Oral controlled drug delivery systems, optimization of release ...

162 Recent Patents on Drug Delivery & Formulation 2009, 3, 162-173
1872-2113/09 $100.00+.00 © 2009 Bentham Science Publishers Ltd.
Recent Trends in Oral Drug Delivery: A Review
Himanshu Gupta1*, Dinesh Bhandari
2 and Aarti Sharma
1
1Department of Pharmaceutics, Faculty of Pharmacy, Jamia Hamdard, New Delhi, India;
2D. J. College of Pharmacy,
Modinagar, U.P., India
Received: January 22, 2009; Accepted: February 12, 2009; Revised: February 14, 2009
Abstract: There are many ways to deliver drugs into the body, viz oral (through swallowing), sub mucosal (through
buccal and sublingual mucosa), parenteral (through injection), transdermal (through skin), pulmonary (through inhalation)
etc. Among these deliveries oral delivery (by swallowing) is widely accepted. In oral drug delivery, many scientific
challenges and breakthrough technologies are required to generate novel dosage forms raising drug delivery to higher
level. Some are self emulsifying systems, solid self nanoemulsion, polymeric micelles, spray freezing, pH controlled
systems, time delayed system, osmotic pumps, prodrugs etc. This paper reviews recent patents, technologies and products
with their importance, manufacturing and novel approaches implemented till date to overcome the challenges in oral drug
delivery systems.
Keywords: Orally disintegrating tablets, controlled drug delivery, gastroretentive formulation, colon targeting, osmotic pumps, prodrugs, nanoemulsion, microemulsion.
INTRODUCTION
Oral drug delivery formulation and technologies are mainly focused on the following areas of gastro intestinal tract (GIT) Fig. (1). [1].
Various challenges associated with
oral route include:
• Pill-swallowing difficulty
• Irritant and unpalatable drugs are not given by this route
• Gastrointestinal (GI) destruction of labile molecules
• Low levels of macromolecular absorption; absorption of drugs may be affected by food in the stomach
• Slow onset of action
• Very little control over release of the drug; non-specific delivery site & side effects
As oral drug delivery is simple, most convenient, safest, noninvasive and most economical, it continues to be the preferential route of administration and researchers are seeking ways to incorporate various technologies in oral formulations; even small improvements in drug delivery technology can make significant differences in enhancing patient compliance and drug bioavailability.
A. Oral Delivery, Ease of Administration
The most facing challenges in oral delivery are to overcome problems like pill-swallowing difficulty, delivery of unpalatable drugs, and reducing dosing frequency. Pill-swallowing difficulty primarily affects the patients having dysphagia, geriatric and pediatric populations; besides these a middle-aged woman undergoing radiation therapy for breast cancer may find it too nauseous to swallow a H2-
*Address correspondence to this author at the Department of Pharmaceutics,
Faculty of Pharmacy, Jamia Hamdard (Hamdard University), New Delhi-110062, India; Tel: 91-11-23828282; Fax: 91-11-23828282;
E-mail: [email protected]; [email protected]
Code Area Property
A Oral pH 6.8, small surface area, lipophilic, neutral
and basic drugs absorbed directly into the
systemic circulation
B Stomach pH 1-3, not too large surface area, lipophilic,
neutral and acidic drugs absorbed but lesser
than that from intestine
C Small
Intestine
pH 5-7.5, very large surface area, major site
for absorption of all types of drugs(lipophilic,
neutral, acidic or basic drugs)
D Colon pH 7.9-8, small surface area, all types of drugs
are absorbed but to a lesser extent
Fig. (1). Different components of GIT (Gastro Intestinal Tract).

Oral Drug Delivery Recent Patents on Drug Delivery & Formulation, 2009, Vol. 3, No. 2 163
blocker) [2]. To eliminate these problems associated with swallowing difficulties, a new dosage form, known as fast-dissolving tablets (FDT), has been developed. The fast-dissolving tablets are also called fast-melt or fast disinte-grating or orally disintegrating tablets (ODT). The orally disintegrating tablets (ODT) are of optimal mechanical strength and disintegrate within 60 seconds in the oral cavity [3]. Oral disintegrating tablets (ODT) are gaining popularity over conventional tablets due to their convenience in administration and suitability for patients having dysphagia [4]. Moreover no water is required for swallowing the tablets and hence suitable for geriatric, pediatric and traveling patients.
In 1986, the first lyophilized fast-dissolving tablet Zydis®
were introduced by Cardinal, after that there was a continuous growth in names by different companies, now a number of fast-dissolving formulations are in market and the technology is still improving. Table 1, different ODT tablets were prepared by different technologies. Zydis
® is manu-
factured by lyophilisation whereas AdvaTab tablets are compressed using a patented external lubrication system in which the lubricant is only applied to the tablet surface. As a result, the tablets are hard and durable, do not require high compression forces during manufacturing and do not repel liquids such as saliva when ingested. Consequently, AdvaTab tablets are robust, disintegrate rapidly in the oral cavity and the tablet compression step does not lead to breakage of the drug particles [5]. Akina has developed a new fast-melting tablet technology, called "Frosta
®." A
Frosta tablet melts in about 10 seconds after placing it in the mouth. The Frosta
® approach utilizes conventional wet
granulation processing for cost-effective production [6]. SPI Pharma’s Pharmaburst is a co-processed excipient system with specific excipients, which allows rapid disintegration and low adhesion to punch faces while another Advantol platform uses proprietary co-processing technology of directly compressible excipient system [7]. It can be used to develop a “soft-chew” or a “quick-melt” solid dosage form for nutraceutical applications. The next in the series is WOWTAB™. It is the first patented, without water quick-dissolve tablet technology of Yamanouchi Pharma, accom-modating both hydrophilic and hydrophobic drugs. WOWTAB™ technology uses a solution of a high-
moldability saccharide (a saccharide that produces a hard compact) to granulate a low-moldability saccharide (poorly compressible saccharide) which provides quick, convenient and consistent dosing [8]. Despite of patented products, many other relevant researches include development of rapidly disintegrating tablets as an oral dosage form for elderly patients with impaired swallowing using agar powders (AG) or treated agar powders (TAG) [9]. OraSolv technology (Cima Labs Inc.) is an oral dosage form, which combines taste-masked drug ingredients with a fast-dissolving system that contains a low level of effervescence. The ‘OraSolv’ tablet dissolves quickly without chewing and need for water. Taste-masking coatings coats the active compound, permitting the active ingredients to be swallowed before they comes in contact with taste buds [10].
DuraSolv (Cima Labs Inc.) is a highly compact, more durable, solid oral dosage system formulated to achieve the primary benefits of the OraSolv fast-dissolve dosage form, but capable of being packaged in conventional packaging such as foil pouches or bottles at much higher production rates and with lower packaging costs. DuraSolv is an appropriate technology for drug products requiring lower levels of active ingredient [11]. The taste of the active ingredient in these products (OraSolv & DuraSolv) is masked by the use of an appropriate coating over the drug particles in conjunction with effective flavoring agents and an artificial sweetener.
Ozeki and coworkers in 2003 examined acidified brewers yeast cell wall (AYC) with respect to novel applications as it can be used as an aqueous coating material for tablets and granules. AYC is a unique pharmaceutical additive posses-sing opposing functions with respect to binding and disintegration [12]. Fukami et al. in 2005 formulated a fast disintegrating compressed tablet using amino acids, such as L-lysine HCl, L-alanine, glycine and L-tyrosine as disinteg-ration accelerator [13]. In another study a rapid disinteg-ration tablet in the oral cavity was prepared using glycine as a disintegrant. It was also suggested in the study that carboxymethylcellulose [14] (NS-300) possessed excellent wetting nature and resulted in the rapid disintegration of tablet [15]. Directly compressible rapidly disintegrating tablets of fenoverine with lower friability, acceptable taste, and shorter disintegration times were obtained using cros-
Table 1. List of Currently Available Fast-Melting Tablet Technologies
Formulation Key Attributes Company
Zydis® Freeze-dried wafers RP Scherer (Cardinal)
Advatab® Direct compression using external lubrication system Eurand
Frosta® Direct compression of granules Akina
Pharmabrust Direct compression of powder mixture SPI Pharma
Advantol™ 200 Directly compressible excipient system SPI Pharma
WOWTAB® High- and low-moldability saccharides Yamanouchi Pharma
OraSolv Low compression force and an effervescent couple as a water-soluble disintegrating agent Cima Labs Inc.
DuraSolv Direct compression using water-soluble excipients Cima Labs Inc.

