Synthesis of 4-azapodophyllotoxins by multicomponent reactions – a review

PHARMACEUTICS, PREFORMULATION ANDDRUG DELIVERY
Cyclodextrin Multicomponent Complexation andControlled Release Delivery Strategies to Optimizethe Oral Bioavailability of Vinpocetine
LAURA S.S. RIBEIRO,1 AMILCAR C. FALCAO,2 JOAO A.B. PATRICIO,3 DOMINGOS C. FERREIRA,4
FRANCISCO J.B. VEIGA1
1Laboratory of Pharmaceutical Tecnology, Faculty of Pharmacy, University of Coimbra, Rua do Norte, 3000-295 Coimbra,Portugal
2Laboratory of Pharmacology, Faculty of Pharmacy, University of Coimbra, Rua do Norte , 3000-295 Coimbra, Portugal
3Laboratory of Experimental Investigation, University Hospital of Coimbra, Avenida Bissaya Barreto, 3000-075,Coimbra, Portugal
4Laboratory of Pharmaceutical Technology, Faculty of Pharmacy, University of Porto, Porto, Portugal
Received 5 September 2004; revised 7 November 2004; accepted 23 November 2004
Published online in Wiley InterScience (www.interscience.wiley.com). DOI 10.1002/jps.20294
ABSTRACT: In the present work, to maintain a suitable blood level of vinpocetine (VP)for a long period of time, VP-cyclodextrin-tartaric acid multicomponent complexeswere prepared and formulated in hydroxypropylmethylcellulose matrix tablets.In vitro and in vivo performances of these formulations were investigated over a VPimmediate release dosage form. Solubility studies were performed to evaluate the drugpH solubilization profile and to assess the effect of multicomponent complexation on VPsolubility. The drug release process was investigated using United States Pharmacopeiaapparatus 3 and a comparative oral pharmacokinetic study was subsequently under-taken in rabbits. Solubility studies denoted the pH-solubility dependence of VP andsolubility improvement attained by complexation. Dissolution results showed controlledand almost complete release behavior of VP over a 12-h period from complex hydro-xypropylmethylcellulose-based formulations. A clear difference between the pharmaco-kinetic patterns of VP immediate release and VP complex-based formulations wasrevealed. The area under the plasma concentration-time curve after oral administrationof complex-based formulations was 2.1–2.9 times higher than that for VP immediaterelease formulation. Furthermore, significant differences found formean residence time,elimination half-life, and elimination rate constant values corroborated prolongedrelease of VP from complex-based formulations. These results suggest that the oralbioavailability of VP was significantly improved by both multicomponent complexationand controlled release delivery strategies. � 2007 Wiley-Liss, Inc. and the American
Pharmacists Association J Pharm Sci 96:2018–2028, 2007
2018 JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 96, NO. 8, AUGUST 2007
Correspondence to: Francisco J.B. Veiga (Telephone: 351-23983-7850; Fax: 351-23983-7731; E-mail: [email protected])
Journal of Pharmaceutical Sciences, Vol. 96, 2018–2028 (2007)� 2007 Wiley-Liss, Inc. and the American Pharmacists Association

Keywords: vinpocetine; cyclodextrins; multicomponent complexes; controlledrelease; bioavailability
INTRODUCTION
Vinpocetine (VP) is a vincamine derivative thathas been used in clinical practice in Europe fornearly three decades for the treatment of disordersarising from cerebrovascular and cerebral degen-erative diseases.1,2 VP is thought to increase thecerebral flow in the ischemic areas of patientswith cerebrovascular disease, decrease plateletaggregability in patients with transient ischemicattack or stroke, increase red blow cell deform-ability in stroke patients, and have neuroprotec-tive abilities and a protective effect against brainischemia.3,4
VP is mainly used as immediate oral dosageforms containing 5 mg of the active ingredient,with a daily dosage regimen that can vary between5mg� 3/day to 20mg� 3/day.5Unfortunately, thevery limited aqueous solubility and wettability ofVP can give rise to problems of both formulationand low bioavailability (�6.7%).6 Indeed, VP is apoorly water-soluble base-type drug that presentspH-dependent solubility. As the solubility anddissolution relationships in the gastrointestinaltract can be critical for the oral bioavailability ofpoorly soluble weak bases as VP, because of thepossibility of drug precipitation upon entry intothe small intestine thatmayalso affect the amountof drug available for uptake through the intestinalmucosa,7 the use of cyclodextrin (CD) multicom-ponent complexation with bCD, sulfobutyl ether(SBE)bCD, tartaric acid (TA), and polyvinylpyrro-lidone (PVP) has been attempted to overcome suchVP solubility and dissolution drawbacks.
One potential method of optimizing the efficacyof drug activity is through the use of rationallydesigned carriermaterials such asCDs. In thepastdecades, CD complexation has been extensivelyapplied to enhance the solubility, dissolution, andbioavailability of poorly water-soluble drugs.Therefore, the use of CDs as excipients in differentdosage forms has receivedmuch attention becauseupon complex formation advantages such as im-proved bioavailability, reduction of unwanted sideeffects, or improved stability have often beenclaimed. The improvement in absorption rate ofdrugs administered in solid dosage forms has beenrelated to the increase in both solubility anddissolution rates of the complexes as comparedwith pure drug.8More recently, a concernwith theamount of CDs in dosage forms due to toxicological
considerations, formulation bulk, and productioncosts encouraged the development ofmethods thatcould enhance the complexation efficiency of CDs,by using a third additive such as water-solublepolymers9,10 and hydroxy acids.11
In previous works, we have reported the com-bined use of both bCDand SBEbCD,water-solublepolymers [PVPandhydroxypropylmethylcellulose(HPMC)], and hydroxy acids (TA) to improve VPsolubility.12,13 Considering that VP has an elim-ination half-life of 2–6 h14 and requires chronicadministration, a controlled release dosage formcould provide increased clinical value over con-ventional formulations, as a result of improvedtherapeutic effect and patient compliance by re-ducing dosing frequency, a more constant orprolonged therapeutic effect, and possible en-hanced bioavailability. Then, in an effort to reachbetter dissolution properties as well as controlledrelease rate of VP, we have prepared VP-CD-TAmulticomponent complexes and an optimal for-mulation was subsequently designed by the com-bination of these complexes into HPMC-basedhydrophilic tablet dosage forms (Ribeiro et al.,submitted for publication).
