Past, present, and future technologies for oral delivery of therapeutic proteins
27
Past, Present, and Future Technologies for Oral Delivery of Therapeutic Proteins RAJESH SINGH, 1 SHAILESH SINGH, 1 JAMES W. LILLARD 1,2 1 Department of Microbiology & Immunology, University of Louisville, Louisville, Kentucky 40202 2 Department of Microbiology, Biochemistry & Immunology, Morehouse School of Medicine, Atlanta, Georgia 30310 Received 5 June 2007; accepted 1 August 2007 Published online in Wiley InterScience (www.interscience.wiley.com). DOI 10.1002/jps.21183 ABSTRACT: Biological drugs are usually complex proteins and cannot be orally deliv- ered due to problems related to degradation in the acidic and protease-rich environment of the gastrointestinal (GI) tract. The high molecular weight of these drugs often results in poor absorption into the periphery when administered orally. The most common route of administration for these therapeutic proteins is injection. Most of these proteins have short serum half-lives and need to be administered frequently or in high doses to be effective. So, difficulties in the administration of protein-based drugs provides the motivation for developing drug delivery systems (DDSs) capable of maintaining therapeutic drug levels without side effects as well as traversing the deleterious mucosal environment. Employing a polymer as an entrapment matrix is a common feature among the different types of systems currently being pursued for protein delivery. Protein release from these matrices can occur through various mechanisms, such as diffusion through or erosion of the polymer matrix, and sometimes a combination of both. Encapsulation of proteins in liposomes has also been a widely investigated technology for protein delivery. All of these systems have merit and our worthy of pursuit. ß 2007 Wiley-Liss, Inc. and the American Pharmacists Association J Pharm Sci 97:2497– 2523, 2008 Keywords: nanoparticles; nanospheres; microparticles; microspheres; poly(lactic/ glycolic) acid (PLGA or PLA); polymeric drug delivery systems; oral drug delivery; protein delivery; vaccine delivery; mucosal delivery INTRODUCTION Proteins perform many important physiological and biological processes of the host. Protein ligands bind their receptors that result in actions or changes due to sometime distant signals. Enzymes are involved in many biotransforma- tional reactions or de novo generation or catalysis of a multitude of substrates. Antibodies can actively participate in neutralizing toxins or host factors (e.g., TNF-a). The knowledge and transla- tion of the human genome, has greatly increased the desire to discover new proteins and under- stand their function, usefulness as a therapy as well as to devise drug delivery systems (DDSs) for these molecules. While designing novel DDSs is not essential for successful and efficacious protein drug delivery, effective DDSs would enable these therapeutic proteins to be delivered via mucosal routes to increase efficacy and patient compliance Correspondence to: James W. Lillard (Telephone: 502 852 2174; Fax: 502 852 3842.; E-mail: [email protected]) Journal of Pharmaceutical Sciences, Vol. 97, 2497–2523 (2008) ß 2007 Wiley-Liss, Inc. and the American Pharmacists Association JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008 2497
-
Upload
nagpuruniversity -
Category
Documents
-
view
1 -
download
0
Transcript of Past, present, and future technologies for oral delivery of therapeutic proteins

Past, Present, and Future Technologies for Oral Delivery ofTherapeutic Proteins
RAJESH SINGH,1 SHAILESH SINGH,1 JAMES W. LILLARD1,2
1Department of Microbiology & Immunology, University of Louisville, Louisville, Kentucky 40202
2Department of Microbiology, Biochemistry & Immunology, Morehouse School of Medicine, Atlanta, Georgia 30310
Received 5 June 2007; accepted 1 August 2007
Published online in Wiley InterScience (www.interscience.wiley.com). DOI 10.1002/jps.21183
Corresponde2174; Fax: 502 8
Journal of Pharm
� 2007 Wiley-Liss
ABSTRACT: Biological drugs are usually complex proteins and cannot be orally deliv-ered due to problems related to degradation in the acidic and protease-rich environmentof the gastrointestinal (GI) tract. The high molecular weight of these drugs often resultsin poor absorption into the periphery when administered orally. The most commonroute of administration for these therapeutic proteins is injection. Most of these proteinshave short serum half-lives and need to be administered frequently or in high doses tobe effective. So, difficulties in the administration of protein-based drugs providesthe motivation for developing drug delivery systems (DDSs) capable of maintainingtherapeutic drug levels without side effects as well as traversing the deleterious mucosalenvironment. Employing a polymer as an entrapment matrix is a common featureamong the different types of systems currently being pursued for protein delivery.Protein release from these matrices can occur through various mechanisms, suchas diffusion through or erosion of the polymer matrix, and sometimes a combinationof both. Encapsulation of proteins in liposomes has also been a widely investigatedtechnology for protein delivery. All of these systems have merit and our worthy ofpursuit. � 2007 Wiley-Liss, Inc. and the American Pharmacists Association J Pharm Sci 97:2497–
2523, 2008
Keywords: nanoparticles; nanosphere
s; microparticles; microspheres; poly(lactic/glycolic) acid (PLGA or PLA); polymeric drug delivery systems; oral drug delivery;protein delivery; vaccine delivery; mucosal deliveryINTRODUCTION
Proteins perform many important physiologicaland biological processes of the host. Proteinligands bind their receptors that result in actionsor changes due to sometime distant signals.Enzymes are involved in many biotransforma-
nce to: James W. Lillard (Telephone: 502 85252 3842.; E-mail: [email protected])
aceutical Sciences, Vol. 97, 2497–2523 (2008)
, Inc. and the American Pharmacists Association
JOURNAL O
tional reactions or de novo generation or catalysisof a multitude of substrates. Antibodies canactively participate in neutralizing toxins or hostfactors (e.g., TNF-a). The knowledge and transla-tion of the human genome, has greatly increasedthe desire to discover new proteins and under-stand their function, usefulness as a therapy aswell as to devise drug delivery systems (DDSs) forthese molecules. While designing novel DDSs isnot essential for successful and efficacious proteindrug delivery, effective DDSs would enable thesetherapeutic proteins to be delivered via mucosalroutes to increase efficacy and patient compliance
F PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008 2497
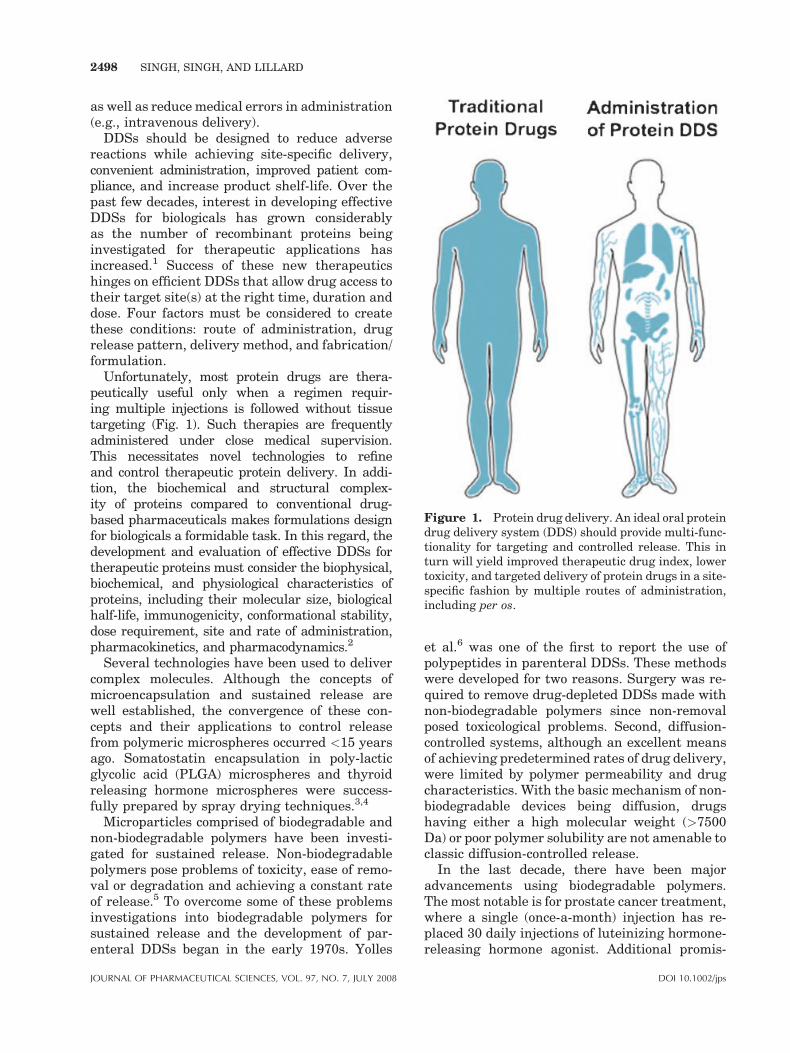
Figure 1. Protein drug delivery. An ideal oral proteindrug delivery system (DDS) should provide multi-func-tionality for targeting and controlled release. This inturn will yield improved therapeutic drug index, lowertoxicity, and targeted delivery of protein drugs in a site-specific fashion by multiple routes of administration,including per os.
2498 SINGH, SINGH, AND LILLARD
as well as reduce medical errors in administration(e.g., intravenous delivery).
DDSs should be designed to reduce adversereactions while achieving site-specific delivery,convenient administration, improved patient com-pliance, and increase product shelf-life. Over thepast few decades, interest in developing effectiveDDSs for biologicals has grown considerablyas the number of recombinant proteins beinginvestigated for therapeutic applications hasincreased.1 Success of these new therapeuticshinges on efficient DDSs that allow drug access totheir target site(s) at the right time, duration anddose. Four factors must be considered to createthese conditions: route of administration, drugrelease pattern, delivery method, and fabrication/formulation.
Unfortunately, most protein drugs are thera-peutically useful only when a regimen requir-ing multiple injections is followed without tissuetargeting (Fig. 1). Such therapies are frequentlyadministered under close medical supervision.This necessitates novel technologies to refineand control therapeutic protein delivery. In addi-tion, the biochemical and structural complex-ity of proteins compared to conventional drug-based pharmaceuticals makes formulations designfor biologicals a formidable task. In this regard, thedevelopment and evaluation of effective DDSs fortherapeutic proteins must consider the biophysical,biochemical, and physiological characteristics ofproteins, including their molecular size, biologicalhalf-life, immunogenicity, conformational stability,dose requirement, site and rate of administration,pharmacokinetics, and pharmacodynamics.2
Several technologies have been used to delivercomplex molecules. Although the concepts ofmicroencapsulation and sustained release arewell established, the convergence of these con-cepts and their applications to control releasefrom polymeric microspheres occurred <15 yearsago. Somatostatin encapsulation in poly-lacticglycolic acid (PLGA) microspheres and thyroidreleasing hormone microspheres were success-fully prepared by spray drying techniques.3,4
Microparticles comprised of biodegradable andnon-biodegradable polymers have been investi-gated for sustained release. Non-biodegradablepolymers pose problems of toxicity, ease of remo-val or degradation and achieving a constant rateof release.5 To overcome some of these problemsinvestigations into biodegradable polymers forsustained release and the development of par-enteral DDSs began in the early 1970s. Yolles
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008
et al.6 was one of the first to report the use ofpolypeptides in parenteral DDSs. These methodswere developed for two reasons. Surgery was re-quired to remove drug-depleted DDSs made withnon-biodegradable polymers since non-removalposed toxicological problems. Second, diffusion-controlled systems, although an excellent meansof achieving predetermined rates of drug delivery,were limited by polymer permeability and drugcharacteristics. With the basic mechanism of non-biodegradable devices being diffusion, drugshaving either a high molecular weight (>7500Da) or poor polymer solubility are not amenable toclassic diffusion-controlled release.
In the last decade, there have been majoradvancements using biodegradable polymers.The most notable is for prostate cancer treatment,where a single (once-a-month) injection has re-placed 30 daily injections of luteinizing hormone-releasing hormone agonist. Additional promis-
DOI 10.1002/jps

ORAL DELIVERY OF BIOLOGICALS 2499
ing treatments for cancer, viral and bacterialinfections, birth control, and AIDS are being in-vestigated.7–12 Indeed, the delivery of geneticallyengineered products as vaccines, for example,soluble recombinant human immunodeficiencyvirus (HIV) proteins, has lead to increased efficacyby entrapment of vaccine antigens in PLGAmicrospheres.13 Therapeutic proteins, e.g., recom-binant erythropoietin,14 have also been encapsu-lated in PLGA microspheres as well as plasmidDNA, antisense oligos, and synthetic double-stranded DNA.15–18
A variety of synthetic and naturally occurringbiodegradable polymers have been studied overthe past 30 years, including polyesters, polyanhy-drides, polyorthoesters, polyphosphazenes, andpseudo amino acids, out of which polyestershave found more widespread use. A number ofother studies have used natural polymers forDDSs that have centered-around proteins (e.g.,collagen, gelatin, albumin) and polysaccharides(e.g., starch, dextran, insulin, cellulose, hyaluro-nic acid). Despite many advantages of polyesters,like PLGA, these polymers have inherent short-comings. By in large, polymers are more hydro-phobic compared with most of the proteins to beencapsulated. Indeed, a lack of protein polymercompatibility leads to stability problems duringstorage or under in vivo release conditions.Hydration and degradation of polyesters areprerequisites for the release of protein duringthe bioerosion phase; however, this can result inan acidic microenvironment (due to formation oflactic and glycolic acids), which might denatureencapsulated proteins. One approach to improveprotein polymer compatibility is by co-encapsulat-ing buffer salts and stabilizers for proteins, whichare thought to modify the internal pH of micro-spheres. Another way might be realized bymodifying the polymer structure itself.
CHALLENGES IN THERAPEUTICPROTEIN DELIVERY
Physiological Obstacles
A major obstacle for the oral absorption ofmacromolecules is their vulnerability to proteo-lytic degradation in the GI tract. Many macro-molecules, especially protein and peptide drugs,are susceptible to rapid degradation by digestiveenzymes. The proteolytic activity is highest in thestomach and duodenum, and is significantly
DOI 10.1002/jps J
reduced in the mouth, pharynx, esophagus, ileum,and colon. Degradation of proteins during theirtransit via the mouth, pharynx and esophagusis minimal. Saliva contains mucus, amylaseand lysozyme and digestion is limited to poly-saccharides hydrolysis by amylase. Indeed, noabsorption of food material occurs in the mouth.The secretions present in the esophagus areentirely mucoid in character and maintain awell-lubricated esophageal lumen. Movement offood through the pharynx and esophagus takesbetween 6 and 10 s. After traveling through theesophagus, food reaches the stomach where it isstored and digested. Based on anatomical andhistological characteristics, the stomach is dividedinto the fundus body and antrum. These regionscoordinate and control the motility function of thestomach.
The digestive juices of the stomach are secretedby gastric (or oxyntic) glands. These glands areresponsible for secretion of hydrochloric acid,pepsinogen and mucus along with other compo-nents. The pyloric (exocrine) glands secrete mucusand some pepsinogen. In this regard, pepsinogenis converted into pepsin by hydrogen chloridesecreted by the oxyntic glands. Pepsin is active atlow pH, but is rapidly inactivated above pH 5.0.Pepsin is most efficient at cleaving bonds betweenaromatic amino acids: phenylalanine, tryptophan,and tyrosine. No absorption of food takes placethrough the stomach. Additional digestion andthe majority of absorption occur in the smallintestine.
The duodenum, jejunum and ileum of the smallintestine have disparate secretion and uptakephysiologies. While small intestinal cells secreteenzymes, e.g., aminopeptidase, this part of the GItract does not significantly contribute to thedigestive process. The exocrine glands in thesmall intestine largely secrete mucus that linesthe inside of the intestinal wall. In fact, proteindigestion in the small intestine mainly occurs dueto pancreatic secretions of amylase and lipase.Pancreatic secretion also contains sodium bicar-bonate that neutralizes the acidity of the contentsemptied by the stomach. The pancreatic proteo-lytic enzyme secretions contain trypsinogen,chymotrypsinogen and procarboxypeptidase. Try-psinogen is converted by an autocatalytic reactionto its active form, trypsin, by an enzyme calledenterokinase present in the wall of the duodenum.Trypsin converts chymotrypsin and procarboxy-peptidases into their active analogues. Theseenzymes act on specific amino acid linkages and
OURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008

