . . . . . . 111 4.2.2 Activity assay and kinetic studies . . . . . . . . . . . . . . . 112 4.2.3 Crystallization and data collection . . . . . . . . . . . . . . 112 4.2.4 Structure solution and refinement . . . . . . . . . . . . . . 113 4.2.5 Domain motion analysis . . . . . . . . . . . . . . . . . . 115
4.3 Results and discussion . . . . . . . . . . . . . . . . . . . . 116 4.3.1 Cloning, overexpression and purification . . . . . . . . . . . . 116 4.3.2 Biochemical studies on TdcD . . . . . . . . . . . . . . . . . 116 4.3.3 Crystallization and structure determination . . . . . . . . . . . 117 4.3.4 Model quality . . . . . . . . . . . . . . . . . . . . . . . 121 4.3.5 Overall structure of propionate kinase . . . . . . . . . . . . . 121 4.3.6 Dimer interface . . . . . . . . . . . . . . . . . . . . . . 125 4.3.7 Nucleotide-binding site . . . . . . . . . . . . . . . . . . . 126 4.3.8 Proposed propionate-binding site . . . . . . . . . . . . . . . 130 4.3.9 Five conserved motifs and proposed catalytic residues . . . . . . 132 4.3.9.1 Phosphate 1 and phosphate 2 motifs . . . . . . . . . . . 132 4.3.9.2 Adenosine motif . . . . . . . . . . . . . . . . . . 133 4.3.9.3 Connect 1 and connect 2 motifs . . . . . . . . . . . . 133 4.3.10 Role of conserved residues . . . . . . . . . . . . . . . . . 134 4.3.11 Analysis of domain movement . . . . . . . . . . . . . . . 134 4.4 Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . 139 5 Crystal structures of Salmonella typhimurium propionate kinase (TdcD) and its complex with Ap4A: Crystallographic evidence for novel Ap4A synthetic activity . . . . . . . . . . . . . . . . . . . . . . . . . 141 5.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . 142
5.1.1 Ap4A and other related dinucleotide polyphosphates . . . . . . 142
xiv

Contents
5.1.2 Enzymes specific for Ap4A . . . . . . . . . . . . . . . . 145 5.1.3 Probable role of Ap4A . . . . . . . . . . . . . . . . . . 147 5.1.4 Ap4A in Salmonella typhimurium . . . . . . . . . . . . . . . 147
5.2 Materials and methods . . . . . . . . . . . . . . . . . . . . . 148 5.2.1 Crystallization and data collection . . . . . . . . . . . . . 148 5.2.2 Structure determination and refinement . . . . . . . . . . . 149 5.2.3 Phosphorylation of TdcD with [γ-32P] ATP . . . . . . . . . . 150
5.3 Results and discussion . . . . . . . . . . . . . . . . . . . . . 150 5.3.1 Crystallization . . . . . . . . . . . . . . . . . . . . . 150 5.3.2 Structure determination . . . . . . . . . . . . . . . . . . 150 5.3.3 Model quality . . . . . . . . . . . . . . . . . . . . . . 155 5.3.4 Native TdcD structure . . . . . . . . . . . . . . . . . . 155 5.3.5 TdcD-Ap4A structure and Ap4A binding . . . . . . . . . . . 156
5.3.5.1 Adenosine A . . . . . . . . . . . . . . . . . . 159 5.3.5.2 α, β, γ and δ phosphates . . . . . . . . . . . . . . 159 5.3.5.3 Adenosine B . . . . . . . . . . . . . . . . . . 160
5.3.6 Formation of Ap4A by TdcD . . . . . . . . . . . . . . . . 162 5.4 Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . 163 6 Crystal structures of Salmonella typhimurium 2-methylisocitrate lyase (PrpB) and its complex with pyruvate and Mg2+ . . . . . . . . . . . 165 6.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . 166 6.2 Materials and methods . . . . . . . . . . . . . . . . . . . . . 168
6.2.1 Cloning, overexpression and purification . . . . . . . . . . . 168 6.2.2 Crystallization and data collection . . . . . . . . . . . . . 169 6.2.3 Structure determination . . . . . . . . . . . . . . . . . 171
6.3 Results and discussion . . . . . . . . . . . . . . . . . . . . . 173 6.3.1. Cloning, overexpression, purification and crystallization . . . . 173 6.3.2 Structure determination . . . . . . . . . . . . . . . . . . 174 6.3.2.1 Multiple isomorphous replacement . . . . . . . . . . 175 6.3.2.2 Molecular replacement and structure refinement . . . . . 178 6.3.3 Overall structure of native PrpB . . . . . . . . . . . . . . 180 6.3.4 Structural comparison with E. coli PrpB . . . . . . . . . . . . 180 6.3.5 Structural comparison with bacterial isocitrate lyase . . . . . . 185 6.3.6 Overall structure of pyruvate/Mg2+ bound PrpB . . . . . . . . 187 6.3.7 Catalytic mechanism . . . . . . . . . . . . . . . . . . . 190
6.4 Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . 191 Future perspectives . . . . . . . . . . . . . . . . . . . . . . . . 193
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . 196
Appendix . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 218
xv

Abbreviations
Å Angstrom
A. niger Aspergillus niger
α Alpha
Ack Acetate kinase
ADP Adenosine diphosphate
AMP Adenosine monophosphate
AMPPNP 5'-adenylyl imidodiphosphate
Ap4A Diadenosine tetraphosphate
ASKHA Acetate and sugar kinase-heat shock cognate 70-actin
ATP Adenosine triphosphate
ATPγS Adenosine 5'-O-(3-thiotriphosphate)
AU Asymmetric unit
β Beta
bp base pair
CMP Cytosine monophosphate
CoA Coenzyme A
Da Dalton
DNA Deoxyribo nucleic acid
DTT Dithiothreitol
E. coli Escherichia coli
EDTA Ethylenediamine tetra acetic acid
Hsc70 Heat shock cognate 70
ICL Isocitrate lyase
IlvA Biosynthetic threonine deaminase
IPTG Isopropyl-β-D-thiogalactopyranoside
kb kilo bases
kDa kilo Daltons
l liter
LB Luria Bertani broth
m milli, meter
µ Micron

Abbreviations
M Molar
MAK Acetate kinase from Methanosarcina thermophila
M. thermophila Methanosarcina thermophila
µl Micro liter
min minute
MW Molecular weight
MR Molecular replacement
MIR Multiple isomorphous replacement
M. tuberculosis Mycobacterium tuberculosis
NADH Nicotinamide adenine dinucleotide phosphate
Ni-NTA Nickel-nitrilotriacetic acid
OD Optical density
ORF Open reading frame
PCMBS para-chloro-mercuribenzene sulphonic acid
PCR Polymerase chain reaction
PDB Protein data bank
PEG Polyethylene glycol
PLP Pyridoxal 5’-phosphate
PrpB 2-methylisocitrate lyase
PrpC 2-methylcitrate synthase
rmsd root-mean-square deviation
rpm revolutions per minute
S. typhimurium Salmonella typhimurium
SD Serine deaminase
SDS-PAGE Sodium dodecyl sulfate polyacrylamide gel electrophoresis
sec second
TB Terrific broth
TdcB Biodegradative threonine deaminase
TdcD Propionate kinase
Tris Tris-(hydroxymethyl)-amino methane
3D Three-dimensional
2

Abstract
Following acetate, propionate is the second most abundant low molecular mass
carbon compound found in the soil. Many aerobic microorganisms, bacteria and fungi as
well as some anaerobes are able to grow on propionate as their sole carbon and energy
source. In the presence of glucose, propionate and other short chain fatty acids inhibit
microbial growth, which has made them useful as preservatives in the food industry. But
the mechanism through which propionate exerts antimicrobial activity is poorly
understood. This compound appears to affect the function at multiple targets within the
cell.
Propionate is mainly produced during the β-oxidation of odd-numbered carbon-
chain fatty acids, fermentation of carbohydrates, oxidative degradation of the branched-
chain amino acids valine and isoleucine, and from the carbon skeletons of threonine,
methionine, thymine and cholesterol. Studies on the degradation of these amino acids to
propionate have revealed several enzymes that utilize diverse catalytic mechanisms. In
Escherichia coli and Salmonella typhimurium, L-threonine can be anaerobically degraded
to propionate via 2-ketobutyrate. Although the enzymatic conversion of L-threonine to
propionate had been demonstrated as early as 1963 in cell-free extracts of Clostridium
tetanomorphum, the metabolic fate of 2-ketobutyrate remained enigmatic for a long time.
In 1987, Van Dyk & La Rossa proposed that 2-ketobutyrate is oxidatively decarboxylated
to propionyl-CoA in S. typhimurium by a thiamine-dependent enzyme. Later Hesslinger
et al showed that L-threonine can be non-oxidatively cleaved to propionate via 2-
ketobutyrate in E. coli by biodegradative threonine deaminase (TdcB), 2-ketobutyrate
formate lyase (TdcE), phosphotransacetylase (Pta) and propionate kinase (TdcD).
Enzymes involved in the anaerobic degradation pathways of L-serine and L-threonine to
acetate and propionate, respectively, are encoded by the anaerobically regulated tdc
operon.
The catabolism of propionate in prokaryotes has been extensively investigated. It
has been demonstrated that propionate is metabolized to pyruvate via 2-methylcitric acid
cycle in S. typhimurium as well as in E. coli. A prp locus required for the catabolism of
propionate has been identified in S. typhimurium. The prp locus is comprised of two
transcribed units. One unit contains four genes (prpBCDE) organized as an operon, that

Abstract
encodes four distinct enzymes, which are required for the catabolism of propionate. The
other unit contains only one gene, prpR, which encodes a σ54-dependent transcriptional
activator. In this pathway, prpE, a propionyl-CoA synthetase forms propionyl-CoA from
propionate and coenzyme A and prpC, a 2-methylcitrate synthase forms 2-methylcitrate
by combining propionyl-CoA and oxaloacetate. The 2-methylcitrate thus formed is
converted to 2-methylisocitrate by two separate enzymes prpD, a 2-methylcitrate
dehydratase and either of the two aconitases present in S. typhimurium. The last step of
this cycle, the cleavage of 2-methylisocitrate to succinate and pyruvate is catalyzed by
prpB, a 2-methylisocitrate lyase.
The thesis begins with a review of the current literature on propionate metabolism
in S. typhimurium (chapter I). Enzymes involved in anaerobic degradation of L-
threonine to propionate and in the 2-methylcitrate pathway which catabolizes propionate
into pyruvate and succinate are described in this chapter. All the common experimental
and computational methods used during the course of investigations are described in
chapter II, as most of these are applicable to all the structure determinations and
analyses. The experimental procedures described include cloning, overexpression,
purification, crystallization, intensity data collection, etc. Computational methods
covered include details of various programs used during data processing, structure
solution, refinement, model building, validation and analysis.
In chapter III, cloning, expression, purification, crystallization and X-ray
crystallographic studies on the biodegradative threonine deaminase and its complex with
the activator molecule CMP is presented and is followed by detailed structure-function
analysis. TdcB from S. typhimurium was cloned and overexpressed in E. coli. In the
presence of AMP or CMP, the recombinant enzyme was converted to tetrameric form
accompanied by significant enzyme activation. To provide insights into ligand-mediated
oligomerization and enzyme activation, crystal structures of S. typhimurium TdcB and its
complex with CMP were determined. In the native structure, TdcB is in a dimeric form
whereas in the TdcB-CMP complex, it exists in a tetrameric form with 222 symmetry and
appears as a dimer of dimers. Tetrameric TdcB binds to four molecules of CMP, two at
each of the dimer interfaces. Comparison of the dimer structure in the ligand (CMP) free
and bound forms suggests that the changes induced by ligand binding at the dimer
4

Abstract
interface are essential for tetramerization. The differences observed in the tertiary and
quaternary structures of TdcB in the absence and presence of CMP appear to account for
enzyme activation and increased binding affinity for L-threonine. Comparison of TdcB
with related PLP-dependent enzymes points to structural and mechanistic similarities.
Propionate kinase (TdcD; EC 2.7.2.15) catalyses the last step in the anaerobic
degradation of L-threonine to propionate by enabling the conversion of propionyl
phosphate and ADP to propionate and ATP. To provide insights into the substrate-
binding pocket and catalytic mechanism of TdcD, crystal structures of the enzyme from
Salmonella typhimurium in complex with ADP and AMPPNP have been determined to
resolutions of 2.2 Å and 2.3 Å, respectively, by molecular replacement using
Methanosarcina thermophila acetate kinase as the model. These and related results are
described in chapter IV. Propionate kinase, like acetate kinase, contains a fold with the
topology βββαβαβα, similar to those of glycerol kinase, hexokinase, heat shock cognate
70 (Hsc70) and actin, the superfamily of phosphotransferases. The structure consists of
two domains with the active site contained in a cleft at the domain interface. Examination
of the active site pocket revealed a plausible structural rationale for the greater specificity
of the enzyme towards propionate than acetate. This was further confirmed by kinetic
studies with the purified enzyme, which showed about ten times lower KM for propionate
(2.3 mM) than for acetate (26.9 mM). Comparison of TdcD complex structures with
those of acetate and sugar kinase/Hsc70/actin obtained with different ligands has
permitted the identification of catalytically essential residues involved in substrate
binding and catalysis, and points to both structural and mechanistic similarities. In the
well-characterized members of this superfamily, ATP phosphoryl transfer or hydrolysis is
coupled to a large conformational change in which the two domains close around the
active site cleft. The significant amino acid sequence similarity between TdcD and
thermophilic acetate kinase has facilitated study of domain movement, which indicates
that the conformation assumed by the two domains in the nucleotide-bound structure of
TdcD may represent an intermediate point in the pathway of domain closure.
In chapter V, structures of TdcD in the native form as well as in complex with
diadenosine 5’,5”’-P1,P4-tetraphosphate (Ap4A) obtained after co-crystallization with
ATP as well as Ap4A is discussed. On the basis of these structures, we provide evidence
5

Abstract
for a novel Ap4A synthetic activity in propionate kinase from Salmonella typhimurium.
Crystals of TdcD obtained in the presence of ATP clearly showed Ap4A bound in the
active site pocket of the enzyme. Presence of Ap4A and its binding to the enzyme was
further confirmed by structure determination of TdcD-Ap4A complex obtained after co-
crystallization of TdcD with commercially available Ap4A. Out of various enzymatic
reactions identified so far involving the enzymatic synthesis of Ap4A, the activity of
dinucleotide polyphosphate phosphorylases (EC 2.7.7.53) could explain the formation of
Ap4A by the TdcD. In the TdcD-Ap4A complex structure, Ap4A is present in an extended
conformation with one adenosine moiety in the nucleotide binding site and other in the
proposed propionate binding site. These observations tend to support the stereochemical
evidence that phosphoryl transfer in this enzyme is direct.
In the 2-methylcitrate pathway, the cleavage of 2-methylisocitrate to succinate
and pyruvate is catalyzed by prpB, a 2-methylisocitrate lyase. In the last chapter (chapter
VI), structural studies carried out on 2-methylisocitrate lyase is presented. 2-
methylisocitrate lyase (molecular weight 32 kDa) has been cloned and overexpressed in
E. coli with a C-terminal polyhistidine affinity tag and purified and crystallized under
different crystallization conditions using the hanging drop and sitting drop vapour
diffusion techniques. The X-ray crystal structure of the native and pyruvate/Mg2+ bound
PrpB from S. typhimurium has been determined at 2.1 and 2.3 Å, respectively. The
structure closely resembles that of the E. coli enzyme. Unlike the E. coli PrpB, Mg2+
could not be located in the native Salmonella PrpB. Only in pyruvate bound PrpB
structure, Mg2+ was found coordinated with pyruvate. Binding of pyruvate to PrpB seems
to induce movement of Mg2+ by 2.5 Å from its position found in E. coli native PrpB. In
the native enzyme and pyruvate/Mg2+ bound forms, the active site loop is completely
disordered. Examination of the pocket in which pyruvate and glyoxylate bind to 2-
methylisocitrate lyase and isocitrate lyase, respectively, reveals plausible rationale for
different substrate specificities of these two enzymes. Structural similarities in substrate
and metal atom binding site as well as presence of similar residues in the active site
suggest possible similarities in the reaction mechanism. The thesis concludes with a brief
discussion on the future prospects of the work.
6

Abstract
The following manuscripts have been published or will be communicated for
publication based on the results presented in the thesis:
Simanshu DK, Satheshkumar PS, Parthasarathy S, Savithri HS, Murthy MR. Cloning, expression, purification and preliminary X-ray crystallographic studies of 2-methylisocitrate lyase from Salmonella typhimurium. Acta Crystallogr Sect D. 2002; 58(12): 2159-61.
Simanshu DK, Satheshkumar PS, Savithri HS, Murthy MR. Crystal structures of
Salmonella typhimurium 2-methylisocitrate lyase (PrpB) and its complex with pyruvate and Mg2+. Biochem Biophys Res Commun. 2003; 311(1): 193-201.
Simanshu DK, Murthy MR. Cloning, expression, purification, crystallization and
preliminary X-ray diffraction analysis of propionate kinase (TdcD) from Salmonella typhimurium. Acta Crystallogr Sect F. 2005; 61(1): 52-5.
Simanshu DK, Savithri HS, Murthy MR. Crystal structures of ADP and AMPPNP-
bound propionate kinase (TdcD) from Salmonella typhimurium: comparison with members of acetate and sugar kinase/heat shock cognate 70/actin superfamily. J Mol Biol. 2005; 352(4): 876-92.
Simanshu DK, Chittori S, Savithri HS, Murthy MR. Crystallization and preliminary
X-ray crystallographic analysis of biodegradative threonine deaminase (TdcB) from Salmonella typhimurium. Acta Crystallogr Sect F. 2006; 62(3): 275-8.
Simanshu DK, Savithri HS, Murthy MR. Crystal structures of Salmonella
typhimurium biodegradative threonine deaminase (TdcB) and its complex with CMP provide structural insights into ligand induced oligomerization and enzyme activation. J. Biol. Chem. 2006; (Manuscript under revision).
Simanshu DK, Savithri HS, Murthy MR. Crystal structures of Salmonella
typhimurium propionate kinase (TdcD) and its complex with Ap4A: Evidence for novel Ap4A synthetic activity. (Manuscript under preparation).
7

Introduction
1

Chapter 1 Introduction
1.1 Salmonella Salmonella is named after Daniel Elmer Salmon, an American veterinary
pathologist who, together with Theobald Smith, first discovered the Salmonella
bacterium in the year 1885 from pigs with hog cholera (Salmon and Smith 1884-1886).
Salmonellosis ranges clinically from the common Salmonella gastroenteritis (diarrhea,
abdominal cramps and fever) to enteric fevers (including typhoid fever) which are life-
threatening febrile systemic illnesses requiring prompt antibiotic therapy. The most
common form of salmonellosis is a self-limited, uncomplicated gastroenteritis. They also
infect many animal species besides humans. Animals, especially poultry and swine, are
the main reservoirs of Salmonella species. Environmental sources of the organism
include water, soil, insects, food processing factory, kitchen surfaces, animal faeces, raw
meat, raw poultry and raw sea foods, to name only a few.
Figure 1.1 Salmonella typhimurium (reproduced from www-instruct.nmu.edu/cls/lriipi/micro/)
1.1.1 General characteristics
Salmonella is a Gram-negative, non-spore forming, obligate parasite, which
appears as straight rods of 0.7-1.5 mμ in width and 2-5 mμ in length (Fig. 1.1). Most are
motile by means of peritrichous flagella. Colonies in a typical culture media are 2-4 mm
in diameter. Salmonella has simple nutritional requirements, grows on routine
bacteriological growth media without enrichment. The organisms are facultative
anaerobes and the optimum growth temperature is 37ºC. They do not produce indole and
ferment glucose by the mixed acid fermentation. Salmonella genome is very similar to
that of Escherichia coli and contains a single circular DNA molecule consisting of about
4 X 106 base pairs and a total length of 1.4 mm.
9

Chapter 1 Introduction
1.1.2 Classification
Salmonella is a genus of bacterium that belongs to the family
“Enterobacteriaceae”. The genus Salmonella consists of two species, Salmonella enterica
(formerly called Salmonella choleraesuis) and Salmonella bongori (previously
subspecies V). Salmonella enterica is divided into six subspecies:
Subspecies I - Salmonella enterica
Subspecies II - Salmonella salamae
Subspecies IIIa - Salmonella arizonae
Subspecies IIIb - Salmonella diarizonae
Subspecies IV - Salmonella houtenae
Subspecies V – obsolete (now forms the Salmonella bongori)
Subspecies VI - Salmonella indica
Salmonella isolates are most usually classified according to serology. There are
numerous (totaling over 2500) serovars within both species, which are found in a
disparate variety of environments and which are associated with many different diseases.
The Salmonellae possess 3 major antigens called somatic O antigen, flagellar H antigen
and surface Vi antigen. The vast majority of human isolates (>99.5%) are subspecies of
Salmonella enterica. An example of a serovar is Salmonella typhi, causes systemic
infections and typhoid fever, which is very host specific and will only infect humans,
killing an estimated 500,000 per year worldwide. On the other side of the spectrum,
Salmonella typhimurium causes gastroenteritis and can infect many mammalian species.
For the sake of simplicity, Salmonella species are referred to only by their genus
and serovar: e.g., Salmonella typhimurium instead of the more correct designation,
Salmonella enterica subspecies enterica serovar Typhimurium.
1.1.3 Salmonella enterica serovar Typhimurium
Salmonella enterica subspecies enterica serovar Typhimurium (Salmonella
typhimurium) is a leading cause of human gastroenteritis and is used as a mouse model of
human typhoid fever. Salmonella typhimurium strain LT2 was isolated in the 1940s and
used in the first studies on phage-mediated transduction. Complete genome sequence of
10

Chapter 1 Introduction
Salmonella enterica serovar Typhimurium LT2 which contains 4,857-kilobase (kb)
chromosome and 94-kb virulence plasmid (McClelland et al. 2001) was published in
2001. A total of 4,330 complete open reading frames (ORFs) from the S. typhimurium
LT2 genome were amplified. Genomic comparisons among the four completed genomes
(S. typhimurium LT2, S. typhi, E. coli K12 and E. coli O157:H7) reveal that they are
collinear for most genes except for inversions over the terminus of replication (TER). For
the present study, Salmonella enterica serovar Typhimurium strain IFO 12529 has been
used, which was generously provided by Professor Toru Nagasawa of Okayama
University, Japan.
Short chain fatty acids (SCFAs) are common by-products of bacterial
fermentations and are produced in abundance in the gastrointestinal tracts of mammals.
Although SCFAs are a good source of carbon and energy for prokaryotes, they also
inhibit cell growth (Cherrington et al. 1991). The growth inhibitory properties of SCFAs
have made them useful as preservatives in the food industry (Kabara and Eklund 1991).
Propionate (a three-carbon SCFA) is one of the most abundant fermentation by-products,
and it is extensively used in the food industry to protect baked goods against microbial
contamination.
Studies on propionate and other SCFAs have shown that they are major
determinants in the ability of Salmonella species to cause disease. SCFAs produced by
fermentative bacteria in mice and chickens can greatly increase resistance to Salmonella
infections (Barnes et al. 1979; Bohnhoff and Miller 1962; Meynell 1963; Meynell and
Subbaiah 1963). These properties have prompted researchers to test the ability of
propionate to inhibit Salmonella growth in animal feeds. The addition of propionic acid to
chicken feeds at levels of 60-100 mM greatly reduced the population of Salmonella
species and limited the ability of Salmonella to colonize the chicken cecum (Hume et al.
1993; Matlho et al. 1997; McHan and Shotts 1992; Thompson and Hinton 1997).
Our understanding of the mechanisms through which propionate exerts
antimicrobial activity is limited. This compound appears to affect the function of multiple
targets within the cell. For example, SCFAs are known to dissipate the proton-motive
force of cell membranes by entering the cell as undissociated molecules and then
dissociating in the cytoplasm (Blankenhorn et al. 1999; Kabara and Eklund 1991;
11

Chapter 1 Introduction
Salmond et al. 1984). Other lines of evidence suggest that the intracellular accumulation
of the SCFA anions blocks metabolic pathways, arresting cell growth. Reports in the
literature suggest that propionyl-coenzyme A (propionyl-CoA) is an important contributor
to the observed toxicity of propionate. In Rhodobacter sphaeroides, propionyl-CoA was
found to inhibit pyruvate dehydrogenase (Maruyama and Kitamura 1985) and in
Escherichia coli, propionyl-CoA is a competitive inhibitor of citrate synthase (Man et al.
1995). Support for the idea that propionyl-CoA is a problem for the cell was also obtained
in Aspergillus niger, where experiments blocking propionyl-CoA formation relieved
propionate sensitivity (Sealy-Lewis and Fairhurst 1998). However, recent experiments
suggest that the inhibitory effects of propionate in A. niger may be caused by 2-
methylcitrate accumulation (Brock et al. 2000).
The mode of action of propionate was studied using Aspergillus nidulans as a
model organism, which also suggested that the growth inhibiting effect of propionate
may be due to the accumulation of propionyl-CoA. Elevated levels of propionyl-CoA can
lead to an unbalanced acetyl-CoA/propionyl-CoA ratio, which might competitively
inhibit other house-keeping enzymes dealing with CoA-esters as substrate or product.
This assumption is supported by the fact that spore colour development is disturbed in the
presence of propionate in Aspergillus nidulans strain with a 2-methylcitrate synthase
negative genetic background. The spore colour derives from the polyketide naphtopyrone
and its synthesis is strongly dependent on acetyl-CoA. In addition, the propionate
concentration required for growth inhibition is decreased in such a strain by a factor of
five compared to wild-type A. nidulans (Brock et al. 2000). Therefore, structural
knowledge of enzymes involved in the propionate metabolism is desirable, since specific
inhibition of any of these enzymes is likely to result in a lower concentration of
propionate necessary for food preservation.
1.2 Propionate metabolism All living organisms must have continuous supply of energy and matter.
Metabolic controls ensure that living cells or organisms get adequate supply of energy
and biomass. Metabolism usually consists of a series of consecutive enzymatic reactions,
also called metabolic pathways, that produce specific products (Voet and Voet 2004). In
12

Chapter 1 Introduction
a metabolic pathway, reactants, intermediates and products are referred to as metabolites.
Since an organism utilizes many metabolites, it has a large number of metabolic
pathways inter-connected with each other forming a complex network inside the cell. To
understand the complex metabolic network, it is important to study individual metabolic
pathways and their connectivity with other metabolic pathways.
Short-chain fatty acids play a major role in nature’s carbon cycle. Fermentative
bacteria produce acetate, propionate, butyrate, and a few other straight and branched
mono-carboxylates that are oxidized to CO2 either aerobically or anaerobically. The
formation and degradation of most of these fatty acids involve an oxidation pathway; a
remarkable exception is propionate, for which possibly several pathways exist.
1.3 Anaerobic degradation of L-threonine to propionate Propionate, following acetate, is the second most abundant low molecular-mass
carbon compound found in the soil. Many aerobic microorganisms, bacteria and fungi as
well as some anaerobes are able to grow on propionate as their sole carbon and energy
source. Propionate is mainly formed during the β-oxidation of odd-numbered carbon-
chain fatty acids, the fermentation of carbohydrates, the oxidative degradation of the
branched-chain amino acids valine and isoleucine and from the carbon skeletons of
threonine, methionine, thymine and cholesterol. Studies on the degradation of these
amino acids in E. coli have revealed that several enzymes that utilize diverse catalytic
mechanisms are involved in these pathways. The anaerobically regulated tdc operon has
been shown to encode enzymes involved in the metabolic pathways for the degradation
of L-serine and L-threonine to acetate and propionate, respectively (reviewed in Sawers
1998).
The hydroxy-amino acid L-threonine can serve as a precursor, directly or
indirectly, to various amino acids and metabolites. L-threonine and L-serine can be
derived from one another through the common intermediate glycine. These two amino
acids have a major bearing on the metabolism of bacteria such as E. coli and other
enterobacteria (Sawers 1998). During formation of L-isoleucine from L-threonine, the
involvement of 2-ketobutyrate as a precursor to L-isoleucine synthesis has been well
studied (Umbarger 1996). Working with extracts derived from Clostridium
13

Chapter 1 Introduction
tetanomorphum, Tokushige et al. demonstrated in vitro that 2-ketobutyrate can be
catabolized to propionate via a propionyl phosphate intermediate (Tokushige et al. 1963).
However, the route of degradation of L-threonine to propionate remained enigmatic until
Van Dyk and LaRossa working with Salmonella typhimurium, demonstrated that
phosphotransacetylase and acetate kinase are involved in the anaerobic degradation of 2-
ketobutyrate, indicating that propionyl-CoA is an intermediate (Van Dyk and LaRossa
1987). In a later study, they proposed that 2-ketobutyrate is converted to propionyl-CoA
by a thymine pyrophosphate-dependent enzyme (LaRossa and Van Dyk 1989). In 1998,
Hesslinger et al showed that a gene tdcE present in the tdc operon has 2-ketobutyrate
formate-lyase activity and a newly identified gene, tdcD, immediately upstream of tdcE
encodes an enzyme with propionate kinase activity (Hesslinger et al. 1998) (Fig. 1.2).
Based on these findings, it was shown that the extended tdc operon (tdcABCDEFG)
encodes components of an anaerobically inducible, catabolite-repressible pathway, which
generates one molecule of ATP from the degradation of L-threonine and L-serine.
Figure 1.2 Pathway for anaerobic degradation of L-threonine to propionate via 2-ketobutyrate. 1.3.1 tdc operon
The anaerobically regulated tdcABCDEFG operon of E. coli and S. typhimurium
encodes proteins involved in the transport and fermentation of L-serine and L-threonine
(Hesslinger et al. 1998; Sawers 1998) (Fig. 1.3). Metabolism of these substrates provides
14

Chapter 1 Introduction
the cell with a source of energy (Merberg and Datta 1982). The importance of the Tdc
enzymes for the metabolism of E. coli stems from their ability to produce energy-rich
keto acids that are subsequently catabolized to produce ATP via substrate-level
phosphorylation (Hesslinger et al. 1998; Sawers 1998). Functional roles have been
assigned, albeit tentatively to most of the gene products: TdcA is a trans-acting positive
regulator (Ganduri et al. 1993; Sawers 2001), TdcB is a biodegradative threonine
dehydratase (Datta et al. 1987), TdcC is an integral membrane protein implicated in the
transport of L-serine and L-threonine into the cell (Sumantran et al. 1990), TdcD is a
propionate kinase (Hesslinger et al. 1998), TdcE is a 2-ketobutyrate formate lyase
(Hesslinger et al. 1998) and TdcG is a L-serine dehydratase (Hesslinger et al. 1998). The
tdcF gene (formerly yhaR) encodes a protein whose function in unknown.
tdcR tdcA tdcB tdcC tdcD tdcE tdcF tdcG
Transcriptionalregulators
Threoninedeaminase
Ser/Thrpermease
Propionatekinase
L-serinedeaminase
?2-ketobutyrateFormate-lyase
E. coli chromosome
Figure 1.3 Genetic organization of the tdc operon in E. coli (Hesslinger et al. 1998; Sawers 1998).
The function of the gene product is shown below the respective gene. The question mark (?)
indicates that the physiological function of TdcF is unknown.
Catabolism of hydroxy-amino acids L-serine and L-threonine in E. coli and S.
typhimurium proceed via pyruvate and 2-ketobutyrate, respectively (Fig. 1.4). L-
threonine can be non-oxidatively cleaved to propionate by biodegradative threonine
deaminase (TdcB), 2-ketobutyrate formate lyase (TdcE), phosphotransacetylase (Pta) and
propionate kinase (TdcD) whereas L-serine can be anaerobically degraded to acetate by
L-serine deaminase (Sda I and Sda II or TdcG), pyruvate formate lyase (Pfl), phosphor-
transacetylase (Pta) and acetate kinase (Ack). With the exception of L-serine dehydratase
(Sda I and Sda II), the other enzymes of these two pathways can accept substrates
15

Chapter 1 Introduction
involved in either L-threonine or L-serine catabolism. This cross-pathway substrate
specificity greatly increases the metabolic flexibility of the organism.
Figure 1.4 Anaerobic degradative pathways for L-serine and L-threonine (reproduced from
Sawers 1998). (ACK, acetate kinase; PFL, pyruvate formate lyase; Pta, phosphotransacetylase;
SDA, serine deaminase; TdcB, threonine deaminase; TdcD, propionate kinase; TdcE 2-
ketobutyrate formate lyase and TdcG, L-serine dehydratase isoenzyme III.
1.3.2 Regulation of the tdc operon
Even though regulation of the tdc operon has been analyzed in detail, there are
still many unanswered questions regarding the underlying mechanisms controlling tdc
operon. Induction of the tdc operon occurs under anaerobic conditions. Five
transcriptional regulators that interact with the DNA regulatory region of the operon have
been identified. These include the cAMP-receptor protein (CRP) and the DNA bending
and binding proteins IHF (integration host factor) and HU (histone like protein), together
with two transcription factors encoded by the tdc locus, TdcR and TdcA (Ganduri et al.
1993; Schweizer and Datta 1989; Wu and Datta 1992; 1995; Wu et al. 1992) (Fig. 1.5).
16

Chapter 1 Introduction
Figure 1.5 Regulation of the tdc operon in Escherichia coli (reproduced from Sawers 1998). An
arrow pointing upwards signifies positive regulation, while an arrow pointing downward
signifies negative regulation. Abbreviations for global transcriptional regulators are as follows:
FNR, fumarate and nitrate reductase activator; ArcA, a member of the two-component ArcBA
sensor-regulator system; cAMP-CRP, cAMP-receptor protein; IHF, integration host factor; HU,
histone-like protein.
It is likely that either TdcA or TdcR responds to the levels of the amino acids
threonine, serine, valine and isoleucine, all of which are necessary for maximal operon
expression. It appears that the LysR-like regulator, TdcA may have a role in fine-tuning
tdc operon expression (Sawers 2001). Anaerobic regulation of this operon is mediated by
the FNR (fumarate and nitrate reductase activator) and ArcA (a member of the two-
component ArcBA sensor-regulator system) proteins; however, they appear to function
indirectly, presumably by modulating the levels of a key metabolite that subsequently is
sensed by TdcA/ TdcR or by another, as yet unidentified regulator (Chattopadhyay et al.
1997). Expression of the operon is also subject to strong catabolite repression by glucose
and pyruvate (Sawers 2001; Wu et al. 1992). Previous studies have revealed that this
catabolite repression can be overcome by the activity of a small histone-like protein from
Clostridium pasteurianum ((Sawers et al. 1998). This alleviation of catabolite repression
may be related in some way to a protein-induced alteration in the topological status of the
DNA (Sawers 2001). The requirement for expression of these genes during anaerobic
growth probably reflects the fact that the catabolism of serine and threonine generates
ATP without the necessity of using oxidative enzymatic reactions and, as such,
epitomizes fermentation.
1.3.3 Enzymes encoded by the tdc operon and phosphotransacetylase
Various enzymes encoded by the tdc operon and phosphotransacetylase that
functions in the anaerobic degradation of L-threonine to propionate are discussed below:
17

Chapter 1 Introduction
1.3.3.1 TdcB: Biodegradative threonine deaminase
Biodegradative threonine deaminase catalyzes the first reaction in the anaerobic
breakdown of L-threonine to propionate. Two distinctly different pyridoxal 5’-phosphate
(PLP) containing threonine deaminases (EC 4.3.1.19), one biosynthetic and the other
biodegradative, are present in E. coli and S. typhimurium (Luginbuhl et al. 1974;
Umbarger 1996). Both the enzymes catalyze the deamination of L-threonine to yield 2-
ketobutyrate and ammonia. Unlike serine deaminase, which is specific to L-serine,
threonine deaminases can also use L-serine as a substrate although serine deamination
has been shown to inactivate the enzyme (Niederman et al. 1969). One of these, encoded
by the gene ilvA, is expressed constitutively under normal growth conditions and is
allosterically inhibited by the end-product of the pathway, L-isoleucine, and activated by
the product of a parallel pathway, L-valine (Eisenstein 1991). This form of threonine
deaminase has been studied as the model system for investigation of feedback inhibition
and allosteric regulation (Monad et al. 1965; Umbarger 1996). Since this enzyme
catalyzes the first reaction in the L-isoleucine biosynthesis from L-threonine, it was
called biosynthetic threonine deaminase (Fig. 1.6).
A second threonine deaminase encoded by the gene tdcB has been shown to be
synthesized inside the cell when the organism is grown anaerobically in a medium
containing high concentrations of amino acids and no glucose (Wood and Gunsalus
1949). In E. coli and S. typhimurium, biodegradative (catabolic) threonine deaminase
(TdcB) is induced anaerobically in a tryptone-yeast extract medium. Unlike biosynthetic
threonine deaminase, this enzyme is insensitive to L-isoleucine and is activated by
adenosine 5’-monophosphate (AMP) (Wood and Gunsalus 1949). TdcB shares 34%
sequence identity with the N-terminal domain of biosynthetic threonine deaminase of E.
coli and does not contain the sequence corresponding to the C-terminal regulatory
domain (Gallagher et al. 1998) (Fig. 1.7). Like biosynthetic threonine deaminase,
biodegradative threonine deaminase is expected to have a fold typical to those of the β-
family of PLP dependent enzymes (Datta et al. 1987).
18

Chapter 1 Introduction
ATP
Figure 1.6 Metabolic breakdown of L-threonine in Salmonella typhimurium. The first reaction in
the pathway of L-isoleucine biosynthesis is catalyzed by biosynthetic threonine deaminase (IlvA),
which is feedback inhibited by L-isoleucine and activated by L-valine. During anaerobic
breakdown of L-threonine to propionate, the first reaction in the pathway is catalyzed by
biodegradative threonine deaminase (TdcB), which is activated by AMP, CMP and to a lower
extent by other nucleotide monophosphates.
Studies on TdcB from E. coli and S. typhimurium have shown multiple modes of
its regulation. In the presence of AMP, the enzymatic activity is enhanced due to a large
decrease in KM for L-threonine and an apparent increase in Vmax (Bhadra and Datta 1978;
Gerlt et al. 1973; Shizuta et al. 1973). Using various analogs of AMP and other natural
nucleotides, the functional atoms or groups of AMP, which are involved in the ligand-
binding and activation of enzymatic activity have been identified (Nakazawa et al. 1967;
Whanger et al. 1968). Among other mononucleotide phosphates, CMP showed
19

Chapter 1 Introduction
significant enzyme activation compared to those of GMP, UMP, and IMP. Further, no
enzymatic activation was observed in the presence of ATP whereas ADP showed a slight
activation. In the absence of AMP, TdcB exists in monomer-dimer equilibrium (Gerlt et
al. 1973; Shizuta et al. 1973; Whanger et al. 1968). This equilibrium shifts towards the
tetrameric form as the concentration of TdcB is increased. Even at low concentrations of
TdcB, presence of AMP induces oligomerization from monomer to tetramer.
Figure 1.7 Amino acid sequence alignment of biodegradative threonine deaminase (TdcB_ST)
and biosynthetic threonine deaminase (IlvA_ST) from Salmonella typhimurium. Black and gray
boxes indicate identical and similar residues, respectively. Sequence alignment indicates absence
of amino acids corresponding to the C-terminal regulatory domain of IlvA.
1.3.3.2 TdcC: Threonine/Serine transport protein
Most catabolic pathways, especially those involved in the utilization of various
amino acids and sugars, include a specific protein(s) for transport of metabolites into the
cell and some catabolic operons, such as lac and tna, harbor genes coding for their
respective permeases lacY and tnaB (Neidhardt et al. 1996; Stewart and Yanofsky 1985).
For the tdc operon, both threonine and serine are required for expression of the tdc genes.
These considerations led to the notion that one of the tdc genes may be involved in the
transport of the amino acids threonine and serine. Later on, it was demonstrated that the
20

Chapter 1 Introduction
tdcC gene encodes a permease that is induced during anaerobic incubation of E. coli in
amino acid-rich medium (Sumantran et al. 1990). Competition experiments revealed that
both L-threonine and L-serine shared the same transport system. In analogy with other
permeases, the TdcC polypeptide appears to be an integral membrane protein with
several hydrophobic domains exhibiting a polytopic feature. This type of genetic
organization, in which a single transcriptional unit contains genes coding for both a
permease and an enzyme needed for catabolism of a specific metabolite, as in lac and tna
operons, provides a biochemical mechanism for the coordinate expression of the gene
products for efficient regulation of cellular metabolism (Sumantran et al. 1990).
1.3.3.3 TdcD: Propionate kinase
In E. coli, immediately upstream of the tdcE gene and separated from it by an
intergenic region of 33 bp is a gene encoding a 402 amino acid protein sharing 44%
identity over the complete length of the protein with the E. coli acetate kinase
(Matsuyama et al. 1989). This gene has been termed tdcD, and encodes an enzyme with
propionate kinase activity (Hesslinger et al. 1998). S. typhimurium TdcD exhibits 42%
identity with acetate kinase from S. typhimurium (Fig. 1.8) and 38% identity with well-
characterized acetate kinase from Methanosarcina thermophila (Latimer and Ferry 1993).
In E. coli, propionate kinase has been shown to also possess acetate kinase
activity (Hesslinger et al. 1998). Propionate kinase exhibits significant amino acid
identity with acetate kinases. Therefore, these two enzymes are likely to have similar
structures and catalytic mechanisms. The only available crystal structure of acetate kinase
is from a thermophilic organism, Methanosarcina thermophila (Buss et al. 2001). Despite
the absence of sequence identity, the fold of acetate kinase contains a core that is similar
to those of the glycerol kinase/hexokinase/ Hsc70/actin, the ASKHA superfamily of
phosphotransferases. The fold consists of a duplicated βββαβαβα secondary structure
with insertions of subdomains between particular β-sheet elements. The ASKHA
phosphotransferase family has undergone extensive divergent evolution. Nevertheless, a
number of elements appear to have been conserved: (i) a two-domain structure containing
duplicated core secondary structure elements, (ii) residues that bind the catalytic
magnesium ion and the nucleotide α-phosphate (the unusual conformation of the residue
21

Chapter 1 Introduction
preceding the glycine that binds the α-phosphate and the following turn of a helix are also
conserved), and (iii) points of insertion of secondary-structure elements into the core fold
(Bork et al. 1992; Buss et al. 2001; Hurley 1996).
The literature provides evidence for two contrasting mechanisms of acetate kinase
function. Acetate kinase becomes phosphorylated in the presence of acetyl phosphate or
ATP and the stable phosphoenzyme can be isolated (Anthony and Spector 1971; 1972;
Fox et al. 1986). The phosphoenzyme is able to transfer the phosphoryl group to either
ADP or acetate, suggesting that the enzyme-linked covalent intermediate is involved in
catalysis. On the other hand, it has been demonstrated that the phosphoryl group is
transferred by E. coli acetate kinase with inversion of configuration (Blattler and Knowles
1979). Such data are typically taken as evidence for a direct in-line transfer of phosphate
from substrate to product without an enzyme-linked covalent intermediate.
Figure 1.8 Amino acid sequence alignment of propionate kinase (TdcD_ST) and acetate kinase
(Ack_ST) from Salmonella typhimurium. Black and gray boxes indicate identical and similar
residues, respectively.
1.3.3.4 TdcE: 2-Ketobutyrate formate lyase
It has been shown in E. coli that 2-ketobutyrate can be non-oxidatively cleaved to
propionyl-CoA and formate by a newly identified keto acid formate lyase, TdcE, which
exhibits 82% amino acid sequence identity to pyruvate formate lyase (Fig. 1.9). When E.
coli grows anaerobically, pyruvate formate lyase (PFL) catalyzes the non-oxidative
dissimilation of pyruvate to acetyl-coenzyme A (acetyl-CoA) and formate. Both TdcE
22

Chapter 1 Introduction
and PFL are functional only during anaerobic growth of the organism and use a glycyl
radical as part of their catalytic mechanism. PFL enzyme is inter-convertible between
inactive and active forms. Activation of PFL to the radical-bearing species occurs only
anaerobically and is catalyzed by an iron-sulfur protein called PFL-activating enzyme.
Introduction of the protein-based radical into TdcE is also catalyzed by PFL-activating
enzyme (Sawers et al. 1998).
Figure 1.9 Amino acid sequence alignment of 2-ketobutyrate formate lyase (TdcE_ST) and
pyruvate formate lyase (PflB_ST) from Salmonella typhimurium. Black and gray boxes indicate
identical and similar residues, respectively.
1.3.3.5 TdcF: A protein of unknown function
Immediately downstream of tdcE lies a gene encoding a 128-amino-acid protein
TdcF, which has high sequence similarity to the YjgH family of proteins (Burland et al.
1995), the function of which is unknown. It is possible that TdcF may have a role in the
23

Chapter 1 Introduction
anaerobic catabolism of serine and threonine, given that its gene is co-transcribed within
an operon containing genes implicated in this process. Recently, crystallization and
preliminary X-ray crystallographic studies on this enzyme from E. coli have been
published (Burman et al. 2003).
1.3.3.6 TdcG: L-serine deaminase
It was thought for a long time that only two serine dehydratase (SdaA and SdaB)
enzymes are present in E. coli and S. typhimurium, but studies involving tdc operon have
revealed the existence of a third enzyme encoded by the tdcG gene (Hesslinger et al.
1998). Although it is yet to be demonstrated categorically that TdcG is a serine
dehydratase, it is nonetheless clear that the gene is transcribed and that the deduced
amino acid sequence shows over 74% amino acid sequence identity with both SdaA and
SdaB (Fig. 1.10).
Figure 1.10 Amino acid sequence alignment of putative L-serine deaminase (TdcG_ST) and L-
serine deaminase I (SdaA_ST) and L-serine deaminase II (SdaB_ST) from Salmonella typhimurium.
Black and gray boxes indicate identical and similar residues, respectively.
24

Chapter 1 Introduction
Unlike threonine deaminases (dehydratases) or the D-serine dehydratase of E.
coli, which are PLP-dependent enzymes, SdaA, SdaB and TdcG are PLP-independent
enzymes. Studies on the L-serine dehydratase from Peptostreptococcus asaccharolyticus
have demonstrated that instead of PLP, the enzyme contains an oxygen sensitive 4Fe-4S
cluster, which is essential for catalysis. The difference in the prosthetic groups used by
serine and threonine dehydratases reflects the different chemistry of elimination of
hydroxyl from a primary alcohol (serine) versus a secondary alcohol (threonine).
1.3.3.7 Phosphotransacetylase (Pta)
During anaerobic degradation of L-threonine to propionate, conversion of
propionyl-CoA to propionyl phosphate is catalyzed by an enzyme called
phosphotransacetylase (Pta; EC 2.3.1.8) which is not present in the tdc operon
(Hesslinger et al. 1998; Sawers 1998). Pta also catalyzes the transfer of the acetyl group
from acetyl-CoA to inorganic phosphates, forming acetyl-phosphate and coenzyme A
during the anaerobic catabolism of L-serine to acetate. Hence, it plays a major role in
acetate metabolism.
Crystal structures of Pta from Bacillus subtilis and Methanosarcina thermophila
in the native form and in complex with acetyl-phosphate as well as coenzyme A have
been determined (Iyer et al. 2004; Lawrence et al. 2006; Xu et al. 2005). The enzyme
folds into α/β architecture with two domains separated by a prominent cleft. Crystal
structure of Pta from Methanosarcina thermophila in complex with the substrate CoA
revealed one CoA bound in the proposed active site cleft and an additional CoA (CoA2)
bound at the periphery of the cleft (Lawrence and Ferry 2006). In Bacillus subtilis, Pta-
acetyl phosphate complex structure revealed a few potential substrate binding sites (Xu et
al. 2005). Two of them were located in the middle of the interdomain cleft; each one is
surrounded by a region of strictly and highly conserved residues. High structural
similarities were found with 4-hydroxythreonine-4-phosphate dehydrogenase (PdxA),
and isocitrate and isopropylmalate dehydrogenases; these enzymes utilize NADP+ as their
cofactor, which binds in the inter-domain cleft. This suggested that the CoA is likely to
bind to the inter-domain cleft of Pta in a similar way as NADP+ binds to these three
enzymes.
25

