
MAP Kinases and Their Roles in Pancreatic β-Cells
10
Copyright 2004 by Humana Press Inc. All rights of any nature whatsoever reserved. 1085-9195/04/Supplement/40/191-200/$25.00 MAP Kinases and Their Roles in Pancreatic 13-Cells Shih Khoo 1,3 Tara Beers Gibson I, Don Arnette I, Michael Lawrence 1, Bridgette January 1,4, Kathleen McGlynn 1, Colleen A. Vanderbilt 1, Steven C. Griffen 2,5, Michael S. German z, and Melanie H. Cobb 1,* 1Department of Pharmacology, University of Texas SouthwesternMedical Centerat Dallas, Dallas, TX; 2Department of Research Administration, Singapore; 3Department of Internal Medicine, Endocrine Division, UC Davis, Davis, CA; 4Hormone Research Institute, University of California, San Francisco, CA; 5Mary Kay, Global Research and Development Group, Dallas, TX. Abstract We discuss our work examining regulation and functions of mitogen-activated protein kinases, particularly ERK1 and EILK2, in pancreatic [~-cells. These enzymes are activated by glucose, other nutrients, and insulinogenic hormones. Their activation by these agents is calcium-dependent. A number of other stimuli also activate ERK1/2, but by mechanisms distinct from those involved in nutrient sensing. Inhibition of ERK1/2 has no apparent effect on insulin secretion measured after 2 h. On the other hand, ERK1/2 activity is required for maximal glucose-dependent activation of the insulin gene promoter. The primary effort has focused on INS-1 cell lines, with supporting and con- firmatory studies in intact islets and other [3-celllines, indicating the generality of our findings in [3- cell function. Thus ERK1/2 participate in transmitting glucose-sensinginformation to [3-cell functions. These kinases most likely act directly and indirectly on multiple pathways that regulate [3-cellfunc- tion and, in particular, to transduce an elevated glucose signal into insulin gene transcription. Index Entries: Insulin; transcription; cAMP; Rsk; phorbol ester. INTRODUCTION We are interested in defining signal transduc- tion events that control homeostatic mecha- *Author to whom all correspondence and reprint requests should be addressed. E-mail: mcobb@ mednet.swmed.edu. nisms and that maintain the differentiated prop- erties of pancreatic [3-cells. Pancreatic a-cells produce insulin, the major hormone controlling glucose utilization in mammals. The release of insulin by a-cells is regulated by an array of nutrient and hormonal cues, the most important of which is glucose itself (1). Glucose regulates not only insulin secretion, but also translation of the large pool of presynthesized proinsulin Cell Biochemistn d and Biophysics 1 91 Supplement, 2004
Transcript of MAP Kinases and Their Roles in Pancreatic β-Cells

�9 Copyright 2004 by Humana Press Inc. All rights of any nature whatsoever reserved. 1085-9195/04/Supplement/40/191-200/$25.00
MAP Kinases and Their Roles in Pancreatic 13-Cells
Shih Khoo 1,3 Tara Beers Gibson I, Don Arnette I, Michael Lawrence 1, Bridgette January 1,4, Kathleen McGlynn 1,
Colleen A. Vanderbilt 1, Steven C. Griffen 2,5, Michael S. German z, and Melanie H. Cobb 1,*
1Department of Pharmacology, University of Texas Southwestern Medical Center at Dallas, Dallas, TX; 2Department of Research Administration, Singapore;
3Department of Internal Medicine, Endocrine Division, UC Davis, Davis, CA; 4Hormone Research Institute, University of California, San Francisco, CA;
5Mary Kay, Global Research and Development Group, Dallas, TX.
Abstract
We discuss our work examining regulation and functions of mitogen-activated protein kinases, particularly ERK1 and EILK2, in pancreatic [~-cells. These enzymes are activated by glucose, other nutrients, and insulinogenic hormones. Their activation by these agents is calcium-dependent. A number of other stimuli also activate ERK1/2, but by mechanisms distinct from those involved in nutrient sensing. Inhibition of ERK1/2 has no apparent effect on insulin secretion measured after 2 h. On the other hand, ERK1/2 activity is required for maximal glucose-dependent activation of the insulin gene promoter. The primary effort has focused on INS-1 cell lines, with supporting and con- firmatory studies in intact islets and other [3-cell lines, indicating the generality of our findings in [3- cell function. Thus ERK1/2 participate in transmitting glucose-sensing information to [3-cell functions. These kinases most likely act directly and indirectly on multiple pathways that regulate [3-cell func- tion and, in particular, to transduce an elevated glucose signal into insulin gene transcription.
Index Entries: Insulin; transcription; cAMP; Rsk; phorbol ester.
I N T R O D U C T I O N
We are interested in defining signal transduc- tion events that control homeostatic mecha-
*Author to whom all correspondence and reprint requests should be addressed. E-mail: mcobb@ mednet.swmed.edu.
nisms and that maintain the differentiated prop- erties of pancreatic [3-cells. Pancreatic a-cells produce insulin, the major hormone controlling glucose utilization in mammals. The release of insulin by a-cells is regulated by an array of nutrient and hormonal cues, the most important of which is glucose itself (1). Glucose regulates not only insulin secretion, but also translation of the large pool of presynthesized proinsulin
Cell Biochemistn d and Biophysics 1 91 Supplement, 2004

192 Khoo et al.
mRNA and transcription of the insulin gene. Although all of these events are initiated rapidly, they have distinct impacts on insulin production, in keeping with their response time scales (2-8). Thus insulin secretion is the acute response to an increase in circulating glucose concentration, translation of mRNA replenishes secreted hormone, and insulin gene transcrip- tion provides the mechanism for long-term sta- bility of insulin production. Each of these processes may be potentiated or suppressed by hormones and other nutrients.