Because dissolution rate of poorly soluble drugsis a function of their water-solubility, enhance-ment of drug solubility is expected to improve itsbioavailability. Recently, SBEbCD, a chemicallymodified CD, became a more interesting optionthan bCD to achieve complexation because ofbetter physicochemical properties and improvedinclusion behavior. Studies had proven that thisCDderivative is able to increasedrug oral bioavail-ability.15,16 Thereby, the present studywas under-taken to evaluate, by means of in vivo absorptionstudies in the rabbit model, the feasibility of usingVP-bCD-TA and VP-SBEbCD-TA multicompo-nent complexes in association with HPMC matrixtablets as strategies to improve VP oral bioavail-ability and also to obtain a prolonged therapeuticeffect of the drug as compared with an immediaterelease formulation.
EXPERIMENTAL
Materials
VP was purchased from Covex (Madrid, Spain).bCD [Kleptose1; molecular weight (MW) 1135]
ORAL BIOAVAILABILITY OF VINPOCETINE 2019
DOI 10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 96, NO. 8, AUGUST 2007

and (sulfobutyl ether)7M (SBE)bCD (CaptisolTM;total degrees of substitution 6.8, MW 2160) werekindly offered by Roquette (Lestrem, France) andCydex (Kansas City, MO). PolyvinylpyrrolidoneK30 (PVP) and TA were purchased from SigmaChemical Co. (St. Louis, MO). For the preparationof the matrix tablets, Methocel1 HPMC K15MCR (Dow Chemical, Midland, MI), monohydratelactose, and magnesium stearate of pharmaceuti-cal grade were used. All solvents and chemicalsused were of high-performance liquid chromato-graphy (HPLC) or analytical grade. Milli-Q waterwas used to prepare buffer solutions and otheraqueous solutions used in this study.
Preparation of VP-CD-TAMulticomponent Complexes
Solid multicomponent complexes, VP-bCD-TAand VP-SBEbCD-TA, with and without PVP,were prepared by the lyophilization method asdescribed in a previous work.12 Briefly, equimolaramounts of CDs and VP were dissolved in waterand in 1.5% (w/v) TA solution, respectively. Thetwo solutions were sonicated for 15min andmixedfor 2 h at 508C. After filtration through a 0.45-mmmembrane filter (PVDF, Tracer1; Teknokroma,Barcelona, Spain), the resulting clear solutionwas frozen by immersion in an ethanol bath (ShellFreezer, Labconco, Freezone1 model 79490) at�508C and then the frozen solution was lyophi-lized in a freeze-dryer (Lyph-lock 6 apparatus;Labconco) for 72 h. In the case of VP-CD-TA-PVPmulticomponent complexes, equimolar amountsof CDs and VP were respectively dissolved in0.25% (w/v) PVP and in 1.5% (w/v) TA solution.The resulting solution was mixed and sonicatedfor 15 min and then heated in an autoclave at1208C for 20 min. After an equilibrium period of72 h at room temperature, the resulting solutionwas filtrated through a 0.45-mm membrane filterand clear solution was frozen by immersion in anethanol bath at �508C, and subsequently lyophi-lized in a freeze-dryer for 72 h.
VP Solubility Studies
The pH solubilization profile of VP was deter-mined in the pH range found in physiologicalfasted conditions of the human gastrointestinaltract. The buffer solutions used for the pH solubi-lization profile were the simulated gastric fluid
without enzymes, pH 1.2 [United States Pharma-copeia (USP) XXV], 0.05 M phosphate buffer,pH 4.5 (European Pharmacopeia, 4th ed.), 0.05 Mphosphate buffer, pH 6.0 (European Pharmaco-peia, 4th ed.), simulated intestinal fluid withoutenzymes, pH 6.8 (USP XXV), and 0.05 M phos-phate buffer, pH 8.0 (European Pharmacopeia,4th ed.). VP solubility studies were conducted bythe shake-flask method. Briefly, an excess of VPwas added to glass flasks containing 5 mL of eachbuffer solution described above. The glass flaskswere sealed and mechanically stirred at ambienttemperature (22� 18C) for 48 h. The pH of thesamples was checked periodically during theexperiment to ensure maintenance of buffer pHvalues. All resulting suspensions were filteredthrough a 0.45-mm membrane filter (La-Pha-Pack1; Langerwehe, Germany) and VP concentra-tions analyzed spectrophotometrically (UV-1603;Shimadzu, Japan) at 316 nm. All experimentswere performed in triplicate.
VP aqueous solubility was evaluated in com-plexed and uncomplexed forms. Samples wereprepared in triplicate by adding 5 mL of deionizedwater and excess of solid powders into 20-mLglass flasks which were sealed and mechanicallystirred at ambient temperature for 48 h. Result-ing suspensions were filtered through a 0.45-mmmembrane filter and VP concentrations analyzedspectrophotometrically at 316 nm.
Preparation of HPMC Matrix Tablets
HPMCmatrix tablets were manufactured by directcompression of the formulation mixtures pre-sented in Table 1 under a pressure of 1000 (F-A)and 4000 kg/cm2 (F-B to F-D). The respectivepowders (VP, multicomponent complexes, HPMCK15 M CR, lactose, and magnesium stearate)were blended thoroughly with a mortar and apestle. Formulation mixtures were weighed andfed manually into the die of an instrumentedsingle-punch tablet press (Specac Ltd., Kent, UK)fitted with 8-mm flat-faced punches. The tabletweight was kept constant, at 200 and 300 mg forthe formulations incorporating pure drug or bCDand SBEbCD multicomponent complexes, respec-tively, by adjusting the amount of lactose usedin each formula. The weight variation of tabletformulations is due to MW differences of bothCDs, because SBEbCD has a MW of 2160, a signi-ficantly higher value than that of bCD (MW 1135).All tablet formulations contained 20 mg of VP orits equivalent.