Chapter 1 Introduction
1.4 Propionate catabolism: 2-methylcitrate pathway Plants, insects and microorganisms make use of a number of alternate pathways
for propionate breakdown. Extensive analyses of propionic acid catabolism in various
organisms have elucidated at least seven different pathways (reviewed by Textor et al.
1997). All known degradation pathways of propionate start with the activation of the fatty
acid as propionyl-CoA, which may undergo α-oxidation, β-oxidation, α-carboxylation,
reductive carboxylation or Claisen condensations.
Propionate degradative pathways proposed to exist in E. coli and S. typhimurium
(Horswill and Escalante-Semerena 1999b) are described below (Fig. 1.11):
α-hydroxyglutarate pathway: Reeves and Ajl reported the synthesis of 2-
hydroxyglutarate from propionyl-CoA and glyoxylate catalyzed by cell-free
extracts from E. coli grown on propionate (Reeves and Ajl 1962).
Citramalate pathway: In Clostridium tetanomorphum, the formation of 3-
methylmalate, dehydration to mesaconate (methylfumarate) and then to
citramalate (2-methylmalate), followed by cleavage to acetate and pyruvate
could be a plausible pathway for α-oxidation of propionate to pyruvate
(Buckel and Bobi 1976). The reoxidation of acetate to glyoxylate could
proceed via the glyoxylate cycle.
Methylmanonyl-CoA pathway: The most common and the best established
pathway of propionate degradation is the α-carboxylation of propionyl-CoA
to (S)-methyl-malonyl-CoA, which racemizes to the (R)-enantiomer. The
latter rearranges in a coenzmye-B12-dependent reaction to succinyl-CoA
(Wegener et al. 1968). This pathway is widespread among aerobic and
anaerobic bacteria and in animal and human mitochondria. Vitamin B12
cannot be synthesized de novo by E. coli and S. typhimurium. It has been
reported that in the presence of Vitamin B12, E. coli also uses this pathway
(Evans et al. 1993). The fact that E. coli and S. typhimurium can grow on
propionate in the absence of vitamin B12 suggests the existence of another
pathway.
26

Chapter 1 Introduction
Acryloyl-CoA pathway: α-oxidation is initiated by dehydrogenation of
propionyl-CoA to acryloyl-CoA, followed by α-hydration to lactoyl-CoA
(Evans et al. 1993).
2-methylcitrate pathway: This pathway was initially discovered in the yeast
Candida lipolytica, in which the partner of propionyl-CoA in the Claisen
condensation is oxaloacetate rather than glyoxylate. The condensation product
2-methylcitrate isomerizes to 2-methylisocitrate (2-hydroxy-2-methyl-3-
carboxyglutarate), which is cleaved to succinate and pyruvate (Tabuchi et al.
1974; Tabuchi and Uchiyama 1975; Uchiyama and Tabuchi 1976).
Oxaloacetate is regenerated by oxidation of succinate in the Krebs cycle.
Figure 1.11 Proposed propionate breakdown pathways in E. coli and S. typhimurium (reproduced
from Horswill and Escalante-Semerena 1999b). Pathways: 1, α-hydroxyglutarate; 2, citramalate; 3,
methyl-malonyl-CoA; 4, acryloyl-CoA; 5, 2-methylcitric acid cycle.
27

Chapter 1 Introduction
In E. coli, 14CO2 evolution experiments and 13C-nuclear magnetic resonance (13C-
NMR) spectroscopic studies led researchers to conclude that propionate was oxidized to
pyruvate through acryloyl-CoA and lactoyl-CoA intermediates (Kay 1972; Wegener et al.
1967). Furthermore, 13C-NMR studies and crude enzyme assays indicated that E. coli
could also be catabolized to propionate through the 2-hydroxyglutarate, citramalate, and
methylmalonyl-CoA pathways (Evans et al. 1993; Wegener et al. 1968). Crude enzyme
assays by Fernandez-Briera and Garrido-Pertierra suggested that S. typhimurium also
possessed the acrylate pathway for propionate breakdown (Fernandez-Briera and
Garrido-Pertierra 1988). Unfortunately, till last decade experiments to fully characterize
the genes and/or enzymes for any of these oxidation pathways in E. coli or
S. typhimurium were not performed. 1.4.1 prp operon and 2-methylcitrate pathway in S. typhimurium
In the year 1996, a locus on the S. typhimurium chromosome was identified which
was required for growth on propionate (Hammelman et al. 1996). Genetic and molecular
characterization of this locus identified five genes, prpRBCDE, all of which are involved
in propionate oxidation (Horswill and Escalante-Semerena 1997). Studies on prpRBCDE
operon using a combination of in vivo and in vitro 13C-NMR experiments in
S. typhimurium led to the identification of the propionate breakdown intermediates,
which were consistent with propionate breakdown through the 2-methylcitric acid cycle
(Tabuchi and Hara 1974; Tabuchi and Serizawa 1975). These results ruled out the
presence of acryloyl-CoA or crotonyl-CoA pathways in S. typhimurium (Horswill and
Escalante-Semerena 1999b). Earlier to these studies, 2-methylcitric acid cycle was
thought to occur only in yeasts and molds. 2-methylcitrate pathway was initially
postulated by studies with mutant strains of Candida lipolytica, in which accumulation of
either 2-methylcitrate or 2-methylisocitrate was observed during growth on odd-chain
fatty acids, which were degraded via propionyl-CoA (Tabuchi et al. 1974).
The genes required for the catabolism of propionate in Salmonella typhimurium
are organized as two transcriptional units (prpR and prpBCDE) that are independently
transcribed from one another. One unit contains four genes (prpBCDE) organized as an
operon that encodes four distinct enzymes, which are required for the catabolism of
28

Chapter 1 Introduction
propionate (Fig. 1.12). The other unit contains only one gene, prpR that encodes a
predicted protein with homology to members of the σ54-dependent family of
transcriptional activators (Horswill and Escalante-Semerena 1997). The region between
the two transcriptional units is 264 nucleotides long and contains a σ70-promoter for prpR,
putative ribosome-binding sites (RBSs) for both transcription units, and a consensus
RpoN σ70-binding region 5' to prpBCDE (Fig. 1.12). Sequence analysis has suggested the
involvement of PrpR and σ54 in the transcription of this operon.
Figure 1.12 Schematic representation of the prpRBCDE locus of Salmonella typhimurium
(reproduced from Palacios and Escalante-Semerena 2000). Genes are shown to scale. The
expanded region shows the sequences required for regulation of prpBCDE and prpR expression.
Ribosome binding sites are indicated in bold face. The -10 and -35 regions of the σ70 promoter of
prpR and the -12 and -24 regions of the σ54 promoter of the prpBCDE operon are underlined.
Arrows show the direction of transcription and identify the translation initiation codon of prpR
and prpB.
29

Chapter 1 Introduction
In 2-methylcitrate pathway, PrpE, a propionyl-CoA synthetase (Horswill and
Escalante-Semerena 1999a), forms propionyl-CoA from propionate and coenzyme A and
PrpC, a 2-methylcitrate synthase (Horswill and Escalante-Semerena 1999b), forms 2-
methylcitrate by combining propionyl-CoA and oxaloacetate. The 2-methylcitrate thus
formed is converted to 2-methylisocitrate by two separate enzymes, PrpD, a 2-
methylcitrate dehydratase, and either of the two aconitases, aconitase A or aconitase B
(AcnA or AcnB), present in S. typhimurium (Horswill and Escalante-Semerena 2001).
The last step of this cycle, the cleavage of 2-methylisocitrate to succinate and pyruvate, is
catalyzed by PrpB, a 2-methylisocitrate lyase (Fig. 1.13). Succinate is further oxidized to
oxaloacetate for condensation with propionyl-CoA, forming 2-methylcitrate and
completing the cycle, whereas pyruvate can be used for energy metabolism and synthesis
of biomass. 2-methylcitric acid cycle resembles the glyoxylate cycle responsible for α-
oxidation of acetate to glyoxylate (Fig. 1.14).
Figure 1.13 2-methylcitrate pathway for propionate catabolism in S. typhimurium. The catabolic
functions of Prp enzymes and the aconitases, AcnA and AcnB are indicated.
30

Chapter 1 Introduction
Isocitratelyase
Figure 1.14 2-Methylcitric acid cycle resembles a part of the glyoxylate and TCA Cycle. 1.5.2 prp operon and 2-methylcitrate pathway in E. coli
2-methylcitrate pathway was also identified in E. coli as the route of propionate
breakdown (London et al. 1999; Textor et al. 1997). In E. coli, Textor et. al., have shown
that extracts of propionate-grown cells contained a specific enzyme that catalyses the
condensation of propionyl-CoA with oxaloacetate, most probably to 2-methylcitrate. The
enzyme was purified and identified as the 2-methylcitrate synthase or citrate synthase II.
By sequence identities, the gene coding for 2-methylcitrate synthase was identified in the
region of 8 minutes on the E. coli chromosome and shows high similarities to several
citrate synthases and to the prpC gene product (96% identity), which is part of a
propionate catabolism operon in S. typhimurium. In E. coli, the gene coding for 2-
methylcitrate synthase belongs to a cluster of four genes, the products of which have high
similarities to those of the propionate catabolism operon in S. typhimurium (Fig. 1.15):
the deduced amino acid sequence of an ORF present upstream of 2-methylcitrate
synthase gene shows 85% identity with PrpB of S. typhimurium and with
31

Chapter 1 Introduction
carboxyphosphonoenolpyruvate phosphono mutases and several isocitrate lyases.
Downstream of the 2-methylcitrate synthase gene, there are two genes that on the protein
level show 89% identity with PrpD and 76% identity with PrpE of S. typhimurium,
respectively. The gene prpE is similar to that of several acetyl-CoA synthetases. An
interesting difference between E. coli and S. typhimurium prpBCDE operons is the gap
between the prpB and prpC genes. The gap distance is 439 bp in E. coli and only 122 bp
in S. typhimurium. Analysis of this gap shows that the first 82 bp are 34% identical
between these bacteria while the last 40 bp are 73% identical. The missing 318 bp in
S. typhimurium are actually part of four 91 bp repeats in the E. coli sequence. In E. coli,
this region has been proposed to contain an open reading frame (ORF) that is transcribed
in the opposite direction to the prp operon (Blattner et al. 1997). However, the presence
of these repeats suggests that this region may have undergone some rearrangements, and
in fact, it does not contain an ORF. It is possible that these repeats negatively affect the
expression of the E. coli prpBCDE operon (Horswill and Escalante-Semerena 1999b).
Figure 1.15 The prp operon present in Escherichia coli shows high similarities to the prp operon of
Salmonella typhimurium (reproduced from Textor et al. 1997). The data in percent are identities on
the amino acid level.
1.4.3 Enzymes encoded by the prp operon and AcnA/AcnB
PrpR, a probable σ54 transcription factor and enzymes encoded by the prpBCDE
operon and aconitases (AcnA or AcnB) involved in the 2-methylcitrate pathway are
discussed below:
32

Chapter 1 Introduction
1.4.3.1 PrpR: A member of σ-54 family of transcriptional activators
prpR gene encodes a putative member of the σ54-family (RpoN) of transcriptional
activators (Shingler 1996) needed for activation of prpBCDE transcription. PrpR is a
sensor of 2-methylcitrate, an intermediate of the 2-methylcitric acid cycle used by this
bacterium to convert propionate to pyruvate. PrpR was unresponsive to citrate (a close
structural analogue of 2-methylcitrate) and to propionate, suggesting that 2-methylcitrate,
not propionate, is the metabolite that signals the presence of propionate in the
environment to S. typhimurium (Palacios and Escalante-Semerena 2004). σ54-dependent
transcriptional activators belong to the AAA+ superfamily of mechanochemical ATPases
(Zhang et al. 2002). These activators display a clear three-domain structure, with a highly
variable N-terminal signal-sensing “A domain”, a catalytic “C domain” with ATPase
activity, and a C-terminal DNA-binding “D domain”. Experiments have shown that in
PrpR, the N-terminal A domain is most likely the sensing domain to which 2-
methylcitrate, the proposed co-activator of PrpR, binds (Palacios and Escalante-Semerena
2000) (Fig. 1.16).
Figure 1.16 Modular representation of PrpR as an example of a σ54-dependent activator
(reproduced from Palacios and Escalante-Semerena 2004).
A medium-copy-number plasmid carrying the lacZ gene under the control of the
native σ54-promoter of prpBCDE was used to study prpBCDE operon expression.
Transcription of the lacZ reporter in prpR, ntrA, and ihfB (integration host factor, a global
regulatory protein) mutants was 85-, 83-, and 15-fold lower than the level of transcription
measured in strains carrying the wild-type allele of the gene tested. These data indicated
that PrpR, integration host factor and transcription sigma factor RpoN were required for
the expression of the prpBCDE operon (Palacios and Escalante-Semerena 2000).
33

Chapter 1 Introduction
1.4.3.2 PrpE: Propionyl-CoA synthetases
Propionyl-CoA synthetase (PrpE) enzyme of Salmonella typhimurium catalyzes
the first step of propionate catabolism, i.e., the activation of propionate to propionyl-CoA
(Horswill and Escalante-Semerena 1999a). The PrpE protein is required for growth of S.
typhimurium on propionate and can substitute for the acetyl-CoA synthetase (Acs)
enzyme during growth on acetate. In S. typhimurium, the PrpE shows 36% amino acid
sequence identity with acetyl-CoA synthetase (Fig 1.17). Acetyl-CoA synthetases can
compensate for the lack of propionyl-CoA synthetase activity in prpE mutants. PrpE and
Acs appear to be the only propionyl-CoA synthetase activities in the cell capable of
supporting growth on propionate. Cell-free extracts enriched for PrpE catalyzed the
formation of propionyl-CoA in a propionate, ATP, Mg2+ and HS-CoA dependent manner.
Acetate substituted for propionate in the reaction at 48% of the rate of the latter.
Figure 1.17 Amino acid sequence alignment of propionyl-CoA synthetase (PrpE_ST) and acetyl-
CoA synthetase (Acs_ST) from Salmonella typhimurium. Black and gray boxes indicate identical
and similar residues, respectively.
Acetyl-CoA synthetases and propionyl-CoA synthetases are the primary route for
34

Chapter 1 Introduction
fatty acid activation. These enzymes activate acyl groups in a two-step reaction
mechanism, which proceeds through an acyl-adenylate (acyl-AMP) intermediate (Berg
1956) as shown in figure 1.18.
Figure 1.18 The acyl-CoA synthetase reaction. This reaction proceeds through an acyl-AMP
intermediate. In case of PrpE, the R group would be CH3-CH2- for propionate (reproduced from
Horswill and Escalante-Semerena 2002).
1.4.3.3 PrpC: 2-methylcitrate synthase
The first key enzyme specific for this pathway is 2-methylcitrate synthase, which
catalyses the condensation of propionyl-CoA and oxaloacetate to 2-methylcitrate. PrpC
also catalyzed the synthesis of citrate from acetyl-CoA and oxaloacetate, demonstrating
that the enzyme has citrate synthase activity as well. Glyoxylate did not substitute for
oxaloacetate in the reaction (Horswill and Escalante-Semerena 1999b). In E. coli, 2-
methylcitrate synthase shows 27% amino acid sequence identity to citrate synthase.
Citrate synthase is a hexametric enzyme produced constitutively, whereas 2-methylcitrate
synthase is a dimeric protein induced during growth on propionate. Citrate synthase does
not possess 2-methylcitrate synthase activity (Gerike et al. 1998). To identify the
preferred substrate for PrpC enzyme, bi-substrate kinetic experiments were performed
with propionyl-CoA or acetyl-CoA as the substrate. The kcat/KM values were about 30:1 in
favor of propionyl-CoA (Horswill and Escalante-Semerena 1999b). The citrate synthases
of the psychrotolerant eubacterium DS2-3R, and of the thermophilic archaea
Thermoplasma acidophilum and Pyrococcus furiosus, are approximately 40% identical in
sequence to the E. coli 2-methylcitrate synthase (Fig. 1.19) and also possess 2-
methylcitrate synthase activity (Gerike et al. 1998).
Aspergillus nidulans has been used as a model organism to investigate fungal
propionate metabolism and the mechanism of growth inhibition by propionate. A 2-
methylcitrate synthase (mscA) deletion strain of A. nidulans was unable to grow on
35

Chapter 1 Introduction
propionate. The inhibitory growth effect of propionate on glucose medium was enhanced
in this strain, which led to the assumption that trapping of the available CoA as
propionyl-CoA and/or the accumulating propionyl-CoA itself interferes with other
biosynthetic pathways such as fatty acid and polyketide syntheses (Brock et al. 2000). In
the wild type, propionyl-CoA is metabolized via the 2-methylcitrate cycle and thus does
not accumulate.
Figure 1.19 Amino acid sequence alignment of 2-methylcitrate synthase with various citrate
synthases. PrpC_ST, 2-methylcitrate synthase from S. typhimurium and GltA_DS, GltA_PF and
GltA_ST denotes citrate synthases from Antarctic bacterium DS2-3R, Pyrococcus furiosus and S.
typhimurium. Black and gray boxes indicate identical and similar residues, respectively.
1.4.3.4 PrpD: 2-methylcitrate dehydratase
Previous studies showed that 2-methylcitrate accumulated in prpD mutant strains
of S. typhimurium during growth on propionate (Horswill and Escalante-Semerena
1999b), suggesting the involvement of the PrpD protein in the conversion of 2-
methylcitrate to 2-methylisocitrate. This reaction is reminiscent of citrate to isocitrate
36

Chapter 1 Introduction
conversion catalyzed by aconitase. Later, it was shown that prpD gene of the prpBCDE
operon encodes a protein with 2-methylcitrate dehydratase enzyme activity (Brock et al.
2002; Horswill and Escalante-Semerena 2001). Homogeneous PrpD enzyme does not
contain an iron-sulfur center, displays no requirements for metal cations or reducing
agents for activity, and does not catalyze the hydration of 2-methyl-cis-aconitate to 2-
methylisocitrate. The DNA sequence of the prpD gene and the predicted amino acid
sequence of its gene product show no similarity to known genes or their encoded
proteins. This observation is surprising considering the large number of known
dehydratases with substrates similar to 2-methylcitrate. PrpD shows striking sequence
similarity to a number of hypothetical proteins such as YqiQ from Bacillus subtilis and
PDH1 from Saccharomyces cerevisiae. Among the organisms with prpD homologues,
only E. coli and S. cerevisiae have been shown to catabolize propionate via the 2-
methylcitric acid cycle. The uniqueness of the PrpD sequence could be used to search for
the 2-methylcitric acid cycle in genomes of organisms, although some prokaryotes with
prp operons do not have the prpD gene (Fig. 1.20).
1.4.3.5 AcnA or AcnB: Aconitase A or Aconitase B
If PrpD did not hydrate 2-methyl-cis-aconitate, a gene outside of the prp operon
in S. typhimurium and E. coli must encode the missing hydratase enzyme. Aconitases
from mammals and fungi have been shown to catalyze the hydration of 2-methyl-cis-
aconitate, which suggested that in the S. typhimurium, aconitase(s) may do the same.
Analysis of bacterial genome databases identified the presence of orthologues of the acnA
gene (encodes aconitase A) in a number of putative prp operons (Horswill and Escalante-
Semerena 2001) (Fig. 1.20). Homogeneous preparation of AcnA protein in S.
typhimurium showed strong aconitase activity and catalyzed the hydration of the 2-
methyl-cis-aconitate to yield 2-methylisocitrate. The purification of this enzyme allows
the complete reconstitution of the 2-methylcitric acid cycle in vitro using homogeneous
preparations of the PrpE, PrpC, PrpD, AcnA, and PrpB enzymes.
However, inactivation of the acnA gene did not block growth of S. typhimurium
on propionate as carbon and energy source. The existence of a redundant aconitase
activity (encoded by acnB) was postulated to be responsible for the lack of a phenotype
37

Chapter 1 Introduction
in acnA mutant strains. Consistent with this hypothesis, homogeneous preparation of
AcnB protein from S. typhimurium also showed strong aconitase activity and catalyzed
the conversion of 2-methyl-cis-aconitate into 2-methylisocitrate. To address the
involvement of AcnB in propionate catabolism, an acnA and acnB double mutant was
constructed, and this mutant strain could not grow on propionate even when
supplemented with glutamate. The phenotype of this double mutant indicated that the
aconitase enzymes are required for the 2-methylcitric acid cycle during propionate
catabolism (Horswill and Escalante-Semerena 2001). In E. coli W3350 cells grown on
propionate as sole carbon and energy source, AcnB was the only enzyme that displayed
activity as a 2-methylisocitrate dehydratase (Brock et al. 2002). The absence of the acnA
gene in the prpBCDE operon of S. typhimurium and E. coli suggests that AcnA may have
other functions, and thus their synthesis cannot be restricted to growth conditions where
propionate is available in the environment. Any additional functions that AcnA may have
are currently unknown.
Figure 1.20 A comparison of prp operons from a number of bacteria (reproduced from Horswill
and Escalante-Semerena 2001). The operons are grouped by similarities in gene organization,
although minor differences exist between each operon in a group. ORFs were not drawn exactly
to the scale due to differences in gene sizes between each species. Predicted ORFs from S. typhi, B.
pertussis, and P. putida KT2440 are from unfinished genomes. Sequence information from E. coli,
N. meningitidis MC58, P. aeruginosa PAO1, and V. cholerae was obtained from published sources
(Altschul et al. 1997; Blattner et al. 1997; Kunst et al. 1997; Stover et al. 2000). ORFs were named
after the S. typhimurium gene designations except ybhH, yfcA, and ackA (acetate kinase), which
were named first in E. coli (Blattner et al. 1997). The putative ackA gene of N. meningitidis also
shows sequence similarity to the propionate kinase tcdD of E. coli.
38

Chapter 1 Introduction
1.4.3.6 PrpB: 2-methylisocitrate lyase
The last step of 2-methylcitric acid cycle, the cleavage of 2-methylisocitrate to
succinate and pyruvate is catalyzed by 2-methylisocitrate lyase. In Saccharomyces
cerevisiae, 2-methylisocitrate lyase activity is induced when threonine is used as a
nitrogen source (Luttik et al. 2000). 2-methylisocitrate lyase (PrpB) of E. coli in a
BLAST search shows significant sequence identity to carboxyphosphonoenolpyruvate
phosphono mutases (CPEP mutases), phosphonoenolpyruvate mutases (PEP mutases)
and with the isocitrate lyases (ICLs) of bacterial and eukaryotic origin. The mutases are
anabolic enzymes used for the production of secondary metabolites and the only enzymes
known to create a carbon-phosphorus bond (Hidaka et al. 1990; Ogawa et al. 1973),
while the lyases are catabolic enzymes (Horswill and Escalante-Semerena 1999b; Luttik
et al. 2000; Vanni et al. 1990). Therefore, the presence of amino acid sequence identity
of 35 % between PrpB from S. typhimurium and CPEP mutase Streptomyces
hygroscopicus is very surprising (Fig. 1.21). Despite the divergence in the chemistry that
it catalyzes, the PEP mutase active site conserves the catalytic scaffold observed in
isocitrate lyase, including the flexible active site loop, which regulates solvent access and
all these enzymes are proposed to proceed via mechanisms that stabilize enol(ate)
intermediates (Seidel and Knowles 1994).
Figure 1.21 Amino acid sequence alignment of 2-methylisocitrate lyase from Salmonella
typhimurium (PrpB_ST) and carboxyphosphoenolpyruvate phosphono mutase from Streptomyces
hygroscopicus (CPEPM_SH). Triangles indicate the consensus sequence around the active-site
cysteine. Black and gray boxes indicate identical and similar residues, respectively.
39

Chapter 1 Introduction
In E. coli and S. typhimurium, 2-methylisocitrate lyase has 35% lower molecular
mass than the isocitrate lyases and shares 25% amino acid sequence identity with the
latter (Fig. 1.22). Therefore, the PrpB protein appears to be one of the smallest proteins of
the isocitrate lyase family and seems to have all essential amino acids conserved.
Figure 1.22 Amino acid sequence alignment of 2-methylisocitrate lyase (PrpB_ST) and isocitrate
lyase (ICL_ST) from Salmonella typhimurium. Triangles indicate the consensus sequence around
the active-site cysteine. Black and gray boxes indicate identical and similar residues, respectively.
The alignment reflects the significantly shorter length of PrpB when compared to ICL.
The KKCGH sequence found around Cys195 in the catalytic site of isocitrate
lyase from E. coli is conserved in all other known isocitrate lyases, whereas PrpB from
E. coli, S. typhimurium, the deduced 2-methylisocitrate lyase from S. cerevisiae (Luttik et
al. 2000), carboxyphosphoenolpyruvate phosphono mutase from Streptomyces
hygroscopicus (Hidaka et al. 1990; Pollack et al. 1992) and five proteobacterial
homologues of PrpB contain the slightly modified sequence KRCGH around Cys123
(Fig. 1.23). Cys195 of isocitrate lyase from E. coli has been shown to be modified by
bromopyruvate (Ko and McFadden 1990). Therefore, it appears likely that the
corresponding Cys123 in 2-methylisocitrate lyase from S. typhimurium is the target for
40

Chapter 1 Introduction
bromopyruvate. These conserved cysteine residues are involved in inactivation by air and
reactivation by dithiothreitol.
Figure 1.23 Alignment of the amino-acid sequences around the active site cysteine in isocitrate
lyases, 2-methylisocitrate lyases and related proteins (reproduced from Brock et al. 2001). ScICL1,
AniICL and Ecoicl denote isocitrate lyases from Saccharomyces cerevisiae, A. nidulans and E. coli,
respectively. EcprpB and SaccII denote 2-methylisocitrate lyases from E. coli and S. cerevisiae,
Saltyp, Pseuae and Neimen represent putative 2-methylisocitrate lyases from Salmonella
typhimurium, Pseudomonas aeruginosa and Neisseria menengitis, SHcpep represents
carboxyphosphoenolpyruvate phosphono mutase from Streptomyces hygroscopicus, respectively.
GenBank accession numbers are shown in parentheses.
1.4.4 Presence of the 2-methylcitrate pathway among prokaryotes
Previous studies on the 2-methylcitric acid cycle demonstrated that it is widely
distributed among fungi (Miyakoshi et al. 1987). The presence of 2-methylcitric acid
cycle in both S. typhimurium and E. coli (Horswill and Escalante-Semerena 1999b;
Textor et al. 1997) has prompted various groups to investigate the prevalence of this
pathway among prokaryotes. With the recent sequencing of many prokaryotic genomes,
homologues of the Prp enzymes have been identified in genomes such as Mycobacterium
tuberculosis, Salmonella typhi, Bacillus pertussis, Pseudomonas putida, Neisseria
meningitidis, Pseudomonas aeruginosa, and Vibrio cholerae (Fig. 1.20). As more
genome sequences become available and prokaryotes are tested for activities of the 2-
methylcitric acid cycle, we may find that this pathway is as widely distributed among
prokaryotes as among fungi.
41

Chapter 1 Introduction
1.5 Objectives of the present study The sequencing of microbial genomes containing several thousand genes is the
underlying driving force for studies towards understanding metabolic networks at a
deeper level of complexity. Advances in molecular biology techniques and genomic data
jointly have helped in identifying new metabolic pathways present in various
microorganisms. Many of these metabolic pathways utilize a large number of enzymes
with diverse catalytic mechanisms. With the availability of sequence as well as probable
role of many such enzymes in a metabolic pathway, our objective was to carry out
structure-function studies on enzymes involved in the metabolism of an important
metabolite, propionate.
Propionate is the second most abundant low-molecular-mass carbon compound
found in the soil. In last decade, propionate metabolic pathways and the enzymes
involved have been identified in Escherichia coli as well as Salmonella typhimurium.
These pathways include anaerobic degradation of L-threonine to propionate and
propionate catabolism by 2-methylcitrate pathway. Since these two pathways have been
well characterized in Salmonella typhimurium, we used this as the model organism.
When we started this work, no biochemical or structural data were available for most of
the enzymes involved in these two pathways. In many cases, only putative functions had
been assigned on the basis of amino acid sequence alignment. One of the most powerful
ways to analyze a protein is to determine its three-dimensional structure. Therefore, our
main objective was to try and solve the structure of these proteins and follow structural
studies by functional characterization.
Out of the four enzymes involved in the anaerobic degradation of L-threonine to
propionate, our focus was mainly on the first and the last enzymes, biodegradative
threonine deaminase and propionate kinase. Structural information on close homologues
of the other two enzymes in this pathway was available. Biodegradative threonine
deaminase (TdcB) is the only well characterized enzyme in this pathway, which catalyzes
the deamination of L-threonine to α-ketobutyrate. Earlier studies had shown that TdcB,
unlike the biosynthetic threonine deaminase, is insensitive to L-isoleucine and is
activated by AMP. Even at low concentrations of TdcB, it was shown that the presence of
AMP induces oligomerization from monomer to tetramer. Extensive biochemical studies
42

Chapter 1 Introduction
carried out on this enzyme had not revealed the exact site of AMP binding and its role in
AMP induced oligomerization and enzyme activation. There was no structural data on
this enzyme from any source. The present study was aimed to provide not only the
structural information but also to provide an insight into ligand induced oligomerization
and enzyme activation. A comparative study between biosynthetic and biodegradative
threonine deaminase was also planned.
Propionate kinase is proposed to catalyze the last step in the catabolism of L-
threonine to propionate by enabling the conversion of propionyl phosphate and ADP to
propionate and ATP. No biochemical or structural data was available on this enzyme
from any source. However, this enzyme was shown to possess 38-44 % sequence identity
with various acetate kinases. The work on propionate kinase was aimed not only to solve
crystal structures of propionate kinase and its complex with various ligands but also to
carry out biochemical studies to demonstrate its propionate kinase activity. After
structure determination, a detailed structural comparison with the well-studied acetate
kinase was also planned. While carrying out structural studies on propionate kinase, we
observed a novel Ap4A synthetic activity in this enzyme. We further extended our work
in this direction to demonstrate the presence of such enzymatic activity in propionate
kinase. The work on biodegradative threonine deaminase and propionate kinase
constitutes the major portion of this thesis.
Oxidation of propionate to pyruvate and succinate by 2-methylcitric acid cycle
resembles the part of the glyoxylate cycle in which acetate is oxidized to glyoxylate. Four
different enzymes encoded by the prp operon and a σ54-transcriptional activator were
selected for structural studies. There was no structural information on any of these
enzymes. In only one or two cases, basic biochemical information was available. Our
studies on Prp enzymes were aimed to provide structural as well as functional insights.
An extensive structural comparison was also planned with glyoxylate enzymes which
catalyze similar reactions but shares low sequence identities with Prp enzymes.
Besides overall structural information, these studies were expected to provide
insights into the active site pocket, substrate specificity and catalytic mechanism of these
enzymes. The remainder of the thesis deals with the work carried out in this direction.
43

Materials and methods 2

Chapter 2 Materials and methods
2.1 Introduction In this chapter, detailed descriptions of experimental and computational
procedures used during the course of investigations are described. The first part of this
chapter discusses experiments such as cloning, expression, purification and
crystallization. This is followed by details of data collection, data processing, structure
solution, refinement, model building, validation and analysis. Experimental procedures
unique to specific enzymes are described in the respective chapters.
2.2 Materials 2.2.1 Chemicals used in the study
The fine chemicals routinely used in the laboratory for the biochemical and
molecular biology experiments such as agarose, ampicillin, IPTG, DTT, Tris, Imidazole,
Triton X-100, Ni-NTA etc were purchased from Sigma-Aldrich, Novagen and
Calbiochem. Restriction endonucleases, DNA modifying enzymes and polymerases were
purchased from MBI Fermentas and New England Biolabs (NEB). Crystallization
screens, paraffin oil and silicone oil required for microbatch experiments were obtained
from Hampton Research. The 24-well multicavity plates used for crystallization were
from Laxbro and 72-well microbatch plates were from Greiner. [γ-32P] ATP was obtained
from New England Nuclear. Most of the other chemicals used in the study were of
analytical grade, purchased from local chemical companies.
2.2.2 Plasmids used in the study
Two plasmids pRSET C (Invitrogen) and pETBlue-2 (Novagen) were used for
cloning the genes of interest with a hexa-histidine tag at N- and C-terminals, respectively
(Fig 2.1). The pRSET vectors are pUC-derived expression vectors designed for high-
level protein expression and purification from cloned genes in E. coli. It includes an ATG
translation initiation codon and a hexa-histidine tag. Expression of the gene of interest
from pRSET is controlled by the strong phage T7 promoter. T7 RNA polymerase
specifically recognizes this promoter. To facilitate cloning, the pRSET vector is provided
in three different reading frames (pRSET A, B & C). They differ only in the spacing
between the sequences that code for the N-terminal peptide and the multiple cloning site.
45

Chapter 2 Materials and methods
Nhe
I
(a)
(b)
Figure 2.1 Map of (a) pRSET C and (b) pETBlue-2 vectors.
The pETBlue-2 vector has been designed to identify recombinants by traditional
blue/white screening while also allowing T7 lac promoter based expression of target
genes. This vector features an expanded multiple cloning site (MCS) and optional C-
terminal HSV-tag and His-tag sequences.
46

Chapter 2 Materials and methods
2.2.3 Bacterial strains used in the study
E. coli strain DH5α (BRL) cells were used for propagation of plasmids for
cloning experiments. The protein expressions were carried out in E. coli BL21(DE3) or
BL21(DE3)pLysS cells. The features associated with these two strains are given in table
2.1. Table 2.1 Host strain features
Expression strain Induction method Advantages Disadvantages
BL21(DE3) strain
IPTG induction of T7 polymerase from lacUV5 promoter
High-level expression
Leaky expression of T7 polymerase can lead to
uninduced expression of potentially toxic proteins
BL21(DE3)pLysS strain IPTG induction of T7 Ease of
induction polymerase
Slight inhibition of induced expression when compared
with BL21(DE3)
2.3 Methods 2.3.1 Preparation of E. coli competent cells and transformation
E. coli DH5α, BL21(DE3) and BL21(DE3)pLysS cells were made competent for
the uptake of DNA by manganese chloride method (Alexander 1987). For the preparation
of competent cells, 2 X LB broth medium was used. Initially, preinoculum (10 ml) of the
culture was grown by inoculating a single colony. One ml of this preinoculum was
inoculated into 100 ml of 2 X LB broth medium and was grown at 30ºC in a shaker until
the OD of the culture reached 0.5 at 600 nm. The culture was chilled for 30 min. Cells
were pelleted in autoclaved tubes at 3000 rpm in a SS34 rotor (Sorvall RC5B) at 4ºC for
15 min. The pellet was resuspended in 20 ml of filter-sterilized ice-cold acid-salt buffer
(40 mM sodium acetate buffer pH 5.5 containing 100 mM CaCl2 and 70 mM MnCl2) and
incubated again on ice for the next 45 min. The cells were pelleted once again at 3000
rpm for 15 min at 4ºC and the pellet was resuspended in 2.5 ml of acid-salt buffer
containing 20% glycerol. 50 μl of the cell suspension was aliquoted into different tubes
and stored at -70ºC. Transformations were performed by the addition of approximately
200 ng of DNA into each aliquot of competent cells. It was then incubated on ice for 30
min. The cells were given heat shock for 5 min at 37ºC and then plated in LB agar plate
containing ampicillin.
47

Chapter 2 Materials and methods
2.3.2 Cloning and overexpression
The DNA encoding the open reading frame for the gene of interest was amplified
using high fidelity KOD HiFi DNA polymerase (Novagen) or Deep Vent DNA
polymerase (NEB) from Salmonella enterica serovar Typhimurium strain IFO 12529
genomic DNA using polymerase chain reaction (PCR). Primers were designed to
introduce NheI and NcoI restriction sites at the 5' end and BamHI and XhoI at the 3' end
of the gene. Restriction sites NheI and BamHI were used to clone the gene into pRSET C
with an N-terminal His tag whereas NcoI and XhoI were used to clone the gene into
pETBlue-2 with a C-terminal His tag. After amplification of the target gene, the PCR-
amplified fragment was digested with restriction enzymes and then cloned into the
pRSET C or pETBlue-2 vector encoding a polypeptide with a hexa-histidine tag to
facilitate its purification using Ni-NTA affinity column chromatography. The sequence of
the cloned gene was determined by nucleotide sequencing and confirmed by comparing it
with the respective gene of Salmonella typhimurium LT2.
The plasmid was then transformed into E. coli strain BL21(DE3) or
BL21(DE3)pLysS and transformants were selected on LB agar plates containing
100 µg ml-1 ampicillin. Bacteria were grown overnight at 37ºC in 25 ml LB broth
containing ampicillin. The bacterial suspension that resulted was then diluted into fresh
2 l terrific broth (TB) medium containing ampicillin and grown at 310 K. When the
culture optical density (OD) at 600 nm reached 0.6-0.7, protein expression was induced
with 0.3 mM IPTG and cells were grown for an additional 6 h at 30ºC before being
harvested by centrifugation.
2.3.3 Purification of the recombinant enzyme
For the preparation of soluble protein fractions, cells obtained from 2 l culture
were resuspended in 100 ml cold lysis buffer containing 50 mM Tris-HCl pH 8.0 and
200 mM NaCl. The suspension was then lysed by sonication on ice. All the following
purification steps were performed at 4ºC. The lysate was centrifuged to remove the cell
debris. The clear supernatant containing soluble proteins was gently mixed with Ni-NTA
resin using an end-to-end rotator for 2 h and then loaded onto a glass column.
Contaminating proteins were washed from the column using 50 mM Tris-HCl pH 8.0,
48

Chapter 2 Materials and methods
200 mM NaCl and 15-25 mM imidazole. Protein was eluted from the column using 200-
250 mM imidazole along with 50 mM Tris-HCl pH 8.0 and 200 mM NaCl. To remove
imidazole, the protein was extensively dialyzed against 25 mM Tris-HCl pH 8.0, 50 mM
NaCl, and 1 mM DTT. The purity of the protein was estimated using SDS-PAGE
(Laemmli 1970). Dialyzed protein was concentrated to approximately 10-20 mg ml-1
using a 10 kDa molecular weight cut-off Amicon Ultra-15 Centrifugal Filter Unit
(Millipore) for crystallization experiments. The protein concentration was determined
with Lowry’s or Bradford reagent using bovine serum albumin as the standard protein
(Bradford 1976; Lowry et al. 1951). The protein concentration was also determined
routinely by measuring OD at 280 nm using a theoretically calculated molar extinction
coefficient obtained using ProtParam server (Wilkins et al. 1999).
2.3.4 Crystallization
Crystallization trials were carried out using hanging drop vapor diffusion, sitting
drop vapor diffusion and microbatch techniques. Hanging drop and microbatch methods
were used during screening whereas sitting drop method was used during optimization of
the crystallization condition.
2.3.4.1 Hanging drop vapor diffusion method
In the hanging drop vapor diffusion technique a drop composed of a mixture of
protein and reagent (crystallization condition) is placed in vapor equilibration with a
reservoir of reagent. Typically the drop contains a lower reagent concentration than the
reservoir. As a result, initially the droplet of protein solution contains an insufficient
concentration of reagent for crystallization, but as water vaporizes from the drop and
transfers to the reservoir, the reagent concentration in the drop increases to a level
sufficient for crystallization. Since the system is in equilibrium, these optimum
conditions are maintained until the crystallization is complete.
2.3.4.2 Sitting drop vapor diffusion method
Sitting drop vapor diffusion technique also works on the same principle as the
hanging drop vapor diffusion technique. In this technique, a small droplet (typically 5 to
10 μl) of the sample (protein) mixed with the crystallization reagent is kept on a
depression slide in vapor diffusion with the reagent present inside the Petri dishes. In this
49

Chapter 2 Materials and methods
technique, the drop size can be much larger than in the hanging drop technique. The
disadvantage of the sitting drop technique is that crystal can adhere to the sitting drop
surface making its removal difficult. For hanging drop and sitting drop vapor diffusion
techniques, cover slips and depression slides were siliconized using Aqua Sil (Hampton
Research) or silane (Sigma) providing a hydrophobic surface.
2.3.4.3 Microbatch method
In the microbatch technique, a small drop of the protein combined with the
crystallization reagent of choice is placed under a layer of oil. In this technique, diffusion
of water takes place from the drop through the oil layer, which results in an increase of
concentration of the protein and the reagents in the drop. The oil used was generally a 1:1
mixture of paraffin oil and silicone oil. The mineral oil used is of branched chain
paraffins in the C20+ range and allows for little to no diffusion of water through the oil.
Various commercially available crystallization screening kits such as Crystal
Screen I and II, Index, SaltRx, PEG/Ion screen available from Hampton Research were
used for initial crystallization trials. Based on a published report (Majeed et al. 2003), a
set of crystallization conditions (precipitant synergy reagent formulations) were also
prepared and used for initial crystallization screening experiments. Initial crystallization
conditions were further optimized by using buffers of different pH ranges and by varying
the concentration of precipitant and organic solvent present in the initial crystallization
condition. Various additives were also used in the initial crystallization conditions for
optimization.
2.3.5 Intensity data collection
Most of the intensity datasets were collected using the in-house X-ray facility for
Structural Biology at the Molecular Biophysics Unit, Indian Institute of Science. The
facility at Molecular Biophysics Unit consists of three MAR research image plate
detectors, two mounted on Rigaku RU-200 X-ray generators and third mounted on a
FR91 Bruker-Nonius generator. The X-ray beams were focused with multilayer mirrors.
Two datasets discussed in this thesis were collected at the synchrotron beamlines
BL44XU and BL26B1, SPring-8, Hyogo, Japan.
50

Chapter 2 Materials and methods
Most of the datasets described in this thesis were collected at 100K using a single
crystal. The crystals were transferred to a cryoprotectant composed of reservoir solution
with 20% glycerol or ethylene glycol prior to mounting. Collecting optimum X-ray
diffraction data involves a number of choices and compromises, including crystal to
detector distance, exposure time, oscillation angle, redundancy, resolution, etc (Dauter
1999). During data collection, crystal to detector distance was adjusted so that it could
record and resolve the diffraction maxima on the image plate to the resolution limit of the
crystal. These parameters depend on the unit cell dimensions and the mosaic spread of
the crystal. The longer the crystal to detector distance, the better the separation between
the reflections in the recorded diffraction pattern. If one of the unit cell dimensions is
very large that setting the detector distance to maximal diffraction resolution lead to
significant overlap of reflection profiles, it is better to sacrifice the resolution of the data
and set the distance further so that reflection profiles separate. Since large oscillation
angles tend to decrease the signal to noise ratio and the accuracy in the estimated
reflections profiles due to higher background, relatively small oscillation angle in the
range of 0.4º to 0.75º was used for better data quality. Exposure time was set long enough
to give reasonable statistics at the highest resolution, but not so long as to overload the
detector with the strong low-angle spots. The choice of exposure time was decided on the
basis of size and diffraction quality of the crystal and the oscillation range. The image
stored in the imaging plate tends to decay with time; reflections collected at the beginning
of the exposure time tend to be underestimated compared to those recorded at the end of
the exposure time. Therefore, for exposure times greater than 5 min, more than one
oscillation was used to minimize these errors. Most of the datasets were collected with
high redundancy because it produces more accurate data and allows for reliable rejection
of outliers. The maximum resolution of a data set was decided after examination of data
reduction statistics. However during the data collection, the detector was positioned a
little closer than the apparent maximum resolution, provided that the spots were resolved.
2.3.6 Data processing
X-ray diffraction data processing proceeds through indexing, pre-refinement of
detector parameters and crystal orientation, intensity integration, post-refinement and
scaling. All the datasets were indexed, integrated and scaled using DENZO and
51

Chapter 2 Materials and methods
SCALEPACK as implemented in the HKL suite of programs (Otwinowsky 1997). The
analysis and reduction of single crystal diffraction data on an image plate detector
consists of following steps:
Visualization and preliminary analysis of the original, unprocessed data.
Autoindexing: requires a peak-picking procedure, followed by an analysis of the
position of the peaks to determine unit cell dimensions, Bravais lattice and crystal
orientation.
Pre-refinement of the detector parameters (crystal to detector distance, unit cell
parameters, position of the direct beam on the image, detector tilt away from
being normal to the X-ray beam), crystal orientation and effective mosaic spread
(actual mosaic spread convoluted with beam divergence).
Intensity integration by profile fitting, assuming reflection position as calculated
from the pre-refined detector and crystal parameters.
Conversion of the data to a common scale.
Symmetry determination and merging of the symmetry related reflections.
Statistical summary and estimation of errors.
The initial four steps are carried out by DENZO and XDISPLAYF and the last
three steps are carried out using the program SCALEPACK. After each cycle of
refinement, DENZO updates the display and prints the new values for the refined
parameters and shift in their values. The output gives the χ2 value for the X and Y
positions of the predicted spots. A good refinement is expected to have χ2 close to one.
Very large value for the χ2 indicates some serious error with indexing, refinement or
detector parameters. At the end of the refinement for each individual image, DENZO
outputs a list of hkl’s and unscaled intensities of reflections.
The program SCALEPACK is used to scale the intensities output by DENZO.
The program calculates single isotropic scale and B-factors for each of the processed
frames that are input. The output gives scaled, merged data corrected for Lorentz and
polarization factors. The high resolution limit of the scaled data is assessed on two
important counts. The first is the mean ratio of the intensity to the error (I/σI). The second
is the agreement between the symmetry related reflections, Rmerge. The first criterion is a
52

Chapter 2 Materials and methods
better indicator for data quality whereas the Rmerge is an unweighted statistic, which
depends on the data redundancy as well as the number of parameters used for scaling the
frames.
Rmerge = ∑j∑ |Ihj − ⟨Ih⟩ | / ∑j∑h Ihj (2.1) where Ihj is the jth measurement of the intensity of reflection h and ⟨Ih⟩ is the average
intensity.
Low redundancy, use of large cut-offs in the datasets can lead to artificially low
Rmerge values. The high resolution limit for most of the datasets was decided on the basis
of average I/σI value greater than 2.5 and Rmerge value around 40% in the highest
resolution shell. The scaled data were converted into the suitable format for CNS suite of
programs using scalepack2cns.f and for CCP4 suite of programs using
SCALEPACK2MTZ and TRUNCATE.
2.3.7 Structure solution
One of the major barriers in solving a macromolecular crystal structure is the
solution of the phase problem, i.e. the reconstruction of the phases of the diffracted X-
rays. In macromolecular crystallography, there are basically three approaches for
recovering the phase information of a diffraction experiment, namely multiple
isomorphous replacement (MIR), multiple-wavelength anomalous dispersion (MAD) and
molecular replacement (MR).
2.3.7.1 Multiple isomorphous replacement
The method which has been used from the early days of macromolecular
crystallography is that of multiple isomorphous replacement. In this method, phase
information is retrieved by recording the X-ray diffraction data from a protein crystal and
a few of its isomorphous heavy atom derivatives. The positions of heavy atoms can be
determined by difference Patterson methods or direct methods using difference between
native and derivative amplitudes. Various steps are used to find and refine the heavy
atom positions. If the positions of bound heavy atoms are known, then the phase of each
diffracted X-ray can be calculated by solving a set of simultaneous equations. Similar
strategy is employed in other methods such as single isomorphous replacement (SIR) and
53

