
Wnt-Related Molecules and Signaling Pathway Equilibrium in Hematopoiesis

Role of TAZ as Mediatorof Wnt SignalingLuca Azzolin,1,4 Francesca Zanconato,1,4 Silvia Bresolin,2 Mattia Forcato,3 Giuseppe Basso,2 Silvio Bicciato,3
Michelangelo Cordenonsi,1,* and Stefano Piccolo1,*1Department of Biomedical Sciences, University of Padua School of Medicine, viale Colombo 3, 35126 Padua, Italy2Department of Woman and Child Health, University of Padova, via Giustiniani 3, 35128 Padova, Italy3Center for Genome Research, Department of Biomedical Sciences, University of Modena and Reggio Emilia, via G. Campi 287,41100 Modena, Italy4These authors contributed equally to this work
*Correspondence: [email protected] (M.C.), [email protected] (S.P.)
http://dx.doi.org/10.1016/j.cell.2012.11.027
SUMMARY
Wnt growth factors are fundamental regulators of cellfate, but how the Wnt signal is translated into biolog-ical responses is incompletely understood. Here, wereport that TAZ, a biologically potent transcriptionalcoactivator, serves as a downstream element ofthe Wnt/b-catenin cascade. This function of TAZ isindependent from its well-established role as medi-ator of Hippo signaling. In the absence of Wntactivity, the components of the b-catenin destructioncomplex—APC, Axin, and GSK3—are also requiredto keep TAZ at low levels. TAZ degradation dependson phosphorylated b-catenin that bridges TAZ to itsubiquitin ligase b-TrCP. Upon Wnt signaling, escapeof b-catenin from the destruction complex impairsTAZ degradation and leads to concomitant accumu-lation of b-catenin and TAZ. At the genome-widelevel, a substantial portion of Wnt transcriptionalresponses is mediated by TAZ. TAZ activation is ageneral feature of Wnt signaling and is functionallyrelevant to mediate Wnt biological effects.
INTRODUCTION
The Wnt signaling pathway has prominent and widespread roles
in development, tissue homeostasis, and cancer (MacDonald
et al., 2009). For example, Wnts coordinate proliferation and
differentiation during organ growth and serve as extrinsic factors
to regulate stem cells for tissue maintenance and regeneration
(Clevers, 2006; Moon et al., 2004; Niehrs and Acebron, 2012).
Aberrant activation of the Wnt cascade is a major theme in
cancer biology, leading to stem cell expansion and disturbed
tissue architecture. Thismay be caused bymutations of pathway
components or by autocrine signaling due to constitutive Wnt
production by tumor cells (Clevers, 2006).
A key step in this pathway is the regulation of cytosolic b-cat-
enin levels (MacDonald et al., 2009). In the absence of Wnt, a
multiprotein complex phosphorylates b-catenin and constantly
C
targets it for degradation. Key elements of this ‘‘destruction’’
complex are scaffold proteins, such as adenomatous polyposis
coli (APC) and Axin, and the b-catenin phosphorylating kinases
glycogen synthase kinase 3 (GSK3) and casein kinase 1 (CK1)
(MacDonald et al., 2009). GSK3 phosphorylation of b-catenin is
critical for recognition by the F box protein b-TrCP and its asso-
ciated E3 ubiquitin ligase complex (Liu et al., 2002). Wnt signal-
ing inhibits b-catenin phosphorylation through sequestration of
GSK3 into multivesicular compartments and other mechanisms.
This causes b-catenin to escape from b-TrCP recognition, lead-
ing to its accumulation and formation of a nuclear complex with
the DNA-binding transcription factors TCF/Lef (Li et al., 2012;
Niehrs and Acebron, 2012; Taelman et al., 2010).
Wnt/TCF/b-catenin target genes account for important as-
pects of Wnt biology, an example being upregulation of c-myc
or cyclin-D1 in some tumors. Yet, it is also increasingly evident
that the highly temporal- and cell-specific expression of the
knownWnt/TCF/b-catenin targets can hardly capture the perva-
siveness and complexity of Wnt/b-catenin biology (Barolo, 2006;
Niehrs and Acebron, 2012). This raises the possibility that other
factors, in addition to b-catenin/TCF, may contribute to Wnt-
induced biological responses.
Independently of Wnt signaling, the transcriptional coactiva-
tors TAZ and YAP have recently emerged at the centerpiece of
poorly understood mechanisms that control tissue growth and
organ size (Pan, 2010). TAZ and YAP promote cell proliferation
and inhibit differentiation, particularly in stem cells and organ-
specific progenitors, and therefore, their activity must be
finely tuned in order to avoid loss of tissue regeneration and, at
the other extreme, overgrowth and emergence of tumors (Ra-
mos and Camargo, 2012). Furthermore, TAZ has been recently
proposed to endow self-renewal capacity to cancer stem cells
(Bhat et al., 2011; Cordenonsi et al., 2011).
TAZ and YAP are well known for their regulation by the Hippo
pathway; the activation of two kinases, MST1/2 (homolog of
Drosophila Hippo) and LATS1/2, leads to LATS-dependent
phosphorylation of TAZ and YAP, limiting their stability, nuclear
localization, and transcriptional activity (Pan, 2010). Recent find-
ings have emphasized the roles of cell polarity or cell-cell adhe-
sion as upstream regulators of the Hippo kinases (Cordenonsi
et al., 2011; Varelas et al., 2010b). Moreover, TAZ and YAP can
ell 151, 1443–1456, December 21, 2012 ª2012 Elsevier Inc. 1443

Figure 1. Wnt Signaling Promotes TAZ Stabilization and Activation
(A) HEK293 cells were transfected with 8xGTIIC-Lux and either with control (siCo, lanes 1–3) or TAZ siRNAs (siTAZ, lane 4). After transfection, cells were exposed
to control- (Co) or Wnt3A-conditioned medium. Where indicated, XAV939 (1 mM, lane 3) was added. Top: luciferase assay of the indicated samples. Data are
normalized to lane 1 and are presented as mean +SD. Bottom: representative western blots for TAZ, YAP, b-catenin, and GAPDH (loading control) in the same
extracts used for the luciferase assay. See also Figure S1B for the validation of Wnt activity in conditioned media and of XAV939 efficacy on a b-catenin/TCF-
luciferase reporter.
(B) Wnt-induced transcriptional activation of 8xGTIIC-Lux (as in [A]) is independent of b-catenin.
(C) Top: luciferase assay on 8xGTIIC-Lux reporter recording TAZ-dependent transcriptional activity in HEK293 cells transfected with control (lanes 1 and 2),
GSK3 (lanes 3 and 4), APC (lanes 5 and 6), or Axin (lanes 7 and 8) siRNAs, either alone or with TAZ siRNA, as indicated. Data are normalized to lane 1 and are
presented as mean +SD. See Figures S1E, S1F, S1I, and S1K for siRNA validations. Bottom: western blot for TAZ in the same extracts used for the luciferase
assay.
(legend continued on next page)
1444 Cell 151, 1443–1456, December 21, 2012 ª2012 Elsevier Inc.

be regulated independently from the Hippo kinases by mechan-
ical and architectural cues, such as cytoskeletal tension and cell
shape (Dupont et al., 2011; Halder et al., 2012).
Interestingly, there are—at least superficially—clear elements
of overlap between b-catenin and TAZ or YAP; biochemically, all
are short-lived proteins in the cytoplasm, as they are degraded
by the same b-TrCP ubiquitin ligase complex (Liu et al., 2010;
MacDonald et al., 2009; Zhao et al., 2010). Biologically, they
appear to control overlapping processes in a number of epithelial
and nonepithelial contexts. Although this hints at some form of
joined regulation, the possibility that Wnt could signal through
TAZ or YAP stabilization has not been explored.
Here, we report the characterization of TAZ as a downstream
component of Wnt/b-catenin signaling cascade. We show that
Wnt induces TAZ protein stabilization and transcriptional activity
in multiple cell types and that this is essential for distinct biolog-
ical effects induced by Wnt signaling. We found that the
same destruction complex responsible for b-catenin degrada-
tion also restrains TAZ protein levels. Yet, TAZ does not require
direct phosphorylation by GSK3; rather, it is GSK3-phosphory-
lated b-catenin that serves as a scaffold for TAZ association
with b-TrCP/E3 ubiquitin-ligase complex. By inhibiting b-catenin
phosphorylation, Wnt promotes not only b-catenin stabilization
but also TAZ accumulation and activation of TAZ-dependent
gene responses.
RESULTS
Wnt Activates TAZThis study was initiated by the discovery that treatment of
HEK293 cells with Wnt3A triggered a remarkable increase of
TAZ protein levels (Figure 1A, bottom). Because TAZ mRNA
levels were not affected by Wnt3A (Figure S1A available online)
and because TAZ has been shown to be an unstable pro-
tein (Liu et al., 2010; data not shown), this suggested that
Wnt3A promoted TAZ protein stabilization. In contrast, Wnt3A
had no overt effect on YAP steady-state protein levels. Impor-
tantly, Wnt3A also triggered a robust induction of transfected
8xGTIIC-Lux (Figure 1A, compare lanes 1 and 2), a synthetic
luciferase sensor containing multimerized responsive elements
of TEAD, the main DNA-binding cofactor of TAZ or YAP (Dupont
et al., 2011). This induction was dependent on stabilization of
TAZ, as depletion of endogenous TAZ was sufficient to abolish
the effect of Wnt3A (Figure 1A, compare lanes 2 and 4), prompt-
ing us to investigate the link between Wnt and TAZ. Knockdown
(D) Western blots for TAZ and YAP inMII human breast cancer cells transfected wi
GSK3 or APC siRNAs. TAZ mRNA levels remain unchanged (see Figures S1G, S
(E) qRT-PCR for TAZ target gene CTGF, normalized to GAPDH expression, in
mean +SD. See Figure S1L for an independent shRNA targeting TAZ.
(F) Confocal images of TAZ (left) in MII cells transfected as indicated in (D). Nucle
same fields.
(G) Pie chart of U133plus2 Affymetrix probesets upregulated in MII cells transfect
area of the graph represents the fraction of probesets whose upregulation by AP
(H) Upregulation of TAZ protein levels upon knockdown of GSK3, APC, or Axin i
(I) Western blot analysis of mouse ES cells, cultured in the absence or presence
(J–L) TAZ levels and expression of TAZ target genes in control and APC-deficient
deleted livers. (K and L) Average expression of TAZ target genes Cyr61 (K) and C
See also Figure S1.
C
of b-catenin had no effect on the induction of 8xGTIIC-Lux by
Wnt, ruling out that Wnt regulates TAZ stability indirectly, for
example, through b-catenin target genes (Figure 1B). We also
tested the possibility that Wnt might regulate TAZ by inhibiting
the Hippo kinase cascade. As shown in Figure S1C, treatment
with Wnt3A ligand could still induce TAZ in LATS1/2-depleted
cells, indicating that LATS activity is not crucial for TAZ regula-
tion by Wnt. We also expressed in HEK239 cells a phosphomu-
tant TAZ protein lacking all LATS phosphorylation sites (TAZ
4SA); as expected, TAZ 4SA is more stable than wild-type
TAZ, and yet it remains sensitive to Wnt regulation (Figure S1D).
Taken together, these data suggested that Wnt signaling stabi-
lizes and activates TAZ by mechanisms other than regulation
of the Hippo pathway.
Wnt ligands are transduced intracellularly through the inacti-
vation of the b-catenin destruction complex, which includes
GSK3, APC, and Axin (MacDonald et al., 2009). We sought to
determine whether Wnt regulates TAZ through the same path-
way. We thus reactivated the b-catenin destruction complex in
Wnt-treated cells by using XAV939, a small molecule that pro-
motes Axin stabilization (Huang et al., 2009). XAV939 inhibited
the effects of Wnt3A on TAZ transcriptional activity and stability
(Figure 1A, compare lanes 2 and 3). Conversely, depletion of
Axin, GSK3, or APC induced TAZ stabilization and activity (Fig-
ure 1C), phenocopying Wnt stimulation. As a control, depletion
of LATS was irrelevant for TAZ regulation by GSK3 or APC
(data not shown), again indicating that activation of TAZ by the
Wnt signaling cascade does not rely on Hippo pathway inhibi-
tion. Thus, Wnt signaling regulates TAZ in a way that depends
on the b-catenin destruction complex.
Next, we tested the validity of this conclusion in a different
cellular model system. In transformed mammary epithelial cells
(MCF10A-T1k or MII cells), knockdown of GSK3, APC, or Axin
caused robust TAZ stabilization (Figures 1D, S1F, S1H, and
S1J) and promoted TAZ-dependent upregulation of CTGF (Fig-
ures 1E and S1L), an established endogenous readout of TAZ
activity (Cordenonsi et al., 2011). This was paralleled by nuclear
accumulation of endogenous TAZ (Figure 1F).
Next, we wished to investigate to what extent Wnt gene re-
sponses are mediated by TAZ on the transcriptomic scale. For
this, we compared the Affymetrix gene expression profiles of
MII cells transfected with control small interfering RNA (siRNA),
APC siRNA, or the combination of APC and TAZ siRNAs. We
first identified a list of 739 probesets upregulated more than
two times after APC depletion, thus entailing Wnt-induced target
th control, GSK3, APC, or Axin siRNAs. See Figures S1F and S1H for alternative
1I, and S1K).
MII-shGFP or MII-shTAZ#3 transfected with the indicated siRNAs. Bars are
i are stained with Hoechst (right). See Figure S1M for b-catenin staining in the
ed with APC siRNA compared to control siRNA-transfected cells. Dark colored
C depletion is reverted by cotransfection of TAZ siRNA.
n HeLa cells, HaCaT cells, and human mesenchymal stem cells (hMSC).
of the GSK3 inhibitor CHIR99021 (CHIR, 3 mM) for 24 hr.
mouse livers (n = 3 for each genotype). (J) Upregulation of TAZ protein in APC-
TGF (L), evaluated by qRT-PCRs. Data are presented as mean +SD.
ell 151, 1443–1456, December 21, 2012 ª2012 Elsevier Inc. 1445

