
proteins - BiomedicaHelp
543
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of proteins - BiomedicaHelp


PROTEINSSTRUCTURE AND FUNCTION


PROTEINSSTRUCTURE AND FUNCTION
David Whitford
John Wiley & Sons, Ltd

Copyright 2005 John Wiley & Sons Ltd, The Atrium, Southern Gate, Chichester,West Sussex PO19 8SQ, England
Telephone (+44) 1243 779777
Email (for orders and customer service enquiries): [email protected] our Home Page on www.wiley.com
All Rights Reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or byany means, electronic, mechanical, photocopying, recording, scanning or otherwise, except under the terms of the Copyright,Designs and Patents Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency Ltd, 90 Tottenham CourtRoad, London W1T 4LP, UK, without the permission in writing of the Publisher. Requests to the Publisher should be addressedto the Permissions Department, John Wiley & Sons Ltd, The Atrium, Southern Gate, Chichester, West Sussex PO19 8SQ,England, or emailed to [email protected], or faxed to (+44) 1243 770620.
This publication is designed to provide accurate and authoritative information in regard to the subject matter covered. It is sold onthe understanding that the Publisher is not engaged in rendering professional services. If professional advice or other expertassistance is required, the services of a competent professional should be sought.
Other Wiley Editorial Offices
John Wiley & Sons Inc., 111 River Street, Hoboken, NJ 07030, USA
Jossey-Bass, 989 Market Street, San Francisco, CA 94103-1741, USA
Wiley-VCH Verlag GmbH, Boschstr. 12, D-69469 Weinheim, Germany
John Wiley & Sons Australia Ltd, 33 Park Road, Milton, Queensland 4064, Australia
John Wiley & Sons (Asia) Pte Ltd, 2 Clementi Loop #02-01, Jin Xing Distripark, Singapore 129809
John Wiley & Sons Canada Ltd, 22 Worcester Road, Etobicoke, Ontario, Canada M9W 1L1
Wiley also publishes its books in a variety of electronic formats. Some content that appearsin print may not be available in electronic books.
British Library Cataloguing in Publication Data
A catalogue record for this book is available from the British Library
ISBN 0-471-49893-9 HBISBN 0-471-49894-7 PB
Typeset in 10/12pt Times by Laserwords Private Limited, Chennai, IndiaPrinted and bound by Graphos SpA, Barcelona, SpainThis book is printed on acid-free paper responsibly manufactured from sustainable forestryin which at least two trees are planted for each one used for paper production.

For my parents,
Elizabeth and Percy Whitford,
to whom I owe everything


Contents
Preface xi
1 An Introduction to protein structure and function 1A brief and very selective historical perspective 1The biological diversity of proteins 5Proteins and the sequencing of the human and other genomes 9Why study proteins? 9
2 Amino acids: the building blocks of proteins 13The 20 amino acids found in proteins 13The acid–base properties of amino acids 14Stereochemical representations of amino acids 15Peptide bonds 16The chemical and physical properties of amino acids 23Detection, identification and quantification of amino acids and proteins 32Stereoisomerism 34Non-standard amino acids 35Summary 36Problems 37
3 The three-dimensional structure of proteins 39Primary structure or sequence 39Secondary structure 39Tertiary structure 50Quaternary structure 62The globin family and the role of quaternary structure in modulating activity 66Immunoglobulins 74Cyclic proteins 81Summary 81Problems 83
4 The structure and function of fibrous proteins 85The amino acid composition and organization of fibrous proteins 85Keratins 86Fibroin 92Collagen 92Summary 102Problems 103

viii CONTENTS
5 The structure and function of membrane proteins 105The molecular organization of membranes 105Membrane protein topology and function seen through organization of the
erythrocyte membrane 110Bacteriorhodopsin and the discovery of seven transmembrane helices 114The structure of the bacterial reaction centre 123Oxygenic photosynthesis 126Photosystem I 126Membrane proteins based on transmembrane β barrels 128Respiratory complexes 132Complex III, the ubiquinol-cytochrome c oxidoreductase 132Complex IV or cytochrome oxidase 138The structure of ATP synthetase 144ATPase family 152Summary 156Problems 159
6 The diversity of proteins 161Prebiotic synthesis and the origins of proteins 161Evolutionary divergence of organisms and its relationship to protein
structure and function 163Protein sequence analysis 165Protein databases 180Gene fusion and duplication 181Secondary structure prediction 181Genomics and proteomics 183Summary 187Problems 187
7 Enzyme kinetics, structure, function, and catalysis 189Enzyme nomenclature 191Enzyme co-factors 192Chemical kinetics 192The transition state and the action of enzymes 195The kinetics of enzyme action 197Catalytic mechanisms 202Enzyme structure 209Lysozyme 209The serine proteases 212Triose phosphate isomerase 215Tyrosyl tRNA synthetase 218EcoRI restriction endonuclease 221Enzyme inhibition and regulation 224Irreversible inhibition of enzyme activity 227Allosteric regulation 231Covalent modification 237Isoenzymes or isozymes 241Summary 242Problems 244

CONTENTS ix
8 Protein synthesis, processing and turnover 247Cell cycle 247The structure of Cdk and its role in the cell cycle 250Cdk–cyclin complex regulation 252DNA replication 253Transcription 254Eukaryotic transcription factors: variation on a ‘basic’ theme 261The spliceosome and its role in transcription 265Translation 266Transfer RNA (tRNA) 267The composition of prokaryotic and eukaryotic ribosomes 269A structural basis for protein synthesis 272An outline of protein synthesis 273Antibiotics provide insight into protein synthesis 278Affinity labelling and RNA ‘footprinting’ 279Structural studies of the ribosome 279Post-translational modification of proteins 287Protein sorting or targeting 293The nuclear pore assembly 302Protein turnover 303Apoptosis 310Summary 310Problems 312
9 Protein expression, purification and characterization 313The isolation and characterization of proteins 313Recombinant DNA technology and protein expression 313Purification of proteins 318Centrifugation 320Solubility and ‘salting out’ and ‘salting in’ 323Chromatography 326Dialysis and ultrafiltration 333Polyacrylamide gel electrophoresis 333Mass spectrometry 340How to purify a protein? 342Summary 344Problems 345
10 Physical methods of determining the three-dimensional structure ofproteins 347Introduction 347The use of electromagnetic radiation 348X-ray crystallography 349Nuclear magnetic resonance spectroscopy 360Cryoelectron microscopy 375Neutron diffraction 379Optical spectroscopic techniques 379Vibrational spectroscopy 387Raman spectroscopy 389

x CONTENTS
ESR and ENDOR 390Summary 392Problems 393
11 Protein folding in vivo and in vitro 395Introduction 395Factors determining the protein fold 395Factors governing protein stability 403Folding problem and Levinthal’s paradox 403Models of protein folding 408Amide exchange and measurement of protein folding 411Kinetic barriers to refolding 412In vivo protein folding 415Membrane protein folding 422Protein misfolding and the disease state 426Summary 435Problems 437
12 Protein structure and a molecular approach to medicine 439Introduction 439Sickle cell anaemia 441Viruses and their impact on health as seen through structure and function 442HIV and AIDS 443The influenza virus 457p53 and its role in cancer 470Emphysema and α1-antitrypsin 475Summary 478Problems 479
Epilogue 481
Glossary 483
Appendices 491
Bibliography 495
References 499
Index 511

Preface
When I first started studying proteins as an undergrad-uate I encountered for the first time complex areas ofbiochemistry arising from the pioneering work of Paul-ing, Sumner, Kendrew, Perutz, Anfinsen, together withother scientific ‘giants’ too numerous to describe atlength in this text. The area seemed complete. Howwrong I was and how wrong an undergraduate’s per-ception can be! The last 30 years have seen an explo-sion in the area of protein biochemistry so that my 1975edition of Biochemistry by Albert Lehninger remains,perhaps, of historical interest only. The greatest changehas occurred through the development of molecularbiology where fragments of DNA are manipulated inways previously unimagined. This has enabled DNAto be sequenced, cloned, manipulated and expressedin many different cells. As a result areas of recom-binant DNA technology and protein engineering haveevolved rapidly to become specialist disciplines in theirown right. Almost any protein whose primary sequenceis known can be produced in large quantity via theexpression of cloned or synthetic genes in recombinanthost cells. Not only is the method allowing scien-tists to study some proteins for the first time but theincreased amount of protein derived from recombinantDNA technology is also allowing the application ofnew and continually advancing structural techniques.In this area X-ray crystallography has remained at theforefront for over 40 years as a method of determin-ing protein structure but it is now joined by nuclearmagnetic resonance (NMR) spectroscopy and morerecently by cryoelectron microscopy whilst other meth-ods such as circular dichroism, infrared and Ramanspectroscopy, electron spin resonance spectroscopy,mass spectrometry and fluorescence provide more lim-ited, yet often vital and complementary, structural data.In many instances these methods have become estab-lished techniques only in the last 20 years and are
consequently absent in many of those familiar text-books occupying the shelves of university libraries.
An even greater impact on biochemistry hasoccurred with the rapid development of cost-effective,powerful, desktop computers with performance equiv-alent to the previous generation of supercomput-ers. Many experimental techniques relied on the co-development of computer hardware but software hasalso played a vital role in protein biochemistry. Wecan now search databases comparing proteins at thelevel of DNA or amino acid sequences, building uppatterns of homology and relationships that provideinsight into origin and possible function. In additionwe use computers routinely to calculate properties suchas isoelectric point, number of hydrophobic residues orsecondary structure – something that would have beenextraordinarily tedious, time consuming and problem-atic 20 years ago. Computers have revolutionized allaspects of protein biochemistry and there is little doubtthat their influence will continue to increase in theforthcoming decades. The new area of bioinformaticsreflects these advances in computing.
In my attempt to construct an introductory yet exten-sive text on proteins I have, of necessity, been circum-spect in my description of the subject area. I have oftenrelied on qualitative rather than quantitative descrip-tions and I have attempted to minimise the introduc-tion of unwieldy equations or formulae. This doesnot reflect my own interests in physical biochemistrybecause my research, I hope, was often quantitative.In some cases particularly the chapters on enzymesand physical methods the introduction of equations isunavoidable but also necessary to an initial descrip-tion of the content of these chapters. I would be failingin my duty as an educator if I omitted some of theseequations and I hope students will keep going at these‘difficult’ points or failing that just omit them entirely

xii PREFACE
on first reading this book. However, in general I wishto introduce students to proteins by describing princi-ples governing their structure and function and to avoidover-complication in this presentation through rigorousand quantitative treatment. This book is firmly intendedto be a broad introductory text suitable for undergrad-uate and postgraduate study, perhaps after an initialexposure to the subject of protein biochemistry, whilstat the same time introducing specialist areas prior tofuture advanced study. I hope the following chapterswill help to direct students to the amazing beauty andcomplexity of protein systems.
Target audienceThe present text should be suitable for all introductorymodes of biochemistry, molecular biology, chemistry,medicine and dentistry. In the UK this generally meansthe book is suitable for all undergraduates betweenyears 1 and 3 and this book has stemmed from lecturesgiven as parts of biochemistry courses to students ofbiochemistry, chemistry, medicine and dentistry in all3 years. Where possible each chapter is structuredto increase progressively in complexity. For purelyintroductory courses as would occur in years 1 or 2it is sufficient to read only the first parts, or selectedsections, of each chapter. More advanced courses mayrequire thorough reading of each chapter together withconsultation of the bibliography and secondly the listof references given at the end of the book.
The world wide webIn the last ten years the world wide web (WWW)has transformed information available to students. Itprovides a new and useful medium with which todeliver lecture notes and an exciting and new teach-ing resource for all. Consequently within this bookURLs direct students to learning resources and a listof important addresses is included in the appendix.In an effort to exploit the power of the internetthis book is associated with ‘web-based’ tutorials,problems and content and is accessed from the followingURL http://www.wiley.com/go/whitfordproteins. These‘pages’ are continually updated and point the interestedreader towards new areas as they emerge. The Bibli-ography points interested readers towards further study
material suitable for a first introduction to a subjectwhilst the list of references provides original sourcesfor many areas covered in each of the twelve chapters.
For the problems included at the end of each chapterthere are approximately 10 questions that aim to buildon the subject matter discussed in the preceding text.Often the questions will increase in difficulty althoughthis is not always the case. In this book I have limitedthe bibliography to broad reviews or accessible journalpapers and I have deliberately restricted the number of‘high-powered’ (difficult!) articles since I believe thisorganization is of greater use to students studying thesesubjects for the first time. To aid the learning processthe web edition has multiple-choice questions for useas a formative assessment exercise. I should certainlylike to hear of all mistakes or omissions encounteredin this text and my hope is that educators and studentswill let me know via the e-mail address at the end ofthis section of any required corrections or additions.
Proteins are three-dimensional (3D) objects that areinadequately represented on book pages. Consequentlymany proteins are best viewed as molecular imagesusing freely available software. Here, real-time manip-ulation of coordinate files is possible and will provehelpful to understanding aspects of structure and func-tion. The importance of viewing, manipulating andeven changing the representation of proteins to com-prehending structure and function cannot be underesti-mated. Experience has suggested that the use of com-puters in this area can have a dramatic effect on stu-dent’s understanding of protein structures. The abilityto visualize in 3D conveys so much information – farmore than any simple 2D picture in this book couldever hope to portray. Alongside many figures I havewritten the Protein DataBank files (e.g. PDB: 1HKO)used to produce diagrams. These files can be obtainedfrom databases at several permanent sites based aroundthe world such as http://www.rscb.org/pdb or one ofthe many ‘mirrors’ that exist (for example, in theUK this data is found at http://pdb.ccdc.cam.ac.uk).For students with Internet access each PDB file canbe retrieved and manipulated independently to pro-duce comparable images to those shown in the text.To explore these macromolecular images with reason-able efficiency does not require the latest ‘all-powerful’desktop computer. A computer with a Pentium III (orlater) based processor, a clock speed of 200 MHz or

PREFACE xiii
greater, 32–64 MB RAM, hard disks of 10 GB, agraphics video card with at least 8 MB memory anda connection to the internet are sufficient to view andstore a significant number of files together with rep-resentative images. Of course things are easier with acomputer with a surfeit of memory (>256 MB) anda high ‘clock’ speed (>2 GHz) but it is not obliga-tory to see ‘on-line’ content or to manipulate molecularimages. This book was started on a 700 MHz PentiumIII based processor equipped with 256 MB RAM and16 MB graphics card.
Organization of this bookThis book will address the structure and function ofproteins in 12 subsequent chapters each with a defini-tive theme. After an initial chapter describing why onewould wish to study proteins and a brief historicalbackground the second chapter deals with the ‘buildingblocks’ of proteins, namely the amino acids togetherwith their respective chemical and physical proper-ties. No attempt is made at any point to describe themetabolism connected with these amino acids and thereader should consult general textbooks for descriptionsof the synthesis and degradation of amino acids. Thisis a major area in its own right and would have length-ened the present book too much. However, I wouldlike to think that students will not avoid these areasbecause they remain an equally important subject thatshould be covered at some point within the under-graduate curriculum. Chapter 3 covers the assemblyof amino acids into polypeptide chains and levels oforganizational structure found within proteins. Almostall detailed knowledge of protein structure and func-tion has arisen through studies of globular proteins butthe presence of fibrous proteins with different struc-tures and functional properties necessitated a separatechapter devoted to this area (Chapter 4). Within thisclass the best understood structures are those belongingto the collagen class of proteins, the keratins and theextended β sheet structures such as silk fibroin. Thedivision between globular proteins and fibrous proteinswas made at a time when the only properties one couldcompare readily were a protein’s amino acid compo-sition and hydrodynamic radius. It is now apparentthat other proteins exist with properties intermediatebetween globular and fibrous proteins that do not lend
themselves to simple classification. However, the ‘old’schemes of identification retain their value and serveto emphasize differences in proteins.
Membrane proteins represent a third group withdifferent composition and properties. Most of theseproteins are poorly understood, but there have beenspectacular successes from the initial low-resolutionstructure of bacteriorhodopsin to the highly definedstructure of bacterial photosynthetic reaction centres.These advances paved the way towards structuralstudies of G proteins and G-protein coupled receptors,the respiratory complexes from aerobic bacteria and thestructure of ATP synthetases.
Chapter 6 focuses both on experimental and com-putational methods of comparing proteins where insilico methods have become increasingly important asa vital tool to assist with modern protein biochemistry.Chapter 7 focuses on enzymes and by discussing basicreaction rate theories and kinetics the chapter leads toa discussion of enzyme-catalysed reactions. Enzymescatalyse reactions through a variety of mechanismsincluding acid–base catalysis, nucleophilic drivenchemistry and transition state stabilization. These andother mechanisms are described along with the princi-ples of regulation, active site chemistry and binding.
The involvement of proteins in the cell cycle,transcription, translation, sorting and degradation ofproteins is described in Chapter 8. In 50 years wehave progressed from elucidating the structure ofDNA to uncovering how this information is convertedinto proteins. The chapter is based around the struc-ture of two macromolecular systems: the ribosomedevoted towards accurate and efficient synthesis andthe proteasome designed to catalyse specific proteoly-sis. Chapter 9 deals with the methods of protein purifi-cation. Very often, biochemistry textbooks describetechniques without placing the technique in the correctcontext. As a result, in Chapter 9 I have attempted todescribe equipment as well as techniques so that stu-dents may obtain a proper impression of this area.
Structural methods determine the topology or foldof proteins. With an elucidation of structure at atomiclevels of resolution comes an understanding of bio-logical function. Chapter 10 addresses this area bydescribing different techniques. X-ray crystallographyremains at the forefront of research with new variationsof the basic principle allowing faster determination of

xiv PREFACE
structure at improved resolution. NMR methods yieldstructures of comparable resolution to crystallographyfor small soluble proteins. In ideal situations thesemethods provide complete structural determination ofall heavy atoms but they are complemented by otherspectroscopic methods such as absorbance and fluores-cence methods, mass spectrometry and infrared spec-troscopy. These techniques provide important ancillaryinformation on tertiary structure such as the helical con-tent of the protein, the proportion and environment ofaromatic residues within a protein as well as secondarystructure content.
Chapter 11 describes protein folding and stabil-ity – a subject that has generated intense research inter-est with the recognition that disease states arise fromaberrant folding or stability. The mechanism of proteinfolding is illustrated by in vitro and in vivo studies.Whilst the broad concepts underlying protein fold-ing were deduced from studies of ‘model’ proteinssuch as ribonuclease, analysis of cell folding path-ways has highlighted specialised proteins, chaperones,with a critical function to the overall process. TheGroES–GroEL complex is discussed to highlight theintegrated process of synthesis and folding in vivo.
The final chapter builds on the preceding 11 chaptersusing a restricted set of well-studied proteins (casestudies) with significant impact on molecular medicine.These proteins include haemoglobin, viral proteins,p53, prions and α1-antitrypsin. Although still a youngsubject area this branch of protein science will expandin the next few years and will rely on the techniques,knowledge and principles elucidated in Chapters 1–11.The examples emphasize the impact of protein scienceand molecular medicine on the quality of human life.
AcknowledgementsI am indebted to all research students and post-docswho shared my laboratories at the Universities of Lon-don and Oxford during the last 15 years in many casesacting as ‘test subjects’ for teaching ideas. I shouldlike to thank Drs Roger Hewson, Richard Newboldand Susan Manyusa whose comments throughout myresearch and teaching career were always valued. Iwould also like to thank individuals, too numerousto name, with whom I interacted at King’s CollegeLondon, Imperial College of Science, Technology and
Medicine and the University of Oxford. In this con-text I should like to thank Dr John Russell, formerlyof Imperial College London whose goodwill, humourand fantastic insight into the history of science, thescientific method and ‘day to day’ experimentation pre-vented absolute despair.
During preparation of this book many individu-als read and contributed valuable comments to themanuscript’s content, phrasing and ideas. In particular Iwish to thank these unnamed and some times unknownindividuals who read one or more of the chapters of thisbook. As is often said by most authors at this pointdespite their valuable contributions all of the remain-ing errors and deficiencies in the current text are myresponsibility. In this context I could easily have spentmore months attempting to perfect the current text.I am very aware that this text has deficiencies but Ihope these defects will not detract from its value. Inaddition my wish to try other avenues, other roads nottaken, dictates that this manuscript is completed with-out delay.
Writing and producing a textbook would not bepossible without the support of a good publisher. Ishould like to thank all the staff at John Wiley & Sons,Chichester, UK. This exhaustive list includes particu-larly Andrew Slade as senior Publishing Editor whohelped smooth the bumpy route towards production ofthis book, Lisa Tickner who first initiated events lead-ing to commissioning this book, Rachel Ballard whosupervised day to day business on this book, replacingevery form I lost without complaint and monitoringtactfully and gently about possible completion dates,Robert Hambrook who translated my text and diagramsinto a beautiful book, and the remainder of the pro-duction team of John Wiley and Sons. Together weinched our way towards the painfully slow productionof this text, although the pace was entirely attributableto the author.
Lastly I must also thank Susan who tolerated theprotracted completion of this book, reading chaptersand offering support for this project throughout whilstcoping with the arrival of Alexandra and Ethaneffortlessly (unlike their father).
David WhitfordApril 2004

1An Introduction to protein structure
and function
Biochemistry has exploded as a major scientificendeavour over the last one hundred years to rival pre-viously established disciplines such as chemistry andphysics. This occurred with the recognition that livingsystems are based on the familiar elements of organicchemistry (carbon, oxygen, nitrogen and hydrogen)together with the occasional involvement of inorganicchemistry and elements such as iron, copper, sodium,potassium and magnesium. More importantly the lawsof physics including those concerning thermodynam-ics, electricity and quantum physics are applicable tobiochemical systems and no ‘vital’ force distinguishesliving from non-living systems. As a result the lawsof chemistry and physics are successfully applied tobiochemistry and ideas from physics and chemistryhave found widespread application, frequently revolu-tionizing our understanding of complex systems suchas cells.
This book focuses on one major component of allliving systems – the proteins. Proteins are found inall living systems ranging from bacteria and virusesthrough the unicellular and simple eukaryotes tovertebrates and higher mammals such as humans.Proteins make up over 50 percent of the dry weightof cells and are present in greater amounts thanany other biomolecule. Proteins are unique amongstthe macromolecules in underpinning every reaction
occurring in biological systems. It goes without sayingthat one should not ignore the other components ofliving systems since they have indispensable roles, butin this text we will consider only proteins.
A brief and very selective historicalperspective
With the vast accumulation of knowledge about pro-teins over the last 50 years it is perhaps surprising todiscover that the term protein was introduced nearly170 years ago. One early description was by GerhardusJohannes Mulder in 1839 where his studies on the com-position of animal substances, chiefly fibrin, albuminand gelatin, showed the presence of carbon, hydro-gen, oxygen and nitrogen. In addition he recognizedthat sulfur and phosphorus were present sometimes in‘animal substances’ that contained large numbers ofatoms. In other words, he established that these ‘sub-stances’ were macromolecules. Mulder communicatedhis results to Jons Jakob Berzelius and it is suggestedthe term protein arose from this interaction where theorigin of the word protein has been variously ascribedto derivation from the Latin word primarius or fromthe Greek god Proteus. The definition of proteins wastimely since in 1828 Friedrich Wohler had shown that
Proteins: Structure and Function by David Whitford 2005 John Wiley & Sons, Ltd

2 AN INTRODUCTION TO PROTEIN STRUCTURE AND FUNCTION
(NH4)OCN C
O
H2N NH2
Figure 1.1 The decomposition of ammonium cyanateyields urea
heating ammonium cyanate resulted in isomerism andthe formation of urea (Figure 1.1). Organic compoundscharacteristic of living systems, such as urea, couldbe derived from simple inorganic chemicals. For manyhistorians this marks the beginning of biochemistry andit is appropriate that the discovery of proteins occurredat the same period.
The development of biochemistry and the study ofproteins was assisted by analysis of their compositionand structure by Heinrich Hlasiwetz and Josef Haber-mann around 1873 and the recognition that proteinswere made up of smaller units called amino acids.They established that hydrolysis of casein with strongacids or alkali yielded glutamic acid, aspartic acid,leucine, tyrosine and ammonia whilst the hydrolysisof other proteins yielded a different group of products.Importantly their work suggested that the properties ofproteins depended uniquely on the constituent parts – atheme that is equally relevant today in modern bio-chemical study.
Another landmark in the study of proteins occurredin 1902 with Franz Hofmeister establishing the con-stituent atoms of the peptide bond with the polypep-tide backbone derived from the condensation of freeamino acids. Five years earlier Eduard Buchner rev-olutionized views of protein function by demonstrat-ing that yeast cell extracts catalysed fermentation ofsugar into ethanol and carbon dioxide. Previously itwas believed that only living systems performed thiscatalytic function. Emil Fischer further studied biolog-ical catalysis and proposed that components of yeast,which he called enzymes, combined with sugar to pro-duce an intermediate compound. With the realizationthat cells were full of enzymes 100 years of researchhas developed and refined these discoveries. Furtherlandmarks in the study of proteins could include Sum-ner’s crystallization of the first enzyme (urease) in1926 and Pauling’s description of the geometry of the
peptide bond; however, extensive discussion of theseadvances and many other important discoveries in pro-tein biochemistry are best left to history of sciencetextbooks.
A brief look at the award of the Nobel Prizesfor Chemistry, Physiology and Medicine since 1900highlighted in Table 1.1 reveals the involvement ofmany diverse areas of science in protein biochemistry.At first glance it is not obvious why William andLawrence Bragg’s discovery of the diffraction ofX-rays by sodium chloride crystals is relevant, butdiffraction by protein crystals is the main route towardsbiological structure determination. Their discovery wasthe first step in the development of this technique.Discoveries in chemistry and physics have beenimplemented rapidly in the study of proteins. By 1958Max Perutz and John Kendrew had determined the firstprotein structure and this was soon followed by thelarger, multiple subunit, structure of haemoglobin andthe first enzyme, lysozyme. This remarkable advancein knowledge extended from initial understanding ofthe atomic composition of proteins around 1900 tothe determination of the three-dimensional structure ofproteins in the 1960s and represents a major chapterof modern biochemistry. However, advances havecontinued with new areas of molecular biology provingequally important to understanding protein structureand function.
Life may be defined as the ordered interactionof proteins and all forms of life from viruses tocomplex, specialized, mammalian cells are based onproteins made up of the same building blocks oramino acids. Proteins found in simple unicellularorganisms such as bacteria are identical in structureand function to those found in human cells illustratingthe evolutionary lineage from simple to complexorganisms.
Molecular biology starts with the dramatic eluci-dation of the structure of the DNA double helix byJames Watson, Francis Crick, Rosalind Franklin andMaurice Wilkins in 1953. Today, details of DNA repli-cation, transcription into RNA and the synthesis of pro-teins (translation) are extensive. This has establishedan enormous body of knowledge representing a wholenew subject area. All cells encode the information con-tent of proteins within genes, or more accurately theorder of bases along the DNA strand, yet it is the

A BRIEF AND VERY SELECTIVE HISTORICAL PERSPECTIVE 3
Table 1.1 Selected landmarks in the study of protein structure and function from 1900–2002 as seen by the awardof the Nobel Prize for Chemistry, Physiology or Medicine
Date Discoverer + Discovery
1901 Wilhelm Conrad Rontgen ‘in recognition of the . . . discovery of the remarkable rays subsequentlynamed after him’
1907 Eduard Buchner ‘cell-free fermentation’
1914 Max von Laue ‘for his discovery of the diffraction of X-rays by crystals’
1915 William Henry Bragg and William Lawrence Bragg ‘for their services in the analysis of crystalstructure by . . . X-rays’
1923 Frederick Grant Banting and John James Richard Macleod ‘for the discovery of insulin’
1930 Karl Landsteiner ‘for his discovery of human blood groups’
1946 James Batcheller Sumner ‘for his discovery that enzymes can be crystallized’.
John Howard Northrop and Wendell Meredith Stanley ‘for their preparation of enzymes and virusproteins in a pure form’
1948 Arne Wilhelm Kaurin Tiselius ‘for his research on electrophoresis and adsorption analysis, especiallyfor his discoveries concerning the complex nature of the serum proteins’
1952 Archer John Porter Martin and Richard Laurence Millington Synge ‘for their invention of partitionchromatography’
1952 Felix Bloch and Edward Mills Purcell ‘for their development of new methods for nuclear magneticprecision measurements and discoveries in connection therewith’
1954 Linus Carl Pauling ‘for his research into the nature of the chemical bond and . . . to the elucidation of. . . complex substances’
1958 Frederick Sanger ‘for his work on the structure of proteins, especially that of insulin’
1959 Severo Ochoa and Arthur Kornberg ‘for their discovery of the mechanisms in the biological synthesisof ribonucleic acid and deoxyribonucleic acid’
1962 Max Ferdinand Perutz and John Cowdery Kendrew ‘for their studies of the structures of globularproteins’
1962 Francis Harry Compton Crick, James Dewey Watson and Maurice Hugh Frederick Wilkins ‘for theirdiscoveries concerning the molecular structure of nucleic acids and its significance for informationtransfer in living material’
1964 Dorothy Crowfoot Hodgkin ‘for her determinations by X-ray techniques of the structures of importantbiochemical substances’
1965 Francois Jacob, Andre Lwoff and Jacques Monod ‘for discoveries concerning genetic control ofenzyme and virus synthesis’
1968 Robert W. Holley, Har Gobind Khorana and Marshall W. Nirenberg ‘for . . . the genetic code and itsfunction in protein synthesis’
1969 Max Delbruck, Alfred D. Hershey and Salvador E. Luria ‘for their discoveries concerning thereplication mechanism and the genetic structure of viruses’
(continued overleaf )

4 AN INTRODUCTION TO PROTEIN STRUCTURE AND FUNCTION
Table 1.1 (continued)
Date Discoverer + Discovery
1972 Christian B. Anfinsen ‘for his work on ribonuclease, especially concerning the connection betweenthe amino acid sequence and the biologically active conformation’ Stanford Moore and William H.Stein ‘for their contribution to the understanding of the connection between chemical structure andcatalytic activity of . . . ribonuclease molecule’
1972 Gerald M. Edelman and Rodney R. Porter ‘for their discoveries concerning the chemical structure ofantibodies’
1975 John Warcup Cornforth ‘for his work on the stereochemistry of enzyme-catalyzed reactions’. VladimirPrelog ‘for his research into the stereochemistry of organic molecules and reactions’
1975 David Baltimore, Renato Dulbecco and Howard Martin Temin ‘for their discoveries concerning theinteraction between tumour viruses and the genetic material of the cell’
1978 Werner Arber, Daniel Nathans and Hamilton O. Smith ‘for the discovery of restriction enzymes andtheir application to problems of molecular genetics’
1980 Paul Berg ‘for his fundamental studies of the biochemistry of nucleic acids, with particular regard torecombinant-DNA’ Walter Gilbert and Frederick Sanger ‘for their contributions concerning thedetermination of base sequences in nucleic acids’
1982 Aaron Klug ‘development of crystallographic electron microscopy and structural elucidation ofnucleic acid–protein complexes’
1984 Robert Bruce Merrifield ‘for his development of methodology for chemical synthesis on a solidmatrix’
1984 Niels K. Jerne, Georges J.F. Kohler and Cesar Milstein ‘for theories concerning the specificity indevelopment and control of the immune system and the discovery of the principle for production ofmonoclonal antibodies’
1988 Johann Deisenhofer, Robert Huber and Hartmut Michel ‘for the determination of the structure of aphotosynthetic reaction centre’
1989 J. Michael Bishop and Harold E. Varmus ‘for their discovery of the cellular origin of retroviraloncogenes’
1991 Richard R. Ernst ‘for . . . the methodology of high resolution nuclear magnetic resonancespectroscopy’
1992 Edmond H. Fischer and Edwin G. Krebs ‘for their discoveries concerning reversible proteinphosphorylation as a biological regulatory mechanism’
1993 Kary B. Mullis ‘for his invention of the polymerase chain reaction (PCR) method’ and Michael Smith‘for his fundamental contributions to the establishment of oligonucleotide-based, site-directedmutagenesis’
1994 Alfred G. Gilman and Martin Rodbell ‘for their discovery of G-proteins and the role of these proteinsin signal transduction’

THE BIOLOGICAL DIVERSITY OF PROTEINS 5
Table 1.1 (continued)
Date Discoverer + Discovery
1997 Paul D. Boyer and John E. Walker ‘for their elucidation of the enzymatic mechanism underlying thesynthesis of adenosine triphosphate (ATP)’. Jens C. Skou ‘for the first discovery of anion-transporting enzyme, Na+, K+-ATPase’
1997 Stanley B. Prusiner ‘for his discovery of prions – a new biological principle of infection’
1999 Gunter Blobel ‘for the discovery that proteins have intrinsic signals that govern their transport andlocalization in the cell’
2000 Arvid Carlsson, Paul Greengard and Eric R Kandel ‘signal transduction in the nervous system’
2001 Paul Nurse, Tim Hunt and Leland Hartwill ‘for discoveries of key regulators of the cell cycle’
2002 Kurt Wuthrich, ‘for development of NMR spectroscopy as a method of determining biologicalmacromolecules structure in solution.’ John B. Fenn and Koichi Tanaka ‘for their development ofsoft desorption ionization methods for mass spectrometric analyses of biological macromolecules’.Sydney Brenner, H. Robert Horvitz and John E. Sulston ‘for their discoveries concerning geneticregulation of organ development and programmed cell death’
conversion of this information or expression into pro-teins that represents the tangible evidence of a livingsystem or life.
DNA −→ RNA −→ protein
Cells divide, synthesize new products, secrete unwantedproducts, generate chemical energy to sustain these pro-cesses via specific chemical reactions, and in all ofthese examples the common theme is the mediationof proteins.
In 1944 the physicist Erwin Schrodinger posed thequestion ‘What is Life?’ in an attempt to understand thephysical properties of a living cell. Schrodinger sug-gested that living systems obeyed all laws of physicsand should not be viewed as exceptional but insteadreflected the statistical nature of these laws. Moreimportantly, living systems are amenable to study usingmany of the techniques familiar to chemistry andphysics. The last 50 years of biochemistry have demon-strated this hypothesis emphatically with tools devel-oped by physicists and chemists rapidly employed inbiological studies. A casual perusal of Table 1.1 showshow quickly methodologies progress from discovery toapplication.
The biological diversity of proteins
Proteins have diverse biological functions ranging fromDNA replication, forming cytoskeletal structures, trans-porting oxygen around the bodies of multicellularorganisms to converting one molecule into another.The types of functional properties are almost end-less and are continually being increased as we learnmore about proteins. Some important biological func-tions are outlined in Table 1.2 but it is to be expectedthat this rudimentary list of properties will expandeach year as new proteins are characterized. A for-mal demarcation of proteins into one class should notbe pursued too far since proteins can have multipleroles or functions; many proteins do not lend them-selves easily to classification schemes. However, forall chemical reactions occurring in cells a protein isinvolved intimately in the biological process. Theseproteins are united through their composition based onthe same group of 20 amino acids. Although all pro-teins are composed of the same group of 20 aminoacids they differ in their composition – some containa surfeit of one amino acid whilst others may lackone or two members of the group of 20 entirely.It was realized early in the study of proteins that

6 AN INTRODUCTION TO PROTEIN STRUCTURE AND FUNCTION
Table 1.2 A selective list of some functional roles for proteins within cells
Function Examples
Enzymes or catalytic proteins Trypsin, DNA polymerases and ligases,Contractile proteins Actin, myosin, tubulin, dynein,Structural or cytoskeletal proteins Tropocollagen, keratin,Transport proteins Haemoglobin, myoglobin, serum albumin, ceruloplasmin,
transthyretinEffector proteins Insulin, epidermal growth factor, thyroid stimulating hormone,Defence proteins Ricin, immunoglobulins, venoms and toxins, thrombin,Electron transfer proteins Cytochrome oxidase, bacterial photosynthetic reaction centre,
plastocyanin, ferredoxinReceptors CD4, acetycholine receptor,Repressor proteins Jun, Fos, Cro,Chaperones (accessory folding proteins) GroEL, DnaKStorage proteins Ferritin, gliadin,
variation in size and complexity is common and themolecular weight and number of subunits (polypep-tide chains) show tremendous diversity. There is nocorrelation between size and number of polypeptidechains. For example, insulin has a relative molecu-lar mass of 5700 and contains two polypeptide chains,haemoglobin has a mass of approximately 65 000 andcontains four polypeptide chains, and hexokinase isa single polypeptide chain with an overall mass of∼100 000 (see Table 1.3).
The molecular weight is more properly referred toas the relative molecular mass (symbol Mr). This isdefined as the mass of a molecule relative to 1/12ththe mass of the carbon (12C) isotope. The mass ofthis isotope is defined as exactly 12 atomic massunits. Consequently the term molecular weight orrelative molecular mass is a dimensionless quantityand should not possess any units. Frequently in thisand many other textbooks the unit Dalton (equivalentto 1 atomic mass unit, i.e. 1 Dalton = 1 amu) is usedand proteins are described with molecular weights of5.5 kDa (5500 Daltons). More accurately, this is theabsolute molecular weight representing the mass ingrams of 1 mole of protein. For most purposes thisbecomes of little relevance and the term ‘molecular
Table 1.3 The molecular masses of proteins togetherwith the number of subunits. The term ‘subunit’ issynonymous with the number of polypeptide chainsand is used interchangeably
Protein Molecularmass
Subunits
Insulin 5700 2Haemoglobin 64 500 4Tropocollagen 285 000 3Subtilisin 27 500 1Ribonuclease 12 600 1Aspartate
transcarbamoylase310 000 12
Bacteriorhodopsin 26 800 1Hexokinase 102 000 1
weight’ is used freely in protein biochemistry and inthis book.
Proteins are joined covalently and non-covalentlywith other biomolecules including lipids, carbohydrates,

THE BIOLOGICAL DIVERSITY OF PROTEINS 7
nucleic acids, phosphate groups, flavins, heme groupsand metal ions. Components such as hemes or metalions are often called prosthetic groups. Complexesformed between lipids and proteins are lipoproteins,those with carbohydrates are called glycoproteins,whilst complexes with metal ions lead to metallo-proteins, and so on. The complexes formed betweenmetal ions and proteins increases the involvement ofelements of the periodic table beyond that expectedof typical organic molecules (namely carbon, hydro-gen, nitrogen and oxygen). Inspection of the periodictable (Figure 1.2) shows that at least 20 elements havebeen implicated directly in the structure and functionof proteins (Table 1.4). Surprisingly elements such asaluminium and silicon that are very abundant in theEarth’s crust (8.1 and 25.7 percent by weight, respec-tively) do not occur in high concentration within cells.Aluminium is rarely, if ever, found as part of proteins
whilst the role of silicon is confined to biomineralizationwhere it is the core component of shells. The involve-ment of carbon, hydrogen, oxygen, nitrogen, phospho-rus and sulfur is clear although the role of other ele-ments, particularly transition metals, has been difficultto establish. Where transition metals occur in proteinsthere is frequently only one metal atom per mole of pro-tein and led in the past to a failure to detect metal. Otherelements have an inferred involvement from growthstudies showing that depletion from the diet leads toan inhibition of normal cellular function. For metallo-proteins the absence of the metal can lead to a loss ofstructure and function.
Metals such as Mo, Co and Fe are often foundassociated with organic co-factors such as pterin,flavins, cobalamin and porphyrin (Figure 1.3). Theseorganic ligands hold metal centres and are often tightlyassociated to proteins.
Table 1.4 The involvement of trace elements in the structure and function of proteins
Element Functional role
Sodium Principal intracellular ion, osmotic balancePotassium Principal intracellular ion, osmotic balanceMagnesium Bound to ATP/GTP in nucleotide binding proteins, found as structural component of
hydrolase and isomerase enzymesCalcium Activator of calcium binding proteins such as calmodulinVanadium Bound to enzymes such as chloroperoxidase.Manganese Bound to pterin co-factor in enzymes such as xanthine oxidase or sulphite oxidase. Also
found in nitrogenase and as component of water splitting enzyme in higher plants.Iron Important catalytic component of heme enzymes involved in oxygen transport as well as
electron transfer. Important examples are haemoglobin, cytochrome oxidase andcatalase.
Cobalt Metal component of vitamin B12 found in many enzymes.Nickel Co-factor found in hydrogenase enzymesCopper Involved as co-factor in oxygen transport systems and electron transfer proteins such as
haemocyanin and plastocyanin.Zinc Catalytic component of enzymes such as carbonic anhydrase and superoxide dismutase.Chlorine Principal intracellular anion, osmotic balanceIodine Iodinated tyrosine residues form part of hormone thyroxine and bound to proteinsSelenium Bound at active centre of glutathione peroxidase

8 AN INTRODUCTION TO PROTEIN STRUCTURE AND FUNCTION
Per
iodi
c ta
ble
of th
e ch
emic
al e
lem
ents
and
thei
r in
volv
emen
t with
pro
tein
s
1 2
3 4
5 6
7 8
9 10
11
12
13
14
15
16
17
18
1s
1
H
Hyd
roge
n
2
He
Hel
ium
s
blo
ck
p b
lock
2s
3
LiLi
thiu
m
4
Be
Ber
yliu
m
5
B
Bor
on
6
C
Car
bon
7
N
Nitr
ogen
8
O
Oxy
gen
9
F
Flu
orin
e
10 N
e N
eon
3s
2p 3p11
Na
Sod
ium
12 M
gM
agne
sium
3p
d b
lock
(tr
ansi
tio
n m
etal
s)
13 A
l A
lum
iniu
m
14
Si
Sili
con
15
P
Pho
spho
rus
16
S
Sul
fur
17 C
l C
hlor
ine
18 A
r A
rgon
4s
19
K
Pot
assi
um
20 C
a C
alc i
um
3 d 21
Sc
Sca
ndiu
m
22
Ti
Tita
nium
23
V
Va
nadi
um 24
Cr
Chr
o miu
m
25 M
n M
anga
nese
26 Fe
Iro
n
27 C
o C
obal
t
28
Ni
Nic
kel
29 C
u C
oppe
r
30
Zn
Zin
c
31 G
a G
alli u
m
32 G
e G
erm
aniu
m
33 A
s A
rsen
ic
34 S
e S
e len
ium
35 B
r B
rom
ine
36 K
r K
ryp t
on5s
37
Rb
Rub
idiu
m 38
Sr
Str
ontiu
m
4 d 39
Y
Yttr
ium
40
Zr
Zirc
oniu
m
41 N
b N
iobi
um
42
Mo
Mol
ybde
num
43 T
c T
echn
etiu
m 44
Ru
Rut
heni
um 45
Rh
Rho
dium
46
Pd
Pal
ladi
um 47
Ag
Silv
er
48
Cd
Cad
miu
m
49
InIn
dium
50 S
n T
in
51 S
b A
ntim
ony
52 Te
Te
lluriu
m
53
I Io
dine
54 X
e X
enon
6s
55
Cs
Cae
sium
56 B
a B
ariu
m
5 d 71
LuLu
tetiu
m
72
Hf
Haf
nium
73 Ta
T
anta
lum
74
W
Tun
gste
n
75 R
e R
heni
um
76 O
s O
smiu
m
77
IrIri
dium
78
Pt
Pla
tinum
79 A
u G
old
80
Hg
Mer
cury
81
Tl
Tellu
rium
82 P
b Le
ad
83 B
i B
ism
uth
84 P
o P
olo
nium
85 A
t A
stat
ine
86 R
n R
adon
7s
87
Fr
Fra
nciu
m
88 R
a R
adiu
m
6 d 10
3
LrLa
wre
nciu
m
Met
al ↔
No
n-m
etal
s
f bl
ock
(la
nth
anid
es a
nd
act
inid
es)
4f
57 La
Lant
hanu
m 58
Ce
Cer
ium
59
Pr
Pra
esod
ymiu
m
60
Nd
Neo
dym
ium
61
Pm
P
rom
ethi
um 62
Sm
S
amar
ium
63
Eu
Eur
opiu
m
64
Gd
Gad
olin
ium
65
Tb
Terb
ium
66
Dy
Dys
pros
ium
67
Ho
Hol
miu
m
68
Er
Erb
ium
69
Tm
T
huliu
m
70
Yb
Ytte
rbiu
m5f
89
Ac
Act
iniu
m
90 T
h T
horiu
m
91
Pa
Pro
actin
ium
92
Ur
Ura
niu
m
93
Np
Nep
tuni
um 94
Pu
Put
oniu
m
95 A
m
Am
eric
um
96 C
m
Cur
ium
97
Bk
Ber
keliu
m
98
Cf
Cal
iforn
ium
99
Es
Eis
tein
ium
100
Fm
Fe
rmiu
m
101
Md
Men
dele
vium
102
No
Nob
eliu
m
Figu
re1.
2Th
epe
riod
icta
ble
show
ing
the
elem
ents
high
light
edin
red
know
nto
have
invo
lvem
ent
inth
est
ruct
ure
and/
orfu
ncti
onof
prot
eins
.Th
ein
volv
emen
tof
som
eel
emen
tsis
cont
enti
ous
tung
sten
and
cadm
ium
are
clai
med
tobe
asso
ciat
edw
ith
prot
eins
yet
thes
eel
emen
tsar
eal
sokn
own
tobe
toxi
c

WHY STUDY PROTEINS? 9
N
CNH2
O
R
N
NN
NH2N
O
N
N
N
N O
O
H3C
H3C
R
P
P
Fe
NN
N N
M
V
M
M
M
V
R1
O
M
MgNN
N N
M
CH2CH3
M
CH2
M
V
O
OM
CH2
Figure 1.3 Organic co-factors found in proteins. These co-factors are pterin, the isoalloxine ring found as part offlavin in FAD and FMN, the pyridine ring of NAD and its close analogue NADP and the porphyrin skeletons of hemeand chlorophyll. R represents the remaining part of the co-factor whilst M and V signify methyl and vinyl side chains
Proteins and the sequencing of thehuman and other genomes
Recognition of the diverse roles of proteins in biolog-ical systems increased largely as a result of the enor-mous amount of sequencing information generated viathe Human Genome Mapping project. Similar schemesaimed at deciphering the genomes of Escherichia coli,yeast (Sacharromyces cerevisiae), and mouse providedrelated information. With the completion of the firstdraft of the human genome mapping project in 2001human chromosomes contain approximately 25–30 000genes. This allows a conservative estimate of the num-ber of polypeptides making up most human cells as∼25 000, although alternative splicing of genes andvariations in subunit composition increase the num-ber of proteins further. Despite sequencing the humangenome it is an unfortunate fact that we do not knowthe role performed by most proteins. Of those thou-sands of polypeptides we know the structures of only asmall number, emphasizing a large imbalance between
the abundance of sequence data and the presence ofstructure/function information. An analysis of proteindatabases suggests about 1000 distinct structures orfolds have been determined for globular proteins. Manyproteins are retained within cell membranes and weknow virtually nothing about the structures of theseproteins and only slightly more about their functionalroles. This observation has enormous consequences forunderstanding protein structure and function.
Why study proteins?
This question is often asked not entirely without reasonby many undergraduates during their first introductionto the subject. Perhaps the best reply that can be givenis that proteins underpin every aspect of biologicalactivity. This is particularly important in areas whereprotein structure and function have an impact onhuman endeavour such as medicine. Advances inmolecular genetics reveal that many diseases stem fromspecific protein defects. A classic example is cystic

10 AN INTRODUCTION TO PROTEIN STRUCTURE AND FUNCTION
Figure 1.4 The shape of erythrocytes in normal and sickle cell anemia arises from mutations to haemoglobin foundwithin the red blood cell. (Reproduced with permission from Voet, D, Voet, J.G and Pratt, C.W. Fundamentals ofBiochemistry. John Wiley & Sons Inc.)
fibrosis, an inherited condition that alters a protein,called the cystic fibrosis transmembrane conductanceregulator (CFTR), involved in the transport of sodiumand chloride across epithelial cell membranes. Thisdefect is found in Caucasian populations at a ratioof ∼1 in 20, a surprisingly high frequency. With 1in 20 of the population ‘carrying’ a single defectivecopy of the gene individuals who inherit defectivecopies of the gene from each parent suffer from thedisease. In the UK the incidence of cystic fibrosis isapproximately 1 in 2000 live births, making it oneof the most common inherited disorders. The diseaseresults in the body producing a thick, sticky mucusthat blocks the lungs, leading to serious infection, andinhibits the pancreas, stopping digestive enzymes fromreaching the intestines where they are required to digestfood. The severity of cystic fibrosis is related to CFTRgene mutation, and the most common mutation, foundin approximately 65 percent of all cases, involves thedeletion of a single amino acid residue from the proteinat position 508. A loss of one residue out of a totalof nearly 1500 amino acid residues results in a severedecrease in the quality of life with individuals sufferingfrom this disease requiring constant medical care andsupervision.
Further examples emphasize the need to understandmore about proteins. The pioneering studies of VernonIngram in the 1950s showed that sickle cell anemiaarose from a mutation in the β chain of haemoglobin.Haemoglobin is a tetrameric protein containing 2α
and 2β chains. In each of the β chains a mutation
is found that involves the change of the sixth aminoacid residue from a glutamic acid to a valine. Thealteration of two residues out of 574 leads to a drasticchange in the appearance of red blood cells from theirnormal biconcave disks to an elongated sickle shape(Figure 1.4).
As the name of the disease suggests individualsare anaemic showing decreased haemoglobin contentin red blood cells from approximately 15 g per100 ml to under half that figure, and show frequentillness. Our understanding of cystic fibrosis and ofsickle cell anaemia has advanced in parallel withour understanding of protein structure and functionalthough at best we have very limited and crude meansof treating these diseases.
However, perhaps the greatest impetus to understandprotein structure and function lies in the hope ofovercoming two major health issues confronting theworld in the 21st century. The first of these is cancer.Cancer is the uncontrolled proliferation of cells thathave lost their normal regulated cell division often inresponse to a genetic or environmental trigger. Thedevelopment of cancer is a multistep, multifactorialprocess often occurring over decades but the preciseinvolvement of specific proteins has been demonstratedin some instances. One of the best examples is aprotein called p53, normally present at low levels incells, that ‘switches on’ in response to cellular damageand as a transcription factor controls the cell cycleprocess. Mutations in p53 alter the normal cycle ofevents leading eventually to cancer and several tumours

WHY STUDY PROTEINS? 11
including lung, colorectal and skin carcinomas areattributed to molecular defects in p53. Future researchon p53 will enable its physicochemical properties tobe thoroughly appreciated and by understanding thelink between structure, folding, function and regulationcomes the prospect of unravelling its role in tumourformation and manipulating its activity via therapeuticintervention. Already some success is being achievedin this area and the future holds great promise for‘halting’ cancer by controlling the properties of p53and similar proteins.
A second major problem facing the world todayis the estimated number of people infected with thehuman immunodeficiency virus (HIV). In 2003 theWorld Health Organization (WHO) estimated thatover 40 million individuals are infected with thisvirus in the world today. For many individuals,particularly those in the ‘Third World’, the prospectof prolonged good health is unlikely as the virusslowly degrades the body’s ability to fight infectionthrough damage to the immune response mechanismand in particular to a group of cells called cytotoxicT cells. HIV infection encompasses many aspects ofprotein structure and function, as the virus enters cellsthrough the interaction of specific viral coat proteinswith receptors on the surface of white blood cells. Onceinside cells the virus ‘hides’ but is secretly replicatingand integrating genetic material into host DNA throughthe action of specific enzymes (proteins). Halting thedestructive influence of HIV relies on understandingmany different, yet inter-related, aspects of proteinstructure and function. Again, considerable progresshas been made since the 1980s when the causativeagent of the disease was recognized as a retrovirus.These advances have focussed on understanding the
structure of HIV proteins and in designing specificinhibitors of, for example, the reverse transcriptaseenzyme. Although in advanced health care systemsthese drugs (inhibitors) prolong life expectancy, theeradication of HIV’s destructive action within thebody and hence an effective cure remains unachieved.Achieving this goal should act as a timely reminderfor all students of biology, chemistry and medicinethat success in this field will have a dramatic impacton the quality of human life in the forthcomingdecades.
Central to success in treating any of the above dis-eases are the development of new medicines, manybased on proteins. The development of new therapieshas been rapid during the last 20 years with the list ofnew treatments steadily increasing and including min-imizing serious effects of different forms of cancer viathe use of specific proteins including monoclonal anti-bodies, alleviating problems associated with diabetesby the development of improved recombinant ‘insulins’and developing ‘clot-busting’ drugs (proteins) for themanagement of strokes and heart attacks. This highlyselective list is the productive result of understandingprotein structure and function and has contributed toa marked improvement in disease management. Forthe future these advances will need to be extendedto other diseases and will rely on an extensive andthorough knowledge of proteins of increasing size andcomplexity. We will need to understand the structureof proteins, their interaction with other biomolecules,their roles within different biological systems and theirpotential manipulation by genetic or chemical meth-ods. The remaining chapters in this book represent anattempt to introduce and address some of these issuesin a fundamental manner helpful to students.


2Amino acids:
the building blocks of proteins
Despite enormous functional diversity all proteins con-sist of a linear arrangement of amino acid residuesassembled together into a polypeptide chain. Aminoacids are the ‘building blocks’ of proteins and in orderto understand the properties of proteins we must firstdescribe the properties of the constituent 20 aminoacids. All amino acids contain carbon, hydrogen, nitro-gen and oxygen with two of the 20 amino acidsalso containing sulfur. Throughout this book a colourscheme based on the CPK model (after Corey, Paulingand Kultun, pioneers of ‘space-filling’ representationsof molecules) is used. This colouring scheme showsnitrogen atoms in blue, oxygen atoms in red, carbonatoms are shown in light grey (occasionally black), sul-fur is shown in yellow, and hydrogen, when shown, iseither white, or to enhance viewing on a white back-ground, a lighter shade of grey. To avoid unnecessarycomplexity ‘ball and stick’ representations of molecu-lar structures are often shown instead of space-fillingmodels. In other instances cartoon representations ofstructure are shown since they enhance visualization oforganization whilst maintaining clarity of presentation.
The 20 amino acids found in proteinsIn their isolated state amino acids are white crystallinesolids. It is surprising that crystalline materials form the
building blocks for proteins since these latter moleculesare generally viewed as ‘organic’. The crystallinenature of amino acids is further emphasized by theirhigh melting and boiling points and together theseproperties are atypical of most organic molecules.Organic molecules are not commonly crystalline nor dothey have high melting and boiling points. Compare,for example, alanine and propionic acid – the formeris a crystalline amino acid and the other is a volatileorganic acid. Despite similar molecular weights (89and 74) their respective melting points are 314 ◦Cand −20.8 ◦C. The origin of these differences and theunique properties of amino acids resides in their ionicand dipolar nature.
Amino acids are held together in a crystallinelattice by charged interactions and these relativelystrong forces contribute to high melting and boilingpoints. Charge groups are also responsible for electricalconductivity in aqueous solutions (amino acids areelectrolytes), their relatively high solubility in waterand the large dipole moment associated with crystallinematerial. Consequently amino acids are best viewedas charged molecules that crystallize from solutionscontaining dipolar ions. These dipolar ions are calledzwitterions. A proper representation of amino acidsreflects amphoteric behaviour and amino acids arealways represented as the zwitterionic state in this
Proteins: Structure and Function by David Whitford 2005 John Wiley & Sons, Ltd
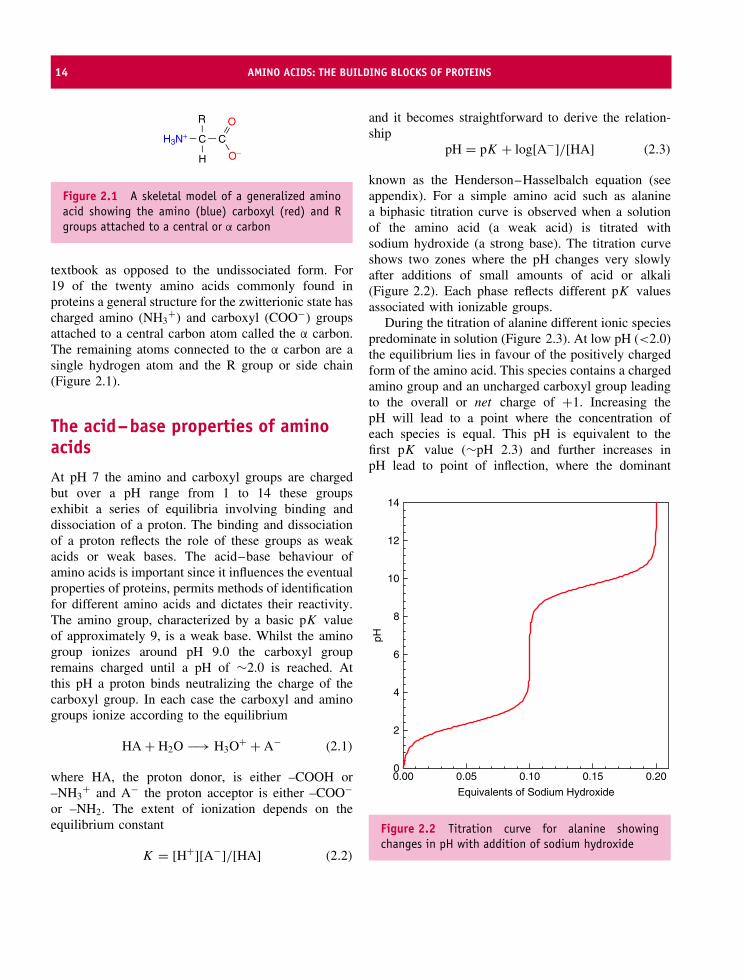
14 AMINO ACIDS: THE BUILDING BLOCKS OF PROTEINS
C
O
O−H
CH3N+
R
Figure 2.1 A skeletal model of a generalized aminoacid showing the amino (blue) carboxyl (red) and Rgroups attached to a central or α carbon
textbook as opposed to the undissociated form. For19 of the twenty amino acids commonly found inproteins a general structure for the zwitterionic state hascharged amino (NH3
+) and carboxyl (COO−) groupsattached to a central carbon atom called the α carbon.The remaining atoms connected to the α carbon are asingle hydrogen atom and the R group or side chain(Figure 2.1).
The acid–base properties of aminoacidsAt pH 7 the amino and carboxyl groups are chargedbut over a pH range from 1 to 14 these groupsexhibit a series of equilibria involving binding anddissociation of a proton. The binding and dissociationof a proton reflects the role of these groups as weakacids or weak bases. The acid–base behaviour ofamino acids is important since it influences the eventualproperties of proteins, permits methods of identificationfor different amino acids and dictates their reactivity.The amino group, characterized by a basic pK valueof approximately 9, is a weak base. Whilst the aminogroup ionizes around pH 9.0 the carboxyl groupremains charged until a pH of ∼2.0 is reached. Atthis pH a proton binds neutralizing the charge of thecarboxyl group. In each case the carboxyl and aminogroups ionize according to the equilibrium
HA + H2O −→ H3O+ + A− (2.1)
where HA, the proton donor, is either –COOH or–NH3
+ and A− the proton acceptor is either –COO−or –NH2. The extent of ionization depends on theequilibrium constant
K = [H+][A−]/[HA] (2.2)
and it becomes straightforward to derive the relation-ship
pH = pK + log[A−]/[HA] (2.3)
known as the Henderson–Hasselbalch equation (seeappendix). For a simple amino acid such as alaninea biphasic titration curve is observed when a solutionof the amino acid (a weak acid) is titrated withsodium hydroxide (a strong base). The titration curveshows two zones where the pH changes very slowlyafter additions of small amounts of acid or alkali(Figure 2.2). Each phase reflects different pK valuesassociated with ionizable groups.
During the titration of alanine different ionic speciespredominate in solution (Figure 2.3). At low pH (<2.0)the equilibrium lies in favour of the positively chargedform of the amino acid. This species contains a chargedamino group and an uncharged carboxyl group leadingto the overall or net charge of +1. Increasing thepH will lead to a point where the concentration ofeach species is equal. This pH is equivalent to thefirst pK value (∼pH 2.3) and further increases inpH lead to point of inflection, where the dominant
Equivalents of Sodium Hydroxide
0.00 0.05 0.10 0.15 0.20
pH
0
2
4
6
8
10
12
14
Figure 2.2 Titration curve for alanine showingchanges in pH with addition of sodium hydroxide

STEREOCHEMICAL REPRESENTATIONS OF AMINO ACIDS 15
H3N+ C C
OCH3
OHH
H3N+ C C
OCH3
O−H
H2N C C
OCH3
O−H
low pH ~ 2
pH ~ 7
high pH ~ 12
+1
0
−1
Figure 2.3 The three major forms of alanineoccurring in titrations between pH 1 and 14
species in solution is the zwitterion. The zwitterion,although dipolar, has no overall charge and at thispH the amino acid will not migrate towards either theanode or cathode when placed in an electric field. ThispH is called the isoelectric point or pI and for alaninereflects the arithmetic mean of the two pK values pI =(pK1 + pK2)/2. Continuing the pH titration still furtherinto alkaline conditions leads to the loss of a proton
from the amino group and the formation of a speciescontaining an overall charge of −1. The R group maycontain functional groups that donate or accept protonsand this leads to more complex titration curves. Aminoacids showing additional pK values include aspartate,glutamate, histidine, argininine, lysine, cysteine andtyrosine (see Table 2.1).
Amino acids lacking charged side chains showsimilar values for pK1 of about 2.3 that are significantlylower than the corresponding values seen in simpleorganic acids such as acetic acid (pK1 ∼4.7). Aminoacids are stronger acids than acetic acid as a resultof the electrophilic properties of the α amino groupthat increase the tendency for the carboxyl hydrogento dissociate.
Stereochemical representationsof amino acids
Although an amino acid is represented by the skeletaldiagram of Figure 2.1 it is more revealing, andcertainly more informative, to impose a stereochemicalview on the arrangement of atoms. In these views anattempt is made to represent the positions in spaceof each atom. The amino, carboxyl, hydrogen and Rgroups are arranged tetrahedrally around the central α
carbon (Figure 2.4).
Table 2.1 The pK values for the α-carboxyl, α-amino groups and side chains found in the individual amino acids
Amino acid pK1 pK2 pKR Amino acid pK1 pK2 pKR
Alanine 2.4 9.9 – Leucine 2.3 9.7 –Arginine 1.8 9.0 12.5 Lysine 2.2 9.1 10.5Asparagine 2.1 8.7 – Methionine 2.1 9.3 –Aspartic Acid 2.0 9.9 3.9 Phenylalanine 2.2 9.3 –Cysteine 1.9 10.7 8.4 Proline 2.0 10.6 –Glutamic Acid 2.1 9.5 4.1 Serine 2.2 9.2 –Glutamine 2.2 9.1 – Threonine 2.1 9.1 –Glycine 2.4 9.8 – Tyrosine 2.2 9.2 10.5Histidine 1.8 9.3 6.0 Tryptophan 2.5 9.4 –Isoleucine 2.3 9.8 – Valine 2.3 9.7 –
Adapted from Dawson, R.M.C, Elliot, W.H., & Jones, K.M. 1986 Data for Biochemical Research, 3rd edn. Clarendon Press Oxford.

16 AMINO ACIDS: THE BUILDING BLOCKS OF PROTEINS
Figure 2.4 The spatial arrangement of atoms in theamino acid alanine
The nitrogen atom (blue) is part of the amino(–NH3
+) group, the oxygen atoms (red) are part ofthe carboxyl (–COO−) group. The remaining groupsjoined to the α carbon are one hydrogen atom and theR group.
The R group is responsible for the differentproperties of individual amino acids. As amino acidsmake up proteins the properties of the R groupcontribute considerably to the physical properties ofproteins. Nineteen of the 20 amino acids found inproteins have the arrangement shown by Figure 2.4 butfor the remaining amino acid, proline, an unusual cyclicring is formed by the side chain bonding directly to theamide nitrogen (Figure 2.5).
N CH
H2C CH2
H2C
C
O−
O
H2
+
Figure 2.5 The structure of proline – an unusualamino acid containing a five-membered pyrroli-dine ring
A glance at the structures of the 20 different sidechains reveals major differences in, for example, size,charge and hydrophobicity although the R group isalways attached to the α carbon (C2 carbon). From theα carbon subsequent carbon atoms in the side chainsare designated as β, γ, δ, ε and ζ. In some databases ofprotein structures the Cβ is written as CB, the Cδ as CD,Cζ as CZ, etc. Both nomenclatures are widely used.The nomenclature is generally unambiguous but careneeds to be exercised when describing the atoms of theside chain of isoleucine. Isoleucine has a branched sidechain in which the Cγ or CG is either a methyl groupor a methylene group. In this instance the two groupsare distinguished by the use of a subscript 1 and 2, i.e.CG1 and CG2. A similar line of reasoning applies tothe carbon atoms of aromatic rings. In phenylalanine,for example, the aromatic ring is linked to the Cβ atomby the Cγ atom and contains two Cδ and Cε atoms (Cδ1
and Cδ2, Cε1 and Cε2) before completing ring at the Cζ
(or CZ) atom.
Peptide bonds
Amino acids are joined together by the formationof a peptide bond where the amino group of onemolecule reacts with the carboxyl group of the other.The reaction is described as a condensation resultingin the elimination of water and the formation of adipeptide (Figure 2.6).
Three amino acids are joined together by two pep-tide bonds to form a tripeptide and the sequence
NH2 CH
CH3
COOHH2N CH2 COOH
H2N CH2 CO NH CH
CH3
COOH
+
Glycylalanine
H2O
Figure 2.6 Glycine and alanine react together toform the dipeptide glycylalanine. The importantpeptide bond is shown in red

PEPTIDE BONDS 17
continues with the formation of tetrapeptides, pen-tapeptides, and so on. When joined in a series ofpeptide bonds amino acids are called residues todistinguish between the free form and the formfound in proteins. A short sequence of residuesis a peptide with the term polypeptide applied tolonger chains of residues usually of known sequenceand length. Within the cell protein synthesis occurson the ribosome but today peptide synthesis ispossible in vitro via complex organic chemistry.However, whilst the organic chemist struggles tosynthesize a peptide containing more than 50 residuesthe ribosome routinely makes proteins with over1000 residues.
All proteins are made up of amino acid residueslinked together in an order that is ultimately derivedfrom the information residing within our genes. Someproteins are clearly related to each other in that theyhave similar sequences whilst most proteins exhibita very different composition of residues and a verydifferent order of residues along the polypeptide chain.From the variety of side chains a single amino acidcan link to 19 others to create a total of 39 differentdipeptides. Repeating this for the other residues leadsto a total of 780 possible dipeptide permutations.If tripeptides and tetrapeptides are considered thenumber of possible combinations rapidly reachesa very large figure. However, when databases ofprotein sequences are studied it is clear that aminoacid residues do not occur with equal frequency inproteins and sequences do not reflect even a smallpercentage of all possible combinations. Tryptophanand cysteine are rare residues (less than 2 percent of allresidues) in proteins whilst alanine, glycine and leucineoccur with frequencies between 7 and 9 percent (seeTable 2.2).
Amino acid sequences of proteins are read fromleft to right. This is from the amino or N terminalto the carboxyl or C terminal. The individual aminoacids have three-letter codes, but increasingly, in orderto save space in the presentation of long proteinsequences, a single-letter code is used for each aminoacid residue. Both single- and three-letter codes areshown alongside the R groups in Table 2.2 togetherwith some of the relevant properties of each side chain.Where possible the three-letter codes for amino acidswill be used but it should be stressed that single letter
codes avoid potential confusion. For example Gly,Glu and Gln are easily mistaken when rapidly readingprotein sequences but their single letter codes of G, Eand Q are less likely to be misunderstood.
Joining together residues establishes a proteinsequence that is conveniently divided into main chainand side chain components. The main chain, orpolypeptide backbone, has the same composition in allproteins although it may differ in extent – that is thenumber of residues found in the polypeptide chain. Thebackbone represents the effective repetition of peptidebonds made up of the N, Cα and C atoms, with pro-teins such as insulin having approximately 50 residueswhilst other proteins contain over 1000 residues andmore than one polypeptide chain (Figure 2.7). Whilstall proteins link atoms of the polypeptide backbonesimilarly the side chains present a variable componentin each protein.
Properties of the peptide bond
The main chain or backbone of the polypeptide chain isestablished by the formation of peptide bonds betweenamino acids. The backbone consists of the amideN, the α-carbon and the carbonyl C linked together(Figure 2.8).
N CH C
OR
NH3 CH C
OR
N C
O
R
CHO−
H
Hn
+
Figure 2.7 Part of a polypeptide chain formed bythe covalent bonding of amino acids where n is often50–300, although values above and below these limitsare known.
C
O
NCHN
CHC
O
H
H
residue i residue i + 1
Figure 2.8 The polypeptide backbone showingarrangement of i, i + 1 residues within a chain.

18 AMINO ACIDS: THE BUILDING BLOCKS OF PROTEINS
Table 2.2 The frequencies with which amino acid residues occur in proteins
Amino acid Property of individual aminoacid residues
Ball and stick representationof each amino acid
AlanineAAlaMr 71.09
Non-polar side chain.Small side chain volume.Van der Waals volume = 67 A3∗
Frequency in proteins = 7.7 %Surface area = 115 A2
Unreactive side chain
ArginineRArgMr 156.19
Positively charged side chain at pH 7.0. pKfor guanidino group in proteins ∼12.0
Van der Waals volume = 167 A3
Frequency in proteins = 5.1 %Surface area = 225 A2
Participates in ionic interactions withnegatively charged groups
AsparagineNAsnMr 114.11
Polar, but uncharged, side chainVan der Waals volume = 148 A3
Frequency in proteins = 4.3 %Surface area = 160 A2
Polar side chain will hydrogen bondRelatively small side chain volume leads to
this residue being found relativelyfrequently in turns
AspartateDAspMr 115.09
Negatively charged side chainpK for side chain of ∼4.0Van der Waals volume = 67 A3
Frequency in proteins = 5.2 %Surface area = 150 A2
Charged side chain exhibits electrostaticinteractions with positively charged groups.
PEPTIDE BONDS 19
Table 2.2 (continued)
Amino acid Property of individual aminoacid residues
Ball and stick representationof each amino acid
CysteineCCysMr 103.15
Side chain contains thiol (SH) group.Van der Waals volume = 86 A3
Frequency in proteins = 2.0 %Surface area = 135 A2
Thiol side chain has pK in isolated aminoacid of ∼8.5 but in proteins varies 5–10
Thiol group is very reactive
GlutamineQGlnMr 128.12
Polar but uncharged side chainVan der Waals volume = 114 A3
Frequency in proteins = 4.1 %Surface area = 180 A2
Polar side chain can hydrogen bond
GlutamateEGluMr 129.12
Negatively charged side chain.Van der Waals volume = 109 A3
Frequency in proteins = 6.2 %Surface area = 190 A2
Side chain has pK of ∼4.5.
GlycineGGlyMr 57.05
Uncharged, small side chain.Often found in turn regions of proteins or
regions of conformational flexibilityNo chiral centre; due to two hydrogens
attached to Cα centreVan der Waals volume = 48 A3
Frequency in proteins = 7.4 %Surface area = 75 A2
(continued overleaf )
20 AMINO ACIDS: THE BUILDING BLOCKS OF PROTEINS
Table 2.2 (continued)
Amino acid Property of individual aminoacid residues
Ball and stick representationof each amino acid
HistidineHHisMr 137.14
Imidazole side chainVan der Waals volume = 118 A3
Frequency in proteins = 2.3 %Surface area = 195 A2
The side chain exhibits a pK ∼ 6.0 in modelpeptides but in proteins can vary from4–10
IsoleucineIIleMr 113.16
Hydrophobic side chain exhibiting non-polarbased interactions but generally unreactive
Van der Waals volume = 124 A3
Frequency in proteins = 5.3 %Surface area = 175 A2
LeucineLLeuMr 113.16
Hydrophobic side chainVan der Waals volume = 124 A3
Frequency in proteins = 8.5 %Surface area = 170 A2
LysineKLysMr 128.17
Positively charged side chainVan der Waals volume = 135 A3
Frequency in proteins = 5.9 %Surface area = 200 A2
Side chain is basic with pK of ∼10.5.Shows ionic interactions

PEPTIDE BONDS 21
Table 2.2 (continued)
Amino acid Property of individual aminoacid residues
Ball and stick representationof each amino acid
MethionineMMetMr 131.19
Sulfur containing hydrophobic side chainThe sulfur is unreactive especially when
compared with thiol group of cysteineVan der Waals volume = 124 A3
Frequency in proteins = 2.4 %Surface area = 185 A2
PhenylalanineFPheMr 147.18
Hydrophobic, aromatic side chainPhenyl ring is chemically unreactive in
proteins. Exhibits weak optical absorbancearound 280 nm
Van der Waals volume = 135 A3
Frequency in proteins = 4.0 %Surface area = 210 A2
ProlinePProMr 97.12
Cyclic ring forming hydrophobic side chainThe cyclic ring limits conformational
flexibility around N-Cα bondIn a polypeptide chain lacks amide hydrogen
and cannot form backbone hydrogen bondsVan der Waals volume = 90 A3
Frequency in proteins = 5.1 %Surface area = 145 A2
SerineSSerMr 87.08
Polar but uncharged side chain. Containshydroxyl group (–OH) that hydrogen bonds
Oxygen atom can act as potent nucleophile insome enzymes
Van der Waals volume = 73 A3
Frequency in proteins = 6.9 %Surface area = 115 A2
(continued overleaf )

22 AMINO ACIDS: THE BUILDING BLOCKS OF PROTEINS
Table 2.2 (continued)
Amino acid Property of individual aminoacid residues
Ball and stick representationof each amino acid
ThreonineTThrMr 101.11
Polar but uncharged side chain. Containshydroxyl group (–OH)
Hydrogen bonding side chainVan der Waals volume = 93 A3
Frequency in proteins = 5.9 %Surface area = 140 A2
TryptophanWTrpMr 186.21
Large, hydrophobic and aromatic side chainAlmost all reactivity is based around the
indole ring nitrogenResponsible for majority of near uv
absorbance in proteins at 280 nmVan der Waals volume = 163 A3
Frequency in proteins = 1.4 %Surface area = 255 A2
TyrosineYTyrMr 163.18
Aromatic side chainVan der Waals volume = 141 A3
Frequency in proteins = 3.2 %Surface area = 230 A2
Phenolic hydroxyl group ionizes at pH valuesaround pH 10
Aromatic ring more easily substituted thanthat of phenylalanine
ValineVValMr 99.14
Hydrophobic side chainVan der Waals volume = 105 A3
Frequency in proteins = 6.6 %Surface area = 155 A2
From Jones, D.T. Taylor, W.R. & Thornton, J.M. (1991) CABIOS 8, 275–282. Databases of protein sequences are weighted towards globularproteins but with the addition of membrane proteins to databases a gradual increase in the relative abundance of hydrophobic residuessuch as Leu, Val, Ile, Phe, Trp is expected. The surface area was calculated for an accessible surface of residue X in the tripeptide G-X-G(Chothia, C. (1975) J. Mol. Biol., 105, 1–14). Volumes enclosed by the van der Waals radii of atoms as described by Richards, F.M. (1974)J. Mol. Biol. 82, 1–14.∗1 A = 0.1 nm.

THE CHEMICAL AND PHYSICAL PROPERTIES OF AMINO ACIDS 23
The linear representation of the polypeptide chaindoes not convey the intricacy associated with thebond lengths and angles of the atoms making up thepeptide bond. The peptide bond formed between thecarboxyl and amino groups of two amino acids is aunique bond that possesses little intrinsic mobility. Thisoccurs because of the partial double bond character(Figure 2.9)–a feature associated with the peptide bondand resonance between two closely related states.
One of the most important consequences of reso-nance is that the peptide bond length is shorter thanexpected for a simple C–N bond. On average a peptidebond length is 1.32 A compared to 1.45 A for an ordi-nary C–N bond. In comparison the average bond lengthassociated with a C=N double bond is 1.25 A, empha-sizing the intermediate character of the peptide bond.More importantly the partial double bond between car-bon and nitrogen atoms restricts rotation about thisbond. This leads to the six atoms shown in Figure 2.9being coplanar; that is all six atoms are found within asingle imaginary plane. For any polypeptide backbonerepresented by the sequence –N–Cα–C–N–Cα–C–onlythe Cα–C and N–Cα bonds exhibit rotational mobil-ity. As a result of restricted motion about the peptidebond two conformations related by an angle of 180◦
are possible. The first occurs when the Cα atoms aretrans to the peptide bond whilst the second and lessfavourable orientation occurs when the Cα atoms arecis (Figure 2.10).
N
Ca
O Ca
H
N
Ca
−O Ca
H
+
Figure 2.9 The peptide bond may be viewed as apartial double bond as a result of resonance
N
Ca
O Ca
H
N
Ca
O H
Catrans cis
Figure 2.10 Cis and trans configurations are possibleabout the rigid peptide bond
C
O
NCHN
CHC
O
H
H
residue i residue i + 1
118.2119.5
121.9 115.6
123.2121.1
0.105 nm 0.123 nm
0.145 nm0.152 nm0.132 nm
0.38 nm
Figure 2.11 Detailed bond lengths and angles foratoms of the polypeptide backbone
The trans form is the more favoured state because inthis arrangement repulsion between non-bonded atomsconnected to the Cα centre are minimized. For mostpeptide bonds the ratio of cis to trans configurationsis approximately 1:1000. However, one exception tothis rule is found in peptide bonds where the followingresidue is proline. Proline, unusual in having a cyclicside chain that bonds to the backbone amide nitrogen,has less repulsion between side chain atoms. This leadsto an increase in the relative stability of the cis peptidebond when compared with the trans state, and forpeptide bonds formed between Xaa and proline (Xaais any amino acid) the cis to trans ratio is 1:4.
The dimensions associated with the peptide bondhave generally been obtained from crystallographicstudies of small peptides and besides the peptide bondlength of 0.132 nm other characteristic bond lengthsand angles have been identified (Figure 2.11). Oneof the most important dimensions is the maximumdistance between corresponding atoms in sequentialresidues. In the trans peptide bond and a fully extendedconformation this distance is maximally 0.38 nmalthough in proteins it is often much less.
The chemical and physical propertiesof amino acids
Chemical reactions exhibited by the 20 amino acidsare extensive and revolve around the reactivity ofamino and carboxyl groups. In proteins, however, thesegroups are involved in peptide bonds and the definingproperties of amino acids are those associated withside chains.

24 AMINO ACIDS: THE BUILDING BLOCKS OF PROTEINS
Glycine
Glycine is the simplest amino acid containing twohydrogen atoms attached to the central α carbon.Consequently it lacks an asymmetric centre anddoes not occur as R/S isomers (see below). Moreimportantly the absence of any significant functionalgroup means that glycine possesses little intrinsicchemical reactivity. However, the absence of a largeside chain results in conformational flexibility aboutthe N–Cα and Cα–C bonds in a polypeptide chain. Thisfact alone has important consequences for the overallstructure of proteins containing significant numbers ofglycine residues.
Aliphatic side chains: alanine, valine,isoleucine and leucine
Alanine, like glycine, has a very simple side chain – asingle methyl group (CH3) that is chemically inert andinactive (Figure 2.12). It is joined in this remarkablenon-reactivity by the side chains of valine, leucineand isoleucine (Figure 2.13). The side chains of theseamino acids are related in containing methine (CH)or methylene (–CH2) groups in branched aliphaticchains terminated by methyl groups. However, onevery important property possessed by these side chainsis their unwillingness to interact with water – theirhydrophobicity. The side chains interact far morereadily with each other and the other non-polarside chains of amino acids such as tryptophan orphenylalanine. In later chapters on tertiary structure,protein stability and folding the important role ofthe weak hydrophobic interaction in maintaining the
Figure 2.12 The side chain of alanine is the un-reactive methyl group (–CH3)
Figure 2.13 The structures of the side chains ofleucine (left) and isoleucine (right)
Figure 2.14 The side chain of valine is branchedat the CB (–CH or methine) group and leads to twomethyl groups
native state of proteins is described. Alanine, valine,leucine and isoleucine will all exhibit hydrophobicinteractions. Isoleucine and leucine as their namessuggest are isomers differing in the arrangement ofmethylene and methyl groups. The first carbon atomin the side chain (CB) is a methylene (CH2) groupin leucine whilst in isoleucine it is a methine (–CH–)moiety. The CB of leucine is linked to a methine group(CG) whilst in isoleucine the CB atom bonds to amethyl group and a methylene group. The side chainof valine (Figure 2.14) has one methylene group lessthan leucine but is otherwise very similar in property.