Chapter 2 Materials and methods
single isomorphous replacement and anomalous scattering (SIRAS), where the
anomalous dispersion due to heavy atom is also considered in resolving the phase
ambiguity. 2.3.7.1.1 Preparation of heavy atom derivative
The first step in the MIR method is the preparation of isomorphous heavy atom
derivatives. Attempts to prepare various heavy atom derivatives were made by soaking
the protein crystal in solutions of compounds of various heavy atoms such as platinum,
gold, mercury and uranium. Binding of heavy atoms to the protein is facilitated by the
presence of large solvent channels in protein crystals. The addition of one or more heavy
atoms to a macromolecule introduces differences in the diffraction intensities of the
derivative relative to that of the native. A perfect isomorphous derivative is the one in
which a few heavy atoms have been incorporated, without any change in the unit cell
parameters and in the relative orientation of the protein molecules within the unit cell.
2.3.7.1.2 Scaling between native and derivative datasets
As a first step towards heavy atom analysis, the derivative data are scaled with
respect to the native dataset. The scale factor can be calculated using least-squares
procedure by minimizing the sum of weighted squares of isomorphous differences
Σw (k |FPH| - |FP|)2 (2.2) with respect to the unknown scale factor k, where FPH and FP are the structure factor
amplitudes of the derivative and native crystals. The scaling of the derivative data sets
was performed using the program SCALEIT, a part of the CCP4 suite of programs.
2.3.7.1.3 Identifying heavy atom positions by difference Patterson
In order to obtain phase information from isomorphous replacement, it is
necessary to locate heavy atom positions. After scaling the datasets, the presence of
heavy atom in the derivatives is identified using isomorphous difference Patterson map. It
is the most common method of identifying heavy atom positions and does not require any
phase information. It requires only structure factor amplitudes.
54

Chapter 2 Materials and methods
2.3.7.1.4 Refinement of heavy atom parameters
Subsequent to the estimation of heavy atom parameters they must be refined to
their best values before being used for phase calculation. In recent years, the principle of
maximum likelihood has been used successfully to refine the heavy atom parameters
without introducing any bias. Maximum likelihood phase refinement is a method wherein
a probability distribution for values of the protein phase αbest is found. This is followed by
the minimization of the weighted differences between observed and calculated values of
FPH generated with this phase using the heavy atom parameters. The parameters which
are refined are as follows:
Relative scale and temperature factor between native and derivative data
Coordinates of heavy atoms
Occupancies of heavy atoms
Atomic displacement factors (either isotropic or anisotropic) of heavy atoms
Error estimates that are used when finding the appropriate weight for each
observation.
The function minimized is
Rj = Σ Σ w(α)(|FPHj|obs – |FPHj(α)|cal)2 (2.3) α hkl
where j denotes the number of heavy atom derivatives. The parameters are updated and
another cycle of refinement is carried out. This method of refinement is implemented in
the program MLPHARE, which refines heavy atom parameters and calculates αbest.
2.3.7.2 Multiple-wavelength anomalous dispersion
In the recent past, the multiple-wavelength anomalous dispersion method has
become increasingly popular in deriving phases with the availability of tunable
synchrotron radiation sources (Hendrickson et al. 1990; Hendrickson et al. 1989). This
method relies on the differences in the intensities of Friedel pairs of reflections caused by
the breakdown of Freidel’s law near the absorption edges of heavy atoms (anomalous
scatterer) present in the protein molecule. Heavy atoms often behave anomalously within
the wavelength range accessible in synchrotrons. Such anomalous scatterers can be
introduced by replacing the amino acids methionines and cysteines amino acids in the
55

Chapter 2 Materials and methods
protein by selenomethionines and selenocysteins, respectively, using molecular biology
techniques.
2.3.7.3 Molecular replacement
Molecular replacement encompasses techniques which are used in
macromolecular crystallography to determine the orientation and position of a molecule
in the unit cell using a previously solved structure as the 'search model'. The goal in MR
is to orient and position the search model, such that it coincides with the position of the
unknown protein in the crystal (Rossmann 1990). The model can then provide phase
information for the unknown structure. The search and target molecules must have
reasonable sequence identity (> 35 %) between them to have a good chance of success.
As the database of solved structures (PDB) gets larger and larger, this method will be
useful for a larger fraction of new structures. Of course, it is also useful for studies of
protein-ligand complexes of previously-determined structures.
Two molecular replacement programs AMoRe and MOLREP were used for all
structure solutions (Navaza 1994; Vagin and Teplyakov 1997). The theoretical aspects of
MR and details of these two programs have been discussed briefly in the following
sections. In MR, the goal is to find the six parameters, three rotational and three
translational, which would describe how the search molecule is to be placed in the unit
cell. Rossmann and Blow realized that this six-dimensional search could be reduced to a
sequence of two three-dimensional searches in which first the orientation (rotation
function) and then the position (translation function) of the search molecule is determined
(Rossmann and Blow 1962).
The rotation function involves looking for agreement between Patterson functions
of the model and the target structure as a function of their relative orientation. To
evaluate this agreement index, a function R is defined as
R = ∫u P1(x)P2(Cx)dV (2.4)
where P1 and P2 are Patterson functions, C is the rotation operator that rotates the
Patterson function P2 with respect to P1, and u is the spherical volume of integration
centered at the origin. A maximum in the rotation function indicates the correct
orientation for the search molecule.
56

Chapter 2 Materials and methods
The translation [T] of the molecule X with an orientation [R] relative to the model
[M] involves the maximization of the function
T(t) = ∫cell P2(u,t) P1(u)du (2.5)
where P1(u) is the observed Patterson of the unknown cell, P2(u,t) is the Patterson
corresponding to a homologous model rotated using the results of cross-rotation function
search to an orientation corresponding to that of the unknown molecule and positioned at
t from origin (Crowther and Blow 1967).
The three-dimensional structure solution can be obtained from the combined
result of rotation and translation function searches.
X = [R] M + [T] (2.6)
where [R] is the appropriate rotation and [T] the required translation to correctly orient
and position the search model in the target unit cell.
2.3.7.3.1 AMoRe (Automated Molecular Replacement)
AMoRe is a package of programs, which facilitates an efficient solution to the
molecular replacement problem (Navaza 1994; Navaza and Saludjian 1997). The main
characteristics of this package are:
Many potential solutions are examined by novel and fast algorithms. It is
important because the best rotation function solution may not be the correct
solution. Other rotation function solutions might lead to the best solution after
the translation function calculations.
The information from the already positioned models is automatically
incorporated into the search procedure for subsequent molecules.
High degree of automation leading to ease of use.
A more sensitive index of correct solution, correlation coefficient, makes
selection of correct solution reliable.
Various sub-programs in this suite are:
1. Program SORTING sorts, packs and assesses the quality of the experimentally
measured data, and is run as the first step.
2. Program PATTING calculates the Patterson function.
57

Chapter 2 Materials and methods
3. Program TABLING calculates continuous Fourier coefficients from the model
placed in the artificial cell.
4. Program ROTING calculates spherical-harmonics expansions of the crystal and
model Patterson functions and computes rotation functions.
5. Program TRAING computes n-body fast translation functions. The output is for
each orientation and lists the correlation coefficients and R-factors of the top
peaks of fast translation functions.
6. Program FITTING performs least-squares fast rigid-body refinement. It is used to
refine the rotational and positional parameters of the molecules corresponding to
different potential solutions.
2.3.7.3.2 MOLREP (Molecular Replacement)
MOLREP is a fully automated program for molecular replacement which utilizes
effective new approaches in data processing and rotational and translational searching
such as an automatic choice of all parameters, scaling by Patterson origin peaks and soft
resolution cut-off (Vagin and Teplyakov 1997). One of the cornerstones of the program is
an original full-symmetry translation function combined with a packing function.
Information from the model already placed in the cell is incorporated in both translation
and packing functions. It is also possible to calculate rotation function (RF), translation
function (TF) and rigid-body refinement separately using three independent sub-
programs which allow manual input of a number of parameters.
2.3.8 Structure refinement
Structure refinement aims at improving the agreement of an atomic model with
both observed diffraction data and chemical restraints. Crystal structures reported in this
thesis were refined using CNS (Crystallography and NMR System) (Brunger et al. 1998)
and/or CCP4 (Collaborative Computing Project Number 4) (CCP4 1994) suite of
programs. The various standard options used during the refinement are discussed below:
Cross-validation: R and Rfree
The quality of the fit of a model to the diffraction data is given by the R-factor,
which measures the discrepancy between the observed (Fo) and calculated (Fc) structure
58

Chapter 2 Materials and methods
factor amplitudes:
R = ∑| |Fo| -|Fc| | / ∑|Fo| (2.7)
This value can be made arbitrarily low by increasing the number of adjustable parameters
used to describe the model. The method of cross-validation is important for avoiding
over-interpretation of the data. For cross-validation, the diffraction data are divided into
two sets: a large working set (usually comprising of 90-95 % of the data) and a small
complementary test set (comprising the remaining of 5-10 % of the data). The diffraction
data present in the working set is used for refinement whereas the test data are not. The
cross-validated R or Rfree computed with the test set is a more faithful indicator of model
quality (Brunger 1992; 1993). It provides a more objective guide during model building
and refinement process than the conventional R-factor. If the model is correct and errors
are statistical, Rfree is expected to be close to R-factor.
Target functions
Crystallographic refinement can be formulated as a search for the global
minimum of the target function (Jack and Levitt 1978).
Etotal = Echem + wxrayExray (2.8)
The term Echem is a function of all atomic positions describing covalent (bond lengths,
bond angles, dihedral angles, chiral centers, planarity) and non-covalent (van der Waals,
hydrogen bonding and electrostatic) interactions. Exray takes into account the differences
between observed and calculated diffraction data. wxray is a weight chosen to balance the
contributions from Echem and Exray. The choice of wxray can be critical. If wxray is too large,
the refined structure will have large deviations from the ideal geometry and if wxray is too
small, there will be large discrepancy between the refined structure and the experimental
data. Automated procedures to calculate initial estimates for optimal weighting is
available in CNS, but cross-validation must be used to obtain the best possible weight for
the diffraction data.
Least squares and maximum likelihood
Energy minimization using least-square method can improve the model, but
results in the accumulation of systematic errors in the model by fitting noise in the
diffraction data. An improved target for macromolecular refinement is maximum
59

Chapter 2 Materials and methods
likelihood function (Pannu and Read 1996). The objective of this method is to maximize
the likelihood of a model, given the estimates of the errors in the model and the measured
intensities.
h EML = Π P (|Fo|; |Fc|) (2.9)
where P represents the probability of observing the measurement |Fo|, given the
calculated structure factor amplitude |Fc|.
The quantity minimized in traditional least-square refinement programs is
Q = ∑σ (|Fo| - |Fc|) 2 (2.10)
where σ is the weight associated with the measurement |Fo|. It has been shown that with
appropriate redefinition of σ2, Fc and derivatives of Fc with respect to each of the refined
parameters, the conventional program for refinement could be used for maximum
likelihood refinement. The conventional least-squares residual is a limiting case of the
maximum-likelihood theory and only justified if the model is nearly complete and error-
free.
Constraints and restraints
When a parameter is given limited freedom, it is said to be restrained and when
held at an exact value it is said to be constraints. In refining macromolecular structures, it
is necessary to supplement the diffraction data with chemical information in the form of
restraints or constraints. To improve observation to parameter ratio, some groups of
atoms can be restrained or constrained during the course of refinement. Typical restraints
include bond lengths, bond angles and van der Waals contact distances. The restraints are
entered as terms in the refinement target, and are weighted so that the deviations from
ideal values match the deviations found in databases of high resolution structures. For
some purposes such as rigid group refinement, occupancy of atoms in the disordered side
chains, riding hydrogen atoms etc, constraints may be more appropriate than restraints.
Bulk solvent scattering
A correction for the bulk solvent is very important for the refinement of a
macromolecular structure, particularly when the resolution limit of the experimental data
is low. Generally the solvent content in protein crystals is ~50% of the total crystal
60

Chapter 2 Materials and methods
volume. It is possible to include all experimental measurements in the refinement by
taking into account a bulk solvent model to compensate for scattering at low resolution.
The volume that the solvent should fill is identified by demarcating a solvent-accessible
volume outside the van der Waals exclusion zone of the protein.
2.3.8.1 CNS
CNS uses molecular dynamics methods to probe the conformational space of the
molecule while minimizing the differences between the observed and calculated structure
factors (Brunger et al. 1998). CNS can be used for the structure solution, refinement, map
calculations and analysis of the structures determined by X-ray crystallography and NMR
(nuclear magnetic resonance). The coordinates of the initial model is converted to CNS
format using the CNS task file “generate.inp”. This script generates two files: a PDB file
and an MTF file (this contains the molecular topology information which describes the
covalent topology of the molecule). Cross validation is used to monitor the subsequent
refinement of the model and also to reduce model bias in map calculations. The cross
validation information is added to the reflection file using the CNS task file
“make_cv.inp”. In CNS, for structure refinement, there are options for rigid body
refinement, positional refinement, restrained and unrestrained B-factor refinement, group
B-factor refinement, occupancy refinement etc. In addition, there are options to perform
simulated annealing, both in Cartesian and torsion angle conformational space. Various
programs used during structure refinement from the CNS suite (version CNS_solve1.1)
are discussed in the following sections.
2.3.8.1.1 Rigid body refinement
CNS provides the possibility of refinement of several rigid groups. This procedure
minimizes the differences in the observed and calculated structure factors by refining the
three rotational and three translational degrees of freedom of the user defined “rigid”
groups. Each rigid group is treated as a continuous mass distribution located at the center
of mass. Parts of the molecule that are not specified in any rigid group remain fixed. The
rigid body refinement is performed with the CNS task file “rigid.inp”.
61

Chapter 2 Materials and methods
2.3.8.1.2 B-factor refinement
The type of B-factor refinement depends on the resolution of the data. Up to about
2.5Å, individual B-factor refinement whereas from 2.5 to 3.5 Å grouped B-factor
refinement is done. The restrained and individual B-factor refinement was performed
with the CNS task file “bindividual.inp”. The grouped (2-group per residue) and
unrestrained B-factor refinements are carried out by the CNS task file “bgroup.inp”.
2.3.8.1.3 Simulated annealing
As its name implies, the simulated annealing (SA) exploits an analogy between
the way in which a metal cools and freezes into a minimum energy crystalline structure
(the annealing process) and the search for a minimum in a more general system. By
defining the Etotal to be equivalent of the potential energy, the annealing process can be
simulated (Brunger et al. 1999). Following rigid body refinement, simulated annealing
using torsion angle dynamics is used to improve the model. The use of torsion angle
dynamics reduces the number of parameters being refined and hence reduces the degree
of overfitting of the data. Compared to conjugate gradient minimization, where such
directions must follow the gradient, simulated annealing achieves more optimal solution
by allowing motion against the gradient. The SA algorithm in CNS uses the molecular
dynamics simulation mechanism to create a Boltzmann distribution at a given
temperature T. For an initial model with relatively large errors (due to manual building or
misplaced atoms) a starting temperature of 5000K is recommended. In order to decrease
the computational time required the cooling rate can be increased from 25K to 50K. The
simulated annealing refinement task file includes energy minimization both before and
after the simulated annealing. The simulated annealing refinement is performed with the
CNS task file “anneal.inp”. 2.3.8.1.4 Positional refinement
Routines are available in CNS to carry out conventional positional refinement
where energy minimization is carried out by the use of a conjugate gradient minimization
algorithm (Powell 1977). The algorithm requires the value of energy and its first
derivative and uses conjugate gradient descent minimization for convergence. The
positional refinement is performed with the CNS task file “minimize.inp”.
62

Chapter 2 Materials and methods
2.3.8.1.5 Automatically locating solvent molecules
During structure refinement, potential sites of solvent molecules were identified
by the automatic water-picking algorithm. The B-factors and coordinates for the water
molecules were also refined in the water-picking script. The positions of these
automatically picked water molecules were checked manually and a few more water
molecules were identified manually on the basis of electron density contoured at 1.0 σ in
the 2Fo−Fc map and 3.0 σ in the Fo−Fc map. Few rounds of this procedure were carried
out until most of the density in the maps expected for solvent molecules was accounted
for. Water molecules are automatically picked using the CNS task file “water_pick.inp”.
2.3.8.1.6 Reducing model bias with omit maps
In order to reduce the effects of model bias, a simulated annealing omit map is
calculated. In the case of a molecular replacement solution, it is not certain which parts of
the model are in error and therefore an omit map that covers the entire molecule is most
useful. During this process, small regions of the model are systematically excluded and a
small map is computed covering the omitted region (Bhat 1988; Vellieux and Dijkstra
1997). These small maps are accumulated and written out as a continuous map covering
the whole molecule (or the defined region). Simulated annealing refinement and
minimization are used to remove the bias from the omitted region. A composite omit,
cross-validated, sigma-A weighted map is calculated using the CNS task file
“composite_omit_map.inp”.
2.3.8.1.7 Electron density map calculation
CNS has options to calculate sigma-A weighted maps where the structure factor
amplitudes are weighted in order to reduce the model bias of an incomplete or partially
incorrect structure. The Fourier coefficients are given by:
Fmap = (mFo – DFc) exp(iαc) (2.7)
where m is the figure of merit and D is a measure of the error in the coordinates of the
model, as defined by Luzzati (Luzzati 1952). After each round of refinement cycle, cross-
validated, sigma-A weighted 2Fo-Fc and Fo-Fc maps are calculated using the CNS task
file “model_map.inp” with options for 2Fo-Fc and Fo-Fc (u=1, v=1 and u=2, v=1,
63

Chapter 2 Materials and methods
respectively). Fo-Fc is a difference map which is calculated to locate either missing parts
of the model or incorrectly placed atoms. 2.3.8.2 CCP4
The CCP4 suite is a collection of disparate programs covering all aspects of
macromolecular crystallography, collected and developed under the auspices of the
Collaborative Computing Project Number 4 in protein crystallography (CCP4 1994). The
CCP4 suite of programs used during the structure refinement and heavy atom location are
described below:
2.3.8.2.1 SCALEPACK2MTZ
This program converts merged data from SCALEPACK (part of the HKL suite)
into an MTZ format. After the conversion, TRUNCATE is used to convert the intensities
into structure factors. Finally, CAD is run to sort the data and transfer reflections into the
correct asymmetric unit for CCP4. 2.3.8.2.2 TRUNCATE
The standard use of this program is to read a file of averaged intensities (output
from SCALEPACK2MTZ) and write a file containing mean amplitudes and the original
intensities. The amplitudes are put on an approximate absolute scale using the scale factor
taken from a Wilson plot (French and Wilson 1978). There are two ways in TRUNCATE
to calculate the amplitudes from the intensities. The simplest is just to take the square
root of the intensities, setting any negative ones to zero (keyword TRUNCATE NO).
Alternatively, the "truncate" procedure (keyword TRUNCATE YES, the default)
calculates a “best” estimate of F from I, σ(I), and the distribution of intensities in
resolution shells. This has the effect of forcing all negative observations to be positive,
and inflating the weakest reflections (less than about 3σ), because an observation
significantly smaller than the average intensity is likely to be underestimated.
2.3.8.2.3 CAD
The program collects and sorts crystallographic reflection data from several files,
to generate a single set. It also places output data in the CCP4 asymmetric unit, and sorts
64

Chapter 2 Materials and methods
it into a standard order. This is an important step when importing data from other
packages.
2.3.8.2.4 SCALEIT
The program SCALEIT calculates and applies a derivative to native scaling
function using either (a) an overall scale factor, (b) a scale and isotropic temperature
factor or (c) a scale and anisotropic temperature factors. SCALEIT would normally be
run after the merged datasets of native and derivatives have been combined into one file
with CAD, and before beginning the search for heavy atom sites. In addition, SCALEIT
also performs a normal probability analysis.
2.3.8.2.5 FFT
The FFT (Fast Fourier Transform) program is used to calculate Fouriers,
difference Fouriers, double difference Fouriers, Pattersons and difference Pattersons from
reflection data (Ten 1973). The type of map is controlled by the input flags. The input file
will contain h, k, l, Fi, SIGFi, Phases, weights and the required Fourier coefficients will
be generated from these.
2.3.8.2.6 MLPHARE
This program is used to refine heavy atom parameters and error estimates, then
use these refined parameters to generate phase information. This program refines heavy
atoms without significant bias. The incorporation of the concepts of maximum likelihood
ensures that the refinements of parameters are more robust, and the generation of phases
and figures of merit is more realistic. The program was originally written for MIR, but
may also be used for phasing from MAD data, where the different wavelengths are
interpreted as different "derivatives".
2.3.8.2.7 REFMAC5
REFMAC5 is a macromolecular refinement program which has been integrated
into the CCP4 suite. The maximum likelihood approach has been implemented in
REFMAC5 (Murshudov et al. 1997). Refinement with REFMAC5 requires reflection
data, a set of model coordinates and a library defining the restraints for standard groups.
If a non-standard group is encountered, the user has to build a set of restraints through the
65

Chapter 2 Materials and methods
use of SKETCHER/LIBCHECK. It minimizes the model parameters to satisfy a
maximum likelihood residual. It can carry out rigid-body, restrained and unrestrained
refinement against X-ray data or idealization of a macromolecular structure. The key
features of REFMAC5 are
It can carry out TLS (translation/libration/screw-rotation) refinement (Winn et
al. 2001; Winn et al. 2003). This is particularly useful when there is
significant anisotropy, but the resolution does not warrant refinement of
individual anisotropic displacement parameters.
Restraints are calculated within the main program. A large dictionary of
standard geometries is included.
A bulk solvent correction is calculated within the program.
For atomic resolution data, full anisotropic refinement can be performed with
refinement of anisotropic displacement parameters for some or all atoms.
REFMAC5 also produces an MTZ output file containing weighted co-
efficients for sigma-weighted mFo-DFcalc and 2mFo-DFcalc maps, where
missing data have been restored.
2.3.8.2.8 LIBCHECK
LIBCHECK generates and manages the library files which provide complete
chemical and geometric descriptions of residues and ligands used in the refinement by
REFMAC5. Restraints can be created for novel ligands using Monomer Library Sketcher
(an interface of the LIBCHECK program) in CCP4i.
2.3.9 Model Building
Examination and interpretation of the electron density maps calculated using CNS
or CCP4 suite of programs were done using COOT and O packages. The 2Fo-Fc map was
usually contoured at 1.0 σ and the Fo-Fc map was at 3.0 σ and -3.0 σ, where σ refers to
the rms deviation in the density in electrons/Å3 in the maps. Regions with poor electron
density were examined with the maps contoured at a lower level. For high resolution
data, map improvement by atom update and refinement was carried out using the
automated model building program ARP/wARP.
66

Chapter 2 Materials and methods
2.3.9.1 COOT
COOT (Crystallographic Object-Oriented Toolkit) is a software package for
model building and validation (Emsley and Cowtan 2004). COOT displays maps and
models and allows model manipulations such as idealization, real space refinement,
manual rotation/translation, rigid-body fitting, ligand search, solvation, mutations,
rotamers, Ramachandran plots etc. COOT is under development as part of the CCP4
molecular graphics project (Potterton et al. 2004). 2.3.9.2 O
O is a graphics interactive program for visualizing and manipulating 3D models
and electron density maps of macromolecular structures. O has been principally
developed by Alwyn Jones (Jones et al. 1991). It is built on top of a versatile database
system. All molecular data are kept in this database, in a predefined structure. The
powerful macro facility of O is mainly aimed at facilitating protein crystallography i.e.
structural manipulations, bringing several new tools which ease the building of models
into the electron density, allowing it to be done faster and correctly. Some new auto-build
options greatly enhance the speed of building and rebuilding molecular models.
2.3.9.3 ARP/wARP
The aim of ARP/wARP is improved automation of model building and refinement
in macromolecular crystallography. In the case of molecular replacement, ARP/wARP
offers three options (Perrakis et al. 2001) to automate model building to varying extents:
(i) autobuilding of a completely new model based on phases calculated from the
molecular replacement solution, (ii) updating the initial model by atom addition and
deletion to obtain an improved map and (iii) docking of a structure onto a new (or
mutated) sequence, followed by rebuilding and refining the side chains in real space.
2.3.10 Structure validation and deposition
A number of excellent validation tools have been developed during the last
decade for checking the geometry and stereochemistry of a set of coordinates against a
target library and the expected parameters derived there from. These tools have been
widely accepted by the crystallographic community and are generally applied before
67

Chapter 2 Materials and methods
submission of data to the PDB. However, these tools do not attempt to check the
coordinates against the experimental data. Some of the validation and deposition tools
which were used extensively during the course of investigation are described below:
2.3.10.1 PROCHECK
The PROCHECK suite of programs provides a detailed check on the
stereochemistry of a protein structure (Laskowski et al. 1993) and is a part of the CCP4
suite. These give an assessment of both the overall quality of the structure, as compared
with well-refined structures of the same resolution, and also highlight regions that may
need further investigation. It compares and assesses the quality of the model vis-à-vis
other structures at comparable resolutions. A very important evaluation criterion is the
Ramachandran plot (Ramachandran and Sasisekharan 1968), where the distribution of the
backbone φ, ψ angles of a given protein structure is compared to that in high-quality
structures. The PROCHECK program was used to validate the model and assess its
quality after every round of refinement in the final stages of model building.
2.3.10.2 SFCHECK
SFCHECK is a software package that features a unified set of procedures for
evaluating the structure factor data obtained from X-ray diffraction experiments and for
assessing the agreement of the atomic coordinates with these data (Vaguine et al. 1999).
The program requires one or two input files, with the coordinates of the model (in PDB
format) and structure factors (MTZ or CIF format), and runs completely automatically.
The program summarizes relevant information on the deposited structure factors and
evaluates their quality using criteria such as data completeness, structure factor
uncertainty and the optical resolution computed from the Patterson origin peak. It also
gives information about R-factor, correlation, Luzzati plot, Wilson plot, B-overall,
pseudo-translation, twinning, local error estimation by residues etc.
2.3.10.3 MolProbity
MolProbity is a general-purpose web service offering quality validation for 3D
structures of proteins, nucleic acids and complexes (Davis et al. 2004). It offers features
including the ability to “flip” Asn, Gln, and His residues for better hydrogen bonding
68

Chapter 2 Materials and methods
throughout the protein structure. Looking at the density alone, this can be challenging,
but the algorithm used in this program looks at neighboring residues as well as solvent
and calculates a score by which it decides how the greatest number of bonding can occur
in a structure. Additionally, it is possible to visualize clashes between atoms after running
the tools available on the site. It also offers Ramachandran plots, rotamer analysis and Cβ
deviations.
2.3.10.4 PDB: ADIT
The Auto Dep Input Tool (ADIT) was developed by the RCSB for depositing
structures to the Protein Data Bank (http://pdbdep.protein.osaka-u.ac.jp/validate/). ADIT
also allows the user to check the format of coordinates and structure factor files and to
perform a variety of validation tests on a structure prior to deposition in the database. The
advantage of performing the data format precheck and structure validation is to determine
any potential changes required to standardize the structure before it is deposited.
2.3.11 Structure visualization
There are many programs available for structure visualization, each with different
features and capabilities. Out of all these, some of them which have been extensively
used during the course of investigation are described below:
2.3.11.1 PyMOl
PyMOL is an open-source, user-sponsored, molecular visualization system
created by W. L. DeLano and commercialized by DeLano Scientific LLC (DeLano
2002). It has an embedded Python interpreter designed for real-time visualization and
rapid generation of high-quality molecular graphics images and animations. It is well
suited to produce high quality 3D images of small molecules and biological
macromolecules such as proteins.
2.3.11.2 MolScript and BobScript
MolScript is a molecular graphics program that draws high quality static images
of molecular structures, especially proteins (Kraulis 1991). MolScript is operated using a
command language rather than a graphical interface. Input consists of a PDB format
69

Chapter 2 Materials and methods
molecule file and a user-written MolScript command file. Output possibilities include
PostScript, Encapsulated PostScript, and a Raster3D command file. BobScript adds -
amongst many others - the ability to render electron density directly, as well as many and
sundry correctly implemented methods of colour ramping (Esnouf 1999).
2.3.11.3 Raster3D
The Raster3D molecular graphics package consists of a core program render and a
number of ancillary programs that read atomic coordinates from PDB files to produce
scene descriptions for input to render (Merritt and Murphy 1994). It uses an efficient
software Z-buffer algorithm which is independent of any graphics hardware. Raster3D
can also be used to render pictures composed in MolScript. 2.3.11.4 GRASP
GRASP (Graphical Representation and Analysis of Structural Properties) is a
molecular visualization and analysis program (Nicholls 1992). GRASP's surface can be
molecular or accessible and can be color coded by electrostatic potential derived from its
internal Poisson-Boltzmann solver or external programs such as DelPhi. This
representation has become a standard tool in assessing electrostatic character of large,
typically protein molecules.
2.3.11.5 TopDraw TopDraw is a sketchpad for drawing topology cartoons of proteins. It is a part
of CCP4 suite of programs (Bond 2003). It does not calculate topology from a PDB file,
but merely allows sketching a topology derived from another source.
2.3.12 Sequence and Structure analysis
A number of programs used for sequence and structure analysis, to gain structural
as well as functional insights, are described below:
2.3.12.1 ProtParam
ProtParam is a tool which allows the computation of various physical and
chemical parameters for a given protein stored in Swiss-Prot or TrEMBL or for a user
entered amino acid sequence (Wilkins et al. 1999). The computed parameters include the
70

Chapter 2 Materials and methods
molecular weight, theoretical pI, amino acid composition, atomic composition, molar
extinction coefficient, estimated half-life, instability index, aliphatic index and grand
average of hydropathicity (GRAVY).
2.3.12.2 CLUSTAL-W
CLUSTAL-W is a general purpose multiple sequence alignment program for
DNA or proteins (Higgins et al. 1994). It calculates the best match for the selected
sequences, and lines them up so that the identities, similarities and differences can be
seen. Evolutionary relationships can be assessed by viewing Cladograms or Phylograms.
2.3.12.3 ESPript
The program ESPript (Easy Sequencing in PostScript) allows the rapid
visualization, via PostScript output, of sequences aligned with programs such as
CLUSTAL-W. It can read secondary structure files to produce an illustration having both
sequence and structural informations. ESPript can be run via a command file or a friendly
html-based user interface (Gouet et al. 2003). The program calculates a homology score
by columns of residues and can sort this calculation by groups of sequences. It offers a
palette of markers to highlight important regions in the alignment. It can also paste
information on residue conservation into coordinate files, for subsequent visualization
with a graphics program.
2.3.12.4 DSSP
The DSSP program was designed by Wolfgang Kabsch and Chris Sander (Kabsch
and Sander 1983). The DSSP program defines secondary structure, geometrical features
and solvent exposure of proteins, given atomic coordinates in Protein Data Bank format.
2.3.12.5 PROMOTIF
PROMOTIF is a suite of programs, which analyzes a protein coordinate file and
provides details regarding the structural motifs in the protein (Hutchinson and Thornton
1996). The program currently analyzes the following structural features: beta- and
gamma-turns; helical geometry and interactions; β-strands and β-sheet topology; β-
bulges; β-hairpins; β-α-β units and ψ-loops; disulphide bridges; and main-chain
hydrogen-bonding patterns.
71

Chapter 2 Materials and methods
2.3.12.6 ALIGN
ALIGN performs an iterative refinement of the superposition of two coordinate
sets (Cohen 1997). The program operates on a set of coordinate pairs that represent the
two structures to be aligned. The program provides information that facilitates the
understanding of transformations between structurally homologous subunits of proteins.
2.3.12.7 NACCESS
NACCESS calculates the atomic accessible area when a probe is rolled around
the van der Waal's surface of a macromolecule (Hubbard and Thornton 1993). The
program uses the Lee & Richards method, whereby a probe of a given radius is rolled
around the surface of the molecule, and the path traced out by its centre is the accessible
surface (Lee and Richards 1971). Typically, the probe has the same radius as water (1.4
Å) and hence the surface described is often referred to as the solvent accessible surface.
The calculation makes successive thin slices through the 3D molecular volume to
calculate the accessible surface of individual atoms. For calculating the buried surface
area between two subunits, one will have to run 3 separate calculations. Once for subunit
A, once for subunit B and a third time for the AB complex. Subtraction of the AB surface
from the A + B surfaces gives the buried surface area of the complex.
2.3.12.8 CONTACT
This is a program for computing various types of contacts in protein structures, a
part of the CCP4 suite (CCP4 1994). It can also analyze water hydrogen bonding. The
program uses a bricking algorithm in which atoms are segregated into 6x6x6 Å boxes and
contact searching is limited to neighboring boxes. "***" indicates the strong possibility
of a hydrogen bond (distance < 3.3 Å), whereas "*" indicates a weak possibility (distance
> 3.3 Å). Blank indicates that the program considers no possibility of a hydrogen bond.
2.3.12.9 BAVERAGE
This is a simple program to read a PDB file, tabulate the average B values residue
by residue (main chain and side chain separately) and the rms deviation of the B values
from the mean. It also outputs a PDB file with outlying B factors reset to lie within the
given range. It is a part of the CCP4 suite of programs (CCP4 1994).
72

Chapter 2 Materials and methods
2.3.12.10 DynDom
DynDom is a program to determine domains, hinge axes and hinge bending
residues in proteins for which two conformations are available (Hayward and Lee 2002;
Lee et al. 2003). The analysis of domain movements only makes sense if the interdomain
deformation is at least comparable to the intradomain deformation. DynDom determines
domains by looking for clusters of rotation vectors that describe the rotational aspect of
the conformational change. These rotation vectors can also be viewed using RasMol
(Sayle and Milner-White 1995). Clusters form dynamic domains are then analyzed for
their interdomain motions to give hinge axes. Finally the residues involved in the hinge
bending are determined. The application of DynDom provides a view of the
conformational change that is easily understood. The conformational change may be
complicated in detail, but by using DynDom one can visualize it as a consequence of the
movement of domains as quasi-rigid bodies.
2.3.12.11 VOIDOO
This program is used for the detection of cavities in macromolecular structures
(Kleywegt and Jones 1994). VOIDOO can handle three different types of cavity: van der
Waals cavities (the complement of the molecular van der Waals surface), probe-
accessible cavities (the cavity volume that can be occupied by the centers of probe atoms)
and MS-like probe-occupied cavities (the volume that can be occupied by probe atoms).
2.3.12.12 DALI
The Dali server is a network service for comparing protein structures in 3D (Holm
and Sander 1995). Comparing 3D structures may reveal biologically interesting
similarities that are not detectable by comparing sequences. The database consists of all
representative structures from the PDB with less than 90% sequence identity to each
other. The classification and alignments are automatically maintained and continuously
updated using the Dali search engine.
73

Crystal structures of Salmonella typhimurium biodegradative threonine deaminase (TdcB) and its complex
with CMP provide structural insights into ligand induced oligomerization and enzyme activation
3

Chapter 3 Biodegradative threonine deaminase (TdcB)
3.1 Introduction Enzymes that use pyridoxal 5’-phosphate (PLP) as a cofactor catalyze many
important reactions involving amino acids such as transamination, decarboxylation, β- or
γ-replacement/elimination and racemization (Eliot and Kirsch 2004). On the basis of the
carbon atom subjected to covalent changes, PLP-dependent enzymes are classified into at
least three distinct families α, β and γ (Alexander et al. 1994). The α- and γ-families
might be distantly related, but are clearly not homologous to the β-family. The β-family
members whose structures are known include tryptophan synthase (Hyde et al. 1988),
biosynthetic threonine deaminase (Gallagher et al. 1998), O-acetylserine sulfhydralase
(Burkhard et al. 1998), cystathionine β-synthase (Meier et al. 2001), serine dehydratase
(Yamada et al. 2003), threonine synthase (Thomazeau et al. 2001) and 1-
aminocyclopropane-1-carboxylate deaminase (Yao et al. 2000). All these enzymes
exhibit fold type II, which is characteristic of the β-family of PLP-dependent enzymes. In
this family of enzymes, each subunit is formed by two distinct domains, both having
typical open α/β architecture. The active sites of these enzymes are composed of residues
from only one subunit. Their PLP-binding lysine residue is positioned in the N-terminal
segment of the polypeptide chain.
Escherichia coli and Salmonella typhimurium are known to possess two distinct
PLP-containing threonine deaminases (EC 4.3.1.19), one biosynthetic and the other
biodegradative (Luginbuhl et al. 1974; Umbarger and Brown 1957). Both the enzymes
catalyze the deamination of L-threonine to yield 2-ketobutyrate and ammonia (Fig. 3.1).
Biosynthetic threonine deaminase (IlvA) encoded by the gene ilvA, is constitutively
expressed under normal conditions and catalyzes the first reaction in the isoleucine
biosynthesis pathway. L-isoleucine and L-valine act as allosteric inhibitor and activator,
respectively. IlvA provided one of the earliest examples of feedback inhibition
(Umbarger 1956). Biodegradative (catabolic) threonine deaminase (TdcB), encoded by
the gene tdcB in E. coli and S. typhimurium, is induced anaerobically and catalyzes the
first reaction in the degradation of L-threonine to propionate. Unlike IlvA, this enzyme is
insensitive to L-isoleucine and L-valine and is activated by AMP. The feedback resistant
TdcB is more suited for the industrial production of L-isoleucine when compared to the
feedback inhibited IlvA (Guillouet et al. 1999).
75

Chapter 3 Biodegradative threonine deaminase (TdcB)
Figure 3.1 Reaction catalyzed by threonine deaminase
Studies on TdcB from E. coli and S. typhimurium have shown that the enzymatic
activity is enhanced in the presence of AMP due to a large decrease in KM for L-threonine
and apparent increase in Vmax (Bhadra and Datta 1978; Dunne and Wood 1975; Shizuta
and Hayaishi 1976). Using various analogs of AMP and other natural nucleotides, the
functional atoms or groups of AMP, which are involved in the ligand-binding and
activation of enzymatic activity have been identified (Nakazawa et al. 1967; Whanger et
al. 1968). Among other mononucleotide phosphates, CMP showed significant enzyme
activation compared to GMP, UMP, and IMP. Further, no enzymatic activation was
observed in the presence of ATP whereas ADP showed slight activation.
In the absence of AMP, TdcB exists in monomer-dimer equilibrium at low
concentration (Gerlt et al. 1973; Shizuta et al. 1973; Whanger et al. 1968). This
equilibrium shifts towards the tetrameric form as the concentration of TdcB is increased.
Even at low concentrations of TdcB, presence of AMP induces oligomerization from
monomer to tetramer. A number of other biochemical properties of this enzyme including
the capacity and nature of its binding to PLP, kinetic studies and molecular behavior in
the presence and absence of AMP, substrate specificity, spectral changes upon addition of
L-threonine, inhibition by the reaction product α-ketobutyrate and other α-keto acids,
inactivation by certain intermediary metabolites of the tricarboxylic acid cycle and
reaction mechanism have been studied extensively (Bhadra and Datta 1978; Dunne and
Wood 1975; Niederman et al. 1969; Park and Datta 1979; Shizuta and Hayaishi 1976;
Shizuta et al. 1973; Xu et al. 2005). However, these investigations did not reveal the
exact site of AMP binding and its role in the formation of oligomers or enzyme
activation. It is believed that the regulation of enzymatic reaction by an effector involves
conformational changes associated with its binding. In oligomeric proteins, these changes
may involve relative rearrangement of subunits as well as subtle changes in the
76

Chapter 3 Biodegradative threonine deaminase (TdcB)
conformation of individual subunits. Three-dimensional structure of TdcB and local and
global conformational changes associated with AMP binding are important to understand
the mechanism by which the activity of TdcB is regulated by AMP.
In this chapter, crystal structures of Salmonella typhimurium biodegradative
threonine deaminase with PLP bound to the active site Lys58 (as an internal aldimine) in
two different crystal forms I and II and in complex with CMP in another crystal form
(crystal form III) have been described. This is the first structural description of the
biodegradative threonine deaminase. Apart from the overall structural features, the
structures of TdcB reveal the mode of PLP binding and its relationship to the expected
binding site of L-threonine. Comparison of TdcB structures with IlvA, serine dehydratase
and other related β-family of PLP-dependent enzymes has revealed the differences as
well as similarities in the overall structure and reaction mechanism. Analysis of the
tertiary and quaternary structural changes observed in the presence of CMP has provided
a structural basis for understanding CMP directed oligomerization and enzyme activation.
3.2 Materials and methods 3.2.1 Cloning, overexpression and purification
The DNA encoding the open reading frame for biodegradative threonine
deaminase (tdcB) was amplified using KOD HiFi DNA polymerase (Novagen) from
Salmonella enterica serovar Typhimurium strain IFO 12529 genomic DNA. After PCR-
amplification of the target gene by sense 5' GCTAGCCATATGCACATTACATACG
ATCTCCC 3' and antisense 5' GGATCCTTACTCGAGAGCGTCAACTAAACCCG 3'
primers, it was digested with NheI and BamHI and then cloned into the pRSET C vector
(Invitrogen) encoding a polypeptide with an N- terminal hexa-histidine tag to facilitate its
purification using Ni-NTA affinity column chromatography. The final polypeptide
contains the amino acid sequence MRGSHHHHHHGMAS from the vector followed by
the amino acid sequence of TdcB. The sequence of the tdcB gene was determined by
nucleotide sequencing and confirmed by comparing it with the tdcB gene of
Salmonella typhimurium LT2. TdcB was purified to homogeneity using Ni-NTA affinity
column chromatography. The protein concentration was determined with Bradford
reagent, using bovine serum albumin as the standard protein (Bradford 1976).
77

Chapter 3 Biodegradative threonine deaminase (TdcB)
3.2.2 Activity assay and kinetic studies
Enzymatic activity of TdcB was routinely examined by measuring directly the
formation of α-ketobutyrate at 310 nm (Shizuta et al. 1973) on a Jasco UV-Visible
spectrophotometer model V-530 (Japan Spectroscopic Company) at 25°C. The standard
reaction mixture (0.2 ml) contained 100 μM of potassium phosphate buffer pH 8.0, 50
μM of L-threonine and rate-limiting concentration of TdcB. The reactions were carried
out in the presence and absence of 5 mM CMP or AMP. α-ketobutyrate produced was
determined assuming molar absorbance of 25.6 M-1 at 310 nm. For kinetic studies, the
concentration of AMP/CMP was held in excess at 5 mM. Experiments were repeated at
least three times from three independent purifications. Kinetic parameters were calculated
by fitting the initial velocity versus substrate concentration to the Michaelis-Menten
equation, v = (Vmax[S])/(KM +[S]), using the non-linear regression analysis option of the
GraphPad Prism software.
3.2.3 Gel filtration chromatography and glutaraldehyde cross-linking
Gel filtration analysis was carried out using Sephacryl S-200 column (Amersham
Pharmacia) previously equilibrated with 100 mM phosphate buffer pH 8.0 at 4°C for the
native TdcB and with additional 5 mM AMP/CMP in case of TdcB-AMP or TdcB-CMP
complex. The column was calibrated with molecular standards containing aldehyde
dehydrogenase (150 kDa), bovine serum albumin (66 kDa), carbonic anhydrase (29 kDa)
and cytochrome C (12 kDa). Elution was monitored by measuring absorption at 280 nm
as well as at 400 nm (peak corresponding to the internal aldimine).
Purified TdcB in the native form and in the presence of AMP and CMP was
incubated with 0.04 % (v/v) glutaraldehyde in 50 mM phosphate buffer pH 8.0, 50 mM
NaCl and 2 mM DTT at 4°C in dark for various time intervals. The protein samples were
then mixed with an equal volume of 2 X SDS-PAGE loading buffer and immersed in a
boiling water bath for 5 min. Cross-linked adducts were resolved using SDS-PAGE
followed by Coomassie blue R-250 staining. 3.2.4 Crystallization and data collection
Purified protein was concentrated to approximately 30 mg/ml using a 10 kDa
78

Chapter 3 Biodegradative threonine deaminase (TdcB)
molecular weight cut-off Amicon Ultra-15 Centrifugal Filter Units (Millipore) for
crystallization experiments. TdcB was crystallized using the hanging-drop vapour-
diffusion technique. During screening, small crystals were obtained in crystallization
conditions containing 0.1 M citrate buffer pH 5.6, 20% polyethylene glycol (PEG) 4000
and 20% 2-propanol (condition No. 40, Hampton Crystal Screen I) and 0.1 M HEPES
buffer pH 7.5, 20% PEG 4000 and 10% 2-propanol (condition No. 41, Hampton Crystal
Screen I). Further optimization of these two conditions was carried out by varying the pH
and the molecular weight of the PEG and using various organic solvents. 10-15% 2-
propanol along with citrate buffer in the pH range 5.6-6.5 and 20-25% PEG 3350 or PEG
4000 gave good diffraction-quality crystals. Both PEG 3350 and PEG 4000 gave
apparently equivalent results. Of the various organic solvents screened, substitution of 2-
propanol by either 10-15% t-butanol or 0.6-0.9 M 1,6-hexanediol gave diffraction-quality
crystals of different crystal forms. Crystals obtained in the presence of 10-15% t-butanol
diffracted better than those obtained in the presence of 2-propanol. The two crystal forms
of native TdcB for which diffraction data are reported in this chapter were obtained from
conditions containing 0.1 M citrate buffer pH 6.0, 20% PEG 3350 and 10% 2-propanol
(crystal form I) and 0.1 M citrate buffer pH 6.0, 20% PEG 3350 and 15% t-butanol
(crystal form II). These crystals appeared after 5 d of equilibration against the
crystallization solution and grew to full size in 10 d. Diffraction data for these two crystal
forms were collected using 20% (v/v) ethylene glycol as a cryoprotectant on an in-house
X-ray source.
Despite extensive efforts, diffraction quality crystals of TdcB in complex with
various L-threonine analogs could not be obtained. Attempts to co-crystallize TdcB with
AMP and CMP were also made. Crystals of TdcB obtained in the presence of AMP
diffracted poorly whereas crystals of TdcB obtained in the presence of CMP diffracted to
a resolution of 3-3.5 Å at in-house X-ray source. A complete dataset to a resolution of 2.5
Å was collected on a TdcB crystal obtained in the presence of CMP and the substrate
analog O-methylthreonine on beamline BL44XU at SPring-8, Hyogo, Japan using a
DIP2040 image plate at a wavelength 0.93 Å. The data were processed and scaled using
the program DENZO and SCALEPACK of the HKL suite (Otwinowsky 1997). The
crystal parameters and the data collection statistics are summarized in table 3.1.
79

Chapter 3 Biodegradative threonine deaminase (TdcB)
3.2.5 Structure solution and refinement
Amino acid sequence alignment shows an identity of 34% between TdcB and the
N-terminal domain of IlvA. The sequence corresponding to the C-terminal regulatory
domain of IlvA is absent in TdcB. Therefore, a model consisting of the atomic
coordinates of the N-terminal domain of IlvA from E. coli (PDB code 1TDJ) (Gallagher
et al. 1998) with non-identical residues converted to alanine was used as the search
model for structure solution of TdcB by molecular replacement with the program AMoRe
(Nawaja 1994). As the quality of the data was better for crystal form II, reflections in the
resolution range 15.0-3.0 Å of this crystal form were used for the rotation and translation
searches. The highest peak of the translation search had a correlation coefficient of 47.2
% and an R-factor of 55.6 %. Refinement was initiated with the program REFMAC5
(Winn et al. 2001). The 2Fo–Fc map calculated after initial positional refinement showed
good fit for the majority of the main chain. Map improvement by atom update and
refinement was carried out using the program ARP/wARP (Morris et al. 2003). The
quality of the electron density map obtained from ARP/wARP was sufficient to allow
unambiguous assignment and building of most amino acid residues using the program
COOT (Emsley and Cowtan 2004). In the final stages of refinement, inspection of
difference (Fo−Fc) map showed strong positive density for the expected ligand PLP. This
was followed by identification of potential sites of solvent molecules by automatic water-
picking algorithm of COOT (Emsley and Cowtan 2004). The positions of these
automatically picked waters were manually checked and a few more waters were
manually identified on the basis of electron density contoured at 1.0 σ in the 2Fo−Fc map
and 3.0 σ in the Fo−Fc map. Electron density peaks which were significantly higher than
those of water molecules and were within the coordination distance from the surrounding
atoms were assigned as sodium ions, since the crystals were grown in presence of sodium
ions (100 mM tri-sodium citrate buffer and 50 mM NaCl in the crystallization condition).
The four subunits of TdcB in the asymmetric unit were labeled A, B, C and D as shown
in figure 3.7(b), thus forming AB and CD dimers. During the final stages of model
building, large stretches of amino acids in the D subunit showed probable alternate
conformations. Careful examination indicated that the entire D subunit was present in
two conformations. Based on the difference (Fo−Fc) map, occupancies for the two
80

