
Repression of Stat3 activity by activation of mitogen-activated protein kinase (MAPK
11
Repression of Stat3 activity by activation of mitogen-activated protein kinase (MAPK) Neeraj Jain, Tong Zhang, Siok Lyn Fong, Cheh Peng Lim and Xinmin Cao Signal Transduction Laboratory, Institute of Molecular and Cell Biology, National University of Singapore, Singapore 117609 STAT proteins are activated by phosphorylation at specific tyrosine residue at the carboxy-terminus which is required for dimer-formation, nuclear translocation, DNA binding and transcriptional activity in cells treated with cytokines and growth factors. Recent studies have indicated that STATs are also phosphorylated by MAPK, or extracellular signal-regulated kinase (ERK) on serine. We investigated the role of ERK on the regulation of STAT activity. Here, we report that ERK2 activated by its upstream kinase, MEK1, represses Stat3 transcriptional activity induced by Src or Jak-2. To unravel the mechanism of repression, we further showed that Stat3 DNA binding activity and its tyrosine phosphorylation are also inhibited under the same conditions. ERK2 phosphorylates Stat3 on three serine- containing peptides and decreases its tyrosine phosphor- ylation induced by EGF treatment. We also detected an association of ERK2 and Stat3 in vivo which is modulated positively by activation of ERK2, but negatively by Jak2. We propose that MAP kinase cascade may negatively regulate Stat3 activities by decreasing its tyrosine phosphorylation and also possibly by association. Keywords: repression; Stat3 transactivation; ERK2; phosphorylation; association Introduction STATs, act as signal transducers and transcriptional activators in the cytokine and growth factor treated cells (reviewed in Darnell et al., 1994; Ihle, 1996). Janus protein tyrosine kinases (JAKs) associate with cytokine receptors which lack intrinsic tyrosine kinase activity and become activated upon cytokine stimulation. Activated JAKs phosphorylate the target tyrosine residues in the receptors (Silvennoinen et al., 1993; Ihle et al., 1994; Stahl et al., 1994) where STATs are selectively recruited through their SH2 domain and then phosphorylated by JAKs on a conserved tyrosine residue at the carboxy terminus (Heim et al., 1995; Stahl et al., 1995). Such tyrosine phosphorylation is required for the formation of homo- or heterodimers through their phosphorylated tyrosine residues and SH2 domains, nuclear translocation, binding to cognate DNA response elements and transactivation of gene expression (Darnell et al., 1994; Ihle, 1996). Stat1 was the first STAT protein identified in the interferon (IFN) signaling pathways and was later found to be activated by other cytokines and growth factor EGF (Schindler et al., 1992; Shuai et al., 1992; Fu and Zhang, 1993). Stat3 (also known as acute- phase response factor APRF) which shares 52.5% amino acid identity with Stat1 and has similar size and structure, is mainly induced by growth factors, such as platelet-derived growth factor (PDGF), colony-stimu- lating factor-1 (CSF-1), EGF, and a number of cytokines, including IFN-a and interleukin-6 (IL-6) (Akira et al., 1994; Zhong et al., 1994). Based on sequence alignment with Stat1, which is phosphory- lated on Tyr-701 in IFN-g-treated cells (Shuai et al., 1993). Tyr-705 of Stat3 was identified as the phosphorylation site for JAKs in the cytokine signaling (Kaptein et al., 1996). However, Jaks may not be required for the activation of Stat3 by growth factors such as EGF and PDGF which bind to their receptors with intrinsic tyrosine kinase activity. Instead, tyrosine kinase activity of the receptors are important for Stat3 activation (Leaman et al., 1996). Furthermore, it has been reported that Stat3 is constitutively phosphorylated on tyrosine in the Src- transformed cells, and Src is required for the activation of Stat3 by CSF-1, suggesting that Src may be specifically involved in Stat3 activation during growth factor signaling (Yu et al., 1995; Cao et al., 1996, Chaturvedi et al., 1997). The Ras/Raf/MAPK pathway is a major signaling pathway of growth factors and cytokines (reviewed in Marshall, 1994; Hill and Triesman, 1995). The cascade involves transient formation of the ras-GTP and activation of raf kinase at the cell membrane to activate MAPK kinase (MAPKK or MEK) which in turn activates MAPK through dual phosphorylation on threonine and tyrosine residues (Leevers et al., 1994; Marshall, 1994; Stokoe et al., 1994). Activated MAPK then translocates into the nucleus and phosphorylates the ternary complex factor TCF (Shaw et al., 1989) which activates the expression of immediate-early genes, such as c-fos and egr-1 (reviewed in Triesman, 1995). Recently, it has been reported that phosphor- ylation of STATs on serine may also be important for their functions (Zhang et al., 1995). A conserved phosphorylation site of MAPK/ERK is identified within the COOH-terminus of Stat1a, Stat3, Stat4 and Stat5 (Wen et al., 1995) and Stat3 is shown to be phosphorylated by ERK on serine 727 in response to growth factors (Chung et al., 1997). These reports suggest that MAPK cascade may be involved in regulating the STAT function. Since the MAPK cascade is a major signaling pathway involved in cell growth and dierentiation, and both the MAPK and the Stat3 are activated by a series of overlapping Correspondence: X Cao Received 6 February 1998; revised 22 June 1998; accepted 22 June 1998 Oncogene (1998) 17, 3157 – 3167 ª 1998 Stockton Press All rights reserved 0950 – 9232/98 $12.00 http://www.stockton-press.co.uk/onc
-
Upload
independent -
Category
Documents
-
view
6 -
download
0
Transcript of Repression of Stat3 activity by activation of mitogen-activated protein kinase (MAPK

Repression of Stat3 activity by activation of mitogen-activated proteinkinase (MAPK)
Neeraj Jain, Tong Zhang, Siok Lyn Fong, Cheh Peng Lim and Xinmin Cao
Signal Transduction Laboratory, Institute of Molecular and Cell Biology, National University of Singapore, Singapore 117609
STAT proteins are activated by phosphorylation atspeci®c tyrosine residue at the carboxy-terminus whichis required for dimer-formation, nuclear translocation,DNA binding and transcriptional activity in cells treatedwith cytokines and growth factors. Recent studies haveindicated that STATs are also phosphorylated byMAPK, or extracellular signal-regulated kinase (ERK)on serine. We investigated the role of ERK on theregulation of STAT activity. Here, we report that ERK2activated by its upstream kinase, MEK1, represses Stat3transcriptional activity induced by Src or Jak-2. Tounravel the mechanism of repression, we further showedthat Stat3 DNA binding activity and its tyrosinephosphorylation are also inhibited under the sameconditions. ERK2 phosphorylates Stat3 on three serine-containing peptides and decreases its tyrosine phosphor-ylation induced by EGF treatment. We also detected anassociation of ERK2 and Stat3 in vivo which ismodulated positively by activation of ERK2, butnegatively by Jak2. We propose that MAP kinasecascade may negatively regulate Stat3 activities bydecreasing its tyrosine phosphorylation and also possiblyby association.
Keywords: repression; Stat3 transactivation; ERK2;phosphorylation; association
Introduction
STATs, act as signal transducers and transcriptionalactivators in the cytokine and growth factor treatedcells (reviewed in Darnell et al., 1994; Ihle, 1996). Janusprotein tyrosine kinases (JAKs) associate with cytokinereceptors which lack intrinsic tyrosine kinase activityand become activated upon cytokine stimulation.Activated JAKs phosphorylate the target tyrosineresidues in the receptors (Silvennoinen et al., 1993;Ihle et al., 1994; Stahl et al., 1994) where STATs areselectively recruited through their SH2 domain andthen phosphorylated by JAKs on a conserved tyrosineresidue at the carboxy terminus (Heim et al., 1995;Stahl et al., 1995). Such tyrosine phosphorylation isrequired for the formation of homo- or heterodimersthrough their phosphorylated tyrosine residues andSH2 domains, nuclear translocation, binding tocognate DNA response elements and transactivationof gene expression (Darnell et al., 1994; Ihle, 1996).