genes. Strikingly, 54% of these probesets were upregulated in
a TAZ-dependent manner (Figure 1G); if we restricted our anal-
yses to genes upregulated more than five times by APC knock-
down, out of 132 probesets, 91 (70%) were TAZ dependent.
These results indicate that TAZ is a relevant mediator of Wnt-
induced transcriptional responses in this cellular context.
We also found that Wnt pathway activation, either by stimula-
tion with Wnt3A or by intracellular inhibition of the destruction
complex, led to TAZ stabilization and induction of TAZ-depen-
dent gene responses in a variety of cell types, such as in HeLa
cervical carcinoma cells, HaCaT immortalized keratinocytes,
primary human mesenchymal stem cells, mouse embryonic
stem (ES) cells, and P19 mouse embryonic carcinoma cells
(Figures 1H, 1I, and S1N). In sum, TAZ activation by Wnt is a
general phenomenon irrespective of cell transformation, epithe-
lial or mesenchymal status, and embryonic lineage.
Finally, to extend our finding to in vivo conditions, we crossed
mice bearing APC floxed alleles (APCfl/fl) with transgenic mice
carrying the inducible Ah-Cre recombinase, in which robust
Cre expression in liver, gut, and other organs can be induced
by the drug b-naphtophlavone (see Extended Experimental
Procedures). In liver extracts, genetic inactivation of APC trig-
gered, as expected, the induction of the b-catenin/TCF target
Cyclin-D1 (Figure S1O) and also triggered a strong upregulation
of TAZ protein levels and of the TAZ targetsCyr61 andCTGF, but
not of TAZ mRNA (Figures 1J–1L and S1P).
Taken together, the data indicate that Wnt activity is a general
inducer of TAZ stabilization and TAZ-dependent transcription,
that TAZ regulation accounts for a significant portion of Wnt-
regulated genes, and that TAZ levels are kept low by the same
set of core proteins—Axin, APC, and GSK3—that continually
keep b-catenin levels in check.
TAZ Mediates Wnt Biological ResponsesFinding that the b-catenin destruction complex regulates TAZ
carries a corollary: in APC mutant cells, TAZ should be already
at the maximal level allowed by this regulation and thus should
be insensitive to GSK3 inactivation. To test this hypothesis, we
used the SW480 colorectal cancer (CRC) cell line, carrying a
homozygous nonsense mutation in APC (Faux et al., 2004).
Indeed, depletion of GSK3 had no effect on TAZ stability in
SW480 cells (Figure 2A, compare lanes 1 and 2). However, add-
ing back APC to SW480 cells not only dramatically downregu-
lated TAZ protein levels and the expression of TAZ target genes
(Figure 2A, compare lanes 1 and 3) but also restored sensitivity to
GSK3 inhibition (Figure 2A, compare lanes 3 and 4). Thus, aber-
rant Wnt signaling in CRC cells entails TAZ stabilization.
b-catenin is a key player downstream of oncogenic Wnt
signaling for the growth of CRC cells (van de Wetering et al.,
2002). Here, we asked whether TAZ might also have important
functions in this context. Knockdown of either TAZ or b-catenin
reduced the proliferation of parental SW480 cells to a similar
extent, with dual depletion of b-catenin and TAZ being required
for a complete growth arrest (Figure 2B, lanes 2–4). APC recon-
stitution led to a severe growth suppression (Figure 2B, compare
lanes 1 and 5), as previously reported (Faux et al., 2004); forced
expression of TAZ rescued proliferation of APC-reconstituted
SW480 cells (lane 6). Of note, manipulation of the levels of TAZ
1446 Cell 151, 1443–1456, December 21, 2012 ª2012 Elsevier Inc.
(by gain or loss of function) does not influence b-catenin levels
(Figure S2B), suggesting that TAZ does not operate through
b-catenin stabilization. Furthermore, loss of TAZ does notmodify
the expression of Axin2 and Cyclin-D1, two of the most estab-
lished readouts of the TCF/b-catenin complex, ruling out that
TAZ is required for TCF-dependent transcription (Figure 2C).
Taken together, these data suggest that TAZ is essential and
instrumental for cell proliferation downstream of aberrant Wnt
signaling, cooperating with the well-established function of
b-catenin in this process.
Parental SW480 cells are highly transformed and able to grow
as clonal ‘‘spheroids’’ in Matrigel (Figure 2E). Consistently with
the role of Wnt signaling as promoter of clonogenic potential
(Niehrs and Acebron, 2012), we found that the status of APC
also contributes to the capacity of SW480 cells to grow under 3D
conditions. Indeed, APC-reconstituted SW480 cells formed a
much-reduced number of colonies than parental SW480 cells
(Figures 2D, 2E, and 2H). Remarkably, TAZ depletion in parental
SW480 cells impaired spheroid formation, whereas knockdown
of b-catenin did not (Figures 2E–2H). In sum, APC deficiency
empowers 2D cell growth in a manner that is dependent both
on b-catenin and on TAZ, aswell as clonogenic properties, which
are prevalently dependent on TAZ stabilization.
We then wished to assess, beyond any specific functional
assay, what is the relative contribution of b-catenin and TAZ
to the global Wnt gene response in CRC cells. For this, we
compared the Affymetrix gene expression profiles of paren-
tal, TAZ-depleted, b-catenin-depleted, and APC-reconstituted
SW480 cells. Importantly, we found that b-catenin and TAZ
accounted for one-third and one-sixth of the �3,000 probesets
modulated by APC reconstitution, respectively (p values <
10�32). This indicates that TAZ targets represent a substantial
fraction of gene expression driven by oncogenic APC.
TAZ Is Required for Wnt-Dependent Differentiation ofMesenchymal Stem CellsWe next addressed the role of TAZ as a downstreammediator of
Wnt signals during Wnt-dependent differentiation of mesen-
chymal stem cells (MSC). Wnt signaling is indeed critical for
bone formation in mammals, and, at least in part, this is due to
the requirement of Wnts to promote an osteogenic lineage in
mesenchymal progenitors (Leucht et al., 2008). To address the
role of TAZ in this process, we used ST-2 murine bone marrow
stromal cells and primary human MSC.
In mouse ST-2 cells, treatment with recombinant Wnt3A
caused a robust increase in alkaline phosphatase (ALP), amarker
of bone differentiation (Figure 3A, top, compare lanes 1 and 3).
Notably, this occurred with a parallel rise of b-catenin and TAZ
protein levels (Figure 3A, bottom). Strikingly, TAZ depletion had
no effect on b-catenin levels but was sufficient to impair ALP
induction by Wnt3A (lane 4), indicating that TAZ is an essential
mediator of bone differentiation induced by Wnt in this cellular
context.
Human MSC can be induced to differentiate into the osteo-
blast lineage by switching their culturing condition from regular
growth medium to osteogenic differentiation medium. Addition
of recombinant Dkk-1, an extracellular Wnt antagonist, severely
impaired bone differentiation (Figures 3B–3D), a finding in line

Figure 2. Role of TAZ in Growth and Clonogenic Potential of APC Mutant Colon Cancer Cells
(A)Western blots for TAZ, CTGF, andGSK3 in parental (APCmutant) and APC-reconstituted SW480 cells, transfectedwith control (lanes 1 and 3) or GSK3 siRNAs
(lanes 2 and 4).Wild-type APC expression effectively reduced b-catenin levels and activity (Figure S2A and see [C] below), indicating the effective reconstitution of
the destruction complex. Other TAZ target genes, such as Ankyrin-D1 and Cyr61, were equally downregulated by APC reconstitution (data not shown).
(B) Growth assay in SW480 cells transfected with control (siCo), TAZ (siTAZ), b-catenin (sibcat), or both TAZ and b-catenin (sibcat siTAZ) siRNAs. In lane 6, APC-
reconstituted SW480 cells were stably transduced with TAZ-expressing lentiviral vector. Bars display the number of SW480 cells counted 1 day after seeding
(light blue bars) or after 4 days (dark blue bars). Data are shown as mean +SD. See Figure S2B for TAZ and b-catenin expression in these samples. See Figures
S2C–S2F for growth curves of parental SW480 cells transfected with independent TAZ or b-catenin siRNAs.
(C) Bars are expression values of Axin2 and Cyclin-D1 mRNAs in parental or APC-reconstituted SW480 cells transfected with the indicated siRNAs. Data are
presented as mean +SD.
(D–G) Representative pictures of Matrigel-embedded spheres formed by SW480+APC (D) or SW480 (E–G) cells transfected with the indicated siRNAs.
(H) Bars represent the number of spheres formed by the indicated cells. Data are shown as mean +SD.
See also Figure S2.
with the notion that autocrine Wnt ligands are essential for this
process. Interestingly, we found that TAZ protein levels rose
upon switching cells in osteogenicmedium, but this was blocked
by concomitant addition of Dkk-1 (Figure 3F, compare lanes 1, 2,
and 3). If, as previously implicated in ST-2 cells, TAZ induction is
instrumental for bone differentiation downstream of Wnt, then
raising TAZ levels should bypass the extracellular block posed
by Dkk-1. For this experiment, we sustained TAZ expression
by transducing MSC with a TAZ lentiviral vector (Figure 3F,
lane 4). Crucially, reconstitution of TAZ restored bone dif-
ferentiation of Dkk1-treated hMSC (Figure 3E; see quantifica-
tions in Figure S3B). Interestingly, the TAZ targets CTGF and
Cyr61 were induced during osteogenic differentiation of MSC
in a manner that was TAZ dependent, opposed by Dkk-1, and
rescued by exogenous TAZ (Figures 3A and 3F). Taken together,
the data reveal that Wnt promotes MSC differentiation into oste-
oblasts through TAZ stabilization.
Contrary to bone differentiation, adoption of an adipogenic
fate by MSC is inhibited by Wnt (Ross et al., 2000), and loss of
C
Axin, GSK3, or APC severely blunted adipocytic differentiation
(Figures 3G, 3H, and S3C–S3G). This inhibition was mediated
by upregulation of endogenous TAZ as double depletion of
Axin, and TAZ restored adipocyte differentiation (Figures 3I
and 3J). Similar results were obtained by double knockdowns
of TAZ and GSK3 or TAZ and APC (data not shown). These
epistatic relationships reinforce the notion that regulation of
TAZ by the Wnt pathway is functionally relevant for MSC
differentiation.
Role of GSK3 in TAZ DegradationNext, we sought to determine by which mechanism Wnt sig-
naling promotes TAZ activity. The results described so far sug-
gested that stabilization of b-catenin and TAZ are both down-
stream of Wnt signaling because both proteins are degraded
by the same destruction complex. The view that TAZ follows
the same steps of b-catenin on its route to degradation is sug-
gested by the fact that b-TrCP is required for both TAZ and
b-catenin degradation (MacDonald et al., 2009; Liu et al., 2010)
ell 151, 1443–1456, December 21, 2012 ª2012 Elsevier Inc. 1447

