
Life Cycle Analysis of the Bioethanol Production from Food ...
Upload
independentCategory
view
3download
0
Review
Recent Advances in Production of Bioethanolfrom Lignocellulosic Biomass
Ethanol is considered the most potential next generation automotive fuel becauseit is carbon-neutral and could be produced from renewable resources like ligno-cellulosic biomass. There are some technological barriers such as pretreatment,saccharification of cellulose and hemicellulose matrix, and simultaneous fermen-tation of hexose and pentose sugars which needs to be addressed for efficient con-version of lignocellulosic biomass to bioethanol. This paper reviews the variousprocess options and kinetic models adopted towards resolving the technologicalchallenges to develop a low-cost commercial process.
Keywords: Acid hydrolysis, Bioethanol, Enzymatic hydrolysis, Lignocellulosic biomass, SSF,Thermophiles
Received: September 03, 2008; revised: October 20, 2008; accepted: October 22, 2008
DOI: 10.1002/ceat.200800442
1 Introduction
Ethanol has attracted worldwide attention because of its po-tential use as an alternative automotive fuel. It has immenseimportance for countries such as India which depends heavilyon import of crude oil, spending a huge sum of its annualbudget. There are numerous advantages of blending ethanolwith gasoline that emits lower quantities of carbon mono ox-ide (CO), nitrogen oxides (NOx), and hydrocarbon after com-bustion compared to that of gasoline alone because ethanolacts as oxidizing agent [1–3].
The traditional feed stocks like molasses, sugarcane juice,corn etc. are used for ethanol production but have social andeconomical barriers. Apart from these feed stocks, lignocellu-losic biomass, which is the most abundant on earth [4], is analternative feed stock for bioethanol production. This biomassincluding forest residues such as wood; agricultural residuessuch as sugarcane bagasse, corn cob, corn stover, wheat andrice straw; industrial residues such as pulp and paper process-ing waste and municipal solid wastes; energy crops such asswitch grass are the most potential feed stocks for fuel ethanol[3–7].
Lignocellulosic biomasses comprise cellulose (20–50 %),hemicellulose (20–35 %), polyphenolic lignin (10–35 %), andother components [2, 3, 7]. The biological process for convert-ing lignocellulose to fuel ethanol requires: (1) delignificationto liberate cellulose and hemicellulose; (2) depolymerization of
carbohydrate polymers to produce free sugars; and (3) fermen-tation of mixed hexose and pentose sugars to produce ethanol[8]. Hydrolysis of these polysaccharides (cellulose and hemicel-lulose) is usually accomplished by acid and/or enzymatic treat-ment. The utilization of both cellulose and hemicellulosicsugars like hexose, pentose etc. present in a typical biomass hy-drolysate is essential for the economical production of ethanol[9, 10]. Therefore, microorganisms which are able to fermentboth glucose and xylose are required for an efficient bioconver-sion of biomass to ethanol [11].
Bioethanol is produced by fermentation of monomeric su-gars mostly by mesophiles at 25–37 °C. These mesophiles havecertain limitations in fermenting pentose sugars producedfrom lignocellulosic biomass. However, bioethanol productionfrom lignocellulosic biomass by thermophiles has certain ad-vantages over mesophiles which could be exploited for ethanolproduction. Solvent tolerance, energy savings through reducedcooling costs, higher saccharification and fermentation rates,continuous ethanol removal and the reduced risk of contami-nation have stimulated the search for thermophilic or thermo-tolerant yeasts [12]. Less energy is required for mixing andproduct recovery in thermophilic fermentations because oflower viscosity, surface tension, higher vapor pressure, and in-creased solubility of organic compounds [8].
In spite of several breakthroughs reported on bioethanolproduction from lignocellulosic feed stocks yet the price of cel-lulosic ethanol remains around at $ 2.65 per gallon [13] due toseveral constraints such as feed stock cost, cost of hydrolysis,detoxification cost, ethanol recovery cost etc. (Tab. 1). Theconversion of by-products to produce value-added chemicalssuch as xylitol from xylose, methyl fuorate from furfural, andplastic from hydroxylmethylfurfural may further reduce thecost of ethanol production.
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim http://www.cet-journal.com
Sachin Kumar1,3
Surendra P. Singh2
Indra M. Mishra3
Dilip K. Adhikari1
1 Biotechnology Area, IndianInstitute of Petroleum,Dehradun, India.
2 Department of PaperTechnology, Indian Institute ofTechnology, Roorkee,Saharanpur Campus, India.
3 Department of ChemicalEngineering, Indian Instituteof Technology, Roorkee, India.
–Correspondence: Dr. Dilip K. Adhikari ([email protected]), Biotech-nology Area, Indian Institute of Petroleum, Dehradun – 248 005, India.
Chem. Eng. Technol. 2009, 32, No. 4, 517–526 517

In the present paper, the current status of ethanol produc-tion using lignocellulosic raw materials is reviewed. Thisencompasses pretreatment required for enzymatic and acidsaccharification of different types of biomass, fermentation bymesophiles and thermophiles, and improvement of the ethanolfermentation process for lignocellulosic bioethanol produc-tion.
2 Saccharification of LignocellulosicBiomass
Acid hydrolysis and enzymatic hydrolysis are two basic meth-ods for saccharification of lignocellulosic biomass. Acid hydro-lysis is one of the oldest methods used in saccharification oflignocellulosic biomass in which sulfuric acid is most com-monly used since it is usually the least expensive acid.
2.1 Acid Hydrolysis
Biomass can be hydrolyzed using different acids to producexylose, arabinose, glucose, and acetic acid by cleavage of theb-1,4 linkage of glucose or xylose monomers, acetyl groups,and other products in cellulose and hemicellulose componentsof biomass. The overall fermentable sugar available by acidhydrolysis may be 90 % of the theoretical value of the sugarpresent in cellulosic biomass [14, 15].
Dilute acid processes are conducted under high tempera-tures of 120 to 200 °C and high pressures of 15 psi to 75 psi,and have reaction times in the range of 30 min to 2 h bycontinuous processes [2, 4]. In dilute acid hydrolysis, thefeed stock is generally pressed before pretreatment either indewatering presses or in compression screw feeders found inseveral commercially available continuous hydrolysis reactors[2, 4].
The concentrated acid processes havebeen somewhat more successful, producinghigher yields of sugar. These processes typ-ically involve the use of 60 to 90 % sulfuricacid, mild temperatures, and moderatepressures created by pumping materialsfrom one vessel to another vessel for effec-tive hydrolysis. The primary advantage ofthe concentrated acid process is the highsugar recovery efficiency, which can be onthe order of > 90 % for both xylose andglucose sugars [2]. The concentrated aciddisrupts the hydrogen bonding betweencellulose chains, converting it to a com-pletely amorphous state. Once cellulosehas been decrystallized, it forms a homoge-neous gel with the acid [16].
