
Mitogen-activated protein kinases activation in T lymphocytes of patients with acute coronary...
13
ORIGINAL CONTRIBUTION Mitogen-activated protein kinases activation in T lymphocytes of patients with acute coronary syndromes Ciro Indolfi • Cosimo Gasparri • Carla Vicinanza • Daniela De Serio • Duino Boncompagni • Annalisa Mongiardo • Carmen Spaccarotella • Valter Agosti • Daniele Torella • Antonio Curcio Received: 28 July 2010 / Revised: 17 February 2011 / Accepted: 9 March 2011 / Published online: 20 March 2011 Ó Springer-Verlag 2011 Abstract Current available biomarkers cannot identify myocardial ischemia without necrosis. To overcome this issue and to increase diagnostic power, we evaluated the activation of the three MAPK pathways, ERK1/2, JNK and p38, in T lymphocytes of patients with acute coronary syndromes (ACS). We included sixty consecutive patients affected by either unstable angina (UA, N = 22), Non- ST- segment elevation MI (NSTEMI, N = 19) or ST-segment elevation MI (STEMI, N = 19). Two separate groups of patients were matched as controls: healthy subjects (CTRL, N = 20) and patients with stable coronary artery disease (CAD, N = 21). MAPK activation in T lymphocytes, measured by phospho-ERK1/2, phospho-JNK and phos- pho-p38 levels, was assessed by flow cytometry analysis which revealed significantly increased phosphorylated levels of ERK1/2 in patients with UA, compared to con- trols. In UA patients no significant changes were detected for phospho-JNK compared to both control groups. NSTEMI and STEMI groups showed a statistically significant increase in both phospho-ERK1/2 and phospho- JNK, compared to control groups. All ACS groups demonstrated significantly increased phosphorylation of p38 compared to CTRL, but not CAD. ROC curves showed that a cut-off value of 22.5 intensity of fluorescence for phospho-ERK1/2 was able to significantly discriminate UA patients from patients with stable angina with 78% sensi- tivity and 90% specificity. Therefore, a differential MAPK activation in T lymphocytes denotes patients with ACS. Indeed, patients with unstable angina are identified with high specificity by activated ERK1/2 and normal JNK levels. These data could represent a valuable new molec- ular signature to be used as specific biomarkers for the diagnosis of unstable angina within ACS. Keywords Acute coronary syndromes Á Biomarkers Á T lymphocytes Á Mitogen-activated protein kinases Introduction Acute coronary syndromes (ACS) are one of the major causes of morbidity and mortality in developed countries [26]. Early recognition of myocardial ischemia and accu- rate diagnosis of ACS represent critical steps for selecting and evaluating the response to therapeutic interventions [1, 36]. A combination of accurate history taking, physical examination, measurement of circulating levels of cardiac troponins (cTn) and the MB isoform of creatine kinase (CK), EKG and echocardiography helps physicians in identifying patients with ACS presenting to the emergency department [37, 39]. However, the current approach is still lacking of a diagnostic test able to identify without delay patients with less-typical symptoms but yet at significant risk for complications with ACS. Systemic inflammation can trigger a cascade that amplifies immune cell infiltration, platelet activation and adhesion, plaque rupture and intermittent arterial occlu- sion, hence representing a preeminent factor to the C. Indolfi (&) Á C. Gasparri Á C. Vicinanza Á D. De Serio Á D. Boncompagni Á A. Mongiardo Á C. Spaccarotella Á D. Torella Á A. Curcio Division of Cardiology, Laboratory of Molecular and Cellular Cardiology, University Magna Graecia, Viale Europa, Campus ‘‘S. Venuta’’ di Germaneto, 88100 Catanzaro, Italy e-mail: indolfi@unicz.it V. Agosti Department of Experimental and Clinical Medicine, Laboratory of Molecular Oncology, University Magna Graecia, Viale Europa, Campus ‘‘S. Venuta’’ di Germaneto, 88100 Catanzaro, Italy 123 Basic Res Cardiol (2011) 106:667–679 DOI 10.1007/s00395-011-0172-1
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Mitogen-activated protein kinases activation in T lymphocytes of patients with acute coronary...

ORIGINAL CONTRIBUTION
Mitogen-activated protein kinases activation in T lymphocytesof patients with acute coronary syndromes
Ciro Indolfi • Cosimo Gasparri • Carla Vicinanza • Daniela De Serio •
Duino Boncompagni • Annalisa Mongiardo • Carmen Spaccarotella •
Valter Agosti • Daniele Torella • Antonio Curcio
Received: 28 July 2010 / Revised: 17 February 2011 / Accepted: 9 March 2011 / Published online: 20 March 2011
� Springer-Verlag 2011
Abstract Current available biomarkers cannot identify
myocardial ischemia without necrosis. To overcome this
issue and to increase diagnostic power, we evaluated the
activation of the three MAPK pathways, ERK1/2, JNK and
p38, in T lymphocytes of patients with acute coronary
syndromes (ACS). We included sixty consecutive patients
affected by either unstable angina (UA, N = 22), Non- ST-
segment elevation MI (NSTEMI, N = 19) or ST-segment
elevation MI (STEMI, N = 19). Two separate groups of
patients were matched as controls: healthy subjects (CTRL,
N = 20) and patients with stable coronary artery disease
(CAD, N = 21). MAPK activation in T lymphocytes,
measured by phospho-ERK1/2, phospho-JNK and phos-
pho-p38 levels, was assessed by flow cytometry analysis
which revealed significantly increased phosphorylated
levels of ERK1/2 in patients with UA, compared to con-
trols. In UA patients no significant changes were detected
for phospho-JNK compared to both control groups.
NSTEMI and STEMI groups showed a statistically
significant increase in both phospho-ERK1/2 and phospho-
JNK, compared to control groups. All ACS groups
demonstrated significantly increased phosphorylation of
p38 compared to CTRL, but not CAD. ROC curves showed
that a cut-off value of 22.5 intensity of fluorescence for
phospho-ERK1/2 was able to significantly discriminate UA
patients from patients with stable angina with 78% sensi-
tivity and 90% specificity. Therefore, a differential MAPK
activation in T lymphocytes denotes patients with ACS.
Indeed, patients with unstable angina are identified with
high specificity by activated ERK1/2 and normal JNK
levels. These data could represent a valuable new molec-
ular signature to be used as specific biomarkers for the
diagnosis of unstable angina within ACS.
Keywords Acute coronary syndromes � Biomarkers �T lymphocytes � Mitogen-activated protein kinases
Introduction
Acute coronary syndromes (ACS) are one of the major
causes of morbidity and mortality in developed countries
[26]. Early recognition of myocardial ischemia and accu-
rate diagnosis of ACS represent critical steps for selecting
and evaluating the response to therapeutic interventions [1,
36]. A combination of accurate history taking, physical
examination, measurement of circulating levels of cardiac
troponins (cTn) and the MB isoform of creatine kinase
(CK), EKG and echocardiography helps physicians in
identifying patients with ACS presenting to the emergency
department [37, 39]. However, the current approach is still
lacking of a diagnostic test able to identify without delay
patients with less-typical symptoms but yet at significant
risk for complications with ACS.
Systemic inflammation can trigger a cascade that
amplifies immune cell infiltration, platelet activation and
adhesion, plaque rupture and intermittent arterial occlu-
sion, hence representing a preeminent factor to the
C. Indolfi (&) � C. Gasparri � C. Vicinanza � D. De Serio �D. Boncompagni � A. Mongiardo � C. Spaccarotella �D. Torella � A. Curcio
Division of Cardiology, Laboratory of Molecular and Cellular
Cardiology, University Magna Graecia, Viale Europa,
Campus ‘‘S. Venuta’’ di Germaneto, 88100 Catanzaro, Italy
e-mail: [email protected]
V. Agosti
Department of Experimental and Clinical Medicine,
Laboratory of Molecular Oncology, University Magna Graecia,
Viale Europa, Campus ‘‘S. Venuta’’ di Germaneto,
88100 Catanzaro, Italy
123
Basic Res Cardiol (2011) 106:667–679
DOI 10.1007/s00395-011-0172-1

instability of atherosclerotic plaques in acute patients [30].
