
Notch signaling drives stemness and tumorigenicity of esophageal adenocarcinoma

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
Role of mitogen activated protein kinases in skin tumorigenicity of Patulin
Neha Saxena, Kausar M. Ansari, Rahul Kumar, Bhushan P. Chaudhari, Premendra D. Dwivedi, Mukul Das ⁎Food, Drug and Chemical Toxicology Group, CSIR-Indian Institute of Toxicology Research (CSIR-IITR), Mahatma Gandhi Marg, P.O. Box#80, Lucknow-226001, India
a b s t r a c ta r t i c l e i n f o
Article history:Received 7 June 2011Revised 15 September 2011Accepted 15 September 2011Available online 22 September 2011
Keywords:MycotoxinPatulinSkin cancerProliferation
WHO has highlighted the need to evaluate dermal toxicity of mycotoxins including Patulin (PAT), detected inseveral fruits. In this study the skin carcinogenic potential of topically applied PAT was investigated. Singletopical application of PAT (400 nmol) showed enhanced cell proliferation (~2 fold), along with increasedgeneration of ROS and activation of ERK, p38 and JNK MAPKs, in mouse skin. PAT exposure also showedactivation of downstream target proteins, c-fos, c-Jun and NF-κB transcription factors. Further, single topicalapplication of PAT (400 nmol) followed by twice weekly application of TPA resulted in tumor formation after14 weeks, indicating the tumor initiating activity of PAT. However no tumors were observed when PAT wasused either as a complete carcinogen (80 nmol) or as a tumor promoter (20 nmol and 40 nmol) for 25 weeks.Histopathological findings of tumors found in PAT/TPA treated mice showed that these tumors were of squa-mous cell carcinoma type and similar to those found in the positive control group (DMBA/TPA) along withsignificant increase of lipid peroxidation and decrease in free sulfydryls, catalase, superoxide dismutaseand glutathione reductase activities. The results suggest the possible role of free radicals in PAT mediateddermal tumorigenicity involving MAPKs.
© 2011 Elsevier Inc. All rights reserved.
Introduction
Patulin (PAT), a secondary metabolite of fungi, mainly Penicilliumspecies, (Steiman et al., 1989) is a frequent contaminant of ripe applesas well as other fruits such as pear, oranges and grapes (Prieta et al.,1993; Harrison, 1989). There are studies showing PAT induced DNAdamage, chromosome aberration, and micronuclei formation in mam-malian cells, indicating its genotoxicity (Alves et al., 2000; Liu et al.,2003). In vivo administration of PAT reveals kidney, liver, intestinal tis-sue, and immune system as its target organs (Speijers et al., 1998;Wichmann et al., 2002). Other investigators have also shown its chron-ic toxic effects, indicating PAT as carcinogenic, embryotoxic, and tera-togenic (Osswald et al., 1978; Smith et al., 1993; Ciegler et al., 1976).Considering the adverse effects of PAT several regulatory agencieshave imposed the permissible levels of 50 ppb in foodstuffs (Codex,2003; USFDA, 2004). Exceedingly high levels of this contaminant(285 to 733 ppb) in commercialized products have been reported invarious survey studies (Ritieni, 2003; Yurdun et al., 2001). This in-cludes our previous study wherein high patulin content (mean:845 μg/l) was analyzed in apple juice samples (Saxena et al., 2008).
Themitogen-activated protein kinases (MAPKs) are serine/threoninekinases that transduce signals from the plasma membrane to the cellnucleus and thus play a central role in the cellular responses to var-ious extracellular stimuli. In mammals there are three major MAPKssubfamilies, extracellular signal-regulated kinase (ERK), p38 kinase,and c-Jun N-terminal kinase (JNK). The ERK pathway is predomi-nantly activated by mitogens and has been linked to cell survivaland cell proliferation, whereas p38 kinase and JNK are preferentiallyactivated by stress stimuli and are linked to the induction of diversephenomenon such as apoptosis, inflammation, differentiation andcell proliferation (Chang and Karin, 2001; Johnson and Lapadat,2002). However, which of these multiple roles of MAPKs are in-volved at any one time is primarily dependent on the stimulus, celltype, specific isoform, and the duration and strength of signal.These MAPKs have been shown to regulate activation of one of themost extensively investigated transcription factors NF-κB and AP-1by diverse mechanisms (Surh and Kundu, 2005). In resting cells,NF-κB is normally sequestered in the cytoplasm as an inactive com-plex. Toxic stimulus may initiate a signal that possibly by turningon MAPKs, leads to rapid phosphorylation of NF-κB. The activatedfree dimer then translocates to the nucleus, where it binds tothe NF-κB element located in the promoter or enhancer regions ofcycloxygenase-2 (COX-2) and other target genes, controlling theirexpression of cell proliferation (Canty et al., 1999; Wong et al.,2003).
Although oral and inhalation are the most prevalent routes ofexposure for mycotoxin, nonetheless the cutaneous route cannotbe ignored due to manual labor practice during pre and post harvest
Toxicology and Applied Pharmacology 257 (2011) 264–271
Abbreviations: PAT, Patulin; MAPK, mitogen activated protein kinase; ERK, Extra-cellular signal-related kinases; JNK, jun-amino terminal kinases; TPA, 12-tetradecanoylphorbol myristate acetate; DMBA, dimethylbenzanthracene; DCFDA, 2′,7′-Dichloro-fluorescin diacetate.⁎ Corresponding author. Fax: +91 0522 2628227.
E-mail addresses: [email protected], [email protected] (M. Das).
0041-008X/$ – see front matter © 2011 Elsevier Inc. All rights reserved.doi:10.1016/j.taap.2011.09.012
Contents lists available at SciVerse ScienceDirect
Toxicology and Applied Pharmacology
j ourna l homepage: www.e lsev ie r .com/ locate /ytaap

Author's personal copy
stages of agriculture especially in developing countries. Thus, WHOhas highlighted the need to evaluate dermal exposure risk due tomycotoxins (WHO, 1998). In this regard, our earlier study showedthe induction of DNA damage and apoptosis in mouse skin topicallyapplied with PAT, involving a cascade of events including cell cyclearrest (Saxena et al., 2009). These observations led us to hypothesizethat during cell cycle arrest by PAT, some of the cells may undergoDNA repair. However, if this DNA repair is faulty in nature, the re-peated insult of PAT may proliferate the mutated cells. Therefore inthe present study, an attempt has been made to gain a better under-standing of the cellular events leading to PAT mediated toxicity andmore particularly to investigate whether PAT causes induction of cellproliferation via activation of MAPKs pathway that may further leadto tumor formation. Also to gain an insight of an impact of long termexposure of PAT, two-stage mouse skin carcinogenesis model wasundertaken which is supposed to be an ideal system to study a num-ber of biochemical alterations, changes in cellular functions and his-tological changes that may occur during chemical carcinogenesis(Conti et al., 1989).
Materials and methods
Chemicals and antibodies. Patulin (PAT), dithiothreteiol (DTT),phenylmethylsulfonyl fluoride (PMSF), 2-mercaptoethanol, 7,12-dimethylbenzanthracene (DMBA), methylcholanthrene (MCA), 12-O-tetradecanoylphorbol-acetate (TPA), diphenylamine (DPA), proteaseinhibitor cocktail set-I, ethylenediamine tetraacetic acid (EDTA) dis-odium salt, Tris buffer, triton X-100 and bovine serum albumin (BSA)were obtained from Sigma Chemicals Co. (St. Louis, MO). 3H-thymidinewas purchased fromAmersham Biosciences (Searle, Chicago, IL). Rabbitpolyclonal antibodies against phospho-ERk1/2, ERK1/2, phospho-p38,phospho-JNK, phospho-c-jun, phospho-c-fos, phospho-NfкB and tubulinwere procured from Santa Cruz Biotechnology (Santa Cruz, CA). Horse-radish peroxidase (HRP) conjugated goat anti-rabbit IgG secondaryantibody was obtained from Bangalore Genei (Bangalore, India) andPD98059 [2-amino-3-methoxyflavone], SB203580 [4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)imidazole], SP600125 [anthrax(1,9-cd)pyrazol-6(2H)-one], DCFDA (2′,7′-Dichlorofluorescin diacetate)and HRP conjugated β-actin were procured from Sigma Chemicals Co.(St. Louis, MO). All other chemicals used were of highest purity commer-cially available.
