
New treatments compared to established treatments in randomized trials
Upload
independentCategory
view
0download
0
Carcinogenesis vol.23 no.3 pp.499–510, 2002
Silymarin inhibits growth and causes regression of establishedskin tumors in SENCAR mice via modulation of mitogen-activatedprotein kinases and induction of apoptosis
Rana P.Singh1,2, Anil K.Tyagi1,2, Jifu Zhao2 andRajesh Agarwal1–4
1Department of Pharmaceutical Sciences, School of Pharmacy, University ofColorado Health Sciences Center, Denver, CO 80262, USA, 2AMC CancerResearch Center, Denver, CO 80214, USA and 3University of ColoradoCancer Center, University of Colorado Health Sciences Center, Denver,CO 80262, USA4To whom correspondence should be addressedEmail: [email protected]
This study reports in vivo therapeutic efficacy of silymarinagainst skin tumors with mechanistic rationale. 7,12-Dimethylbenz[a]anthracene–12-O-tetradecanoyl-phorbol-13-acetate (DMBA–TPA)-induced established skin papil-loma (tumor)-bearing SENCAR mice were fed with 0.5%silymarin in AIN-93M-purified diet (w/w), and both tumorgrowth and regression were monitored during 5 weeks offeeding regimen. Silymarin feeding significantly inhibited(74%, P < 0.01) tumor growth and also caused regression(43%, P < 0.01) of established tumors. Proliferating cellnuclear antigen and terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling immunohistochemicalstaining of tumors showed that silymarin decreases prolif-eration index by 48% (P < 0.001) and increases apoptoticindex by 2.5-fold (P < 0.001), respectively. Skin tumorgrowth inhibition and regression by silymarin were alsoaccompanied by a strong decrease (P < 0.001) in phospho-ERK1/2 levels in tumors from silymarin-fed mice comparedwith controls. In the studies evaluating bioavailability andphysiologically achievable level of silymarin (as silibinin)in plasma, skin tumor, skin, liver, lung, mammary glandand spleen, we found 10, 6.5, 3.1, 13.7, 7.7, 5.9 and 4.4 µgsilibinin/ml plasma or per gram tissue, respectively. In anattempt to translate these findings to human skin cancerand to establish biological significance of physiologicallyachievable level, effect of plasma concentration of silibininwas next examined in human epidermoid carcinoma A431cells. Silibinin treatment of cells in culture at 12.5, 25(plasma level) and 50 µM doses resulted in 30–74% (P <0.01–0.001) growth inhibition and 7–42% death of A431cells in a dose- and time-dependent manner; apoptosis wasidentified as a cell death response by silibinin. Similarsilibinin treatments also resulted in a significant decreasein phospho-mitogen-activated protein kinase/extracellularsignal-regulated protein kinase 1/2 (MAPK/ERK1/2) levels,but an up-regulation of stress-activated protein kinase/jun
Abbreviations: DAB, 3,3�-diaminobenzidine; DMBA, 7,12-dimethyl-benz[a]anthracene; DMEM, Dulbecco’s modified Eagle medium; HPLC,high performance liquid chromatography; MAPK/ERK1/2, mitogen-activatedprotein kinase/extracellular signal-regulated protein kinase 1/2; MTT, 3-[4,5-diamethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide; PCNA, proliferatingcell nuclear antigen; PI, propidium iodide; p38 MAPK, p38 mitogen-activated protein kinase; SAPK/JNK1/2, stress-activated protein kinase/junNH2-terminal kinase; TdT, terminal deoxynucleotidyl transferase; TPA, 12-O-tetradecanoyl-phorbol-13-acetate; TUNEL, terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling.
© Oxford University Press 499
NH2-terminal kinase (SAPK/JNK1/2) and p38 mitogen-activated protein kinase (p38 MAPK) activation in A431cells. The use of MEK1 inhibitor, PD98059, showed thatinhibition of ERK1/2 signaling, in part, contributes tosilibinin-caused cell growth inhibition. Together, the datasuggest that an inhibition of ERK1/2 activation and anincreased activation of JNK1/2 and p38 by silibinin couldbe possible underlying molecular events involved in inhibi-tion of proliferation and induction of apoptosis in A431cells. These data suggest that silymarin and/or its majoractive constituent silibinin could be an effective agent forboth prevention and intervention of human skin cancer.
Introduction
The role of dietary modification in the reduction of cancerrisk has recently drawn widespread attention, because thedifferences in worldwide human cancer mortality often dependon lifestyle and dietary habits (1–3). As a result of theunavoidable presence of mutagens and carcinogens in ourenvironment and also in the human diet, it has been suggestedthat dietary intake of phytochemicals, including antioxidants,could be a useful strategy against the deleterious effects ofthese mutagenic and carcinogenic agents (4). Diets rich innaturally occurring polyphenolic flavonoids have beenassociated with the reduced incidence of certain human cancers(1–10). Among these, silymarin, isolated from the fruits ofmilk thistle, Silybum marianum L. Gaertn. (11), is being usedclinically as an anti-hepatotoxic agent for the treatment ofvarious liver diseases in Europe and Asia (9,11) and recently,has also been marketed in the USA and Europe as a dietarysupplement. Silymarin is composed mainly of silibinin withsmall amounts of other silibinin stereoisomers, namely,isosilybin, dihydrosilybin, silydianin and silychristin (12).Recent studies with silymarin showed that it could be usedagainst a wide range of liver and gall bladder diseases,including hepatitis and cirrhosis as well as dermatologicalconditions (13–15). Studies on various animal models usingdifferent modes of administration of silymarin showed thatit is non-toxic and largely free of adverse side effects insubchronic and chronic tests even at large doses, and alsothere is no known LD50 in laboratory animals (16). Severalstudies in rodents and cell cultures have shown that silymarinis a strong antioxidant and scavenges both free radicals andreactive oxygen species, and thereby provides significantprotection against different cancers of epithelial origin (17–19). It has also been shown that silymarin inhibits the 12-O-tetradecanoyl-phorbol-13-acetate (TPA)-induced anchorage-independent growth of JB6 mouse epidermal cells (20), 7,12-dimethylbenz[a]anthracene (DMBA)–TPA-induced mammarylesion formation in organ culture (21) and formation oftransformed rat tracheal epithelial cell colonies induced byexposure to benzo[a]pyrene (22).
We have recently demonstrated the strong cancer chemo-

R.P.Singh et al.
preventive effects of silymarin using different long-term skintumorigenesis animal protocols as well as short-term cellculture systems (5,23–26). Studies in our laboratory haveshown that silymarin strongly inhibits ornithine decarboxylaseactivity and mRNA expression as well as TNF-α mRNAexpression induced by structurally different tumor promoters,including free radical-generating compounds, in mouse epi-dermis (24,26). In another study we have shown that silymarinaffords strong protection against UV-B radiation-induced tumorinitiation, tumor promotion and complete carcinogenesis inSKH-1 hairless mouse skin (23). A mechanistic study hasshown that silymarin inhibits TPA-caused induction of cyclo-oxygenase (COX) 2 and interleukin-1α expression in SENCARmouse epidermis (25). Recently, we showed that the skincancer preventive effect of silymarin involves the impairmentof epidermal growth factor receptor (EGFR)-mediated sig-naling (27,28).
After establishing that silymarin is strongly effective ininhibiting the initial stages of skin tumor development, theaim of the present study was to ascertain its anticancer effecton DMBA–TPA-induced established skin papillomas (tumors)in SENCAR mice, and possible implications of such findingsfor human skin cancer intervention at actinic keratosis stage,a benign skin cancer condition. First, we studied in vivotherapeutic effect of silymarin in DMBA–TPA-induced estab-lished mouse skin tumors and associated molecularmechanisms. Next, we examined whether the mechanism-based anticancer effect of silymarin (as its major activeconstituent silibinin) is operative in human epidermoidcarcinoma A431 cells. We also examined the levels of dietaryadministered silymarin (as silibinin) in plasma, skin tumors,skin and some other organs which are susceptible to thedevelopment of cancer to evaluate its bio-distribution, andto determine pharmacologically and physiologically effectivedoses for further advanced studies. Here, we present theevidence that silymarin effectively suppresses tumor growthand causes regression of established skin tumors in vivo,possibly via inhibition of mitogen-activated protein kinase/extracellular signal-regulated protein kinase 1/2 (MAPK/ERK1/2) activation leading to a decrease in proliferation andan induction of apoptosis in tumor cells. Furthermore, it showsa strong anticancer effect against A431 cells via similarmechanisms together with an activation of stress-activatedprotein kinase/jun NH2-terminal kinase (SAPK/JNK1/2) andp38 mitogen-activated protein kinase (p38 MAPK), making itan excellent candidate for an anticancer agent against humanskin cancer.
Materials and methods
Animals, diets and chemicals
Six-week-old female SENCAR mice were obtained from the National CancerInstitute (Frederick Cancer Research and Development Center, Frederick,MD) and housed at standard laboratory conditions (temperature 24 � 2°C,relative humidity 50 � 10% and 12 h room light/12 h dark cycle). They wereacclimatized for 1 week before starting the present study, and fed with Purinachow diet and water ad libitum. DMBA was from Aldrich Chemical Co.(Milwaukee, WI) and silymarin, silibinin and TPA were from Sigma ChemicalCo. (St Louis, MO). The silymarin dose used in this study was 0.5% (w/w)in AIN-93M-purified rodent diet (pelleted) prepared by Dyets Inc. (Bethlehem,PA). All other chemicals were obtained in the purest form available commer-cially.
Cell line and reagents
Human epidermal carcinoma cell line A431 was from the American TypeCulture Collection (Manassas, VA). Cells were grown in Dulbecco’s modified
500
Eagle medium (DMEM; Gibco BRL, Grand Island, NY) with 10% fetalbovine serum (Hyclone, Logan, UT) and 1% penicillin–streptomycin (GibcoBRL) at standard culture conditions. Phospho- (and regular) MAPK/ERK1/2(Thr202/Tyr204) antibodies were from New England Biolabs (Beverly, MA).Phospho- (and regular) SAPK/JNK1/2 (Thr183/Tyr185) and p38 MAPK(Thr180/Tyr182) antibodies were purchased from Cell Signaling Technology(Fremont, CA). Anti-rabbit immunoglobulin horseradish peroxidase-conjug-ated secondary antibody was purchased from Santa Cruz Biotechnology (SantaCruz, CA). PD98059 was from Alexis Biochemicals (San Diego, CA). ECLdetection system was from Amersham (Arlington Heights, IL).