Stat1 was the ®rst STAT protein identi®ed in theinterferon (IFN) signaling pathways and was laterfound to be activated by other cytokines and growthfactor EGF (Schindler et al., 1992; Shuai et al., 1992;Fu and Zhang, 1993). Stat3 (also known as acute-phase response factor APRF) which shares 52.5%amino acid identity with Stat1 and has similar size andstructure, is mainly induced by growth factors, such asplatelet-derived growth factor (PDGF), colony-stimu-lating factor-1 (CSF-1), EGF, and a number ofcytokines, including IFN-a and interleukin-6 (IL-6)(Akira et al., 1994; Zhong et al., 1994). Based onsequence alignment with Stat1, which is phosphory-lated on Tyr-701 in IFN-g-treated cells (Shuai et al.,1993). Tyr-705 of Stat3 was identi®ed as thephosphorylation site for JAKs in the cytokinesignaling (Kaptein et al., 1996). However, Jaks maynot be required for the activation of Stat3 by growthfactors such as EGF and PDGF which bind to theirreceptors with intrinsic tyrosine kinase activity.Instead, tyrosine kinase activity of the receptors areimportant for Stat3 activation (Leaman et al., 1996).Furthermore, it has been reported that Stat3 isconstitutively phosphorylated on tyrosine in the Src-transformed cells, and Src is required for the activationof Stat3 by CSF-1, suggesting that Src may bespeci®cally involved in Stat3 activation during growthfactor signaling (Yu et al., 1995; Cao et al., 1996,Chaturvedi et al., 1997).The Ras/Raf/MAPK pathway is a major signaling
pathway of growth factors and cytokines (reviewed inMarshall, 1994; Hill and Triesman, 1995). The cascadeinvolves transient formation of the ras-GTP andactivation of raf kinase at the cell membrane toactivate MAPK kinase (MAPKK or MEK) which inturn activates MAPK through dual phosphorylationon threonine and tyrosine residues (Leevers et al., 1994;Marshall, 1994; Stokoe et al., 1994). Activated MAPKthen translocates into the nucleus and phosphorylatesthe ternary complex factor TCF (Shaw et al., 1989)which activates the expression of immediate-earlygenes, such as c-fos and egr-1 (reviewed in Triesman,1995). Recently, it has been reported that phosphor-ylation of STATs on serine may also be important fortheir functions (Zhang et al., 1995). A conservedphosphorylation site of MAPK/ERK is identi®edwithin the COOH-terminus of Stat1a, Stat3, Stat4and Stat5 (Wen et al., 1995) and Stat3 is shown to bephosphorylated by ERK on serine 727 in response togrowth factors (Chung et al., 1997). These reportssuggest that MAPK cascade may be involved inregulating the STAT function. Since the MAPKcascade is a major signaling pathway involved in cellgrowth and di�erentiation, and both the MAPK andthe Stat3 are activated by a series of overlapping
Correspondence: X CaoReceived 6 February 1998; revised 22 June 1998; accepted 22 June1998
Oncogene (1998) 17, 3157 ± 3167ã 1998 Stockton Press All rights reserved 0950 ± 9232/98 $12.00
http://www.stockton-press.co.uk/onc

growth factors and cytokines, we investigated the roleof MAPK on Stat3 activity. Here we report that ERK2negatively regulates Stat3 transcription activity, DNAbinding, and its tyrosine phosphorylation. We furthershow that ERK2 associates with Stat3 which ismodulated positively by the activation of ERK2, butnegatively by tyrosine phosphorylation of Stat3. ERK2phosphorylates Stat3 on three serine-containing pep-tides and decreases its tyrosine phosphorylation. Wepropose that the MAP kinase cascade negativelyregulates Stat3 activities by regulating its phosphoryla-tion as well as by association.
Results
Repression of Stat3 transcription activity by ERK2activated by MEK1
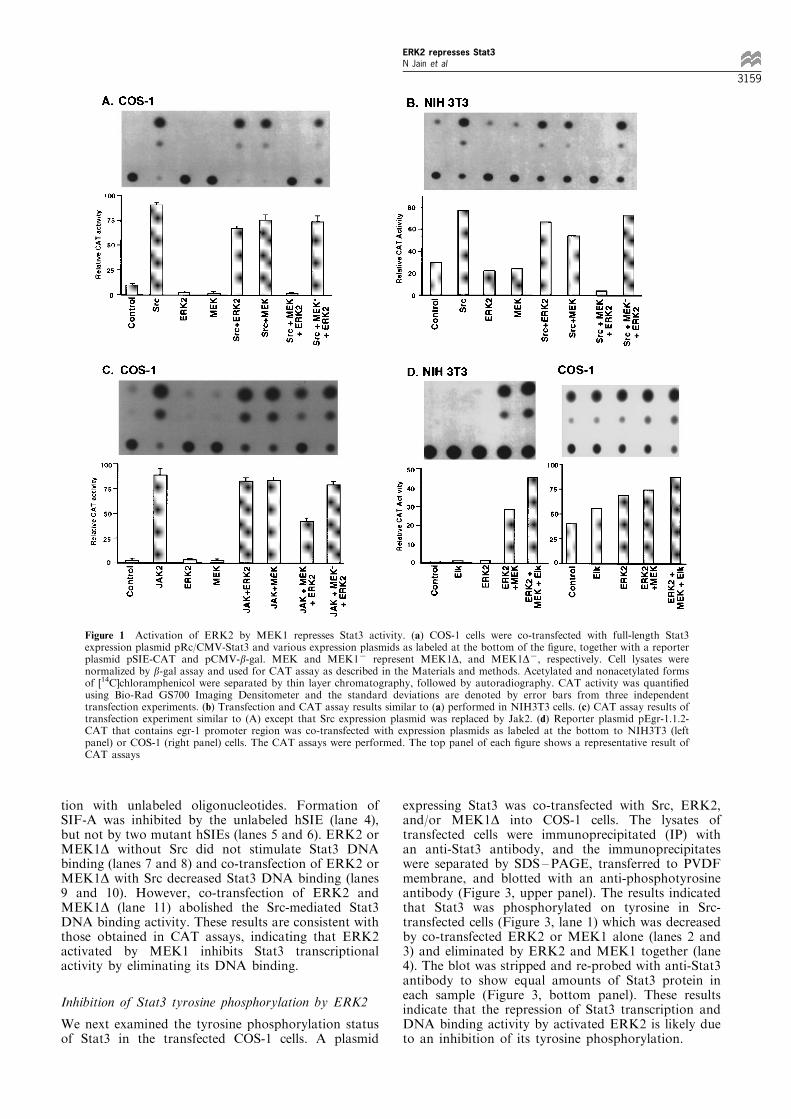
A consensus sequence of MAPK phosphorylation,PMSP, was observed at Ser-727 in the COOH-terminus of Stat3, and mutation of this site decreasedStat3 transcriptional activity (Wen et al., 1995). Tofurther investigate the e�ect of ERK2 on Stat3function, we ®rst examined how ERK2 a�ected Stat3transactivation. Since tyrosine phosphorylation is aprerequisite for STAT activities (Darnell et al., 1994;Schindler and Darnell, 1995; Ihle, 1996) and thetyrosine kinase Src speci®cally increases Stat3 tyrosinephosphorylation and its DNA binding (Yu et al., 1995;Cao et al., 1996), we ®rst tested whether Src canstimulate Stat3 transcriptional activity and how ERK2a�ects such activity with transient transfectionexperiments. COS-1 cells, which express very lowendogenous Stat3 (Wen et al., 1995), were co-transfected with a CAT reporter plasmid containingthree copies of a high a�nity binding site of Stat3,hSIE, together with a Stat3 expression plasmid in theabsence or presence of Src expression plasmid. Thecells were lysed and the CAT activity assayed. TheCAT activity was low in the cells transfected withStat3 without Src, but increased by ninefold in thepresence of Src (Figure 1a), indicating that Src indeedstimulates Stat3 transcriptional activity. It is knownthat ERK2 is phosphorylated and activated by itsupstream kinase MEK1. To examine the e�ect ofERK2 on Stat3 transactivation stimulated by Src,plasmids expressing ERK2 (pCDNA3.p42) andMEK1D, a constitutively activated MEK1 (Mansouret al., 1994), were co-transfected either alone ortogether, in the absence or presence of Src. Theresults showed that the expression of ERK2 orMEK1D without Src did not stimulate Stat3 activityas expected. Surprisingly, the Src-stimulated Stat3activity was decreased by ERK2 or MEK1 alone,and completely inhibited by co-transfection of ERK2and MEK1 together (Figure 1a). The repression wasrecovered more than 75% when ERK2 was co-transfected with a mutant MEK1 (MEK1D7) whichlacks the kinase activity (Figure 1a). To exclude thepossibility that the repression was due to the highlyactivated MEK1D, a normal MEK1 was examined andthe repression on Stat3 transcriptional activityremained (data not shown). These results demonstratethat activated ERK2 by MEK1 represses Stat3transcriptional activity speci®cally.
To con®rm these results, similar experiments wereperformed in NIH3T3 cells. Although the basal level ofStat3 activity was higher than that in the COS-1 cells,Stat3 activity was stimulated by Src and repressed byco-expression of MEK1D and ERK2 (Figure 1b) in asimilar pattern to that in the COS-1 cells, indicatingthat the inhibition is not cell type restricted.Since JAKs are the upstream kinases for STAT
protein tyrosine phosphorylation in response to certaincytokines, we also examined the e�ect of ERK2 onStat3 transactivation stimulated by Jak2. Src wasreplaced by Jak2 in transient transfection experi-ments. The results showed that the Jak2 stimulatedStat3 activity was dramatically reduced, but notcompletely inhibited (approximately 60% inhibition)by ERK2 and MEK1D together (Figure 1c). Thesedata demonstrate that ERK2 activated by MEK1negatively regulates Stat3 transcriptional activitymediated by either Src or Jak2.To exclude the possibility of a non-speci®c
inhibitory e�ect of MEK1D and ERK2, another setof e�ector and reporter genes were examined. Theternary complex factor TCF is one of the main targetsof Ras/MAP kinase pathway (Shaw et al., 1989; Hilland Triesman, 1995). ERK2 is phosphorylated byMEK1, and in turn phosphorylates TCF on serineresidues. The active TCF binds to SRF and SRE toform a ternary complex, and induces the expression ofimmediate-early genes such as c-fos and egr-1. Sinceegr-1 contains six SREs in its promoter region and isinducible by various growth factors (Sukhatme et al.,1988; Tsai-Morris et al., 1988), we tested the e�ect ofERK2 and MEK1 on egr-1 transcription. pEgr-1.1.2-CAT containing the 1.2 kb promoter sequence of egr-1(Tsai-Morris et al., 1988) upstream of CAT gene wasused as a reporter. ERK2, MEK1D and a member ofthe TCF family, Elk-1, (Marais et al., 1993), wereeither transfected alone or co-transfected in variouscombinations with the reporter into NIH 3T3 cells, andthe CAT activities were assayed. Very low CATactivity was observed with ERK2 alone, but increasedby 39-fold in cells co-transfected with ERK2 andMEK1D together, and further increased by 62-foldwhen the exogenous Elk-1 was included (Figure 1d, leftpanel). The activation of egr-1 promoter by MEK1Dand ERK2 in the absence or the presence of Elk-1 wasalso observed in transfected COS-1 cells (Figure 1d,right panel). These results excluded the possibility of anon-speci®c inhibitory e�ect of MEK1D and ERK2 onStat3 SIE-reporter in these cells.