Farone and Cuzens [16] claimed Arke-nol’s process in which the biomass was de-crystallized by 70–77 % sulfuric acid at50 °C followed by adding water to dilutethe acid to 20–30 %, and heating at 100 °C
for 1 h resulted in the release of sugars. The residual solidswere subjected to a second hydrolysis step. At least 98 % sugarwas recovered by a chromatographic separation technique withless than 3 % loss of acid which was recycled for the hydrolysisstep [17].
Some researchers used nitric acid and phosphoric acid atvarying concentrations, reaction time, and temperature to hy-drolyze the sugar cane bagasse [18, 19]. Hydrochloric acid wasfound to be less effective for degradation of xylose than sulfu-ric acid [14].
Acid hydrolysis processes have several disadvantages due toformation of toxic compounds, such as furfural, hydroxyl-methylfurfural, acetic acid, formic acid, levulinic acid etc.,which inhibit the fermentation. Removal of these compoundsincreases the additional costs. The use of lime to neutralizeacid has the disadvantage of significant loss of sugar in thegypsum. However, such processes could be replaced by highlyeconomical chromatographic separations with acid recycling.
2.1.1 Kinetics of Acid Hydrolysis of Lignocellulosic Biomass
The kinetic model was developed by Saeman [20] for cellulosehydrolysis by sulfuric acid involving two consecutive first-or-der reactions:
Cellulose → Glucose → Decomposition products (1)
Some researchers applied this model to other polysaccha-rides, such as xylan, mannan, arabinan etc., which can be gen-eralized as [18, 19]:
Polymers ��k1Monomers ��k2
Decomposition products (2)
where k1 and k2 are the first-order reaction rate constants formonomer release and decomposition, respectively, both having
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim http://www.cet-journal.com
Table 1. Costs of ethanol production from corn and cellulosic feed stock (currently andby 2010-12) targeted by DOE [13].
Corn ethanol cost$ per gallon
Cellulosic ethanolcost
currently$ per gallon
Cellulosic ethanolcost
2010-12$ per gallon
Feed stock 1.17(Available at $3.22
per bushel)
1.00(Available at $60 per
dry ton)
0.33(Available at $30 per
dry ton)
Enzymes 0.04 0.40 0.10
Other costs1) 0.62 0.80 0.22
Capital costs 0.20 0.55 0.54
Total costs 2.03 2.75 1.19
By- product –0.38 –0.10 –0.09
Net costs 1.65 2.65 1.10
1) includes preprocessing, fermentation, labor costs.
518 Sachin Kumar et al. Chem. Eng. Technol. 2009, 32, No. 4, 517–526

units of reciprocal time. The polymers can be cellulose, hemi-cellulose; monomers can be glucose, xylose, arabinose etc., anddecomposition products can be furfural, hydroxylmethylfur-fural, formic acid, levulinic acid, etc. Solving the differentialequations for an isothermal batch reactor and assuming thatthe initial monomer concentration is negligible; the followingequation is obtained for the concentration of monomers:
M � P0k1
k2 � k1e�k1t � e�k2t� �
(3)
where Po is the initial concentration of polymers and M is theconcentration of monomers released at reaction time t.
Assuming the reaction rate constant, k1, to have Arrhenius-type temperature dependence [21]:
k1 � A1 exp � E1
RT1
� �(4)
where E1 is the activation energy (kJ mol–1), R = 8.3143 · 10–3
(kJ mol–1 K–1), T1 is the temperature (K), and A1 is the pre-ex-ponential factor (min–1).
Lignocellulosic biomass can be categorized in a fast fractionand slow fraction based on the rateof hydrolysis of hemicellulose andcellulose components [14, 19]. Ro-dríguez-Chong et al. [18] and Ga-mez et al. [19] evaluated the two-fraction model for such type ofpolymers, where one fraction wasconsidered to be a susceptible orfast fraction and the other a lesssusceptible or slow fraction, andfound that this model fitted better.The two fractions are related to aparameter a, the mass fraction ofthe susceptible polymer in the rawmaterial. The simplest case for thetwo-fraction model is when the lesssusceptible fraction does not reactand always remains in the solidphase. For this condition, the ki-netic equation giving the concen-tration of monomers is [14, 19]:
M � P0 ak1
k2 � k1e�k1t � e�k2t� �
(5)
2.2 Enzymatic Hydrolysis
Hydrolysis of cellulose to glucosein aqueous media catalyzed by thecellulase enzyme suffers from slowreaction rates due to the highlycrystalline structure of cellulosewhich makes the penetration of en-zymes to the active sites very diffi-
cult [22]. This necessitates a pretreatment to break the crystal-line structure of the lignocellulose and removal of the lignin toexpose the cellulose and hemicellulose molecules to enzymaticaction [2]. Typically, enzymatic hydrolysis yields sugars < 20 %of theoretical quantity without pretreatment, whereas sugars> 90 % of theoretical quantity are obtained with pretreatment[23]. Therefore, the pretreatment is necessary for obtaining themaximum yield of sugar by enzymatic hydrolysis.
The pretreatment processes (physical and chemical), whichare effective for the conversion of lignocellulosic biomass in-clude organosolv process [24, 25], dilute acids [10, 25–27],H2O2 [28], ultrasonic irradiation [29], and SO2-catalyzedsteam explosion [25, 30]. The pretreatments required for enzy-matic processes and sugar yields are summarized in Tab. 2.
Addition of polyethylene glycol (PEG) facilitates the use of ahigher process temperature (50 °C), resulting in shorter processtimes [31, 32]. The disruption of the cellulosic structure usingan ionic liquid (IL), 1-n-butyl-3-methylimidazolium chloride,has also been tried. This accelerates the rate of the subsequenthydrolysis reaction [22]. The atmospheric aqueous glycerolautocatalytic organosolv process pretreatment (AAGAOP),using the crude glycerol from oleochemical industry, was suit-able for enhancing enzymatic hydrolysis of lignocellulosic bio-
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim http://www.cet-journal.com
Table 2. Required pretreatments and sugar yield in the enzymatic hydrolysis for different feedstocks.
S. No. Feedstock
Pretreatment Enzymes Hydrolysisconditions
Sugar yield Ref.
