
Kip's Role as Mediator in Michael Ondaatje's The English ...
Upload
khangminh22Category
view
1download
0
CLARIFICATION OF THE ROLE OF HOST DNA AS A MEDIATOR OF
VACCINE ADJUVANT ACTIVITY
by
LAURA EVELYN NOGES
B.S., University of Richmond, 2009
A thesis submitted to the
Faculty of the Graduate School of the
University of Colorado in partial fulfillment
of the requirements for the degree of
Doctor of Philosophy
Immunology Program
2015

ii
This thesis for the Doctor of Philosophy degree by
Laura Evelyn Noges
has been approved for the
Immunology Program
by
Roberta Pelanda, Chair
Holger Eltzschig
Rachel Friedman
Peter Henson
Claudia Jakubzick
Philippa Marrack, Advisor
Date 08/14/2015

iii
Noges, Laura Evelyn (Ph.D., Immunology)
Clarification of the Role of Host DNA as a Mediator of Vaccine Adjuvant Activity
Thesis directed by Professor Philippa Marrack.
ABSTRACT
Aluminum salt (alum) adjuvants are widely used in human vaccination
because they are safe and effective at generating antibody-mediated protective
immunity. Though it is well known that alum induces T helper 2 responses and
antigen-specific IgG1 and IgE antibody production in mice, the mechanism by
which alum elicits these effects is poorly understood. Recently, others and our
group reported that alum immunization results in the release of host DNA, which
mediates adjuvant activity by acting as an endogenous immunostimulatory
signal. These conclusions were based, in part, on experiments that combined
alum vaccines with DNase I enzymes to study loss-of-function scenarios.
Curiously, DNase only inhibited CD4 T cell priming and antibody responses to
alum; CD8 T cell activation was not affected. As such, further clarification was
needed regarding the role of host DNA in alum adjuvant activity.
Here I employed a mouse model system and injectable DNase treatments
to study the role of host DNA in mediating immune responses to intramuscular
alum vaccines. To ensure that my studies pertained solely to the effects of host
DNA within alum biology, I performed comprehensive analysis of relevant DNase
reagents that had been used for alum studies in previous publications.
Unfortunately, my data indicated that commercial DNase reagents are prone to

iv
contain impurities and contaminants including digestive proteases. When alum
studies were repeated using pure DNase, I found that alum-associated DNA is
actually dispensable in the generation of effective cellular and humoral immune
responses by alum vaccines. Furthermore, when proteases were co-injected
with alum vaccines, the effects on immune responses mimicked those observed
following co-injection with impure DNase reagents.
In conclusion, we suggest that host DNA is not a major contributor to the
adjuvant effect of alum when vaccines are administered to the muscle and that
contaminated reagents may have misled interpretations of past experiments
involving DNase reagents. This correction of the literature may help improve
future studies to elucidate the mechanism of aluminum salts as vaccine
adjuvants, an endeavor that remains paramount to understanding why alum
vaccines have successfully protected hundreds of millions of people from
disease and how they may be improved for future vaccination strategies.
The form and content of this abstract are approved. I recommend its publication.
Approved: Philippa Marrack

v
I dedicate this dissertation to the incredible mentors I’ve had throughout my
education. I hope this work will contribute to the integrity, persistence, and
betterment of science for our world.

vi
ACKNOWLEDGEMENTS
I am deeply indebted to many people who provided advice, support, and
encouragement throughout my graduate career. Above all, I want to thank Jan
and Paul Noges, Joel Crandall, Bailey Gross, Nicole Desch, Bryan Wee, Raul
Torres, and Janice White for always being there when I needed them.
I would like to thank past and present members of my thesis committee,
Roberta Pelanda, Holger Eltzschig, Rachel Friedman, Laurent Gapin, Dirk
Homann, Peter Henson, and Claudia Jakubzick, for their guidance, input, and
encouragement throughout my thesis project.
I thank all of the current and past members of the Kappler/Marrack lab for
scientific discussion, experimental advice, and technical help. Specifically, I want
to thank Janice White for her generous benchside help and practical advice on all
problems science and otherwise, Fran Crawford for her careful explanations of
protein chemistry and her camaraderie in fighting ‘I got mine,’ and Megan
MacLeod for her patience in explaining science and techniques, willingness to
contribute ideas to my projects, and lightning fast email responses from the other
side of the world. My officemates Matt Phillips and Harsha Krovi provided me
with moral support, scientific insight, and countless jokes – thank you both.
Finally, I will thank my doctorate advisor, Dr. Pippa Marrack. Pippa
nurtured me from an infantile graduate student to an independent, conscientious,
and confident scientist. Through the highs and lows of bench science, graduate
school, and life, Pippa has dedicated her time and energy to guide my
development as a young woman and scientist.

vii
TABLE OF CONTENTS
CHAPTER
I. INTRODUCTION 1
Natural Protective Immune Responses 2
Primary immune responses (overview) 3
Initiation 3
Contraction and establishment of immune memory 4
Optimizing immune responses 4
Pathogen detection by antigen presenting cells 5
Immunization 6
History of immunizations 7
Immunity 7
Variolation 7
Vaccination 8
Immunization 8
Adjuvants 8
Types of immunizations 9
Live attenuated 9
Inactivated 11
Subunit 11
Adjuvants used in subunit vaccines 12
Aluminum Salts as Vaccine Adjuvants 13
Discovery and use in vaccines 13

viii
Mechanism of action 13
Generation of protective antibodies 15
Innate immune cell activation 19
Stimulating T cell responses 23
Alum’s cytotoxicity and host DNA as an induced self adjuvant 25
Immunogenicity of Heat Aggregated Proteins 27
II. MATERIALS AND METHODS 29
Mice 29
Reagents, Antigens, Tetramers, and Antibodies 29
Reagents 29
Model antigens 30
Tetramers 31
Antibodies 31
DNases 32
Preparation of Soluble or Aggregated Protein Antigens (Chapter V) 34
Soluble protein 34
Aggregated protein 34
Assessing Protein Conformation and Aggregation (Chapter V) 34
Size exclusion chromatography 34
Micro-flow imaging 35
Infrared spectroscopy 35
Measuring surface hydrophobicity 35

ix
Immunizations 36
Assessment of Antigen-Specific T Cell Priming 36
Antigen Presenting Cell Analyses 37
RNA Sequencing: Cell Preparation and Analysis 39
Antibody Detection by ELISA 42
Assessing DNase Reagent Purity 42
SDS-PAGE 42
Protease activity assay 43
Mass spectrometry 43
DNase Activity Assay 44
Antigen Destruction Assay 45
Generation of 3NP311-2 CD4 T Cell Hybridomas 45
Statistical Analyses 46
III. HOST DNA IS DISPENSIBLE IN ALUM RESPONSES 47
Introduction 47
Results 49
Creating model antigens: OVA-NP and mutant nucleoprotein 49
Roche DNase I grade II does not affect adaptive immune responses to epitopes within intact protein antigens 50
Primary T cell responses 50
Primary antibody responses and secondary CD4 T cell responses 51
Commercial DNase I reagents are contaminated with active proteases 55

x
Proteases are predominantly responsible for the effects of contaminated DNases on CD4 T cell responses to alum 60
DNase enzymatic activity does not impair T cell responses to alum 64
STING may be dispensable in alum vaccine responses 67
Discussion 69
IV. EFFECTS OF ALUM ON ANTIGEN PRESENTING CELLS 71
Introduction 71
Results 74
Technical considerations when using fluorescent antigens 74
Choice of fluorophore for antigen tracking affects detection of antigen-loaded cell subsets 74
Artifacts of flow cytometry: possible B cell + iMono doublets 76
Alum adjuvant effect on antigen-loaded APCs 76
RNA sequencing identifies alum-induced inflammatory pathways 79
Discussion 85
V. SINGLE DOSES OF HEAT AGGREGATED PROTEINS ARE NOT IMMUNOGENIC 88
Introduction 88
Results 89
Characterizing protein antigens: soluble versus heat aggregated OVA 89
Soluble ovalbumin must be filtered and can be stored for at least two weeks at 4°C 89

xi
Heat aggregation of ovalbumin at concentrations >1 mg/ml alters its surface hydrophobicity, conformation, and structure 90
Heat aggregated proteins do not prime T cells 93
Discussion 96
VI. DISCUSSION AND FUTURE DIRECTIONS 99
Alum’s Mechanism: A Working Model + Unanswered Questions 100
Step 1: Damage and inflammation at the site of injection 101
Step 2: Activation of antigen presenting cells 104
Step 3: Priming adaptive immune cells 106
Step 4: Resolution and boosting 107
Beyond the Model: Additional Unresolved Questions 108
T cell responses to alum 108
B cells as alum/antigen presenters 112
Adjuvant Action of Host DNA 113
Reproducibility in Science and Implications of DNase Contamination 116
The Future of Vaccines 117
Future Directions 119
Concluding Remarks 121
REFERENCES 122

xii
LIST OF TABLES
TABLE
1.1: Common vaccines in the United States 10
1.2: Vaccines that contain aluminum salt adjuvants 14
1.3: Effector functions and alum induction of human and mouse antibody isotypes 18
2.1: Tetramers used for flow cytometry 31
2.2: Antibodies used for flow cytometry 31
2.3: DNase reagents used in Chapter III 33
2.4: Sorted cell populations submitted for RNA sequencing 40
4.1: APC subset phenotypes 77
4.2: Functional gene clusters that have altered expression in response to alum 85
6.1: Signaling pathways that involve Tbk1 and Irf3 114

xiii
LIST OF FIGURES
FIGURE
1.1: Primary immune response schematic 5
1.2: Human vaccine adjuvants 12
1.3: Efficacy of vaccines in the United States 15
1.4: Conventional dendritic cell and monocyte differentiation in mice 22
1.5: CD11b+ cDCs and iMonos are the predominant APC subsets that respond to intramuscular alum injections 23
2.1: Antigen-specific CD4 and CD8 T cell gating strategy 38
2.2: Gating scheme for antigen-loaded APC sorting prior to RNA sequencing 40
3.1: MutNP and NP stimulate equivalent alum immune responses 50
3.2: Added BSA has no effect on the magnitude of NP311-325-specific CD4 T cell responses to alum immunization 52
3.3: DNase treatment does not affect CD4 T cell responses to intact proteins 53
3.4: DNase treatment does not impair secondary CD4 T cell responses 52
3.5: Commercial DNases are contaminated with active proteases 57
3.6: Roche recombinant DNase has no protease activity 59
3.7: Contaminating proteases rapidly destroy NP311-325 peptide on OVA-NP but not within NP 59
3.8: Worthington DNase, wtDNase, and mutDNase are all pure and appropriately active or inactive 61
3.9: Proteases impair CD4 T cell responses to alum + OVA-NP 62
3.10: Trypsin and chymotrypsin cleavage sites in NP311-325 and SIINFEKL peptides 64
3.11 MutDNase is inactive up to 1 mg/ml concentration 65

xiv
3.12 DNase activity does not impair T cell responses to alum + OVA-NP 66
3.13: STING is dispensable in adaptive immune responses to alum 68
4.1: Choice of fluorescent marker affects the detection of antigen-bearing cells by flow cytometry 75
4.2: Alum increases numbers of antigen-loaded APCs, but does not affect loaded cell type of degree of antigen uptake 78
4.3: Inflammatory monocytes capture the most alum-adsorbed antigen per cell 81
4.4: Antigen arrives to dLNs rapidly via iMonos, but later time points reveal more antigen-bearing migDCs (DCs) 82
4.5: Alum induces vast changes in gene expression profiles within exposed APC subsets 84
5.1: Resuspended lyophilized OVA is predominantly monomeric after filtration and ultracentrifugation 89
5.2: Filtering is required to rid resuspended OVA of particulates 91
5.3: Heating OVA alters its structural conformation 92
5.4: Heat treatment alters the secondary structure of OVA 93
5.5: Single doses of heat aggregated proteins are not immunogenic when administered i.p. or s.c. 95
6.1: Working model of alum’s mechanism 102

xv
LIST OF ABBREVIATIONS
A488 Alexa Fluor 488 A647 Alexa Fluor 647 ADCC Antibody-dependent cellular cytotoxicity Alum Aluminum hydroxide ANS 1-Anilinonaphthalene-8-sulfonic acid APC Professional antigen presenting cell BCR B cell antigen receptor BSA Bovine serum albumin CD Circular dichroism CDC Complement-dependent cytotoxicity cDC Conventional dendritic cell CLR C-type lectin receptor CTL Cytotoxic T lymphocyte CTM Complete Tumor Medium DAMP Damage/danger-associated molecular pattern DAVID Database for annotation, visualization, and integrated discovery DC Dendritic cell dLN Draining lymph node ELISA Enzyme-linked immunosorbent assay FPLC Fast protein liquid chromatography FTIR Fourier transfer infrared spectroscopy g Gravitational-force (acceleration) HMGB1 High mobility group box 1 proteins HPLC High performance liquid chromatography iDC Inflammatory monocyte-derived dendritic cell ICAM1 Intercellular adhesion molecule 1 Ig Immunoglobulin IL Interleukin iMono Inflammatory monocyte (monocyte-derived dendritic cell) IR Infrared spectroscopy Irf3 Interferon regulatory transcription factor 3 LFA1 Lymphocyte function-associated antigen 1 LN Lymph node LPS Lipopolysaccharide MFI Mean fluorescence intensity MHC Major histocompatibility complex migDC Migratory dendritic cell MSU Monosodium urate NLR NOD-like receptor NOD Nucleotide-binding oligomerization domain NP Influenza A virus nucleoprotein NP311-325 NP peptide (QVYSLIRPNENPAHK) NP366-374 NP peptide (ASNENMETM) OVA Chicken ovalbumin

xvi
OVA-NP Chicken ovalbumin with covalently conjugated NP311-325 peptide PAMP Pathogen-associated molecular pattern pMHCI Peptide-loaded major histocompatibility complex class I pMHCII Peptide-loaded major histocompatibility complex class II PRR Pattern recognition receptor resDC Lymphoid resident dendritic cell RIG-I Retinoic acid-inducible gene I RLR RIG-I-like receptor STING Stimulator of interferon genes Tbk1 Tumor necrosis activating factor-associated NFκB activator
(TANK)-binding kinase 1 TCR T cell antigen receptor TH Helper T lymphocyte TH1 Type 1 helper T lymphocyte TH2 Type 2 helper T lymphocyte TLR Toll-like receptor

1
CHAPTER I
INTRODUCTION
“Nothing in biology makes sense except in the light of evolution.”
–Theodosius Dobzhansky
This statement is the title of a 1973 essay by Ukrainian-born Theodosius
Dobzhansky1. The piece brazenly criticized anti-evolution creationism and
implored readers to come to their senses and accept the intellectually satisfying
and even inspiring phenomenon of evolution. With ample biological examples
and logic, Dobzhansky argued that evolution plays a central and unifying role in
the science of biology. This statement resonates deeply with me so I’ve begun
my dissertation with an explanation of how I mentally approach biology and my
research projects in light of evolution.
Dobzhansky’s outlook applies directly to the field of immunology: evolution
drove the development of host immune systems over a multimillion-year-old arms
race between pathogens and hosts. Though some of the most primitive
components of immune systems date back to the origin of multicellularity, the
adaptive arm of the immune system appeared within vertebrates some 450
million years ago2. The purpose of an organism’s immune system is to react to
infection by mounting biological responses that both resolve the primary infection
and establish protective immunological memory that prevents future reinfection
with the same pathogen. This body system evolved in response to endless

2
infection and parasitism of multicellular hosts by microbes, viruses, and
parasites. Therefore, the immune system is functionally diverse and able to
make refined responses to limitless types of infectious organisms. As the battle
between pathogens and hosts rages on, human beings have developed an
ingenious technological edge: vaccination.
This dissertation includes several research projects having to do with how
vaccines work. The first two studies examine how aluminum salt adjuvants
stimulate protective immune responses. Chapter III focuses on alum induction
of adaptive immune responses while Chapter IV focuses on alum activation of
innate immune cells. Last, the immunogenicity of a separate vaccine platform is
explored in Chapter V: heat aggregated proteins.
Natural Protective Immune Responses
Before discussing vaccinations in great detail, I must first introduce the
natural mechanisms of protective immune responses. The mammalian immune
system is comprised of both innate and adaptive immune mechanisms. As can
be expected, there are also immune mechanisms that seem to have qualities of
both innate and adaptive immunity, such as memory natural killer cells3,4. As a
rule, innate immune components detect and respond to pathogens in a general
manner and do not confer antigen-specific protective immunity to the host.
Adaptive immunity, however, involves a system of highly specialized cells that
respond in an antigen-specific manner. Moreover, after an initial response to a

3
given pathogen, immunological memory is created and subsequent encounters
with the same pathogen are met with greatly enhanced immune responses that
generally prevent symptomatic disease. This memory is the basis of acquired
protective immunity and it is the central goal of vaccination.
Primary immune responses (overview)
Initiation. The initiation of an immune response to an infection requires
collaboration between innate immune cells and T lymphocytes of the adaptive
immune system. The activation of T cells depends on their interactions with
professional antigen presenting cells (APCs), which are specialized innate
immune cells that are directly activated by the pathogens that they engulf or
otherwise encounter5. Upon traveling to lymphoid organs such as lymph nodes,
APCs process and present pathogens to T cells via peptide-loaded major
histocompatibility complex proteins (MHC) on their cell surfaces5. Activated T
cells proliferate, mobilize, and orchestrate specific immune responses that are
tailored to optimally protect the body from various types of infections.
Specifically, T cells activate other immune cells and also become cytotoxic
themselves in order to kill off infected cells within the body. Among the immune
cells activated by T cells, antibody-producing B lymphocytes are one of the most
important subsets. B cells are capable of producing a variety of isotypes of
antibody molecules, each of which are designed to protect against different
infections5. For example, IgA antibodies protect against mucosal pathogens.
The bottom line is that innate and adaptive immune cells are capable of

4
chemically detecting the qualities of an offending pathogen and work together to
mount the most effective immune counterattack to combat the infection.
Contraction and establishment of immune memory. At the end of an
immune response, the majority of activated T and B cells undergo apoptosis.
However, a small percentage remain alive because they have differentiated into
memory cells that stay primed and ready to respond rapidly if the host is ever re-
exposed to the same pathogen6,7. Mimicking natural responses, vaccines must
be able to prime antigen-specific T and B cells so that some of them remain in
the body as long lived memory cells.
Optimizing immune responses. Moreover, the best vaccines aim to initiate
the proper type of immune response that will best protect against a particular
pathogen. Naturally, the body has ways to distinguish between different types of
pathogens and respond accordingly. The different ways in which the immune
system can respond to antigen often revolve around the characteristics of the
pathogen (intra/extra-cellular, viral/bacterial/parasitic, etc) that are detected by
APCs and communicated to either or both of the major classes of T cells: helper
CD4 T cells (TH) and cytotoxic CD8 T cells (CTL) (Fig. 1.1). The qualities of
subsequent immune responses depend on this pathogen recognition by innate
immune cells. For example, detection of a viral infection results in CTL killing of
infected cells and antibody neutralization of extracellular virions (Fig. 1.1).
Alternatively, some bacterial or parasitic infections require CD4 T cells to secrete
cytokines that activate particular groups of innate immune cells (Fig. 1.1).

5
Figure 1.1. Primary immune response schematic, reprinted from McKee et al., BMC Biology, 2010 (Fig. 1)8.
The next section will focus on the ways in which APCs detect different
pathogenic features upon infection, since vaccines endeavor to imitate this
process.
Pathogen detection by antigen presenting cells
APCs must distinguish between different kinds of pathogens in order to
mount the proper immune response for each type of threat. Innate immune cells
can achieve this by recognizing generally conserved pathogen-associated
molecular patterns (PAMPs) on microorganisms5. PAMPs include molecules
derived from bacterial cell walls, viral RNA genetic codes, CpG-rich DNA, and
other common features of microbes. In turn, innate immune cells express pattern
recognition receptors (PRRs) that specifically detect types of microbial patterns5.
Toll-like receptors (TLRs), nucleotide-binding domain and leucine-rich-repeat-
containing family (NLRs), retinoic acid-inducible gene I (RIG-I)-like receptors

6
(RLRs), and C-type lectin receptors (CLRs) are among the best known PRRs9,10.
TLR9, for example, is expressed within cell endosomes and detects CpG-rich
DNA.
Host cell recognition of PAMPs at the site of infection promotes the
recruitment of innate immune cells, including APCs. Signals from PAMP-
activated cells as well as direct PAMP recognition activates APCs, leading to
increased antigen uptake, expression of activation-associated cell surface
molecules, and secretion of soluble chemical mediators that promote T cell
activation5. Together, these effects influence the magnitude and quality of T and
B cell responses, which subsequently affect the generation of memory
lymphocytes that are produced. PRR activation of innate immune cells is a
critical first step toward effective immune responses because it serves to warn
surrounding cells of an infection (reducing collateral damage), activates adaptive
immune cells, and guides the type of immune response that develops.
Immunization
Immunization is a process by which an individual is protected against a
disease through vaccination. Vaccination (interchangeably called immunization)
refers to the controlled introduction of all or a piece of pathogen for the purpose
of generating enhanced immunity to subsequent encounters with the pathogen.
The discovery and use of immunization to protect against particularly deadly
infectious diseases is regarded as the single most influential biomedical

7
advancement to date11. The goal of immunization is to generate long-lived
protective immunity against specific pathogens without causing sickness in order
to protect an individual against future infection. This protective immunity is
achieved by stimulating adaptive immune responses that are directed against
pathogenic antigens.
History of immunizations
Immunity: The concept of immunity has existed since ancient times. For
example, the first European mention of immunity was recorded by Thucydides in
Athens in 430 BC. He describes that only those who had recovered from the
plague could nurse the sick because they could not contract the disease a
second time. Allegedly, in gratitude for their tending to the sick, nurses were
exempt from paying municipal taxes: they were immunis.
Variolation: As the notion of acquired immunity was observed and
accepted, a variety of societies around the world began manipulating this
phenomenon. The most widely known early attempts to induce active immunity
were aimed at containing the deadly disease smallpox. Both in China by 1000
AD and Europe within the 15th century, the practice of variolation, also called
inoculation, became common: scratching the skin or inhaling powdered material
derived from smallpox lesions to induce a mild case of smallpox disease12,13. If
variolated subjects recovered from their illness, they would be protected from
future, more deadly cases of the smallpox infection.

8
Vaccination: A much safer form of smallpox immunization was reported in
1798 by Edward Jenner12. Jenner deliberately infected a young boy with pus
from the lesions of a cowpox-infected milkmaid. Once the boy recovered from
the cowpox infection, Jenner then challenged his acquired immunity to smallpox
by infecting him with smallpox. The boy suffered no symptoms or disease and
was thus declared immune. Jenner termed this cowpox inoculation procedure
‘vaccination’ as ‘vacca’ is the Latin term for cow. Now, the terms vaccination and
immunization are used interchangeably.
Immunization: As the science of immunology developed and infectious
diseases were better understood in the years following Jenner’s discovery,
scientists began formulating additional vaccines. By 1885, a rabies vaccine had
been devised by two French scientists, Louis Pasteur and Emile Roux, that
employed attenuated rabies virus as an immunogen14. Within the twentieth
century, a variety of successful vaccines were developed as scientists found
ways to produce safe immunogens for vaccination15,16. Many disease-causing
microorganisms were inactivated or attenuated toward this end. Additionally,
toxins from bacteria such as diphtheria and tetanus were inactivated into ‘toxoids’
that were effective immunogens.
Adjuvants: Underlying each successful vaccine was a common theme: a
safe and effective adjuvant must accompany target antigens in order to mount
robust immune responses. An adjuvant is any biochemical substance, organic or
inorganic, that augments immune responses in a way that promotes
immunological memory. Early vaccines employed natural adjuvants that were

9
provided by pathogens themselves: bacterial or viral components. These
methods are still used today. The acknowledgement of adjuvants as necessary
components within vaccines was one of the driving forces behind the
development of subunit vaccines, vaccines that deliver only part of an agent
rather than the whole entity. In the early 1920s, Gaston Ramon discovered that
addition of extra substances to antigenic vaccines could enhance immune
responses17. Soon thereafter, Alexander Glenny utilized an inorganic adjuvant to
promote immune responses that would open up a new realm of possibilities for
vaccine development: aluminum salts18.
Types of immunizations
Currently, three main categories of vaccines are used in humans: live
attenuated vaccines composed of a virus or bacterium that is similar but less
pathogenic than the wild form; inactivated vaccines that are heat-killed or
otherwise chemically inactivated forms of the wild pathogen; and subunit
vaccines that are a combination of components of a pathogen (such as surface
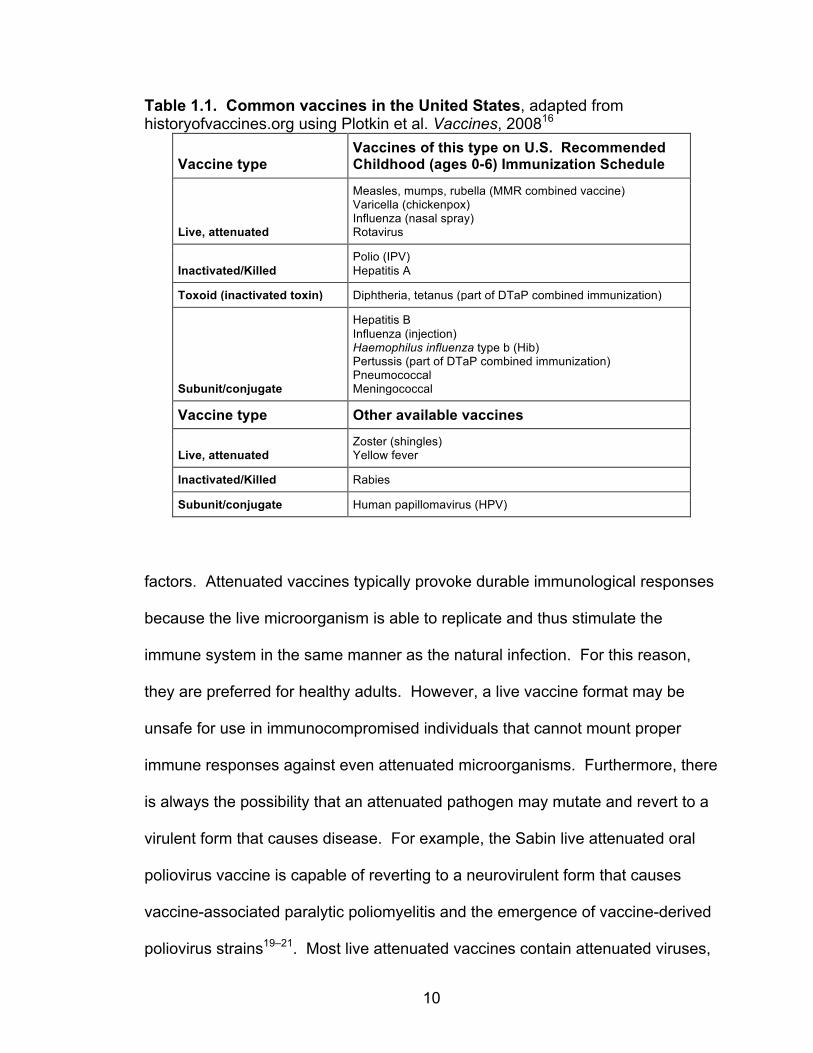
proteins) and biochemical immunostimulants called adjuvants. Table 1.1 lists by
type common vaccines given in the United States.
Live attenuated: Vaccines may contain live but weakened, or attenuated,
microbes that are able to replicate within the body but are no longer virulent.
Immune responses to these agents are broad enough to cross-react with the
more dangerous strains that cause disease. Attenuation of viruses or bacteria is
often achieved by culturing under conditions that promote the loss of virulence
10
Table 1.1. Common vaccines in the United States, adapted from historyofvaccines.org using Plotkin et al. Vaccines, 200816
Vaccine type Vaccines of this type on U.S. Recommended Childhood (ages 0-6) Immunization Schedule
Live, attenuated
Measles, mumps, rubella (MMR combined vaccine) Varicella (chickenpox) Influenza (nasal spray) Rotavirus
Inactivated/Killed Polio (IPV) Hepatitis A
Toxoid (inactivated toxin) Diphtheria, tetanus (part of DTaP combined immunization)
Subunit/conjugate
Hepatitis B Influenza (injection) Haemophilus influenza type b (Hib) Pertussis (part of DTaP combined immunization) Pneumococcal Meningococcal
Vaccine type Other available vaccines
Live, attenuated Zoster (shingles) Yellow fever
Inactivated/Killed Rabies
Subunit/conjugate Human papillomavirus (HPV)
factors. Attenuated vaccines typically provoke durable immunological responses
because the live microorganism is able to replicate and thus stimulate the
immune system in the same manner as the natural infection. For this reason,
they are preferred for healthy adults. However, a live vaccine format may be
unsafe for use in immunocompromised individuals that cannot mount proper
immune responses against even attenuated microorganisms. Furthermore, there
is always the possibility that an attenuated pathogen may mutate and revert to a
virulent form that causes disease. For example, the Sabin live attenuated oral
poliovirus vaccine is capable of reverting to a neurovirulent form that causes
vaccine-associated paralytic poliomyelitis and the emergence of vaccine-derived
poliovirus strains19–21. Most live attenuated vaccines contain attenuated viruses,

11
such as measles, mumps, rubella, yellow fever, or poliovirus (Sabin version).
However, there are a few bacterial attenuated vaccines, including those for
typhoid fever, Yersinia pestis, and tuberculosis.
Inactivated: Alternatively, vaccines may contain viruses or bacteria that
are entirely inactivated or otherwise ‘dead.’ Pathogenic microbes may be killed
by chemicals, heat, or radiation. Inactivated vaccines are more stable and safer
than live vaccines because dead microbes have no possibility of mutating back to
their disease-causing state. However, most inactivated vaccines struggle to
stimulate strong immune responses compared to live vaccines and individuals
may require multiple booster shots until protective immunity is built.
Subunit: Instead of vaccinating with the entire microbe, subunit vaccines
include only the antigens that best stimulate the immune system. These
vaccines generally contain one or several protein antigens along with an adjuvant
that stimulates the immune system. The antigens are selected because they are
common antibody targets and/or they stimulate strong T cell responses. These
proteins are either harvested from the microbes themselves or they are
manufactured via recombinant DNA technology. A limited number of vaccine
adjuvants have been approved for human use. By far, aluminum-containing salts
such as aluminum hydroxide (alum) are the most widely used adjuvants in
human subunit vaccines11. Though alum has been used successfully in human
vaccinations for over 80 years, its mechanism of action as an immunological
adjuvant remains unclear.

12
Adjuvants used in subunit vaccines
Adjuvants promote immune responses by recruiting APCs to the
vaccination site, increasing antigen uptake by APCs, and/or by activating APCs
to produce immunostimulatory cytokines that affect T cells and other immune
cells. Figure 1.2 summarizes adjuvants that are currently in use or being tested
for use in human vaccines. The class of adjuvant that boasts the longest
historical use in human vaccines is aluminum salts, referred to as alum. To
create an alum subunit vaccine, proteins sourced from a given pathogen are
adsorbed onto alum crystals, creating a slurry suspension that is injected
intramuscularly. Despite its long-standing and widespread use in human
vaccines, it is still not clear exactly how alum adjuvants work. The following
section is devoted to reviewing alum as an adjuvant, including proposed
mechanisms by which it stimulates immune responses.
Figure 1.2. Human vaccine adjuvants, adapted from Rappuoli et al., Nat Rev Immunol, 201122.

