
The contribution of coral reef-derived dimethyl sulfide to ...
Upload
independentCategory
view
5download
0
©2004 FASEB The FASEB Journal express article10.1096/fj.04-2279fje. Published online September 15, 2004.
Hydrogen sulfide-induced apoptosis of human aorta smooth muscle cells via the activation of mitogen-activated protein kinases and caspase-3 Guangdong Yang, Xianfeng Sun, and Rui Wang
Department of Physiology, College of Medicine, The Cardiovascular Research Group, University of Saskatchewan, Saskatoon, SK, Canada S7N 5E5
Corresponding author: Rui Wang, Department of Physiology, University of Saskatchewan, 107 Wiggins Road, Saskatoon, SK, Canada S7N 5E5. E-mail: [email protected]
ABSTRACT
The endogenous production of hydrogen sulfide (H2S) and its physiological functions, including membrane hyperpolarization and smooth muscle cell relaxation, position this gas well in the family of gasotransmitters together with nitric oxide (NO) and carbon monoxide (CO). In this study, we demonstrate that H2S at physiologically relevant concentrations induced apoptosis of human aorta smooth muscle cells (HASMCs). Exposure of HASMCs to H2S did not induce necrosis as verified with Trypan blue exclusion and LDH release analysis. After inhibiting endogenous H2S production, exogenous H2S induced much more significant apoptosis, which was not altered by the presence of albumin or glutathione. H2S treatment increased the activities of ERK and p38 mitogen-activated protein kinase (MAPK), but not c-Jun N-terminal kinase activity. Suppression of extracellular signal-regulated kinase (ERK) activity, but not of p38 activity, inhibited the H2S-induced apoptosis of HASMCs. The activation of ERK by H2S in HASMCs was accompanied by increased caspase-3 activity. Inhibition of caspase-3 by AC-DEVD-CHO attenuated the H2S-induced cell apoptosis. Inhibition of ERK by U0126 decreased caspase-3 activity, whereas AC-DEVD-CHO did not alter ERK activity. In conclusion, exogenous H2S induces apoptosis of HASMCs, which is significantly affected by the endogenous H2S level. Of the three investigated MAPKs, only ERK played an active role in mediating H2S-induced apoptosis of HASMCs by activating caspase-3. These findings may help reveal novel mechanisms for many diseases linked to H2S-related abnormal cellular proliferation and apoptosis.
Key words: blood vessel • ERK • gasotransmitter • p38 • remodeling
ydrogen sulfide (H2S) has been traditionally known as a toxic gas with the smell of rotten eggs. Recently, the physiological importance of H2S has gained increasing recognition (1–5). H2S is endogenously generated from L-cysteine by pyridoxal-5′-
phosphate-dependent enzymes, including cystathionine β-synthase (CBS) and/or cystathionine γ-lyase (CSE) in mammalian cells (6). In vascular smooth muscle cells (SMCs), only the expression of CSE has been detected (3). Production of H2S in different organs and tissues as well as circulatory concentration of H2S have been elucidated (2, 3). Physiological concentrations of H2S in plasma have been reported between 45 µM (3) and 300 µM (7, 8). The
H
Page 1 of 21(page number not for citation purposes)

production of H2S is up-regulated by nitric oxide (NO) (3). H2S at physiologically relevant concentrations hyperpolarizes cell membranes, relaxes SMCs, and modulates neuronal excitability (1, 3, 4). The endogenous metabolism and physiological functions of H2S position this gas well in the novel family of gasotransmitters together with NO and carbon monoxide (CO) (1).
Mounting evidence indicates that both NO and CO are important modulators of cellular apoptosis (9–11). NO and CO can promote or inhibit apoptosis depending on cell types (12–14). The antiproliferative effect of H2S on T lymphocytes has been investigated in relation to the therapeutic mechanism of sulfurous water bath (15). However, little is known about the physiological effects of H2S on cell proliferation and growth in general and on apoptosis of vascular SMCs in particular.
Apoptosis, a genetically programmed process to eliminate unwanted cells, is manifested with characteristic morphologic changes, such as cellular shrinkage, nuclear condensation, and chromatin fragmentation (16). Apoptosis of vascular SMCs is involved in angiogenesis and several vascular pathologies, including atherosclerosis, intimal hyperplasia following vascular injury, and vascular remodeling (17, 18). Understanding the mechanism of apoptosis has important implications in prevention and treatment of many vascular diseases. Mitogen-activated protein kinase (MAPK) superfamily is significantly involved in cellular survival and apoptotic responses. Mammalian MAPK family consists of three major members, including extracelluar signal-regulated kinase (ERK); c-Jun N-terminal kinase (JNK), also called stress-activated protein kinase); and p38 MAPK (19, 20). Each of these MAPKs has a unique role in the regulation of intracellular metabolism and gene expression related to growth and development, apoptosis, and cellular responses to external stresses (20). Phosphorylation of MAPK results in protein conformational change and an increase in kinase activity (16). Once phosphorylated, MAPK activates a number of cytosolic components through its proline directed serine/threonine activity. The activated MAPK can also translocate to the nucleus where it phosphorylates and activates a number of transcription factors involved in immediate early gene expression (21, 22). The activated caspases, comprising a family of cysteine proteases (23, 24), are also implicated in apoptosis by specifically cleaving several cellular substrates with an aspartic acid in the P1 position (25). For example, caspase-3 is activated by various apoptosis inducers and is seemingly essential for the execution and completion of apoptosis in many types of cells (26, 27). Multiple lines of evidence also suggest the involvement of Bcl-2 family members in cell apoptosis (13, 28), which can either inhibit or promote apoptosis. The interactions of H2S with these apoptosis-related kinases and enzymes have not been reported.
The present study sought to elicit the physiological role of H2S in apoptotic process of vascular SMCs and to examine the underlying cellular signaling transduction pathways. Using multifaceted approaches, we provide evidence for the first time that H2S induces apoptosis of human vascular SMCs, which may be mediated by increased ERK activity. The activation of caspase-3 is the key link between the increased ERK activity and H2S-induced cellular apoptosis. An important physiological role of H2S as an apoptotic modulator and the underlying mechanisms are thus elucidated.
Page 2 of 21(page number not for citation purposes)

MATERIALS AND METHODS
Cell culture
Human aorta smooth muscle cells (HASMCs) (ATCC, CRL-1999) were cultured with HAM’s F-12K medium supplemented with 10% fetal bovine serum (FBS) (Gibco-BRL, Life Technologies, Gaithersburg, MD), 100 U/ml penicillin and 100 µg/ml streptomycin, 30 mg/l ECGS, 10 mg/l insulin, 10 mg/l transferrin, 50 mg/l ascorbic acid, 10 mM HEPES, 10 mM TES, and 10 µg/l sodium selenite at 37°C in a humidified chamber containing 5% CO2. The experiments were performed when the cells reached 70–80% confluence between passages 16 and 22. In all studies, cells were incubated in the serum-free medium for 12 h. In certain selective experiments, cells were subsequently incubated in 10% serum.
Necrosis assays
Cell viability was determined by exclusion of trypan blue (29). At the indicated times after treatment with H2S, cells were detached with trypsin, pelleted, and resuspended in culture medium. After staining with trypan blue, viable cells in five random fields of view were counted and quantified with a hemocytometer. The percentage of trypan blue-exclusive viable cells was determined as a percentage of the total number of cells. The extent of necrosis was also evaluated using lactate dehydrogenase (LDH) release in culture medium (30). LDH activity (absorbance at 340 nm) of treated cells was determined using a Multiskan spectrum microplate spectrophotometer (Thermo Labsystems, Boston, MA). Results were presented as the percentage of maximum LDH release, which was determined by complete lysis of cells.
Hoechst 33258 staining
HASMCs (1×105) were plated onto 18-mm2 coverslips in 35-mm dishes and cultured with complete medium. After they were treated with H2S, the cells were fixed with 4% formaldehyde for 20 min at room temperature and were then washed with PBS. Cold methanol was added for another 20 min at room temperature followed by washes with PBS three times. The membrane-permeable fluorescent dye Hoechst 33258 (2 µg/ml), which binds to chromatin of cells, was added to the fixed cells, and the cells were examined by an inverted Olympus IX70 microscope (Tokyo, Japan). Apoptotic cells were identified by condensation and fragmentation of nuclei. Percentage of apoptotic cells in all cells was calculated. For each experiment, nuclei from 10 random fields of each coverslip were examined at ×200 magnification.