Animals. Female Swiss albino mice (5–6 weeks old) obtained from an-imal breeding colony of Indian Institute of Toxicology Research (IITR),Lucknow, India, were acclimatized under standard laboratory con-ditions and were provided free access to commercial pellet diet(Ashirwad, Chandigarh, India) and water ad libitum for 1 week priorto the experiment. The dorsal side of mice skin was shaved using anelectric clipper and only those animals in the resting phase of thehair cycle were used in all experiments.
Thymidine uptake. The excised skin from vehicle control and PATtreated (400 nmol) mice for 24–48 h were lysed and processedaccording to the modified method of Gupta and Mehrotra, (Guptaand Mehrotra, 1992). This dose was chosen as an outcome of ODCactivity result from our earlier study (Saxena et al., 2009). Further,3H-thymidine (30 μci per animal) was given intraperitonially to eachanimal 2 h before termination. In brief, the skin sample was homoge-nized with Ultra Turax Polytron (Janke & Kunkel, IKA-Labortechnik,Staufen, Germany) in ice cold water and slightly vortexed in the pres-ence of 60% perchloric acid (PCA). The lysate was then kept in ice for20 min and the samples were centrifuged at 10,000×g for 30 min at4 °C. The pellet obtained was suspended in cold 0.2 M PCA and re-suspended in absolute alcohol after washing by centrifugation at10,000×g for 10 min. The DNA pellet obtained was dissolved in 2 ml0.5 M PCA and hydrolyzed at 90 °C for 15 min. DNA was estimated in
acid hydrolysate samples using diphenyl amine reagent and the in-corporated 3H-thymidine was counted in a scintillation counter andexpressed as counts per minute (CPM)/μg DNA.
ROS measurement. The animals were distributed randomly into eightgroups, with three mice per group. The animals in the first groupreceived a single topical application of 0.2 ml acetone, and theremaining seven groups received a single topical dose of 400 nmolPAT/0.2 ml acetone per mouse. The animals were euthanized after 1,3, 6, 12, 24, 48, and 72 h of PAT treatment and 2 cm2 of treated skinwere excised from each animal and single cell suspensions were pre-pared as described earlier (Saxena et al., 2009), and cell viability wasdetermined by trypan blue exclusion assay. For estimation of ROS,1×105 cells were incubated in the presence of 100 μM H2-DCFDAfor 60 min at 37 °C in a black 96-well plate, and relative fluorescenceintensity was measured in SYNERGY-HT multiwell plate reader (Bio-Tek, Winooski, VT) using KC4 software at excitation and emissionwavelengths of 485 and 528 nm, respectively.
Preparation of whole cell extract. For short term biomarker studies, an-imals were divided into two groups containing 3 animals each: Firstgroup was that of acetone, that served as vehicle control and the sec-ond group of PAT (400 nmol) that was applied topically to the dorsalshaved area. The animals were maintained in controlled atmosphereof 12 h dark/light, 22±2 °C and 50–60% humidity, fasted overnightafter last dose and euthanized after 1, 3, 6, 12, 24, 48 and 72 h by cer-vical dislocation with minimal suffering following the rules laid downby Animal Welfare Committee of IITR.
The mouse skin from PAT treated and acetone treated animals wasexcised, rinsed in normal saline and fat was removed by scrapping.The skin was homogenized in ice cold-RIPA buffer (50 mM Tris PH:7.5, 150 mM Nacl, 1% triton ×100, 0.5% sodium deoxycholate, 0.1%SDS, 0.2 mM PMSF) with freshly added protease inhibitor. The celllysate was kept on ice for 10 min, and then centrifuged at 16,000×gfor 20 min at 4 °C. The clear supernatant (whole cell extract) wasused for the detection of phosphorylated proteins.
Preparation of nuclear extract. The cells were suspended in cytoplas-mic lysis buffer (20 mM HEPES pH 7.5, 10 mM NaCl, 1.5 mM MgCl2.0.2 mM EDTA, 1 mM dithiothreitol, 0.5 mM phenylmethylsulfonylfluoride, 1% Triton X-100, Protease Inhibitor Cocktail) for 10 min onice and then vortexed for 10 s. Nuclei were pelleted by centrifugationat 12,000×g for 20 s, supernatant was collected as cytoplasmic ex-tract for detection of c-jun and c-fos, and pellets were resuspendedin nuclear lysis buffer (20 mM HEPES pH: 7.6, 20% glycerol, 0.5 MNaCl, 0.2 mM EDTA, 1 mM dithiothreitol, 1% Triton X-100, 0.5 mMphenylmethylsulfonyl fluoride) for 30 min on ice. The supernatantscontaining nuclear proteins were collected after centrifugation at12,000×g for 2 min and stored at −80 °C for detection of phosphory-lation of NF-κB.
Western blotting. Eighty micrograms of proteins from the whole cellextract samples was boiled in 1× sodium dodecyl sulfate (SDS) sam-ple-loading buffer for 5 min before electrophoresis on a 10% SDS-polyacrylamide gel. After transferring the protein to methanol soakedpolyvinylidene difluoride membrane for 2 h at 48 mA with a semidryelectroblotting assembly (Amersham Life Sciences, Inc, Arlington Hts,IL), the membranes were blocked with 5% fat-free dry milk in 1× PBSTbuffer (phosphate-buffered saline containing 0.1% Tween 20) for 1 hat room temperature (RT). The blocked membranes were incubatedwith 1:1000 dilutions of primary antibodies for phospho-ERK, pho-spho-p38, phospho-JNK, c-fos, c-jun (overnight at 4 °C) and with1:500 dilutions of primary antibody of phospho-NF-κB (overnight at4 °C). Each blot was washed with 1× PBST for 25 min, then incubatedwith 1:2000 dilutions of anti-rabbit or anti-mouse horseradish perox-idase conjugated-secondary antibodies for 1 h at RT and again
265N. Saxena et al. / Toxicology and Applied Pharmacology 257 (2011) 264–271

Author's personal copy
washed three times with 1× PBST for 5 min each. Thereafter the anti-bodies were stripped for 30 min at RT with Stripping buffer (62.5 mMTris pH: 6.8, 2% SDS, 100 mM 2-mercaptoethanol) and re-probed with1:2000 dilutions of anti-actin antibody and anti-ERK for 4 h at RT. Thebound antibody on the membrane was detected using an enhancedchemiluminescence detection kit (Amersham Pharmacia Biotech,Amersham, UK) according to the manufacturer's protocol.
Skin-tumorigenic assessment of PAT. For tumor studies, six to sevenweek old female Swiss albino mice (20±3 g) were randomly allocat-ed into 7 groups of 15 animals each. The following experimental pro-tocol was adopted:
Group 1: Control (non-treated).Group 2: Vehicle Control (100 μl, acetone treated twice weekly).Group 3: Twiceweekly application ofMCA (1.8 μmol/0.1 mL of acetone).Group 4: Twiceweekly application of PAT (80 nmol/0.1 mL of acetone).Group 5: Single dose of DMBA (120 nmol/0.1 mL of acetone) fol-
lowed by twice weekly application of TPA (4 nmol/0.1 mLof acetone) after a week of initiation.