Glucose activates a panoply of signaling pathways and second messengers to regulate [3- cell function. Among these are the mitogen-acti- vated protein (MAP) kinases ERK1 and ERK2 (9-11). ERK1 and ERK2 are the most intensely studied MAP kinases and have been implicated in many signal transduction pathways. Their nearly universal involvement in responses to ligands underscores the capacity of these pro- tein kinases, in concert with other signal trans- ducers, to change cellular functions (12,13). Among responses modulated by these enzymes are the activities of membrane enzymes such as phospholipase A2 (14), cell attachment and cell motility (15,16), gene transcription (12), and cell proliferation (17). Although generally regarded as linked to cell proliferation, ERK1 and ERK2 are highly expressed in the nervous system in postmitotic neurons. They are believed to con- tribute significantly to long-term potentiation, a relatively slow process of synaptic modification (18-21). Likewise, in neuroendocrine cells, such as [3-cells, ERK1/2 are relatively insensitive to serum factors. Instead, these kinases are stimu- lated by glucose and other nutrients, presum- ably to mediate the actions of these nutrients on the differentiated functions of ~l-cells (9-11).
To contribute to an understanding of how glu- cose-dependent signals are transduced in [3-cells, we wish to define the mechanisms of activation of ERK1/2 by nutrients and hormones and the functions of the ERK1/2-regulated signaling pathways that are glucose-sensitive. Our studies suggest little effect of this kinase cascade on insulin secretion. Subcellular localization plays an important part in determining the functions
of ERKs and other MAP kinases (22). We showed previously that exposure of the [3-cell line INS-1 to glucose activates ERK1 and ERK2 in the cytosol and in nuclei and causes their nuclear accumulation (10). Thus we have examined potential actions of ERK1/2 in the nucleus and have found that these kinases promote tran- scription of the insulin gene. Our data are con- sistent with the hypothesis that ERK1/2 help to integrate information describing the state of pan- creatic [3-cells over a relatively long time scale to maintain contextually appropriate insulin pro- duction and other [3-cell functions.
MATERIALS AND METHODS
Materials
The MAP/ERK kinase (MEK) inhibitor U0126 was initially provided by J. Trzaskos (Dupont-Merck) and then obtained from Calbiochem. The MEK inhibitor PD98059 was from Calbiochem. Inhibitors were dissolved in dimethyl sulfoxide and diluted so that the sol- vent concentration in medium was no more than 0.1%. Replication-deficient adenoviruses were generated as previously described or gen- erously provided by L. Klesse (23,24).
Cell Growth and Harvest
INS-1 cells or subclones selected for increased insulin secretion (25) were grown and harvested as described (10). The activities of ERK1/2 were analyzed by immunoblotting lysates with anti- bodies (antiphosphoERK1/2 antibodies were obtained from Promega, Biosource, and then Sigma as these studies progressed) that recog- nized the activated, doubly phosphorylated forms of these kinases (26). Anti-phosphoRsk antibodies were from Cell Signaling.
Reporter Assays
The rat insulin I promoter (-410/+1 bp) and the multimerized E2A3/4 (minienhancer) dri- ven luciferase and chloramphenicol acetyltrans- ferase reporter constructs were as described
Cell Biochemistry and Biophysics Supplement, 2004
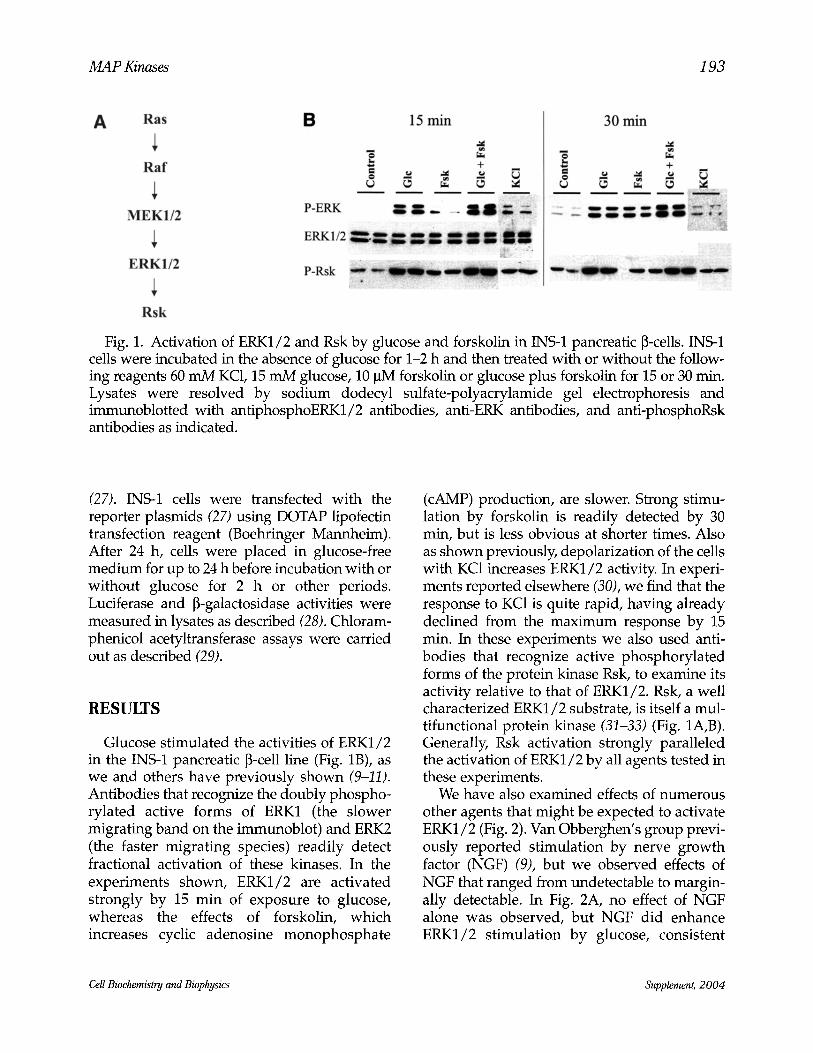
MAP Kinases 193
A R a $
Raf
MEN1/2
EIlN1/2
Rsk
B 15 rnin
+
P-ERK " ' - . a - . m a ~ , , -
P-Rsk " ~ - - ' ~ , ~ ~
30 min
o +
