
Reduced sarcolemmal dystrophin distribution and upregulation of utrophin in the cardiac and skeletal...
20
(OCopyright 1997by Humana Press Inc. All rights of any nature, whatsoever,reserved. 1044-7393/97/3102-187 $13.00 Reduced Sarcolernrnal Dystrophin Distribution and Upregulation of Utrophin in the Cardiac and Skeletal Muscles of CHF-146 Dystrophic Hamsters SYAMAL K. BHATTACHARYA, 1'2'3'4 PATTI L. JOHNSON, *'1'2 HUA-JU El, 1'2 RAJ K. HANDA, ~'~ AND THOMAS A. ADAMEC 5 ~Edward Dana Mitchell Surgical Research Laboratories, and Chemistry and Nutrient Data Output Laboratory, Departments of2Surgery, 3Anatomy & Neurobiology, 4Pharmaceutical Sciences, and 5Pathology, The University of Tennessee Medical Center, 956 Court Ave., Ste. B220 Memphis, TN 38163 Received September 27, 1996; Revised December 15, 1996; Accepted January 27, 1997 ABSTRACT Abnormalities in the dystrophic gene product, dystrophin, have been implicated in initiating the primary membrane defect and exces- sive intracellular calcium accumulation (EICA), which play fundamen- tal pathogenetic roles in hereditary muscular dystrophy (HMD). Two other cytoskeletal proteins, spectrin and utrophin, bear remarkable structural and functional homologies to dystrophin. CHF-146 strain dys- trophic hamsters (DH), like patients with Duchenne muscular dystro- phy (DMD), die prematurely from cardiopulmonary insufficiency, focal myonecrosis, and progressive degeneration of the cardiac and skeletal muscles with EICA. Although DH present a suitable model for HMD, there are controversies concerning their dystrophin and utrophin status. Using immunocytochemistry and Western blotting, we studied dys- trophin, spectrin, and utrophin anomalies in the cardiac and skeletal muscles of 6-To-old male DH. Age- and sex-matched CHF-148 strain albino normal hamsters (NH) served as controls. Sarcolemmal dys- *Author to whom all correspondence and reprint requests should be addressed. E-mail: pljohnson@utmem 1.utmem.edu Molecular and Chemical Neuropathology 187 Vol. 31, 1997
Transcript of Reduced sarcolemmal dystrophin distribution and upregulation of utrophin in the cardiac and skeletal...

(OCopyright 1997 by Humana Press Inc. All rights of any nature, whatsoever, reserved. 1044-7393/97/3102-187 $13.00
Reduced Sarcolernrnal Dystrophin Distribution and Upregulation
of Utrophin in the Cardiac and Skeletal Muscles of CHF-146
Dystrophic Hamsters SYAMAL K. BHATTACHARYA, 1'2'3'4 PATTI L. JOHNSON, *'1'2
HUA-JU El, 1'2 RAJ K. HANDA, ~'~ AND THOMAS A . ADAMEC 5
~Edward Dana Mitchell Surgical Research Laboratories, and Chemistry and Nutrient Data Output Laboratory, Departments
of2Surgery, 3Anatomy & Neurobiology, 4Pharmaceutical Sciences, and 5Pathology, The University of Tennessee Medical
Center, 956 Court Ave., Ste. B220 Memphis, TN 38163
Received September 27, 1996; Revised December 15, 1996; Accepted January 27, 1997
ABSTRACT
Abnormalities in the dystrophic gene product, dystrophin, have been implicated in initiating the primary membrane defect and exces- sive intracellular calcium accumulation (EICA), which play fundamen- tal pathogenetic roles in hereditary muscular dystrophy (HMD). Two other cytoskeletal proteins, spectrin and utrophin, bear remarkable structural and functional homologies to dystrophin. CHF-146 strain dys- trophic hamsters (DH), like patients with Duchenne muscular dystro- phy (DMD), die prematurely from cardiopulmonary insufficiency, focal myonecrosis, and progressive degeneration of the cardiac and skeletal muscles with EICA. Although DH present a suitable model for HMD, there are controversies concerning their dystrophin and utrophin status. Using immunocytochemistry and Western blotting, we studied dys- trophin, spectrin, and utrophin anomalies in the cardiac and skeletal muscles of 6-To-old male DH. Age- and sex-matched CHF-148 strain albino normal hamsters (NH) served as controls. Sarcolemmal dys-
*Author to whom all correspondence and reprint requests should be addressed. E-mail: pljohnson@utmem 1.utmem.edu
Molecular and Chemical Neuropathology 187 Vol. 31, 1997

188 Bhattacharya et al.
trophin staining was much weaker ,-rod interruptive in the DH. The den- sitometric analysis of the immunoblots revealed that dystrophin is reduced in DH by 83% in cardiac muscle (p < 0.0001), and by 50% in skeletal muscle (p < 0.0001). We conclude that sarcolemmal dystrophin distribution is markedly reduced and discontinuous in the cardiac and skeletal muscles of DH, with simultaneous upregulation of utrophin and a varied degree of spectrin labeling. This observation suggests that reduced sarcolemmal dystrophin is associated with membrane hyper- permeability, which leads to progressive muscle degeneration via EICA and segmental necrosis in DH. As in DMD, utrophin appears to play an important compensatory role in hamster dystrophinopathy.
Index Entries: Dystrophin-related protein; immunocytochem- istry; Western blotting; dystrophinopathy; hereditary muscular dys- trophy; hypertrophic cardiomyopathy.
Abbreviations: EICA, excessive intracellular calcium accumula- tion; HMD, hereditary muscular dystrophy; DMD, Duchenne muscu- lar dystrophy; BMD, Becker muscular dystrophy; DH, dystrophic hamsters; NH, normal hamsters; DAG, dystrophin-associated glyco- proteins; DRP, dystrophin-related protein; CBB, Coomassie brilliant blue; MAb, monoclonal antibody; PAb, polyclonal antibody.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is an early childhood heredi- tary muscular dystrophy (HMD) with no effective treatment (Worton and Burghes, 1988), in which a defect in the 2000-kb DMD gene is localized on the Xp 21 chromosome (Koenig et al., 1987; Zubrzycka-Gaarn et al., 1988; Bieber and Hoffman, 1990). DMD is associated with membrane-mediated excessive intracellular calcium accumulation (EICA), chronic cardiac and skeletal muscle degeneration, morpholo~c abnormalities, and 75-80% reduced life-span (Sanyal, 1980; Bertorini et al., 1982, 1984; Bhattacharya et al., 1989). Lack of the dystrophic gene product dystrophin, a 427-kDa sub- sarcolemmal protein, in DMD (Hoffman et al., 1987; Koenig et al., 1987; Arahata et al., 1988; Bonilla et al., 1988) and Becker muscular dystrophy (BMD) (Hoffman et al., 1988; Sklar et al., 1990; Morandi et al., 1990; Bulman, 1991; Byers et al., 1992), has revolutionized our concept of the likely patho- genetic mechanisms, involving primary membrane dysfunction and EICA, which are responsible for progressive muscle degeneration and premature death in t IMD, such as DMD, BMD, and severe childhood autosomal reces- sive muscular dystrophy (SCARMD).
Over the last two decades, many dystrophic animal models, i.e., chickens (Hudecki et al. 1984), C57BI./6J dy2J/dy2j dystrophic mice (Noireaud and L6oty 1984), mdx-mice (Turner et al., 1988; Partridge et al., 1989; Anderson et al., 1990; Menke and Jockusch, 1991), hamsters (Bajusz et al., 1969; Bhattacharya et al., 1982; Johnson and Bhattacharya, 1993;
Molecular and Chemical Neuropalhology Vol. 31, 1997

