
Towards an oral influenza vaccine: Comparison between intragastric and intracolonic delivery of...
192
THE DEVELOPMENT OF STABLE INFLUENZA VACCINE POWDER FORMULATIONS FOR NEW NEEDLE-FREE DOSAGE FORMS Jean-Pierre Amorij
Transcript of Towards an oral influenza vaccine: Comparison between intragastric and intracolonic delivery of...

THE DEVELOPMENT OF STABLE INFLUENZA VACCINE POWDER FORMULATIONS FOR
NEW NEEDLE-FFREE DOSAGE FORMS
Jean-PPierre Amorij

Paranimfen: Armand AmorijMicha HartenhofLutea de Jong
Printing of this thesis was supported by generous contributions from the University ofGroningen, Faculty of Mathematics and Natural Sciences of the University of Groningen,and the graduate school GUIDE.
© 2007 Jean-Pierre Amorij. All rights reserved.
No part of this book may be reproduced or transmitted in any form or by any means wit-hout prior written permission of the author.
ISBN: 978-90-367-3238-3 (electronic version)978-90-367-3239-0 (hardcopy)
Cover: Kaleidoscopic presentation of a particle of spray-freeze dried subunit vaccine powder (using a Scanning Electron Micrograph with a magnification of 5000x).
Lay-out design: Jean-Pierre AmorijCover design: Jean-Pierre AmorijPrinting: Gildeprint B.V., Enschede

THE DEVELOPMENT OF STABLE INFLUENZAVACCINE POWDER FORMULATIONS FOR
NEW NEEDLE-FFREE DOSAGE FORMS
PPrrooeeffsscchhrriifftt
ter verkrijging van het doctoraat in deWiskunde en Natuurwetenschappenaan de Rijksuniversiteit Groningen
op gezag van deRector Magnificus, dr. F. Zwarts,in het openbaar te verdedigen op
vrijdag 4 januari 2008om 16.15 uur
door
Jean-PPierre Amorij
geboren op 5 december 1978te Zaanstad
RRIIJJKKSSUUNNIIVVEERRSSIITTEEIITT GGRROONNIINNGGEENN

Promotores Prof. dr. H.W. FrijlinkProf. dr. J.C. Wilschut
Copromotores Dr. W.L.J. HinrichsDr. A. Huckriede
Beoordelingscommissie Prof. dr. H.J. HaismaProf. dr. W. JiskootProf. dr. A.D.M.E. Osterhaus

Contents
CHAPTER I Introduction 1
CHAPTER II Development of stable influenza vaccine powder formulations 7for non-parenteral dosage forms: challenges and possibilities.Review(s): in preparation.
CHAPTER III Rational design of an influenza subunit vaccine powder with 65sugar-glass-technology: Preventing conformational changes of haemagglutinin during freezing and freeze-drying.Vaccine 25 (2007): 6447-6457.
CHAPTER IV Inulin sugar glasses preserve the structural integrity and bio- 87logical activity of influenza virosomes during freeze-drying and storage.European Journal of Pharmaceutical Sciences 32 (2007): 33-44.
CHAPTER V Towards an oral influenza vaccine: Comparison between intra- 109gastric and intracolonic delivery of influenza subunit vaccine in a murine model.Vaccine (2007): in press.
CHAPTER VI Pulmonary delivery of an inulin-stabilized influenza subunit 131vaccine prepared by spray-freeze drying induces systemic, mucosal humoral as well as cell-mediated immune responses in BALB/c mice.Vaccine (2007): in press.
CHAPTER VII General discussion 155
Summary 169
Nederlandse samenvatting 175
Dankwoord 179
Curriculum Vitae 183
TABLE OF CONTENTS
page


Chapter I
Introduction

Few infectious diseases cause such a huge annual toll of morbidity, mortality, and eco-nomic loss as influenza. Each year influenza affects millions of people [1]. Although anti-viral drugs can be used for prophylaxis and therapy of influenza infections, vaccinationis recognized as the most cost-effective method for controlling the disease. Current influ-enza vaccines are mostly formulations composed of whole inactivated virus, virosomes,split virus or subunit antigen, i.e. purified haemagglutinin (HA) and neuraminidase(NA), which are administered parenterally [2]. However, currently also a cold-adaptedlive influenza vaccine is marketed for nasal administration [3].
Stabilization of influenza vaccinesToday's influenza vaccines are all formulated as liquids. In the aqueous environment,however, they are subjected to physical and chemical degradation processes that maylead to loss of activity. Elevated temperatures increase the rate of degradation of the vaccine compounds [4, 5], while temperatures below the freezing point of the disper-sion cause formation of ice and solute concentration, processes that also may damage theantigen [5, 6]. Therefore (inactivated) influenza vaccines have to be stored within thenarrow temperature range of 2 to 8°C. This relatively narrow temperature range requi-res a well-controlled cold chain, which makes the process of distribution and storagecomplicated and expensive [7]. An influenza vaccine that is stable at ambient tem-peratures and not sensitive to freezing stresses would reduce the dependency on cold-chain facilities and would therefore allow the integration of the vaccine logistics withgeneral drug distribution; especially in developing countries this would be attractive.Moreover, this would reduce the risk of vaccine losses caused by "off-label" storage.Overall this would result in enormous annual savings. In addition, a stable vaccine for-mulation would facilitate stockpiling of potential vaccines against pandemic viruses,which provides an immediate availability and simple distribution of vaccine in a pan-demic situation.
A potentially successful strategy to stabilize biopharmaceuticals, such as proteins,vaccines and gene delivery systems, is to convert them into a dry-powder formulation.However, during drying and subsequent storage stabilizers are required to prevent da-mage to these substances. It is well known that sugars can stabilize various biopharma-ceuticals during drying [8-15]. If dried properly, the biopharmaceutical is incorporatedin a glassy matrix of amorphous sugar and thereby stabilized during subsequent storage.
Only limited research has been done on the stabilization of influenza vaccines byincorporating them in a glassy matrix of amorphous sugar. Therefore, more research isrequired to get a complete understanding of the formulation and process design.Especially the dependence of the integrity and stability of the (dried) vaccine on factorssuch as type of vaccine, used excipients and the drying process are unknown.
2

Needle-ffree dosage formsCurrent inactivated influenza vaccines are generally administered via the intramuscular(i.m.) or subcutaneous (s.c.) route using needles and syringes. Despite its common use,needle-based immunization has several disadvantages. Needle-phobia along with limitedease of use for vaccination programs are typical shortcomings of injections [16]. Needle-free delivery, such as mucosal delivery via the respiratory or gastro-intestinal tract, mayprovide several potential advantages in vaccine delivery, such as eliminated pain at theinjection site, easier and faster vaccine distribution and administration, and reduced costs[17-19]. In addition, an important and promising advantage of mucosal vaccination isthat it, in contrast to i.m. vaccines, may result in a local immune response in the respi-ratory tract. As a result antibodies in the respiratory tract might give protection againstinfluenza infection at the port of entry. In addition, since mucosal IgA responses havebeen shown to exhibit cross-protective immunity against antigenically distinct viruses[20, 21], such a mucosal immune response might offer broader protection against drifted,heterologous strains. Unfortunately, despite these potential advantages, until now muco-sal vaccination approaches have suffered from several limitations or practical problemsrelated to the use of inadequate or old-fashioned delivery technologies, and thus havefrequently resulted in inadequate antibody responses or even in a state of immunologi-cal tolerance [22]. Therefore, marketed influenza vaccines, being in the liquid state, arestill mainly administered through injection.
However, recent developments in the area of vaccine formulation and deliverytechnologies now allow efficient delivery of vaccines to specific sites in the human bodyand therefore provide new opportunities for the use of alternative needle-free dosageforms of influenza vaccines. This thesis addresses some of the issues involved in thisdevelopment.
Scope of this thesisThe first objective of the studies described in this thesis was to study and develop stableinfluenza vaccine formulations. Dry-powder formulations, which are not dependent ona cold chain, of two vaccine types (a subunit and a virosomal vaccine) were investigated.The second objective of the studies described in this thesis was to study administrationstrategies for the development of needle-free dosage forms of influenza vaccines.Administration strategies that may lead to needle-free dosage forms of influenza vac-cines via the oral or pulmonary route were investigated.
Outline of this thesisIn Chapter II, several aspects of influenza vaccine formulation and administration werereviewed: the different vaccine types used today; the rationale and need for stabilizedvaccines; strategies by which influenza vaccines can be stabilized; the current status ofstabilized solid vaccines and the current developments in the field of needle-free dosageforms for influenza vaccination. This review also contains a brief discussion of theresearch described in the following chapters.
INTRODUCTION
3

In Chapter III, a stable dry-powder subunit vaccine formulation was developed.Influenza subunit vaccine was incorporated in a glassy matrix of carbohydrate usinglyophilization. The influence of freezing rate, buffer composition, and type of carbo-hydrate (disaccharide, oligosaccharide or polysaccharide) on the structure and antigenicactivity of HA after freezing and freeze-drying respectively was studied. The structuralintegrity of HA was investigated with a proteolytic assay, fluorescence spectroscopy andcircular dichroism spectroscopy. The antigenic properties of the subunit vaccine powderwere determined by a single radial immunodiffusion assay in vitro. The antigenic pro-perties of the subunit vaccine powder after reconstitution were further evaluated in anin vivo study in BALB/c mice.
In Chapter IV, the aim of the presented work was to develop a stable dry-powder formu-lation of influenza virosomes with the objective to preserve the structural integrity andbiological activity of the virosomes. To this end virosomes and pDNA-virosomes werelyophilized with inulin as a stabilizer. The physical and functional properties of freeze-dried virosomes were studied and followed during storage. Freeze-dried virosomal vac-cine was evaluated for the preservation of its immunological properties, while freeze-dried pDNA-virosomes were investigated for preservation of their delivery properties bystudying their fusion capacity and transfection efficiency.
In Chapter V, a strategy for an oral influenza vaccine is presented. It was assessed towhich part of the gastro-intestinal (GI) tract (the upper part or the lower part) an oralinfluenza vaccine should be targeted to result in an effective immune response. For thispurpose BALB/c mice were immunized with a liquid influenza subunit vaccine via dif-ferent routes. Intragastric delivery was used to target to the upper GI-tract and intra-colonic delivery was used to target to the lower GI-tract. Furthermore, the effect of anadjuvant, E.coli heat-labile enterotoxin, on the immune responses elicited by the dif-ferently delivered vaccines was investigated.
In Chapter VI, pulmonary vaccination with an inulin stabilized influenza subunit vac-cine powder, produced by spray-freeze drying, was evaluated. Immune responses afterpulmonary vaccination of BALB/c mice with vaccine powder were determined and com-pared to those induced by intramuscular vaccination with a conventional liquid subunitvaccine and pulmonary administered liquid subunit vaccine.
In Chapter VII, the findings of this thesis and the perspectives of solid influenza vac-cines and needle-free delivery strategies are discussed.
4

REFERENCES
[1] WHO. WHO Media Influenza Factsheet N°211. 2003.[2] Wilschut J, McElhaney JE, Palache AM. Rapid Reference Influenza. 2nd ed. London: Mosby/Elsevier
Science, 2006.[3] Abramson JS. Intranasal, cold-adapted, live, attenuated influenza vaccine. Pediatr Infect Dis J
1999;18(12):1103-4.[4] Coenen F, Tolboom JT, Frijlink HW. Stability of influenza sub-unit vaccine. Does a couple of days out-
side the refrigerator matter? Vaccine 2006;24(4):525-31.[5] WHO Department of Immunization Va, Biologicals. Temperature sensitivity of vaccines. 2006.[6] Luykx DM, Casteleijn MG, Jiskoot W, Westdijk J, Jongen PM. Physicochemical studies on the stability
of influenza haemagglutinin in vaccine bulk material. Eur J Pharm Sci 2004;23(1):65-75.[7] Zweig SE. Advances in vaccine stability monitoring technology. Vaccine 2006;24(33-34):5977-85.[8] Randolph TW. Phase separation of excipients during lyophilization: effects on protein stability.
J Pharm Sci 1997;86(11):1198-203.[9] Slade L, Levine H. Non-equilibrium behavior of small carbohydrate-water systems. Pure Appl Chem
1988;60:1841-64.[10] Hinrichs WL, Prinsen MG, Frijlink HW. Inulin glasses for the stabilization of therapeutic proteins. Int
J Pharm 2001;215(1-2):163-74.[11] Sun WQ, Leopold AC, Crowe LM, Crowe JH. Stability of dry liposomes in sugar glasses. Biophys J
1996;70(4):1769-76.[12] Crowe JH, Crowe LM, Carpenter JF, Aurell Wistrom C. Stabilization of dry phospholipid bilayers and
proteins by sugars. Biochem J 1987;242(1):1-10.[13] Levy JA, Fieldsteel AH. Freeze-drying is an effective method for preserving infectious type C retro-
viruses. J Virol Methods 1982;5(3-4):165-71.[14] Bieganski RM, Fowler A, Morgan JR, Toner M. Stabilization of active recombinant retroviruses in an
amorphous dry state with trehalose. Biotechnol Prog 1998;14(4):615-20.[15] Croyle MA, Roessler BJ, Davidson BL, Hilfinger JM, Amidon GL. Factors that influence stability of
recombinant adenoviral preparations for human gene therapy. Pharm Dev Technol 1998;3(3):373-83.[16] Mitragotri S. Immunization without needles. Nat Rev Immunol 2005;5(12):905-16.[17] Giudice EL, Campbell JD. Needle-free vaccine delivery. Adv Drug Deliv Rev 2006;58(1):68-89.[18] Freytag LC, Clements JD. Mucosal adjuvants. Vaccine 2005;23(15):1804-13.[19] Holmgren J, Czerkinsky C. Mucosal immunity and vaccines. Nat Med 2005;11(4 Suppl):S45-53.[20] Liew FY, Russell SM, Appleyard G, Brand CM, Beale J. Cross-protection in mice infected with influ-
enza A virus by the respiratory route is correlated with local IgA antibody rather than serum antibody or cytotoxic T cell reactivity. Eur J Immunol 1984;14(4):350-6.
[21] Tumpey TM, Renshaw M, Clements JD, Katz JM. Mucosal delivery of inactivated influenza vaccine induces B-cell-dependent heterosubtypic cross-protection against lethal influenza A H5N1 virus infec-tion. J Virol 2001;75(11):5141-50.
[22] Smith DJ, Bot S, Dellamary L, Bot A. Evaluation of novel aerosol formulations designed for mucosal vaccination against influenza virus. Vaccine 2003;21(21-22):2805-12.
5
INTRODUCTION

6

J-P. Amorij1, W.L.J. Hinrichs1, A. Huckriede2, J. Wilschut2, H.W. Frijlink1
Chapter II
Development of stable influenza vaccine powderformulations for non-pparenteral dosage forms:challenges and possibilities.
Reviews: - Development of influenza vaccine powder formulations. (in preparation)- Development of non-parenteral influenza vaccines. (in preparation)
1 Department of Pharmaceutical Technology and Biopharmacy, University of Groningen, Groningen, The Netherlands.
2 Department of Medical Microbiology, Molecular Virology Section, University Medical Center Groningen and University of Groningen, Groningen, The Netherlands.

Contents
Introduction 9Influenza virus 11
Structure 11Entry and replication 12Immunity, antigenic drifts and shifts 13
Influenza vaccines 15Split / subunit vaccine 15Whole inactivated virus vaccine 16Virosomal vaccines 17Live (attenuated) influenza vaccines 18Vaccine stability 18
Rationale for stabilized vaccine formulations 19Cold chain 19Stock piling 20
Stabilization by incorporation in sugar glasses 21Strategies for stabilization of influenza vaccines and vaccine integrity 22
Freeze drying and spray-freeze drying 24Spray drying 28 Vacuum drying and desiccating 31Supercritical drying 33
Storage stability of dried influenza vaccines 34New needle-ffree influenza vaccine dosage forms 37
Epidermal and transcutanous immunization 37Penetration of the stratum corneum 37Jet injectors 38Patches 38Microneedles 40
The mucosal route 41The mucosal immune system 41Mucosal immunity at the port of entry of influenza 42Mucosal vaccines 42Nasal delivery 43Pulmonary delivery 45Oral delivery 48Oral-cavity delivery 51
Summary 52
Abbreviations 53References 54
8

Introduction
Yearly recurrent influenza epidemics and the threat of an influenza pandemic remainmajor public health concerns. Few infectious diseases cause such a huge annual toll ofmorbidity, mortality, and economic loss as influenza. Each year, influenza affects mil-lions of people (estimates go up to 5-15% of the world population [1]). The symptoms inhumans range from typical influenza-like effects, like fever, cough, sore throat and mus-cle aches, to eye infections, pneumonia, acute respiratory distress, viral pneumonia, andother severe and potentially life-threatening complications [2, 3]. For epidemic influ-enza strains this is especially true for the elderly and other high-risk populations where-as pandemic strains may be very dangerous for all age groups. One method of influenzaprevention and/or treatment is the use of antiviral drugs like M2 proton channel inhi-bitors (amantadine and rimantadine) and the neuraminidase (NA) inhibitors (zanamivirand oseltamivir).
Although these antiviral drugs can be used for prophylaxis and therapy of influ-enza virus infections, vaccination is recognized as the most cost-effective method forcontrolling the disease. Vaccination represents the cornerstone for influenza prevention.Many countries recommend influenza vaccination against epidemic influenza strains forpersons who are at increased risk of influenza complications, persons older than 65 years,residents of nursing homes and health-care workers [4]. However, in a pandemic situa-tion influenza vaccines are expected to form the main prophylactic measure to preventall age groups against pandemic influenza [5]. Until today, the induction of an adequatelevel of virus-neutralizing antibodies in the serum is considered the assurance for influ-enza vaccine efficacy. These antibodies are mainly directed against haemagglutinin (HA)and NA.
Current influenza vaccines are mostly inactivated formulations composed of wholeinactivated virus, split virus or subunit antigen, i.e. purified HA and NA. However,recently also a cold-adapted live influenza vaccine has entered the market. Today's influ-enza vaccines are all formulated as liquids, in the aqueous environment they are subjec-ted to physical and chemical degradation that may lead to inactivation. Examples of phy-sical degradation processes are:
- aggregation,- denaturation,- loss of the spring-loaded conformation of HA,- hydrolyses, or- oxidation.
Elevated temperatures increase the rate of inactivation of the vaccine compounds, whiletemperatures below the freezing point of the suspension cause formation of ice and so-lute concentration, processes that both may damage the antigen. Therefore (inactivated)influenza vaccines have to be stored within the narrow temperature range of 2 to 8 °C.This relatively narrow temperature range requires a well-controlled cold chain, whichmakes the process of distribution and storage complicated and expensive. An influenza
CHAPTER II
9

vaccine that is stable at ambient temperatures and not sensitive to freezing stresses wouldreduce the dependency on cold-chain facilities and would therefore be attractive for theintegration of the vaccine logistics with general drug distribution, especially in develo-ping countries. Moreover this would reduce the risk of vaccine losses caused by "of-label"storage. Overall this would result in enormous annual savings. In addition, a stable vac-cine formulation would facilitate stockpiling of potential vaccines against pandemicviruses, which provides an immediate availability and simple distribution of vaccine in apandemic situation in both Western as well as developing countries. A commonly usedmethod to stabilize biologically active macromolecules, such as proteins, vaccines andgene delivery systems, is to convert them into a dry-powder formulation.
Current inactivated influenza vaccines are generally administered via the intramus-cular (i.m.) route using needles and syringes. Despite its common use, needle basedimmunization has several disadvantages. Needle-phobia along with limited ease of usefor vaccination programs are typical shortcomings of injections [6]. In addition, paren-teral immunizations with influenza vaccines have some limitations in inducing immu-nity. Current i.m. influenza vaccines induce the production of virus-neutralizing anti-bodies in the serum but no cellular and mucosal humoral immune responses at relevantmucosal sites, such as the respiratory tract. As a result, they do not prevent against theinitial replication of the virus in the airways.
Needle-free delivery may provide several significant potential advantages in vac-cine delivery: eliminated pain at the injection site; mucosal immune responses (e.g. in therespiratory tract); better efficacy (improved immune responses); easier and faster vac-cine distribution and administration; and reduced costs [7-9]. Therefore, alternative vac-cine delivery systems are in development that delivers the vaccine via epidermal, trans-cutaneous or mucosal routes [10]. However, up till now these approaches suffer fromseveral limitations or practical problems that frequently result in inadequate antibodyresponses or even in a state of immunological tolerance [11]. As a result, marketed influ-enza vaccines, being in the liquid state, are still mainly administered through injection.
In the development of new needle-free dosage forms, dried influenza vaccine for-mulations are an interesting tool, that offers the opportunity of a (more) stable product,combined with the facilitation of new or improved targeting strategies of the vaccinecompound.
This paper intends to provide an up-to-date perspective on the development of solidinfluenza vaccines (against epidemic as well as pandemic influenza strains), covering itschallenges, possibilities and potential applications, including the recent developmentsand achievements in this field. After a general brief introduction on the influenza virusand its pathogenesis, four interrelated topics are discussed sequentially: (i) types of influ-enza vaccines, (ii) rationales for the development of dry vaccine formulations, (iii) dry-ing methods for different influenza vaccines and (iv) the development of new needle-free influenza vaccines. The advantages of needle-free solid dosage forms will be revie-wed. However, because also many needle-free liquid state dosage forms have been eva-luated, these approaches will also be discussed.
10

Influenza virus
StructureInfluenza is a respiratory pathogen belonging to the family of the Orthomyxoviridae[12]. There are three types of influenza (A, B, C) distinguished by the antigenic differen-ces in two of their internal proteins, i.e. nucleoprotein and matrix protein. These threetypes of viruses differ in their pathogenicity and genome organization. Influenza A andB viruses are the types that most common cause human disease. Influenza A viruses aresubdivided further into subtypes based on the surface antigens, HA and NA. In influ-enza A viruses 16 subtypes of HA (H1-H16) and 9 subtypes of NA (N1-N9) have beenfound.
Influenza A (Fig.1) and B contain negative-stranded segmented RNA (8 segments).Each RNA segment is encapsulated by the nucleoprotein to form a ribonucleotide-
CHAPTER II
11
Fig. 1 A schematic drawing of the influenza virus.
nucleoprotein complex. These complexes are surrounded by a shell of matrix protein(M1), which is enveloped by a lipid bilayer. Besides the two surface glycoproteins, HAand NA, the envelope contains a proton channel (M2 in influenza A and NB in influen-za B).
= HA
= NA
= M1
= M2
= Ribonucleotide-nucleoproteincomplex
= Lipid bilayer

Fig. 2 The three-dimensional structure of the influenza HA. The HA monomer (A) and trimer (B).Adapted from: (http://fig.cox.miami.edu/~cmallery/255/255prot/mcb3.7.HA.jpg)
HA and NA are the major antigenic determinants of influenza A viruses and as suchserve as the basis for subtype classification. HA, the major surface glycoprotein of theinfluenza virus, is responsible for both attachment of the virus to sialic acid containingreceptors on the host cell surface and fusion of the viral and endosomal membrane. HAis a trimer (±225 kD) of three identical monomers (±75 kD) (Fig. 2).
Each monomer consists of the polypeptides HA1 (±50 kD) and HA2 (±25 kD). Thesepolypeptides are linked by two intra-monomer disulfide bridges. The three monomersare assembled into a central α-helical coiled-coil that forms the stem-like domain, andthree globular heads containing sialic acid-binding sites. Each globular domain consistsexclusively of HA1 folded in highly variable loops of eight antiparallel β-strands. The glo-bular heads contain both the receptor binding sites and the antigenic epitopes [13, 14].The NA cleaves sialic acid and plays an important role in transport of the virus particlesthrough the mucin layer lining the respiratory tract (virus entry) and mediates therelease of newly assembled virus particles (virus release). NA is a tetrameric glycoprotein(±240 kD) consisting of a hydrophobic stalk and a globular head that contains the enzy-matic and antigenic sites.
Entry and replicationThe influenza virus uses the HA-induced membrane fusion strategy to deliver its ge-nome to the cytosol of target cells (Fig.3). In humans, the primary targets for the influ-enza virus are epithelial cells in the respiratory tract.
12
A B
Fibrous domain
C
N
N
Viralmembrane
DISTAL
PROXIMAL
HA1
HA2
Globulardomain
Sialic acid

Fig. 3 Virus entry and genome release in the cytosol.
The virus attaches to host cells through binding of HA to sialic acid residues of gly-coproteins or glycolipids on the cell surface [15]. Human influenza viruses preferential-ly bind to sialic acid linked to galactose by an α2 6 linkage, while avian viruses bindmainly to sialic acid linked to galactose by an α2 3 linkage [16]. After receptor-binding,virus particles are engulfed by the host cell plasma membrane (endocytosis). In the endo-some (after fusion with the lysosome) the acidification of the environment enables thefusion of the viral membrane with the membrane of the endosome, after which thenucleocapsid can be delivered to the cytoplasm. [17-19]. The endosomal content is aci-dified by proton pumps embedded in the endosomal membrane. This acidification is away to disconnect internalized compounds from the endosomal receptors [20]. Theinfluenza virus uses this low pH inside the endosome (pH 5-6) to trigger the fusion reac-tion between the viral envelope and the endosomal membrane by a conformationalchange of HA [17]. After fusion, the viral ribonucleotide-nucleoprotein complexes arereleased into the cytoplasm from where they can migrate to the nucleus to initiate repli-cation of the virus [21, 22].
Immunity, antigenic drift and shiftNew (drifted) influenza strains are constantly formed by changes in HA and NA.Influenza viruses lack proof-reading mechanisms and are therefore unable to repair(RNA-) errors that occur during replication. These mutations accumulate within theviral genome, resulting in replacement of the existing strain by a new antigenic variant.This mechanism of acquiring new mutations is known as antigenic drift. These muta-tions are seen in each of the gene products of the virus. However, it is most pronouncedin amino acid changes in the surface proteins HA and NA.
In contrast to the antigenic drift, that constantly occurs, the antigenic shift occursat irregular intervals with more dramatic changes in the viral proteins. Antigenic shiftstarts by either direct introduction of new avian influenza viruses into the human popu-lation or reassortment between human and avian viruses, which is believed to occur viaintermediate hosts such as pigs [23]. Aquatic birds are the natural reservoir of all known
CHAPTER II
13

subtypes of the influenza A virus. These birds are highly mobile and are known to carryviruses over great distances. In addition, they transfer viruses to other birds via theexcretion of large quantities of virus in their faeces. While remaining perfectly healthythese birds form a mobile pool from which an influenza pandemic can arise [3, 24]. It ispossible that an avian influenza virus changes, by antigenic shift or drift, so that it is ableto infect humans. Because these viruses do not commonly infect humans, there is littleor no immune protection against them in the human population. Consequently, if anavian virus is able to infect people, it can spread easily from person to person, by whichan influenza pandemic, a global outbreak, could arise [25, 26].Once in several decades an influenza pandemic occurs. An influenza pandemic occurswhen an influenza strain with a novel HA subtype (with or without a novel NA sub-type) appears and spreads in the human population, which has little or no immunity tothe novel HA. There have been three such pandemics in the twentieth century: in 1918,1957, and 1968. At this moment there is for birds a new highly pathogenic influenza sub-type (H5N1), which forms potentially a high risk-factor for a new pandemic [24]. Thesenew viruses may cause pandemics since few or no people have had prior immunologicexposure to their surface proteins [27].
14

Influenza vaccines
The currently used vaccines are (mainly inactivated) formulations containing the twosurface antigens, HA and NA. There are four different types of inactivated influenza vac-cines; split, subunit, whole inactivated influenza vaccines and virosomal influenza vac-cines. Recently also a live attenuated influenza vaccine reached the market [28]. The vac-cines are trivalent, containing the antigens from two subtypes of influenza A and oneinfluenza B as recommended by the World Health Organization (WHO) for each hemis-phere. To ensure an antigenic match with new circulating influenza viruses, the compo-sition of these trivalent vaccines is updated, on basis of WHO's worldwide surveillanceof new influenza strains twice a year. Following vaccination with influenza A, around90% of normal subjects achieve serum haemagglutination inhibition (HI) titers higherthan 40, a level generally associated with protection of about 50% of the population. Asa result, implemented criteria for vaccine immunogenicity are based on the induction ofan adequate level of virus-neutralizing antibodies [29].
Influenza virus for vaccines is generally produced by propagation of virus in embry-onated hen's eggs, although recent developments include vaccine virus production incultured cells, such as MDCK (Madin-Darby-Canine-Kidney), Vero cells (derived fromhuman embryonic lung fibroblasts) or Per.C6 cells (human fetal retinoblast immorta-lized upon transfection with an E1 minigene of adenovirus type 5) [30-33]. The virus (inallantoic fluid) is harvested, concentrated and (highly) purified. The virus is subsequent-ly inactivated with formaldehyde or β-propiolactone and processed to the vaccine typeof choice.
Split / subunit vaccineMost inactivated influenza vaccines are supplied as split vaccines, produced from thechemically disrupted influenza virus, or as subunit vaccines containing predominantlypurified HA and NA. Millions of doses of these influenza vaccines are administered byintramuscular or subcutaneous injection throughout the world each year. These vaccinestrigger the humoral immune system to produce serum antibodies directed against HAand NA. These serum antibodies play a role in both resistance to and recovery frominfluenza infection [4]. The resistance to influenza infection, especially protectionagainst severe viral pneumonia, is caused by transudation of haemagglutinin-specificserum antibodies from the blood into the lungs [34, 35]. The overall rate of adverse reac-tions of split and subunit vaccine is very low. As a result of this low incidence of adver-se effects, the use of split or subunit preparations is first choice in children younger than9 years. However, the overall efficacy of current vaccinations is not optimal in the elder-ly [29]. This appears to be primarily related to a diminishing T cell activity with age [36, 37]. Moreover, the presentation form of conventional split and subunit vaccines issuboptimal for stimulation of cell-mediated immunity, cytotoxic T lymphocyte (CTL)activity in particular, which is of crucial importance for the destruction of virus-infectedcells and thus the clearance of influenza virus infections [37].
CHAPTER II
15

Whole inactivated virus vaccineIn contrast to split and subunit vaccines who are made from disrupted viruses, wholeinactivated virus (WIV) vaccines contain inactivated influenza virus particles (virions)retaining the receptor binding and membrane fusion activity of the native virus.Although limited evidence is present today about the exact interaction of WIV with cellsof the immune system, some hypothetical principles were described. Fig. 4 shows a sche-matic hypothesis how WIV may interact with cells (like B lymphocytes and dendriticcells (DCs)) of the immune system.
The HA and NA spikes protruding from the WIV membrane can be recognized bymembrane-associated immunoglobulin receptor molecules on B lymphocytes. The repe-titive arrangement of the antigens on the WIV surface presumably enables cross-linkingof these immunoglobulin receptors on the B cells [29] which is known to be an enormousstrong activation signal [38].
By virtue of their membrane fusion activity, these WIV vaccines activate not onlythe humoral but also the cellular arm of the immune system [29]. Although cell-media-ted immunity does not seem to contribute significantly in preventing infection, it playsa role in the recovery from influenza infection and may prevent influenza-associatedcomplications [4]. WIV vaccines activate the cellular arm of the immune system by vir-tue of their membrane fusion activity. As for the native influenza virus, binding of WIVto the sialic acid receptors will initiate uptake of WIV by the receptor-mediated endocy-tosis. In the lumen of the endosome the WIV membrane fuses with the endosomal mem-brane resulting in release of the nucleocapsid compounds (RNA, nucleoprotein andmatrix protein) into the cytosol of the antigen presenting cell (APC). However, no repli-cation occurs, since replication is blocked due to modified nucleic bases (mainly purines)in the viral genome caused by the virus-inactivation step with formaldehyde or β-pro-piolactone [39].
Subsequently, the nucleocapsid compounds may be processed and presented via themajor histocompatibility (MHC) class I presentation route resulting in CTL activation[40-42]. A part of the WIV is already degraded within the endosome (or lysosome) resul-ting in generation of peptides derived from the viral (surface as well as encapsulated)proteins. These peptides can be processed and presented via the MHC class II presenta-tion route resulting in strong T helper responses which play an essential role in the sti-mulation of CTLs as well as in the support of antibody-forming B cells. Therefore, vac-cination with WIV activates both the humoral and cellular arm of the adaptive immuneresponse and has been shown to be more immunogenic than split or subunit vaccines [4,42-46]. Moreover, WIV vaccines are expected to induce more subtype cross-reactive cel-lular responses directed against conserved epitopes in internal influenza proteins [4, 41,47, 48]. However, WIV vaccines are associated with frequent local and systemic adverseeffects, like pain and redness at the injection site or fever. Therefore they are less suita-ble for the use in young children. The use of WIV vaccines is limited and in many coun-tries they are unlicensed [49].
16
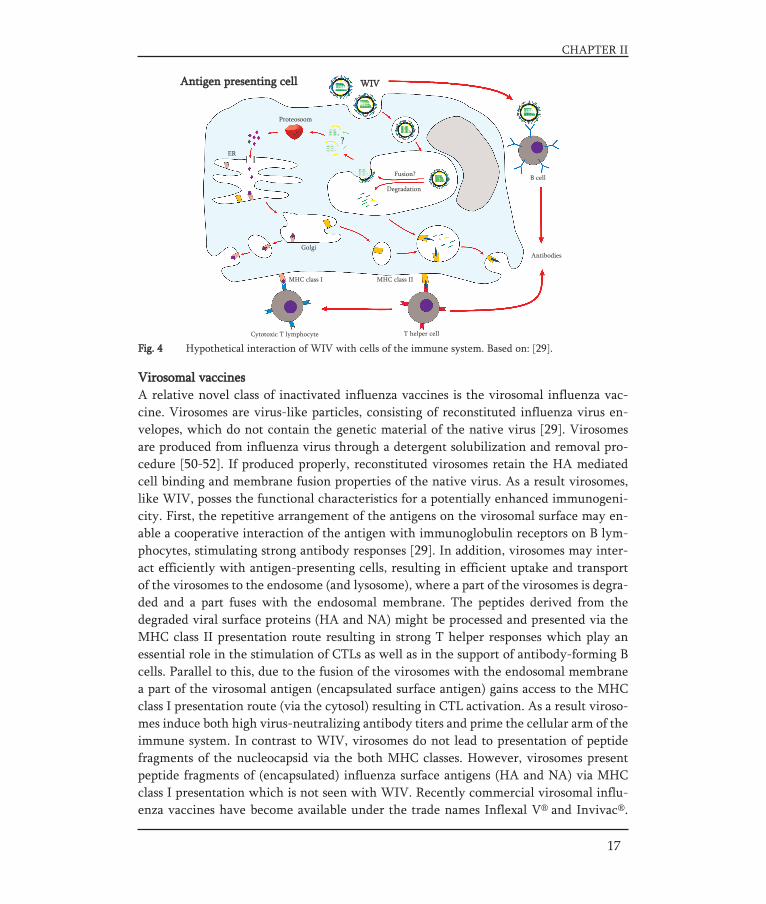
Fig. 4 Hypothetical interaction of WIV with cells of the immune system. Based on: [29].
Virosomal vaccinesA relative novel class of inactivated influenza vaccines is the virosomal influenza vac-cine. Virosomes are virus-like particles, consisting of reconstituted influenza virus en-velopes, which do not contain the genetic material of the native virus [29]. Virosomesare produced from influenza virus through a detergent solubilization and removal pro-cedure [50-52]. If produced properly, reconstituted virosomes retain the HA mediatedcell binding and membrane fusion properties of the native virus. As a result virosomes,like WIV, posses the functional characteristics for a potentially enhanced immunogeni-city. First, the repetitive arrangement of the antigens on the virosomal surface may en-able a cooperative interaction of the antigen with immunoglobulin receptors on B lym-phocytes, stimulating strong antibody responses [29]. In addition, virosomes may inter-act efficiently with antigen-presenting cells, resulting in efficient uptake and transportof the virosomes to the endosome (and lysosome), where a part of the virosomes is degra-ded and a part fuses with the endosomal membrane. The peptides derived from thedegraded viral surface proteins (HA and NA) might be processed and presented via theMHC class II presentation route resulting in strong T helper responses which play anessential role in the stimulation of CTLs as well as in the support of antibody-forming Bcells. Parallel to this, due to the fusion of the virosomes with the endosomal membranea part of the virosomal antigen (encapsulated surface antigen) gains access to the MHCclass I presentation route (via the cytosol) resulting in CTL activation. As a result viroso-mes induce both high virus-neutralizing antibody titers and prime the cellular arm of theimmune system. In contrast to WIV, virosomes do not lead to presentation of peptidefragments of the nucleocapsid via the both MHC classes. However, virosomes presentpeptide fragments of (encapsulated) influenza surface antigens (HA and NA) via MHCclass I presentation which is not seen with WIV. Recently commercial virosomal influ-enza vaccines have become available under the trade names Inflexal V® and Invivac®.
CHAPTER II
17
T helper cellCytotoxic T lymphocyte
MHC class I MHC class II
Golgi
ER
Proteosoom
?
WWIIVVAntigen presenting cell
Fusion?
Degradation
Antibodies
B cell

Compared to conventional inactivated subunit influenza vaccine, local reactions arereported at a lower frequently with virosomal vaccines, whereas the reporting rates ofsystemic side effects are comparable between the two vaccine types [53].
Live (attenuated) influenza vaccinesBesides the marketed vaccines, recent developments resulted in the design and authori-zation (in the USA) of a live attenuated influenza vaccine (LAIV), Flumist®. In contrastto the inactivated influenza vaccines which are administered by intramuscular or subcu-taneous injection, this LAIV is administered intranasally (0.25 ml in each nostril). TheLAIV is administered via a spray device that produces aerosols with large droplets whichare deposited in the nasopharynx [28]. LAIV, a 6/2 reassortant, contains the genes enco-ding the 6 internal segments from the attenuated donor strain (PB1, PB2, PA, M, NP andNS) and the 2 surface proteins (HA and NA) of the wild-type virus. The donor strain iscold-adapted (CA) by genetic modification and as such capable to grow in human nasalcavities (±32°C) but not in internal organs such as the lungs (>37°C) [28]. The underly-ing idea of vaccination with LAIV via the upper respiratory tract (nose) is to induce asecretory and systemic immune response that more closely resembles the immune res-ponse observed after natural infection. LAIVs induce a broad mucosal and systemicimmune response. This in contrast, to the current inactivated influenza vaccines thatbecause of their systemic administration only stimulate the systemic (higher HI titersthan LAIV) and not the mucosal immune system [4]. However, LAIV nasal vaccines andinactivated i.m. vaccines are found to be similarly efficacious in preventing influenza ill-ness from homologous virus infections [54, 55]. In contrast to injected inactivated influ-enza vaccines, LAIVs are believed to provide broader immunity against circulating hete-rologous virus strains. This broader immunity might be the result of mucosal IgA and thehigh MHC class I presentation induced by high presentation of viral compounds into thecytosol of the APC, especially the nucleoprotein and matrix proteins produced by repli-cation after infection. The feature of CA virus to replicate, the possibility to prime theimmune system of naïve persons, may result in a vaccine that is more immunogenic inyoung children than inactivated vaccines [4]. To date, it is unclear whether CA vaccinesare also safe in immunocompromised patients [4].
Vaccine stabilityIn the liquid state the stability of influenza vaccine is limited. The stability of new vac-cine compositions is tested by the manufacturer in order to support regulatory filing andfor GMP compliance. The stability is determined by investigation of a.o. the content ofHA antigen, presence of NA, pH, content of preservative (if applicable) and appearance.The result of the most sensitive parameter, in general the HA content (HA potency),determines the shelf-life of the product [56]. Stability studies are generally performedaccording to ICH guidelines [57, 58]. Stability depends among others on vaccine strain[59], pH, addition of stabilizers such as gelatin or polysorbate, compatibility of the pro-duct with container and closure and preparative treatments needed to reduce adsorptionor interaction with the container [56]. Most inactivated influenza vaccines are stable for
18

Fig. 5 Visual observation of freeze-induced damage caused by accidental storage below 0°C. The left bott-le contains the original subunit vaccine. The right bottle contains the freeze-damaged subunit vaccine. Thefreeze-thaw cycle resulted in a turbid, less opalescent vaccine solution
Rationale for the development of stabilized vaccine formulations
Cold chainInactivated influenza vaccines are temperature sensitive and must be stored at 2 to 8°C.Elevated temperatures can cause inactivation of the vaccine antigens, while temperatu-res below freezing result in formation of ice and solute concentration that may causedenaturation of the antigen [59-61]. The narrow temperature range makes the process ofdistribution and storage complicated, fragile and costly. Although it was demonstratedthat the influenza subunit vaccine could be stored for a couple of days outside the refri-gerator at room temperature [59], vaccine distribution remains one of the greatest riskfor vaccine quality, especially when the vaccine passes the central storage depots. Duringtransport to and storage at local level (general practitioners) the risk of storage outsidethe temperature range (2-8°C) increases. Freeze-sensitive vaccines are still carried withfrozen ice packs and/or improperly conditioned ice packs risking that they will be expo-sed to freezing temperatures. Sometimes improper storage can be detected. For example,time-temperature indicators (VVMs=vaccine vial monitors) can prevent usage of vacci-nes that were stored too long at elevated temperatures. In certain cases freezing-induceddamage can be detected visually (Fig. 5), with or without a shake-test. However in manycases no clear visual changes are observed [62].
CHAPTER II
19
about one year in the refrigerator (2 to 8°C). In contrast to inactivated influenza vacci-nes, live attenuated influenza vaccines must be stored frozen (-15°C to -25°C). To over-come the freezing stresses, the three live attenuated influenza virus reassortants ofFluMist® are stabilized with phosphate glutamate buffer, containing sucrose. Before usethe vaccine must be thawed (for up to 60 hours at 2 to 8°C) and should not be refrozen.However, a refrigerator stable formulation is in development [56].

An influenza vaccine that is stable at room temperature, or even somewhat highertemperatures (up to 35°C), and is not sensitive to freezing stresses would reduce thedependency on cold-chain facilities. Such a vaccine would considerably simplify vac-cine distribution and enable the integration of vaccine logistics with general drug distri-bution, especially in developing countries. Moreover, this would reduce vaccine losses.Both aspects would result in enormous annual cost savings.
Stock pilingRecent outbreaks of highly pathogenic avian influenza A virus infections in poultry andhumans have raised concerns that a new influenza pandemic might occur in the nearfuture. The key preventive method to protect the population against a pandemic virus isan influenza vaccine [63]. In the most extreme scenario, adequate pandemic prepared-ness would mean the availability of 13 billion doses of vaccine (2 doses for 6.5 billionpeople) to vaccinate the world population. However, today's global production capacityof trivalent influenza vaccine is around 400 million doses per year and the time neededuntil the first vaccine dose can be used is at least 4 months [12, 64]. As a result the WHOhas encouraged every member state to produce a pandemic preparedness plan. Such aplan would guarantee fair distribution of vaccine and pre-organized vaccine supply in aclearly defined manner.
A new feature discussed is stockpiling of vaccine from different strains assuringimmediate availability [63, 64]. Although such vaccines would not be a perfect match toa newly emerged influenza A H1-H16 virus, some intertypic immunity and immunolo-gical priming would be expected to ameliorate the effects of the initial pandemic wave.Current seasonal (inactivated) influenza vaccines have a shelf-life claim of one year only.In contrast, stable vaccine formulations of the H1-H16 subtypes would not only reducethe dependency on the cold chain, but could also increase the shelf-life of the vaccines.As a result such a stable vaccine formulation would facilitate stockpiling of potential vac-cines against pandemic viruses and provide in immediately available and simple to dis-tribute vaccine in a pandemic situation. In the ideal situation this would imply that forexample from 3 HA types (e.g. H5, H7 and H9) a powder amount equivalent to about 12 billion doses is stockpiled in bottles containing for example 100-200 doses. In case ofemergency such bottles do only have to be distributed and reconstituted before use.
20

Stabilization of biopharmaceuticals by incorporation in sugar glasses
The most commonly used method to stabilize biologically active macromolecules, suchas proteins, vaccines and gene delivery systems, is to convert them to dry powders. Ingeneral biopharmaceuticals are more stable in the solid state than in the liquid state. Thisis believed to be related to mobility of the biopharmaceutical and the absence or reduc-tion of certain degradation pathways such as hydrolysis. However, depending on thedrying method, freezing and/or drying stresses may affect the structural integrity and/oractivity of the biopharmaceutical. Accordingly, appropriate stabilizers are required forpreservation of these properties. It is well known that sugars can stabilize proteins [65-70], liposomes, lipoplexes [71-76] and various viruses [77-80] during drying and subse-quent storage. If dried properly, the active substance, complex or vesicle is incorporatedin a matrix consisting of amorphous sugar in its glass state. The stabilizing effect of thesesugar glasses has been explained by the formation of a sugar matrix which acts as a phy-sical barrier between particles (particle isolation) and strongly reduces diffusion andmolecular mobility (vitrification). Both the physical barrier [81] and the lack of mobili-ty provided by the glass matrix [82], prevent aggregation and degradation of the biophar-maceutical. Moreover, during the drying process, the sugar replaces the water moleculesin the hydrogen-bonding interaction with the active material, such that the structuralintegrity of the drug is preserved [83]. Under dry conditions, the glass matrix is maintai-ned as long as the temperature is kept below the glass transition temperature (Tg), whichis characteristic for the stabilizing sugar used. In summary carbohydrates are suitable excipients because:
- most carbohydrates contain hydroxyl groups that can form hydrogen bonds with the active compound;
- some carbohydrates possess a high Tg (e.g. trehalose or inulin), and - in general sugars are considered as safe.
CHAPTER II
21

Strategies for stabilization of influenza vaccines and vaccine integrity
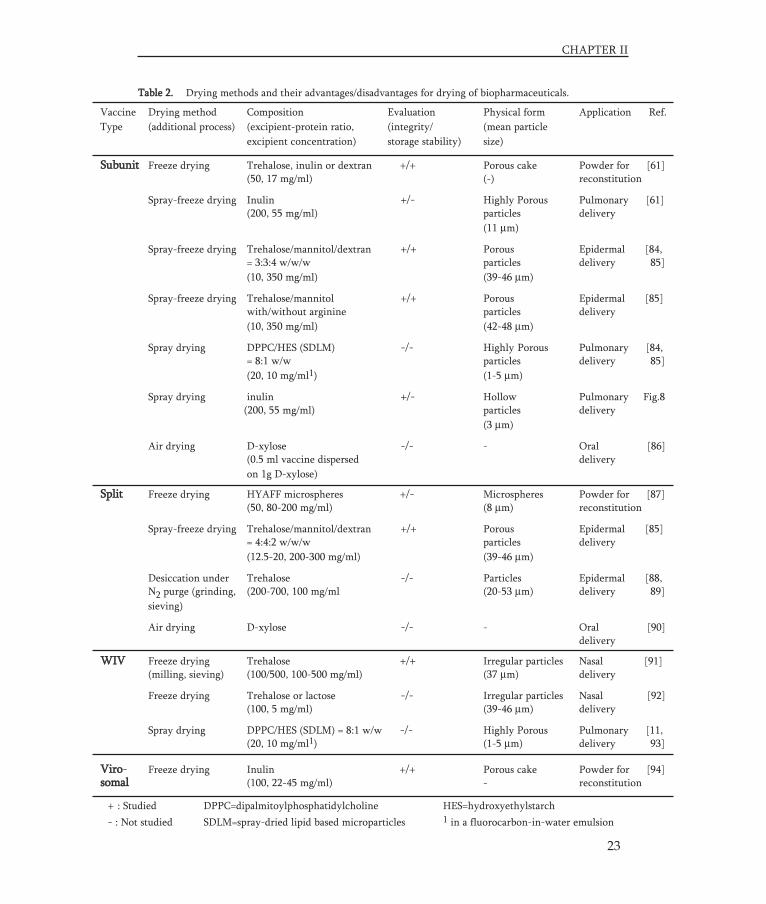
Potentially there are a number of different drying methods that can be used to dry theinfluenza vaccine (with the required excipients) to a stable powder. The major methodswith their advantages and disadvantages are given in table 1.
22
Atomiza- Freezing Heating Process Crystalliza- Particle Capacity Costs Industrialtionstress stress stress speed tion risk design experience
Already at first glance it is clear that not all drying methods are suitable and theideal method has certainly not been found yet. Each of the methods has typical advanta-ges as well as drawbacks, of which the relevance and magnitude may furthermore bedetermined by the applied process conditions as well as the formulation (excipientsused). There are a number of relevant aspects that determine the suitability of a specificdrying method. Most important in this respect are process stresses, crystallization risk,process speed, ease to design particles, capacity, recovery, costs and current (industrial)experience (Table 1).
In the past decades several papers have been published in which dried influenzavaccines were used/described (Table 2). Most of these dried influenza vaccines were pro-duced in order to facilitate new needle-free dosage forms for nasal, pulmonary or epider-mal delivery. The integrity and stability of the influenza vaccine compound after dryingis examined in only a limited number of the published articles. From these studies it isdifficult to draw definite conclusions with regard to the influences of process and formu-lation parameters on vaccine integrity and stability. However, it is possible to extra-polate general concepts described in literature concerning drying of biopharmaceuticalsas discussed in the proceding paragraphs.
Table 1. Drying methods and their advantages/disadvantages for drying of biopharmaceuticals.
Freeze drying + - + - + - +/- - +
Spray-freeze drying - + + +/- + + +/- - -
Spray drying - + - + - + + + +
Vacuum drying + + +/- - - - +/- + +
Supercritical - + +/- + +/- + + - -fluid drying
+ : Favorable - : Unfavorable
CHAPTER II
23
Vaccine Drying method Composition Evaluation Physical form Application Ref. Type (additional process) (excipient-protein ratio, (integrity/ (mean particle
excipient concentration) storage stability) size)
Table 2. Drying methods and their advantages/disadvantages for drying of biopharmaceuticals.
Subunit Freeze drying Trehalose, inulin or dextran +/+ Porous cake Powder for [61](50, 17 mg/ml) (-) reconstitution
Spray-freeze drying Inulin +/- Highly Porous Pulmonary [61](200, 55 mg/ml) particles delivery
(11 μm)
Spray-freeze drying Trehalose/mannitol/dextran +/+ Porous Epidermal [84,= 3:3:4 w/w/w particles delivery 85](10, 350 mg/ml) (39-46 μm)
Spray-freeze drying Trehalose/mannitol +/+ Porous Epidermal [85]with/without arginine particles delivery(10, 350 mg/ml) (42-48 μm)
Spray drying DPPC/HES (SDLM) -/- Highly Porous Pulmonary [84,= 8:1 w/w particles delivery 85](20, 10 mg/ml1) (1-5 μm)
Spray drying inulin +/- Hollow Pulmonary Fig.8(200, 55 mg/ml) particles delivery
(3 μm)
Air drying D-xylose -/- - Oral [86](0.5 ml vaccine dispersed deliveryon 1g D-xylose)
+ : Studied DPPC=dipalmitoylphosphatidylcholine HES=hydroxyethylstarch - : Not studied SDLM=spray-dried lipid based microparticles 1 in a fluorocarbon-in-water emulsion
Split Freeze drying HYAFF microspheres +/- Microspheres Powder for [87](50, 80-200 mg/ml) (8 μm) reconstitution
Spray-freeze drying Trehalose/mannitol/dextran +/+ Porous Epidermal [85]= 4:4:2 w/w/w particles delivery(12.5-20, 200-300 mg/ml) (39-46 μm)
Desiccation under Trehalose -/- Particles Epidermal [88,N2 purge (grinding, (200-700, 100 mg/ml (20-53 μm) delivery 89] sieving)
Air drying D-xylose -/- - Oral [90]delivery
WIV Freeze drying Trehalose +/+ Irregular particles Nasal [91](milling, sieving) (100/500, 100-500 mg/ml) (37 μm) delivery
Freeze drying Trehalose or lactose -/- Irregular particles Nasal [92](100, 5 mg/ml) (39-46 μm) delivery
Spray drying DPPC/HES (SDLM) = 8:1 w/w -/- Highly Porous Pulmonary [11,(20, 10 mg/ml1) (1-5 μm) delivery 93]
Viro- Freeze drying Inulin +/+ Porous cake Powder for [94]somal (100, 22-45 mg/ml) - reconstitution
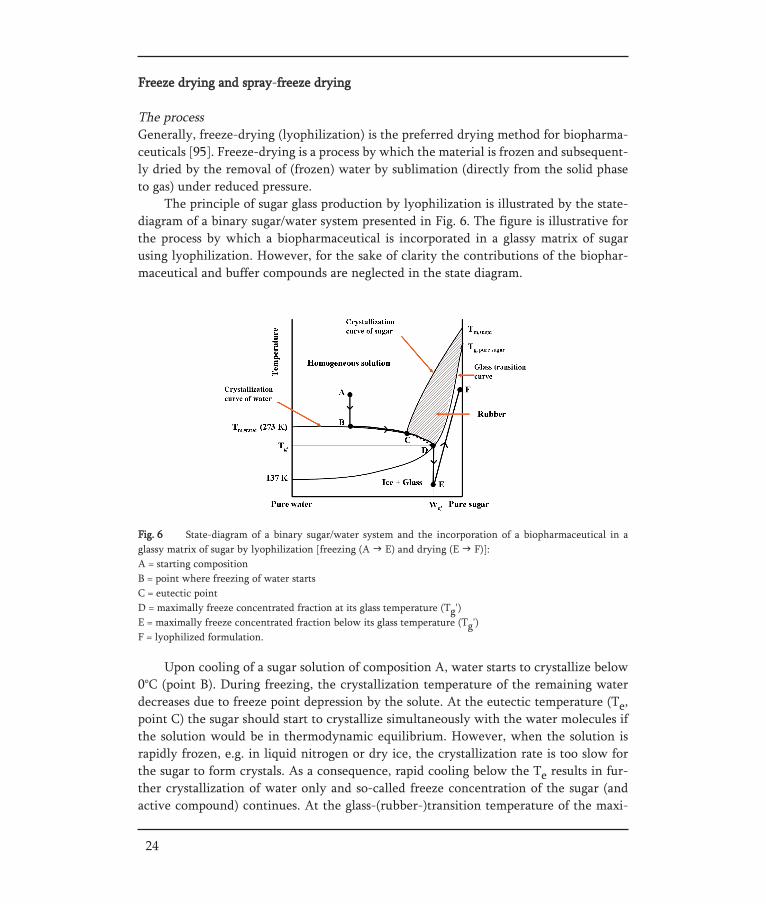
Fig. 6 State-diagram of a binary sugar/water system and the incorporation of a biopharmaceutical in a glassy matrix of sugar by lyophilization [freezing (A E) and drying (E F)]:A = starting compositionB = point where freezing of water startsC = eutectic pointD = maximally freeze concentrated fraction at its glass temperature (Tg')E = maximally freeze concentrated fraction below its glass temperature (Tg') F = lyophilized formulation.
Upon cooling of a sugar solution of composition A, water starts to crystallize below0°C (point B). During freezing, the crystallization temperature of the remaining waterdecreases due to freeze point depression by the solute. At the eutectic temperature (Te,point C) the sugar should start to crystallize simultaneously with the water molecules ifthe solution would be in thermodynamic equilibrium. However, when the solution israpidly frozen, e.g. in liquid nitrogen or dry ice, the crystallization rate is too slow forthe sugar to form crystals. As a consequence, rapid cooling below the Te results in fur-ther crystallization of water only and so-called freeze concentration of the sugar (andactive compound) continues. At the glass-(rubber-)transition temperature of the maxi-
24
Freeze drying and spray-ffreeze drying
The processGenerally, freeze-drying (lyophilization) is the preferred drying method for biopharma-ceuticals [95]. Freeze-drying is a process by which the material is frozen and subsequent-ly dried by the removal of (frozen) water by sublimation (directly from the solid phaseto gas) under reduced pressure.
The principle of sugar glass production by lyophilization is illustrated by the state-diagram of a binary sugar/water system presented in Fig. 6. The figure is illustrative forthe process by which a biopharmaceutical is incorporated in a glassy matrix of sugarusing lyophilization. However, for the sake of clarity the contributions of the biophar-maceutical and buffer compounds are neglected in the state diagram.