Chapter 3 Biodegradative threonine deaminase (TdcB)
conformations were manually fixed to 80% and 20%, respectively, which implied that
one conformation was major (D’) and the other was minor (D”). Final rounds of
refinement were performed using REFMAC5 (Winn et al. 2001), incorporating TLS
restraints (Winn et al. 2001). The final model of native TdcB in crystal form II contains
four subunits of TdcB, four PLP molecules (bound to Lys58), two sodium ions and 782
water molecules. The final refinement statistics are given in table 3.1.
Structures of TdcB in crystal forms I and III were solved by molecular
replacement using the atomic coordinates of protein atoms from the A subunit of TdcB
crystal form II as the search model. For TdcB crystal form I, molecular replacement
solution using the program MOLREP (Vagin and Teplyakov 1997) had a correlation
coefficient of 67.1 % and an R-factor of 41.8% in the resolution range 30-2.5 Å. This was
followed by model building and refinement using COOT and REFMAC5, following a
protocol similar to the one described for crystal form II. The final model of native TdcB
in crystal form I contains two subunits of TdcB, two PLP molecules (bound to Lys58),
one sodium ion and 274 water molecules. For TdcB co-crystallized with CMP and O-
methylthreonine (crystal form III), the molecular replacement solution using the program
MOLREP had a correlation coefficient of 58.4% and R-factor of 43.9% in the resolution
range 30-3.5 Å. The initial difference Fourier map indicated alternate positions for a few
stretches of residues in the polypeptide chain and electron density for CMP, which were
incorporated into the model. No electron density corresponding to O-methylthreonine
was observed. The final model of TdcB in complex with CMP (here-after referred as
TdcB-CMP) includes one subunit of TdcB, one molecule of PLP (bound to Lys58), CMP,
sodium ion and 62 water molecules. The final refinement statistics are given in table 3.1.
3.3 Results and discussion 3.3.1 Cloning, overexpression and purification
TdcB from Salmonella typhimurium was cloned in pRSET C vector and over-
expressed in E. coli BL21(DE3)pLysS along with an N-terminal hexa-histidine tag. TdcB
was purified to homogeneity using Ni-NTA affinity column chromatography. The
purified enzyme showed a single polypeptide band in SDS-PAGE, corresponding to a
subunit molecular mass of 36 kDa (Fig. 3.2).
81

Chapter 3 Biodegradative threonine deaminase (TdcB)
Figure 3.2 SDS-PAGE analysis of TdcB during purification. Lane 1, crude cell lysates after 0.3 mM
IPTG induction; lane 2, clear supernatant; lane 3, purified TdcB after Ni-NTA affinity column
chromatography; lane M, molecular weight markers.
3.3.2 Biochemical studies on TdcB
Enzymatic activity was measured by monitoring increase in absorbance at 310 nm
at 25ºC due to the product α-ketobutyrate, formed by deamination of L-threonine. Using
the purified S. typhimurium TdcB, the KM value for L-threonine was estimated as 123 ± 7
mM. In the presence of 5 mM AMP and CMP, the KM values for L-threonine were 16 ± 2
mM and 32 ± 5 mM, respectively. Thus, AMP and CMP cause a decrease in KM for L-
threonine by approximately 7.7- and 3.5-fold. The ratios of the observed Vmax values of
the enzyme in the presence of AMP and CMP to that of native TdcB were approximately
9 and 3, respectively. Thus, interaction of TdcB with AMP/CMP results in the activation
of the enzyme.
Gel filtration experiments with TdcB in the absence and presence of CMP showed
shift in the apparent molecular mass of the enzyme from 45 kDa for native TdcB to 100
kDa in the presence of CMP and 120 kDa in the presence of AMP (Fig. 3.3). These
results are indicative of rapid equilibrium between various forms of the enzyme in
solution as observed previously in E. coli as well as in S. typhimurium (Bhadra and Datta
82

Chapter 3 Biodegradative threonine deaminase (TdcB)
1978; Gerlt et al. 1973; Shizuta et al. 1973). Gel filtration studies on the E. coli enzyme
has also indicated a monomeric form at low concentration of the enzyme or in the
absence of AMP and approaching tetrameric form in the presence of AMP or at higher
enzyme concentrations (Whanger et al. 1968). Previous studies involving sucrose density
ultracentrifugation have shown that the sedimentation velocity of the enzyme increases
smoothly from 4.8 S to 7.6 S with increasing enzyme concentration or even at low
concentrations of the enzyme in the presence of AMP/CMP (Whanger et al. 1968). The
increase in the S value from 4.8 S to 7.6 S corresponded to a shift of dimeric to tetrameric
form of TdcB.
Fraction number (in ml)
10 15 20 25 30 35 40 45 50 55 60 65 70 75 80 85 90
AU
0.00
0.01
0.02
0.03
0.04
0.05
TdcB-CMPTdcB-AMPTdcB-NativeMarkers
1 2 3
Figure 3.3 Gel filtration analysis of TdcB in the native form and in complex with CMP and AMP.
Gel filtration experiments were carried out in an analytical pre-packed Sephacryl S-200 column
calibrated with standard molecular weight markers at 4ºC. The positions of peak fraction of
molecular weight (MW) markers are indicated using arrows. 1, alcohol dehydrogenase (MW 150
kDa); 2, bovine serum albumin (MW 66 kDa); 3, carbonic anhydrase (MW 29 kDa).
TdcB was cross-linked in the absence and presence of AMP and CMP with 0.04%
(v/v) glutaraldehyde and was analyzed using SDS-PAGE. The cross-linked enzyme in all
the three cases showed four polypeptide bands corresponding to one each for monomeric
and tetrameric forms and two bands near the expected dimeric form of TdcB (Fig. 3.4).
83

Chapter 3 Biodegradative threonine deaminase (TdcB)
Previous studies involving cross-linking of the native TdcB and in the presence of AMP
with dimethyl suberimidate followed by reduction and alkylation showed three
polypeptides corresponding to the molecular weights of monomer, dimer and tetramer
(Bhadra and Datta 1978). Thus, the results obtained by various other groups and the
present gel filtration and glutaraldehyde cross-linking experiments strongly suggest that
in the presence of AMP or CMP or at high concentration of the enzyme, TdcB is in a
tetrameric form.
1 2 3 4 5kDa
116
66.2
45
25
35
Monomer
Dimer
Tetramer
Figure 3.4 SDS-PAGE analysis showing glutaraldehyde cross-linking of TdcB in the native form.
Similar results were obtained when TdcB was cross-linked in the presence of CMP and AMP.
Lane 1, TdcB-native control; lane 2, 3 and 4, lane 5, molecular weight markers (kDa).
3.3.3 Crystallization and structure solution
The purified native TdcB was crystallized in the two forms (Fig. 3.5). Diffraction
data for these two crystal forms I and II were collected to resolutions of 2.2 and 1.7 Å,
respectively. Both the crystal forms belonged to the space group P1. The volumes of the
asymmetric units in these two crystal forms were compatible with two and four subunits
of TdcB, respectively. Matthews coefficient (VM) and solvent content of both forms were
2.1 Å3 Da−1 and 41.7% (v/v), respectively (Matthews 1968). The unit cell volume of
crystal form II was almost twice that of crystal form I. The structure of TdcB was solved
84

Chapter 3 Biodegradative threonine deaminase (TdcB)
by molecular replacement using the N-terminal domain (residues 5-333) of IlvA from E.
coli (PDB code 1TDJ) as the search model (Gallagher et al. 1998). Because of higher
resolution and better quality, crystal form II data was initially used to solve the structure
of TdcB. In the crystal form II, a subunit in one of the two dimers of the asymmetric unit
was present in two conformations. Occupancies for these two alternate conformations
were manually adjusted on the basis of difference Fourier maps. The final model of TdcB
form II has been refined to R and Rfree of 19.0 % and 22.1 %, respectively. The structure
of TdcB form I was solved using protein atoms of a monomer of the TdcB form II, and
was finally refined to R and Rfree of 21.0 % and 25.3%, respectively.
(a) (b) (c)
Figure 3.5 Crystals of TdcB obtained in three different crystal forms in the presence of
crystallization conditions containing (a) 2-propanol (crystal form I) (b) t-butanol (crystal form II)
and (c) 1,6-hexanediol (crystal form III).
We also attempted co-crystallization of S. typhimurium biodegradative threonine
deaminase with AMP/CMP and L-threonine analogs. Crystals of TdcB obtained in
presence of the AMP or L-threonine analogs were small in size and diffracted poorly
when examined at the home source. However it was possible to collect a complete dataset
to a resolution of 2.5 Å on a TdcB crystal obtained in the presence of CMP and O-
methylthreonine (Fig. 3.5). TdcB-CMP complex (crystal form III) belonged to the space
group P622. The asymmetric unit of the crystal was compatible with one subunit of TdcB
with a VM of 3.6 Å3Da-1 and a solvent content of 65.6% (Matthews 1968). The structure
was solved by molecular replacement using protein atoms from a monomer of TdcB form
II as the search model. Inspection of difference electron density maps showed strong
85

Chapter 3 Biodegradative threonine deaminase (TdcB)
positive density for the CMP and PLP bound to Lys58. The final model of TdcB in
complex with CMP was refined to R and Rfree of 19.7% and 22.6%, respectively.
Table 3.1 Crystal parameters and statistics on data collection, refinement and model quality.
Values in parentheses correspond to the highest resolution shell.
Data set TdcB-Crystal form I TdcB-Crystal form II TdcB-CMP-Crystal form III
Crystal parameters Unit cell parameters a, b, c (Å) 46.32, 55.30, 67.24 56.68, 76.83, 78.50 161.15, 161.15, 68.72 α, β, γ (°) 103.09, 94.70, 112.94 66.12, 89.16, 77.08 90.00, 90.00, 120.00 Space group P1 P1 P622 Temperature (K) 100 100 100 Wavelength (Å) 1.542 1.542 0.93
Data collection Resolution range (Å) 30.0-2.2 (2.28-2.20) 30.0-1.7 (1.76 -1.70) 30.0-2.50 (2.59-2.50) No. of monomers per AU 2 4 1 Total no. of reflections 360350 2012989 733136 No. of unique reflections 29556 128680 18646 Data redundancy 12.19 15.64 39.32 VM (Å3Da-1) 2.1 2.1 3.6 I/σ(I) 22.18 (4.98) 26.58 (2.76) 53.41 (7.56) Completeness (%) 94.7 (84.5) 93.1 (86.9) 99.8 (100.0) Rmerge (%) 5.3 (22.2) 4.1 (42.6) 5.5 (38.3) Refinement R (%) 21.0 (22.9) 19.0 (31.5) 19.7 (23.8) Rfree (%) 25.3 (29.6) 22.1 (36.0) 22.6 (24.1) No. of atoms Protein atoms 4765 9665 2417 Ligand atoms 31 77 37 Solvent atoms 274 782 62 Model quality RMS deviation from ideal values Bond length (Å) 0.008 0.007 0.007 Bond angle (deg.) 1.025 1.031 1.062 Dihedral angles (deg.) 5.470 5.106 5.360 Wilson B factor (Å2) 31.42 26.0 66.0 Average B factors (Å2) Protein atoms 35.8 24.8 66.1 Ligand 31.4 22.5 62.0 Water 37.4 32.5 66.1 Residues in Ramachandran plot (%) Most favoured region 89.6 91.4 90.4 Allowed region 9.6 8.3 8.9 Generously allowed region 0.5 0.1 0.7 Disallowed region 0.2 0.2 0.0
86

Chapter 3 Biodegradative threonine deaminase (TdcB)
3.3.4 Model quality
In all the three crystal forms, electron density is of good quality throughout the
polypeptide chain except for a few surface residues. In crystal forms I and II, relatively
poorer density is observed for the side chains of residues ranging from 108-121 and 125-
135. Some of the residues in these regions as well as a few other surface residues have
been truncated according to the extent of density observed for their side chains. In all the
three structures, besides the N-terminal hexa-histidine tag, a few residues at the N- and C-
termini were not included in the model due to absence of well-defined electron density.
The most favored and additionally allowed regions of the Ramachandran plot (Laskowski
et al. 1993; Ramachandran and Sasisekharan 1968) contained 89-91% and 7-8%,
respectively, of non-glycine and non-proline residues. In all the three structures, 2-3
residues were present in the boundary region between generously allowed and disallowed
regions of the Ramachandran map. These residues are either in a region of poor electron
density or near the CMP binding pocket. One of the common outliers in all the three
structures is Ser121. This residue with weak electron density in crystal forms I and II is
situated next to Tyr120, which is hydrogen bonded to the CMP molecule in TdcB-CMP
complex.
3.3.5 Tertiary structure of TdcB
As expected, TdcB exhibits fold type II, characteristic of the β-family of PLP-
dependent enzymes (Fig. 3.6). Each subunit comprises nine β-strands (13.5%), sixteen α-
helices (48.8%) and four 310-helices (3.1%). The tertiary structure and the topology
diagram of the secondary structural elements of TdcB as defined by the program DSSP
(Kabsch and Sander 1983) are shown in figure 3.6. The structure consists of a small and a
large domain. Residues from Ile59 to Tyr153 form the small domain whereas the large
domain is formed by two parts of the polypeptide chain, from the N-terminus (Met1) to
Lys58 and from Asp154 to the last C-terminal residue (Asp328). The small domain is an
open twisted α/β structure with four parallel β-strands (β5, β2, β3 and β4) surrounded by
five α-helices (α4, α5, α6, α7 and α8). The large domain also assumes an α/β structure
with an open twisted β-sheet formed by five parallel β-strands (β1, β9, β6, β7 and β8).
87

Chapter 3 Biodegradative threonine deaminase (TdcB)
These β-strands are strongly twisted so that the fifth β-strand is nearly perpendicular to
the first β-strand.
(b) (a)
Figure 3.6 (a) Ribbon diagram of a subunit of TdcB with PLP bound to Lys58. The secondary
structural elements in the subunit are colored differently. (b) Topology diagram of the secondary
structural elements of TdcB. Arrows represent β-strands, while cylinders represent α-helices. β-
strands and α-helices are colored cyan and green, respectively. The residue numbers at both ends
of structural elements are given. The upper and lower parts of the figure correspond to the small
and large domains, respectively.
Unlike the other members of fold type II or β-family of PLP-dependent enzymes,
the central β-sheet in the large domain is made up of only parallel strands. The small
amino terminal β-strand present in the large domain, which is antiparallel in most of the
members of the β-family, is absent in TdcB. In the large domain, the central β-sheet is
surrounded by six α-helices (α1, α2, α9, α10, α11 and α12) on one side and by four α-
helices (α3, α13, α14 and α15) on the other side. The C-terminal helix α16 protrudes
away from this domain towards the solvent. The corresponding helix in IlvA connects the
88

Chapter 3 Biodegradative threonine deaminase (TdcB)
N-terminal catalytic domain with the C-terminal regulatory domain. Unlike most of the
other members of the β-family of PLP-dependent enzymes, the first β-strand in the small
domain in TdcB (β2) is preceded by an α-helix (α5). Two helices, α14 and α15, present
in TdcB form a single long helix in most of the other members of the β-family of PLP
dependent enzymes. Between the two domains, there is a large internal gap that provides
space for the active site. The PLP cofactor is covalently bound as a Schiff base to the ε-
amino group of Lys58 present at the N-terminal end of the helix α4 yielding an internal
aldimine.
3.3.6 Dimeric TdcB
Presence of one dimer (AB dimer) in the asymmetric unit of crystal form I and
two dimers (AB and CD dimers) in the asymmetric unit of crystal form II of native TdcB
(Fig. 3.7) allows for a comparison of three independent models of the dimeric TdcB in
different crystallographic environments and therefore provides a measure of structural
variability of the protein.
(a) (b)
Figure 3.7 Unit cell of dimeric TdcB in crystal form (a) I and (b) II showing one (AB) and two (AB
and CD) dimers of TdcB, respectively. In the TdcB crystal form II, the D subunit has been built in
two conformations (D’ and D”).
89

Chapter 3 Biodegradative threonine deaminase (TdcB)
Figure 3.8 Electron density map corresponding to the two conformations of D subunit (D’ and
D”) in TdcB crystal form II. The electron density map corresponding to the D’ subunit (yellow)
from 2Fo−Fc map (gray) is contoured at 1.5 σ and for the D” subunit (cyan) from Fo−Fc map
(magenta) is contoured at 2.0 σ.
Careful examination of both crystal forms I and II indicated that differences occur
mainly due to the presence of the D subunit in two different conformations (D’ and D”)
in crystal form II. The electron density map associated with both the conformations is
shown in figure 3.8. Most of the crystal packing interactions of the two different
conformations of the D subunit are similar. However, there is enough space to
accommodate D’ and D” subunits with subtle differences at the unit cell interface. In
crystal form II, the interactions between the AB and CD dimers are much weaker than
those at A and B or C and D interface (Fig. 3.7). Structural superposition of D’ and D”
subunits indicated no significant structural differences between the two conformations.
Careful examination of CD’ and CD” dimers indicated that the two conformations of the
D subunit result from slightly altered dimer interfaces formed between C and D’ and C
and D” subunits. The maximum deviation observed between corresponding Cα atoms of
the two conformations of the D subunit is 4.8Å. In the TdcB crystal form II,
transformation of AB dimer by a matrix that superposes A and C subunits transforms the
B subunit such that its new position is related to the D’ subunit by a rotation of 9.9º (Fig.
3.9). The dimer formed by C and D” subunit is similar to the AB dimer. In crystal form
90

Chapter 3 Biodegradative threonine deaminase (TdcB)
I, a sodium ion has been modeled between one of the subunits in the unit cell and the
subunit in the next unit cell. Similarly, two sodium ions have been modeled in crystal
form II, one at the interface of the two dimers in the asymmetric unit and another
between dimers of neighboring unit cells.
Figure 3.9 Structural superposition of AB and CD’ dimers of TdcB crystal form II. The AB and
CD’ dimers are shown in green and violet, respectively. Structural superposition was carried out
by transforming the A subunit of the AB dimer on to the C subunit of the CD dimer.
3.3.7 Inter-subunit interactions in the dimeric TdcB
In the dimeric TdcB, inter-subunit interactions (≤4.0Å) between residues in the
AB and CD’ dimers of crystal form II have been analyzed. In each subunit, the large
domain contributes to the dimer formation. Accessible surface area calculations show that
a single subunit of TdcB with bound PLP has a surface area ranging from 12,368 to
12,671Ų. In the AB dimer, the total surface area buried on dimerization is 1046 Ų
(8.3%) per subunit, of which 63.2% (661.4 Ų) is non-polar and 36.8 % (385.0 Ų) is
polar. In the CD’ dimer, the total surface area buried on dimerization is 978 Ų (7.8%)
per subunit, of which 62.4 % (610.9 Ų) is non-polar and 37.6% (367.7 Ų) is polar.
Atoms of 19-21 residues of one subunit make hydrophobic and polar interactions (≤4.0
Å) with the atoms of the other subunit. Residues that mainly contribute to the interface in
both the dimers are 26-29, 31-38, 48-53, 274-280 and 313-324, which are on the helices
91

Chapter 3 Biodegradative threonine deaminase (TdcB)
α3, α13 and α16 and the loops preceding α3, α4, α14 and α16 helices. The interface
between the two subunits is mainly formed by hydrophobic interactions and hydrogen
bonds. There is a salt bridge between Glu38 and Arg53 at the interfaces of AB and CD’
dimers. There are 8 and 11 hydrogen bonds (cut-off value of 3.5Å) across the dimer
interfaces of AB and CD’ dimers respectively. Hydrogen bonds which are common to
interfaces of both AB and CD’ dimers are between Lys278-Gly313, Lys278-Ile280 and
their dyad symmetry mates. Other hydrogen bonds which are unique to AB interface are
between Asn34-Met51, Asn34-Arg53 and their dyad symmetry mates. Hydrogen bonds
which are unique to CD’ interface are between Lys27-Asn34, Lys27-Arg32, Glu38-
Tyr26, and Arg276-Tyr120. The contacts at the dimer interface of native TdcB are not as
extensive as in other members of the β-family of PLP-dependent enzymes. This may lead
to energetically inexpensive movement of the subunits leading to slightly altered dimer
interface as observed in the case of CD’ dimer. In this dimer, the D’ subunit is stabilized
by a salt bridge between Arg75 from the D’ subunit and Asp264 from the B subunit of
the neighboring unit cell. Therefore, AB dimers of crystal form I and II and CD” dimer of
crystal form II are likely to represent the physiological dimeric state.
3.3.8 Tetrameric TdcB in complex with CMP
Analysis of subunit packing in the TdcB-CMP structure shows a homotetrameric
arrangement of subunits related by 222 symmetry as shown in figure 3.10. The four
subunits of the tetramer are designated A, B, C and D (Fig. 3.11). The arrangement of
subunits in the tetrameric TdcB structure is similar to the tetrameric association observed
in the crystal structure of IlvA from E. coli (Gallagher et al. 1998). Superposition of the
A subunit of tetrameric TdcB with the A subunit of the dimeric TdcB structure using the
program ALIGN (Cohen 1997) gave an rms deviation of 0.39 Å between corresponding
Cα atoms. This difference is mainly due to alternate tracing of main chain atoms for
residues 104-121, 126-146, 274-277 and residues at the N- and C-termini (Fig 3.12).
Most of these changes are associated with either the residues of the small domain forming
the entry to the active site pocket or the residues involved in inter-subunit interactions. In
comparison with the dimeric TdcB structure, residues Leu126-Gly146 of tetrameric
TdcB-CMP (present in the helix α8 and the loop preceding it) lining the entrance to the
92

Chapter 3 Biodegradative threonine deaminase (TdcB)
active site pocket move away from the active site, thus broadening the area available for
substrate entry to the active site pocket. Between the dimeric and tetrameric TdcB, the
largest deviation (3.42 Å) is observed for the Cα atom of Phe131 (Fig. 3.12).
Figure 3.10 Crystal packing diagram for TdcB-CMP complex in the hexagonal form. Each
tetramer of TdcB in the unit cell is shown in a different color. There is a large void at each of the
6-folds with a diameter of approximately 80 Å. Unit cell is shown in black color.
3.3.9 Inter-subunit interactions in the tetrameric TdcB
In the tetrameric TdcB structure, each subunit interacts with only two other
subunits. The contacts between A and B subunits are most extensive while presence of
only sparse interactions between A and C and no contacts between A and D subunits
gives a dimer of dimers appearance to the tetramer (Fig. 3.11). Accessible surface area
calculations show that a single subunit in the tetrameric TdcB has a surface area of
13357.1Å2. The total surface area buried on dimerization is 1124.3 Å2 (8.4%) per subunit,
which is 78 Å2 more than that of the AB dimer of dimeric TdcB. Two molecules of CMP
in the dimer interface interact with residues from both the subunits. The electrostatic
surface representation of a dimer showing CMP binding pocket in the tetrameric TdcB-
CMP complex is shown in figure 3.13. Besides CMP, atoms from 20 residues of one
subunit make hydrophobic and polar interactions (≤4.0 Å) with the atoms of the other
93

Chapter 3 Biodegradative threonine deaminase (TdcB)
subunit. In TdcB-CMP structure, residues that mainly contribute to the interface in both
the dimers are 34-35, 50-53, 271-280 and 313-325, which are in helices α3, α13 and α16
and in the loops preceding α4, α14 and α16. There are 20 hydrogen bonds across the
dimer interface of TdcB-CMP dimer formed by A and B or C and D subunits and CMP.
Each CMP molecule forms three hydrogen bonds with Asn34, Gln275 and Lys278 of
another subunit of the dimer. Hydrogen bonds between Lys278-Gly313, Lys278-Ile280
and their dyad symmetry mates are common to the interface of AB dimer of dimeric and
tetrameric TdcB. Hydrogen bonds unique to tetrameric TdcB-CMP dimer interface are
between Asn34-Arg53, Lys278-Asn314, Ser321-Thr324, Ser321-Gly325 and their dyad
symmetry mates.
Figure 3.11 The quaternary structure of tetrameric TdcB-CMP complex. Ribbon representation of
the tetrameric TdcB is shown in an orientation such that two of the three perpendicular 2-folds of
the tetramer are along the plane of the paper. The tetramer with subunits A, B, C and D appears
as a “dimer of dimers” in which each subunit interacts with two other subunits along different
subunit interfaces. The CMP molecule at the dimer interface is shown in stick model. The black
spheres represent sodium ions present at the dimer-dimer interface.
94

Chapter 3 Biodegradative threonine deaminase (TdcB)
Figure 3.12 Stereo view of superposition of the A subunit from dimeric and tetrameric TdcB
showing the tertiary structural changes due to the binding of CMP. The subunits are in the same
color in regions of good structural alignment and in different colors (TdcB-CMP, cyan; TdcB
crystal form II, magenta) in regions where tertiary structural changes were observed. In these two
structures, PLP is shown to indicate the active site pocket.
Figure 3.13 The electrostatic surface representation of a dimer of tetrameric TdcB-CMP complex
showing the CMP binding pocket. Two molecules of CMP are at the dimer interface. PLP bound
to Lys58 is in the cleft between the two domains in each subunit. Positively charged residues are
shown in blue, negatively charged residues in red and neutral residues in white.
95

Chapter 3 Biodegradative threonine deaminase (TdcB)
There is a small region of contact between subunits at the dimer-dimer interface
(between A and C or B and D subunits), which involves hydrophobic as well as polar
interactions. These contacts are formed mainly by residues in two stretches of the
polypeptide chain between Leu167-Tyr174 at the C-terminal region of helix α9 and
Ile199-Thr202 at the C-terminal region of helix α10 (Fig. 3.14). In IlvA, residues
involved in dimer-dimer contacts are also at the C-terminal region of α-helices
corresponding to α9 and α10 of TdcB (Gallagher et al. 1998). The solvent accessible
surface area buried at this interface is 877.2 Å2 per dimer, (corresponding to 438.6 Å2
(3.3%) per subunit), out of which 69.3% (607.6 Å2) is non-polar and 30.7% (269.6 Å2) is
polar. The dimer-dimer interface contains four hydrogen bonds formed by Ile199-Asn200
and Ile199-Thr202 and their dyad symmetry mates. Other residues at this interface are
involved in van der Waals contacts and hydrophobic interactions. A sodium ion bound to
the main chain oxygen atom of Glu271 from both the subunits has been modeled at the 2-
fold axis.
Figure 3.14 Stereo view of the inter-subunit interactions at the dimer-dimer interface (AC and BD
interface) in the tetrameric TdcB. A and C subunits are shown in two colors and hydrogen bonds
formed between the residues (≤ 3.5 Å) from these two subunits are shown as broken lines. The
blue sphere represent sodium ion present at the dimer-dimer interface.
To investigate the possibility of tetramer formation by native (dimeric) TdcB, the
structures of dimers in dimeric and tetrameric forms of TdcB were compared. A
tetrameric form of native TdcB was generated by superposing two copies of native AB
dimer on AB and CD dimers of the tetrameric TdcB-CMP (Fig. 3.15) by two separate
transformations. In the first transformation, the A subunit of native TdcB was made to
96

Chapter 3 Biodegradative threonine deaminase (TdcB)
superpose on the A subunit of TdcB-CMP while in the second transformation, the A
subunit of the native dimer was superposed on the C subunit of TdcB-CMP. The residual
rotation and translation between the transformed B subunit of native TdcB and the B or D
subunits of TdcB-CMP were 21.3º and 0.05 Å, respectively. Similar results were
obtained when B subunit of native TdcB was used for structural superposition. The native
TdcB tetramer so generated had a large number of short-contacts (≤2.2Å) at the interface
of the transformed B subunits (Fig. 3.15). Atoms from 17 residues (Met170, Leu173-
Asn198, Lys197-Ile203 and Asn303-Lys305) from each B subunit are involved in the
short-contacts at this interface. From the above analysis, it can be concluded that the
conformational changes accompanying the binding of CMP at the dimer interface are
essential to facilitate the tetramerization of TdcB.
Figure 3.15 Hypothetical tetrameric form of native TdcB. This was generated by superposing two
copies of native AB dimer on AB and CD dimers of tetrameric TdcB-CMP (see text for details).
The native TdcB tetramer so generated had a large number of short-contacts (≤2.2Å). Enlarged
view of the residues involved in the short-contacts is shown. The figure is shown in the same
orientation as in figure 3.11. All four subunits of the tetrameric TdcB-CMP complex are shown in
light gray whereas the two AB dimers of native TdcB used for the generation of the tetramer are
shown in green and magenta. CMP molecules are shown in red as stick models.
97

Chapter 3 Biodegradative threonine deaminase (TdcB)
3.3.10 Active site pocket
The active sites of TdcB are situated in clefts between the small and large
domains of each subunit. The coenzyme PLP is deeply buried in the active site and is
accessible only via a narrow channel. The 2Fo-Fc electron density map corresponding to
PLP bound to Lys58 and the associated hydrogen-bonding network are shown in figure
3.16.
(a)
(b)
Figure 3.16 PLP in the active site pocket of TdcB. (a) Stereo view of electron density
corresponding to PLP bound to Lys58 from a 2Fo−Fc map contoured at 1.5 σ. (b) Stereo diagram
of the active site region showing hydrogen bonding network of PLP. Hydrogen bonds formed by
PLP (≤ 3.5 Å) with protein atoms and water molecules are shown as broken lines.
The pyridine ring is sandwiched between Phe57 on the side facing the protein
interior and Gly237, Cys238, and Ala284 on the other side. The aromatic ring of Phe57 is
almost perpendicular to the pyridine ring of PLP. The N1 atom of the pyridine ring of
PLP forms a hydrogen bond to the OG of Ser311 (2.66 Å). The 3’ hydroxyl group of PLP
is hydrogen bonded to the ND2 atom of Asn85 (2.83 Å) and to the NZ atom of Lys58
(2.54 Å). The side chain amide group of Asn85 is coplanar with the pyridine ring. This
98

Chapter 3 Biodegradative threonine deaminase (TdcB)
will allow the expected pyridine ring tilt during transaldimination without any steric
hindrance and loss of hydrogen bond. The semi-circular tetra-glycine loop formed by
Gly184, Gly185, Gly186, Gly187 and the residues Leu188 and Ile189 form the binding
site for the phosphate moiety of PLP. The phosphate group forms six hydrogen bonds
with the main chain amides of the semicircular loop and three hydrogen bonds with water
molecules. These water molecules interact with Pro152, Gln162 and Ile189 present in the
active site pocket. The positive side of the helix dipole of α10 present at the carboxyl end
of the semicircular loop is close to the phosphate group of PLP and compensates for its
negative charge. All the amino acid residues which are hydrogen bonded to PLP are well
conserved in TdcB and IlvA (Fig. 3.17).
Figure 3.17 Amino acid sequence alignment of biodegradative threonine deaminase (TdcB) from
Salmonella typhimurium (TdcB_ST) and biosynthetic threonine deaminase (IlvA) from Escherichia
coli (IlvA_EC). The residues conserved in both are shaded in yellow color. The residues forming
hydrogen bonds with PLP are shown in cyan boxes whereas residues forming hydrogen bonds
with CMP are shown in red boxes. The secondary structure elements of TdcB_ST and IlvA_EC
are represented by arrows (β-strands) and spirals (α-helices) at the top and the bottom of the
aligned sequences, respectively.
99

Chapter 3 Biodegradative threonine deaminase (TdcB)
TdcB and IlvA structures have not been determined in the presence of the
substrate, L-threonine or its analogs. To find the probable substrate binding site, we have
carried out cavity calculations using the program VOIDOO (Kleywegt and Jones 1994)
of the Uppsala program package. This revealed a small cavity in the active site pocket
near PLP. The wall of this cavity is formed mainly by His86, Pro152, Tyr153, Val158
and Gln162. All these residues are also conserved in IlvA, providing further evidence of
their role in substrate binding or catalysis. In IlvA, auxotrophic mutants of some of these
residues have been shown to affect enzyme activity either by decreasing the substrate
affinity or by destabilizing the catalytic intermediates (Fisher and Eisenstein 1993). 3.3.11 CMP binding site
Previous studies carried out using various structural analogs of AMP and other
natural nucleotides had indicated that considerable alteration in the adenine base could be
tolerated, mainly in the imidazole portion of the ring (Nakazawa et al. 1967; Whanger et
al. 1968). Substitution at N6 position of AMP appeared to decrease the binding affinity.
These studies had also shown that the atoms at 2' and 3' hydroxyl groups of ribose moiety
and 5'-phosphate group of AMP are of primary importance, both for activation and
binding. Structural information obtained from the present work fully explains these
observations. Two molecules of CMP in the dimer interface help in the formation of a
tight dimer, which in turn interacts with another dimer forming a tetramer. The 2Fo-Fc
electron density map associated with CMP and the hydrogen-bonding interactions with
the two subunits at the dimer interface are shown in figure 3.18. CMP molecules interact
with residues from both the subunits. The ribose sugar is in the C2' endo form. The
cytosine base is in the anti conformation with respect to the ribose sugar. CMP forms 14
hydrogen bonds with the protein atoms and 4 hydrogen bonds with water molecules. The
cytosine base is sandwiched between Ala116 of one subunit and Arg276 of the other
subunit. The N4 atom of cytosine base is hydrogen bonded to the side chain oxygen atom
of Asp119 and the O2 atom is hydrogen bonded to the side chain oxygen of Gln275.
100

Chapter 3 Biodegradative threonine deaminase (TdcB)
(a)
(b)
Figure 3.18 CMP in the inter-subunit interface in the tetrameric TdcB-CMP complex. (a) Stereo
view of electron density corresponding to CMP from a 2Fo−Fc map contoured at 1.0 σ. (b) Stereo
diagram of the CMP binding region showing bound CMP. Residues interacting with CMP from
the two subunits of tetrameric TdcB are shown in different colour. Hydrogen bonds formed by
CMP (≤ 3.5 Å) with protein atoms are shown as broken lines.
Superposition of AMP on the bound CMP indicates that, N3 and N6 atoms of
adenine are expected to correspond to N4 and O2 atoms of cytosine and would form
hydrogen bonds with Gln275 and Asp119, respectively. Since the base region of CMP
faces the solvent, there is enough space for the purine ring of AMP to bind at this site.
Presence of the bulkier adenine base is expected to further strengthen the interaction
between the two subunits. In the sugar, O2' is hydrogen bonded to the side chain atom of
Gln88, Asn314 and main chain oxygen of Thr54, all from the same subunit. The O3'
101

Chapter 3 Biodegradative threonine deaminase (TdcB)
atom of the ribose forms a strong hydrogen bond with Asn314 from one subunit and with
NZ atom of Lys278 from the other subunit. The O5' atom is hydrogen bonded to the
phenolic hydroxyl group of Tyr120. The oxygen atoms of the phosphate group bind with
guanidium group of Arg53, phenolic hydroxyl group of Tyr120, main chain oxygen atom
of Thr54 from one subunit and with the side chain nitrogen atom of Asn34 from the other
subunit. Thus, all the three moieties - base, sugar and phosphate form one hydrogen bond
each with the atoms from the second subunit of the dimer. Previous studies to identify
AMP binding site in E. coli TdcB by photolabeling using photo-reactive AMP analog, 8-
azido-AMP followed by tryptic digestion revealed one tryptic peptide Thr230-Arg242
with radioactivity (Patil and Datta 1988). In this work, this region is not present near the
CMP binding pocket, which indicates that 8-azido-AMP might have bound non-
specifically to the enzyme. 3.3.12 Role of CMP in oligomerization and enzyme activation
Comparative analysis of the crystal structures of dimeric and tetrameric forms of
TdcB provides direct structural insight on the ligand induced oligomerization, and the
framework needed for understanding the significant increase in the affinity of substrate
binding and Vmax. In the absence of CMP, TdcB exists in a dimeric form, the structure of
which is different from the dimer structure observed in the CMP-induced tetrameric
TdcB-CMP. In the structure of TdcB-CMP complex, the movement of the residues from
the small domain away from the active site pocket lead to increase in the size of the
channel for the entry of substrate to the active site pocket, which in turn can increase the
rate of the reaction. However, residues at the active site pocket of the tetrameric TdcB-
CMP structure do not show significant structural changes from those of the dimeric TdcB
structure. Presence of CMP in the dimer interface, far from the active site pocket,
supports the previous observation that the CMP binding does not have a direct role in the
enzyme activation per se.
The present results can also be interpreted in terms of the allosteric model with a
low activity T state and a high activity R state. The enzyme exists mainly in the T state in
the absence of AMP. The T↔R equilibrium is shifted to the R state in the presence of
AMP or at high concentrations of TdcB. Dimeric TdcB structure can represent the T state
102

Chapter 3 Biodegradative threonine deaminase (TdcB)
whereas the dimer structure observed in the TdcB-CMP tetrameric form may correspond
to the R state of the enzyme. However, the absence of sigmoidal kinetics or allosteric
behavior suggests that TdcB has some atypical properties. In this respect, TdcB differs
from Clostridium tetanomorphum ADP-activated threonine deaminase, which has similar
subunit structure and undergoes a dimer-tetramer transition by either the activator ADP
or high levels of threonine (Tokushige et al. 1963) and exhibits sigmoidal kinetics in the
absence of ADP and Michaelis-Menten kinetics in the presence of ADP (Tokushige et al.
1963). Studies on AMP-activated TdcB in S. typhimurium and E. coli and ADP-activated
threonine deaminase from Clostridium tetanomorphum suggest that energy production by
controlled catabolism of L-threonine by allosteric effectors may be a regulatory
mechanism during anaerobic condition. It is likely that during periods in which level of
energy in the cells is low, the higher concentration of AMP provides a signal for
activation and conversion of L-threonine to propionate and ATP. In E. coli and S.
typhimurium, propionate can be further catabolized into pyruvate and succinate by 2-
methylcitric acid cycle.
3.3.13 Structural comparison with related enzymes
A comparison between TdcB and a representative subset of structures from the
Protein Data Bank using the DALI server (Holm and Sander 1993) revealed that the
structures most similar to TdcB were within the fold type II or β-family of PLP
dependent enzymes. The first nine crystal structures with the highest Z scores are IlvA,
serine racemase, O-acetylserine sulfhydralase, cystathionine β-synthase, β-subunit of
tryptophan synthase, serine dehydratase, O-phosphoserine sulfhydrylase, threonine
synthase, and 1-aminocyclopropane-1-carboxylate deaminase (Table 3.2). Most of these
enzymes can catalyze the cleavage of the Cβ-O bond of serine or threonine. Structural
alignment of TdcB with these structures resulted in rms deviations of 1.6-3.1Å and Z
scores ranging from 43.6 to 22.0 for 265-314 aligned Cα residues. Superposition of all
these structures indicates that besides overall structural similarity, structral equivalence
extends to the PLP-binding pocket and the regions related to reaction specificity (Fig.
3.19). Structural conservation observed among these enzymes provides strong evidence
of their phylogenetic relationship.
103

Chapter 3 Biodegradative threonine deaminase (TdcB)
104
Figure 3.19 Structural superposition of TdcB
with the four other structures obtained using
DALI server with rms deviations less than 2.5
Å: biosynthetic threonine deaminase (IlvA), O-
acetylserine sulfhydralase (OASS), serine
dehydratase (SD) and cystathionine β-synthase
(CBS). TdcB is drawn in magenta, IlvA in cyan,
OASS in green, SD in yellow and CBS in pink.
For clarity, only one monomer is shown for
each structure. In these structures, PLP bound
to the active site lysine is shown in red.
Table 3.2 DALI structural superposition statistics for proteins with rmsd lower than 3.0 Å.
1334127723.82.91F2DHansenula saturnus1-aminocyclopropane-1-carboxylate deaminase
1839230131.92.91KFKSalmonella typhimurium
Tryptophan synthase (β-subunit)
1943828024.12.81E5XArabidopsis thalianaThreonine synthase
1250929725.82.81Kl7SaccharomycescerevisiaeThreonine synthase
2134829733.42.41JBQHomo sapiensCystathionine β-synthase
2331527931.02.41PWERattus norvegicusSerine dehydratase
2031529032.92.11OASSalmonella typhimurium
O-acetyl serinesulfhydralase
3831830743.31.71V71Schizosaccharomyces pombeSerine racemase*
3549431443.61.61TDJEscherichia coliBiosynthetic threonine deaminase
Identity (%)
Sequence length
Length of alignment
Z score
RMSD(Å)
PDB codeOrganismEnzyme
*M. Goto, unpublished result.

Chapter 3 Biodegradative threonine deaminase (TdcB)
3.3.14 Mechanistic considerations
The general catalytic mechanism of PLP-dependent enzymes has been well
studied (Eliot and Kirsch 2004). In the TdcB structure, N1 atom of the PLP is hydrogen
bonded to the OG of Ser311. In TdcB, as seen in IlvA, serine dehydratase, β-subunit of
tryptophan synthase, O-acetylserine sulfhydralase, cystathionine β-synthase and
threonine synthase, the hydrogen bond partner of N1 atom of PLP is a neutral amino acid
(Ser, Cys or Thr), and hence the N1 atom is not protonated in these enzymes. The
N1····H-O neutral hydrogen bond induces a weak electron-withdrawing effect on the PLP
pyridine ring which acts as an electron sink. Unlike aminotransferases, in serine
dehydratase, the carboxyl group of the substrate is less polarized due to the presence of a
neutral environment around it (Yamada et al. 2003). Even in the TdcB structure, presence
of neutral amino acids near the expected threonine binding site suggests a neutral
environment for the carboxyl group of the substrate. Therefore, it is more difficult to
abstract the Cα hydrogen from the substrate in the external aldimine. Therefore, it is
expected that the reaction proceeds via the carbanion intermediate rather than the
quinonoid intermediate.
The probable reaction mechanism for TdcB is shown in figure 3.20 as a group of
partial reactions in which the last two steps occur in solution non-enzymatically. During
catalysis, an external aldimine is formed between PLP and L-threonine by
transaldimination. It was proposed in the case of serine dehydratase that the phosphate
group of PLP abstracts a proton from the α-amino group of the substrate during the
formation of external aldimine and then acts as a strong general acid by donating a proton
to the OG of serine. The hydrogen atom from the Cα carbon atom of serine is then
abstracted by the active site lysine in a concerted fashion (Yamada et al. 2003). Similar
catalytic reaction in TdcB will result in α, β elimination of water from the PLP-Thr
leading to the formation of PLP-aminocrotonate. A second transaldimination reaction
results in the release of aminocrotonate leading to the formation of PLP-Lys Schiff base.
Non-enzymatic tautomerization of aminocrotonate forms α-iminobutyrate, which is then
spontaneously hydrolyzed into α-ketobutyrate and ammonia. Because of the similarities
in the architecture of the active site, substrate structure and chemistry performed, it might
105

Chapter 3 Biodegradative threonine deaminase (TdcB)
be appropriate to assume that threonine and serine deaminases have similar catalytic
mechanisms.
Figure 3.20 Probable reaction mechanism of PLP-dependent deamination of L-threonine. The
partial reactions are (1) transaldimination of the PLP-Lys to similar linkage with the L-threonine
amino group (2) removal of α-hydrogen atom from the substrate to form a carbanion (3) β-
elimination of the hydroxyl group (4) transaldimination with the enzyme to yield the PLP-Lys
and aminocrotonate (5) non-enzymatic tautomerization to the imine and (6) non-enzymatic
hydrolysis to form α-ketobutyrate and ammonia.
106

Chapter 3 Biodegradative threonine deaminase (TdcB)
3.4 Conclusions In this chapter, crystal structures of TdcB and its complex with the activator
molecule, CMP, have been presented. Structural comparison of native TdcB and its
complex with CMP has revealed interesting differences. In the native structure, TdcB is
in a dimeric form whereas in complex with CMP, it forms a tetramer, which appears as a
dimer of dimers. Tetrameric TdcB binds to four molecules of CMP, two molecules at
each of the dimer interfaces. CMP interacts with residues from two different subunits and
results in the formation of a tight dimer. In the absence of CMP, TdcB forms a relatively
loose dimer which seems to explain the monomer-oligomer equilibrium observed in the
solution at low enzyme concentration. Structural superposition of dimers of dimeric and
tetrameric TdcB shows differences in the arrangement of the two subunits. Structural
analysis has shown that this difference in the arrangement of the two subunits is essential
for the tetramerization of TdcB. Most of the tertiary structural changes observed in the
TdcB-CMP complex are associated with either the residues of the small domain lining
the entry to the active site pocket or the residues at the dimer interface. The differences
observed at the dimer interface and in the tertiary and quaternary structures of TdcB in
the absence and presence of CMP appear to account for the enzyme activation and
increased binding affinity for L-threonine. Most of the residues in the active site pocket
involved in PLP and substrate binding are conserved in TdcB, IlvA and serine
dehydratase suggesting a similar catalytic mechanism for these enzymes.
107

Crystal structures of ADP- and AMPPNP-bound propionate kinase (TdcD) from Salmonella typhimurium:
Comparison with members of acetate and sugar Kinase/ heat shock cognate 70/actin superfamily
4

Chapter 4 Propionate kinase (TdcD)
4.1 Introduction Kinases are a ubiquitous group of enzymes central to many metabolic processes.
The transfer of the terminal phosphoryl group from a nucleotide to a small molecule or to
a protein substrate is a fundamental process in signal transduction, energy transfer and
gene regulation. The X-ray crystal structures of kinase-substrate complexes have revealed
structural details of substrate-binding sites and provided insights into the mode of
phosphoryl transfer. On the basis of structural folds and sequence similarities, kinases
have been classified into three distinct families (Matte et al. 1998). The classical P-loop-
containing kinases share a common loop with a conserved amino acid sequence between
a β-strand and an α-helix (Matte et al. 1998). A second group consists of mainly protein
kinases that have a conserved loop connecting two antiparallel β-strands involved in
phosphate binding (Bossemeyer 1994). The third group, which includes acetate and sugar
kinase-heat shock cognate 70-actin (ASKHA), uses two β-hairpins to bind the phosphate
groups of ATP (Bork et al. 1992; Buss et al. 2001).
In Escherichia coli and Salmonella typhimurium, it has been shown that L-
threonine can be cleaved non-oxidatively into propionate via 2-ketobutyrate by
biodegradative threonine deaminase, 2-ketobutyrate formate lyase, phosphotransacetylase
and propionate kinase (Dyk and LaRossa 1987; Hesslinger et al. 1998; Sawers 1998).
The final reaction during the anaerobic degradation of L-threonine to propionate is
catalyzed by propionate kinase encoded by the gene tdcD, which enables the conversion
of propionyl phosphate and ADP to propionate and ATP (Fig. 4.1).
Figure 4.1 Reaction catalyzed by propionate kinase (TdcD).
Propionate kinase (TdcD) exhibits significant amino acid sequence identity with
acetate kinase. In E. coli, propionate kinase has been shown to also possess acetate kinase
activity (Hesslinger et al. 1998). Therefore, these two enzymes are likely to have similar
structures and catalytic mechanisms. The only available crystal structure of acetate kinase
109

Chapter 4 Propionate kinase (TdcD)
is from a thermophilic organism, Methanosarcina thermophila (Buss et al. 2001). The
polypeptide fold of M. thermophila acetate kinase (MAK) contains a core similar to those
of glycerol kinase/hexokinase/Hsc70/actin, the ASKHA superfamily of
phosphotransferases (Buss et al. 2001). This superfamily is characterized by two
domains, each with the topology βββαβαβα, separated by an active site cleft (Hurley
1996). In the members of this superfamily, phosphoryl transfer is coupled to a large
conformational change in which the two domains close around the nucleotide. It is now
generally believed that these proteins have undergone divergent evolution from a
common ancestor (Bork et al. 1992; Buss et al. 2001).
Previous studies on E. coli acetate kinase have shown an inversion of
configuration during phosphoryl transfer (Blattler and Knowles 1979), suggesting either a
direct in-line transfer or a covalent triple displacement mechanism involving two
phosphoenzyme intermediates as two distinct possibilities for the catalytic mechanism.
The triple displacement mechanism (Spector 1980) was proposed when a
phosphoenzyme was isolated after incubating E. coli acetate kinase with radiolabeled
ATP or acetyl phosphate and the isolated phosphoenzyme was able to transfer the
phosphoryl group to ADP or acetate (Anthony and Spector 1971; 1972). However, the
ability of E. coli acetate kinase to phosphorylate enzyme I of the phosphotransferase
system (Fox et al. 1986) and CheY in vitro (Dailey and Berg 1993) was thought to
indicate the role of the phosphoenzyme in sugar transport rather than in phosphorylation
of acetate or ADP. Further, the direct in-line transfer mechanism is supported by steady-
state kinetics as well as stereochemical evidence. Recently, inhibition and structural
studies carried out on a probable transition state analog of MAK formed by AlF3, acetate
and ADP provided strong additional support for the direct in-line transfer of the
phosphoryl group from ATP to acetate (Gorrell et al. 2005; Miles et al. 2002).
In this chapter, three-dimensional structures of S. typhimurium propionate kinase
(TdcD) in complex with ADP and AMPPNP, a non-hydrolysable analog of ATP, have
been described. This is the first structural description of a propionate kinase from any
source and a homologue of acetate kinase from a mesophile. The structures of TdcD in
complex with AMPPNP and ADP revealed intact nucleotides. This is in contrast to
MAK, where only ADP and a sulphate ion were observed in the MAK co-crystallized
110