Dystrophin Deficiency in Dystrophic Hamsters 189
Iwata et al., 1993; Roberds et al., 1993; Yamanouchi et al., 1994; Mizuno et al., 1995), Xmd-dogs (Kornegay et al., 1988; Valentine et al., 1988), and cats (Carpenter et al., 1989), have been investigated in search of a suitable experimental model for DMD. The mdx-mice reveal primary deficiencies of sarcolemmal dystrophin and dystrophin-associated gly- coproteins (DAG) (Ervasti et al., 1990), but remain asymptomatic with- out significant functional deficits or shorter life-span (Johnson et al., 1991). In contrast, BIO-14.6 and CHF-146 strain dystrophic hamsters (DH), both of which are phenotypically and genotypically identical, dis- play a vast majority of the salient dystrophic features characteristic of DMD, including significant structural and functional deficits, voluntary muscle weakness, membrane-mediated EICA, cardiac involvement with electrocardiographic abnormalities, and premature death (Bhattacharya et al., 1987, 1989; Johnson and Bhattacharya, 1993). Most DMD patients, as well as BIO-14.6 and CHF-146 strains of DH, die of cardiopulmonary insufficiency owing to myocardial degeneration accompanied by membrane-mediated EICA, cardiomyopathy, myocytolysis, and sporadic necrosis (Bajusz et al., 1969; Paterson et al., 1972; Bhattacharya et al., 1987, 1989). Although CHF-146 strain DH have been shown to be a suit- able model for HMD (Hunter et al., 1984; Bhattacharya et al., 1987, 1993; Johnson and Bhattacharya, 1993), there are controversies concerning the relative abundance and distribution pattern of sarcolemmal dystrophin, spectrin (235 kDa) and utrophin, a -420-kDa dystrophin-related protein (DRP), in this model. Spectrin and utrophin are the two most important cytoskeletal proteins, bearing remarkable structural and functional homologies to dystrophin (Pearce et al., 1993). Two independent groups have suggested that utrophin is upregulated in DMD and BMD to com- pensate for inherent sarcolemmal dystrophin loss (Karpati et al., 1993; Mizuno et al., 1993, 1994). Dystrophin anomalies in DH, without any ref- erence to spectrin or utrophin, have been reported in the heart by Iwata et al. (1993), and in the cardiac and skeletal muscles by Roberds et al. (1993). Yamanouchi et al. (1994) reported a subtle reduction in dystrophin with no appreciable change in utrophin, but with a significant decrease in the 50- (adhalin), 43- (A3a), and 35-kDa DAG, in the skeletal muscle of 6- to 12-mo-old NSJ-my/my strain DH, which may be phenotypically different from the original BIO-14.6 stock with a likely mutation in some secondary loci. Later, Mizuno et al. (1995) characterized the dystroglycan and sarcoglycan subcomplexes in the cardiac and skeletal muscle of NSJ- my /my strain DH, describing changes in DAG, with a minute reduction in dystrophin, but utrophin and spectrin were not studied.
The CHF-146 strain DH express a phenotypicaUy recessive, and clin- ically progressive HMD, with severe involvement of the ventricular myocardium inducing hypertrophic cardiomyopathy and cardiopul- monary failure, EICA and fibrosis, identical to those commonly seen in DMD (Sanyal et al., 1980; Bhattacharya et al., 1989; Johnson and Bhat- tacharya, 1993). To overcome controversies of whether dystrophinopathy
Molecular and Chemical Neuropathology Vol. 31, 1997

190 Bhattacharya et al.
exists in DH, we investigated the staining pattern and relative abundance of sarcolemmal dystrophin, spectrin, and utrophin in the cardiac and skeletal muscles of 6-mo-old CHF-146 strain DH and corresponding CHF-148 strain normal albino hamsters (NH), by light microscopic immunocytochemistry and densitometric analysis of the Western blots using several monoclonal (MAbs) and polyclonal antibodies (PAbs).
MATERIALS AND METHODS
Animals
Six-month-old male, CHF-146 strain DH, as well as age- and sex- matched CHF-148 strain albino NH, were procured from Canadian Hybrid Farms, Nova Scotia, Canada. These animals were derived by selective inbreeding of the BIO-14.6 cardiomyopathic hamsters (Bio Research, Cam- bridge, MA), originally developed by Bajusz et al. (1969), and are genotyp- ically and phenotypically different from golden Syrian hamsters. The CHF-146 strain DH are homologous to the BIO-14.6 strain, and sponta- neously express HMD and hypertrophic cardiomyopathy, beginning as early as 22-25 d of age (Hunter et al., 1984; Johnson et al., 1994). Hamsters were maintained on Purina chow 5001 and tap water ad libitum, and kept in individual cages on a rigid 10-h dark and 14-h light cycle, in our air- conditioned vivarium. The experimental protocol involving animals adhered to the strict NIH guidelines, and was approved by the Institutional Animal Care and Use Committee of the University of Tennessee.
Materials
DYS1 (mid-rod domain), DYS2 (C-terminus), and DYS3 (N-termi- nus) antidystrophin monoclonal antibodies (MAb), raised against the specific domains of human dystrophin, as well as SPEC2 (MAb for spec- trin) and DRP1 (MAb for utrophin), were purchased from Novocastra Laboratories, Newcastle upon Tyne, UK. We also used two polyclonal antibodies (PAb) to dystrophin in this study, recognizing the fusion pro- teins that contain the 30- and 60-kDa peptide sequences of human dys- trophin (Hoffman et al., 1987). These antibodies were gifts from Dr. Louis Kunkel's laboratory, Children's Hospital, Boston, MA. All other chemi- cals used for immunocytochemical studies, sodium dodecyl sulfate- polyacrylamide gel electrophoresis (SDS-PAGE), and Western blotting were analytical grade, most of which were procured from Sigma (St. Louis, MO) and Bio-Rad (Hercules, CA).
Experimental Protocol
Upon arrival, hamsters were allowed to acclimate for several days. The animals were anesthetized by ip injection of sodium pentobarbital
Molecular and Chemical Neuropathology Vol. 31, 1997

Dystrophin Deficiency in Dystrophic Hamsters 191
(Abbott Laboratories, Chicago, IL), 80 mg/kg body wt. Biopsies were obtained from the ventricular myocardium and rectus femoris of anes- thetized hamsters, and immediately frozen in liquid nitrogen for the quantitation of dystrophin by densitometric analysis of the Western blots. Separate fresh biopsies were also obtained at sacrifice and snap-frozen in 2-methylbutane prechilled with liquid nitrogen for immunocytochemical localization of sarcolemmal dystrophin, spectrin, and utrophin in the ser- ial cryosections.
Light Microscopic Immunocytochemistry for Dystrophin Six-micrometer-thick serial transverse cryosections of snap-frozen
biopsies from the cardiac and skeletal muscles were recovered using a Tissue-Tek Microtome/Cryostat (Miles Laboratory, Elkhart, IN), and later thawed on poly-L-lysine (Sigma) coated slides for light microscopy. Phos- phate buffered saline (PBS) at pH 7.4, containing 10~ fetal calf serum (FCS), was used to block the sections for 15 min to reduce nonspecific binding. Three MAbs with different molecular specificities to dystrophin, DYS1 (mid-rod segment), DYS2 (C-terminus) and DYS3 (N-terminus), were used to distinguish the irnmunolabeling patterns of sarcolemmal dystrophin at the three strategically important, biologically active, spe- cific regions of the molecule. Each of the three MAbs was applied indi- vidually at a 1:20 dilution as the primary antibody to 2-4 serial cryosections on separate slides, and incubated for 1 h at ambient room temperature. The PAbs against dystrophin were used at a 1:200 dilution. Following five washes in PBS, the sections were reincubated with a biotinylated goat anti- mouse IgG secondary antibody (Sigma) for 45 rain, and a final 45-rain incu- bation with a 200-gL aliquot of 1:500 dilution of labeled streptavidin-Texas Red conjugate (Gibco-BRL, Baltimore, MD) for fluorescent color develop- ment. The sections were washed five times with PBS between incubations and mounted in 50% glycerol. Immunostained sections were examined and photographed using a Leitz Ortholux II Fluorescence microscope equipped with a Texas Red filter and a Nikon camera system.
Light Microscopic immunocytochemistry for Spectrin and Utrophin To distinguish between the nonspecific membrane damage stemming
from generalized myofibrillar degeneration with myocytolysis, and that specifically related to dystrophin deficiency, we employed a double stain- ing technique for assessing the dystrophin and spectrin distribution pat- terns simultaneously on the same section. Serial transverse cryosections, from fresh frozen biopsies of cardiac and skeletal muscles from Nt I and DH, immunostained with the MAbs and PAbs for dystrophin, were suc- cessively double stained with the MAb against spectrin. SPEC2 with bl~)ad
Molecular and Chemical Neuropathology Vol. 3I, 1997

