
Action Knolwedge and Symbolic Knowledge: The Computer as Mediator
Upload
univ-lille1Category
view
1download
0
Journal of Structural Biology 174 (2011) 245–251
Contents lists available at ScienceDirect
Journal of Structural Biology
journal homepage: www.elsevier .com/locate /y jsbi
Structure Report
NMR structure of the human Mediator MED25 ACID domain
François Bontems a,⇑, Alexis Verger b, Frédérique Dewitte b, Zoé Lens b, Jean-Luc Baert b, Elisabeth Ferreira b,Yvan de Launoit c, Christina Sizun a, Eric Guittet a, Vincent Villeret b,⇑, Didier Monté b
a CNRS UPR 2301, Chimie et biochimie structurales, Institut de Chimie des Substances Naturelles, Centre de recherche de Gif-sur-Yvette, 91190 Gif-sur-Yvette, Franceb CNRS USR 3078, Institut de Recherche Interdisciplinaire, Université de Lille 1 – Université de Lille 2, Parc CNRS de la Haute Borne, 50 avenue de Halley – BP 70478, 59658Villeneuve d’Ascq Cedex, Francec CNRS UMR 8161, Institut de Biologie de Lille, Université de Lille – Nord de France, Institut Pasteur de Lille, BP447, 1 rue Calmette, 59021 Lille, France
a r t i c l e i n f o
Article history:Received 27 August 2010Received in revised form 20 October 2010Accepted 20 October 2010Available online 23 October 2010
Keywords:NMRMediatorMED25PTOVVP16Transcriptional activation domain
1047-8477/$ - see front matter � 2010 Elsevier Inc. Adoi:10.1016/j.jsb.2010.10.011
Abbreviations: TAD, Transcriptional activation doActivator interacting domain; PTOV, Prostate tumouparalog and ortholog C-terminal.⇑ Corresponding authors.
E-mail addresses: [email protected]@iri.univ-lille1.fr (V. Villeret).
a b s t r a c t
MED25 (ARC92/ACID1) is a 747 residues subunit specific to higher eukaryote Mediator complex, anessential component of the RNA polymerase II general transcriptional machinery. MED25 is a target ofthe Herpes simplex virus transactivator protein VP16. MED25 interacts with VP16 through a centralMED25 PTOV (Prostate tumour overexpressed)/ACID (Activator interacting domain) domain of unknownstructure. As a first step towards understanding the mechanism of recruitment of transactivationdomains by MED25, we report here the NMR structure of the MED25 ACID domain. The domain architec-ture consists of a closed b-barrel with seven strands (D1–D7) and three a-helices (H1–H3), an architectureshowing similarities to that of the SPOC (Spen paralog and ortholog C-terminal domain) domain-likesuperfamily. Preliminary NMR chemical shift mapping showed that VP16 H2 (VP16C) interacts withMED25 ACID through one face of the b-barrel, defined by strands B4–B7–B6.
� 2010 Elsevier Inc. All rights reserved.
1. Introduction
Mediator is an essential component of the general transcrip-tional machinery and plays a crucial part in the activation andrepression of eukaryotic mRNA synthesis (Taatjes, 2010). Mediatorwas first discovered in Saccharomyces cerevisiae. Yeast Mediator iscomposed of 20 subunits, which are present in three distinct Medi-ator subdomains referred to as ‘‘head’’, ‘‘middle’’ and ‘‘tail mod-ules’’. An additional module, which includes a kinase–cyclin pair,is associated with a subset of yeast Mediator complexes and hasbeen implicated in repression of a subset of genes (Blazek et al.,2005; Taatjes, 2010). The topology and the subunit organizationof these modules have been characterized by cryo-EM, direct inter-action studies and partial structural data (Cai et al., 2010; Chadickand Asturias, 2005). Despite the fact that some Mediator com-plexes are only found in metazoan, cross-species comparisons havedetected metazoan counterparts for nearly all yeast Mediator sub-units (Bourbon, 2008). Mammalian Mediator-like complexes con-
ll rights reserved.
main; MED, Mediator; ACID,r overexpressed; SPOC, Spen
r (F. Bontems), vincent.viller-
tain several additional subunits (named MED23 to MED30)compared to the yeast Mediator. The isolated mammalian Media-tor complexes include distinct, but overlapping sets of subunits.Despite their crucial roles in transcriptional regulation, the individ-ual functions of Mediator subunits are still obscure and deservefurther studies. Most Mediator subunit sequences are highly vari-able and contain almost no predicted functional structured do-mains but rather many predicted disordered regions (Toth-Petroczy et al., 2008). There are only a few reports on structuresof Mediator proteins either in complex or individually. To date,structural studies mainly described interaction motifs betweenMediator subunits parts (Baumli et al., 2005; Cai et al., 2010; Kos-chubs et al., 2009; Lariviere et al., 2006).