Group 6: Single dose of PAT (400 nmol/0.1 mL of acetone) followedby twice weekly application of TPA (4 nmol/0.1 mL of ace-tone) after a week of initiation.
Group 7: Single dose of DMBA (120 nmol/0.1 mL of acetone) fol-lowed by twice weekly application of PAT (20 nmol/0.1 mLof acetone) after a week of initiation.
Skin tumor formation and bodyweight were recorded twiceweek-ly for 24 weeks and tumors greater than 1 mm in diameter were in-cluded in the cumulative total only if they persisted for 2 weeks ormore. Cumulative tumors were defined as the total number of tumorsobserved on each mouse during the experimental protocol of25 weeks. The number of cumulative tumors for animals that diedwas included in all tumor counts for all remaining weeks of the study.
Histopathological processing. The animals were euthanized by cervicaldislocation at the termination of the experiment. Skin and liver wereimmediately removed, washed in cold normal saline solution, soakdried over filter paper and weighed. A portion of the organs wasfixed in 10% buffered formalin and embedded in paraffin afterprocessing. Sections of 5 μm thickness were cut and stained with he-matoxylin and eosin for microscopic examination.
Homogenization and enzyme preparation. A weighed portion of skinwas homogenized in chilled 0.1 M phosphate buffer pH 7.4 con-taining 0.15 M potassium chloride using Potter Elvehjem glass Teflonhomogenizer and volume adjusted to give 10% w/v homogenate. Incase of skin, fat towards the dermal side was removed by scrappingand 10% w/v homogenate was prepared using Ultra Turrax Polytron.The homogenates were centrifuged at 10,000 g for 20 min at 4 °C toisolate the post mitochondrial supernatant and stored at −80 °C forthe enzymatic assays.
Oxidative stress markers and antioxidant enzymes. The rate of lipidperoxidation (LPO) was determined according to the method ofUtley et al. (1967) by estimating malondialdehyde (MDA) formedwith 2-thiobarbituric acid (TBA) and expressed as nanomoles ofmalondialdehyde formed per hour per g tissue. Free sulfydryl (−SH)content was measured according to the method of Ellman (1959) andexpressed as μmoles GSH per g tissue. Glutathione-S-transferase (GST)activity was assayed by the method of Habig et al. (1974) with CDNBas substrate. Specific activity was expressed as nmoles CDNB conjugateformed per minute per mg protein. Catalase (CAT) activity was assayedby the method of Sinha (1972) and calculated in terms of mmol H2O2
consumed/min/mg protein. Superoxide dismutase (SOD) activity wasdetermined by the method based on the ability of SOD to inhibit theauto oxidation of epinephrine at pH 10.2 (Misra and Fridovich, 1972).
Glutathione reductase (GR) and glutathione peroxidise (GPx) activitieswere assayed according to the method of Carlberg and Mannervik(1985) and Rotruck et al. (1973) and expressed as nmol NADPHoxidized/min/mg protein and mmol GSH consumed/min/mg protein,respectively.
Estimation of protein. Protein in the samples was determined usingbovine serum albumin as standard (Lowry et al., 1951).
Statistical analysis. Values are presented as mean±SE. Statistical eval-uation was carried with one-way ANOVA (Snedecor and Cochran,1967). In all the cases, P values less than 0.05 were considered signif-icant when compared to controls.
Results
PAT induced thymidine uptake
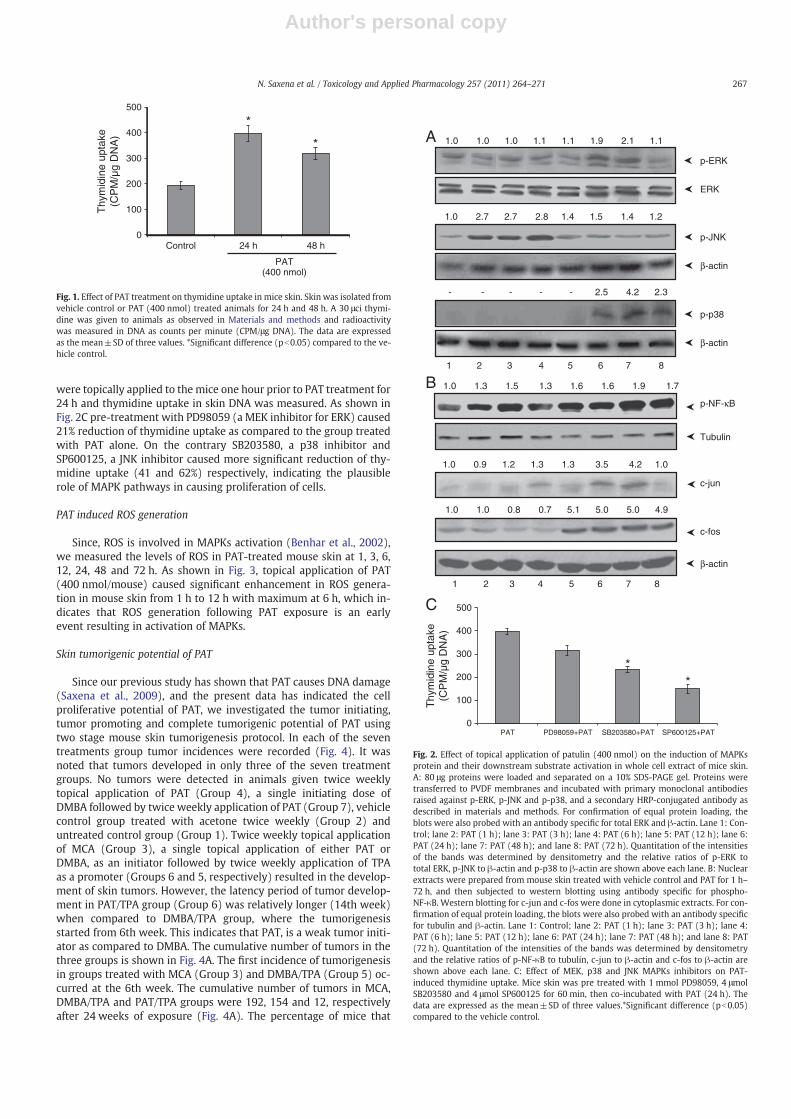
Induction of cell proliferation may be one of the key mechanismby which PAT may cause skin tumorigenesis, therefore, we investigat-ed the effect of single topical application of PAT on [3H] thymidine up-take. The induction of thymidine uptake, an assay used to determineDNA synthesis, (a marker of cell proliferation) following PAT applica-tion in mouse skin is depicted in Fig. 1. Our observation showed thatPAT caused 2.0 and 1.5 fold enhanced uptake of thymidine after 24and 48 h.
PAT induced MAPKs Phosphorylation and activation of c-jun, c-fos andNF-κB protein
MAPKs are serine/threonine-specific kinases that respond toextra- as well as intra-cellular stimuli and regulate various cellular ac-tivities including cell proliferation (Pearson et al., 2001). The ability ofPAT to activate the major MAPKs (ERK, JNK and p38) was investigatedby exposing the skin of mice to PAT at different time points. Mice skinwas exposed to PAT for 1, 3, 6, 12, 24, 48 and 72 h respectively, andthe whole cell extract were subjected to Western blotting using spe-cific antibodies. As shown in Fig. 2A, PAT causes activation of phos-phorylated forms of all the three MAPK proteins in mice skin. PATadministration significantly elevated the levels of phospho-ERK andphospho-p38 in mouse skin at 24 h which remained detectable for72 h in p38. However the signals of phospho-JNK were enhanced atan early time point of 1 h that persisted for 6 h and then graduallysubsides at further time points. Similarly a consistent signal of totalERK and β-actin was obtained in both p38 and JNK to ensure equalloading of proteins.