192 Bhattacharya et al.
species reactivity to sarcolemmal spectrin, which immunostains well with rodent muscle, was used at a 1:100 dilution. For fluorescent labeling and light microscopic examination of slides, the protocols as described earlier for dystrophin were employed, except that a 200-gL aliquot of a 1:500 dilution of streptavidin-fluorescein isothiocyanate-Texas Red conjugate (Gibco-BRL) was used for fluorescent color development. Likewise, we investigated the utrophin labeling patterns in the dystrophin-prestained serial cryosections using DRP1 to delineate the compensatory role of utrophin in mitigating dystrophin-mediated myofibriUar damage. Since we employed the double immunostaining technique to simultaneously label sarcolemmal dystrophin and spectrin, adjacent cryosections from the same biopsy block were used for simultaneous characterization of utrophin and dystrophin. The DRP1 MAb was used at a 1:10 dilution.
Sample Preparation for Western Blotting Liquid nitrogen-frozen biopsies of cardiac and skeletal muscles were
processed individually by grinding with a prechilled mortar and pestle on dry ice at -70~ The pulverized tissue was homogenized in Buffer-1 (50 mM Tris-HC1, pH 7.4, containing 0.6 M KCI, 0.165 M sucrose, and protease inhibitors including 1 mM iodoacetamide, 1 mM benzemethonium chlo- ride, 0.5 mM phenylmethylsulfonyl fluoride, and 0.05 mM pepstain A). The homogenate was centrifuged at 10,000g for 30 rain, using a J-21 Beckman refrigerated centrifuge with a JA-20 rotor; and the microsomal fraction was harvested after centrifuging the post 10,000g supernatant at 105,000g for 1 h, using an L8-M60 Beckman ultracentrifuge. The resulting microsomal pel- let was resuspended in Buffer-1 with the addition of 1% digitonin and 0.5 M NaC1, according to Ervasti et al. (1991b). Protein levels were quanti- tated using the modified microtechnique as described by Peterson (1977), and the sample was diluted to 4 mg/mL concentration. Each sample was then rediluted with an equal volume of 2X sample buffer (125 mM Tris- HC1, pH 6.8, 4% SDS [w/v], 20% glycerol, 10% ]3-mercaptoethanol, and 0.002% bromophenol blue) according to Laemmli (1970). Samples were boiled for 3 rain, cooled, and centrifuged at 3000g for 10 rain. Then 25-~tL aliquots of the supernatant, containing 50 ~tg of sarcolemmal protein, were applied to SDS-PAGE gels.
SDS-PA GE Microsomal proteins were resolved electrophoretically on a 1.5-ram-
thick 3-15% SDS-PAGE gradient gel (Laemmli 1970). Samples were applied in duplicate on two separate gels in the same sequence and run simultane- ously, under identical experimental conditions. To assure equivalent load- ing of protein on the gel from NH and DH, one of the electrophoresed gels was stained with Coomassie brilliant blue (CBB) to determine the relative abundance of myosin. Lyophilized rabbit skeletal muscle dystropbin, iso- lated and purified in our laboratory according to Ervasti et al. (1991b), and
Molecular and Chemical Neuropathology Vol. 31, I997

Dystrophin Deficiency in Dystrophic Hamsters 193
heavy molecular weight marker proteins containing myosin (from Sigma), were used as markers. Myosin served as the internal protein standard for normal and dystrophic preparations. The second gel was Western blotted to estimate the amotmt of dystrophin by densitometric analysis.
Densitometric Analysis of Dystrophin by lmmunoblot t ing
Electroblotting of proteins on nitrocellulose membranes was accom- plished at 100 V, or with 2.0 Amp of maximal current, for 8 h, according to rlbwbin et al. (1979), as modified by Otter et al. (1987). Enhanced chemiluminescence (ECL) detection of dystrophin was achieved by blocking the membrane with 10% FCS for 2 h, followed by incubation overnight at 4~ with 1:300 diluted I)YS2. The nitrocellulose blot was reincubated for 1 h with 1:15,000 diluted peroxidase conjugated goat antimouse IgG (HVL) secondary antibody. After each incubation, the blot was thoroughly washed 4-6 times for 5-rain durations with PBS-Tween (80 mM Na2HPO4, 20 mM NaH2PO4, 100 mM NaC1, and 0.05% Tween- 20). Following this, the membrane was incubated for 1 rain with an equal volume of Western blotting detection reagents 1 and 2 (Amersham) in the dark and exposed on Kodak XAR-5 X-ray film. The film was developed using an Ecomat-400 film developer. Dystrophin and myosin were quan- titated densitometrically on the ECL developed immunoblots and the CBB stained dry gel, respectively. The percentage of dystrophin in the cardiac and skeletal muscles of DH was calculated from their respective optical density values, assuming the optical density of dystrophin in the corresponding control muscles from NH as 100%. Computer-assisted densitometric scanning of the immunoblots was accomplished using a pdi Discovery Series Gel Scanner (pdi Systems, Huntington Station, NY).
Statistical Analysis Group data on optical density were expressed as mean + SEM. Sta-
tistical significance of the differences between the means, obtained from the NH and DH, was determined by the two-tailed unpaired Student's t-test. A minimum level of significance was set at p < 0.05.
RESULTS
Animals It is important to point out that neither the dystrophic nor normal
control hamsters used in this investigation were golden Syrian hamsters. Color photographs of 6-mo-old male CHF-146 strain DH (white coat with black ears and darker eyes) and CHF-148 strain albino NH (white coat with pink ears and eyes), used Jn the study, are presented in Fig. 1. (Shown on next page)
Molecular and Chemical Neuropathology Vol. 31, 1997
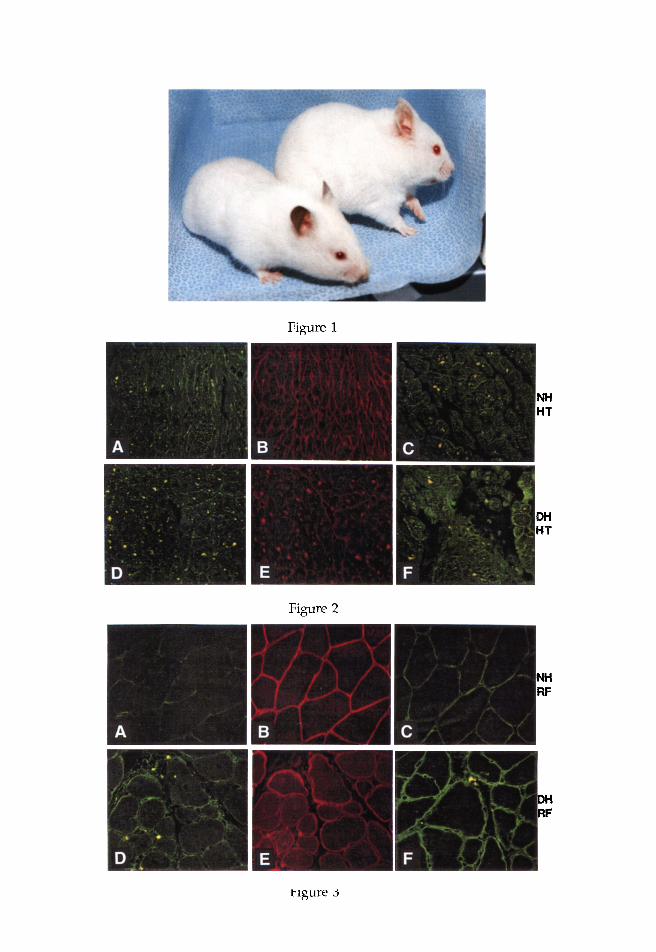
Figure 1
Nil HT
DH HT
Figure 2
NH RF
DH RF
Figure 3