DNA fragmentation
Internucleosomal DNA fragmentation was examined according to Peyot et al. (31). The cells were lysed in a buffer solution (10 mM Tris-HCl with pH 8.0, 5 mM EDTA, 100 mM NaCl, 0.5% SDS, and 10 µg/ml proteinase K) at 37°C for 3 h. NaCl was then added to the reaction mixture to a final concentration of 1 M, and the reaction was continued for 1 h at 4°C with occasional shaking. After centrifugation at 12,000g, DNA in the supernatant was extracted with an equal volume of 25:24:1 phenol/chloroform/isoamyl alcohol and precipitated overnight with isopropanol at –20°C. The DNA pellet was obtained after centrifugation, resuspended in water, and digested with 1 mg/ml RNase for 30 min at 37°C. DNA electrophoresis was performed on
Page 3 of 21(page number not for citation purposes)

1.8% agarose gel containing 0.5 µg/ml ethidium bromide, and DNA fragments were visualized under UV light.
TUNEL assay
Terminal deoxyribonucelotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) was carried out to detect DNA fragmentation using an in situ cell death detection kit, according to the manufacturer (Roche Molecular Biochemicals, Laval, Quebec, Canada). Briefly, after different treatments, 1 × 105 cells grown on coverslips were rinsed three times with PBS and fixed by adding freshly prepared 4% paraformaldehyde in PBS for 1 h at 15–25°C. Afterward, they were incubated with blocking solution (3% H2O2 in methanol) for 10 min at 15–25°C and were then permeabilized in freshly prepared 0.1% Triton X-100/0.1% sodium citrate for 2 min on ice. The permeablized cells were then incubated with TUNEL reaction mixture at 37°C in a humidified atmosphere in the dark for 1 h. Converter-POD and DAB substrate were then added to incubate cells in a humidified chamber at 37°C for 30 min and at 15–25°C for 10 min, respectively. Between each step, cells were rinsed with PBS. A reaction blank control containing cells and the fluorescence dye, but no TdT enzyme, was carried out under the same conditions. Apoptotic cells were observed and analyzed under an inverted Olympus IX70 microscope.
Caspase-3 activity assay
Caspase-3 activity was quantified using Fluorometric Immunosorbent Enzyme Assay system, according to manufacturer’s instructions (Roche). This assay is based on the generation of free AC-DEVD-AFC fluorescence when the provided substrate is cleaved by caspase-3. Briefly, the cultured HASMCs (3×106) after different treatments were harvested in lysis buffer (×1 DTT), and cell extracts were centrifuged to eliminate cellular debris. Aliquots (100 µl) of the cell extracts were incubated at 37°C for 1 h in the presence of the fluorescence substrate. Free AC-DEVD-AFC is determined fluorometrically with excitation at 355 nm and emission at 527 nm. The developed fluorochrome is proportional to the concentration of activated caspase-3.
Western immunoblotting
Cultured cells were harvested, washed twice in ice-cold PBS, and lysed in a lysis buffer (0.5 M EDTA; 1 M Tris-Cl, pH 7.4; 0.3 M sucrose; 1 µg/ml antipain hydrochloride; 1 mM benzamidine hydrochloride hydrate; 1 µg/ml leupeptin hemisulfate; 1 mM 1,10-phenanthroline monohydrate; 1 µM pepstatin A; 0.1 mM plenylmethylsulfonyl fluoride; and 1 mM iodoacetamide) using a polytron homogenizer. The supernatants containing crude cellular proteins were collected by centrifugation at 14,000g for 15 min at 4°C and used for Western blot studies. The supernatant samples were resolved on a 10% SDS-PAGE gel and transferred onto the PVDC nitrocellulose membrane. The membrane was blocked with 3% nonfat dry milk solution in PBS at room temperature for 2 h and rinsed three times with PBS before incubating with primary antibody (diluted 1:1000 in blocking buffer for phosphorylated or total ERK, p38 MAPK, or JNK; diluted 1:500 in blocking buffer for Bcl-2 and Bax; and diluted 1:5000 in blocking buffer for β-actin). After the incubation, the membrane was washed three times with the blocking solution for 5 min and incubated with the horseradish (HRP)-conjugated secondary antibody (1:5000) for 1 h at room temperature on a shaker. Finally, the membrane was washed with blocking solution and PBS consecutively three times. The immunoreactions were visualized by ECL and exposed to X-
Page 4 of 21(page number not for citation purposes)

ray film (X-omat Blue XB-1, Kodak, Rochester, NY). Phosphorylated ERK, p38 MAPK, and JNK were normalized to the total amount of ERK, p38 MAPK, and JNK detected in the same membrane. Membranes were stripped by incubating in a buffer containing 100 mM β-mercaptoethanol, 2% SDS, and 62.5 mM Tris-HCl (pH 6.8).
Reagents and chemicals
H2S stock solution was freshly prepared by directly bubbling distilled water with pure H2S gas (Praxair, Danbury, CT) to make the saturated H2S solution (0.09 M at 30°C) (3–5). H2S stock solution was diluted to different concentrations into cell culture medium, and the pH of medium was adjusted to 7.4.
All chemicals were from Sigma (St. Louis, MO) or New England Biolabs (Beverly, MA). The caspase-3 inhibitor AC-DEVD-CHO was obtained from BioSource International (Camarillo, CA). Different specific antibodies were from New England Biolabs or Trevigen (Gaithersburg, MD). HRP-conjugated goat anti-rabbit IgG antibody was from Bio-Rad (Hercules, CA).
Statistical analysis
All data are expressed as the mean ± SE and represent at least three independent experiments. Statistical comparisons were made using Student’s t test or one-way ANOVA followed by a post hoc analysis (Tukey test) where applicable. Significance level was set at P < 0.05.
RESULTS
H2S-induced apoptosis of HASMCs
Using the trypan blue exclusion test and LDH release assay, we found that H2S treatment did not induce significant cell necrotic death (Fig. 1). Whether H2S induced HASMC apoptosis was subsequently examined. The nuclei of untreated control cells were uniformly stained by Hoechst 33258, and the cells exhibited normal morphology (Fig. 2A). When cells were incubated with 500 µM H2S, the number of condensed apoptotic nuclei increased drastically (Fig. 2C, 2D), and the cells showed the morphological changes typical of apoptosis (Fig. 2B). Furthermore, after they were incubated with various concentrations of H2S, the cells were processed for the TUNEL assay. The number of TUNEL-positive cells increased after exposure to H2S (Fig. 2E, 2F). H2S-induced apoptosis in HASMCs was concentration-dependent, with significant apoptosis detected at concentrations of 200 µM and higher (Fig. 3A). The kinetics of H2S-induced apoptosis was examined (Fig. 3B). Morphological characteristics of apoptosis were apparent within 2 h of H2S application. A higher incidence of apoptosis (10.4±0.6%) was observed when the cells were incubated with 200 µM H2S for 12 h.
Induction of HASMC apoptosis by H2S was further confirmed by internucleosomal DNA fragmentation. In the presence of 200 µM or 500 µM H2S for 12 h, oligonucleosomal DNA fragments formed a ladder, which became visible with 20 and 50 µM H2S and was very evident with 200 and 500 µM H2S (Fig. 3C).
Page 5 of 21(page number not for citation purposes)

H2S-activated ERK and p38 MAPK signaling pathways
To determine whether MAPKs were phosphorylated and activated by H2S in HASMCs, lysates obtained at various times from H2S-treated cells were subjected to Western blot analysis using anti-phospho-MAPK antibodies. The same blots were afterward stripped and reprobed with an antibody that recognizes both phosphorylated and unphosphorylated forms of MAPK. Treatment with H2S resulted in strong activation of ERK and p38 MAPK, but not of JNK (Fig. 4A). ERK activation appeared during the first 15 min of H2S treatment and peaked at 2 h followed by a slow decline. A similar but delayed stimulatory effect of H2S was observed on p38 MAPK. H2S maximally activated p38 MAPK within 2 h of H2S application. JNK activity, however, was constant during the 12 h period of H2S treatment. The total amount of MAPK protein remained unchanged with H2S stimulation. The activation of ERK and p38 MAPK by H2S in HASMCs was also concentration-dependent. At a concentration as low as 20 µM, H2S activated ERK and p38 MAPK (Fig. 4B). U0126 (a selective inhibitor of the MEK/ERK signaling pathway) and SB203580 (a p38 MAPK inhibitor) were used in the following experiments. As shown in Figure 5A, 20 µM U0126 suppressed H2S-dependent increases in ERK activity. SB203580 at 40 µM completely inhibit H2S-induced phosphorylation of p38 MAPK.