Figure 3. Role of TAZ in Wnt-Controlled Mesenchymal Stem Cell Differentiation
(A) Wnt3A promotes bone differentiation in a TAZ-dependent manner in ST-2 cells. ST-2 cells were transfected with control (lanes 1 and 3) or TAZ (lanes 2 and 4)
siRNAs. Where indicated, cells were exposed to recombinant Wnt3A (lanes 3 and 4). Top: alkaline phosphatase (ALP) activity. Data are presented as mean +SD.
Bottom: western blots for TAZ, Cyr61, and b-catenin in the corresponding extracts. GAPDH serves as loading control. Wnt3A does not affect TAZmRNA levels
(Figure S3A).
(B–F) TAZ rescues bone differentiation in Dkk1-treated hMSC. hMSC, transducedwith lentiviral vectors encoding for control (empty vector) or TAZ, were cultured
either in growth medium (GM) or osteogenic differentiation medium (ODM); where indicated, Dkk-1 protein (500 ng/ml) was added to the ODM. (B–E) Repre-
sentative fields of hMSC stained for ALP. See Figure S3B for quantifications of ALP-positive areas. (F) Western blots for TAZ, Cyr61, CTGF, and b-catenin in the
samples described above. b-catenin was still inhibited by Dkk-1 in TAZ-transduced MSCs (lanes 3 and 4).
(G–J) hMSCwere transfected with control or Axin siRNAs, alone or with TAZ siRNA (siTAZ), and induced to differentiate in adipocytes. (G–I) Representative fields
of hMSC, stained with oil red to visualize lipid droplets. Nuclei are stained with Hoechst. (J) Quantification of oil-red-positive areas. Values are mean +SD.
See also Figure S3.
and was further supported by the following evidence: (1) TAZ
protein levels increased after depletion of b-TrCP (Figures 4A
and S4A); (2) loss of b-TrCP also promoted potent upregulation
of TAZ transcriptional responses (Figure S4A); (3) similar to
b-TrCP depletion, interfering with Cullin1, an essential adaptor
of the b-TrCP E3 ubiquitin ligase complex, caused upregulation
of TAZ (Figure S4A).
The interaction of b-catenin with b-TrCP occurs upon phos-
phorylation by GSK3 (MacDonald et al., 2009). We then tested
whether GSK3 phosphorylation and b-TrCP recognition also
drive TAZ degradation. We first tested this hypothesis by
assaying whether the interaction of TAZ with b-TrCP required
GSK3. Indeed, endogenous b-TrCP failed to associate to TAZ
in coimmunoprecipitation assays from lysates of GSK3-depleted
MII cells (Figure 4B). Moreover, GSK3 kinase activity is essential
1448 Cell 151, 1443–1456, December 21, 2012 ª2012 Elsevier Inc.
for TAZ degradation; knockdown of GSK3 stabilized TAZ in
MII cells (Figure 4C, lanes 1 versus 2 and 4 versus 5), and re-
constitution with wild-type GSK3 (Figure 4C, lane 3), but not
with kinase-dead GSK3 (Figure 4C, lane 6), rescued TAZ
levels and activity. Similar results were obtained in HEK293
cells (Figure S4B). These findings are in line with the effect of
CHIR99021, a small-molecule inhibitor of GSK3 enzymatic
activity, leading to TAZ stabilization in a variety of cell types (Fig-
ure 1I; data not shown).
GSK3 regulates the stability of b-catenin and other proteins
through phosphorylation of a DSGXXS motif, called ‘‘phospho-
degron’’ (Taelman et al., 2010; Kim et al., 2009; Xu et al.,
2009). Phosphorylation of the two serines in the phosphodegron
potentially tags a given protein for b-TrCP recognition, ubiquiti-
nation, and proteosomal degradation. Regulation of TAZ stability

Figure 4. The Kinase Activity of GSK3 Is Essential for TAZ Degradation by b-TrCP
(A) Endogenous TAZ is degraded by b-TrCP. b-TrCP depletion leads to TAZ stabilization in MII cells, as revealed by western blot. GAPDH serves as loading
control.
(B) Endogenous TAZ binds to b-TrCP in a GSK3-dependent manner. Coimmunoprecipitation/western blot analysis of MII cell lysates shows that endogenous
b-TrCP is pulled down specifically by TAZ (lane 1 versus 2) only in the presence of GSK3 (lane 2 versus 3).
(C) GSK3 kinase activity is essential to dampen TAZ protein levels and function. MII cells were engineered to express doxycycline-inducible/siRNA-insensitive
human GSK3b, either wild-type (wt; lanes 1–3) or kinase-dead mutant (KD; lanes 4–6). After depletion of endogenous GSK3 by siRNA transfection (lanes 2, 3, 5,
and 6), cells were exposed to 0.5 mg/ml doxycycline to induce the expression of either wt GSK3b (lane 3) or KDGSK3b (lane 6). Lanes 1 and 4 are cells transfected
with control siRNA. Top: quantitative real-time PCRs forCTGF (mean +SD). Bottom: western blots for TAZ and GSK3. Lane 1 versus 2 and lane 4 versus 5: TAZ is
stabilized and activated (CTGF induction) upon GSK3 depletion; lane 3: reconstitution with wt GSK3b rescues the effect of GSK3 depletion; lane 6: reconstitution
with KD GSK3b has no effect.
(D) GSK3 regulates TAZ independently of its phosphodegron. Lanes 1 and 2: western blot for endogenous TAZ shows its stabilization upon GSK3 depletion.
Lanes 3–6:MII cells were transfectedwith TAZ siRNA (to avoid interference from regulations of endogenous TAZ) and reconstitutedwith siRNA-insensitivemouse
TAZ, either wt (lanes 3 and 4), or S58/62Amutant (lanes 5 and 6). Both wild-type and S58/62A TAZ are sensitive to GSK3 siRNA. See Figures S4D and S4E for TAZ
mRNA levels.
(E)Wnt activates TAZ independently of its GSK3-phosphodegron. HEK293 cells were transfectedwith the synthetic 8xGTIIC-Lux reporter. Lanes 1 and 2:Wnt3A-
conditioned medium activates the reporter. Lanes 3 and 4: knockdown of endogenous TAZ abolishes the effect of Wnt. Lanes 5–8: cells were transfected with
TAZ siRNA to avoid interference from regulations of endogenous TAZ and were reconstituted with either mouse wt TAZ (lanes 5 and 6) or TAZ S58/62A (lanes
7 and 8). The phosphodegron is irrelevant for TAZ activation by Wnt (lanes 6 and 8). Bars are mean +SD.
See also Figure S4.
by GSK3 has been previously documented downstream of
PI3K/AKT signaling through phosphorylation of an N-terminal
phosphodegron motif, entailing S58 and S62 (Huang et al.,
2012) (see Figure S4C). This raised the possibility that, in analogy
to b-catenin, GSK3 might promote TAZ degradation by phos-
phorylating these two sites also in the context of the Wnt
cascade. If this were the case, mutation of those two residues
from serine to alanine should render TAZ resistant to GSK3-
mediated degradation. In stark contrast with this prediction,
both wild-type and S58/62A TAZ were similarly stabilized by
GSK3 knockdown (Figure 4D). Moreover, the activity of TAZ
S58/62A could still be induced by Wnt3A (Figure 4E). We
conclude that GSK3 keeps TAZ levels low irrespective of integ-
rity of the N-terminal TAZ phosphodegron.
Taken together, the results indicated that, although GSK3
kinase activity is essential for TAZ association to b-TrCP, a
mechanism entailing direct GSK3 phosphorylation of TAZ was
C
unlikely. We thus considered that the effect of GSK3 on TAZ
stability was primarily indirect, that is, through modification of
an intermediary protein serving as adaptor for TAZ degradation.
And, in the context of Wnt signaling, the most likely culprit was
b-catenin itself, whose degradation is well known to require
direct GSK3 phosphorylation.
b-Catenin Promotes TAZ DegradationBy coimmunoprecipitation, we found that b-catenin and TAZ
form a complex at endogenous protein levels (Figures 5A and
S5A), prompting us to test whether b-catenin was required for
TAZ association to b-TrCP and TAZ degradation. Intriguingly,
depletion of b-catenin by siRNA revealed that b-catenin was
required for the endogenous association of TAZ to b-TrCP in
MII cells (Figure 5B). Furthermore, knockdown of b-catenin by
independent siRNAs caused a robust stabilization of TAZ pro-
tein, induction of TAZ transcriptional activity, and TAZ nuclear
ell 151, 1443–1456, December 21, 2012 ª2012 Elsevier Inc. 1449

Figure 5. Role of b-Catenin for TAZ Degradation
(A and B) b-catenin associates with TAZ at endogenous protein levels (A) and is required for TAZ interaction with b-TrCP (B). Endogenous TAZ was immuno-
precipitated from lysates of MII cells transfected with control, b-catenin, or TAZ siRNAs by using anti-TAZ antibody, and coprecipitating proteins were detected
by western blot.
(C) Endogenous b-catenin is required for TAZ degradation. MII cells were transfected with control or b-catenin siRNAs. Top: quantitative real-time PCRs for TAZ
target CTGF. Bars are mean +SD, normalized to lane 1. Bottom: western blots for TAZ and b-catenin. GAPDH serves as loading control. TAZmRNA expression
wasn’t affected by b-catenin depletion (data not shown).
(D) Luciferase assay on 8xGTIIC reporter in HEK293 cells transfected with control or b-catenin siRNAs, with or without TAZ siRNA. Data are normalized to control
siRNA-transfected cells, and bars are mean +SD.
(E) Representative confocal images of TAZ (top) in MII cells transfected with control or b-catenin siRNAs. Nuclei are stained with Hoechst (bottom). See also
Figure S5B.
(F) The WW domain of TAZ is required for association to b-catenin and b-TrCP. TAZ was immunoprecipitated from lysates of MII cells stably expressing Flag-
tagged wild-type or DWW TAZ by using anti-Flag antibody, and co-precipitating proteins were detected by Western blot.
(G) Luciferase assay on 8xGTIIC reporter recording the transcriptional activity of wild-type or DWW TAZ transiently transfected in HEK293 cells in the absence
(Co) or presence (Wnt3A) of Wnt stimulation. Cells were transfected with TAZ siRNA to avoid interference from regulations of endogenous TAZ. Data are
normalized to lane 1 and are presented as mean +SD. Bottom: western blots for TAZ in the same extracts used for the luciferase assay.
See also Figure S5.
accumulation (Figures 5C–5E and S5B). Of note, loss of b-cate-
nin was not accompanied by events known to regulate TAZ
through the Hippo pathway (Cordenonsi et al., 2011), that is,
downregulation of cadherin adherens junctions, epithelial-mes-
enchymal transition, disturbed apico-basal polarity, or changes
in the levels of active LATS (Figures S5C and S5D).
Our results thus suggested a model whereby b-catenin plays
a central role in TAZ inhibition by bridging TAZ to the b-TrCP
complex (Figure 6A, left). In this scenario, a TAZ mutant that
cannot interact with b-catenin should not bind b-TrCP, nor
should it be regulated by Wnt signaling. To validate this notion,
we carried out mapping studies by using recombinant GST-
TAZ deletion constructs and in-vitro-translated b-catenin. The
1450 Cell 151, 1443–1456, December 21, 2012 ª2012 Elsevier Inc.
results revealed that the ‘‘WW’’ domain of TAZ was required
for association to b-catenin (Figure S5E). Once expressed in
human cells not experiencing Wnt signaling, TAZ deleted of the
WW domain (TAZDWW) was more stable and basally more
active than wild-type TAZ (Figures 5G and S5F), correlating
with the incapacity of TAZDWW to associate to endogenous
b-catenin and b-TrCP (Figure 5F). As predicted, TAZDWW could
not be further stabilized nor activated by Wnt signaling (Fig-
ure 5G). Critically, the hyperactive behavior of TAZDWW could
not be ascribed to an escape from the Hippo pathway, as
TAZDWW is still phosphorylated on the leading LATS target
S306 and can be further stabilized by LATS1/2 depletion (Figures
S5G and S5H). We conclude from these structure-function