2500 SINGH, SINGH, AND LILLARD
convert peptide fragments into small peptides andamino acids.
Size and Charge of Particles
The low oral bioavailability of macromolecules isdue primarily to their large molecular weight andvariable solubility. Bioavailability is essentiallyindependent of molecular mass for drugs <700Daltons (Da); however, bioavailability decreasessharply when molecular mass increases beyondthis threshold. A minimum level of hydrophobicityis also needed for macromolecules to permeate theepithelium and to be transcellularly absorbedthrough passive diffusion. Without this minimumdegree of lipophilicity, no passive absorption cantake place unless it is through the paracellularpathway, which is restricted to relatively smallcompounds (<200 Da). Unfortunately, mostmacromolecules being evaluated as biotherapeu-tics are typically >700 Da and hydrophilic. Thisposes a major obstacle for DDS formulationmethods.
Challenges in Formulation Methods
Out of the host of microencapsulation techniques,the most commonly used methods of micro-encapsulation of proteins are spray drying, multi-ple emulsion, and phase separation methods. Thedifficulties associated with developing effectiveformulations for proteins have been discussed invarious articles.19–23 Despite many attractivefeatures, proteins as therapeutic agents havesome serious limitations. Proteins are relativelylarge molecules with often-complex structures.Unlike low-molecular weight drugs, they possesssecondary, tertiary, and in some cases, quatern-ary structures with labile bonds and side chains ofchemically reactive groups. Disruption of thesestructures or modification of side chains can leadto loss of immunogenicity or activity.
The fragile nature of protein therapeuticsrequires the processes involved in the fabricationof DDSs must not damage the protein, reduce itsbiological activity, or render the protein immuno-genic. For example, aggregated human growthhormone (hGH) has less biological activity than itsnative monomeric form.24 Recombinant therapeu-tic proteins hGH can undergo non-chemicalchanges such as folding and unfolding (denatura-tion), which leads to loss of native structure andenables the protein to interact with its surround-
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008
ings, which might lead to surface absorption oraggregation.25 Aggregation of insulin has beenwell characterized and depends on the unfoldingof insulin.26 Chemical degradation may also occurat many points during formulation and deliveryprocesses. The most common adverse chemicalmodification associated with DDSs is oxidation.24
Deamidation contributes to a reduction in thecatalytic activity of lysozyme and ribonuclease athigh temperatures.27 This reaction is also widelyobserved in therapeutic proteins. Peptide bondhydrolysis results in the loss of activity whenproteins are heated.25 Aggregation of lyophilizedformulations of bovine serum albumin, b-lacto-globulin, and glucose oxidase are attributed todisulphide bond interchange.28
It is important to devise formulation strategiesto preserve protein stability. These approachesinclude adding stabilizing agents and developingfabrication processes for delivery systems thatare benign to proteins. Stabilizing additivesused in the formulation of proteins are diverseand include proteins, sugars, polyols, amino acids,chelating agents, and inorganic salts. Theseadditives can stabilize proteins in solution andalso in frozen and dried states, although not alladditives confer stability under all three condi-tions. For example, carbohydrates in particularhave the ability to stabilize dried proteins.29
Sugars such as trehalose, sucrose, maltose, andglucose are used as collagen, ribonuclease, andovalbumin stabilizers.30 Cyclodextrins have alsobeen used as stabilizing excipients in proteinformulations.31,32 In particular, this dextrinprotects growth hormones from thermal andinterfacial denaturation.33 Heparin stabilizesacidic fibroblast growth factor by increasing itsunfolding temperature by >158C.34
Surfactants have also been used as proteinstabilizers. Nutropin1 (recombinant hGH) con-tains surfactant polysorbates as stabilizers.24
Polysorbate 20 was found to be useful in stabiliz-ing hGH incorporated in a PLG polymer matrix. Itis presumed that these surfactants protect pro-teins against denaturation during several stagesfrom formulation to release at the site of delivery.Certain transition metals have also been shown toconfer protein stability. Zinc stabilizes the hGHagainst urea-induced denaturation.35 Zinc-hGHcomplex was more stable in PLG microspherescompared with hGH alone.
Lyophilization or spray drying also increasesthe storage stability of proteins.36 Freeze dryingitself exposes the protein to destabilizing stresses,
DOI 10.1002/jps

ORAL DELIVERY OF BIOLOGICALS 2501
therefore suitable excipients are included infreeze drying formulations.37 Freeze-drying pro-tectants such as dextran, glycols, glycerol, andcyclodextrin have been found to minimize instabil-ity in freeze-dried formulations of luteinizinghormone release hormone (LHRH),38 monoclonalantibodies,39 and tumor necrosis factor.37
The incorporation of therapeutic proteins intosolid delivery matrices exposes them to a highsurface-to-volume environment creating ampleopportunity for absorption to the delivery device,which limits the amount of free unabsorbedprotein available for release. Incorporating sur-face-active agents to compete for protein bindingsites might reduce protein retention by sustaineddelivery systems. For example, adding albumin toan insulin solution was found to reduce theabsorption of the latter to solid surfaces. In manysustained delivery matrices, protein drugs areexposed to changing environments as the deliverymatrix degrades over time. This degradationmight lead to the generation of acidic oligomers(lactic/glycolic acids), resulting in increasedacidity making the protein prone to degradation.40
To overcome the potential of acidic microen-vironment in the DDSs, basic salts such assodium bicarbonate or magnesium hydroxidemay be incorporated as buffering agents intothe matrix.
APPROACHES FOR MUCOSAL DELIVERYOF THERAPUTIC PROTEINS
Besides parenteral delivery, which is the mostwidely followed route for delivery of proteins,considerable emphasis has gone into exploringnon-injectable methods of protein delivery in-cluding oral,41 rectal,42 buccal,43 transdermal,44
nasal,45 and ocular46 routes. One obstacle as-sociated with oral delivery of protein-baseddrugs, as discussed earlier, is the poor permea-tion across biological barriers, such as theintestinal lumen (Fig. 2). Since the lumen is linedwith proteases and peptidases, this can lead toprotein degradation. The tight junctions, orzonula occludens,47 across the intestinal epithe-lium is another physiologic barrier against para-cellular diffusion of large molecules, aberrantcharge or hydrophilic nature. These character-istics generally lead to low oral bioavailabi-lities (<1%) and short in vivo half lives(<30 min).48,49
DOI 10.1002/jps J
Rectal Delivery of Protein Drugs and Vaccines
In contrast to the oral route of administration,rectal delivery of proteins provides the advantageof greater systemic bioavailability.2 Variousabsorption enhancers like surfactants, bile acids,and sodium salicylate have been used to enhancethe uptake of insulin, gastrin, lysozyme, andheparin following rectal administration.50 How-ever, the absorption rate of rectal administeredmacromolecules is relatively poor when comparedto parenteral routes of administration. Moreover,rectal delivery of biologicals has poor culturalacceptance in several countries.
Ocular Administration
The ocular route may be used for systemic deliveryof therapeutic proteins. Absorption occurs mainlythrough the nasolacrimal system. In addition toinsulin, thyrotropin-releasing hormone, LHRH,encephalin, calcitonin, and glucagon have beenadministered via the ocular route. However, theavailability of ocular administered proteins is stillexpected to be significantly lower than that ofconventional small drug molecules because oftheir unfavorable molecular size, hydrophilicity,and susceptibility to degradation by peptidases invarious compartments of the eye. Fortunately, thesystemic absorption by this route is relatively fast.Absorbed proteins also bypass portal circulation tothe liver thus avoiding first pass metabolism.Even though using the ocular route for systemicdelivery is acceptable it may not be possible.Ophthalmic administration of particles can resultin irritation and can induce lachrymation withpossible consequences of reducing drug bioavail-ability. However, the topical use of growth factorsto heal eye injury is promising, as these injuriesheal very slowly from a lack of blood supply.
Buccal Delivery
The intestinal mucosa has greater permeabilityand perfusion than the skin, while the oral cavityprovides an environment almost free from theacidity and protease activity encountered else-where in the mucosa.51 However, recent studiesrevealed the presence of aminopeptidase activityalong the buccal mucosa, which could be inhibitedby enzyme inhibitors.52 In addition, blood vesselsof the oral mucosa drain directly into the jugularvein avoiding ‘‘first pass extraction’’ by the liver.There is also regional variation in drug perme-
OURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008
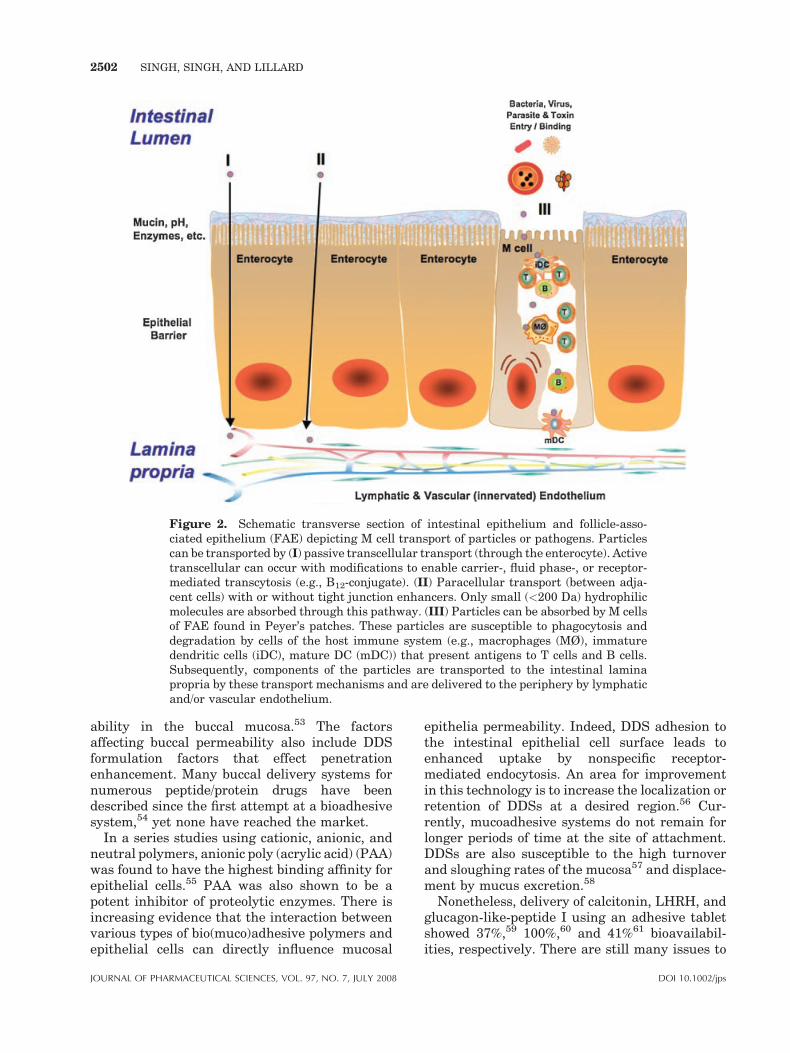
Figure 2. Schematic transverse section of intestinal epithelium and follicle-asso-ciated epithelium (FAE) depicting M cell transport of particles or pathogens. Particlescan be transported by (I) passive transcellular transport (through the enterocyte). Activetranscellular can occur with modifications to enable carrier-, fluid phase-, or receptor-mediated transcytosis (e.g., B12-conjugate). (II) Paracellular transport (between adja-cent cells) with or without tight junction enhancers. Only small (<200 Da) hydrophilicmolecules are absorbed through this pathway. (III) Particles can be absorbed by M cellsof FAE found in Peyer’s patches. These particles are susceptible to phagocytosis anddegradation by cells of the host immune system (e.g., macrophages (MØ), immaturedendritic cells (iDC), mature DC (mDC)) that present antigens to T cells and B cells.Subsequently, components of the particles are transported to the intestinal laminapropria by these transport mechanisms and are delivered to the periphery by lymphaticand/or vascular endothelium.
2502 SINGH, SINGH, AND LILLARD
ability in the buccal mucosa.53 The factorsaffecting buccal permeability also include DDSformulation factors that effect penetrationenhancement. Many buccal delivery systems fornumerous peptide/protein drugs have beendescribed since the first attempt at a bioadhesivesystem,54 yet none have reached the market.
In a series studies using cationic, anionic, andneutral polymers, anionic poly (acrylic acid) (PAA)was found to have the highest binding affinity forepithelial cells.55 PAA was also shown to be apotent inhibitor of proteolytic enzymes. There isincreasing evidence that the interaction betweenvarious types of bio(muco)adhesive polymers andepithelial cells can directly influence mucosal
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008
epithelia permeability. Indeed, DDS adhesion tothe intestinal epithelial cell surface leads toenhanced uptake by nonspecific receptor-mediated endocytosis. An area for improvementin this technology is to increase the localization orretention of DDSs at a desired region.56 Cur-rently, mucoadhesive systems do not remain forlonger periods of time at the site of attachment.DDSs are also susceptible to the high turnoverand sloughing rates of the mucosa57 and displace-ment by mucus excretion.58
Nonetheless, delivery of calcitonin, LHRH, andglucagon-like-peptide I using an adhesive tabletshowed 37%,59 100%,60 and 41%61 bioavailabil-ities, respectively. There are still many issues to
DOI 10.1002/jps

ORAL DELIVERY OF BIOLOGICALS 2503
resolve before this effective and convenient routeof drug delivery can be thoroughly and safelyutilized. Challenges still remain for reproducibleand maximal bioavailability of protein drugdelivery across the buccal mucosa.
In contrast, others studies of protein absorptionand permeability through the buccal mucosa wereshown to present a number of problems. Althougha higher dose of protein could be administered viathe buccal route than parenteral, the resultingplasma levels of the therapeutic were much lowerthan when systemically delivered.62 It is believedthat these limitations were due to reduce perme-ability of proteins delivered by this route. Forexample, calcitonin with a molecular weight of3500 Da is not able to permeate through the buccalmucosa. Similarly, protein delivery via thetransdermal route is also limited. Without che-micals to alter skin permeability the mere size andcharge of proteins prevents passive absorptionthrough the skin.63
Thus, poor absorption and low bioavailability oftherapeutic proteins delivered by these non-injectable routes, has forced biologicals to beparenterally administered by subcutaneous orintramuscular injection. However, the half-livesof parenterally injected proteins are only a fewhours in most cases, necessitating multipleinjections per week for therapeutic effectiveness.As a result, patient compliance is a concern withsystemically administered drugs. This problemcould be resolved by sustained release of proteinsto obtain well-defined pharmacokinetic profiles.Due to rapid clearance by the mononuclearphagocytic system (MPS) and because site-specifictargeting of intravenously administered particu-late drug carriers is not yet possible, it isextremely difficult to maintain DDSs in thebloodstream. Future DDSs should avoid theMPS and possess optimal surface characteristicsto minimize interactions with opsonins that leadto phagocytosis. The main parameters governingthese interactions are surface charge64 andhydrophobicity.65
POLYMERS AND GELS USEDIN PROTEIN DDSS
Polyethylene Glycol
Strategies to circumvent the MPS include graftingpolyethylene glycol (PEG) to the surface of theDDS or protein. PEG is considered to be a nontoxic
DOI 10.1002/jps J
hydrophilic polymer with FDA approval. PEG-grafting results in a steric barrier at the surface ofDDSs that reduces absorption of various proteinsand diminishes complement activation.66 Thechain length and density of PEG domains aremajor parameters that determine the extent ofuptake or lack thereof by the MPS. The presenceof hydrophilic chains reduces the influence ofserum proteins on particle internalization bymonocytes.
While PEG-conjugated proteins may not be amicro- or nano-particle per se, PEGylation ofprotein drugs was developed and commercializedby Dr. Abraham Abuchowski. Three FDA-approved protein drugs: ADAGEN1, ONCAS-PAR1, and PEG-INTRON1 were among the firstto use this technology. Currently, the PEGylatedbiological market is approximately $1 billon(USD) per year. Industry analysts expect thismarket will grow to over $6 billion by 2008.
PEGylation technology was also exploited toextend the product (i.e., patent) lifecycle of app-roved protein therapeutics. For example, Scher-ing Plough’s PEG-Intron1 is a PEGylated versionof Intron and Amgen’s NEULASTA1 isPEGylated Neupogen. PEGASYS1 (PEGylatedinterferon) is another PEGylated interferonmarketed by Roche. Various other PEG-graftedproteins include: soluble TNF receptor-type I(PEG-sTNF-RI), synthetic thrombopoietin, argi-nine deaminase, anti-growth factor receptor anti-body fragment, anti-IL-1b antibody fragment andanti-PDGF b-receptor antibody fragment.
Polyesters
In addition to their biocompatibility, thermoplasticity, high tensile strength, stability, con-trolled degradation rates, adjustable hydrophili-city/hydrophobicity, tailored release rates, andproven non-toxic biodegradable polymers areuniquely suited for incorporation in DDSs. Avariety of synthetic and naturally occurringpolymers have been intensively studied over thelast 30 years. Of these, polyesters have found themost widespread use.67
Features that attracted investigations to usingpolyesters in protein formulation include pre-existing toxicological and chemical data as well astheir biocompatibility, predictable biodegradationkinetics, ease of fabrication, versatility, commer-cial availability and perhaps most importantly—regulatory track record.68 A broad spectrum of
OURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008