Chapter 4 Propionate kinase (TdcD)
with ATP (MAK-ATP), and ADP and a sulphate ion in the A subunit and AMP and
thiopyrophosphate in the B subunit were seen in MAK co-crystallized with adenosine 5′-
[γ-thio]phosphate (ATPγS) (MAK-ATPγS) (Buss et al. 2001; Gorrell et al. 2005). Apart
from overall features, the structures of the TdcD complexes reveal the mode of nucleotide
binding and its relationship to the proposed binding site of propionate. Comparison of the
TdcD complex structures with members of the ASKHA superfamily (Bennett and Steitz
1980; Bystrom et al. 1999; Flaherty et al. 1990; Kabsch et al. 1990), mainly with that of
acetate kinase (Buss et al. 2001; Gorrell et al. 2005) obtained with different ligands,
along with results obtained from various site-directed mutagenesis experiments with
MAK (Gorrell et al. 2005; Ingram-Smith et al. 2000; Miles et al. 2001; Singh-Wissmann
et al. 1998; Singh-Wissmann et al. 2000) and hexokinase (Zeng et al. 1996), has
permitted identification of catalytically essential residues involved in substrate binding
and catalysis, and has highlighted the unique features of TdcD. The liganded structures of
TdcD and MAK have been compared to obtain insights into the structural basis of the
increased dimeric thermostability of MAK and differences in the activities of these two
enzymes. The significant sequence similarity between TdcD and MAK has facilitated
study of domain movement observed in these and other members of the ASKHA
superfamily.
4.2 Materials and methods 4.2.1 Cloning, overexpression and purification
The tdcD gene was PCR-amplified from S. enterica serovar Typhimurium
genomic DNA using KOD HiFi DNA polymerase (Novagen). After amplification of the
target gene by sense 5'-CATGCCATGGCTAGCAATGAATTTCCGGTCG-3' and
antisense 5'-CGGGATCCTTACTCGAGTGCAAATTCCACTGG-3' primers, the PCR-
amplified fragment was digested with NheI and BamHI and then cloned into the vector
pRSET C (Invitrogen) encoding a polypeptide with an N-terminal hexa-histidine tag.
TdcD was purified to homogeneity using Ni-NTA affinity column chromatography. The
purity of the protein was estimated using SDS-PAGE and was found to be nearly
homogeneous (Fig 4.3).
111

Chapter 4 Propionate kinase (TdcD)
4.2.2 Activity assay and kinetic studies
Propionate kinase activity was examined routinely by coupled-enzyme
spectrophotometric assay in which the reaction was monitored by coupling the formation
of ADP to the oxidation of NADH by pyruvate kinase and lactate dehydrogenase. The
reaction mixture contained the following components in a final volume of 0.1 ml: 50 mM
HEPES-NaOH (pH 7.5), 5 mM propionate, 3 mM ATP, 12.5 mM MgCl2, 3 mM
phosphoenolpyruvate, 0.25 mM reduced NADH, 2.5 units of lactate dehydrogenase, 2
units of pyruvate kinase. The reaction was started by the addition of propionate kinase.
The progress of the reaction was followed by measuring the formation of NAD+ at 25°C,
which results in a decrease in absorbance at 340 nm. The concentration of TdcD was
determined by Lowry's method (Lowry et al. 1951) using bovine serum albumin as the
standard.
Substrate specificities of the homogeneous enzyme were determined with ATP,
propionate and acetate. For kinetic studies, the concentration of MgCl2 was held in excess
at 25 mM in the typical assay system. The kinetic constants for propionate and acetate
were determined using purified TdcD in the presence of an excess of ATP (5 mM),
whereas the concentration of propionate was held in excess at 50 mM for the
determination of KM for ATP. Enzymatic activity using acetate as substrate was measured
in exactly the same way, except that acetate replaced propionate in the assay. Kinetic
parameters were determined using at least seven different concentrations of substrates.
The concentration ranges used for ATP, propionate and acetate were 0.025-6 mM, 0.25-
40 mM and 2-350 mM, respectively. All enzyme assays were conducted at 25°C, and
experiments were repeated at least three times from independent purifications. Kinetic
parameters were calculated by fitting the initial velocity versus substrate concentration to
the Michaelis-Menten equation, v = (Vmax[S])/(KM+[S]), using the non-linear regression
analysis option of the GraphPad Prism software. The kcat values were determined on the
basis of molecular mass corresponding to the dimer of TdcD.
4.2.3 Crystallization and data collection
Prior to crystallization, the protein was concentrated to 30 mg ml-1. The
complexes were prepared by incubating native protein (10 mg/ml) with 10 mM ADP or
112

Chapter 4 Propionate kinase (TdcD)
10 mM AMPPNP overnight at 4°C. Diffraction-quality crystals were obtained at 291 K
in condition No. 57 [0.05 M ammonium sulfate, 0.05 M Bis-Tris pH 6.5, 30% penta-
erythritol ethoxylate (15/4 EO/OH)] and condition No. 46 [0.1 M Bis-Tris pH 6.5, 20%
polyethylene glycol monomethyl ether (PEG MME) 5000] of the Index Screen from
Hampton Research. Further optimization of the latter condition with different molecular
weights of PEG MME at various concentrations gave good diffraction-quality crystals
using 12% PEG MME 5000, 18% PEG MME 2000 or 20% PEG MME 550 in the
presence of 0.1 M Bis-Tris pH 6.5. Crystals of the TdcD-ADP and TdcD-AMPPNP
complexes for which diffraction data are described in this chapter were obtained from
conditions containing 0.1 M Bis-Tris (pH 6.5), 18% (w/v) polyethylene glycol
monomethyl ether 2000 and 0.1 M Bis-Tris (pH 6.5), 35% (v/v) pentaerythritol
ethoxylate (15/4 EO/OH), respectively. These crystals appeared after 5 d equilibration
against the crystallization solution and grew to full size in 10 d (Fig 4.5). Diffraction data
were collected to resolutions of 2.2 Å and 2.3 Å for ADP and AMPPNP complexes,
respectively, using 20% (v/v) ethylene glycol as a cryoprotectant. Systematic absences
indicated that the crystals belonged to the space group P3121 or P3221. The volume of the
asymmetric unit of the crystals was compatible with only one subunit, with a volume per
unit molecular mass of the protein of 2.7 Å3 Da−1 and a calculated solvent content of
53.2% (v/v) (Matthews 1968). The crystal parameters and the data collection statistics are
summarized in table 4.1.
4.2.4 Structure solution and refinement
The structure of TdcD-ADP was solved by molecular replacement (MR) using a
monomer of MAK-ATP (PDB:1G99) as the search model (Buss et al. 2001). TdcD
shares a sequence identity of 38% with MAK. Since, in the A and B subunits of MAK,
domain I and II are closed to different extents (Fig. 4.2), both the subunits, with non-
identical residues mutated to alanine, were used separately as search models for the
structure solution using the program AMoRe (Nawaja 1994). The highest peak of the
translation search using the A subunit as the search model had a correlation coefficient of
30% and a R-factor of 47%, and using the B subunit as the search model resulted in a
correlation coefficient of 37% and a R-factor of 47% for the resolution range of 15.0 Å to
113

Chapter 4 Propionate kinase (TdcD)
3.5 Å in the space group P3121. MR solutions obtained with the B subunit as the search
model were used for further refinement. Examination of the top solution revealed good
crystal packing and no clash between symmetry-related molecules. Refinement using the
CNS_solve1.1 (Brunger et al. 1998) program was initiated, with a randomly chosen
subset of 5% of reflections for the calculation of Rfree as a monitor of refinement. The
2Fo-Fc map calculated after initial positional refinement showed good fit for the majority
of the main-chain and revealed major changes at places corresponding to insertions and
deletions.
A subunit B subunit
Figure 4.2 The tertiary structure of A and B subunit of Methanosarcina thermophila acetate kinase
co-crystallized with ATP (Buss et al. 2001) illustrating the different degrees of domain closure in
A and B subunits.
Initially, towards the N-terminal end, no electron density was observed for
residues 27-87. After rigid body refinement, the model was subjected to one round of
simulated annealing with torsion angle dynamics, in which the protein atoms were heated
to 5000 K and then cooled in steps of 25 K to the final temperature of 300 K. The σA
weighted 2Fo-Fc and Fo-Fc maps were then calculated, and visualized using the
interactive model building program O (Jones et al. 1991), which showed no improvement
in the map for the missing region. In the rest of the model, the position of the main-chain
was adjusted manually in several places, and side-chains were added to the model in
places where the density was clear. Density modification techniques using the program
114

Chapter 4 Propionate kinase (TdcD)
CNS_solve1.1 (Brunger et al. 1998) were applied to reduce the model dependency and to
improve the electron density map. After several rounds of model building and refinement,
it was possible to build most of the side-chains into the electron density. From the
resulting improved model, 2Fo-Fc and Fo-Fc electron density maps were calculated,
which allowed the completion of all missing parts of the model. Ambiguities in the model
were resolved by setting the weights of the atoms concerned to zero, until the density
could be interpreted clearly. An omit map was calculated in order to remove the model
bias by omitting 5% of the model in each cycle. The structure of TdcD in complex with
AMPPNP (TdcD-AMPPNP) was solved using the protein atoms from the TdcD-ADP
structure. In the final stages of refinement, inspection of difference electron density maps
showed strong positive density for the expected ligands AMPPNP and ADP. The
topology and the parameters for the ADP and AMPPNP were evaluated using the web-
based program HIC-UP (Kleywegt and Jones 1998). This was followed by identification
of potential sites of solvent molecules by the automatic water-picking algorithm of
CNS_solve1.1 (Brunger et al. 1998). The positions of these automatically picked water
molecules were checked manually and a few more water molecules were identified
manually on the basis of electron density contoured at 1.0 σ in the 2Fo-Fc map and 3.0 σ
in the Fo-Fc map. The model was then subjected to 20 steps of individual B-factor
refinement. The final refinement statistics and assessment of model quality of the refined
structures are summarized in table 4.1.
4.2.5 Domain motion analysis
The program DynDom (Hayward and Lee 2002) and DOMOV
(http://bioinfo1.mbfys.lu.se/cgi-bin/Domov/domov.cgi) were used to analyze the domain
motion. DynDom identifies dynamic domains by clusters of rotation vectors
corresponding to main-chain segments, which may be composed of several regions
moving in a concerted fashion without necessarily forming a globular unit. This program
was used to identify fixed and moving domains and hinge residues at the transition points
between the two domains. DOMOV, a server to detect the domain movement from two
homologous protein structures, was used to cross-check the domain movement. It was
also used to calculate rotation and translation of the moving domain relative to the fixed
domain.
115

Chapter 4 Propionate kinase (TdcD)
4.3 Results and Discussion 4.3.1 Cloning, overexpression and purification
TdcD was successfully cloned in the pRSET C vector (Invitrogen) with an N-
terminal hexa-histidine tag and was purified using Ni-NTA affinity column
chromatography. The recombinant enzyme was obtained with a final yield of 25 mg per
liter of cell culture. The purified enzyme showed a single polypeptide band in SDS-
PAGE, corresponding to a subunit molecular mass of 44 kDa (Fig 4.3). Gel filtration
chromatography experiments suggested that TdcD is a dimer in solution.
Figure 4.3 SDS-PAGE analysis of TdcD during purification. Proteins were analyzed on 10% SDS-
PAGE and stained with Coomassie blue. Lane 1, crude cell lysates after 0.3 mM IPTG induction;
lane 2, clear supernatant; lane 3, purified TdcD after Ni-NTA affinity column chromatography;
lane M, molecular-weight markers (kDa).
4.3.2 Biochemical studies on TdcD
Enzymatic activity was measured by a coupled-enzyme assay based on the
measurement of ADP produced during the phosphorylation of propionate. Activity was
also monitored for acetate, an analog of propionate reported to be a phosphate acceptor
(Hesslinger et al. 1998). The KM value for ATP using the purified enzyme was 112
116

Chapter 4 Propionate kinase (TdcD)
±12 μM. The KM values for propionate and acetate were 2.33 ±0.35 mM and 26.92 ±
4.70 mM, respectively, showing that TdcD has about ten times higher affinity for
propionate than for acetate. The KM values of ATP and propionate in TdcD were
comparable to those of ATP and acetate in MAK determined using a similar coupled-
enzyme assay (Gorrell et al. 2005). In TdcD, kcat values for propionate (100 ± 8 s−1) and
acetate (125 ± 9 s−1) were similar. Comparison of kcat/KM values for propionate
(4.64 mM−1 s−1) and acetate (42.91 mM−1 s−1) shows clearly that TdcD is more specific
towards propionate than acetate. In the case of MAK, the KM value for acetate was
reported to be about ten times lower than that of propionate (Ingram-Smith et al. 2005).
These results demonstrate that propionate and acetate are the preferred substrates for
TdcD and MAK, respectively.
Recent studies have revealed a gene, pduW, encoding another propionate kinase
(PduW) in S. typhimurium, whose activity is important for the synthesis of propionyl-
CoA during growth on 1,2-propanediol (Palacios et al. 2003). In S. typhimurium, a cell
extract enriched for PduW and TdcD showed levels of propionate kinase activity ~11-
and 45-fold higher than background, respectively (Palacios et al. 2003). These two
proteins share 42% amino acid sequence identity. TdcD and PduW exhibit amino acid
sequence identities of 38% and 45%, respectively, with acetate kinase from
S. typhimurium. Figure 4.4 shows the alignment of the amino acid sequences of
propionate and acetate kinase enzymes exhibiting propionate/acetate kinase activity in
S. typhimurium and M. thermophila. TdcD and MAK share 38% amino acid sequence
identity, which suggested that the latter could serve as a good starting model for
determination of the structure of TdcD. Most of the residues involved in the substrate
binding and catalysis are well conserved in these enzymes. The presence of significant
levels of sequence identity and conservation of most of the active site residues among
these enzymes suggest similar structures and catalytic mechanisms.
4.3.3 Crystallization and structure determination
Attempts were made to crystallize S. typhimurium propionate kinase as well as its
complexes with the nucleotide or its analog. Diffraction-quality crystals were obtained
only for TdcD bound with ADP and AMPPNP. Crystals of TdcD-ADP and TdcD-
117

Chapter 4 Propionate kinase (TdcD)
AMPPNP complexes were obtained in crystallization condition containing 0.1 M Bis-
Tris (pH 6.5), 18% (w/v) polyethylene glycol monomethyl ether 2000 and in 0.1 M Bis-
Tris (pH 6.5), 35% (v/v) pentaerythritol ethoxylate (15/4 EO/OH), respectively (Fig. 4.5).
Diffraction data were collected to resolutions of 2.2 Å and 2.3 Å for TdcD-ADP and
TdcD-AMPPNP complexes, respectively.
Figure 4.4 Amino acid sequence alignment of propionate kinase and acetate kinase from
S. typhimurium and M. thermophila. The residues conserved in all four molecules are shown in
black boxes. TdcD_ST and PduW_ST denote propionate kinases encoded by TdcD and PduW in
S. typhimurium, whereas AK_ST and AK_MT denote acetate kinases present in S. typhimurium
and M. thermophila, respectively. The secondary structure elements of ADP bound TdcD are
represented at the top of the aligned sequences by arrows (β-strands) and spirals (310 and α-
helices). The red triangles below the aligned sequences show the probable residues involved in
substrate binding or catalytic mechanism.
118

Chapter 4 Propionate kinase (TdcD)
Figure 4.5 Crystals of S. typhimurium TdcD obtained in complex with ADP and AMPPNP.
The structure of TdcD-ADP complex was solved by molecular replacement using
M. thermophila acetate kinase (MAK-ATP) structure as the search model (Buss et al.
2001). The best molecular replacement solution was obtained using the B subunit of
MAK-ATP as a search model in the space group P3121. Since TdcD has an appreciable
level of sequence identity with MAK, further refinement of the structure was made
assuming that the molecular replacement solutions were correct. The initial 2Fo-Fc and
Fo-Fc maps showed reasonably good density for most of the regions, except for residues
ranging from 27 to 87 at the N-terminus. Improved electron density maps obtained after
building the rest of the model helped in tracing these missing residues in subsequent
rounds of model building. Further analysis carried out after the structure solution
suggested that these difficulties resulted from large differences in the organization of
domains between TdcD and MAK. Addition of ligand and water molecules to the model
resulted in further decrease in R and Rfree for the model. The final model of TdcD-ADP
has been refined to R and Rfree of 19.3% and 21.9%, respectively. The structure of TdcD
in complex with AMPPNP was solved using protein atoms of TdcD-ADP complex as the
119

Chapter 4 Propionate kinase (TdcD)
model, and was finally refined to R and Rfree of 19.7% and 22.8%, respectively (Table
4.1).
Table 4.1 Crystal parameters and statistics on data collection, refinement and model quality.
Values in parentheses correspond to the highest resolution shell.
AMPPNP bound TdcD ADP bound TdcD Crystal parameters Unit cell parameters (Å)
a = b 111.27 110.52 c 66.81 66.54
Space group P3121 P3121 Temperature (K) 100 100 Data collection Resolution range (Å) 35.0-2.3 (2.38-2.3) 35.0-2.2 (2.28-2.2) No. of monomers per AU 1 1 No. of reflections measured 699205 424150 No. of unique reflections 21467 24118 Data redundancy 32.57 17.58 I/σ(I) 23.37 (2.84) 21.55 (2.88) Completeness (%) 99.5 (96.7) 99.3 (93.4) Rmerge (%) 6.2 (45.8) 7.0 (40.1) Refinement R (%) 19.7 (29.9) 19.3 (30.4) Rfree (%) 22.8 (31.6) 21.9 (36.1) No. of atoms
Protein atoms 2934 2929 Solvent atoms 153 152 Ligand atoms 35 27
Model quality Average B factors (Å2)
Protein atoms 47.89 45.81 Water 46.39 45.67 Ligand 62.35 77.77
Coordinate error Luzzati (working/test) 0.26/0.31 0.25/0.29 Sigmaa (working/test) 0.33/0.37 0.33/0.35
RMS deviation from ideal values Bond length (Å) 0.006 0.006 Bond angle (deg.) 1.2 1.2 Dihedral angle (deg.) 22.8 22.7
Residues in Ramachandran plot (%) Most favored region 89.9 89.6 Allowed region 9.8 10.4 Generously allowed region 0.3 0.0
120

Chapter 4 Propionate kinase (TdcD)
4.3.4 Model quality
In both the ligand-bound TdcD structures, the electron density is of good quality
throughout the polypeptide chain, except in the residue ranges 39-42 and 49-58, where
poor density is observed. The electron density maps were not clear enough in these
locations to permit stereo-chemically reasonable interpretation of side-chains of most of
the residues. Therefore, most of the residues in this region have been truncated to alanine.
This region is fully exposed to the solvent and is expected to undergo a large movement
during substrate-induced domain closure, as seen in other members of the ASKHA
superfamily. Unlike MAK complexes (Buss et al. 2001; Gorrell et al. 2005), in liganded
TdcD structures, residues present in this region are not involved in crystal contacts. No
electron density could be observed for the N-terminal hexa-histidine tag. The initial three
residues at the N-terminus and final five residues at the C-terminus were not included in
the model due to the absence of well-defined electron density. These residues are
presumably disordered. Both the structures are well restrained to standard bond distances
and angles and the main-chain torsion angles correspond well with the expected values
(Table 4.1). The structures have around 89-90% of the residues in the most favored
regions of the Ramachandran map calculated with the program PROCHECK (Laskowski
et al. 1993). In both the structures, none of the residues is present in a disallowed region
and residue Asn48, which occurs in the generously allowed region in the TdcD-AMPPNP
complex, is in a region of poor electron density. 4.3.5 Overall structure of propionate kinase
One subunit of TdcD comprises of 14 β-strands, 15 α-helices and five 310 helices
(Fig. 4.6), as indicated by the program PROMOTIF (Hutchinson and Thornton 1996).
These elements of regular secondary structure comprise 62.1% of the polypeptide chain.
Each subunit comprises two domains of unequal size, a small N-terminal domain and a
large C-terminal domain, denoted as domains I and II, respectively. The propionate and
nucleotide-binding sites are present in a cleft between the two domains (Fig. 4.6). Both
the domains contain a core of secondary structure βββαβαβα (Fig. 4.7), which is similar
to that of acetate kinase/glycerol kinase/hexokinase/Hsc70/actin (Hurley 1996). These
proteins differ from each other in subdomains inserted between particular secondary
structural elements.
121

Chapter 4 Propionate kinase (TdcD)
(a) (b)
Figure 4.6 The tertiary structure of propionate kinase. (a) Ribbon diagram of a subunit of
propionate kinase bound with AMPPNP and ethylene glycol. The secondary structure elements
in the subunit are colored differently. (b) Surface representation of the TdcD-AMPPNP showing
the binding site of AMPPNP and ethylene glycol near the proposed binding site of propionate.
Positively charged residues are shown in blue, negatively charged residues are shown in red and
neutral residues are shown in white. The ligands AMPPNP and ethylene glycol present in the
active site cleft are drawn as ball and stick models.
The α-helices present at the C-terminal ends of both the domains extend and form
part of the other domain. Domain I comprises of a mixed eight-stranded β-sheet (β1, β2,
β3, β4, β5, βa, βb, βc) packed on one side by three α-helices (α1, α2, αa) and on the other
side by two small helices (αb, αc) as well as by the C-terminal helix α3* emanating from
domain II. The domain II comprises of a mixed six-stranded β-sheet (β1*, β2*, β3*, β4*,
β5*, βa*) and nine α-helices. The α-helix extending from domain I (α3) forms one side of
the central β-sheet, whereas the other side is covered by eight α-helices (αa*, αb*, αc*,
αd*, αe*, αf*, α1*, α2*), most of which constitute the dimer interface. Superposition of
all Cα atoms of TdcD complexed to ADP and AMPPNP using the program ALIGN
(Cohen 1997) gave an average rmsd of 0.15Å , indicating that there is no major
conformational change between these two TdcD complexes.
122

Chapter 4 Propionate kinase (TdcD)
Figure 4.7 Topology diagram of the secondary structural elements of TdcD. Arrows represent β-
strands, while small and large cylinders represent 310 and α-helices, respectively. β-strands and α-
helices, colored cyan and green, respectively, constitute the core secondary structure βββαβαβα
in both the domains, while insertions of sub-domains between particular secondary structural
elements are colored yellow. The core secondary structure elements βββαβαβα are numbered as
β1, β2, β3, α1, β4, α2, β5, α3 in domain I and as β1*, β2*, β3*, α1*, β4*, α2*, β5*, α3* in domain II.
Secondary structural elements present in the inserted subdomains are numbered as βa, βb and βc
for β-strands, αa, αb and αc for α-helices in domain I, and βa* for β-strand and αa*, αb*, αc*, αd*,
αe* and αf* for α-helices in domain II. The 310 helices are numbered as 3a, 3b, 3c, 3d, 3e and 3f.
Different biological functions performed by acetate kinase/glycerol
kinase/hexokinase/Hsc70/actin are mediated by insertions of distinct subdomains at
different topological positions. In the members of the ASKHA superfamily (Hurley
1996), the two core subdomains and the insertions together constitute each subunit.
According to the nomenclature first proposed for Hsc70 (Flaherty et al. 1990), in
domains I and II, the conserved subdomains are denoted as IA and IIA, and the divergent
subdomains are labeled as IB and IIB. Both the domains of propionate kinase, as in other
123

Chapter 4 Propionate kinase (TdcD)
members of the ASKHA superfamily, contain subdomains IB and IIB between the third
β-strand and the first α-helix of the core secondary structure βββαβαβα (Fig. 4.7). In
domain I, the insertion corresponding to subdomain IB is between β3 and α1, and
consists of two anti-parallel β-strands (βa and βb), which extend the central β-sheet. The
βb strand is short compared to that of acetate kinase and electron density for this strand is
poor. An insertion (subdomain IIB) at this position (between β3* and α1*) in domain II
comprises of four α-helices (αa*-αd*) constituting a major part of the dimer interface. An
insertion in domain I, which forms part of the dimer interface, occurs before α3 and
contains a few small helices. This insertion is unique to propionate and acetate kinases.
Another insertion that is unique to propionate and acetate kinases is between α2* and β5*
of domain II, which consists of an additional β-strand (βa*) extending the central β-sheet,
an α-helix (αf*) and two 310 helices (3e and 3f). As observed in acetate kinase, glycerol
kinase and hexokinase, an insertion in propionate kinase between β4 and α2 contains an
additional β-strand (βc), which borders the central β-sheet, and an additional α-helix (αa).
Analysis of the subunit packing within the crystal shows that one subunit is related to a
second subunit by a 2-fold crystallographic axis, forming a plausible dimer (Fig. 4.8).
These subunits are referred to as A and B.
Figure 4.8 The quaternary structure of homodimeric propionate kinase in complex with ADP,
with each subunit colored differently. The molecular 2-fold axis is along the plane of the paper.
The ADP present in the active site cleft is drawn as ball and stick model.
124

Chapter 4 Propionate kinase (TdcD)
4.3.6 Dimer interface
The crystal structures of liganded TdcD have revealed an interface characteristic
of a tight dimer. In each subunit, domain II contributes to a large part of the dimeric
association. Extensive complementarity exists in the surface features of the two subunits
at the dimer interface. Because the structure of TdcD dimer has a 2-fold axis of symmetry
that coincides with crystallographic symmetry, all interactions between the two subunits
occur in a symmetric fashion. Accessible surface area calculations (Hubbard and
Thornton 1993) show that a single subunit of TdcD in the TdcD-ADP complex structure
has an accessible surface area of 17,516.3 Å2. The total surface area buried on
dimerization is 3080.6 Å2 (17.6 %) per subunit, out of which 72.2% (2224.9 Å2) is non-
polar and 27.8% (855.6 Å2) is polar. Residues that mainly contribute to the interface are
113, 153-171, 214-272, 292-318 and 341-344. Atoms from 70 residues of one subunit
make hydrophobic and polar interactions (≤4.0 Å) with the atoms of the other subunit.
The dimer interface is extensive and predominantly non-polar (Fig. 4.9). A large number
of hydrophobic residues occur at the dimer interface. Polar interactions including salt
bridges and hydrogen bonds are present at the interface.
Figure 4.9 Electrostatic surface representation illustrating the charge distribution in TdcD at the
dimer interface.
125

Chapter 4 Propionate kinase (TdcD)
There are 4 salt bridges (cut-off value of 4.0 Å) and 31 hydrogen bonds (cut-off
value of 3.5 Å) across the dimer interface of TdcD. In contrast, at the same cut-off values,
there are 10 salt bridges and 26 hydrogen bonds across the dimer interface in MAK. As
seen in other thermostable proteins, large number of salt bridges present at the interface
in MAK structure may provide greater thermostability to the dimeric protein from the
thermophile. Salt bridges present at the dimer interface of TdcD are between Arg306 and
Asp220 and between Glu260 and Lys250. Equivalent residues at these positions in MAK
structure also form salt bridges. A few water molecules that are present at the dimer
interface contribute to the stability of TdcD dimer via water-mediated hydrogen bonds. 4.3.7 Nucleotide-binding site
Inspection of difference electron density map during refinement showed a strong
positive density near the expected nucleotide-binding site. As anticipated, AMPPNP and
ADP could be built into the electron density maps obtained for the corresponding
complexes of TdcD (Fig. 4.10 and 4.11). The adenine base is in the anti conformation
with respect to the ribose sugar. The sugar ring in both AMPPNP and ADP complexes is
in C2′-endo form with its O5′ in the trans, gauche conformation. The average B-factors
of the bound nucleotides AMPPNP and ADP, refined with full occupancy, are lower for
the adenine ring (54.2 Å2 and 70.4 Å2 for the AMPPNP and ADP complexes,
respectively) and the ribose sugar (60.0 Å2 and 76.0 Å2, respectively) and higher for the
phosphate groups in the order of B(α)< B(β)<B(γ). The average B-factors for the di- and
tri-phosphate moieties are 89.0 Å2 and 74.6 Å2 in the ADP and AMPPNP complex
structures, respectively. This pattern for atomic B-factors along the nucleotide molecule,
observed also in the case of MAK, can be interpreted as the result of increased flexibility
and correlates to the varying levels of solvent exposure and interactions with protein
atoms. In hexokinase, in the absence of glucose, the phosphate groups and the metal ion
of the metal-nucleotide complex are proposed to be disordered, possibly to reduce the
inherent ATPase activity of the enzyme (Steitz et al. 1981; Zeng et al. 1996).
126
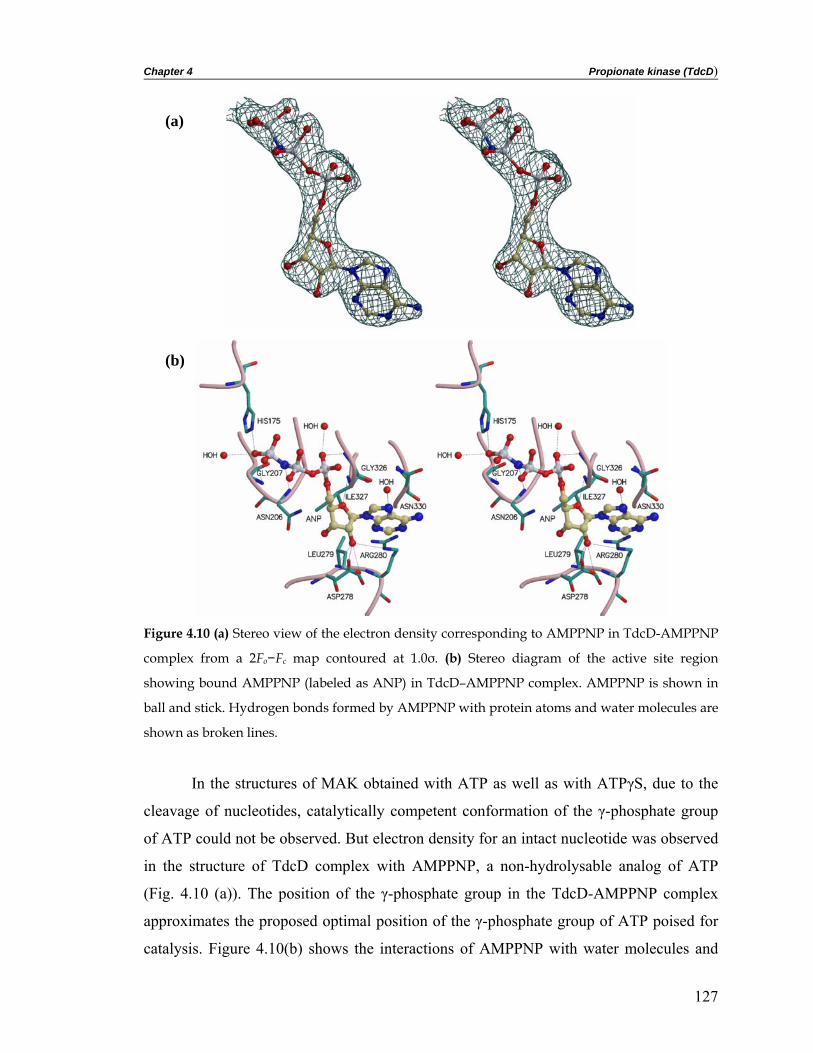
Chapter 4 Propionate kinase (TdcD)
(a)
(b)
Figure 4.10 (a) Stereo view of the electron density corresponding to AMPPNP in TdcD-AMPPNP
complex from a 2Fo−Fc map contoured at 1.0σ. (b) Stereo diagram of the active site region
showing bound AMPPNP (labeled as ANP) in TdcD–AMPPNP complex. AMPPNP is shown in
ball and stick. Hydrogen bonds formed by AMPPNP with protein atoms and water molecules are
shown as broken lines.
In the structures of MAK obtained with ATP as well as with ATPγS, due to the
cleavage of nucleotides, catalytically competent conformation of the γ-phosphate group
of ATP could not be observed. But electron density for an intact nucleotide was observed
in the structure of TdcD complex with AMPPNP, a non-hydrolysable analog of ATP
(Fig. 4.10 (a)). The position of the γ-phosphate group in the TdcD-AMPPNP complex
approximates the proposed optimal position of the γ-phosphate group of ATP poised for
catalysis. Figure 4.10(b) shows the interactions of AMPPNP with water molecules and
127

Chapter 4 Propionate kinase (TdcD)
protein residues. The adenine base is present in a hydrophobic pocket formed by residues
Ile327, Leu279, the aliphatic chain of Arg280 and Cβ of Asn330. The N7 and N6 nitrogen
atoms of the adenine ring make hydrogen bonds with water molecules. There is no direct
hydrogen bond between the adenine ring and the protein atoms. The C2′ oxygen atom of
the ribose ring is hydrogen bonded to oxygen atoms of the carboxyl group of conserved
Asp278 and the main-chain N atom of Leu279. The α-phosphate oxygen atom is bound to
the amide nitrogen atom of Gly326. Interaction of the α-phosphate oxygen atom with
equivalent glycine residues has been observed in other members of this superfamily. This
glycine residue is preceded by glycine in propionate kinases and alanine in acetate
kinases. The φ, ψ angles for the residue preceding the glycine residue, obtained from the
program PROCHECK (Laskowski et al. 1993), is found to fall in the epsilon region of
the Ramachandran plot. First observed in the structure of acetate kinase (Buss et al.
2001), all members of the ASKHA superfamily have this unusual conformation for the
residue that precedes the α-phosphate-bound glycine residue. The α-phosphate group is
also hydrogen bonded to a water molecule. The β-phosphate group of AMPPNP is
exposed to the solvent and only N forms a hydrogen bond with the main-chain amide of
Asn206, which is present in the turn between strands β1* and β2*. The γ-phosphate
group forms hydrogen bonds with His175 and a water molecule present above the side-
chain of conserved Arg236. Since the phosphate moiety of the nucleotide binds between
the two domains, the binding site is incomplete until the expected inter-domain motion
results in the closure of the domains.
In the structure of TdcD complexed with ADP, the nucleotide base and ribose are
positioned in the active site at the same place as seen for TdcD-AMPPNP complex
structure. However, substantial differences in the orientation of α- and β-phosphate
groups of ADP are apparent. The β-phosphate group of ADP points towards His203 and
Gly325 present in the domain II (Fig. 4.11). The α-phosphate group is hydrogen bonded
to the main-chain N atom of Gly326 and a water molecule. The β-phosphate group is
hydrogen bonded to a water molecule present nearby and there is no direct hydrogen
bonding with the protein atoms. Electron density for the β-phosphate group in ADP was
poor and it is probably oriented in a non-productive conformation (in view of the direct
in-line transfer mechanism), as in the case of the A subunit of all MAK complexes (Buss
128

Chapter 4 Propionate kinase (TdcD)
et al. 2001; Gorrell et al. 2005) except that the β-phosphate group of ADP points in the
opposite direction, towards the solvent-exposed region. The Mg2+ bound to nucleotide,
observed in structures of some of the members of this superfamily, was not seen in either
model of TdcD.
(a)
(b)
Figure 4.11 (a) Stereo view of the electron density corresponding to ADP in the TdcD–ADP
complex from a 2Fo−Fc map contoured at 1.0 σ. (b) Stereo diagram of the active site region
showing ADP bound to TdcD in the TdcD-ADP complex structure. ADP is shown in ball and
stick. Hydrogen bonds formed by ADP with protein atoms and water molecules are shown as
broken lines.
129

Chapter 4 Propionate kinase (TdcD)
4.3.8 Proposed propionate-binding site
Crystallization trials with propionate and ADP/AMPPNP showed no electron
density for the propionate. In the recently determined structure of MAK with the
components of the putative transition state analog containing AlF3, ADP and acetate
(MAK-AlF3) (Gorrell et al. 2005; Ingram-Smith et al. 2005), acetate is present in the
active site cleft formed by Val93, Leu122, Phe179 and Pro232. Surprisingly, ethylene
glycol used as the cryoprotectant during data collection of TdcD-AMPPNP crystal was
found to be present in the pocket, corresponding to acetate in MAK-AlF3 structure, which
is expected to be the binding site of propionate in TdcD. Figure 4.12 shows the
interactions of ethylene glycol (EDO) with the neighboring residues. The oxygen atoms
(O1 and O2) of ethylene glycol form hydrogen bonds with a water molecule and the NE2
atom of His118. Aliphatic carbon atoms of ethylene glycol are present in a hydrophobic
pocket formed by Ala88, Leu117, His118, Phe174 and Pro227.
Figure 4.12 Stereo diagram of the active site showing ethylene glycol (labeled as EDO) bound
near the proposed binding site for propionate in TdcD. The figure shows the electron density
corresponding to ethylene glycol and a water molecule from a 2Fo-Fc map contoured at 1.0 σ.
Ethylene glycol is shown in ball and stick. Hydrogen bonds between EDO and nearby atoms are
shown as broken lines.
However, electron density for the side-chain of the conserved Leu117 (not shown
in Fig. 4.12) is absent in both the models. Interestingly, Val93 of MAK is replaced by
130

Chapter 4 Propionate kinase (TdcD)
Ala88 in TdcD to accommodate the bulkier propionate. The hydrophobic binding pocket
is proposed to accept the aliphatic group of the propionate and hence orient the
phosphoryl acceptor oxygen atom of the carboxyl group at the mouth of the hydrophobic
pocket pointing towards the position of the terminal γ-phosphate seen in the TdcD-
AMPPPNP complex. A less bulky residue, glycine (Gly76), is present in all the butyrate
kinase sequences, and a more bulky group, valine (Val83), is found in all acetate kinases
at positions equivalent to Ala88, present at the bottom of the hydrophobic pocket in
propionate kinase (Fig. 4.13). These observations, along with the acetate-binding pocket
in MAK-AlF3 structure (Ingram-Smith et al. 2005), suggest that the size of the
hydrophobic pocket is a determinant of substrate specificity in acetate, propionate and
butyrate kinases. The lower KM value for propionate than for acetate in TdcD can be
explained on the basis of these differences in the size of hydrophobic pocket.
Acetate kinase Val 93
Acetate
Propionate kinaseAla 88
EDO
Butyrate kinaseGly 76
(a) (b) (c)
Figure 4.13 Substrate binding pocket in the acetate, propionate and butyrate kinases. (a) Acetate
bound in the M. thermophila acetate kinase, (b) ethylene glycol present in the proposed binding
pocket of propionate in S. typhimurium propionate kinase and (c) proposed butyrate binding
pocket in Thermotoga maritima butyrate kinase. Val93 in acetate kinase, Ala88 in propionate kinase
and Gly76 in butyrate kinase are expected to provide specificity in these enzymes for acetate,
propionate and butyrate, respectively.
131

Chapter 4 Propionate kinase (TdcD)
4.3.9 Five conserved motifs and proposed catalytic residues
Even though propionate and acetate kinases show no overall significant sequence
similarity with other ASKHA phosphotransferases (Bennett and Steitz 1980; Bystrom et
al. 1999; Flaherty et al. 1990; Kabsch et al. 1990), they share five motifs (Fig. 4.14)
composed of secondary structural elements and conserved amino acid residues involved
in catalysis, nucleotide and metal binding. These five conserved motifs, first identified by
Bork et al. (Bork et al. 1992), correspond to two phosphate (phosphate 1 and phosphate
2) and one base (adenosine) binding regions and two other regions connecting domain I
and domain II (connect 1 and connect 2). Each of these five motifs was deduced on the
basis of structure-based multiple sequence alignment and plausible roles of amino acids
present at particular positions in the well-characterized members of the ASKHA
superfamily (Fig. 4.14).
Phosphate 1 Phosphate 2 Adenosine Connect 1 Connect 2
TdcD 8 LVINCGSSSIKFSVL 198 LIVAHLGNGASICAV 319 GIIFTGGIGENSVLI 138 QVAVFDTSFHQTLAP 381 EEKMIALDAIHLGNV
MAK 4 LVINAGSSSLKYQLI 204 IITCHLGNGSSITAV 325 AVVFTAGIGENSASI 143 MVIVFDTAFHQTMPP 384 EELAIARETKEIVET
HK 83 LAIDLGGTNLRVVLV 228 KMGVIFGTGVNGAYY 413 HIAADGSVYNRYPGF 206 VALINDTTGTLVASY 458 DGSGAGAAVIAALAQ
GK 7 VALDQGTTSSRAVVM 260 MAKNTYGTGCFMLMN 406 LRVDGGAVANNFLMQ 240 SGIAGDQQAALFGQL 437 EVTALGAAYLAGLAV
Actin 8 LVCDNGSGLVKAGFA 150 GIVLDSGDGVTHNVP 296 NNVMSGGTTMYPGIA 132 MYVAIQAVLSLYASG 337 YSVWIGGSILASLST
Hsc70 7 VGIDLGTTYSCVGVF 195 VLIFDLGGGTFDVSI 333 DIVLVGGSTRIPKIQ 170 LRIINEPTAAAIAYG 367 EAVAYGAAVQAAILS
Figure 4.14 Alignments of the five putative motifs based on structural equivalence in the well-
characterized members of the ASKHA superfamily. Residues shown in bold in each motif
correspond to either the conserved catalytic residues or those that have a structurally equivalent
role in metal/nucleotide binding. TdcD, propionate kinase from S. typhimurium; MAK, acetate
kinase from M. thermophila; HK, hexokinase from yeast; GK, glycerol kinase from E. coli; actin,
actin from rabbit skeletal muscle; Hsc70, heat shock cognate 70 from Bos taurus.
4.3.9.1 Phosphate 1 and phosphate 2 motifs
The phosphate 1 motif formed by residues present in the turn between the β1- and
β2-strands of subdomain Ia plays a role in nucleotide binding by interacting with the β-
phosphate group. The primary structure of this turn is nearly identical in most of the
members of this superfamily. The conserved Asp/Asn present in the β1-strand has been
shown to interact with the Mg2+ bound to ATP. Besides this, a basic residue from the β2-
strand binds the β-phosphate group of ADP in glycerol kinase (Arg17) and actin (Lys18).
Even in the TdcD-AMPPNP structure, the closest residue from the phosphate 1 motif to
132

Chapter 4 Propionate kinase (TdcD)
the nucleotide is Lys14 in the β2-strand, which is expected to stabilize the β-phosphate
group of the nucleotide upon domain closure. The phosphate 2 motif, consisting of
residues present in β1*-β2* turn in subdomain IIa, interacts with the γ-phosphate group
of the nucleotide via main-chain amides. The residues that form this motif are DXG
(where X is any amino acid) in actin/Hsc70, GTG in hexokinase/glycerol kinase and
GNG in acetate and propionate kinases. Conserved residues present in this motif in TdcD
complexes bind to α- and β-phosphate groups of the nucleotide.
4.3.9.2 Adenosine motif
The adenosine motif contains conserved glycine residues in the ASKHA
superfamily, including TdcD. The adenosine base is bound by diverse interactions. It is in
the anti conformation, as seen in most of the enzyme-bound nucleotides, and binds
primarily through hydrophobic interactions with residues only from domain II. The N6
atom of the adenine ring is solvent-exposed in most of the members of this superfamily.
4.3.9.3 Connect 1 and connect 2 motifs
In connect 1 and connect 2 motifs, conserved alanine and glycine residues have
been proposed to form a close contact between these two motifs. Residues present in
these two motifs have been shown to have properties appropriate for an interdomain
hinge. In TdcD also, residues present in these two motifs were found to be a part of the
hinge bending residues. In the connect 1 motif of acetate kinase, conserved Asp148 was
found to be essential for activity (Miles et al. 2001). The carboxyl group of the equivalent
aspartate residue in members of the ASKHA superfamily is proposed to function in base
catalysis or metal binding. The equivalent residue in TdcD is also aspartate (Asp143) and
is expected to play a similar role in catalysis. Glu384 present in the connect 2 motif was
implicated in domain movement or catalysis in acetate kinase (Singh-Wissmann et al.
1998). The presence of an equivalent residue (Glu/Asp) in other members of the ASKHA
superfamily including TdcD (Glu381) suggests that it probably plays a similar role in
domain movement or catalysis.
133

Chapter 4 Propionate kinase (TdcD)
4.3.10 Role of conserved residues
Earlier work carried out on the members of the ASKHA superfamily, mainly
acetate kinase, has been taken into consideration to discuss the role of proposed catalytic
residues in TdcD. The kinetics and various site-directed mutagenesis experiments have
led to the identification of catalytically essential residues in MAK (Ingram-Smith et al.
2000; Miles et al. 2001; Singh-Wissmann et al. 1998; Singh-Wissmann et al. 2000). The
equivalent residues in the case of propionate and butyrate kinases were found to be
largely conserved. The equivalent catalytic residues in propionate kinase are Asn11,
Arg86, Asp143, His175, Arg236 and Glu381. Previous studies on acetate kinase have
suggested that phosphoryl transfer occurs either by a direct in-line transfer (Blattler and
Knowles 1979) or by a covalent triple displacement mechanism involving two
phosphoenzyme intermediates (Spector 1980). In the structure of acetate kinase with
putative transition state components AlF3, ADP and acetate (Gorrell et al. 2005), close
proximity of the carboxyl group of acetate and the β-phosphate group of ADP to AlF3,
which seems to mimic the meta-phosphate intermediate, provided strong support for
direct in-line phosphoryl transfer. Because of the similarities in the architecture of the
active site, substrate structure and chemistry performed, it might be appropriate to assume
that propionate and acetate kinases have similar catalytic mechanisms. Structural details
obtained from the TdcD complex structures indicate that once the substrate binds in the
cleft, the proposed substrate-induced domain movement occurs, which positions the
reactants, brings the active site residues present in both the domains close to the reactants,
and shields the active site pocket from the surrounding solvent. In parallel with the direct
in-line transfer reaction mechanism proposed for the acetate kinase (Gorrell et al. 2005),
in propionate kinase, Arg86 and Arg236 probably interact with the carboxyl group of
propionate, while Asn11 and Glu381 interact with magnesium bound to ATP in a bi-
dentate coordination. Evidence from the structure of MAK-AlF3 suggests that His175 and
Arg236 mainly provide stabilization of the transition state in TdcD.
4.3.11 Analysis of the domain movement
All well-characterized members of this superfamily, such as hexokinase (Bennett
and Steitz 1980), glycerol kinase (Bystrom et al. 1999), Hsc70 (Harrison et al. 1997) and
134