experiments that, in Wnt-OFF cells, b-catenin bridges TAZ to
b-TrCP (Figure 6A, left). Activation of the Wnt pathway reduces
such association of b-catenin to b-TrCP and, consequently,
dampens TAZ degradation (Figure 6A, middle).
In the Wnt-OFF world, cytoplasmic b-catenin is recruited to
b-TrCP after being phosphorylated by GSK3, whose kinase
activity is also critical for TAZ degradation (Figure 4). Is the
GSK3-phosphorylated pool of b-catenin the ultimate mediator
of TAZ degradation? For this, we analyzed TAZ stability and
CTGF induction in two MII cell lines, which were depleted of
endogenous b-catenin and reconstituted at near-to-endoge-
nous levels with either wild-type b-catenin or a phosphomutant
b-catenin (Figure 6B). This mutant harbors N-terminal point
mutations (serine to alanine, S/A) that prevent GSK3 phosphor-
ylation and b-TrCP recognition (Liu et al., 2002). As above,
knockdown of b-catenin promoted TAZ stabilization in both MII
derivatives (Figure 6B, lanes 1 and 2 and lanes 4 and 5); adding
back wild-type b-catenin rescued TAZ degradation (Figure 6B,
lane 3). However, reconstitution with S/A phosphomutant b-cat-
enin had no effect (Figure 6B, lane 6), indicating that TAZ degra-
dation relies on b-catenin phosphorylation by GSK3. In these
experiments, changes in TAZ protein levels were consistently
paralleled by changes in its activity, as indicated by the expres-
sion of its target CTGF (Figure 6B).
Further support to the model proposed in Figure 6A was
provided by experiments in HepG2 hepatoma cells. These cells
carry two different b-catenin alleles: one encoding wild-type
b-catenin and the other encoding a constitutively active b-cate-
nin bearing a natural deletion in the N terminus encompassing
the GSK3 phosphorylation sites (Figure 6C). We designed
a siRNA oligonucleotide (#4) that specifically binds within the
deletion and, as such, only targets the wild-type and not the
mutated allele. By using this siRNA, we found that the sole
wild-type b-catenin is responsible for TAZ degradation (Fig-
ure 6D, compare lanes 1 and 5), as depletion of both isoforms
does not further increase TAZ stability (Figure 6D, compare lanes
1, 3, and 5). Conversely, b-catenin-dependent induction of one
of its transcriptional targets, Cyclin-D1, is almost exclusively
supported by the mutant allele (Figure S6C). Thus, the HepG2
model system allows uncoupling of the two functions of b-cate-
nin: nuclear/transcriptional activity, a function almost exclusively
mediated by the stabilized b-catenin, and TAZ degradation,
which requires a phosphorylatable b-catenin.
We then asked whether the role of b-catenin as TAZ inhibitor
is functionally relevant for TAZ activity. TAZ and Wnt have
been independently proposed as critical factors in sustaining
the self-renewal of normal and transformed mammary epithelial
cells, as assessed in mammosphere assay (Cordenonsi et al.,
2011; Scheel et al., 2011). For example, TAZ depletion impairs
formation of mammospheres in cell populations enriched of
prospective cancer stem cells, whereas gain of TAZ promotes
mammosphere potential in otherwise non-stem-cell populations
(Cordenonsi et al., 2011). In line with b-catenin depletion leading
to TAZ stabilization, knockdown of b-catenin in MII cells caused
an increase in mammosphere formation that was blunted upon
concomitant depletion of TAZ (Figure 6E).
We next sought to determine to what extent b-catenin serves
as endogenous TAZ inhibitor in cells lacking Wnt stimulation or
C
oncogenic activation of the pathway. For this, we first monitored
by microarray profiling the global transcriptional effects of b-cat-
enin depletion in MII cells and then asked to what extent these
were dependent on TAZ upregulation. We found that b-catenin
depletion led to induction of about 350 probesets; a remarkable
74%of this list was TAZ dependent, as induction of these probe-
sets was reverted by concomitant loss of TAZ (Figure 6F). Inter-
estingly, >90% of the TAZ-dependent probesets upregulated by
b-catenin depletion were also upregulated by APC depletion in
a TAZ-dependent manner (Figure 1G) and, thus, represented
targets of the following pathway:
Wnt signaling C APC/Phospho-b-catenin C TAZ
The results collectively indicate that APC-regulated cytoplasmic
b-catenin, so far considered transcriptionally irrelevant and only
meant to be degraded, is in fact a potent repressor of the TAZ
transcriptional program.
g-Catenin Participates in TAZ Regulationg-catenin is a homolog of b-catenin, and the two proteins are
thought to operate in an overlapping manner in several con-
texts: they have redundant functions in cell adhesion and share
Wnt-dependent regulation through phosphorylation by GSK3
and b-TrCP-mediated degradation (Xu et al., 2009). Importantly,
TAZ associates with g-catenin at endogenous protein levels
(Figure 6G), and, similarly to b-catenin, this interaction is depen-
dent on the integrity of the TAZ WW domain (Figure S6F). This
prompted us to test whether g-catenin also shares with b-cate-
nin the capacity to regulate TAZ. To examine this, we monitored
TAZ activity in HEK293 cells transfected with siRNAs against
b-catenin, g-catenin, or both. Depletion of g-catenin led to
TAZ stabilization (Figure 6H) and TAZ-dependent transcriptional
activity (Figure 6I, lane 5). Interestingly, concomitant depletion
of both b- and g-catenin resulted in even stronger transcrip-
tional activation (Figure 6I). This indicates that these factors
play independent and additive roles in TAZ inhibition.
Finally, we tested whether depletion of g-catenin is sufficient
to sustain the biological functions of TAZ even in the absence
of Wnt stimulation. For this, we chose to monitor osteogenic
differentiation of bone marrow cells that—as shown above (Fig-
ure 3)—is promoted by Wnt through TAZ stabilization. Indeed,
depletion of g-catenin in murine bone marrow stromal cells
induced a robust increase of TAZ protein and TAZ-dependent
activation of the osteogenic program (Figure 6J). This activity
closely resembles the effects of Wnt ligands and suggests that
b-catenin and its homolog g-catenin play a relevant role in the
control of TAZ stability and activity.
DISCUSSION
In this work, we have presented evidence indicating that TAZ is
downstream of Wnt signaling and is critical for some established
Wnt-dependent biological responses.
Inhibition of theb-catenin destruction complex is central toWnt
signaling; this results in b-catenin stabilization. In this classic
view, b-catenin is the last known element of the Wnt signaling
cascade. Here, we reveal the existence of a new downstream
ell 151, 1443–1456, December 21, 2012 ª2012 Elsevier Inc. 1451

Figure 6. TAZ Regulation by the Wnt/b-Catenin Pathway
(A) A model depicting the proposed mechanism for TAZ regulation by Wnt pathway and b-catenin. See Discussion.
(B) Phospho-b-catenin mediates TAZ degradation. MII cells were engineered to express doxycycline-inducible siRNA-insensitive b-catenin, either wt (lanes 1–3)
or phosphomutant (S/A, lanes 4–6). After depletion of endogenous b-catenin by siRNA (lanes 2, 3, 5, and 6), cells were treated with 0.5 mg/ml doxycycline to
induce the expression of wt (lane 3) or S/A b-catenin (lane 6). Lanes 1 and 4were cells transfectedwith control siRNA. Panels are western blots for TAZ, CTGF, and
b-catenin. Lane 1 versus 2 and lane 4 versus 5: TAZ is stabilized and activated upon b-catenin depletion; lane 3: reconstitution with wt b-catenin rescues TAZ
inhibition; lane 6: S/A b-catenin cannot revert the effect of b-catenin siRNA. Both wt and S/A b-catenin are equally able to localize to the plasma membrane (see
Figure S6A).
(C) Scheme of the two transcripts (and protein products) encoded by the wt (black) and exon3/exon4-deleted (del ex3-4, blue) b-catenin alleles of HepG2 cells.
The phosphodegron site on wild-type b-catenin, missing in the mutant protein, is in orange. The regions of b-cateninmRNA targeted by siRNAs are indicated in
red; as the sequence targeted by siRNA#4 is within the deletion, this siRNA only hits the wild-type transcripts, whereas siRNA#3 is used to deplete both isoforms.
(legend continued on next page)
1452 Cell 151, 1443–1456, December 21, 2012 ª2012 Elsevier Inc.

layer in this cascade, revealing a key role for b-catenin in
regulating the stability of another potent transcriptional coactiva-
tor, TAZ.
TAZ as a Downstream Mediator of Wnt SignalingThe results shed new light on the modalities by which the Wnt
pathway controls gene expression. Prior to this work, we could
have equated the Wnt signal only to the genes controlled by
b-catenin through its association to TCF/Lef. Here, we show
that formation of the b-catenin/TCF complex is not the sole
modality by which the Wnt/b-catenin pathway can regulate
gene expression, as we found that TAZ protein stabilization
and TAZ-mediated transcription are also a general feature of
the Wnt response in a variety of cellular model systems. It
is noteworthy that, when we challenged by gene expression
profiling the global relevance of TAZ downstream of Wnt sig-
naling, that is, in a manner that goes beyond by the use of
specific bioassays or genetic sensors, the regulation of a very
significant fraction of Wnt target genes turned out to be TAZ
dependent both in mammary epithelial cells and in colorectal
cancer cells.
We propose that Wnt signaling has thus two consequences:
to turn on the transcriptional functions of b-catenin, as so far
acknowledged, and to turn on the transcriptional functions of
TAZ. The specific relevance of these two aspects will likely
depend on the cellular context. In some cell types, or experi-
mental conditions, the effects of Wnt may primarily depend
on TCF-dependent transcription. However, we reason that the
most likely situation will be one in which both b-catenin- and
TAZ-dependent biological effects are occurring at the same
time in response to Wnt pathway activation. For example, in
colorectal cancer cells, the presence of oncogenic mutations
in APC potently stabilizes both b-catenin and TAZ, with both
factors sustaining cell proliferation. Still, even in this oncogenic
setup, we could highlight a specific role of TAZ in endowing
clonogenic potential to CRC cells, a function not shared by stabi-
lized b-catenin. Similarly, TAZ plays essential roles in the regula-
tion of mesenchymal stem cell differentiation, likely in concert
with, and complementary to, b-catenin.
Makita et al. (2008) reported that a minor fraction of TAZ
mutant mice develop to term, suggesting that TAZ may primarily
(D) HepG2 cells were transfected with b-catenin siRNAs indicated in (C), with or wi
lane 1. Bottom: western blots for TAZ and b-catenin. Depletion of the sole wild-ty
stability and activity without affecting TAZ mRNA expression or the transcription
(E) Quantification of mammospheres formed by MII cells transfected with con
mean +SEM. See Figure S6D for independent b-catenin siRNAs.
(F) Pie chart of U133plus2 Affymetrix probesets upregulated in MII cells transfec
colored area of the graph represents the fraction of probesets whose upregulati
(G) Coimmunoprecipitation between endogenous TAZ and g-catenin in MII cell l
(H) Western blots for TAZ and g-catenin in extracts from HEK293 cells transfected
mediated phosphorylation of LATS (Figure S6G).
(I) Luciferase assay on 8xGTIIC reporter in HEK293 cells transfected with control, b
siRNA. Data are normalized to control siRNA-transfected cells, and bars are m
interfering sequences (Figure S6H; data not shown).
(J) Effects of g-catenin downregulation on TAZ expression and on osteoblastic d
and TAZ siRNAs as indicated. Left: ALP activity (mean +SEM). Right: western b
g-catenin siRNA (data not shown).
See also Figure S6.
C
contribute to Wnt responses in adult tissues. It must be also
noticed, however, that the vast majority of TAZ mutants die in
utero with an as-yet-uncharacterized phenotype (Makita et al.,
2008); in fact, we have been unable to obtain any TAZ�/�
newborn in our colony (L.A. and S.P., unpublished data). This
leaves open the potential of an overlap between Wnt and TAZ
also in embryonic development, perhaps consistently with the
severe phenotype of TAZ-deficient zebrafish embryos (Hong
et al., 2005).
TAZ and its related protein YAP are functionally redundant
during embryonic development, with double knockouts failing
to form the blastocyst (Nishioka et al., 2009). In this work, we
have focused on TAZ because of its overt stabilization by Wnt
signaling and functional relevance in Wnt bioassays. A question
is whether YAP may be also controlled by Wnt through similar or
unrelated mechanisms; of note, a complex between b-catenin
and YAP has been previously reported (Imajo et al., 2012).
Although we failed to detect changes in YAP protein steady-
state levels upon Wnt signaling, YAP stability may be regulated
more finely or at the level of nuclear activity and nucleo-cyto-
plasmic localization. Dedicated studies are required to dissect
these possibilities.
A New Role for b-CateninHere, we propose a distinct perspective on Wnt signaling
whereby b-catenin plays active roles both in the presence and
absence of Wnt ligands (Figure 6A). In the absence of Wnt
signaling (i.e., Wnt OFF state), the pool of phospho-b-catenin
is an essential element for continuous TAZ degradation, as it
serves as critical scaffold for TAZ recognition by the b-TrCP E3
ubiquitin ligase. This configures an unexpected role for cyto-
plasmic b-catenin: besides being recruited to the destruction
complex for its own degradation, b-catenin works as yet another
intermediary of the Wnt cascade upstream of TAZ. Much evi-
dence supports this conclusion, including (1) TAZ and b-catenin
form a complex at endogenous protein levels in unstimulated
cells; (2) b-catenin phosphorylation is required for TAZ degrada-
tion; and (3) depletion of both GSK3 and b-catenin impairs TAZ/
b-TrCP interaction.
One possible interpretation of these results would be that
TAZ could only bind the phosphorylated form of b-catenin.
thout TAZ siRNA. Top: qRT-PCRs forCTGF. Bars are mean +SD, normalized to
pe pool of b-catenin (with siRNA#4, lanes 5 and 6) is sufficient to promote TAZ
al activity of the mutant b-catenin pool (see Figures S6B and S6C).
trol or b-catenin siRNAs, with or without TAZ siRNA. Data are presented as
ted with b-catenin siRNA compared to control siRNA-transfected cells. Dark
on by b-catenin depletion is reverted by cotransfection of TAZ siRNA.
ysates.
with control or g�catenin siRNAs. Depletion of g-catenin didn’t reduce MST-
-catenin (sibcat), or g-catenin (sigcat) siRNAs as indicated, with or without TAZ
ean +SD. Similar results were obtained with different b-catenin or g-catenin
ifferentiation of ST-2 cells. ST-2 cells were transfected with control, g-catenin,
lots for TAZ and g-catenin. Similar results were obtained with an alternative
ell 151, 1443–1456, December 21, 2012 ª2012 Elsevier Inc. 1453