Mediator activates transcription, at least in part, via direct inter-actions with DNA-binding transcriptional activators bound at up-stream promoter elements and enhancers, with pol II and, mostlikely, with one or more of the general initiation factors bound atthe core promoter. Importantly, different Mediator subunits arethe targets for interaction with transcriptional activation domains(TAD) of different DNA-binding transcriptional activators, withsome TAD interacting with more than one subunit. Previous stud-ies have revealed that for example p53 binds to MED1 and MED17(Meyer et al., 2010). Direct activator–Mediator interactions notonly recruit and stabilize Mediator at the gene promoter but alsoinduce significant structural shifts within the Mediator as assessed

246 F. Bontems et al. / Journal of Structural Biology 174 (2011) 245–251
by cryo-EM experiments (Chadick and Asturias, 2005; Meyer et al.,2010). Thus, the huge surface area provided by the Mediator forprotein–protein interactions can be modulated differently upontranscription factor binding, exposing new specific protein–proteininteraction areas. The conformational shifts are thus probably animportant feature of the regulatory mechanism of Mediatorfunctions.
The MED25 (ARC92/ACID1) subunit was first identified as a tar-get of the Herpes simplex virus transactivator protein VP16 (Leeet al., 2007; Mittler et al., 2003; Yang et al., 2004). MED25comprises 747 residues and includes two potential foldeddomains: an N-terminal VWA (von Willebrand type A) relateddomain (residues 15–233) implicated in the recruitment of theMediator complex and a central ACID (Activator-interactingdomain) domain (residues 392–548) of unknown structure. Theintervening region between these two folded domains is predictedto be unstructured. The ACID domain appears to impose a criticalcontrol on transcription activation by VP16 in mammalian cells,
Fig. 1. HSQC spectra and stereo view of 10 superimposed structures. (A) Superimposition293 K (in green). The peaks in the second HSQC correspond to HN involved in hydrogencore (indicated as yellow spheres on the ribbon view of the structure). (B) Superimpositioregular regions are in blue, red and green, respectively. The side-chains are in light blue
uncovering a novel molecular activation pathway of VP16 throughtranscriptional complexes containing the MED25 subunit. More re-cently, in addition to the activation domain of the viral proteinVP16 (H1 and H2), the TAD from IE62 and LANA-1 have also beenreported to interact with the MED25 ACID domain (Roupelievaet al., 2010; Yamamoto et al., 2009). As a first step towards under-standing the mechanism of recruitment of transactivation domainsby MED25, we solved the NMR structure of its ACID domain andwe mapped its interaction surface with the VP16 H2 (VP16C)peptide.
2. Protein expression and purification
The human MED25 ACID domain (residues 391 to 548) wasamplified by PCR and cloned in phase into the NcoI and XhoI sitesof the pET24d vector, resulting in the pET24d-DA-6xHis expressionplasmid. This plasmid was transformed into Escherichia coli BL21
of two 15N-HSQC spectra recorded in H2O (in magenta) and in D2O after one hour atbonds between the strands of the b-barrel forming MED25 ACID domain structuraln of the 10 lowest energy structures. The backbone of the b-strands, helices and non-. The hydrogen atoms were omitted to simplify the view.

Table 1Structural statistics.
Experimental restraintsNOEs 2594
Intra-residual and sequential 1294Medium range (1 < |i�j| < 5) 381Long range 919
Dihedral angles 226Hydrogen bonds 33
ViolationsCYANA target function 1.21 ± 0.16Upper limits > 0.3 Å 0Upper limits 1–10
Mean of the maximal violations 0.2 ÅRmsd of the upper limits 0.005 ± 0.0008 Å
Angles > 5� 0Mean of the maximal violations 1.9�Rmsd of the angle constraints 0.21 ± 0.08
Ramachandran plotMost favoured 74.5%Additionally allowed 24.7%Generously allowed 0.4%Disallowed 0.4%
Structural dispersion (amino acids 10–150)Backbone rmsd to mean 0.88 ± 0.23 ÅHeavy atoms rmsd to mean 1.30 ± 0.20 Å
CYANA working in dihedral angle space, there is no deviation of the structuregeometry.
F. Bontems et al. / Journal of Structural Biology 174 (2011) 245–251 247
(DE3) and bacteria were grown in LB medium at 37 �C to an opticaldensity of 0.6 at 600 nm. Expression was induced with 0.1 mMIPTG for 18 h at 20 �C. For protein purification, cells were washedin PBS and lyzed using a high pressure homogenizer (Avestin) inbuffer NPI-10 (50 mM sodium phosphate, 300 mM NaCl, 10 mMimidazole pH 8). After centrifugation and filtration on 0.45 lm fil-ter, sample was then loaded onto a 1 ml Ni–NTA column (Qiagen)equilibrated with buffer NPI-10. The column was extensivelywashed with buffer NPI-20 (50 mM sodium phosphate, 300 mMNaCl, 20 mM imidazole pH 8) and eluted with buffer NPI-250(50 mM sodium phosphate, 300 mM NaCl, 250 mM imidazolepH 8). The sample was then further purified by cation exchangechromatography on a S-TRAP (GE) equilibrated in E-buffer (Tris20 mM pH 7, 25 mM NaCl, 1 mM DTT). The C-terminally taggedprotein was then eluted with a linear gradient from 25 mM to1 M NaCl. After concentration, the sample was desalted on aPD10 column equilibrated with NMR buffer (10 mM sodium phos-phate pH 6.75, 100 mM NaCl, 5 mM DTT), resulting in typical 0.3 to0.5 mM samples. Desalting column was also used to prepare theD2O samples. For labelling experiments, LB was replaced by mini-mal M9 medium supplemented with 15NH4Cl and/or 13C-glucose.The VP16 H2 456–490 (VP16C) peptide was purified as previouslydescribed (Nedialkov et al., 2003).
