
An Electrochemical Study of the Dissolution of Pure Gold And ...

Oxidative UO2 Dissolution Induced by Soluble Mn(III)Zimeng Wang,*,† Wei Xiong,† Bradley M. Tebo,‡ and Daniel E. Giammar†
†Department of Energy, Environmental and Chemical Engineering, Washington University in St. Louis, St. Louis, Missouri 63130,United States‡Division of Environmental and Biomolecular Systems, Oregon Health and Science University, Portland, Oregon 97239, UnitedStates
*S Supporting Information
ABSTRACT: The stability of UO2 is critical to the success ofreductive bioremediation of uranium. When reducing con-ditions are no longer maintained, Mn redox cycling maycatalytically mediate the oxidation of UO2 and remobilizationof uranium. Ligand-stabilized soluble Mn(III) was recentlyrecognized as an important redox-active intermediate in Mnbiogeochemical cycling. This study evaluated the kinetics ofoxidative UO2 dissolution by soluble Mn(III) stabilized bypyrophosphate (PP) and desferrioxamine B (DFOB). TheMn(III)−PP complex was a potent oxidant that induced rapidUO2 dissolution at a rate higher than that by a comparableconcentration of dissolved O2. However, the Mn(III)−DFOBcomplex was not able to induce oxidative dissolution of UO2.The ability of Mn(III) complexes to oxidize UO2 was probably determined by whether the coordination of Mn(III) with ligandsallowed the attachment of the complexes to the UO2 surface to facilitate electron transfer. Systematic investigation into thekinetics of UO2 oxidative dissolution by the Mn(III)−PP complex suggested that Mn(III) could directly oxidize UO2 withoutinvolving particulate Mn species (e.g., MnO2). The expected 2:1 reaction stoichiometry between Mn(III) and UO2 was observed.The reactivity of soluble Mn(III) in oxidizing UO2 was higher at lower ratios of pyrophosphate to Mn(III) and lower pH, whichis probably related to differences in the ligand-to-metal ratio and/or protonation states of the Mn(III)−pyrophosphatecomplexes. Disproportionation of Mn(III)−PP occurred at pH 9.0, and the oxidation of UO2 was then driven by both MnO2 andsoluble Mn(III). Kinetic models were derived that provided excellent fits of the experimental results.
■ INTRODUCTIONAs an in situ remediation strategy for uranium contamination insoil and groundwater, microbial metabolism stimulated byorganic electron donors can reduce soluble U(VI) to U(IV),producing biogenic UO2 and other low solubility U(IV)species.1−4 The longevity of UO2 with respect to oxidativedissolution is critical to the success of uranium bioremediation.Once the supply of electron donor ceases, UO2 can bereoxidized by subsurface oxidants that include O2,
5 oxidizednitrogen species,6 and metal oxides (e.g., Fe(III)7,8 and Mn9,10
oxides). Therefore, an understanding of the kinetics andmechanisms of UO2 oxidation and dissolution is necessary forthe optimal design of bioremediation strategies and the long-term stewardship of spent nuclear fuel.1
There is increasing evidence that U(IV) stability can beinfluenced by Mn biogeochemical cycles through both solid andsoluble forms of oxidized Mn. Mn is present at appreciableconcentrations at several U-contaminated sites (e.g., up to 20μM in groundwater at a site in Rifle, Colorado11,12). OxidizedMn species can exert major geochemical impact even if they areonly present as minor constituents in the subsurface.13 Thenatural occurrence of Mn oxides (e.g., MnO2) is largely drivenby Mn-oxidizing microorganisms in a process that occurs
rapidly even at extremely low dissolved O2 concentration.14−16
The introduction of Mn(II) into microoxic subsurface regionsmay provide conditions ideal for microbial Mn oxidationcoupled to the abiotic oxidation of UO2. A recent laboratorystudy found that active Mn bio-oxidation under low O2conditions dramatically promoted UO2 dissolution comparedwith O2 alone.17 Because Mn(II) that is oxidized can beregenerated from the reaction between UO2 and Mn oxide,only small amounts of Mn(II) were needed to sustain thecoupled redox processes as long as a supply of O2 (even low)was available. U(IV) oxidation mediated by Mn is intuitivelyhypothesized to involve the reaction between UO2 and Mnoxide, which appears to require direct contact due to the lowsolubilities of the two minerals.18,19 In addition to the solid−solid interaction, reaction pathways of U(IV) oxidationmediated by soluble Mn species may be important in thepresence of certain ligands and in the process of in situ Mn bio-oxidation.18
Received: August 21, 2013Revised: November 21, 2013Accepted: November 28, 2013
Article
pubs.acs.org/est
© XXXX American Chemical Society A dx.doi.org/10.1021/es4037308 | Environ. Sci. Technol. XXXX, XXX, XXX−XXX

Soluble Mn(III) species can play important roles in variousbiogeochemical processes in subsurface environments.20 It haslong been known that free Mn(III) ions rapidly dispropor-tionate to form Mn(II) and MnO2, although soluble Mn(III)can be stable at extremely low pH or through complexationwith high affinity ligands.21 Studies at environmentally relevantconditions suggested that ligand-stabilized Mn(III) species mayoccur as intermediates of Mn oxidation22−24 or reduction25,26
and as ligand promoted dissolution products of Mn(III)-bearing minerals (e.g., MnOOH).27−29 These ligands, such aspyrophosphate,30 siderophores28,31 oxalate, and citrate arewidely present in natural aquatic systems, particularly inrhizosphere soil environments. Because many of the Mn(III)ligands have biological origins, their presence in organicelectron donor stimulated bioremediation sites may also bepossible. More recent evidence of aqueous Mn(III) in suboxicregions of oceanic environments32 and estuary sedimentporewaters33,34 demonstrated that Mn(III) can constitute alarge fraction of the dissolved Mn pool at oxic/anoxic interfacesin a wide range of environmental settings.Although Mn(III) species have been anticipated as a
potential environmental oxidant, their reactivity and kineticcharacteristics are not fully appreciated or understood. SolidMn oxides that contain more Mn(III) were found to be morereactive oxidants for Cr(III) and organics than those oxideswith less or no Mn(III).35−37 A recent study38 demonstratedthat the oxidative decomposition of carbadox (C11H10N4O4) byMnO2 could be dramatically accelerated by addition of oxalate,in which the redox reaction between MnO2 and oxalateproduced soluble Mn(III) stabilized by excess oxalate.Conversely, other studies suggested that formation of Mn(III)
passivated the surface of MnO2 and made it less reactive.39,40
Although studies have shown that soluble Mn(III) can oxidizeFe(II), sulfide, organic compounds,41,42 and Cr(III),43 otherstudies have suggested that soluble Mn(III) pyrophosphatecomplexes could not readily oxidize Cr(III) unless MnO2 wasformed from disproportionation.44−46
The objectives of this study were to (1) evaluate and quantifythe kinetics of UO2 oxidative dissolution by soluble Mn(III) atenvironmentally relevant conditions in the presence of naturallyoccurring ligands (pyrophosphate and desferrioxamine B), and(2) assess the effect of pH and ligand-to-metal ratio on thereactivity of Mn(III) with respect to UO2 oxidation. Knowledgeof the kinetics of Mn(III)−UO2 reaction can also provideinsights into Mn-mediated oxidation of other redox-activecontaminants and nutrients in subsurface environments.