Treatment of HASMCs with different concentrations of H2S for 12 h did not significantly alter the expression of Bax protein (Fig. 5B). Additionally, we observed no difference in the expression of Bcl-2 protein between H2S-treated and nontreated HASMCs (not shown).
Caspase-3 activation in H2S-induced apoptosis
Caspase-3 is an effector caspase and one of the main enzymes involved in the apoptotic process. H2S induced a dramatic increase in caspase-3 activity in a concentration-dependent manner (Fig. 6A). Addition of AC-DEVD-CHO (10 µM), a specific caspase-3 inhibitor, completely inhibited the activation of caspase-3 by H2S in HASMCs (Fig. 6B). H2S-induced caspase-3 activation was partly and significantly decreased by U0126 (20 µM), but not by SB203580 (Fig. 6B). Pretreatment of HASMCs with AC-DEVD-CHO (10 µM) for 1 h did not alter the effects of H2S on the activation of ERK and p38 MAPK (Fig. 7A). Taken together, these results demonstrate that H2S first activated ERK, and the latter stimulated caspase-3 in HASMCs.
Blockade of H2S-induced apoptosis by inhibiting ERK and caspase-3 activation
Inhibition of caspase-3 with AC-DEVD-CHO (10 µM) alone had no effect on cell apoptosis. This treatment, however, significantly reduced the H2S-induced apoptosis of HASMCs by 57.0±0.3% (P<0.05). Application of U0126 (20 µM) also inhibited the H2S-induced apoptosis of HASMCs by 52.1±0.3% (P<0.05) (Fig. 7B). Inhibiting p38 MAPK activation with SB203580 (40 µM), on the other hand, did not alter the apoptotic effect of H2S (Fig. 7B). These observations were in agreement with the inhibitory effects of U0126 and AC-DEVD-CHO on caspase-3 activation shown in Fig. 6B.
Endogenous H2S level modulated exogenous H2S-induced apoptosis of HASMCs
To determine whether apoptosis of HASMCs induced by exogenously applied H2S was modulated by the endogenous levels of H2S, we first treated HASMCs with DL-propargylglycine
Page 6 of 21(page number not for citation purposes)

(PPG) to inhibit the activity of CSE (3–5). PPG treatment alone (10–20 mM) did not induce any apoptotic changes. Interestingly, a 1 h pretreatment with PPG significantly enhanced the apoptotic effect of exogenously applied H2S. H2S at 50 µM started to induce apoptosis of HASMCs in the presence of PPG, and this effect of H2S became significant at 100 µM (Fig. 8A). Exogenous H2S at 100 µM induced 5.2 ± 0.7% apoptosis of HASMCs in the absence of PPG treatment (P>0.05), whereas after PPG treatment exogenous H2S at 100 µM induced a significant 11.7 ± 0.7% apoptosis (P<0.05). Furthermore, the H2S-induced apoptosis of HASMCs was not altered by either albumin or glutathoine (GSH) at millimolar concentrations (Fig. 8B, 8C).
DISCUSSION
H2S can be generated in blood vessels in a reaction catalyzed mainly by CSE. Abnormal metabolism and functions of endogenous H2S may impact on vascular contractile status and structural remodeling of blood vessel walls under different pathophysiological conditions (32). Proliferation and apoptosis of vascular SMCs are important cellular events of vascular remodeling. Whether H2S plays a physiologically important role in the modulation of SMC proliferation and apoptosis has been unknown.
HASMCs have been used extensively in the investigation of the regulation of cell proliferation, differentiation, and apoptosis as well as the involved signal transduction pathways (31–33). In this study, for the first time, we describe the novel physiological effect of H2S on apoptosis of human vascular SMCs. The four most significant and novel findings follow. First, we demonstrated that H2S induced apoptosis of HASMCs at concentrations ≥200 µM. It has been demonstrated that physiological concentrations of H2S in plasma are between 45 (3) and 300 µM (7, 8). Therefore, the pro-apoptotic effect of H2S reported in this study is physiologically relevant. Since the pH of H2S-containing culture media has been tightly controlled at 7.4, exogenous H2S-induced apoptosis of HASMCs could not be ascribed to the nonspecific effects such as pH change. Second, after inhibiting endogenous H2S production, exogenous H2S induced even more significant apoptosis, which was not altered by the presence of albumin or glutathione. Therefore, the endogenous H2S level is critical for the apoptotic process of SMCs. A desensitization mechanism of apoptotic signaling system resulting from the endogenous basal level of H2S may exist. By minimizing the endogenous level of H2S, one may unmask the real sensitivity of HASMCs to H2S stimulation. Third, we presented evidence that the pro-apoptotic effect of H2S was mediated by the activation of ERK, but not that of p38 MAPK or JNK. Thus, this study reveals the novel mechanism underlying the vascular effects of a novel physiological endogenous gas. Fourth, we showed that the activation of ERK by H2S was accompanied by increased caspase-3 activity. Inhibition of caspase-3 attenuated the H2S-induced cell apoptosis. This discovery further elucidates the downstream targets of H2S-ERK interaction.
The MAPK family represents important signal transduction machinery and occupies a central position in cell growth, differentiation, and programmed cell death (20). Different MAPKs are activated by different stimuli and target at different downstream molecules and, therefore, perform different functions (21). In the present study, we provided evidence that activation of ERK is important for the H2S-induced apoptosis of HASMCs. Both ERK and p38 MAPK were activated at an early stage of H2S incubation, whereas the activity of JNK had little change during the 12 h incubation with H2S. Down-regulation of ERK activity inhibited H2S-induced apoptosis (Fig. 7). These findings are in line with a previous report that ERK and p38 MAPKs
Page 7 of 21(page number not for citation purposes)

were also involved in H2S-induced cell cycle entry of nontransformed intestinal epithelial cells (IEC-18) (34). However, inhibition of p38 kinases did not alter the apoptotic sensitivity of HASMCs to H2S. Thus, of the three MAPKs only ERK is involved in the apoptosis of H2S-treated HASMCs. The protective effect of ERK inhibitor U0126 against H2S suggests that ERK plays a major role in the induction of apoptosis evoked by H2S. Involvement of ERK in the process of cell apoptosis has been shown in many cell types (35–39). Given that U0126 also inhibits MEK, a tyrosine/threonine kinase that phosphorylates and activates ERK, it is possible that MEK and further upstream molecules such as Raf and Ras may be subjected to H2S modulation.
Some apoptosis inducers have been shown to activate caspase-3 (40, 41), which is an important effector caspase. Caspase-3 can modulate MAPK activation in TNF-α- or Fas-induced apoptosis (42), and caspase inhibitors attenuate MAPK activation (43), indicating a close causative relationship between MAPK and caspase activation. Here, we report that activation of ERK in H2S-treated HASMCs was accompanied by caspase-3 activation. Our observations that the caspase-3 inhibitor, AC-DEVD-CHO, partly blocked H2S-induced programmed cell death, but not ERK activation (Fig. 7), suggest that ERK activity may precede the activation of caspase-3 in H2S-induced cell death. This hypothesis is rationalized since the inhibition of ERK leads to a decreased caspase-3 activity. The occurrences of apoptosis accompanied by the activations of ERK and caspase-3 have been reported (44). Conversely, caspase-3 and apoptosis were inhibited by the activation of the ERK pathway in hippocampal HN2-5 cells (45). Further experiments are needed to investigate the mechanisms by which ERK activates caspase-3 in the process of apoptosis of HASMCs.
In conclusion, our study for the first time demonstrates that endogenous H2S is not only a vasorelaxant or a messenger in neuronal systems (1, 3, 5), but is also an important endogenous modulator of cellular apoptosis via the activation of the MAPK pathway. It is hypothesized that H2S may first activate ERK through various mechanisms. The phosphorylated and activated ERK then transduces the apoptotic signal to its downstream enzyme cascades, eventually activating caspase-3 (Fig. 9). The identification of the missing links in this cascade will lead to a refined understanding of the cellular and molecular mechanisms underlying the H2S-induced cellular apoptosis. These findings will help advance our understanding of H2S biology and physiology as well as novel mechanisms for many diseases linked to H2S-related abnormal cellular proliferation and apoptosis.