Dystrophin Deficiency in Dystrophic Hamsters 195
l m m u n o c y t o c h e m i c a l Localization of Sarco l emmal Dystrophin, Spectrin, and Utrophin
The three MAbs (DYS1, DYS2, and DYS3) from Novocastra lJaborato- ties, and the two PAbs from Dr. Kunkel's Laboratory~ were used for dys- trophin labeling. DYS1 and DYS3, which target the mid-rod domain and N-terminus regions of the dystrophin molecule, respectively, did not yield consistent data. These antibodies also failed to produce acceptable negative staining when used on cryosections of skeletal muscle from mdx-mice with primary dystrophin deficiency. However, the PAbs produced a uniform dystrophin staining pattern to those obtained by DYS2, both in quality as well as in degree of fluoroscence intensity; and they generated excellent negative controls with the cardiac and skeletal muscles from mdx-mice, compared to the intensely stained corresponding biopsy sections from the C57BI~-6J normal mice (not shown). Therefore, to conserve space, the immunocytochemical data presented are limited to those obtained with DYS2, which targets the C-terminus of the dystrophin molecule.
Fig. 1. Color photographs of 6-too-old, male, CHF-146 strain dystrophic hamsters (white coat with black ears and darker eyes) and CHF-148 strain albino normal hamsters (white coat with pink ears and eyes) are presented.
Fig. 2. Immunocytochemical localization of sarcolemmal spectrin (A), dys- trophin (B), and utrophin {C) distribution in the cardiac muscle (HT) from 6-mo- old, male, CHF-148 strain albino NH are presented in the upper panel. The lower panel {D-F) shows the corresponding slides of HT from the age- and sex- matched CHF-146 strain DII. The immunocytochemistry for spectrin was obtained by double immunostaining the same slide using SPEC2 as a primary MAb, previously stained with DYS2, to colocalize dystrophin and spectrin to evaluate dystrophin deficiency in HT. Compared to NH (B), dystrophin staining in the hearts of D] 1 (E) is markedly reduced, whereas utrophin labeling is more intense in the DH (F) than NH (C). Spectrin localization in DH (D) appears to be varied throughout the slide, and there are regions where spectrin labeling is either somewhat weaker or more intense, compared to NH (A).
Fig. 3. Immunocytochemical localization of sarcolemmal spectrin (A), dys- trophin (B), and utrophin (C) distribution in the rectus femoris muscle (RF) from 6-too-old, male, albino NH are presented in the upper panel. The lower panel (D-F) shows the corresponding slides of RF from the age- and sex-matched DH. The immunocytochemistry for spectrin was obtained by double immunostaining the same slide using SPEC2 as a primary MAb, previously stained with DYS2, to colocalize dystrophin and spectrin to evaluate dystrophin deficiency in RF. Compared to NH (B), dystrophin staining in the RF of DH (E) is significantly reduced and discontinuous in places, whereas utrophin labeling is clearly more intense in the DH (F) than NH (C). However, spectrin staining pattern in DH (D) appears to be somewhat stronger and varied throughout the slide, compared to NH (A). Nuclei appear as black dots, many of which are centrally located in the dystrophic muscle (D).
Molecular and Chemical Neuropathology Vol. 31, 1997

196 Bhattacharya et al.
Cardiac Muscle
In the upper panel of Fig. 2 shown on page 8 (center), immunocyto- chemical staining of spectrin (A), dystrophin (B), and utrophin (C) are presented from the control myocardium, obtained from 6-too-old male NH. The sarcolemmal dystrophin distribution in the cardiac muscle from the age- and sex-matched DH was very weak and discontinuous (Fig. 2E) compared to NH (Fig. 2B). However, the spectrin staining pattern, obtained via double immunostaining of the same slide from DH (Fig. 2D), revealed numerous segments where spectrin labeling was basically normal compared to NH (Fig. 2A). Strikingly similar to DMD and BMD (Mizuno et al., 1993, 1994), the utrophin labeling in the DH hearts (Fig. 2F) was significantly more intense compared to NH (Fig. 2C).
Skeletal Muscle
Immunocytochemical localization of sarcolemmal spectrin (Fig. 3A), dystrophin (Fig. 3B), and utrophin (Fig. 3C) from the control rectus femoris muscle, obtained from NH, are presented in the upper panel of Fig. 3 shown on page 8 (bottom). Corresponding to our data in dystrophic hearts (Fig. 2), the reduced and weaker staining pattern for sarcolemmal dys- trophin in the rectus femoris muscle from age- and sex-matched DH is shown in Fig. 3E, clearly presenting evidence that dystrophin distribution is markedly reduced in DH. The spectrin labeling pattern, obtained via double immunostaining of the same slide, is presented in Fig. 3D. This pho- tomicrograph depicts multiple sarcolemmal segments in the skeletal mus- cle from DH (Fig. 3D), where spectrin labeling patterns appear to be either similar or somewhat intense compared to NH (Fig. 3A), thus revealing the presence of intact sarcolemma. The black dots (shown by arrows) on the photomicrographs of spectrin irnmunocytochemistry represent nuclei that are mostly centronucleated in the DH (Fig. 3D). Intriguinglj6 the utrophin labeling in the dystrophic skeletal muscle (Fig. 3F) was more intense com- pared to the control (Fig. 3C).
Confirmation of Dystrophin Deficiency by Western Blotting
The significantly reduced sarcolemmal dystrophin staining in the dystrophic myocardium (Fig. 2E) and rectus femoris muscle (Fig. 3E), observed immunocytochemically, was confirmed by Western blotting upon resolving the sarcolemmal proteins on a SDS-PAGE gradient slab gel. The immunoblots of cardiac muscle (Fig. 4) and skeletal muscle (Fig. 5) showed a marked depletion of dystrophin in DH compared to NH. The computer-assisted densitometric scanning of the immunoblots revealed that dystrophin in dystrophic myocardium and rectus femoris muscles was reduced by 83% (p < 0.0001) and 50% (p < 0.0001), respec- tively, relative to their age- and sex-matched controls. However, as
Molecular aad Chemical Neuropathology Vol. 31, 1997

Dystrophin Deficiency in Dystrophic Hamsters 197
Dyst DH NH DH NH DH NH Myosin 1 2 3 4 5 6 7 8
Fig. 4. Immunoblot of cardiac muscle shows a virtual absence of dys- trophin (upper bands) in two of the three 6-mo-old, male, DH (lanes 2 and 6), compared to the age- and sex-matched albino NH (lanes 3, 5, and 7). A 25-gL aliquot of microsomal preparation containing 50 l~g protein was applied to each lane. The microsomal extracts from the heart of three individual DH were applied to lanes 2, 4, and 6; whereas, lanes 3, 5, and 7 contained the heart sam- ples from three individual NH. Lanes 1 and 8 show dystrophin and myosin standard, respectively. The lower bands, showing a relative abundance of myosin content in each of the heart samples from NH and DH, were transposed from the CBB stained SDS-PAGE gel from the same run. The myosin band served as the internal standard for dystrophin analysis, confirming an equiva- lent amount of protein loading in each lane.
shown in Table 1, the optical density of myosin bands was essentially the same in both NH and DH. Myosin bands served as the internal stan- dards for dystrophin analysis, ensuring an equal amount of protein load- ing on the gel for each of the samples derived from multiple sets of NH and DH.
DISCUSSION
Abnormal sarcolemmal dystrophin expression is intimately related to the molecular pathogenesis of DMD and BMD (Koenig et al., 1987; Hoffman et al., 1987, 1988; Arahata et al., 1988; Bonilla et al., 1988; Wor- ton and Burghes, 1988; Zubrzycka-Gaarn et al., 1988; Bulman et al., 1991), including symptomatic DMD carriers (Arahata et al., 1989; Morandi et al., 1990). It is known that utrophin, an autosomal homologue of dystrophin that is encoded by the DMD gene (Pearce et al., 1993), is upregulated in the dystrophin-deficient muscle of DMD, BMD, and in DMD carriers (Karpati et al., 1993; Mizuno et al., 1993, 1994). Dystrophin deficiency has been linked to cellular hyperpermeabili ty (Menke and Jockusch, 1991); higher intracellular [Ca 2+] with decreased mean closed-time and increased
Molecular and Chemical Neuropathology Vol. 31, 1997