Fig. 1. Activation of ERK1/2 and Rsk by glucose and forskolin in INS-1 pancreatic ~-cells. INS-1 cells were incubated in the absence of glucose for 1-2 h and then treated with or without the follow- ing reagents 60 mM KC1, 15 mM glucose, 10 pM forskolin or glucose plus forskolin for 15 or 30 min. Lysates were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblotted with antiphosphoERK1/2 antibodies, anti-ERK antibodies, and anti-phosphoRsk antibodies as indicated.
(27). INS-1 cells were transfected with the reporter plasmids (27) using DOTAP lipofectin transfection reagent (Boehringer Mannheim). After 24 h, cells were placed in glucose-free medium for up to 24 h before incubation with or without glucose for 2 h or other periods. Luciferase and ~-galactosidase activities were measured in lysates as described (28). Chloram- phenicol acetyltransferase assays were carried out as described (29).
RESULTS
Glucose stimulated the activities of ERK1/2 in the INS-1 pancreatic [3-cell line (Fig. 1B), as we and others have previously shown (9-11). Antibodies that recognize the doubly phospho- rylated active forms of ERK1 (the slower migrating band on the immunoblot) and ERK2 (the faster migrating species) readily detect fractional activation of these kinases. In the experiments shown, ERK1/2 are activated strongly by 15 min of exposure to glucose, whereas the effects of forskolin, which increases cyclic adenosine monophosphate
(cAMP) production, are slower. Strong stimu- lation by forskolin is readily detected by 30 min, but is less obvious at shorter times. Also as shown previously, depolarization of the cells with KC1 increases ERK1/2 activity. In experi- ments reported elsewhere (30), we find that the response to KC1 is quite rapid, having already declined from the maximum response by 15 rain. In these experiments we also used anti- bodies that recognize active phosphorylated forms of the protein kinase Rsk, to examine its activity relative to that of ERK1/2. Rsk, a well characterized ERK1/2 substrate, is itself a mul- tifunctional protein kinase (31-33) (Fig. 1A,B). Generally, Rsk activation strongly paralleled the activation of ERK1/2 by all agents tested in these experiments.
We have also examined effects of numerous other agents that might be expected to activate ERK1/2 (Fig. 2). Van Obberghen's group previ- ously reported stimulation by nerve growth factor (NGF) (9), but we observed effects of NGF that ranged from undetectable to margin- ally detectable. In Fig. 2A, no effect of NGF alone was observed, but NGF did enhance ERK1/2 stimulation by glucose, consistent
Cell Biochemistry and Biophysics Supplement, 2004

194 Khoo et al.
A
P-ERK
ERK1/2
"~ + +
" - p = l l z
B i
-" P-ERK1/2
ERK1/2
Fig. 2. Activation of ERK1/2 by other agents in INS-1 cells. Cells incubated in the absence of glucose as in Fig. 1 were untreated, exposed to glucose or treated with: (A) 10 ng/mL NGF or 1 ~tM phorbol ester, each without or with 15 mM glucose for 15 min; (B) 1 ~tM insulin; (C) 1 ~tM insulin or 1 ~tM phorbol ester plus or minus 1 p~M staurosporine; and (D) bradykinin. Lysates were resolved by sodium dodecyl sul- fate-polyacrylamide gel electrophoresis and immunoblotted with anti-phosphoERK1/2 antibodies, anti-ERK antibodies, and anti- phosphoAkt antibodies as indicated.
with an effect on the pathway. Phorbol ester elicited readily detectable ERK1/2 activity and also potentiated the effect of glucose. Effects of insulin on ERK1/2 activity were also variable (compare Fig. 2B,C), ranging from unde- tectable to clear but small effects, in spite of the fact that activation of a key insulin target, Akt (Fig. 2C, bottom panel), was readily observed. Bradykinin, a strong ERK1/2 stimulus in PC12
cells (34), did not activate ERK1/2 in INS-1 cells (Fig. 2D).