ACKNOWLEDGMENTS
This study has been supported by Natural Sciences and Engineering Research Council of Canada. G. Yang and X. Sun have been supported by postdoctoral fellowship awards of Saskatchewan Health Research Foundation, Canada. R. Wang has been supported by an Investigator Award of Canadian Institutes of Health Research.
REFERENCES
1. Wang, R. (2002) Two’s company, three’s a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J. 16, 1792–1798
Page 8 of 21(page number not for citation purposes)

2. Zhao, W., Ndisang, J. F., and Wang, R. (2003) Modulation of endogenous production production of H2S in rat tissues. Can. J. Physiol. Pharmacol. 81, 848–853
3. Zhao, W., Zhang, J., Lu, Y., and Wang, R. (2001) The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 20, 6008–6016
4. Wang, R. (2003) The gasotransmitter role of hydrogen sulphide. Antioxid. Redox Signal. 5, 493–501
5. Zhao, W., and Wang, R. (2002) H2S-induced vasorelaxant and the underlying cellular and molecular mechanisms. Am. J. Physiol. Heart Circ. Physiol. 283, 474–480
6. Levonen, A. L., Lapatto, R., Saksela, M., and Raivio, K. O. (2000) Human cystathionine gamma-lyase: developmental and in vitro expression of two isoforms. Biochem. J. 347, 291–295
7. Zhang, Q., Du, J., Zhou, W., Yan, H., Tang, C., and Zhang, C. (2004) Impact of hydrogen sulfide on carbon monoxide/heme oxygenase pathway in the pathogenesis of hypoxic pulmonary hypertension. Biochem. Biophys. Res. Commun. 317, 30–37
8. Zhong, C., Du, J., Bu, D., Yan, H., Tang, X., and Tang, C. (2003) The regulatory effect of hydrogen sulfide on hypoxic pulmonary hypertension in rats. Biochem. Biophys. Res. Commun. 302, 810–816
9. Kim, Y. M., Bombeek, C. A., and Billiar, T. R. (1999) Nitric Oxide as a biofunctional regulator of apoptosis. Circ. Res. 84, 253–256
10. Brouard, S., Otterbein, L. E., Anrather, J., Tobiasch, E., Bach, F. H., Choi, A. M., and Soares, M. P. (2000) Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J. Exp. Med. 192, 1015–1026
11. Otterbein, L. E., Bach, F. H., Alam, J., Soares, M., Tao, L. H., Wysk, M., Davis, R. J., Flavell, R. A., and Choi, A. M. (2000) Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med. 6, 422–428
12. Peyton, K. J., Reyna, S. V., Chapman, G. B., Ensenat, D., Liu, X. M., Wang, H., Schafer, A. I., and Durante, W. (2002) Hemeoxygenase-1–derived carbon monoxide is an autocrine inhibitor of vascular smooth muscle cell growth. Blood 99, 4443–4448
13. Kim, Y. M., Kim, T. H., Seol, D. W., Talanian, R. V., and Billiar, T. R. (1998) Nitric oxide suppression of apoptosis occurs in association with an inhibition of Bcl-2 cleavage and cytochrome c release. J. Biol. Chem. 273, 31437–31441
14. Chiche, J. D., Schlutsmeyer, S. M., Bloch, D. B., de la Monte, S. M., Roberts, J. D., Jr., Filippov, G., Janssens, S. P., Rosenzweig, A., and Bloch, K. D. (1998) Adenovirus-mediated gene transfer of cGMP-dependent protein kinase increases the sensitivity of cultured vascular smooth muscle cells to the antiproliferative and pro-apoptotic effects of nitric oxide/cGMP. J. Biol. Chem. 273, 34263–34271
Page 9 of 21(page number not for citation purposes)

15. Valitutti, S., Castellino, F., and Musiani, P. (1990) Effect of sulfurous (thermal) water on T lymphocyte proliferative response. Ann. Allergy 65, 463–468
16. Hengartner, M. O. (2000) The biochemistry of apoptosis. Nature 407, 770–776
17. Gibbons, G. H., and Dzau, V. J. (1994) The emerging concept of vascular remodelling. N. Engl. J. Med. 330, 1431–1438
18. Bochaton-Piallat, M. L., Gabbiani, F., Redard, M., Desmouliere, A., and Gabbiani, G. (1995) Apoptosis participates in cellularity regulation during rat aortic intimal thickening. Am. J. Pathol. 146, 1059–1064
19. Xia, Z., Dickens, M., Raingeaud, J., Davis, R. J., and Greenberg, M. E. (1995) Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270, 1326–1331
20. Hershenson, M. B., Naureckas, E. T., and Li, J. (1997) Mitogen-activated signaling in cultured airway smooth muscle cells. Can. J. Physiol. Pharmacol. 75, 898–910
21. Schaeffer, H. J., and Weber, M. J. (1999) Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol. Cell. Biol. 19, 2435–2444
22. Chen, Y. R., and Tan, Y. H. (2000) The c-Jun N-terminal kinase pathway and apoptotic signaling (review). Int. J. Oncol. 16, 651–662
23. Hatai, T., Matsuzawa, A., Inoshita, S., Mochida, Y., Kuroda, T., Sakamaki, K., Kuida, K., Yonehara, S., Ichijo, H., and Takeda, K. (2000) Execution of apoptosis signal-regulating kinase 1 (ASK1)-induced apoptosis by the mitochondria-dependent caspase activation. J. Biol. Chem. 275, 26576–26581
24. Enomoto, A., Suzuki, N., Morita, A., Ito, M., Liu, C. Q., Matsumoto, Y., Yoshioka, K., Shiba, T., and Hosoi, Y. (2003) Caspase-mediated cleavage of JNK during stress-induced apoptosis. Biochem. Biophys. Res. Commun. 306, 837–842
25. Alnemri, E. S., Livingston, D. J., Nicholson, D. W., Salvesen, G., Thornberry, N. A., Wong, W. W., and Yuan, J. (1996) Human ICE/CED-3 protease nomenclature. Cell 87, 171–175
26. Varghese, J., Khandre, N. S., and Sarin, A. (2003) Caspase-3 activation is an early event and initiates apoptotic damage in a human leukemia cell line. Apoptosis 8, 363–370