However, TAZ and b-catenin proteins can associate in vitro,
arguing against a strict requirement of b-catenin phosphoryla-
tion. Another equally plausible interpretation is that, irrespec-
tively of b-catenin phosphorylation per se, TAZ may effectively
associate to b-catenin only after cytoplasmic b-catenin is
captured by the destruction complex, a scenario that is compat-
ible with other proteins of the destruction complex either rein-
forcing the b-catenin/TAZ interaction or escorting the b-cate-
nin/TAZ/b-TrCP recognition.
The relevance of b-catenin as endogenous inhibitor of TAZ
was validated at the genome-wide level in mammary epithelial
cells by microarray experiments. These analyses uncovered
that APC-regulated cytoplasmic b-catenin, per se transcrip-
tionally irrelevant, does nevertheless actively control gene ex-
pression, as it restrains the TAZ-dependent gene expression
program (Figure 6F). As proof of principle of the functional rele-
vance of this inhibition, we used an established TAZ biological
assay (mammosphere assay; Cordenonsi et al., 2011) and
confirmed that b-catenin depletion enhances TAZ activity in
mammary epithelial cells.
g-catenin represents yet another variation on this scenario;
this b-catenin-related protein is regulated by the Wnt/GSK3/
b-TrCP axis, but its relevance for TCF-dependent transcription
is still debated (Ben-Ze’ev, 1999). Nevertheless, here we show
that g-catenin is able to regulate TAZ-dependent transcriptional
responses by promoting TAZ instability in concert, or redun-
dantly, with b-catenin. From this ‘‘TAZ perspective,’’ g-catenin
may effectively be part of the Wnt response.
We propose that, in cells experiencing Wnt activity, b-catenin,
per se largely dissociated from b-TrCP, is incapable of carrying
out any adaptor function for TAZ and, as such, is irrelevant for
TAZ regulation (Figure 6A). In this view, it is the loss of the asso-
ciation of phospho-b-catenin to b-TrCP that causes TAZ stabili-
zation; we found that TAZ is indeed stabilized not only by Wnt
signaling but also by experimental removal of b-catenin (Fig-
ure 6A, middle and right). It also follows that removing b-catenin
from cells where the fraction of phospho-b-catenin is negli-
gible—such as Wnt-treated cells or cells carrying oncogenic
APC—is inconsequential for an already stabilized TAZ protein.
In fact, b-catenin knockdown cannot further increase TAZ levels
in APC-deficient SW480 colon cancer cells (Figure S2B, lane 3)
nor TAZ-dependent transcription in Wnt-stimulated HEK293
cells (Figure 1B, lane 3).
The functions of the two pools of b-catenin could be visualized
at the endogenous level and within the same cellular context by
using HepG2 cells, which contain both wild-type and a nonphos-
phorylatable mutant b-catenin. In line with our model, only wild-
type b-catenin is relevant to oppose TAZ; mutant-b-catenin is
unable to bring TAZ to degradation, whereas it mediates the
b-catenin/TCF transcriptional responses.
TAZ Stability at the Crossroad between Wnt and HippoSignalingCell-cell adhesion and polarity cues activate the Hippo/LATS
kinases, leading to TAZ phosphorylation and sequestration in
the cytoplasm (Cordenonsi et al., 2011; Pan, 2010). More
recently, a largely LATS-independent modality to regulate TAZ
stability and activity has been also shown to occur as conse-
1454 Cell 151, 1443–1456, December 21, 2012 ª2012 Elsevier Inc.
quence of changes in cell shape and cytoskeletal reorganization
induced by mechanical cues, such as rigidity of the extracellular
matrix (Dupont et al., 2011). Here, we further expanded the land-
scape of TAZ regulation by showing that TAZ is controlled by
a major family of secreted growth factors in a manner largely
independent fromHippo signaling. This network accommodates
self-regulating feedback loops; TAZ was demonstrated to nega-
tively regulate the Wnt pathway by inhibiting Dishevelled, a posi-
tive Wnt regulator (Varelas et al., 2010a). This may provide a
ceiling to the levels of TAZ activation by Wnts and a mechanism
to turn off TAZ-dependent responses.
One point worth discussing is that TAZ is phosphorylated by
LATS and CK1 to generate a C-terminal phosphodegron that
has been shown to be relevant for TAZ stability and to promote
direct association to b-TrCP, at least under conditions of protein
overexpression (Liu et al., 2010). This is apparently at odds with
the present findings indicating that, at endogenous protein
levels, TAZ and b-TrCP associate indirectly through a b-catenin
bridge. In fact, these two scenarios are perfectly compatible, as
these represent formally independent modalities of regulating
TAZ stability by the Hippo and Wnt cascades, respectively.
Indeed, here we show that LATS inhibition or mutation of the
LATS or CK1 phosphorylation sites are irrelevant for TAZ regula-
tion by Wnt (Figures S1C and S1D; data not shown); conversely,
a TAZ mutant unable to bind b-catenin is insensitive to Wnt but
remains under LATS1/2 control. Taken together, these findings
open very intriguing possibilities for TAZ as a hub integrating
different physiological inputs.
Finally, having established TAZ as Wnt effector opens up
many therapeutic possibilities. Targeting TAZ could curb aber-
rant Wnt signaling in a variety of cancers and other disorders.
Conversely, targeting the Wnt pathway by restoring the integ-
rity of the destruction complex would effectively inhibit, at
once, two of the most potent oncogenic drivers of human
malignancies.
EXPERIMENTAL PROCEDURES
Luciferase Assays
Luciferase reporters (50 ng/cm2) were transfected together with CMV-b-gal
(75 ng/cm2) to normalize for transfection efficiency with CPRG (Roche) colori-
metic assay. For luciferase assays in siRNA-depleted cells, cells were first
transfected with the indicated siRNAs and, after 24 hr, were transfected with
plasmid DNA. Where indicated, cells were exposed to control- or Wnt3A-
conditioned medium for 24 hr before harvesting. Each sample was transfected
in duplicate, and each experiment was repeated at least three times indepen-
dently. siRNA sequences are listed in Table S1.
MSC Differentiation Assays
For bone differentiation of ST-2 mouse cells, cells were cultured in the pres-
ence of 100 mg/ml ascorbic acid (Sigma) and 5 mM b-glicerophosphate
(Sigma), alone or in combination with 100 ng/ml recombinant Wnt3A (Pepro-
tech) for 72 hr. Cells were then harvested in 0.5% Triton X-100, and alkaline
phosphatase (ALP) activity was assessed by measuring spectroscopycally
at 405 nm the hydrolysis of p-nitrophenyl phosphate (Sigma) to p-nitrophenol
(expressed as nmol/min/mg protein) after a 10 min incubation at room temper-
ature. Data were normalized to the total protein content, determined by
Bradford. For bone differentiation of hMSC, cells were switched from growth
medium (GM) to osteogenic differentiation medium (ODM) 24 hr after seeding;
medium was then renewed every 2 days for a total of 10 days of differen-
tiation. MSC were then fixed and assayed by ALP staining kit (Sigma 85L2).

