
Identification of a Novel Thermostable Alkaline Protease from ...
Upload
independentCategory
view
0download
0
Vol. 44 ı No. 6 ı 2008 www.biotechniques.com ı BioTechniques ı 787
Research Reports
INTRODUCTION
Proteins or peptides required for structural and biochemical study are usually produced via bacterial expression systems when posttransla-tional modifications are not required (1,2). In many cases, however, poor expression, low yield, instability, and difficulty in purification are complica-tions (2,3). Furthermore, production of heterogeneous proteins in Escherichia coli often results in the accumulation of targets in insoluble aggregates, forming inclusion bodies. Recovery of the biological activity of the target protein by an in vitro refolding process is a difficult task (4).
Protein fusion technology is widely used to improve expression, purification, and solubility of recombinant protein expressed in E. coli. Several proteins such as maltose-binding protein (MBP), thioredoxin (TRX), transcription pausing factor L (NusA), thiol-disulfide oxido-reductase, glutathione S-transferase (GST), and others have been reported to enhance the solubility of fused protein (2,5,6). However, no system or tool respresents a complete solution, and improvement is always necessary. For example, among these proteins, MBP
was found more effective in solubilizing fused proteins than GST and TRX, but it was unable to promote proper folding of the fused proteins, and in many cases, it could not enhance expression of difficult-to-express proteins (6–8).
Recent reports have described the use of some thermostable proteins as fusion partners to improve the stability and purification of targets fused to them (9). In this context, we examined several E. coli thermostable proteins that might be capable of enhancing solubility of target proteins or peptides in order to find improved fusion partners. By identifying an appropriate protein for use as a fusion partner, target protein and peptides that form inclusion bodies in the conven-tional expression system were success-fully expressed and purified without the need for solubilization and recovery.
MATERIALS AND METHODS
Identification of Thermostable Proteins from E. coli
Whole cell lysate was prepared fromE. coli DH5α cells grown at 37°C overnight by 4 to 5 passages through a French pressure cell at 18,000 lb/in2 in
lysis buffer A (20 mM Tris-Cl, pH 8.0, 10 mM NaCl, 0.1 mM phenylmethane-sulphonylfluoride, 0.1 mM EDTA, and 1 mM 2-mercaptoethanol) and centrifu-gation at 47,000× g for 1 h. The soluble fraction (cell extract) was boiled for 10 min, centrifuged at 16,500× g for 30 min, and the soluble fraction was again recovered. Proteins in the fraction were resolved by native 10% polyacrylamide gel electrophoresis (PAGE). Then, the gel portion containing proteins was excised and denatured by boiling in the presence of 0.1% 2-mercaptoethanol for 10 min, followed by sodium dodecyl sulfate PAGE (SDS-PAGE). The selected protein spots were analyzed by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF; ProteomeTech. Inc., Seoul, Korea) and identified by National Center for Biotechnology Information (NCBI) BLAST search on the basis of probability.
Cloning of Thermostable Proteins
Genes encoding the identified thermostable proteins were PCR-amplified using E. coli DH5α genomic DNA as a template and cloned in NdeI and BamHI sites (or NdeI/ HindIII for FK-506-binding protein, FKBP) of the
Purification of inclusion body−forming peptides and proteins in soluble form by
fusion to Escherichia coli thermostable proteinsEscherichia coli thermostable proteinsEscherichia coli
Arjun Thapa1, Md. Shahnawaz1, Pratap Karki1, Giri Raj Dahal1, Md. Golam Sharoar1, Song Yub Shin1, Jung Sup Lee1, Byungyun Cho2, and Il-Seon Park1
BioTechniques 44:787-796 (May 2008)doi 10.2144/000112728
Proteins and peptides expressed in the prokaryotic system often form inclusion bodies. Solubilization and refolding procedures can be used for their recovery, but this process remains difficult. One strategy for improving the solubility of a protein of interest is to fuse it to a highly soluble protein. To select a suitable fusion partner capable of solubilizing the aggregation-prone (inclusion body−forming) proteins and peptides, −forming) proteins and peptides, − Escherichia coli thermostable proteins were identified and tested. Among them, trigger factor (TF) protein was selected because of its high expression and stability. Using an expression system based on fusion to TF, selected proteins and peptides that otherwise form inclusion bodies were expressed in soluble state and were purified like other soluble pro-teins. This system provides a convenient method for production of aggregation-prone proteins and peptides.
1Chosun University, Gwanju and 2Yonsei University, Wonju, Republic of Korea

788 ı BioTechniques ı www.biotechniques.com Vol. 44 ı No. 6 ı 2008
Research Reports
pET21b expression system (Novagen, Madison, WI, USA). The primers used for cloning DnaK, trigger factor (TF), MBP, D-ribose periplasmic protein (RbsB), putative EscN protein (EscN), GroES, FKBP, Adenylate kinase (AKN), and NusA are shown in Table 1.
Expression and Heat Stability of the Prospective Thermostable Cloned Genes
Expression of proteins in E. coliBL21 (DE3) pLysS transformed with each plasmid was induced with 0.4 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at an A600 of ∼0.6, after which the cells were cultured at 37°C for an additional 4 h. Cells were harvested, lysed in the lysis buffer A, disrupted by a brief sonication, and centrifuged at 16,500× g for 1 h. SDS-PAGE analysis of the supernatants before and after boiling for 10 min was carried out to examine the expression and thermosta-bility of the proteins.