13
Aluminum Salts as Vaccine Adjuvants
Discovery and use in vaccines
In 1926, Alexander T. Glenny and colleagues reported that superior
antibody responses resulted from soluble protein immunizations if the protein
antigen was first precipitated onto insoluble particles of aluminum potassium
sulfate (potash alum)18. This was the first study to indicate that aluminum salts
have adjuvant properties. Following this discovery, aluminum salts were used in
vaccine preparations with tetanus and diphtheria toxoids to protect against C.
tetani and C. diphtheria, respectively. Today, various alum species are used in a
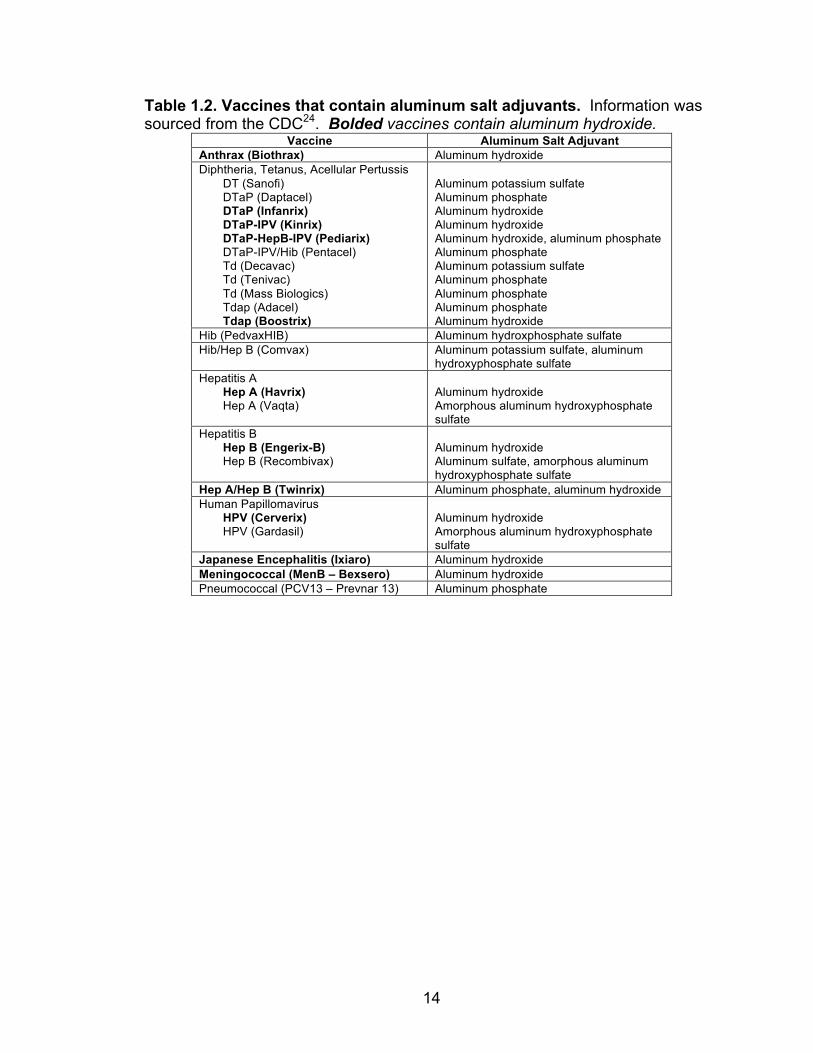
variety of safe and effective human vaccines worldwide (Table 1.2) that have
saved millions of lives, as illustrated in an infographic of vaccine efficacy within
the United States23 (Fig. 1.3).
Mechanism of action
Glenny proposed that aluminum salts were effective adjuvants because
they created antigen depots within the body and that, upon injection, alum
particles slowly released antigen over a long period of time. He reasoned that
this slow antigen release would promote prolonged and effective stimulation of
the immune system25, an effect referred to as the ‘depot effect.’ This explanation
was accepted as dogma for more than 60 years as there was little academic
interest in exploring the mechanism of alum adjuvant effect. In the past few
decades, however, interest in alum adjuvants has reignited and many research
14
Table 1.2. Vaccines that contain aluminum salt adjuvants. Information was sourced from the CDC24. Bolded vaccines contain aluminum hydroxide.
Vaccine Aluminum Salt Adjuvant Anthrax (Biothrax) Aluminum hydroxide Diphtheria, Tetanus, Acellular Pertussis
DT (Sanofi) DTaP (Daptacel) DTaP (Infanrix) DTaP-IPV (Kinrix) DTaP-HepB-IPV (Pediarix) DTaP-IPV/Hib (Pentacel) Td (Decavac) Td (Tenivac) Td (Mass Biologics) Tdap (Adacel) Tdap (Boostrix)
Aluminum potassium sulfate Aluminum phosphate Aluminum hydroxide Aluminum hydroxide Aluminum hydroxide, aluminum phosphate Aluminum phosphate Aluminum potassium sulfate Aluminum phosphate Aluminum phosphate Aluminum phosphate Aluminum hydroxide
Hib (PedvaxHIB) Aluminum hydroxphosphate sulfate Hib/Hep B (Comvax) Aluminum potassium sulfate, aluminum
hydroxyphosphate sulfate Hepatitis A
Hep A (Havrix) Hep A (Vaqta)
Aluminum hydroxide Amorphous aluminum hydroxyphosphate sulfate
Hepatitis B Hep B (Engerix-B) Hep B (Recombivax)
Aluminum hydroxide Aluminum sulfate, amorphous aluminum hydroxyphosphate sulfate
Hep A/Hep B (Twinrix) Aluminum phosphate, aluminum hydroxide Human Papillomavirus
HPV (Cerverix) HPV (Gardasil)
Aluminum hydroxide Amorphous aluminum hydroxyphosphate sulfate
Japanese Encephalitis (Ixiaro) Aluminum hydroxide Meningococcal (MenB – Bexsero) Aluminum hydroxide Pneumococcal (PCV13 – Prevnar 13) Aluminum phosphate

15
Figure 1.3. Efficacy of vaccines in the United States, adapted from an infographic created by Leon Farrant, based on a study by Roush et al., JAMA, 200723.
efforts have been directed at understanding the precise adjuvant actions of these
compounds. Alum’s mechanism of action remains largely mysterious as a
surprising number of immune pathways have been implicated as necessary and
later dismissed as dispensable, including the depot effect26,27.
Generation of protective antibodies: As it is their essential use, alum
adjuvants are well known to generate long-lived protective antibody responses
against their adsorbed antigens. Antibodies act by rapidly binding to pathogens
(or pathogenic products such as toxins) within the serum, mucosa, and other
tissues. Upon binding, antibodies may neutralize pathogens, opsonize them for
Alum%

16
future destruction, and/or activate complement cascades that directly kill the
bound pathogen. Antibody responses are considered protective when they are
present in high enough concentrations to effectively neutralize threatening
pathogens before they cause damage or disease in the host.
As an effective adjuvant, alum stimulates the activation and differentiation
of B cells into long-lived plasma cells that home to bone marrow and continue
secreting antibodies for months to years, maintaining a stable serum titer of
protective antigen-specific antibodies28–30. However, alum vaccines generally do
not induce antibody responses quite as robust as those generated by live
attenuated vaccines. For example, the alum-containing tetanus vaccine must be
re-administered every 10 to 15 years to ensure protection because the induced
tetanus toxoid-specific plasma cells eventually die off within vaccinees31.
The discoveries that class-switched antibody production depends on T cell
help32 and that different infections induce different TH cell subsets with disparate
functions33 led to an effort to determine which TH subsets are induced by alum
adjuvants. Alum was found to preferentially induce TH2 responses34 that direct
activated B cells to secrete TH2-associated antibody isotypes IgG1 and IgE in
mice. In humans and rhesus macaques, alum also induces potent IgG1-
dominated antibody responses, with smaller inductions of IgG3 and sometimes a
little bit of IgG4 isotypes35–41. However, mouse IgG1 and human IgG1 are
dissimilar in function, so inferences regarding alum-induced human antibody
responses from mouse studies must be done with great care.

17
At this point, I will discuss some of the issues that arise when mouse
research is translated into human biology when studying antibody isotypes within
protective immunity. In mammals, there are five classes of antibodies (IgM, IgD,
IgG, IgE, and IgA) with distinct structures, biological functions, and distributions
throughout the body. In humans there are four subclasses of IgG: IgG1, IgG2,
IgG3, and IgG4. IgG subclass nomenclature is independent among species so
murine IgG subclasses do not correspond to human. However, within a given
species, there is always a variety of antibody isotypes and subclasses that have
different functional abilities to fix complement or bind Fc receptors. For example,
human IgG1 and IgG3 excel at mediating antibody-dependent cellular cytotoxicity
(ADCC) and complement-dependent cytotoxicity (CDC) effector functions.
Accordingly, vaccines generally aim to induce these IgG subclasses to
confer immunity against most viral and bacterial pathogens. The ADCC and
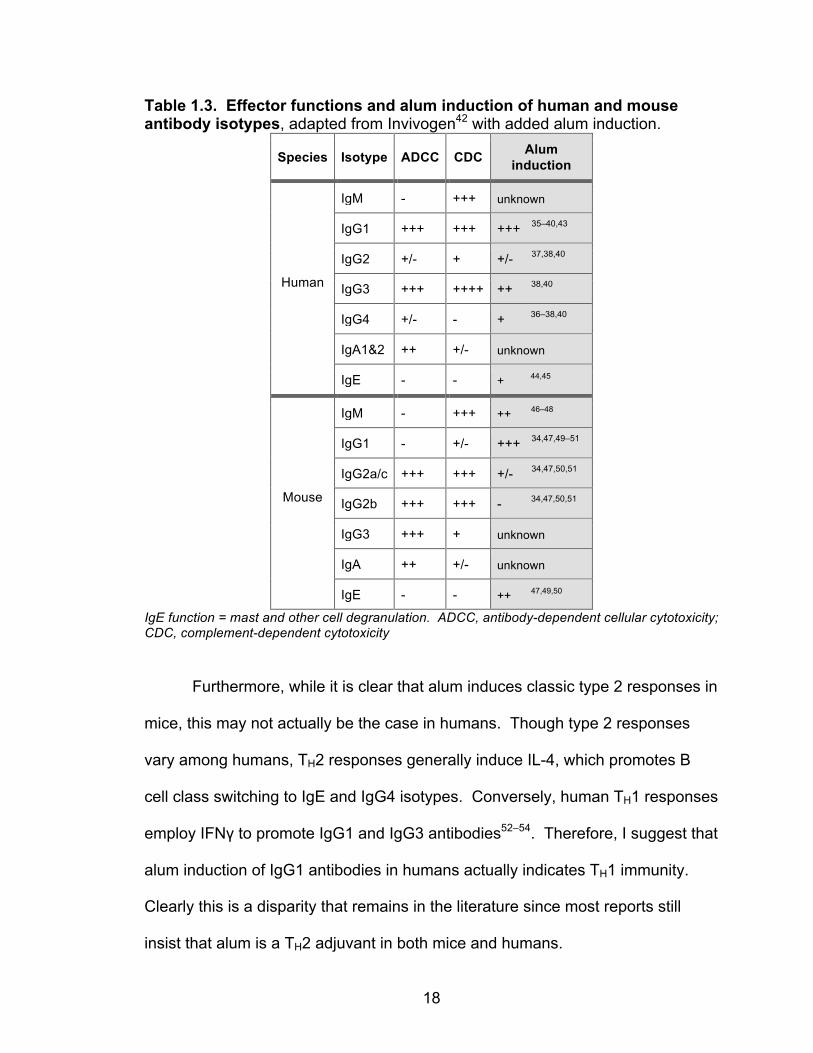
CDC capabilities of human and mouse IgG subclasses are compared in Table
1.3. Alarmingly, alum predominantly induces antibodies with different functions
in humans (strong ADCC and CDC abilities) and mice (poor ADCC and CDC
abilities) (Table 1.3). This disparity suggests that mouse models are limited in
their ability to mirror alum responses in humans and should therefore be used
cautiously.
18
Table 1.3. Effector functions and alum induction of human and mouse antibody isotypes, adapted from Invivogen42 with added alum induction.
Species Isotype ADCC CDC Alum induction
Human
IgM - +++ unknown
IgG1 +++ +++ +++ 35–40,43
IgG2 +/- + +/- 37,38,40
IgG3 +++ ++++ ++ 38,40
IgG4 +/- - + 36–38,40
IgA1&2 ++ +/- unknown
IgE - - + 44,45
Mouse
IgM - +++ ++ 46–48
IgG1 - +/- +++ 34,47,49–51
IgG2a/c +++ +++ +/- 34,47,50,51
IgG2b +++ +++ - 34,47,50,51
IgG3 +++ + unknown
IgA ++ +/- unknown
IgE - - ++ 47,49,50
IgE function = mast and other cell degranulation. ADCC, antibody-dependent cellular cytotoxicity; CDC, complement-dependent cytotoxicity
Furthermore, while it is clear that alum induces classic type 2 responses in
mice, this may not actually be the case in humans. Though type 2 responses
vary among humans, TH2 responses generally induce IL-4, which promotes B
cell class switching to IgE and IgG4 isotypes. Conversely, human TH1 responses
employ IFNγ to promote IgG1 and IgG3 antibodies52–54. Therefore, I suggest that
alum induction of IgG1 antibodies in humans actually indicates TH1 immunity.
Clearly this is a disparity that remains in the literature since most reports still
insist that alum is a TH2 adjuvant in both mice and humans.

19
Regardless of the confusion surrounding which type of immune response
alum induces, the plain truth remains that alum is an effective and safe vaccine
adjuvant. One possible reason for its efficacy as a hepatitis B vaccine is that it
induces the same IgG subclasses as hepatitis B virus. Naturally, both chronic
and acute infections with hepatitis B virus induce mostly IgG1 antibody
responses, with contributions from IgG3 and IgG4 and little to no IgG238–40,55.
Therefore, it is fortunate that alum stimulates IgG1 antibodies in humans.
Innate immune cell activation: Antigen presenting cells (APCs) are central
to adaptive immune defenses – they bridge the gap between innate and adaptive
immunity by acquiring foreign antigens and presenting them to T lymphocytes.
They are also pivotal in initiating immune responses to alum vaccines. Aluminum
salts are known to generally activate APCs in vivo: promoting antigen uptake and
presentation, increasing surface expression of activation molecules, and
encouraging migration to lymphoid organs 56–61. However, alum only variably
induces these effects in vitro, as there have been conflicting reports of effect and
no effect of alum on APC maturation in vitro62,63. Additionally, the mechanism(s)
by which alum achieves APC activation is still unclear.
Though many natural adjuvants such as PAMPs stimulate TLRs, studies
in the literature agree that alum does not activate APCs via TLR signaling, since
MyD88 and TRIF signaling molecules are dispensable in alum responses
(reviewed in 56). Alum stimulates inflammasome activation within APCs, though
there is disagreement about whether or not this pathway is required for alum
adjuvant activity47,59,64–68. Furthermore, it is unclear whether inflammasome

20
activation by alum is direct or indirect. Both models agree that phagocytosis by
innate cells is required. In the direct activation model, phagocytic cells would
directly engage and engulf alum particles, leading to lysosomal damage, followed
by the release of stimulatory endosomal contents into the cytosol64,69. There is
no identified surface receptor that is specific for alum particles, though alum may
be detected by lipid sorting, similar to the detection of uric acid-derived
monosodium urate (MSU) crystals70,71. Alternatively, alum could indirectly
stimulate inflammasome activation by acting as a cytotoxin that causes the
release of endogenous DAMPs (including uric acid) from dying cells59. Uric acid
and resulting MSU crystals are known to activate the inflammasome pathway.
The cytotoxicity of alum will be discussed further in its own section.
In summary, all identified immunostimulatory actions of alum have been
deemed nonessential for the adjuvant activity of alum, except for the recently
identified innate mechanism of APC activation following lipid sorting upon
interaction of the plasma membrane with alum crystals. Alum is likely detected
by this innate mechanism that causes broad downstream inflammation.
Subsequent general inflammation seems to employ redundant pathways; it is
affected little by the loss of any one mechanism, making reductionist alum
research attempts difficult.
Many cells can act as antigen presenters, but the most efficient APCs are
conventional dendritic cells (cDCs), inflammatory monocytes (iMonos),
macrophages, and B cells. Though phenotypically and functionally very similar
(discussed in detail in the next sections), cDCs and iMonos differentiate from

21
different progenitor cells72,73 (Fig. 1.4). In the context of intramuscular injection,
alum-adsorbed antigen is acquired and presented to T cells by a variety of APC
subsets, the most prominent being the CD11b+ subset of cDCs and
iMonos58,59,61.
Though there are many specialized subsets of dendritic cells, conventional
DCs (cDCs) can be grouped into two general categories: 1) lymphoid tissue
resident DCs (resDCs) that live within secondary lymphoid tissues and 2) tissue-
resident DCs that reside in the parenchyma of nonlymphoid tissues and, upon
taking up antigen, migrate to draining lymph nodes (dLNs), where they are called
migratory DCs (migDCs). In the dLNs, migDCs excel in activating antigen-
specific naive T cells. cDCs express CD11c and MHC class II (MHC II)
molecules and can be categorized as CD8α+ or CD11b+ cDCs, a dichotomy that
accounts for phenotypic, developmental, and functional attributes74–76. Tissue
(muscle) resident CD11b+ cDCs are activated by i.m. alum vaccination. They are
the most numerous APC subset to take up alum-adsorbed antigen and migrate to
dLNs to present antigen to T cells (illustrated in Fig. 1.5, pink). Upon migration to
dLNs, they can be identified as CD11b+ migDCs by their very high MHC II
expression.
Monocytes exist in two subsets (Ly6Chigh classical blood monocytes and
Ly6Clow non-classical monocytes73,77) that mainly circulate within the blood,
though they have been found within various tissues during steady state as well.
In inflammatory conditions such as those caused by intramuscular alum
immunization, classical blood monocytes extravasate into tissues and locally

22
Figure 1.4. Conventional dendritic cell and monocyte differentiation in mice. Adapted from Geissmann et al. Science 2010 (Fig. 2)72 and Guilliams et al. Nat Rev Immunol 2014 (Fig. 2)73. Abbreviations: BATF3, basic leucine zipper transcriptional factor ATF-like 3; cDC1, classical type 1 DC; cDC2, classical type 2 DC; CDP, common DC precursor; cMop, common monocyte progenitor; FLT3, FMS-like tyrosine kinase 3; HSC, haematopoietic stem cell; IRF4, interferon-regulatory factor 4; LP, lymphoid precursor; MDP, monocyte, macrophage, and DC precursors; MP, myeloid precursor; PDC, plasmacytoid DC.
develop into CD11b+ CD11c− MHCII+ macrophages and CD11b+ CD11c+ MHCII+
inflammatory monocytes (iMonos). iMonos have also been called inflammatory
DCs or monocyte-derived DCs. Though iMonos and CD11b+ cDCs express
many of the same surface activation markers (namely CD11c, MHC II, and
CD11b), iMonos can be distinguished from CD11b+ cDCs by their expression of
CD64, the high affinity IgG receptor FcγRI61. Alum-containing immunizations
stimulate the recruitment and differentiation of classical blood monocytes into
CD64+ iMonos at the site of injection59 (illustrated in Fig. 1.5, yellow).
cMoP
?
Monocyte-derived cells
iMono
FLT3 ligand
CD11b+
CD8α+
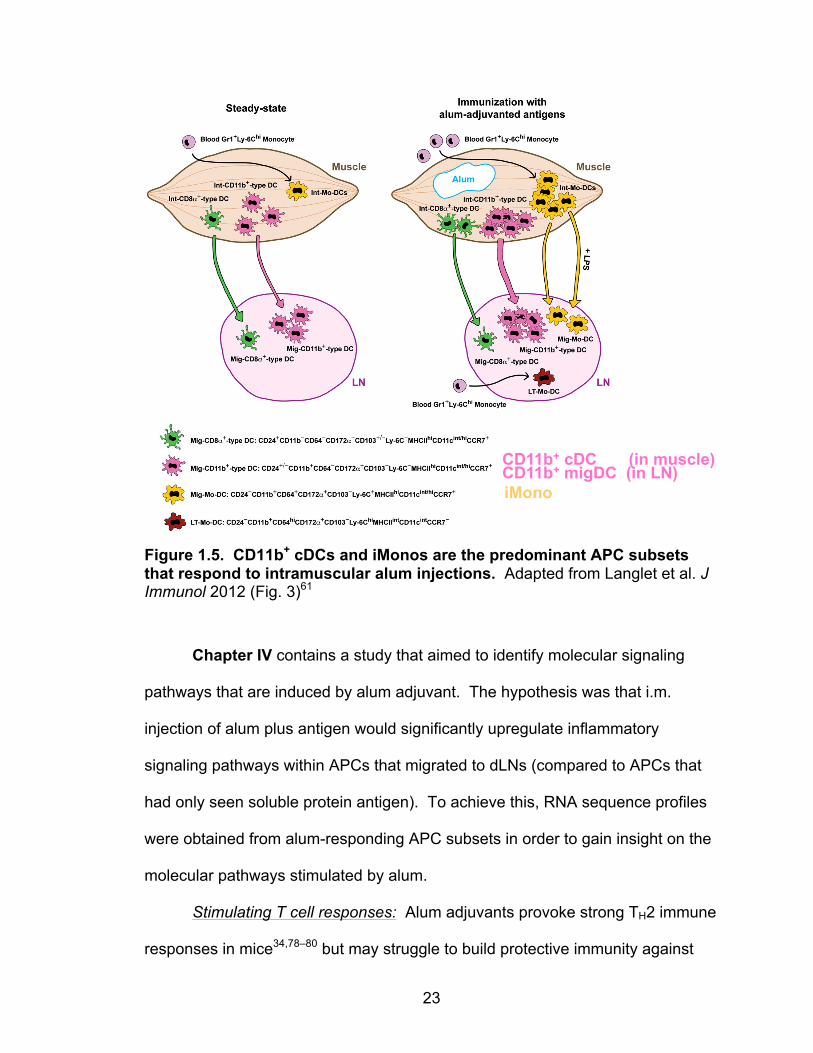
23
Figure 1.5. CD11b+ cDCs and iMonos are the predominant APC subsets that respond to intramuscular alum injections. Adapted from Langlet et al. J Immunol 2012 (Fig. 3)61
Chapter IV contains a study that aimed to identify molecular signaling
pathways that are induced by alum adjuvant. The hypothesis was that i.m.
injection of alum plus antigen would significantly upregulate inflammatory
signaling pathways within APCs that migrated to dLNs (compared to APCs that
had only seen soluble protein antigen). To achieve this, RNA sequence profiles
were obtained from alum-responding APC subsets in order to gain insight on the
molecular pathways stimulated by alum.
Stimulating T cell responses: Alum adjuvants provoke strong TH2 immune
responses in mice34,78–80 but may struggle to build protective immunity against
CD11b+ cDC (in muscle) CD11b+ migDC (in LN) iMono

24
pathogens that require TH1 and cell-mediated immunity for clearance. TH2
responses are thought to have evolved to protect against multi-cellular parasites
and, now in many Western countries, drive allergies and atopic illnesses81.
Notably, alum responses can be skewed toward TH1 when alum is combined with
a toll-like receptor (TLR) 4 agonist such as monophosphoryl lipid A (MPL) as in
the Adjuvant System 04 (AS04) created by GlaxoSmithKline Biologicals82.
Though still up for debate, alum induction of TH2 responses has been attributed
to inflammasome activation62,65,83, early IL-4-producing eosinophil
recruitment46,48,50,59,84, and production of several other TH2-inducing cytokines
(IL-25, IL-1β, IL-6, and prostaglandin E2)68,78,85–87.
Furthermore, alum induces robust TFH differentiation in mice80,88, which
may explain why it is so effective at generating antibody responses since TFH are
critical helpers in B cell germinal centers89. Alum has also been reported to
induce TH17 responses in addition to TH2. In fact, one study found that IL-1
signaling promoted alum induction of TH17 cells and claimed that TH2 cells were
dispensable while TH17 cells were required for protective immunity against
pertussis51.
Alum is widely recognized as a poor inducer of CTL responses90. One
study even suggested that alum skews immune responses away from producing
CTLs since such strong antibody responses are initiated91. However, antigen-
specific CD8 T cells can be detected, though in small numbers, after alum
immunization92,93.

25
To summarize, alum induces robust TH2 responses in mice that
orchestrate long-lived antibody production; this is the basis for alum-induced
immune protection. Alum also induces protective antibody responses in humans,
though the ‘type’ of immune response may not be strictly TH2. A significant
contribution of TH17 responses has been suggested51, but needs to be verified by
other groups. Studies to verify alum’s effect on T cell differentiation in humans
are lacking, though our lab is working to remedy this gap in knowledge.
Alum’s cytotoxicity and host DNA as an induced self adjuvant: It has long
been known that alum crystals exert some level of cytotoxicity when injected as
vaccine adjuvants94. They are known to cause cell death at the site of injection47
and dying cells can release molecules that act as endogenous danger signals or
DAMPs that may activate innate immune cells95,96. Our group previously
reported that alum particles become entrapped by host chromatin upon i.m.
injection26, the site of administration for most human vaccines. Since this finding,
another group and we have suggested that DNA released by host cells at the site
of injection contributes to alum’s adjuvant activity47,49.
In 2011, Marichal et al. reported that host DNA is required for IgG1 and
IgE induction by alum vaccination47. Host DNA’s mechanism for promoting IgG1
remains a mystery, but IgE depended on stimulation of DNA-sensing pathways
involving [Tumor necrosis activating factor-associated NFκB activator (TANK)]-
binding kinase 1 (Tbk1) and interferon regulatory transcription factor 3 (Irf3).
DNA sensing led to increased local production of IL-12p80 and activation of
inflammatory DCs and monocytes that promoted downstream adaptive immune

26
responses. In 2013, our lab further contributed to the theory that DNA mediates
alum activity by reporting that alum-associated host DNA augments CD4 T cell
priming by enhancing MHC II antigen presentation by APCs and prolonging DC/T
cell interactions in draining LNs49. These effects were dependent on stimulator of
interferon genes (STING), a molecule upstream of Tbk1 and Irf3 in an
intracellular DNA-sensing pathway. Together, these studies defined an important
role for host DNA as an inducible, endogenous adjuvant that mediates alum
activity.
I began the main project of my thesis research with the goal of further
investigating the role of host DNA as a mediator of alum responses. Upon close
inspection, I noticed that the model systems and approaches used in the two
aforementioned studies had some caveats. First, they employed transgenic CD4
T cell adoptive transfer experiments that can be flawed in their ability to reflect
endogenous, wild-type biology. Second, both groups combined alum vaccines
with DNase I enzymes purchased from Roche Diagnostics Corporation to
examine loss-of-function scenarios that lacked extracellular host DNA as a
stimulus; there could be off-target effects of injected DNase enzymes. Third,
both groups curiously made no mention of DNA’s effects on CD8 T cell
responses, though it is not surprising that they focused on CD4 T cell responses
given alum’s preferential priming of TH2 cells. These caveats, especially the use
of DNase I as a vaccine treatment, will be discussed in great detail in Chapter III
as they turned out to be significant oversights in this field.

27
Chapters III and IV contain studies that aimed to better define the role of
host DNA in alum biology. Chapter III focuses on determining how DNA
stimulates T cell responses. In the early stages of this study, some of the results
from the Marichal et al. and McKee et al. papers were unrepeatable.
Consequently, my objective shifted from extending the research to reconciling
the disparities. It turns out that DNA is actually not necessary for alum
responses to certain antigens. Chapter IV examines the effect of alum on innate
immune cells, as mentioned above, though the study was originally launched to
explore the effects of host DNA on APCs.
Immunogenicity of Heat Aggregated Proteins
Chapter V contains data from my first few years of graduate study that
was spent focusing on the adjuvant activity of aggregated proteins. This
scientific topic is particularly relevant in clinical settings because many
therapeutic proteins that are administered to patients are partially aggregated
due to production or storage conditions.
Protein aggregation can cause a variety of diseases and conditions such
as Alzheimer’s disease, systemic amyloidoses, inappropriate neutralization of
administered therapeutic proteins, and even allergies97–100. Inappropriate
aggregation of endogenous proteins is usually prevented by complex cellular
quality control mechanisms such as heat shock protein chaperones during

28
protein folding as well as the unfolded protein response101. It is widely
recognized that protein aggregates are immunogenic and can induce specific
adaptive immune responses98–100,102–105. Most endogenous protein aggregates
are formed by hydrophobic interactions between damaged or misfolded proteins
that expose regions of their hydrophobic cores106–109. It is not clear how foreign
or endogenous protein aggregates are recognized as harmful entities by the
immune system. This project aimed to determine how protein aggregates
activate innate immune sensors and induce adaptive immune responses.
Given the wide, albeit vague, body of literature (explained further in the
Introduction to Chapter V) that suggests aggregated proteins are
immunogenic, I hypothesized that heat denatured protein aggregates are more
immunogenic than soluble proteins because dendritic cells can more efficiently
present them to prime specific T cell responses. Much of this project was left
unfinished as Pippa and I refocused my efforts on the alum project (Chapters III
and IV). In fact, after developing several reagents with which to test my
hypothesis, I only succeeded in testing the effect of aggregated proteins on T cell
responses before this project was moved to the back burner. Our long-term goal
was to understand how T cell responses to protein aggregates could be
manipulated for therapeutic purposes. Promoting immune responses to specific
protein epitopes would be instrumental in the effective treatment of viral diseases
and cancer, while suppressing immunity will mitigate unwanted immunity against
harmless protein antigens.

29
CHAPTER II
MATERIALS AND METHODS
Mice
C57BL/6.J mice were purchased from The Jackson Laboratory at 5 weeks
of age. STING-deficient mice were generated and provided by John Cambier at
National Jewish Health (NJH). STING-deficient mice had their genotypes
reconfirmed by PCR at the time of sacrifice. In all experiments, C57BL/6.J mice
were used unless specifically indicated otherwise. All mice were age matched
and between 6-18 weeks of age at the time of first immunization. Animals were
housed and maintained in the Biological Resource Center within NJH in
accordance with the research guidelines of the NJH Institutional Animal Care and
Use Committee.
Reagents, Antigens, Tetramers, and Antibodies
Reagents
Alhydrogel® aluminum hydroxide (Brenntag) was purchased from
Accurate Chemical. The following reagents were purchased from Sigma-Aldrich:
BSA (A8806), TPCK Trypsin (T-1426), a-Chymotrypsin (C-4129), and LPS (from
E. coli). The Kedl lab provided Poly(I:C) (GE Healthcare) and anti-CD40
antibody (FGK-45, BioXcell). Complete Tumor Medium (CTM) was created by

30
adding 10% FBS and KM tumor cocktail (containing nutrients and antibiotics) to
Minimal Essential Medium for suspension cultures (GibcoTM).
Model antigens
Chicken ovalbumin (OVA) was purchased from Sigma-Aldrich (grade VII,
A7641). Fluorescent antigens were either purchased from InvitrogenTM
Molecular Probes® (OVA-A488, OVA-A647, OVA-FITC) or conjugated in-house
using life technologiesTM molecular probes® conjugation kits (OVA-A647, OVA-
A488, NP-A647, NP-A488).
OVA-NP was generated using Imject Maleimide-Activated Ovalbumin Kit
(Pierce Biotechnology) and cysteine-linked influenza A nucleoprotein peptide
NP311-325 (QVYSLIRPNENPAHKGGGC) purchased from CHI Scientific.
PR8 influenza A nucleoprotein (NP) was produced as previously
described92. Briefly, Hi-5 insect cells were infected with a baculovirus expression
vector containing Influenza A nucleoprotein (PR8) with a 6-Histadine tag. After 3
days, infected cells were lysed, treated with DNase and RNase, and NP was
purified by nickel column (Ni++-NTA-Agarose beads, Qiagen) and, in some cases,
size exclusion chromatography using “Aurora,” a HiLoadTM 26/60 SuperdexTM
200 prep grade column (GE Healthcare). Mutant NP (mutNP), containing five
arginine residues mutated to alanines at positions 74, 75, 174, 175, and 221, was
cloned and produced in the same manner using a baculovirus expression vector.
31
Tetramers
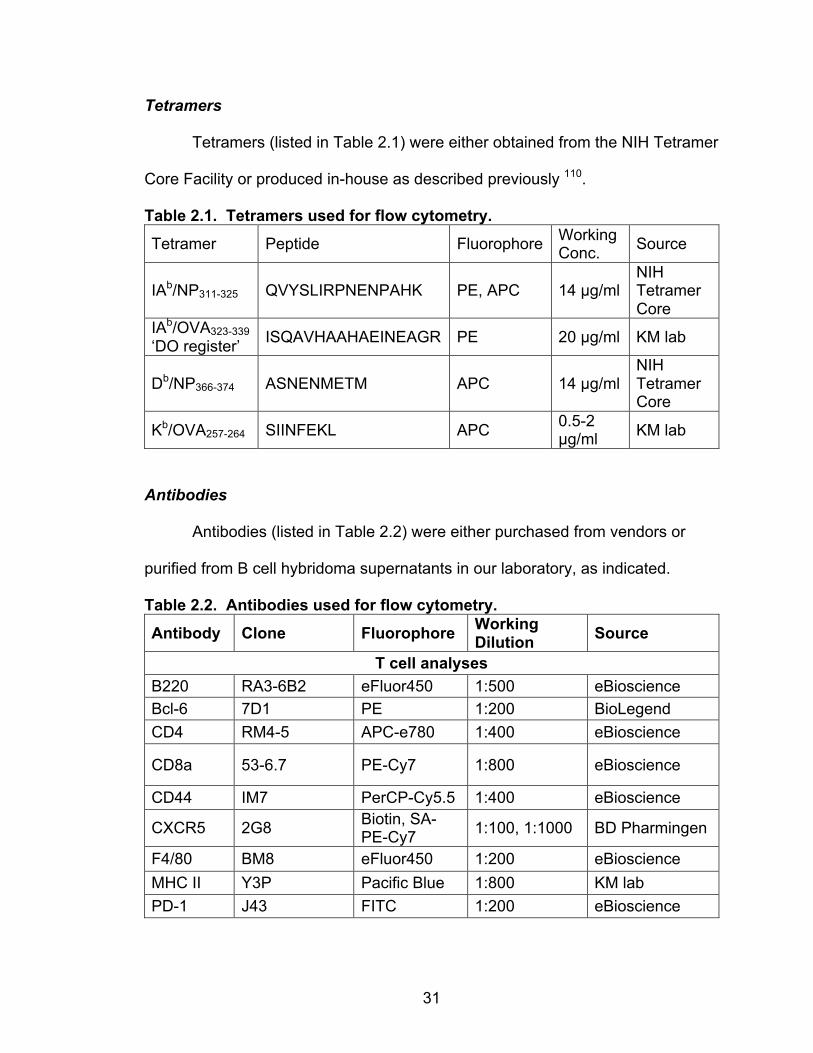
Tetramers (listed in Table 2.1) were either obtained from the NIH Tetramer
Core Facility or produced in-house as described previously 110.
Table 2.1. Tetramers used for flow cytometry.
Tetramer Peptide Fluorophore Working Conc. Source
IAb/NP311-325 QVYSLIRPNENPAHK PE, APC 14 µg/ml NIH Tetramer Core
IAb/OVA323-339 ‘DO register’ ISQAVHAAHAEINEAGR PE 20 µg/ml KM lab
Db/NP366-374 ASNENMETM APC 14 µg/ml NIH Tetramer Core
Kb/OVA257-264 SIINFEKL APC 0.5-2 µg/ml KM lab
Antibodies
Antibodies (listed in Table 2.2) were either purchased from vendors or
purified from B cell hybridoma supernatants in our laboratory, as indicated.
Table 2.2. Antibodies used for flow cytometry.
Antibody Clone Fluorophore Working Dilution Source
T cell analyses B220 RA3-6B2 eFluor450 1:500 eBioscience Bcl-6 7D1 PE 1:200 BioLegend CD4 RM4-5 APC-e780 1:400 eBioscience
CD8a 53-6.7 PE-Cy7 1:800 eBioscience
CD44 IM7 PerCP-Cy5.5 1:400 eBioscience
CXCR5 2G8 Biotin, SA-PE-Cy7 1:100, 1:1000 BD Pharmingen
F4/80 BM8 eFluor450 1:200 eBioscience MHC II Y3P Pacific Blue 1:800 KM lab PD-1 J43 FITC 1:200 eBioscience

32
Table 2.2. Antibodies used for flow cytometry.
Antibody Clone Fluorophore Working Dilution Source
Antigen presenting cell analyses
B220 RA3-6B2 FITC, PE, PE-Cy7, PerCP-Cy5.5
1:100-500 BD Pharmingen, eBioscience
CD11c N418 PE-Cy7 1:400 eBioscience
CD11b M1/70 APC-A750, PerCP-Cy5.5, PB
1:400-500 BioLegend, eBioscience,
CD64 X54-5/7.1.1 PE 1:100-200 BD Pharmingen
Ly6C HK1.4 eFluor450, PerCP-Cy5.5 1:200 eBioscience
MHC II (IA/IE) M5/114.15.2 FITC, APC,
APC-Cy7 1:500-1000 eBioscience
DNases
DNase preparations were either purchased or produced in-house, as
indicated in Table 2.3. To produce wtDNase and mutDNase, plasmids
containing wild-type or mutated (R111A, D212A, H252A) bovine DNase I
nucleotide sequences with 6x Histidine tags that were optimized for Homo
sapiens translation were ordered from Integrated DNA Technologies, Inc. Janice
White digested and ligated these genes into the CMVR-VRC01-L plasmid (NIH
AIDS Reagent Program) in place of the immunoglobulin genes it came with,
using Sal1 and BamH1 cut sites. Plasmids were transformed into XL1-Blue E.
coli (Agilent) and selected on kanamycin agar plates. Colonies were picked,
Minipreps were made, and plasmids were sequenced using the following

33
primers: 6187 DNase fwd, 6188 DNase rev, 2844 CMV fwd. Suitable plasmids
were: p2127-2 = wtDNase; p2128-1 = mutDNase.
Transfection of FreeStyleTM 293-F cells (Life Technologies, derived from
293 human embryonic kidney cells) with these plasmids was outsourced to Lori
Sherman at the UCCC Protein Production Core Facility. She followed a standard
PEI transfection protocol, using 750 µg plasmid DNA per liter of cells.
Supernatants were collected 6 days after transfection, bound to Ni-NTA
agarose (QIAGEN), washed, and eluted with an increasing gradient of imidazole
elution buffer. DNase-containing fractions were grouped, buffer exchanged to
PBS by Amicon Ultra® centrifugal filtration, flash frozen, and stored at -20°C.
Table 2.3. DNase reagents used in Chapter III Alias within Thesis Product Name Vendor Source
Catalog Number
Lot(s) used
Roche DNaseGII DNase I, grade II Roche bovine pancreas 10104159001 10172600, 13814800
Roche Recomb DNase DNase I recombinant Roche
from bovine pancreas, expressed in Pichia pastoris grade I
04 536 282 001 10392800
Sigma DNase Deoxyribonuclease I, from bovine pancreas
Sigma-Aldrich bovine pancreas D4513-1VL 040M7012,
070M7008V
Worth DNase Deoxyribonuclease I, Ribonuclease & Protease Free
Worthington Biochemical Corporation
bovine pancreas LS006334 52N13773
wtDNase -------- -------- WT DNase I from bovine pancreas + 6xHis tag, expressed in 293F cells
-------- --------
mutDNase -------- -------- mutant DNase I (R111A, D212A, H252A) from bovine pancreas + 6xHis tag, expressed in 293F cells
-------- --------

34
Preparation of Soluble or Aggregated Protein Antigens (Chapter V)
Soluble protein
OVA (Sigma-Aldrich, grade VII, lot 066K7020) or NP was filtered through
a 0.22 µm polyethersulfone membrane (Millex® or Thermo Scientific) and, if
indicated, ultracentrifuged at 200,000 x g for 2 hours (OptimaTM L-100 XP
ultracentrifuge, Beckman Coulter). Only the top portion of a sample was
collected after ultracentrifugation. Protein preparations were stored at 4°C.
Aggregated protein
OVA or NP (at indicated concentration in PBS) was filtered (0.22 µm) and
then heated at 80°C for 10 min or 96-98°C for 5-10 min, as indicated.
Alternatively, NP was cross-linked with gluteraldehyde (0.5%) for 2 hours, then
buffer exchanged to PBS.
Assessing Protein Conformation and Aggregation (Chapter V)
Size exclusion chromatography
Monomers and aggregates were distinguished within soluble OVA
preparations by size exclusion chromatography on “Fiona,” a SuperdexTM 200
10/300 GL column (GE Healthcare). Fran Crawford oversaw this procedure.