2504 SINGH, SINGH, AND LILLARD
performance characteristics with these polymerscan be obtained by careful manipulation of fourkey variables: monomer stereochemistry, co-monomer ratio, polymer chain linearity, andpolymer molecular weight. Hence, different poly-mers can modulate the structure, range ofhydrophilic behavior, and solubility of DDSs.Together, these factors ultimately affect thebiodegradation and release profile of the resultingDDS. For example, crystalline domains and stereoirregularity inhibit the degradation of the poly-mer; hence, stereo irregularity in lactides candetermine degradation time.
Poly L � lactide > Poly DL � lactide > PolyglycolideðCrystalline; stereoirregularÞ > ðAmorphous; stereoirregularÞ > ðCrystalline; stereoregularÞ
Varying co-polymer ratios results in differentcrystallinities, transition temperature and hydro-philicities (or hydrophobicities), which affectbiodegradation profiles. Polymer chain linearityaffects the hydrophilicity of the polymer, which inturn affects its degradation rate. The extent ofblock or random structure in the copolymer alsoaffects hydration rate and the degradation profile.Polyesters are commercially available in a widerange of molecular weights. Higher molecularweight polyesters have higher viscosities thataffect entrapment efficiency as well as sphere sizeand shape. For example, the size of calcitonin-encapsulated microspheres increased when poly-mers with higher viscosities where use in theirformulation.69
Degradation of aliphatic polyesters occurs byrandom, non-enzymatic, and/or hydrolytic clea-vage of ester linkages. The nature of degradationcan be heterogeneous or homogeneous. Hetero-geneous degradation is confined to the surface ofthe polymeric carrier where it is interfaced withthe physiological microenvironment. The externaldegradation rate is constant, while the non-degraded carrier core retains its chemical integ-rity. As expected, carriers possessing highersurface to volume ratios undergo faster degrada-tion.
Homogeneous degradation occurs in bulk,where erosion takes place throughout the DDSand the rate of water penetration is greater thanits conversion to water-soluble fragments. Initi-ally during this process, there is random removalof hydrogen bonds, due to hydration, followed bycleavage of covalent bonds. PLA and PLGA spheredegrade by this process where their chains arecleaved to monomeric acids, for example, lactic
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008
and glycolic acids, that can be metabolized via theKrebs cycle. The mass of these polymers decreasesdue to continuous cleavage and solubilization oflow molecular weight fragments and their absorp-tion. However, the role of enzymes in the bio-degradation of these polymers is not clear.
Natural Polymers
The use of natural biodegradable polymers todeliver drugs continues to be an area of activeresearch despite the advent of synthetic biode-
gradable polymers. In light of the benefits ofpolyesters, these polymers remain attractiveprimarily because they are ‘‘natural’’ products ofliving organisms that are readily available,inexpensive, and capable of a multitude ofchemical modifications. Most investigations usingnatural polymers as matrices in DDSs havecentered on proteins (e.g., collagen, gelatin andalbumin) and polysaccharides (e.g., starch, dex-tran, cellulose, etc.).
Collagen has been extensively tested in DDSsbecause of its unique structural properties. It hasbeen fabricated into a wide variety of formsincluding: cross-linked films, meshes, fibers, andsponges. Collagen, as a biomaterial, offers severaladvantages. It is biocompatible and non-toxic inmost tissues. It can be easily isolated and purifiedin large quantities and has well-documentedstructural chemical and immunological proper-ties.70 However, certain properties of collagenhave adversely influenced its use as a DDS. Theseproperties include: poor dimensional stability dueto swelling; low mechanical strength and elasti-city; anti-collagen immune responses; tissueirritation due to residual aldehyde crosslinkingagents; poor patient tolerance (e.g., ocularinserts); and variability in drug release kinetics.
Albumin, gelatin, casein, and fibrinogen in theform of microspheres and nanoparticles continueto be exploited as DDSs. Albumin microsphereshave been extensively used in diagnostic nuclearmedicine for the evaluation of organ function andcirculatory studies following administration by avariety of routes. The exploitable features ofalbumin include its biodegradation into naturalbyproducts, lack of toxicity and non-antigenicity,and availability. Although the literature contains
DOI 10.1002/jps

ORAL DELIVERY OF BIOLOGICALS 2505
many examples of albumin microsphere use, thereare few reports describing gelatin systems.Gelatin offers several advantages for use in DDSs.For example, this protein weakly interacts withother proteins or drugs, which reduces the chanceof encapsulated molecules being altered or aggre-gated. Moreover, this protein is considered to havelow antigenicity.
Hence, natural polymers particularly in theform of microspheres have an important role inDDSs and targeting to selective sites. Yet manyconcerns must be addressed before they will havewidespread use. Among these issues are a betterunderstanding of the kinetics of drug release,more effective ways to control natural polymerDDS, greater understanding of drug–polymerinteractions and their effect on shelf life stability.The field would also benefit from additionalanimal studies to determine host responses tonatural polymers, tissue adsorption, biodegrada-tion, and metabolic rates. Perhaps most impor-tantly, well-designed clinical studies arenecessary to assess efficacy in relation to currenttherapies.
Hydrogels
Much attention has focused on developing stimulisensitive hydrogels that exhibit dramatic changesin network structure or swelling behavior inresponse to change in pH, temperature, electricfield or ionic strength.71 Most of these systems relyon the sensitive nature of specific interpolymericinteractions within the hydrogel. By exploitingthe sensitive nature of these gels, external(magnetic field, light etc.) or internal triggers(pH, enzymes, etc.) can be used for temporal and/or spatial delivery of biomolecules in the host.Delivery systems can also be designed to releasemacromolecules in response to increased concen-tration of a specific compound or changes in thesurrounding environment.72,73
These polymer complexes are prepared by freeradical solution74 or dispersion75 polymerizationmethods. For example, methacrylic acid (MAA)and methoxy-terminated PEG mono methacrylateadded with tetra-ethylene glycol (EG) dimetha-crylate crosslinks the gel matrix. These materialsexhibit pH-dependent swelling behavior due tothe formation and dissociation of interpolymercomplexes.76,77 Hydrogels comprising of MAA andEG in equimolar amounts exhibit maximumchange in the mesh size or the correlation length
DOI 10.1002/jps J
of the network due to the pH shift. With anincrease in the amount of MAA in the network, theaverage mesh size in the acidic media increasesdue to the steady decrease in the number of MAA–EG interactions. Depending on the pH of thesurrounding medium, the average mesh size in anetwork with MAA:EG ratio of 1:1 changes by afactor of 3-fold between swelling states, whichcorresponds to a 10-fold change in the effectivearea for diffusion of the encapsulated drug. Thisresults in changes in the diffusion coefficient ofthe drug.78
Thus, hydrogels are ideal for the oral delivery ofpeptides and proteins due to their large change innetwork structure over a small pH range. Hence,in the acidic environment of the stomach, drugswould be entrapped in the collapsed gel andprotected from degradation. However, in the nearneutral environment of the intestine, whereprotein drugs can be better absorbed, the peptidesand proteins could be released albeit susceptibleto digestive enzymes. In addition, the polymerstructure and composition can be altered bychanging parameters such as the crosslinker,crosslinker density and relative amounts ofmonomers added to achieve controlled deliveryof proteins and peptides of therapeutic interest.
PREPARATION OF MICROSPHERES
Biodegradable polymers can be used to preparemicrospheres by several methods, each withadvantages and disadvantages. It is essential toselect an encapsulation process fulfilling therequirements of the desired DDS. The require-ments to consider are optimal protein loading,high yield of microspheres, stability of the encap-sulated protein, batch uniformity and inter-batchreproducibility, adjustable release profiles, lowburst effect, and free-flowing or non-aggregatingmicrospheres.
The encapsulation efficiency of the formulationprocess should be high so that the contents arenot wasted. The protein:polymer ratio shouldbe as large as possible to reduce the mass ofthe material to be administered. The processof encapsulation should generate high yield ofparticles of the desired size, depending on thetissue target and route of administration. Impor-tantly, the biological activity of the encapsulatedprotein should be maintained throughout theformulation process. It is desirable to use a processwhere exposure to potentially denaturing solvents
OURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008

2506 SINGH, SINGH, AND LILLARD
or heat is low. The process should be simple,reproducible, and scaleable so that differentbatches of the DDS have the same propertiesand release characteristics. The encapsulationmethod used should ideally produce free-flowingmicrospheres that do not aggregate. This will helpto produce uniform, reproducible bioavailabilityand uptake.
As with all parenteral products, microspheresneed to be sterile. This can be ensured by aterminal sterilization step or through asepticprocessing. Further, in relation to safety require-ments, the excipients and processing solvent usedshould either be nontoxic or removed from thefinal product. There are many procedures forpreparing lactide–glycolide microspheres for pro-tein delivery like phase separation–coacervation,double emulsion, spray drying, interfacial deposi-tion, phase inversion microencapsulation, in situpolymerization, chemical and thermal crosslink-ing, to name a few. The most widely used tech-niques for microsphere preparation of proteinsare: spray drying, double emulsion, and phaseseparation–coacervation.
Spray Drying
In principle, the biodegradable polyester is dis-solved in a volatile organic solvent, such asdichloromethane or acetone, the drug in solidform is dispersed in polymer solution by high-speed homogenization, and this dispersion isatomized in a stream of heated air. As the dropletsform, the solvent evaporates instantaneouslyyielding microspheres typically 1–100 mmdepending on conditions. The microspheres arecollected from the airstream by a cyclone separa-tor and residual solvents can be removed byvacuum drying. Spray drying in a nitrogenatmosphere is technically feasible. Importantadvantages of this technique over other encapsu-lation methods are reproducibility, well-definedcontrol of particle size, control of drug releaseproperties of resulting microspheres, and theprocess is quite tolerant to small changes ofpolymer specifications. The disadvantages includehigh capital investment, encapsulation requireslyophilization of protein before dispersion, andhomogenization in the organic polymer solution.These process conditions are likely to induceaggregation and denaturation to sensitive pro-teins and antigens. Hence, stability of micro-
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008
encapsulated proteins during processing, release,and storage becomes a major concern.
Double Emulsion Method
In this process, protein in an aqueous solvent isemulsified with a non-miscible organic solution ofpolymer to form a water in oil emulsion. Theorganic solvent dichloromethane is frequentlyused while the homogenization step is carriedout using either high-speed homogenizers orsonicators. This primary emulsion is rapidlytransferred to an excess of an aqueous medium,containing a stabilizer, usually polyvinyl alcohol.Again homogenization or intensive stirring isnecessary to initially form a double emulsion ofwater-oil-water. Subsequent removal of organicsolvents by heat, vacuum or both results in phaseseparation of the polymer and core to producemicrospheres. Instead of solvent evaporation,solvent extraction can also be undertaken yieldingmicrospheres containing protein. The advantagesof this method are that the proteins can beencapsulated from an aqueous solution, and highyields and encapsulation efficiencies are obtained.The disadvantages include a complex process,protein sensitivity to polymer and solvents,limited control of release profiles of drug frommicrospheres, limited shelf life and stability ofthis DDS.
Phase Separation
Protein is dispersed in solid form into solutioncontaining dichloromethane and polymer. Oil (e.g.,silicon) is added to this dispersion at a defined rate,reducing solubility of polymer in its solvent. Thepolymer-rich liquid phase (coacervation) encapsu-lates the dispersed drug particles and ‘embryonic’microspheres are subjected to hardening andwashing steps. This process is quite sensitive topolymer properties and residual solvents.
EFFECT OF PARTICLE SIZE, CHARGEAND HYDROPHOBICITY ONMUCOSAL UPTAKE
Particle Size
Particle size charge and size distribution arearguably the most important characteristics ofmucosal DDSs. They determine the in vivo dis-tribution, biological fate, toxicity, uptake and tissuetargeting. In addition, they can also influence the
DOI 10.1002/jps

ORAL DELIVERY OF BIOLOGICALS 2507
drug loading, release kinetics and stability ofparticles. Many studies have demonstrated thatparticles of sub-micron size have a number ofadvantages over microparticles as a DDS.79 Gen-erally, nanoparticles have relatively higher intra-cellular uptake compared to microparticles and areavailable to a wider range of biological targets dueto their small size and relative mobility. Desai et al.found that 100 nm nanoparticles had a 2.5-foldgreater uptake than 1mm microparticles, and 6-foldgreater uptake than 10 mm microparticles by Caco-2 cell line.80 Indeed, nanoparticles penetrate thesubmucosal layers of rat intestinal loops, whilemicroparticles were predominantly localized to theepithelial lining.81
Drug release is also affected by particle size.Smaller particles have a larger surface area;therefore, most of the protein drug is associated ator near the particle surface leading to faster drugrelease. Whereas, larger particles have largecores, that allow more drug to be encapsulated,but might take longer to release.82 However,nanoparticles can have a greater risk of aggrega-tion during storage and dispersion. DDS degrada-tion can also be affected by size. For instance, therate of PLGA polymer degradation was found toclimb with increasing particle size.83 It wasthought that encapsulated contents of smallerPLGA particles, can diffuse more readily; largeparticles have degradation products that remainwith the DDS matrix longer, to cause autocata-lytic degradation of the polymer material. How-ever, PLGA particles of different sizes were shownto have similar polymer degradation ratesin vitro.84
Many studies regarding size effects on nanopar-ticle absorption by intestinal epithelia have beenperformed using polystyrene standard particlesuspensions of defined size distributions. Particleswith mean diameters of 50 and 100 nm showed ahigher uptake in the rat intestine than largerparticles.85,86 The nanoparticle uptake was followedby its appearance in the systemic circulation anddistribution to different tissues. After administra-tion of equivalent doses 33% of the 50 nm and 26% ofthe 100 nm particles were detected in the intestinalmucosa and, in the case of 500 nm particles only 10%were localized in intestinal tissues. Particles >1 mmin diameter were exclusively localized in Peyer’spatches. Although particles >3 mm were foundoccasionally in follicle-associated epithelia andshowed no passage to associated lymphoid tissues.
Summarizing numerous absorption studies ofpolystyrene particles in intestinal tissues reveals
DOI 10.1002/jps J
important facts that should be considered whendesigning an oral DDS. Particles <100 nm showhigher rates of uptake by absorptive enterocytesthan particles >300 nm. The uptake of particles<100 nm by the follicle-associated epithelia ismore efficient than uptake via absorptive enter-ocytes. Uptake of particles >500 nm by absorptiveenterocytes is an unlikely event and only particles<500 nm reach the general circulation. The size-dependent particle passage to mesenteric lymphnodes is still the subject of controversy. Theuptake of 100 nm PLGA nanoparticles in the ratintestine was significantly increased compared tolarger particles of 1–10 mm. Nearly identicaluptake rates were observed in Peyer’s patchregions and enterocytes for 100 nm size particles,while particles >100 nm were only detected in thePeyer’s patches. In summary, size is an importantparameter controlling the internalization of DDSsby the GI tract. As a rule of thumb, sizes <500 nmwill be required for optimal uptake and bioavail-ability.
Hydrophobicity and Surface Charge
Apart from particle size, DDS surface propertiesalso influence how they are taken up by intestinalepithelia. Uptake of particles prepared fromhydrophobic polymers seems to be higher thanthose with more hydrophilic surfaces. Poloxamercoating of polystyrene nanoparticles caused adecrease in GI uptake in vivo. Moreover, hydro-phobic polystyrene nanoparticles seem to have ahigher affinity for M cells than for absorptiveepithelia. Less hydrophobic PLGA particles showinteractions with both cell types.87 These resultsare in accordance with observations by Norris andSinko88 who investigated the in vitro mucuspermeability of particles consisting of polymerswith varying hydrophobic/hydrophilic character-istics. They found that in contrast to morehydrophilic particles, hydrophobic beads showedpoor mucus penetration.
The affinity of charged carriers to intestinaltissues is a subject of great interest. Carboxylatedpolystyrene beads have significantly lower affinityfor intestinal epithelia, especially to M cells, thancompared to positively charged or unchargedpolystyrene beads.85 Coincidentally, nanoparti-cles consisting of negatively charged polyanhy-dride copolymers of fumaric and sebacic acid werehighly adhesive to the cell surfaces.89 Afteradministration, these particles were detected inparacellular spaces, enterocytes and Peyer’s
OURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008