THE CHEMICAL AND PHYSICAL PROPERTIES OF AMINO ACIDS 25
The hydroxyl-containing residues: serineand threonine
The side chains of serine and threonine are charac-terized by the presence of a polar hydroxyl (–OH)group (Figure 2.15). Their side chains are generallysmall (Table 2.2). Although small the side groups ofboth serine and threonine hydrogen bond with otherresidues in proteins whilst in an isolated state their reac-tivity is confined to that expected for a primary alcohol,with esterification representing a common reaction withorganic acids. One of the most important reactions to aserine or threonine side chain is the addition of a phos-phate group to create phosphoserine or phosphothreo-nine. This occurs as a post-translational modificationin the cell after protein synthesis on the ribosome andis important in protein–protein interactions and intra-cellular signalling pathways.
In one group of enzymes – the serine proteases – theactivity of a single specific serine side chain isenhanced by its proximity to histidine and aspartyl sidechains. By itself the side chain of Ser is a weak nucle-ophile but combined with His and Asp this triad ofresidues becomes a potent catalytic group capable ofsplitting peptide bonds.
The acidic residues: aspartate and glutamate
Aspartic acid and glutamic acid have side chains witha carboxyl group (Figure 2.16). This leads to the sidechain having a negative charge under physiological
Figure 2.15 The side chains of serine and threonineare aliphatic containing hydroxyl groups
Figure 2.16 The acidic side chains of aspartate andglutamate
conditions and hence to their description as acidicside chains. For this reason the residues are normallyreferred to as aspartate and glutamate reflecting theionized and charged status under most cellular con-ditions. The two side chains differ by one methylenegroup (CH2) but are characterized by similar pK values(range 3.8–4.5).
The side chains behave as typical organic acids andexhibit a wide range of chemical reactions includingesterification with alcohols or coupling with amines.Both of these reactions have been exploited in chemicalmodification studies of proteins to alter one or moreaspartate/glutamate side chains with a loss of thenegatively charged carboxyl group. As expected theside chains of aspartate and glutamate are potentchelators of divalent metal ions and biology exploitsthis property to bind important ions such as Ca2+ inproteins such as calmodulin or Zn2+ in enzymes suchas carboxypeptidase.
The amide-containing residues: asparagineand glutamine
Asparagine and glutamine residues are often confusedwith their acidic counterparts, the side chains of aspar-tate and glutamate. Unlike the acidic side chains thefunctional group is an amide – a generally unreactivegroup that is polar and acts as hydrogen bond donorand acceptor (see Figure 2.17). The amide group islabile at alkaline pH values, or extremes of temperature,being deamidated to form the corresponding acidic side

26 AMINO ACIDS: THE BUILDING BLOCKS OF PROTEINS
Figure 2.17 The uncharged side chains of asparagineand glutamine
chain. This reaction can occur during protein isolationand occasionally leads to protein sequences containingthe three letter code Glx or Asx where the identity of aresidue, either Gln or Glu and Asp or Asn, is unclear.
The sulfur-containing residues: cysteineand methionine
Sulfur occurs in two of the 20 amino acids: cysteine andmethionine. In cysteine the sulfur is part of a reactivethiol group whilst in methionine sulfur is found aspart of a long, generally unreactive, side chain. Theside chain of methionine is non-polar (Figure 2.18)and is larger than that of valine or leucine althoughit is unbranched. Its properties are dominated by thepresence of the sulfur atom, a potent nucleophile undercertain conditions.
The sulfur atom is readily methylated using methyliodide in a reaction that is often used to introducea ‘label’ onto methionine residue via the use of 13Clabelled reactant. In addition the sulfur of methionineinteracts with heavy metal complexes particularly thoseinvolving mercury and platinum such as K2PtCl4 or
Figure 2.18 The side chain of methionine
CH2 CH2 S CH3
CH2 CH2 S CH3
O
CH2 CH2 S CH3
O
O
Figure 2.19 Oxidation of methionine side chains bystrong oxidizing agents
HgCl2 and these have proved extremely useful in theformation of isomorphous heavy atom derivatives inprotein crystallography (see Chapter 10). The sulfuratom of methionine can be oxidized to form first asulfoxide and finally a sulfone derivative (Figure 2.19).This form of oxidative damage is known to occur inproteins and the reaction scheme involves progressiveaddition of oxygen atoms.
One of the most important reactions of methionineinvolves cyanogen bromide – a reagent that breaks thepolypeptide chain on the C-terminal side of methionineresidues by sequestering the carbonyl group of the nextpeptide bond in a reaction involving water and leadingto formation of a homoserine lactone (Figure 2.20).This reaction is used to split polypeptide chainsinto smaller fragments for protein sequencing. Whencompared with cysteine, however, the side chain ofmethionine is less reactive undergoing comparativelyfew important chemical reactions.
The functional group of cysteine is the thiol (–SH)group, sometimes called the sulfhydryl or mercaptogroup. It is the most reactive side chain foundamongst the 20 naturally occurring amino acids under-going many chemical reactions with diverse reagents(Figure 2.21). Some enzymes exploit reactivity byusing a conserved cysteine residue at their active sitesthat participates directly in enzyme-catalysed reactions.
The large sulfur atom as part of the thiol group influ-ences side chain properties significantly, with disulfidebonds forming between cysteine residues that are close

THE CHEMICAL AND PHYSICAL PROPERTIES OF AMINO ACIDS 27
N C C
OCH2
CH2
S
CH3
N
HH H
Br
C
N
N C C
OCH2
CH2
S+
CH3
N
H H
N
H
N C C
OH2C
N+
HH H
CH2
SH3C C N
N C C
OH2C
H H
CH2
O
H2N
Br−
H2O
Peptide+ +
Figure 2.20 The reaction of cyanogen bromide withmethionine residues
Figure 2.21 The thiol group in the side chain ofcysteine is the most reactive functional group foundin amino acid residues
together in space. This forms a strong covalent bondand exercises considerable conformational restraint onthe structure adopted in solution by polypeptides. For-mation of a disulfide bridge is another example ofpost-translational modification (Figure 2.22).
The thiol group ionizes at alkaline pH values (∼pH8.5) to form a reactive thiolate anion (S−). The
CH2CH
NH
CO
S S CH2 CH
NH
CO
Figure 2.22 Formation of a disulfide bridge betweentwo thiol side chains
R1
S−
SR2
R2S
R2
S−
SR2
R1S
+ +
Figure 2.23 The formation of mixed disulfides viathe interaction of thiolate groups. A thiolate anionreacts with other symmetrical disulfides to form amixed disulfide formed between R1 and R2
thiolate anion reacts rapidly with many compounds butthe most important includes other thiols or disulfidesin exchange type reactions occurring at neutral toalkaline pH values. A general reaction scheme for thiol-disulfide exchange is given in Figure 2.23.
A common reaction of this type is the reactionbetween cysteine and Ellman’s reagent (dithionitroben-zoic acid, DTNB; Figure 2.24). The aromatic disulfideundergoes exchange with reactive thiolate anions form-ing a coloured aromatic thiol – nitrothiobenzoate. Thebenzoate anion absorbs intensely at 416 nm allowingthe concentration of free thiol groups to be accuratelyestimated in biological systems.
Thiols are also oxidized by molecular oxygen inreactions catalysed by trace amounts of transition met-als, including Cu and Fe. More potent oxidants suchas performic acid oxidize the thiol group to a sul-fonate (SO3
2−) and this reaction has been exploitedas a method of irreversibly breaking disulfide bridgesto form two cysteic acid residues. More frequentlythe disulfide bridge between cysteine residues is bro-ken by reducing agents that include other thiols suchas mercaptoethanol or dithiothreitol (Figure 2.25) aswell as more conventional reductants such as sodiumborohydride or molecular hydrogen. Dithiothreitol,

28 AMINO ACIDS: THE BUILDING BLOCKS OF PROTEINS
S
S
NO2
O2N
O
O−
O
O−
CH2 S−
CH2 S S NO2
O
O−
S−O2N
O
O−
+
+
DTNB
NTB
Figure 2.24 The reaction between the thiolate anionof cysteine and Ellman’s reagent
SS
OHHO
RSHR'SH
RS S
R′
CH2CH2
SH SH
OH
H OH
H
+
+
+
Figure 2.25 The reaction between dithiothreitol anddisulfide groups leads to reduction of the disulfide andthe formation of two thiols
sometimes called Cleland’s reagent, reacts with disul-fide groups to form initially a mixed disulfide inter-mediate but this rapidly rearranges to yield a stablesix-membered ring and free thiol groups (–SH). The
equilibrium constant for the reaction lies over to theright and is largely driven by the rapid formation ofcyclic disulfide and its inherent stability.
The basic residues: lysine and arginine
The arginine side chain (Figure 2.26) contains threemethylene groups followed by the basic guanidino(sometimes called guanadinium) group, which isusually protonated, planar and with the carbon atomexhibiting sp2 hybridization.
The guanidino group is the most basic of the sidechains found in amino acids with a pK of 12 andunder almost all conditions the side chain retains a netpositive charge (Figure 2.27). The positive charge isdistributed over the entire guanidino group as a resultof resonance between related structures
Lysine possesses a long side chain of four methy-lene groups terminated by a single ε amino group(Figure 2.28). The amino group ionizes with a pK
of approximately 10.5–11.0 and is very basic. Asexpected the side chain interacts strongly in proteinswith oppositely charged side chains but will alsoundergo methylation, acetylation, arylation and acyla-tion. Many of these reactions are performed at highpH (above pH 9.0) since the unprotonated nitrogenis a potent nucleophile reacting rapidly with suitablereagents. One of the most popular lysine modifications
Figure 2.26 The side chain of arginine is very basicand contains a guanidino group
+NH2
NH2+
H N+
CNH2
NH2
N
CH2N+
NH2
HN
C
H N
CNH2
NH2+
H
Figure 2.27 Charge delocalization and isomerization within the guanidino group

THE CHEMICAL AND PHYSICAL PROPERTIES OF AMINO ACIDS 29
Figure 2.28 The lysine side chain is basic
Cl C
O
O−
O2N
O2N
(CH2)4 NH2
(CH2)4 N C
O
O−
O2N
O2N
+
H
Figure 2.29 Reaction of lysine side chains withdinitrobenzene derivatives
involves adding a nitrobenzene derivative to theε-amino group. The nitrobenzene group is coloured andthe rate of the reaction is therefore comparatively easilyfollowed spectrophotometrically. In the example shownin Figure 2.29 4-chloro-3, 5-dinitrobenzoic acid reactswith lysine side chains to form a negatively chargeddinitrophenol derivative. Methylation, unlike the ary-lation reaction described above, preserves the positivecharge on the side chain and in some systems, partic-ularly fungi, trimethylated lysine residues are found asnatural components of proteins.
One of the most important reactions occurring withlysine side chains is the reaction with aldehydes toform a Schiff base (see Figure 2.30). The reaction isimportant within the cell because pyridoxal phosphate,a co-factor derived from vitamin B6, reacts with the ε
amino group of lysine and is found in many enzyme
N
CH3HO
C
H2C
P
O
CH2 NH24
N
CH3HO
C
H2C
P
CH2 N
H
+
Schiff base
Lysine
Pyridoxal phosphate
4
H
Figure 2.30 Reaction between pyridoxal phosphateand lysine side chains results in formation of acovalent Schiff base intermediate. The aldehyde groupof pyridoxal phosphate links with the ε amino groupof a specific lysine residue at the active site ofmany enzymes
active sites. Pyridoxal phosphate is related to vitaminB6, pyridoxine.
Proline
The side chain of proline is unique in possessing a sidechain that covalently bonds with the backbone nitrogenatom to form a cyclic pyrrolidine ring with groupslacking reactivity. One of the few reactions involvingprolyl side chains is enzyme-catalysed hydroxylation.The cyclic ring imposes rigid constraints on the N-Cα
bond leading to pronounced effects on the configurationof peptide bonds preceding proline. In addition thering is puckered with the Cγ atom displaced by 0.5 Afrom the remaining atoms of the ring which showapproximate co-planarity.
Histidine
The imidazole side chain of histidine (Figure 2.31) isunusual and alone amongst the side chains of aminoacids in exhibiting a pK around 7.0. In its ionized

30 AMINO ACIDS: THE BUILDING BLOCKS OF PROTEINS
Figure 2.31 The side chain of histidine showingnomenclature for side chain atoms. The IUPAC schemeencourages the use of pros (meaning ‘near’ andabbreviated with the symbol π) and tele (meaning‘far’, abbreviated with the symbol τ) to show theirposition relative to the main chain
state the side chain has a positive charge whilst inthe unionized state the side chain remains neutral.However, protonation not only modulates the chargeand acid–base behaviour of the side chain but alsoalters nucleophilic and electrophilic properties of thering (Figure 2.32).
The protonated nitrogen, shown in red in Figure 2.32,is called the NE2 or ε2. Experimental evidence suggeststhat the hydrogen atom is usually located on the NE2nitrogen but upon further protonation the structure onthe right is formed with the ND1 nitrogen now bindinga proton. The charge cannot be accurately assigned toone of the nitrogen atoms and resonance structures exist(Figure 2.33).
+ H+
CH2
N
C
CH
NH
CH2
HN+
C
CH
NH
HH
Figure 2.32 Protonation of the imidazole side chainleads to a positively charge
CH2
HN
C
CH
NH
CH2
HN
C
CH
NH++
H H
Figure 2.33 Delocalization of charge in protonatedimidazole side chains of histidine is possible
The unprotonated nitrogen of the uncharged imida-zole ring is a potent nucleophile and has a capacity forhydrogen bonding.
The aromatic residues: phenylalanine,tyrosine and tryptophan
The aromatic side chains have a common propertyof absorbing in the ultraviolet region of the electro-magnetic spectrum. Table 2.3 shows the spectroscopicproperties of Phe, Tyr and Trp. As a result Phe, Tyrand Trp are responsible for the absorbance and flu-orescence of proteins frequently measured between
Table 2.3 Spectroscopic properties of the aromatic amino acids
Amino acid Absorbance Fluorescence
λmax (nm) ε (M−1 cm−1) λmax (nm) Quantum yield
Phenylalanine 257.4 197 282 0.04Tyrosine 274.6 1420 303 0.21Tryptophan 279.8 5600 348 0.20
λ, wavelength; ε, molar absorptivity coefficient.

THE CHEMICAL AND PHYSICAL PROPERTIES OF AMINO ACIDS 31
250 and 350 nm. In this region the molar extinctioncoefficients of Phe, Tyr and Trp, an indication of howmuch light is absorbed at a given wavelength, are notequal. Tryptophan exhibits a molar extinction coeffi-cient approximately four times that of tyrosine and over28 times greater than phenylalanine. Almost all spec-trophotometric measurements of a protein’s absorbanceat 280 nm reflect the intrinsic Trp content of that pro-tein. At equivalent molar concentrations proteins witha high number of tryptophan residues will give a muchlarger absorbance at 280 nm when compared with pro-teins possessing a lower Trp content.
Phenylalanine
The aromatic ring of phenylalanine is chemically inertand has generally proved resistant to chemical modi-fication (Figure 2.34). Only with genetic modificationof proteins have Phe residues become routinely altered.The numbering scheme of atoms around the aromaticbenzene ring is shown for carbon and hydrogens. Alter-native nomenclatures exist for the ring proteins withlabels of HD, HE or HZ (Hδ, Hε or Hζ in some schemes)being the most common. In many proteins rotation ofthe aromatic ring around the CB–CG axis leads to theprotons at the 2,6 positions (HD) and the 3,5 positions(HE) being indistinguishable.
Although the aromatic ring is inert chemically itremains hydrophobic and prefers interactions in pro-teins with other non-polar residues. With other aromatic
CZ/HZ
CE/HE
CD/HD
CB/HB
CG
Figure 2.34 The aromatic side chain of phenylalanine
rings Phe forms important π–π interactions derivedfrom the delocalized array of electrons that arises frombonding between the ring carbon atoms. In a simplealkene such as ethene each carbon links to two hydro-gens and a single carbon. This is achieved by sp2
hybridization where the normal ground state of car-bon 1s22s22px
12py1 is transformed by elevating one
of the 2s electrons into the vacant pz orbital yield-ing 1s22s12px
12py12pz
1. The sp2 hybrids, made fromrearrangement of the 2s orbital and two of the three2p orbitals, are orientated at an angle of 120◦ to eachother within a plane. The remaining 2pz orbital is atright angles to these hybrids. Formation of three bondsleaves the pz orbital vacant and a similar situation existsin benzene or the aromatic ring of phenylalanine; eachsp2 hybridized carbon is bonded to two carbon atomsand a single hydrogen and has a single unpaired elec-tron. This electron lies in the vacant pz orbital proximalto and overlapping with another carbon atom possess-ing an identical configuration. The extensive overlapproduces a system of π bonds where the electrons arenot confined between two carbon atoms but delocal-ize around the entire ring. Six delocalized electronsgo into three π molecular orbitals – two in each. Theinteraction of these π molecular orbitals by the closeapproach of two aromatic rings leads to further delocal-ization. The coplanar orientation is the basis for π–π
interactions and may account for the observation thataromatic side chains are found in proteins in closeproximity to other aromatic groups. π–π interactionsare expected to make favourable and significant con-tributions to the overall protein stability. Studies withmodel compounds suggest that an optimal geometryexists with the aromatic rings perpendicular to eachother and leading to positively charged hydrogens onthe edge of one ring interacting favourably with theπ electrons and partially negatively charged carbonsof the other. For proteins, as opposed to model com-pounds, this type of interaction is less common than aco-planar orientation of aromatic rings.
Tyrosine
Tyrosine is more reactive than phenylalanine due tothe hydroxyl group (Figure 2.35) substituted at thefourth (CZ) aromatic carbon, often called the para

32 AMINO ACIDS: THE BUILDING BLOCKS OF PROTEINS
Figure 2.35 The side chain of tyrosine contains areactive hydroxyl group
position. The hydroxyl group leaves the aromatic ringsusceptible to substitution reactions.
Nucleophiles such as nitrating agents or activatedforms of iodide react with tyrosine side chains inproteins and change the acid–base properties of thering. Reaction with nitrating agents yields mononitrotyrosine derivatives at the 3,5 position followedby dinitro derivatives. Normally the hydroxyl group isa weak acid with a pK ∼ 11.0 but the addition of oneor more nitro groups causes the pK to drop by up to4 pH units. The tyrosine interacts with other aromaticrings but the presence of the OH group also allowshydrogen bonding.
Tryptophan
The indole side chain (Figure 2.36) is the largest sidechain occurring in proteins and is responsible for mostof the intrinsic absorbance and fluorescence. As a crudeapproximation the molar extinction coefficient of a pro-tein at 280 nm may be estimated by adding up thenumber of Trp residues found in the sequence andmultiplying by 5800 (see Table 2.3). However, the rel-atively low frequency of Trp residues in proteins meansthat this approach is not always accurate and in somecases (for proteins lacking Trp) will be impossible.
The side chain is hydrophobic and does not undergoextensive chemical reactions. Exceptions to the rule ofunreactivity include modifying reagents such as iodine,N -bromosuccinimide and ozone although their use inchemical modification studies of proteins is limited.Universally the weakest or most sensitive part of theindole ring is the pyrrole nitrogen atom.
CH2
NH
H
HH
H
H
CB/HB
C2/H2
C4/H4
C7/H7
C5/H5
C6/H6
C3C3a
C7a
Figure 2.36 The indole ring of tryptophan. Thenomenclature of the ring protons names the ringprotons as H2, H4, H5, H6 and H7. The carbons withprotons attached are similarly named i.e C2, C3 . . . C7.The carbon centres lacking protons are C3, C3a andC7a. The nitrogen is properly termed the N1 centrebut is frequently called the indole, or NE1, nitrogen.In addition Greek symbols are often applied to thesecarbon and protons with the following symbols usedCξ2 = C7, Cη2 = C6, Cξ3 = C5, Cη3 = C4
Detection, identification andquantification of amino acidsand proteinsTo quantify protein isolation requires a means ofestimating protein concentration and most methodsuse the properties of amino acid side chains. Proteinsabsorb around 280 nm principally because of therelative contributions of tryptophan, phenylalanine andtyrosine. If their occurrence in proteins is knownthen, in theory at least, the concentration of a proteinsolution is calculated using the data of Table 2.4 andBeer–Lamberts law where
A280 = ε280c l (2.4)
(where A is absorbance, ε is molar absorptivitycoefficient, c is the concentration in moles dm−3 andl is the light path length normally 1 cm). In practicethis turns out to be a very crude method of measuringprotein concentration and of limited accuracy. Thenumber of Trp, Tyr or Phe residues is not alwaysknown and more importantly the absorbance of proteinsin their native state does not usually equate with asimple summation of their individual contributions.Moreover the presence of any additional componentsabsorbing at 280 nm will lead to inaccuracies andthis could include the presence of impurities, disulfide

DETECTION, IDENTIFICATION AND QUANTIFICATION OF AMINO ACIDS AND PROTEINS 33
bridges, or additional co-factors such as heme or flavin.Performing spectrophotometric measurements underdenaturing conditions allows improved estimation ofprotein concentration from the sum of the Trp, Tyr andPhe components. The contribution of disulfide bridges,although weak around 280 nm, can be eliminated bythe addition of reducing agents, and under denaturingconditions co-factors are frequently lost.
An alternative method of determining protein con-centration exploits the reaction between the thiol groupof cysteine and Ellman’s reagent. The reaction pro-duces the nitrothiobenzoate anion and since the molarabsorptivity coefficient for this product at 410 nm isaccurately known (13 600 M−1 cm−1) the reagent offersone route of determining protein concentration if thenumber of thiol groups is known beforehand. In gen-eral, protein concentration may be determined by anyreaction with side chains that lead to coloured prod-ucts that can be quantified by independent methods.Other reagents with applications in estimating protein,peptide or amino acid concentration include ninhy-drin, fluorescamine, dansyl chloride, nitrophenols andfluorodinitrobenzene. They have the common theme ofpossessing a reactive group towards certain side chainscombined with a strong chromophore. A commonweakness is that all rely on reactions with a restrictednumber of side chains to provide estimates of total pro-tein concentration. A better approach is to devise assaysbased on the contribution of all amino acids.
Historically the method of choice has been the biuretreaction (Figure 2.37) where a solution of copper(II)sulfate in alkaline tartrate solution reacts with peptidebonds to form a coloured (purple) complex absorbingaround 540 nm. The structure of the complex formedis unclear but involves the coordination of copper ions
CHR
CO
HN
HN
NH
R
NH
CO
HCCu2+
Figure 2.37 The complexation of cupric ions bypeptide bonds in the biuret method
by the amide hydrogen of peptide bonds. An importantaspect of this assay is that it is based around theproperties of the polypeptide backbone. Reduction ofCu(II) to Cu(I) is accompanied by a colour changeand if the absorbance of the unknown protein iscompared with a calibration curve then concentrationis accurately determined. Ideally the standard proteinused to construct a calibration curve will have similarproperties, although frequently bovine serum albumin(BSA) is used.
Another commonly used method of estimatingprotein concentration is the Folin–Lowry method.The colour reaction is enhanced by the additionof Folin–Ciocalteau’s reagent to a protein solu-tion containing copper ions. The active ingredientof Folin–Ciocalteau’s reagent is a complex mixtureof phosphotungstic and phosphomolybdic acids. Thereduced Cu(I) generated in the biuret reaction formsa number of reduced acid species in solution eachabsorbing at a wavelength maximum between 720 and750 nm and leading to an obvious blue colour. Estima-tion of protein concentration arises by comparing theintensity of this blue colour to a ‘standard’ protein ofknown concentration at wavelengths between 650 and750 nm.
Coomassie Brilliant Blue (Figure 2.38) does notundergo chemical reactions with proteins but forms
CH2
NC2H5
SO3 H2C
NC2H5
HN
R R
SO3
OC2H5
− −
Figure 2.38 Coomassie blue dyes are commonlyused to estimate protein concentration (R250 = H;G250 = CH3)

34 AMINO ACIDS: THE BUILDING BLOCKS OF PROTEINS
stable complexes via non-covalent interactions.Coomassie blue (G250) is used to estimate the amountof protein in solution because complex formation isaccompanied by a shift in absorbance maxima from465 to 595 nm. The mechanism of complex forma-tion between dye and protein is unclear but from thestructure of the dye involves non-polar interactions.
StereoisomerismUp to this point the α carbon of amino acids has beendescribed as an asymmetric centre and little attentionhas been paid to this characteristic property of tetrahe-dral carbon centres. One of the most important conse-quences of the asymmetric α carbon is that it gives riseto a chiral centre and the presence of two isomers. Thetwo potential arrangements of atoms about the centralcarbon are shown for alanine (Figure 2.39) and whilstsuperficially appearing identical a closer inspection ofthe arrangement of atoms and the relative positionsof the amino, carboxyl, hydrogen and methyl groupsreveals that the two molecules can never be exactlysuperimposed.
The molecules are mirror images of each other orstereoisomers. Most textbooks state that these isomersare termed the L and D isomers without resorting tofurther explanation. All naturally occurring amino acids
Figure 2.39 Two stereoisomers of alanine. As viewedthe C–H bond of the α-carbon is pointing away fromthe viewer (down into the page) and superpositionof the remaining groups attached this chiral centreis impossible. Each molecule is a mirror image ofthe other known variously as enantiomers, opticalisomers or stereoisomers.
Table 2.4 The specific optical rotation of selectedamino acids
L-Amino
Acid
[α]D(H2O) L-Amino
Acid
[α]D(H2O)
Alanine +1.8 Isoleucine +12.4
Arginine +12.5 Leucine −11.0
Cysteine −16.5 Phenylalanine −34.5
Glutamic Acid +12.0 Threonine −28.5
Histidine −38.5 Tryptophan −33.7
found in proteins belong to the L absolute configuration.However, at the same time the asymmetric or chiralcentre is also described as an optically active centre.An optically active centre is one that rotates plane-polarized light. Amino acids derived from the hydrol-ysis of proteins under mild conditions will, with theexception of glycine, rotate plane-polarized light in asingle direction. If this direction is to the right theamino acids are termed dextrorotatory (+) whilst rota-tion to the left is laevorotatory (−). Values for isolatedamino acids are known, with L-amino acids rotatingplane-polarized light in different directions and to dif-fering extents (Table 2.4).
The terms dextro- and levorotatary must not beconfused with the L and D isomers, which for historicalreasons are related to the arrangement of atoms in L-and D-glyceraldehyde. L-Trp is laevorotatory whilst L-Glu is dextrorotatory. The D and L stereoisomers ofany amino acid have identical properties with twoexceptions: they rotate plane-polarized light in oppositedirections and they exhibit different reactivity withasymmetric reagents. This latter point is important inprotein synthesis where D amino acids are effectiveinhibitors and in enzymes where asymmetric activesites discriminate effectively between stereoisomers.
The Cahn–Ingold–Prelog scheme has been devisedto avoid confusion and provides an unambiguousmethod of assigning absolute configurations particularlyfor molecules containing more than one asymmetriccentre. The amino acids isoleucine and threonine eachcontain two asymmetric centres, four stereoisomers,and potential confusion in naming isomers using theDL system. Using threonine as an example we can see

NON-STANDARD AMINO ACIDS 35
that the α carbon and the β carbon are each attachedto four different substituents. For the β carbon theseare the hydroxyl group, the methyl group, the hydrogenatom and the α carbon. The RS system is based onranking the substituents attached to each chiral centreaccording to atomic number. The smallest or lowestranked group is arranged to point away from the viewer.This is always the C–H bond for the Cα carbon and theremaining three groups are then viewed in the directionof decreasing priority. For biomolecules the functionalgroups commonly found in proteins are ranked in orderof decreasing priority:
SH > P > OR > OH > NH3 > COO > CHO
> CH2OH > C6H5 > CH3 > H
The priority is established initially on the basis ofatomic number so H (atomic no. = 1) has the lowestpriority whilst groups containing sulfur (atomic no. =16) are amongst the highest. To identify RS isomersone first identifies the asymmetric or stereogenic centreand in proteins this involves an sp3 carbon centrewith a tetrahedral arrangement containing four differentgroups. With the C–H bond (lowest priority) pointingaway from the viewer the three remaining groupsare ranked according to the scheme shown above. Inthe case where two carbon atoms are attached to anasymmetric centre then one moves out to the nextatom and applies the same selection rule at this point.This leads to CH3 being of lower priority than C2H5
which in turn is lower than COO−. Where two isotopesare present and this is most commonly deuterium andhydrogen the atomic number rule is applied leadingto D having a greater priority than H. Having rankedthe groups we now assess if the priority (from highto low) is in a clockwise or anticlockwise direction.A clockwise direction leads to the R isomer (Latin:rectus = right) whilst an anticlockwise direction leadsto the S isomer (Latin: sinister = left).
In L-threonine at the first asymmetric centre(Cα) when looking down the Cα–H bond (pointingaway from the viewer in the arrangement shownin Figure 2.40) the direction of decreasing order ofpriority (NH3
+, COO− and CH–OH–CH3) is counterclockwise. The Cα, the second carbon, is therefore con-figuration S. A similar line of reasoning for the asym-metric centre located at the third carbon (CG) yields
Figure 2.40 The asymmetric Cα centre of Thr is inthe S configuration. The Cα is shown on green whilstthe other atoms have their normal CPK colours
Figure 2.41 The asymmetric CG centre of threonine isin the R configuration. The CG atom is shown in greenand the C–H bond is pointing away from the viewer
a decreasing order of priority in a clockwise directionand hence the R configuration. Combining these twoschemes yields for L-threonine the nomenclature [2S-3R] threonine (Figure 2.41).
Non-standard amino acids
The 20 amino acids are the building blocks of pro-teins but are also precursors for further reactions thatproduce additional amino acids. Amino acids withunusual stereochemistry about the Cα carbon oftencalled D amino acids are comparatively common inmicro-organisms. Prominent examples are D-alanineand D-isoglutamate in the cell wall of the Gram-positivebacterium Staphylococcus aureus. In other bacteria

36 AMINO ACIDS: THE BUILDING BLOCKS OF PROTEINS
NH3+
C
CO
C
O−
H
N
HH
H C
CO
O
O−
NH3+
CH
CO
CH2CH2ONHC
NH
H2N
O−
Figure 2.42 Canavanine and β-N-oxalyl L-α, β-diamino propionic acid (ODAP)
small peptide molecules known as ionophores formchannels in membranes through the use of proteins con-taining D amino acid residues. A well-known exampleis the 15 residue peptide Gramicidin A containing aseries of alternating D and L amino acids. In each casethe use of these unusual or non-standard amino acidsmay be viewed as a defensive mechanism. Plants andmicroorganism use non-standard amino acids as pro-tective weapons. Canavanine, a homologue of argininecontaining an oxygen instead of a methylene group atthe δ position, accumulates as a storage protein in alfafaseeds where it acts as a natural defence against insectpredators (Figure 2.42).
A serious occurrence of an unusual amino acid is thepresence of β-N -oxalyl L-α, β-diamino propionic acid(ODAP) in the legume Lathyrus sativus. Ingestion ofseeds containing this amino acid is harmless to humansin small quantities but increased consumption leads to
a neurological disorder (lathyrism) resulting in irre-versible paralysis of leg muscles. ODAP is the culpritand acts by mimicking glutamate in biological systems.
Amino acids undergo a wide range of metabolicconversions as part of the cell’s normal syntheticand degradative pathways. However, in a few casesamino acids are converted to specific products. Insteadof undergoing further metabolism these modifiedamino acids are used to elicit biological response.The most important examples of the conversion ofamino acids into other modified molecules includehistamine, dopamine, thyroxine and γ-amino butyricacid (GABA) (Figure 2.43). GABA is formed from theamino acid glutamate as a result of decarboxylationand is an important neurotransmitter; thyroxine is aniodine-containing hormone stimulating basal metabolicrate via increases in carbohydrate metabolism invertebrates, whilst in amphibians it plays a role inmetamorphosis; histamine is a mediator of allergicresponse reactions and is produced by mast cells aspart of the body’s normal response to allergens.
Summary
Twenty different amino acids act as the building blocksof proteins. Amino acids are found as dipolar ionsin solutions. The charged properties result from thepresence of amino and carboxyl groups and lead tosolubility in water, an ability to act as electrolytes, acrystalline appearance and high melting points.
NH3+
CH2CH2
CH2C
O
O−
NH3+
CH2
CH2N
NH
H3N+
CH
CH2
HO
C
HO O
O−
H3N+
CH
CH2
I
CI
O O
O−
I
I
O
Figure 2.43 Biologically active amino acid derivatives. Clockwise from top left are GABA, dopamine, histamineand thyroxine

PROBLEMS 37
Of the 20 amino acids found in proteins 19 have acommon structure based around a central carbon, theCα carbon, in which the amino, carboxyl, hydrogenand R group are arranged tetrahedrally. An exceptionto this arrangement of atoms is proline. The Cα carbonis asymmetric with the exception of glycine and leadsto at least two stereoisomeric forms.
Amino acids form peptide bonds via a condensationreaction and the elimination of water in a processthat normally occurs on the ribosomes found in cells.The formation of one peptide bond covalently linkstwo amino acids forming a dipeptide. Polypeptides orproteins are built up by the repetitive formation ofpeptide bonds and an average sized protein may contain1000 peptide bonds.
The peptide bond possesses hybrid characteristicswith properties between that of a C–N single bondand those of a C=N double bond. These propertiesresult in decreased peptide bond lengths compared to aC–N single bond, a lack of rotation about the peptidebond and a preferred orientation of atoms in a transconfiguration for most peptide bonds. One exception tothis rule is the peptide bond preceding proline residueswhere the cis configuration is increased in stabilityrelative to the trans configuration.
The side chains dictate the chemical and phys-ical properties of proteins. Side chain propertiesinclude charge, hydrophobicity and polarity and under-pin many aspects of the structure and functionof proteins.
Problems1. Why are amino acids white crystalline solids and
how does this account for their physicochemicalproperties?
2. Using Figures 2.13 and 2.14 identify methine, me-thene and methyl groups in the side chains ofleucine, isoleucine and valine. Label each carbonatom according to the usual nomenclature. Repeat theexercise of nomenclature for Figure 2.18.
3. Translate the following sequence into a sequencebased on the single letter codes.Ala-Phe-Phe-Lys-Arg-Ser-Ser-Ser-Ala-Thr-Leu-Ile-Val-Thr-Lys-Lys-Gln-Gln-Phe-Asn-Gly-Gly-Pro-Asp-Glu-Val-Leu-Arg-Thr-Ala-Ser-Thr-Lys-Ala-Thr-Asp.
4. What is the average mass of an amino acid residue?Why is such information useful yet at the sametime limited?
5. Which of the following peptides might be expected tobe positively charged, which are negatively chargedand which carry no net charge?Ala-Phe-Phe-Lys-Arg-Ser-Ser-Ser-Ala-Thr-Leu-Ile-Val-Ala-Phe-Phe-Lys-Arg-Ser-Glu-Asp-Ala-Thr-Leu-Ile-Val-Ala-Phe-Phe-Lys-Asp-Ser-Ser-Asp-Ala-Thr-Leu-Ile-Val-Ala-Phe-Phe-Lys-Asp-Ser-Ser-His-Ala-Thr-Leu-Ile-Val-
Ala-Phe-Phe-Lys-Asp-Ser-Glu-His-Ala-Thr-Leu-Ile-Val-Are there any contentious issues?
6. The reagent 1-fluoro 2,4-dinitrobenzene has beenused to identify amino acids. Describe the groupsand residues you would expect this reagent to reactwith preferentially. Under what conditions would youperform the reactions.
7. Histidine has three ionizable functional groups. Drawthe structures of the major ionized forms of histidineat pH 1.0, 5.0, 8.0 and 13.0. What are the respectivecharges at each pH?
8. The pK1 and pK2 values for alanine are 2.34 and9.69. In the dipeptide Ala-Ala these values are 3.12and 8.30 whilst the tripeptide Ala-Ala-Ala has valuesof 3.39 and 8.00. Explain the trends for the values ofpK1 and pK2 in each peptide.
9. A peptide contains the following aromatic residues3 Trp, 6 Tyr and 1 Phe and gives an absorbanceat 280 nm of 0.8. Having added 1 ml of a solutionof this peptide to a cuvette of path length 1 cmlight calculate from the data provided in Table 2.4the approximate concentration of the peptide (youcan assume the differences in ε between 280 nmand those reported in Table 2.4 are negligible). Themolecular mass of the peptide is 2670 what was theconcentration in mg ml−1.

38 AMINO ACIDS: THE BUILDING BLOCKS OF PROTEINS
10. Citrulline is an amino acid first isolated fromthe watermelon (Citrullus vulgaris). Determine theabsolute (RS) configuration of citrulline for anyasymmetric centre. Isoleucine has two asymmetriccentres. Identify these centres and determine theconfiguration at each centre. How many potentialoptical isomers exist for isoleucine?

3The three-dimensional structure
of proteins
Amino acids linked together in a flat ‘two-dimensional’representation of the polypeptide chain fail to conveythe beautiful three-dimensional arrangement of pro-teins. It is the formation of regular secondary structureinto complicated patterns of protein folding that ulti-mately leads to the characteristic functional propertiesof proteins.
Primary structure or sequenceThe primary structure is the linear order of amino acidresidues along the polypeptide chain. It arises fromcovalent linkage of individual amino acids via peptidebonds. Thus, asking the question ‘What is the primarystructure of a protein?’ is simply another way of asking‘What is the amino acid sequence from the N to Cterminals?’ To read the primary sequence we simplytranslate the three or single letter codes from left toright, from amino to carboxyl terminals. Thus in thesequence below two alternative representations of thesame part of the polypeptide chain are given, startingwith alanine at residue 1, glutamate at position 2 andextending to threonine as the 12th residue (Figure 3.1).
Every protein is defined by a unique sequenceof residues and all subsequent levels of organization(secondary, super secondary, tertiary and quaternary)rely on this primary level of structure. Some proteins
are related to one another leading to varying degreesof similarity in primary sequences. So myoglobin,an oxygen storage protein found in a wide range oforganisms, shows similarities in human and whalesin the 153 residue sequence (Figure 3.2). Most ofthe sequence is identical and it is easier to spot thedifferences. When a change occurs in the primarysequence it frequently involves two closely relatedresidues. For example, at position 118 the humanvariant has a lysine residue whilst whale myoglobinhas an arginine residue. Reference to Table 2.2 willshow that arginine and lysine are amino acids thatcontain a positively charged side chain and this changeis called a conservative transition. In contrast in a fewpositions there are very different amino acid residues.Consider position 145 where asparagine (N) is replacedby lysine (K). This transition is not conservative; thesmall, polar, side chain of asparagine is replaced by thelarger, charged, lysine. Regions, or residues, that neverchange are called invariant.
Secondary structurePrimary structure leads to secondary structure; thelocal conformation of the polypeptide chain or thespatial relationship of amino acid residues that are closetogether in the primary sequence. In globular proteins
Proteins: Structure and Function by David Whitford 2005 John Wiley & Sons, Ltd

40 THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
NH3-Ala-Glu-Glu-Ser-Ser-Lys-Ala-Val-Lys-Tyr-Tyr-Thr-………
NH3---A---E---E---S----S----K----A---V---K---Y----Y----T……..
Figure 3.1 Single- and three-letter codes for aminoterminal of a primary sequence
the three basic units of secondary structure are theα helix, the β strand and turns. All other structuresrepresent variations on one of these basic themes.This chapter will focus on the chemical and physicalproperties of polypeptides that permit the transitionfrom randomly oriented polymers of amino acidresidues to regular repeating secondary structure. With20 different amino acid residues found in proteins thereare 780 possible permutations for a dipeptide. In anaverage size polypeptide of ∼100 residues the numberof potential sequences is astronomical. In view ofthe enormous number of possible conformations manyfundamental studies of protein secondary structure havebeen performed on homopolymers of amino acids forexample poly-alanine, poly-glutamate, poly-proline orpoly-lysine.
Homopolymers have the advantage that either allthe residues are identical or in some cases simplerepeating units such as poly Ala-Gly, where Ala-Gly dipeptides are repeated along the length of thepolymer. In comparison with polypeptides derivedfrom proteins these polymers have the advantage of
consistent conformations. This is not to imply that theconformation of homopolymers is regular and ordered.In some instances these polymers are unstructured,however, the common theme is that the polymersare uniform in their conformations and hence aremuch more attractive candidates for initial studiesof structure.
Studies of homopolymers have been advanced byhost–guest studies where a polymer of alanine residuesis modified by an introduction of a single differentresidue in the middle of the polypeptide. By measuringchanges in stability, solubility, or helical propertiesthese studies allow the effect of the new or guestresidue to be accurately defined. Indeed, much ofthe data reported in tables throughout this book wereobtained by the introduction of a single amino acidresidue into a polymer containing just one type ofresidue. The host peptide is usually designed to bemonomeric (i.e. non-aggregating), to be soluble andnot more than 15 residues in length. By systematicallyreplacing residue 8 with any of the other 19 aminoacids these studies have elucidated many of theproperties of residues within polypeptide chains. Oneproperty that has been extensively studied using thisapproach is the relative helical tendency of amino acidresidues in a peptide of poly-alanine. Results suggest,unsurprisingly, that alanine is the most stable residueto substitute into a poly-alanine peptide whilst prolineis the most destabilizing. More revealing is the relative
* 20 * 40Human Mb : GLSDGEWQLVLNVWGKVEADIPGHGQEVLIRLFKGHPETL : 40Whale Mb : VLSEGEWQLVLHVWAKVEADVAGHGQDILIRLFKSHPETL : 40
* 60 * 80Human Mb : EKFDKFKHLKSEDEMKASEDLKKHGATVLTALGGILKKKG : 80Whale Mb : EKFDRFKHLKTEAEMKASEDLKKHGVTVLTALGAILKKKG : 80
* 100 * 120Human Mb : HHEAEIKPLAQSHATKHKIPVKYLEFISECIIQVLQSKHP : 120Whale Mb : HHEAELKPLAQSHATKHKIPIKYLEFISEAIIHVLHSRHP : 120
* 140 *Human Mb : GDFGADAQGAMNKALELFRKDMASNYKELGFQG : 153Whale Mb : GDFGADAQGAMNKALELFRKDIAAKYKELGYQG : 153
Figure 3.2 The primary sequences of human and sperm whale myoglobin. Identical residues are shown in blue,the regions in yellow show conserved substitutions whilst the red regions show non-conservative changes. Astericksindicate every tenth residue. Single letter codes for residues are used
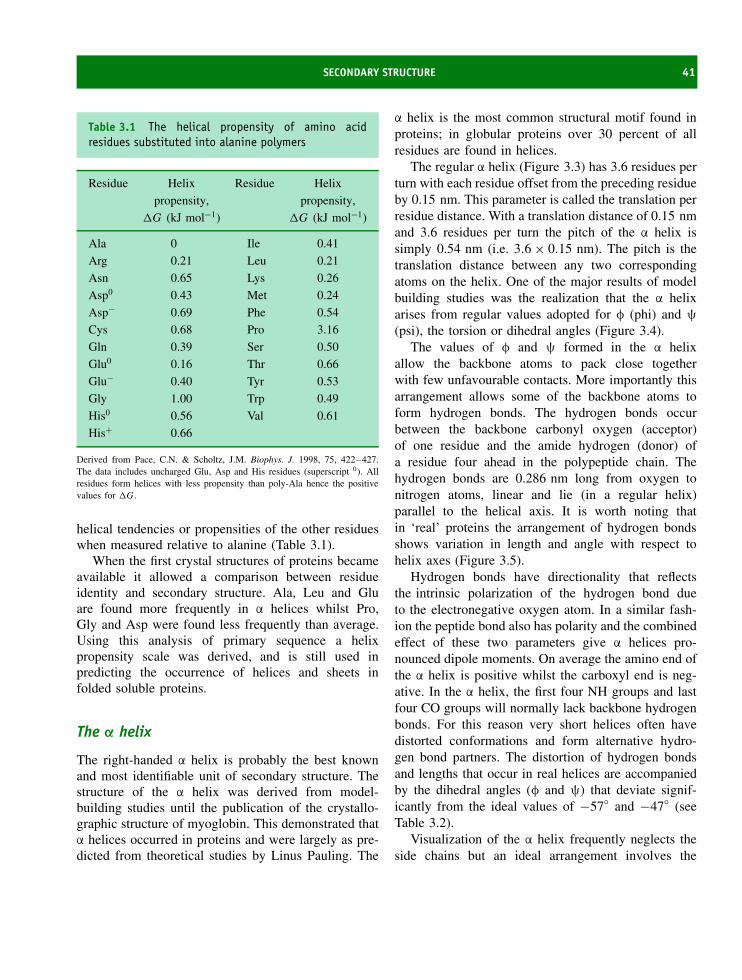
SECONDARY STRUCTURE 41
Table 3.1 The helical propensity of amino acidresidues substituted into alanine polymers
Residue Helix
propensity,
�G (kJ mol−1)
Residue Helix
propensity,
�G (kJ mol−1)
Ala 0 Ile 0.41
Arg 0.21 Leu 0.21
Asn 0.65 Lys 0.26
Asp0 0.43 Met 0.24
Asp− 0.69 Phe 0.54
Cys 0.68 Pro 3.16
Gln 0.39 Ser 0.50
Glu0 0.16 Thr 0.66
Glu− 0.40 Tyr 0.53
Gly 1.00 Trp 0.49
His0 0.56 Val 0.61
His+ 0.66
Derived from Pace, C.N. & Scholtz, J.M. Biophys. J. 1998, 75, 422–427.The data includes uncharged Glu, Asp and His residues (superscript 0). Allresidues form helices with less propensity than poly-Ala hence the positivevalues for �G .
helical tendencies or propensities of the other residueswhen measured relative to alanine (Table 3.1).
When the first crystal structures of proteins becameavailable it allowed a comparison between residueidentity and secondary structure. Ala, Leu and Gluare found more frequently in α helices whilst Pro,Gly and Asp were found less frequently than average.Using this analysis of primary sequence a helixpropensity scale was derived, and is still used inpredicting the occurrence of helices and sheets infolded soluble proteins.
The α helix
The right-handed α helix is probably the best knownand most identifiable unit of secondary structure. Thestructure of the α helix was derived from model-building studies until the publication of the crystallo-graphic structure of myoglobin. This demonstrated thatα helices occurred in proteins and were largely as pre-dicted from theoretical studies by Linus Pauling. The
α helix is the most common structural motif found inproteins; in globular proteins over 30 percent of allresidues are found in helices.
The regular α helix (Figure 3.3) has 3.6 residues perturn with each residue offset from the preceding residueby 0.15 nm. This parameter is called the translation perresidue distance. With a translation distance of 0.15 nmand 3.6 residues per turn the pitch of the α helix issimply 0.54 nm (i.e. 3.6 × 0.15 nm). The pitch is thetranslation distance between any two correspondingatoms on the helix. One of the major results of modelbuilding studies was the realization that the α helixarises from regular values adopted for φ (phi) and ψ
(psi), the torsion or dihedral angles (Figure 3.4).The values of φ and ψ formed in the α helix
allow the backbone atoms to pack close togetherwith few unfavourable contacts. More importantly thisarrangement allows some of the backbone atoms toform hydrogen bonds. The hydrogen bonds occurbetween the backbone carbonyl oxygen (acceptor)of one residue and the amide hydrogen (donor) ofa residue four ahead in the polypeptide chain. Thehydrogen bonds are 0.286 nm long from oxygen tonitrogen atoms, linear and lie (in a regular helix)parallel to the helical axis. It is worth noting thatin ‘real’ proteins the arrangement of hydrogen bondsshows variation in length and angle with respect tohelix axes (Figure 3.5).
Hydrogen bonds have directionality that reflectsthe intrinsic polarization of the hydrogen bond dueto the electronegative oxygen atom. In a similar fash-ion the peptide bond also has polarity and the combinedeffect of these two parameters give α helices pro-nounced dipole moments. On average the amino end ofthe α helix is positive whilst the carboxyl end is neg-ative. In the α helix, the first four NH groups and lastfour CO groups will normally lack backbone hydrogenbonds. For this reason very short helices often havedistorted conformations and form alternative hydro-gen bond partners. The distortion of hydrogen bondsand lengths that occur in real helices are accompaniedby the dihedral angles (φ and ψ) that deviate signif-icantly from the ideal values of −57◦ and −47◦ (seeTable 3.2).
Visualization of the α helix frequently neglects theside chains but an ideal arrangement involves the

42 THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Pitch ~ 0.54 nm
Pitch ~ 0.54 nm
C terminal direction
N terminal direction
Ca
Ca
Ca
Figure 3.3 A regular α helix. Only heavy atoms (C, N and O, but not hydrogen) are shown and the side chains areomitted for clarity
N
N
O
O
y
f
Ca
Ca
C′
C′
Figure 3.4 φ is defined by the angle between theC′–N–Cα –C′ atoms whilst ψ is defined by the atomsN–Cα –C′–N
Figure 3.5 Arrangement of backbone hydrogenbonds in a real α helix from myoglobin, showsdeviations from ideal geometry
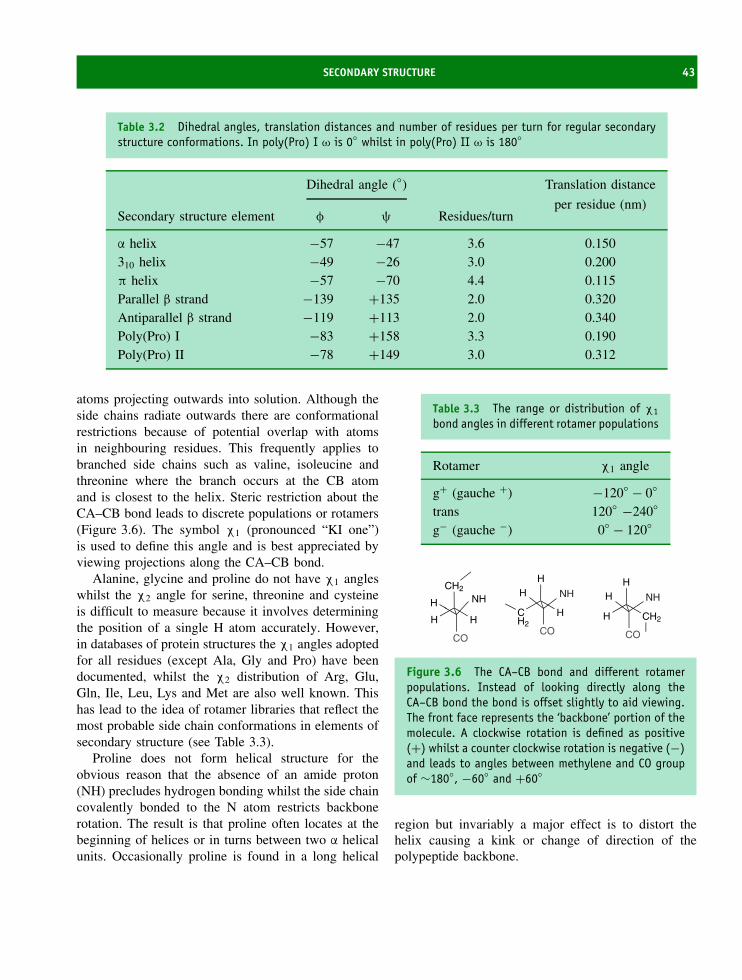
SECONDARY STRUCTURE 43
Table 3.2 Dihedral angles, translation distances and number of residues per turn for regular secondarystructure conformations. In poly(Pro) I ω is 0◦ whilst in poly(Pro) II ω is 180◦
Dihedral angle (◦) Translation distance
per residue (nm)Secondary structure element φ ψ Residues/turn
α helix −57 −47 3.6 0.150310 helix −49 −26 3.0 0.200π helix −57 −70 4.4 0.115Parallel β strand −139 +135 2.0 0.320Antiparallel β strand −119 +113 2.0 0.340Poly(Pro) I −83 +158 3.3 0.190Poly(Pro) II −78 +149 3.0 0.312
atoms projecting outwards into solution. Although theside chains radiate outwards there are conformationalrestrictions because of potential overlap with atomsin neighbouring residues. This frequently applies tobranched side chains such as valine, isoleucine andthreonine where the branch occurs at the CB atomand is closest to the helix. Steric restriction about theCA–CB bond leads to discrete populations or rotamers(Figure 3.6). The symbol χ1 (pronounced “KI one”)is used to define this angle and is best appreciated byviewing projections along the CA–CB bond.
Alanine, glycine and proline do not have χ1 angleswhilst the χ2 angle for serine, threonine and cysteineis difficult to measure because it involves determiningthe position of a single H atom accurately. However,in databases of protein structures the χ1 angles adoptedfor all residues (except Ala, Gly and Pro) have beendocumented, whilst the χ2 distribution of Arg, Glu,Gln, Ile, Leu, Lys and Met are also well known. Thishas lead to the idea of rotamer libraries that reflect themost probable side chain conformations in elements ofsecondary structure (see Table 3.3).
Proline does not form helical structure for theobvious reason that the absence of an amide proton(NH) precludes hydrogen bonding whilst the side chaincovalently bonded to the N atom restricts backbonerotation. The result is that proline often locates at thebeginning of helices or in turns between two α helicalunits. Occasionally proline is found in a long helical
Table 3.3 The range or distribution of χ1
bond angles in different rotamer populations
Rotamer χ1 angle
g+ (gauche +) −120◦ − 0◦
trans 120◦ −240◦
g− (gauche −) 0◦ − 120◦
CO
CH2
H H
NHHNHH
CO
H
CH2
H
NHH
CO
H
H CH2
Figure 3.6 The CA–CB bond and different rotamerpopulations. Instead of looking directly along theCA–CB bond the bond is offset slightly to aid viewing.The front face represents the ‘backbone’ portion of themolecule. A clockwise rotation is defined as positive(+) whilst a counter clockwise rotation is negative (−)and leads to angles between methylene and CO groupof ∼180◦, −60◦ and +60◦
region but invariably a major effect is to distort thehelix causing a kink or change of direction of thepolypeptide backbone.

44 THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Figure 3.7 Wireframe and space-filling representa-tions of one end of a regular α helix
The α helix is frequently portrayed in textbooksincluding this one with a ‘ball and stick’ or ‘wireframe’representation (Figure 3.7). A better picture is providedby ‘space-filling’ representations (Figure 3.7) whereatoms are shown with their van der Waals radii.In the ‘end-on’ view of the α helix the wireframerepresentation suggests a hollow α helix. In contrast,the space filling representation emphasizes that littlespace exists anywhere along the helix backbone.Other representations of helices include cylindersshowing the length and orientation of each helixor a ribbon representation that threads through thepolypeptide chain. The preceding section views thehelix as a stable structure but experiments withsynthetic poly-amino acids suggest that very fewpolymers fold into regular helical conformation.
Other helical conformations
The 310 helix is a structural variation of the α helixfound in proteins (Figure 3.8). The 310 helices areoften found in proteins when a regular α helix isdistorted by the presence of unfavourable residues,near a turn region or when short sequences fold intohelical conformation. In the 310 helix the dominanthydrogen bonds are formed between residues i, i + 3 incontrast to (i, i + 4) bonds seen in the regular α helix.The designation 310 refers to the number of backboneatoms located between the donor and acceptor atoms(10) and the fact that there are three residues per
4
3
2
1
Figure 3.8 A 310 helix shows many similarities toa regular α helix. Closer inspection of the hydrogenbond donor and acceptor reveals i, i + 3 connectivity,three residues per turn and dihedral angle values of∼−50 and −25 reflect a more tightly coiled helix (seeTable 3.2)
turn. With three residues per turn the 310 helix is atighter, narrower structure in which the potential forunfavourable contacts between backbone or side chainatoms is increased.
Whilst the 310 helix is a narrower structure thanthe α helix a third possibility is a more loosely coiledhelix with hydrogen bonds formed between the CO andNH groups separated by five residues (i, i + 5). Thisstructure is the π helix and at one stage it was thoughtnot to occur naturally. However, examples of this helixstructure have been described in proteins. Soyabeanlipoxygenase has a 43-residue helix containing regionsessential to enzyme function and stability runningthrough the centre of the molecule. Three turns of anexpanded helix with eight (i, i + 5) hydrogen bondsand 4.4 residues per turn make it an example of a π
helix containing more than one turn found in a protein(Figure 3.9).
The rarity of this form of secondary structure arisesfor a number of reasons. One major limitation is thatthe φ/ψ angles of a π helix lie at the edge of theallowed, minimum energy, region of the Ramachandran

SECONDARY STRUCTURE 45
Figure 3.9 The π helix showing i, i + 5 hydrogenbonds and the end on view of the helix
plot. The large radius of the π helix means thatbackbone atoms do not make van der Waals contactacross the helix axis leading to the formation of ahole down the middle of the helix that is too smallfor solvent occupation.
The β strand
The β strand, so called because it was the secondunit of secondary structure predicted from the model-building studies of Pauling and Corey, is an extendedconformation when compared with the α helix. Despiteits name the β strand is a helical arrangement althoughan extremely elongated form with two residues per turnand a translation distance of 0.34 nm between similaratoms in neighbouring residues. Although less easyto recognize, this leads to a pitch or repeat distanceof nearly 0.7 nm in a regular β strand (Figure 3.10).A single β strand is not stable largely because ofthe limited number of local stabilizing interactions.However, when two or more β strands form additionalhydrogen bonding interactions a stable sheet-likearrangement is created (Figure 3.11). These β sheetsresult in significant increases in overall stability and
~ 0.68 nm
φ
ψ
0.34 nmTranslation distance
Figure 3.10 The polypeptide backbone of a single β
strand showing only the heavy atoms
N C
C N
Inter-strandhydrogen bond
Figure 3.11 Two adjacent β strands are hydrogen bonded to form a small element of β sheet. The hydrogen bondsare inter-strand between neighbouring CO and NH groups. Only the heavy atoms are shown in this diagram for clarity

46 THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
are stabilized by the formation of backbone hydrogenbonds between adjacent strands that may involveresidues widely separated in the primary sequence.
Adjacent strands can align in parallel or antiparallelarrangements with the orientation established by deter-mining the direction of the polypeptide chain from theN- to the C-terminal. ‘Cartoon’ representations of β
strands make establishing directions in molecular struc-tures easy since β strands are often shown as arrows;the arrowheads indicate the direction towards the Cterminal. These cartoons take different forms but all‘trace’ the arrangement of the polypeptide backboneand summarize secondary structural elements foundin proteins without the need to show large numbersof atoms.
Polyamino acid chains do not form β sheets whendispersed in solution and this has hindered study ofthe formation of such structures. However, despite thisobservation many proteins are based predominantlyon β strands, with chymotrypsin a proteolytic enzymebeing one example (see Figure 3.12).
Unlike ideal representations strands found in pro-teins are often distorted by twisting that arises from
a systematic variation of dihedral angles (φ and ψ)towards more positive values. The result is a slight, butdiscernable, right hand twists in the polypeptide chain.In addition when strands hydrogen bond together toform sheets further distortions occur especially withmixtures of anti-parallel and parallel β strands. Onaverage β sheets containing antiparallel strands aremore common than sheets made up entirely of par-allel strands. Anti-parallel sheets often form from justtwo β strands running in opposite directions whilst itis observed that at least four β strands are required toform parallel sheets. β strands associate spectacularlyinto extensive curved sheets known as β barrels. Theβ barrel is found in many proteins (Figure 3.13) andconsists of eight parallel β strands linked together byhelical segments.
Turns as elements of secondary structure
As more high-resolution structures are deposited inprotein databases it has allowed turns from differentproteins to be defined and compared in terms of
Figure 3.12 Representation of the elements of β strands and α helices found in the serine protease chymotrypsin.(PDB:2CGA). The strands shown in cyan have arrows indicating a direction leading from the N > C terminus, thehelices are shown in red and yellow with turns in grey. There is no convention describing the use of colours to aparticular element of secondary structure

SECONDARY STRUCTURE 47
Figure 3.13 The β barrel of triose phosphate isomerase seen in three different views. The eight β strandsencompassed by helices are shown in the centre with two different views of the eight parallel β strands shown in theabsence of helical elements. Eight strands are arranged at an angle of approximately 36◦ to the barrel axis (runningfrom top to bottom in the right most picture) with each strand offset from the previous one by a constant amount
residue composition, angles and bond distances. Insome proteins the proportion of residues found in turnscan exceed 30 percent and in view of this high value itis unlikely that turns represent random structures. Turnshave the universal role of enabling the polypeptide tochange direction and in some cases to reverse backon itself. The reverse turns or bends arise from thegeometric properties associated with these elements ofprotein structure.
Analysis of the amino acid composition of turnsreveals that bulky or branched side chains occur atvery low frequencies. Instead, residues with small sidechains such as glycine, aspartate, asparagine, serine,cysteine and proline are found preferentially. An anal-ysis of the different types of turns has established thatperhaps as many as 10 different conformations existon the basis of the number of residues making up theturn and the angles φ and ψ associated with the centralresidues. Turns can generally be classified accordingto the number of residues they contain with the mostcommon number being three or four residues.
A γ turn contains three residues and frequently linksadjacent strands of antiparallel β sheet (Figure 3.14).The γ turn is characterized by the residue in themiddle of the turn (i + 1) not participating in hydrogenbonding whilst the first and third residues can formthe final and initial hydrogen bonds of the antiparallelβ strands. The change in direction of the polypeptidechain caused by a γ turn is reflected in the values of φ
and ψ for the central residue. As a result of its size and
CαCα
Cα
Cα
Cα
Cβ
CβCα
Cα
C′ O
O
O
C
CC′
O
O
i + 2
γ turn-three residues β turn-four residues
i + 3
i + 1
i + 1
ii
i + 2
N
N
N
N
N
Figure 3.14 Arrangement of atoms in γ and β turnsconnected to strands of an antiparallel β sheet. γ
turns contain three residues whilst β turns have fourresidues. A large number of variations on this basictheme exist in proteins. Hydrogen bonds are shown by adotted line whilst only heavy atoms of the polypeptidebackbone together with the CB atom are shown
conformational flexibility glycine is a favoured residuein this position although others are found.
More commonly found in protein structures are fourresidue turns (β turns). Here the middle two residues(i + 1, i + 2) are never involved in hydrogen bondingwhilst residues i and i + 3 will participate in hydrogen

48 THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
bonding if a favourable arrangement forms betweendonor and acceptor. Analysis of structures depositedin protein databases reveals strong residue preferences.In the relatively common type 1β turn any residue canoccur at position i, i + 3 with the exception that Pro isnever found. More significantly glycine is often foundat position i + 3 whilst proline predominates at i + 1.Asn, Asp, Cys and Ser are also found frequently inβ-turns as the first residue.
Additional secondary structure
Both glycine and proline are associated with unusualconformational flexibility when compared with theremaining 18 residues. In the case of glycine thereis very little restriction to either φ/ψ as a result ofthe small side chain (H). In the case of proline theopposite situation applies with φ, the angle defined bythe N–Cα bond, restricted by the five-membered cyclic(pyrrolidine) ring. Proline residues are not suited toeither helical or strand arrangements but are found athigh frequency in turns or bends.
Additionally polyproline chains adopt unique andregular conformations distinct from helices, turns orstrands. Two recognized conformations are calledpoly(Pro) I and poly(Pro) II. Proline is unique amongstthe twenty residues in showing a much higher propor-tion of cis peptide bonds. The two forms of poly(Pro)therefore contain all cis (I) or all trans (II) peptidebonds. The value of φ is restricted by bond geometryto −83◦ (I) and −78◦ (II) and the torsion angle restric-tions create poly(Pro) I as a right-handed helix with 3.3residues per turn, whilst poly(Pro) II is a left-handedhelix with three residues per turn. Poly(Gly) chains alsoadopt regular conformation and presents the oppositesituation to poly(Pro) chains. Glycine has extreme con-formational flexibility. In the solid state poly(Gly) hasbeen shown to adopt two regular conformations des-ignated I and II. State I has an extended conformationlike a β strand whilst state II has three residues per turnand is similar to poly(Pro).
The Ramachandran plot
The peptide bond is planar as a result of resonanceand its bond angle, ω, has a value of 0 or 180◦. A
N O
CH3
N
O
H3C
HN
O
CH3
N
OH3C
H
cistrans
Figure 3.15 Cis and trans peptide bonds
peptide bond in the trans (Figure 3.15) conformation(ω = 180◦) is favoured over the cis (Figure 3.15)arrangement (ω = 0◦) by a factor of ∼1000 becausethe preferential arrangement of non-bonded atoms leadsto fewer repulsive interactions that otherwise decreasestability. In the cis peptide bond these non-bondedinteractions increase due to the close proximity ofside chains and Cα atoms with the preceding residueand hence results in decreased stability relative tothe trans state. Peptide bonds preceding proline arean exception to this trend with a trans/cis ratio ofapproximately 4.
The peptide bond is relatively rigid, but far greatermotion is possible about the remaining backbonetorsion angles. In the polypeptide backbone C–N–Cα–Cdefines the torsion angle φ whilst N–Cα–C–N definesψ. In practice these angles are limited by unfavourableclose contacts with neighbouring atoms and thesesteric constraints limit the conformational space that issampled by polypeptide chains. The allowed values forφ and ψ were first determined by G.N. Ramachandranusing a ‘hard sphere model’ for the atoms and thesevalues are indicated on a two-dimensional plot of φ
against ψ that is now called a Ramachandran plot(Figure 3.16).
In the Ramachandran plot shown in Figure 3.16the freely available conformational space is shaded ingreen. This represents ideal geometry and is exhibitedby regular strands or helices. Analysis of crystal struc-tures determined to a resolution of <2.5 A showedthat over 80 percent of all residues are found in thisregion of the Ramachandran plot. The yellow regionindicates areas that although less favourable can beformed with small deviations from the ideal angularvalues for φ or ψ. The yellow and green regions include95 percent of all residues within a protein. Finally, thepurple coloured region, although much less favourable,

SECONDARY STRUCTURE 49
180
120
60
0
−60
−120
−180−180 −120 −60 0
φ
ψ ψ
60 120 180
left handedhelix
strand
helicalregion
−180 −120 −60 0
φ60 120 180
180
120
60
0
−60
−120
−180
Figure 3.16 Ramachandran plots showing favourable conformational parameters for different φ/ψ values. Left:Ramachandran plot showing the φ/ψ angles exhibited by regular α helix and β strands. In addition the left-handedhelix has a region of limited stability and although not widely shown in protein structures isolated residues do adoptthis conformation. Right: Ramachandran plot derived from the crystal structure of bovine pancreatic trypsin inhibitorPDB: 1BPI. The residue numbers for glycine are shown. In BPTI some of these residues do not exhibit typical φ/ψangles
will account for 98 percent of all residues in proteins.All other regions are effectively disallowed with theminor exception of a small region representing left-handed helical structure. In total only 30 percent of thetotal conformational space is available suggesting thatthe polypeptide chain itself imposes severe restrictions.
One exception to this rule is glycine. Glycine lacks aCβ atom and with just two hydrogen atoms attached tothe Cα centre this residue is able to sample a far greaterproportion of the space represented in the Ramachan-dran plot (Figure 3.17). For glycine this leads to asymmetric appearance for the allowed regions. Asexpected residues with large side chains are more likelyto exhibit unfavourable, non-bonded, interactions thatlimit the possible values of φ and ψ. In the Ramachan-dran plot the allowed regions are smaller for residueswith large side chains such as phenylalanine, trypto-phan, isoleucine and leucine when compared with, forexample, the allowed regions for alanine.
Similar plots of χ1 versus χ2 describe their distri-bution in proteins and it was found that the distribution
180
120
60
0
−60
−120
−180−180 −120 −60 0
f
y
60 120 180
Figure 3.17 Ramachandran plot showing conforma-tional space ‘sampled’ by glycine residues

50 THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
of χ1 and χ2 side-chain torsion angles followed simpleenergy-based calculations with preferences for valuesof approximately +60◦, 180◦ and −60◦ for χ1 and χ2
in aliphatic side chains, and +90◦ and −90◦ for theχ2 torsion angle of aromatic residues. Inspection ofχ1/χ2 distribution reveals that several residues displaypreferences for certain combinations of torsion angles.Leucine residues prefer the combinations −60◦/180◦
and 180◦/+60◦. This approach has led to the derivationof a library of preferred, but not obligatory, side-chainrotamers. These libraries are useful in the refinementof protein structures and include residue-specific pref-erences for combinations of side-chain torsion angles.Valine, for example, shows the greatest preference forone rotamer (χ1 is predominantly t) and is unique inthis respect amongst the side chains.
Tertiary structure
At the beginning of 2004 ∼22 000 sets of atomiccoordinates were deposited in databases such as theProtein Data Bank (PDB) and this value increases dailywith the deposition of new structures from increasinglydiverse organisms (see Figure 3.18). This database is
currently maintained at Rutgers University with severalmirrors (identical sites) found at other sites acrossthe world (http://www.rcsb.org/pdb). Whilst some ofthese proteins are duplicated or related (there are over50 structures of T4 lysozyme and over 200 globinstructures) the majority represent the determination ofnovel structures using mainly X-ray diffraction andnuclear magnetic resonance (NMR) spectroscopy. Over1000 different protein folds have been discovered todate with undoubtedly more to follow.
PDB files contain information of the positions inspace of the vast majority of atoms making up aprotein. By identifying the x, y and z coordinate ofeach atom we define the whole molecule. However,besides the x, y and z coordinates of atoms PDB filesalso contain header information describing the primarysequence, the method used to determine the structure,the organism from which this protein was derived, theelements of secondary structure, any post-translationalmodifications as well as the authors. As a consequenceof the great development in bioinformatics PDB filesare undergoing homologization to enable easy compar-ison between structures and to permit widespread usewith different computer software packages. Figure 3.19shows a representative PDB file.
35000
30000
25000
20000
15000
10000
5000
0
1972
1974
1976
1978
1980
1982
1984
1986
1988
1990
1992
1994
1996
1998
2000
2002
2004
Figure 3.18 Data produced by the Protein Data Bank on the deposition of structures. From Berman, H.M. et al. TheProtein Data Bank. Nucl. Acids Res. 2000 28, 235–242. See http://www.rcsb.org/pdb for latest details. Red blocksindicate total structures, purple blocks the number of submissions per year

TERTIARY STRUCTURE 51
HEADER HYDROLASE(ZYMOGEN) 16-JAN-87 2CGA 2CGA 3 COMPND CHYMOTRYPSINOGEN *A 2CGA 4 SOURCE BOVINE (BOS $TAURUS) PANCREAS 2CGA 5 AUTHOR D.WANG,W.BODE,R.HUBER 2CGA 6 >>>>>> >>>>>> JRNL AUTH D.WANG,W.BODE,R.HUBER 2CGA 8 JRNL TITL BOVINE CHYMOTRYPSINOGEN *A. X-RAY CRYSTAL STRUCTURE 2CGA 9 >>>>>> REMARK 1 2CGA 14 REMARK 1 REFERENCE 1 2CGA 15 >>>>>> SEQRES 1 A 245 CYS GLY VAL PRO ALA ILE GLN PRO VAL LEU SER GLY LEU 2CGA 71 SEQRES 2 A 245 SER ARG ILE VAL ASN GLY GLU GLU ALA VAL PRO GLY SER 2CGA 72 SEQRES 3 A 245 TRP PRO TRP GLN VAL SER LEU GLN ASP LYS THR GLY PHE 2CGA 73 >>>>>> >>>>>> SEQRES 17 B 245 LEU VAL GLY ILE VAL SER TRP GLY SER SER THR CYS SER 2CGA 106 SEQRES 18 B 245 THR SER THR PRO GLY VAL TYR ALA ARG VAL THR ALA LEU 2CGA 107 SEQRES 19 B 245 VAL ASN TRP VAL GLN GLN THR LEU ALA ALA ASN 2CGA 108 >>>>>> CRYST1 59.300 77.100 110.100 90.00 90.00 90.00 P 21 21 21 8 2CGA 113 ORIGX1 1.000000 0.000000 0.000000 0.00000 2CGA 114 ORIGX2 0.000000 1.000000 0.000000 0.00000 2CGA 115 ORIGX3 0.000000 0.000000 1.000000 0.00000 2CGA 116 SCALE1 .016863 0.000000 0.000000 0.00000 2CGA 117 SCALE2 0.000000 .012970 0.000000 0.00000 2CGA 118 SCALE3 0.000000 0.000000 .009083 0.00000 2CGA 119 MTRIX1 1 .987700 .155000 .017700 6.21700 1 2CGA 120 MTRIX2 1 .022800 -.031400 -.999200 115.61600 1 2CGA 121 MTRIX3 1 -.154300 .987400 -.034600 -3.74800 1 2CGA 122 ATOM 1 N CYS A 1 -10.656 55.938 41.808 1.00 11.66 2CGA 123 ATOM 2 CA CYS A 1 -10.044 57.246 41.343 1.00 11.66 2CGA 124 ATOM 3 C CYS A 1 -10.076 58.323 42.431 1.00 11.66 2CGA 125 ATOM 4 O CYS A 1 -10.772 58.097 43.448 1.00 11.66 2CGA 126 ATOM 5 CB CYS A 1 -10.807 57.718 40.066 1.00 11.66 2CGA 127 >>>>>>>>>>>>>>>>>>ATOM 744 N ASN A 100 -13.152 77.724 22.378 1.00 8.65 2CGA 866 ATOM 745 CA ASN A 100 -14.213 76.940 23.011 1.00 8.65 2CGA 867 ATOM 746 C ASN A 100 -14.134 75.441 22.693 1.00 8.65 2CGA 868 ATOM 747 O ASN A 100 -13.706 75.062 21.563 1.00 8.65 2CGA 869 >>>>>> >>>>>> ATOM 1461 N VAL A 200 -9.212 70.793 39.923 1.00 9.30 2CGA1583 ATOM 1462 CA VAL A 200 -9.875 69.689 40.639 1.00 9.30 2CGA1584 ATOM 1463 C VAL A 200 -10.634 70.148 41.868 1.00 9.30 2CGA1585 ATOM 1464 O VAL A 200 -10.151 70.985 42.657 1.00 9.30 2CGA1586 >>>>>> HETATM 3601 O HOH 601 -20.008 66.224 26.138 1.00 26.69 2CGA3723 HETATM 3602 O HOH 602 -21.333 66.182 28.756 1.00 18.10 2CGA3724 HETATM 3603 O HOH 603 -18.000 68.022 22.774 1.00 34.03 2CGA3725 MASTER 60 3 0 0 0 0 0 9 3927 2 0 38 2CGAA 6 END 2CGA4053
Figure 3.19 An abbreviated version of a representative PDB file. The file 2CGA refers to the chymotrypsin. In theabove example the structure was determined by X-ray crystallography and the initial lines (header and remarks) of aPDB file give the authors, important citations, source of the protein and other useful information. Later the primarysequence is described and this is followed by crystallographic data on the unit cell dimensions and space group.The important lines are those beginning with ATOM since collectively these lines list all heavy atoms of the proteintogether with their respective x, y and z coordinates. A line beginning HETATM lists the position of hetero atomswhich might include co-factors but more frequently lists water molecules found in the crystal structure. The symbol������� is used here to denote the omission of many lines of text. Where more than one chain exists thecoordinates will be listed as chain A, chain B, etc.
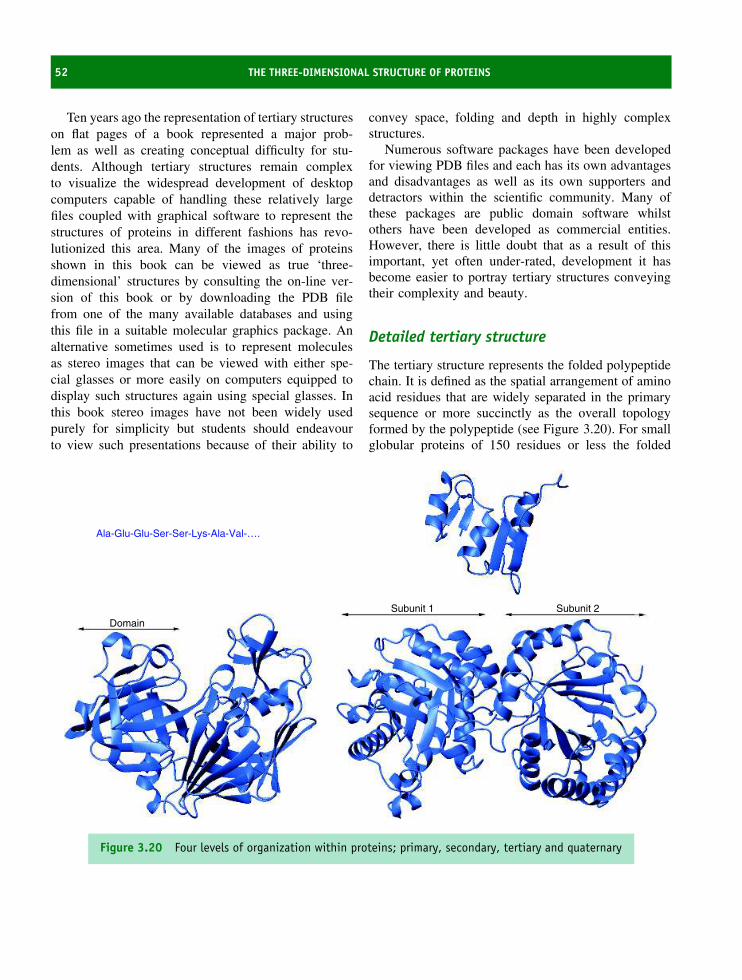
52 THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Ten years ago the representation of tertiary structureson flat pages of a book represented a major prob-lem as well as creating conceptual difficulty for stu-dents. Although tertiary structures remain complexto visualize the widespread development of desktopcomputers capable of handling these relatively largefiles coupled with graphical software to represent thestructures of proteins in different fashions has revo-lutionized this area. Many of the images of proteinsshown in this book can be viewed as true ‘three-dimensional’ structures by consulting the on-line ver-sion of this book or by downloading the PDB filefrom one of the many available databases and usingthis file in a suitable molecular graphics package. Analternative sometimes used is to represent moleculesas stereo images that can be viewed with either spe-cial glasses or more easily on computers equipped todisplay such structures again using special glasses. Inthis book stereo images have not been widely usedpurely for simplicity but students should endeavourto view such presentations because of their ability to
convey space, folding and depth in highly complexstructures.
Numerous software packages have been developedfor viewing PDB files and each has its own advantagesand disadvantages as well as its own supporters anddetractors within the scientific community. Many ofthese packages are public domain software whilstothers have been developed as commercial entities.However, there is little doubt that as a result of thisimportant, yet often under-rated, development it hasbecome easier to portray tertiary structures conveyingtheir complexity and beauty.
Detailed tertiary structure
The tertiary structure represents the folded polypeptidechain. It is defined as the spatial arrangement of aminoacid residues that are widely separated in the primarysequence or more succinctly as the overall topologyformed by the polypeptide (see Figure 3.20). For smallglobular proteins of 150 residues or less the folded
Ala-Glu-Glu-Ser-Ser-Lys-Ala-Val-….
Domain
Subunit 1 Subunit 2
Figure 3.20 Four levels of organization within proteins; primary, secondary, tertiary and quaternary

TERTIARY STRUCTURE 53
structure involves a spherical compact molecule com-posed of secondary structural motifs with little irregularstructure. Disordered or irregular structure in proteinsis normally confined to the N and C terminals or morerarely to loop regions within a protein or linker regionsconnecting one or more domains. Asking the ques-tion ‘what is the tertiary structure of a protein?’ issynonymous with asking ‘what is the protein fold?.’The fold arises from linking together secondary struc-tures forming a compact globular molecule. Elementsof secondary structure interact via hydrogen bonds,as in β sheets, but also depend on disulfide bridges,electrostatic interactions, van der Waals interactions,hydrophobic contacts and hydrogen bonds betweennon-backbone groups.
Interactions stabilizing tertiary structure
To form stable tertiary structure proteins must clearlyform more attractive interactions than unfavourableor repulsive ones. The formation of stable tertiaryfolds relies on interactions that differ in their relativestrengths and frequency in proteins.
Disulfide bridges
Disulfide bridges dictate a protein fold by formingstrong covalent links between cysteine side chains thatare often widely separated in the primary sequence.A disulfide bridge cannot form between consecutivecysteine residues and it is normal for each cysteineto be separated by at least five other residues.The formation of a disulfide bridge restrains theoverall conformation of the polypeptide and in bovinepancreatic trypsin inhibitor (BPTI; Figure 3.21), asmall protein of 58 residues, there are three disulfidebridges formed between residues 5–55, 14–38 and30–51. The effect is to bring the secondary structuralelements closer together. Disulfide bonds are onlybroken at high temperatures, acidic pH or in thepresence of reductants. In BPTI reduction of thedisulfide bonds leads to decreased protein stability thatis mirrored by other disulfide-rich proteins.
The hydrophobic effect
The importance of the hydrophobic effect was for along time underestimated. Charged interactions and
Figure 3.21 Structure of BPTI (PDB: 5PTI) showingthe protein fold held by three disulfide bridges (5–55,14–38 and 30–51)
hydrogen bonds are not strong intramolecular forcesbecause water molecules compete significantly withthese effects. However, water is a very poor solventfor many non-polar molecules and this is exemplifiedby dissolving an organic solute such as cyclohexanein water. Non-polar molecules cannot form hydrogenbonds with water and this prevents molecules suchas cyclohexane dissolving extensively in aqueoussolutions. As a consequence interactions between waterand non-polar molecules are weakened and may bevirtually non-existent. The result is an enhancementof interactions between non-polar molecules and theformation of hydrophobic clusters within water. Theenhanced interactions between non-polar moleculesin the presence of water are the basis for thehydrophobic effect. Since the side chains of manyamino acid residues are hydrophobic it is clear thatthe hydrophobic effect may contribute significantly tointramolecular interactions. The hydrophobic effect canbe restated as the preference of non-polar atoms fornon-aqueous environments.