Formation of established skin papillomas in SENCAR mice and treatmentwith silymarin
For the formation of established skin papillomas in SENCAR mice, arecently published protocol by Lu et al. was used (29). Briefly, SENCARmice were treated with a single topical application of DMBA (200 nmol) in200 µl acetone on to the shaved dorsal skin of each mouse. After 1 week,tumor initiated skin was promoted with 5 nmol TPA (in 200 µl acetone) permouse twice a week for 24 weeks, at which point 100% of mice showedestablished skin papillomas (29), hereafter referred to as skin tumors. Todetermine the effect of silymarin on established skin tumor growth at 24weeks, mice were transferred from a regular chow diet to a more definedAIN-93M-purified diet for 5 days before starting silymarin treatment. Micehaving six to eight tumors were randomly divided into two groups of nine to10 animals each and one group of animals was continued on the AIN-93Mdiet and served as control whereas the other group of animals was fed withthe AIN-93M diet containing 0.5% silymarin (w/w) for 5 weeks. Body weightwas recorded throughout the experiment, and tumor sizes were measuredtwice weekly and tumor volume was calculated by: 4πr3/3, where ‘r’ is themean radius of two dimensions of each tumor. With regard to tumor study,data from the control or silymarin-fed group of mice were analyzed for: (i)tumor volume/mouse that represents total tumor burden in terms of volume/mouse; (ii) tumor volume/tumor that represents the average volume of eachtumor from all the tumors in each mouse; and (iii) a comparison betweenoverall change in tumor volume/mouse at the start (0 week) and end (5 weeks)of dietary feeding.
At the end of the experiment, blood was collected intracardiacally for highperformance liquid chromatography (HPLC) analysis of silymarin level inplasma samples. Additionally, tumors, skin, liver, lung, mammary gland andspleen were also collected to estimate bio-distribution of dietary fed silymarin.All tumors were stored as desired and processed for molecular and immuno-histochemical studies as needed.
Immunohistochemical detection of proliferating cell nuclear antigen in skintumors
Briefly, tumor samples were fixed in 10% buffered formalin for 24 h andprocessed conventionally. Paraffin-embedded tumor sections (5 µm thick)were heat immobilized, and deparaffinized using xylene and rehydrated in agraded series of ethanol with a final wash in distilled water. Antigen retrievalwas carried out in 10 mM citrate buffer (pH 6.0) in a microwave for 2 and18 min at full and 20% of power levels, respectively. Endogenous peroxidaseactivity was blocked by immersing the sections in 3% H2O2 in methanol(v/v) followed by three changes in 10 mM phosphate-buffered saline (PBS,pH 7.4). Sections were then incubated with mouse monoclonal anti-prolifer-ating cell nuclear antigen (PCNA) antibody IgG2a (Dako, Carpinteria, CA),1:400 in PBS for 1 h at 37°C in humidity chamber. Negative controls weretreated only with PBS under identical conditions. Sections were then incubatedwith biotinylated rabbit anti-mouse antibody IgG (1:200 in 10% normal rabbitserum from Dako) for 30 min at room temperature. Thereafter, following washwith PBS, sections were incubated with conjugated horseradish peroxidasestreptavidin (Dako), 1:1000 in PBS for 30 min at room temperature in ahumidity chamber. Sections were then incubated with 3,3�-diaminobenzidine(DAB; Sigma) working solution for 10 min at room temperature and counter-stained with diluted Harris hematoxylin for 2 min, and rinsed in Scott’s water.Slides were then dehydrated, mounted, viewed and photographed using Zeisslight microscope (Germany). Proliferating cells were quantified by countingPCNA-positive cells and total number of cells at 10 arbitrarily selected fieldsat 400-fold magnification in a double-blinded manner. The proliferation index(per 400-fold microscope field) was determined as number of PCNA-positivecells�100/total number of cells.
In situ apoptosis detection in skin tumors by terminal deoxynucleotidyltransferase-mediated dUTP nick end-labeling staining
First, formalin-fixed and paraffin-embedded 5 µm thick sections of tumorsamples (those used for PCNA staining) were used for conventional H&Estaining to observe the cellular morphological changes. Next, in order toidentify early as well as late apoptotic cells, tumor samples were also studiedby terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling

Silymarin inhibits growth of established skin tumors
(TUNEL) staining. DNA fragmentation in the individual apoptotic cell wasvisualized by detection of biotinylated nucleotides incorporated onto the free3�-hydroxyl residues of these DNA fragments by Tumor TACS In SituApoptosis Detection Kit (R & D Systems, Minneapolis, MN). Briefly, tumorsamples were first fixed in 10% buffered formalin for 8 h to prevent the lossof low molecular weight DNA fragments; then paraffin-embedded 5 µm thicksections were cleared in xylene and rehydrated in graded concentrations ofethanol. Slides were rinsed with Ca2�, Mg2� and DNAase-free PBS (10 mMPBS, pH 7.4) and permeabilized with proteinase K at room temperature tomake DNA accessible to labeling enzyme. For positive control, sections wereincubated with TACS-nuclease for 30 min, which generated DNA strandbreaks in virtually every cell. Endogenous peroxidase activity was quenchedusing 5% H2O2 (in methanol, v/v) for 5 min and sections were incubated withTdT (terminal deoxynucleotidyl transferase) labeling buffer for 5 min beforestarting the labeling reaction. Sections were then incubated with TdT enzymeand biotinylated nucleotides (for negative control, labeling buffer was usedinstead of TdT enzyme) for 1 h at 37°C in humidified chamber. The reactionwas stopped by adding TdT stop buffer for 5 min. Sections were incubatedwith streptavidin-conjugated horseradish peroxidase for 10 min. Brown colorwas developed by incubation in DAB working solution (1:5:5000; 30%H2O2:DAB:1�PBS) for 7 min at room temperature. Slides were counter-stained in 1% methyl green for 1 min and visualized and scored under a lightmicroscope. Apoptosis was evaluated by counting positive cells (brownstained) as well as total number of cells at 10 arbitrarily selected fields at400-fold magnification in a double-blinded manner. The apoptotic index (per400-fold microscope field) was calculated as number of apoptotic cells�100/total number of cells.
MAPK/ERK1/2 study in skin tumorsBriefly, one piece of tumor, collected from each mouse at the termination ofthe experiment, was frozen in liquid nitrogen and powdered by mortar andpestle. Tumor samples (100 mg each) were added with 200 µl of lysisbuffer (10 mM Tris–HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, 1 mMEDTA, 1 mM EGTA, 0.3 mM phenyl methyl sulfonyl fluoride, 0.2 mMsodium orthovanadate, 0.5% NP-40, 5 U/ml aprotinin), homogenized at 4°Cand kept on ice for 30 min followed by centrifugation at 15 000 r.p.m. for30 min. Protein concentration was determined in clear supernatants (tumorlysates) using Bio-Rad DC (Hercules, CA) protein assay kit. For westernimmunoblotting, 70 µg protein/tumor lysate from both control- and silymarin-fed groups were denatured with 2� sample buffer and subjected to SDS–PAGE on a 12% gel. Separated proteins were transferred on to nitrocellulosemembrane, and probed with anti-phospho-MAPK/ERK1/2 and anti-MAPK/ERK1/2 antibodies followed by peroxidase-conjugated appropriate secondaryantibody and visualization by ECL detection system.
HPLC analysis for bio-distribution of dietary administered silymarin (assilibinin) in different organs and plasma of miceAt the end of 5 weeks of dietary feeding of silymarin to the mice withestablished skin tumors, all the mice were killed and the blood was collectedintracardiacally to harvest plasma samples. Additionally, to assess the bio-distribution of silymarin, different organs (skin tumor, skin, liver, lung,mammary gland and spleen) were also collected and frozen in liquid nitrogenand stored at –80°C until further processing. For the extraction of totalsilibinin (free and conjugated forms), 100–200 mg of tissue samples werethoroughly homogenized in 3 vol of 50 mM Tris–HCl (pH 7.4) at 4°C usingan Polytron PT-10 homogenizer (VWR Scientific, Painfield, NJ). Thereafter,each tissue homogenate or plasma sample (100 µl) was mixed with 10 µl ofan ascorbate–EDTA solution (20% ascorbic acid and 0.01% EDTA dissolvedin a 0.4 M sodium phosphate buffer, final pH 3.6), and a mixture ofβ-glucuronidase (250 U) and sulfatase (20 U). The reaction mixture was thenincubated at 37°C for 45 min and extracted twice with ethyl acetate. Theethyl acetate extracts were pooled and evaporated to dryness in a vacuumcentrifuge concentrator. The residues obtained were re-dissolved in 20 µl of10% acetonitrile aqueous solution and subjected to centrifugation. The resultantclear supernatants were stored at –80°C until HPLC analysis.
Standard silibinin and the ethyl acetate extracts of plasma and tissue sampleswere analyzed on HPLC system (ESA, Bedford, MA) using C18 reversedphase analytical column (3 µm, 4.6�250 mm). HPLC mobile phase used wassolvent A (0.05% v/v acetic acid in distilled water) and solvent B (100%acetonitrile). The linear gradient system, employed at room temperature, was:0–5 min, 100% solvent A; 5–10 min, 100% solvent A to 70% solvent A and30% solvent B; 10–30 min, 70% solvent A and 30% solvent B to 30% solventA and 70% solvent B; 32 min, stop of run. The solvent flow rate throughoutthe run was 1 ml/min. The column eluate was monitored by UV absorbanceat 270 nm. The silibinin peak in plasma, and other tissue samples, wasidentified by comparing their HPLC retention time with authentic standardsilibinin under identical conditions.
Before the analysis and quantification of silibinin extracted from plasma
501
and tissue samples, different concentrations of standard silibinin were analyzedby HPLC to find a quantitative linear range for UV detection. Silibinin insamples was quantified on the basis of peak area under the curve calculationsand comparison with standard silibinin. The recovery of silibinin afterextraction from plasma and tissue homogenates was checked by adding aknown amount of silibinin to plasma and each tissue homogenate, andfollowing vigorous mixing, its extraction and quantification by HPLC asdetailed above.