164 Recent Patents on Drug Delivery & Formulation, 2009, Vol. 3, No. 2 Gupta et al.
povidone and other excipients at optimum concentrations to assist patients palatability of any age group [16]. As compared to direct compression tablets, Kollidon CL excipient base was observed to have maximum drug release and minimum disintegration time showing the superiority of the spray dried excipient base technique [17]. Direct com-pression has been applied to prepare mouth-dissolving tablets of Metformin HCl using various superdisintegrants and tablet containing crosscarmellose and crosspovidone at 5% w/w concentration showed disintegration time of less than 60 seconds along with rapid in vitro dissolution (95% drug release in 5 min.) [18].
Oral Controlled Release Drug Delivery Systems (OCRDDS)
OCRDDS is considered as the solution to second challenge in oral delivery i.e. for reducing dosing frequency. OCRDDS have many advantages over a conventional dosage form like improved patient compliance; reduction in the steady state level of drug hence better control of disease condition and reduced level of local or systemic side effects. It also increases the safety margin of high potency drugs due to better control of plasma levels, increased bioavailability and therefore reduction in total amount of dose administered. This leads to shorter treatment period, meanwhile reduction in the personnel time to dispense, administer and monitor patients etc. which ultimately leads to reduction in health care costs through this improved therapy like Geomatrix™ technology [19]. It allows producing tablets with a wide range of predictable and reproducible drug release profiles, to achieve customized levels of controlled release of specific drugs or it can achieve simultaneous release of two different drugs at different rates from a single tablet. It can deliver an initial burst of a drug for fast therapeutic effect, followed by controlled release at a constant rate, or deliver a drug to specific sites within the patient's digestive system. The controlled release is achieved by constructing a multilayered tablet having a water soluble, swelling as well as erosion occurs simultaneously and both of them contribute to the overall drug release [20]. Polymer matrix such as hydroxyl-propyl methylcellulose (HPMC) can be used. Povidone can be included into the coating dispersion as stabilizer pH of polymers also possess an important property when drug is to be delivered to the targeted area e.g. Gastrointestinal tract or in the colon [21]. Barrier layers delay the interaction between core and dissolution medium [22], and hence, in general, linear release profiles can be obtained by applying hydrophilic barrier layers on both faces of a hydrophobic matrix tablet or vice-versa [23]. Some Geomatrix™ products in the market are, Sular
®, ZYFLO CR™, Coruno
®, diclo-
fenac-ratiopharm-uno®
and Madopar DR®
[19].
B. Stomach
One of the challenge in oral drug delivery is to develop gastric retention platforms for long-term (ranging from 6 to 24 h) delivery of drugs by oral administration.
Need for Gastro-Retentive Drug Delivery
A controlled drug delivery system with prolonged residence time in the stomach is of particular interest for drugs which are locally active in the stomach (e.g., miso-
prostol, antacids, antibiotics against Helicobacter pylori), have an absorption window in the stomach or in the upper small intestine, (e.g. L-DOPA, p-aminobenzoic acid, furosemide, riboflavin etc), unstable in the intestinal or colonic environment (e.g., captopril), exhibit low solubility at high pH values (e.g., diazepam, chlordiazepoxide, vera-pamil HCl). Currently, there are a number of drugs that can be delivered using once-daily formulations. This is possible only for drugs that are well absorbed throughout the GI tract or typically have long elimination half-lives. There are a large number of drugs that are not absorbed to the same extent once they pass the upper small intestine, and thus, once-daily formulations are elusive for these drugs. To overcome the fast GI transit, maintenance of dosage form in the stomach for extended periods of time is development of gastric retention platforms. Various attempts have been made to develop gastroretentive delivery systems. There are 3 ways by which this can be achieved, altering the density of drug particles, use of mucoadhesive polymers and altering the size of dosage form [24]. The composition of the coated controlled release polymer particles includes a polymer core encapsulating the active agent and a mucoadhesive coating around the core [25]. The density of GI fluid is around 1.4 g/cc. Use of drug pellets having density greater than this value, preferable above 1.6 g/cc, results in prolonged GI residence that is unaffected by food. Iron oxide, titanium dioxide and barium sulphate have been used to increase the density of drug pellets. The drug is coated on the heavy core and then covered by a diffusion controlled membrane [24]. Intestinal Protective Drug Absorption System (IPDAS
®
Technology) is a high density multiparticulate tablet tech-nology, intended for use with GI irritant compounds. The IPDAS
® technology is composed of numerous high density
controlled release beads, which are compressed into a tablet form. Once an IPDAS
® tablet is ingested, it rapidly
disintegrates and disperses drug containing beads in the stomach, which subsequently pass into the duodenum and resides in the gastrointestinal tract in a controlled and gradual manner, independent of the feeding state [26].
Low Density pellets can also be called as hydro-dynamically balanced systems, such pellets having density less than that of GI fluids, float on the gastric juice for an extended period of time while slowly releasing the drug. Globular shells such as that of poprice and cellulose have been used to lower the density of system. A swellable gum like HPMC was also used [24]. Manufacturing process include granulating a drug with 20 to 80% of hydrogel such as HPMC, HEC and HPC. On contact with the GI fluids, the tablet swells and forms a diffusible gel barrier that lowers the density of system to less than 1 which allows it to float [26]. Lipophilic polymers such as silicon elastomer can also be modified to have swelling properties. This is achieved by impregnating a water miscible liquid such as glycerol or a water-soluble salt such as sodium chloride in the lipophilic matrix. On contact with aqueous medium, the modified lipophilic polymer swells due to absorption of water by the hydrophilic additives in the matrix. A gas filled flotation chamber can be attached to a membrane coated tablet for making it buoyant [26]. Scintigraphic studies in non-fasting human volunteers confirms that the intra gastric buoyancy of the floating forms is the main process determining their

Oral Drug Delivery Recent Patents on Drug Delivery & Formulation, 2009, Vol. 3, No. 2 165
prolonged gastro retentive time (GRT) and protecting them from random gastric emptying related to antral peristaltism [27].
Retention time of the drugs can be increased to more
than 12 hours by utilizing floating or hydrodynamically controlled drug delivery systems, such as using cellulose acetate microspheres as floating depot systems increases gastric retention of antidiabetic drug [28].
These
formulations, with their reduced frequency of administration and better control over drug disposition, may provide an economic benefit to the user as compared to market available products. Microspheres based mucoadhesive formulation are extensively evaluated and characterized to determine diffu-sion parameters and drug retention in the stomach mem-brane. Such as chitosan microspheres appear, technically promising mucoadhesive drug delivery systems for delivering clarithromycin to treat stomach ulcers [29].
It
demonstrates good mucoadhesion of the microspheres with the stomach mucosa as well as higher accumulation of drug in the stomach membrane. Microspheres also exhibited sustained release of drug. Amoxicillin mucoadhesive micros-pheres, pH-sensitive excipient composition microspheres were prepared containing chitosan [30] (used as polymer) and glutaraldehyde as a cross-linking agent by simple emulsification phase separation technique. These micro-spheres were systematically evaluated and showed that they were able to reside in the gastrointestinal tract for an extended period of time for a more effective H. pylori eradication and hence treating gastric and duodenal ulcers which were associated with Helicobacter pylori. The results showed a more complete H. pylori clearance effect in case of amoxicillin mucoadhesive microspheres as compared to amoxicillin powder. In conclusion, the prolonged gastroin-testinal residence time and enhanced amoxicillin stability [31]. The use of Chitosan, thiolated chitosan, Carbopol 71G and Methocel K15M as mucoadhesive microspheres prolon-ged the delivery of acyclovir with significant improvement in oral bioavailability due to enhanced retension in upper GI tract [32].
Size based systems relies on the fact that gastric emptying of a dosage form can be delayed in the fed state if its size is greater than 2mm. Dosage form of size 2.5 cm or large is often required to delay emptying time long enough to allow once daily dosing. Such forms are however difficult to swallow [26].
C. Small Intestine
Challenges related to delivery to small intestine include development of formulations of poorly soluble and high molecular weight drugs and delivering of protein peptides due to their instability in GI environment. Solubility of drug is an important factor concerned with development of con-trolled release formulations. The first step in drug absorption is dissolution of a drug in an aqueous environment. Drugs with low solubility are those requiring more than 250 mL of water to dissolve the highest dose. Volume of 250 mL is not easy to find in the intestine. Only a limited number of com-mercial formulations are available for poorly water soluble drug.