3. NMR measurements and structure calculation
All NMR experiments were carried out at 293 K on a BrukerAvance II 600 MHZ, a Bruker Avance III 800 MHz and a BrukerAvance III 950 MHz spectrometer equipped with triple resonanceTXI cryoprobes. The data were processed on-line by using topspin2.1. The spectra were analyzed using the CCPN NMR Analysis pack-age (Vranken et al., 2005).
The resonance frequency assignment of the backbone atoms(HN, Ha, N, C0, Ca, Cb) was obtained by recording and analyzingHNCO, HNCA, HN(CO)CA, HNCACB, CBCA(CO)NH, HNHA and COCAtriple-resonance experiments recorded on 15N/13C U-labelled sam-ple in 90/10% H2O/D2O. The aliphatic side-chain proton and carbonresonance frequencies were further assigned by the means of a
CCH-TOCSY and an HCCH-TOCSY (12 ms spin-lock time) recordedon a 13C U-labelled sample in 100% D2O. Distance constraints andaromatic frequencies assignment were obtained from the analysisof 15N-NOESY-HSQC (15N U-labelled sample in 90/10% H2O/D2O),13Caliphatic-NOESY-HSQC and 13Caromatic-NOESY-HSQC (13C U-la-belled sample in 100% D2O) spectra recorded with 80 ms mixingtime. Finally, the slowly exchangeable amide protons were identi-fied by recording a set of HSQC spectra on a 15N U-labelled sampleexchanged against the D2O buffer at low temperature and frozenuntil it was put in the spectrometer.
The structure was calculated using CYANA 2.1 software (Herr-mann et al., 2002). Three peak lists were derived from the analysisof the three NOESY–HSQC spectra (15N-, 13Caliphatic- and 13Caromatic-). They were associated to two assignment files (one for the 15N-NOESY-HSQC spectrum recorded in H2O, the other for the two13C-NOESY-HSQC spectra recorded in D2O) obtained from the iden-tification of the intraresidual NOESY correlations. The evaluation ofthe phi and psi dihedral angles provided by the Dangle function ofAnalysis was used to generate 226 dihedral angle restraints corre-sponding to residues clearly identified as belonging to the b-strands and a-helices of the protein. Finally, the characterizationof the slowly exchangeable protons led to the identification of 33hydrogen bonds, all located in the b-barrel of the protein (Fig. 1).Accordingly, a set of 33 HN–O (2.2 Å) and 33 N–O (3.2 Å) upper lim-it restraints was introduced in the final runs of calculation. ARamachandran plot of the final 20 structures, calculated usingPROCHECK NMR (Laskowski et al., 1996), showed 74.5%, 24.7%,0.4% and 0.4% of the residues in the most favoured, additionally al-lowed, generously allowed, and disallowed regions, respectively.The coordinates of the 20 structures (over 100) with the lowest en-ergy obtained at the end of the last calculation run have beendeposited in the Protein Data Bank (PDB code: 2L23).
The interaction between VP16 H2 (VP16C) peptide and MED25ACID domain was probed by recording HSQC spectra of a 15N U-la-belled sample of the ACID domain in the presence of increasingamount of non-labelled peptide. Seven HSQC were recorded inthe same conditions, corresponding to peptide/protein ratios of 0(reference spectrum), 0.1, 0.2, 0.3, 0.5, 1.0 and 1.25. Practically,an NMR protein sample of 0.5 mM in 200 lL was prepared. In par-allel, the peptide was dissolved in pure H20 and its pH was ad-justed to 6.8. The peptide solution was then dispatched infraction corresponding to 1/10 or 2.5/10 of the protein amountpresent in the NMR sample and each fraction was lyophilized. Afterrecording the reference spectra, the following experiments wererealized by removing the protein sample from the NMR tube, add-ing it to the desired amount of lyophilized peptide and putting itagain in the tube. The absence of pH variation was verified at theend of the titration.
4. Structural determination of the MED25 ACID domain
The three-dimensional structure of the MED25 ACID domainwas determined, using standard heteronuclear multidimensionalNMR. By using all experiments, we were able to assign more than90% of all protons, including 70% of the aromatic. The missing res-onances concern essentially the N- and C-terminus of the protein,some lysine and aromatic side chains, and the Pro501, His502,Cys506, Glu507 and Arg509 spin system (Fig. 1). In fact, the assign-ment of this last region, which corresponds to an apparently disor-dered loop, was complicated by the existence of a slow equilibrium(at the NMR time scale) between at least three conformations,likely due to the presence of the two prolines 501 and 505. Indeed,the very characteristic signature of the Thr503–Ala504 chainingwas found three times in the HNCACB spectrum. It is noteworthythat chemical shifts of the Cb and Cc carbons of the other prolines

248 F. Bontems et al. / Journal of Structural Biology 174 (2011) 245–251
(414, 428, 445, 454, 463 and 527) and of the form we assigned forthe proline 505 were indicative of a trans conformation, which wasimposed during the structure calculation.
