
Selective concomitant inhibition of mTORC1 and mTORC2 activity in estrogen receptor negative breast...
11
Selective concomitant inhibition of mTORC1 and mTORC2 activity in estrogen receptor negative breast cancer cells by BN107 and oleanolic acid Ruth Chu 1 , Xiaoyue Zhao 1 , Chandi Griffin 2 , Richard E. Staub 1 , Mark Shoemaker 1 , Joan Climent 3 , Dale Leitman 2 , Isaac Cohen 1 , Emma Shtivelman 1 and Sylvia Fong 1 1 Bionovo Inc., Emeryville, CA 2 Department of Obstetrics, Gynecology and Reproductive Sciences, University of California at San Francisco, San Francisco, CA 3 Cancer Research Institute, University of California at San Francisco, San Francisco, CA Hormonal, targeted and chemotherapeutic strategies largely depend on the expression of their cognate receptors and are often accompanied by intolerable toxicities. Effective and less toxic therapies for estrogen receptor negative (ER2) breast cancers are urgently needed. Here, we present the potential molecular mechanisms mediating the selective pro-apoptotic effect induced by BN107 and its principle terpene, oleanolic acid (OA), on ER2 breast cancer cells. A panel of breast cancer cell lines was examined and the most significant cytotoxic effect was observed in ER2 breast lines. Apoptosis was the major cellular pathway mediating the cytotoxicity of BN107. We demonstrated that sensitivity to BN107 was correlated to the status of ERa. Specifically, the presence of functional ERa protected cells from BN107-induced apoptosis and absence of ERa increased the sensitivity. BN107, an extract rich in OA derivatives, caused rapid alterations in cholesterol homeostasis, presumably by depleting cholesterol in lipid rafts (LRs), which subsequently interfered with signaling mediated by LRs. We showed that BN107 or OA treatment in ER2 breast cancer cells resulted in rapid and specific inhibition of LR-mediated survival signaling, namely mTORC1 and mTORC2 activities, by decreasing the levels of the mTOR/FRAP1, RAPTOR and RICTOR. Cotreatment with cholesterol abolished the proapoptotic effect and restored the disrupted mTOR activities. This is the first report demonstrating possible concomitant inhibition of both mTORC1 and mTORC2 activities by modulating the levels of protein constituents present in these signaling complexes, and thus provides a basis for future development of OA-based mTOR inhibitors. Despite the favorable advances that treatment options have had on survival, current regimens still lead to toxic side effects and are mostly ineffective against ER metastatic breast cancer. Currently, patients with ER/progesterone re- ceptor negative (PR)/HER2 negative (Her2) tumors still represent a therapeutic challenge for oncologists. Novel and effective therapies with minimal toxicities are urgently needed for this patient population. The anticancer effect of the fruit of Gleditsia sinensis Lam (G. sinensis) has been attributed to the induction of cytotox- icity. 1–5 BN107 is an aqueous extract of G. sinensis that has been shown to exhibit antiproliferative activity on a panel of Key words: oleanolic acid, mTORC1, mTORC2, ER, breast cancer, lipid raft Abbreviations: 4E-BP: eukaryotic initiation factor 4E binding protein; Akt: v-akt murine thymoma viral oncogene homolog 1; CHL: cholesterol; CAV-1: caveolin 1; ER: estrogen receptor negative; ERa: estrogen receptor a; FOH: farnesol; GGOH: geranylgeraniol; GM-1: monosialotetrahexosylganglioside; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; Her2: v-erb-b2 erythroblastic leukemia viral oncogene homolog 2; LacZ: b-galactosidase containing plasmid; LRs: lipid rafts; non LR PM: non-lipid raft plasma membrane; LDL: low- density lipoprotein; mTOR/FRAP1: mammalian target of rapamycin/ FK506 binding protein 12-rapamycin associated protein 1; mTORC1/2: mammalian target of rapamycin complex 1/2; MTP: mitochondrial transmembrane potential; MbCD: methyl-b-cyclodextrin; OA: oleanolic acid; p70S6K: ribosomal p70S6 kinase; PI3K: phosphoinositide-3-kinase; PR: progesterone receptor; Raptor: regulatory associated protein of mTOR; Rictor: rapamycin-insensitive companion of mTOR; SGK1: serum glucocorticoid-induced protein kinase 1; TsA: trichostatin A; TR: transferrin receptor Additional Supporting Information may be found in the online version of this article. Conflict of interest: Ruth Chu, Xiaoyue Zhao, Richard E. Staub, Mark Shoemaker, Isaac Cohen, Emma Shtivelman and Sylvia Fong are employees of Bionovo Inc. that supported the work carried out in this article. DOI: 10.1002/ijc.25116 History: Received 13 May 2009; Accepted 26 Nov 2009; Online 21 Dec 2009 Correspondence to: Sylvia Fong, Bionovo Inc. Emeryville, CA 94608, USA, Tel.: 510-420-4167, Fax: 510-420-4197, E-mail: [email protected] Cancer Therapy Int. J. Cancer: 127, 1209–1219 (2010) V C 2009 UICC International Journal of Cancer IJC
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Selective concomitant inhibition of mTORC1 and mTORC2 activity in estrogen receptor negative breast...

Selective concomitant inhibition of mTORC1 and mTORC2activity in estrogen receptor negative breast cancer cells byBN107 and oleanolic acid
Ruth Chu1, Xiaoyue Zhao1, Chandi Griffin2, Richard E. Staub1, Mark Shoemaker1, Joan Climent3, Dale Leitman2,
Isaac Cohen1, Emma Shtivelman1 and Sylvia Fong1
1 Bionovo Inc., Emeryville, CA2 Department of Obstetrics, Gynecology and Reproductive Sciences, University of California at San Francisco, San Francisco, CA3 Cancer Research Institute, University of California at San Francisco, San Francisco, CA
Hormonal, targeted and chemotherapeutic strategies largely depend on the expression of their cognate receptors and are
often accompanied by intolerable toxicities. Effective and less toxic therapies for estrogen receptor negative (ER2) breast
cancers are urgently needed. Here, we present the potential molecular mechanisms mediating the selective pro-apoptotic
effect induced by BN107 and its principle terpene, oleanolic acid (OA), on ER2 breast cancer cells. A panel of breast cancer
cell lines was examined and the most significant cytotoxic effect was observed in ER2 breast lines. Apoptosis was the major
cellular pathway mediating the cytotoxicity of BN107. We demonstrated that sensitivity to BN107 was correlated to the status
of ERa. Specifically, the presence of functional ERa protected cells from BN107-induced apoptosis and absence of ERa
increased the sensitivity. BN107, an extract rich in OA derivatives, caused rapid alterations in cholesterol homeostasis,
presumably by depleting cholesterol in lipid rafts (LRs), which subsequently interfered with signaling mediated by LRs. We
showed that BN107 or OA treatment in ER2 breast cancer cells resulted in rapid and specific inhibition of LR-mediated
survival signaling, namely mTORC1 and mTORC2 activities, by decreasing the levels of the mTOR/FRAP1, RAPTOR and RICTOR.
Cotreatment with cholesterol abolished the proapoptotic effect and restored the disrupted mTOR activities. This is the first
report demonstrating possible concomitant inhibition of both mTORC1 and mTORC2 activities by modulating the levels of
protein constituents present in these signaling complexes, and thus provides a basis for future development of OA-based
mTOR inhibitors.
Despite the favorable advances that treatment options havehad on survival, current regimens still lead to toxic sideeffects and are mostly ineffective against ER� metastaticbreast cancer. Currently, patients with ER�/progesterone re-ceptor negative (PR�)/HER2 negative (Her2�) tumors stillrepresent a therapeutic challenge for oncologists. Novel and
effective therapies with minimal toxicities are urgently neededfor this patient population.
The anticancer effect of the fruit of Gleditsia sinensis Lam(G. sinensis) has been attributed to the induction of cytotox-icity.1–5 BN107 is an aqueous extract of G. sinensis that hasbeen shown to exhibit antiproliferative activity on a panel of
Key words: oleanolic acid, mTORC1, mTORC2, ER�, breast cancer, lipid raft
Abbreviations: 4E-BP: eukaryotic initiation factor 4E binding protein; Akt: v-akt murine thymoma viral oncogene homolog 1; CHL:
cholesterol; CAV-1: caveolin 1; ER�: estrogen receptor negative; ERa: estrogen receptor a; FOH: farnesol; GGOH: geranylgeraniol; GM-1:
monosialotetrahexosylganglioside; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; Her2: v-erb-b2 erythroblastic leukemia viral
oncogene homolog 2; LacZ: b-galactosidase containing plasmid; LRs: lipid rafts; non LR PM: non-lipid raft plasma membrane; LDL: low-
density lipoprotein; mTOR/FRAP1: mammalian target of rapamycin/ FK506 binding protein 12-rapamycin associated protein 1; mTORC1/2:
mammalian target of rapamycin complex 1/2; MTP: mitochondrial transmembrane potential; MbCD: methyl-b-cyclodextrin; OA: oleanolic
acid; p70S6K: ribosomal p70S6 kinase; PI3K: phosphoinositide-3-kinase; PR: progesterone receptor; Raptor: regulatory associated protein of
mTOR; Rictor: rapamycin-insensitive companion of mTOR; SGK1: serum glucocorticoid-induced protein kinase 1; TsA: trichostatin A; TR:
transferrin receptor
Additional Supporting Information may be found in the online version of this article.