Approaches to Enhancing Delivery of Poorly Water-
Soluble Drugs
Solid dispersion refers to the dispersion of one or more active ingredients in an inert carrier or matrix in solid state prepared by the hot melt extrusion, co-preciptation or the melting-solvent method [33]. A controlled release phar-maceutical composition for oral use can be comprised of a solid dispersion of an active substance, a polymer that has plasticizing properties, a stabilizing agent, then at least one active substance having a limited water solubility [34]. It has also been defined as the product formed by converting a fluid drug carrier combination to the solid state[35]. Solid dispersions can be classified as Solid solutions; glass solutions of suspensions (A glass solution is a homogeneous system in which a glassy or a vitreous form of the carrier solubilizes drug molecules); compound or complex for-mation between the drug and the carriers; amorphous precipitations of drug in crystalline carrier.
Solid solutions consist of a solid solute dissolved in a solid solvent. The particle size in solid solution is reduced to molecular level. Successful solubilization of Itraconazole has been achieved using solid solution technique [35]. Solid solutions of lower drug concentrations generally give faster dissolution rate. Drug dissolution improves considerably with an increase in molecular weight of a water soluble polymer such as polyethylene glycol. Gris-PEG
® (Pedinol
Inc.) is another example of solid dispersion which contains ultramicrosize griseofulvin, colloidal silicon dioxide, polyethylene glycol 400 and 8000 [36]. The technique of solid dispersion is used for increasing the dissolution performance of glyburide tablets in polyethylene glycol (high hydrophilic carrier). The possibility of developing solid dispersion of glyburide tablets (a poorly water-soluble oral hypoglycemic agent) with fast, efficient, reproducible, and markedly higher drug dissolution rate and dissolution efficiency; clearly better than those from various commercial tablets [37]. Compound or complex formation between the drug and the carriers system is characterized by com-plexation of two components in a binary system during solid dispersion preparation. The availability of a drug from the complex is dependent on the solubility, dissociation constant and the intrinsic absorption rate of the complex. and CD (cyclodextrine) in combination with polyethylene glycol (PEG) 6000 have been used to formulate such systems.
Amorphous precipitation occurs when the drug precipitates as an amorphous form in the inert carrier. The high-energy state of the drug in this system generally produces much greater dissolution rates than the corresponding crystalline forms of the drug [35].
Microemulsions are liquid dispersions of water and oil that are made homogenous, transparent, and stable by the addition of relatively large amounts of a surfactant and co-surfactants [38]. Micelles can solubilize hydrophobic therapeutic agents effectively and are safe for human use. Micelles formed from amphiphilic block copolymers such as poly(ethylene glycol) as a hydrophile and poly(propylene sulfide) (PPS) as a hydrophobe, using the immunosup-pressive drug cyclosporin A (a highly hydrophobic drug) was developed. These micelles demonstrate interesting solubilization characteristics, due to the low glass transition

166 Recent Patents on Drug Delivery & Formulation, 2009, Vol. 3, No. 2 Gupta et al.
temperature of the PPS block and low melting temperature of the block polymer, highly hydrophobic nature, and good solvent properties of it enable the use of such a simple drug encapsulation technique [39]. Amphiphilic ABC triblock copolymers composed of monomethoxy-capped poly (ethylene glycol) (MPEG), poly(2-(dimethylamino)ethyl methacrylate) (DMA), and poly(2-(diethylamino)ethyl methacrylate) (DEA) have been used for pH-induced micellization for solubilization and controlled release of a hydrophobic drug dipyridamole [40].
Self-Emulsifying Systems, a novel self-emulsifying drug delivery system (SEDDS) is useful for the adminis-tration of a water-insoluble drug. The SEDDS comprises a hydrophilic surfactant with hydrophilic-lipophilic balance (HLB) value greater than 10 [41]. These formulations have also attracted interest because they can improve the bioavailability of compounds that fall into Class II of the biopharmaceutical classification system (BCS). Class II compounds are poor water soluble and highly permeable [42].
The IDD
®-SE
(Self-Emulsifying) technology (Skyepharma) constitutes a special class of the IDD
® systems due to their production.
The particles of IDD®
-P and IDD®
-D formulations result from application of a physical or mechanical process. On the other hand, surface stabilised micrometre to submicrometre sized particles or droplets are self generated when a dosage form containing IDD
®-SE formulation is exposed to an
aqueous medium such as those present in gastrointestinal or vascular compartments [43]. A self-microemulsifying system (SMES) consisting of ethyl oleate, Cremophor
® RH 40 and
Labrasol®
with hydroxy-propylmethylcellulose (HPMC) is used to control the in vitro release of poorly soluble drugs from solid oral dosage forms [44].
The control of the release
of water-insoluble drugs from pellet formulations by incorporating them into self-emulsifying systems, which enhances their rate of release and then, by applying a water-insoluble polymer containing water soluble plasticizer and talc, reduce the rate of drug release. A sample of pellets containing methyl paraben in the self-emulsifying system was pre-coated with a film of hydroxypropylmethyl cellulose from an aqueous solution and then coated. The application of a sub-coating of hydroxypropylmethyl cellulose was able to reduce the release rate of methyl paraben self-emulsifying pellet system coated with ethyl cellulose. Thus, the formulation approach offers the possibility of formulating and controlling the in vitro release of water-insoluble drugs from solid oral dosage forms [45].
Solid Self-Nanoemulsifying Systems (SSNES)
Solidification of these liquid systems to yield solid self-nanoemulsifying systems (SSNES) offer a possible solution over Conventional SNES which consist of liquid forms filled in hard or soft gelatin capsules, which are least preferred due to leaching and leakage phenomenon, interaction with capsule shell components, handling difficulties, machina-bility, and stability problems, and that is why these systems have attracted wide attention [46]. Surfactants are molecules with well defined polar and non-polar regions that allow them to aggregate in solution to form micelles. Non-polar drugs can partition into these micelles and can be made solubilized. Depending on the nature of the polar area, surfactants can be non-ionic (eg, polyethylene glycol),
anionic (eg, sodium dodecyl sulfate), cationic (eg, trialkyl-ammonium) and zwitterionic (eg, glycine and proteins) [47]. Among these the most commonly used ones are the anionic and non-ionic surfactants. Since the process of solubilization occurs due to presence of micelles, generally high concen-trations of surfactants are needed to significantly improve drug solubility. One example of surfactant based solution is Taxol (paclitaxel), an anticancer drug that is solubilized in 50% solution of Cremophor. Other examples include Valru-bicin in 50% Cremophor and Cyclosporin in 65% Cremophor [35].
Wet milling reduces the particle size to nano-sized through attrition in the presence of some stabilizing agents [48]. Various technologies such as Elan's NanoCrystal
®
Technology is a drug enablement and optimization tech-nology applicable to poorly water-soluble compounds. Similarly, NanoCrystal
® Particles are very small particles of
drug substance, of the order of less than 1,000 nanometers (nm) in diameter, particles of this diameter are produced by milling the drug substance using a proprietary, wet-milling technique. The NanoCrystal
® particles of the drug are
stabilized against agglomeration by stabilizers such as surface adsorption of selected GRAS (Generally Regarded As Safe) stabilizers. The result is the formation of NanoCrystal Colloidal Dispersion
TM solution, which can be
processed into finished dosage forms for all routes of administration [49].
Nano and Microparticle Approach
Recently, the nano and microparticle approach [50] has become popular. A decrease in particle size results in increased surface area, resulting in faster dissolution, normally by a small order of magnitude. In some cases, this may be enough to result in increased bioavailability. Eurand's novel Biorise
® technology can be applied to a wide
range of compounds to enhance bioavailability and solu-bility. Biorise is based on the fact that the solubility of nanocrystalline and amorphous forms of drugs is higher than that of unmodified or even micronized forms. Therefore, if drug permeability is sufficient, the absorption rate and absolute bioavailability of a drug can be increased by enhancing solubility and solubilization. Biorise creates New Physical Entities (NPEs) by physically breaking down a drug's crystal lattice. This results in drug nanocrystals and/or amorphous drug, which are then stabilized with biologically inert carriers. The carriers used in the Biorise system are biocompatible and readily disperse in the body's GI fluids. The final product is a free-flowing powder that can be incorporated into a variety of dosage forms to achieve the most effective delivery [51].
The insoluble drug delivery
(IDD®) platform (Skye pharma) consists technologies to
address formulation and delivery problems originating from poor solubility of biologically active compounds [52]. These IDD
® technologies feature easily dispersible narrow particle
size distribution dosage forms derived from surface modified micrometer to sub micrometer sized particles or droplets stabilized by surface modifiers, preferably phospholipids. The IDD
® platform is different to other drug delivery
systems as its effectiveness is dependant on the physi-cochemical properties of the drug rather than the chemical structure and reactivity [43].