The manual picking of the three 3D-NOESY-HSQC spectra(15N-, 13Caliphatic-, 13Caromatic-) gives rise to the identification of4127 non-trivial correlations (not corresponding to pairs of pro-tons belonging to rigid elements). These peaks were converted, atthe end of the CYANA calculation process, in 2594 non-ambiguousconstraints (Table 1). The 469 non-assigned remaining peaks werechecked. Most of them correspond to noise or miss-positionedpeaks (located, for example, in the centre of a peak due in fact tothe coalescence of two close correlations). The calculation ended
A
B
C
Fig. 2. Backbone organization of the domain and sequence comparison. (A) Superimposimolecule on the right side is rotated by 70� around the horizontal axis. The b-barrel is inB1 and B2 are antiparallel and B1 and B3 are parallel. B3 is followed by the first helix H1Greek-key motif. The closure of the b-barrel is insured by the antiparallel interactions bethe program PYMOL. (B) Schematic overview of human MED25 and human PTOV1 protebox) and the localization of the ACID domain in MED25 and PTOV1 proteins are indicaMED25 and PTOV1 proteins. Hu: Homo sapiens sapiens; Xe: Xenopus tropicalis; Da: Danioconserved and equivalent (I() L()M() V; F() Y) residues are in red and orangebarrel or in the interactions between the loops or a-helices and the b-barrel. All the coexcepting the glycine 436 in the B2-B3 loop. Sequence alignment was carried out using
with a set of 20 structures presenting no geometrical deformation(as CYANA works in dihedral angle space and keeps constant thebond, angle and improper) and no distant violation larger than0.3 Å (Table 1).
5. Description of the structure
The MED25 ACID domain is folded into a single compact do-main �40 � 45 � 35 Å3. The domain architecture consists of a b-barrel with seven strands D1–D7 (B1, 399–409; B2, 424–432; B3,447–454; B4, 469–475; B5, 494–499; B6, 511–516; B7, 521–526)
tion of the ribbon representation of the 10 structures with the lowest energies. Theblue, the a-helices in red. Strands B1, B2 and B3 form a first mixed b-sheet in which, the fourth strand B4 and the second helix H2. The B4, B5, B6 and B7 strands form atween B3 and B5. The H3 helix is packed against H1. This figure was prepared usingins. The localization of the von Willebrand factor A domain (VWA) in MED25 (greyted (green boxes). (C) Sequence alignment of the ACID domains of representativererio; Fu: Fugu rubripes; Dr: Drosophila melanogaster; Mu: Mus musculus. The strictly, respectively. The residues highlighted in yellow are involved in the core of the b-nserved and equivalent residues are involved in the stabilization of the structure,CLUSTAL_X (Larkin et al., 2007).

A
B C D
Fig. 3. Structural comparison of the b-barrel of MED25, Sharp and Ku70. (A) Structural alignment of MED25, Sharp and Ku b-barrel sequences, after a manually inspection andcorrection of the one provided by DALI (Holm and Rosenstrom, 2010). Part of the large B2–B3 extension present in Ku has been omitted (the corresponding sequence isreplaced by //). The residues in bold have equivalent positions in MED25 and at least in one of the two other structures, the others belong to structurally divergent regions.Structurally conserved residues correspond to the seven b-strands, to helix H2 and to the beginning of helix H1. As in Fig. 2, residues in red and orange correspondrespectively to the identical and equivalent residues of the structurally conserved regions. Residues highlighted in yellow correspond to the residues of the structuralhydrophobic core. The positions of the structurally equivalent residues are reported in red on the structure of the b-barrel domains of (B) MED25, (C) Sharp (PDB ID code1OW1) and (D) Ku (PDB ID code 1JEY).
F. Bontems et al. / Journal of Structural Biology 174 (2011) 245–251 249
and three a-helices H1–H3 (H1, 455–465; H2, 481–492; H3, 529–543) (Fig. 2A). The b-barrel is closed and has the following topol-ogy: B1–B3–B5–B6–B7–B4–B2–B1. Helices H1 and H3 are locatedclose to each other and are facing strands B3, B5 and B6 while helixH2 is nearly perpendicular to the b-barrel and connects strands B4and B5. Helix H1 which is defined from residues Q455 to F465 ishighly distorted as a result of the presence of a Gly–Pro motif inpositions 462 and 463. This distortion of helix H1 contributes tothe formation of a groove located between this a-helix and theb-strands B4–B7–B6–B5. The surface composed by strandsB4–B7–B6 is rather hydrophobic and from these conformationalfeatures could play a role in protein–protein interactions. StrandsB4 and B7 contain a methionine residue at positions 470 and 523which are accessible at the surface of the protein and for whichthe sulfur atoms are only separated by 4.9 Å.