Several studies have reported that MAPKs play a critical role in thetranscriptional activation of NF-κB and AP-1 transcription factors inmouse skin (Surh and Kundu, 2005). We therefore studied the effectsof single topical application of PAT (400 nmol) on the expression ofc-fos, and c-jun proteins in cytoplasmic extract as well as activationof NF-κB in nuclear extract of mouse skin (Fig. 2B). It was observedthat over-expressed levels of c-jun, a downstream substrate of JNKat 24 and 48 h that dramatically reduced further. However skin topi-cally treated with PAT showed a persistent activation of c-fos proteinfrom 12 to 72 h wherein more intense signals were obtained. How-ever the phosphorylation of NF-κB protein, a transcription factor in-volved in regulation of genes responsible to proliferation was alsoobserved in nucleus of PAT exposed mouse for 1–72 h. The activationof NF-κB protein was detected after 3 h exposure and remained acti-vated upto 72 h.
MAPKs Inhibitors suppressed PAT induced thymidine uptake
To provide further evidence that MAPKs are involved in PAT in-duced cell proliferation, different inhibitors of MAPKs pathways
266 N. Saxena et al. / Toxicology and Applied Pharmacology 257 (2011) 264–271
Author's personal copy
were topically applied to the mice one hour prior to PAT treatment for24 h and thymidine uptake in skin DNA was measured. As shown inFig. 2C pre-treatment with PD98059 (a MEK inhibitor for ERK) caused21% reduction of thymidine uptake as compared to the group treatedwith PAT alone. On the contrary SB203580, a p38 inhibitor andSP600125, a JNK inhibitor caused more significant reduction of thy-midine uptake (41 and 62%) respectively, indicating the plausiblerole of MAPK pathways in causing proliferation of cells.
PAT induced ROS generation
Since, ROS is involved in MAPKs activation (Benhar et al., 2002),we measured the levels of ROS in PAT-treated mouse skin at 1, 3, 6,12, 24, 48 and 72 h. As shown in Fig. 3, topical application of PAT(400 nmol/mouse) caused significant enhancement in ROS genera-tion in mouse skin from 1 h to 12 h with maximum at 6 h, which in-dicates that ROS generation following PAT exposure is an earlyevent resulting in activation of MAPKs.
Skin tumorigenic potential of PAT
Since our previous study has shown that PAT causes DNA damage(Saxena et al., 2009), and the present data has indicated the cellproliferative potential of PAT, we investigated the tumor initiating,tumor promoting and complete tumorigenic potential of PAT usingtwo stage mouse skin tumorigenesis protocol. In each of the seventreatments group tumor incidences were recorded (Fig. 4). It wasnoted that tumors developed in only three of the seven treatmentgroups. No tumors were detected in animals given twice weeklytopical application of PAT (Group 4), a single initiating dose ofDMBA followed by twice weekly application of PAT (Group 7), vehiclecontrol group treated with acetone twice weekly (Group 2) anduntreated control group (Group 1). Twice weekly topical applicationof MCA (Group 3), a single topical application of either PAT orDMBA, as an initiator followed by twice weekly application of TPAas a promoter (Groups 6 and 5, respectively) resulted in the develop-ment of skin tumors. However, the latency period of tumor develop-ment in PAT/TPA group (Group 6) was relatively longer (14th week)when compared to DMBA/TPA group, where the tumorigenesisstarted from 6th week. This indicates that PAT, is a weak tumor initi-ator as compared to DMBA. The cumulative number of tumors in thethree groups is shown in Fig. 4A. The first incidence of tumorigenesisin groups treated with MCA (Group 3) and DMBA/TPA (Group 5) oc-curred at the 6th week. The cumulative number of tumors in MCA,DMBA/TPA and PAT/TPA groups were 192, 154 and 12, respectivelyafter 24 weeks of exposure (Fig. 4A). The percentage of mice that
0
100
200
300
400
500
Control 24 h 48 h
Thy
mid
ine
upta
ke(C
PM
/µg
DN
A)
PAT(400 nmol)
*
*
Fig. 1. Effect of PAT treatment on thymidine uptake in mice skin. Skin was isolated fromvehicle control or PAT (400 nmol) treated animals for 24 h and 48 h. A 30 μci thymi-dine was given to animals as observed in Materials and methods and radioactivitywas measured in DNA as counts per minute (CPM/μg DNA). The data are expressedas the mean±SD of three values. *Significant difference (pb0.05) compared to the ve-hicle control.
p-ERK
ERK
p-JNK
β-actin
p-p38
- - - - -
1.0 1.0 1.0 1.1 1.1 1.9 2.1 1.1
1.0 2.7 2.7 2.8 1.4 1.5 1.4 1.2
2.5 4.2 2.3
1 2 3 4 5 6 7 8
β-actin
p-NF-κB
Tubulin
1.0 1.3 1.5 1.3 1.6 1.6 1.9 1.7
c-jun
1.0 0.9 1.2 1.3 1.3 3.5 4.2 1.0
c-fos
β-actin
1.0 1.0 0.8 0.7 5.1 5.0 5.0 4.9
1 2 3 4 5 6 7 8
0
100
200
300
400
500
PAT PD98059+PAT SB203580+PAT SP600125+PAT
Thy
mid
ine
upta
ke(C
PM
/µg
DN
A)
**
A
B
C
Fig. 2. Effect of topical application of patulin (400 nmol) on the induction of MAPKsprotein and their downstream substrate activation in whole cell extract of mice skin.A: 80 μg proteins were loaded and separated on a 10% SDS-PAGE gel. Proteins weretransferred to PVDF membranes and incubated with primary monoclonal antibodiesraised against p-ERK, p-JNK and p-p38, and a secondary HRP-conjugated antibody asdescribed in materials and methods. For confirmation of equal protein loading, theblots were also probed with an antibody specific for total ERK and β-actin. Lane 1: Con-trol; lane 2: PAT (1 h); lane 3: PAT (3 h); lane 4: PAT (6 h); lane 5: PAT (12 h); lane 6:PAT (24 h); lane 7: PAT (48 h); and lane 8: PAT (72 h). Quantitation of the intensitiesof the bands was determined by densitometry and the relative ratios of p-ERK tototal ERK, p-JNK to β-actin and p-p38 to β-actin are shown above each lane. B: Nuclearextracts were prepared from mouse skin treated with vehicle control and PAT for 1 h–72 h, and then subjected to western blotting using antibody specific for phospho-NF-κB. Western blotting for c-jun and c-fos were done in cytoplasmic extracts. For con-firmation of equal protein loading, the blots were also probed with an antibody specificfor tubulin and β-actin. Lane 1: Control; lane 2: PAT (1 h); lane 3: PAT (3 h); lane 4:PAT (6 h); lane 5: PAT (12 h); lane 6: PAT (24 h); lane 7: PAT (48 h); and lane 8: PAT(72 h). Quantitation of the intensities of the bands was determined by densitometryand the relative ratios of p-NF-κB to tubulin, c-jun to β-actin and c-fos to β-actin areshown above each lane. C: Effect of MEK, p38 and JNK MAPKs inhibitors on PAT-induced thymidine uptake. Mice skin was pre treated with 1 mmol PD98059, 4 μmolSB203580 and 4 μmol SP600125 for 60 min, then co-incubated with PAT (24 h). Thedata are expressed as the mean±SD of three values.*Significant difference (pb0.05)compared to the vehicle control.