Chapter 4 Propionate kinase (TdcD)
actin (Page et al. 1998), undergo a large conformational change, resulting from inter-
domain motion during the reaction cycle. The domain movement in TdcD has been
analyzed by comparison with the subunit structures of the homologous acetate kinase.
However, precise analysis of the domain movements is not possible because of
substitutions, insertions and deletions in the primary structure of the proteins. Knowledge
of the structure of TdcD in the absence of any ligand or in complex with propionate could
help to confirm the deduced domain movements, but suitable crystals are not available.
As seen in the structure of glycerol kinase in complex with the non-hydrolysable ATP
analog β,γ-difluoromethylene adenosine 5′-triphosphate (AMPPCF2P) (Bystrom et al.
1999), an interesting observation in all crystal structures of MAK with various
nucleotide/ligand complexes (Buss et al. 2001; Gorrell et al. 2005) is that the dimer in the
crystal is asymmetric, with subunit A in a relatively closed conformation and the second
subunit B in an open conformation. Here, open and closed conformations of domains
have been defined on the basis of the distances between the residues present in domain I
and domain II at the farthest point from the hinge residues. In the two subunits of the
MAK-ATP complex structure (Buss et al. 2001), ADP and SO4 are bound in different
orientations, and the terminal β-strand (βb) of the central β-sheet present in domain I
interacts with the same β-strand of a symmetry-related molecule. In TdcD, the subunits
are related by a crystallographic 2-fold axis. Thus, no difference between the subunits
could exist. The moving domain in the liganded TdcD structures is not involved in crystal
packing interactions. The evidence currently available from proteins that undergo
significant domain motion seems to suggest that both open and closed states are only
slightly different in energy, and they are in dynamic equilibrium at room temperature.
Therefore, even relatively weak crystal packing forces can stabilize various possible
structures. Like glycerol kinase (Bystrom et al. 1999), acetate and propionate kinase
structures differ in the organization of domains, even though all the structures were
obtained in the presence of nucleotides. This suggests that in TdcD and MAK, binding of
propionate/acetate deeper inside the cleft or a later catalytic step rather than nucleotide
binding might be required for complete closure of the domains. Although the actual
driving force for domain motion remains to be resolved, the cleft between the two
135

Chapter 4 Propionate kinase (TdcD)
domains is expected to close upon propionate/acetate binding, leading to the positioning
of both the substrate and the nucleotide in orientations optimal for catalysis.
The programs DynDom (Hayward and Lee 2002) and DOMOV
(http://bioinfo1.mbfys.lu.se/cgi-bin/Domov/domov.cgi) have been used to analyze
differences in the organization of domains in the structures of TdcD and MAK.
According to the results of DynDom, in TdcD and MAK, the moving domain (domain I),
the fixed domain (domain II) and the hinge residues present at the interface between the
two domains comprise of several segments of non-sequential residues (Table 4.2). The
residues that comprise the fixed and moving domains and the hinge region are largely
equivalent in propionate and acetate kinases. In the A and B subunits of the MAK-ATP
structure, the motion of the moving domain is a hinge rotation of 27° and a translation of
-0.8 Å relative to the fixed domain (Fig. 4.15). The ratio of inter-domain to intra-domain
displacement is 3.0 for the A and B subunits of the MAK-ATP structure.
Figure 4.15 Structural superposition of the A and B subunit of MAK-ATP structure showing the
movement of domain I (moving domain) relative to the domain II (fixed domain). Inter-domain
rotation axis within the A and B subunits of the MAK–ATP complex structure has been
determined by the program DynDom (Hayward and Lee 2002). Arrow indicates the direction of
rotation of the moving domain by the right-hand rule.
136

Chapter 4 Propionate kinase (TdcD)
(a) (b)
(c)
(d)
Figure 4.16 Structural superposition of the A subunit of TdcD and the (a) A (b) B and (c) A and B
subunits of the MAK–ATP structure, showing the movement of domain I (moving domain)
relative to the fixed domain (domain II). The A subunit of TdcD and the A and B subunits of the
MAK–ATP structure are shown in purple, yellow and green, respectively. (d) Structural
superposition of the moving domain and hinge residues of the A subunit of TdcD and the A and
B subunits of the MAK–ATP structure, indicating that the domain motion is a result of rigid body
movement across hinge residues (shown in red).
137

Chapter 4 Propionate kinase (TdcD)
Structural superposition of both A and B subunits of MAK-ATP and A subunit of
TdcD-ADP show that domain I in TdcD structure occupies an intermediate position
compared to its position in A and B subunits of the MAK-ATP structure (Fig. 4.16). In
TdcD, the domain motion corresponds to a rotation of 20.95° and a translation of -0.13 Å
relative to the A subunit of the MAK-ATP structure and a rotation of 20.81° and a screw
translation of 0.06 Å relative to the B subunit of the MAK-ATP structure. Superposition
of the Cα atoms of domain I/domain II of TdcD individually with the respective domains
of the A and B subunits of MAK-ATP using the program ALIGN (Cohen 1997) gave
rmsd values ranging from 0.8 Å to 1.1Å, indicating that domain repositioning is a result
of rigid body movement (Fig. 4.16(d)). These finding suggest that the conformation
assumed by domains I and II in the nucleotide-bound structure of TdcD might represent
an intermediate point in the pathway of domain closure.
Table 4.2 Residue range in the fixed and moving domains and in the hinge regions of TdcD and
MAK.
TdcD Residues
Fixed domain: 86-96, 101-123, 142-382
Moving domain: 4-85, 97-100, 124-141, 383-395
Hinge region: 84-86, 96-101, 123-124, 141-142, 382-383
MAK Residues
Fixed domain: 91-101, 106-128, 147-385
Moving domain: 3-90, 102-105, 129-146, 386-396
Hinge region: 89-91, 101-106, 128-129, 146-47, 385-386
In both the liganded TdcD structures, two stretches of residues have relatively
high B-factors and poor electron density compared to the rest of the polypeptide, which is
indicative of intrinsic structural mobility of these residues. When mapped on the tertiary
structure, these residues were found to be present at the edge of domain I and II away
from the hinge region, which is indicative of domain motion in TdcD (Fig. 4.17).
138

Chapter 4 Propionate kinase (TdcD)
Figure 4.17 Mapping the high B-factor regions observed in propionate kinase structures on the
tertiary structures of propionate and acetate kinases.
The expected domain motions in propionate kinase and acetate kinase are larger
compared to the values reported for glycerol kinase, hexokinase, Hsc70 and actin. The
superposition of glycerol kinase (Bystrom et al. 1999) domains is achieved by a rotation
of 6.0° and a 0.2 Å translation along a unique axis compared to a 12° rotation and a 0.3Å
translation for hexokinase (Bennett and Steitz 1980; Bystrom et al. 1999). The hinge
motions between the major domains of actin and Hsc70 were found to be ~9.6° and 14°,
respectively (Harrison et al. 1997; Page et al. 1998). The domain motions observed in the
structures of propionate kinase and acetate kinase, which need to be corroborated by
further studies, is another crystallographic example of domain movement observed in
proteins of the ASKHA superfamily.
4.4 Conclusions The structures of TdcD in complex with ADP and AMPPNP are the first of
propionate kinase structures, and the first structural description for an acetate kinase
homologue from a mesophilic organism. The overall structures of TdcD complexes are
similar to the structures of acetate kinase solved with various ligands. However, structural
comparisons of TdcD and MAK complexes revealed a few interesting differences. The
major difference was observed in the arrangement of the two domains and is indicative of
139

Chapter 4 Propionate kinase (TdcD)
the large domain motion in these two kinases. The domain movement is known to play a
role in catalysis and resembles to those of the other members of the ASKHA superfamily,
where also domain movement is required to bring the active site residues from the
moving domain into close proximity to the substrate, position the reactants and shield the
reaction intermediates from the surrounding solvent. The other difference observed in the
TdcD-AMPPNP complex structure is the presence of intact AMPPNP. This is in contrast
to MAK, where the structure with ATP as well as the non-hydrolysable analog ATPγS
yielded cleaved nucleotides. The structure of TdcD-AMPPNP approximates the optimal
position of the γ-phosphate group poised for catalysis. The cryoprotectant ethylene glycol
observed in the proposed propionate-binding site in the AMPPNP-bound TdcD complex
structure probably reflects the mode of propionate binding. The differences in the size of
the hydrophobic pocket that binds the substrate, particularly the replacement of Val93 of
MAK by Ala88 in TdcD, could account for the observed greater affinity of TdcD towards
propionate rather than acetate. The larger numbers of salt bridges present at the dimer
interface of MAK explains the integrity of the dimeric enzyme at the high growth
temperature of the thermophile. Most of the catalytically essential residues identified
using kinetics and mutagenesis experiments in acetate kinase were found to be conserved
in propionate kinase, suggesting a similar active site pocket and catalytic mechanism for
both the enzymes.
140

Crystal structures of Salmonella typhimurium propionate kinase and its complex with Ap4A: Crystallographic
evidence for novel Ap4A synthetic activity
5

Chapter 5 Propionate kinase in complex with Ap4A 5.1 Introduction
L-threonine is anaerobically degraded to propionate via 2-ketobutyrate by
biodegradative threonine deaminase, 2-ketobutyrate formate lyase, phosphotransacetylase
and propionate kinase (Hesslinger et al. 1998). The last step in this process, conversion of
propionyl phosphate and ADP to propionate and ATP is catalyzed by propionate kinase
(TdcD; EC 2.7.1.15). As described in the previous chapter, propionate kinase from
Salmonella typhimurium has been cloned and overexpressed in Escherichia coli and the
crystal structures of the recombinant enzyme in complex with ADP and non-hydrolysable
ATP analog AMPPNP have been determined. The polypeptide fold of TdcD contains a
core similar to those of acetate and sugar kinase, Hsc70 and actin, the ASKHA
superfamily of phosphotransferases. This superfamily is characterized by two domains,
each with the topology βββαβαβα separated by an active site cleft (Hurley 1996). In the
members of this superfamily, phosphoryl transfer is coupled with a large conformational
change in which the two domains close around the active site pocket. The structures of
TdcD complexes have permitted the identification of catalytically essential residues
involved in the substrate binding and catalysis. The structure of TdcD-AMPPNP revealed
the optimal position for the γ-phosphate poised for catalysis. The cryoprotectant ethylene
glycol was observed in the proposed propionate binding site reflecting the mode of
propionate binding. The present work provides crystallographic evidence that the
propionate kinase from S. typhimurium can synthesize diadenosine 5',5'''-P1,P4-
tetraphosphate (Ap4A) from ATP. Crystals of TdcD obtained in the presence of ATP
clearly showed Ap4A bound to the active site pocket. Further co-crystallization trials of
TdcD with commercially available Ap4A confirmed its binding to the enzyme.
5.1.1 Ap4A and other related dinucleotide polyphosphates
Dinucleoside 5,5-P1,Pn-polyphosphates (NpnN’s; where N and N’ are nucleosides
and n represents the number of phosphate residues in the polyphosphate chain that links
N with N’) have been found in various organisms (Garrison and Barnes 1992) (Fig. 5.1).
Their normal, submicromolar levels increase dramatically during cellular stress, reaching
submillimolar concentration in some cases (Coste et al. 1987; Lee et al. 1983; Palfi et al.
1991). Ap3A and Ap4A appear to be the most prominent but Ap5A (Pintor et al. 1992a)
142

Chapter 5 Propionate kinase in complex with Ap4A and Ap6A (Pintor et al. 1992b) as well as Ap7A (Jankowski et al. 1999; Luo et al. 1999)
have been detected in some mammalian cells. Ap4A and Ap3A are generally present in
prokaryotic and eukaryotic organisms at basal concentrations of 10-8 to 10-6 M. They are
present in the secretory granules of platelets, adrenal chromaffin cells and some neurons
at higher concentrations (Garrison and Barnes 1992). Ap5A and Ap6A have also been
reported to be present in some of these cell types.
Figure 5.1 Structure of diadenosine 5’,5”‘-P1, P4-tetraphosphate (reproduced from Guranowski 2003).
Ap4A is predominantly a by-product of protein synthesis, specifically by
aminoacyl-tRNA synthetases (Goerlich et al. 1982). Synthesis of Ap4A was first
demonstrated during protein synthesis in a backward reaction consisting of ATP, Mg2+,
L-lysine and purified E. coli lysyl-tRNA synthetase (Zamecnik et al. 1966). This
dinucleotide is produced when ATP replaces PPi in the back reaction of amino acid
activation and is adenylylated by the enzyme (Fig. 5.2). In fact, it has been found that
many nucleoside 5'-di- and 5'-triphosphates can be adenylylated when they substitute for
PPi in this reversible pyrophosphate exchange catalyzed by the synthetase (Randerath et
al. 1966; Rapaport et al. 1975; Zamecnik 1983). A variety of aminoacyl-tRNA
synthetases from both prokaryotic and eukaryotic cells can synthesize Ap4A in vitro,
although some tRNA synthetases may not have this capability (Goerlich et al. 1982;
Plateau et al. 1981; Zamecnik 1983).
143

Chapter 5 Propionate kinase in complex with Ap4A
Figure 5.2 Synthesis of adenylated nucleotides by aminoacyl-tRNA synthetases in the absence of
cognate tRNA (reproduced from Lee et al. 1983).
In vitro studies have suggested that the following enzymes may be responsible for
the formation of the adenine-containing NpnN’s in vivo (reviewed in Guranowski 2003):
certain aminoacyl-tRNA synthetases (EC 6.1.1.X) (Zamecnik et al. 1966),
Ap4A phosphorylase (EC 2.7.7.53) (Guranowski et al. 1988),
firefly luciferase (EC 1.13.12.7) (Guranowski et al. 1990),
acyl-CoA synthetase (EC 6.2.1.8) (Fontes et al. 1998),
HIV-1 reverse transcriptase (EC 2.7.7.49) (Meyer et al. 1998),
DNA ligases (EC 6.5.1.1) (Gunther et al. 2002; Madrid et al. 1998),
RNA ligase (EC 6.5.1.3) (Atencia et al. 1999),
non-ribosomal peptide synthetase (Dieckmann et al. 2001) and
coumarate-CoA synthetase (EC 6.2.1.2) (Pietrowska-Borek et al. 2003).
These enzymes are particularly effective in the synthesis of dinucleoside
tetraphosphates Np4N’s: diadenosine tetraphosphate (AppppA or Ap4A) and diguanosine
tetraphosphate (GppppG or Gp4G), respectively. They are also able to synthesize NpnN’s
in which n>4. Not all of them, however, can produce Np3N’s. Johnstone and Farr used a
144

Chapter 5 Propionate kinase in complex with Ap4A radio/affinity labeled Ap4A analog ([32P]-8-azido-Ap4A) to identify at least twelve E. coli
Ap4A binding proteins in the cell lysate. These included stress proteins GroEL (Cpn60),
DnaK (Hsp70) and ClpB (Hsp100) (Johnstone and Farr 1991).
tRNA
aa-tRNA + AMP + aminoacyl-tRNA synthetase
Figure 5.3 Specific and non-specific enzymes involved in the synthesis and degradation of Ap4A
(reproduced from Baxi and Vishwanatha 1995).
5.1.2 Enzymes specific for Ap4A
The levels of Ap4A inside the cell can be precisely regulated by numerous
enzymes present in the cell (Guranowski 2000) (Fig. 5.3). In addition to the non-specific
ones, there are a few specific enzymes which act on Ap4A, which are discussed below:
Symmetric Ap4A hydrolase (EC 3.6.1.17): All prokaryotes studied so far
possess an Ap4A (symmetrical) pyrophosphohydrolase of ~33 kDa
(Guranowski et al. 1983; Plateau et al. 1985).
Ap4A → ADP + ADP
145

Chapter 5 Propionate kinase in complex with Ap4A
Asymmetric Ap4A hydrolase (EC 3.6.1.17): Higher eukaryotes (vertebrates,
invertebrates and plants) have a small, 17-21 kDa Ap4A (asymmetrical)
pyrophosphohydrolase (Cameselle et al. 1982; Jakubowski and Guranowski
1983; Prescott et al. 1989).
Ap4A → ATP + AMP
A reversible Ap4A phosphorylase (EC 2.7.7.53): Among the lower
eukaryotes, however, the phylogenetic picture is not very clear: the acellular
slime mould Physarum polycephalum has a symmetrical hydrolase (Barnes
and Culver 1982), the fission yeast Schizosaccharomyces pombe has a dimeric
(49 kDa) asymmetrical hydrolase (Robinson et al. 1993), and the budding
yeast Saccharomyces cerevisiae and the photosynthetic protozoan Euglena
gracilis have a quite distinct enzyme, a reversible Ap4A phosphorylase
(Guranowski and Blanquet 1985; Guranowski et al. 1988).
Ap4A + Pi ↔ ATP + ADP
S. cerevisiae has in fact two phosphorylases (I and II, both 36-38 kDa),
both of which have been cloned and shown to be distinct gene products,
sharing 60 % sequence identity (Plateau et al. 1990). These enzymes are
reversible and capable of a low rate of Ap4A synthesis at physiological pH
(Brevet et al. 1987). The phylogenetic and functional significance of this
cellular variation in the enzymes of Ap4A catabolism is not yet clear. Ap4A
phosphorylase is expected to be a valuable candidate for the production of
Ap4A in vivo. Ap4A phosphorolytic activity has not been detected in
prokaryotes so far.
In the recent past, an asymmetrically-acting Ap4A hydrolase related to the higher
eukaryotic enzyme has been detected in several bacteria (Bessman et al. 2001; Cartwright
et al. 1999; Conyers and Bessman 1999; Lundin et al. 2003). The asymmetrical Ap4A
hydrolases belong to the “nudix” protein family, comprising enzymes that hydrolyze
nucleotides in which a nucleoside diphosphate is attached to one of various groups
146

Chapter 5 Propionate kinase in complex with Ap4A assigned as x (Bessman et al. 1996; Bessman et al. 2001). These nudix proteins have a
conserved amino acid sequence that directly participates in catalysis (Harris et al. 2000;
Maksel et al. 2001). A search for nudix proteins in various genomes has led to the
discovery of hydrolases that prefer Ap5A and/or Ap6A as substrates in budding yeast (S.
cerevisiae) (Cartwright and McLennan 1999), fission yeast (Schizosaccharomyces
pombe) (Ingram et al. 1999) and humans (Safrany et al. 1999).
5.1.3 Probable role of Ap4A
In many cases, the concentrations of Ap4A and related dinucleotide
polyphosphates such as Ap4G and Ap3G increase after exposure of cells to various forms
of metabolic stress such as heat, oxidative, nutritional and DNA damage. Therefore, they
have been implicated in the regulation of cellular response to such stresses (Baker and
Jacobson 1986; Bochner et al. 1984; Farr et al. 1989; Gilson et al. 1988). Other evidence
has supported a role of Ap4A in DNA replication in eukaryotes (Vishwanatha and Wei
1992; Zamecnik 1983). Studies involving Ap4A have also indicated its role in cell
proliferation, DNA repair, cardiovascular system, neurotransmission and apoptosis (Baxi
and Vishwanatha 1995; Flores et al. 1999; Grummt et al. 1979; Nishimura 1998;
Rapaport and Zamecnik 1976; Vartanian et al. 1999). However, definitive role of Ap4A
in these situations is still unknown.
5.1.4 Ap4A in Salmonella typhimurium
Studies carried out by Lee et. al., showed that the bacterium Salmonella
typhimurium accumulates a family of adenylylated nucleotides at intracellular
concentrations of up to 100 µM following exposure to a bacteriostatic quinone, ACDQ
(6-amino-7-chloro-5,8-dioxoquinoline) but not under variety of other metabolic stresses
(Bochner et al. 1984; Lee et al. 1983). Characterization of these compounds established
the structures of the two major nucleotides as Ap4A (AppppA) and Ap3Gp2 (ApppGpp).
Preliminary evidence indicates that the other compounds are Ap4G (AppppG), Ap3G
(ApppG), and Ap3A (ApppA). The induced intracellular concentrations of Ap4A and the
other adenylylated nucleotides in S. typhimurium were found to be approximately 100-
fold higher than those found in eukaryotic cells. Based on these experiments, it was
147

Chapter 5 Propionate kinase in complex with Ap4A proposed that these dinucleotides are alarmones, signal molecules, such as ppGpp and
cAMP, which alert the cell to the onset of particular metabolic stresses (Lee et al. 1983;
Varshavsky 1983).
In this chapter, the crystal structures of native TdcD and its complexes with Ap4A
obtained after co-crystallization with ATP as well as Ap4A are presented. Structure of
unliganded TdcD has provided the information about the conformational change which
takes place in the active site pocket upon ligand binding. The present work provides
crystallographic evidence for a novel Ap4A synthetic activity of TdcD. Structure of TdcD
in complex with Ap4A tends to support direct in-line transfer of phosphoryl group during
the kinase activity.
5.2 Materials and methods 5.2.1 Crystallization and data collection
TdcD from Salmonella typhimurium was cloned in pRSET C vector and over-
expressed in E. coli BL21(DE3)pLysS along with an N-terminal hexa-histidine tag. The
recombinant protein was purified to homogeneity using Ni-NTA affinity column
chromatography. The protein-ligand complexes were prepared by incubating 10 mg/ml of
the protein with the ligand in the molar ratio of 1: 50 at 4°C for at least 6 h. The ligands
(ATP or Ap4A) used for the crystallization were purchased from Sigma Chemical Co.
Crystallization experiments were carried out by the hanging drop vapour-diffusion as
well as microbatch methods. Variations of crystallization conditions, in which crystals of
TdcD-ADP and TdcD-AMPPNP complex were obtained, were used for crystallization
trials. Efforts to obtain crystals of unliganded TdcD were successful using microbatch
method after repeated trials. Diffraction data for native TdcD and with Ap4A complex
were collected at 100K to resolutions of 2.6 Å and 2.4 Å, respectively, using 20% (v/v)
ethylene glycol as a cryoprotectant using a MAR345 image plate detector system
mounted on a Rigaku RU200 X-ray generator equipped with a 300 μm focal cup. A
complete dataset to a resolution of 2.0 Å was collected on a TdcD crystal obtained in the
presence of ATP on beamline BL26B1 at SPring-8, Hyogo, Japan. The data were
processed and scaled using the program DENZO and SCALEPACK of the HKL suite
(Otwinowsky 1997).
148

Chapter 5 Propionate kinase in complex with Ap4A 5.2.2 Structure determination and refinement
Structures of native TdcD and its complex with ATP and Ap4A were determined
using the atomic coordinates of protein atoms from a monomer of TdcD-ADP structure
by difference Fourier analysis. Randomly chosen subsets of 5% of the reflections were
reserved for the calculation of Rfree. The 2Fo–Fc map calculated after initial restrained
refinement using REFMAC5 (Murshudov et al. 1997) showed good fit for the majority of
the main chain. This was followed by model building and restrained refinement using
COOT (Emsley and Cowtan 2004) and REFMAC5 (Murshudov et al. 1997), until R and
Rfree converged. Ambiguities in the model were resolved by setting the occupancies of the
atoms concerned to zero until the density could be interpreted clearly. In the final stages
of refinement, inspection of difference electron density maps of complexes showed
strong positive density for the ligand in the active site pocket. In the TdcD dataset
obtained in the presence of ATP, difference Fourier computed at this stage showed
continuous density beyond γ-phosphate of ATP. Careful examination of this extra
electron density suggested that the density fits a molecule of diadenosine tetraphosphate
(Ap4A) very well. In the TdcD dataset obtained in the presence of Ap4A (TdcD-Ap4A),
difference Fourier clearly revealed Ap4A in the active site pocket. Electron density
corresponding to ethylene glycol was observed near the active site pocket of TdcD
structures. To eliminate effects of phase bias originating from the starting model,
simulated annealed Fo−Fc omit maps were calculated using the program CNS_solve1.1
with the ligand removed from the model (Bhat 1988; Brunger et al. 1998). Addition of
ligand was followed by identification of potential sites of solvent molecules by automatic
water-picking algorithm of COOT (Emsley and Cowtan 2004). The positions of these
automatically picked waters were manually checked and a few more waters were
manually identified on the basis of electron density contoured at 1.0 σ in the 2Fo−Fc map
and 3.0 σ in the Fo−Fc map. Addition of ligand and water molecules to the model resulted
in further decrease in R and Rfree. During the last refinement cycles, riding hydrogen
atoms were introduced for the protein residues.
149

Chapter 5 Propionate kinase in complex with Ap4A 5.2.3 Phosphorylation of TdcD with [γ-32P] ATP
The purified enzyme was phosphorylated by incubation with [γ-32P] ATP and then
re-fractionated by gel filtration (Anthony and Spector 1971). An incubation volume of
100 μl contained (in micromoles): magnesium chloride (0.60), dithiothreitol (0.40),
triethanolamine-HCl, pH 7.5 (5.0), [γ-32P] ATP (0.050) and purified propionate kinase
(0.001). The enzyme mixture was incubated at room temperature for 45 min prior to the
addition of ATP to start the reaction. After the addition of ATP, reaction mixture was
incubated for 30 minutes and then applied to a column (1.0 X 50 cm) of Sephadex G-25
(fine) which was equilibrated at 4°C with 10 mM potassium pyrophosphate (pH 7.0)
containing 10 mM dithiothreitol. Potassium pyrophosphate buffer was used to prevent the
formation of TdcD-[γ-32P] ATP complex. Elution was conducted with the same buffer at
4°C. Fractions (0.50 ml) were collected and were quantified by liquid scintillation.
Alternate fractions were checked on SDS-PAGE to confirm that the peak fractions
contained phosphorylated TdcD.
5.3 Results and discussion 5.3.1 Crystallization
Crystals of TdcD in the presence of ATP and Ap4A were obtained in 0.1 M Bis-
Tris (pH 6.5), 17% (w/v) polyethylene glycol monomethyl ether 5000 and in 0.1 M Bis-
Tris (pH 6.5), 45% (v/v) pentaerythritol ethoxylate (15/4 EO/OH), 100 mM ammonium
sulfate, respectively, by the hanging drop method. Crystals of native TdcD were obtained
by the microbatch method in 0.1 M Bis-Tris (pH 6.5), 30% (v/v) pentaerythritol
ethoxylate (15/4 EO/OH). All the three crystals belonged to the space group P3121 and
the volume of the asymmetric unit of the crystals was compatible with only one subunit,
with a Matthews coefficient of 2.7 Å3Da−1 and a calculated solvent content of 53.2%
(v/v) (Matthews 1968).
5.3.2 Structure determination
The structures of TdcD in the native form and in the presence of ATP have been
determined using the protein atoms of TdcD-ADP complex as model. Surprisingly, in the
structure of TdcD obtained in the presence of ATP, when ATP was fitted into the electron
150

Chapter 5 Propionate kinase in complex with Ap4A density present in the active site pocket, a blob of continuous electron density was present
beyond the γ-phosphate. Attempts were made to build various other ligands into this
density such as ATP and Mg2+, Ap4 (adenosine tetraphosphate), etc. In the case of ATP
and Mg2+, absence of proper coordination for Mg2+ as well as relatively big blob of
density corresponding to Mg2+ ruled out presence of any metal ion whereas in case of
adenosine tetraphosphate, extra electron density was present even beyond the δ-
phosphate. Finally, a diadenosine tetraphosphate (Ap4A) was built in this electron
density. Ap4A fitted very well in the electron density, which could be successfully refined
and accounted for the total electron density (Fig. 5.4). Electrospray ionisation mass
spectrometry (ESI-MS) of ATP, used for crystallization experiments, did not show any
presence of Ap4A. To confirm the ability of Ap4A to bind into the active pocket of TdcD,
structure of TdcD was solved in the presence of commercially available Ap4A. The
electron density of Ap4A in TdcD datasets obtained in the presence of ATP (TdcD-
Ap4A(ATP)) as well as in presence of Ap4A (TdcD-Ap4A) was sufficiently clear so that
its conformation and orientation could be explicitly determined. The simulated annealed
Fo−Fc omit maps for Ap4A calculated using the program CNS_solve1.1 (Brunger et al.
1998) is shown in figure 5.5.
Ethylene glycol used as the cryoprotectant during data collection was bound at a
site distinct from the propionate binding site in the TdcD structures complexed with
Ap4A. Mg2+ used in the crystallization experiment could not be located in the model
which suggests that divalent ions are required for the enzymatic reaction and not for the
binding of nucleotides to the enzyme. Presence of ammonium sulphate in the
crystallization condition led to very poor ligand density in the active site pocket in the
datasets of TdcD obtained in the presence of ATP. Only in the absence of ammonium
sulphate in the crystallization condition, TdcD dataset obtained in the presence of ATP
showed good electron density for the Ap4A. Presence or absence of ammonium sulfate
during co-crystallization of TdcD with Ap4A did not show any such effect on the electron
density of Ap4A.
151

Chapter 5 Propionate kinase in complex with Ap4A Table 5.1 Crystal parameters and statistics of data collection, refinement and model quality.
Values in parentheses correspond to the highest resolution shell.
Data set TdcD-native TdcD-Ap4A(ATP) TdcD-Ap4A
Crystal parameters Unit cell parameters a= b (Å) 110.80 110.48 110.56 c (Å) 66.53 66.61 66.61 α, β, γ (deg.) 90, 90, 120 90, 90, 120 90, 90, 120 Space group P3121 P3121 P3121 Temperature (K) 100 100 100 Data collection Resolution range (Å) 30.0-2.6(2.69-2.60) 50.0-1.98(2.05-1.98) 30.0-2.4 (2.49-2.40) No. of monomers per AU 1 1 1 No. of reflections measured 497420 457493 590394 No. of unique reflections 14818 32959 18755 Data redundancy 33.6 13.8 31.5 VM (Å3Da-1) 2.62 2.62 2.62 Solvent content (%) 53.1 53.1 53.1 I/σ(I) 15.0 (2.2) 24.4 (2.2) 22.1 (3.1) Completeness (%) 98.3 (84.3) 99.9 (100.0) 99.2 (95.2) Rmerge (%) 10.1 (45.7) 7.3 (49.4) 7.4 (41.3) Refinement R (%) 20.2 (31.5) 19.2 (24.8) 20.0 (26.7) Rfree (%) 25.2 (36.6) 22.1 (31.8) 25.2 (37.1) No. of atoms Protein atoms 2944 2955 2938 Ligand atoms 57 57 Solvent atoms 104 189 152 Model quality RMS deviation from ideal values Bond length (Å) 0.007 0.009 0.008 Bond angle (deg.) 0.937 1.182 1.145 Dihedral angles (deg.) 5.676 5.715 5.859 Wilson B factor (Å2) 57.29 29.35 53.8 Average B factors (Å2) Protein atoms 43.0 32.2 48.1 Ligand 44.9 52.6 Water 37.9 37.6 44.6 Residues in Ramachandran plot (%) Most favoured region 88.7 91.3 89.3 Allowed region 10.7 8.7 10.7 Generously allowed region 0.6 0.0 0.0 Disallowed region 0.0 0.0 0.0
152

Chapter 5 Propionate kinase in complex with Ap4A
(a) ATP (b) ATP & Mg2+
(c) Ap4 (d) Ap4A
Figure 5.4 Various ligands build into the electron density map (Fo - Fc map contoured at 3 σ)
observed in the active site pocket of propionate kinase obtained after co-crystallization with ATP.
These ligands were (a) ATP; (b) ATP and Mg2+; (c) Ap4, adenosine tetraphosphate and (d) Ap4A,
diadenosine tetraphosphate.
153

Chapter 5 Propionate kinase in complex with Ap4A
(a)
(b)
Figure 5.5 Stereo view of the electron density corresponding to Ap4A in the (a) TdcD-Ap4A(ATP)
and (b) TdcD-Ap4A structure from a simulated annealed Fo−Fc omit map contoured at 2.7 σ.
154

Chapter 5 Propionate kinase in complex with Ap4A 5.3.3 Model quality
The structures of TdcD-native (2.6 Å), TdcD-Ap4A(ATP) (2.0 Å) and TdcD-
Ap4A (2.4 Å) have been determined with good stereo-chemical parameters. In all the
three structures, the electron density is of good quality for the major part of the
polypeptide chain, except in the residue ranges 39-42 and 49-58, where poor density is
observed. Besides N-terminal His tag, initial three residues at the N-terminus and final
five residues at the C-terminus were not included in the model due to the absence of well-
defined electron density. The structures have 89-90% of the residues in the most favored
regions of the Ramachandran map calculated with the program PROCHECK (Laskowski
et al. 1993). The crystal parameters and data collection and refinement statistics are
summarized in table 5.1. 5.3.4 Native TdcD structure
Superposition of Cα atoms of native TdcD and its complex with Ap4A(ATP),
Ap4A, ADP and AMPPNP on each other using the program ALIGN (Cohen 1997) gave
an rmsd values ranging from 0.15 to 0.33 Å, indicating that there are no major main-
chain alterations between these TdcD structures. Even though the structure of native
TdcD has similar conformation for most of the amino acids in the active site pocket as
seen in the structures of TdcD obtained in presence of various nucleotides, significant
conformational changes were observed in Gly205, Asn206 and Gly207 present in the turn
between the first and second β-strands of the C-terminal domain. These residues form the
binding site for α- and β-phosphate group of the nucleotide, as seen in the TdcD-
AMPPNP complex structure. In the native TdcD structure, in the absence of any
nucleotide, backbone atoms of these three residues and the side chain of Asn206 move
towards the site for the phosphate group of the nucleotide, in the direction of His175 and
His203 (Fig. 5.6). Comparison of relative positions of N- and C-terminal domains in
native TdcD as well as in complex with Ap4A, ADP and AMPPNP did not reveal any
domain motion which is expected to occur during catalysis. However, the high B factors
and relatively poor electron density of the residues present in the N- and C-terminal
domains away from the hinge region in all the TdcD structures are indicative of plausible
155

Chapter 5 Propionate kinase in complex with Ap4A domain motion in TdcD similar to that observed in the other members of the ASKHA
superfamily (Fig. 4.17).
Figure 5.6 Stereo view of the conformational changes that occur at the nucleotide binding site in
the TdcD-native dataset. Superposed structures of TdcD-native and TdcD-AMPPNP complex are
shown in magenta and cyan, respectively.
Figure 5.7 Superposition of Ap4A observed in TdcD-Ap4A(ATP) and TdcD-Ap4A structures,
colored in cyan and yellow, respectively.
5.3.5 TdcD-Ap4A structure and Ap4A binding
Binding of Ap4A to TdcD does not induce any significant conformational changes
in the active site pocket or in the tertiary structure of the enzyme. The binding modes of
156

Chapter 5 Propionate kinase in complex with Ap4A Ap4A in the active site of TdcD-Ap4A(ATP) and TdcD-Ap4A structures are almost
identical (Fig 5.7). The electrostatic surface potential representation of the enzyme with
Ap4A present in the active site pocket is shown in figure 5.8. Ap4A binds in a single,
stable conformation facilitating detailed structural analysis and comparison with related
structures. Atom numbering scheme followed for Ap4A is shown in figure 5.9. Ap4A
present in the active site pocket extends from the nucleotide binding site to the propionate
binding site. The adenosines present in the nucleotide and propionate binding sites are
designated as A and B, respectively. Unlike TdcD-AMPPNP structure, where ethylene
glycol is present in the propionate binding site, presence of adenine B at the propionate
binding site in the structure of TdcD complexed with Ap4A displaces the ethylene glycol
towards the phosphate groups. Figure 5.10 shows the interactions of Ap4A with amino
acid residues and with water molecules present within a radius of 4Å in the active site
pocket.
(a) (b)
Figure 5.8 The electrostatic surface representation of the TdcD-Ap4A complex structure showing
(a) the binding site of Ap4A (shown in the left panel) and (b) the enlarged view of the active site
pocket with Ap4A present in it (shown in the right panel). Positively charged residues are shown
in blue, negatively charged residues are shown in red and neutral residues are shown in white.
157

Chapter 5 Propionate kinase in complex with Ap4A
Figure 5.9 Atom-numbering scheme for Ap4A.
Figure 5.10 Stereo diagram of the active site region showing Ap4A bound to TdcD in the TdcD-
Ap4A(ATP) structure. Protein atoms and Ap4A are colored yellow and magenta, respectively.
Water molecules present in the active site pocket are shown as red spheres. Hydrogen bonds
formed by Ap4A with protein atoms and water molecules are shown as broken lines.
Most of the hydrogen bonds formed by Ap4A are common to these structures
except those formed with water molecules in the TdcD-Ap4A(ATP) structure. These
water molecules are missing in the TdcD-Ap4A structure, perhaps due to its lower
resolution compared to that of TdcD-Ap4A(ATP). Hydrogen bonds formed by Ap4A in
TdcD-Ap4A(ATP) and TdcD-Ap4A datasets are listed in table 5.2. Both the adenine rings
158

Chapter 5 Propionate kinase in complex with Ap4A A and B are in the anti conformation with respect to the ribose sugar, which is in the C2′-
endo form. The adenosine present in the nucleotide binding site shows stronger electron
density compared to the one present in the propionate binding site due to the tighter
binding of adenine ring A.
5.3.5.1 Adenosine A
In the structures of TdcD complexed with Ap4A, adenosine A forms interactions
similar to those seen in the structures of TdcD complexed with ADP and AMPPNP. The
adenine A ring is present in a pocket formed by residues Leu279, Arg280, Glu283,
Gly326, Asn330 and Ser331. It is sandwiched between side chains of Arg280 and
Asn330. No aromatic residue is sufficiently close to stack with the base. There are no
direct hydrogen bonds between the base and the protein atoms suggesting low specificity
of the enzyme towards nucleotide bases. Only AN7 atom of the adenine ring makes a
hydrogen bond with a water molecule. The ribose A sugar forms hydrogen bonds with
protein atoms via AO2*, AO3* and AO4*. The AO2* is hydrogen bonded to the oxygen
atoms of the carboxyl group of Asp278 and the main chain N of Leu279 whereas AO3*
and AO4* are hydrogen bonded to the main chain N of Leu279 and Ile327, respectively.
5.3.5.2 α, β, γ and δ phosphates
In contrast to the adenosine moieties, the interactions of the four phosphates of
Ap4A with protein atoms are much stronger. The α-phosphate forms three hydrogen
bonds, one each with the main chain N of Asn206, Gly326 and with the side chain ND2
atom of Asn206. It also forms one hydrogen bond with a water molecule present nearby.
The β-phosphate is poorly connected with the enzyme and forms just one hydrogen bond
with ND2 atom of Asn206. All the three oxygen atoms of γ-phosphate are hydrogen
bonded to the protein atoms or water molecules. Out of the five hydrogen bonds formed
by the γ-phosphate, two are with water molecules and the other three are with main chain
atoms of Asn206, Gly207 and NE2 of His203. The δ-phosphate forms two hydrogen
bonds, one with NE2 of His175 and another with the oxygen atom of ethylene glycol
(EDO) present nearby.
159

Chapter 5 Propionate kinase in complex with Ap4A 5.3.5.3 Adenosine B
The ribose B forms 5 hydrogen bonds, 3 with water molecules and one each with
ND2 of Asn11 present in the N-terminal domain and O1 of ethylene glycol. The adenine
B ring is present in the proposed propionate binding site in a pocket formed by Ala88,
His118, Phe174, Pro227, Arg236 and ethylene glycol present nearby. In the acetate
kinase, acetate binds in the pocket formed by these amino acids. Hence, propionate in
TdcD is expected to bind in this pocket. Aromatic amino acid His118 forms stacking
interactions with the base. Adenine B forms hydrogen bonds with the side chain atom of
conserved His118 and Arg236. It also forms three hydrogen bonds with water molecules
in TdcD-Ap4A(ATP) structure and two hydrogen bonds in TdcD-Ap4A structure.
Only two other protein structures in the Protein Data Bank have Ap4A bound as a
ligand. In human adenylate kinase 2, Ap4A is present in a folded conformation with two
phosphates and one adenosine built in alternate conformation (PDB code 2C9Y;
unpublished result). In the structure of eosinophil-derived neurotoxin in complex with
Ap4A, the second adenosine moiety is disordered (Baker et al. 2006). Thus, TdcD
complexed with Ap4A is the first structure in which Ap4A is in an extended conformation
and fully ordered (Fig. 5.11).
Figure 5.11 Comparison of conformation of Ap4A observed in the structure of TdcD and
adenylate kinase. The TdcD bound conformation of Ap4A is shown in magenta whereas human
adenylate kinase 2 bound conformation (PDB entry 2C9Y) is shown in cyan. The adenine A rings
of the two nucleotides are superposed. In human adenylate kinase 2, the γ- and δ-phosphates and
the adenine B ring are present in two conformations.
160

Chapter 5 Propionate kinase in complex with Ap4A Table 5.2 List of hydrogen bonds (≤ 3.5 Å) formed by Ap4A with protein atoms and water
molecules in TdcD-Ap4A(ATP) and TdcD-Ap4A complex.
TdcD-Ap4A(ATP) TdcD-Ap4A
Ap4A AN7 HOH-80 (59) O 3.14 3.02
Ap4A AO2* ASP-278 OD2 3.09 3.34
ASP-278 OD1 2.95 2.84
LEU-279 N 3.25 3.06
Ap4A AO3* LEU-279 N 3.36 3.26
Ap4A AO4* ILE-327 N 3.38 3.20
Ap4A O1A HOH-168 (126) O 2.73 2.65
GLY-326 N 2.87 2.77
Ap4A O3A ASN-206 N 3.37 3.44
ASN-206 ND2 3.52 3.41
Ap4A O2B ASN-206 ND2 3.16 3.03
Ap4A O1G ASN-206 N 2.69 2.68
GLY-207 N 2.78 2.92
Ap4A O2G HOH-171(129) O 2.63 2.62
HIS-203 NE2 2.80 2.87
Ap4A O3G HOH-171 O 3.49 --
Ap4A O2D EDO-401 O2 2.51 2.62
HIS-175 NE2 2.90 2.84
Ap4A BO3* ASN-11 ND2 3.01 2.85
HOH-160 (120) O 2.58 2.65
HOH-183 (137) O 3.32 3.26
Ap4A BO4* HOH-159 (120) O 3.19 3.47
EDO-401 O1 3.21 3.09
Ap4A BN7 HIS-118 NE2 3.12 2.82
ARG-236 NH2 3.19 --
HOH-(144) O -- 2.82
Ap4A BN6 HOH-170 O 3.36 --
Ap4A BN3 HOH-159 O 2.78 --
Ap4A BN1 HOH-170 (128) O 2.69 3.03
Numbers in parentheses correspond to the water molecules present in the TdcD-Ap4A dataset. Hyphens
indicate missing hydrogen bonds. EDO, Ethylene glycol; HOH, water.
161

Chapter 5 Propionate kinase in complex with Ap4A 5.3.6 Formation of Ap4A by TdcD
Out of various enzymatic reactions identified so far involving the enzymatic
formation of Ap4A, the activity of dinucleotide polyphosphate phosphorylases (EC
2.7.7.53) could explain the formation of Ap4A by the TdcD enzyme (Baxi and
Vishwanatha 1995; Guranowski 2000). Dinucleotide polyphosphate phosphorylases form
Ap4A in the presence of ATP and ADP. In the case of TdcD, initially ATP might undergo
hydrolysis to yield ADP and phosphoenzyme as observed in case of acetate kinase
(Anthony and Spector 1972). The ADP formed in this reaction could in turn react with
excess ATP present in the crystallization mixture and can lead to the formation of Ap4A
in the presence of Mg2+.
Enzyme + ATP ↔ Enzyme-P + ADP
ADP + ATP ↔ Ap4A + Pi
Further experiments were carried out to examine the formation of ADP and
phosphoenzyme in the case of TdcD. Incubation of TdcD with [γ-32P] ATP followed by
gel filtration experiment showed the transfer of labeled γ-phosphate from ATP to the
enzyme resulting in the formation of phosphoenzyme and ADP (Fig. 5.12), as seen in the
case of acetate kinase (Anthony and Spector 1972). Formation of ADP and
phosphoenzyme argues in the favor of dinucleotide polyphosphate phosphorylase activity
in TdcD. Further biochemical studies are being carried out to confirm the presence of
such enzymatic activity in case of TdcD.
Control
(b)
(a)
Figure 5.12 Phosphorylation of
TdcD. (a) Sephadex G-25 gel
filtration of TdcD
phosphorylated with [γ-32P]
ATP. (b) After gel filtration,
alternate fractions were checked
on SDS-PAGE.
162

Chapter 5 Propionate kinase in complex with Ap4A Structure of adenylate kinase complexed with the inhibitor diadenosine
pentaphosphate (Ap5A), mimicking the two substrates (ATP and AMP), has allowed the
construction of a stabilized transition state, which supports the view that these kinases
mediate direct transfer of phosphoryl groups from ATP to acceptors rather than acting by
a double displacement mechanism (Muller and Schulz 1992). Similarly, presence of
Ap4A in the active site pocket of TdcD seems to mimic the presence of its substrates
(either propionyl phosphate and ADP or propionate and ATP) in the active site pocket
and is suggestive of direct in-line transfer mechanism. Further, the direct in-line transfer
mechanism is supported by stereo-chemical evidence and steady state kinetics (Blattler
and Knowles 1979; Gorrell et al. 2005; Miles et al. 2002).
The ability of E. coli acetate kinase (a homologue of propionate kinase) to
phosphorylate enzyme I of the phosphotransferase system (Fox et al. 1986) and CheY in
vitro (Dailey and Berg 1993) was thought to indicate the role of the phosphorylated
acetate kinase in sugar transport rather than in phosphorylation of acetate or ADP. A
similar role might also be applicable to the phosphoenzyme form of propionate kinase.
5.4 Conclusions In this chapter, the structure of propionate kinase in the native form as well as in
complex with diadenosine 5',5'''-P1,P4-tetraphosphate (Ap4A) has been described. On the
basis of these structures, crystallographic evidence is provided for a novel Ap4A synthetic
activity in propionate kinase from Salmonella typhimurium. Crystals of TdcD obtained in
the presence of ATP clearly showed Ap4A bound in the active site pocket of the enzyme.
Presence of Ap4A and its binding to the enzyme was further confirmed by the structure of
TdcD-Ap4A complex obtained after co-crystallization of TdcD with commercially
available Ap4A. Out of various enzymatic reactions identified so far involving the
enzymatic synthesis of Ap4A, the activity of dinucleotide polyphosphate phosphorylases
(EC 2.7.7.53) could explain the formation of Ap4A by the TdcD. Formation of ADP and
phosphoenzyme argues in the favor of the presence of dinucleotide polyphosphate
phosphorylase activity in TdcD. In the TdcD-Ap4A complex structure, Ap4A is present in
an extended conformation with one adenosine moiety present in the nucleotide binding
163

Chapter 5 Propionate kinase in complex with Ap4A site and other in the proposed propionate binding site. These observations tend to support
the stereo chemical evidence that phosphoryl transfer in this enzyme is direct.
Following the early discovery by Zamecnik et al that Ap4A is generated as a by-
product by aminoacyl t-RNA synthetases (Zamecnik et al. 1966), there has been
considerable interest in the possible role of Ap4A inside the cell. Considering its role in
various metabolic processes, diadenosine polyphosphates and their synthetic analogs are
being evaluated for their potential as pharmacological agents. Since, induced expression
of enzymes involved in the anaerobic breakdown of L-threonine takes place in the
absence of glucose and oxygen in the medium during which energy level inside the cell is
low, formation of Ap4A and its binding to TdcD could be of additional interest in view of
its proposed role as a metabolic regulator. The present work provides a new direction to
investigate several unanswered questions regarding the formation of Ap4A by TdcD and
its roles in the metabolic processes.
164

Crystal structures of Salmonella typhimurium 2-methylisocitrate lyase (PrpB) and its
complex with pyruvate and Mg2+
6

Chapter 6 2-methylisocitrate lyase (PrpB)
6.1. Introduction In Escherichia coli and Salmonella typhimurium, propionate is metabolized to
pyruvate and succinate via 2-methylcitric acid cycle (Horswill and Escalante-Semerena
1999b). This pathway was initially postulated by studies with mutant strains of Candida
lipolytica, in which accumulation of either 2-methylcitrate or 2-methylisocitrate was
observed during growth on odd-chain fatty acids, which were degraded via propionyl-
CoA (Tabuchi and Hara 1974; Tabuchi and Serizawa 1975). Genes required for the
catabolism of propionate in these bacteria were first identified in S. typhimurium and are
referred as `prp' genes (Horswill and Escalante-Semerena 1999b). The prp locus
comprises of two transcriptional units. One unit contains four genes (prpBCDE)
organized as an operon that encodes four distinct enzymes required for the catabolism of
propionate. The other unit contains only one gene, prpR, which encodes a σ54-dependent
transcriptional activator (Horswill and Escalante-Semerena 1997). In this pathway, PrpE,
a propionyl-CoA synthetase (Horswill and Escalante-Semerena 1999a), forms propionyl-
CoA from propionate and coenzyme A and PrpC, a 2-methylcitrate synthase (Horswill
and Escalante-Semerena 1999b), forms 2-methylcitrate by combining propionyl-CoA and
oxaloacetate. The 2-methylcitrate thus formed is converted to 2-methylisocitrate by two
separate enzymes, PrpD, a 2-methylcitrate dehydratase, and either of the two aconitases
(AcnA or AcnB) present in S. typhimurium (Horswill and Escalante-Semerena 2001).
The last step of this cycle, the cleavage of 2-methylisocitrate to succinate and pyruvate, is
catalyzed by PrpB (Fig. 6.1), a 2-methylisocitrate lyase. Succinate is further oxidized to
oxaloacetate for condensation with propionyl-CoA, forming 2-methylcitrate and
completing the cycle, whereas pyruvate can be used for energy metabolism and synthesis
of biomass.
Figure 6.1 Reaction catalyzed by 2-methylisocitrate lyase (PrpB) leading to the cleavage of 2-
methylisocitrate into succinate and pyruvate.
166