6. Structure-based sequence analysis of ACID domains
Previous works have revealed that Prostate tumour overexpres-sed (PTOV-1) is the only protein with sequence homology withMED25 ACID domain (Benedit et al., 2001; Mittler et al., 2003;Yang et al., 2004). PTOV-1 contains two ACID domains in tandem(PTOV-1A and PTOV-1B), found after a short N-terminal extension.The domain organization of PTOV-1 is shown in Fig. 2B. This pro-tein was identified during a differential display screening for geneexpression in prostate cancer (Benedit et al., 2001). Although itsfunction is currently unknown, PTOV-1 is overexpressed in 71%of prostate carcinoma and barely detectable in normal prostateepithelium. Each of the two ACID domains of PTOV-1 shows a veryhigh sequence homology with the ACID domain of MED25 (se-quence identity of respectively 79.7% and 71.8 %). We establisheda structure-based sequence alignment of representative ACIDdomains (Fig. 2C). Strictly conserved residues are highlighted in
yellow. These hydrophobic residues are involved in the b-barrelinterior (Trp408, Leu452, Leu486, Leu513, Tyr515), in the stabiliza-tion of the B2–B3 and B5–B6 loops (Trp402, Trp444, Pro445,Phe500 and Cys506) and in the interface between helices H1, H3and the b-barrel (Leu458, Phe465, Cys497, Leu514 and Phe533).
7. Search for structural homologues
A structural similarity search using DALI (Holm and Rosen-strom, 2010) indicates that the architecture of the MED25 ACID do-main shows similarities to that of the SPOC domain (Spen paralogand ortholog C-terminal domain: PDB ID code 1OW1) of SHARP(SMRT/HDAC1-associated repressor protein) (Ariyoshi and Schwa-be, 2003) and to that of the b-barrel domains of the heterodimericDNA repair protein Ku70–Ku80 (PDB ID code 1JEY) (Walker et al.,2001) (Fig. 3). These two proteins have no functional link withthe MED25 ACID domain. The SPOC domain of SHARP is presumedto mediate interactions with SMRT/NCoR corepressors, whereasthe Ku70–Ku80 heterodimer binds to DNA. Despite the low se-quence identity, the seven-stranded b-barrel and the helices H1and H2 of MED25 ACID superimpose on the corresponding regionsof SPOC and Ku70 with a root mean square deviation (rmsd) ofrespectively 3.2 and 3.6 Å over 112 Ca atoms (Fig. 3). The SPOC do-main has also a seven stranded b-barrel and comprises six a heli-ces, two of which being at equivalent positions to helices H1 andH2 in the ACID domain. Helix H3 is not present in the SPOC do-main. In SHARP and other proteins that contain a SPOC domain,the regions surrounding this domain show little evidence of struc-tured regions, similarly to what is observed in the MED25 subunit.On the other hand, the b-barrel domains in Ku70 and Ku80 are onepart of a larger structure and thus makes many protein–proteininteractions with other structured regions of the Ku70–Ku80 het-erodimer such as the C-terminal alpha-helical arms of the subunits
250 F. Bontems et al. / Journal of Structural Biology 174 (2011) 245–251
and DNA encircling insertions (Walker et al., 2001). Thus this do-main in Ku70 and Ku80 is not a functional autonomous domainlike the SPOC and ACID domains.
8. Characterization of the MED25 ACID/VP16 H2 (VP16C)interface
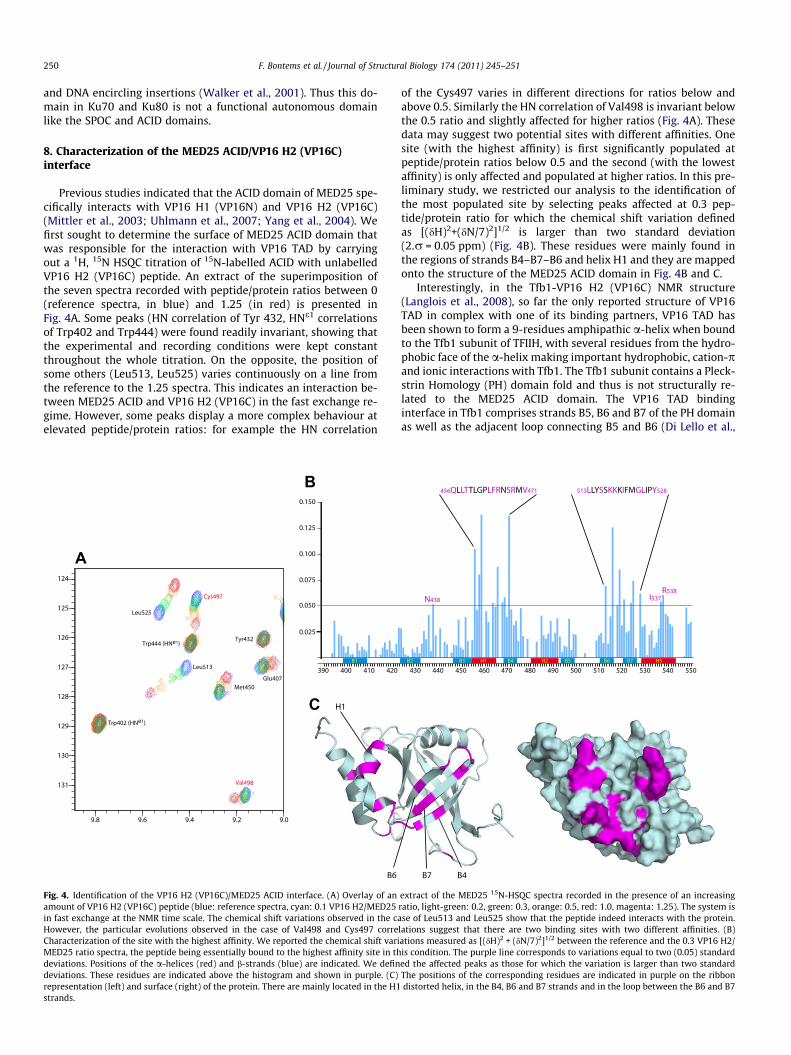