Conflict of interest: Ruth Chu, Xiaoyue Zhao, Richard E. Staub, Mark Shoemaker, Isaac Cohen, Emma Shtivelman and Sylvia Fong are
employees of Bionovo Inc. that supported the work carried out in this article.
DOI: 10.1002/ijc.25116
History: Received 13 May 2009; Accepted 26 Nov 2009; Online 21 Dec 2009
Correspondence to: Sylvia Fong, Bionovo Inc. Emeryville, CA 94608, USA, Tel.: 510-420-4167, Fax: 510-420-4197,
E-mail: [email protected]
Can
cerTherapy
Int. J. Cancer: 127, 1209–1219 (2010) VC 2009 UICC
International Journal of Cancer
IJC

human breast cancer lines.4 G. sinensis contains several terpe-noidal saponins that possess a similar base structure to olea-nolic acid (OA).5 These oleanane saponins have been shownto exhibit differential cytotoxicities against tumor cells, whichdepend greatly on the presence and position of the oligosac-charide moieties and the monoterpene units.5 In addition,OA and its synthetic derivatives have been shown to inducestrong antitumor activity in a wide variety of tumor cells inculture and in animal models.5–7
The physiological activity of terpenoidal saponins is usu-ally associated with their ability to complex with plasmamembrane cholesterol.8 It is now well established that choles-terol is important for the functions of lipid rafts (LRs) andthat agents which bind to and/or extract cholesterol from therafts alter the localization and function of the raft-associatedproteins.9–11 LRs are sites where cell surface receptors andsignaling molecules are concentrated and which spatiallyorganize signal transduction at the cell surface.12 Some pro-teins selectively partition into the LRs, including glycosyl-phosphatidylinositol-anchored proteins, myristoylated or pal-mitoylated proteins (such as Akt, flotillin),13,14 doublyacylated proteins (such as Src-family kinases), phospholipidbound proteins (such as annexins), and cholesterol-boundtransmembrane proteins (such as caveolins).12 In addition,LRs have been shown to provide a ‘‘platform’’ for properassembly of functional protein complexes: mTOR activitieswere shown to be dependent on the presence of the complexcomponents on LRs.15,16
The Akt/mammalian target of rapamycin (mTOR) path-way is the prototypic survival mechanism that is aberrantlyactivated in many types of cancer, including ER� breast can-cer.17 This pathway is central in the transmission of thegrowth regulatory and survival signals originating from cellsurface receptors.18,19 Growth factors and cytokines activateAkt via phosphatidylinositol-3-kinase (PI3K), which phos-phorylates phosphatidylinositol-4,5-bisphosphate [PI(4,5)P2]to generate [PI(1,4,5)P3] that binds to the PH domain of Aktand phosphoinositide-dependent protein kinase 1 (PDK1),recruiting them to the plasma membrane. Once in the mem-brane, Akt is phosphorylated by PDK1 at Thr 308 and bymTORC2 at Ser 473.20 When Akt is fully activated, signalingthrough activated Akt can be propagated to a diverse array ofsubstrates, including mTORC1,21 a key regulator of proteintranslation.19
mTOR/FRAP1 is an atypical serine/threonine kinase thatbelongs to the PI3K-like kinase family and regulates a varietyof cellular activities that are sensitive to environmentalstress.18,22,23 In mammalian cells, two mTOR/FRAP1-contain-ing complexes have been identified, mTORC1 and mTORC2.mTORC1 is comprised of mTOR/FRAP1, RAPTOR andmLST8, while mTORC2 contains mTOR/FRAP1, RICTOR,mLST8, sin1 and protor.23–25 mTORC1 phosphorylatesp70S6K (ribosomal p70S6 kinase) and 4E-BP (eukaryotic ini-tiation factor 4E binding protein), both regulators of proteintranslation.22 Activated-mTORC2 regulates the actin skeleton
and phosphorylates the hydrophobic motif of SGK1 atSer42226,27 and Akt at Ser473,23,28 which in conjunction withPDK1-mediated phosphorylation (Thr308) drives full activa-tion of Akt.29,30 It has been proposed that mTOR/FRAP1polypeptide and other complex components of mTORC1 andmTORC2 reside on the LRs and mTOR/FRAP1 polypeptideis shared between mTORC1 and mTORC2 complexes.16
Recently, Copp et al. showed that mTOR/FRAP1 is phospho-rylated differentially when associated with mTORC1 andmTORC2 complexes. Specifically, mTORC1 contains pre-dominately phosphorylated mTOR/FRAP1 on Ser2448;whereas mTORC2 contains mTOR/FRAP1 phosphorylatedpredominantly on Ser2481.31
Given that aberrant mTOR activities are critical for tumorcells to grow and survive, efforts have been directed towarddeveloping potential cancer therapies which inhibit mTOR ac-tivity.18,30 mTOR inhibitors, rapamycin and its derivatives(rapalogs) have been developed to target mTORC1 complex.Some rapalogs have recently demonstrated significant activityin the treatment of metastatic renal cell carcinoma.32 Activitiesagainst other solid tumors, including breast, are not as impres-sive.18,30 The molecular mechanisms responsible for these dif-ferences in sensitivity have not yet been clearly identified.Rapalogs blocking mTORC1 were shown to exert direct anti-proliferative effects. However, the overall antitumour effects ofrapalogs appear to be more complex than just inhibition oftumour growth and could be a result of their apoptotic-induc-ing effects in carcinoma cells along with the inhibition oftumour angiogenesis. Despite extensive cognitive research, it isdifficult to appraise which of those mechanisms is predomi-nant in patients.33 Evidence shows that mTORC1 inhibitioncan lead to pathway reactivation: abrogation of the negative-feedback loop which is normally initiated by the directmTORC1 substrate p70S6k that can lead to strong PI3K-Aktreactivation (chemoresistance). Moreover, rapalogs cannotefficiently inhibit mTORC2 in certain cell types.34 Together,this would suggest that pathway activation and reactivationcould be avoided by agents that lead to concomitant inhibitionof mTORC1 and mTORC2.35 To this end, we report here themolecular mechanism through which BN107 and OA induceapoptosis selectively in ER� breast cancer cells by simultane-ous inhibition of both mTORC1 and mTORC2 complexes.
Material and MethodsReagents and antibodies
BN107 is an aqueous preparation of the grounded fruit ofGleditsia sinensis (Sichuan Medicines and Health Products,Chengdu, China).4 All chemicals were purchased from Sigma(St Louis, MO) unless noted otherwise. The following anti-bodies were purchased from Cell Signal except noted : phos-pho-mTOR, total mTOR, RICTOR, RAPTOR, phosho-4EBP,total 4EBP, pS6 kinase, total S6 kinase, phospho-AKT, totalAKT (Cruz Technology, Santa Cruz, CA), Caveolin 1 (BDBiosciences, San Jose, CA), CD44 (Epitomics, Burlingame,
Can
cerTherapy
1210 Concomitant inhibition of mTORC1/C2 activities
Int. J. Cancer: 127, 1209–1219 (2010) VC 2009 UICC

CA), ERa (Upstate Biotechnology, Temecula, CA) and cyto-chrome C (Biovision, Mountain View, CA). GM-1 wasdetected by using subunits of cholera toxin (CT) subunit Bconjugated to HRP (Invitrogen, Carlsbad, CA).
Cell cultures and treatments
All the cell lines used were purchased from ATCC (Manassas,VA). Cells were treated with 70 lg/ml of BN107, 110 lM ofOA, 500 lM of cholesterol or as indicated in the text and/orlegends to Figures.
ERa transduction
MDA-MB-231 cells were transduced with adenovirus par-ticles (ViraQuest, North Liberty, IA) expressing LacZ or ERaon day 0. Infected cells (300,000) were trypsinized and platedin the presence of 10 nM estradiol per well in 6-well plate onDay 1. Treatment of these cells began 8 hr later on Day 1and continued for 16 hr.