1 wheatstraw
0.75 % (v/v)H2SO4
cellulase,b-glucosidase,
xylanase, esterase
45 °C, pH 5.0,72 h
56.5 % [10]
2 wheatstraw
2.15 %(v/v)H2O2
cellulase,a-glucosidase,
xylanase
45 °C, pH 5.0,120 h
67.2 % [28]
3 wheatstraw
diluteH2SO4
cellulases,xylanases,
recombinantferuloyl esterase
50 °C, pH 4.8,24 h
51.4 % [26]
4 5 %(w/w)
cellulose
1-n-butyl-3-methylimida
zolium chloride
cellulase 50 °C, pH 4.8,12 h
72 % [22]
5 yellowpoplar
ethanolorganosolv
pulping process
cellulase 50 °C, pH 5.0,24 h
glucose yield92 %
[24]
6spruce steam
cellulase,b-glucosidase,polyethylene
glycol
50 °C, 48 h glucose yield82 %
[32]
7 cornstover
steam and 3 %(w/w) SO2
cellulase,b-glucosidase,
xylanase
45 °C, 72 h glucose yield96 %, xyloseyield 86 %
[30]
8 olivetree
1 % H2SO4,190°C
cellulase,b-glucosidase
50 °C, pH 4.8,72 h
36.3 % [27]
9 switchgrass
0.1 g g–1
alkali, 190°Ccellulase,
b-glucosidasepH 4.8, 50 °C 58.7 % [81]
Chem. Eng. Technol. 2009, 32, No. 4, 517–526 Lignocellulosic biomass 519

mass [33, 34]. The AAGAOP technique disrupted the physicalstructure and reduced the average size of fiber [34].
Hydrolysis of lignocellulosic biomass by enzymes is an effec-tive method. Cellulases, the most commonly used enzymes fordepolymerization of cellulose to glucose, consist of three majorclasses: endoglucanases, exoglucanases, and b-glucosidases[23, 35] whereas xylanases hydrolyze hemicellulose to xylose,arabinose, mannose, acetate etc. Biomass processing by enzy-matic or microbial hydrolysis commonly involves four biologi-cally mediated transformations: (i) production of saccharolyticenzymes (cellulases and hemicellulases); (ii) hydrolysis ofcarbohydrate components present in pretreated biomass tosugars; (iii) fermentation of hexose sugars (glucose, mannose,and galactose); and (iv) fermentation of pentose sugars (xyloseand arabinose) [36]. These four transformations occur in asingle step in a process configuration called Consolidated Bio-Process (CBP).
Tabka et al. [26] investigated the enzymatic saccharificationof wheat straw which was pretreated by dilute acid followed bysteam explosion. They used a combination of enzymes such as10 U/g of cellulases and 3 U/g of xylanases from Trichodermareesei, 10 U/g of recombinant feruloyl esterase from Aspergillusniger, and oxidoreductases from Pycnoporus cinnabarinus. Acombination of cellulases from Trichoderma viride and Asper-gillus niger was used by Imai et al. [29]. They found that a rela-tively high weight fraction of cellulase from A. niger in themixed enzyme system afforded rapid initiation of the hydroly-sis reaction, while a cellulase from T. viride weight fraction ofca. 0.3 was optimal in enhancing the maximum rate constant,Vmax without increasing the half saturation constant, Km.
An efficient fermentation process could be achieved unless ahigh concentration of fermentable sugars is present in thehydrolysate stream. Enzymatic saccharification of biomasshaving solids of 20 % consistency results in a glucose solutionof 52 g L–1 with a sugar yield of 64 %. A delignification step per-formed on pretreated materials increases glucose concentrationbut, in turn, reduces sugar yields at high concentrations of bio-mass, due to end product inhibition [37]. The results showedthat cellulose and hemicellulose conversion decreased almostlinearly with increasing dry mass (2–40 % w/w) [38].
However, enzymatic hydrolysis is a promising alternativethat generally provides hydrolysates with a lower inhibitoryimpact on the fermentation step, but it is more costly thanchemical hydrolysis [39, 40] due to the high cost of enzymes[41]. The use of less expensive enzymes holds several advan-tages, such as the quite high efficiency, low by-product forma-tion, less expensive materials for construction due to mildprocess conditions, and low process energy requirement [2].
2.2.1 Kinetics for Enzymatic Hydrolysis of LignocellulosicBiomass
Kadam et al. [42] developed a multireaction kinetic model forclosed-system enzymatic hydrolysis of lignocellulosic biomass.Three hydrolysis reactions were modeled: (a) two heteroge-neous reactions for breakdown of cellulose to cellobiose andglucose, and (b) one homogeneous reaction for hydrolyzing cel-lobiose to glucose. It has been found that the various mass
transfer steps are quite fast with very low resistance and theoverall hydrolysis rate is mainly controlled by the surface ki-netics promoted by the adsorbed enzyme. It has also been foundthat the initial hydrolysis rate strongly depends on the initialextent of soluble protein adsorption and the effectiveness of theadsorbed soluble protein to promote hydrolysis [43].
2.2.1.1 Enzyme Adsorption
The adsorption of cellulase enzymes on the lignocellulosic resi-due as well as on cellulose must be taken into account in thedevelopment of hydrolysis kinetics. The equilibrium adsorp-tion of an enzyme on cellulose and on the lignocellulosicresidue could be represented by the Langmuir-type isotherm[42, 44–46]:
E� �ad�A� �max S� �total E� �free
Kd � E� �free
(6)
where [E]ad is the concentration of adsorbed cellulase (mg cel-lulase/mL); [E]free is the concentration of cellulase in solution(mg cellulase/mL); [S]total is the total substrate concentration(mg cellulose/mL); and Kd is an equilibrium constant(mg mL–1).
2.2.1.2 Saccharification of Lignocellulosic Biomass
The initial hydrolysis rate, v0, can be expressed as a function ofthe initial enzyme concentration, [E0]. Accordingly, it is conve-nient to define a maximal velocity, Vemax, and a correspondinghalf saturation constant, Ke, represented by the Michaelis-Menten-type kinetics [47–49]:
v0 � Vemax E0� �Ke � E0� � (7)
With an increase in the concentration of products, such ascellobiose and glucose in enzymatic hydrolysis, the enzymeactivity is inhibited. The inhibition pattern depends on the cel-lulase binding constant, enzyme concentration, maximumadsorption of the enzyme (cellulose surface area accessible tothe enzyme), the range in which the substrate concentrationvaries, and b-glucosidase activity.
The rate equations based on a competitive mode of sugar in-hibition are developed using the following assumptions: (1)the enzyme adsorption follows a Langmuir-type isotherm withthe first-order reactions occurring on the cellulose surface; (2)the cellulose matrix is uniform in terms of its susceptibility toenzymatic attack; (3) the enzyme activity remains constant;and (4) the conversion of cellobiose to glucose occurs in solu-tion and follows the classical Michaelis-Menten kinetics [42].These equations are shown below.