To examine potential functions of ERK1/2 we have used pharmacological inhibitors and adenoviruses that express inhibitory mutants of proteins in the ERK1/2 cascade (see Fig. 1A for the cascade components). Given the wide variety of signaling events they might regulate, we initially examined a possible requirement of ERKs in insulin secretion. One possible func- tion of ERKs in secretion might be through their capacity to activate phospholipase A2 (14) to facilitate vesicle fusion. As we found earlier, the inhibitor PD98059, which prevents activa- tion of the kinases upstream of ERK1/2 (i.e., MEK1 and MEK2; Fig. 1A), blocks ERK1/2 stimulation but has no effect on insulin secre- tion after 2 h (10). Adenoviruses that intro- duced inhibitory forms of components of the cascade, either the small G protein H-Ras or ERK2 itself, did not cause a decrease in insulin secretion elicited by glucose plus forskolin dur- ing a 2-h static incubation (Fig. 3A). Consistent with these results, adenoviruses that intro- duced inhibitory or constitutively active forms of MEK1 neither increased nor decreased secre- tion of insulin caused by glucose plus forskolin (Fig. 3B). In each case expression of these het- erologous proteins caused the expected increase or decrease in ERK1/2 activity as con- firmed by immunoblotting.
We then used inhibitors and transfection of inhibitory mutants to examine the roles of ERK1/2 in insulin gene transcription (Fig. 4). Based on earlier work (35), we focused on reporter activity driven not only by the full- length insulin gene promoter, but also by a multimerized form of the E2A3/4 elements, referred to as the minienhancer, from the insulin gene promoter. These are well-charac- terized glucose-sensitive elements (27,36). Inhibitory mutants of Raf-1 (Raf C4B, an N-ter- minal fragment), the MAP kinase for the ERK1/2 pathway (Fig. 1A) and H-Ras (A15), inhibited basal transcription driven by the full- length insulin gene promoter and E2A3/4, as well as blocking glucose-stimulated transcrip- tion from both promoter constructs (Fig. 4A,B).
Cell Biochemistry and Biophysics Supplement, 2004

M A P Kinases 195
A 4 .~.
�9 ~ 3- e ~
"~ 2- .=_. =.
.=. 1-
[] 0mMglucose [] 15mMglucose
+forskolin [] 15mMglucose
control Ras ERK2 ERK2 dn dn wt
B
.= "==
4 3~~ ~ F 2" l -
0 control MEK1 MEK1 MIK1
wt CA dn
D 0 m M g l u c o s e
[]15mMglucose +forskolin
[~15mMglucose
Fig. 3. Inhibition of ERK1/2 does not reduce total insulin secreted in 2 h. Cells were infected with recombinant adenoviruses expressing the indicated proteins for 48 h. After an overnight incu- bation in the absence of glucose, the cells were exposed to 15 mM glucose with or without forskolin for an additional 2 h. Medium was harvested and insulin was measured by radioim- munoassay. (A) Ras-dominant negative (dn), ERK2-dominant negative (dn), ERK2 wild-type (wt), and (B) MEK1 wild-type (wt), constitutively active (CA), dominant negative (dn).
A ~ = = 2 .<
0
B ==2
< ~9
~ 0
I (n=2) I l
vector V12 A15 vector V12 A15 Ras Ras Ras Ras
rat insulin I promoter
minienhancer
C 5
2
0 vector ERK2-
MEK1 ERK2- MEK1LA
minienhancer
0 mM �9 glucose
�9 15 mM glucose
vector Raf Raf vector Raf Raf BXB C4B BXB C4B
rat insulin I minienhancer promoter
Fig. 4. Effects of inhibition and activation of the ERK1/2 pathway on transcription from the insulin gene promoter and the minienhancer. INS-1 cells were transfected with the rat insulin I pro- moter or the minienhancer along with vector and active V12 Ras or dominant interfering A15 Ras (A) or with active RafBXB or the inhibitory Raf fragment C4B (B). (C) INS-1 cells were transfected with the E2A3/4-driven luciferase construct plus vector, or ERK2-MEK1, or ERK2-MEK1LA. Dual luciferase assays were performed. Data presented are averages of three independent experiments.