35
Micro-flow imaging
Particulates between 1-1000 µm in size were counted (per ml) for various
soluble OVA preparations. Samples were prepared at 5 mg/ml in PBS and
analyzed by micro-flow imaging (MFITM DPA 4100, ProteinSimple®) at 0.1
ml/minute flow rate.
Infrared spectroscopy
Soluble OVA preparations were assessed for secondary structure by mid-
infrared Fourier transform infrared spectroscopy (FTIR). Soluble and heat
aggregated OVA samples were prepared at 20 mg/ml in PBS and analyzed using
an MB-series FTIR spectrometer (ABB Bomem Inc). Amide I region spectra
were corrected and second-derivatives were compared between samples using
BGRAMSTM software (Galactic Industries).
Measuring surface hydrophobicity
OVA samples were diluted to 0.1 mg/ml in PBS and 1-anilinonaphthalene-
8-sulfonic acid (ANS) was added to a final concentration of 20 µM. ANS is a
fluorescent stain that binds to hydrophobic regions on proteins. Upon binding to
hydrophobic surfaces, ANS fluorescence was detected using a FeliX32TM
spectrofluorometer (Photon Technology International). Samples were
maintained at 23°C, excited at 350 nm, and scanned for emission between 400-
500 nm.

36
Immunizations
Mice were anesthetized with 2.5% (vol/vol) isoflurane and injected i.m. in
each calf muscle with a total vol of 50µl per calf, unless another immunization
route is indicated. For T cell studies, all vaccines consisted of 10 µg protein
antigen (OVA, OVA-NP, NP, or mutNP) that was fully adsorbed to 200 µg alum
(or combined with other adjuvants, as indicated) and suspended in endotoxin-
free PBS (Cellgro). For APC studies, vaccines contained dose of antigen
between 5-20 µg of OVA-A647, OVA-A488, OVA-FITC, NP-A647, or NP-A488.
When indicated, additional reagents (BSA, various DNases, trypsin, and/or
chymotrypsin) were added as treatments to vaccines immediately before
injection. The endotoxin content of each vaccine component was <1
EU/injection, as measured by Limulus Amoebocyte Lysate Assay (Lonza).
Assessment of Antigen-Specific T Cell Priming
Popliteal LNs were harvested into ice-cold balanced salt solution and
disrupted through nylon mesh to create single cell suspensions. Cells were
counted using a Z1 Coulter® Particle Counter (Beckman Coulter) and then
stained with tetramers (Table 2.1) for 2 hours at 37°C (25 µl volume in CTM +
heat inactivated normal mouse serum and 2.4G2 blocking antibody). Antibodies
for CD4, CD44, CD8a, B220, MHC II, and F4/80 were added and the cells were
further incubated for at least 30 min on ice or at 4°C. Cells were washed and

37
analyzed on a CyAnTM ADP Analyzer (Beckman Coulter) flow cytometer using
Summit Software (DakoCytomation) and then FlowJo software (TreeStar).
IAb/peptide tetramer-positive cells were defined after gating on live,
singlet, CD4+ CD44hi cells that were negative for CD8a, B220, MHC II, and
F4/80 (Fig. 2.1). Kb/peptide or Db/peptide tetramer-positive cells were similarly
defined, but were gated on CD8a+ and CD4– (Fig. 2.1). Total numbers of
tetramer+ cells per organ were calculated by multiplying the percentage of
tetramer+ cells within the FlowJo live gate by the total cells counted per organ by
Coulter® Counter.
Antigen Presenting Cell Analyses
Draining LNs (dLNs) were harvested from mice 6-72 hours (as indicated)
after immunization into ice cold Collagenase D solution: HBSS (without calcium
or magnesium, GibcoTM) + Collagenase D (2.5 mg/ml, Roche) + 1% FCS that
was pretreated with 0.02 mM EDTA + 100 µg/ml DNase I (Roche, DNaseGII).
dLNs were minced with two 25G needles, incubated 37°C for 30 min, then placed
on ice and collagenase digestion was stopped with EDTA addition to 10-50 mM.
Digested tissues were mechanically disrupted by vigorous Pasteur pipetting,
filtered (100µm), washed, and then stained with antibodies (listed in Table 2.2)
for 45-60 min while on ice or at 4°C. Cells were washed and analyzed on a
CyAnTM ADP Analyzer (Beckman Coulter) flow cytometer using Summit Software
(DakoCytomation). Data were analyzed using FlowJo software (TreeStar).

38
Live
CD
4+ d
ump-
C
D4+
CD
8-
sing
lets
C
D44
+ te
t+
CD
8+ d
ump-
C
D8+
CD
4-
sing
lets
C
D44
+ te
t+
Figu
re 2
.1.
Ant
igen
-spe
cific
CD
4 an
d C
D8
T ce
ll ga
ting
stra
tegy
. C
ells
wer
e ga
ted
on li
ve, c
o-re
cept
or+
(CD
4 or
C
D8)
, B22
0-, M
HC
II-,
F4/8
0-, a
nd s
ingl
ets
by p
ulse
wid
th.
Antig
en-s
peci
fic T
cel
ls w
ere
iden
tifie
d as
CD
44+
tetra
mer
+ (IA
b for C
D4,
Kb o
r Db fo
r CD
8).
Tota
l tet
ram
er+
cell
num
bers
per
org
an w
ere
calc
ulat
ed b
y m
ultip
lyin
g th
e pe
rcen
tage
of t
etra
mer
+ ce
lls w
ithin
the
live
gate
of F
low
Jo b
y th
e to
tal c
ells
cou
nted
per
org
an (b
y C
oulte
r®
Cou
nter
) im
med
iate
ly fo
llow
ing
harv
est a
nd s
ingl
e ce
ll su
spen
sion
.

39
RNA Sequencing: Cell Preparation and Analysis
Mice were immunized with 10 µg OVA-A647 ± 200 µg alum + 1 mg BSA
or Roche DNaseGII. Popliteal dLNs were harvested 24 hours after
immunization, grouped from 10-20 mice per treatment group, and processed and
stained as described above for antigen presenting cells. Samples were
resuspended in sorting diluent (1X PBS + 1 mM EDTA + 1% FCS + 25 mM
HEPES pH 7.0) and antigen-loaded A647+ cells were sorted into B220+ (B cell),
CD11chigh (DC), or CD11bhigh (iMono) populations (Fig. 2.2 and Table 2.4),
collected in CTM. Samples were sorted using a MoFloTM XDP-70 (Beckman
Coulter) high speed cell sorter by Alistaire (Lester) Acosta at the University of
Colorado Cancer Center Flow Cytometry Shared Resource (Anschutz Medical
Campus location). Sorted cells were washed once with PBS, spun, resuspended
in QIAzol Lysis Reagent (QIAGEN), flash frozen with dry ice, and stored at -80°C
before shipment on dry ice to Expression Analysis, Inc. (Durham, NC) for RNA
sequencing analysis.
Expression Analysis, Inc., performed RNA sequencing on these sorted cell
populations. RSEM expression counts were normalized within samples by upper
quartile (75th percentile) normalization to 1000:
Normalized count genei samplex =
expression count genei x 1000 75th percentile (samplex)
Normalized gene expression was compared between treatment groups (antigen
± alum + BSA or DNase) within given cell populations (B cell, iMono, or DC) by
40
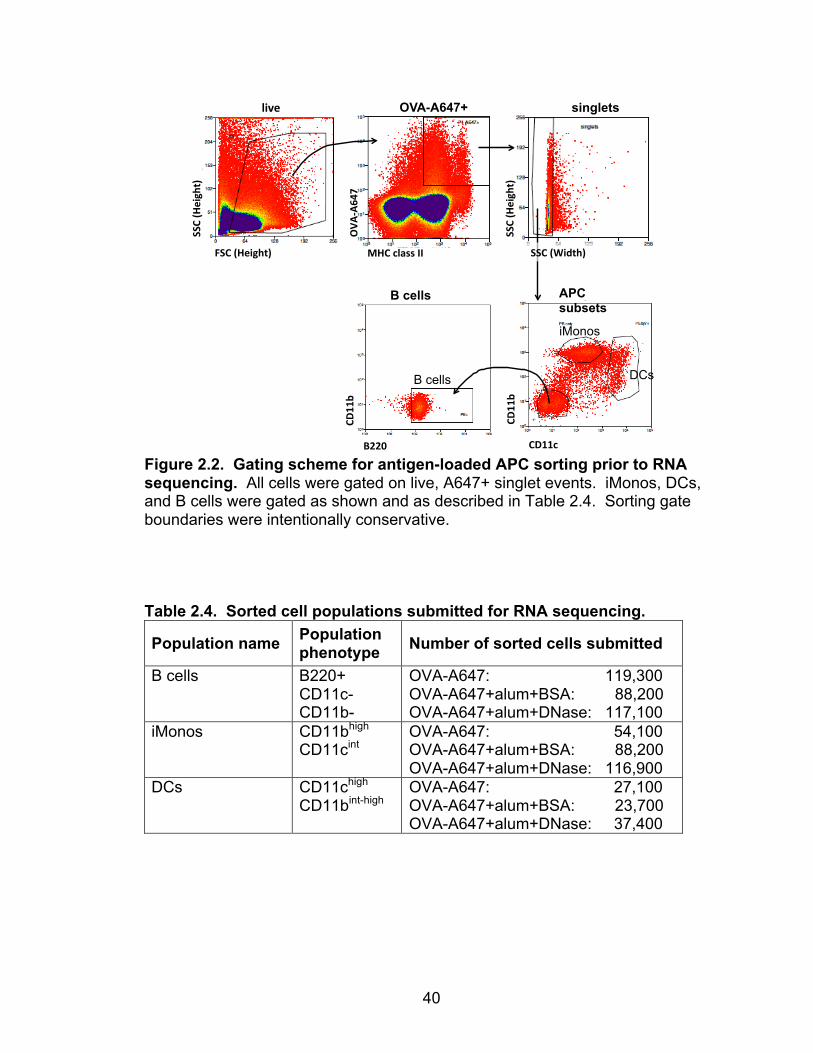
Figure 2.2. Gating scheme for antigen-loaded APC sorting prior to RNA sequencing. All cells were gated on live, A647+ singlet events. iMonos, DCs, and B cells were gated as shown and as described in Table 2.4. Sorting gate boundaries were intentionally conservative. Table 2.4. Sorted cell populations submitted for RNA sequencing.
Population name Population phenotype Number of sorted cells submitted
B cells B220+ CD11c- CD11b-
OVA-A647: 119,300 OVA-A647+alum+BSA: 88,200 OVA-A647+alum+DNase: 117,100
iMonos CD11bhigh CD11cint
OVA-A647: 54,100 OVA-A647+alum+BSA: 88,200 OVA-A647+alum+DNase: 116,900
DCs CD11chigh CD11bint-high
OVA-A647: 27,100 OVA-A647+alum+BSA: 23,700 OVA-A647+alum+DNase: 37,400
live%
MHC%class%II%
CD11b%
CD11c%
FSC%(Height)%
SSC%(Height)%
APC subsets
OVA-A647+ singlets
B cells
SSC%(Height)%
OVA
:A647%
CD11b%
B220%
SSC%(Width)%
B cells
iMonos
DCs

41
calculating fold-change differences. Fold-change of normalized gene expression
between experimental groups was calculated by dividing one group’s gene
expression value by another’s. To avoid mathematical issues, if a gene’s
normalized expression value was equal to 0, its value was changed to 1 for fold-
change calculations because 1 represented a low value within the normalized
gene expression counts. There were few normalized counts that were positive
values less than 1. Also, for each fold-change comparison between groups, if
the numerator value was zero for a given gene, that gene was excluded from the
analysis. Fold-change calculations were performed in excel, but the general
equation is described here in an example to calculate genes that alum treatment
increased, compared to antigen alone:
Fold-change (antigen+alum+BSA
antigen) =
Norm. expression (genei (antigen+alum+BSA), if not = 0)Norm. expression (genei (antigen), if = 0, then change to = 1)
Sonia Leach at National Jewish Health generated a heat map of gene
cluster expression within each of the alum-treated or untreated APC subsets.
Relative expression values of genes were clustered by heat map with respect to
respective mean expression for each gene across all samples. Thus warmer
colors (red) indicate expression higher than the gene's mean across all samples
while cooler colors (blue) indicate expression lower than the gene's mean.
Genes with greater than 2-fold-change difference in their expression
between treatments were identified for each cell population and analyzed by
Database for Annotation, Visualization, and Integrated Discovery (DAVID) v6.7
bioinformatics resources web program (NIAID, NIH). Analyses included Kegg

42
Pathways, Swiss-Prot and Protein Information Resource Keywords, and Gene
Ontologies of cellular components, molecular functions, and biological
processes. Functional gene clusters were analyzed if the DAVID program
calculated their enrichment score to be ≥1.
Antibody Detection by ELISA
For OVA-, mutNP-, and NP-specific IgG1 detection, we incubated serially-
diluted sera from immunized mice on 96-well Immulon plates (Thermo Scientific)
coated with OVA (grade VII, Sigma-Aldrich) at 100 µg/ml or mutNP or NP at 10
µg/ml. We detected bound IgG1 using alkaline phosphatase-conjugated anti-
mouse IgG1 antibodies (BD Pharmingen) followed by incubation with p-
nitrophenyl phosphate and measurement by spectrophotometry. To determine
relative units, we used positive control serum samples from B6 mice that
contained OVA-specific or NP-specific antibodies.
Assessing DNase Reagent Purity
SDS-PAGE
Samples (>8mg/ml) were reduced, heat denatured, and applied to a SDS
PhastGelTM Homogenous 12.5% polyacrylamide gel (GE Healthcare) as
recommended by the manufacturer. Low Molecular Weight standards (GE

43
Healthcare) were applied alongside the samples. Gels were stained with
Coomassie® Brilliant Blue G-250 (Bio-Rad).
Protease activity assay
Pierce Protease Assay Kit (Thermo Scientific) was used according to the
manufacturer’s instructions. TPCK trypsin standard was serially diluted by 5-fold
while samples were serially diluted by 2-fold.
Mass spectrometry
Samples were prepared in PBS and reduced, denatured, alkylated, and
digested overnight at 37°C with Trypsin Gold (Promega). Peptides within
samples were chromatographically resolved on-line using a C18 column and
1260 series high performance liquid chromatography (HPLC, Agilent
Technologies) and analyzed using a 6550 LCMS QTOF mass spectrometer
(Agilent Technologies, Palo Alto, CA) in the National Jewish Health Proteomics
Facility. Raw data was extracted and searched using the Spectrum Mill search
engine (Rev B.04.00.127, Agilent Technologies, Palo Alto, CA). “Peak picking” is
performed within SpectrumMill with the following parameters: signal-to-noise set
at 15, maximum charge state of 4 was allowed (z=4), and the program was
directed to find a precursor charge state. During searching the following
parameters were applied: searched the SwissProt Bovine database,
carbamidomethylation as a fixed modification, oxidized methionine and
deamidated asparagine as variable modifications, collected spectra were

44
compared to tryptic peptides in the database, maximum of 2 missed cleavages,
precursor mass tolerance +/- 20 PPM, product mass tolerance +/- 50 PPM, and
maximum ambiguous precursor charge = 3. Data were evaluated and protein
identifications were considered significant if the following confidence thresholds
were met: minimum of 3 peptides per protein, protein score > 10, individual
peptide scores of at least 6, and Scored Percent Intensity (SPI) of at least 60%.
The SPI provides an indication of the percent of the total ion intensity that
matches the peptide’s MS/MS spectrum. Standards were run at the beginning of
each day of analyses for quality control purposes.
DNase Activity Assay
DNase reagents were serially diluted in 96-well round bottomed tissue
culture plates and exposed to 800 ng genomic mouse DNA within PBS + 25 mM
MgCl2 and 5 mM CaCl2 for 30 min at 37°C. Enzymatic activity was stopped by
heat denaturation at 95°C for 5 min and samples were immediately placed on ice
and assessed for double-stranded DNA content using a Qubit® dsDNA Broad
Range Assay Kit. Alternatively, samples were ran on a 5% agarose gel (50V, 50
min) alongside untreated DNA (800 ng) and HyperLadderTM V (25bp, Bioline)
molecular weight marker for determination of size of DNA fragments. DNA
fragments were visualized by staining the gel with ethidium bromide and imaging
under ultraviolet light.

45
Antigen Destruction Assay
Protein antigens (OVA-NP or NP) were prepared at 200 µg/ml in PBS + 25
mM MgCl2 and exposed to treatments for 0, 1, or 21 hours at 37°C. To mimic
vaccine preparations, treatments were as follows: BSA (20 mg/ml), Roche
DNaseGII (20 mg/ml), trypsin (200 µg/ml), or chymotrypsin (200 µg/ml). OVA-NP
or NP mixtures were then added in duplicate to tissue culture wells containing
Chb-2.4.4 antigen-presenting B cells and ‘3NP311-2’ NP311-325-specific T cell
hybridomas. Cells were incubated for 24 hours at 37°C then supernatant were
tested for IL-2 by sandwich ELISA. Supernatants were serially diluted on 96-well
Immulon plates (Thermo Scientific) coated with anti- IL-2 (eBioscience). Bound
IL-2 was detected with biotin-conjugated anti-IL-2 (eBioscience) followed by
HRP-conjugated streptavidin (Jackson Immunoresearch) and subsequent
incubation with 1-step Ultra TMB substrate (Thermo Scientific) and 2M H2SO4
stopping solution. Plates were measured by spectrophotometry.
Generation of 3NP311-2 NP311-325-specific CD4 T Cell Hybridomas
Female C57BL/6 mice were immunized i.m. with 10 µg OVA-NP + 200 µg
alum. Draining LNs were harvested on day 7 and stimulated for 4 days in vitro
with 100 µg/ml NP311-325 peptide (CHI Scientific) in Click’s medium + 1% normal
mouse serum. Antigen-specific T cell blasts were grown for 3 more days in CTM
+ IL-2 and then fused to BW5147α-β- (AKR thymoma) cells and cultured in CTM

46
with HAT (hypoxanthine-aminopterin-thymidine) added after 1 day. Colonies of
hybrids were picked and grown up in CTM. The colonies were not cloned
because they were mathematically clonal from fusion. Hybrids (including
3NP311-2) were tested for their ability to respond (IL-2 secretion) to the following
proteins/peptides presented by Chb cells: OVA (no response), OVA-NP (positive
response), and NP311-325 peptide (positive response).
Statistical Analyses
All statistical analyses were conducted using GraphPad Prism software.
All statistics shown were generated using two-tailed pairwise Student’s t tests
unless otherwise specified. Asterisks (*) were used to indicate significant
differences observed when comparing indicated groups: * p < 0.05; ** p < 0.01;
*** p < 0.001. Comparisons that were not significant were marked ‘ns’, in which
p ≥ 0.05.

47
CHAPTER III
HOST DNA IS DISPENSABLE IN ALUM RESPONSES1
Introduction
For over 80 years, insoluble aluminum salts (alum) have been safely used
in subunit vaccines to generate protective immunity in hundreds of millions of
people. Alum, in a variety of forms, is a component of many commonly
administered vaccines (listed in Table 1.2 and Fig. 1.3 of Chapter I). Despite its
extensive and continuous use, the mechanism by which alum promotes effective
immune responses eludes complete understanding. In mouse models of
vaccination, alum is widely known to induce T helper 2 (TH2) responses and
subsequent production of IgG1 and IgE antigen-specific antibodies (reviewed in
56). However, the immune mechanisms triggered by alum to cause these effects
are controversial or still unknown.
Our lab has previously reported that alum particles become entrapped by
host chromatin upon intramuscular injection26. Since reporting this finding,
another group and we have shown that DNA released by host cells at the site of
injection can mediate adaptive immune responses to alum vaccines47,49. In these
studies, it was concluded that alum-associated DNA acted as an endogenous
immunostimulatory signal that triggered activation of the DNA-sensing pathway
involving Stimulator of Interferon Genes (STING) within innate immune cells such
1 This chapter is under review for publication in The Journal of Immunology.

48
as inflammatory monocyte-derived dendritic cells (iDCs). Activated iDCs then
facilitated CD4 T cell priming and subsequent IgG1 and IgE antibody responses.
These studies were based, in part, on transgenic CD4 T cell adoptive transfer
experiments as well as loss-of-function experiments that combined alum
vaccines with DNase I enzymes purchased from Roche Diagnostics Corporation.
Notably, there were no reported effects of DNA on alum-induced CD8 T cell
responses. These reports defined a possible role for host DNA in alum biology
that needed further clarification.
The objective of this study was to reconcile the differences observed
between the effects of alum-associated DNA on CD4 versus CD8 T cell
responses to alum vaccines. I began by scrutinizing the quality of commonly
used DNase I reagents that were used to study deficiencies within alum-induced
T cell responses mounted in the absence of host DNA stimuli. Here I report that
DNase treatment impairs T cell responses to alum due to activity of
contaminating proteases within DNase reagents and not DNase enzyme activity.
Furthermore, I observed normal immune responses to alum in mice deficient in
the DNA-sensing molecule STING. We conclude that DNA and DNA-sensing
molecules are not major contributors to adaptive immune responses mounted
against alum vaccines administered to the muscle.

49
Results
Creating model antigens: OVA-NP and mutant nucleoprotein
Others and we recently proposed that co-injected DNase I treatment
impairs primary CD4 T cell priming following immunization with alum adjuvant
because endogenous DNA danger signals are eliminated47,49. To elucidate the
mechanisms behind this phenomenon as it pertains to single doses of
intramuscularly administered human vaccines that contain alum, we analyzed T
cell responses in mice to intramuscular (i.m.) vaccinations containing alum, with
and without DNase treatment, and two model antigens: chicken ovalbumin (OVA)
and influenza A nucleoprotein (NP). OVA is well known to generate robust CD8
T cell responses in C57BL/6 mice to its immunodominant CD8 epitope:
SIINFEKL. However, OVA-specific CD4 T cell responses are inconsistent
among individual C57BL/6 mice, so we instead created an altered OVA antigen
(OVA-NP) that consists of the NP311-325 peptide (an immunodominant epitope of
influenza A nucleoprotein in mice expressing IAb MHC class II molecules)
chemically conjugated to OVA protein. CD4 T cells in C57BL/6 mice respond
consistently to NP311-325 and their response is easily detectable with IAb tetramers
5-9 days after immunization92,111.
Since influenza NP binds influenza RNA, it was possible that NP-bound
RNA could act as a confounding adjuvant within alum + NP vaccines. To ensure
that NP antigen was adjuvant-free, we produced a mutant version of NP (mutNP)
that cannot bind RNA112. The five alanine mutations within mutNP and its
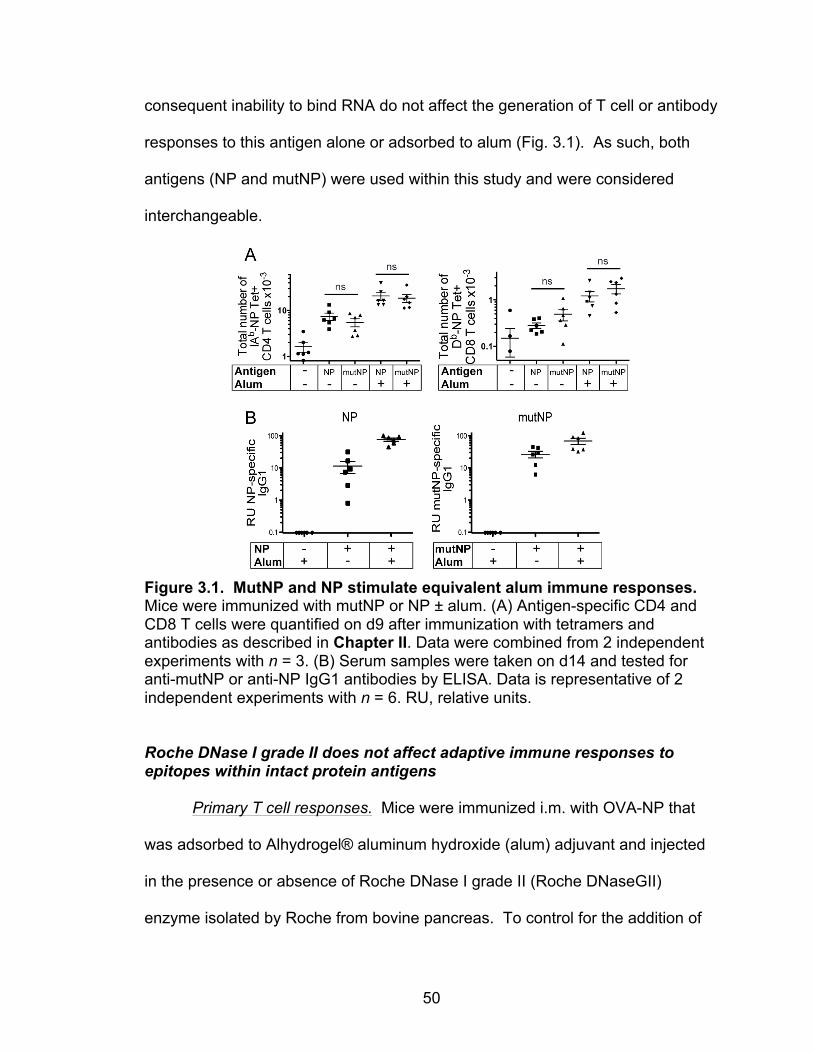
50
consequent inability to bind RNA do not affect the generation of T cell or antibody
responses to this antigen alone or adsorbed to alum (Fig. 3.1). As such, both
antigens (NP and mutNP) were used within this study and were considered
interchangeable.
Figure 3.1. MutNP and NP stimulate equivalent alum immune responses. Mice were immunized with mutNP or NP ± alum. (A) Antigen-specific CD4 and CD8 T cells were quantified on d9 after immunization with tetramers and antibodies as described in Chapter II. Data were combined from 2 independent experiments with n = 3. (B) Serum samples were taken on d14 and tested for anti-mutNP or anti-NP IgG1 antibodies by ELISA. Data is representative of 2 independent experiments with n = 6. RU, relative units.
Roche DNase I grade II does not affect adaptive immune responses to epitopes within intact protein antigens
Primary T cell responses. Mice were immunized i.m. with OVA-NP that
was adsorbed to Alhydrogel® aluminum hydroxide (alum) adjuvant and injected
in the presence or absence of Roche DNase I grade II (Roche DNaseGII)
enzyme isolated by Roche from bovine pancreas. To control for the addition of

51
Roche DNaseGII, which could potentially compete with OVA-NP either as a
foreign antigen or for binding to alum, we added BSA instead of DNase to the
immunizations administered to control mice. The addition of BSA to the alum
vaccines did not significantly affect antigen-specific responses (Fig. 3.2).
Antigen-specific T cell responses were quantified in immunized mice 7 days after
injection. Co-injection of Roche DNaseGII with alum + OVA-NP resulted in a 4-
fold reduction in numbers of NP311-325-specific CD4 T cells in draining popliteal
lymph nodes (Fig. 3.3A). However, Roche DNaseGII did not affect SIINFEKL-
specific CD8 T cell responses (Fig. 3.3B). Unexpectedly, when intact
nucleoprotein (NP) was used as an alum vaccine antigen, we found that CD4
and CD8 T cell responses were unaffected by Roche DNaseGII (Fig 3.3C-D).
Primary antibody responses and secondary CD4 T cell responses. Unlike
past reports on this subject 47,49, IgG1 responses to alum + OVA or mutNP were
resistant to Roche DNaseGII treatment (Fig 3.3E-F). Since alum vaccines are
used to generate protective memory responses, I determined if Roche DNaseGII
treatment altered the effectiveness of alum vaccines to generate protective
immunity by evaluating secondary recall responses upon challenge. Mice were
primed with OVA-NP + alum ± Roche DNaseGII and then later challenged with
mutNP + alum. The change of antigen from OVA-NP to mutNP removed the
effects of anti-OVA-NP neutralizing antibodies from the challenge environment
while retaining the NP311-325 epitope for CD4 T cells. Secondary CD4 T cell
responses were assessed on day 5 after challenge. Roche DNaseGII treatment
of the priming immunization did not affect secondary CD4 T cell responses (Fig.

52
3.4). This indicated that the reduced total numbers of CD4 T cells in the primary
response (reported in Fig. 3.3A) were not impaired from becoming memory cells
capable of mounting robust secondary responses upon challenge.
Figure 3.2. Added BSA has no effect on the magnitude of NP311-325-specific CD4 T cell responses to alum immunization. Mice were immunized with OVA-NP ± alum ± 1 mg BSA or Roche DNaseGII. Antigen-specific CD4 T cells were quantified on d7 after immunization with tetramer and antibodies as described in Chapter II. Data is representative of 2 independent experiments each with n = 3 or 5. Error bars show means ± SEM for each group. Statistical differences were determined using an unpaired Student’s t test; ns indicates p > 0.05.
Figure 3.4. DNase treatment does not impair secondary CD4 T cell responses. Mice were challenged with 10 µg mutNP + 200 µg Alhydrogel® alum at least 25 weeks after primary immunization with OVA-NP ± alum ± Roche DNaseGII or BSA. Cells from dLNs were analyzed 5 days after challenge immunization, as described in Chapter II. Data are cumulative of two independent experiments each with n = 4. Error bars show means ± SEM for each group. Statistical differences were determined using an unpaired Student’s t test; ns indicates p > 0.05.
1
10
ns**
Tota
l num
ber o
fIA
b -N
P T
et+
CD
4 T
cells
x10
-3
Priming immunization:
All groups challenged with mutNP + alum
Treatment − − BSA Roche DNaseGII
Alum − − + + OVA-NP311 − + + +

53
Figure 3.3. DNase treatment does not affect CD4 T cell responses to intact proteins

54
Figure 3.3. DNase treatment does not affect CD4 T cell responses to intact proteins. Mice were immunized with the indicated combinations of antigen ± adjuvant ± 1 mg of Roche DNaseGII or BSA. (A-D,G) Antigen-specific CD4 and CD8 T cells were quantified on d7 after immunization with tetramer and antibodies as described in Chapter II. (E,F) Serum samples were taken 21 d after immunization and tested for the presence of anti-OVA or anti-mutNP IgG1 antibodies by ELISA. RU, relative units. Data in: (A-D) were combined from 4-10 independent experiments each with n = 3-4, (E,F) were combined from two independent experiments each with n = 5-8, and (G) were combined from 2-3 independent experiments each with n = 3-4. Error bars show means ± SEM for each group. Statistical differences were determined using an unpaired Student’s t test; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ns indicates p > 0.05.