Oral Drug Delivery Recent Patents on Drug Delivery & Formulation, 2009, Vol. 3, No. 2 167
Some insoluble drug delivery approaches are, IDD®-P
(MicroParticle) a microparticulate variation of the IDD®
drug delivery system, which consists of a pure solid drug in the core of the particle [43].
IDD
®-D formulations
(MicroDroplet), the core is constituted of essentially liquid drug substance. IDD
®-P and IDD
®-D formulations are
produced by application of high shear, cavitations or impaction (e.g. attrition, homogenization, microfluidisation, milling, ultrasonication, etc.) to reduce the drug particle size in the presence of phospholipids (and/or other surface modifiers) that associate at the freshly generated drug surface. A particle size reduction from approximately 100-200 m to about1 m is achieved resulting in a very homo-geneous and stable formulation [43]. Flamel’s Medusa
®
delivery system is a versatile nanotechnology which can accommodate the physicochemical specifications and the delivery therapeutic-requirements of a wide variety of protein drugs. Medusa
® nanocarrier is also suitable for the
efficient delivery of insoluble (i.e. hydrophobic) small molecule drugs and could be applied to peptides [53]. Polymeric nanoparticles and nanoparticles-in-microsphere oral system (NiMOS) are useful for systemic and oral gene therapy, respectively [54].
Particle engineering technologies [55] includes tech-
nologies from BioAqueous solubilization services (Dow pharma) modify drug substances, changing particle size, surface area or morphology to yield stabilized, isolated redispersible powders with enhanced dissolution charac-teristics. These powders can be readily formulated into final dosage forms. Some of these techniques are reviewed briefly as Controlled Precipitation, a technology that creates crys-talline nanostructured drug particles with rapid dissolution rates [56]. With this technology, the drug is dissolved in a water-miscible solvent and dispersed into water. Nano-structured drug particles are formed from the solvent droplets as the solvent dissolves into the water phase. Stabi-lizers are added to the water and/or solvent to control particle growth and to stabilize the final particles. The stabilized particles are isolated as dry powders by standard techniques [55]. Template Emulsion, a technology that creates nanostructured drug particles with controlled particle size distribution and yielding rapid dissolution performance [57]. With this technology, an oil-in-water emulsion is prepared then swelled with a nonaqueous solution of the drug and stabilizers. The particle size distribution of the drug particles is a direct result of the size of the emulsion droplets prior to loading with drug, a property which can be controlled and optimized in this process. Furthermore, through selected use of solvents and stabilizers, emulsion stability is achieved with no or suppressed ostwald ripening. Subsequently, the solvent and water are removed, and the stabilized nanostructured drug particles are recovered. Various particle morphologies can be achieved by appropriate control of processing conditions [55]. Spray Freezing into Liquid
(SFL), [58] a novel, cryogenic technology to create nano-structured drug particles with greatly enhanced surface area. This technology consists of an organic or organo-aqueous solution of a drug with stabilizers, which is injected into a cryogenic liquid, such as liquid nitrogen. The droplets of the drug solution freeze at a rate sufficient to minimize crystal-lization and particle growth, thus forming nano-structured
particles. The choice of solvent system and processing conditions, determines particle morphology of particles. In the isolation step, the nitrogen and solvent were removed to avoid agglomeration or ripening of the drug particles [55].
Evaporative Precipitation into Aqueous Solutions (EPAS), a technology that creates nanostructured drug particles with rapid dissolution performance. For this technology, the drug is dissolved in a hydrophobic solvent and atomized into water at a temperature above the boiling point of the solvent. The rapid expansion and atomization of the solvent in the water phase cause the formation of nano-structured drug particles. The solvent and/or water phase contains the stabilizers for the particles. The evaporated solvent is recovered, and the stabilized particles are isolated from the aqueous phase by standard techniques [55]. RR01, a new highly lipophilic drug showing extremely low water solubility and poor oral bioavailability, has been incor-porated into pH-dependent dissolving particles made of a poly(methacrylic acid-co-ethylacrylate) copolymer these pH-dependent dissolving particles have a great potential as oral delivery systems for drugs with low water solubility and acceptable permeation properties [59]. A novel cubic liquid crystalline phase is described within the pseudoternary system comprising lauric acid, monolaurin, and simulated endogenous intestinal fluid (SEIF). This cubic phase may potentially be of benefit in the delivery of poorly water-soluble compounds due to its high loading capacity and potential for sustained release [60].
Polymeric micelles are Amphiphilic polymers assemble into nanoscopic supramolecular core-shell structures, which are under extensive study for drug delivery [61]. Potential of polymeric micelles for the solubilization and delivery of STAT3 inhibitor cucurbitacins in solid tumors
has been
shown by Molavi O et al. [62].
A very low water soluble
drug Propofol (99.7%) was added directly to an aqueous solution of poly(N-vinyl-2-pyrrolidone)-block-poly(d,l-lactide) copo-lymers (PVP-PLA) block copolymers and stirred in order to obtain a clear solution. This formulation was filtered sterile and then lyophilized to its solid form Propofol-PM (propofol polymeric micelle) which recons-titutes to a propofol 1%w/v (10 mg ml
-1) clear aqueous
solution of 30-60 nm propofol-containing micelles [63].
Some patented technologies Table 2 are Polymeric Nano-Delivery System™ (PNDS) technologies (Labopharm) are an excellent complement to Contramid because they allow optimized delivery of water-insoluble drugs with poor bioavailability or safety profiles. PNDS are a library of block copolymers designed to address the unmet needs of insoluble drug delivery. Comprising hydrophobic and hydrophilic GRAS (Generally Recognized As Safe) subunits fused as novel molecular entities, PNDS can be simply manufactured to form stable nano-vehicles or micelles that entrap insoluble drugs, making them available for oral or parenteral adminis-tration in safe and convenient forms [64].
OraVescent
technology to improve the transport of poorly absorbed and large molecule active drugs across mucosal membranes in the oral cavity or the gastrointestinal tract has been deve-loped by Cima Lab [65].
.Akina Inc. has developed concepts
of hydrotropic polymer (Hytrop®
), hydrotropic hydrogel (HyTrogel
®), and hydrotropic polymer micelle (Hytro
Cell™) for delivery of poorly water-soluble drugs. Hytrops,

168 Recent Patents on Drug Delivery & Formulation, 2009, Vol. 3, No. 2 Gupta et al.
HyTrogels, and HytroCelles are able to increase the aqueous solubilities of poorly soluble drugs by 2-4 orders of magnitudes. Hydrotropic polymer micelle (HytroCell™); HytroCelles based on PEG-b-poly (2-(4-vinylbenzyloxy)-N,N-diethylnicotinamide) (PEG-b-PVBDENA) can dissolve paclitaxel more than 30%wt and yet they are stable in aqueous solution for weeks. Such HytroCelles can be formulated for oral administration of paclitaxel [66].
Lip'ral
is a patented technology (Lipocine Inc) based on lipidic compositions which form the optimal dispersed phase in the gastrointestinal environment for improved absorption of the insoluble drug. Lip'ral presents insoluble drugs efficiently to the intestinal absorption site, thus bringing the absorption process under formulation control and making the product robust to physiological variables such as dilution, pH, and food effects.
Lip'ral can be extended with Lipocine's
Synchronized Solubilizer Release technology, Lip'ral-SSR, to enable controlled release of insoluble drugs and drugs with pH-sensitive solubility [67]
Approaches to Enhancing Delivery of Highly Water
Soluble, Poorly Permeable Drugs
Hydroance technology (Lipocine Inc.) enables oral delivery of compounds that are currently delivered only by the invasive route, such as conventional polar organics (MW<1000), Polysaccharides, Peptides, Peptidomimetics, Oligonucleotides, Genetic materials, Cytokines, and Proteins (vaccines). Hydroance customizes lipid based carriers for the optimal availability of the drug and absorption at the site, thus enabling absorption of the drug across the epithelial cell membranes of the intestinal tract [67].
Delivery of Proteinous Drugs
A major challenge with large molecules such as proteins is the vast number of functional groups that can be the subject of chemical degradation as well as the complex structure of the individual proteins. Generally, it is important to develop efficient techniques for characterization and profiling of these complex protein samples. Even a small conformational change may have a detrimental effect on the function of the protein. Therefore, efforts to correctly characterize as well as creating a suitable and stable dosage form of each therapeutic protein have to be made such as microparticles of protein, polypeptide and peptide drugs [68].