■ MATERIALS AND METHODSMaterials. Synthetic uraninite (UO2) was prepared by
heating studtite (UO2O2(H2O)2·2H2O) in a continuous flow ofpure hydrogen at 400 °C as described by Ulrich et al.5 As arepresentative inorganic complexant, pyrophosphate (P2O7
4−,PP) was selected as the primary Mn(III) ligand used in thisstudy. Pyrophosphate is relevant to soil and subsurfaceenvironments with biological origin30,47 and is redox-inert.42
Desferrioxamine B (C25H46N5O8NH3+, DFOB), which is an
environmentally relevant and widely used biogenic trihydrox-amate siderophore,23,48 was also used in selected experiments.The preparation of the Mn(III)−PP stock solution followed theprocedures in Kostka et al.41 and Trouwborst et al.41 and isbriefly described here. Sodium pyrophosphate decahydrate(>99%, Sigma-Aldrich) was dissolved in water to reach a
Table 1. Summary of Batch Experiments Using Pyrophosphate as a Mn(III) Liganda
expb pHc Mn(III) (μM) UO2 (μM) Mn(II) (μM) PP (mM) PP:Mn(III) ratio rate constant k1d (L·mol−1·h−1)
Variation of Mn(III) Concentrations1 7.5 300 300 0 2 6.7 37.0 ± 3.42 7.5 200 300 0 2 10 31.6 ± 2.63 7.5 100 300 0 2 20 25.9 ± 2.54 7.5 50 300 0 2 40 17.4 ± 0.95 7.5 25 300 0 2 80 14.5 ± 0.6
Variation of Pyrophosphate (PP) Concentrations6 7.5 200 300 0 1.2 6 55.4 ± 6.27 7.5 200 300 0 4 20 18.6 ± 1.0
Variation of pH8 6.5 200 300 0 2 10 112 ± 13.79 9.0 200 300 0 2 10 23.5 ± 3.1e
Control Experiments10 6.5 0 300 0 2 N.A.g N.A.11 7.5 0 300 0 2 N.A. N.A.12 9.0 0 300 0 2 N.A. N.A.13 6.5 0 0 200 2 N.A. N.A.14 9.0 200 0 0 2 10 N.A.15f 7.5 0 300 0 2 N.A. N.A.
aThis table only summarizes the experiments involving pyrophosphate as the Mn(III) ligand. Note that there are some experiments (shown inFigure 1b) involving DFOB. For the consistency and conciseness of this table, DFOB experiments are not numbered throughout the paper.bExperiments were performed in duplicate. cThe pH was monitored during the experiments and varied by less than 0.1. dThe rate constant for theUO2−Mn(III) reaction was obtained by fitting the experimental dissolved U and Mn(III) data using the kinetic rate expression of eqs 5 and 6. Theuncertainty indicates the range of the rate constant that yielded sum of square of residuals within 10% of the value obtained by the optimized rateconstant. eThe rate constant for the Mn(III)−UO2 reaction at pH 9.0 was estimated using a model that considered Mn(III) disporportionation (seedetails in text). The model also included a rate constant (k2) for UO2 reaction with MnO2 that was generated from disproportionation. fA controlexperiment of UO2 dissolution in a fully oxic condition (PO2 = 0.21 bar). Experiments were conducted on a laboratory bench outside the anaerobicchamber. The reactor was an open system which maintained the dissolved O2 concentration at 280 μm in equilibrium with ambient air. gN.A.: Notapplicable.
Environmental Science & Technology Article
dx.doi.org/10.1021/es4037308 | Environ. Sci. Technol. XXXX, XXX, XXX−XXXB
concentration of 50 mM with pH adjusted to 8.2. Manganese-(III) acetate dihydrate (>97%, Sigma-Aldrich) solid was slowlyadded to the sodium pyrophosphate solution during vigorousstirring to form a bright purple solution with 10 mM Mn(III)and 50 mM PP. The final pH was adjusted to 7.0. Theconcentration and purity (>98%) of the Mn(III)−PP stocksolution was confirmed by inductively coupled plasma massspectrometry (ICP-MS) for total Mn and by an iodometrictitration for oxidation state.32 The preparation of the Mn(III)−DFOB stock solution followed the procedures in Faulkner etal.,49 which were also used in several recent studies.23,33 Briefly,desferrioxamine mesylate (>92.5%, Sigma-Aldrich) solid wasdissolved in water to reach a concentration of 5 mM.Concentrated manganese(II) chloride solution was added toreach a Mn(II) concentration of 5 mM. The pH of the solutionwas adjusted to 9.0. The solution was aerated for 12 h, whichresulted in Mn(II) oxidation to Mn(III) and yielded a cleargreen solution. This solution was filtered (0.02-μm poly-ethersulfone, Tisch Environmental, OH) to remove anypossible Mn precipitates. The yield of the Mn(III)−DFOBsynthesis was higher than 90%.23 Both Mn(III)−PP andMn(III)−DFOB stock solutions were freshly made and usedwithin three weeks in this study. H2-sparged anoxic ultrapurewater (resistivity >18.2 MΩ·cm) was used to prepare allsolutions. For the Mn(III)−DFOB solution, the dissolved O2introduced in the preparation process was removed by spargingwith a 95% N2/5% H2 gas mixture overnight. HNO3 andNaOH were used for pH adjustment.Batch Experiments. Batch experiments of UO2 oxidation
and dissolution by soluble Mn(III) were performed in ananaerobic chamber (Coy Lab Products Inc., MI) with acomposition of O2 controlled below 1 ppm by circulating anatmosphere of 95% N2/5% H2 over a heated Pd catalyst (Table1). PP was always in excess of Mn(III) and also served as thepH buffer (pKa = 0.9, 2.0, 6.6, and 9.450). For the experimentsinvolving DFOB (pKa = 8.4, 9.4, and 10.1 for the threehydroxyl groups and 11.7 for the terminating amine group23), 5mM MOPS (3-(N-morpholino)propanesulfonic acid, pKa =7.2) was added as the buffer for pH 7.5. Dissolved inorganiccarbon (DIC) was added as NaHCO3 at a concentration of 1.0
mM for all experiments, which increased the solubility of U(VI)produced by UO2 oxidation and allowed dissolved U to reflectthe total extent of UO2 oxidation. One experiment (exp 15 inTable 1) was performed under ambient oxic condition in theabsence of Mn(III), which contained a fixed concentration ofdissolved O2 at 280 μM. The details of the experimentalprocedure of preparing the batch reactors are described in theSupporting Information (SI). During the experiments, pH wasmonitored and samples were periodically collected and filteredwith 0.02-μm polyethersulfone syringe filters (Tisch Environ-mental, OH). Half of the filtrate was preserved in 2% HNO3 fortotal dissolved U and Mn analysis by ICP-MS. The other halfwas immediately used for Mn(III) analysis. Selected solidsamples were collected for X-ray diffraction (XRD) analysis toidentify possible secondary precipitates.
Analytical Methods. Dissolved U and Mn concentrationswere measured by ICP-MS (Agilent 7500ce). Although thedissolved uranium concentration obtained from ICP-MS couldnot differentiate between U(VI) and U(IV), Mn(III)-freecontrol experiments confirmed that the primary form of thedissolved uranium was U(VI) as long as oxidation occurred.Dissolved Mn is the sum of Mn(II) and Mn(III). Decreases intotal dissolved Mn were attributed to the formation of insolubleMnO2 unless other secondary precipitation was noted. ForMn(III) analysis in the PP experiments, the absorbance offiltered samples at 258 nm was measured on a UV−visspectrophotometer (PerkinElmer-Lambda XLS), following amethod previously used to measure Mn(III) intermediatesduring the bacterial oxidation of Mn in the presence of PP.22
This method was selected for its simplicity and low detectionlimit (<5 μM, with ε258 nm > 6000 M−1 cm−1). Although theabsorbance was linearly proportional to the concentration ofMn(III) concentration, adding U(VI) (as UO2(NO3)2) incalibration standards increased absorbance, probably due to theUV-range absorbance of U(VI)−PP51 and U(VI)−CO3complexes. We performed a double-variable calibration bymeasuring the absorbance of a set of standards with differentcombinations of Mn(III) and U(VI) concentrations (Figure S1of the SI). The fact that all data points fell on a plane suggestedthat the absorbances induced by Mn(III) and U(VI) at 258 nm
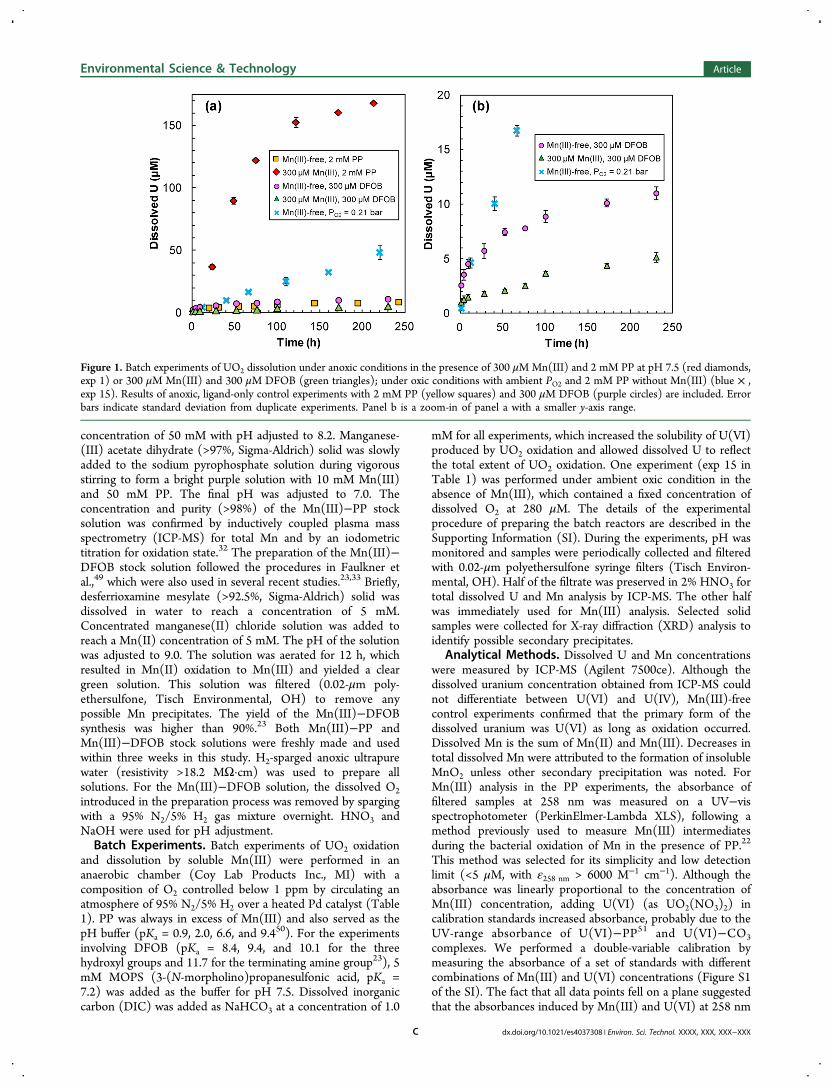
Figure 1. Batch experiments of UO2 dissolution under anoxic conditions in the presence of 300 μMMn(III) and 2 mM PP at pH 7.5 (red diamonds,exp 1) or 300 μM Mn(III) and 300 μM DFOB (green triangles); under oxic conditions with ambient PO2 and 2 mM PP without Mn(III) (blue × ,exp 15). Results of anoxic, ligand-only control experiments with 2 mM PP (yellow squares) and 300 μM DFOB (purple circles) are included. Errorbars indicate standard deviation from duplicate experiments. Panel b is a zoom-in of panel a with a smaller y-axis range.