Indeed, the investigation of novel circulating serum and
plasma biomarkers in patients with ACS has recently
unmasked the relationship between the inflammatory bio-
marker C-reactive protein (CRP) and the outcome in acute
patients [38]. Activated T lymphocytes are frequently
increased in peripheral blood of subjects affected by car-
diovascular diseases [9]. These evidences suggest that the
inflammatory component might be systemically detectable
in patients with ACS [2, 3, 8, 27]. However, even though
biomarkers of inflammation may provide unique informa-
tions to the clinician apart from that provided by bio-
markers of myocyte necrosis, the prognostic significance of
these new tools is still debated with regard to their potential
to enhance the care of patients [4, 25, 29, 34, 35].
Inflammation and different forms of cellular stress are
capable to elicit biochemical responses [10, 46] and
subsequently trigger molecular cascades such as mitogen-
activated protein kinases (MAPK), which act by phos-
phorylating various intracellular substrates including
transcription factors, hence regulating signal transduction
and specific genetic responses to extracellular stimuli
[19–21]. The MAPK family includes three major sub-
groups, extracellular signal-regulated kinase-1 and -2
(ERK1/2) and two stress-activated protein kinase path-
ways, c-Jun NH2-terminal kinase (JNK) and p38 MAPK
[43]. Specifically, in cardiovascular system ERK1/2 has
been involved in several serine/threonine phosphorylation
cascades that control cellular proliferation, differentiation,
survival, and motility in response to diverse extracellular
stimuli including mitogens, growth factors, and cytokines
[13], whereas in the heart JNK activation has been dem-
onstrated by hyperosmotic shock, low concentrations of
protein synthesis inhibitors such as anisomycin, the car-
diotoxic agent daunomycin, hypoxia/reoxygenation and
reactive oxygen species but can also occur in response to
mechanical stretch or pacing [31].
Moreover, the activation of p38 has been demonstrated
in several pathological conditions affecting the heart such
as experimental ischemic preconditioning in rat [23], rabbit
[18] and pig [40, 41] models. More recently, inhibition of
p38 enhanced the number of circulating vasculogenic cells
and improved the functional capacity of different pro-
angiogenic cells, thus reducing atherosclerotic disease
progression [42].
To date, it is unknown whether MAPK proteins are
activated within circulating inflammatory cells in response
to acute myocardial ischemia in patients with acute coro-
nary syndromes. Furthermore, it has never been evaluated
whether a differential activation of one or more kinases
involved in this cascade could be elicited by the different
clinical subsets of ACS. If MAPK activation was propor-
tional to the extent of myocardial ischemia ranging from a
partial MAPK activation in unstable angina on one hand to
a complete MAPK activation in NSTEMI and STEMI
patients on the other, this differential molecular activation
could prove very useful as diagnostic testing to discern
between the different ACS conditions.
Therefore, the aim of this study was to assess MAPK
activity in T lymphocytes of patients with ACS in an
independent manner from antiplatelet, antithrombotic
and/or HMG-CoA reductase inhibitors treatment.
Methods
Patient population
Eighty-one consecutive patients who were admitted at the
Division of Cardiology of the University of Magna Graecia
were enrolled in the study. All patients gave written an
informed consent to the study that was carried out in
accordance with the principles of the Declaration of Hel-
sinki and approved by local ethical committee. All patients
were assigned to different groups depending on clinical,
laboratory and angiographic features.
Group I included 21 patients with stable angina pectoris
(coronary artery disease, CAD), clinical evidence of
Canadian Cardiovascular Society class II and III, and at
least 1 coronary artery stenosis detected at the angiographic
examination ([75% reduction of lumen diameter). Group
II included 22 patients with unstable angina (UA) admitted
to our Hospital. All patients with UA had experienced chest
pain at rest within the preceding 12 h without evidence of
myocardial infarction (MI) as shown by the absence of
elevation of CK-MB or cardiac troponin I (cTnI) blood
levels. In all UA patients, a culprit lesion was confirmed at
the coronary angiography. Group III included 19 patients
with Non ST-segment elevation MI (NSTEMI) who pre-
sented within 12 h of the onset of pain, elevated cTnI blood
levels and angiographic evidence of coronary lesions.
Finally, patients with ST-segment elevation MI (STEMI,
N = 19), documented with an increase of CKMB and cTnI,
admitted within 12 h from the onset of symptoms were
included in Group IV. All patients underwent coronary
angiography and coronary revascularization, if needed,
during hospitalization. Additional healthy volunteers
(CTRL, N = 20) with no clinical signs of CAD and
without coronary risk factors were recruited from the blood
donors bank.
Exclusion criteria were as follows: previous MI within
6 months, previous revascularization procedures, inflam-
matory conditions likely to be associated with an acute
phase response, autoimmune disease, neoplastic disease,
advanced liver disease, renal failure or severe heart failure
(NYHA class III-IV).
668 Basic Res Cardiol (2011) 106:667–679
123

In the groups I–IV, the blood samples for MAPK ana-
lysis were obtained immediately at the patients admission.
Cardiac TnI and CK measurements were performed
according to the current guidelines, as routinely performed
in our institution.
Isolation of peripheral blood mononuclear cells
and flow cytometry analysis
Peripheral blood samples were obtained from all patients
and collected in a Vacutest Kima tube with K3EDTA.
Isolation of mononuclear cells was performed by Ficoll
(biocoll separating solution; Biochrom AG) density gradi-
ent centrifugation. Mononuclear cells were resuspended in
cold incubation buffer (PBS 19, BSA 0.5%) (Becton–
Dickinson) and were incubated with 10 ll of CD3-PE or
CD19-PE antibody (markers of T and B lymphocytes,
respectively) at 4�C for 30 min in the dark. Then, after
antibody wash-out in the incubation buffer, cells were fixed
with paraformaldehyde 2% (Sigma-Aldrich) for 10 min at
37�C, and permeabilized with cold methanol 90% (Sigma-
Aldrich) for 30 min at 4�C. Hence, cells were resuspended
with incubation buffer and subsequently incubated with
phospho-ERK1/2, phospho-p38 and phospho-JNK anti-
bodies (Cell Signaling Technology) for 1 h at room tem-
perature followed by monoclonal antibodies anti-rabbit
FITC conjugated (Jackson ImmunoResearch Laboratories).
A total of 30,000 events in a specific gate were analyzed on
a FACS Calibur flow cytometer with CellQuest acquisition
analysis software (Becton–Dickinson).
Immunoblotting analysis
In order to confirm the flow cytometry analysis, immuno-
blotting analysis was carried out including additional 28
patients. Briefly, proteins were extracted from T lympho-
cytes of CTRL subjects (N = 5) and both chronic and
acute coronary disease patients (CAD, N = 5; UA, N = 6;
NSTEMI, N = 6; STEMI, N = 6) obtained as above
described. Forty micrograms of total proteins were sepa-
rated by SDS-PAGE, transferred onto a nitrocellulose filter
through semi-dry trans-blot system (Biorad) and hybridized
with the different primary antibodies for activated MAPK:
phospho-ERK1/2, phospho-JNK, phospho-p38, and nor-
malized by antibodies recognizing the total ERK1/2, JNK
and p38 (Cell Signaling). The nitrocellulose filters were
blocked with 5% bovine serum albumin and the primary
antibodies were used at the dilution of 1:1,000 according to
the manufacturer’s recommendations (Cell Signaling
Technology). Specific HRP-conjugated secondary anti-
bodies were used according to the manufacturer’s recom-
mendations (Santa Cruz Biotechnology). Specific protein
bands were detected by chemiluminescence using the
Chemidoc XRS system (BioRad). Western blots for the
expression of total ERK1/2, JNK and p38 (antibodies from
Cell Signaling Technology) were performed to normalize
the phosphorylated levels of each protein kinase.
Statistical analysis
Significance between all groups was determined in multiple
comparisons by the analysis of variance (ANOVA). Bon-
ferroni’s post hoc method was used to locate the differences.