27. Wang, X., Martindale, J. L., and Holbrook, N. J. (2000) Requirement for ERK activation in cisplatin-induced apoptosis. J. Biol. Chem. 275, 39435–39443
28. Yang, J., Liu, X., Bhalla, K., Kim, C. N., Ibrado, A. M., Cai, J., Peng, T., Jones, D. P., and Wang, X. (1997) Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275, 1129–1132
Page 10 of 21(page number not for citation purposes)

29. Tamango, E., Aragno, M., Parola, M., Parola, S., Brignardello, E., Boccuzzi, G., and Danni, O. (2000) NT2 neurons, a classical model for Alzheimer's disease, are highly susceptible to oxidative stress. Neuroreport 11, 1865–1869
30. Decker, T., and Lohmann-Matthes, M. L. (1988) A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J. Immunol. Methods 115, 61–69
31. Peyot, M. L., Gadeau, A. P., Dandre, F., Belloc, I., Dupuch, F., and Desgranges, C. (2000) Extracellular adenosine induces apoptosis of human arterial smooth muscle cells via A(2b)-purinoceptor. Circ. Res. 86, 76–85
32. Negre-Aminou, P., van Leeuwen, R. E., van Thiel, G. C., van den IJssel, P., de Jong, W. W., Quinlan, R. A., and Cohe, L. H. (2002) Differential effect of simvastatin on activation of Rac(1) vs. activation of the heat shock protein 27-mediated pathway upon oxidative stress, in human smooth muscle cells. Biochem. Pharmacol. 64, 1483–1491
33. Schmid, F. X., Bielenberg, K., Schneider, A., Haussler, A., Keyser, A., and Birnbaum, D. (2003) Ascending aortic aneurysm associated with bicuspid and tricuspid aortic valve: involvement and clinical relevance of smooth muscle cell apoptosis and expression of cell death-initiating proteins. Eur. J. Cardiothorac. Surg. 23, 537–543
34. Deplancke, B., and Gaskins, H. R. (2003) Hydrogen sulfide induces serum-independent cell cycle entry in nontransformed rat intestinal epithelial cells. FASEB J. 17, 1310–1312
35. Shiota, M., Sugai, N., Tamura, M., Yamaguchi, R., Fukushima, N., Miyano, T., and Miyazaki, H. (2003) Correlation of mitogen-activated protein kinase activities with cell survival and apoptosis in porcine granulosa cells. Zoolog. Sci. 201, 193–201
36. Choi, Y. J., Lim, S. Y., Woo, J. H., Kim, Y. H., Kwon, Y. K., Suh, S. I., Lee, S. H., Choi, W. Y., Kim, J. G., Lee, I. S., et al. (2003) Sodium orthovanadate potentiates EGCG-induced apoptosis that is dependent on the ERK pathway. Biochem. Biophys. Res. Commun. 305, 176–185
37. Najib, S., and Sanchez-Margalet, V. (2002) Human leptin promotes survival of human circulating blood monocytes prone to apoptosis by activation of p42/44 MAPK pathway. Cell. Immunol. 220, 143–149
38. Cunningham, K. A., Chapman, N. M., and Carson, S. D. (2003) Caspase-3 activation and ERK phosphorylation during CVB3 infection of cells: influence of the coxsackievirus and adenovirus receptor and engineered variants. Virus Res. 92, 179–186
39. Guyton, K. Z., Liu, Y., Gorospe, M., Xu, Q., and Holbrook, N. J. (1996) Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J. Biol. Chem. 271, 4138–4142
Page 11 of 21(page number not for citation purposes)

40. Roulston, A., Reinhard, C., Amiri, P., and Williams, L. T. (1998) Early activation of c-Jun N-terminal kinase and p38 kinase regulate cell survival in response to tumor necrosis factor alpha. J. Biol. Chem. 273, 10232–10239
41. Zhang, X., Shan, P., Otterbein, L. E., Alam, J., Flavell, R. A., Davis, R. J., Choi, A. M., and Lee, P. J. (2003) Carbon monoxide inhibition of apoptosis during ischemia-reperfusion lung injury is dependent on the p38 mitogen-activated protein kinase pathway and involves caspase 3. J. Biol. Chem. 278, 1248–1258
42. Sarker, K. P., Obara, S., Nakata, M., Kitajima, I., and Maruyama, I. (2000) Anandamide induces apoptosis of PC-12 cells: involvement of superoxide and caspase-3. FEBS Lett. 472, 39–44
43. Ozaki, I., Tani, E., Ikemoto, H., Kitagawa, H., and Fujikawa, H. (1999) Activation of stress-activated protein kinase/c-Jun NH2-terminal kinase and p38 kinase in calphostin C-induced apoptosis requires caspase-3-like proteases but is dispensable for cell death. J. Biol. Chem. 274, 5310–5317
44. Frese, S., Pirnia, F., Miescher, D., Krajewski, S., Borner, M. M., Reed, J. C., and Schmid, R. A. (2003) PG490-mediated sensitization of lung cancer cells to Apo2L/TRAIL-induced apoptosis requires activation of ERK2. Oncogene 22, 5427–5435
45. Adayev, T., Ray, I., Sondhi, R., Sobocki, T., and Banerjee, P. (2003) The G protein-coupled 5-HT1A receptor causes suppression of caspase-3 through MAPK and protein kinase Calpha. Biochim. Biophys. Acta 1640, 85–96
Received May 20, 2004; accepted July 26, 2004.
Page 12 of 21(page number not for citation purposes)
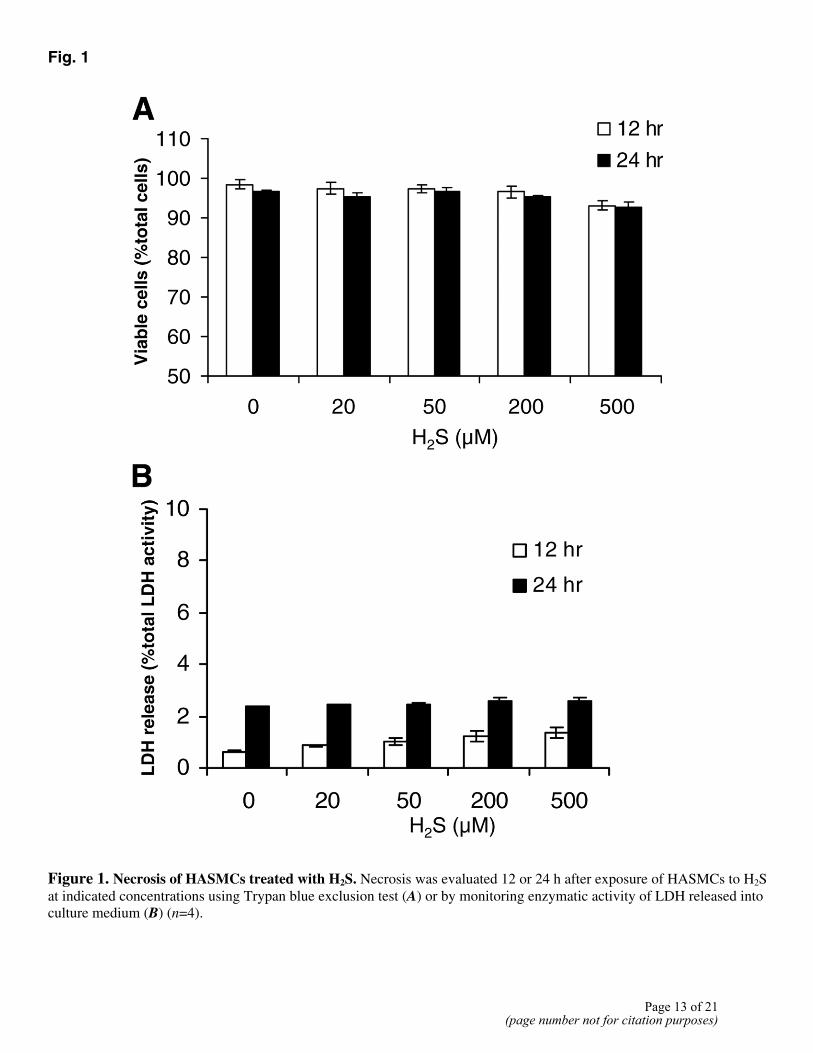
Fig. 1
Figure 1. Necrosis of HASMCs treated with H2S. Necrosis was evaluated 12 or 24 h after exposure of HASMCs to H2S at indicated concentrations using Trypan blue exclusion test (A) or by monitoring enzymatic activity of LDH released into culture medium (B) (n=4).
Page 13 of 21(page number not for citation purposes)
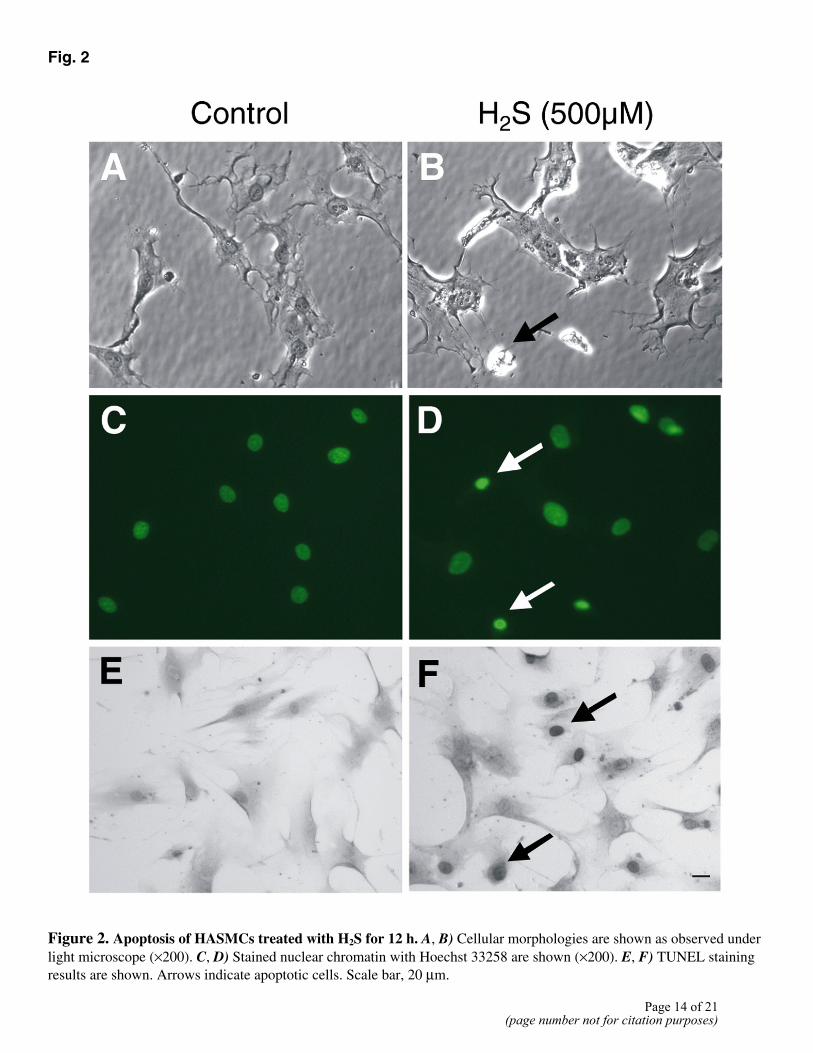
Fig. 2
Figure 2. Apoptosis of HASMCs treated with H2S for 12 h. A, B) Cellular morphologies are shown as observed under light microscope (×200). C, D) Stained nuclear chromatin with Hoechst 33258 are shown (×200). E, F) TUNEL staining results are shown. Arrows indicate apoptotic cells. Scale bar, 20 µm.