198 Bhattacharya et al.
NH DH NH DH NH DH 1 2 3 4 5 6
Dyst
Myosin
Fig. 5. Immunoblot of rectus femoris muscle (RF) shows a significant depletion of dystrophin (upper bands) in 6-mo-old, male, DH (lanes 2, 4, and 6) compared to the age- and sex-matched albino NH (lanes 1, 3, and 5). A 25-laL aliquot of microsomal preparation containing 50 lag protein was applied to each lane. The microsomal extracts from the RF of three individual DH were applied to lanes 2, 4, and 6; whereas, lanes 1, 3, and 5 contained the RF samples from three individual NH. The lower bands, showing a relative abundance of myosin content in each of the RF samples from NH and DH, were transposed from the CBB stained SDS-PAGE gel from the same run. The myosin band served as the internal standard for dystrophin analysis, confirming an equivalent amount of protein loading in each lane.
Table 1 Optical Density of Dystrophin and Myosin Bands from Immunoblot
and Dried PAGE Slab Gel a
Group
Ventricular myocardium
Dystrophin (N) Myosin (N)
Rectus femoris muscle
Dystrophin (N) Myosin (N)
NH 0.74 + 0.06 (4) 0.75 +_ 0.03 (4) 0.14 +_ 0.01 (6) 1.35 +_ 0.01 (6) DH 0.13 + 0.02 (4) 0.74 + 0.04 (4) 0.07 _+ 0.01 (6) 1.30 + 0.04 (6) %A in DH 83 % $ 1% $ 50% $ 4% ,/, Significance p < 0.0001 ns p < 0.0001 ns
aData are g iven as m e a n + SEM; N = n u m b e r of prepara t ions f rom different animals. p va lues _> 0.05 are cons idered nonsignif icant (ns).
open probabil i ty of voltage-gated slow Ca2+-channels (Turner et al., 1988, 1991; Fong et al., 1990); and a greater propens i ty for myonecrosis in mdx- mice (Weller et al., 1990) and in DMD and BMD (Morandi et al., 1990).
To emphas ize the clinical relevance of the present findings, it is impor tan t to point out that CHF-146 strain DH and BIO-14.6 strain DH have been p roposed as a suitable mode l for SCARMD because of the selective deficiency of 50- (adhalin), 43-, and 35-kDa DAG, which are also k n o w n as ~-, 13-, and 7-sarcoglycans, respectively (McNally et al., 1996). However , the recent characterization of o~-, 13-, and 7-sarcoglycan genes in
Molecular and Chemical Neuropathology Vol. 31, 1997

Dystrophin Deficiency in Dystrophic Hamsters 199
this strain of DH have been found to be normal, suggesting a possible mutation in yet a putative fourth gene (McNally et al., 1996). Roberds and Campbell (1995) have shown that adhalin mICNA and cDNA sequence are also normal in the BIO-14.6 DH. There are contradictory reports on the immunostaining patterns and immunoblot intensities of dystrophin and utrophin in the various strains and muscle types of DH, although some of these differences could very well be attributed to their genetic heterogeneity, i.e., those seen in NSJ-my/my DH (Mizuno et al., 1995; Yamanouchi et al., 1994) versus the pure BIO-14.6 strain DH (Roberds et al., 1993; Johnson et al., 1994). Furthermore, the normal coun- terparts used in most of the other studies have been the commercially available golden Syrian hamsters of various strains with different genetic pools, compared to the CHF-148 strain albino NH used in this study.
~Ihe present immunocytochemical findings in the ventricular myocardium and rectus femoris from DH and NH (Figs. 2 and 3) suggest that sarcolemmal dystrophin distribution is unequivocally diminished and discontinuous in the CHF-146 strain DH. Densitometric analysis of the immunoblots, originating from different sets of DH and NH, confirmed that sarcolemmal dystrophin is significantly reduced in the cardiac and skeletal muscles (Table 1). ]he present observations of higher relative dys- trophin loss in the cardiac muscle, compared to skeletal muscle, corroborate well with our previous findings of greater EICA in the heart than in rectus femoris of the BIO-14.6 and CHF-146 strain DH, compared to their normal counterparts (Bhattacharya et al., 1982, 1987, 1993; Johnson and Bhat- tacharya, 1993). Furthermore, the linear relationship between increased sar- colemmal dystrophin loss and greater membrane-mediated EICA in the dystrophic hearts implicates an important pathogenetic role of dystrophin in initiating myonecrosis and progressive muscle degeneration. Our hypothesis is also supported by the observation of Weller et al. (1990) that dystrophin-deficient mdx myofibers are potentially more vulnerable to membrane-mediated EICA and necrosis. Similarly, our immunocytochemi- cal findings that utrophin is more intensely stained in the cardiac (Fig. 2F) and skeletal muscles (Fig. 3F) of DH, in the presence of significant dys- trophin deficiency, are compatible with those reported in patients with DMD and BMD, and in DMD carriers (Karpati et al., 1993; Mizuno et al., 1994), but do not corroborate well with those seen in the NJS-my/my DH (Yamanouchi et al., 1994). This inverse relationship suggests a putative mechanistic role of sarcolemmal dystrophin and utrophin in the regulation of transmembrane slow Ca2-channels, and the maintenance of overall intra- cellular Ca 2+ homeostasis. Our hypothesis also offers an intriguing thera- peutic strategy in patients with DMD and BMD that slow Ca2+-channel blockers, such as diltiazem, may provide multiple salutary effects, includ- ing the enhancement of sarcolemmal dystrophin and utrophin levels, reduction of membrane-mediated EICA and sporadic myonecrosis, and perhaps the regression of premature death (Johnson and Bhattacharya, 1993). The preliminary observations of increased sarcolemmal dystrophin
Molecular and Chemical Neuropathology Vol. 31, 1997

200 Bhattacharya et al.
distribution with significantly reduced EICA and plasma enzymes, together with reduced morbidity and mortality rate in diltiazem-treated DH, tend to support these contentions (Johnson et al., 1994).
Cardiac Muscle
Our immunocytochemical observation of markedly reduced sar- colemmal dystrophin distribution in DH hearts (Fig. 2E), and the confir- mation of 83% reduced dystrophin content (Table 1) by densitometric analysis of the corresponding Western blots (Fig. 4), are not only in agreement with those in DMD (Hoffman et al., 1987; Koenig et al., 1987; Arahata et al., 1988; Bonilla et al., 1988) and BMD (Hoffman et al., 1988; Morandi et al., 1990; Byers et al., 1992), but are also supported by the recent reports of secondary dystrophin abnormalities in DH by Iwata et al. (1993) and Roberds et al. (1993). Furthermore, the striking inverse rela- tionship between reduced sarcolemmal dystrophin distribution, and cor- responding upregulation of utrophin observed in the cardiac muscles of DH, suggests a potential compensatory role of utrophin in HMD in response to dystrophin loss.
Skeletal Muscle
Similar to our findings in the heart, dystrophin distribution in the rectus femoris was also reduced as noted by immunocytochemistry (Fig. 3Ii) and confirmed by densitometric analysis of the immunoblots (Fig. 5, Table 1). These data provide the first quantitative evidence that dys- trophin is reduced by 50% in the rectus femoris of DH. However, Yamanouchi et al. (1994) and Mizuno et al. (1995) failed to confirm the dystrophin deficiency and utrophin upregulation in the skeletal muscle of NSJ-my/my strain DH. Perhaps these differences were due, in part, to inherent genetic heterogeneity with the CHF-146 DH, which are homol- ogous to BIO-14.6 DH (Hunter et al., 1984), the types of skeletal muscle used, the different age group of animals studied, and the use of MAbs and PAbs to dystrophin and utrophin of diverse origin. The inverse rela- tionship observed in the heart, between reduced sarcolemmal dystrophin and upregulation of utrophin, is also present in the skeletal muscle, and lends additional support for a compensatory role of utrophin in hamster dystrophinopathy.
Structural and Functional Implications of Sarcolemmal Dystrophin and Dystrophin-Associated Glycoproteins, and Their Potential Roles in the Regulation of Transmembrane Slow Ca2§
Campbell and coworkers have reported cooperative interactions of dystrophin with the family of perisarcolemmal (35-, 43~, 50-kDa) DAG,
Molecular and Chemical Neuropathology Vol. 31, 1997