Bovine serum albumin (BSA) as a protein model drug was loaded in Thiolated Eudragit-coated CMs (TECMs) to study the release character of the delivery system. Results indicated that TECMs have potentials to be an oral protein drug carrier [69]. Emisphere’s broad-based oral drug delivery technology platform, known as the eligen
® tech-
nology, is based on the use of proprietary, synthetic chemical compounds, known as EMISPHERE
® delivery agents, or
“carriers.” These delivery agents facilitate or enable trans-port of therapeutic macromolecules across biological mem-branes such as those of the gastrointestinal tract, allowing the therapeutic molecules to exert their desired pharmacological effect. The delivery agents have no known pharmacological activity themselves at the intended clinical dose levels. Emisphere’s eligen
® technology makes it possible to orally
deliver a therapeutic molecule without altering its chemical
form or biological integrity [70]. The eligen®
technology is applicable to a diverse group of drug molecules (e.g., pro-teins, peptides and other poorly absorbed compounds). Molecules ranging in size from 500 Daltons to >150,000 Daltons can be delivered easily [70].
D. Targeting to the Colon
Targeted delivery of drugs to the colon become popular in recent years to achieve one or more of four objectives. (a) to reduce dosing frequency; (b) to achieve high local concentrations of drug in the treatment of diseases of the distal gut e.g. ulcerative colitis or Chron’s disease; (c) to delay delivery to a time appropriate to treat acute phases of disease (chronotherapy); and finally, (d) to deliver drug to a region that is less hostile metabolically, e.g. to facilitate absorption of acid and enzymatically labile materials, especially peptides. Colon is also rich in lymphoid tissue so uptake of antigens into the mast cells of the colonic mucosa produces rapid local production of antibodies thus helping in vaccine delivery [71].
For low molecular weight drugs, aqueous solubility plays an important role. Drugs with high solubility may prematurely release before reaching the colon. Preventing premature release would require a very thick coating which may result in incomplete drug release in the colon. Drugs with poor solubility may not dissolve in the colon where there is not as much fluid as in the upper portion of the GI tract. The current technologies are mainly focused on delayed release, i.e. no release until the dosage form reaches the colon. The time-delay approach relies on drug release following a predetermined lag time and would therefore be less dependable. Combinations of time-delay and enzyme-degradable systems would seem be more reliable. Common approaches used for colon targeting are listed in Table 3.
pH Controlled system relies on the fact that the pH in the terminal ileum and colon (except ascending colon) is higher than in any other region of the GI tract. Thus a dosage form that disintegrates preferentially at high pH levels has good potential for site-specific delivery into this region. A rapidly disintegrating tablets coated with a model pH depedent polymer, Eudragit S
®, have been investigated
in vivo, using gamma scintigraphy, to assess its suitability for use in colonic drug delivery [72]. In spite of the limitation of change in luminal pH due to disease state, such pH dependent systems are still very commonly investigated for colon targeting. A system for colonic targeting of metronidazole was developed by coating the drug-containing capsules with azoaromatic and pH-sensitive polymers that are prone to selective degradation in the colon environment. These polymers were characterized for physical attributes, namely film-forming properties, effect of colon bacterial flora, and pH [73]. A novel formulation for oral adminis-tration using pH-enzyme Di-dependent chitosan mciro-spheres (MS) and 5-Fu as a model drug has been inves-tigated for colon-specific drug delivery by the emulsifi-cation/chemical cross-linking and coating technique, respectively and results clearly demonstrated that the pH-enzyme Di-dependent chitosan MS is potential system for
Oral Drug Delivery Recent Patents on Drug Delivery & Formulation, 2009, Vol. 3, No. 2 169
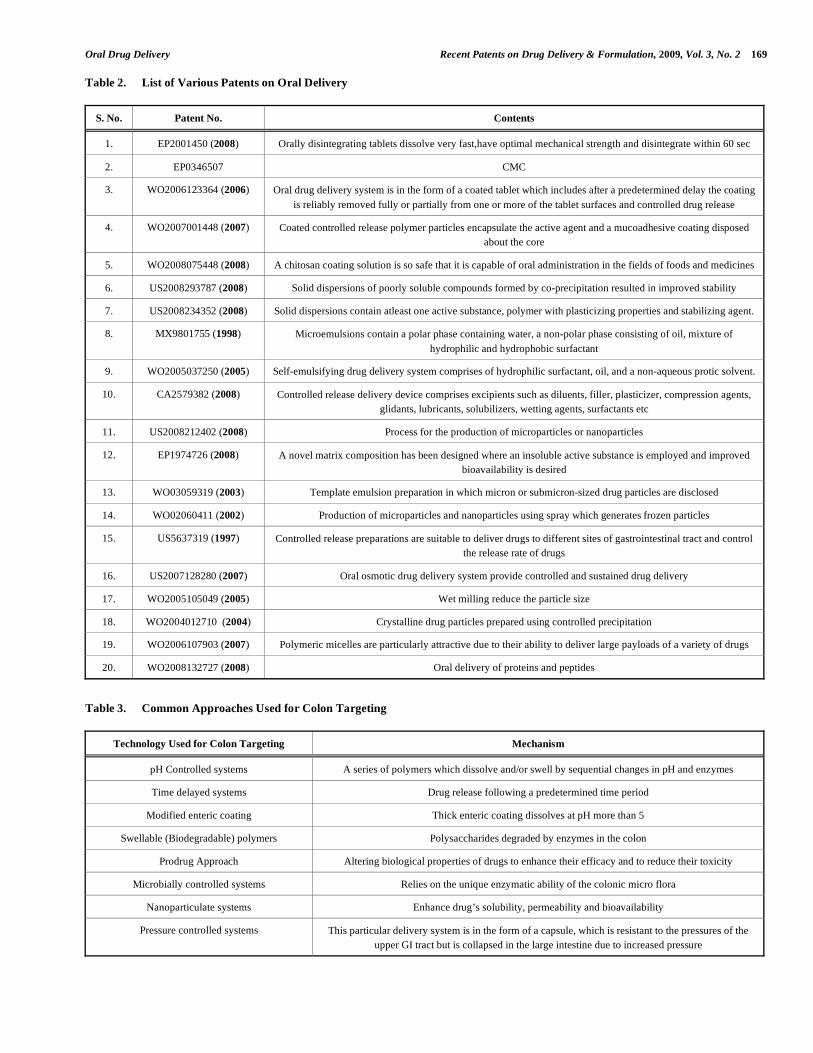
Table 2. List of Various Patents on Oral Delivery
S. No. Patent No. Contents
1. EP2001450 (2008) Orally disintegrating tablets dissolve very fast,have optimal mechanical strength and disintegrate within 60 sec
2. EP0346507 CMC
3. WO2006123364 (2006) Oral drug delivery system is in the form of a coated tablet which includes after a predetermined delay the coating
is reliably removed fully or partially from one or more of the tablet surfaces and controlled drug release
4. WO2007001448 (2007) Coated controlled release polymer particles encapsulate the active agent and a mucoadhesive coating disposed
about the core
5. WO2008075448 (2008) A chitosan coating solution is so safe that it is capable of oral administration in the fields of foods and medicines
6. US2008293787 (2008) Solid dispersions of poorly soluble compounds formed by co-precipitation resulted in improved stability
7. US2008234352 (2008) Solid dispersions contain atleast one active substance, polymer with plasticizing properties and stabilizing agent.
8. MX9801755 (1998) Microemulsions contain a polar phase containing water, a non-polar phase consisting of oil, mixture of
hydrophilic and hydrophobic surfactant
9. WO2005037250 (2005) Self-emulsifying drug delivery system comprises of hydrophilic surfactant, oil, and a non-aqueous protic solvent.