Environmental Science & Technology Article
dx.doi.org/10.1021/es4037308 | Environ. Sci. Technol. XXXX, XXX, XXX−XXXC

were independent of each other. Consequently, a double-variable linear model was a reliable Mn(III) analytical method,given that U(VI) concentration could be measured independ-ently by ICP-MS. This method was verified to be insensitive tothe variation of pH (from 6.5 to 9.0) and concentrations of PPand DIC (from 1 to 5 mM). The quantification of theMn(III)−DFOB concentration was enabled by the measure-ment of absorbance at 310 nm (ε310 nm≈ 2000 M−1 cm−1). Itwas verified that the presence of U(VI) did not affect theabsorbance. Solid samples were viewed with a JEOL JSM07001FLV scanning electron microscope equipped with an Oxfordenergy dispersive X-ray spectrometer (EDXS). X-ray diffraction(XRD) was performed on a Rigaku Geigerflex D-MAX/Adiffractometer using Cu Ka radiation.Kinetic and Equilibrium Modeling. The kinetics of the
UO2−Mn(III) reaction were modeled with a spreadsheet solverfor determining optimal parameters in the rate equationsdiscussed below. Experimental data fitting and parameteroptimization followed the approach of residual sum of squares(RSS) minimization. Chemical equilibrium modeling wasperformed using MINEQL+,52 in which pyrophosphate wasadded as a stable basis component species because itshydrolysis is slow at environmentally relevant conditions inabiotic systems.30
■ RESULTS AND DISCUSSIONUO2 Dissolution: Mn(III)−PP vs Mn(III)−DFOB. The
rates of UO2 dissolution in the presence of Mn(III)−PP,Mn(III)−DFOB, and dissolved O2 can be compared in Figure1. The rapid dissolution of UO2 in the presence of Mn(III)−PPcompared with the negligible dissolution in the PP-only anoxiccontrol experiment indicated that soluble Mn(III)−PP was apotent oxidant for UO2. The UO2 dissolution rate with aninitial Mn(III)−PP concentration of 300 μM was six timeshigher than in a control experiment with 280 μM dissolved O2(i.e., PO2 = 0.21 bar) and an equal concentration of PP. Thepresent comparison was made on the basis of moles instead ofelectron-equivalents of oxidants. The faster oxidation byMn(III)−PP occurred even with Mn(III) having only 1/4 thetotal oxidizing capacity of dissolved O2. Moreover, thecomparison was made between a limited initial concentrationof Mn(III) and a fixed concentration of dissolved O2maintained by the equilibrium with the ambient air.In contrast to the Mn(III)−PP complex, the Mn(III)−
DFOB complex was not able to induce oxidative dissolution ofUO2. UO2 dissolution in the presence of Mn(III)−DFOB wasmuch less than for Mn(III)−PP or dissolved O2. This was alsoconfirmed by the negligible change of the Mn(III)−DFOBconcentration (data not shown). Interestingly, the dissolved Uconcentration was higher for the experiment with DFOB onlythan for the one with both DFOB and Mn(III). As a U(IV)ligand, DFOB can promote UO2 dissolution.
53 In the presenceof an equimolar concentration of Mn(III), DFOB may bepreferentially bound to aqueous Mn(III), which limits its abilityto facilitate ligand-promoted dissolution of UO2.The contrasting results suggest that the redox activity of
soluble Mn(III) is strongly dependent on the donor groupidentity and ligand structure (see TOC Art), which determinesthe coordination environments of Mn(III) and theirthermodynamic and kinetic favorability with respect to U(IV)oxidation.31 The structure of PP only allows it to formbidentate coordination with Mn(III) through two hydroxylgroups on the two phosphorus atoms (i.e., a single PP ligand
cannot occupy all six Mn(III) coordination spots). Thestructure of DFOB determined that the Mn(III)−DFOBcomplex at circumneutral pH is a hexadentate complex(Mn(III)HDFOB+) with three five-membered chelate ringswith the deprotonated hydroxamate groups.23,54 This stronghexadentate coordination may limit the ability of the Mn(III)complex to associate with UO2 surface to enable electrontransfer. Frazier et al. found that a Fe(III)−DFOB complex alsocould not promote UO2 dissolution, and they attributed that tothe diminishing reduction potential of the Fe(III)/Fe(II) pairinduced by the strong complexation effect.53 Stewart et al.recently reported a similar inhibitory effect of strong Fe(III)chelators on UO2 oxidation by ferrihydrite.55 Thermodynamicconstraints of UO2 oxidation by Fe(III) have previouslyestablished that the energetic favorability of UO2 oxidation byFe(III) oxides is very sensitive to the solution chemistry andthe crystalline phases of ferric oxides.56 Our comparativeanalysis of the redox couples of UO2 and various Mn oxidesindicated that the redox reaction of UO2 and Mn(III, IV)oxides is thermodynamically favorable over a much wider rangeof environmental conditions than for the reaction between UO2and ferric oxides.18 Electrochemical and equilibrium analysis byDuckworth and Sposito23 reported a large stability constant ofthe Mn(III)HDFOB+
+ ⇌ =− + +HDFOB Mn Mn(III)HDFOB LogK 29.92 3
(1)
and a standard reduction potential
+ ⇌
=
+ −Mn(III)HDFOB e Mn(II)HDFOB
Eh 0.176 V
aq
o (2)
Using the protonation and stability constants reportedtherein, the dominant Mn(III), Mn(II), and free DFOB speciesat pH 7.5 are predicted to be Mn(III)HDFOB+, Mn2+, andH4DFOB
+, respectively (Figure S2 of the SI). DFOB is such astrong complexant for Mn(III) so that the dominant Mn(III)species is Mn(III)HDFOB+. Therefore, the standard reductionpotential of the half reaction written in terms of the majorspecies is
+ + ⇌ +
=
+ + − + +Mn(III)HDFOB 3H e Mn H DFOB
Eh 1.368 V
24
o (3)
The Eh calculated for pH 7.5, 150 μM Mn2+, 150 μMMn(III)HDFOB+, and 150 μM H4DFOB
+ (corresponding to50% conversion of the initial 300 μM Mn(III)−DFOB) was0.266 V. This value is still much higher than the calculated Ehof 0.01 V for the redox pair UO2(CO3)2
2−/UO2(s) at pH 7.5with 75 μM U(VI) and 1 mM DIC, where UO2(CO3)2
2− andHCO3
− represent the dominant U(VI) and inorganic carbonspecies. Therefore, the inability of Mn(III)−DFOB to promoteUO2 dissolution observed in the present study may beattributed to a combined effect of the structural hindrance ofthe coordination of DFOB-surrounded Mn(III) with the UO2surface, the diminishing thermodynamic driving force, and thestronger affinity of DFOB to aqueous Mn(III) than to surfaceU(IV). Because the dissolution of UO2 was not accelerated byMn(III)−DFOB and would also not be anticipated to occurover a range of conditions, the subsequent discussion focuseson the effects of water chemistry on UO2 oxidation by Mn(III)pyrophosphate.