2508 SINGH, SINGH, AND LILLARD
patches demonstrating increased absorption ratesof encapsulated dicoumarol, insulin and plasmidDNA. In summary, these results showed thatuncharged or positively charged nanoparticlesconsisting of hydrophobic polystyrene have anaffinity for follicle-associated epithelium as wellas absorptive enterocytes, whereas negativelycharged polystyrene nanoparticles show onlylow affinity for intestinal tissues. Negativelycharged nanoparticles comprised of hydrophilicpolymers show high bioadhesive properties andare readily absorbed by both M cells andabsorptive enterocytes. Hence, surface charge incombination with hydrophilicity of the DDSmatrix material affects GI uptake of particles.
When nanoparticles are administered intrave-nously, they are easily recognized by the hostimmune system and frequently cleared by phago-cytes. Apart from the size of nanoparticles, theirsurface hydrophobicity determines the amount ofadsorbed blood components, mainly opsonins thatbind and influence DDS bioavailability.90 Theassociation of protein drug with conventionalcarriers leads to modification of the proteintherapeutic biodistribution profile, as it is mainlydelivered to the mononuclear-phagocyte system(MPS). Specifically, the MPS might direct DDSs totheliver,spleen, lungsand/orbonemarrow.Indeed,once in the bloodstream, non-modified nanoparti-cles are rapidly opsonized and cleared by macro-phages. Hence, DDS formulation should minimizethe potential of particle opsonization to prolongtheir bioavailability. This can be achieved bysurface coating with hydrophilic polymers/surfac-tants. Alternatively, biodegradable copolymerswith hydrophilic segments such as polyethyleneglycol (PEG), polyethylene oxide, polyoxamer,poloxamine and polysorbate 80 (Tween-80) can begrafted tonanoparticles to reduce interactions withthe MPS. PEG surfaces in brush-like and inter-mediate configurations reduce phagocytosis andcomplement activation, whereas PEG surfaces inbranched conformations activate complement andfavor phagocytosis.91,92 Hence, this versatile poly-mer can be used to modulate mucosal uptake,bioavailability and tissue targeting.
APPROACHES FOR OPTIMIZING UPTAKEAND BIOAVAILABILITY
Enzyme Inhibition
The strategy of employing enzyme inhibitor(s) andabsorption enhancers to protect the DDS contents
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008
from various enzymatic actions is a popularapproach for oral protein drug delivery.22 Due tothe nature of enzyme distribution and quantities,the use of digestive protease inhibitor(s) for oraldelivery of therapeutic protein would be difficult.However, some successful results have beenreported for insulin administration with sodiumglycocholate, camostat mesilate, and bacitracin torats93 and FK-448 with other protease inhibi-tors.20,94 Indeed, CYP3A4 showed a markedincrease in the oral bioavailability of cyclospor-ine.95 The absorption of large peptides, cholecys-tokinin/enkephalin analogs and protein drugs wasimproved by using protease inhibitor cocktails withattention to specific absorption sites.96–98
A new and interesting method for oral deliveryof protein drugs makes use of a polymer-enzymeinhibitor conjugates that protect the therapeuticprotein from enzymatic degradation.99 Chitosanand its derivatives showed multiple effects onenhancement of insulin, calcitonin, and buserelinabsorption, following oral administration byexploiting enzyme inhibitors and their mucoadhe-sion properties.100 In similar work, pepstatinanalogs were covalently joined to mucoadhesivepolymers to inhibit proteolysis of model proteindrugs.101 This system provided some advantagesby increasing contact-time with the mucosa andmaintaining a controlled as well as sustained drugrelease. It also reduced toxic effects of theinhibitor because of its attachment to the non-absorbable polymer backbone. However, from apractical point of view, the utility of this approachmay be limited by high manufacturing costs.102
Carrier or Uptake Enhancers
Carrier molecules and permeability agents hasalso been used to increase the mucosal absorptionof protein drugs.102,103 This approach was suc-cessfully used to orally deliver hGH, IFN a-2b,and insulin.103,104 Hence, enhancement of intest-inal permeability increased serum concentrationsof hGH, IFN, and insulin as much as 800%. Oralabsorption of granulocyte colony stimulatingfactor (G-CSF) and erythropoietin (EPO) wasachieved by covalently coupling DDSs withvitamin B12.105,106 In this case, uptake occuredvia receptor-mediated endocytosis. This systemhas possible disadvantages since vitamin B12-mediated delivery is limited by its active transportmechanism along with interference from freevitamin B12 in the host.107
DOI 10.1002/jps

ORAL DELIVERY OF BIOLOGICALS 2509
Prodrugs and Analogs
Altering physico-chemical properties of DDSsseems to be the easiest approach to increaseefficacy, but it requires the synthesis of newchemical entities.102,108,109 Changes can be madein lipophilicity, charge, molecular size, solubility,configuration, isoelectric point, chemical stabilityand affinity to carriers to enhance absorption andsystemic circulation. Two specific approacheswere made for oral peptide delivery usingchemical modification of peptide amide bond toenhance intestinal permeability and the design ofcompounds bearing nonpeptide templates.108
Targeting Optimal Absorption Sites
Targeting specific absorption site(s) and dosagevia DDS modification (e.g., lipid vesicles, colloidalcarrier systems with and without mucoadhesivepolymers) are other approaches to improveprotein absorption. It has also been suggestedthat identification of the optimal absorption sitefor a given peptide or protein is the first steptoward the design of a DDS to maximize uptake.56
Regional variations in intestinal penetrationbarriers to peptides may result in regionaldifferences in absorption. For instance, M cellslocated on the dome epithelium of gut-associatedlymphoid tissue are known to sample macromo-lecules from the ileum through an endocyticpathway.110 Controlling the characteristics ofDDS to deliver proteins and large peptide drugsto M cells has been attempted. However, thismethod has had variable success.96–98
Targeting Intestinal Transporters
Recent advances in molecular biology techniqueshave made it possible to study the structure,function and distribution of cellular transportersof the mucosa. Based on molecular characteriza-tions of membrane transporter specificities, andkinetics, the modification and targeting of specifictransporter(s) is a promising strategy for DDSs toimprove bioavailability and tissue distribu-tion.111,112 However, the utility of these transpor-ters are limited by the size of molecules that can bedelivered.106 For example, the most permissivetransporter, the bile acid transporter, is limited topeptides <400 Da. Hence, strategies to improvethe interaction of nanoparticles with adsorptiveenterocytes and M cells of Peyer’s patches can be
DOI 10.1002/jps J
classified into those utilizing specific binding toligands or receptors and nonspecific adsorptivemechanisms.
Adhesive Carrier Systems
Various colloidal systems have been studied forabsorption of DDSs, such as sub-micron emul-sions, lipid suspensions, liposomes, and polymericnano- and micro-particles. Controversy still existson the factors that govern GI uptake, includingsize, size distribution, consistency, hydrophobi-city, and surface properties of colloidal carriers.107
Prolonged contact of nanoparticles with absorp-tive cells may be achieved using bioadhesivematerials. Bioadhesion has also been followed byparticle uptake in a second step.113 Hence,biomaterials with both adhesive and protectiveproperties might be desirable for oral protein drugdelivery to insure drug stability and bioavail-ability.
In general, GI tract absorption of macromole-cules and particulate materials involves eitherparacellular or endocytic pathways. The para-cellular route of absorption is accessible in <1% ofthe mucosal surface area. Using polymers suchas chitosan,114 starch57 or polyacrylate115 canincrease the paracellular permeability of macro-molecules. Endocytic absorption occurs by eitherreceptor-mediated or adsorptive endocytosis. Thelater process is initiated by an unspecific cellsurface interaction due to electrostatic forces.116
Hence, adsorptive endocytosis depends primarilyon the size and surface properties of the DDS. Ifthe surface charge is positive or uncharged, then itwill provide an affinity to adsorptive enterocytesthrough hydrophobic interactions. Whereas if theDDS has a negative surface charged and ishydrophilic, it will have an even greater affinityfor adsorptive enterocytes and M cells.
Liposomes
The use of liposomes has been largely abandonedas oral DDSs due to poor stability under thediverse physiological conditions typically found inthe GI tract.117 While homogeneous lamellar100 nm liposomes were taken up by M cells,118
similar studies have led to the overall conclusionthat liposomes are ineffective as vehicles for oralvaccines. However, mucoadhesive liposomal sys-tems prepared by coating negatively (phosphati-dyl choline) or positively charged (salicylic acid)
OURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008

2510 SINGH, SINGH, AND LILLARD
lipid suspensions with mucoadhesive polymersolutions, such as chitosan and carbopol, showedsome success in intestinal absorption of proteindrugs, for example, insulin and calcitonin.119
Nanoparticles
The surface area of the human mucosa extends to�200 times that of skin. The histological archi-tecture of the mucosa is designed to efficientlyprevent uptake of particulate matter from theenvironment. One important strategy to overcomethe GI barrier is to deliver therapeutic proteins ina DDS, such as nanoparticles, that are capable ofenhancing interaction and uptake by the epithe-lium of the GI tract. Nanoparticles, as defined bysolid particles, with size in the range of <200 nm,allow encapsulation of the protein drugs inside amatrix that protects them from enzymatic andhydrolytic degradation. Various biomaterials ofpolymers, lectins, etc, can be employed to makenanoparticles using techniques of emulsionpolymerization, interfacial polymerization, em-ulsification evaporation, solvent displacement,desalting, emulsification and diffusion.120 Solventdisplacement and salting-out have received in-creasing attention because they provide lessstress to protein drugs. The physicochemicalproperties of nanoparticles and their behavioron exposure to physiological media are mediatedby their chemical structures and surface char-acteristics.107
The development of suitable nanoparticle car-riers remains a challenge due to the fact that thebioavailability of these molecules is limited by thephysiology of the epithelial barriers of the GI tractand susceptibility to digestive enzymes. Fortu-nately, the formulation of polymeric nanoparticlesallow for encapsulation of bioactive molecules thatprotects against enzymatic and hydrolytic degra-dation. For instance, it has been found thatinsulin-loaded nanoparticles preserve insulinactivity and produce blood glucose reduction forup to 14 days following the oral administration.
MACRO- AND NANO-PARTICLE INDUCERSOF IMMUNITY AND TOLERANCE
Oral Vaccines
Peptide and protein encapsulation in DDSs hasbeen applied to several oral vaccination applica-tions. Moreover, mucosal vaccination, and more
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008
specifically oral vaccination would lead to lowerproduction and administration costs. Compared tosystemic administration, mucosal vaccinationtargets the common mucosal immune systemsand avoids pain as well as the many of the risksassociated with injections. Vaccination at the siteof the potential infection is highly desirable toobtain a local mucosal defense. Indeed,>95% of allthe pathogens enter via mucosal routes. Locallyproduced secretory IgA constitutes >80% of allantibodies produced in the host and are consid-ered to be among the most important protectivehumoral immune factors.121,122 Furthermore, afascinating feature of the common mucosalimmune system is that administration of anantigen at one mucosal site can lead to thegeneration of immune responses at distantmucosal sites. Finally, mucosal immunizationhas the potential to elicit an immune responseagainst infectious diseases for which currentparenteral vaccines either have a low efficiencyor minimally effective, such as vaccines againstHIV and tuberculosis.123,124 In the scope of oralvaccination, it is particularly interesting to favorthe uptake of antigen-loaded DDSs by M cells.There is a consensus that Peyer’s patch M cellsrepresent a key portal site for some bacteria,viruses and prions to subsequently initiatemucosal immunity. To this end, several strategieshave been employed to deliver vaccines by thisroute.
Eldridge et al.125 asserted that microspheres <5mm in diameter were transported by M cells formucosal immunization. As a result, numerousmicroparticulate systems were developed for oralimmunization. Many of the polymeric biodegrad-able microparticles have been composed of PLA orPLGA. Many vaccine antigens have been success-fully encapsulated in PLGA microparticles with-out altering their structural and immunologicintegrity.126,127 In general, ovalbumin, peptides,bacterial toxoids, inactivated bacteria and, morerecently, plasmid DNA entrapped in PLGAmicroparticles has been shown to induce bothmucosal and systemic immune responses follow-ing oral or intragastric administration.128–132
While many antigens have been successfullydelivered, it is important to mention that proteindenaturation can occur during encapsulation inPLA or PLGA polymers, due largely to exposure toorganic solvents, elevated temperatures andaqueous organic interfaces.133 Latex and PLGAparticles (<500 nm) may be taken up better thanthe particles 1–5 mm.133 However, there is no
DOI 10.1002/jps