54 THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
The magnitude of the hydrophobic effect has proveddifficult to estimate but has been accomplished bymeasuring the free energy associated with transferof a non-polar solvent into water from the gaseous,liquid or solid states. The thermodynamics governingthe transfer of non-polar molecules between phasesare complicated but it is worth remembering thatthe enthalpy change, �H , represents changes in non-covalent interactions in going between the two phaseswhilst the entropy change (�S) reflects differencesin the order of each system. A summation of thesetwo terms gives �Gtr, the free energy of transfer ofa non-polar or hydrophobic molecule from one phaseto another, via the relationship �G = �H − T �S,where T is temperature. The overall process involvesthe transfer of a solute molecule into the aqueousphase by (i) creating a cavity in the water, (ii) addingsolute to the cavity, and (iii) maximizing favourableinteractions between solute molecules and betweensolvent molecules.
In ice water molecules are arranged in a regularcrystal lattice that maximizes hydrogen bonding withother water molecules, forming on average fourhydrogen bonds. In the liquid phase these hydrogenbonds break and form rapidly with an estimated half-life of less than 1 ns. This leads to each watermolecule forming an average of 3.4 hydrogen bonds.The capacity for hydrogen bonding and intermolecularattraction accounts for the high boiling point of waterand the relatively large amounts of energy requiredto break these interactions, especially when comparedwith the interactions between non-polar liquids such ascyclohexane (Figure 3.22). Here the C–H bonds showlittle tendency to hydrogen bond and this is true formost of the non-polar side chains of amino acids inproteins. The hydrophobic interaction does not derivefrom the interaction between non-polar molecules orfrom the interaction between water and non-polarsolutes because hydrogen bonds do not form. Thedriving force for the formation of hydrophobic clustersis the tendency for water molecules to hydrogenbond with each other. Water forms hydrogen bondednetworks around non-polar solutes to become moreordered and one extreme example of this effect is theobservation of clathrates where water forms an orderedcage around non-polar solutes. Around the cage water
∆H = −8T∆S = −6
∆G = −2∆Cp = +12
∆H = −8T∆S = −12
∆G = +4∆Cp = +120
∆H = +2T∆S = −3∆G = +5
∆G = +6∆Cp = 108
∆H = 0T∆S = −6
∆H = −2T∆S = −3∆G = +1
Liquid
Solid
Gas
Aqueoussolution
Figure 3.22 Transfer of a non-polar solute (cyclo-hexane) from gas, liquid and solid phases to aqueoussolution around room temperature. The hydrophobicinteraction is temperature dependent and the respec-tive enthalpy (�H), entropy (T�S) and free energy(�G) of transfer are given in kcal mol−1. (Repro-duced courtesy of Creighton, T.E. Proteins Structure &Molecular Properties, 2nd edn. W.H. Freeman, 1993)
forms with maximum hydrogen bonding but each bondhas less than optimal geometry.
The energetics of the hydrophobic interaction cannow be explored in more detail to provide a physicalbasis for the phenomena in protein structure. Themost interesting transitions are those occurring fromliquid phases involving the transfer of a non-polarsolute from a non-polar liquid to water. The adverseordering of water around the solute leads to anunfavourable decrease in entropy, whilst the �Htr
term is approximately zero. It is the entropic factorthat dominates in the transfer of a non-polar soluteinto aqueous solutions with enthalpy terms reflectingincreased hydrogen bonding.
The temperature dependence of the hydrophobicinteraction provides still more clues concerning theprocess. As the temperature is increased water around

TERTIARY STRUCTURE 55
non-polar solutes is disrupted by breaking hydrogenbonds and becomes more like bulk water. Thisprocess is reflected by a large heat capacity (�Cp)that accompanies the hydrophobic interaction and ischaracteristic of this type of interaction. The largechange in heat capacity underpins the temperaturedependency of the hydrophobic interaction throughits effect on both enthalpic and entropic terms. Themagnitude of Cp is related to the non-polar surfacearea of the solute exposed to water along with manyother thermodynamic parameters. As a result a largenumber of correlations have been made betweenaccessible surface area, solubility of non-polar solutesin aqueous solutions and the energetics associated withthe hydrophobic interaction. The hydropathy indexestablished for amino acids is just one manifestationof the hydrophobic interaction described here.
Charge–charge interactions
These interactions occur between the side chains ofoppositely charged residues as well as between theNH3
+ and COO− groups at the ends of polypeptide
ARG 154
GLU 21
Figure 3.23 An example of electrostatic or charge–charge interactions occurring in proteins. The exampleshown is from the protease chymotrypsin (PDB: 2CGA).Here the interaction is between arginine and glutamateside chains. The donor atom is the hydrogen of the NEnitrogen whilst the acceptor groups are both oxygenatoms, OE1 and OE2. Reproduced courtesy Creighton,T.E. Proteins Structure & Molecular Properties, 2ndedn. W.H. Freeman, 1993
chains (Figure 3.23). Of importance to charge–chargeinteractions1 are the side chains of lysine, arginine andhistidine together with the side chains of aspartate,glutamate and to a lesser extent tyrosine and cysteine.
As a result of their charge the side chains ofthese residues are found on the protein surfacewhere interactions with water or solvent moleculesdramatically weaken these forces. In view of their lowfrequency and solvated status these interactions do notusually contribute significantly to the overall stabilityof a protein fold. Coulomb’s law describes the potentialenergy (V ) between two separated charges (in a perfectvacuum) according to the relationship
V = q1 q2/ε 4π r2 (3.1)
where q1 and q2 are the magnitude of the charges (nor-mally +1 and −1), r is the separation distance betweenthese charges and ε is the permittivity of free space. Inother media, such as water this equation is modified to
V = q1 q2/εo 4π r2 (3.2)
where εo is the permittivity of the medium and is relatedto the dielectric constant (εr) by
εr = ε/εo (3.3)
When the medium is water this has a value ofapproximately ∼80 whilst methanol has a value of ∼34and a hydrophobic solvent such as benzene has a valueof ∼2 (a perfect vacuum has a value of 1 by definition).It has been estimated that each charge–pair interactionlocated on the surface of a protein may contribute lessthan 5 kJ mol−1 to the overall stability of a proteinand this must be compared with the much strongerdisulfide bridge (∼100–200 kJ mol−1). Occasionallycharge interactions exist within non-polar regions inproteins and under these conditions with a low dielectricmedium and the absence of water the magnitude of thecharge–charge interaction can be significantly greater.
A variation occurring in normal charge–chargeinteractions are the partial charges arising from hydro-gen bonding along helices. Hydrogen bonding betweenamide and carbonyl groups gives rise to a net dipolemoment for helices. In addition the peptide bondspointing in the same direction contribute to the accumu-lative polarization of helices. As a result longer helices
1Sometimes called electrostatic, ionic or salt-bridge interactions.

56 THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
have a greater macrodipole. The net result is that asmall positive charge is located at the N-terminal endof a helix whilst a negative charge is associated withthe C-terminal end. For a helix of average length this isequivalent to +0.5–0.7 unit charge at the N-terminusand −0.5–0.7 units at the C terminus. Consequently,to neutralize the overall effect of the helix macrodipoleacidic side chains are more frequently located at thepositive end of the helix whilst basic side chains canoccur more frequently at the negative pole.
Hydrogen bonding
Hydrogen bonds contribute significantly to the stabilityof α helices and to the interaction of β strands toform parallel or antiparallel β sheets. As a result suchhydrogen bonds contribute significantly to the overallstability of the tertiary structure or folded state. Thesehydrogen bonds are between main chain NH and COgroups but the potential exists with protein folding toform hydrogen bonds between side chain groups andbetween main chain and side chain. In all cases thehydrogen bond involves a donor and acceptor atom andwill vary in length from 0.26 to 0.34 nm (this is thedistance between heavy atoms, i.e. between N and O ina hydrogen bond of the type N–H- - - O=C) and maydeviate in linearity by ±40◦. In proteins other typesof hydrogen bonds can occur due to the presence ofdonor and acceptor atoms within the side chains. Thisleads to hydrogen bonds between side chains as wellas side chain–main chain hydrogen bonds. Particularlyimportant in hydrogen bond formation are the sidechains of tyrosine, threonine and serine containingthe hydroxyl group and the side chains of glutamineand asparagine with the amide group. Frequently, sidechain atoms hydrogen bond to water molecules trappedwithin the interior of proteins whilst at other timeshydrogen bonds appear shared between two donor oracceptor groups. These last hydrogen bonds are termedbifurcated. Table 3.4 shows examples of the differenttypes of hydrogen bond.
Van der Waals interactions
There are attractive and repulsive van der Waals forcesthat control interactions between atoms and are very
Table 3.4 Examples of hydrogen bonds betweenfunctional groups found in proteins
Residues involved inhydrogen bonding
Nature of interactionand typical distancebetween donor and
acceptor atoms (nm)
Amide–carbonyl
Gln/Asn -backbone
N H O C~0.29 nm
Amide-hydroxyl.
Backbone-Ser N H O C
H
H2~0.3 nmAsn/Gln -Ser
Amide-imidazole.
Asn/Gln -His
Backbone -His
N H N NH
H2C
~0.31 nm
Hydroxyl-carbonyl
Ser/Thr/Tyr–backboneO
H
O C
~0.28 nmSer-Asn/Gln
Thr- Asn/Gln
Tyr- Asn/Gln
Hydroxyl-hydroxyl
Thr,Ser and TyrO
H
H O
~0.28 nmTyr-Ser
important in protein folding. These interactions occurbetween adjacent, uncharged and non-bonded atomsand arise from the induction of dipoles due to fluctuat-ing charge densities within atoms. Since atoms are con-tinually oscillating the induction of dipoles is a constantphenomena. It is unwise to view van der Waals forces

TERTIARY STRUCTURE 57
as simply the interaction between temporary dipoles;the interaction occurs between uncharged atoms andinvolves several different types. Unlike the electrostaticinteractions described above they do not obey inversesquare laws and they vary in their contributions to theoverall intermolecular attraction. Three attractive con-tributions are recognized and include in order of dimin-ishing strength: (i) the orientation effect or interactionbetween permanent dipoles; (ii) the induction effect orinteraction between permanent and temporary dipoles;(iii) the dispersion effect or London force, which is theinteraction between temporary, induced, dipoles.
The orientation effect is the interaction energybetween two permanent dipoles and depends on theirrelative orientation. For a freely rotating molecule itmight be expected that this effect would average outto zero. However, very few molecules are absolutelyfree to rotate with the result that preferred orienta-tions exist. The energy of interaction varies as theinverse sixth power of the interatomic separation dis-tance (i.e. ∝ r−6) and is inversely dependent uponthe temperature. The induction effect also varies asr−6 but is independent of temperature. The magni-tude of this effect depends on the polarizability ofmolecules. The dispersion effect, sometimes called theLondon force, involves the interactions between tem-porary and induced dipoles. It arises from a temporarydipole inducing a complementary dipole in an adja-cent molecule. These dipoles are continually shifting,depend on the polarization of the molecule and resultin a net attraction that varies as r−6.1
When atoms approach very closely repulsion be-comes the dominant and unfavourable interaction. Therepulsive term is always positive but drops awaydramatically as the distance between the two atomsincreases. In contrast it becomes very large at shortatomic distances and is usually modelled by r−12 dis-tance dependency, although there are no experimentalgrounds for this relationship. The combined repulsiveand attractive van der Waals terms are described byplots of the potential energy as a function of inter-atomic separation distance (Figure 3.24). The energyof the van der Waals reactions is therefore described
1The dispersion effect is so called because the movement of electronsunderlying the phenomena cause a dispersion of light.
Repulsiveregion
Attractiveregion
Repulsive interaction
Net interaction
Attractive interaction
r
Figure 3.24 The van der Waals interaction as afunction of interatomic distance r. The repulsive termincreases rapidly as atoms get very close together
by the difference between the attractive (r−6) and repul-sive terms (r−12)
Evdw ∝ −A/r6 + B/r12 (3.4)
Evdw = � E [(rm/r)12 − 2(rm/r)6] (3.5)
where the van der Waals potential (Evdw) is the sumof all interactions over all atoms, E is the depth of thepotential well and rm is the minimum energy interactiondistance. This type of interaction is often called a6–12 interaction, or the Leonard Jones potential.Although van der Waals forces are extremely weak,especially when compared with other forces influencingprotein conformation, their large number arranged closetogether in proteins make these interactions significantto the maintenance of tertiary structure.
A protein’s folded state therefore reflects the sum-mation of attractive and repulsive forces embodied bythe summation of the electrostatic, hydrogen bond-ing, disulfide bonding, van der Waals and hydrophobicinteractions (see Table 3.5). It is often of consider-able surprise to note that proteins show only marginalstability with the folded state being between 20 and80 kJ mol−1 more stable than the unfolded state. Therelatively small value of �G reflects the differencesin non-covalent interactions between the folded and

58 THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Table 3.5 Distance dependence and bond energy forinteractions between atoms in proteins
Bond Distancedependence
Approximatebond energy(kJ mol−1)
Covalent No simpledependence
∼200
Ionic ∝ 1/r2 <20Hydrogen bond No simple
expression<10
van der Waals ∝ 1/r6 <5Hydrophobic No simple
expression<10
unfolded states. This arises because an unfolded pro-tein normally has an identical covalent structure tothe folded state and any differences in protein stabilityarise from the favourable non-covalent interactions thatoccur during folding.
The organization of proteins into domains
For proteins larger than 150 residues the tertiarystructure may be organized around more than onestructural unit. Each structural unit is called a domain,although exactly the same interactions govern itsstability and folding. The domains of proteins interacttogether although with fewer interactions than thesecondary structural elements within each domain.These domains can have very different folds ortertiary structures and are frequently linked by extendedrelatively unstructured regions of polypeptide.
Three major classes of domains can be recognized.These are domains consisting of mainly α helices,domains containing mainly β strands and domains thatare mixed by containing α and β elements. In thislast class are structures containing both alternating α/βsecondary structures as well as proteins made up ofcollections of helices and strands (α + β). Within eachof these three groups there are many variations of thebasic themes that lead to further classification of proteinarchitectures (see Figure 3.25).
Protein domains arise by gene duplication andfusion. The result is that a domain is added ontoanother protein to create new or additional properties.An example of this type of organization is seen in thecytochrome b5 superfamily. Cytochrome b5 is a smallglobular protein containing both α helices and β strandsfunctioning as a soluble reductant to methaemoglobinin the erythrocyte or red blood cell. The cytochromecontains a non-covalently bound heme group inwhich the iron shuttles between the ferric (Fe3+)and ferrous (Fe2+) states (Figure 3.26). The reversibleredox chemistry of the iron is central to the role ofthis protein as an electron carrier within erythrocytes.When united with a short hydrophobic chain thisprotein assumes additional roles and participates inthe fatty acyl desaturase pathway. In mitochondria theenzyme sulfite oxidase converts sulfite to sulfate aspart of the dissimilatory pathway for sulfur withincells. Sulfite oxidase contains cytochrome b5 linkedto a molybdenum-containing domain. Further use ofthe b5-like fold occurs in nitrate reductase where aflavin binding domain is linked to the cytochrome. Theresult is a plethora of new proteins containing differentdomains that allow the basic redox role of cytochromeb5 to be exploited and enhanced to facilitate thecatalysis of new reactions.
A rigorous definition of a domain does not exist.One acceptable definition is the presence of anautonomously folding unit within a protein. Alterna-tively a domain may be defined as a region of a pro-tein showing structurally homology to other proteins.However, in all cases domains arise from the foldingof a single polypeptide chain and are distinguished fromquaternary structure (see below) on this basis.
Super-secondary structure
The distinctions between secondary structure andsuper-secondary structure or between tertiary structureand super-secondary structure are not well defined.However, in some proteins there appears to bean intermediate level of organization that reflectsgroups of secondary structural elements but does notencompass all of the structural domain or tertiaryfold. The β barrel found in enzymes such as triosephosphate isomerase could form an element of supersecondary structure since it does not represent all of the

TERTIARY STRUCTURE 59
l repressor
Plastocyanin
Cytochrome b562
cis-trans proline isomerase
Thioredoxin
γ-crystallin
Figure 3.25 The secondary structure elements found in monomeric proteins. The λ repressor protein (PDB: 1LMB)contains the helix turn helix (HTH) motif; cytochrome b-562: (PDB: 256b) is a four-helix bundle heme bindingdomain; human thioredoxin: (PDB: 1ERU), a mixed α/β protein containing a five-stranded twisted β sheet. spinachplastocyanin: (PDB: 1AG6) a single Greek key motif binds Cu (shown in green); human cis-trans proline isomerase(PDB: 1VBS), a small extensive β domain containing a collection of strands that fold to form a ‘sandwich’; humanγ-crystallin: (PDB: 2GCR), two domains each of which is an eight-stranded β barrel type structure composed of twoGreek key motifs
folded domain yet represents a far greater proportionof the structure than a simple β strand. Amongst otherelements of super-secondary structure recognized inproteins are the β-α-β motif, Rossmann fold, four-helixbundles, the Greek-key motif and its variants, and theβ meander. The cartoon representations in Figure 3.27demonstrate the arrangement of β strands in some ofthese motifs and the careful viewer may identify thesemotifs in some figures in this chapter.
The β meander motif is a series of antiparallelβ strands linked by a series of loops or turns. Inthe β meander the order of strands across the sheetreflects their order of appearance along the polypeptide
sequence. A variation of this design is the so-calledGreek key motif, and it takes its name from thedesign found on many ancient forms of pottery orarchitecture. The Greek key motif shown here linksfour antiparallel β strands with the third and fourthstrands forming the outside of the sheet whilst strands1 and 2 form on the inside or middle of the sheet.The Greek key motif can contain many more strandsranging from 4 to 13. The Cu binding metalloproteinplastocyanin contains eight β strands arranged in aGreek key motif.
A β sandwich forms normally via the interactionof strands at an angle and connected to each other

60 THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Mo
Mo
Figure 3.26 The heme binding domain of cytochrome b5 (PDB:3B5C), a schematic representation of the duplicationof this domain (red) to form part of multi-domain proteins by linking it to FAD domains, Mo-containing domains orhydrophobic tails. The arrangement of domains in proteins such as nitrate reductase, yeast flavocytochrome b2 andsulfite oxidase is shown (left). The structure of sulfite oxidase (bottom) (PDB: 1SOX) shows the cytochrome domainin the foreground linked to a larger molybdenum-pterin binding domain
via short loops. In some cases these strands canoriginate from a different polypeptide chain but theemphasis is on two layers of β strands interactingtogether within a globular protein. The layers of thesandwich can be aligned with respect to each otheror arranged orthogonally. An example of the secondarrangement is shown in human cis-trans prolinepeptidyl isomerase.
The Rossmann fold, named after its discovererMichael Rossmann, is an important super-secondarystructure element and is an extension of the β-α-βdomain. The Rossmann fold consists of three parallelβ strands with two intervening α helices i.e. β-α-β-α-β. These units are found together as a dimer – sothe Rossmann fold contains six β strands and fourhelices – and this collection of secondary structure

TERTIARY STRUCTURE 61
Greek-keymotif
(a) (b) (c)
Figure 3.27 Cartoon representations of (a) the β meander, (b) Greek key and (c) Swiss or Jelly roll motifs
neuraminidase
Lactate dehydrogenase
P22 tail spike protein
Figure 3.28 The structure of complex motifs involving β strands. Native influenza neuraminidase showingcharacteristic β propellor (PDB:1F8D). A β helix found in the tailspike protein of bacteriophage P22 (PDB:1TSP). Theβ-α-β-α-β motif is shown in the Rossmann fold of Lactate dehydrogenase with each set of three strands shown indifferent colours (PDB:1LDH)

62 THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
frequently forms a nucleotide-binding site. Nucleotidebinding domains are found in many enzymes and inparticular, dehydrogenases, where the co-factor nicoti-namide adenine dinucleotide is bound at an active site.Examples of proteins or enzymes containing the Ross-mann fold are lactate dehydrogenase, glyceraldehyde-3-phosphate dehydrogenase, alcohol dehydrogenase,and malate dehydrogenase. However, it is clear thatthis fold is found in other nucleotide proteins besidedehydrogenases, including glycogen phosphorylase andglyceroltriphosphate binding proteins.
Elements of super secondary structure are frequentlyused to allow protein domains to be classified bytheir structures. Most frequently these domains areidentified by the presence of characteristic folds. A foldrepresents the ‘core’ of a protein domain formed from acollection of secondary structures. In many cases thesefolds occur in more than one protein allowing structuralrelationships to be established. These characteristicfolds include four-helix bundles (cytochrome b562 inFigure 3.25), helix turn helix motifs (the λ repressorin Figure 3.25), β barrels, and the β sandwich aswell as more complicated structures such as the β
propellor and β helix (see Figure 3.28). The β helixis an unusual arrangement of secondary structure – β
strands align in a parallel manner one above anotherforming inter-strand hydrogen bonds but collectivelytwisting as a result of the displacement of successivestrands. The strands all run in the same direction andthe displacement result in the formation of a helix. Bothleft-handed and right-handed β helix proteins have beendiscovered, and a prominent example of a right handedβ helix occurs in the tailspike protein of bacteriophageP22. In this protein the tailspike protein is actuallya trimer containing three interacting β helices. Thearrangement of strands within a β helix gives anysubunit with this structural motif a very elongatedappearance and leads to the hydrophobic cores beingspread out along the long axis as opposed to a typicalglobular packing arrangement.
Quaternary structure
Many proteins contain more than one polypeptidechain. The interaction between these chains under-scores quaternary structure. The interactions are exactly
the same as those responsible for tertiary structure,namely disulfide bonds, hydrophobic interactions,charge–pair interactions and hydrogen bonds, withthe exception that they occur between one or morepolypeptide chains. The term subunit is often usedinstead of polypeptide chain.
Quaternary structure can be based on proteinswith identical subunits or on non-identical subunits(Figure 3.29). Triose phosphate isomerase, HIV pro-tease and many transcription factors function ashomodimers. Haemoglobin is a tetramer containing twodifferent subunits denoted by the use of Greek letters,α and β. The protein contains two α and two β subunitsin its tetrameric state and for haemoglobin this is nor-mally written as α2β2. Although it might be thoughtthat aggregates of subunits are an artefact resultingfrom crystal packing it is abundantly clear that correctfunctional activity requires the formation of quater-nary structure and the specific association of subunits.Subunits are held together predominantly by weak non-covalent interactions. Although individually weak theseforces are large in number and lead to subunit assemblyas well as gains in stability.
Quaternary structure is shown by many proteinsand allows the formation of catalytic or binding sitesat the interface between subunits. Such sites areimpossible for monomeric proteins. Further advantagesof oligomeric proteins are that ligand or substrate-binding causes conformational changes within thewhole assembly and offer the possibility of regulatingbiological activity. This is the basis for allostericregulation in enzymes.
In the following sections the quaternary organizationof transcription factors, immunoglobulins and oxygen-carrying proteins of the globin family are describedas examples of the evolution of biological function inresponse to multiple subunits. The presence of higherorder or quaternary structure allows greater versatilityof function, and by examining the structures of some ofthese proteins insight is gained into the interdependenceof structure and function. A common theme linkingthese proteins is that they all bind other molecules suchas nucleic acid in the case of transcription factors, smallinorganic molecules such as oxygen and bicarbonate bythe globins and larger proteins or peptides in the caseof immunoglobulins.
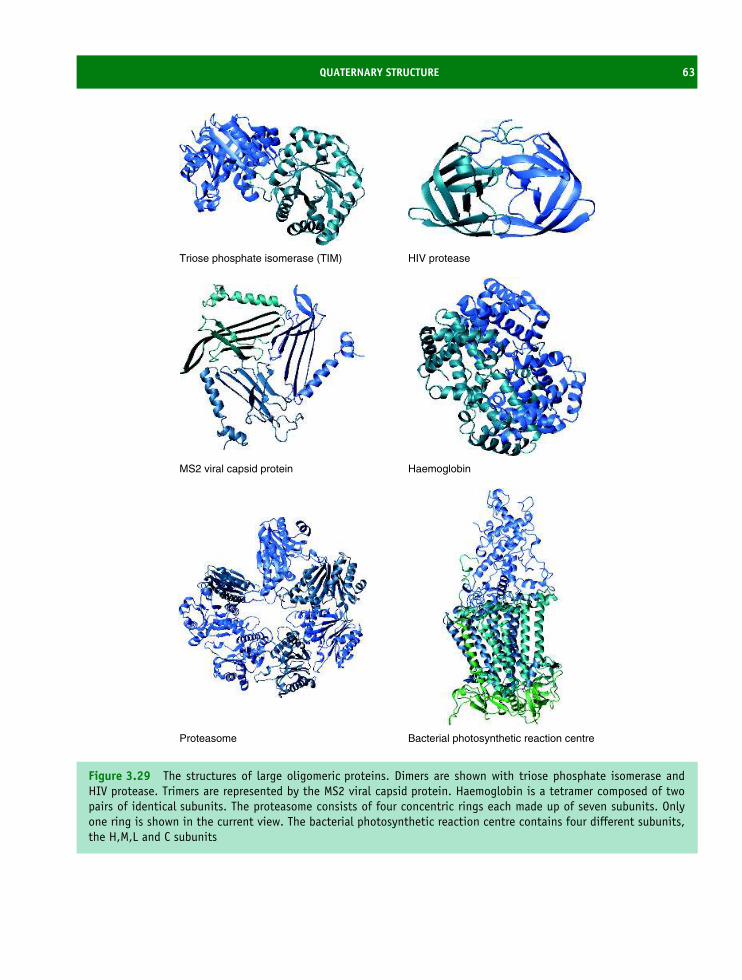
QUATERNARY STRUCTURE 63
Triose phosphate isomerase (TIM) HIV protease
MS2 viral capsid protein Haemoglobin
Proteasome Bacterial photosynthetic reaction centre
Figure 3.29 The structures of large oligomeric proteins. Dimers are shown with triose phosphate isomerase andHIV protease. Trimers are represented by the MS2 viral capsid protein. Haemoglobin is a tetramer composed of twopairs of identical subunits. The proteasome consists of four concentric rings each made up of seven subunits. Onlyone ring is shown in the current view. The bacterial photosynthetic reaction centre contains four different subunits,the H,M,L and C subunits

64 THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Dimeric DNA-binding proteins
Many dimeric proteins exist in the proteomes of cellsand one of the most common occurrences is the useof two subunits and a two-fold axis of symmetry tobind DNA. DNA binding proteins are a very largegroup of proteins typified by transcription factors.Transcription factors bind to promoter regions ofDNA sequences called, in eukaryotic systems, theTATA box, and in prokaryotes a Pribnow box. TheTATA box is approximately 25 nucleotides upstreamof the transcription start site and as their namesuggests these factors mediate the transcription of DNAinto RNA by promoting RNA polymerase bindingto this region of DNA in a pre-initiation complex(Figure 3.30).
In prokaryotes the mode of operation is simplerwith fewer modulating elements. Prokaryotic promotercontains two important zones called the −35 regionand −10 region (Pribnow box). The −35 bp regionfunctions in the initial recognition of RNA polymeraseand possesses a consensus sequence of TTGACAT. The−10 region has a consensus sequence (TATAAT) andoccurs about 10bp before the start of a bacterial gene.
The cI and cro proteins from phage λ wereamongst the first studied DNA binding proteins andact as regulators of transcription in bacteriophagessuch as 434 or λ. The life cycle of λ bacteriophageis controlled by the dual action of the cI and croproteins in a complicated series of reactions whereDNA binding close to initiation sites physicallyinterferes with gene transcription by RNA polymerase
Figure 3.30 Transcription starts at the initiation site(+1) but is promoted by binding to the consensusTATA box sequence (−25) of the RNA polymerasecomplex that includes transcription factors. UpstreamDNA sequences that facilitate transcription have beenrecognized (CAT and GC boxes ∼−80 and ∼−90bases upstream of start site) whilst downstreamelements lack consensus sequences but exist for sometranscription systems
Figure 3.31 The control of transcription in bacterio-phage λ by cro and cI repressor proteins. The cI dimermay bind to any of three operators, although the orderof affinity is O1 ≈ O2 > O3. In the absence of cI pro-teins, the cro gene is transcribed. In the presence ofcI proteins only the cI gene may be transcribed. HighcI concentrations prevents transcription of both genes
(Figure 3.31). For this reason these proteins arealso called repressors. When phage DNA enters abacterial host cell two outcomes are possible: lyticinfection results in the production of new viralparticles or alternatively the virus integrates intothe bacterial genome lying dormant for a periodknown as the lysogenic phase. Lytic and lysogenicphases are initiated by phage gene expression butcompetition between the cro and cI repressor proteinsdetermines which pathway is followed. Cro andcI repressor compete for control by binding to anoperator region of DNA that contains at least threesites that influence the lytic/lysogenic switch. Thebacteriophage remains in the lysogenic state if cIproteins predominate. Conversely the lytic cycle isfollowed if cro proteins dominate.
Determining the structure of the cI repressor in thepresence of a DNA containing a consensus bindingsequence uncovered detailed aspects of the mechanismof nucleic acid binding. The mechanism of DNAbinding is of considerable interest in view of thewidespread occurrence of transcription factors in allcells and the growing evidence of their involvementin many disease states. In the absence of DNA a

QUATERNARY STRUCTURE 65
Figure 3.32 The monomer form of the cI repressor(PDB: 1LMB)
structure for the cI repressor revealed a polypeptidechain containing five short helices within a domain of∼70 residues (Figure 3.32). Far more revealing wasthe structure in the presence of DNA and the use ofhelices 2 and 3 as a helix-turn-helix or HTH motif tofit precisely into the major groove (Figure 3.33).
The N- and C-terminal domains of one subunit areseparated by mild proteolysis. Under these conditionstwo isolated N-terminal domains form a less stabledimer that has lowered affinity for DNA whencompared with the complete repressor. The HTH motifis 20 residues in length and is formed by helices2 and 3 found in the N terminal domain. Althoughthe HTH motif is often described as a ‘domain’ itshould be remembered it is part of a larger proteinand does not fold into a separate, stable element ofstructure. Helix 3 of the HTH motif makes a significantnumber of interactions with the DNA. It is calledthe recognition helix with contact points involvingGln33, Gln44, Ser45, Gly46, Gly48, and Asn52. Theseresidues contain a significant proportion of polar sidechains and bind directly into the major groove foundin DNA. In comparison helix 2 makes fewer contactsand has a role of positioning helix 3 for optimalrecognition. Of the remaining structure helices 4 and 5
Figure 3.33 The structure of the N terminal domainof the λ repressor in the presence of DNA. The fifthhelix forms part of the dimerization domain that allowstwo monomer proteins to function as a homodimer. Ineach case helices 2 and 3 bind in the major groove ofDNA since the spatial separation of each HTH motif iscomparable with the dimensions of the major groove
form part of a dimer interface whilst the C terminaldomain is involved directly in protein dimerization.Dimerization is important since it allows HTH motifsto bind to successive major grooves along a sequenceof DNA. Determination of the structure of the crorepressor protein showed a very similar two-domainstructure to the cI repressor with comparable modesof DNA binding (see Figure 3.34). The overwhelmingsimilarity in structure between these proteins suggesteda common mechanism of DNA binding and one thatmight extend to the large number of transcriptionfactors found in eukaryotes.
Comparison of the sequences of HTH motifs fromλ and cro repressors showed sequence conservation(Figure 3.35). The first seven residues of the 20 residueHTH motif form helix 2, a short 4 residue turn extendsfrom positions 8 to 11 and the next (recognition) helixis formed from residues 12 to 20. The sequences of

66 THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Figure 3.34 The cro repressor protein from bacterio-phage 434 bound to a 20-mer DNA helix (PDB:4CRO)
Cro : QTKTAKDLGVYQSAINKAIH434Cro : QTELATKAGVKQQSIQLIEAcI : QESVADKMGMGQSGVGALFN
Figure 3.35 The sequence homology in the cro andcI repressors. Residues highlighted in red are invariantwhilst those shown in yellow illustrate conservativechanges in sequence
the HTH motif in the cro and λ repressors helpeddefine structural constraints within this region. Residue9 is invariant and is always a glycine residue. It islocated in the ‘turn’ region and larger side chains arenot easily accommodated and would disrupt the turnand by implication the positioning of the DNA bindinghelix. Residues 4 and 15 are completely buried fromthe solvent whilst residues 8 and 10 are partially buried,non-polar side chains charged side chains unfavourableat these locations. Proline residues are never foundin HTH motifs. Further structural constraints wereapparent at residue 5 located or wedged between thetwo helices. Large or branched side chains would causea different alignment of the helices and destroy the
primary function of HTH motif. DNA binding wascritically dependent on the identity of side chainsfor residues 11–13, 16–17 and 20. Residues withpolar side chains were common and were importantin forming hydrogen bonds to the major groove.
The globin family and the role ofquaternary structure in modulatingactivity
Myoglobin occupies a pivotal position in the historyof protein science. It was the first protein structure tobe determined in 1958. Myoglobin and haemoglobin,whose structure was determined shortly afterwards,have been extensively studied particularly in relation tothe inter-dependence of structure and function. Manyof the structure–function relationships discovered formyoglobin have proved to be of considerable impor-tance to the activity of other proteins.
The evolutionary development of oxygen carryingproteins was vital to multicellular organisms wherelarge numbers of cells require circulatory systems todeliver oxygen to tissues as well as specialized proteinsto carry oxygen around the body. In vertebratesthe oxygen-carrying proteins are haemoglobin andmyoglobin. Haemoglobin is located within red bloodcells and its primary function is to convey oxygen fromthe lungs to regions deficient in oxygen. This includesparticularly the skeletal muscles of the body whereoxygen consumption as a result of mechanical workrequires continuous supplies of oxygen for optimalactivity. In skeletal muscle myoglobin functions as anoxygen storage protein.
For both haemoglobin and myoglobin the oxygen-carrying capacity arises through the presence of aheme group. The heme group is an organic carbonskeleton called protoporphyrin IX made up of fourpyrrole groups that chelate iron at the centre of thering (Figure 3.36). The heme group gives myoglobinand haemoglobin a characteristic colour (red) thatis reflected by distinctive absorbance spectra thatshow intense peaks around 410 nm due to the Fe-protoporphyrin IX group. The iron group is foundpredominantly in the ferrous state (Fe2+) and onlythis form binds oxygen. Less frequently the irongroup is found in an oxidized state as the ferric iron

THE GLOBIN FAMILY AND THE ROLE OF QUATERNARY STRUCTURE IN MODULATING ACTIVITY 67
N N
NN
M
V
M
M
M
V
P
P
P
P
Fe
NN
N N
M
V
M
M
M
V
HH
Figure 3.36 The structure of protoporphyrin IX andheme (Fe-protoporphyrin-IX)
15
10
500 550 600
5
Figure 3.37 The absorbance spectra of oxy, met anddeoxyhaemoglobin. The dotted line is the spectrum ofdeoxyHB, the solid line is metHb whilst the dashedline is oxyHb. DeoxyHb shows a single maximum at550 nm that shifts upon oxygen binding to give twopeaks at 528 and 563 nm. The extinction coefficientsassociated with these peaks are ε555 = 12.5 mM cm−1,ε541 = 13.5 mM cm−1, and ε576 = 14.6 mM cm−1
(Fe3+), a state not associated with oxygen binding.The binding of oxygen (and other ligands) to thesixth coordination site of iron in the heme ring isaccompanied by distinctive changes to absorbancespectra (Figure 3.37).
The structure of myoglobin
Detailed crystallographic studies of myoglobin pro-vided the first three-dimensional picture of a proteinand established many of the ground rules governingsecondary and tertiary structure described earlier inthis chapter. Myoglobin was folded into an extremelycompact single polypeptide chain with dimensions of∼4.5 × 3.5 × 2.5 nm. There was no free space on theinside of the molecule with the polypeptide chainfolded efficiently by changing directions regularly tocreate a compact structure. Approximately 80 percentof all residues were found in α helical conformations.The structure of myoglobin (Figure 3.38) provided thefirst experimental verification of the α helix in pro-teins and confirmed that the peptide group was planar,found in a trans configuration and with the dimensionspredicted by Pauling.
Eight helices occur in myoglobin and these helicesare labelled as A, B, C etc up to the eighth helix,
Figure 3.38 The structure of myoglobin showingα helices and the heme group. Helix A is in theforeground along with the N terminal of the protein.The terminology used for myoglobin labels the helicesA–H and residues within helices as F8, A2 etc.

68 THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
helix H. Frequently, although not in every instance, thehelices are disrupted by the presence of proline residuesas for example occurs between the B and C helices, theE and F helices and the G and H helices. Even in low-resolution structures of myoglobin (the first structureproduced had a resolution of ∼6 A) the position of theheme group is easily discerned from the location of theelectron-dense iron atom.
The heme group was located in a crevice surroundedalmost entirely by non-polar residues with the excep-tion of two heme propionates that extended to thesurface of the molecule making contact with solventand two histidine residues, one of which was in con-tact with the iron and was termed the proximal (F8)histidine (Figure 3.39). The imidazole side chain of F8provided the fifth ligand to the heme iron. A second his-tidine, the distal histidine (E7), was more distant fromthe Fe centre and did not provide the sixth ligand. Anobvious result of this arrangement was an asymmetricconformation for the iron where it was drawn out ofthe heme plane towards the proximal histidine.
The structure of myoglobin confirmed the parti-tioning of hydrophobic and hydrophilic side chains.The interior of the protein and the region surroundingthe heme group consisted almost entirely of non-polarresidues. Here leucine, phenylalanine and valine werecommon whilst hydrophilic side chains were locatedon the exterior or solvent accessible surface.
CH2
N
N
CH2N
N
Fe
O2
Heme plane
Figure 3.39 Diagram of the proximal and distalhistidine side chains forming part of the oxygenbinding site of myoglobin with the ferrous iron pulledout of the plane of the heme ring. The vacant sixthcoordination position is the site of oxygen binding
There are approximately 200 structures, includingmutants, of myoglobin deposited in the PDB butthe three most important structures of relevance tomyoglobin are those of the oxy and deoxy formstogether with ferrimyoglobin (metmyoglobin) wherethe iron is present as the ferric state. The structuresof all three forms turned out to be remarkably similarwith one exception located in the vicinity of the sixthcoordination site. In oxymyoglobin a single oxygenmolecule was found at the sixth coordination site. Inthe deoxy form this site remained vacant whilst inmetmyoglobin water was found in this location. Theunique geometry of the iron favours its maintenancein the reduced state but oxygen binding resulted inmovement of the iron approximately 0.2 A towardsthe plane of the ring (Figure 3.40). An analysis ofthe heme binding site emphasizes how the propertiesof the heme group are modulated by the polypeptide.The identical ferrous heme group in cytochromesundergoes oxidation in the presence of oxygen toyield the ferric (Fe3+) state, other enzymes such ascatalase or cytochrome oxidase convert the oxygeninto hydrogen peroxide and water respectively but inglobins the oxygen is bound with the iron remaining inthe ferrous state.
Myoglobin has a very high affinity for oxygen andthis is revealed by binding curves or profiles that recordthe fractional saturation versus the concentration ofoxygen (expressed as the partial pressure of oxygen,pO2). The oxygen-binding curve of myoglobin is
CH2
NH
N
CH2N
N
Fe O
O
Heme plane
Figure 3.40 Binding of oxygen in oxymyoglobin andthe movement of the iron into the approximate hemeplane
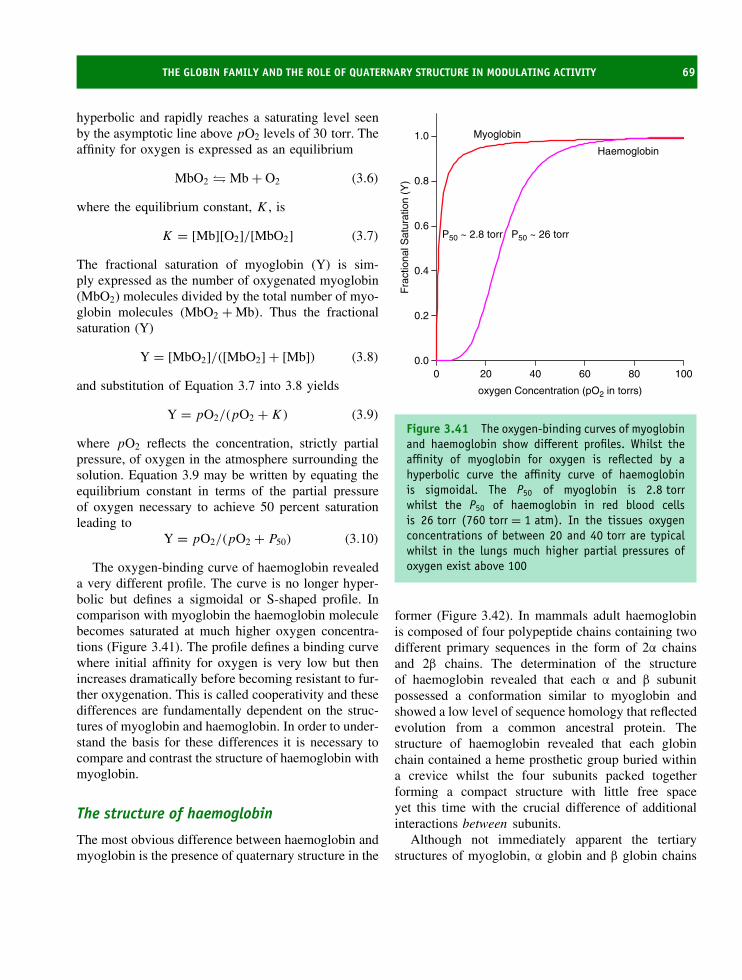
THE GLOBIN FAMILY AND THE ROLE OF QUATERNARY STRUCTURE IN MODULATING ACTIVITY 69
hyperbolic and rapidly reaches a saturating level seenby the asymptotic line above pO2 levels of 30 torr. Theaffinity for oxygen is expressed as an equilibrium
MbO2 � Mb + O2 (3.6)
where the equilibrium constant, K , is
K = [Mb][O2]/[MbO2] (3.7)
The fractional saturation of myoglobin (Y) is sim-ply expressed as the number of oxygenated myoglobin(MbO2) molecules divided by the total number of myo-globin molecules (MbO2 + Mb). Thus the fractionalsaturation (Y)
Y = [MbO2]/([MbO2] + [Mb]) (3.8)
and substitution of Equation 3.7 into 3.8 yields
Y = pO2/(pO2 + K) (3.9)
where pO2 reflects the concentration, strictly partialpressure, of oxygen in the atmosphere surrounding thesolution. Equation 3.9 may be written by equating theequilibrium constant in terms of the partial pressureof oxygen necessary to achieve 50 percent saturationleading to
Y = pO2/(pO2 + P50) (3.10)
The oxygen-binding curve of haemoglobin revealeda very different profile. The curve is no longer hyper-bolic but defines a sigmoidal or S-shaped profile. Incomparison with myoglobin the haemoglobin moleculebecomes saturated at much higher oxygen concentra-tions (Figure 3.41). The profile defines a binding curvewhere initial affinity for oxygen is very low but thenincreases dramatically before becoming resistant to fur-ther oxygenation. This is called cooperativity and thesedifferences are fundamentally dependent on the struc-tures of myoglobin and haemoglobin. In order to under-stand the basis for these differences it is necessary tocompare and contrast the structure of haemoglobin withmyoglobin.
The structure of haemoglobin
The most obvious difference between haemoglobin andmyoglobin is the presence of quaternary structure in the
oxygen Concentration (pO2 in torrs)
0 20 40 60 80 100
Fra
ctio
nal S
atur
atio
n (Y
)
0.0
0.2
0.4
0.6
0.8
1.0
Haemoglobin
Myoglobin
P50 ~ 26 torrP50 ~ 2.8 torr
Figure 3.41 The oxygen-binding curves of myoglobinand haemoglobin show different profiles. Whilst theaffinity of myoglobin for oxygen is reflected by ahyperbolic curve the affinity curve of haemoglobinis sigmoidal. The P50 of myoglobin is 2.8 torrwhilst the P50 of haemoglobin in red blood cellsis 26 torr (760 torr = 1 atm). In the tissues oxygenconcentrations of between 20 and 40 torr are typicalwhilst in the lungs much higher partial pressures ofoxygen exist above 100
former (Figure 3.42). In mammals adult haemoglobinis composed of four polypeptide chains containing twodifferent primary sequences in the form of 2α chainsand 2β chains. The determination of the structureof haemoglobin revealed that each α and β subunitpossessed a conformation similar to myoglobin andshowed a low level of sequence homology that reflectedevolution from a common ancestral protein. Thestructure of haemoglobin revealed that each globinchain contained a heme prosthetic group buried withina crevice whilst the four subunits packed togetherforming a compact structure with little free spaceyet this time with the crucial difference of additionalinteractions between subunits.
Although not immediately apparent the tertiarystructures of myoglobin, α globin and β globin chains

70 THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Figure 3.42 The quaternary structure of haemoglobinshowing the four subunits each with a heme grouppacking together. The heme groups are shown in redwith the α globin chain in purple and the β globinchain in green
are remarkably similar despite differences in primarysequence (Figure 3.43). With similar tertiary structuresfor myoglobin and the α and β chains of haemoglobinit is clear that the different patterns of oxygenbinding must reflect additional subunit interactions inhaemoglobin. In other words cooperativity arises as aresult of quaternary structure.
The pattern of oxygenation in haemoglobin is per-fectly tailored to its biological function. At highconcentrations of oxygen as would occur in thelungs (pO2 > 100 torr or ∼0.13 × atmospheric pres-sure) both myoglobin and haemoglobin become satu-rated with oxygen. Myoglobin would be completelyoxygenated carrying 1 molecule of oxygen per pro-tein molecule. Complete oxygenation of haemoglobinwould result in the binding of 4 oxygen molecules.However, as the concentration of oxygen decreasesbelow 50 torr myoglobin and haemoglobin react differ-ently. Myoglobin remains fully saturated with oxygenwhilst haemoglobin is no longer optimally oxygenated;sites on haemoglobin are only 50 percent occupied at∼26 torr whereas myoglobin exhibits a much lower P50
of ∼3 torr.
Figure 3.43 Superposition of the polypeptide chainsof myoglobin, α globin and β globin. Structuralsimilarity occurs despite only 24 out of 141 residuesshowing identity. The proximal and distal histidinesare conserved along with several residues involved inthe heme pocket structure such as leucine (F4) and aphenylalanine between C and D helices
The physiological implications of these bindingproperties are profound. In the lungs haemoglobinis saturated with oxygen ready for transfer aroundthe body. However, when red blood cells (contain-ing high concentrations of haemoglobin) reach theperipheral tissues, where the concentration of oxygenis low, unloading of oxygen from haemoglobin occurs.The oxygen released from haemoglobin is immediatelybound by myoglobin. The binding properties of myo-globin allow it to bind oxygen at low concentrationsand more importantly facilitate the transport of oxy-gen from lungs to muscles or from regions of highconcentrations to tissues with much lower levels.
The cooperative binding curve of haemoglobinarises as a result of structural changes in conformationthat occur upon oxygenation. In the deoxy form thesixth coordination site is vacant and the iron is drawnout of the heme plane towards the proximal histidine.

THE GLOBIN FAMILY AND THE ROLE OF QUATERNARY STRUCTURE IN MODULATING ACTIVITY 71
Figure 3.44 The spin state changes that occur in thedeoxy and oxy ferrous forms of haemoglobin showingthe population of the d orbitals (xy, xz, yz, z2, x2-y2) by electrons. The deoxy form is high spin (S = 2)ferrous state with an ionic radius too large to fitbetween the four tetra pyrrole ring nitrogens. The lowspin (S = 1/2) state of ferrous iron with a differentdistribution of orbitals has a smaller radius and movesinto the plane of the heme
Oxygenation results in conformational change medi-ated by oxygen binding to the iron. The events aretriggered by changes in the electronic structure of theiron as it shifts from a high spin ferrous centre inthe deoxy form to a low spin state when oxygenated.Changes in spin state are accompanied by reorgani-zation of the orbital structure and decreases in ionicradius that allows the iron to move closer to the planeof the heme macrocycle (Figure 3.44).
Changes at the heme site are accompanied by reori-entation of helix F, the helix containing the proximalhistidine, and results in a shift of approximately 1 Ato avoid unfavourable contact with the heme group.The effect of this conformational change is transmittedthroughout the protein but particularly to interactionsbetween subunits. Oxygen binding causes the disrup-tion of ionic interactions between subunits and triggersa shift in the conformation of the tetramer from thedeoxy to oxy state. By examining the structure ofhaemoglobin in both the oxy and deoxy states manyof these interactions have been highlighted and anothersignificant milestone was a structural understanding for
the cooperative oxygenation of haemoglobin proposedby Perutz in 1970.
The mechanism of oxygenation
Perutz’s model emphasized the link between coopera-tivity and the structure of the tetramer. The cooperativecurve derives from conformational changes exhibitedby the protein upon oxygenation and reflects the effectof two ‘competing’ conformations. These conforma-tions are called the R and T states and the termsderive originally from theories of allosteric transitionsdeveloped by Monod, Wyman and Changeaux (MWC).According to this model haemoglobin exists in an equi-librium between the R and T states where the R statesignifies a ‘relaxed’ or active state whilst the T signifiesa ‘tense’ or inactive form.
The deoxy state of haemoglobin is resistant tooxygenation and is equated with the inactive, T state.In contrast the oxy conformation is described as theactive, R state. In this model haemoglobin is anequilibrium between R and T states with the observedoxygen-binding curve reflecting a combination ofthe binding properties of each state. The shift inequilibrium between the R and T states is envisagedas a two state or concerted switch with only weakand strong binding states existing and intermediatestates containing a mixture of strong and weak bindingsubunits specifically forbidden. This form of the MWCmodel could be described as an ‘all or nothing’ schemebut is more frequently termed the ‘concerted’ model. Ingeneral detailed structural analysis has retained manyof the originally features of the MWC model.
At a molecular level the relative orientation betweenthe α1β1 and α2β2 dimers differed in the oxy and deoxystates. Oxygenation leads to a shift in orientation of∼15◦ and a translation of ∼0.8 A for one pair ofα/β subunits relative to the other. It arises as a resultof changes in conformation at the interfaces betweenthese pairs of subunits. One of the most importantregions involved in this conformational switch centresaround His97 at the boundary between the F and Ghelices of the β2 subunit. This region of the β subunitmakes contact with the C helix of the α1 subunitand lies close to residue 41. In addition the foldingof the β2 subunit leads to the C terminal residue,His146, being situated above the same helix (helix C)

72 THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Figure 3.45 Interactions at the α1β2 interface in deoxyhaemoglobin and the changes that occur after oxygenation.In the T state His97 of the β2 subunit fits next to Thr41 of the α1 chain. In the R state conformation changesresult in this residue lying alongside Thr38. Although a salt bridge between Asp99 and Tyr42 is broken in the T > Rtransition a new interaction between Asn102 and Asp94 results. There is no detectable intermediate between theT and R states and the precisely interacting surfaces allow the two subunits to move relative to each other easily.(Reproduced with permission from Voet, D., Voet, J.G & Pratt, C.W., Fundamentals of Biochemistry. Chichester, JohnWiley & Sons, 1999.)
and it forms a series of hydrogen bonds and saltbridges with residues in this region (38–44). In thedeoxy state His 146 is restrained by an interactionwith Asp94. As a result of symmetry an entirelyanalogous set of interactions occur at the α2β1 interface(Figure 3.45).
In the T state the iron is situated approximately0.6 A out of the plane of the heme ring as part ofa dome directed towards HisF8. Oxygenation causesa shortening of the iron–porphyrin bonds by 0.1 Adue to electron reorganization and causes the ironto move into the plane dragging the proximal Hisresidue with it. Movement of the His residue by 0.6 Ais unfavourable on steric grounds and as a resultthe helix moves to compensate for changes at theheme centre. Helix movement triggers conformationalchanges throughout the subunits and more importantlybetween the subunits. Other important conformationalswitches include disruption of a network of ionpairs within and between subunits. In particular ionicinteractions of the C terminal residue of the α subunit
(Arg141) with Asp126 and the amino terminal of the α1subunit and that of the β subunit (His146) with Lys40(α) and Asp94(β) are broken upon oxygenation. Sincethese interactions stabilize the T state their removaldrives the transition towards the R state.
Cooperativity arises because structural changeswithin one subunit are coupled to conformationalchanges throughout the tetramer. In addition the natureof the switch means that intermediate forms cannotoccur and once one molecule of oxygen has bound toa T state subunit converting it to the R state all othersubunits are transformed to the R state. As a resultthe remaining subunits rapidly bind further oxygenmolecules, leading to the observed shape of the oxygenbinding curve seen in Figure 3.46.
Allosteric regulation and haemoglobin
The new properties of haemoglobin extend furtherthan the simple binding of oxygen to the protein.Haemoglobin’s affinity for oxygen is modulated by

THE GLOBIN FAMILY AND THE ROLE OF QUATERNARY STRUCTURE IN MODULATING ACTIVITY 73
Oxygen concentration (pO2 in torrs)
0 20 40 60 80 100
Fra
ctio
nal S
atur
atio
n (Y
)
0.0
0.2
0.4
0.6
0.8
1.0
Hamoglobin
T form
R form
Tissues Lungs
Figure 3.46 Oxygen binding profiles of the R andT states of haemoglobin. The R or active formof haemoglobin binds oxygen readily and shows ahyperbolic curve. The T or inactive state is resistantto oxygenation but also shows a hyperbolic bindingcurve with a higher P50. The observed binding profileof haemoglobin reflects the sum of affinities of the Rand T forms for oxygen and leads to the observed P50
of ∼26 torr for isolated haemoglobin. Shown in yellowis the approximate range of partial pressures of oxygenin tissues and lungs
small molecules (effectors) as part of an importantphysiological control process. This mode of regulationis called allostery. An allosteric modulator binds toa protein altering its activity and in the case ofhaemoglobin the effector alters oxygen binding. Themost important allosteric regulator of haemoglobin is2,3-bisphosphoglycerate (2,3 BPG) (Figure 3.47).1
The allosteric modulator of haemoglobin, 2,3 BPG,raises the P50 for oxygen binding in haemoglobinfrom 12 torr in isolated protein to 26 torr observed forhaemoglobin in red blood cells (Figure 3.48). Indeedthe existence of an allosteric modulator of haemoglobinwas first suspected from careful comparisons of theoxygen-binding properties of isolated protein with thatfound in red blood cells. In erythrocytes 2,3 BPG
1Sometimes called 2,3 DPG = 2,3-diphosphoglycerate.
C
O
H H
C
C
H O
OO−
P
P
O−
O
O−
O
O−O−
Figure 3.47 2,3-Bisphosphoglycerate is a negativelycharged molecule
Oxygen Affinity
0 20 40 60 80 100
Fra
ctio
nal S
atur
atio
n
0.0
0.2
0.4
0.6
0.8
1.0
+2,3 BPG
−2,3 BPG
Figure 3.48 Modulation of oxygen binding propertiesof haemoglobin by 2,3 BPG. The 2,3 BPG modulatesoxygen binding by haemoglobin raising the P50 from12 torr in isolated protein to 26 torr observed forhaemoglobin in red blood cells. This is shown by therespective oxygen affinity curves
is found at high concentrations as a result of highlyactive glycolytic pathways and is formed from thekey intermediates 3-phosphoglycerate or 1,3 bispho-sphoglycerate. The result is to create concentrations of2,3 BPG approximately equimolar with haemoglobinwithin cells. In the erythrocyte this results in 2,3 BPGconcentrations of ∼5 mM.

74 THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
The effect arises by differential binding – theassociation constant (Ka) for 2,3 BPG for deoxy-haemoglobin is ∼4 × 104 M−1 compared with a valueof ∼300 M−1 for the oxy form. In other words it sta-bilizes the T state, which has low affinity for oxygenwhilst binding to the oxy state is effectively inhibitedby the presence of bound oxygen. The effect of 2,3BPG on the oxygen-binding curve of haemoglobin isto shift the profile to much higher P50 (Figure 3.48).Detailed analysis of the structures of the oxy anddeoxy states showed that 2,3 BPG bound in the cen-tral cavity of the deoxy form and allowed a molecularinterpretation for its role in allostery. In the centre ofhaemoglobin between the four subunits is a small cav-ity lined with positively charged groups formed fromthe side chains of His2, His143 and Lys82 of the β
subunits together with the two amino groups of thefirst residue of each of these chains (Val1). The resultof this charge distribution is to create a strong bind-ing site for 2,3 BPG. In oxyhaemoglobin the bindingof oxygen causes conformational changes that lead toa closure of the allosteric binding site. These changesarise as a result of subunit movement upon oxygenationand decrease the cavity size to prevent accommodationof 2,3 BPG. Both oxygen and 2,3 BPG are reversiblybound ligands yet each binds at separate sites and leadto opposite effects on the R→T equilibrium.
2,3 BPG is not the only modulator of oxygenbinding to haemoglobin. Oxygenation of haemoglobincauses the disruption of many ion pairs and leads to therelease of ∼0.6 protons for each oxygen bound. Thiseffect was first noticed in 1904 by Christian Bohr and isseen by increase oxygen binding with increasing pH ata constant oxygen level (Figure 3.49). The Bohr effectis of utmost importance in the physiological delivery ofoxygen from the lungs to respiring tissues and also inthe removal of CO2 produced by respiration from thesetissues and its transport back to the lungs. Within theerythrocyte CO2 is carried as bicarbonate as a resultof the action of carbonic anhydrase, which rapidlycatalyses the slow reaction
CO2 + H2O � H+ + HCO−3 (3.11)
In actively respiring tissues where pO2 is low theprotons generated as a result of bicarbonate formationfavour the transition from R > T and induce the
0 20 40 60 80 100F
ract
iona
l Sat
urat
ion
(Y)
0.0
0.2
0.4
0.6
0.8
1.0
Increasing pH
pO2 (torr)
Figure 3.49 The Bohr effect. The oxygen affinity ofhaemoglobin increases with increasing pH
haemoglobin to unload its oxygen. In contrast inthe lungs the oxygen levels are high and binding tohaemoglobin occurs readily with the disruption of Tstate ion pairs and the formation of the R state. Afurther example of the Bohr effect is seen in veryactive muscles where an unavoidable consequence ofhigh rates of respiration is the generation of lactic acid.The production of lactic acid will cause a loweringof the pH and an unloading of oxygen to thesedeficient tissues. Carbon dioxide can also bind directlyto haemoglobin to form carbamates and arises as aresult of reaction with the N terminal amino groupsof subunits. The T form (or deoxy state) binds morecarbon dioxide as carbamates than the R form. Againthe physiological consequences are clear. In capillarieswith high CO2 concentrations the T state is favouredleading to an unloading of oxygen from haemoglobin.
Immunoglobulins
The immunoglobulins are a large group of proteinsfound in vertebrates whose unique function is tobind foreign substances invading a host organism. By

IMMUNOGLOBULINS 75
breaking the physical barrier normally represented byskin or mucous membranes pathogens enter a system,but once this first line of defence is breached animmune response is normally started. In most instancesthe immune response involves a reaction to bacteriaor viruses but it can also include isolated proteinsthat break the skin barrier. Collectively these foreignsubstances are termed antigens.
The immune response is based around two systems.A circulating antibody system based on B cellssometimes called by an older name of humoral immuneresponse. The cells were first studied in birds wherethey matured from a specialized pocket of tissueknown as the bursa of Fabricius. However, there isno counterpart in mammals and B cells are derivedfrom the bone marrow where the ‘B’ serves equallywell to identify its origin. B cells have specificproteins on their surfaces and reaction with antigenactivates the lymphocyte to differentiate into cellscapable of antibody production and secretion. Thesecells secrete antibodies binding directly to antigensand acting as a marker for macrophages to destroy theunwanted particle.
This system is supported by a second cellularimmune response based around T lymphocytes. The Tcells are originally derived from the thymus gland andthese cells also contain molecules on their membranesurfaces that recognize specific antigens and assist intheir destruction.
Early studies of microbial infection identified thatall antigens are met by an immune response thatinvolves a collection of heterogeneous proteins knownas antibodies. Antibodies are members of a largerimmunoglobulin group of proteins and an importantfeature of the immune response is its versatility inresponding to an enormous range of antigens. Inhumans this response extends from before birth andincludes our lifelong ability to fight infection. Onefeature of the immune response is ‘memory’. Thisproperty is the basis of childhood vaccination and arisesfrom an initial exposure to antigen priming the systemso that a further exposure leads to the rapid productionof antibodies and the prevention of disease. From abiological standpoint the immune response representsan enigma. It is a highly specific recognition systemcapable of identifying millions of diverse antigens yetit retains an ability to ‘remember’ these antigens over
considerable periods of time (years). The operation ofthe immune system was originally based around theclassic observations reported by Edward Jenner at theend of the 18th century. Jenner recognized that milkmaids were frequently exposed to a mild disease knownas cowpox, yet rarely succumbed to the much moreserious smallpox. Jenner demonstrated that cowpoxinfection, a relatively benign disease with completerecovery taking a few days, conferred immunityagainst smallpox. Exposure to cowpox triggered animmune response that conferred protection againstthe more virulent forms of smallpox, at that timerelatively common in England. It was left to othersto develop further the ideas of vaccination, notablyLouis Pasteur, but for the last 100 years vaccinationhas been a key technique in fighting many diseases.The basis to vaccination lies in the working of theimmune system.
The mechanism of specific antibody productionremained puzzling but a considerable advance inthis area arose with the postulation of the clonalselection theory by Neils Jerne and Macfarlane Burnetin 1955. This theory is now widely supported andenvisages stem cells in the bone marrow differentiatingto become lymphocytes each capable of producing asingle immunoglobulin type. The immunoglobulin isattached to the outer surfaces of B lymphocytes andwhen an antigen binds to these antibodies replicationof the cell is stimulated to produce a clone. The result isthat only cells experiencing contact with an antigen arestimulated to replicate. Within the group of cloned Bcells two distinct populations are identified. EffectorB cells located in the plasma will produce solubleantibodies. These antibodies are comparable to thosebound to the surface of B cells but lack membrane-bound sequences that anchor the antibodies to thelipid bilayer. The second group within the cloned Bcell populations are called ‘memory cells’. These cellspersist for a considerable length of time even after theremoval of antigen and allow the rapid production ofantibodies in the event of a second immune reaction.The clonal selection theory was beautiful in that itexplained how individuals distinguish ‘self’ and ‘non-self’. During embryonic development immature B cellsencounter ‘antigens’ on the surfaces of cells. These Bcells do not replicate but are destroyed thereby remov-ing antibodies that would react against the host’s own

76 THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
proteins. At birth the only B cells present are those thatare capable of producing antibodies against non-selfantigens.
Immunoglobulin structure
Despite the requirement to recognize enormousnumbers of potential antigens all immunoglobulinmolecules are based around a basic pattern that waselucidated by the studies of Rodney Porter and GeraldEdelman. Porter showed that immunoglobulin G (IgG)with a mass of ∼150 000 could be split into threefragments each retaining biological activity throughthe action of proteolytic enzymes such as papain.Normal antibody contains two antigen-binding sites(Figure 3.50) but after treatment with papain twofragments each binding a single antigen molecule wereformed, along with a fragment that did not bind antigenbut was necessary for biological function. The twofragments were called the Fab (F = fragment, ab =antigen binding) and Fc (c = crystallizable) portions.The latter’s name arose from the fact that itshomogeneous composition allowed the fragment to becrystallized in contrast to the Fab fragments.
The IgG molecule is dissociated into two distinctpolypeptide chains of different molecular weight viathe action of reducing agents. These chains were calledthe heavy (H) and light (L) polypeptides (Figure 3.51).
IgG
Antigen
Figure 3.50 Schematic diagram showing bivalentinteraction between antigen and antibody (IgG) andextended lattice
S
S-S
L LH H
S-S
S
Figure 3.51 The subunit structure of IgG is H2L2
Further studies by Porter revealed that IgG wasreconstituted by combining 2H and 2L chains, anda model for immunoglobulin structure was proposedenvisaging each L chain binding to the H chain viadisulfide bridges, whilst the H chains were similarlylinked to each other. The results of papain digestionwere interpretable by assuming cleavage of the IgGmolecule occurred on the C terminus side of thedisulfide bridge linking H and L chains. The Fab
region therefore contained both L chains and the aminoterminal region of 2 H chains whilst the Fc regioncontained only the C terminal half of each H chain.
The amino acid sequence revealed not only theperiodic repetition of intrachain disulfide bonds in theH and L chains but also the existence of regions ofsequential homology between the H and L chains andwithin the H chain itself. Portions of the molecule, nowknown as the variable regions in the H and L chains,showed considerable sequence diversity, whilst constantregions of the H chain were internally homologous,showing repeating units. In addition the constant regionswere homologous to regions on the L chain. Thispattern of organization immediately suggested that

IMMUNOGLOBULINS 77
immunoglobulins were derived from simpler antibodymolecules based around a single domain of ∼100residues, and by a process of gene duplication thestructure was assimilated to include multiple domainsand chains.
Although the organization of IgG molecules isremarkably similar antigen binding is promoted throughthe use of sequence variability. These differences arenot distributed uniformly throughout the H and L chainsbut are localized in specific regions (Figure 3.53). Theregions of greatest sequence variability lie in the Nterminal segments of the H and L chains and are calledthe VL or VH regions. In the L chain the region extendsfor ∼110 residues and is accompanied by a more highlyconserved region of the same size called the constantregion of CL. In the H chain the VH region extends for
Figure 3.52 The immunoglobulin fold containing asandwich formed by two antiparallel sets of β strands.Normally seven β strands make up the two sets. In thisdomain additional sheet structures occur and smallregions of a helix are found shown in blue. Theimportant loop and turn regions are shown in grey.In the IgG light chain this fold would occur twice;once in the variable region and once again in theconstant region. In the heavy chain it occurs fourtimes
∼110 residues whilst the remainder of the sequence isconserved, the CH region. The CH region is subdividedinto three homologous domains: CH1, CH2 and CH3.
Fractionation studies identified antigen-binding sitesat the N terminal region of the H and L chainsin a region composed of the VH and VL domainswhere the ability to recognize antigens is based on thesurface properties of these folds. Within the VH and VL
domains the sequences of highest variability are threeshort segments known as hypervariable sequences andcollectively known as complementarity determiningregions (CDRs). They bind antigen and by arrangingthese CDRs in different combinations cells generatevast numbers of antibodies with different specificity.
The light chain consists of two discrete domainsapproximately 100–120 residues in length whilst theheavy chain is twice the size and has four suchdomains. Each domain is characterized by commonstructural topology known as the immunoglobulinfold and is repeated within the IgG molecule. Theimmunoglobulin fold is a sandwich of two sheets ofantiparallel β strands each strand linked to the nextvia turns or large loop regions, with the sheets usuallylinked via a disulfide bridge. The immunoglobulin foldis found in other proteins operating within the immunesystem but is also noted in proteins with no obviousfunctional similarity (Figure 3.52).
The hypervariable regions are located in turns orloops of variable length that link together the differentβ strand elements. Collectively these regions formthe antigen-binding site at the end of each arm ofthe immunoglobulin molecule and insight into theinteraction between antibody and antigen has beengained from crystallization of an Fab fragment with henegg white lysozyme.
Raising antibodies against lysozyme generated sev-eral antibody populations – each antibody recognizinga different site on the surface of the protein. These sitesare known as epitopes or antigenic determinants. Bystudying one antibody–antigen complex further bind-ing specificity was shown to reside in the contact ofthe VL and VH domains with lysozyme at the Fab
tip (Figure 3.54). Interactions based around hydrogenbonding pairs involved at least five residues in the VL
domain (Tyr32, Tyr50, Thr53, Phe91, Ser93) and fiveresidues in the VH domain (Gly53, Asp54, Asp100,Tyr101, Arg102). These donor and acceptors hydrogen

78 THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Figure 3.53 The structure and organization of IgG. The location of VL, VH and CH within the H and L chains of anantibody together with a crystal structure determined for the isolated Fab region (PDB: 7FAB). The H chain is shownwith yellow strands whilst the L chain has strands shown in green. In each chain two immunoglobulin folds are seen
bonded to two groups of residues on lysozyme with theVL domain interacting with Asp18, Asn19 and Gln121whilst those on the VH domain formed hydrogen bondswith 7 residues on the surface of lysozyme (Gly22,Ser24, Asn27, Gly117, Asp119, Val120, and Gln121).
The epitope on the surface of lysozyme is made upof two non-contiguous groups of residues (18–27 and117–125) found on the antigen’s surface. All six hyper-variable regions participated in epitope recognition, buton the surface of lysozyme Gln121 was particularlyimportant protruding away from the surface and form-ing a critical residue in the formation of a high-affinity
antibody–antigen complex. The antibody forms a cleftthat surrounds Gln121 with a hydrogen bond formedbetween the side chain and Tyr101 on the antibody(Figure 3.55). Elsewhere, van der Waals, hydrophobicand electrostatic interactions play a role in binding theother regions of the interacting surfaces between anti-body and antigen.
Five different classes of immunoglobulins (Ig)have been recognized called IgA, IgD, IgE, IgG andIgM. These classes of immunoglobulins differ in thecomposition of their heavy chains whilst the light chainis based in all cases around two sequences identified

IMMUNOGLOBULINS 79
Figure 3.54 Interaction between monoclonal antibody fragment Fab and lysozyme. The lysozyme is shown in blueon right and the prominent side chain of Gln121 is shown in red (PDB:1FDL)
Figure 3.55 Interaction between Gln121 and residuesformed by a pocket on the surface of the antibody ina complex formed between lysozyme and a monoclonalantibody
by the symbols κ and λ. The heavy chains are calledα, δ, ε, γ and µ by direct analogy to the parent protein(Table 3.6).
As a consequence of remarkable binding affini-ties exemplified by dissociation constants between104 –1010 M antibodies have been used extensively as
‘probes’ to detect antigen. In the area of clinical diag-nostics this is immensely valuable in detecting infec-tions and many diseases are routinely identified viacross-reaction between antibodies and serum contain-ing antigen. Antibodies are routinely produced todayby injecting purified protein into a subject animal. Theanimal recognizes a protein as foreign and producesantibodies that are extracted and purified from sera.One extension of the use of antibodies as medical andscientific ‘tools’ was pioneered by Cesar Milstein andGeorges Kohler in the late 1970s and has proved pop-ular and informative. This is the area of monoclonalantibody production, and although not described indetail here, the methods are widely used to identifydisease or infection, to specifically identify a singleantigenic site and increasingly as therapeutic agents inthe battle against cancer and other disease states.
The humoral immune response involves the secre-tion of antibodies by B cells and the aggregationof antigen. Aggregation signals to macrophages thatdigestion should occur along with the destruction ofpathogen. The cellular immune response to antigeninvolves different cells and a very different mech-anism. On the surface of cytotoxic T lymphocytessometimes more evocatively called ‘killer T cells’ areproteins whose structure and organization resemblesthe Fab fragments of IgG. T cells identify antigenicpeptide fragments that are bound to a surface pro-tein known as the major histocompatibility complex

80 THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Table 3.6 Chain organization within the different Ig classes together with their approximate mass and biological roles
Class Heavychain
Lightchain
Organization Mass(kDa)
Role
IgA α κ or λ (α2κ2)n or (α2λ2)n 360–720 Found mainly in secretionsIgD δ κ or λ δ2κ2 or δ2λ2 160 Located on cell surfacesIgE ε κ or λ ε2κ2 or ε2λ2 190 Found mainly in tissues. Stimulates
mast cells to release histaminesIgG γ κ or λ γ2κ2 or γ2λ2 150 Activates complement system.
Crosses membranesIgM µ κ or λ (µ2κ2)5 or (µ2λ2)5 950 Early appearance in immune
reactions. Linked to complementsystem and activates macrophages
Cytotoxic T cell
C domain V domain
Membrane
CH
CH
CH
CHCL
VL VH
CL CH
VH VH
CH
CH
VH
CL
CL
VL
VLB lymphocyte cell
Membrane
C
C
C
C
VL
CH
Figure 3.56 The use of the immunoglobulin fold in cell surface receptors by B and T lymphocytes

SUMMARY 81
(MHC). Cytotoxic or killer T cells recognize anti-gen through receptors on their surface (Figure 3.56)and release proteins that destroy the infected cell. TheMHC complex is based around domains carrying theimmunoglobulin fold.
Cyclic proteins
Until recently it was thought that cyclic proteins werethe result of unusual laboratory synthetic reactionswithout any counterparts in biological systems. This isnow known to be untrue and several systems have beenshown to possess cyclic peptides ranging in size fromapproximately 14 residues to the largest cyclic proteincurrently known a highly basic 70 residue proteincalled AS-48 isolated from Enterococcus faecalis S-48. Cyclic proteins are a unique example of tertiarystructure but employ exactly the same principle aslinear proteins with the exception that their endsare linked together (Figure 3.57). In view of thefact that many globular proteins have the N andC terminals located very close together in spacethere is no conceptual reason why the amino andcarboxy terminals should not join together in a furtherpeptide bond.
Cyclic proteins are distinguished from cyclic pep-tides such as cyclosporin. These small peptides havebeen known for a long time and are synthesized bymicroorganisms as a result of multienzyme complexreactions. The latter products contain unusual aminoacid residues often extensively modified and are notthe result of transcription. In contrast, cyclic proteinsare known to be encoded within genomes and appearto have a broad role in host defence mechanisms.
Many cyclic proteins are of plant origin and acommon feature of these proteins appears to betheir size (∼30 residues), a cyclic peptide backbonecoupled with a disulfide rich sequence containing sixconserved cysteine residues and three disulfide bonds.Unusually the disulfides cross to form a knot likearrangement and the cyclic backbone coupled withthe cysteine knot has lead to the recognition of anew structural motif called the CCK motif or cycliccysteine knot. The term cyclotides has been applied tothese proteins. A common feature of all cyclotides istheir derivation from longer precursor proteins in steps
that involve both cleavage and cyclization. Althoughthe gene sequences for the precursors are known theputative cleaving and cyclizing enzymes have not yetbeen reported. Many of these proteins have assumedenormous importance with the demonstration thatseveral are natural inhibitors of enzymes such as trypsinwhilst others are seen as possible lead compoundsin the development of new pharmaceutical productsdirected against viral pathogens, and in particularanti-HIV activity. Whilst the cyclotide family (seeTable 3.7) appear to have a common structural themebased around the CCK motif and the presence of threeβ strands other cyclic proteins such as bacteriocin AS-48 contain five short helices connected by five shortturn regions that enclose a compact hydrophobic core.Despite different tertiary structure, all of the cyclicproteins are characterized by high intrinsic stability(denaturation only at very high temperatures) as wellas resistance to proteolytic degradation.
Summary
Proteins fold into precise structures that reflect theirbiological roles. Within any protein three levelsof organization are identified called the primary,secondary and tertiary structures, whilst proteins withmore than one polypeptide chain exhibit quaternarylevels of organization.
Primary structure is simply the linear order of aminoacid residues along the polypeptide chain from theN to C terminals. Long polymers of residues cannotfold into any shape because of restrictions placed onconformational flexibility by the planar peptide bondand interactions between non-bonded atoms.
Conformational flexibility along the polypeptidebackbone is dictated by φ and ψ torsion angles.Repetitive values for φ and ψ lead to regular structuresknown as the α helix and β strand. These are elementsof secondary structure and are defined as the spatialarrangement of residues that are close together in theprimary sequence.
The α helix is the most common element of sec-ondary structure found in proteins and is characterizedby dimensions such as pitch 5.4 A, the translation dis-tance 1.5 A, and the number of residues per turn (3.6).A regular α helix is stabilized by hydrogen bonds

82 THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Figure 3.57 The topology of naturally occurringcircular proteins. Three-dimensional structures ofnaturally occurring proteins. Clockwise from the upperleft the proteins are: bacteriocin AS-48 (PDB:1E68)from Enterococcus faecalis, microcin J25 (PDB:1HG6)from E. coli, MCoTI-II (PDB:1HA9, 1IB9) from bittermelon seeds, RTD-1 (PDB:1HVZ) from the leukocytesof Rhesus macaques, kalataB1 (PDB:1KAL) from severalplants of the Rubiaceae and Violaceae plant families,and SFTI-1 (PDB:1SFI, 1JBL) from the seeds of thecommon sunflower. Disulfide bonds are shown inyellow. (Reproduced with permission from Trabi, M., &Craik, D.J. Trends Biochem. Sci. 2002, 27, 132–138.Elsevier)
orientated parallel to the helix axis and formed betweenthe CO and NH groups of residues separated by fourintervening residues.
In contrast the β strand represents an extendedstructure as indicated by the pitch distance ∼7 A,the translation distance 3.5 A and fewer residues perturn (2). Strands have the ability to hydrogen bondwith other strands to form sheets – collections of β
strands stabilized via inter-strand hydrogen bonding.Numerous variations on the basic helical and strandstructures are found in proteins and truly regular orideal conformations for secondary structure are rare.
Tertiary structure is formed by the organizationof secondary structure into more complex topology
or folds by interaction between residues (side chainand backbone) that are often widely separated in theprimary sequence.
Several identifiable folds or motifs exist withinproteins and these units are seen as substructures withina protein or represent the whole protein. Examplesinclude the four-helix bundle, the β barrel, the β helix,the HTH motif and the β propeller. Proteins can now beclassified according to their tertiary structures and thishas led to the description of proteins as all α, α + β andα/β. The recognition that proteins show similar tertiarystructures has led to the concept of structural homologyand proteins grouped together in related families.
Tertiary structure is maintained by the magnitudeof favourable interactions outweighing unfavourableones. These interactions include covalent and morefrequently non-covalent interactions. A covalent bondformed between two thiol side chains results in adisulfide bridge, but more common stabilizing forcesinclude charged interactions, hydrophobic forces, vander Waals interactions and hydrogen bonding. Theseinteractions differ significantly in their strength andnumber.
Quaternary structure is a property of proteinswith more than one polypeptide chain. DNA bindingproteins function as dimers with dimerization the resultof specific subunit interaction. Haemoglobin is theclassic example of a protein with quaternary structurecontaining 2α,2β subunits.
Proteins with more than one chain may exhibitallostery; a modulation of activity by smaller effectormolecules. Haemoglobin exhibits allostery and this isshown by sigmoidal binding curves. This curve isdescribed as cooperative and differs from that shownby myoglobin. Oxygen binding changes the structure ofone subunit facilitating the transition from deoxy to oxystates in the remaining subunits. Historically the studyof the structure of haemoglobin provided a platformfrom which to study larger, more complex, proteinstructures together with their respective functions.
One such group of proteins are the immunoglobu-lins. These proteins form the body’s arsenal of defencemechanisms in response to foreign macromolecules andare collectively called antibodies. All antibodies arebased around a Y shaped molecule composed of twoheavy and two light chains held together by covalentand non-covalent interactions.
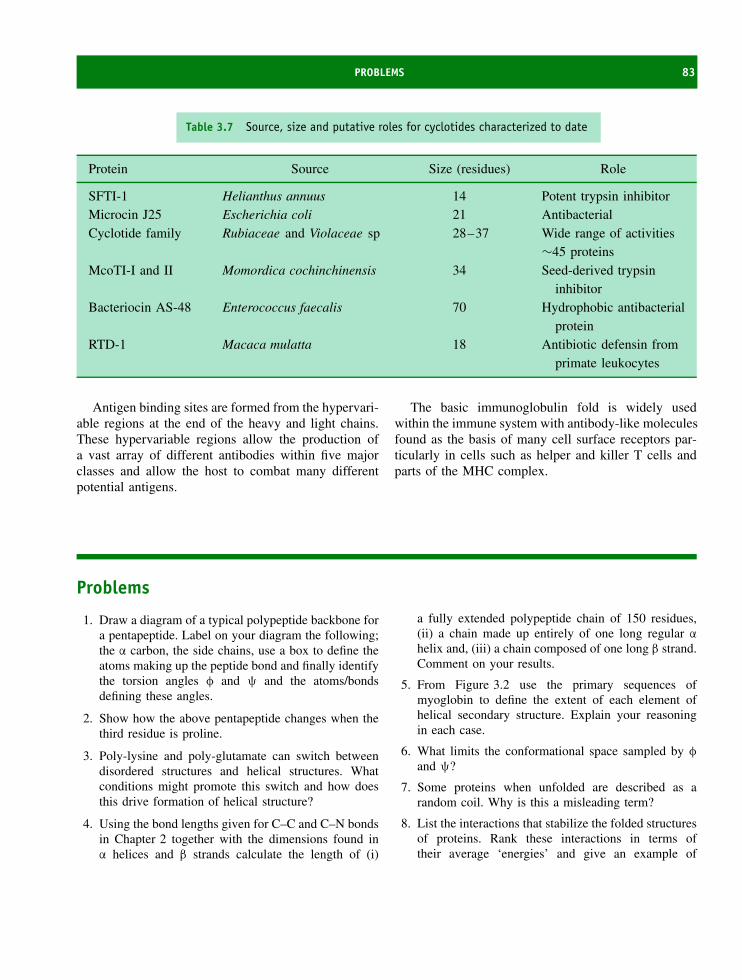
PROBLEMS 83
Table 3.7 Source, size and putative roles for cyclotides characterized to date
Protein Source Size (residues) Role
SFTI-1 Helianthus annuus 14 Potent trypsin inhibitorMicrocin J25 Escherichia coli 21 AntibacterialCyclotide family Rubiaceae and Violaceae sp 28–37 Wide range of activities
∼45 proteinsMcoTI-I and II Momordica cochinchinensis 34 Seed-derived trypsin
inhibitorBacteriocin AS-48 Enterococcus faecalis 70 Hydrophobic antibacterial
proteinRTD-1 Macaca mulatta 18 Antibiotic defensin from
primate leukocytes
Antigen binding sites are formed from the hypervari-able regions at the end of the heavy and light chains.These hypervariable regions allow the production ofa vast array of different antibodies within five majorclasses and allow the host to combat many differentpotential antigens.
The basic immunoglobulin fold is widely usedwithin the immune system with antibody-like moleculesfound as the basis of many cell surface receptors par-ticularly in cells such as helper and killer T cells andparts of the MHC complex.
Problems
1. Draw a diagram of a typical polypeptide backbone fora pentapeptide. Label on your diagram the following;the α carbon, the side chains, use a box to define theatoms making up the peptide bond and finally identifythe torsion angles φ and ψ and the atoms/bondsdefining these angles.
2. Show how the above pentapeptide changes when thethird residue is proline.
3. Poly-lysine and poly-glutamate can switch betweendisordered structures and helical structures. Whatconditions might promote this switch and how doesthis drive formation of helical structure?
4. Using the bond lengths given for C–C and C–N bondsin Chapter 2 together with the dimensions found inα helices and β strands calculate the length of (i)
a fully extended polypeptide chain of 150 residues,(ii) a chain made up entirely of one long regular α
helix and, (iii) a chain composed of one long β strand.Comment on your results.
5. From Figure 3.2 use the primary sequences ofmyoglobin to define the extent of each element ofhelical secondary structure. Explain your reasoningin each case.
6. What limits the conformational space sampled by φ
and ψ?
7. Some proteins when unfolded are described as arandom coil. Why is this a misleading term?
8. List the interactions that stabilize the folded structuresof proteins. Rank these interactions in terms oftheir average ‘energies’ and give an example of

84 THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
each interaction as it occurs between residues withinproteins.
9. How would you identify turn regions within thepolypeptide sequence of proteins? Why do turnregions occur more frequently on protein surfaces?What distinguishes a turn from a loop region?
10. What are the advantages of more than one subunitwithin a protein? How are multimeric proteinsstabilized?