Construction of Thermostable Protein Ubiquitin Target Fusion Plasmids
Amyloid β(1–42) [Aβ(1–42)] was PCR-amplified with appropriate primers (labeled Aβ42 in Table 1) using the Aβ(1–42) gene as a template and cloned in the BamHI and XhoI sites of pET28b (Novagen). Ubiquitin (kindly provided by Rohan T. Baker, Australian National University) was cloned upstream of the Aβ(1–42) gene using NdeI/BamI/BamI/ HI sites. Then, the ubiquitin-Aβ(1–42) fusion was PCR-amplified (primers: Ub-Aβ42 in Table 1) after removal of the BamHI site by site-directed mutagenesis [primers: Ub-Aβ42 (BamHI) in Table 1] and cloned into the plasmid containing the thermostable protein to be tested. This step used the BamHI and XhoI sites, placing the ubiquitin-Aβ(1–42)
fusion downstream of the thermo-stable protein. The ubiquitin-Aβ(1–40)
construct was prepared by insertion of a termination codon two amino acid residues from the end of the Aβ(1–42)
sequence (primers: Ub-Aβ40 in Table 1). The ubiquitin-Aβ(42–1) [a reverse form of Aβ(1–42)] fusion was synthe-sized in a multistep process using the
primers shown in Table 1 [Ub-Aβ42 forward and Aβ(42–1)], after which it was subcloned after the TF fusion protein. Humanin peptide (10,11) was synthesized using the primers listed in Table 1 and subcloned as described above. To construct reverse-caspase-2(12), the small and large subunit of the protein were PCR-amplified separately (primers: Casp-2 SS and LS in Table 1). The small subunit of the full-length caspase-2 was cloned in pET21b downstream of the large subunit. Finally, the whole structure was subcloned into the BamHI/XhoHI/XhoHI/ I sites of a newly constructed TF-ubiquitin plasmid. The plasmid was
constructed by PCR amplification with appropriate primers (listed as reverse-caspase-2 in Table 1) and then cloned into the NdeI/BamI/BamI/ HI sites of the TF-ubiquitin pET21b expression system, after removing the BamHI site located between TF and ubiquitin.
Purification of Aββ(1–42) and Other Targets
Aβ(1–42) as a fusion with TF and ubiqitin was overexpressed, and the cell extract was prepared as described in the identification of thermostable proteins section above. The soluble fraction
Figure 1. Identification and overexpression of thermostable Escherichia coli proteins. (A) Two-di-mensional polyacrylamide gel electrophoresis (PAGE) analysis of thermostable E. coli proteins. E. colicell extract was boiled for 10 min, and the supernatant after centrifugation was saved. The native gel por-tion containing the supernatant was excised, denatured, and subjected to sodium dodecyl sulfate PAGE (SDS-PAGE). The indicated spots were identified by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) as follows: 1, DnaK; 2, transcription pausing factor L (NusA); 3, 4, 5, trigger factor (TF); 6, maltose-binding protein (MBP); 7, galactose glucose-binding protein; 8, FK-506-binding protein (FKBP); 9, D-ribose periplasmic protein (RbsB); 10, adenylate kinase (AKN); 11, outer mem-brane lipoprotein carrier protein; 12, ribosomal protein S19 (RS19); 13, putative EscN protein (EscN); 14, GroES; 15, ribosomal protein S19. Relative molecular weights are indicated on the left. (B) SDS-PAGE analysis of the selected proteins that were overexpressed in E. coli. Each cell extract was boiled for 10 min, and the supernatant recovered after centrifugation was analyzed. For TF, spot 5 was used. Each pair of lanes consists of the equivalent amount of proteins before (-) and after (+) boiling. Ten times as much of MBP, FKBP, AKN, RS19, and EscN were loaded compared with the other proteins.
A B
Figure 2. Selection of fusion proteins for the vector construction. (A) Representation of the construct used for soluble expression of target protein. Parts of the amino acid sequence of ubiquitin are indicated. (B) Sodium dodecyl sul-fate polyacrylamide gel electrophoresis (SDS-PAGE) analysis of the selected fusion proteins. The cell extract from each construct was centri-fuged, and soluble (S) and insoluble (P) fractions were analyzed on SDS-PAGE to examine if they conferred solubility to amyloid β(1–42) [Aβ(1–42)]. An equal amount of soluble and insoluble frac-tion was loaded in each pair of lanes. The arrows indicate the expressed fusion proteins. Relative molecular weights are indicated on the left. TF, trigger factor; MBP, maltose-binding protein; EscN, putative EscN protein.
A
B

was applied to a Ni-NTA column (Amersham Biosciences, Piscataway, NJ, USA), washed with five column volumes of buffer A (50 mM Tris-Cl, pH 8.0, 150 mM NaCl, and 1 mM 2-mercaptoethanol), and the protein was eluted with buffer B (buffer A plus 250 mM imidazole). Peak fractions of the fusion protein were pooled, quantified, diluted 2-fold, and incubated with a deubiquitylating enzyme, Usp2-cc (13), in 1:100 enzyme-protein molar ratio at 37°C for 2 h. The samples were then subjected to preparative reversed-phase high-perfor-mance liquid chromatography (RP-HPLC; C18 column, 25 mm × 250 mm × 8 μm; Grace Vydac, Hesperia, CA, USA). Aβ(1–42) was purified with a solvent system buffer C (30 mM ammonium acetate, pH 10.0, and 2% acetonitrile) and buffer D (70% acetonitrile in water) at a flow rate 10 mL/min using a 20%–40% linear gradient of buffer D over 35 min. The same protocol was used for purification of Aβ(1–40) and Aβ(42–1). Humanin was purified in a silica-based column, with buffer E [0.1% trifluoroacetic acid (TFA) in water] and buffer F (buffer E plus 80% acetonitrile), using a 5%–60% linear gradient of buffer F over 40 min. The purified peptides were solubilized in 100% 1,1,1,3,3,3,-hexafluoro-2-propanol and dried under nitrogen flow and subsequently under vacuum for 30 min. Then they were stored at -20°C. Immediately before use, the peptides Aβ(1–42), Aβ(1–40), and Aβ(42–1) were dissolved in 0.1%NH4OH at a concentration of 1 mg/mL, followed by bath sonication for 10 min, and resuspended in phosphate-buffered saline (PBS; 10 mM phosphate buffer, pH 7.4, 137 mM NaCl, and 2 mM KCl). Humanin was dissolved in 10% glacial acetic acid and subsequently diluted five times in PBS.