Dystrophin Deficiency in Dystrophic Hamsters 201
the 156-kDa extracellular DAG, and the 59-kDa intracellularly localized DRP to form the dystrophin-glycoprotein complex (DGC) (Campbell et al., 1989, 1991; Ervasti et al., 1990, 1991a; Ohlendieck et al., 1991). The dystrophin dimers have been proposed to link the cytoskeleton with the glycoprotein complex by binding to actin through the amino terminus, and to the 50-, 43-, and 35-kDa DAG through the carboxyl terminus (Ervasti et al., 1991a). Focused studies on the characterization and purifi- cation of the various subunits--i.e., 0~1 and 0% as well as other crucial membrane peptides that form the components of the dihydropyridine- sensitive slow Ca2+-channels (Campbell et al., 1988; Knudson et al., 1989; Jay et al., 1991)--will shed new light on the missing pathogenetic links between the abnormalities in DGC, dystro- and sarcoglycan subcom- plexes, and membrane-mediated EICA in HMD. It seems highly plausi- ble that dystrophin and the family of DAG and DRP may participate in the formation and regulation of transmembrane slow Ca2+-channels, and that their functional deficits may render the dystrophic sarcolemma hyperpermeable to Ca 2+, thereby augmenting membrane-mediated EICA, progressive muscle degeneration, and segmental myonecrosis (Weller et al., 1990; Menke and Jockusch, 1991; Johnson and Bhattacharya, 1993).
There are reports suggesting that the interplay among sarcolemmal dystrophin, DGC, and dystro- and sarcoglycan subcomplexes is more complex than previously thought (Ervasti et al., 1991a; Ohlendieck et al., 1991; Zubrzycka-Gaarn et al., 1991; Mizuno et al., 1995). Steinhardt and coworkers have shown that myoplasmic-free [Ca2+i] is significantly increased in the dystrophin-deficient myotubes from mdx-mice and DMD patients (Fong et al., 1990; Turner et al., 1991), and that DAGs are degraded at an abnormally high rate in the dystrophic myofibers (Turner et al., 1988). It is plausible that lack of functional integrity in the DGC, dystro-, and sarcoglycan complexes, as well as increased degradation of DAG, may exacerbate the clinical manifestations in patients with DMD and BMD, causing hyperpermeability of dystrophic sarcolemma, mem- brane-mediated EICA, and myonecrosis (Bertorini et al., 1982, 1984; Bhat- tacharya et al., 1989). Furthermore, EICA is invariably present in dystrophic animals with either primary or secondary dystophin defi- ciency, such as DH (Bhattacharya et al., 1987), mdx-mice (Fong et al., 1990; Weller et al., 1990), Xmd-dogs (Kornegay et al., 1988; Valentine et al., 1988), and dystrophic cats (Carpenter et al., 1989). Thus, sarcolemmal deficits of dystrophin and DAG may likely initiate the final common pathogenetic pathway in HMD, leading to progressive muscle degenera- tion via membrane-mediated EICA, and consequent cell death (Bhat- tacharya et al., 1989).
The crucial observation by Vater et al. (1992) on the fate of sar- colemmal dystrophin distribution in a nondystrophic rodent--i.e., its dis- appearance and reappearance during degeneration and regeneration of the denervated rat soleus muscle, lacking any genetic dystrophin abnor- malities-broadens our understanding of the wider physiologic roles of
Molecular and Chemical Neuropathology Vol. 31, 1997

202 Bhattacharya et al.
dystrophin and its potential interactions in the regulation of voltage- gated slow Ca2+-channels, and overall intracellular Ca2+-homeostasis. These temporal changes in sarcolemmal dystrophin in nondystrophic muscle, together with the fact that dystrophin lacking mdx-mice do not display any obvious dystrophic features or suffer from noticeable func- tional deficits (Dunn et al., 1991; Johnson et al., 1991)--and that a very mild form of muscular dystrophy is manifested with the deletion of 46% dystrophin in humans (England et al., 1990)--congruently suggest that dystrophin deficiency may not be solely responsible for the expression of severe HMD, and that sarcolemmal dystrophin may be autoregulated during regenerative processes (Vater et al., 1992). It is therefore tempting to postulate that high Ca2+i-induced degradation of membrane-bound DAG, together with primary or secondary dystrophin loss, may cause transitory deregulation of transmembrane slow Ca2+-channels in HMD.
We have shown in two separate studies--a 55-d trial of oral diltiazem, a slow Caa+-channel blocker (Bhattacharya et al., 1982); and a 6-too trial for comparative efficacy of oral diltiazem, nifedipine, and verapamil (Johnson and Bhattacharya, 1993)--that chronic diltiazem therapy produces a signif- icant reduction in EICA and corresponding improvement in the biochemi- cal, structural, and functional aberrations in DH. Since diltiazem imparts multiple salutary effects toward the normalization of membrane-mediated dystrophic pathobiology and high mortality in DH, it is also likely to improve the membrane integrity by enhancing sarcolemmal dystropl~n and utrophin distribution, either by retardation of Ca2+-induced autodegra- dation of these proteins or by upregulation of utrophin, via a hitherto tmknown protective mechanism (Johnson et al., 1994).
In summary, sarcolemmal dystrophin distribution is markedly reduced and discontinuous in the cardiac and skeletal muscles of CHF-146 DH, with upregulation of utrophin and a varied degree of spectrin labeling. As in DMD and BMD, utrophin also appears to play a compensatory role in hamster dystrophinopathy. Furthermore, reduced dystrophin distribution is associated with sarcolemmal hyperpermeability and membrane-mediated EICA, leading to chronic muscle degeneration and segmental necrosis. Therefore, any therapeutic strategy, e.g., slow Ca2+-channel blocker therap36 that mitigates EICA may simultaneously enhance sarcolemmal dystrophin and utrophin distribution with improved membrane integrity, overall qual- ity of life, and longevity in DMD and BMD.
ACKNOWLEDGMENTS
Dr. Raj K. Handa was a postdoctoral fellow partially supported by the University of Tennessee CAN-DO Laboratory Research Funds. This work was supported, in part, by grants to SKB from the National Insti- tute of Arthritis and Musculoskeletal and Skin Diseases (USPHS #R01-
Molecular and Chemical Neuropathology Vol. 31, 1997