Inhibition of DNA binding activity of Stat3 by ERK2
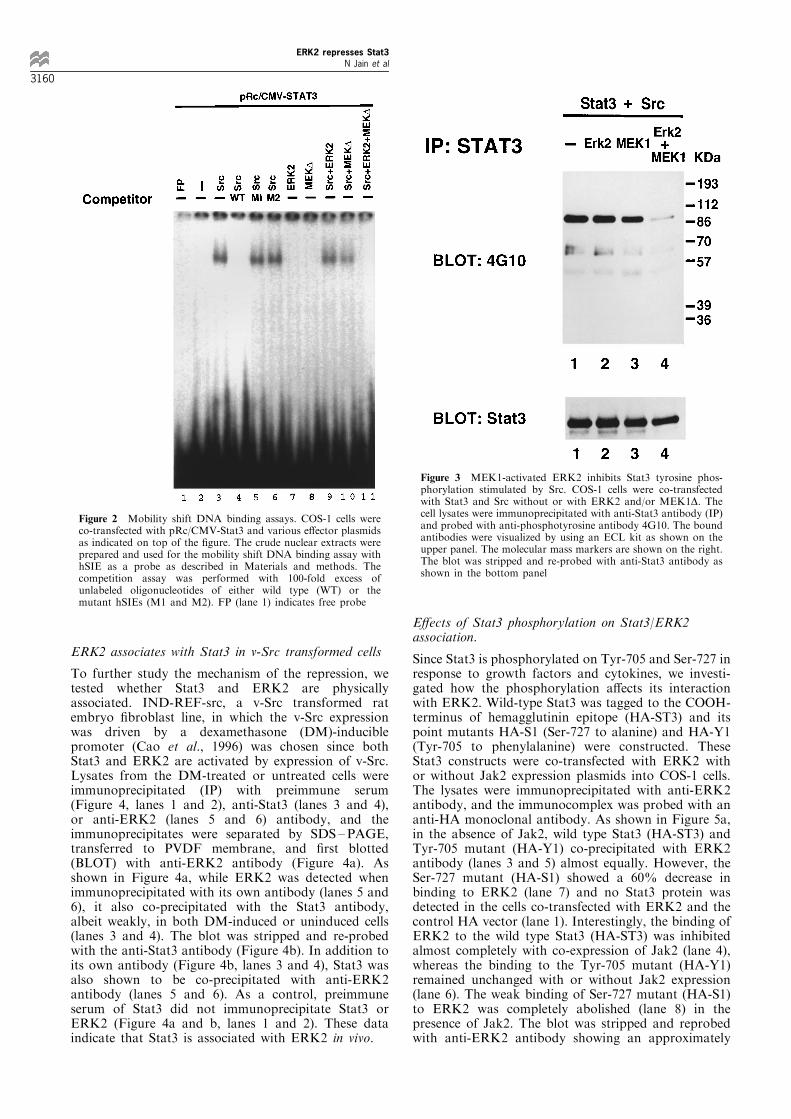
In order to understand the mechanism of therepression, we tested whether ERK2 activated byMEK1D a�ects the Stat3 DNA binding activity.COS-1 cells were transfected as described in Figure1a without the reporter plasmid. The nuclear extractswere prepared and the mobility shift assay wasperformed using hSIE as a probe. Activated Stat3and Stat1 form three complexes with SIE (Sadowski etal., 1993) containing Stat3 (SIF-A) and Stat1 (SIF-C)homodimers and the Stat1/Stat3 heterodimer (SIF-B).As shown in Figure 2, SIF-A was observed in the cellsco-transfected with Stat3 and Src (lane 3). Thespeci®city of the binding was determined by competi-
ERK2 represses Stat3N Jain et al
3158
tion with unlabeled oligonucleotides. Formation ofSIF-A was inhibited by the unlabeled hSIE (lane 4),but not by two mutant hSIEs (lanes 5 and 6). ERK2 orMEK1D without Src did not stimulate Stat3 DNAbinding (lanes 7 and 8) and co-transfection of ERK2 orMEK1D with Src decreased Stat3 DNA binding (lanes9 and 10). However, co-transfection of ERK2 andMEK1D (lane 11) abolished the Src-mediated Stat3DNA binding activity. These results are consistent withthose obtained in CAT assays, indicating that ERK2activated by MEK1 inhibits Stat3 transcriptionalactivity by eliminating its DNA binding.
Inhibition of Stat3 tyrosine phosphorylation by ERK2
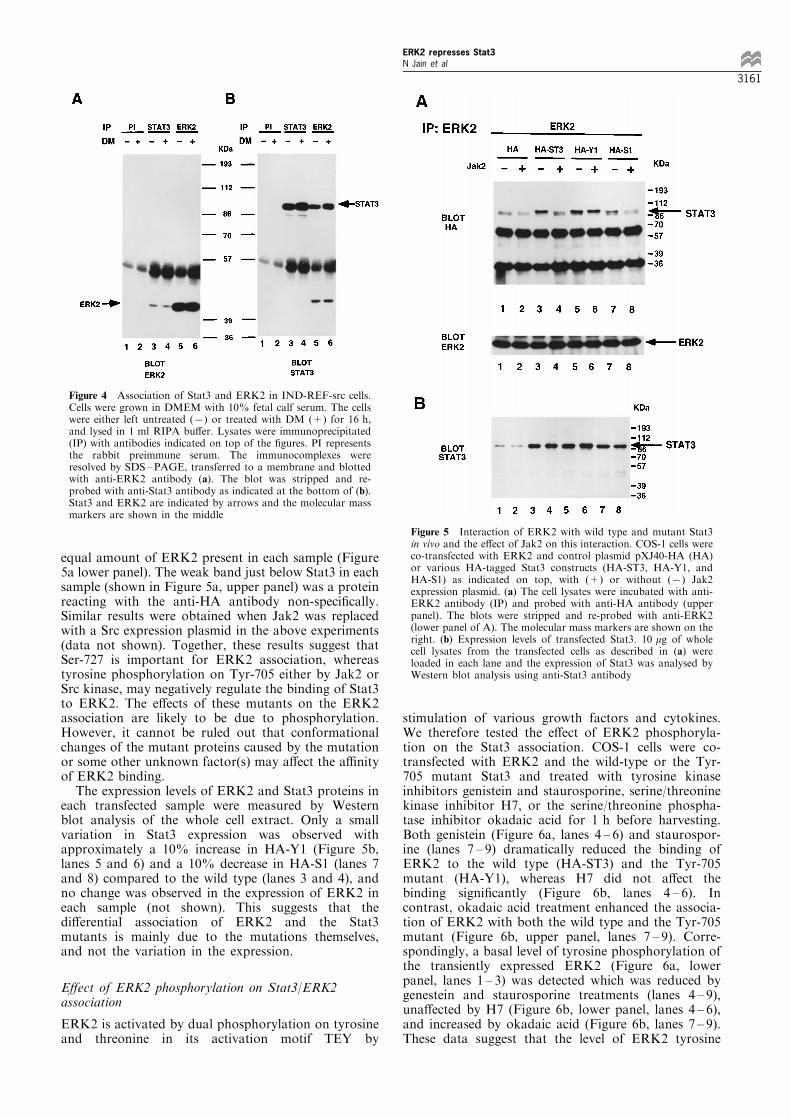
We next examined the tyrosine phosphorylation statusof Stat3 in the transfected COS-1 cells. A plasmid
expressing Stat3 was co-transfected with Src, ERK2,and/or MEK1D into COS-1 cells. The lysates oftransfected cells were immunoprecipitated (IP) withan anti-Stat3 antibody, and the immunoprecipitateswere separated by SDS ±PAGE, transferred to PVDFmembrane, and blotted with an anti-phosphotyrosineantibody (Figure 3, upper panel). The results indicatedthat Stat3 was phosphorylated on tyrosine in Src-transfected cells (Figure 3, lane 1) which was decreasedby co-transfected ERK2 or MEK1 alone (lanes 2 and3) and eliminated by ERK2 and MEK1 together (lane4). The blot was stripped and re-probed with anti-Stat3antibody to show equal amounts of Stat3 protein ineach sample (Figure 3, bottom panel). These resultsindicate that the repression of Stat3 transcription andDNA binding activity by activated ERK2 is likely dueto an inhibition of its tyrosine phosphorylation.