267N. Saxena et al. / Toxicology and Applied Pharmacology 257 (2011) 264–271

Author's personal copy
developed tumors during the treatment schedule is presented inFig. 4B. In MCA and DMBA/TPA treated groups, 100% of the mice de-veloped tumors by the 12th and 16th week of treatment, respectively.Nearly 40% of mice developed tumors in the PAT/TPA treated groupafter 20th week, which remained consistent until the completion of24 weeks treatment (Fig. 4B). The number of tumors per tumor bear-ing mouse in the three groups is depicted in Fig. 4C. A total of 21, 12and 2 tumors per tumor bearing mouse were observed in MCA,DMBA/TPA and PAT/TPA treated groups, respectively after 24 weeksof treatment (Fig. 4C). The data, when expressed as mean numberof tumors per mouse, showed 12.7, 9.4 and 2.4 in MCA, DMBA/TPAand PAT/TPA treated groups, respectively after 24 weeks (Table 1).
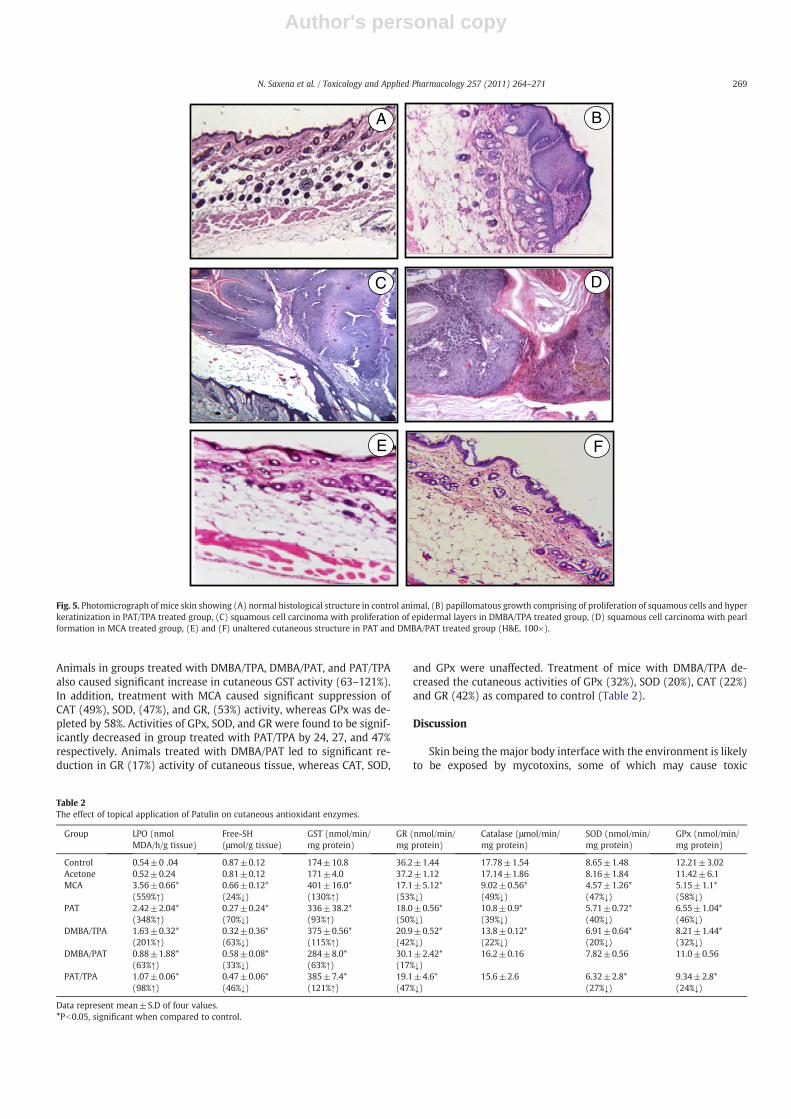
Fig. 5 depicts the histopathological features of skin in theuntreated control, MCA, DMBA/TPA, and PAT/TPA treated mice. Theuntreated control mice (Group 1) showed normal appearance ofboth the epidermis and dermis of skin (Fig. 5A). Acetone treatedgroup (Group 2) showed histopathological features similar to thatof untreated control (data not shown). Mice initiated with DMBAfollowed by twice weekly application of PAT at a dose of 20 nmol(Group 7) and with PAT (80 nmol/0.1 ml) as complete carcinogendid not show any papillomatous growth and showed almost normalepidermis and dermis (Fig. 5E and F). In a complete carcinogenicprotocol, where MCA (Group 3) was used as positive control, charac-teristic skin lesions comprising squamous cell carcinoma (SCC), pearlformation and basaloid type lesions projecting deep into the dermisinvolving hair follicles and sebaceous glands were observed(Fig. 5D). In a two-stage skin tumor protocol where DMBA/TPA wasused as positive control (Group 5), squamous cell carcinoma withmarked proliferation of the epidermal layers was seen (Fig. 5C). Theskin of mice treated with a single initiating dose (400 nmol/0.1 ml)of PAT followed by twice weekly application of TPA demonstratedpapillomatous growth comprising of proliferation of squamous cellsand hyper keratinization (Fig. 5B). These histopathological findingsare in agreement with our tumor data, which suggests only a tumorinitiating property for PAT.
Oxidative stress markers and enzymatic antioxidants
As single exposure of PAT showed enhanced generation of ROSwhich is known to be involved in tumor progression (Storz, 2005),we investigated the marker enzymes of oxidative stress in the ani-mals at the termination of tumorigenesis experiment. Table 2 showsthe effect of topically applied PAT on oxidative stress markers andFree-SH and lipid peroxidation in cutaneous tissue. Twice weeklytopical application of MCA and PAT for 24 weeks caused significantdecrease in cutaneous (24 and 131%) glutathione content. Animals
treated with DMBA/TPA, DMBA/PAT and PAT/TPA also led to signifi-cant reduction in glutathione content (33–63%). LPO was found tobe substantially (Pb0.05) enhanced in cutaneous (348 and 559%)tissue of PAT and MCA treated groups. Lesser enhancement, thoughsignificant was observed in DMBA/TPA and DMBA/PAT groups,where as treatment with PAT/TPA resulted in an increase of 98% ascompared to control. Twice weekly application of PAT and MCAresulted in significant elevation of skin GST (93 and 130%) activity.
0
2
4
6
8
10
12
14
Control 1 h 3 h 6 h 12 h 24 h 48 h 72 h
Flu
ores
cenc
e U
nits
(X 1
0000
) * *
*
*
PAT(400 nmol)
Fig. 3. Measurement of ROS generation in mouse skin following PAT (400 nmol) treat-ment at different time intervals. Single cell suspensions from vehicle/PAT treated miceskin were prepared and 1×105 cells were incubated with 100 μMH2-DCFDA for 60 minat 37 °C and relative fluorescence intensity was determined by spectrofluorimetry(λex=488 nm, λem=520 nm). The data are expressed as the mean±SD of threevalues. *Pb0.05, significant with respect to control group.