ERa knockdown
Chemical knockdown—a selective estrogen receptor down-regulator ICI182,780 (1 lM) was used to induce degradationof ERa protein in MCF7 cells in RPMI1640 media containingregular fetal bovine serum for 3 days. The cells were thentreated for additional 3 days in 4% charcoal-stripped serumcontaining phenol-red free RPMI-1640 media to deplete es-tradiol, followed by 16 hr of BN107 treatment. Cells wereharvested and subjected to Annexin V/PI analysis. Smallhairpin RNA (shRNA) stable knockdown—various shRNAconstructs in the pGIPZ vector specifically targeting ERawere purchased from Open Biosystem (ThermoScientific,Huntsville, AL). The following shRNAs targeting the follow-ing sequences of ERa were used: shRNA 2-CTTATTGTCTG-TAATTGAA, shRNA4-TTACAGACAATAAGGTCAC andshRNA9-GACTTACTGATAATTTACT. Lentivirus was pro-duced as described.36 Transduced-MCF7 cells were selectedin 2.5 lg/ml of puromycin for one week to allow efficientknockdown. Two populations of transduced-MCF7 cellsexhibited significant ERa knockdown (shRNA 2 and shRNA4) and 1 shRNA 9, did not produce appreciable ERa know-down. These 3 MCF7 pooled populations along with aselected population infected with vector only lentivirus weredepleted of estradiol and treated with BN107 as describedearlier. Cell viability was assessed using MTT assays after 16hr of treatment.
Cell death/apoptosis measurement
Apoptosis was quantified using flow cytometry analysis ofAnnexinV/PI-stained cells (Invitrogen, Carlsbad, CA). MTPwas determined using JC-1 loading (Invitrogen, Carlsbad,CA) of live cells and analyzed using flow cytometry. Caspase3 and 9 activities were measured using specific caspase pep-tide inhibitors (Calbiochem, San Diego, CA) conjugated toFITC followed by measurement with fluorescence platereader (SpectraMax, Sunnyville, CA). Cytosolic cytochrome C
release was examined in cytosol versus mitochondrial frac-tions (Biovision kit, Biovision, Mountain View, CA) followedby immunoblotting. DNA fragmentation was analyzed fol-lowing manufacturer’s instruction (DNA laddering Kit,Roche, Indianapolis, IN). MTT assay was used to assess cellsurvival according to manufacturer’s instruction (Invitrogen).
Immunofluorescence
MDA-MB-231 cells were plated in 8-chamber slides on day0. The cells were treated with BN107 for 4 hr and fixed withmethanol: acetone (1:1) at �20�C for 5 min. Cells wererinsed in phosphate-buffered-saline (PBS) and blocked in 2%bovine serum albumin (BSA) in PBS for 1 hr before applyinganti-caveolin (1/1000) or anti-CD44 (1:250) in 2% BSA inPBS overnight at 4�C. The chamber slides were rinsed withPBS and incubated with appropriate Alexa 488 conjugatedsecondary antibody for one hour. The nuclei were stainedwith 1 lg/ml Hoechst 33258 for 5 min, and the slides weremounted with Fluoromount-G (Southern Biotech, Birming-ham, AL) before viewing.
Cholesterol determination
Total cellular cholesterol levels were determined by lysingcells in lysis buffer (50 mM TrisHCl pH 7.4, 150 mM NaCl,1 mM ethylenediaminetetraacetic acid (EDTA), 1% sodiumdeoxycholate, 0.1% sodium dodecyl sulfate (SDS), 1% triton)and extracted using chloroform (3 times). Cholesterol contentin the LRs was determined by analyzing fractions enriched inGM-1, which were subjected to chloroform extraction (3times). The pooled organic phase was dried down and sub-jected to vacuum drying. The Amplex Red cholesterol assaykit was used to quantitate the amount of cholesterol and cho-lesterol ester in the samples (Invitrogen).
Lipid raft isolation
A modified procedure for density gradient centrifugation usingNycodenz was used to fractionate Triton X-100-solubilized celllysate.33 The fractions were dialyzed against PBS to removethe gradient sugars and concentrated using Amicon Ultra 4centrifugal filter device (Millipore, Jeffrey, New Hampshire)before protein quantitation (BCA reagent, ThermoFisher, Pitts-burgh, PA). The enrichment of GM1 in fractions was testedusing horseradish peroxidase-conjugated cholera toxin B subu-nit (Invitrogen, Carlsbad, CA) and dot blot analysis.
ResultsBN107 selectively induces apoptosis in ER2 breast
cancer cells
We analyzed the cytotoxicity of BN107 on a panel of humanbreast cell lines and investigated whether there was a correla-tion between genotypic characteristics of the cells and theirsensitivity to BN107. Table 1 shows that cells lacking ERexpression were highly sensitive to BN107, while cells con-taining functional ER were relatively insensitive to BN107 atthe same dose (70 lg/ml). Cytotoxicity from BN107 in ER�
Can
cerTherapy
Chu et al. 1211
Int. J. Cancer: 127, 1209–1219 (2010) VC 2009 UICC

lines was apoptotic and primarily mediated by the mitochon-drial pathway, as evidenced by mitochondrial transmembranepotential (MTP) dissipation, caspases 3 and 9 activation,cytochrome C release into cytosol, Annexin V binding andDNA fragmentation (Figs. 1a–1c, and Supporting Informa-tion Figs. 1a and b).
To evaluate if functional ER plays a protective role inBN107-induced apoptosis, we expressed ERa via adenoviraltransduction in MDA-MB-231 cells that are null for ERexpression and highly sensitive to BN107. Figure 2a showsERa protein expression after transduction and Supporting In-formation. Figure 2a shows expression of RNA of WISP2, anER responsive gene that is indicative of functional ER status.Figure 2b shows that ERa versus control lacZ expression inMDA-MB-231 cells conferred significant protection fromBN107-induced apoptosis.
We treated parental MDA-MB-231 cells with differentiat-ing agent trichostatin A (TsA) in an attempt to reverse theirmesenchymal phenotypes and induce expression of the epi-thelial markers. For example, MDA-MB-231 cells have beenshown to re-express ERa, E-cadherin and CD24, and todownregulate CD44, caveolin and vimentin expression onprolonged treatment with low-dose TsA (50 nM), which doesnot cause cell death.37 We examined the levels of RNA andprotein expression for these genes in MDA-MB-231 cells af-ter treatment with TsA for 2 days and confirmed that thesecells indeed express ERa and CD24 and downregulate CD44(Supporting Information Table 1, and Supporting Informa-tion Figs. 2b and 2c). We then treated these TsA-differenti-ated MDA-MB-231 cells with BN107 and showed that thesecells were more resistant to BN107 (Fig. 2c), consistent withour hypothesis that ERa plays a protective role againstBN107-induced apoptosis.
To further examine the role of ERa in protecting breastcancer cells from BN107-induced death, we investigated the
effect of knocking down ERa expression in ERþ MCF7 cells.We approached this question using 2 experimental strategies.First, we attempted to eliminate ERa protein using ICI182,780, which has been shown to specifically degrade ERaRNA and protein.38 This approach yielded results that showelimination of ERa protein followed by removal of estradiolfrom the medium leads to enhanced sensitivity of MCF7 toBN107 (Figs. 3a and 3b).
The second approach involved stable knockdown of ERausing shRNAs specifically targeting ERa. MCF7 cells wereinfected with lenti-virus expressing shRNA constructs specifi-cally targeting ERa and selected to generate transduced popu-lations of MCF7 cells. We analyzed the ERa expression afterone week of selection since ERa protein was shown to be rel-atively stable (data not shown and Ref. 39). To rule out off-target effect, we used 2 different hairpin sequences targetingERa. MCF7 ERa shRNA transduced pools 2 and 4 showedspecific knockdown of ERa protein (Fig. 3c). In addition, we
Figure 1. BN107 induces apoptosis in ER� breast cancer cells,
assessed by (a) mitochondrial transmembrane potential (MTP)
assessed by JC-1 staining, (b) activation of caspases 3 and 9, and
(c) Western blot showing cytosolic Cytochrome C release. All cells
(Hs578T, MDA-MB-231 and MCF7) were treated with BN107 (70
lg/ml) and harvested after 6 (a), 16 (b) or indicated hours (c) of
treatment. Data are represented as mean 6 SEM (b). [Color figure
can be viewed in the online issue, which is available at
www.interscience.wiley.com.]
Table 1. Cells without ER are more sensitive to BN107-inducedapoptosis, while Her2 status appears not correlative withBN107 sensitivity
Annexin V PI staining ER Her2
SKBr3 þþþ � þHs578T þþþþ � þMDA-MB-468 þþ � �MDA-MB-231 þþþþ � �MDA-MB-453 þþþ � þMCF10A þþþ � �MDA-MB-361 �/þ þ þBT474 �/þ þ þMCF7 � þ �
Cells were treated with BN107 and harvested after 18 hr for analysis ofAnnexin V/PI binding (þþþþ > 40%, þþþ 30–40%, þþ 10–30%,�/þ 5–10%, � <5%). The summary shown is a result of 3independent experiments.