2020 RIBEIRO ET AL.
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 96, NO. 8, AUGUST 2007 DOI 10.1002/jps
In Vitro Dissolution Testing
Dissolution tests were performed using USPapparatus 3 (Bio-Dis III extended release tester,VankelTM, Cary, NC), with a pH gradient method,to simulate the conditions of the fasted humangastrointestinal tract. Buffer solutions used fordissolution testing were the same as describedabove. The pH of the dissolutionmedia and corres-ponding dissolution durations were set as follow:pH 1.2 for 1 h, pH 4.5 for 0.5 h, pH 6.0 for 2.5 h,and pH 6.8 for 8 h. The pH values and residencetimes in each row were selected on the basis ofliterature information of the pH values foundin different parts of the human gastrointestinaltract in the fasted state.17–20 Dissolution testingwas performed at 37� 0.28C. The vessels werefitted with 250 mL of media and tablet formula-tions, corresponding to 20 mg of VP, were placedin the dipping tubes which contained a polypro-pylene bottom screen of 420-mm mesh size. Themesh size of the top screens was also fixed at420 mm. A standard dip rate of 15 per minute wasused and dipping tubes were drained for 1 minbefore moving to the following media. Samplesolutions (3 mL) were collected at specified timeintervals from dissolution vessels, using a plasticsyringe coupled with a polypropylene tube, whichwas inserted inside the vessel, and an equal volumeof fresh test medium was replaced. Samples werefiltered through membrane filters of 0.45-mm poresize (La-Pha-Pack1, Langerwehe, Germany) andanalyzed for UV absorption (UV-1603; Shimadzu,Kyoto, Japan) at 316 nm. The cumulative percentof drug released was calculated according tocalibration curves for each pH buffer solutionand a correction was applied for the cumulativedilution caused by replacement of the sample withan equal volume of freshmedium. All experimentswere made in triplicate.
Fit factors (f1 and f2) were used for comparingthe dissolution patterns because they are verypopular methods used to compare dissolution
profile data and are recommended for use in anumber of FDA guidance documents.21 For curvesto be considered similar, f1 value should be close to0 and f2 value to 100. Generally, f1 values�15 andf2 values �50, which means an average differenceof no more than 10% at the sample time points,ensures equivalence of the curves and thus of theperformance of the test and reference products.22
In Vivo Study Design
In vivo study was designed as a two-way rando-mized crossover study, with 2 weeks’ dosinginterval, with a single oral dosing administration(in six male New Zealand rabbits with an averageweight of 3.3 kg) to minimize any residual orcumulative effects of the preceding dose. Thestudy was approved by the local Committee ofLaboratory Animal Care from the UniversityHospital of Coimbra in accordance with the rulesand guidelines concerning the care and use oflaboratory animals. Rabbits were housed indivi-dually in cages under environmentally controlledconditions, on a 12-h light/dark cycle, and weregiven free access to food and water. The day beforeeach experiment, rabbits were fasted for 14–16 hand provided water ad libitum. They were cathe-terized in the marginal ear vein for blood samplingbefore drug administration. All rabbits receivedtwo tablet formulations corresponding to 40 mg ofVP as a single dose. The relatively high dose wasneeded to ensure blood levels appropriate foranalytical detection and pharmacokinetic anal-ysis.15 Tablet formulations were administeredorally by gently compelling the opening of therabbit mouth and depressing of the tongue withpincers. Tablets were swallowed intact by push-ing them to the back of the pharynx with a flexiblegastric tube. After tablet administration, approxi-mately 10 mL of water was given orally with asyringe to facilitate swallowing and to prevent thetablet from sticking to the rabbit throat. Serial
Table 1. Composition of VP Tablet Formulations
FormulationComplex or
Pure VP (mg) HPMC (mg) Lactose (mg) Mg Stearate (mg) Total Weight (mg)
F-Aa 20.0 — 178.0 2.0 200.0F-Bb 99.6 60.0 38.4 2.0 200.0F-Cb 158.2 90.0 48.8 3.0 300.0F-Db 120.6 90.0 86.4 3.0 300.0
aImmediate release formulation of VP.bHPMC-based matrix tablet formulations of VP: B) VP-bCD-TA, C) VP-SBEbCD-TA, D) VP-SBEbCD-TA-PVP, multicomponent
complexes.
ORAL BIOAVAILABILITY OF VINPOCETINE 2021
DOI 10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 96, NO. 8, AUGUST 2007

blood samples (1.2–1.5 mL) were drawn from themarginal ear vein before drug administration andat 0.5, 1, 2, 3, 4, 6, 8, 10, 12, and 24 h after drugadministration, to spiked ethylenediaminetetraa-cetic acid tubes to prevent clotting and centri-fuged (3500 rpm/15 min) immediately to obtainplasma. Plasma samples were then stored at�208C until analysis.
Determination of VP Plasma Levels
Frozen samples were allowed to thaw at roomtemperature. Rabbit plasma (0.5 mL), test sampleor free plasma spiked with known amounts of VP,along with 50 mL of 0.35 mg/mL hydroxyprogester-one acetate solution [internal standard, (IS)],were deproteinized by the addition of 1.5 mL ofacetonitrile following vortex mixing at high speedfor 30 s and centrifugation for 10min at 5300 rpm.The clear supernatant was transferred to a newglass tube having 150 mL of 1 N hydroxide sodiumsolution. After equilibrating, 3.5 mL of n-hexanewas added and the extraction was performed byvortex mixing for 1 min. The tubes were centri-fuged (5300 rpm/3 min) and the organic layer wastransferred to another set of clean tubes to beback-extracted with 1.5 mL of 0.05 N sulfuric acidsolution (vortex mixing for 1 min). Tubes werecentrifuged (5300 rpm/3 min), the organic layerwas discarded and the aqueous phase transferredto a clean tube containing 100 mL of 1 N hydroxidesodium solution and back-extracted with 3.5 mLof n-hexane. After mixing (1 min) and centrifu-ging (5300 rpm/3 min), the organic phase wasrecovered to a clean tube to be evaporated todryness at 408C under vacuum. The dried residuewas reconstituted with 120 mL of mobile phaseand 100 mL was injected into the chromatographicsystem. The extraction efficiency of VP was deter-mined by comparing the peak height of extractedsamples to those of nonprocessed quality control(QC) samples at three different concentrationlevels of 10, 50, and 100 ng/mL (n¼ 5) and wasfound to be in the range of 90.7–98.3%.