mally freeze-concentrated fraction (Tg', point D), the viscosity increases dramaticallyresulting in immobilization of the sugar and water (and further components), and a glassis formed. In this glass the sugar molecules are randomly orientated (amorphous state)and form a vitrified matrix in which water and biopharmaceutical are captured. Due tothe high viscosity of the amorphous matrix the composition of the glass remains the same(also water molecules do not crystallize) upon further cooling (D E).
To obtain the biopharmaceutical in a dry amorphous glass, the frozen sample is keptunder vacuum and water is removed by sublimation. During primary drying, the firststage of the sublimation process, the ice formed during freezing of the sample is remo-ved. The temperature during this primary drying must be held below the Tg'. This isessential because above this temperature the sugar glass turns into the rubbery state inwhich the molecular mobility is considerably increased and crystallization of the sugaror phase separation may occur. This is detrimental for the stabilization of the incorpora-ted biopharmaceutical compound since hydrogen bonds or other stabilizing interactionswith the sugar are lost and the translational freedom of the biopharmaceutical increases,which could cause aggregation. In addition, the mechanical forces induced by crystalli-zation of the sugar may damage the structure of the biopharmaceutical compound,which in turn may lose functional activity.
The remaining water molecules captured in the glassy matrix upon rapid cooling areremoved during the secondary drying when the sugar surface is free of ice. During thissecondary drying the temperature can slowly be increased as long as the temperature isbelow the Tg of the water-containing product (E F). After removal of all water, the bio-pharmaceutical compound is incorporated in a dry sugar glass with a Tg depending onthe composition and on the sugar used. To assure a long shelf-life, the dry formulationshould be stored below its Tg to avoid transition into the rubbery state (which couldresult in crystallization). Moreover, generally a highly porous cake with a high specificsurface area is obtained after lyophilization, which can be easily reconstituted. However,this porous cake also easily absorbs water. As a result the product should be kept at a lowrelative humidity (adequate packaging required), since water decreases the Tg of the for-mulation (see glass transition curve as function of water content).
Spray-freeze drying (SFD) is a relative new drying process to produce biopharma-ceutical powders that combines atomization, generating a cloud of small droplets (lea-ding to rapid freezing), and lyophilization. The state-diagram of incorporation of a bio-pharmaceutical in a glassy matrix of sugar by SFD is almost the same as that of normallyophilization (Fig. 6). A liquid solution containing a biopharmaceutical compound andstabilizer(s) is atomized into a cryogenic medium, in general liquid nitrogen, to vitrifythe droplets (A E), followed by removal of ice and water molecules (captured in theglassy matrix) by lyophilization (E F)]. A main advantage of SFD over normal freezedrying is the extremely rapid vitrification (fast A E traject) due to the enormous surfa-ce area for heat transfer generated during the atomization (the spraying process) and adirect contact of the liquid droplets with the freezing medium. This is important sincerapid vitrification prevents phase separation. Moreover, the large surface area allows arapid drying. Another advantage of SFD is the capability to produce spherical particles.
CHAPTER II
25

Freeze drying and spray-freeze drying of influenza vaccinesFreeze drying and spray-freeze drying have been used for drying of influenza vaccines.During drying, various factors can affect the integrity of the vaccine compound. Thesefactors are mostly dependent on the drying method used. Lyophilization, for exampleraises concerns related to typical stresses induced during freezing and drying. In respon-se to freezing, multiple ice crystals of various sizes grow and interact with the freeze con-centrated fraction (the highly viscous fluid phase containing non-crystalline componentsand the remaining non-frozen water). During this process, an enormous ice/liquid inter-face is created that presents a surface area for protein adsorption. This may result in con-formational changes and disruption of the vaccine compound. Moreover, the solute con-centration of the non-frozen fraction increases during freezing, resulting in accelerationof reaction kinetics [96] and changed physical properties such as ionic strength and rela-tive composition of the solution, which may further destabilize the vaccine [95, 97].
HA in influenza subunit vaccines is susceptible to freezing stresses, especially at lowfreezing rates (higher freezing temperatures), as revealed by various spectroscopic tech-niques [60, 61]. However, Amorij et al. showed that sugars (trehalose, inulin and dex-tran) can prevent freeze induced damage as revealed by tryptophan fluorescence spec-troscopy, circular dichroism spectroscopy and a proteolytic assay [61].
In the development of lyophilized influenza vaccines (as for several other biophar-maceuticals) the choice of buffer type has been shown to be of major importance [61, 98].Phosphate buffered saline (PBS) is an often used buffer for biopharmaceutical com-pounds. However, the use of PBS during lyophilization of subunit vaccine results in pHchanges leading to conformational changes of HA [61]. This is a typical example of com-position changes during freezing that may destabilize a vaccine compound. Due to thecrystallization of sodium or potassium dibasic phosphate during freezing, the formula-tion pH of the PBS shows a downshift. By nature, HA within the influenza virus posses-ses a spring-loaded conformation that changes upon acidification in the endosome (pH ±5) in order to mediate fusion of the virus (see Entry and replication). As a result HAis sensitive to acidification like pH changes caused by freezing of PBS. The conformatio-nal changes of HA during freezing could be prevented by the use of another buffer, hepesbuffered saline (HBS). Hepes does not crystallize and consequently no strong pH changesoccur during freezing [61]. Moreover, a fast freezing rate and the use of sugars, like tre-halose, inulin or dextran, are aspects that may result in a vaccine powder with conserva-tion of the vaccine with full maintenance of structure [61].
Different influenza vaccines have been dried by lyophilization. Subunit and splitvaccines have been successfully (spray-) freeze dried with conservation of the antigen'smolecular structure, potency and/or antigenicity in mice [61, 84, 85].
Maa et al. produced a vaccine powder of a trivalent split vaccine by SFD using com-binations of sugars. They found that combinations of trehalose, mannitol and dextran atdifferent solid contents in the feed solution (20-30 %w/v) were capable to preserve theantigen's potency as well as immunogenicity in vivo during SFD [85].
26

Whole inactivated virus vaccine has been successfully lyophilized by Huang et al.[91]. They lyophilized a mixture of whole inactivated virus and trehalose in a ratio of1:500 (10 μg virus/5 mg trehalose) from sterile saline. After lyophilization, subsequentmilling and reconstitution the whole virions retained their haemagglutination capacitywith chicken erythrocytes [91].
Influenza virosomes were lyophilized by De Jonge et al. [94]. Virosomes (225-450μg/ml) prepared by a fast-reconstitution method from A/Panama virus were lyophilizedwith inulin 1.8 kD (22.5-45 mg/ml) in a ratio of 1:100 (w/w). These lyophilized viroso-mes retained their fusogenic properties in vitro and antigenicity in mice. In contrast,lyophilization of virosomes without protectant resulted in reduced fusogenic propertiesand disruption of the vesicular structure of the virosomes.
CHAPTER II
27
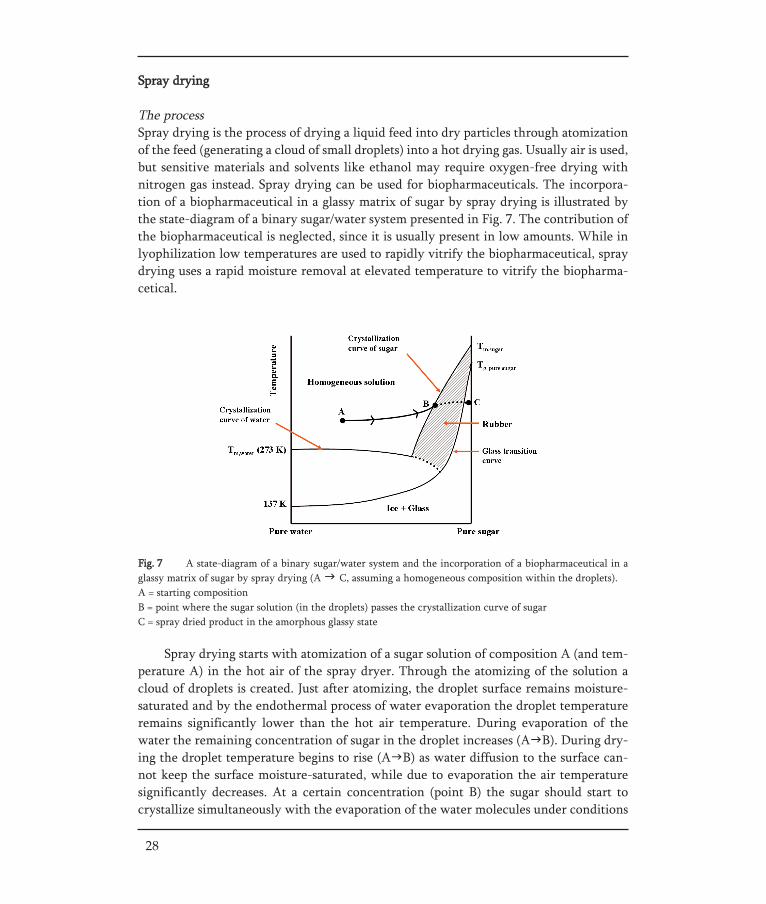
Fig. 7 A state-diagram of a binary sugar/water system and the incorporation of a biopharmaceutical in aglassy matrix of sugar by spray drying (A C, assuming a homogeneous composition within the droplets).A = starting compositionB = point where the sugar solution (in the droplets) passes the crystallization curve of sugarC = spray dried product in the amorphous glassy state
Spray drying starts with atomization of a sugar solution of composition A (and tem-perature A) in the hot air of the spray dryer. Through the atomizing of the solution acloud of droplets is created. Just after atomizing, the droplet surface remains moisture-saturated and by the endothermal process of water evaporation the droplet temperatureremains significantly lower than the hot air temperature. During evaporation of thewater the remaining concentration of sugar in the droplet increases (A B). During dry-ing the droplet temperature begins to rise (A B) as water diffusion to the surface can-not keep the surface moisture-saturated, while due to evaporation the air temperaturesignificantly decreases. At a certain concentration (point B) the sugar should start tocrystallize simultaneously with the evaporation of the water molecules under conditions
28
Spray drying
The processSpray drying is the process of drying a liquid feed into dry particles through atomizationof the feed (generating a cloud of small droplets) into a hot drying gas. Usually air is used,but sensitive materials and solvents like ethanol may require oxygen-free drying withnitrogen gas instead. Spray drying can be used for biopharmaceuticals. The incorpora-tion of a biopharmaceutical in a glassy matrix of sugar by spray drying is illustrated bythe state-diagram of a binary sugar/water system presented in Fig. 7. The contribution ofthe biopharmaceutical is neglected, since it is usually present in low amounts. While inlyophilization low temperatures are used to rapidly vitrify the biopharmaceutical, spraydrying uses a rapid moisture removal at elevated temperature to vitrify the biopharma-cetical.

that the solution is in thermodynamic equilibrium. However, when the evaporation rateof water is fast enough, the glass transition temperature of the dried formulation is abovethe outlet temperature and the crystallization rate too slow for the sugar to form cryst-als. Consequently, the sugar will pass through rubbery state (B C) and turns into the dryamorphous glassy state.
However, spray drying is not a process without concerns. First, the vaccine com-pound may suffer from heat denaturation by hot air. Although the droplet temperatureonly increases marginally as a result of heat absorption by the evaporated water, it is wiseto use process parameters, like a relatively low inlet air temperature, in order to assurethe outlet temperature is not too high and consequently reduce the risk of denaturati-on/degradation of the vaccine compound. Secondly, air-water interfacial stresses andshear stresses induced by the atomization of the feed solution may lead to degradation ofthe biopharmaceutical. Biopharmaceuticals, vaccine compounds such as HA, NA andlipids, being (amphiphilic) membrane components, all posses surface active properties.As a result, the biopharmaceuticals tend to be adsorbed to the air water interface (thefine droplets have a high specific surface area) where the large surface free energy maycause the biopharmaceutical to be disrupted and to expose its hydrophobic regions resul-ting in aggregation.
Again the use of sugars may prevent deterioration by increases unfolding tempera-tures of the (proteinous) biopharmaceutical. In general, sugars that easily crystallize andhave a relative low Tg are not suitable for spray-drying. However, formulations thatcrystallize less easily and have a high Tg can be made [99-102].
Spray drying of influenza vaccinesSpray drying has been used to prepare dried influenza subunit and WIV vaccines.Various sugars can be used for the spray drying of proteinous compounds. However,during spray drying mono- and disaccharides with a low Tg have the tendency to crys-tallize resulting in degradation of the biopharmaceutical [103-106]. Crucial in spray dry-ing of proteins is the tendency of proteins, being amphiphilic, to adsorp at the air-liquidinterface of droplets in the aerosol, which may result in unfolding and aggregation [107].Therefore, the addition of surface-acting agents (surfactants) to the mixture before spraydrying has been used to remove proteins from this interface and consequently improvetheir stability [96, 104, 108].
Recently, we developed a strategy to prepare an influenza subunit vaccine powderby spray drying using the oligosaccharide inulin (inulin 4 kD, Tg of 156°C). A solution ofsubunit vaccine (A/Panama H3N2; 275 μg/ml) and inulin (55 mg/ml) in PBS was spraydried using a Mini Spray Dryer. The vaccine powder obtained after spray drying consis-ted of spherical and smooth particles with an average particle size of 3 μm. Moreover,the process stresses did not have an adverse effect on the antigen's immunogenicity invivo (Fig. 8).
CHAPTER II
29

Fig. 8 Vaccine antigenicity of spray dried influenza subunit vaccine. The vaccine was dried with inulin asstabilizer, using a 5.5 % w/v sugar solution at a ratio sugar/HA = 200. Subunit antigen-specific IgG serum titers(black bars) and serum HI titers (grey bars) in BALB/c mice. Animals were immunized i.m (on day 0, 14 and28) with 5 μg subunit antigen (A/Panama) from unprocessed (n=4) or reconstituted spray dried subunit vacci-ne (n=8). On day 52 mice were sacrificed and titers were determined according to [61]. The results are expres-sed as the geometric mean titer ± standard deviation for each group.
Spray-dried lipid-based microparticles (SDLM) have been used to encapsulate subu-nit and WIV vaccine in microparticles in order to target APC in the respiratory tract [11,93]. Besides vaccine these microparticles consisted of lipid-surfactants, DPPC and DSPC(75-85% w/w; both surfactants occur in the lung), and a polysaccharide, hydroxyethyl-starch (HES). The release of antigens (pharmaceutical availability) from these SDLMswas limited, but co-formulation with the biocompatible surfactant tyloxapol improvedthe immune profile of these particles [11]. However, it is unclear whether the structuralintegrity of the vaccine compounds was affected by the drying process, since only a bio-assay based on peptide recognition and SDS-treatment was used to determine the antigen content.
30
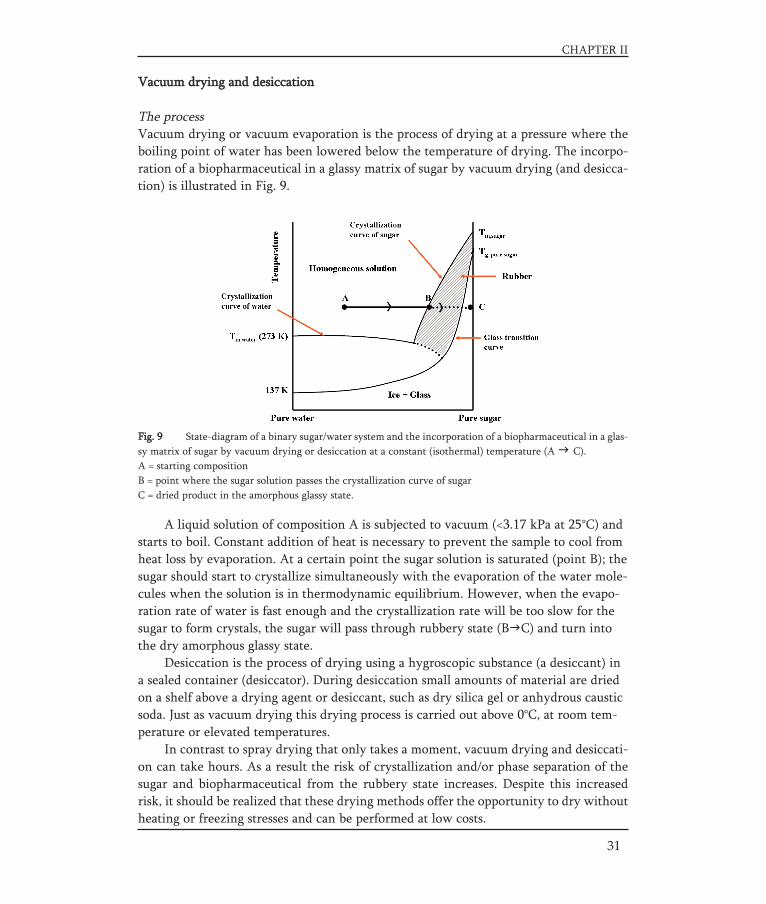
Fig. 9 State-diagram of a binary sugar/water system and the incorporation of a biopharmaceutical in a glas-sy matrix of sugar by vacuum drying or desiccation at a constant (isothermal) temperature (A C).A = starting compositionB = point where the sugar solution passes the crystallization curve of sugarC = dried product in the amorphous glassy state.
Vacuum drying and desiccation
The processVacuum drying or vacuum evaporation is the process of drying at a pressure where theboiling point of water has been lowered below the temperature of drying. The incorpo-ration of a biopharmaceutical in a glassy matrix of sugar by vacuum drying (and desicca-tion) is illustrated in Fig. 9.
CHAPTER II
31
A liquid solution of composition A is subjected to vacuum (<3.17 kPa at 25°C) andstarts to boil. Constant addition of heat is necessary to prevent the sample to cool fromheat loss by evaporation. At a certain point the sugar solution is saturated (point B); thesugar should start to crystallize simultaneously with the evaporation of the water mole-cules when the solution is in thermodynamic equilibrium. However, when the evapo-ration rate of water is fast enough and the crystallization rate will be too slow for thesugar to form crystals, the sugar will pass through rubbery state (B C) and turn intothe dry amorphous glassy state.
Desiccation is the process of drying using a hygroscopic substance (a desiccant) ina sealed container (desiccator). During desiccation small amounts of material are driedon a shelf above a drying agent or desiccant, such as dry silica gel or anhydrous causticsoda. Just as vacuum drying this drying process is carried out above 0°C, at room tem-perature or elevated temperatures.
In contrast to spray drying that only takes a moment, vacuum drying and desiccati-on can take hours. As a result the risk of crystallization and/or phase separation of thesugar and biopharmaceutical from the rubbery state increases. Despite this increasedrisk, it should be realized that these drying methods offer the opportunity to dry withoutheating or freezing stresses and can be performed at low costs.

Vacuum drying/desiccation of influenza vaccinesAir drying and desiccation have been used for preparing dry subunit and split influenzavaccines. In different laboratories, subunit vaccine has been air dried after dispersing 0.5ml vaccine on 1 g D-xylose [86, 90]. Chen et al prepared dried split vaccine powder bydesiccation a vaccine in trehalose (100 mg/ml) solution overnight using an N2 purge [88,89]. Although these researchers performed immunization studies it was not investigatedwhether the integrity of the vaccine compound in their dried product was affected. Theintegrity of the vaccine is a concern with these specific drying techniques. These dryingprocesses need a long drying time during which the sugar vaccine mixture passes relati-ve slowly the intermediate rubbery state [109]. Consequently sugar crystallization andsubsequent diminished stability of the vaccine compound are a risk. Especially for mono-and disaccharides, like D-xylose and trehalose, that may easily crystallize, this riskshould be taken seriously [105, 109-111].
32
Fig. 10 A pressure-temperature diagram for pure CO2.

Supercritical drying
A relative new drying method is supercritical fluid (SCF) drying (reviewed in: [112] and[113]). SCF drying makes use of a fluid that is supercritical i.e. when both pressure andtemperature are above the critical pressure (Pc) and critical temperature (Tc), respective-ly (Fig. 10). Above the critical temperature, it is not possible to convert a gas in the liquidstate by increasing the pressure. However, the density of the gas increases continuouslywith increasing pressure. The supercritical fluid has gas-like physical properties, such asa high diffusity and low viscosity [114]. The application of SCFs by pharmaceutical com-panies is restricted to supercritical CO2 (SC-CO2), because this SCF is generally regardedas safe by the FDA, available in large quantities at high purity, inexpensive, has a low Tc(31,5°C) etc.. However, until now no influenza vaccines have been dried by supercriticaldrying. There are two main principles of SCF drying which may be applicable for dryingand formulation of influenza vaccines.
The first concept is spray drying in a supercritical fluid. A vaccine sugar solution issprayed by atomization into a vessel containing SC-CO2. Although SC-CO2 is not com-pletely miscible with water it dissolves in the vaccine sugar solution. However, the vac-cine compound and sugar are poorly soluble in SC-CO2 (antisolvent). As a result the sol-vent in the vaccine sugar droplets loses solvent power and becomes supersaturated. Thisin combination with the water transfer from the supersaturated droplets to the SC-CO2(extraction) leads to the incorporation of the vaccine compound in a glassy matrix ofsugar. Critical in this process is the mass transfer of water to ensure a rapid dehydrationin order to prevent crystallization of the stabilizing sugar. The mass transfer can beimproved by decreasing the droplet size, decreasing the relative velocity between thedroplets and SC-CO2 and increasing the SC-CO2/vaccine sugar solution ratio [113].Another critical issue in the drying process is the pH drop (final pH 2.5-3) due to thedissolution of CO2 in the water phase [113]. Since the structure of HA changes below apH of approximately 5, it is essential to use a sufficiently buffered vaccine solution.
In the second concept, the SC-CO2 is dissolved at high pressure in the solution con-taining the vaccine compound and sugar and sprayed to atmospheric conditions. Uponspraying, the CO2 expands and droplets break up in smaller droplets, which are thendried by a flow of nitrogen. This process is a spray-drying process at relative low-tempe-rature (20-50°C, but usually somewhat above Tc (32°C) [113]), using the SC-CO2 as effer-vescent and extraction agent to enhance the atomization process and water transfer the-reby shortening the drying process.
CHAPTER II
33

Storage stability of dried influenza vaccines
Although the drying process may give a stable vaccine, it does not guarantee long-termstability. In the dry state, the long-term stability of the influenza vaccine may still belimited, especially at elevated storage temperatures. The stability of the dried vaccine ismerely dependent on the formulation (composition), the structure in which the vaccineis incorporated in the formulation and, of course, the storage conditions. It should be rea-lized that even a successful drying process does not assure vaccine stability in the solidstate.
Amorij et al. showed that the storage stability of lyophilized influenza subunit vac-cine was dependent on the type of carbohydrate, type of buffer and storage conditions[61]. Subunit vaccine lyophilized with trehalose, inulin 0.9 kD, inulin 1.8 kD have beenshown to be stable for at least 26 weeks at room temperature. In contrast, vaccine incor-porated in a glassy matrix of dextran 56 kD lost its potency during storage for 26 weeks.When influenza subunit vaccine lyophilized with inulin 0.9 kD or 1.8 kD was stored at45°C, the potency of the vaccine was almost completely lost within 4 weeks. In contrast,when trehalose was used as stabilizer the subunit vaccine retained its potency at thistemperature for at least 26 weeks. The poor stabilization of HA by dextran might be dueto phase separation (during freezing) and/or the bulkiness of dextran (steric hindrance),preventing efficient hydrogen bonding with the protein [65, 95, 115]. This in contrast tothe small disaccharide trehalose. The less efficient stabilization of HA at elevated tempe-ratures by the oligosaccharide inulin compared to trehalose might be due to a lowerextent and intimacy of hydrogen bond formation [95].
In the study of Amorij it was also found that HBS was superior to PBS to preservethe in vitro immunological properties of HA in the carbohydrate formulation upon freeze-drying and storage. The antigen activity of the powders decreased more readilywhen PBS was used instead of HBS. Reasons for this could be an improper inclusion inthe glassy matrix due to the pH shift during freezing with PBS and the capability of HBSto form an amorphous matrix [75] that acts as a stabilizer during freeze-drying and storage.
Maa et al evaluated the stability of trivalent influenza subunit powder (containingA/Panama(H3N2), A/New Caledonia(H1N1) and B/Yamanashi strains) produced byspray-freeze drying using highly concentrated feed solutions (35.64 mg HA/ml and35%w/v carbohydrate) [85]. Vaccine formulations of different compositions were eva-luated for stability:
- A: Trehalose/Mannitol/Dextran 10 kD = 3:3:4 (10% vaccine)- B: Trehalose/Mannitol/Arginine glutamate = 4:2:4 (10% vaccine;0.5% polysorbate80)- C: Trehalose /Mannitol = 7:3 (10% vaccine; 5% poloxamer 188)
During storage for 12 weeks at 40°C in sealed glass vials, formulation A and B possessed unchanged HA potency whereas formulation C suffered continuous potency
34

loss. This might be due to decrease of the Tg by poloxamer. Although the HA potencywas unaffected, formulation B lost its uniform particle properties (shape/flowability)during storage due to crystallization of arginine glutamate. In an additional study formu-lation A was found to be resistant to humidities of 10 and 40 % relative humidity (RH)for 8 weeks at 40°C, but gradually lost potency upon storage at 75% RH.
The WIV vaccine powder stabilized with trehalose (trehalose/virus, 500:1) as produced by Huang et al. showed improved stability compared to liquid WIV vaccine[91]. While the liquid formulation lost more than 50% of its haemagglutination activityupon storage for 2 weeks at 4°C/25% RH and 25°C/25% RH the powder formulationretained full haemagglutination activity up to 12 weeks. However, the powder formu-lation showed an almost instant drop in stability upon storage at 40°C/75% RH. This wasprobably due to crystallization of trehalose resulting from a decrease in Tg by the highhumidity (Tg(trehalose+water) < 40°C) resulting in an increase in molecular mobility [69].
Inulin sugar glasses have been shown to preserve the structural integrity and bio-logical activity of influenza virosomes during storage [94]. Virosomes (225-450 μg/ml)lyophilized with inulin 1.8 kD (22.5-45 mg/ml) retained HA potency upon storage for 12weeks at 20°C. Upon storage at 42°C the inulin lyophilized virosomes gradually decrea-sed in HA potency, but to a lower extent than the virosomes lyophilized without sugar.Moreover, virosome aggregation upon storage and subsequent rehydration was observedwhen virosomes were lyophilized without sugar, but did not occur when they werelyophilized with inulin. The preservation of the vesicular structure in the presence ofsugars, in particular inulin is believed to be related to the fact that the oligosaccharideinteracts with membrane lipids and as such may preserve the structural and functionalfeatures of membrane vesicles during dehydration [71, 116, 117].
Although only limited research has been done on the storage stability of differentinfluenza vaccine types an interesting trend can be noticed. The most successful sta-bilization studies have been performed with subunit preparations, which are relativelystable even at elevated temperatures (40°C). In contrast, WIV and virosomes incorpora-ted in a glassy sugar matrix showed to be less stable at comparable conditions. The maindifference between subunit (and split) vaccine compared to WIV and virosomes is thevesicular structure consisting of lipids, which may explain the less efficient stabilizationof the WIV and virosomes.
Lipids have a melting point (Tm) above which they are in the liquid crystallinephase; below this Tm they are in the gel phase. In the hydrated state, for example the Tmfor POPC (1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) is about -1°C and rises toabout 70°C when it is dried without sugars [118]. This is due to increase in Van der Waalforces between lipids upon removal of water molecules from the lipid headgroup region.As a result the bilayer undergoes a phase transition during rehydration, which may leadto disturbance of the vesicle (Fig. 11). However, certain carbohydrates can prevent thisphase transition, according to the water replacement hypothesis formulated by Crow etal. [73]. Carbohydrates containing many hydroxyl-groups can take over hydrogen bon-ding to a certain extent, thereby forcing themselves in between the lipid headgroups.This results in more space between the lipid molecules and reduction in Van der Waals
CHAPTER II
35

Fig. 11 Water replacement hypothesis for lipid bilayers. The diagram shows how trehalose is thought to stabilize dry lipid bilayers (adopted from Crowe et al. [73]).
36
forces, leading to lower phase transition temperatures. For example, the Tm for POPC inthe dry state in the presence of trehalose is lowered to -20°C [118]. In addition carbohy-drates that vitrify in between the bilayers (form glasses) tend to keep the membrane inthe phase it was at the moment of vitrification [119-121] and prevent aggregation/fusiondue to particle isolation [82].
Disaccharides and oligosaccharides like trehalose and inulin, respectively, are capa-ble of stabilizing lipid bilayers, when present on both sides of the lipid bilayer [73, 116-118, 120]. However, in the studies performed on stabilization of WIV and virosomes nosugar was inside the vaccine particle giving the change of (partial) reorganization of thelipids and subsequent phase transitions. As a result WIV and virosomes incorporated insugar glasses might have a reduced shelf-life compared to sugar glass incorporated subu-nit and split vaccines.
LLyyoopphhiilliizzaattiioonn RReehhyyddrraattiioonn((rroooomm tteemmppeerraattuurree))
DDeehhyyddrraattiioonn
DDeehhyyddrraattiioonnwwiitthh ttrreehhaalloossee
Hydrated
Damage
Retention

New needle-ffree influenza vaccine dosage forms
In this section, various needle-free delivery systems for influenza vaccines will be discus-sed. The development of most needle-free dosage forms are approached with use ofliquid influenza vaccines. In addition to the stability increase of dried vaccine formula-tions, the development of certain needle-free dosage forms requires the use of a dry-powder vaccine formulation. A dry vaccine powder for needle-free delivery can be idealfor several reasons. However, thus far the application of dry-powder vaccine in needle-free delivery technologies is limited. Therefore also needle-free delivery technologiesbased on liquid vaccine forms are discussed in this review. The needle-free delivery of vaccines can be divided by the route of administration: viathe skin (intradermal or transcutaneous) and via mucosal routes (nasal, pulmonary, oralcavity and oral). Each of these routes has their own advantages/disadvantages related tothe ease of vaccination and the type of immune response elicited.
Epidermal and transcutaneous immunizationUntil recently, the skin was considered as a barrier through which vaccines could not bedelivered. Currently, however, the skin is seen as a highly immune competent organbeing suitable for vaccine delivery by transcutaneous or epidermal immunization. Theskin is divided into three layers: the stratum corneum consisting of cornified keratinocy-tes, the epidermis consisting of live keratinocytes and an extensive network of immunecells; and the underlying dermis, which supports the epidermis with blood vessels, lym-phatics and other structures. The primary antigen-presenting cell (APC) type found inthe epidermis, the Langerhans cell, is a bone-marrow-derived DC. Langerhans cells areefficient APCs covering about 25% of the dermal boundary [122]. Upon encountering anantigen in the skin, epidermal Langerhans cells take the antigen up, migrate to a draininglymph node and induce a systemic immune response [123]. In addition activatedLangerhans cells produce cytokines that induce strong antigen-specific responses by B-and T-lymphocytes. Epidermal and transcutaneous immunization with influenza vacci-nes has also been shown to elicit mucosal immune responses (both IgG and IgA) at mul-tiple sites including the lungs and nasal cavity [84, 124, 125]. This is of particular impor-tance since antibodies at these sites give rise to protection at the port of entry of theinfluenza virus.
Penetration of the stratum corneumUnder normal circumstances, the stratum corneum acts as an effective barrier to thepenetration of foreign molecules. From literature it is evident that transdermal deliveryof most drugs and bioactive molecules larger than 500 Da to the blood vessels in the der-mis is not possible by diffusion and that delivery beyond the skin with needles is nee-ded. However, vaccine antigens merely require delivery to the more superficial epider-mis. So with merely penetration of the stratum corneum (recombinant) antigens as largeas 1 mD are capable to reach the epidermis and elicit immune responses [126].
CHAPTER II
37

Jet injectorsOne method to deliver vaccine is needle-free injection by delivery to epidermal (but alsodeeper subcutaneous or intramuscular) tissue using jet injectors. This method is especi-ally suitable for mass vaccination. Although the older multi-use nozzle jet injectors wereassociated with rare inadvertent transmission of blood-borne infections, recently twodisposable-cartridge jet injectors, Biojector®2000 (Bioject Medical Technologies,Bedminister, NJ) and Injex™ (Equidyne Systems, Tustin, CA), are approved by the FDA[127]. These devices have the same efficacy as the traditional influenza vaccinationmethod with needle and syringe [128] and make use of liquid vaccine. Recently a pow-der jet injector, PowderJect (PowderJect Pharmaceuticals PLC, Oxford, UK), has beendeveloped to deliver powder particles in the epidermis [129]. When the device is acti-vated one dose vaccine powder is accelerated to such a speed that the particles perforatethe stratum corneum and thus reach the epidermis [88]. Powders for epidermal immu-nization have been made by both desiccation/grinding/sieving and spray-freeze drying.Important factors for the design of these powders were a high particle density and a par-ticle size of 20-70 μm for effective penetration to the epidermis [130].
Animal studies in mice have demonstrated that epidermal powder immunizationwith a PowderJect device induces both antibody responses and protective immunityagainst homologous and heterologous challenge [88, 89]. In addition, upon co-admini-stration of adjuvant (like CTB and LT-mutants with reduced toxicity) a significantlylower dose of influenza vaccine was required to induce the same antibody response inboth mice and monkeys when compared with conventional i.m. injection [84, 89].
In a phase I clinical trial conducted by Dean et al. it was found that a powdered tri-valent influenza vaccine (as designed in: [85]) delivered by epidermal powder immuni-zation using the PowderJect ND5.2 delivery system (Fig. 12) is safe and elicits humoralimmune responses in humans [131]. This phase I clinical trial showed that seroconver-sion (75-100%) and geometric mean titers were equivalent or higher in epidermal pow-der immunized groups than in groups who received the vaccine by normal liquid injec-tion. However, since only studies with limited numbers of subjects have been performedmore data and clinical trials are needed to prove the efficacy and safety of this method.
PatchesAnother method to deliver antigens into the skin is transcutaneous immunization ortopical immunization using patches. In transcutaneous immunization penetration of thestratum corneum is facilitated by means of hydration and/or mechanical disruption.Hydration by occlusion or wetting the skin results in swelling of keratinocytes and poo-ling of fluid in the intracellular spaces, which allows antigens to pass through the stra-tum corneum more easily [123]. Glenn et al. demonstrated the concept of transcutane-ous immunization with a wet patch in humans. In a phase I trial vaccination of volun-teers with wet gauze containing E.Coli heat-labile enterotoxin (LT) under an adhesivepatch resulted in robust LT-antibody responses [132].
Topical delivery has also been facilitated by physical means of skin disruption suchas abrasion, tape stripping or electroporation [7]. The effect of pretreatment of the sha-
38

Fig. 12 PowderJect ND5.2 delivery system. The prototype single-use, disposable delivery device used forepidermal powder immunization. Powdered vaccine is housed in the cassette between polycarbonate membra-nes. After actuation the release of helium from the microcylinder ruptures the membranes of the vaccine cas-sette and propels the vaccine particles from the cassette through the nozzle. The particles are acceleratedto high velocity with enough force to penetrate the epidermal layer of the skin. (Reprinted from [131] withpermission from Elsevier)
ved skin on the immune response induced by a LT adjuvanted split-virus influenza vac-cine (A/Panama) was investigated in mice. The serum antibody titers to A/Panama wereincreased 100- to 300-fold by the combination of hydration and stratum corneum disrup-tion by emery paper, D-Squame tape or 3M tape compared to hydration alone [123]. Ina similar mouse study, transcutaneous immunization with WIV after mechanical skindisruption induced immune responses that protected against challenge [125]. In the samestudy, it was found that the penetration enhancers/immunomodulators, i.e. oleic acidand retinoic acid, enhanced protection and differentially affected the pattern of cyto-kine production upon stimulation with WIV.
Recently, a novel patch delivery technology, PassPortTMSystem, showed to be sui-table for transcutaneous immunization [133]. The system consists of an array of metallicfilaments, that creates 80 micropores within 1 cm2 area by thermal energy, which iscovered with a vaccine reservoir patch. The patch system loaded with a recombinant H5haemagglutinin vaccine formulation was used for transcutaneous immunization via theabdominal skin of mice. This immunization induced protective immunity against chal-lenge with an H5N1 virus.
In a clinical study it was evaluated whether transcutaneous immunization with anon-replicative adenovirus-vectored influenza vaccine expressing HA could induce animmune response [134]. The abdominal skin was pretreated by shaving with a razor andsubsequently 30 strokes with a toothbrush. The vaccine applied to the pretreated abdo-minal skin of healthy adults induced serum antibodies, but the responses were lowerthan those induced by intranasal administration.
Besides the use for direct vaccination with antigen, patches have also been appliedto stimulate or modulate the immune response of subcutaneous or intramuscularly admi-nistered influenza vaccines [123, 135-137]. An immunostimulant LT containing patchimproved the immune response to influenza vaccination in the elderly [137]. Elderlyreceived an i.m. injection with virosomal influenza vaccine proximal to a LT-containingpatch applied at skin which was disrupted by emery paper (after hydration). The reci-
CHAPTER II
39

pients of the LT-immunostimulating patch (IS™ patch, IOMAI) showed improved sero-conversion and seroprotection compared to elderly receiving vaccine alone.
MicroneedlesBesides the use for i.m. injection, traditional needles also have been used for penetrationof the skin to allow delivery into the dermis for a variety of vaccines [138]. Recently thisapproach has been shown to improve the immunogenicity of influenza vaccines and toallow reduction to 20% of the administered dose [139-141]. Special designed(micro)needles allow a controlled depth of penetration of the skin (painless), and havebeen found suitable for intradermal vaccination with influenza vaccines in preclinicalstudies [142]. Although this concept of targeting to the Langerhans cells showed attrac-tive results, there are (technical) challenges to ensure successful delivery into the appro-priate intradermal site.
In contrast to the use of one single needle, systems composed of arrays of pointedmicroneedles (as short as 25 μm) have been developed that can penetrate the stratumcorneum. These arrays are designed to target Langerhans cells in the epidermis, but donot reach the nerves in the underlying tissue [143]. As a result these microneedle arraysinduce minimal sensation and no pain as shown in preliminary clinical studies [144]. Anadditional advantage is the ease of administration, since these patches can be applied bypersons with minimal medical training [7]. Recently, a micro-projection array has beenused for the topical application of a recombinant anthrax antigen in rabbits and providedcomplete protection against inhalation challenge [145].
Recently micro-projection arrays have been developed to deliver solid antigens[147, 148]. A simple controllable method can be used to coat microneedles (Fig. 13) withdrugs, proteins, DNA, viruses and microparticles for delivery into the skin [146]. Tworecent studies have evaluated the ability of microneedles to deliver solid state vaccinesinto the skin of hairless guinea pigs. In the first, microneedles of a micro-projection arraysystem coated with a dry film of ovalbumin antigen penetrated the skin to an averagedepth of 100 μm which resulted in antibody responses [147]. In an additional study it wasshown that by adjustment of the array architecture and antigen coating procedure, thepenetration and amount of antigen delivered can be controlled [146]. The microneedlearray was coated with ovalbumin using aqueous solutions containing 2-20% ovalbumin,30-48% sucrose and 0.2% polysorbate 20. An overall delivery efficiency of 48-58% anti-gen could be reached using microneedles of 225-600 μm. Moreover, when the immuneresponse elicited by i.m. injection was compared to that of a microneedle array on theskin, the microneedle array technology demonstrated significantly higher antibody titersagainst ovalbumin. This technology might be applicable to deliver an influenza vaccineinto the skin by a dry, sugar-glass-stabilized, microneedle array patch.
40

Fig. 13 Out-of-plane microneedle array uniformly coated with model-vaccin (colored yellow). Imaging bybrightfield microscopy shows coating of microneedle shafts of an array of 50 microneedles (Image: HarvinderGill, adapted from: http://www.newswise.com/articles/view/533982/#imagetop ).
The mucosal routeMucosal surfaces of the body are highly immunologic organs, which perform constantsurveillance for foreign antigens. As a result all mucosal surfaces including nasal, pulmo-nary, oral, rectal and vaginal mucosa are considered as potential sites for vaccination.However, most research focus on nasal, pulmonary and oral delivery due to practical rea-sons and/or expected lack of cultural acceptance of rectal and vaginal delivery.
The mucosal immune systemThe mucosal membranes are the port of entry and main site of infection for most patho-gens and several mechanisms protect these surfaces from pathogens. Mucus forms a phy-sical barrier and different enzymes kill or inhibit the growth of pathogens. Moreover,mucosal-associated lymphoid tissue (MALT) protects the mucosa by discriminatingbetween harmless antigens (e.g. food) and pathogens. MALT is situated along the muco-sa of the nose, lungs and gastrointestinal tracts and includes nasopharyngeal-associatedlymphoid tissue (NALT), bronchus-associated lymphoid tissue (BALT) and gut-associa-ted lymphoid tissues (GALT, including Peyer's patches), respectively.
After invasion of pathogens via inhalation, ingestion or sexual contact the mucosalimmune system is stimulated at the mucosal inductive sites (also known as IgA induc-tive sites) by uptake of antigens through a unique set of antigen-sampling cells. Thesemicrofold cells (M-cells) are located on mucosal surfaces overlying the MALT. After suc-cessful uptake at the inductive site, the antigens are immediately processed and presen-ted by macrophages and DCs resulting in the development of effector and memory B andT cells. These antigen-specific B and T cell populations than migrate from the inductivesite via lymphatic drainage, circulate through the bloodstream and most of them returnto the area from which they originated. However a part of the antigen-specific B and Tcell populations home to distant mucosal effector sites including the lamina propriaregions of the respiratory, intestinal and reproductive tracts [149-151]. At the originaltissues and the more diffuse mucosal tissues, antigen-specific IgA-committed B, Th1 andTh2 cells interact resulting in generation of IgA at the mucosal site. Consequently, secre-
CHAPTER II
41

ted IgA acts as the first line of protection at mucosal surfaces. It is generally assumed thatIgA is the main antibody isotype and effector molecule in host defense at mucosal surfa-ces [152].
In addition to the elicited mucosal responses, parts of the systemic immune respon-se cascade can be stimulated, including the production of serum antibodies, lymphocyteproliferation, cytokine production and CTL activity, thereby providing protectionagainst systemic infection upon the next challenge.
Mucosal immunity at the port of entry of influenzaIn humans, the primary targets for influenza virus are the mucosa in the upper and lowerrespiratory tract. After infection with influenza virus, the immune system reacts to repli-cation of the virus in the respiratory tract by both innate and adaptive immune respon-ses. Antibody and T cell responses are triggered against epitopes located on the virusenvelope such as HA [153]. The antibodies at the mucosal tissues, locally produced IgAand IgG transudated from serum, together with local IgA-committed memory cells maygive rise to mucosal immunity [4]. This mucosal immunity in the (upper) respiratorytract is of major importance as it provides the first line of defense against homologousvirus [25] and possibly also drift virus variants [154-158].
Most currently marketed vaccines are administered by i.m. or s.c. injection bywhich protective immunity in the systemic compartment is induced. In addition, virus-neutralizing antibodies from the circulation can protect against severe pneumonia as aresult of transudation from the blood into the lungs [34, 35]. However, these vaccines failto stimulate the mucosal immune system of the respiratory tract, the main port of entryfor influenza virus, which provides the first line of defense against infection [4, 159-162].Especially secretion of mucosal IgA in the nose (upper respiratory tract) is desired, as itmay prevent infection by incoming viruses. Induction of nasal immunity might improvethe efficacy of current (parenterally delivered) vaccines and protect against infection atthe site of invasion.
Mucosal vaccinesInduction of mucosal immunity generally requires direct application of vaccine to themucosal surface for stimulation of the specific local immune mechanisms of the MALT.Mucosal influenza vaccines aim at the induction of mucosal, systemic humoral as well ascell-mediated immune responses. A mucosal influenza vaccine could be ideal for sever-al reasons. First mucosal influenza vaccines may provide several potential advantages,such as eliminated pain at the injection site, easier and faster vaccine distribution andadministration, and reduced costs [6, 7, 163]. Moreover, mucosal vaccination can resultin a mucosal immune response in the respiratory tract, which might give protectionagainst influenza infection at the port of entry. Finally, since mucosal IgA responses havebeen shown to exhibit cross-protective immunity against antigenically distinct viruses[154, 157], such a mucosal immune response induced by mucosal vaccination might offerbroader protection against drifted, heterologous strains.
42

Nasal deliveryUntil now vaccination via the intranasal route (i.n.), targeting the NALT, is the onlymucosal route that has been successfully applied for influenza vaccines. Today the onlyintranasal vaccine in the market is a live cold-adapted trivalent influenza intranasal vac-cine approved by the FDA in 2003 (Flumist™). This vaccine is used in healthy personsfrom 5 to 49 years and administered via a pre-filled single use device that sprays vaccineinto the nostrils. Flumist™ has shown to induce both mucosal and systemic immunity inhumans. However, this vaccine induces lower serum antibodies than conventionalparenteral inactivated vaccines [28, 54].
A disadvantage of live cold-adapted influenza vaccines is that there are a number of(hypothetical) safety concerns, e.g. their safety is undetermined in immunocompromi-sed patients, the attenuated vaccine strain may reassort with other influenza viruses, pos-sible risk of vaccine-induced central nervous system complication [54]. The use of inac-tivated intranasal vaccines may be an alternative which combines the convenience ofintranasal administration and the safety of inactivated intramuscular vaccine [54].
A disadvantage of intranasally delivered pure influenza antigens (HA + NA) is thatthey are poorly immunogenic without the use of special delivery systems and/or muco-sal adjuvants [164]. Therefore, in recent preclinical and clinical studies investigators ex-amined the suitability of a number of candidate vaccines which are adjuvanted or spe-cially formulated. The most widely used mucosal adjuvants in animal studies are chole-ra toxin (CT) from Vibrio Cholerae and the closely related E. coli heat-labile enterotoxin(LT). These toxins consist of a pentameric B subunit oligomer linked to a single A sub-unit. In particular the A subunit is associated with both toxicity and adjuvant effects. Thetoxins increase permeability of the epithelium, leading to enhanced uptake and presen-tation of co-administered antigen, promotion of isotype differentiation in B cells leadingto an increased IgA formation and complex stimulatory as well as inhibitory effects onT-cell proliferation and cytokine production [10]. The adjuvant effect occurs only whenCT or LT is administered simultaneously with an antigen and by the same route [165].Although LT and CT are closely related molecules, important differences exist in theadjuvant effects that they exhibit. Use of CT as an adjuvant generally results in the induc-tion of antigen-specific Th2 cytokine production, while LT elicits both Th1 and Th2cytokines [8, 166].
Enterotoxins have been applied for nasal vaccination with inactivated influenzavaccines. In preclinical and clinical trials LT has been shown to improve the immune res-ponse of subunit vaccines [155, 167], virosomes [168, 169] or antigen admixed with hya-luronic acid biopolymer [87]. In 2001, NasalFlu® (Berna, Switzerland), a nasal virosomalvaccine containing LT was approved in Switzerland [168, 169]. However, soon afterintroduction this vaccine had to be withdrawn due to serious side effects [29]. Duringpost marketing surveillance, an association between the nasal vaccine and Bell's palsy, anadverse neurological event, was found [170]. It has been suggested that LT was redirec-ted into the brain resulting in the involvement of the central nervous system in Bell'spalsy [171, 172].
Since CT and LT are considered to be toxic for humans, non-toxic variants of CT and
CHAPTER II
43

LT are currently in development [10]. Isolated B subunits of CT and LT (CTB and LTB)have been explored for their ability to augment immune responses against co-administe-red antigens [173-176]. Although, their capacity as mucosal adjuvants was much lessthan that of holotoxins [10], some studies showed that addition of detoxified forms of LTcould improve the immunogenicity of influenza vaccine upon nasal vaccination withvaccine alone [155, 177] or in combination with a novel biovector (a nanoparticulate bio-adhesive delivery system with a positively charged polysaccharide core enclosed by aphospholipid-cholesterol double layer) [178].
In addition to the use of toxins, other approaches for nasal vaccination involve theuse of glycolipids [179, 180], proteasome [181], immunostimulating complexes (ISCOMs)and chitosan. Youn et al. showed that a single intranasal immunization of mice withinactivated influenza virus and α-galactosylceramide induces long-term immunity thatgives protection against challenge with live influenza virus. In addition, α-galactosyl-ceramide did not redirect antigens (both WIV and ovalbumin) into the brain as revealedby tracking studies using antigens labeled with the chemoluminescent-tag Acridium[180]. Intranasal administration of proteasomes containing HA and purified outer mem-brane proteins extracted from N. Meningitidis have been shown to be well tolerated inadults and capable to induce both nasal IgA as well as serum antibodies [181]. Coulter etal. showed that it may be possible to induce effective immunity to influenza usingISCOMs intranasally [182]. ISCOM complexes are 40 nm cage-like structures built upfrom subunits formed by the interaction of Quil-A with cholesterol. By addition of phos-pholipids, hydrophobic and amphiphilic antigens can be incorporated into ISCOMs byhydrophobic interactions during the assembly of the subunits [183]. In mice and sheepintranasal delivery of inactivated influenza vaccine mixed with ISCOMATRIX® (emptyISCOM) adjuvant, was able to induce serum HI titers higher than those obtained withnon-adjuvanted vaccine delivered subcutaneously. Furthermore, the ISCOMATRIX®
adjuvanted vaccine delivered intranasally induced mucosal IgA responses in the lung andnasal passages. Various studies have been performed using chitosan or chitosan deriva-tives, a mucoadhesive biopolymer, as nasal adjuvant for inactivated influenza vaccine[184-187]. Chitosan is supposed to have mucoadhesive properties enhancing the resi-dence time in the nose and enhance the penetration capacity across mucosal barriers[188]. The use of such mucoadhesive polymers in nasal dosage forms that increase theresidence time within the nasal cavity provides the opportunity for sustained nasal drugdelivery [188]. In addition, nanoparticulate dispersions based on chitosan are believed totarget the lymphoid tissue by the M-cells, resulting in an appropriate immune response[186, 189]. In clinical trials it was shown that sufficient seroprotection and serocon-version rates are possible by nasally vaccination with a combination of chitosan and tri-valent inactivated influenza [184, 187]. Although many adjuvants showed in some casespromising results there is still a search for new, improved and safe adjuvants.
Besides liquid formulations, also powders have been developed for nasal delivery ofvaccines [92, 190-192]. In addition to the fact that powders offer the advantage of impro-ved vaccine stability [7, 193], certain powder formulations also have demonstrated in-creased residence time in the nasal cavity compared with liquid [194, 195], which may
44

translate into higher bioavailability and immune responses. Scintigraphic evaluation inrabbits of nasal drug delivery systems based on carbopol 971p and carboxymethylcellu-lose showed increased residence times upon addition of these mucoadhesive polymers[196].
A range of devices have been developed for intranasal delivery of powders [190]. Aprototype system for delivery of a lyophilized powder has been described that offers theadvantage that a powder can be metered as a liquid and lyophilized directly in the de-vice [197]. When particles larger than 50 μm are used, intranasally delivery is highlyreproducible and independent of the vaccinee's control of breathing, because depositionis governed by inertial impaction [198]. However, due to nasal anatomy and physiology,with a non-ciliated area in the anterior part of the nasal cavity and a ciliated region inthe more posterior part of the nose, the site of deposition is of importance in view ofmucociliary clearance of a formulation from the nose. In a study of Tafaghodi et al. it wasshown that the limiting step for nasal clearance of particulate systems is their disloca-tion from the initial site of deposition [199]. The site of deposition depends on severalparameters which are related to the delivery device and formulation, such as velocity ofthe delivered particles and particle size of the formulation [197]. Therefore, the rightcombination of formulation (particle characteristics) and delivery device is important todeposit the particles at the desired location.
A few studies report the development of dry-powder vaccine formulations for intra-nasal delivery. However, dry-powder formulations for intranasal delivery conductedthus far could only elicit a sufficient immune response when they were co-formulatedwith a mucoadhesive polymer or mucosal adjuvant [91]. Huang et al. described an intra-nasal powder formulation of WIV and a novel intranasal delivery device. A dry-powderof WIV and trehalose was prepared by lyophilization and the lyophilized cake was re-duced to sizes suitable for nasal delivery. The final dry-powder formulation consisted ofvaccine powder and a mucoadhesive compound in the size range of 45 to 125 μm [92].The powder-formulated vaccine, elicited a significant serum antibody response in ratsthat was at least as strong as that provided by the liquid vaccine administered intranasal-ly or via i.m. injection. Moreover, significant nasal IgA responses were observed afterintranasal delivery [91].
Pulmonary deliveryPulmonary delivery of influenza vaccine is a potential alternative for today's influenzavaccination. The lungs are highly vascularized, have a large absorptive surface area [101,107, 190, 200] and contain BALT [9]. Furthermore, local APCs like different types ofmacrophages and DCs are ideally located for antigen sampling and subsequent presenta-tion to T cells [201-203]. As a result, the combination of the unique anatomical/physio-logical features of the lungs and the possibility to expose a relative large mucosal surfaceto vaccine facilitates an effective uptake of antigen by many different types of APCs.Therefore, pulmonary delivery of an influenza vaccine is an interesting strategy. In addi-tion, pulmonary immunization by targeting the BALT can offer the advantage of indu-cing both systemic immunity as well as local immunity in the respiratory tract [151].
CHAPTER II
45

Pulmonary delivery of vaccines against measles has been extensively investigated.The effectiveness of measles vaccination by inhalation has been shown in a number ofclinical studies, with pioneering studies performed already in the 1960s [204]. Severalclinical studies have shown that attenuated measles vaccines administered by inhalationwere at least as effective as vaccine given via the conventional parenteral route [205].
Pulmonary delivery of vaccines against influenza has already been investigated in anumber of older clinical studies. Although most of the studies lack efficient targeting ofsolely the lungs, some general trends can be seen. The research group of Waldman eva-luated aerosol immunization with a classical inactivated influenza vaccine [206-208].The vaccine was sprayed into the posterior oropharynx during rapid, deep inspirationfollowed by spraying 0.125 ml liquid vaccine into each nostril, with the use of an atomi-zer which emits droplets ranging in size from 1 to 100 μm. In one of their studies, theseinfluenza vaccine aerosols gave a lower over-all protection rate compared to injectionadministration [206]. A booster dose, however, seemed to have a marked effect.
In follow-up studies it was shown that respiratory immunization prevented illnessin humans [209]. In addition, aerosol immunization in humans with inactivated influ-enza vaccine induced higher levels of respiratory secretion antibody than subcutaneousimmunization [207, 210]. Interestingly, these respiratory antibodies were also morecross-reactive with heterologous viruses [211].
The efficacy of influenza immunization by inhalation is further supported by stu-dies of Haigh et al. [212, 213]. A group of volunteers who received WIV by inhalationusing a pressurized metered dose inhaler (361 subjects) had the same incidence of influ-enza illness compared to the group receiving WIV by hypojet gun (706 subjects), whichwas significantly lower than the incidence in the control group (361 subjects) [213]. Thisresult was confirmed in another study performed by Waldman et al. [214]. A further cli-nical study showed that inhaled inactivated vaccine can provide protection against aheterologous variant [212]. The incidence of influenza-related sickness was assessed in1007 male subjects immunized by inhaled inactivated vaccine and 1007 unimmunizedsubjects. The vaccine contained A2/Aichi/68 whereas the circulating virus was exclu-sively A/England/42/72. The protection rate of the trial was 47% as opposed to approxi-mately 60% in previous trials using the same vaccine but in which no variant was knownto have been encountered. These results confirm that cross-reactivity of the respiratoryantibodies elicited by inhaled influenza can be achieved.
Recently, the pulmonary route has been re-explored for vaccination against influ-enza in various pre-clinical studies. A comparison between pulmonary immunization(intratracheal) with nasal immunization using WIV and influenza subunit vaccine show-ed that pulmonary immunization was more effective in inducing local and systemicimmune responses [11]. A more recent study by Minne et al. assessed the impact of anti-gen distribution within the respiratory tract on the immune response to a monovalentinfluenza split virus vaccine administered to mice that were previously intranasally pri-med with WIV [215]. By variation of the administration technique the liquid vaccinewas targeted to different sites of the mouse respiratory tract, i.e. the nasal cavity, theupper or central airways, or the deep lungs. Delivery of the vaccine to the different res-
46

piratory regions generated systemic, mucosal (local) humoral and cellular virus-specificimmune responses which increased upon increasing the depth of vaccine deposition.Deep-lung vaccination resulted in systemic neutralizing antibodies similar to intra-muscular vaccination. In addition, immunization via the deep-lung in stead of i.m. resul-ted in Th1 skewing of the cellular immune response. Minne et al. explained the higherefficacy of deep-lung vaccination by the prolonged residence time of the vaccine in thelungs.
A few studies report the development of dry-powder vaccine formulations for pul-monary delivery of influenza vaccine. A spray-dried lipid formulation containing WIVor split influenza vaccine has been evaluated for mucosal vaccination via the pulmonarytract. The inhalation powder contained the two lipophilic surfactants (lipids), DPPC andDSPC, and the hydrophilic surfactant tyloxapol for targeting phagocytic APCs andenhancing antigen release, respectively [11]. Pulmonary delivery of the powders contai-ning WIV in naive rats induced substantial systemic immune responses, but no mucosalIgA in the respiratory tract. In contrast to microparticles containing WIV, microparticlescontaining the subunit vaccine powder showed enhanced serum antibody levels uponpulmonary delivery that were even higher than those induced by i.m. vaccination.
Recently, pulmonary vaccination with a new influenza subunit vaccine powder wasevaluated in mice [216]. Vaccine powder was produced by spray-freeze drying (SFD)using inulin as stabilizer. The powder possessed a large particle size distribution in orderto target both central and deeper lungs. Pulmonary vaccination of mice with the vac-cine powder induced mucosal, systemic humoral as well as cell-mediated immune res-ponses. These responses were superior to those elicited by conventional i.m. vaccinationor pulmonary vaccination with a liquid aerosolized subunit vaccine. The superiority ofthe SFD vaccine powder compared to the aerosolized liquid subunit vaccine can beexplained by:
- the lower respiratory tract deposition of the powder and resulting increased residence time in the lungs,
- the inulin may increase viscosity at the site of deposition which increases the residence time, and
- the solid inulin may have an adjuvant effect.
The adjuvant activity of inulin was mentioned in several papers [217-219]. In thesepublications it was explicitly the inulin gamma-crystal that was assigned to have adju-vant activity. However, in the system used by Amorij et al. [216] amorphous inulin par-ticles were used. In addition alveolar deposition, lacks mucociliair clearance, which mayhave resulted in an increased residence time of the vaccine [215].
From the studies described above it can be concluded that pulmonary immuniza-tion is a highly promising route for vaccination. Vaccination via the pulmonary route hasbeen shown to induce broad immune responses in preclinical studies. However, clinicaltrials are needed to prove the efficacy and safety of pulmonary vaccination in humans. These clinical trials should be performed using recently designed inhalers which guaran-tee high and reproducible lung deposition of the vaccine. There are three types of inha-
CHAPTER II
47