Environmental Science & Technology Article
dx.doi.org/10.1021/es4037308 | Environ. Sci. Technol. XXXX, XXX, XXX−XXXD
Stoichiometry and Reaction Kinetics. For experimentsusing pyrophosphate (Table 1), the anticipated electronequivalent balance predicts a 2:1 ratio between the con-sumption of Mn(III) and production of U(VI).
+ = +U 2Mn U 2MnIV(s)
III(aq)
VI(aq)
II(aq) (4)
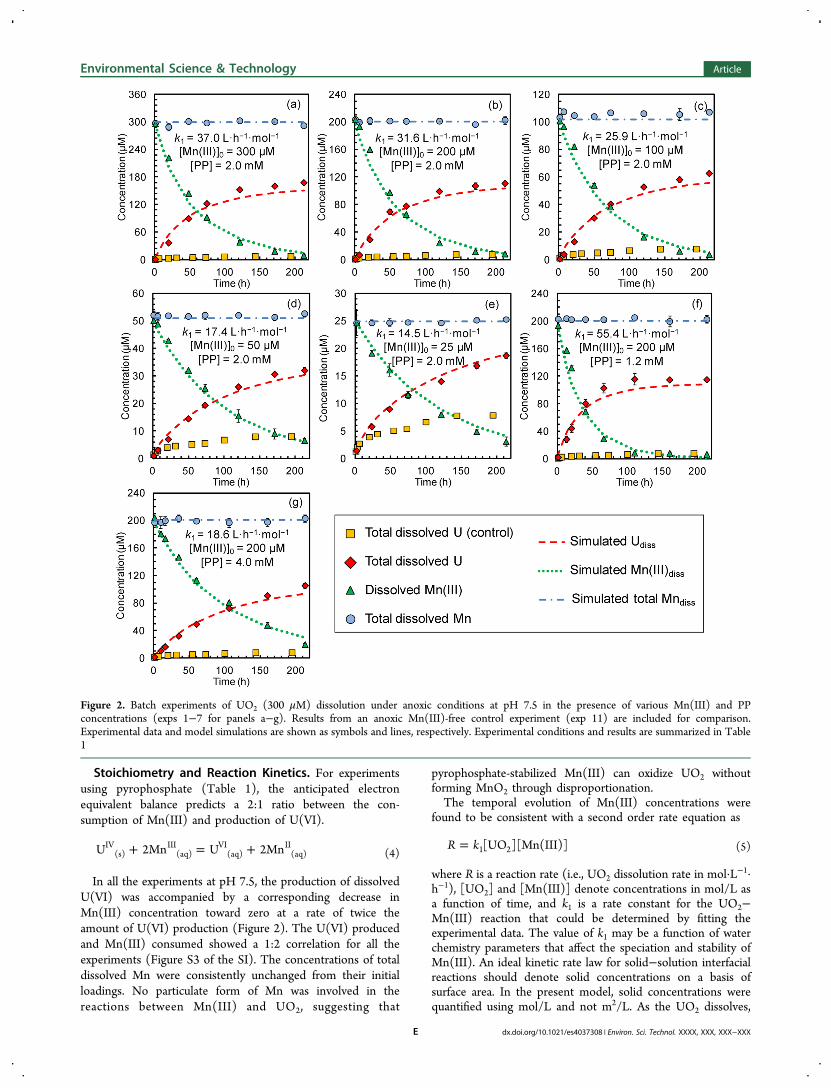
In all the experiments at pH 7.5, the production of dissolvedU(VI) was accompanied by a corresponding decrease inMn(III) concentration toward zero at a rate of twice theamount of U(VI) production (Figure 2). The U(VI) producedand Mn(III) consumed showed a 1:2 correlation for all theexperiments (Figure S3 of the SI). The concentrations of totaldissolved Mn were consistently unchanged from their initialloadings. No particulate form of Mn was involved in thereactions between Mn(III) and UO2, suggesting that
pyrophosphate-stabilized Mn(III) can oxidize UO2 withoutforming MnO2 through disproportionation.The temporal evolution of Mn(III) concentrations were
found to be consistent with a second order rate equation as
=R k [UO ][Mn(III)]1 2 (5)
where R is a reaction rate (i.e., UO2 dissolution rate in mol·L−1·h−1), [UO2] and [Mn(III)] denote concentrations in mol/L asa function of time, and k1 is a rate constant for the UO2−Mn(III) reaction that could be determined by fitting theexperimental data. The value of k1 may be a function of waterchemistry parameters that affect the speciation and stability ofMn(III). An ideal kinetic rate law for solid−solution interfacialreactions should denote solid concentrations on a basis ofsurface area. In the present model, solid concentrations werequantified using mol/L and not m2/L. As the UO2 dissolves,
Figure 2. Batch experiments of UO2 (300 μM) dissolution under anoxic conditions at pH 7.5 in the presence of various Mn(III) and PPconcentrations (exps 1−7 for panels a−g). Results from an anoxic Mn(III)-free control experiment (exp 11) are included for comparison.Experimental data and model simulations are shown as symbols and lines, respectively. Experimental conditions and results are summarized in Table1
Environmental Science & Technology Article
dx.doi.org/10.1021/es4037308 | Environ. Sci. Technol. XXXX, XXX, XXX−XXXE

both the mol/L and m2/L of UO2 decrease. The decreases arenot completely proportional, but the deviation from linearityover the conditions studied was verified to be negligible withrespect to the change of the corresponding molar concen-trations. In the absence of Mn(III) disproportionation (as forthe results shown in Figure 2), the stoichiometry links thechanges in concentrations of U(VI) and Mn(III)
= = −Rt t
d[U(VI)]d
12
d[Mn(III)]d (6)
The constraint of stoichiometry with known initialconditions allowed the separation of eq 6 as two independentlinear differential equations for solving [U(VI)] and [Mn(III)]as functions of time
= −
−t
kd[U(VI)]
d([UO ] [U(VI)])([Mn(III)]
2[U(VI)])
1 2 0 0
(7)
= − −−⎛
⎝⎜⎞⎠⎟t
kd[Mn(III)]
d2 [UO ]
[Mn(III)] [Mn(III)]2
[Mn(III)]
1 2 00
(8)
where [UO2]0 and [Mn(III)]0 denote initial concentrations(mol/L). Analytical solutions are given as
=− −
− −k t
k t
[U(VI)][Mn(III)] [UO ] (exp( ([Mn(III)] 2[UO ] )) 1)
[Mn(III)] exp( ([Mn(III)] 2[UO ] )) 2[UO ]0 2 0 1 0 2 0
0 1 0 2 0 2 0 (9)
=−
− −k t
[Mn(III)](2[UO ] [Mn(III)] )[Mn(III)]
2[UO ] exp( (2[UO ] [Mn(III)] )) [Mn(III)]2 0 0 0
2 0 1 2 0 0 0 (10)
Equations 9 and 10 were used to simulate the experimentaldata of U(VI) and Mn(III) simultaneously with k1 as the onlyoptimization parameter. The fitting accounted for theproduction of U(VI) that could occur in the absence ofMn(III) as determined from Mn(III)-free control experiments.Because the change in Mn(III) concentration as it was reducedto Mn(II) was twice the change in U(VI) concentration due tooxidation of U(IV), a weighting factor of 1/4 was applied in thecalculation of the residual sum of squares for [Mn(III)] results.The excellent fit of both [U(VI)] and [Mn(III)] data (Figure2) validated the capability of the simple kinetic model todescribe UO2 oxidative dissolution by Mn(III) complexed bypyrophosphate. Fitting the experimental data using a kineticmodel was helpful for comparing the reactivity of PP-stabilizedMn(III) for UO2 oxidation at different water chemistryconditions.Effect of PP:Mn(III) Ratio. Unlike DFOB whose molecular
structure constrains the 1:1 coordination with Mn(III), theratio of PP to Mn(III) is expected to change the speciation(stoichiometry and/or protonation level) of Mn(III)−PPcomplexes. An increase in the ratio of PP to Mn(III) appearedto decrease the reactivity of soluble Mn(III) in oxidizing UO2.Although the kinetic model (eq 5) could fit the individual datasets from experiments at different initial Mn(III) concen-trations (Figures 2a−e), the second order rate constantdetermined for each data set was systematically lower atlower initial Mn(III) concentrations (i.e., higher PP:Mn(III)ratios) (Figure 3). In addition to the experiments with fixed PPloading and varied Mn(III) concentrations, the results from
experiments with varied PP concentrations (Figure 2f and g)also indicated that the rate constants were influenced by thePP:Mn(III) ratio (Figure 3). When the PP:Mn(III) ratio waslarger than 40, the rate constant became insensitive to theligand-to-metal ratio; even at this great excess of stabilizingligand, Mn(III) complexed by pyrophosphate was still aneffective oxidant for UO2. When the PP:Mn(III) ratiodecreased from 40 to 6, the reactivity of Mn(III) increaseddramatically with the rate constant tripling. The reactivity ofMn(III) with lower PP:Mn(III) could not be experimentallyevaluated due to the need for some excess ligand to keepMn(III) in solution.27