Cell growth and viability assays
Human epidermoid carcinoma A431 cells were plated at 5000 cells/cm2 in35 mm dishes under the culture conditions described above. At 25–30%confluency, cells were fed with fresh medium and treated with either DMSOalone (control) or different concentrations (12.5, 25 and 50 µM) of silibininin DMSO. At 24, 48 and 72 h after the treatment, both floating and attachedcells were collected by trypsinization, and counted in duplicate with ahemocytometer. Trypan blue dye exclusion was used to determine the cellviability. Each treatment and time point had two plates. The representativedata shown in this study were reproducible in two independent experiments.
Silibinin treatment of A431 cells, and MAPK/ERK1/2, SAPK/JNK1/2 and p38MAPK studies
A431 cells were grown in 100 mm dishes, as described above, and at40–50% confluency were treated with either DMSO alone or differentconcentrations (12.5, 25 and 50 µM) of silibinin in DMSO. After 24 and48 h of these treatments, medium was aspirated and cells were washed twicewith ice-cold PBS. Two hundred microliters of lysis buffer (used in skintumor study) was added in each dish and kept at 4°C for 15 min. Cell lysateswere scraped and kept on ice for an additional 20 min followed by clearingof lysate and protein estimation as described above. For western immunoblot-ting, 50 µg protein/sample lysate was used as described in the skin tumor study.
FACS analysis for apoptotic death of A431 cells
Briefly, at ~30% confluency (in culture conditions described above) cells weretreated with either DMSO alone (control) or varying concentrations (12.5, 25and 50 µM) of silibinin. After 48 and 72 h of these treatments, both floatingand attached cells were collected, and a quantitative apoptotic death assaywas performed by annexin V and propidium iodide (PI) staining followed byFACS analysis using Vybrant Apoptosis Assay Kit 2 (Molecular Probes,Eugene, OR) and vendor’s protocol as described recently (30). The kit containsrecombinant annexin V conjugated to fluorophores and Alexa Fluro 488 dyehaving almost perfect spectral match to fluorescein and comparatively greatersensitivity. In apoptotic cells, annexin V binds to phospatidylserine, which istranslocated from inner to outer leaflet of the plasma membrane.
3-[4,5-Diamethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide assay
Briefly, 5000 A431 cells/well were plated in 96 well plates under the cultureconditions described above, and treated either with vehicle (0.1% DMSOv/v), or with silibinin (50 µM), or MEK1 inhibitor PD98059 (50 µM) or bothfor 24 h. At the end of the treatment, cells were washed twice with 1�PBS and incubated with 1 mg 3-[4,5-diamethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT)/ml of serum-free media for 2 h. Finally, treatmentmedium was aspirated out and DMSO was added in each plate, and absorbanceof developed color was taken at 590 nm against DMSO using a 96 well-microplate reader. Similar silibinin and PD98059 doses were also used forcell growth assay as described above.
Densitometric analysis and statistical evaluation
Autoradiograms of western immunoblots were scanned with Adobe Photoshop(Adobe Systems, San Jose, CA), and adjusted for brightness and contrast forminimum background. Mean density of each band was analyzed by ScanimageProgram (NIH, Bethesda, MD). In each case, densitometric analysis data forphospho-ERK1/2, JNK1/2 and p38 blots were corrected for loading with thedensity of total ERK1/2, JNK1/2 and p38 blots, respectively. For statisticalevaluation, data were analyzed using Jandel Scientific SigmaStat 2.0 softwarepackage. For all measurements, Student’s two-tailed t-test was employed toassess statistical significance of difference between control and silymarin (orsilibinin)-treated groups. A statistically significant difference was consideredto be present at P � 0.05.
Results
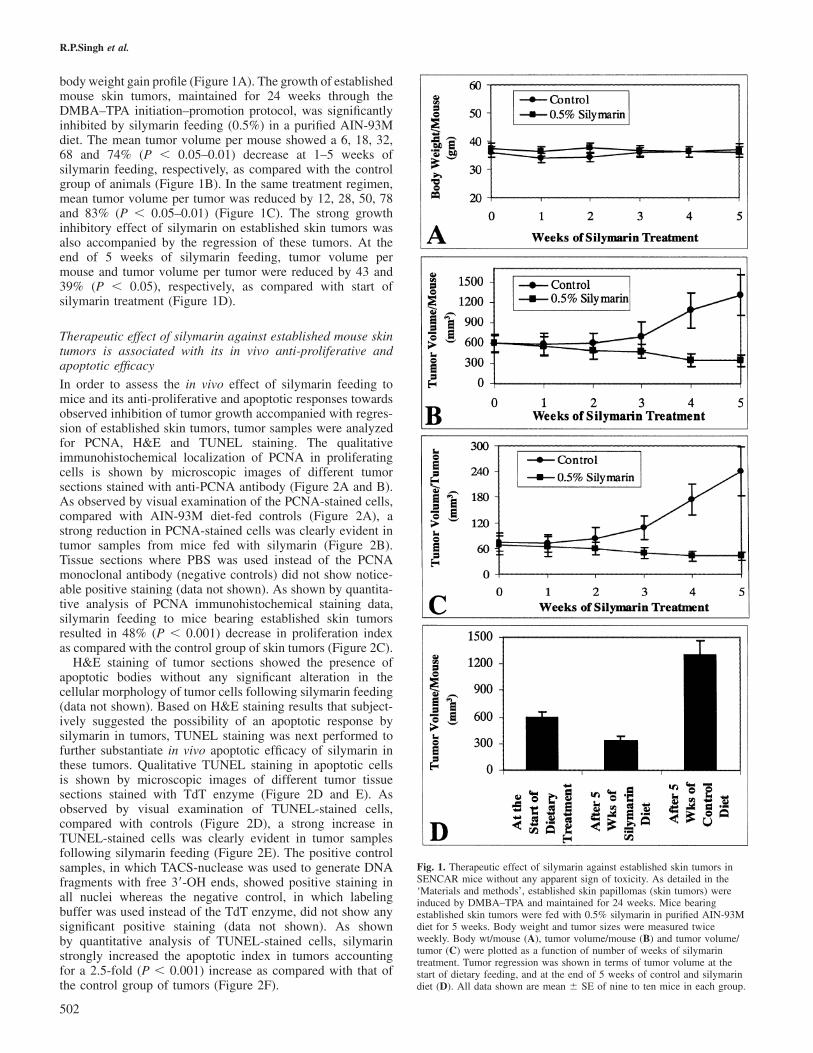
Growth inhibition and regression of established skin tumorsin SENCAR mice by silymarinThe important observation was that dietary feeding of silymarinat 0.5% (w/w) dose to mice bearing established skin tumorsdid not show any apparent sign of toxicity as monitored by
R.P.Singh et al.
body weight gain profile (Figure 1A). The growth of establishedmouse skin tumors, maintained for 24 weeks through theDMBA–TPA initiation–promotion protocol, was significantlyinhibited by silymarin feeding (0.5%) in a purified AIN-93Mdiet. The mean tumor volume per mouse showed a 6, 18, 32,68 and 74% (P � 0.05–0.01) decrease at 1–5 weeks ofsilymarin feeding, respectively, as compared with the controlgroup of animals (Figure 1B). In the same treatment regimen,mean tumor volume per tumor was reduced by 12, 28, 50, 78and 83% (P � 0.05–0.01) (Figure 1C). The strong growthinhibitory effect of silymarin on established skin tumors wasalso accompanied by the regression of these tumors. At theend of 5 weeks of silymarin feeding, tumor volume permouse and tumor volume per tumor were reduced by 43 and39% (P � 0.05), respectively, as compared with start ofsilymarin treatment (Figure 1D).
Therapeutic effect of silymarin against established mouse skintumors is associated with its in vivo anti-proliferative andapoptotic efficacy
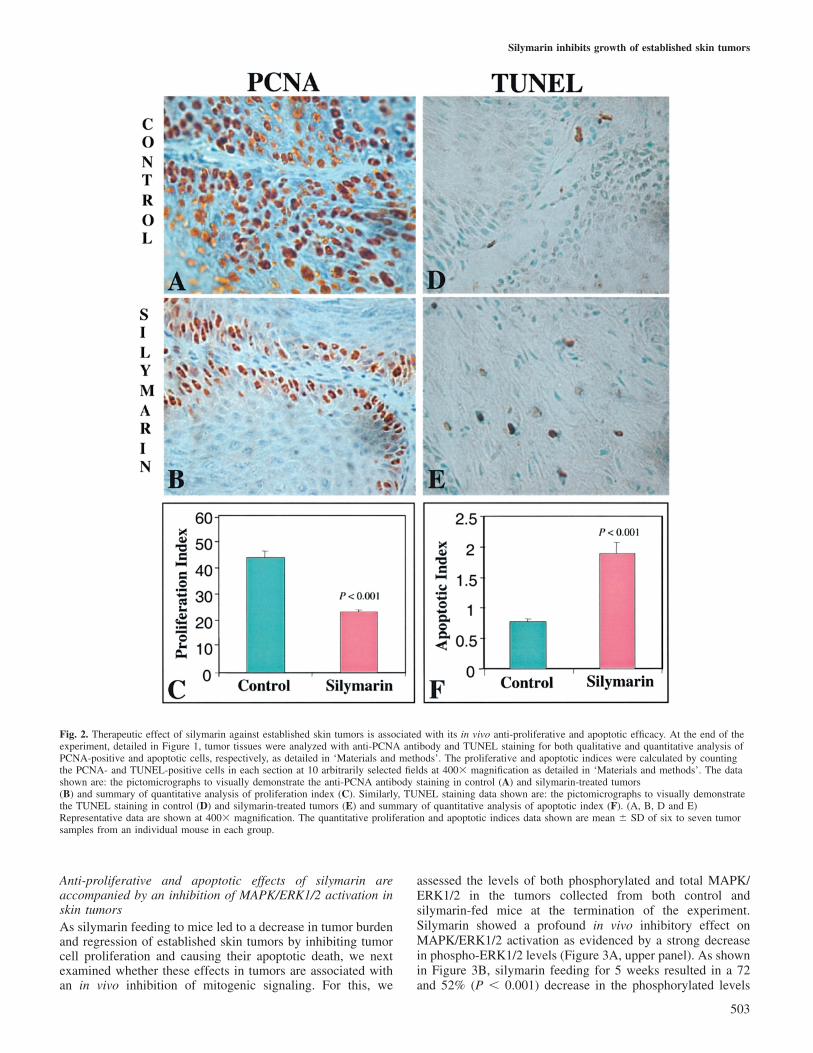
In order to assess the in vivo effect of silymarin feeding tomice and its anti-proliferative and apoptotic responses towardsobserved inhibition of tumor growth accompanied with regres-sion of established skin tumors, tumor samples were analyzedfor PCNA, H&E and TUNEL staining. The qualitativeimmunohistochemical localization of PCNA in proliferatingcells is shown by microscopic images of different tumorsections stained with anti-PCNA antibody (Figure 2A and B).As observed by visual examination of the PCNA-stained cells,compared with AIN-93M diet-fed controls (Figure 2A), astrong reduction in PCNA-stained cells was clearly evident intumor samples from mice fed with silymarin (Figure 2B).Tissue sections where PBS was used instead of the PCNAmonoclonal antibody (negative controls) did not show notice-able positive staining (data not shown). As shown by quantita-tive analysis of PCNA immunohistochemical staining data,silymarin feeding to mice bearing established skin tumorsresulted in 48% (P � 0.001) decrease in proliferation indexas compared with the control group of skin tumors (Figure 2C).