Receiver-operating characteristic (ROC) analysis was
also performed on the levels of phospho-ERK1/2, phospho-
JNK and phospho-p38 activation for UA, NSTEMI and
STEMI ACS. This analysis plots the true-positive fraction
(sensitivity) against the false-positive fraction (1-specifi-
city) by changing the cut-off value for the test. Areas under
the ROC curves indicate the relative accuracy of diagnostic
tests. Values of p \ 0.05 were considered statistically
significant.
Results
The baseline characteristics of all enrolled patients were well
matched among groups and are summarized in Table 1.
MAPK are activated in T but not B lymphocytes
in ACS patients
T lymphocytes are abnormally activated during the acute
phases of coronary atherosclerotic disease and can be
considered among putative novel biomarkers for early
diagnosis [16]. Then, we first ascertained whether MAPK
activation was confined to T lymphocytes or involved also
B lymphocytes in patients with ACS (UA, n = 5; NSTE-
MI, n = 5; STEMI, n = 5). Mononuclear cells freshly
isolated from blood samples were stained with antibodies
specific for the phosphorylated forms of ERK1/2, JNK and
p38 and either with CD3 antibody (recognizing T lym-
phocytes) or CD19 antibody (recognizing B lymphocytes)
and analyzed by fluorescent activated cell sorting (FACS).
Importantly, in all ACS patients ERK1/2, JNK and p38
were phosphorylated only in T lymphocytes while they
were mostly un-phosphorylated in B lymphocytes (Fig. 1).
Thus, we constrained the analysis of MAPK activation
in T lymphocytes isolated through CD3 immunomagnetic
bead cell sorting from mononuclear cells.
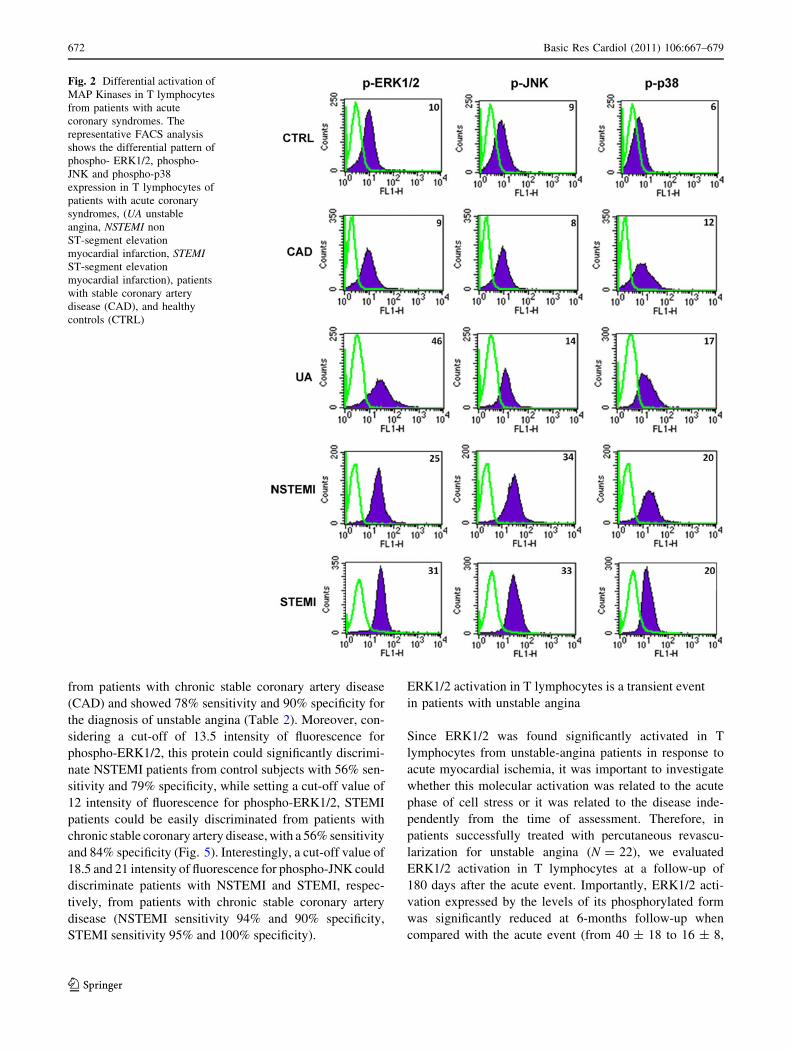
ERK1/2 is activated in T lymphocytes in all the subsets
of ACS patients
We first assessed ERK1/2 activation by FACS analysis and
reported the level of ERK1/2 phosphorylation (phospho-
Basic Res Cardiol (2011) 106:667–679 669
123

ERK1/2) as intensity of fluorescence for the specific
antibody. Importantly, ERK1/2 was significantly phos-
phorylated in UA, NSTEMI and STEMI patients (phospho-
ERK1/2 in UA = 40 ± 18, in NSTEMI = 26 ± 15 and in
STEMI = 34 ± 23) when compared with control subjects
(phospho-ERK1/2 in CTRL = 6 ± 3; p \ 0.05 vs. UA,
NSTEMI and STEMI). Of note, ERK1/2 was not signifi-
cantly activated in patients with chronic coronary artery
disease (phospho- ERK1/2 in CAD = 16 ± 8; p = NS vs.
UA, NSTEMI, STEMI and CTRL, Figs. 2, 3). Thus, these
data indicate that MAPK ERK1/2 is generally activated in
T lymphocytes of patients with ACS in response to myo-
cardial ischemia regardless of the presence of myocyte
necrosis.
JNK is activated in T lymphocytes only in ACS patients
with myocardial necrosis
A statistically significant increase of phospho-JNK, com-
pared to patients with stable angina pectoris, was observed
only in ACS patients with myocardial infarction (phospho-
JNK = 36 ± 19 in NSTEMI patients and 40 ± 28 in
STEMI patients; p \ 0.05 vs. UA, CAD and CTRL),
whereas JNK was not significantly activated in UA
(phospho-JNK = 12 ± 7) compared to CAD (10 ± 5;
p = NS vs. UA), or control patients (CTRL: 4 ± 2,
p = NS vs. UA and CAD, Figs. 2, 3).
Therefore, in T lymphocytes from ACS patients, in
contrast to ERK1/2, JNK activation could distinguish
patients with myocardial infarction in whom JNK is sig-
nificantly phosphorylated from patients with unstable
angina in whom JNK is un-activated.
p38 is activated in T lymphocytes in both acute
and chronic myocardial ischemia
Finally, FACS analysis showed that p38 was significantly
phosphorylated in T lymphocytes isolated from patients
with both chronic and acute myocardial ischemia (Figs. 2,
3). Indeed, a statistically significant increase of the phos-
phorylated form of p38 was detected in patients with
ST-segment elevation myocardial infarction (34 ± 27),
non ST-segment elevation myocardial infarction
(21 ± 20), unstable angina (15 ± 13) and patients with
chronic coronary artery disease (16 ± 13) compared to
healthy control subjects (p \ 0.05, Figs. 2, 3). Importantly,
Table 1 Clinical characteristics and biological parameters of the study populations
CTRL CAD UA NSTEMI STEMI
Patients, n 20 21 22 19 19
Age (years) 59 ± 13 61 ± 7 61 ± 8 60 ± 13 60 ± 11
Male sex, n (%) 16 (80%) 16 (76%) 17 (77%) 15 (79%) 15 (79%)
Risk Factors, n (%)
Hypertension 2 (10%) 5 (25%)* 6 (26%)* 5 (26%)* 5 (26%)*
Diabetes mellitus 0 (0%) 1 (3%)* 1 (3%)* 1 (5%)* 1 (5%)*
Smoking 0 (0%) 5 (23%)* 6 (25%)* 4 (23%)* 5 (28%)*
Hypercholesterolemia 0 (0%) 4 (18%)* 4 (18%)* 4 (19%)* 4 (20%)*
Familial history of CAD 2 (10%) 6 (27%)* 7 (30%)* 5 (28%)* 6 (29%)*
Single-vessel disease 0 (0%) 10 (48%)* 11 (50%)* 10 (53%)* 9 (47%)*
Multi-vessels disease 0 (0%) 11 (52%)* 11 (50%)* 9 (47%)* 10 (53%)*
BMI 26.5 ± 4.2 26.7 ± 2.0 27.1 ± 5.0 26.7 ± 2.9 27.8 ± 3.5
EF (%) 55 ± 10 50 ± 12 52 ± 7 50 ± 7 42 ± 16*,#,?
hs CRP (mg/l) 0.9 ± 0.9 1.9 ± 0.6* 8.4 ± 3.4*,# 25.0 ± 8.3*,#,? 21.1 ± 8.6*,#,?