The effect of PP:Mn(III) ratio on Mn(III) reactivity isrelated to Mn(III) speciation with variations in charge,protonation state, and coordination number of the Mn(III)−PP complexes. However, the currently available thermodynamicdatabases57,58 for Mn(III)−PP complexes were obtained fromdata at highly acidic conditions and have discrepancies42 thatpreclude a conclusive quantitative interpretation at environ-mentally relevant pH.27,59 Regardless of the lack of quantitativeinformation, it was speculated27,60 that at circumneutral pHMn(III)−PP complexes are likely to be in fully deprotonatedforms (i.e., MnP2O7
−, Mn(P2O7)25−, and possibly Mn-
(P2O7)39−). Higher PP:Mn(III) ratios favor the complexes
with larger ligand-to-metal stoichiometries, which may have lessaffinity for UO2 surfaces to form the transition state complexesthat are necessary to facilitate electron transfer. Mn(III)−oxalate complexation, which is a better-studied system, alsoshowed stepwise addition of the ligand group to the Mn(III)speciation as oxalate-to-metal ratio inceases.27,61 The effect ofPP:Mn(III) ratio on the reactivity of soluble Mn(III) inoxidizing UO2 was consistent with a recent study on thereactivity of Mn(III) as an intermediate of permanganateoxidation of bisphenol A in the presence of PP.59 That studyobserved that PP addition to low concentrations could increasethe exposure of bisphenol A to soluble Mn(III) and enhance
Figure 3. Second order rate constants for Mn(III)−UO2 reaction atdifferent PP:Mn(III) ratios. The PP:Mn(III) is expected to affect thereactivity of Mn(III) with respect to UO2 oxidation by changing thespeciation of Mn(III)−PP complexes. The included results are fromexperiments at pH 7.5. Error bars indicate the ranges of the rateconstants that yielded sum of square of residuals within 10% of theoptimized value.
Environmental Science & Technology Article
dx.doi.org/10.1021/es4037308 | Environ. Sci. Technol. XXXX, XXX, XXX−XXXF
the overall oxidation rates but that further increases of PPconcentration decreased the reactivity of Mn(III). Strongdependence of Mn(III) reactivity on ligand-to-metal ratio maybe anticipated for other low-molecular-mass phosphonate- andcarboxylate-based chelating agents.Effect of pH. The oxidative UO2 dissolution by PP-
stabilized Mn(III) is critically affected by pH. The results of theexperiments at pH 7.5 described above suggested that solubleMn(III) could oxidize UO2 without involving any solid form ofMn. However, the time variation of total soluble Mn at pH 6.5and pH 9.0 contained features that can be explained by twodifferent types of Mn precipitation (Figure 4). In addition,different kinetic behaviors were observed for both U(VI)production and Mn(III) consumption at pH 6.5 and 9.0 than at7.5. For pH 6.5, the kinetic model (eqs 5 and 6) yieldedexcellent fits of [U(VI)] and [Mn(III)] results with a rateconstant that is larger than the one for pH 7.5 by a factor of 4.This is consistent with a recent study62 of estradiol oxidation byMn(III)−PP complexes at varied pH. Based on the existingthermodynamic information of PP and Mn(III) (i.e., Mn-(P2O7)x
−(4x‑3), x = 1−3), lower pH was qualitatively expected tofavor the formation of Mn(III)-PP complexes with smaller PP-to-Mn(III) stoichiometry that would allow larger amounts ofuncomplexed or weakly complexed Mn(III) due to the
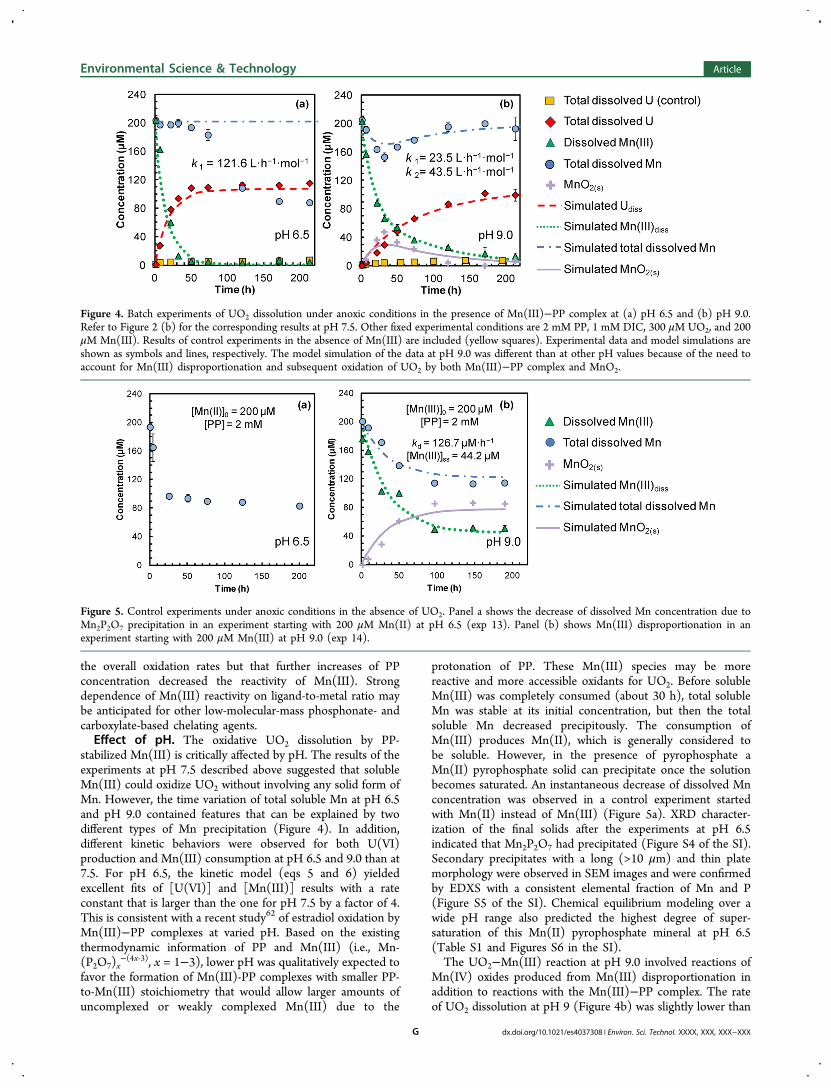
protonation of PP. These Mn(III) species may be morereactive and more accessible oxidants for UO2. Before solubleMn(III) was completely consumed (about 30 h), total solubleMn was stable at its initial concentration, but then the totalsoluble Mn decreased precipitously. The consumption ofMn(III) produces Mn(II), which is generally considered tobe soluble. However, in the presence of pyrophosphate aMn(II) pyrophosphate solid can precipitate once the solutionbecomes saturated. An instantaneous decrease of dissolved Mnconcentration was observed in a control experiment startedwith Mn(II) instead of Mn(III) (Figure 5a). XRD character-ization of the final solids after the experiments at pH 6.5indicated that Mn2P2O7 had precipitated (Figure S4 of the SI).Secondary precipitates with a long (>10 μm) and thin platemorphology were observed in SEM images and were confirmedby EDXS with a consistent elemental fraction of Mn and P(Figure S5 of the SI). Chemical equilibrium modeling over awide pH range also predicted the highest degree of super-saturation of this Mn(II) pyrophosphate mineral at pH 6.5(Table S1 and Figures S6 in the SI).The UO2−Mn(III) reaction at pH 9.0 involved reactions of
Mn(IV) oxides produced from Mn(III) disproportionation inaddition to reactions with the Mn(III)−PP complex. The rateof UO2 dissolution at pH 9 (Figure 4b) was slightly lower than
Figure 4. Batch experiments of UO2 dissolution under anoxic conditions in the presence of Mn(III)−PP complex at (a) pH 6.5 and (b) pH 9.0.Refer to Figure 2 (b) for the corresponding results at pH 7.5. Other fixed experimental conditions are 2 mM PP, 1 mM DIC, 300 μM UO2, and 200μM Mn(III). Results of control experiments in the absence of Mn(III) are included (yellow squares). Experimental data and model simulations areshown as symbols and lines, respectively. The model simulation of the data at pH 9.0 was different than at other pH values because of the need toaccount for Mn(III) disproportionation and subsequent oxidation of UO2 by both Mn(III)−PP complex and MnO2.