A
B
C
0
30
60
90
120
150
180
210
0 2 4 6 8 10 12 14 16 18 20 22 24
Cum
ulat
ive
num
ber
of tu
mor
s
Number of weeks
MCADMBA/TPAPAT/TPA
0
15
30
45
60
75
90
105
7.5
5
2.5
0
10
12.5
15
17.5
20
0 2 4 6 8 10 12 14 16 18 20 22 24
22.5
0 2 4 6 8 10 12 14 16 18 20 22 24
% o
f mic
e w
ith tu
mor
sTu
mor
per
tum
or b
earin
g m
ice
Number of weeks
Number of weeks
MCA
DMBA/TPA
PAT/TPA
MCADMBA/TPAPAT/TPA
Fig. 4. Evaluation of skin tumorigenic potential of topically applied PAT in Swiss albinomice: Comparison with twice weekly topical application of MCA and single applicationof DMBA followed by twice weekly application of TPA to animals. No tumors werefound in the following groups: control; twice weekly topical application of PAT; andsingle topical application of DMBA followed by twice weekly application of PAT.
Table 1The incidence of skin tumorigenesis in terms of tumor per mice induced by MCA,PAT/TPA and DMBA/TPA groups using two stage skin carcinogenesis model.
Group MCA PAT/TPA DMBA/TPA
8th week 0.47±0.27 NTF 0.73±0.3612th week 4.60±1.39 NTF 3.73±0.7918th week 12.8±2.88 1.8±0.60 6.80±1.1424th week 12.7±2.78 2.4±0.58 9.40±1.57
Data represent mean±S.E of 15 animals at different time points.NTF — no tumor found.
268 N. Saxena et al. / Toxicology and Applied Pharmacology 257 (2011) 264–271
Author's personal copy
Animals in groups treated with DMBA/TPA, DMBA/PAT, and PAT/TPAalso caused significant increase in cutaneous GST activity (63–121%).In addition, treatment with MCA caused significant suppression ofCAT (49%), SOD, (47%), and GR, (53%) activity, whereas GPx was de-pleted by 58%. Activities of GPx, SOD, and GR were found to be signif-icantly decreased in group treated with PAT/TPA by 24, 27, and 47%respectively. Animals treated with DMBA/PAT led to significant re-duction in GR (17%) activity of cutaneous tissue, whereas CAT, SOD,
and GPx were unaffected. Treatment of mice with DMBA/TPA de-creased the cutaneous activities of GPx (32%), SOD (20%), CAT (22%)and GR (42%) as compared to control (Table 2).
Discussion
Skin being the major body interface with the environment is likelyto be exposed by mycotoxins, some of which may cause toxic
F
DC
E
BA
Fig. 5. Photomicrograph of mice skin showing (A) normal histological structure in control animal, (B) papillomatous growth comprising of proliferation of squamous cells and hyperkeratinization in PAT/TPA treated group, (C) squamous cell carcinoma with proliferation of epidermal layers in DMBA/TPA treated group, (D) squamous cell carcinoma with pearlformation in MCA treated group, (E) and (F) unaltered cutaneous structure in PAT and DMBA/PAT treated group (H&E, 100×).
Table 2The effect of topical application of Patulin on cutaneous antioxidant enzymes.
Group LPO (nmolMDA/h/g tissue)
Free-SH(μmol/g tissue)
GST (nmol/min/mg protein)
GR (nmol/min/mg protein)
Catalase (μmol/min/mg protein)
SOD (nmol/min/mg protein)
GPx (nmol/min/mg protein)
Control 0.54±0 .04 0.87±0.12 174±10.8 36.2±1.44 17.78±1.54 8.65±1.48 12.21±3.02Acetone 0.52±0.24 0.81±0.12 171±4.0 37.2±1.12 17.14±1.86 8.16±1.84 11.42±6.1MCA 3.56±0.66* 0.66±0.12* 401±16.0* 17.1±5.12* 9.02±0.56* 4.57±1.26* 5.15±1.1*
(559%↑) (24%↓) (130%↑) (53%↓) (49%↓) (47%↓) (58%↓)PAT 2.42±2.04* 0.27±0.24* 336±38.2* 18.0±0.56* 10.8±0.9* 5.71±0.72* 6.55±1.04*
(348%↑) (70%↓) (93%↑) (50%↓) (39%↓) (40%↓) (46%↓)DMBA/TPA 1.63±0.32* 0.32±0.36* 375±0.56* 20.9±0.52* 13.8±0.12* 6.91±0.64* 8.21±1.44*
(201%↑) (63%↓) (115%↑) (42%↓) (22%↓) (20%↓) (32%↓)DMBA/PAT 0.88±1.88* 0.58±0.08* 284±8.0* 30.1±2.42* 16.2±0.16 7.82±0.56 11.0±0.56
(63%↑) (33%↓) (63%↑) (17%↓)PAT/TPA 1.07±0.06* 0.47±0.06* 385±7.4* 19.1±4.6* 15.6±2.6 6.32±2.8* 9.34±2.8*
(98%↑) (46%↓) (121%↑) (47%↓) (27%↓) (24%↓)
Data represent mean±S.D of four values.*Pb0.05, significant when compared to control.
269N. Saxena et al. / Toxicology and Applied Pharmacology 257 (2011) 264–271

Author's personal copy
manifestation in cutaneous as well as in extracutaneous tissues fol-lowing percutaneous absorption. In this regard our earlier studieshave been shown that PAT resulted in enhanced activity of ODC, thefirst and the rate limiting enzyme in the biosynthesis of polyaminesin the single topically exposed mouse skin (Saxena et al., 2009). It iswell known that ODC plays an important role in the regulation ofcell proliferation, and studies claim that hyperproliferation andaltered differentiation in skin keratinocytes have been linked withderegulation of MAPK signaling pathways. This in turn contributesto malignant transformation in human cancers, including those ofthe cutaneous epithelium (Bedogni et al., 2004; Chung et al., 2000;Takahashi et al., 2002).
In the present investigation topical application of PAT at a dose of400 nmol to mice is sufficient to detect the activated forms of ERK,JNK and p38 MAPK, indicating that PAT causes activation of all thethreeMAPKs, nonetheless the time of activation and the signal variesin each case. They are linked with distinct biological responses andregulate various cell fate decisions in diverse biological system. Ourin vivo dermal studies are in agreement with those of previousin vitro studies where PAT was shown to activate MAPKs signalingcascade in human embryonic kidney (HEK293) cells, human periph-eral blood mononuclear (PBMCs) cells, and Madin-Darby caninekidney (MDCK) cells (Wu et al., 2005; Liu et al., 2006). Further, ourstudy shows that specific inhibitors viz SP600125 and SP203580of MAPKs pathways were able to suppress cell proliferation signifi-cantly, which suggest that possibly p38 and JNK pathway might beinvolved in PAT-induced cell proliferation. In this regard, it is wellestablished that SP600125, an inhibitor of JNK, reduces COX-2expression, which is known to regulate cell proliferation via activa-tor protein 1 (AP-1) complex (Cui and Douglas, 1997). Phosphorylat-ed JNK undergoes dimerization and translocates into the nucleus tophosphorylate certain transcriptional factors such as c-jun, whereinc-jun and c-fos homodimers bind to the AP-1 consensus sequence(Halazonetis et al., 1988). AP-1 in turn controls a number of cellularprocesses including cell proliferation, via upregulation of COX-2 ex-pression. It is well documented that activation of the p38 kinasemay induce either cell death or cell survival in different cell typesunder different conditions (Anderson, 1997). The present findingsalso showed signs of cell proliferation following single treatment ofPAT, which emphasizes the involvement of multiple MAPK signalingcascades.
The observed early activation of JNK and NF-κB (1–3 h) appears tobe a direct response to PAT while later activation of ERK, p38, c-jun,c-fos and NF-κB may be due to enhanced generation of ROS. Unequiv-ocal evidences suggest that ROS within cells act as a double edgedsword by virtue of their ability to elicit cell proliferative response,survival, cellular migration, and also by inducing DNA damage leadingto mutations that initiate tumorigenicity and sustain subsequenttumor progression (Storz, 2005). Thus, present study provide evi-dence that PAT induced ROS act as second messengers in intracellularsignaling cascades and may mediate cell proliferation by activatingErk, and p38 along with activation of downstream targets viz. c-jun,c-fos and NF-κB (Benhar et al., 2002).