55
To examine these disparities further, I tested the effects of Roche
DNaseGII on CD4 T cell responses to two other common vaccine adjuvants:
lipopolysaccharide (LPS) and combined anti-CD40 + poly(I:C). Roche DNaseGII
treatment reduced CD4 T cell responses to OVA-NP combined with either of
these adjuvants (Fig. 3.3E). These were surprising results because neither of
these adjuvants is thought to act via host DNA. My findings suggested that the
Roche DNaseGII preparation might be affecting the immune response in some
unforeseen way.
Commercial DNase I reagents are contaminated with active proteases
To determine if other commercial DNase products had the same effects as
Roche DNaseGII did on alum-induced immune responses, we tested two other
DNase preparations: Sigma-Aldrich DNase I (Sigma DNase, from bovine
pancreas) and Roche DNase I recombinant (Roche Recomb DNase, from Pichia
pastoris). DNase treatments were administered as 2000 Kunitz units per mouse
based on vendor-reported Kunitz units of activity.
These three DNases had different effects on CD4 T cell responses to
alum + OVA-NP (Fig. 3.5A). Sigma DNase had the same effect as Roche
DNaseGII: leading to a more than 4-fold reduction in response. Roche Recomb
DNase, on the other hand, did not significantly impair CD4 T cell responses.
This DNase was recombinantly produced in yeast, however, and is probably
glycosylated with high mannose sugars since it contains two possible N-linked
glycosylation sites. High mannose sugars are known to have adjuvant properties

56
that stimulate T cell responses in mice113,114. Therefore we cannot tell whether
the Roche Recomb DNase simply had no effect on CD4 T cell priming or
possessed confounding adjuvant properties.
My first step in investigating why the DNases had variable effects on alum
T cell priming was to analyze the protein content of each DNase preparation by
SDS-PAGE. The smear of bands that appear in both the Roche DNaseGII and
Sigma DNase lanes indicated that these products are highly impure (Fig. 3.5B).
The lane containing Roche Recomb DNase, on the other hand, had only two
heterogeneous bands at the estimated weight for glycosylated DNase: 31-34
kDa115. In agreement with the gel analyses, mass spectrometry revealed that
Roche DNaseGII and Sigma DNase preparations contained trypsin,
chymotrypsin and other contaminants in addition to DNase while Roche Recomb
DNase mainly contained DNase (Fig. 3.5C). It must be noted that the peptides
found in these samples could not distinguish between chymotrypsinogen and
chymotrypsin. Regardless, chymotrypsinogen is activated by trypsin cleavage
followed by trans-proteolysis and, since trypsin was also present in the
contaminated preps, we assume that the samples contain active chymotrypsin.
I used a broad protease activity assay to confirm that the proteases found
within Roche DNaseGII and Sigma DNase were indeed proteolytically active
(Fig. 3.5D). As expected, Roche Recomb DNase had no protease activity (Fig.
3.6). Next, I created an in vitro assay (Fig. 3.7A) to determine if these active
proteases could destroy the NP311-325 antigenic epitope when it was either
conjugated to OVA or present within intact NP. OVA-NP or NP antigens were

57
Figure 3.5. Commercial DNases are contaminated with active proteases. (A) Mice were immunized with OVA-NP ± alum ± 2000 Kunitz units DNase I reagent from indicated sources. Antigen-specific CD4 T cells were quantified on d7 as before. Data in (A) were combined from 2-3 independent experiments each with n = 3-4. Error bars show means ± SEM. Statistical differences are with respect to the positive control group (treated with OVA-NP + alum + BSA) and were determined using an unpaired t test; ***p ≤ 0.001; ns indicates p > 0.05. (B) SDS-PAGE was performed on the indicated DNAse preparations. Gels were stained with Coomassie® Brilliant Blue dye. (C) DNase products were analyzed by mass spectrometry, as described in Chapter II. Spectra were compared to SwissProt databases to identify protein contents within samples (tan: DNase I, red: protease). Database matches (+) were listed when a sample contained at least 3 distinct peptides from the protein. (D) DNase samples were assessed for protease activity at indicated concentrations using the Pierce Protease Assay Kit. The dotted line indicates the lowest level of detection. Per mg of protein, Roche DNaseGII and Sig-DNase contained protease activities equal to approximately 10 µg and 1 µg trypsin, respectively. Data are representative of (C) 1 or (B,D) 2 independent experiments.

58
incubated for 0, 1, or 21 hours with or without Roche DNaseGII or control BSA.
The antigen mixtures were then fed to antigen-presenting Chb-2.4.4 cells, which
bear IAb, and ‘3NP311-2’ T cell hybridomas specific for IAb/NP311-325. Production
of IL-2 within 24 hours indicated the presence of intact NP311-325 peptide ligand.
The Roche DNaseGII preparation rapidly destroyed NP311-325 when it was
conjugated to OVA (Fig. 3.7B) but not when it was contained in its natural
context, intact NP protein (Fig. 3.7C). Notably, Roche DNaseGII destroyed the
epitope even at the 0 h time point, meaning that the DNase was mixed with OVA-
NP and immediately added to the cell culture. One possible explanation for this
result is that Roche DNase GII preparation destroyed the epitope within the very
short amount of time between when they were combined and when the
combination was fed to cells in culture. Alternatively, perhaps the cell culture
medium (containing 10% FBS) did not fully quench the protease activity within
the Roche DNaseGII preparation, allowing for continued antigen cleavage during
the 24 hours in cell culture. These observations suggest that NP311-325-specific
CD4 T cell impairment by Roche DNaseGII was due to rapid antigen destruction
by contaminating proteases rather than any DNase effect on alum biology.
In conclusion, none of these three commercial DNase reagents (Roche
DNaseGII, Roche Recomb DNase, and Sigma DNase) can be used as
unequivocal measurements of the effects of DNase enzyme either because they
contain contaminating proteases or they include high mannose glycosylations
that act as confounding adjuvants. As such, careful examination of DNase
reagents should be done prior to their use in certain experiments.
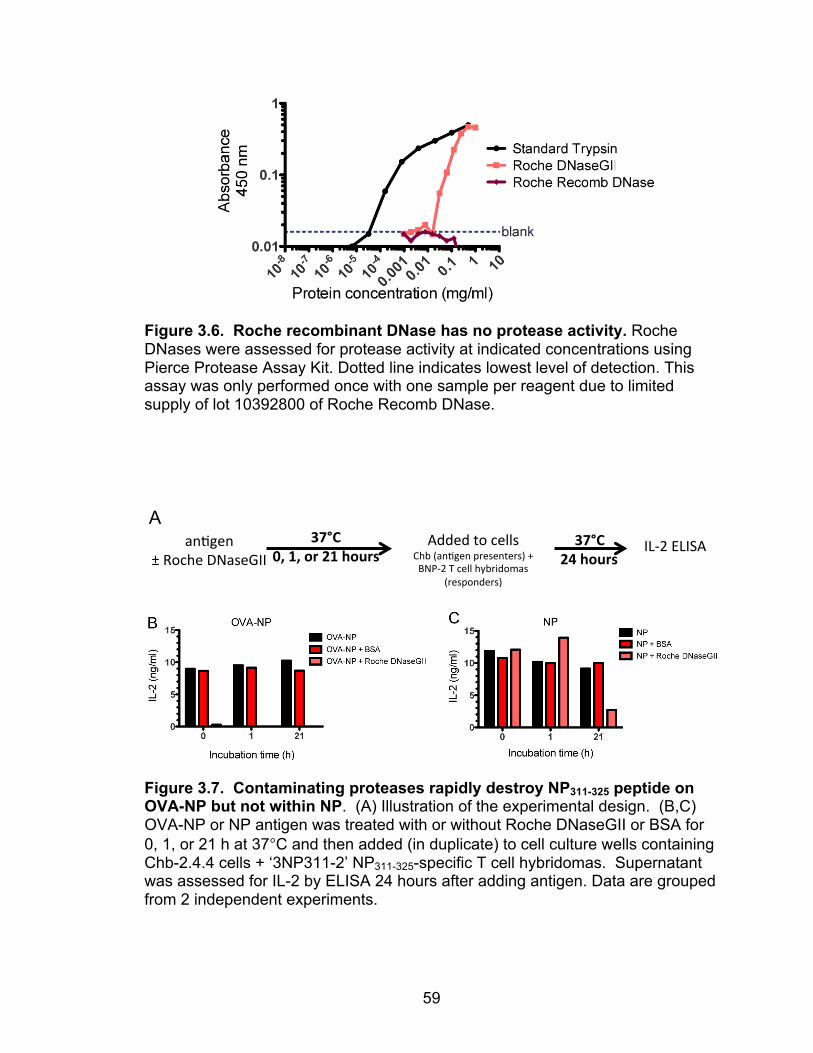
59
Figure 3.6. Roche recombinant DNase has no protease activity. Roche DNases were assessed for protease activity at indicated concentrations using Pierce Protease Assay Kit. Dotted line indicates lowest level of detection. This assay was only performed once with one sample per reagent due to limited supply of lot 10392800 of Roche Recomb DNase.
Figure 3.7. Contaminating proteases rapidly destroy NP311-325 peptide on OVA-NP but not within NP. (A) Illustration of the experimental design. (B,C) OVA-NP or NP antigen was treated with or without Roche DNaseGII or BSA for 0, 1, or 21 h at 37°C and then added (in duplicate) to cell culture wells containing Chb-2.4.4 cells + ‘3NP311-2’ NP311-325-specific T cell hybridomas. Supernatant was assessed for IL-2 by ELISA 24 hours after adding antigen. Data are grouped from 2 independent experiments.
an#gen&±&Roche&DNaseGII&
37°C%0,%1,%or%21%hours%
Added&to&cells&Chb&(an#gen&presenters)&+&BNP>2&T&cell&hybridomas&
(responders)&
37°C%24%hours%
IL>2&ELISA&
A

60
Proteases are predominantly responsible for the effects of contaminated DNases on CD4 T cell responses to alum
Upon extending my search for a commercially available mammalian-
produced and protease-free DNase I, I found that Worthington Protease- and
Ribonuclease-Free DNase I (Worth DNase, from bovine pancreas) is
satisfactorily pure as established by SDS-PAGE (Fig. 3.8C), mass spectrometry
(Fig. 3.8F), and protease activity (Fig. 3.8E). As with the other commercial
DNases from before, I compared the effects of Worth DNase and Roche
DNaseGII on the vaccine adjuvant activity of alum. For this comparison,
however, I did not administer the DNases based on vendor-reported Kunitz units
of activity. Instead, I measured their enzymatic activities myself by measuring
their abilities to cleave mouse genomic DNA within 30 minutes at 37°C. I found
that while Worth DNase and Roche DNaseGII were nearly equivalent in DNase
activity on a per milligram of protein basis (823 µg Worth = 1 mg Roche) (Fig.
3.8B), the activity in vendor-reported Kunitz units per milligram was surprisingly
different: 8000 Worthington Kunitz units were equal to 2263 Roche Kunitz units.
When co-injected with alum + OVA-NP, Worth DNase only marginally impaired
the CD4 T cell response by 1.6-fold, whereas Roche DNaseGII reduced the CD4
T cell response 5-fold as before (Fig 3.9A). Worth DNase did not affect CD8 T
cell responses (Fig. 3.9B).
My previous experiment established that each milligram of Roche
DNaseGII was contaminated with proteases that have cumulative activities
equivalent to either 10 µg trypsin or chymotrypsin (Fig. 3.5D). To determine if
this contamination was sufficient to reduce CD4 T cell responses, alum vaccines
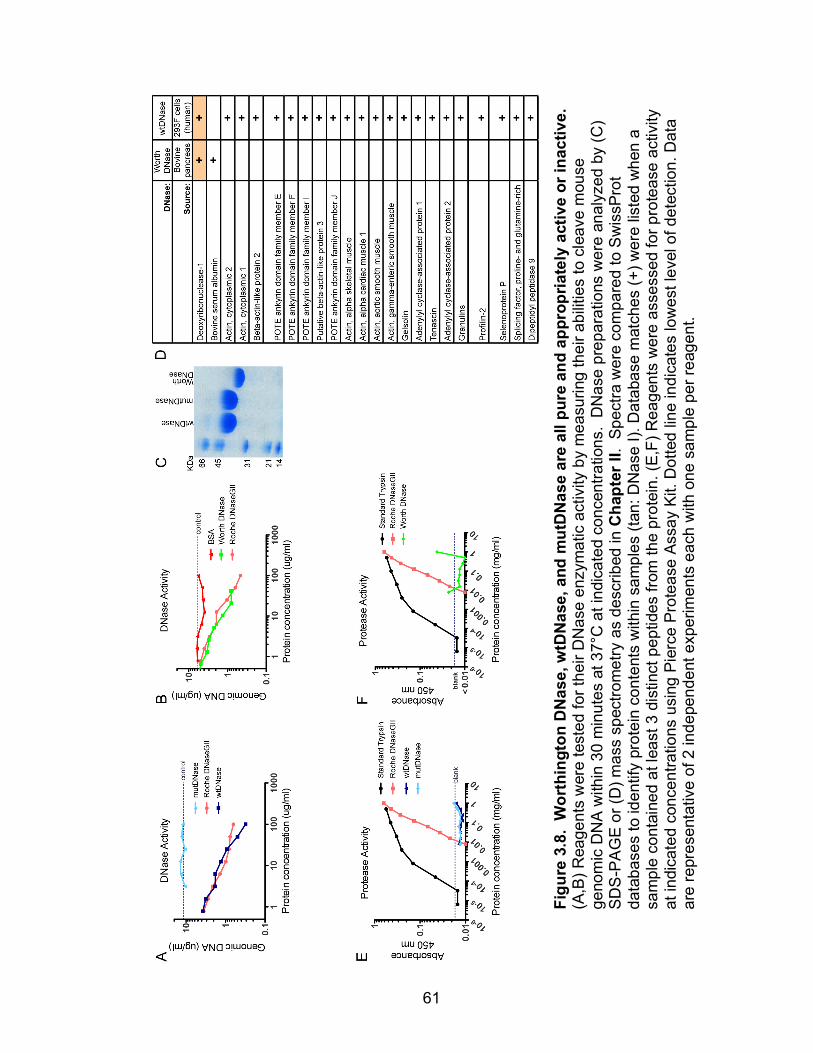
61
Figu
re 3
.8.
Wor
thin
gton
DN
ase,
wtD
Nas
e, a
nd m
utD
Nas
e ar
e al
l pur
e an
d ap
prop
riate
ly a
ctiv
e or
inac
tive.
(A
,B) R
eage
nts
wer
e te
sted
for t
heir
DN
ase
enzy
mat
ic a
ctiv
ity b
y m
easu
ring
thei
r abi
litie
s to
cle
ave
mou
se
geno
mic
DN
A w
ithin
30
min
utes
at 3
7°C
at i
ndic
ated
con
cent
ratio
ns.
DN
ase
prep
arat
ions
wer
e an
alyz
ed b
y (C
) SD
S-P
AG
E or
(D) m
ass
spec
trom
etry
as
desc
ribed
in C
hapt
er II
. Sp
ectra
wer
e co
mpa
red
to S
wis
sPro
t da
taba
ses
to id
entif
y pr
otei
n co
nten
ts w
ithin
sam
ples
(tan
: DN
ase
I). D
atab
ase
mat
ches
(+) w
ere
liste
d w
hen
a sa
mpl
e co
ntai
ned
at le
ast 3
dis
tinct
pep
tides
from
the
prot
ein.
(E,F
) Rea
gent
s w
ere
asse
ssed
for p
rote
ase
activ
ity
at in
dica
ted
conc
entra
tions
usi
ng P
ierc
e Pr
otea
se A
ssay
Kit.
Dot
ted
line
indi
cate
s lo
wes
t lev
el o
f det
ectio
n. D
ata
are
repr
esen
tativ
e of
2 in
depe
nden
t exp
erim
ents
eac
h w
ith o
ne s
ampl
e pe
r rea
gent
.
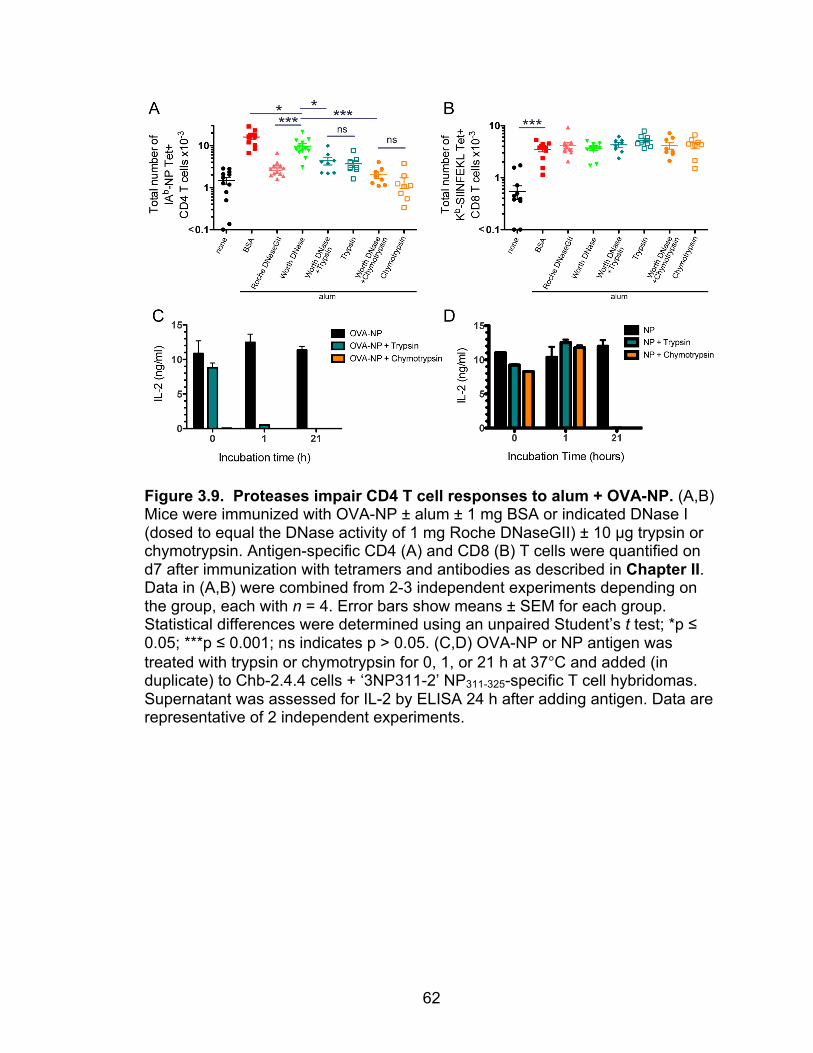
62
Figure 3.9. Proteases impair CD4 T cell responses to alum + OVA-NP. (A,B) Mice were immunized with OVA-NP ± alum ± 1 mg BSA or indicated DNase I (dosed to equal the DNase activity of 1 mg Roche DNaseGII) ± 10 µg trypsin or chymotrypsin. Antigen-specific CD4 (A) and CD8 (B) T cells were quantified on d7 after immunization with tetramers and antibodies as described in Chapter II. Data in (A,B) were combined from 2-3 independent experiments depending on the group, each with n = 4. Error bars show means ± SEM for each group. Statistical differences were determined using an unpaired Student’s t test; *p ≤ 0.05; ***p ≤ 0.001; ns indicates p > 0.05. (C,D) OVA-NP or NP antigen was treated with trypsin or chymotrypsin for 0, 1, or 21 h at 37°C and added (in duplicate) to Chb-2.4.4 cells + ‘3NP311-2’ NP311-325-specific T cell hybridomas. Supernatant was assessed for IL-2 by ELISA 24 h after adding antigen. Data are representative of 2 independent experiments.

63
were given with 10 µg trypsin or chymotrypsin, with or without Worth DNase. I
found that both of these proteases could independently reduce NP311-325-specific
CD4 T cell responses whether or not the Worth DNase was also present (Fig.
3.9A). Notably, chymotrypsin was the more potent treatment. None of the
preparations affected CD8 T cell responses (Fig 3.9B).
In vitro antigen-destruction studies confirmed that trypsin treatment was
less efficient than chymotrypsin at destroying the NP311-325 epitope within OVA-
NP (Fig. 3.9C). Chymotrypsin could destroy the NP311-325 epitope by cleaving
after Y313, a putative anchor residue for binding the peptide to IAb, and/or after
L315 (Fig. 3.10). It is unclear whether or not trypsin can destroy the epitope by
cleaving after R317 because the relevant bond may or may not be protected by
the following proline residue116,117. Trypsin cleavage would, however, completely
sever NP311-325 peptides from OVA molecules by cleaving on the C-terminal side
of the K325 (Fig. 3.10). This would result in disassociated, but nonetheless
intact, peptide antigen that could potentially be loaded into MHC II. Interestingly,
the NP311-325 epitope within intact NP protein was protected from protease
cleavage for at least one hour at 37°C (Fig. 3.9D). Theoretically, the CD8-
stimulating SIINFEKL epitope from OVA could be attacked by either trypsin or
chymotrypsin (Fig. 3.10). However, CD8 T cell responses to SIINFEKL within
intact OVA were not impaired by protease treatments (Fig. 3.9B). Therefore, the
SIINFEKL peptide within intact OVA must be resistant to protease cleavage
within the timeframe and biological environment of i.m. vaccinations.

64
Figure 3.10. Trypsin and chymotrypsin cleavage sites in NP311-325 and SIINFEKL peptides.
Taken together, my data offer an explanation for how impure Roche
DNaseGII and Sigma DNase affect CD4 T cell priming by alum: contaminating
proteases destroy or remove the CD4 epitope of interest. We observed only a
slight inhibitory effect of protease-free Worth DNase treatment within alum
vaccines on CD4 T cell responses, suggesting that DNase itself might indeed
have a small effect on alum-containing vaccines.
DNase enzymatic activity does not impair T cell responses to alum
To test if this DNase effect was due to the protein’s enzymatic activity, I
produced enzymatically active (wtDNase) and inactive (mutDNase) recombinant
bovine DNase I proteins using an HEK293 mammalian expression system with
help from Lori Sherman at the UCCC Protein Production Core. MutDNase
differed from wtDNase by three substitutions: R111A, D212A, and H252A118. I
confirmed that the wtDNase enzymatic activity per milligram protein was
QVYSLIRPNENPAHK-GGGC-OVA
Underlined = known or predicted IAb or Kb binding sites
?
flexible'linker'with'cysteine'NP3115325'pep9de'
Trypsin cleavage sites
Chymotrypsin cleavage sites
primary'amine'groups'(lysine'residues)'on'OVA'
OVA-SI INFEKL-OVA

65
equivalent to that of Roche DNaseGII (Fig. 3.8A) and that the mutDNase had no
enzymatic activity at 100 µg/ml (Fig. 3.8A) and 1 mg/ml (Fig. 3.11). As before, I
assessed the purity of these homemade DNases by SDS-PAGE (Fig. 3.8C),
mass spectrometry (Fig. 3.8D), and protease activity (Fig. 3.8E). The
mammalian-expressed recombinant wtDNase and mutDNase both had higher
molecular weights than bovine pancreas-sourced DNases (Fig. 3.8C). We
hypothesize that this difference is caused by variations in protein glycosylation
patterns used by different mammalian cell lines119: HEK293 may glycosylate
proteins differently than primary bovine pancreas cells.
Figure 3.11. MutDNase is inactive up to 1 mg/ml concentration. DNases were assessed for enzymatic activity on 800 ng of mouse genomic DNA within 30 minutes at 37°C at indicated concentrations (2-fold dilutions from 1 mg/ml). Data are representative of 3 independent experiments each with single replicates.

66
Co-injection of wtDNase or mutDNase with alum vaccines yielded the
same numbers of antigen-specific CD4 or CD8 T cells (Fig. 3.12). Curiously, both
the control mutDNase and the active wtDNase treatments reduced CD4 T cell
responses by approximately 2-fold when compared with responses induced in
the absence of added protein or with added BSA. This dampening effect was
generally consistent between wtDNase, mutDNase, and Worth DNase, though
there were a few experiments in which Worth DNase did not significantly reduce
CD4 T cell priming (not shown). We hypothesize that the small effect of DNase
may be caused not by enzymatic activity, but by some biochemical property of
the DNase proteins, such as altered competition with the model antigen for
adherence to alum. Regardless of this issue, the comparison of T cell responses
in mutDNase- and wtDNase-treated mice indicates that DNase enzymatic activity
does not affect T cell responses to alum plus antigen.
Figure 3.12. DNase activity does not impair T cell responses to alum + OVA-NP. Mice were immunized with OVA-NP ± alum ± 1 mg indicated treatment. Antigen-specific CD4 (A) and CD8 (B) T cells were quantified on d7 as described in Chapter II. Data were combined from 2-4 independent experiments each with n = 4-5. Error bars show means ± SEM for each group. Statistical differences were determined using an unpaired Student’s t test; ***p ≤ 0.001; ns indicates p > 0.05.

67
STING may be dispensable in alum vaccine responses
In another approach to measure the effects of host DNA on alum’s
adjuvant activity, I evaluated the role of STING, a cytoplasmic DNA-sensing
molecule, on alum vaccination. Previous experiments had suggested that
defects in the STING signaling pathway reduced alum’s ability to prime CD4 T
cell responses to OVA or the 3K peptide47,49. Host DNA is thought to attach to
and travel with alum crystals as they gain entrance into cytosolic spaces by
damaging and ultimately rupturing lysosomes, a phenomenon reported by
Hornung et al. in 200864. Cytoplasmic host DNA would then activate the
intracellular DNA-sensing pathway involving STING, Tbk1, and Irf3, resulting in
inflammatory responses such as type I interferon production and NFκB
activation64,120–122.
I repeated the alum vaccination experiments with STING-deficient mice.
Contrary to previous reports47,49, I found that both CD4 and CD8 T cells in
STING-deficient mice respond normally to the OVA-NP plus alum vaccine (Fig.
3.13A-B). Alum is known to induce substantial T follicular helper (TFH)
differentiation within CD4 cells80,88,123–125, an effect that is assumed to be critical
for the generation of protective antibodies by alum vaccines. Since both quantity
and quality of responding T cells contributes to alum’s effectiveness as an
adjuvant, I tested STING’s effect on T cell TFH differentiation. I found that alum-
primed CD4 T cells in STING-deficient mice differentiate into TFH cells at the
same frequency as those in WT mice (Fig. 3.13C). Lastly, I confirmed that IgG1
responses to alum are also STING-independent (Fig. 3.13D), as others have

68
reported. Overall, these findings suggest that the STING signaling pathway is
dispensable for adaptive immune responses to certain antigens, such as OVA-
NP, when delivered with alum.
Figure 3.13. STING is dispensable in adaptive immune responses to alum. WT or STING-deficient mice were immunized with OVA-NP + 1 mg BSA ± alum. Antigen-specific CD4 (A) and CD8 (B) T cells were quantified on d7 as described in Chapter II. (D) Antigen-specific CD4 T cells were assessed for frequency of TFH phenotype (PD1+ Bcl6+), compared to TFH frequency observed in CD44high CD4 T cells within PBS control mice. Data were combined from 5 (A) or 2 (B,C) independent experiments each with n = 3-4. (D) Serum samples were taken on d21 after immunization and tested for the presence of anti-OVA IgG1 antibodies by ELISA. RU, relative units. Data are from 1 experiment, n = 6. Error bars show means ± SEM for each group. Statistical differences were determined using an unpaired Student’s t test; ns indicates p > 0.05.
D C

69
Discussion
Findings in this study contrast those reported in previous publications that
examine the role for DNA and DNA-sensing molecules in alum biology. Though
extracellular host DNA is known to be an inflammatory danger-associated
molecular pattern, we found that it is dispensable in the generation of cellular and
humoral immune responses following i.m. alum immunization. Given the
conflicting results now published on the subject, the role of host DNA and the
STING pathway in alum-induced T cell activation remains unclear, similar to
alum’s relationships with several other inflammatory pathways.
The release of host DNA by alum is attributed to the massive cell death
observed at the site of i.m. injection47. The immune system recognizes necrotic
cell death by detecting the release of intracellular contents and rapidly mobilizing
an ensuing inflammatory response. The intracellular contents such as genomic
dsDNA, heat-shock proteins, uric acid, and high mobility group box 1 (HMGB1)
proteins act as endogenous adjuvants that have been called damage-associated
molecular patterns (DAMPs) (reviewed in 95). Our data suggest that alum
responses do not depend on DAMP signaling from host DNA, even though
genomic dsDNA has been shown to have adjuvant activity126. However, alum-
induced cell death releases a variety of DAMPs in addition to dsDNA. We
previously showed that alum nodules contain host chromatin26 which, in addition
to nucleic acid, contains DAMPs such as HMGB1. We show that host DNA is not
required for alum responses, but perhaps alum is releasing DAMPs that stimulate
the immune system redundantly even if one particular stimulus is absent.

70
Alum is known to induce many inflammatory pathways including the
creation of an antigen depot within the body27,127,128, activation of APCs
(reviewed in 56), inflammasome activation64–67, and release of endogenous
danger signals such as uric acid and host DNA26,47,59. Many of these pathways
have been implicated in or dismissed as contributors to alum’s adjuvant activities,
depending on the vaccine scenario. It is likely that the biochemical properties of
antigens, antigen dose, immunization schedule, and differences in precursor T
cell frequencies for given epitopes all affect which types of immunostimulatory
pathways triggered by alum vaccines ultimately contribute to the immune
response129–133. In this case, since the C57BL/6 murine CD4 T cell response to
NP311-325 within the OVA-NP antigen is quite robust, perhaps alum no longer
requires the STING pathway to be operative to mount a sufficient immune
response. Alternatively, responses to OVA (without NP peptide) and alum need
stronger instigation in the form of STING pathway activation.
In conclusion, we suggest that DNA may not play a prominent role in
mediating adaptive immune responses to alum and that contaminated reagents
may have led to incorrect interpretations of past experiments involving DNase
reagents. The correction of the literature that is presented here may help improve
future studies to elucidate the mechanism of aluminum salts as vaccine
adjuvants, an endeavor that remains paramount to understanding why alum
vaccines have successfully protected hundreds of millions of people from
disease and how they may be improved for future vaccination strategies.

71
CHAPTER IV
EFFECTS OF ALUM ON ANTIGEN PRESENTING CELLS
Introduction
The experiments in Chapter III demonstrated that Roche DNaseGII is
contaminated with active proteases that destroy the CD4 epitope on OVA-NP.
This discovery altered the interpretations of all Roche DNaseGII experiments
conducted before the contamination was discovered (years of work). This caveat
nullified many experiments because they are uninterpretable; these experiments
were excluded from this thesis. However, some were still of some use if the
following argument is acceptable. Since some of the peptide epitopes
(SIINFEKL and NP311-325) within intact proteins (OVA and NP, respectively) were
resistant to protease digestion within the relevant conditions of i.m. vaccination,
this means that alum vaccines administered with Roche DNaseGII indeed
delivered alum + antigen, as intended. Only analyses of CD4 T cell responses to
conjugated peptides (NP311-325) were compromised due to possible protease
destruction of their protease-susceptible epitope. Therefore, if we focused on
immune responses other than NP311-325-specific CD4 T cells, experiments using
Roche DNaseGII can still be cautiously interpreted regarding the effects of
DNase enzymic activity on immune responses to alum vaccines. This chapter
focuses on antigen presenting cell activation by alum and the effect of DNase
treatment on this process.

72
Antigen presenting cells (APCs) are instrumental in the initiation of
adaptive immune responses. They educate lymphocytes differently depending
on the type of foreign threat that is detected, ultimately guiding different adaptive
immune responses. For this reason, it is imperative to understand which APCs
react to aluminum salt (alum) adjuvants in order to understand how alum works
to generate protective immune responses.
CD11b+ conventional dendritic cells (cDCs) and inflammatory monocytes
(iMonos) have been implicated as the primary APC subsets that facilitate
immune responses to intramuscularly administered alum plus antigen58–61.
CD11b+ cDCs were found to be the most numerous alum antigen presenters
because they migrate easily from the muscular injection site to the dLN61. At the
same time, while only a small fraction of iMonos migrate from the muscle to the
draining lymph nodes (dLNs), iMonos were found to be more potent to induce
IFNγ-producing T cells on a per cell basis61.
Though these APC subsets have been identified, it is still unclear how
alum activates these cells within the muscle, since a variety of suggested
inflammatory pathways have been found to be dispensable for alum responses
(discussed in Chapter III). A specific receptor for which alum is a ligand has not
been identified. However several groups have proposed that alum is detected by
APCs through strong interactions between alum crystals and APC plasma
membranes. This can occur in two ways. Flach et al. reported that alum crystals
directly interact with dendritic cell plasma membranes and subsequent lipid
sorting, abortive phagocytosis, and antigen uptake may be responsible for DC

73
activation64,71,134. Alternatively, Kool et al. found that, when injected i.p., alum
increases levels of uric acid in the intraperitoneal space, which was important for
iMono migration to dLNs59. Uric acid forms monosodium urate (MSU) crystals
when released into extracellular spaces, and MSU crystals have been shown to
interact strongly with cholesterol and other lipids on the plasma membrane,
resulting in dendritic cell activation70.
In this study, I aimed to identify molecular stimuli and signaling pathways
that are induced by alum adjuvant within antigen-loaded APC subsets. My
approach was to use fluorescent antigen to identify and sort antigen-bearing
APCs 24 hours after i.m. immunization with antigen alone or antigen plus alum.
The RNA within these APC subsets were then sequenced and compared to gain
insight regarding the molecular pathways that are activated by alum. I found
that, curiously, alum does not seem to affect the types or ratios of APCs loaded
with antigen when compared to APCs that took up soluble antigen alone.
However, there were substantial up and down regulations of inflammation-
associated mRNA sequences among alum-treated and -untreated APCs.
Overall, alum appears to act as an inflammatory stimulus through a variety of
pathways, resulting in wide scale immune activation.