Drug concentration levels were determinedusing a reversed-phase HPLC method. Briefly,quantitative analysis was performed on an HPLCchromatograph (model 1050; Hewlett-Packard)equipped with an injection valve of 20-mL sampleloop (model 7125; Rheodyne, Cotati, CA), a stati-onary phase of LiChrospher 100 RP18 (250�4.6 mm, 5 mm; Merck, Darmsdadt, Germany)fitted with a LiChrospher RP18 guard column
(4� 4 mm, 5 mm; Merck). The mobile phase con-sisted of a mixture of acetonitrile–0.01 M sodiumphosphate buffer solution containing 5 mMheptane-1-sulfonic acid sodium, and 0.2% (v/v)triethylamine, for which pH was adjusted to 5.5with 1% (v/v) phosphoric acid solution (55:45).Themobile phasewasdegassedbypassing througha 0.45-mm membrane filter (PVDF, Tracer1,Teknokroma) before use. The volume injectedwas 100 mL which was eluted isocratically with aflow rate of 1.0 mL/min at 308C, with detectionperformed with ultraviolet absorption at a wave-length of 230 nm. The extraction and HPLCmethod resulted in symmetrical peak shape andgood baseline resolution for both VP and IS, withretention times of approximately 8.5 and 10.3min,respectively.
Stock solutions (0.1 mg/mL) of VP and IS wereprepared by dissolution in acetonitrile and kept at48C. Standard solutions (n¼ 4) of VP in humanplasma (5, 10, 25, 50, 100, and 150 ng/mL) wereprepared daily from stock solutions by spiking thesuitable volume of various working solutionsdiluted in the mobile phase and extracted andanalyzed as described above. Peak height ratios ofeach VP to IS were measured and the calibrationcurves of three consecutive days were obtainedfrom the least-squares linear regression. Theaverage regression line was used to calculate therespective concentrations of VP in the plasmasamples. The linearity of detector response wasassessed for extracted plasma standards over therange of 5–150 ng/mL. The calibration curve of VPexhibited an excellent linearity and a correlationcoefficient of 0.9989.
The intra-day precision of the method (coeffi-cient of variation) was assessed, on a single day,by extracting and analyzing free plasma spikedwith known amounts of VP appropriate workingsolutions yielding concentrations of 10, 50, and100 ng/mL (QC samples, n¼ 5). The intra-dayprecision was 6.3% (10 ng/mL), 3.4% (50 ng/mL),and 1.5% (100 ng/mL). The inter-day precision(coefficient of variation) was determined for thesameQCsamples (n¼ 5) on three consecutive daysand was found to be 7.2% (10 ng/mL), 3.9% (50 ng/mL), and 2.1% (100 ng/mL). Accuracy, expressedas the mean % [(mean measured concentration)/(theoretical concentration)]� 100, was determin-ed simultaneously as intra- and inter-day preci-sion in the sameQCsamples (n¼ 5). Themeasuredvalues varied between 97.4–104.5% (intra-day)and 93.1–106.2% (inter-day). These results con-firmed the precision and accuracy of the method.
2022 RIBEIRO ET AL.
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 96, NO. 8, AUGUST 2007 DOI 10.1002/jps

Pharmacokinetics and Statistical Data Analysis
The pharmacokinetic parameters extracted fromthe plasma data were calculated using noncom-partmental analysis (WinNonlin1, version 1.1)and included the maximum plasma concentration(Cmax), the time to reach the maximum plasmaconcentration (tmax), the whole area under thedrug plasma concentration-time curve (AUC0–1),the mean residence time (MRT), the eliminationhalf-life (t1/2), and elimination rate constant (lz).The relative bioavailability of formulations B, C,and D was calculated taking as reference for-mulation A, through the ratio of the respectiveAUC0–1 values. Data are presented as meanvalues� standard deviation.
Statistical analysis of the pharmacokinetic para-meters (Cmax, tmax, t1/2,MRT, lz, andAUC0–1) wasperformed using GraphPad Prism1 version 4.0software, by one-way analysis of variance andfollowed by Tukey’s post test for multiple compar-ison analysis. A value of p� 0.05 defined thestatistical equivalence.
RESULTS AND DISCUSSION
VP Solubility Studies
Solubility and pH-solubility profiles are particu-larly useful means of identifying compoundslikely to have absorption and distribution pro-blems,23 especially if drugs present pKa values inthe physiological range, because the apparentsolubility may change greatly with changes in pHof the environment.18 Because the rate and extentof drug release from most controlled releasesystems are influenced by the pH of the dissolu-tion medium for drugs with pH-dependent solu-bility and this dependency of drug release on pHmay lead to additional inter and intra-subjectvariability in drug absorption,24 VP solubility wasexamined as a function of pH over the rangetypically encountered under fasted physiologicalconditions of the human gastrointestinal tract.
In the pH range studied (1.2–8.0), VP showedan approximately linear relationship betweenthe logarithm of the solubility and pH (Fig. 1).Over this range, the solubility was significantlyenhanced by a reduction in pH, as expected,because of the weak base character (pKa¼ 7.31)of the drug and a drastic decreased solubility wasobserved with higher pH values. Indeed, theestimated solubility values for each pH solutionstudied were 24.16� 0.04 mg/mL at pH 1.2,
199.14� 4.49 mg/mL at pH 4.5, 24.23� 0.25 mg/mL at pH 6.0, 5.38� 0.53 mg/mL at pH 6.8, and1.39� 0.15 mg/mL at pH 8.0. Hence, VP solubilitywas increased by a factor of approximately 17,400considering both extreme pH values tested.
The solubility studies reported clearly illustratethe impact of pH on VP solubility and dissolution.From the solubility values obtained, it is predict-able that the extent of VP dissolution in the gastricenvironment will be high. Oppositely, under thepH values generally found in the upper regionsof the gastrointestinal tract, the solubility anddissolution of pure VP will not be sufficient forcomplete dissolution of the doses habituallyadministered. However, it is expected that thisdrawback may be overcome by multicomponentcomplexation of VP with CDs and TA, becauseresulting complexes have demonstrated to havebetter solubility and dissolution performances insimulated intestinal fluid (pH 6.8) (Ribeiro et al.,submitted for publication).
VP solubility measurements in deionized waterare schematically represented in Figure 2. As
Figure 1. VP pH solubilization profile in the pHrange of 1.2–8.0. Each point represents mean values�SD.
Figure 2. VP aqueous solubility obtained from theuncomplexed form and lyophilized multicomponentcomplexes. Results are presented as mean values.