Chapter 6 2-methylisocitrate lyase (PrpB)
Oxidation of propionate via 2-methylcitric acid cycle resembles the part of the
glyoxylate cycle in which acetate is oxidized to glyoxylate. Isocitrate lyase, involved in
glyoxylate cycle, is related to 2-methylisocitrate lyase with respect to catalytic reaction.
As deduced from its primary structure, PrpB belongs to the isocitrate lyase protein family
comprising isocitrate lyases of bacterial and eukaryotic origin as well as
phosphonoenolpyruvate mutases (PEP mutases) and carboxyphosphonoenolpyruvate
phosphono mutases (CPEP mutases) (Brock et al. 2001). The PEP mutase and CPEP
mutase belong to families of enzymes catalyzing reactions of carbon-phosphorous bonds,
which function in phosphonate biosynthesis. Despite the divergence in the chemistry that
it catalyzes, the PEP mutase active site conserves the same catalytic scaffold observed in
isocitrate lyase, including the flexible active site loop, which regulates solvent access and
they are all proposed to proceed via mechanisms that stabilize enol(ate) intermediates.
The PrpB enzyme from E. coli has been shown to be functionally dependent on
Mg2+ with an apparent KM of 35 µM (Brock et al. 2001). In vitro synthesis of 2-
methylisocitrate using the purified enzyme indicated formation of only the threo-
stereoisomer of 2-methylisocitrate. In the presence of 2 mM Mg2+, the KM for threo-2-
methylisocitrate was determined to be 19 µM; the catalytic efficiency was estimated as
V/KM = 6.4 × 105 M-1 s-1 (Brock et al. 2001).
Studies carried out on PrpB enzyme from S. typhimurium (Grimek et al. 2003)
has shown that KM of 2-methylisocitrate is 19 µM, with a kcat of 105 s-1. Optimal 2-
methylisocitrate lyase activity was measured at pH 7.5 and 50°C, and the reaction
required Mg2+ ions; the same concentration of Mn2+ ions was a poor substitute for Mg2+
(28% specific activity). Dithiothreitol (DTT) or reduced glutathione (GSH) was required
for optimal activity; the role of DTT or GSH was apparently not to reduce disulfide
bonds, since the disulfide-specific reducing agent Tris (2-carboxyethyl) phosphine
hydrochloride failed to substitute for DTT or GSH. Site-directed mutagenesis
experiments carried out on S. typhimurium PrpB enzyme have shown that the
PrpB(Lys121Ala), PrpB(His125Ala), and PrpB(Arg122Lys) mutant enzymes had 1,050-,
750-, and 2-fold decreased kcat for 2-methylisocitrate lyase activity, respectively. The
PrpB(Asp58Ala) and PrpB(Cys123Ala) proteins displayed no detectable 2-methyl-
isocitrate lyase activity indicating that both of these residues are essential for catalysis.
167

Chapter 6 2-methylisocitrate lyase (PrpB)
Based on the proposed mechanism of the closely related isocitrate lyases, Cys123 is
proposed to serve as the active site base and Asp58 is believed to be critical for the
coordination of a required Mg2+ ion in PrpB.
When cultured in the presence of glucose, propionate and other short chain fatty
acids have the additional property of inhibiting cell growth. The growth inhibiting effect
of propionate, in the presence of glucose, may be due to the accumulation of propionyl-
CoA which leads to competition among other house-keeping genes dealing with CoA-
esters as substrates or products (Brock et al. 2000). Sensitivity against propionate has
been found to increase drastically when 2-methylcitrate pathway is blocked in Salmonella
suggesting that intermediates of this pathway may have a more profound negative impact
on cell growth compared to propionate (Horswill et al. 2001).
In this chapter, crystal structures of Salmonella typhimurium 2-methylisocitrate
lyase and its complex with pyruvate and Mg2+ have been described. Examination of the
active site pocket revealed a plausible structural rationale for the specificity of 2-
methylisocitrate lyase and isocitrate lyase towards their substrates pyruvate and
glyoxylate, respectively. Structure of Salmonella PrpB has been compared with E. coli
PrpB and with bacterial isocitrate lyases. The results reveal the differences between these
enzymes and provide further insights into the structure-function relationships.
6.2. Materials and methods 6.2.1. Cloning, overexpression and purification
The prpB gene was PCR-amplified from Salmonella typhimurium genomic DNA
using Deep Vent polymerase (NEB). After amplification of target gene by sense (5’
CATGCCATGGCTAGCTCTTTACATTCGCCGGGG 3’) and antisense (5’ CGGGATC
CTTACTCGAGCGATTTTTTATTCCTGTACAG 3’) primers, the PCR-amplified
fragment was digested with NcoI and XhoI. It was then cloned into pETBlue-2 vector
(Novagen), an expression vector that incorporates a hexa-histidine tag at the carboxyl
terminal of the recombinant protein to facilitate purification. After purification, protein
was concentrated by several cycles of low-speed centrifugation using a 10 kDa molecular
weight cut-off Centricon (Amicon) until a final concentration of 20 mg/ml was reached.
Protein concentration was estimated by Lowry's method (Lowry et al. 1951). The
168

Chapter 6 2-methylisocitrate lyase (PrpB)
sequence of the prpB gene was determined by nucleotide sequencing and confirmed by
comparing it with the prpB gene of S. typhimurium LT2.
6.2.2 Crystallization and data collection
Crystallization was carried out with the hanging-drop vapour-diffusion method
using Crystal Screen I and II from Hampton Research (Jancarik and Kim 1991) as well as
a few crystallization screens prepared in the lab. Droplets containing 4 μl of protein
solution were mixed with 4 μl of crystallization solution. During the initial crystallization
screening, crystals were observed under following conditions.
1. 4 M Sodium formate
2. 0.2 M Lithium sulfate monohydrate, 0.1 M Tris pH 8.5, 30% PEG 4000
3. 2 M Sodium formate, 0.1 M Sodium acetate trihydrate
4. 2 M NaCl, 10% PEG 6000
5. 0.1 M Imidazole pH 6.5, 1.0 M Sodium acetate trihydrate
6. 0.1 M Sodium cacodylate pH 6.0, 1.4 M Sodium acetate trihydrate
7. 0.1 M Tris pH 8.5, 8 % PEG 8000
8. 0.1 M HEPES pH 7.5, 2 % PEG 400, 2 M Ammonium sulfate
9. 0.1 M MES pH 6.5, 10% PEG 6000
10. 1.6 M Ammonium sulfate, 0.1 M MES pH 6.5, 10% Dioxane
11. 0.1 M HEPES pH 7.5, 10 % PEG 6000, 1.5 M Ammonium sulfate
12. 0.1 M HEPES pH 7.5, 10% PEG 8000, 8% Ethylene glycol
13. 0.1 M NaCl, 0.1 M HEPES pH 7.5, 1.6 M Ammonium sulfate,
14. 0.2 M MgCl2. 6H2O, 0.1 M Tris pH 8.5, 30 % PEG 4000
However, good diffraction quality crystals were obtained at 291K only in the
conditions 2, 3, 4 and 5 (mentioned above). These crystals appeared after 6 d of
equilibration against the crystallization solution and grew to full size in 10 d. For co-
crystallization experiments, PrpB was incubated overnight with 50 mM of pyruvic acid.
On the basis of the proposed reaction mechanism in isocitrate lyases (Sharma et al.
2000), in PrpB, pyruvate is expected to bind first followed by binding of succinate.
Therefore, a crystal obtained in the presence of pyruvate was soaked in 50 mM succinate
169

Chapter 6 2-methylisocitrate lyase (PrpB)
for 3 h before data collection. The crystals were transferred to a cryoprotectant composed
of reservoir solution with 20% glycerol prior to mounting. Complete X-ray diffraction
data were collected for the native PrpB and the pyruvate bound PrpB extending to a
resolution of 2.1 and 2.3 Å, respectively, at 100 K (Table 6.1).
Table 6.1 Crystal parameters and data collection statistics for native and pyruvate/Mg2+ bound
PrpB at 100K. Values in parentheses correspond to the highest resolution shell.
Native PrpB Pyruvate/Mg2+ bound PrpB Crystal parameters Space group P212121 P212121
Unit cell parameters (Å) a 62.81 62.95 b 99.09 99.68 c 201.57 202.40
Data collection No. of monomers per AU 4 4 Resolution range (Å) 20.0 - 2.10 (2.10 - 2.18) 20.0 - 2.30 (2.30 - 2.38) No. of reflections measured 391895 273290 No. of unique reflections 70807 53974 Multiplicity 5.5 5.1 Completeness (%) 95.4 (97.2) 94.0 (99.5) I/σ (I) 15.5 (5.2) 17.0 (5.6) Rmerge (%) 6.1 (42.2) 6.4 (28.8)
Extensive trials were also made to crystallize PrpB with various other ligands
such as:
3-bromopyruvate (forms a covalent adduct with the active site Cys123),
pyruvate and nitropropionate (succinate analogue),
glyoxylate (pyruvate analog) and succinate,
isocitrate (2-methylisocitrate analog) and
2-phosphoglycerate (known to bind to isocitrate lyases).
Co-crystallization of PrpB with these ligands in the concentration range from 5 to
50 mM were tried out using various crystallization screens and in the crystallization
condition optimized for the native PrpB. Crystals obtained from these co-crystallization
170

Chapter 6 2-methylisocitrate lyase (PrpB)
experiments were used for data collection. Datasets were processed and scaled using the
program DENZO and SCALEPACK of the HKL suite (Otwinowsky 1997).
6.2.3 Structure determination
Attempts to solve the structure of PrpB using the closest structural homologue
phosphoenolpyruvate mutase (PDB code: 1PYM) (shares 24 % sequence identity with
PrpB) were unsuccessful. Further attempts were made to solve the structure by multiple
isomorphous replacement. Towards this end, a mercury derivative was prepared by
soaking the PrpB crystal in 5mM para-chloro-mercuribenzene sulphonic acid (PCMBS)
for 24 hours before data collection. X-ray diffraction dataset for PCMBS derivative was
collected at room temperature to a resolution of 3.5 Å which was isomorphous to another
native PrpB data collected at room temperature to a resolution of 2.5 Å (Table 6.2). The
isomorphous differences between the two datasets were used for MIR phasing. Most of
the other datasets collected after soaking in various other heavy atom compounds led to
either non-isomorphism or poor or excessive binding of heavy atoms.
Table 6.2 Crystal parameters and data collection statistics for PrpB and its mercury derivative
collected at room temperature. Values in parentheses correspond to the highest resolution shell.
Mercury derivative PrpB-native
Crystal parameters Space group P212121 P212121 Unit cell parameters (Å)
a 63.40 63.60 b 101.00 100.67 c 205.18 204.75
Temperature (K) 293 293 Data collection No. of monomers per AU 4 4 Resolution range (Å) 20.0 - 3.5 (3.5 -3.62) 20.0-2.5(2.5-2.54) No. of reflections measured 81437 400726 No. of unique reflections 17412 46499 Multiplicity 4.7 8.62 Completeness in % 91.8 (87.4) 98.4 (98.1) I/σ(I) 8.87 (3.87) 11.7(3.9) Rmerge (%) 11.4 (23.9) 9.1 (36.1)
171

Chapter 6 2-methylisocitrate lyase (PrpB)
Around this time, the structure of 2-methylisocitrate lyase (PrpB) from E. coli
was published (Grimm et al. 2003). The orientation and position of the tetrameric 2-
methylisocitrate lyase molecule in the P212121 asymmetric unit could easily be
determined using the protomer of E. coli PrpB enzyme (PDB code:1MUM) as the starting
model using the molecular replacement program AMoRe (Navaza 1994; Navaza and
Saludjian 1997). The PrpB model corresponding to the molecular replacement solution
was subjected to refinement using CNS_solve1.1 (Brunger et al. 1998). The data between
20.0 and 2.1 Å were used for the refinement, setting aside 5% of the reflections for cross-
validation. After initial rigid body and positional refinements, sigma-A weighted 2Fo-Fc
and Fo-Fc maps were calculated and visualized using the interactive model building
program O (Jones et al. 1991). Model building was alternated with iterations of positional
and individual temperature factor refinements. This was followed by identification of
potential sites of solvent molecules, first by automatic water-picking algorithm of
CNS_solve1.1 (Brunger et al. 1998) and then manually on the basis of 2Fo-Fc and Fo-Fc
maps. Positional and B-value parameters of all the atoms, including protein and water
were further refined. The structures of ligand bound PrpB were solved using native PrpB
as the starting model. In the PrpB dataset obtained in the presence of pyruvate and
succinate, the resulting difference Fourier map revealed electron density only for the
bound pyruvate-Mg2+ in all the four PrpB subunits present in the asymmetric unit. No
electron density was observed for succinate. The structure was refined at 2.3 Å with the
program CNS_solve1.1 using a protocol similar to the one used for the native structure.
In all other datasets of PrpB obtained after co-crystallization with various other
ligands, no electron density was observed corresponding to the ligand near the active site
pocket. Only in the case of PrpB data obtained in the presence of 3-bromopyruvate, a
different crystal form was obtained with unit-cell parameters a = 81.46, b = 129.1, c =
147.04 Å and α = β = γ = 90º. Systematic absences showed that the crystal belongs to the
space group P212121. The value of the Matthews constant (Matthews 1968) is 2.97 Å3 Da-
1 assuming four molecules of PrpB (molecular weight 32 kDa) in the asymmetric unit
(Table 6.3). Structure was solved by molecular replacement using the program AMoRe
with a protomer of native PrpB as a search model (Navaza 1994).
172

Chapter 6 2-methylisocitrate lyase (PrpB)
Table 6.3 Crystal parameters and data collection statistics for PrpB data (different crystal form)
obtained in the presence of 3-bromopyruvate. Values in parentheses correspond to the highest
resolution shell.
PrpB-bromopyruvate Crystal parameters Space group P212121 Unit cell parameters (Å)
a 81.467 b 127.089 c 147.046
Temperature (K) 100 Data collection No. of monomers per AU 4 Resolution range (Å) 20.0-2.40 (2.40-2.48) No. of reflections measured 401033 No. of unique reflections 60466 Multiplicity 6.63 Completeness in % 98.6 (97.9) I/σ(I) 13.8 (2.6) Rmerge (%) 8.7 (33.9)
6.3. Results and discussion 6.3.1 Cloning, overexpression, purification and crystallization
PrpB was successfully cloned in the pETBlue-2 vector (Novagen) with a C-
terminal hexa-histidine tag (Fig. 6.2(a)). After transforming the plasmid into E. coli strain
BL21(DE3)pLysS, the recombinant protein was overexpressed by IPTG induction. The
recombinant protein was purified using Ni-NTA affinity column chromatography. Pure
PrpB enzyme was obtained with a final yield of 35 mg per liter of cell culture. The purity
of the protein was estimated using SDS-PAGE and was found to be nearly homogeneous
(Fig. 6.2(b)). Prior to crystallization, the protein was concentrated to 20 mg per ml.
Crystallization trials were performed using various crystallization screens available in the
lab, which produced well diffracting crystals in a few conditions (mentioned in materials
and methods section). Crystals obtained from various crystallization conditions were of
the same crystal form (Fig. 6.3). Crystals of native PrpB used for X-ray diffraction data
collection were obtained at 291K from solutions containing 0.1M imidazole pH 6.5, and
1.0 M Sodium acetate trihydrate. Crystals of pyruvate bound PrpB appeared in 2 M NaCl
and 10% PEG 6000.
173

Chapter 6 2-methylisocitrate lyase (PrpB)
1 2 (a) (b)
Figure 6.2 (a) Cloning of prpB gene in the pETBlue-2 vector. Lane 1, pETBlue-2 vector and lane 2,
pETBlue-2 vector containing prpB gene. (b) SDS-PAGE of PrpB during purification. Proteins were
analyzed on 12% SDS-PAGE and stained with Coomassie blue. Lane 1, crude cell lysates before
IPTG induction; lane 2, crude cell lysates after 0.3 mM IPTG induction; lane 3, clear supernatant;
lane 4, purified PrpB after Ni-NTA affinity column chromatography; M, molecular weight
markers.
Figure 6.3 Crystals of PrpB obtained in various crystallization conditions.
6.3.2 Structure determination
Systematic absences showed that the crystals belong to the space group P212121.
The value of the Matthews constant (Matthews 1968) is 2.56 Å3 Da-1 assuming four
174

Chapter 6 2-methylisocitrate lyase (PrpB)
molecules of PrpB (molecular weight 32 kDa) in the asymmetric unit. The crystal
structures of the members of the isocitrate lyase protein family that are known so far
comprise two bacterial isocitrate lyases, namely from M. tuberculosis and E. coli, and
isocitrate lyase from the filamentous fungus Aspergillus nidulans and
phosphoenolpyruvate mutase from the mussel Mytilus edulis. Bacterial isocitrate lyases
consist of about 430 residues and eukaryotic isocitrate lyases of about 540 residues. Like
mussel PEP mutase, Salmonella typhimurium PrpB consists of 295 amino acid residues.
Thus, these two enzymes constitute one of the smallest categories of proteins of the
isocitrate lyase family. Amino acid sequence alignment of 2-methylisocitrate lyase with
the members of isocitrate lyase family with known structures shows higher sequence
alignment (sequence identity of 24%) with PEP mutase than with isocitrate lyases.
Secondary structure prediction by the program PSIPRED (McGuffin et al. 2000) showed
exactly the same pattern of secondary structure as in the crystal structure of PEP mutase.
However, attempts to determine the crystal structure of 2-methylisocitrate lyase by the
molecular replacement method using PEP mutase as the search model (PDB code:
1PYM) were not successful, presumably because of the low sequence identity of 24%.
Further attempts were made to determine the crystal structure of PrpB by multiple
isomorphous replacement.
6.3.2.1 Multiple isomorphous replacement
With the view of determining the structure by multiple isomorphous replacement,
a mercurial derivative dataset was collected after soaking the PrpB crystal in solution
containing 5 mM para-chloro-mercuribenzene sulphonic acid (PCMBS). The native and
mercury derivative datasets were combined together using the program CAD and then
scaled using the SCALEIT program in the CCP4 program suite (CCP4 1994). After
scaling, the quality of the derivative data was assessed based on the values of Riso, which
gives an estimate about the degree of substitution of heavy atom in the derivative crystal.
The presence of anomalous signal was discerned from the values of Rano. Isomorphous
and anomalous difference Patterson maps were calculated for this derivative using the
program FFT of CCP4 program suite (CCP4 1994) and Harker sections for the space
group P212121 were plotted. The isomorphous difference Patterson maps indicated the
175

Chapter 6 2-methylisocitrate lyase (PrpB)
presence of heavy atoms in the derivative data whereas the anomalous Patterson maps
indicated absence of any useful anomalous signal in the derivative data. Harker sections
(Harker 1956) of the isomorphous difference Patterson maps calculated using native and
derivative datasets are illustrated in figure 6.4. The positions of the heavy atoms in the
derivative dataset were identified manually and confirmed using the program RSPS of the
CCP4 suite (CCP4 1994). The fractional coordinates of all the four sites, their
occupancies and anomalous occupancies were refined using MLPHARE to a resolution
of 3.5 Å. The final parameters of the refined heavy atoms are given in table 6.4.
However, an electron density map based on this single derivative was not interpretable.
While efforts were being made to obtain few more derivatives, the structure of E. coli 2-
methylisocitrate lyase (PrpB) was published (Grimm et al. 2003). Finally, we solved the
structure of S. typhimurium PrpB by molecular replacement using E. coli PrpB as a
search model.
Table 6.4 Heavy atom refinement statistics (15 - 4.0 Å resolution).
(a)
Fractional coordinates
Site Heavy atom x y z Occupancy B-factor
1 HG 0.511 0.637 0.124 0.606 20.005 2 HG 0.982 0.352 0.079 0.634 21.979 3 HG 0.008 0.682 0.132 0.568 24.924 4 HG 0.400 0.311 0.163 0.590 22.527 (b) Phasing power (acentric) b 2.09 R_Cullis (acentric) a 0.61Phasing power (centric) 1.64 R_Cullis (centric) 0.57
a R_Cullis = Σ||FPh ± FP| – |Fh| calc| / Σ|FPh ± FP| b Phasing Power = [Σ Fh
2 / Σ(|FPh obs| – |FPh calc|)2]1/2, where FPh and Fh are the derivative and
calculated structure factors respectively.
176

Chapter 6 2-methylisocitrate lyase (PrpB)
(a)
(b)
(c)
Figure 6.4 Harker sections of the isomorphous difference Patterson map computed using 15 - 4.0
Å resolution data from the native PrpB and a mercury derivative crystal at (a) x = 0.5, (b) y = 0.5
and (c) z = 0.5.
177

Chapter 6 2-methylisocitrate lyase (PrpB)
6.3.2.2 Molecular replacement and structure refinement
The standard procedure of molecular replacement using the program AMoRe
(Navaza 1994) was used to solve the structure of S. typhimurium PrpB using E. coli PrpB
as the starting model (Grimm et al. 2003). In the molecular replacement solution, the
correlation coefficient and R-factor was 53.0 % and 38.7 %, respectively, for the
reflections in the resolution range of 15-3.5 Å. The final model was refined at 2.1 Å to R
and Rfree of 20.5% and 23.2 %, respectively. In the final map, the electron density is of
good quality for most of the model. However, significant and unambiguous density was
not found in three different places: first 3 to 4 residues at the N-terminus, the active site
loop between the fourth β-strand and the sixth α-helix (residues 118 to residue 129), and
the last 6 to 13 C-terminal residues in different subunits. The structure has acceptable
stereochemistry with 92.3 % of all residues in the most favored region of the
Ramachandran plot (Fig. 6.5) calculated using the program PROCHECK (Laskowski et
al. 1993). Only the main-chain torsion angles of Asp87 within each subunit fall at the
border between the generously allowed and disallowed regions of the Ramachandran
plot. This residue is directly involved in binding of Mg2+ and located in the active site.
Inspection of the Ramachandran plots of the known ligand-free structures of isocitrate
lyases shows that the corresponding residues display similar main-chain torsion angles.
The structure of PrpB in complex with pyruvate and Mg2+ was solved using only
the protein atoms of native PrpB structure. In the final stages of refinement, inspection of
difference electron density maps showed strong positive density for the pyruvate and
Mg2+. No electron density was observed for the succinate. The topology and the
parameters for the pyruvate were evaluated using the web-based program HIC-UP
(Kleywegt and Jones 1998). In the final map, the electron density is of good quality for
most of the protein atoms. Breaks in the electron density occur at equivalent regions to
breaks observed in the native PrpB structure in each subunit. The final model was refined
at 2.3 Å to R and Rfree of 20.3% and 25.2 %, respectively. The structure shows good
stereochemistry with 91.4 % of all residues in the most favored regions of the
Ramachandran plot calculated with the program PROCHECK (Laskowski et al. 1993).
The refinement and model quality statistics of PrpB and its complex with pyruvate and
Mg2+ are given in table 6.5.
178

Chapter 6 2-methylisocitrate lyase (PrpB)
Table 6.5 Refinement and model quality statistics for native and pyruvate/Mg2+ bound PrpB (at
100K). Values in parentheses correspond to the highest resolution shell.
Native PrpB Pyruvate/Mg+2 bound PrpB Refinement R (%) 20.5 (24.3) 20.3 (23.1) Rfree (%) 23.2 (27.6) 25.2 (29.1) No. of atoms
Protein atoms 8220 8208 Solvent atoms 565 553 Metal atoms 0 4 Ligand 0 24
Model quality RMS deviation from ideal values Bond length (Å) 0.006 0.006 Bond angle (deg.) 1.10 1.10 Average B factors (Å2)
Overall 38.8 40.7 Protein 38.67 40.74 Water 40.18 39.9 Mg2+ 0 41.34 Pyruvic acid 0 38.60 Residues in Ramachandran plot (%) Most favored region 92.3% 91.4% Allowed region 7.3% 8.2 % Generously allowed region 0.3% 0.2% Disallowed region 0.1% 0.2%
Figure 6.5 Ramachandran plot for the A, B, C and D subunits of native PrpB.
179

Chapter 6 2-methylisocitrate lyase (PrpB)
In all the other datasets of PrpB obtained after co-crystallization with various
ligands, no electron density was observed corresponding to the ligand and the active site
loop was found to be disordered. Even in the new crystal form obtained after co-
crystallization of PrpB with 3-bromopyruvate, no electron density was observed for the
3-bromopyruvate as well as for the active site loop. 3-bromopyruvate is expected to bind
to the active site Cys123 and in the absence of electron density corresponding to the
active site loop, it was not clear if it had bound to Cys or not. It might be possible that
even after binding to Cys123, active site loop is highly flexible in the crystal.
6.3.3 Overall structure of native PrpB
The structures of the native and the ligand bound 2-methylisocitrate lyase (PrpB)
have been determined to a resolution of 2.1 and 2.3 Å, respectively. The asymmetric unit
contains a tetramer, the active oligomeric state, with 222 symmetry (Fig. 6.6). These
subunits are designated as A, B, C and D in the following discussions. Like all members
of the isocitrate lyase family, which have been biochemically characterized so far
(Britton et al. 2000; Britton et al. 2001; Brock et al. 2001; Sharma et al. 2000), PrpB
forms a homotetramer (Grimek et al. 2003). In the crystal structure, contacts across one
of the 2-folds (AB dimer) are extensive in comparison with other 2-folds (AC and AD
dimers) giving predominantly a dimer of dimers appearance to the tetramer.
Figure 6.6 The quaternary structure of
homotetrameric 2-methylisocitrate
lyase (dimer of dimers), with each
subunit colored differently. The four
subunits are related by 222 symmetry.
180

Chapter 6 2-methylisocitrate lyase (PrpB)
(b) (a)
Figure 6.7 Comparison of the tertiary structure of (a) PrpB from S. typhimurium and (b) triose-
phosphate isomerase from Plasmodium falciparum. The α-helices and the β-strands are shown in
sky blue and green, respectively. The C-terminal eighth helix in PrpB subunit is not part of the
(β/α)8 barrel of the same subunit. Therefore, the overall fold in PrpB consists of (β/α)7β.
Figure 6.8 Topology diagram of the secondary structural elements of 2-methylisocitrate lyase.
Arrows represent β-strands, while cylinders represent α-helices. β-strands and α-helices are
colored cyan and green, respectively.
181

Chapter 6 2-methylisocitrate lyase (PrpB)
The subunit structure of PrpB consists of a (β/α)8 barrel motif, reminiscent of
TIM (triosephosphate isomerase) barrels (Fig. 6.7). The inner strands are longer when
compared to the canonical triosephosphate isomerase strands (Velanker et al. 1997). As
in E. coli PrpB and other isocitrate lyases, the carboxyl-terminal eighth helix protrudes
away from the subunit and is not a part of the (β/α)8 barrel of the same subunit. It fills a
similar cavity in the (β/α)8 barrel of the neighboring subunit in a dimer. Thus, the overall
fold consists of (β/α)7β as shown in figure 6.8. Exchange of the eighth helix between
symmetry related subunits has been called “helix swapping”. This exchange leads to the
formation of a partial “knot” like structure such that the two subunits in a dimer cannot be
separated from each other without conformational changes in a loop region between the
two helical segments of the swapped region (Fig. 6.9). Helix swapping is expected to
provide a mechanism for oligomeric assembly concomitant with folding.
Figure 6.9 Illustration of the “helix swapping” interaction between A/B and C/D subunits in 2-
methylisocitrate lyase (PrpB).
182

Chapter 6 2-methylisocitrate lyase (PrpB)
6.3.4 Structural comparison with E. coli PrpB
Because of high sequence identity (89%) between PrpB from S. typhimurium and
E. coli, we expected almost identical structures. However, some differences were indeed
observed. Further, these differences were found in and around the active site, in particular
the electron density for the active site loop and in mode of binding of Mg2+. In E. coli
PrpB as well as other isocitrate lyases, the active site loop between the fourth β-strand
and the sixth α-helix of the (β/α)7β barrel is expected to move from an “open” state in the
ligand free enzyme to a “closed” state upon ligand binding. The open conformation of
this loop in the E. coli PrpB enzyme is distinct from the corresponding conformation in
isocitrate lyases reflecting the conformational variability of this segment (Grimm et al.
2003). The active site loop is found to be completely disordered in Salmonella PrpB. The
open conformation adopted by the active site loop of E. coli PrpB differs completely
from the open conformation observed in the three known structures of substrate-free
isocitrate lyases. The conformation of this loop in E. coli PrpB mainly results from its
interaction with the carboxyl-terminal helix of the second subunit within the same dimer,
although slight contacts of this loop to both α-helix 4 and the loop connecting β-strand 3
and α-helix 3 of a third subunit within the same tetramer are also observed. In contrast, in
the structures of ligand-free isocitrate lyases the active site loops interact in their open
form with a β-sheet which is inserted between the fifth β-strand and the fifth α-helix of
the (β/α)8 barrel, which is absent in PrpB. In addition, about 17 carboxyl-terminal
residues of isocitrate lyase which also undergo a conformational change upon substrate
binding in order to lock the active site loop in its closed conformation are missing in
PrpB. Therefore, the conformational change taking place in PrpB upon ligand binding
must essentially be restricted to the movement of the active site loop (Grimm et al. 2003).
In a PrpB dimer, since C-terminal end of one subunit comes close to the active
site loop of the other subunit; we suspected that the hexa-histidine tag present at the C-
terminal end in the recombinant enzyme may be responsible for the disorder of the loop.
To rule out any such possibility, PrpB was cloned in pRSET C vector with an N-terminal
hexa-histidine tag and the recombinant enzyme was purified and crystallized. Crystals of
PrpB with the N-terminal His tag diffracted poorly when compared to that of the PrpB
with the C-terminal His tag. Structure of the N-terminal His tag PrpB was solved to a
183

Chapter 6 2-methylisocitrate lyase (PrpB)
resolution of 3Å using a protomer of PrpB with the C-terminal His tag as a model (data
not shown). Even in the N-terminal His tag PrpB structure, the active site loop was found
to be disordered in all the four subunits. This indicated that the active site is inherently
more flexible in S. typhimurium compared to other members of isocitrate lyase family.
In the enzymes of isocitrate lyase family, the catalysis is strictly dependent on
Mg2+. Accordingly, a Mg2+ is observed within the active site of E. coli PrpB (Grimm et
al. 2003) and all isocitrate lyase crystal structures determined so far, regardless of
whether Mg2+ was added during crystallization or not (Britton et al. 2000; Britton et al.
2001; Brock et al. 2001; Sharma et al. 2000). But in the present case, where Mg2+ was
not added during the purification and crystallization experiments, electron density for the
Mg2+ couldn’t be located in the native PrpB. However, there is some evidence of bound
Mg2+ in the A and B subunits. In C subunit, electron density for Asp87 is very poor
whereas in D subunit, no electron density was found for Asp87 side chain. The low
occupancy of Mg2+ probably has led to a degree of order in Asp87 in the A and B
subunits. Despite the absence of Mg2+ in the present case, Asp87, which is directly
coordinated to Mg2+ in the E. coli native PrpB, is present at the border between the
generously allowed and disallowed regions of the Ramachandran plot (Fig. 6.10).
Figure 6.10 Stereo view of the fit of Asp87 having main chain torsion angles in the unfavorable
conformation to the 2Fo-Fc electron density map contoured at 1.0σ.
184

Chapter 6 2-methylisocitrate lyase (PrpB)
Another minor difference between the two structures is in the peptide bond of
Pro18 in Salmonella PrpB which was found in the cis-configuration in all the four
subunits in both the native and in the complex structures of PrpB, and the associated
electron density is very clear (Fig. 6.11). The corresponding residue Pro19 in E. coli PrpB
has been built in the trans-configuration.
Figure 6.11 Stereo view of the fit of Pro18 cis-peptide to the 2Fo-Fc electron density map contoured
at 1.0 σ.
6.3.5 Structural comparison with bacterial isocitrate lyase
2-methylisocitrate lyase from S. typhimurium has a molecular mass of
approximately 32 kDa per subunit, which is 35% lower than the molecular mass of
bacterial isocitrate lyase that is approximately 48 kDa per subunit. The PrpB protein is
therefore one of the smallest members of the isocitrate lyase family and seems to have all
essential amino acids conserved. The KKCGH sequence at the catalytic site of isocitrate
lyase from E. coli is conserved in all other known isocitrate lyases. In contrast, PrpB
from S. typhimurium and E. coli, the deduced 2-methylisocitrate lyase from S. cerevisiae
(Luttik et al. 2000), carboxyphosphoenolpyruvate phosphono mutase from Streptomyces
hygroscopicus (Hidaka et al. 1990) and five proteobacterial homologues of PrpB contain
the slightly modified sequence KRCGH in the catalytic site (Brock et al. 2001).
185

Chapter 6 2-methylisocitrate lyase (PrpB)
Comparison of structures of S. typhimurium PrpB and E. coli isocitrate lyase
shows a number of insertions, present only in the latter enzyme (Fig. 6.12). In both the
enzymes the N-terminal end is shielded from the solvent by an α-helix. In E. coli
isocitrate lyase, two more α-helices, which are absent in PrpB, precede this helix. The
eighth helix present at the C-terminal, which is involved in helix swapping, is prolonged
by 15-20 residues in bacterial isocitrate lyases compared to PrpB. Insertion of 25-30
residues between sixth β-strand (PrpB) and seventh α-helix (PrpB) in the case of
isocitrate lyases form an extra pair of β-strands resulting in the formation of an
antiparallel β-sheet. The sixth α-helix in isocitrate lyase, corresponding to fourth α-helix
of PrpB, is longer by 10-12 residues and is followed by an extended loop.
Figure 6.12 Structural superposition of 2-methylisocitrate lyase (shown in sky blue) from S.
typhimurium and isocitrate lyase (shown in green) from E. coli depicting regions conserved in
PrpB, the smallest member of the isocitrate lyase family. Regions that are absent in PrpB but
present in isocitrate lyase are colored in coral.
186

Chapter 6 2-methylisocitrate lyase (PrpB)
6.3.6 Overall structure of pyruvate/Mg2+ bound PrpB Unlike native PrpB, Mg2+ could be easily located in the pyruvate bound PrpB
model (Fig. 6.13). In the Fo-Fc map of pyruvate bound PrpB model, weak electron
density is present near Asp 87 which seems to correspond to the Mg2+ position as seen in
native PrpB from E. coli (Grimm et al. 2003). However, confident assignment was not
possible due to the very low occupancy. Binding of pyruvate seems to trigger the
movement of Mg2+ away from Asp87. This movement is approximately 2.5 Å towards
pyruvate (Fig. 6.15). The pyruvic acid binds with the two carboxylate groups
coordinating to Mg2+, which in turn interacts with two (A and C subunits) or three (B and
D subunits) water molecules and with the carboxylate group of Asp85 (Fig. 6.14). The
coordination geometries of Mg2+ ions are octahedral in B and D subunits and square
bipyramidal in A and C subunits. In the model, cation-ligand distances have slightly
larger deviation from the target values, probably due to the presence of Mg2+ with partial
occupancy at two different places, one close to pyruvate as in PrpB-pyruvate model and
another close to Asp87 as seen in E. coli PrpB.
Figure 6.13 Stereo view of the electron
density corresponding to pyruvic acid
and Mg2+ in the Fo–Fc map. The contours
are drawn at 3 σ.
In pyruvate bound PrpB model, pyruvate-protein interactions occur mainly
through Ser45OG, Gly47N and Arg158NH1 of the protein (Fig. 6.16(a)). These residues
are conserved in isocitrate lyases, which bind glyoxylate. However in the ligand bound
isocitrate lyase from Mycobacterium tuberculosis, glyoxylate binds to the atoms
Ser91OG, Gly92N, Trp93N and Arg228NH2 of the protein (Fig. 6.16(b)) (Sharma et al.
2000). Methyl group of pyruvate in the present case is placed in a hydrophobic
depression formed by Phe186, Leu234 and Pro236. In all known isocitrate lyases (ICLs),
these three residues are replaced by Trp283, Phe345 and Thr347.
187

Chapter 6 2-methylisocitrate lyase (PrpB)
Figure 6.14 Stereo diagram of the electron density corresponding to the Mg2+ coordination in the
active site from the 2Fo-Fc electron density map contoured at 1.0 σ.
Figure 6.15 Substrate binding pocket of PrpB from S. typhimurium. Residues forming the pocket
and pyruvate present near the active site are shown as ball and stick models. Mg2+ present in the
active site is shown as a sphere colored in magenta.
188

Chapter 6 2-methylisocitrate lyase (PrpB)
Replacement of glyoxylate by pyruvate in the glyoxylate binding pocket of
isocitrate lyase brings the side-chain methyl group of Thr347 into close contact (with in
2.4Å) with the methyl group of pyruvate (Fig. 6.17). The residue Thr347 in the isocitrate
lyase is replaced by Pro236 in PrpB. Here proline participates in the formation of a
hydrophobic depression accommodating the methyl group of pyruvate not present in
glyoxylate. Therefore, it seems that the presence of these three residues, Phe186, Leu234
and Pro236 in PrpB; Trp283, Phe345 and Thr347 in isocitrate lyases, provide substrate
specificity for pyruvate and glyoxylate, respectively. Accordingly, sequence comparison
between all known bacterial 2-methylisocitrate lyases and isocitrate lyases shows that
these three residues, Phe186, Leu234 and Pro236 in case of PrpB and Trp283, Phe345
and Thr347 in case of isocitrate lyases, are strictly conserved (Grimm et al. 2003).
(b)
(a)
Figure 6.16 Stereo view of (a) interaction of Mg2+ and pyruvate with Salmonella typhimurium 2-
methylisocitrate lyase and (b) interaction of Mg2+ and glyoxylate with isocitrate lyase from
Mycobaterium tuberculosis. Residues Phe186, Leu234 and Pro236 in PrpB and Trp283, Phe345 and
Thr347 in isocitrate lyase appear to provide substrate specificity for pyruvate and glyoxylate,
respectively.
189

Chapter 6 2-methylisocitrate lyase (PrpB)
Figure 6.17 Glyoxylate replaced by the pyruvate in the ligand-binding pocket of M. tuberculosis
isocitrate lyase. Methyl group of pyruvate comes unfavorably close to the Thr347 side-chain
methyl group of isocitrate lyase (indicated by a red dotted line).
6.3.7 Catalytic mechanism
Most of the residues thought to be involved in the catalysis of isocitrate lyase are
conserved in 2-methylisocitrate lyase. Since these two enzymes catalyze similar reactions
with the substrates differing in only one methyl group, 2-methylisocitrate instead of
isocitrate (in the forward reaction) or pyruvate instead of glyoxylate (in the reverse
reaction), they are expected to follow similar reaction mechanisms. In the recent past,
site-directed mutagenesis experiments on Salmonella PrpB have shown that Cys123,
present in 2-methylisocitrate lyase signature sequence KRCGH, and Asp58 are critical to
catalysis (Grimek et al. 2003). As seen in the E. coli native PrpB structure, Cys123 is
proposed to serve as the active site base and residue Asp58 is critical for the coordination
to the Mg2+.
Sharma et. al., have proposed a reaction mechanism for the isocitrate lyase,
deduced from an analysis of crystal structures of native and inhibitor bound forms of
isocitrate lyase (Sharma et al. 2000). In the reverse reaction catalyzed by isocitrate lyase,
in which glyoxylate and succinate are condensed to isocitrate, glyoxylate binds first and
is followed by the succinate to form a ternary complex. This is based on the observation
that glyoxylate is buried deeper in the active site than succinate and loop closure requires
succinate binding. The key step in the reaction is the deprotonation from the Cα atom of
the carboxylate of succinate to a base, most probably to Cys191 (ICL numbering) in the
case of isocitrate lyase and Cys123 (PrpB numbering) in the case of 2-methylisocitrate
190

Chapter 6 2-methylisocitrate lyase (PrpB)
lyase. A general acid, probably Glu295 (ICL numbering) in isocitrate lyase, protonates
the carboxylate adjacent to the bond formed. It has been proposed that the negative
charge on the aldehyde oxygen atom of glyoxylate is stabilized by Mg2+ and two other
basic residues (Sharma et al. 2000). In agreement with these observations, negative
charge of pyruvate is stabilized by Mg2+ in the case of pyruvate bound PrpB model.
According to Sharma et. al., binding of succinate appears to trigger the movement
of the Mg2+ by 2.5 Å towards glyoxylate, allowing Lys in the active site to form
electrostatic interactions within this region and facilitating closure of the active site loop
over the bound substrates. Surprisingly, in pyruvate/ Mg2+ bound PrpB model, even in the
absence of succinate, Mg2+ appears to have moved by approximately 2.5 Å leading to
stabilization of the negative charge of pyruvate (Fig. 6.15). A complete understanding of
the mechanism of PrpB and its differences, if any, from that of isocitrate lyase, therefore,
will require the structure of PrpB-2-methylisocitrate or PrpB-pyruvate-succinate complex
with the active site loop in the closed conformation.
6.4. Conclusions
Analysis of the structure of Salmonella typhimurium 2-methylisocitrate lyase and
its complex with pyruvate/Mg2+ has provided important information on the active site
region involved in binding of pyruvate and metal ion in the protein. Even in the absence
of succinate, Mg2+ seems to move towards pyruvate from its position in the E. coli native
PrpB. In the present case, the active site loop, which includes the critical cysteine residue
that acts as the base during catalysis, is completely disordered in both native and
pyruvate/Mg2+ bound PrpB.
Comparison of Salmonella typhimurium 2-methylisocitrate lyase with the known
bacterial isocitrate lyase reveals regions within the isocitrate lyase that are not present in
PrpB, one of the smallest members of the family. These regions mainly comprise of
amino and carboxyl terminal ends as well as specific insertions within loops connecting
β-strands and α-helices of the (β/α)7β barrel.
Sequence comparison among all known bacterial 2-methylisocitrate lyases and
isocitrate lyases shows that Phe186, Leu234 and Pro236 residues, which bind pyruvate in
the case of 2-methylisocitrate lyase, and Trp283, Phe345 and Thr347, which bind
191

Chapter 6 2-methylisocitrate lyase (PrpB)
glyoxylate in the case of isocitrate lyases, are strictly conserved, which seems to confer
substrate specificity in these two enzymes. Similarities and differences in the structure of
substrates, the active site region and the residues that interact with the ligands in these
enzymes suggest similar catalytic mechanisms in isocitrate lyases and 2-methylisocitrate
lyases.
192

Future perspectives

Future perspectives
The present work on biodegradative threonine deaminase (TdcB) has provided
structural insights on ligand-induced oligomerization, and the framework needed for
understanding the significant increase in the affinity of substrate binding and Vmax. The
following extension of the work is planned to enhance the scope of the present study.
Mutational analysis of residues interacting with CMP molecule at the dimer interface in
tetrameric TdcB-CMP complex may further increase our understanding of the role of
CMP in enzyme activation. To understand if tetramerization is important for enzyme
activation, some of the key residues present at the dimer-dimer interface of TdcB could
be mutated so that TdcB fails to form tetramers even in the presence of AMP/CMP. It
will be interesting to see if these dimeric TdcB molecules obtained in the presence of
activator molecule AMP/CMP will still be able to show significant enzyme activation.
Apart from overall structural features, the present work on propionate kinase has
revealed the mode of nucleotide binding and its relationship to the proposed binding site
of propionate. In future, the main aim of the study on propionate kinase would be to
understand the reaction mechanism and role of domain motion in catalytic process. To
achieve this, extensive biochemical and structural studies on a large number of active site
mutants of propionate kinase would be carried out. Attempts would be made to determine
the structures of propionate kinase with compounds mimicking reaction intermediates
such as AlF3, ADP and propionate.
To prove conclusively that Ap4A synthetic activity is present in propionate kinase,
it is important to demonstrate the formation of Ap4A in solution by incubating ATP with
propionate kinase. Towards this end, attempts will be made to show the presence of Ap4A
in a reaction mixture containing propionate kinase, ATP and Mg2+. Liquid
chromatography electrospray ionisation mass spectrometry (LC-ESI-MS) will be
employed to separate the nucleotides from the reaction mixture after incubation for
different time intervals. We also plan to employ thin-layer chromatography to separate
the mixture of nucleotides after incubating propionate kinase and Mg2+ with α-32P labeled
ATP for different time intervals. Due to the presence of radiolabeled 32P at the α-
phosphate, all the nucleotides in the reaction mixture will be labeled, which can be
observed on a chromatogram.
194

Future perspectives
Our efforts on the four different Prp enzymes have led so far to the structure of
only PrpB. Since PrpB has been extensively studied by four different research groups
around the world, in future we plan to focus on PrpC (2-methylisocitrate synthase), an
enzyme whose structure is not available from any source. We have already achieved
initial crystallization hits on this protein, which we hope to improve in the future.
Attempts will be made to prevent extensive precipitation of PrpC during crystallization
by optimizing the buffer as well as additives.
195

References

References
Alexander DC (1987) An efficient vector-primer cDNA cloning system. Methods Enzymol 154, 41-64. Alexander FW, Sandmeier E, Mehta PK, Christen P (1994) Evolutionary relationships among pyridoxal-5'-phosphate-dependent enzymes. Regio-specific alpha, beta and gamma families. Eur J Biochem 219, 953-960. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389-3402. Anthony RS, Spector LB (1971) Exchange reactions catalyzed by acetate kinase. J Biol Chem 246, 6129-6135. Anthony RS, Spector LB (1972) Phosphorylated acetate kinase. Its isolation and reactivity. J Biol Chem 247, 2120-2125. Atencia EA, Madrid O, Gunther Sillero MA, Sillero A (1999) T4 RNA ligase catalyzes the synthesis of dinucleoside polyphosphates. Eur J Biochem 261, 802-811. Baker JC, Jacobson MK (1986) Alteration of adenyl dinucleotide metabolism by environmental stress. Proc Natl Acad Sci U S A 83, 2350-2352. Baker MD, Holloway DE, Swaminathan GJ, Acharya KR (2006) Crystal structures of eosinophil-derived neurotoxin (EDN) in complex with the inhibitors 5'-ATP, Ap3A, Ap4A, and Ap5A. Biochemistry 45, 416-426. Barnes EM, Impey CS, Stevens BJ (1979) Factors affecting the incidence and anti-salmonella activity of the anaerobic caecal flora of the young chick. J Hyg (Lond) 82, 263-283. Baxi MD, Vishwanatha JK (1995) Diadenosine polyphosphates: their biological and pharmacological significance. J Pharmacol Toxicol Methods 33, 121-128. Berg P (1956) Acyl adenylates; an enzymatic mechanism of acetate activation. J Biol Chem 222, 991-1013. Bessman MJ, Frick DN, O'Handley SF (1996) The MutT proteins or "Nudix" hydrolases, a family of versatile, widely distributed, "housecleaning" enzymes. J Biol Chem 271, 25059-25062. Bessman MJ, Walsh JD, Dunn CA, Swaminathan J, Weldon JE, Shen J (2001) The gene ygdP, associated with the invasiveness of Escherichia coli K1, designates a Nudix hydrolase, Orf176, active on adenosine (5')-pentaphospho-(5')-adenosine (Ap5A). J Biol Chem 276, 37834-37838.
197