Figure 1 Activation of ERK2 by MEK1 represses Stat3 activity. (a) COS-1 cells were co-transfected with full-length Stat3expression plasmid pRc/CMV-Stat3 and various expression plasmids as labeled at the bottom of the ®gure, together with a reporterplasmid pSIE-CAT and pCMV-b-gal. MEK and MEK17 represent MEK1D, and MEK1D7, respectively. Cell lysates werenormalized by b-gal assay and used for CAT assay as described in the Materials and methods. Acetylated and nonacetylated formsof [14C]chloramphenicol were separated by thin layer chromatography, followed by autoradiography. CAT activity was quanti®edusing Bio-Rad GS700 Imaging Densitometer and the standard deviations are denoted by error bars from three independenttransfection experiments. (b) Transfection and CAT assay results similar to (a) performed in NIH3T3 cells. (c) CAT assay results oftransfection experiment similar to (A) except that Src expression plasmid was replaced by Jak2. (d) Reporter plasmid pEgr-1.1.2-CAT that contains egr-1 promoter region was co-transfected with expression plasmids as labeled at the bottom to NIH3T3 (leftpanel) or COS-1 (right panel) cells. The CAT assays were performed. The top panel of each ®gure shows a representative result ofCAT assays
ERK2 represses Stat3N Jain et al
3159
ERK2 associates with Stat3 in v-Src transformed cells
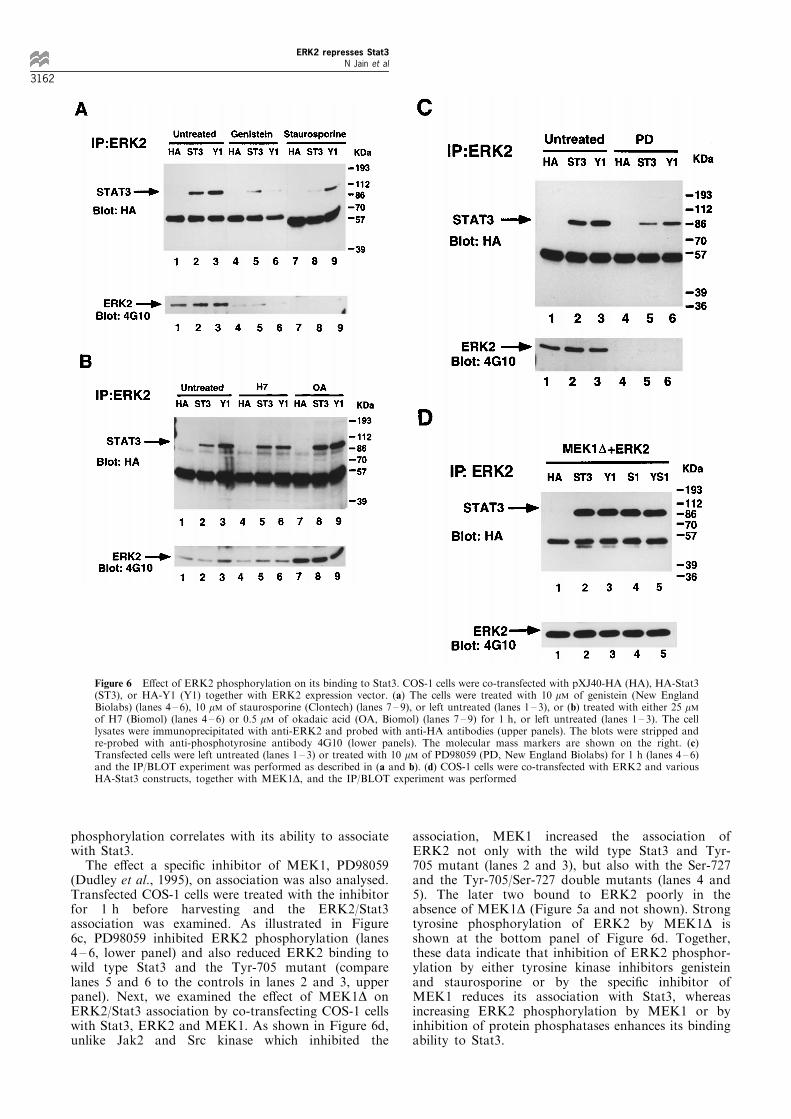
To further study the mechanism of the repression, wetested whether Stat3 and ERK2 are physicallyassociated. IND-REF-src, a v-Src transformed ratembryo ®broblast line, in which the v-Src expressionwas driven by a dexamethasone (DM)-induciblepromoter (Cao et al., 1996) was chosen since bothStat3 and ERK2 are activated by expression of v-Src.Lysates from the DM-treated or untreated cells wereimmunoprecipitated (IP) with preimmune serum(Figure 4, lanes 1 and 2), anti-Stat3 (lanes 3 and 4),or anti-ERK2 (lanes 5 and 6) antibody, and theimmunoprecipitates were separated by SDS ±PAGE,transferred to PVDF membrane, and ®rst blotted(BLOT) with anti-ERK2 antibody (Figure 4a). Asshown in Figure 4a, while ERK2 was detected whenimmunoprecipitated with its own antibody (lanes 5 and6), it also co-precipitated with the Stat3 antibody,albeit weakly, in both DM-induced or uninduced cells(lanes 3 and 4). The blot was stripped and re-probedwith the anti-Stat3 antibody (Figure 4b). In addition toits own antibody (Figure 4b, lanes 3 and 4), Stat3 wasalso shown to be co-precipitated with anti-ERK2antibody (lanes 5 and 6). As a control, preimmuneserum of Stat3 did not immunoprecipitate Stat3 orERK2 (Figure 4a and b, lanes 1 and 2). These dataindicate that Stat3 is associated with ERK2 in vivo.
E�ects of Stat3 phosphorylation on Stat3/ERK2association.
Since Stat3 is phosphorylated on Tyr-705 and Ser-727 inresponse to growth factors and cytokines, we investi-gated how the phosphorylation a�ects its interactionwith ERK2. Wild-type Stat3 was tagged to the COOH-terminus of hemagglutinin epitope (HA-ST3) and itspoint mutants HA-S1 (Ser-727 to alanine) and HA-Y1(Tyr-705 to phenylalanine) were constructed. TheseStat3 constructs were co-transfected with ERK2 withor without Jak2 expression plasmids into COS-1 cells.The lysates were immunoprecipitated with anti-ERK2antibody, and the immunocomplex was probed with ananti-HA monoclonal antibody. As shown in Figure 5a,in the absence of Jak2, wild type Stat3 (HA-ST3) andTyr-705 mutant (HA-Y1) co-precipitated with ERK2antibody (lanes 3 and 5) almost equally. However, theSer-727 mutant (HA-S1) showed a 60% decrease inbinding to ERK2 (lane 7) and no Stat3 protein wasdetected in the cells co-transfected with ERK2 and thecontrol HA vector (lane 1). Interestingly, the binding ofERK2 to the wild type Stat3 (HA-ST3) was inhibitedalmost completely with co-expression of Jak2 (lane 4),whereas the binding to the Tyr-705 mutant (HA-Y1)remained unchanged with or without Jak2 expression(lane 6). The weak binding of Ser-727 mutant (HA-S1)to ERK2 was completely abolished (lane 8) in thepresence of Jak2. The blot was stripped and reprobedwith anti-ERK2 antibody showing an approximately
Figure 2 Mobility shift DNA binding assays. COS-1 cells wereco-transfected with pRc/CMV-Stat3 and various e�ector plasmidsas indicated on top of the ®gure. The crude nuclear extracts wereprepared and used for the mobility shift DNA binding assay withhSIE as a probe as described in Materials and methods. Thecompetition assay was performed with 100-fold excess ofunlabeled oligonucleotides of either wild type (WT) or themutant hSIEs (M1 and M2). FP (lane 1) indicates free probe
Figure 3 MEK1-activated ERK2 inhibits Stat3 tyrosine phos-phorylation stimulated by Src. COS-1 cells were co-transfectedwith Stat3 and Src without or with ERK2 and/or MEK1D. Thecell lysates were immunoprecipitated with anti-Stat3 antibody (IP)and probed with anti-phosphotyrosine antibody 4G10. The boundantibodies were visualized by using an ECL kit as shown on theupper panel. The molecular mass markers are shown on the right.The blot was stripped and re-probed with anti-Stat3 antibody asshown in the bottom panel
ERK2 represses Stat3N Jain et al
3160
equal amount of ERK2 present in each sample (Figure5a lower panel). The weak band just below Stat3 in eachsample (shown in Figure 5a, upper panel) was a proteinreacting with the anti-HA antibody non-speci®cally.Similar results were obtained when Jak2 was replacedwith a Src expression plasmid in the above experiments(data not shown). Together, these results suggest thatSer-727 is important for ERK2 association, whereastyrosine phosphorylation on Tyr-705 either by Jak2 orSrc kinase, may negatively regulate the binding of Stat3to ERK2. The e�ects of these mutants on the ERK2association are likely to be due to phosphorylation.However, it cannot be ruled out that conformationalchanges of the mutant proteins caused by the mutationor some other unknown factor(s) may a�ect the a�nityof ERK2 binding.The expression levels of ERK2 and Stat3 proteins in
each transfected sample were measured by Westernblot analysis of the whole cell extract. Only a smallvariation in Stat3 expression was observed withapproximately a 10% increase in HA-Y1 (Figure 5b,lanes 5 and 6) and a 10% decrease in HA-S1 (lanes 7and 8) compared to the wild type (lanes 3 and 4), andno change was observed in the expression of ERK2 ineach sample (not shown). This suggests that thedi�erential association of ERK2 and the Stat3mutants is mainly due to the mutations themselves,and not the variation in the expression.