Previous studies indicated that the ACID domain of MED25 spe-cifically interacts with VP16 H1 (VP16N) and VP16 H2 (VP16C)(Mittler et al., 2003; Uhlmann et al., 2007; Yang et al., 2004). Wefirst sought to determine the surface of MED25 ACID domain thatwas responsible for the interaction with VP16 TAD by carryingout a 1H, 15N HSQC titration of 15N-labelled ACID with unlabelledVP16 H2 (VP16C) peptide. An extract of the superimposition ofthe seven spectra recorded with peptide/protein ratios between 0(reference spectra, in blue) and 1.25 (in red) is presented inFig. 4A. Some peaks (HN correlation of Tyr 432, HNe1 correlationsof Trp402 and Trp444) were found readily invariant, showing thatthe experimental and recording conditions were kept constantthroughout the whole titration. On the opposite, the position ofsome others (Leu513, Leu525) varies continuously on a line fromthe reference to the 1.25 spectra. This indicates an interaction be-tween MED25 ACID and VP16 H2 (VP16C) in the fast exchange re-gime. However, some peaks display a more complex behaviour atelevated peptide/protein ratios: for example the HN correlation
A
B
C
Fig. 4. Identification of the VP16 H2 (VP16C)/MED25 ACID interface. (A) Overlay of anamount of VP16 H2 (VP16C) peptide (blue: reference spectra, cyan: 0.1 VP16 H2/MED25in fast exchange at the NMR time scale. The chemical shift variations observed in the caHowever, the particular evolutions observed in the case of Val498 and Cys497 correCharacterization of the site with the highest affinity. We reported the chemical shift variMED25 ratio spectra, the peptide being essentially bound to the highest affinity site in thdeviations. Positions of the a-helices (red) and b-strands (blue) are indicated. We defindeviations. These residues are indicated above the histogram and shown in purple. (C)representation (left) and surface (right) of the protein. There are mainly located in the H1strands.
of the Cys497 varies in different directions for ratios below andabove 0.5. Similarly the HN correlation of Val498 is invariant belowthe 0.5 ratio and slightly affected for higher ratios (Fig. 4A). Thesedata may suggest two potential sites with different affinities. Onesite (with the highest affinity) is first significantly populated atpeptide/protein ratios below 0.5 and the second (with the lowestaffinity) is only affected and populated at higher ratios. In this pre-liminary study, we restricted our analysis to the identification ofthe most populated site by selecting peaks affected at 0.3 pep-tide/protein ratio for which the chemical shift variation definedas [(dH)2+(dN/7)2]1/2 is larger than two standard deviation(2.r = 0.05 ppm) (Fig. 4B). These residues were mainly found inthe regions of strands B4–B7–B6 and helix H1 and they are mappedonto the structure of the MED25 ACID domain in Fig. 4B and C.
Interestingly, in the Tfb1-VP16 H2 (VP16C) NMR structure(Langlois et al., 2008), so far the only reported structure of VP16TAD in complex with one of its binding partners, VP16 TAD hasbeen shown to form a 9-residues amphipathic a-helix when boundto the Tfb1 subunit of TFIIH, with several residues from the hydro-phobic face of the a-helix making important hydrophobic, cation-pand ionic interactions with Tfb1. The Tfb1 subunit contains a Pleck-strin Homology (PH) domain fold and thus is not structurally re-lated to the MED25 ACID domain. The VP16 TAD bindinginterface in Tfb1 comprises strands B5, B6 and B7 of the PH domainas well as the adjacent loop connecting B5 and B6 (Di Lello et al.,
extract of the MED25 15N-HSQC spectra recorded in the presence of an increasingratio, light-green: 0.2, green: 0.3, orange: 0.5, red: 1.0, magenta: 1.25). The system isse of Leu513 and Leu525 show that the peptide indeed interacts with the protein.