The use of dry-powder inhalers seems to be the most suitable option. First, dry-powder inhalation would overcome instability problems when a suitable stable dry-pow-der formulation is used [221, 222]. In addition, dry-powder inhalation would be prefer-red over administration of liquid aerosols, since dry-powder inhalation has been shownto be more reproducible and efficient. Furthermore, a dry-powder would provide theopportunity to design aerosols, particles, with the desired properties for inhalation toensure efficient targeting of the lung areas of interest. Some recently developed inhalersmight be suitable for vaccination purposes. For example De Boer et al. recently develo-ped a single-use disposable inhaler, the TwincerTM (Fig. 14), that combines low costs,good dispersion of high powder doses and good moisture protection of the powder for-mulation [223].
Oral deliveryOral delivery of influenza vaccines is an attractive mode of immunization because oraldosage forms are easy to administer and well accepted by the population. Orally delive-red vaccines aim at targeting the mucosal inductive site of the gastrointestinal (GI) tract,the GALT, which consists of Payer's patches, appendix, solitary lymphoid nodules and
48
Fig. 14 An example of a disposable DPI, the TwincerTM.
lation devices: nebulizers, medical metered dose inhalers (MDIs) and dry-powder inha-lers (DPIs). However, not all systems seem suitable for vaccination purposes. Nebulizersare associated with a time consuming administration, high drug losses and a very poordrug delivery to the respiratory tract and they use high shear stresses which can lead toprotein denaturation. MDIs utilize propellants (chlorofluorocarbons and increasinglyhydrofluoroalkane) and atomization of the vaccine solution. Vaccine compounds mightbecome susceptible to denaturation or degradation when they come into contact withthese propellants or with the large air-liquid interfaces during aerosolization. The use ofso-called "soft mist inhalers" might be an interesting strategy since they apply the con-cept of low-velocity aerosol in combination with the generation of monodisperse aero-sols [75, 220]. Moreover, in contrast to pressurized metered dose inhalers, soft mist inha-lers can directly aerosolize the aqueous vaccine formulation without the use of propel-lants that may affect the integrity of the influenza vaccine [75]. However, next to the cli-nical trials in humans that are needed to prove efficacy, it will first have to be investi-gated whether the vaccine is resistant against the aerosolization.

isolated lymphoid follicles. The surface of Payer's patches is covered by follicle-associtedepithelium (FAE) enriched with specialized antigen-sampling cells, microfold or mem-branous cells (M cells). These M cells are supposed to deliver intact antigen to the under-lying APC. After presentation of processed antigen by DCs, Th cells become Th1 and/orTh2 cells. These matured Th cells are subsequently responsible for the induction andregulation of antigen-specific cell-mediated immunity and IgA responses.
Today, only a few vaccines are administered orally and most of them are based onlive attenuated pathogens [6]. The most prominent oral vaccine is the Sabin oral poliovaccine, that was brought on the market in the early 1960s, and had an important rolein the program for global eradication of polio [6]. However, live vaccines have a theo-retical risk of virulence, and occasionally they have other undesired vaccine-related sideeffects. For example, the use of live attenuated polio vaccine was discontinued in theUnited States because of rare cases of vaccine-associated paralytic polio and was replacedby inactivated poliovirus vaccine [6].
Until now no effective vaccine for oral influenza vaccination has been licensed.However, over the past decades, a number of clinical studies with oral influenza vac-cines have been performed.
Moldoveanu et al. immunized healthy adults with 10 doses (total 150 μg HA/strain)of trivalent split inactivated influenza vaccine on five consecutive mornings [90]. Toensure that the vaccine was delivered to the small intestine, the vaccine was air-driedwith D-xylose and packed into enteric-coated gelatin capsules. Following oral immuni-zation, serum antibodies of all isotypes were only slightly increased 21 days after in-gestion of the vaccine. However, in external secretions (saliva and nasal lavage), antigen-specific IgA and IgG responses were detected.
In another study it was shown that oral immunization of 5 volunteers with enteric-coated alum-absorbed WIV vaccine on day 1, 3, 5, 8, 10 and 12 (6x4 μg HA) resulted ina significant rise of IgA-specific antibodies in tears, saliva and nasal secretion [224]. Thesecretory antibody response was slow and reached a maximum 5-7 weeks after com-pletion of immunization. However, serum antibody titers were not increased.
Oral immunization with split inactivated influenza vaccine formulated as an emul-sion was investigated in humans by Avtushenko et al. [225]. Volunteers were immuni-zed once or twice (at an interval of 14 days) with 140 μg HA. Only after one oral admi-nistration the vaccine induced reliable increases in the level of secretory IgA in nasalsecretions and saliva of volunteers. After 2 immunizations however, this response wasnot found. The authors explained this remarkable effect with oral tolerance caused byrepeated ingestion of significant doses of antigen. It was concluded that the dose of anti-gen needs to be optimized to ensure a maximal and safe immune response.
Although in most of the clinical studies relatively high doses of antigen were orallyapplied [86, 90, 225] in order to overcome limited absorption by M cells. These immuni-zations all resulted in IgG responses below detection level. In contrast, most studiesdemonstrate a significant increase in IgA antibodies in both saliva and nasal lavage fluids.It is unknown whether these IgA antibodies alone could provide adequate protectionagainst influenza infection in humans. Renegar et al. demonstrated in a preclinical study
CHAPTER II
49

in pre-challenged mice that intranasal instillation of anti-influenza IgA but not anti-IgGor anti-IgM antiserum (for neutralization of the specific antibodies in the nasal secre-tions) could abrogate the immunity against re-challenge with influenza virus [226]. IgAwas the major (if not the sole) mediator that provided protection against nasal challengewith influenza virus. Moreover, influenza resistance of an organism has been shown todepend on the titer of secretory antibodies rather than on the titer of serum antibodies[154, 156, 157]. Therefore, sufficient protection might be achieved by mucosal IgA indu-ced by oral immunization. However, to our knowledge, no clinical study has beendesigned so far to reveal the level of protection provided by orally-induced local IgAimmune responses in humans.
Regardless of the possibility to induce effective mucosal immune responses, influen-za vaccines have to evoke an adequate level of virus-neutralizing antibodies in the serumto fulfill current regulatory requirements for vaccine immunogenicity [29, 227]. In orderto achieve satisfying serum antibody levels current preclinical research on oral immuni-zation with influenza vaccines mainly focus on the use of adjuvants and complex vac-cine formulations. Studies in mice have demonstrated that live recombinant vaccines andWIV vaccines represent potentially promising oral vaccines [228, 229]. These oral vac-cines were co-formulated with adjuvants, such as CT and LT [175, 176, 230]. Besides theaddition of adjuvants, also more complex vaccine formulations have been developed[231, 232]. Carrier systems like bilosomes [233, 234], chicken erythrocyte "ghosts" [185,235], CT/CTB conjugated liposomes [236] and biodegradable microparticles [90, 237]have been demonstrated to improve an orally induced immune response directed againstinfluenza. However, as discussed before, there is still a need for new improved adjuvantsthat can safely be used in humans.
A few studies on oral influenza vaccine development paid attention to the deliverysite of the vaccine. Meitin et al. reported that intrajejunal administration of a live recom-binant vaccine virus consistently induced immunity in mice, while intragastric admini-stration was much less reliable in inducing this immune response [228]. Also for a sub-unit vaccine it was found that the delivery site within the gastro-intestinal tract had dif-ferent impacts on the immune response [238]. Moreover, co-administrating LT enhan-ced the immune responses in different ways. Intracolonic administration of vaccine withLT resulted in enhanced cellular immune responses and the desired Th1-skewing ofthese responses. Although intragastric administration of the adjuvanted vaccine also in-creased T-helper responses, no Th1-skewing was present. It was concluded that the rightcombination of a strong mucosal adjuvant and proper antigen delivery site within thegastro-intestinal tract might result in effective vaccination via the oral route. Until nowno clinical trials have been assessed to reveal the most optimal delivery site for vaccinein the gastro-intestinal tract. For these assessments tablets or capsules containing driedvaccine and supplied with special coatings that enable targeting to different GI-sites[239-242] should be developed. For example, Schellekens et al. developed a pH-control-led pulsatile delivery system in order to target specific sites in the gastro-intestinal tract[241, 242]. The pH-controlled pulsatile release system comprised a pH-sensitive coatingmaterial wherein a swellable agent is embedded. This method may improve vaccines that
50

are degraded and/or poorly immunogenic in the upper part of the GI-tract by specificpulsatile delivery to the colon, partly because of the lower levels of digestive enzymes ascompared with the small intestine.
Oral-cavity deliveryDelivery via the sublingual, buccal or gingival route, all aim at targeting the mucosa-associated lymphoid tissue of the oral cavity. In comparison to the gastrointestinal tract,the oral cavity provides an environment that is almost free from acidity and proteaseactivity which reduces risks for degradation of the vaccine compound [243]. The oralcavity has a full complement of immune cells, both intra-epithelially and submucosally[244]. The lymphoid tissues in the oral cavity are like the nasopharyngeal tonsils part ofthe Waldeyer's ring (palatine, lingual tonsils and adenoids). Despite decades of worktowards understanding immune mechanisms in the blood and secondary lymphoid tis-sues, only limited information is available about the immunological processes within theoral cavity [245, 246]. In particular the importance of the palatine and lingual tonsils inthe induction and contribution of specific antibody responses in humans is not clear[247].
In spite of this, the sublingual administration for allergen-specific immunotherapyhas raised attention. This route was investigated in a number of clinical trials [248].These studies indicated that the oral cavity might be a suitable target site for vaccines totrigger the immune system. In addition tonsillar immunization of rabbits with killedCandida albicans has been shown to induce salivary antibodies [249]. Recently, oralspray immunization was evaluated in a phase I/II trial [250]. Healthy adult volunteerswere immunized four times at 1-week intervals with WIV, i.e. 4 x 90 μg HA. The vac-cine was administered by a spray (100 μl) into a wide open mouth using a conventionalspray bottle. Although no significant increase in salivary IgA antibodies was found, theoral spray immunization induced serum HI antibodies in 75% of the volunteers alreadyafter 2 doses. A critical note with respect to this study is that the authors did not ensurethat no aerosol particles were inhaled. Therefore, it cannot be excluded that a part of thefound immune response is caused by pulmonary deposited WIV. However, delivery ofan influenza vaccine in the oral cavity, e.g. by sublingual or buccal tablets or patches,might be an interesting alternative route for vaccine delivery. In addition delivery in theoral cavity offers the opportunity to use mucosal adjuvants that are not yet suitable fornasal and/or pulmonary use.
CHAPTER II
51

Summary
This paper reviews the perspectives of current influenza vaccines and its administration.Attention is given to the different influenza vaccine types used today, the rational andneed for stabilized influenza vaccines and strategies by which influenza vaccines can bestabilized. Furthermore the status of stabilized/solid influenza vaccines and the currentstatus of various needle-free dosage forms for influenza vaccination are reviewed.
From the limited investigations performed so far it was concluded, that the integri-ty and stability of the dried vaccine depends on the type of vaccine, the used excipientsand the production (drying) process. However, more research is needed to get a com-plete understanding of the formulation and process design that provides successful vac-cine stabilization. Currently a variety of strategies that potentially may lead to needle-free dosage forms are under investigation. However, for most of these strategies a lot ofadditional (clinical) research is required before any definite statements on their useful-ness in man can be made.
52

CHAPTER II
53
ABBREVIATIONS
APC antigen presenting cellBALT bronchus-associated lymphoid tissueCA cold-adaptedCT cholera toxin from Vibrio CholeraeCTB isolated B unit of CTCTL cytotoxic T lymphocyteDC dendritic cellDPI dry-powder inhalerFAE follicle-associated epitheliumGALT gut-associated lymphoid tissueGI-tract gastrointestinal tractHA haemagglutininHI haemagglutination-inhibitionHES hydroxyethylstarchi.m. intramusculari.n. intra nasalISCOM immunostimulating complexICHLAIV live attenuated influenza vaccineLT heat-labile enterotoxin of E.ColiLTB isolated B unit of LTM1 matrix protein of influenzaM2 proton channel of influenza AMALT mucosal-associated lymphoid tissueM cell microfold cell, membranous cellMDCK Madin-Darby-Canine-KidneyMDI medical metered dose inhalerMHC major histocompatibilityNA neuraminidaseNALT nasopharyngeal-associated lymphoid tissueNB protonchannel of influenza BPBS phosphate buffered salinePc critical pressurePOPC 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine RH relative humiditySDLM spray dried lipid based micro particlesSC-CO2 supercritical CO2SCF supercritical fluidSFD spray-freeze dryingTc critical temperatureTg glass transition temperature Tg' glass transition temperature of the maximal freeze-
concentrated fractionTm melting temperature, melting pointTe eutectic temperatureVVM vaccine vial monitorWHO World Health OrganizationWIV whole inactivated virus

REFERENCES
[1] WHO. WHO Media Influenza Factsheet N°211, 2003.[2] Suzuki Y. Sialobiology of influenza: molecular mechanism of host range variation of influenza viruses.
Biol Pharm Bull 2005;28(3):399-408.[3] Stephenson I, Nicholson KG, Wood JM, Zambon MC, Katz JM. Confronting the avian influenza threat:
vaccine development for a potential pandemic. Lancet Infect Dis 2004;4(8):499-509.[4] Cox RJ, Brokstad KA, Ogra P. Influenza virus: immunity and vaccination strategies. Comparison of the
immune response to inactivated and live, attenuated influenza vaccines. Scand J Immunol 2004;59(1):1-15.
[5] The European Agency for the Evaluation of Medicinal Products (EMEA) Committee for Proprietary Medicinal Products (CPMP). Guideline on dossier structure and content for pandemic influenza vac-cine marketing authorisation application. CPMP/VEG/4717/03, 2004.
[6] Mitragotri S. Immunization without needles. Nat Rev Immunol 2005;5(12):905-16.[7] Giudice EL, Campbell JD. Needle-free vaccine delivery. Adv Drug Deliv Rev 2006;58(1):68-89.[8] Freytag LC, Clements JD. Mucosal adjuvants. Vaccine 2005;23(15):1804-13.[9] Holmgren J, Czerkinsky C. Mucosal immunity and vaccines. Nat Med 2005;11(4 Suppl):S45-53.[10] Holmgren J, Czerkinsky C, Eriksson K, Mharandi A. Mucosal immunisation and adjuvants: a brief
overview of recent advances and challenges. Vaccine 2003;21 Suppl 2:S89-95.[11] Smith DJ, Bot S, Dellamary L, Bot A. Evaluation of novel aerosol formulations designed for mucosal
vaccination against influenza virus. Vaccine 2003;21(21-22):2805-12.[12] Wilschut J, McElhaney JE, Palache AM. Rapid Reference Influenza. 2nd ed. London: Mosby/Elsevier
Science, 2006.[13] Wilson IA, Cox NJ. Structural basis of immune recognition of influenza virus hemagglutinin. Annu Rev
Immunol 1990;8:737-71.[14] Wiley DC, Wilson IA, Skehel JJ. Structural identification of the antibody-binding sites of Hong Kong
influenza haemagglutinin and their involvement in antigenic variation. Nature 1981;289(5796):373-8.[15] Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemag-
glutinin. Annu Rev Biochem 2000;69:531-69.[16] Ito T, Couceiro JN, Kelm S, Baum LG, Krauss S, Castrucci MR, et al. Molecular basis for the generation
in pigs of influenza A viruses with pandemic potential. J Virol 1998;72(9):7367-73.[17] Matlin KS, Reggio H, Helenius A, Simons K. Infectious entry pathway of influenza virus in a canine
kidney cell line. J Cell Biol 1981;91(3 Pt 1):601-13.[18] Lakadamyali M, Rust MJ, Zhuang X. Endocytosis of influenza viruses. Microbes Infect 2004;6(10):929-
36.[19] Smith AE, Helenius A. How viruses enter animal cells. Science 2004;304(5668):237-42.[20] Helenius A, Mellman I, Wall D, Hubbard A. Endosomes. Trends in Biochemical Sciences
1983;8(7):245-50.[21] Earp LJ, Delos SE, Park HE, White JM. The many mechanisms of viral membrane fusion proteins. Curr
Top Microbiol Immunol 2005;285:25-66.[22] Jardetzky TS, Lamb RA. Virology: a class act. Nature 2004;427(6972):307-8.[23] Amano Y, Cheng Q. Detection of influenza virus: traditional approaches and development of biosensors.
Anal Bioanal Chem 2005;381(1):156-64.[24] Ligon BL. Avian influenza virus H5N1: a review of its history and information regarding its potential to
cause the next pandemic. Semin Pediatr Infect Dis 2005;16(4):326-35.[25] Wong SS, Yuen KY. Avian influenza virus infections in humans. Chest 2006;129(1):156-68.[26] Luke CJ, Subbarao K. Vaccines for pandemic influenza. Emerg Infect Dis 2006;12(1):66-72.[27] Fauci AS. Pandemic influenza threat and preparedness. Emerg Infect Dis 2006;12(1):73-7.[28] Abramson JS. Intranasal, cold-adapted, live, attenuated influenza vaccine. Pediatr Infect Dis J
1999;18(12):1103-4.[29] Huckriede A, Bungener L, Stegmann T, Daemen T, Medema J, Palache AM, et al. The virosome con-
54

CHAPTER II
55
cept for influenza vaccines. Vaccine 2005;23 Suppl 1:S26-38.[30] Brands R, Visser J, Medema J, Palache AM, van Scharrenburg GJ. Influvac: a safe Madin Darby Canine
Kidney (MDCK) cell culture-based influenza vaccine. Dev Biol Stand 1999;98:93-100; discussion 11.[31] Kistner O, Barrett PN, Mundt W, Reiter M, Schober-Bendixen S, Dorner F. Development of a mamma-
lian cell (Vero) derived candidate influenza virus vaccine. Vaccine 1998;16(9-10):960-8.[32] Palache AM, Scheepers HS, de Regt V, van Ewijk P, Baljet M, Brands R, et al. Safety, reactogenicity
and immunogenicity of Madin Darby Canine Kidney cell-derived inactivated influenza subunit vac-cine. A meta-analysis of clinical studies. Dev Biol Stand 1999;98:115-25; discussion 33-4.
[33] Pau MG, Ophorst C, Koldijk MH, Schouten G, Mehtali M, Uytdehaag F. The human cell line PER.C6 provides a new manufacturing system for the production of influenza vaccines. Vaccine 2001;19(17-19):2716-21.
[34] Hobson D, Curry RL, Beare AS, Ward-Gardner A. The role of serum haemagglutination-inhibiting antibody in protection against challenge infection with influenza A2 and B viruses. J Hyg (Lond) 1972;70(4):767-77.
[35] Small PA, Jr., Waldman RH, Bruno JC, Gifford GE. Influenza infection in ferrets: role of serum anti-body in protection and recovery. Infect Immun 1976;13(2):417-24.
[36] Boon AC, Fringuelli E, Graus YM, Fouchier RA, Sintnicolaas K, Iorio AM, et al. Influenza A virus specific T cell immunity in humans during aging. Virology 2002;299(1):100-8.
[37] McElhaney JE. The unmet need in the elderly: designing new influenza vaccines for older adults. Vaccine 2005;23 Suppl 1:S10-25.
[38] Bachmann MF, Zinkernagel RM, Oxenius A. Immune responses in the absence of costimulation: viruses know the trick. J Immunol 1998;161(11):5791-4.
[39] Budowsky EI, Smirnov Yu A, Shenderovich SF. Principles of selective inactivation of viral genome. VIII. The influence of beta-propiolactone on immunogenic and protective activities of influenza virus. Vaccine 1993;11(3):343-8.
[40] Fleischer B, Becht H, Rott R. Recognition of viral antigens by human influenza A virus-specific T lym-phocyte clones. J Immunol 1985;135(4):2800-4.
[41] Kees U, Krammer PH. Most influenza A virus-specific memory cytotoxic T lymphocytes react with antigenic epitopes associated with internal virus determinants. J Exp Med 1984;159(2):365-77.
[42] Reiss CS, Schulman JL. Cellular immune responses of mice to influenza virus vaccines. J Immunol 1980;125(5):2182-8.
[43] Barry DW, Mayner RE, Staton E, Dunlap RC, Rastogi SC, Hannah JE, et al. Comparative trial of influ-enza vaccines. I. Immunogenicity of whole virus and split product vaccines in man. Am J Epidemiol 1976;104(1):34-46.
[44] Gross PA, Ennis FA, Gaerlan PF, Denson LJ, Denning CR, Schiffman D. A controlled double-blind comparison of reactogenicity, immunogenicity, and protective efficacy of whole-virus and split-product influenza vaccines in children. J Infect Dis 1977;136(5):623-32.
[45] Cox RJ, Hovden AO, Brokstad KA, Szyszko E, Madhun AS, Haaheim LR. The humoral immune respon-se and protective efficacy of vaccination with inactivated split and whole influenza virus vaccines in BALB/c mice. Vaccine 2006;24(44-46):6585-7.
[46] Jennings R, Pemberton RM, Smith TL, Amin T, Potter CW. Demonstration of an immunosuppressive action of detergent-disrupted influenza virus on the antibody response to inactivated whole virus vac-cine. J Gen Virol 1987;68 ( Pt 2):441-50.
[47] Lu X, Edwards LE, Desheva JA, Nguyen DC, Rekstin A, Stephenson I, et al. Cross-protective immunity in mice induced by live-attenuated or inactivated vaccines against highly pathogenic influenza A (H5N1) viruses. Vaccine 2006;24(44-46):6588-93.
[48] Ennis FA, Rook AH, Qi YH, Schild GC, Riley D, Pratt R, et al. HLA restricted virus-specific cytotoxic T-lymphocyte responses to live and inactivated influenza vaccines. Lancet 1981;2(8252):887-91.
[49] Stephenson I, Nicholson KG. Influenza: vaccination and treatment. Eur Respir J 2001;17(6):1282-93.[50] Stegmann T, Morselt HW, Booy FP, van Breemen JF, Scherphof G, Wilschut J. Functional reconstitu-
tion of influenza virus envelopes. Embo J 1987;6(9):2651-9.

[51] Bron R, Ortiz A, Dijkstra J, Stegmann T, Wilschut J. Preparation, properties, and applications of recon-stituted influenza virus envelopes (virosomes). Methods Enzymol 1993;220:313-31.
[52] de Jonge J, Schoen P, ter Veer W, Stegmann T, Wilschut J, Huckriede A. Use of a dialyzable short-chain phospholipid for efficient solubilization and reconstitution of influenza virus envelopes. Biochim Biophys Acta 2006;1758(4):527-36.
[53] de Bruijn IA, Nauta J, Cramer WC, Gerez L, Palache AM. Clinical experience with inactivated, viro-somal influenza vaccine. Vaccine 2005;23 Suppl 1:S39-49.
[54] Beyer WE, Palache AM, de Jong JC, Osterhaus AD. Cold-adapted live influenza vaccine versus inacti-vated vaccine: systemic vaccine reactions, local and systemic antibody response, and vaccine efficacy. A meta-analysis. Vaccine 2002;20(9-10):1340-53.
[55] Clements ML, Betts RF, Tierney EL, Murphy BR. Serum and nasal wash antibodies associated with resistance to experimental challenge with influenza A wild-type virus. J Clin Microbiol 1986;24(1):157-60.
[56] WHO Department of Immunization, Vaccines and Biologicals. Temperature sensitivity of vaccines., 2006.
[57] ICH. Stability testing guidelines: stability testing of new drug substances and products. CPMP/ICH/2736/99.
[58] ICH. Stability testing of biotechnological/biological products. CPMP/ICH/138/95.[59] Coenen F, Tolboom JT, Frijlink HW. Stability of influenza sub-unit vaccine. Does a couple of days out-
side the refrigerator matter? Vaccine 2006;24(4):525-31.[60] Luykx DM, Casteleijn MG, Jiskoot W, Westdijk J, Jongen PM. Physicochemical studies on the stability
of influenza haemagglutinin in vaccine bulk material. Eur J Pharm Sci 2004;23(1):65-75.[61] Amorij JP, Meulenaar J, Hinrichs WL, Stegmann T, Huckriede A, Coenen F, et al. Rational design of an
influenza subunit vaccine powder with sugar glass technology: Preventing conformational changes of haemagglutinin during freezing and freeze-drying. Vaccine 2007.
[62] Zweig SE. Advances in vaccine stability monitoring technology. Vaccine 2006;24(33-34):5977-85.[63] Oxford JS, Manuguerra C, Kistner O, Linde A, Kunze M, Lange W, et al. A new European perspective
of influenza pandemic planning with a particular focus on the role of mammalian cell culture vaccines. Vaccine 2005;23(46-47):5440-9.
[64] Osterhaus AD. Pre- or post-pandemic influenza vaccine? Vaccine 2007;25(27):4983-4.[65] Randolph TW. Phase separation of excipients during lyophilization: effects on protein stability. J
Pharm Sci 1997;86(11):1198-203.[66] Schebor C, Burin L, Buera MP, Aguilera JM, Chirife J. Glassy state and thermal inactivation of inver-
tase and lactase in dried amorphous matrices. Biotechnol Prog 1997;13(6):857-63.[67] Slade L, Levine H. Non-equilibrium behavior of small carbohydrate-water systems. Pure Appl Chem
1988;60:1841-64.[68] Slade L, Levine H. Water and the glass transition: dependance of the glass transition on the composi-
tion and chemical structure: special implications for flour functionality in cooky baking. J Food Eng 1995;24:431-509.
[69] Hinrichs WL, Prinsen MG, Frijlink HW. Inulin glasses for the stabilization of therapeutic proteins. Int J Pharm 2001;215(1-2):163-74.
[70] Eriksson HJ, Hinrichs WL, van Veen B, Somsen GW, de Jong GJ, Frijlink HW. Investigations into the stabilisation of drugs by sugar glasses: I. Tablets prepared from stabilised alkaline phosphatase. Int J Pharm 2002;249(1-2):59-70.
[71] Hincha DK, Zuther E, Hellwege EM, Heyer AG. Specific effects of fructo- and gluco-oligosaccharides in the preservation of liposomes during drying. Glycobiology 2002;12(2):103-10.
[72] Sun WQ, Leopold AC, Crowe LM, Crowe JH. Stability of dry liposomes in sugar glasses. Biophys J 1996;70(4):1769-76.
[73] Crowe LM, Crowe JH, Rudolph A, Womersley C, Appel L. Preservation of freeze-dried liposomes by trehalose. Arch Biochem Biophys 1985;242(1):240-7.
[74] Crowe JH, Crowe LM, Carpenter JF, Aurell Wistrom C. Stabilization of dry phospholipid bilayers and
56

proteins by sugars. Biochem J 1987;242(1):1-10.[75] Hinrichs WL, Sanders NN, De Smedt SC, Demeester J, Frijlink HW. Inulin is a promising cryo- and
lyoprotectant for PEGylated lipoplexes. J Control Release 2005;103(2):465-79.[76] Hinrichs WL, Mancenido FA, Sanders NN, Braeckmans K, De Smedt SC, Demeester J, et al. The choice
of a suitable oligosaccharide to prevent aggregation of PEGylated nanoparticles during freeze thawing and freeze drying. Int J Pharm 2006;311(1-2):237-44.
[77] Yannarell DA, Goldberg KM, Hjorth RN. Stabilizing cold-adapted influenza virus vaccine under vari-ous storage conditions. J Virol Methods 2002;102(1-2):15-25.
[78] Levy JA, Fieldsteel AH. Freeze-drying is an effective method for preserving infectious type C retro-viruses. J Virol Methods 1982;5(3-4):165-71.
[79] Bieganski RM, Fowler A, Morgan JR, Toner M. Stabilization of active recombinant retroviruses in an amorphous dry state with trehalose. Biotechnol Prog 1998;14(4):615-20.
[80] Croyle MA, Roessler BJ, Davidson BL, Hilfinger JM, Amidon GL. Factors that influence stability of recombinant adenoviral preparations for human gene therapy. Pharm Dev Technol 1998;3(3):373-83.
[81] Allison SD, Molina MC, Anchordoquy TJ. Stabilization of lipid/DNA complexes during the freezing step of the lyophilization process: the particle isolation hypothesis. Biochim Biophys Acta 2000;1468(1-2):127-38.
[82] Molina MC, Armstrong TK, Zhang Y, Patel MM, Lentz YK, Anchordoquy TJ. The stability of lyophi-lized lipid/DNA complexes during prolonged storage. J Pharm Sci 2004;93(9):2259-73.
[83] Carpenter JF, Crowe JH. An infrared spectroscopic study of the interactions of carbohydrates with dried proteins. Biochemistry 1989;28(9):3916-22.
[84] Chen D, Endres R, Maa YF, Kensil CR, Whitaker-Dowling P, Trichel A, et al. Epidermal powder immunization of mice and monkeys with an influenza vaccine. Vaccine 2003;21(21-22):2830-6.
[85] Maa YF, Ameri M, Shu C, Payne LG, Chen D. Influenza vaccine powder formulation development: spray-freeze-drying and stability evaluation. J Pharm Sci 2004;93(7):1912-23.
[86] Lazzell V, Waldman RH, Rose C, Khakoo R, Jacknowitz A, Howard S. Immunization against influenza in humans using an oral enteric-coated killed virus vaccine. J Biol Stand 1984;12(3):315-21.
[87] Singh M, Briones M, O'Hagan DT. A novel bioadhesive intranasal delivery system for inactivated influ-enza vaccines. J Control Release 2001;70(3):267-76.
[88] Chen D, Endres RL, Erickson CA, Weis KF, McGregor MW, Kawaoka Y, et al. Epidermal immuniza-tion by a needle-free powder delivery technology: immunogenicity of influenza vaccine and protection in mice. Nat Med 2000;6(10):1187-90.
[89] Chen D, Endres RL, Erickson CA, Maa YF, Payne LG. Epidermal powder immunization using non-toxic bacterial enterotoxin adjuvants with influenza vaccine augments protective immunity. Vaccine 2002;20(21-22):2671-9.
[90] Moldoveanu Z, Clements ML, Prince SJ, Murphy BR, Mestecky J. Human immune responses to influ-enza virus vaccines administered by systemic or mucosal routes. Vaccine 1995;13(11):1006-12.
[91] Huang J, Garmise RJ, Crowder TM, Mar K, Hwang CR, Hickey AJ, et al. A novel dry powder influenza vaccine and intranasal delivery technology: induction of systemic and mucosal immune responses in rats. Vaccine 2004;23(6):794-801.
[92] Garmise RJ, Mar K, Crowder TM, Hwang CR, Ferriter M, Huang J, et al. Formulation of a dry powder influenza vaccine for nasal delivery. AAPS PharmSciTech 2006;7(1):E19.
[93] Bot AI, Smith DJ, Bot S, Dellamary L, Tarara TE, Harders S, et al. Receptor-mediated targeting of spray-dried lipid particles coformulated with immunoglobulin and loaded with a prototype vaccine. Pharm Res 2001;18(7):971-9.
[94] de Jonge J, Amorij JP, Hinrichs WL, Wilschut J, Huckriede A, Frijlink HW. Inulin sugar glasses pre-serve the structural integrity and biological activity of influenza virosomes during freeze-drying and storage. Eur J Pharm Sci 2007.
[95] Wang W. Lyophilization and development of solid protein pharmaceuticals. Int J Pharm 2000;203(1-2):1-60.
[96] Abdul-Fattah AM, Truong-Le V, Yee L, Pan E, Ao Y, Kalonia DS, et al. Drying-induced variations in
CHAPTER II
57

physico-chemical properties of amorphous pharmaceuticals and their impact on Stability II: stability of a vaccine. Pharm Res 2007;24(4):715-27.
[97] Korte T, Ludwig K, Huang Q, Rachakonda PS, Herrmann A. Conformational change of influenza virus hemagglutinin is sensitive to ionic concentration. Eur Biophys J 2007;36(4-5):327-35.
[98] Eriksson JH, Hinrichs WL, de Jong GJ, Somsen GW, Frijlink HW. Investigations into the stabilization of drugs by sugar glasses: III. The influence of various high-pH buffers. Pharm Res 2003;20(9):1437-43.
[99] Elversson J, Millqvist-Fureby A. Particle size and density in spray drying-effects of carbohydrate pro-perties. J Pharm Sci 2005;94(9):2049-60.
[100] Liao YH, Brown MB, Nazir T, Quader A, Martin GP. Effects of sucrose and trehalose on the preser-vation of the native structure of spray-dried lysozyme. Pharm Res 2002;19(12):1847-53.
[101] Bosquillon C, Rouxhet PG, Ahimou F, Simon D, Culot C, Preat V, et al. Aerosolization properties, sur-face composition and physical state of spray-dried protein powders. J Control Release 2004;99(3):357-67.
[102] Branchu S, Forbes RT, York P, Petren S, Nyqvist H, Camber O. Hydroxypropyl-beta-cyclodextrin inhibits spray-drying-induced inactivation of beta-galactosidase. J Pharm Sci 1999;88(9):905-11.
[103] Costantino HR, Andya JD, Nguyen PA, Dasovich N, Sweeney TD, Shire SJ, et al. Effect of mannitol crystallization on the stability and aerosol performance of a spray-dried pharmaceutical protein, re-combinant humanized anti-IgE monoclonal antibody. J Pharm Sci 1998;87(11):1406-11.
[104] Millqvist-Fureby A, Malmsten M, Bergenstahl B. Spray-drying of trypsin - surface characterisation and activity preservation. Int J Pharm 1999;188(2):243-53.
[105] Surana R, Pyne A, Suryanarayanan R. Effect of preparation method on physical properties of amor-phous trehalose. Pharm Res 2004;21(7):1167-76.
[106] Forbes RT, Davis KG, Hindle M, Clarke JG, Maas J. Water vapor sorption studies on the physical sta-bility of a series of spray-dried protein/sugar powders for inhalation. J Pharm Sci 1998;87(11):1316-21.
[107] Shoyele SA, Cawthorne S. Particle engineering techniques for inhaled biopharmaceuticals. Adv Drug Deliv Rev 2006;58(9-10):1009-29.
[108] Adler M, Unger M, Lee G. Surface composition of spray-dried particles of bovine serum albumin/tre-halose/surfactant. Pharm Res 2000;17(7):863-70.
[109] Aksan A, Toner M. Isothermal desiccation and vitrification kinetics of trehalose-dextran solutions. Langmuir 2004;20(13):5521-9.
[110] Orford PD, Parker R, Ring SG. Aspects of the glass transition behaviour of mixtures of carbohydrates of low molecular weight. Carbohydr Res 1990;196:11-8.
[111] Saleki-Gerhardt A, Zografi G. Non-isothermal and isothermal crystallization of sucrose from the amor-phous state. Pharm Res 1994;11(8):1166-73.
[112] Kompella UB, Koushik K. Preparation of drug delivery systems using supercritical fluid technology. Crit Rev Ther Drug Carrier Syst 2001;18(2):173-99.
[113] Jovanovic N, Bouchard A, Hofland GW, Witkamp GJ, Crommelin DJ, Jiskoot W. Stabilization of pro-teins in dry powder formulations using supercritical fluid technology. Pharm Res 2004;21(11):1955-69.
[114] Reverchon E. Supercritical antisolvent precipitation of micro-and nano-particles. J Supercrit Fluids 1999;15:1-21.
[115] Allison SD, Chang B, Randolph TW, Carpenter JF. Hydrogen bonding between sugar and protein is responsible for inhibition of dehydration-induced protein unfolding. Arch Biochem Biophys 1999;365(2):289-98.
[116] Vereyken IJ, Chupin V, Hoekstra FA, Smeekens SC, de Kruijff B. The effect of fructan on membrane lipid organization and dynamics in the dry state. Biophys J 2003;84(6):3759-66.
[117] Vereyken IJ, Chupin V, Islamov A, Kuklin A, Hincha DK, de Kruijff B. The effect of fructan on the phospholipid organization in the dry state. Biophys J 2003;85(5):3058-65.
[118] Crowe JH, Crowe LM, Oliver AE, Tsvetkova N, Wolkers W, Tablin F. The trehalose myth revisited: introduction to a symposium on stabilization of cells in the dry state. Cryobiology 2001;43(2):89-105.
[119] Koster KL, Webb MS, Bryant G, Lynch DV. Interactions between soluble sugars and POPC (1-palmi-toyl-2-oleoylphosphatidylcholine) during dehydration: vitrification of sugars alters the phase behavior
58

of the phospholipid. Biochim Biophys Acta 1994;1193(1):143-50.[120] Koster KL, Lei YP, Anderson M, Martin S, Bryant G. Effects of vitrified and nonvitrified sugars on
phosphatidylcholine fluid-to-gel phase transitions. Biophys J 2000;78(4):1932-46.[121] Wolfe J, Bryant G. Freezing, drying, and/or vitrification of membrane- solute-water systems.
Cryobiology 1999;39(2):103-29.[122] Yu RC, Abrams DC, Alaibac M, Chu AC. Morphological and quantitative analyses of normal epidermal
Langerhans cells using confocal scanning laser microscopy. Br J Dermatol 1994;131(6):843-8.[123] Glenn GM, Kenney RT, Hammond SA, Ellingsworth LR. Transcutaneous immunization and immuno-
stimulant strategies. Immunol Allergy Clin North Am 2003;23(4):787-813.[124] Chen D, Periwal SB, Larrivee K, Zuleger C, Erickson CA, Endres RL, et al. Serum and mucosal immune
responses to an inactivated influenza virus vaccine induced by epidermal powder immunization. J Virol 2001;75(17):7956-65.
[125] Skountzou I, Quan FS, Jacob J, Compans RW, Kang SM. Transcutaneous immunization with inacti-vated influenza virus induces protective immune responses. Vaccine 2006;24(35-36):6110-9.
[126] Glenn GM, Kenney RT. Mass vaccination: solutions in the skin. Curr Top Microbiol Immunol 2006;304:247-68.
[127] CDC National Immunization Program. Needle-free injection Technology. www.cdc.gov/nip/dev/jetinject.htm, 2007.
[128] Jackson LA, Austin G, Chen RT, Stout R, DeStefano F, Gorse GJ, et al. Safety and immunogenicity of varying dosages of trivalent inactivated influenza vaccine administered by needle-free jet injectors. Vaccine 2001;19(32):4703-9.
[129] Sarphie DF, Johnson B, Cormier M, Burkoth TL, Bellhouse BL. Bioavailabillity following transdermal powdered delivery (TDP) of radiolabeled insulin to hairless guinea pigs. J Control Release 1997;47:61-69.
[130] Maa YF, Shu C, Ameri M, Zuleger C, Che J, Osorio JE, et al. Optimization of an alum-adsorbed vaccine powder formulation for epidermal powder immunization. Pharm Res 2003;20(7):969-77.
[131] Dean HJ, Chen D. Epidermal powder immunization against influenza. Vaccine 2004;23(5):681-6.[132] Glenn GM, Taylor DN, Li X, Frankel S, Montemarano A, Alving CR. Transcutaneous immunization: a
human vaccine delivery strategy using a patch. Nat Med 2000;6(12):1403-6.[133] Garg S, Hoelscher M, Belser JA, Wang C, Jayashankar L, Guo Z, et al. Needle-free Skin Patch Delivery
of a Pandemic Influenza Vaccine Protects Mice from Lethal Viral Challenge. Clin Vaccine Immunol 2007.
[134] Van Kampen KR, Shi Z, Gao P, Zhang J, Foster KW, Chen DT, et al. Safety and immunogenicity of adenovirus-vectored nasal and epicutaneous influenza vaccines in humans. Vaccine 2005;23(8):1029-36.
[135] Guebre-Xabier M, Hammond SA, Epperson DE, Yu J, Ellingsworth L, Glenn GM. Immunostimulant patch containing heat-labile enterotoxin from Escherichia coli enhances immune responses to injected influenza virus vaccine through activation of skin dendritic cells. J Virol 2003;77(9):5218-25.
[136] Guebre-Xabier M, Hammond SA, Ellingsworth LR, Glenn GM. Immunostimulant patch enhances immune responses to influenza virus vaccine in aged mice. J Virol 2004;78(14):7610-8.
[137] Frech SA, Kenney RT, Spyr CA, Lazar H, Viret JF, Herzog C, et al. Improved immune responses to influenza vaccination in the elderly using an immunostimulant patch. Vaccine 2005;23(7):946-50.
[138] Hunsaker BD, Perino LJ. Efficacy of intradermal vaccination. Vet Immunol Immunopathol 2001;79(1-2):1-13.
[139] Chiu SS, Peiris JS, Chan KH, Wong WH, Lau YL. Immunogenicity and safety of intradermal influenza immunization at a reduced dose in healthy children. Pediatrics 2007;119(6):1076-82.
[140] Belshe RB, Newman FK, Cannon J, Duane C, Treanor J, Van Hoecke C, et al. Serum antibody responses after intradermal vaccination against influenza. N Engl J Med 2004;351(22):2286-94.
[141] Kenney RT, Frech SA, Muenz LR, Villar CP, Glenn GM. Dose sparing with intradermal injection of influenza vaccine. N Engl J Med 2004;351(22):2295-301.
[142] Alarcon JB, Hartley AW, Harvey NG, Mikszta JA. Preclinical evaluation of microneedle technology for
CHAPTER II
59

intradermal delivery of influenza vaccines. Clin Vaccine Immunol 2007;14(4):375-81.[143] McAllister DV, Wang PM, Davis SP, Park JH, Canatella PJ, Allen MG, et al. Microfabricated needles
for transdermal delivery of macromolecules and nanoparticles: fabrication methods and transport studies. Proc Natl Acad Sci U S A 2003;100(24):13755-60.
[144] Mikszta JA, Alarcon JB, Brittingham JM, Sutter DE, Pettis RJ, Harvey NG. Improved genetic immu-nization via micromechanical disruption of skin-barrier function and targeted epidermal delivery. Nat Med 2002;8(4):415-9.
[145] Mikszta JA, Sullivan VJ, Dean C, Waterston AM, Alarcon JB, Dekker JP, 3rd, et al. Protective immu-nization against inhalational anthrax: a comparison of minimally invasive delivery platforms. J Infect Dis 2005;191(2):278-88.
[146] Gill HS, Prausnitz MR. Coating formulations for microneedles. Pharm Res 2007;24(7):1369-80.[147] Matriano JA, Cormier M, Johnson J, Young WA, Buttery M, Nyam K, et al. Macroflux microprojection
array patch technology: a new and efficient approach for intracutaneous immunization. Pharm Res 2002;19(1):63-70.
[148] Gill HS, Prausnitz MR. Coated microneedles for transdermal delivery. J Control Release 2007;117(2):227-37.
[149] McDermott MR, Bienenstock J. Evidence for a common mucosal immunologic system. I. Migration of B immunoblasts into intestinal, respiratory, and genital tissues. J Immunol 1979;122(5):1892-8.
[150] Neutra MR, Pringault E, Kraehenbuhl JP. Antigen sampling across epithelial barriers and induction of mucosal immune responses. Annu Rev Immunol 1996;14:275-300.
[151] Holmgren J, Czerkinsky C, Lycke N, Svennerholm AM. Mucosal immunity: implications for vaccine development. Immunobiology 1992;184(2-3):157-79.
[152] Brandtzaeg P. Overview of the mucosal immune system. Curr Top Microbiol Immunol 1989;146:13-25.[153] Couch RB, Kasel JA. Immunity to influenza in man. Annu Rev Microbiol 1983;37:529-49.[154] Liew FY, Russell SM, Appleyard G, Brand CM, Beale J. Cross-protection in mice infected with influ-
enza A virus by the respiratory route is correlated with local IgA antibody rather than serum antibody or cytotoxic T cell reactivity. Eur J Immunol 1984;14(4):350-6.
[155] Haan L, Verweij WR, Holtrop M, Brands R, van Scharrenburg GJ, Palache AM, et al. Nasal or intra-muscular immunization of mice with influenza subunit antigen and the B subunit of Escherichia coli heat-labile toxin induces IgA- or IgG-mediated protective mucosal immunity. Vaccine 2001;19(20-22):2898-907.
[156] Perkins JC, Tucker DN, Knopf HL, Wenzel RP, Kapikian AZ, Chanock RM. Comparison of protective effect of neutralizing antibody in serum and nasal secretions in experimental rhinovirus type 13 illness. Am J Epidemiol 1969;90(6):519-26.
[157] Tumpey TM, Renshaw M, Clements JD, Katz JM. Mucosal delivery of inactivated influenza vaccine induces B-cell-dependent heterosubtypic cross-protection against lethal influenza A H5N1 virus infec-tion. J Virol 2001;75(11):5141-50.
[158] Asahi-Ozaki Y, Yoshikawa T, Iwakura Y, Suzuki Y, Tamura S, Kurata T, et al. Secretory IgA antibodies provide cross-protection against infection with different strains of influenza B virus. J Med Virol 2004;74(2):328-35.
[159] Tamura S, Kurata T. Defense mechanisms against influenza virus infection in the respiratory tract mucosa. Jpn J Infect Dis 2004;57(6):236-47.
[160] Potter CW, Jennings R. Intranasal immunization with inactivated influenza vaccine. Pharm Sci Technol Today 1999;2(10):402-08.
[161] Sweet C, Smith H. Pathogenicity of influenza virus. Microbiol Rev 1980;44(2):303-30.[162] Boyce TG, Hsu HH, Sannella EC, Coleman-Dockery SD, Baylis E, Zhu Y, et al. Safety and immu-
nogenicity of adjuvanted and unadjuvanted subunit influenza vaccines administered intranasally to healthy adults. Vaccine 2000;19(2-3):217-26.
[163] Fine PE. Poliomyelitis: very small risks and very large risks. Lancet Neurol 2004;3(12):703.[164] Atmar RL, Keitel WA, Cate TR, Munoz FM, Ruben F, Couch RB. A dose-response evaluation of inacti-
vated influenza vaccine given intranasally and intramuscularly to healthy young adults. Vaccine 2007.
60

[165] Jackson RJ, Fujihashi K, Xu-Amano J, Kiyono H, Elson CO, McGhee JR. Optimizing oral vaccines: induction of systemic and mucosal B-cell and antibody responses to tetanus toxoid by use of cholera toxin as an adjuvant. Infect Immun 1993;61(10):4272-9.
[166] Arrington J, Braun RP, Dong L, Fuller DH, Macklin MD, Umlauf SW, et al. Plasmid vectors encoding cholera toxin or the heat-labile enterotoxin from Escherichia coli are strong adjuvants for DNA vac-cines. J Virol 2002;76(9):4536-46.
[167] Hashigucci K, Ogawa H, Ishidate T, Yamashita R, Kamiya H, Watanabe K, et al. Antibody responses in volunteers induced by nasal influenza vaccine combined with Escherichia coli heat-labile enterotoxin B subunit containing a trace amount of the holotoxin. Vaccine 1996;14(2):113-9.
[168] Gluck U, Gebbers JO, Gluck R. Phase 1 evaluation of intranasal virosomal influenza vaccine with and without Escherichia coli heat-labile toxin in adult volunteers. J Virol 1999;73(9):7780-6.
[169] Glueck R. Pre-clinical and clinical investigation of the safety of a novel adjuvant for intranasal immu-nization. Vaccine 2001;20 Suppl 1:S42-4.
[170] Mutsch M, Zhou W, Rhodes P, Bopp M, Chen RT, Linder T, et al. Use of the inactivated intranasal influenza vaccine and the risk of Bell's palsy in Switzerland. N Engl J Med 2004;350(9):896-903.
[171] Kanerva M, Mannonen L, Piiparinen H, Peltomaa M, Vaheri A, Pitkaranta A. Search for Herpesviruses in cerebrospinal fluid of facial palsy patients by PCR. Acta Otolaryngol 2007;127(7):775-9.
[172] van Ginkel FW, Jackson RJ, Yoshino N, Hagiwara Y, Metzger DJ, Connell TD, et al. Enterotoxin-based mucosal adjuvants alter antigen trafficking and induce inflammatory responses in the nasal tract. Infect Immun 2005;73(10):6892-902.
[173] de Haan L, Holtrop M, Verweij WR, Agsteribbe E, Wilschut J. Mucosal immunogenicity of the Escherichia coli heat-labile enterotoxin: role of the A subunit. Vaccine 1996;14(4):260-6.
[174] de Haan L, Verweij WR, Feil IK, Lijnema TH, Hol WG, Agsteribbe E, et al. Mutants of the Escherichia coli heat-labile enterotoxin with reduced ADP-ribosylation activity or no activity retain the immu-nogenic properties of the native holotoxin. Infect Immun 1996;64(12):5413-6.
[175] Lu X, Clements JD, Katz JM. Mutant Escherichia coli heat-labile enterotoxin [LT(R192G)] enhances protective humoral and cellular immune responses to orally administered inactivated influenza vac-cine. Vaccine 2002;20(7-8):1019-29.
[176] Barackman JD, Ott G, Pine S, O'Hagan DT. Oral administration of influenza vaccine in combination with the adjuvants LT-K63 and LT-R72 induces potent immune responses comparable to or stronger than traditional intramuscular immunization. Clin Diagn Lab Immunol 2001;8(3):652-7.
[177] McCluskie MJ, Weeratna RD, Clements JD, Davis HL. Mucosal immunization of mice using CpG DNA and/or mutants of the heat-labile enterotoxin of Escherichia coli as adjuvants. Vaccine 2001;19(27):3759-68.
[178] Stephenson I, Zambon MC, Rudin A, Colegate A, Podda A, Bugarini R, et al. Phase I evaluation of intranasal trivalent inactivated influenza vaccine with nontoxigenic Escherichia coli enterotoxin and novel biovector as mucosal adjuvants, using adult volunteers. J Virol 2006;80(10):4962-70.
[179] Halperin SA, Smith B, Clarke K, Treanor J, Mabrouk T, Germain M. Phase I, randomized, controlled trial to study the reactogenicity and immunogenicity of a nasal, inactivated trivalent influenza virus vaccine in healthy adults. Hum Vaccin 2005;1(1):37-42.
[180] Youn HJ, Ko SY, Lee KA, Ko HJ, Lee YS, Fujihashi K, et al. A single intranasal immunization with inactivated influenza virus and alpha-galactosylceramide induces long-term protective immunity without redirecting antigen to the central nervous system. Vaccine 2007;25(28):5189-98.
[181] Treanor J, Nolan C, O'Brien D, Burt D, Lowell G, Linden J, et al. Intranasal administration of a proteo-some-influenza vaccine is well-tolerated and induces serum and nasal secretion influenza antibodies in healthy human subjects. Vaccine 2006;24(3):254-62.
[182] Coulter A, Harris R, Davis R, Drane D, Cox J, Ryan D, et al. Intranasal vaccination with ISCOMATRIX adjuvanted influenza vaccine. Vaccine 2003;21(9-10):946-9.
[183] Morein B, Bengtsson KL. Immunomodulation by iscoms, immune stimulating complexes. Methods 1999;19(1):94-102.
[184] Read RC, Naylor SC, Potter CW, Bond J, Jabbal-Gill I, Fisher A, et al. Effective nasal influenza vaccine
CHAPTER II
61

delivery using chitosan. Vaccine 2005;23(35):4367-74.[185] Bacon A, Makin J, Sizer PJ, Jabbal-Gill I, Hinchcliffe M, Illum L, et al. Carbohydrate biopolymers
enhance antibody responses to mucosally delivered vaccine antigens. Infect Immun 2000;68(10):5764-70.
[186] Amidi M, Romeijn SG, Verhoef JC, Junginger HE, Bungener L, Huckriede A, et al. N-Trimethyl chito-san (TMC) nanoparticles loaded with influenza subunit antigen for intranasal vaccination: Biological properties and immunogenicity in a mouse model. Vaccine 2006.
[187] Illum L, Jabbal-Gill I, Hinchcliffe M, Fisher AN, Davis SS. Chitosan as a novel nasal delivery system for vaccines. Adv Drug Deliv Rev 2001;51(1-3):81-96.
[188] Ugwoke MI, Agu RU, Verbeke N, Kinget R. Nasal mucoadhesive drug delivery: background, applica-tions, trends and future perspectives. Adv Drug Deliv Rev 2005;57(11):1640-65.
[189] Illum L. Nanoparticulate systems for nasal delivery of drugs: a real improvement over simple systems? JPharm Sci 2007;96(3):473-83.
[190] Sullivan VJ, Mikszta JA, Laurent P, Huang J, Ford B. Noninvasive delivery technologies: respiratory delivery of vaccines. Expert Opin Drug Deliv 2006;3(1):87-95.
[191] Jiang G, Joshi SB, Peek LJ, Brandau DT, Huang J, Ferriter MS, et al. Anthrax vaccine powder formula-tions for nasal mucosal delivery. J Pharm Sci 2006;95(1):80-96.
[192] Huo Z, Sinha R, McNeela EA, Borrow R, Giemza R, Cosgrove C, et al. Induction of protective serum meningococcal bactericidal and diphtheria-neutralizing antibodies and mucosal immunoglobulin A in volunteers by nasal insufflations of the Neisseria meningitidis serogroup C polysaccharide-CRM197 conjugate vaccine mixed with chitosan. Infect Immun 2005;73(12):8256-65.
[193] Dean HJ. Alternative routes of influenza vaccine delivery. Expert Opin Drug Deliv 2006;3(5):557-61.[194] Soane RJ, Frier M, Perkins AC, Jones NS, Davis SS, Illum L. Evaluation of the clearance characteristics
of bioadhesive systems in humans. Int J Pharm 1999;178(1):55-65.[195] McInnes FJ, Thapa P, Baillie AJ, Welling PG, Watson DG, Gibson I, et al. In vivo evaluation of nico-
tine lyophilised nasal insert in sheep. Int J Pharm 2005;304(1-2):72-82.[196] Ugwoke MI, Agu RU, Vanbilloen H, Baetens J, Augustijns P, Verbeke N, et al. Scintigraphic evaluation
in rabbits of nasal drug delivery systems based on carbopol 971p((R)) and carboxymethylcellulose. J Control Release 2000;68(2):207-14.
[197] Vidgren MT, Kublik H. Nasal delivery systems and their effect on deposition and absorption. Adv Drug Deliv Rev 1998;29(1-2):157-77.
[198] Newman SP, Pitcairn GR, Dalby RN. Drug delivery to the nasal cavity: in vitro and in vivo assessment. Crit Rev Ther Drug Carrier Syst 2004;21(1):21-66.
[199] Tafaghodi M, Abolghasem Sajadi Tabassi S, Jaafari MR, Zakavi SR, Momen-Nejad M. Evaluation of the clearance characteristics of various microspheres in the human nose by gamma-scintigraphy. Int J Pharm 2004;280(1-2):125-35.
[200] Groneberg DA, Paul H, Welte T. Novel strategies of aerosolic pharmacotherapy. Exp Toxicol Pathol 2006;57 Suppl 2:49-53.
[201] Lambrecht BN, Prins JB, Hoogsteden HC. Lung dendritic cells and host immunity to infection. Eur Respir J 2001;18(4):692-704.
[202] von Garnier C, Filgueira L, Wikstrom M, Smith M, Thomas JA, Strickland DH, et al. Anatomical loca-tion determines the distribution and function of dendritic cells and other APCs in the respiratory tract. J Immunol 2005;175(3):1609-18.
[203] Holt PG. Pulmonary dendritic cells in local immunity to inert and pathogenic antigens in the respira-tory tract. Proc Am Thorac Soc 2005;2(2):116-20.
[204] Cutts FT, Clements CJ, Bennett JV. Alternative routes of measles immunization: a review. Biologicals 1997;25(3):323-38.
[205] Pulmonary drug delivery. Aulendorf: Editio Cantor Verlag für Medizin und Naturwissenschaften GmbH, 2007.
[206] Waldman RH, Mann JJ, Kasel JA. Influenza virus neutralizing antibody in human respiratory secre-tions. J Immunol 1968;100(1):80-5.
62

[207] Mann JJ, Waldman RH, Togo Y, Heiner GG, Dawkins AT, Kasel JA. Antibody response in respiratory secretions of volunteers given live and dead influenza virus. J Immunol 1968;100(4):726-35.
[208] Kasel JA, Fulk RV, Togo Y, Hornick RB, Heiner GG, Dawkins AT, Jr., et al. Influenza antibody in human respiratory secretions after subcutaneous or respiratory immunization with inactivated virus. Nature 1968;218(5141):594-5.
[209] Waldman RH, Mann JJ, Small PA, Jr. Immunization against influenza. Prevention of illness in man by aerosolized inactivated vaccine. Jama 1969;207(3):520-4.
[210] Waldman RH, Jurgensen PF, Olsen GN, Ganguly R, Johnson JE, 3rd. Immune response of the human respiratory tract. I. Immunoglobulin levels and influenza virus vaccine antibody response. J Immunol 1973;111(1):38-41.
[211] Waldman RH, Wigley FMSPA, Jr. Specificity of respiratory secretion antibody against influenza virus. J Immunol 1970;105(6):1477-83.
[212] Haigh W, Howell RW. The efficacy of the A2/Aichi/68 strain in inhaled influenza immunisation against the A/England/42/72 variant. J Soc Occup Med 1973;23(4):125-7.
[213] Haigh W, Howell RW, Meichen FW. A comparative trial of influenza immunization by inhalation and hypojet methods. Practitioner 1973;211(263):365-70.
[214] Waldman RH, Wood SH, Torres EJ, Small PA, Jr. Influenza antibody response following aerosal admi-nistration of inactivated virus. Am J Epidemiol 1970;91(6):574-85.
[215] Minne A, Louahed J, Mehauden S, Baras B, Renauld JC, Vanbever R. The delivery site of a monovalent influenza vaccine within the respiratory tract impacts on the immune response. Immunology 2007.
[216] Amorij JP, Saluja V, Hinrichs WL, Huckriede A, Frijlink HW. Pulmonary delivery of an SFD influenza subunit vaccine (Chapter 5). Submitted 2007.
[217] Cooper PD, Steele EJ. The adjuvanticity of gamma inulin. Immunol Cell Biol 1988;66 ( Pt 5-6):345-52.[218] Silva DG, Cooper PD, Petrovsky N. Inulin-derived adjuvants efficiently promote both Th1 and Th2
immune responses. Immunol Cell Biol 2004;82(6):611-6.[219] Petrovsky N. Novel human polysaccharide adjuvants with dual Th1 and Th2 potentiating activity.
Vaccine 2006;24 Suppl 2:S2-26-9.[220] Dalby R, Spallek M, Voshaar T. A review of the development of Respimat Soft Mist Inhaler. Int J
Pharm 2004;283(1-2):1-9.[221] Frijlink HW, De Boer AH. Dry powder inhalers for pulmonary drug delivery. Expert Opin Drug Deliv
2004;1(1):67-86.[222] Laube BL. The expanding role of aerosols in systemic drug delivery, gene therapy, and vaccination.
Respir Care 2005;50(9):1161-76.[223] de Boer AH, Hagedoorn P, Westerman EM, Le Brun PP, Heijerman HG, Frijlink HW. Design and in
vitro performance testing of multiple air classifier technology in a new disposable inhaler concept (Twincer) for high powder doses. Eur J Pharm Sci 2006;28(3):171-8.
[224] Bergmann KC, Waldman RH, Tischner H, Pohl WD. Antibody in tears, saliva and nasal secretions fol-lowing oral immunization of humans with inactivated influenza virus vaccine. Int Arch Allergy Appl Immunol 1986;80(1):107-9.
[225] Avtushenko SS, Sorokin EM, Zoschenkova NY, Zacharova NG, Naichin AN. Clinical and immuno-logical characteristics of the emulsion form of inactivated influenza vaccine delivered by oral immu-nization. J Biotechnol 1996;44(1-3):21-8.
[226] Renegar KB, Small PA, Jr. Immunoglobulin A mediation of murine nasal anti-influenza virus immu-nity. J Virol 1991;65(4):2146-8.
[227] Eriksson HJ, Verweij WR, Poelstra K, Hinrichs WL, de Jong GJ, Somsen GW, et al. Investigations into the stabilisation of drugs by sugar glasses: II. Delivery of an inulin-stabilised alkaline phosphatase in the intestinal lumen via the oral route. Int J Pharm 2003;257(1-2):273-81.
[228] Meitin CA, Bender BS, Small PA, Jr. Enteric immunization of mice against influenza with recombinant vaccinia. Proc Natl Acad Sci U S A 1994;91(23):11187-91.
[229] Chen KS, Burlington DB, Quinnan GV, Jr. Active synthesis of hemagglutinin-specific immunoglobulin A by lung cells of mice that were immunized intragastrically with inactivated influenza virus vaccine. J
CHAPTER II
63

Virol 1987;61(7):2150-4.[230] Katz JM, Lu X, Young SA, Galphin JC. Adjuvant activity of the heat-labile enterotoxin from entero-
toxigenic Escherichia coli for oral administration of inactivated influenza virus vaccine. J Infect Dis 1997;175(2):352-63.
[231] Kersten G, Hirschberg H. Antigen delivery systems. Expert Rev Vaccines 2004;3(4):453-62.[232] Yuki Y, Kiyono H. New generation of mucosal adjuvants for the induction of protective immunity. Rev
Med Virol 2003;13(5):293-310.[233] Conacher M, Alexander J, Brewer JM. Oral immunisation with peptide and protein antigens by formu-
lation in lipid vesicles incorporating bile salts (bilosomes). Vaccine 2001;19(20-22):2965-74.[234] Mann JF, Ferro VA, Mullen AB, Tetley L, Mullen M, Carter KC, et al. Optimisation of a lipid based oral
delivery system containing A/Panama influenza haemagglutinin. Vaccine 2004;22(19):2425-9.[235] Pang GT, Clancy RL, O'Reilly SE, Cripps AW. A novel particulate influenza vaccine induces long-term
and broad-based immunity in mice after oral immunization. J Virol 1992;66(2):1162-70.[236] Harokopakis E, Childers NK, Michalek SM, Zhang SS, Tomasi M. Conjugation of cholera toxin or its B
subunit to liposomes for targeted delivery of antigens. J Immunol Methods 1995;185(1):31-42.[237] Chattaraj SC, Rathinavelu A, Das SK. Biodegradable microparticles of influenza viral vaccine: compa-
rison of the effects of routes of administration on the in vivo immune response in mice. J Control Release 1999;58(2):223-32.
[238] Amorij JP, Westra TA, Hinrichs WL, Huckriede A, Frijlink HW. Towards an oral influenza vaccine (Chapter 4). Submitted 2007.
[239] Maroni A, Zema L, Cerea M, Sangalli ME. Oral pulsatile drug delivery systems. Expert Opin Drug Deliv 2005;2(5):855-71.
[240] Gazzaniga A, Maroni A, Sangalli ME, Zema L. Time-controlled oral delivery systems for colon tar-geting. Expert Opin Drug Deliv 2006;3(5):583-97.
[241] Schellekens RC, Frijlink HW, inventors; PH-controlled pulsatile delivery system, methods for prepara-tion and use thereof. patent WO 2007/013794 A1. 2007.
[242] Schellekens RC, Stuurman FE, van der Weert FH, Kosterink JG, Frijlink HW. A novel dissolution method relevant to intestinal release behaviour and its application in the evaluation of modified release mesalazine products. Eur J Pharm Sci 2007;30(1):15-20.
[243] Senel S, Kremer M, Nagy K, Squier C. Delivery of bioactive peptides and proteins across oral (buccal) mucosa. Curr Pharm Biotechnol 2001;2(2):175-86.
[244] Challacombe S, Coogan M, Williams D, Greenspan J. Overview and research agenda arising from the 5th World Workshop on Oral Health and Disease in AIDS. Adv Dent Res 2006;19(1):5-9.
[245] Otten K, Wang HC, Wyde PR, Klein JR. Modulation of gamma delta T cells in mouse buccal epithe-lium following antigen priming. Biochem Biophys Res Commun 2002;294(3):626-9.
[246] Gebert A, Pabst R. M cells at locations outside the gut. Semin Immunol 1999;11(3):165-70.[247] Childers NK, Powell WD, Tong G, Kirk K, Wiatrak B, Michalek SM. Human salivary immunoglobulin
and antigen-specific antibody activity after tonsillectomy. Oral Microbiol Immunol 2001;16(5):265-9.[248] Canonica GW, Passalacqua G. Noninjection routes for immunotherapy. J Allergy Clin Immunol
2003;111(3):437-48; quiz 49.[249] Fukuizumi T, Nagamatsu H, Kojo T, Inoue H. Induction of salivary antibodies to inhibit Candida albi-
cans adherence to human epithelial cells by tonsillar immunization in rabbits. FEMS Immunol Med Microbiol 2006;47(3):398-404.
[250] Bakke H, Samdal HH, Holst J, Oftung F, Haugen IL, Kristoffersen AC, et al. Oral spray immunization may be an alternative to intranasal vaccine delivery to induce systemic antibodies but not nasal muco-sal or cellular immunity. Scand J Immunol 2006;63(3):223-31.
64

J-P. Amorij1, J. Meulenaar1, W.L.J. Hinrichs1, T. Stegmann2, A. Huckriede3, F. Coenen4, H.W. Frijlink1
Chapter III
Rational design of an influenza subunit vaccine powder with sugar-gglass-ttechnology:Preventing conformational changes of haemag-glutinin during freezing and freeze-ddrying.
Vaccine 25 (2007): 6447-6457.
1 Department of Pharmaceutical Technology and Biopharmacy, University of Groningen, Groningen, The Netherlands.
2 Virosome Biologicals, Leiden, The Netherlands.3 Department of Medical Microbiology, Molecular Virology Section, University Medical Center Groningen and University of Groningen, Groningen, The Netherlands.
4 Solvay Pharmaceuticals, Weesp, The Netherlands.