74
Results
Technical considerations when using fluorescent antigens
Choice of fluorophore for antigen tracking affects detection of antigen-
loaded cell subsets. The first step to achieve the goals of this project was to sort
antigen-loaded APCs from dLNs of alum-immunized mice. To make sure the
reports in the literature were repeatable in my hands, I began by immunizing
mice with fluorescent antigen ± alum and analyzing antigen-bearing cells by flow
cytometry. Through the course of this study, I used every combination of OVA or
NP protein antigen conjugated to FITC, Alexa Fluor 488 (A488), or Alexa Fluor
647 (A647).
It is important to note that choice of fluorophore for antigen-tracking
experiments seems to greatly affect the distribution of cell populations that
appear to be antigen+ and the relative brightness of fluorescent antigen within
antigen-bearing cells (Fig. 4.1). OVA-FITC was difficult to rely upon because it
was very dim within antigen-bearing cells when analyzed by flow cytometry (Fig.
4.1, top) and there is often high background within the FL1 FITC channel (488
nm excitation, emission: 530 nm) due to cellular autofluorescence of flavin
nucleotides, which are excited by and emit similar wavelengths of light
(excitation: 430-500 nm, emission: 530-550 nm). OVA- or NP-A488 was very
bright, but similar high background issues were a concern. OVA- and NP-A647
seemed to be the most reliable reagents because loaded cells were quite bright
(shifted up to three decades) and there was minimal background. The most

75
concerning analytical difference observed when using different fluorophores was
that B cells were (with A488 and A647) or were not (with FITC) appearing to
make up a substantial population of antigen-loaded cells in dLN 24 hours after
alum immunization (Fig. 4.1). B cells (B220+) make up the majority of the
CD11b- population in the right-most panels. As discussed in the next sections, B
cells are generally loaded with low amounts of antigen, compared to DCs or
iMonos. Since FITC was quite dim as a fluorescent marker, it is possible that
minimally-loaded B cells were registering as FITC- instead of FITClow.
Figure 4.1. Choice of fluorescent marker affects the detection of antigen-bearing cells by flow cytometry. Cells from dLNs were analyzed by flow cytometry 24 hours after mice were immunized i.m. with PBS or 5-20 µg OVA or NP that was conjugated to indicated fluorophores (FITC, A488, or A647) ± 200 µg alum ± 1 mg BSA. After gating on live cell singlet events, MHC II+ fluorophore+ cells were selected and plotted by CD11b and MHC II to distinguish general APC subsets: DCs (CD11b+ MHC IIhigh), iMonos (CD11b+), and B cells (CD11b-). Data are representative of at least 2 independent experiments per fluorescent marker, n = 2-3.
0 101 102 103
0
101
102
103
16 alum ova647 BSA.fcs…OVA647
<FL 1 Log>: FITC-MHCII
<FL
6 Lo
g>: P
B-C
D11
b
8.22
56
32.8
0 101 102 103
<FL 8 Log>: APC-MHCII
0
101
102
103
<FL
1 Lo
g>: F
ITC
-OV
A
0.11
0 101 102 103
<FL 8 Log>: APC-MHCII
0
101
102
103
<FL
1 Lo
g>: F
ITC
-OV
A
0.16
0 101 102 103
<FL 8 Log>: APC-MHCII
0
101
102
103
<FL
1 Lo
g>: F
ITC
-OV
A
0.23
PBS Antigen Antigen
+ alum ± BSA
0 101 102 103
<FL 8 Log>: APC-MHCII
0
101
102
103
<FL
1 Lo
g>: F
ITC
-OV
A48
8
1.28
0 101 102 103
<FL 8 Log>: APC-MHCII
0
101
102
103
<FL
1 Lo
g>: F
ITC
-OV
A48
8
6.34
0 101 102 103
<FL 8 Log>: APC-MHCII
0
101
102
103
<FL
1 Lo
g>: F
ITC
-OV
A48
8
6.24
0 101 102 103
<FL 1 Log>: FITC-MHCII
0
101
102
103
<FL
8 Lo
g>: A
PC
-ova
647
1.2
0 101 102 103
<FL 1 Log>: FITC-MHC II
0
101
102
103
<FL
8 Lo
g>: A
PC
-NP
647
0.93
0 101 102 103
<FL 1 Log>: FITC-MHC II
0
101
102
103
<FL
8 Lo
g>: A
PC
-NP
647
0.084
MHC$class$II$
An,g
en/Fluor$
FITC
A488
A647
0 101 102 103
<FL 8 Log>: APC-MHCII
0
101
102
103
<FL
6 Lo
g>: P
B-C
D11
b
3.14
14.5
73.4
0 101 102 103
0
101
102
103
5 4C OVA488alum.fcs…NP488+
<FL 8 Log>: APC-MHCII
<FL
6 Lo
g>: P
B-C
D11
b
46.8
8.52
29.9
CD11b$
Antigen+ subsets

76
Artifacts of flow cytometry: possible B cell + iMono doublets. Another
technical difficulty was that often up to 40% of iMonos (identified as described in
Table 4.1) stained positive for B220, the CD45R isoform that exists on B cell
surfaces. This substantial proportion of B220+ iMonos has never been reported,
so I am hesitant to believe they are biologically real cells. Instead, they are likely
B cell/iMono doublets, especially because they are loaded with an intermediate
amount of fluorescent antigen compared to iMonos (higher) and B cells (lower)
(Fig. 4.2C). Doublets were excluded to the greatest extent possible without
sacrificing singlet events (as shown in Fig. 4.3A), but perhaps the small size of
coupled B cells + iMonos was within this singlet gate. This exemplifies a
limitation of using flow cytometry to analyze heterogeneous cell populations.
Bearing in mind the technical caveats described in this section, Figures
4.2 and 4.3 are generally representative of all the APC experiments performed
with either OVA or NP antigen, conjugated to A488, A647, or FITC (10
independent experiments).
Alum adjuvant effect on antigen-loaded APCs
Previous reports have established that peak numbers of antigen-loaded
APCs appear in dLNs 24 hours after i.m. alum vaccination60,61. Using flow
cytometry, I identified APC subsets within dLN of mice that were injected with
PBS or OVA-A647 ± alum ± BSA. APC subsets can vary in nomenclature
throughout the literature, so the phenotypes and corresponding APC subset
names that are used within this section are listed in Table 4.1.
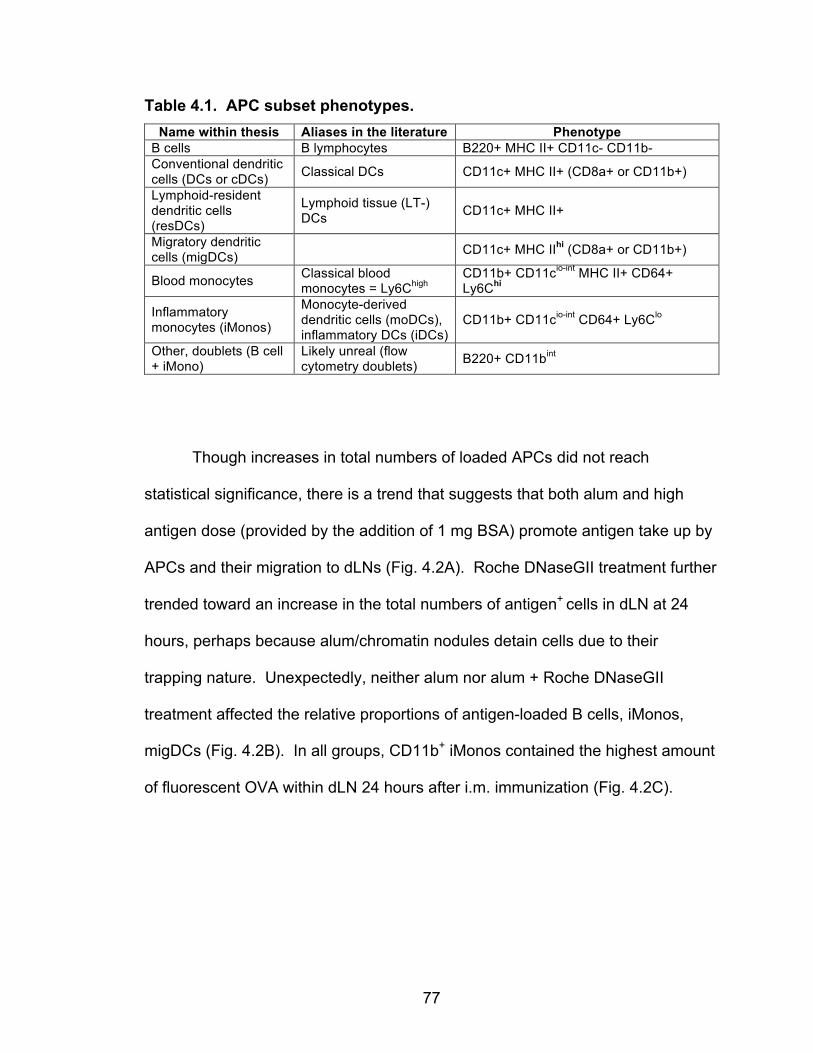
77
Table 4.1. APC subset phenotypes. Name within thesis Aliases in the literature Phenotype
B cells B lymphocytes B220+ MHC II+ CD11c- CD11b- Conventional dendritic cells (DCs or cDCs) Classical DCs CD11c+ MHC II+ (CD8a+ or CD11b+)
Lymphoid-resident dendritic cells (resDCs)
Lymphoid tissue (LT-) DCs CD11c+ MHC II+
Migratory dendritic cells (migDCs) CD11c+ MHC IIhi (CD8a+ or CD11b+)
Blood monocytes Classical blood monocytes = Ly6Chigh
CD11b+ CD11cio-int MHC II+ CD64+ Ly6Chi
Inflammatory monocytes (iMonos)
Monocyte-derived dendritic cells (moDCs), inflammatory DCs (iDCs)
CD11b+ CD11cio-int CD64+ Ly6Clo
Other, doublets (B cell + iMono)
Likely unreal (flow cytometry doublets) B220+ CD11bint
Though increases in total numbers of loaded APCs did not reach
statistical significance, there is a trend that suggests that both alum and high
antigen dose (provided by the addition of 1 mg BSA) promote antigen take up by
APCs and their migration to dLNs (Fig. 4.2A). Roche DNaseGII treatment further
trended toward an increase in the total numbers of antigen+ cells in dLN at 24
hours, perhaps because alum/chromatin nodules detain cells due to their
trapping nature. Unexpectedly, neither alum nor alum + Roche DNaseGII
treatment affected the relative proportions of antigen-loaded B cells, iMonos,
migDCs (Fig. 4.2B). In all groups, CD11b+ iMonos contained the highest amount
of fluorescent OVA within dLN 24 hours after i.m. immunization (Fig. 4.2C).

78
Figure 4.2. Alum increases numbers of antigen-loaded APCs, but does not affect loaded cell type or degree of antigen uptake. Cells from dLNs were analyzed by flow cytometry 24 hours after mice were immunized i.m. with 10 µg OVA-A647 ± 200 µg alum ± 1 mg BSA or Roche DNaseGII, as described in Chapter II. Panels show (A) total number of OVA-A647+ cells, (B) relative proportions of antigen-loaded APCs by cell phenotype, and (C) MFI of OVA-A647 within indicated cell subsets. Data are representative of 2 independent experiments, n = 3. Error bars show means ± SEM for each group. Inflammatory monocytes take up the most antigen upon intramuscular injection
Similarly, when using NP-A647 fluorescent antigen, I found that iMonos
contained the most antigen on a per cell basis (Fig. 4.3C-D) and that both B cells
and iMonos were the most numerous antigen-loaded subsets within the dLN at
24 hours (Fig. 4.3E). It should be noted that Fig. 4.3E is comprised of cumulative
data from three independent experiments in which it varied whether B cells or
iMonos were the largest antigen-bearing population. Given the NP-A647+ gate
position, it is possible that antigen non-bearing B cells were wrongly included
# Ag+ cells
PBS
OVA647
OVA647 +
alum
OVA647 +
alum
+ BSA
OVA647 +
alum
+ DNas
e
0
100
200
300
400
Tota
l num
ber o
fO
VA-A
647+
cel
lsin
dLN
x10
-3
B220+%36%%
CD11b+%22%%
CD11b+%B220+%17%%
CD11c+%6%%
other%19%%
OVA8A647%
B220+%38%%
CD11b+%23%%
CD11b+%B220+%15%%
CD11c+%7%%
other%17%%
OVA8A647%+%alum%+%BSA%
Treatment:% −% −% −% BSA$ Roche$DNaseGII$
Adjuvant:% −% −% alum$ alum$ alum$
OVA8A647:% −% +$ +$ +$ +$
B220+%CD11b+%CD11b+%B220+%CD11chi%Total%anIgen+%cells%
OVA6A647$
A B
C
B220+%37%%
CD11b+%25%%
CD11b+%B220+%12%%
CD11c+%8%%
other%18%%
OVA8A647%+%alum%+%Roche%DNaseGII%
OVA6647$ OVA6647$+$alum$+$BSA$ OVA6647$+$alum$+$Roche$DNaseGII$

79
even though the gate was set comfortably above background fluorescence within
PBS treated samples (not shown).
Though OVA-FITC was dim for identifying fluorescent antigen ex vivo, this
reagent was used for a time course to identify when and which antigen-loaded
APC subsets appeared in the dLN after i.m. alum injection. This experiment
suggests that iMonos are the cells that rapidly (within 6 hours after injection)
acquire antigen and traffic to the dLN, while migDCs are slower to appear in
dLNs (Fig. 4.4). An important caveat is that my experimental approach did not
allow me to distinguish between cells that took up antigen at the site of injection
and brought it to the dLN from cells that may have acquired the antigen while in
the dLN. As time progressed after vaccination, there were fewer antigen-loaded
iMonos and more loaded migDCs within dLNs (Fig. 4.4). It is possible that
antigen is being passed off among APC subsets within the dLN or that DCs are
slower to migrate from the site of injection to dLNs than iMonos.
RNA sequencing identifies alum-induced inflammatory pathways
I next sought to identify inflammatory pathways affected by alum in an
unbiased and broad manner by using RNA sequencing to examine changes in
gene expression after alum injection. Groups of mice (n = 10-20) were
immunized i.m. with fluorescent antigen (OVA-A647) either alone or adsorbed to
alum. Antigen-loaded APCs (iMonos, DCs, and B cells) were sorted 24 hours
after injection and their mRNA was sent to Expression Analysis, Inc., for RNA
sequencing. One drawback of this study was that samples were sent without
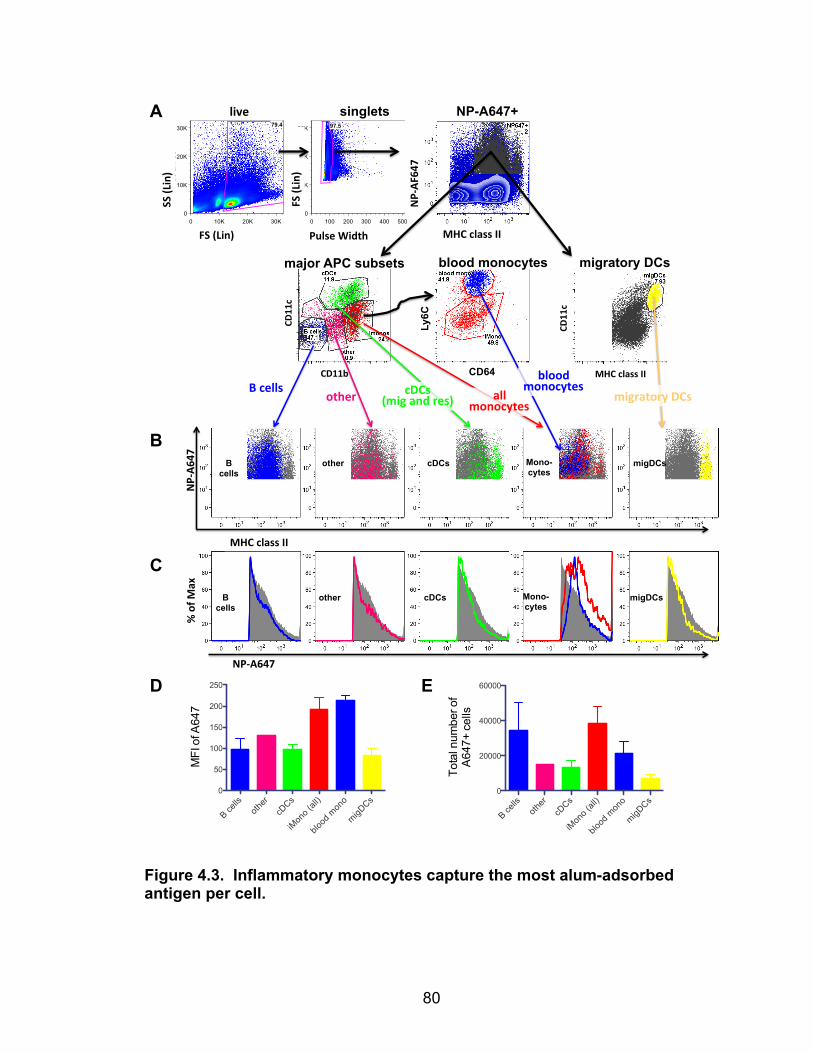
80
Figure 4.3. Inflammatory monocytes capture the most alum-adsorbed antigen per cell.
live%
SS%(Lin)%
NP-A647+ singlets A
MHC%class%II%
NP4A6
47%
NP4A647%
%%of%M
ax%
B
C
D
B cells
Mono-cytes
cDCs migDCs other
E
Mono-cytes
Ungatedsample monos.fcsEvent Count: 3003939
0 10K 20K 30KFS Lin: FS
0
10K
20K
30K
SS
Lin
: SS
79.4
livesample monos.fcsEvent Count: 2384056
0 100 200 300 400 500Pulse Width: Pulse Width
0
10K
20K
30K
FS L
in: F
S
97.5
singlesample monos.fcsEvent Count: 2324169
0 101 102 103
<FL 1 Log>: FITC-MHC II
0
101
102
103
<FL
8 Lo
g>: A
PC
-NP
647
2.67
NP647+sample monos.fcsEvent Count: 62106
0 101 102 103
<FL 9 Log>: APC-Cy7-CD11b
0
101
102
103
<FL
5 Lo
g>: P
E-C
y7-C
D11
c
53.3
11.7
18.5
11.6
NP647+sample monos.fcsEvent Count: 62106
0 101 102 103
<FL 1 Log>: FITC-MHC II
0
101
102
103
<FL
5 Lo
g>: P
E-C
y7-C
D11
c
7.69
imonossample monos.fcsEvent Count: 11509
0 101 102 103
<FL 2 Log>: PE-CD64
0
101
102
103
<FL
6 Lo
g>: P
B-L
y6C
50.2
41.1
MHC%class%II%
NP4AF
647%
Pulse%Width%FS%(Lin)%
FS%(Lin)%
CD11c%
CD11c%
Ly6C
CD11b% CD64 MHC%class%II%
migratory%DCs%all%monocytes%
cDCs%(mig%and%res)%other%
B%cells%blood%
monocytes%
B cells
cDCs migDCs other
major APC subsets blood monocytes migratory DCs
# of NP647 higher ag gate
B cells
other
cDCs
iMon
o (all
)
blood
mon
o
migDCs
0
20000
40000
60000
Tota
l num
ber o
fA
647+
cel
ls
MFI of NP647 higher ag gate
B cells
other
cDCs
iMon
o (all
)
blood
mon
o
migDCs
0
50
100
150
200
250
MFI
of A
647

81
Figure 4.3. Inflammatory monocytes capture the most alum-adsorbed antigen per cell. Mice were immunized i.m. with PBS or 200 µg alum + 5-20 µg NP-A647 ± 1 mg BSA. Popliteal dLNs were harvested 24 hours after immunization, grouped into one sample (n = 3), and analyzed as described in Chapter II. (A) Gating scheme to identify antigen-loaded (A647+) cell subsets: B cells (blue), conventional DCs (cDCs), migratory DCs (migDCs), inflammatory monocytes, blood monocytes, and ‘other’ cells that displayed an indeterminate phenotype. All subsets were gated on live cells, singlet events, and MHC II+ A647+. A647+ gate was set based on PBS control mice. Subsets were then compared to the cumulative population of A647+ cells (grey) and analyzed for their (B) MHCII x A647 phenotype and (C) A647 fluorescence intensity by % of max. (D) Mean fluorescence intensity (MFI) of A647 per APC subset. (E) Total number of A647+ cells per APC subset. Data are representative (A-C) or cumulative (D-E) of 3 independent experiments. Error bars show means ± SEM for each group.
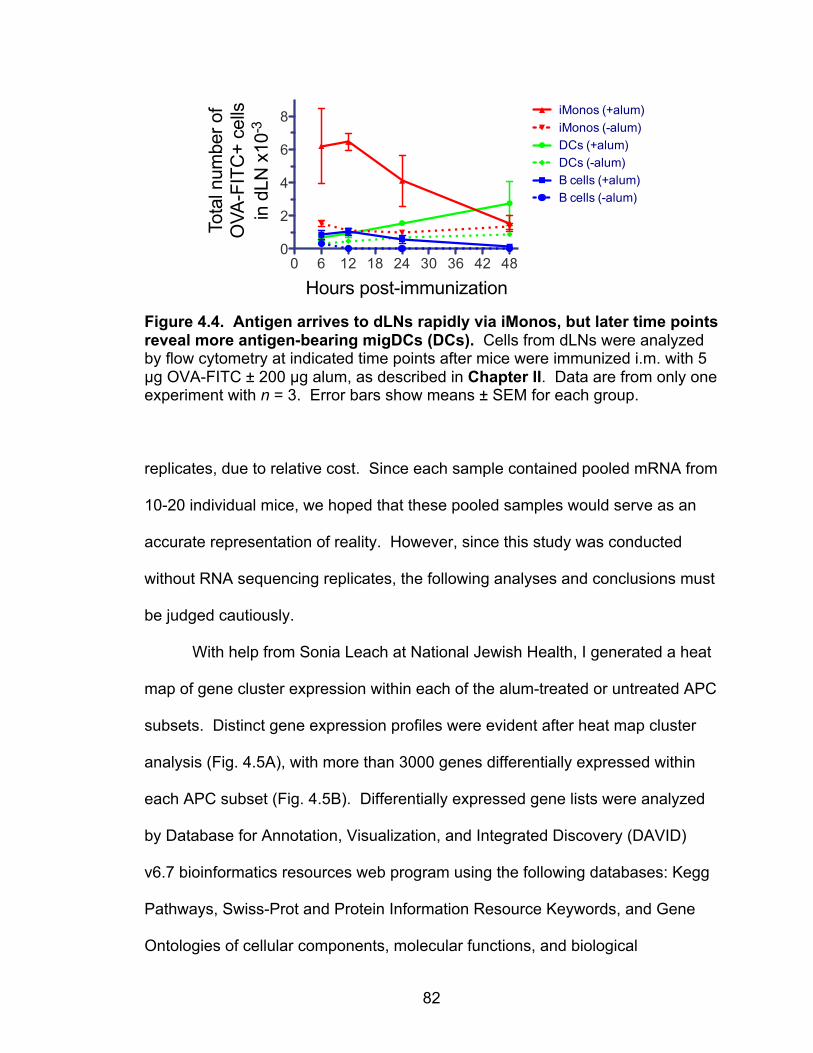
82
Figure 4.4. Antigen arrives to dLNs rapidly via iMonos, but later time points reveal more antigen-bearing migDCs (DCs). Cells from dLNs were analyzed by flow cytometry at indicated time points after mice were immunized i.m. with 5 µg OVA-FITC ± 200 µg alum, as described in Chapter II. Data are from only one experiment with n = 3. Error bars show means ± SEM for each group. replicates, due to relative cost. Since each sample contained pooled mRNA from
10-20 individual mice, we hoped that these pooled samples would serve as an
accurate representation of reality. However, since this study was conducted
without RNA sequencing replicates, the following analyses and conclusions must
be judged cautiously.
With help from Sonia Leach at National Jewish Health, I generated a heat
map of gene cluster expression within each of the alum-treated or untreated APC
subsets. Distinct gene expression profiles were evident after heat map cluster
analysis (Fig. 4.5A), with more than 3000 genes differentially expressed within
each APC subset (Fig. 4.5B). Differentially expressed gene lists were analyzed
by Database for Annotation, Visualization, and Integrated Discovery (DAVID)
v6.7 bioinformatics resources web program using the following databases: Kegg
Pathways, Swiss-Prot and Protein Information Resource Keywords, and Gene
Ontologies of cellular components, molecular functions, and biological
OVA-FITC+ loaded cells following immunization
0 6 12 18 24 30 36 42 480
2
4
6
8 iMonos (+alum)
DCs (+alum)
B cells (+alum)
iMonos (-alum)
DCs (-alum)
B cells (-alum)
Hours post-immunization
Tota
l num
ber o
fO
VA-F
ITC
+ ce
llsin
dLN
x10
-3

83
processes. Functional annotation gene clusters were counted if the DAVID
program calculated their enrichment score to be ≥1 and quantified by APC
subset (Fig. 4.5C).
Though there were too many gene clusters to analyze fully within the
scope of this thesis, I have summarized the functional theme of each cluster that
had a DAVID enrichment score above 5 (highly significant) in Table 4.2.
Enrichment scores rank the biological significance of gene clusters based on
overall modified p-values (EASE scores) of all genes within the cluster. As could
be expected, alum treatment both up-regulated and down-regulated RNA
associated with a broad category labeled: protein changes, cell signaling, and
secreted proteins. This cluster is so broad that little can be interpreted without
analyzing specific genes. Similarly, cell adhesion genes were both up regulated
and down regulated across the board, indicating that closer inspection of the
specific genes in this functional cluster is needed to identify distinct biological
effects. The iMono gene profiles indicate a pan-inflammatory effect of alum, as
alum-treated gene expression is increased in clusters associated with the
immune response, inflammatory response, and lysosomes. Curiously, genes
implicated in sugar and carbohydrate binding were down regulated in iMonos, but
up regulated in B cells at this time point. Lastly, two clusters were preferentially
down regulated in alum-treated iMonos and DCs: cytokine and cytokine
receptors and extracellular matrix proteins. These alum effects can be followed
up by analyzing the regulation of specific genes within these functional groups.
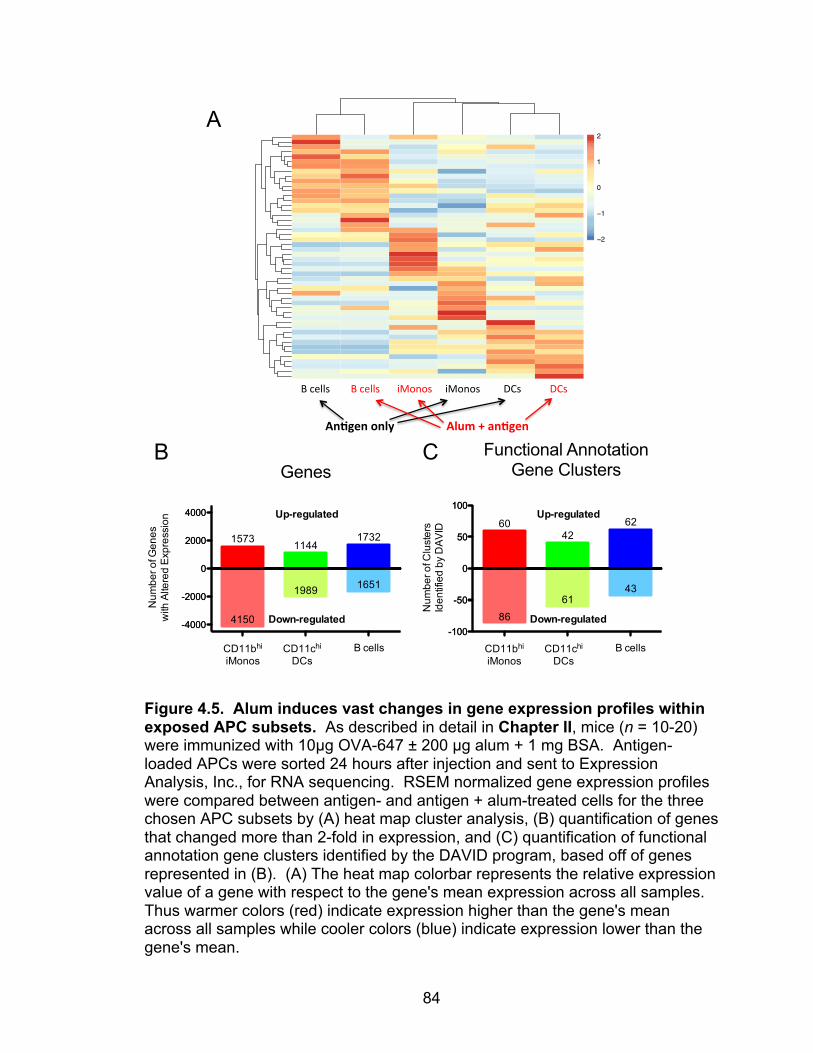
84
Figure 4.5. Alum induces vast changes in gene expression profiles within exposed APC subsets. As described in detail in Chapter II, mice (n = 10-20) were immunized with 10µg OVA-647 ± 200 µg alum + 1 mg BSA. Antigen-loaded APCs were sorted 24 hours after injection and sent to Expression Analysis, Inc., for RNA sequencing. RSEM normalized gene expression profiles were compared between antigen- and antigen + alum-treated cells for the three chosen APC subsets by (A) heat map cluster analysis, (B) quantification of genes that changed more than 2-fold in expression, and (C) quantification of functional annotation gene clusters identified by the DAVID program, based off of genes represented in (B). (A) The heat map colorbar represents the relative expression value of a gene with respect to the gene's mean expression across all samples. Thus warmer colors (red) indicate expression higher than the gene's mean across all samples while cooler colors (blue) indicate expression lower than the gene's mean.
Ag_B220
alumBSA_B220
alumBSA_C
D11b_hi
Ag_CD11b_hi
Ag_CD11c_hi
alumBSA_C
D11c_hi
−2
−1
0
1
2
B"cells""""""""B"cells """"iMonos"""""iMonos"""""""""DCs """"""DCs"""""
An#gen&only& &&&&&&&&&&&&Alum&+&an#gen&
Genes
CD11b hi iMonosCD11c hi DCs B cells
-4000
-2000
0
2000
4000 Up-regulated
CD11bhi
iMonosCD11chi
DCsB cells
15731144
1732
Num
ber o
f Gen
esw
ith A
ltere
d E
xpre
ssio
n
CD11b hi iMonosCD11c hi DCs B cells
-4000
-2000
0
2000
4000
4150
1989 1651
Down-regulated
CD11b hi iMonosCD11c hi DCs B cells
-100
-50
0
50
100
86
6143
Down-regulated
Functional AnnotationGene Clusters
CD11b hi iMonosCD11c hi DCs B cells
-100
-50
0
50
100Up-regulated
CD11bhi
iMonosCD11chi
DCsB cells
6042
62
Num
ber o
f Clu
ster
sId
entif
ied
by D
AV
ID
A
B C

85
Overall, the functional gene clustering analysis helps to confirm that alum
has many effects on APCs, but lacks the specificity to identify distinct immuno-
stimulatory pathways that are affected. Furthermore, replicate data are needed
before thorough analysis of this RNA sequencing experiment is warranted.
Table 4.2. Functional gene clusters that have altered expression in response to alum
iMonos DCs B cells Increased Expression After Alum Treatment
• Protein changes, signaling proteins
• Immune response • Inflammatory defense response • Cell surface proteins • Lysosome • Positive regulation of immune
response
• Protein changes, secreted proteins, signaling proteins
• Cell adhesion
• Protein changes, signaling proteins
• Secreted protein signals • Cell surface proteins • Extracellular matrix • Cell adhesion • Angiogenesis • Plasma membrane • Sugar binding • Carbohydrate binding • Inflammatory response
Decreased Expression After Alum Treatment • Protein changes, signaling
proteins • Secreted proteins • Cell surface markers • Cell adhesion • Sugar binding • Extracellular matrix • Cytokine & cytokine receptors • Cell-cell signaling • Carbohydrate binding
• Protein changes, signaling proteins, secreted protein signals
• Extracellular matrix • Cell surface proteins • Cell adhesion • Cytokine & cytokine receptor • Synapse
• Protein changes, signaling proteins, secreted protein signals
• Extracellular matrix • Cell junction & synapse • Bone development • Cell adhesion
*Clusters are arranged in descending order of DAVID enrichment score; only clusters with enrichment scores ≥ 5 are listed.
Discussion
The data in this chapter suggest that intramuscularly injected alum does
not greatly alter how the mouse immune system takes up foreign antigen and
shuttles it to dLNs. Compared to soluble protein alone, it was unexpected that
alum adjuvant does not more significantly skew the APCs subsets that acquire

86
antigen. This suggests that the APC subsets others59–61 and I have identified to
respond to i.m. alum injection may simply specialize in patrolling skeletal
musculature and, consequently, pick up all forms of injected antigen regardless
of whether or not alum is present. However, it is clear from the RNA sequencing
data that alum has immunostimulatory effects on all involved APC subsets.
This study is a valuable extension to another report that focused solely on
APCs within steady state and alum-treated skeletal muscle, along with their
migratory counterparts in dLNs61. My study added the condition of soluble
protein injected without alum, which elicited the same antigen-loaded APC
subsets within dLNs as did alum-adsorbed antigen. Furthermore, my use of
fluorescent antigen enabled the quantification of antigen acquisition by distinct
APC subsets. I determined that antigen is most concentrated in iMonos, followed
by CD11b+ DCs, and lastly B cells. This may explain why previous reports have
found that iMonos are the most efficient subset at priming IFNγ-producing T
cells61.
However, there is a notable difference between the other studies and
mine. Concerning the relative frequencies of migratory APCs involved in
detecting intramuscular vaccine antigens, I found a large proportion of iMonos
and B cells to be fluorescent antigen-loaded in the dLNs while other authors
emphasized a large role for CD11b+ migDCs60,61. Some of this discrepancy is
due to variable nomenclature for DC-like APCs that either does or does not
distinguish between monocyte-derived and DC-derived CD11b+ CD11c+ cells.
Furthermore, reports that did not observe much iMono migration to dLNs used

87
small volume injections (15 µl) whereas my injections were larger per muscle (50
µl). This difference may not be trivial as the increased volume of liquid draining
from the muscle may influence the appearance of different APC subsets in the
dLNs. Lastly, my data are the first to raise the possibility that antigen-loaded B
cells may constitute a substantial population of APCs within the dLNs after alum
injection. Alternatively, perhaps B cells are antigen-loaded merely because they
must present linked antigen to TFH cells in order to receive help within germinal
centers. It is still unclear where B cells acquire this antigen, though it is likely
acquired within the dLN because few B cells are found in steady state skeletal
muscle. Antigen-loaded B cells may have been previously overlooked and/or
discounted due to their low antigen load and consequent difficult nature to detect
by fluorescent antigen tracking. B cell APCs may be an important factor in alum
biology because they have been reported to promote type 2 immune
responses135,136, as is elicited by alum. Additionally, B cells may have a
tolerogenic role when stimulated through TLR9 by DNA complexes associated
with dexamethasone-treated apoptotic cells137.
In conclusion, this study confirms other reports that iMonos and CD11b+
DCs are important alum/antigen APCs and also suggests a new role for B cells
as alum-induced APCs. Furthermore, the preliminary RNA sequencing data also
suggest that alum may affect a variety of immunostimulatory pathways within
APCs. However, these findings must be repeated before they can be relied
upon.