Dystrophin Deficiency in Dystrophic Hamsters 203
AR-38540). Presented in part at the 24th Annual Meeting of the Society for Neuroscience, Miami Beach, Florida; November 15, 1994. This article is dedicated to Maurice H. Palmieri in loving appreciation of his exem- plary life and for his kind cooperation with our studies; he greatly inspired each of us with his perseverance, dedication and sacrifice by liv- ing with Duchenne muscular dystrophy for 26 years.
REFERENCES
Anderson J. E., Kao L., Bressler B. H., and Gruenstein E. (1990) Analysis of dystrophin in fast and slow-twitch skeletal muscles from mdx and dy2j mice at different stages. Muscle Nerve 13, 6-11.
Arahata K., Ishiura S., Ishiguro T., Tsukahara T., Suhara Y., Eguchi C., Ishihara T., Non- aka I., Ozawa E., and Sugita H. (1988) Immtmostaining of skeletal and cardiac mus- cle surface membrane with antibody against Duchenne muscular dystrophy peptide. Nature 333, 861-863.
Arahata K., lshihara T., Kamakura K., Tsukahara T., lshiura S., Baba C., Matsumoto T., Nonaka i., and Sugita It. (1989) Mosaic expression of dystrophin in symptomatic car- riers of Duchenne muscular dystrophy. N. Engl. J. Med. 320, 138-142.
Bajusz E., Baker J. R., Nixon C. W., and Homburger E (1969) Spontaneous hereditary myocardial degeneration and congestive heart failure in a strain of Syrian hamsters. Ann. N.Y. Acad. Sci. 156, 105-129.
Bertorini T. E., Bhattacharya S. K., Palmieri G. M. A., Chesney C. M., Pifer D., and Baker B. (1982) Muscle calcium and magnesium content in Duchenne muscular dystrophy. Neu rology 32, 1088-1092.
Bertorini T.E., Cornelio F., Bhattacharya S. K., Palmieri G. M. A., Dones I., Dworzak E, and Brambati B. (1984) Calcium and magnesium content in fetuses at risk and pre- necrotic Duchenne muscular dystrophy. Neurology 34, 1436-1440.
Bhattacharya S. K., Palmieri G. M. A., Bertorini T. E., and Nutting D. F. (1982) The effect of diltiazem in dystrophic hamsters. Muscle Nerve 5, 73-78.
Bhattacharya S. K., Crawford A. J., and Pate J. W. (1987) Electrocardiographic, biochemi- cal, and morphologic abnormalities in dystrophic hamsters with cardiomyopathy. Muscle Nerve 10, 168-176.
Bhattacharya S. K., Crawford A. J., Thakar J. H., and Johnson P. L. (1989) Pathogenetic roles of intracellular calcium and magnesium in membrane-mediated progressive muscle degeneration in Duchenne muscular dystrophy, in: Cell Calcium Metabolism- 87: Physiology, Biochemistry, Pharmacology and Clinical Implications (Fiskum G., ed.), Plenum, New York, pp. 513-525.
Bhattacharya S. K., Johnson P. L., and Thakar J. H. (1993) Reversal of impaired oxidative phosphorylation and calcium overloading in the in vitro cardiac mitochondria of CHF-146 dystrophic hamsters with hereditary muscular dystrophy. J. Neurol. Sci. 120, 180-186.
Bieber F. R. and Hoffman E. P. (1990) Duchenne and Becker muscular dystrophies: Genet- ics, prenatal diagnosis, and future prospects. Clinics in Perinatology 17, 845-863.
Bonilla E., Samitt C. E., Miranda A. F., Hays A. P., Salviati G., DiMauro S., Kunkel L. M., Hoffman E. P., and Rowland L. P. (1988) Duchenne muscular dystrophy: Deficiency of dystrophin at the muscle cell surface. Cell 54, 447-452.
Bulman D. E., Murphy E. G., Zubrzycka-Gaarn E. E., Worton R. G., and Ray P. N. (1991) Differentiation of Duchenne and Becker muscular dystrophy phenotypes with amino- and carboxy-terminal antisera specific for dystrophin. Am. J. Hum. Genet. 48, 295-304.
Molecular and Chemical Neuropathology Vol. 31, 1997

204 Bhattacharya et al.
Byers T. J., Neumann P. E., Beggs A. H., and Kunkel L. M. (1992) ELISA quantitation of dystrophin for the diagnosis of Duchenne and Becker muscular dystrophies. Neurol- ogy 42, 570-576.
Campbell K. P., Leung A. T., and Sharp A. H. (1988) The biochemistry and molecular biol- ogy of the dihydropyridine-sensitive calcium channel. Trends in NeuroscL 11, 425-430.
Campbell K. P. and Kahl S. D. (1989) Association of dystrophin and an integral membrane glycoprotein. Nature 338, 259-262.
Campbell K. P., Ervasti J. M., Ohlendieck K. and Kahl S. D. (1991) The dystrophin- glycoprotein complex: Identification and biochemical characterization, in Frontiers in Muscle Research (Ozawa E., Masaki T., and Nabeshima Y. eds.), Elsevier, Amsterdam, pp. 321-340.
Carpenter J. L., Hoffman E. P., Ramanul E C. A., Kunkel L. M., Rosales R. K., Ma N. S. F., Dasbach J. J., Rae J. F., Moore F. M., McAfee M. B., and Pearce L. K. (1989) Feline muscular dystrophy with dystrophin deficiency. Am. ]. Pathol. 135, 909-919.
Dunn J. F., Frostick S., Brown G., and Radda G. K. (1991) Energy status of cells lacking dystrophin: an in vivo/in vitro study of mdx mouse skeletal muscle. Biochim. Bio- phys. Acta 1096, 115-120.
England S. B., Nicholson L. V. B., Johnson M. A., Forrest S. M., Love D. R., Zubrzycka- Gaarn E. E., Bulman D. E., Harris J. B., and Davies K. E. (1990) Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature 343, 180-182.
Ervasti J. M., Ohlendieck K., Kahl S. D., Gaver M. G., and Campbell K. P. (1990) Defi- ciency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 345, 315-319.
Ervasti J. M. and Campbell K. P. (1991a) Membrane organization of the dystrophin- glycoprotein complex. Cell 66, 1121-1131.
Ervasti J. M., Kahl S. D., and Campbell K. P. (1991b) Purification of dystrophin from skeletal muscle. J. Biol. Chem. 266, 9161-9165.
Fong P., Turner P. R., Denetclaw W. F., and Steinhardt R. A. (1990) Increased activity of calcium leak channels in myotubes of Duchenne human and mdx mouse origin. Sci- ence 250, 673-676.
Hoffman E. P., Brown R. H., and Kunkel L. M. (1987) Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 51, 919-928.
Hoffman E. P., Fischbeck K. H., Brown R. H., Johnson M., Medori R., Loike J. D., Harris J. B., Waterston R., Brooke M., Specht L., Kupsky W., Chamberlain J., Caskey T., Shapiro F., and Kunkel L. M. (1988) Characterization of dystrophin in muscle-biopsy specimens from patients with Duchenne or Becker's muscular dystrophy. N. Engl. J. Med. 318, 1363-1368.
Hudecki M. S., Pollina C. M., and Heffner R. R. (1984) In vivo effects of three calcium blockers on chickens with inherited muscular dystrophy. Exp. Neurol. 84, 512-523.
Hunter E. G., Hughes V., and White J. (1984) Cardiomyopathic hamsters, CHF 146 and CHF 147: a preliminary study. Can. J. Physiol. Pharmacol. 62, 1423-1428.
Iwata Y., Nakamura H., Fujiwara K., and Shigekawa M., (1993) Altered membrane- dystrophin association in the cardiomyopathic hamster heart muscle. Biochem. Bio- phys, Res. Comm. 190, 589-595.
Jay S. D., Sharp A. H., Kahl S. D., Vedvick T. S., Harpold M. M., and Campbell K. P. (1991) Structural characterization of the dihydropyridine-sensitive calcium channel c~2 sub- unit and the associated peptides. J. Biol. Chem. 266, 3287-3293.
Johnson P. L., Bhattacharya S. K., Handa R. K., Gupta M. P., Adamec T. A., and Shanklin D. R. (1991) Biochemical and morphological studies in the cardiac and skeletal mus- cles of mdx mice: Relevance as a model for Duchenne muscular dystrophy. Soc. Neu- rosci. Abstr. 17, 160.
Johnson P. L. and Bhattacharya S. K. (1993) Regulation of membrane-mediated chronic muscle degeneration in dystrophic hamsters by calcium-chal~nel blockers: Diltiazem, Nifedipine and Verapamil. J. NeuroL Sci. 115, 76-90.
Molecular and Chemical Neuropathology Vol. 31, 1997