Cell Biochemistry and Biophysics Supplement, 2 0 0 4
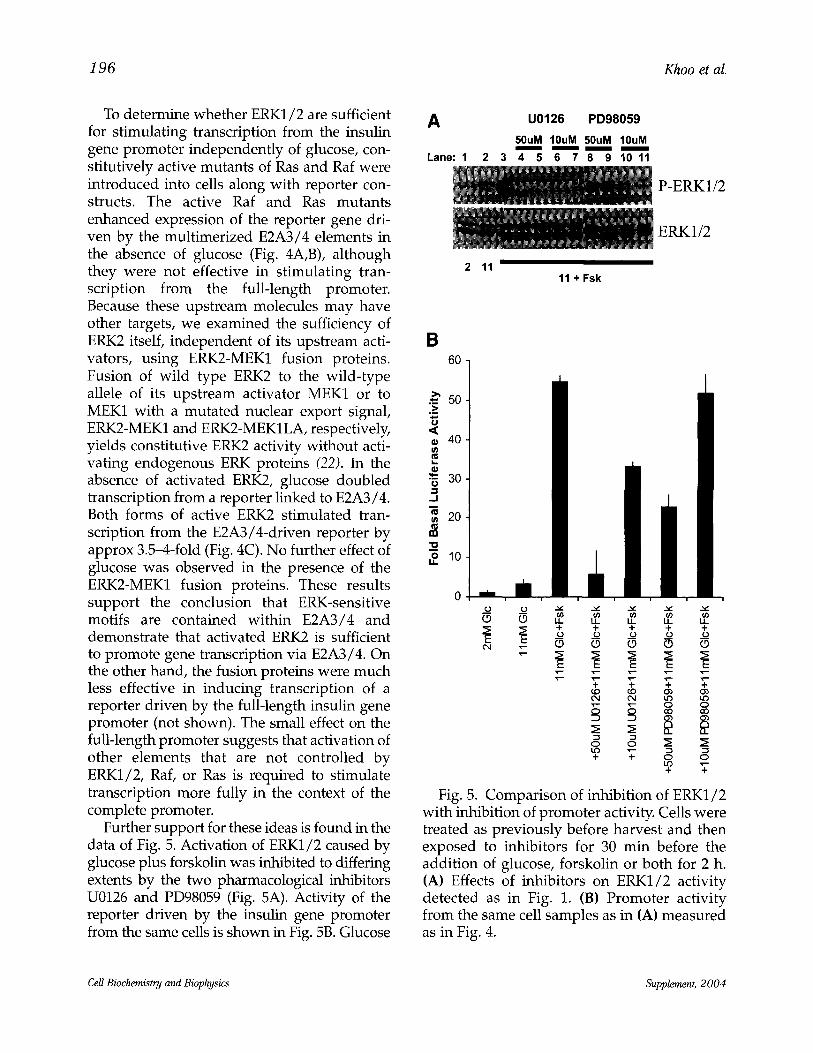
196 Khoo et al.
To determine whether ERK1/2 are sufficient for stimulating transcription from the insulin gene promoter independently of glucose, con- stitutively active mutants of Ras and Raf were introduced into cells along with reporter con- structs. The active Raf and Ras mutants enhanced expression of the reporter gene dri- ven by the multimerized E2A3/4 elements in the absence of glucose (Fig. 4A,B), although they were not effective in stimulating tran- scription from the full-length promoter. Because these upstream molecules may have other targets, we examined the sufficiency of ERK2 itself, independent of its upstream acti- vators, using ERK2-MEK1 fusion proteins. Fusion of wild type ERK2 to the wild-type allele of its upstream activator MEK1 or to MEK1 with a mutated nuclear export signal, ERK2-MEK1 and ERK2-MEKILA, respectively, yields constitutive ERK2 activity without acti- vating endogenous ERK proteins (22). In the absence of activated ERK2, glucose doubled transcription from a reporter linked to E2A3/4. Both forms of active ERK2 stimulated tran- scription from the E2A3/4-driven reporter by approx 3.5-4-fold (Fig. 4C). No further effect of glucose was observed in the presence of the ERK2-MEK1 fusion proteins. These results support the conclusion that ERK-sensitive motifs are contained within E2A3/4 and demonstrate that activated ERK2 is sufficient to promote gene transcription via E2A3/4. On the other hand, the fusion proteins were much less effective in inducing transcription of a reporter driven by the fulMength insulin gene promoter (not shown). The small effect on the full-length promoter suggests that activation of other elements that are not controlled by ERK1/2, Raf, or Ras is required to stimulate transcription more fully in the context of the complete promoter.
Further support for these ideas is found in the data of Fig. 5. Activation of ERK1/2 caused by glucose plus forskolin was inhibited to differing extents by the two pharmacological inhibitors U0126 and PD98059 (Fig. 5A). Activity of the reporter driven by the insulin gene promoter from the same cells is shown in Fig. 5B. Glucose
A U0126 PD98059
50uM 10uM 50uM 10uM
Lane: 1 2 3 4 5 6 7 8 9 10 11
2 11 " 11 + Fsk
P-ERK1/2
ERK1/2
B 60
50 ._~
,< 40
t~
�9 ~ 30 '-,t
_1
,~ 20
,9 lO
_o _~
_9.o ,._ (.9
v - x--
+ + + +
o o ~ o
u u u u
~ O O O D
o o ~ N
+ + O O
+ u
Fig. 5. Comparison of inhibition of ERK1/2 with inhibition of promoter activity. Cells were treated as previously before harvest and then exposed to inhibitors for 30 min before the addition of glucose, forskolin or both for 2 h. (A) Effects of inhibitors on ERK1/2 activity detected as in Fig. 1. (B) Promoter activity from the same cell samples as in (A) measured as in Fig. 4.
Cell Biochemistry and Biophysics Supplement, 2004

MAP Kinases 19 7
caused a small increase in reporter activity, which was greatly increased by the addition of forskolin. The inhibitors reduced reporter activ- ity, with the condition causing the greatest inhi- bition of ERK1/2 also resulting in the largest reduction in promoter activity. Overall, there was a reasonable correlation between promoter activity and ERK1/2 activity.