10. CA2579382 (2008) Controlled release delivery device comprises excipients such as diluents, filler, plasticizer, compression agents,
glidants, lubricants, solubilizers, wetting agents, surfactants etc
11. US2008212402 (2008) Process for the production of microparticles or nanoparticles
12. EP1974726 (2008) A novel matrix composition has been designed where an insoluble active substance is employed and improved
bioavailability is desired
13. WO03059319 (2003) Template emulsion preparation in which micron or submicron-sized drug particles are disclosed
14. WO02060411 (2002) Production of microparticles and nanoparticles using spray which generates frozen particles
15. US5637319 (1997) Controlled release preparations are suitable to deliver drugs to different sites of gastrointestinal tract and control
the release rate of drugs
16. US2007128280 (2007) Oral osmotic drug delivery system provide controlled and sustained drug delivery
17. WO2005105049 (2005) Wet milling reduce the particle size
18. WO2004012710 (2004) Crystalline drug particles prepared using controlled precipitation
19. WO2006107903 (2007) Polymeric micelles are particularly attractive due to their ability to deliver large payloads of a variety of drugs
20. WO2008132727 (2008) Oral delivery of proteins and peptides
Table 3. Common Approaches Used for Colon Targeting
Technology Used for Colon Targeting Mechanism
pH Controlled systems A series of polymers which dissolve and/or swell by sequential changes in pH and enzymes
Time delayed systems Drug release following a predetermined time period
Modified enteric coating Thick enteric coating dissolves at pH more than 5
Swellable (Biodegradable) polymers Polysaccharides degraded by enzymes in the colon
Prodrug Approach Altering biological properties of drugs to enhance their efficacy and to reduce their toxicity
Microbially controlled systems Relies on the unique enzymatic ability of the colonic micro flora
Nanoparticulate systems Enhance drug’s solubility, permeability and bioavailability
Pressure controlled systems This particular delivery system is in the form of a capsule, which is resistant to the pressures of the
upper GI tract but is collapsed in the large intestine due to increased pressure

170 Recent Patents on Drug Delivery & Formulation, 2009, Vol. 3, No. 2 Gupta et al.
colon-specific drug delivery of 5-Fu [74]. Eudragit-coated
pectin microspheres are promising controlled release carriers for colon-targeted delivery of 5-fluorouracil (FU). The release profile of FU from Eudragit-coated pectin microspheres was pH dependent. In acidic medium, the release rate was much slower; however, the drug was released quickly at pH 7.4 [75]. The formulation containing locust bean gum and chitosan in the ratio of 4:1 held a better dissolution profile, higher bioavailability and hence a potential carrier for drug targeting to colon [76]. A novel colon targeted tablet formulation was developed using pectin as carrier and diltiazem HCl and indomethacin as model drugs. These tablets were coated with inulin followed by shellac. It was evaluated that this coating system limited the drug release in stomach and small intestinal environment and released maximum amount of drug in the colonic environ-ment. The study revealed that polysaccharides as carriers and inulin and shellac as a coating material can be used effectively for colon targeting of both water soluble and insoluble drugs [77].
One of the example of time delayed system is the OCAS
™ (Oral Controlled Absorption System) (Yamanouchi
pharma) drug delivery technology enables the gradual release of drug active as the tablet system travels through the digestive tract, including the colon, where drug release is difficult to achieve [78]. Modified enteric coating, allows for a small portion of a drug dose to be released into the stomach, with the remainder of release occurring rapidly upon passage of the dosage form into the small intestine. This release profile is particularly suited to drugs which demonstrate site specific absorption in the upper portion of the GI tract (duodenum, jejunum), or where high dose drugs are being delivered which have narrow therapeutic indices. These high volume formulations can result in prolonged gastric retention times, which can then release drug close to the time when the next dose is delivered, thereby resulting in potential overdose of patients. Such a release profile can be achieved through the use of hydrophilic pore formers in pH dependent enteric coatings. Modified enteric coating is thus another approach of colon targeting, colon targeting of ATL-2502 (Prednisolone sodium metasulphobenzoate) is achieved by using colal
TM coating [79].
Swellable (Biodegradable) polymers, based on this approach, various polysaccharides have been investigated for colon-specific drug release. These polysaccharides include pectin, guar gum, amylose, inulin, dextran, chitosan, and chondroitin sulphate. This family of natural polymers has an appeal to drug delivery as it is comprised of polymers with a large number of derivatizable groups, a wide range of molecular weights, varying chemical compositions, and, for the most part, low toxicity and biodegradability yet high stability. The most favorable property of these materials is their approval as pharmaceutical excipients [80]. The for-mation of colonic digestible films of amylose and ethyl-cellulose from aqueous dispersions at temperatures below 37
0C. This technique also demonstrated that the ethyl-
cellulose and the amylose were not miscible. The ability of faecal slurry to digest the films formed at low temperatures was confirmed by the use of a batch fermenter. The extent of digestion was directly related to the amylose content of the films, ensuring the potential to provide films, which could
function as colon specific coatings [81].
Amylose-ethylcellulose film coatings obtained from organic-based solvents were investigated as potential vehicles for colonic drug delivery [82]. Enzyme controlled release pellets were prepared by a fluid bed coater,using ethylcellulose aqueous dispersion(Surlease) and Amylose as the controlled coating meterials,using diethyl phthalate as the plasticizer [83].
A
novel colon targeted tablet formulation was developed using natural polysaccharides such as chitosan and guar gum as carriers and diltiazem hydrochloride as model drug. The prepared blend of polymer-drug tablets were coated with two layers, inulin as an inner coat followed by shellac as outer coat. The study revealed that polysaccharides as carriers and inulin and shellac as coating materials can be used effectively for colon targeting of drugs for treating local as well as systemic disorders [84].
A novel tablet of protein
drug matrix for colon targeting was developed using resistant starch as a carrier prepared by pre-gelatinization and crosslinking of starch [85]. A novel formulation for oral administration using coated calcium alginate gel beads-entrapped liposome and bee venom peptide as a model drug has been investigated for colon-specific drug delivery in vitro. The results clearly demonstrated that the coated calcium alginate gel beads-entrapped liposome is a potential system for colon-specific drug delivery [86]. System which consists of ketoprofen-loaded Zn-pectinate gel (ZPG) microparticles together with pectin/dextran mixtures in a tablet form has been investigated, in vitro and this approach suggests that ZPG microparticles and their modified-release formulations are promising as useful controlled-release carriers for colon-targeted delivery of drugs [87]. Novel formulation consisting of cross-linked microspheres of guar gum has been investigated for colon-targeted delivery of metronidazole. An emulsification method involving the dispersion of aqueous solution of guar gum in castor oil was used to prepare spherical microspheres [88]. Prodrug
Approach includes chemical modification of a biologically active compound that will liberate the active compound in vivo by enzymatic or chemical processes. The primary purposes for designing prodrugs are improving physicoche-mical properties of drugs to overcome formulation problems, and altering biological properties to enhance their efficacy and to reduce their toxicity. Azo-prodrugs such as sulphasalazine, olsalazine and balsalazide, have long been used in managing inflammatory disorders of colon. After entry into the large intestine, the active species, 5-ASA, is released from the prodrug after the cleavage of the azo-linkage [89]. Flurbiprofen beta-cyclodextrin (Fbp-beta-CyD) prodrug was found beneficial in treatment of inflammatory bowel disease [90]. The experimental results showed the good colon targeting property of dexamethasone succinate dextran (DSD) prodrug compared with free dexamethasone [91].
One strategy for targeting orally administered drugs to
the colon exploits carriers that are degraded specifically by colonic bacteria and utilizes microbially degradable polymers/drugs, especially azo-cross-linked polymers/drugs. Prodrugs utilizing azo linkages are sulfasalazine, ipsalazine, balsalazine, and olsalazine. These were developed for delivery of 5-amino salicylic acid to the colon for localized chemotherapy of inflammatory bowl disease. The azo-conju-gation approach utilizes the ability of the colonic environ-ment to cleave these conjugates and protects the drug from

Oral Drug Delivery Recent Patents on Drug Delivery & Formulation, 2009, Vol. 3, No. 2 171
absorption or degradation in the upper gastrointestinal tract. It is believed that flavin mediators present in the colon and azo-reductase enzymes released from colonic bacteria are responsible for the degradation of azo-aromatic compounds for site-specific delivery of the drug to the colon [92]. Microbially controlled systems includes CODES™ (Yamanouchi pharma), a new drug delivery technology enabling targeted release of proteins/peptides and other drug compounds in the colon, an area where drug release is difficult to achieve. By including lactulose (which is degraded when exposed to the colon's microflora) in the drug core formulation, drug is released in the colon subsequent to the formation of organic acids. The drug core is prevented from degradation/absorption prior to the colon by first an enteric coating and then a cationic polymer coating for passage through the small intestine to the cecum [93]. Nanoparticulate systems is the latest approach in colon targeting, using colon cancer cell lines as surrogate biomar-kers, the feasibility of preparing a colon-targeted aspirin and folic acid loaded nanoparticulate drug delivery system with a high encapsulation efficiency and narrow size range was shown. Preliminary in vitro drug release studies with aspirin loaded nanoparticles show a controlled pattern of drug release over a 48-96h period. Thus, demonstrate the capa-bility of drug-loaded nanoparicles to be targeted directly to the colon at low doses for prolonged chemopreventive action [94].