Dystrophin Deficiency in Dystrophic Hamsters 205
Johnson E L., Li H. J., and Bhattacharya S. K. (1994) Characterization of sarcotemmal dys- trophin, spectrin and utrophin in the cardiac and skeletal muscles of Ct IF-146 dys- trophic hamsters. Soc. Neurosci. Abstr. 20, 831.
Karpati G., Carpenter S., Morris G. E., Davies K. E., Guerin C., and Holland P. (1993) Local- ization and quantitation of the chromosome 6-encoded dystrophin-related protein in normal and pathological human muscle. J. Neuropathol. Exp. Neurol. 52, 119-128.
Knudson C. M., Chaudhari N., Sharp A. I t., Powell J. A., Beam K. G., and Campbell K. P. (1989) Specific absence of the al subunit of the dihydropyridine receptor in mice with muscular dysgenesis. J. Biol. Chem. 264, 1345-1348.
Koenig M., Hoffman E. P., Bertelson C. J., Monaco A. P., Freener C. and Kunkel L. M. (1987) Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individ- uals. Cell 50, 509-517.
Koenig M. and Kunkel L. M. (1990) Detailed analysis of the repeat domain of dystrophin reveals four potential hinge segments that may confer flexibility. J. Biol. Chem. 265, 4560-4566.
Kornegay J. N., Tuler S. M., Miller D. M., and Levesque D. C. (1988) Muscular dystrophy in a litter of golden retriever dogs. Muscle Nerve 11, 1056-1064.
Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680--685.
McNally E. M., B6nnemann C. G., Kunkel L. M., and Bhattacharya S. K. (1996) Deficiency of adhalin in a patient with muscular dystrophy and cardiomyopathy. N. Engl. J. Med. 334, 1610, 1611.
Menke A. and Jockusch H. (1991) Decreased osmotic stability of dystrophin-less muscle cells from the mdx mouse. Nature 349, 69-71.
Mizuno Y., Noguchi S., Yamamoto H., Yoshida M., Nonaka I., Hirai S., and Ozawa E. (1995) Sarcoglycan complex is selectively lost in dystrophic hamster muscle. Am. J. Pathol. 146, 530-536.
Mizuno Y., Nonaka I., Hirai S., and Ozawa E. (1993) Reciprocal expression of dystrophin and utrophin in muscles of Duchenne muscular dystrophy patients, female DMD- carriers and control subjects. J. Neurol. Sci. 119, 43--52.
Mizuno Y., Yoshida M., Nonaka I., Hirai S., and Ozawa E. (1994) Expression of utrophin (Dystrophin-related protein) and dystrophin-associated glycoproteins in muscles from patients with Duchenne muscular dystrophy. Muscle Nerve 17, 206--216.
Morandi L., Mora M., Gussoni E., Tedeschi S., and Cornelio E (1990) Dystrophin analy- sis in Duchenne and Becker muscular dystrophy carriers: correlation with intracellu- lar calcium and albumin. Ann. Neurol. 28, 674-679.
Noireaud J. and L6oty C. (1984) A comparative study of contractile responses in diaphragm muscles of normal and dystrophic (C57BL/6J dy2}/dy2l) mice. Exp. Neu- rol. 83, 589-603.
Ohlendieck K., Ervasti J. M., Snook J. B., and Campbell K. P. (1991) Dystrophin-glyco- protein complex is highly enriched in isolated skeletal muscle sarcolemma. J. Cell Biol. 112, 135-148.
Otter T., King S. M., and Witman G. B. (1987) A two-step procedure for efficient electro- transfer of both high-molecular-weight (>400,000) and low-molecular-weight (<20,000) proteins. Anal. Biochem. 162, 370-377.
Partridge T. A., Morgan J. E., Coulton G. R., Hofflnan E. P., and Kunkel L. M. (1989) Con- version of mdx myofibers from dystrophin-negative to -positive by injection of nor- mal myoblasts. Nature 337, 176--179.
Paterson R. A., Layberry R. A., and Nadkarni B. B. (1972) Cardiac failure in the hamster: A biochemical and electron microscopic study. Lab. Invest. 26, 755-766.
Pearce M., Blake D. J., Tinsley J. M., Byth B. C., Campbell L., Monaco A. P., and Davies K. E. (1993) The utrophin and dystrophin genes share similarities in genomic struc- ture. Human Mol. Gen. 2, 1765-1772.
Molecular and Chemical Neuropathology Vol. 31, 1997

206 Bhattacharya et al.
Peterson G. L. (1977) A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal. Biochem. 83, 346-356.
Roberds S. L., Ervasti J. M., Anderson R. D., Ohlendieck K., Kahl S. D., Zoloto D., and Campbell K. P. (1993) Disruption of the dystropbin-glycoprotein complex in the car- diomyopathic hamster. J. Biol. Chem. 268, 11,496-11,499.
Roberds S. L. and Campbell K. P. (1995) Adhalin mRNA and cDNA sequence are normal in the cardiomyopathic hamster. FEBS Lett. 364, 245-249.
Sanyal S. K., Jotmson W. W., Dische M. R., Pitner S. E., and Beard C. (1980) Dystrophic degeneration of papillary muscle and ventricular myocardium: a basis for mitral valve prolapse in Duchenne's muscular dystrophy. Circulation 62, 430-438.
Sklar R. M., Beggs A. H., Lev A. A., Specht L., Shapiro F., and Brown R. H. (1990) Defec- tive dystrophin in Duchenne and Becker dystrophy myotubes in cell ctdture. Neurol- ogy 40, 1854- 1858.
Towbin H., Staehelin T., and Gordon J. (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedures and some applications. Proc. Natl. Acad. Sci. USA 76, 4350-4354.
Turner P. R., Westwood T., Regen C. M., and Steinhardt R. A. (1988) Increased protein degradation results from elevated free calcium levels found in muscle from mdx mice. Nature 335, 735--738.
Turner P. R., Fong P., Denetclaw W. F., and Steinhardt R. A. (1991) Increased calcium influx in dystrophic muscle. J. Cell Biol. 115, 1701-1712.
Valentine B. A., Cooper B. J., de Lahunta A., O'Quinn R., and Blue J. T. (1988) Canine X- linked muscular dystrophy. An animal model of Duchenne muscular dystrophy: Clinical studies. ]. Neurol. Sci. 88, 69-81.
Vater R., Cullen M. J., Nicholson L. V. B., and Harris J. B. (1992) The fate of dystrophin during the degeneration and regeneration of the soleus muscle of the rat. Acta Neu- ropathol. 83, 140-148.
Weller B., Karpati G., and Carpenter S. (1990) Dystrophin-deficient mdx muscle fibers are preferentially vulnerable to necrosis induced by experimental lengtherling contrac- tions. [. Neurol. Sci. 100, 9-13.
Worton R. G. and Burghes A. M. M. (1988) Molecular genetics of Duchenne and Becker muscular dystrophy. Int. Rev. Neurobiol. 29, 1-76.
Yamanouchi Y., Mizuno Y., Yamamoto H., Takemitsu M., Yoshida M., Nonaka I., and Ozawa E. (1994) Selective defect in dystrophin-associated glycoproteins 50DAG (A2) and 35DAG (A4) in the dystrophic hamster: an animal model for severe childhood autosomal recessive muscular dystrophy (SCARMD). Neuromusc. Disord. 4, 49-54.
Zubrzycka-Gaarn E. E., Bulman D. E., Karpati G., Burghes A. H. M., Belfall B., Klamut H. J., Talbot J., Hodges R. S., Ray P. N., and Worton R. G. (1988) The Duchenne mus- cular dystrophy gene product is localized in sarcolemma of human skeletal muscle. Nature 333, 466-469.
Zubrzycka-Gaarn E. E., Hutter O. E, Karpati G., Klamut H. J., Bulman D. E., Hodges R. S., Worton R. G., and Ray P. N. (1991) Dystrophin is tightly associated with the sar- colemma of mammalian skeletal muscle fibers. Exp. Cell Res. 192, 278-288.
Molecular and Chemical Neuropathology VoL 31, 1997




