4The structure and function of fibrous
proteins
Historically a division into globular and fibrous pro-teins has been made when describing chemical andphysical properties. This division was originally madeto account for the very different properties of fibrousand globular proteins as well as the different roleseach group occupied within cells. Today it is prefer-able to avoid this distinction and to treat proteins asbelonging to families that exhibit structural or sequen-tial homology (see Chapter 6). However, despite verydifferent properties the structures of fibrous proteinswere amongst the first to be studied because of theiraccumulation in bodies such as hair, nails, tendons andligaments. These proteins demonstrate new aspects ofbiological design not shown by globular proteins thatrequires separate description.
Fibrous proteins were named because they werefound to make up many of the ‘fibres’ found in thebody. Here, fibrous proteins had a common role inconferring strength and rigidity to these structures aswell as physically holding them together. Subsequentstudies have shown that these proteins are morewidely distributed than previously supposed, beingfound in cells as well as making up connectivetissues such as tendons or ligaments. More importantly,these proteins occupy important biological roles thatarise from their wide range of chemical and physicalproperties. These properties are distinct from globularproteins and arise from the individual amino acid
sequences. A common feature of most fibrous proteinsis their long, drawn-out, or filamentous structure.Essentially, these proteins tend to occur as ‘rod-like’structures extended more in two out of the threepossible dimensions and lacking the compactness ofglobular proteins. As a result fibrous proteins tend topossess architectures based around regular secondarystructure with little or no folding resulting from long-range interactions. In other words they lack truetertiary structure.
The second large class of proteins distinct fromglobular proteins are the membrane proteins. Membersof this group of proteins probably make up the vastmajority of all proteins found in cells. For manyyears the purification of these proteins remained verydifficult and limited our knowledge of their structureand function. However, slowly membrane proteinshave become amenable to biochemical characterizationand Chapter 5 will deal with the properties of thisimportant group to highlight in successive sectionsglobular, fibrous and membrane proteins.
The amino acid compositionand organization of fibrous proteinsIn fibrous proteins at least three different structuralplans or designs have been recognized in construction.
Proteins: Structure and Function by David Whitford 2005 John Wiley & Sons, Ltd

86 THE STRUCTURE AND FUNCTION OF FIBROUS PROTEINS
These designs include: (i) structure composed of‘coiled-coils’ of α helices and represented by the α
keratins; (ii) structures made up of extended antipar-allel β sheets and exemplified by silk fibroin a col-lection of proteins made by spiders or silkworms; and(iii) structures based on a triple helical arrangement ofpolypeptide chains and shown by the collagen fam-ily of proteins. The structures of each class of fibrousproteins will be described in the following sections,highlighting how the structure is suitable for its partic-ular role and also emphasizing how defects in fibrousproteins can lead to serious and life threatening condi-tions.
An analysis of the amino acid composition of typ-ical fibrous proteins (Table 4.1) reveals considerabledifferences in their constituent amino acids to that
Table 4.1 The amino acid composition of threecommon classes of fibrous proteins in mole percent
Amino acid Fibroin (silk) α-keratin Collagen
Gly 44.6 8.1 32.7Ala 29.4 5.0 12.0Ser 12.2 10.2 3.4Glx 1.0 12.1 7.7Cys 0 11.2 0Pro 0.3 7.5 22.1Arg 0.5 7.2 5.0Leu 0.5 6.9 2.1Thr 0.9 6.5 1.6Asx 1.3 6.0 4.5Val 2.2 5.1 1.8Tyr 5.2 4.2 0.4Ile 0.7 2.8 0.9Phe 0.5 2.5 1.2Lys 0.3 2.3 3.7Trp 0.2 1.2 0His 0.2 0.7 0.3Met 0 0.5 0.7
In collagen considerable amounts of hydroxylated lysine and prolineare found. Adapted from Biochemistry, 3rd edn. Mathews, van Holde& Ahern (eds). Addison Wesley Longman, London.
described earlier for globular proteins (see Table 2.2,Chapter 2). More significantly the amino acid compo-sitions of fibrous proteins differ with each group. Forexample, collagen has a proline content in excess of20 percent whilst in silk fibroin this value is below 1percent. Similarly in α keratin the cysteine content is11.2 percent but in collagen and silk fibroin the levelsof cysteine are essentially undetectable. In each casethe amino acid composition influences the secondarystructure formed by fibrous proteins.
Keratins
Keratins are the major class of proteins found inhair, feathers, scales, nails or hooves of animals. Ingeneral the keratin class of proteins are mechani-cally strong, designed to be unreactive and resistantto most forms of stress encountered by animals. Atleast two major groups of keratins can be identified;the α keratins are typically found in mammals andoccur as a large number of variants whilst β ker-atins are found in birds and reptiles as part of feathersand scales containing a significantly higher proportionof β sheet. The β keratins are analogous to the silkfibroin structures produced by spiders and silkwormsand described later in this chapter. The α keratinsare a subset of a much larger group of filamentousproteins based on coiled-coils called intermediate fila-ments (IF). The distribution of intermediate filamentsis not restricted to mammals but appears to extend tomost animal cells as major components of cytoskele-tal structures.
In mammals approximately 30 different variantsof keratin have been identified with each appearingto be expressed in cells in a tissue-specific manner.In each keratin the ‘core’ structure is similar andis based around the α helix so that the followingdiscussion of the conformation applies equally wellto all proteins within this group. Although the basicunit of keratin is an α helix this structure is slightlydistorted as a result of interactions with a second helixthat lead to the formation of a left handed coiled-coil. The most common arrangement for keratin isa coiled-coil of two α helices although three helicalstranded arrangements are known for extracellularkeratins domains whilst in insects four stranded coiled

KERATINS 87
coils have been found. In 1953 Francis Crick postulatedthat the stability of α helices would be enhanced ifpairs of helices interacted not as straight rods butin a simple coiled-coil arrangement (Figure 4.1). Thiscoiled coil arrangement is sometimes called a superhelix with in this instance α keratin found as aleft-handed super helix. Detailed structural studies ofcoiled-coils confirmed this arrangement of helices anddiffraction studies showed a periodicity of 1.5 A and5.1 A.
The coiled-coil is formed by each helix interact-ing with the other and by burying their hydropho-bic residues away from the solvent interface. Thehydrophobic or non-polar side chains are not ran-domly located within the primary sequence but occurat regular intervals throughout the chain. This non-random distribution of hydrophobic residues is alsoaccompanied by a preference for residues with chargedside chains at positions within helices that are incontact with solvent. As a result of this periodic-ity a repeating unit of seven residues occurs alongeach chain or primary sequence. This is called theheptad repeat and the residues within this unit arelabelled a, b, c, d, e, f and g. To facilitate identi-fication of each residue within a helix the positionsare frequently represented by a helical wheel dia-gram (Figure 4.2).
Although a regular α helix has 3.6 residues per turn,the coiled-coil arrangement leads to a slight decreasein the number of residues/turn to 3.5. Each helix is
NC
C
C
C
N
NN
Parallel coiled coil
Anti-parallel coiled coil
Figure 4.1 Two individual α helices distorted alongtheir longitudinal axes as a result of twisting. The twohelices can pack together to form coiled-coils
inclined at an angle of approximately 18◦ towards theother helix and this allows for the contacting sidechains to make a precise inter-digitating surface. Italso leads to a slightly different pitch for the helixof 5.1 A compared with 5.4 A in a regular α helix.The hydrophobic residues located at residues a and dof each heptad form a hydrophobic surface interact-ing with other hydrophobic surfaces. The hydrophobicresidues form a seam that twists about each helix.By interacting with neighbouring hydrophobic surfaceshelices are forced to coil around each other formingthe super helix or coiled-coil. The interleaving of sidechains has been known as ‘knobs-into-holes’ pack-ing and in other proteins as a leucine zipper arrange-ment, although this last term is slightly misleading. Acoiled coil arrangement not only enhances the stabilityof the intrinsically unstable single α- helix but alsoconfers considerable mechanical strength in a man-ner analogous to the intertwining of rope or cable.Further aggregation of the coiled-coils occurs andleads to larger aggregates with even greater strengthand stability. Besides the high content of hydropho-bic residues α keratins also have significant propor-tions of cysteine (see Table 4.1). The cysteine residuesparticipate in disulfide bridges that cross-link neigh-bouring coiled-coils to build up a filament or bundleand ultimately the network of protein constituting hairor nail.
The coiled-coil containing many heptad repeatsextends on average for approximately 300–330 residuesand is flanked by amino and carboxy terminal domains.These amino and carboxy domains vary greatly in size.In some keratins very small domains of approximately10 residues can exist whilst in other homologousproteins, such as nestin, much larger domains in excessof 500 residues are found. More significantly theseregions show much greater sequence variability whencompared with the coiled-coil regions suggesting thatthese domains confer specificity on the individualkeratins and are tailored towards increasing functionalspecificity.
The keratins are often classified as components ofthe large group of filamentous proteins making up thecytoskeletal system and in particular they are classifiedas IF (Table 4.2). IF are generally between 8 and10 nm in diameter and are more common in cellsthat have to withstand stress or extreme conditions.
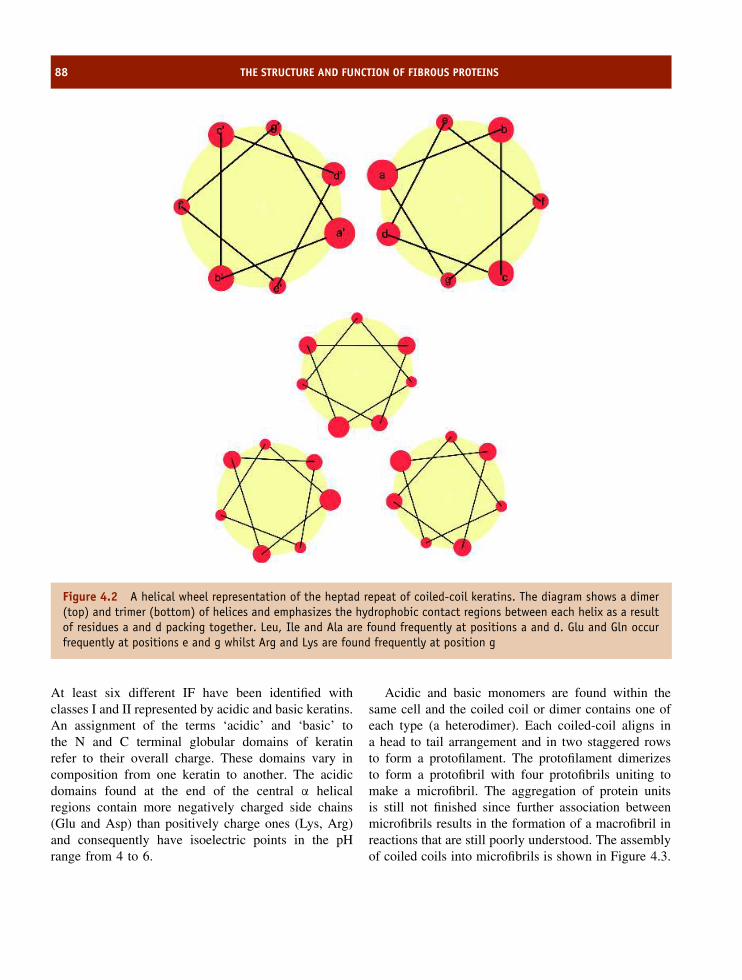
88 THE STRUCTURE AND FUNCTION OF FIBROUS PROTEINS
Figure 4.2 A helical wheel representation of the heptad repeat of coiled-coil keratins. The diagram shows a dimer(top) and trimer (bottom) of helices and emphasizes the hydrophobic contact regions between each helix as a resultof residues a and d packing together. Leu, Ile and Ala are found frequently at positions a and d. Glu and Gln occurfrequently at positions e and g whilst Arg and Lys are found frequently at position g
At least six different IF have been identified withclasses I and II represented by acidic and basic keratins.An assignment of the terms ‘acidic’ and ‘basic’ tothe N and C terminal globular domains of keratinrefer to their overall charge. These domains vary incomposition from one keratin to another. The acidicdomains found at the end of the central α helicalregions contain more negatively charged side chains(Glu and Asp) than positively charge ones (Lys, Arg)and consequently have isoelectric points in the pHrange from 4 to 6.
Acidic and basic monomers are found within thesame cell and the coiled coil or dimer contains one ofeach type (a heterodimer). Each coiled-coil aligns ina head to tail arrangement and in two staggered rowsto form a protofilament. The protofilament dimerizesto form a protofibril with four protofibrils uniting tomake a microfibril. The aggregation of protein unitsis still not finished since further association betweenmicrofibrils results in the formation of a macrofibril inreactions that are still poorly understood. The assemblyof coiled coils into microfibrils is shown in Figure 4.3.
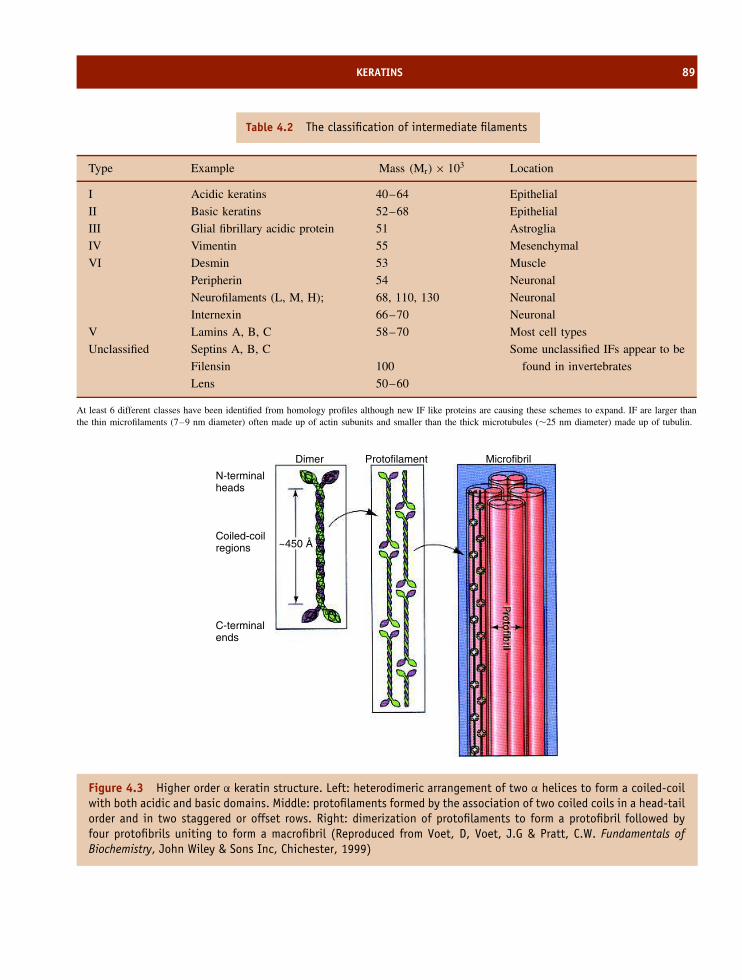
KERATINS 89
Table 4.2 The classification of intermediate filaments
Type Example Mass (Mr) × 103 Location
I Acidic keratins 40–64 Epithelial
II Basic keratins 52–68 Epithelial
III Glial fibrillary acidic protein 51 Astroglia
IV Vimentin 55 Mesenchymal
VI Desmin 53 Muscle
Peripherin 54 Neuronal
Neurofilaments (L, M, H); 68, 110, 130 Neuronal
Internexin 66–70 Neuronal
V Lamins A, B, C 58–70 Most cell types
Unclassified Septins A, B, C
Filensin
Lens
100
50–60
Some unclassified IFs appear to be
found in invertebrates
At least 6 different classes have been identified from homology profiles although new IF like proteins are causing these schemes to expand. IF are larger thanthe thin microfilaments (7–9 nm diameter) often made up of actin subunits and smaller than the thick microtubules (∼25 nm diameter) made up of tubulin.
N-terminalheads
Dimer Protofilament Microfibril
~450 ÅCoiled-coilregions
C-terminalends
Figure 4.3 Higher order α keratin structure. Left: heterodimeric arrangement of two α helices to form a coiled-coilwith both acidic and basic domains. Middle: protofilaments formed by the association of two coiled coils in a head-tailorder and in two staggered or offset rows. Right: dimerization of protofilaments to form a protofibril followed byfour protofibrils uniting to form a macrofibril (Reproduced from Voet, D, Voet, J.G & Pratt, C.W. Fundamentals ofBiochemistry, John Wiley & Sons Inc, Chichester, 1999)

90 THE STRUCTURE AND FUNCTION OF FIBROUS PROTEINS
Figure 4.4 Examples of coiled coil motifs occurring in non-keratin based proteins. In a clockwise direction fromthe top left the diagrams show part of the leucine zipper DNA binding protein GCN4 (PDB:1YSA); a heterodimer ofthe c-jun proto-oncogene (the transcription factor Ap-1) dimerized with c-fos and complexed with DNA (PDB:1FOS);and two views of the gp41 core domain of the simian immunodeficiency virus showing the presence of six α helicescoiled together as two trimeric units – an inner trimer often called N36 and an outer trimer called C34 (PDB:2SIV)
Crystallographic studies have shown that the coiled-coil motif occurs in many other proteins as a recogniz-able motif. It occurs in viral membrane-fusion proteinsincluding the gp41 domain found as part of the humanand simian immunodeficiency virus (HIV/SIV), in thehaemagglutinin component of the influenza virus aswell as transcription factors such as the leucine zipperprotein GCN4. It is also found in muscle proteins suchas tropomyosin and is being identified with increasingregularity in proteins of diverse function and cellu-lar location. Although first recognized as part of longfibrous proteins the coiled-coil is now established as astructural element of many other proteins with widely
differing folds (Figure 4.4). In the coiled-coil structuresthe α helices do not have to run in the same directionfor hydrophobic interactions to occur. Although a par-allel conformation is the most common arrangementantiparallel orientations, where the chains run in oppo-site directions, occur in dimers but are very rare inhigher order aggregates.
Mutations in the genes coding for keratin leadto impaired protein function. In view of the almostubiquitous distribution of keratin these genetic defectscan have severe consequences on individuals. Defectsprove particularly deleterious to the integrity of skinand several inherited disorders are known where

KERATINS 91
cell adhesion, motility and proliferation are severelydisturbed. Since many human cancers arise in epithelialtissues where keratins are prevalent such defectsmay predispose individuals to more rapid tumourdevelopment.
Keratins, the most abundant proteins in epithelialcells, are encoded by two groups of genes designatedtype I and type II. There are >20 type I and >15type II keratin genes occurring in clusters at separateloci in the human genome. A distinction is often madeon the type of cell from which the keratin is derivedor linked. This leads to type II keratin proteins fromsoft epithelia labelled as K1–K8 whilst those derivedfrom hard epithelia (such as hair, nail, and parts ofthe tongue) are designated Hb1–Hb8. Similarly typeI keratins are comprised of K9–K20 in soft epitheliaand Ha1–Ha10 in hard epithelia. All α keratins are richin cysteine residues that form disulfide bonds linkingadjacent polypeptide chains. The term ‘hard’ or ‘soft’refers to the sulfur content of keratins. A high cysteine(i.e. sulfur) content leads to hard keratins typical ofnails and hair and is resistant to deformation whilst alow sulfur content due to a lower number of cysteineresidues will be mechanically less resistant to stress.
In vitro the combination of any type I and type IIkeratins will produce a fibrous polymer when mixedtogether but in vivo the pairwise regulation of type Iand II keratin genes in a tissue specific manner givesrise to ‘patterns’ that are very useful in the study ofepithelial growth as well as disease diagnosis. Thedistribution of some of these keratins is described inTable 4.3 and with over 30 different types of keratinidentified there is a clear preferential location forcertain pairs of keratins.
In all complex epithelia a common set of keratingenes are transcribed consisting of the type II K5 andthe type I K14 genes (along with variable amountsof K15 or K19, two additional type I keratins). Post-mitotic, suprabasal cells in these epithelia transcribeother pairs of keratin genes, the identity of whichdepends on the differentiation route of these cells. Thusthe K1 and K10 pair is characteristic of cornifyingepithelia such as the epidermis, whilst the K4 andK13 pair is expressed in epithelia found lining the oralcavity, the tongue and the oesophagus, and the K3 andK12 pair is found in the cornea of the eye.
Table 4.3 Distribution of type I and II keratin indifferent cells
Type I(acidic)
Type II(basic)
Location
K10 K1 Suprabasal epidermalkeratinocytes
K9 K1 Suprabasal epidermalkeratinocytes
K10 K2e Granular layer of epidermisK12 K3 Cornea of eyeK13 K4 Squamous epithelial layersK14 K5 Basal layer keratinocytesK15 K5 Basal layer of non-keratinizing
epitheliaK16 K6a Outer sheath of hair root, oral
epithelial cells,hyperproliferativekeratinocytes
K17 K6b NailsK7 Seen in transformed cells
K18 K8 Simple epitheliaK19 Follicles, simple epitheliaK21 Intestinal epithelia
In skin diseases such as psoriasis and atopic dermatitis it is noticedthat keratin 6 and 16 predominate whilst a congenital blisteringdisease, epidermolysis bullosa simplex, arises from gene defectsaltering the structure of keratins 5 and 14 at the basal layer.
A property of disulfide bridges between cysteineresidues is their relative ease of reduction withreducing agents such as dithiothreitol, mecaptoethanolor thioglycolate. In general these reagents are calledmercaptans, and for hair the use of thioglycolate allowsthe reduction of disulfide bridges and the relaxingof hair from a curled state to a straightened form.Removal of the reducing agent and the oxidation ofthe thiol groups allows the formation of new disulfidebridges and in this way hair may be reformed in a new‘curled’, ‘straightened’ or ‘permed’ conformation. Thespringiness of hair is a result of the extensive numberof coiled-coils and their tendency in common with a

92 THE STRUCTURE AND FUNCTION OF FIBROUS PROTEINS
conventional spring to regain conformation after initialstretching. The reduction of disulfide bridges in hairallows a keratin fibre to stretch to over twice theiroriginal length and in this very extended ‘reduced’conformation the structure of the polypeptide chainsshift towards the β sheet conformations found infeathers or in the silk-like sheets of fibroin. In the1930s W.T. Astbury showed that a human hair gavea characteristic X-ray diffraction pattern that changedupon stretching the hair, and it was these two formsthat were designated α and β.
Fibroin
A variation in the structure of fibrous proteins is seenin the silk fibroin class made up of an extended arrayof β strands assembled into a β sheet. Insect andspiders produce a variety of silks to assist in theproduction of webs, cocoons, and nests. Fibroin isproduced by cultivated silkworm larvae of the mothBombyx mori and has been widely characterized. Silkconsists of a collection of antiparallel β strands withthe direction of the polypeptide backbone extendingalong the fibre axis. The high content of β strandsleads to a microcrystalline array of fibres in a highlyordered structure. The polypeptide backbone has theextended structure typical of β strands with the sidechains projecting above and below the plane of thebackbone. Of great significance in silk fibroin are thelong stretches of repeating composition. A six residuerepeat of (Gly-Ser-Gly-Ala-Gly-Ala)n is observed tooccur frequently and it is immediately apparent thatthis motif lacks large side chains. These three residuesappear to represent over 85 percent of the total aminoacid composition with approximate values for theindividual fractions being 45 percent Gly, 30 percentAla and 15 percent Ser in silk fibroin (see Table 4.1).The sequence of six residues is part of a largerrepeating unit
-(Gly-Ala)2-Gly-Ser-Gly-Ala-Ala-Gly-
(Ser-Gly-Ala-Gly-Ala)8-Tyr-
that may be repeated up to 50 times leading to massesfor silk polypeptides between 300 000 and 400 000(see Figure 4.5). Glycine, alanine and serine are the
three smallest side chains in terms of their molecularvolumes (see Table 2.3) and this is significant in termsof packing antiparallel strands. More importantly theorder of residues in this repeating sequence placesthe glycine side chain (simply a hydrogen) on oneside of the strand whilst the Ser and Ala side chainsproject to the other side. This arrangement of sidechains leads to a characteristic spacing between strandsthat represents the interaction of Gly residues on onesurface and the interaction of Ala/Ser side chains on theother. The interaction between Gly surfaces yields aninter-sheet spacing or regular periodicity of 0.35 nmwhilst the interaction of the Ser/Ala rich surfacesgives a spacing of 0.57 nm between the strands insilk fibroin. Larger amino acid side chains wouldtend to disrupt the regular periodicity in the spacingof strands and they tend to be located in regionsforming the links between the antiparallel β strands.The structure of these linker regions has not beenclearly defined.
Silk has many remarkable properties. Weight-for-weight it is stronger than metal alloys such as steel, it ismore resilient than synthetic polymers such as Kevlar,yet is finer than a human hair. It is no exaggerationto say that silk is nature’s high-performance polymerfine-tuned by evolution over several hundred millionyears. As a result of these desirable properties therehave been many attempts to mimic the properties of silkwith the development of new materials in the area ofbiomimetic chemistry. Silk is extremely strong becausein the fully extended conformation of β strands anyfurther extension would require the breakage of strongcovalent bonds. However, this strength is coupled withsurprisingly flexibility that arises as a result of theweaker van der Waals interactions that exist betweenthe antiparallel β strands. These desirable physicalproperties are very difficult to reproduce in mostsynthetic polymers.
CollagenCollagen is a major component of skin, tendons, lig-aments, teeth and bones where it performs a widevariety of structural roles. Collagen provides the frame-work that holds most multicellular animals together andconstitutes a major component of connective tissue.Connective tissue performs many functions including

COLLAGEN 93
0.35 nm
0.57 nm
Figure 4.5 The interaction of alternate Gly and Ala/Ser rich surfaces in antiparallel β strands in the silk fibroinstructure from Bombyx mori. The spacing between strands alternates between 0.35 and 0.57 nm as shown in thebottom figure. The structures of silk were generated for an alternating series of Gly/Ala polymers (PDB: 2SLK).Top left: the arrangement of strands in the antiparallel β sheet structure showing backbone only. Top right: Theantiparallel β strands showing the interaction between Ala residue (shown by cyan colour) in an end on view wherethe backbone is running in a direction proceeding into and out of the page. In the bottom diagram the strands arerunning laterally and the Ala/Ser rich interface shown by the cyan side chains is wider than the smaller interfaceformed between glycine residues
binding together body structures and providing supportand protection. Connective tissue is the most abun-dant tissue in vertebrates and depends for its structuralintegrity primarily on collagen. In vertebrates connec-tive tissue appears to account for approximately 30percent of the total mass. Although collagen is oftendescribed as the single most abundant protein it hasmany diverse biological roles and to date at least 30distinct types of collagen have been identified from therespective genes with each showing subtle differencesin the amino acid sequence along their polypeptidechains. Collagen is usually thought of as a proteincharacteristic of vertebrates such as mammals but it isknown to occur in all multicellular animals. Sequencingof the nematode (flatworm) genome of Caenorhabdi-tis elegans has revealed over 160 collagen genes. In
this nematode collagen proteins are the major structuralcomponent of the exoskeleton with the collagen genesfalling into one of three major gene families.
All collagens have the structure of a triple helixdescribed in detail below and are assembled from threepolypeptide chains. Since these chains can be combinedin more than one combination a great many distinctcollagens can exist and several have been identifiedas occurring predominantly in one group of vertebratetissues. In humans at least 19 different collagens areassembled from the gene products of approximately30 distinct and identified genes. Within these 19structural types four major classes or groupings aregenerally identified. These groupings are summarizedin Table 4.4. Type I collagen consists of two identicalchains called α1(I) chains and a third chain called α2.

94 THE STRUCTURE AND FUNCTION OF FIBROUS PROTEINS
Table 4.4 The major collagen groups
Type Function and location
Type I The chief component of tendons,ligaments, and bones
Type II Represents over 50 % of the protein incartilage. It is also used to build thenotochord of vertebrate embryos
Type III Strengthens the walls of hollowstructures like arteries, the intestine,and the uterus
Type IV Forms the basal lamina (sometimescalled a basement membrane) ofepithelia. For example, a network oftype IV collagens provides the filterfor the blood capillaries and theglomeruli of the kidneys
In contrast Type II collagen contain three identicalα1 chains.
In a mature adult collagen fibres are extremelyrobust and insoluble. The insolubility of collagen wasfor many years a barrier to its chemical characterizationuntil it was realized that the tissues of younger animalscontained a greater proportion of collagen with highersolubility. This occurred because the extensive cross-linking of collagen characteristic of adults is lackingin young animals and it was possible to extract thefundamental structural unit called tropocollagen.
The structure and function of collagen
Tropocollagen is a triple helix of three similarly sizedpolypeptide chains each on average about 1000 aminoacid residues in length. This leads to an approximateMr of 285 000, an average length of ∼300 nm and adiameter of ∼1.4 nm. A comparison of the dimensionsof tropocollagen with an average globular proteinis useful and instructive. The length of collagen isapproximately 100 times greater than myoglobin yetits diameter is only half that of the globular proteinemphasizing its extremely elongated or filamentous
nature. The polypeptides of tropocollagen are unusualin their amino acid composition and are defined byhigh proportions of glycine residues (see Table 4.2)as well as elevated amounts of proline. Collagen hasa repetitive primary sequence in which every thirdresidue is glycine. The sequence of the polypeptidechain can therefore be written as
-Gly-Xaa-Yaa−Gly-Xaa-Yaa-Gly-Xaa-Yaa-
where Xaa and Yaa are any other amino acid residue.However, further analysis of collagen sequencesreveals that Xaa and Yaa are often found to be theamino acids proline or lysine. Many of the proline andlysine residues are hydroxylated via post-translationalenzymatic modification to yield either hydroxyproline(Hyp) or hydroxylysine (Hyl) (see Chapter 8). Thesequence Gly-Pro-Hyp occurs frequently in collagen.The existence of repetitive sequences is a feature ofcollagen, keratin and fibroin proteins and is in markedcontrast to globular proteins where repetitive sequencesare the exception.
Each polypeptide chain intertwines with the remain-ing two chains to form a triple helix (Figure 4.6). Thehelix arrangement is very different to the α helix andshows most similarity with the poly-proline II helicesdescribed briefly in Chapter 3. Each chain has thesequence Gly-Xaa-Yaa and forms a left-handed superhelix with the other two chains. This leads to thetriple helix shown in Figure 4.6. When viewed ‘end-on’ the super helix can be seen to consist of left handedpolypeptide chain supercoiled in a right handed mannerabout a common axis.
The rise or translation distance per residue for eachchain in the triple helix is 0.286 nm whilst the numberof residues per turn is 3.3. Combining these twofigures yields a value of ∼0.95 nm for the helix pitchand reveals a more extended conformation especiallywhen compared with α helices or 310 helices (pitch∼0.5–0.54 nm).
Glycine lacking a chiral centre and possessing con-siderable conformational flexibility presents a signif-icant contrast to proline. In proline conformationalrestraint exists as a result of the limited variation inthe torsion angle (φ) permitted by a cyclic pyrroli-done ring. The presence of large amounts of prolinein tropocollagen is also significant because the absence

COLLAGEN 95
Figure 4.6 The basic structure of the triple helix ofcollagen. Space filling representation of the triple helixand a simpler wireframe view of the different chains(orange, dark green and blue chains). Careful analysisof the wireframe view reveals regular repeating Proresidues in all three chains
of the amide hydrogen (HN) eliminates any poten-tial hydrogen bonding with suitable acceptor groups.As a result of the presence of both glycine andproline in high frequency in the collagen sequencesthe triple helix is forced to adopt a different strat-egy in packing polypeptide chains. Since the glycineresidues are located at every third position and makecontact with the two remaining polypeptide chainsit is clear that only a very small side chain (i.e.glycine) can be accommodated at this position. Anyside chain bigger than hydrogen would disrupt theconformation of the triple helix. As a result there isvery little space along the helix axis of collagen andglycine is always the residue closest to the helix axis(Figure 4.7). The side chains of proline residues alongwith lysine and other residues are on the outside ofthe helix.
Figure 4.7 The interaction of glycine residues at thecentre of the collagen helix. Note the side chainsof residues Xaa and Yaa are located towards theoutside of the helix on each chain where they remainsterically unhindered. In each of the three chainsproline and hydroxyproline side chains are shown inyellow with the remaining atoms shown in their usualCPK colour scheme. Only heavy atoms are shown inthis representation. The arrows indicate the hydrogenbonds from the glycine NH to the CO of residue Xaain the neighbouring chain. Each chain is staggered sothat Gly and Xaa and Yaa occur at approximately thesame level along the axis of the triple helix (derivedfrom PDB:1BKV)
The close packing of chains clearly stabilizesthe triple helix through van der Waals interactions,but in addition extensive hydrogen bonding occursbetween polypeptide chains. The hydrogen bondsform between the amide (NH) group of one glycineresidue and the backbone carbonyl (C=O) groupof residue Xaa on adjacent chains. The directionof the hydrogen bonds are transverse or across thelong axis of the helix. Interactions within the triplehelix are further enhanced by hydrogen bondingbetween amide groups and the hydroxyl group of Hypresidues. An indication of the importance of hydrogenbonding interactions in collagen helices has beenobtained through constructing synthetic peptides anddetermining the melting temperature of the collagentriple helix. The melting temperature is the temperature

96 THE STRUCTURE AND FUNCTION OF FIBROUS PROTEINS
−20 0 20 40 60 80 1000.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
Fra
ctio
n of
Col
lage
n as
trip
le h
elix
Temperature (°C)
Figure 4.8 Thermal denaturation curve for collagen. In normal collagens the transition midpoint temperature or Tm
is related to the normal body temperature of the organism and for mammals is above 40 ◦C as shown by the blue linein the above graph
at which half the helical structure has been lost and ischaracterized by a curve showing a sharp transitionat a certain temperature reflecting the loss of orderedstructure. The loss of structural integrity reflectsdenaturation of the triple helix and is accompanied bya progressive loss of function.
The importance of hydroxyproline to the transitiontemperature is shown by synthetic peptides of (Gly-Pro-Pro)n and (Gly-Pro-Hyp)n. The former has atransition mid point temperature (Tm) of 24 ◦C whilstthe latter exhibits a much higher Tm of ∼60 ◦C(Figure 4.8). This experiment strongly supports theidea of triple helix stabilization through hydroxylationof proline (Hyp) and the formation of hydrogen bondswith neighbouring chains. Heating of collagen formsgelatin a disordered state in which the triple helix hasdissociated. Although cooling partially regenerates thetriple helix structure much of the collagen remainsdisordered. The reasons underlying this observationwere not immediately apparent until the route ofcollagen synthesis was studied in more detail. Collagenis synthesized as a precursor termed procollagen inwhich additional domains at the N and C terminal
specifically modulate the folding process. Maturecollagen lacks these domains so any unfolding ordisordering that occurs remains difficult to reverse.
Although hydrogen bonds and van der Waals inter-actions impart considerable stability to the tropocol-lagen triple helix and underpin its use as a structuralcomponent of many cells further strength arises fromthe association of tropocollagen molecules together aspart of a collagen fibre. Each tropocollagen moleculeis approximately 300 nm in length and packs togetherwith neighbouring molecules to produce a characteris-tic banded appearance of fibres in electron micrographs.The banded appearance arises from the overlappingof each triple helix by approximately 64 nm therebyproducing the striated appearance of collagen fib-rils. This pattern of association relies on furthercross-linking both within individual helices, known asintramolecular cross-links, as well as bonds betweenhelices where they are called intermolecular cross-links. Both cross-links are the result of covalentbond formation.
The covalent cross-links among collagen moleculesare derived from lysine or hydroxylysine and involve

COLLAGEN 97
NH
HC
C O
CH2 CH2 CH2 CH2 NH3+
Lysyl oxidase
NH
HC
C O
CH2 CH2 CH2 CH
O
Figure 4.9 Oxidation of ε-amino group of lysine toform aldehyde called allysine
the action of an enzyme called lysyl oxidase. This cop-per dependent enzyme oxidizes the ε-amino group of alysine side chain and facilitates the formation of a crosslink with neighbouring lysine residues (Figure 4.9).Lysine sidechains are oxidized to an aldehyde calledallysine and this promotes a condensation reactionbetween two chains forming strong covalent cross-links (Figure 4.10). A reaction between lysine residuesin the same collagen fibre results in an intramolecu-lar cross-link whilst reaction between different triplehelices results in intermolecular bridges. In view ofthe presence of hydroxylysine and lysine in colla-gen these reactions can occur between two lysine,two hydroxylysines or between one hydroxylysine andone lysine. The products are called hydroxylysinonor-leucine or lysinonorleucine. Further cross-linking canform trifunctional cross-links and a hydroxypyridino-line structure.
Cross-linking of collagen is a progressive processbut does not occur in all tissues to the same extent. Ingeneral, younger cells have less cross-linking of theircollagen than older cells, with a visible manifestationof this process being the increase in the appearance ofwrinkled skin in the elderly, especially when comparedwith that found in a newborn baby. It is also thereason why meat from older animals is tougher thanthat derived from younger individuals.
Collagen biosynthesis
Collagen is a protein that undergoes significant post-translational modification and serves to introduce a
NH
HC
C
CH2 CH2 CH2 CH
O
NH
HC
C O
CH2 CH2 C
CH
O
CH
NH
CH
CO
CH2CH2CH2
O
NH
CH
CO
CH2CH2CH2CH
O
+
Allysine aldol condensation
Allysine Allysine
Further cross-linking reactions
Figure 4.10 Outline of the reaction pathway leadingto the formation of lysine crosslinks in collagen fibres.The first oxidative step results in the deamination ofthe ε-amino group and the formation of allysine (orhydroxyallysine). Two allysine residues condense toform a stable cross-link which can undergo furtherreactions that heighten the complexity of the cross-link. The allysine route predominates in skin whilst thehydroxyallysine route occurs in bone and cartilage
subject that is described in more detail in Chapter 8.The initial translation product synthesized at theribosome is very different to the final product andwithout these subsequent modifications it is extremelyunlikely that the initial translation product couldperform the same biological role as mature collagen.It is also true to say that any process interfering withmodification of collagen tends to result in severe formsof disease.
The biosynthesis of collagen is divided into dis-crete reactions that differ not only in the nature of themodification but their cellular location. Step 1 is the ini-tial formation of preprocollagen, the initial translationproduct formed at the ribosome. In this state the col-lagen precursor contains a signal sequence that directsthe protein to the endoplasmic reticulum membrane and

98 THE STRUCTURE AND FUNCTION OF FIBROUS PROTEINS
Colla
gen
mRN
A
Sign
al s
eque
nce
N te
rmin
al
C
term
inal
pept
ide
p
eptid
e
OH
OH
O
H
OH
OH
OH
O
H
OH
OH
OH
O
H
OH
5 4 3 2 1
6O
H
OH
OH
OH
OH
OH
O
H
OH
OH
S S S S
S S
OH OH OH
OH OH OH
S S S S
7
Tran
spor
t ves
icle
En
do
cyto
sis
Pla
sma
me
mb
ran
e
Ext
race
llula
r re
gio
n
Tro
po
colla
ge
n w
ith N
an
dC
te
rmin
al p
ep
tide
s re
move
d
Col
lage
n m
olec
ules
cov
alen
tly c
ross
-link
ed to
fibr
il11 10 9 8
Trop
ocol
lage
n
Exo
cyto
sis
Exo
cyto
sis
Figu
re4.
11Pr
oces
sing
ofpr
ocol
lage
nre
mov
esN
and
Cte
rmin
alex
tens
ions
and
allo
ws
asso
ciat
ion
into
stag
gere
d,cr
oss-
linke
dfib
rils
.1.
Synt
hesi
son
ribo
som
es.E
ntry
ofch
ains
into
lum
enof
endo
plas
mic
reti
culu
moc
curs
wit
hfir
stpr
oces
sing
reac
tion
rem
ovin
gsi
gnal
pept
ide
2.Co
llage
npr
ecur
sor
wit
hN
and
Cte
rmin
alex
tens
ions
.3.
Hyd
roxy
lati
onof
sele
cted
prol
ine
and
lysi
nes
4.Ad
diti
onof
Asn-
linke
dol
igos
acch
arid
esto
colla
gen
5.In
itia
lgly
cosy
lati
onof
hydr
oxyl
ysin
ere
sidu
es6.
Alig
nmen
tof
thre
epo
lype
ptid
ech
ains
and
form
atio
nof
inte
r-ch
ain
disu
lfide
brid
ges
7.Fo
rmat
ion
oftr
iple
helic
alpr
ocol
lage
n8.
Tran
sfer
byen
docy
tosi
sto
tran
spor
tve
sicl
e9.
Exoc
ytos
istr
ansf
ers
trip
lehe
lixto
extr
acel
lula
rph
ase
10.Re
mov
alof
Nan
dC
term
inal
prop
epti
des
bysp
ecifi
cpe
ptid
ases
11.La
tera
lass
ocia
tion
ofco
llage
nm
olec
ules
coup
led
toco
vale
ntcr
oss
linki
ngcr
eate
sfib
ril

COLLAGEN 99
facilitates its passage into the lumen where the signalpeptide is cleaved by the action of specific proteases.However, while still associated with the ribosome, thispolypeptide is hydroxylated from the action of pro-lyl hydroxylase and lysyl hydroxylase, resulting in theformation of hydroxyproline and hydroxylysine, andis followed by transfer of the polypeptide into thelumen of the endoplasmic reticulum. Here, a third stepinvolves glycosylation of the collagen precursor andthe attachment of sugars, chiefly glucose and galac-tose, occurs via the hydroxyl group of Hyl. Frequently,the sugars are added as disaccharide units. In this statethe pro-α-chains join forming procollagen whilst the N-and C-terminal regions form inter-chain disulfide bondsand the central regions pack into a triple helix. In thisstate the collagen is termed procollagen and it is trans-ported to the Golgi system prior to secretion from thecell. Procollagen peptidases remove the disulfide-richN and C terminal extensions leaving the triple helicalcollagen in the extracellular matrix (Figure 4.11) whereit can then associate with other collagen moleculesto form staggered, parallel arrays (Figure 4.12). Thesearrays undergo further modification by the formationof cross-links through the action of lysyl oxidase, asdescribed above.
Collagenases degrade collagen and have been shownto be one member of a large group of enzymes calledmatrix metalloproteinases (MMPs). These enzymes
Collagen
~300 nm‘‘hole’’ betweencollagen fibres.
staggered by ~40 nm
Figure 4.12 Association of procollagen into stag-gered parallel arrays making up collagen fibres. Eachcollagen fibre is approximately 300 nm in length andis staggered by ∼40 nm from adjacent parallel fibres.This ‘hole’ can represent the site of further attachmentof extracellular proteins and can become filled withcalcium phosphate
degrade the extracellular matrix. Abnormal metallopro-teinase expression leads to premature degradation ofthe extracellular matrix and is implicated in diseasessuch as atherosclerosis, tumour invasion and rheuma-toid arthritis.
Disease states associated with collagen defects
The widespread involvement of collagen in not onlytendons and ligaments, but also the skin and bloodvessels, means that mutations in collagen genesoften result in impaired protein function and severedisorders affecting many organ systems. Mutationsin the 30 collagen genes discovered to date giverise to a large variety of defects in the protein.In addition defects have also been found in theenzymes responsible for the assembly and matura-tion of collagen creating a further group of dis-ease states. In humans defects in collagen as aresult of gene mutation lead to osteogenesis imper-fecta, hereditary osteoporosis and familial aorticaneurysm.
Several hereditary connective tissue diseases havebeen identified as arising from mutations in genesencoding collagen chains. Most common are sin-gle base mutations that result in the substitutionof glycine by a different residue thereby destroy-ing the characteristic repeating sequence of Gly-Xaa-Yaa. A further consequence of these muta-tions is the incorrect assembly or folding of col-lagen. Two particularly serious diseases attributableto defective collagen are osteogenesis imperfecta andEhlers–Danlos syndrome. The molecular basis forthese diseases in relation to the structure of collagenwill be described.
Osteogenesis imperfecta is a genetic disorder char-acterized by bones that break comparatively easilyoften without obvious cause. It is sometimes calledbrittle bone disease. At least four different types ofosteogenesis imperfecta are recognized by clinicians(Table 4.5) although all appear to arise from mutationof the collagen genes coding for either the α1 or α2
chains of type I collagen.Osteogenesis imperfecta is caused by a mutation
in one allele of either the α1 or α2 chains of themajor collagen in bone, type I collagen. Type I col-lagen contains two α1 chains together with a single

100 THE STRUCTURE AND FUNCTION OF FIBROUS PROTEINS
Table 4.5 Classification of osteogenesis imperfecta.Currently, diagnosis of type II form of osteogenesisimperfecta is determined in utero whilst diagnosis ofother forms of the disease can only be made antenatal
Type Inheritance Description
I Dominant Mild fragility, slightdeformity, short stature.Presentation at youngage (2–6)
II Recessive Lethal: death at pre- orperinatal stage
III Dominant Severe, progressivedeformity of limbs andspine
IV Dominant Skeletal fragility andosteoporosis, bowing oflimbs. Most mild formof disease
α2 chain and the presence of one mutant collagenallele is sufficient to produce a defective collagen fibre.The incidence of all forms of osteogenesis imperfectais estimated to be approximately 1 in 20 000; fromscreening individuals it was found that glycine sub-stitution was a frequently observed event with cys-teine, aspartate and arginine being the most frequentreplacement residues. The disruption of the Gly-Xaa-Yaa repeat has pathological consequences that varyconsiderably, as shown by Table 4.5 where four differ-ent types of osteogenesis imperfecta ranging in severityfrom lethal to mild are observed. One consequence ofglycine substitution is defective folding of the collagenhelix and this is accompanied by increased hydrox-ylation of lysine residues N-terminal to the mutationsite. Since hydroxylation occurs on unfolded chainsone possible effect of mutation is to inhibit the rateof triple helix formation rendering the proteins sus-ceptible to further modification or interaction withmolecular chaperones or enzymes of the endoplasmicreticulum that alter their processing or secretion. Afurther sequela of mutant collagen fibres is incorrect
processing by N -propeptide peptidases (see sectionon collagen biosynthesis) and where mutant collagenmolecules are incorporated into fibrils there is evi-dence of poor mineralization. It seems very likely thatthe exact site of the mutation of α1 or α2 chainswill influence the overall severity of the disease withsome evidence suggesting mutations towards the C-terminal produce more severe phenotypes. However,the precise relationship between disease state, muta-tion site and perturbation of collagen structure remainto be elucidated.
A second important disease arising from mutationsin a single collagen gene is Ehlers–Danlos syndrome.This syndrome results in widely variable phenotypes,some relatively benign whilst others are life threat-ening. Classically the syndrome has been recognizedby physicians from the presentation of patients withjoint hypermobility as well as extreme skin extensi-bility, although many other diagnostic traits are nowrecognized including vascular fragility. Many differ-ent types of the disease are recognized both medi-cally and at the level of the protein. The variabilityarises from mutations at different sites and in differ-ent types of collagens leading to the variety of phe-notypes. These mutations can lead to changes in thelevels of collagen molecules, changes in the cross-linking of fibres, a decreased hydroxylysine contentand a failure to process collagen correctly by removalof the N-terminal regions. The common effect ofall mutations is to create a structural weakness inconnective tissue as a result of a molecular defectin collagen.
Related disorders characterized at amolecular level
Marfan’s syndrome is an inherited disorder of connec-tive tissue affecting multiple organ systems includingthe skeleton, lungs, eyes, heart and blood vessels. Fora long time Marfan’s syndrome was believed to becaused by a defect in collagen but this defect is nowknown to reside in a related protein that forms part ofthe microfibrils making up the extracellular matrix thatincludes collagen. The condition affects both men andwomen of all ethnic groups with an estimated incidenceof ∼1 per 20 000 individuals throughout the world.
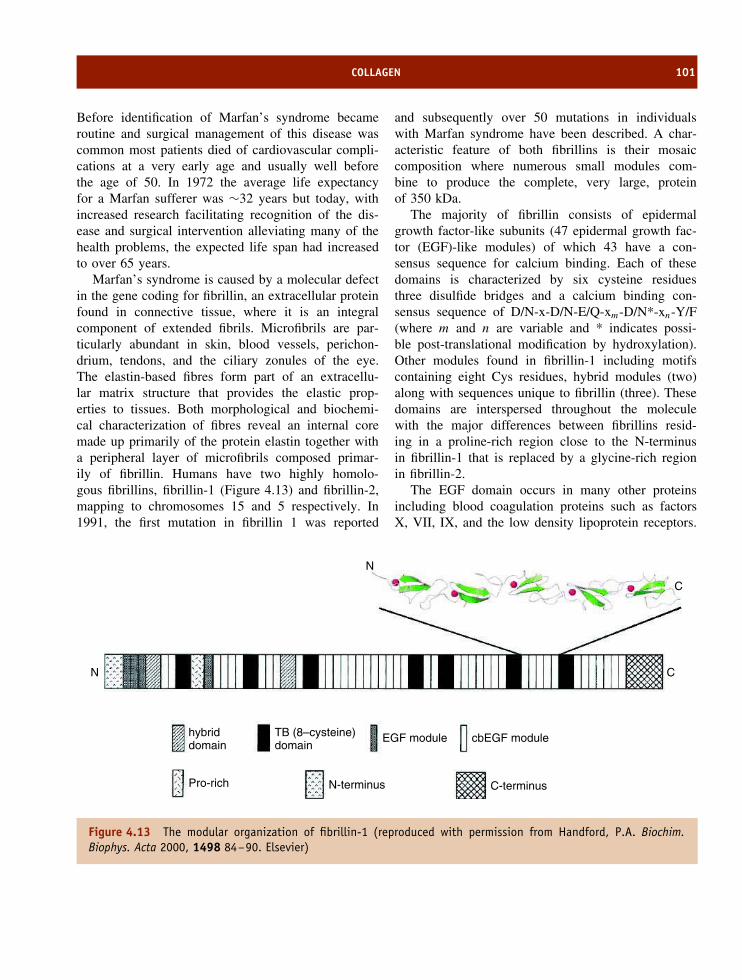
COLLAGEN 101
Before identification of Marfan’s syndrome becameroutine and surgical management of this disease wascommon most patients died of cardiovascular compli-cations at a very early age and usually well beforethe age of 50. In 1972 the average life expectancyfor a Marfan sufferer was ∼32 years but today, withincreased research facilitating recognition of the dis-ease and surgical intervention alleviating many of thehealth problems, the expected life span had increasedto over 65 years.
Marfan’s syndrome is caused by a molecular defectin the gene coding for fibrillin, an extracellular proteinfound in connective tissue, where it is an integralcomponent of extended fibrils. Microfibrils are par-ticularly abundant in skin, blood vessels, perichon-drium, tendons, and the ciliary zonules of the eye.The elastin-based fibres form part of an extracellu-lar matrix structure that provides the elastic prop-erties to tissues. Both morphological and biochemi-cal characterization of fibres reveal an internal coremade up primarily of the protein elastin together witha peripheral layer of microfibrils composed primar-ily of fibrillin. Humans have two highly homolo-gous fibrillins, fibrillin-1 (Figure 4.13) and fibrillin-2,mapping to chromosomes 15 and 5 respectively. In1991, the first mutation in fibrillin 1 was reported
and subsequently over 50 mutations in individualswith Marfan syndrome have been described. A char-acteristic feature of both fibrillins is their mosaiccomposition where numerous small modules com-bine to produce the complete, very large, proteinof 350 kDa.
The majority of fibrillin consists of epidermalgrowth factor-like subunits (47 epidermal growth fac-tor (EGF)-like modules) of which 43 have a con-sensus sequence for calcium binding. Each of thesedomains is characterized by six cysteine residuesthree disulfide bridges and a calcium binding con-sensus sequence of D/N-x-D/N-E/Q-xm-D/N*-xn-Y/F(where m and n are variable and * indicates possi-ble post-translational modification by hydroxylation).Other modules found in fibrillin-1 including motifscontaining eight Cys residues, hybrid modules (two)along with sequences unique to fibrillin (three). Thesedomains are interspersed throughout the moleculewith the major differences between fibrillins resid-ing in a proline-rich region close to the N-terminusin fibrillin-1 that is replaced by a glycine-rich regionin fibrillin-2.
The EGF domain occurs in many other proteinsincluding blood coagulation proteins such as factorsX, VII, IX, and the low density lipoprotein receptors.
N
N
C
C
hybriddomain
TB (8–cysteine)domain
EGF module cbEGF module
Pro-rich N-terminus C-terminus
Figure 4.13 The modular organization of fibrillin-1 (reproduced with permission from Handford, P.A. Biochim.Biophys. Acta 2000, 1498 84–90. Elsevier)

102 THE STRUCTURE AND FUNCTION OF FIBROUS PROTEINS
Figure 4.14 The structure of a pair of calciumbinding EGF domains from fibrillin-1 (PDB:1EMN)
The structure of a single EGF like domain and apair of calcium-binding domains confirmed a rigidrod-like arrangement stabilized by calcium bindingand hydrophobic interactions (Figure 4.14). Mutationsknown to result in Marfan’s syndrome lead todecreased calcium binding to fibrillin, and this seems toplay an important physiological role. The importance ofthe structure determined for the modules of fibrillin wasthat it offered an immediate explanation of the defectat a molecular level. The structural basis for Marfan’ssyndrome resides in the disruption of calcium bindingwithin fibrillin as a result of single site mutation.
Marfan’s syndrome is an autosomal dominant dis-order affecting the cardiovascular, skeletal, and ocu-lar systems. The major clinical manifestations areprogressive dilatation of the aorta (aortic dissection),mitral valve disorder, a tall stature frequently associ-ated with long extremities, spinal curvature, myopiaand a characteristic thoracic deformity leading to a
‘pigeon-chest’ appearance. The majority of the muta-tions known today are unique (i.e. found only inone family) but at a molecular level it results in thesubstitution of a single amino acid residue that dis-rupts the structural organization of individual EGF-like motifs.
Flo Hyman, a famous American Olympian volley-ball player, was a victim of Marfan’s syndrome anda newspaper extract testifies to the sudden onset ofthe condition in apparently healthy or very fit athletes.“During the third game, Hyman was taken out in a rou-tine substitution. She sat down on the bench. Secondslater she slid silently to the floor and lay there, still. Shewas dead”. Many victims of Marfan’s disease are tallerthan average. As a result basketball and volleyball play-ers are routinely screened for the genetic defect whichdespite phenotypic characteristics such as pigeon chest,enlarged breastbone, elongated fingers and tall statureoften remains undiagnosed until a sudden and earlydeath. It has also been speculated largely on the basis oftheir physical appearances that Abraham Lincoln (16th
president of the United States, 1809–1865) and thevirtuoso violinist Niccolo Paganini (1782–1840) weresuffering from connective tissue disorders.
SummaryFibrous proteins represent a contrast to the normaltopology of globular domains where compact foldedtertiary structures exist as a result of long-range inter-actions. Fibrous proteins lack true tertiary structure,showing elongated structures and interactions confinedto local residues.
The amino acid composition of fibrous proteinsdeparts considerably from globular proteins but alsovaries widely within this group. This variation reflectsthe different roles of fibrous proteins.
Three prominent groups of fibrous proteins are thecollagens, silk fibroin and keratins and all occupypivotal roles within cells.
Collagen, in particular, is very abundant in verte-brates and invertebrates where the triple helix providesa platform for a wide range of structural roles in theextracellular matrix delivering strength and rigidity toa wide range of tissues.
The triple helix is a repetitive structure containingthe motif (Gly-Xaa-Yaa) in high frequency with Xaa

PROBLEMS 103
and Yaa often found as proline and lysine residues.Repeating sequences of amino acids are a feature ofmany fibrous proteins and help to establish the topologyof each protein. In collagen the presence of glycine atevery third residue is critical because its small sidechain allows it to fit precisely into a region that formsfrom the close contact of three polypeptide chains.Larger side chains would effectively disrupt this regionand perturb the triple helical structure.
Although based around a helical design collagendiffers considerably in dimensions to the typical α
helix. The triple helix of collagen undergoes consider-able post-translational modification to increase strengthand rigidity.
Keratins make up a considerable proportion of hairand nails and contain polypeptide chains arranged inan α helical conformation. The helices interact viasupercoiling to form coiled-coils. Of importance to
coiled-coils are specific interactions between residues indifferent helices via non-polar interactions that confersignificant stability. The basis of this interaction is aheptad of repeating residues along the primary sequence.
A heptad repeat possesses leucine or other residueswith hydrophobic side chains arranged periodicallyto favour inter-helix interactions. Helices are usuallyarranged in pairs. Significantly, this mode of organiza-tion is found in other intracellular proteins as well asin viral proteins. Several DNA binding proteins containcoiled-coil regions and the hydrophobic-rich domainsare often called leucine zippers.
In view of their widespread distribution in all animalcells mutations in fibrous proteins such as keratins orcollagens lead to serious medical conditions. Manydisease states are now known to arise from inheriteddisorders that lead to impaired structural integrity inthese groups of proteins.
Problems
1. Describe the different amino acid compositions ofcollagen, silk and keratins?
2. Explain why glycine and proline are found inhigh frequency in the triple helix of collagen butare not found frequently within helical regions ofglobular proteins?
3. Why is silk both strong and flexible?
4. Why is wool easily stretched or shrunk? Why is silkmore resistant to these deformations.
5. Poly-L-proline is a synthetic polypeptide that adoptsa helical conformation with dimensions comparableto those of a single helix in collagen. Why doespoly-L–proline fail to form a triple helix. Will thesequence poly-(Gly-Pro-Pro)n form a triple helix andhow does the stability of this helix compare withnative collagen. Does poly-(Gly-Pro-Gly-Pro)n forma collagen like triple helix?
6. A mutation is detected in mRNA in a region in framewith the start codon (AUG) CCCUAAAUG. . . . . ..
GGACCCAAAGGACCUAAGUGUCCAUCUGGUCCGAAGGGGUCCAACGGACCCAAGGGU. . . . . .
Establish the identity of the peptide and describe thepossible consequences of this mutation.
7. Describe four post-translational modifications of col-lagen and list these modifications in their orderof occurrence?
8. Gelatin is primarily derived from collagen, the proteinresponsible for most of the remarkable strengthof connective tissues in tendons and other tissues.Gelatin is usually soft and floppy and generallylacking in strength. Explain this observation.
9. Ehlers–Danlos syndromes are a group of collagenbased diseases characterized by hyperextensible jointsand skin. What is the most probable cause of thisdisorder in terms of the structure of collagen?
10. Keratin is based on a seven residue or heptad repeat.Describe the properties of this sequence that favourcoiled-coil structures.


5The structure and function of membrane
proteins
By 2004 the Protein Data Bank of structures con-tained over 22 000 submissions. Almost all of thesefiles reflect the atomic coordinates of soluble proteinsand less than 1 percent of all deposited coordinatesbelong to proteins found within membranes. It is clearfrom this small fraction that membrane proteins presentspecial challenges towards structure determination.
Many of these challenges arise from the difficultyin isolating proteins embedded within lipid bilayers.For many decades membrane proteins have been rep-resented as amorphous objects located in a sea oflipid with little consideration given to their organiza-tion. This is an unfortunate state of affairs becausethe majority of proteins encoded by genomes arelocated in membranes or associated with the membraneinterface. Slowly, however, insight has been gainedinto the different types of membrane proteins, theirdomain structure and more recently the three dimen-sional structures at atomic levels of resolution. Thischapter addresses the structural and functional proper-ties of membrane proteins.
The molecular organizationof membranesAlthough this book is primarily about proteins itis foolish to ignore completely the properties of
membranes since the lipid bilayers strongly influenceprotein structure and function. The term lipid refers toany biomolecule that is soluble in an organic solvent,and extends to quinones, cholesterol, steroids, fattyacids, triacylglycerols and non-polar vitamins such asvitamin K3. All of these components are found inbiological membranes. However, for most purposes theterm lipid refers to the collection of molecules formedby linking a long-chain fatty acid to a glycerol-3-phosphate backbone; these are the major componentsof biological membranes.
Two long chain fatty acids are esterified to theC1 and C2 positions of the glycerol backbone whilstthe phosphate group is frequently linked to anotherpolar group. Generically all of these molecules aretermed glycerophospholipids (Figure 5.1). Phospha-tidic acid occurs in low concentrations within mem-branes and the addition of polar head groups suchas ethanolamine, choline, serine, glycerol, and inositolforms phosphatidylethanolamine (PE), phosphatidyl-choline (PC, also called lecithin), phosphatidylserine(PS), phosphatidylglycerol (PG) and phosphatidylinos-itol (PI), the key components of biological membranes(Table 5.1). Fatty acyl chains usually containing 16or 18 carbon atoms occupy the R1 and R2 posi-tions although chain length can range from 12 to24 carbons.
Proteins: Structure and Function by David Whitford 2005 John Wiley & Sons, Ltd

106 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
C CO
O
H
H
C O
P O−
O
O
C O
O
H
C
X
R1
R2
H2
Figure 5.1 The structure of glycerophospholipidsinvolves the esterification of two long-chain fattyacids to a glycerol-3-phosphate backbone with furtheradditions of polar groups occurring at the phosphategroup. When X=H the parent molecule phosphatidicacid is formed
In animal membranes the C1 position is typicallya saturated lipid of 16–18 carbons whilst the C2
position often carries an unsaturated C16 –C20 fattyacid. Unsaturated acyl chains (Table 5.2) contain oneor more double bonds usually found at carbons 5,6, 8, 9, 11, 12, 14 and 15. Where more than onedouble bond occurs the sites are separated by threecarbons so that, for example in linolenic acid the doublebonds are at 9, 12 and 15, whilst in arachidonic acidthe four double bonds are located at carbons 5, 8,11 and 14. The double bonds are found in the cisstate with trans isomers rarely found. With a largenumber of possible head groups coupled to a diversenumber of fatty acyl chains (each potentially differingin their degree of saturation) the number of possiblecombinations of non-polar chain/polar head group isenormous.
Naming phospholipids follows the identity of thefatty acid residues; so a glycerophospholipid con-taining two 16 carbon saturated fatty acyl chainsand a choline head group is commonly referred toas dipalmitoylphosphatidylcholine (DPPC). Although
Table 5.1 The common head groups attached to the glycerol-3-phosphatebackbone of glycerophospholipids
Donor Head group Phospholipid
Water –H Phosphatidic acid
Ethanolamine CH2 CH2 NH3+
Phosphatidylethanolamine
Choline
CH2 CH2 N+
CH3
CH3
CH3
Phosphatidylcholine
Serine CH2 CH C
O
O−
NH3+
Phosphatidylserine
Inositol
OH
OH
OH OH
OH
Phosphatidylinositol
Glycerol CH2 CH CH2
OH
OH Phosphatidylglycerol

THE MOLECULAR ORGANIZATION OF MEMBRANES 107
Table 5.2 Common acyl chains found in lipids of bio-logical membranes
Name Numeric
representation
Name Numeric
representation
Lauric 12:0 Stearic 18:1
Myristic 14:0 Linoleic 18:2
Palmitic 16:0 Linolenic 18:3
Palmitoleic 16:1 Arachidic 20:0
Oleic 18:0 Arachidonic 20:4
For molecules such as linolenic acid the position of the three double bonds aredenoted by the nomenclature �9,12,15 with carbon 1 starting from the carboxylend. The IUPAC name for this fatty acid is 9,12,15 octadecenoic acid. A thirdalternative identifies the number of carbon atoms in the acyl chain, the numberof double bonds and the geometry and carbon number associated with eachbond. Thus, linolenic acid is 18:3 9-cis, 12-cis, 15-cis
Figure 5.2 The diagram shows a wireframe and spacefilling model of two acyl chains, one 16 carbons inlength, the other 18 carbons in length containing acis double bond. A phosphatidylcholine head groupis linked to a glycerol backbone. Protons have beenomitted to increase clarity
glycerophospholipids are the most common lipidsof biological membranes other variants occur suchas sphingolipids, sulfolipids and galactolipids. In
plant membranes sulfolipids and galactolipids occuralongside phospholipids, increasing the variety oflipids.
A phospholipid such as DPPC is described as amphi-pathic, containing polar and non-polar regions. Spacefilling models (Figure 5.2) show that introducing a sin-gle cis double bond produces a ‘kink’ in the acylchain and an effective volume increase. This has pro-nounced effects on physical properties of lipids such astransition temperatures, fluidity and structures formedin solution.
When dispersed in aqueous solutions lipid moleculesminimize contact between the hydrophobic acyl chainand water by forming structures where the polarhead groups interact with aqueous solvents whilst thenon-polar ‘tails’ are buried away from water. Thesize and shape of the lipid assembly is a complexcombination of factors, including polar head groupvolume, charge, degree of unsaturation and length.Thus lipids with short acyl chains form micellesand pack efficiently into a spherical shape wherethe large volume of the hydrated polar head groupexceeds that of the tail and favours micellar structure.Lipids with a single acyl chain, as well as detergents,adopt this shape. Longer acyl chains, particularlythose with one or more double bonds, cannot packefficiently into a spherical micelle and produce bilayersin most cases although alternative structures, suchas inverted micelles where the polar head group isburied on the inside, can be formed by some ofthe common lipids. Additionally lipid aggregates aremodulated by ionic strength or pH, but a detaileddiscussion of these states is beyond the scope ofthis text.
The ability of lipids or detergents to aggregate insolution is often expressed in terms of their criticalmicelle concentration (CMC). At concentrations abovetheir CMC lipids are found as aggregated structureswhilst below this concentration monomeric speciesexist in solution. The CMC of dodecylmaltoside(a single 12 carbon fatty acyl chain linked to amaltose group) is estimated at ∼150 µM whilst thecorresponding value for DPPC is 0.47 nM and meansthat this polar lipid is always found as an aggregatedspecies. An isolated bilayer dispersed in a polarsolvent is unlikely to be stable since at some pointthe ends of the bilayer will be exposed leading

108 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
Ele
ctro
n de
nsity
ChainsWater WaterHead Head
DHH
Figure 5.3 Electron density profile determined for a single bilayer. The electron dense regions correspond to thehead group region whilst the non-polar domain are less electron dense. Such data established the basic dimensionsof a bilayer (DHH) at around 40 nm and the profile is shown alongside a diagram of a bilayer showing relativepositions of head groups and acyl chains
to unfavourable interactions between non-polar andpolar surfaces. As a result, extended bilayers inaqueous solution often form closed, sealed, solvent-filled structures known as liposomes or single lamellarvesicles that have proved to be good models forbiological membranes.
Since integral membrane proteins are located inbilayers the properties and arrangement of lipidsdictates the structure of any protein residing in thisregion. The structure of a lipid bilayer has beenexamined by neutron and X-ray diffraction to confirma periodic arrangement for the distribution of electrondensity. The polar head groups are electron densewhilst the acyl chains represent comparatively ‘diffuse’areas of electron density (Figure 5.3).
Fluidity and phase transitions are importantproperties of lipid bilayers
An important property of lipid bilayers is fluid-ity. Membrane fluidity depends on lipid composition,temperature, pH and ionic strength. At low tempera-tures the fluidity of a membrane is reduced with theacyl chains showing lower thermal mobility and pack-ing into a ‘frozen’ gel state. At higher temperaturesincreased translational and rotational mobility results ina less ordered fluid state, sometimes called the liquidcrystal (Figure 5.4). Transitions from the gel to fluidstates are characterized by phase transition tempera-tures (Table 5.3). Short chains have lower transitiontemperatures when compared with longer lipids. The

THE MOLECULAR ORGANIZATION OF MEMBRANES 109
transition temperature increases with chain length sincehydrophobic interactions increase with the number ofnon-polar atoms. The transition temperature decreaseswith increasing unsaturation and is reflected by greaterinherent disorder packing.
Biological membranes exhibit broad phase transitiontemperatures over a wide temperature range. Allbiological membranes exist in the fluid state andorganisms have membranes whose lipid compositionsexhibit transition temperatures significantly below theirlowest body temperature. Thus, fish living in the depths
Gel Liquid crystalline
Figure 5.4 The gel to fluid (liquid crystalline) tran-sition in lipid bilayers. In the gel phase the headgroups are tightly packed together, the tails have aregular conformation and the bilayer width is thickerthan the liquid crystalline state
Table 5.3 Phase transition temperatures of commondiacylphospholipids
Diacylphospholipid Transitiontemperature
◦C (Tm)
C22 phosphatidyl choline 75C18 phosphatidyl choline 55C16 phosphatidyl choline 41C14 phosphatidyl choline 24C18:1 phosphatidyl choline −22C16 phosphatidyl serine (low pH) 72C16 phosphatidyl serine (high pH) 55C16 phosphatidyl ethanolamine 60C14 phosphatidyl ethanolamine 50
Phase transition temperatures are measured by differential scanningcalorimetry (DSC) where heat is absorbed (�H increases) as thelipid goes through a phase transition
of the ocean where temperatures may drop below−20 ◦C have membranes rich in unsaturated long-chainfatty acids such as linolenic and linoleic acid.
Real membranes are not homogeneous dispersionsof lipids but contain proteins, often in considerablequantity. The lipid:protein ratio varies dramatically,with membranes such as the myelin sheath possess-ing comparatively little protein (Table 5.4). In con-trast membranes concerned with energy transductionhave larger amounts of protein and exhibit lowerlipid:protein ratios.
The fluid mosaic model
Any model of membrane organization must account forthe properties of lipids, such as self-association intostable structures, mobility of lipids within monolay-ers, and the impermeability of cell membranes to polarmolecules. In addition models must account for thepresence of proteins both integral and peripheral andat a wide variety of ratios with the lipid component. In1972, to account for these and many other observations,S.J. Singer and G. Nicolson proposed the fluid mosaicmodel; a model that has with only minor amendmentsstood the test of time. In this model proteins were orig-inally viewed as floating like icebergs in a sea of lipidbut in view of the known lipid:protein ratios found insome membranes this picture is unrealistic and repre-sentations should involve higher proportions of protein.
Table 5.4 Protein and lipid content of differentmembranes
Membrane Protein Lipid
Myelin sheath (Schwann cells) 21 79
Erythrocyte plasma membrane 49 43
Bovine retinal rod 51 49
Mitochondrial outer membrane 52 48
Mitochondrial inner membrane 76 24
Sarcoplasmic reticulum (muscle cells) 67 33
Chloroplast lamellae 70 30
The figures represent the percentage by weight. The figures do not alwaysadd up to 100 % reflecting different contents of carbohydrate within eachmembrane (derived from Guidotti, G. Ann. Rev. Biochem. 1972, 41, 731–752)
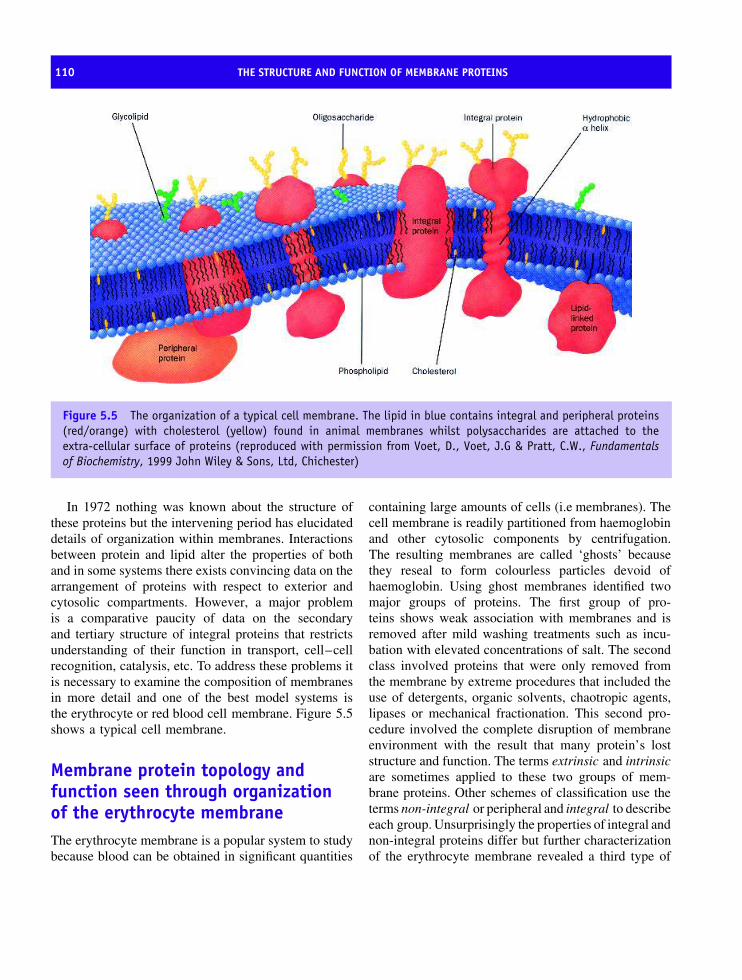
110 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
Figure 5.5 The organization of a typical cell membrane. The lipid in blue contains integral and peripheral proteins(red/orange) with cholesterol (yellow) found in animal membranes whilst polysaccharides are attached to theextra-cellular surface of proteins (reproduced with permission from Voet, D., Voet, J.G & Pratt, C.W., Fundamentalsof Biochemistry, 1999 John Wiley & Sons, Ltd, Chichester)
In 1972 nothing was known about the structure ofthese proteins but the intervening period has elucidateddetails of organization within membranes. Interactionsbetween protein and lipid alter the properties of bothand in some systems there exists convincing data on thearrangement of proteins with respect to exterior andcytosolic compartments. However, a major problemis a comparative paucity of data on the secondaryand tertiary structure of integral proteins that restrictsunderstanding of their function in transport, cell–cellrecognition, catalysis, etc. To address these problems itis necessary to examine the composition of membranesin more detail and one of the best model systems isthe erythrocyte or red blood cell membrane. Figure 5.5shows a typical cell membrane.
Membrane protein topology andfunction seen through organizationof the erythrocyte membraneThe erythrocyte membrane is a popular system to studybecause blood can be obtained in significant quantities
containing large amounts of cells (i.e membranes). Thecell membrane is readily partitioned from haemoglobinand other cytosolic components by centrifugation.The resulting membranes are called ‘ghosts’ becausethey reseal to form colourless particles devoid ofhaemoglobin. Using ghost membranes identified twomajor groups of proteins. The first group of pro-teins shows weak association with membranes and isremoved after mild washing treatments such as incu-bation with elevated concentrations of salt. The secondclass involved proteins that were only removed fromthe membrane by extreme procedures that included theuse of detergents, organic solvents, chaotropic agents,lipases or mechanical fractionation. This second pro-cedure involved the complete disruption of membraneenvironment with the result that many protein’s loststructure and function. The terms extrinsic and intrinsicare sometimes applied to these two groups of mem-brane proteins. Other schemes of classification use theterms non-integral or peripheral and integral to describeeach group. Unsurprisingly the properties of integral andnon-integral proteins differ but further characterizationof the erythrocyte membrane revealed a third type of

MEMBRANE PROTEIN TOPOLOGY AND FUNCTION SEEN THROUGH ORGANIZATION OF THE ERYTHROCYTE MEMBRANE 111
6
4
321
65
59
9
10
11
C
C
N N
8
8
1234 10
7
7
Figure 5.6 Structure of band 3 monomer. The secondary structure distribution (left) and a ribbon diagram of band3 monomer (right). Regions shown in blue correspond to the peripheral protein binding domain and the red regionsto the dimerization arm (reproduced with permission from Zhang, D., Kiyatkin, A., Bolin, J.T. & Low, P.S. Blood 2000,96, 2925–2933. The American Society of Hematology)
protein. This integral protein was attached firmly to thebilayer yet had substantial proportions of its sequenceaccessible to the aqueous phase.
An intrinsic protein of the erythrocytemembrane: band 3 proteinThe name ‘band 3’ originally arose from sodium dode-cyl sulfate–polyacrylamide gel electrophoresis studiesof the protein composition of erythrocyte membraneswhere it was the subunit with the third heaviest molec-ular mass. It is the most abundant protein found inthe erythrocyte membrane facilitating anion exchangeand the transfer of bicarbonate (HCO3
−) and chloride(Cl−) ions. Band 3 protein is hydrophobic with manynon-polar side chains and is released from the mem-brane only after extraction with detergents or organicsolvents. As such it is typical of many membrane pro-teins and until recently there was little detail on theoverall tertiary structure.
Band 3 consists of two independent domains(Figure 5.6). The N-terminal cytoplasmic domain linksthe membrane to the underlying spectrin–actin-basedcytoskeleton using ankyrin and protein 4.1 as bridg-ing proteins. The C-terminal domain mediates anionexchange across the erythrocyte membrane. As aresult of its cytoplasmic location the structure of
the N-terminal domain has been established bycrystallography to a resolution of 2.6 A but the mem-brane C-terminal domain remains structurally problem-atic. The cytoplasmic domain contains 11 β strandsand 10 helical segments and belongs to the α + β foldclass. Eight β-strands assemble into a central β-sheetof parallel and antiparallel strands whilst two of theremaining strands (β6 and β7, residues 176–185) forma β-hairpin that constitutes the core of the ankyrin bind-ing site.
The approach of studying large extracellular regionsbelonging to membrane proteins is widely used to pro-vide information since it represents in many instancesthe only avenue of structural analysis possible forthese proteins.
Membrane proteins with globular domains
Integral membrane proteins are distinguished from sol-uble domains by their high content of hydrophobicresidues. Residues such as leucine, isoleucine andvaline often occur in blocks or segments of approxi-mately 20–30 residues in length. Consequently, it ispossible to identify membrane proteins by the distribu-tion of residues with hydrophobic side chains through-out a primary sequence. Hydropathy plots reflectthe preference of amino acid side chains for polar

112 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
and non-polar environments and as such numericalparameters are assigned to each residue (Table 5.5).The hydropathy values reflect measurements of thefree energy of transfer of an amino acid from non-polar to polar solvents. By assigning numerical valuesto each residue along a sequence and then averagingthese values over a pre-defined window size a hydropa-thy profile is established. For a small window sizethe precision is not great but for a window size ofbetween 18 and 22 accurate prediction of transmem-brane regions is possible. Increasingly sophisticatedalgorithms allow transmembrane region recognitionand this approach is routinely performed on new,unknown, proteins.
Transmembrane regions occur as peaks in thehydropathy profile. An analysis of band 3 proteinreveals a primary sequence containing many hydropho-bic residues arranged in at least 13 transmembranedomains. This was therefore consistent with its roleas an integral membrane protein. In contrast the ‘inte-gral’ protein glycophorin also found in the erythrocytehas a low content of non-polar residues compatiblewith a single transmembrane helix (Figure 5.7). This
Table 5.5 Kyte and Doolittle scheme of rankinghydrophobicity of side chains
Aminoacid
Parameter Aminoacid
Parameter
Ala 1.80 Leu 3.80Arg −4.50 Lys −3.90Asn −3.50 Met 1.90Asp −3.50 Phe 2.80Cys 2.50 Pro −1.60Gln −3.50 Ser −0.80Glu −3.50 Thr −0.70Gly −0.40 Tyr −0.90His −3.20 Trp −1.30Ile 4.50 Val 4.20
was unexpected in view of its location and shows thedramatic variation of integral protein organization andsecondary structure.
7
6
5
4
3
2
1
00 100 200 300 400 500
Residue Number
Pro
file
Sco
re
600 700 800 900 1000
5
4
3
2
1
00 20 40 60 80 100
Residue Number
Pro
file
Sco
re
120 140
Figure 5.7 The putative transmembrane domains of band 3 and glycophorin. The red bars indicate the positions ofthe transmembrane domains

MEMBRANE PROTEIN TOPOLOGY AND FUNCTION SEEN THROUGH ORGANIZATION OF THE ERYTHROCYTE MEMBRANE 113
Figure 5.8 Sequence and postulated structure of glycophorin A. The external N terminal region has many polarresidues and is extensively glycosylated. There is no glycosylation of the hydrophilic domain on the cytoplasmicside of the membrane. The non-polar region stretches for approximately 20 residues. A common genetic variant ofglycophorin occurs at residues 1 and 5 with Ser and Gly replaced by Leu and Glu
Careful inspection of the hydropathy plot for gly-cophorin reveals that a single hydrophobic domainextends for approximately 20–25 residues and that lessthan 20 % of the protein’s 131 residues are compat-ible with membrane locations. This observation wassupported by amino acid sequencing of glycophorin,the first membrane protein to be sequenced, showingsignificant numbers of polar residues at the N and Cterminals (Figure 5.8).
A combination of biochemical techniques estab-lished the arrangement of glycophorin – large hydro-
philic domains protruded from either side of theerythrocyte membrane. One approach was to usemembrane-impermeant agents that reacted with specificresidues to yield fluorescent or radioactive labels thatfacilitated subsequent detection. Using this approachonly the N-terminal domain was labelled and it wasdeduced that this region protruded out from the cell’ssurface. Rupturing the erythrocyte membrane labelledC-terminal domains but the transmembrane regionwas never labelled. Detailed studies established an N-terminal region of approximately 70 residues (1–72)

114 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
with the hydrophobic domain extending from 73–92and the C-terminal region occupying the remainingfraction of the polypeptide from residues 92–131.In vivo the hydrophobic domain promotes dimeriza-tion and studies have shown that fusion of this pro-tein sequence to ‘foreign’ proteins not only promotesmembrane localization but also favours associationinto dimers. Dimerization is sequence dependent withmutagenesis of critical residues completely disruptingprotein association. However, although considerabledetail has been acquired on the organization of proteinswithin the erythrocyte membrane a description at anatomic level was largely lacking. In part this stemmedfrom the difficulty in isolating proteins in forms suit-able for analysis by structural methods.
What was required was a different approach. Anew approach came with two very different integralmembrane proteins. The first was the purple protein,bacteriorhodopsin, found in the bacterium Halobac-terium halobium. This membrane protein functions asa light-driven proton pump. The second system wasa macromolecular photosynthetic complex from purplenon-sulfur bacteria that ultimately converted light intochemical energy. The results from studies of these pro-teins would revolutionize our understanding of mem-brane protein structure.
Bacteriorhodopsin and the discoveryof seven transmembrane helices
The study of the protein bacteriorhodopsin represents alandmark in membrane protein structure determinationbecause for the first time it allowed an insight into theorganization of secondary and tertiary structure, albeitat low resolution. Bacteriorhodopsin, a membrane pro-tein of 247 residues (Mr∼26 kDa), accumulates withinthe membranes of H. halobium – a halophilic (salt lov-ing) bacterium that lives in extreme conditions and isrestricted to unusual ecosystems. The cell membrane ofH. halobium contains patches approximately 0.5 µmwide consisting predominantly of bacteriorhodopsinalong with a smaller quantity of lipid. These patchescontribute to a purple appearance of membranes andcontain protein arranged in an ordered two-dimensionallattice whose function is to harness light energy for theproduction of ATP via light driven proton pumps. This
NH
CHC
OCH2
CH2
CH2CH2
N+
CH3
CH3H3C CH3CH3H
retinal cofactor Lysine 216
Figure 5.9 The retinal chromophore of bacteri-orhodopsin linked to lysine 216 of bacteriorhodopsin.The retinal is responsible for the purple colour observedin the membranes of H. halobium
process is assisted by the presence of retinal co-factorlinked covalently to lysine 216 in bacteriorhodopsin(Figure 5.9).
Bacteriorhodopsin is the simplest known protonpump and differs from those found in other systemsin not requiring associated electron transfer systems.By dispersing purified purple membranes containingbacteriorhodopsin in lipid vesicles along with bovineheart ATP synthetase (the enzyme responsible for syn-thesizing ATP) Efraim Racker and Walter Stoecke-nius were able to demonstrate that light-driven protonfluxes resulted in ATP synthesis. Not only was this aconvincing demonstration of the light-driven pump ofbacteriorhodopsin but it was also an important mile-stone in emphasizing the role of proton gradients inATP formation.
Richard Henderson and Nigel Unwin used thetwo-dimensional patches of protein to determinethe structure of bacteriorhodopsin using electroncrystallography a technique (see Chapter 10) analo-gous to X-ray crystallography. By a process of recon-struction Henderson and Unwin were able to producelow-resolution structural images of bacteriorhodopsinin 1975 (Figure 5.10). This was the first time the struc-ture of a membrane protein had been experimentallydetermined at anything approaching an atomic levelof resolution.
The structure of bacteriorhodopsin revealed seventransmembrane helices and it was assumed that theseregions were linked by short turns that projectedinto the aqueous phase on either side of the purplemembrane. Shortly after initial structure determinationthe primary sequence of this hydrophobic proteinwas determined. This advance allowed a correlation

BACTERIORHODOPSIN AND THE DISCOVERY OF SEVEN TRANSMEMBRANE HELICES 115
Figure 5.10 A model proposed for the structure ofbacteriorhodopsin derived using electron microscopy(after Henderson, R. & Unwin, P.N.T Nature 1975, 257,28–32). The view shown lies parallel to the membranewith the helices running across the membrane.Although clearly ‘primitive’ and of low resolutionthis representation of seven transmembrane heliceshad a dramatic effect on perceptions of membraneprotein structure
between structural and sequence data. Hydrophobicityplots of the amino acid sequence identified sevenhydrophobic segments within the polypeptide chainand this agreed with the transmembrane helices seenin electron density maps. The primary sequence ofbacteriorhodopsin was pictured as a block of seventransmembrane helices (Figure 5.11).
Subsequently, determination of the structure of bac-teriorhodospin by X-ray crystallography to a resolutionof 1.55 A confirmed the organization seen by elec-tron diffraction and showed that the seven transmem-brane helices were linked by extra- and intracellularloops as originally proposed by Henderson and Unwin(Figure 5.12). Although not apparent from the earlylow-resolution structures a short antiparallel β strand is
found in a loop between helices B and C. More signifi-cantly this structure allowed insight into the mechanismof signal transduction and ion pumping, the major bio-logical roles of bacteriorhodopsin.
The light-driven pump is energized by light absorp-tion by retinal and leads to the start of a photocyclewhere the conversion of retinal from an all-trans iso-mer to a 13-cis isomer is the trigger for subsequentreactions. Photobleaching of pigment leads to the vec-torial release of a proton on the extracellular side ofthe membrane and a series of reactions characterizedby spectral intermediates absorbing in different regionsof the UV–visible spectrum.
The discovery that bacteriorhodopsin containedseven transmembrane helices arranged transverselyacross the membrane has assisted directly in under-standing the structure and function of a diverse rangeof proteins. Of direct relevance is the presence ofrhodopsin in visual cycle of the eye in vertebrates andinvertebrates as well as a large family of proteins basedon seven transmembrane helices.
Seven transmembrane helices and thediscovery of G-protein coupled receptors
These receptors are embedded within membranes andare not easily isolated let alone studied by structuralmethods. However, cloning cDNA sequences for manyputative receptors allowed their amino acid sequencesto be defined with the recognition of similaritywithin this large class of receptors. The proteinswere generally 400–480 residues in length withsequences containing seven conserved segments orblocks of hydrophobic residues. These seven regions ofhydrophobic residues could only mean, in the light ofthe work on bacteriorhodopsin, transmembrane heliceslinked by cytoplasmic or extracellular loops. Amongstthe first sequences to be recognized with this topologywas human β2 adrenergic receptor (Figure 5.13). Seventransmembrane helices approximately 24 residues inlength were identified together with a glycosylated N-terminal domain projecting out from the extracellularsurface and on the other side of the membrane a muchlarger C-terminal domain.
Prior to sequence analysis receptors such as the β2
adrenergic receptors had been subjected to enormousexperimental study. It was shown that receptors bind

116 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
Figure 5.11 The primary sequence of bacteriorhodopsin superimposed on a seven transmembrane helix model.Charged side chains are shown in either red (negative) or blue (positive). Charged residues are usually found either inthe loop regions or at the loop–helix interface. The presence of charged residues within a helix is rare and normallyindicates a catalytic role for these side chains. This is seen in helix C and G of bacteriorhodopsin
ligands or hormones with kinetics resembling thoseexhibited by enzymes where the hormone (substrate)bound tightly with dissociation constants measured inthe picomolar range (10−12 M). Binding studies alsohelped to establish the concept of agonist and antag-onist. The use of structural analogues or agonists tothe native hormone yielded a binding reaction andelicited a normal receptor response. In contrast antago-nists compete for hormone binding site on the receptorinhibiting the normal biological reaction. One impor-tant hormone antagonist is propranolol – a member ofa group of chemicals widely called β blockers – thatcompetes with adrenaline (epinephrine) for binding
sites on adrenergic receptors. These compounds havebeen developed as drugs to control pulse rate and bloodpressure. Not only have useful drugs emerged fromagonist and antagonist binding studies of adrenergicreceptors but four major groups of adrenergic receptorhave been recognized in the membranes of vertebrates(α1, α2 β1 and β2).
These receptors play a role in signal transductionand a major discovery in 1977 was the distinctionbetween β adrenergic receptors and adenylate cyclase.Adenylate cyclase was shown in classic research onhormonal control of glycogen levels to be a membranebound receptor for adrenaline. This led to the incorrect

BACTERIORHODOPSIN AND THE DISCOVERY OF SEVEN TRANSMEMBRANE HELICES 117
Figure 5.12 High resolution structure of bacteri-orhodopsin (PDB:1C3W) showing seven transmembranehelices linked by extra and intracellular loops
idea that receptors were adenylate cyclase – a roleshown later to be performed by G protein coupledreceptors. Both adenylate cyclase and membrane boundreceptors catalyse the activation or inhibition ofcyclic AMP synthesis. A crucial discovery was theobservation that transduction of the hormonal signalto adenylate cyclase involved not only a membranebound receptor but a separate group of proteins calledG proteins. G proteins are so called because they bindguanine nucleotides such as GTP/GDP. As a resultthe overall picture of signal transduction mechanismschanged to involve a hormone receptor, adenylatecyclase and a G protein. All of these proteins areassociated with the membrane.
Studies of bacteriorhodopsin played a key role inunderstanding rhodopsin, the photoreceptive pigmentat the heart of the visual cycle and responsible for thetransduction of light energy in the eyes of vertebratesand invertebrates. Rhodopsin is the best characterizedreceptor and much of our current knowledge ofsignal transduction arose from studies of G proteinsand G protein-coupled receptors in the visual cycle.The model of signal transduction receptors as seventransmembrane helices with extra and intracellular
Extracellularsurface
Cytoplasm
Figure 5.13 The amino acid sequence of the β2
adrenergic receptor defines the organization ofthe protein into seven transmembrane helices. Theadrenergic receptors are so called because they interactwith the hormone adrenaline (after Dohlman et al.Biochemistry 1987, 26, 2660–2666). The receptorinteracts with cellular proteins in the C-terminalregion and interaction is controlled by the reversiblephosphorylation of Ser/Thr residues
loops was in part possible because of the initialstructural studies of bacteriorhodopsin – at first glancea completely unrelated protein.
Within the retina of the eye rhodopsin is themajor protein of the disc membrane of rod cellsand by analogy to the bacterial protein it containsa chromophore 11-cis retinal that binds to opsin(the protein). Rhodopsin absorbs strongly betweenwavelengths of 400 and 600 nm and absorption oflight triggers the conversion of the bound chromophoreto the all-trans form. Several conformational changeslater metarhodopsin II is formed as an importantintermediate in the multiple step process of visualtransduction. The release of trans-retinal from theprotein and the conversion of retinal back to the initial11-cis state by the action of retinal isomerase signalsthe end of the cycle (Figure 5.14).