Abstract
The development of a stable influenza subunit vaccine in the dry state was investigated.The influence of various carbohydrates, buffer types and freezing rates on the integrityof haemagglutinin after freeze-thawing or freeze-drying was investigated with a range ofanalytical and immunological methods. The use of fast freezing, Hepes buffer and carbo-hydrates (trehalose, inulin or dextran) as cryo- and lyoprotectants resulted in a sig-nificant reduction or even absence of conformational changes of haemagglutinin (HA) asrevealed by the used methods. The subunit vaccine in the powder was shown to remainimmunogenic in an in vivo-study in mice, using reconstituted powder. Moreover the HApotency of the influenza subunit vaccine powder was stable for at least 26 weeks at roomtemperature.
Keywords
Influenza vaccine; buffer; lyophilization.
66

Introduction
Every year millions of people are infected with influenza resulting in a high morbi-dity and mortality rate for the elderly and high-risk populations [1]. To control the disease, vaccination with influenza vaccines is used.
The currently used vaccines contain two surface antigens, haemagglutinin (HA) andneuraminidase (NA). Haemagglutinin, the major surface glycoprotein of the influenzavirus, is a trimer (225 kD) of three identical monomers (75 kD). Each monomer consistsof the polypeptides HA1 (50 kD) and HA2 (25 kD). These polypeptides are linked by twointramonomer disulfide bridges. The three monomers are assembled into a central α-helical coiled-coil that forms the stem-like domain, and three globular heads containingsialic acid-binding sites. Each globular domain consists exclusively of HA1 folded inhighly variable loops of eight antiparallel β-strands. The globular heads contain both thereceptor binding sites and the antigenic epitopes [2, 3].
Haemagglutinin mediates binding of the influenza virus to the cell surface as well asthe subsequent fusion of viral and cellular membranes. The change of the spring-loaded conformation of HA during fusion follows two stages as reported by Stegmann etal. [4, 5]. The initial phase is the exposure of the hydrophobic N-termini of the HA2 poly-peptides and is pH dependent. In the second phase, which is less pH dependent andoccurs after a lag period, major refolding takes place, involving extended coiled coil for-mation and dissociation of the membrane-distal domains of the protein, exposing clea-vage sites for trypsin among others.
Vaccination is a key strategy in the prevention of influenza infection. Vaccines aregenerally composed of whole inactivated or attenuated influenza or of (purified) mem-brane proteins (split vaccine, subunit vaccine). The virus proteins of the influenza vac-cine are produced in a solution in which they have a limited physical and chemical sta-bility. Storage at temperatures below zero, at room temperature as well as at elevatedtemperature among others decreases the antigenic potency of haemagglutinin [6, 7]. Carret al. found that the conformation change of HA described above can occur in vitro notonly at low pH but also at neutral pH, upon exposure of HA to either heat or the dena-turant urea [8, 9]. This conformational change results in a reduced or diminished immu-ne response after vaccination [10].
Currently, influenza vaccines are administered by intramuscular injection. In orderto further stimulate vaccination there is a need for new improved vaccine formulationsthat make vaccine delivery easier and safer, decrease dependency on the cold chainand/or reduce the number of immunization interventions [11]. To overcome the inci-dence of iatrogenic infections upon immunization of a predominantly healthy populati-on the development of a needle-free vaccination method, such as jet-injectors, oral,nasal, aerosol, or patch delivery is required. Furthermore, non-invasive dosage formssimplify the logistics of immunization, thus improving immunization coverage.
CHAPTER III
67

One of the keystones in the improvement of vaccine formulations is obtaining stable antigen-proteins in the dry state. The most commonly used method for preparingsolid proteinaceous drugs is freeze-drying (lyophilization). However, during freeze-drying the proteinaceous drug is subjected to freezing and drying stress by which its acti-vity can be lost. Therefore a protective agent is required to prevent damaging effects oflyophilization. From literature, it is well known that carbohydrates can protect varioustypes of drug substances like proteins [12-14] and vaccines [15-17] during freezing, dry-ing and subsequent storage. If dried properly, the proteinaceous drug is incorporated ina matrix consisting of carbohydrate in the amorphous glassy state. The stabilizing effectof these sugar glasses has been explained by the formation of a matrix which stronglyreduces diffusion and molecular mobility (vitrification) and acts as a physical barrierbetween particles or molecules (particle/molecule isolation). Both the lack of mobility[18, 19] and the physical barrier [12, 20] provided by the glass matrix, prevent aggrega-tion and degradation of the dried material. Moreover, during the lyophilization process,the water molecules that form H-bonds with the active material are replaced by thehydroxyl groups of the carbohydrate, by which the 3D-structure/structural integrity ofthe active compound is maintained [21].
The aim of this research is to design an influenza subunit vaccine powder with fullmaintenance of structure and functionality using sugar glass technology. The influenceof freezing rate, buffer composition, and type of carbohydrate on the structure and anti-genic activity of HA after freezing and freeze-drying respectively is studied. The carbo-hydrates investigated are a disaccharide (trehalose), an oligosaccharide (inulin) and apolysaccharide (dextran).
Maa et al. also incorporated subunit vaccine in sugar glasses [17]. However, theyused a trivalent vaccine (containing A/Panama(H3N2), A/New Caledonia(H1N1) andB/Yamanashi strains) and predominantly focussed on powder properties and physicalstability, while less attention was paid to structure and functionality of the differentHA's. In the present study, the structural integrity of haemagglutinin (A/Panama(H3N2)strain) is investigated with a proteolytic assay, fluorescence spectroscopy and circulardichroism spectroscopy. The antigenic properties of the subunit vaccine powder aredetermined by a single radial immunodiffusion assay (SRID) and compared to unproces-sed subunit vaccine. The antigenic properties of the produced influenza powder afterreconstitution are further evaluated in an in vivo study in mice.
68

Materials and Methods
MaterialsInfluenza sub-unit vaccine of strain A/Panama (H3N2) (monovalent bulk batch numberHPR 40, containing 437 μg/ml HA) was kindly provided by Solvay Pharmaceuticals(Weesp, The Netherlands). Two different types of inulin, inulin 0.9 kD (SC95; Tg=109°C)and inulin 1.8 kD (HD0011111; Tg=148°C), were generous gifts from Sensus (Roosendaal,The Netherlands). Dextran 56 kD (Tg=225°C) from Leuconostoc mesenteroides, was pur-chased from Fluka Biochemika (Buchs, Switzerland). Trypsin from Bovine Pancreas andtrehalose (Tg=121°C) were from Sigma-Aldrich (Steinheim, Germany). Trypsin-inhibitorfrom egg white was purchased from Roche Diagnostics (Darmstadt, Germany). All otherchemicals were of reagent or analytical grade and purchased from commercial suppliers.
DialysisThe influenza vaccine was dialyzed with Slide-A-Lyzer Dialysis Cassettes (Pierce) witha molecular weight cut-off of 10 kD to remove thiomersal and to replace buffer and salts.Four different batches were produced, differing in buffer components (Hepes or phosphate buffered) and acid-treatment. Acid-induced change in HA conformation wasprovoked by dialyzing the protein against 30 mM citrate buffer, pH 4.75 for 8 hours at 37°C.
The dialyzed samples were then transferred to glass vials and stored in a refrigera-tor at 4°C until use. The dialysis resulted in batches consisting of:1. HA in 2.0 mM Hepes buffered saline (HBS), containing 125 mM NaCl, 0.9 mM
CaCl2*2H2O and 0.5 mM MgCl2*6H2O pH 7.4;2. Acid-treated HA in HBS;3. HA in phosphate buffered saline (PBS) containing 2.0 mM Na2HPO4*2H2O,
0.4 mM KH2PO4, 125 mM NaCl, 0.9 mM CaCl2*2H2O and 0.5 mM MgCl2*6H2O pH 7.4;
4. Acid-treated HA in PBS.
Freeze-ddrying and freeze-tthawingFreeze-drying was carried out in a Christ Alpha 1-4 freeze-dryer (Salm en Kipp,Breukelen, The Netherlands). In a typical experiment, 4 ml glass vials were charged with0.48 ml aqueous solutions of 360 μg/ml HA and 1.7 % w/v carbohydrate. The solutionswere frozen in the refrigerator for 24 hrs at -20°C or in liquid nitrogen for 5-10 minutesand subsequently lyophilized or thawed at room temperature. Frozen at -20°C or inliquid nitrogen will be denoted as slow and rapid freezing, respectively. The freeze-dryerwas set at a shelf temperature of -35°C, a condenser temperature of -55°C and a pressureof 0.220 mbar. After 24 h the pressure was lowered to 0.060 mbar, and the shelf tem-perature was gradually increased to 20°C, which was maintained for 24 h. The dry sam-ples were transferred to a vacuum desiccators at room temperature containing orangesilica gel (EMERGO BV, The Netherlands), where they were kept for at least 2 days. The
CHAPTER III
69

thawed samples were stored in a refrigerator at 4°C until further analysis.
PH during freezing The buffer solutions (HBS and PBS, 1.00 ml, pH 7.4), were mixed with universal indi-cator (20 μl). The universal indicator contained 0.02% w/v methyl red, 0.02% w/v phe-nolphthalein, 0.04% w/v bromthymol blue, and 0.04% w/v thymol blue in ethanol [22].The color of the solutions before and after slow and rapid freezing was noted. Also, buf-fer solutions containing 1.7 % w/v of carbohydrate and indicator solution were frozen inorder to elucidate the influence of carbohydrate on the pH of the respective buffer so-lutions during freezing.
Proteolytic assay with trypsinIn order to evaluate the appearance of an acid-induced conformational change of HAafter freezing and freeze-drying a proteolytic assay was performed. Proteolysis of dilutedHA samples (90 μg/ml) was carried out for 1 hr at pH 7.4 and 37°C with 100 μg/ml tryp-sin. After 1 hr proteolysis was stopped with 200 μg/ml trypsin-inhibitor. Each sample(content 3.6 μg HA) was boiled with loading buffer for 5 min. After loading on 10% poly-acrylamide gels (Fermentas), the (virus) proteins and protein fragments were separatedby SDS-PAGE under non-reducing conditions. Proteins were stained with PAGE bluestaining solution (Fermentas).
Fluorescence SpectroscopyFluorescence spectroscopy was performed using a modified version of the method des-cribed by Luykx et al. [6]. Fluorescence spectra were obtained with a SLM-Aminco AB2spectrofluorometer at 37°C using cylindrical quartz cuvettes with sample volumes of 700 μl containing 90 μg/ml HA. An excitation wavelength of 295 nm was used, with a band pass of 2.0 nm for the excitation monochromator and 4.0 nm for the emissionmonochromator. When excited at 295 nm, the fluorescence emission spectrum is indi-cative of the localisation within the HA molecule and the degree of solvent exposure ofthe 12 tryptophan residues of A/Panama HA [6]. Data were recorded at 1 nm intervalsover the range 300-400 nm. Fluorescence spectra were the average of 5 scans and cor-rected for background signal caused by buffer, salts or carbohydrate.
Circular dichroism spectroscopyCD spectra from 90 μg/ml HA samples were recorded on an Aviv 62A DS CD spectro-meter at 37°C, using a 0.1 cm path length 200 μl quartz cuvette with continuously N2-flushing of the sample compartment. Spectra were recorded from 250 to 200 nm (far UV)with an instrumental time constant of 5 s using a bandwidth of 1 nm and a step reso-lution of 1 nm. The spectra shown are the average of 12 scans. All spectra are presentedas molar absorbance difference, Δε, based on a mean residue weight of 110 Da and takinginto account that HA has a Mw of 220 kD including 19% glycosylated-carbohydrate byweight [3].
70

SRIDA single radial immunodiffusion (SRID) method was used to test the potency of the freeze-dried HA immediately after freeze-drying and after freeze-drying and storage.Samples were stored up to 26 weeks at 20°C/0%RH (using a silica containing vacuum dis-siccator in climate controlled room) or at 45°C/11±2%RH in a climate cabinet. The testis a quantitative immunodiffusion technique and is used as recommended by theEuropean Pharmacopoeia [23, 24].
ImmunizationAnimal experiments were conducted according to the guidelines provided by the DutchAnimal Protection Act, and were approved by the Committee for AnimalExperimentation (DEC) of the University of Groningen. For all experiments 6-8 weeksold female BALB/c mice (Harlan, Zeist, Netherlands) were used. The treated groups consisted of 8 mice each and the control group consisted of 3 mice. Mice were injectedintramuscularly in the hind-right leg with a 30 μl sample. Mice received 5 μg unproces-sed subunit antigen or 5 μg reconstituted inulin-stabilized subunit antigen (as deter-mined according to the precipitation method of Lowry with trichloro acetic acid [25,26]). This reconstituted vaccine was previously freeze-dried from an inulin 1.8 kD con-taining HBS and the powder was stored for 2 weeks at 20°C. The mice of the controlgroup received only HBS. On day 21 mice were sacrificed. Mice were bled by an orbitapunction under isofluran/oxygen anesthesia. Serum samples were obtained by centrifu-gation at 11.000 x g. Until the antibody assay, sera were stored at -20°C.
Antibody assays - ELISAs Influenza subunit antigen-specific antibody responses were determined by ELISA [27].ELISA plates (96-well flat bottom, Microlon®600, Greiner, Alphen a/d Rijn,Netherlands) were coated with 200 ng of influenza subunit antigen per well. Appropriatedilutions of sera of each individual mouse were applied to the plates, serially diluted twofold in PBS/Tween and then incubated for 1 h at 37°C. Subsequently, plates werewashed and incubated with horseradish peroxidase-conjugated goat antibodies directedagainst mouse IgG (1:5000). After incubation of the plates with mouse Ig-isotype conju-gate for 1 h at 37°C, plates were washed twice with PBS/Tween, and once with PBS.Antibodies were detected using substrate buffer (0.02% 1,2-phenylendiamin-dihydro-chlorid in 50 mM phosphate buffer pH 5.6, containing 0.006 % H2O2). Plates were developed in the dark for 30 min at room temperature after which the reaction was stop-ped by addition of 50 μl 2 M H2SO4 per well. The absorbance at 490 nm was read witha Benchmark Microplate reader (BioRad, Hercules, CA). Titers are given as the recipro-cal of the calculated sample dilution corresponding with an A490 = 0.2 after backgroundcorrection.
Haemagglutination-inhibition assayFor determination of haemagglutination-inhibi-tion (HI) titers in serum, 75 μl of serumwas first inactivated by incubation for 30 min at 56°C [27]. In order to reduce non-spe-
CHAPTER III
71

cific haemagglutination, 225 μl of a 25% kaolin suspension was added. After centri-fuga-tion for 2 min at 1400 x g, 50 μl of the super-natant was transferred in duplicate to 96-well round-bottom plates (Greiner, Alphen a/d Rijn, Netherlands) and serially dilutedtwofold in PBS. Next 50 μl PBS containing 4 haemagglutination units (HAU) ofA/Panama influenza virus was added to each well. After mixing the contents of each wellgently with a micropipette, the plates were incubated for 40 min at room temperature.Finally, 50 μl of a 1% guinea pig erythrocyte suspen-sion was added to each well and haemagglutination was allowed to proceed for at least 2 h at room temperature. Thehighest serum dilution capable of preventing haemagglutination was scored as the HI-titer. The results are expressed as the geometric mean titer ± standard deviation for eachgroup.
Statistical analysis Comparisons between experimental groups were made using Student's t-test. Probability(P) values < 0.05 were considered significant.
72
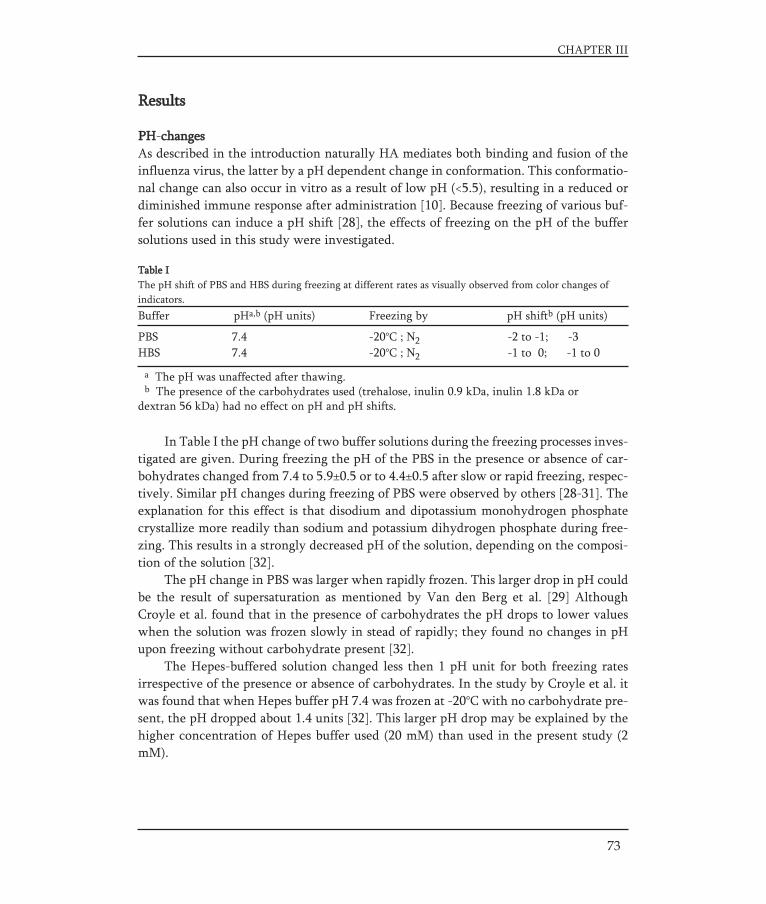
Results
PH-cchangesAs described in the introduction naturally HA mediates both binding and fusion of theinfluenza virus, the latter by a pH dependent change in conformation. This conformatio-nal change can also occur in vitro as a result of low pH (<5.5), resulting in a reduced ordiminished immune response after administration [10]. Because freezing of various buf-fer solutions can induce a pH shift [28], the effects of freezing on the pH of the buffersolutions used in this study were investigated.
In Table I the pH change of two buffer solutions during the freezing processes inves-tigated are given. During freezing the pH of the PBS in the presence or absence of car-bohydrates changed from 7.4 to 5.9±0.5 or to 4.4±0.5 after slow or rapid freezing, respec-tively. Similar pH changes during freezing of PBS were observed by others [28-31]. Theexplanation for this effect is that disodium and dipotassium monohydrogen phosphatecrystallize more readily than sodium and potassium dihydrogen phosphate during free-zing. This results in a strongly decreased pH of the solution, depending on the composi-tion of the solution [32].
The pH change in PBS was larger when rapidly frozen. This larger drop in pH couldbe the result of supersaturation as mentioned by Van den Berg et al. [29] AlthoughCroyle et al. found that in the presence of carbohydrates the pH drops to lower valueswhen the solution was frozen slowly in stead of rapidly; they found no changes in pHupon freezing without carbohydrate present [32].
The Hepes-buffered solution changed less then 1 pH unit for both freezing ratesirrespective of the presence or absence of carbohydrates. In the study by Croyle et al. itwas found that when Hepes buffer pH 7.4 was frozen at -20°C with no carbohydrate pre-sent, the pH dropped about 1.4 units [32]. This larger pH drop may be explained by thehigher concentration of Hepes buffer used (20 mM) than used in the present study (2mM).
Table IThe pH shift of PBS and HBS during freezing at different rates as visually observed from color changes ofindicators.
CHAPTER III
73
Buffer pHa,b (pH units) Freezing by pH shiftb (pH units)
PBS 7.4 -20°C ; N2 -2 to -1; -3HBS 7.4 -20°C ; N2 -1 to 0; -1 to 0
a The pH was unaffected after thawing.b The presence of the carbohydrates used (trehalose, inulin 0.9 kDa, inulin 1.8 kDa or
dextran 56 kDa) had no effect on pH and pH shifts.

Proteolytic assayTo evaluate whether or not the pH-induced conformational change of HA occurred afterfreeze-thawing and freeze-drying, a proteolytic assay with trypsin was performed. In thenative conformation, HA is resistant to digestion by trypsin, but in the acid-induced con-formation, HA is digested into small fragments. The digestion of HA is readily resolvedby PAGE, providing a convenient assay for the detection of the conformational changein HA (Fig. 1, A and B). This assay is especially capable in detecting the pH-dependentconformational change of HA after neutralization [33].
Slow freezing of HA in PBS (Fig. 1, C) and freeze-drying of HA in PBS (Fig. 1, E-F)resulted in a conformational change of HA, although less than observed for the acidinduced change. Slow freezing resulted in increased susceptibility of HA for trypsin clea-vage compared to rapid freezing. After freeze-drying more HA became susceptible fortrypsin cleavage indicating that during drying a further change in conformation occur-red. Clearly the presence of carbohydrates resulted in preservation of the spring-loaded conformation (Fig. 1, G-J). The use of carbohydrates thus prevented the conformationchange of HA during freeze-drying.
When PBS was replaced by HBS, no trypsin digestion was observed, neither after freeze-thawing nor after freeze-drying in HBS a pH-induced conformational change ofHA occured (data not shown).
Fluorescence spectroscopyChanges in the tertiary structure of A/Panama HA in PBS or in HBS as a result of freezing and freeze-drying were monitored by tryptophan fluorescence spectroscopy(Fig. 2A and Fig. 2B).
The intrinsic tryptophanyl fluorescence of HA exposed to freezing in PBS revealedslightly lower fluorescence intensities than was observed for the untreated HA, indepen-dent of the freezing rate. Slow and fast freezing of HA in HBS instead of PBS showed lesspronounced changes in tryptophanyl fluorescence. Compared to freezing alone, freeze-drying introduced a more pronounced change in the tryptophan fluorescence spectrumof HA independent of the buffer used.
Fig. 1 Susceptibility of HA to trypsin digestion. HA was treated as indicated below and analyzed by non-reducing SDS-PAGE. Shown are the HA monomer bands (75 kD) after different treatments. "Native HA" (A)and "Acid treated HA" (B) are used as negative and positive control for the trypsin digestion, respectively. Theeffect of freeze-thawing and freeze-drying using different freezing rates and PBS is shown in C-F: HA freeze-thawed using slow freezing (C), freeze-thawed using rapid freezing (D), freeze-dried using slow freezing (E)and freeze-dried using rapid freezing (F). The effect of the different sugars using a rapid freezing rate and PBSis shown in G-J: HA freeze-dried with trehalose (G), inulin 0.9 kD (H), inulin 1.8 kD (I) and dextran 56 kD (J).
74
A B C D E F G H I J

CHAPTER III
75
Fig. 2 The effect of freeze-thawing and freeze-drying on the tryptophanyl fluorescence of A/Panama HAprocessed in (A) PBS or (B) HBS. Fluorescence spectra of HA untreated (bold solid line), freeze-thawed at -20°C(solid grey line), freeze-thawed in liquid N2 (solid line), freeze-dried after freezing at -20°C (dashed line) andfreeze-dried after freezing in liquid N2 (dotted line).
The influence of carbohydrates on the freeze-drying damage of A/Panama HA pro-cessed with PBS is shown in Figure 3. Freeze-drying in HBS buffer showed the similarresults (data not shown). In contrast to freeze-drying in the absence of carbohydrates,freeze-drying in the presence of carbohydrates did not result in any significant differen-ces in fluorescence intensity.
Fig. 3 The influence of sugars on the freeze-drying damage of A/Panama HA processed in PBS using rapidfreezing. Fluorescence spectra were recorded of HA untreated (bold solid line), freeze-dried without sugar(dotted line), freeze-dried with trehalose (solid grey line), freeze-dried with inulin 0.9 kD (solid line), freeze-dried with inulin 1.8 kD (dashed line) and freeze-dried with dextran 56 kD (dashed grey line).
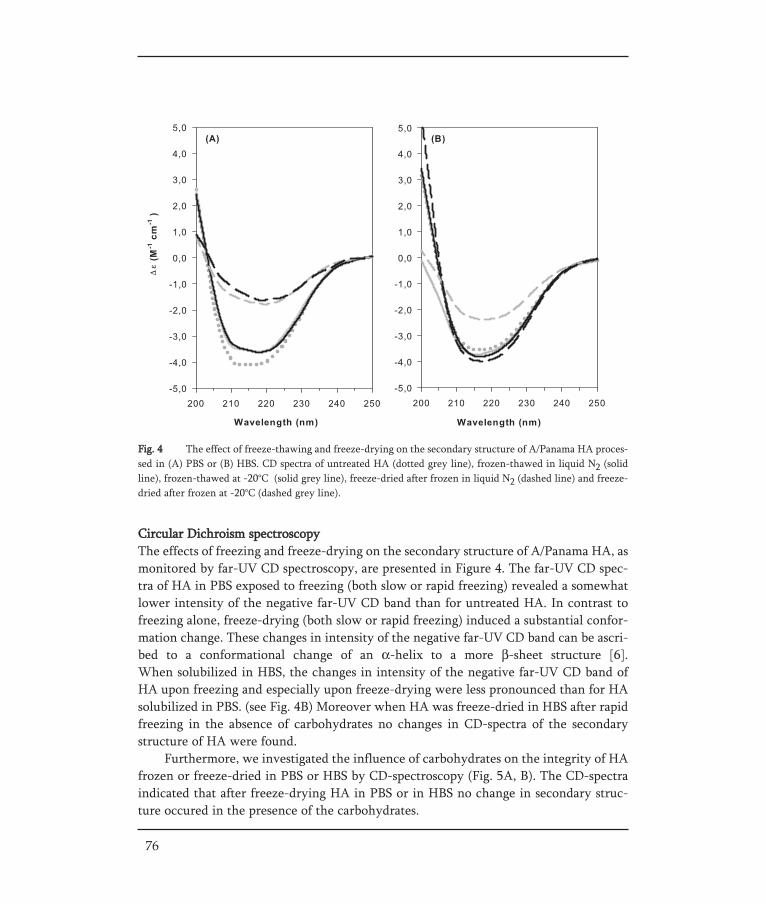
Fig. 4 The effect of freeze-thawing and freeze-drying on the secondary structure of A/Panama HA proces-sed in (A) PBS or (B) HBS. CD spectra of untreated HA (dotted grey line), frozen-thawed in liquid N2 (solidline), frozen-thawed at -20°C (solid grey line), freeze-dried after frozen in liquid N2 (dashed line) and freeze-dried after frozen at -20°C (dashed grey line).
76
Circular Dichroism spectroscopyThe effects of freezing and freeze-drying on the secondary structure of A/Panama HA, asmonitored by far-UV CD spectroscopy, are presented in Figure 4. The far-UV CD spec-tra of HA in PBS exposed to freezing (both slow or rapid freezing) revealed a somewhatlower intensity of the negative far-UV CD band than for untreated HA. In contrast tofreezing alone, freeze-drying (both slow or rapid freezing) induced a substantial confor-mation change. These changes in intensity of the negative far-UV CD band can be ascri-bed to a conformational change of an α-helix to a more β-sheet structure [6].When solubilized in HBS, the changes in intensity of the negative far-UV CD band ofHA upon freezing and especially upon freeze-drying were less pronounced than for HAsolubilized in PBS. (see Fig. 4B) Moreover when HA was freeze-dried in HBS after rapidfreezing in the absence of carbohydrates no changes in CD-spectra of the secondarystructure of HA were found.
Furthermore, we investigated the influence of carbohydrates on the integrity of HAfrozen or freeze-dried in PBS or HBS by CD-spectroscopy (Fig. 5A, B). The CD-spectraindicated that after freeze-drying HA in PBS or in HBS no change in secondary struc-ture occured in the presence of the carbohydrates.
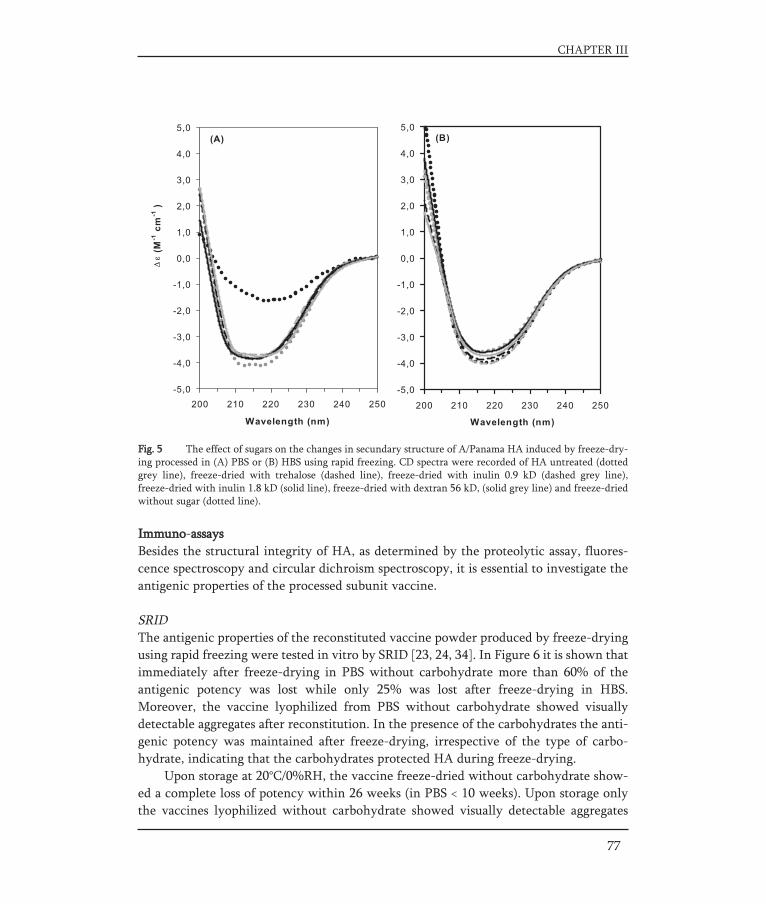
77
CHAPTER III
Immuno-aassaysBesides the structural integrity of HA, as determined by the proteolytic assay, fluores-cence spectroscopy and circular dichroism spectroscopy, it is essential to investigate theantigenic properties of the processed subunit vaccine.
SRIDThe antigenic properties of the reconstituted vaccine powder produced by freeze-dryingusing rapid freezing were tested in vitro by SRID [23, 24, 34]. In Figure 6 it is shown thatimmediately after freeze-drying in PBS without carbohydrate more than 60% of theantigenic potency was lost while only 25% was lost after freeze-drying in HBS.Moreover, the vaccine lyophilized from PBS without carbohydrate showed visuallydetectable aggregates after reconstitution. In the presence of the carbohydrates the anti-genic potency was maintained after freeze-drying, irrespective of the type of carbo-hydrate, indicating that the carbohydrates protected HA during freeze-drying.
Upon storage at 20°C/0%RH, the vaccine freeze-dried without carbohydrate show-ed a complete loss of potency within 26 weeks (in PBS < 10 weeks). Upon storage onlythe vaccines lyophilized without carbohydrate showed visually detectable aggregates
Fig. 5 The effect of sugars on the changes in secundary structure of A/Panama HA induced by freeze-dry-ing processed in (A) PBS or (B) HBS using rapid freezing. CD spectra were recorded of HA untreated (dottedgrey line), freeze-dried with trehalose (dashed line), freeze-dried with inulin 0.9 kD (dashed grey line), freeze-dried with inulin 1.8 kD (solid line), freeze-dried with dextran 56 kD, (solid grey line) and freeze-driedwithout sugar (dotted line).
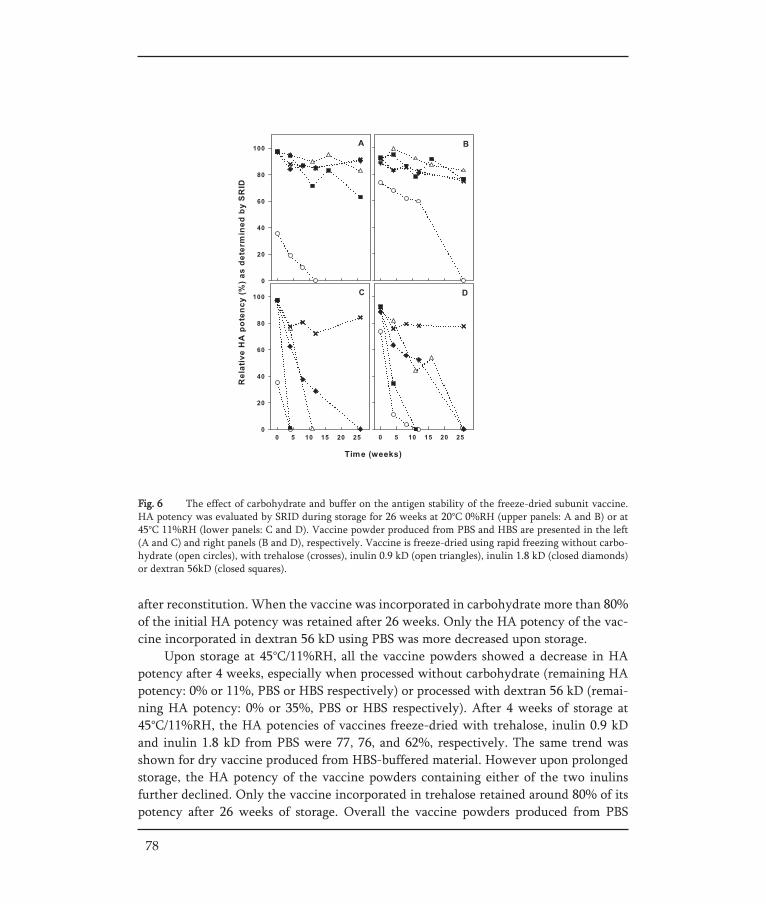
after reconstitution. When the vaccine was incorporated in carbohydrate more than 80%of the initial HA potency was retained after 26 weeks. Only the HA potency of the vac-cine incorporated in dextran 56 kD using PBS was more decreased upon storage.
Upon storage at 45°C/11%RH, all the vaccine powders showed a decrease in HApotency after 4 weeks, especially when processed without carbohydrate (remaining HApotency: 0% or 11%, PBS or HBS respectively) or processed with dextran 56 kD (remai-ning HA potency: 0% or 35%, PBS or HBS respectively). After 4 weeks of storage at45°C/11%RH, the HA potencies of vaccines freeze-dried with trehalose, inulin 0.9 kDand inulin 1.8 kD from PBS were 77, 76, and 62%, respectively. The same trend wasshown for dry vaccine produced from HBS-buffered material. However upon prolongedstorage, the HA potency of the vaccine powders containing either of the two inulins further declined. Only the vaccine incorporated in trehalose retained around 80% of itspotency after 26 weeks of storage. Overall the vaccine powders produced from PBS
78
Fig. 6 The effect of carbohydrate and buffer on the antigen stability of the freeze-dried subunit vaccine.HA potency was evaluated by SRID during storage for 26 weeks at 20°C 0%RH (upper panels: A and B) or at45°C 11%RH (lower panels: C and D). Vaccine powder produced from PBS and HBS are presented in the left(A and C) and right panels (B and D), respectively. Vaccine is freeze-dried using rapid freezing without carbo-hydrate (open circles), with trehalose (crosses), inulin 0.9 kD (open triangles), inulin 1.8 kD (closed diamonds)or dextran 56kD (closed squares).
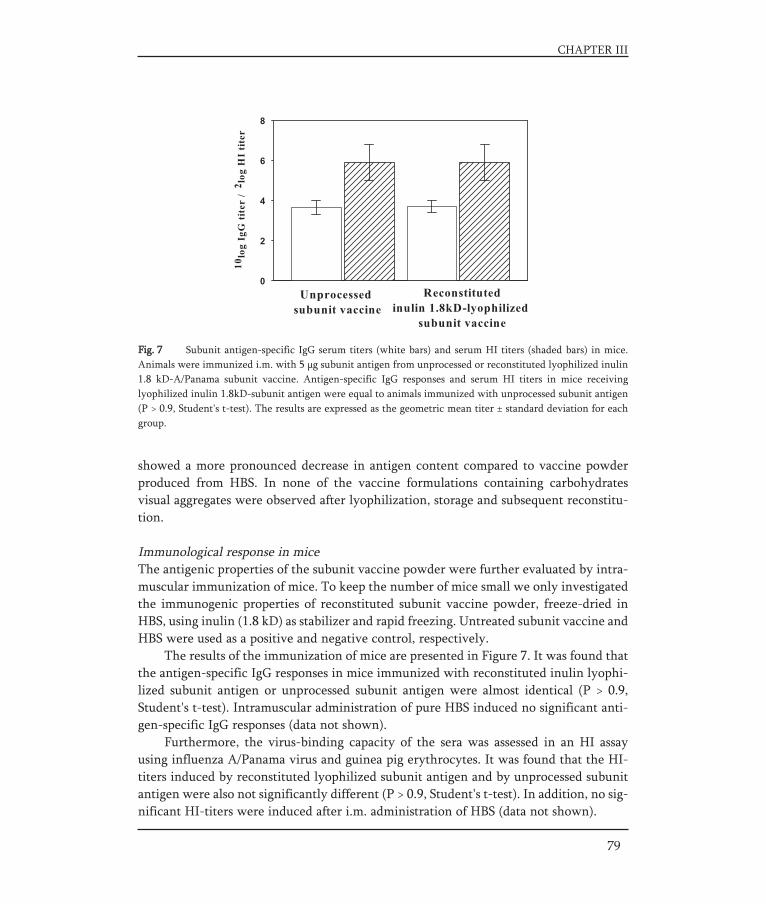
Fig. 7 Subunit antigen-specific IgG serum titers (white bars) and serum HI titers (shaded bars) in mice.Animals were immunized i.m. with 5 μg subunit antigen from unprocessed or reconstituted lyophilized inulin1.8 kD-A/Panama subunit vaccine. Antigen-specific IgG responses and serum HI titers in mice receiving lyophilized inulin 1.8kD-subunit antigen were equal to animals immunized with unprocessed subunit antigen (P > 0.9, Student's t-test). The results are expressed as the geometric mean titer ± standard deviation for eachgroup.
showed a more pronounced decrease in antigen content compared to vaccine powderproduced from HBS. In none of the vaccine formulations containing carbohydrates visual aggregates were observed after lyophilization, storage and subsequent reconstitu-tion.
Immunological response in miceThe antigenic properties of the subunit vaccine powder were further evaluated by intra-muscular immunization of mice. To keep the number of mice small we only investigatedthe immunogenic properties of reconstituted subunit vaccine powder, freeze-dried inHBS, using inulin (1.8 kD) as stabilizer and rapid freezing. Untreated subunit vaccine andHBS were used as a positive and negative control, respectively.
The results of the immunization of mice are presented in Figure 7. It was found thatthe antigen-specific IgG responses in mice immunized with reconstituted inulin lyophi-lized subunit antigen or unprocessed subunit antigen were almost identical (P > 0.9,Student's t-test). Intramuscular administration of pure HBS induced no significant anti-gen-specific IgG responses (data not shown).
Furthermore, the virus-binding capacity of the sera was assessed in an HI assayusing influenza A/Panama virus and guinea pig erythrocytes. It was found that the HI-titers induced by reconstituted lyophilized subunit antigen and by unprocessed subunitantigen were also not significantly different (P > 0.9, Student's t-test). In addition, no sig-nificant HI-titers were induced after i.m. administration of HBS (data not shown).
CHAPTER III
79

Discussion
This study clearly shows that with the proper choice of buffer, lyoprotectant and processconditions, intact influenza subunit vaccine can be obtained as a powder in which HAmaintained its structure and functionality according to the analytical and immunologi-cal methods used.
The structural integrity of haemagglutininDuring freeze-drying HA is exposed to freezing and dehydration stresses. To evaluatewhether possible deterioration of the protein is due to freezing or dehydration stressesduring the freezing or drying step of the cycle, the structure of HA after freeze-thawingor freeze-drying was investigated. The combination of the proteolytic assay, fluorescen-ce spectroscopy and circular dichroism spectroscopy was used because they are capableto indicate changes in the spring-loaded conformation, tertiary structure and secondarystructure of HA, respectively.
The effect of freeze-thawing on the structural integrity of HA was found to dependon the type of buffer and freezing rate. While dissolved in HBS the structure of HAchanged only slightly, the secondary structure and tertiary structure of HA were moreaffected upon freezing in PBS. This difference is explained by the fact that freezing ofHBS resulted in a much smaller decrease of the pH than freezing of PBS. As a consequen-ce, the acid-induced conformation change of HA during freezing occurred only in PBS.
Surprisingly, a less pronounced acid-induced conformational change of HA in PBSwas found after rapid freezing than after slow freezing, although the pH of the PBS dec-reased more strongly. Two reasons may explain this result. First, the pH drop after rapidfreezing is misleading due to supersaturation [29]. Secondly, the system is rapidly vitri-fied during rapid freezing which may prevent the conformational change to occur.
Slow freezing in PBS caused structural changes of HA (exposed trypsin cleavagesites), despite the moderate pH-shift (not below 5.5). Moreover, these structural changeswere not prevented by the low temperature, which in general slows down the pH-indu-ced conformation change of HA [4, 5, 35]. This can be ascribed to the increase in soluteconcentration of the non-frozen fraction during freezing, resulting in acceleration ofreaction kinetics [36] and changed physical properties such as ionic strength and relati-ve composition of the solution, which may destabilize the protein [12].
In addition fluorescence and CD-spectroscopy of HA after freeze thawing indicatethat the conformational changes of HA after freezing are not induced by a low pH alone,but also by freezing related stresses. Upon accidification of HA fluorescence and CDspectra changes, as reported in [6, 37, 38] and [6, 35], respectively. However, these spec-tral changes were found to be reversible after subsequent neutralization [35, 38]. We alsofound no decrease in intrinsic tryptophanyl fluorescence after acidification of HA fromA/Panama at 37°C (pH 4.75) and subsequent neutralization (data not shown).Consequently the reversibility of a pH related conformational change of HA would indi-cate that structural changes of HA revealed by fluorescence and CD-spectroscopy are not
80

the result of a pH-shift (alone). This is further supported by the study of Luykx et al. [6]who found similar decreased intrinsic tryptophanyl fluorescence upon freeze-thawingwhile no pH change appeared upon freezing. In contrast, the lack of changes in thesecondary and tertiary structure of HA freeze-thawed in HBS may indicate that Hepesacts as a cryoprotectant.
We conclude that freezing can induce structural changes of HA, depending on thetype of buffer and freezing rate. The structural integrity of HA during freeze-thawing isbest maintained when HA is dissolved in HBS and when the solution is frozen rapidly.In most cases, freeze-drying induced more pronounced changes in the structure of HAthan freezing alone, indicating that besides the freezing stresses also the drying stressescan deteriorate the structure of HA. During drying in PBS, the secondary and tertiarystructure of HA changed to a further extent resulting in increased susceptibility for tryp-sin cleavage. However freeze-drying of HA in HBS instead of PBS resulted in a less pro-nounced altered conformation of HA. The tertiary structure of HA in HBS changed to afurther extent during drying, while the secondary structure changed only after slowfreezing and subsequent drying. In conclusion, deterioration of HA during freeze-dryingalso depends on the composition of the buffer and the freezing rate.
In order to further protect HA from deterioration during freeze-drying differentcarbohydrates were investigated. Differential scanning colorimetry revealed that afterfreeze-drying HA was incorporated in a carbohydrate matrix in the amorphous glassystate (data not shown). First we investigated the influence of carbohydrates on the free-zing and drying induced damage of HA processed in PBS. After freeze-drying in the pre-sence of carbohydrates using PBS as buffer no changes in HA structure were found. Thisindicates that the carbohydrates prevented the conformational changes of HA inducedby a decrease in pH and lyophilization stress. The carbohydrates also protected HAduring freeze-drying in HBS since no changes in HA structure were found. So, the car-bohydrates protected the structural integrity of HA from the lyophilization stresses,irrespective of the type of carbohydrate used.
Immuno-aassaysBesides the maintenance of the structural integrity of the haemagglutinin, it is alsoimportant to investigate whether the antigenic properties of the subunit vaccine areaffected during processing. These properties have been evaluated both in vitro and invivo.
The antigenic properties evaluated in vitro are in line with the results concerningthe structural integrity of HA. HBS was found to be superior to PBS and the carbohydra-tes used preserved the in vitro antigenic properties of HA upon freeze-drying, indepen-dently of the type of carbohydrate.
Furthermore the vaccine powders produced with carbohydrate, except dextran 56kD, were able to retain their HA potency during storage for at least 26 weeks at20°C/0%RH. Upon storage at more challenging conditions (45°C/11%RH) the stabilizati-on of HA was the best for trehalose and the worst for dextran 56 kD. The poor stabiliza-
CHAPTER III
81

tion of HA by dextran might be due to phase separation (during freezing) and/or the bul-kiness of the dextran molecule (steric hindrance), preventing efficient hydrogen bondingwith the protein [12, 39]. This is in contrast to the small disaccharide trehalose. The lessefficient stabilization of HA at elevated temperatures by the oligosaccharide inulin com-pared to trehalose might be due to a lower extent and intimacy of hydrogen bond forma-tion [12]. Overall, the antigen activity of the powders decreased more readily when PBSwas used instead of HBS. Reasons for this could be an improper inclusion in the glassmatrix due to the pH shift during freezing when PBS is used and the capability of HBSto form an amorphous matrix [40] that acts as a stabilizer during freeze-drying and sto-rage.
The immunogenic properties of the subunit vaccine powder were evaluated byintramuscular immunization of mice. HBS was chosen because HA dissolved in this buf-fer without carbohydrates displayed the least structural changes after freeze-drying.Furthermore, despite of the better stabilization capacity of trehalose during storage atelevated temperatures, inulin 1.8 kD was chosen because of its excellent physicochemi-cal characteristics such as a high glass transition temperature and high process resistan-ce which makes it suitable for the development of formulations [13, 14, 41, 42]. Theimmunization study revealed that the subunit vaccine in the powder remained immuno-genic, as determined by IgG-titer and virus-neutralizing capacity of sera.
82

Conclusions
Subunit influenza vaccine can be obtained in the dry state with conservation of thestructural integrity and antigenic properties of HA. When using freeze-drying as dryingmethod the use of HBS is preferred over PBS. Furthermore preference should be givento a high freezing rate. Carbohydrates (trehalose, inulin and dextran) appeared to be sui-table lyoprotectants to retain the antigenic properties of HA after drying. Finally thisresearch shows that it is possible to produce an influenza subunit vaccine powder as astable powder for reconstitution. In addition, the powder may also facilitate the devel-opment of patient-friendly, non-invasive, dosage forms.
Acknowledgements
The authors wish to thank T. Meijerhof (Virosome Biologicals, Groningen, TheNetherlands) and W. ter Veer, M. Holtrop and dr. L. Bungener (Department of MedicalMicrobiology, Molecular Virology Section, University Medical Center Groningen,Groningen, The Netherlands) for their technical support. QC Influenza from SolvayPharmaceuticals, The Netherlands, is acknowledged for performing the SRID measure-ments.
CHAPTER III
83

REFERENCES
[1] Simonsen L, Clarke MJ, Schonberger LB, Arden NH, Cox NJ, Fukuda K. Pandemic versus epidemic influenza mortality: a pattern of changing age distribution. J Infect Dis 1998;178(1):53-60.
[2] Wilson IA, Cox NJ. Structural basis of immune recognition of influenza virus hemagglutinin. Annu Rev Immunol 1990;8:737-71.
[3] Wiley DC, Wilson IA, Skehel JJ. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 1981;289(5796):373-8.
[4] Stegmann T, White JM, Helenius A. Intermediates in influenza induced membrane fusion. Embo J 1990;9(13):4231-41.
[5] Stegmann T, Booy FP, Wilschut J. Effects of low pH on influenza virus. Activation and inactivation of the membrane fusion capacity of the hemagglutinin. J Biol Chem 1987;262(36):17744-9.
[6] Luykx DM, Casteleijn MG, Jiskoot W, Westdijk J, Jongen PM. Physicochemical studies on the stability of influenza haemagglutinin in vaccine bulk material. Eur J Pharm Sci 2004;23(1):65-75.
[7] Coenen F, Tolboom JT, Frijlink HW. Stability of influenza sub-unit vaccine Does a couple of days outside the refrigerator matter? Vaccine 2006;24(4):525-31.
[8] Carr CM, Chaudhry C, Kim PS. Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc Natl Acad Sci U S A 1997;94(26):14306-13.
[9] Epand RM, Epand RF. Thermal denaturation of influenza virus and its relationship to membrane fusion. Biochem J 2002;365(Pt 3):841-8.
[10] Mills KH, Skehel JJ, Thomas DB. Conformational-dependent recognition of influenza virus hemaggluti-nin by murine T helper clones. Eur J Immunol 1986;16(3):276-80.
[11] Friede M, Aguado MT. Need for new vaccine formulations and potential of particulate antigen and DNAdelivery systems. Adv Drug Deliv Rev 2005;57(3):325-31.
[12] Wang W. Lyophilization and development of solid protein pharmaceuticals. Int J Pharm 2000;203(1-2):1-60.
[13] Hinrichs WL, Prinsen MG, Frijlink HW. Inulin glasses for the stabilization of therapeutic proteins. Int J Pharm 2001;215(1-2):163-74.
[14] Eriksson HJ, Hinrichs WL, van Veen B, Somsen GW, de Jong GJ, Frijlink HW. Investigations into the stabilisation of drugs by sugar glasses: I. Tablets prepared from stabilised alkaline phosphatase. Int J Pharm 2002;249(1-2):59-70.
[15] Greiff D, Rightsel WA, Schuler EE. Effects of Freezing, Storage at Low Temperatures, and Drying bySublimation in Vacuo on the Activities of Measles Virus. Nature 1964;202:624-5.
[16] Huang J, Garmise RJ, Crowder TM, Mar K, Hwang CR, Hickey AJ, et al. A novel dry powder influenza vaccine and intranasal delivery technology: induction of systemic and mucosal immune responses in rats.Vaccine 2004;23(6):794-801.
[17] Maa YF, Ameri M, Shu C, Payne LG, Chen D. Influenza vaccine powder formulation development: spray-freeze-drying and stability evaluation. J Pharm Sci 2004;93(7):1912-23.
[18] Hagen SJ, Hofrichter J, Eaton WA. Protein reaction kinetics in a room-temperature glass. Science 1995;269(5226):959-62.
[19] Duddu SP, Zhang G, Dal Monte PR. The relationship between protein aggregation and molecular mobi-lity below the glass transition temperature of lyophilized formulations containing a monoclonal antibody. Pharm Res 1997;14(5):596-600.
[20] Molina MC, Armstrong TK, Zhang Y, Patel MM, Lentz YK, Anchordoquy TJ. The stability of lyo-philized lipid/DNA complexes during prolonged storage. J Pharm Sci 2004;93(9):2259-73.
[21] Carpenter JF, Crowe JH. An infrared spectroscopic study of the interactions of carbohydrates with dried proteins. Biochemistry 1989;28(9):3916-22.
[22] Van Alpen J, De Vries KA. Tabellenboekje ten dienste van laboratoria. Hilversum: D.B. Centens uit-geversmaatschappij, 1962.
[23] Influenza vaccine (surface antigen, inactivated). In: European Pharmacopoeia 5th edition. Strasbourg, Council of Europe, 2004: 673-74.
84

[24] Immunochemical methods (2.7.1). In: European Pharmacopoeia, 5th edition. Strasbourg, Council of Europe, 2004: 187.
[25] Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem 1951;193(1):265-75.
[26] Bensadoun A, Weinstein D. Assay of proteins in the presence of interfering materials. Anal Biochem 1976;70(1):241-50.
[27] Haan L, Verweij WR, Holtrop M, Brands R, van Scharrenburg GJ, Palache AM, et al. Nasal or intra-muscular immunization of mice with influenza subunit antigen and the B subunit of Escherichia coli heat-labile toxin induces IgA- or IgG-mediated protective mucosal immunity. Vaccine 2001;19(20-22):2898-907.
[28] Eriksson JH, Hinrichs WL, de Jong GJ, Somsen GW, Frijlink HW. Investigations into the stabilization of drugs by sugar glasses: III. The influence of various high-pH buffers. Pharm Res 2003;20(9):1437-43.
[29] Van den Berg L. Effect of freezing on the pH and composition of sodium and potassium phosphate solutions: the reciprocal system KH2PO4-Na2HPO4-H20. Arch Biochem Biophys 1959;81:319-29.
[30] Gomez G, Pikal MJ, Rodriguez-Hornedo N. Effect of initial buffer composition on pH changes during far-from-equilibrium freezing of sodium phosphate buffer solutions. Pharm Res 2001;18(1):90-7.
[31] Orii Y. Measurement of the pH of frozen buffer solutions by using pH indicators. J Biochem 1977;81(1):163-68.
[32] Croyle MA, Roessler BJ, Davidson BL, Hilfinger JM, Amidon GL. Factors that influence stability of recombinant adenoviral preparations for human gene therapy. Pharm Dev Technol 1998;3(3):373-83.
[33] Wharton SA, Skehel JJ, Wiley DC. Studies of influenza haemagglutinin-mediated membrane fusion. Virology 1986;149(1):27-35.
[34] Buxton Bridges C, Fukuda K, Holman RC, De Guzman AM, Hodder RA, Gomolin IH, et al. Decreased antibody response among nursing home residents who received recalled influenza vaccine and results of revaccination, 1996-97. Vaccine 2000;18(11-12):1103-9.
[35] Ruigrok RW, Martin SR, Wiley DC, Bayley PM, Wharton SA, Skehel JJ. Conformation changes in the haemagglutinin of influenza virus which accompany heat-induced fusion of virus with liposomes. Virology 1986;155(2):484-97.
[36] Pikal MJ. Mechanisms of protein stabilization during freeze-drying and storage: The relative importance of thermodynamic stabilization and glassy state relaxation dynamics Freeze-Drying/Lyophilization of Pharmaceutical and Biological Products. 1999;96:161-98.
[37] Remeta DP, Krumbiegel M, Minetti CA, Puri A, Ginsburg A, Blumenthal R. Acid-induced changes in thermal stability and fusion activity of influenza hemagglutinin. Biochemistry 2002;41(6):2044-54.
[38] Krumbiegel M, Herrmann A, Blumenthal R. Kinetics of the low pH-induced conformational changes and fusogenic activity of influenza hemagglutinin. Biophys J 1994;67(6):2355-60.
[39] Allison SD, Chang B, Randolph TW, Carpenter JF. Hydrogen bonding between sugar and protein is responsible for inhibition of dehydration-induced protein unfolding. Arch Biochem Biophys 1999;365(2):289-98.
[40] Hinrichs WL, Sanders NN, De Smedt SC, Demeester J, Frijlink HW. Inulin is a promising cryo- and lyo-protectant for PEGylated lipoplexes. J Control Release 2005;103(2):465-79.
[41] Eriksson HJ, Verweij WR, Poelstra K, Hinrichs WL, de Jong GJ, Somsen GW, et al. Investigations into the stabilisation of drugs by sugar glasses: II. Delivery of an inulin-stabilised alkaline phosphatase in the intestinal lumen via the oral route. Int J Pharm 2003;257(1-2):273-81.
[42] Van Drooge DJ, Hinrichs WL, Dickhoff BH, Elli MN, Visser MR, Zijlstra GS, et al. Spray freeze drying to produce a stable Delta(9)-tetrahydrocannabinol containing inulin-based solid dispersion powder suitable for inhalation. Eur J Pharm Sci 2005;26(2):231-40.
CHAPTER III
85

86

J. de Jonge1 *, J-P. Amorij2 *, W.L.J. Hinrichs2, J. Wilschut1, A. Huckriede1, H.W. Frijlink2
Chapter IV
European Journal of Pharmaceutical Sciences 32 (2007): 33-44.
1 Department of Medical Microbiology, Molecular Virology Section, University Medical Center Groningen and University of Groningen, Groningen, The Netherlands.
2 Department of Pharmaceutical Technology and Biopharmacy, University of Groningen The Netherlands.
* These authors contributed equally to this work.
Inulin sugar glasses preserve the structuralintegrity and biological activity of influenzavirosomes during freeze-ddrying and storage.