DISCUSSION
In spite of considerable effort, the mecha- nism of activation of ERK1/2 by glucose is still undefined. We demonstrated that glucose metabolism was required for activation of ERK1/2 (10). Work from several laboratories has confirmed the requirement for calcium in ERK1/2 activation (10,11), although the site and mechanism of action of calcium in the ERK1/2 cascade both remain unknown. Work we describe elsewhere indicates that conventional protein kinase C isoforms are not involved, and that events in addition to entry of calcium through L-type voltage-gated calcium channels participate in the process (30). An important issue that has not been resolved is whether the activation of ERK1/2 occurs solely as a conse- quence of calcium signaling induced by glu- cose, or if other glucose-induced events independent of calcium are also involved in activating ERK1/2. Metabolic signals generated from glucose, as well as from other agents, may also input to ERK1/2; and the calcium-depen- dent and metabolic signals may confer upon ERK1/2 the property of coincidence monitor in the nutrient-sensing process.
A few targets of ERK1/2 in pancreatic [~-cells have been identified. These include several transcription factors implicated in insulin gene transcription (35) and the protein kinase Rsk (Gibson, T. B, Lawrence, M., Arnette, D., McGlynn, K., Vanderbilt, C. A., and Cobb, M. H., submitted). Rsk was the second substrate ever identified for ERK1/2 (31). In other cell types Rsk has been found to phosphorylate the cAMP-response element binding protein CREB (32) and the serum response factor (33). Thus
Rsk itself may directly regulate transcription factors in ~-cells downstream of ERK1/2. Undoubtedly many other protein targets of ERK1/2 will be found in these cells.
ERK1/2 are not required for glucose-stimu- lated insulin secretion measured in static 2-h assays, but their inhibition reduces the insulin content of cultured [~-cells and reduces the glu- cose-stimulated transcription of reporters dri- ven by the insulin gene promoter and the glucose-sensitive E2A3/4 elements in both cul- tured [3-cells and freshly isolated islets. Active ERK2 also stimulates transcription from reporters linked to E2A3/4 elements. Evidence from other laboratories also indicates that ERK1/2 are required for glucose-induced gene transcription (37). ERK1/2 probably have sev- eral sites of action that alter transcription of the insulin gene. Some of these effects are direct, such as phosphorylation of transcriptional reg- ulators by ERK1/2, and some indirect, acting through kinases that are activated downstream of ERK1/2, such as Rsk.
Glucose-sensing mechanisms must regulate acute events in ~-cells and support continued ~-cell survival and their differentiated proper- ties. The first steps in glucose sensing are glu- cose uptake and metabolism. The kinetic properties of the Glut2 glucose transporter are essential, as is a glucokinase isoform with a Km for glucose in the mM range (38-40). These proteins are the proximal sensors that allow ~- cells to take up and phosphorylate glucose in proportion to its concentration in the circula- tion. The sensing of glucose metabolism by the [~-cell is more complex. The adenosine 5'-diphosphate/adenosine triphosphate ratio as interpreted by the adenosine triphosphate- sensitive potassium channel is an important part, but not all, of the process (41). Gene dis- ruption experiments in mice indicate that ani- mals that do not assemble this channel, either due to lack of protein or of the Surl sulfony- lurea binding subunit, are not diabetic unless exposed to stress (42). Glycolytic products such as succinate are thought to play important roles in mediating glucose sensing at the level of proinsulin biosynthesis (7). As a consequence
Cell Biochemistn d and Biophysics Supplement, 2004

1 9 8 Khoo et al.
of these early events, glucose brings about the activation of a host of effectors that regulate 13- cell functions.
ERK1/2 are components of this assembly of glucose effectors. ERK1/2 are sensitive to con- centrations of glucose, other nutrients, and hormones that potentiate insulin secretion. Therefore, we suggest that their activities are modulated in a manner that reflects the secre- tory state of 13-cells. The fact that microarray experiments indicate that ERK2 mRNA is prominent ly regulated by glucose supports this theme (43). Because ERK1/2 activity is required for glucose-stimulated insulin tran- scription, it appears that ERK1/2 offer one mechanism to link nutrient sensing to insulin transcription. The activation of ERK1/2 by glu- cose and other nutrients, and the modest basal activity of ERK1/2 at low physiological glu- cose concentrations also support the idea that ERK1/2 are components of the signaling mech- anisms that integrate long- and short-term nutrient-sensing information at the level of the insulin promoter.
ACKNOWLEDGMENTS
We thank the members of the Cobb labora- tory and many colleagues for their comments and Dionne Ware for her administrative assis- tance. These studies were supported by grants from the National Institutes of Health (DK55310 to MHC and DK46993 to MSG) and the Juvenile Diabetes Foundation (to MHC). Tara Beers Gibson's research was supported by an individual NIH postdoctoral fellowship from the National Institute of Diabetes & Digestive & Kidney Diseases (NIDDK), and Michael Lawrence's studies were sup- ported by an American Diabetes Association mentor-based postdoctoral fellowship.
REFERENCES
1. Newgard, C. B. and McGarry, J. D. (1995) Metabolic coupling factors in pancreatic ~-cell
signal transduction. Annu. Rev. Biochem. 64, 689-719.
2. Welsh, M., Scherberg, N., Gilmore, R., and Steiner, D. F. (1986) Translational control of insulin biosynthesis. Evidence for regulation of elongation, initiation and signal-recognition- particle-mediated translational arrest by glu- cose. Biochem. J. 235, 459-467.