Can
cerTherapy
1212 Concomitant inhibition of mTORC1/C2 activities
Int. J. Cancer: 127, 1209–1219 (2010) VC 2009 UICC
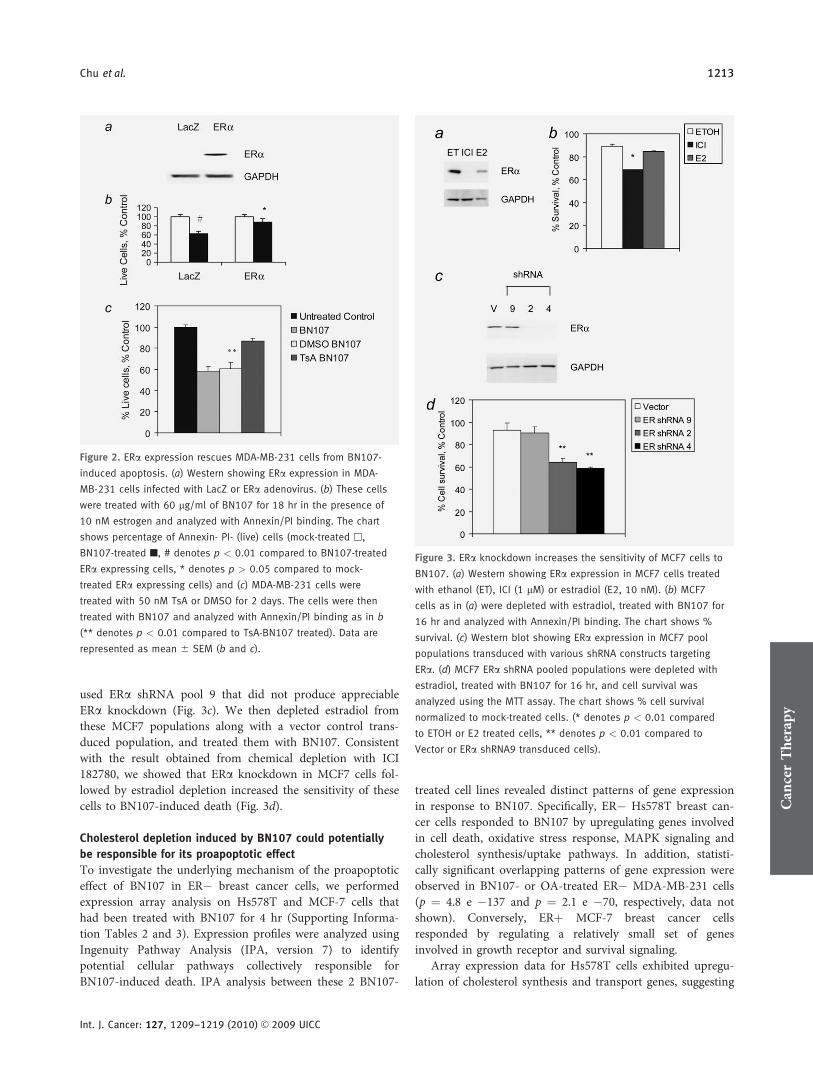
used ERa shRNA pool 9 that did not produce appreciableERa knockdown (Fig. 3c). We then depleted estradiol fromthese MCF7 populations along with a vector control trans-duced population, and treated them with BN107. Consistentwith the result obtained from chemical depletion with ICI182780, we showed that ERa knockdown in MCF7 cells fol-lowed by estradiol depletion increased the sensitivity of thesecells to BN107-induced death (Fig. 3d).
Cholesterol depletion induced by BN107 could potentially
be responsible for its proapoptotic effect
To investigate the underlying mechanism of the proapoptoticeffect of BN107 in ER� breast cancer cells, we performedexpression array analysis on Hs578T and MCF-7 cells thathad been treated with BN107 for 4 hr (Supporting Informa-tion Tables 2 and 3). Expression profiles were analyzed usingIngenuity Pathway Analysis (IPA, version 7) to identifypotential cellular pathways collectively responsible forBN107-induced death. IPA analysis between these 2 BN107-
treated cell lines revealed distinct patterns of gene expressionin response to BN107. Specifically, ER� Hs578T breast can-cer cells responded to BN107 by upregulating genes involvedin cell death, oxidative stress response, MAPK signaling andcholesterol synthesis/uptake pathways. In addition, statisti-cally significant overlapping patterns of gene expression wereobserved in BN107- or OA-treated ER� MDA-MB-231 cells(p ¼ 4.8 e �137 and p ¼ 2.1 e �70, respectively, data notshown). Conversely, ERþ MCF-7 breast cancer cellsresponded by regulating a relatively small set of genesinvolved in growth receptor and survival signaling.
Array expression data for Hs578T cells exhibited upregu-lation of cholesterol synthesis and transport genes, suggesting
Figure 3. ERa knockdown increases the sensitivity of MCF7 cells to
BN107. (a) Western showing ERa expression in MCF7 cells treated
with ethanol (ET), ICI (1 lM) or estradiol (E2, 10 nM). (b) MCF7
cells as in (a) were depleted with estradiol, treated with BN107 for
16 hr and analyzed with Annexin/PI binding. The chart shows %
survival. (c) Western blot showing ERa expression in MCF7 pool
populations transduced with various shRNA constructs targeting
ERa. (d) MCF7 ERa shRNA pooled populations were depleted with
estradiol, treated with BN107 for 16 hr, and cell survival was
analyzed using the MTT assay. The chart shows % cell survival
normalized to mock-treated cells. (* denotes p < 0.01 compared
to ETOH or E2 treated cells, ** denotes p < 0.01 compared to
Vector or ERa shRNA9 transduced cells).
Figure 2. ERa expression rescues MDA-MB-231 cells from BN107-
induced apoptosis. (a) Western showing ERa expression in MDA-
MB-231 cells infected with LacZ or ERa adenovirus. (b) These cells
were treated with 60 lg/ml of BN107 for 18 hr in the presence of
10 nM estrogen and analyzed with Annexin/PI binding. The chart
shows percentage of Annexin- PI- (live) cells (mock-treated h,
BN107-treated n, # denotes p < 0.01 compared to BN107-treated
ERa expressing cells, * denotes p > 0.05 compared to mock-
treated ERa expressing cells) and (c) MDA-MB-231 cells were
treated with 50 nM TsA or DMSO for 2 days. The cells were then
treated with BN107 and analyzed with Annexin/PI binding as in b
(** denotes p < 0.01 compared to TsA-BN107 treated). Data are
represented as mean 6 SEM (b and c).
Can
cerTherapy
Chu et al. 1213
Int. J. Cancer: 127, 1209–1219 (2010) VC 2009 UICC
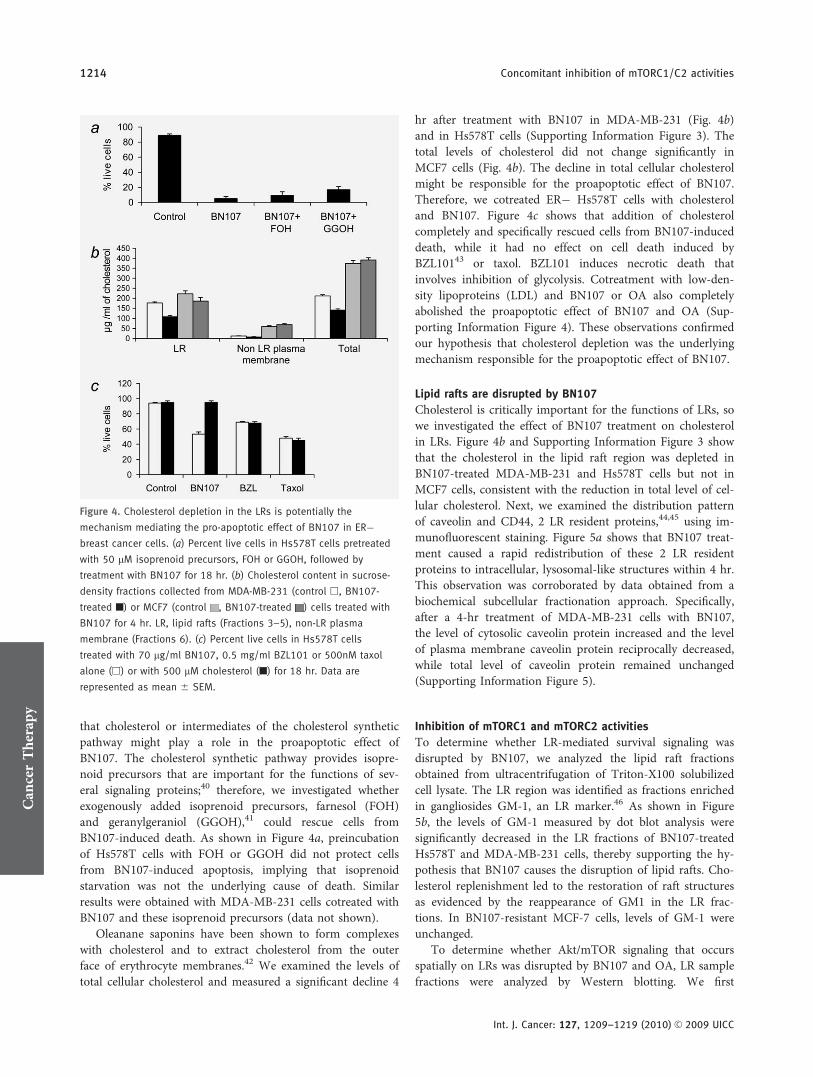
that cholesterol or intermediates of the cholesterol syntheticpathway might play a role in the proapoptotic effect ofBN107. The cholesterol synthetic pathway provides isopre-noid precursors that are important for the functions of sev-eral signaling proteins;40 therefore, we investigated whetherexogenously added isoprenoid precursors, farnesol (FOH)and geranylgeraniol (GGOH),41 could rescue cells fromBN107-induced death. As shown in Figure 4a, preincubationof Hs578T cells with FOH or GGOH did not protect cellsfrom BN107-induced apoptosis, implying that isoprenoidstarvation was not the underlying cause of death. Similarresults were obtained with MDA-MB-231 cells cotreated withBN107 and these isoprenoid precursors (data not shown).