Adipogenic differentiation of hMSC and quantifications of ALP-positive or
oil-red-positive areas were carried out as in Dupont et al. (2011).
Western Blot, Immunoprecipitation, and qPCR
MII cells were grown for 2 days at high density. Extracts and total RNA were
prepared and analyzed as in Cordenonsi et al. (2011). Immunoprecipitations
were carried out as in Cordenonsi et al. (2011) by using the anti-TAZ mono-
clonal antibody from BD Biosciences, the anti-b-catenin monoclonal antibody
(E-5) from Santa Cruz, or the anti-Flag-tag monoclonal antibody (M2) from
Sigma.
See also Extended Experimental Procedures.
ACCESSION NUMBERS
The GEO accession number for the Affymetrix gene expression data used in
this paper is GSE39907.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Extended Experimental Procedures, six
figures, and one table and can be found with this article online at http://dx.
doi.org/10.1016/j.cell.2012.11.027.
ACKNOWLEDGMENTS
We thank O. Wessely and S. Dupont for comments; S. Zanotti for ST-2 differ-
entiation protocol; G. Martello, M.C. Faux, A.R. Clarke, H. Miyoshi, and L. Nal-
dini for gifts of reagents; and Tito Panciera for helping with RNA preparations.
Plasmids purchased from Addgene were kindly deposited by K. Hochedlinger
and W. Harper. This work is supported by ‘‘Young Italian Researchers’’ grants
from the Italian Welfare Ministry and AIRC-MFAG to M.C.; from Fondazione
Citta della SperanzaGrant to G.B.; fromAIRC Special ProgramMolecular Clin-
ical Oncology ‘‘5 per mille’’ to S.P. and S.B.; and from HSFP, Excellence-IIT,
and Epigenetics Flagship project CNR-Miur grants to S.P.
Received: August 2, 2012
Revised: October 6, 2012
Accepted: November 11, 2012
Published online: December 13, 2012
REFERENCES
Barolo, S. (2006). Transgenic Wnt/TCF pathway reporters: all you need is Lef?
Oncogene 25, 7505–7511.
Ben-Ze’ev, A. (1999). The dual role of cytoskeletal anchor proteins in cell adhe-
sion and signal transduction. Ann. N Y Acad. Sci. 886, 37–47.
Bhat, K.P., Salazar, K.L., Balasubramaniyan, V., Wani, K., Heathcock, L., Hol-
lingsworth, F., James, J.D., Gumin, J., Diefes, K.L., Kim, S.H., et al. (2011).
The transcriptional coactivator TAZ regulates mesenchymal differentiation in
malignant glioma. Genes Dev. 25, 2594–2609.
Clevers, H. (2006). Wnt/b-catenin signaling in development and disease. Cell
127, 469–480.
Cordenonsi, M., Zanconato, F., Azzolin, L., Forcato, M., Rosato, A., Frasson,
C., Inui, M., Montagner, M., Parenti, A.R., Poletti, A., et al. (2011). The Hippo
transducer TAZ confers cancer stem cell-related traits on breast cancer cells.
Cell 147, 759–772.
Dupont, S., Morsut, L., Aragona, M., Enzo, E., Giulitti, S., Cordenonsi, M., Zan-
conato, F., Le Digabel, J., Forcato, M., Bicciato, S., et al. (2011). Role of YAP/
TAZ in mechanotransduction. Nature 474, 179–183.
Faux, M.C., Ross, J.L., Meeker, C., Johns, T., Ji, H., Simpson, R.J., Layton,
M.J., and Burgess, A.W. (2004). Restoration of full-length adenomatous poly-
posis coli (APC) protein in a colon cancer cell line enhances cell adhesion.
J. Cell Sci. 117, 427–439.
Halder, G., Dupont, S., and Piccolo, S. (2012). Transduction of mechanical
and cytoskeletal cues by YAP and TAZ. Nat. Rev. Mol. Cell Biol. 13, 591–600.
C
Hong, J.H., Hwang, E.S., McManus, M.T., Amsterdam, A., Tian, Y., Kalmu-
kova, R., Mueller, E., Benjamin, T., Spiegelman, B.M., Sharp, P.A., et al.
(2005). TAZ, a transcriptional modulator of mesenchymal stem cell differentia-
tion. Science 309, 1074–1078.
Huang, S.M., Mishina, Y.M., Liu, S., Cheung, A., Stegmeier, F., Michaud,
G.A., Charlat, O., Wiellette, E., Zhang, Y., Wiessner, S., et al. (2009). Tankyr-
ase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461,
614–620.
Huang, W., Lv, X., Liu, C., Zha, Z., Zhang, H., Jiang, Y., Xiong, Y., Lei, Q.Y., and
Guan, K.L. (2012). The N-terminal phosphodegron targets TAZ/WWTR1
protein for SCFb-TrCP-dependent degradation in response to phosphatidyli-
nositol 3-kinase inhibition. J. Biol. Chem. 287, 26245–26253.
Imajo, M., Miyatake, K., Iimura, A., Miyamoto, A., and Nishida, E. (2012). A
molecular mechanism that links Hippo signalling to the inhibition of Wnt/b-cat-
enin signalling. EMBO J. 31, 1109–1122.
Kim, N.G., Xu, C., and Gumbiner, B.M. (2009). Identification of targets of the
Wnt pathway destruction complex in addition to b-catenin. Proc. Natl. Acad.
Sci. USA 106, 5165–5170.
Leucht, P., Minear, S., Ten Berge, D., Nusse, R., and Helms, J.A. (2008). Trans-
lating insights from development into regenerative medicine: the function of
Wnts in bone biology. Semin. Cell Dev. Biol. 19, 434–443.
Li, V.S., Ng, S.S., Boersema, P.J., Low, T.Y., Karthaus, W.R., Gerlach, J.P.,
Mohammed, S., Heck, A.J., Maurice, M.M., Mahmoudi, T., and Clevers, H.
(2012). Wnt signaling through inhibition of b-catenin degradation in an intact
Axin1 complex. Cell 149, 1245–1256.
Liu, C., Li, Y., Semenov, M., Han, C., Baeg, G.H., Tan, Y., Zhang, Z., Lin, X., and
He, X. (2002). Control of b-catenin phosphorylation/degradation by a dual-
kinase mechanism. Cell 108, 837–847.
Liu, C.Y., Zha, Z.Y., Zhou, X., Zhang, H., Huang, W., Zhao, D., Li, T., Chan,
S.W., Lim, C.J., Hong, W., et al. (2010). The hippo tumor pathway promotes
TAZ degradation by phosphorylating a phosphodegron and recruiting the
SCFb-TrCP E3 ligase. J. Biol. Chem. 285, 37159–37169.
MacDonald, B.T., Tamai, K., and He, X. (2009). Wnt/b-catenin signaling:
components, mechanisms, and diseases. Dev. Cell 17, 9–26.
Makita, R., Uchijima, Y., Nishiyama, K., Amano, T., Chen, Q., Takeuchi, T., Mi-
tani, A., Nagase, T., Yatomi, Y., Aburatani, H., et al. (2008). Multiple renal cysts,
urinary concentration defects, and pulmonary emphysematous changes in
mice lacking TAZ. Am. J. Physiol. Renal Physiol. 294, F542–F553.
Moon, R.T., Kohn, A.D., De Ferrari, G.V., and Kaykas, A. (2004). WNT and
b-catenin signalling: diseases and therapies. Nat. Rev. Genet. 5, 691–701.
Niehrs, C., and Acebron, S.P. (2012). Mitotic and mitogenic Wnt signalling.
EMBO J. 31, 2705–2713.
Nishioka, N., Inoue, K., Adachi, K., Kiyonari, H., Ota, M., Ralston, A., Yabuta,
N., Hirahara, S., Stephenson, R.O., Ogonuki, N., et al. (2009). The Hippo
signaling pathway components Lats and Yap pattern Tead4 activity to distin-
guish mouse trophectoderm from inner cell mass. Dev. Cell 16, 398–410.
Pan, D. (2010). The hippo signaling pathway in development and cancer. Dev.
Cell 19, 491–505.
Ramos, A., and Camargo, F.D. (2012). The Hippo signaling pathway and stem
cell biology. Trends Cell Biol. 22, 339–346.
Ross, S.E., Hemati, N., Longo, K.A., Bennett, C.N., Lucas, P.C., Erickson, R.L.,
and MacDougald, O.A. (2000). Inhibition of adipogenesis by Wnt signaling.
Science 289, 950–953.
Scheel, C., Eaton, E.N., Li, S.H., Chaffer, C.L., Reinhardt, F., Kah, K.J., Bell, G.,
Guo,W., Rubin, J., Richardson, A.L., andWeinberg, R.A. (2011). Paracrine and
autocrine signals induce andmaintainmesenchymal and stem cell states in the
breast. Cell 145, 926–940.
Taelman, V.F., Dobrowolski, R., Plouhinec, J.L., Fuentealba, L.C., Vorwald,
P.P., Gumper, I., Sabatini, D.D., and De Robertis, E.M. (2010). Wnt signaling
requires sequestration of glycogen synthase kinase 3 inside multivesicular en-
dosomes. Cell 143, 1136–1148.
van de Wetering, M., Sancho, E., Verweij, C., de Lau, W., Oving, I., Hurlstone,
A., van der Horn, K., Batlle, E., Coudreuse, D., Haramis, A.P., et al. (2002).
ell 151, 1443–1456, December 21, 2012 ª2012 Elsevier Inc. 1455

The b-catenin/TCF-4 complex imposes a crypt progenitor phenotype on
colorectal cancer cells. Cell 111, 241–250.
Varelas, X., Miller, B.W., Sopko, R., Song, S., Gregorieff, A., Fellouse, F.A., Sa-
kuma, R., Pawson, T., Hunziker, W., McNeill, H., et al. (2010a). The Hippo
pathway regulates Wnt/b-catenin signaling. Dev. Cell 18, 579–591.
Varelas, X., Samavarchi-Tehrani, P., Narimatsu, M., Weiss, A., Cockburn, K.,
Larsen, B.G., Rossant, J., and Wrana, J.L. (2010b). The Crumbs complex
1456 Cell 151, 1443–1456, December 21, 2012 ª2012 Elsevier Inc.
couples cell density sensing to Hippo-dependent control of the TGF-
b-SMAD pathway. Dev. Cell 19, 831–844.
Xu, C., Kim, N.G., and Gumbiner, B.M. (2009). Regulation of protein stability by
GSK3 mediated phosphorylation. Cell Cycle 8, 4032–4039.
Zhao, B., Li, L., Tumaneng, K., Wang, C.Y., and Guan, K.L. (2010). A coordi-
nated phosphorylation by Lats and CK1 regulates YAP stability through
SCF(b-TRCP). Genes Dev. 24, 72–85.

Supplemental Information
EXTENDED EXPERIMENTAL PROCEDURES
Reagents and PlasmidsXAV939, b-naphthoflavone and doxycycline were from Sigma. CHIR99021 (GSK3 kinase inhibitor) was from Axon MedChem. Re-
combinant Wnt3A and Dkk-1 were from Peprotech.
The constructs for shGFP and shTAZ#2 and #3 were as in Cordenonsi et al. (2011).
The retroviral constructs coding for Flag-mTAZ S58/62A, Flag-mTAZ S306A, Flag-mTAZ S306/309A and Flag-mTAZDWW
(deleted of residues 110-159) were generated by mutagenesis from pBABEHygro-mTAZWT (Cordenonsi et al., 2011). Flag-mTAZ
4SA was described in Dupont et al., 2011. All TAZ cDNA sequences were also subcloned in pCS2 for transient expression in
HEK293 cells. For expression of TAZ in SW480 cells and hMSC, TAZ cDNAs were subcloned in lentiviral vector pCSII-CMV-
MCS-IRES2-Bsd (kind gift of H. Miyoshi). For GST-pull down experiments WT full-length mouse TAZ was cloned in pGEX4T1;
from this deletion constructs of GST-TAZ were generated by enzymatic digestion and self-ligation. Mouse TAZ cDNA is insensitive
to the siRNAs used to target the human transcript.
rtTA cDNA was subcloned from FUdeltaGW-rtTA (Addgene #19780, Maherali et al., 2008) to pBABE-hygro. This construct was
used to generate MII-rtTA cells used in Figures 4C, 6B, and S6A. Human myc-tagged GSK3b wt or kinase-dead (KD) mutant
(K85A) cDNAs were subcloned in pCS2 and made insensitive to GSK3b siRNA#1 by introducing silent mutations within the siRNA
targeting sequence by PCR. For inducible expression of GSK3b in MII cells, siRNA-insensitive GSK3b variants were subcloned in
a doxycycline-inducible retroviral expression vector (pSTC-Puro), and retroviral particles were used to transduce MII-rtTA cells.
cDNAs for human b-catenin, wild-type and phospho mutant (S33/37/41A, corresponding to the GSK3 sites and the CK1 priming
site S45A), were subcloned in pCSP1 and were made insensitive to b-catenin siRNA#4 by introducing silent mutations within the
siRNA targeting sequence by PCR. For inducible expression of b-catenin in MII cells, both siRNA-insensitive b-catenin variants
were subcloned in pSTC-Puro, and retroviral particles were used to transduce MII-rtTA cells. pcDNA3-DN-hCUL1-FLAG (#15818)
was purchased from Addgene (Jin et al., 2005). All constructs were confirmed by sequencing.
Cell LinesHEK293 cells, HeLa cells, HaCaT cells, P19 cells, Control-L-cells (ATCC #CRL-2648) and Wnt3a-L-cells (ATCC #CRL-2647) were
cultured in DMEM (GIBCO, Life Technologies) supplemented with 10% (20% in the case of P19 cells) FBS, glutamine and antibiotics.
Conditioned media from L-cells were harvested according to the ATCC protocol. MII cells were cultured in DMEM/F12 (GIBCO, Life
Technologies) with 5% HS, glutamine and antibiotics, freshly supplemented with insulin, EGF, hydrocortisone, and cholera toxin.
Mouse embryonic stem (ES) cell lines were a kind gift of Graziano Martello, and were cultured on gelatin-coated plates in GMEM
(Sigma) supplemented with 10% FBS, glutamine, nonessential amino acids, pyruvate, mouse LIF, b-mercaptoethanol. SW480 cells
were cultured in RPMI 1640 with 10% FBS, glutamine and antibiotics, freshly supplemented with insulin, hydrocortisone, 1-thyogli-
cerol (traces). SW480 cells stably expressing empty vector or APC were a kind gift of M.C. Faux (Faux et al., 2004) and were main-
tained with 1.5 mg/ml G418. ST-2 stromal mouse cells were purchased from DSMZ (Braunschweig, Germany) and were cultured in
MEM alpha (GIBCO, Life Technologies) supplemented 10%FBS, glutamine and antibiotics. Primary humanmesenchymal stem cells
(hMSC), their growth and differentiation media were from Lonza. HepG2 cells were cultured in MEM (GIBCO, Life Technologies) sup-
plemented with 10% FBS, glutamine, antibiotics and nonessential amino acids.
TransfectionssiRNA transfections were done with Lipofectamine RNAi-MAX (Life technologies) in antibiotics-free medium according to manufac-
turer instructions. Sequences of siRNAs are provided in Table S1. DNA transfections were donewith TransitLT1 (Mirus Bio) according
to manufacturer instructions. Lentiviral particles were prepared by transiently transfecting HEK293T cells with lentiviral vectors
together with packaging vectors (pMD2-VSVG and psPAX2). Retroviral particles were prepared and infections were carried out
as in (Martello et al., 2010).
Luciferase AssaysLuciferase assays were performed in HEK293 cells with the established YAP/TAZ-responsive reporter 8xGTIIC-Lux (Dupont et al.,
2011) or with the b-catenin/TCF-responsive reporter BAT-Lux (Maretto et al., 2003). Luciferase reporters (50 ng/cm2) were trans-
fected together with CMV-b-gal (75 ng/cm2) to normalize for transfection efficiency with CPRG (Roche) colorimetic assay.
GSK3b, TAZ and DN-Cul1 plasmids were co-transfected at 100, 50 and 100 ng/cm2 respectively. DNA content in all samples was
kept uniform by adding pBluescript plasmid up to 250 ng/cm2. For luciferase assays in siRNA-depleted cells, cells were first trans-
fected with the indicated siRNAs and, after 24 hr, washed from transfection media, transfected with plasmid DNA, and harvested
48 hr later. Where indicated, cells were exposed to Control- or Wnt3A-conditioned medium for 24 hr before harvesting. Each sample
was transfected in duplicate and each experiment was repeated at least three times independently.
Growth Assay105 SW480 cells were plated in a 35 mm dish (day 0) in triplicate for each time point. Cells were counted with Scepter 2.0 handheld
automated cell counter (Millipore) after detachment with trypsin. Each experiment was repeated at least three times independently.
Cell 151, 1443–1456, December 21, 2012 ª2012 Elsevier Inc. S1