118 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
CH
NH
+
CH
NCHO
CH
NH
+
H3N+
H3N+
Light
CHO
all trans retinal rhodopsin
Metarhodopsin II
H+
Opsin
Opsin
rhodopsin
retinal isomerasecatalyses trans to cisconversion11-cis retinal
all trans retinal
Figure 5.14 The basic visual cycle involving chemical changes in rhodopsin. The critical 11-cis bond is shown inred with retinal combining with opsin to form rhodopsin. Light converts the rhodopsin into the trans-retinal followedby the formation of metarhodopsin II. This intermediate reacts with transducin before dissociating after ∼1 s intotrans-retinal and opsin
Photochemically induced conformational changes inmetarhodopsin II lead to interactions with a secondprotein transducin found in the disc membrane of reti-nal rods and cones. Transducin is a heterotrimericguanine nucleotide binding protein (G protein) con-taining three subunits designated as α, β and γ ofmolecular weight 39 kDa, 36 kDa and 8 kDa. Pho-toexcited rhodopsin interacts with the α subunit oftransducin promoting GTP binding in place of GDPand a switch from inactive to active states. Thetransducin α subunit is composed of two domains.One domain contains six β strands surrounded bysix helices whilst the other domain is predominantlyhelical. Although in the visual cycle the extracellu-lar stimulus and biochemical endpoint are different tohormone action the transmembrane events are remark-ably similar.
Interaction between rhodopsin and G protein causesdissociation with the Gα subunit separating from the
Gβγ dimer (Figure 5.15). The transducin Gα –GTPcomplex activates a specific phosphodiesterase enzymeby displacing its inhibitory domain and thus trigger-ing subsequent events in the signalling pathway. Thephosphodiesterase cleaves the cyclic nucleotide guano-sine 3′, 5′-monophosphate (cyclic GMP) in a processanalogous to the production of cyclic AMP in hor-mone action and this in turn stimulates visual signalsto the brain.
As a signalling pathway in humans the rhodopsin–transducin system contributed significantly to ourunderstanding of other G proteins and G-protein cou-pled receptors. Subsequent work has suggested that allG proteins share a common structure of three sub-units. The identification of human cDNA encoding24 α proteins, 5 β proteins and 6 γ proteins testifiesto the large number of G protein catalysed reactionsand their importance within cell signalling pathways.The α subunits of G proteins are one group of an

BACTERIORHODOPSIN AND THE DISCOVERY OF SEVEN TRANSMEMBRANE HELICES 119
Figure 5.15 The structure of the Gα subunit oftransducin. Two domains are shown in dark green(helical) and blue (α + β) together with a GTP analogue(magenta) bound at the active site (PDB: 1TND)
important class of nucleotide binding proteins whoseactivity is enhanced by GTP binding and diminishedby GDP binding. Other members with this group ofG proteins include the elongation factors involved inprotein synthesis and the products of oncogenes suchas Ras.
Addressing hormone action is an entirely analogousprocess and is best visualized through inspection ofthe structure of the β2 adrenergic receptor. Hormonebinding at the extracellular surface promotes confor-mational change and the interaction of the C-terminalregion with the Gα subunit. This stimulates GDP/GTPexchange and in its active state the Gα subunit bindsto adenylate cyclase causing the formation of cAMPwhich functions downstream in protein phosphoryla-tion reactions that stimulate or inhibit metabolic pro-cesses (Figure 5.16). G proteins are further sub-dividedinto two major classes: Gs and Gi. The former stimu-late adenylate cyclase whilst the latter inhibit althoughboth interact with a wide range of receptors and interactwith other proteins besides adenylate cyclase.
Rhodopsin and β2 adrenergic receptors are seventransmembrane receptor proteins that activate het-erotrimeric GTP binding proteins and are groupedunder the term G-protein coupled receptors (GPCRs).
GPCRs are part of an even larger group of seven trans-membrane receptors and represent the fourth largestsuperfamily found in the human genome. It is esti-mated that within the human genome there are over 600genes encoding seven transmembrane helices receptors.Within this family there are three major structural sub-divisions with the rhodopsin-like family representingthe largest class. In almost all cases there is a com-mon mechanism involving G protein activation and twotypes of intracellular or secondary messengers, calcium(Ca2+) and cyclic AMP (cAMP). Approximately 70 %of GPCRs signal through the cAMP pathway and 30 %signal through the Ca2+ pathway.
Detailed structural data on GPCRs remains lim-ited and this is unfortunate because many proteinswithin this family are important and lucrative phar-maceutical targets. Many drugs in clinical use inhumans are directed at seven transmembrane recep-tors. Despite overwhelming sequence data and biophys-ical data the structure of a non-microbial rhodopsinhas only recently been achieved. The crystal structureconfirmed, perhaps belatedly, the eponymous seventransmembrane helical model but more importantly thisstructure provided a model for other GPCRs that allowsinsight into receptor–G protein complex formation andan understanding of the mechanism by which agonist-receptor binding leads to G protein activation.
The photosynthetic reaction centreof Rhodobacter viridis
Studies of bacteriorhodopsin defined a basic topologyof helices traversing the membrane but did not revealsufficient detail of helical arrangement with respectto each other and the membrane. Detailed analysisof membrane proteins required the use of X-raycrystallography, a technique whose resolution is oftensuperior to electron crystallography. However, thisexperimental approach requires the formation of three-dimensional crystals of suitable size and this hadproved extremely difficult for non-globular proteins.Bacteriorhodopsin forms a two-dimensional array ofmolecules naturally within the membrane but foralmost all other membrane proteins the formation ofcrystals is a major hurdle to be overcome beforeacquisition of structural information.

120 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
Figure 5.16 The adenylate cyclase signalling system. Binding of hormone to either receptor caused a stimulatory(Rs) or inhibitory (Ri) activity of adenylate cyclase. These receptors undergo conformational changes leading tobinding of the G protein and the exchange of GDP/GTP in the Gα subunit. The action of the Gα subunit instimulating or inhibiting adenylate cyclase continues until hydrolysis of GTP occurs. Adenylate cyclase forms cAMPwhich activates a tetrameric protein kinase by promoting the dissociation of regulatory and catalytic subunits(reproduced with permission from Voet, C, Voet, J.G & Pratt, C.W. Fundamentals of Biochemistry John Wiley & Sons,Ltd, Chichester, 1999).
Removing membrane proteins from their environ-ment as a prelude to crystallization was frequentlyunsuccessful since the lipid bilayer plays a majorrole maintaining conformation. Moreover, exposureof hydrophobic residues that were normally in con-tact with non-polar regions of the bilayer to aqueoussolutions resulted in many proteins unfolding with aconcomitant loss of activity. One approach adoptedin an attempt to improve methods of membrane pro-tein crystallization was to substitute detergents that
mimicked the structure and function of lipids. In manyinstances these detergents failed to preserve structureor function satisfactorily. The situation changed drasti-cally in 1982 when Hartmut Michel prepared highlyordered crystals of the photosynthetic reaction cen-tre from Rhodobacter viridis (originally Rhodopseu-domonas viridis).
The reaction centre is a macromolecular com-plex located in heavily pigmented membranes ofphotosynthetic organisms (Figure 5.17). Within most

BACTERIORHODOPSIN AND THE DISCOVERY OF SEVEN TRANSMEMBRANE HELICES 121
2hu
LH2 LH1 RC
QA QB QH2 QH2
2 QH2
Fe2S2
Q
Cytoplasm
Periplasm
cyt bc1 complex
cyt c1
cyt bL
cyt bH
cyt c2
2H+
4H+ 2 e−
2 e−
2 e−
2H+
Figure 5.17 The organization of reaction centres and the photosynthetic electron transfer system in purple bacteriasuch as R. sphaeroides. In R. viridis a bound cytochrome is linked to the reaction centres but is absent in mostspecies. Bacteriochlorophyll molecules are represented by blue diamonds, bacteriopheophytin molecules by purplediamonds and hemes by red diamonds. The red arrows represent excitation transfer from light harvesting chlorophyllcomplexes and the black and blue arrows correspond to electron and proton transfer, respectively (reproduced withpermission Vermeglio, A & Joliot, P. Trends Microbiol. 1999, 7, 435–440. Elsevier).
bacterial membranes the centre is part of a cyclicelectron transfer pathway using light to generateATP production.
Absorption of a photon by light-harvesting com-plexes (LH1 and LH2) funnels energy to the reac-tion centre initiating primary charge separation wherean electron is transferred from the excited primarydonor, a bacteriochlorophyll dimer, to a quinone accep-tor (QB) via other pigments. After a second turnover,the reduced quinone picks up two protons from thecytoplasmic space to form a quinol (QH2) and is oxi-dized by cytochrome c2, a reaction catalysed by thecytochrome bc1 complex. This reaction releases protonsto the periplasmic space for use in ATP synthesis andthe cyclic electron transfer pathway is completed by thereduction of the photo-oxidized primary electron donor.
Fractionation studies of R. viridis membranes usingdetergents such as lauryl dimethylamine oxide (LDAO)yield a ‘core’ reaction centre complex containing fourpolypeptides lacking the light-harvesting chlorophylland cytochrome bc1 complexes normally associatedwith these centres. The subunits (H, M, and L)
possessed all of the components necessary for primaryphotochemistry and were associated with a membranebound cytochrome.
Analysis of the photosynthetic complex showed thatits ability to perform primary photochemical reactionswas due to the presence of chromophores that includedfour bacteriochlorophyll molecules, two molecules ofa second related pigment called bacteriopheophytin,carotenoids, a non-heme iron centre, and two quinonemolecules (ubiquinone and menaquinone). Using spec-troscopic techniques such as circular dichroism, laserflash photolysis and electron spin resonance the orderand time constants associated with electron transferprocesses was established (Figure 5.18). In all caseselectron transfer was characterized by extremely rapidreactions. The light harvesting complexes act as anten-nae and transfer energy to a pair of chlorophyllmolecules (called P870, and indicating the wavelengthassociated with maximum absorbance) in the reactioncentre located close to the periplasmic membrane sur-face. Absorption of a photon causes an excited stateand at room temperature the photo-excited pair reduces

122 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
Figure 5.18 The sequence of donors and acceptors in the reaction centres of R. viridis. QA is menaquinone and QB
is ubiquinone. Rapid back reactions occur at all stages and the primary photochemical reactions occur very rapidly(ps timescale) presenting numerous technical difficulties in accurate measurement
a pheophytin molecule within 3 ps. The initial pri-mary charge separation generates a strong reductantand reduces the primary acceptor bacteriophaeophytin.Within a further 200 ps the electron is transferred frombacteriopheophytin to a quinone molecule. These reac-tions are characterized by high quantum efficiency andthe formation of a stable charge separated state. Nor-mally the quinone transfers an electron to a secondquinone within 25 µs before further electron transfer toother acceptors located within membrane protein com-plexes. In isolated reaction centres electrons reachingthe quinone recombine with a special pair of chloro-phyll by reverse electron transfer.
The electron transfer pathway was defined by spec-troscopic techniques but the arrangement of chro-mophores together with the topology of proteinsubunits remained unknown until Michel obtainedordered crystals of reaction centres.
Crystallization requires the ordered precipitation ofproteins. Membrane proteins have both hydrophilic andhydrophobic segments due to their topology, with theresult that the proteins are rarely soluble in eitheraqueous buffers or organic media. By partitioning the
membrane protein in detergent (LDAO) micelles itwas possible to purify subunits but it proved verydifficult to crystallize reaction centres from LDAO.As a general rule macromolecular protein complexeswill only aggregate into ordered crystalline arrays iftheir exposed polar surfaces can approach each otherclosely. The use of detergents, particularly if theyformed micelles of large size, tended to limit the closeapproach of polar surfaces preventing crystallization.The major advance of Michel was to introduce smallamphiphilic molecules such as heptane 1,2,3 triol intothe detergent mixture. Although the precise mechanismof action of these amphiphiles is unclear they arethought to displace large detergent molecules fromthe crystal lattice, thereby assisting aggregation. Inaddition they do not form micelles themselves butlower the effective concentration of detergent bybecoming incorporated within micelles and limitingprotein denaturation. Lastly, amphiphiles interact withprotein but do not prevent close proximity of solvent-exposed polar regions.
The structure of the reaction centre revealed thetopology of structural elements within the H,M,L
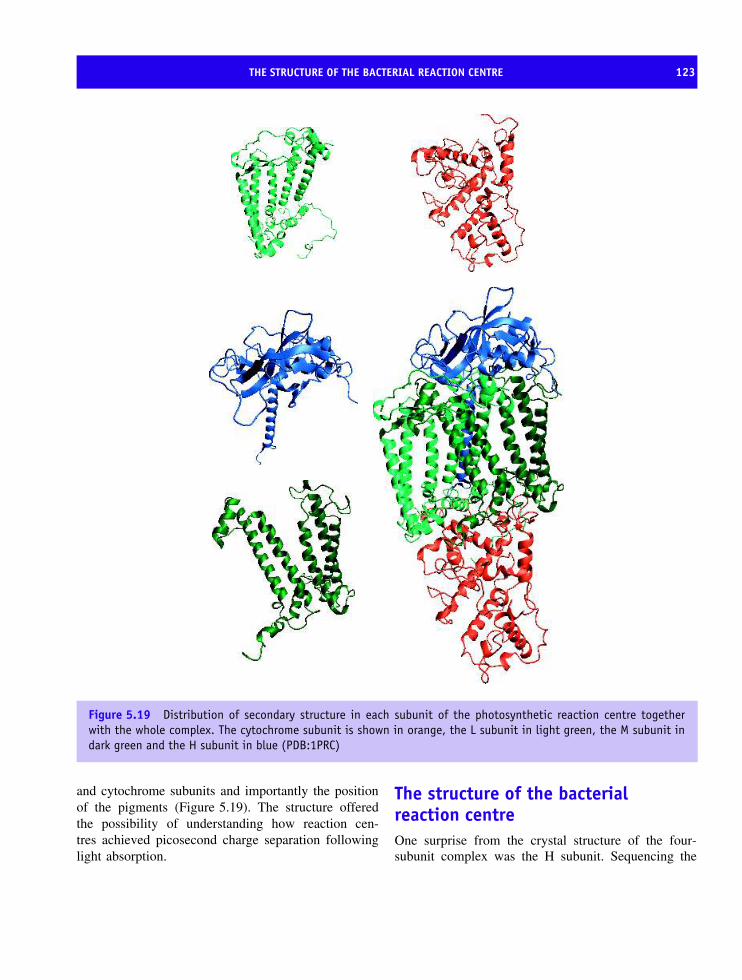
THE STRUCTURE OF THE BACTERIAL REACTION CENTRE 123
Figure 5.19 Distribution of secondary structure in each subunit of the photosynthetic reaction centre togetherwith the whole complex. The cytochrome subunit is shown in orange, the L subunit in light green, the M subunit indark green and the H subunit in blue (PDB:1PRC)
and cytochrome subunits and importantly the positionof the pigments (Figure 5.19). The structure offeredthe possibility of understanding how reaction cen-tres achieved picosecond charge separation followinglight absorption.
The structure of the bacterialreaction centreOne surprise from the crystal structure of the four-subunit complex was the H subunit. Sequencing the

124 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
gene for subunit H revealed that it was the smallestsubunit in terms of number of residues. Its migration onSDS gels was anomalous when compared to the L andM subunits leading to slightly inaccurate terminology.Careful inspection of Figure 5.19 reveals that the Hsubunit is confined to the membrane by a singletransmembrane α helix (shown in blue) whilst mostof the protein is found as a cytoplasmic domain. Thephysiological role of the H subunit is unclear sinceit lacked all pigments. The structure of subunit His divided into three distinct segments; a membrane-spanning region, a surface region from residues 41to 106, and a large globular domain formed by theremaining residues. The membrane-spanning regionof the H subunit is a single regular α helix fromresidues 12 to 35 with some distortion near thecytoplasmic membrane surface. Following the singletransmembrane helix are ∼65 residues forming asurface region interacting with the L and M subunitsand based around a single α helix and 2 β strands. In thecytoplasmic domain of subunit H an extended system
of antiparallel and parallel β strands are found togetherwith a single α helix from residues 232 to 248.
The L and M subunits each contain five transmem-brane helices (Table 5.6) but more significantly eachsubunit is arranged with an approximate two-fold sym-metry that allows the superposition of atoms from onesubunit with those on the other. The superposition issurprising and occurs with low levels of sequence iden-tity (∼26 percent). In each subunit the five helices aredesignated A–E; three (A, B and D) are extremelyregular and traverse the membrane as straight heliceswhilst C and E are distorted. Helix E in both subunitsshows a kink caused by a proline residue that dis-rupts regular patterns of hydrogen bonding. Althoughthe helices show remarkably regular torsion angles andhydrogen bonding patterns they are inclined at anglesof ∼11◦ with respect to the membrane. Elements ofβ strand were absent and the results confirmed thoseobtained with bacteriorhodopsin. In the reaction centrebetween 22 and 28 residues were required to cross themembrane each helix linked by a short loop.
Figure 5.20 The arrangement of heme, bacteriochlorophyll, bacteriopheophytin, quinone and non heme iron in thehydrophobic region between L and M subunits. Electron transfer proceeds from the hemes shown on the left throughto the quinones shown on the right

THE STRUCTURE OF THE BACTERIAL REACTION CENTRE 125
Table 5.6 The transmembrane helices found in theL/M subunits of the reaction centre
Helical segments in subunits L and M
Region and number of residues in brackets
Helix Subunit L Subunit M
A L33–L53 (21) M52–M76 (25)
B L84–L111 (28) M111–M137 (27)
C L116–L139 (24) M143–M166 (24)
D L171–L198 (28) M198–M223 (26)
E L226–L249 (24) M260–M284 (25)
For the L and M subunits the five transmem-brane helices defined the bilayer region of themembrane. Surprisingly, direct interactions betweensubunits are limited by the presence of chromophoresbetween the subunits and most of the subunit con-tacts were restricted to periplasmic surfaces close tothe H subunit.
The cytochrome subunit contained 336 residueswith a total of nine helices and four heme groupscovalently linked to the polypeptide via two thioetherbridges formed with cysteine residues. The fifth andsix ligands to the hemes were methionine and histidinefor three of the centres whilst the fourth showed bis-histidine ligation. The heme groups have differentredox potentials and serve to shuttle electrons tothe special pair chlorophyll after excitation. Althoughpredominantly in the aqueous phase the cytochromeremains membrane bound by a covalently linkeddiglyceride attached to the N-terminal Cys residue.
It is, however, the arrangement of pigments that pro-vides the most intriguing picture and sheds most lighton biological function. The active components (4 ×BChl, 2 × BPh, 2 × Q + Fe) are confined to a regionbetween the L and M subunits in a hydrophobic corewith each chromophore bound via specific side chainligands. The special pair of chlorophyll molecules islocated on the cytoplasmic side of the membrane closeto one of the heme groups of the cytochrome. Strippingthe L and M polypeptides reveals the two-fold sym-metry of the chromophores (see Figures 5.20 & 5.21).The structural picture agrees well with the order of
Figure 5.21 Arrangement of chromophores in thebacterial reaction centre. The cytochrome redox co-factors have been removed
electron transfer established earlier using kinetic tech-niques as
B(Chl)2 −→ BPh −→ QA −→ QB −→ other acceptors
The additional chlorophylls found in the reaction centredo not participate directly in the electron transferprocess and were called ‘voyeur’ chlorophylls.
The co-factors between the L and M subunits arearranged as two arms diverging from the special pairof chlorophylls. A single ‘voyeur’ chlorophyll togetherwith a pheophytin molecule lies on each side andthey lead separately to menaquinone and ubiquinonemolecules with a single non-heme iron centre locatedbetween these acceptors. It was therefore a considerablesurprise to observe that despite the high degree ofinherent symmetry in the arrangement of co-factorsthe left hand branch (L side) was the more favouredelectron transfer route by a ratio of 10:1 over the righthand or M route.
The underlying reasons for asymmetry in electrontransfer are not clear but rates of electron transferare very dependent on free energy differences betweenexcited and charge separated states as well as the dis-tance between co-factors. Both of these parameters areinfluenced by the immediate protein surroundings ofeach co-factor and studies suggest that rates of elec-tron transfer are critically coupled to the propertiesof the bacteriochlorophyll monomer (the voyeur) that

126 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
lies between the donor special pair and bacteriopheo-phytin acceptor.
The X-ray crystal structure of the R. viridis reactioncentre was determined in 1985 and was followed bythe structure of the R. sphaeroides reaction centrethat confirmed the structural design and pointed tofold conservation. Research started with crystallizationof the bacterial photosynthetic resulted in one of theoutstanding achievements of structural biology andHartmut Michel, Johan Diesenhofer, and Robert Huberwere rewarded with the Nobel Prize for Chemistry in1988. Over the last two decades this membrane proteinhas been extensively studied as a model system forunderstanding the energy transduction, the structureand assembly of integral membrane proteins, and thefactors that govern the rate and efficiency of biologicalelectron transfer.
Oxygenic photosynthesis
The photosynthetic apparatus of most bacteria issimpler than that found in algae and higher plants. Heretwo membrane-bound photosystems (PSI and PSII) actin series splitting water into molecular oxygen andgenerating ATP/NADPH via photosynthetic electrontransfer (Figure 5.22). The enzymatic splitting of wateris a unique reaction in nature since it is the onlyenzyme to use water as a principal substrate. Oxygenis one of the products and the evolution of thisreaction represents one of the most critical events inthe development of life. It generated an atmosphererich in oxygen that supported aerobic organisms.The geological record suggests the appearance ofoxygen in the atmosphere fundamentally changedthe biosphere.
The water splitting reaction is carried out by PSIIand involves Mn ions along with specific proteins.Within PSII are two polypeptides called D1 and D2that show a low level of homology with the L andM subunits of anoxygenic bacteria. The structure ofPSII from the thermophilic cyanobacterium (some-times called blue–green algae) Synechococcus elon-gatus confirmed structural homology with bacterialreaction centres. PSII is a large membrane-bound com-plex comprising of at least 17 protein subunits, 13redox-active co-factors and possibly as many as 30
Photosystem II Photosystem I
PQ
Volts
0.0
−1.0
1.0 P680
P680∗P700∗
P700
Q
Ph
A0
A1
A/BFd
Pc
Reiske
Cyt f
H2O
O2
Cyt b
Figure 5.22 The oxygenic photosynthetic systemfound in cyanobacteria and eukaryotic algae and higherplants. Two photosystems acting in series generatereduced NADPH and ATP via the absorption of light.Photosystem II precedes photosystem I
accessory chlorophyll pigments. Of these subunits 14are located within the photosynthetic membrane andinclude most significantly the reaction centre proteinsD1 (PsbA) and D2 (PsbD). The observation that D1and D2 each contained five transmembrane helicesarranged in two interlocked semicircles and related bythe pseudo twofold symmetry axis emphasized the stag-gering similarity to the organization of the L and Msubunits (Figure 5.23). Moreover the level of structuralhomology between D1/D2 and L/M subunits suggestedthat all photosystems evolved from common ances-tral complexes.
Photosystem I
Oxygenic photosynthesis requires two photosystemsacting in series and the structure of the cyanobacterialPSI reaction centre further confirmed the design patternof heterodimers (Figure 5.24). Two subunits, PsaA

PHOTOSYSTEM I 127
Figure 5.23 Structure of PSII with assignment of protein subunits and co-factors. Arrangement of transmembraneα helices and co-factors in PSII. Chl a and hemes are indicated by grey wireframe representations. The direction ofview is from the lumenal side, perpendicular to the membrane plane. The α helices of D1, D2 are shown in yellowand enclosed by an ellipse. Antennae chlorophylls in subunits CP43 and CP47 are enclosed by circles. Unassigned α
helices are shown in grey (reproduced with permission from Zouni, A, et al. Nature 2001 409 739–743. Macmillan)
and PsaB make up a reaction centre that containseleven transmembrane helices. By now this soundsvery familiar, and five helices from each subunittraverse the membrane and together with a thirdprotein, PsaC, contain all of the co-factors participatingin the primary photochemistry. A further six subunitscontribute transmembrane helices to the reaction centrecomplex whilst two subunits (PsaD and E) bind closelyto the complex on the stromal side of the membrane.
Support for the hypothesis that reaction centresevolved from a common ancestor were strengthenedfurther by the observation that the five C-terminal
helices of PsaA and PsaB in PSI showed homology tothe D1/D2 and L/M heterodimers described previously.Reaction centres are based around a special pair ofchlorophyll molecules that transfer electrons via inter-mediary monomeric states of chlorophyll and quinonemolecules to secondary acceptors that are quinones intype II reaction centres and iron–sulfur proteins in typeI reaction centres. Figure 5.25 summarizes the organi-zation of reaction centre complexes.
Oxygenic photosynthesis is the principal energyconverter on Earth converting light energy into biomassand generating molecular oxygen. As a result of the

128 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
D
A B
L
16
I F J K M
5 83 83 15 5 9 4P700
A0
FX
A1
ferredoxin andother soluble proteins
Stroma
Lumen
ThylakoidMembrane
Soluble cytochrome/plastocyanin donor
C E
915 8FA FB
Figure 5.24 The organization of PSI showingsubunit composition and co-factors involved inelectron transfer
success of membrane protein structure determinationwe are beginning to understand the molecular mecha-nisms underlying catalysis in complexes vital for con-tinued life on this planet.
Membrane proteins based ontransmembrane β barrels
So far membrane proteins have been characterized bythe exclusive use of the α helix to traverse membranes.However, some proteins form stable membrane proteinstructure based on β strands. Porins are channel-forming proteins found in the outer membrane ofGram-negative bacteria such as Escherichia coli wherethey facilitate the entry of small polar molecules intothe periplasmic space. Porins are abundant proteinswithin this membrane with estimates of ∼100 000
Figure 5.25 A summary of the distribution of photosynthetic reaction centre complexes in pro- and eukaryotes.Oxygenic photosynthesis is based around two reaction centre complexes, PSI and PSII, while anoxygenicphotosynthesis uses only one reaction centre. This reaction centre can be either type II or type I. Type-IIreaction centres are found in purple non-sulfur bacteria, green filamentous bacteria and oxygenic PSII and type-Ireaction centres are typical of green sulfur bacteria, heliobacteria and oxygenic PSI

MEMBRANE PROTEINS BASED ON TRANSMEMBRANE β BARRELS 129
copies of this protein per cell. Porins have a size limitof less than 600 for transport by passive diffusionthrough pores. Although most porins form non-specific channels a few display substrate-specificitysuch as maltoporin.
The OmpF porin from the outer membrane of E.coli contains 16 β strands arranged in a barrel-likefold (Figure 5.26). The significance of this topology isthat it overcomes the inherent instability of individualstrands by allowing inter-strand hydrogen bonding.OmpF appears to be a typical E. coli porin and willserve to demonstrate structural properties related tobiological function. Although shown as a monomer thefunctional unit for porins is more frequently a trimericcomplex of three polypeptide barrels packed closelytogether within the bilayer.
The individual OmpF monomer contains 362residues within a subunit of 39.3 kDa where the lengthof the monomer (5.5 nm) is sufficient to span a lipidbilayer and to extrude on each side into the aqueousphase. In the barrel hydrophobic groups, particularlyaromatic side chains, occupy regions directly in contact
Figure 5.26 The arrangement of strands in OmpFfrom the outer membrane of E. coli. OmpF stands forouter membrane protein f. The strands make an angleof between 35 and 50◦ with the barrel axis with thebarrel often described as having a rough end, marked bythe presence of extended loop structures and a smoothend where shorter loop regions define the channel
with the lipid bilayer (Figure 5.27). As a consequenceporins have a ‘banded’ appearance when the distribu-tion of hydrophobic residues is plotted over the sur-face of the molecule. The barrel fold creates a cavityon the inside of this protein that is lined with polargroups. These groups located away from the hydropho-bic bilayer form hydrogen bonds with water found inthis cavity.
The cavity itself is not of uniform diameter – thepore range is 1.1 nm at its widest but it is constrictednear the centre with the diameter narrowing to∼0.7 nm. In this cavity are located side chains thatinfluence the overall pore properties. For example,OmpF shows a weak cation selectivity whilst highlyhomologous porins such as PhoE phosphoporin (63percent identity) shows a specificity towards anions.The molecular basis for this charge discrimination hasbecome clearer since the structures of both porins havebeen determined. A region inside the barrel contributesto a constriction of the channel and in most porinsthis region is a short α helix. In OmpF the short helixextends from residues 104 to 112 and is followedby a loop region that, instead of forming the hairpin
Figure 5.27 Distribution of hydrophobic residuesaround the barrel of OmpF porin. The space fillingmodel of porin shows the asymmetric distribution ofhydrophobic side chains (Gly, Ala, Val, Ile, Leu, Phe,Met, Trp and Pro) in a relatively narrow band. This banddelineates the contact region with the lipid bilayer

130 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
Figure 5.28 Two views of the OmpF monomer showing the L3 loop region (yellow) located approximately half waythrough the barrel and forming a constriction site
Figure 5.29 The biological unit of OmpF (and other porins) is a trimer of barrels with interaction occurring via thehydrophobic surfaces of each monomer. The three monomers are shown in a side view (viewed from a position withinthe membrane) and a top view
structure between two strands, projects into the channel(Figure 5.28). In the vicinity of this constriction buton the opposite side of the barrel are charge sidechains that influence ion selectivity. The organizationand properties of OmpF is shared by the PhoE porinbut also by evolutionary more distant porins fromRhodobacter and Pseudomonas species.
Porins are stable proteins because the 16-strandedantiparallel β-barrel is closed by a salt bridge formedbetween the N- and C-terminal residues (Ala and
Phe, respectively) whilst the biological unit packstogether as a trimeric structure enhanced in stabilityby hydrophobic interactions between the sides of thebarrel (Figure 5.29).
Another important example of a membrane proteinbased on a β barrel is α-haemolysin, a channel-forming toxin, exported by the bacterium Staphylococ-cus aureus. Unusually this protein is initially found asa water soluble monomer (Mr∼33 kDa) but assemblesin a multistep process to form a functional heptamer on

MEMBRANE PROTEINS BASED ON TRANSMEMBRANE β BARRELS 131
Figure 5.30 The structure of α-haemolysin showingthe complex arrangement of β strands. The heptameris mushroom shaped as seen from the above figurewith the transmembrane domain residing in the‘stalk’ portion of the mushroom and each protomercontributing two strands of the 14-stranded β barrel.This arrangement is not obvious from the abovestructure but becomes clearer when each monomeris shown in a different colour and from an alteredperspective (Figure 5.31)
the membranes of red blood cells. Once assembled andfastened to the red blood cell membrane α-haemolysincauses haemolysis by creating a pore or channel in themembrane of diameter ∼2 nm. If left untreated infec-tion will result in severe illness.
The monomeric subunit associates with membranescreating a homoheptameric structure of ∼230 kDathat forms an active transmembrane pore that allowsthe bilayer to become permeable to ions, water andsmall solutes. The three-dimensional structure of α-haemolysin (Figures 5.30 and 5.31) shows a structurecontaining a solvent-filled channel of length 10 nm thatruns along the seven-fold symmetry axis of the pro-tein. As with porins the diameter of this channel variesbetween 1.4 and 4.6 nm.
Porins and the α-haemolysins make up a large classof membrane proteins based on β barrels. Previouslythought to be just an obscure avenue of foldingfollowed by a few membrane proteins it is nowclear that this class is increasing in their numberand distribution. Porins are found in a range ofbacterial membranes as well as the mitochondrial andchloroplast membranes. The OmpF structure is basedon 16 β strands and porins from other membranes
Figure 5.31 The structure of α-haemolysin showing the contribution of two strands from each protomer to theformation of a 14-stranded β barrel representing the major transmembrane channel. The channel or pore is clearlyvisible from a top view looking down on the α-haemolysin (PDB: 7AHL)

132 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
contain between 8 and 22 strands (in all cases thenumber of strands is even) sharing many structuralsimilarities.
Respiratory complexesStructure determination of bacterial reaction centresprovided enormous impetus to solving new membraneprotein structures. In the next decade success wasrepeated for membrane complexes involved in respi-ration.
Respiration is the major process by which aerobicorganisms derive energy and involves the transfer ofreducing equivalents through series of electron carri-ers resulting in the reduction of dioxygen to water. Ineukaryotes this process is confined to the mitochon-drion, an organelle with its own genome, found inlarge numbers in cells of metabolically active tissuessuch as flight muscle or cardiac muscle. The mito-chondrion is approximately 1–2 µm in length althoughit may sometimes exceed 10 µm, with a diameter of∼0.5–1 µm and contains a double membrane. Theouter membrane, a semipermeable membrane, con-tains porins analogous to those described in previoussections. However, the inner mitochondrial membraneis involved in energy transduction with protein com-plexes transferring electrons in steps coupled to thegeneration of a proton gradient. The enzyme ATP syn-thetase uses the proton gradient to make ATP fromADP in oxidative phosphorylation.
Purification of mitochondria coupled with fraction-ation of the inner membrane and the use of spe-cific inhibitors of respiration established an orderof electron transfer proteins from the oxidation ofreduced substrates to the formation of water. The innermitochondrial membranes called cristae are highlyinvaginated with characteristic appearances in elec-tron micrographs and are the location for almost allenergy transduction. This process is dominated by fourmacromolecular complexes that catalyse the oxida-tion of substrates such as reduced nicotinamide ade-nine dinucleotide (NADH) or reduced flavin adeninedinucleotide (FADH2) through the action of metallo-proteins such as cytochromes and iron sulfur proteins(Figure 5.32).
Some complexes of mitochondrial respiratory chainsare amenable to structural analysis and although
structures of complex I (NADH-ubiquinone oxidore-ductase; see Figure 5.33) and complex II (succinatedehydrogenase) are not available, advances in definingcomplex III (ubiquinol-cytochrome c oxidoreductase)and the final complex of the respiratory chain, complexIV or cytochrome oxidase, have assisted understandingof catalytic mechanisms.
Complex III, the ubiquinol-cytochromec oxidoreductase
The cytochrome bc family developed early in evolutionand universally participates in energy transduction. Allbut the most primitive members of this family ofprotein complexes contain a b type cytochrome, a ctype cytochrome1 and an iron sulfur protein (ISP).In addition there are often proteins lacking redoxgroups with non-catalytic roles controlling complexassembly. In all cases the complex oxidizes quinolsand transfers electrons to soluble acceptors such ascytochrome c.
Two-dimensional crystals of low resolution (∼20 A)defined the complex as a dimer with ‘core’ proteinslacking co-factors projecting out into solution. Muchlater the structure of the extrinsic domains of theiron–sulfur protein and the bound cytochrome werederived by crystallography but the main body of thecomplex, retained within the confines of the lipidbilayer, remained structurally ‘absent’. At the begin-ning of the 1990s several groups obtained diffractionquality crystals of the cytochrome bc1 complex frombovine and chicken mitochondria. An extensive struc-ture for the cytochrome bc1 complex from bovineheart mitochondria was produced by S. Iwata andco-workers in 1998 at a resolution of 3 A after sev-eral groups had produced structures for the major-ity of the complex. The structures showed similarorganization although small structural differences werenoticed and appeared to be of functional importance(see below).
Isolated cytochrome bc1 complexes from eukary-otic organisms contain 10 (occasionally 11) subunits
1In plants and algae the c type cytochrome is sometimes calledcytochrome f but in all other respects it has analogous structureand function. The f refers to the latin for leaf, frons

COMPLEX III, THE UBIQUINOL-CYTOCHROME C OXIDOREDUCTASE 133
NADH
UQ pool
Succinate-ubiquinoneoxidoreductase
Outermembrane
Innermembrane
Cristae
Matrix
NADH-ubiquinoneoxidoreductase
Ubiquinol-cytochrome coxidoreductase
Cytochrome c
Cytochrome oxidase
I II
III
IV
FADH2
Figure 5.32 The organization of the respiratory chain complexes within the inner mitochondrial membrane. Thetraditional view of mitochondria (centre) was derived from electron microscopy of fixed tissues
including a b type cytochrome with two heme centres,an iron–sulfur protein (called the Reiske protein afterits discoverer) and a mono heme c type cytochrome.DNA sequences for all subunits are known and con-siderable homology exists between the three meta-zoan branches represented by bovine, yeast and potato.Detailed UV-visible absorbance studies combined withpotentiometric titrations established that the two hemesof cytochrome b had slightly different absorbance max-ima in the reduced state (α bands at 566 and 562 nm)
and different equilibrium redox potentials (Em∼120and −20 mV respectively). This led to each heme ofcytochrome b being identified as bH or bL for high andlow potential.
Further understanding of electron transfer in thebc1 complex came from detailed spectroscopic inves-tigations coupled with the use of inhibitors thatblock electron (and proton) transfer at specific sitesby binding to different protein subunits. One com-monly used inhibitor is an anti-fungal compound called
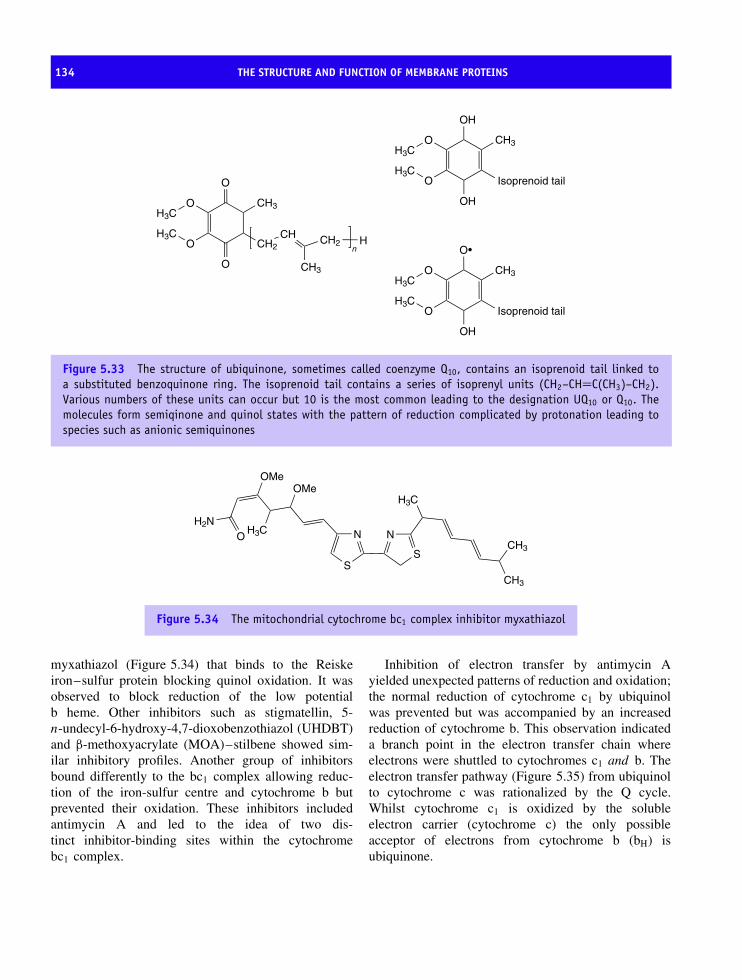
134 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
O
O
O
O
CH2
CH3H3C
H3C CH
CH3
CH2 Hn
O
O
OH
OH
CH3H3C
H3CIsoprenoid tail
O
O
O•
OH
CH3H3C
H3CIsoprenoid tail
Figure 5.33 The structure of ubiquinone, sometimes called coenzyme Q10, contains an isoprenoid tail linked toa substituted benzoquinone ring. The isoprenoid tail contains a series of isoprenyl units (CH2–CH=C(CH3)–CH2).Various numbers of these units can occur but 10 is the most common leading to the designation UQ10 or Q10. Themolecules form semiqinone and quinol states with the pattern of reduction complicated by protonation leading tospecies such as anionic semiquinones
S
N
S
N
H3C
CH3
CH3
H3C
OMeOMe
H2NO
Figure 5.34 The mitochondrial cytochrome bc1 complex inhibitor myxathiazol
myxathiazol (Figure 5.34) that binds to the Reiskeiron–sulfur protein blocking quinol oxidation. It wasobserved to block reduction of the low potentialb heme. Other inhibitors such as stigmatellin, 5-n-undecyl-6-hydroxy-4,7-dioxobenzothiazol (UHDBT)and β-methoxyacrylate (MOA)–stilbene showed sim-ilar inhibitory profiles. Another group of inhibitorsbound differently to the bc1 complex allowing reduc-tion of the iron-sulfur centre and cytochrome b butprevented their oxidation. These inhibitors includedantimycin A and led to the idea of two dis-tinct inhibitor-binding sites within the cytochromebc1 complex.
Inhibition of electron transfer by antimycin Ayielded unexpected patterns of reduction and oxidation;the normal reduction of cytochrome c1 by ubiquinolwas prevented but was accompanied by an increasedreduction of cytochrome b. This observation indicateda branch point in the electron transfer chain whereelectrons were shuttled to cytochromes c1 and b. Theelectron transfer pathway (Figure 5.35) from ubiquinolto cytochrome c was rationalized by the Q cycle.Whilst cytochrome c1 is oxidized by the solubleelectron carrier (cytochrome c) the only possibleacceptor of electrons from cytochrome b (bH) isubiquinone.

COMPLEX III, THE UBIQUINOL-CYTOCHROME C OXIDOREDUCTASE 135
Figure 5.35 Outline of electron transfer reactions in cytochrome bc1 complex. UQ/UQH2 is ubiquinone/ubiquinol;ISP, Reiske iron sulfur protein; Cyt c1, cytochrome c1; bL and bH low and high potential hemes of cytochrome b
The Q cycle accounts for the fact that ubiquinolis a 2e/2H+ carrier whilst the bc1 complex transferselectrons as single entities. Quinones have roles asoxidants and reductants. Quinol is oxidized at a Qo
site within the complex and one electron is transferredto the high-potential portion of the bc1 complex whilstthe other electron is diverted to the low-potential part(cytochrome b) of the pathway. The high-potentialchain, consisting of the Reiske iron–sulfur protein,cytochrome c1 and the natural acceptor of the inter-membrane space (cytochrome c) transfers the firstelectron from quinol through to the final complexof the respiratory chain, cytochrome oxidase. Thelow-potential portion consists of two cytochrome bhemes and forms a separate pathway for transferringelectrons across the membrane from the Qo site to a Qi
site. The rate-limiting step is quinol (QH2) oxidationat the Qo site and it is generally supposed that anintermediate semiquinone is formed at this site. In orderto provide two electrons at the Qi site for reductionof quinone, the Qo site oxidizes two molecules ofquinol in successive reactions. The first electron atthe Qi site generates a semiquinone that is furtherreduced to quinol by the second electron from hemebH. The overall reaction generates four protons inthe inter-membrane space, sometimes called the P-side, and the formation of quinol uses up two protonsfrom the mitochondrial matrix. The whole reaction is
summarized by the scheme
QH2 + 2cyt c(Fe3+) + 2H+ −→ Q
+ 2cyt c(Fe2+) + 4H+
Cytosolic side
Matrix side
ISP cyt c1
Q pool
2 QH2
2 Q
bHQ
Q.−
QH2
cyt c
e e
4H+
2H+
Antimycin A
Myxathiazol
bL
Figure 5.36 The Q cycle and the generation of avectorial proton gradient
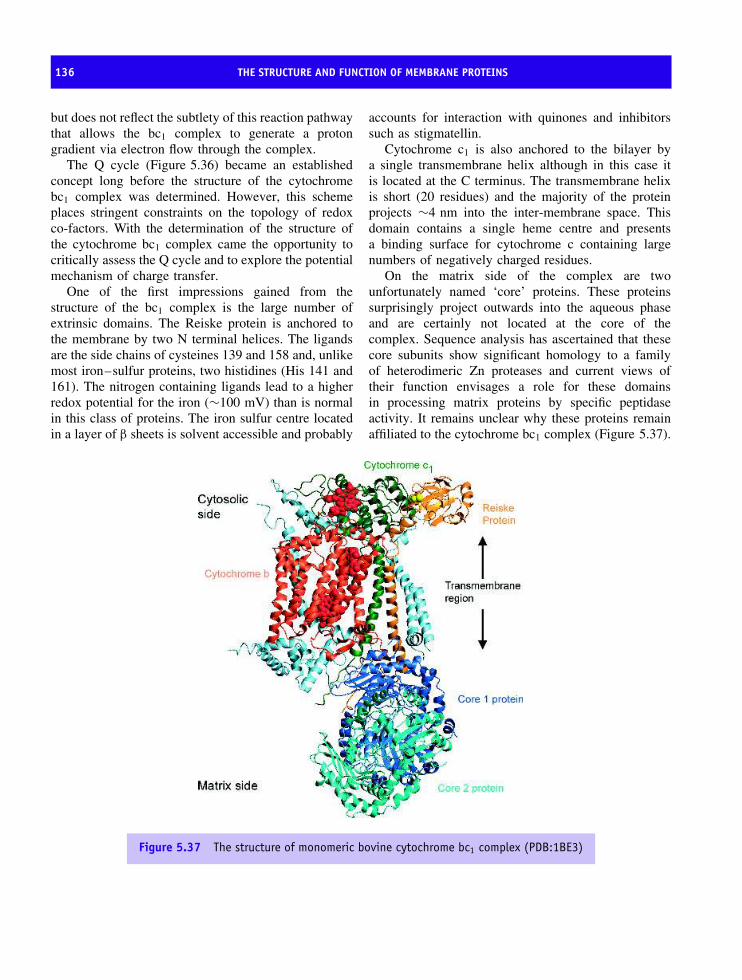
136 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
but does not reflect the subtlety of this reaction pathwaythat allows the bc1 complex to generate a protongradient via electron flow through the complex.
The Q cycle (Figure 5.36) became an establishedconcept long before the structure of the cytochromebc1 complex was determined. However, this schemeplaces stringent constraints on the topology of redoxco-factors. With the determination of the structure ofthe cytochrome bc1 complex came the opportunity tocritically assess the Q cycle and to explore the potentialmechanism of charge transfer.
One of the first impressions gained from thestructure of the bc1 complex is the large number ofextrinsic domains. The Reiske protein is anchored tothe membrane by two N terminal helices. The ligandsare the side chains of cysteines 139 and 158 and, unlikemost iron–sulfur proteins, two histidines (His 141 and161). The nitrogen containing ligands lead to a higherredox potential for the iron (∼100 mV) than is normalin this class of proteins. The iron sulfur centre locatedin a layer of β sheets is solvent accessible and probably
accounts for interaction with quinones and inhibitorssuch as stigmatellin.
Cytochrome c1 is also anchored to the bilayer bya single transmembrane helix although in this case itis located at the C terminus. The transmembrane helixis short (20 residues) and the majority of the proteinprojects ∼4 nm into the inter-membrane space. Thisdomain contains a single heme centre and presentsa binding surface for cytochrome c containing largenumbers of negatively charged residues.
On the matrix side of the complex are twounfortunately named ‘core’ proteins. These proteinssurprisingly project outwards into the aqueous phaseand are certainly not located at the core of thecomplex. Sequence analysis has ascertained that thesecore subunits show significant homology to a familyof heterodimeric Zn proteases and current views oftheir function envisages a role for these domainsin processing matrix proteins by specific peptidaseactivity. It remains unclear why these proteins remainaffiliated to the cytochrome bc1 complex (Figure 5.37).
Figure 5.37 The structure of monomeric bovine cytochrome bc1 complex (PDB:1BE3)

COMPLEX III, THE UBIQUINOL-CYTOCHROME C OXIDOREDUCTASE 137
The major transmembrane protein is cytochrome band the protein has two hemes arranged to facilitatetransmembrane electron transfer exactly as predictedby Q cycle schemes. Each heme is ligated by twohistidine residues and these side chains arise fromjust two (out of a total of eight) transmembranehelices found in cytochrome b. The two hemes areseparated by 20.7 A (Fe–Fe centre distances) with theclosest approach reflected by an edge–edge distanceof 7.9 A – a distance compatible with rapid inter-hemeelectron transfer. Cytochrome b is exclusively α helicaland lacks secondary structure based on β strands.The helices of cytochrome b delineate the membrane-spanning region and are tilted with respect to the planeof the membrane. The remaining subunits of the bc1
complex (see Table 5.7) are small with masses below10 kDa with undefined roles.
Although all of the structures produced fromdifferent sources are in broad agreement it was noticedthat significant differences existed in the position ofthe Reiske iron–sulfur protein with respect to theother redox co-factors. This different position reflectedcrystallization conditions such as the presence ofinhibitors and led to a position for the iron–sulfurprotein that varied from a location close to thesurface of cytochrome b to a site close to cytochromec1. In the presence of the inhibitor stigmatellin the
Table 5.7 The conserved subunits of bovine mito-chondrial cytochrome bc1
Subunit Identity No ofresidues
Mr
1 Core 1 446 536042 Core 2 439 465243 Cyt b 379 427344 Cyt c1 241 272875 Reiske 196 216096 Su6 111 134777 QP-Cb 82 97208 Hinge 78 91259 Su10 62 719710 Su 11 56 6520
The colour shown in the first column identifies the relevant subunitin the structure shown in Figure 5.37.
iron–sulfur protein shifted towards a position proximalto cytochrome b and suggested intrinsic mobility.The concept of motion suggested that the Reiskeprotein might act as a ‘gate’ for electron transfermodulating the flow of electrons through the complex(Figure 5.38).
Cytochrome c1 Cytochrome c1 Cytochrome c1
Figure 5.38 The Reiske iron–sulfur protein acts as a redox gate. The schematic diagrams show the Reiske Fe-Scentre associated with the cytochrome b, in an intermediate state and associated with cytochrome c1. Motion at theReiske Fe-S centre is believed to control electron and proton transfer at the cytochrome bc1 complex

138 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
Complex IV or cytochrome oxidase
Cytochrome c oxidase is the final complex of therespiratory chain catalyzing dioxygen reduction towater. For many years it has occupied biochemiststhoughts as they attempted to unravel the secretsof its mechanism. Isolation of cytochrome oxidasehas two heme groups (designated a and a3) togetherwith 2 Cu centres (called CuA and CuB). Tech-niques such as electron spin resonance and mag-netic circular dichroism alongside conventional spec-trophotometric methods showed that heme a is a lowspin iron centre, whilst heme a3 was a high spinstate compatible with a five-coordinate centre anda sixth ligand provided by molecular oxygen. Thecatalytic function of cytochrome oxidase is summa-rized as
4 Cyt c(Fe2+) + O2 + 8H+ −→ 4 Cyt c(Fe3+)
+ 2H2O + 4H+
where oxidation of ferrous cytochrome c leads tofour-electron reduction of dioxygen and the generationof a proton electrochemical gradient across the innermembrane. Armed with knowledge of the overallreaction spectroscopic studies established an order forelectron transfer of
Cyt c −→ CuA −→ heme a −→ heme a3–CuB
CuB and heme a3 were located close together mod-ulating their respective magnetic properties and thiscluster participated directly in the reduction of oxy-gen with inhibitors such as cyanide binding tightlyto heme a3–CuB and abolishing respiration. Transientintermediates are detected in the reduction of oxy-gen and included the formation of dioxygen adjunct(Fe2+–O2), the ferryl oxide (Fe4+–O) and the hydrox-ide (Fe3+–OH). Other techniques established histidineside chains as the ligands to hemes a and a3–CuB onsubunit I and CuA as a binuclear centre.
The catalytic cycle occurs as discrete steps involvingelectron transfer from cytochrome c via CuA, heme aand finally to the heme a3–CuB site binuclear complex(Figure 5.39). The initial starting point for catalysisinvolves a fully oxidized complex with ferric (Fe3+)and cupric centres (Cu2+).
Complex IV, with oxidized heme a3–CuB centres,receives two electrons in discrete (1e) steps from thepreceding donors. Both centres are reduced and bindtwo protons from the mitochondrial matrix. In this stateoxygen binds to the binuclear centre and is believed tobridge the Fe and Cu centres. Reduction of dioxygenforms a stable peroxy intermediate that remains bound(Fe3+– O−=O−–Cu2+) and further single electron(from cytochrome c) and proton transfers lead to theformation of hydroxyl and then ferryl (Fe4+) states.Rearrangement at the catalytic centre during the lasttwo stages completes the cycle forming two moleculesof water for every oxygen molecule reduced.
In the absence of a structure for cytochrome oxidasethe scheme proposed would need to consider the routeand mechanism of proton uptake for the reduction ofoxygen from the matrix side of the enzyme to thecatalytic site, the mechanism and route of substrate(O2) binding to the active site and finally the routefor product removal from the catalytic site. A furtherdifficulty in understanding the catalytic mechanism ofcytochrome oxidase lies with the observation that inaddition to the protons used directly in the reductionof water it appears from measurements of H+/eratios that up to four further protons are pumpedacross the membrane to the inter-membrane space. Itseems likely that conformational changes facilitatingelectron and proton transfer lie at the heart of oxidasefunctional activity.
Cytochrome oxidase from denitrifying bacteria suchas Paracoccus denitrificans has a simpler subunit com-position with only three subunits: all of the redox com-ponents are found on the two heaviest polypeptides(subunits I and II). In eukaryotic cytochrome oxidaseat least 13 subunits are present with subunits I–III,comparable to those observed in prokaryotic oxidases,encoded by the mitochondrial genome. The crystalliza-tion of the complete 13 subunit complex of bovinecytochrome oxidase by Yoshikawa and colleagues in1995 resolved or suggested answers to several out-standing questions concerning the possible electrontransfer route through the complex, a mechanism andpathway of proton pumping and the organization of thedifferent subunits.
The mitochondrially coded subunits are the largestand most hydrophobic subunits and contain four redoxcentres. Surprisingly the oxidase binds two additional
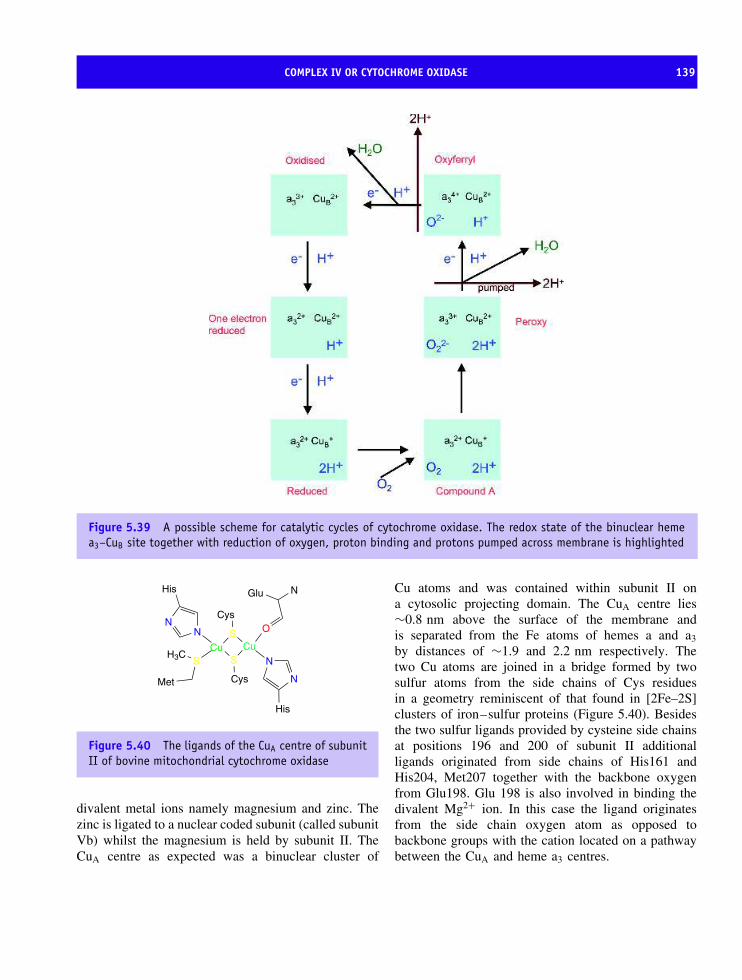
COMPLEX IV OR CYTOCHROME OXIDASE 139
Figure 5.39 A possible scheme for catalytic cycles of cytochrome oxidase. The redox state of the binuclear hemea3–CuB site together with reduction of oxygen, proton binding and protons pumped across membrane is highlighted
SCu
SCu
Cys
Cys
His
N
N
His
NN
Met
SH3C
N
O
Glu
Figure 5.40 The ligands of the CuA centre of subunitII of bovine mitochondrial cytochrome oxidase
divalent metal ions namely magnesium and zinc. Thezinc is ligated to a nuclear coded subunit (called subunitVb) whilst the magnesium is held by subunit II. TheCuA centre as expected was a binuclear cluster of
Cu atoms and was contained within subunit II ona cytosolic projecting domain. The CuA centre lies∼0.8 nm above the surface of the membrane andis separated from the Fe atoms of hemes a and a3
by distances of ∼1.9 and 2.2 nm respectively. Thetwo Cu atoms are joined in a bridge formed by twosulfur atoms from the side chains of Cys residuesin a geometry reminiscent of that found in [2Fe–2S]clusters of iron–sulfur proteins (Figure 5.40). Besidesthe two sulfur ligands provided by cysteine side chainsat positions 196 and 200 of subunit II additionalligands originated from side chains of His161 andHis204, Met207 together with the backbone oxygenfrom Glu198. Glu 198 is also involved in binding thedivalent Mg2+ ion. In this case the ligand originatesfrom the side chain oxygen atom as opposed tobackbone groups with the cation located on a pathwaybetween the CuA and heme a3 centres.

140 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
Heme a is coordinated by the side chains of His61and His378 of subunit I. The iron is a low spinferric centre lying in the plane of the heme withtwo imidazole ligands – a geometry entirely consistentwith the spin properties. The heme plane is positionedperpendicularly to the membrane surface with thehydroxyethylfarnesyl sidechain of the heme pointingtowards the membrane surface of the matrix side in afully extended conformation. The remaining two redoxcentres heme a3 and CuB make up the oxygen reductionsite and lie very close to heme a. Hemes a and a3 arewithin 0.4 nm of each other and this short distance isbelieved to facilitate rapid electron transfer.
Of some considerable surprise to most observers wasthe location in the oxidase structure of the fifth ligandto heme a3. The side chain of histidine 376 providesthe fifth ligand to this heme group and lies extremelyclose to one of the ligands to heme a (Figure 5.41). Inthe oxidase structure the CuB site is close to heme a3
(∼0.47 nm) and has three imidazole ligands (His240,290 and 291) together with a strong possibility thata fourth unidentified ligand to the metal ion exists.It is generally believed that trigonal coordination isunfavourable and a more likely scenario will involve atetrahedral geometry around the copper.
A hydrogen-bond network exists between the CuA
and heme a and includes His204, one of the ligandsto CuA, a peptide bond between Arg438 and Arg439and the propionate group of heme a. Althoughdirect electron transfer between these centres has notbeen observed and they are widely separated thearrangement of bonds appears conducive for facileelectron transfer. In addition, a network connects CuA
and heme a3 via the magnesium centre and mayrepresent an effective electron transfer path directlybetween these centres. Direct electron transfer betweenCuA and heme a3 has never been detected with kineticmeasurements, suggesting that electron transfer fromCuA to heme a is at least two orders of magnitudegreater than the rate from CuA to heme a3. From thearrangement of the intervening residues in subunit Ithe structural reasons underlying this quicker electrontransfer from CuA −→ heme a than CuA −→ heme a3
are not obvious.Before crystallization the composition and arrange-
ment of subunits within eukaryotic oxidases was
heme a3
Tyr 244
His 240
His 290
His 376
His 291
CuB
Figure 5.41 The heme a3–CuB redox centre ofcytochrome oxidase. The dotted line denotes ahydrogen bond and broken lines denote coordinationbonds. Heme a3 is shown in blue, CuB in pink and aminoacid residues in green. (Reproduced with permissionfrom Yoshikawa, S. Curr. Opin. Struct. Biol. 1997, 7,574–579)
unclear. This has changed dramatically with the crys-tallization of oxidases from bovine heart mitochondriaand P. denitrificans so that there is now precise infor-mation on the arrangement of subunits (Table 5.8). Inthe crystal structure of bovine cytochrome oxidase theenzyme is a homodimer with each monomer contain-ing 13 polypeptides and six co-factors. The assemblyof the 13 subunits into a complex confirmed that thesepolypeptides are intrinsic constituents of the activeenzyme and not co-purified contaminants. The crystalstructure confirmed the presence of Zn and Mg atomsas integral components albeit of unknown function.
In the mammalian enzyme the 13 subunits ofcytochrome oxidase contain 10 transmembrane proteinsthat contribute a total of 28 membrane-spanninghelices. The three subunits encoded by mitochondrialgenes form a core to the monomeric complex with the10 nuclear-coded subunits surrounding these subunitsand seven contributing a single transmembrane helix.The remaining three nuclear coded subunits (subunitsVa, Vb, VIb) do not contain transmembrane regions.