The cellulose-to-cellobiose reaction with competitive glu-cose, cellobiose, and xylose inhibition can be written as:
r1 � k1rE1BRSS
1 � G2
K1IG2
� G
K1IG
� X
K1IX
(8)
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim http://www.cet-journal.com
520 Sachin Kumar et al. Chem. Eng. Technol. 2009, 32, No. 4, 517–526

The cellulose-to-glucose reaction with competitive glucose,cellobiose, and xylose inhibition can be written as:
r2 � k2r E1B � E2B� �RSS
1 � G2
K2IG2
� G
K2IG
� X
K2IX
(9)
The cellobiose-to-glucose reaction with competitive glucoseand xylose inhibition can be written as:
r3 � k3rE2FG2
K3M 1 � G
K3IG
� X
K3IX
� �� G2
(10)
where r1, r2, and r3 are the reaction rates (g kg–1h–1), k1r, k2r,
and k3r are the reaction rate constants (kg g–1h–1), E1B is thebound concentration of cellobiohydrolase and endoglucanase(g kg–1), E2B is the bound concentration of b-glucosidase(g kg–1), E2F is the concentration of b-glucosidase in solution(g kg–1), Rs is the substrate reactivity, S is the substrate concen-tration (g kg–1), G is the glucose concentration (g kg–1), G2 isthe cellobiose concentration (g kg–1), X is the xylose concentra-tion (g kg–1), K1IG, K2IG, and K3IG are the inhibition constantsfor glucose (g kg–1), K1IG2 and K2IG2 are the inhibition con-stants for cellobiose (g kg–1), K1IX, K2IX, and K3IX are the inhi-bition constants for xylose (g kg–1), and K3M is the substrate(cellobiose) saturation constant (g kg–1). It is assumed that theenzyme and sugar concentrations in free form in the liquid aretaken in terms of cellulose as solid substrate.
Assuming that the normalized initial saccharification ratesduring secondary hydrolysis of the residual substrates areindicative of substrate reactivity, the following correlation isproposed [42]:
RS � aS
S0(11)
where RS is a substrate reactivity parameter (dimensionless), ais a constant (dimensionless), S is the substrate concentrationat a given time (g kg–1), S0 is the initial substrate concentration(g kg–1), and S/S0 is the dimensionless substrate concentrationat a given time.
3 Fermentation of Biomass Hydrolysate toEthanol
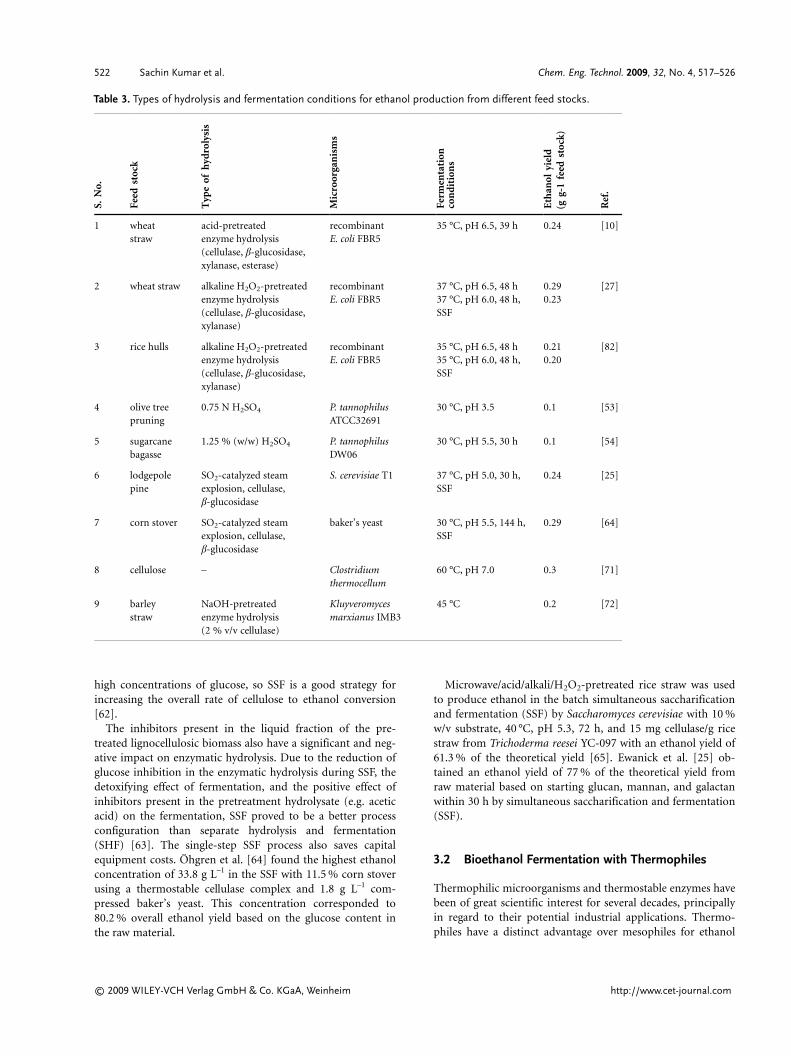
Lignocellulose-based ethanol production involves mixed sugarfermentation in the presence of inhibiting compounds, such aslow-molecular-weight organic acids, furan derivatives, pheno-lics, and inorganic compounds released and formed duringpretreatment and/or hydrolysis of the raw material [50].Bioethanol produced from different lignocellulosic biomassfeed stocks with different hydrolysis processes and various mi-croorganisms is summarized in Tab. 3.
Ethanol fermentation of pentose sugars (xylose, arabinose)is not abundently performed by most of the microorganismsas that of hexose sugars. A limited number of bacteria, yeasts,
and fungi can convert hemicellulose or its monomers (xylose,arabinose, mannose, and galactose) into ethanol with a satis-factory yield and productivity [51]. The yield of ethanol rangesfrom 20 to 51 % of the substrate weight [52]. Consequently,most research efforts have been devoted to the development ofefficient microorganisms that are capable to ferment glucoseand xylose separately or simultaneously [50].
The fermentation of olive tree pruning hydrolysate contain-ing xylose by Pachysolen tannophilus was found to give an etha-nol yield of 0.38 g g–1 at 30 °C and pH 3.5 [53]. Sugarcane ba-gasse hemicellulose hydrolysates were fermented to ethanolusing the xylose-fermenting yeast Pachysolen tannophilusDW06 giving 21 g L–1 ethanol, the yield being 0.35 g g–1 sugarand the productivity 0.59 g L–1 h–1 at 30 °C [54]. Surprisingly,the fungus Mucor indicus was able to produce ethanol fromglucose but xylitol under anaerobic conditions and ethanolunder aerobic conditions from xylose [55].