ORAL DELIVERY OF BIOLOGICALS 2511
compelling evidence that nanoparticles are moreeffective than microparticles in oral delivery ofvaccines. Up to now, only a few studies haveexamined the capacity of biodegradable nanopar-ticles to induce mucosal immunity after oraladministration.
Jung et al.130 used poly vinyl alcohol-co-PLGAto reach a high level of tetanus toxoid (TT) loadingby adsorption. Particles given per os to miceinduced significant TT-specific IgG and IgAimmune responses, when compared to intraperitoneal administration of antigen. Particle sizewas found to significantly affect the induction ofantibody production; smaller particles inducedhigher titers. In addition, cholera toxin B subunit(CTB) entrapped in submicron particles (�400nm) caused comparable immunogenicity than thepotent oral adjuvant, cholera toxin.134 The influ-ence of particle size on immune response after oraldelivery of BSA entrapped in 200, 500, and1000 nm PLGA particles have also been studied.Despite the literature showing extensive intest-inal absorption of nanoparticles, high antigen-specific serum IgG antibody levels are routinelyobserved following oral administration of 1 mmparticles, compared with particles 200–500 nm insize.
PLGA nanoparticles containing Helicobacterpylori lysates stimulate antigen-specific mucosaland systemic immune responses and induce Th2-type responses.131 However, antibody titers ofgroups immunized with H. pylori-loaded PLGAnanoparticles were lower than particles immu-nized containing free H. pylori protein associatedwith the CTB. Fattal et al.135 showed protection ofmice against following oral administration ofSalmonella typhimurium antigen-encapsulatedPLGA particles. Oral administration of Bordetellapertussis antigen-entrapped PLGA microparticlesor nanoparticles was shown to protect againstrespiratory challenge.136 This study demon-strated that a single oral dose of encapsulatedB. pertussis fimbria could confer protection.
However promising, there are few commerciallyavailable oral vaccines. Although particle uptakeby M cell has been repeatedly demonstrated inrodents, it remains uncertain whether this will bethe case in man.137 Two rather disappointingPhase I oral vaccine trials using PLGA have beenconducted the last 10 years. Orally administratedE. coli colonization factor antigen P (CFA P)entrapped in PLGA microspheres add only 30%efficacy.138 A significant increase of anti-CFA P
IgA and IgG antibody secreting cells in human
DOI 10.1002/jps J
volunteers, following oral administration of waterE. coli CFA II-entrapped PLGA microspheres.139
Group B Streptococcus vaccine (GBS) is theleading bacterial cause of neonatal sepsis andmeningitis. Although antibiotics have decreasedthe severity of this infection, the best long-termsolution lies in the development of effectivevaccines. The GBS capsular polysaccharide is amajor target of antibody-mediated immunity. Thefeasibility of producing a GBS vaccine having theability to produce both a local (i.e., IgA) immuneresponses and systemic humoral responses thatare capable of active and transplacental passiveimmunization was investigated using an oralDDS. Inactivated GBS antigen was encapsulatedin poly (D,L-lactic-co-glycolic acid) by a water-in-oil-in-water (w/o/w) multiple emulsion techniquealong with immunostimulatory cytosine phos-phate guanosine (CpG) motif adjuvants.140 Immu-nization of female mice with these microparticlescontaining GBS type III polysaccharide and CpGadjuvant resulted in significantly higher GBSantibody responses, as compared to nonencapsu-lated GBS polysaccharide or PLGA-encapsulatedGBS polysaccharide vaccine without the additionof the CpG.
Tetanus, caused by tetanus toxin, is considereda significant health problem worldwide, withapproximately one million new cases occurringeach year. Mice were immunized with TT-encapsulated sulfobutylate-grafted PLGA nano-particles by oral and nasal route.130 EncapsulatedTT and Haemophilus influenzae type b capsularpolysaccharide conjugated to TT (Hib-T) in PLGAmicrospheres were evaluated for their humoralimmunogenicity in mice. A single injection ofthese microencapsulated vaccines elicited highantibody levels, which persisted for severalmonths. The antibody levels were similar orsuperior to those elicited by conventional formu-lations of Alum-adsorbed TT or soluble Hib-Tconjugate vaccine.141
Diphtheria is a communicable disease caused byCorynebacterium diphtheriae, which colonizes themucosa and forms a pseudomembrane at theinfection site. This pathogen produces diphtheriatoxin, which is responsible for the typical systemictoxemia. Fortunately, anti-diphtheria toxoid (DT)antibodies can protect against diphtheria. DT hasbeen encapsulated in various types of PLA andPLGA microspheres by spray drying and coacer-vation.142 Recently, poly-epsilon-caprolactone(PCL)-PLGA blend and co-polymer nanoparticleswere used to orally immunize against diphtheria
OURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008

2512 SINGH, SINGH, AND LILLARD
by encapsulating DT.143 In vitro studies usingCaco-2 cells revealed higher uptake of therelatively hydrophobic PCL nanoparticles incomparison to polymeric PLGA, PLGA-PCL blendor co-polymer nanoparticles. Hydrophobic DTnanoparticles induced the highest serum IgGantibody responses when delivered by intranasalroute.
Cholera is an acute intestinal infection causedby Vibrio cholerae and produces an enterotoxincausing copious, painless, watery diarrhea thatcan quickly lead to severe dehydration and death.Inactivated V. cholerae was successfully entrapp-ed in the PLG microspheres by double emulsionmethod with trapping efficiencies up to 98%.144
The immunogenic potential of V. cholerae-loadedmicrospheres was evaluated in adult mice byoral immunization in comparison to V. choleraesolution. Results indicated that following oralco-administration of these microspheres, Vibrio-specific serum antibody responses were inducedwith vibriocidal activity.
Oral DNA-Based Vaccines
Plasmid DNA can be encapsulated in nanoparti-cles with significant retention of biological func-tion, after oral delivery in polymers can elicitsystemic and mucosal antibody responses toencoded antigens. Oral administration of chitosannanoparticles (200 nm) complexed with DNAcoding for a dominant peanut allergen elicitedantigen-specific secretory IgA and serum IgG2atiters.145 Similarly, oral feeding of DNA-loadedchitosan nanoparticles can raise immuneresponses against native dust mite allergens inmice, whereas intramuscular immunization alonedid not. Nanoparticles might also facilitatemucoadhesion and DNA uptake by host cells toenhance transfection efficiency. Bivas-Benitaet al. compared the potential of chitosan nano-particles (�500 nm) loaded with Toxoplasmagondii GRA1 encoding DNA plasmid (pDNA) orchitosan microparticles loaded with recombinantGRA-1 protein to elicit GRA-1-specific immuneresponses after intragastric administration usingdifferent prime/boost regimens.145,146 Interest-ingly, the GRA1 DNA vaccine resulted in higheranti-GRA1 antibody levels. These results showedthat oral delivery of DNA-based vaccines usingchitosan carriers efficiently induced immuneresponses to expressed protein. The type ofimmune response, however, may largely depend
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008
on the prime/boost regimen and the type ofvaccine used. A single oral immunization of PLGAnanoparticles (�500 nm) containing rotavirusVP6 DNA was sufficient to elicit rotavirus-specificserum IgG and IgM as well as intestinal IgAresponses.147,148
Antigen-Loaded Particles Induce Oral Tolerance
Some polymers are able to elicit an immuneresponse alone when administrated orally, suchas chitosan, which support a Th2/Th3-biasedmicroenvironment at the mucosal level, inabsence of antigen.149 Systemic unresponsivenessto orally delivered antigens (oral tolerance) mayadversely affect oral vaccination or, conversely,could be used as a therapy for individuals thatrespond to innocuous antigens (e.g., food aller-gens, transplantation antigens or commensalbacteria).150 Oral administration of free antigenshas been recognized as a method to induceantigen-specific peripheral tolerance.151 Oral tol-erance is mediated by two mechanisms thatdepend on the dose of administrated anti-gens.152,153 Repeated administrations of low dosesof antigen can induce active suppression. Thismechanism functions by expanding antigen-spe-cific regulatory T cell population that activelysuppress T helper responses to antagonize pro-inflammatory responses.154 In contrast, higherdoses of antigen induce T cell clonal deletion and/or anergy, characterized by both antibody andcell-mediated immune response inhibition.155
Biodegradable microparticles, first promotedfor vaccine development, now appear attractive asinducers of oral tolerance. Kim et al.156 showedthat a single administration of PLGA nanoparti-cles containing type P collagen (CP) could induceoral tolerance more efficiently than repeated oraladministrations of intact CP. Type II collagen-encapsulated in 300 nm PLGA particles wasdetectable in Peyer’s patches, by microscopy 14days after stable oral administration.156 Thisregimen significantly reduced the incidence andseverity of arthritis, serum IgG anti-CII anti-bodies, and CII-specific T cell proliferation ascompared with controls. Similarly, newborns areprone to milk allergies that can be prevented byinducing oral tolerance to b-lactoglobulin. Thismajor allergenic protein was encapsulated in PLGmicrospheres by w/o/w multiple emulsion techni-que. Oral administration of these microspheresdrastically reduced the amount of protein
DOI 10.1002/jps

ORAL DELIVERY OF BIOLOGICALS 2513
required to reduce specific anti-b-lactaglobulinIgE response.135 A single feeding of 5 mgof encapsulated b-lactoglobulin tolerized BALB/cmice to subsequent challenge. The tolerogenicdose was 10000-fold less than the dose of solubleantigen alone.
ORAL DELIVERY OF THERAPEUTICPEPTIDES AND PROTEINS
Insulin
Insulin is the most important regulatory hormonein the control of glucose homeostasis. WHOreports indicated that more than 50 millionpeople around the world suffering from diabetesrequire daily parenteral injections of insulin. Forthe treatment of Type I diabetes, insulin isadministered typically by three injections perday. An insulin DDS for long-term therapy of thisdisease would be well received, as this systemcould alleviate daily injections and possibleimprove patient compliance. Insulin has beenincorporated into the hydrogel microparticles fororal delivery.157 Upon exposure to an acidicenvironment, <10% of insulin is released fromthe microparticles. However, when the pH of thesurrounding medium rises to physiological pH inthe small intestine, the insulin trapped insidethe gel network is rapidly released. PEG chains inthis network serve to maintain the biologicalactivity of the insulin by preventing binding tothe ionizable backbone of the encapsulationmatrix.78,158 The effectiveness of this system fordelivering insulin was evident from the improv-ed hypoglycemic effect on oral administration.159
Oral administration of insulin-loaded in polyisobutylcyanoacrylate nanocapsules caused adramatic reduction of blood glycaemia in diabeticrats.160 It was later shown that these nanocap-sules were absorbed by intestinal epithelialcells.161 However, much of the nanocapsules weredegraded upon transport across M cells. Adding tothis disappointment, Cournarie et al.162 under-lined the high variability in insulin transportacross the intestinal barrier.
Oral administration of nanosphere-based insu-lin delivery systems comprised of polyfumaricanhydride and PLG maintained normoglycemiain the face of a glucose challenge.163 Oraladministration of chitosan–insulin nanoparticles(50 U or 100 U/kg) were effective at loweringserum glucose levels of streptozotocin-induced
DOI 10.1002/jps J
diabetic rats.164 Most likely, this effectiveness canbe attributed to the local effect of insulinavailability in the intestine.165 Finally, Alonsoco-workers tested the efficacy of insulin-loadedchitosan–glucomannan nanoparticles followingoral administration to normal rats. This carriersystem was able to elicit a delayed hypoglycemicresponse, 14 h post-administration.
Perhaps the delay in clinical studies of the oralinsulin delivery systems have been deferred bythe high variability of the insulin concentrationdelivered to the blood. Moreover, high doses ofinsulin will be required due to its low bioavail-ability for oral delivery. To increase availability,insulin has been encapsulated in blends of PEGalong with PLA and PLG by a w/o/w multipleemulsion technique with entrapment efficienciesof 56 and 48% for PLG/PEG and PLA/PEG,respectively.166 Insulin-loaded microspheres werecapable of controlling the release of insulin for28 days with in vitro delivery rates of 0.94 and0.65 mg of insulin per mg of particle per day in4 days and with a steady release rate of 0.4 and0.43 mg of insulin per mg of particle per day over4 weeks, respectively. In addition, the extensivedegradation of PLG/PEG microspheres over4 weeks as compared to PLA/PEG blends resultedin stable particle morphology along with reduc-ed fragmentation and aggregation of associatedinsulin.
These studies may not translate to clinical oraldelivery. Only a small portion of insulin orallyadministered has been shown to reach the bloodstream, mainly due to extensive degradation ofthe protein in the GI tract. Further, insulin’s sizeand hydrophilicity limits its transport across theintestinal epithelium. No specific transportmechanism is present for the passage of insulincross the intestinal cell monolayer. For manyyears, researchers have tried to find a solution tothese problems and in effect increase the oralbioavailability of insulin. The use of permeationenhancers, protease inhibitors chitosan coatingsto stabilize the protein and improve cellularpermeability, entrapment of insulin within micro-particles and protein modification to resist pro-teolytic attack are additional creative approachesto orally deliver insulin.157,167–172
Other Peptide and Protein Drugs
Calcitonin has been delivered using oral DDSs.When calcitonin was incorporated in nanoparti-
OURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008

2514 SINGH, SINGH, AND LILLARD
cles, oral absorption was enhanced in rats andconsequently calcium concentration in blood wasdecreased, compared to oral administration of acalcitonin solution.107 Mucoadhesive (chitosan)polymeric nanospheres containing calcitonin werealso shown to reduce blood calcium 12 h afteradministration at doses of 125 IU/Kg and 250 IU/Kg.119,173 Chitosan nanoparticles have also beenused to deliver hydrophobic peptides such asciclosporin A.174 Chitosan nanoparticles orallyadministered to beagle dogs provided an improvedabsorption compared to the currently availableciclosporin A microemulsion (Neoral1). Finally,chitosan particles were used to encapsulate theerythropoietin gene resulted in a rapid increase ofhematocrit that was sustained for >1 week.175
Das and Lin176 developed a double-coated(Tween-80 and PEG 20000) poly butylcyanoacry-late (PBCA) DDS for oral delivery of dalargin.While the mechanism of PBCA nanoparticletransport of dalargin from the GI tract andrelease to brain was not elucidated, the studynoted a significant dalargin-induced analgesiawith double-coated PBCA nanoparticles comparedto single-coated particles. The lack of comparativestudies makes it difficult to determine the mostefficient formulation to orally deliver therapeuticpeptides or proteins. To this end, the core ofnanocapsules (i.e., liquid vs. solid) has no effect onoral bioavailability of calcitonin.173,177
Chitosan-coated nanoparticles resulted in less(27%) serum calcitonin levels, when compared touncoated particles. Chitosan-modified with PEGimproved the stability of nanocapsules in GI fluidsand reduced nanocapsule cytotoxicity. Therefore,modulating the degree of chitosan (i.e., charge)PEGylation modified the stability, cytotoxicity,and enhanced absorption of the particles. Thisnew carrier seems to be close to the ideal carriercombining an adequate size (160–250 nm), mod-erate encapsulation efficiency (44–50%), stabilityand mucoadhesion. The translation of theseanimal studies to human application may not beso straightforward. Unfortunately, the inaccuracyof the dose delivered, and bioavailability greatlylimit using DDSs for oral delivery of peptides.
Prolidase deficiency results in chronic intract-able skin ulcerations, particularly of lower limbs.To counter this problem, recombinant prolidasewas encapsulated in PLGA microspheres by w/o/wmultiple emulsion technique.178 Microencapsula-tion stabilizes enzymatic activity and resulted inactive peptidase release in vitro and in vivo.Although this was not orally delivered this opens
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008
the doors for enzyme replacement therapythrough oral protein DDSs.
Interferon a (IFNa) is used in the treatment ofchronic hepatitis C virus infection. A novelmicrosphere delivery system was developed toencapsulate recombinant IFNa in calcium algi-nate cores surrounded by poly DL-lactide-poly-ethylene glycol (PLPEG) by w/o/w multipleemulsion technique.179 Core-coated microspheresstabilized IFNa in the PLPEG matrix. Impor-tantly, the extent of burst release reduced to 14%in core-coated microspheres from 31% in conven-tional microspheres, highlighting this newapproach for water-soluble macromolecular drugdelivery. Perhaps this formulation approach canbe used in the future for orally delivery of proteintherapeutics.
Orally Administered Antibodies
There is evidence to indicate that a fraction oforally administered antibodies can survive pas-sage through the human gastrointestinal tractand retain structural characteristics and immu-nologic activity. This raises the possibility oftreating GI infections by passive immunizationusing orally administered immunoglobulin. GIinfections are important causes of morbidity andmortality, particularly in developing nations.Passive immunization is currently under experi-mental and clinical evaluation, and results areencouraging.
Infectious agents such as rotavirus, E. coli,V. cholerae, Clostridium parvum, and H. pyloriare known to cause gastroenteritis. Severe diar-rhea is a major complication and if untreated islife threatening. Evidence seems to indicate thatgastroenteritis of most microbial origins may betreated by passive immunization and may notnecessarily require attenuated or killed patho-gens for vaccination. Guarino et al.,180 success-fully treated rotavirus-induced diarrhea ininfants using a nonspecific human serum immu-noglobulin. In addition, the shorter duration ofdiarrhea was associated with a shorter length ofrotavirus shedding. Similar results have beenshown using rotavirus-specific immunoglobulinisolated from bovine colostrums rather thannonspecific immunoglobulin to treat cases ofinfant gastroenteritis.181
In other studies, several HIV-infected patientswith C. parvum-induced diarrhea were success-fully treated with specific bovine colostral immu-
DOI 10.1002/jps