Oleanane saponins have been shown to form complexeswith cholesterol and to extract cholesterol from the outerface of erythrocyte membranes.42 We examined the levels oftotal cellular cholesterol and measured a significant decline 4
hr after treatment with BN107 in MDA-MB-231 (Fig. 4b)and in Hs578T cells (Supporting Information Figure 3). Thetotal levels of cholesterol did not change significantly inMCF7 cells (Fig. 4b). The decline in total cellular cholesterolmight be responsible for the proapoptotic effect of BN107.Therefore, we cotreated ER� Hs578T cells with cholesteroland BN107. Figure 4c shows that addition of cholesterolcompletely and specifically rescued cells from BN107-induceddeath, while it had no effect on cell death induced byBZL10143 or taxol. BZL101 induces necrotic death thatinvolves inhibition of glycolysis. Cotreatment with low-den-sity lipoproteins (LDL) and BN107 or OA also completelyabolished the proapoptotic effect of BN107 and OA (Sup-porting Information Figure 4). These observations confirmedour hypothesis that cholesterol depletion was the underlyingmechanism responsible for the proapoptotic effect of BN107.
Lipid rafts are disrupted by BN107
Cholesterol is critically important for the functions of LRs, sowe investigated the effect of BN107 treatment on cholesterolin LRs. Figure 4b and Supporting Information Figure 3 showthat the cholesterol in the lipid raft region was depleted inBN107-treated MDA-MB-231 and Hs578T cells but not inMCF7 cells, consistent with the reduction in total level of cel-lular cholesterol. Next, we examined the distribution patternof caveolin and CD44, 2 LR resident proteins,44,45 using im-munofluorescent staining. Figure 5a shows that BN107 treat-ment caused a rapid redistribution of these 2 LR residentproteins to intracellular, lysosomal-like structures within 4 hr.This observation was corroborated by data obtained from abiochemical subcellular fractionation approach. Specifically,after a 4-hr treatment of MDA-MB-231 cells with BN107,the level of cytosolic caveolin protein increased and the levelof plasma membrane caveolin protein reciprocally decreased,while total level of caveolin protein remained unchanged(Supporting Information Figure 5).
Inhibition of mTORC1 and mTORC2 activities
To determine whether LR-mediated survival signaling wasdisrupted by BN107, we analyzed the lipid raft fractionsobtained from ultracentrifugation of Triton-X100 solubilizedcell lysate. The LR region was identified as fractions enrichedin gangliosides GM-1, an LR marker.46 As shown in Figure5b, the levels of GM-1 measured by dot blot analysis weresignificantly decreased in the LR fractions of BN107-treatedHs578T and MDA-MB-231 cells, thereby supporting the hy-pothesis that BN107 causes the disruption of lipid rafts. Cho-lesterol replenishment led to the restoration of raft structuresas evidenced by the reappearance of GM1 in the LR frac-tions. In BN107-resistant MCF-7 cells, levels of GM-1 wereunchanged.
To determine whether Akt/mTOR signaling that occursspatially on LRs was disrupted by BN107 and OA, LR samplefractions were analyzed by Western blotting. We first
Figure 4. Cholesterol depletion in the LRs is potentially the
mechanism mediating the pro-apoptotic effect of BN107 in ER�breast cancer cells. (a) Percent live cells in Hs578T cells pretreated
with 50 lM isoprenoid precursors, FOH or GGOH, followed by
treatment with BN107 for 18 hr. (b) Cholesterol content in sucrose-
density fractions collected from MDA-MB-231 (control h, BN107-
treated n) or MCF7 (control , BN107-treated ) cells treated with
BN107 for 4 hr. LR, lipid rafts (Fractions 3–5), non-LR plasma
membrane (Fractions 6). (c) Percent live cells in Hs578T cells
treated with 70 lg/ml BN107, 0.5 mg/ml BZL101 or 500nM taxol
alone (h) or with 500 lM cholesterol (n) for 18 hr. Data are
represented as mean 6 SEM.
Can
cerTherapy
1214 Concomitant inhibition of mTORC1/C2 activities
Int. J. Cancer: 127, 1209–1219 (2010) VC 2009 UICC
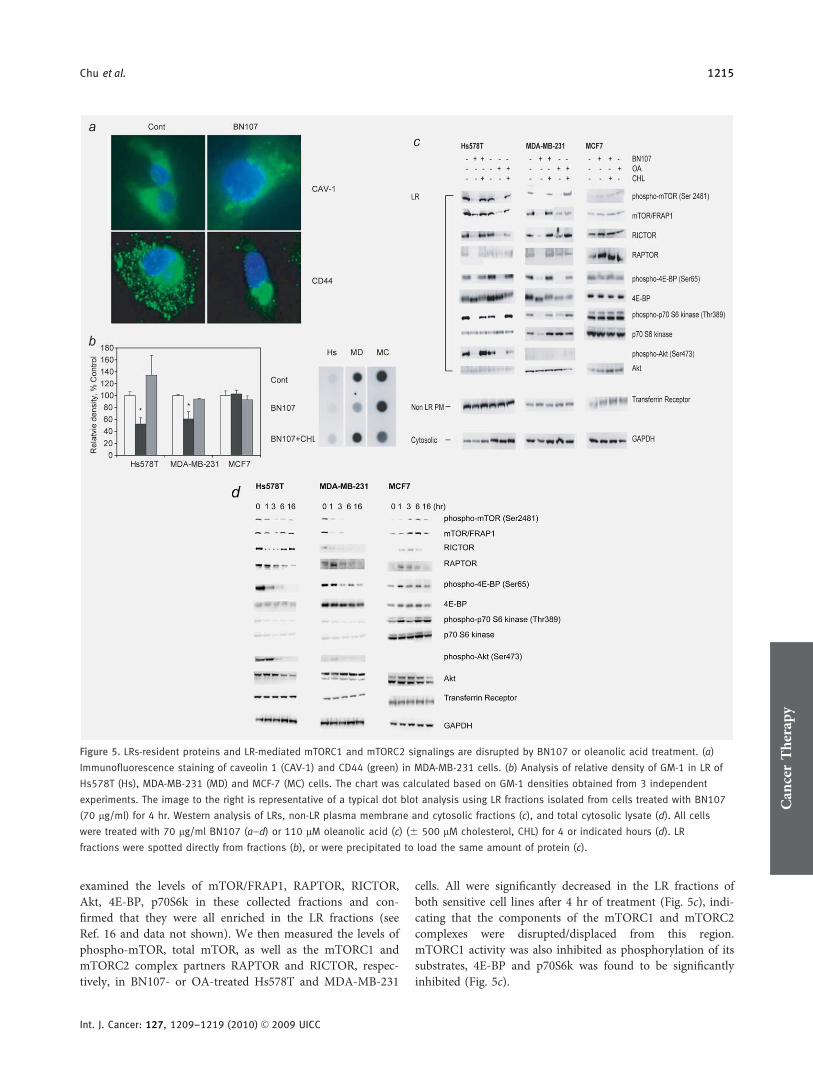
examined the levels of mTOR/FRAP1, RAPTOR, RICTOR,Akt, 4E-BP, p70S6k in these collected fractions and con-firmed that they were all enriched in the LR fractions (seeRef. 16 and data not shown). We then measured the levels ofphospho-mTOR, total mTOR, as well as the mTORC1 andmTORC2 complex partners RAPTOR and RICTOR, respec-tively, in BN107- or OA-treated Hs578T and MDA-MB-231
cells. All were significantly decreased in the LR fractions ofboth sensitive cell lines after 4 hr of treatment (Fig. 5c), indi-cating that the components of the mTORC1 and mTORC2complexes were disrupted/displaced from this region.mTORC1 activity was also inhibited as phosphorylation of itssubstrates, 4E-BP and p70S6k was found to be significantlyinhibited (Fig. 5c).
Figure 5. LRs-resident proteins and LR-mediated mTORC1 and mTORC2 signalings are disrupted by BN107 or oleanolic acid treatment. (a)
Immunofluorescence staining of caveolin 1 (CAV-1) and CD44 (green) in MDA-MB-231 cells. (b) Analysis of relative density of GM-1 in LR of
Hs578T (Hs), MDA-MB-231 (MD) and MCF-7 (MC) cells. The chart was calculated based on GM-1 densities obtained from 3 independent
experiments. The image to the right is representative of a typical dot blot analysis using LR fractions isolated from cells treated with BN107
(70 lg/ml) for 4 hr. Western analysis of LRs, non-LR plasma membrane and cytosolic fractions (c), and total cytosolic lysate (d). All cells
were treated with 70 lg/ml BN107 (a–d) or 110 lM oleanolic acid (c) (6 500 lM cholesterol, CHL) for 4 or indicated hours (d). LR
fractions were spotted directly from fractions (b), or were precipitated to load the same amount of protein (c).