In recent years, metabolic engineering concepts have beenused for the production of fuel ethanol [56]. Ethanologenicrecombinant bacteria such as Escherichia coli, Klebsiella oxyto-ca, and Zymomonas mobilis and the yeast Saccharomyces cerevi-siae were successfully used in fermentation of mixed sugars ob-tained from biomass containing glucose, xylose, arabinose,and galactose to produce ethanol [57]. The genetically engi-neered microbes Zymomonas mobilis AX101, Escherichia coliKO11, Klebsiella oxytoca P2, and Saccharomyces cerevisiae areconsidered for commercial scale-up [58]. The recombinantyeast strain Saccharomyces cerevisiae MT8-1 was found to fer-ment xylose and cello-oligosaccharides by integrating genesfrom Pichia stipitis that express xylose reductase and xylitoldehydrogenase, and xylulokinase from Saccharomyces cerevisiaeand a gene for displaying b-glucosidase from Aspergillus aclea-tus on the cell surface [59].
Encapsulated Saccharomyces cerevisiae was able to fermentdilute acid lignocellulosic hydrolysate to ethanol at dilutionrates up to 0.5 h–1 with an ethanol yield of 0.44 g g–1 and a spe-cific productivity of 0.14–0.17 g g–1 h–1 [60]. The encapsula-tion of microbes increases the capacity for in-situ detoxifica-tion of the hydrolysate and protects them against theinhibitors present in the hydrolysate. An ethanol productionprocess using the flocculating yeast Saccharomyces cerevisiaestrain KF-7 with acid hydrolysate of wood biomass as feed wasinvestigated in continuous fermentation with a high ethanolproductivity of > 20 g L–1 h–1 at a dilution rate of 0.3 h–1 with apH of 4.5 at 35 °C [61].
3.1 Simultaneous Saccharification and Fermentation(SSF)
The enzymes cellulases (endoglucanase, exoglucanase, andb-glucosidase) responsible for enzymatic hydrolysis of pre-treated cellulosic biomass are strongly inhibited by the hydro-lysis products glucose, xylose, cellobiose, and other oligosac-charides. Simultaneous saccharification and fermentation(SSF) plays an effective role to overcome enzyme inhibition.SSF combines enzymatic hydrolysis with ethanol fermentationto keep the concentration of glucose low. The accumulation ofethanol in the fermenter does not inhibit cellulases as much as
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim http://www.cet-journal.com
Chem. Eng. Technol. 2009, 32, No. 4, 517–526 Lignocellulosic biomass 521
high concentrations of glucose, so SSF is a good strategy forincreasing the overall rate of cellulose to ethanol conversion[62].
The inhibitors present in the liquid fraction of the pre-treated lignocellulosic biomass also have a significant and neg-ative impact on enzymatic hydrolysis. Due to the reduction ofglucose inhibition in the enzymatic hydrolysis during SSF, thedetoxifying effect of fermentation, and the positive effect ofinhibitors present in the pretreatment hydrolysate (e.g. aceticacid) on the fermentation, SSF proved to be a better processconfiguration than separate hydrolysis and fermentation(SHF) [63]. The single-step SSF process also saves capitalequipment costs. Öhgren et al. [64] found the highest ethanolconcentration of 33.8 g L–1 in the SSF with 11.5 % corn stoverusing a thermostable cellulase complex and 1.8 g L–1 com-pressed baker’s yeast. This concentration corresponded to80.2 % overall ethanol yield based on the glucose content inthe raw material.
Microwave/acid/alkali/H2O2-pretreated rice straw was usedto produce ethanol in the batch simultaneous saccharificationand fermentation (SSF) by Saccharomyces cerevisiae with 10 %w/v substrate, 40 °C, pH 5.3, 72 h, and 15 mg cellulase/g ricestraw from Trichoderma reesei YC-097 with an ethanol yield of61.3 % of the theoretical yield [65]. Ewanick et al. [25] ob-tained an ethanol yield of 77 % of the theoretical yield fromraw material based on starting glucan, mannan, and galactanwithin 30 h by simultaneous saccharification and fermentation(SSF).
3.2 Bioethanol Fermentation with Thermophiles
Thermophilic microorganisms and thermostable enzymes havebeen of great scientific interest for several decades, principallyin regard to their potential industrial applications. Thermo-philes have a distinct advantage over mesophiles for ethanol
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim http://www.cet-journal.com
Table 3. Types of hydrolysis and fermentation conditions for ethanol production from different feed stocks.S.
No.
Feed
stoc
k
Typ
eof
hyd
roly
sis
Mic
roor
gan
ism
s
Ferm
enta
tion
con
dit
ion
s
Eth
anol
yiel
d(g
g-1
feed
stoc
k)
Ref
.
1 wheatstraw
acid-pretreatedenzyme hydrolysis(cellulase, b-glucosidase,xylanase, esterase)
recombinantE. coli FBR5
35 °C, pH 6.5, 39 h 0.24 [10]
2 wheat straw alkaline H2O2-pretreatedenzyme hydrolysis(cellulase, b-glucosidase,xylanase)
recombinantE. coli FBR5
37 °C, pH 6.5, 48 h37 °C, pH 6.0, 48 h,SSF
0.290.23
[27]
3 rice hulls alkaline H2O2-pretreatedenzyme hydrolysis(cellulase, b-glucosidase,xylanase)
recombinantE. coli FBR5
35 °C, pH 6.5, 48 h35 °C, pH 6.0, 48 h,SSF
0.210.20
[82]
4 olive treepruning
0.75 N H2SO4 P. tannophilusATCC32691
30 °C, pH 3.5 0.1 [53]
5 sugarcanebagasse
1.25 % (w/w) H2SO4 P. tannophilusDW06
30 °C, pH 5.5, 30 h 0.1 [54]
6 lodgepolepine
SO2-catalyzed steamexplosion, cellulase,b-glucosidase
S. cerevisiae T1 37 °C, pH 5.0, 30 h,SSF
0.24 [25]
7 corn stover SO2-catalyzed steamexplosion, cellulase,b-glucosidase
baker’s yeast 30 °C, pH 5.5, 144 h,SSF
0.29 [64]
8 cellulose – Clostridiumthermocellum
60 °C, pH 7.0 0.3 [71]
9 barleystraw
NaOH-pretreatedenzyme hydrolysis(2 % v/v cellulase)
Kluyveromycesmarxianus IMB3
45 °C 0.2 [72]
522 Sachin Kumar et al. Chem. Eng. Technol. 2009, 32, No. 4, 517–526

production in terms of increased solubility of substrates, im-proved mass transfer due to decreased viscosity, increased dif-fusion rates, high bioconversion rates, their ability to use avariety of inexpensive biomass feed stocks, low risk of contam-ination, and facilitated product recovery [66–68].