Lymphocyte count (9103/ll) 2.1 ± 0.9 2.1 ± 0.6 2.3 ± 0.6 2.0 ± 0.5 2.0 ± 0.5
cTnI (ng/ml) \0.01 ± 0 \0.01 ± 0 \0.01 ± 0 0.88 ± 0.87*,#,? 1.6 ± 4*,#,?
CK (U/l) 89.0 ± 57.0 99.0 ± 87.0 154.2 ± 26.4* 266.3 ± 23.8*,#,? 2085.0 ± 999.0*,#,?,§
Pro-BNP (pg/ml) 198.3 ± 13.6 213.7 ± 140.7 216.0 ± 120.0 1800.2 ± 230.0*,#,? 5650.0 ± 521.0*,#,?,§
All values are expressed as mean ± SD or percentage
Statistical comparisons were performed by applying ANOVA test. For hs CRP evaluation, a Student’s t test was applied with Bonferroni
correction
CTRL healthy volunteers from donors bank, CAD stable coronary artery disease, UA unstable angina, NSTEMI non-ST-elevation myocardial
infarction, STEMI ST-elevation myocardial infarction, EF ejection fraction, hs CRP high sensitivity C-reactive protein, cTnI cardiac isoform of
the I subunit of the troponin complex, CK creatine kinase, Pro-BNP precursor of the brain natriuretic peptide
* p \ 0.05 versus CTRL, #p \ 0.05 versus CAD, ?p \ 0.05 versus UA, §p \ 0.05 versus NSTEMI
670 Basic Res Cardiol (2011) 106:667–679
123

p38 activation was higher in STEMI patients compared to
all the other groups.
Immunoblotting analysis confirms differential
activation of MAPK in ACS patients
We further validated the data by western blotting analysis
of activated MAPK in T lymphocytes protein extracts
obtained from additional patients in all the groups included
in the study. In agreement with the FACS data, western
blots showed that phospho-ERK1/2 was significantly
increased in all ACS patients when compared to CAD and
CTRL groups (Fig. 4). Furthermore, JNK was found to be
significantly phosphorylated in both ST- and non-ST-ele-
vation MI when compared to UA, CAD and CTRL patients
(Fig. 4). Finally, p38 was significantly phosphorylated in
all ischemic patients when compared to control subjects
and higher levels of phospho-p38 were detected in T
lymphocytes of STEMI patients.
These data strongly suggest that the acute and intense
myocardial stress produced by coronary occlusion is scouted
by T lymphocytes activating ERK1/2. The latter molecular
switch turns into the concomitant JNK activation when
ischemia time lasts enough to induce myocardial necrosis.
Moreover, according to above results, both acute and chronic
myocardial ischemia activates p38 in T lymphocytes
whereby the highest levels of phospho-p38 are associated
with more intense myocardial damage in STEMI patients.
Sensitivity and specificity
Given a cut-off value of 22.5 intensity of fluorescence for
phospho-ERK1/2 by FACS analysis in T lymphocytes, this
activated kinase could significantly discriminate UA patients
Fig. 1 Increased amounts of
ERK1/2-, JNK-, and
p38-activated forms in T but not
B lymphocytes during ACS.
The representative fluorescent
activated cell sorting (FACS)
analysis shows that ERK1/2,
JNK and p38 are specifically
activated (phosphorylated) in
CD3? T lymphocytes while
they are practically un-
phosphorylated in CD19? B
lymphocytes from patients with
acute coronary syndrome (in
particular, the data presented are
from a patient with NSTEMI)
Basic Res Cardiol (2011) 106:667–679 671
123
from patients with chronic stable coronary artery disease
(CAD) and showed 78% sensitivity and 90% specificity for
the diagnosis of unstable angina (Table 2). Moreover, con-
sidering a cut-off of 13.5 intensity of fluorescence for
phospho-ERK1/2, this protein could significantly discrimi-
nate NSTEMI patients from control subjects with 56% sen-
sitivity and 79% specificity, while setting a cut-off value of
12 intensity of fluorescence for phospho-ERK1/2, STEMI
patients could be easily discriminated from patients with
chronic stable coronary artery disease, with a 56% sensitivity
and 84% specificity (Fig. 5). Interestingly, a cut-off value of
18.5 and 21 intensity of fluorescence for phospho-JNK could
discriminate patients with NSTEMI and STEMI, respec-
tively, from patients with chronic stable coronary artery
disease (NSTEMI sensitivity 94% and 90% specificity,
STEMI sensitivity 95% and 100% specificity).
ERK1/2 activation in T lymphocytes is a transient event
in patients with unstable angina
Since ERK1/2 was found significantly activated in T
lymphocytes from unstable-angina patients in response to
acute myocardial ischemia, it was important to investigate
whether this molecular activation was related to the acute
phase of cell stress or it was related to the disease inde-
pendently from the time of assessment. Therefore, in
patients successfully treated with percutaneous revascu-
larization for unstable angina (N = 22), we evaluated
ERK1/2 activation in T lymphocytes at a follow-up of
180 days after the acute event. Importantly, ERK1/2 acti-
vation expressed by the levels of its phosphorylated form
was significantly reduced at 6-months follow-up when
compared with the acute event (from 40 ± 18 to 16 ± 8,
Fig. 2 Differential activation of
MAP Kinases in T lymphocytes
from patients with acute
coronary syndromes. The
representative FACS analysis
shows the differential pattern of
phospho- ERK1/2, phospho-
JNK and phospho-p38
expression in T lymphocytes of
patients with acute coronary
syndromes, (UA unstable
angina, NSTEMI non
ST-segment elevation
myocardial infarction, STEMIST-segment elevation
myocardial infarction), patients
with stable coronary artery
disease (CAD), and healthy
controls (CTRL)
672 Basic Res Cardiol (2011) 106:667–679
123

p \ 0.05, Fig. 6a, b), returning at values similar to coro-
nary artery disease patients. Moreover, considering an
intermediate follow-up, such as 60 days, it may be argued
that the reduction in ERK1/2 activation levels is a pro-
gressive phenomenon which starts after a successful
revascularization proceeding until complete normalization
after 6 months. Thus, the latter indicates that ERK1/2 is
specifically activated in T lymphocytes during acute
myocardial ischemia in the setting of UA. Indeed, the acute
administration of antiplatelet therapy, such as clopidogrel,
did not affect phospho-ERK1/2 levels in UA patients
0
1
2
3
4
5
42 kDa
42 kDa
46 kDa
46 kDa
40 kDa
40 kDa
Phospho-ERK 1/2
Total-ERK 1/2
Phospho-JNK
Total-JNK
Phospho-p38
Total-p38C
AD
STE
MI
NS
TEM
I
UA
CTR
L
T Lymphocytes
Fo
ld in
du
ctio
n (
ove
r co
ntr
ol)
CA
D
STE
MI
NS
TEM
I
UA
CA
D
STE
MI
NS
TEM
I
UA
CA
D
STE
MI
NS
TEM
I
UA
Phospho-ERK 1/2
Phospho-JNK
Phospho-p38
*
*
*
* * ***
(A)
(B)
Fig. 4 Immunoblotting analysis confirms differential activation of
MAPK in T lymphocytes from ACS patients. a Representative
western blot analysis showing ERK1/2 phosphorylation and the
MAPK levels in patients with acute coronary syndromes (UA, N = 6;
NSTEMI, N = 6; STEMI, N = 6), stable angina pectoris (CAD,
N = 5) and control subjects (CTRL, N = 5). b Densitometric anal-
ysis of activated forms of ERK1/2, JNK, and p38 (*p \ 0.05 vs.
stable angina pectoris)
Fig. 3 ERK1/2, JNK and p38 activation in T lymphocytes from
patients with acute coronary syndromes. Phospho-ERK1/2 fluores-
cence intensity was significantly increased in patients with unstable
angina, non-ST-elevation and ST-elevation myocardial infarction but
not in patients with stable angina pectoris and in control subjects
(*p \ 0.05 vs. CTRL). A statistically significant increase in fluores-
cence intensity of phospho-JNK was observed only in patients with
acute coronary syndromes associated with an increase of biomarkers
of necrosis (p \ 0.05 vs. CTRL), whereas P-JNK levels in UA were
not significantly elevated in comparison to CAD and control subjects.