H&E staining of tumor sections showed the presence ofapoptotic bodies without any significant alteration in thecellular morphology of tumor cells following silymarin feeding(data not shown). Based on H&E staining results that subject-ively suggested the possibility of an apoptotic response bysilymarin in tumors, TUNEL staining was next performed tofurther substantiate in vivo apoptotic efficacy of silymarin inthese tumors. Qualitative TUNEL staining in apoptotic cellsis shown by microscopic images of different tumor tissuesections stained with TdT enzyme (Figure 2D and E). Asobserved by visual examination of TUNEL-stained cells,compared with controls (Figure 2D), a strong increase inTUNEL-stained cells was clearly evident in tumor samplesfollowing silymarin feeding (Figure 2E). The positive controlsamples, in which TACS-nuclease was used to generate DNAfragments with free 3�-OH ends, showed positive staining inall nuclei whereas the negative control, in which labelingbuffer was used instead of the TdT enzyme, did not show anysignificant positive staining (data not shown). As shownby quantitative analysis of TUNEL-stained cells, silymarinstrongly increased the apoptotic index in tumors accountingfor a 2.5-fold (P � 0.001) increase as compared with that ofthe control group of tumors (Figure 2F).
502
Fig. 1. Therapeutic effect of silymarin against established skin tumors inSENCAR mice without any apparent sign of toxicity. As detailed in the‘Materials and methods’, established skin papillomas (skin tumors) wereinduced by DMBA–TPA and maintained for 24 weeks. Mice bearingestablished skin tumors were fed with 0.5% silymarin in purified AIN-93Mdiet for 5 weeks. Body weight and tumor sizes were measured twiceweekly. Body wt/mouse (A), tumor volume/mouse (B) and tumor volume/tumor (C) were plotted as a function of number of weeks of silymarintreatment. Tumor regression was shown in terms of tumor volume at thestart of dietary feeding, and at the end of 5 weeks of control and silymarindiet (D). All data shown are mean � SE of nine to ten mice in each group.
Silymarin inhibits growth of established skin tumors
Fig. 2. Therapeutic effect of silymarin against established skin tumors is associated with its in vivo anti-proliferative and apoptotic efficacy. At the end of theexperiment, detailed in Figure 1, tumor tissues were analyzed with anti-PCNA antibody and TUNEL staining for both qualitative and quantitative analysis ofPCNA-positive and apoptotic cells, respectively, as detailed in ‘Materials and methods’. The proliferative and apoptotic indices were calculated by countingthe PCNA- and TUNEL-positive cells in each section at 10 arbitrarily selected fields at 400� magnification as detailed in ‘Materials and methods’. The datashown are: the pictomicrographs to visually demonstrate the anti-PCNA antibody staining in control (A) and silymarin-treated tumors(B) and summary of quantitative analysis of proliferation index (C). Similarly, TUNEL staining data shown are: the pictomicrographs to visually demonstratethe TUNEL staining in control (D) and silymarin-treated tumors (E) and summary of quantitative analysis of apoptotic index (F). (A, B, D and E)Representative data are shown at 400� magnification. The quantitative proliferation and apoptotic indices data shown are mean � SD of six to seven tumorsamples from an individual mouse in each group.
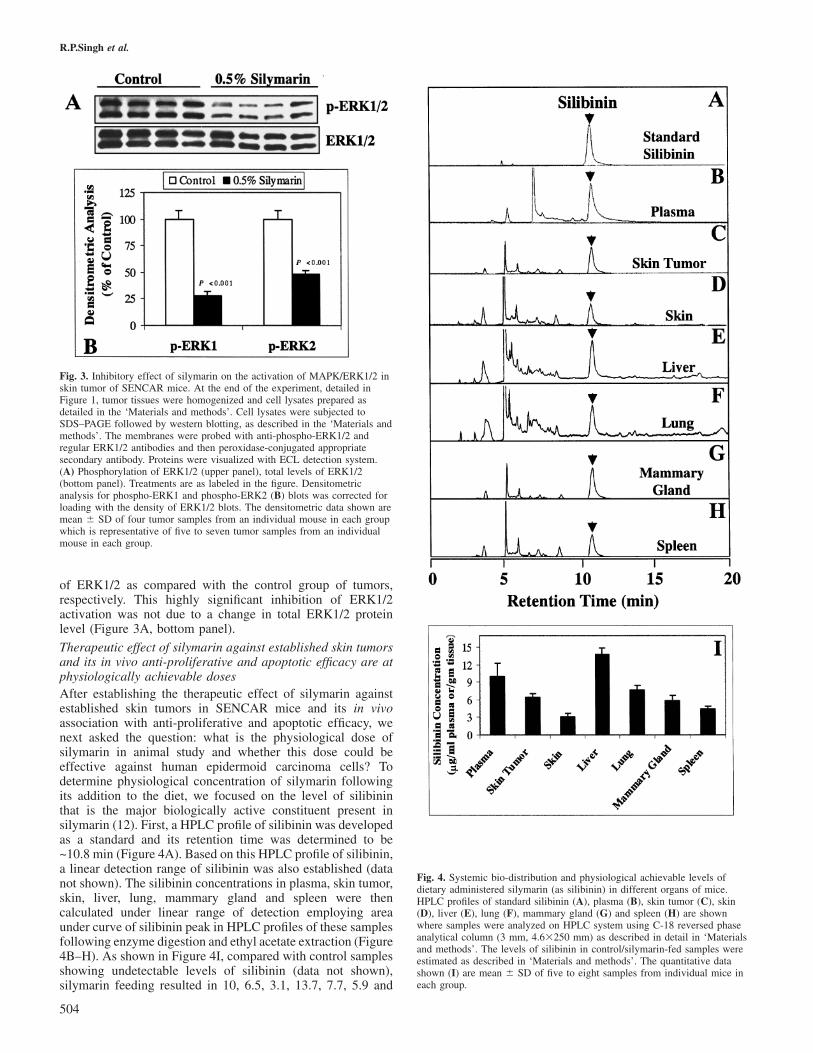
Anti-proliferative and apoptotic effects of silymarin areaccompanied by an inhibition of MAPK/ERK1/2 activation inskin tumorsAs silymarin feeding to mice led to a decrease in tumor burdenand regression of established skin tumors by inhibiting tumorcell proliferation and causing their apoptotic death, we nextexamined whether these effects in tumors are associated withan in vivo inhibition of mitogenic signaling. For this, we
503
assessed the levels of both phosphorylated and total MAPK/ERK1/2 in the tumors collected from both control andsilymarin-fed mice at the termination of the experiment.Silymarin showed a profound in vivo inhibitory effect onMAPK/ERK1/2 activation as evidenced by a strong decreasein phospho-ERK1/2 levels (Figure 3A, upper panel). As shownin Figure 3B, silymarin feeding for 5 weeks resulted in a 72and 52% (P � 0.001) decrease in the phosphorylated levels
R.P.Singh et al.
Fig. 3. Inhibitory effect of silymarin on the activation of MAPK/ERK1/2 inskin tumor of SENCAR mice. At the end of the experiment, detailed inFigure 1, tumor tissues were homogenized and cell lysates prepared asdetailed in the ‘Materials and methods’. Cell lysates were subjected toSDS–PAGE followed by western blotting, as described in the ‘Materials andmethods’. The membranes were probed with anti-phospho-ERK1/2 andregular ERK1/2 antibodies and then peroxidase-conjugated appropriatesecondary antibody. Proteins were visualized with ECL detection system.(A) Phosphorylation of ERK1/2 (upper panel), total levels of ERK1/2(bottom panel). Treatments are as labeled in the figure. Densitometricanalysis for phospho-ERK1 and phospho-ERK2 (B) blots was corrected forloading with the density of ERK1/2 blots. The densitometric data shown aremean � SD of four tumor samples from an individual mouse in each groupwhich is representative of five to seven tumor samples from an individualmouse in each group.
of ERK1/2 as compared with the control group of tumors,respectively. This highly significant inhibition of ERK1/2activation was not due to a change in total ERK1/2 proteinlevel (Figure 3A, bottom panel).