Thermophilic anaerobic bacteria are of current interest dueto their ability to ferment components of cellulosic biomassinto chemicals and fuels. Many thermophilic bacteria producecellulase and hemicellulase enzymes and have the ability to fer-ment biomass to ethanol without the addition of externalhydrolytic enzymes, which is a substantial cost advantage forthe biomass conversion process [69]. These bacteria also meta-bolize both pentose and hexose sugars found in lignocellulosichydrolysates [68]. Thermoanaerobacterium saccharolyticumJW/SL-YS485, a thermophilic anaerobic bacterium, grows in atemperature range of 45–65 °C and a pH range of 4.0 to 6.5and is able to ferment hemicellulose and xylan polymers di-rectly, as well as primary sugars found in cellulosic biomass,including cellobiose, glucose, xylose, mannose, galactose, andarabinose [69].
Thermoanaerobacter ethanolicus is an extremely thermophi-lic non-spore-forming ethanol-producing anaerobic bacteriumwhich produces ethanol at 69 °C [70]. The thermophilic anae-robic bacterial strain Thermoanaerobacter BG1L1 was able toferment undetoxified corn stover hydrolysate in a continuousimmobilized reactor system at 70 °C with an ethanol yield of0.39–0.42 g g–1 sugars consumed. Nearly 89–98 % xylose incorn stover hydrolysate was utilized to produce ethanol by thebacteria [68]. The anaerobic and thermophilic bacterium Clos-tridium thermocellum was able to ferment cellulose to ethanoldirectly with a yield of about 0.3 g g–1 of consumed celluloseat 60 °C and pH 7.0 [71].
The thermotolerant, ethanol-producing yeast strain Kluyver-omyces marxianus IMB3 produced 12 g L–1 ethanol fromNaOH-pretreated barley straw, supplemented with 2 % (v/v)cellulase at 45 °C with an ethanol yield of 20 g ethanol/100 gstraw [72]. Immobilized on Kissiris (mineral glass foam de-rived from lava) in column-packed reactors, the yeast Kluyver-omyces marxianus IMB3 produced 68.6 and 55.9 g L–1 ethanolon 200 g molasses under continuous culture conditions attemperatures of 40 °C and 50 °C, respectively [73].
4 Improvement of the EthanolFermentation Process
4.1 Two-Stage Bioreactor with Recycling forContinuous Ethanol Production
In order to achieve higher ethanol productivity, many re-searchers [74–79] used two or more bioreactors or packed col-umns in series with or without recycling the cells to maintainhigh dilution rates. Nishiwaki and Dunn [74–76] demonstrat-ed a two-stage bioreactor with recycling the cells by a cellseparator at each stage (Fig. 1). Chaabane et al. [78] studied atwo-stage continuous bioreactor by recycling the cells wherethe first reactor was used for cell growth and the second reac-tor for ethanol production. Higher ethanol productivity (up to
41 g L–1 h–1) was obtained with complete conversion of glucoseto ethanol in the two-stage system. Bai et al. [79] observedhigh fluctuations in fermentation parameters in three tubularbioreactors arranged in series (Fig. 2). These fluctuations wereattenuated when the tubular bioreactors were packed with In-tlox ceramic saddle.
The kinetic parameters were obtained for a continuousglucose-to-ethanol fermentation with Saccharomyces cerevisiaein recycling of cells using the membrane fermenter by Groot[74–77].
For specific growth rate under product inhibition:
l � lmS
KS � S1 � P
Pm
� �n
(12)
For specific glucose consumption rate:
qS � lm
YX�S(13)
For specific ethanol production rate:
t = YX/S qS (14)
where l is the specific growth rate (h–1), lm is the maximumspecific growth rate (h–1), S is the substrate concentration(g L–1), KS is the saturation constant (g L–1), P is the productconcentration (g L–1), Pm is the maximum product concentra-tion (g L–1), qS is the specific consumption rate of the substrate(g g–1 h), t is the specific production rate of ethanol (g g–1 h–1),and YX/S is the yield of cells based on the glucose consumed(g g–1).
Bio-Process Innovation (BPI), Inc. [80] developed andtested a continuous cascade fermentation system for produc-tion of ethanol from lignocellulose waste. The biomass feed isintroduced continuously into the first of three to five stirredreactors placed in series, with the outflow of one reactor intothe next reactor (Fig. 3). The liquid stream that moves from
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim http://www.cet-journal.com
Figure 1. Two-stage fermenter with cell recyling at each stage[74].
Chem. Eng. Technol. 2009, 32, No. 4, 517–526 Lignocellulosic biomass 523

reactor to reactor is contacted with a stripping gas to removethe ethanol. A low-energy solvent absorption/extractive distil-lation system extracts ethanol from the stripping gas and re-
covers the gas for reuse. Removal of ethanol from the fermen-tation broth facilitates the high rate of ethanol production byminimizing its inhibitory action on the microorganisms.
5 Summary
Minimization of the costs of ethanol production isthe prime objective of the current research pro-gram. In this direction, reducing the costs of sac-charolytic enzymes, developments of fermentativemicroorganisms by genetic recombination that arecapable of fermenting hexose and pentose sugarsto ethanol, and improvement of yield and produc-tivity by designing novel bioreactors are the majorachievements to reduce the costs of ethanol from$ 2.65 per gallon to $ 1.1 per gallon. However,further cost compensation could be achieved bysynthesizing value- added products from furfural,hydroxymethylfurfural etc.
Ethanol production at high temperature usingthermophiles would facilitate the in-situ removalof ethanol either by gas striping or by solvent ex-traction. In addition, thermophiles can reduce thecosts of ethanol production from lignocellulosebiomass as they can convert glucose and xylose toethanol efficiently with high fermentation rate andproductivity.
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim http://www.cet-journal.com
Figure 2. Process diagram for continuous ethanol fermentation using a combined bioreactor system. 1,substrate storage tank; 2, peristaltic pump; 3, stirred tank fermenter; 4, pH controlling unit; 5, tempera-ture controlling unit; 6, tubular fermenters; 7, thermostat water inlet; 8, thermostat water outlet; 9, brothstorage tank; 10, exhaust gas washing tank; 11, air flowmeters; 12, air humidifiers; 13, sampling parts[79].
Figure 3. Continuous cascade fermentation system for producing ethanol fromwaste sugars [80].
524 Sachin Kumar et al. Chem. Eng. Technol. 2009, 32, No. 4, 517–526

Acknowledgement
We thank Dr. M. O. Garg, Director IIP, Dehradun, for his valu-able suggestions and encouragement to write this review paper.One of the authors (Sachin Kumar) gratefully acknowledgesthe Senior Research Fellowship awarded by the Council ofScientific and Industrial Research (CSIR), India.