A statistical increase in fluorescence intensity of phospho-p38 was
detected in all patients with coronary atherosclerosis (acute and stable
patients) compared to control subjects (*p \ 0.05 vs. CTRL)
b
Basic Res Cardiol (2011) 106:667–679 673
123

compared to additional evaluations performed 24 h after
clopidogrel intake (42 ± 9 vs. 39 ± 11, p = NS).
HMG-CoA reductase inhibition decreases MAPK
activation in activated T lymphocytes in vitro
The role of HMG-CoA reductase inhibitors is mandatory
in stabilizing atherosclerotic plaques, and this issue has
been addressed from several studies. Moreover, published
data from our lab already demonstrated that statin
administration can reduce vascular smooth muscle cell
proliferation in vitro and in vivo [22], suggesting that the
pleiotropic effects of these medications is involved also in
migration of the cells into the plaque. In order to inves-
tigate the effective role of pharmacological treatment on
T lymphocytes activation long after the acute event, T
lymphocytes were harvested from healthy subjects and
MAPK signaling pathway was assessed by immunoblot-
ting. Briefly, in order to mimic an inflammatory state,
cultured T cells were treated with 1,000 UI/ml interleu-
kin-2 (IL-2) at 37�C, 5% CO2 for 15 h; subsequently,
10 lmol rosuvastatin (Rosu) was administered in a single-
shot and cells were collected after 15 min or at the end of
the stimulation. IL-2 administration markedly increased
ERK1/2 activation levels (Fig. 6c), while Rosu treatment
inhibited the phosphorylation levels of ERK1/2 in cul-
tured T lymphocytes in a time-dependent manner, indi-
cating that ERK1/2 activation in T lymphocytes is a
continuous mechanism leading to worsening of the
inflammatory state, and mostly, that inflammation affects
circulating T lymphocytes both acutely and chronically,
therefore suggesting that standard optimal medical ther-
apy, including statins, should be started as soon as pos-
sible in acute patients.
Discussion
The major finding of the present study is that MAPK are
differently activated in T lymphocytes of patients with
ACS depending on the clinical presentation. In particular,
in all patients with ACS, regardless of the presence of
myocyte necrosis, we documented a significant increase of
phospho-ERK1/2, whereas in ACS, phospho-JNK was
activated only in patients with acute myocardial infarction
but not in unstable angina patients. Finally, phospho-p38
was found activated in all patients with atherosclerosis,
including ACS and CAD. Of note, these findings were not
correlated with the extension of coronary artery disease, in
terms of angiographic findings considering neither single-
nor multi-vessels disease (see Table 1), since no significant
difference was found among the groups.
Biomarkers are integral to the care of patients with
cardiovascular disease, in particular those presenting with
ACS with chest discomfort, for whom biomarkers are
central to establishing a diagnosis and guiding evidence-
based therapy [5]. The I subunit of cardiac troponin com-
plex (cTnI) established a paradigm for the modern clinical
use of a biomarker for identifying high-risk patients for
whom specific therapeutic interventions could be imple-
mented to modify the associated risk [33]. Indeed, in the
clinical setting, once it is established that no cTnI has been
released, the patient with suspected ACS may be consid-
ered to have experienced unstable angina. On the other
hand, the diagnosis of myocardial infarction (MI) is
established if a biomarker, i.e. cTnI, is released in the
bloodstream [25]. Moreover, a previous work from another
laboratory reported that beta2-integrin activation of T cells
is increased in patients with UA and severe CAD inde-
pendent of cardiac specific troponin levels [24], suggesting
that evidences that precede myocardial necrosis are needed
in these patients.
Therefore, from a clinical point of view, the diagnosis of
NSTEMI or STEMI is relatively straightforward by mea-
suring the markers of myocyte necrosis, with the caveat of
the delay of their raise in the bloodstream up to several
hours after the onset of ischemic chest pain [2, 3, 8, 9, 37,
38]. However, unstable angina still represents a difficult
diagnosis in a substantial percentage of patients. In the
United States each year, 5.3 million patients present to
emergency departments with chest discomfort and related
symptoms. Ultimately, 1.4 million individuals are hospi-
talized for unstable angina and NSTEMI. Due to the lack of
a biomarker for detecting myocardial ischemia in absence
of necrosis, the correct diagnosis and consequent treatment
of UA patients represent a significant challenge for phy-
sicians [15, 16].
It is well recognized that thrombosis underlies most
acute complications of atherosclerosis, notably UA and
Table 2 Diagnostic performances of the assays
Phospho-ERK1/2 Phospho-JNK
UA
Cut-off 22.5 10.5
Sensitivity 78 67
Specificity 90 60
NSTEMI
Cut-off 13.5 18.5
Sensitivity 56 94
Specificity 79 90
STEMI
Cut-off 12 21
Sensitivity 56 95
Specificity 84 100
The cut-off, sensitivity and specificity of phospho-ERK1/2 and
phospho-JNK in patients with unstable angina (UA), non-ST-eleva-
tion myocardial infarction (NSTEMI) and STEMI
674 Basic Res Cardiol (2011) 106:667–679
123

acute MI. A consensus has emerged that inflammation
plays a key role in the pathophysiology of these acute
thrombotic events [15]. Indeed, patients with cardiovas-
cular risk factors, such as tobacco smoke, have a reduced
number of circulating endothelial progenitor cells and a
high rate of arterial thrombotic complications [11]. To this
regard, it has been proven that the effect of glutathione
S-transferase gene polymorphism was most marked among
smokers with CAD [44]. Accordingly, smoking-induced
chronic inflammation is sufficient to increase activated
levels of all MAPK, but our study demonstrated for the first
time that only when an ACS with unstable angina occurs,
we detect significantly higher levels of phospho-ERK1/2
compared to CAD group.
Active investigation has brought forward an increas-
ingly large number of novel candidate markers of inflam-
mation in the setting of myocardial ischemia; however,
these markers have yet to be incorporated into routine
clinical use [5]. Non-invasive indicators of separate
pathobiologically diverse contributors to the progression of
cardiovascular disease, such as inflammation and throm-
bosis, could add complementary informations to the
available biomarkers of myocardial ischemia [32]. In fact,
novel biomarkers of inflammation, such as C-reactive
Fig. 5 Sensitivity and specificity data obtained with the ROC curves.
Receiver-operating characteristic (ROC) curves of phospho-ERK1/2
and phospho-JNK for diagnosis of ACS (a–c) and ACS without ST
elevation (d) among consecutive patients undergoing coronary
angiography. True-positive fraction (sensitivity as y-axis) was plotted
versus false-positive fraction (1-specificity as x-axis) by changing cut-
off values for the test
Basic Res Cardiol (2011) 106:667–679 675
123

protein (CRP) and myeloperoxidase, and of pathways for
thrombosis, such as soluble CD40 ligand and von Wille-
brand factor, have been shown to add independent prog-
nostic information in a variety of clinical settings,
including those involving stable and unstable ischemic
heart disease [5, 33]. Also, serum lectin-like oxidized LDL
receptor-1 (LOX-1) levels were found to be significantly
high in ACS [16]. Additionally, we have recently shown
through the use of proteomic serum analysis that vitamin D
binding protein is significantly increased in the serum of
STEMI patients [14].