Figure 3. Purification of amyloid ββ(1–42) [Aββ(1–42)] and other peptides. (A) Tricine polyacrylamide gel electrophoresis (PAGE; polyacrylamide: 13.5%) anal-ysis of Aβ(1–42) and its fusion protein. Lanes: 1, TF-ubiquitin-Aβ(1–42) fusion pro-tein purified using Ni-NTA column; 2, the fusion protein digested by Usp2-cc; 3, Aβ(1–42) purified by C18 reverse-phase column chromatography. Expected molecu-lar weights are indicated on the left. (B) Aβ(1–42) elution profile of C18 reverse-phase column chromatography. (C) Tricine PAGE (polyacrylamide: 13.5%) analysis of Aβ(42–1) (lane 1), Aβ(1–40) (lane 2), and humanin (lane 3) fractions. The peptides were overexpressed and purified by the same method as Aβ(1–42) using trigger fac-tor (TF)-based vector construct as described. The arrows in panels A and C indicate the purified peptides.
A
B
www.RayBiotech.com 1-888-494-8555 [email protected]
R
Screening or multiplex ELISA arrays; chemiluminescence or fluorescence detection
Cell-cultured media, lysates, serum, plasma, urine, CSF, BAL, tears and more
RayBiotech’s Antibody Arrays are revolutionizing biomedical research!
High Density
Multiple Sample Types
Flexible Format
Proven TechnologyExtensive validation and hundreds of publications
Alzheimer's Arthritis Cancer Cardio Disease Diabetes Infectious Disease Inflammation Obesity
Cytokines Chemokines Growth Factors Angiogenic Factors Signal Transduction Metalloproteinases Soluble Receptors Adipokines
What are you waiting for?
Key FactorsBiomarkers Drug Targets
High ThroughputAccelerated biomarker and drug discovery processes
Screen for up to 507 proteins or quantify 40 proteins in a single aliquot
RayBiotech, Inc.the protein array pioneer company
Circle Reader Service No. 190

790 ı BioTechniques ı www.biotechniques.com Vol. 44 ı No. 6 ı 2008
Research Reports
TF-ubiquitin-reverse-caspase-2 was overexpressed for 4 h at 30°C with 0.4 mM IPTG. Cells were lysed in lysis buffer B (20 mM Tris-Cl, pH 8.0, 10 mM NaCl, 0.1 mM EDTA, and 1 mM 2-mercaptoethanol), disrupted by French pressure cell, and centrifuged at 47,000× g for 1 h. The cell extract was injected into a Ni-NTA column, washed with five column volumes of buffer G (20 mM Tris-Cl, pH 8.0, 10 mM NaCl, and 1 mM 2-mercaptoethanol), and eluted by a linear gradient of buffer H (buffer G plus 250 mM imidazole). The 2-fold diluted fusion protein fractions were digested with Usp2-cc (13) and purified by HiTrap Q column chroma-tography (Amersham Biosciences) using buffer I (20 mM HEPES-NaOH, pH 7.5, 10 mM NaCl, 0.1 mM EDTA, 10% glycerol, and 1 mM dithiothreitol) and buffer J (buffer I plus 1 M NaCl). Pooled fractions were dialyzed to remove NaCl and loaded on a HiTrap CM column (Amersham Biosciences) for final purification with buffer K (20 mM HEPES-NaOH, pH 7.0, 10 mM NaCl, 0.1 mM EDTA, 10% glycerol, and 1 mM dithiothreitol) and buffer L (buffer K plus 1 M NaCl). The purified enzymes were dialyzed in buffer K plus 10 mM NaCl and stored at -80°C.
Fibrillogenesis Assay
Aβ(1–42) (20 μM) and Aβ(1–40) (40 μM) were incubated in a final volume of 300 μL. A 20-μL aliquot of the reaction was mixed with 80 μL freshly prepared 5 μM thioflavin T (ThT). Fluorescence was measured on a microplate spectro-fluoremeter (SpectraMax GeminiXS; Molecular Devices, Sunnyvale, CA, USA) using an excitation of 445 nm and emission of 490 nm (14).
Circular Dichroism Spectroscopy
For circular dichroism (CD) analysis, 20 μM Aβ(1–42) in PBS were incubated at 37°C for 0 or 24 h. CD spectra were obtained using a Jasco J-810 spectropolarimeter (Jasco, Tokyo, Japan) at 25°C. Far UV CD spectra were recorded using a cuvet with a 1-mm path length at 0.5-nm intervals between 190–250 nm. For each sample, five accumulative readings were averaged and acquired with a 0.1 nm
resolution, a 0.5 s response time, and 50 nm/min scan speed (15).