Abstract
Influenza virosomes are reconstituted influenza virus envelopes that may be used as vac-cines or as carrier systems for cellular delivery of therapeutic molecules. Here we presenta procedure to generate influenza virosomes as a stable dry-powder formulation by freeze-drying (lyophilization) using an amorphous inulin matrix as a stabilizer. In thepresence of inulin the structural integrity and fusogenic activity of virosomes were fullypreserved during freeze-drying. For example, the immunological properties of the virosomes, i.e. the HA potency in vitro and the immunogenic potential in vivo, weremaintained during lyophilization in the presence of inulin. In addition, compared tovirosomes dispersed in buffer, inulin-formulated virosomes showed substantially prolon-ged preservation of the HA potency upon storage. Also the capacity of virosomes tomediate cellular delivery of macromolecules was maintained during lyophilization in thepresence of inulin and upon subsequent storage. Specifically, when dispersed in buffer,virosomes with encapsulated plasmid DNA lost their transfection activity completelywithin 6 weeks, whereas their transfection activity was fully preserved for at least 12 weeks after incorporation in an inulin matrix. Thus, in the presence of inulin as a sta-bilizing agent, the shelf-life of influenza virosomes with and without encapsulatedmacromolecules was considerably prolonged. Formulation of influenza virosomes as adry-powder is advantageous for storage and transport and offers the possibility to de-velop needle-free dosage forms e.g. for oral, nasal, pulmonal, or dermal delivery.
Keywords
Delivery vehicle; influenza vaccine; gene delivery; stabilization; lyophilization; formulation of virosomes.
88

Introduction
Virosomes are reconstituted viral membrane vesicles that are prepared from envelopedviruses. They consist of the viral spike glycoproteins embedded in a bilayer formed bythe viral membrane lipids but are devoid of the genetic material of the virus. Upon pro-per reconstitution, the structural integrity and biological activity of the viral spike gly-coproteins are preserved and consequently, virosomes exhibit receptor-binding andmembrane-fusion properties similar to those of the virus they are derived from.
Influenza virus has been one of the viruses most commonly used for the generationof virosomes [1-3]. The surface of the influenza virus is decorated with two spike pro-teins, haemagglutinin (HA) and neuraminidase (NA). HA is involved in receptor-bindingand membrane fusion. Fusion is induced by a conformational change, which is triggeredby the low pH encountered after uptake of the virus particles in Endosomes [4]. One ofthe roles of NA is to release the virus from the cell surface during the budding process.HA constitutes the major antigen of influenza virus and induces a strong humoral immu-ne response during infection [4].
Previously, we published on methods for the reconstitution of influenza HA in viro-somes [2, 5, 6]. When prepared according to these methods, influenza virosomes aresimilar in morphology and size to native influenza virus. Moreover, these virosomesexhibit HA-mediated fusion with target membranes, comparable to native virus [2, 5, 6]. Because of their structural resemblance with native virus, influenza virosomes are suita-ble as vaccines [7, 8]. Currently, virosomal influenza vaccines are marketed by BernaBiotech and Solvay Pharmaceuticals under the trade names Inflexal V® and Invivac®.These vaccines have been evaluated for potency in several clinical studies and have beenfound to be safe and to induce high and long-lasting antibody titers [7, 9, 10].
Due to their fusogenic properties, virosomes can be utilized for cellular delivery ofvarious compounds including peptides, proteins and nucleic acids as reviewed in [11].For this purpose, peptides and proteins have been encapsulated passively into the lumenof virosomes [12, 13]. Early procedures to deliver nucleic acids with the help of viroso-mes relied on binding of the molecules to the surface of virosomes by means of cationiclipids co-reconstituted in the virosomal membrane [14, 15]. Recently, we developed aprocedure to encapsulate nucleic acids (such as small interfering RNA (siRNA) [16] andplasmid DNA (pDNA) (unpublished results)) in the lumen of virosomes. Using this pro-cedure the encapsulated nucleic acids are completely protected from nuclease activitywhile they are efficiently transported to the cytosol of target cells. Virosomes withencapsulated DNA or RNA may pose as an alternative for currently used viral and non-viral delivery systems applied for gene therapy and RNA interference.
The clinical use of virosomes could be further facilitated by improving their phar-maceutical properties. Since virosomes are produced in aqueous solution, they are inhe-rently prone to chemical and physical degradation as has been observed for a variety ofother biopharmaceuticals [17]. Degradation might affect the immunological properties ofHA as was shown for influenza HA in a subunit vaccine formulation [18]. Moreover, the
CHAPTER IV
89

structural integrity of virosomes and thereby most likely also the fusion activity of HA,both being essential for cellular delivery of virosome-encapsulated compounds, may belost upon storage in aqueous solution.
The limited shelf-life of virosomes calls for the development of formulations thatprovide adequate stability during manufacturing, transport and storage. The most com-monly used method to stabilize biologically active macromolecules, such as proteins,vaccines and gene delivery systems, is to convert them to a dry-powder formulation.Generally, freeze-drying (lyophilization) is the preferred drying method for pharmaceu-tical products. However, during the freeze-drying procedure, freezing and drying stres-ses may affect the structural integrity and/or activity of the product. Accordingly, lyop-rotectants are required for the preservation of these properties.
It is well known that sugars can stabilize proteins [19-24], liposomes, lipoplexes [25-30] and various viruses [31-34] during freeze-drying and subsequent storage. If driedproperly, the active substance, complex or vesicle is incorporated in a matrix consistingof sugar in its glass state. The stabilizing effect of these sugar glasses has been explainedby the formation of a sugar matrix which acts as a physical barrier between particles(particle isolation) and strongly reduces diffusion and molecular mobility (vitrification).Both the physical barrier [35] and the lack of mobility [36] provided by the glass matrix,prevent aggregation and degradation of the dried material. Moreover, during the lyophi-lization process, the sugar replaces the water molecules in the hydrogen-bonding inter-action with the active material, such that the structural integrity of the drug is preserved[37]. Under dry conditions, the glass matrix is maintained as long as the temperature iskept below the glass transition temperature (Tg), which is characteristic for the stabili-zing sugar used. Above the Tg, the glass state turns into a rubber state, which has a hig-her degree of mobility.
The oligosaccharide inulin is a potent lyoprotectant, since it possesses physicoche-mical properties essential for a stabilizing agent, such as high Tg, high Tg of the maximal-ly freeze-concentrated fraction, low number of reducing groups and a low crystallizati-on rate [23]. In several studies, inulin has been successfully used for the stabilization ofproteins, liposomes, lipoplexes and polyplexes [23, 29, 30].
The aim of this study was to develop a stable dry-powder formulation of influenzavirosomes with the objective to preserve the structural integrity and biological activityof the virosomes. To this end, inulin was used as a lyoprotectant and stabilizer. To assessthe lyoprotecting capacity of inulin, physical and functional properties of virosomeswere studied before and after freeze-drying with and without inulin and were followedduring storage. This study shows that during freeze-drying in the presence of inulin, thestructural integrity of virosomes is retained. Moreover, the immunogenic properties andtransfecting capacities of the virosomes are preserved during both lyophilization andsubsequent storage.
90

Materials and Methods
Materials1,2-dihexanoyl-sn-glycero-3-phosphocholine (DCPC) and 1,2-dioleoyl-3-trimethylam-monium-propane (DOTAP) were obtained from Avanti Polar Lipids, Inc (Alabaster, AL,USA). 1-hexadecanoyl-2-(1-pyrenedecanoyl)-sn-glycero-3-phosphocholine (pyrene-PC)was obtained from Molecular Probes (Eugene, OR, USA). Influenza virus(A/Panama/2007/99) was kindly provided by Solvay Pharmaceuticals B.V. (Weesp, theNetherlands). Inulin 1.8 kD (type HD001111) was a generous gift from Sensus(Roosendaal, The Netherlands). The pCMV-EGFP plasmid, encoding the enhanced greenfluorescent protein was a kind gift of Alida Weeke-Klimp (Department of Cell Biology,Liposome Research Section, University Medical Center Groningen). All other chemicalswere of reagent or analytical grade.
Preparation of virosomesThe influenza virus membrane proteins and lipids were isolated by solubilization of theviral membrane components with 200 mM DCPC followed by removal of the nucleo-capsid by ultracentrifugation as described previously [6]. The supernatant containing thedissolved membrane components was used for the reconstitution of the viral membranes.
Virosomes to be used as influenza vaccines were generated by the fast-reconstitu-tion method as described before [6]. In brief, the supernatant containing the isolatedviral lipids and membrane proteins was instantly diluted 5 times with HEPES bufferedsaline (HBS; 5 mM HEPES, 0.15 M NaCl, pH 7.4) and subsequent reconstitution of theviral membrane was achieved through removal of DCPC by dialysis against HBS. Thepreparation was further purified on a discontinuous sucrose gradient and then dialysedagainst HBS.
DNA-virosomes were prepared based on a previously published protocol for thepreparation of virosomes with encapsulated siRNA with adaptations [16]. ThepCMV/EGFP plasmid (65 μg in 130 μL) was added to a dried film of DOTAP cationiclipid (0.808 μmol) and incubated for 15 min. The supernatant containing the isolatedviral membrane components (1.5 μmol of phospholipids) was added to the cationiclipid/pDNA mixture and incubated for 30 min. Subsequently, DNA-virosomes wereobtained by dialysis, purified on a discontinuous sucrose gradient and then dialysedagainst HBS as described before [16].
Biochemical analysisVirosomes were analyzed for protein content by a modified Lowry assay [38] and forpDNA content by the PicoGreen assay [39]. The phospholipid content was establishedby phosphate determination [40] and when appropriate, corrected for phosphate presentin the pDNA.
CHAPTER IV
91

Freeze-ddryingFreeze-drying was carried out in a Christ Alpha 1-4 freeze dryer (Salm en Kipp,Breukelen, The Netherlands) according to a method described previously [23]. In typicalexperiments, 150-300 μL aqueous dispersions containing 225-450 μg/mL virosomes (sum-mation of protein, phospholipid and where appropriate pDNA concentrations) and 22.5-45 mg/L carbohydrate (carbohydrate/virosome ratio of 100 w/w) were frozen in 4 mLglass vials in liquid nitrogen for 10 minutes and subsequently lyophilized. The freeze-dryer was set at a shelf temperature of -35°C, a condenser temperature of -55°C and apressure of 0.220 mbar. After 24 h the pressure was lowered to 0.060 mbar, and the shelftemperature was gradually increased to 20°C, which was maintained for 24 h.
The dry samples were either rehydrated or stored in a desiccator containing driedsilica gel at 4, 20 or 42°C for the stability study. Control virosomes dispersed in bufferwere supplemented with 0.01 % (w/v) sodium azide to prevent bacterial and fungalgrowth during storage.
Sucrose density gradient analysisThe virosome preparations were analyzed for co-migration of phospholipid and proteinon a linear sucrose density gradient (10-60% sucrose (w/v) in HBS). The sucrose gra-dients were centrifuged at 300.000 g for 65 h at 4°C (SW55Ti). Fractions were analyzedfor protein and phosphate content as described above.
Electron microscopyVirosomes were dialyzed against ammonium acetate buffer (75 mM ammonium acetate,2.5 mM Hepes, pH 7.4) overnight at 4°C. Subsequently, the virosome suspensions wereapplied to glow-discharged 200 mesh grids covered with a Formvar film. Virosomes werestained with freshly prepared 3% ammonium molybdate, pH 7.2 and analyzed on aPhilips CM 12 transmission electron microscope.
Membrane fusionFor determination of the fusogenic properties of DNA-virosomes 10 mol% pyrene-PCrelative to the amount of viral phospholipids was incorporated in the virosomal mem-brane during production. For this purpose, the required amount of pyrene-PC togetherwith the cationic lipid was dried down as a film and virosomes were then prepared asdescribed above. To study the fusion characteristics of the virosomes, pH-induced fusionwith erythrocyte ghosts as target membranes was measured, and fusion inactivation byexposure to low pH was performed, as described before [41, 42].
Single radial immunodiffusionThe single radial immunodiffusion (SRID) method was used to evaluate HA potency ofvirosomes. The test is a quantitative immunodiffusion technique and was used as des-cribed in the European Pharmacopoeia [43, 44].
92

Immunization studiesAnimal experiments were conducted according to the guidelines provided by the DutchAnimal Protection Act, and were approved by the Committee for AnimalExperimentation (DEC) of the University of Groningen. For all experiments femaleBALB/c mice of age 6-8 weeks (Harlan, Zeist, Netherlands) were used. Mice (8 per group)were immunized intramuscularly in the right hind leg with 5 μg unprocessed virosomesor rehydrated inulin-stabilized virosomes in 30 μL, based on protein content. The miceof the control group (n=3) received only HBS. On day 21, mice were bled by an orbitapunction under isofluran/oxygen anesthesia for collection of blood and then sacrificed.Serum was isolated by centrifugation at 11.000 x g and stored at -20°C until further ana-lysis.
Influenza antigen-specific antibody responses were determined using an ELISA asdescribed previously [45], using A/Panama HA and horseradish peroxidase-conjugatedgoat antibodies directed against mouse IgG (Southern Biotech Associate; dilution 1:5000)as antigen and secondary antibody, respectively. Titers are given as the reciprocal of thecalculated sample dilution corresponding to an A490 = 0.2 after background correction.
Haemagglutination-inhibition (HI) titers of sera were determined by a haemagglu-tination test as described by de Haan et al. [45] using 4 haemagglutination units (HAU)of A/Panama influenza virus and a 1% guinea pig erythrocyte suspension. The highestdilution of serum capable of preventing haemagglutination was scored as the HI-titer.
Student's t-test was used to compare the different experimental groups. Probability(P) values < 0.05 were considered significant.
TransfectionBHK-21 cells were grown at 37°C and 5% CO2 in BHK-21 medium (Glasgow MEM,Invitrogen, Paisly, Scotland), supplemented with 5% FCS (Bodinco BV, Alkmaar, TheNetherlands), 20 mM HEPES (Invitrogen), 10% tryptose phosphate broth (Invitrogen),100 U/mL penicillin and 100 μg/mL streptomycin. The cells (5x104) were seeded in 24 well-plates containing 1 mL culture medium. After 24 h, DNA-virosomes containing750 ng pCMV/EGFP plasmid were added to the cells. To examine whether virosome-mediated transfection was pH- and thus HA-dependent, NH4Cl (final concentration of20 mM) was added to the BHK-21 cells 20 min prior to transfection. This treatment inhi-bits acidification of endosomes [46]. Alternatively, experiments were performed withDNA-virosomes that had been fusion-inactivated by low-pH treatment prior to transfec-tion [42]. After 48 h, cells were analyzed on a Zeiss Axiovert microscope equipped withepifluorescence. Subsequently, total EGFP fluorescence per well was determined on aFL500 microplate fluorescence reader (Bio-Tek instruments) with excitation filters set at485 nm (bp 20 nm) and emission filters set at 530 nm (bp 25 nm) and finally cells werewashed, trypsinized and analyzed for EGFP expression by flow cytometry (Epics Elite,Coulter, Miami, USA).
CHAPTER IV
93
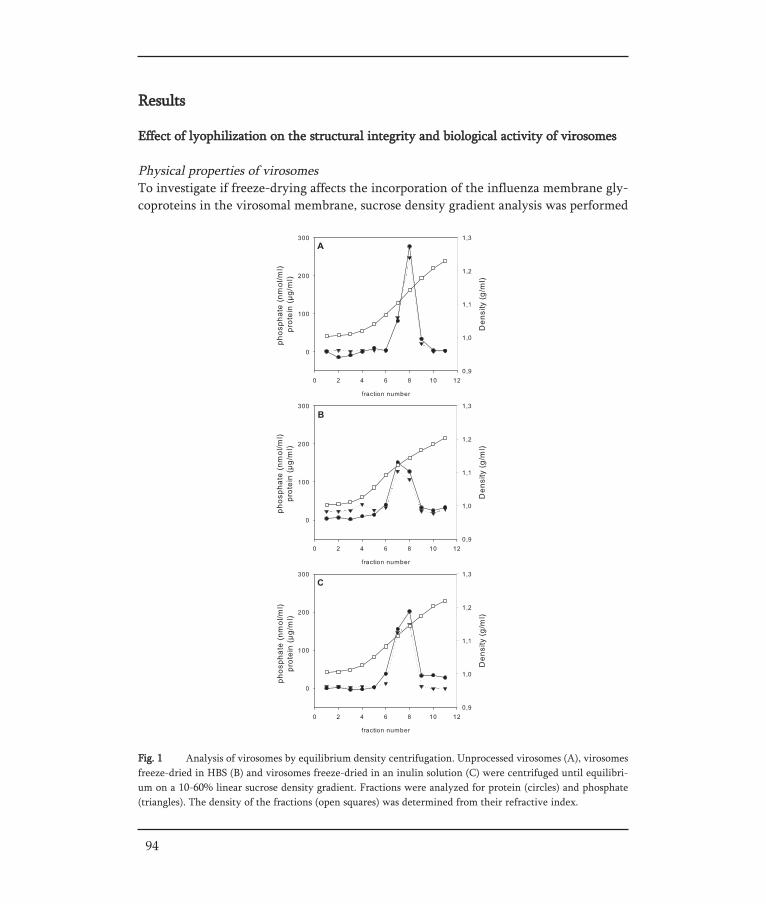
Fig. 1 Analysis of virosomes by equilibrium density centrifugation. Unprocessed virosomes (A), virosomesfreeze-dried in HBS (B) and virosomes freeze-dried in an inulin solution (C) were centrifuged until equilibri-um on a 10-60% linear sucrose density gradient. Fractions were analyzed for protein (circles) and phosphate(triangles). The density of the fractions (open squares) was determined from their refractive index.
Results
Effect of lyophilization on the structural integrity and biological activity of virosomes
Physical properties of virosomesTo investigate if freeze-drying affects the incorporation of the influenza membrane gly-coproteins in the virosomal membrane, sucrose density gradient analysis was performed
94

Fig. 2 Analysis of virosomes by negative stain transmission electron microscopy. Unprocessed virosomes(A), virosomes freeze-dried in HBS (B) and virosomes freeze-dried in an inulin solution (C). Arrowheads in-dicate spike proteins and arrows indicate protein and phospholipid aggregates. Bar represents 100 nm.
CHAPTER IV
95
on untreated virosomes or virosomes freeze-dried either in HBS or in an inulin solution.During centrifugation to equilibrium, phospholipid and protein of the untreated viroso-mes co-migrated as a single band in the sucrose gradient (Fig. 1A), which indicates thatthese components are physically associated. A similar migration pattern was observed forvirosomes freeze-dried in an inulin solution (Fig. 1C). When virosomes were freeze-dried without a lyoprotectant, protein and phospholipids were also detected in identicalfractions (Fig. 1B). Apparently, freeze-drying either in buffer or in an inulin solutiondoes not affect the physical association of protein and phospholipids.
Particulate nature of virosomesTo investigate the effect of freeze-drying on virosome morphology, samples of treatedand untreated virosomes were analyzed by transmission electron microscopy. Analysis ofuntreated virosomes revealed vesicles of approximately 100-150 nm in diameter with
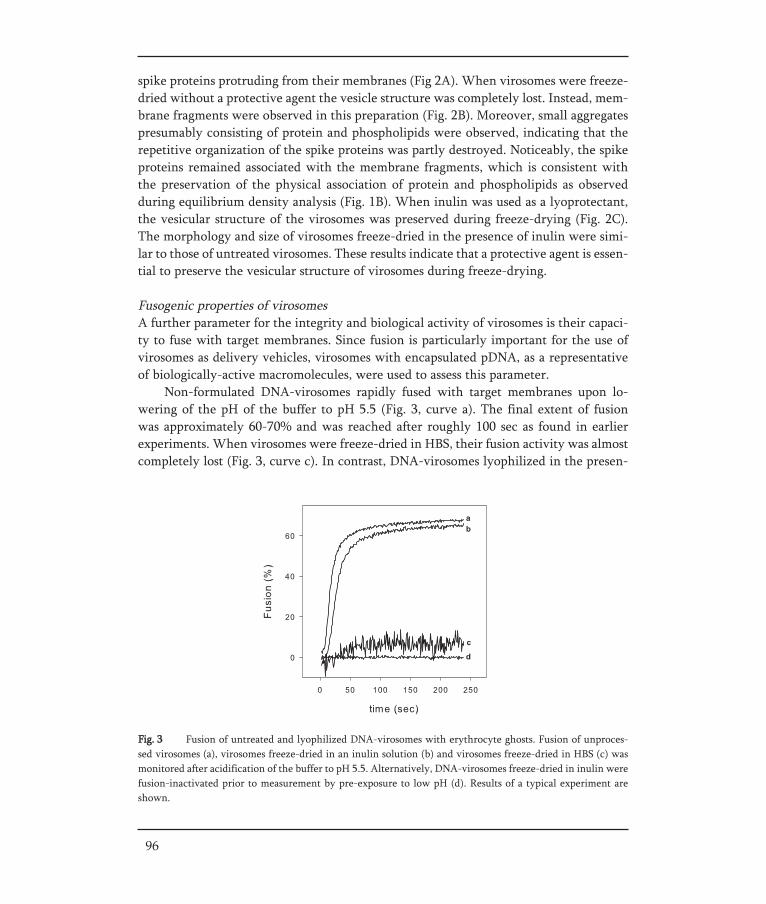
Fig. 3 Fusion of untreated and lyophilized DNA-virosomes with erythrocyte ghosts. Fusion of unproces-sed virosomes (a), virosomes freeze-dried in an inulin solution (b) and virosomes freeze-dried in HBS (c) wasmonitored after acidification of the buffer to pH 5.5. Alternatively, DNA-virosomes freeze-dried in inulin werefusion-inactivated prior to measurement by pre-exposure to low pH (d). Results of a typical experiment areshown.
spike proteins protruding from their membranes (Fig 2A). When virosomes were freeze-dried without a protective agent the vesicle structure was completely lost. Instead, mem-brane fragments were observed in this preparation (Fig. 2B). Moreover, small aggregatespresumably consisting of protein and phospholipids were observed, indicating that therepetitive organization of the spike proteins was partly destroyed. Noticeably, the spikeproteins remained associated with the membrane fragments, which is consistent withthe preservation of the physical association of protein and phospholipids as observedduring equilibrium density analysis (Fig. 1B). When inulin was used as a lyoprotectant,the vesicular structure of the virosomes was preserved during freeze-drying (Fig. 2C).The morphology and size of virosomes freeze-dried in the presence of inulin were simi-lar to those of untreated virosomes. These results indicate that a protective agent is essen-tial to preserve the vesicular structure of virosomes during freeze-drying.
Fusogenic properties of virosomesA further parameter for the integrity and biological activity of virosomes is their capaci-ty to fuse with target membranes. Since fusion is particularly important for the use ofvirosomes as delivery vehicles, virosomes with encapsulated pDNA, as a representativeof biologically-active macromolecules, were used to assess this parameter.
Non-formulated DNA-virosomes rapidly fused with target membranes upon lo-wering of the pH of the buffer to pH 5.5 (Fig. 3, curve a). The final extent of fusion was approximately 60-70% and was reached after roughly 100 sec as found in earlierexperiments. When virosomes were freeze-dried in HBS, their fusion activity was almostcompletely lost (Fig. 3, curve c). In contrast, DNA-virosomes lyophilized in the presen-
96
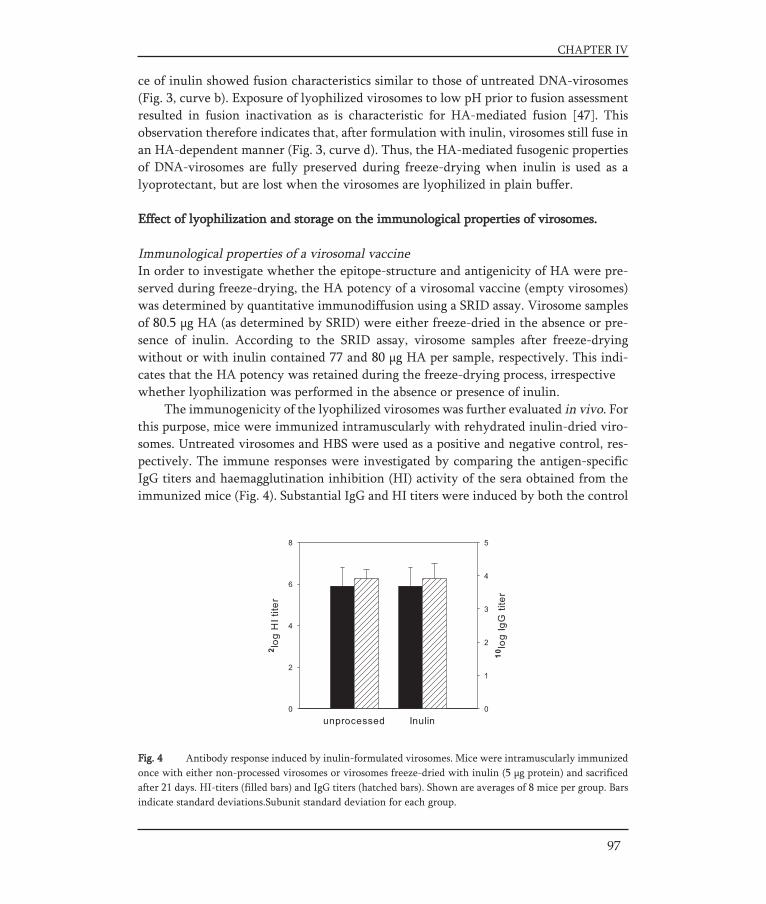
Fig. 4 Antibody response induced by inulin-formulated virosomes. Mice were intramuscularly immunizedonce with either non-processed virosomes or virosomes freeze-dried with inulin (5 μg protein) and sacrificedafter 21 days. HI-titers (filled bars) and IgG titers (hatched bars). Shown are averages of 8 mice per group. Barsindicate standard deviations.Subunit standard deviation for each group.
ce of inulin showed fusion characteristics similar to those of untreated DNA-virosomes(Fig. 3, curve b). Exposure of lyophilized virosomes to low pH prior to fusion assessmentresulted in fusion inactivation as is characteristic for HA-mediated fusion [47]. Thisobservation therefore indicates that, after formulation with inulin, virosomes still fuse inan HA-dependent manner (Fig. 3, curve d). Thus, the HA-mediated fusogenic propertiesof DNA-virosomes are fully preserved during freeze-drying when inulin is used as alyoprotectant, but are lost when the virosomes are lyophilized in plain buffer.
Effect of lyophilization and storage on the immunological properties of virosomes.
Immunological properties of a virosomal vaccineIn order to investigate whether the epitope-structure and antigenicity of HA were pre-served during freeze-drying, the HA potency of a virosomal vaccine (empty virosomes)was determined by quantitative immunodiffusion using a SRID assay. Virosome samplesof 80.5 μg HA (as determined by SRID) were either freeze-dried in the absence or pre-sence of inulin. According to the SRID assay, virosome samples after freeze-drying without or with inulin contained 77 and 80 μg HA per sample, respectively. This indi-cates that the HA potency was retained during the freeze-drying process, irrespective whether lyophilization was performed in the absence or presence of inulin.
The immunogenicity of the lyophilized virosomes was further evaluated in vivo. Forthis purpose, mice were immunized intramuscularly with rehydrated inulin-dried viro-somes. Untreated virosomes and HBS were used as a positive and negative control, res-pectively. The immune responses were investigated by comparing the antigen-specificIgG titers and haemagglutination inhibition (HI) activity of the sera obtained from theimmunized mice (Fig. 4). Substantial IgG and HI titers were induced by both the control
CHAPTER IV
97
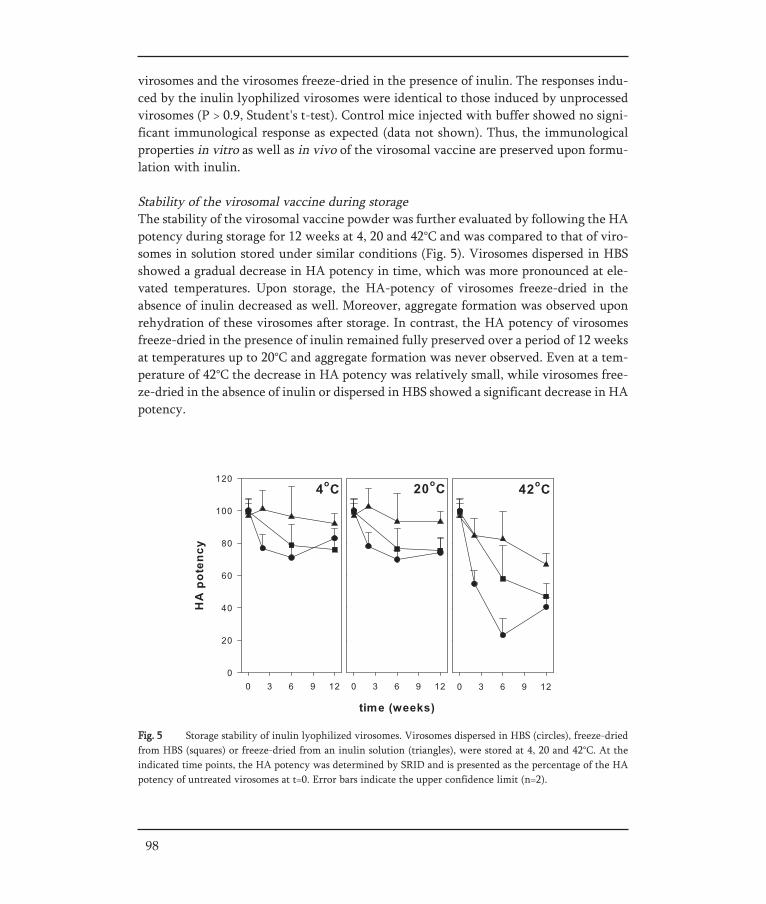
98
virosomes and the virosomes freeze-dried in the presence of inulin. The responses indu-ced by the inulin lyophilized virosomes were identical to those induced by unprocessedvirosomes (P > 0.9, Student's t-test). Control mice injected with buffer showed no signi-ficant immunological response as expected (data not shown). Thus, the immunologicalproperties in vitro as well as in vivo of the virosomal vaccine are preserved upon formu-lation with inulin.
Stability of the virosomal vaccine during storageThe stability of the virosomal vaccine powder was further evaluated by following the HApotency during storage for 12 weeks at 4, 20 and 42°C and was compared to that of viro-somes in solution stored under similar conditions (Fig. 5). Virosomes dispersed in HBSshowed a gradual decrease in HA potency in time, which was more pronounced at ele-vated temperatures. Upon storage, the HA-potency of virosomes freeze-dried in theabsence of inulin decreased as well. Moreover, aggregate formation was observed uponrehydration of these virosomes after storage. In contrast, the HA potency of virosomesfreeze-dried in the presence of inulin remained fully preserved over a period of 12 weeksat temperatures up to 20°C and aggregate formation was never observed. Even at a tem-perature of 42°C the decrease in HA potency was relatively small, while virosomes free-ze-dried in the absence of inulin or dispersed in HBS showed a significant decrease in HApotency.
Fig. 5 Storage stability of inulin lyophilized virosomes. Virosomes dispersed in HBS (circles), freeze-driedfrom HBS (squares) or freeze-dried from an inulin solution (triangles), were stored at 4, 20 and 42°C. At theindicated time points, the HA potency was determined by SRID and is presented as the percentage of the HApotency of untreated virosomes at t=0. Error bars indicate the upper confidence limit (n=2).

Fig. 6 Microscopical analysis of BHK-21 cells transfected with DNA-virosomes. BHK-21 cells (5x104) weregrown for 24 h and exposed to unprocessed DNA-virosomes (A,B), DNA-virosomes freeze-dried in HBS (C,D)and inulin-lyophilized DNA-virosomes (E,F), all containing 750 ng of encapsulated CMV/EGFP reporter pla-smid. Cells were analyzed after 48 h by phase contrast microscopy (lefts panels) and fluorescence microscopy(right panels). Bar represents 100 μm.
Effect of lyophilization and storage on the cellular delivery properties of virosomes
Transfection properties of DNA-virosomesTo evaluate whether the cellular delivery capacity of virosomes was preserved duringlyophilization, the ability of virosomes with encapsulated pDNA to transfect culturedcells was assessed prior to or after freeze-drying either with or without inulin. For thispurpose, BHK-21 cells (5 x 104) were grown for 24 h and subsequently exposed for 48 hto either non-processed or processed virosomes containing 750 ng of encapsulated pDNAencoding the EGFP reporter gene.
Analysis of the cells by light microscopy revealed that control DNA-virosomes andDNA-virosomes lyophilized in the presence of inulin did not induce any obvious toxiceffects (Fig. 6A,E). In contrast, incubation of the cells with DNA-virosomes lyophilizedfrom HBS induced extensive cell detachment and cell death (Fig. 6C). Non-processedDNA-virosomes efficiently mediated transfection resulting in a high percentage of cellsexpressing large amounts of EGFP, as observed by fluorescent microscopy (Fig. 6B). Onthe other hand, DNA-virosomes freeze-dried in HBS, only transfected a small number ofcells (Fig. 6D). In contrast, DNA-virosomes lyophilized in inulin induced similar trans-fection percentages and EGFP expression levels as control DNA-virosomes, which indi-cates that their transfection capacity was maintained during freeze-drying (Fig. 6F).
CHAPTER IV
99
Fig. 9 Storage stability of inulin-lyophilized DNA-virosomes. DNA-virosomes freeze-dried in an inulinsolution were stored at 4°C (triangles) and 20°C (squares) and compared with DNA-virosomes dispersed in HBSstored at 4°C (circles). At the indicated time points, the transfection percentage (see Fig 7) was determined induplicate. Duplicates of the means shown varied maximally by 4%.
100
Fig. 7 Quantitative analysis of transfected BHK-21 cells. BHK-21 cells were transfected with DNA-viro-somes as described in Fig. 6. After 48 h, total EGFP fluorescence, expressed as percentage fluorescence ofunprocessed DNA-virosomes, was determined using a fluorescence platereader (grey bars) and the percentageof transfected cells within the vital population was determined by flow cytometry (black bars). Error bars indicate standard deviations (n=3).
Fig. 8 HA-mediated transfection of BHK-21 cells. To study if transfection was dependent on the fusogenicproperties of HA, virosomes were either left untreated (black bars; n=3; error bars represent standard devia-tions) or were pH-inactivated prior to addition to cells (hatched bars; n=2; error bars represent higher/lowervalues). Alternatively, transfection was performed with BHK-21 cells which were grown in the presence of theendosomal acidification blocker NH4Cl (empty bars; n=2; error bars represent higher/lower values).Transfections were performed as described in Fig. 6 and cells were analyzed after 48 hr by flow cytometry.

CHAPTER IV
101
Total EGFP expression of BHK-21 cells incubated with non-processed and proces-sed DNA-virosomes was quantified by fluorescence determination. DNA-virosomeslyophilized in buffer induced only 35% of the EGFP expression obtained with non-pro-cessed virosomes, while the EGFP expression levels obtained with DNA-virosomes for-mulated with inulin were similar to those of control virosomes (Fig. 7).
Flow-cytometric analysis of the BHK-21 cells revealed that 80% of the cells weretransfected when incubated with control DNA-virosomes or inulin-formulated freeze-dried DNA-virosomes (Fig. 7). For DNA-virosomes freeze-dried in HBS, approximately40% of the vital cells were transfected (Fig. 7), while many cells were lost due to severetoxic effects as described above (Fig. 6C) and could not be analyzed. Thus, the transfec-tion efficiency in terms of total EGFP expression and percentage of transfected cells waspreserved when DNA-virosomes were freeze-dried in the presence of inulin.
To study if transfection by the inulin-formulated DNA-virosomes was still media-ted by HA-induced membrane fusion, the HA of the inulin-formulated DNA-virosomeswas pH-inactivated prior to transfection. Alternatively, DNA-virosomes were added toBHK-21 cells, which were grown in the presence of NH4Cl, which is known to inhibitacidification of endosomes [46]. For both conditions, transfection by control DNA-viro-somes and inulin-formulated DNA-virosomes was almost completely blocked (Fig. 8).This indicates that transfection is due to the fusogenic properties of HA and that theseproperties are not affected by lyophilization using inulin.
Stability of DNA-virosomes during storageThe functional performance of DNA-virosomes freeze-dried in the presence of inulinwas evaluated by monitoring their transfection efficiencies (Fig. 9) during storage at 4°Cand 20°C, using untreated virosomes dispersed in HBS and stored at 4°C as a reference.In these reference samples, precipitates were visible by eye within a few days, indicatingextensive virosome aggregation.
The transfection capacity of untreated DNA-virosomes was completely lost after 6 weeks of storage at 4°C. In contrast, inulin-lyophilized DNA-virosomes largely retai-ned their transfection activity over a period of up to 12 weeks when stored at 4°C. At amore challenging condition (20°C), the transfection properties of inulin freeze-driedDNA-virosomes were preserved over a period of at least 6 weeks.
Assessment of membrane fusion showed that the fusogenic properties followed thesame trend as the transfection properties being lost rapidly in untreated DNA virosomesand preserved for extended time periods in inulin-stabilized samples (data not shown).These results indicate that application of inulin sugar glass technology increases theshelf-life of DNA-virosomes stored at low as well as ambient temperatures.

Discussion
Here we demonstrate that the structural integrity and functional properties of influenzavirosomes are preserved during freeze-drying in the presence of inulin. Inclusion of viro-somes in the amorphous inulin glass matrix does not affect the immunological propertiesof influenza virosomal vaccine during freeze-drying as demonstrated by the preserva-tion of the HA potency and the ability of the vaccine to induce an immune response inmice. Similarly, the capacity of virosomes to mediate cellular delivery of plasmid DNAwas maintained during lyophilization, as shown by efficient transfection of cultured cellsby virosomes with encapsulated DNA after freeze-drying and reconstitution. Moreover,we demonstrate that formulation with inulin greatly improves the stability of the viro-somes during storage.
Freeze-drying is a technique frequently applied for stabilization of biopharmaceuti-cal products. In addition to simple peptides and proteins, also more complex structuressuch as liposomes and lipoplexes (lipid/DNA complexes) have been successfully lyophi-lized [29, 30, 48]. Viruses consisting of a viral capsid (containing the protein-coated viralDNA or RNA) and optionally a lipid membrane with inserted glycoproteins are amongthe most complex structures stabilized by freeze-drying [31-34]. Virosomes when usedas vaccines or transport vehicles for encapsulated cargo are comparable in complexity toenveloped viruses. To our knowledge we are the first to describe a procedure for lyophi-lization of virosomes compatible with their use for vaccination and for delivery pur-poses.
The choice of the buffer system is essential in the process of freeze-drying. In thisstudy, HBS was used as buffer system instead of the more commonly applied PBS, sincea pH shift of PBS during freezing [49-51] might affect the fusion properties of the viro-somes. Additionally, in a previous study it was found that HBS provides better protec-tion of an HA subunit vaccine during lyophilization and subsequent storage than PBS(Amorij, submitted). Indeed, in this latter study it was shown that HBS is a reasonablelyoprotectant for the preservation of the immunological properties of HA, However,another stabilizer is required for storage.
When virosomes are to be used for influenza vaccination, preservation of the immu-nological properties of the viral HA is essential. Moreover, it has been suggested that therepetitive arrangement of the HA antigen might contribute to the high immunogenicityof virosomal influenza vaccines [8] and therefore the preservation of the particulatenature of the virosomes might be of importance.
Our results show that the process of freeze-drying in the presence of inulin had little effect on the HA potency of the virosomal vaccine nor on its capacity to induce HA-specific antibodies upon immunization of mice. Interestingly, for the preservation of theHA potency during freeze-drying, a lyoprotectant was not required. This is in contrastto observations made on influenza subunit vaccine, the HA potency of which decreasedsubstantially when freeze-dried without protection (about 25% decrease, (Chapter II)).Possibly, embedding of HA in the natural environment of the viral lipids as achieved in
102

virosomes provides extra stability to HA and/or prevents aggregation of HA moleculesduring lyophilization. However, the presence of inulin was essential for preservation ofthe HA potency during storage. Virosomes lyophilized without a sugar formed aggre-gates upon rehydration and showed a decrease in HA potency. Moreover, the HA poten-cy of virosomes incorporated in inulin was preserved over a longer period of time thanthat of virosomes dispersed in buffer. This increased shelf-life may be highly beneficialfor the widespread use of virosomes as influenza vaccines.
For preservation of the particulate nature of virosomes, the presence of inulinduring freeze-drying was essential. Freeze-drying in buffer resulted in disintegration ofthe vesicles to small membrane fragments and thus in the loss of the virosome structureand the organized arrangement of the viral HA. The preservation of the vesicle struc-ture in the presence of inulin is in line with previous studies [25, 52, 53], which showthat the oligosaccharide interacts with membrane lipids and as such may preserve thestructural and functional features of membrane vesicles during dehydration.
Besides their application as vaccines, virosomes may also be used as delivery systemsfor various compounds encapsulated in the virosome lumen including toxins, peptides,protein antigens, plasmid DNA and siRNA. Cellular delivery of virosome-encapsulatedmaterial relies on the binding of virosomes to cellular receptors and especially on HA-mediated fusion of the virosomal and the endosomal membrane after virosome inter-nalization through receptor-mediated endocytosis [12, 16, 54]. Successful freeze-dryingof virosomes intended for use as delivery devices obviously demands preservation of thevesicle structure since loss of the structural integrity of the virosomal vesicles would leadto loss of the encapsulated material. As discussed above, virosomes maintain their vesicle structure only when freeze-dried in the presence of inulin.
With respect to fusion activity, our study shows that freeze-drying in the presenceof inulin preserves the fusogenic properties of virosomes. In contrast, freeze-drying inbuffer resulted in immediate and complete loss of fusion activity. Storage of virosomes inan aqueous dispersion was accompanied by a relatively sudden loss of fusion activity.Thus, the incorporation in an amorphous inulin-glass preserves the biological activity ofHA to such a degree that even the complex pH-induced refolding of the protein re-quired for membrane fusion is unimpaired after reconstitution.
The transfection capacity of the virosomes largely paralleled their fusogenic proper-ties. DNA-virosomes stored in soluble form lost their transfection capacity within 6weeks. However, virosomes freeze-dried in buffer showed some transfection despiteabsence of any fusion activity. Interestingly, these preparations induced abundant celldeath. Possibly, upon rehydration lipoplex-like aggregates were formed, which areknown to be toxic under certain conditions [55]. Only when DNA-virosomes were lyo-philized in the presence of inulin their fusogenic as well as transfection properties weremaintained. Additionally, the use of inulin during freeze-drying prevented the formati-on of toxic complexes. Moreover, using inulin as a stabilizer, the stability during storagewas considerably improved, since the transfection activity was maintained for at least 12 weeks at 4°C, while also storage at elevated temperatures was possible. These resultsare in agreement with the preservation of the transfection activity of other gene deli-
CHAPTER IV
103

very systems using a stabilizer. For example, when lipoplexes were freeze-dried in thepresence of certain sugars, transfection activity was almost completely recovered [35, 36,48, 56]. Also upon storage, sugars delayed the loss of transfection efficiency [36].Furthermore, the infectivity of recombinant viral vectors freeze-dried in the presence ofa sugar was largely preserved and their half-life was substantially extended [33, 34, 57].
104

Conclusions
Inulin serves as an excellent stabilizer for virosomes during freeze-drying and sub-sequent storage. Virosomes could be formulated as a stable powder with retention oftheir structural integrity and biological activity, as demonstrated by the preservation ofvirosomal delivery capacities and immunological properties. Thus, stabilization by sugarglass technology can prolong the shelf-life of virosomes and allows storage at ambienttemperatures. Formulation of virosomes as a dry-powder can reduce the storage andtransport costs of virosome-based pharmaceuticals, as the use of a cold-chain is notnecessary. Moreover, it opens the possibility to develop novel needle-free dosage forms,e.g. for oral, nasal, pulmonal, or dermal delivery, thus improving bioavailability of vaccines as well as virosome-delivered compounds.
Acknowledgements
This work was supported by The Netherlands Organization for Scientific Research(NWO), under the auspices of the Chemical Foundation (CW) and the TechnologyFoundation (STW, Grant 790.33.571). We thank Solvay Pharmaceuticals Weesp, TheNetherlands for generous supplies of influenza virus. Klaas Sjollema, Department ofEukaryotic Microbiology, University of Groningen and Bert Dontje, Department of CellBiology, University Medical Center Groningen are acknowledged for excellent technicalhelp with electron microscopy and animal experiments, respectively. Marijke Holtrop ofthe Department of Medical Microbiology, Molecular Virology Section, UniversityMedical Center Groningen is acknowledged for her helpful assistance with the ELISAassay.
CHAPTER IV
105

REFERENCES
[1] Almeida JD, Edwards DC, Brand CM, Heath TD. Formation of virosomes from influenza subunits and liposomes. Lancet 1975;2(7941):899-901.
[2] Stegmann T, Morselt HW, Booy FP, van Breemen JF, Scherphof G, Wilschut J. Functional reconstitu-tion of influenza virus envelopes. Embo J 1987;6(9):2651-9.
[3] Gluck R, Mischler R, Brantschen S, Just M, Althaus B, Cryz SJ, Jr. Immunopotentiating reconstituted influenza virus virosome vaccine delivery system for immunization against hepatitis A. J Clin Invest 1992;90(6):2491-5.
[4] Lamb RA, Krug RM. Orthomyxoviridae: The viruses and their replication. Philadelphia: Lippincott Williams & Wilkins; 2001.
[5] Bron R, Ortiz A, Dijkstra J, Stegmann T, Wilschut J. Preparation, properties, and applications of re-constituted influenza virus envelopes (virosomes). Methods Enzymol 1993;220:313-31.
[6] de Jonge J, Schoen P, ter Veer W, Stegmann T, Wilschut J, Huckriede A. Use of a dialyzable short-chain phospholipid for efficient solubilization and reconstitution of influenza virus envelopes. Biochim Biophys Acta 2006;1758(4):527-36.
[7] Gluck U, Gebbers JO, Gluck R. Phase 1 evaluation of intranasal virosomal influenza vaccine with and without Escherichia coli heat-labile toxin in adult volunteers. J Virol 1999;73(9):7780-6.
[8] Huckriede A, Bungener L, ter Veer W, Holtrop M, Daemen T, Palache AM, et al. Influenza virosomes: combining optimal presentation of hemagglutinin with immunopotentiating activity. Vaccine 2003;21(9-10):925-31.
[9] de Bruijn IA, Nauta J, Gerez L, Palache AM. Virosomal influenza vaccine: a safe and effective influenza vaccine with high efficacy in elderly and subjects with low pre-vaccination antibody titers. Virus Res 2004;103(1-2):139-45.
[10] de Bruijn IA, Nauta J, Cramer WC, Gerez L, Palache AM. Clinical experience with inactivated, viro-somal influenza vaccine. Vaccine 2005;23 Suppl 1:S39-49.
[11] Daemen T, de Mare A, Bungener L, de Jonge J, Huckriede A, Wilschut J. Virosomes for antigen and DNA delivery. Adv Drug Deliv Rev 2005;57(3):451-63.
[12] Bron R, Ortiz A, Wilschut J. Cellular cytoplasmic delivery of a polypeptide toxin by reconstituted influenza virus envelopes (virosomes). Biochemistry 1994;33(31):9110-7.
[13] Bungener L, Huckriede A, de Mare A, de Vries-Idema J, Wilschut J, Daemen T. Virosome-mediated delivery of protein antigens in vivo: efficient induction of class I MHC-restricted cytotoxic T lympho-cyte activity. Vaccine 2005;23(10):1232-41.
[14] Waelti ER, Gluck R. Delivery to cancer cells of antisense L-myc oligonucleotides incorporated in fusogenic, cationic-lipid-reconstituted influenza-virus envelopes (cationic virosomes). Int J Cancer 1998;77(5):728-33.
[15] Schoen P, Chonn A, Cullis PR, Wilschut J, Scherrer P. Gene transfer mediated by fusion protein hemagglutinin reconstituted in cationic lipid vesicles. Gene Ther 1999;6(5):823-32.
[16] de Jonge J, Holtrop M, Wilschut J, Huckriede A. Reconstituted influenza virus envelopes as an efficient carrier system for cellular delivery of small-interfering RNAs. Gene Ther 2006;13(5):400-11.
[17] Wang W. Instability, stabilization, and formulation of liquid protein pharmaceuticals. Int J Pharm 1999;185(2):129-88.
[18] Coenen F, Tolboom JT, Frijlink HW. Stability of influenza sub-unit vaccine Does a couple of days out-side the refrigerator matter? Vaccine 2006;24(4):525-31.
[19] Randolph TW. Phase separation of excipients during lyophilization: effects on protein stability. J Pharm Sci 1997;86(11):1198-203.
[20] Schebor C, Burin L, Buera MP, Aguilera JM, Chirife J. Glassy state and thermal inactivation of invertase and lactase in dried amorphous matrices. Biotechnol Prog 1997;13(6):857-63.
[21] Slade L, Levine H. Non-equilibrium behavior of small carbohydrate-water systems. Pure Appl Chem 1988;60:1841-64.
106

[22] Slade L, Levine H. Water and the glass transition: dependance of the glass transition on the composition and chemical structure: special implications for flour functionality in cooky baking. J Food Eng 1995;24:431-509.
[23] Hinrichs WL, Prinsen MG, Frijlink HW. Inulin glasses for the stabilization of therapeutic proteins. Int J Pharm 2001;215(1-2):163-74.
[24] Eriksson HJ, Hinrichs WL, van Veen B, Somsen GW, de Jong GJ, Frijlink HW. Investigations into the stabilisation of drugs by sugar glasses: I. Tablets prepared from stabilised alkaline phosphatase. Int J Pharm 2002;249(1-2):59-70.
[25] Hincha DK, Zuther E, Hellwege EM, Heyer AG. Specific effects of fructo- and gluco-oligosaccharides in the preservation of liposomes during drying. Glycobiology 2002;12(2):103-10.
[26] Sun WQ, Leopold AC, Crowe LM, Crowe JH. Stability of dry liposomes in sugar glasses. Biophys J 1996;70(4):1769-76.
[27] Crowe LM, Crowe JH, Rudolph A, Womersley C, Appel L. Preservation of freeze-dried liposomes by tre-halose. Arch Biochem Biophys 1985;242(1):240-7.
[28] Crowe JH, Crowe LM, Carpenter JF, Aurell Wistrom C. Stabilization of dry phospholipid bilayers and proteins by sugars. Biochem J 1987;242(1):1-10.
[29] Hinrichs WL, Sanders NN, De Smedt SC, Demeester J, Frijlink HW. Inulin is a promising cryo- and lyo-protectant for PEGylated lipoplexes. J Control Release 2005;103(2):465-79.
[30] Hinrichs WL, Mancenido FA, Sanders NN, Braeckmans K, De Smedt SC, Demeester J, et al. The choice of a suitable oligosaccharide to prevent aggregation of PEGylated nanoparticles during freeze thawing and freeze drying. Int J Pharm 2006;311(1-2):237-44.
[31] Yannarell DA, Goldberg KM, Hjorth RN. Stabilizing cold-adapted influenza virus vaccine under various storage conditions. J Virol Methods 2002;102(1-2):15-25.
[32] Levy JA, Fieldsteel AH. Freeze-drying is an effective method for preserving infectious type C retro-viruses. J Virol Methods 1982;5(3-4):165-71.
[33] Bieganski RM, Fowler A, Morgan JR, Toner M. Stabilization of active recombinant retroviruses in an amorphous dry state with trehalose. Biotechnol Prog 1998;14(4):615-20.
[34] Croyle MA, Roessler BJ, Davidson BL, Hilfinger JM, Amidon GL. Factors that influence stability of recombinant adenoviral preparations for human gene therapy. Pharm Dev Technol 1998;3(3):373-83.
[35] Allison SD, Molina MC, Anchordoquy TJ. Stabilization of lipid/DNA complexes during the freezing step of the lyophilization process: the particle isolation hypothesis. Biochim Biophys Acta 2000;1468(1-2):127-38.
[36] Molina MC, Armstrong TK, Zhang Y, Patel MM, Lentz YK, Anchordoquy TJ. The stability of lyophi-lized lipid/DNA complexes during prolonged storage. J Pharm Sci 2004;93(9):2259-73.
[37] Carpenter JF, Crowe JH. An infrared spectroscopic study of the interactions of carbohydrates with dried proteins. Biochemistry 1989;28(9):3916-22.
[38] Peterson GL. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem 1977;83(2):346-56.
[39] Ferrari ME, Nguyen CM, Zelphati O, Tsai Y, Felgner PL. Analytical methods for the characterization of cationic lipid-nucleic acid complexes. Hum Gene Ther 1998;9(3):341-51.
[40] Böttcher CJF, van Gent CM, Pries C. A rapid and sensitive sub-micro phosphorus determination. AnalChimActa 1961;24:203-04.
[41] Stegmann T, Schoen P, Bron R, Wey J, Bartoldus I, Ortiz A, et al. Evaluation of viral membrane fusion assays. Comparison of the octadecylrhodamine dequenching assay with the pyrene excimer assay. Biochemistry 1993;32(42):11330-7.
[42] Bungener L, Serre K, Bijl L, Leserman L, Wilschut J, Daemen T, et al. Virosome-mediated delivery of protein antigens to dendritic cells. Vaccine 2002;20(17-18):2287-95.
[43] Immunochemical methods (2.7.1) In: European Pharmacopoeia, 5th edition. Strasbourg, Council of Europe, 2004: 187.
[44] Influenza vaccine (surface antigen, inactivate) In: European Pharmacopoeia, 5th edition. Strasbourg, Council of Europe, 2004: 673-74.
CHAPTER IV
107

[45] Haan L, Verweij WR, Holtrop M, Brands R, van Scharrenburg GJ, Palache AM, et al. Nasal or intra-muscular immunization of mice with influenza subunit antigen and the B subunit of Escherichia coli heat-labile toxin induces IgA- or IgG-mediated protective mucosal immunity. Vaccine 2001;19(20-22):2898-907.
[46] Ohkuma S, Poole B. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc Natl Acad Sci U S A 1978;75(7):3327-31.
[47] Puri A, Booy FP, Doms RW, White JM, Blumenthal R. Conformational changes and fusion activity of influenza virus hemagglutinin of the H2 and H3 subtypes: effects of acid pretreatment. J Virol 1990;64(8):3824-32.
[48] Li B, Li S, Tan Y, Stolz DB, Watkins SC, Block LH, et al. Lyophilization of cationic lipid-protamine-DNA (LPD) complexes. J Pharm Sci 2000;89(3):355-64.
[49] Gomez G, Pikal MJ, Rodriguez-Hornedo N. Effect of initial buffer composition on pH changes during far-from-equilibrium freezing of sodium phosphate buffer solutions. Pharm Res 2001;18(1):90-7.
[50] Van Den Berg L, Rose D. Effect of freezing on the pH and composition of sodium and potassium phosphate solutions; the reciprocal system KH2PO4-Na2-HPO4-H2O. Arch Biochem Biophys 1959;81(2):319-29.
[51] Eriksson JH, Hinrichs WL, de Jong GJ, Somsen GW, Frijlink HW. Investigations into the stabilization of drugs by sugar glasses: III. The influence of various high-pH buffers. Pharm Res 2003;20(9):1437-43.
[52] Vereyken IJ, Chupin V, Hoekstra FA, Smeekens SC, de Kruijff B. The effect of fructan on membrane lipid organization and dynamics in the dry state. Biophys J 2003;84(6):3759-66.
[53] Vereyken IJ, Chupin V, Islamov A, Kuklin A, Hincha DK, de Kruijff B. The effect of fructan on the phospholipid organization in the dry state. Biophys J 2003;85(5):3058-65.
[54] Arkema A, Huckriede A, Schoen P, Wilschut J, Daemen T. Induction of cytotoxic T lymphocyte acti-vity by fusion-active peptide-containing virosomes. Vaccine 2000;18(14):1327-33.
[55] Dass CR. Lipoplex-mediated delivery of nucleic acids: factors affecting in vivo transfection. J Mol Med 2004;82(9):579-91.
[56] Anchordoquy TJ, Carpenter JF, Kroll DJ. Maintenance of transfection rates and physical characteriza-tion of lipid/DNA complexes after freeze-drying and rehydration. Arch Biochem Biophys 1997;348(1):199-206.
[57] Cruz PE, Silva AC, Roldao A, Carmo M, Carrondo MJ, Alves PM. Screening of novel excipients for improving the stability of retroviral and adenoviral vectors. Biotechnol Prog 2006;22(2):568-76
108

J-P. Amorij1, T.A. Westra1, W.L.J. Hinrichs1, A. Huckriede2, H.W. Frijlink1
Chapter V
Towards an oral influenza vaccine:Comparison between intragastric and intra-colonic delivery of influenza subunit vaccinein a murine model.
Vaccine (2007): in press.
1 Department of Pharmaceutical Technology and Biopharmacy, University of Groningen, Groningen, The Netherlands.
2 Department of Medical Microbiology, Molecular Virology Section, University Medical Center Groningen and University of Groningen, Groningen, The Netherlands.