Can
cerTherapy
Chu et al. 1215
Int. J. Cancer: 127, 1209–1219 (2010) VC 2009 UICC

As mTORC1 and mTORC2 complexes may share thesame pool of mTOR/FRAP1 polypeptide,16 and levels of RIC-TOR were also decreased in the LRs of BN107- or OA-treated Hs578T and MDA-MB231 cells (Fig. 5c), we investi-gated whether mTORC2 activity is inhibited. Given therecent discovery that mTORC2 is one of the main kinasesphosphorylating Akt at Ser473, and that Akt signaling wasshown to take place on LRs,13 we analyzed the level ofSer473 phosphorylated Akt in LR fractions as a read-out formTORC2 activity. We showed that BN107 or OA treatmentstrongly decreased the level of Ser473-phosphorylated Aktparticularly in Hs578T cells that harbor high levels of endog-enous phospho-Akt (Ser473), while it had minor effect incells containing low (MDA-MB-231) or undetectable (MCF7)endogenous levels of phospho-Akt on LRs (Fig. 5c). Theaddition of exogenous cholesterol restored the signalingevents disrupted by BN107 or OA (Fig. 5c). None of thesedecreases were observed in the resistant MCF-7 cells (rightpanel, Fig. 5c). The levels of transferrin receptor marking thenon-LR plasma membrane region (Non LR PM) andGAPDH marking the cytosolic fractions were not affected inthese cell lines treated with BN107 or OA.
As mTORC1 and mTORC2 complexes are also found inthe cytosol, we examined the changes of relevant proteins intotal cytosolic extracts after BN107 treatment. Consistent withthe data for the lipid raft fractions, levels of cytosolic mTOR/FRAP1, phospho-mTOR, RAPTOR and RICTOR were alldecreased within 1 hr of BN107 treatment, resulting in mini-mal mTORC1 and mTORC2 activities to phosphorylate 4E-BP, p70S6k and Ser473-Akt, respectively (Fig. 5d). Thedecrease in total protein levels of mTOR/FRAP1, RAPTORand RICTOR occurred posttranscriptionally since the levels oftheir corresponding mRNAs were not modulated by BN107or OA treatment in ER� Hs578T cells (Supporting Informa-tion Figure 6). Also consistent with data for LRs, cytosolicSer-473 phosphorylated Akt was markedly decreased; whilelevels of total cytosolic Akt were slightly decreased at latertime points. These data collectively indicated that LRs andLR-mediated mTORC1 and mTORC2 signaling were specifi-cally disrupted by BN107 and OA in ER� breast cancer cells.These effects were presumably due to their cholesterol deplet-ing effect on LRs, as addition of exogenous cholesterolreversed these changes (Figs. 5c, 6a and 6b). The cytosolic lev-els of phospho-mTOR, total mTOR/FRAP1, RICTOR andRAPTOR were increased in a time-dependent manner in theresistant MCF-7 cells treated with BN107 (Fig. 5d).
DiscussionGene-expression profiling of primary tumors has identifieddifferent subtypes of breast cancers. The basal/mesenchymal-like subtype does neither express the ER or PR nor do theyhave amplified HER2. These tumors are less differentiated,more aggressive and do not respond to available targetedtherapies. Instead they are treated with conventional chemo-therapies that have limited efficacy and many side effects.
Therefore, it is critically important to identify alternativetherapeutic strategies for these patients.
In our study, we showed that BN107 and oleanolic acidspecifically targeted mesenchymal-like, ER� breast cancercells, while ERþ cells were not as sensitive to these treat-ments. Consistent with this observation, we clustered thepublically available expression profiles of various breastlines47 according to their sensitivity to BN107 and found thatexpression of many ER down-stream targets was associatedwith insensitivity to BN107 (not shown). At the same time,the proapoptotic effect induced by BN107 showed no correla-tion with Her2 status (Table 1). We further demonstratedthat when ERa status was restored in breast cancer cells lack-ing functional ER by forced expression with adenovirus orinduced expression with TsA, the sensitivity to BN107 inthese cells was significantly decreased (Fig. 2). Moreover, wehave eliminated ERa protein expression in MCF7 cells usinga selective estrogen receptor downregulator, ICI 182780,48 orshRNA-mediated knockdown approaches. Both experimentsshow that elimination of ERa protein followed by removal ofestradiol from the medium leads to enhanced sensitivity toBN107 (Fig. 3). Collectively, these findings demonstrated thatfunctional ER status played a protective role in BN107-induced apoptosis.
To elucidate the mechanism mediating the selective proa-poptotic effect of BN107 on ER� breast cancer cells, we
Figure 6. Cholesterol specifically restores mTOR signaling disrupted
by BN107 (a) or OA (b) induced apoptosis. Western analysis of the
total levels of phospho-4E-BP or phospho-p70S60 kinase and
phosphor-Akt(ser 473) in BN107- or OA-treated Hs578T, MDA-MB-
231 or MCF7, as a read out for mTORC1 and mTORC2 activities,
respectively. All cells were treated with 70 lg/ml BN107 (6500
lM CHL) or 125 lM OA (6500 lM CHL) for 4 hr.
Can
cerTherapy
1216 Concomitant inhibition of mTORC1/C2 activities
Int. J. Cancer: 127, 1209–1219 (2010) VC 2009 UICC

performed expression profiling analysis comparing theexpression patterns in the sensitive (ER�) versus insensitive(ERþ) cell lines. Using IPA analysis, we identified that genesinvolved in cholesterol synthesis/uptake pathway were over-represented. Similar results were obtained when ER� cellswere treated with OA suggesting that cholesterol or cholesterolsynthetic pathway intermediates might be involved in apopto-sis induced by BN107. We demonstrated that depletion of LRcholesterol, but not isoprenoid precursors, appeared to be re-sponsible for BN107-induced apoptosis, since pretreatmentwith exogenous cholesterol or cholesterol equivalents (i.e.,LDL) protected cells from treatment with BN107 and OA. Theintegrity of lipid rafts is very dependent upon the presence ofcholesterol.9 The loss of cholesterol from lipid rafts after treat-ment with cholesterol-sequestering agents [methyl-b-cyclodex-trin (MbCD)], through increased sterol oxidation or by inhibi-ting its de novo synthesis leads to loss of LR-associatedproteins and decreased cell survival.13,14,49
We further showed that BN107- or OA-induced apoptosiswas based on decreased mTORC1 and mTORC2 activities inthe LRs of ER� breast cancer cells. This presumably led tosubsequent inhibition of Akt activity. We cannot rule outthat a decrease in the level of phosphatidylinositol phosphatesin LRs from cholesterol depletion might lead to reduction ofAkt membrane recruitment and phosphorylation. However,the total amount of Akt proteins remained relativelyunchanged in the LRs, albeit the level of total cytosolic Aktprotein decreased slightly at later time points (Fig. 5d).Therefore, Akt inactivation was likely a consequence ofmTORC2 inhibition, due to decrease in mTOR/FRAP1 andRICTOR components. Aberrant activation of the Akt/mTORpathway has been shown to exist in many cancers, includingER� breast cancer,17 allowing malignant tumor cells to pro-liferate and evade death signaling or become resistant to vari-ous therapies. Despite numerous efforts directed at develop-ing therapeutics that target mTOR, most clinical testing hasnot shown promising results against solid tumor cancers.18
The clinical data point to our incomplete understanding ofthe regulation of mTOR pathways, especially regardingmTORC2 activity.35 Recent data have implicated mTORC2 asthe major kinase that phosphorylates Ser473 on Akt, and,along with PDK1, facilitates the full activation of Akt.28
Rapamycin showed a minimal acute inhibitory effect onmTORC2, as compared to mTORC1, and only prolongedincubation with high dose can lead to some inhibition onmTORC2.34 Rapamycin-induced mTORC1 inhibition alonecan lead to PI3K/Akt pathway reactivation. Conversely, dis-ruption of mTORC2 activity alone might also lead toincreased mTORC1 activity.16 Altogether, these observationswould suggest that pathway activation and reactivation couldbe avoided by treatments that lead to concomitant mTORC1and mTORC2 inhibition resulting in subsequent inactivationof Akt.35 Indeed, recent studies reported the development ofATP-competitive inhibitors that specifically bind to the ATP-binding site of mTOR/FRAP1 kinase and inhibit the catalytic
activity of both mTORC1 and mTORC2.50,51 These ATP-competitive mTOR inhibitors showed significantly strongeranti-proliferative activity on cells than rapamycin.50,51
The inhibition of mTORC1 and mTORC2 activities byBN107 or OA appeared to be based on disruption of themTORC1 and mTORC2 complex formation on LRs in theER� breast cancer cells. This is likely a consequence of thelower levels of complex components mTOR/FRAP1, RAP-TOR and RICTOR components present in LRs of theBN107- or OA-treated cells. The disruption of the complexeslikely leads to degradation of these proteins, as the roles ofRAPTOR and RICTOR are mostly structural and the totalcellular levels of them also showed a concomitant decrease.The decreased levels of mTOR/FRAP1, RAPTOR and RIC-TOR were not caused by downregulation in their steady-statemRNA levels. It is possible that BN107 or OA specificallydownregulates the levels of these proteins posttranscription-ally which results in decreased mTORC1 and mTORC2 activ-ities on LRs. Furthermore, since we also observed that theactivities of 2 important protein translation regulators,p70S6k and 4E-BP, were decreased, it is possible that thetranslation of the mTOR components was inhibited in sensi-tive breast cancer cells. Interestingly, we observed a time-de-pendent increase in the cytosolic levels of phospho-mTOR,mTOR/FRAP1, RICTOR and RAPTOR in the ERþ MCF7cells treated with BN107, which produced increased phospho-rylation of 4E-BP and p70 S6 kinase (Fig. 5d). It is temptingto postulate that the MCF-7 cells’ relative resistance toBN107 is in part a result of the increased activity ofmTORC1 and mTORC2 complexes caused by increases inthese protein levels. It is also possible that BN107 or OAtreatment disrupts the homeostasis of phosphatidic acid onthe plasma membrane, which is required for the stabilizationof mTORC1 and mTORC2 complexes.52–54 To our knowl-edge, this is the first report demonstrating downregulation ofthe activities for both mTOR complexes concomitantly bytreating cells with agents that decrease the levels of mTORCscomponents on LRs, as well as the total cytosolic level.