Figure 5. Control experiments under anoxic conditions in the absence of UO2. Panel a shows the decrease of dissolved Mn concentration due toMn2P2O7 precipitation in an experiment starting with 200 μM Mn(II) at pH 6.5 (exp 13). Panel (b) shows Mn(III) disproportionation in anexperiment starting with 200 μM Mn(III) at pH 9.0 (exp 14).
Environmental Science & Technology Article
dx.doi.org/10.1021/es4037308 | Environ. Sci. Technol. XXXX, XXX, XXX−XXXG

that at pH 7.5 (Figure 2b). The total dissolved Mnconcentration decreased immediately with formation ofMnO2 as brown particles. As the reaction proceeded, thetotal dissolved Mn concentration gradually recovered to itsinitial levels with the brown color fading out. This trend can beexplained by the generation of MnO2 from Mn(III)disporportionation that was subsequently reduced to solubleMn(II) by UO2. A UO2-free control experiment with 200 μMMn(III) at pH 9.0 (Figure 5b) confirmed the disproportiona-tion of Mn(III), in which dissolved Mn(III) and total dissolvedMn decayed exponentially and eventually leveled off, which isalso consistent with an earlier study.42
Modeling the reaction involving UO2 and Mn(III) at pH 9.0accounted for additional reactions of Mn(III) disproportiona-tion
= +2Mn Mn MnIII(aq)
IV(s)
II(aq) (11)
in which two moles of Mn(III) yield one mole of MnO2, andthe subsequent UO2−MnO2 reaction
+ = +U Mn U MnIV(s)
IV(s)
VI(aq)
II(aq) (12)
The stoichiometry of Mn(III) disproportionation wasconfirmed by the results of the control experiment (Figure5b), and the disproportionation rate (Rd) could be modeledusing the following rate expression with parameters obtained bysimultaneously fitting the concentrations of Mn(III) andMnO2.
=
−≥
<
⎧
⎨⎪⎪⎪
⎩⎪⎪⎪
⎛⎝⎜
⎞⎠⎟
R
k[Mn(III)]
[Mn(III)]1 if [Mn(III)]
[Mn(III)]
0 if [Mn(III)][Mn(III)]
d
d
ssss
ss (13)
where kd (126.7 μM·h−1) is the disproportionation rateconstant and [Mn(III)]ss (44.2 μM) is the observedconcentration at which Mn(III) leveled off in the controlexperiment. The disproportionation reaction was assumed to beirreversible. The rate of the UO2−MnO2 solid−solid reactioncould be assumed to be k2[UO2][MnO2] (k2 is a second orderrate constant in L·h−1·mol−1) given that the MnO2 concen-tration was small relative to UO2.
18 A set of nonlineardifferential equations was then used to model the temporalvariation of [Mn(III)], [U(VI)], and [MnO2].
= − −t
R kd[Mn(III)]
d2 [UO ][Mn(III)]d 1 2 (14)
= +t
k kd[U(VI)]
d[UO ][Mn(III)] [UO ][MnO ]1 2 2 2 2
(15)
= −t
R kd[MnO ]
d12
[UO ][MnO ]2d 2 2 2 (16)
The equations were solved numerically with k1 and k2 variedsimultaneously to fit the experimental data of [Mn(III)],[U(VI)], and [MnO2]. The model yielded excellent simulationsof the experimental results, including the rapid accumulationand subsequent disappearance of MnO2 (Figure 4b). Althoughthe single data set might not enable conclusive determination ofthe rate constants k1 and k2, the present analysis illustrates therole and dynamics of Mn(III) disproportionation coupled with
oxidation of UO2. An improved understanding of the speciationand thermodynamics of ligand-stabilized Mn(III) would bevaluable for predicting its kinetic behavior as an environmentaloxidant.
Environmental Implications. With the emerging evidenceof its presence in environments with oxic/anoxic interfaces,soluble Mn(III) may be important in the coupling of Mn redoxcycling to the oxidation of U(IV). Given that biologicallyproduced U(IV) species are more susceptible to oxidation thantheir chemogenic counterparts,63 soluble Mn(III) is anticipatedto be able to oxidize U(IV) at bioremediation sites. The resultsof the present study highlight the delicate balance that governsthe significance of soluble Mn(III) in biogeochemical redoxprocesses. On the one hand, ligands can facilitate themobilization of Mn(III), which may circumvent the physicalcontact barrier separating UO2 and Mn oxides.18 As anintermediate of Mn bio-oxidation and bioreduction, solubleMn(III) complexed with pyrophosphate might be kineticallymore reactive than dissolved O2, which may explain theenhancement of UO2 oxidation in the presence of activemicrobial Mn oxidation.17 Furthermore, the UO2−MnO2reaction may be passivated by the adsorption of reactionproducts to MnO2,
18 which can retard the transport of U(VI)and mitigate the antagonistic effect of Mn on U stability.64 Incomparison, soluble Mn(III) could directly oxidize UO2without involving any solid forms of Mn that could adsorbthe U(VI) produced. On the other hand, too strong of Mn(III)complexation, e.g., by siderophore compounds, can make theMn(III) inert with respect to heterogeneous electron transfer.The contrasting behaviors of Mn(III) reactivity are dictated bythe different binding strengths of the ligands.The present results also highlight the general value of
improving our knowledge of the environmental occurrence,chemical speciation, redox activity, and biological roles ofaqueous Mn(III) complexes. Solution pH is a sensitive andcritical factor that governs the oxidizing reactivity and stabilityof Mn(III) species. For Mn(III)−pyrophosphate complexes,higher pH (above 9.0) favors disproportionation and lower pH(e.g, pH 6.0) may form more reactive oxidizing species. Forredox active organic ligands, low pH (e.g., below pH 7 forDFOB) can induce intramolecular electron transfer from theorganic ligand to the Mn(III) center, producing Mn(II) and anoxidized form of the ligand.23,31 Mn(III) coordinated bystronger ligand is also expected to persist up to higher pHwithout disproportion occurring (e.g., previously reporteddisproportionation pH of Mn(III)−DFOB was above 11.3).In addition to pyrophosphate and DFOB, Mn(III) ligands innatural and engineered aquatic environments include a largefamily of biogenic siderophore compounds and their break-down products, and a variety of phosphonate- and carboxylate-based synthetic chelators that may be introduced as scaleinhibitors, pharmaceuticals, and herbicides.20,27 DFOB andpyrophosphate are representative strong and moderate Mn(III)ligands, respectively. The present results provided insights forfuture efforts toward comprehensive characterization ofaqueous Mn(III) species.
■ ASSOCIATED CONTENT
*S Supporting InformationAdditional information, including six figures, one table, andadditional details of the experimental procedure. This materialis available free of charge via the Internet at http://pubs.acs.org.
Environmental Science & Technology Article
dx.doi.org/10.1021/es4037308 | Environ. Sci. Technol. XXXX, XXX, XXX−XXXH

■ AUTHOR INFORMATIONCorresponding Author* Phone: 63130 314-935-9437; e-mail: [email protected]; mail: Campus Box 1180, One Brookings Drive, St. Louis,MO 63130, USA.
NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSWe acknowledge valuable discussions with Alan Stone, Sung-Woo Lee, John Bargar, Jeff Catalano, and Rizlan Bernier-Latmani. Laboratory assistance provided by Qingyun Li and YinWang is appreciated. Funding for this research was provided bythe U.S. Department of Energy, Subsurface BiogeochemicalResearch Program under Grant DE-SC0005324. Commentsand suggestions of Associate Editor Stephan Hug and threeanonymous reviewers were helpful for improving an earlyversion of this paper.