References
[1] C. M. Takahashi, K. G. C. Lima, D. F. Takahashi, F. Alter-thum, World J. Microb. Biotechnol. 2000, 16, 829. DOI:10.1023/A:1008987103701
[2] P. C. Badger, in Trends in New Crops and New Uses (Eds: J. Ja-nick, A. Whipkey), ASHS Press, Alexandria, VA 2002.
[3] J. R. Mielenz, Curr. Opin. Microbiol. 2001, 4 (3), 324. DOI:10.1016/S1369-5274 (00)00211-3
[4] K. H. Kim, M. P. Tucker, Q. A. Nguyen, Biotechnol. Prog.2002, 18 (3), 489. DOI: 10.1021/bp025503i
[5] K. L. Kadam, J. D. McMillan, Bioresour. Technol. 2003, 88, 17.DOI: 10.1016/S0960-8524 (02)00269-9
[6] A. Demirbas, Energy Sources, Part A 2008, 30 (1), 27. DOI:10.1080/00908310600626705
[7] M. Knauf, M. Moniruzzaman, Int. Sugar J. 2004, 106 (1263),147.
[8] J. Lee, J. Biotechnol. 1997, 56 (1), 1. DOI: 10.1016/S0168-1656 (97)00073-4
[9] R. Purwadi, C. Niklasson, M. J. Taherzadeh, J. Biotechnol.2004, 114, 187. DOI: 10.1016/j.jbiotec.2004.07.006
[10] B. C. Saha, L. B. Iten, M. A. Cotta, Y. V. Wu, Proc. Biochem.2005, 40, 3693. DOI: 10.1016/j.procbio.2005.04.006
[11] C. Martín et al., Enzyme Microb. Technol. 2002, 31 (3), 274.DOI: 10.1016/S0141-0229 (02)00112-6
[12] I. M. Banat et al., World J. Microbiol. Biotechnol. 1998, 14 (6),809. DOI: 10.1023/A:1008802704374
[13] K. Collins, EIA Energy Outlook, Modeling, and Data Confer-ence, Washington, DC 2007.
[14] B. P. Lavarack, G. J. Griffin, D. Rodman, Biomass Bioenergy2002, 23 (5), 367. DOI: 10.1016/S0961-9534 (02)00066-1
[15] W. J. Frederick Jr. et al., Bioresour. Technol. 2008, 99 (11),5051. DOI: 10.1016/j.biortech.2007.08.086
[16] W. A. Farone, J. E. Cuzens, US Patent 5 562 777, 1996.[17] W. A. Farone, J. E. Cuzens, US Patent 5 580 389, 1996.[18] A. Rodríguez-Chong, J. A. Ramírez, G. Garrote, M. Vázquez,
J. Food Eng. 2004, 61, 143.DOI: 10.1016/S0260-8774 (03)00080-3
[19] S. Gámez et al., J. Food Eng. 2006, 74, 78.DOI: 10.1016/j.jfoodeng.2005.02.005
[20] J. F. Saeman, Ind. Eng. Chem. 1945, 37 (1), 43.DOI: 10.1021/ie50421a009
[21] S. C. Yat, A. Berger, D. R. Shonnard, Bioresour. Technol. 2008,99, 3855. DOI: 10.1016/j.biortech.2007.06.046
[22] A. P. Dadi, S. Varanasi, C. A. Schall, Biotechnol. Bioeng. 2006,95 (5), 904. DOI: 10.1002/bit.21047
[23] P. Ghosh, T. K. Ghose, in Advances in Biochemical Engineer-ing/Biotechnology (Ed: T. Scheper), Vol. 85, Springer, NewYork 2003. DOI: 10.1007/3-540-36466-8
[24] A. Berlin et al., Biotechnol. Bioeng. 2006, 93 (5), 880. DOI:10.1002/bit.20783
[25] S. M. Ewanick, R. Bura, J. N. Saddler, Biotechnol. Bioeng.2007, 98 (4), 737. DOI 10.1002/bit.21436
[26] M. G. Tabka et al., Enzyme Microb. Technol. 2006, 39, 897.DOI: 10.1016/j.enzmictec.2006.01.021
[27] C. Cara et al., Bioresour. Technol. 2008, 99, 1869. DOI:10.1016/j.biortech.2007.03.037
[28] B. C. Saha, M. A. Cotta, Biotechnol. Prog. 2006, 22, 449. DOI:10.1021/bp050310r
[29] M. Imai, K. Ikari, I. Suzuki, Biochem. Eng. J. 2004, 17, 79.DOI: 10.1016/S1369-703X(03)00141-4
[30] K. Öhgren, R., Bura, J. Saddler, G. Zacchi, Bioresour. Technol.2007, 98, 2503. DOI: 10.1016/j.biortech.2006.09.003
[31] J. Börjesson, M. Engqvist, B. Sipos, F., Tjerneld, Enzyme Mi-crob. Technol. 2007, 41, 186.DOI: 10.1016/j.enzmictec.2007.01.003
[32] J. Börjesson, R. Peterson, F. Tjerneld, Enzyme Microb. Tech-nol. 2007, 40, 754. DOI: 10.1016/j.enzmictec.2006.06.006
[33] F. Sun, H. Chen, Bioresour. Technol. 2007, 99 (13), 5474.DOI: 10.1016/j.biortech.2007.11.001
[34] F. Sun, H. Chen, Bioresour. Technol. 2008, 99 (14), 6156.DOI: 10.1016/j.biortech.2007.12.027
[35] J. Medve, J. Karlsson, D. Lee, F. Tjerneld, Biotechnol. Bioeng.1998, 59 (5), 621. DOI: 10.1002/(SICI)1097-0290 (19980905)59:5<621::AID-BIT13>3.0.CO;2-C
[36] L. R. Lynd, W. H. Zyl, J. E. McBride, M. Laser, Curr. Opin.Biotechnol. 2005, 16, 577. DOI: 10.1016/j.copbio.2005.08.009
[37] C. Cara et al., Proc. Biochem. 2007, 42, 1003.DOI: 10.1016/j.procbio.2007.03.012
[38] H. Jørgensen, J. Vibe-Pedersen, J. Larsen, C. Felby, Biotech-nol. Bioeng. 2007, 96 (5), 862. DOI: 10.1002/bit.21115
[39] M. Galbe, G. Zacchi, Appl. Microbiol. Biotechnol. 2002, 59,618. DOI: 10.1007/s00253-002-1058-9
[40] T. Brandberg, N. Sanandaji, L. Gustafsson, C. J. Franzén, Bio-technol. Prog. 2005, 21 (4), 1093. DOI: 10.1021/bp050006y
[41] R. R. Singhania, R. K. Sukumaran, A. Pandey, Appl. Biochem.Biotechnol. 2007, 142, 60. DOI: 10.1007/s12010-007-0019-2
[42] K. L. Kadam, E. C. Rydholm, J. D. McMillan, Biotechnol.Prog. 2004, 20 (3), 698. DOI: 10.1021/bp034316x
[43] Y. H. Lee, L. T. Fan, Biotechnol. Bioeng. 1982, 24 (11), 2383.DOI: 10.1002/bit.260241107
[44] G. Beldman et al., Biotechnol. Bioeng. 1987, 30 (2), 251. DOI:10.1002/bit.260300215
[45] H. Ooshima, D. S. Burns, A. O. Converse, Biotechnol. Bioeng.1990, 36 (5), 446. DOI: 10.1002/bit.260360503
[46] B. Yang, D. M. Willies, C. E. Wyman, Biotechnol. Bioeng.2006, 94 (6), 1122. DOI: 10.1002/bit.20942
[47] C. J. Bailey, Biochem. J. 1989, 262, 1001.[48] F. Carrillo et al., Proc. Biochem. 2003, 39, 257.