Experimental and epidemiological evidence suggested thatcholesterol may play a promotional role in cancer develop-ment and progression.55,56 Others have proposed that pro-gressive increases in membrane cholesterol contribute to theexpansion of rafts, which may potentiate oncogenic pathways(i.e., Akt).11,57,58 These findings collectively suggest thatagents interfering with cholesterol homeostasis in LRs, suchas BN107 and OA, represent a novel approach to disrupttumor cell survival signaling.
A major concern connected to the potential clinical appli-cation of raft-ablating chemicals is that these agents may alsononselectively alter LRs and interfere with the function ofvital organs. MbCD derivatives, however, are widely utilizedas carriers for water-insoluble drugs for parenteral use,59
implying that lower doses of these compounds do not ulti-mately exert marked systemic toxicity. Even though the Gle-ditsia saponins have been shown to strip plasma membrane
Can
cerTherapy
Chu et al. 1217
Int. J. Cancer: 127, 1209–1219 (2010) VC 2009 UICC

cholesterol from erythrocytes in vitro, antitumor doses of OAhave exhibited minimal toxicity in animals.6 It must be notedthat distinct types of LRs have been identified that differ intheir biochemical composition, compartmentalization andfunctions.60 Many studies have shown that depletion of cho-lesterol from cells leads to the disruption of LRs and therelease of raft constituents into the bulk plasma membrane;60
however, not all LRs appear to be equally sensitive to choles-terol depletion. Rajendran et al. showed that in Jurkat andU937 cells, several raft proteins, including lck, lyn and LAT,were released from rafts by treatment with MbCD, but flotil-lins remained in low-density, detergent-resistant domains.61
These findings suggest that there is heterogeneity in the LRpopulation based on its dependence on or interaction withcholesterol. Consistent with this notion, Ostapkowicz et al.showed that lipid rafts undergo significant structural reorgan-ization during transition from ERþ breast cancer cells to themore aggressive ER� genotype.37 Potentially only a specific
subset or composition of LRs supports mTOR signaling thatwas inhibited by BN107 or OA in the ER� breast cancercells. How ERa contributes to the protection of BN107-induced lipid raft disruption and apoptosis remains unknownand requires further investigation as well as detailed charac-terization of the specific interactions between BN107/OA andvarious LRs components that will facilitate the developmentof drugs selectively targeting raft components associated withAkt/mTOR signaling.
In summary, our findings demonstrate for the first timethat BN107 and OA are strong inhibitors of both mTORC1and mTORC2 activities. Unlike allosteric mTORC1 inhibitorssuch as rapalogs, BN107 and oleanolic acid inhibit mTORactivities through decreasing the protein components presentin the complexes. This approach may negate development ofchemoresistance and growth factor driven activation of Aktand potentially provide better clinical outcomes for ER�breast cancers.
References
1. Cheung F, Chui CH, Chan AS, Lau FY,Cheng GY, Wong RS, Kok SH, Teo IT,Cheng CH, Tang JC. Inhibition ofproteasome activity in Gleditsia sinensisfruit extract-mediated apoptosis on humancarcinoma cells. Int J Mol Med 2005;16:925–9.
2. Chui CH, Lau FY, Chan AS, Cheng GY,Wong RS, Lai KB, Kok SH, Yeung TT, TeoIT, Yau MY, Cheung F, Cheng CH, et al.Gleditsia sinensis fruit extract-inducedapoptosis involves changes of reactiveoxygen species level, mitochondrialmembrane depolarization and caspase 3activation. Int J Mol Med 2005;15:539–43.
3. Pak KC, Lam KY, Law S, Tang JC. Theinhibitory effect of Gleditsia sinensis oncyclooxygenase-2 expression in humanesophageal squamous cell carcinoma. Int JMol Med 2009;23:121–9.
4. Shoemaker M, Hamilton B, Dairkee SH,Cohen I, Campbell MJ. In vitro anticanceractivity of twelve Chinese medicinal herbs.Phytother Res 2005;19:649–51.
5. Zhong L, Li P, Han J, Qu G, Guo D.Structure-activity relationships of saponinsfrom Gleditsia sinensis in cytotoxicity andinduction of apoptosis. Planta. Med. 2004;70:797–802.
6. Hsu HY, Yang JJ, Lin CC. Effects ofoleanolic acid and ursolic acid oninhibiting tumor growth and enhancing therecovery of hematopoietic systempostirradiation in mice. Cancer Lett 1997;111:7–13.
7. Liby KT, Yore MM, Sporn MB.Triterpenoids and rexinoids asmultifunctional agents for the preventionand treatment of cancer. Nat Rev Cancer2007;7:357–69.
8. Melzig MF, Bader G, Loose R.Investigations of the mechanism ofmembrane activity of selected triterpenoidsaponins. Planta Med 2001;67:43–8.
9. Simons K, Toomre D. Lipid rafts andsignal transduction. Nat Rev Mol Cell Biol2000;1:31–9.
10. Zidovetzki R, Levitan I. Use ofcyclodextrins to manipulate plasmamembrane cholesterol content: evidence,misconceptions and control strategies.Biochim Biophys Acta 2007;1768:1311–24.
11. Patra SK. Dissecting lipid raft facilitatedcell signaling pathways in cancer. BiochimBiophys Acta 2008;1785:182–206.
12. Anderson RG Transendothelial movementand caveolae. Nat Biotechnol 2008;26:380–1; author reply 1–2.
13. Adam RM, Mukhopadhyay NK, Kim J, DiVizio D, Cinar B, Boucher K, Solomon KR,Freeman MR. Cholesterol sensitivity ofendogenous and myristoylated Akt. CancerRes 2007;67:6238–46.
14. Fedida-Metula S, Elhyany S, Tsory S, SegalS, Hershfinkel M, Sekler I, Fishman D.Targeting lipid rafts inhibits protein kinaseB by disrupting calcium homeostasis andattenuates malignant properties ofmelanoma cells. Carcinogenesis 2008;29:1546–54.
15. Asaki C, Usuda N, Nakazawa A, KametaniK, Suzuki T. Localization of translationalcomponents at the ultramicroscopic level atpostsynaptic sites of the rat brain. BrainRes 2003;972:168–76.
16. Partovian C, Ju R, Zhuang ZW, MartinKA, Simons M. Syndecan-4 regulatessubcellular localization of mTORComplex2 and Akt activation in a
PKCalpha-dependent manner inendothelial cells. Mol Cell 2008;32:140–9.
17. Akkiprik M, Nicorici D, Cogdell D, Jia YJ,Hategan A, Tabus I, Yli-Harja O, Y D,Sahin A, Zhang W. Dissection of signalingpathways in fourteen breast cancer celllines using reverse-phase protein lysatemicroarray. Technol Cancer Res Treat 2006;5:543–51.
18. Strimpakos AS, Karapanagiotou EM, SaifMW, Syrigos KN. The role of mTOR inthe management of solid tumors: anoverview. Cancer Treat Rev 2008.
19. Steelman LS, Stadelman KM, ChappellWH, Horn S, Basecke J, Cervello M,Nicoletti F, Libra M, Stivala F, MartelliAM, McCubrey JA. Akt as a therapeutictarget in cancer. Expert Opin Ther Targets2008;12:1139–65.