ORAL DELIVERY OF BIOLOGICALS 2515
noglobulin.180,182,183 Those who are pre-disposedto gastrointestinal infections, such as childrenand adults with severe immunodeficiencydiseases184 and recipients of bone marrow trans-plants185 may also benefit from passive immu-nization. Interestingly, the degradation ofimmunoglobulin in the GI tract of patients withbone marrow transplants is impaired because ofthe destruction of the intestinal mucosa resultingfrom the transplant preparation regimen, highgastric pH, rapid intestinal transit, limited oralintake and antibacterial therapy which reducesflora.185,186
REGULATORY CONSIDERATIONS FORORAL PROTEIN DELIVERY
Preclinical and toxicological studies must beperformed in accordance with guidelines set bythe FDA to eliminate formulations that are tootoxic for human or animal use and to indicatewhether an oral DDS, for example, biodegradablenanoparticles or microspheres containing proteindrugs are effective and safe. To gain FDA approvalfor any oral DDS formulation, it is necessary toconsider the presence of residual solvents andpolymers that might remain after delivery as wellas preclinical and toxicological studies. Virtuallyall DDS processes require the use of an organicsolvent such as dichloromethane or ethyl acetatefor maintaining polymer solubility during fabrica-tion. These solvents may pose significant healthrisks for long-term exposure. Acceptable residualamounts of these solvents may vary amongregulatory agencies. For example, the Interna-tional Conference on Harmonization (ICH) guide-line for permissible dichloromethane is 6 mg/dayunless it can be shown that the residual solvent isreleased in a sustained fashion for several days.
The FDA requires the safety and biocompat-ibility of all polymeric materials used for medicaland dental applications to be established prior touse. The tests used to establish safety will dependon the type of device, the drug to be delivered, andits application. In vivo and in vitro testing ofpolymeric materials should be designed to inves-tigate the polymer mucosal interface reactions,effects on subsurface tissue, and systemic effects.After oral delivery, bioabsorption studies wouldbegin with animals at predetermined time periodsalong with mucosal tissue isolation and prepara-tion for immunohistochemical or cytochemical
DOI 10.1002/jps J
analysis. Scoring systems have been used basedon the number of specific cell types within aspecific area of detected DDS to effectivelycompare the biocompatibility of polymers.187–189
Skin patch tests are common tests for delayedtype hypersensitivity evaluation.190 Hence, acutetoxicity could generally be measured by applying atest material comprised of the encapsulatedcontents onto shaved intact or abraded skin.Various biological parameters such as bodyweight, mortality, and gross pathological evalua-tion would also be assessed includes multipledoses over longer periods of time to recognize bothacute and chronic toxicity of DDS components.191
Evaluation of biocompatibility of polymers viatissue culture techniques are based on analysisof cellular growth, division, enzyme levels, andsynthesis of important macromolecules.192,193
CONCLUSIONS AND FUTURE PROSPECTS
The GI tract has formidable physiological andchemical barriers that will pose several challengesfor oral DDS. However, significant progress hasbeen made with each of these obstacles. Thedevelopment of composite formulation methods,which improves bioavailability yet meets regula-tory requirements for reproducibility, intra-andinter-subject variability, and manufacturing costwill be difficult; however, the potential of thisemerging field is promising.
Despite considerable research efforts andimpressive progress made in recent years, thefeasibility of biodegradable DDSs for therapeuticprotein or vaccine delivery systems remains opento debate. Micro and nano encapsulation techni-ques have evolved to allow for the incorporation ofsensitive proteins able to resist the harsh envir-onment of the mucosa. It seems that the w/o/wmultiple emulsion technique is the mostadvanced. Indeed, biodegradable polymers (e.g.PLGA) have been used for such DDSs, with well-known degradation properties. An area requiringstudy is analytical characterization of encapsu-lated proteins. Advanced methods for proteincharacterization will be needed to definitivelysolve real and perceived problems of DDS proteinstabilization in DDSs. Further, the developmentof methods to correlate in vitro with in vivo proteinrelease would advance the field and increase therate of development of new DDSs. More colla-borative interactions between immunologists,
OURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008

2516 SINGH, SINGH, AND LILLARD
biochemists pharmacologists, and physiology spe-cialists are required to understand protein releaseand uptake. The future success of biodegradableoral DDSs will primarily depend on the commit-ment of academia and industry to develop newstrategies to orally deliver therapeutic proteins inefficient and cost effective DDSs.
ACKNOWLEDGMENTS
The content of this manuscript benefited fromediting by Andrew Marsh and many fruitful con-versations with members of the Morehouse Schoolof Medicine and the University of Louisville. Thisstudy was supported by funds from the Smith &Lucille Gibson Endowment and National Instituteof Health Grants AI057808, DK58967, MD00525,and RR03034.
REFERENCES
1. Cohen S, Yoshioka T, Lucarelli M, Hwang LH,Langer R. 1991. Controlled delivery systems forproteins based on poly(lactic/glycolic acid) micro-spheres. Pharm Res 8:713–720.
2. Lee VHL. 1986. Peptide and protein drug delivery:Opportunities and challenges. Pharm Int 7:208–212.
3. Hermann J, Bodmeier R. 1995. The effect of par-ticle micro structure on the somatostatin releasefrom poly(lactide)microspheres prepared by a w/o/w solvent evaporation method. J Control Release36:63–71.
4. Takada S, Uda Y, Toguchi H, Ogawa Y. 1995.Application of a spray drying technique in produc-tion of TRH-containing injectable sustainedrelease microparticles of biodegradable polymers.J Pharm Sci Technol 49:180–184.
5. Jalil R, Nixon JR. 1990. Biodegradable poly(lacticacid) and poly(lactide-co-glycolide) microcapsules:Problems associated with preparative techniquesand release properties. J Microencapsul 7:180–184.
6. Yolles S, Eldridge JE, Woodland JHR. 1971. Sus-tained delivery of drugs from polymer drug mix-tures. Polym News 1:9–12.
7. Sanders LM, McRae GI, Vitale KM, Kell BA. 1985.Controlled delivery of LHRH analogues from bio-degradable injectable microspheres. J ControlRelease 2:187–195.
8. Rogers JA, Ownsu-Ababio G. 1993. Formulations ofantibiotics in polymeric microcapsules: Ciprofloxa-cin. In: The 20th International Meeting of the Con-trolled Release Society, Washington DC, 24–30.
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008
9. Brannon-Peppas L, Grosvenor AL, Smith BS.1994. Drug delivery of penicillin and b-estradiolfrom biodegradable microparticles within degrad-able and nondegradable films. in: The 21st Inter-national Meeting of the Controlled ReleaseSociety, Nice, France, 27–30.
10. Fujita SM, Sherman JM, Godowski KC, Tipton AJ.1992. Delivery of amikacin from an aerosolid bio-degradable film. In: The 7th Annual Meeting of theAmerican Association of Pharmaceutical Science,San Antonio, Texas, 15–20.
11. Cowsar DR, Tice TR, Giley RM, English JP. 1985.Poly(lactide-co-glycolide) microcapsules for con-trolled release of steroids. Methods Enzymol112:101–116.
12. Eldridge JH, Staas JK, Chen D, Marx PA, Tice TR,Gilley RM. 1993. New advances in vaccine deliverysystems. Semin Hematol 30:16–24.
13. Moore A, McGuirk P, Adams S, Jones WC, McGeeJP, O’Hagon DT, Mills KH. 1995. Immunizationwith a soluble recombinant HIV protein entrappedin biodegradable microparticles induces HIV-spe-cific CD 81cytoxic T lymphocytes and CD41 Th1cells. Vaccine 13:1741–1749.
14. Bittner B, Morlock M, Koll H, Winter G, KisselT. 1998. Recombinant human erythropoietin(rHEPO) loaded poly(lactide-co-glycolide) micro-spheres: Influence of the encapsulation techniqueand polymer purity on microspheres characteris-tics. Eur J Pharm Biopharm 45:295–305.
15. Ando S, Putnam D, Pack DW, Langer R. 1999.PLGA micro-spheres containing plasmid DNA:Preservation of supercoiled DNA via cryoprepara-tion and carbohydrate stabilization. J Pharm SciTechnol 88:126–130.
16. Wang D, Robinson DR, Kown GS, Samuel J. 1999.Encapsulation of plasmid DNA in biodegradablepoly(D,L-lactic-coglycolic acid) microspheres asa novel approach for immunogene delivery.J Control Release 57:9–18.
17. Fattal E, Vauthier C, Aynie I, Nakada Y, LambertG, Malvy C, Convreur P. 1998. Biodegradable polyalkylcyanoacrylate nanoparticles for the deliveryof oligonucleotides. J Control Release 53:137–143.
18. Lewis KJ, Irwin WJ, Akhtar S. 1998. Developmentof a sustained release biodegradable polymerdelivery system for site-specific delivery of oligo-nucleotides: Characterization of P(LA-GA) copoly-mer microspheres in vitro. J Drug Target 5:291–302.
19. Humphrey MJ, Ringrose PS. 1986. Peptide andrelated drugs: A review of their absorption meta-bolism and excretion. Drug Metab Rev 17:283–310.
20. Lee VHL. 1991. Protease inhibitors and penetra-tion enhancers as approaches to modify peptideabsorption. J Control Release 13:213–223.
DOI 10.1002/jps

ORAL DELIVERY OF BIOLOGICALS 2517
21. Merkle HP. 1994. New aspects of pharmaceuticaldosage forms for controlled drug delivery of pep-tides and proteins. Eur J Pharm Sci 2:19–21.
22. Lehr CM. 1994. Bioadhesion technologies for thedelivery of peptide and protein drugs to the gas-trointestinal tract. Crit Rev Ther Drug CarrierSyst 11:119–160.
23. Sarciaux JM, Acar L, Sado PA. 1995. Using micro-emulsion formulations for oral drug delivery oftherapeutic peptides. Int J Pharm 120:27–136.
24. Pearlman R, Bewley TA. 1993. Stability and char-acterization of human growth hormone. In: WangYJ, Pearlman R, editors. Stability and character-isation of protein and peptide drugs: Case his-tories. New York: Plenum. pp 1–57.
25. Manning MC, Patel K, Borchardt RT. 1989. Sta-bility of protein pharmaceuticals. Pharm Res6:903–918.
26. Volkin DB, Middaugh CR. 1992. Protein solubi-lity. In: Ahern TJ, Manning MC, editors. Stabilityof protein pharmaceuticals. Part A: Chemicaland physical pathways of protein degradation.New York: Plenum. pp 109–134.
27. Zale SE, Klibanov AM. 1986. Why does ribonu-clease irreversibly inactivate at high tempera-tures? Biochemistry 25:5432–5443.
28. Johnson OL, Tracy MA. 1999. Peptide and proteindrug delivery. In: Mathiowitz E, editor. Encyclo-pedia of controlled drug delivery 2. New York:Wiley. pp 816–833.
29. Carpenter JF, Crowe JH. 1989. An infrared spec-troscopic study of the interactions of carbohy-drates with dried proteins. Biochemistry 28:3916–3922.
30. Arakawa T, Kita Y, Carpenter JF. 1991. Proteinsolvent interactions in pharmaceutical formula-tions. Pharm Res 8:285–291.
31. Loftsson T, Brewster ME. 1996. Pharmaceuticalapplications of cyclodextrins. 1. Drug solubilisa-tion and stabilization. J Pharm Sci 85:1017–1025.
32. Rajewski RA, Stella VJ. 1996. Pharmaceuticalapplications of cyclodextrins. 2. In vivo drug deliv-ery. J Pharm Sci 85:1142–1169.
33. Charman SA, Mason KL, Charman WN. 1993.Techniques for assessing the effects of pharma-ceutical excipients on the aggregation of porcinegrowth hormone. Pharm Res 10:954–962.
34. Tsai PK, Volkin DB, Dabora JM, Thompson KC,Bruner MW, Gress JO, Matuszewska B, KeoganM, Bondi JV, Middaugh CR. 1993. Formulationdesign of acidic fibroblast growth factor. PharmRes 10:649–659.
35. Cunningham BC, Mulkerrin MG, Wells JA. 1991.Dimerization of human growth hormone by zinc.Science 253:545–548.
36. Hanson MA, Rouan SKE. In: Ahern TJ, ManningMC, editors. 1992. Stability of protein pharmaceu-ticals. Part B: In vivo pathways of degradation and
DOI 10.1002/jps J
strategies for protein stabilization. New York:Plenum. pp 209–233.
37. Hora MS, Rana RK, Smith FW. 1992. Lyophilisedformulations of recombinant tumor necrosis fac-tor. Pharm Res 9:33–36.
38. Powell MF, Sanders LM, Rogerson A, Si V. 1991.Parenteral peptide formulations: Chemical andphysical properties of native luteinizing hor-mone-releasing hormone (LHRH) and hydropho-bic analogues in aqueous solution. Pharm Res8:1258–1263.
39. Ressing ME, Jiskoot W, Talsma H, van Ingen CW,Bwuvery C, Crommelin DJA. 1992. The influenceof sucrose, dextran, and hydroxypropyl-beta-cyclo-dextrin as lyoprotectants for a freeze-dried mouseIgG2a monoclonal antibody(MN12). Pharm Res9:226–270.
40. Zhu G, Hallery SR, Schwendemem SP. 2000. Sta-bilisation of proteins encapsulated in injectablepoly(lactide-co-glycolide) polymers. Nat Biotech18:52–57.
41. Wang W. 1996. Oral protein drug delivery. J DrugTarget 4:195–232.
42. DeBore AG, Hoogdalem EJ, van Breimer DD.1992. Rate controlled rectal peptide drug absorp-tion. Adv Drug Deliv Rev 8:237–251.
43. Ho NFH, Barshun CL, Burton PS, Merkle HP.1992. Mechanistic insights to buccal delivery ofproteinaceous substances. Adv Drug Deliv Rev8:197–235.
44. Cullander C, Guy RH. 1992. Transdermal deliveryof peptides and proteins. Adv Drug Deliv Rev 8:291–329.
45. Hirai S, Yashiki T, Mima H. 1981. Effects ofsurfactants on the nasal absorption of insulin inrats. Int J Pharm 9:165–172.
46. Lee VHL. 1987. Ophthalmic delivery of peptidesand proteins. Pharm Technol 11:26–38.
47. Madara JL, Dharmsathaphorn K. 1985. Occludingjunction structure-function relationships in a cul-tured epithelial monolayer. J Cell Biol 101: 2124–2133.
48. Modi NB. 1994. Pharmacokinetics and pharmaco-dynamics of recombinant proteins and peptides.J Control Release 29:269–281.
49. Zhou XH. 1994. Overcoming enzymatic andabsorption barriers to non-parenterally adminis-tered protein and peptide drugs. J Control Release29:239–252.
50. Deim Z, Deim T, Acarturk F, Erdoan D, Ozoul C,Koksal M. 2005. Rectal and vaginal administra-tion of insulin-chitosan formulations: An experi-mental study in rabbits. J Drug Target 13:563–572.
51. Senel S, Kremer M, Nagy K, Squier C. 2001.Delivery of bioactive peptides and proteins acrossoral (buccal) mucosa. Curr Pharm Biotechnol 2:175–186.
OURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008