E�ect of ERK2 phosphorylation on Stat3/ERK2association
ERK2 is activated by dual phosphorylation on tyrosineand threonine in its activation motif TEY by
stimulation of various growth factors and cytokines.We therefore tested the e�ect of ERK2 phosphoryla-tion on the Stat3 association. COS-1 cells were co-transfected with ERK2 and the wild-type or the Tyr-705 mutant Stat3 and treated with tyrosine kinaseinhibitors genistein and staurosporine, serine/threoninekinase inhibitor H7, or the serine/threonine phospha-tase inhibitor okadaic acid for 1 h before harvesting.Both genistein (Figure 6a, lanes 4 ± 6) and staurospor-ine (lanes 7 ± 9) dramatically reduced the binding ofERK2 to the wild type (HA-ST3) and the Tyr-705mutant (HA-Y1), whereas H7 did not a�ect thebinding signi®cantly (Figure 6b, lanes 4 ± 6). Incontrast, okadaic acid treatment enhanced the associa-tion of ERK2 with both the wild type and the Tyr-705mutant (Figure 6b, upper panel, lanes 7 ± 9). Corre-spondingly, a basal level of tyrosine phosphorylation ofthe transiently expressed ERK2 (Figure 6a, lowerpanel, lanes 1 ± 3) was detected which was reduced bygenestein and staurosporine treatments (lanes 4 ± 9),una�ected by H7 (Figure 6b, lower panel, lanes 4 ± 6),and increased by okadaic acid (Figure 6b, lanes 7 ± 9).These data suggest that the level of ERK2 tyrosine
Figure 4 Association of Stat3 and ERK2 in IND-REF-src cells.Cells were grown in DMEM with 10% fetal calf serum. The cellswere either left untreated (7) or treated with DM (+) for 16 h,and lysed in 1 ml RIPA bu�er. Lysates were immunoprecipitated(IP) with antibodies indicated on top of the ®gures. PI representsthe rabbit preimmune serum. The immunocomplexes wereresolved by SDS±PAGE, transferred to a membrane and blottedwith anti-ERK2 antibody (a). The blot was stripped and re-probed with anti-Stat3 antibody as indicated at the bottom of (b).Stat3 and ERK2 are indicated by arrows and the molecular massmarkers are shown in the middle
Figure 5 Interaction of ERK2 with wild type and mutant Stat3in vivo and the e�ect of Jak2 on this interaction. COS-1 cells wereco-transfected with ERK2 and control plasmid pXJ40-HA (HA)or various HA-tagged Stat3 constructs (HA-ST3, HA-Y1, andHA-S1) as indicated on top, with (+) or without (7) Jak2expression plasmid. (a) The cell lysates were incubated with anti-ERK2 antibody (IP) and probed with anti-HA antibody (upperpanel). The blots were stripped and re-probed with anti-ERK2(lower panel of A). The molecular mass markers are shown on theright. (b) Expression levels of transfected Stat3. 10 mg of wholecell lysates from the transfected cells as described in (a) wereloaded in each lane and the expression of Stat3 was analysed byWestern blot analysis using anti-Stat3 antibody
ERK2 represses Stat3N Jain et al
3161
phosphorylation correlates with its ability to associatewith Stat3.The e�ect a speci®c inhibitor of MEK1, PD98059
(Dudley et al., 1995), on association was also analysed.Transfected COS-1 cells were treated with the inhibitorfor 1 h before harvesting and the ERK2/Stat3association was examined. As illustrated in Figure6c, PD98059 inhibited ERK2 phosphorylation (lanes4 ± 6, lower panel) and also reduced ERK2 binding towild type Stat3 and the Tyr-705 mutant (comparelanes 5 and 6 to the controls in lanes 2 and 3, upperpanel). Next, we examined the e�ect of MEK1D onERK2/Stat3 association by co-transfecting COS-1 cellswith Stat3, ERK2 and MEK1. As shown in Figure 6d,unlike Jak2 and Src kinase which inhibited the
association, MEK1 increased the association ofERK2 not only with the wild type Stat3 and Tyr-705 mutant (lanes 2 and 3), but also with the Ser-727and the Tyr-705/Ser-727 double mutants (lanes 4 and5). The later two bound to ERK2 poorly in theabsence of MEK1D (Figure 5a and not shown). Strongtyrosine phosphorylation of ERK2 by MEK1D isshown at the bottom panel of Figure 6d. Together,these data indicate that inhibition of ERK2 phosphor-ylation by either tyrosine kinase inhibitors genisteinand staurosporine or by the speci®c inhibitor ofMEK1 reduces its association with Stat3, whereasincreasing ERK2 phosphorylation by MEK1 or byinhibition of protein phosphatases enhances its bindingability to Stat3.
Figure 6 E�ect of ERK2 phosphorylation on its binding to Stat3. COS-1 cells were co-transfected with pXJ40-HA (HA), HA-Stat3(ST3), or HA-Y1 (Y1) together with ERK2 expression vector. (a) The cells were treated with 10 mM of genistein (New EnglandBiolabs) (lanes 4 ± 6), 10 mM of staurosporine (Clontech) (lanes 7 ± 9), or left untreated (lanes 1 ± 3), or (b) treated with either 25 mMof H7 (Biomol) (lanes 4 ± 6) or 0.5 mM of okadaic acid (OA, Biomol) (lanes 7 ± 9) for 1 h, or left untreated (lanes 1 ± 3). The celllysates were immunoprecipitated with anti-ERK2 and probed with anti-HA antibodies (upper panels). The blots were stripped andre-probed with anti-phosphotyrosine antibody 4G10 (lower panels). The molecular mass markers are shown on the right. (c)Transfected cells were left untreated (lanes 1 ± 3) or treated with 10 mM of PD98059 (PD, New England Biolabs) for 1 h (lanes 4 ± 6)and the IP/BLOT experiment was performed as described in (a and b). (d) COS-1 cells were co-transfected with ERK2 and variousHA-Stat3 constructs, together with MEK1D, and the IP/BLOT experiment was performed
ERK2 represses Stat3N Jain et al
3162
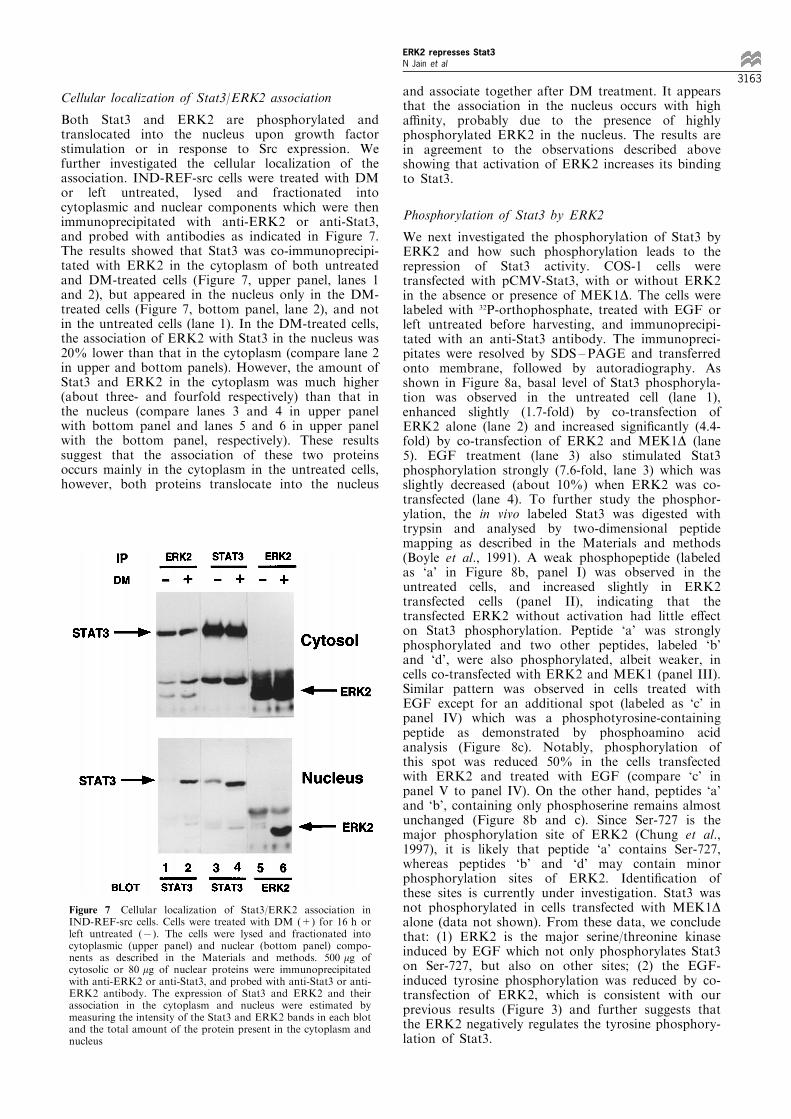
Cellular localization of Stat3/ERK2 association
Both Stat3 and ERK2 are phosphorylated andtranslocated into the nucleus upon growth factorstimulation or in response to Src expression. Wefurther investigated the cellular localization of theassociation. IND-REF-src cells were treated with DMor left untreated, lysed and fractionated intocytoplasmic and nuclear components which were thenimmunoprecipitated with anti-ERK2 or anti-Stat3,and probed with antibodies as indicated in Figure 7.The results showed that Stat3 was co-immunoprecipi-tated with ERK2 in the cytoplasm of both untreatedand DM-treated cells (Figure 7, upper panel, lanes 1and 2), but appeared in the nucleus only in the DM-treated cells (Figure 7, bottom panel, lane 2), and notin the untreated cells (lane 1). In the DM-treated cells,the association of ERK2 with Stat3 in the nucleus was20% lower than that in the cytoplasm (compare lane 2in upper and bottom panels). However, the amount ofStat3 and ERK2 in the cytoplasm was much higher(about three- and fourfold respectively) than that inthe nucleus (compare lanes 3 and 4 in upper panelwith bottom panel and lanes 5 and 6 in upper panelwith the bottom panel, respectively). These resultssuggest that the association of these two proteinsoccurs mainly in the cytoplasm in the untreated cells,however, both proteins translocate into the nucleus
and associate together after DM treatment. It appearsthat the association in the nucleus occurs with higha�nity, probably due to the presence of highlyphosphorylated ERK2 in the nucleus. The results arein agreement to the observations described aboveshowing that activation of ERK2 increases its bindingto Stat3.