■ REFERENCES(1) Bargar, J. R.; Bernier-Latmani, R.; Giammar, D. E.; Tebo, B. M.Biogenic uraninite nanoparticles and their importance for uraniumremediation. Elements 2008, 4 (6), 407−412.(2) Bargar, J. R.; Williams, K. H.; Campbell, K. M.; Long, P. E.;Stubbs, J. E.; Suvorova, E. I.; Lezama-Pacheco, J. S.; Alessi, D. S.; Stylo,M.; Webb, S. M.; Davis, J. A.; Giammar, D. E.; Blue, L. Y.; Bernier-Latmani, R. Uranium redox transition pathways in acetate-amendedsediments. Proc. Natl. Acad. Sci., U. S. A. 2013, 110 (12), 4506−4511.(3) Bernier-Latmani, R.; Veeramani, H.; Vecchia, E. D.; Junier, P.;Lezama-Pacheco, J. S.; Suvorova, E. I.; Sharp, J. O.; Wigginton, N. S.;Bargar, J. R. Non-uraninite products of microbial U(VI) reduction.Environ. Sci. Technol. 2010, 44 (24), 9456−9462.(4) Fletcher, K. E.; Boyanov, M. I.; Thomas, S. H.; Wu, Q. Z.;Kemner, K. M.; Loffler, F. E. U(VI) reduction to mononuclear U(IV)by Desulf itobacterium species. Environ. Sci. Technol. 2010, 44 (12),4705−4709.(5) Ulrich, K. U.; Ilton, E. S.; Veeramani, H.; Sharp, J. O.; Bernier-Latmani, R.; Schofield, E. J.; Bargar, J. R.; Giammar, D. E. Comparativedissolution kinetics of biogenic and chemogenic uraninite underoxidizing conditions in the presence of carbonate. Geochim.Cosmochim. Acta 2009, 73 (20), 6065−6083.(6) Senko, J. M.; Suflita, J. M.; Krumholz, L. R. Geochemical controlson microbial nitrate-dependent U(IV) oxidation. Geomicrobiol. J. 2005,22 (7−8), 371−378.(7) Ginder-Vogel, M.; Stewart, B.; Fendorf, S. Kinetic andmechanistic constraints on the oxidation of biogenic uraninite byferrihydrite. Environ. Sci. Technol. 2010, 44 (1), 163−169.(8) Sani, R. K.; Peyton, B. M.; Dohnalkova, A.; Amonette, J. E.Reoxidation of reduced uranium with iron(III) (hydr)oxides undersulfate-reducing conditions. Environ. Sci. Technol. 2005, 39 (7), 2059−2066.(9) Fredrickson, J. K.; Zachara, J. M.; Kennedy, D. W.; Liu, C.; Duff,M. C.; Hunter, D. B.; Dohnalkova, A. Influence of Mn oxides on thereduction of uranium(VI) by the metal-reducing bacterium Shewanellaputrefaciens. Geochim. Cosmochim. Acta 2002, 66 (18), 3247−3262.(10) Liu, C. X.; Zachara, J. M.; Fredrickson, J. K.; Kennedy, D. W.;Dohnalkova, A. Modeling the inhibition of the bacterial reduction ofU(VI) by β-MnO2(s). Environ. Sci. Technol. 2002, 36 (7), 1452−1459.(11) Campbell, K. M.; Veeramani, H.; Urich, K. U.; Blue, L. Y.;Giammar, D. E.; Bernier-Latmani, R.; Stubbs, J. E.; Suvorova, E.;Yabusaki, S.; Lezama-Pacheco, J. S.; Mehta, A.; Long, P. E.; Bargar, J.R. Oxidative dissolution of biogenic uraninite in groundwater at OldRifle, CO. Environ. Sci. Technol. 2011, 45 (20), 8748−8754.(12) Zachara, J. M.; Long, P. E.; Bargar, J.; Davis, J. A.; Fox, P.;Fredrickson, J. K.; Freshley, M. D.; Konopka, A. E.; Liu, C.; McKinley,J. P.; Rockhold, M. L.; Williams, K. H.; Yabusaki, S. B. Persistence of
uranium groundwater plumes: Contrasting mechanisms at two DOEsites in the groundwater−river interaction zone. J. Contam. Hydrol.2013, 147 (0), 45−72.(13) Borch, T.; Kretzschmar, R.; Kappler, A.; Van Cappellen, P.;Ginder-Vogel, M.; Voegelin, A.; Campbell, K. Biogeochemical redoxprocesses and their impact on contaminant dynamics. Environ. Sci.Technol. 2010, 44 (1), 15−23.(14) Morgan, J. J. Kinetics of reaction between O2 and Mn(II)species in aqueous solutions. Geochim. Cosmochim. Acta 2005, 69 (1),35−48.(15) Tebo, B. M.; Bargar, J. R.; Clement, B. G.; Dick, G. J.; Murray,K. J.; Parker, D.; Verity, R.; Webb, S. M. Biogenic manganese oxides:Properties and mechanisms of formation. Annu. Rev. Earth Planet. Sci.2004, 32 (1), 287−328.(16) Clement, B. G.; Luther, G. W.; Tebo, B. M. Rapid, oxygen-dependent microbial Mn(II) oxidation kinetics at sub-micromolaroxygen concentrations in the Black Sea suboxic zone. Geochim.Cosmochim. Acta 2009, 73 (7), 1878−1889.(17) Chinni, S.; Anderson, C. R.; Ulrich, K. U.; Giammar, D. E.;Tebo, B. M. Indirect UO2 oxidation by Mn(II)-oxidizing spores ofBacillus sp Strain SG-1 and the effect of U and Mn concentrations.Environ. Sci. Technol. 2008, 42 (23), 8709−8714.(18) Wang, Z.; Lee, S.-W.; Kapoor, P.; Tebo, B. M.; Giammar, D. E.Uraninite oxidation and dissolution induced by manganese oxide: Aredox reaction between two insoluble minerals. Geochim. Cosmochim.Acta 2013, 100 (1), 24−40.(19) Plathe, K. L.; Lee, S.-W.; Tebo, B. M.; Bargar, J. R.; Bernier-Latmani, R. Impact of microbial Mn oxidation on the remobilization ofbioreduced U(IV). Environ. Sci. Technol. 2013, 47 (8), 3606−3613.(20) Duckworth, O. W.; Bargar, J. R.; Sposito, G. Coupledbiogeochemical cycling of iron and manganese as mediated bymicrobial siderophores. BioMetals 2009, 22 (4), 605−613.(21) Johnson, K. S. Manganese redox chemistry revisited. Science2006, 313 (5795), 1896−1897.(22) Webb, S. M.; Dick, G. J.; Bargar, J. R.; Tebo, B. M. Evidence forthe presence of Mn(III) intermediates in the bacterial oxidation ofMn(II). Proc. Natl. Acad. Sci., U. S. A. 2005, 102 (15), 5558−5563.(23) Duckworth, O. W.; Sposito, G. Siderophore-manganese(III)interactions. I. Air-oxidation of manganese(II) promoted by desferriox-amine B. Environ. Sci. Technol. 2005, 39 (16), 6037−6044.(24) Butterfield, C. N.; Soldatova, A. V.; Lee, S.-W.; Spiro, T. G.;Tebo, B. M. Mn(II,III) oxidation and MnO2 mineralization by anexpressed bacterial multicopper oxidase. Proc. Natl. Acad. Sci., U. S. A.2013, 110 (29), 11731−11735.(25) Ehrlich, H. L. Manganese oxide reduction as a form of anaerobicrespiration. Geomicrobiol. J. 1987, 5 (3−4), 423−431.(26) Lin, H.; Szeinbaum, N. H.; DiChristina, T. J.; Taillefert, M.Microbial Mn(IV) reduction requires an initial one-electron reductivesolubilization step. Geochim. Cosmochim. Acta 2012, 99 (0), 179−192.(27) Wang, Y.; Stone, A. T. Phosphonate- and carboxylate-basedchelating agents that solubilize (hydr)oxide-bound MnIII. Environ. Sci.Technol. 2008, 42 (12), 4397−4403.(28) Duckworth, O. W.; Sposito, G. Siderophore−manganese(III)interactions. II. Manganite dissolution promoted by desferrioxamine B.Environ. Sci. Technol. 2005, 39 (16), 6045−6051.(29) Pena, J.; Duckworth, O. W.; Bargar, J. R.; Sposito, G.Dissolution of hausmannite (Mn3O4) in the presence of thetrihydroxamate siderophore desferrioxamine B. Geochim. Cosmochim.Acta 2007, 71 (23), 5661−5671.(30) Gunary, D. Pyrophosphate in soil; Some physico-chemicalaspects. Nature 1966, 210 (5042), 1297−1298.(31) Harrington, J. M.; Parker, D. L.; Bargar, J. R.; Jarzecki, A. A.;Tebo, B. M.; Sposito, G.; Duckworth, O. W. Structural dependence ofMn complexation by siderophores: Donor group dependence oncomplex stability and reactivity. Geochim. Cosmochim. Acta 2012, 88(0), 106−119.(32) Trouwborst, R. E.; Clement, B. G.; Tebo, B. M.; Glazer, B. T.;Luther, G. W. Soluble Mn(III) in suboxic zones. Science 2006, 313(5795), 1955−1957.