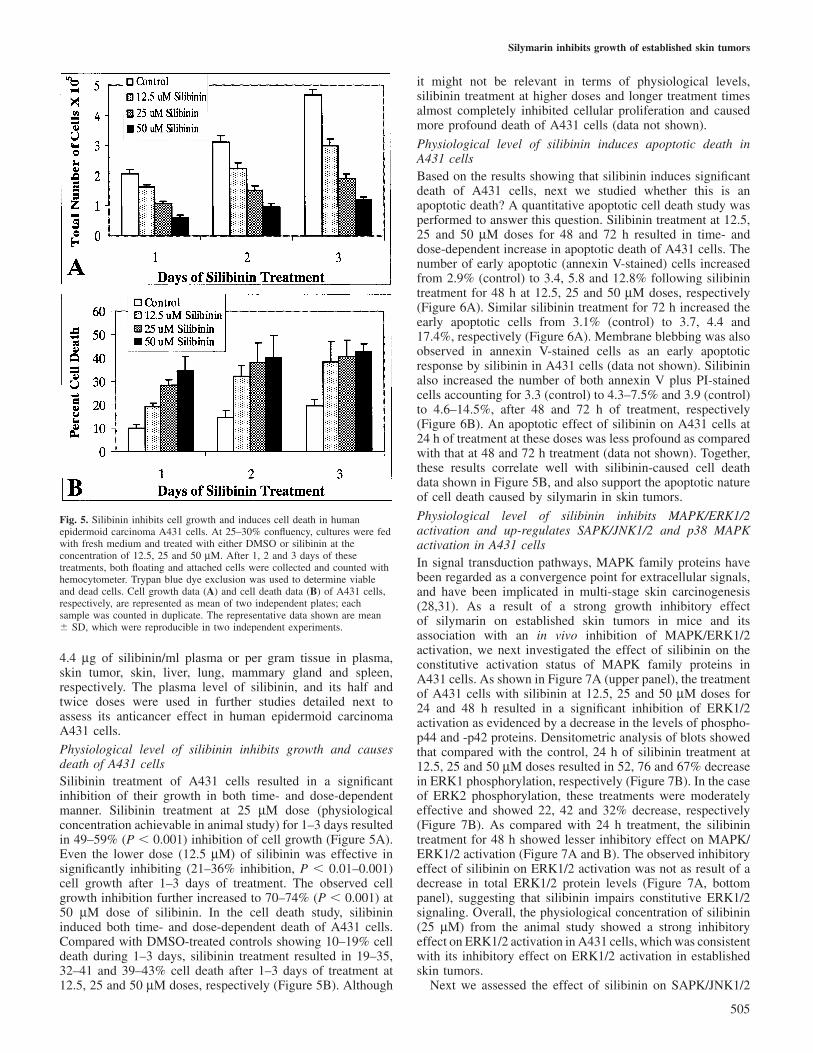
Therapeutic effect of silymarin against established skin tumorsand its in vivo anti-proliferative and apoptotic efficacy are atphysiologically achievable dosesAfter establishing the therapeutic effect of silymarin againstestablished skin tumors in SENCAR mice and its in vivoassociation with anti-proliferative and apoptotic efficacy, wenext asked the question: what is the physiological dose ofsilymarin in animal study and whether this dose could beeffective against human epidermoid carcinoma cells? Todetermine physiological concentration of silymarin followingits addition to the diet, we focused on the level of silibininthat is the major biologically active constituent present insilymarin (12). First, a HPLC profile of silibinin was developedas a standard and its retention time was determined to be~10.8 min (Figure 4A). Based on this HPLC profile of silibinin,a linear detection range of silibinin was also established (datanot shown). The silibinin concentrations in plasma, skin tumor,skin, liver, lung, mammary gland and spleen were thencalculated under linear range of detection employing areaunder curve of silibinin peak in HPLC profiles of these samplesfollowing enzyme digestion and ethyl acetate extraction (Figure4B–H). As shown in Figure 4I, compared with control samplesshowing undetectable levels of silibinin (data not shown),silymarin feeding resulted in 10, 6.5, 3.1, 13.7, 7.7, 5.9 and
504
Fig. 4. Systemic bio-distribution and physiological achievable levels ofdietary administered silymarin (as silibinin) in different organs of mice.HPLC profiles of standard silibinin (A), plasma (B), skin tumor (C), skin(D), liver (E), lung (F), mammary gland (G) and spleen (H) are shownwhere samples were analyzed on HPLC system using C-18 reversed phaseanalytical column (3 mm, 4.6�250 mm) as described in detail in ‘Materialsand methods’. The levels of silibinin in control/silymarin-fed samples wereestimated as described in ‘Materials and methods’. The quantitative datashown (I) are mean � SD of five to eight samples from individual mice ineach group.
Silymarin inhibits growth of established skin tumors
Fig. 5. Silibinin inhibits cell growth and induces cell death in humanepidermoid carcinoma A431 cells. At 25–30% confluency, cultures were fedwith fresh medium and treated with either DMSO or silibinin at theconcentration of 12.5, 25 and 50 µM. After 1, 2 and 3 days of thesetreatments, both floating and attached cells were collected and counted withhemocytometer. Trypan blue dye exclusion was used to determine viableand dead cells. Cell growth data (A) and cell death data (B) of A431 cells,respectively, are represented as mean of two independent plates; eachsample was counted in duplicate. The representative data shown are mean� SD, which were reproducible in two independent experiments.
4.4 µg of silibinin/ml plasma or per gram tissue in plasma,skin tumor, skin, liver, lung, mammary gland and spleen,respectively. The plasma level of silibinin, and its half andtwice doses were used in further studies detailed next toassess its anticancer effect in human epidermoid carcinomaA431 cells.
Physiological level of silibinin inhibits growth and causesdeath of A431 cellsSilibinin treatment of A431 cells resulted in a significantinhibition of their growth in both time- and dose-dependentmanner. Silibinin treatment at 25 µM dose (physiologicalconcentration achievable in animal study) for 1–3 days resultedin 49–59% (P � 0.001) inhibition of cell growth (Figure 5A).Even the lower dose (12.5 µM) of silibinin was effective insignificantly inhibiting (21–36% inhibition, P � 0.01–0.001)cell growth after 1–3 days of treatment. The observed cellgrowth inhibition further increased to 70–74% (P � 0.001) at50 µM dose of silibinin. In the cell death study, silibinininduced both time- and dose-dependent death of A431 cells.Compared with DMSO-treated controls showing 10–19% celldeath during 1–3 days, silibinin treatment resulted in 19–35,32–41 and 39–43% cell death after 1–3 days of treatment at12.5, 25 and 50 µM doses, respectively (Figure 5B). Although
505
it might not be relevant in terms of physiological levels,silibinin treatment at higher doses and longer treatment timesalmost completely inhibited cellular proliferation and causedmore profound death of A431 cells (data not shown).
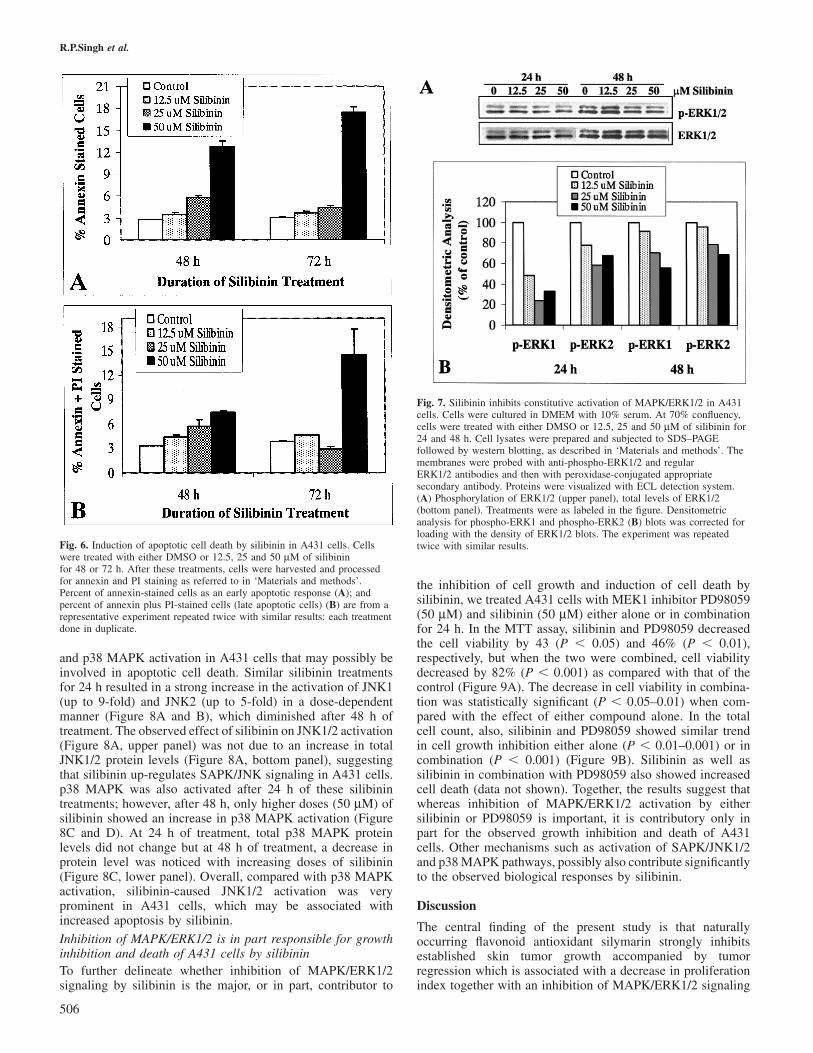
Physiological level of silibinin induces apoptotic death inA431 cellsBased on the results showing that silibinin induces significantdeath of A431 cells, next we studied whether this is anapoptotic death? A quantitative apoptotic cell death study wasperformed to answer this question. Silibinin treatment at 12.5,25 and 50 µM doses for 48 and 72 h resulted in time- anddose-dependent increase in apoptotic death of A431 cells. Thenumber of early apoptotic (annexin V-stained) cells increasedfrom 2.9% (control) to 3.4, 5.8 and 12.8% following silibinintreatment for 48 h at 12.5, 25 and 50 µM doses, respectively(Figure 6A). Similar silibinin treatment for 72 h increased theearly apoptotic cells from 3.1% (control) to 3.7, 4.4 and17.4%, respectively (Figure 6A). Membrane blebbing was alsoobserved in annexin V-stained cells as an early apoptoticresponse by silibinin in A431 cells (data not shown). Silibininalso increased the number of both annexin V plus PI-stainedcells accounting for 3.3 (control) to 4.3–7.5% and 3.9 (control)to 4.6–14.5%, after 48 and 72 h of treatment, respectively(Figure 6B). An apoptotic effect of silibinin on A431 cells at24 h of treatment at these doses was less profound as comparedwith that at 48 and 72 h treatment (data not shown). Together,these results correlate well with silibinin-caused cell deathdata shown in Figure 5B, and also support the apoptotic natureof cell death caused by silymarin in skin tumors.