3. Brunstedt, J. and Chan, S. J. (1982) Direct effect of glucose on the preproinsulin mRNA level in isolated pancreatic islets. Biochem. Biophys. Res. Commun. 106, 1383-1389.
4. Giddings, S., Chirgwin, J., and Permutt, M. A. (1982) Effects of glucose on proinsulin messen- ger RNA in rats in vivo. Diabetes 31, 624-629.
5. Nielsen, D. A., Welsh, M., Casadaban, M. J., and Steiner, D. E (1985) Control of insulin gene expression in pancreatic beta-cells and in an insulin-producing cell line, RIN-5F cells. I. Effects of glucose and cyclic AMP on the tran- scription of insulin mRNA. J. Biol. Chem. 260, 13585-13589.
6. Leibiger, B., Moede, T., Schwarz, T., Brown, G. R., Kohler, M., Leibiger, I. B., et al. (1998) Short- term regulation of insulin gene transcription by glucose. Proc. Natl. Acad. Sci. USA 95, 9307-9312.
7. Alarcon, C., Wicksteed, B., Prentki, M., Corkey, B. E., and Rhodes, C. J. (2002) Succinate is a preferential metabolic stimulus-coupling signal for glucose-induced proinsulin biosynthesis translation. Diabetes 51, 2496-2504.
8. Bratanova-Tochkova, T. K., Cheng, H., Daniel, S., Gunawardana, S., Liu, Y. J., Mulvaney-Musa, J., et al. (2002) Triggering and augmentation mechanisms, granule pools, and biphasic insulin secretion. Diabetes 51, $83-$90.
9. Fr6din, M., Sekine, N., Roche, E., Filloux, C., Prentki, M., Wollheim, C. B., et al. (1995) Glucose, other secretagogues, and nerve growth factor stimulate mitogen-activated protein kinase in the insulin-secreting B-cell line, INS-1. J. Biol. Chem. 270, 7882-7889.
10. Khoo, S. and Cobb, M. H. (1997) Activation of MAP kinase by glucose is not required for insulin secretion. Proc. Natl. Acad. Sci. USA 94, 5599-5604.
11. Benes, C., Roisin, M. E, Van Tan, H., Creuzet, C., Miyazaki, J., and Fagard, R. (1998) Rapid activa- tion and nuclear translocation of mitogen-acti- vated protein kinases in response to physiological concentration of glucose in the MIN6 pancreatic beta cell line. J. Biol. Chem. 273, 15507-15513.
Cell Biochemistry and Biophysics Supplement, 2004

M A P Kinases 1 9 9
12. Lewis, T. S., Shapiro, P. S., and Ahn, N. G. (1998) Signal transduction through MAP kinase cas- cades. Adv. Cancer. Res. 74, 49-139.
13. Chen, Z., Gibson, T.B., Robinson, E, Silvestro, L., Pearson, G., Xu, B., Wright, A., et al. (2001) MAP kinases. Chem. Rev. 101, 2449-2476.
14. Lin, L. L., Wartmann, M., Lin, A. Y., Knopf, J. L., Seth, A., and Davis, R. J. (1993) cPLA2 is phos- phorylated and activated by MAP kinase. Cell 72, 269-278.
15. Hughes, P. E., Renshaw, M. W., Pfaff, M., Forsyth, J., Keivens, V. M., Schwartz, M. A., et al. (1997) Suppression of integrin activation: a novel function of a Ras/Raf-initiated MAP kinase pathway. Cell 88, 521-530.
16. Klemke, R. L., Cai, S., Giannini, A. L., Gallagher, P. J., de Lanerolle, P., and Cheresh, D. A. (1997) Regulation of cell motility by mitogen-activated protein kinase. J. Cell. Biol. 137, 481-492.
17. Sontag, E., Federov, S., Robbins, D., Cobb, M., and Mumby, M. (1993) The interaction of SV40 small tumor antigen with protein phosphatase 2A stimulates the MAP kinase pathway and induces cell proliferation. Cell 75, 887-897.
18. English, J. D. and Sweatt, J. D. (1996) Activation of p42 mitogen-activated protein kinase in hip- pocampal long term potentiation. J. Biol. Chem. 271, 24329-24332.
19. Rossi-Arnaud, C., Grant, S. G., Chapman, P. F., Lipp, H. P., Sturani, E., and Klein, R. (1997) A role for the Ras signalling pathway in synaptic transmission and long-term memory. Nature 390, 281-286.
20. Martin, K. C., Michael, D., Rose, J. C., Barad, M., Casadio, A., Zhu, H., et al. (1997) MAP kinase translocates into the nucleus of the presynaptic cell and is required for long-term facilitation in Aplysia. Neuron 18, 899-912.
21. Xia, Z. G., Dudek, H., Miranti, C. K., and Greenberg, M. E. (1996) Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J. Neurosci. 16, 5425-5436.
22. Robinson, M. J., Stippec, S. A., Goldsmith, E., White, M. A., and Cobb, M. H. (1998) Constitutively active ERK2 MAP kinase is suffi- cient for neurite outgrowth and cell transforma- tion when targeted to the nucleus. Curr. Biol. 8, 1141-1150.