ORAL BIOAVAILABILITY OF VINPOCETINE 2023
DOI 10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 96, NO. 8, AUGUST 2007

expected from previous work,13 VP solubility inmulticomponent form was found to be higher thanpure VP. In fact, VP solubility in uncomplexedform (&5 mg/mL) increased to approximately80 mg/mL in VP-SBEbCD-TA-PVP multicompo-nent complex, which corresponds to a 16,000-fold-higher increase on VP aqueous solubility. Theimproved VP solubility observed as a consequenceof multicomponent complexation was greater forVP-SBEbCD-TA lyophilized complexes comparedwith VP-bCD-TA lyophilized complex, clearlydenoting the superior solubilizationand complexa-tion efficiency of SBEbCD. In addition, solubilityimprovement of VP may be associated with thehigh energetic amorphous state after complexa-tion in VP from VP-SBEbCD-TA lyophilized com-plexes as confirmedpreviously byX-ray diffractionstudies performed.12
In Vitro Dissolution Testing
In vitro dissolution testing serves as an impor-tant tool for characterizing the biopharmaceuticalquality of a product at different stages in itslife cycle. In early drug development, in vitrodissolution properties are supportive for choosingbetween different alternative formulation candi-dates for further development and for evaluationof active ingredients.25 The therapeutic benefit ofa number of drugs administered in a traditionaldosage form is sometimes limited by physiologicalbarriers, undesirable physiochemical drug prop-erties, or issues of drug toxicology. In such cases,the development of drug delivery systems thatproduce a modified in vivo drug release is a com-mon research aim. By manipulating the release ofdrugs from its dosage form, such restrictions maybe overcome and an improvement in therapeuticeffect observed.26 The present in vitro dissolutionstudy was designed to theoretically investigatethe influence of drug release properties fromimmediate and controlled release formulations,the dissolution of the different dosage forms, andthe release performance of drug particles frommulticomponent complexes through the hydratedgel layer of HPMC tablet formulations.
The release profiles of VP from an immediaterelease formulation (A) and from hydrophilicHPMC-based matrix formulations (B, C, D) arepresented in Figure 3. Because of the basic natureof VP, the release of the drug from formulation Awas immediate in the first hour of the dissolutiontesting (pH 1.2) and thus there was no more VPavailable to proceed on the following steps of
the dissolution experiment. Based on previoussolubility studies, a drastic decrease was expectedon VP solubility and dissolution at higher pHvalues; therefore, multicomponent complexationwas attempted to improve the dissolution perfor-mance of the drug over a pH range that simulatesthe one found in the fasted gastrointestinal tract.Hence, hydrophilic HPMC-based matrix formu-lationsB,C, andD, containing the drug in the formof VP-CD-TA multicomponent complexes, weredesigned to overcome the VP solubility drawbackat higher pH values and to extend drug releasefor a longer period of time without the risk ofprecipitation. The dissolution profiles of theseformulations clearly indicated a controlled releasepattern over 12 h of the experiment, becauseHPMC tablet formulations swelled upon contactwith the dissolution media and a gel layer wasformed on their surface. This gel retarded furtheringress of fluid and subsequent drug release.Nevertheless, drug release was nearly completein the end of the dissolution experiment, almostcertainly because of higher solubility and disso-lution performances of VP after multicomponentcomplexation. Consequently, there existed a cleardifference in the in vitro drug release character-istics of hydrophilic HPMC-based matrix formula-tions B, C, and D as compared with VP immediaterelease formulationA,which in turn,was expectedto provide a different in vivo drug release.
The results of the mathematical comparison ofhydrophilic HPMC-based matrix formulations B,C, andD, byapplying f1 and f2 fit factors, revealed aconvergence of the dissolution profiles of theseformulations (Table 2) and demonstrated the
Figure 3. Dissolution profiles of the immediaterelease formulation (F-A) and multicomponent com-plex-based HPMC matrix formulations (F-B, F-C, andF-D) of VP. Each point represents mean values�SD.
2024 RIBEIRO ET AL.
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 96, NO. 8, AUGUST 2007 DOI 10.1002/jps
equivalence of in vitro performances of formula-tions containing VP-bCD-TA, VP-SBEbCD-TA,and VP-SBEbCD-TA-PVP multicomponent com-plexes (f1� 15 and f2� 50). This fact may haverelevant pharmaceutical usefulness, in the caseof formulation C and D containing respectivelyVP-SBEbCD-TA and VP-SBEbCD-TA-PVP multi-component complexes, because the latter complexincorporates a higher amount of VP, due to theenhanced complexation efficiency of SBEbCDtoward VP in the presence of PVP, and conse-quently a decrease of approximately 13% ofSBEbCD in the solid dosage form.12,13,27
Pharmacokinetics and Bioavailability Studies
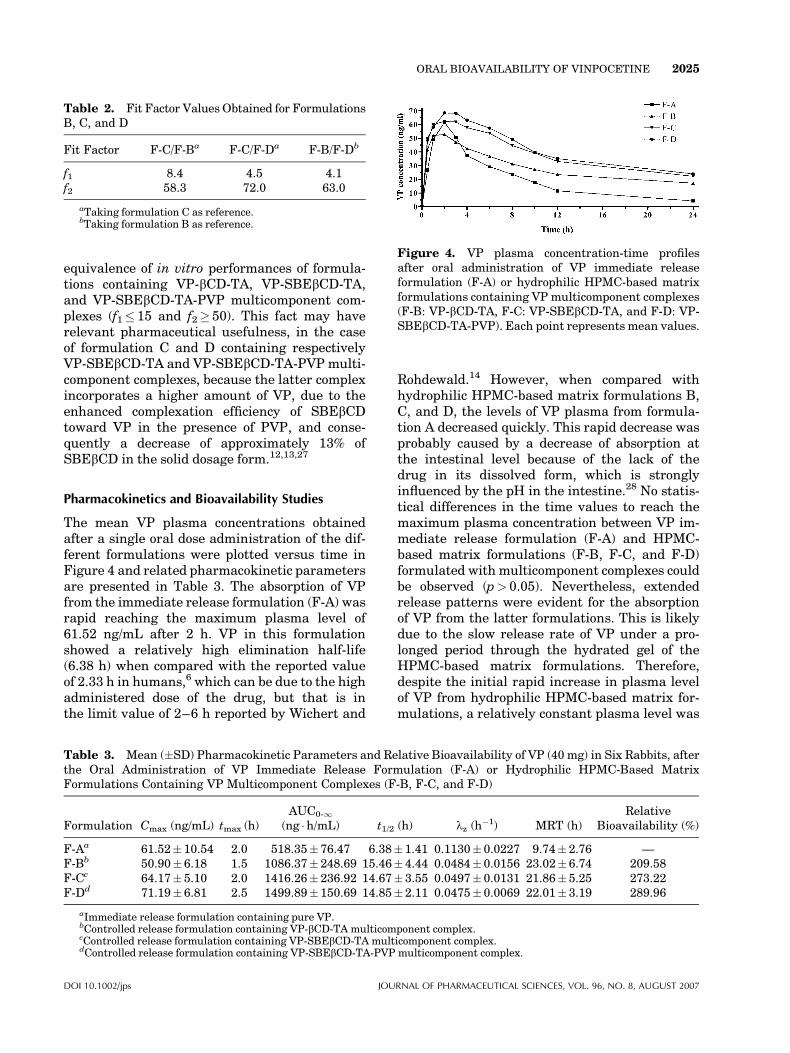