lations suggest that there are two binding sites with two different affinities. (B)ations measured as [(dH)2 + (dN/7)2]1/2 between the reference and the 0.3 VP16 H2/is condition. The purple line corresponds to variations equal to two (0.05) standarded the affected peaks as those for which the variation is larger than two standardThe positions of the corresponding residues are indicated in purple on the ribbondistorted helix, in the B4, B6 and B7 strands and in the loop between the B6 and B7

F. Bontems et al. / Journal of Structural Biology 174 (2011) 245–251 251
2005; Langlois et al., 2008). The three b-strands comprise twostructurally adjacent methionines (at positions 59 and 88) thatcontribute to sulfur-p interactions with Phe475 and Phe479 fromVP16 TAD (Langlois et al., 2008). It is noteworthy that the surface(strands B4–B7–B6) displaying significant chemical shift changesupon formation of the MED25 ACID/VP16C complex has featureswhich are reminiscent of the Tfb1 VP16 binding site. The TAD fromVP16 is acidic and positively charged residues in Tfb1 are involvedin TAD binding (Arg61 and Arg86). In MED25 ACID, three adjacentlysines at positions 518, 519 and 520 (in the b turn betweenstrands B7 and B6) are solvent exposed (Fig. 4B and C) and inagreement with our titration experiments have been shown by sitedirected mutagenesis to alter VP16 TAD binding (Santolin, 2006).Taken together, these results point towards an involvement ofthe B4–B7–B6 groove in VP16 H2 (VP16C) binding.
9. Conclusion
We report here the first structure of an ACID domain, that of thehuman Mediator subunit MED25. ACID domain is organizedaround a seven stranded b-barrel of complex architecture, whichbelongs to the SPOC domain-like superfamily (Ariyoshi and Schw-abe, 2003). Our structural analysis, combined to NMR chemicalshift mapping suggests a VP16 H2 (VP16C) binding site on one faceof the b-barrel, defined by strands B4–B7–B6. This interface showsfeatures also observed in the Tfb1 VP16 binding site, the onlycomplex of VP16 with one of its binding partner reported so far(Langlois et al., 2008). Our structure is also representative of theACID domains found in the PTOV-1 protein (Benedit et al., 2001),which is mainly composed of two ACID domains arranged in tan-dem, and highly homologous to the MED25 domain. Little informa-tion is so far available about the function and binding partners ofPTOV-1 but such similarities between one domain of the Mediatorsubunit MED25 and PTOV-1 are intriguing and deserve furthercharacterization.
Although the mechanisms by which VP16 and other transacti-vation domains bind to the MED25 ACID domain remain to be elu-cidated, the structure reported here will be useful for futurestudies aiming to decipher the complex mechanism of recruitmentof transactivation domains by MED25.
Acknowledgments
We thank René Wintjens for helpful discussion and HervéDrobecq for mass spectrometry. Financial support from the TGERMN THC Fr3050 for conducting the research is gratefullyacknowledged. This work was supported by the ‘Fondation pourle Recherche Médicale’ (FRM, Comité Nord-Pas de Calais, France),the ‘Association pour la Recherche contre le Cancer’ (ARC, France),the ‘Ligue contre le Cancer’ (Comité Nord, France), The Ministry ofHigher Education and Research, Nord-Pas de Calais Regional Coun-cil and FEDER through the’ Contrat de Projets Etat Region (CPER)2007–2013’.
References
Ariyoshi, M., Schwabe, J.W., 2003. A conserved structural motif reveals the essentialtranscriptional repression function of Spen proteins and their role indevelopmental signaling. Genes Dev. 17, 1909–1920.
Baumli, S., Hoeppner, S., Cramer, P., 2005. A conserved mediator hinge revealed inthe structure of the MED7.MED21 (Med7.Srb7) heterodimer. J. Biol. Chem. 280,18171–18178.
Benedit, P., Paciucci, R., Thomson, T.M., Valeri, M., Nadal, M., Caceres, C., de Torres, I.,Estivill, X., Lozano, J.J., Morote, J., Reventos, J., 2001. PTOV1, a novel proteinoverexpressed in prostate cancer containing a new class of protein homologyblocks. Oncogene 20, 1455–1464.
Blazek, E., Mittler, G., Meisterernst, M., 2005. The mediator of RNA polymerase II.Chromosoma 113, 399–408.
Bourbon, H.M., 2008. Comparative genomics supports a deep evolutionary origin forthe large, four-module transcriptional mediator complex. Nucleic Acids Res. 36,3993–4008.
Cai, G., Imasaki, T., Yamada, K., Cardelli, F., Takagi, Y., Asturias, F.J., 2010. Mediatorhead module structure and functional interactions. Nat. Struct. Mol. Biol. 17,273–279.
Chadick, J.Z., Asturias, F.J., 2005. Structure of eukaryotic Mediator complexes.Trends Biochem. Sci. 30, 264–271.
Di Lello, P., Nguyen, B.D., Jones, T.N., Potempa, K., Kobor, M.S., Legault, P.,Omichinski, J.G., 2005. NMR structure of the amino-terminal domain from theTfb1 subunit of TFIIH and characterization of its phosphoinositide and VP16binding sites. Biochemistry 44, 7678–7686.