Environmental Science & Technology Article
dx.doi.org/10.1021/es4037308 | Environ. Sci. Technol. XXXX, XXX, XXX−XXXI

(33) Madison, A. S.; Tebo, B. M.; Luther, G. W. Simultaneousdetermination of soluble manganese(III), manganese(II) and totalmanganese in natural (pore)waters. Talanta 2011, 84 (2), 374−381.(34) Madison, A. S.; Tebo, B. M.; Mucci, A.; Sundby, B.; Luther, G.W., III Abundant porewater Mn(III) is a major component of thesedimentary redox system. Science 2013, 341 (6148), 875−878.(35) Weaver, R. M.; Hochella, M. F. The reactivity of seven Mn-oxides with Cr3+ aq: A comparative analysis of a complex,environmentally important redox reaction. Am. Mineral. 2003, 88(11−12), 2016−2027.(36) Nico, P. S.; Zasoski, R. J. Importance of Mn(III) availability onthe rate of Cr(III) oxidation on δ-MnO2. Environ. Sci. Technol. 2000,34 (16), 3363−3367.(37) Nico, P. S.; Zasoski, R. J. Mn(III) center availability as a ratecontrolling factor in the oxidation of phenol and sulfide on δ-MnO2.Environ. Sci. Technol. 2001, 35 (16), 3338−3343.(38) Chen, W.-R.; Liu, C.; Boyd, S. A.; Teppen, B. J.; Li, H.Reduction of carbadox mediated by reaction of Mn(III) with oxalicacid. Environ. Sci. Technol. 2013, 47 (3), 1357−1364.(39) Lafferty, B. J.; Ginder-Vogel, M.; Zhu, M. Q.; Livi, K. J. T.;Sparks, D. L. Arsenite oxidation by a poorly crystalline manganese-oxide. 2. Results from X-ray absorption spectroscopy and X-raydiffraction. Environ. Sci. Technol. 2010, 44 (22), 8467−8472.(40) Nesbitt, H. W.; Canning, G. W.; Bancroft, G. M. XPS study ofreductive dissolution of 7Å-birnessite by H3AsO3, with constraints onreaction mechanism. Geochim. Cosmochim. Acta 1998, 62 (12), 2097−2110.(41) Kostka, J. E.; Luther, G. W.; Nealson, K. H. Chemical andbiological reduction of Mn(III)-pyrophosphate complexes - Potentialimportance of dissolved Mn(III) as an environmental oxidant.Geochim. Cosmochim. Acta 1995, 59 (5), 885−894.(42) Klewicki, J. K.; Morgan, J. J. Kinetic behavior of Mn(III)complexes of pyrophosphate, EDTA, and citrate. Environ. Sci. Technol.1998, 32 (19), 2916−2922.(43) Rophael, M. Kinetics of the oxidation of chromium (III) bymanganese (III) in sulphuric acid media. Chem. Scr. 1982, 20, 171−173.(44) Murray, K. J.; Tebo, B. M. Cr(III) is indirectly oxidized by theMn(II)-oxidizing bacterium Bacillus sp strain SG-1. Environ. Sci.Technol. 2007, 41 (2), 528−533.(45) Weaver, R. M.; Hochella, M. F., Jr.; Ilton, E. S. Dynamicprocesses occurring at the CrIIIaq-Manganite (γ-MnOOH) interface:Simultaneous adsorption, microprecipitation, oxidation/reduction, anddissolution. Geochim. Cosmochim. Acta 2002, 66 (23), 4119−4132.(46) Perez-Benito, J. F.; Arias, C. The pyrophosphate-assistedreduction of chromium(VI) by manganese(II) and its reverse reaction.New J. Chem. 2001, 25 (11), 1438−1446.(47) Turner, B. L.; Newman, S. Phosphorus cycling in wetland soils.J. Environ. Qual. 2005, 34 (5), 1921−1929.(48) Bi, Y.; Hesterberg, D. L.; Duckworth, O. W. Siderophore-promoted dissolution of cobalt from hydroxide minerals. Geochim.Cosmochim. Acta 2010, 74 (10), 2915−2925.(49) Faulkner, K. M.; Stevens, R. D.; Fridovich, I. Characterization ofMn(III) complexes of linear and cyclic desferrioxamines as mimics ofsuperoxide dismutase activity. Arch. Biochem. Biophys. 1994, 310 (2),341−346.(50) McElroy, W. D.; Glass, B. Phosphorus Metabolism; JohnsHopkins University Press: Baltimore, MD, 1951.(51) Wazer, J. R. V.; Campanella, D. A. Structure and properties ofthe condensed phosphates. IV. Complex ion formation in poly-phosphate solutions. J. Am. Chem. Soc. 1950, 72 (2), 655−663.(52) Schecher, W. D.; McAvoy, D. C. MINEQL+: A chemicalequilibrium modeling system, version 4.6; Environmental ResearchSoftware: Hallowell, ME, 2007.(53) Frazier, S. W.; Kretzschmar, R.; Kraemer, S. M. Bacterialsiderophores promote dissolution of UO2 under reducing conditions.Environ. Sci. Technol. 2005, 39 (15), 5709−5715.
(54) Duckworth, O. W.; Bargar, J. R.; Sposito, G. Quantitativestructure−activity relationships for aqueous metal−siderophorecomplexes. Environ. Sci. Technol. 2009, 43 (2), 343−349.(55) Stewart, B. D.; Girardot, C.; Spycher, N.; Sani, R. K.; Peyton, B.M. Influence of chelating agents on biogenic uraninite reoxidation byFe(III) (hydr)oxides. Environ. Sci. Technol. 2013, 47 (1), 364−371.(56) Ginder-Vogel, M.; Criddle, C. S.; Fendorf, S. Thermodynamicconstraints on the oxidation of biogenic UO2 by Fe(III) (hydr)oxides.Environ. Sci. Technol. 2006, 40 (11), 3544−3550.(57) Gordienko, V. I.; Sidorenko, V. I.; Mikhailyuk, Y. I.Amperometric investigation of Mn(III) pyrophosphate complexes.Russ. J. Inorg. Chem. 1970, 15, 1241−1244.(58) Ciavatta, L.; Palombari, R. On the equilibria of complexformation between manganese(III) and pyrophosphate ions. Gazz.Chim. Ital. 1983, 113, 557−552.(59) Jiang, J.; Pang, S. Y.; Ma, J. Role of ligands in permanganateoxidation of organics. Environ. Sci. Technol. 2010, 44 (11), 4270−4275.(60) Kolthoff, I.; Watters, J. Polarographic determination ofmanganese as tridihydrogen pyrophosphatomanganiate. Ind. Eng.Chem. Anal. Ed. 1943, 15 (1), 8−13.(61) Taube, H. Catalysis by manganic ion of the reaction of bromineand oxalic acid. Stability of manganic ion complexes. J. Am. Chem. Soc.1948, 70 (11), 3928−3935.(62) Jiang, J.; Pang, S.-Y.; Ma, J.; Liu, H. Oxidation of phenolicendocrine disrupting chemicals by potassium permanganate insynthetic and real waters. Environ. Sci. Technol. 2011, 46 (3), 1774−1781.(63) Cerrato, J. M.; Ashner, M. N.; Alessi, D. S.; Lezama Pacheco, J.S.; Bernier-Latmani, R.; Bargar, J.; Giammar, D. E. Relative reactivityof biogenic and chemogenic uraninite and biogenic non-crystallineU(IV). Environ. Sci. Technol. 2013, 47 (17), 9756−9763.(64) Wang, Z.; Lee, S.-W.; Catalano, J. G.; Lezama-Pacheco, J. S.;Bargar, J. R.; Tebo, B. M.; Giammar, D. E. Adsorption of uranium(VI)to manganese oxides: X-ray absorption spectroscopy and surfacecomplexation modeling. Environ. Sci. Technol. 2013, 47 (2), 850−858.
Environmental Science & Technology Article
dx.doi.org/10.1021/es4037308 | Environ. Sci. Technol. XXXX, XXX, XXX−XXXJ
Copyright © 2022 FDOKUMEN




