Cell Culture and Cell Death Assay
Human neuroblastoma SH-SY5Y cells were maintained in Dulbecco’s modified Eagle’s medium and Ham’s F12 (1:1) supplemented with 10%fetal bovine serum and 1% antibiotics (Jeil Biotech Services, Daegu, Korea) at 37°C under 5% CO2. Cells were seeded at a density of 2 × 104 cells/well on the 96-well plates (Nunc, Roskilde, Denmark) and cultured for 24 h. The cell death assay with purified Aβ(1–42)
or Aβ(1–40) was assessed by a MTT reduction test. Briefly, 20 μL 5 mg/mL MTT solution were added to 100 μL culture medium in a 96-well plate, incubated for 2 h in a CO2 incubator, and then 100 μL 20% SDS solution in 50% (4,5-dimethylthiazol-2yl) 2,5-diphenyl-tetarzolium bromide (MTT) and N,N-dimethylformamide (DMF; pH 4.7) were added. After 16 h of incubation, absorbance was recorded using a microplate reader (SpectraMax 190 Spectrophotometer; Molecular Devices) at 570 nm.
DEVDase Activity Assay and Activity of the Purified Reverse-Caspase-2
To measure DEVDase (caspase-3/7-like) activity, treated cells were washed twice with ice-cold PBS. Then 40 μL cell lysis buffer C (20 mM HEPES-NaOH, pH 7.0, 1 mM EDTA, 1 mM EGTA, 20 mM NaCl, 1 mM dithioth-reitol, 1 mM phenylmethanesulpho-nylfluoride, 10 μg/mL leupeptin, 5 μg/mL pepstatin A, 2 μg/mL aprotinin, and 25 μg/mL ALLN) were added into each well and incubated on ice for 20 min. The enzyme activity assay was performed in 100 μL caspase assay buffer (10 mM HEPES-KOH, pH 7.0, 10 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, and 10 mM dithio-threitol) using 10 μM N-acetyl Asp-Glu-Val-Asp-amino methyl coumarin (Ac-DEVD-AMC) as a substrate at 30°C. Enzymatic activity of reverse-caspase-2 was examined using 10 μM N-acetyl Val-Asp-Val-Ala-Asp-amino methyl coumarin (Ac-VDVAD-AMC). The fluorescence was monitored at excitation and emission wavelengths of 360 and 480 nm, respectively, using the SpectraMax GeminiXS.
Figure 4. Characterization of purified peptide. The purified amyloid β (Aβ) peptides were tested for their biophysical and biochemical properties. (A) Time-dependent fibrillogenesis curve of Aβ(1–42)
(closed circle) and Aβ(1–40) (open circle) obtained by thioflavin T (ThT) binding assay. (B) Circular di-chroism (CD) spectra showing random coil conformation of freshly dissolved Aβ(1–42) (broken line) and β-sheet structure when aggregated for 24 h (solid line). (C) MTT assay showing dose-dependent toxicity of Aβ(1–42) (close bar) and Aβ(1–40) (open bar) in SH-SY5Y cells. The error bars indicate mean ± standard deviation obtained from three independent experiments. (D) DEVDase (caspase-3/7-like) activity analy-sis in cells exposed to 20 μM Aβ(1–42) (closed circle) and 20 μM Aβ(1–40) (open circle) or in untreated cells (thin line) with 10 μM DEVD-AMC as a substrate. RFU, relative fluorescence unit.
A B
C D

792 ı BioTechniques ı www.biotechniques.com Vol. 44 ı No. 6 ı 2008
Research Reports
Table 1. Primers Used for PCR
Protein Primers
DnaK Forward 5′-GAATTCCATATGGGTAAAATAATTGGTATC-3′
Reverse 5′- GAAGAAGTCAAAGACAAAAAAGGATCCCG-3′
TF Forward 5′- GGAATTCCATATGCAAGTTTCAGTTGAAAC-3′
Reverse 5′- GAGCTGATGAACCAGCAGGCGGGATCCCG-3′
MBP Forward 5′- GGAATTCCATATGAAAATAAAAACAGGTG-3′
Reverse 5′- CGCAGACTCGTATCACCAAGGTGGATCCCG-3′
RbsB Forward 5′-GGAATTCCATATGAACATGAAAAAACTGGC-3′
Reverse 5′-CTGAAACTGGTTGTTAAGCAGATGGATCCC-3′
EscN Forward 5′-GGAATTCCATATGACAACGTCAGACCGTCC-3′
Reverse 5′-AAGTCCGCTCTATTCTTGATGCGGATCCCG-3′
FKBP Forward 5′-GGAATTCCATATGAAATCACTGTTTAAAG-3′
Reverse 5′-CCGCAGATTCTGCTAAAAAAAAGCTTGGG-3′
AKN Forward 5′-GGAATTCCATATGGTGGTATCGTTTATCG-3′
Reverse 5′-GCTCTGGAAAAAATTCTCGGCGGATCCCG-3′
NusA Forward 5′-GGAATTCCATATGAACAAAGAAATTTTGGCTG-3′
Reverse 5′-GCTGGTTCGGTGACGAAGCGTGGGATCCCG-3′
GroES Forward 5′-GGAATTCCATATGAATATTCGTCCATTGCA-3′
Reverse 5′-ATTCTGGCAATTGTTGAAGCGGGATCCGCG-3′
Aβ42 Forward 5′-CGGGATCCAGATGAAGTTGATGATCGGAAATTTC-3′
Reverse 5′-CCGCTCGAGTCACGCAATCACCACGCCGCCCAC-3′
Ub-Aβ42 Forward 5′-CGCGGATCCCAGATCTTTGTGAAGAC-3′
Reverse 5′-CCGCTCGAGTCACGCTATGACAACACCGCC-3′
Ub-Aβ42(BamHI)
Forward 5′-CTCCGCGGTGGAGATGCAGGATTC-3′,
Reverse 5′-GGATCCTGCATCTCCACCGCGGAG-3′
Ub-Aβ40 Forward 5′-GGGGGAGTATGAATCGCCCTC-3′
Reverse 5′-GAGGCCGATTCATACTCCCCC-3′
Aβ(42–1) Forward 5′-CGCGGATCCCAGATCTTTGTGAAGAC-3′
Reverse1 5′-ATCACGCCACCAACGACAATCGCTCCACCGCGGAGGCGCAAGAC-3′
Reverse2 5′-TGAGTTTTTACCTGCGATAATTCCGAGCATCACGCCACCAACGAC-3′
Reverse3 5′-CAACACGAAAAATGCTTCATCCACAACTGAGTTTTTAACTGCGAT-3′