2518 SINGH, SINGH, AND LILLARD
52. Walker GF, Langoth N, Bemkop-Schnurch A.2002. Peptidase activity on the surface of theporcine buccal mucosa. Int J Pharm 21:141–147.
53. Kurosaki Y, Kimura T. 2000. Regional variation inoral mucosal drug permeability. Crit Rev TherDrug Carrier Syst 17:467–508.
54. Nagai T. 1988. Drug delivery systems by con-trolled release. Yakugaku Zasshi 108:613–624.
55. Park K, Robinson JR. 1984. Bioadhesive polymersas platforms for oral-controlled drug-delivery:Method to study bioadhesion. lnt J Pharm 19:107–127.
56. Kompella UB, Lee VH. 2001. Delivery systems forpenetration enhancement of peptide and proteindrugs: Design considerations. Adv Drug Deliv Rev46:211–245.
57. Lehr CM, Bouwstra JA, Tukker JJ, Junginger HE.1990. Intestinal transit of bioadhesive micro-spheres in an in situ loop in the rat. A compara-tive study with polymers and blends based onpoly(acrylic acid). J Control Release 13:51–62.
58. Allen A, Cuncliffe WJ, Peerson JP, Sellers LA,Ward R. 1984. Studies on gastrointestinal mucus.Scan J Gastroenterol 19:101–113.
59. Alur HH, Beal JD, Pather SI, Mitra AK, JohnstonTP. 1999. Evaluation of a novel, natural oligosac-charide gum as a sustained-release and mucoad-hesive component of calcitonin buccal tablets.J Pharm Sci 88:1313–1319.
60. Nakane S, Kakumoto M, Yukimatsu K, Chien YW.1996. Oramucosal delivery of LHRH: Pharmaco-kinetic studies of controlled and enhanced trans-mucosal permeation. Pharm Dev Technol 1:251–259.
61. Gutniak MK, Larsson H, Heiber SJ, JuneskansOT, Holst JJ, Ahren B. 1996. Potential therapeuticlevels of glucagon-like peptide I achieved inhumans by a buccal tablet. Diabetes Care 19:843–848.
62. Webber W. 1999. Mucosal drug delivery: Buccal.In: Mathiowitz E, editor. Encyclopedia of con-trolled drug delivery. Wiley: New York. pp. 553–563.
63. Potts RO, Bommannan D, Wong O, Tamada JA,Riviere JE, Monteiro-Riviere NA. In: Sanders LM,Hendren RW, editors. 1997. Transdermal peptidedelivery using electroporation in protein delivery.New York, London: Plenum. pp 213–228.
64. Porter CJ, Davies MC, Davis SS, Illum L.1994. Microparticulate system for site-specifictherapy-bone marrow targeting. In: Domb A, edi-tor. Site specific pharmacotherapy. New York:Wiley. pp. 157–203.
65. Illum L, Davis SS. 1983. Effect of the nonionicsurfactant poloxamer 338 on the fate and deposi-tion of polystyrene microspheres following intra-venous administration. J Pharm Sci 72:1086–1089.
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008
66. Torchilin VP, Papisov MI. 1994. Hypothesis: Whydo PEG coated liposomes circulate so long?J Liposome Res 4:725–739.
67. Murthy RSR. 1997. Biodegradable polymers. In:Jain NK, editor. Controlled and novel drug deliv-ery. New Delhi, India: CBS Publishers and Dis-tributors. pp 27–51.
68. Lewis D, Hi CM, Langer R, editors. 1990. Biode-gradable polymers as drug delivery systems, drugsand pharmaceutical sciences, vol. 45. New York:Marcel Dekker. pp. 1–41.
69. Jeyanthi R, Mehta RC, Thanoo BC, DeLuca PP.1997. Effect of processing parameters on the prop-erties of peptide-containing PLGA microspheres.J Microencapsul 14:163–174.
70. Timpi Ri GN, Reddi AH. editors. 1976. Biochem-istry of collagen. New York: Plenum. pp. 55–78.
71. Qiu Y, Park K. 2001. Environment-sensitivehydrogels for drug delivery. Adv Drug Deliv Rev53:321–339.
72. Goldraich M, Kost J. 1993. Glucose-sensitive poly-meric matrices for controlled drug delivery. ClinMater 13:135–142.
73. Podual K, Doyle FJ III, Peppas NA. 2000.Dynamic behavior of glucose oxidase-containingmicroparticles of poly(ethylene glycol)-graftedcationic hydrogels in an environment of chang-ing pH. Biomaterials 21:1439–1450.
74. Madsen F, Peppas NA. 1999. Complexation graftcopolymer networks: Swelling properties, calciumbinding and proteolytic enzyme inhibition. Bioma-terials 20:1701–1708.
75. Torres-Lugo M, Garcia M, Record R, Peppas NA.2002. Physicochemical behavior and cytotoxiceffects of p(methacrylic acid-g-ethylene glycol)nanospheres for oral delivery of proteins.J Control Release 80:197–205.
76. Lowman AM, Peppas NA. 1997. An analysis of thecomplexation/decomplexation phenomena in graftcopolymer network. Macromolecules 30:4959–4965.
77. Peppas NA, Klier J. 1991. Controlled release byusing poly(methacrylic acid-g-ethylene glycol)hydrogels. J Control Release 16:203–214.
78. Peppas NA, Lowman AM. 1998. Protein deliveryfrom novel bioadhesive complexation hydrogels.In: Frokjaer S, Christrup L, Krogsgaard-LarsenP, editors. Peptide and protein delivery, AlfredBenzon symposium, vol. 43. Copenhagen: Munks-gaard. pp. 206–216.
79. Panyam J, Labhasetwar V. 2003. Biodegradablenanoparticles for drug and gene delivery tocells and tissue. Adv Drug Deliv Rev 55:329–347.
80. Desai MP, Labhasetwar V, Walter E, Levy RJ,Amidon GL. 1997. The mechanism of uptake ofbiodegradable microparticles in Caco-2 cells is sizedependent. Pharm Res 14:1568–1573.
DOI 10.1002/jps

ORAL DELIVERY OF BIOLOGICALS 2519
81. Desai MP, Labhasetwar V, Amidon GL, Levy RJ.1996. Gastrointestinal uptake of biodegradablemicroparticles: Effect of particle size. Pharm Res13:1838–1845.
82. Redhead HM, Davis SS, Illum L. 2001. Drugdelivery in poly(lactide-co-glycolide) nanoparticlessurface modified with poloxamer 407 and poloxa-mine 908: In vitro characterisation and in vivoevaluation. J Control Release 70:353–363.
83. Dunne M, Corrigan I, Ramtoola Z. 2000. Influenceof particle size and dissolution conditions on thedegradation properties of polylactide-co-glycolideparticles. Biomaterials 21:1659–1668.
84. Panyam J, Dali MM, Sahoo SK, Ma W, Chakra-varthi SS, Amidon GL, Levy RJ, Labhasetwar V.2003. Polymer degradation and in vitro release of amodel protein from poly(D,L-lactide-co-glycolide)nano- and microparticles. J Control Release 92:173–187.
85. Jani P, Halbert GW, Langridge J, Florence AT.1989. The uptake and translocation of latex nano-spheres and microspheres after oral administra-tion to rats. J Pharm Pharmacol 41:809–812.
86. Jani P, Halbert GW, Langridge J, Florence AT.1990. Nanoparticle uptake by the rat gastrointest-inal mucosa: Quantitation and particle size depen-dency. J Pharm Pharmacol 42:821–826.
87. Jepson MA, Simmons NL, O’Hagan DT, Hirst BH.1993. Comparison of poly(DL-lactide-co-glycolide)and polystyrene microsphere targeting to intest-inal M cells. J Drug Target 1:245–249.
88. Norris S, Crosbie O, McEntee G, Traynor O, NolanN, McCann S, Hegarty J. 1997. Orthotopic livertransplantation for veno-occlusive disease compli-cating autologous bone marrow transplantation.Transplantation 63:1521–1524.
89. Mathiowitz E, Jacob JS, Jong YS, Carino GP,Chickering DE, Chaturvedi P, Santos CA, Vijayar-aghavan K, Montgomery S, Bassett M, Morrell C.1997. Biologically erodable microspheres as poten-tial oral drug delivery systems. Nature 386:410–414.
90. Brigger I, Dubernet C, Couvreur P. 2002. Nano-particles in cancer therapy and diagnosis. AdvDrug Deliv Rev 54:631–651.
91. Bhadra D, Bhadra S, Jain P, Jain NK. 2002.Pegnology: A review of PEG-ylated systems. Phar-mazie 57:5–29.
92. Olivier JC. 2005. Drug transport to brain withtargeted nanoparticles. NeuroRx 2:108–119.
93. Yamamato A, Taniguchi T, Rikyuu K, Tsuji T,Fujita T, Murakami M, Muranishi J. 1994. Effectsof various protease inhibitors on the intestinalabsorption and degradation of insulin in rats.Pharm Res 11:1496–1500.
94. Fuiji S, Yakohama T, Ikegaya K, Sato E, Yohoo N.1991. Promoting effect of the new chymotrypsininhibitor FK-448 on the intestinal absorption of
DOI 10.1002/jps J
insulin in rats and dogs. J Control Release 13:213–223.
95. Foradori A, Mezzano S, Videla C, Pefaur J, ElbergA. 1998. Modification of the pharmacokinetics ofcyclosporine A and metabolites by the concomitantuse of Neoral and diltiazem or ketoconazol instable adult kidney transplants. Transplant Proc30:1685–1687.
96. Su SF, Amidon GL, Lee HJ. 2002. Intestinal meta-bolism and absorption of cholecystokinin analogsin rats. Biochem Biophys Res Commun 292:632–638.
97. Lee HJ, Amidon GL. 2002. The effect of enzymeinhibitor and absorption site following [D-ala2, D-leu5]enkephalin oral administration in rats. Bio-pharm Drug Dispos 23:131–141.
98. Lee HJ, Lee MG. 2002. Controlling absorption site,metabolism, and membrane permeability in theintestine to develop strategies for protein drug oraldelivery. Abstract submitted for the 16th annualmeeting of the AAP5 to be held in Toronto, Canadain November, 2002.
99. Bemkop-Schnurch A, Schwarz GH, Kratzel M.1997. Modified mucoadhesive polymers for theperoral administration of mainly elastase degrad-able therapeutic (poly)peptides. J Control Release47:113–121.
100. Bernkop-Schnurch A. 2000. Chitosan and its deri-vatives: Potential excipients for peroral peptidedelivery systems. Int J Pharm 194:1–13.
101. Kratzel M, Hiessbock R, Bernkop-Schnurch A.1998. Auxiliary agents for the peroral administra-tion of peptide and protein drugs: Synthesis andevaluation of novel pepstatin analogues. J MedChem 41:2339–2344.
102. Leone-Bay A, Paton DR, Weidner JJ. 2000. Thedevelopment of delivery agents that facilitate theoral absorption of macromolecular drugs. Med ResRev 20:169–186.
103. Leone-Bay A, Ho KK, Agarwal R, Baughman RA,Chaudhary K, DeMorin F, Genoble L, McInnes C,Lercara C, Milstein S, O’Toole D, Sarubbi D, Var-iano B, Paton DR. 1996. 4-[4-[(2-Hydroxybenzoy-l)amino]phenyl]butyric acid as a novel oraldelivery agent for recombinant human growthhormone. J Med Chem 39:2571–2578.
104. Stoll BR, Leipold HR, Milstein S, Edwards DA.2000. A mechanistic analysis of carrier-mediatedoral delivery of protein therapeutics. J ControlRelease 64:217–228.
105. Russell-Jones GJ, Arthur L, Walker H. 1999. Vita-min B12-mediated transport of nanoparticlesacross Caco-2 cells. Int J Pharm 179:247–255.
106. Russell-Jones GJ. 1998. Use of vitamin B12 con-jugates to deliver protein drugs by the oral route.Crit Rev Ther Drug Carrier Syst 15:557–586.
107. Jung T, Kamm W, Breitenbach A, Kaiserling E,Xiao JX, Kissel T. 2000. Biodegradable nanopar-
OURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008

2520 SINGH, SINGH, AND LILLARD
ticles for oral delivery of peptides: Is there a rolefor polymers to affect mucosal uptake? Eur JPharm Biopharm 50:147–160.
108. Samanen J, Wilson G, Smith PL, Lee CP, Bondi-nell W, Ku T, Rhodes G, Nichols A. 1996. Chemicalapproaches to improve the oral bioavailability ofpeptidergic molecules. J Pharm Pharmacol 48:119–135.
109. Woodley JF. 1994. Enzymatic barriers for GI pep-tide and protein delivery. Crit Rev Ther DrugCarrier Syst 11:61–95.
110. Keljo DJ, Hamilton JR. 1983. Quantitative deter-mination of macromolecular transport rate acrossintestinal Peyer’s patches. Am J Physiol 244:G637–G644.
111. Wacher VJ, Salphati L, Benet LZ. 2001. Activesecretion and enterocytic drug metabolism bar-riers to drug absorption. Adv Drug Deliv Rev 46:89–102.
112. Tsuji A, Tamai I. 1996. Carrier-mediated intest-inal transport of drugs. Pharm Res 13:963–977.
113. Sakuma S, Hayashi M, Akashi M. 2001. Design ofnanoparticles composed of graft copolymers fororal peptide delivery. Adv Drug Deliv Rev 47:21–37.
114. Schipper NG, Varum KM, Stenberg P, Ocklind G,Lennernas H, Artursson P. 1999. Chitosans asabsorption enhancers of poorly absorbable drugs.3: Influence of mucus on absorption enhancement.Eur J Pharm Sci 8:335–343.
115. Bjork E, Isaksson U, Edman P, Artursson P. 1995.Starch microspheres induce pulsatile delivery ofdrugs and peptides across the epithelial barrier byreversible separation of the tight junctions. J DrugTarget 2:501–507.
116. Florence AT, Hussain N. 2001. Transcytosis ofnanoparticle and dendrimer delivery systems:Evolving vistas. Adv Drug Deliv Rev 50:69–89.
117. Kreuter J. 1991. Peroral administration of nano-particles. Adv Drug Deliv Rev 7:71–86.
118. DiBiase MD, Morrel EM. 1997. Oral delivery ofmicroencapsulated proteins. Pharm Biotechnol10:255–288.
119. Takeuchi H, Yamamoto H, Kawashima Y. 2001.Mucoadhesive nanoparticulate systems for pep-tide drug delivery. Adv Drug Deliv Rev 47:39–54.
120. Quintanar-Guerrero D, Allemann E, Fessi H,Doelker E. 1998. Preparation techniques andmechanisms of formation of biodegradable nano-particles from preformed polymers. Drug Dev IndPharm 24:1113–1128.
121. McGhee JR, Mestecky J, Dertzbaugh MT, EldridgeJH, Hirasawa M, Kiyono H. 1992. The mucosalimmune system: From fundamental concepts tovaccine development. Vaccine 10:75–88.
122. Mestecky J, McGhee JR. 1987. Immunoglobulin A(IgA): Molecular and cellular interactions involved
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008
in IgA biosynthesis and immune response. AdvImmunol 40:153–245.
123. Illum L, Jabbal-Gill I, Hinchcliffe M, Fisher AN,Davis SS. 2001. Chitosan as a novel nasal deliverysystem for vaccines. Adv Drug Deliv Rev 51:81–96.
124. Regnstrom K, Ragnarsson EGE, Koping-HoggardM, Torstensson E, Nyblom H, Artursson P. 2003.PEI—A potent, but not harmless, mucosalimmuno-stimulator of mixed T-helper cell re-sponse and FasL-mediated cell death in mice.Gene Ther 10:1575–1583.
125. Eldridge JH, Meulbroek JA, Staas JK, TiceTR, Gilley RM. 1989. Vaccine-containing bio-degradable microspheres specifically enter thegut-associated lymphoid tissue following oraladministration and induce a disseminated muco-sal immune response. Adv Exp Med Biol 251:191–202.
126. Jones DH, McBride BW, Thornton C, O’Hagan DT,Robinson A, Farrar GH. 1996. Orally adminis-tered microencapsulated Bordetella pertussis fim-briae protect mice from B. pertussis respiratoryinfection. Infect Immun 64:489–494.
127. Kofler N, Ruedl C, Rieser C, Wick G, Wolf H. 1997.Oral immunization with poly-(D,L-lactide-co-gly-colide) and poly-(L-lactic acid) microspheres con-taining pneumotropic bacterial antigens. Int ArchAllergy Immunol 113:424–431.
128. Challacombe SJ, Rahman D, O’Hagan DT. 1997.Salivary, gut, vaginal and nasal antibodyresponses after oral immunization with biode-gradable microparticles. Vaccine 15:169–175.
129. Esparza I, Kissel T. 1992. Parameters affectingthe immunogenicity of microencapsulated tetanustoxoid. Vaccine 10:714–720.
130. Jung T, Kamm W, Breitenbach A, Hungerer KD,Hundt E, Kissel T. 2001. Tetanus toxoid loadednanoparticles from sulfobutylated poly(vinyl alco-hol)-graft-poly(lactide-co-glycolide): Evaluation ofantibody response after oral and nasal applicationin mice. Pharm Res 18:352–360.
131. Kim SY, Doh HJ, Jang MH, Ha YJ, Chung SI,Park HJ. 1999. Oral immunization with Helico-bacter pylori-loaded poly(D, L-lactide-co-glycolide)nanoparticles. Helicobacter 4:33–39.
132. Maloy KJ, Donachie AM, O’Hagan DT, MowatAM. 1994. Induction of mucosal and systemicimmune responses by immunization with ovalbu-min entrapped in poly(lactide-co-glycolide) micro-particles. Immunology 81:661–667.
133. Shakweh M, Ponchel G, Fattal E. 2004. Particleuptake by Peyer’s patches: A pathway for drug andvaccine delivery. Expert Opin Drug Deliv 1:141–163.
134. Gutierro I, Hernandez RM, Igartua M, Gascon AR,Pedraz JL. 2002. Influence of dose and immuniza-tion route on the serum IgG antibody response to
DOI 10.1002/jps