In patients with ACS, the role of systemic inflammation
and lymphocyte activation has been previously reported
[34]. In fact, several laboratories demonstrated an increase
in circulating activated T cells and IgM in patients with
unstable angina. Patients with unstable angina have sig-
nificant higher admission percentage of circulating
T-helper (CD41) cells than controls. Activated T lympho-
cytes are also frequently found in peripheral blood of
patients with ACS [9, 26, 28, 30]. A previous report has
shown the usefulness of tailored interventions in ACS
patients, such as intravascular ultrasound with virtual his-
tology technique for preventing periprocedural coronary
microembolization in distal territories after PCI, thus
reinforcing the importance of appropriate diagnosis and
treatment [6, 7]. To this regard, the specificity in treating
inflammatory plaque rupture exerted by statins and anti-
platelet agents is translated in reduction of thrombus bur-
den release [17]. Indeed, our experimental study
demonstrates for the first time that ERK1/2 phosphorylated
levels measured at fluorescent activated cell sorting could
represent an important marker in stratifying risk for acute
patients and no correlation was found with previous phar-
macological therapy; in fact, FACS spectra have been
compared analyzing twice the same patients before anti-
platelet agents administration at the time of first admission
for chest pain and 24 h after clopidogrel intake for treat-
ment of ACS according to current guidelines. No signifi-
cant difference was found in sera 24 h after clopidogrel
administration, since phospho-ERK1/2 levels in UA with-
out clopidogrel versus same patients evaluated 24 h after
clopidogrel administration were 42 ± 9 vs. 39 ± 11
(p = NS). On the other hand, the majority of patients
admitted for an ACS were already under treatment with
different types and doses of statins, hence making the
impact of statins on MAPK difficult to assess. However,
HMG-CoA reductase inhibitors may affect MAPK in acute
coronary syndromes, since our in vitro data from cultured T
lymphocytes demonstrate that statin administration abo-
lished interleukin 2-induced ERK1/2 activation.
It is well known that the mitogen-activated protein
(MAP) kinase family plays a critical role in intracellular
signal transduction and regulation. These proteins form
complex signalling networks that can be induced by a large
array of external stimuli and can achieve highly specific
cellular effects through multitudes of regulatory mecha-
nisms [31]. Inflammation as well as different cellular
stresses activate MAP kinases proteins and specifically the
three different pathways, JNK, ERK1/2 and p38 [10, 22].
The prototypic ERK1/2 pathway is found to be responsive
mainly to stimulation of growth signalling as well as in
response to inflammatory cytokines. JNK and p38 are
collectively called stress-activated MAP kinases because of
their selective response to physical, chemical and different
stressors (such as ultraviolet rays, osmotic shocks, infection
and cytokines).
CADUA
Phospho-ERK1/2
15 m
in
15 h
rs
IL-2
CON
IL-2+Rosu
Phospho-ERK1/2
Total-ERK1/2
Ph
osp
ho
-ER
K1/
2 fl
uo
resc
ence
inte
nsi
ty in
T ly
mp
ho
cyte
s
0
20
40
60
80
UA
UA-FU
2m 6m CAD CAD6mFU
*
UA-FU
6m2mCAD
6mFU
(A)
(B)
(C)
Fig. 6 In vivo and in vitro transient MAP kinases activation in T
lymphocytes. a Representative immunoblot comparing phospho-
ERK1/2 in unstable angina (UA) and in stable coronary artery
disease (CAD), together with respective follow-up evaluations. b The
bar graphs display phospho-ERK1/2 fluorescence intensity in T
lymphocytes of twenty-two patients at the time of presentation for UA
and after 2 and 6 months (40 ± 18; 35 ± 11; 16 ± 8, respectively,
p \ 0.05). No significant changes in levels of phospho-ERK1/2 were
observed in CAD population. c T lymphocytes were harvested
from healthy subjects and cultured in the absence (CON) or with
1,000 UI/ml interleukin-2 (IL-2); ERK 1/2 activation was assessed
after 15 min and 15 h of rosuvastatin (Rosu, 10 lmol) administration
676 Basic Res Cardiol (2011) 106:667–679
123

The evaluation of MAPK in patients with ACS before
and after PCI was not accompanied by a significant change
in phospho-ERK1/2 activity. This finding has been previ-
ously underlined in other studies, which demonstrated that
the liver-X-receptors activation inhibits chemokine-
induced migration of CD4-positive lymphocytes without
abolishing atherogenesis [45].
Recently, increased levels of activated MAPK have
been found in white blood cells isolated from hypertensive
patients with uncontrolled blood pressure values, therefore
corroborating the hypothesis that in several human cardiac
diseases, such as uncontrolled hypertension, MAPK can be
used as early biomarkers [12]. Thus, we set to investigate
whether MAPK proteins are activated within circulating T
lymphocytes in response to acute myocardial ischemia in
patients with ACS.
Phosphorylated ERK1/2 in T lymphocytes was remark-
ably higher in UA, NSTEMI and STEMI compared with
stable CAD and control subjects. On the other hand, phos-
pho-ERK1/2 was not significantly activated in patients with
stable CAD compared to normal healthy controls. More
interestingly, JNK was significantly phosphorylated in T
lymphocytes from patients with ACS associated with an
increase of biomarkers of necrosis, i.e. NSTEMI and
STEMI, whereas phospho-JNK was not significantly acti-
vated in UA when compared to CAD, NSTEMI or STEMI
patients. A mechanistical explanation for differential MAPK
activation might be due to different triggers in the settings of
either unstable angina or myocardial infarction. Some per-
tinent studies already demonstrated that hypoxia, ischemia
and ischemia/reperfusion induce mitogen activated protein
kinase and transcriptional changes in cardiac myocytes.
Cellular stresses, including infarction, activate p38 and
JNK, whereas the activation of ERK by ischemia–reperfu-
sion is still controversial. In the present study, the phos-
phorylation of all MAPK was increased by myocardial
infarction, whereas ERK1/2 was increased in unstable
angina with normal JNK. In this setting, we found that the
temporal window of ERK1/2 and p38MAPK activation was
different from JNK. ERK1/2 and p38MAPK activities
increased more rapidly than that of JNK. Perhaps, JNK is
activated by proinflammatory cytokines and environmental
stress, such as a tumor necrosis factor and ultraviolet irra-
diation, and is not mediated via a Ras-dependent pathway.
In contrast, ERK1/2 is activated by growth factors via a
Ras-dependent signal-transduction pathway. Interestingly,
although the signal pathways of JNK is very similar to
p38MAPK in the majority of in vitro studies, the timing of
JNK activation has been reported to be distinct from
p38MAPK activation in a rat model of coronary artery
ligation. Myocardial infarction causes inflammation in the
infarcted region and this process may induce JNK activa-
tion, hence the assumption can be made that each MAPK
family may play a different role during myocardial
ischemia.
In conclusion, in the present study, the evaluation of
MAPK activation in T lymphocytes from ACS patients was
able to discriminate acute myocardial ischemia in absence
of myocyte necrosis. Indeed, patients with unstable angina
presented with T lymphocytes showing ERK1/2 but not
JNK phosphorylation. On the other hand, STEMI and
NSTEMI patients had T lymphocytes with increased levels
of both phosphorylated forms of ERK1/2 and JNK.
Study limitations
The present study was performed to assess the molecular
activation of T lymphocytes in a relative small selected
population of patients with ACS. Further studies, however,
should be performed in a larger population to establish the
potential role of MAP kinases activation as biomarker for
the diagnosis and perhaps risk stratification of acute coro-
nary syndromes. Additional studies should also be per-
formed to assess the exact time-course of MAPK activation
in T lymphocytes during ACS and its potential role to
direct timely the appropriate therapy.
Acknowledgments This study was supported, in part, by Grants for
Scientific Research from the Ministry of the Education, University and
Research of Italy (PRIN2003065977_002, RBAU018WWP_001), by
Ricerca sanitaria finalizzata 2007—Programma Strategico APICE
‘‘Activity of Platelets after Inhibition and Cardiovascular Events’’, and
by GENECOR, a non-profit organization.