Reverse4 5′-TGATCCATATTCAACATGATGTTGTTTCAACACGAAAAATGCTTC-3′
Reverse5 5′-GAGTCAATCTGCTTCGAAGCGATGGTCTGATCCATATTCAACATG-3′
Reverse6 5′-CCGCTCGAGTCAATCTGCTTCGAA-3′
Humanin Forward 5′-CGCGGATCCCAGATCTTTGTGAAGAC-3′
Reverse1 5′-AAAACCACGCGGCGCCATTCCACCGCGGAGGCGCAA-3′,
Reverse2 5′-ATCAATTTCACCGGTCAGCAGCAGCAGGCAGCTAAAACCACGCGGCGCCA-3′
Reverse3 5′-CCGCTCCAGTCACGCACGACGTTTCACCGGCAGATCAATTTCACCGGTCA-3′
Casp-2 SSa Forward 5′-GGATTCCATATGGCCGGTAAGAAAAGTTG-3′
Reverse 5′-CGGATCCTGTGGGAGGGTGTCCTGG-3′
Casp-2 LSb Forward 5′-CGGATCCGGTCCTGTCTGCCTTCAAG-3′
Reverse 5′-GCGTTTTTTGCGGCCATCTTGTTGGTCAACCCC-3′
Reverse-caspase-2 Forward 5′-GAATTCCATATGCAAGTTTCAGTTGAAAC-3′
Reverse 5′-CGCGGATCCACCGCGGAGGCGCAACAACAG-3′
aSmall subunit. bLarge subunit.TF, trigger factor; MBP, maltose-binding protein; RbsB, D-ribose periplasmic protein; EscN, putative EscN protein; FKBP, FK-506-binding protein; AKN, adenylate kinase; NusA, transcription pausing factor L; Aβ(1–42), amyloid β(1–42).

RESULTS AND DISCUSSION
Identification of E. coli Thermostable Proteins
Many proteins and peptides overexpressed in E. coli form insoluble inclusion bodies (16), often causing difficulties in solubilization and recovery of the biological activity of the target proteins (4). Fusion of the target proteins or peptides to a protein that makes them soluble might be a way to solve these difficulties (17). In this study, we speculated that a highly soluble and stable protein could assist the proper folding and solubilization of heterogenous targets and that a thermostable protein might be a good choice for this purpose. Accordingly, we examined several endogenous E. coli thermostable proteins to see if they could make aggre-gation-prone peptides and proteins soluble.
To isolate thermostable proteins from the E. coliproteome, the cell extract was boiled for 10 min, and the soluble fraction was saved after extensive centrifugation. This fraction was subjected to two-dimensional (2-D) PAGE, which revealed about 15 proteins that were subse-quently analyzed by MALDI-TOF analysis (Figure 1A). Among the candidates, genes encoding 10 proteins were arbitrarily selected for cloning, expression, and testing of overexpression and thermostability. The DnaK, NusA, TF, FKBP, AKN, GroES, and EscN proteins were highly expressed and thermostable, whereas MBP partially precip-itated upon boiling (Figure 1B) despite its being expressed well (18). RbsB was also highly expressed, but for unknown reasons, two bands were observed. On the other hand, ribosomal protein S19 was poorly expressed (Figure 1B). After exclusion of some proteins that were not expressed well or that had already been tested before (5,19), DnaK, TF, FKBP, AKN, EscN, and MBP were selected for the following experiments.
Construction of a Plasmid Vector with E. coliThermostable Proteins
Aβ(1–42) peptide was chosen for initial testing because it had previously been reported to form inclusion bodies in E. coli (20) and had been purified from them (21). A plasmid vector was designed and constructed to contain the thermostable protein to be tested, ubiquitin sequences to allow specific cleavage of the target from the fusion protein by the deubiquitylating enzyme Usp2-cc (13), and Aβ(1–42)
(Figure 2A). Each construct with a thermostable protein was expressed in E. coli and examined for its overexpression and solubility. DnaK and TF fusions were highly soluble, whereas most (>70%) of the EscN fusion was found as in inclusion bodies (Figure 2B). MBP has been described in other studies as a fusion partner that promotes solubility, but its fusion with Aβ(1–42) was largely insoluble (Figure 2B). For unknown reasons, the FKBP and AKN fusion proteins were not expressed. On the other hand, the DnaK (22) and TF (23) fusion proteins were expressed well and were mostly soluble (Figure 2B). However, in some cases, DnaK complicated the evaluation of the expression level of fused proteins (data not shown) because this endogenous protein is highly expressed
www.syngene.com
USA Tel: 800-686-4407/301-662-2863 UK Tel: +44 (0)1223 727123
By combining Dyversity, our 16 bit
CCD based 2D gel imaging system
with Dymension, our rapid 2D analysis
software you’ll get the fastest 2D
imaging and analysis ever, without
compromising accuracy. So whether
you want to image gels stained with Coomassie Blue,
Deep Purple, Pro-Q Diamond, Silver Stain, SYPRO Ruby
or Cy dyes, the only difference you’ll see is how
much quicker you’ll get results.