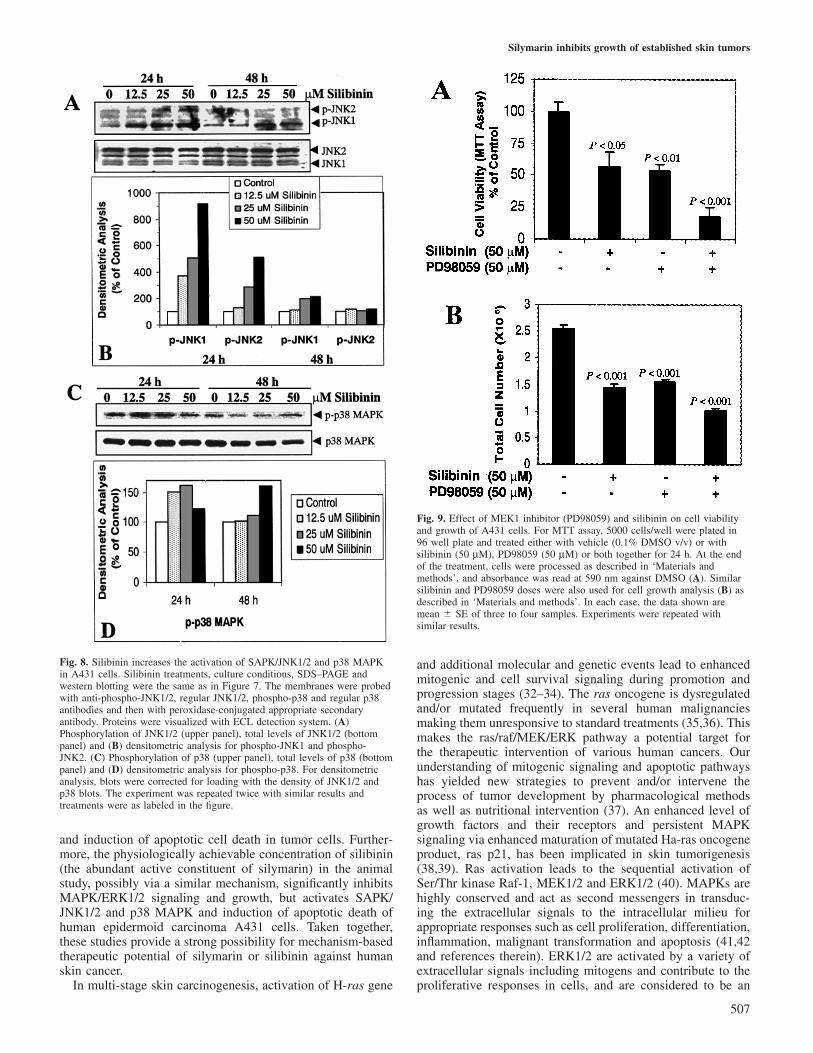
Physiological level of silibinin inhibits MAPK/ERK1/2activation and up-regulates SAPK/JNK1/2 and p38 MAPKactivation in A431 cellsIn signal transduction pathways, MAPK family proteins havebeen regarded as a convergence point for extracellular signals,and have been implicated in multi-stage skin carcinogenesis(28,31). As a result of a strong growth inhibitory effectof silymarin on established skin tumors in mice and itsassociation with an in vivo inhibition of MAPK/ERK1/2activation, we next investigated the effect of silibinin on theconstitutive activation status of MAPK family proteins inA431 cells. As shown in Figure 7A (upper panel), the treatmentof A431 cells with silibinin at 12.5, 25 and 50 µM doses for24 and 48 h resulted in a significant inhibition of ERK1/2activation as evidenced by a decrease in the levels of phospho-p44 and -p42 proteins. Densitometric analysis of blots showedthat compared with the control, 24 h of silibinin treatment at12.5, 25 and 50 µM doses resulted in 52, 76 and 67% decreasein ERK1 phosphorylation, respectively (Figure 7B). In the caseof ERK2 phosphorylation, these treatments were moderatelyeffective and showed 22, 42 and 32% decrease, respectively(Figure 7B). As compared with 24 h treatment, the silibinintreatment for 48 h showed lesser inhibitory effect on MAPK/ERK1/2 activation (Figure 7A and B). The observed inhibitoryeffect of silibinin on ERK1/2 activation was not as result of adecrease in total ERK1/2 protein levels (Figure 7A, bottompanel), suggesting that silibinin impairs constitutive ERK1/2signaling. Overall, the physiological concentration of silibinin(25 µM) from the animal study showed a strong inhibitoryeffect on ERK1/2 activation in A431 cells, which was consistentwith its inhibitory effect on ERK1/2 activation in establishedskin tumors.
Next we assessed the effect of silibinin on SAPK/JNK1/2
R.P.Singh et al.
Fig. 6. Induction of apoptotic cell death by silibinin in A431 cells. Cellswere treated with either DMSO or 12.5, 25 and 50 µM of silibininfor 48 or 72 h. After these treatments, cells were harvested and processedfor annexin and PI staining as referred to in ‘Materials and methods’.Percent of annexin-stained cells as an early apoptotic response (A); andpercent of annexin plus PI-stained cells (late apoptotic cells) (B) are from arepresentative experiment repeated twice with similar results: each treatmentdone in duplicate.
and p38 MAPK activation in A431 cells that may possibly beinvolved in apoptotic cell death. Similar silibinin treatmentsfor 24 h resulted in a strong increase in the activation of JNK1(up to 9-fold) and JNK2 (up to 5-fold) in a dose-dependentmanner (Figure 8A and B), which diminished after 48 h oftreatment. The observed effect of silibinin on JNK1/2 activation(Figure 8A, upper panel) was not due to an increase in totalJNK1/2 protein levels (Figure 8A, bottom panel), suggestingthat silibinin up-regulates SAPK/JNK signaling in A431 cells.p38 MAPK was also activated after 24 h of these silibinintreatments; however, after 48 h, only higher doses (50 µM) ofsilibinin showed an increase in p38 MAPK activation (Figure8C and D). At 24 h of treatment, total p38 MAPK proteinlevels did not change but at 48 h of treatment, a decrease inprotein level was noticed with increasing doses of silibinin(Figure 8C, lower panel). Overall, compared with p38 MAPKactivation, silibinin-caused JNK1/2 activation was veryprominent in A431 cells, which may be associated withincreased apoptosis by silibinin.
Inhibition of MAPK/ERK1/2 is in part responsible for growthinhibition and death of A431 cells by silibininTo further delineate whether inhibition of MAPK/ERK1/2signaling by silibinin is the major, or in part, contributor to
506
Fig. 7. Silibinin inhibits constitutive activation of MAPK/ERK1/2 in A431cells. Cells were cultured in DMEM with 10% serum. At 70% confluency,cells were treated with either DMSO or 12.5, 25 and 50 µM of silibinin for24 and 48 h. Cell lysates were prepared and subjected to SDS–PAGEfollowed by western blotting, as described in ‘Materials and methods’. Themembranes were probed with anti-phospho-ERK1/2 and regularERK1/2 antibodies and then with peroxidase-conjugated appropriatesecondary antibody. Proteins were visualized with ECL detection system.(A) Phosphorylation of ERK1/2 (upper panel), total levels of ERK1/2(bottom panel). Treatments were as labeled in the figure. Densitometricanalysis for phospho-ERK1 and phospho-ERK2 (B) blots was corrected forloading with the density of ERK1/2 blots. The experiment was repeatedtwice with similar results.
the inhibition of cell growth and induction of cell death bysilibinin, we treated A431 cells with MEK1 inhibitor PD98059(50 µM) and silibinin (50 µM) either alone or in combinationfor 24 h. In the MTT assay, silibinin and PD98059 decreasedthe cell viability by 43 (P � 0.05) and 46% (P � 0.01),respectively, but when the two were combined, cell viabilitydecreased by 82% (P � 0.001) as compared with that of thecontrol (Figure 9A). The decrease in cell viability in combina-tion was statistically significant (P � 0.05–0.01) when com-pared with the effect of either compound alone. In the totalcell count, also, silibinin and PD98059 showed similar trendin cell growth inhibition either alone (P � 0.01–0.001) or incombination (P � 0.001) (Figure 9B). Silibinin as well assilibinin in combination with PD98059 also showed increasedcell death (data not shown). Together, the results suggest thatwhereas inhibition of MAPK/ERK1/2 activation by eithersilibinin or PD98059 is important, it is contributory only inpart for the observed growth inhibition and death of A431cells. Other mechanisms such as activation of SAPK/JNK1/2and p38 MAPK pathways, possibly also contribute significantlyto the observed biological responses by silibinin.
Discussion
The central finding of the present study is that naturallyoccurring flavonoid antioxidant silymarin strongly inhibitsestablished skin tumor growth accompanied by tumorregression which is associated with a decrease in proliferationindex together with an inhibition of MAPK/ERK1/2 signaling
Silymarin inhibits growth of established skin tumors
Fig. 8. Silibinin increases the activation of SAPK/JNK1/2 and p38 MAPKin A431 cells. Silibinin treatments, culture conditions, SDS–PAGE andwestern blotting were the same as in Figure 7. The membranes were probedwith anti-phospho-JNK1/2, regular JNK1/2, phospho-p38 and regular p38antibodies and then with peroxidase-conjugated appropriate secondaryantibody. Proteins were visualized with ECL detection system. (A)Phosphorylation of JNK1/2 (upper panel), total levels of JNK1/2 (bottompanel) and (B) densitometric analysis for phospho-JNK1 and phospho-JNK2. (C) Phosphorylation of p38 (upper panel), total levels of p38 (bottompanel) and (D) densitometric analysis for phospho-p38. For densitometricanalysis, blots were corrected for loading with the density of JNK1/2 andp38 blots. The experiment was repeated twice with similar results andtreatments were as labeled in the figure.
and induction of apoptotic cell death in tumor cells. Further-more, the physiologically achievable concentration of silibinin(the abundant active constituent of silymarin) in the animalstudy, possibly via a similar mechanism, significantly inhibitsMAPK/ERK1/2 signaling and growth, but activates SAPK/JNK1/2 and p38 MAPK and induction of apoptotic death ofhuman epidermoid carcinoma A431 cells. Taken together,these studies provide a strong possibility for mechanism-basedtherapeutic potential of silymarin or silibinin against humanskin cancer.