Page 14 of 21(page number not for citation purposes)
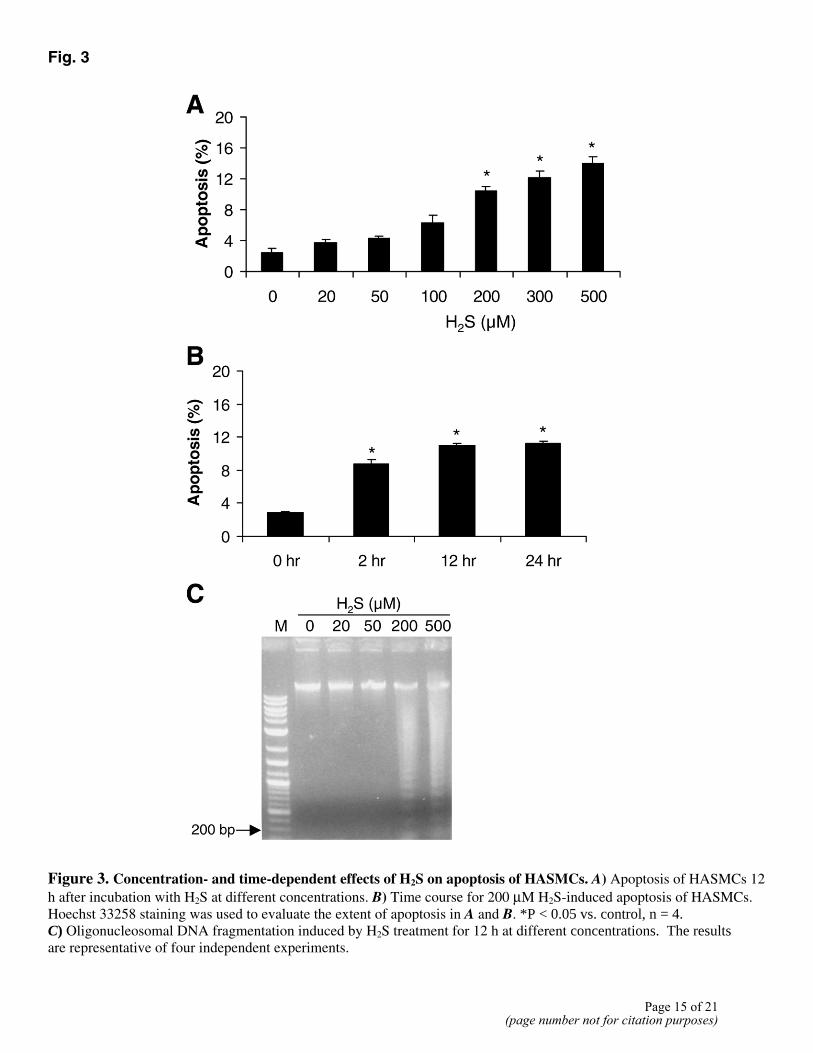
Fig. 3
Figure 3. Concentration- and time-dependent effects of H2S on apoptosis of HASMCs. A) Apoptosis of HASMCs 12 h after incubation with H2S at different concentrations. B) Time course for 200 µM H2S-induced apoptosis of HASMCs. Hoechst 33258 staining was used to evaluate the extent of apoptosis in A and B. *P < 0.05 vs. control, n = 4. C) Oligonucleosomal DNA fragmentation induced by H2S treatment for 12 h at different concentrations. The results are representative of four independent experiments.
Page 15 of 21(page number not for citation purposes)
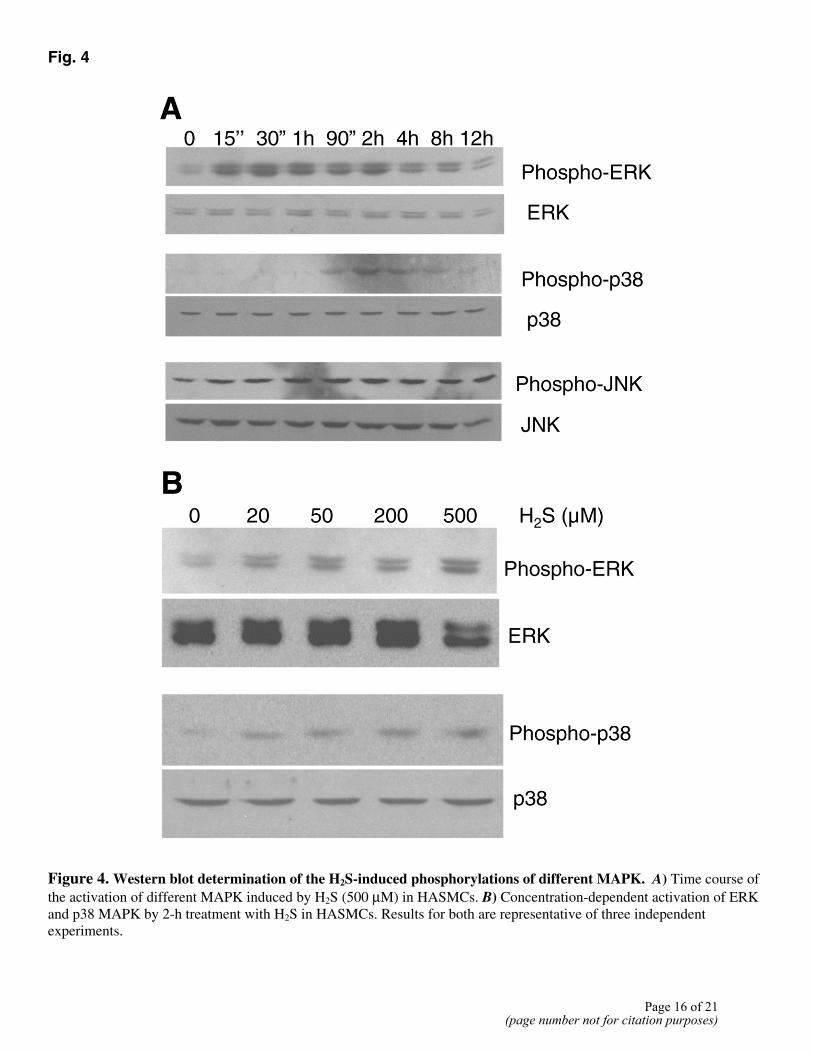
Fig. 4
Figure 4. Western blot determination of the H2S-induced phosphorylations of different MAPK. A) Time course of the activation of different MAPK induced by H2S (500 µM) in HASMCs. B) Concentration-dependent activation of ERK and p38 MAPK by 2-h treatment with H2S in HASMCs. Results for both are representative of three independent experiments.