Mammosphere and Matrigel Clonogenic AssaysMII mammosphere assays were carried out as in Cordenonsi et al. (2011). The clonogenic potential of parental or APC-reconstituted
SW480 cells was analyzed in 3D non-adherent culture conditions using Growth Factor Reduced Matrigel matrix (BD Biosciences).
SW480 cells were counted, resuspended in 50% Matrigel Matrix + 50% growth medium in ice and plated on Matrigel-coated
chamber slides (Nunc) (500 cells per well). After solidification, gels were supplemented with an appropriate volume of growth
medium, which was changed every 2 days during the experiment. 10 days after seeding, representative fields of the colonies
were photographed. The number of spheres within eachwell was counted after recovery fromMatrigel, using the Cell Recovery Solu-
tion from BD Biosciences. Statistical analyses were done using the Prism software (Graph Pad).
AntibodiesFor Western blot: anti-YAP/TAZ, anti-GSK3, anti-b-catenin and anti-g-catenin monoclonal antibodies and anti-CTGF and anti-Cyr61
polyclonal antibodies were fromSanta Cruz. anti-GAPDHmonoclonal antibody was fromMillipore. anti-b-TrCP, anti-LATS1 and anti-
phospho-S909 LATS1 polyclonal antibodies were from Cell Signaling, and anti-b-catenin polyclonal antibody was from Sigma.
Antiphospho-S306 TAZ polyclonal antiserum was obtained from one rabbit (out of two) immunized with a synthetic peptide
(Covance).
For immunofluorescence: anti-Scribble (Santa Cruz; 1:100, blocking with BSA), anti-TAZ (BDBiosciences, 1:100, blocking in BSA),
anti-E-Cadherin (BD Biosciences, 1:5000, blocking with GS), anti-b-catenin (Sigma; 1:5000, blocking with GS). Immunofluores-
cences were carried out as described in Cordenonsi et al. (2011).
GST Pull-DownFor GST pull-downs, beads with purified proteins were incubated with 35S-methionine labeled in vitro-translated b-catenin for 3 hr in
Binding Buffer (25mMHEPES (pH 7.9), 0.4MKCl, 0.4%NP40, 5mMEDTA, 1mMDTT, 10%glycerol with protease inhibitors). After 4
washes in binding buffer, copurified proteins were analyzed by SDS-page and autoradiography, using Cyclone Plus Phospho-imager
(Perkin Elmer).
Quantitative Real-Time PCRCells or tissues were harvested in Trizol (Invitrogen) for total RNA extraction, and contaminant DNA was removed by DNase treat-
ment. qRT-PCR analyses were carried out on retrotranscribed cDNAs with Rotor-Gene Q (Quiagen) thermal cycler and analyzed
with Rotor-Gene Analysis6.1 software. Experiments were performed at least three times, with duplicate replicates. Expression levels
are always given relative toGAPDH. PCR oligo sequences for human samples not listed in Dupont et al. (2011) and Cordenonsi et al.
(2011) are: APC, for: GCCCCTGACCAAAAAGGAAC; rev: TGGCAGCAACAGTCCCACTA; Axin1, for: AGCCGTGTCGGACATGGA;
rev: AAGTAGTACGCCACAACGATGCT; Axin2, for: TGTGAGGTCCACGGAAACTG; rev: CGTCAGCGCATCACTGGATA; Cyclin-
D1, for: TCAAATGTGTGCAGAAGGAGGT; rev: GACAGGAAGCGGTCCAGGTA.
PCR oligo sequences for mouse samples are: GAPDH, for: ATCCTGCACCACCAACTGCT; rev: GGGCCATCCACAGTCTTCTG;
TAZ, for: ATGAATCCGTCCTCGGTGC; rev: GAGTTGAAGAGGGCTTCGAG; Flag-mouse TAZ, for: ATGGACTACAAAGACGAT
GACG; rev: GAGTTGAAGAGGGCTTCGAG; CTGF, for: CTGCCTACCGACTGGAAGAC; rev: CATTGGTAACTCGGGTGGAG;
Cyr61, for: GCTCAGTCAGAAGGCAGACC; rev: GTTCTTGGGGACACAGAGGA; Cyclin-D1, for: GACCTTTGTGGCCCTCTGTG;
rev: AAAGTGCGTTGTGCGGTAGC.
Microarray ExperimentsFor microarray experiments, GeneChips Human Genome U133 Plus 2.0 (Affymetrix, Santa Clara, CA, USA) were used. Total RNA
from MII or SW480 cells (four replicas for each experimental condition) was extracted using TRIZOL (Invitrogen) and treated with
DNaseI (Ambion). RNA quality and purity were assessed on the Agilent Bioanalyzer 2100 (Agilent Technologies, Waldbronn,
Germany); RNA concentration was determined using the NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies Inc.).
In vitro transcription, hybridization and biotin labeling were performed according to Affymetrix One Cycle Target Labeling protocol
(Affymetrix). As control of effective gene modulation and of the whole procedure, we monitored the expression levels of TAZ, b-cat-
enin, APC - and of known TAZ or b-catenin target genes - by qRT-PCR prior to microarray hybridization and in the final microarray
data.
All data analyses were performed in R (version 2.14.2) using Bioconductor libraries (BioC 2.9) and R statistical packages. Probe
level signals were converted to expression values using robust multi-array average procedure RMA (Irizarry et al., 2003) of Bio-
conductor affy package. Differentially expressed genes were identified using Significance Analysis of Microarray algorithm coded
in the samr R package (Tusher et al., 2001). In SAM, we estimated the percentage of false positive predictions (i.e., False Discovery
Rate, FDR) with 100 permutations.
To identify genes upregulated by APC or b-catenin depletion in MII cells (Figures 1G and 6F, respectively), we set the lower limit for
fold change induction to 2. All selected probesets had an estimated percentage of false positive predictions equal to 0. To identify
TAZ-dependent genes, we compared the expression profiles of APC-depleted/TAZ-depleted cells with APC-depleted cells, and of
b-catenin-depleted/TAZ-depleted with b-catenin-depleted cells and selected those probesets with a negative fold change and an
FDR equal to 0.
S2 Cell 151, 1443–1456, December 21, 2012 ª2012 Elsevier Inc.

To identify genes whose expression is modified by APC mutation in SW480 cells, we selected those probesets whose estimated
percentage of false positive predictions was lower than 5%. To identify TAZ- or b-catenin-dependent genes, we refined this selection
comparing the expression profiles TAZ- or b-catenin-depleted cells with SW480 controls and selecting those probesets whose FDR
was lower than 5% and expression level consistently regulated by APC mutation.
MiceAh-Cremice and Apcfl/fl mice were a kind gift of Alan R. Clarke (Sansom et al., 2004). Ah-Cremice were crossed with mice carrying
the Apc floxed allele to yield Ah-Cre;Apcfl/fl mice. For induction of the Ah promoter, Ah-Cre;Apc+/+ control and Ah-Cre;Apcfl/fl mice
received three intraperitoneal injections of 10 mg/kg each of the cytochrome p450 inducer b-naphthoflavone (Sigma) dissolved in
corn oil (10 mg/ml). After 4 days, mice were sacrificed and livers were collected for further analysis. Mice were genotyped by
PCR on tail genomic DNA extracted by standard procedures (Morsut et al., 2010). Effective recombination of the APC locus was veri-
fied in the livers of mice injected with b-naphthoflavone.
SUPPLEMENTAL REFERENCES
Irizarry, R.A., Hobbs, B., Collin, F., Beazer-Barclay, Y.D., Antonellis, K.J., Scherf, U., and Speed, T.P. (2003). Exploration, normalization, and summaries of high
density oligonucleotide array probe level data. Biostatistics 4, 249–264.
Jin, J., Ang, X.L., Shirogane, T., and Wade Harper, J. (2005). Identification of substrates for F-box proteins. Methods Enzymol. 399, 287–309.
Maherali, N., Ahfeldt, T., Rigamonti, A., Utikal, J., Cowan, C., andHochedlinger, K. (2008). A high-efficiency system for the generation and study of human induced
pluripotent stem cells. Cell Stem Cell 3, 340–345.
Maretto, S., Cordenonsi, M., Dupont, S., Braghetta, P., Broccoli, V., Hassan, A.B., Volpin, D., Bressan, G.M., and Piccolo, S. (2003). Mapping Wnt/beta-catenin
signaling during mouse development and in colorectal tumors. Proc. Natl. Acad. Sci. USA 100, 3299–3304.
Martello, G., Rosato, A., Ferrari, F., Manfrin, A., Cordenonsi, M., Dupont, S., Enzo, E., Guzzardo, V., Rondina, M., Spruce, T., et al. (2010). A MicroRNA targeting
dicer for metastasis control. Cell 141, 1195–1207.
Morsut, L., Yan, K.P., Enzo, E., Aragona, M., Soligo, S.M., Wendling, O., Mark, M., Khetchoumian, K., Bressan, G., Chambon, P., et al. (2010). Negative control of
Smad activity by ectodermin/Tif1gamma patterns the mammalian embryo. Development 137, 2571–2578.
Sansom, O.J., Reed, K.R., Hayes, A.J., Ireland, H., Brinkmann, H., Newton, I.P., Batlle, E., Simon-Assmann, P., Clevers, H., Nathke, I.S., et al. (2004). Loss of Apc
in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 18, 1385–1390.
Tusher, V.G., Tibshirani, R., and Chu, G. (2001). Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 98,
5116–5121.
Cell 151, 1443–1456, December 21, 2012 ª2012 Elsevier Inc. S3