COMPLEX IV OR CYTOCHROME OXIDASE 141
Table 5.8 The polypeptide composition of eukaryoticand prokaryotic oxidases
Subunit Prokaryotic Eukaryotic
I 554, 620112 514, 57032II 279, 29667 227, 26021III 273, 30655 261, 29919IV 49,∗ 5370 147, 19577Va Absent 109, 12436Vb 98, 10670VIa 97, 10800VIb 85, 10025VIc 73, 8479VIIa 80, 9062VIIb 88, 10026VIIc 63, 7331VIII 70, 7639
∗Paracoccus denitrificans has a fourth subunit that is unique toprokaryotes with no obvious homology to that found in eukaryotes.Three of the subunits of the eukaryotic enzyme, VIa, VIIa, and VIII,have two isoforms. Of the two isoforms one is expressed in heart andskeletal muscle and the other is expressed in the remaining tissues.The masses and length of the peptides includes signal sequences fornuclear-coded proteins.
In addition to the three metal centre (heme a, hemea3 and CuB) subunit I has 12 transmembrane α helicesand this was consistent with structural predictionsbased on the amino acid sequence. Subunit I is confinedto the bilayer region with few loops extending out intothe soluble phase. The subunit is the largest found incytochrome oxidase containing just over 510 residues(Mr ∼57 kDa) (Figure 5.42).
Subunits II and III both associate with transmem-brane regions of subunit I without forming any directcontact with each other. From the distribution ofcharged residues on exposed surfaces of subunit IIan ‘acidic’ patch faces the inter-membrane space anddefines a binding site for cytochrome c. Unsurprisinglyin view of its role as the point of entry of electronsinto the oxidase subunit II has just two transmembranehelices interacting with subunit I, but has a much largerpolar domain projecting out into the inter-membranespace. This domain consists of a 10-stranded β barrel
Figure 5.42 The monomeric ‘core’ of bovinecytochrome oxidase showing subunits I and II. SubunitI is shown in gold whilst subunit II is shown in blue.The limited number of transmembrane helices in sub-unit II is clear from the diagram in contrast to subunitI. The Cu atoms are shown in green, Mg in purple andthe hemes in red (PDB: 1OCC)
with some structural similarities to class I copper pro-teins, although it is a binuclear Cu centre. The magne-sium ion is located at the interface between subunits Iand II.
Subunit III lacks redox co-factors but remains asignificant part of the whole enzyme complex. Ithas seven α helices and is devoid of large extra-membrane projections. The seven helices are arrangedto form a large V-shaped crevice that opens towardsthe cytosolic side where it interacts with subunit VIb(Figure 5.43).
Subunits Va and Vb (Figure 5.44) are found on thematrix side and both are predominantly located in thesoluble phase. Subunit Va is extensively helical withsubunit Vb possessing both helical- and strand-richdomains and binding a single Zn ion within a series

142 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
Figure 5.43 To the monomeric core of subunits I andII the remaining mt DNA coded subunit (III) has beenadded along with subunit IV. Subunit IV (red) has asingle helix crossing the membrane with a helix richdomain projecting into the matrix phase. Subunit III(green) does not make contact with subunit II andconsists almost exclusively of transmembrane helices
of strands linked by turns that form a small barrel-likestructure.
Subunit VIa contributes a single transmembranehelix and is located primarily towards the cytosolic sideof the inner mitochondrial membrane. Subunit VIb islocated on the same side of the membrane but doesnot contribute transmembrane helices to the oxidasestructure. It is affiliated primarily with subunit III. Sub-unit VIc contributes a single transmembrane helix andinteracts predominantly with subunit II (Figure 5.45).
Subunits VIIa, VIIb and VIIc, together with subunitVIII (Figure 5.46), are small subunits with less than 90residues in each of the polypeptide chains. Their bio-logical function remains unclear. Unlike complex III afar greater proportion of cytochrome oxidase is retainedwith the boundaries imposed by the lipid bilayer.
The structural model for cytochrome oxidase pro-vides insight into the mechanism of proton pump-ing, a vital part of the overall catalytic cycle ofcytochrome oxidase. Proton pumping results in thevectorial movement of protons across membranes and
Figure 5.44 The structure of cytochrome oxidasemonomer (PDB: 1OCC). Subunits Va (orange) and Vb(aquamarine) have been added. Subunit Vb binds Zn(shown in purple) found in the crystal structure

COMPLEX IV OR CYTOCHROME OXIDASE 143
Figure 5.45 The three-dimensional structure ofcytochrome oxidase monomer (PDB: 2OCC). The sevensmaller subunits VIa (grey) VIb (purple), VIc (lightblue), VIIa (pink), VIIb (beige), VIIc (brown) and VIII(black) have been added to the structure shown inFigure 5.44. Subunits VIIb, VIIc, and VIII are largelyhidden by the larger subunits (I and III) in this view
in bacteriorhodopsin occurred via strategically placedAsp and Glu residues facilitating proton movementfrom one side of the membrane to another. This pic-ture, together with that provided by ion channels suchas porins, suggests a similar mechanism in cytochromeoxidase. The exact mechanism of proton pumping ishotly debated but it was of considerable interest toobserve that the bovine crystal structure contained twopossible proton pathways that were shared by simplerbacterial enzymes (Figure 5.47).
The oxygen binding site is located in the hydropho-bic region of the protein approximately (35 A) fromthe M side. Pathways were identified from hydrogenbonds between side chains, the presence of internalcavities containing water molecules, and structuresthat could form hydrogen bonds with small pos-sible conformational changes to side chains. Twopathways called the D- and K-channels and namedafter residues Asp91 and Lys319 of bovine sub-unit I were identified. (In Paracoccus subunit Ithese residues are Asp124 and Lys354.) Despite sig-nificant advances the proton pumping mechanismof cytochrome oxidase remains largely unresolved.This arises mainly from difficulties associated withresolving proton pumping events during dioxygen
Figure 5.46 The homodimer of bovine cytochrome oxidase using previous colour schemes. This view shows subunitsVIIb, VIIc, and VIII in more detail and is from the opposite side to that of the preceding figures

144 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
Figure 5.47 The proton pathways in bovine and Paracoccus denitrificans cytochrome oxidase. Residues implicatedin proton translocation in bovine cytochrome c oxidase are shown in red and in blue for P. denitrificans oxidase. Thestructures of helices, heme a3 and metal centres such as the Mg2+ binding site are based on PDB: 1ARI (reproducedwith permission from Abramson, S et al. Biochim. Biophys. Acta 2001, 1544, 1–9. Elsevier)
reduction and it is currently unclear whether pump-ing precedes oxygen reduction and what structuralrearrangements in the reaction cycle occur. In bac-teriorhodopsin intermediate structures were detectedand associated with proton movements by combin-ing genetic, spectroscopic and structural studies, andit is likely that such methods will resolve the mecha-nism of proton transfer in cytochrome oxidase in thenear future.
From the study of different membrane proteins acommon mode of proton (or cation) transfer involvesspecifically located aspartate and glutamate residuesto shuttle protons across membranes. Both bacte-riorhodopsin and cytochrome oxidase provide com-pelling evidence in their structures for this method ofproton conduction across membranes.
The structure of ATP synthetaseIt has been estimated that even the most inactiveof humans metabolizes kg quantities of ATP in anormal day. This results from repeated phosphorylationreactions with on average each molecule of ADP/ATPbeing ‘turned over’ about 1000 times per day. Theenzyme responsible for the synthesis of ATP is the F1Fo
ATPase, also called the ATP synthase or synthetase.Dissection of the enzyme complex and genetic studiesshow the synthase to contain eight different subunitsin E. coli whilst a greater number of subunits arefound in mammalian enzymes. Despite variationsin subunit number similarity exists in their ratiosand primary sequences with most enzymes having acombined mass between 550 and 650 kDa. With a

THE STRUCTURE OF ATP SYNTHETASE 145
central role in energy conservation and a presence inbacterial membranes, the thylakoid membranes of plantand algae chloroplasts and the mitochondrial innermembrane of plants and animals the enzyme is oneof the most abundant complexes found in nature.
The link between electron transfer and protontransfer was the basis of the chemiosmotic theoryproposed in its most accessible form by Peter Mitchellat the beginning of the 1960s. One strong line ofevidence linking electron and proton transfer was thedemonstration that addition of chemicals known as‘uncouplers’ dissipates proton gradients and halts ATPsynthesis. Many uncouplers were weak, membranepermeable, acids and a major objective involvedelucidating the link between transmembrane protongradients and the synthesis of ATP from ADP andinorganic phosphate.
Fractionation and reconstitution studies showed thatATP synthases contained two distinct functional unitseach with different biological and chemical properties.In electron micrographs this arrangement was charac-terized by the appearance of the enzyme as a largeglobular projection extending on stalks from the surfaceof cristae into the mitochondrial matrix (Figure 5.48).A hydrophobic, membrane-binding, domain called Fo
interacted with a larger globular domain peripherallyassociated with the membrane. Fo stands for ‘factor
Figure 5.48 An electron micrograph of cristae fromisolated mitochondria showing the F1 units projectinglike ‘lollipops’ from the membrane and into the matrix(reproduced with permission from Voet, D., Voet, J.G &Pratt, C.W. Fundamentals of Biochemistry. John Wiley& Sons, Chichester, 1999)
oligomycin’ referring to the binding of this antibi-otic to the hydrophobic portion whilst F1 is simply“factor one”. Fo is pronounced ‘ef-oh’. The periph-eral protein, F1, was removed from membranes bywashing with solutions of low ionic strength or thosecontaining chelating agents. This offered a convenientmethod of purification. Although not strictly a mem-brane protein it is described in this section because itsphysiological function relies intimately on its associa-tion with the Fo portion of ATP synthetase, a typicalmembrane protein. In an isolated state the F1 unithydrolysed ATP but could not catalyse ATP synthe-sis. For this reason the F1 domain is often describedas an ATPase reflecting this discovery. A picture ofATP synthesis evolved where Fo formed a ‘channel’that allowed a flux of protons and their use by F1 inATP synthesis.
In E. coli Fo is extremely hydrophobic and consistsof three subunits designated a, b and c (Table 5.9). InE. coli a ratio of a1, b2, c9 – 14 exists with the a and csubunits making contact and appearing to form a protonchannel. Within both subunits are residues essentialfor proton translocation and amongst those to havebeen identified from genetic and chemical modificationstudies are Arg210, His245, Glu196, and Glu219 ofthe a subunit and Asp61 of the c subunit. Since all ofthese residues possess side chains that bind protons theconcept of a proton channel formed by these groups isnot unrealistic.
Table 5.9 The composition of ATP synthetases
Subunits in
F1 region
Eukaryotic (bovine)
α3β3γδε
α 509 residues, Mr∼55,164
β 480, 51,595
γ 272, 30,141
δ 190, 20,967
ε 146, 15,652
Subunits in Fo region Prokaryotic (E. coli ) a,b2,c9 – 14
a 271, 30,285
b 156, 17,202
c 79, 8,264

146 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
The much larger F1 unit contains five different sub-units (α–ε) occurring with a stoichiometry α3β3γ1δ1ε1.The total mass of the F1Fo ATPase from bovine mito-chondria is ∼450 kDa with the F1 unit having a mass of∼370 kDa and arranged as a spheroid of dimensions8 × 10 nm wide supported on a stem or stalk 3 nmin length. Chemical modification studies identified cat-alytic sites on the β subunits with affinity labellingand mutagenesis studies highlighting the importance ofLys155 and Thr156 in the E. coli β subunit sequence149Gly-Gly-Ala-Gly-Val-Gly-Lys-Thr-Ala157.
A scheme for ATP synthesis envisaged three dis-crete stages:
1. Translocation of protons by the Fo
2. Catalysis of ATP synthesis via the formation of aphosphoanhydride bond between ADP and Pi by F1
3. A coupling between synthesis of ATP and thecontrolled dissipation of the proton gradient via thecoordinated action and interaction of F1 and Fo.
The mechanism of ATP synthesis
A mechanism for ATP synthesis was proposed byPaul Boyer before structural data was available andhas like all good theories stood the test of time andproved to be compatible with structural models derivedsubsequently for F1/Fo ATPases. Boyer envisaged theF1 ATPase as three connected, interacting, sites eachbinding ADP and inorganic phosphate. The modelembodied known profiles for the kinetics of catalysedreactions and the binding of ATP and ADP byisolated F1. Each catalytic site was assumed to existin a different conformational state although inhibitionof ATP synthesis by modification of residues inthe β subunit suggested that these sites interactedcooperatively.
Rate-limiting steps were substrate binding or prod-uct release (i.e ATP release or ADP binding) but notthe formation of ATP. Careful examination of the ATPhydrolysis reaction suggested three binding sites forATP each exhibiting a different Km. When ATP isadded in sub-stoichiometric amounts so that only oneof the three catalytic sites is occupied substrate bind-ing is very tight (low Kd) and ATP hydrolysis occursslowly. Addition of excess ATP leads to binding at all
three sites but with lowered affinity at the second andthird sites. The Kd for binding ATP at the first site is<1 nM whereas values for sites 2 and 3 are ∼1 µM
and ∼30 µM. Upon occupancy of the third site, therate of overall ATP hydrolysis increases by a factor of∼104 –105.
As long as the results from hydrolysis of ATPcan be related directly to that of the synthetic reac-tion and isolated F1 behaves comparably to the com-plete F1Fo complex then these results point towardskey features of the catalytic mechanism. IsolatedF1 exhibits negative cooperativity with respect tosubstrate binding where ATP binding at the sitewith the lowest Km makes further binding of ATPmolecules difficult. This occurs because the Km forother sites is effectively increased. In contrast theenzyme displays positive cooperativity with respect tocatalytic activity with the rate increasing massively ina fully bound state. To explain these unusual prop-erties, Boyer proposed the ‘binding change’ mecha-nism (Figure 5.49).
A central feature of this hypothesis are threecatalytic sites formed by the three α/β subunit pairseach with a different conformation at any one time.One site is open and ready for substrate binding(ATP or ADP + Pi) whilst the second and third sitesare partly open and closed, respectively. Substratebinding results in the closure of the open site andproduces a cooperative conformational change in theother two sites; the closed one becomes partly openand the partly open one becomes fully open. In thismanner the sites alternate between conformationalstates. Obvious questions from this model arise. Howare conformational changes required in the bindingchange mechanism propagated? How are these eventslinked to proton translocation and the Fo part ofthe ATPase?
From a consideration of the organization of Fo G.B.Cox independently suggested a rotary motor involvedthe c subunit ring turning relative to the a and bsubunits. This idea was adopted and incorporated asa major refinement of the binding change mechanismwith the rotary switch causing a sequential change inbinding. At the time (1984) the concept of enzymesusing rotary motion to catalyse reactions was unusual.The cooperative kinetics of the enzyme (negative forATP binding and positive for catalysis) supported the

THE STRUCTURE OF ATP SYNTHETASE 147
Figure 5.49 The binding change hypothesis and catalytic site activity at the β subunit. Each catalytic site cyclesthrough three states: T, L and O. ATP binds to the O (open and empty) site converting it into a T (tight andATP-occupied) site. After bond cleavage at this site the T site is converted into the L (loose and ADP-occupied) sitewhere the products escape to recover the O state. The concerted switching of states in each of the sites results inthe hydrolysis of one ATP molecule. Sub-steps in the hydrolysis of one ATP molecule based on kinetic and inhibitorstudies are shown for a three-site mechanism. Thus, ATP binding in the open site (top) leads to transition stateformation and then bond cleavage in a closed site (bottom), followed by release from a partly open site as it opensfully and releases ADP (middle) (reproduced with permission from Trends Biochem. Sci 2002, 27, 154–160. Elsevier)
binding-change mechanism. However, the idea of arotary switch was so novel that there were few seriousattempts to test it until over a decade later when in1995 the first structures for bovine F1 ATPase producedby John Walker and co-workers appeared to supportBoyer’s model.
The structure of the F1 ATPase
As a peripheral membrane protein the water-solubleF1 unit is easily purified and the structure showed analternate arrangement of three α and three β subunitsbased around a central stalk formed by two α helicesrunning in opposite directions derived from the γ
subunit. These two helices form a coiled-coil at thecentre of F1 and the remaining part of the γ subunitprotrudes from the (αβ)3 assembly and interacts withpolar regions of the c subunit of Fo (Figure 5.50).
The α and β subunits exhibit sequence identity of∼20 percent and show similar overall conformations
βTP
βDP
αDP βE
αE
βE
βDP
αDP
αE
αTP
Figure 5.50 A space filling model of bovine F1-ATPase from a side view (left) and from the membrane(right) showing α subunits (red) and β subunits(yellow) and the attached central stalk containingthe γ, δ and ε subunits (blue, green and magenta,respectively). The βTP, βDP and βE subunits had AMP-PNP, ADP and no nucleotide bound, respectively. The α
subunits are named according to the catalytic interfaceto which they contribute

148 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
O− P
O−
O
H P
O−
O
O P O
O−
O
ON N
NN
NH2
OH OH
N
Figure 5.51 The structure of AMP-PNP – a non-hydrolysable derivative of ATP
arranged around the central stalk made up of the γ,δ and ε subunits. F1 was co-crystallized with ADPand AMP-PNP (5′-adenylyl-imidophosphate, a non-hydrolysed analogue of ATP; Figure 5.51). Only fiveof the six nucleotide binding sites were occupied bysubstrate. Each of the α subunits bound AMP-PNP andexhibited identical conformation.
In contrast one β subunit bound AMP-PNP (βTP),another bound ADP (βDP) and the third subunitremained empty (βE). The structure provided com-pelling evidence for three different modes of nucleotidebinding as expected from the Boyer model. Althoughboth α and β subunits have nucleotide binding sitesonly those on the β subunits are catalytically important.
Each α and β subunit contains three distinct domains(Figure 5.52). A β barrel located at the N-terminalis followed by a central nucleotide-binding domaincontaining a Rossmann fold. The β barrels form acrown that sits on top of the central nucleotide-bindingdomain. At the C terminal is a helix rich domain. Theconformation of the β subunit changes with nucleotide-binding forming states associated with high, mediumand low nucleotide affinities. These three states aremost probably related to release of product (ATP),binding of substrates (ADP and inorganic phosphate),and formation of ATP, respectively. In an isolated statethe structure of the β subunit has the open form withnucleotide binding causing transition to closed forms.On the β subunit the principal residues involved inADP binding have been determined by examining thecrystal structure of this subunit in the presence of ADP-fluoroaluminate (ADP-AlF3) (Figure 5.53). Positivelycharged side chains of arginine and lysine have a majorrole in binding ADP-fluoroaluminate whilst the side
Figure 5.52 An individual β subunit showing thethree distinct domains together with the boundnucleotide – in this case ADP. The ADP is shownin yellow, the barrel structure in dark green, thenucleotide binding region in blue and the helix richregion in violet (PDB:1BMF)
chain of Glu188 binds a water molecule and the amideof Gly159 binds the second phosphate group of ADP.Arginine 373 is part of the α subunit and contributesto the binding site for ADP. ADP-fluoroaluminatemimics the transition state complex with fluorine atomsoccupying similar conformations to the extra phosphategroup of ATP but acting as a potent inhibitor ofATP synthesis. Significantly, the structure adopted bythe β subunit in the presence of ADP-AlF3 is verysimilar to the catalytically active conformation forthis subunit (βTP). These results further suggest thatonce the substrate is bound only small conformationalchanges are required to promote synthesis of ATP.

THE STRUCTURE OF ATP SYNTHETASE 149
Al FF
F
O P O
O
OAMP
Mg2+
NH
Gly159NH3
+
Lys162
H2N
H2N
NH2
Arg373
O
O Glu188
NH2
Arg189
OH2
Figure 5.53 Schematic representation of the nu-cleotide-binding site of the β subunit of theAlF3 –F1 complex, showing the coordination ofthe aluminofluoride group. Possible hydrogen-bondinteractions are shown by dotted lines
The stoichiometry of the bovine mitochondrial F1
ATPase means that the single γ, δ and ε subunits lacksymmetry, and in the case of the γ subunit the intrinsicasymmetry causes a different interaction with each ofthe three catalytic β subunits of F1. This interactionendows each β subunit with different nucleotide affinityand arises from rotation of the central stalk. Rotationof the γ subunit driven by proton flux through theFo region drives ATP synthesis by cycling throughstructural states corresponding to the tight, open andloose conformations.
Initial structures of F1 ATPase showed disorder forthe protruding end of the γ subunit from the (αβ)3
hexamer. Examining enzyme inhibited with dicyclo-hexylcarbodiimide (DCCD), a known stoichiometricinhibitor of ATP synthesis that reacts specifically withGlu 199 of the β subunit, resulted in improved orderand confirmed the orientation of the central pair ofhelices of the γ subunit, the structures of the δ andε subunits and their interaction with the Fo region atthe end of the central stalk. The protruding part of thecentral stalk is composed of approximately half of theγ subunit and the entire δ and ε subunits. The over-all length of the central stalk from the C-terminus ofthe γ subunit to the foot of the protruding region is∼11.4 nm. It extends ∼4.7 nm from the (αβ)3 domainthereby accounting for ‘lollipop’ appearance in electronmicrographs (Figure 5.48). The central stalk is the key
rotary element in the catalytic mechanism. The γ sub-unit interacts directly with the ring of c subunits locatedin the membrane of Fo and in the absence of covalentbonding between c subunits and the stalk region thisinteraction must be sufficiently robust to permit rota-tion. In the γ subunit, three carboxyl groups (γAsp 194,γAsp 195 and γAsp 197) are exposed on the lower faceof the foot suggesting that they may interact with basicresidues found in loop regions of the c-ring.
It is established that all species contain a, band c subunits within the Fo part of the enzymealthough there is currently no clear agreement on thetotal number of subunits. In yeast ATP synthetaseat least thirteen subunits are found in the purifiedenzyme. As well as the familiar α–ε subunits ofF1 and the a, b and c subunits of Fo additionalsubunits called f and d are found in Fo whilst OSCP,ATP8 and h occur in F1. Using the yeast enzymethe F1 region was co-crystallized with a ring of10 c subunits and although the loss of c subunitsduring preparation cannot be eliminated definitivelythe lack of symmetry relations between the hexamericcollection of α/β subunits and the 10 c subunits wassurprising. The structure of the c subunit monomerwas established independently by NMR spectroscopy(Figure 5.55) and showed two helical regions that wereassumed to be transmembrane segments linked byan extramembranous loop region. These loop regionsmake contact with γ and δ subunits and represent sitesof interaction (Figure 5.56). Only the a and c subunitsare essential for proton translocation (Figure 5.57) withArg210, His245, Glu196, and Glu219 (a subunit) andAsp61 of the c subunit implicated as essential residues.
Further refinement of the ATP synthetase structurehas shown a second, peripheral, stalk connecting theF1 and Fo domains in the enzyme (Figure 5.58). Thesecond stalk visible using cryoelectron microscopy ofdetergent solubilized F1Fo from E. coli, chloroplast andmitochondrial ATPases is located around the outsideof the globular F1 domain and may act as a stator tocounter the tendency of the (αβ)3 domain to follow therotation of the central stalk.
With the structural model derived from X-raycrystallography in place for the F1 subunit a numberof elegant experiments established that rotation is thelink between proton flux and ATP synthesis. The

150 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
Figure 5.54 The structures of the central stalk assembly with the individual subunits shown in isolation alongside.The γ subunit contains the long α helical coiled coil region that extends through the (αβ)3 assembly (top right). Thebovine ε subunit is very small (∼50 residues in length) with one major helix together with a poorly organized helix(bottom right). The δ subunit contains an extensive β sheet formed from seven β strands together with a distincthelix turn helix region (bottom left)

THE STRUCTURE OF ATP SYNTHETASE 151
idea of rotary switches is now no longer hereticalbut is accepted as another solution by proteins tothe mechanistic problem of harnessing energy fromproton gradients.
Covalently linking the γ subunit of F1 to the Cterminal domain of the β subunit blocked activitywhilst cross-linking to the α subunit was without effect.Complementary studies introducing cysteine residuesinto the structure of β and γ subunits with the formationof thiol cross links inhibited activity but reducing thesebonds followed by further cross-linking resulted inthe modification of a different β subunit. This couldonly occur if rotation occurred within ATPases. Finally,covalently joining the γ, ε and c subunit ring did notinhibit ATP synthesis and suggested that the subunitsact in unison as a rotating unit. Direct observation ofrotation in the synthetase was obtained using actinfilaments attached to the γ subunit. By followingthe fluorescence of covalently linked fluorophoresin a confocal microscope it was shown that ATPhydrolysis led to a 360◦ rotation of the filament in three
Figure 5.55 The structure of the c ring monomerdetermined using NMR spectroscopy is a simple HTHstructure (PDB:1A91)
Figure 5.56 The arrangement of the c ring with the F1 catalytic domain in yeast ATP synthetase. The c ring of 10monomers is shown in blue, the (αβ)3 unit is shown in red and orange and the γ subunit in dark green. A view ofthe F1 Fo ATPase from the inter-membrane space looking through the membrane region is shown on the right
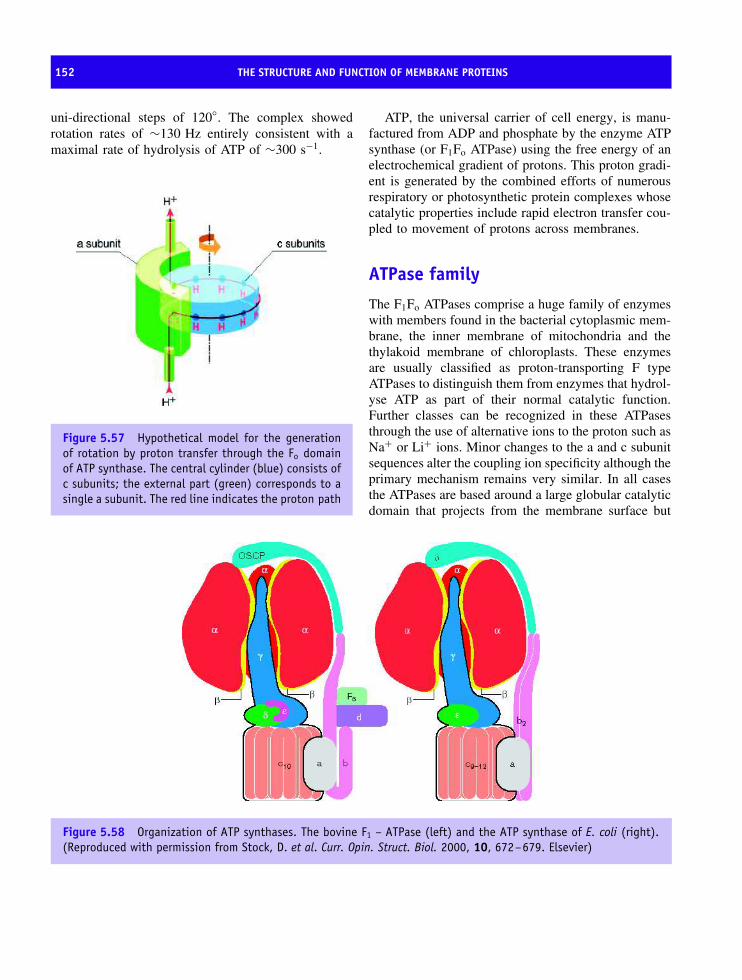
152 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
uni-directional steps of 120◦. The complex showedrotation rates of ∼130 Hz entirely consistent with amaximal rate of hydrolysis of ATP of ∼300 s−1.
Figure 5.57 Hypothetical model for the generationof rotation by proton transfer through the Fo domainof ATP synthase. The central cylinder (blue) consists ofc subunits; the external part (green) corresponds to asingle a subunit. The red line indicates the proton path
ATP, the universal carrier of cell energy, is manu-factured from ADP and phosphate by the enzyme ATPsynthase (or F1Fo ATPase) using the free energy of anelectrochemical gradient of protons. This proton gradi-ent is generated by the combined efforts of numerousrespiratory or photosynthetic protein complexes whosecatalytic properties include rapid electron transfer cou-pled to movement of protons across membranes.
ATPase family
The F1Fo ATPases comprise a huge family of enzymeswith members found in the bacterial cytoplasmic mem-brane, the inner membrane of mitochondria and thethylakoid membrane of chloroplasts. These enzymesare usually classified as proton-transporting F typeATPases to distinguish them from enzymes that hydrol-yse ATP as part of their normal catalytic function.Further classes can be recognized in these ATPasesthrough the use of alternative ions to the proton such asNa+ or Li+ ions. Minor changes to the a and c subunitsequences alter the coupling ion specificity although theprimary mechanism remains very similar. In all casesthe ATPases are based around a large globular catalyticdomain that projects from the membrane surface but
Figure 5.58 Organization of ATP synthases. The bovine F1 – ATPase (left) and the ATP synthase of E. coli (right).(Reproduced with permission from Stock, D. et al. Curr. Opin. Struct. Biol. 2000, 10, 672–679. Elsevier)

ATPASE FAMILY 153
interacts with a hydrophobic complex that controls iontranslocation via rotary motion.
In all cells there are membrane proteins whosefunction relies on the hydrolysis of ATP and thecoupling of this energy to the movement of ions suchas Na+, K+ or Ca2+ across membranes. Transportproteins represent a major category of membraneproteins and a number of different mechanisms exist fortransport. Transport across membranes is divided intomediated and non-mediated processes. Non-mediatedtransport involves the diffusion of ions or smallmolecules down a concentration gradient. Transportis closely correlated with solute solubility in thelipid phase with steroids, anaesthetics, narcotics andoxygen diffusing down a concentration gradient andacross membranes. Mediated transport is divided intotwo categories. Passive-mediated transport also calledfacilitated diffusion relies on the transfer of a specificmolecule from high concentration to low concentration.Transport proteins involved in this mechanism oftransport have already been described in this chapterand are exemplified by the β barrel porin proteins.Unfortunately these proteins cannot transport substratesagainst concentration gradients; this requires activetransport and the hydrolysis of ATP.
Na+–K+ ATPase is one member of a class ofmembrane bound enzymes that transfer cations withATP hydrolysis providing the driving force. TheNa+–K+ ATPases, a highly conserved group of integralproteins, are widely expressed in all eukaryotic cellsand are very active with a measure of their importanceseen in the estimate that ∼25 percent of all cytoplasmicATP is hydrolysed by sodium pumps in resting humancells. In nerve cells, a much higher percentage ofATP is consumed (>70 percent) to fuel sodium pumpscritical to neuronal function.
The Na+–K+ ATPase is a transmembrane proteincontaining two types of subunit. An α subunit of∼110 kDa contains the ion-binding site and is respon-sible for catalytic function whilst a β subunit, glyco-sylated on its exterior surface, is of less clear func-tion. Sequence analysis suggests that the α subunitcontains eight transmembrane helices together withtwo cytoplasmic domains. In contrast the β subunit(Mr∼55 kDa) has a single transmembrane helix anda much larger domain projecting out into the extra-cellular environment where glycosylation accounts for
∼20 kDa of the total mass of the protein. The β sub-unit is required for activity and it is thought to playa role in membrane localization and activation of theα subunit. In vivo the Na+–K+ ATPase functions as atetramer of composition α2β2.
The Na+–K+ ATPase pumps sodium out of the celland imports potassium with the hydrolysis of ATP atthe intracellular surface of the complex. The overallstoichiometry of the reaction results in the movementof 3 Na+ ions outward and 2 K+ ions inwards and isrepresented by the equation
3Na+(in) + 2K+
(out) + ATP + H2O � 3Na+(out)
+ 2K+(in) + ADP + PI
The ATPase is described as an antiport where there isthe simultaneous movement of two different moleculesin opposite directions, and leads to the net movementof positive charge to the outside of the cell. A uniportinvolves the transport of a single molecule whilsta symport simultaneously transports two differentmolecules in the same direction (Figure 5.59).
In the presence of Na+ ions ATP phosphorylates aspecific aspartate residue on the α subunit triggeringconformational change. In the presence of high levelsof K+ the dephosphorylation reaction is enhanced andsuggests two distinct conformational states for theNa+–K+ ATPase. These states, designated E1 and E2,differing in conformation show altered affinity andspecificity. On the inside of the cell the Na+–K+ATPase (in the E1 state) binds three sodium ions atspecific sites followed by ATP association to yieldan E1 –ATP–3Na+ complex. ATP hydrolysis occurs toform an aspartyl phosphate intermediate E1 ∼ P–3Na+that undergoes a conformational change to form theE2 state (E2 ∼ P–3Na+) and the release of boundNa outside the cell. The presence of a ‘high energy’phosphoryl intermediate (denoted by ∼P) has led tothis class of membrane proteins being called P typeATPases. As an E2 –P state the enzyme binds twoK+ ions from the outside to form the E2 –P–2K+complex. Hydrolysis of the phosphate group to reformthe original aspartate side chain causes conformationchange with the result that 2K+ ions are released on theinside of the cell. The ATPase ion pump, upon releaseof the potassium ions, forms the E1 state capable

154 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
Figure 5.59 Diagrammatic representation of uniport, symport and antiport systems used in processes of activetransport across membranes
of binding three Na+ ions and restarting the cycleagain.
Study of the function of Na+–K+ ATPases has beenaided by the use of cardiac glycosides, natural productsthat increase the intensity of muscular contractionin impaired heart muscle. Abnormalities have beenidentified in the function of Na+–K+ ATPases andare involved in several pathologic states includingheart disease and hypertension. Several types of heartfailure are associated with significant reductions inthe myocardial concentration of Na+–K+ ATPaseor impaired activity. Similarly excessive renal re-absorption of sodium due to oversecretion of thehormone aldosterone is associated with forms of
hypertension. Oubain is a naturally occurring steroidand is still prescribed as a cardiac drug. Its action isto block the efflux of Na+ ions leading to an increasein their intracellular concentration (Figure 5.60). Theresulting increase in Na+ concentration causes astimulation of secondary active transport systems incardiac muscle such as the Na+–Ca2+ antiport system.This pump removes Na+ ions from the cell andtransports Ca2+ to the inside. Transient increasesin cytosolic Ca2+ ion concentration often triggersmany intracellular responses one of which is muscularcontraction. The Ca2+ ions stimulate cardiac musclecontraction producing a larger than normal responseto aid cardiovascular output. Although of major
Figure 5.60 The active transport of Na+ and K+ ions by the Na+–K+ ATPase. Oubain (pronounced ‘wabane’) is acardiac glycoside promoting increased intracellular Na+ concentration

ATPASE FAMILY 155
Figure 5.61 Scheme for active transport of Ca2+ by SR Ca ATPase. The inside refers to the cytosol and the outsiderepresents the lumen of the sarcoplasmic reticulum of skeletal muscle cells. Similar Ca pumps exist in the plasmamembranes and ER
importance to the physiology of all cells detailedstudies of the Na+–K+ ATPase protein and ultimatelythe development of improved drugs is hampered by theabsence of a well-defined structure.
Fortunately, another prominent member of the Ptype ATPases, the Ca2+ –ATPase from the sarcoplas-mic reticulum (SR) of muscle cells, has been crys-tallized to reveal some aspects of the organizationof transport proteins. SR Ca2+ ATPase is amongstthe simplest proteins in this large family consistingof a single polypeptide chain of ∼110 kDa. Its bio-logical function is to transport 2Ca2+ ions againsta concentration gradient for every molecule of ATPhydrolysed (Figure 5.61). Within the cytosol the con-centration of Ca2+ is ∼0.1 µM – about 10000 timeslower than the extracellular or lumenal concentration(∼1.0–1.5 mM). When muscle contracts large amountsof Ca2+ stored in the SR are released into the cytosolof muscle cells. In order to relax again after contrac-tion the Ca concentration must be reduced by pump-ing ions back into the SR against a concentrationgradient.
As an ion pump the Ca2+ ATPase works compara-tively slowly with a turnover number of ∼60 Ca2+ ionsper second. Pumps such as the Na+–K+ ATPase trans-fer ions at rates of 106 s−1 and in order to overcome
this rate deficiency for Ca2+ transfer the cell compen-sates by accumulating large amounts of enzyme withinthe SR membrane. The accumulation of enzyme in thismembrane leads to ∼60 percent of the total proteinbeing Ca2+ ATPase.
The structure of the SR Ca2+ ATPase reveals twoCa2+ ions bound in a transmembrane domain of 10α helical segments. In addition to the transmembraneregion three cytosolic regions were identified as phos-phorylation (P), nucleotide binding (N) and actuator(A) domains. This last domain transmits conforma-tional change through the protein leading to movementof Ca across membranes. The organization of thesedomains can be seen in the beautiful structure of Cabound SR-ATPase where ten transmembrane α helices(M1–M10) segregate into two blocks (M1–M6 andM7–M10), a structural organization consistent with thelack of M7–M10 in simpler bacterial P-type ATPases(Figure 5.62).
The lengths and inclination of the ten helices differsubstantially; helices M2 and M5 are long and straightwhilst others are unwound (M4 and M6) or kinked(M10) in the middle of the membrane. The Ca ions arelocated side by side surrounded by four transmembranehelices (M4, M5, M6 and M8), with the ligands toCa2+ provided at site I by side-chain oxygen atoms

156 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
Figure 5.62 The structure of SR Ca-ATPase deter-mined to a resolution of A. (PDB: 1EUL). The fourdistinct domains are shown in different colours; trans-membrane domain (blue), Actuator or A domain (yel-low), phosphorylation or P domain (magenta) and thenucleotide binding or N domain (green). Two bound Caions are shown in red. In this view the lumen of theSR is at the bottom whilst the cytosol is found at thetop containing the A, P and N domains
derived from Asn768, Glu771 (M5), Thr799, Asp800(M6) and Glu908 (M8). Site II is formed almostentirely by helix M4 with backbone oxygen atoms ofVal304, Ala305 and Ile307 together with the side-chainoxygens of Asn796, Asp800 (M6) and Glu309 (M4)providing the key ligands. The coordinating ligandsprovided by residues 304, 305, 307 and 309 occurin residues located either sequentially or very closetogether in the primary sequence. Such an arrangementis not compatible with regular helical conformationsand is only possible as a result of local unfolding ofthe M4 helix in this region. In this context a prolineresidue at position 308 almost certainly contributes
to unfolding whilst the sequence PEGL is a keymotif in all ATPases. Although the position of theCa2+ is clearly seen within the transmembrane regionthe structure of the SR-CaATPase does not offerany clues as to the route of ion conduction. Therewere no obvious vestibules in the bound state, asseen in porins, and conformational changes facilitatetransport of Ca ions across the membrane by couplingevents in the transmembrane domain with those inthe cytoplasmic domains. This involves Ca bindingand release, nucleotide binding as well as Asp351phosphorylation and dephosphorylation.
Summary
Following the determination of the structure of thebacterial photosynthetic reaction centre there has been aprogressive increase in the number of refined structuresfor membrane bound.
Compared with the large number of structuresexisting for globular domains in the protein databankit is clear that the number of highly resolved (<5 A)structures available for membrane proteins is muchsmaller. This will change rapidly as more membraneproteins become amenable to methods of structuredetermination.
Peripheral or extrinsic proteins are associated withthe membrane but are generally released by mild dis-ruptive treatment. In contrast, integral membrane pro-teins remain firmly embedded within the hydrophobicbilayer and removal from this environment frequentlyresults in a loss of structure and function.
Crystallization of membrane proteins requires con-trolled and ordered association of subunits via theinteraction of exposed polar groups on the surfaces ofproteins. The process is assisted by the introduction ofamphiphiles that balance conflicting solvent require-ments of membrane proteins.
The vast majority of proteins rely on the foldingof the primary sequence into transmembrane helicalstructures. These helices have comparable geometry tothose occurring in soluble proteins exhibiting similarbond lengths and torsion angles.
The major differences with soluble proteins liein the relative distribution of hydrophobic aminoacid residues.

SUMMARY 157
Table 5.10 Some of the membrane protein structures deposited in PDB; Most structures were determined usingX-ray crystallography
Protein PDB Resolution (A) Function
Rhodopsin family and G-protein coupled receptorsBacteriorhodopsin (EM) 2BRD 3.5 Light driven proton pump(X-ray) 1C3W 1.55Halorhodopsin 1E12 1.8Rhodopsin 1F88 2.8 Visual cycle transducing light energy
into chemical signalsSensory rhodopsin II 1H68 2.1
Photosynthetic and light harvesting complexes
R. viridis reaction centre 1PRC 2.3 Primary charge separation initiated by aphoton of light
R. sphaeroides reaction centre 1PSS 3.0Light harvesting chlorophyll
complexes from R. acidophila1 KZU 2.5 Gathering light energy and focussing
photons to the photosyntheticreaction centre
Photosystem I from S. elongatus 1JB0 2.5 Primary photochemistryPhotosystem II from S. elongatus 1FE1 3.8 Oxygenic photosynthesis
Bacterial and mitochondrial respiratory complexes
Cytochrome oxidase (aa3) from P.denitrificans
1AR1 2.8 Catalyses terminal step of aerobicrespiration
Cytochrome oxidase (ba3) from T.thermophilus
1EHK 2.4
Cytochrome oxidase from bovineheart mitochondria
1OCC 2.8
Bovine heart mitochondria bc1
complex1BGY 2.8 Electron transfer complex that oxidizes
ubiquinonols and reducescytochrome c
Chicken heart cytochrome bc1
complex1BCC 3.2
S. cerevisiae cytochrome bc1
complex1EZV 2.3
(continued )
158 THE STRUCTURE AND FUNCTION OF MEMBRANE PROTEINS
Table 5.10 (continued)
Protein PDB Resolution (A) Function
Other energy transducing membrane proteinsATP synthase (F1 c10) S. cerevisiae 1E79 3.9 Membrane portion forms proton
channelFumarate reductase complex from
Wolinella succinogenes1QLA 2.2
β barrel membrane proteins of porin family
Porin from R. capsulatus 2POR 1.8 Semi-selective pore proteinsPorin from R. blastica 1PRN 2.0OmpF from E. coli. 2OMF 2.4PhoE from E. coli. 1PHO 3.0Maltoporin from S. typhimurium 2MPR 2.4Maltoporin from E. coli 1MAL 3.1OmpA from E. coli. (NMR) 1G90(X-ray) 1BXW 2.5
Ion and other channel proteins
Calcium ATPase from SR of rabbit 1EUL 2.6 Controls Ca flux in sarcoplasmicreticulum
Toxins
α-haemolysin from S. aureus 7AHL 1.9 Channel forming toxinLukF from S. aureus 3LKF 1.9 Homologous in structure and function
to α-haemolysin
Membrane proteins such as toxins or pore proteinsare based on β strands assembling into compact, bar-rel structures that assume great stability via inter-strandhydrogen bonding.
Sequencing proteins assists in the definition of trans-membrane domains via identification of hydrophobicside chains. Proteins containing high proportions ofresidues with non-polar side chains such as Leu, Val,Ile, Phe, Met and Trp in blocks of ∼20–30 residuesare probably membrane bound.
The structures of bacteriorhodopsin revealed seventransmembrane helices of ∼23 residues in lengthcorrelating with seven ‘blocks’ in the primary sequence
composed predominantly of residues with non-polarside chains.
The seven transmembrane helix structure is acommon architecture in membrane proteins with G-protein coupled receptors sharing these motifs.
Methods developed for crystallization of the bacte-rial reaction centre from R. viridis proved applicable toother classes of membrane proteins including respira-tory complexes found in mitochondrial membranes andin aerobic bacteria. These complexes generate protongradients vital for the production of ATP and representthe ‘power stations’ providing energy for all cellu-lar processes.

PROBLEMS 159
The structure of Ca ATPase reveals the coordinatedaction of cytosolic and membrane located domains inCa transport across the membrane and points the wayto understanding the movement of charged ions acrossimpermeant lipid bilayers.
ATP is produced by ATP synthase a large enzymecomposed of a peripheral F1 complex together with a
smaller membrane bound region (Fo). These domainsform a rotary motor where the flux of protonsthrough Fo drives rotation of c subunits via trans-mission to a central stalk (γ, δ and ε subunits)that mediates conformational changes at the cat-alytic centres in (αβ)3 subunits promoting ATPformation.
Problems
1. Having determined the nucleotide sequence of a puta-tive membrane protein describe how you would usethis data to obtain further information on this protein.
2. List the post-translational modifications added toproteins to retain them within or close to thelipid bilayer. Give an example protein for eachmodification.
3. Find the membrane-spanning region in the followingsequence: . . . . . . . . .GELHPDDRSKITKPSESIITTIDSNPSWWTNWLIPAISALFVALIYHLYTSEN. . . . . ...
4. How many residues are required to span a lipid bilayerif they are all form within a regular α helix? Howmany turns of a regular α helix are required to crossthe membrane once?
5. Using the crystal structure for the bacterial reactioncentre estimate the average length of the transmem-brane helices. Are there any differences and if so howdo you account for them?
6. Describe the advantages and disadvantages of usingthe erythrocyte membrane as a model for studyingmembrane protein structure and organization.
7. Use protein databases to find membrane proteinstructures determined at resolution below 0.3 nm andthose between 0.3 and 0.5 nm.
8. Gramicidin is a small polypeptide of 15 residues thatadopts a helical conformation. This molecule has beenobserved to act as an ionophore allowing K+ ions tocross the membrane. How might this occur?


6The diversity of proteins
The incredible diversity shown by the living worldranges from bacteria and viruses to unicellular organ-isms, eventually culminating with complex multicel-lular systems of higher plants, including gymnospermsand angiosperms, and animals such as those of the ver-tebrate kingdom. However, the living world is basedaround proteins made up of the same 20 amino acids.There is no fundamental difference between the aminoacids and proteins making up a bacterium such asEscherichia coli to those found in higher vertebrates.As a result the principles governing protein structureand function are equally applicable to all living sys-tems. This pattern of similarity is not surprising ifone considers that all living systems are related bytheir evolutionary origin to primitive ancestors that hadacquired the basic 20 amino acids to use in the syn-thesis of proteins. Higher levels of complexity wereacquired by evolutionary divergence that led to a sub-tle alteration in primary sequence and the generationof new or altered functional properties. Although theexact nature of the ‘first’ ancestral cell is unclear alongwith details of self-replicating systems advances havebeen made in understanding the origin of proteins.
Prebiotic synthesis and the originsof proteinsThe origin of life represents one of the greatest puzzlesfacing scientists today. What originally seemed to be
an impossible problem has gradually become betterunderstood via experimentation. In order to synthesizeproteins it is first necessary to make amino acidscontaining carbon, hydrogen, oxygen and nitrogen, andoccasionally sulfur. The source of all carbon, hydrogen,oxygen and nitrogen would have been the originalatmospheric gases of carbon dioxide, nitrogen, watervapour, ammonia, and methane, but not atmosphericoxygen since this was almost certainly lacking in earlyevolutionary periods. When this series of events isplaced in a time span we are describing reactions thatoccurred more than 3.6 billion years ago.
In a carefully designed experiment Stanley Millerand Harold Urey showed that simulating the primitiveconditions present on the Earth around 4 billion yearsago could result in the production of biomolecules. Thisstudy involved adding inorganic molecules to a closedsystem under a reducing atmosphere (lacking oxygen).The gaseous mixture of mainly ammonia, hydrogen andmethane simulated the early Earth’s atmosphere. Thewhole mixture was refluxed in a closed evacuated sys-tem with the water phase representative of the ‘oceans’of the early earth analysed at the end of an experimentlasting several days (Table 6.1). Subjecting the systemto electrical discharge (lightning) and high amounts ofultraviolet light (Sun) formed biomolecules, includingthe amino acids glycine, alanine and aspartate. The for-mation of hydrogen cyanide, aldehydes and other cyano
Proteins: Structure and Function by David Whitford 2005 John Wiley & Sons, Ltd

162 THE DIVERSITY OF PROTEINS
+CH
RO
Aldehyde
OH2N
CC
R
HOH
Amino acid
HCN H2O+
Hydrogen Cyanide
Figure 6.1 Prebiotic synthesis of amino acids fromsimple organic molecules
Table 6.1 Yields of biomolecules from simulatingprebiotic conditions using a mixture of methane,ammonia, water and hydrogen
Biomolecule Approximate yield (%)
Formic acid 4.0Glycine 2.1Glycolic acid 1.9Alanine 1.7Lactic acid 1.6β-Alanine 0.76Propionic acid 0.66Acetic acid 0.51Iminodiacetic acid 0.37α-Hydroxybutyric acid 0.34Succinic acid 0.27Sarcosine 0.25Iminoaceticpropionic acid 0.13N -Methylalanine 0.07Glutamic acid 0.051N -Methylurea 0.051Urea 0.034Aspartic acid 0.024α-Aminoisobutyric acid 0.007
Shown in red are constituents of proteins (after Miller, S.J. & Orgel,L.E. The Origins of Life on Earth. Prentice-Hall, 1975).
compounds was also important since these simple com-pounds undergo a wide range of further reactions. Thegeneral reaction is summarized by a simple additionreaction between aldehydes and hydrogen cyanides inthe presence of water (Figure 6.1), although in practice
Figure 6.2 An example of the apparatus used byUrey and Miller to demonstrate prebiotic synthesis oforganic molecules
the reaction may form a nitrile derivative in the atmo-sphere followed by hydrolysis in the ‘ocean’ to yieldsimple amino acids.
Performing this type of experiment with dif-ferent mixtures of starting materials yielded addi-tional biomolecules, including adenine. Variations onthis basic theme have suggested that although theEarth’s early atmosphere lacked oxygen the pres-ence of gases such as CO, CO2 and H2S werevital for prebiotic synthesis. The presence of sul-fur enhances the number and type of reactions thatcould occur. More recently, examination of oceanfloors has revealed the presence of deep sea ventssometimes called ‘smokers’ or fumaroles. These ventsrelease hot gases and minerals from the Earth’scrust into the ocean and are also prime sites fororganic synthesis. This suggests that many potential

EVOLUTIONARY DIVERGENCE OF ORGANISMS AND ITS RELATIONSHIP TO PROTEIN STRUCTURE AND FUNCTION 163
sites and sources of energy were available for prebi-otic synthesis.
The next barrier to the evolution of life involvedthe formation of polymers from precursors. The geneticsystems found in cells today are specialized polymers.They are able to direct the synthesis of proteins frommessenger ribonucleic acid (mRNA), the latter rep-resenting the information present in deoxyribonucleicacid (DNA). In addition these polymers are capableof directing their own synthesis in a macromolecularworld of DNA/RNA and protein.
DNA −→ RNA −→ protein
These systems are self-replicating and their evolutionrepresents one of the greatest hurdles to be overcomein the development of living systems. Replicating DNArequires proteins to assist in the overall process whilstthe scheme above demonstrates that protein synthesisrequires DNA and RNA. This creates a paradox oftencalled the ‘chicken and egg’ puzzle of molecularbiology of which came first.
The recent demonstration that RNA molecules havecatalytic function analogous to conventional enzymeshas revolutionized views of prebiotic synthesis. Cat-alytic forms of RNA called ribozymes mean that RNAmolecules, in theory, at least have the means to directtheir own synthesis and to catalyse a limited num-ber of chemical reactions. For this reason a preva-lent view of molecular evolution involves a worlddominated by RNA molecules that gradually evolvedinto a system in which proteins carried out catalysis,whilst nucleic acid performed a role of informationstorage, transfer and control. The details of this transi-tion remain far from complete but supportive lines ofevidence include: (i) the existence of different formsof RNA such as rRNA, mRNA, tRNA and genomicRNA; (ii) molecules such as nicotinamide adeninedinucleotide (NAD), adenosine tri- and diphosphate(ATP/ADP), and flavin adenine dinucleotide (FAD)found universally throughout cells are composed ofadenine units analogous to those occurring in RNA;(iii) tRNAs have a tertiary structure; (iv) ribosomesrepresent hybrid RNA–protein systems where cataly-sis is RNA based; and (v) the enzyme RNaseP from E.coli catalyses the degradation of polymeric RNA intosmaller nucleotide units in a reaction where RNA isthe active component.
A major objection to the view of an ‘RNA world’has been the comparative instability of RNA (especiallywhen compared to DNA). RNA is easily degradedand it is difficult to see how stable systems capa-ble of replication and catalysis evolved. Additionallythe synthesis of polymers of RNA under conditionssimilar to those found early in the earth’s historyhas proved remarkably difficult. Despite these prob-lems most researchers view RNA as a likely inter-mediate between the ‘primordial soup’ and the sys-tems of replication and catalysis found in moderncell types.
The fossil record evidence shows that bacteria-likeorganisms were present on earth 3.6 billion years ago.This implies that the systems of replication presenttoday in living cells had already evolved. The ‘RNA-directed world’ was therefore a comparatively shorttime interval of ∼0.5 billion years!
Having evolved a primitive replication and catalysissystem based on RNA the simple amino acids couldbe used in protein biosynthesis. It is very unlikelythat all amino acids were present in the primordialsoup since some of the amino acids are relativelyunstable especially under acidic conditions, and thisincludes the side chains of asparagine, glutamine,and histidine. In addition the amino acids found inproteins represent a very small subset of the totalnumber of amino acids known to exist. This mightsuggest that the present class of amino acids evolvedover millions of years to reflect a blend of chemicaland physical properties required by proteins althoughit was essentially complete and intact 3.6 billionyears ago.
Evolutionary divergence of organismsand its relationship to proteinstructure and function
The precise details of the origin of life involving thegeneration of a self-replicating system and its evolu-tion into the complex multicellular structures foundin higher plants and animals are unclear but the fos-sil record shows that primitive bacteria were presenton earth in the pre-Cambrian period nearly 3.6 bil-lion years ago. These fossilized cells resemble aclass of bacteria found on present day Earth called

164 THE DIVERSITY OF PROTEINS
cyanobacteria. Although it is surprising that bacterialeave fossil records cyanobacteria often form a signif-icant cell wall together with layered structures calledstromatolites (Figure 6.3). These structures form a matas cyanobacteria grow trapping sediment and helpingthe fossilization process. Cyanobacteria are prokary-otic cells, lacking a nucleus and internal membranes,capable of both photosynthetic and respiratory growthand are represented today by genera such as Nostoc,Anacystis and Synechococcus.
Less ambiguously and more importantly the fos-sil record demonstrates progressive increases in com-plexity from the simple prokaryotic cell lacking anucleus to more complicated structures similar to mod-ern eukaryotic cells. Eukaryotic cells became multi-cellular and evolved by specialization towards specificcellular functions (Figure 6.4). These cells increased instructural complexity by internal compartmentalizationwith genome organization becoming more complexalong with the variety of biochemical reactions catal-ysed within these cells. Whilst the fossil record aidedour understanding of evolution one of the best methodsof deciphering evolutionary pathways has come fromcomparing protein and DNA sequences.
20
0
10 mm
Figure 6.3 Microfossil of filamentous bacterial cells.The fossil shown alongside an interpretive drawing isfrom Western Australia and rocks dated at ∼3.4 × 109
years (Reproduced with permission from Voet, D., Voet,J.G. and Pratt, C.W. Fundamentals of Biochemistry. JohnWiley & Sons, Ltd, Chichester, 1999)
Plasma membrane
Mitochondrion
Filamentous cytoskeleton Centrioles
Lysosome
Small membranevesicle
Golgi vesicles
Rough endoplasmic reticulumFilamentous cytoskeleton
Small membrane vesicle
Golgi vesicles
Leukoplast
Smooth endoplasmic reticulum
Smoothendoplasmicreticulum
Rough endoplasmic reticulum
Nucleus
Nucleus
Cell wall
Chloroplast
Mitochondrion
Plasmodesmeta
Plasmamembrane
PeroxisomeCilia
Peroxisome
NucleolusNucleolus
Vacuole
Figure 6.4 Generalized eukaryotic cells (plant and animal) (reproduced courtesy of Darnell, J.E. et al. Molecular CellBiology. Scientific American, 1990)

PROTEIN SEQUENCE ANALYSIS 165
In a protein of 100 amino acid residues there are20100 unique or possible sequences. It is clear that bio-logical sequences represent only a small fraction ofthe total number of permutations. Protein sequencesoften show similarities with these relationships gov-erned by evolutionary lineage. Over millions of yearsa sequence can change but it cannot cause completeloss of function unless gene duplication has occurred.If mutation provides new enhanced functional activitiesany selective advantage conferred on the host organismpossessing this protein will lead to improved survivaland gene perpetuation.
Protein sequence analysisProtein sequencingThe sequencing of DNA has advanced so rapidly thatthis method is now by far the most common andeffective way of determining the sequence of a protein.By translating the order of nucleotide bases along aDNA sequence one can simply derive the sequenceof amino acid residues. However, there are occasionswhen it becomes important to sequence a proteindirectly and this might include determining the extentof post-translational processing or arranging peptidefragments in a linear order.
Protein sequencing is an automated techniquecarried out using sophisticated instruments (sequena-tors) and based on methods devised by Pehr Edman(it is often called Edman degradation). The unknownpolypeptide is reacted under alkaline conditions (pH∼9) with phenylisothiocyanate (PTC) where the freeamino group at the N-terminal forms a phenylthio-carbamoyl derivative, which is hydrolysed from theremaining peptide using anhydrous trifluoroacetic acid(Figures 6.5 and 6.6). PTC makes the first peptidebond less stable and easily hydrolysed. Residue rear-rangement in aqueous acidic solution yield a phenylth-iohydantoin (PTH) derivative of the N-terminal aminoacid that is identified using chromatography or massspectrometry (Figure 6.7). The significance of thisseries of reactions is that the N-terminal amino acid is‘tagged’ by attaching PTC but the remaining polypep-tide chain (now containing n − 1 residues) remainsintact and can undergo further reactions with PTC atits new N-terminal residue. The Edman degradationis a repetitive, cyclical, series of reactions, although
N C S N C peptideH2N C C
OR1 H
H
R2
H
N C
S
N N C peptide
R2
C C
OR1 H
HH
+
~pH 9.0
H
Figure 6.5 The reaction of the N-terminal amino acidresidue with phenylisothiocyanate in the first step ofthe Edman degradation
HN C
S
N N C peptide
R2
C C
OR1 H
HH
H2N C peptide
R2
H
HN C S
NH CC
O
R1 H
trifluoroacetic acid
+
available for nextcycle reaction with PTC
H
+
Figure 6.6 Hydrolysis of the phenylthiocarbamoylderivative of the peptide to yield a protein of n − 1residues and a free ‘labelled’ amino acid
as with most repetitive procedures, errors accumu-late and progressively degrade the accuracy of thewhole process. Errors include: random breakage ofthe polypeptide chain producing a second free aminoterminal residue; incomplete reaction between PTCand the N-terminal amino acid leading to its appear-ance in the next reactive cycle; and side reactionsthat compete with the reaction between PTC andthe polypeptide chain. Sequenators are very sensitive

166 THE DIVERSITY OF PROTEINS
HN C S
NH+ C O
R1 H
N C S
NHCO
R1H
PTH-amino acid
~1M HCl
Figure 6.7 Re-arrangement of the PTC-derivative toform a phenylthiohydantoin (PTH) derivative of theN-amino acid
instruments capable of sequencing picomole amountsof polypeptide. Usually an upper limit on the length ofthe polypeptide chain that can be sequenced directlyis about 70 residues. Since most proteins containfar more than 70 residues the sequencing proceduresrelies on ‘chopping’ the polypeptide chain into aseries of smaller fragments that are each sequencedindependently.
Generation of smaller peptide fragments involvesusing hydrolytic enzymes that cleave the polypeptide
Table 6.2 Enzymes or reagents for generating peptidefragments suitable for sequencing
Enzyme/reagent Cleavage site
Trypsin -Arg -↑-Yaa or Lys -↑-Yaa-
Endoprotease Arg-C -Arg -↑-Yaa
Chymotrypsin -Phe -↑-Yaa, -Tyr -↑-Yaa, -Trp
-↑-Yaa
Clostripain -Arg -↑-Yaa
Asp-N -Xaa-↑-Asp
Thermolysin -Xaa-↑-Leu, -Xaa-↑-Ile,
-Xaa-↑-Val, -Xaa-↑-Met,
V-8 protease -Asp -↑-Yaa, -Glu -↑-Yaa
Cyanogen bromide
(CNBr)
-Met -↑-Yaa
In many cases the above enzymes show wider specificity. Forexample chymotrypsin will cleave other large side chains particularlyLeu and care needs to be exercised in interpreting the results ofproteolytic cleavage. In other instances the identity of Xaa/Yaacan influence whether cleavage occurs. For example Lys-Pro is notcleaved using trypsin.
at specific sequences or by the use of cyanogenbromide that splits polypeptide chains after methionineresidues (Table 6.2).
After purification of the individual fragments theshorter peptides are sequenced, although the majorproblem is now to deduce the respective order of each
Table 6.3 Fragments derived by digestion of unknownprotein with Asp-N and trypsin
Digestion with trypsin
Mass Peptide sequence
4905.539 ITKPSESIITTIDSNPSWW
TNWLIPAISALFVALIYHLYTSEN
2205.928 EQAGGDATENFEDVGHSTDAR
1511.749 FLEEHPGGEEVLR
1412.717 TFIIGELHPDDR
1186.599 YYTLEEIQK
1160.646 STWLILHYK
738.403 VYDLTK
650.299 AEESSK
599.290 HNNSK
476.271 ELSK
317.218 AVK
234.145 SK
Digestion with Asp-N
Mass Peptide Sequence
4100.113 AEESSKAVKYYTLEEIQK
HNNSKSTWLILHYKVY
3621.805 DSNPSWWTNWLIPAISA
LFVALIYHLYTSEN
2411.184 DLTKFLEEHPGGEEVLREQAGG
1825.981 DARELSKTFIIGELHP
1789.007 DRSKITKPSESIITTI
825.326 DATENFE
615.273 DVGHST
134.045 D

PROTEIN SEQUENCE ANALYSIS 167
peptide. This problem is resolved by repeating thedigestion of the intact protein with a second enzymethat reacts at different sites producing fragments whoserelationship to the first set is established by sequencingto determine an unambiguous order of residues alonga polypeptide chain. The principle of this methodis demonstrated with an ‘unknown’ protein of 133residues and its digestion with two enzymes; trypsinand Asp-N (Table 6.3 and Figure 6.8). Trypsin showssubstrate specificity for lysine and arginine residuescleaving peptide bonds on the C terminal side of theseresidues whilst Asp-N is a protease that cleaves beforeaspartate residues.
To verify the primary sequence a total amino acidanalysis is usually performed on the unknown proteinby complete hydrolysis of the protein into individualamino acid residues. Quite clearly the total amino acidcomposition must equate with the combined number ofamino acids derived from the primary sequence.
Amino acid analysis consists of three steps: (i)hydrolysis of the protein into individual amino acids;(ii) separation of the amino acids in this mixture; and(iii) identification of amino acid type and its quan-tification. Hydrolysis of the protein is normally com-plete after dissolving a small amount of the sam-ple in 6 M HCl and heating the sample in a vac-uum at 110 ◦C for 24 hours. The peptide bonds arebroken leaving a mixture of individual amino acids.This approach destroys the amino acid tryptophancompletely whilst cysteine residues may be oxidized
and partially destroyed by these conditions. Simi-larly, acid hydrolysis of glutamine and asparagineside chains can form aspartate and glutamate and itis not usually possible to distinguish Asn/Asp andGlu/Gln in protein hydrolysates. For this reason proteinsequences may be written as Glx or Asx represent-ing the combined number of glutamine/glutamate andasparagine/aspartate residues.
Separation is achieved by cation exchange chro-matography (Figure 6.9) using resins, supported withinstainless steel or glass columns, containing nega-tively charge groups such as sulfonated polystyrenes.The negatively charge amino acids such as aspartateand glutamate elute rapidly whilst the flow of pos-itively charged amino acids through the column isretarded due to interaction with the resin. By alter-ing the polarity of the eluting solvent the interac-tion of hydrophobic amino acids with the column isenhanced. Amino acids with large hydrophobic sidechains, for example phenylalanine and isoleucine, elutemore slowly than smaller amino acids such as alanineand glycine.
To enhance detection the amino acids are reactedwith a colored reagent such as ninhydrin, fluores-cein, dansyl chloride or PTC. If this procedure is per-formed prior to column separation the absorbance ofderivatized amino acids as they elute from the col-umn is readily recorded at ∼540 nm or from theirfluorescence. Due to the high reproducibility of these
* 2 0 * 4 0 *Unknown : A E E S S K A V K Y Y T L E E I Q K H N N S K S T W L I L H Y K V Y D L T K F L E E H P G G E E V L : 5 0Unknown : A E E S S K A V K Y Y T L E E I Q K H N N S K S T W L I L H Y K V Y D L T K F L E E H P G G E E V L : 5 0
6 0 * 8 0 * 1 0 0Unknown : R E Q A G G D A T E N F E D V G H S T D A R E L S K T F I I G E L H P D D R S K I T K P S E S I I T : 1 0 0Unknown : R E Q A G G D A T E N F E D V G H S T D A R E L S K T F I I G E L H P D D R S K I T K P S E S I I T : 1 0 0
* 1 2 0 *Unknown : T I D S N P S W W T N W L I P A I S A L F V A L I Y H L Y T S E N : 133Unknown : T I D S N P S W W T N W L I P A I S A L F V A L I Y H L Y T S E N : 133
Figure 6.8 The figure shows the unknown protein whose primary sequence can be deduced from sequencing smallerfragments derived by digestion with trypsin (top line) and Asp-N (bottom line). For clarity alternate fragmentsderived using trypsin are shown in red and purple whilst the Asp-N fragments are shown in magenta and green

168 THE DIVERSITY OF PROTEINS
Rel
ativ
e flu
ores
cenc
e
Ser
Asp
Glu
CM
-Cys
Asn G
ln
2-N
H2-
buty
ric
His
Gly Thr
Met V
alP
helle Le
u
Lys
Arg A
la
Tyr
0 4 8
Retention time (min)
12 15
Figure 6.9 Elution of protein hydrolysate from a cation exchange column. The amino acids were derivatized firstwith a fluorescent tag to aid detection (after Hunkapiller, M.W. et al. Science 1984, 226, 339–344)
profiles the amino acids of each type can be identifiedand their relative numbers quantified.
DNA sequencing
DNA sequencing methods are now routine and therecent completion of genomic sequencing projects tes-tifies to the efficiency and accuracy of these techniques.The first genome sequencing projects were completedin the early 1980s for viruses and bacteriophages suchas φX174 as well as organelles such as mitochondriathat contain small circular DNA genomes. Howeverin the last decade massive DNA sequencing projects
were initiated and have resulted in vast numbers ofprimary sequences. In 2001 the human genome projectwas completed as a ‘first draft’ and the genomesof many other prokaryotic and eukaryotic organismshave been sequenced. This list includes the com-pletion of the genome of the fruit fly Drosophilamelanogaster as well as the genomes of many bac-teria including pathogenic strains such as Haemophilusinfluenzae, Helicobacter pylori, Yersinia pestis, Pseu-domonas aeruginosa, Campylobacter jejuni, as wellas E. coli strain K-12. This has been complementedby completion of the genomes of Saccharomycespombe and cerevisiae (the fission and baker’s yeast,

PROTEIN SEQUENCE ANALYSIS 169
respectively), the nematode worm Caenorhabditis ele-gans and the plant genome of Arabidopsis thaliana.Unfortunately it remains largely true that we have noidea about the structure or function of many of theproteins encoded within these sequenced genomes.
Nowadays it is common to determine the order ofbases along a gene and in so doing deduce the primarysequence of a protein. After locating the relevantstart codon (ATG) it is straightforward to use thegenetic code to translate the remaining triplet of basesinto amino acids and thereby determine the primarysequence of the protein. There are many computerprograms that will perform these tasks using theprimary sequence data to derive additional propertiesabout the protein. These properties can include overallcharge, isoelectric point (pI), hydrophobicity andsecondary structure elements and are part of thebioinformatics revolution that has accompanied DNAand protein sequencing.
For eukaryotes translation of the order of basesalong a gene is complicated by the presence ofintrons or non coding sequences. The recognitionof these sites or the use of cDNA derived fromprocessed mRNA allows protein sequences to beroutinely translated. In databases it is normal for DNAsequences of eukaryotic cells to reflect the codingsequence although occasionally sequences containingintrons are deposited.
Gene sequencing is an automated technique thatinvolves determining the order of only four components(adenine, cytosine, guanine, and thymine, i.e. A, C,G and T) compared with 20 different amino acids.With fewer components it becomes critical to correctlyestablish the exact order of bases since a mistake,such as an insertion or deletion, will result in acompletely different translated sequence. DNA isamplified to produce many complementary copies ofthe template using the polymerase chain reaction (PCR)a process that exploits the activity of thermostableDNA polymerases. The PCR technique is dividedinto three steps: denaturation, annealing and extension(Figure 6.10). Each step is optimized with respectto time and temperature. However, the process isgenerally performed at three different temperatures of∼96, ∼55 and ∼72 ◦C with each phase lasting for about30, 30 and 120 s, respectively.
Figure 6.10 General principle of primer-directed DNAsynthesis by polymerases. The PCR extends this processin a cyclical series of reactions since the polymerase isthermostable and withstands repeated cycles of hightemperature
DNA polymerase activity extends new strands fromprimers (15–25 bases in length) that are complemen-tary to the two template strands (Figure 6.11). A poly-merase commonly used in these studies is that iso-lated from Thermus aquaticus and often referred toas Taq polymerase. The double helix is dissociated athigh temperatures (∼96 ◦C) and on cooling the sus-pension primers anneal to each DNA strand (∼55 ◦C).With a suitable supply of nucleotide triphosphates(dNTPs, Figure 6.12) new DNA strand synthesis bythe polymerase occurs rapidly at 72 ◦C forming twocomplementary strands. After one cycle the PCR dou-bles the number of copies and the beauty of the processis that it can be repetitively cycled to provide largeamounts of identical DNA. It can be seen that start-ing with one copy of DNA leads after thirty roundsof replication to 230 copies of DNA – all identical tothe initial starting material.1 This procedure is used toamplify DNA fragments as part of a cloning, muta-genesis or forensic study but it can also be used tosequence DNA.
If PCR techniques are carried out with dNTPs anda small amount of dideoxy (ddNTPs) nucleotides thenchain termination will result randomly along the newDNA strand. Dideoxynucleotides were first introducedby Frederick Sanger as part of a sequencing strategybased on the absence of an oxygen atom at the
1Only with ‘proofreading’ DNA polymerases. Taq polymerase is notproof reading and exhibits an error rate of ∼1 in 400 bases.

170 THE DIVERSITY OF PROTEINS
Figure 6.11 The use of PCR-based methods for amplification and replication of template DNA
NO
N
N
HN
NH2
HO
HO
O
HO
N
N
N
N
NH2
Figure 6.12 The base and sugar components of NTPs.In this case the deoxyadenosine and dideoxyadenosine(note absence of hydroxyl at C3′ position)
C3′ position of the ribose ring. The effect of themissing oxygen is to prevent further elongation of thenucleotide chain in the 5′ –3′ direction via an inhibitionof phosphodiester bond formation.
When small amounts of the four nucleotides arepresent as ddNTPs random incorporation will result inchain termination (Figure 6.13). On average the PCRwill result in a series of DNA fragments truncatedat every nucleotide, each fragment differing fromthe previous one by just one base in length. Quiteclearly if we can establish the identity of the lastbase we can gradually establish the DNA sequence.The second step of DNA sequencing separates these
DNA fragments. To aid identification each ddNTP isalso labelled with a fluorescent probe based on thechromophores fluoroscein and rhodamine 6 G. Eachof the four dideoxy nucleotides is tagged with adifferent fluorescent dye that has an emission maxima(λmax) that allows the final base to be discriminated.Dye-labelled DNA fragments are separated accordingto mass by running through polyacrylamide gelsor capillaries. Today the most sophisticated DNAsequencing systems use capillary electrophoresis withthe advantages of high separation efficiency, fastseparations at high voltages, ease of use with smallsample volumes (∼1 µ l), and high reproducibility.‘Labelled’ DNA exits the capillaries and laser-inducedfluorescence detected by a charge coupled device(CCD) is interpreted as a fluorescence profile by acomputer leading to routine sequencing of over 1000nucleotides with very low error rates (Figure 6.14).
Sequence homology
Advances in both DNA and protein sequencing havegenerated enormous amounts of data. This data repre-sents either the order of amino acids along a polypeptidechain or the order of bases along a nucleotide chain andcontains within this ‘code’ significant supplementaryinformation on proteins. With the introduction of power-ful computers sequences can be analysed and comparedwith each other. In particular computational analysis has

PROTEIN SEQUENCE ANALYSIS 171
5' 3'
primer
GT
AA
C
C
CA
AA
T
T
G
GG
Sequencing mixture of DNA polymerase, template, dNTPs, plus 'fluorescent' ddATP, ddCTP, ddGTP, ddTTP.
Figure 6.13 Chain termination of an extending DNA sequence by incorporation of dideoxynucleotide triphosphates(ddNTPs)
allowed the recognition of sequence similarity. For anysequence there is a massive number of permutations andsimilarity does not arise by chance. Instead sequencesimilarity may indicate evolutionary links and in thiscontext the term homology is used reservedly. Proteinsequences can be similar without needing to invoke anevolutionary link but the term homology implies evo-lutionary lineage from a common ancestor. Both DNAand protein sequences can show homology. In order
to establish that protein sequences are homologous wehave to establish rules governing this potential similarity.Consider the partial sequences
-D-E-A-L-V-S-V-A-F-T-S-I-V-G-G-
-D-E-A-F-T-S-I-V-G-G-M-D-D-P-G-
This represents a small section of the polypeptidechain (15 residues) in each of two sequences. Are they
T T A A A A A A A A AC C CT T T TG G G60 70
G G G G
Figure 6.14 A small section of the computer-interpreted fluorescence profile of a DNA sequence. A is denoted bythe green trace, C in blue, G in black and T by the red trace. Sequence anomalies arise when ‘clumps’ of bases arefound together, for example, a block of T residues and may require user intervention

172 THE DIVERSITY OF PROTEINS
similar and if so how similar or are they different?An initial inspection ‘by eye’ might fail to establishany relationship. However, closer examination wouldreveal that the two sequences could be ‘aligned’ byintroducing a gap of seven residues into the secondsequence. When the sequences are re-written as
D-E-A-L-V-S-V-A-F-T-S-I-V-G-G-D-E- ∗ -∗ - ∗ -∗ - ∗ - ∗ -∗ -T-S-I-V-G-G-M-D-D-P-G
a good alignment exists for some of the residues andsignificantly these residues are identical. Clearly tosome observers this is significant similarity (8 out of15 residues are identical) whilst to others it could beviewed as a major difference (7 residues are missingand information is lacking about the similarity of thelast 5 residues in the second sequence!). What is neededis a way of quantifying this type of problem.
Alignment of protein sequences is the first steptowards quantifying similarity between one or moresequences. As a result of point mutations or largermutational events sequences change giving proteinscontaining different residues. This obscures relation-ships between proteins and one reason for comparingand aligning sequences is to deduce these relationships.For newly determined sequences this allows identifica-tion to previously characterized proteins and highlightsa shared common origin.
In Chapter 3 the tertiary structures of haemoglobin α
and β chains were superimposed to reveal little differ-ence in respective fold (Figure 3.43). This suggestedan evolutionary relationship as a result of ancestralgene duplication. Structural homology, supported bysignificant sequence homology between α and β chains,reveals a level of identity between the two chains ofover 40 percent (62/146) (Figure 6.15).
Domains are key features of modular or mosaicproteins. Sequence alignments reveal that gene dupli-cation leads to a proliferation of related domains indifferent proteins. As a result, proteins are relatedby the presence of similar domains – one example isthe occurrence of the SH3 domain in proteins thatshare little else in common except the presence of thismotif. SH3 (or Src Homology 3) domains are small,non-catalytic, modules of 50–70 residues that medi-ate protein–protein interactions by binding to proline-rich peptide sequences. The domain was discovered intyrosine kinase as one module together with SH1 (tyro-sine kinase) and SH2 (phosphotyrosine binding). Thedomain is found in kinases, lipases, GTPases, structuralproteins and viral regulatory proteins.
Alignment methods offer a way of pictorially repre-senting similarity between one or more ‘test’ sequencesand a library of ‘known’ sequences derived fromdatabases. In addition alignment methods offer a routetowards quantifying the extent of this similarity byincorporating ‘scoring’ schemes. A number of differentapproaches exist for aligning sequences. A prevalentapproach is called a ‘pairwise similarity’ and involvescomparing each sequence in the database (library)with a ‘test’ sequence (Figure 6.16). The observa-tion of ‘matches’ indicates sequence similarity. A sec-ond level of comparison involves comparing familiesof sequences with libraries to establish relationships(Figure 6.17). This approach establishes ‘profiles’ forthe initial family and then attempts to fit this profile toother members of the database.
A third approach is to use known motifs foundwithin proteins. These motifs are invariant or highlyconserved blocks of residues characteristic of a proteinfamily. These motifs are used to search databasesfor other sequences bearing the corresponding motif.
* 20 * 40 * 60 *: -VLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHF-DLS-----HGSAQVKGHGKKVADALTNAVA: VHLTPEEKSAVTALWGKV--NVDEVGGEALGRLLVVYPWTQRFFESFGDLSTPDAVMGNPKVKAHGKKVLGAFSDGLA
80 * 100 * 120 * 140 *: HVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTSKYR-----:
ab
ab HLDNLKGTFATLSELHCDKLHVDPENFRLLGNVLVCVLAHHFGKEFTPPVQAAYQKVVAGVANALAHKYH-----
Figure 6.15 The sequence of the α and β chains of haemoglobin. Identical residues are shown in red whilstconserved residues are shown in yellow

PROTEIN SEQUENCE ANALYSIS 173
Figure 6.16 Pairwise similarity search of the databank using single ‘query’ sequence
Figure 6.17 The use of a profile established from aligned proteins to improve quality of matches
Table 6.4 A selection of the characteristic motifs used to identify proteins
Motif sequence Protein family Example
CX2CH Class I soluble c type cytochromes Bacterial cytochrome c551
F(Y)L(IVMK)X2HPG(A)G Cytochrome b5 family Nitrate reductase
CX7L(FY)X6F(YW)XR(K)X8CXCX6C Ribosomal proteins L3 protein
A(G)X4GKS(T). ATPases ATP synthetase
CX2CX3L(IVMFYWC)X8HX3H Zn finger-DNA binding proteins TFIIIA
LKEAExRAE Tropomyosin family Tropomyosin
Motifs are simply short sequences of amino acid residues within a polypeptide chain that facilitate the identification of related proteins. The residues inparenthesis are alternative residues that make up the consensus motif. The X indicates the number of intervening residues lacking any form of consensus. Thenumber of intervening residues can show minor variations in length.

174 THE DIVERSITY OF PROTEINS
Query sequence Domain database or library Domain identification in proteins
Figure 6.18 A query sequence is used with a domain-based database to identify similar proteins containingrecognized domains
Diagnostic motifs occur regularly in proteins with wholedatabases devoted to their identification (Table 6.4).Many protein families have conserved sequence motifsand it is often sufficient to ‘query’ the database only withthis motif to identify related proteins (Figure 6.18).
The best forms of sequence alignment are derivedvia computational methods known as dynamic program-ming that detect optimal pairwise alignment betweentwo or more proteins. The details of computational pro-gramming are beyond the scope of this book but theprocedure compares two or more sequences by look-ing for individual characters, or a series of characterpatterns, that are in the same order in each sequence.Identical or similar characters are placed in the same col-umn, and non-identical characters can be placed in thesame column either as a mismatch or opposite a ‘gap’ inthe other sequences. Irrespective of the approach used toderive alignments a query sequence is compared with allsequences within a selected database to yield a ‘score’that indicates a level of similarity. An optimal alignmentwill result in the arrangement of non-identical charactersand gaps minimized to yield identical or similar charac-ters as a vertical register. The art of these methods is tocreate a feasible scoring scheme, and the identity matrixor matrices based on physicochemical properties oftenfail to establish relationships, even for proteins knownto be related.
The most important improvement in scoring schemescame from methods based on the observed changes
in amino acid sequence in homologous proteins. Thisallowed mutation rates to be derived from their evo-lutionary distances and was pioneered by MargaretDayhoff in the 1970s. The study measured the fre-quencies with which residues are changed as a resultof mutation during evolution and involved carefullyaligning (by eye) all proteins recognized to be withina single family. The process was repeated for differ-ent families and used to construct phylogenetic treesfor groups of proteins. This approach yielded a tableof relative frequencies describing the rate of residuereplacement by each of the nineteen other residuesover an evolutionary period. By combining this tablewith the relative frequency of occurrence of residues inproteins a family of scoring matrices were computedknown as point accepted mutation (PAM) matrices.The PAM matrices are based on estimated mutationrates from closely related proteins and are effective at‘scoring’ similarities between sequences that divergedwith evolutionary time. In the PAM 250 matrix thedata reflects aligned protein sequences extrapolated toa level of 250 amino acid replacements per 100 residuesper 100 million years. A score above 0 indicates thatamino acids replace each other more frequently thanexpected from their distribution in proteins and thisusually means that such residues are functionally equiv-alent. As expected mutations involving the substitutionof D > E or D > N are relatively common whilst tran-sitions such as D > R or D > L are rare.

PROTEIN SEQUENCE ANALYSIS 175
An alternative approach is the BLOCKS databasebased on ungapped multiple sequence alignments thatcorrespond to conserved regions of proteins. These‘blocks’ were constructed from databases of familiesof related proteins (such as Pfam, ProDom, InterProor Prosite). This has yielded approximately 9000blocks representing nearly 2000 protein families. TheBLOCKS database is the basis for the BLOSUMsubstitution matrices that form a key component ofcommon alignment programs such as BLAST, FASTA,etc. (BLOSUM is an acronym of BLOcks SUbstitutionMatrices.) These substitution matrices are widely usedfor scoring protein sequence alignments and are basedon the observed amino acid substitutions in a largeset of approximately 2000 conserved amino acidpatterns, called blocks. The blocks act as identifiersof protein families and are based around a greaterdataset than that used in the PAM matrices. TheBLOSUM matrices detect distant relationships andproduce alignment that agrees well with subsequentdetermination of tertiary structure. In general if a testsequence shares 25–30 percent identity with sequencesin the database it is likely to represent a homologousprotein. Unfortunately, when the level of similarity fallsbelow this level of identity it proves difficult to drawfirm conclusions about homology.
Structural homology arising from sequencesimilarity
With large numbers of potential sequences for proteinsthe number of different folded conformations mightalso be expected to be large. However, the tertiary
or folded conformations of proteins are less diversethan would be expected from the total number ofsequences. Protein folds have been conserved duringevolution with conservation of structure occurringdespite changes in primary sequence.
In most cases structural similarities arise as aresult of sequence homology. However, in a fewinstances structural homology has been observed wherethere is no obvious evolutionary link. One familyof proteins that clearly indicates both sequence andstructural homology is the cytochrome c family.Vertebrate cytochrome c isolated from mitochondria,such as those from horse and tuna, share very similarprimary sequences (only 17 out of 104 residues aredifferent) and this is emphasized by comparable tertiarystructures (Figure 6.20). The conservation of structureis expected as both horse and tuna proteins occupysimilar functional roles. Yeast cytochrome c alsoexhibits homology and a similar tertiary structure buta reduced level of sequence identity to either horseor tuna cytochrome c (∼59 out of 104 residues areidentical) reflects a more distant evolutionary lineage(hundreds of millions of years). In bacteria a wide rangeof c type cytochromes are known all containing theheme group covalently ligated to the polypeptide viatwo thioether bridges derived from cysteine residues.The proteins are soluble, contain between 80 and 130residues, and function as redox carriers.
If the sequences of cytochrome c2 from Rhodobacterrubrum, cytochrome c-550 from Paracoccus denitrifi-cans and the mitochondrial cytochromes c from yeast,tuna and horse are compared only 18 residues remaininvariant (Figure 6.19). These residues include His
Horse : GDVEKGKKIFVQKCAQCHTVE--------KGGKHKTGPNLHGLFGRKTGQAPGFTYTDA---NKNKG--ITWKEETuna : GDVAKGKKTFVQKCAQCHTVE--------NGGKHKVGPNLWGLFGRKTGQAEGYSYTDA---NKSKG--IVWNENYeast : GSAKKGATLFKTRCLQCHTVE--------KGGPHKVGPNLHGIFGRHSGQAEGYSYTDA---NIKKN--VLWDENP.denitrif : GDAAKGEKEF-NKCKACHMVQAPDGTDIVKGG--KTGPNLYGVVGRKIASEEGFKYGDGILEVAEKNPDLVWTEAR.rubrum : GDAAAGEKV-SKKCLACHTFD--------QGGANKVGPNLFGVFENTAAHKDNYAYSESYTEMKAKG--LTWTEA
Horse : TLMEYLENPKKYI------PG--TKMIFAGIKKKTEREDLIAYLKKATNETuna : TLMEYLENPKKYI------PG--TKMIFAGIKKKGERQDLVAYLKSATS-Yeast : NMSEYLTNPKKYI------PG--TKMAFGGLKKEKDRNDLITYLKKACE-P.denitrif : DLIEYVTDPKPWLVEKTGDSAAKTKMTFK-LGKNQA--DVVAFLAQNSPDR.rubrum : NLAAYVKNPKAFVLEKSGDPKAKSKMTFK-LTKDDEIENVIAYLKTLK--
Figure 6.19 Sequence homology between the cytochromes c of horse, tuna, yeast, R. rubrum and P. denitrificans.Shown in red are identical residues between the sequences whilst yellow residues highlight those residues that areconserved within the horse, tuna and yeast eukaryotic sequences. Only 18 out of 104 residues show identity

176 THE DIVERSITY OF PROTEINS
(a) (b)
(c)
(e)
(d)
Figure 6.20 The structures of cytochrome c from different source organisms. The structures shown for fivecytochrome c were obtained from (a) tuna (PDB: 3CYT), (b) yeast – the iso-1 form (PDB:1YCC), (c) P. denitrificans(PDB:155C), (d) R. rubrum cytochrome c2 (PDB:1C2R), and (e) horse (PDB:1CRC). The insertions of residues in thebacterial sequences can be seen in the additional backbone structure shown in the bottom left region of the molecule

PROTEIN SEQUENCE ANALYSIS 177
18, Met80, Cys14 and Cys17 and are critical to thefunctional role of cytochrome c in electron transfer.Consequently their conservation is expected. There isvery little sequence similarity between cytochrome c2
and horse cytochrome c but an evolutionary lineage isdefined by tracking progressive changes in sequencethrough micro-organisms, animal and plants. In thismanner it is clear that cytochrome c represents the sys-tematic evolution of a protein designed for biologicalelectron transfer from a common ancestral protein. Amore thorough analysis of the sequences reveals thatalthough changes in residue occur at many positionsthe majority of transitions involve closely related aminoacid residues. For example, near the C terminus of eachcytochrome c a highly conserved Phe residue is usu-ally found but in the sequence of P. denitrificans thisresidue is substituted with Tyr.
Irrespective of the changes in primary sequence forthese cytochromes c considerable structural homologyexists between all of these proteins. This homologyextends from the protein found in the lowliest prokary-ote to that found in man. It is only by comparing inter-mediate sequences between cytochrome c2 and horsecytochrome c that evolutionary links are establishedbut structural homology is a strong indicator that theproteins are related. As a caveat although structuralhomology is normally a good indicator of relationshipsit is not always true (see below) and must be supportedby sequence analysis.
The serine proteases are another example of struc-tural homology within an evolutionarily related groupof proteins. This family of enzymes includes famil-iar proteins from higher organisms such as trypsin,chymotrypsin, elastase and thrombin and they havethe common function of proteolysis. If the sequencesof trypsin, chymotrypsin and elastase are compared(Figure 6.21) they exhibit an identity of ∼40 percentand this is comparable to the identity shown betweenthe haemoglobin α and β chains. The three-dimensionalstructure of all of these enzymes are known and theyshare similar folded conformations with invariant Hisand Ser residues equivalent to positions 57 and 195in chymotrypsin located in the active sites along withthe third important residue of Asp 102. Together theseresidues make up the catalytic triad and from theirrelative positions other members of the family of ser-ine proteases have been identified. The similarity in
Figure 6.21 The structures of chymotrypsin, trypsinand elastase shown with elements of secondarystructure superimposed. Chymotrypsin (blue, PDB:2CGA), elastase (magenta, PDB: 1QNJ) and trypsin(green, PDB: 1TGN). Shown in yellow in a cleft oractive site are the catalytic triad of Ser, His and Asp(from left to right) that are a feature of all serineprotease enzymes
sequence and structure between chymotrypsin, elastaseand trypsin indicates that these proteins arose fromgene duplication of an ancestral protease gene withsubsequent evolution accounting for individual differ-ences.
By following changes in sequence for different pro-teins in a family an average mutation rate is calcu-lated and reveals that ‘house-keeping’ proteins such ashistones, enzymes catalysing essential metabolic path-ways, and proteins of the cytoskeleton evolve at veryslow rates. This generally means the sequences incor-porate between 1 and 10 mutations per 100 residuesper 100 million years. Consequently to obscure allevolutionary information, which generally requires∼250–350 substitutions per 100 residues, takes a con-siderable length of time. As a result of this slow

178 THE DIVERSITY OF PROTEINS
Table 6.5 Rate of evolution for different proteins (adapted from Wilson, A.C. Ann. Rev. Biochem. 1977, 46, 573–639)
Protein Accepted pointmutations/100
residues/108 years
Protein Accepted pointmutations/100
residues/108 years
Histone H2 0.25 Insulin 7Collagen α1 2.8 Glucagon 2.3Cytochrome c 6.7 Triose phosphate isomerase 5.3Cytochrome b5 9.1 Lactate dehydrogenase M chain 7.7Lysozyme 40 α-lactalbumin 43Ribonuclease A 43 Immunoglobulin V region 125Myoglobin 17 Haemoglobin α 27Histone H1 12 Haemoglobin β 30
rate of evolution ‘house-keeping’ proteins are excel-lent tools with which to trace evolutionary relation-ships over hundreds of millions of years. Higher ratesof evolution are seen in proteins occupying less criti-cal roles.
Mutations arising in DNA are not always convertedinto changes in protein primary sequence. Somemutations are silent due to the degeneracy inherent inthe genetic code. A nonsense mutation results fromthe insertion of a stop codon within the open readingframe of mRNA and gives a truncated polypeptidechains. The generation of a stop codon close to thestart codon will invariably lead to a loss of proteinactivity whilst a stop codon located relatively near tothe original ‘stop’ sight may well be tolerated by theprotein. Missense mutations change the identity of aresidue by altering bases within the triplet coding foreach amino acid. From the standpoint of evolutionaryanalysis it is these mutations that are detected viachanges in primary sequences.
Proteins with different structures and functionsevolve at significantly different rates. This is seen mostclearly by comparing proteins found in Homo sapiensand Rhesus monkeys. For cytochrome c the respectiveprimary sequences differ by less than 1 percent of theirresidues but for the α and β chains of haemoglobinthese differences are at a level of 3–5 percent whilstfor fibrinopeptides involved in blood clotting the differ-ences are ∼30 percent of residues. This emphasizes the
need to use families of proteins to extrapolate rates ofprotein evolution. Systematic studies have determinedrates of evolution for a wide range of proteins of dif-ferent structure and function (Table 6.5) with mutationrates expressed as the number of point mutations per100 residues per 108 years.
Conotoxins are a family of small peptides derivedfrom the Conus genus of predatory snails. These snailshave a proboscis containing a harpoon-like organ thatis capable of injecting venom into fish, molluscs andother invertebrates causing rapid paralysis and death.The venom contains over 75 small toxic peptides thatare between 13 and 35 residues in length and disulfiderich. The toxins have been purified and exhibit vary-ing degrees of toxicity on the acetylcholine receptor ofvertebrate neurones. The peptides represent a molec-ular arms race whereby rapid evolution has allowedConus to develop ‘weapons’ in the form of toxins ofdifferent sequence and functional activity. As potentialprey adapt to the toxins Conus species evolve new tox-ins based around the same pattern. Whereas proteinssuch as histones show evolutionary rates of changeof ∼0.25 point mutations/100 residues/108 years theconotoxins have much higher rates of change estimatedat ∼60–180 point mutations/100 residues/108 years.This pattern is supported by analysis of other toxinssuch as those from snake venom where typical muta-tion rates are ∼100 point mutations/100 residues/108
years.