Page 16 of 21(page number not for citation purposes)
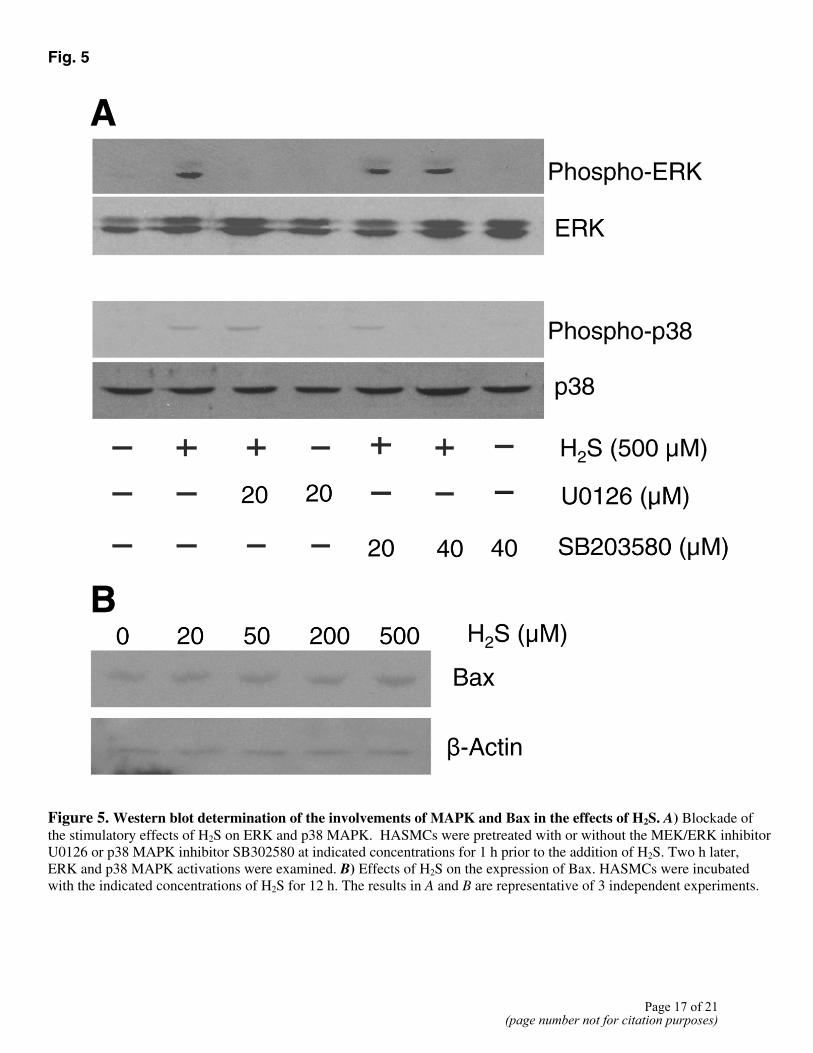
Fig. 5
Figure 5. Western blot determination of the involvements of MAPK and Bax in the effects of H2S. A) Blockade of the stimulatory effects of H2S on ERK and p38 MAPK. HASMCs were pretreated with or without the MEK/ERK inhibitor U0126 or p38 MAPK inhibitor SB302580 at indicated concentrations for 1 h prior to the addition of H2S. Two h later, ERK and p38 MAPK activations were examined. B) Effects of H2S on the expression of Bax. HASMCs were incubated with the indicated concentrations of H2S for 12 h. The results in A and B are representative of 3 independent experiments.
Page 17 of 21(page number not for citation purposes)
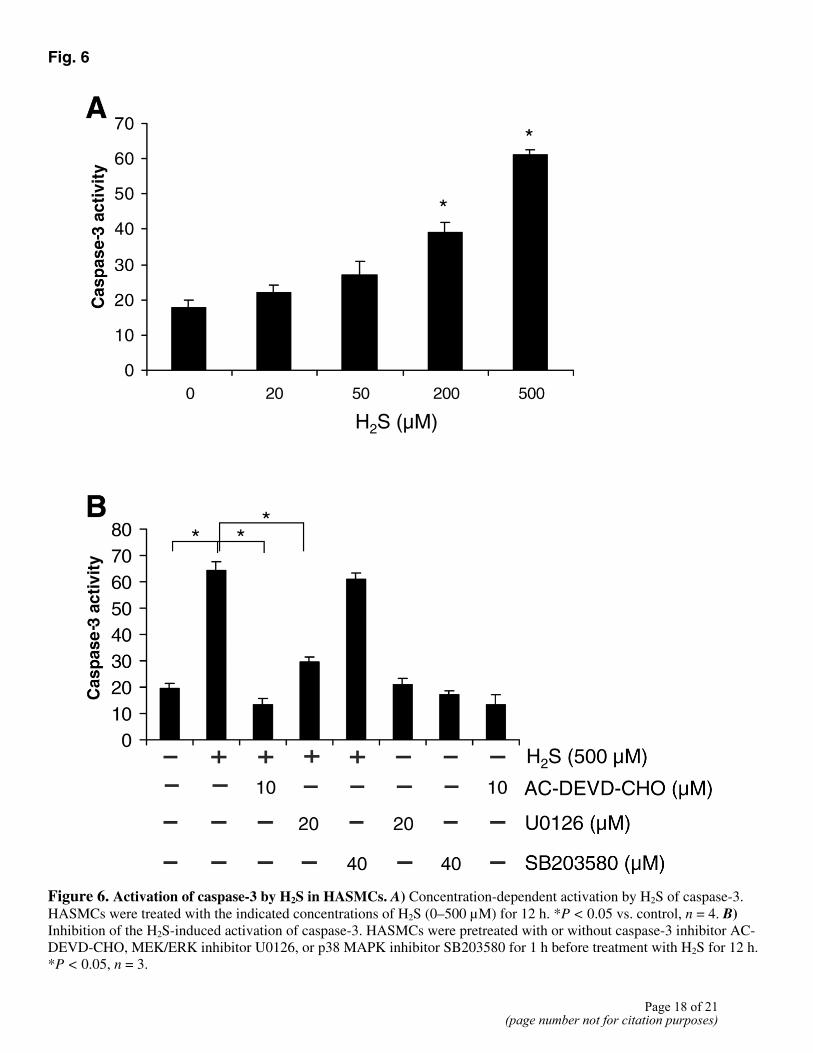
Fig. 6
Figure 6. Activation of caspase-3 by H2S in HASMCs. A) Concentration-dependent activation by H2S of caspase-3. HASMCs were treated with the indicated concentrations of H2S (0–500 µM) for 12 h. *P < 0.05 vs. control, n = 4. B) Inhibition of the H2S-induced activation of caspase-3. HASMCs were pretreated with or without caspase-3 inhibitor AC-DEVD-CHO, MEK/ERK inhibitor U0126, or p38 MAPK inhibitor SB203580 for 1 h before treatment with H2S for 12 h. *P < 0.05, n = 3.
Page 18 of 21(page number not for citation purposes)
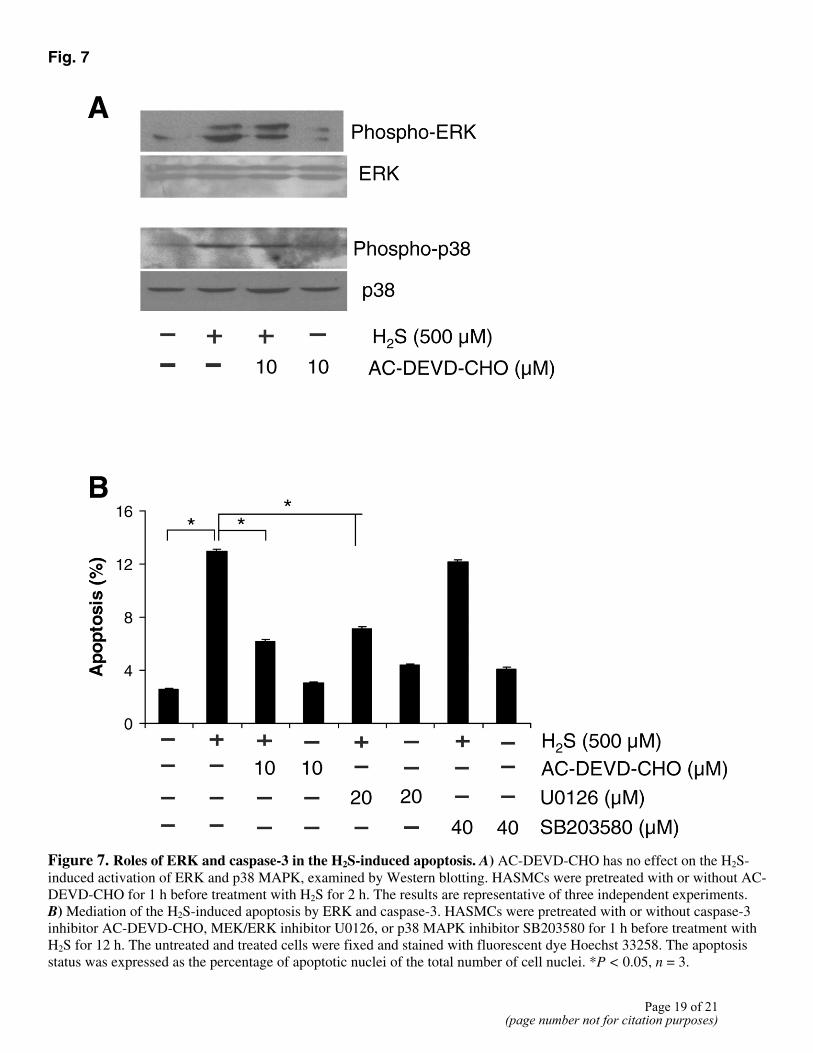
Fig. 7
Figure 7. Roles of ERK and caspase-3 in the H2S-induced apoptosis. A) AC-DEVD-CHO has no effect on the H2S-induced activation of ERK and p38 MAPK, examined by Western blotting. HASMCs were pretreated with or without AC-DEVD-CHO for 1 h before treatment with H2S for 2 h. The results are representative of three independent experiments. B) Mediation of the H2S-induced apoptosis by ERK and caspase-3. HASMCs were pretreated with or without caspase-3 inhibitor AC-DEVD-CHO, MEK/ERK inhibitor U0126, or p38 MAPK inhibitor SB203580 for 1 h before treatment with H2S for 12 h. The untreated and treated cells were fixed and stained with fluorescent dye Hoechst 33258. The apoptosis status was expressed as the percentage of apoptotic nuclei of the total number of cell nuclei. *P < 0.05, n = 3.