Figure S1. Wnt Signaling Promotes TAZ Activity, Related to Figure 1
(A) qRT-PCR for TAZ mRNA expression in HEK293 cells left untreated (control conditioned medium, Co) or treated with Wnt3A-conditioned medium for 24 hr.
Data were normalized to control cells; bars are mean + SD.
(B) Validation of Wnt activity in Wnt3A-conditioned media, and validation of XAV939 efficacy, using the b-catenin/TCF-luciferase reporter (BAT-Lux). HEK293
cells were transfected with BAT-Lux andwith Control siRNA (siCo, lanes 1, 2, 3) or b-catenin siRNA (sibcat, lane 4). After transfection, cells were treated as in (A) or
treated with Wnt3A + the Wnt inhibitor XAV939 (XAV, 1 mM, lane 3). Data are normalized to lane 1 and are presented as mean + SD.
(C) Luciferase assay on 8xGTIIC reporter in HEK293 cells transfected with Control siRNA (siCo, lanes 1, 3) or LATS siRNAs (siLATS, lanes 2, 4). After transfection,
cells were either left untreated (control conditioned medium, Co) or treated withWnt3A-conditioned medium for 24 hr. Top panel: Luciferase reporter assay of the
indicated samples. Data are normalized to lane 1 and are presented as mean + SD Bottom panel: representative Western blots for TAZ, and GAPDH (loading
control) in the same extracts used for the luciferase assay.
(D) Luciferase assay on 8xGTIIC reporter recording the transcriptional activity of mouse wild-type TAZ (lanes 1 and 2) or TAZ 4SA (lanes 3 and 4) transiently
transfected in HEK293 cells, in the absence (Co, lanes 1 and 3) or presence (Wnt3A, lanes 2 and 4) of Wnt stimulation. Cells were transfected with TAZ siRNA to
avoid interference from regulations of endogenous TAZ. Top panel: Luciferase reporter assay of the indicated samples. Data are normalized to lane 1 and are
presented as mean + SD Bottom panel: representative Western blots for TAZ, and GAPDH (loading control) in the same extracts used for the luciferase assay.
(E) Luciferase assay on BAT-Lux reporter recording the b-catenin transcriptional activity in HEK293 cells transiently transfectedwith Control siRNA (lanes 1 and 2)
or siRNAs targeting GSK3 (lanes 3 and 4), APC (lanes 5 and 6) or Axin (lanes 7 and 8), either alone (black bars) or in combination with siRNA against b-catenin (gray
bars). Data are normalized to lane 1 and are presented as mean + SD.
(F–K) Validation of siRNAs against GSK3, APC and Axin in MII human breast cancer cells. (F) Western blots for TAZ and GSK3 expression in MII cells transfected
with two independent sets of siRNAs targeting both GSK3a andGSK3b. GAPDH serves as loading control. (G) qRT-PCR for TAZmRNA expression in the samples
described in (F). (H)Western blots for TAZ and b-catenin expression inMII cells transfectedwith two independent siRNAs targeting APC. (I) qRT-PCR for APC and
TAZ mRNAs expression in the samples described in (H). (J) Western blots for TAZ expression in MII cells transfected with a set of siRNAs targeting Axin1 and
Axin2. (K) qRT-PCR for Axin1, Axin2 and TAZ mRNA expression in the samples described in (J). Bars in (G), (I), (K) are mean + SD.
(L) qRT-PCR analysis for the TAZ target gene CTGF in MII-shGFP or MII-shTAZ#2 transfected with the indicated siRNAs. Bars are mean + SD.
(M) Confocal images of b-catenin (top panels) in the same fields of MII cells shown in Figure 1F. Nuclei are stained with Hoechst (bottom panels).
(N) Luciferase assay on TAZ/TEAD-reporter 8xGTIIC in P19mouse teratocarcinoma cells transfected either with Control (siCo) or TAZ siRNAs (siTAZ), and treated
with Control (Co)- or Wnt3A-conditioned media. Data are presented as mean + SD.
(O and P) Average expression, evaluated by qRT-PCR, of b-catenin target gene Cyclin-D1 (O) and TAZ (P) mRNA in livers from Ah-Cre;Apc+/+ mice and Ah-
Cre;Apcfl/fl mice, intraperitoneally injected with b-naphthoflavone to induce Cre expression. Data are presented as mean + SD.
S4 Cell 151, 1443–1456, December 21, 2012 ª2012 Elsevier Inc.
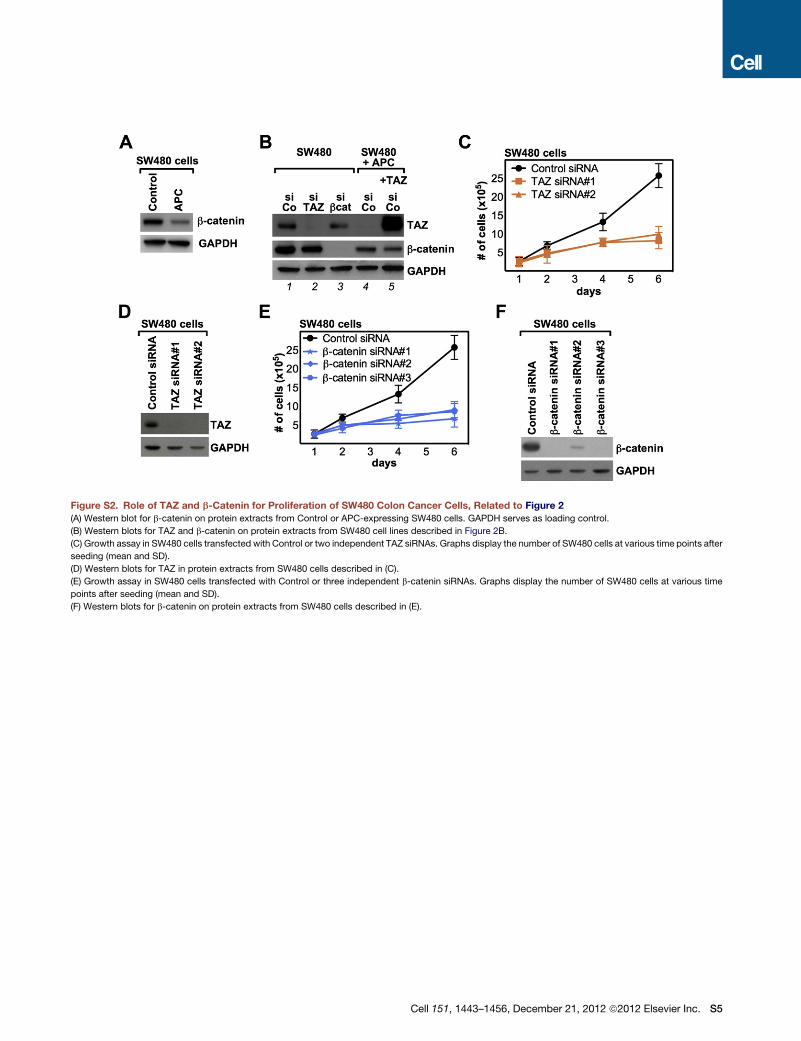
Figure S2. Role of TAZ and b-Catenin for Proliferation of SW480 Colon Cancer Cells, Related to Figure 2
(A) Western blot for b-catenin on protein extracts from Control or APC-expressing SW480 cells. GAPDH serves as loading control.
(B) Western blots for TAZ and b-catenin on protein extracts from SW480 cell lines described in Figure 2B.
(C) Growth assay in SW480 cells transfected with Control or two independent TAZ siRNAs. Graphs display the number of SW480 cells at various time points after
seeding (mean and SD).
(D) Western blots for TAZ in protein extracts from SW480 cells described in (C).
(E) Growth assay in SW480 cells transfected with Control or three independent b-catenin siRNAs. Graphs display the number of SW480 cells at various time
points after seeding (mean and SD).
(F) Western blots for b-catenin on protein extracts from SW480 cells described in (E).
Cell 151, 1443–1456, December 21, 2012 ª2012 Elsevier Inc. S5

Figure S3. TAZ Acts Downstream of Wnt to Determine Mesenchymal Stem Cell Fate, Related to Figure 3
(A) qRT-PCR for TAZmRNA levels on ST-2 cells transfectedwith Control or TAZ siRNAs. Cells were exposed for 72 hr to recombinantWnt3A (100 ng/ml). Bars are
mean + SD.
(B) Quantification of alkaline phosphatase (ALP)-positive areas (mean + SD) in hMSC treated as described in Figures 3B–3F.
(C–F) Representative pictures of hMSC stained with Oil Red to visualize lipid droplets. hMSC were transfected with Control, GSK3, APC or Axin siRNAs and
induced to differentiate into adipocytes. Nuclei are stained with Hoechst.
(G) Quantification of Oil Red-positive areas (mean + SD) in hMSC treated as described in (C–F).
S6 Cell 151, 1443–1456, December 21, 2012 ª2012 Elsevier Inc.

Figure S4. Effects of GSK3 and b-TrCP Depletion on TAZ, Related to Figure 4
(A) Top panel: Luciferase assay on 8xGTIIC reporter recording TAZ transcriptional activity in HEK293 cells transfected with Control siRNA (lanes 1, 2, 7 and 8) or
two independent siRNAs targeting b-TrCP (lanes 3 and 4 and lanes 5 and 6, respectively), with or without TAZ siRNA (siTAZ). Where indicated, a construct
encoding a dominant-negative version of human Cullin1 was transfected together with the reporter (lanes 7 and 8). Data are normalized to lane 1 and bars are
mean + SD Bottom panel: Western blot for TAZ expression in the same extracts used for the luciferase assay. GAPDH serves as loading control.
(B) Top panel: Luciferase assay on 8xGTIIC reporter in HEK293 cells transiently transfected with Control siRNA (lanes 1) or with siRNAs targeting GSK3 (lanes
2–4). Where indicated, constructs encoding siRNA-insensitive GSK3b wt (lane 3) or GSK3b KD (lane 4) were transfected together with the reporter. Data are
normalized to lane 1 and bars are mean + SD Bottom panel: Western blots for TAZ and GSK3 expression in the same extracts used for the luciferase assay.
(C) Comparison of the GSK3-dependent phosphodegrons of TAZ, b-catenin and other proteins known to be regulated by GSK3 and b-TrCP. Amino acids
essential for binding to b-TrCP are indicated in red.
(D and E) qRT-PCRs for endogenous human TAZ (D) or exogenous mouse Flag-TAZ (E) mRNA levels in the samples described in Figure 4D, assuring equal
expression of the two TAZ variants as well as effective depletion of endogenous TAZ. Data are normalized to lane 1 in (D) and to lane 3 in (E); bars are mean + SD.
Cell 151, 1443–1456, December 21, 2012 ª2012 Elsevier Inc. S7

Figure S5. Effect of b-Catenin Depletion on TAZ Localization, Related to Figure 5
(A) TAZ associates with b-catenin at endogenous protein levels. MII cells were transfected with Control or b-catenin siRNAs, cultured at high cell density, and then
harvested for protein extraction. Endogenous b-catenin was immunoprecipitated from the cell lysates, and co-precipitating TAZ was visualized by Western blot.
(B) Representative immunofluorescence pictures for TAZ (top panels) in MII cells transfected with Control siRNA or independent b-catenin siRNAs. Nuclei are
stained with Hoechst (bottom panels). Compendium of Figure 5E.
(C) Representative immunofluorescence pictures for the cell polarity determinant Scribble (see Cordenonsi et al., 2011) or for E-cadherin in MII cells transfected
with Control siRNA or independent b-catenin siRNAs. Nuclei are stained with Hoechst.
(D)Western blots for total and Serine 909-phosphorylated LATS1 inMII cells transfectedwith Control or independent b-catenin siRNAs. GAPDH serves as loading
control. Samples are the same as in Figure 5C.
(E) Autoradiography of in vitro translated 35S b-catenin pulled-down by the indicated GST-TAZ deletion constructs immobilized on a glutathione-resin. GST
protein was used as a negative control. The lane labeled as ‘‘Input’’ represents 1/100 of 35S b-catenin used for the pull down experiments. On the left, a schematic
representation of the TAZ constructs here used. TB: TEAD binding domain; TA: Transcriptional Activation domain; PB: PDZ-binding motif.
(F) Western blot for TAZ inMII cells depleted of endogenous TAZ and transducedwithwild-type orDWWTAZ. TAZDWW ismore stable than thewild-type protein,
despite the fact that both the N-terminal (encompassing S58 and S62) and the C-terminal (encompassing S309) phosphodegrons are not affected by the deletion
of the WW domain, as shown in the scheme above the blot (abbreviations are as in [E]).
(G) TAZ DWW is phosphorylated at serine 306. MII cells were depleted of endogenous TAZ and transduced with mouse (siRNA insensitive) wild-type, S306A or
DWW TAZ. TAZ was immunoprecipitated with an anti-TAZ antibody and phosphorylation on S306 was detected on the immunoprecipitated protein using
a specific antiserum raised in rabbit. TAZ S306A mutant was used as negative control for the antiserum.
(H) TAZ DWW is sensitive to LATS inhibition. MII cells depleted of endogenous TAZ were transduced with wild-type, DWW or 4SA TAZ, and transfected with
Control (siCo) or LATS siRNAs (siLATS). TAZ levels were evaluated by Western blot. Note that mutation of the four LATS phosphorylation sites renders TAZ
insensitive to LATS depletion, whereas deletion of the WW domain does not.
S8 Cell 151, 1443–1456, December 21, 2012 ª2012 Elsevier Inc.

Figure S6. TAZ Is Inhibited by b- and g-Catenin, Related to Figure 6
(A) Confocal immunofluorescence images of b-catenin (upper panels) and E-cadherin (middle panels) in MII cell derivatives, engineered to express either
a doxycycline-inducible siRNA-insensitive b-catenin wild-type (wt; 1-3) or phospho mutant (S/A; 4-6). Cells were depleted of endogenous b-catenin and induced
with doxycycline as described in Figure 6B. Upon reconstitution, both wild-type and S/A b-catenin localize to the E-cadherin junctional complexes. Nuclei are
stained with Hoechst (lower panels).
(B) qRT-PCR for TAZ mRNA expression, confirming efficient depletion of TAZ in the same samples shown in Figure 6D. Note that b-catenin-depletion does not
change TAZ levels. Data are presented as mean +SD.
(C) mRNA expression of the b-catenin target geneCyclin-D1 in the same samples shown in Figure 6D.Cyclin-D1 is downregulated by depletion of both b-catenin
transcripts (siRNA#3), but is not affected by depletion of the sole wild-type b-catenin with siRNA#4. Data are presented as mean +SD.
(D) Quantification of mammospheres formed by MII cells transfected with Control siRNA or two independent siRNAs targeting b-catenin, alone or in combination
with TAZ siRNAs (mean + SEM).
(E) qRT-PCRs for Axin2 and Cyclin-D1 mRNA expression in MII cells transfected with Control siRNA (siCo) or b-catenin siRNA (sibcat). Data are presented as
mean + SD.
(F) The WW domain of TAZ is required for association to g-catenin. MII cells stably expressing Flag-tagged wild-type or DWW TAZ were cultured at high cell
density and then harvested for protein extraction. TAZ was immunoprecipitated from the cell lysates using anti-Flag antibody, and co-precipitating proteins were
detected by Western blot.
(G) Western blots for total and Serine 909-phosphorylated LATS1 in HEK293 cells transfected with Control or g-catenin siRNA. GAPDH serves as loading control.
Samples are the same as in Figure 6H.
(H) Luciferase assay on 8xGTIIC reporter in HEK293 cells transfected with Control siRNA or siRNAs targeting b-catenin (bcat siRNA#2), g-catenin (gcat siRNA) or
both. Where indicated, cells were also transfected with TAZ siRNAs (gray bars). Data are normalized to Control siRNA-transfected cells and bars are mean +SD.
Cell 151, 1443–1456, December 21, 2012 ª2012 Elsevier Inc. S9
Copyright © 2022 FDOKUMEN




