Abstract
In this paper we investigated to which part of the gastro-intestinal (GI) tract, the upperor lower part, an oral influenza vaccine should be targeted to result in an effective immu-ne response in mice. Our study demonstrates that without adjuvant substantial systemicbut low respiratory mucosal immune responses were induced in mice after delivery ofinfluenza subunit vaccine to the upper GI-tract (intragastric) as well as the lower GI-tract (intracolonically). When the vaccine was adjuvanted with E.Coli heat-labileenterotoxin (LT) these responses were significantly enhanced. Interestingly, intracolonicadministration of vaccine with adjuvant also resulted in enhanced cellular immune res-ponses and the desired Th1-skewing of these responses. Intragastric administration of theadjuvanted vaccine also increased Thelper responses. However Th1-skewing was absent.In conclusion, the right combination of strong mucosal adjuvant (e.g. LT) and antigendelivery site (e.g. the lower part of the gastro-intestinal tract) might result in effectivevaccination via the oral route.
Keywords
Oral vaccine; heat-labile enterotoxin; Th1-skewing.
110

Introduction
Yearly recurrent influenza epidemics and the threat of an influenza pandemic remainmajor public health concerns. Influenza can cause high morbidity and mortality rates.For epidemic influenza strains this is especially true for the elderly and other high-riskpopulations whereas pandemic strains may be very dangerous for all age groups [1].Immunization against influenza virus is recognized as the most effective method for con-trolling influenza in epidemic as well as in pandemic situations [2].
Current inactivated influenza vaccines are generally administered by intramuscularinjection. Yet, for mass vaccination against mucosally contracted infections as influenzaother administration routes, especially oral vaccine administration, might be preferrablefor several reasons. First, an oral vaccine, like other non-invasive vaccines, drasticallysimplifies the logistics of immunization and improves the immunization coverage as hasbeen shown by the success of the oral polio vaccine [3]. The ease of use and cheap ad-ministration in vaccination programs of the oral (live) polio vaccine, parameters especi-ally important in developing countries, resulted in the successful polio reduction all overthe world. Use of the oral route will make administration of vaccines safer during vac-cination programs, since iatrogenic infections due to the use of unsterile needles or needle-stick injuries, which are especially a high risk in third-world countries, cannotoccur [4, 5].
Moreover, oral vaccination can result in a mucosal immune response (IgA) in therespiratory tract [6-9], which might give protection against influenza infection at theport of entry [10]. This is in contrast to current parenteral vaccinations, that only pro-vide virus-neutralizing antibodies (IgG) in the lungs as a result of transudation from thecirculation [11, 12]. Finally, since mucosal IgA responses have been shown to exhibitcross-protective immunity against antigenically distinct viruses [13, 14], such a mucosalimmune response induced by oral vaccination might offer broader protection againstdrifted, heterologous strains.
Over the past decades, a number of clinical studies with oral influenza vaccines(such as a water in oil emulsion or enterically coated capsules containing dried vaccine)have been performed [6-9]. Unfortunately, these immunizations resulted in IgG respon-ses below detection level. In contrast, most studies demonstrate a significant increase inIgA antibodies in both saliva and nasal lavage fluids. It is unknown whether these IgAantibodies alone could provide adequate protection against influenza infection inhumans. There is sufficient pre-clinical as well as clinical evidence that secretory IgA canneutralize invading influenza virus by itself, independently from the presence of serumIgG [13, 15, 16]. For example, Renegar et al. demonstrated in a preclinical study in micethat passive transfer of anti-influenza IgA alone into nasal secretions provided protec-tion against nasal challenge with influenza virus [17]. Moreover, Belshe et al. showed ina clinical study that nasal wash IgA induced by live-attenuated cold-adapted intranasalvaccine was a stronger correlate for protection from experimental challenge than serum
CHAPTER V
111

antibody. Thus, secretory IgA can be seen as another correlate for protection [16, 18].Therefore possibly sufficient protection by mucosal IgA induced by oral immunizationmight be achieved. However, to our knowledge, no clinical studies have been designedso far to reveal the level of protection provided by the orally-induced local IgA immuneresponses in humans.
Regardless of the possibility to induce effective mucosal immune responses,influenza vaccines have to evoke an adequate level of virus-neutralizing antibodies in theserum to fulfil current regulatory requirements for vaccine immunogenicity [19, 20]. Inorder to achieve satisfying serum antibody levels preclinical research on oral immuniza-tion with influenza vaccines is mainly focussed on the use of adjuvants [21-23] and com-plex vaccine formulations [24-34]. Only few studies on oral influenza vaccine develop-ment paid attention to the delivery site of the vaccine. Meitin et al. reported that intra-jejunal administration of a live recombinant vaccinia virus consistently induced immu-nity, while intragastric administration was much less reliable in doing so [25]. When thedelivery site of the vaccine in the gastro-intestinal (GI) tract is an important factor, newtechnologies, like special coatings, might enable targeting to these sites [35-38] and thusimprove vaccine efficacy.
The aim of the present study was to evaluate to which part of the GI-tract, theupper part (stomach-duodenum) or lower part (colon-rectum), an oral influenza vaccineshould be targeted to result in an effective immune response in mice. For this purposeBALB/c mice were immunized with an influenza subunit vaccine via different routes.Intragastric delivery was used to target to the upper GI-tract and intracolonic deliverywas used to target to the lower GI-tract. Standard i.m. immunization of mice was used asa control. Systemic and mucosal antibody responses elicited by the vaccination via thedifferent routes were compared. Additionally, cell-mediated immunity induced by thedifferent immunization routes was monitored by measuring the frequency of influenza-specific IFNγ-, IL2- and IL4- producing spleen cells. Furthermore, the effect of a strongmucosal adjuvant on the immune responses elicited by the differently delivered vaccineswas investigated. In this study E.Coli heat-labile enterotoxin (LT) was used as a modelfor a strong mucosal adjuvant. LT is known to increase the permeability of the epitheli-um, leading to enhanced uptake and presentation of co-administered antigen and promo-tion of isotype differentiation in B cells. As a result adjuvanting with LT can lead to anincreased IgA production and complex stimulatory as well as inhibitory effects on T-cellproliferation and cytokine production [39].
112

Materials and Methods
VaccinePurified monovalent A/Panama (H3N2) subunit antigen was a gift from SolvayPharmaceuticals (Weesp, the Netherlands). The protein concentration of the vaccine(which consists mainly of haemagglutinin (HA)) was determined with a Lowry assay[40].
Adjuvant production For the production of wild-type LT an in house pROFIT-LT vector [41, 42], containinga temperature-inducible λPR promotor [43] was used. The E.coli host strain, MC1061[44], was transformed with this plasmid. LT expression was obtained by shifting the cul-ture temperature of a log phase culture of E. coli strain MC1061, harbouring the aboveplasmid, from 28°C to 42°C. Purification of LT was carried out according to the methoddescribed by Uesaka et al. [45]. In brief, sonificated crude periplasmic LT preparationswere applied to an immobilized-D-galactose column (Pierce) and the column was rinsedwith TEAN buffer (50 mM Tris, 200 mM NaCl, 1 mM EDTA and 0.02% w/v sodiumazide) overnight. The purified LT was eluted with 0.3 M galactose in TEAN buffer.Appropriate elution fractions were pooled on basis of A280 and dialysed against PBS. TheLT concentration was determined using a Lowry assay [40].
AnimalsAnimal experiments were conducted according to the guidelines provided by the DutchAnimal Protection Act, and were approved by the Committee for AnimalExperimentation (DEC) of the University of Groningen. For all experiments 6-8 weeksold female BALB/c mice (Harlan, Zeist, Netherlands) were used.
Vaccination protocolFive groups of 8 female mice were vaccinated 3 times (day 0, 14, 28) intragastrically (i.g.),intracolonically (i.c.), or intramuscularly (i.m.) with immunogen preparations as descri-bed below. Mice were fasted 12 h prior to each immunization to minimize the risk thatagents in the food inhibit the efficacy of the delivered immunogens [46]. Vaccine wasadministered to the mice during the light period.
Intramuscular immunization was carried out by injection of 50 μl PBS containing 5μg HA divided over the posterior thigh muscles, while mice were anesthetized by isoflu-ran/O2.
For intragastric immunization 200 μl of sodium bicarbonate solution (3.2% w/v)containing 20 μg HA or 20 μg HA + 2 μg LT was administered intragastrically withoutanaesthesia. Intragastric administration was performed using a 0.9 x 38 mm stainless steelfeeding needle with silicone tip (Scanbur BK, Sollentuna, Sweden) connected to a 1-mlsyringe. In this way, damage to the oesophagus and introduction of the needle into thetrachea were prevented. The oral vaccine was buffered with sodium hydrogen carbo-
CHAPTER V
113

nate in order to neutralize the acidic environment of the stomach [21] thereby preven-ting the acid-induced conformation change of HA (HA inactivation) [47].
For intracolonic immunization each mouse was installed upside-down at an angle of45° and 100 μl vaccine (containing 20 μg HA or 20 μg HA + 2 μg LT) was administeredintracolonically under isofluran anaesthesia. Intracolonic administration was preformedwith a specially designed 1.2 x 38 mm flexible teflon feeding needle with soft silicone tip(Scanbur BK, Sollentuna, Sweden) attached to a 1-ml syringe. After intracolonic admini-stration of the vaccine the mice were held upside-down under isofluran/O2 anaesthesiafor 10 minutes to prevent leakage. During this procedure no internal bleedings wereobserved. In addition, after sacrifice of the animals no abnormalities or leakage of thecolon were detected.
During the immunization period all mice showed a normal gain in body-weight.
Sample collectionBefore the first and last vaccine administration, blood samples (approx. 40 μl) were takenfrom the mice by an orbita punction under isofluran/O2 anaesthesia. Four weeks after thelast vaccination, the animals were sacrificed, by draining the abdominal aorta under iso-fluran/O2 anaesthesia with a syringe and needle.
The spleens were harvested immediately after sacrifice of the mice and stored untilfurther analysis in ice-cold IMDM Glutamax medium (Life Technologies, Paisley, UK)supplemented with 10% w/v FCS (PAA Laboratories, Linz, Austria), 100 U/ml penicillin(Life Technologies), 100 μg/ml streptomycin (Life Technologies) and 50 mM β-mercaptoethanol.
Next, nasal and lung lavages were performed by cannulating the trachea via a smallincision using a 20G Insyte Autoguard catheter (Becton Dickinson BV, Alphen aan deRijn, The Netherlands) connected to a 1-ml syringe and rapid rinsing. Nasal lavage fluidwas obtained by flushing twice through the nasopharynx and lung lavage was performedby repeated flushing of the lungs, with 1 ml of PBS (Fluka) for each lavage.
Next, the gastro-intestinal tract was prepared for intestine lavage. Colon/rectumlavage was performed by intubating via the rectum using a 1.2 x 38 mm flexible teflonfeeding needle with silicone tip (Scanbur BK, Sollentuna, Sweden). Before lavage, thecolon (posterior of the caecum) was closed with a ligature. Next, a 1-ml syringe was at-tached to the feeding needle and the lavage was performed by repeated flushing of thecolon/rectum with 1 ml of PBS (Fluka).
Duodenum/jejunum lavage was performed by intubating the duodenum via an in-cision posterior to the stomach using a 1.2 x 38 mm flexible teflon feeding needle withsilicone tip (Scanbur BK, Sollentuna, Sweden). Before lavage, the jejunum was closedanterior of the ileum with a ligature. Next, a 1-ml syringe was attached to the feedingneedle and the lavage was performed by repeated flushing of the duodenum/jejunumwith 1 ml of PBS (Fluka).
Immediately after each sample collection, the lavage was mixed with 10 μl stocksolution (1 tablet/2ml PBS) of Complete® protease inhibitor cocktail (Roche DiagnosticsGmbH, Penzberg, Germany) and lavages were kept on ice until further preparation.
114

Serum samples were obtained by centrifugation at 11.000 x g for 30 min. and serawere stored at -20°C until further analysis. Lavage samples were centrifuged at 11.000 xg for 15 min, and supernatants were collected and stored at 4°C until further analysis.
Haemagglutination inhibition (HI) assayHI-titers of sera were determined by a standard HI-microtiter assay as described before[48] using 4 haemagglutinating units of A/Panama influenza virus and 1% w/v guinea pigerythrocyte solution. The HI titers are defined as the highest serum dilution capable ofpreventing haemagglutination.
ELISAInfluenza subunit antigen-specific antibody responses were determined by ELISA as de-scribed before [48]. In brief, ELISA plates (96-well flat bottom, Microlon®600, Greiner,Alphen a/d Rijn, The Netherlands) were coated with 200 ng of influenza subunit antigenper well. After incubation with serially diluted sera and mucosal lavage fluids, plateswere incubated with horseradish peroxidase-conjugated goat antibodies or rat antibody(for IgE only) directed against the appropriate mouse antibody (dilution 1:5000;Southern Biotech, Birmingham, Alabamba, USA). Sera were analyzed for IgG, IgA, IgG1,IgG2a, IgG2b, while lavages were analyzed for IgG and IgA. Serum samples were testedin se-rial dilution with a minimal dilution of 40. In addition, as a positive control for theIgE determination, plates were coated with 100 ng of goat antibody directed againstmouse IgE, and subsequently incubated with mouse myeloma IgE. After incubation ofthe plates with mouse Ig-isotype conjugate, antibodies were detected using 1,2-pheny-len-diamin-dihydrochlorid as substrate and a Benchmark Microplate reader equippedwith a 492 nm filter (BioRad, Hercules, CA). Titers are given as the reciprocal of the cal-culated sample dilution corresponding to A492 = 0.2 after background correction (wellswithout serum). Prevaccination titers were always below detection levels and are there-fore not shown.
ELISPOT assaysELISPOT analyses were performed according to a protocol adapted from the proceduredescribed by Miyahira et al. [49]. ELISA plates (Microlon 600, Greiner, Alphen a/d Rijn,The Netherlands) were coated with purified anti-mouse IFNγ, anti-mouse IL2, or anti-mouse IL4 (BD Pharmingen, Erembodegem, Belgium). After overnight incubation at 4°Cplates were washed three times with sterile PBS/Tween and incubated with blockbuffer(PBS + 4% w/v BSA) at 37°C for 1h. Spleen cells were isolated and plated in differentquantities (0.125 - 2.0 x106 cells) in IMDM-medium containing 10% w/v FCS and incu-bated overnight at 37°C with or without 500 ng of influenza subunit antigen.
After overnight stimulation, cells were lysed in ice-cold water for 15 min. Afterwashing the plates 5 times with PBS/Tween, IFNγ, IL2, or IL4 were detected using bio-tinylated anti-mouse antibodies and streptavidin-alkaline phosphatase (dilution 1:1000,BD Pharmingen, Erembodegem, Belgium). 100 μl Substrate solution (1 mg/ml 5-bromo-4-chloro-3-indolylphosphate in 0.6% w/v agarose solution containing 0.92% w/v 2-
CHAPTER V
115

amino-2-methyl-1-propanol and 0.08 μl/ml Triton X-405), was added to each well andplates were put on ice. When the agarose was solidified, plates were incubated at 37°C toaccelerate spot formation. Spots were subsequently counted using a dissection micro-scope. The numbers of influenza-specific IFNγ, IL2, or IL4-secreting cells were obtainedby substracting the background (number of spots after incubation without antigen) fromthe number of spots observed in wells containing spleen cells incubated with influenzasubunit antigen.
Statistical analysesThe titers are given as the geometric mean ± SD, unless stated otherwise. Differencesbetween antibody levels were analyzed using Student's t-test at a 95% confidence level.A probability factor of less then 0.05 (P<0.05) was considered to represent a statisticallysignificant difference. Statistically significant differences in the figures are givenbetween the gastro-intestinal immunized groups and the i.m. immunized group.Increases are denoted by *, while decreases are denoted by #. Increases in significance aregiven by increasing numbers of symbols: one symbol = P<0.05; two symbols = P <0.01and three symbols = P<0.001.
116
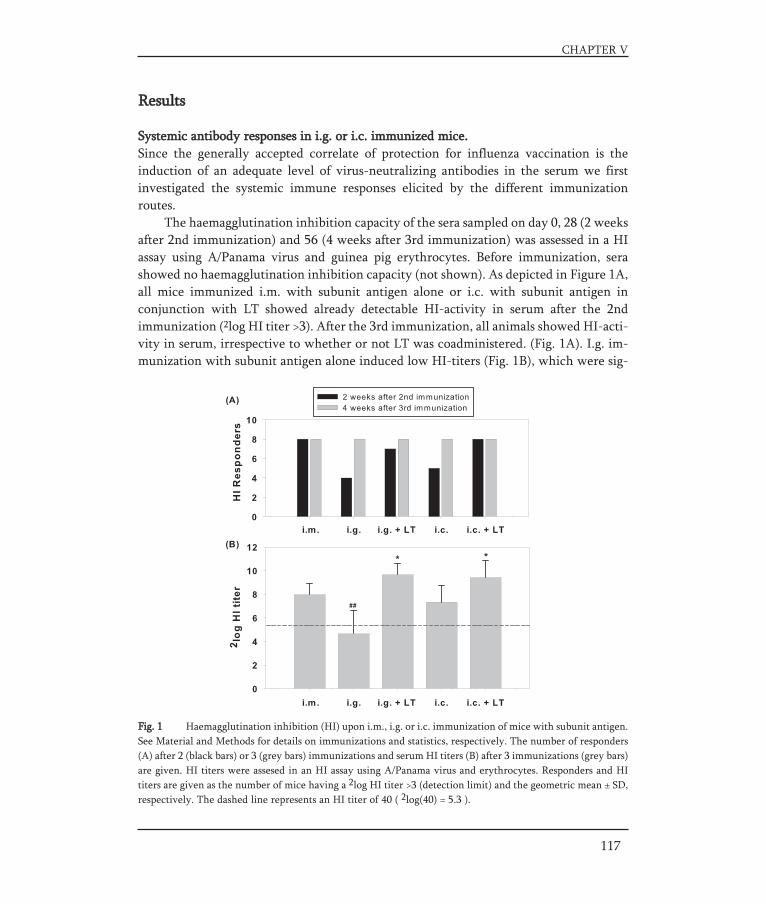
Fig. 1 Haemagglutination inhibition (HI) upon i.m., i.g. or i.c. immunization of mice with subunit antigen.See Material and Methods for details on immunizations and statistics, respectively. The number of responders(A) after 2 (black bars) or 3 (grey bars) immunizations and serum HI titers (B) after 3 immunizations (grey bars)are given. HI titers were assesed in an HI assay using A/Panama virus and erythrocytes. Responders and HItiters are given as the number of mice having a 2log HI titer >3 (detection limit) and the geometric mean ± SD,respectively. The dashed line represents an HI titer of 40 ( 2log(40) = 5.3 ).
Results
Systemic antibody responses in i.g. or i.c. immunized mice.Since the generally accepted correlate of protection for influenza vaccination is theinduction of an adequate level of virus-neutralizing antibodies in the serum we firstinvestigated the systemic immune responses elicited by the different immunization routes.
The haemagglutination inhibition capacity of the sera sampled on day 0, 28 (2 weeksafter 2nd immunization) and 56 (4 weeks after 3rd immunization) was assessed in a HIassay using A/Panama virus and guinea pig erythrocytes. Before immunization, serashowed no haemagglutination inhibition capacity (not shown). As depicted in Figure 1A,all mice immunized i.m. with subunit antigen alone or i.c. with subunit antigen in conjunction with LT showed already detectable HI-activity in serum after the 2ndimmunization (2log HI titer >3). After the 3rd immunization, all animals showed HI-acti-vity in serum, irrespective to whether or not LT was coadministered. (Fig. 1A). I.g. im-munization with subunit antigen alone induced low HI-titers (Fig. 1B), which were sig-
CHAPTER V
117
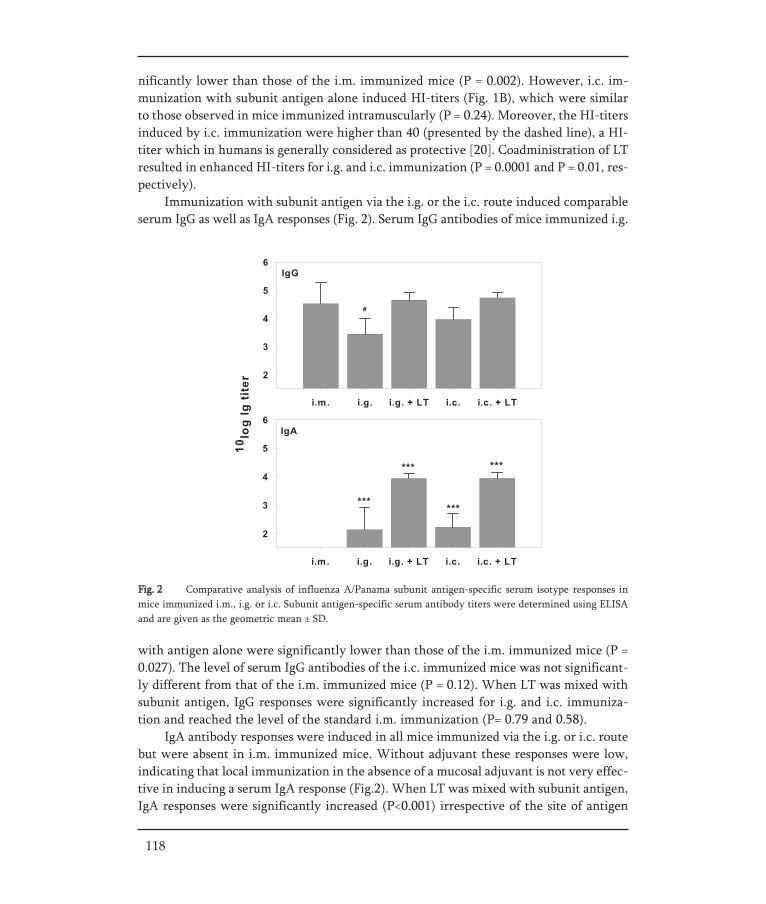
with antigen alone were significantly lower than those of the i.m. immunized mice (P =0.027). The level of serum IgG antibodies of the i.c. immunized mice was not significant-ly different from that of the i.m. immunized mice (P = 0.12). When LT was mixed withsubunit antigen, IgG responses were significantly increased for i.g. and i.c. immuniza-tion and reached the level of the standard i.m. immunization (P= 0.79 and 0.58).
IgA antibody responses were induced in all mice immunized via the i.g. or i.c. routebut were absent in i.m. immunized mice. Without adjuvant these responses were low,indicating that local immunization in the absence of a mucosal adjuvant is not very effec-tive in inducing a serum IgA response (Fig.2). When LT was mixed with subunit antigen,IgA responses were significantly increased (P<0.001) irrespective of the site of antigen
Fig. 2 Comparative analysis of influenza A/Panama subunit antigen-specific serum isotype responses inmice immunized i.m., i.g. or i.c. Subunit antigen-specific serum antibody titers were determined using ELISAand are given as the geometric mean ± SD.
118
nificantly lower than those of the i.m. immunized mice (P = 0.002). However, i.c. im-munization with subunit antigen alone induced HI-titers (Fig. 1B), which were similarto those observed in mice immunized intramuscularly (P = 0.24). Moreover, the HI-titersinduced by i.c. immunization were higher than 40 (presented by the dashed line), a HI-titer which in humans is generally considered as protective [20]. Coadministration of LTresulted in enhanced HI-titers for i.g. and i.c. immunization (P = 0.0001 and P = 0.01, res-pectively).
Immunization with subunit antigen via the i.g. or the i.c. route induced comparableserum IgG as well as IgA responses (Fig. 2). Serum IgG antibodies of mice immunized i.g.
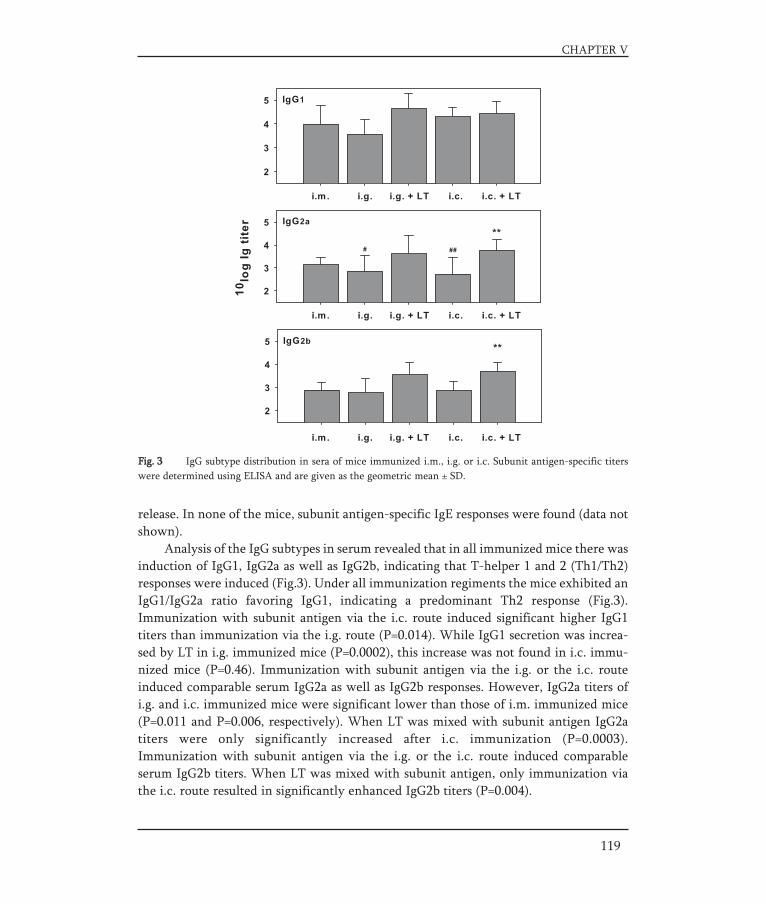
119
CHAPTER V
Fig. 3 IgG subtype distribution in sera of mice immunized i.m., i.g. or i.c. Subunit antigen-specific titerswere determined using ELISA and are given as the geometric mean ± SD.
release. In none of the mice, subunit antigen-specific IgE responses were found (data notshown).
Analysis of the IgG subtypes in serum revealed that in all immunized mice there wasinduction of IgG1, IgG2a as well as IgG2b, indicating that T-helper 1 and 2 (Th1/Th2)responses were induced (Fig.3). Under all immunization regiments the mice exhibited anIgG1/IgG2a ratio favoring IgG1, indicating a predominant Th2 response (Fig.3).Immunization with subunit antigen via the i.c. route induced significant higher IgG1titers than immunization via the i.g. route (P=0.014). While IgG1 secretion was increa-sed by LT in i.g. immunized mice (P=0.0002), this increase was not found in i.c. immu-nized mice (P=0.46). Immunization with subunit antigen via the i.g. or the i.c. routeinduced comparable serum IgG2a as well as IgG2b responses. However, IgG2a titers ofi.g. and i.c. immunized mice were significant lower than those of i.m. immunized mice(P=0.011 and P=0.006, respectively). When LT was mixed with subunit antigen IgG2atiters were only significantly increased after i.c. immunization (P=0.0003).Immunization with subunit antigen via the i.g. or the i.c. route induced comparableserum IgG2b titers. When LT was mixed with subunit antigen, only immunization viathe i.c. route resulted in significantly enhanced IgG2b titers (P=0.004).

Upon co-administration with LT the number of mice with mucosal IgA in the respirato-ry tract was considerably enhanced. However, still not all animals showed detectable IgAresponses in the respiratory tract. I.m. immunization with subunit antigen alone did notresult in detectable IgA responses in the respiratory tract (nose and lungs) in any of thevaccinated mice.
Since the intestinal tract was the induction site to prime antigen-specific B cells wealso investigated the local generation of IgA antibodies in the intestinal tract (duodenumand colon). In mice that were immunized by i.g. and i.c. administration we expected alocal IgA response in the gastro-intestinal tract, which was indeed revealed. Surprisingly,also upon i.m. immunization we found a subunit antigen-specific IgA response in theduodenum, which was similar to that found upon i.g. and i.c. immunization with anti-
120
Fig. 4 Subunit antigen-specific IgA titers in mucosal lavage fluids of i.m., i.g. or i.c. immunized mice. IgAtiters were determined using ELISA and are given as the geometric mean ± SD of IgA-positive lavages. Thenumbers above the columns indicate the number of responders and group size.
Mucosal antibody responses in i.g. or i.c. immunized mice.Next, the induction of mucosal IgA antibody responses upon immunization with antigenvia the different delivery routes was evaluated (Fig. 4). I.g. and i.c. immunization withsubunit antigen alone could elicit IgA responses, however, infrequently.

Fig. 5 Subunit antigen-specific IgG titers in nasal and lung lavage fluids of i.m., i.g. or i.c. immunized miceas determined by ELISA. Geometric means ± SD are given. The numbers above the columns indicate the num-ber of responders and group size.
gen alone or supplemented with LT (Fig.4). In the colon, subunit antigen-specific IgAresponses were not found when mice received i.m. or i.g. immunization with antigenalone. In contrast, i.c. immunization with and without LT and i.g. immunization with LTresulted in IgA responses in the colon.
Next, we assessed the IgG responses in the respiratory tract of the immunized ani-mals. Figure 5 shows that oral immunization with subunit antigen alone infrequently eli-cited IgG responses in the nasal cavity, which was slightly improved by coadministra-tion of LT. Similarly, only 2 out of the 8 i.m. immunized mice developed nasal IgG titers.Despite the poor IgG responses in nasal fluids lung lavage fluids of the i.g., i.c, and i.m.immunized mice contained substantial levels of subunit antigen-specific IgG antibodies.
Cell-mmediated immunity in i.g. or i.c. immunized mice.The cell-mediated immunity activated by immunization with subunit antigen was eva-luated by determining the frequency of IL2-, IFNγ- and IL4-producing cells in spleens ofmice immunized i.m., i.g., or i.c. with subunit antigen in an ELISPOT assay.
Immunization with subunit antigen alone induced similar numbers of IL2- pro-ducing spleen cells irrespective of the route of administration (Fig. 6A), indicating T cellactivation after the different immunizations. LT enormously increased these numbers.I.g., i.c., and i.m. immunization with subunit antigen alone resulted in equal, but lownumbers of IFNγ-producing spleen cells (Fig. 6B). Again, LT showed a strong stimula-tory effect on the activation of this T cell subset. The number of IL4-producing spleencells after immunization with antigen alone (Fig. 6C) depended on the route of immu-nization (i.m. > i.c.> i.g.). LT strongly increased the frequency of IL4-producing cells inmice receiving i.g. subunit antigen. In contrast, the frequency of IL4-producing spleencells of mice immunized i.c. was similar for subunit- and subunit/LT-immunized mice (P = 0.1) and remained lower than in i.m. immunized mice (P= 0.015).
CHAPTER V
121

Fig. 6 Cytokine release profile of the effector cell population from mice i.m., i.g. or i.c. immunized withsubunit antigen alone or in conjunction with LT. Freshly isolated splenocytes of immunized mice were incu-bated overnight with or without subunit antigen and the number of IL2, IFNγ and IL4-secreting spleen cellswere calculated by subtracting the number of spots obtained without stimulation from the number of spotsobtained with antigen stimulation. Results of (A) IL2, (B) IFNγ and (C) IL4-producing spleen cells are presen-ted as the mean ± SEM.
122

Fig. 7 The IFNγ/IL4 ratio of the effector cell population from mice i.m., i.g. or i.c. immunized with subu-nit antigen alone or in conjunction with LT. Results are presented as the ratio of the mean of the IFNγ-produ-cing spleen cells (Fig. 6B) and the mean of the IL4-producing spleen cells (Fig. 6C).
Since the frequencies of cytokine-producing spleen cells for the different immu-nization routes shifted when LT was used, the IFNγ / IL4 ratios were calculated as anindicator of the Th1/Th2 balance (Figure 7). The i.m. immunization resulted in an immu-ne response strongly dominated by the Th2 type (IL4). Also, the i.g. and i.c. immuniza-tion with subunit antigen alone resulted in an immune response having a strong Th2bias. The addition of LT upon i.g. immunization resulted in the same Th2 bias as wasfound upon i.g. immunization with antigen alone. Interestingly, after i.c. immunizationwith antigen in conjunction with LT a clear Th1 shift occurred, resulting in a balancedTh1/Th2 response.
CHAPTER V
123

Discussion
In this paper we approached the question which site of antigen delivery in the GI-tractis most suitable to obtain an effective immune response in mice after oral administra-tion of influenza vaccine.
Since the standard criterium for current influenza vaccines is the induction of virus-neutralizing antibodies in serum [19, 20], serum antibody levels and HI titers induced bythe different routes of immunization were compared. Although IgG titers were not sig-nificantly different, immunization without adjuvant via the lower GI-tract resulted insignificantly higher HI titers than immunization via the upper GI-tract. Moreover, theseHI-titers were higher than 40, which in humans is considered as protective [20].
In order to enhance the immune responses of the i.g. and i.c. immunizations weused LT as a model for a strong adjuvant. LT enhanced the immune responses considera-bly, resulting in HI-titers higher than and IgG titres comparable to those of the standardi.m. immunization. These results are in agreement with observations by others [21-23]and underline that LT has potent adjuvant activity when orally administered to mice inconjunction with influenza antigen. When administered via the i.c. route, the adjuvantactivity of LT was so effective that all the animals already developed high HI-titers after2 immunizations. Besides serum immunity, LT enhanced mucosal and cell-mediatedimmunity. Unresolved toxicity issues exclude the use of native LT in humans [19].However, genetically detoxified mutant proteins have been developed and are currentlyexplored in preclinical and clinical experiments [21, 22, 41, 42, 50, 51].
Our study also demonstrates that immunization via the GI-tract with influenza anti-gen in conjunction with a potent adjuvant induced a local immune response at the portof entry (respiratory tract). Antigen-specific IgA antibodies, although in moderate titers,were detected in nose and lungs. In contrast, i.m. immunization induced no detectableIgA response in the respiratory tract. Upon i.g. immunization of mice with influenza vac-cine, also others found antigen-specific IgA antibodies in the respiratory tract , however,only after immunization in the presence of a potent adjuvant [21-23]. In contrast, clini-cal studies with oral influenza vaccines revealed that no adjuvant was needed to elicitantigen-specific IgA in both saliva and nasal lavage fluids of humans [6-9]. This appea-rance of IgA antibodies at distant mucosal sites can be explained by cellular migration ofprimed antigen-specific B cells from the site of induction [52-54]. It is generally assumedthat IgA is the main antibody isotype and effector molecule in host defence at mucosalsurfaces [55]. Moreover, nasal IgA appears to mediate protection of the upper respira-tory tract from virus infection [13, 15, 17, 56].
On the other hand, some studies have shown that mucosal IgG, derived from serumby a mechanism of transudation or locally produced, can mediate protection of the res-piratory tract [48, 57, 58]. From this point of view, our data for IgG titers in mucus indi-cate that immunization via the gastro-intestinal route contributes to IgG-mediated pro-tection in the lower respiratory tract. However, in the upper respiratory tract mucosalIgG was only found in some animals.
124

The commercially available trivalent parenteral vaccines derived from inactivatedviruses, induce poor cell-mediated immunity [10]. This is partly caused by the presenta-tion form of conventional split and subunit vaccines, which is not optimal for stimula-tion of cell-mediated immunity, in particular not for cytotoxic T lymphocyte activity[19]. Moreover, diminishing T cell activity with age appears to result in suboptimal ef-ficacy of current inactivated influenza vaccines in the elderly [59]. To improve currentvaccines significant efforts have been made to enhance cell-mediated immunity andmimic the natural immune response against influenza infection [60-63]. Our data showthat gastro-intestinal immunization of BALB/c mice with antigen alone resulted in aTh1/Th2 response dominated by a Th2 response. Moreover, moderate cell-mediatedimmunity comparable to that of the standard immunizations (i.m.) was induced by gastro-intestinal immunization without adjuvant. The addition of LT upon GI-immu-nization, however, enhanced the cellular immunity. Interestingly, addition of adjuvantto the i.c. administered vaccine resulted in the desired Th1-skewing of the cellularimmune responses. This balanced Th1/Th2 response was mainly caused by the fact thatcolonic immunization with LT showed no increase in frequency of IL4-producing T cellswhile strongly increasing IFNγ-producing T cells. As a result the inhibitory effect of IL4on both primary and secondary antiviral immune responses [64] might be prevented byimmunization via the lower GI-tract.
The differences in cellular immunity between the LT-adjuvanted groups reflectedthe IgG subtype profile observed (Fig. 3), since IL4 stimulates secretion of IgG1 [65].While IgG1 secretion was increased by LT in i.g. immunized mice, this increase was notfound in i.c. immunized mice. Thus, adjuvant effects of LT depend not only on the na-ture of the co-administered antigen but also on the route of administration as also repor-ted by Cheng et al, who compared the intragastric route with the intranasal route [51].
CHAPTER V
125

Conclusions
The present study demonstrates that an oral influenza vaccine should be targeted to thelower GI-tract to result in an effective immune response in mice. Without adjuvant, thelower GI-tract seems favourable over the upper GI-tract on basis of HI titers. A potentadjuvant, however, is needed to obtain an immune response comparable to that observedafter i.m. vaccination. Finally, the adjuvant effect of LT depends on the site of admini-stration/delivery. The right combination of adjuvant and antigen delivery site, e.g. LTand the lower part of the GI-tract, respectively, could result in effective influenza vac-cination via the oral route.
Acknowledgements
We thank Marijke Holtrop (Department of Medical Microbiology, Molecular VirologySection, University Medical Center Groningen, University of Groningen, TheNetherlands) for her technical assistance with the LT-production and Natasha Broersma(University Medical Center Groningen, University of Groningen, The Netherlands) forbiotechnical assistance with the animal experiments.
126

REFERENCES
[1] Simonsen L, Clarke MJ, Schonberger LB, Arden NH, Cox NJ, Fukuda K. Pandemic versus epidemic influenza mortality: a pattern of changing age distribution. J Infect Dis 1998;178(1):53-60.
[2] UNICEF. press release "Low investment in immunization and vaccines threatens global health". 2002.[3] Fine PE. Poliomyelitis: very small risks and very large risks. Lancet Neurol 2004;3(12):703.[4] Giudice EL, Campbell JD. Needle-free vaccine delivery. Adv Drug Deliv Rev 2006;58(1):68-89.[5] Mitragotri S. Immunization without needles. Nat Rev Immunol 2005;5(12):905-16.[6] Bergmann KC, Waldman RH, Tischner H, Pohl WD. Antibody in tears, saliva and nasal secretions fol-
lowing oral immunization of humans with inactivated influenza virus vaccine. Int Arch Allergy Appl Immunol 1986;80(1):107-9.
[7] Avtushenko SS, Sorokin EM, Zoschenkova NY, Zacharova NG, Naichin AN. Clinical and immunologi-cal characteristics of the emulsion form of inactivated influenza vaccine delivered by oral immuniza-tion. J Biotechnol 1996;44(1-3):21-8.
[8] Moldoveanu Z, Clements ML, Prince SJ, Murphy BR, Mestecky J. Human immune responses to influ-enza virus vaccines administered by systemic or mucosal routes. Vaccine 1995;13(11):1006-12.
[9] Lazzell V, Waldman RH, Rose C, Khakoo R, Jacknowitz A, Howard S. Immunization against influenza in humans using an oral enteric-coated killed virus vaccine. J Biol Stand 1984;12(3):315-21.
[10] Cox RJ, Brokstad KA, Ogra P. Influenza virus: immunity and vaccination strategies. Comparison of the immune response to inactivated and live, attenuated influenza vaccines. Scand J Immunol 2004;59(1):1-15.
[11] Hobson D, Curry RL, Beare AS, Ward-Gardner A. The role of serum haemagglutination-inhibiting antibody in protection against challenge infection with influenza A2 and B viruses. J Hyg (Lond) 1972;70(4):767-77.
[12] Small PA, Jr., Waldman RH, Bruno JC, Gifford GE. Influenza infection in ferrets: role of serum anti-body in protection and recovery. Infect Immun 1976;13(2):417-24.
[13] Liew FY, Russell SM, Appleyard G, Brand CM, Beale J. Cross-protection in mice infected with influ-enza A virus by the respiratory route is correlated with local IgA antibody rather than serum antibody or cytotoxic T cell reactivity. Eur J Immunol 1984;14(4):350-6.
[14] Tumpey TM, Renshaw M, Clements JD, Katz JM. Mucosal delivery of inactivated influenza vaccine induces B-cell-dependent heterosubtypic cross-protection against lethal influenza A H5N1 virus infec-tion. J Virol 2001;75(11):5141-50.
[15] Renegar KB, Small PA, Jr. Immunoglobulin A mediation of murine nasal anti-influenza virus immu-nity. J Virol 1991;65(4):2146-8.
[16] Belshe RB, Gruber WC, Mendelman PM, Mehta HB, Mahmood K, Reisinger K, et al. Correlates of immune protection induced by live, attenuated, cold-adapted, trivalent, intranasal influenza virus vac-cine. J Infect Dis 2000;181(3):1133-7.
[17] Renegar KB, Small PA, Jr. Passive transfer of local immunity to influenza virus infection by IgA anti-body. J Immunol 1991;146(6):1972-8.
[18] Beyer WE, Palache AM, de Jong JC, Osterhaus AD. Cold-adapted live influenza vaccine versus inacti-vated vaccine: systemic vaccine reactions, local and systemic antibody response, and vaccine efficacy. A meta-analysis. Vaccine 2002;20(9-10):1340-53.
[19] Huckriede A, Bungener L, Stegmann T, Daemen T, Medema J, Palache AM, et al. The virosome con-cept for influenza vaccines. Vaccine 2005;23 Suppl 1:S26-38.
[20] The European Agency for the Evaluation of Medicinal Products (EMEA) Committee for Proprietary Medicinal Products (CPMP). Note for Guidance on Harmonization of Requirements for Influenza Vaccines. CPMP/BWP/214/96, London, March 12, 1997.
[21] Barackman JD, Ott G, Pine S, O'Hagan DT. Oral administration of influenza vaccine in combination with the adjuvants LT-K63 and LT-R72 induces potent immune responses comparable to or stronger
CHAPTER V
127

than traditional intramuscular immunization. Clin Diagn Lab Immunol 2001;8(3):652-7.[22] Lu X, Clements JD, Katz JM. Mutant Escherichia coli heat-labile enterotoxin [LT(R192G)] enhances
protective humoral and cellular immune responses to orally administered inactivated influenza vac-cine. Vaccine 2002;20(7-8):1019-29.
[23] Katz JM, Lu X, Young SA, Galphin JC. Adjuvant activity of the heat-labile enterotoxin from entero-toxigenic Escherichia coli for oral administration of inactivated influenza virus vaccine. J Infect Dis 1997;175(2):352-63.
[24] Chen KS, Burlington DB, Quinnan GV, Jr. Active synthesis of hemagglutinin-specific immunoglobulin A by lung cells of mice that were immunized intragastrically with inactivated influenza virus vaccine. J Virol 1987;61(7):2150-4.
[25] Meitin CA, Bender BS, Small PA, Jr. Enteric immunization of mice against influenza with recombinant vaccinia. Proc Natl Acad Sci U S A 1994;91(23):11187-91.
[26] Kersten G, Hirschberg H. Antigen delivery systems. Expert Rev Vaccines 2004;3(4):453-62.[27] Yuki Y, Kiyono H. New generation of mucosal adjuvants for the induction of protective immunity. Rev
Med Virol 2003;13(5):293-310.[28] Conacher M, Alexander J, Brewer JM. Oral immunisation with peptide and protein antigens by formu-
lation in lipid vesicles incorporating bile salts (bilosomes). Vaccine 2001;19(20-22):2965-74.[29] Mann JF, Ferro VA, Mullen AB, Tetley L, Mullen M, Carter KC, et al. Optimisation of a lipid based oral
delivery system containing A/Panama influenza haemagglutinin. Vaccine 2004;22(19):2425-9.[30] Sizer PJ. Towards an oral influenza vaccine. Trends Biotechnol 1997;15(8):282-5.[31] Pang GT, Clancy RL, O'Reilly SE, Cripps AW. A novel particulate influenza vaccine induces long-term
and broad-based immunity in mice after oral immunization. J Virol 1992;66(2):1162-70.[32] Harokopakis E, Childers NK, Michalek SM, Zhang SS, Tomasi M. Conjugation of cholera toxin or its B
subunit to liposomes for targeted delivery of antigens. J Immunol Methods 1995;185(1):31-42.[33] Chattaraj SC, Rathinavelu A, Das SK. Biodegradable microparticles of influenza viral vaccine: compa-
rison of the effects of routes of administration on the in vivo immune response in mice. J Control Release 1999;58(2):223-32.
[34] Moldoveanu Z, Novak M, Huang WQ, Gilley RM, Staas JK, Schafer D, et al. Oral immunization with influenza virus in biodegradable microspheres. J Infect Dis 1993;167(1):84-90.
[35] Maroni A, Zema L, Cerea M, Sangalli ME. Oral pulsatile drug delivery systems. Expert Opin Drug Deliv 2005;2(5):855-71.
[36] Gazzaniga A, Maroni A, Sangalli ME, Zema L. Time-controlled oral delivery systems for colon tar-geting. Expert Opin Drug Deliv 2006;3(5):583-97.
[37] Schellekens RC, Stuurman FE, van der Weert FH, Kosterink JG, Frijlink HW. A novel dissolution method relevant to intestinal release behaviour and its application in the evaluation of modified release mesalazine products. Eur J Pharm Sci 2007;30(1):15-20.
[38] Schellekens RC, Frijlink HW, inventors; PH-controlled pulsatile delivery system, methods for prepara-tion and use thereof. patent WO 2007/013794 A1. 2007.
[39] Holmgren J, Czerkinsky C, Eriksson K, Mharandi A. Mucosal immunisation and adjuvants: a brief overview of recent advances and challenges. Vaccine 2003;21 Suppl 2:S89-95.
[40] Bensadoun A, Weinstein D. Assay of proteins in the presence of interfering materials. Anal Biochem 1976;70(1):241-50.
[41] de Haan L, Verweij WR, Feil IK, Lijnema TH, Hol WG, Agsteribbe E, et al. Mutants of the Escherichia coli heat-labile enterotoxin with reduced ADP-ribosylation activity or no activity retain the immuno-genic properties of the native holotoxin. Infect Immun 1996;64(12):5413-6.
[42] de Haan L, Holtrop M, Verweij WR, Agsteribbe E, Wilschut J. Mucosal immunogenicity of the Escherichia coli heat-labile enterotoxin: role of the A subunit. Vaccine 1996;14(4):260-6.
[43] van der Linden MP, Mottl H, Keck W. Cytoplasmic high-level expression of a soluble, enzymatically active form of the Escherichia coli penicillin-binding protein 5 and purification by dye chromato-graphy. Eur J Biochem 1992;204(1):197-202.
128

[44] Meissner PS, Sisk WP, Berman ML. Bacteriophage lambda cloning system for the construction of direc-tional cDNA libraries. Proc Natl Acad Sci U S A 1987;84(12):4171-5.
[45] Uesaka Y, Otsuka Y, Lin Z, Yamasaki S, Yamaoka J, Kurazono H, et al. Simple method of purification of Escherichia coli heat-labile enterotoxin and cholera toxin using immobilized galactose. Microb Pathog 1994;16(1):71-6.
[46] de Aizpurua HJ, Russell-Jones GJ. Oral vaccination. Identification of classes of proteins that provoke an immune response upon oral feeding. J Exp Med 1988;167(2):440-51.
[47] Stegmann T, Booy FP, Wilschut J. Effects of low pH on influenza virus. Activation and inactivation of the membrane fusion capacity of the hemagglutinin. J Biol Chem 1987;262(36):17744-9.
[48] Haan L, Verweij WR, Holtrop M, Brands R, van Scharrenburg GJ, Palache AM, et al. Nasal or intra-muscular immunization of mice with influenza subunit antigen and the B subunit of Escherichia coli heat-labile toxin induces IgA- or IgG-mediated protective mucosal immunity. Vaccine 2001;19(20-22):2898-907.
[49] Miyahira Y, Murata K, Rodriguez D, Rodriguez JR, Esteban M, Rodrigues MM, et al. Quantification of antigen specific CD8+ T cells using an ELISPOT assay. J Immunol Methods 1995;181(1):45-54.
[50] Stephenson I, Zambon MC, Rudin A, Colegate A, Podda A, Bugarini R, et al. Phase I evaluation of intranasal trivalent inactivated influenza vaccine with nontoxigenic Escherichia coli enterotoxin and novel biovector as mucosal adjuvants, using adult volunteers. J Virol 2006;80(10):4962-70.
[51] Cheng E, Cardenas-Freytag L, Clements JD. The role of cAMP in mucosal adjuvanticity of Escherichia coli heat-labile enterotoxin (LT). Vaccine 1999;18(1-2):38-49.
[52] McDermott MR, Bienenstock J. Evidence for a common mucosal immunologic system. I. Migration of B immunoblasts into intestinal, respiratory, and genital tissues. J Immunol 1979;122(5):1892-8.
[53] Neutra MR, Pringault E, Kraehenbuhl JP. Antigen sampling across epithelial barriers and induction of mucosal immune responses. Annu Rev Immunol 1996;14:275-300.
[54] Holmgren J, Czerkinsky C, Lycke N, Svennerholm AM. Mucosal immunity: implications for vaccine development. Immunobiology 1992;184(2-3):157-79.
[55] Brandtzaeg P. Overview of the mucosal immune system. Curr Top Microbiol Immunol 1989;146:13-25.[56] Clements ML, Betts RF, Tierney EL, Murphy BR. Serum and nasal wash antibodies associated with
resistance to experimental challenge with influenza A wild-type virus. J Clin Microbiol 1986;24(1):157-60.
[57] Mbawuike IN, Pacheco S, Acuna CL, Switzer KC, Zhang Y, Harriman GR. Mucosal immunity to influ-enza without IgA: an IgA knockout mouse model. J Immunol 1999;162(5):2530-7.
[58] Zhang Y, Pacheco S, Acuna CL, Switzer KC, Wang Y, Gilmore X, et al. Immunoglobulin A-deficient mice exhibit altered T helper 1-type immune responses but retain mucosal immunity to influenza virus. Immunology 2002;105(3):286-94.
[59] McElhaney JE, Hooton JW, Hooton N, Bleackley RC. Comparison of single versus booster dose of influenza vaccination on humoral and cellular immune responses in older adults. Vaccine 2005;23(25):3294-300.
[60] Roman M, Martin-Orozco E, Goodman JS, Nguyen MD, Sato Y, Ronaghy A, et al. Immunostimulatory DNA sequences function as T helper-1-promoting adjuvants. Nat Med 1997;3(8):849-54.
[61] Mohamadzadeh M, Olson S, Kalina WV, Ruthel G, Demmin GL, Warfield KL, et al. Lactobacilli acti-vate human dendritic cells that skew T cells toward T helper 1 polarization. Proc Natl Acad Sci U S A 2005;102(8):2880-5.
[62] Pollock KG, Conacher M, Wei XQ, Alexander J, Brewer JM. Interleukin-18 plays a role in both the alum-induced T helper 2 response and the T helper 1 response induced by alum-adsorbed interleu-kin-12. Immunology 2003;108(2):137-43.
[63] Palladino G, Scherle PA, Gerhard W. Activity of CD4+ T-cell clones of type 1 and type 2 in generation of influenza virus-specific cytotoxic responses in vitro. J Virol 1991;65(11):6071-6.
[64] Moran TM, Isobe H, Fernandez-Sesma A, Schulman JL. Interleukin-4 causes delayed virus clearance in influenza virus-infected mice. J Virol 1996;70(8):5230-5.
CHAPTER V
129

130
[65] Stevens TL, Bossie A, Sanders VM, Fernandez-Botran R, Coffman RL, Mosmann TR, et al. Regulation of antibody isotype secretion by subsets of antigen-specific helper T cells. Nature 1988;334(6179):255-8.