In multi-stage skin carcinogenesis, activation of H-ras gene
507
Fig. 9. Effect of MEK1 inhibitor (PD98059) and silibinin on cell viabilityand growth of A431 cells. For MTT assay, 5000 cells/well were plated in96 well plate and treated either with vehicle (0.1% DMSO v/v) or withsilibinin (50 µM), PD98059 (50 µM) or both together for 24 h. At the endof the treatment, cells were processed as described in ‘Materials andmethods’, and absorbance was read at 590 nm against DMSO (A). Similarsilibinin and PD98059 doses were also used for cell growth analysis (B) asdescribed in ‘Materials and methods’. In each case, the data shown aremean � SE of three to four samples. Experiments were repeated withsimilar results.
and additional molecular and genetic events lead to enhancedmitogenic and cell survival signaling during promotion andprogression stages (32–34). The ras oncogene is dysregulatedand/or mutated frequently in several human malignanciesmaking them unresponsive to standard treatments (35,36). Thismakes the ras/raf/MEK/ERK pathway a potential target forthe therapeutic intervention of various human cancers. Ourunderstanding of mitogenic signaling and apoptotic pathwayshas yielded new strategies to prevent and/or intervene theprocess of tumor development by pharmacological methodsas well as nutritional intervention (37). An enhanced level ofgrowth factors and their receptors and persistent MAPKsignaling via enhanced maturation of mutated Ha-ras oncogeneproduct, ras p21, has been implicated in skin tumorigenesis(38,39). Ras activation leads to the sequential activation ofSer/Thr kinase Raf-1, MEK1/2 and ERK1/2 (40). MAPKs arehighly conserved and act as second messengers in transduc-ing the extracellular signals to the intracellular milieu forappropriate responses such as cell proliferation, differentiation,inflammation, malignant transformation and apoptosis (41,42and references therein). ERK1/2 are activated by a variety ofextracellular signals including mitogens and contribute to theproliferative responses in cells, and are considered to be an

R.P.Singh et al.
essential common element of mitogenic signaling (41–43).Their constitutive expression causes cell transformation andplays a putative role in the carcinogenesis process (43–46).Accordingly, the observed effect of silymarin on tumor cellgrowth inhibition and apoptosis induction under both in vivoand in vitro systems together with a decrease in phospho-ERK1/2 levels suggest a possibility that as an initial responsesilymarin modulates ERK1/2 activation that leads to inhibitionof tumor growth and regression by apoptotic cell death.
During mammalian cell growth and proliferation, in mostcell types, the mitogenic signals are transduced to the nucleusby the nuclear translocation of ERK1 and 2, resulting in theactivation of various transcription factors including c-Myc andElk1 (47–49). Recent studies have shown that these Ser/Thrprotein kinases play a determinant role in the carcinogenesisprocess and their activation is sufficient to stimulate early genetranscription and to reduce growth factor requirement for DNAsynthesis (47,49). Enhanced expression of PCNA, a 36 kDaco-factor of DNA polymerase δ, is one of the downstreameffects of the activation of MAPK/ERK1/2 signaling and wellcorrelated to the status of cell proliferation. PCNA is apotentially useful molecular biomarker of cellular proliferationkinetics and fits to the expected biological mechanisms whereit can be correlated to the decreased/increased cancer incidence(50). Based on a series of epidemiological and experimentalstudies in recent years, it is clearly evident that changes indietary and/or nutritional patterns might have profound andimmediate impact on reducing the cancer incidence. Accord-ingly, the results of our present study, showing stronggrowth inhibition of established skin tumors accompanyingsignificant decrease in proliferation index and inhibition ofMAPK/ERK1/2 activation, and similar anticancer effects ofsilibinin on human epidermoid carcinoma A431 cells viasimilar mechanism, provide a potential strategy of nutritionalintervention of skin cancer. SAPK/JNK1/2 and p38 MAPKsignal transduction pathways have been shown to mediatevarious forms of cellular stress, such as damage repairmechanisms, cell growth arrest and apoptotic cell death (51,52).It has been demonstrated that the activation of JNK and p38with concurrent inhibition of ERK is critical for the inductionof apoptosis in abnormal cells including cancer cells (53and references therein). Therefore, the results of our presentinvestigation showing inhibition of ERK activation andsimultaneous increase in JNK and p38 activation, suggest theinvolvement of these pathways in silibinin-caused apoptosisin A431 cells, and that similar mechanisms may also beoperative in in vivo conditions. Additional studies are neededto support this suggestion, and are in fact the major focus ofour ongoing work in this area. As recent clinical trials havedemonstrated the potential of signal transduction modulatorsin cancer treatment (54 and references therein), our presentfindings demonstrate the strong potential of silymarin fortargeting both mitogenic and survival pathways in cancer cells.The present data combined with our earlier studies suggestthat more mechanistic studies are needed to further substantiatethe therapeutic efficacy of silymarin or silibinin againsthuman cancers.
The induction of apoptotic cell death was identified asanother possible anticarcinogenic mechanism of silymarinagainst skin cancer. The results from TUNEL analysis of skintumor samples, and annexin V and PI staining of A431 cellsshowed that apoptosis predominantly contributes to tumor cellkilling caused by silymarin or silibinin. It is well established
508
that apoptosis and, the associated cell-signaling pathways andcellular events controlling it, have a profound effect on theprogression of benign to malignant phenotype, and that theycan be targeted for the therapy of various malignanciesincluding skin cancer (55–57). The molecular mechanism ofapoptosis induction in skin tumor by silymarin is yet to beexplored. Studies are in progress to identify the signalingpathways and molecular events associated with silymarin-caused apoptotic cell death in skin tumor under both in vivoand cell culture conditions.
In the present study, we also observed that dietaryadministration of silymarin results in its distribution in severalimportant organs of the body, which are generally susceptibleto cancer development. These systemic bio-distribution resultsof silymarin, and the fact that tissue specificity and histo-pathological course of tumor development in mice is remark-ably similar to that observed in several human epithelialmalignancies (58), suggest that silymarin could be a novelcompound for both prevention (based on earlier studies) andtherapy of different epithelial cancers. This suggestion couldat least in part be supported by a series of recent cell culturestudies showing an anticancer potential of silymarin and/orsilibinin against human skin, prostate, breast and cervicalcancers (28,59–61).
In summary, based on the results of the present study andthose reported recently, investigational clinical trials withcollaborative laboratory studies are needed to develop silymarinas both preventive and therapeutic agent against skin and otherepithelial cancers in humans. A positive outcome of suchstudies could specifically be beneficial as silymarin is alreadyin clinical use as an anti-hepatotoxic agent and consumed asa dietary supplement around the world including the USA,and is devoid of any untoward toxicity and side effects.
Acknowledgement
This work was supported by USPHS grant CA 64514 from the NationalCancer Institute, NIH.
References
1.Wargovich,M.J. (1999) Nutrition and cancer: the herbal revolution. Curr.Opin. Clin. Nutr. Metab. Care, 2, 421–424.
2.Stolzenberg-Solomon,R.Z., Albanes,D., Nieto,F.J., Hartman,T.J.,Tangrea,J.A., Rautalahti,M., Sehlub,J., Virtamo,J. and Taylor,P.R. (1999)Pancreatic cancer risk and nutrition-related methyl-group availabilityindicators in male smokers. J. Natl Cancer Inst., 91, 535–541.
3.Hill,M.J. (1997) Nutrition and human cancer. Ann. N.Y. Acad. Sci., 833,68–78.
4.Sporn,M.B. and Suh,N. (2000) Chemoprevention of cancer. Carcinogenesis,21, 525–530.
5.Lahiri-Chatterjee,M., Katiyar,S.K., Mohan,R.R. and Agarwal,R. (1999) Aflavonoid antioxidant, silymarin, affords exceptionally high protectionagainst tumor promotion in the SENCAR mouse skin tumorigenesis model.Cancer Res., 59, 622–632.
6.Franke,A.A., Cooney,R.V., Custer,L.J., Mordan,L.J. and Tanaka,Y. (1998)Inhibition of neoplastic transformation and bioavailability of dietaryflavonoid agents. Adv. Exp. Med. Biol., 439, 237–248.
7.Reddy,B.S., Hirose,Y., Cohen,L.A., Simi,B., Cooma,I. and Rao,C.V. (2000)Preventive potential of wheat bran fractions against experimental coloncarcinogenesis: implications for human colon cancer prevention. CancerRes., 60, 4792–4797.
8.Wenzel,U., Kuntz,S., Brendel,M.D. and Daniel,H. (2000) Dietary flavoneis a potent apoptosis inducer in human colon carcinoma cells. CancerRes., 60, 3823–3831.
9.Dragsted,L.O. (1998) Natural antioxidants in chemoprevention. Arch.Toxicol., 20 (suppl.), 209–226.
10.Fotsis,T., Pepper,M.S., Aktas,E., Breit,S., Rasku,S., Aldercreutz,H.,Wahala,K., Montesano,R. and Schweigerer,L. (1997) Flavonoids, dietary-

Silymarin inhibits growth of established skin tumors
derived inhibitors of cell proliferation and in vitro angiogenesis. CancerRes., 57, 2916–2921.
11.Mereish,K.A., Bunner,D.L., Ragland,D.R. and Creasia,D.A. (1991)Protection against microcystin-LR-induced hepatotoxicity by silymarin:biochemistry, histopathology and lethality. Pharm. Res., 8, 273–277.
12.Wagner,V.H., Diesel,P. and Seitz,M. (1974) Chemistry and analysis ofsilymarin from Silybum marianum Gaertn. Arzneimittelforschung, 24,466–471.
13.Ely,H. (1989) Dermatologic therapies you’ve probably never heard of.Dermatol. Clin., 7, 19–35.
14.Luper,S. (1998) A review of plants used in the treatment of liver disease:part 1. Altern. Med. Rev., 3, 410–421.
15.Pares,A., Planas,R., Torres,M., Caballeria,J., Viver,J.M., Acero,D., Panes,J.,Rigau,J., Santos,J. and Rodes,J. (1998) Effects of silymarin in alcoholicpatients with cirrhosis of the liver: results of a controlled, double-blind,randomized and multicenter trial. J. Hepatol., 28, 615–621.
16.Vogel,G., Trost,W. and Braatz,R. (1975) Studies on the pharmacodynamics,including site and mode of action, of silymarin: the antihepatotoxicprinciple from Silybum marianum (L) Gaertn. Arzneim. Forsch., 25, 82–89.
17.Racz,K., Feher,J., Csomos,G., Varga,I., Kiss,R. and Glaz,E. (1990) Anantioxidant drug, silibinin, modulates steroid secretion in humanpathological adrenocortical cells. J. Endocrinol., 124, 341–345.
18.Valenzula,A., Guerra,R. and Videla,L.A. (1986) Antioxidant properties ofthe flavonoids silybin and (�)-cyanidanol-3: comparison with butylatedhydroxyanisole and butylated hydroxytoluene. Planta Med., 5, 438–440.
19.Muzes,G., Deak,G., Lang,I., Nekam,K., Gergely,P. and Feher,J. (1991)Effect of the bioflavonoid silymarin on the in vitro activity and expressionof superoxide dismutase (SOD) enzyme. Acta Physiol. Hung., 78, 3–9.
20.Rudd,C.J., Suing,K.D., Pardo,K. and Kelloff,G. (1990) Evaluation ofpotential chemopreventive agents using a mouse epidermal cell line, JB6.Proc. Am. Assoc. Cancer Res., 31, 127.