88
CHAPTER V
SINGLE DOSES OF HEAT AGGREGATED PROTEINS
ARE NOT IMMUNOGENIC
Introduction
Many diseases and conditions stem from inappropriate adaptive immune
responses mounted against protein aggregates99,103–105,138. Surprisingly, T cell
responses to aggregates are not well characterized, though one study suggested
that heat aggregated proteins specifically elicit CTL responses91. Furthermore, it
has been previously reported that heat denatured OVA enhances production of
OVA-specific TH1-dependent IgG2a class-switched antibodies104. Others also
showed that pathogen-sized particulate proteins are typically more TH1-
stimulating than soluble proteins139,140. These scattered reports within the
literature needed clarification by a direct study on the effect of aggregated
proteins on T cell responses.
Consequently, the objective of this study was to determine the magnitude
and functionality of primary T cell responses to heat aggregated proteins.
Though this project was discontinued, my preliminary data are presented in this
chapter and they suggest that heat aggregated proteins do not stimulate T cell
responses in mice. This inability of protein aggregates to induce cellular immune
responses after one immunization raises concern about their value as vaccine
components.
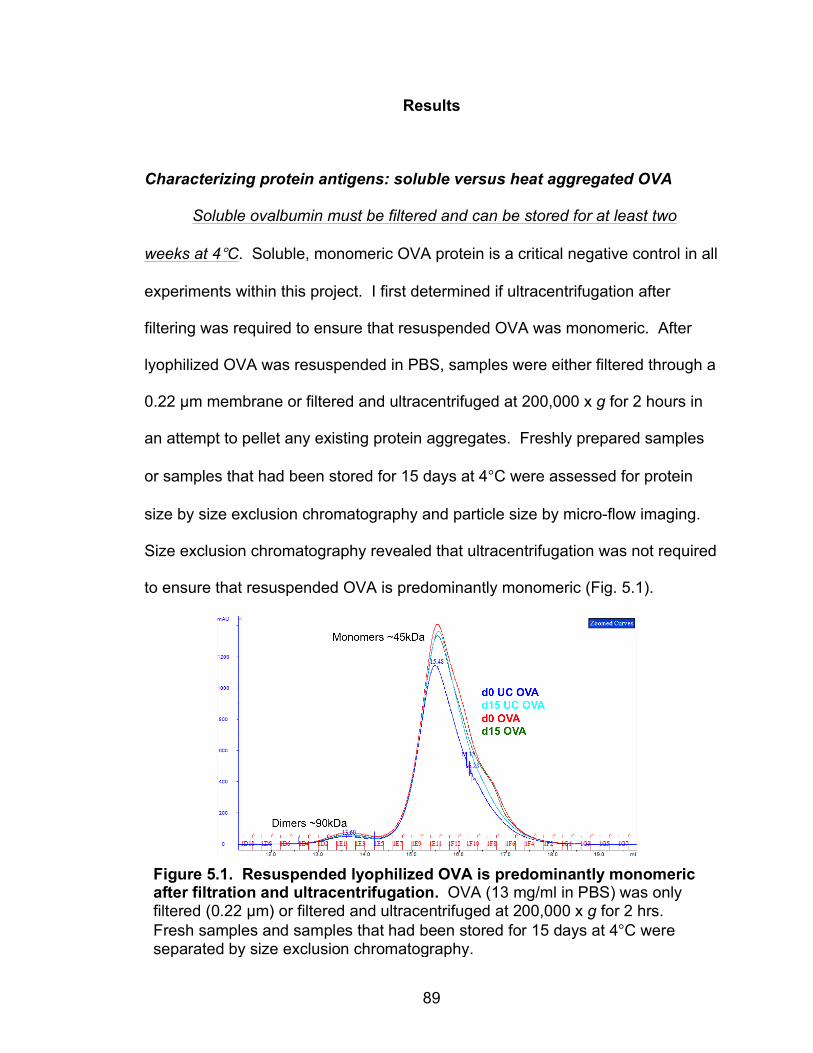
89
Results
Characterizing protein antigens: soluble versus heat aggregated OVA
Soluble ovalbumin must be filtered and can be stored for at least two
weeks at 4°C. Soluble, monomeric OVA protein is a critical negative control in all
experiments within this project. I first determined if ultracentrifugation after
filtering was required to ensure that resuspended OVA was monomeric. After
lyophilized OVA was resuspended in PBS, samples were either filtered through a
0.22 µm membrane or filtered and ultracentrifuged at 200,000 x g for 2 hours in
an attempt to pellet any existing protein aggregates. Freshly prepared samples
or samples that had been stored for 15 days at 4°C were assessed for protein
size by size exclusion chromatography and particle size by micro-flow imaging.
Size exclusion chromatography revealed that ultracentrifugation was not required
to ensure that resuspended OVA is predominantly monomeric (Fig. 5.1).
Figure 5.1. Resuspended lyophilized OVA is predominantly monomeric after filtration and ultracentrifugation. OVA (13 mg/ml in PBS) was only filtered (0.22 µm) or filtered and ultracentrifuged at 200,000 x g for 2 hrs. Fresh samples and samples that had been stored for 15 days at 4°C were separated by size exclusion chromatography.

90
However, ultracentrifugation seemed to rid the OVA samples of a small portion of
degraded protein that was less than 45 kDa in size. When compared to
unfiltered OVA (Fig 5.2A), filtered OVA (Fig. 5.2B) contained very few particles,
as assessed by micro-flow imaging. Furthermore, filtered OVA can be stored at
4°C for at least 15 days without protein aggregation or particle accumulation (Fig.
5.1 and 5.2B). These results indicate that “soluble OVA” (native and monomeric)
could be prepared by resuspending the lyophilized OVA in PBS, filtering, and
storing at 4°C until use.
Heat aggregation of ovalbumin at concentrations >1 mg/ml alters its
surface hydrophobicity, conformation, and structure. To generate aggregated
OVA antigen, I first turned to heat denaturation because this is the simplest
approach. Alternative approaches included urea denaturation, chemical
conjugation, or gluteraldehyde crosslinking. To confirm that heating OVA
produced denatured protein aggregates, I compared the secondary and tertiary
structures, surface hydrophobicity, and tryptophan fluorescence of soluble OVA
and heat aggregated OVA. Hydrophobicity has been proposed as an
immunogenic factor of unfolded or otherwise denatured proteins141. Heating
OVA at either 1 mg/ml or 10 mg/ml in PBS resulted in increased surface
hydrophobicity of the proteins, as measured by 8-anilinonaphthalene-1-sulfonate
(ANS) fluorescence staining and spectrofluorometry (Fig. 5.3A). Tryptophan
peak fluorescence, an indication of protein conformation, was increasingly
altered (blue-shifted) as OVA was heated at higher concentrations (1 mg/ml
versus 10 mg/ml) (Fig. 5.3B,C). Infrared (IR) spectroscopy was used to assess
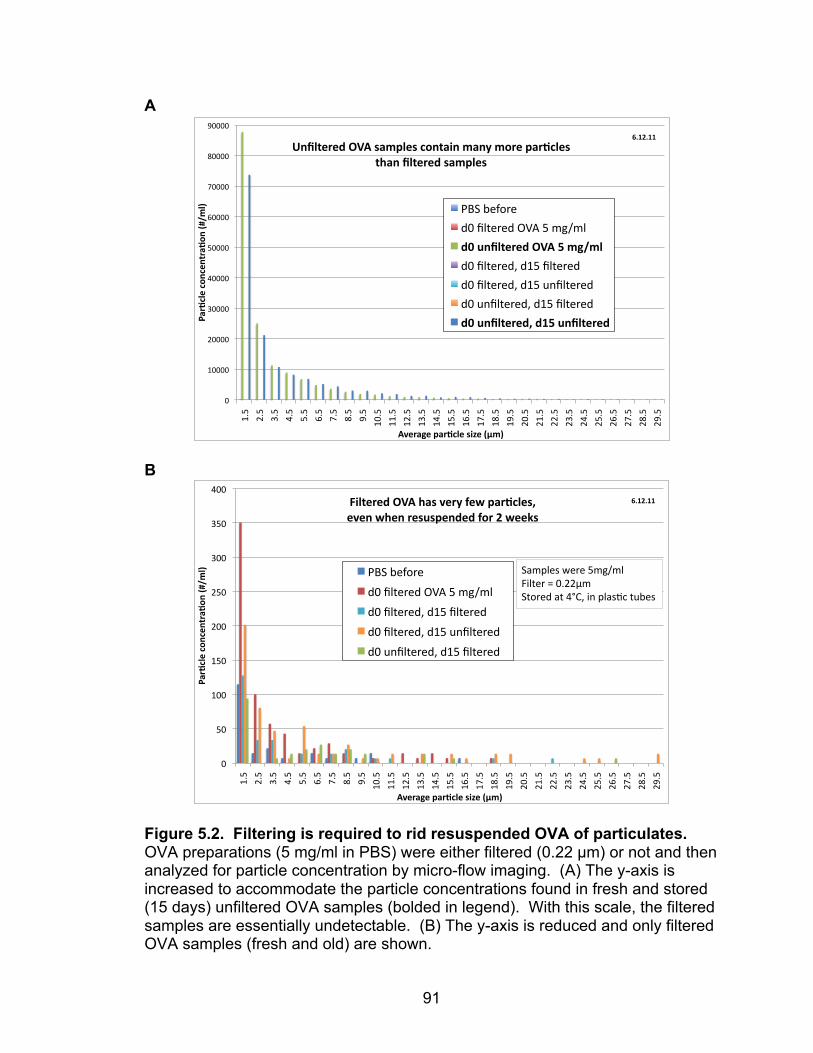
91
A
B
Figure 5.2. Filtering is required to rid resuspended OVA of particulates. OVA preparations (5 mg/ml in PBS) were either filtered (0.22 µm) or not and then analyzed for particle concentration by micro-flow imaging. (A) The y-axis is increased to accommodate the particle concentrations found in fresh and stored (15 days) unfiltered OVA samples (bolded in legend). With this scale, the filtered samples are essentially undetectable. (B) The y-axis is reduced and only filtered OVA samples (fresh and old) are shown.
!"
#!!!!"
$!!!!"
%!!!!"
&!!!!"
'!!!!"
(!!!!"
)!!!!"
*!!!!"
+!!!!"
#,'"
$,'"
%,'"
&,'"
','"
(,'"
),'"
*,'"
+,'"
#!,'"
##,'"
#$,'"
#%,'"
#&,'"
#','"
#(,'"
#),'"
#*,'"
#+,'"
$!,'"
$#,'"
$$,'"
$%,'"
$&,'"
$','"
$(,'"
$),'"
$*,'"
$+,'"
!"#$%&'(%)*%'*+#"$)*(,-./
&0(
12'#"3'(4"#$%&'(567'(,8/0(
9*:&+'#';(<=1(5"/4&'5(%)*+"6*(/"*>(/)#'(4"#$%&'5(
+?"*(:&+'#';(5"/4&'5(
-./"012341"
5!"6781415"9:;"'"<=><7"
;@(A*:&+'#';(<=1(B(/3./&(
5!"6781415?"5#'"6781415"
5!"6781415?"5#'"@A6781415"
5!"@A6781415?"5#'"6781415"
;@(A*:&+'#';C(;DB(A*:&+'#';(
EFDGFDD(
!"
#!"
$!!"
$#!"
%!!"
%#!"
&!!"
&#!"
'!!"
$(#"
%(#"
&(#"
'(#"
#(#"
)(#"
*(#"
+(#"
,(#"
$!(#"
$$(#"
$%(#"
$&(#"
$'(#"
$#(#"
$)(#"
$*(#"
$+(#"
$,(#"
%!(#"
%$(#"
%%(#"
%&(#"
%'(#"
%#(#"
%)(#"
%*(#"
%+(#"
%,(#"
!"#$%&'(%)*%'*+#"$)*(,-./
&0(
12'#"3'(4"#$%&'(567'(,8/0(
96&+'#':(;<1(="5(2'#>(?'@(4"#$%&'5A(
'2'*(@='*(#'5B54'*:':(?)#(C(@''D5(
-./"012341"
5!"6781415"9:;"#"<=><7"
5!"6781415?"5$#"6781415"
5!"6781415?"5$#"@A6781415"
5!"@A6781415?"5$#"6781415"
EFGCFGG(
/B<C71D"E141"#<=><7"
FG7814"H"!(%%I<"
/83415"B8"'JK?"GA"C7BDLM"8@01D"
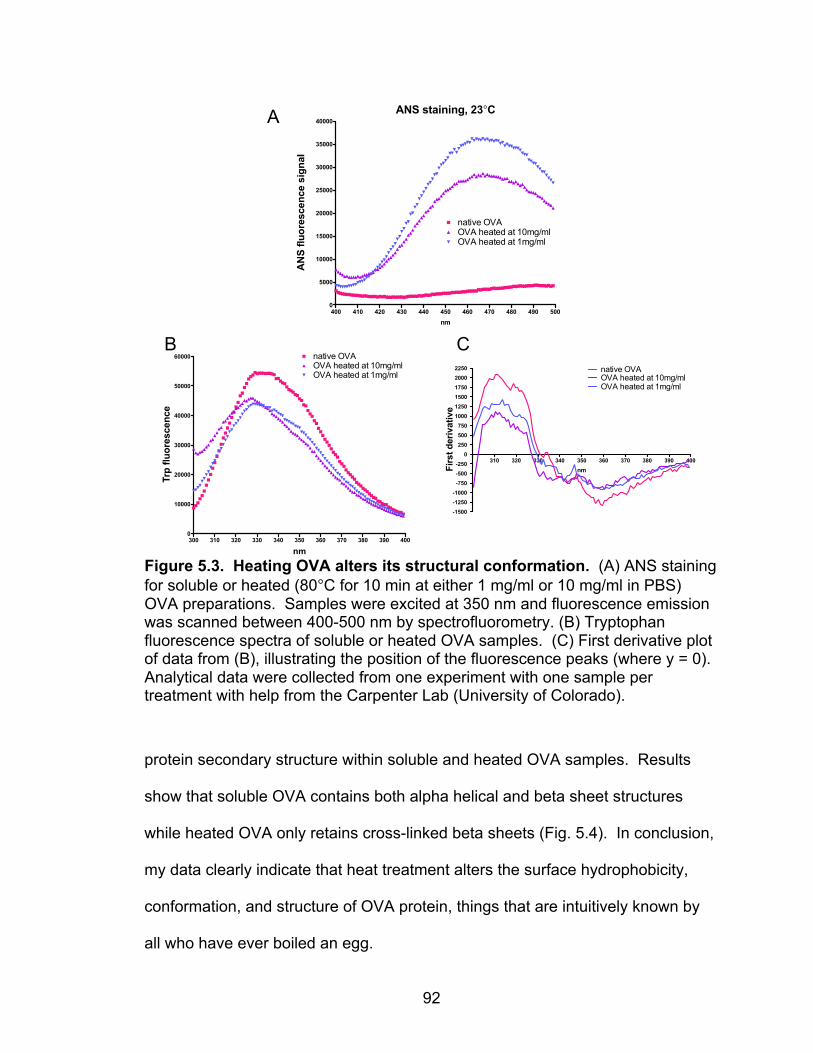
92
Figure 5.3. Heating OVA alters its structural conformation. (A) ANS staining for soluble or heated (80°C for 10 min at either 1 mg/ml or 10 mg/ml in PBS) OVA preparations. Samples were excited at 350 nm and fluorescence emission was scanned between 400-500 nm by spectrofluorometry. (B) Tryptophan fluorescence spectra of soluble or heated OVA samples. (C) First derivative plot of data from (B), illustrating the position of the fluorescence peaks (where y = 0). Analytical data were collected from one experiment with one sample per treatment with help from the Carpenter Lab (University of Colorado).
protein secondary structure within soluble and heated OVA samples. Results
show that soluble OVA contains both alpha helical and beta sheet structures
while heated OVA only retains cross-linked beta sheets (Fig. 5.4). In conclusion,
my data clearly indicate that heat treatment alters the surface hydrophobicity,
conformation, and structure of OVA protein, things that are intuitively known by
all who have ever boiled an egg.
400 410 420 430 440 450 460 470 480 490 5000
5000
10000
15000
20000
25000
30000
35000
40000
native OVAOVA heated at 10mg/mlOVA heated at 1mg/ml
ANS staining, 23°C
nm
AN
S fl
uore
scen
ce s
igna
l
300 310 320 330 340 350 360 370 380 390 4000
10000
20000
30000
40000
50000
60000 native OVAOVA heated at 10mg/mlOVA heated at 1mg/ml
nm
Trp
fluor
esce
nce
310 320 330 340 350 360 370 380 390 400
-1500
-1250
-1000
-750
-500
-250
0
250
500
750
1000
1250
1500
1750
2000
2250 native OVAOVA heated at 10mg/mlOVA heated at 1mg/ml
nmFirs
t der
ivat
ive
A
B C
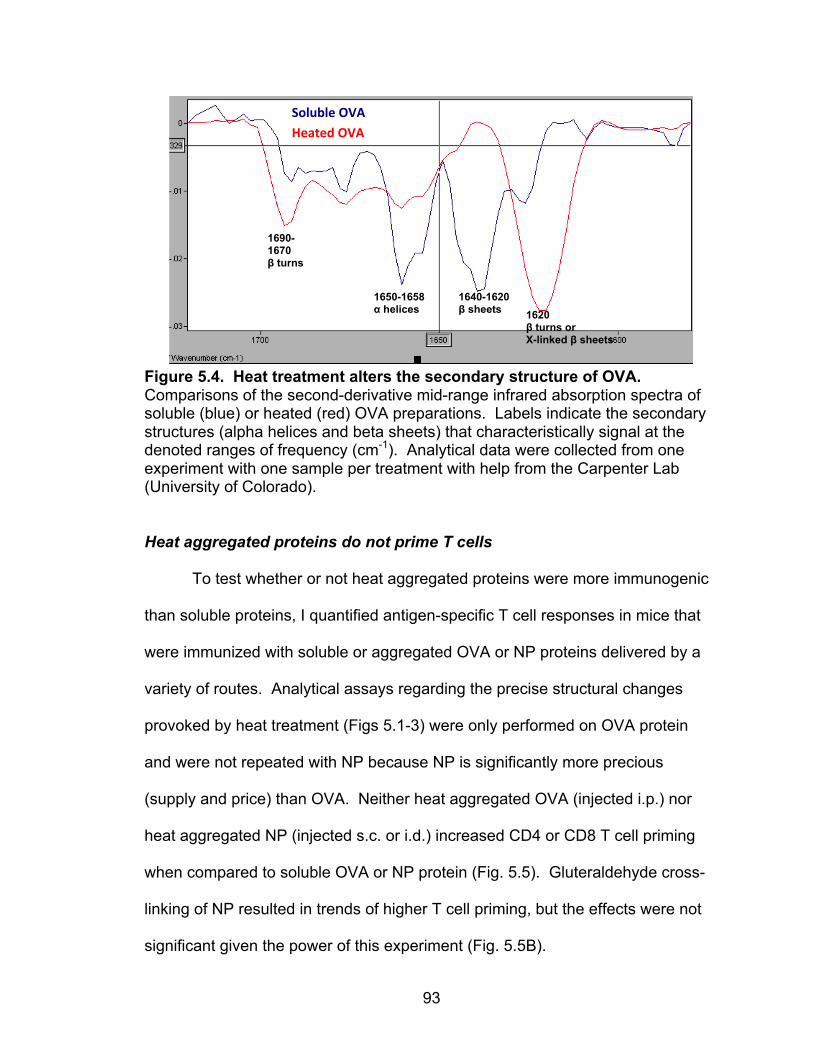
93
Figure 5.4. Heat treatment alters the secondary structure of OVA. Comparisons of the second-derivative mid-range infrared absorption spectra of soluble (blue) or heated (red) OVA preparations. Labels indicate the secondary structures (alpha helices and beta sheets) that characteristically signal at the denoted ranges of frequency (cm-1). Analytical data were collected from one experiment with one sample per treatment with help from the Carpenter Lab (University of Colorado). Heat aggregated proteins do not prime T cells
To test whether or not heat aggregated proteins were more immunogenic
than soluble proteins, I quantified antigen-specific T cell responses in mice that
were immunized with soluble or aggregated OVA or NP proteins delivered by a
variety of routes. Analytical assays regarding the precise structural changes
provoked by heat treatment (Figs 5.1-3) were only performed on OVA protein
and were not repeated with NP because NP is significantly more precious
(supply and price) than OVA. Neither heat aggregated OVA (injected i.p.) nor
heat aggregated NP (injected s.c. or i.d.) increased CD4 or CD8 T cell priming
when compared to soluble OVA or NP protein (Fig. 5.5). Gluteraldehyde cross-
linking of NP resulted in trends of higher T cell priming, but the effects were not
significant given the power of this experiment (Fig. 5.5B).
1650-1658 α helices
1640-1620 β sheets 1620
β turns or X-linked β sheets
1690- 1670 β turns
Soluble'OVA'Heated'OVA!
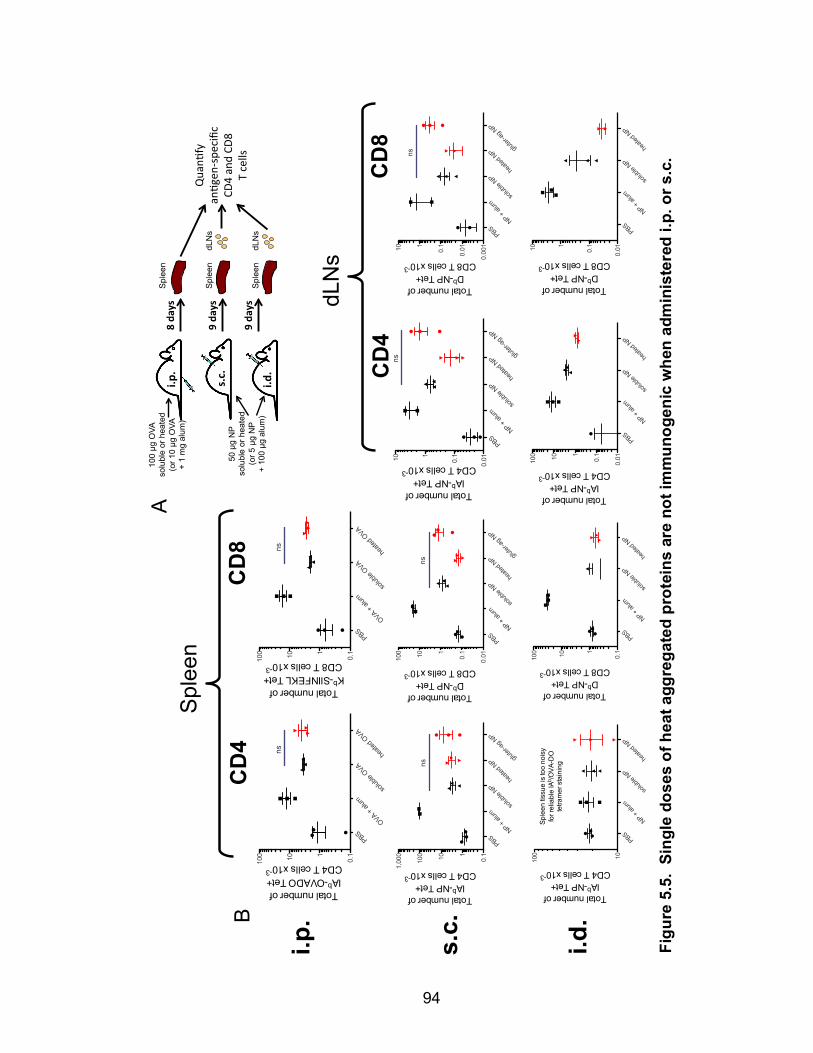
94
PBSOVA +
alum
solub
le OVA
heate
d OVA
0.1110100
ns
Total number ofIAb-OVADO Tet+CD4 T cells x10-3
PBSOVA +
alum
solub
le OVA
heate
d OVA
0.1110100
ns
Total number ofKb-SIINFEKL Tet+CD8 T cells x10-3
i.p.
PBSNP +
alum
solub
le NP
heate
d NP glu
ter-ag
NP
0.010.1110
Total number ofIAb-NP Tet+
CD4 T cells x10-3
ns
PBSNP +
alum
solub
le NP
heate
d NP glu
ter-ag
NP
0.00
1
0.010.1110
Total number ofDb-NP Tet+
CD8 T cells x10-3
ns
PBSNP +
alum
solub
le NP
heate
d NP glu
ter-ag
NP
0.1110100
1,00
0
Total number ofIAb-NP Tet+
CD4 T cells x10-3
ns
PBSNP +
alum
solub
le NP
heate
d NP glu
ter-ag
NP
0.010.1110100
Total number ofDb-NP Tet+
CD8 T cells x10-3
ns
s.c.
i.d.
PBS
NP + alu
m
solub
le NP
heate
d NP
0.010.1110100
Total number ofIAb-NP Tet+
CD4 T cells x10-3
PBS
NP + alu
m
solub
le NP
heate
d NP
0.010.1110
Total number ofDb-NP Tet+
CD8 T cells x10-3
PBS
NP + alu
m
solub
le NP
heate
d NP
10100
Sple
en ti
ssue
is to
o no
isy
for r
elia
ble
IAb /O
VA-D
Ote
tram
er s
tain
ing
Total number ofIAb-NP Tet+
CD4 T cells x10-3
PBS
NP + alu
m
solub
le NP
heate
d NP
0.1110100
Total number ofDb-NP Tet+
CD8 T cells x10-3
Spl
een
CD
4 C
D8
dLN
s
CD
4 C
D8
s.c.$
9$da
ys$
Quan%
fy(
an%g
en+spe
cific(
CD4(and(CD
8(T(cells(
i.d.$
i.p.$
9$da
ys$
8$da
ys$
Spl
een
dLN
s S
plee
n
Spl
een
dLN
s
100 µg
OVA
so
lubl
e or
hea
ted
(or 1
0 µg
OVA
+
1 m
g al
um)
50 µ
g N
P so
lubl
e or
hea
ted
(or 5
µg
NP
+ 10
0 µg
alu
m)
A
B
Figu
re 5
.5.
Sing
le d
oses
of h
eat a
ggre
gate
d pr
otei
ns a
re n
ot im
mun
ogen
ic w
hen
adm
inis
tere
d i.p
. or s
.c.

95
Figure 5.5. Single doses of heat aggregated proteins are not immunogenic when administered i.p. or s.c. (A) Experimental design. Mice were immunized via the indicated route with indicated doses of soluble OVA or NP, heat aggregated OVA or NP (red), gluteraldehyde aggregated NP (red), or Alhydrogel® alum + soluble OVA or NP. OVA was heated at 96°C for 5 min at 50 mg/ml in PBS. NP (1 mg/ml in PBS) was aggregated by heat (98°C for 10 min) or gluteraldehyde treatment (0.5% for 2 hrs, then buffer exchanged to PBS). (B) OVA-or NP-specific CD4 and CD8 T cells were quantified on d8 or d9 after immunization with tetramers and antibodies as described in Chapter II. Data are from experiments that were only conducted once each, n = 3. Error bars show means ± SEM for each group. Statistical differences were determined using an unpaired Student’s t test; ns indicates p > 0.05.

96
Discussion
The current literature contains largely correlative accounts of antigen-
specific immunity to protein aggregates that have been reported since the 1960s
when David Dresser first reported that aggregated proteins can be immunogenic
142–144,102,104,100. Few direct studies to illuminate the immunological
consequences of protein aggregate introduction into the body have been
published. To the best of my knowledge, there is only one published study from
1997 that directly reported the ability of heat denatured proteins to prime CTL
responses when injected intraperitoneally into mice91. However, their
experimental approach was limited due to the techniques and reagents available
at that time. Furthermore, their data contradict other more recent reports in that
they show that heated protein aggregates stimulate little to no antibody
responses91, unlike reports of anti-therapeutic protein responses in human
patients97,99,145. My study set out to provide clarity about how protein aggregates
become immunogenic.
Unlike the aforementioned study91, the data presented in this chapter
suggest that heat aggregated proteins are not immunogenic for T cells when
given in a single dose. However, the limited experimental conditions in this study
were unable to fully test the hypothesis that aggregated proteins can be
immunogenic. Single dose exposures to aggregated proteins may not be
sufficient to prime immune responses, though single doses seemed to be
sufficient for one group of researchers to elicit antigen-specific CTLs in 199791,

97
perhaps due to reagent contaminants. The discrepancy here may also be due to
a difference in assay sensitivity used for each study. Perhaps re-stimulated ex
vivo CTL ability to carry out specific lysis is more sensitive than MHC tetramer
identification of antigen-specific T cells within directly ex vivo.
It is possible that aggregated proteins, such as those that form within vials
of therapeutic proteins97, only become immunogenic when administered routinely
over a long period of time. Chronic exposure of aggregated antigen may be
essential for the stimulation of adaptive immune responses. It is difficult to draw
conclusions from these preliminary data because this study is incomplete.
It is well established that particles containing repetitive arrangements of
antigens have their own adjuvant activity that is capable of stimulating innate
immunity. This observation has led to the design of vaccines that employ virus-
like particles or virosomes to deliver antigens as well as the use of protein
aggregation in some vaccines138,141,146,147. While VLPs and virosomes seem to
stimulate immunity by mimicking the physical characteristics of pathogens, it is
unclear how simple native or denatured protein aggregates stimulate innate
immune sensors. Since T cell responses rely upon stimulation by APCs, an
important part of this study would be to determine how aggregates are detected
by and activate APCs.
To extend this study, I would recommend augmenting the delivery routes
and immunization schedules used to administer protein aggregates. Most
endogenous protein aggregates are formed by hydrophobic interactions between
damaged or misfolded proteins that expose regions of their hydrophobic

98
cores106–109. However, aggregates composed of predominantly native proteins
can also be immunogenic148–150. One study directly compared native and
denatured aggregates and found that native aggregated are more immunogenic
than their denatured counterparts151. This suggests that the nativity of the
proteins affects the immunogenic mechanism of aggregate species. In both
cases, it is not clear how foreign, endogenous, native, or denatured protein
aggregates stimulate our innate immune cells.
Understanding how aggregates activate the innate immune system and
induce adaptive responses will greatly improve our understanding of protein
immunogenicity. This insight will aid clinical progress toward the deliberate
manipulation of immune responses to protein aggregates, namely those mounted
against protein therapeutics or even destructive anti-protein autoimmune
responses such as Alzheimer’s disease. Targeting immunostimulatory pathways
that are triggered by offending protein aggregates could be used to treat protein
aggregate-associated conditions and diseases. At the same time, understanding
the primary features of aggregate immunogenicity will encourage more deliberate
use or avoidance of protein aggregates as vaccine components. Specific
exploitation of the immunogenic or tolerogenic strengths of denatured or native
protein aggregates could lead to the generation of aggregate-based vaccines
that are easily stored and transported due to their stability. Potent vaccines with
these qualities would greatly improve global public health.