J-P. Amorij1, V. Saluja1, A.H. Petersen2, W.L.J. Hinrichs1, A. Huckriede3, H.W. Frijlink1
Chapter VI
Pulmonary delivery of an inulin-sstabilized influenza subunit vaccine prepared by spray-ffreeze drying induces systemic, mucosalhumoral as well as cell-mmediated immune responses in BALB/c mice.
Vaccine (2007): in press.
1 Department of Pharmaceutical Technology and Biopharmacy, University of Groningen, Groningen, The Netherlands.
2 Department of Pathology and Laboratory Medicine, Medical Biology Section, University Medical Center Groningen, Groningen, The Netherlands.
3 Department of Medical Microbiology, Molecular Virology Section, University Medical Center Groningen and University of Groningen, Groningen, The Netherlands.

Abstract
In this study pulmonary vaccination with a new influenza subunit vaccine powder wasevaluated. Vaccine powder was produced by spray-freeze drying (SFD) using the oligos-accharide inulin as stabilizer. Immune responses after pulmonary vaccination of BALB/cmice with vaccine powder were determined and compared to those induced by intra-muscular and pulmonary vaccination with a conventional liquid subunit vaccine. Allvaccinations were performed without adjuvant. Pulmonary vaccination with liquidsubunit vaccine resulted in systemic humoral (IgG) immune responses similar to intra-muscular immunization. In contrast, the vaccine powder delivered by the pulmonaryroute, induced not only systemic humoral (IgG) responses, but also cell-mediated (Il-4,IFNγ) and mucosal immune responses (IgA, IgG). This study demonstrates that the com-bination of pulmonary antigen delivery and antigen powder production by SFD impro-ves the immunogenic potential of (influenza subunit) antigen. In conclusion, vaccinati-on with a non-adjuvanted SFD subunit vaccine powder by inhalation might be feasibleand could be an alternative to conventional parenteral vaccine administration.
Keywords
Influenza vaccine; Respiratory tract; Dry powder formulation.
132

Introduction
Influenza is a highly contagious disease. During annual influenza epidemics 5-15% of theworld population is affected [WHO fact sheet 2003], resulting in extensive morbidity anda substantial number of deaths especially in the elderly and other high-risk populations.In humans, the primary targets for the influenza virus are epithelial cells in the upperand lower respiratory tract. After infection with influenza virus, the immune systemreacts to replication of the virus in the respiratory tract by both innate and adaptiveimmune responses [1]. These immune reactions include antibody and T cell responsestriggered against epitopes located on virus envelope antigens such as heamagglutinin(HA). The antibodies on the mucosal tissues (IgA produced locally and IgG transudatingfrom serum) may give rise to mucosal immunity [2]. This mucosal immunity in the(upper) respiratory tract is of major importance as it provides the first line of defenseagainst homologous virus infection [3] and might result in cross protection against infec-tions by viruses that display antigenic drift [4-6].
Immunization against the flu is recognized as the most cost-effective method forcontrolling the disease. However, most currently marketed vaccines are administeredparenterally and evoke a systemic immune response without a mucosal IgA response inthe respiratory tract [7-9]. In addition, these vaccines have the disadvantages that theyhave to be administered by trained personnel and that they have to be transported via anexpensive cold chain.
Recently, vaccination against influenza via the intranasal route has drawn consi-derable attention, since it can be applied by self administration and has the potential toelicit a local immune response in addition to the systemic immune response [9-13].However, in many cases, increased local but moderate systemic immune responses werefound after intranasal vaccination. A further disadvantage of most intranasal influenzavaccines is that they are unable to elicit immune responses without an adjuvant [9].Besides the use of adjuvants [14-17] other approaches, like the use of nanoparticles andmicrospheres [18, 19] or cold adapted live influenza virus [20, 21], have been demonstra-ted to improve the efficacy of intranasally administered influenza vaccine. However, inmany cases safety and regulatory issues associated with these approaches negativelyinterfered with their development [22, 23]. As a result, there is still need for a non-adju-vanted vaccine that complies with safety and regulatory requirements.
Several studies report on the development of dry powder vaccine formulations forintranasal delivery. Besides facilitating a more convenient vaccine administration a drypowder vaccine precludes restrictions and problems, like sterility and stability, asso-ciated with liquid formulations [24, 25]. Improved stability of influenza vaccine powderwas shown for vaccine formulations dried with carbohydrates like inulin and trehalose[26, 27]. In addition, a dry powder formulations for intranasal delivery could be formu-lated with trehalose. However, dry powder vaccines for intranasal delivery used thus farcould only elicited a satisfying immune response when they were co-formulated with amucoadhesive polymer or mucosal adjuvant [28].
CHAPTER VI
133

Pulmonary delivery may be an alternative to intranasal delivery of an influenza vac-cine. The lungs are highly vascularized, have a larger absorptive surface area than thenose [29, 30] and contain mucosa-associated lymphoid tissue [15]. Furthermore, localantigen presenting cells like different types of macrophages and dendritic cells are ideal-ly located for antigen sampling and subsequent presentation to T cells [31-33]. In additi-on, dry powder inhalation has proven (for decennia) to be very useful for to the induc-tion of local as well as systemic effects. In general, dry powder inhalers and powders forinhalation are designed to guarantee a high deposition in the lower airways. The smallerthe particles the further the particles travel through the airways during inhalation.Particles with an aerodynamic size in the range of 1-5 μm can reach the alveoli in thelower airways, while particles of 5-10 μm will be deposited in the upper airways, like themain bronchioles. Particle enginering techniques, like spray drying and spray freeze dry-ing (SFD), can be used to produce particles with a broad size distribution which can beused to target both the lower airways as well as the main bronchioles. A vaccine powderhaving these characteristics offers the opportunity to expose a relatively large surfacewithin the entire lung to (high doses of) antigen. Therefore, pulmonary delivery of aninfluenza vaccine is an interesting strategy, that can offer the advantage of inducing bothsystemic and local immunity [34].
We investigated whether pulmonary delivery of inulin stabilized influenza subunitvaccine powder, produced by SFD, could induce an immune response in mice withoutthe use of an adjuvant. For pulmonary immunization with vaccine powder, the powderwas delivered by endotracheal insufflation into the lungs of BALB/c mice. Subsequently,systemic and mucosal antibody responses were determined and compared to those ofmice immunized by pulmonary delivery or intramuscular injection of a conventionalliquid subunit vaccine formulation. Additionally, cell-mediated immunity induced bythe different immunization routes was monitored by measuring the frequency of IFNγand IL-4 producing spleen cells.
134

Materials and Methods
VaccineMonovalent influenza subunit vaccine (A/Panama/2007/99, H3N2) was a gift of SolvayPharmaceuticals (Weesp, the Netherlands). The subunit vaccine was dialyzed againstHBS (2 mM Hepes, 150 mM NaCl, pH 7.4) and subsequently stored at 4°C until furtheruse. The concentration of antigen, HA, was determined by single radial immunodiffusi-on assay [35] .
Formulation of subunit vaccine powder for inhalationThe subunit vaccine powder was produced by SFD. A solution of 275 μg/ml HA and 5.5%w/v inulin 4kD (Sensus, Roosendaal, Netherlands) in HBS, was sprayed into a vessel ofliquid nitrogen with a two-fluid nozzle (diameter 0.5 mm) of the Büchi 190 Mini SprayDryer. The nozzle was placed approximately 5 cm above the surface of the liquid nitro-gen and the vaccine solution was atomized with an air flow of 700 ln/h. After the spray-ing was completed, the vessel with liquid nitrogen and vaccine was transferred to aChrist model Epsilon 2-4 freeze dryer (Salm & Kipp, Breukelen, The Netherlands) pre-cooled at a shelf temperature of -40°C. After liquid nitrogen was evaporated, drying wasperformed at a pressure of 0.220 mbar with a condensor temperature of -85°C while theshelf temperature was gradually increased from -40°C to 5°C over a time period of 32hours. Next, the pressure was reduced to 0.055 mbar. In addition the shelf temperaturewas further gradually increased to 20°C over a time period of 11 h. The obtained pow-ders were stored at room temperature in a vacuum dessicator containing silicagel untilfurther use.
Aerosolization of vaccine solution and powderAerosolization of vaccine solution was performed using a MicroSprayerTM (Model IA-1C, 1¼" long, Penn-Century Inc., Philadelphia, USA). For the powder a dry powderinsufflator (DP-4M-model, Penn-Century Inc., Philadelphia, USA), suitable for mice wasused. The devices previously showed improved delivery efficiency compared to passiveaerosol inhalation [36] and did not cause adverse effects after pulmonary delivery ofliquid [37] or SFD powder [38].
Aerosol droplet / particle size and morphology of SFD particlesThe geometric particle size distributions of the SFD vaccine powder and liquid aerosolswere measured with a Sympatic HELOS compact model KA laser diffraction apparatus(Sympatec GmbH, Clausthal-Zellerfeld, Germany). Small quantities of SFD vaccine pow-der were dispersed at a pressure of 0.5 bar using the RODOS dispersing system or the drypowder insufflator. Liquid aerosols were dispersed using the MicroSprayerTM. A 100-mmlens was used and calculations were based on the Fraunhofer theory. Data reported arethe mean of at least three measurements.
CHAPTER VI
135

For evaluation of the morphology of the produced powder, scanning electron micro-graphs (SEM) were recorded with a JEOL JSM 6301-F microscope (JEOL, Japan). Powderwas dispersed on top of double-sided adhesive carbon tape on a metal disk. Subsequently,particles were coated with 80 nm of gold/palladium using a Balzers 120B sputtering de-vice (BlazerUNION, Liechtenstein). Micrographs were taken at a magnification of 500 or5000.
Aerosol characterizationIn vitro deposition of the aerosols dispersed from the insufflator or microsprayer was tes-ted by cascade impactor analysis with a multi-stage liquid impinger (MSLI) of the Astratype (Erweka, Heusenstamm, Germany) as described in the Pharmacopoeia [39]. Thistest was used to determine the fine particle characteristics of the aerosol clouds genera-ted during inhalation. The insufflator or microsprayer was attached to the inductionport. A flow rate of 30 l/min was used during each dispersion. Stages of the cascadeimpactor were filled with 25 ml of demineralized water and in the final stage, a dry glassfilter (Pall Corp., Michigan, US) was used for the retention of particles that passed thefourth stage. In each experiment 10 or 5 insufflations were performed using a dose of 2mg vaccine powder or 50 μl 5% glucose solution (as model) per insufflation, respective-ly. Each dose (one insufflation) in the DP-4-insufflator was administered using 10 airpuffs of 200 μl. The deposition (determined as inulin or glucose) at each stage as well aslosses in the insufflator and the induction port were determined using the anthrone assayas described earlier [40]. All the experiments were performed under controlled ambientconditions (20°C/50% relative humidity). The data reported are the mean of three inde-pendent experiments and in each experiment the recovery was above 95%.
Biochemical integrity and in vittro antigenicityTo ensure that the conformation and antigenicity of HA in the vaccine was unaffectedduring the powder production process, powder samples were reconstituted in ultra purewater and subjected to a proteolytic assay and a single radial immunodiffusion assay(SRID), respectively. In the proteolytic assay HA is subjected to trypsine treatment;when its conformation is changed trypsin cleavage sites become exposed and HA is di-gested into small fragments resulting in decreased recovery of the HA monomer bandafter SDS-PAGE under non-reducing conditions. The proteolytic assay with trypsin wasperformed according to Amorij et al. [26] and the SRID analysis was performed accor-ding to European Pharmacopoeia guidelines [35].
Animal experimentsAnimal experiments were conducted according to the guidelines provided by the DutchAnimal Protection Act and were approved by the Committee for AnimalExperimentation of the University of Groningen, The Netherlands. For all experimentsfemale BALB/c mice (6-8 weeks old) were obtained from Harlan (Zeist, TheNetherlands). All the animals were immunized 3 times i.e. on day 0, 14 and 28 with 5 μgHA. From previous research it is known that i.m. immunization of BALB/c mice accor-
136

ding to this immunization scheme (3 times 5 μg HA) results in a robust immune respon-se [14].
IIn vivo immunogenicityTo test the effect of the SFD process on the in vivo potency of the vaccine, vaccine pow-der was reconstituted and the immunogenicity was determined in mice after i.m. injec-tion (3 times with 5 μg HA; day 0, 14 and 28). One group of mice (n=8) received recon-stituted SFD powder and one group of mice (n=4) received the unprocessed vaccine.Animals were anaesthesized by an i.p injection with 10 ml/kg FFM (Fentanyl, fluani-sone-midazolam) and injected i.m. with 50 μl of the vaccine divided over the posteriorthigh muscles.
Pulmonary immunizationAnimals were anaesthetized by i.p. injection with 10 ml/kg FFM (Fentanyl, fluanisone-midazolam). In order to reduce mucus secretion during intubation animals were s.c.injected with 0.04 mg/kg atropine 5 minutes prior to intubation. Then the animals wereplaced in a supinely position at an angle of 45° using a platform according to a methodderived from Bivas-Benita et al. [41]. Mice were oropharyngeally intubated using amodified 20G Insyte Autoguard catheter (Becton Dickinson BV, Alphen aan de Rijn, TheNetherlands). BALB/c mice were subsequently immunized by pulmonary delivery ofliquid vaccine aerosols (l.i. = liquid insufflation) or vaccine powder (p.i. = powder insuf-flation). The animals were immunized 3 times i.e. on day 0, 14 and 28 with 5 μg HA.
For liquid aerosol insufflation (l.i.), the MicroSprayerTM connected to a pre-filledhigh-pressure syringe (Model FMJ-250, Penn-Century Inc., Philadelphia, USA) wasinserted through the oropharyngeal tube. Subsequently, 50 μl vaccine solution con-taining 5 μg HA was insufflated.
For powder aerosol insufflation (p.i.), the pre-filled dry powder insufflator wasinserted through the oropharyngeal tube ensuring that the tip of the insufflator was 5mm outside the tube at carina height for proper aerosolization of the powder. For eachadministration the insufflator was filled with 1 mg of vaccine powder and 10 air puffs of200 μl were used for aerosolization of the powder. After administration of the powderthe insufflator was taken out and refilled for the subsequent inhalations. A total of 5 μgHA was given. The insufflator was weighed before and after filling as well as after eachadministration to ensure accurate dosing of each mouse.
Sample collectionBefore the first and last vaccine administration blood samples (approx. 40 μl) were takenfrom the mice by an orbita punction under isofluran/O2 anaesthesia. Two weeks after thelast vaccination (day 42), the animals were sacrificed. For determination of serum andmucosal antibody responses mice were bled under isofluran anaesthesia by draining theabdominal aorta. Serum samples were obtained by centrifugation at 1.200 x g for 5 minand were stored at -20°C until further analysis.
The spleens were harvested immediately after sacrification of the mice and stored
CHAPTER VI
137

until further analysis in ice-cold IMDM Glutamax medium (Life Technologies, Paisley,UK) supplemented with 10% w/v FCS (PAA Laboratories, Linz, Austria), 100 U/ml peni-cillin (Life Technologies), 100 μg/ml streptomycin (Life Technologies) and 50 mM β-mercaptoethanol.
Next, nasal and lung lavages were performed by cannulating the trachea via a smallincision using a 20G Insyte Autoguard catheter (Becton Dickinson BV, Alphen aan deRijn, The Netherlands) connected to a 1 ml syringe. Nasal lavage fluids were obtained byflushing twice with the same 1 ml PBS (Fluka) through the nasopharynx. Lung lavageswere performed by repeated flushing of the lungs with the same 1 ml PBS (Fluka).
Immediately after each sample collection, the lavage was mixed with 10 μl stocksolution (1 tablet/2ml PBS) of Complete® protease inhibitor cocktail (Roche DiagnosticsGmbH, Penzberg, Germany). The nasal and lung lavages were stored at -20°C until ana-lysis.
ELISAInfluenza subunit antigen-specific antibody responses were determined by ELISA as de-scribed previously [26]. In brief, ELISA plates (96-well flat bottom, Microlon®600,Greiner, Alphen a/d Rijn, The Netherlands) were coated with 200 ng of influenza subu-nit antigen per well. Appropriate dilutions of sera and mucosal lavage fluids of each indi-vidual mouse were applied to the plates. Sera were analyzed for IgG, IgA, IgE, IgG1 andIgG2a, while lavages were analyzed for IgG and IgA. In addition, as a positive control forthe IgE determination, plates were coated with 100 ng of goat antibody directed againstmouse IgE, and subsequently incubated with mouse myeloma IgE. Antigen-specific IgG-IgA-, IgG1-, IgG2a- and IgE-responses were determined using horseradish peroxidase-conjugated goat antibodies or rat antibody (for IgE only) directed against mouse (1:5000;Southern Biotech, Birmingham, Alabama, USA) and 1,2-phenylendiamin-dihydrochlo-rid. Absorbances at 492 nm were measured with a Benchmark Microplate reader(BioRad, Hercules, CA). Titers are given as the reciprocal of the calculated sample dilu-tion corresponding to an A492 = 0.2 after background correction.
Haemagglutination inhibition (HI) assayThe haemagglutination inhibition titers in serum were determined as described pre-viously [26]. Briefly, test sera were inactivated at 56°C for 30 min. One part of test serawas mixed with three parts of a 25% w/v kaolin stock solution and incubated for 20 minat room temperature. After incubation sera were centrifuged at 1.200 x g for 2 min toremove from kaolin. 50 μl containing 4 hemagglutinating units (HAU) of influenza viruswas then added to wells containing 50 μl of serially diluted test sera, mixed and incu-bated at room temperature. After 40 minutes, 50 μl 1% v/v guinea pig erythrocyte solu-tion was added to each well and plates were incubated at room temperature for 1-2 hoursallowing haemagglutination to occur. The HI titers are defined as the highest serum dilu-tion capable of preventing haemagglutination.
138

ELISPOTFor determination of the cytokine production of spleen cells from the immunized micean ELISPOT assay was performed according to a protocol, which was a combination ofthe procedures described by Bungener et al. [42] and McCutcheon et al. [43]. 96 micro-well plates with a Nylon 6,6 BiodyneTM B membrane bottom (Nunc Silent Screen Plates,Nunc, USA) were coated with purified anti-mouse IFN-γ or anti-mouse IL-4 (BDPharmingen, Erembodegem, Belgium), in a concentration of 5 μg/ml. After overnightincubation at 4°C plates were washed three times with sterile PBS containing 0.02% w/vTween80 and incubated with blocking buffer (PBS + 4% w/v BSA) at 37°C for 1h. Spleencells were isolated in IMDM-medium containing 10% w/v FCS, plated in different quan-tities (0.125 - 2.0 x106 cells) in 96 wells and incubated overnight at 37°C with or without500 ng of influenza subunit antigen/well. After overnight stimulation, cells were lysedby incubating them on ice for 15 min. After washing the plates 5 times with PBS/Tween,IFN-γ or IL-4 were detected using biotinylated anti-mouse antibodies and streptavidin-alkaline phosphatase (1:1000, BD Pharmingen). For the development of spots, 100 μl sub-strate solution, (1 mg/ml 5-bromo-4-chloro-3-indolylphosphate, 0.92% w/v 2-amino-2-methyl-1-propanol and 0.08 μl/ml Triton X-405 in water) was added to each well andplates were incubated at room temperature. Spots were subsequently counted on anElispot reader (A.EL.VIS Elispot reader). The number of IFN-γ or IL-4-secreting cellswere obtained by subtracting the background (number of spots after incubation withoutantigen) from the number of spots observed in wells containing spleen cells incubatedwith influenza subunit antigen.
Statistical analysisThe statistical analyses were performed using Student's t-test with P<0.05 as the minimallevel of significance. The results are presented as mean ± standard error unless indicatedotherwise.
CHAPTER VI
139

Fig. 1 Analysis of the structural integrity of HA in the spray freeze dried vaccine by the proteolytic assay.Shown are the HA monomer (75 kDa) bands revealed by SDS-PAGE (under non-reducing conditions) afterincubation with trypsine. A) Positive control: unprocessed subunit vaccine; B) Negative control: acid treatedsubunit vaccine; C) SFD subunit vaccine.
Fig. 2 Analysis of the immunogenicity of spray freeze dried vaccine. Mice were i.m. immunized threetimes at day 0, 14 and 28 with 5 μg HA of either unprocessed subunit vaccine (4 mice) or reconstituted SFDvaccine powder (8 mice) and sacrificed after 42 days. IgG titers (black bars) and HI-titers (grey bars) are shown.
140
Results
Biochemical integrity and immunogenicity.
To ensure that the conformation and antigenicity of HA in the vaccine was unaffectedduring the powder production process used in the present study, i.e. the combination ofspraying and lyophilization, the biochemical and antigenic properties of the vaccinepowder were evaluated. The structural integrity of HA in the SFD powder was testedusing a proteolytic assay. In the native conformation, HA is resistant to digestion by tryp-sin, but when trypsin cleavage sites of HA are exposed due to a conformational change,HA is digested into small fragments. The digestion of HA can be readily resolved byPAGE. In Fig.1 it is shown that the protein band (A) associated with the intact HAmonomer (75kDa), was identical to that of the sample of the SFD powder (C). In con-trast, in an acid-treated sample, which was used as a negative control, no intact HAmonomer was present after trypsin digestion (B).
The antigenic integrity of HA was investigated in vitro by measuring the HApotency by SRID analysis. SRID analysis revealed a HA potency recovery of 104%. TheHA potency of SFD-vaccine and the unprocessed vaccine did not significantly differ
indicating that the potency of HA was not affected by the SFD process. The immunogenicity of the SFD powder was assessed by i.m. immunization of
BALB/c mice with reconstituted SFD vaccine powder or unprocessed influenza subunitvaccine. As shown in Fig.2 both vaccines elicited equivalent serum IgG titers and HItiters. These results demonstrate that SFD of subunit vaccine using inulin as a stabilizerdid neither affect the biochemical integrity nor the immunogenicity of the vaccine.
Particle size, morphology and aerosol characterizationThe particle size distributions of the SFD vaccine powder and of the aerosols of the vac-cine powder and liquid vaccine are given in table I.
The geometrical mean particle size of subunit vaccine powder as determined bylaser diffraction analysis was in the range of 10-11 μm. Moreover, the vaccine powderpossessed a relative large size distribution (Span = 1.88) when the RODOS dispersing sys-tem was used and even larger after dispersion from the Insufflator (Span = 2.3). Scanningelectron micrographs (Fig. 3A and 3B) revealed that the SFD powder consisted of parti-cles that were spherical and highly porous. The size of the SFD particles revealed by SEMcorresponded with the size measured by laser diffraction. Liquid aerosols possessed alarger geometrical mean particle size (table I), in the range of 25 μm, but a smaller sizedistribution (Span = 1.21).
Table I Analysis of the geometric particle size of SFD powder, powder- and liquid vaccine aerosols by laserdiffraction.
CHAPTER VI
141
X10 X50 X90 Span1
(μm ± sd) (μm ± sd) (μm ± sd) (μm ± sd)
SFD vaccine powder from RODOS dispersing system 4.44 ± 0.03 10.36 ± 0.04 23.93 ± 0.30 1.88 ± 0.03
SFD vaccine powder from Insufflator 4.20 ± 0.05 11.05 ± 0.11 29.66 ± 1.45 2.30 ± 0.12
Liquid vaccine from MicrosprayerTM 13.77 ± 1.31 24.57 ± 1.88 42.02 ± 3.60 1.21 ± 1.55
1 Span = (X90-X10)/X50.

Fig. 3 Analysis of spray-freeze dried subunit vaccine powder by scanning electron microscopy. A) Magnification 500; B) Magnification 5000. Bars represent 50 μm and 5 μm, respectively.
142
rp
geoaero dd ρρ=
Fig. 4 Aerosol characterization by cascade impactor analysis. Dispersion efficiency of spray freeze driedsubunit vaccine powder from the DP4-Insufflator. Recovery as percentage of the insufflated dose. FPF is theweight fraction of the insufflated dose having an aerodynamic diameter < 5 μm.
Dispersion efficiencies from the insufflator and microsprayer, respectively, wereassessed using cascade impactor analysis. (Fig. 4) The cascade impactor analysis revealedthat the vaccine powder aerosolized by the insufflator had a fine particle fraction (FPF)
CHAPTER VI
143
of 38% and an aerodynamic diameter (daero) of 5.3 μm. As described by Concessio andHickey et al.[44], the aerodynamic diameter (daero) of spherical particles is determinedby the geometric diameter (dgeo), the density of the particle (ρp) and the reference den-sity (ρr ; the density of water taken as 1000 kg/m3) [45] :
As a result, the difference between the measured aerodynamic mean particle size (daero)and the geometric mean particle size (dgeo) can be explained by the low density of theparticles (ρp ± 230 kg/m3 ; ρp << ρr ) due to their high porosity as revealed by the SEMmicrographs.
In contrast to the aerosolized vaccine powder, the aerosols from aerosolized conven-tional liquid vaccine were too large in size (X50 = 24.57±1.88) to pass the first tube andto perform proper cascade impactor analysis.

Fig. 6 Serum IgG. Comparative analysis of influenza A/Panama subunit antigen-specific serum IgG res-ponses in mice immunized i.m., l.i. or p.i. at 0, 28 and 42 days after the first immunization. Standard errors <0.15. * l.i. vs. i.m.: P<0.05 ; ** p.i. vs. i.m.: P<0.01 ; *** p.i. vs. l.i.: P<0.001.
Fig. 5 Haemagglutination inhibiting activity of sera. The HI titer of sera was assessed in an HI assay usingA/Panama virus and guinea pig erythrocytes. * l.i. vs. i.m.: P<0.05 ; ** p.i. vs. i.m.: P<0.02 ; *** p.i. vs. l.i.: P<0.001.
144
Systemic antibody responses in i.g. or i.c. immunized mice.In order to evaluate the potential of pulmonary delivery of the SFD vaccine powder,mice were immunized 3 times with 5 μg HA (day 0, 14 and 28) without adjuvant by thedifferent administration routes. The immune responses induced after pulmonary vac-cination with aerosolized conventional liquid vaccine (l.i. = liquid insufflation) or SFDvaccine powder (p.i. = powder insufflation) were compared with those induced afterintramuscular (i.m.) vaccination with conventional liquid vaccine.
First, the HI-titers of the sera were assessed in an HI assay (Fig.5). I.m. immuniza-tion with conventional vaccine resulted in high HI-titers. Also l.i. immunization with
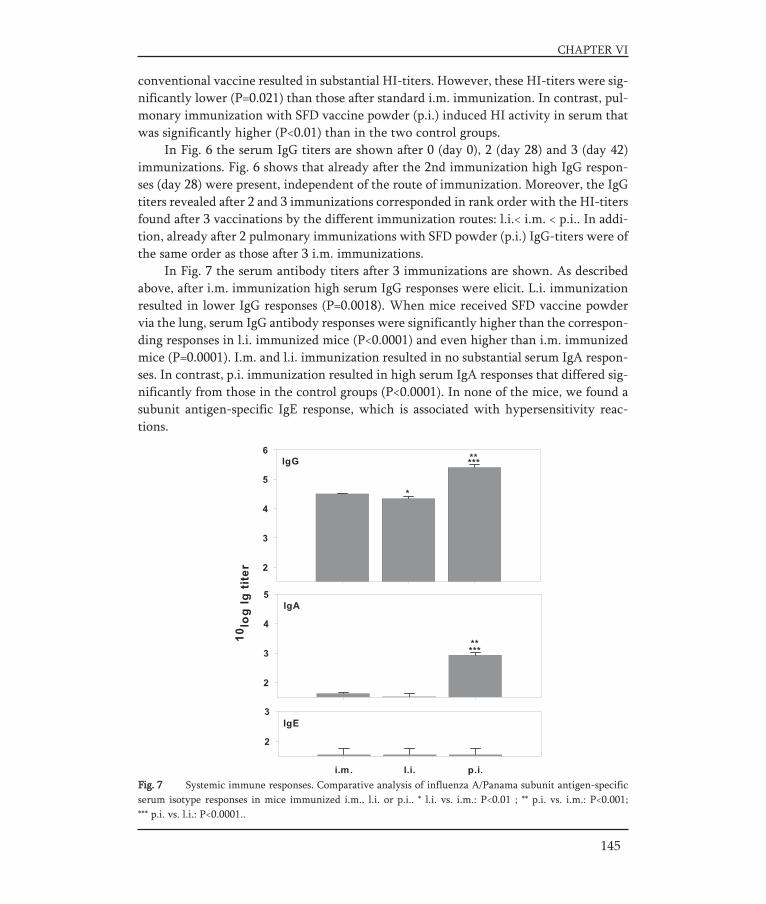
Fig. 7 Systemic immune responses. Comparative analysis of influenza A/Panama subunit antigen-specificserum isotype responses in mice immunized i.m., l.i. or p.i.. * l.i. vs. i.m.: P<0.01 ; ** p.i. vs. i.m.: P<0.001; *** p.i. vs. l.i.: P<0.0001..
conventional vaccine resulted in substantial HI-titers. However, these HI-titers were sig-nificantly lower (P=0.021) than those after standard i.m. immunization. In contrast, pul-monary immunization with SFD vaccine powder (p.i.) induced HI activity in serum thatwas significantly higher (P<0.01) than in the two control groups.
In Fig. 6 the serum IgG titers are shown after 0 (day 0), 2 (day 28) and 3 (day 42)immunizations. Fig. 6 shows that already after the 2nd immunization high IgG respon-ses (day 28) were present, independent of the route of immunization. Moreover, the IgGtiters revealed after 2 and 3 immunizations corresponded in rank order with the HI-titersfound after 3 vaccinations by the different immunization routes: l.i.< i.m. < p.i.. In addi-tion, already after 2 pulmonary immunizations with SFD powder (p.i.) IgG-titers were ofthe same order as those after 3 i.m. immunizations.
In Fig. 7 the serum antibody titers after 3 immunizations are shown. As describedabove, after i.m. immunization high serum IgG responses were elicit. L.i. immunizationresulted in lower IgG responses (P=0.0018). When mice received SFD vaccine powdervia the lung, serum IgG antibody responses were significantly higher than the correspon-ding responses in l.i. immunized mice (P<0.0001) and even higher than i.m. immunizedmice (P=0.0001). I.m. and l.i. immunization resulted in no substantial serum IgA respon-ses. In contrast, p.i. immunization resulted in high serum IgA responses that differed sig-nificantly from those in the control groups (P<0.0001). In none of the mice, we found asubunit antigen-specific IgE response, which is associated with hypersensitivity reac-tions.
CHAPTER VI
145

Fig. 8 Mucosal immune responses. Comparative analysis of influenza A/Panama subunit antigen-specificIgG and IgA antibodies in nasal (black bars) and lung lavages (grey bars) of immunized mice. The numbersabove the columns indicate the number of responders per group. * p.i. vs. i.m.: P<0.02 ; ** p.i. vs. i.m.: P<0.0001*** p.i. vs. l.i.: P<0.0001; **** p.i. vs. i.m.: P<0.01; **** p.i. vs. l.i.: P<0.01.
146
Mucosal immune response.In order to evaluate whether any of the immunization regimens induced a mucosalimmune response, both influenza antigen specific IgG and IgA antibodies in nasal andlung lavages of immunized mice were assessed. Nasal IgG was found in all of the i.m. and1 out of 8 of the l.i. immunized mice, but titers were very low. In the p.i. immunizedgroup in 6 out of 8 mice nasal IgG was found. Moreover, after p.i. immunization the nasalIgG titers were significantly higher than after i.m. and l.i. immunization (P<0.05).
Lung IgG titers were found in all immunized mice and they were higher than nasalIgG. The IgG titers in the lungs of p.i. immunized mice were about fifty fold higher thanin the l.i. or i.m. groups. Moreover, the lung IgG titers corresponded in rank order withthe IgG titers found in serum by the different immunization routes: l.i.< i.m. < p.i..
While i.m and l.i. immunization of mice induced only minor IgA titers in nasal andlung lavages, pulmonary immunization with SFD influenza vaccine powder induced sub-stantial/higher influenza specific IgA titers both in the nose (P<0.01) and lungs (P<0.001)in 7 and 8 mice, respectively. Furthermore, preliminary data showed that the lung lava-ges of mice pulmonarily immunized with powder were capable of inhibiting heamagglu-tination of virus (data not shown).

Fig. 9 Cell-mediated immune responses. Cytokine release profile of the effector cell population from micei.m., l.i. or p.i. immunized with subunit antigen. Results of (A) IFNγ− and (B) IL-4-producing spleen cells arepresented as the geometric mean ± SEM. * vs. i.m.: P<0.05; ** vs. i.m.: P<0.02; *** vs. l.i.: P<0.05.
Cell-mmediated immune response. In order to evaluate whether the immunization regimens induced cell-mediated im-mune response, both the antigen specific frequency of IFNγ− and IL-4 producing spleencells of immunized mice was assessed (Fig. 9). Intramuscular immunization induced noIFN-γ producing spleen cells but substantial frequencies of IL-4 producing spleen cells,indicating a predominant T-helper 2 (Th2) response. Pulmonary immunization withliquid subunit vaccine resulted in low frequencies of both IFNγ− and IL-4 producingspleen cells, indicating a poor but balanced Th1 and Th2 response. In contrast, pulmo-nary immunization with SFD influenza vaccine powder not only induced a balanced Th1and Th2 response, but also high frequencies of both IFNγ− and IL-4 producing spleencells.
CHAPTER VI
147

Fig. 10 Serum IgG subtype distribution in sera of mice immunized i.m., l.i. or p.i. Subunit antigen-specificserum IgG1 responses (black bars) were significantly (P<0.001) dominant over IgG2a (grey bars) in all immu-nized mice, resulting in IgG1/IgG2a ratio's > 1. * vs. i.m.: P<0.01; ** vs. i.m.: P<0.001.
Next, the IgG subtype profile in the sera of the differently immunized mice was ana-lysed (Fig.10). In all immunized mice there was induction of both IgG1 and IgG2a. L.i.immunization induced lower IgG1 titers (P=0.009), but comparable IgG2a titers (P=0.88)than i.m. immunization. In contrast, p.i. immunization induced both higher IgG1(P=0.0004) as well as higher IgG2a titers (P=0.0002) than the i.m. immunization. Allimmunized mice exhibited an IgG1/IgG2a ratio favoring IgG1 (P<0.001), indicating apredominant Th2 response. Yet, the IgG1/Ig2a ratio after p.i. immunization was signifi-cant lower than after i.m. immunization (P=0.017), indicating a more balanced Th1/Th2response.
148

Discussion
The present study demonstrates that pulmonary administration of an influenza subunitvaccine powder, prepared by SFD can induce a strong immune response in BALB/c miceeven when administered without adjuvant.
The new powder formulation of subunit influenza vaccine used in this study, i.e.SFD formulation, has the potential to be used for vaccination by dry powder inhalationin humans. First, it is shown that the vaccine antigen, HA, retained its structural andantigenic properties after SFD using inulin as stabilizer. Secondly, the SFD formulationis suitable for vaccine delivery into the lungs by inhalation. SFD has been described as asuitable process to produce a powder that possesses the required characteristics neededfor dry powder inhalation [46, 47]. Our SFD formulation consisted of large porous parti-cles having a geometric diameter of approximately 11 μm with a broad size distribution(4-24 μm) facilitating vaccine deposition over the large surface area of the lungs. In gene-ral, the larger the particles are, the higher their site of deposition in the airways. Our SFDformulation, consisted of highly porous particles with an average aerodynamic diameterof 5.3 μm and a broad size distribution. As a result the SFD particles are capable to bedeposited throughout the entire lung, including the lower airways. This is supported bythe FPF of the SFD formulation (38%) measured with cascade impactor analysis and pre-vious experience with SFD formulations for inhalation [47, 48]. Also Maa et al. produceda stable influenza subunit powder using SFD [27]. However, they used a solution with ahigh solid content (35%wt excipient) for SFD in order to produce high density particlesin the range of 30-60 μm suitable for epidermal immunization.
Pulmonary delivery of the SFD formulation in BALB/c mice (p.i.) induced strongcell-mediated as well as systemic and mucosal humoral immune responses. With respectto the cell-mediated immune responses, pulmonary delivery of the SFD formulationresulted in higher numbers of IFNγ- and IL4-producing T-helper cells than the conven-tional i.m. injection of influenza subunit vaccine. Moreover, the phenotype of theseimmune responses was more balanced. A relative high contribution of Th1 cells isthought to be important, since they are superior to Th2 cells in providing protectionagainst viral infection and can provide a certain degree of cross-protective immunity [49,50] .
In general, the development of influenza vaccines aims at the induction of an ade-quate level of virus-neutralizing antibodies in the serum due to current criteria for vac-cine immunogenicity, indicated by an HI titer higher than 40 in humans [51]. However,additional induction of mucosal antibodies in the respiratory tract is considered highlyadvantageous because these antibodies can provide protection against influenza virus atthe port of entry [52, 53]. IgA is regarded as the main effector antibody that providesmucosal immunity. However, also mucosal IgG has shown to correlate with protectionduring challenge experiments in mice [54, 55]. Pulmonary delivery of the SFD formu-lation not only resulted in enhanced systemic humoral as well as cell-mediated immune
CHAPTER VI
149

responses but also in strong mucosal IgA and IgG responses in the respiratory tract, inparticular in the lungs. Moreover, significant levels of antigen-specific IgA antibodiesappeared in the nose, while the powder was administered to the lungs. This generationof IgA antibodies at distant mucosal sites can be explained by cellular migration of pri-med antigen-specific B cells from the original mucosal induction site [56, 57]. Theseresults suggest that effective targeting of lung-associated lymphoid tissue results in de-velopment of immune effector cells locally, that successfully colonize the secondarylymphoid organs throughout the body [58].
The induction of strong mucosal immune responses in the respiratory tract by pul-monary vaccination with SFD vaccine as demonstrated here is in contrast to a study per-formed by Smith et al.. These authors found increased serum IgG antibody levels, but nomucosal IgA antibodies in the respiratory tract upon pulmonary immunization of ratswith an influenza subunit vaccine powder [58]. This difference may be explained by thedifference in solubility and particle size distribution of the formulations and the animalmodel used. Smith et al. used a spray-dried formulation suitable for delivery to the lowerairways (particles of 1-5 μm) consisting of 1,2-dipalmitoyl-sn-glycero-3-phosphocholine(DPPC), which decreases the dissolution rate of the formulation. Although DPPC is con-sidered to decrease the particle uptake by macrophages [59, 60], the particulate nature ofthe spray-dried formulation could promote phagocytic clearance from the lung by alve-olar macrophages (AM) [60], which are considered as rather inefficient stimulators ofimmune responses [61-65]. In contrast to the microparticles used by Smith et al., our SFDparticles have a broader size distribution, are hygroscopic [66] and highly porous; andtherefore will instantaneously dissolve in the lung fluids. This will result in release ofantigen within the entire lung and subsequent persistence of antigen on the lung surfa-ces. As a result, SFD particles escape the phagocytic clearance from the lung and releasethe antigen efficiently thus promoting effective targeting of lung-associated lymphoidtissue (within the entire lung) such that mucosal and systemic humoral immune respon-ses can be initiated.
Aerosolized liquid vaccine (l.i.) induced substantial systemic (both IgG- and HItiters) but low mucosal humoral immune responses. Differences in immune responsebetween the aerosolized liquid vaccine and the SFD vaccine powder may be explainedby differences in targeting of the (lower) respiratory tract as result of aerosol behavior.After aerosolization with the MicrosprayerTM, the liquid vaccine aerosols had a size of20-25 μm, a size that could not pass the inlet tube during cascade impactor analysis. Asa result, it is expected that after insufflation the vaccine would reach mainly the upperparts of the lungs of mice. In contrast, the SFD vaccine powder consisted of porous par-ticles with a mean aerodynamic particle size of 5.3 μm and a broad size distribution sui-table to expose a relatively large surface within the entire lung to (high doses of) antigen.In addition, the inulin in the SFD vaccine powder might facilitate antigen uptake by hin-dering mucociliary clearence of vaccine (that is not deposited in the alveoli) by increa-sing local viscosity after dissolution at the lung mucosa.
150

Conclusion
This study demonstrates that pulmonary vaccination with an SFD influenza subunit vac-cine powder induces mucosal, systemic humoral as well as cell-mediated immune res-ponses even in the absence of adjuvant. These responses were superior to those elicitedby conventional i.m. vaccination or pulmonary vaccination with a liquid aerosolizedvaccine. Our results imply that vaccination with a non-adjuvanted SFD subunit influen-za vaccine powder by inhalation might be feasible.
Acknowledgements
We thank T. Westra for (biotechnical) assistance and A. Eissens for making the SEM-micrographs. A.H de Boer and P. Hagendoorn are acknowledged for helpful instructionsconcerning the particle and aerosol measurements. (Department of PharmaceuticalTechnology and Biopharmacy, University of Groningen)
CHAPTER VI
151

REFERENCES
[1] Couch RB, Kasel JA. Immunity to influenza in man. Annu Rev Microbiol 1983;37:529-49.[2] Cox RJ, Brokstad KA, Ogra P. Influenza virus: immunity and vaccination strategies. Comparison of the
immune response to inactivated and live, attenuated influenza vaccines. Scand J Immunol 2004;59(1):1-15.
[3] Wong SS, Yuen KY. Avian influenza virus infections in humans. Chest 2006;129(1):156-68.[4] Liew FY, Russell SM, Appleyard G, Brand CM, Beale J. Cross-protection in mice infected with influ-
enza A virus by the respiratory route is correlated with local IgA antibody rather than serum antibody or cytotoxic T cell reactivity. Eur J Immunol 1984;14(4):350-6.
[5] Tumpey TM, Renshaw M, Clements JD, Katz JM. Mucosal delivery of inactivated influenza vaccine induces B-cell-dependent heterosubtypic cross-protection against lethal influenza A H5N1 virus infec-tion. J Virol 2001;75(11):5141-50.
[6] Asahi-Ozaki Y, Yoshikawa T, Iwakura Y, Suzuki Y, Tamura S, Kurata T, et al. Secretory IgA antibodies provide cross-protection against infection with different strains of influenza B virus. J Med Virol 2004;74(2):328-35.
[7] Tamura S, Kurata T. Defense mechanisms against influenza virus infection in the respiratory tract mucosa. Jpn J Infect Dis 2004;57(6):236-47.
[8] Sweet C, Smith H. Pathogenicity of influenza virus. Microbiol Rev 1980;44(2):303-30.[9] Potter CW, Jennings R. Intranasal immunization with inactivated influenza vaccine. Pharm Sci
Technol Today 1999;2(10):402-08.[10] Cassetti MC, Katz JM, Wood J. Report of a consultation on role of immunological assays to evaluate
efficacy of influenza vaccines. Initiative for Vaccine Research and Global Influenza Programme, World Health Organization, Geneva, Switzerland, 25 January 2005. Vaccine 2006;24(5):541-3.
[11] Chen D, Endres RL, Erickson CA, Weis KF, McGregor MW, Kawaoka Y, et al. Epidermal immuniza-tion by a needle-free powder delivery technology: immunogenicity of influenza vaccine and protection in mice. Nat Med 2000;6(10):1187-90.
[12] Joseph A, Itskovitz-Cooper N, Samira S, Flasterstein O, Eliyahu H, Simberg D, et al. A new intranasal influenza vaccine based on a novel polycationic lipid--ceramide carbamoyl-spermine (CCS) I. Immunogenicity and efficacy studies in mice. Vaccine 2006;24(18):3990-4006.
[13] Read RC, Naylor SC, Potter CW, Bond J, Jabbal-Gill I, Fisher A, et al. Effective nasal influenza vaccine delivery using chitosan. Vaccine 2005;23(35):4367-74.
[14] Haan L, Verweij WR, Holtrop M, Brands R, van Scharrenburg GJ, Palache AM, et al. Nasal or intra-muscular immunization of mice with influenza subunit antigen and the B subunit of Escherichia coli heat-labile toxin induces IgA- or IgG-mediated protective mucosal immunity. Vaccine 2001;19(20-22):2898-907.
[15] Holmgren J, Czerkinsky C. Mucosal immunity and vaccines. Nat Med 2005;11(4 Suppl):S45-53.[16] McCluskie MJ, Weeratna RD, Clements JD, Davis HL. Mucosal immunization of mice using CpG DNA
and/or mutants of the heat-labile enterotoxin of Escherichia coli as adjuvants. Vaccine 2001;19(27):3759-68.
[17] Stephenson I, Zambon MC, Rudin A, Colegate A, Podda A, Bugarini R, et al. Phase I evaluation of intranasal trivalent inactivated influenza vaccine with nontoxigenic Escherichia coli enterotoxin and novel biovector as mucosal adjuvants, using adult volunteers. J Virol 2006;80(10):4962-70.
[18] Davis SS, Illum L. Absorption enhancers for nasal drug delivery. Clin Pharmacokinet 2003;42(13):1107-28.
[19] Amidi M, Romeijn SG, Verhoef JC, Junginger HE, Bungener L, Huckriede A, et al. N-Trimethyl chi-tosan (TMC) nanoparticles loaded with influenza subunit antigen for intranasal vaccination: Biological properties and immunogenicity in a mouse model. Vaccine 2006.
[20] Boyce TG, Hsu HH, Sannella EC, Coleman-Dockery SD, Baylis E, Zhu Y, et al. Safety and immuno-genicity of adjuvanted and unadjuvanted subunit influenza vaccines administered intranasally to heal-thy adults. Vaccine 2000;19(2-3):217-26.
152

[21] Belshe RB, Gruber WC, Mendelman PM, Cho I, Reisinger K, Block SL, et al. Efficacy of vaccination with live attenuated, cold-adapted, trivalent, intranasal influenza virus vaccine against a variant (A/Sydney) not contained in the vaccine. J Pediatr 2000;136(2):168-75.
[22] Fujihashi K, Koga T, van Ginkel FW, Hagiwara Y, McGhee JR. A dilemma for mucosal vaccination: efficacy versus toxicity using enterotoxin-based adjuvants. Vaccine 2002;20(19-20):2431-8.
[23] McCarthy MW, Kockler DR. Trivalent intranasal influenza vaccine, live. Ann Pharmacother 2004;38(12):2086-93.
[24] Giudice EL, Campbell JD. Needle-free vaccine delivery. Adv Drug Deliv Rev 2006;58(1):68-89.[25] Dean HJ. Alternative routes of influenza vaccine delivery. Expert Opin Drug Deliv 2006;3(5):557-61.[26] Amorij JP, Meulenaar J, Hinrichs WL, Stegmann T, Huckriede A, Coenen F, et al. Rational design of an
influenza subunit vaccine powder with sugar glass technology: preventing conformational changes of haemagglutinin during freezing and freeze-drying. Vaccine 2007;25(35):6447-57.
[27] Maa YF, Ameri M, Shu C, Payne LG, Chen D. Influenza vaccine powder formulation development: spray-freeze-drying and stability evaluation. J Pharm Sci 2004;93(7):1912-23.
[28] Huang J, Garmise RJ, Crowder TM, Mar K, Hwang CR, Hickey AJ, et al. A novel dry powder influenza vaccine and intranasal delivery technology: induction of systemic and mucosal immune responses in rats. Vaccine 2004;23(6):794-801.
[29] Shoyele SA, Slowey A. Prospects of formulating proteins/peptides as aerosols for pulmonary drug delivery. Int J Pharm 2006;314(1):1-8.
[30] Sullivan VJ, Mikszta JA, Laurent P, Huang J, Ford B. Noninvasive delivery technologies: respiratory delivery of vaccines. Expert Opin Drug Deliv 2006;3(1):87-95.
[31] Lambrecht BN, Prins JB, Hoogsteden HC. Lung dendritic cells and host immunity to infection. Eur Respir J 2001;18(4):692-704.
[32] von Garnier C, Filgueira L, Wikstrom M, Smith M, Thomas JA, Strickland DH, et al. Anatomical loca-tion determines the distribution and function of dendritic cells and other APCs in the respiratory tract. J Immunol 2005;175(3):1609-18.
[33] Holt PG. Pulmonary dendritic cells in local immunity to inert and pathogenic antigens in the respira-tory tract. Proc Am Thorac Soc 2005;2(2):116-20.
[34] Holmgren J, Czerkinsky C, Lycke N, Svennerholm AM. Mucosal immunity: implications for vaccine development. Immunobiology 1992;184(2-3):157-79.
[35] Immunochemical methods (2.7.1). In: European Pharmacopoeia, 5th edition. Strasbourg, Council of Europe, 2004: 187.
[36] Beck SE, Laube BL, Adams RJ, Chesnut K, Flotte TR, Guggino WB. Deposition of aerosolized AAV vec-tors in the lungs of rhesus macaques. Ped Pulm 1999;Suppl:228-29.
[37] Century TJ. A new intrapulmonary aerosol delivery device. Respiratory Drug Delivery 2000;VII:1-4.[38] Loebenberg R, Finlay WH, Roa WH, Ely L, inventors; Effervescent powders for inhalation patent
490331. 2007.[39] Preparations for inhalation: aerodynamic assessment of fine particles (2.9.18).
In: European Pharmacopoeia, 5th edition. Strasbourg, Council of Europe, 2004: 244-51.[40] Eriksson HJ, Verweij WR, Poelstra K, Hinrichs WL, de Jong GJ, Somsen GW, et al. Investigations into
the stabilisation of drugs by sugar glasses: II. Delivery of an inulin-stabilised alkaline phosphatase in the intestinal lumen via the oral route. Int J Pharm 2003;257(1-2):273-81.
[41] Bivas-Benita M, Zwier R, Junginger HE, Borchard G. Non-invasive pulmonary aerosol delivery in mice by the endotracheal route. Eur J Pharm Biopharm 2005;61(3):214-8.
[42] Bungener L, Huckriede A, de Mare A, de Vries-Idema J, Wilschut J, Daemen T. Virosome-mediated delivery of protein antigens in vivo: efficient induction of class I MHC-restricted cytotoxic T lympho-cyte activity. Vaccine 2005;23(10):1232-41.
[43] McCutcheon M, Wehner N, Wensky A, Kushner M, Doan S, Hsiao L, et al. A sensitive ELISPOT assay to detect low-frequency human T lymphocytes. J Immunol Methods 1997;210(2):149-66.
[44] Concessio NM, Hickey AJ. Descriptors of irregular particle morphology and powder properties. Adv Drug Deliv Rev 1997;26(1):29-40.
CHAPTER VI
153

[45] Edwards DA, Hanes J, Caponetti G, Hrkach J, Ben-Jebria A, Eskew ML, et al. Large porous particles for pulmonary drug delivery. Science 1997;276(5320):1868-71.
[46] Irngartinger M, Camuglia V, Damm M, Goede J, Frijlink HW. Pulmonary delivery of therapeutic pepti-des via dry powder inhalation: effects of micronisation and manufacturing. Eur J Pharm Biopharm 2004;58(1):7-14.
[47] Maa YF, Nguyen PA, Sweeney T, Shire SJ, Hsu CC. Protein inhalation powders: spray drying vs spray freeze drying. Pharm Res 1999;16(2):249-54.
[48] Zijlstra GS, Hinrichs WL, de Boer AH, Frijlink HW. The role of particle engineering in relation to for-mulation and de-agglomeration principle in the development of a dry powder formulation for inhala-tion of cetrorelix. Eur J Pharm Sci 2004;23(2):139-49.
[49] Bot A, Bot S, Bona CA. Protective role of gamma interferon during the recall response to influenza virus. J Virol 1998;72(8):6637-45.
[50] Swain SL, Agrewala JN, Brown DM, Jelley-Gibbs DM, Golech S, Huston G, et al. CD4+ T-cell memory: generation and multi-faceted roles for CD4+ T cells in protective immunity to influenza. Immunol Rev 2006;211:8-22.
[51] Committee for Proprietary Medicinal Products (CPMP).Note for Guidance on Harmonisation of Requirements for Influenza Vaccines (CPMP/BWP/214/96)
[52] Clements ML, Betts RF, Tierney EL, Murphy BR. Serum and nasal wash antibodies associated with resistance to experimental challenge with influenza A wild-type virus. J Clin Microbiol 1986;24(1):157-60.
[53] Renegar KB, Small PA, Jr. Passive transfer of local immunity to influenza virus infection by IgA anti-body. J Immunol 1991;146(6):1972-8.
[54] Mbawuike IN, Pacheco S, Acuna CL, Switzer KC, Zhang Y, Harriman GR. Mucosal immunity to influ-enza without IgA: an IgA knockout mouse model. J Immunol 1999;162(5):2530-7.
[55] Zhang Y, Pacheco S, Acuna CL, Switzer KC, Wang Y, Gilmore X, et al. Immunoglobulin A-deficient mice exhibit altered T helper 1-type immune responses but retain mucosal immunity to influenza virus. Immunology 2002;105(3):286-94.
[56] McDermott MR, Bienenstock J. Evidence for a common mucosal immunologic system. I. Migration of B immunoblasts into intestinal, respiratory, and genital tissues. J Immunol 1979;122(5):1892-8.
[57] Neutra MR, Pringault E, Kraehenbuhl JP. Antigen sampling across epithelial barriers and induction of mucosal immune responses. Annu Rev Immunol 1996;14:275-300.
[58] Smith DJ, Bot S, Dellamary L, Bot A. Evaluation of novel aerosol formulations designed for mucosal vaccination against influenza virus. Vaccine 2003;21(21-22):2805-12.
[59] Reynolds HY. Respiratory host defenses--surface immunity. Immunobiology 1994;191(4-5):402-12.[60] Geiser M. Morphological aspects of particle uptake by lung phagocytes. Microsc Res Tech
2002;57(6):512-22.[61] Brain JD. Lung macrophages: how many kinds are there? What do they do? Am Rev Respir Dis
1988;137(3):507-9.[62] Ferro TJ, Kern JA, Elias JA, Kamoun M, Daniele RP, Rossman MD. Alveolar macrophages, blood mono-
cytes, and density-fractionated alveolar macrophages differ in their ability to promote lymphocyte pro-liferation to mitogen and antigen. Am Rev Respir Dis 1987;135(3):682-7.
[63] Gong JL, McCarthy KM, Rogers RA, Schneeberger EE. Interstitial lung macrophages interact with den-dritic cells to present antigenic peptides derived from particulate antigens to T cells. Immunology 1994;81(3):343-51.
[64] de Haan A, Groen G, Prop J, van Rooijen N, Wilschut J. Mucosal immunoadjuvant activity of liposo-mes: role of alveolar macrophages. Immunology 1996;89(4):488-93.
[65] Thepen T, Hoeben K, Breve J, Kraal G. Alveolar macrophages down-regulate local pulmonary immune responses against intratracheally administered T-cell-dependent, but not T-cell-independent antigens. Immunology 1992;76(1):60-4.
[66] Hinrichs WL, Prinsen MG, Frijlink HW. Inulin glasses for the stabilization of therapeutic proteins. Int J Pharm 2001;215(1-2):163-74.
154