Further, earlier reports have revealed that long-term exposure ofPAT is tumorigenic to Wistar rats, leading to sarcoma at the injectionsite on subcutaneous administration (Dickens and Jones, 1961) andalso causes benign tumors of the forestomach and glandular stomachin Sprague–Dawley rats after gavage treatment (Osswald et al., 1978).Nonetheless, there is still limited literature regarding the dermal tox-icity through long term exposure of PAT, the need of which is alsohighlighted by WHO (WHO, 1998). We have already explored theDNA damaging ability of PAT in skin cells which is in agreementwith other in vitro findings where PAT is shown to cause oxidativeDNA damage in few mammalian cells (Pfeiffer et al., 1998; Alveset al., 2000; Liu et al., 2003; Saxena et al., 2009). Since PAT is shownto possess DNA damaging as well as cell proliferative potential via
generation of free radicals, further attempts were made to study thetumorigenic effect of PAT on mice skin.
The formation of tumors and histological evaluation of the micetreated topically with PAT at a dose of 400 nmol suggests that PATacts as a tumor initiator in a two-stage mouse skin tumor protocol.Studies of Imaida et al. (1982) have also suggested that PAT hashepatic tumor initiating potential in rat liver. In our study the eval-uation of PAT as either a complete carcinogen (80 nmol) or a tumorpromoter (20 nmol) failed to produce any skin tumors, which wasconfirmed by histological studies of the skin. Significant increase ofLPO accompanied with decrease in free sulfydryls, catalase, SOD,and GR activities suggest the production of free radicals in skin trea-ted with PAT as initiator. The increase in LPO in cutaneous tissuefollowing topical application of PAT tested as complete carcinogenand as promoter also showed alteration of oxidative stress markersthereby implicating oxidative damage. This observation is consistentwith earlier studies which show that PAT mediates oxidative dam-age through generation of ROS (Barhoumi and Burghardt, 1996;Liu et al., 2007). Further, it can be argued that the concentrationof PAT may not be at the threshold levels to elicit tumor formation incomplete carcinogen (80 nmol) or promotion (20 nmol) protocol ascompared to PAT (400 nmol) used as initiator. Though our in vitroobservations showed induction of cell proliferation in epidermis by sin-gle topical treatment of PAT at a dose of 400 nmol yet it failed to possesstumor promoting potential at the tested dose (20 nmol). In this regardit has been suggested that induction of cell proliferation is necessary butnot a sufficient condition for tumor promotion in mouse skin model(Hennings and Yuspa, 1985). Several chemicals including acetic acid,mezerein, ethyl phenyl propiolate, could produce cell proliferationafter a single application and yet these compounds exhibited onlypoor papilloma promoting ability (Slaga, 1984). Thus, it is clear thatthough we could not detect PAT as a tumor promoter at the testeddose (20 nmol), it may be possible that prolonged exposure to PAT ata higher dose may induce tumor promotion and cause further toxico-logical manifestations.
In conclusion our result suggests that topical application of PAT tomice resulted in cell proliferation, which is mediated by ROS inducedMAPKs signaling pathway leading to transcriptional activation ofdownstream target proteins c-fos, c-jun and transcription factorNFκB and that PAT has dermal tumorigenic potential.
Conflict of interest
The authors have declared no conflict of interest.
Acknowledgments
We are grateful to the director of our institute, for his keen interestin this present study. This work was supported by funds from Depart-ment of Science and Technology (Govt of India) and CSIR NetworkingProject (NWP-17), New Delhi. N.S. and R.K. are also thankful to Uni-versity grants commission and Council of Scientific and Industrial Re-search, New Delhi for the award of Senior Research Fellowship. Themanuscript is IITR communication #2977.
References
Alves, I., Oliveira, N.G., Laires, A., Rodrigues, A.S., Rueff, J., 2000. Induction of micronu-clei and chromosomal aberrations by the mycotoxin patulin in mammalian cells:role of ascorbic acid as a modulator of patulin clastogenicity. Mutagenesis 15,229–234.
Anderson, P., 1997. Kinase cascades regulating entry into apoptosis. Microbiol. Mol.Biol. Rev. 61, 33–46.
Barhoumi, R., Burghardt, R.C., 1996. Kinetic analysis of the chronology of patulin andgossypol-induced cytotoxicity in vitro. Fundam. Appl. Toxicol. 30, 290–297.
Bedogni, B.O'., Neill, M.S., Welford, S.M., Bouley, D.M., Giaccia, A.J., Denko, N.C., Powell,M.B., 2004. Topical treatment with inhibitors of the phosphatidylinositol 3′-kinase/Akt and Raf/mitogen-activated protein kinase kinase/extracellular signal-
270 N. Saxena et al. / Toxicology and Applied Pharmacology 257 (2011) 264–271

Author's personal copy
regulated kinase pathways reduces melanoma development in severe combinedimmunodeficient mice. Cancer Res. 64, 2552–2560.
Benhar, M., Engelberg, D., Levitzki, A., 2002. ROS, stress-activated kinases and stresssignaling in cancer. EMBO Rep. 3 (5), 420–425.
Canty Jr., T.G., Boyle Jr., E.M., Farr, A., Morgan, E.N., Verrier, E.D., Pohlman, T.H., 1999.Oxidative stress induces NF-κB nuclear translocation without degradation ofIκBα. Circulation 100 (suppl II), II–361-II-364.
Carlberg, I., Mannervik, B., 1985. Glutathione reductase. Methods Enzymol. 113, 484–490.Chang, L., Karin, M., 2001. MammalianMAP kinase signaling cascades. Nature 410, 37–40.Chung, J.H., Kang, S., Varani, J., Lin, J., Fisher, G.J., Voorhees, J.J., 2000. Decreased extra-
cellular signal regulated kinase and increased stress activated MAP kinase activi-ties in aged human skin in vivo. J. Invest. Dermatol. 115, 177–182.
Ciegler, A., Becwith, A.C., Jackson, L.K., 1976. Teratogenicity of Patulin and Patulin ad-ducts formed with cysteine. Appl. Environ. Microbiol. 31, 664–667.
Codex Alimentarius Commission, 2003. Maximum level for Patulin in apple juice andapple juice ingredients and other beverages. No: 235, 30 June–5 July, 26th session,Rome, Italy.
Conti, C.J., Slaga, T.J., Klein-Szanto, A.J.P., 1989. Cellular and Molecular MechanismsInvolved in Multistage Skin Carcinogenesis in Carcinogenesis, A ComprehensiveSurvey, Skin Tumors: Experimental and Clinical Aspects. Raven Press, New York.
Cui, X.L., Douglas, J.G., 1997. Arachidonic acid activates c-jun N-terminal kinasethrough NADPH oxidase in rabbit proximal tubular epithelial cells. Proc. Natl.Acad. Sci. USA 94 (8), 3771–3776.
Dickens, F., Jones, H.E.H., 1961. Carcinogenic activity of a series of reactive lactones andrealted substances. Br. J. Cancer 15, 85–100.
Ellman, G.L., 1959. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 82, 70–77.Gupta, K.P., Mehrotra, N.K., 1992. Status of ornithine decarboxylase activity and DNA
synthesis in mancozed-exposed mouse skin. Carcinogenesis 13, 131–133.Habig, W.H., Pabst, M.J., Jakoby, W.B., 1974. Glutathione S-transferases. The first enzy-
matic step in mercapturic acid formation. J. Biol. Chem. 249, 7130–7139.Halazonetis, T.D., Georgopoulos, K., Greenberg, M.E., Leder, P., 1988. C-Jun dimerizes
with itself and with c-Fos, forming complexes of different DNA binding affinities.Cell 55, 917–924.