DOI: 10.1016/S0032-9592 (03)00066-9[49] F. Carrill et al., Proc. Biochem. 2005, 40, 3360.
DOI: 10.1016/j.procbio.2005.03.003[50] B. Hahn-Hägerdal et al., Trends Biotechnol. 2006, 24 (12),
549. DOI: 10.1016/j.tibtech.2006.10.004[51] P. Sommer, T. Georgieva, B. K. Ahring, Biochem. Soc. Trans.
2004, 32, 283. (www.biochemsoctrans.org)[52] J. D. Murphy, K. McCarthy, Appl. Energy 2005, 82, 148. DOI:
10.1016/j.apenergy.2004.10.004
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim http://www.cet-journal.com
Chem. Eng. Technol. 2009, 32, No. 4, 517–526 Lignocellulosic biomass 525

[53] I. Romero et al., Biochem. Eng. J. 2007, 36, 108. DOI:10.1016/j.bej.2007.02.006
[54] K. K. Cheng et al., Biotechnol. Lett. 2007, 29, 1051. DOI:10.1007/s10529-007-9361-2
[55] K. Karimi, G. Emtiazi, M. J. Taherzadeh, Proc. Biochem.2006, 41, 653. DOI: 10.1016/j.procbio.2005.08.014
[56] T. W. Jeffries, Y. S. Jin, Appl. Microbiol. Biotechnol. 2004, 63,495. DOI: 10.1007/s00253-003-1450-0
[57] R. J. Bothast, N. N. Nichols, B. S. Dien, Biotechnol. Prog.1999, 15, 867. DOI: 10.1021/bp990087w
[58] B. S. Dien, M. A. Cotta, T. W. Jeffries, Appl. Microbiol. Bio-technol. 2003, 63, 258. DOI: 10.1007/S00253-003-1444-y
[59] S. Katahira, A. Mizuike, H. Fukuda, A. Kondo, Appl. Micro-biol. Biotechnol. 2006, 72, 1136.DOI: 10.1007/s00253-006-0402-x
[60] F. Talebnia, M. J. Taherzadeh, J. Biotechnol. 2006, 125, 377.DOI: 10.1016/j.jbiotec.2006.03.013
[61] Y. Tang et al., Proc. Biochem. 2006, 41, 909.DOI: 10.1016/j.procbio.2005.09.008
[62] D. R. Shonnard, CM4710 Biochem. Processes, Course, Michi-gan Technology University, Michigan 2003.
[63] K. Öhgren et al., Proc. Biochem. 2007, 42, 834.DOI: 10.1016/j.procbio.2007.02.003
[64] K. Öhgren et al., Enzyme Microb. Technol. 2007, 40, 607.DOI: 10.1016/j.enzmictec.2006.05.014
[65] Z. Shengdong et al., Chem. Eng. Commun. 2006, 193 (5),639. DOI: 10.1080/00986440500351966
[66] O. Holst et al., Comp. Biochem. Physiol. 1997, 118A (3), 415.DOI: 10.1016/S0300-9629 (97)00002-9
[67] B. L. Knutson et al., J. Supercrit. Fluids 1999, 16 (2), 149.DOI: 10.1016/S0896-8446 (99)00026-1
[68] T. I. Georgieva, B. K. Ahring, Appl. Microbiol. Biotechnol.2007, 77, 61. DOI: 10.1007/s00253-007-1149-8
[69] A. J. Shaw, F. E. Jenney Jr., M. W. W. Adams, L. R. Lynd, En-zyme Microb. Technol. 2008, 42, 453.DOI: 10.1016/j.enzmictec.2008.01.005
[70] V. Kannan, R. Mutharasan, Enzyme Microb. Technol. 1985, 7(2), 87. DOI: 10.1016/S0960-8524 (02)00269-9
[71] L. Zertuche, R. R. Zall, Biotechnol. Bioeng. 1982, 24, 57. DOI:10.1002/bit.260240106
[72] M. Boyle, N. Barron, A. P. McHale, Biotechnol. Lett. 1997, 19(1), 49. DOI: 10.1023/A:1018315003916
[73] P. Nigam et al., World J. Microbiol. Biotechnol. 1997, 13 (3),283. DOI: 10.1023/A:1018578806605
[74] A. Nishiwaki, I. J. Dunn, Chem. Eng. Commun. 1998, 168(1), 207. DOI: 10.1080/00986449808912715
[75] A. Nishiwaki, I. J. Dunn, Chem. Eng. Commun. 1997, 162,179. DOI: 10.1080/00986449708936638
[76] A. Nishiwaki, I. J. Dunn, Biochem. Eng. J. 1999, 4 (1), 37.DOI: 10.1016/S1369-703X(99)00029-7
[77] W. J. Groot et al., Bioprocess Eng. 1992, 8, 39.DOI: 10.1007/BF00369262
[78] F. B. Chaabane et al., Bioprocess Biosyst. Eng. 2006, 29, 49.DOI: 10.1007/s00449-006-0056-1
[79] F. W. Bai, L. J. Chen, W. A. Anderson, M. Moo-Young, Bio-technol. Bioeng. 2004, 88 (5), 558. DOI: 10.1002/bit.20221
[80] M. C. Dale, Inventions and Innovation Annual Report, US De-partment of Energy, Washington, DC 2005.
[81] Z. Hu, Z. Wen, Biochem. Eng. J. 2008, 38, 369.DOI: 10.1016/j.bej.2007.08.001
[82] B. C. Saha, M. A. Cotta, Enzyme Microb. Technol. 2007, 41,528. DOI: 10.1016/j.enzmictec.2007.04.006
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim http://www.cet-journal.com
526 Sachin Kumar et al. Chem. Eng. Technol. 2009, 32, No. 4, 517–526
Copyright © 2022 FDOKUMEN




