20. Sarbassov DD, Guertin DA, Ali SM,Sabatini DM. Phosphorylation andregulation of Akt/PKB by therictor-mTOR complex. Science 2005;307:1098–101.
21. Nave BT, Ouwens M, Withers DJ, AlessiDR, Shepherd PR. Mammalian target ofrapamycin is a direct target for proteinkinase B: identification of a convergencepoint for opposing effects of insulin andamino-acid deficiency on proteintranslation. Biochem J 1999;344 Pt 2:427–31.
22. Reiling JH, Sabatini DM. Stress andmTORture signaling. Oncogene 2006;25:6373–83.
23. Sabatini DM. mTOR and cancer: insightsinto a complex relationship. Nat RevCancer 2006;6:729–34.
24. Huang J, Manning BD. A complexinterplay between Akt. TSC2 and the two
Can
cerTherapy
1218 Concomitant inhibition of mTORC1/C2 activities
Int. J. Cancer: 127, 1209–1219 (2010) VC 2009 UICC

m. TOR complexes. Biochem Soc Trans2009;37:217–22.
25. Pearce LR, Huang X, Boudeau J,Pawlowski R, Wullschleger S, Deak M,Ibrahim AF, Gourlay R, Magnuson MA,Alessi DR. Identification of Protor as anovel Rictor-binding component ofmTOR complex-2. Biochem J 2007;405:513–22.
26. Garcia-Martinez JM, Alessi DR. mTORcomplex 2 (mTORC2) controlshydrophobic motif phosphorylation andactivation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). BiochemJ 2008;416:375–85.
27. Alessi DR, Pearce LR, Garcia-Martinez JM.New insights into mTOR signaling:mTORC2 and beyond. Sci Signal 2009;2:pe27.
28. Sarbassov DD, Ali SM, Sabatini DM.Growing roles for the mTOR pathway.Curr Opin Cell Biol 2005;17:596–603.
29. Bhaskar PT, Hay N. The two TORCs andAkt. Dev Cell 2007;12:487–502.
30. Guertin DA, Sabatini DM. Defining therole of mTOR in cancer. Cancer Cell 2007;12:9–22.
31. Copp J, Manning G, Hunter T. TORC-specific phosphorylation of mammaliantarget of rapamycin (mTOR): phospho-Ser2481 is a marker for intact mTORsignaling complex 2. Cancer Res 2009;69:1821–7.
32. Rini BI, Campbell SC, Escudier B. Renalcell carcinoma. Lancet 2009;28:1119–32.
33. Le Tourneau C, Faivre S, Serova M,Raymond E. mTORC1 inhibitors: istemsirolimus in renal cancer telling us howthey really work? Br J Cancer 2008;99:1197–203.
34. Sarbassov DD, Ali SM, Sengupta S, SheenJH, Hsu PP, Bagley AF, Markhard AL,Sabatini DM. Prolonged rapamycintreatment inhibits mTORC2 assembly andAkt/PKB. Mol Cell 2006;22:159–68.
35. Maira SM, Stauffer F, Schnell C, Garcia-Echeverria C. PI3K inhibitors for cancertreatment: where do we stand? BiochemSoc Trans 2009;37:265–72.
36. King FW, Fong S, Griffin C, Shoemaker M,Staub R, Zhang YL, Cohen I, Shtivelman ETimosaponin AIII is preferentiallycytotoxic to tumor cells through inhibitionof mTOR and induction of ER stress. PloSone 2009;4:e7283.
37. Ostapkowicz A, Inai K, Smith L, Kreda S,Spychala J. Lipid rafts remodeling inestrogen receptor-negative breast cancer isreversed by histone deacetylase inhibitor.Mol Cancer Ther 2006;5:238–45.
38. Zwart W, Rondaij M, Jalink K, Sharp ZD,Mancini MA, Neefjes J, Michalides R.Resistance to antiestrogen arzoxifene ismediated by overexpression of cyclin D1.Mol Endocrinol 2009;23:1335–45.
39. Petrel TA, Brueggemeier RW. Increasedproteasome-dependent degradation ofestrogen receptor-alpha by TGF-beta1 inbreast cancer cell lines. J Cell Biochem2003;88:181–90.
40. Konstantinopoulos PA, Karamouzis MV,Papavassiliou AG. Post-translationalmodifications and regulation of the RASsuperfamily of GTPases as anticancertargets. Nat Rev Drug Discov 2007;6:541–55.
41. Ownby SE, Hohl RJ. Farnesol andgeranylgeraniol: prevention and reversionof lovastatin-induced effects in NIH3T3cells. Lipids 2002;37:185–92.
42. Mitsumasa Haruna M, Sugimoto T, KojimaR, Suzuki Y, Konoshima T, Kozuka M, ItoK. Alteration of Naþ permeability inhuman erythrocytes as studied by 23Na-NMR and inhibition of the kidneyNaþ,Kþ-ATPase activities with saponins:interaction of gleditsia saponins withhuman erythrocyte membranes. BioorgMed Chem Lett 1995;5:827–30.
43. Fong S, Shoemaker M, Cadaoas J, Lo A,Liao W, Tagliaferri M, Cohen I,Shtivelman E. Molecular mechanismsunderlying selective cytotoxic activity ofBZL101, an extract of Scutellaria barbata,towards breast cancer cells. Cancer BiolTher 2008;7:577–86.
44. Lee JL, Wang MJ, Sudhir PR, Chen JY.CD44 engagement promotes matrix-derived survival through the CD44-SRC-integrin axis in lipid rafts. Mol Cell Biol2008;28:5710–23.
45. Minshall RD, Sessa WC, Stan RV,Anderson RG, Malik AB. Caveolinregulation of endothelial function. Am JPhysiol Lung Cell Mol Physiol 2003;285:L1179–83.
46. Michel V, Bakovic M. Lipid rafts in healthand disease. Biol Cell 2007;99:129–40.
47. Neve RM, Chin K, Fridlyand J, Yeh J,Baehner FL, Fevr T, Clark L, Bayani N,Coppe JP, Tong F, Speed T, Spellman PT,et al. A collection of breast cancer cell linesfor the study of functionally distinct cancersubtypes. Cancer Cell 2006;10:515–27.
48. McClelland RA, Gee JM, Francis AB,Robertson JF, Blamey RW, Wakeling AE,Nicholson RI Short-term effects of pureanti-oestrogen ICI 182780 treatment onoestrogen receptor, epidermal growthfactor receptor and transforming growth
factor-alpha protein expression in humanbreast cancer. Eur J Cancer 1996;32A:413–6.
49. Pike LJ. Lipid rafts: heterogeneity on thehigh seas. Biochem J 2004;378:281–92.
50. Feldman ME, Apsel B, Uotila A, LoewithR, Knight ZA, Ruggero D, Shokat KM.Active-site inhibitors of mTOR targetrapamycin-resistant outputs of mTORC1and mTORC2. PLoS Biol 2009;7:e38.
51. Thoreen CC, Kang SA, Chang JW, Liu Q,Zhang J, Gao Y, Reichling LJ, Sim T,Sabatini DM, Gray NS. An ATP-competitive mammalian target ofrapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J BiolChem 2009;284:8023–32.
52. Fang Y, Vilella-Bach M, Bachmann R,Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTORsignaling. Science 2001;294:1942–5.
53. Foster DA, Toschi A. Targeting mTORwith rapamycin: one dose does not fit all.Cell Cycle 2009;8:1026–9.
54. Toschi A, Lee E, Xu L, Garcia A, Gadir N,Foster DA. Regulation of mTORC1 andmTORC2 complex assembly byphosphatidic acid: competition withrapamycin. Mol Cell Biol 2009;29:1411–20.
55. Di Vizio D, Solomon KR, Freeman MR.Cholesterol and cholesterol-richmembranes in prostate cancer: an update.Tumori 2008;94:633–9.
56. Swyer G. The cholesterol content ofnormal and enlarged prostates. Cancer Res1942;2:372–5.
57. Freeman MR, Cinar B, Lu ML. Membranerafts as potential sites of nongenomichormonal signaling in prostate cancer.Trends Endocrinol Metab 2005;16:273–9.
58. Maxfield FR, Tabas I. Role of cholesteroland lipid organization in disease. Nature2005;438:612–21.
59. Szejtli J, Szente L. Elimination of bitter,disgusting tastes of drugs and foods bycyclodextrins. Eur J Pharm Biopharm 2005;61:115–25.
60. Jacobson K, Mouritsen OG, Anderson RG.Lipid rafts: at a crossroad between cellbiology and physics. Nat Cell Biol 2007;9:7–14.
61. Rajendran L, Masilamani M, Solomon S,Tikkanen R, Stuermer CA, Plattner H,Illges H. Asymmetric localization offlotillins/reggies in preassembled platformsconfers inherent polarity to hematopoieticcells. Proc Natl Acad Sci USA 2003;100:8241–6.
Can
cerTherapy
Chu et al. 1219
Int. J. Cancer: 127, 1209–1219 (2010) VC 2009 UICC




