The mean VP plasma concentrations obtainedafter a single oral dose administration of the dif-ferent formulations were plotted versus time inFigure 4 and related pharmacokinetic parametersare presented in Table 3. The absorption of VPfrom the immediate release formulation (F-A) wasrapid reaching the maximum plasma level of61.52 ng/mL after 2 h. VP in this formulationshowed a relatively high elimination half-life(6.38 h) when compared with the reported valueof 2.33 h in humans,6 which can be due to the highadministered dose of the drug, but that is inthe limit value of 2–6 h reported by Wichert and
Rohdewald.14 However, when compared withhydrophilic HPMC-based matrix formulations B,C, and D, the levels of VP plasma from formula-tion A decreased quickly. This rapid decrease wasprobably caused by a decrease of absorption atthe intestinal level because of the lack of thedrug in its dissolved form, which is stronglyinfluenced by the pH in the intestine.28 No statis-tical differences in the time values to reach themaximum plasma concentration between VP im-mediate release formulation (F-A) and HPMC-based matrix formulations (F-B, F-C, and F-D)formulated with multicomponent complexes couldbe observed (p> 0.05). Nevertheless, extendedrelease patterns were evident for the absorptionof VP from the latter formulations. This is likelydue to the slow release rate of VP under a pro-longed period through the hydrated gel of theHPMC-based matrix formulations. Therefore,despite the initial rapid increase in plasma levelof VP from hydrophilic HPMC-based matrix for-mulations, a relatively constant plasma level was
Table 3. Mean (�SD) Pharmacokinetic Parameters and Relative Bioavailability of VP (40 mg) in Six Rabbits, afterthe Oral Administration of VP Immediate Release Formulation (F-A) or Hydrophilic HPMC-Based MatrixFormulations Containing VP Multicomponent Complexes (F-B, F-C, and F-D)
Formulation Cmax (ng/mL) tmax (h)AUC0-1
(ng �h/mL) t1/2 (h) lz (h�1) MRT (h)
RelativeBioavailability (%)
F-Aa 61.52� 10.54 2.0 518.35� 76.47 6.38� 1.41 0.1130� 0.0227 9.74� 2.76 —F-Bb 50.90� 6.18 1.5 1086.37� 248.69 15.46� 4.44 0.0484� 0.0156 23.02� 6.74 209.58F-Cc 64.17� 5.10 2.0 1416.26� 236.92 14.67� 3.55 0.0497� 0.0131 21.86� 5.25 273.22F-Dd 71.19� 6.81 2.5 1499.89� 150.69 14.85� 2.11 0.0475� 0.0069 22.01� 3.19 289.96
aImmediate release formulation containing pure VP.bControlled release formulation containing VP-bCD-TA multicomponent complex.cControlled release formulation containing VP-SBEbCD-TA multicomponent complex.dControlled release formulation containing VP-SBEbCD-TA-PVP multicomponent complex.
Figure 4. VP plasma concentration-time profilesafter oral administration of VP immediate releaseformulation (F-A) or hydrophilic HPMC-based matrixformulations containing VPmulticomponent complexes(F-B: VP-bCD-TA, F-C: VP-SBEbCD-TA, and F-D: VP-SBEbCD-TA-PVP). Each point represents mean values.
Table 2. Fit Factor Values Obtained for FormulationsB, C, and D
Fit Factor F-C/F-Ba F-C/F-Da F-B/F-Db
f1 8.4 4.5 4.1f2 58.3 72.0 63.0
aTaking formulation C as reference.bTaking formulation B as reference.
ORAL BIOAVAILABILITY OF VINPOCETINE 2025
DOI 10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 96, NO. 8, AUGUST 2007

maintained for a longer time, reflecting thein vitro release characteristics.
Despite similar in vitro release profiles of VPfrom formulations B, C, and D, the in vivo studyrevealed differences in the values of Cmax betweenthe formulations. Indeed, the Cmax value obtainedfor formulation B was significantly lower than theones obtained for formulations C (p< 0.05) and D(p< 0.001); this difference was probably related tothe distinct solubility performances of bCD andSBEbCD complexes, discussed previously. Con-versely, MRT, t1/2, and lz values were comparablefor all three formulations (p> 0.05) and signifi-cantly different from the same parameters offormulation A (p< 0.001). Formulations B, C,and D produced a 2.2- to 2.4-fold increase in MRTvalues compared with formulation A, clearlysuggesting a longer maintenance of VP in plasmaand hence prolonged release in the rabbit model.Upon comparing t1/2 values, we found that forcontrolled release matrices, the values were morethan double those for the immediate releaseformulation (F-A). Because t1/2 values should besimilar for the same substance, the differencesencountered here are due to a prolonged absorp-tion phase in the controlled release matrix for-mulations (F-B, F-C, and F-D), where there is aprolonged continuous introduction of VP into thebloodstream.29
Compared with the VP immediate releaseformulation (F-A), the administration of all hydro-philic HPMC-based matrix formulations in-creased the AUC0–1 values of VP, reflecting agreater systemic exposure and, consequently,higher oral bioavailability of the drug. The ratioof mean AUC0–1 ranged from 2.1 to 2.9 and thedifference in AUC0–1 values between the formu-lations was found to be statistically different(p< 0.001). The more than twofold increase inthemeanAUC0–1values of formulationsB,C, andD, was afforded by a greater VP aqueous solubilityand dissolution rate from multicomponent com-plexes. In turn, this made possible the design anduse of controlled release solid dosage forms, withsmaller risk of drug precipitation in the upperregions of the gastrointestinal tract, and, conse-quently, in association to HPMC-based matrixformulations, a maintenance of a more constantVP blood level.