PROTEIN SEQUENCE ANALYSIS 179
The rapid development of molecular cloningtechniques, DNA sequencing methods, sequence com-parison algorithms and powerful yet affordable com-puter workstations has revolutionized the importanceof protein sequence data. Thirty years ago proteinsequence determination was often one of the final stepsin the characterization of a protein, whilst today onepremise of the human genome mapping project is thatsequencing all of the genes found in man will uncovertheir function via data analysis. There is no doubt thatthis premise is beginning to yield rich rewards with theidentification of new homologues as well as new openreading frames (ORFs), but in many cases sequencedata has remained impervious to analysis.
The above examples highlight structural homol-ogy that has persisted from a common ancestralprotein despite the slow and gradual divergence ofprotein sequences by mutation. Occasionally structuralhomology is detected where there is no discernablerelationship between proteins. This is called convergentevolution and arises from the use of similar structuralmotifs in the absence of sequence homology. In sub-tilisin and serine carboxypeptidase II a catalytic triadof Ser-Asp-His is observed (Figure 6.22) that mightimply a serine protease but the arrangement of residueswithin their primary sequences are different to chy-motrypsin and these proteins differ in overall structure.It is therefore very unlikely that these three proteins
Figure 6.22 The positions of the catalytic triadtogether with hydrophobic residues involved in sub-strate binding in unrelated serine proteases. Subtilisin,chymotrypsin and serine carboxypeptidase II
could have arisen from a common ancestor of chy-motrypsin or a related serine protease since the order
Table 6.6 Potential examples of convergent evolution in proteins
Protein 1 Protein 2 Functional motif
Chymotrypsin Subtilisin
Carboxypeptidase II
Catalytic triad of Ser/Asp/His residues in the
proteolysis of peptides
Triose phosphate
isomerase
As many as 17 different groups of enzymes
possess β barrel. Luciferase, pyruvate kinase
and ribulose 1,5 bisphosphate
carboxylase-oxygenase (rubisco)
TIM barrel fold of eight parallel β strands forming
a cylinder connected by eight helices arranged
in an outer layer of the protein
Carbonic
anhydrase (α)
Carbonic anhydrase (β) form plants. Zn ion ligated to protein and involved in catalytic
conversion of CO2 to HCO3−
Thermolysin Carboxypeptidase A Zn ion ligated to two imidazole side chains and
the carboxyl side chain of Glu. Also
coordinated by water molecule playing a crucial
role in proteolytic activity.

180 THE DIVERSITY OF PROTEINS
of residues in the catalytic triad differs as do otherstructural features present in each of the enzymes. Thisphenomenon is generally viewed as an example of con-vergent evolution where nature has discovered the samecatalytic mechanism on more than one occasion (seeTable 6.6).
The term convergent evolution has been appliedto the Rossmann fold found in nucleotide bindingdomains consisting of three β strands interspersedwith two α helices with a common role of bind-ing ligands such as NAD+ or ADP. In many pro-teins these structural elements are identified, but lit-tle sequence homology exists between nucleotide-binding proteins. The sequences reflect the productionof comparable topological structures for nucleotide-binding domains by different permutations of residues.Are these domains diverged from a common ances-tor or do they result from convergent evolution? Thelarge number of domains found in proteins capableof binding ATP/GTP or NAD/NADP suggests that itis unlikely that proteins have frequently and inde-pendently evolved a nucleotide-binding motif domain.It is now thought more likely that the Rossmannfold evolved with numerous variations arising as aresult of divergent evolution from a common andvery distant ancestral protein. The β barrel exem-plified by triose phosphate isomerase is also widelyfound in proteins and may also represent a simi-lar phenomenon.
Protein databases
A number of important databases are based on thesequence and structure information deposited andarchived in the Protein Data Bank. These databasesattempt to order the available structural informa-tion in a hierarchical arrangement that is valuablefor an analysis of evolutionary relationships as wellas enabling functional comparisons. In the SCOP(Structural Classification of Proteins) database all ofthe deposited protein structures are sorted accord-ing to their pattern of folding. The folds are eval-uated on the basis of their arrangement and in par-ticular the composition and distribution of secondarystructural elements such as helices and strands. Fre-quently construction of this hierarchical system is
based on manual classification of protein folds andas such may be viewed as subjective. Currently,non-subjective methods are being actively pursued inother databases in an attempt to remove any biasin classification of protein folds. The levels of orga-nization are hierarchical and involve the classes offolds, superfamilies, families and domains (the indi-vidual proteins).
Domains within a family are homologous and have acommon ancestor from which they have diverged. Thehomology is established from either sequence and/orfunctional similarity. Proteins within a superfamilyhave the same fold and a related function and thereforealso probably have a common ancestor. However,within a superfamily the protein sequence compositionor function may be substantially different leadingto difficulty in reaching a conclusive decision aboutevolutionary relationships. At the next level, the fold,the proteins have the same topology, but there isno evidence for an evolutionary relationship except alimited structural similarity.
The CATH database attempts to classify proteinfolds according to four major hierarchical divisions;Class, Architecture, Topology and Homology (seeFigure 6.23 for an example). It also utilizes algo-rithms to establish definitions of each hierarchicaldivision. Class is determined according to the sec-ondary structure composition and packing within aprotein structure. It is assigned automatically for moststructure (>90 percent) with manual inspection usedfor ‘difficult’ proteins. Four major classes are recog-nized; mainly α, mainly β and α–β protein domainstogether with domains that have a very low secondarystructure content. The mixed α–β class can be fur-ther divided into domains with alternating α/β struc-tures and domains with distinct α rich and β richregions (α + β). The architecture of proteins (A-levelhierarchy) describes the overall shape of the domainand is determined by the orientations of the indi-vidual secondary structural elements. It ignores theconnectivity between these secondary structures andis assigned manually using descriptions of secondarystructure such as β barrel or β–α–β sandwich. Sev-eral well-known architectures have been described inthis book and include the β propellor, the four-helixbundle, and the helix-turn-helix motif. Structures aregrouped into fold families or topologies at the next

SECONDARY STRUCTURE PREDICTION 181
C
TIM barrel
flavodoxin(4fxn)
b–lactamase(1mbIA1)
Sandwich Roll
a a + b b
A
T
Figure 6.23 The distinguishing of flavodoxin and β
lactamase; two α/β proteins based around a β sandwichstructure
level of organization. Assignment of topology dependson the overall shape and connectivity of the secondarystructures and has started to be automated via algo-rithm development. A number of topologies or foldfamilies have been identified, with some, such as theβ two-layer sandwich architecture and the α–β three-layer sandwich structures, being relatively common.The H level of classification represents the homolo-gous superfamily and brings together protein domainsthat share a common ancestor. Similarities are iden-tified first by sequence comparisons and subsequentlyby structural comparisons. Hierarchical levels of orga-nization are shown at the C, A and T states for α/βcontaining proteins.
Gene fusion and duplication
The preceding sections highlight that some proteinsshare similar sequences reflected in domains of similarstructure. The α + β fold of cytochrome b5 firstevolved as an electron transfer protein and thenbecame integrated as a module found in larger multi-domain proteins. The result was that ligated to ahydrophobic tail the protein became a membrane-bound component of the endoplasmic reticulum fattyacid desaturase pathway. When this domain was joinedto a Mo-containing domain an enzyme capable ofconverting sulfite into sulfate was formed, sulfiteoxidase. Joining the cytochrome to a flavin-containingdomain formed the enzyme, nitrate reductase. Proteinssharing homologous domains arose as a result ofgene duplication and a second copy of the gene.This event is advantageous to an organism since largegenetic variation can occur in this second copy withoutimpairing the original gene.
The globin family of proteins has arisen by geneduplication. Haemoglobin contains 2α and 2β chainsand each α and β chain shows homology and issimilar to myoglobin. This sequence homology reflectsevolutionary origin with a primitive globin functioningsimply as an oxygen storage protein like myoglobin.Subsequent duplication and evolution allowed thesubtle properties of allostery as well as the differencesbetween the α and β subunits. During embryogenesisother globin chains are observed (ξ and ε chains)and fetal haemoglobin contains a tetramer made upof α2γ2 subunits that persists in adult primates at alevel of ∼1 percent of the total haemoglobin. The δ
chain is homologous to the β subunit and leads to theevolution of the globin chains in higher mammals as agenealogical tree, with each branch point representinggene duplication that gives rise to further globinchains (Figure 6.24).
Secondary structure prediction
One of the earliest applications of using sequenceinformation involved predicting elements of secondarystructure. If sufficient numbers of residues can beplaced in secondary structure then, in theory at least,it is possible to generate a folded structure based on

182 THE DIVERSITY OF PROTEINS
Figure 6.24 Evolution of the globin chains inprimates. Each branch point represents a geneduplication event with myoglobin evolving early inthe evolutionary history of globin subunit
the packing together of helices, turns and strands.Secondary structure prediction methods are often basedon the preference of amino acid residues for certainconformational states.
One algorithm for secondary structure predictionwas devised by P.Y. Chou and G.D. Fasman, andalthough superior methods and refinements exist todayit proved useful in defining helices, strands and turnsin the absence of structural data. It was derived fromdatabases of known protein structures by estimatingthe frequency with which a particular amino acid wasfound in a secondary structural element divided by thefrequency for all other residues. A value of 1 indicates arandom distribution for a particular amino acid whilsta value greater than unity suggests a propensity forfinding this residue in a particular element of secondarystructure. A series of amino acid preferences wereestablished (Table 6.7).
The numbers in the first three columns, Pα, Pβ Pt,are equivalent to preference parameters for the 20amino acids for helices, strands and reverse turnsrespectively. From this list one can assign the residueson the basis of the preferences; Ala, Arg, Glu, Gln,His, Leu, Lys, and Met are more likely to be found inhelices; Cys, Ile, Phe, Thr, Trp, Tyr and Val are likelyto be found in strands while Asn, Asp, Gly, Pro andSer are more frequently located in turns. The algorithm
Table 6.7 Propensity for a given amino acid residueto be found in helix, strand or turn
Residue Pα Pβ Pt Residue Pα Pβ Pt
Ala 1.41 0.72 0.82 Leu 1.34 1.22 0.57
Arg 1.21 0.84 0.90 Lys 1.23 0.84 0.90
Asn 0.76 0.48 1.34 Met 1.30 1.14 0.52
Asp 0.99 0.39 1.24 Phe 1.16 1.33 0.65
Cys 0.66 1.40 0.54 Pro 0.34 0.31 1.34
Gln 1.27 0.98 0.84 Ser 0.57 0.96 1.22
Glu 1.59 0.52 1.01 Thr 0.76 1.17 0.90
Gly 0.43 0.58 1.77 Trp 1.02 1.35 0.65
His 1.05 0.80 0.81 Tyr 0.74 1.45 0.76
Ile 1.09 1.67 0.47 Val 0.90 1.87 0.41
(After Chou, P.Y. & Fasman, G.D. Ann. Rev. Biochem 1978, 47, 251–276;and Wilmot, C.M. & Thornton, J.M. J. Mol. Biol. 1988, 203, 221–232).A value of 1.41 observed for alanine in helices implies that alanine occurs 41percent more frequently than expected for a random distribution
of Chou and Fasman pre-dated the introduction ofinexpensive computers but contained a few simplesteps that could easily be calculated (Table 6.8). Inpractice the Chou–Fasman algorithm has a success rateof ∼50–60 percent, although later developments havesucceeded in reaching success rates above 70 percent.A major failing of the Chou–Fasman algorithm isthat it considers only local interactions and neglectslong-range order known to influence the stability ofsecondary structure. In addition the algorithm makesno distinction between types of helices, types of turnsor orientation of β strands.
The availability of large families of homologoussequences (constructed using the algorithms describedabove) has revolutionized methods of secondary struc-ture prediction. Traditional methods, when applied to afamily of proteins rather than a single sequence haveproved much more accurate in identifying secondarystructure elements. Today the combination of sequencedata with sophisticated computing techniques such asneural networks has given accuracy levels in excess of70 percent. Besides the use of advanced computationaltechniques recent approaches to the problem ofsecondary structure prediction have focussed on theinclusion of additional constraints and parameters toassist precision. For example, the regular periodicity of

GENOMICS AND PROTEOMICS 183
Table 6.8 Algorithm of Chou and Fasman
Step Procedure
1 Assign all of the residues in the peptidethe appropriate set of parameters
2 Scan through the peptide and identifyregions where four out of 6 consecutiveresidues have Pα > 1.0
This region is declared to be an α helix3 Extend the helix in both directions until a
set of four residues yield an averagePα < 1.00. This represents the end ofthe helix
4 If the segment defined in step 3 is longerthan 5 residues and the average value ofPα > Pβ for this segment it can also beassigned as a helix
5 Repeat this procedure to locate all helicalregions
6 Identify a region of the sequence where 3out of 5 residues have a value ofPβ > 1.00. These residues are in a β
strand7 Extend the sheet in both directions until a
set of four contiguous residues yieldingan average Pβ < 1.00 is reached. Thisrepresents the end of the β strands
8 Any segment of the region located by thisprocedure is assigned as a β-strand ifthe average Pβ > 1.05 and the averagevalue of Pβ > Pα for the same region
9 Any region containing overlapping helicaland strand assignments are taken to behelical if the average Pα > Pβ for thatregion. If the average Pβ > Pα for thatregion then it is declared a β strand
the α helix of 3.6 residues per turn means that in pro-teins many regular helices are amphipathic with polar
residues on the solvent side and non-polar residuesfacing the inside of the protein. Recognition of a peri-odicity of i, i + 3, i + 4, i + 7, etc. in hydrophobicresidues has proved particularly effective in predictinghelical regions in membrane proteins.
Genomics and proteomics
The completion of the human genome sequencingproject has provided a large amount of data concerningthe number and distribution of proteins. It seems likelythat the human genome contains over 25 000 differentpolypeptide chains, and most of these are currentlyof unknown structure and function. Genomics reflectsthe wish to understand more about the complexityof living systems through an understanding of geneorganization and function. Advances in genomicshave provided information on the number, size andcomposition of proteins encoded by the genome.It has highlighted the organization of genes withinchromosomes, their homology to other genes, thepresence of introns together with the mechanismand sites of gene splicing and the involvementof specific genes in known human disease states.The latter discovery is heralding major advances inunderstanding the molecular mechanisms of diseaseparticularly the complex interplay of genetic andenvironmental factors. Genomics has stimulated thediscovery and development of healthcare products byrevealing thousands of potential biological targets fornew drugs or therapeutic agents. It has also initiated thedesign of new drugs, vaccines and diagnostic DNA kits.So, although genomic based therapeutic agents includetraditional ‘chemical’ drugs, we are now seeing theintroduction of protein-based drugs as well as the veryexciting and potentially beneficial approach of genetherapy.
However, as the era of genomics reaches maturityit has expanded from a simple definition referring tothe mapping, sequencing, and analysis of genomesto include an emphasis on genome function. Toreflect this shift, genome analysis can now be dividedinto ‘structural genomics’ and ‘functional genomics’.Structural genomics is the initial phase of genomeanalysis with the end point represented by the high-resolution genetic maps of an organism embodied

184 THE DIVERSITY OF PROTEINS
by a complete DNA sequence. Functional genomicsrefers to the analysis of gene expression and the useof information provided by genome mapping projectsto study the products of gene expression. In manyinstances this involves the study of proteins and amajor branch of functional genomics is the new andexpanding area of proteomics.
Proteomics is literally the study of the proteomevia the systematic and global analysis of all proteinsencoded within the genome. The global analysis ofproteins includes specifically an understanding ofstructure and function. In view of the potentiallylarge number of polypeptides within genomes thisrequires the development and application of large-scale, high throughput, experimental methodologies toexamine not only vast numbers of proteins but alsotheir interaction with other proteins and nucleic acids.Proteomics is still in its infancy and current scientificresearch is struggling with developing methods to dealwith the vast amounts of information provided by thegenomic revolution. One approach that will be usedin both genomic and proteomic study is statistical andcomputational analysis usually called bioinformatics.
Bioinformatics
Bioinformatics involves the fusion of biology withcomputational sciences. Almost all of the areas
described in the previous sections represent part ofthe expanding field of bioinformatics. This new andrapidly advancing subject allows the study of biologicalinformation at a gene or protein level. In general, bioin-formatics deals with methods for storing, retrieving andanalysing biological data. Most frequently this involvesDNA, RNA or protein sequences, but bioinformat-ics is also applied to structure, functional properties,metabolic pathways and biological interactions. With awide range of application and the huge amount of ‘raw’data derived from sequencing projects and deposited indatabanks (Table 6.9) it is clear that bioinformatics as amajor field of study will become increasingly importantover the next few decades.
Bioinformatics uses computer software tools fordatabase creation, data management, data storage, datamining and data transfer or communication. Advancesin information technology, particularly the use of theInternet allows rapid access to increasing amounts ofbiological information. For example, the sequencedgenomes of many organisms are widely available atmultiple web sites throughout the world. This allowsresearchers to download sequences, to manipulatethem or to compare sequences in a large number ofdifferent ways.
Central to the development of bioinformatics hasbeen the explosion in the size, content and popularity
Table 6.9 A selection of databases related to protein structure and function; some have been used frequentlythroughout this book to source data
Database/repository/resource URL (web address)
The Protein DataBank. http://www.rcsb.org/pdbExpert Protein Analysis System (EXPASY) http://www.expasy.ch/European Bioinformatics Institute http://www2.ebi.ac.ukProtein structure classification (CATH) http://www.biochem.ucl.ac.uk/bsm/cathStructural classification of proteins (SCOP) http://scop.mrc-lmb.cam.ac.uk/scopAtlas of proteins side chain interactions. http://www.biochem.ucl.ac.uk/bsm/sidechainsHuman genome project (a tour!) http://www.ncbi.nlm.nih.gov/TourAn online database of inherited diseases http://www.ncbi.nlm.nih.gov/OmimRestriction enzyme database http://rebase.neb.comAmerican Chemical Society http://pubs.acs.org

GENOMICS AND PROTEOMICS 185
of the world wide web, the most recognized componentof the internet.
Initially expected to be of use only to scien-tists the world wide web is a vast resource allow-ing data transfer in the form of pages containingtext, images, audio and video content. Pages, linkedby pointers, allow a computer on one side of theworld to access information anywhere in the worldvia series of connected networks. The pointers referto URL’s or ‘uniform resource locators’ and are thebasic ‘sites’ of information. So, for example, the pro-tein databank widely referred to in this book has aURL of http://www.rcsb.org. The ‘http’ part of a webaddress simply refers to the method of transferringdata and stands for hypertext transfer protocol. Hyper-text is the language of web pages and all web pagesare written in a specially coded set of instructionsthat governs the appearance and delivery of pagesknown as hypertext markup language (HTML). Finally,the web pages are made comprehensible (interpreted)by ‘browsers’ – software that reads HTML and dis-plays the content. Browsers include Internet Explorer,Netscape and Opera and all can be used to view ‘on-line content’ connected with this book. In 10 years theweb has become a familiar resource.
The development of computers has also beenrapid and has seen the introduction of faster ‘chips’,increased memory and expanded disk sizes to providea level of computational power that has enabled thedevelopment of bioinformatics. In a trite formulationof a law first commented upon by Gordon Moore,and hence often called Moore’s law, the clock speed(in MHz) available on a computer will double every18 months. So in 2003 I am typing this bookon a 1.8 GHz-based computer. Although computerspeeds will undoubtedly increase there are grounds forbelieving rates of increase will slow. Searching andmanipulating large databases, coupled with the use ofcomplex software tools to analyse or model data, placeshuge demands on computer resources. It is likely that inthe future single powerful computer workstations witha very high clock speed, enormous amounts of memoryand plentiful storage devices will become insufficientto perform these tasks. Instead bioinformatics will haveto harness the power of many computers working eitherin parallel or in grid-like arrays. These computerscumulatively will allow the solution to larger and more
complex problems. However, today bioinformatics isan important subject in its own right that directsbiological studies in directions likely to result infavourable outcomes.
One of the results of genome sequencing projects isthat software tools can be developed to compare andcontrast genetic information. One particular avenue thatis being pursued actively at the moment is the pre-diction of protein structure from only sequence infor-mation. The determination of three dimensional pro-tein structures is an expensive, time consuming andformidable task usually involving X-ray crystallogra-phy or NMR spectroscopy. Consequently any methodthat allows a ‘by-pass’ of this stage is extremely attrac-tive especially to pharmaceutical companies where theprime objective is often product development. Oneapproach is to compare a protein sequence to otherproteins since sequential homology (identity >25 per-cent) will always be accompanied by similar topology.If the structure of a sequentially homologous proteinis known then the topology of the new protein canbe deduced with considerable certainty. A more likelyscenario, however, is that the sequence may show rel-atively low levels of identity and one would like toknow how similar, or different, the three dimensionalstructures might be to each other. This is a far moredifficult problem. Comparative modelling will workwhen sequentially homologous proteins are comparedbut may fail when the levels of identity fall below abenchmark of ∼25 percent. Additionally some proteinsshow very similar structures with very low levels ofsequence homology.
A second approach is the technique of ‘threading’.This approach attempts to compare ‘target sequences’against libraries of known structural templates. Acomparison produces a series of scores that are rankedand the fold with the best score is assumed to be the oneadopted by the unknown sequence. This approach willfail when a new ‘fold’ or tertiary structure is discoveredand it relies on a representative database of structuresand sequences.
The ab initio approach (Figure 6.25) ignoressequence homology and attempts to predict the foldedstate from fundamental energetics or physicochemicalproperties associated with the constituent residues.This involves modelling physicochemical parametersin terms of force fields that direct the folding of the

186 THE DIVERSITY OF PROTEINS
Figure 6.25 The ab initio approach to fold prediction
primary sequence from an initial randomized structureto one satisfying all constraints. These constraintswill reflect the energetics associated with charge,hydrophobicity and polarity with the aim being to find asingle structure of low energy. The resulting structuresshould have very few violations with respect to bondangles and length and can be checked for consistencyagainst the Ramachandran plot. Ab initio protocols donot utilize experimental constraints but depend on thegeneration of structure from fundamental parametersin silico.
This approach is based on the thermodynamicargument that the native structure of a protein is theglobal minimum in the free energy profile. Ab initiomethods place great demands on computational powerbut have the advantage of not using peripheral informa-tion. Despite considerable technical difficulties successis being achieved in this area as a result of regular‘contests’ held to judge the success of ab initio pro-tocols. These proceedings go under the more formalname of critical assessment of techniques for proteinstructure prediction (CASP). As well as involving thecomparative homology based methods CASP involvesthe use of ab initio methods to predict tertiary struc-tures for ‘test’ sequences (Figure 6.26). The empha-sis is on prediction as opposed to “postdiction” andinvolves a community wide attempt to determine struc-tures for proteins that have been assessed independently(but are not available in public domain databases).Independent structural knowledge allows an accurateassessment of the eventual success of the different ab
Figure 6.26 One good model predicted for targetprotein (T0091) in CASP4. The native structure ofT0091 is shown on the left and the predicted model isshown on the right. Structurally equivalent residues aremarked in yellow (reproduced with permission Sippl,M.J. et al. Proteins: Structure, Function, and Genetics,Suppl. 5, 55–67. John Wiley & Sons, Chichester, 2001)
initio approaches. Generally the results are expressedas rmsd (root mean square deviations), reflecting thedifference in positions between corresponding atoms inthe experimental and calculated (predicted) structures.In successful predictions rmsd values below 0.5 nmwere seen for small proteins (<100 residues) usingab initio approaches. Although the agreement betweenpredicted and experimentally determined structures isstill relatively poor the resolution allows in favourable

PROBLEMS 187
circumstances the backbone of ‘target proteins’ to bedefined with reasonable accuracy and for overall foldto be identified. For larger proteins the refinement ofstructure is worse but ab initio methods are becomingsteadily better and offer the possibility of true proteinstructure prediction in the future.
Summary
Despite the incredible diversity of living cells allorganisms are made up of the same 20 amino acidresidues linked together to form proteins. This arisesfrom the origin of the amino acid alphabet very earlyin evolution before the first true cells. All subsequentforms of life evolved using this basic alphabet.
Prebiotic synthesis is the term applied to non-cellularbased methods of amino acid synthesis that existed over3.6 × 109 years ago. The famous experiment of Ureyand Miller demonstrated formation of organic moleculessuch as adenine, alanine and glycine that are theprecursors today of nucleic acids and protein systems.
A major development in molecular evolution wasthe origin of self-replicating systems. Today this roleis reserved for DNA but the first replicating systemswere based on RNA a molecule now known to havecatalytic function. An early prebiotic system involvingRNA molecules closely associated with amino acids isthought to be most likely.
The fossil record shows primitive prokaryotic cellsresembling blue-green cyanobacteria evolved 3.6 bil-lion years ago. Evolution of these cells through com-partmentalization, symbiosis and specialization yieldedsingle cell and metazoan eukaryotes.
Protein sequencing is performed using the Edmanprocedure and involves the labelling and identificationof the N-terminal residue with phenylisothiocyanatein a cyclic process. The procedure can be repeated
∼50–80 times before cumulative errors restrict theaccuracy of sequencing.
Nucleic acid sequencing is based on dideoxy chaintermination procedures. The result of efficient DNAsequencing methods is the completion of genomesequencing projects and the prevalence of enormousamounts of bio-information within databases.
Databases represent the ‘core’ of the new areaof bioinformatics – a subject merging the disciplinesof biochemistry, computer science and informationtechnology together to allow the interpretation ofprotein and nucleic acid sequence data.
One of the first uses of sequence data was toestablish homology between proteins. Sequence homol-ogy arises from a link between proteins as a resultof evolution from a common ancestor. Serine pro-teases show extensive sequence homology and this isaccompanied by structural homology. Chymotrypsin,trypsin and elastase share homologous sequences andstructure.
Structural homology will also result when sequencesshow low levels of sequence identity. The c typecytochromes from bacteria and mitochondria exhibitremarkably similar folds achieved with low overallsequence identity. The results emphasize that proteinsevolve with the retention of the folded structure andthe preservation of functional activity.
The bioinformatics revolution allows analysis ofprotein sequences at many different levels. Com-mon applications include secondary structure predic-tion, conserved motif recognition, identification of sig-nal sequences and transmembrane regions, determina-tion of sequence homology, and structural predictionab initio.
In the future bioinformatics is likely to guide thedirections pursued by biochemical research by allowingthe formation of new hypotheses to be tested viaexperimental methods.
Problems1. Use the internet to locate some or all of the web
pages/databases listed in Table 6.9. Find any webpage(s) describing this book.
2. A peptide containing 41 residues is treated withcyanogen bromide to liberate three smaller peptideswhose sequences are

188 THE DIVERSITY OF PROTEINS
(i) Phe-Leu-Asn-Ser-Val-Thr-Val-Ala-Ala-Tyr-Gly-Gly-Pro-Ala-Lys-Pro-Ala-Val-Glu-Asp-Gly-Ala-Met
(ii) Ala-Ser-Ser-Glu-Glu-Lys-Gly-Met and
(iii) Val-Ser-Thr-Asn-Glu-Lys-Ala-Ala-Val-Phe
Trypsin digestion of the same 41 residue peptide yieldsthe sequences:
(i) Ala-Ala-Val-Phe
(ii) Gly-Met-Phe-Leu-Asn-Ser-Val-Thr-Val-Ala-Ala-Tyr-Gly-Gly-Pro-Ala-Lys-Pro-Ala-Val-Glu-Asp-Gly-Ala-Met-Val-Ser-Thr-Asn-Glu-Lys and
(iii) Ala-Ser-Ser-Glu-Glu-Lys
Can the sequence be established unambiguously?
3. Obtain the amino acid sequences of human α-lactalbumin and lysozyme from any suitable database.Use the BLAST or FASTA tool to compare theamino acid sequences of human α-lactalbumin andlysozyme. Are the sequences sufficiently similar tosuggest homology?
4. Identify the unknown protein of Table 6.3.
5. Download the following coordinate files from theprotein databank 1CYO and 1NU4. In each caseuse your molecular graphic software to highlight thenumber of Phe residues in each protein, any co-factorspresent in these proteins and the number of chargedresidues present in each protein.
6. What is the EC number associated with the enzyme β-galactosidase. Locate the enzyme derived from E. coli
in a protein or sequence database. How many domainsdoes this protein possess? Are these domains related?Describe the structure of each domain. Estimate theresidues encompassing each domain. Identify theactive site and any conserved residues.
7. Find a program on the internet that allows theprediction of secondary structure. Use this programto predict the secondary structure content of myo-globin. Does this value agree with the known sec-ondary structure content from X-ray crystallogra-phy? Repeat this trial with your ‘favourite’ protein?Now repeat the analysis with a different secondarystructure prediction algorithm. Do you get the sameresults?
8. Identify proteins that are sequentially homologous to(a) α chain of human haemoglobin, (b) subtilisin Efrom Bacillus subtilis, (c) HIV protease. Commenton your results and some of the implications?
9. The β barrel structure is a common topology foundin many proteins. An example is triose phosphateisomerase. Use databases to find other proteins withthis structural motif. Do these sequences exhibithomology? Comment on the evolutionary relationshipof β barrels.
10. Assuming a point accepted DNA mutation rate of20 (20 PAMs/100 residues/108 years) establish thedegree of DNA homology for two proteins eachof 200 residues that diverged 500 million yearsago. What is the maximal level of amino acidresidue homology between proteins and what is thelowest level?

7Enzyme kinetics, structure, function,
and catalysis
One important function performed by proteins is theability to catalyse chemical reactions. Catalytic func-tion was amongst the first biological roles recognizedin proteins through the work of Eduard Buchner andEmil Fischer. They identified and characterized theability of some proteins to convert reactants into prod-ucts. These proteins, analogous to chemical catalysts,increased rates of reaction but did not shift the equi-librium formed between products and reactants. Thebiological catalysts were named enzymes – the namederived from the Greek for ‘in yeast’ – ‘en’ ‘zyme’.
Within all cells every reaction is regulated bythe activity of enzymes. Enzymes catalyse metabolicreactions and in their absence reactions proceed atkinetically insignificant rates incompatible with living,dynamic, systems. The presence of enzymes resultsin reactions whose rates may be enhanced (catalysed)by factors of 1015 although enhancements in therange 103 –109 are more typical. Enzymes participatein the catalysis of many cellular processes rangingfrom carbohydrate, amino acid and lipid synthesis,their breakdown or catabolic reactions (see Figure 7.1),DNA repair and replication, transmission of stimulithrough neurones, programmed cell death or apoptosis,the blood clotting cascade reactions, the degradation ofproteins, and the export and import of proteins acrossmembranes. The list is extensive and for each reaction
the cell employs a unique enzyme tailored via millionsof years of evolution to catalyse the reaction withunequalled specificity.
This chapter explores how enzyme structure allowsreactions to be catalysed with high specificity andrapidity. In order to appreciate enzyme-catalysed reac-tions the initial sections deal with simple chem-ical kinetics introducing the terms applicable tothe study of reaction rates. Analysis of chemicalkinetics proved to be very important in unravel-ling mechanisms of enzyme catalysis. Kinetic stud-ies uncovered the properties of enzymes, productsand reactants, as well as the steps leading to theirformation. In vivo enzyme activity differs from thatobserved in vitro by its modulation by other pro-teins or ligands. The modulation of enzyme activ-ity occurs by several mechanisms but is vital tonormal cell function and homeostasis. In principle,regulation allows cells to respond rapidly to environ-mental conditions, turning on or off enzyme activityto achieve metabolic control under fluctuating con-ditions. When conditions change and the enzyme isno longer required activity is reduced without wastingvaluable cell resources. Feedback mechanisms con-trol enzyme activity and are a feature of allostery.Allosteric enzymes exist within the biosynthetic path-ways of all cells.
Proteins: Structure and Function by David Whitford 2005 John Wiley & Sons, Ltd

190 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
Pyruvate
O
O
CH3
CH3
CH2
CH2
HO
HO C
CH2
H
1/2‡COO−
1/2‡COO−
1/2‡COO−
1/2‡COO−
1/2‡COO−
‡COO−
CH2
CH2
C
S CoA
O
1/2‡COO−
CH2
CH2
CH
HC
C COO−
CoASH
1. citrate synthase
8. malate dehydrogenase
Citric acidcycle
6. succinate dehydrogenase
7. fumarase
H2O
L-Malate
Citrate
2. aconitase
Isocitrate
3. isocitrate dehydrogenase
a-Ketoglutarate
4. a-Ketoglutarate dehydrogenase
C‡
S CoA
COO−
COO−
‡COO−
*COO−
‡COO−
‡COO−
*COO−
CH2
CH2
C O
*COO−
CH2
C
CHO H
H COO−
CO2
CH2
H2O
*COO−
C O
CoASH + NAD+
CO2 + NADH
pyruvate dehydrogenase
Acetyl-CoA
NADH
NAD+
H+
NAD+
NADH
NAD+
NADH H++
H++
+
Oxaloacetate
Fumarate FADH2
FAD
Succinate
GTP+GDP Pi
5. succinyl-CoA synthetase
CoASH
Succinyl-CoA
CoASH*CO2
C‡
Figure 7.1 The conversion of pyruvate to acetate from the reactions of glycolysis lead to the Krebs citricacid cycle. Reproduced from Voet et al. (1999) by permission of John Wiley & Sons, Ltd. Chichester

ENZYME NOMENCLATURE 191
Table 7.1 Examples of enzymes belonging to the six major classes
Enzyme class Typical example Reaction catalysed
Oxidoreductases Lactate dehydrogenase (EC1.1.1.27) Reduction of pyruvate to lactate with thecorresponding formation of NAD
Transferases Hexokinase (EC 2.7.1.1) Transfer of phosphate group to glucosefrom ATP
Hydrolases Acetylcholinesterase (EC 3.1.1.7) Hydrolysis of acetylcholine to choline andacetate
Lyases Phenylalanine Ammonia Lyase (EC 4.3.1.5) Splits phenylalanine into ammonia andtrans-cinnamate
Isomerases Triose phosphate isomerase (EC 5.3.1.1) Reversible conversion betweendihydroxyacetone phosphate andglyceraldehyde-3-phosphate
Ligases T4 DNA ligase (EC 6.5.1.1) Phosphodiester bond linkage andconversion of ATP → AMP
Before reviewing these areas it is necessary to estab-lish the basis of enzyme nomenclature and to establishthe broad groups of enzyme-catalysed reactions occur-ring within all cells. For each catalysed reaction aunique enzyme exists; in glycolysis the conversion of3-phosphoglycerate phosphate to 3-phosphoglycerate iscatalysed by phosphoglycerokinase (PGK).
3-phosphoglyceroyl phosphate + ADP −−−⇀↽−−−3-phosphoglycerate + ATP
No other enzyme catalyses this reaction within cells.
Enzyme nomenclatureThe naming of enzymes gives rise to much confusion.Some enzymes have ‘trivial’ names such as trypsinwhere the name provides no useful clue to biologicalrole. Many enzymes are named after their codinggenes and by convention the gene is written initalic script whilst the protein is written in normaltype. Examples are the Escherichia coli gene polAand its product DNA polymerase I; the lacZ geneand β-galactosidase. To systematically ‘label’ enzymesand to help with identification of new enzymes the
International Union of Biochemistry and MolecularBiology (IUBMB) devised a system of nomenclaturethat divides enzymes into six broad classes (Table 7.1gives examples of these). Further division into sub-classes groups enzymes sharing functional properties.The major classes of enzyme are:
1. Oxidoreductases: catalyse oxidation–reduction (re-dox) reactions.
2. Transferases: catalyse transfer of functional groupsfrom one molecule to another.
3. Hydrolases: perform hydrolytic cleavage of bonds.
4. Lyases: remove groups from (or add a group to)a double bond or catalyse bond scission involvingelectron re-arrangement.
5. Isomerases: catalyse intramolecular rearrangementof atoms.
6. Ligases: joins (ligates) two molecules together.
Each enzyme is given a four-digit number written,for example, as EC 5.3.1.1. The first number indi-cates an isomerase whilst the second and third numbersidentify further sub-classes. In this case the second digit

192 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
(3) indicates an isomerase that acts as an intramolecu-lar isomerase whilst the third digit (1) indicates thatthe substrates for this enzyme are aldose or ketosecarbohydrates. The fourth number is a serial num-ber that frequently indicates the order in which theenzyme was recognized. The enzyme EC 5.3.1.1 istriose phosphate isomerase found in the glycolytic path-way catalysing inter-conversion of glyceraldehyde 3phosphate and dihydroxyacetone phosphate. A com-plete list of enzyme classes is given in the Appendix.At the end of 2003 the structures of over 8000 enzymeswere deposited in the PDB database whilst otherdatabases recognized over 3700 different enzymes(http://www.expasy.ch/enzyme/). These lists are con-tinuing to be updated and grow with the completion offurther genome sequencing projects.
Until recently it was thought that all biological cat-alysts (enzymes) were proteins. This view has beenrevised with the observation that RNA molecules(ribozymes) catalyse RNA processing and protein syn-thesis. The observation of catalytic RNA raises aninteresting and perplexing scenario where RNA syn-thesis is catalysed by proteins (e.g. RNA polymerase)but protein synthesis is catalysed by RNA (the ribo-some). This chapter will focus on the properties andmechanisms of protein based enzymes, in other wordsthose molecules synonymous with the term enzyme.
Enzyme co-factorsMany enzymes require additional co-factors to catalysereactions effectively. These co-factors, also called co-enzymes, are small organic molecules or metal ions(Figure 7.2). The nature of the interaction between co-factor and enzyme is variable, with both covalent andnon-covalent binding observed in biological systems.
A general scheme of classifying co-factors recognizesthe strength of their interaction with enzymes. Forexample, some metal ions are tightly bound at activesites and participate directly in the reaction mechanismwhilst others bind weakly to the protein surface andhave remote roles (Table 7.2). Co-factors are vital toeffective enzyme function and many were recognizedoriginally as vitamins. Alongside vitamins metal ionsmodulate enzyme activity; in the apo (metal-free)state metalloenzymes often exhibit impaired catalyticactivity or decreased stability. The metal ions, normallydivalent cations, exhibit a wide range of biologicalroles (Table 7.3). In carboxypeptidase Zn2+ stabilizesintermediates formed during hydrolysis of peptide bondswhilst in heme and non-heme enzymes iron participatesin redox reactions.
Chemical kineticsIn order to understand the details of enzyme actionit is worth reviewing the basics of chemical kineticssince these concepts underpin biological catalysis. Fora simple unimolecular reaction where A is convertedirreversibly into B
A → B
the rate of reaction is given by the rate of disappearanceof reactant or the rate of appearance of product
−d[A]/dt or d[B]/dt (7.1)
The disappearance of reactants is proportional to theconcentration of A
−d[A]/dt = k[A] (7.2)
where k is a first order rate constant (units s−1).Rearranging this equation and integrating between
Figure 7.2 Different co-factors and their interactions with proteins
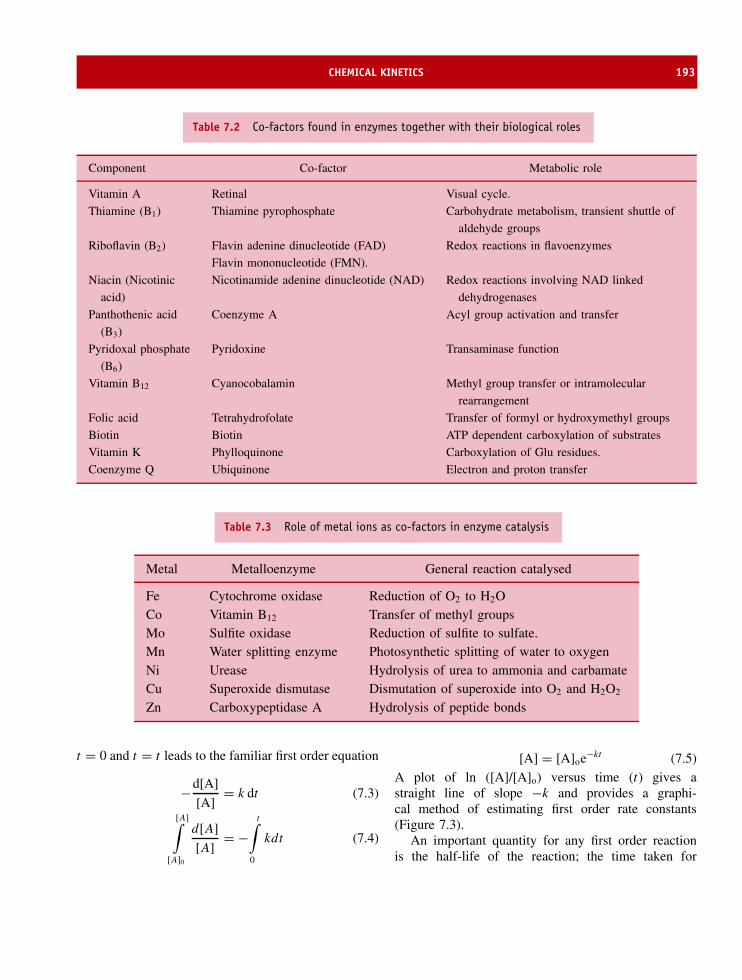
CHEMICAL KINETICS 193
Table 7.2 Co-factors found in enzymes together with their biological roles
Component Co-factor Metabolic role
Vitamin A Retinal Visual cycle.
Thiamine (B1) Thiamine pyrophosphate Carbohydrate metabolism, transient shuttle of
aldehyde groups
Riboflavin (B2) Flavin adenine dinucleotide (FAD) Redox reactions in flavoenzymes
Flavin mononucleotide (FMN).
Niacin (Nicotinic
acid)
Nicotinamide adenine dinucleotide (NAD) Redox reactions involving NAD linked
dehydrogenases
Panthothenic acid
(B3)
Coenzyme A Acyl group activation and transfer
Pyridoxal phosphate
(B6)
Pyridoxine Transaminase function
Vitamin B12 Cyanocobalamin Methyl group transfer or intramolecular
rearrangement
Folic acid Tetrahydrofolate Transfer of formyl or hydroxymethyl groups
Biotin Biotin ATP dependent carboxylation of substrates
Vitamin K Phylloquinone Carboxylation of Glu residues.
Coenzyme Q Ubiquinone Electron and proton transfer
Table 7.3 Role of metal ions as co-factors in enzyme catalysis
Metal Metalloenzyme General reaction catalysed
Fe Cytochrome oxidase Reduction of O2 to H2OCo Vitamin B12 Transfer of methyl groupsMo Sulfite oxidase Reduction of sulfite to sulfate.Mn Water splitting enzyme Photosynthetic splitting of water to oxygenNi Urease Hydrolysis of urea to ammonia and carbamateCu Superoxide dismutase Dismutation of superoxide into O2 and H2O2
Zn Carboxypeptidase A Hydrolysis of peptide bonds
t = 0 and t = t leads to the familiar first order equation
−d[A]
[A]= k dt (7.3)
[A]∫
[A]0
d[A]
[A]= −
t∫
0
kdt (7.4)
[A] = [A]oe−kt (7.5)A plot of ln ([A]/[A]o) versus time (t) gives astraight line of slope −k and provides a graphi-cal method of estimating first order rate constants(Figure 7.3).
An important quantity for any first order reactionis the half-life of the reaction; the time taken for
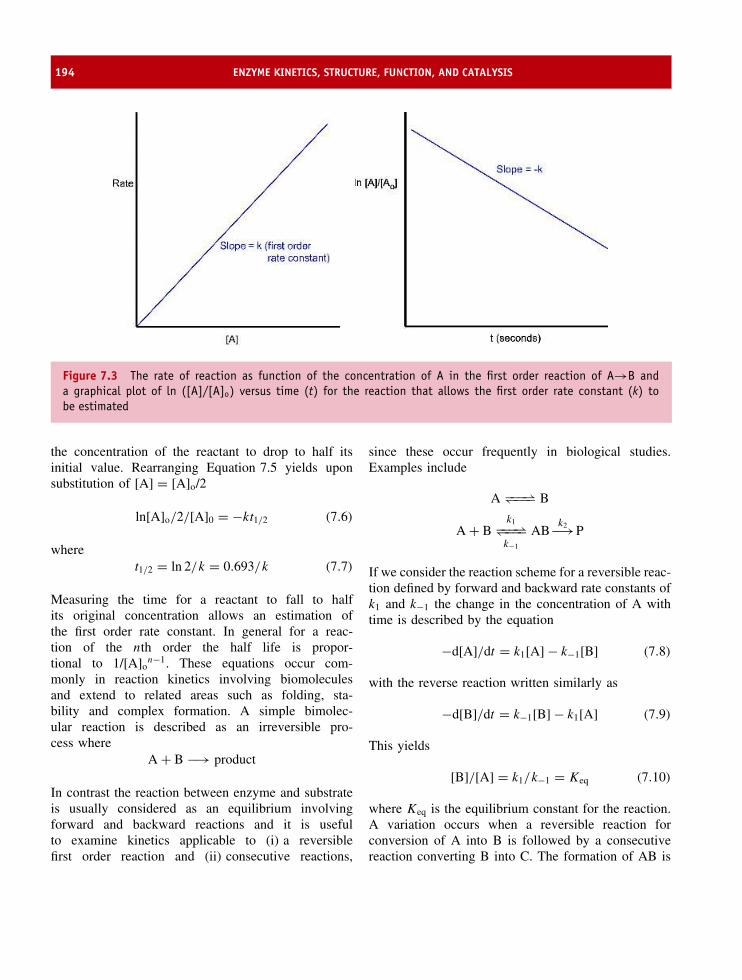
194 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
Figure 7.3 The rate of reaction as function of the concentration of A in the first order reaction of A→B anda graphical plot of ln ([A]/[A]o) versus time (t) for the reaction that allows the first order rate constant (k) tobe estimated
the concentration of the reactant to drop to half itsinitial value. Rearranging Equation 7.5 yields uponsubstitution of [A] = [A]o/2
ln[A]o/2/[A]0 = −kt1/2 (7.6)
wheret1/2 = ln 2/k = 0.693/k (7.7)
Measuring the time for a reactant to fall to halfits original concentration allows an estimation ofthe first order rate constant. In general for a reac-tion of the nth order the half life is propor-tional to 1/[A]o
n−1. These equations occur com-monly in reaction kinetics involving biomoleculesand extend to related areas such as folding, sta-bility and complex formation. A simple bimolec-ular reaction is described as an irreversible pro-cess where
A + B −→ product
In contrast the reaction between enzyme and substrateis usually considered as an equilibrium involvingforward and backward reactions and it is usefulto examine kinetics applicable to (i) a reversiblefirst order reaction and (ii) consecutive reactions,
since these occur frequently in biological studies.Examples include
A −−−⇀↽−−− B
A + Bk1−−−⇀↽−−−k−1
ABk2−→P
If we consider the reaction scheme for a reversible reac-tion defined by forward and backward rate constants ofk1 and k−1 the change in the concentration of A withtime is described by the equation
−d[A]/dt = k1[A] − k−1[B] (7.8)
with the reverse reaction written similarly as
−d[B]/dt = k−1[B] − k1[A] (7.9)
This yields
[B]/[A] = k1/k−1 = Keq (7.10)
where Keq is the equilibrium constant for the reaction.A variation occurs when a reversible reaction forconversion of A into B is followed by a consecutivereaction converting B into C. The formation of AB is

THE TRANSITION STATE AND THE ACTION OF ENZYMES 195
a bimolecular reaction between A and B whilst thebreakdown of AB to yield P is unimolecular. Theeasiest way to deal algebraically with this reactionis to describe the formation of AB. This leads tothe equation
d[AB]/dt = k1[A][B] − k−1[AB] − k2[AB] (7.11)
Under conditions where either k2 � k1 or k−1 � k1
the concentration of the encounter complex [AB]remains low at all times. This assumption leads to theequality d[AB]/dt ≈ 0 and is known as the steady stateapproximation where
k1[A][B] = k−1[AB] + k2[AB] (7.12)
= (k−1 + k2)[AB] (7.13)
[AB] = k1[A][B]/(k−1 + k2) (7.14)
The rate of formation of product P is described by therate of breakdown of AB
d[P]/dt = k2[AB] (7.15)
= k2k1[A][B]/(k−1 + k2) (7.16)
By assimilating the terms k2k1/(k−1 + k2) into aconstant called kobs the bimolecular reaction A + B →AB → P can be described as a one step reactiongoverned by a single rate constant (kobs).
In the above equation two interesting boundary con-ditions apply. When the dissociation of AB reflectedby the constant k−1 is much smaller than the rate k2
(k−1 � k2) then the overall reaction kobs approximatesto k1 and is the diffusion-controlled rate. The secondcondition applies when k2 � k−1 and is known as thereaction-controlled rate leading to an observed rate con-stant of
kobs = k2k1/k−1 (7.17)
orkobs = Keqk2 (7.18)
The transition state and the actionof enzymes
Understanding enzyme action was assisted by transitionstate theory developed by Henry Eyring from the 1930s
onwards. For a bimolecular reaction involving threeatoms there exists an intermediate state of high energythat reflects the breaking of one covalent bond and theformation of a new one. For the reaction of a hydrogenatom with a molecule of hydrogen this state equates tothe breakage of the HA–HB bond and the formation ofa new HB–HC bond.
HA–HB + HC −→ HA + HB–HC
An intermediate called the transition state is denotedby the symbol “‡”. The free energy associated withthe transition state, �G‡, is higher than the freeenergy associated with either reactant or products(Figure 7.4).
Eyring formulated the transition state in quantitativeterms where reactants undergo chemical reactionsby their respective close approach along a path ofminimum free energy called the reaction coordinate.Reactions are expressed by transition state diagramsthat reflect changes in free energy as a function ofreaction coordinate. Favourable reactions have negativevalues for �G(�G < 0) and are called exergonic.Other reactions require energy to proceed and aredescribed as endergonic (�G > 0). The free energychanges as the reaction progresses from reactantsto product. For the reaction R → P we identifyaverage free energies associated with R and P, as GR
and GP, respectively, with the free energy associatedwith the products being lower than that of thereactants; the change in free energy is negative andthe reaction is favourable. However, although thereaction is favourable the profile does not revealanything about the rate of reaction nor does it offerany clues about why some reactions occur fasterthan others.
The concept of activation energy recognized byArrhenius in 1889 from the temperature dependenceof reactions follows the relationship
k = A exp−Ea/RT (7.19)
where k is a rate constant, A is a frequency factorand Ea is the activation energy. Eyring realizedthat a new view of chemical reactions could beobtained by postulating an intermediate, the transitionstate, occurring between products and reactants. The

196 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
Figure 7.4 Transition state diagrams. In the absence of a transition state the pathway from reactants to productsis obscure but one can recognize that the products have a lower free energy than the reactants (GP < GR)
transition state exists for approximately 10−13 –10−14 sleading to a small percentage of reactants being foundin this conformation at any one time. The formationof product required crossing the activation barrierotherwise the transition state decomposed back to thereactants. With this view of a chemical reaction therate-limiting step became the decomposition of thetransition state to products or reactants involving a non-conventional equilibrium established between reactantsand the transition state. For the reaction
A + B −−−⇀↽−−− X‡ −→ products
the equilibrium constant (K‡) is expressed in terms ofthe activated complex (X‡) and the reactants (A and B).
K‡ = X‡/[A][B] (7.20)
The rate of the reaction is simply the product of theconcentration of activated complex multiplied by thefrequency of crossing the activation barrier (ν).
Rate = ν[X‡] = ν[A][B]K‡ (7.21)
Since the rate of reaction is also
= k[A][B] (7.22)
this leads tok = νK‡ (7.23)
Although evaluation of k depends on our ability todeduce ν and K‡ this can be achieved via statisticalmechanics to leave the result
k = kBT /h K‡ (7.24)
where kB on the right-hand side of the equation isthe Boltzmann constant, T is temperature and h isPlanck’s constant. Equation 7.24 embodies thermody-namic quantities (�G, �H and �S) since
�G‡ = −RT ln K‡ (7.25)
and the rate constant can be expressed as
k = kBT /h e−�G‡/RT (7.26)
where � G‡ is the free energy of activation. Since theparameter kBT /h is independent of A and B the rate ofreaction is determined solely by �G‡, the free energyof activation. Since
�G‡ = �H ‡ − T �S‡ (7.27)
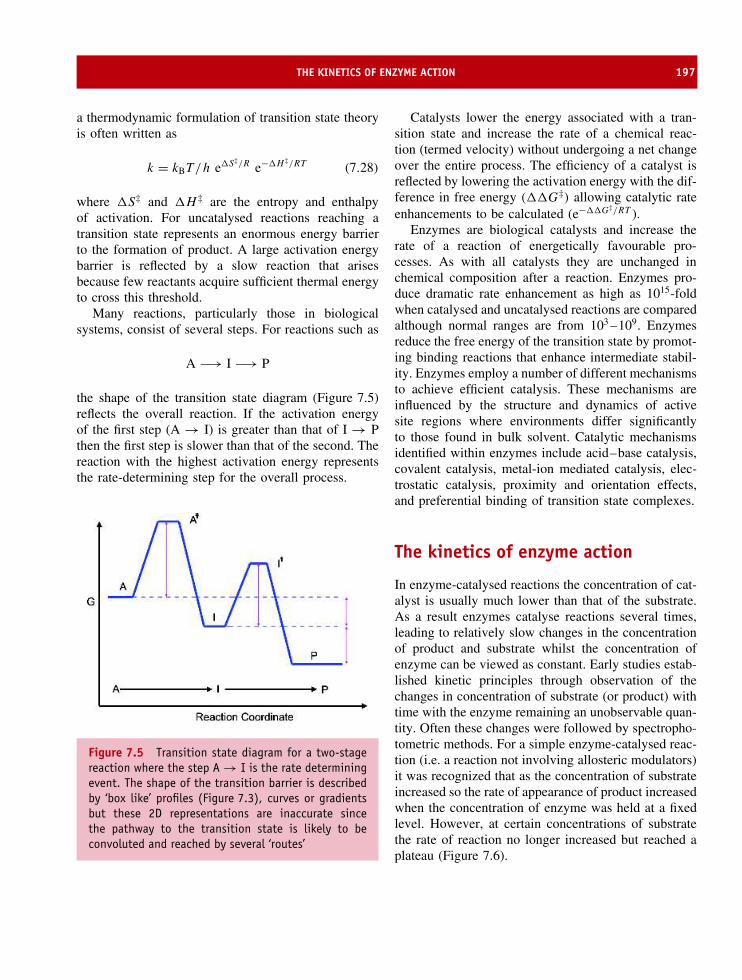
THE KINETICS OF ENZYME ACTION 197
a thermodynamic formulation of transition state theoryis often written as
k = kBT /h e�S‡/R e−�H ‡/RT (7.28)
where �S‡ and �H ‡ are the entropy and enthalpyof activation. For uncatalysed reactions reaching atransition state represents an enormous energy barrierto the formation of product. A large activation energybarrier is reflected by a slow reaction that arisesbecause few reactants acquire sufficient thermal energyto cross this threshold.
Many reactions, particularly those in biologicalsystems, consist of several steps. For reactions such as
A −→ I −→ P
the shape of the transition state diagram (Figure 7.5)reflects the overall reaction. If the activation energyof the first step (A → I) is greater than that of I → Pthen the first step is slower than that of the second. Thereaction with the highest activation energy representsthe rate-determining step for the overall process.
Figure 7.5 Transition state diagram for a two-stagereaction where the step A → I is the rate determiningevent. The shape of the transition barrier is describedby ‘box like’ profiles (Figure 7.3), curves or gradientsbut these 2D representations are inaccurate sincethe pathway to the transition state is likely to beconvoluted and reached by several ‘routes’
Catalysts lower the energy associated with a tran-sition state and increase the rate of a chemical reac-tion (termed velocity) without undergoing a net changeover the entire process. The efficiency of a catalyst isreflected by lowering the activation energy with the dif-ference in free energy (��G‡) allowing catalytic rateenhancements to be calculated (e−��G‡/RT ).
Enzymes are biological catalysts and increase therate of a reaction of energetically favourable pro-cesses. As with all catalysts they are unchanged inchemical composition after a reaction. Enzymes pro-duce dramatic rate enhancement as high as 1015-foldwhen catalysed and uncatalysed reactions are comparedalthough normal ranges are from 103 –109. Enzymesreduce the free energy of the transition state by promot-ing binding reactions that enhance intermediate stabil-ity. Enzymes employ a number of different mechanismsto achieve efficient catalysis. These mechanisms areinfluenced by the structure and dynamics of activesite regions where environments differ significantlyto those found in bulk solvent. Catalytic mechanismsidentified within enzymes include acid–base catalysis,covalent catalysis, metal-ion mediated catalysis, elec-trostatic catalysis, proximity and orientation effects,and preferential binding of transition state complexes.
The kinetics of enzyme action
In enzyme-catalysed reactions the concentration of cat-alyst is usually much lower than that of the substrate.As a result enzymes catalyse reactions several times,leading to relatively slow changes in the concentrationof product and substrate whilst the concentration ofenzyme can be viewed as constant. Early studies estab-lished kinetic principles through observation of thechanges in concentration of substrate (or product) withtime with the enzyme remaining an unobservable quan-tity. Often these changes were followed by spectropho-tometric methods. For a simple enzyme-catalysed reac-tion (i.e. a reaction not involving allosteric modulators)it was recognized that as the concentration of substrateincreased so the rate of appearance of product increasedwhen the concentration of enzyme was held at a fixedlevel. However, at certain concentrations of substratethe rate of reaction no longer increased but reached aplateau (Figure 7.6).

198 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
24
× 103
20
16
12
8
4
1 2 3Substrate concentration (mM)
Rat
e (s
−1)
4 5
Figure 7.6 Three different enzyme-catalysed reactions showing different Km values but similar Vmax values. See textfor discussion of Vmax and Km. Each Vmax is estimated from the asymptote approximating to the curve’s maximumvalue and despite appearances these curves actually reach the same value at very high concentrations of substrate
The rate of reaction (ν) is proportional to the rateof removal of substrate or alternatively the rate offormation of product.
v = −d[S]/dt = d[P]/dt (7.29)
At low substrate concentrations a linear or firstorder dependence on [S] exists whilst at highersubstrate concentrations this shifts to zero order orsubstrate independent. The profile suggests that theenzyme-catalysed conversion of substrate into productinvolved the formation of an intermediate called theenzyme–substrate complex (Figure 7.7).
From a minimal reaction scheme, together with twoassumptions originally formulated by Leonor Michaelisand Maud Menten, we can derive important enzyme
E + S ES E + Pk1 k2
k−1
Figure 7.7 The basic enzyme-catalysed reactionconverts substrate into product
kinetic relationships. The two assumptions made byMichaelis and Menten were that the enzyme existseither as a free form [E] or as enzyme substratecomplex [ES] and secondly that the enzyme substratecomplex [ES] is part of an equilibrium formed betweenE and S during the time scale of the experiment orobservation. These assumptions require that k2, therate of breakdown of the enzyme substrate complex,is much smaller than the remaining two rate constants.The total concentration of enzyme active sites (ET) isexpressed as the sum of the free enzyme concentration(E) plus that found in the enzyme substrate complex.The initial velocity of an enzyme-catalysed reaction is
v = −d[S]/dt d = [P]/dt = k2[ES] (7.30)
with a maximal velocity achieved at high substrateconcentrations when the enzyme is saturated withsubstrate. Defining this maximal velocity as Vmax leadsto the following equalities
[ES] = [ET] ⇒ Vmax = k2[ET] = kcat[ET] (7.31)

THE KINETICS OF ENZYME ACTION 199
The enzyme substrate complex is called the Michaeliscomplex and the equilibrium formed with E and S isdescribed by a constant called the Michaelis constant(Km). From the scheme above the Michaelis constantKm is equal to
Km = (k2 + k−1)/k1 = ([ET] − [ES])[S]/[ES](7.32)
and since [ET] − [ES] = [E] reorganization of thisequation leads to
Km[ES] = [ET][S] − [ES][S] (7.33)
Kmv/k2 = Vmax/k2[S] − v/k2[S] (7.34)
A final re-arrangement of the above equation leadsto the velocity of an enzyme-catalysed reaction, v,expressed as a function of substrate concentration–thefamous Michaelis–Menten equation
v = Vmax[S]/Km + [S] (7.35)
The velocity is related to substrate concentrationthrough two parameters (Vmax and Km) that are specificand characteristic for a given enzyme under a definedset of conditions (pH, temperature, ionic strength, etc.).From the above equation it can be seen that when
[S] � Km v = Vmax[S]/Km
[S] � Km v = Vmax
[S] = Km v = Vmax/2 (7.36)
The Michaelis–Menten equation is usually analysedby linear plots to derive the parameters Km andVmax. Most frequently used in enzyme kinetics is theLineweaver–Burk plot of 1/v versus 1/[S] (Figure 7.8).
Although widely used the Lineweaver–Burk plotcan be unsatisfactory because it fails to adequatelydefine deviations away from linearity at high andlow substrate concentrations. This problem can beneglected with the use of line fitting or linear regressionanalysis methods. More satisfactory in terms of dataanalysis is the Eadie–Hofstee plot where rearrangingthe Michaelis–Menten equation yields
v = −Km ν/[S] + Vmax (7.37)
Experimental data are usually derived by measuringthe initial velocity of the enzyme-catalysed reactionat a range of different substrate concentrations beforesubstantial substrate depletion or product accumulation.
The steady state approximation and thekinetics of enzyme action
An alternative approach to enzyme kinetic analysisproposed by G.E. Briggs and J.B.S. Haldane showed
Lineweaver–Burk Eadie–Hofstee
vo
1/Vmax
1/[S]1/Kmvo/[S]
1/vo
Figure 7.8 Lineweaver–Burk and Eadie–Hofstee plots derived from the Michaelis–Menten equation. The slopesassociated with each plot are Km/Vmax and −Km respectively

200 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
E + S ES E + Pk1 k2
k−1
Figure 7.9 The basic scheme for an enzyme-catalysedreaction
that it was unnecessary to assume that enzyme andsubstrate are in thermodynamic equilibrium with theES complex (Figure 7.9).
After mixing together enzyme and substrate theconcentration of ES rises rapidly reaching a constantlevel called the ‘steady state’ that persists untilsubstrate depletion. Most rate measurements are madeduring the steady state period after the initial build upof ES called the pre-steady state and before substratedepletion. The ‘steady state approximation’ leads to thefollowing relationships
d[ES]/dt = 0 = k1[E][S] − (k2 + k−1)[ES] (7.38)
d[E]/dt = 0 = k2 + k−1[ES] − k1[E][S] (7.39)
The rate of formation of product is proportional to theconcentration of ES
d[P]/dt = k2[ES] (7.40)
and from the steady state relationships defined abovewe have
k1[E][S] = (k2 + k−1)[ES] (7.41)
This leads with the substitution of [E] = [ET] − [ES] to
[ET] = (k2 + k−1)[ES]/k1[S] + [ES] (7.42)
= [ES](1 + (k2 + k−1)/k1[S]) (7.43)
The Michaelis constant Km is defined as (k2 + k−1)/k1
and allows the above equation to be further simplified
[ES] = [ET]/(1 + Km/[S]) (7.44)
Having defined the concentration of the ES complexwe can now relate the formation of product (d[P]/dt)to substrate concentration since
d[P]/dt = k2[ES] = k2[E]T/(1 + Km/[S]) (7.45)
= k2[E]T[S]/([S] + Km) (7.46)
E + S ES EP E + Pk1
k−1
k2
k−2
k3
k−3
Figure 7.10 A more detailed reaction scheme thatdescribes an enzyme catalysed reaction
A second assumption made in the Michaelis–Mententheory was that the enzyme exists in two forms:the enzyme and the enzyme–substrate complex.Observations derived from kinetic studies indicateseveral forms of enzyme–substrate complex andenzyme–product complex co-exist with the freeenzyme. This allows the simple kinetic schemeto be redefined including reversible reactions andenzyme–product complex formed before product re-lease (Figure 7.10).
Km is the initial substrate concentration that allowsthe reaction to proceed at the half maximal velocity.Km is sometimes defined as the substrate dissociationconstant (Ks = k−1/k1) although this applies only undera restricted set of conditions when the catalyticconstant kcat is much smaller than either k1 or k−1.Under these conditions, and only these conditions, theparameter Km describes the affinity of an enzyme forits substrate.
Km discriminates between hexokinase and glucok-inase; a rare example of two enzymes that catal-yse the phosphorylation of glucose. The Km forglucokinase is ∼10 mM whilst the Km for hexok-inase is ∼100 µM. The Km of each enzyme dif-fers by a factor of ∼100 and although catalysingthe same reaction these enzymes have different bio-logical functions. Hexokinase is distributed through-out all cells and is the normal route for glucoseconversion into glucose-6-phosphate. In contrast glu-cokinase is found in the liver and is used whenblood glucose levels are high, for example after acarbohydrate-rich meal. It seems likely that enzymespossess Km values reflecting intracellular levels ofsubstrate to ensure turnover at rates close to maxi-mal velocity.
More rigorous descriptions of enzyme activityinvolve the use of kcat and Km. Km is a mea-sure of the stability of the enzyme–substrate [ES]complex whilst kcat is a first order rate constantdescribing the breakdown of the ES complex into

THE KINETICS OF ENZYME ACTION 201
Table 7.4 The efficiency of enzymes; kcat/Km ratios or specificity constants show a wide range of values
Enzyme Substrate Km (mM) kcat (s−1) kcat/Km (M−1 s−1)
Carbonic anhydrase CO2 12.0 1.0 × 106 8.3 × 107
Catalase H2O2 25.0 4.0 × 105 1.5 × 107
Pepsin Phe-Gly of peptide chain 0.3 0.5 1.7 × 103
Ribonuclease (RNA)n 7.9 7.9 × 102 1.0 × 105
Tyrosl-tRNA synthetase tRNA 0.9 7.6 8.4 × 103
Glucose isomerase Glucose 140.0 1.04 × 103 7.4Fumarase Fumarate 0.005 8.0 × 102 1.6 × 108
Lysozyme (NAG-NAM)3 6 0.5 83DNA polymerase I dNTPs 0.025 1 4.0 × 104
Acetylcholinesterase Acetylcholine 0.095 1.4 × 104 1.5 × 108
products. The magnitude of kcat varies enormouslywith enzymes such as superoxide dismutase, car-bonic anhydrase and catalase exhibiting high kcat val-ues (>105 s−1) although values between 102 –103 aremore typical of enzymes such as trypsin, pepsin orribonuclease. By comparing the ratio of the turnovernumber, kcat, to the Km a measure of enzymeefficiency is obtained (Table 7.4). Some enzymesachieve high kcat/Km values by maximizing catalyticrates but others enzymes achieve high efficiencyby binding substrate at low concentrations (Km <
10 µM).Further insight into the significance of kcat/Km val-
ues is obtained by considering the Michaelis–Mentenequation at low substrate concentration. Under condi-tions where [S] � Km most of the enzyme is free anddoes not contain bound substrate, i.e. [E]t ≈ [E]. Theequation becomes
ν ≈ kcat/Km[E][S] (7.49)
The term kcat/Km is a second order rate constantfor the reaction between substrate and free enzymeand is a true estimate of enzyme efficiency since itmeasures binding and catalysis under conditions whensubstrate and enzyme are not limiting. These valuesare particularly useful for comparing the efficiency ofenzymes with different substrates. Proteolytic enzymessuch as chymotrypsin show a wide range of substrate
Table 7.5 The side chain preference of chymotrypsinin the hydrolysis of different amino acid methyl esters
Amino acidanalogue
Side chain kcat/Km
Glycine H 0.13
Norvaline CH2 CH2 CH3360
Norleucine CH2 CH2 CH3CH23000
Phenylalanine CH2 100 000
specificity, but measuring kcat/Km values for differentsubstrates demonstrates a clear preference for cleavageof polypeptide chains after aromatic residues suchas phenylalanine (Table 7.5). Using the amino acidmethyl esters (Figure 7.11) as substrates the secondorder rate constant (kcat/Km) is greater by a factor of∼107 when the side chain is phenylalanine as opposedto glycine.
The kcat/Km ratio for enzymes such as fumarase,catalase, carbonic anhydrase and superoxide dismutaserange from 107 to 109 M−1 s−1 and are close to thediffusion-controlled limit where rates are determined

202 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
NH C CO
R
O
CO
H3C
CH3H
Figure 7.11 A general structure for N-acetyl aminoacid methyl esters
by diffusion through solution and all collisions resultin productive encounters. For an average-sized enzymethis value is ∼108 –109 M−1 s−1 and such reactionsare said to be ‘diffusion controlled’. Many enzymesapproach diffusion-controlled rates and molecular evo-lution has succeeded in generating catalysts of max-imum efficiency. With knowledge about the kineticsand efficiency of enzymes it is relevant to look at thestructural basis for enzyme activity yielding near per-fect biological catalysts.
Catalytic mechanismsWhy do enzymes catalyse reactions more rapidlyand with higher specificity than normal chemicalprocesses? Answering this question is the basis ofunderstanding catalysis although it is interesting tonote that biology employs exact the same atomsand molecules as chemistry. Enzymes have ‘finetuned’ catalysis by binding substrates specifically andwith sufficient affinity in an active site that containsall functional groups. Enzymes are potent catalystswith ‘perfection’ resulting from a wide range ofmechanisms that include acid–base catalysis, covalentcatalysis, metal ion catalysis, electrostatic mechanisms,proximity and orientation effects and preferentialbinding of the transition state complex. To reachcatalytic perfection many enzymes combine one ormore of these mechanisms.
Acid–base catalysis
Acid or base catalysis is a common method used toenhance reactivity. In acid-catalysed reactions partialproton transfer from a donor (acid) lowers the freeenergy of a transition state. One example of thisreaction is the keto-enol tautomerization reactions(Figure 7.12) that occur frequently in biology. The
uncatalysed reaction is slow and involves the transferof a proton from a methyl group to the oxygen via ahigh energy carbanion transition state.
Proton donation to the electronegative oxygen atomdecreases the carbanion character of the transition stateand increases rates of reaction whilst a similar effect isachieved by the proximity of a base and the abstractionof a proton. In biological reactions both processes canoccur as concerted acid–base catalysis. Many reactionsare susceptible to acid or base catalysis and in theside chains of amino acid residues enzymes have awide choice of functional group. The side chains ofArg, Asp, Cys, Glu, His, Lys and Tyr are knownto function as acid–base catalysts with active sitescontaining several of these side chains arranged aroundthe substrate to facilitate catalysis.
Many enzymes exhibit acid–base catalysis althoughRNase A (Figure 7.13) from bovine pancreas is usedhere as an illustrative example. The digestive enzymesecreted by the pancreas into the small intestinedegrades polymeric RNA into component nucleotides.Careful analysis of the pH dependency of catalysisof RNA hydrolysis observed two ionizable groupswith pKs of 5.4 and 6.4 that governed overall ratesof reaction. Chemical modification studies suggestedthese groups were imidazole side chains of histidineresidues. The active site of RNase A contains twohistidines (12 and 119) with the side chains actingin a concerted fashion to catalyse RNA hydrolysis.A key event towards understanding mechanism wasthe observation that 2′ –3′ cyclic nucleotides couldbe isolated from reaction mixtures indicating theirpresence as intermediates.
Hydrolysis is a two-step process; in the firststage His12 acts as a general base abstracting aproton from the 2′–OH group of the ribose ring ofRNA. This event promotes nucleophilic attack by theoxygen on the phosphorus centre. His119 acts as anacid catalyst enhancing bond scission by donating aproton to the leaving group (the remaining portion ofthe polynucleotide chain) (Figure 7.14). The resulting2′ –3′ cyclic nucleotide is the identified intermediate.The cyclic intermediate is hydrolysed in a reactionwhere His119 acts as a base abstracting a proton fromwater whilst His12 acts as an acid. The completion ofstep 2 leaves the enzyme in its original state and readyto repeat the cycle for the next catalytic reaction.

CATALYTIC MECHANISMS 203
H
H
Hd+
Hd+
Hd+H+
H+
H
HH
(top)
(centre)
(bottom)
Figure 7.12 Mechanism of keto-enol tautomerization. Top: uncatalysed reaction; centre: general acid-catalysedreaction; bottom: base-catalysed reaction. HA represents an acid B: represents a base. The transition state is enclosedin brackets to indicate ‘instability’
Figure 7.13 The active site of RNase A within theprotein structure. The enzyme is based predominantlyaround two collections of β sheet and four disulfidebridges (yellow). The active site histidines side areshown in red (PDB: 1FS3)
Covalent catalysis
Transient formation of covalent bonds between enzymeand substrate accelerates catalysis. One method ofachieving covalent catalysis is to utilize nucleophilicgroups on an enzyme and their reaction with centreson the substrate. In enzymes the common nucleophilesare the oxygen of hydroxyl groups, the sulfur atomof sulfhydryl groups, unprotonated amines as wellas unprotonated imidazole groups (Figure 7.15). Thecommon link is that all groups have at least oneset of unpaired electrons available to participate incatalysis.
Electrophiles include groups with unfilled shellsfrequently bonded to electronegative atoms such asoxygen. In substrates the most common electrophiles(Figure 7.16) are the carbon atoms of carbonyl groupsalthough the carbon in cationic imine groups (Schiffbase) are also relevant to catalysis.

204 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
PO− O
Base
O
OCH2
H
HHH
O
ONN+
H
NH
NH
OH
His12
His119
OPO−
O
Base
OO
OCH2
H
HHH
H
O
OPO−
O
Base
OO
OCH2
H
HHH
H
O
NN
NH
N
H
His12
His119
OPO−
O
Base
OO
OCH2
H
HHH
H
OH
H2O
P
O
O
Base
O
OCH2
H
HHH
O
O
H
O−H
NN
NH
N
His12
His119
Figure 7.14 The acid–base catalysis by His12 and His119 in bovine pancreatic RNaseA occurs in two discrete stepsinvolving the alternate action of His12 first as a base and second as an acid. His119 acts in the opposite order
Metal ion catalysis
Metalloenzymes use ions such as Fe2+/Fe3+, Cu+/Cu2+,Zn2+, Mn2+ and Co2+ for catalysis. Frequently, themetal ions have a redox role but additional importantcatalytic functions include directing substrate binding
and orientation via electrostatic interactions. Carbonicanhydrase, a zinc metalloenzyme found widely incells (Figure 7.17), catalyses the reversible hydration ofcarbon dioxide
CO2 + H2O −−−⇀↽−−− HCO3− + H+

CATALYTIC MECHANISMS 205
CH2N
N
CH2HS
CH2HO
CH2CH2
CH2CH2
H2N
Figure 7.15 Important nucleophiles found in pro-teins include the imidazole, thiol, amino andhydroxyl groups
R′C O
R
R′C NH
R
carbonyl carbon atom carbon of cationic imine
+
Figure 7.16 Biologically important electrophilicgroups
In red blood cells this reaction is of major importanceto gaseous exchange involving transfer of CO2 fromhaemoglobin. At the heart of carbonic anhydrase is adivalent Zn cation essential for catalytic activity lyingat the bottom of a cleft, ∼1.5 nm deep, coordinated ina tetrahedral environment to three invariant histidineresidues and a bound water molecule. Enzyme kineticsshow pH-dependent catalysis increasing at high pHand modulated by a group with a pKa of ∼7.0.Water molecules bound close to a metal ion tend tobe stronger proton donors than bulk water with theresult that in carbonic anhydrase the Zn not onlyorients the water at the active site but assists in basecatalysis by removing a proton. This generates a boundhydroxide ion, a potent nucleophile, even at pH 7.0.This model is supported by the observation that anionaddition inhibits enzyme activity by competing withOH− anions at the Zn site.
At least 10 isozymes of carbonic anhydrase areidentified in human cells with carbonic anhydrase II ofparticular pharmaceutical interest because its activity islinked to glaucoma via increased intra-ocular pressure.Uncontrolled increases in pressure in the eye damagesthe optic nerve leading to a loss of peripheral vision
and ‘blind spots’ in the field of sight. Drugs (enzymeinhibitors) combat glaucoma by binding to the activesite. Acetazolamide, a member of the sulfonamidegroup of antibacterial agents, is the prototypal inhibitorand displaces the hydroxide ion at the Zn active siteby ligation through the ionized nitrogen of the primarysulfonamide group (Figure 7.18).
Electrostatic catalysis
Substrate binding to active sites is often mediatedvia charged groups located in pockets lined withhydrophobic side chains. Any charged side chainlocated in this region will exhibit a pKa shiftedaway from its normal ‘aqueous’ value and exhibitsin theory at least, a massive potential for electrostaticbinding and catalysis. Direct involvement of chargedresidues in catalysis has proved harder to demonstrate,in contrast to many studies showing that substratebinding is enhanced by charge distribution. The aimof electrostatic catalysis is to use the distribution ofcharged groups at the active site of enzymes to enhancethe stability of the transition state. This aspect ofelectrostatic enhancement of catalysis has been termedthe ‘circe’ effect by W.P. Jencks and encompasses theuse of attractive forces to ‘lure’ the substrate into theactive site destabilizing the reactive group undergoingchemical transformation.
Orotidine 5′-monophosphate decarboxylase(ODCase) catalyses the conversion of orotidine 5′-monophosphate to uridine 5′-monophosphate, the laststep in the biosynthesis of pyrimidine nucleotides(Figure 7.19). ODCase catalyses exchange of CO2 for aproton at the C6 position of uridine 5′-monophosphatein a reaction that is exceptionally slow in aque-ous solution but is accelerated 17 orders of magni-tude by enzyme. The mechanism underlying enhance-ment has fascinated chemists and biochemists alikeand in the active site of the β barrel are a seriesof charged side chains (Lys42-Asp70-Lys72-Asp75)found close together. Although definitive demonstra-tion of the role of these residues in the activity ofODCase requires further study the frequent presenceof charge groups in non-polar active sites points to theinvolvement of electrostatics in catalysis.