99
CHAPTER VI
DISCUSSION AND FUTURE DIRECTIONS
Aluminum salt adjuvants have greatly impacted global health by providing
protective immunity against deadly diseases to hundreds of millions of
people11,23. Since its adjuvant effect was first discovered in 1926, it has been
unclear how alum achieves such effective immunostimulation. Recently, it was
proposed that alum works by causing a release of host DNA that acts as an
endogenous adjuvant26,47,49. My thesis work contributes to the growing body of
knowledge about alum mechanism by providing an alternative opinion: DNA is
dispensable in alum responses.
The study in Chapter III exhaustively demonstrated that host DNA does
not play a prominent role in mediating adaptive immune responses to alum,
contradicting the aforementioned literature. Furthermore, this study highlighted
the importance of thoroughly evaluating the purity of commercial DNase reagents
prior to use, as active protease contaminants can have off target effects. Here I
will discuss how this study affects both the working model for how alum functions
as a vaccine adjuvant and other fields of research, as well as touch on issues of
reproducibility in published science.
Data presented in Chapter IV confirmed published reports that iMonos
and CD11b+ DCs are important alum-reactive APCs and also suggested a new
role for B cells as a substantial population of alum-induced APCs. These data

100
will be incorporated to the current working model of alum adjuvant action and I
will briefly discuss the importance of B cells as antigen presenters.
Last, the experiments and discussion presented in Chapter V emphasized
how very little is known about the immunogenic mechanisms triggered by protein
aggregates. More research is warranted to clarify this subject, as it is highly
relevant to the clinical use of protein therapeutics in human patients. This study
is incomplete and was appropriately discussed at the end of Chapter V, so I will
not mention it further in this section.
Alum’s Mechanism: A Working Model + Unanswered Questions
My work presented in Chapter III serves to correct the literature about
DNA’s role in mediating alum adjuvant action: i.e. it is not required for immune
responses to alum vaccines. It is important to note that these data do not
discount the fact that alum stimulates the extracellular release of host chromatin,
which in turn stimulates several inflammatory pathways. (The subject of DNA as
an inducible endogenous adjuvant will be discussed in the next section).
However, since I’ve demonstrated that DNA-sensing is not the key to alum’s
success as an adjuvant, the question remains: how does alum work? There is a
trend within the alum field that inflammatory pathways will be proposed as
responsible for alum’s adjuvant effect, only to be dismissed shortly thereafter
when other labs do not find them essential. I think there is a reason for this: alum
triggers many redundant inflammatory pathways, each of which may be

101
individually dispensable. In this section I will integrate my findings with what is
known from current publications to generate a working model for how alum
functions as an adjuvant, illustrated in Fig. 6.1. Furthermore, I will emphasize the
gaps in the literature where questions about alum’s mechanism remain
unanswered.
Step 1: Damage and inflammation at the site of injection
In the literature, all signs point to the fact that alum crystals cause
substantial inflammatory damage at the site of injection. Since its discovery,
alum vaccines are known to form “nodules” at the site of injection25. Thorough
examinations of these nodules revealed that they contain dead or dying cells,
damage-associated factors such as host chromatin, and a myriad of
inflammatory innate cells26,47,128. Abundant cell death has been documented at
the site of alum injection, owing to alum’s cytotoxic properties47,94. Dying cells
release their intracellular contents, including uric acid, chromatin, and heat shock
proteins, that can act as immunostimulatory damage-associated molecular
patterns (DAMPs)59,95,96,126,152,153. In fact, one group suggested that alum elicits
increased heat shock protein 70 in DCs154, suggesting that alum causes stress
signaling in innate immune cells. The profile of innate cells that are recruited to
alum nodules is extensive, including eosinophils, mast cells, neutrophils,
macrophages, inflammatory monocytes, NK cells, and dendritic cells among
others26,68. Adding to the chromatin content of alum nodules, neutrophils also
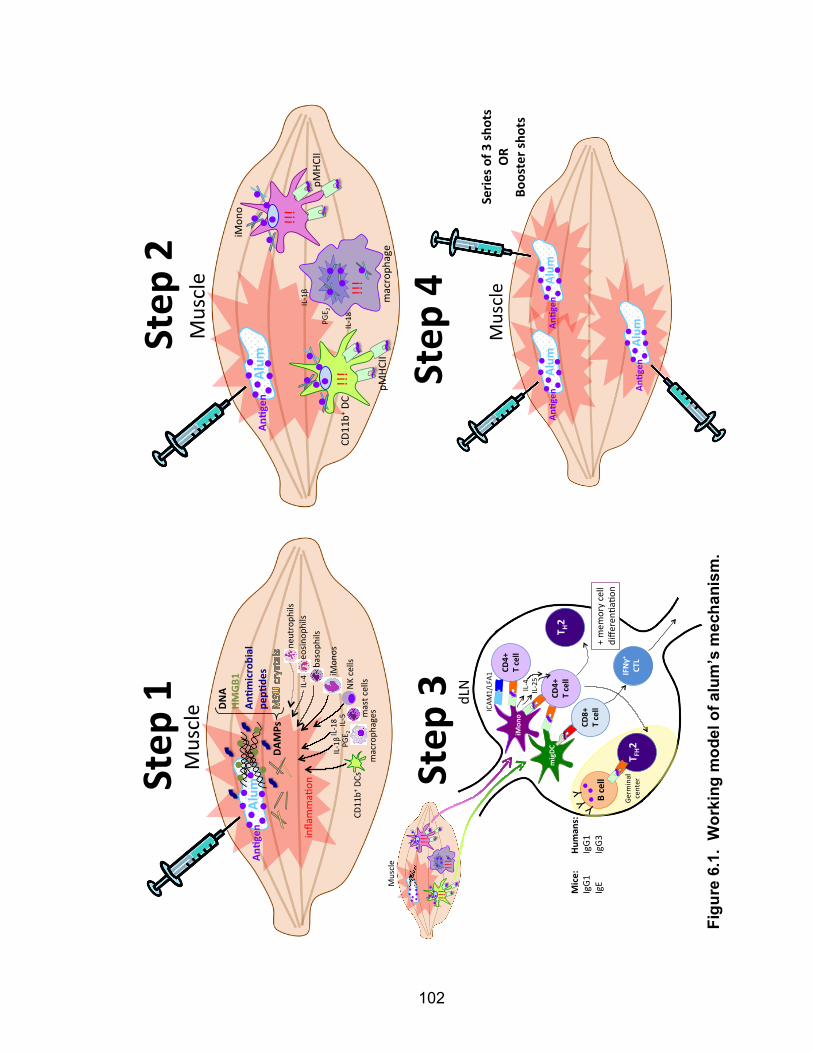
102
Alum
%Muscle' DN
A%HM
GB1%
An.m
icrobial%
pep.
des%
inflamma-
on'
An.g
en%
DAMPs%
neutroph
ils'
eosin
ophils'
basoph
ils'
iMon
os'
NK'cells'
mast'cells'
macroph
ages'
CD11
b+'DCs'
IL=18'
IL=1β''
IL=5'
PGE 2'
IL=4'
migDC
&
iMon
o&
dLN$
CD4+&
T&cell&
Muscle$
!!!$
!!!$
!!!$
T H2&
T FH2&
B&cell&
CD8+&
T&cell&
CD4+&
T&cell&
Germ
inal$
center$
IFNγ+
&
CTL&
ICAM
1/LFA1
$
IL94$
IL925$
Mice:
&Hum
ans:&
IgG1
$$IgG1
$IgE
$$IgG3
$Y$Y$
Y$
+$mem
ory$cell$
diffe
renE
aEon
$
Muscle'
Alum
%An
'gen
%Alum
%An
'gen
%
Alum
%An
'gen
%
Serie
s%of%3
%shots%
OR%
Booster%sho
ts%
Step
%1%
Step
%2%
Step
%3%
Step
%4%
Alum
%Muscle'
An'g
en%
iMon
o'
CD11
b+'DC'
IL218'
IL21β''
PGE 2' m
acroph
age'
!!!'
!!!'
!!!'
pMHC
II'
pMHC
II'
Figu
re 6
.1.
Wor
king
mod
el o
f alu
m’s
mec
hani
sm.

103
Figure 6.1. Working model of alum’s mechanism. Step 1: Alum causes inflammation at the site of injection, causing release of DAMPs and recruitment of inflammatory innate immune cells. Step 2: Alum crystals themselves and/or induced DAMPs cause activation and antigen uptake by APCs, predominantly iMonos and CD11b+ DCs. Step3: Activated APCs migrate to dLNs and prime CD4 and CD8 T cells via pMHCII/TCR and pMHCI/TCR interactions, respectively. APC/CD4 T cell interactions are stabilized by ICAM1/LFA1 molecules. Cytokine signals (IL-4, IL-25, etc.) promote TH2 differentiation of CD4 T cells. TFH2 cells aid B cells in class switch recombination to IgG1 and IgG3 in humans (IgG1 and IgE in mice) and differentiation into long lived plasma cells. Memory T cells are also formed through unclear mechanisms. Step 4: Protective memory is generated after a series of 3 alum immunizations or is maintained through the administration of booster shots throughout life. DC, dendritic cell; iMono, inflammatory monocyte; HMGB1, high mobility group box 1; MSU, monosodium urate; IL, interleukin; pMHC, peptide-loaded MHC; PGE2, prostaglandin E2; ICAM1, intercellular adhesion molecule 1; LFA1, lymphocyte function-associated antigen 1; IFNγ, interferon gamma.

104
release neutrophil extracellular traps containing granule proteins and, of course,
chromatin26,155, though neutrophils are not required for alum nodule formation26.
Several types of recruited innate immune cells (especially mast cells and
macrophages) secrete inflammatory IL-1β and IL-18 due to inflammasome
activation68. Though not required for alum responses, the inflammasome has
been proposed to be activated by alum in a number of ways, all of which
probably occur in vivo. These pathways include detection of alum crystals
themselves or damage-induced uric acid crystals59, lysosomal rupture by
engulfed alum crystals and subsequent release of cathepsin B protein into the
cytosol64, and even host DNA sensing156. In addition to inflammasome-
associated molecules that are secreted by a number of innate immune cell
subsets, eosinophils rapidly produce IL-4 and mast cells produce IL-5 at the site
of alum injection50,68 and macrophages produce prostaglandin E286,87.
Cumulatively, these inflammatory signals seem to promote the development of
type 2 immune responses in mice.
Step 2: Activation of antigen presenting cells
The most important consequence of the damage and inflammation caused
by alum is the activation of professional antigen presenting cells. As discussed
and demonstrated in Chapter IV, conventional dendritic cells (cDCs) and
inflammatory monocytes (iMonos) are the most prominent players in alum
antigen presentation to T cells. Activation of these and other subsets is critical

105
for the initiation of adaptive immune responses and generation of protective
immune memory.
There seems to be disagreement about whether or not alum crystals are
phagocytosed by immune cells. One group used macrophage cell lines to show
that alum can be engulfed and caused lysosomal destabilization that leads to
inflammasome activation64. Conversely, another group found that alum does not
enter cells; it instead delivers soluble antigen across the plasma membrane71. In
this latter study, Flach et al. reported that alum directly engages lipids in the
plasma membrane of dendritic cells, leading to lipid sorting that initiates abortive
phagocytosis through spleen tyrosine kinase (Syk) and phosphoinositide 3-
kinase (PI3K) signaling71 as well as DC activation. Furthermore, they claimed
the inflammasome is not required for this lipid sorting-mediated DC activation.
Both of these studies used in vitro models to make these discoveries. It is
unknown how the in vivo cell milieu affects these processes, but I am inclined to
think that different immune cell subsets deal with alum differently: engulfing the
crystals or not. Since the route by which APCs take up particulate antigen (pino-,
endo-, or phago-cytosis) affects their ability to present or cross-present antigen to
T cells157, alum uptake or lack thereof by APCs likely affects how and which T
cells the APCs prime in dLNs. Flow cytometry studies, such as mine, that use
fluorescent antigen with alum cannot distinguish whether a cell has internalized
the antigen/alum or has it on its cell surface. Immunohistochemistry must be
done to parse this difference out.

106
Regardless of whether or not alum is engulfed by APCs, alum
vaccinations cause widespread APC activation at the site of injection. Several
studies agree that detection of alum leads to increased antigen uptake, DC
maturation and upregulation of associated cell surface molecules, migration to
draining lymph nodes, and increased peptide presentation on MHC II57–60,62,83,158.
In conclusion, it seems likely that alum triggers a number of redundant
inflammation pathways that all contribute to broad type 2-biased APC activation.
Step 3: Priming adaptive immune cells
Upon activation, APCs migrate to draining lymph nodes to initiate adaptive
immune responses against the offending threat. Antigen-loaded APCs prime
CD4 T cell responses through pMHC II/TCR interactions. These APC/T cell
interactions are stable, mediated by co-stimulatory and adhesion molecules such
as intracellular adhesion molecule-1 (ICAM-1) and lymphocyte function-
associated antigen-1 (LFA-1)49,71. Additionally, primed CD4 T cells receive
chemical signals (such as IL-4 and IL-25) from alum-activated APCs that bias
them toward TH2 and TFH2 differentiation in mice78,79. To prime CD8 T cells,
APCs must cross-present antigen. It is not clear which APCs are responsible for
CD8 T cell priming, though I hypothesize that cells which took up alum crystals
and suffered from lysosomal rupture would be best at presenting antigen on
MHC I because antigen has leaked into their cytosols and thus could be easily
loaded onto MHC I molecules. Primed CD8 T cells differentiate into IFNγ-
producing CTLs79,93. Importantly, alum-primed APCs promote strong enough T

107
cell activation in both CD4 and CD8 T effector subsets to generate protective
memory T cell populations in mice92,125,159 and humans160, though the process by
which memory cell differentiation occurs remains unclear.
Germinal center B cells must acquire and present antigen to TFH cells to
receive ‘help’, so these antigen-bearing cells contribute to (but don’t fully explain)
the large number of antigen-positive B cells in dLNs after alum vaccination
(Chapter IV). It is unknown when, where, or how B cells encounter alum-
adsorbed antigen, nor did my experiments determine if B cells had indeed
engulfed the fluorescent antigen. Activated TFH cells use their upregulated
CXCR5 receptor to migrate into B cell zone germinal centers to aid antigen-
specific B cells with class switching to IgG1 and IgE isotypes (in mice)56 or IgG1
and IgG3 (in humans)40. To the best of my knowledge, the degree of IgE
induction by alum vaccines in humans has not been formally reported. Alum-
activated B cells differentiate into long-lived plasma cells that home to bone
marrow (or alum nodules128) and continue secreting antibodies for months to
years. This maintains a stable serum titer of antigen-specific antibodies that
protect the individual from future infection by pathogens that contain the vaccine
antigens28–30.
Step 4: Resolution and boosting
The nodules that form upon alum injection are long-lived; they can be
recovered from injected animals more than 7 weeks following immunization and
still contain antigen and immunogenic properties at that point127. However, alum

108
nodules are not required for the establishment of an alternative antigen depot,
but this is less well understood26,27. A recent study by Tamburini et al. proposed
that viral or vaccine-elicited inflammation can lead to ‘antigen archiving’ by
lymphatic endothelial cells: these cells capture and harbor antigens involved in
robust immune responses161. Lymphatic endothelial cell-mediated antigen
archiving may provide an explanation for how alum forms an antigen depot aside
from the nodules it forms.
Though alum stimulates effective antibody responses, these responses
are generally not as robust as those generated by live attenuated vaccines or
infections themselves. The long-lived antigen-specific plasma cells eventually
die off, so people require booster shots (every 10 to 15 years in the case of the
tetanus vaccine31) or a priming series of three vaccinations (as with the hepatitis
B vaccine24). Even with this drawback, alum vaccines have revolutionized public
health by providing an adaptable platform with which to provide people with
prophylactic protective immunity against a variety of deadly diseases11,23.
Beyond the Model: Additional Unresolved Questions
T cell responses to alum
It is clear that mice mount TH2 responses to alum, but the evidence for
TH2-bias in humans is much less convincing162. There is a paucity of studies on
human T cell responses to alum, but the few available suggest responses driven
by mixed populations of TH1, TH2, and possibly other CD4 effector subtypes.

109
Infants immunized with alum + Bordatella pertussis antigens generated mixed
TH1/TH2 cytokine profiles upon restimulation of peripheral blood mononuclear
cells44. Furthermore, alum is often used as an adjuvant in allergen preparations
for desensitization of patients with allergies163. A possible explanation for the
effectiveness of this treatment is that alum shifts immune responses away from
TH2 responses and induces IgE-blocking IgG antibodies and regulatory T cells164.
The issue of mouse immunology failing to reflect human immunology is not new
or particularly surprising. However, it must be emphasized and dealt with
carefully within the field of alum research because, at this point, the literature is
too far skewed toward mouse-only studies. It is difficult to evaluate the projected
success or failure of alum with new pathogenic antigens (for vaccines for
emerging diseases, per se) when too little is known about human T cell
responses to this adjuvant. After all, CD4 T cells are critical players in the
development of immune responses.
My experimental approach in Chapter III focused mainly on CD4 and CD8
T cell priming, with little attention paid to effector T cell functionality. It was
recently proposed that TH17 cells are responsible for the effectiveness of alum
vaccines in mice51,165, while TH2 cells are irrelevant. Although I phenotyped
responding CD4 T cells for TFH characteristics in WT and STING-deficient mice
immunized with alum, I did not look for TH17 cells. Mostly, my studies to evaluate
DNA’s role in mediating CD4 T cell priming by alum vaccines leaned heavily on
total numbers of primed CD4 T cells. In future studies, human peripheral blood

110
mononuclear cells should be examined for antigen-specific TH phenotype and
functionality shortly after alum vaccine administrations.
To further complicate matters, one study reported that the particular
antigen adsorbed to alum can alter which TH immune response is mounted in
humans. When humans subjects were immunized with Keyhole Limpet
Hemocyanin (KLH) plus alum, they mounted TH2-biased responses involving IL-
4, -5, -13, and -10 as well as induction of IgG4 antibodies166. However, people
who had received alum vaccinations with tetanus toxoid mounted TH1 responses
indicated by high IFNγ production and little to no IL-13 production in tetanus
toxoid-stimulated peripheral blood mononuclear cells166. This antigen effect
strongly suggests that alum research should be conducted with biomedically
relevant antigens rather than model antigens like chicken ovalbumin. It is
important to discern how alum acts when it has particular pathogenic antigens
adsorbed to it, such as hepatitis B surface antigen, tetanus toxoid, inactivated
hepatitis A virus, human papillomavirus L1 protein, etc. Lastly, this issue leads to
another question: How well do the characteristics of alum responses correlate to
the natural responses elicited by the pathogens against which alum vaccines
protect? A comprehensive study on this question would contribute greatly to our
understanding on alum efficacy. In conclusion, there are discrepancies between
how alum acts in mice versus humans and the body of alum literature is lacking
when it comes to human studies and characterizations of CD4 T cell responses.
It is important to patch these gaps in knowledge to understand why alum is an
effective human vaccine adjuvant.

111
Alum is widely thought to struggle with induction of CD8 CTL
responses162. One group proposed that the robust antibody responses elicited
by alum actually prevent strong CTL priming91. Very little is known about the
quality, homing, or memory induction of CD8 T cells by alum in humans. Our lab
previously reported that, in mice, alum can induce CD8 memory T cells that can
protect against influenza challenge93. However, alum-activated CD8 T cells
produce IFNγ but fail to differentiate into CTLs68,79,93. IFNγ secretion, and not
CTL activity, has been identified as an important aspect of immune responses
against select pathogens, such as Mycobacterium tuberculosis167. Perhaps alum
could stimulate effective CD8 T cells for protection against these sorts of
diseases. Importantly, our group is beginning a study to quantify and examine
antigen-specific CD8 T cells in peripheral blood samples from people who have
previously received the alum-containing hepatitis B vaccine series. This will be
the first study to evaluate CD8 T cell memory responses to an alum vaccine in
human beings. If possible, we will also analyze primary CD8 T cell responses in
recently-vaccinated subjects. In summary, there is a large gap in our knowledge
about the qualities of CD8 T cells stimulated by alum vaccines. Though alum is
recognized for its ability to generate antibody-mediated protection against a
variety of antigens, its ability to stimulate protective CD8 T cell responses may be
underestimated. Using this well-tolerated vaccine adjuvant in new vaccines that
protect against intracellular pathogens (that require CD8 T cell responses) could
be a powerful tool to better global health.

112
B cells as alum/antigen presenters
In Chapter IV, I observed that a large population of B cells was antigen-
loaded within dLN 24 hours after alum immunization. In addition to dendritic cells
and macrophages, antigen-specific B cells are also considered professional
APCs. There is disagreement about whether or not B cells are important in
and/or capable of primary T cell priming168,169. B cells very efficiently present
specific antigens to TFH CD4 T cells in order to obtain help for the production of
high-affinity antibodies. B cells can also present nonspecific antigens derived
from endogenous and pinocytosed proteins, but the outcome is T cell
tolerance169–171. Furthermore, B cells as APCs have been reported to promote
type 2 immune responses because they produce IL-4 in response to IL-12-
producing DCs136. Alum has been documented to induce local IL-12p80 cytokine
concentration when injected intraperitoneally47 in mice, suggesting that IL-12-
induced B cell production of IL-4 could contribute to TH2 skewing in alum
responses. Alternatively, if they secrete IL-4 in an autocrine manner, this IL-4
production could purely be used to promote self-stimulation within an
inflammatory setting since IL-4 is an anti-apoptotic B cell survival and
differentiation factor89,172–174. More studies are required to discern the role of
antigen-loaded B cells in alum immunogenicity.

113
Adjuvant Action of Host DNA
Data in Chapter III indicate that host DNA does not play a major role in
mediating the adjuvant activity of alum in mice. Amy McKee and I both used
STING-deficient animals to test STING’s importance to alum responses as its
main known function is to mediate intracellular DNA-sensing pathways. Using
OVA-3K as a model antigen, she found that STING was necessary for 3K-
specific CD4 T cell responses49, but did not affect CD8 responses (unpublished
data). Using OVA-NP as a model antigen, I found that STING was dispensable
for both NP311-325-specific CD4 and SIINFEKL-specific CD8 T cell responses.
Beside genetic drift within STING-deficient and C57BL/6 animals, this
discrepancy can only be accounted for by the difference in CD4 T cell epitope
used (conjugated to OVA). Since the C57BL/6 murine CD4 T cell response to
NP311-325 within the OVA-NP antigen is quite robust, perhaps alum no longer
requires the STING pathway to be operative to mount a sufficient immune
response. Alternatively, responses to OVA (without NP peptide) or 3K peptide
and alum need stronger instigation in the form of STING pathway activation.
While I found that STING-deficient animals respond normally to alum
vaccines, Marichal et al. reported that IgE responses to alum depend on Tbk1
and Irf3 signaling47, two molecules that are downstream of STING upon
intracellular DNA-sensing. If we assume that alum requires DNA to trigger this
pathway for immunostimulatory effects, my data directly contradict those reported
by Marichal and colleagues. However, STING (and host DNA) may be

114
dispensable while Tbk1 and Irf3 signaling are still required because these two
molecules are involved in more pathways than just STING-associated DNA
sensing. Tbk1 and Irf3 are implicated in a variety of known intracellular signaling
pathways175 (listed in Table 6.1) and, of course, may be activated by yet
unidentified mechanisms. For these reasons, it is still possible that Tbk1 and Irf3
are required for efficient IgE induction upon alum immunization.
Table 6.1. Signaling pathways that involve Tbk1 and Irf3175.
PRR PRR full name Known ligands
Molecule that interacts with Tbk1
TLR3 Toll-like receptor 3 dsRNA TRIF (TIR-domain-containing adapter-inducing interferon-β)
TLR4 Toll-like receptor 4 LPS, gp96176 TRIF RIG-I Retinoic acid-inducible gene 1 dsRNA RIG-I
MDA5 Melanoma differentiation-associated protein 5 dsRNA MDA5
DAI DNA-dependent activator of IFN-regulatory factors dsDNA DAI
NODs (1 and 2)
Nucleotide-binding oligomerization domain containing proteins (1 and 2)
Peptidoglycan RIPK2 (Receptor-interacting serine-threonine kinase 2)
Even though I report that DNA is non-essential for alum responses, I do
not discount that host DNA can still contribute to alum responses via STING
activation and (likely) other mechanisms. As I discussed in my working model of
alum activity, alum is capable of triggering many inflammatory pathways, most of
which have been shown to be dispensable for alum’s overall effect as an
adjuvant. This redundancy may explain why inflammatory detection of chromatin
release by dying cells and neutrophils is not required for alum responses. DNA
has been shown to be immunostimulatory for a variety of cells in a variety of

115
settings95,96,126,177. In fact, mammals have evolved to express a large number of
dsDNA sensing molecules, implying that dsDNA sensing is an important cellular
function178. As an added adjuvant, genomic DNA was shown to activate iMonos,
promote antigen-specific T cell proliferation, and induce IgG1 and IgE antibody
production47. Self DNA can be transported into cells such as plasmacytoid DCs
and monocytes, leading to production of type I IFNs via the STING/Tbk1/Irf3
pathway179. DNA can also activate the inflammasome in an ASC- (apoptosis-
associated speck-like protein containing a caspase activation and recruitment
[CARD] domain) and caspase-1-dependent, but not NLRP3-dependent,
manner156. Furthermore, one study reported that T cells can directly sense
genomic DNA, which is released from dead cells, and promotes the induction of
TH2 differentiation180. This DNA sensing was independent of known nucleic acid
sensors, including TLRs, RLRs, inflammasomes, and STING-dependent sensors.
It is clear that host DNA can indeed stimulate immune responses, but my findings
show that it is not essential for alum responses.
Another way to infer if host DNA plays a role in immune responses is to
examine pathogenic virulence factors for proteins that interact with DNA. Indeed,
Streptococcus pneumoniae escapes from neutrophil extracellular traps
(comprised of a DNA backbone with embedded antimicrobial peptides and
enzymes181) by expressing its own DNase called Sda1182,183. Thus the Sda1
bacterial virulence factor specifically counteracts NETs, implying that NETs
impede infection by this bacterial pathogen. It is important to note that NETs
contain host chromatin, not just host DNA, and chromatin-associated proteins

116
such as HMGB1 are also known to be inflammatory DAMPs95. In conclusion,
release of chromatin as a danger signal from dying cells and within NETs is an
important innate mechanism of immune activation.
Reproducibility in Science and Implications of DNase Contamination
It is no secret that reproducibility (or lack thereof) is a considerable issue
in scientific research184. Our discovery that bovine-sourced commercial DNase
reagents can be contaminated with active proteases (Chapter III) has far-
reaching implications, the first of which is that reported quality assessments of
commercial products may be faulty (i.e. Sigma reported zero protease activity,
though the product contained substantial activity). Though unreproducible
studies are caused by a variety of factors, both innocent and fraudulent, the use
of reagents that have contaminants with off target effects is one possible cause
of this problem. I hope my example can serve as a warning to other researchers
that reagents must be thoroughly analyzed for quality and purity before use.
More importantly, unsuspected protease contamination within the
commonly used Roche DNaseGII and Sigma DNase products has potentially
affected a substantial number of experiments in the last decade or more. Use of
contaminated DNases in any procedure that aims to purify or retain proteins
while degrading DNA would have potentially digested the proteins of interest. At-
risk procedures include chromatin immunoprecipitation, recombinant protein
purification, tissue digestion for isolation of antigen presenting cells, preparation

117
of cell samples for flow cytometric analysis, and unknown numbers of other
procedures. In fact, I used Roche DNaseGII in the purification of recombinant
NP and mutNP, used in Chapters III-V, which raises uncertainty about both
proteins’ intactness when used as vaccine model antigens. Of course, the
experimental conditions during which DNase is used would affect the kinetics of
protease digestion of proteins within samples. However, many protocols allow
DNase digestion to occur for approximately 30 minutes either at room
temperature or 37°C. I demonstrated that this is ample time for active proteases
to cause off target damage (Chapter III).
In conclusion, protease contamination within DNase reagents may have
unintentionally altered the results or interpretations of published scientific studies,
particularly those that are not repeatable. This contamination was present within
at least two lots of a very commonly used commercial product, Roche DNaseGII,
and therefore this discovery could have widespread effects on laboratory
protocols across the nation.
The Future of Vaccines
Finally, I want to briefly share my thoughts on the future of vaccines. Alum
has been reported to cause substantial injury to muscle fibers, necrosis,
microabscesses, and histopathological lesions within injected muscle
tissue94,185,186. Additionally, I already mentioned that antigen-containing alum
nodules remain at the site of injection for at least 7 weeks after immunization127.

118
Are these aspects of alum immunization acceptable? Arguably, damaged
muscle and lingering alum nodules are a relatively small price to pay for
protection against deadly infections such as pertussis, tetanus, or diphtheria or
cancer-causing viruses like HPV16, but is there a better way? Furthermore, it
has been known for quite some time that intradermal administration of vaccines
is generally more effective than intramuscular injection187. Perhaps we should
consider tailoring our standard routes of administration to encourage the highest
efficacy possible for each vaccine? Additionally, different routes of administration
and components within vaccines may be desirable for patients of different age
groups, immune status, or genetic backgrounds. In the past few decades, there
has been a flurry of progress toward the intelligent design of vaccines,
specifically regarding the deliberate selection of antigens and adjuvants for
effective subunit vaccines. For example, simultaneous stimulation of several
TLRs188 and/or targeted activation of specific dendritic cell subsets to achieve
desired immune outcomes189 can drastically affect immune responses to
vaccines.
Lastly, returning to the subject of alum as a versatile vaccine adjuvant,
additional adjuvants (such as TLR4-stimulating LPS derivatives) have been
added to alum to enhance immune responses82,93. Additionally, altered forms of
antigen have been adsorbed to alum, which can also alter the ensuing immune
responses. For example, if mildly heat denatured allergens are adsorbed to alum
instead of soluble protein allergens, IgE-binding epitopes are largely destroyed
but IgG2a antibody responses are augmented, which helps skew allergic

119
responses away from TH2104. This and other studies indicate that either antigen
properties and/or whole particle size affects immune responses190–192. In
summary, there are many known and unknown factors that play into the efficacy
of human vaccines. In my opinion, we should leverage what we know to
generate the most effective vaccines against the most diseases possible, without
getting bogged down in precedent, paperwork, or politics. We have yet to design
effective prophylactic vaccines against HIV or malaria and therapeutic vaccines
against many cancers, but I believe these goals will soon be in reach.
Future Directions
My thesis concludes that DNA plays only a minor role in alum biology,
contrary to two recent publications. Thus, the future of this investigation is no
longer concerned with DNA and alum. Interesting but incomplete studies
presented in this thesis should be followed up. As described in Chapter IV, RNA
sequencing of alum-treated or untreated APCs should be repeated in order to
more confidently identify genes that have altered expression after alum
exposure. Once genes are identified, experiments are needed to validate that
RNA expression alterations correlate with differences in protein expression.
Finally, the importance of these proteins in alum sensing and inflammation may
be verified by performing loss-of-function and phenotype rescue experiments.
This extension of Chapter IV studies could prove useful in validating or
discovering new inflammatory pathways stimulated by alum. Particularly, this

120
approach may validate the lipid sorting57 and lysosomal destabilization49 theories
of alum recognition by APCs. However, there is a large, if incongruent, body of
literature that focuses on how alum stimulates innate immune cells in mice.
Instead of studying alum stimulation of mouse APCs further, there is a great
need for better characterization of T cell and antibody responses to alum in
humans.
Given the expertise of the Kappler/Marrack lab in T cell biology, I propose
that this project move toward better characterizing human T cell responses to
alum vaccines. A reasonable next study would be to examine primary and
memory CD4 and CD8 T cell populations in peripheral blood of alum-vaccinated
human subjects. Quantification of antigen-specific T cells is warranted, though
this requires the creation of human peptide/HLA tetramers that can recognize T
cells specific for alum-adsorbed antigens of interest. Obtaining or designing
these reagents is no small feat. Ideally, functional tests on human T cells would
also be conducted to determine which cytokines they secrete and thus infer
which type(s) of immune response they facilitate. This would help settle the
issue of whether or not alum induces TH2 responses in humans. If possible,
several (or all) currently licensed and used alum vaccines should be examined to
determine if alum behaves similarly or differently based on the type of protein
antigen(s) used and presence of other vaccine components. Information from
such a study should provide definitive information about the adjuvant mechanism
of alum in humans, possibly improving vaccine practices.

121
Concluding Remarks
Alum is a critical component in subunit vaccines that have greatly helped
mankind avoid illness and death from a variety of infectious diseases. Though it
is widely used, alum’s mechanism of action is still mysterious in some ways. My
thesis project contributes to alum research by questioning the theory that DNA
mediates alum activity. I found that DNA is not essential to this process. My
work serves to correct the literature and hopefully redirect other labs’ research
efforts on this subject. This knowledge will improve future studies to elucidate
the mechanism of aluminum salts in future vaccination strategies. Additionally, I
have revealed an important contamination issue within current DNase reagents,
a discovery that has broad implications for scientific protocols in many fields of
research. I hope my thesis work contributes to biomedical literature in a way that
assists other researchers in their goals.

122
REFERENCES 1. Dobzhansky, T. Nothing in Biology Makes Sense except in the Light of Evolution.
Am. Biol. Teach. 35, 125–129 (1973). 2. Travis, J. On the Origin of The Immune System. Science 324, 580–582 (2009). 3. Paust, S. & von Andrian, U. H. Natural killer cell memory. Nat. Immunol. 12, 500–
508 (2011). 4. Marcus, A. & Raulet, D. H. Evidence for Natural Killer Cell Memory. Curr. Biol. CB
23, (2013). 5. Murphy, K. M., Travers, P. & Walport, M. Janeway’s Immunobiology. (Garland
Science, 2007). 6. Sprent, J. & Surh, C. D. T cell memory. Annu. Rev. Immunol. 20, 551–579 (2002). 7. Ahmed, R. & Gray, D. Immunological memory and protective immunity:
understanding their relation. Science 272, 54–60 (1996). 8. McKee, A. S., MacLeod, M. K., Kappler, J. W. & Marrack, P. Immune mechanisms
of protection: can adjuvants rise to the challenge? BMC Biol 8, 37 (2010). 9. Creagh, E. M. & O’Neill, L. A. TLRs, NLRs and RLRs: a trinity of pathogen
sensors that co-operate in innate immunity. Trends Immunol 27, 352–7 (2006). 10. Geijtenbeek, T. B. H. & Gringhuis, S. I. Signalling through C-type lectin receptors:
shaping immune responses. Nat. Rev. Immunol. 9, 465–479 (2009). 11. Clements, C. J. & Griffiths, E. The global impact of vaccines containing aluminium
adjuvants. Vaccine 20, Supplement 3, S24–S33 (2002). 12. Riedel, S. Edward Jenner and the history of smallpox and vaccination. Proc. Bayl.
Univ. Med. Cent. 18, 21–25 (2005). 13. Choices, N. H. S. The history of vaccination - vaccinations - NHS Choices. (2014).
at <http://www.nhs.uk/conditions/vaccinations/pages/the-history-of-vaccination.aspx>
14. Stern, A. M. & Markel, H. The History Of Vaccines And Immunization: Familiar
Patterns, New Challenges. Health Aff. (Millwood) 24, 611–621 (2005). 15. History of Vaccines — A Vaccine History Project of The College of Physicians of
Philadelphia. at <http://www.historyofvaccines.org/> 16. Plotkin, S. A., Orenstein, W. A. & Offit, P. A. Vaccines. (Elsevier Health Sciences,
2008). 17. Ramon, G. Sur l’augmentation anormale de l’antitoxine chez chevaux producteurs
de serum antidiphtérique. Bull Soc Centr Med Vet 101, 227–234 (1925).