In corroboration with our previous in vitroresults, there was no significant difference be-tween AUC0–1 values of formulations C and D(p> 0.05) and therefore similar oral bioavailabilityin rabbits was observed with both formulations.
These formulations were obtained respectivelywith VP-SBEbCD-TA and VP-SBEbCD-TA-PVPmulticomponent complexes. In the latter complex,PVP increased the solubility and complexationefficiency of SBEbCD toward VP and as a resultthe amount of SBEbCD in solid dosage forms wasreduced approximately 13%.13 Such observationsuggests the possibility of decreasing the amountof CDs in solid dosage forms, by the addition ofwater-soluble polymers to the aqueous complexa-tion media, thereby contributing to the reductionof formulation bulks, toxicity, and costs with CDs,without injury of their pharmaceutical potential,as previously stated by other authors.30
Significantly higher AUC0–1 values wereobtainedwithboth formulationsCandDrelativelyto formulation B. The differences stated reflectthe same trend observed for Cmax values for theseformulations and hence are probably related todifferent solubility and efficiency complexationperformances ofVP observedwith bothCDs. Thus,the use of VP-SBEbCD-TA multicomponent com-plexes in the design of controlled release soliddosage forms seems to be a better approach thanthat of VP-bCD-TA multicomponent complexes.
CONCLUSIONS
The present study demonstrates the suitability ofthe combined CD multicomponent complexationand controlled release delivery strategies to im-prove and maintain the oral absorption of VP for alonger period of time.
Solubility is an important property that influ-ences drug liberation and absorption and hencehas an important role in drug bioavailability,because for a drug to be absorbed it must bepresent in the form of an aqueous solution inthe absorption site.31 Therefore, in an effort toachieve better solubility and dissolution proper-ties through all regions of the gastrointestinaltract, controlled release rate, as well as improvedoral bioavailability of VP, we have prepared VP-CD-TA multicomponent complexes and then opti-mal formulations were designed by the com-bination of these complexes into HPMC-basedhydrophilic tablet dosage forms. A comparativein vitro dissolution study of the resulting formula-tions with an immediate release formulation ofthe drug clearly showed controlled and almostcomplete release behavior of VP over a 12-h periodin the case of HPMC formulations. Conversely,complete dissolution of VP was observed for the
2026 RIBEIRO ET AL.
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 96, NO. 8, AUGUST 2007 DOI 10.1002/jps

immediate release formulation in the first hour ofthe dissolution experiment, because of the basicnature and pH-solubility dependence of the drug.
Further biopharmaceutics studies demon-strated that controlled release multicomponentcomplex-based formulations could dramaticallyimprove VP bioavailability over an immediaterelease formulation, after oral administration inrabbits. The lower oral bioavailability of VP fromthe immediate release formulation was attributedto poor solubility at intestinal pH, leading to pre-cipitation of the drug from the dosage form,followed by a slow process of dissolution and hencea limiting step for absorption.However, increasingthe solubility of drug such as through multi-component complexation with CDs and hydroxyacids, as used in the present study, ensured betterVP dissolution in the upper regions of the gastro-intestinal tract and resulted in higher extent ofbioavailability. Furthermore, the administrationof these multicomponent complexes assisted bytheir formulation in HPMC matrix tablets re-sulted in the maintenance of higher plasma druglevels, thus supporting that not only multicom-ponent complexes but also additional propertiesof the carrier material may be responsible for thecontrolled release and improved oral bioavail-ability of VP.
The use of experimental animals such asrabbits is common to predict pharmacokineticparameters of drugs administered by the oralroute in humans.16,32–34 Although VP solubilityand dissolution studies were performed in condi-tions that intended to simulate the pH range foundin physiological conditions of the human fastedgastrointestinal tract and in vivo studies wereperformed in the rabbit model, we believe thatinterspecies comparisons with humans are feasi-ble because the pH values found in the gastro-intestinal tract of rabbit vary between 1.9 in thestomach to 6.0–8.0 in the intestine.35 Consideringthe relatively high pKa value of VP (7.31), suchdifferences should not reflect an important varia-bility of VP dissolved in the different regions of thegastrointestinal tract in both species.
ACKNOWLEDGMENTS
This work was financially supported by a grant(Praxis XXI/BD/21455/99) from FCT (Fundacaopara a Ciencia e a Tecnologia, Portugal). Theauthors acknowledge Cydex L. C. (Kansas City,MO) and Roquette (Lestrem, France) for their
support in providing SBEbCD and bCD, respec-tively. The authors also thank Miss Maria deLurdes Silva and Mr. Carlos Silva for their valu-able technical assistance in the realization ofanimal studies.
REFERENCES
1. Subhan Z, Hindmarch I. 1985. Psychopharmacolo-gical effects of vinpocetine in normal healthyvolunteers. Eur J Clin Pharmacol 28:567–571.
2. Bonoczk P, Panczel G, Nagy Z. 2002. Vinpocetineincreases blood flow oxygenation in stroke patients:A near infrared spectroscopy and transcranialdoppler study. Eur J Ultrasound 15:85–91.
3. Feigin VL, Doronin BM, Popova TF, GribatchevaEV, Tchernov DV. 2001. Vinpocetine treatment inacute ischaemic stoke: A pilot single-blind rando-mized clinical trial. Eur J Neurol 8:81–85.
4. McDaniel MA, Maier S, Einstein GO. 2002. ‘‘Brain-specific’’ nutrients: A memory cure? Psychol SciPub Int 3:12–38.
5. Szatmari SZ, Whitehouse PJ. 2003. Vinpocetine forcognitive impairment and dementia. CochraneDatabase Syst Rev 1:CD003119.
6. Grandt R, Beitinger H, Schaltenbrand R, BraunW.1989. Vinpocetine pharmacokinetics in elderlysubjects. Arzneimittelforschung 39:1599–1602.
7. Kostewicz ES, Wunderlich M, Brauns U, Becker R,Bock T, Dressman JB. 2004. Predicting the pre-cipitation of poorly soluble weak bases upon entryin the small intestine. J Pharm Pharmacol 56:43–51.
8. Frijlink HW, Eissens AC, Schoonen AJM, Lerk CF.1990. The effect of cyclodextrin on drug absorption.II. In vivo observations. Int J Pharm 64:195–205.