Phosphorylation of Stat3 by ERK2
We next investigated the phosphorylation of Stat3 byERK2 and how such phosphorylation leads to therepression of Stat3 activity. COS-1 cells weretransfected with pCMV-Stat3, with or without ERK2in the absence or presence of MEK1D. The cells werelabeled with 32P-orthophosphate, treated with EGF orleft untreated before harvesting, and immunoprecipi-tated with an anti-Stat3 antibody. The immunopreci-pitates were resolved by SDS ±PAGE and transferredonto membrane, followed by autoradiography. Asshown in Figure 8a, basal level of Stat3 phosphoryla-tion was observed in the untreated cell (lane 1),enhanced slightly (1.7-fold) by co-transfection ofERK2 alone (lane 2) and increased signi®cantly (4.4-fold) by co-transfection of ERK2 and MEK1D (lane5). EGF treatment (lane 3) also stimulated Stat3phosphorylation strongly (7.6-fold, lane 3) which wasslightly decreased (about 10%) when ERK2 was co-transfected (lane 4). To further study the phosphor-ylation, the in vivo labeled Stat3 was digested withtrypsin and analysed by two-dimensional peptidemapping as described in the Materials and methods(Boyle et al., 1991). A weak phosphopeptide (labeledas `a' in Figure 8b, panel I) was observed in theuntreated cells, and increased slightly in ERK2transfected cells (panel II), indicating that thetransfected ERK2 without activation had little e�ecton Stat3 phosphorylation. Peptide `a' was stronglyphosphorylated and two other peptides, labeled `b'and `d', were also phosphorylated, albeit weaker, incells co-transfected with ERK2 and MEK1 (panel III).Similar pattern was observed in cells treated withEGF except for an additional spot (labeled as `c' inpanel IV) which was a phosphotyrosine-containingpeptide as demonstrated by phosphoamino acidanalysis (Figure 8c). Notably, phosphorylation ofthis spot was reduced 50% in the cells transfectedwith ERK2 and treated with EGF (compare `c' inpanel V to panel IV). On the other hand, peptides `a'and `b', containing only phosphoserine remains almostunchanged (Figure 8b and c). Since Ser-727 is themajor phosphorylation site of ERK2 (Chung et al.,1997), it is likely that peptide `a' contains Ser-727,whereas peptides `b' and `d' may contain minorphosphorylation sites of ERK2. Identi®cation ofthese sites is currently under investigation. Stat3 wasnot phosphorylated in cells transfected with MEK1Dalone (data not shown). From these data, we concludethat: (1) ERK2 is the major serine/threonine kinaseinduced by EGF which not only phosphorylates Stat3on Ser-727, but also on other sites; (2) the EGF-induced tyrosine phosphorylation was reduced by co-transfection of ERK2, which is consistent with ourprevious results (Figure 3) and further suggests thatthe ERK2 negatively regulates the tyrosine phosphory-lation of Stat3.
Figure 7 Cellular localization of Stat3/ERK2 association inIND-REF-src cells. Cells were treated with DM (+) for 16 h orleft untreated (7). The cells were lysed and fractionated intocytoplasmic (upper panel) and nuclear (bottom panel) compo-nents as described in the Materials and methods. 500 mg ofcytosolic or 80 mg of nuclear proteins were immunoprecipitatedwith anti-ERK2 or anti-Stat3, and probed with anti-Stat3 or anti-ERK2 antibody. The expression of Stat3 and ERK2 and theirassociation in the cytoplasm and nucleus were estimated bymeasuring the intensity of the Stat3 and ERK2 bands in each blotand the total amount of the protein present in the cytoplasm andnucleus
ERK2 represses Stat3N Jain et al
3163
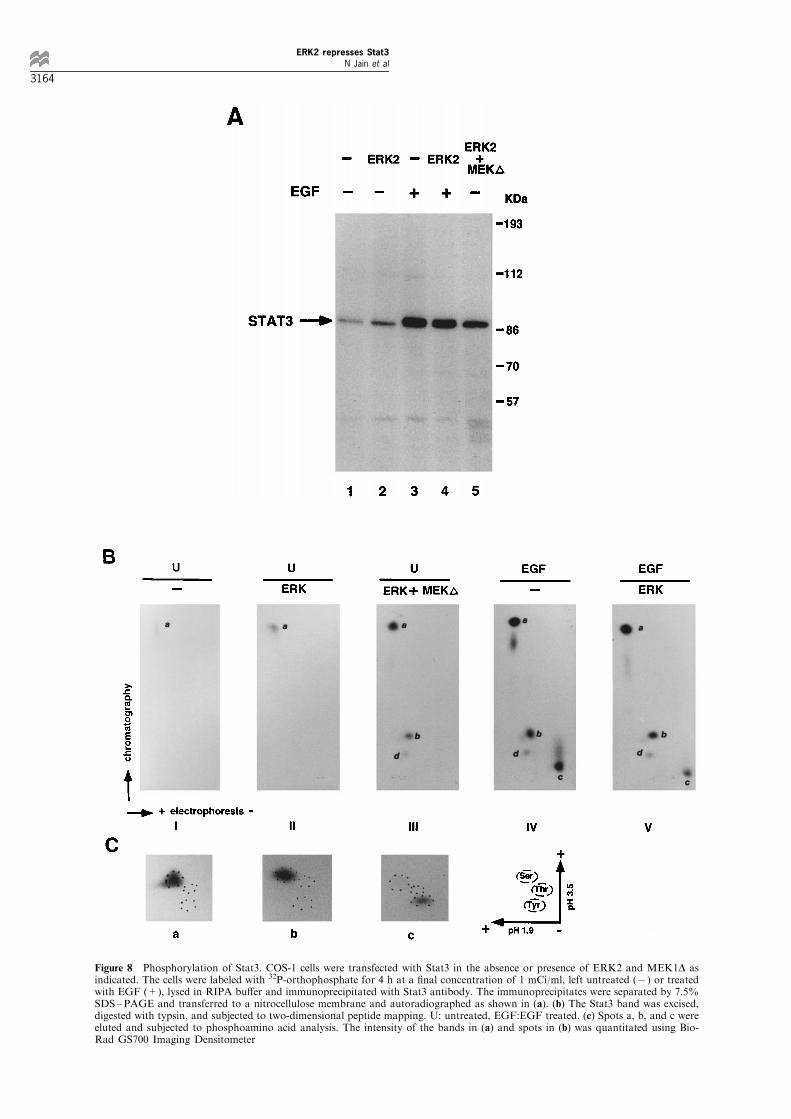
Figure 8 Phosphorylation of Stat3. COS-1 cells were transfected with Stat3 in the absence or presence of ERK2 and MEK1D asindicated. The cells were labeled with 32P-orthophosphate for 4 h at a ®nal concentration of 1 mCi/ml, left untreated (7) or treatedwith EGF (+), lysed in RIPA bu�er and immunoprecipitated with Stat3 antibody. The immunoprecipitates were separated by 7.5%SDS±PAGE and transferred to a nitrocellulose membrane and autoradiographed as shown in (a). (b) The Stat3 band was excised,digested with typsin, and subjected to two-dimensional peptide mapping. U: untreated, EGF:EGF treated. (c) Spots a, b, and c wereeluted and subjected to phosphoamino acid analysis. The intensity of the bands in (a) and spots in (b) was quantitated using Bio-Rad GS700 Imaging Densitometer
ERK2 represses Stat3N Jain et al
3164

Discussion
The role of serine phosphorylation on Stat1 and Stat3activities was ®rst reported by Zhang et al. (1995)showing the requirements of such phosphorylation forthe DNA binding. Subsequently, the consensusphosphorylation site of MAPK in STATs (Ser-727 inboth Stat1 and Stat3) was demonstrated to be requiredfor the maximum transcription but not for the DNAbinding activity through mutational studies on this site(Wen et al., 1995; Wen and Darnell, 1997). Co-precipitation of Stat1 with ERK2 in the IFN-b-induced cells and reduction of IFN-b-induced genetranscription by dominant negative mutant ERK2 werealso reported (David et al., 1995). These resultssuggested that MAPK acts as a positive signal in theregulation of STAT activity. Since both Stat3 andMAPK are phosphorylated and activated by growthfactors, such as PDGF, EGF, and CSF-1, weinvestigated the role of MAPK phosphorylation onStat3 function by testing the e�ect of ERK2 on Stat3transcriptional activity. Unexpectedly, we found thatexpression of MAPK activated by its upstream kinase,MEK1, represses Stat3 transcriptional activity.Furthermore, its DNA binding and tyrosine phosphor-ylation were also inhibited. These data suggest MAPKnegatively regulates Stat3 activity. Recently, tworeports addressing the role of MAPK and the serinephosphorylation on STATs have been published.Chung et al. (1997) reported that Stat3 serinephosphorylation is mediated through an ERK-depen-dent pathway in response to EGF and negativelymodulates its tyrosine phosphorylation. Our results areconsistent with the report and further demonstrate thatthe negative e�ect on tyrosine phosphorylation resultsin the inhibition of its DNA binding and transcrip-tional activity. Serine phosphorylation of Stat3 in theIL-6-induced cells, however, was found to be ERK-independent as reported by the same group (Chung etal., 1997). Another report by Zhu et al. (1997)indicated that MAPK is weakly activated in responseto IFN-g and is not responsible for Stat1 serinephosphorylation. These data suggest the e�ect ofserine phosphorylation on STATs could be acomplicated issue. The individual STAT protein maybe phosphorylated by di�erent kinases in a ligand-dependent manner. Furthermore, di�erent levels of theserine phosphorylation may lead to opposite e�ects onSTAT activity. The precise e�ects of serine phosphor-ylation on STATs requires further studies.To elucidate the possible mechanisms of the
repression, we investigated Stat3 phosphorylation byERK2 using two-dimensional peptide mapping. Threepeptides were found to be phosphorylated on serine(`a', `b' and `d' in Figure 8b) by activated ERK2 orEGF stimulation. The strong phosphorylated peptide`a' probably represents Ser-727 since it has beendemonstrated to be the major phosphorylation site byERK (Chung et al., 1997) and the identity of the othertwo is unknown. Peptide `c' is the only tyrosinephosphorylated peptide which is likely to containTyr-705 identi®ed previously (Kaptein et al., 1996).Expression of transfected ERK2 in the EGF-treatedcells did not further increase the serine phosphoryla-tion, but instead, it reduced the tyrosine phosphoryla-tion (Figure 8b, panels IV and V). Therefore, the
repression of Stat3 activity by ERK2 is probably theresult of decreased tyrosine phosphorylation, and notby directly increasing the serine phosphorylation.To further address the relation between serine and