Example of pressure-controlled system for colonic
delivery includes only one invention related to the develop-ment of pressure-controlled system for colonic delivery [95-96]. This particular delivery system is in the form of a capsule, which is resistant to the pressures of the upper GI tract but is collapsed in the large intestine due to increased pressure. The capsule shells are fabricated from ethyl-cellulose and the collapse time of the capsule in the large intestine can be controlled by adjusting the thickness of the capsule shell wall [95]. The preferred thickness of the capsule wall is about 35-60 μm [96].
Oral Osmotic Drug Delivery System is used to provide controlled deliver of drug which includes a drug layer, an osmotic layer, and an outer coating surrounding the drug layer and osmotic layer, where the outer coating includes at least one opening therein that is provided adjacent the drug layer [97].
CURRENT & FUTURE DEVELOPMENTS
Although oral delivery meets the need for self-adminis-tered drugs but targeted, sustained release and increased bioavailability present the areas of difficulty in meeting the emerging value proposition. To address this difficulty, companies are developing micro-fabricated drug delivery systems. Technologies such as nano-pore membranes and micro-particles enable the drug to survive stomach acids and released at specific targeted areas of the gastrointestinal tract. The presented technologies are being developed to provide more efficient drug absorption and enhanced bioavailability. In future more research work can be carried out for further developments in oral drug delivery systems.
CONFLICT OF INTEREST
Authors do not have any conflict of interest for this article.
REFERENCES
[1] Brahmankar DM, Jaiswal SB. Absorption of drugs. In: Biopharmaceutics and pharmacokinetics a treatise. Delhi, Vallabh
Prakashan 2006; 51. [2] http://www.hughes-medical.com/products/fast-dissolving-
tablets2.htm [3] Pilgaonkar, P.S., Rustomjee, M.T., Gandhi, A.S., Bagde, P.,
Morvekar, H.N.: EP2001450 (2008). [4] Dina NM, Madhu B, Shailendra KS, Sengodan GVK. Spray dried
excipient base-a novel technique for the formulation of orally disintegrating tablets. Chem Pharm Bull 2006; 54: 99-102.
[5] http://www.eurand.com/Technologies/Dosage-Form Technology/ AdvaTab/
[6] http://www.akinainc.com/frosta.htm [7] http://www.spipharma.com/downloads/Products/DDS/Advantol_20
0/Advantol_Tech_Bulletin.pdf [8] http://books.google.co.in/books?id=mw9W82MLYZ8C&pg=PA21
5&lpg=PA215&dq=WOWTAB%2Bfirst+patented%2Bhigh moldability+saccharide%2BYamanouchi+Pharma&source=web&ot
s=_phBQ_THjL&sig=ftrfOeOE6uD6Bi8B7oPUx58iOds&hl=en&sa=X&oi=book_result&resnum=1&ct=result
[9] Ito A, Sugihara M. Development of oral dosage form for elderly patients-use of agar as base of rapidly disintegrating oral tablets.
Chem Pharm Bull (Tokyo) 1996; 44:2132-2136. [10] http://www.cimalabs.com/technology/orasolv
[11] http://www.cimalabs.com/technology/durasolv [12] Ozeki T, Yasuzawa Y, Katsuyama H, et al. Design of rapidly
disintegrating oral tablets using acid treated yeast cell wall: A technical note. AAPS Pharm Sci Tech 2003; 4: 70.
[13] Fukami J, Ozawa A, Yoshihashi Y, Yonemochi E, Terada K. Development of fast disintegrating compressed tablets using amino
acid as disintegration accelerator. Chem Pharm Bull (Tokyo) 2005; 53:1536-1539.
[14] Brown, R.M. Jr.: EP0346507 (1989). [15] Rios M. Polymers for controlled release formulation follows
function. Pharmatech Com Pharm Technol 2005; 6: 9. [16] http://www.expresspharmaonline.com/20080731/research03.shtml
[17] Chidambaram N, William P, Kolette F, Yihong Q. Formulation and characterization of new layered diffusional matrices for zero-order
sustained release. J Control Release 1998; 52:149-158. [18] Ravi K, Shiirwaikar AA, Shirwaikar A, Mahalaxmi R, Parba LS.
Formulation and evaluation of mouth dissolving tablets of metformin HCl. Int J Pharm Excipients 2006; 5: 86.
[19] http://www.skyepharma.com/technology/oral-technology/ geomatrix/
[20] Dharmadhikari, N.B., Zala, Y.R.: WO2006123364 (2006). [21] Fukami J, Yonemochi E, Yoshihashi Y, Terada K. Evaluation of
rapidly disintegrating tablets containing glycine and carboxy-methylcellulose. Int J Pharm 2006; 310:101-109.
[22] Battu SK, Repka MA, Majumdar S, Madhusudan RY. Formulation and evaluation of rapidly disintegrating fenoverine tablets: Effect of
superdisintegrants. Drug Dev Ind Pharm 2007; 33:1225-1232. [23] Mishra DN, Bindal M, Singh SK, Vijaya KSG. Spray dried
excipient base: A novel technique for the formulation of orally disintegrating tablets. Chem Pharm Bull (Tokyo) 2006; 54: 99-102.
[24] Brahmankar DM, Jaiswal SB. Controlled release medication. In Biopharmaceutics and pharmacokinetics a treatise. Delhi, Vallabh
Prakashan 2006; 355-356. [25] Jon, S., Farokhazd, O.C., Langer, R.S., Cheng, J.: WO2007001448
(2007). [26] http://www.elan.com/EDT/oral_controlled_release_technology/
ipdas [27] Moës AJ. Gastroretentive dosage forms. Crit Rev Ther Drug Carrier
Syst 1993; 10:143-195.

172 Recent Patents on Drug Delivery & Formulation, 2009, Vol. 3, No. 2 Gupta et al.
[28] Choudhury PK, Kar M, Chauhan CS. Cellulose acetate
microspheres as floating depot systems to increase gastric retention of antidiabetic drug: Formulation, characterization and in vitro-in
vivo evaluation. Drug Dev Ind Pharm 2008; 34:349-354. [29] Majithiya RJ, Murthy RS. Chitosan-based mucoadhesive
microspheres of clarithromycin as a delivery system for antibiotic to stomach. Curr Drug Deliv 2005; 2:235-242.
[30] Ito, K., Mizobata, K., Kamimura, M., Shobu, T.: WO2008075448 (2008).
[31] Patel JK, Patel MM. Stomach specific anti-helicobacter pylori therapy: Preparation and evaluation of amoxicillin-loaded chitosan
mucoadhesive microspheres. Curr Drug Deliv 2007; 4: 41-50. [32] Dhaliwal S, Jain S, Singh HP, Tiwary AK. Mucoadhesive
microspheres for gastroretentive delivery of acyclovir: in vitro and in vivo Evaluation. AAPS J 2008; 10:322-330.
[33] Fischer, G., Bar-Shalom, D., Slot, L., Lademann, A.M., Jensen, C.: US2008234352 (2008).
[34] Chatterji, A., Dong, Z., Sandhu, H.K., Shah, N.H.: US2008293787 (2008).
[35] Pardeep KG. Solution and phase equilibria; In: Remington the science and practice of pharmacy. New Delhi, Wolters Kluwer
India 2007; 211-230. [36] http://www.pedinol.com/GrisPEG/Gris-PEG%20(about%20
grispeg).htm [37] Valleri M, Mura P, Maestrelli F, Cirri M, Ballerini R. Development
and evaluation of glyburide fast dissolving tablets using solid dispersion technique. Drug Dev Ind Pharm 2004; 30: 525-534.
[38] Velluto D, Demurtas D, Hubbell JA. PEG-b-PPS Diblock copolymer aggregates for hydrophobic drug solubilization and
release: Cyclosporin a as an example. Mol Pharm 2008; 5: 632-642. [39] Corswant, C.V.: MX9801755 (1998).
[40] Tang Y, Liu SY, Armes SP, Billingham NC. Solubilization and controlled release of a hydrophobic drug using novel micelle-
forming ABC triblock copolymers. Biomacromolecules 2003; 4: 1636-1645.
[41] Ong, J., Stetsko, G., Levy, O.E., Ghosh, S.S.: WO2005037250 (2005).
[42] Chi-Yuan W, Leslie ZB. Predicting drug disposition via application of BCS. Pharm Res 2005; 22:11-23.