123
18. Glenny, A. T., Pope, C. G., Waddington, H. & Wallace, U. The antigenic value of toxoid precipitated by potassium alum. J. Pathol. Bacteriol. 29, 38–45 (1926).
19. Cann, A. J. et al. Reversion to neurovirulence of the live-attenuated Sabin type 3
oral poliovirus vaccine. Nucleic Acids Res. 12, 7787–7792 (1984). 20. Kew, O. M., Sutter, R. W., de Gourville, E. M., Dowdle, W. R. & Pallansch, M. A.
Vaccine-derived polioviruses and the endgame strategy for global polio eradication. Annu. Rev. Microbiol. 59, 587–635 (2005).
21. Pliaka, V., Kyriakopoulou, Z. & Markoulatos, P. Risks associated with the use of
live-attenuated vaccine poliovirus strains and the strategies for control and eradication of paralytic poliomyelitis. Expert Rev. Vaccines 11, 609–628 (2012).
22. Rappuoli, R., Mandl, C. W., Black, S. & De Gregorio, E. Vaccines for the twenty-
first century society. Nat. Rev. Immunol. 11, 865–872 (2011). 23. Roush SW, Murphy TV & Vaccine-Preventable Disease Table Working Group a.
HIstorical comparisons of morbidity and mortality for vaccine-preventable diseases in the united states. JAMA 298, 2155–2163 (2007).
24. Pinkbook | Home | Epidemiology of Vaccine Preventable Diseases | CDC. at
<http://www.cdc.gov/vaccines/pubs/pinkbook/index.html> 25. Glenny, A. T., Buttle, G. a. H. & Stevens, M. F. Rate of disappearance of
diphtheria toxoid injected into rabbits and guinea - pigs: Toxoid precipitated with alum. J. Pathol. Bacteriol. 34, 267–275 (1931).
26. Munks, M. W. et al. Aluminum adjuvants elicit fibrin-dependent extracellular traps
in vivo. Blood 116, 5191–9 (2010). 27. Hutchison, S. et al. Antigen depot is not required for alum adjuvanticity. FASEB J.
Off. Publ. Fed. Am. Soc. Exp. Biol. 26, 1272–1279 (2012). 28. DiLillo, D. J. et al. Maintenance of Long-Lived Plasma Cells and Serological
Memory Despite Mature and Memory B Cell Depletion during CD20 Immunotherapy in Mice. J. Immunol. 180, 361–371 (2008).
29. Ahuja, A., Anderson, S. M., Khalil, A. & Shlomchik, M. J. Maintenance of the
plasma cell pool is independent of memory B cells. Proc. Natl. Acad. Sci. U. S. A. 105, 4802–4807 (2008).
30. Wrammert, J. & Ahmed, R. Maintenance of serological memory. Biol. Chem. 389,
537–539 (2008). 31. Simonsen, O., Kjeldsen, K. & Heron, I. Immunity against tetanus and effect of
revaccination 25-30 years after primary vaccination. Lancet 2, 1240–1242 (1984). 32. Claman, H. N., Chaperon, E. A. & Triplett, R. F. Immunocompetence of
transferred thymus-marrow cell combinations. J. Immunol. Baltim. Md 1950 97, 828–832 (1966).

124
33. Mosmann, T. R., Cherwinski, H., Bond, M. W., Giedlin, M. A. & Coffman, R. L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. Baltim. Md 1950 136, 2348–2357 (1986).
34. Grun, J. L. & Maurer, P. H. Different T helper cell subsets elicited in mice utilizing
two different adjuvant vehicles: the role of endogenous interleukin 1 in proliferative responses. Cell. Immunol. 121, 134–145 (1989).
35. Shokrgozar, M. A. & Shokri, F. Subtype specificity of anti-HBs antibodies
produced by human B-cell lines isolated from normal individuals vaccinated with recombinant hepatitis B vaccine. Vaccine 20, 2215–2220 (2002).
36. Skvaril, F. & Joller-Jemelka, H. IgG subclasses of anti-HBs antibodies in
vaccinated and nonvaccinated individuals and in anti-HBs immunoglobulin preparations. Int. Arch. Allergy Appl. Immunol. 73, 330–337 (1984).
37. Honorati, M. C., Borzì, R. M., Dolzani, P., Toneguzzi, S. & Facchini, A. Distribution
of IgG subclasses after anti-hepatitis B virus immunization with a recombinant vaccine. Int. J. Clin. Lab. Res. 27, 202–206 (1997).
38. Wang, L. et al. Utilizing self-prepared ELISA plates for a cross-population study of
different anti-HBe IgG subclass profiles. J. Med. Virol. 79, 495–502 (2007). 39. Wang, L. et al. Anti-hepatitis B surface antigen IgG1 subclass is predominant in
individuals who have recovered from hepatitis B virus infection, chronic carriers, and vaccinees. Med. Microbiol. Immunol. (Berl.) 194, 33–38 (2005).
40. Gregorek, H. et al. The IgG subclass profile of anti-HBs response in vaccinated
children and children seroconverted after natural infection. Vaccine 18, 1210–1217 (2000).
41. Ruiz, W., McClements, W. L., Jansen, K. U. & Esser, M. T. Kinetics and isotype
profile of antibody responses in rhesus macaques induced following vaccination with HPV 6, 11, 16 and 18 L1-virus-like particles formulated with or without Merck aluminum adjuvant. J. Immune Based Ther. Vaccines 3, 2 (2005).
42. Invivogen. Immunoglobulin G Review. (2011). at <www.invivogen.com> 43. Dinç But, A., Ersoy, Y., Ozerol, I. H. & Firat, M. [Investigation of serum IgG
subclass distributions and anti-HBs response in healthy adults after hepatitis B vaccination]. Mikrobiyoloji Bül. 39, 483–490 (2005).
44. Ryan, M., Gothefors, L., Storsaeter, J. & Mills, K. H. Bordetella pertussis-specific
Th1/Th2 cells generated following respiratory infection or immunization with an acellular vaccine: comparison of the T cell cytokine profiles in infants and mice. Dev. Biol. Stand. 89, 297–305 (1997).
45. Schure, R.-M. et al. T-cell responses before and after the fifth consecutive
acellular pertussis vaccination in 4-year-old Dutch children. Clin. Vaccine Immunol. CVI 19, 1879–1886 (2012).

125
46. Wang, H.-B. & Weller, P. F. Pivotal advance: eosinophils mediate early alum adjuvant-elicited B cell priming and IgM production. J. Leukoc. Biol. 83, 817–821 (2008).
47. Marichal, T. et al. DNA released from dying host cells mediates aluminum
adjuvant activity. Nat Med 17, 996–1002 (2011). 48. Jordan, M. B., Mills, D. M., Kappler, J., Marrack, P. & Cambier, J. C. Promotion of
B cell immune responses via an alum-induced myeloid cell population. Science 304, 1808–1810 (2004).
49. McKee, A. S. et al. Host DNA released in response to aluminum adjuvant
enhances MHC class II-mediated antigen presentation and prolongs CD4 T-cell interactions with dendritic cells. Proc. Natl. Acad. Sci. 110, E1122–E1131 (2013).
50. McKee, A. S. et al. Gr1+IL-4-producing innate cells are induced in response to
Th2 stimuli and suppress Th1-dependent antibody responses. Int. Immunol. 20, 659–669 (2008).
51. Ross, P. J. et al. Relative Contribution of Th1 and Th17 Cells in Adaptive
Immunity to Bordetella pertussis: Towards the Rational Design of an Improved Acellular Pertussis Vaccine. PLoS Pathog. 9, e1003264 (2013).
52. Spellberg, B. & Edwards, J. E. Type 1/Type 2 Immunity in Infectious Diseases.
Clin. Infect. Dis. 32, 76–102 (2001). 53. Lundgren, M. et al. Interleukin 4 induces synthesis of IgE and IgG4 in human B
cells. Eur. J. Immunol. 19, 1311–1315 (1989). 54. Punnonen, J. & Vries, J. E. de. IL-13 induces proliferation, Ig isotype switching,
and Ig synthesis by immature human fetal B cells. J. Immunol. 152, 1094–1102 (1994).
55. Tsai, T.-H. et al. Study of IgG subclass profiles of anti-HBs in populations with
different HBV infection status. Viral Immunol. 19, 277–284 (2006). 56. Marrack, P., McKee, A. S. & Munks, M. W. Towards an understanding of the
adjuvant action of aluminium. Nat Rev Immunol 9, 287–93 (2009). 57. Mannhalter, J. W., Neychev, H. O., Zlabinger, G. J., Ahmad, R. & Eibl, M. M.
Modulation of the human immune response by the non-toxic and non-pyrogenic adjuvant aluminium hydroxide: effect on antigen uptake and antigen presentation. Clin. Exp. Immunol. 61, 143–151 (1985).
58. Ulanova, M., Tarkowski, A., Hahn-Zoric, M. & Hanson, L. Å. The Common
Vaccine Adjuvant Aluminum Hydroxide Up-Regulates Accessory Properties of Human Monocytes via an Interleukin-4-Dependent Mechanism. Infect. Immun. 69, 1151–1159 (2001).
59. Kool, M. et al. Alum adjuvant boosts adaptive immunity by inducing uric acid and
activating inflammatory dendritic cells. J. Exp. Med. 205, 869–882 (2008).

126
60. Calabro, S. et al. Vaccine adjuvants alum and MF59 induce rapid recruitment of neutrophils and monocytes that participate in antigen transport to draining lymph nodes. Vaccine 29, 1812–1823 (2011).
61. Langlet, C. et al. CD64 expression distinguishes monocyte-derived and
conventional dendritic cells and reveals their distinct role during intramuscular immunization. J Immunol 188, 1751–60 (2012).
62. Sokolovska, A., Hem, S. L. & HogenEsch, H. Activation of dendritic cells and
induction of CD4(+) T cell differentiation by aluminum-containing adjuvants. Vaccine 25, 4575–4585 (2007).
63. Sun, H., Pollock, K. G. J. & Brewer, J. M. Analysis of the role of vaccine adjuvants
in modulating dendritic cell activation and antigen presentation in vitro. Vaccine 21, 849–855 (2003).
64. Hornung, V. et al. Silica crystals and aluminum salts activate the NALP3
inflammasome through phagosomal destabilization. Nat Immunol 9, 847–56 (2008).
65. Eisenbarth, S. C., Colegio, O. R., O’Connor, W., Sutterwala, F. S. & Flavell, R. A.
Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 453, 1122–1126 (2008).
66. Franchi, L. & Núñez, G. The Nlrp3 inflammasome is critical for aluminium
hydroxide-mediated IL-1beta secretion but dispensable for adjuvant activity. Eur. J. Immunol. 38, 2085–2089 (2008).
67. Li, H., Nookala, S. & Re, F. Aluminum hydroxide adjuvants activate caspase-1
and induce IL-1beta and IL-18 release. J. Immunol. Baltim. Md 1950 178, 5271–5276 (2007).
68. McKee, A. S. et al. Alum induces innate immune responses through macrophage
and mast cell sensors, but these sensors are not required for alum to act as an adjuvant for specific immunity. J Immunol 183, 4403–14 (2009).
69. Sharp, F. A. et al. Uptake of particulate vaccine adjuvants by dendritic cells
activates the NALP3 inflammasome. Proc Natl Acad Sci U A 106, 870–5 (2009). 70. Ng, G. et al. Receptor-independent, direct membrane binding leads to cell-surface
lipid sorting and Syk kinase activation in dendritic cells. Immunity 29, 807–818 (2008).
71. Flach, T. L. et al. Alum interaction with dendritic cell membrane lipids is essential
for its adjuvanticity. Nat. Med. 17, 479–487 (2011). 72. Geissmann, F. et al. Development of Monocytes, Macrophages, and Dendritic
Cells. Science 327, 656–661 (2010). 73. Guilliams, M. et al. Dendritic cells, monocytes and macrophages: a unified
nomenclature based on ontogeny. Nat. Rev. Immunol. 14, 571–578 (2014).

127
74. Heath, W. R. & Carbone, F. R. Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat. Immunol. 10, 1237–1244 (2009).
75. Helft, J., Ginhoux, F., Bogunovic, M. & Merad, M. Origin and functional
heterogeneity of non-lymphoid tissue dendritic cells in mice. Immunol. Rev. 234, 55–75 (2010).
76. Guilliams, M. et al. From skin dendritic cells to a simplified classification of human
and mouse dendritic cell subsets. Eur. J. Immunol. 40, 2089–2094 (2010). 77. Geissmann, F., Jung, S. & Littman, D. R. Blood monocytes consist of two principal
subsets with distinct migratory properties. Immunity 19, 71–82 (2003). 78. Serre, K. et al. Molecular differences between the divergent responses of
ovalbumin-specific CD4 T cells to alum-precipitated ovalbumin compared to ovalbumin expressed by Salmonella. Mol. Immunol. 45, 3558–3566 (2008).
79. Serre, K. et al. IL-4 directs both CD4 and CD8 T cells to produce Th2 cytokines in
vitro, but only CD4 T cells produce these cytokines in response to alum-precipitated protein in vivo. Mol. Immunol. 47, 1914–1922 (2010).
80. Serre, K. et al. Selective effects of NF-κB1 deficiency in CD4+ T cells on Th2 and
TFh induction by alum-precipitated protein vaccines. Eur. J. Immunol. 41, 1573–1582 (2011).
81. Zhu, J. T helper 2 (Th2) cell differentiation, type 2 innate lymphoid cell (ILC2)
development and regulation of interleukin-4 (IL-4) and IL-13 production. Cytokine (2015). doi:10.1016/j.cyto.2015.05.010
82. Didierlaurent, A. M. et al. AS04, an aluminum salt- and TLR4 agonist-based
adjuvant system, induces a transient localized innate immune response leading to enhanced adaptive immunity. J. Immunol. Baltim. Md 1950 183, 6186–6197 (2009).
83. Kool, M. et al. Cutting Edge: Alum Adjuvant Stimulates Inflammatory Dendritic
Cells through Activation of the NALP3 Inflammasome. J. Immunol. 181, 3755–3759 (2008).
84. Mosca, F. et al. Molecular and cellular signatures of human vaccine adjuvants.
Proc. Natl. Acad. Sci. U. S. A. 105, 10501–10506 (2008). 85. Lambrecht, B. N., Kool, M., Willart, M. A. & Hammad, H. Mechanism of action of
clinically approved adjuvants. Curr. Opin. Immunol. 21, 23–29 (2009). 86. Kuroda, E. et al. Silica crystals and aluminum salts regulate the production of
prostaglandin in macrophages via NALP3 inflammasome-independent mechanisms. Immunity 34, 514–526 (2011).

128
87. Brewer, J. M., Conacher, M., Satoskar, A., Bluethmann, H. & Alexander, J. In interleukin-4-deficient mice, alum not only generates T helper 1 responses equivalent to freund’s complete adjuvant, but continues to induce T helper 2 cytokine production. Eur. J. Immunol. 26, 2062–2066 (1996).
88. Johnston, R. J. et al. Bcl6 and Blimp-1 Are Reciprocal and Antagonistic
Regulators of T Follicular Helper Cell Differentiation. Science 325, 1006–1010 (2009).
89. Crotty, S. Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 29, 621–663
(2011). 90. Kool, M., Fierens, K. & Lambrecht, B. N. Alum adjuvant: Some of the Tricks of the
oldest adjuvant. J Med Microbiol (2011). doi:jmm.0.038943-0 [pii] 10.1099/jmm.0.038943-0
91. Speidel, K. et al. Priming of cytotoxic T lymphocytes by five heat-aggregated
antigens in vivo: conditions, efficiency, and relation to antibody responses. Eur. J. Immunol. 27, 2391–2399 (1997).
92. Macleod, M. K. L. et al. Influenza nucleoprotein delivered with aluminium salts
protects mice from an influenza A virus that expresses an altered nucleoprotein sequence. PloS One 8, e61775 (2013).
93. MacLeod, M. K. et al. Vaccine adjuvants aluminum and monophosphoryl lipid A
provide distinct signals to generate protective cytotoxic memory CD8 T cells. Proc Natl Acad Sci U A 108, 7914–9 (2011).
94. Goto, N. et al. Local tissue irritating effects and adjuvant activities of calcium
phosphate and aluminium hydroxide with different physical properties. Vaccine 15, 1364–1371 (1997).
95. Kono, H. & Rock, K. L. How dying cells alert the immune system to danger. Nat.
Rev. Immunol. 8, 279–289 (2008). 96. Chen, G. Y. & Nuñez, G. Sterile inflammation: sensing and reacting to damage.
Nat. Rev. Immunol. 10, 826–837 (2010). 97. De Groot, A. S. & Scott, D. W. Immunogenicity of protein therapeutics. Trends
Immunol 28, 482–90 (2007). 98. Aguzzi, A. & O’Connor, T. Protein aggregation diseases: pathogenicity and
therapeutic perspectives. Nat Rev Drug Discov 9, 237–48 (2010). 99. Barbosa, M. D. & Celis, E. Immunogenicity of protein therapeutics and the
interplay between tolerance and antibody responses. Drug Discov Today 12, 674–81 (2007).
100. Zaborsky, N. et al. Antigen aggregation decides the fate of the allergic immune
response. J Immunol 184, 725–35 (2010).

129
101. Gregersen, N., Bross, P., Vang, S. & Christensen, J. H. Protein misfolding and human disease. Annu Rev Genomics Hum Genet 7, 103–24 (2006).
102. Bruijn, L. I. et al. Aggregation and motor neuron toxicity of an ALS-linked SOD1
mutant independent from wild-type SOD1. Science 281, 1851–4 (1998). 103. Chiti, F. & Dobson, C. M. Protein misfolding, functional amyloid, and human
disease. Annu Rev Biochem 75, 333–66 (2006). 104. Johansen, P. et al. Heat denaturation, a simple method to improve the
immunotherapeutic potential of allergens. Eur J Immunol 35, 3591–8 (2005). 105. Rosenberg, A. S. Effects of protein aggregates: an immunologic perspective.
AAPS J 8, E501–7 (2006). 106. Croguennec, T., Renault, A., Beaufils, S., Dubois, J. J. & Pezennec, S. Interfacial
properties of heat-treated ovalbumin. J Colloid Interface Sci 315, 627–36 (2007). 107. Tani, F., Shirai, N., Nakanishi, Y. & Kitabatake, N. Analysis of molecular
interactions in heat-induced aggregation of a non-inhibitory serpin ovalbumin using a molecular chaperone. Biosci Biotechnol Biochem 67, 1030–8 (2003).
108. Fink, A. L. Protein aggregation: folding aggregates, inclusion bodies and amyloid.
Fold Des 3, R9–23 (1998). 109. Seong, S. Y. & Matzinger, P. Hydrophobicity: an ancient damage-associated
molecular pattern that initiates innate immune responses. Nat Rev Immunol 4, 469–78 (2004).
110. Crawford, F., Kozono, H., White, J., Marrack, P. & Kappler, J. Detection of
antigen-specific T cells with multivalent soluble class II MHC covalent peptide complexes. Immunity 8, 675–682 (1998).
111. Crowe, S. R. et al. Uneven distribution of MHC class II epitopes within the
influenza virus. Vaccine 24, 457–67 (2006). 112. Ng, A. K. et al. Structure of the influenza virus A H5N1 nucleoprotein: implications
for RNA binding, oligomerization, and vaccine design. FASEB J 22, 3638–47 (2008).
113. Lam, J. S., Mansour, M. K., Specht, C. A. & Levitz, S. M. A model vaccine
exploiting fungal mannosylation to increase antigen immunogenicity. J. Immunol. Baltim. Md 1950 175, 7496–7503 (2005).
114. Luong, M., Lam, J. S., Chen, J. & Levitz, S. M. Effects of fungal N- and O-linked
mannosylation on the immunogenicity of model vaccines. Vaccine 25, 4340–4344 (2007).
115. Abe, A. & Liao, T. H. The immunological and structural comparisons of
deoxyribonucleases I. Glycosylation differences between bovine pancreatic and parotid deoxyribonucleases. J. Biol. Chem. 258, 10283–10288 (1983).

130
116. Keil, B. Specificity of Proteolysis. (Springer-Verlag, 1992). 117. Rodriguez, J., Gupta, N., Smith, R. D. & Pevzner, P. A. Does trypsin cut before
proline? J. Proteome Res. 7, 300–305 (2008). 118. Pan, C. Q., Ulmer, J. S., Herzka, A. & Lazarus, R. A. Mutational analysis of
human DNase I at the DNA binding interface: implications for DNA recognition, catalysis, and metal ion dependence. Protein Sci. Publ. Protein Soc. 7, 628–636 (1998).
119. Croset, A. et al. Differences in the glycosylation of recombinant proteins
expressed in HEK and CHO cells. J. Biotechnol. 161, 336–348 (2012). 120. Ishikawa, H., Ma, Z. & Barber, G. N. STING regulates intracellular DNA-mediated,
type I interferon-dependent innate immunity. Nature 461, 788–792 (2009). 121. Zhang, Z. et al. The helicase DDX41 senses intracellular DNA mediated by the
adaptor STING in dendritic cells. Nat. Immunol. 12, 959–965 (2011). 122. Sun, L., Wu, J., Du, F., Chen, X. & Chen, Z. J. Cyclic GMP-AMP synthase is a
cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013).
123. Poholek, A. C. et al. In vivo regulation of Bcl6 and T follicular helper cell
development. J. Immunol. Baltim. Md 1950 185, 313–326 (2010). 124. Wang, H. et al. The transcription factor Foxp1 is a critical negative regulator of the
differentiation of follicular helper T cells. Nat. Immunol. 15, 667–675 (2014). 125. MacLeod, M. K. L. et al. Memory CD4 T Cells That Express CXCR5 Provide
Accelerated Help to B Cells. J. Immunol. 186, 2889–2896 (2011). 126. Ishii, K. J. et al. Genomic DNA Released by Dying Cells Induces the Maturation of
APCs. J. Immunol. 167, 2602–2607 (2001). 127. Harrison, W. T. Some Observations on the Use of Alum Precipitated Diphtheria
Toxoid. Am. J. Public Health Nations Health 25, 298–300 (1935). 128. White, R. G., Coons, A. H. & Connolly, J. M. Studies on antibody production. III.
The alum granuloma. J. Exp. Med. 102, 73–82 (1955). 129. Anderson, K. P. et al. Effect of dose and immunization schedule on immune
response of baboons to recombinant glycoprotein 120 of HIV-1. J. Infect. Dis. 160, 960–969 (1989).
130. Morokata, T., Ishikawa, J. & Yamada, T. Antigen dose defines T helper 1 and T
helper 2 responses in the lungs of C57BL/6 and BALB/c mice independently of splenic responses. Immunol. Lett. 72, 119–126 (2000).
131. Keck, S. et al. Antigen affinity and antigen dose exert distinct influences on CD4
T-cell differentiation. Proc. Natl. Acad. Sci. U. S. A. 111, 14852–14857 (2014).

131
132. Langley, J. M. et al. A dose-ranging study of a subunit Respiratory Syncytial Virus subtype A vaccine with and without aluminum phosphate adjuvantation in adults > or =65 years of age. Vaccine 27, 5913–5919 (2009).
133. Moon, J. J. et al. Naive CD4(+) T cell frequency varies for different epitopes and
predicts repertoire diversity and response magnitude. Immunity 27, 203–13 (2007).
134. DeFranco, A. L. ‘Dangerous Crystals’. Immunity 29, 670–671 (2008). 135. Stockinger, B., Zal, T., Zal, A. & Gray, D. B cells solicit their own help from T cells.
J. Exp. Med. 183, 891–899 (1996). 136. Skok, J., Poudrier, J. & Gray, D. Dendritic cell-derived IL-12 promotes B cell
induction of Th2 differentiation: a feedback regulation of Th1 development. J. Immunol. Baltim. Md 1950 163, 4284–4291 (1999).
137. Miles, K. et al. A tolerogenic role for Toll-like receptor 9 is revealed by B-cell
interaction with DNA complexes expressed on apoptotic cells. Proc. Natl. Acad. Sci. 109, 887–892 (2012).
138. Kastenmüller, K. et al. Protective T cell immunity in mice following protein-TLR7/8
agonist-conjugate immunization requires aggregation, type I IFN, and multiple DC subsets. J. Clin. Invest. 0, 0–0 (2011).
139. Kovacsovics-Bankowski, M., Clark, K., Benacerraf, B. & Rock, K. L. Efficient major
histocompatibility complex class I presentation of exogenous antigen upon phagocytosis by macrophages. Proc Natl Acad Sci U A 90, 4942–6 (1993).
140. Unanue, E. R. The regulatory role of macrophages in antigenic stimulation. Part
Two: symbiotic relationship between lymphocytes and macrophages. Adv Immunol 31, 1–136 (1981).
141. Bachmann, M. F. & Jennings, G. T. Vaccine delivery: a matter of size, geometry,
kinetics and molecular patterns. Nat Rev Immunol 10, 787–96 (2010). 142. Dresser, D. W. Specific inhibition of antibody production. II. Paralysis induced in
adult mice by small quantities of protein antigen. Immunology 5, 378–88 (1962). 143. Claman, H. N. Tolerance to a Protein Antigen in Adult Mice and the Effect of
Nonspecific Factors. J Immunol 91, 833–9 (1963). 144. Dresser, D. W. Specific inhibition of antibody production. IV. Standardization of
the antigen-elimination test; immunological paralysis of mice previously immunized. Immunology 9, 261–73 (1965).
145. Baker, M. P., Reynolds, H. M., Lumicisi, B. & Bryson, C. J. Immunogenicity of
protein therapeutics. Self Nonself 1, 314–322 (2010). 146. Roy, P. & Noad, R. Virus-like particles as a vaccine delivery system: myths and
facts. Adv Exp Med Biol 655, 145–58 (2009).

132
147. Wilschut, J. Influenza vaccines: the virosome concept. Immunol Lett 122, 118–21 (2009).
148. Dong, A., Jones, L. S., Kerwin, B. A., Krishnan, S. & Carpenter, J. F. Secondary
structures of proteins adsorbed onto aluminum hydroxide: infrared spectroscopic analysis of proteins from low solution concentrations. Anal Biochem 351, 282–9 (2006).
149. Plakoutsi, G. et al. Evidence for a mechanism of amyloid formation involving
molecular reorganisation within native-like precursor aggregates. J Mol Biol 351, 910–22 (2005).
150. St. Clair, N., Shenoy, B., Jacob, L. D. & Margolin, A. L. Cross-linked protein
crystals for vaccine delivery. Proc. Natl. Acad. Sci. U. S. A. 96, 9469–9474 (1999). 151. Hermeling, S. et al. Antibody response to aggregated human interferon alpha2b in
wild-type and transgenic immune tolerant mice depends on type and level of aggregation. J Pharm Sci 95, 1084–96 (2006).
152. Srivastava, P. Interaction of heat shock proteins with peptides and antigen
presenting cells: chaperoning of the innate and adaptive immune responses. Annu Rev Immunol 20, 395–425 (2002).
153. Osterloh, A. & Breloer, M. Heat shock proteins: linking danger and pathogen
recognition. Med Microbiol Immunol 197, 1–8 (2008). 154. Wang, Y., Rahman, D. & Lehner, T. A comparative study of stress-mediated
immunological functions with the adjuvanticity of alum. J. Biol. Chem. 287, 17152–17160 (2012).
155. Brinkmann, V. et al. Neutrophil Extracellular Traps Kill Bacteria. Science 303,
1532–1535 (2004). 156. Muruve, D. A. et al. The inflammasome recognizes cytosolic microbial and host
DNA and triggers an innate immune response. Nature 452, 103–107 (2008). 157. Kamphorst, A. O., Guermonprez, P., Dudziak, D. & Nussenzweig, M. C. Route of
antigen uptake differentially impacts presentation by dendritic cells and activated monocytes. J Immunol 185, 3426–35 (2010).
158. Ghimire, T. R., Benson, R. A., Garside, P. & Brewer, J. M. Alum increases antigen
uptake, reduces antigen degradation and sustains antigen presentation by DCs in vitro. Immunol. Lett. 147, 55–62 (2012).
159. Serre, K. et al. Early simultaneous production of intranodal CD4 Th2 effectors and
recirculating rapidly responding central-memory-like CD4 T cells. Eur. J. Immunol. 39, 1573–1586 (2009).
160. Axelsson, S. et al. Long-lasting immune responses 4 years after GAD-alum
treatment in children with type 1 diabetes. PloS One 6, e29008 (2011).

133
161. Tamburini, B. A., Burchill, M. A. & Kedl, R. M. Antigen capture and archiving by lymphatic endothelial cells following vaccination or viral infection. Nat. Commun. 5, 3989 (2014).
162. HogenEsch, H. Mechanism of Immunopotentiation and Safety of Aluminum
Adjuvants. Front. Immunol. 3, (2013). 163. Francis, J. N. & Durham, S. R. Adjuvants for allergen immunotherapy:
experimental results and clinical perspectives. Curr. Opin. Allergy Clin. Immunol. 4, 543–548 (2004).
164. Eifan, A. O., Shamji, M. H. & Durham, S. R. Long-term clinical and immunological
effects of allergen immunotherapy. Curr. Opin. Allergy Clin. Immunol. 11, 586–593 (2011).
165. Mills, K. H. G., Dungan, L. S., Jones, S. A. & Harris, J. The role of inflammasome-
derived IL-1 in driving IL-17 responses. J. Leukoc. Biol. 93, 489–497 (2013). 166. Spazierer, D. et al. T helper 2 biased de novo immune response to Keyhole
Limpet Hemocyanin in humans. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 39, 999–1008 (2009).
167. Tascon, R. E., Stavropoulos, E., Lukacs, K. V. & Colston, M. J. Protection against
Mycobacterium tuberculosisInfection by CD8+ T Cells Requires the Production of Gamma Interferon. Infect. Immun. 66, 830–834 (1998).
168. Constant, S. L. B lymphocytes as antigen-presenting cells for CD4+ T cell priming
in vivo. J. Immunol. Baltim. Md 1950 162, 5695–5703 (1999). 169. Chen, X. & Jensen, P. E. The role of B lymphocytes as antigen-presenting cells.
Arch. Immunol. Ther. Exp. (Warsz.) 56, 77–83 (2008). 170. Raimondi, G., Zanoni, I., Citterio, S., Ricciardi-Castagnoli, P. & Granucci, F.
Induction of peripheral T cell tolerance by antigen-presenting B cells. I. Relevance of antigen presentation persistence. J. Immunol. Baltim. Md 1950 176, 4012–4020 (2006).
171. Raimondi, G., Zanoni, I., Citterio, S., Ricciardi-Castagnoli, P. & Granucci, F.
Induction of peripheral T cell tolerance by antigen-presenting B cells. II. Chronic antigen presentation overrules antigen-presenting B cell activation. J. Immunol. Baltim. Md 1950 176, 4021–4028 (2006).
172. Paul, W. E. & Ohara, J. B-Cell Stimulatory Factor-1/Interleukin 4. Annu. Rev.
Immunol. 5, 429–459 (1987). 173. Nelms, K., Keegan, A. D., Zamorano, J., Ryan, J. J. & Paul, W. E. THE IL-4
RECEPTOR: Signaling Mechanisms and Biologic Functions. Annu. Rev. Immunol. 17, 701–738 (1999).

134
174. Wurster, A. L., Rodgers, V. L., White, M. F., Rothstein, T. L. & Grusby, M. J. Interleukin-4-mediated Protection of Primary B Cells from Apoptosis through Stat6-dependent Up-regulation of Bcl-xL. J. Biol. Chem. 277, 27169–27175 (2002).
175. Trinchieri, G. Type I interferon: friend or foe? J. Exp. Med. 207, 2053–2063
(2010). 176. Vabulas, R. M. et al. The endoplasmic reticulum-resident heat shock protein Gp96
activates dendritic cells via the Toll-like receptor 2/4 pathway. J Biol Chem 277, 20847–53 (2002).
177. Ablasser, A., Hertrich, C., Waßermann, R. & Hornung, V. Nucleic acid driven
sterile inflammation. Clin. Immunol. Orlando Fla 147, 207–215 (2013). 178. Unterholzner, L. The interferon response to intracellular DNA: Why so many
receptors? Immunobiology 218, 1312–1321 (2013). 179. Chamilos, G. et al. Cytosolic sensing of extracellular self-DNA transported into
monocytes by the antimicrobial peptide LL37. Blood 120, 3699–3707 (2012). 180. Imanishi, T. et al. Nucleic acid sensing by T cells initiates Th2 cell differentiation.
Nat. Commun. 5, 3566 (2014). 181. Wartha, F., Beiter, K., Normark, S. & Henriques-Normark, B. Neutrophil
extracellular traps: casting the NET over pathogenesis. Curr. Opin. Microbiol. 10, 52–56 (2007).
182. Beiter, K. et al. An endonuclease allows Streptococcus pneumoniae to escape
from neutrophil extracellular traps. Curr. Biol. CB 16, 401–407 (2006). 183. Buchanan, J. T. et al. DNase expression allows the pathogen group A
Streptococcus to escape killing in neutrophil extracellular traps. Curr. Biol. CB 16, 396–400 (2006).
184. Yaffe, M. B. Reproducibility in science. Sci. Signal. 8, eg5–eg5 (2015). 185. Goto, N. & Akama, K. Histopathological studies of reactions in mice injected with
aluminum-adsorbed tetanus toxoid. Microbiol. Immunol. 26, 1121–1132 (1982). 186. Verdier, F. et al. Aluminium assay and evaluation of the local reaction at several
time points after intramuscular administration of aluminium containing vaccines in the Cynomolgus monkey. Vaccine 23, 1359–1367 (2005).
187. Abadie, V. et al. Original encounter with antigen determines antigen-presenting
cell imprinting of the quality of the immune response in mice. PloS One 4, e8159 (2009).
188. Kasturi, S. P. et al. Programming the magnitude and persistence of antibody
responses with innate immunity. Nature 470, 543–547 (2011).

135
189. Desch, A. N. et al. Dendritic cell subsets require cis-activation for cytotoxic CD8 T-cell induction. Nat. Commun. 5, 4674 (2014).
190. Xiang, S. D. et al. Pathogen recognition and development of particulate vaccines:
does size matter? Methods 40, 1–9 (2006). 191. Manolova, V. et al. Nanoparticles target distinct dendritic cell populations
according to their size. Eur J Immunol 38, 1404–13 (2008). 192. Rettig, L. et al. Particle size and activation threshold: a new dimension of danger
signaling. Blood 115, 4533–41 (2010).
Copyright © 2022 FDOKUMEN




