9. Loftsson T, Masson M, Sirgurjonsdottir JF. 1999.Methods to enhance the complexation efficiency ofcyclodextrins. STP Pharma Sci 9:237–242.
10. Loftsson T, Fririksdottir H, Sigurdardottir AM,Ueda H. 1994. The effect of water-soluble polymerson drug-cyclodextrin complexation. Int J Pharm110:169–177.
11. Redenti E, Szente L, Szejtli J. 2000. Drug/cyclodex-trin/hydroxy acid multicomponent systems. Pro-perties and pharmaceutical applications. J PharmSci 89:1–8.
12. Ribeiro L, Ferreira D, Veiga F. 2003. Physicochem-ical investigation of the effects of water-solublepolymers on vinpocetine complexation with b-cyclodextrin and its sulfobutyl ether derivative insolution and solid state. Eur J Pharm Sci 20:253–266.
13. Ribeiro L, Carvalho RA, Ferreira DC, Veiga FJB.2005. Multicomponent complex formation be-tween vinpocetine, cyclodextrins, tartaric acid,
ORAL BIOAVAILABILITY OF VINPOCETINE 2027
DOI 10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 96, NO. 8, AUGUST 2007

andwater-soluble polymersmonitored byNMR andsolubility studies. Eur J Pharm Sci 24:1–13.
14. Wichert B, Rohdewald P. 1990. A new method forthe preparation of drug containing polylacticacid microparticles without using organic solvents.J Controlled Release 14:269–283.
15. Savolainen J, Jarvinen K, Matilainen L, JarvinenT. 1998. Improved dissolution and bioavaibilityof phenytoin by sulfobutylether-b-cyclodextrin[(SBE)-b-CD] and hydroxypropyl-b-cyclodextrin.Int J Pharm 165:69–78.
16. Lefeubre C, Le Corre P, Dollo G, Chevanne F,Burgot JL, Verge R. 1999. Biopharmaceutics andpharmacokinetics of 5-phenyl-1,2-dithiole-3-thionecomplexed with sulfobutyl ether7-b-cyclodextrin inrabbits. J Pharm Sci 88:1016–1020.
17. Charman WN, Porter CJH, Mithani S, DressmanJB. 1997. Physicochemical and physiological me-chanisms for the effects of food on drug absorption:The role of lipids and pH. J Pharm Sci 86:269–282.
18. Galia E, Nicolaides E, Horter D, Lobenberg R,Reppas C, Dressman JB. 1998. Evaluation ofvarious dissolution media for predicting in vivoperformance of class I and II drugs. Pharm Res15:698–705.
19. Nicolaides E, Galia E, Efthymiopoulos C, DressmanJB, Reppas C. 1999. Forecasting the in vivoperformance of four low solubility drugs from theirin vitro dissolution data. Pharm Res 16:1876–1882.
20. Kostewicz ES, Brauns U, Becker P, Dressman JB.2002. Forecasting the oral absorption behavior ofpoorly soluble weak bases using solubility anddissolution studies in biorelevant media. PharmRes 19:345–349.
21. O’Hara T, Dunne A, Butler J, Devane J. 1998. Areview of methods used to compare dissolutionprofile data. Pharm Sci Technol Today 1:214–223.
22. Moore JW, Flanner HH. 1996. Mathematical com-parison of curves with an emphasis on in vitrodissolution profiles. Pharm Technol 20:64–74.
23. Morelock MM, Choi LL, Bell GL, Wright JL. 1994.Estimation and correlation of drug water solubilitywith pharmacological parameters required forbiological activity. J Pharm Sci 83:948–952.
24. Rao VM, Engh K, Qiu Y. 2003. Design of pH-independent controlled release matrix tablets foracidic drugs. Int J Pharm 252:81–86.
25. FIP. 1996. FIP guidelines for dissolution testing ofsolid oral products (final drafts, 1995). Drug Inf J30:1071–1084.
26. Bibby DC, Davies NM, Tucker IG. 2000. Mechanismby which cyclodextrins modify drug release frompolymeric drug delivery systems. Int J Pharm197:1–11.
27. Ribeiro L, Loftsson T, Ferreira D, Veiga F. 2003.Investigation and physicochemical characteriza-tion of vinpocetine-sulfobutyl ether b-cyclodextrinbinary and ternary complexes. Chem Pharm Bull51:914–922.
28. Luengo J, Aranguiz T, Sepulveda J, Hernandez L,Von Plessing C. 2002. Preliminary pharmacoki-netic study of different preparations of acyclovirwith b-cyclodextrin. J Pharm Sci 91:2593–2598.
29. Hasan EI, Amro BI, Arafat T, Badwan AA. 2003.Assessment of a controlled release hydrophilicmatrix formulation for metoclopramide HCl. EurJ Pharm Biopharm 55:339–344.
30. Savolainen J, Jarvinen K, Taipale H, Jarho P,Loftsson T, Jarvinen T. 1998. Co-administration ofa water-soluble polymer increases the usefulness ofcyclodextrins in solid oral dosage forms. Pharm Res15:1696–1701.
31. Panchagnula R, Thomas NS. 2000. Biopharmaceu-tics and pharmacokinetics in drug research. Int JPharm 201:131–150.
32. Ngo TH, Quintens I, Roets E, Declerck PJ,Hoogmartens J. 1997. Bioavailability of differentartemisinin tablet formulation in rabbit plasma:Correlation with results obtained by an in vitrodissolution method. J Pharm Biomed Anal 16:185–189.
33. Dollo G, Le Corre P, Guerin A, Chevanne F, BurgotJL, Leverge R. 2003. Spray-dried redispersible oil-in-water emulsion to improve oral bioavailability ofpoorly soluble drugs. Eur J Pharm Sci 19:273–280.
34. Fernandes C, Ramos P, Falcao AC, Veiga F. 2003.Hydrophilic and hydrophobic cyclodextrin in a newsustained release oral formulation of nicardipine:In vitro evaluation and bioavailability studies inrabbits. J Controlled Release 88:127–134.
35. Zwart LL, Rompelberg CJM, Sips AJAM, VanEngelen JGM. 1999. Anatomical and physiologicaldifferences between various species used in studieson the pharmacokinetics and toxicology of xenobio-tics. Bilthoven: RIVMReport 623860610, pp 1–100.
2028 RIBEIRO ET AL.
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 96, NO. 8, AUGUST 2007 DOI 10.1002/jps
Copyright © 2022 FDOKUMEN