To find how the
speed and
accuracy of
Dyversity and
Dymension can
help your
proteomics
research,
please
contact us.
Spot the Difference!Difference!
All trademarks acknowledged
Image from Laser Scanner Scans a gel set Cy2, Cy3 Scans a gel set Cy2, Cy3 and Cy5) at 100 microns.
Image from DyversityCaptures a gel set (Cy2, Cy3 Captures a gel set (Cy2, Cy3 and Cy5) at 98 microns.
Circle Reader Service No. 794
Research Reports
in E. coli. Therefore, TF was selected as the most suitable fusion partner (Figure 2, A and B).
Application of the Plasmid Vector for Purification of Other Peptides
TF-ubiquitin-Aβ(1–42) fusion protein was stable at 4°C for over a month. The initial purification through Ni-NTA column chromatography was followed by treatment of the fractions with Usp2-cc to cleave the peptide (Figure 3A). Then the digested fractions were subjected to conventional C18 column chromatography (Figure 3, A and B). Aβ(1–42) eluted as a single peak (Figure 3B) and was homogeneous in PAGE (Figure 3A, lane 3) and MALDI-TOF mass spectrometry (MS) analysis (measured mass, 4514.6 Da; expected value, 4513.8 Da; data not shown). Approximately 6 mg of the peptide per liter of bacterial culture could be obtained, which is about 1.5 times better than previously described results (21). Using this system, Aβ(1–40), Aβ(42–1), and humanin (24 amino acids) were also successfully purified, and the yield was comparable (∼8, ∼6, and ∼8 mg from 1 L of bacterial culture, respectively) (Figure 3C).
The biophysical and biochemical characteristics of amyloid peptide depend upon how it was produced or
prepared (24,25). The purified Aβ(1–42)
and Aβ(1–40) peptides were examined to see if they had the same characteristics as reported previously. Typical curves of fibrillogenesis (Figure 4A) were observed for both Aβ(1–42) and Aβ(1–40)
in a ThT binding assay, consistent with a previous report (14). In CD spectroscopy, the random coil confor-mation of freshly dissolved Aβ(1–42) and the β-sheet structure of the aggregated peptide were prominent (Figure 4B), demonstrating that the biophysical properties of the purified peptides were preserved (15). Furthermore, cellular toxicity of the two peptides (Figure 4C), with an increase of caspase-3 activity (Figure 4D) in human neuroblastoma SH-SY5Y cells, confirmed that the purified peptides were biologically functional.
Application of the Plasmid Vector for Protein Purification
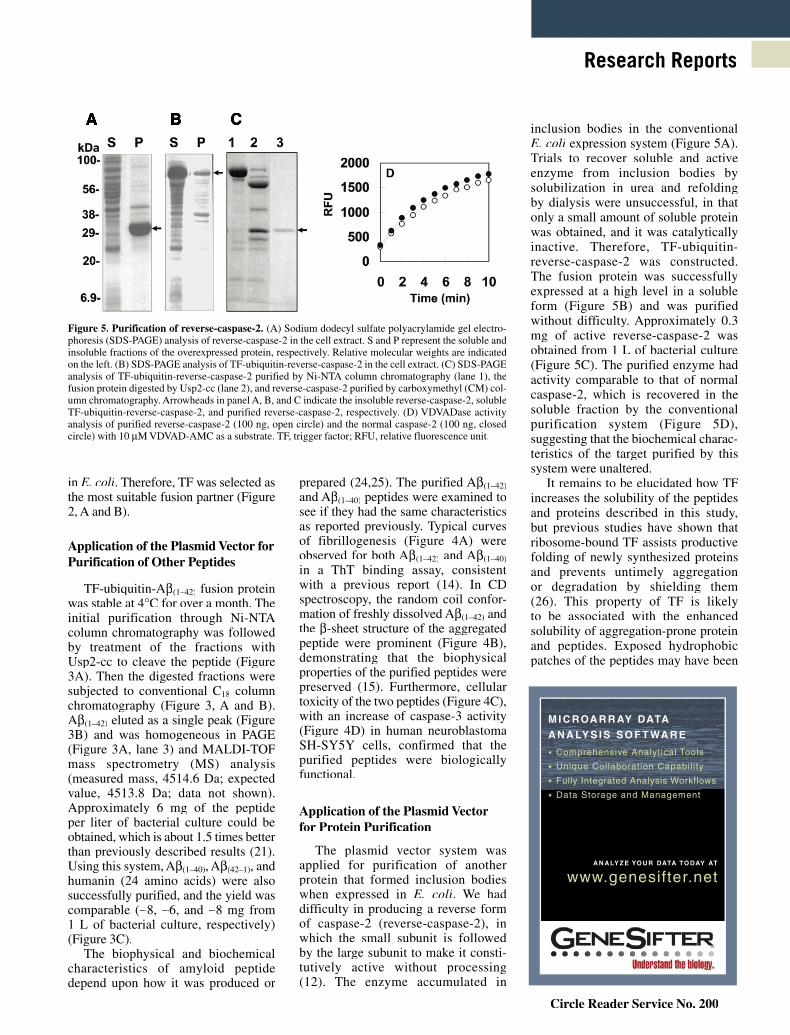
The plasmid vector system was applied for purification of another protein that formed inclusion bodies when expressed in E. coli. We had difficulty in producing a reverse form of caspase-2 (reverse-caspase-2), in which the small subunit is followed by the large subunit to make it consti-tutively active without processing (12). The enzyme accumulated in
inclusion bodies in the conventional E. coli expression system (Figure 5A). Trials to recover soluble and active enzyme from inclusion bodies by solubilization in urea and refolding by dialysis were unsuccessful, in that only a small amount of soluble protein was obtained, and it was catalytically inactive. Therefore, TF-ubiquitin-reverse-caspase-2 was constructed. The fusion protein was successfully expressed at a high level in a soluble form (Figure 5B) and was purified without difficulty. Approximately 0.3 mg of active reverse-caspase-2 was obtained from 1 L of bacterial culture (Figure 5C). The purified enzyme had activity comparable to that of normal caspase-2, which is recovered in the soluble fraction by the conventional purification system (Figure 5D), suggesting that the biochemical charac-teristics of the target purified by this system were unaltered.