Conflict of interest The authors declare that they have no conflict
of interest.
References
1. Antman EM, Sacks DB, Rifai N, McCabe CH, Cannon CP,
Braunwald E (1998) Time to positivity of a rapid bedside assay
for cardiac-specific troponin T predicts prognosis in acute coro-
nary syndromes: a Thrombolysis in Myocardial Infarction (TIMI)
11A substudy. J Am Coll Cardiol 31:326–330. doi:10.1016/
S0735-1097(97)00485-3
2. Berk BC, Weintraub WS, Alexander RW (1990) Elevation of
C-reactive protein in ‘‘active’’ coronary artery disease. Am J
Cardiol 65:168–172. doi:10.1016/0002-9149(90)90079-G
3. Biasucci LM, Vitelli A, Liuzzo G, Altamura S, Caligiuri G,
Monaco C, Rebuzzi AG, Ciliberto G, Maseri A (1996) Elevated
levels of interleukin-6 in unstable angina. Circulation 94:874–877
4. Biasucci LM, D’Onofrio G, Liuzzo G, Zini G, Monaco C, Ca-
ligiuri G, Tommasi M, Rebuzzi AG, Maseri A (1996) Intracel-
lular neutrophil myeloperoxidase is reduced in unstable angina
and acute myocardial infarction, but its reduction is not related to
ischemia. J Am Coll Cardiol 27:611–616. doi:10.1016/0735-
1097(95)00524-2
5. Bonaca MP, Morrow DA (2008) Defining a role for novel bio-
markers in acute coronary syndromes. Clin Chem 54:1424–1431.
doi:10.1373/clinchem.2008.105387
Basic Res Cardiol (2011) 106:667–679 677
123

6. Bose D, von Birgelen C, Zhou XY, Schmermund A, Philipp S,
Sack S, Konorza T, Mohlenkamp S, Leineweber K, Kleinbongard
P, Wijns W, Heusch G, Erbel R (2008) Impact of atherosclerotic
plaque composition on coronary microembolization during per-
cutaneous coronary interventions. Basic Res Cardiol 103:
587–597. doi:10.1007/s00395-008-0745-9
7. Bose D, Leineweber K, Konorza T, Zahn A, Brocker-Preuss M,
Mann K, Haude M, Erbel R, Heusch G (2007) Release of TNF-
alpha during stent implantation into saphenous vein aortocoro-
nary bypass grafts and its relation to plaque extrusion and
restenosis. Am J Physiol Heart Circ Physiol 292:H2295–H2299.
doi:10.1152/ajpheart.01116.2006
8. Buja LM, Willerson JT (1994) Role of inflammation in coronary
plaque disruption. Circulation 89:503–505
9. Caligiuri G, Liuzzo G, Biasucci LM, Maseri A (1998) Immune
system activation follows inflammation in unstable angina:
pathogenetic implications. J Am Coll Cardiol 32:1295–1304. doi:
10.1016/S0735-1097(98)00410-0
10. Cook SA, Sugden PH, Clerk A (1999) Activation of c-Jun
N-terminal kinases and p38-mitogen-activated protein kinases in
human heart failure secondary to ischaemic heart disease. J Mol
Cell Cardiol 31:1429–1434. doi:10.1006/jmcc.1999.0979
11. Dernbach E, Randriamboavonjy V, Fleming I, Zeiher AM,
Dimmeler S, Urbich C (2008) Impaired interaction of platelets
with endothelial progenitor cells in patients with cardiovascular
risk factors. Basic Res Cardiol 103:572–581. doi:10.1007/
s00395-008-0734-z
12. Esposito G, Perrino C, Schiattarella GG, Belardo L, di Pietro E,
Franzone A, Capretti G, Gargiulo G, Pironti G, Cannavo A,
Sannino A, Izzo R, Chiariello M (2010) Induction of mitogen-
activated protein kinases is proportional to the amount of pressure
overload. Hypertension 55:137–143. doi:10.1161/HYPER
TENSIONAHA.109.135467
13. Friedrich EB, Werner C, Walenta K, Bohm M, Scheller B (2009)
Role of extracellular signal-regulated kinase for endothelial
progenitor cell dysfunction in coronary artery disease. Basic Res
Cardiol 104:613–620. doi:10.1007/s00395-009-0022-6
14. Gasparri C, Curcio A, Torella D, Gaspari M, Celi V, Salituri F,
Boncompagni D, Torella M, Gulletta E, Cuda G, Indolfi C (2010)
Proteomics reveals high levels of vitamin D binding protein in
myocardial infarction. Front Biosci 2:796–804. doi:10.2741/E140
15. Gibler WB, Cannon CP, Blomkalns AL, Char DM, Drew BJ,
Hollander JE, Jaffe AS, Jesse RL, Newby LK, Ohman EM,
Peterson ED, Pollack CV (2005) American Heart Association
Council on Clinical Cardiology (Subcommittee on Acute Cardiac
Care); Council on Cardiovascular Nursing, and Quality of Care
and Outcomes Research Interdisciplinary Working Group; Soci-
ety of Chest Pain Centers. Interdisciplinary Working Group, in
Collaboration With the Society of Chest on Cardiovascular
Nursing, and Quality of Care and Outcomes Research Council on
Clinical Cardiology (Subcommittee on Acute Cardiac Care),
Council Department: A Scientific Statement From the American
Heart Association Angina/Non–ST-Segment Elevation Myocar-
dial Infarction in the Emergency. Circulation 111:2699–2710.
doi:10.1161/01.CIR.0000165556.44271.BE
16. Hayashida K, Kume N, Murase T, Minami M, Nakagawa D,
Inada T, Tanaka M, Ueda A, Kominami G, Kambara H, Kimura
T, Kita T (2005) Serum soluble lectin-like oxidized low-density
lipoprotein receptor-1 levels are elevated in acute coronary syn-
drome a novel marker for early diagnosis. Circulation
112:812–818. doi:10.1161/CIRCULATIONAHA.104.468397
17. Heusch G, Kleinbongard P, Bose D, Levkau B, Haude M, Schulz
R, Erbel R (2009) Coronary microembolization: from bedside to
bench and back to bedside. Circulation 120:1822–1836. doi:
10.1161/CIRCULATIONAHA.109.888784
18. Heusch P, Canton M, Aker S, van de Sand A, Konietzka I, Rassaf
T, Menazza S, Brodde OE, Di Lisa F, Heusch G, Schulz R (2010)
The contribution of reactive oxygen species and p38 mitogen-
activated protein kinase to myofilament oxidation and progres-
sion of heart failure in rabbits. Br J Pharmacol 160:1408–1416.
doi:10.1111/j.1476-5381.2010.00793.x
19. Indolfi C, Avvedimento EV, Rapacciuolo A, Di Lorenzo E, Es-
posito G, Stabile E, Feliciello A, Mele E, Giuliano P, Condorelli
G, Chiariello M (1995) Inhibition of cellular ras prevents smooth
muscle cell proliferation after vascular injury in vivo. Nat Med
6:541–545. doi:10.1038/nm0695-541
20. Indolfi C, Chiariello M, Avvedimento EV (1996) Selective gene
therapy for proliferative disorders: sense and antisense. Nat Med
2:634–635. doi:10.1038/nm0696-634
21. Indolfi C, Avvedimento EV, Di Lorenzo E, Esposito G, Rapac-
ciuolo A, Giuliano P, Grieco D, Cavuto L, Stingone AM, Ciullo I,
Condorelli G, Chiariello M (1997) Activation of cAMP-PKA
signaling in vivo inhibits smooth muscle cell proliferation
induced by vascular injury. Nat Med 7:775–779. doi:10.1038/
nm0797-775
22. Indolfi C, Curcio A, Chiariello M (2003) Simvastatin reduces
neointimal thickening after experimental angioplasty. Circulation
107:e25. doi:10.1161/01.CIR.0000050549.85811.9D
23. Jacquet S, Nishino Y, Kumphune S, Sicard P, Clark JE, Kobay-
ashi KS, Flavell RA, Eickhoff J, Cotten M, Marber MS (2008)
The role of RIP2 in p38 MAPK activation in the stressed heart.