ORAL DELIVERY OF BIOLOGICALS 2521
BSA loaded PLGA microspheres. Vaccine 20:2181–2190.
135. Fattal E, Pecquet S, Couvreur P, Andremont A.2002. Biodegradable microparticles for the muco-sal delivery of antibacterial and dietary antigens.Int J Pharm 242:15–24.
136. Conway MA, Madrigal-Estebas L, McClean S,Brayden DJ, Mills KH. 2001. Protection againstBordetella pertussis infection following parenteralor oral immunization with antigens entrapped inbiodegradable particles: Effect of formulation androute of immunization on induction of Th1 andTh2 cells. Vaccine 19:1940–1950.
137. Brayden DJ, Baird AW. 2004. Apical membranereceptors on intestinal M cells: Potential targetsfor vaccine delivery. Adv Drug Deliv Rev 56:721–726.
138. Tacket CO, Reid RH, Boedeker EC, Losonsky G,Nataro JP, Bhagat H, Edelman R. 1994. Enteralimmunization and challenge of volunteers givenenterotoxigenic E. coli CFA/II encapsulated inbiodegradable microspheres. Vaccine 12:1270–1274.
139. Katz DE, DeLorimier AJ, Wolf MK, Hall ER,Cassels FJ, van Hamont JE, Newcomer RL, Dava-chi MA, Taylor DN, McQueen CE. 2003. Oralimmunization of adult volunteers with microen-capsulated enterotoxigenic Escherichia coli(ETEC) CS6 antigen. Vaccine 21:341–346.
140. Hunter SK, Andracki ME, Kreig AM. 2001. Biode-gradable microspheres containing group B Strep-tococcus vaccine: Immune response in mice. Am JObstet Gynecol 185:1174–1179.
141. Gupta RK, Chang AC, Siber GR. 1998. Biodeg-radable polymer microspheres as vaccine adju-vants and delivery systems. Dev Biol Stand 92:63–78.
142. Johansen P, Moon L, Tamber H, Merkle HP,Gander B, Sesardic D. 1999. Immunogenicity ofsingle-dose Diphtheria vaccines based on PLA/PLGA microspheres in guinea pigs. Vaccine 18:209–215.
143. Singh J, Pandit S, Bramwell VW, Alpar HO. 2006.Diphtheria toxoid loaded poly-(epsilon-caprolac-tone) nanoparticles as mucosal vaccine deliverysystems. Methods 38:96–105.
144. Yeh MK, Liu YT, Chen JL, Chiang CH. 2002. Oralimmunogenicity of the inactivated Vibrio choleraewhole cell vaccine encapsulated in biodegradablemicroparticles. J Control Release 82:237–247.
145. Roy K, Mao HQ, Huang SK, Leong KW. 1999. Oralgene delivery with chitosan-DNA nanoparticlesgenerates immunologic protection in a murinemodel of peanut allergy. Nat Med 5:387–391.
146. Bivas-Benita M, Laloup M, Versteyhe S, Dewit J,De Braekeleer J, Jongert E, Borchard G. 2003.Generation of Toxoplasma gondii GRA1 proteinand DNA vaccine loaded chitosan particles: Pre-
DOI 10.1002/jps J
paration, characterization, and preliminary invivo studies. Int J Pharm 266:17–27.
147. Chen SC, Jones DH, Fynan EF, Farrar GH, CleggJC, Greenberg HB, Herrmann JE. 1998. Protec-tive immunity induced by oral immunization witha rotavirus DNA vaccine encapsulated in micro-particles. J Virol 72:5757–5761.
148. Herrmann JE, Chen SC, Jones DH, Tinsley-BownA, Fynan EF, Greenberg HB, Farrar GH. 1999.Immune responses and protection obtained by oralimmunization with rotavirus VP4 and VP7 DNAvaccines encapsulated in microparticles. Virology259:148–153.
149. Porporatto C, Bianco ID, Correa SG. 2005. Localand systemic activity of the polysaccharide chit-osan at lymphoid tissues after oral administration.J Leukoc Biol 78:62–69.
150. Foster N, Hirst BH. 2005. Exploiting receptorbiology for oral vaccination with biodegradableparticulates. Adv Drug Deliv Rev 57:431–450.
151. Weiner HL, Friedman A, Miller A, Khoury SJ, al-Sabbagh A, Santos L, Sayegh M, Nussenblatt RB,Trentham DE, Hafler DA. 1994. Oral tolerance:Immunologic mechanisms and treatment of ani-mal and human organ-specific autoimmune dis-eases by oral administration of autoantigens.Annu Rev Immunol 12:809–837.
152. Garside P, Mowat AM. 2001. Oral tolerance.Semin Immunol 13:177–185.
153. Mowat AM. 2003. Anatomical basis of toleranceand immunity to intestinal antigens. Nature RevImmunol 3:331–341.
154. Masuda K, Horie K, Suzuki R, Yoshikawa T,Hirano K. 2003. Oral-antigen delivery via awater-in-oil emulsion system modulates the bal-ance of the Th1/Th2 type response in oral toler-ance. Pharm Res 20:130–134.
155. Trentham DE, Dynesius-Trentham RA, Orav EJ,Combitchi D, Lorenzo C, Sewell KL, Hafler DA,Weiner HL. 1993. Effects of oral administration oftype II collagen on rheumatoid arthritis. Science261:1727–1730.
156. Kim WU, Lee WK, Ryoo JW, Kim SH, Kim J, YounJ, Min SY, Bae EY, Hwang SY, Park SH, Cho CS,Park JS, Kim HY. 2002. Suppression of collagen-induced arthritis by single administration of poly(-lactic-co-glycolic acid) nanoparticles entrappingtype II collagen: A novel treatment strategy forinduction of oral tolerance. Arthritis Rheum46:1109–1120.
157. Morishita M, Lowman AM, Takayama K, Nagai T,Peppas NA. 2002. Elucidation of the mechanism ofincorporation of insulin in controlled release sys-tems based on complexation polymers. J ControlRelease 81:25–32.
158. Lowman AM, Morishita M, Peppas NA, Nagai T.1998. Novel bioadhesive complexation networksfor oral protein drug delivery. In: McCulloch I,
OURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008

2522 SINGH, SINGH, AND LILLARD
Shalaby SW, editors. Materials for controlledrelease applications. Washington, DC: AmericanChemical Society. pp 156–164.
159. Lowman AM, Morishita M, Kajita M, Nagai T,Peppas NA. 1999. Oral delivery of insulin usingpH-responsive complexation gels. J Pharm Sci88:933–937.
160. Damge C, Michel C, Aprahamian M, Couvreur P.1988. New approach for oral administration ofinsulin with polyalkylcyanoacrylate nanocapsulesas drug carrier. Diabetes 37:246–251.
161. Pinto-Alphandary H, Aboubakar M, Jaillard D,Couvreur P, Vauthier C. 2003. Visualization ofinsulin-loaded nanocapsules: In vitro and in vivostudies after oral administration to rats. PharmRes 20:1071–1084.
162. Cournarie F, Auchere D, Chevenne D, Lacour B,Seiller M, Vauthier C. 2002. Absorption and effi-ciency of insulin after oral administration of insu-lin-loaded nanocapsules in diabetic rats. Int JPharm 242:325–328.
163. Carino GP, Jacob JS, Mathiowitz E. 2000. Nano-sphere based oral insulin delivery. J ControlRelease 65:261–269.
164. Ma Z, Lim TM, Lim LY. 2005. Pharmacolog-ical activity of peroral chitosan-insulin nano-particles in diabetic rats. Int J Pharm 293:271–280.
165. Pan Y, Li YJ, Zhao H-Y, Zheng JM, Xu H, Wei G,Hao JS, Cui FD. 2002. Bioadhesive polysaccharidein protein delivery system: Chitosan nanoparticlesimprove the intestinal absorption of insulinin vivo. Int J Pharm 249:139–147.
166. Yeh MK. 2000. The stability of insulin in bio-degradable microparticles based on blends oflactide polymers and polyethylene glycol. J Micro-encapsul 7:743–756.
167. Clement S, Dandona P, Still JG, Kosutic G. 2004.Oral modified insulin (HIM2) in patients with type1 diabetes mellitus: Results from a phase I/IIclinical trial. Metabolism 53:54–58.
168. Xia CQ, Wang J, Shen WC. 2000. Hypoglycemiceffect of insulin-transferrin conjugate in strepto-zotocin-induced diabetic rats. J Pharmacol ExpTher 295:594–600.
169. Bernkop-Schnurch A. 1998. The use of inhibitoryagents to overcome the enzymatic barrier to pero-rally administered therapeutic peptides and pro-teins. J Control Release 52:1–16.
170. Fasano A, Uzzau S. 1997. Modulation of intestinaltight junctions by Zonula occludens toxin permitsenteral administration of insulin and other macro-molecules in an animal model. J Clin Invest99:1158–1164.
171. Wu ZH, Ping QN, Wei Y, Lai JM. 2004. Hypogly-cemic efficacy of chitosan-coated insulin liposomesafter oral administration in mice. Acta PharmacolSin 25:966–972.
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008
172. Sajeesh S, Sharma CP. 2004. Poly methacrylicacid-alginate semi-IPN microparticles for oraldelivery of insulin: A preliminary investigation.J Biomater Appl 19:35–45.
173. Prego C, Garcia M, Torres D, Alonso MJ. 2005.Transmucosal macromolecular drug delivery.J Control Release 101:151–162.
174. El-Shabouri MH. 2002. Positively charged nano-particles for improving the oral bioavailability ofcyclosporin-A. Int J Pharm 249:101–108.
175. Chen J, Yang WL, Li G, Qian J, Xue JL, Fu SK, LuDR. 2004. Transfection of mEpo gene to intestinalepithelium in vivo mediated by oral delivery ofchitosan-DNA nanoparticles. World J Gastroen-terol 10:112–116.
176. Das D, Lin S. 2005. Double-coated poly (butylcy-nanoacrylate) nanoparticulate delivery systemsfor brain targeting of dalargin via oral adminis-tration. J Pharm Sci 94:1343–1353.
177. Prego C, Torres D, Alonso MJ. 2006. Chitosannanocapsules as carriers for oral peptide delivery:Effect of chitosan molecular weight and type of salton the in vitro behaviour and in vivo effectiveness.J Nanosci Nanotechnol 6:2921–2928.
178. Genta I, Perugini P, Pavanetto F, Maculotti K,Modena T, Casado B, Lupi A, Iadarola P, Conti B.2001. Enzyme loaded biodegradable microspheresin vitro ex vivo evaluation. J Control Release77:287–295.
179. Zhou S, Deng X, He S, Li X, Jia W, Wei D, Zhang Z,Ma J. 2002. Study on biodegradable microspherescontaining recombinant interferon-alpha-2a.J Pharm Pharmacol 54:1287–1292.
180. Guarino A, Canani RB, Russo S, Albano F, CananiMB, Ruggeri FM, Donelli G, Rubino A. 1994. Oralimmunoglobulins for treatment of acute rotaviralgastroenteritis. Pediatrics 93:12–16.
181. Hilpert H, Brussow H, Mietens C, Sidoti J, LernerL, Werchau H. 1987. Use of bovine milk concen-trate containing antibody to rotavirus to treatrotavirus gastroenteritis in infants. J Infect Dis156:158–166.
182. Nord J, Ma P, DiJohn D, Tzipori S, Tacket CO.1990. Treatment with bovine hyperimmune colos-trum of cryptosporidial diarrhea in AIDS patients.Aids 4:581–584.
183. Ungar BL, Ward DJ, Fayer R, Quinn CA. 1990.Cessation of Cryptosporidium-associated diarrheain an acquired immunodeficiency syndromepatient after treatment with hyperimmune bovinecolostrum. Gastroenterology 98:486–489.
184. Losonsky GA, Johnson JP, Winkelstein JA,Yolken RH. 1985. Oral administration of humanserum immunoglobulin in immunodeficient patientswith viral gastroenteritis. A pharmacokinetic andfunctional analysis. J Clin Invest 76:2362–2367.
185. Copelan EA, Avalos BR, Kapoor N, Bechtel T,Tutschka PJ. 1992. Alternate applications of
DOI 10.1002/jps

ORAL DELIVERY OF BIOLOGICALS 2523
immunoglobulin following bone marrow trans-plantation. Semin Hematol 29:96–99.
186. Copelan EA, Bechtel TP, Klein JP, Klein JL,Tutschka P, Kapoor N, Featheringham NC, AvalosBR. 1994. Controlled trial of orally administeredimmunoglobulin following bone marrow transplan-tation. Bone Marrow Transplant 13:87–91.
187. Marchant RE, Miller KM, Anderson JM. 1984.In vivo leukocyte interactions with biomer.J Biomed Mater Res 18:1169–1174.
188. Sewell WR, Wiland J, Craver BN. 1955. A newmethod of comparing sutures of ovine catgut withsutures of bovine catgut in three species. SurgGynecol Obstet 100:483–494.
189. Gourlay SJ, Rice RM, Hegyeli AF, Wade CWR,Dillon JG, Jaffe H, Kulkarni RK. 1978. Biocom-patibility testing of polymers: In vivo implantationstudies. J Biomed Mater Res 12:219–232.
DOI 10.1002/jps J
190. Bronaugh RL, Maibach HI. Evaluation of skinirritation: Correlation between animals andhumans. In: Klingman AM, Leyden JJ, editors.1982. Safety and efficacy of topical drugs andcosmetics. New York: Grune and Stratton. p. 51.
191. Foster GV. 1982. Comparative acute toxicitystudies in female rats with five synthetic muskchemicals. In: Klingman AM, Leyden JJ, editors.Safety and efficacy of topical drugs and cosmetics.New York: Grune and Stratton. p. 99.
192. Autian J. 1983. Toxicological aspects of implanta-ble plastic used in medical and paramedicalexperiments. In: Williams DF, editor. Fundamen-tal aspects of biocompatibility. Boca Raton, FL:CRC Press. p 63.
193. Imai Y, Watanbe A, Masuhara E. 1983. Structure-biocompatibility relationship of condensationpolymers. J Biomed Mater Res 17:905–912.
OURNAL OF PHARMACEUTICAL SCIENCES, VOL. 97, NO. 7, JULY 2008