tyrosine phosphorylation, we examined the time courseof Stat3 phosphorylation in response to EGFtreatment. The phosphoamino acid analysis of invivo-labeled Stat3 showed that phosphorylation ontyrosine appeared at 30 s, increased signi®cantly at 2and 8 min and continued to increase at 15 min,whereas a constitutive serine phosphorylation wasobserved in the untreated cells which increased at8 min and was further enhanced at 15 min (data notshown). These results are consistent to those observedin IL-6-induced HepG2 cells (LuÈ tticken et al., 1995)and IFN-g-induced cells for Stat1 (Zhu et al., 1997),indicating that tyrosine phosphorylation occurs priorto serine. These data support the hypothesis that serinephosphorylation may be involved in the inactivation ofSTAT activity.We further investigated the mechanism of ERK2
inhibition on Stat3 tyrosine phosphorylation by testingthe interaction of ERK2 and Stat3 and demonstratedtheir association in vivo. To further characterize thisassociation, we examined the e�ects of Stat3 andERK2 phosphorylation on their association. MAPKrecognition site on Stat3, Ser-727, is important for theassociation since mutation on this site reduced theassociation drastically. On the other hand, tyrosinephosphorylation by Jak2 or c-Src reduced the wildtype, but not the Tyr-705 mutant of Stat3 binding toERK2. An increase of ERK2 phosphorylationmediated by okadaic acid, enhances its binding toStat3 (Figure 6b). Furthermore, ERK2 phosphoryla-tion by its upstream kinase MEK1 increased not onlyits association with the wild type Stat3, but also withthe Ser-727 mutant which could not associate withERK2 in the absence of MEK1 in the uninduced cells(Figure 6d). This suggests a high a�nity binding ofStat3 with the activated ERK2 which can override therequirement of the serine phosphorylation. In agree-ment with this, we also detected a high a�nityassociation appearing in the nucleus after v-Srcexpression. Therefore, it appears that two types ofbinding exist: unphosphorylated ERK2 binds Stat3with low a�nity in unstimulated cells in the cytoplasmwhich can be replaced by the other Stat3 molecule,whereas phosphorylated ERK2 binds to Stat3 withhigh a�nity, possibly in the nucleus, which negativelymodulates Stat3 activity by decreasing its Tyr-phosphorylation through an as yet unknown mechan-ism. Surprisingly, unlike c-Src and Jak2, v-Srcexpression seems not to a�ect Stat3/ERK2 associationin the v-Src transformed cells. The reason for this isunknown and one possible explanation could be thestrong phosphorylation of ERK2 by v-Src which leadsto an increase in the association.Based on these data, we propose the following
model: ERK2 associates with Stat3 in the untreatedcells and may phosphorylate Stat3 on serine at lowlevel that is required for the full transcriptional activityof Stat3. After growth factor stimulation, the ERK2/Stat3 association is rapidly disrupted by formation ofStat3 dimer in the cytoplasm. Meanwhile, ERK2 itselfis also activated which negatively modulates Stat3activity by further phosphorylation and/or by tight
ERK2 represses Stat3N Jain et al
3165

association with Stat3. Further experiments are beingcarried out to address this hypothesis.
Materials and methods
Construction of expression plasmids
The expression plasmids of Stat3, pRc/CMV-Stat3 (Zhonget al., 1994), and Stat1, pMNC-91 (Shuai et al., 1993), wereobtained from Dr JE Darnell (The Rockefeller University,USA). Glutathione-S-transferase (GST)-Stat3 fusion pro-tein (GST-Stat3) containing an almost full-length Stat3 wasconstructed as described previously (Cao et al., 1996).Point mutations of GST-Stat3 were prepared by using thePCR-based site-directed mutagensis kit ExSiteTM (Strata-gene, La Jolla, CA, USA) following the manufacturer'sinstructions. The resultant plasmids contain mutations atTyr-705 (replaced by Phe), Ser-727 (replaced by Ala), ordouble mutations at both positions and named as GST-Y1,GST-S1, or GST-YS1. All the point mutations werecon®rmed by DNA sequencing. Stat3 expression plasmidsfor transfection were prepared by inserting the EcoRI ±XhoI fragments containing amino acids 28 ± 770 of Stat3from GST-Stat3 or GST-Stat3 mutants into expressionvector pcDNA3 (Invitrogen, San Diego, CA, USA). TheBamHI ±XhoI fragments from pcDNA3-Stat3 constructswere inserted into plasmid pXJ40-HA (a hemagglutininepitope-tagged expression vector from Dr E Manser of ourinstitute), so that the HA was tagged at NH2-terminal ofStat3 and its mutants. The plasmids were named as HA-ST3, HA-Y1, HA-S1 and HA-YS1 respectively. Theexpression vectors containing wild type chicken Src(pSGTsrc K+) and kinase inactive Src (pSGTsrcK7) wereobtained from Dr SA Courtneidge (Sugen Inc., RedwoodCity, CA, USA) as described by Barone and Courtneidge(1995). Wild type Jak2 and the kinase defective mutant ofJak2 expression vectors (Silvennoinen et al., 1993) wereobtained from Dr O Silvennoinen (University of Helsinki,Finland) and the Elk-1 expression plasmid MLV-Elk-1(Marais et al., 1993) was obtained from Dr R Treisman(Imperial Cancer Research Fund, London, UK). TheERK2 expression plasmid pcDNA3.ERK2 was preparedby inserting the ERK2 coding sequence from plasmidpBA4 (Her et al., 1991) into pcDNA3.
The plasmid MEK1D (DN2/S218E/S222D) expresses aconstitutively active MAPK kinase as described by Mansouret al. (1994), and the mutant MEK1D7 which lacks thekinase activity with substitution of lysine 97 by methionine,were obtained from Dr A Whitmarsh (University ofMassachusetts Medical School, Worcester, MA, USA).Plasmid expressing normal MEK1 was obained from Dr.Richard Jove (University of South Florida, Tampa, FL,USA). The reporter plasmid pSIE-CAT for CAT assay wasprepared by inserting three copies of the high a�nity SIEconsensus sequence (TTCCCGTAA) upstream of a c-fosminimal promoter followed by the CAT gene in plasmidpFOSCATD56 (Gilman et al., 1986).
DNA transfection, CAT assay and mobility shift DNA bindingassay
COS-1 and NIH3T3 cells were grown in DMEM with 10%fetal calf serum purchased from Gibco BRL LifeTechnologies (Grand Island, NY, USA) or Hyclone(Logan, UT, USA). Transfection of the plasmids intoCOS-1 cells was performed by the calcium phosphate-DNAcoprecipitation method as described previously (Gorman etal., 1982). For CAT-assays, the cells were co-transfectedwith 4 mg CAT-containing reporter plasmids, 5 mg ofexpression plasmids, and 2 mg of pCMV-b-gal containingbacterial b-galactosidase gene. The total amount of DNA
transfected was kept constant at 20 mg per plate withcontrol plasmid pCMV-5. The cells were maintained inDMEM with 10% fetal calf serum and harvested 45 h aftertransfection. Transfection was also performed withlipofectamine reagent (GIBCO BRL Life Technologies,Gaithersburg, MD, USA) in NIH3T3 cells for CAT assaysfollowing the manufacturer's instruction. The cells werelysed in 0.25 M Tris-HCl (pH 8.0) with three `freeze-thaw'cycles. The lysate was spun and the supernatant wascollected and used for b-galactosidase activity and CATassays. The pellets were resuspended in high salt bu�er(20 mM HEPES (N-2-hydroxyethylpiperazine-N'-2-ethane-sulfonic acid), pH 7.9, 1 mM EDTA, 1 mM EGTA, 420 mM
NaCl, 20% glycerol, 1 mM Na4P2O7, 1 mM Na3VO4, 20 mM
NaF, 1 mM DTT and 0.5 mM PMSF) as crude nuclearextract for DNA mobility shift assay. The amount ofcytoplasmic extract used in each CAT assay was normal-ized with equivalent b-galactosidase activity. Acetylatedand nonacetylated forms of [14C]chloramphenicol wereseparated by thin layer chromatography, followed byautoradiography and quanti®cation using a Bio-RadGS700 Imaging Densitometer. Each transfection andCAT assay was repeated at least three times. The crudenuclear extracts were used for the mobility shift DNAbinding assay with hSIE as a probe under conditionsdescribed previously except in the absence of salt in thebinding bu�er (Cao et al., 1996). The competition assaywas performed with 100-fold excess of unlabeled oligonu-cleotides of either wild type or the mutant hSIEs in whichthe core hSIE sequence TTCCCGTAA was mutated toTTCACGTCA (M1) or TTCCAGGAG (M2).