It remains to be elucidated how TF increases the solubility of the peptides and proteins described in this study, but previous studies have shown that ribosome-bound TF assists productive folding of newly synthesized proteins and prevents untimely aggregation or degradation by shielding them (26). This property of TF is likely to be associated with the enhanced solubility of aggregation-prone protein and peptides. Exposed hydrophobic patches of the peptides may have been
Figure 5. Purification of reverse-caspase-2. (A) Sodium dodecyl sulfate polyacrylamide gel electro-phoresis (SDS-PAGE) analysis of reverse-caspase-2 in the cell extract. S and P represent the soluble and insoluble fractions of the overexpressed protein, respectively. Relative molecular weights are indicated on the left. (B) SDS-PAGE analysis of TF-ubiquitin-reverse-caspase-2 in the cell extract. (C) SDS-PAGE analysis of TF-ubiquitin-reverse-caspase-2 purified by Ni-NTA column chromatography (lane 1), the fusion protein digested by Usp2-cc (lane 2), and reverse-caspase-2 purified by carboxymethyl (CM) col-umn chromatography. Arrowheads in panel A, B, and C indicate the insoluble reverse-caspase-2, soluble TF-ubiquitin-reverse-caspase-2, and purified reverse-caspase-2, respectively. (D) VDVADase activity analysis of purified reverse-caspase-2 (100 ng, open circle) and the normal caspase-2 (100 ng, closed circle) with 10 μM VDVAD-AMC as a substrate. TF, trigger factor; RFU, relative fluorescence unit.
A B C
M I C ROA R R AY DATA
A N A LY S I S S O F T WA R E
* Comprehensive Analytical Tools
* Unique Collaboration Capability
* Fully Integrated Analysis Workflows
* Data Storage and Management
A N A LY Z E YO U R DATA TO DAY AT
www.genesifter.net
Circle Reader Service No. 200

796 ı BioTechniques ı www.biotechniques.com Vol. 44 ı No. 6 ı 2008
Research Reports
stabilized in the hydrophobic fold of TF. Peptidyl-propyl cis/tras isomerase activity has also been reported for TF (27). It is possible that the foldase activity of TF might also play some role in the promotion of solubility of the targets fused to TF.
In summary, a plasmid vector was developed that uses TF as a fusion partner to produce and recover aggre-gation-prone peptides and proteins in a soluble state. The easy purification steps, specific cleavage of the fusion protein, and satisfactory yields make it a promising tool for production of proteins and peptides that form the inclusion bodies in E. coli.
ACKNOWLEDGMENTS
A.T. and Md.S. contributed equally to this work. A.T., P.K., and G.R.D. were sup-ported by a Korea Research Foundation grant through a Ph.D./Master fellowship program for foreigners, 2004–2007 and 2003–2006, respectively. We thank Rohan T. Baker (Australian National University, Canberra, Australia) for providing pHUE and pHUsp2-cc plasmids.
COMPETING INTERESTS STATEMENT
The authors declare no competing interests.
REFERENCES
1. Makrides, S.C. 1996. Strategies for achieving high-level expression of genes in Escherichia coli. Microbiol. Rev. 60:512-538.
2. Catanzariti, A.M., T.A. Soboleva, D.A.Jans, P.G. Bord, and R.T. Baker. 2004. An efficient system for high-level expression and easy purification of authentic recombinant proteins. Protein Sci. 13:1331-1339.
3. Hannig, G. and S.C. Makrides. 1998. Strategies for optimizing heterologous pro-tein expression in Escherichia coli. Trends Biotechnol. 16:54-60.
4. Sorensen, H.P. and K.K. Mortensen. 2005. Advanced genetic strategies for recombinant protein expression in Escherichia coli. J. Biotechnol. 115:113-128.
5. De Marco, V., G. Stier, S. Blandin, and A.de Marco. 2004. The solubility and stabil-ity of recombinant proteins are increased by their fusion to NusA. Biochem. Biophys. Res. Commun. 322:766-771.
6. Kapust, R.B. and D.S. Waugh. 1999. Escherichia coli maltose-binding protein is uncommonly effective at promoting the sol-ubility of polypeptides to which it is fused. Protein Sci. 8:1668-1674.
7. Marblestone, J.G., S.C. Edavettal, Y.Lim, P. Lim, X. Zuo, and T.R. Butt. 2006. Comparison of SUMO fusion technol-ogy with traditional gene fusion systems: enhanced expression and solubility with SUMO. Protein Sci. 15:182-189.