Harrison, M.A., 1989. Presence and stability of patulin in apple products: a review. J.Food Saf. 9, 147–153.
Hennings, H., Yuspa, S.H., 1985. Two-stage tumor promotion in mouse skin: an alterna-tive interpretation. J. Natl. Cancer Inst. 74, 735–740.
Imaida, K., Hirose, M., Ogiso, T., Kurata, Y., Ito, N., 1982. Quantitative analysis of initiat-ing and promoting activities of five mycotoxins in liver carcinogenesis in rats.Cancer Lett. 16, 137–143.
Johnson, G.L., Lapadat, R., 2002. Mitogen-activated protein kinase pathways mediatedby ERK, JNK and p38 protein kinases, a review. Science 298, 1911–1912.
Liu, B.H., Wu, T.S., Yu, F.Y., Su, C.C., 2007. Induction of oxidative stress response by themycotoxin Patulin in mammalian cells. Toxicol. Sci. 95, 340–347.
Liu, B.H., Wu, T.S., Yu, F.Y., Wang, C.H., 2006. Mycotoxin patulin activates the P38 kinase andJNK signaling pathways in human embryonik kidney cells. Toxicol. Sci. 89, 423–430.
Liu, B.H., Yu, F.Y., Wu, T.S., Li, S.Y., Su, M.C., Wang, M.C., Shih, S.M., 2003. Evaluation ofgenotoxic risk and oxidative DNA damage in mammalian cells exposed to myco-toxins, Patulin and citrinin. Toxicol. Appl. Pharmacol. 191, 255–263.
Lowry, O.H., Rosebrough, N.J., Farr, A.L., Randal, R.J., 1951. Protein measurement withfolin phenol reagent. J. Biol. Chem. 193, 265–275.
Misra, H.P., Fridovich, I., 1972. The generation of superoxide radical during the autoxi-dation of hemoglobin. J. Biol. Chem. 247, 6960–6962.
Osswald, H., Frank, H.K., Komitowski, D., Winter, H., 1978. Long-term testing of patulinadministered orally to Sprague–Dawley rats and Swiss mice. Food Cosmet. Toxicol.16, 243–247.
Pearson, G., Robinson, F., Gibson, T.B., Xu, B., Karandikar, M., Berman, K., Cobb, M.H.,2001. Mitogen-activated protein (MAP) kinase pathways: regulation and physio-logical functions. Endocr. Rev. 22 (2), 153–183.
Pfeiffer, E., Gross, K., Metzler, M., 1998. Aneuploidogenic and clastogenic potential ofthe mycotoxins citrinin and patulin. Carcinogenesis 19, 1313–1318.
Prieta, J., Moreno, M.A., Bayo, J., Diaz, S., Suárez, G., Domínguez, L., Canela, R., Sanchis,V., 1993. Determination of patulin by reversed-phase high-performance liquidchromatography with extraction by diphasic dialysis. Analyst 118, 171–173.
Ritieni, A., 2003. Patulin in Italian commercial apple products. J. Agric. Food Chem. 51,6086–6090.
Rotruck, J.T., Pope, A.L., Ganther, H.E., Swanson, A.B., Hafeman, D.G., Hoekstra, W.G.,1973. Selenium: biochemical role as a component of glutathione peroxidase. Sci-ence 179, 588–590.
Saxena, N., Ansari, K.M., Kumar, R., Dhawan, A., Dwivedi, P.D., Das, M., 2009. Patulincauses DNA damage leading to cell cycle arrest and apoptosis through modulationof Bax, p53 and p21/WAF1 proteins in skin of mice. Toxicol. Appl. Pharmacol. 234,192–201.
Saxena, N., Dwivedi, P.D., Ansari, K.M., Das, M., 2008. Incidence of patulin in applejuices and its likely intake in Indian population. Food Addit. Contam. B. 1, 140–146.
Sinha, A.K., 1972. Colorimetric assay of catalase. Anal. Biochem. 47, 389–394.Slaga, T.J., 1984. In: Slaga, T.J. (Ed.), Mechanisms of Tumor Promotion: Tumor Promo-
tion and Skin Carcinogenesis. CRC Press, Boca Raton, Florida, pp. 189–196.Smith, E.E., Duffus, E.A., Small, M.H., 1993. Effect of patulin on postimplantation rat
embryos. Arch. Environ. Contam. Toxicol. 25, 267–270.Snedecor, G.W., Cochran, W.G., 1967. One way classification: analysis of variance.
Statistical Methods. Iowa State University Press, Iowa, pp. 258–296.Speijers, G.J., Franken, M.A., Van Leeuwen, F.X., 1998. Subacute toxicity study of patulin
in the rat: effect on the kidney and the gastrointestinal tract. Food Chem. Toxicol.26, 23–30.
Steiman, R., Seigle-Murandi, F., Sage, L., Krivobok, S., 1989. Production of patulin bymicromycetes. Mycopathologia 105, 129–133.
Storz, P., 2005. Reactive oxygen species in tumor progression. Front. Biosci. 10,1881–1896.
Surh, Y.J., Kundu, J.K., 2005. Signal Transduction network leading to COX-2 induction: aroad map in search of cancer chemopreventives. Arch. Pharm. Res. 28, 1–15.
Takahashi, H., Ibe, M., Nakamura, S., Ishida-Yamamoto, A., Hasimoto, Y., Iizuka, H.,2002. Extracellular regulated kinase and c-Jun N-terminal kinase are activated inpsoriatic involved epidermis. J. Dermatol. Sci. 30, 94–99.
U.S. Food and Drug Administration [USFDA], 2004. Apple juice, apple juice concen-trates, and apple juice products adulteration with patulin. Com-pliance policyguidance for FDA staff. Sec. 510.150.
Utley, H.G., Bernheim, F., Hochstein, P., 1967. Effect of sulfhydryl reagents on peroxida-tion in microsomes. Arch. Biochem. Biophys. 118, 29–32.
WHO, 1998. Safety evaluation of certain mycotoxins in food WHO. 26th Report ofJECFA, Geneva, Switzerland: Technical Report Series, 859, p. 11.
Wichmann, G., Herbaeth, O., Lehmann, I., 2002. The Mycotoxins citrinin, gliotoxin andpatulin affect interferon-gamma rather than interlukin-4 production in humanblood cells. Environ. Toxicol. 17, 211–218.
Wong, B.C., Jiang, X., Fan, X.M., Lin,M.C., Jiang, S.H., Lam, S.K., Kung, H.F., 2003. Suppressionof RelA/p65 nuclear translocation independent of IκB-α degradation bycyclooxygenase-2 inhibitor in gastric cancer. Oncogene 22, 1189–1197.
Wu, T.S., Yu, F.Y., Su, C.C., Kan, J.C., Chung, C.P., Liu, B.H., 2005. Activation of ERKmitogen-activated protein kinase in human cells by the mycotoxin patulin. Toxicol.Appl. Pharmacol. 207, 103–111.
Yurdun, T., Zehra Omurtag, G., Ersov, O., 2001. Incidence of patulin in apple juices mar-kets in Turkey. J. Food Prot. 64, 1851–1853.
271N. Saxena et al. / Toxicology and Applied Pharmacology 257 (2011) 264–271
Copyright © 2022 FDOKUMEN




