J Biol Chem 283:11964–11971. doi:10.1074/jbc.M707750200
24. Konstandin MH, Aksoy H, Wabnitz GH, Volz C, Erbel C, Kir-
chgessner H, Giannitsis E, Katus HA, Samstag Y, Dengler TJ
(2009) Beta2-integrin activation on T cell subsets is an inde-
pendent prognostic factor in unstable angina pectoris. Basic Res
Cardiol 104:341–351. doi:10.1007/s00395-008-0770-8
25. Kovanen PT, Kaartinen M, Paavonen T (1995) Infiltrates of
activated mast cells at the site of coronary atheromatous erosion
or rupture in myocardial infarction. Circulation 92:1084–1088
26. Libby P (2001) Current concepts of the pathogenesis of the acute
coronary syndromes. Circulation 104:365–372
27. Liuzzo G, Biasucci LM, Gallimore JR, Grillo RL, Rebuzzi AG,
Pepys MB, Maseri A (1994) The prognostic value of C-reactive
protein and serum amyloid a protein in severe unstable angina.
N Engl J Med 331:417–424. doi:10.1056/NEJM19940818
3310701
28. Liuzzo G, Biasucci LM, Trotta G, Brugaletta S, Pinnelli M,
Digianuario G, Rizzello V, Rebuzzi AG, Rumi C, Maseri A, Crea
F (2007) Unusual CD4 ? CD28null T lymphocytes and recur-
rence of acute coronary events. J Am Coll Cardiol 50:1450–1458.
doi:10.1016/j.jacc.2007.06.040
29. Mazzone A, De Servi S, Ricevuti G, Mazzucchelli I, Fossati G,
Pasotti D, Bramucci E, Angoli L, Marsico F, Specchia G (1993)
Increased expression of neutrophil and monocyte adhesion mol-
ecules in unstable coronary artery disease. Circulation 88:358–363
30. Methe H, Brunner S, Wiegand D, Nabauer M, Koglin J, Edelman
ER (2005) Enhanced T-helper-1 lymphocyte activation patterns
in acute coronary syndromes. J Am Coll Cardiol 45:1939–1945.
doi:10.1016/j.jacc.2005.03.040
31. Michel MC, Li Y, Heusch G (2001) Mitogen-activated protein
kinases in the heart. Naunyn Schmiedebergs Arch Pharmacol
363:245–266. doi:10.1007/s002100000363
32. Morrow DA, Braunwald E (2003) Future of biomarkers in acute
coronary syndromes: moving toward a multimarker strategy.
Circulation 108:250–252. doi:10.1161/01.CIR.0000078080.
37974.D2
33. Morrow DA, Cannon CP, Jesse RL, Newby LK, Ravkilde J,
Storrow AB, Wu AH, Christenson RH, Apple FS, Francis G,
Tang W (2007) National Academy of Clinical Biochemistry
678 Basic Res Cardiol (2011) 106:667–679
123

Laboratory Medicine Practice Guidelines: clinical characteristics
and utilization of biochemical markers in acute coronary syn-
dromes. Clin Chem 53:552–574. doi:10.1373/clinchem.2006.
084194
34. Mulvihill N, Foley B, Ghaisas N, Murphy R, Crean P, Walsh M
(2000) Evidence of prolonged inflammation in unstable angina
and non Q-wave myocardial infarction. J Am Coll Cardiol
34:1210–1216. doi:10.1016/S0735-1097(00)00824-X
35. Neri Serneri GG, Prisco D, Martini F, Gori AM, Brunelli T,
Poggesi L, Rostagno C, Gensini GF, Abbate R (1997) Acute
T-cell activation is detectable in unstable angina. Circulation
95:1806–1812
36. Ohman EM, Armstrong PW, Christenson RH, Granger CB, Katus
HA, Hamm CW, O’Hanesian MA, Wagner GS, Kleiman NS,
Harrell FE Jr, Califf RM, Topol EJ (1996) Cardiac troponin T
levels for risk stratification in acute myocardial ischemia.
GUSTO IIA Investigators. N Engl J Med 335:1333–1341. doi:
10.1056/NEJM199610313351801
37. Puleo PR, Meyer D, Wathen C, Tawa CB, Wheeler S, Hamburg
RJ, Ali N, Obermueller SD, Triana FJ, Zimmerman JL, Perryman
MB, Roberts R (1994) Use of a rapid assay of subforms of cre-
atine kinase-MB to diagnose or rule out acute myocardial
infarction. N Engl J Med 331:561–566. doi:10.1056/NEJM19940
9013310901
38. Quilici J, Banzet N, Paule P, Meynard JB, Mutin M, Bonnet JL,
Ambrosi P, Sampol J, Dignat-George F (2004) Circulating
endothelial cell count as a diagnostic marker for non–st-elevation
acute coronary syndromes. Circulation 110:1586–1591. doi:
10.1161/01.CIR.0000142295.85740.98
39. Sabatine MS, Morrow DA, de Lemos JA, Gibson CM, Murphy
SA, Rifai N, McCabe C, Antman EM, Cannon CP, Braunwald E
(2002) Multimarker approach to risk stratification in non-ST
elevation acute coronary syndromes simultaneous assessment of
Troponin I, C-reactive protein, and B-type natriuretic peptide.
Circulation 105:1760–1763. doi:10.1161/01.CIR.0000015464.
18023.0A
40. Schulz R, Gres P, Skyschally A, Duschin A, Belosjorow S,
Konietzka I, Heusch G (2003) Ischemic preconditioning pre-
serves connexin 43 phosphorylation during sustained ischemia in
pig hearts in vivo. FASEB J 10:1355–1357. doi:10.1096/
fj.02-0975fje
41. Schulz R, Belosjorow S, Gres P, Jansen J, Michel MC, Heusch G
(2002) p38 MAP kinase is a mediator of ischemic precondition-
ing in pigs. Cardiovasc Res 55:690–700. doi:10.1016/S0008-
6363(02)00319-X
42. Seeger FH, Sedding D, Langheinrich AC, Haendeler J, Zeiher
AM, Dimmeler S (2010) Inhibition of the p38 MAP kinase in
vivo improves number and functional activity of vasculogenic
cells and reduces atherosclerotic disease progression. Basic Res
Cardiol 105:389–397. doi:10.1007/s00395-009-0072-9
43. Sun C, Liang C, Ren Y, Zhen Y, He Z, Wang H, Tan H, Pan X,
Wu Z (2009) Advanced glycation end products depress function
of endothelial progenitor cells via p38 and ERK 1/2 mitogen-
activated protein kinase pathways. Basic Res Cardiol 104:42–49.
doi:10.1007/s00395-008-0738-8
44. Tamer L, Ercan B, Camsari A, Yildirim H, Cicek D, Sucu N,
Ates NA, Atik U (2004) Glutathione S-transferase gene poly-
morphism as a susceptibility factor in smoking-related coronary
artery disease. Basic Res Cardiol 99:223–229. doi:10.1007/
s00395-004-0465-8
45. Walcher D, Vasic D, Heinz P, Bach H, Durst R, Hausauer A,
Hombach V, Marx N (2010) LXR activation inhibits chemokine-
induced CD4-positive lymphocyte migration. Basic Res Cardiol
105:487–494. doi:10.1007/s00395-010-0092-5
46. Wang Y (2007) Mitogen-activated protein kinases in heart
development and diseases. Circulation 116:1413–1423. doi:
10.1161/CIRCULATIONAHA.106.679589
Basic Res Cardiol (2011) 106:667–679 679
123









![Syndromes drépanocytaires atypiques : à propos de deux cas [Atypical sickle cell syndromes: A report on two cases]](https://static.fdokumen.com/doc/165x107/6319e3d265e4a6af371005c0/syndromes-drepanocytaires-atypiques-a-propos-de-deux-cas-atypical-sickle-cell.jpg)