Herrmann, T., Guntert, P., Wuthrich, K., 2002. Protein NMR structure determinationwith automated NOE assignment using the new software CANDID and thetorsion angle dynamics algorithm DYANA. J. Mol. Biol. 319, 209–227.
Holm, L., Rosenstrom, P., 2010. Dali server: conservation mapping in 3D. NucleicAcids Res 38 Suppl: W545–549.
Koschubs, T., Seizl, M., Lariviere, L., Kurth, F., Baumli, S., Martin, D.E., Cramer, P.,2009. Identification, structure, and functional requirement of the Mediatorsubmodule Med7N/31. EMBO J. 28, 69–80.
Langlois, C., Mas, C., Di Lello, P., Jenkins, L.M., Legault, P., Omichinski, J.G., 2008. NMRstructure of the complex between the Tfb1 subunit of TFIIH and the activationdomain of VP16: structural similarities between VP16 and p53. J. Am. Chem.Soc. 130, 10596–10604.
Lariviere, L., Geiger, S., Hoeppner, S., Rother, S., Strasser, K., Cramer, P., 2006.Structure and TBP binding of the Mediator head subcomplex Med8-Med18-Med20. Nat. Struct. Mol. Biol. 13, 895–901.
Larkin, M.A., Blackshields, G., Brown, N.P., Chenna, R., McGettigan, P.A., McWilliam,H., Valentin, F., Wallace, I.M., Wilm, A., Lopez, R., Thompson, J.D., Gibson, T.J.,Higgins, D.G., 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23,2947–2948.
Laskowski, R.A., Rullmannn, J.A., MacArthur, M.W., Kaptein, R., Thornton, J.M., 1996.AQUA and PROCHECK-NMR: programs for checking the quality of proteinstructures solved by NMR. J. Biomol. NMR 8, 477–486.
Lee, H.K., Park, U.H., Kim, E.J., Um, S.J., 2007. MED25 is distinct from TRAP220/MED1in cooperating with CBP for retinoid receptor activation. EMBO J. 26, 3545–3557.
Meyer, K.D., Lin, S.C., Bernecky, C., Gao, Y., Taatjes, D.J., 2010. P53 activatestranscription by directing structural shifts in Mediator. Nat. Struct. Mol. Biol. 17,753–760.
Mittler, G., Stuhler, T., Santolin, L., Uhlmann, T., Kremmer, E., Lottspeich, F., Berti, L.,Meisterernst, M., 2003. A novel docking site on Mediator is critical for activationby VP16 in mammalian cells. EMBO J. 22, 6494–6504.
Nedialkov, Y.A., Shooltz, D.D., Triezenberg, S.J., 2003. Purification and proteininteraction assays of the VP16C transcription activation domain. MethodsEnzymol. 370, 522–535.
Roupelieva, M., Griffiths, S.J., Kremmer, E., Meisterernst, M., Viejo-Borbolla, A.,Schulz, T., Haas, J., 2010. Kaposi’s sarcoma-associated herpesvirus Lana-1 is amajor activator of the serum response element and mitogen-activated proteinkinase pathways via interactions with the Mediator complex. J. Gen. Virol. 91,1138–1149.
Santolin, L., 2006. Functional characterization of the Mediator subunit MED25.Dissertation, University of Munich.
Taatjes, D.J., 2010. The human Mediator complex: a versatile, genome-wideregulator of transcription. Trends Biochem. Sci. 35, 315–322.
Toth-Petroczy, A., Oldfield, C.J., Simon, I., Takagi, Y., Dunker, A.K., Uversky, V.N.,Fuxreiter, M., 2008. Malleable machines in transcription regulation: themediator complex. PLoS Comput. Biol. 4, e1000243.
Uhlmann, T., Boeing, S., Lehmbacher, M., Meisterernst, M., 2007. The VP16activation domain establishes an active mediator lacking CDK8 in vivo. J. Biol.Chem. 282, 2163–2173.
Vranken, W.F., Boucher, W., Stevens, T.J., Fogh, R.H., Pajon, A., Llinas, M., Ulrich, E.L.,Markley, J.L., Ionides, J., Laue, E.D., 2005. The CCPN data model for NMRspectroscopy: development of a software pipeline. Proteins 59, 687–696.
Walker, J.R., Corpina, R.A., Goldberg, J., 2001. Structure of the Ku heterodimer boundto DNA and its implications for double-strand break repair. Nature 412, 607–614.
Yamamoto, S., Eletsky, A., Szyperski, T., Hay, J., Ruyechan, W.T., 2009. Analysis of thevaricella-zoster virus IE62 N-terminal acidic transactivating domain and itsinteraction with the human mediator complex. J. Virol. 83, 6300–6305.
Yang, F., DeBeaumont, R., Zhou, S., Naar, A.M., 2004. The activator-recruitedcofactor/Mediator coactivator subunit ARC92 is a functionally important targetof the VP16 transcriptional activator. Proc. Natl Acad. Sci. USA 101, 2339–2344.
Copyright © 2022 FDOKUMEN




