Chapter VII
General Discussion

Stabilized influenza vaccines
Stabilization of different vaccine-ssubtypesIn this thesis it is shown that influenza vaccines (subunit and virosomes) incorporated insugar glass can be stored at room temperature which reduces the dependency on the coldchain. It was found that the stability of the dried vaccine is dependent on the for-mulation (composition), the sugar in which the vaccine is incorporated and of course storage conditions. The extent of stabilization seems further to be related to the type ofvaccine. Especially the differences in complexity between WIV, virosomes and subunitor split vaccines deserve attention. The sugar-glass stabilization of subunit preparationswas highly successful even at elevated temperatures (40°C). In contrast, virosomes (andalso WIV [1]) incorporated in a glassy sugar matrix showed to be less stable at compara-ble storage conditions. This difference may be explained by the "intact" lipid vesicularstructure of virosomes (and WIV). Lipid vesicles are known to be more difficult to sta-bilize than proteins. An improved stability of vesicular lipid bilayer systems (like lipo-somes, red blood cells, or other mammalian cells) was found when a stabilizer (like trehalose) was present at both sides of the lipid bilayer instead of the outside only [2-6].Lack of a stabilizer at the inside of the vesicle can result in (partial) reorganization of thelipids and subsequent phase transitions. In the studies performed in this thesis on sta-bilization of virosomes, the vaccine dispersion was mixed with a sugar solution beforelyophilization. Consequently, the inside of these vaccine particles lacks the presence of sugar. As a result virosomes (and WIV) incorporated in sugar glasses are less success-fully stabilized than subunit vaccines. Stabilization of lyophilized virosomes may beimproved by incorporating sugar inside the virosome before formulation and lyophiliza-tion. Moreover, also stabilization of WIV vaccines may be improved by loading WIV-particles with sugar from an "extracellular" medium through a combination of osmoticimbalance and phospholipid phase transitions as has been shown successful for stabiliza-tion of red blood cells [6]. In addition this sugar uptake by the WIV-particles may befacilitated by increasing the membrane fluidity with compound such as benzyl alcoholor other weak surfactants [6]. In conclusion, stabilization of each type of vaccine shouldbe optimized individually.
In this thesis, subunit vaccine was brought in the dry state by both freeze drying andspray-freeze drying, while virosomes were dried by freeze drying only. Also other dry-ing methods could have been applied for the incorporation of vaccine compounds insugar glass matrices. However, additional research is needed regarding the use of thesemethods and in particular the comparison between the different drying methods for theproduction of stabilized influenza vaccines. In addition, each vaccine type may possessits own intrinsic sensitivity to different process stresses and have its own limitations. E.g. due to their particulate nature, virosomes and WIV may possess a higher sensitivityto shear stresses during atomization applied by spray (freeze) drying than subunit andsplit vaccines being proteinous vaccines. As a result the incorporation of a vaccine com-
156

pound in sugar glasses should be optimized by both formulation and drying process considerations.
Methods to examine vaccine stability by determining the antigen structureOne of the methods that has been used in this thesis to reveal integrity of HA in vac-cine formulations after the formulation process and/or storage is the single radial immu-nodiffusion (SRID) assay. For several decades, this standardized method has been used todetermine the antigen content (HA potency) of all human inactivated influenza vac-cines, as recommended by the European Pharmacopoeia and the WHO [7, 8]. SRID isbased on the diffusion of viral antigen in an agarose gel containing specific antibodies tothe antigen measured.
However, for the determination of antigen integrity a SRID assay may not be suffi-cient. The SRID assay is based on haemagglutinin binding and does not address the anti-gen's stability. In case no structural alterations in HA are detected by SRID this does notguarantee a complete absence of conformational changes, since the method may notreveal every structural change.
Combining SRID analyses with additional analytical techniques, like fluorescencespectroscopy, circular dichroism spectroscopy, surface plasmon resonance transfer (SPR),asymmetric flow field flow fractionation (AFFF), reversed-phase high performanceliquid chromatography (RP-HPLC), electron microscopy and the use of proteolyticassays may give more complete information on structural state of HA in the product.However, even with the combination of all techniques it is hard (impossible) to detectevery possible structural change in large and complex proteins like HA.
Furthermore, it should be realized that until today only limited information is avai-lable on the effects of structural changes of the vaccine compounds on the final immuneresponse in humans. The effects of low pH, detergents or process stresses on the immu-nogenicity of the vaccine may not be simply represented by changes in HA potencydetermined in vitro. For this purpose well designed studies using appropriate animalmodels or even human volunteers should be conducted. In addition the critical end-points that are to be defined should be based on the desired or expected type of immuneresponse in relation to the presentational form of HA, e.g. subunit, virosomal and WIV.In a study of Babiuk et al it was shown that changes in the production process of influ-enza split vaccine can have remarkable effects on the immunogenicity of the vaccine [9],while no changes in HA potency may be revealed by SRID. Due to a new viral splittingprocedure the amount of un-split virions and aggregates, in the split vaccine were inc-reased. This led to a change in the immune response to a greater Th2 cytokine patternwith potential implications for vaccine safety and efficacy. This shows that solely deter-mining the HA content will not suffice as determinant for the immunity of the vaccine.In addition, criteria should be formulated to address the relevant physical and functionalproperties for each vaccine type. For example WIV and virosomal vaccines might haveimmunological advantages over subunit vaccine related to their particulate form and/orability to deliver material to the cytoplasm of APCs. Therefore appropriate criteriashould be formulated for these functional characteristics like fusion activity. Strikingly,
GENERAL DISCUSSION
157

there is no criterion for vesicular size mentioned in the European Pharmacopoeia forWIV vaccines, while on the other hand the size of virosomes should be between 100 and500 nm [7].
Development of mucosal dosage forms
In this thesis, it has been shown that administration strategies of influenza vaccines viathe oral or pulmonary route induce/promote mucosal and systemic humoral as well ascell-mediated immune responses. In contrast to parenteral vaccination, these mucosalvaccinations induce secretory antibody responses in the respiratory tract. As a resultthese mucosal vaccinations might give protection against influenza infection at the portof entry not only against homologous strains, but also against drifted, heterologousstrains.
Towards an oral influenza vaccineDelivery to the gastrointestinal tract In the development of an oral influenza vaccine various pre-clinical studies addressedthe issue of the use of adjuvants. However, in this thesis we paid attention to the deli-very site of the vaccine in the gastro-intestinal tract. In the murine study presented inChapter V it was found that the delivery site within the gastro-intestinal tract had se-veral effects on the immune response for a subunit vaccine. Moreover, co-administra-tion of the adjuvant LT enhanced the immune responses in different ways depending onthe site of delivery. It was concluded that the right combination of a (strong) mucosaladjuvant and antigen delivery site within the gastro-intestinal tract might result in effec-tive vaccination via the oral route.
Although no clinical trials have been performed to reveal the optimal delivery sitefor vaccine in the gastro-intestinal tract, a number of clinical studies with oral influenzavaccines for delivery in the intestinal tract have been performed. In most of the clinicalstudies relative high doses of antigen were applied to overcome the limited absorption byM cells, all these immunizations resulted in IgG responses below detection level [10-12].Some authors explained the lack of IgG responses with oral tolerance caused by repeatedingestion of significant doses of antigen. It was concluded that the dose of antigen needsto be optimized to ensure a maximal and safe immune response [11]. A critical noteregarding this study is that the antigen integrity in the formulations used (inactivatedvaccines air-dried with D-xylose) was not determined. It should, however, be emphasi-zed that a vaccine powder with known antigenicity is required to perform adequate doseresponse study. For this purpose a well designed influenza vaccine powder may guaran-tee the antigenicity of the vaccine.
On the other hand, most clinical studies demonstrated a significant increase in IgAantibodies in both saliva and nasal lavage fluids. It is unknown whether these IgA anti-bodies alone could provide sufficient protection against influenza infection in humans.Sufficient protection by mucosal IgA induced by oral immunization might be achieved.
158

However, to our knowledge, no clinical study has been designed so far to reveal the levelof protection provided by the orally-induced local IgA immune response in humans.
Since no clinical trials have been performed to reveal the optimal delivery site forvaccine in the gastro-intestinal tract, it would be highly interesting to perform clinicalstudies and/or studies with primates in which specific areas of the gastro-intestinal tractare targeted. For these studies, state of the art technologies, like formulation technolo-gies (tablets with dried vaccine) and special coatings, should be used to enable targetingof specific GI-sites [13-16].Influenza vaccination via the GI-tract may be improved by specific colon delivery of vac-cine that is normally degraded and/or poorly immunogenic in the upper part of the GI-tract. This might be envisaged by a pH-controlled pulsatile delivery system (system com-prising a pH-sensitive coating material wherein a swellable agent is embedded) as deve-loped by Schellekens et al. that enables pulsatile colon-specific release of the antigen [15,16].
Delivery to the oral cavityAlthough the oral cavity was not evaluated in this thesis for vaccination against influ-enza, the oral cavity could be a promising site for induction of immunity. In contrast toGI-administration, formulations for delivery to the oral cavity do not need protection ofthe antigen (e.g. enteric coating) against the harsh environment of the stomach.Moreover, in the oral-cavity many enzymes found in the GI-tract that may degrade theantigen are absent. The approach to use the oral cavity as an access to the immune sys-tem is best explored by research done on allergen specific immunotherapy, especially bysublingual application of allergens. Although the mechanism by which sublingual immu-notherapy exerts its effects remains unclear, this therapy recently resulted in the launchof a tablet-based vaccine against grass pollen allergy (GRAZAX®, ALK-Abelló,Danmark). GRAZAX® is a sublingual tablet of a lyophilisate containing standardizedallergen extract of grass pollen, gelatin (fish source), mannitol and sodium hydroxide.
Recently, oral spray immunization with WIV was evaluated in a phase I/II trial [17].Although no significant increase in salivary IgA antibodies was found, the oral sprayimmunization induced serum HI antibodies in 75% of the volunteers already after 2doses. A critical note with respect to this study is that the authors did not ensure that noaerosol particles were inhaled. Therefore, it cannot be excluded that a part of the foundimmune response is caused by pulmonary deposited WIV. However, delivery of an influ-enza vaccine in the oral cavity, e.g. by sublingual or buccal tablets or patches, might bean interesting alternative route for vaccine delivery. For this approach a lyophilizedinfluenza vaccine powder as presented in Chapters III and IV may be adequate startingpoints. In addition, delivery in the oral cavity might offer the opportunity to use muco-sal adjuvants that are not suitable for nasal and/or pulmonary use.
159
GENERAL DISCUSSION

Towards a pulmonary influenza vaccineFrom the pre-clinical study on pulmonary vaccination with an influenza subunit vac-cine powder in mice as described in this thesis (Chapter VI) it was concluded that pul-monary delivery of influenza vaccines is a promising strategy for vaccination againstinfluenza.
Already, in the 1960s and 1970s pulmonary delivery has been investigated in a num-ber of clinical studies. However, in many of these studies efficient targeting to the lungsis unlikely. First of all atomizers generating aerosols with droplet sizes between 1 and 100?m were used, which is to large for deep lung penetration (since this requires 1 to 5 μmdroplets). Secondly a number of studies were performed with pressurized metered doseinhalers (pMDIs). In these studies it is unclear whether the vaccine was compatible withthe propellant. Moreover, the old pMDIs gave lung deposition below 15% [18, 19]. However, some general trends can be seen:
- Aerosol immunization with inactivated influenza vaccine prevented influenza related illness in humans [20-22].
- Aerosol immunization in humans stimulated higher levels of respiratory secreted antibodies than subcutaneous immunization [23, 24].
- The respiratory antibodies were more cross-reactive with heterologous viruses [25].
- Inhaled inactivated vaccine can provide protection against a heterologousVariant [26].
- These last two results confirm that cross-reactivity of the respiratory antibodieselicited by inhaled influenza can be achieved.
More recently, pre-clinical studies have shown that respiratory delivered influenzavaccine generated systemic, mucosal (local) humoral and cellular virus-specific immuneresponses that increased with increasing the depth of vaccine deposition within the res-piratory tract [27, 28]. The higher efficacy of deep-lung vaccination has been explainedby the prolonged residence time of the influenza vaccine within the lungs [28].Especially, deposition in the alveolar region which lacks mucociliair clearance, is consi-dered to result in an increased residence time of the vaccine [28]. In addition, immuni-zation via the deep-lung instead of i.m. injection resulted in Th1 skewing of the cellularimmune response [28].
These pre-clinical studies suggest that deep-lung deposition may be a critical para-meter for immunogenic efficacy of influenza vaccines in humans. Since current inhala-tion technologies provide a higher deep-lung deposition, it may be expected that pulmo-nary vaccination will be more effective than those from 1960s and 1970s. Therefore itwould be highly interesting to perform clinical trials using modern inhalation devicesand formulations. Especially the use of so-called "soft mist inhalers" would be interestingsince they apply the concept of low-velocity aerosol in combination with the genera-tion of mono-disperse aerosols [19, 29]. In contrast to pressurized metered dose inhalers,soft mist inhalers can directly aerosolize the aqueous vaccine formulation without theuse of propellants that may affect the integrity of the influenza vaccine [29]. However,
160

next to the clinical trials in humans that are needed to prove efficacy, it will first have tobe investigated whether the vaccine is resistant against the aerosolization.
Dry-powder inhalers (DPIs) are potentially the most attractive inhalation systemsfor the delivery of vaccines to the lungs. Advantages such as the stability of the vaccinein the solid state, the high lung depositions that can be generated with simple and cheapdevices, and their robustness, make these systems in principle superior to the liquid inha-lation systems [29-31]. Moreover, they can be disposable systems [32].
The first development studies on formulations suitable for dry-powder inhalation ofinfluenza vaccine are described and discussed in Chapter II. In the pre-clinical study described in Chapter VI, pulmonary vaccination with a new influenza subunit vaccinepowder was evaluated in mice. Vaccine powder was produced by spray-freeze drying(SFD) using inulin as stabilizer. Pulmonary vaccination of mice with the vaccine powderinduced mucosal, systemic humoral as well as cell-mediated immune responses. Theseresponses were superior to those elicited by conventional i.m. vaccination or pulmonaryvaccination with a liquid aerosolized subunit vaccine. The superiority of the SFD vac-cine powder compared to the aerosolized liquid subunit vaccine was ascribed to:
- the deposition in the lower respiratory tract of the powder, resulting in increa-sed residence time in the lungs,
- the increased viscosity at the site of deposition caused by inulin, which increa-ses the residence time.
To confirm these hypothesis, further studies should be designed that address de-position, tracking and immunity, e.g. with labeled particles, with labeled HA and la-beled carbohydrates. In this way the relation between deposition, residence time andimmunological outcome might become clear(er).
After the lessons learned from studies on inhalable measles vaccines, like the use ofless hygroscopic formulations and more purified vaccine compounds in the formulation[33] and addressing the questions related to toxicity, clinical trials should be performedusing recently designed inhalers and dry-powder formulations. These inhalers and for-mulations have to guarantee high and reproducible deposition of the vaccine in the lungarea of interest. Finally, these studies might prove the efficacy and safety of pulmonarydry-powder vaccination against influenza in humans.
Nasal influenza vaccine considerationsAlthough not investigated in this study, vaccination via the intranasal route (i.n.) is theonly mucosal route that has been successfully applied in the form of the live-cold adap-ted trivalent influenza intranasal vaccine (Flumist™). So far a number of (hypothetical)safety concerns exist regarding the live-cold adapted vaccine, e.g. the safety in immuno-compromised patients which has not been established, the attenuated vaccine strain mayreassort with other influenza viruses and the possible risk of vaccine induced central nervous system complications [34].
The use of inactivated intranasal vaccines may be an alternative which combines theconvenience of intranasal administration and the safety of inactivated intramuscular vac-
161
GENERAL DISCUSSION

cine [34]. However, a disadvantage of intranasally delivered inactivated influenza vac-cines is that they are poorly immunogenic without the use of special delivery systemsand/or mucosal adjuvants [35]. Therefore, in recent preclinical and clinical studies in-vestigators examined the suitability of a number of candidate vaccines which are ad-juvanted or specially formulated (as reviewed in Chapter II).
Powder formulations may have additional advantages for nasal delivery of vaccines.In addition to the fact that powders offer the advantages of improved vaccine stability[36, 37], certain powder formulations also have demonstrated increased residence timein the nasal cavity compared with liquid [38, 39], which may translate into higher "bioavailability" and immune responses. A few studies report the development of dry-powder vaccine formulations for intranasal delivery [1, 40-43]. However, the intranasaldelivery of these dry-powder formulations prepared by lyophilization could only elicit asufficient immune response when they were co-formulated with a mucoadhesive polymer or mucosal adjuvant.
The effectiveness of the SFD vaccine powder upon pulmonary delivery, as describedin Chapter VI, was ascribed to site specific deposition; increase in residence time in thelungs due to lower respiratory tract deposition and an inulin-induced local viscosity in-crease at the site of deposition. These characteristics, if proven, may facilitate the effec-tiveness of such a vaccine powder also upon nasal delivery.
Due to nasal anatomy and physiology, with a non-ciliated area in the anterior partof the nasal cavity and a ciliated region in the more posterior part of the nose, the site ofdeposition is of importance for the nasal mucociliary clearance [44]. The site of deposi-tion depends on several parameters which are related to the delivery device and formu-lation, such as velocity of the delivered particles and particle size of the formulation [45].As a result the vaccine powder should be reformulated to obtain deposition of the vac-cine powder at the desired site in the nose. In conclusion, the required (deep) nose depo-sition may be obtained with the right combination of formulation and delivery device,providing relative long vaccine retention in the nose and an effective immune response.
Correlates of protectionDue to current criteria for vaccine immunogenicity, most new developments on influ-enza vaccines aim only at the induction of an adequate level of virus-neutralizing anti-bodies in the serum, indicated by an HI titer higher than 40 in humans [46]. However,other immune responses may be just as or even more valuable (protective) as the induc-tion of HI-titers. For example, secretory antibodies in the respiratory tract induced bymucosal immunization have the potency to provide protection against influenza virus atthe port of entry [47, 48]. In addition, from human studies it was concluded that cel-lular immunity, especially CTL activity, is important for recovery from influenza infec-tion even in the presence of protective antibodies [49, 50].
Ideally, criteria for vaccine efficacy should not be based on the induction of ade-quate virus-neutralizing antibodies in the serum only, but should be based on the levelof protection that is desired. The desired protection may depend upon the route of ad-ministration as well as on the formulation of the vaccine. The induction of mucosal virus
162

neutralizing antibodies (IgA) may for example be a better parameter for protection afternasally administered vaccines, whereas for i.m. injected subunit vaccine clearly the levelof neutralizing antibodies in the serum is more relevant. As a result, parameters for pro-tection, e.g. serum antibodies, mucosal antibodies as well as cellular immunity, should bedetermined (and re-evaluated) for each type of vaccine and route of administration byadequate (post licensing) human studies.
Inulins as adjuvants for needle-ffree inactivated influenza vaccines Today's vaccines may still need adjuvants to improve the immune response elicited bycurrent inactivated vaccines, to facilitate dose sparing vaccination and/or to improvemucosal immunization strategies. Although, investigation of various adjuvants and adju-vanting systems showed in some cases promising results thus far, the search for new,improved and safe adjuvants continues.
In Chapter II, the hypothesis was drawn that inulin may have been acting as anadjuvant in the pre-clinical study on pulmonary delivery of an inulin-stabilized subunitvaccine in mice. The adjuvant activity of inulin was mentioned in several papers as revie-wed in [51]. In these publications inulin gamma-crystals were explicitly assigned to haveadjuvant activity. However, in the system described in Chapter VI amorphous inulinparticles were used, that are believed to dissolve rapidly after deposition at the mucus.However, part of the inulin may be processed by the immune system before dissolutionin the mucosal fluid or after dissolution and possible subsequent crystallization to thegamma-crystal.
Inulin derived adjuvant, based on gamma-inulin, efficiently promotes both Th1 andTh2 immune responses against a variety of antigens (e.g. diphtheria toxoid, whole cellmeningococci, hepatitis B surface antigen, malarial merozoite surface antigen and pro-tein of human papilloma virus) [51]. Pure inulin gamma-crystals given with virus immu-nogen (A/JAP, H2N2) have been shown to induce heterotypic protection against lethalchallenge with live influenza virus (A/WSN, H1N1).[52] BALB/c mice were injectedwith (live/gamma-irradiated) influenza virus alone or combined with gamma-inulin.Whereas, only 3.8 % of the mice primed with virus survived, 50% of the mice primedwith gamma-inulin/virus mixture survived. This improved protection was assigned tocytotoxic T-cell mediated immunity [52].Consequently, it would be interesting to investigate whether amorphous inulin has adju-vant activity or the adjuvant activity should solely be assigned to the gamma-crystal-form of inulin. Finally, dried vaccine formulations based on inulin might be investigatedfor use in epidermal powder, nasal, oral and oro-pharyngeal immunization.
163
GENERAL DISCUSSION

Concluding perspective
In this thesis formulation and delivery strategies are presented, that finally may result ina stabilized influenza vaccine that is administered via a non-parenteral route.
Incorporation of vaccines in amorphous glassy sugar matrices has been shown tohave the potential to solve the problems associated with the cold chain requirement ofliquid vaccines. However, many aspects of the stabilization of influenza vaccines have tobe further investigated. Not only vaccine type dependent formulation and processdesign, but also methods to establish the critical (formulation, process and immunogenic)parameters for vaccine stability should be addressed.
Solid vaccine powders are interesting starting materials for the development of non-parenteral dosage forms for influenza vaccines. Various strategies, such as oral, nasal orpulmonary delivery may evolve in successful non-parenteral dosage forms for influenzavaccines. Critical re-evaluation of old clinical studies, the use of new (up-to-date) deli-very technologies, site specific vaccine delivery, introduction of new criteria for vaccineimmunogenicity and the design of more effective vaccine compounds together with newadjuvants may facilitate the development of such needle-free influenza vaccines.
In this thesis the most promising strategy presented is the development of a pulmo-nary influenza vaccine. The pulmonary delivery of a subunit vaccine powder (inulinbased) in mice was shown to be highly immunogenic. In addition, compared to the con-ventional i.m. immunization pulmonary immunization induced not only higher serumantibodies, but was also capable to induce secretory antibodies in the respiratory tract.The induction of these secretory antibodies, that exhibit cross-reactivity against anti-genically distinct viruses, might offer broader protection against drifted, heterologousstrains.
Finally, cellular-immunity was shown to be increased by this strategy of immuni-zation. Therefore, future developments should aim at pulmonary immunization withinfluenza vaccines. Clinical studies should address the immunogenicity of pulmonarydelivered vaccine aerosols with modern inhalation devices such as "soft mist inhalers".Or even more interesting, clinical studies should be performed to address the immuno-genicity of pulmonary delivered vaccine powders with modern dry-powder inhalers.Such a vaccination strategy, by dry-powder inhalation, may lead not only to influenzavaccinations that provide broader protection against new emerging influenza viruses, butalso would facilitate vaccination of people in "hard to reach" areas with a temperature-resistant self-administerable and needle-free influenza vaccine.
164

165
[1] Huang J, Garmise RJ, Crowder TM, Mar K, Hwang CR, Hickey AJ, et al. A novel dry powder influenza vaccine and intranasal delivery technology: induction of systemic and mucosal immune responses in rats. Vaccine 2004;23(6):794-801.
[2] Crowe LM, Crowe JH, Rudolph A, Womersley C, Appel L. Preservation of freeze-dried liposomes by trehalose. Arch Biochem Biophys 1985;242(1):240-7.
[3] Crowe JH, Crowe LM, Oliver AE, Tsvetkova N, Wolkers W, Tablin F. The trehalose myth revisited: introduction to a symposium on stabilization of cells in the dry state. Cryobiology 2001;43(2):89-105.
[4] Koster KL, Lei YP, Anderson M, Martin S, Bryant G. Effects of vitrified and nonvitrified sugars on phosphatidylcholine fluid-to-gel phase transitions. Biophys J 2000;78(4):1932-46.
[5] Vereyken IJ, Chupin V, Hoekstra FA, Smeekens SC, de Kruijff B. The effect of fructan on membrane lipid organization and dynamics in the dry state. Biophys J 2003;84(6):3759-66.
[6] Satpathy GR, Torok Z, Bali R, Dwyre DM, Little E, Walker NJ, et al. Loading red blood cells with trehalose: a step towards biostabilization. Cryobiology 2004;49(2):123-36.
[7] Influenza vaccine (surface antigen, inactivated) In: European Pharmacopoeia, 5th edition. Strasbourg, Council of Europe, 2004: 673-74.
[8] WHO Department of Immunization Va, Biologicals. Temperature sensitivity of vaccines. 2006.[9] Babiuk S, Skowronski DM, De Serres G, HayGlass K, Brunham RC, Babiuk L. Aggregate content in-
fluences the Th1/Th2 immune response to influenza vaccine: evidence from a mouse model. J Med Virol 2004;72(1):138-42.
[10] Moldoveanu Z, Clements ML, Prince SJ, Murphy BR, Mestecky J. Human immune responses to influ-enza virus vaccines administered by systemic or mucosal routes. Vaccine 1995;13(11):1006-12.
[11] Avtushenko SS, Sorokin EM, Zoschenkova NY, Zacharova NG, Naichin AN. Clinical and immunologi-cal characteristics of the emulsion form of inactivated influenza vaccine delivered by oral immuniza-tion. J Biotechnol 1996;44(1-3):21-8.
[12] Lazzell V, Waldman RH, Rose C, Khakoo R, Jacknowitz A, Howard S. Immunization against influenza in humans using an oral enteric-coated killed virus vaccine. J Biol Stand 1984;12(3):315-21.
[13] Maroni A, Zema L, Cerea M, Sangalli ME. Oral pulsatile drug delivery systems. Expert Opin Drug Deliv 2005;2(5):855-71.
[14] Gazzaniga A, Maroni A, Sangalli ME, Zema L. Time-controlled oral delivery systems for colon tar-geting. Expert Opin Drug Deliv 2006;3(5):583-97.
[15] Schellekens RC, Frijlink HW, inventors; PH-controlled pulsatile delivery system, methods for prepara-tion and use thereof. patent WO 2007/013794 A1. 2007.
[16] Schellekens RC, Stuurman FE, van der Weert FH, Kosterink JG, Frijlink HW. A novel dissolution method relevant to intestinal release behaviour and its application in the evaluation of modified release mesalazine products. Eur J Pharm Sci 2007;30(1):15-20.
[17] Bakke H, Samdal HH, Holst J, Oftung F, Haugen IL, Kristoffersen AC, et al. Oral spray immunization may be an alternative to intranasal vaccine delivery to induce systemic antibodies but not nasal muco-sal or cellular immunity. Scand J Immunol 2006;63(3):223-31.
[18] Newman SP, Pavia D, Moren F, Sheahan NF, Clarke SW. Deposition of pressurised aerosols in the human respiratory tract. Thorax 1981;36(1):52-5.
[19] Dalby R, Spallek M, Voshaar T. A review of the development of Respimat Soft Mist Inhaler. Int J Pharm 2004;283(1-2):1-9.
[20] Waldman RH, Mann JJ, Small PA, Jr. Immunization against influenza. Prevention of illness in man by aerosolized inactivated vaccine. Jama 1969;207(3):520-4.
[21] Waldman RH, Wood SH, Torres EJ, Small PA, Jr. Influenza antibody response following aerosal ad-ministration of inactivated virus. Am J Epidemiol 1970;91(6):574-85.
[22] Haigh W, Howell RW, Meichen FW. A comparative trial of influenza immunization by inhalation and
References
GENERAL DISCUSSION

hypojet methods. Practitioner 1973;211(263):365-70.[23] Mann JJ, Waldman RH, Togo Y, Heiner GG, Dawkins AT, Kasel JA. Antibody response in respiratory
secretions of volunteers given live and dead influenza virus. J Immunol 1968;100(4):726-35.[24] Waldman RH, Jurgensen PF, Olsen GN, Ganguly R, Johnson JE, 3rd. Immune response of the human
respiratory tract. I. Immunoglobulin levels and influenza virus vaccine antibody response. J Immunol 1973;111(1):38-41.
[25] Waldman RH, Wigley FMSPA, Jr. Specificity of respiratory secretion antibody against influenza virus. J Immunol 1970;105(6):1477-83.
[26] Haigh W, Howell RW. The efficacy of the A2/Aichi/68 strain in inhaled influenza immunisation against the A/England/42/72 variant. J Soc Occup Med 1973;23(4):125-7.
[27] Smith DJ, Bot S, Dellamary L, Bot A. Evaluation of novel aerosol formulations designed for mucosal vaccination against influenza virus. Vaccine 2003;21(21-22):2805-12.
[28] Minne A, Louahed J, Mehauden S, Baras B, Renauld JC, Vanbever R. The delivery site of a monovalent influenza vaccine within the respiratory tract impacts on the immune response. Immunology 2007.
[29] Hinrichs WL, Sanders NN, De Smedt SC, Demeester J, Frijlink HW. Inulin is a promising cryo- and lyoprotectant for PEGylated lipoplexes. J Control Release 2005;103(2):465-79.
[30] Frijlink HW, De Boer AH. Dry powder inhalers for pulmonary drug delivery. Expert Opin Drug Deliv 2004;1(1):67-86.
[31] Laube BL. The expanding role of aerosols in systemic drug delivery, gene therapy, and vaccination. Respir Care 2005;50(9):1161-76.
[32] de Boer AH, Hagedoorn P, Westerman EM, Le Brun PP, Heijerman HG, Frijlink HW. Design and in vitro performance testing of multiple air classifier technology in a new disposable inhaler concept (Twincer) for high powder doses. Eur J Pharm Sci 2006;28(3):171-8.
[33] Pulmonary drug delivery. Aulendorf: Editio Cantor Verlag für Medizin und Naturwissenschaften GmbH, 2007.
[34] Beyer WE, Palache AM, de Jong JC, Osterhaus AD. Cold-adapted live influenza vaccine versus in-activated vaccine: systemic vaccine reactions, local and systemic antibody response, and vaccine effi-cacy. A meta-analysis. Vaccine 2002;20(9-10):1340-53.
[35] Atmar RL, Keitel WA, Cate TR, Munoz FM, Ruben F, Couch RB. A dose-response evaluation of in-activated influenza vaccine given intranasally and intramuscularly to healthy young adults. Vaccine 2007.
[36] Giudice EL, Campbell JD. Needle-free vaccine delivery. Adv Drug Deliv Rev 2006;58(1):68-89.[37] Dean HJ. Alternative routes of influenza vaccine delivery. Expert Opin Drug Deliv 2006;3(5):557-61.[38] Soane RJ, Frier M, Perkins AC, Jones NS, Davis SS, Illum L. Evaluation of the clearance characteristics
of bioadhesive systems in humans. Int J Pharm 1999;178(1):55-65.[39] McInnes FJ, Thapa P, Baillie AJ, Welling PG, Watson DG, Gibson I, et al. In vivo evaluation of nico-
tine lyophilised nasal insert in sheep. Int J Pharm 2005;304(1-2):72-82.[40] Sullivan VJ, Mikszta JA, Laurent P, Huang J, Ford B. Noninvasive delivery technologies: respiratory
delivery of vaccines. Expert Opin Drug Deliv 2006;3(1):87-95.[41] Jiang G, Joshi SB, Peek LJ, Brandau DT, Huang J, Ferriter MS, et al. Anthrax vaccine powder formula-
tions for nasal mucosal delivery. J Pharm Sci 2006;95(1):80-96.[42] Huo Z, Sinha R, McNeela EA, Borrow R, Giemza R, Cosgrove C, et al. Induction of protective serum
meningococcal bactericidal and diphtheria-neutralizing antibodies and mucosal immunoglobulin A in volunteers by nasal insufflations of the Neisseria meningitidis serogroup C polysaccharide-CRM197 conjugate vaccine mixed with chitosan. Infect Immun 2005;73(12):8256-65.
[43] Garmise RJ, Mar K, Crowder TM, Hwang CR, Ferriter M, Huang J, et al. Formulation of a dry powder influenza vaccine for nasal delivery. AAPS PharmSciTech 2006;7(1):E19.
[44] Tafaghodi M, Abolghasem Sajadi Tabassi S, Jaafari MR, Zakavi SR, Momen-Nejad M. Evaluation of the clearance characteristics of various microspheres in the human nose by gamma-scintigraphy. Int J Pharm 2004;280(1-2):125-35.
166

167
[45] Vidgren MT, Kublik H. Nasal delivery systems and their effect on deposition and absorption. Adv Drug Deliv Rev 1998;29(1-2):157-77.
[46] (CPMP) TEAftEoMPECfPMP. Note for Guidance on Harmonisation of Requirements for Influenza Vaccines, London; 1997 March 12.
[47] Clements ML, Betts RF, Tierney EL, Murphy BR. Serum and nasal wash antibodies associated with resistance to experimental challenge with influenza A wild-type virus. J Clin Microbiol 1986;24(1):157-60.
[48] Renegar KB, Small PA, Jr. Immunoglobulin A mediation of murine nasal anti-influenza virus immu-nity. J Virol 1991;65(4):2146-8.
[49] Wilschut J, McElhaney JE, Palache AM. Rapid Reference Influenza. 2nd ed. London: Mosby/Elsevier Science, 2006.
[50] McMichael AJ, Gotch FM, Noble GR, Beare PA. Cytotoxic T-cell immunity to influenza. N Engl J Med 1983;309(1):13-7.
[51] Silva DG, Cooper PD, Petrovsky N. Inulin-derived adjuvants efficiently promote both Th1 and Th2 immune responses. Immunol Cell Biol 2004;82(6):611-6.
[52] Cooper PD, Steele EJ. The adjuvanticity of gamma inulin. Immunol Cell Biol 1988;66 ( Pt 5-6):345-52.
GENERAL DISCUSSION

168

Influenza vaccination is the key stone in controlling re-occurring influenza epidemics.The studies described in this thesis evaluated dry-powder formulation of influenza vac-cines and explored administration strategies for the development of needle-free dosageforms of influenza vaccines.
Stabilization of influenza vaccines
The first objective of the studies described in this thesis was to study and developmethods for improved influenza vaccine stability. Current inactivated influenza vaccinesare mostly formulations composed of whole inactivated virus, virosomes, split virus orsubunit antigen, i.e. purified haemagglutinin (HA) and neuraminidase (NA). Today theseliquid vaccines have to be handled under refrigerated conditions (2-8°C), with far-rea-ching consequences like high costs, because of transport and storage issues. Although thelogistics of the vaccines is performed under a cold chain regime, accidental storage at ele-vated temperatures and/or freeze thaw cycles may occur. This may result in deteriorati-on of the vaccine compound. An influenza vaccine that is stable at ambient temperatu-res and not sensitive to freezing stresses would reduce the dependency on cold-chainfacilities and would therefore allow the integration of the vaccine logistics with generaldrug distribution; especially in developing countries this would be highly attractive.Moreover, this would reduce the risk of vaccine losses caused by "off-label" storage.Overall this would result in enormous annual savings. In addition, a stable vaccine for-mulation would facilitate stockpiling of potential vaccines against pandemic viruses,which provides an immediate availability and simple distribution of vaccine in a pande-mic situation. A potentially successful strategy to stabilize biopharmaceuticals, such asproteins, vaccines and gene delivery systems, is to dry them in the presence of sugars. Ifdried properly, the biopharmaceutical is incorporated in a glassy matrix of amorphoussugar and thereby stabilized during subsequent storage. Dry-powder formulations,which are less dependent on a cold chain, of two vaccine types (a subunit and a viroso-mal vaccine) were investigated.
In Chapter III, the design of a stable influenza subunit vaccine in the dry state usinglyophilization as drying method is presented. It was shown that HA in influenza subunitvaccines is susceptible to freezing and drying stresses, especially at low freezing rates.The use of PBS during lyophilization of subunit vaccine resulted in strong pH changes(due to crystallization of sodium or potassium dibasic phosphate during freezing) leadingto conformational changes of HA. The conformational changes of HA during freezingcould be prevented by the use of another buffer, hepes (HBS), that does not crystallizeand consequently does not result in strong pH changes during freezing. Independent ofthe buffer used the use of carbohydrates (trehalose, inulin or dextran) as cryo- and lyop-rotectants prevented or reduced conformational changes of HA. Subunit vaccine lyophi-lized with trehalose and inulin was stable for at least 26 weeks at room temperature(20°C). In contrast, vaccine incorporated in a glassy matrix of dextran 56 kD substantial-ly lost its potency during storage for 26 weeks. At elevated temperatures the subunit vac-
170

cine lyophilized in trehalose was most stable. It was concluded that the use of fast free-zing, hepes buffer and the choice for effective carbohydrates (trehalose or inulin) ascryo- and lyoprotectants enables the preparation of a stable subunit vaccine powder bylyophilization.
In Chapter IV the formulation of influenza virosomes as a stable dry-powder byfreeze-drying (lyophilization) using an amorphous inulin matrix as a stabilizer is presen-ted. In the presence of inulin the structural integrity and fusogenic activity of virosomeswere fully preserved during freeze-drying. For example, the immunologic properties ofthe virosomes, i.e. the HA potency in vitro and the immunogenic potential in vivo, weremaintained during lyophilization in the presence of inulin. In addition, compared tovirosomes dispersed in buffer, inulin-formulated virosomes showed a substantially pro-longed shelf-life and preservation of the HA potency, upon storage. Also the capacity ofvirosomes to mediate cellular delivery of macromolecules (e.g. pDNA) was maintainedduring lyophilization in the presence of inulin and upon subsequent storage. Specifically,when dispersed in buffer, virosomes with encapsulated plasmid DNA lost their transfec-tion activity completely within 6 weeks, whereas their transfection activity was fullypreserved for at least 12 weeks after incorporation in an inulin matrix. It was concludedthat lyophilization in the presence of inulin as a stabilizing agent, considerably prolon-ged the shelf-life of influenza virosomes with and without encapsulated macromolecu-les.
Needle-ffree dosage forms
The second objective of the studies described in this thesis was to study administrationstrategies for the development of needle-free dosage forms of influenza vaccines. Currentinactivated influenza vaccines are generally administered via the intramuscular (i.m.) orsubcutaneous (s.c.) route using needles and syringes. Needle-free delivery, such as muco-sal delivery via the respiratory or gastro-intestinal tract, may provide several potentialadvantages in vaccine delivery, such as eliminated pain at the injection site, easier andfaster vaccine distribution and administration, and reduced costs. In addition, an impor-tant and promising advantage of mucosal vaccination is that it, in contrast to i.m. vacci-nes, may result in a local immune response in the respiratory tract. As a result antibodiesin the respiratory tract might give protection against influenza infection at the port ofentry. In addition, since mucosal IgA responses have been shown to exhibit cross-pro-tective immunity against antigenically distinct viruses, such a mucosal immune respon-se might offer broader protection against drifted, heterologous strains. Unfortunately,despite these potential advantages, until now mucosal vaccination approaches have suf-fered from several limitations or practical problems related to the use of inadequate orold-fashioned delivery technologies, and thus have frequently resulted in inadequateantibody responses or even in a state of immunological tolerance. Therefore, marketedinfluenza vaccines, being in the liquid state, are still mainly administered through injec-tion. However, recent developments in the area of vaccine formulation and delivery
SUMMARY
171

technologies now allow efficient delivery of vaccines to specific sites in the human bodyand therefore provide new opportunities for the use of alternative needle-free dosageforms of influenza vaccines. This thesis addresses some of the issues involved in thisdevelopment.
In Chapter V it was investigated to which part of the gastro-intestinal (GI) tract, theupper part or the lower part, an oral influenza vaccine should be targeted to result in aneffective immune response in mice. Our study demonstrated that without adjuvant sub-stantial systemic but low respiratory mucosal immune responses were induced in miceafter delivery of influenza subunit vaccine to the upper GI-tract (intragastric) or thelower GI-tract (intracolonic). In order to enhance the immune responses of the immuni-zations, E.Coli heat-labile enterotoxin (LT) was added as a model for a strong adjuvant.LT, indeed, enhanced the immune responses of the intragastric and intracolonic immu-nizations significantly. Interestingly, intracolonic administration of vaccine with adju-vant also resulted in enhanced cellular immune responses and the desired Th1-skewingof these responses. Intragastrically administration of the adjuvanted vaccine also increa-sed T-helper responses. However Th1-skewing was absent. The differences in cellularimmunity between the LT-adjuvanted groups were in correspondence with the IgG sub-type profile. While IgG1 secretion was increased by LT in intragastric immunized mice,this increase was not found in intracolonic immunized mice. It was concluded, that theright combination of strong mucosal adjuvant (e.g. LT) and antigen delivery site (e.g. thelower part of the gastro-intestinal tract) might result in effective vaccination via the oralroute.
In Chapter VI pulmonary vaccination with a new influenza subunit vaccine powderwas evaluated. Vaccine powder was produced by spray-freeze drying (SFD) using inulinas stabilizer. The new powder formulation of subunit influenza vaccine described in thischapter, has the potential to be used for vaccination by dry powder inhalation inhumans. First, it was shown that the vaccine antigen, HA, retained its structural andantigenic properties after SFD using inulin as stabilizer. Secondly, the SFD formulationis suitable for vaccine delivery into the lungs by inhalation. The SFD formulation consis-ted of large porous particles having an average aerodynamic diameter of 5.3 μm with abroad size distribution (2-12 μm) facilitating vaccine deposition over the large surfacearea of the lungs. As a result the SFD particles are capable to be deposited throughout theentire lung, including the lower airways, which is supported by the FPF of the SFD for-mulation (38%) measured with cascade impactor analysis.
It was demonstrates that pulmonary administration of the influenza subunit vacci-ne powder, induced strong cell-mediated as well as systemic and mucosal humoralimmune responses in Balb/c mice. These responses were superior to those elicited byconventional i.m. vaccination or pulmonary vaccination with a liquid aerosolized subu-nit vaccine. The superiority of the SFD vaccine powder compared to the aerosolizedliquid subunit vaccine might be ascribed to: the lower respiratory tract deposition of thepowder and increased residence time in the lungs. This increased residence time can bethe result of a lower respiratory tract deposition as well as an increased viscosity at thesite of deposition caused by the inulin.
172

With respect to the cell-mediated immune responses, pulmonary delivery of theSFD formulation resulted in higher numbers of IFNγ- and IL4-producing T-helper cellsthan the conventional i.m. injection of influenza subunit vaccine. Moreover, the pheno-type of these immune responses was more balanced. The relative high contribution ofTh1 cells is important, since they are superior to Th2 cells in providing protection againstviral infection and can provide a certain degree of cross-protective immunity. In conclusion, this study demonstrated that the combination of SFD antigen powder andpulmonary antigen delivery improves the immunogenic potential of (influenza subunit)antigen. Vaccination with an SFD subunit vaccine powder by inhalation might be feasi-ble and could be an alternative to conventional parenteral vaccine administration.
Conclusions
In this thesis formulation and delivery strategies are presented that finally may result ina stabilized influenza vaccine that is administered via a non-parenteral route. Incorporation of vaccines in amorphous glassy sugar matrices has been shown to havethe potential to solve the problems associated with the cold chain requirement of liquidvaccines. These solid vaccine powders are interesting starting materials for the develop-ment of non-parenteral dosage forms for influenza vaccines. Various strategies, such asoral or pulmonary delivery may evolve in successful non-parenteral dosage forms forinfluenza vaccines. Critical re-evaluation of old clinical studies, the use of new (up-to-date) delivery technologies, site specific vaccine delivery together with new adjuvantsmay facilitate the development of such needle-free influenza vaccines. Such vaccination strategies, based on stable influenza vaccine powders, may lead notonly to influenza vaccinations that provide broader protection against new emerginginfluenza viruses, but also would facilitate vaccination of people in "hard to reach" areaswith a temperature-resistant self-administerable and needle-free influenza vaccine.
SUMMARY
173

Summary

174

Nederlandse Samenvatting

"The development of stable influenza vaccine powder formulations for new needle-ffree dosage forms".
Griepvaccinatie is de hoeksteen in de beheersing van de jaarlijks terugkerende griep epidemieën en wordt gezien als één van de belangrijkste wapens tegen een dreigendepandemie. Hedendaagse griepvaccins zijn vloeibare formuleringen gemaakt van (geïnac-tiveerd) influenza virus. Het nadeel van deze vaccins is dat deze getransporteerd en op-geslagen dienen te worden onder de regie van een cold-chain (bij een lage temperatuur:2-8°C) en nog steeds door middel van een injectie worden toegediend.
In dit proefschrift "De ontwikkeling van stabiele influenza vaccin poederformule-ringen voor nieuwe naaldvrije toedieningsvormen" zijn formulerings- en toedienings-strategieën gepresenteerd, welke uiteindelijk kunnen leiden tot een stabiel griepvaccindat niet meer via een injectie hoeft te worden toegediend.
StabilisatieEerst is onderzocht of door middel van vriesdrogen stabielere influenza vaccins kondenworden gemaakt. Door te drogen in de aanwezigheid van suikers, zijn influenza vaccinsingebouwd in een amorfe matrix van suiker (een suikerglas). De integriteit van het vac-cin bleek afhankelijk te zijn van verschillende facetten van het droogproces en de for-mulering. Zo zijn het gebruik van een hoge invriessnelheid, de juiste suiker (trehalose,inuline) en een moeilijk kristalliserende buffer (hepes) noodzakelijk om de werking vanhet vaccin te behouden. Op de juiste wijze gedroogde influenza vaccins bleken tenmin-ste meerdere maanden houdbaar te zijn bij kamertemperatuur.
Naaldvrije toedieningMet behulp van muismodellen zijn vervolgens twee nieuwe strategieën onderzochtwelke mogelijk kunnen leiden tot een naaldvrij griepvaccin:
- In het kader van de ontwikkeling van een oraal vaccin is onderzocht of er een verschilin immuunrespons is wanneer het vaccin in de dunne of dikke darm wordt afgegeven.Aangetoond werd dat de dikke darm de meeste potentie heeft. Het bleek echter dat adju-vantia noodzakelijk zijn om een adequate immuunrespons op te wekken.
- In het kader van de ontwikkeling van een pulmonaal vaccin is pulmonale vaccinatiemet vloeibaar vaccin en gesproei-vriesdroogd vaccin onderzocht. In vergelijking tot destandaard griepprik resulteerde alleen de pulmonale vaccinatie met het vaccinpoeder (ziefiguur 1.) in een verhoogd en verbeterd immuunrespons. Naast de systemische respons(het voorkomen van anti-lichamen in het bloed) werd er ook een respons in de slijmlaagvan de luchtwegen gevonden. De antilichamen in de slijmlaag van de luchtwegen zijnbelangrijk omdat zij het virus al zullen aanpakken wanneer het virus het lichaam bin-nenkomt. Besmetting met het griepvirus komt namelijk vaak door inademing van hetvirus.
176

ConclusiesIn dit proefschrift zijn formulerings- en toedieningsstrategieën gepresenteerd welke uit-eindelijk kunnen leiden tot een stabiel griepvaccin dat niet meer via een injecte hoeft teworden toegediend.
Het inbouwen van vaccins in een suikerglas stabiliseert de vaccins zodanig dat zebuiten de koelkast bewaard kunnen worden. Deze vaste vaccinpoeders zijn interessanteuitgangsmaterialen voor de ontwikkeling van naaldvrije toedieningsvormen.Verschillende strategieën, zoals orale of pulmonale toediening kunnen zich ontwikkelentot succesvolle innovatieve toedieningsvormen voor influenza vaccins.Vaccinatiestrategieën, gebaseerd op deze producten kunnen niet alleen leiden tot griep-vaccins die een betere bescherming bieden tegen (nieuwe) influenza virussen, maarbovendien kunnen zij de vaccinatie bevorderen in moeilijk bereikbare gebieden.Vervoer en opslag onder koude omstandigheden is niet langer nodig en de toedieninghoeft niet meer door medisch geschoold personeel te worden uitgevoerd.
NEDERLANDSE SAMENVATTING
177
Fig. 1 Electronen microscoop foto van gesproei-vriesdroogd influenza vaccin poeder. Vergroting x 5000. Maatstreep komt overeen met 5 μm.

178

Dankwoord

Graag wil ik iedereen bedanken die heeft bijgedragen aan de totstandkoming van ditproefschrift.
Allereerst wil ik mijn promotoren professor dr. H.W. Frijlink (Erik) en professor dr. J.C.Wilschut (Jan) bedanken.Erik, mijn eerste promotor, bedankt voor je enthousiasme, vertrouwen en kritische blik.Jij hebt ervoor gezorgd dat ik mij heb kunnen ontwikkelingen als onderzoeker. Degesprekken tijdens lunches, aan het einde van de dag of onderweg naar werkbezoekenaan Solvay of het RIVM, over onderzoek, universitaire politiek, patent kwesties en meerheb ik zeer gewaardeerd. Ik heb bewondering voor de manier waarop jij mensen kuntenthousiasmeren en tot actie kunt laten komen, dit heeft mij zeker veel geholpen mijnpromotie tot stand te laten komen. Jan, mijn tweede promotor, onze samenwerking is pas laat ontstaan. Jouw input op heteind is van grote waarde. Mede dankzij jou is dit proefschrift een mooi samenhangendgeheel geworden. Ondanks jouw drukke agenda en mijn scherpe "promotieplanning",heb ik het genoegen gehad om van onze korte discussies over de "general discussion" temogen genieten.
Ten tweede wil ik mijn copromotoren dr. W.L.J. Hinrichs (Wouter) en dr. A. Huckriede(Anke) bedanken. Wouter, ik heb van jou veel geleerd over de proefopzet, controle-experimenten en deevaluatie van resultaten. Dit heeft ertoe geleid dat we over het algemeen efficiënt totpubliceerbare resultaten konden komen. Verder heb je me het schrijven van een weten-schappelijk artikel geleerd. Hierbij ben ik je vooral erg dankbaar voor de investering vanjouw tijd (ik heb de weekenden en avonden niet geteld) en de snelheid waarmee ik decorrecties mocht ontvangen. Anke, dank voor het feit dat jouw deur altijd voor mij open stond. Naast het schrijvenvan een "DEC" heb ik enorm veel "virologie" van jou geleerd. Dit heeft geresulteerd ineen aantal mooie publicaties. Het is voor mij dan ook een groot genoegen jou "promotiemoeder" te mogen noemen.
De leden van de leescommissie, prof. dr. H.J. Haisma, prof. dr. W. Jiskoot en prof. dr.A.D.M.E. Osterhaus wil ik bedanken voor de tijd die zij namen om dit proefschrift doorte lezen en voor de aangename (doch kritische) reacties die ik heb mogen ontvangen.
Vervolgens wil ik mijn directe collega's van de vakgroep Farmaceutische Technologie enBiofarmacie bedanken. Joke, Jan, Hans v. D., Hans d. W, Yu San, Vinay, Els, Lida,Marinella, Anko, Klaas, Gerad, Doetie, Parinda, Maarten, Gieta, Anneke, Dirk Jan,Bastiaan, Gerrit, Paul en Anne. Naast de gezelligheid mijn oprechte dank voor debehulpzaamheid en belangstelling die jullie stelden in mijn onderzoek. Een aantal (ex-)collega's wil ik in het bijzonder noemen. Gerrit, mijn sparringpartner, alskamergenoot heb ik (gezamenlijk "de jongens van beneden") veel plezier beleefd aan hetsamen bespreken van de vele onderzoeksopzetten, onderzoeksresultaten en schrijfplan-
180

genoegen samen vol spanning de bioluminiscentie van muizenlongen af te wachten.Arjen, we hebben heel wat dates gehad. Bedankt voor alle intubaties; ik zal je grappenen kopjes koffie in OK-III missen.
Als AIO aan de universiteit heb je het voorrecht kennis te kunnen overdragen aan studenten. Ik wil de studenten Communicatie & Oriëntatie "farmaceutische technologieen biofarmacie" van 2005, 2006 en 2007 bedanken voor hun directe en/of indirecte bij-drage aan mijn promotie periode. Jelte, bedankt voor al het werk dat je hebt verzet bij destabilisatie van het subunit vaccin. Het was een groot genoegen samen met jou tot in dekleine uurtjes door te werken. Het is nog steeds jammer dat de ratten het "niet deden".Tjalke, jou wil ik graag bedanken voor je enorme biotechnologische bijdrage. Ik heb jezelfstandigheid en communicatieve eigenschappen enorm gewaardeerd, door jouw toe-doen ging het werk tijdelijk 2,5 x zo snel.
Dan heb ik nog een aantal vrienden te bedanken die altijd voor een goede afleiding eneen hoop lol hebben gezorgd. De "Tech-boys": Micha, Guus, Willem, Monique, Niels,Marleen en Kuik, bedankt voor de kerstdiners en de mooie nachten. De "Apothekers":Akbar, Ronald, Camilo en Annelies, bedankt voor de gezellige etentjes en opfriscursus-sen. Blauw-Wit 3, 4 en nu 5, bedankt voor de weekendjes weg en de ontspanning op hetvoetbalveld. Christiaan, vriend !! Bedankt voor de vele DVD's en heerlijke rosé.
Mijn paranimfen, Micha en Lutea, jullie wil ik bedanken voor de vele etentjes onder hetgenot van een glas wijn. Dank zij jullie gesprekken heb ik veel kunnen ventileren enrelativeren.
Liefste Joske, jou wil ik graag bedanken voor de vele dingen die je mij hebt gebracht: jegeduld, je vertrouwen, het wederzijdse respect, maar natuurlijk ook je kookkunsten, deknuffeltijd en nog veel meer.
Armand, de liefste broer die ik heb, bedankt dat je m'n broer bent, blijft en wilt zijn! Ikben er trots op dat JIJ mijn broertje bent.
Als laatste wil ik mijn ouders bedanken. Pa en ma door jullie steun, goede zorgen enonvoorwaardelijke vertrouwen, in welk opzicht dan ook, heb ik me kunnen ontwik-kelen tot de persoon die ik nu ben. Ik ben jullie hiervoor echt heel dankbaar.
Mocht ik nog mensen zijn vergeten, bij dezen allen mijn dank,
Nederland, november 2007.
Jean-Pierre
182

nen. Bedankt voor de gesprekken, de potjes tennis en de dagelijkse gezelligheid. Ikbeschouw je als mijn "promotie broer". Bastiaan, ik hoor het je nog zeggen: "het levenvan een AIO gaat niet over rozen". Of dit waar is? In ieder geval kunnen we nu de bloe-metjes buiten zetten. Bedankt voor de interessante discussies, de knallende nachten, hetmeesleuren naar spinning en de avonden kolonisten. Ik hoop dat ik nog vaak van dekookkunsten en gastvrijheid van jou en Ellen mag genieten. DJ, bedankt voor het filoso-feren over de vele mogelijk van toepassing zijnde theorieën. Het is jammer dat ik niet demogelijkheid heb gehad de carboxy-inulines in een artikel te verwerken. Nog bedanktvoor de gezellige tripjes naar Gent en Ware; super relaxed. Dear Vinay, thank you foryour contribution to the paper on the pulmonary vaccinations. I have really appreciatedyour extensive literature searches, your flexibility as well as your perseverant persona-lity. Moreover, I enjoyed our visit to the EUFEPS workshop in Munich. I'm looking forward to see your PhD thesis. Hans d. W., bij dezen wil ik jou bedanken voor het deon-stress-momenten en de mogelijkheid om bij Amicitia een balletje te trappen.
Vervolgens wil ik Virosome Biologicals en de afdeling medische micro biologie (sectiemoleculaire virologie) bedanken voor de gastvrijheid. Toon, jou wil ik graag bedankenvoor de hulp en het delen van je enorme kennis. Tijdens ons eerste contact zette jij evensnel een "klein" voorstel (2 A4-tjes) op papier. Dit heeft ertoe geleid dat ik snel geïntro-duceerd werd met het Haemagglutinine (HA) in het griepvaccin en de zogeheten pH-afhankelijke structuur verandering van HA; mede hierdoor hebben we een gedegen arti-kel over de stabilisatie van influenza subunit vaccin met, zoals jij het noemde, "suiker-melk" hebben kunnen publiceren. JØrgen, na het schrijven van een Business plan voor"Science to the market" zijn we in contact gekomen om het complex systeem van viro-somen te stabiliseren. Ondanks de complexiteit, uiteenlopend in de tijdrovendheid vandeze materie, hebben we veel kunnen lachen. Bedankt voor deze prettige samenwerking.Annechien, Arjen, Felix, Laura, Marijke, Paul, Tjarko, Wouter, wil ik graag bedankenvoor het met mij delen van (praktische) kennis ten aanzien van ELISA's, ELISPOT's, HIanalyses, opzuiveren van WIV en/of de productie van LT.
Tijdens mijn AIO-schap heb ik verscheidene samenwerkingsverbanden gekend en heb-ben verscheidene mensen "van buiten" een bijdrage geleverd aan mijn promotieonder-zoek. Solvay Pharmaceuticals wil ik bedanken voor het aanleveren van vaccin, het doenvan SRID metingen door QC influenza en voor de mogelijkheid mijn resultaten in Weespte presenteren. Het Centrum voor Geneesmiddelen en Medische Technologie (BMT) vanhet RIVM onder leiding van Rogier Bos, mijn dank voor de mogelijkheid gezamenlijkonderzoek te doen aan "de biacoor analyse van influenza subunit vaccin" en "de stabili-satie van het mazelen vaccin". Marcel en Marjolijn, ik vond het altijd erg leuk en gezel-lig om bij jullie in Bilthoven langs te komen. Ik hoop dat de opgestarte studies in de toe-komst de gewenste resultaten opleveren. Almer, onder het motto "more to do withTRAIL", bedankt voor de prettige samenwerking en het mij bijbrengen van enkele far-maceutische en biologische aspecten van deze apoptose inducerende ligand. Go vanDam, bedankt voor de enorm energieke samenwerking. Het is steeds weer een groot
181
DANKWOORD

Curriculum Vitae
Jean-Pierre Amorij

Jean-Pierre Amorij
Phone +31-6-47516553E-mail [email protected]
1991-11997High school, Bertrand Russell College, Krommenie.
1997-22002Pharmacy, University of Groningen.Thesis: "The 'ideal' lactose carrier for inhalation" Qualification: Master of Science in Pharmacy.
1998-22002Pharmaceutical technology, University of Groningen. Qualification: Master of Science in Pharmaceutical technology.
2002-22003Professional training for pharmacist, University of Groningen.Qualification: Doctor of Pharmacy.
2003-22007PhD project, Dept. of Pharmaceutical Technology and Biopharmacy, University of Groningen.Thesis: The development of stable influenza vaccine powder formulations for new needle-free dosage forms. Qualification: Doctor of Philosophy in Mathematics and Natural Sciences.
2007-ppresentPostdoc position, Dept. of Pharmaceutical Technology and Biopharmacy, University of Groningen.
184
CURRICULUM VITAE

Notes





