Page 19 of 21(page number not for citation purposes)
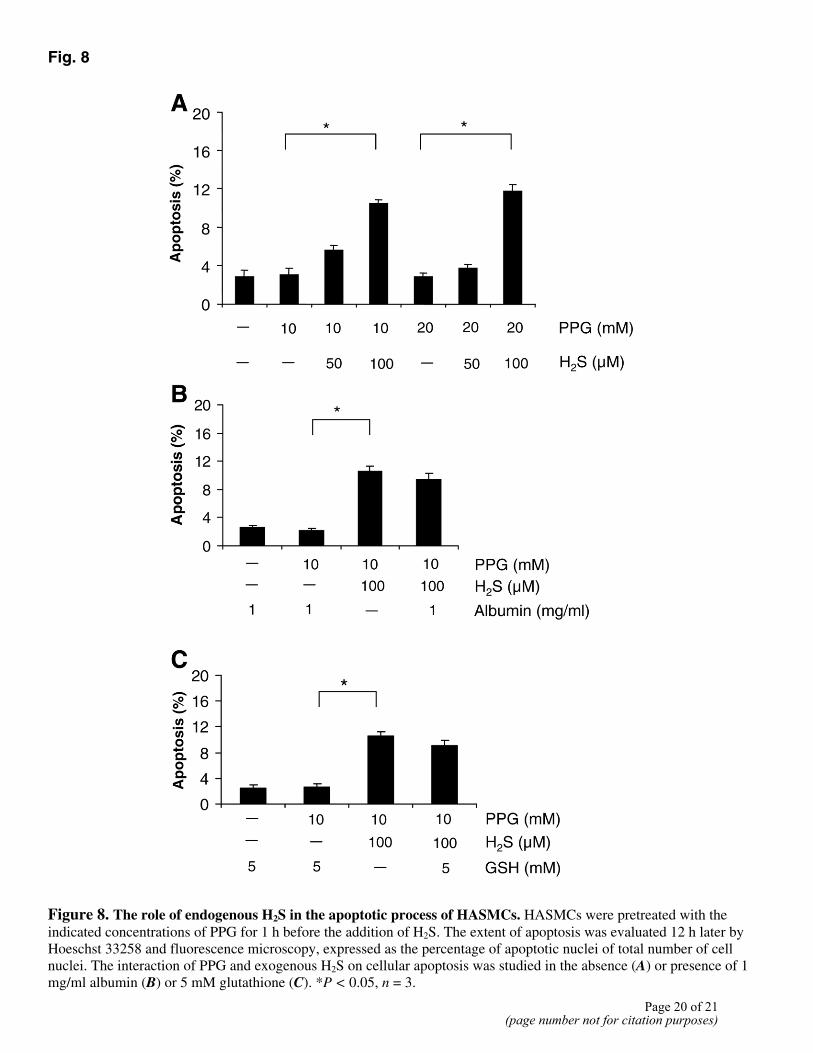
Fig. 8
Figure 8. The role of endogenous H2S in the apoptotic process of HASMCs. HASMCs were pretreated with the indicated concentrations of PPG for 1 h before the addition of H2S. The extent of apoptosis was evaluated 12 h later by Hoeschst 33258 and fluorescence microscopy, expressed as the percentage of apoptotic nuclei of total number of cell nuclei. The interaction of PPG and exogenous H2S on cellular apoptosis was studied in the absence (A) or presence of 1 mg/ml albumin (B) or 5 mM glutathione (C). *P < 0.05, n = 3.
Page 20 of 21(page number not for citation purposes)
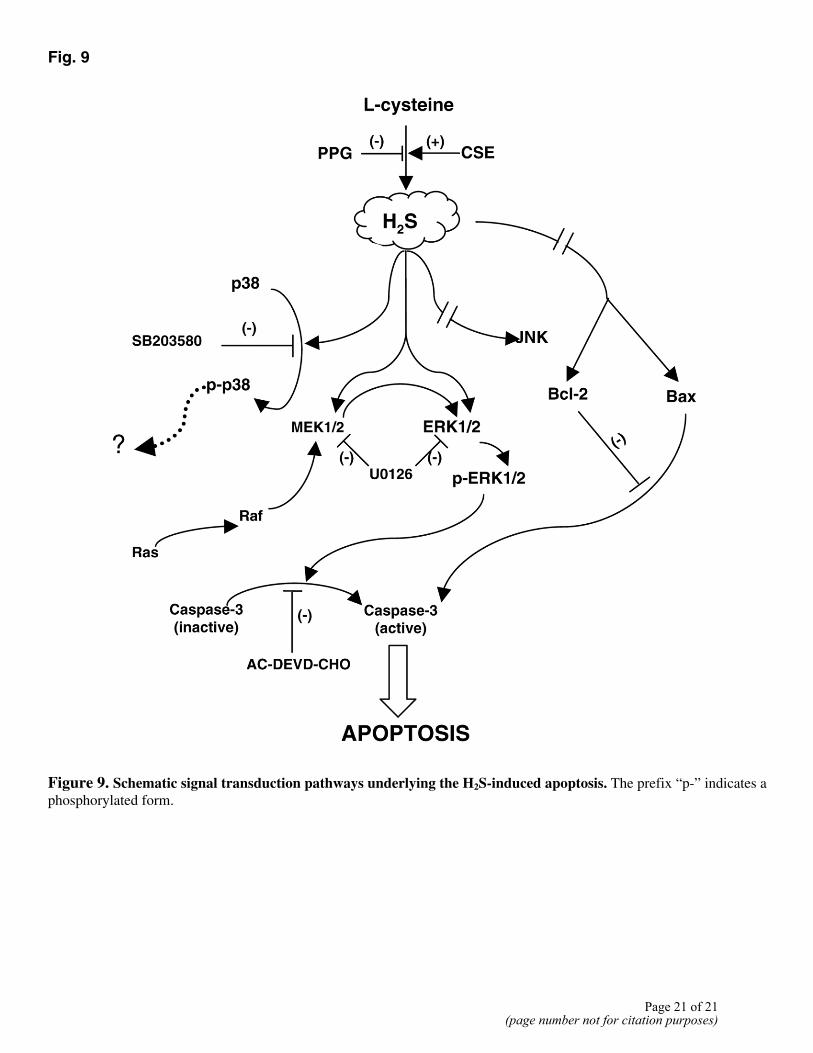
Fig. 9
Figure 9. Schematic signal transduction pathways underlying the H2S-induced apoptosis. The prefix “p-” indicates a phosphorylated form.
Page 21 of 21(page number not for citation purposes)
Copyright © 2022 FDOKUMEN




