Antibodies, immunoprecipitation and immunoblotting
Antibodies against Stat1, Stat3, ERK1, and ERK2 werepurchased from Transduction Laboratories (Lexington,KY), and Santa Cruz Biotechnology (Santa Cruz, CA,USA). Antiphosphotyrosine antibody, 4G10, was pur-chased from Upstate Biotechnology Incorporated (UBI)(Lake Placid, NY, USA). Polyclonal antibody against GSTfusion protein of Stat3 COOH-terminus was produced asdescribed previously (Cao et al., 1996).
The COS-1 cells were co-transfected with 5 mg of Stat3expression plasmids, and 5 mg of plasmids expressing eitherSrc, Jak2 or ERK2. Preparation of total cell lysates,immunoprecipitation (IP) and immunoblotting (Blot) wereperformed as described previously (Cao et al., 1996).
Separation of nuclear and cytoplasmic components
The nuclear and cytoplasmic fractions were separated asdescribed previously (Cao et al., 1993) with minormodi®cations. Cells were washed with PBS three times,resuspended in Bu�er A containing 10 mM HEPES,pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 1 mM
PMSF and 0.5 mM Nonidet P-40 with protease andphosphatase inhibitors, incubated on ice for 10 min, andhomogenized in a Dounce homogenizer with 25 strokes.The nuclei were removed by centrifugation and thesupernatant was collected as cytoplasmic portion. Thenuclear proteins were extracted in RIPA and used for IP/Blot experiments.
32P-orthophosphate labeling, phosphoamino acid analysis andpeptide mapping
COS-1 cells were transfected and labeled for 4 h beforeharvesting as described previously (Jain et al., 1996) exceptthat 32P-orthophosphate was used at a ®nal concentrationof 1 mCi/ml. Cells were lysed in RIPA bu�er withappropriate protease and phosphatase inhibitors. Trans-fected Stat3 was immunoprecipitated from the lysates by
ERK2 represses Stat3N Jain et al
3166

anti-Stat3 antibody and the immunoprecipitates werewashed two times with RIPA bu�er and two times withPBS. The proteins were separated by 7.5% SDS ± PAGEand transferred to a nitrocellulose membrane. The Stat3band, located after an overnight exposure to X-ray ®lm,was excised, digested with trypsin and subjected to peptidemapping. The phosphopeptide was eluted from the TLCplate with 20% acetonitrile and used for phosphoaminoacid analysis as described (Boyle et al., 1991).
AcknowledgementsThe ®rst two authors contributed equally to this work. Weare grateful to Drs SA Courtneidge for Src and
O Silvennoinen for Jak2 expression plasmids, JE DarnellJr and X-Y Fu for Stat3 and Stat1 expression plasmids, JPouysse gur for p42MAPK, MJ Weber for pBA4 plasmid, NGAhn and A Whitmarsh for mutant MEK plasmids, EManser for pXJ40-HA vector, R Treisman for MLV-Elk-1and R Jove for MEK1. We thank Mr F Leong and Mr RTham for photography. This work was supported by theNational Science and Technology Board of Singapore.
References
Akira S, Nishio Y, Inoue M, Wang XJ, Wei S, Matsusaka T,Yoshida K, Sudo T, Naruto M and Kishimoto T. (1994).Cell, 77, 63 ± 71.
Barone MV and Courtneidge SA. (1995). Nature, 378, 509 ±512.
Boyle WJ, van der Geer P and Hunter T. (1991). MethodsEnzymol., 201, 110 ± 149.
Cao X, Mahendren R, Guy GR and Tan YH. (1993). J. Biol.Chem., 268, 16949 ± 16957.
Cao X, Tay A, Guy GR and Tan YH. (1996). Mol. Cell.Biol., 16, 1595 ± 1603.
Chaturvedi P, Sharma S and Reddy EP. (1997). Mol. Cell.Biol., 17, 3295 ± 3304.
Chung J, Uchida E, Grammer TC and Blenis J. (1997). Mol.Cell. Biol., 17, 6508 ± 6516.
Darnell JE Jr, Kerr IM and Stark GR. (1994). Science, 264,1415 ± 1421.
David M, Petricoin E III, Benjamin C, Pine R, Weber MJand Larner AC. (1995). Science, 269, 1721 ± 1723.
Dudley DT, Pang L, Decker SJ, Bridges AJ and Saltiel AR.(1995). Proc. Natl. Acad. Sci. USA, 92, 7686 ± 7689.
Fu X-Y and Zhang J-J. (1993). Cell, 74, 1135 ± 1145.Gilman MZ, Wilson RN and Weinberg RA. (1986). Mol.
Cell. Biol., 6, 4305 ± 4316.Gorman CM, Mo�at LF and Howard BH. (1982).Mol. Cell.
Biol., 2, 1044 ± 1051.Heim MH, Kerr IM, Stark GR and Darnell JE Jr. (1995).
Science, 267, 1347 ± 1349.Her JH, Wu J, Rall TB, Sturgill TW and Weber MJ. (1991).
Nucleic Acids Res., 19, 3743.Hill CS and Treisman R. (1995). Cell, 80, 199 ± 211.Ihle JN, Witthuhn BA, Quelle FW, Yamamoto K,Thierfelder WE, Kreider B and Silvennoinen O. (1994).Trends Biochem. Sci., 19, 222 ± 227.
Ihle JN. (1996). Cell, 84, 331 ± 334.Jain N, Mahendran R, Philip R, Guy GR, Tan YH and CaoX. (1996). J. Biol. Chem., 271, 13530 ± 13536.
Kaptein A, Paillard V and Saunders M. (1996). J. Biol.Chem., 271, 5961 ± 5964.
Leaman DW, Pisharody S, Flickinger TW, Commane MA,Schlessinger J, Kerr IM, Levy DE and Stark GR. (1996).Mol. Cell. Biol., 16, 369 ± 375.
Leevers SJ, Paterson HF and Marshall CJ. (1994). Nature,369, 411 ± 414.
LuÈ tticken C, Co�er P, Yuan J, Schwartz C, Caldenhoven E,Schindler C, Kruijer W, Heinrich PC and Horn F. (1995).FEBS Lett., 360, 137 ± 143.
Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S,Fukasawa K, Vande Woude GF and Ahn NG. (1994).Science, 265, 966 ± 970.
Marais R, Wynne J and Treisman R. (1993). Cell, 73, 381 ±393.
Marshall CJ. (1994). Curr. Opin. Genet. Dev., 4, 82 ± 89.Sadowski HB, Shuai K, Darnell Jr JE and Gilman MZ.(1993). Science, 261, 1739 ± 1744.
Schindler C, Fu X-Y, Improta T, Aebersold R and Darnell JrJE. (1992). Proc. Natl. Acad. Sci. USA, 89, 7836 ± 7839.
Schindler C and Darnell Jr JE. (1995). Ann. Rev. Biochem.,64, 621 ± 651.
Shaw PE, SchroÈ ter H and Nordheim A. (1989). Cell, 56,563 ± 572.
Shuai K, Schindler C, Prezioso VR and Darnell Jr JE. (1992).Science, 258, 1808 ± 1812.
Shuai K, Stark GR, Kerr IM and Darnell Jr JE. (1993).Science, 261, 1744 ± 1746.
Silvennoinen O, Ihle JN, Schlessinger J and Levy DE. (1993).Nature, 366, 583 ± 585.
Stahl N, Boulton TG, Farruggella T, Ip NY, Davis S,Witthuhn BA, Quelle FW, Silvennoinen O, Barbieri G,Pellegrini S, Ihle JN and Yancopoulos GD. (1994).Science, 263, 92 ± 95.
Stahl N, Farruggella TJ, Boulton TG, Zhong Z, Darnell JrJE and Yancopoulos GD. (1995). Science, 267, 1349 ±1353.
Stokoe D, Macdonald SG, Cadwallader K, Symons M andHancock JF. (1994). Science, 264, 1463 ± 1467.
Sukhatme VP, Cao X, Chang LC, Tsai-Morris CH,Stamenkovich D, Ferreira PCP, Cohen DR, EdwardsSA, Shows TB, Curran T, Le BeauMM and Adamson ED.(1988). Cell, 53, 37 ± 43.
Treisman R. (1995). EMBO J., 14, 4905 ± 4913.Tsai-Morris CH, Cao X and Sukhatme VP. (1988). Nucleic
Acids Res., 16, 8835 ± 8846.Wen Z, Zhong Z and Darnell Jr JE. (1995). Cell, 82, 241 ±250.
Wen Z and Darnell Jr JE. (1997). Nucleic Acids Res., 25,2062 ± 2067.
Yu CL, Meyer DJ, Campbell GS, Larner AC, Caster-Su C,Schwartz J and Jove R. (1995). Science, 269, 81 ± 83.
Zhang X, Blenis J, Li H-C, Schindler C and Chen-Kiang S.(1995). Science, 267, 1990 ± 1994.
Zhong Z, Wen Z and Darnell Jr JE. (1994). Science, 264, 95 ±98.
Zhu X, Wen Z, Xu ZL and Darnell Jr JE. (1997). Mol. Cell.Biol., 17, 6618 ± 6623.
ERK2 represses Stat3N Jain et al
3167




