8. Fox, J.D., R.B. Kapust, and D.S. Waugh.2001. Single amino acid substitutions on the surface of Escherichia coli maltose-bind-ing protein can have a profound impact on the solubility of fusion proteins. Protein Sci. 10:622-630.
9. de Marco, A., E. Casatta, S. Savaresi, andA. Geerlof. 2004. Recombinant proteins fused to thermostable partners can be purified by heat incubation. J. Biotechnol. 107:125-133.
10. Shahnawaz, M., A. Thapa, and I.S. Park.2007. Stable activity of a deubiquitylating enzyme (Usp2-cc) in the presence of high concentrations of urea and its application to purify aggregation-prone peptides. Biochem. Biophys. Res. Commun. 359:801-805.
11. Evangelou, A., C. Zikos, E. Livaniou, andG.P. Evangelatos. 2004. High-yield, solid-phase synthesis of humanin, an Alzheimer’s disease associated, novel 24-mer peptide which contains a difficult sequence. J. Pept. Sci. 10:631-635.
12. Baliga, B.C., P.A. Colussi, S.H. Read, M.M.Dias, D.A. Jans, and S. Kumar. 2003. Role of prodomain in importin-mediated nuclear localization and activation of caspase-2. J. Biol. Chem. 278:4899-4905.
13. Baker, R.T., A.M. Catanzariti, Y.Karunasekara, T.A. Soboleva, R.Sharwood, S. Whitney, and P.G. Board.2005. Using deubiquitylating enzymes as research tools. Methods Enzymol. 398:540-554.
14. Naiki, H. and K. Nakakuki. 1996. First-order kinetic model of Alzheimer’s beta-amyloid fibril extension in vitro. Lab. Invest. 74:374-383.
15. Kirkitadze, M.D., M.M. Condron, andD.B. Teplow. 2001. Identification and char-acterization of key kinetic intermediates in amyloid beta-protein fibrillogenesis. J. Mol. Biol. 312:1103-1119.
16. Turner, P., O. Holst, and E.N. Karlsson.2005. Optimized expression of soluble cy-clomaltodextrinase of thermophilic origin in Escherichia coli by using a soluble fusion-tag and by tuning of inducer concentration. Protein Expr. Purif. 39:54-60.
17. Schrodel, A., J. Volz, and A. de Marco.2005. Fusion tags and chaperone co-expres-sion modulate both the solubility and the inclusion body features of the recombinant CLIPB14 serine protease. J. Biotechnol. 120:2-10.
18. Nallamsetty, S. and D.S. Waugh. 2006. Solubility-enhancing proteins MBP and NusA play a passive role in the folding of their fusion partners. Protein Expr. Purif. 45:175-182.
19. Ideno, A., M. Furutani, T. Iwabuchi, T.Iida, Y. Iba, Y. Kurosawa, H. Sakuraba, T.Ohshima, et al. 2004. Expression of foreign proteins in Escherichia coli by fusing with an archaeal FK506-binding protein. Appl. Microbiol. Biotechnol. 64:99-105.
20. Wurth, C., N.K. Guimard, and M.H.Hecht. 2002. Mutations that reduce aggrega-tion of the Alzheimer’s Abeta42 peptide: an unbiased search for the sequence determi-nants of Abeta amyloidogenesis. J. Mol. Biol. 319:1279-1290.
21. Lee, E.K., J.H. Hwang, D.Y. Shin, D.I. Kim, and Y.J. Yoo. 2005. Production of recombi-nant amyloid-beta peptide 42 as an ubiquitin extension. Protein Expr. Purif. 40:183-189.
22. Genevaux, P., F. Keppel, F. Schwager, P.S.Langendijk-Genevaux, F.U. Hartl, and C.Georgopoulos. 2004. In vivo analysis of the overlapping functions of DnaK and trigger factor. EMBO Rep. 5:195-200.
23. Maier, T., L. Ferbitz, E. Deuerling, and N.Ban. 2005. A cradle for new proteins: trig-ger factor at the ribosome. Curr. Opin. Struct. Biol. 15:204-212.
24. Dobeli, H., N. Draeger, G. Huber, P. Jakob,D. Schmidt, B. Seilheimer, D. Stuber, B.Wipf, et al. 1995. A biotechnological method provides access to aggregation competent monomeric Alzheimer’s 1-42 residue amyloid peptide. Biotechnology (NY) 13:988-993.
25. Busciglio, J., A. Lorenzo, and B.A.Yankner. 1992. Methodological variables in the assessment of beta amyloid neurotoxicity. Neurobiol. Aging 13:609-612.
26. Hoffmann, A., F. Merz, A. Rutkowska, B.Zachmann-Brand, E. Deuerling, and B.Bukau. 2006. Trigger factor forms a protec-tive shield for nascent polypeptides at the ri-bosome. J. Biol. Chem. 281:6539-6545.
27. Lyon, W.R. and M.G. Caparon. 2003. Trigger factor-mediated prolyl isomerization influences maturation of the Streptococcus pyogenes cysteine protease. J. Bacteriol. 185:3661-3667.
Received 3 October 2007; accepted 12 December 2007.
Address correspondence to Il-Seon Park, RCPM, Department of Biomaterials Engineering, Cell and Molecular Biology, College of Medicine, Chosun University, Gwanju, 501-759, Republic of Korea. e-mail: [email protected]
To purchase reprints of this article, contact: [email protected]
Copyright © 2022 FDOKUMEN




