References
Bhadra R, Datta P (1978) Allosteric inhibition and catabolite inactivation of purified biodegradative threonine dehydratase of Salmonella typhimurium. Biochemistry 17, 1691-1699. Bhat TN (1988) Calculation of an OMIT map. J. Appl. Cryst. 21, 279-281. Blankenhorn D, Phillips J, Slonczewski JL (1999) Acid- and base-induced proteins during aerobic and anaerobic growth of Escherichia coli revealed by two-dimensional gel electrophoresis. J Bacteriol 181, 2209-2216. Blattler WA, Knowles JR (1979) Stereochemical course of phosphokinases. The use of adenosine [gamma-(S)-16O,17O,18O]triphosphate and the mechanistic consequences for the reactions catalyzed by glycerol kinase, hexokinase, pyruvate kinase, and acetate kinase. Biochemistry 18, 3927-3933. Blattner FR, Plunkett G, 3rd, et al. (1997) The complete genome sequence of Escherichia coli K-12. Science 277, 1453-1474. Bochner BR, Lee PC, Wilson SW, Cutler CW, Ames BN (1984) AppppA and related adenylylated nucleotides are synthesized as a consequence of oxidation stress. Cell 37, 225-232. Bohnhoff M, Miller CP (1962) Enhanced susceptibility to Salmonella infection in streptomycin-treated mice. J Infect Dis 111, 117-127. Bond CS (2003) TopDraw: a sketchpad for protein structure topology cartoons. Bioinformatics 19, 311-312. Bork P, Sander C, Valencia A (1992) An ATPase domain common to prokaryotic cell cycle proteins, sugar kinases, actin, and hsp70 heat shock proteins. Proc Natl Acad Sci U S A 89, 7290-7294. Bossemeyer D (1994) The glycine-rich sequence of protein kinases: a multifunctional element. Trends Biochem Sci 19, 201-205. Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72, 248-254. Brevet A, Coste H, Fromant M, Plateau P, Blanquet S (1987) Yeast diadenosine 5',5'''-P1,P4-tetraphosphate alpha,beta-phosphorylase behaves as a dinucleoside tetraphosphate synthetase. Biochemistry 26, 4763-4768.
198

References
Britton K, Langridge S, Baker PJ, Weeradechapon K, Sedelnikova SE, De Lucas JR, Rice DW, Turner G (2000) The crystal structure and active site location of isocitrate lyase from the fungus Aspergillus nidulans. Structure 8, 349-362. Britton KL, Abeysinghe IS, et al. (2001) The structure and domain organization of Escherichia coli isocitrate lyase. Acta Crystallogr D Biol Crystallogr 57, 1209-1218. Brock M, Darley D, Textor S, Buckel W (2001) 2-Methylisocitrate lyases from the bacterium Escherichia coli and the filamentous fungus Aspergillus nidulans: characterization and comparison of both enzymes. Eur J Biochem 268, 3577-3586. Brock M, Fischer R, Linder D, Buckel W (2000) Methylcitrate synthase from Aspergillus nidulans: implications for propionate as an antifungal agent. Mol Microbiol 35, 961-973. Brock M, Maerker C, Schutz A, Volker U, Buckel W (2002) Oxidation of propionate to pyruvate in Escherichia coli. Involvement of methylcitrate dehydratase and aconitase. Eur J Biochem 269, 6184-6194. Brunger AT (1992) Free R-Value - a novel statistical quantity for assessing the accuracy of crystal-structures. Nature 355, 472-475. Brunger AT (1993) Assessment of phase accuracy by cross validation: the free R value. Methods and applications. Acta Crystallogr D Biol Crystallogr 49, 24-36. Brunger AT, Adams PD, et al. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 54, 905-921. Brunger AT, Adams PD, Rice LM (1999) Annealing in crystallography: a powerful optimization tool. Prog Biophys Mol Biol 72, 135-155. Buckel W, Bobi A (1976) The enzyme complex citramalate lyase from Clostridium tetanomorphum. Eur J Biochem 64, 255-262. Burkhard P, Rao GS, Hohenester E, Schnackerz KD, Cook PF, Jansonius JN (1998) Three-dimensional structure of O-acetylserine sulfhydrylase from Salmonella typhimurium. J Mol Biol 283, 121-133. Burland V, Plunkett G, 3rd, Sofia HJ, Daniels DL, Blattner FR (1995) Analysis of the Escherichia coli genome VI: DNA sequence of the region from 92.8 through 100 minutes. Nucleic Acids Res 23, 2105-2119. Burman JD, Stevenson CE, Hauton KA, Sawers G, Lawson DM (2003) Crystallization and preliminary X-ray analysis of the E. coli hypothetical protein TdcF. Acta Crystallogr D Biol Crystallogr 59, 1076-1078.
199

References
Buss KA, Cooper DR, Ingram-Smith C, Ferry JG, Sanders DA, Hasson MS (2001) Urkinase: structure of acetate kinase, a member of the ASKHA superfamily of phosphotransferases. J Bacteriol 183, 680-686. Bystrom CE, Pettigrew DW, Branchaud BP, O'Brien P, Remington SJ (1999) Crystal structures of Escherichia coli glycerol kinase variant S58-->W in complex with nonhydrolyzable ATP analogues reveal a putative active conformation of the enzyme as a result of domain motion. Biochemistry 38, 3508-3518. Cameselle JC, Costas MJ, Sillero MA, Sillero A (1982) Bis-(5'-guanosyl) tetra-phosphatase in rat tissues. Biochem J 201, 405-410. Cartwright JL, Britton P, Minnick MF, McLennan AG (1999) The IalA invasion gene of Bartonella bacilliformis encodes a (de)nucleoside polyphosphate hydrolase of the MutT motif family and has homologs in other invasive bacteria. Biochem Biophys Res Commun 256, 474-479. Cartwright JL, McLennan AG (1999) The Saccharomyces cerevisiae YOR163w gene encodes a diadenosine 5',5"'-P1,P6-hexaphosphate (Ap6A) hydrolase member of the MutT motif (Nudix hydrolase) family. J Biol Chem 274, 8604-8610. CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50, 760-763. Cherrington CA, Hinton M, Mead GC, Chopra I (1991) Organic acids: chemistry, antibacterial activity and practical applications. Adv Microb Physiol 32, 87-108. Cohen GE (1997) ALIGN: a program to superimpose protein coordinates accounting for insertions and deletions. J. Appl. Crystallog. 30, 1160-1161. Conyers GB, Bessman MJ (1999) The gene, ialA, associated with the invasion of human erythrocytes by Bartonella bacilliformis, designates a nudix hydrolase active on dinucleoside 5'-polyphosphates. J Biol Chem 274, 1203-1206. Coste H, Brevet A, Plateau P, Blanquet S (1987) Non-adenylylated bis(5'-nucleosidyl) tetraphosphates occur in Saccharomyces cerevisiae and in Escherichia coli and accumulate upon temperature shift or exposure to cadmium. J Biol Chem 262, 12096-12103. Crowther RA, Blow DM (1967) A method of positioning a known molecule in an unknown crystal structure. Acta Crystallographica 23, 544-548. Dailey FE, Berg HC (1993) Change in direction of flagellar rotation in Escherichia coli mediated by acetate kinase. J Bacteriol 175, 3236-3239.
200

References
Datta P, Goss TJ, Omnaas JR, Patil RV (1987) Covalent structure of biodegradative threonine dehydratase of Escherichia coli: homology with other dehydratases. Proc Natl Acad Sci U S A 84, 393-397. Dauter Z (1999) Data-collection strategies. Acta Crystallogr D Biol Crystallogr 55, 1703-1717. DeLano WL (2002) The PyMOL Molecular Graphics System. (DeLano Scientific: San Carlos, CA). Dieckmann R, Pavela-Vrancic M, von Dohren H (2001) Synthesis of (di)adenosine polyphosphates by non-ribosomal peptide synthetases (NRPS). Biochim Biophys Acta 1546, 234-241. Eisenstein E (1991) Cloning, expression, purification, and characterization of biosynthetic threonine deaminase from Escherichia coli. J Biol Chem 266, 5801-5807. Eliot AC, Kirsch JF (2004) Pyridoxal phosphate enzymes: Mechanistic, structural, and evolutionary considerations. Annual Review of Biochemistry 73, 383-415. Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126-2132. Esnouf RM (1999) Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr D Biol Crystallogr 55, 938-940. Evans CT, Sumegi B, Srere PA, Sherry AD, Malloy CR (1993) [13C] propionate oxidation in wild-type and citrate synthase mutant Escherichia coli: evidence for multiple pathways of propionate utilization. Biochem J 291, 927-932. Farr SB, Arnosti DN, Chamberlin MJ, Ames BN (1989) An apaH mutation causes AppppA to accumulate and affects motility and catabolite repression in Escherichia coli. Proc Natl Acad Sci U S A 86, 5010-5014. Fernandez-Briera A, Garrido-Pertierra A (1988) A degradation pathway of propionate in Salmonella typhimurium LT2. Biochimie 70, 757-768. Fisher KE, Eisenstein E (1993) An efficient approach to identify ilvA mutations reveals an amino-terminal catalytic domain in biosynthetic threonine deaminase from Escherichia coli. J Bacteriol 175, 6605-6613. Flaherty KM, DeLuca-Flaherty C, McKay DB (1990) Three-dimensional structure of the ATPase fragment of a 70K heat-shock cognate protein. Nature 346, 623-628.
201

References
Flores NA, Stavrou BM, Sheridan DJ (1999) The effects of diadenosine polyphosphates on the cardiovascular system. Cardiovasc Res 42, 15-26. Fontes R, Sillero MA, Sillero A (1998) Acyl coenzyme A synthetase from Pseudomonas fragi catalyzes the synthesis of adenosine 5'-polyphosphates and dinucleoside polyphosphates. J Bacteriol 180, 3152-3158. Fox DK, Meadow ND, Roseman S (1986) Phosphate transfer between acetate kinase and enzyme I of the bacterial phosphotransferase system. J Biol Chem 261, 13498-13503. French S, Wilson K (1978) On the treatment of negative intensity observations. Acta Crystallographica Section A 34, 517-525. Gallagher DT, Gilliland GL, Xiao G, Zondlo J, Fisher KE, Chinchilla D, Eisenstein E (1998) Structure and control of pyridoxal phosphate dependent allosteric threonine deaminase. Structure 6, 465-475. Ganduri YL, Sadda SR, Datta MW, Jambukeswaran RK, Datta P (1993) TdcA, a transcriptional activator of the tdcABC operon of Escherichia coli, is a member of the LysR family of proteins. Mol Gen Genet 240, 395-402. Garrison PN, Barnes LD (1992) Determination of dinucleoside polyphosphates. In Ap4A and other dinucleoside polyphosphates. McLennnan, A. G. (ed) (CRC press: Boca Raton, FL). Gerike U, Hough DW, Russell NJ, Dyall-Smith ML, Danson MJ (1998) Citrate synthase and 2-methylcitrate synthase: structural, functional and evolutionary relationships. Microbiology 144, 929-935. Gerlt JA, Rabinowitz KW, Dunne CP, Wood WA (1973) The mechanism of action of 5'-adenylic acid-activated threonine dehydrase. V. Relation between ligand-induced allosteric activation and the protomeroligomer interconversion. J Biol Chem 248, 8200-8206. Gilson G, Ebel JP, Remy P (1988) Is Ap4A involved in DNA repair processes? Exp Cell Res 177, 143-153. Goerlich O, Foeckler R, Holler E (1982) Mechanism of synthesis of adenosine(5')tetraphospho(5')adenosine (AppppA) by aminoacyl-tRNA synthetases. Eur J Biochem 126, 135-142. Gorrell A, Lawrence SH, Ferry JG (2005) Structural and kinetic analyses of arginine residues in the active site of the acetate kinase from Methanosarcina thermophila. J Biol Chem 280, 10731-10742.
202

References
Gouet P, Robert X, Courcelle E (2003) ESPript/ENDscript: Extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res 31, 3320-3323. Grimek TL, Holden H, Rayment I, Escalante-Semerena JC (2003) Residues C123 and D58 of the 2-methylisocitrate lyase (PrpB) enzyme of Salmonella enterica are essential for catalysis. J Bacteriol 185, 4837-4843. Grimm C, Evers A, Brock M, Maerker C, Klebe G, Buckel W, Reuter K (2003) Crystal structure of 2-methylisocitrate lyase (PrpB) from Escherichia coli and modelling of its ligand bound active centre. Journal of Molecular Biology 328, 609-621. Grummt F, Waltl G, Jantzen HM, Hamprecht K, Huebscher U, Kuenzle CC (1979) Diadenosine 5',5'''-P1,P4-tetraphosphate, a ligand of the 57-kilodalton subunit of DNA polymerase alpha. Proc Natl Acad Sci U S A 76, 6081-6085. Guillouet S, Rodal AA, An G, Lessard PA, Sinskey AJ (1999) Expression of the Escherichia coli catabolic threonine dehydratase in Corynebacterium glutamicum and its effect on isoleucine production. Appl Environ Microbiol 65, 3100-3107. Gunther S, Montes M, de DA, del VM, Atencia EA, Sillero A (2002) Thermostable Pyrococcus furiosus DNA ligase catalyzes the synthesis of (di)nucleoside polyphosphates. Extremophiles 6, 45-50. Guranowski A (2000) Specific and nonspecific enzymes involved in the catabolism of mononucleoside and dinucleoside polyphosphates. Pharmacol Ther 87, 117-139. Guranowski A (2003) Analogs of diadenosine tetraphosphate (Ap4A). Acta Biochim Pol 50, 947-972. Guranowski A, Blanquet S (1985) Phosphorolytic cleavage of diadenosine 5',5'''-P1,P4-tetraphosphate. Properties of homogeneous diadenosine 5',5'''-P1,P4-tetraphosphate alpha, beta-phosphorylase from Saccharomyces cerevisiae. J Biol Chem 260, 3542-3547. Guranowski A, Jakubowski H, Holler E (1983) Catabolism of diadenosine 5',5"'-P1,P4-tetraphosphate in procaryotes. Purification and properties of diadenosine 5',5"'-P1,P4-tetraphosphate (symmetrical) pyrophosphohydrolase from Escherichia coli K12. J Biol Chem 258, 14784-14789. Guranowski A, Just G, Holler E, Jakubowski H (1988) Synthesis of diadenosine 5',5'''-P1,P4-tetraphosphate (AppppA) from adenosine 5'-phosphosulfate and adenosine 5'-triphosphate catalyzed by yeast AppppA phosphorylase. Biochemistry 27, 2959-2964.
203

References
Guranowski A, Sillero MA, Sillero A (1990) Firefly luciferase synthesizes P1,P4-bis(5'-adenosyl)tetraphosphate (Ap4A) and other dinucleoside polyphosphates. FEBS Lett 271, 215-218. Hammelman TA, O'Toole GA, Trzebiatowski JR, Tsang AW, Rank D, Escalante-Semerena JC (1996) Identification of a new prp locus required for propionate catabolism in Salmonella typhimurium LT2. FEMS Microbiol Lett 137, 233-239. Harker D (1956) The determination of the phases of the structure factors of non-centrosymmetric crystals by the method of double isomorphous replacement. Acta Cryst. 9, 1-9. Harris TK, Wu G, Massiah MA, Mildvan AS (2000) Mutational, kinetic, and NMR studies of the roles of conserved glutamate residues and of lysine-39 in the mechanism of the MutT pyrophosphohydrolase. Biochemistry 39, 1655-1674. Harrison CJ, Hayer-Hartl M, Di Liberto M, Hartl F, Kuriyan J (1997) Crystal structure of the nucleotide exchange factor GrpE bound to the ATPase domain of the molecular chaperone DnaK. Science 276, 431-435. Hayward S, Lee RA (2002) Improvements in the analysis of domain motions in proteins from conformational change: DynDom version 1.50. J Mol Graph Model 21, 181-183. Hendrickson WA, Horton JR, LeMaster DM (1990) Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): a vehicle for direct determination of three-dimensional structure. Embo J 9, 1665-1672. Hendrickson WA, Horton JR, Murthy HM, Pahler A, Smith JL (1989) Multiwavelength anomalous diffraction as a direct phasing vehicle in macromolecular crystallography. Basic Life Sci 51, 317-324. Hesslinger C, Fairhurst SA, Sawers G (1998) Novel keto acid formate-lyase and propionate kinase enzymes are components of an anaerobic pathway in Escherichia coli that degrades L-threonine to propionate. Mol Microbiol 27, 477-492. Hidaka T, Imai S, Hara O, Anzai H, Murakami T, Nagaoka K, Seto H (1990) Carboxyphosphonoenolpyruvate phosphonomutase, a novel enzyme catalyzing C-P bond formation. J Bacteriol 172, 3066-3072. Higgins D, Thompson J, Gibson T, Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl. Acids Res 22, 4673-4680.
204

References
Horswill AR, Dudding AR, Escalante-Semerena JC (2001) Studies of propionate toxicity in Salmonella enterica identify 2-methylcitrate as a potent inhibitor of cell growth. J Biol Chem 276, 19094-19101. Horswill AR, Escalante-Semerena JC (1997) Propionate catabolism in Salmonella typhimurium LT2: two divergently transcribed units comprise the prp locus at 8.5 centisomes, prpR encodes a member of the sigma-54 family of activators, and the prpBCDE genes constitute an operon. J Bacteriol 179, 928-940. Horswill AR, Escalante-Semerena JC (1999a) The prpE gene of Salmonella typhimurium LT2 encodes propionyl-CoA synthetase. Microbiology 145 ( Pt 6), 1381-1388. Horswill AR, Escalante-Semerena JC (1999b) Salmonella typhimurium LT2 catabolizes propionate via the 2-methylcitric acid cycle. J Bacteriol 181, 5615-5623. Horswill AR, Escalante-Semerena JC (2001) In vitro conversion of propionate to pyruvate by Salmonella enterica enzymes: 2-methylcitrate dehydratase (PrpD) and aconitase Enzymes catalyze the conversion of 2-methylcitrate to 2-methylisocitrate. Biochemistry 40, 4703-4713. Horswill AR, Escalante-Semerena JC (2002) Characterization of the propionyl-CoA synthetase (PrpE) enzyme of Salmonella enterica: residue Lys592 is required for propionyl-AMP synthesis. Biochemistry 41, 2379-2387. Hubbard SJ, Thornton JM (1993) 'NACCESS, Computer Program, (Department of Biochemistry and Molecular Biology, University College: London). Hume ME, Corrier DE, Ambrus S, Hinton A, Jr., DeLoach JR (1993) Effectiveness of dietary propionic acid in controlling Salmonella typhimurium colonization in broiler chicks. Avian Dis 37, 1051-1056. Hurley JH (1996) The sugar kinase/heat shock protein 70/actin superfamily: implications of conserved structure for mechanism. Annu Rev Biophys Biomol Struct 25, 137-162. Hutchinson EG, Thornton JM (1996) PROMOTIF--a program to identify and analyze structural motifs in proteins. Protein Sci 5, 212-220. Hyde CC, Ahmed SA, Padlan EA, Miles EW, Davies DR (1988) Three-dimensional structure of the tryptophan synthase alpha 2 beta 2 multienzyme complex from Salmonella typhimurium. J Biol Chem 263, 17857-17871. Iyer PP, Lawrence SH, Luther KB, Rajashankar KR, Yennawar HP, Ferry JG, Schindelin H (2004) Crystal structure of phosphotransacetylase from the methanogenic archaeon Methanosarcina thermophila. Structure 12, 559-567.
205

References
Jack A, Levitt M (1978) Refinement of large structures by simultaneous minimization of energy and R factor. Acta Crystallographica Section A 34, 931-935. Jakubowski H, Guranowski A (1983) Enzymes hydrolyzing ApppA and/or AppppA in higher plants. Purification and some properties of diadenosine triphosphatase, diadenosine tetraphosphatase, and phosphodiesterase from yellow lupin (Lupinus luteus) seeds. J Biol Chem 258, 9982-9989. Jankowski J, Tepel M, van der Giet M, Tente IM, Henning L, Junker R, Zidek W, Schluter H (1999) Identification and characterization of -P1,P7-Di(adenosine-5')-heptaphosphate from human platelets. J Biol Chem 274, 23926-23931. Johnstone DB, Farr SB (1991) AppppA binds to several proteins in Escherichia coli, including the heat shock and oxidative stress proteins DnaK, GroEL, E89, C45 and C40. Embo J 10, 3897-3904. Jones TA, Zou JY, Cowan SW, Kjeldgaard (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47 ( Pt 2), 110-119. Kabara JJ, Eklund T (1991) 'Food Preservatives.' (Glasgow Blachie: New York). Kabsch W, Mannherz HG, Suck D, Pai EF, Holmes KC (1990) Atomic structure of the actin:DNase I complex. Nature 347, 37-44. Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577-2637. Kay WW (1972) Genetic control of the metabolism of propionate by Escherichia coli K12. Biochim Biophys Acta 264, 508-521. Kleywegt GJ, Jones TA (1994) Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr D Biol Crystallogr 50, 178-185. Kleywegt GJ, Jones TA (1998) Databases in protein crystallography. Acta Crystallog. D 54, 1119–1131. Ko YH, McFadden BA (1990) Alkylation of isocitrate lyase from Escherichia coli by 3-bromopyruvate. Arch Biochem Biophys 278, 373-380. Kraulis PJ (1991) Molscript - a program to produce both detailed and schematic plots of protein structures. Journal of Applied Crystallography 24, 946-950. Kunst F, Ogasawara N, et al. (1997) The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature 390, 249-256.
206

References
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680-685. LaRossa RA, Van Dyk TK (1989) Leaky pantothenate and thiamin mutations of Salmonella typhimurium conferring suphometuron methyl sensitivity. J Gen Microbiol 135, 2209-2222. Laskowski RA, McArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallog. 26, 283-291. Latimer MT, Ferry JG (1993) Cloning, sequence-analysis, and hyper-expression of the genes encoding phosphotransacetylase and acetate kinase from Methanosarcina-thermophila. Journal of Bacteriology 175, 6822-6829. Lawrence SH, Ferry JG (2006) Steady-state kinetic analysis of phosphotransacetylase from Methanosarcina thermophila. J Bacteriol 188, 1155-1158. Lawrence SH, Luther KB, Schindelin H, Ferry JG (2006) Structural and functional studies suggest a catalytic mechanism for the phosphotransacetylase from Methanosarcina thermophila. J Bacteriol 188, 1143-1154. Lee B, Richards FM (1971) The interpretation of protein structures: estimation of static accessibility. J Mol Biol 55, 379-400. Lee PC, Bochner BR, Ames BN (1983) Diadenosine 5',5"'-P1,P4-tetraphosphate and related adenylylated nucleotides in Salmonella typhimurium. J Biol Chem 258, 6827-6834. Lee RA, Razaz M, Hayward S (2003) The DynDom database of protein domain motions. Bioinformatics 19, 1290-1291. London RE, Allen DL, Gabel SA, DeRose EF (1999) Carbon-13 nuclear magnetic resonance study of metabolism of propionate by Escherichia coli. J Bacteriol 181, 3562-3570. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193, 265-275. Luginbuhl GH, Hofler JG, Decedue CJ, Burns RO (1974) Biodegradative L-threonine deaminase of Salmonella typhimurium. J Bacteriol 120, 559-561. Lundin A, Nilsson C, Gerhard M, Andersson DI, Krabbe M, Engstrand L (2003) The NudA protein in the gastric pathogen Helicobacter pylori is an ubiquitous and
207

References
constitutively expressed dinucleoside polyphosphate hydrolase. J Biol Chem 278, 12574-12578. Luo J, Jankowski J, et al. (1999) Identification and characterization of diadenosine 5',5"'-P1,P2 -diphosphate and diadenosine 5',5"'-P1,P3-triphosphate in human myocardial tissue. Faseb J 13, 695-705. Luttik MA, Kotter P, Salomons FA, van der Klei IJ, van Dijken JP, Pronk JT (2000) The Saccharomyces cerevisiae ICL2 gene encodes a mitochondrial 2-methylisocitrate lyase involved in propionyl-coenzyme A metabolism. J Bacteriol 182, 7007-7013. Luzzati V (1952) Traitement statistique des erreurs dans la determination des structures cristallines. Acta Crystallographica 5, 802-810. Madrid O, Martin D, Atencia EA, Sillero A, Gunther Sillero MA (1998) T4 DNA ligase synthesizes dinucleoside polyphosphates. FEBS Lett 433, 283-286. Majeed S, Ofek G, Belachew A, Huang CC, Zhou T, Kwong PD (2003) Enhancing protein crystallization through precipitant synergy. Structure 11, 1061-1070. Maksel D, Gooley PR, Swarbrick JD, Guranowski A, Gange C, Blackburn GM, Gayler KR (2001) Characterization of active-site residues in diadenosine tetraphosphate hydrolase from Lupinus angustifolius. Biochem J 357, 399-405. Man WJ, Li Y, O'Connor CD, Wilton DC (1995) The binding of propionyl-CoA and carboxymethyl-CoA to Escherichia coli citrate synthase. Biochim Biophys Acta 1250, 69-75. Maruyama K, Kitamura H (1985) Mechanisms of growth inhibition by propionate and restoration of the growth by sodium bicarbonate or acetate in Rhodopseudomonas sphaeroides S. J Biochem (Tokyo) 98, 819-824. Matlho G, Himathongkham S, Riemann H, Kass P (1997) Destruction of Salmonella enteritidis in poultry feed by combination of heat and propionic acid. Avian Dis 41, 58-61. Matsuyama A, Yamamoto H, Nakano E (1989) Cloning, expression, and nucleotide-sequence of the Escherichia coli K12 Acka gene. Journal of Bacteriology 171, 577-580. Matte A, Tari LW, Delbaere LT (1998) How do kinases transfer phosphoryl groups? Structure 6, 413-419. Matthews BW (1968) Solvent content of protein crystals. J Mol Biol 33, 491-497.
208

References
McClelland M, Sanderson KE, et al. (2001) Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 413, 852-856. McGuffin LJ, Bryson K, Jones DT (2000) The PSIPRED protein structure prediction server. Bioinformatics 16, 404-405. McHan F, Shotts EB (1992) Effect of feeding selected short-chain fatty acids on the in vivo attachment of Salmonella typhimurium in chick ceca. Avian Dis 36, 139-142. Meier M, Janosik M, Kery V, Kraus JP, Burkhard P (2001) Structure of human cystathionine beta-synthase: a unique pyridoxal 5'-phosphate-dependent heme protein. Embo J 20, 3910-3916. Merberg D, Datta P (1982) Altered expression of biodegradative threonine dehydratase in Escherichia coli mutants. J Bacteriol 150, 52-59. Merritt EA, Murphy ME (1994) Raster3D Version 2.0. A program for photorealistic molecular graphics. Acta Crystallogr D Biol Crystallogr 50, 869-873. Meyer PR, Matsuura SE, So AG, Scott WA (1998) Unblocking of chain-terminated primer by HIV-1 reverse transcriptase through a nucleotide-dependent mechanism. Proc Natl Acad Sci U S A 95, 13471-13476. Meynell GG (1963) Antibacterial mechanisms of the mouse gut. II. The role of Eh and volatile fatty acids in the normal gut. Br J Exp Pathol 44, 209-219. Meynell GG, Subbaiah TV (1963) Antibacterial mechanisms of the mouse gut. I. Kinetics of infection by Salmonella typhimurium in normal and streptomycin-treated mice studied with abortive transductants. Br J Exp Pathol 44, 197-208. Miles RD, Gorrell A, Ferry JG (2002) Evidence for a transition state analog, MgADP-aluminum fluoride-acetate, in acetate kinase from Methanosarcina thermophila. J Biol Chem 277, 22547-22552. Miyakoshi S, Uchiyama H, Someya T, Satoh T, Tabuchi T (1987) Distribution of the methylcitric acid cycle and beta -oxidation for propionate catabolism in fungi. Agric. Biol. Chem. 51, 2381-2387. Monad J, Wyman J, Changeux JP (1965) On the nature of allosteric transitions: A plausible model. J. Mol. Biol. 12, 88-118. Morris RJ, Perrakis A, Lamzin VS (2003) ARP/wARP and automatic interpretation of protein electron density maps. Methods Enzymol 374, 229-244.
209

References
Muller CW, Schulz GE (1992) Structure of the complex between adenylate kinase from Escherichia coli and the inhibitor Ap5A refined at 1.9 Å resolution. A model for a catalytic transition state. J Mol Biol 224, 159-177. Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53, 240-255. Nakazawa A, Tokushige M, Hayaishi O, Ikehara M, Mizuno Y (1967) Studies on the interaction between regulatory enzymes and effectors. I. Effect of adenosine 5'-monophosphate analogues on threonine deaminase. J Biol Chem 242, 3868-3872. Navaza J, Saludjian P (1997) AMoRe: An automated molecular replacement program package. Macromolecular Crystallography, Pt A 276, 581-594. Nawaja J (1994) AMoRe: an automated package for molecular replacement. Acta Crystallog. A 50, 157-163. Neidhardt FC, Curtiss III R, et al. (1996) ' In Escherichia coli and Salmonella typhimurium: cellular and molecular biology. (American Society for Microbiology, Washington, D.C.). Nicholls A (1992) GRASP: Graphical Representation and Analysis of Surface Properties. Columbia University, New York. Niederman RA, Rabinowitz KW, Wood WA (1969) Allosteric control of biodegradative L-threonine dehydrase: effect of AMP on an early step in the reaction mechanism. Biochem Biophys Res Commun 36, 951-956. Nishimura A (1998) The timing of cell division: Ap4A as a signal. Trends Biochem Sci 23, 157-159. Ogawa Y, Tsuruoka T, Inoue S, Niida. T (1973) Studies on a new antibiotic SF-1293. II. Chemical structure of antibiotic SF-1293. Sci. Rep. Meijii Kaisha 13, 42-48. Otwinowsky ZM, W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307-326. Page R, Lindberg U, Schutt CE (1998) Domain motions in actin. J Mol Biol 280, 463-474. Palacios S, Escalante-Semerena JC (2000) prpR, ntrA, and ihf functions are required for expression of the prpBCDE operon, encoding enzymes that catabolize propionate in Salmonella enterica serovar typhimurium LT2. J Bacteriol 182, 905-910.
210

References
Palacios S, Escalante-Semerena JC (2004) 2-Methylcitrate-dependent activation of the propionate catabolic operon (prpBCDE) of Salmonella enterica by the PrpR protein. Microbiology 150, 3877-3887. Palacios S, Starai VJ, Escalante-Semerena JC (2003) Propionyl coenzyme A is a common intermediate in the 1,2-propanediol and propionate catabolic pathways needed for expression of the prpBCDE operon during growth of Salmonella enterica on 1,2-propanediol. J Bacteriol 185, 2802-2810. Palfi Z, Suranyi G, Borbely G (1991) Alterations in the accumulation of adenylylated nucleotides in heavy-metal-ion-stressed and heat-stressed Synechococcus sp. strain PCC 6301, a cyanobacterium, in light and dark. Biochem J 276, 487-491. Pannu NS, Read RJ (1996) Improved structure refinement through maximum likelihood. Acta Crystallographica Section A 52, 659-668. Patil RV, Datta P (1988) Photoaffinity labeling of the allosteric AMP site of biodegradative threonine dehydratase of Escherichia coli with 8-azido-AMP. Eur J Biochem 177, 569-574. Perrakis A, Harkiolaki M, Wilson KS, Lamzin VS (2001) ARP/wARP and molecular replacement. Acta Crystallogr D Biol Crystallogr 57, 1445-1450. Pietrowska-Borek M, Stuible HP, Kombrink E, Guranowski A (2003) 4-Coumarate:coenzyme A ligase has the catalytic capacity to synthesize and reuse various (di)adenosine polyphosphates. Plant Physiol 131, 1401-1410. Pintor J, Diaz-Rey MA, Torres M, Miras-Portugal MT (1992a) Presence of diadenosine polyphosphates--Ap4A and Ap5A--in rat brain synaptic terminals. Ca2+ dependent release evoked by 4-aminopyridine and veratridine. Neurosci Lett 136, 141-144. Pintor J, Rotllan P, Torres M, Miras-Portugal MT (1992b) Characterization and quantification of diadenosine hexaphosphate in chromaffin cells: granular storage and secretagogue-induced release. Anal Biochem 200, 296-300. Plateau P, Fromant M, Brevet A, Gesquiere A, Blanquet S (1985) Catabolism of bis(5'-nucleosidyl) oligophosphates in Escherichia coli: metal requirements and substrate specificity of homogeneous diadenosine-5',5'''-P1,P4-tetraphosphate pyrophospho-hydrolase. Biochemistry 24, 914-922. Plateau P, Fromant M, Schmitter JM, Blanquet S (1990) Catabolism of bis(5'-nucleosidyl) tetraphosphates in Saccharomyces cerevisiae. J Bacteriol 172, 6892-6899.
211

References
Plateau P, Mayaux JF, Blanquet S (1981) Zinc(II)-dependent synthesis of diadenosine 5', 5"'-P1,P4-tetraphosphate by Escherichia coli and yeast phenylalanyl transfer ribonucleic acid synthetases. Biochemistry 20, 4654-4662. Pollack SJ, Freeman S, Pompliano DL, Knowles JR (1992) Cloning, overexpression and mechanistic studies of carboxyphosphonoenolpyruvate mutase from Streptomyces hygroscopicus. Eur J Biochem 209, 735-743. Potterton L, McNicholas S, et al. (2004) Developments in the CCP4 molecular-graphics project. Acta Crystallogr D Biol Crystallogr 60, 2288-2294. Powell MJD (1977) Restart procedures for the conjugate gradient method. Mathematical Programming 12, 241-254. Prescott M, Milne AD, McLennan AG (1989) Characterization of the bis(5'-nucleosidyl) tetraphosphate pyrophosphohydrolase from encysted embryos of the brine shrimp Artemia. Biochem J 259, 831-838. Ramachandran GN, Sasisekharan V (1968) Conformation of polypeptides and proteins. Advan. Protein Chem 23, 283–438. Randerath K, Janeway CM, Stephenson ML, Zamecnik PC (1966) Isolation and characterization of dinucleoside tetra- and tri-phosphates formed in the presence of lysyl-sRNA synthetase. Biochem Biophys Res Commun 24, 98-105. Rapaport E, Svihovec SK, Zamecnik PC (1975) Relationship of the first step in protein synthesis to ppGpp: formation of A(5')ppp(5')Gpp. Proc Natl Acad Sci U S A 72, 2653-2657. Rapaport E, Zamecnik PC (1976) Presence of diadenosine 5',5''' -P1, P4-tetraphosphate (Ap4A) in mamalian cells in levels varying widely with proliferative activity of the tissue: a possible positive "pleiotypic activator". Proc Natl Acad Sci U S A 73, 3984-3988. Reeves HC, Ajl SJ (1962) α-hydroxyglutaric acid synthetase. J Bacteriol 84, 186-187. Robinson AK, de la Pena CE, Barnes LD (1993) Isolation and characterization of diadenosine tetraphosphate (Ap4A) hydrolase from Schizosaccharomyces pombe. Biochim Biophys Acta 1161, 139-148. Rossmann M (1990) The molecular replacement method. Acta Crystallographica Section A 46, 73-82. Rossmann MG, Blow DM (1962) The detection of sub-units within the crystallographic asymmetric unit. Acta Crystallographica 15, 24-31.
212

References
Safrany ST, Ingram SW, Cartwright JL, Falck JR, McLennan AG, Barnes LD, Shears SB (1999) The diadenosine hexaphosphate hydrolases from Schizosaccharomyces pombe and Saccharomyces cerevisiae are homologues of the human diphosphoinositol polyphosphate phosphohydrolase. Overlapping substrate specificities in a MutT-type protein. J Biol Chem 274, 21735-21740. Salmon DE, Smith T (1884-1886) On a new method of producing immunity from contagious diseases. Proceedings of the Biological Society of Washington, 3, 29-33. Salmond CV, Kroll RG, Booth IR (1984) The effect of food preservatives on pH homeostasis in Escherichia coli. J Gen Microbiol 130, 2845-2850. Sawers G (1998) The anaerobic degradation of L-serine and L-threonine in enterobacteria: networks of pathways and regulatory signals. Arch Microbiol 171, 1-5. Sawers G (2001) A novel mechanism controls anaerobic and catabolite regulation of the Escherichia coli tdc operon. Mol Microbiol 39, 1285-1298. Sawers G, Hesslinger C, Muller N, Kaiser M (1998) The glycyl radical enzyme TdcE can replace pyruvate formate-lyase in glucose fermentation. Journal of Bacteriology 180, 3509-3516. Sayle RA, Milner-White EJ (1995) RASMOL: biomolecular graphics for all. Trends Biochem Sci 20, 374. Schweizer HP, Datta P (1989) Identification and DNA sequence of tdcR, a positive regulatory gene of the tdc operon of Escherichia coli. Mol Gen Genet 218, 516-522. Sealy-Lewis HM, Fairhurst V (1998) Isolation of mutants deficient in acetyl-CoA synthetase and a possible regulator of acetate induction in Aspergillus niger. Microbiology 144, 1895-1900. Seidel HM, Knowles JR (1994) Interaction of inhibitors with phosphoenolpyruvate mutase: implications for the reaction mechanism and the nature of the active site. Biochemistry 33, 5641-5646. Sharma V, Sharma S, Hoener zu Bentrup K, McKinney JD, Russell DG, Jacobs WR, Jr., Sacchettini JC (2000) Structure of isocitrate lyase, a persistence factor of Mycobacterium tuberculosis. Nat Struct Biol 7, 663-668. Shin BS, Choi SK, Park SH (1999) Regulation of the Bacillus subtilis phosphotransacetylase gene. J Biochem (Tokyo) 126, 333-339. Shingler V (1996) Signal sensing by sigma 54-dependent regulators: derepression as a control mechanism. Mol Microbiol 19, 409-416.
213

References
Shizuta Y, Kurosawa A, Inoue K, Tanabe T, Hayaishi O (1973) Regulation of biodegradative threonine deaminase. I. Allosteric inhibition of the enzyme by a reaction product and its reversal by adenosine 5'-monophosphate. J Biol Chem 248, 512-520. Singh-Wissmann K, Ingram-Smith C, Miles RD, Ferry JG (1998) Identification of essential glutamates in the acetate kinase from Methanosarcina thermophila. J Bacteriol 180, 1129-1134. Singh-Wissmann K, Miles RD, Ingram-Smith C, Ferry JG (2000) Identification of essential arginines in the acetate kinase from Methanosarcina thermophila. Biochemistry 39, 3671-3677. Spector LB (1980) Acetate kinase: a triple-displacement enzyme. Proc Natl Acad Sci U S A 77, 2626-2630. Steitz TA, Shoham M, Bennett WS, Jr. (1981) Structural dynamics of yeast hexokinase during catalysis. Philos Trans R Soc Lond B Biol Sci 293, 43-52. Stewart V, Yanofsky C (1985) Evidence for transcription antitermination control of tryptophanase operon expression in Escherichia coli K-12. J Bacteriol 164, 731-740. Stover CK, Pham XQ, et al. (2000) Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature 406, 959-964. Sumantran VN, Schweizer HP, Datta P (1990) A novel membrane-associated threonine permease encoded by the tdcC gene of Escherichia coli. J Bacteriol 172, 4288-4294. Tabuchi T, Hara S (1974) Production of 2-methylcitric acid from n-paraffins by mutants of Candida lipolytica. Agric. Biol. Chem. 38, 1105-1106. Tabuchi T, Serizawa N (1975) A hypothetical cyclic pathway for the metabolism of odd-carbon n-alkanes or propionyl-CoA via seven-carbon tricarboxylic acids in yeasts. Agric. Biol. Chem. 39, 1055-1061. Tabuchi T, Serizawa N, Uchiyama H (1974) A novel pathway for the partial oxidation of propionyl-CoA to pyruvate via seven-carbon tricarboxylic acids in yeasts. Agric Biol Chem 38, 2571–2572. Tabuchi T, Uchiyama H (1975) Methylcitrate condensing and methylisocitrate cleaving enzymes: evidence for the pathway of oxidation of propionyl-CoA to pyruvate via C7-tricarboxylic acids. Agric Biol Chem 39, 2035–2042. Ten L (1973) Crystallographic fast Fourier transforms. Acta Crystallographica Section A 29, 183-191.
214

References
Textor S, Wendisch VF, De Graaf AA, Muller U, Linder MI, Linder D, Buckel W (1997) Propionate oxidation in Escherichia coli: evidence for operation of a methylcitrate cycle in bacteria. Arch Microbiol 168, 428-436. Thomazeau K, Curien G, Dumas R, Biou V (2001) Crystal structure of threonine synthase from Arabidopsis thaliana. Protein Sci 10, 638-648. Thompson JL, Hinton M (1997) Antibacterial activity of formic and propionic acids in the diet of hens on Salmonellas in the crop. Br Poult Sci 38, 59-65. Tokushige M, Whiteley HR, Hayaishi O (1963) Energy-linked regulation of threonine metabolism by ADP. . Biochem Biophys Res Commun 27, 380–385. Uchiyama H, Tabuchi T (1976) Properties of methylcitrate synthase from Candida lipolytica. Agric Biol Chem 40, 1411–1418. Umbarger E, Brown B (1957) Threonine deamination in Escherichia coli II. Evidence for Two l-Threonine Deaminases. J Bacteriol. 73, 105–112. Umbarger HE (1956) Evidence for a negative-feedback mechanism in the biosynthesis of isoleucine. Science 123, 848. Umbarger HE (1996) Biosynthesis of branched-chain amino acids. In ' In Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology.' (Ed. F.C.Neidhardt, Curtiss, III, R., Ingraham, J.L., Lin, E.C.C., Low, K.B., Magasanik, B., et al.) pp. 442-457. (American Society for Microbiology Press: Washington DC). Vagin A, Teplyakov A (1997) MOLREP: an automated program for molecular replacement. Journal of Applied Crystallography 30, 1022-1025. Vaguine AA, Richelle J, Wodak SJ (1999) SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr D Biol Crystallogr 55, 191-205. Van Dyk TK, LaRossa RA (1987) Involvement of ack-pta operon products in alpha-ketobutyrate metabolism by Salmonella typhimurium. Mol Gen Genet 207, 435-440. Vanni P, Giachetti E, Pinzauti G, McFadden BA (1990) Comparative structure, function and regulation of isocitrate lyase, an important assimilatory enzyme. Comp Biochem Physiol B 95, 431-458. Varshavsky A (1983) Diadenosine 5', 5"'-P1, P4-tetraphosphate: a pleiotropically acting alarmone? Cell 34, 711-712.
215

References
Vartanian A, Alexandrov I, Prudowski I, McLennan A, Kisselev L (1999) Ap4A induces apoptosis in human cultured cells. FEBS Lett 456, 175-180. Velanker SS, Ray SS, Gokhale RS, Suma S, Balaram H, Balaram P, Murthy MRN (1997) Triosephosphate isomerase from Plasmodium falciparum: The crystal structure provides insights into antimalarial drug design. Structure 5, 751-761. Vellieux FMD, Dijkstra BW (1997) Computation of Bhat's OMIT maps with different coefficients. Journal of Applied Crystallography 30, 396-399. Vishwanatha JK, Wei Z (1992) Diadenosine tetraphosphate binding protein from human HeLa cells: purification and characterization. Biochemistry 31, 1631-1635. Voet D, Voet JG (2004) 'Biochemistry.' ((John Wiley & sons). Walter KA, Nair RV, Cary JW, Bennett GN, Papoutsakis ET (1993) Sequence and arrangement of two genes of the butyrate-synthesis pathway of Clostridium acetobutylicum ATCC 824. Gene 134, 107-111. Wegener WS, Reeves HC, Ajl SJ (1967) Propionate oxidation in Escherichia coli. Arch Biochem Biophys 121, 440-442. Wegener WS, Reeves HC, Rabin R, Ajl SJ (1968) Alternate pathways of metabolism of short-chain fatty acids. Bacteriol Rev 32, 1-26. Whanger PD, Phillips AT, Rabinowitz KW, Piperno JR, Shada JD, Wood WA (1968) The mechanism of action of 5'-adenylic acid-activated threonine dehydrase. II. Protomer-oligomer interconversions and related properties. J Biol Chem 243, 167-173. Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF (1999) Protein identification and analysis tools in the ExPASy server. Methods Mol Biol 112, 531-552. Winn MD, Isupov MN, Murshudov GN (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr 57, 122-133. Winn MD, Murshudov GN, Papiz MZ (2003) Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol 374, 300-321. Wood WA, Gunsalus IC (1949) Serine and threonine deaminases of Escherichia coli: activators for a cell-free enzyme. J. Biol. Chem. 181, 171-182. Wu YF, Datta P (1992) Integration host factor is required for positive regulation of the tdc operon of Escherichia coli. Journal of Bacteriology 174, 233-240.
216

References
Wu YF, Datta P (1995) Influence of DNA topology on expression of the tdc operon in Escherichia coli K-12. Molecular & General Genetics 247, 764-767. Wu YF, Patil RV, Datta P (1992) Catabolite gene activator protein and integration host factor act in concert to regulate tdc operon expression in Escherichia coli. Journal of Bacteriology 174, 6918-6927. Xu QS, Jancarik J, Lou Y, Kuznetsova K, Yakunin AF, Yokota H, Adams P, Kim R, Kim SH (2005) Crystal structures of a phosphotransacetylase from Bacillus subtilis and its complex with acetyl phosphate. J Struct Funct Genomics 6, 269-279. Yamada T, Komoto J, Takata Y, Ogawa H, Pitot HC, Takusagawa F (2003) Crystal structure of serine dehydratase from rat liver. Biochemistry 42, 12854-12865. Yao M, Ose T, et al. (2000) Crystal structure of 1-aminocyclopropane-1-carboxylate deaminase from Hansenula saturnus. J Biol Chem 275, 34557-34565. Zamecnik P (1983) Diadenosine 5',5"'-P1,P4-tetraphosphate (Ap4A): its role in cellular metabolism. Anal Biochem 134, 1-10. Zamecnik PC, Stephenson ML, Janeway CM, Randerath K (1966) Enzymatic synthesis of diadenosine tetraphosphate and diadenosine triphosphate with a purified lysyl-sRNA synthetase. Biochem Biophys Res Commun 24, 91-97. Zeng C, Aleshin AE, Hardie JB, Harrison RW, Fromm HJ (1996) ATP-binding site of human brain hexokinase as studied by molecular modeling and site-directed mutagenesis. Biochemistry 35, 13157-13164. Zhang X, Chaney M, Wigneshweraraj SR, Schumacher J, Bordes P, Cannon W, Buck M (2002) Mechanochemical ATPases and transcriptional activation. Mol Microbiol 45, 895-903.
217

Appendix

Appendix
The atomic coordinates and structure factors of the following structures described in the thesis have been deposited or will be deposited to Protein Data Bank: Biodegradative threonine deaminase (TdcB)
2GN0 : Crystal structure of dimeric biodegradative threonine deaminase (TdcB) from Salmonella typhimurium at 1.7 Å resolution (Triclinic form with two dimers of TdcB in the asymmetric unit)
2GN1: Crystal structure of dimeric biodegradative threonine deaminase (TdcB) from
Salmonella typhimurium at 2.2 Å resolution (Triclinic form with one dimer of TdcB in the asymmetric unit)
2GN2: Crystal structure of tetrameric biodegradative threonine deaminase from
Salmonella typhimurium in complex with CMP at 2.5 Å resolution (Hexagonal form) Propionate Kinase (TdcD)
1X3M: Crystal structure of ADP-bound propionate kinase (TdcD) from Salmonella typhimurium
1X3N: Crystal structure of AMPPNP-bound propionate kinase (TdcD) from
Salmonella typhimurium
---- : Crystal structure of propionate kinase (TdcD) from Salmonella typhimurium
----: Crystal structure of S. typhimurium propionate kinase (TdcD) in complex with diadenosine tetraphosphate (Ap4A) obtained after co-crystallization with ATP
----: Crystal structure of S. typhimurium propionate kinase (TdcD) in complex with
diadenosine tetraphosphate (Ap4A).
2-methylisocitrate lyase (PrpB) 1UJQ: Crystal structure of 2-methylisocitrate lyase from Salmonella typhimurium.
1O5Q: Crystal structure of pyruvate and Mg2+ bound 2-methylisocitrate lyase (PrpB)
from Salmonella typhimurium
219




