[43] http://www.skyepharma.com/technology/oral-technology/particle-engineering-technologies/insoluble-drug-delivery-platform.html
[44] Yi T, Wan J, Xu H, Yang X. Controlled poorly soluble drug release from solid self-microemulsifying formulations with high viscosity
hydroxypropylmethylcellulose. Eur J Pharm Sci 2008; 34: 274-280. [45] Serratoni M, Newton M, Booth S, Clarke A. Controlled drug
release from pellets containing water-insoluble drugs dissolved in a self-emulsifying system. Eur J Pharm Biopharm 2007; 65: 94-98.
[46] Bansal T, Mustafa G, Khan ZI, Ahmad FJ, Khar RK, Talegaonkar S. Solid self-nanoemulsifying delivery systems as a platform
technology for formulation of poorly soluble drugs. Crit Rev Ther Drug Carrier Syst 2008; 25:63-116.
[47] Odidi, I., Odidi, A.: CA2579382 (2008). [48] Venkatesh, G.M., Qian, K. K., Vangala, S., Clevenger, J.M.,
Guenther, D.: WO2005105049 (2005). [49] http://www.elan.com/EDT/nanocrystal_technology
[50] Yun, S.L.J., Chen, J.F.: US2008212402 (2008). [51] http://www.eurand.com/Technologies/Bioavailability-
enhancement/Biorise/ [52] Bar-Shalom, D., Fischer, G., Slot, L., Lademann, A.M.: EP1974726
(2008). [53] http://www.flamel.com/techAndProd/medusa.shtml
[54] Jabr-Milane L, Van Vlerken L, Devalapally H, et al. Multi-functional nanocarriers for targeted delivery of drugs and genes. J
Control Release 2008; 130:121-128. [55] http://labs.tdl.org/tdl/handle/2152/125
[56] Svenson, S., Tucker, C.J., Hitt, J.E.: WO2004012710 (2004). [57] Svenson, S., Tucker, C.J., Lubetkin, S.D., Evans, J.C., Rosenberg,
S.S.: WO059319 (2003). [58] Williams, R.O.III., Johnston, K.P., Young, T.J., Rogers, T.L.,
Barron, M.K., Yu, Z., Hu, J.: WO060411 (2002). [59] De Jaeghere F, Allémann E, Cerny R, et al. pH-Dependent
dissolving nano and microparticles for improved peroral delivery of a highly lipophilic compound in dogs. AAPS Pharm Sci 2001; 3: 8.
[60] Kossena GA, Charman WN, Boyd BJ, Porter CJ. A novel cubic phase of medium chain lipid origin for the delivery of poorly water
soluble drugs. J Control Release 2004; 99: 217-229.
[61] Breitenkamp, K., Sill, K., Skaff, H., Breintenkamp, R.:
WO2006107903 (2006). [62] Molavi O, Ma Z, Mahmud A, et al. Polymeric micelles for the
solubilization and delivery of STAT3 inhibitor cucurbitacins in solid tumors. Int J Pharm 2008; 347:118-127.
[63] Ravenelle F, Vachon P, Rigby-Jones AE, et al. Anaesthetic effects of propofol polymeric micelle: A novel water soluble propofol
formulation. Br J Anaesth 2008; 101:186-193. [64] http://www.prnewswire.com/cgi-bin/stories.pl
[65] www.cimalab.com [66] http://www.akinainc.com/hytrocell.htm
[67] http://www.lipocine.com/content/view/53/506/ [68] Shimoni, E., Ramon, O., Kopelman, I.J., Mizrahi, S., Salzman, N.,
Nahmias, Y., Oren, A.: WO2008132727 (2008). [69] Quan JS, Jiang HL, Kim EM, et al. pH-Sensitive and mucoadhesive
thiolated eudragit-coated chitosan microspheres. Int J Pharm 2008; 359:205-210.
[70] http://www.emisphere.com/eligen.html [71] Sarasija S, Hota A. Colon-specific drug delivery systems. Indian J
Pharm Sci 2000; 62:1-8. [72] Ashford M, Fell JT, Attwood D, Sharma H, Woodhead PJ. An in
vivo investigation into the suitability of pH-dependent polymers for colonic targeting. Int J Pharm 1993; 95:193-199.
[73] Hita V, Ranjit S, Sanjay KJ. Colonic targeting of metronidazole using azo aromatic polymers: Development and characterization.
Drug Deliv 1997; 4:19-22. [74] Zhao XL, Li KX, Zhao XF, Pang DH, Chen DW. Study on colon-
specific 5-Fu pH-enzyme di-dependent chitosan microspheres. Chem Pharm Bull (Tokyo) 2008; 56: 963-968.
[75] Paharia A, Yadav AK, Rai G, Jain SK, Pancholi SS, Agrawal GP. Eudragit-coated pectin microspheres of 5-fluorouracil for colon
targeting. AAPS Pharm Sci Tech 2007; 8: 12. [76] Chellan VR, Chithambaram M, Joseph AJLJ, Thengungal KR. An
in vitro and in vivo investigation into the suitability of bacterially triggered delivery system for colon targeting. Chem Pharm Bull
2002; 50: 892-895. [77] Ravi V, Pramod KT, Siddaramaiah. Novel colon targeted drug
delivery system using natural polymers. Indian J Pharm Sci 2008; 70: 111-113.
[78] http://www.imagesrising.com/ypt/ocas.shtml [79] http://www.aapspharmsci.org/abstracts/AM_1999/1686.htm
[80] Chourasia MK, Jain SK. Polysaccharides for colon targeted drug delivery. Drug Deliv 2004; 11:129-148.
[81] Leong CW, Newton JM, Basit AW, Podczeck F, Cummings JH, Ring SG. The formation of colonic digestible films of amylose and
ethylcellulose from aqueous dispersions at temperatures below 37°C. Eur J Pharm Biopharm 2002; 54: 291-297.
[82] Siew LF, Basit AW, Newton JM. The potential of organic-based amylose-ethylcellulose film coatings as oral colon-specific drug
delivery systems. AAPS Pharm Sci Tech 2000; 1: 22. [83] http://www.shvoong.com/medicine-and-health/1609118-
preparation-5-amino-salicylic-acid/ [84] Valluru R, Siddaramaiah, Pramod KT. Influence of natural polymer
coating on novel colon targeting drug delivery system. Mater Sci Mater Med 2008; 19: 2131-2136.
[85] Ling C, Xiaoxi L, Yansheng P, Lin L, Ximei Z, Long Y. Resistant starch as a carrier for oral colon-targeting drug matrix system. J
Mater Sci Mater Med 2007; 18: 2199-2203. [86] Xing L, Dawei C, Liping X, Rongqing Z. Oral colon-specific drug
delivery for bee venom peptide: Development of a coated calcium alginate gel beads-entrapped liposome. J Control Release 2003; 93:
293-300. [87] El-Gibaly I. Oral delayed-release system based on zn-pectinate gel
(ZPG) microparticles as an alternative carrier to calcium pectinate beads for colonic drug delivery. Int J Pharm 2002; 232: 99-211.
[88] Chourasia MK, Jain SK. Potential of guar gum microspheres for target specific drug release to colon. J Drug Target 2004; 12: 435-
442. [89] Xuan D, Adam WGA, Joseph RR. Extended-release and targeted
drug; In: Remington the science and practice of pharmacy. New Delhi, Wolters Kluwer India 2007; p. 958.
[90] El-Kamel AH, Abdel-Aziz AA, Fatani AJ, El-Subbagh HI. Oral colon targeted delivery systems for treatment of inflammatory
bowel diseases: Synthesis, in vitro and in vivo assessment. Int J Pharm 2008; 358: 248-255.

Oral Drug Delivery Recent Patents on Drug Delivery & Formulation, 2009, Vol. 3, No. 2 173
[91] Pang YN, Zhang ZR, Pang QJ, Li TL. Study on the enzyme
dependant colon targeting prodrug-dexamethasone succinate dextran and its tablets. Yao Xue Xue Bao Acta Pharm Sin 2001; 36:
625-630. [92] Jain A, Gupta Y, Jain SK. Azo chemistry and its potential for
colonic delivery. Crit Rev Ther Drug Carrier Syst 2006; 23: 349-400.
[93] http://www.imagesrising.com/ypt/codes.shtml
[94] Prabhu S, Kanthamneni N. A targeted nanotechnology-based therapeutic system for the combined chemoprevention of colon
cancer. Nanotech 2007 Conference Program Abstract. [95] Takada, K., Murakami, M.: US20056890547 (2005).
[96] Takada, K.: US5637319 (1997). [97] Patel, H.B.: US2007128280 (2007).
Copyright © 2022 FDOKUMEN