23. Becker, T. C., Noel, R. J., Coats, W. S., Gomez- Foix, A. M., Alam, T., Gerard, R. D., et al. (1994) Use of recombinant adenovirus for metabolic
engineering of mammalian cells. Meth. Cell Biol. 43, A161-A189.
24. Klesse, L. J. and Parada, L. F. (1998) p21 ras and phosphatidylinositol-3 kinase are required for survival of wild-type and NF1 mutant sensory neurons. J. Neurosci. 18, 10420-10428.
25. Hohmeier, H. E., Mulder, H., Chen, G., Henkel- Rieger, R., Prentki, M., and Newgard, C. B. (2000) Isolation of INS-l-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 49, 424-430.
26. Khokhlatchev, A., Xu, S., English, J., Wu, P., Schaefer, E., and Cobb, M. H. (1997) Reconstitution of mitogen-activated protein kinase phosphorylation cascades in bacteria. J. Biol. Chem. 272, 11057-11062.
27. German, M. S. and Wang, J. (t994) The insulin gene contains multiple transcriptional elements that respond to glucose. Mol. Cell. Biol. 14, 4067-4075.
28. Pearson, G., English, J. M., White, M. A., and Cobb, M. H. (2001) ERK5 and ERK2 cooperate to regulate NF-kappaB and cell transformation. J. Biol. Chem. 276, 7927-7931.
29. Swantek, J. L., Cobb, M. H., and Geppert, T. D. (1997) JNK/SAPK is required for lipopolysac- charide stimulation of tumor necrosis factor alpha translation: glucocorticoids inhibit TNF- alpha translation by blocking JNK/SAPK. Mol. Cell. Biol. 17, 6274-6282.
30. Arnette, D., Gibson, T. B., Lawrence, M., January, B., Khoo, S., McGlynn, K., et al. (2003) Regulation of ERK1 and ERK2 by glucose and peptide hormones in pancreatic beta cells. J. Biol. Chem. 278, 32517-32525.
31. Sturgill, T. W., Ray, L. B., Erikson, E., and Maller, J. (1988) Insulin-stimulated MAP-2 kinase phos- phorylates and activates ribosomal protein $6 kinase II. Nature 334, 715-718.
32. Xing, J., Ginty, D. D., and Greenberg, M. E. (1996) Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regu- lated CREB kinase. Science 273, 959-963.
33. Rivera, V. M., Miranti, C. K., Misra, R. P., Ginty, D. D., Chen, R. H., Blenis, J., et al. (1993) A growth factor-induced kinase phosphorylates the serum response factor at a site that regu- lates its DNA-binding activity. Mol. Cell. Biol. 13, 6260-6273.
34. Robbins, D. J., Cheng, M., Zhen, E., Vanderbilt, C., Feig, L. A., and Cobb, M. H. (1992) Evidence for a ras-dependent extracellular signal-regu-
Cell Biochemistry and Biophysics Supplement, 2004

2 0 0 Khoo et al.
lated protein kinase (ERK) cascade. Proc. Natl. Acad. Sci. U S A 89, 6924-6928.
35. Khoo, S., Griffen, S. C., Xia, Y., Baer, R. J., German, M. S., and Cobb, M. H. (2003) Regulation of insulin gene transcription by ERK1 and ERK2 in pancreatic beta cells. J. Biol. Chem. 278, 32969-32977.
36. German, M., Ashcroft, S., Docherty, K., Edlund, H., Edlund, T., Goodison, S., et al. (1995) The insulin gene promoter. Diabetes 44, 1002-1004.
37. Benes, C., Poitout, V., Marie, J.C., Martin- Perez, J., Roisin, M. P., and Fagard, R. (1999) Mode of regulation of the extracellular signal- regulated kinases in the pancreatic beta-cell line MIN6 and their implication in the regula- tion of insulin gene transcription. Biochem. J. 340, 219-225.
38. Hughes, S. D., Quaade, C., Johnson, J. H., Ferber, S., and Newgard, C. B. (1993) Transfection of ATT-20ins cells with GLUT-2 but not GLUT-1 confers glucose-stimulated insulin secretion. Relationship to glucose metabolism. ]. Biol. Chem. 268, 15205-15212.
39. German, M. S. (1993) Glucose sensing in pan- creatic islet beta cells: the key role of glucoki- nase and the glycolytic intermediates. Proc. Natl. Acad. Sci. USA 90, 1781-1785.
40. Efrat, S., Tal, M., and Lodish, H. E (1994) The pancreatic ~-cell glucose sensor. TIBS 19, 535-538.
41. Aguilar-Bryan, L., Bryan, J., and Nakazaki, M. (2001) Of mice and men: K(ATP) channels and insulin secretion. Recent Prog. Horm. Res. 56, 47-68.
42. Seghers, V., Nakazaki, M., DeMayo, F., Aguilar- Bryan, L., and Bryan, J. (2000) Surl knockout mice. A model for K(ATP) channel-independent regulation of insulin secretion. J. Biol. Chem. 275, 9270-9277.
43. Webb, G. C., Akbar, M. S., Zhao, C., and Steiner, D. F. (2000) Expression profiling of pancreatic beta cells: glucose regulation of secretory and metabolic pathway genes. Proc. Natl. Acad. Sci. USA 97, 5773-5778.
Cell Biochemistry and Biophysics Supplement, 2004




