21.Mehta,R.G. and Moon,R.C. (1991) Characterization of effectivechemopreventive agents in mammary gland in vitro using an initiation–promotion protocol. Anticancer Res., 11, 593–596.
22.Steele,V.E., Kelloff,G.J., Wilkinson,B.P. and Arnold,J.T. (1990) Inhibitionof transformation in cultured rat tracheal epithelial cells by potentialchemopreventive agents. Cancer Res., 50, 2068–2074.
23.Katiyar,S.K., Korman,N.J., Mukhtar,H. and Agarwal,R. (1997) Protectiveeffects of silymarin against photocarcinogenesis in a mouse skin model.J. Natl. Cancer Inst., 89, 556–566.
24.Agarwal,R., Katiyar,S.K., Lundgren,D.W. and Mukhtar,H. (1994)Inhibitory effect of silymarin, an anti-hepatotoxic flavonoid, on 12-O-tetradecanoylphorbol-13-acetate-induced epidermal ornithine decarboxyl-ase activity and mRNA in SENCAR mice. Carcinogenesis, 15, 1099–1103.
25.Zhao,J., Sharma,Y. and Agarwal,R. (1999) Significant inhibition by theflavonoid antioxidant silymarin against 12-O-tetradecanoylphorbol 13-acetate-caused modulation of antioxidant and inflammatory enzymes andcyclooxygenase 2 and interleukin-1α expression in SENCAR mouseepidermis: implications in the prevention of stage I tumor promotion. Mol.Carcino., 26, 321–333.
26.Zi,X., Mukhtar,H. and Agarwal,R. (1997) Novel cancer preventive effectsof a flavonoid antioxidant silymarin: inhibition of m RNA expression ofan endogenous tumor promoter TNFa. Biochem. Biophys. Res. Commun.,239, 334–339.
27.Ahmad,N., Gali,H., Javed,S. and Agarwal,R. (1998) Skin cancerchemopreventive effects of a flavonoid antioxidant silymarin are mediatedvia impairment of receptor tyrosine kinase signaling and perturbation incell cycle progression. Biochem. Biophys. Res. Commun., 248, 294–301.
28.Zi,X. and Agarwal,R. (1999) Modulation of mitogen-activated proteinkinase activation and cell cycle regulators by the potent skin cancerpreventive agent silymarin. Biochem. Biophys. Res. Commun., 263, 528–536.
29.Lu,Y.-P., Lu,Y.-R., Xie,J.-G., Yen,P., Huang,M.-T. and Conney,A.H. (1997)Inhibitory effect of black tea on the growth of established skin tumors inmice: effects on tumor size, apoptosis, mitosis and bromodeoxyuridineincorporation into DNA. Carcinogenesis, 18, 2163–2169.
30.Agarwal,C., Sharma,Y. and Agarwal, R. (2000) Anticarcinogenic effect ofa polyphenolic fraction isolated from grape seeds in human prostatecarcinoma DU145 cells: modulation of mitogenic signaling and cell cycleregulators and induction of G1 arrest and apoptosis. Mol Carcino., 28, 1–10.
31.Guyton,K.Z., Gorospe,M., Kensler,T.W. and Holbrook,N.J. (1996)Mitogen-activated protein kinase (MAPK) activation by butylatedhydroxytoluene hydroperoxide: implications for cellular survival and tumorpromotion. Cancer Res., 56, 3480–3485.
32.Yuspa,S.H. (2000) Overview of carcinogenesis: past, present and future.Carcinogenesis, 21, 341–344.
509
33.Hanahan,D. and Weinberg,R.A. (2000) The hallmarks of cancer. Cell, 100,57–70.
34.Bremner,R. and Balmain,A. (1990) Genetic changes in skin tumorprogression: correlation between presence of a mutant ras gene and lossof heterozygosity on mouse chromosome 7. Cell, 61, 407–417.
35. Irani,K., Xia,Y., Zweier,J.L., Sollott,S.J., Der,C.J., Fearon,E.R.,Sundaresan,M., Finkel,T. and Goldschmidt-Clermont,P.J. (1997) Mitogenicsignaling mediated by oxidants in Ras-transformed fibroblasts. Science,275, 1649–1652.
36.Bos,J.L. (1999) ras oncogenes in human cancer: a review. Cancer Res.,49, 4682–4689.
37.Zi,X., Grasso,A.W., Kung,H. and Agarwal,R. (1998) A flavonoidantioxidant, silymarin, inhibits activation of erbB1 signaling and inducescyclin-dependent kinase inhibitors, G1 arrest and anticarcinogenic effectsin human prostate carcinoma DU145 cells. Cancer Res., 58, 1920–1929.
38.Khan,S.G., Saxena,R., Bickers,D.R., Mukhtar,H. and Agarwal,R. (1995)Inhibition of ras p21 membrane localization and modulation of proteinkinase C isozyme expression during regression of chemical carcinogen-induced murine skin tumors by lovastatin. Mol. Carcino., 12, 205–212.
39.Rodriguez-Puebla,M.L., LaCava,M., Gimenez-Conti,I.B., Johnson,D.G.and Conti,C.J. (1998) Deregulated expression of cell-cycle proteinsduring premalignant progression in SENCAR mouse skin. Oncogene, 17,2251–2258.
40.Van-Aelst,L., Barr,M., Marcus,S., Polverino,A. and Wigler,M. (1993)Complex formation between RAS and RAF and other protein kinases.Proc. Natl Acad. Sci. USA, 90, 6213–6217.
41.Seger,R. and Krebs,E.G. (1995) The MAPK signaling cascade. FASEB J.,9, 726–735.
42.Whitmarsh,A.J., Cavanagh,J., Tournier,C., Yasuda,J. and Davis,R.J. (1998)A mammalian scaffold complex that selectively mediates MAP kinaseactivation. Science, 281, 1671–1674.
43.Groom,L.A., Sneddon,A.A., Alessi,D.R., Dowd,S. and Keyse,S.M. (1996)Differential regulation of the MAP, SAP and RK/p38 kinases by Pyst1, anovel cytosolic dual-specificity phosphatase. EMBO J., 15, 3621–3632.
44.Kortenjann,M., Thomae,O. and Shaw,P.E. (1994) Inhibition of v-raf-dependent c-fos expression and transformation by a kinase-defectivemutant of the mitogen-activated protein kinase Erk2. Mol. Cell. Biol., 14,4815–4824.
45.Brunet,A., Pages,G. and Pouyssegur,J. (1994) Constitutively active mutantsof MAP kinase kinase (MEK1) induce growth factor-relaxation andoncogenicity when expressed in fibroblasts. Oncogene, 9, 3379–3387.
46.Cowley,S., Paterson,H., Kemp,P. and Marshall,C.J. (1994) Activation ofMAP kinase kinase is necessary and sufficient for PC12 differentiationand for transformation of NIH 3T3 cells. Cell, 77, 841–852.
47.Marais,R., Wynne,J. and Treisman,R. (1993) The SRF accessory proteinElk-1 contains a growth factor-regulated transcriptional activation domain.Cell, 73, 381–393.
48.Wasylyk,B., Hahn,S.J. and Giovane,A. (1993) The Ets family oftranscription factors. Eur. J. Biochem., 211, 7–18.
49.Coffer,P., de Jonge,M., Mettouchi,A., Binetruy,B., Ghysdael,J. andKruijer,W. (1994) JunB promoter regulation: Ras mediated transactivationby c-Ets-1 and c-Ets-2. Oncogene, 9, 911–921.
50.Hall,P.A., Levison,D.A., Woods,A.L., Yu,C.C.-W., Kellock,D.B. andWatkins,J.A. (1990) Proliferating cell nuclear antigen (PCNA)immunolocalization in paraffin sections: an index of cell proliferation withevidence of deregulated expression in some neoplasms. J. Pathol., 162,285–294.
51.Vantus,T., Keri,G., Krivickiene,Z. et al. (2001) The somatostatin analogueTT-232 induces apoptosis in A431 cells: sustained activation of stress-activated kinases and inhibition of signalling to extracellular signal-regulated kinases. Cell Signal., 13, 717–725.
52. Ichijo,H. (1999) From receptors to stress-activated MAP kinases. Oncogene,18, 6087–6093.
53.Xia,Z., Dickens,M., Raingeaud,J., Davis,R.J. and Greenberg,M.E. (1995)Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis.Science, 270, 1326–1331.
54.Cunningham,C.C., Holmlund,J.T., Schiller,J.H., Geary,R.S., Kwoh,T.H.,Dorr,A. and Nemunaitis,J. (2000) A phase I trial of C-raf kinase antisenseoligonucleotide ISIS 5132 administered as a continuous intravenousinfusion in patients with advanced cancer. Clin. Cancer Res., 6, 1626–1631.
55.Dive,C. and Hickman,J.A. (1991) Drug-target interactions: only the firststep in the commitment to a programmed cell death? Br. J. Cancer, 64,192–196.
56.Thompson,C.B. (1995) Apoptosis in the pathogenesis and treatment ofdisease. Science, 267, 1456–1462.
57.Lowe,S.W. and Lin,A.W. (1990) Apoptosis in cancer. Carcinogenesis, 21,485–495.

R.P.Singh et al.
58.Balmain,A. and Harris,C.C. (2000) Carcinogenesis in mouse and humancells: parallels and paradoxes. Carcinogenesis, 21, 371–377.
59.Zi,X. and Agarwal,R. (1999) Silibinin decreases prostate-specific antigenwith cell growth inhibition via G1 arrest, leading to differentiation ofprostate carcinoma cells: implications for prostate cancer intervention.Proc. Natl Acad. Sci. USA, 96, 7490–7495.
60.Zi,X., Feyes,D.K. and Agarwal,R. (1998) Anticarcinogenic effect of aflavonoid antioxidant, silymarin, in human breast cancer cells MDA-MB
510
468: induction of G1 arrest through an increase in Cip1/p21 concomitantwith a decrease in kinase activity of cyclin-dependent kinases andassociated cyclins. Clin. Cancer Res., 4, 1055–1064.
61.Bhatia,N., Zhao,J., Wolf,D.M. and Agarwal,R. (1999) Inhibition of humancarcinoma cell growth and DNA synthesis by silibinin, an active constituentof milk thistle: comparison with silymarin. Cancer Lett., 147, 77–84.
Received July 5, 2001; revised November 26, 2001;accepted December 12, 2001
Copyright © 2022 FDOKUMEN




