206 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
Zn2+
ImIm
Im
O
HC
O
O
C
O
O
O
H
Zn2+
ImIm
Im
C
O
O
OH
OH2
Zn2+
ImIm
Im
O
H
H+
+ +
+
+
His 64
His 94His 119
Thr 199
HCO3
Gln 92
Zn2+
Figure 7.17 The arrangement of imidazole ligands (Im) around the central Zn cation in carbonic anhydrase favoursabstraction of a proton from water and the nucleophilic attack by the hydroxide ion on carbon dioxide bound nearby.The imidazole ligands arise from His 94, His96 (hidden by Zn), and His119. A zinc bound hydroxide ion is hydrogenbonded to the hydroxyl side chain of Thr199, which in turn hydrogen bonds with Glu106. Catalytic turnover requiresthe transfer of the product proton to bulk solvent via shuttle groups such as His64. Shown in the diagram of theactive site is the bicarbonate ion bound to the zinc
Catalysis through proximityand orientation effects
Biological catalysis is typified by high specificity cou-pled with remarkable speed. The gains in efficiencyfor enzyme-catalysed reactions arise from specific envi-ronments found at active sites that promote chemicalreactions to levels of efficiency the organic chemist canonly dream about. Two parameters that are importantin modulating catalysis are proximity and orientation.
These superficially nebulous terms include the effectsof binding substrates close to specific groups therebyfacilitating chemical reactions coupled with the bindingof substrate in specific, restrictive, orientations.
The potential effects of proximity on rates ofreaction have been demonstrated through experi-ments involving the non-enzymatic hydrolysis of p-bromophenylacetate. The reaction is first performedas a simple bimolecular reaction and compared witha series of intramolecular reactions in which the

CATALYTIC MECHANISMS 207
SS
Y
S
N
O O
X
R
NH
OO
Zn2+
His94His96
His119
Phe 131
Gln 92
Pro 202Leu 198
Thr 199
Glu 106
Figure 7.18 General scheme of sulfonamide inhibitorbinding to carbonic anhydrase. The ‘primary sulfon-amide’ coordinates to the active site zinc ion andhydrogen bonds with Thr199
HN
NO
O
R
COO
HN
N
O
O
R
ODCase
Figure 7.19 The reaction catalysed by orotidine 5′-monophosphate decarboxylase converts orotidine 5′-monophosphate into uridine monophosphate in thefinal step in pyrimidine biosynthesis. R is a ribose5′-phosphate group in both reactant and product
reacting groups are connected by a bridge that exertsprogressively more conformational restriction on themolecule. In the simple bimolecular reaction p-bromophenylacetate is hydrolysed to yield bromophe-nolate anion with the formation of acetic anhydride(Figure 7.20). The rate of hydrolysis of bromophe-nol esters increases dramatically as the reactants areheld in closer proximity and in restricted conforma-tions. The relative rate constants increase from 1 (onan arbitrary scale) to 103, to 105 and finally in themost restricted form to a value approaching 108 times
greater than that exhibited by the bimolecular reac-tion. Although the unimolecular reactions describedhere do not involve true catalysis they emphasize thesignificant rate enhancement that results from con-trolling proximity and orientation. The intramolecularreactions model the binding and positioning of twosubstrates within an enzyme active site and althoughsizeable rate enhancements occur they do not accountfor all of the observed increase in rates seen inenzyme versus uncatalysed reactions (for example oro-tidine 5′-monophosphate decarboxylase enhances rates∼1017-fold). Enzymes have yet further mechanisms toenhance catalysis.
Preferential binding of the transition statecauses large rate enhancements
Acid–base catalysis and covalent catalytic mecha-nisms can perhaps enhance rates by factors of 10–102
whilst proximity and orientation can achieve enhance-ments of up to 108, although frequently it is muchless. To achieve the magnitude of catalytic enhance-ment seen for some enzymes additional mechanismsare required to achieve the ratios of 1014 –1017
seen for urease, alkaline phosphatase and ODCase.One of the most important catalytic mechanismsexhibited by enzymes is the ability to bind transi-tion states with greater affinity than either productsor reactants.
For many organic reactions the concept of strain iswell known and it would not be surprising if enzymesadopted similar mechanisms. Enzymes are envisagedas ‘straining’ or ‘distorting’ their substrates towards thetransition state geometry by the formation of bindingsites that do not allow productive binding of ‘native’substrates. Thus, the rate of a catalysed reaction com-pared with an uncatalysed one is related to the magni-tude of transition state binding. Catalysis results frompreferential transition state binding or the stabilizationof the transition state relative to the substrate. In thismanner a rate enhancement of 106 would require a106 enhancement in transition state binding relative tothat of the substrate. This corresponds at room tem-perature (298 K) to an increased binding affinity of∼34 kJ mol−1. In terms of bond energies this is the

208 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
Br
O
CH3
O O
CH3
O+
OO
CH3
O
CH3
Br
O
+
OO
CH2
Br
O
CH2
O
CH2
O
CH2CH2
O
CH2
OBr
O
+
OO
CH2
Br
O
CH2
O
O
CH2
O O
CH2
Br
O
+
Br
O
O
OO
O
O
O
O
O
BrO +
Figure 7.20 Bimolecular and unimolecular reactions describing the hydrolysis of bromophenol esters. The reactionsillustrate how proximity effects enhance catalysis. A progressive increase in rate constant occurs from a relativevalue of 1 in the top reaction to 103,105 and finally 108
equivalent of a handful of hydrogen bonds. Very largerate enhancements will result from the formation ofnew bonds between transition state and enzyme and thismechanism underpins rate enhancement by enzymes.Additional bonds can involve non-reacting regions
of the substrate and many proteolytic enzymes showsubstrate specificity that depends on residues border-ing the scissile bond at the P4,P3 subsites, for example.The concept of transition state binding also explainswhy some enzymes bind substrates with acceptable

LYSOZYME 209
E + DHAP E-DHAP E-intermediate E-G3P E + G3P
Figure 7.21 Conversion of dihydroxyacetone phosphate (DHAP) into glyceraldehyde-3-phosphate (G3P) by triosephosphate isomerase
affinity but fail to show significant catalytic enhance-ment. These ‘poor’ substrates do not form transitionstates and hence do not exhibit high turnover rates. Asa corollary it is not accurate to say that enzymes bindsubstrates with the highest affinity – this is reserved fortransition state complexes.
The concept of transition state binding receivesfurther support from the observation that transi-tion state analogues, stable molecules that resemblethe transition state, are potent catalytic inhibitors.Many transition state analogues are known andused as enzyme inhibitors. 2-Phosphoglycolate is aninhibitor of triose phosphate isomerase. The reac-tion (Figure 7.21) involves general acid–base cataly-sis and the formation of enediol/enediolate intermedi-ates (Figure 7.22). 2-Phosphoglycolate resembles theintermediate and was observed to bind to the enzymewith greater affinity than either dihydroxyacetone phos-phate or glyceraldehyde-3-phosphate. Increased bind-ing arises from the partially charged oxygen atom
found on 2-phosphoglycolate and the transition statebut not on either substrate.
Enzyme structureOne of the prime reasons for wanting to determinethe structure of enzymes is to verify different catalyticmechanisms. In many instances structural descriptionof enzymes refines or enhances particular catalyticschemes, whilst in a few cases it has dictated a freshlook into enzyme mechanism.
LysozymeNo discussion of enzyme structure would be completewithout a description of lysozyme, a hydrolytic enzymeor hydrolase. It destroys bacterial cell walls byhydrolysing the β(1→4) glycosidic bonds between N -acetylglucosamine (NAG) and N -acetylmuramic acid(NAM) that form the bulk of peptidoglycan cell walls(Figure 7.23). It will also hydrolyse poly-NAG chains
H
CH2
CO
CHOH
O PO3
H
CH2
CO
CHOH
O PO3
H
CH2
COH
CO
O PO3
CH2
CO
O PO3
O
DHAP
2-phosphoglycolate
2− 2− 2−
2−
d−
d−
d−
d+
G3Penediol intermediate
Figure 7.22 The first enediol intermediate in the conversion of dihydroxyacetone phosphate (DHAP) intoglyceraldehyde-3-phosphate (G3P) and the structure of 2-phosphoglycolate, a transition state analogue and aninhibitor of triose phosphate isomerase

210 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
H O
OH
OO
O
H
H NH
NAG NAGNAM NAM
CH3CHCOO−
C
O
CH3
OH
CH2OH
H
H
H
5
11
6
3 2
4
OH
H NH C
O
CH3
OH
CH2OH
H
OH
H NH C
O
CH3
CH2OHLysozymecleavage
H
O
CH3CHCOO−
OH
H NH C
O
CH3
CH2OH
HO
H
H
O
H
H
Figure 7.23 The lysozyme cleavage site after the β(1→4) linkage in alternating NAG–NAM units of the polysaccharidecomponents of cell walls. Reproduced from Voet et al. (1998) by permission of John Wiley & Sons, Ltd. Chichester
Figure 7.24 The three-dimensional structure of lysozyme (PDB: 1HEW). The distribution of secondary structure isshown together with the binding of a poly-NAG trimer. A space filling model from the same view emphasizes thecleft into which the polysaccharide, shown in magenta, fits precisely. The cleft can accommodate six sugar units
such as those in chitin a key extracellular componentof fungal cell walls and the exoskeletons of insects andcrustaceans.
The structure of lysozyme determined by DavidPhillips in 1965 represented the first three-dimensionalstructure for an enzyme (Figure 7.24). The proteinwas elliptical in shape (3.0 × 3.0 × 4.5 nm), with themost striking feature being a prominent cleft thattraversed one face of the molecule. Although a pictureof substrate binding sites had been established fromkinetic and chemical modification studies this featureprovided a potent image of ‘active sites’. Lysozymecontains five helical segments together with a triplestranded anti-parallel β sheet that forms one wall of thebinding site for NAG–NAM polymers and a smallerβ strand region. Models of lysozyme complexed withsaccharides reveal a cleft accommodating six units at
individual sites arranged linearly along the bindingsurface. The sites are designated A–F; NAM binds tosites B, D and F because there is not enough space forthe bulkier side chains at positions A, C and E. Theremaining sites are occupied by NAG.
Sites B and F accommodate NAM without distortionbut at site D the sugar molecule will only fitwith distortion from its normal chair conformationinto a half-chair arrangement (Figure 7.25). Moreover,trimers of NAG bound at sites A–C are not hydrolysedat significant rates, and by performing lysozyme-mediated catalysis in the presence of H2O18 it wasdemonstrated that hydrolysis occurred between the C1
and O atoms of the glycosidic bond located at subsitesD and E (Figure 7.26). At this site two side chains fromGlu35 and Asp52 are located close to the C1 positionof the distorted sugar. These two side chains are the
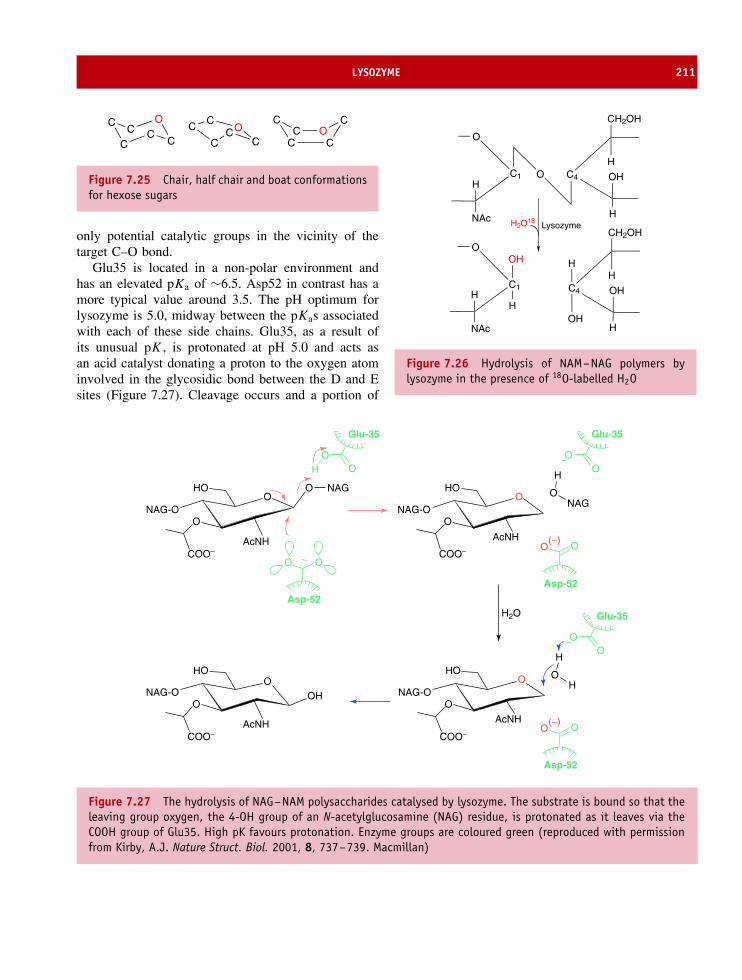
LYSOZYME 211
CC
O
CC
C C C
CCC OC
CC
CO
C
Figure 7.25 Chair, half chair and boat conformationsfor hexose sugars
only potential catalytic groups in the vicinity of thetarget C–O bond.
Glu35 is located in a non-polar environment andhas an elevated pKa of ∼6.5. Asp52 in contrast has amore typical value around 3.5. The pH optimum forlysozyme is 5.0, midway between the pKas associatedwith each of these side chains. Glu35, as a result ofits unusual pK , is protonated at pH 5.0 and acts asan acid catalyst donating a proton to the oxygen atominvolved in the glycosidic bond between the D and Esites (Figure 7.27). Cleavage occurs and a portion of
C4
H
H
OHC1
O
OH
NAc
CH2OH
C1
O
H
NAc
H
OH
C4
H
H
OH
OH
CH2OH
H
LysozymeH2O18
Figure 7.26 Hydrolysis of NAM–NAG polymers bylysozyme in the presence of 18O-labelled H2O
OO NAGHO
NAG-OO
COO−AcNH
OOH
HO
NAG-OO
COO−AcNH
OHO
NAG-OO
COO−
AcNH
O
O(−)
HO
NAG-OO
COO−
AcNH
O
O O
OH
Glu-35
O
O
O
Glu-35
Asp-52
Asp-52
NAGO
H
OO
Glu-35
HO
H
O(−)
O
Asp-52
H2O
Figure 7.27 The hydrolysis of NAG–NAM polysaccharides catalysed by lysozyme. The substrate is bound so that theleaving group oxygen, the 4-OH group of an N-acetylglucosamine (NAG) residue, is protonated as it leaves via theCOOH group of Glu35. High pK favours protonation. Enzyme groups are coloured green (reproduced with permissionfrom Kirby, A.J. Nature Struct. Biol. 2001, 8, 737–739. Macmillan)

212 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
the substrate bound at sites E and F can diffuse out ofthe cleft to be replaced by water. The resulting cleavageleads to the formation of a carbocation which interactswith the carboxylate group of Asp 52 (nucleophile) toform a glycosyl–enzyme intermediate in a concertedSN2-type reaction. The carboxylate group is thendisplaced from the glycosyl–enzyme intermediate by awater molecule that restores the original configurationat the C1 centre.
The serine proteasesThe serine proteases include the common proteolyticenzymes trypsin and chymotrypsin, as well as acetyl-cholinesterase, an enzyme central to the transmission ofnerve impulses across synapses, thrombin (an enzymewith a pivotal role in blood coagulation) and elastase(a protease important in the degradation of connectivefibres). As proteases these enzymes hydrolyse peptidebonds in different target substrates but are linked by thepresence of an invariant serine residue at the enzymeactive site that plays a pivotal role in catalysis.
For trypsin, chymotrypsin, elastase and thrombinthe enzymes cut polypeptides on the C-terminal sideof specific residues. These residues are not random
Residues restrict entryto pocket to small side chains
Interaction with Aspat base of pocket 'fixes'substrate in active site.
Val Thr
Small, non-polar, residues line pocket creating spacefor large substrate.
_Asp
Elastase
Chymotrypsin
Trypsin
Figure 7.28 Representation of substrate specificitypockets for elastase, chymotrypsin and trypsin
but show a substrate specificity that differs widelyfrom one enzyme to another. Trypsin cuts preferentiallyafter positively charged side chains such as arginine orlysine whilst chymotrypsin shows a different specificitycutting after bulking hydrophobic residues with a clearpreference for phenylalanine, tyrosine, tryptophan andleucine. In contrast, elastase cuts only after small andneutral side chains whilst thrombin shows a restrictedspecificity for arginine side chains (Figure 7.28). Thesubstrate specificity arises from the properties of theactive site pocket and in particular the identity ofresidues at subsites making up the substrate bindingregion. The subsites that bind substrate residuespreceding the scissile bond are termed S1, S2, S3,whilst those after this bond are given the names S1′,S2′, S3′, etc. A similar terminology is applied to thepeptide substrate where P3, P2 and P1 sites precede thescissile bond and P1′, P2′, and P3′ continue after it.
Serine proteases show similar structures with theactive site containing three invariant residues arrangedin close proximity. These residues are His57, Asp102and Ser195 and together they form a catalytic triadlinked by a network of hydrogen bonds. In addition tothese three invariant residues the serine proteases pos-sess a pocket, located close to the active site serine thatstrongly influences substrate specificity. In trypsin thepocket is relatively long and narrow with a strategi-cally located carboxylate side chain found at the bot-tom of the pocket. In view of the preferred specificityof trypsin for cleavage after lysine or arginine sidechains this pocket appears to have been tailored to ‘cap-ture’ the substrate very effectively. In chymotrypsin thepocket is larger, wider, lacks charged residues but con-tains many hydrophobic groups. This is consistent withspecificity towards bulky aromatic groups. Elastase, incontrast, has a shallower pocket formed by valine andthreonine side chains. Its specificity for cleavage on theC-terminal side of small, uncharged, side chains againreflects organization of the pocket.
Despite tailoring substrate specificity through theproperties of the pocket all serine proteases showsimilar catalytic mechanisms based around a highlyreactive serine residue identified from labelling andchemical inactivation studies. Agents based aroundorganophosphorus compounds such as di-isopropyl-phosphofluoridate (DIPF) were identified via their actionas potent neurotoxins (Figure 7.29). Neurotoxicity arises

THE SERINE PROTEASES 213
P OF
O
OCH(CH3)2
CH(CH3)2
+
HN
CH CH2
OH
O
C
Ser195
P O
O
OCH(CH3)2
CH(CH3)2
HN
CH CH2 O
O
C
HF+
Figure 7.29 Inactivation of serine proteases by di-isopropylphosphofluoridate with the formation of DIP-enzyme
O
CH3 S N
O
CH C
CH2
CH2Cl
O
Figure 7.30 Tosyl-L-phenylalanine chloromethylke-tone (TPCK)
from modification of an active site serine residue inacetylcholinesterase and indicated the similarity of thisenzyme to other serine proteases such as trypsin andchymotrypsin where DIPF covalently reacts with Ser195to form an adduct lacking catalytic activity. Labellingstudies also identified the importance of a second residueof the catalytic triad, His57. Chymotrypsin binds tosyl-L-phenylalanine chloromethylketone (TPCK, Figure 7.30)as a result of the presence of a phenyl group, a preferredsubstrate, within the analogue. The reactive group formsa stable covalent bond with the imidazole side chain(Figure 7.31).
The structure of chymotrypsin revealed the closeproximity of His57 and Ser195 together with Asp102in the active site despite their separation in the primary
C
CH2
O
R
Cl
CH2
NN
H
CH2
NN CH2 C
O
R
H+ Cl−
His 57Chymotrypsin
Figure 7.31 The reaction of TPCK with His57 ofchymotrypsin. Only the active part of TPCK is shown(red)- alkylation causes enzyme inactivation
Ser195
His57
Asp102
Figure 7.32 The spatial arrangement of residues inthe catalytic triad (His57, Asp102 and Ser195) in theactive site of chymotrypsin (PDB: 2CGA)
sequence (Figure 7.32). The first step of catalysisis substrate binding to the active site region andspecificity is largely dictated by residues at the bottomof a well-defined cleft. Interactions at subsites eitherside of the scissile bond are limited. Specific bindingleads to close proximity between Ser195 and thecarbonyl group of the bond to be cleaved. Undernormal conditions the pKa of the –OH group of serineis very high (>10) but in chymotrypsin the proximityof His57 leads to proton transfer and formation of

214 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
a charged imidazole group. Protonation of His57 isfurther stabilized by the negative charge associatedwith Asp102 – the third member of the catalytictriad – that lies in a hydrophobic environment. Theformation of a strong nucleophile in the activatedserine attacks the carbonyl group as the first stepin peptide bond cleavage. Nucleophile generation
is triggered by small conformational changes uponsubstrate binding that cause Asp102 and His57 toform a hydrogen bond. The influence of this hydrogenbond raises the pK of the imidazole side chainfrom ∼7 to ∼11 increasing its basicity. Electronsdriven towards the second nitrogen atom form astronger base that abstracts a proton from the normally
H2CC
O
O−
Asp102 CH2
N
N:
His57
H
CH2O
Ser195
H
CR′N
O
R
H
Enzyme substrate complex
H2CC
O
O−
Asp102 CH2
N
N
His57
H
N
H
H R CR′
O
CH2O
Ser195
Acyl enzyme intermediate
H2CC
O
O−
Asp102 CH2
N
N
His57
H
CR′
O
CH2O
Ser195
O
H
H
H2CC
O
O−
Asp102 CH2
N
N+
His57
H
H
O
H
CR′
O−
CH2O
Ser195
Second tetrahedral intermediate
H2CC
O
O
Asp102 CH2
N
N
His57
H
O
H
CR′
O
CH2O
Ser195
H
+Release of polypeptide with new C terminus
H2CC
O
O−
Asp102 CH2
N
N+
His57
H
H
R′N
O−
R
H
CH2O
Ser195
First tetrahedral intermediate
Figure 7.33 The catalytic cycle of serine proteases such as chymotrypsin

TRIOSE PHOSPHATE ISOMERASE 215
unreactive side chain of Ser195 creating the potentnucleophile.
Nucleophilic attack results in a tetrahedral transitionstate followed by cleavage of the peptide bond. TheN-terminal region of the peptide substrate remainscovalently linked to the enzyme forming an acylenzyme intermediate. The proton originally attachedto the serine residue and located on the histidine isabstracted by the C-terminal peptide to form a newterminal amino group and this region of the peptidedoes not bind strongly to the enzyme. As a hydrolyticenzyme, water is involved in the overall reactionsummarized in Figure 7.33.
In chymotrypsin water displaces C-terminal pep-tide from the active site and hydrolyses the acyl-enzyme intermediate. The water molecule donates aproton to the imidazole side chain and forms a sec-ond tetrahedral transition state with the acyl inter-mediate. This intermediate is hydrolysed as a resultof proton transfer from His57 back to Ser195 withrestoration of the active site to its original state withthe release of the N-terminal peptide. Serine pro-teases follow a ping-pong mechanism (Figure 7.34);the substrate is first bound to the enzyme, the C-terminal peptide is released followed by binding awater molecule and the formation of an acyl–enzymeintermediate. This is completed by release of the N-terminal peptide from hydrolysis of the acyl–enzyme
intermediate. The type of group transfer where oneor more products are released before all substratesare added is also known as a double-displacementreaction.
It is interesting to note that serine proteases utilizethe whole gamut of catalytic mechanisms. Proximityeffects play a role in generation of the bound substrateswhilst transition state stabilization ensures effectivebinding prior to catalysis. This is complemented bythe use of covalent catalysis (the serine nucleophile)together with acid–base catalysis by histidine to yieldefficient enzyme action.
Triose phosphate isomeraseTriose phosphate isomerase has been widely studiedand catalyses the conversion between DHAP and G3P(Figure 7.35). The equilibrium for the reaction lies infavour of DHAP, but the rapid removal of G3P bysuccessive reactions in glycolysis drives the reactiontowards the formation of G3P.
Triose phosphate isomerase merits further discussionas an example of enzyme catalysis because of twointeresting phenomena: the use of the imidazolate formof histidine in catalysis and the evolution of the enzymetowards a near perfect biological catalyst. Influencingboth of these phenomena is the structure of triosephosphate isomerase with crystallographic structures
Figure 7.34 The ping-pong mechanism for serine proteases. Using the normal convention A and B representsubstrates in the order that they are bound by the enzyme whilst P and Q represent products in the order that theyleave the enzyme. The activity of trypsin and other serine proteases are therefore summarized by a scheme
E + A −−−⇀↽−−− E.A −−−⇀↽−−− E.Q + P
EQ + B −−−⇀↽−−− E-Q.B −−−⇀↽−−− E + Q-B

216 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
C O
C
C
H
H OH
HH
O
P O−
O−
O
C OH
C
C
HH
O
P O−
O−
O
H
H O
TIM
Figure 7.35 The interconversion of dihydroxyace-tone phosphate (left) and glyceraldehyde-3-phosphatecatalysed by triose phosphate isomerase
of the enzyme available in a free state, bound withDHAP and plus 2-phosphoglycolate. Kinetic studies onthe pH dependence of the catalytic process revealedtwo inflection points in the profile correspondingto pH values of ∼6.0 and 9.0. When consideredalongside the structure of triose phosphate isomerase(Figure 7.36) these values implicated the side chains ofglutamate and histidine. Affinity labelling studies usingbromohydroxyacetone phosphate modified a glutamateresidue within a hexapeptide (Ala-Tyr-Glu-Pro-Val-Trp) and this residue was identified as Glu165. The
active site lies near the top of the β barrel with Glu165close to His 95 as well as Lys12, Glu97 and Tyr208.Both Glu165 and His 95 are essential for catalyticactivity and the enzyme provides a cage or reactionvessel holding substrate and protecting intermediatesthrough a series of 11 residues immediately afterGlu165 (residues 166–176) that form a loop and actsas a lid for the active site.
Dihydroxyacetone phosphate binds to the activesite with the carbonyl oxygen forming a hydrogenbond with the neutral imidazole side chain of His95.The glutamate side chain is charged and facilitatesabstraction of a proton from the C1 carbon ofdihydroxyacetone phosphate. In this state the His95forms a strong hydrogen bond with the carbonyloxygen (C2) of the enediolate intermediate that leadsto protonation of this atom. Protonation of the oxygenatom forms an imidazolate side chain on His95 thathas a negative charge and is a strong base. Protonabstraction from the hydroxyl group attached to the C1carbon results in the formation of a second enediolateintermediate with this unstable intermediate convertedto the final product by donation of a proton fromGlu165 to the C2 carbon. These reactions are shownin Figure 7.37.
Kinetic and thermodynamic characterization oftriose phosphate isomerase show measurable binding
Figure 7.36 Structure of triose phosphate isomerase monomer together with the active site residues surroundingsubstrate. Strands of the β barrel are shown in purple, helices in blue. The two critical residues of His95 and Glu165are shown as solid spheres. The enzyme is shown from two orientations

TRIOSE PHOSPHATE ISOMERASE 217
O−P
O O−
O
C O
C
CH2
H
H OHO−O
CH2Glu165
CH2
NN
His95
H
O
O
O−P
O O−
C O−
C
CH2
HOHOH
CH2Glu165
CH2
NN
His95
H
Enediolate intermediate
O
O
O−P
O O−
C O H
C
CH2
HO HOH
CH2Glu165
CH2
N
His 95
N
Enediol intermediate
----
Figure 7.37 The mechanism for conversion of DHAPto G3P in reactions catalysed by triose phosphateisomerase. The enzyme employs general acid–basecatalysis with the uncharged histidine side chain actingas a proton donor whilst the charged imidazolate stateacts as an acceptor. Alternative mechanisms have beenproposed for this reaction including the removal of aproton from the OH group attached to the C1 anddonation to the C2 oxygen by Glu165
O−P
O O−
C O
C
CH2
HO−
O
CH2Glu165
H
CH2
N
His 95
NH
Enediolate intermediate
O
O
O−P
O O−
O
C O
C
CH2
HO
H
O−O
CH2Glu165
CH2
N
His 95
NH
Glyceraldehyde-3-phosphate
H
H
of DHAP, conversion of DHAP to enediol interme-diate, formation of G3P and the release of product(Figure 7.38). The equilibrium lies in favour of DHAPby a factor of 100 with the enzyme showing a highspecificity constant of 4.8 × 108 M−1 s−1. The slowestreaction will have the greatest activation energy (�G‡)
but in the four reactions encompassing conversion ofDHAP to G3P it was observed that each reaction hasapproximately the same activation energy. One reactioncould not be clearly defined as the rate-determining
E + DHAP E-DHAP
E-DHAP E-enediol
E-enediol E-G3P
E-G3P E + G3P
Figure 7.38 The reactions of triose phosphateisomerase in a minimal kinetic scheme

218 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
step. This is unlikely to have arisen by accident andreflects catalytic ‘fine tuning’ to allow maximal effi-ciency. A kcat of 4 × 103 s−1 is not very high but thesuperb enzyme efficiency arises from a combinationof fast turnover, effective tight binding of substrate(Km ∼ 10 µM) and transition state stabilization.
Tyrosyl tRNA synthetaseThe attachment of amino acid residues to tRNAmolecules is a major part of the overall reaction ofprotein synthesis. The reaction involves the additionof activated amino acids by a group of enzymesknown as amino acyl tRNA synthetases, and issummarized as
Amino acid + tRNA + ATP ↼−−−−−−⇁ Aminoacyl-tRNA
+ AMP + PPi
Any error at this stage is disastrous to protein synthesisand would lead to incorrect amino acid incorporationinto a growing polypeptide chain. The error rate formost tRNA synthetases is extremely low and partof this efficiency comes from high discriminationduring binding. Tyrosyl tRNA always binds tyrosinebut never the structurally similar phenylalanine andthe binding constant for tyrosine of 2 × 10−6 M is atleast five orders of magnitude smaller than that ofphenylalanine.
Amino acids attach to tRNA molecules at the accep-tor arm containing a conserved base sequence of-CCA-3′. Tyrosyl tRNA synthetase represents one ofthe simplest catalytic mechanisms known in biologywith all activity deriving from binding interactionsat the active site. The three-dimensional structure oftyrosyl tRNA synthetase from the Bacillus stearother-mophilus is a dimer of subunits each containing 418residues. (A dimer is an exception for most Class ItRNA synthetases which are generally monomeric, seeChapter 8.) The structure of the protein (Figure 7.39)has been determined with tyrosine and tyrosine-adenylate and is predominantly α helical with a centralsix-stranded β sheet forming the core of the enzyme.The enzyme contains the Rossmann fold with tyro-syl adenylate bound at the bottom of the sheet withthe phosphate groups reaching the surface of the pro-tein. Although functionally a dimer the structure shown
Figure 7.39 The structure of tyrosyl tRNA from B.stearothermophilus in a complex with tyrosyl adenylate(PDB: 3TSI). Helices are shown in blue and the sixstranded sheet in green. The bound tyrosyl adenylateis shown in red, with the adenylate portion situated onthe left and the tyrosine pointing away from the viewer
is that of the monomer and represents the first 320residues of the protein with remaining 99 residuesat the C terminal found disordered. The monomercontains 220 residues in an amino terminal domainwith α/β structure together with a helix rich domainextending from residues 248–318 (shown on left ofFigure 7.39).
The reaction catalysed by tyrosyl tRNA synthetaseis divided into the formation of tyrosine adeny-late (Tyr-AMP) and pyrophosphate from the initialreactants of tyrosine and ATP and then in a sec-ond reaction the Tyr-AMP is transferred to a spe-cific tRNA. The structures of the enzyme boundwith tyrosine, ATP and the tyrosyl adenylate inter-mediate are known and interactions between polypep-tide chain and substrates or activated intermediatehave been identified. These structures allowed indi-vidual residues to be identified as important forthe formation of tyrosyl-adenylate – an intermedi-ate stabilized by the enzyme and seen in crys-tal complexes.
Binding of substrate to tyrosyl tRNA synthetaseis a random kinetic process with either tyrosine orATP bound first. However, an examination of bindingconstants for each substrate suggests that ATP bindingis weaker leading to an ordered process in whichtyrosine binds first followed by ATP (Figure 7.40).

TYROSYL TRNA SYNTHETASE 219
E E-Tyrosine-ATP
E-Tyrosine
E-ATP
E-Tyr-AMP-PPi E-Tyr-AMP PPi
ATPTyr
ATP Tyr
+
Figure 7.40 Kinetic schemes for the formation of tyrosyl adenylate from tyrosine and ATP. This reaction representsthe first part of the tyrosyl tRNA synthetase catalysed process. The products of the first reaction are tyrosyl adenylate(enzyme bound) and pyrophosphate (dissociates from ternary complex)
From the crystal structures in the presence oftyrosine and ATP it is apparent that residues Asp78,Tyr169 and Glu173 form a binding site for the α-aminogroup of the tyrosine substrate. Not unexpectedly, theside chain of tyrosine forms hydrogen bonds via thephenolate group with residues Tyr34 and Asp176. Thecrystal structure of the complex with tyrosyl adenylateindicated that Cys35, Thr51 and His48 interact with theribose ring (Figure 7.41).
3435 40
4548
51
78169
173
176
Figure 7.41 Tyrosyl-adenylate binding to tyrosyltRNA synthetase (PDB: 3TSI). Shown in orange is Tyr-AMP together with the side chains of Tyr34, Cys35,Thr40, His45, His48, Thr51, Asp78, Tyr169, Glu173and Asp176
The role of residues in substrate binding was definedusing mutagenesis to perturb the overall efficiency oftyrosyl tRNA synthetase. Cys35 was the first mutatedresidue and although in contact with the ribose ring ofATP and having a role in substrate binding it is remotefrom the critical active site chemistry based around thephosphate groups. Mutation of Cys35 to Gly estimatedthe effect of deleting the side chain hydrogen bondinginteraction and revealed an enzyme operating with alower efficiency by ∼6 kJ mol−1. Modifying Tyr34 toPhe34 decreased the affinity of the enzyme for tyrosine,lowered the stability of the Tyr-AMP enzyme complexand decreased the ability of the enzyme to discriminatebetween tyrosine and phenylalanine. Mutagenesis com-bined with kinetic studies showed that changing Thr40and His45 produced dramatic decreases in rates ofTyr-AMP formation. Changing Thr40 to Ala decreasedTyr-AMP formation by approximately 7000 fold whilstthe mutation His45 to Gly caused a 200-fold decrease.A double mutant, bearing both substitutions, showeda decrease of ∼105. In each case the affinity of theenzyme for substrate was unaltered but the affinity ofthe enzyme for the intermediate (Tyr-AMP..PPi) waslowered suggesting that Thr40 and His45 played keyroles in stabilizing the transition state complex viainteractions with inorganic pyrophosphate groups.
The single most important property of tRNAsynthetases is substrate binding in pockets in restrainedand extended conformation. The geometry lowers theactivation energy barrier for the reaction and allowsformation of the amino acyl adenylate complex. Twocharacteristic motifs with sequences His-Ile-Gly-His(HIGH) and Met-Ser-Lys (MSK) are important to

220 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
formation of the transition state. The close approachof negatively charge carboxyl groups (from the aminoacid substrate) and the α phosphate group of theATP promotes formation of a transition state. Theα phosphate group is stabilized by interaction withHis43 and Lys270 of the HIGH and MSK motifs. Thepyrophosphate acts as a leaving group and the aminoacyl adenylate transition state complex is stabilized byinteractions with specific residues, as described in thepreceding section.
The second half of the reaction, the transfer ofthe amino acid to the correct tRNA, has been exam-ined through crystallization of other class I andclass II tRNA synthetases (Table 7.6) with tRNAbound in a tight complex. The acceptor arm oftRNA and in particular the C74C75A76 base sequencebinds alongside ATP in the active site. The arrange-ment of substrate in the active site of glutaminyltRNA synthetase (Figure 7.42) has been derived byusing glutaminyl-adenylate analogues (Figure 7.43)that co-crystallize with the enzyme defining interac-tions between residues involved in tRNA and aminoacyl binding (Figure 7.44).
Gln tRNA synthetase must avoid selecting eitherglutamate or asparagine – two closely related aminoacids. Discrimination between amino acids is favouredby recognition of both hydrogen atoms of the nitro-gen in the glutamine side chain. Interactions involvethe hydroxyl group of Tyr211 and a water moleculeas obligatory hydrogen-bond acceptors in a network of
Figure 7.42 Gln-tRNA synthetase showing complexof tRNA and ATP (PDB: 1GTG). The tRNA molecule isshown in orange with ATP in yellow. The secondarystructure elements of enzyme are shown in blue(helices), green (strands) and grey (turns)
side chains and water molecules. In molecular recogni-tion discrimination against glutamate and glutamic acidoccurs because they lack one or both hydrogen atomswhilst the side chain of asparagine is too short.
Table 7.6 Principal features of class I and class II aminoacyl tRNA synthetases
Property Class 1 Class II
Conserved substratebinding motifs
HIGHKMSKS
Motif 2 (FRxE)Motif 3 (GxGxGXER)
Dimerization motifs None Motif 1 (little consensus)Active site fold Parallel β sheet of Rossmann fold Antiparallel β sheetATP conformation Extended BentAmino acylation site 2′ OH 3′OHtRNA binding Variable loop faces solvent Variable loop faces protein
Binds to minor groove of acceptor stem Binds to major groove of acceptor stem

ECORI RESTRICTION ENDONUCLEASE 221
POO−
O
NON
N
OO
N
N
OO
NH3+
NH2
O
SOO
NH
NON
N
OO
N
N
OO
NH3+ NH2
O
Glutaminyl aminoacyl phosphate
Transition state analogue
Figure 7.43 Glutaminyl amino acyl-adenylate and itstransition state analogue
EcoRI restriction endonucleaseRestriction enzymes were characterized through the1960s and early 1970s, largely through the efforts ofWerner Arber, Hamilton O. Smith and Daniel Nathans.The discovery of type II restriction endonucleases
represented a key event at the beginning of a newchapter of modern biochemistry. Subsequently, the useof type II restriction enzymes would allow manipula-tion of DNA molecules to create new sequences thatwould form the basis of molecular biology.
In addition to type II restriction endonucleases thereare also type I and type III enzymes. Type I restrictionenzymes have a combined restriction/methylation func-tion within the same protein and cleave DNA randomlyoften at regions remote from recognition sites and ofteninto a large number of fragments. They are not usedin molecular cloning protocols. Type III restrictionenzymes have a combined restriction/methylation func-tion within the same protein but bind to DNA and atspecific recognition sites. Again this class of enzymesis not routinely used in cloning procedures. In type IIenzymes the restriction function is not accompanied bya methylase activity. Type II enzymes do not requireATP for activity although all enzymes appear to requireMg2+ ions.
Currently more that 3000 type II restriction endonu-cleases have been discovered with more than 200different substrate specificities. These enzymes arewidely used in ‘tailoring’ DNA for molecular cloningapplications. The substrate is double stranded DNA
SOO
NH
NON
N
OHOH
N
NH2
OO
NH3NH2
O
ON
H
N
O
O
H
OH H
NO
NH3+
+
O H
H
OH
NHN
NH2
O
OH
OH
A76
OO
Leu261
Tyr211Asp66
Thr230
Arg260
Lys270
Figure 7.44 The interaction between glutaminyl adenylate analogue and the active site pocket of Gln-tRNAsynthetase

222 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
Table 7.7 A selection of restriction endonucleases showing cleavage sequences and sources. The arrows betweenbases indicate the site of cleavage within palindromic sequences
Enzyme Recognition site SourceAvaI C (T/C)GG (A/G)G Anabaena variablis
BamHI G GATCC Bacillus amyloliquefaciens H
EcoRI G AATTC Escherichia coli RY13
HindIII A AGCTT Haemophilus influenzae Rd
NcoI C CATGG Nocardia corallina
PvuI CGAT CG Proteus vulgaris
SmaI CCC GGG Serratia marcescens
XhoI C TCGAG Xanthomonas holcicola
and in the majority of cases restriction endonucleasesrecognize short DNA sequences between 4 and 8base pairs in length although in some instances longersequences are identified. The names of restrictionenzymes derive from the genus, species and strain des-ignations of host bacteria (Table 7.7). So for exampleEcoRI is produced by E. coli strain RY13 whilst NcoIis obtained from Nocardia corallina and HindIII fromHaemophilus influenzae.
Restriction enzymes recognize a symmetrical se-quence of DNA where the top strand is the same asthe bottom strand read backwards (palindromic). WhenEcoRI cuts the motif GAATTC it leaves overhangingchains termed ‘sticky ends’. The uncut regions ofDNA remain base paired together. Sticky ends are anessential part of genetic engineering since they allowsimilarly cut fragments of DNA to be joined together.
EcoRI is one of the most widely used restric-tion enzymes in molecular biology and its struc-ture has been determined by X-ray crystallography(Figure 7.45). The protein is a homodimer with eachsubunit containing a five-stranded β sheet flankedby helices. Subsequent determination of the structure
of other restriction enzymes such as BamHI, BglII,Cfr10I, MunI, and NgoMIV revealed significant foldsimilarity despite relatively low levels of sequenceidentity. This further suggested a common mechanismof catalytic action.1 The EcoRI family share a sub-structure based around five β strands and two α helicescalled the common core motif (CCM). The CCM is theresult of a β meander followed by a binding pocket thatinteracts with recognition site DNA and is formed pre-dominantly by two helices called the outer and innerhelices. The core is readily seen by comparing struc-tures of EcoRI complexed with DNA and devoid of itsrecognition partner.
The above view is, however, slightly misleadingsince whilst it portrays the secondary structure elementsof the CCM it does not accurately reflect the actionof the homodimer of subunits in surrounding DNA.The subunits bind to specific bases (GAATTC) inthe major groove of the DNA helix with the minorgroove exposed to the solvent. The dimers form a
1The other major structural family of restriction endonucleases isbased around the structure of EcoRV.

ECORI RESTRICTION ENDONUCLEASE 223
Figure 7.45 The structure of EcoRI is shown with and without a recognition sequence of DNA. The five-stranded β
sheet making up most of the CCM is shown in the centre of the protein in green
Figure 7.46 Space filling representation of homod-imer of EcoRI bound to DNA. The subunits of theenzyme are shown in blue and green whilst the DNA isshown in orange/yellow. The minor groove is exposedto the solvent
cleft lined with basic residues that associate with sugar-phosphate chains via charge interactions (Figure 7.46).Of more importance are arginine side chains at residues145 and 200, together with a glutamate side chain(Glu144) in each monomer that form hydrogen bondswith base pairs of the recognition motif. This confersspecificity to the binding reaction with DNA andavoids interactions with incorrect, but often closelyrelated, sequences. Sequence specificity is mediatedby 16 hydrogen bonds originating from α helical
recognition regions. Twelve of the hydrogen bondsoccur between protein and purine whilst four existbetween protein and pyrimidine. EcoRI ‘reads’ allsix bases making up the recognition sequence byforming interactions with each of them. Arg200 formstwo hydrogen bonds with guanine while Glu144 andArg145 form four hydrogen bonds to adjacent adenineresidues. In addition to the hydrogen bonds an elaboratenetwork of van der Waals contacts exist betweenbases and recognition helices. It is these interactionsthat discriminate the EcoRI hexanucleotide GAATTCfrom all other hexanucleotide sequences. Changes inbase identity lose hydrogen bonds and effectivelydiscriminate between different sequences. Hydrogenbond formation distorts the DNA helix introducingstrain and a torsional kink in the middle of itsrecognition sequence leading to local DNA unwinding.The unwinding involves movement of DNA by ∼28◦
and groove widening by ∼0.35 nm where ‘strain’ mayenhance catalytic rates.
The presence of divalent cations (Mn2+, Co2+, Zn2+and Cd2+) results in bond scission but in their absenceenzymes shows low activity. The use of electrondense ions pinpoints the metal centre location as closeto Glu111 in co-crystals of protein–DNA–divalention and emphasizes the role of a metal centre incatalysis. Metal ions are coordinated in the vicin-ity of the active site acidic side chains. The obser-vation of mono and binuclear metal centres withinactive sites of restriction endonucleases suggests that

224 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
although the structure of the active site-DNA bind-ing region of many enzymes is similar the catalyticmechanisms employed may differ from one enzyme toanother.
Restriction endonucleases contain a signature se-quence D-(X)n-D/E-Z-K, in which three residues areweakly conserved (D, D/E and K). The two acidicresidues are usually Asp, although Glu occurs in EcoRIand several other restriction endonucleases, includingBamHI. These residues are separated by 9–20 residuesand are followed by a basic lysine side chain pre-ceded by Z, a residue with a hydrophobic side chain.In EcoRI the catalytic residues are Asp91, Glu111 andLys113, whilst Asp59, preceding the conserved clus-ter, also appears to be important to catalysis. Theseresidues make up the active site, whilst in BamHIthe corresponding residues are Asp94, Glu111, andGlu113 with Glu77 preceding the cluster of chargedresidues. BamHI cuts the sequence GGATTC – a hex-anucleotide sequence almost identical to that recog-nized by EcoRI (GAATTC) – yet each enzyme showsabsolute specificity for its own sequence. Both enzymespossess homodimeric structures with a catalytic coreregion of ∼200–250 residues per monomer consist-ing of five strands and two helices. However, themethod of sequence recognition and potential mech-anism of catalysis in two clearly related enzymesappears to differ. The catalytic mechanism for EcoRIinvolves a single metal ion whilst that of BamHIinvolves two cations (Figure 7.47). Similarly, DNA
binding through base contacts in the major grooveuses a set of loops between strands together with β
strands (EcoRI) whilst BamHI lacks these regions anduses the N-terminal end of a helix bundle. Despitea lack of agreement conclusions can be drawn aboutthe requirements for phosphodiester bond cleavagein any restriction enzyme. At least three componentsare required; a general base is needed to activatewater, a Lewis acid is necessary to stabilize nega-tive charge formed in the transition state complex anda general acid is required to protonate the leavinggroup oxygen.
Enzyme inhibition and regulationEnzymes are inhibited and regulated. Studying eachprocess sheds light on the control of catalysis, catalyticpathways, substrate binding and specificity, as well asthe role of specific functional groups in active sitechemistry. Inhibition and regulation are inter-relatedwith cells possessing proteins that inhibit enzymes aspart of a normal cellular function.
Reversible inhibition involves non-covalent bindingof inhibitor to enzyme. This mode of inhibition isshown by serpins, proteins that act as natural trypsininhibitors preventing protease activity. The complexformed between bovine pancreatic trypsin inhibitor(BPTI) and trypsin has extraordinary affinity with alarge association constant (Ka) of ∼1013 M−1. Tightassociation between proteins causes effective enzyme
O
O
O
OO
O
O
OO
H
H
O POO
O
C5'
C3'
O HH
M1 M2
E113
E111
D94
E77
2+ 2+
Reactant binding
O
O
O
OO
O
O
OO
H
O HH
M1 M2
O P
O
O
C5'O
C3'
E113
E111
D94
E77
2+2+
Transition state complex
O
O
O
OO
O
O
OO
HO
P
O
O
C5'
M1 M2
OC3'
H
E113
E111
D94
E77
2+ 2+
Post reaction state
Figure 7.47 A general mechanism of catalysis for type II restriction endonucleases based on BamHI. The reactionoccurs through an SN2 mechanism, with an in-line displacement of the 3′-OH group and an inversion of configurationof the 5′-phosphate group

ENZYME INHIBITION AND REGULATION 225
Figure 7.48 Binary complex formed between trypsinand BPTI showing insertion of Lys15 (red) into thespecificity pocket. Trypsin is shown in blue, BPTI inyellow (PDB: 2PTC)
inhibition and crystallography of the protein complexreveals a tightly packed interface between BPTI andtrypsin formed by numerous intermolecular hydrogenbonds (Figure 7.48). On the surface of BPTI Lys15occupies the specificity pocket of trypsin with aLys–Ala peptide bond representing the scissile region.However, bond cleavage does not occur despite theproximity of Ser195 and the formation of a tetrahedralintermediate. Absence of bond cleavage results fromtight binding between BPTI and enzyme that preventsdiffusion of the leaving group (peptide) away fromthe active site and restricts access of water tothis region.
By measuring reaction kinetics at different con-ditions activity profiles (kcat/Km) are determined forindividual enzymes. Extremes of pH lead to a lossof activity as a result of enzyme denaturation andsolutions of pH 2.0 do not in general support foldedstructure and lead to a loss of activity that is restoredwhen the pH returns to normal values. Reversibility isobserved because covalent modification of the enzymedoes not occur.
Investigating kinetic profiles for enzyme-catalysedreactions in the presence of an inhibitor may identifymechanisms of inhibition and provides important clues
in the absence of structural data of the active site geom-etry and composition. Armed with structural knowl-edge the pharmaceutical industry has produced newinhibitors with improved binding or inhibitory actionthat play crucial roles in the battle against disease. Thestarting point for enzyme inhibitor studies is often iden-tification of a natural inhibitor. When inhibitor datais available with detailed structural knowledge of anenzyme’s active site it becomes possible to enter thearea of rational drug design. This combines structuraland kinetic data to design new improved compoundsand is often performed on computers. With a fam-ily of ‘lead in’ compounds chemical synthesis of arestricted number of favourable targets, followed byassays of binding and inhibition, can save years ofexperimental trials and allows drugs to enter clini-cal trials and reach the marketplace. Such approachescombining classical enzymology with bioinformat-ics and molecular modelling allows development ofnew pharmaceutical arsenals and a new approach tomolecular medicine. Using these techniques enzymeinhibitors have been introduced to counteract emphy-sema, HIV infection, alcohol abuse, inflammation andarthritis.
The most important type of reversible inhibi-tion is that caused by molecules that competefor the active sites of enzymes (Figure 7.49). Thecompetitor is almost always structurally related tothe natural substrate and can combine with theenzyme to form an enzyme–inhibitor complex anal-ogous to that formed between the enzyme and thesubstrate. Occasionally the ‘inhibitor’ is convertedinto product but more frequently inhibitors bindto active sites without undergoing further catalyticreactions.
When the enzyme binds inhibitor it is unableto catalyse reactions leading to products and thisis observed by a decrease in enzyme activity ata given substrate concentration. A usual presenta-tion of competitive inhibition involves Eadie–Hofsteeor Lineweaver–Burk plots, and is marked by adecrease in Km although the overall Vmax remainssimilar between inhibited and uninhibited reactions(Figures 7.50 and 7.51). Vmax is unchanged becauseaddition of substrate in high concentrations willeffectively displace inhibitor from the active site ofan enzyme leading to unimpeded rates of reaction.
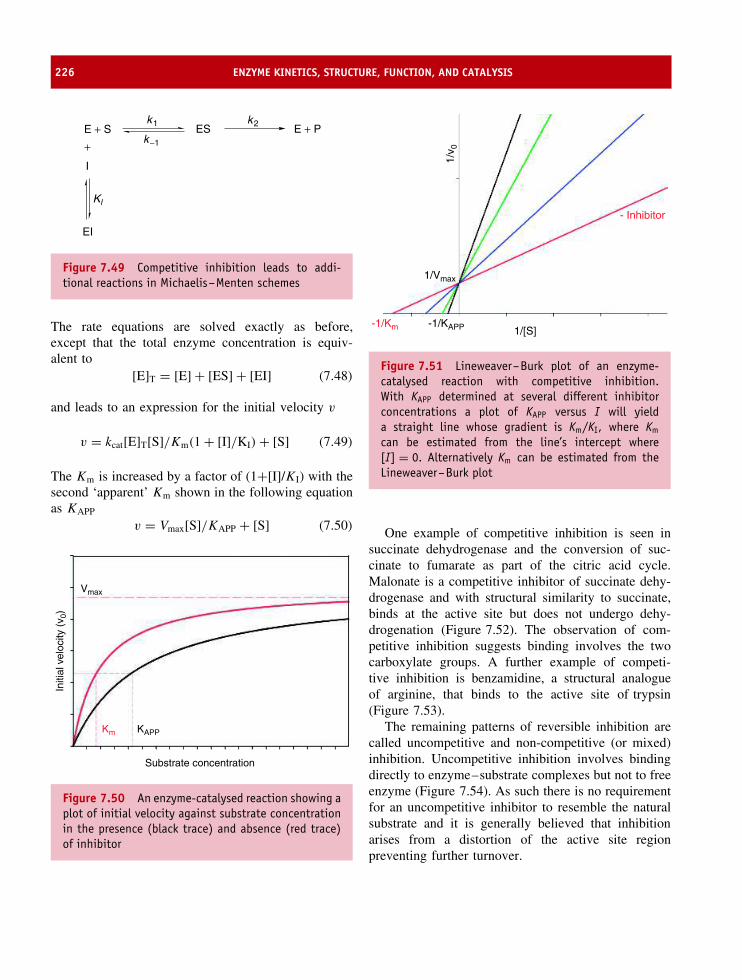
226 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
E + S ES
EI
I
+E + P
k1 k2
k−1
Kl
Figure 7.49 Competitive inhibition leads to addi-tional reactions in Michaelis–Menten schemes
The rate equations are solved exactly as before,except that the total enzyme concentration is equiv-alent to
[E]T = [E] + [ES] + [EI] (7.48)
and leads to an expression for the initial velocity v
v = kcat[E]T[S]/Km(1 + [I]/KI) + [S] (7.49)
The Km is increased by a factor of (1+[I]/KI) with thesecond ‘apparent’ Km shown in the following equationas KAPP
v = Vmax[S]/KAPP + [S] (7.50)
Substrate concentration
Initi
al v
eloc
ity (
v 0)
KAPP
Vmax
Km
Figure 7.50 An enzyme-catalysed reaction showing aplot of initial velocity against substrate concentrationin the presence (black trace) and absence (red trace)of inhibitor
- Inhibitor
-1/Km -1/KAPP 1/[S]
1/v 0
1/Vmax
Figure 7.51 Lineweaver–Burk plot of an enzyme-catalysed reaction with competitive inhibition.With KAPP determined at several different inhibitorconcentrations a plot of KAPP versus I will yielda straight line whose gradient is Km/KI, where Km
can be estimated from the line’s intercept where[I] = 0. Alternatively Km can be estimated from theLineweaver–Burk plot
One example of competitive inhibition is seen insuccinate dehydrogenase and the conversion of suc-cinate to fumarate as part of the citric acid cycle.Malonate is a competitive inhibitor of succinate dehy-drogenase and with structural similarity to succinate,binds at the active site but does not undergo dehy-drogenation (Figure 7.52). The observation of com-petitive inhibition suggests binding involves the twocarboxylate groups. A further example of competi-tive inhibition is benzamidine, a structural analogueof arginine, that binds to the active site of trypsin(Figure 7.53).
The remaining patterns of reversible inhibition arecalled uncompetitive and non-competitive (or mixed)inhibition. Uncompetitive inhibition involves bindingdirectly to enzyme–substrate complexes but not to freeenzyme (Figure 7.54). As such there is no requirementfor an uncompetitive inhibitor to resemble the naturalsubstrate and it is generally believed that inhibitionarises from a distortion of the active site regionpreventing further turnover.

IRREVERSIBLE INHIBITION OF ENZYME ACTIVITY 227
CO
O−
CH2
C
O
O−
O− CO
CH2
CH2
CO−
O
Figure 7.52 The structures of succinate andmalonate
NHCH
CH2
CH2
CH2
NH
CH2N NH2
CO
CH2N NH2+ +
Figure 7.53 Benzamidine and arginine. Competitiveinhibitor and substrate in trypsin
E + S ES
ESI
I
+E + P
k1 k2
k−1
Kl
Figure 7.54 Kinetic scheme illustrating uncompeti-tive inhibition of enzyme-catalysed reaction
Non-competitive inhibition (Figure 7.55) involvesinhibitor binding to free enzyme and enzyme substratecomplexes to form inactive EI or ESI states. It iscomparatively rare but is exhibited by some allostericenzymes. In these cases it is thought that inhibitor bindsto the enzyme causing a change in conformation thatpermits substrate binding but prevents further catalysis.
E + S ES
ESI
I
+E + P
k1 k2
k−1
k1
k−1
Kl
El + S
I
+
Kl
Figure 7.55 Kinetic scheme describing non-compe-titive inhibition. In the above example numerouskinetic simplifications have been introduced. Thecomplex ESI can sometimes exhibit catalytic activityat reduced rates, or the binding of inhibitor may altersubstrate binding and/or kcat. As a result of thesedifferential effects the term mixed inhibition is used
Since a non-competitive inhibitor binds to both E andES removing catalytic competence in the latter theeffect of this is to lower the number of effective enzymemolecules. The result is a decrease in Vmax as a result ofchanges in kcat. Since non-competitive inhibitors bindat sites distinct from the substrate there is not normallyany effect on Km.
Irreversible inhibition of enzymeactivity
Irreversible inhibition can involve the inactivation ofenzymes by extremes of pH, temperature or chem-ical reagents. Most frequently, however, it involvesthe covalent modification of the enzyme in a man-ner that destroys activity. Most substances that causeirreversible inhibition are toxic to cells, although inthe case of aspirin the ‘toxic’ effect is beneficial.Many irreversible inhibitors have been identified; someare naturally occurring metabolites whilst others resultfrom laboratory synthesis (Table 7.8).
The inhibition of enzymes involved in bacterial cellwall synthesis is exploited through the use of penicillinas an antibiotic that binds to bacterial enzymes involvedin cell wall synthesis causing inactivation. The inabilityto extend bacterial cell walls prevents division andleads to the cessation of bacterial growth and limitsthe spread of the infection.

228 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
Table 7.8 Irreversible enzyme inhibitors and sites of action
Compound Common source and site of action
Diisopropylfluorophosphate (DIFP), Sarin Synthetic; inactivate serine residues of serine proteaseenzymes
Ricin/abrin Castor bean; ribosomal inactivating proteins; inhibitribosome function via depurination reactions
N -tosyl-L-phenylalanine chloromethylketone(TPCK)
Synthetic inhibitor; binds to active site histidine inchymotrypsin
Penicillin Naturally occurring antibiotic plus many syntheticvariants; inhibits cell wall biosynthesis enzymesparticularly in Gram-positive bacteria
Cyanide Released during breakdown of cyanogenic glycosidesfound in many plants; inhibits respiration
Aspirin Natural product – methyl salicylate plus syntheticvariants. All bind to active site of cyclo-oxygenaseinhibiting substrate binding
Aspirin: an inhibitor of cyclo-oxygenases
Aspirin’s pain-killing capacity was known to Hip-pocrates in the 5th century BC but provides a mar-velous example of how studies of enzyme inhibitioncombined with structural analysis leads to advances inhealth care.
Prostaglandins are hormones created by cells that actonly in the local or surrounding area before they arebroken down. Unlike most hormones they are not trans-ported systemically around the body but control ‘local’cellular processes such as the constriction of blood ves-sels, platelet aggregation during blood clotting, uterineconstriction during labour, pain transmission and theinduction of inflammation. Although these diverse pro-cesses are controlled by different prostaglandins theyare all created from a common precursor moleculecalled arachidonic acid. Cyclo-oxygenase catalysesthe addition of two oxygen molecules to arachidonicacid in a pathway that leads to the formation ofprostaglandins.
Aspirin-like drugs, sometimes called non-steroidalanti-inflammatory drugs or NSAIDs (Figure 7.56),prevent prostaglandin biosynthesis through inhibitionof cyclo-oxygenases such as COX-1 (Figure 7.57).
O
H3C
O
COO−
Aspirin
O
CH2Br
O
COO−
Bromoaspirin
COO− Ibuprofen
Figure 7.56 Three common non-steroidal anti-inflammatory drugs (NSAIDs)
Crystallization of cyclo-oxygenase by R. Garavitoshowed a membrane protein comprised of three

IRREVERSIBLE INHIBITION OF ENZYME ACTIVITY 229
HOO
OO
HO2C
911
15 HO
O O
HO2C
9
15
11
CO2H
911
13
15O2
reductant
Arachidonic acid Prostaglandin G2(PGG2)
Prostaglandin H2(PGH2)
Figure 7.57 Formation of prostaglandin H2 from arachidonic acid by cyclo-oxygenase
Figure 7.58 The three-dimensional structure of cyclo-oxygenase (PDB:1PRH). On the left the distribution ofsecondary structure is shown for one subunit of the dimer found in the crystal lattice. The space filling modelhighlights the different domains and the buried heme group. (blue, residues 33–72; orange, membrane-bindingregion formed from residues 73–116; and purple the globular domain containing the catalytic heme group)
domains in a chain of 551 residues (Figure 7.58).Residues 33–72 form a small compact module whilsta second domain (residues 73–116) adopts a right-handed spiral of four helical segments along one sideof the protein. This domain binds to the membranevia amphipathic helices. The third domain is thecatalytic heart of the bifunctional enzyme performingcyclo-oxygenation and peroxidation. Cyclo-oxygenaseis a homodimer, and should be more correctly calledprostaglandin H synthase (or PGHS-1) because the
cyclo-oxygenase reaction is only the first of twofunctional reactivities.
The cyclo-oxygenase reaction occurs in a hydropho-bic channel that extends from the membrane-bindingdomain to the catalytic core and a non-covalentlybound heme centre. The arachidonic acid is positionedin this channel in an extended L-shaped conformationand catalysis begins with abstraction of the 13-pro-S-hydrogen to generate a substrate bound radical. At thispoint, a combination of active site residue interactions

230 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
and conformational changes control the regio- andstereospecific additions of molecular oxygen and ringclosures resulting in the product.
Insight into the mechanism of action of aspirinstemmed from crystallization of this protein in thepresence of inhibitors. In the bromoaspirin-inactivatedstructure Ser530 is bromoacetylated (as opposed tosimple acetylation with aspirin) with the salicylateleaving-group bound in the tunnel as part of theactive site (Figure 7.59). The bound salicylate groupblocks substrate access to the heme centre whilst otherNSAIDs do not covalently modify the enzyme butprevent conversion of arachidonic acid in the firstreaction of prostaglandin synthesis. The active site isessentially a hydrophobic channel of highly conservedresidues such as Arg120, Val349, Tyr348, Tyr355,Leu352, Phe381, Tyr385, and Ser530. At the mouth ofthe channel lie Arg120 and Tyr 355 and these residuesinteract with the carboxylate groups of arachidonic acidwith the remainder of the eicosanoid chain extendinginto the channel and binding to hydrophobic residuesclose to Ser530. Although the details of catalysisand inhibition remain to be clarified the heme grouplies at one end of the channel and it is thought
that heme-catalysed peroxidase activity involves theformation of a tyrosyl radical to initiate the process.
Aspirin is an extremely beneficial drug but con-cern exists over the damage caused to the stom-ach and duodenum in significant numbers of patients.About one in three people who take NSAIDs for longperiods develop gastrointestinal bleeding. The under-lying reasons for the unwanted side effects becameclear with the realization that two forms of cyclo-oxygenase (COX-1 and COX-2) exist in vivo. COX-1is the constitutive isoform whilst COX-2 is induced byinflammatory stimuli. The beneficial anti-inflammatoryactions of NSAIDs are due to inhibition of COX-2whereas unwanted side effects such as irritation of thestomach lining arise from inhibition of COX-1. It isclearly desirable to obtain enzyme inhibitors (drugs)that selectively target (inhibit) COX-2.
Despite different physiological roles COX-1 andCOX-2 show similar structures and catalytic functions,with a small number of amino acid substitutions givingrise to subtle differences in ligand interaction betweeneach isoform. These differences form the basis ofdeveloping structure–function relationships in cyclo-oxygenases and allow rational drug design aimed atimproving the desirable properties of aspirin without
Figure 7.59 The active site of cyclo-oxygenase (COX-1) shown by the yellow residues superimposed on the secondarystructure elements. Removing the bulk of the protein shows the approximate position of the channel (arrow) andthe lining of this pocket by conserved residues. Ser530, acetylated by aspirin, lies approximately midway alongthe channel
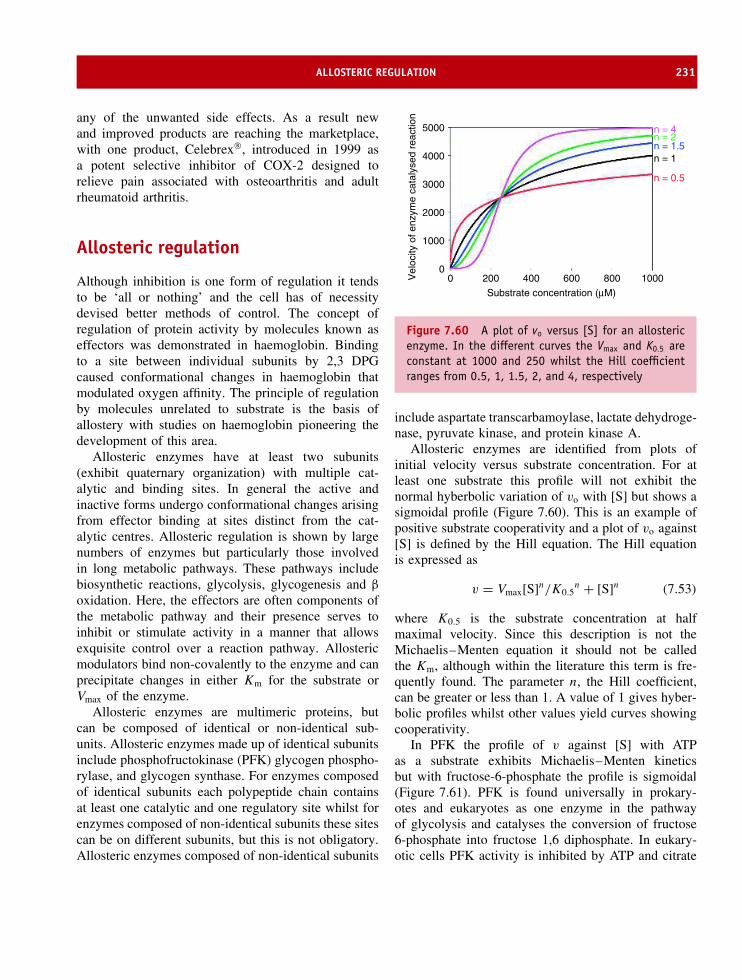
ALLOSTERIC REGULATION 231
any of the unwanted side effects. As a result newand improved products are reaching the marketplace,with one product, Celebrex, introduced in 1999 asa potent selective inhibitor of COX-2 designed torelieve pain associated with osteoarthritis and adultrheumatoid arthritis.
Allosteric regulation
Although inhibition is one form of regulation it tendsto be ‘all or nothing’ and the cell has of necessitydevised better methods of control. The concept ofregulation of protein activity by molecules known aseffectors was demonstrated in haemoglobin. Bindingto a site between individual subunits by 2,3 DPGcaused conformational changes in haemoglobin thatmodulated oxygen affinity. The principle of regulationby molecules unrelated to substrate is the basis ofallostery with studies on haemoglobin pioneering thedevelopment of this area.
Allosteric enzymes have at least two subunits(exhibit quaternary organization) with multiple cat-alytic and binding sites. In general the active andinactive forms undergo conformational changes arisingfrom effector binding at sites distinct from the cat-alytic centres. Allosteric regulation is shown by largenumbers of enzymes but particularly those involvedin long metabolic pathways. These pathways includebiosynthetic reactions, glycolysis, glycogenesis and β
oxidation. Here, the effectors are often components ofthe metabolic pathway and their presence serves toinhibit or stimulate activity in a manner that allowsexquisite control over a reaction pathway. Allostericmodulators bind non-covalently to the enzyme and canprecipitate changes in either Km for the substrate orVmax of the enzyme.
Allosteric enzymes are multimeric proteins, butcan be composed of identical or non-identical sub-units. Allosteric enzymes made up of identical subunitsinclude phosphofructokinase (PFK) glycogen phospho-rylase, and glycogen synthase. For enzymes composedof identical subunits each polypeptide chain containsat least one catalytic and one regulatory site whilst forenzymes composed of non-identical subunits these sitescan be on different subunits, but this is not obligatory.Allosteric enzymes composed of non-identical subunits
5000
4000
3000
2000
1000
00 200 400 600
Substrate concentration (µM)
Vel
ocity
of e
nzym
e ca
taly
sed
reac
tion
800 1000
n = 4n = 2n = 1.5
n = 0.5
n = 1
Figure 7.60 A plot of vo versus [S] for an allostericenzyme. In the different curves the Vmax and K0.5 areconstant at 1000 and 250 whilst the Hill coefficientranges from 0.5, 1, 1.5, 2, and 4, respectively
include aspartate transcarbamoylase, lactate dehydroge-nase, pyruvate kinase, and protein kinase A.
Allosteric enzymes are identified from plots ofinitial velocity versus substrate concentration. For atleast one substrate this profile will not exhibit thenormal hyberbolic variation of vo with [S] but shows asigmoidal profile (Figure 7.60). This is an example ofpositive substrate cooperativity and a plot of vo against[S] is defined by the Hill equation. The Hill equationis expressed as
v = Vmax[S]n/K0.5n + [S]n (7.53)
where K0.5 is the substrate concentration at halfmaximal velocity. Since this description is not theMichaelis–Menten equation it should not be calledthe Km, although within the literature this term is fre-quently found. The parameter n, the Hill coefficient,can be greater or less than 1. A value of 1 gives hyber-bolic profiles whilst other values yield curves showingcooperativity.
In PFK the profile of v against [S] with ATPas a substrate exhibits Michaelis–Menten kineticsbut with fructose-6-phosphate the profile is sigmoidal(Figure 7.61). PFK is found universally in prokary-otes and eukaryotes as one enzyme in the pathwayof glycolysis and catalyses the conversion of fructose6-phosphate into fructose 1,6 diphosphate. In eukary-otic cells PFK activity is inhibited by ATP and citrate

232 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
1000
800
600
400
200
00 200 400 600
Substrate concentration
Rea
ctio
n ve
loci
ty
800 1000
Figure 7.61 The sigmoidal and hyperbolic kineticsfor PFK in the presence of effector (fructose-6-phosphate) and normal substrate (ATP)Fructose-6-phosphate + ATP −−−⇀↽−−− Fructose 1,6dipho-sphate + ADP
whilst activation occurs with ADP, AMP and cyclicAMP. In bacteria the PFK enzymes are structurallysimpler with a diverse array of allosteric modulators.The most studied PFKs are isolated from B. stearother-mophilus and E. coli. These enzymes are inhibited by
phosphoenolpyruvate (an end product of glycolysis)and stimulated by ADP as well as GDP.
PFK is a homotetramer containing polypeptidechains of 320 residues each folded into two domains.These domains are of different structure with domain1 formed by residues 1–141 and residues 256–300containing α and β secondary structure (α/β) anddomain 2 formed by residues 142–255 and the C-terminal sequence forming an α-β-α layered structure(Figure 7.62). Each subunit has 12 α helices and 11β
strands and forms a large contact area with an identi-cal subunit creating a dimer. Dimers associate throughsmaller interfacial contact zones forming the tetramer.Initial studies of the enzyme showed that one of theproducts of the reaction (ADP) together with Mg2+ ionsbind to the larger domain (blue) whilst an allostericregulator (ADP again) is also found attached to thesmaller second domain (orange). In this instance theeffector molecule (ADP) resembles the structure of oneof the substrates (ATP) and represents an example ofhomotypic regulation (Figures 7.63 and 7.64).
The active site for ADP and fructose-6-phosphatebinding is formed between two subunits in a cleft.The T state of PFK has a low affinity for fructose-6-phosphate (substrate) whilst the R form has a muchhigher affinity.
Figure 7.62 The arrangement of domains 1 and 2 within a single subunit of PFK. Shown in blue is the domainencompassing residues 1–138 and 256–305 whilst in orange is the secondary structure of residues 139–255 and306–319

ALLOSTERIC REGULATION 233
Figure 7.63 The arrangement of domains 1 and 2within the PFK dimer showing ADP and Mg2+ ionsbound to the different parts of the enzyme. ADP isshown in yellow, Mg2+ ions in green with the largeand small domains of each subunit shown using thecolour scheme of Figure 7.62. The dimers associateforming the binding site for the effector ADP molecule
Alongside fructose-6-phosphate ATP is also boundat the active site whilst at the allosteric site ATP(inhibitor) and ADP (activator) bind in additionto phosphoenolpyruvate (PEP), a second allostericinhibitor. The allosteric sites are located principallyin the smaller effector domain (orange section inFigures 7.63 and 7.64) but form contacts with theactive site domain of neighbouring dimers. As a resultof this arrangement conformational changes occurringat allosteric sites are easily transmitted to the activesite domains of other subunits.
An analogue of PEP, 2-phosphoglycolate, bindstightly to the allosteric site yielding an inhibitedform of the enzyme equivalent to the T state. Bycomparing the structure of T state enzyme with thatformed in the presence of products (R state) the extentof conformational changes occurring in PFK wereestimated. The crystal structures of PFK determinedin the R and T states suggest the structural differencesreside in rotation of dimers relative to one another.Comparing the inhibited state with the active state
Figure 7.64 The arrangement of domains 1 and 2within a PFK dimer showing fructose-6-bisphosphate(magenta) and ADP/Mg2+ bound to the active siteof the large domain. These are the products of thereaction and the structure is the R state enzyme.ADP/Mg2+ is also bound at the effector site locatedon the smaller domain (PDB:1PFK). Only the dimeris shown and it should be remembered an additionaldimer of subunits is present in the PFK homotetramer
showed that the tetramer twists about its long axis sothat one pair of subunits rotates relative to the otherpair by about 8◦ around one of the molecular dyad axes.This rotation partly closes the binding site for the co-operative substrate fructose-6-phosphate and thereforeexplains its weaker binding to this conformationalstate. A single subunit contains two domains andthe R > T transition also involves rotation of theseregions relative to each other by ∼4.5◦ that furthercloses the active (fructose-6-phosphate) site betweendomains. Another consequence of these movements isto alter the orientation of residues lining the catalyticpocket. In the R state two arginine side chains formionic interactions with fructose-6-phosphate whilst aglutamate side chain is displaced out of the pocket.In the T state movement of dimers results in Arg162pointing away from the binding site and Glu161 facinginto the binding site. The effect of replacing a positivecharge with a negative charge at the active site is

234 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
to decrease fructose 6-phosphate binding by repulsionbetween negatively charged groups. PFK appears torepresent a simple allosteric kinetic scheme in whichthe active and the inhibited conformations differ intheir affinities for fructose-6-phosphate but do not differin their catalytic competence. The presence of twoconformational states is very similar to that previouslydescribed for haemoglobin.
Aspartyl transcarbamoylase (ATCase) is a largecomplex composed of 12 subunits of two differenttypes. The enzyme catalyses formation of N -carbamoylaspartate from aspartate and N -carbamoyl phosphate,and is an early step in a unique and critical biosyntheticpathway leading to the synthesis of pyrimidines (suchas cytosine, uracil and thymine; Figure 7.65).
Both aspartate and carbamoyl phosphate bind coop-eratively to the enzyme giving sigmoidal profiles(Figure 7.66) that are altered in the presence of cyti-dine triphosphate (CTP), a pyrimidine nucleotide, andadenosine triphosphate (ATP), a purine nucleotide.Allosteric inhibition is observed in the presence ofCTP whilst ATP induces allosteric activation. Exper-imentally, a plot of vo against [S] is observed to besigmoidal and to shift to the left in the presence of ATP
O
P
O−
O− O
COH2N
H3N+ CHC
O
CH2
O
O−
O−
CO
NH2
NHCH
CO
CH2
C
O O−
O−
H2PO4
+
Aspartatetranscarbamoylase −
Figure 7.65 Initial reaction of pyrimidine biosyn-thesis involving condensation of aspartate and car-bamoyl phosphate
6
5
4
3
2
1
00 5 10
[Aspartate] mM
Vel
ocity
(m
mol
es p
rodu
ct/h
/mg
prot
ein)
15 20
Figure 7.66 The steady-state kinetic behaviour ofATCase. Carbamoyl aspartate formation at a fixedconcentration of substrate (blue) is increased in thepresence of ATP (green) whilst CTP has the oppositeeffect acting as an inhibitor
and to the right in the presence of CTP. At a givensubstrate concentration CTP diminishes the reactionvelocity whilst ATP increases it. ATP is therefore a pos-itive heterotropic effector, CTP a negative heterotropiceffector, and aspartate is a positive homotropic effector.
The effects of ATP and CTP are not unex-pected; CTP is one product of pyrimidine biosynthe-sis and inhibition of ATCase activity is an exampleof feedback inhibition where end products modulatebiosynthetic reactions to provide effective means ofcontrol (Figure 7.67). Consequently, high intracellularlevels of CTP decrease pyrimidine biosynthesis whilsthigh intracellular levels of ATP indicate an imbalancebetween purine and pyrimidine concentrations and leadto increased rates of pyrimidine biosynthesis.
Structural insight into allosteric regulation ofATCase is provided by crystallographic analysis of theE. coli enzyme by William Lipscomb (Figure 7.68).The enzyme has a composition of c6r6 where c and rrepresent catalytic and regulatory subunits respectivelyarranged as two sets of catalytic trimers complexedwith three sets of regulatory dimers. Each regulatorydimer links together two subunits present in differenttrimers. For both catalytic and regulatory subunits twodomains exist within each polypeptide chain.

ALLOSTERIC REGULATION 235
Figure 7.67 The metabolic pathway leading to UTPand CTP formation – feedback inhibition of ATCase ispart of the normal control process
The catalytic subunit (Figure 7.69) contains onedomain involved in aspartate binding whilst a seconddomain binds carbamoyl phosphate with the active sitelying between domains. In view of their separationthe c subunit must undergo large conformationalchange to bring the sites closer together for productformation. A bifunctional analogue of carbamoylphosphate and aspartate called N -(phosphonacetyl)-L-aspartate (PALA) binds to ATCase, but is unreactiveand has facilitated structural characterization of theenzyme–substrate complex (Figure 7.70). PALA bindsto the R state, and by comparing the structure ofthis form of the enzyme with that obtained in the Tstate (ATCase plus CTP) the conformational changes
Figure 7.68 The quaternary structure of ATCase fromE. coli showing the r2c2 structure (PDB: 1ATI). Tworegulatory units (orange) link to catalytic subunits(green) belonging to different trimers
occurring in transitions from one allosteric form toanother were estimated.
The observed kinetic profiles of enzyme activitywith ATP and CTP provide further insight into thebasis of allosteric regulation. ATP enhances activityand binds to the R state of the enzyme whilst CTPan allosteric inhibitor binds preferentially to the Tor low affinity state. Both ATP and CTP bind tothe regulatory subunit and compete for the samesite. The allosteric binding domain is the largerpart of the r subunit containing a Zn2+ bindingregion that forms the interface between the regulatoryand catalytic subunits. Displacement of the non-covalently bound Zn using mercurial reagents leadsto the absence of binding and the dissociation of thec6r6 complex.
The rate of catalysis in isolated trimer is not mod-ulated by ATP or CTP but exceeds the rate observedin the complete enzyme. This kinetic behaviour indi-cates that r subunits reduce the activity of the c sub-units in the complete enzyme. In view of the differentkinetic properties exhibited by ATCase in the pres-ence of ATP or CTP it is somewhat surprising tofind that both nucleotides bind in similar manner tothe r subunit. This makes a structural explanation of

236 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
Figure 7.69 The structure of a single catalytic subunit and its adjacent regulatory subunit. The catalytic subunit isshown in green and the regulatory subunit in yellow (reproduced with permission from Lipscomb, W.N. Adv. Enzymol.1994, 73, 677–751. John Wiley & Sons)
C
O−
OC
O− O
PO−O
O−
CCH2O
NHCH
CH2
C
O−
O
CO− O
NH3+
CHCH2
PO−O
O−
COO
NH2
Aspartate
N-(Phosphonacetyl)-L-aspartate(PALA)
Carbamoyl phosphate
Figure 7.70 The structure of the bifunctional substrate analogue PALA and its relationship to carbamoyl phosphateand aspartate
their effects difficult to understand and the structuralbasis of allostery in ATCase has proved very elusivedespite structures for the enzyme in the ATCase–CTPcomplex (T state) and the ATCase–PALA (R-state)complex (Figure 7.71). A comparison of these two
structures shows that in the T>R transition theenzyme’s catalytic trimers separate by ∼1.1 nm andreorient their axes relative to each other by ∼5◦. Thetrimers assume an elongated or eclipse-like configura-tion along one axis accompanied by rotation of the r

COVALENT MODIFICATION 237
Figure 7.71 A diagram of the quaternary structure of aspartate transcarbamoylase (ATCase). The quaternarystructure in the T state (left) shows six catalytic subunits and six regulatory subunits. Between each catalytic subunitlies the catalytic site whilst on each regulatory subunit is a single effector site on its outer surface (reproduced withpermission from Lipscomb, W.N. Adv. Enzymol. 1994, 73, 677–751. John Wiley & Sons)
subunits by ∼15◦ about their two-fold rotational sym-metry axis.
Covalent modificationCovalent modification regulates enzyme activity bythe synthesis of inactive precursor enzymes contain-ing precursor sequences at the N terminal. Removal ofthese regions by proteases leads to enzyme activation.Trypsin, chymotrypsin and pepsin are initially synthe-sized as longer, inactive, precursors called trypsino-gen, chymotrypsinogen and pepsinogen or collectivelyzymogens. Zymogens are common for proteolyticenzymes such as those found in the digestive tracts ofmammals where the production of active enzyme couldlead to cell degradation immediately after translation.Acute pancreatitis is precipitated by damage to the pan-creas resulting from premature activation of digestiveenzymes.
Trypsinogen is the zymogen of trypsin and is mod-ified when it enters the duodenum from the pan-creas. In the duodenum a second enzyme enteropep-tidase secreted from the mucosal membrane excises
H3N+
H3N+
Val
(Asp)4
(Asp)4
Lys
Ile
Val
Val LysIle
Val
+
EnteropeptidaseorTrypsin
Figure 7.72 The activation of trypsinogen to trypsin.Limited proteolysis of trypsinogen by enteropeptidaseand subsequently trypsin removes a short hexapeptidefrom the N-terminal
a hexapeptide from the N terminus of trypsinogen toform trypsin (Figure 7.72). Enteropeptidase is itselfunder hormonal control whilst the site of cleavage afterLys6 is important. The product is trypsin with a naturalspecificity for cleavage after Lys or Arg residues and

238 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
the small amount of trypsin produced is able to catalysethe further production of trypsin from the zymogen. Asa result the process is often described as auto-catalytic.
Chymotrypsin is closely related to trypsin and is acti-vated by cleavage of chymotrypsinogen between Lys15and Val16 to form π-chymotrypsin (Figure 7.73). Fur-ther autolysis (self-digestion) to remove residues Ser14-Arg15 and Thr147-Asn148 yields α-chymotrypsin, thenormal form of the enzyme. The three segments ofpolypeptide chain produced by zymogen processingremain linked by disulfide bridges.
Trypsin plays a major role in the activation ofother proteolytic enzymes. Proelastase, a zymogen ofelastase, is activated by trypsin by removal of a shortN terminal region along with procarboxypeptidases Aand B and prophospholipase A2. The pivotal role oftrypsin in zymogen processing requires tight control oftrypsinogen activation. At least two mechanisms existfor tight control. Trypsin catalysed activation occursslowly possibly as a result of the large amount ofnegative charge in the vicinity of the target lysineresidue (four sequential aspartates). This charge repelssubstrate from the catalytic pocket that also contains
aspartate residues. Zymogens are stored in the pancreasin intracellular vesicles known as zymogen granules thatare resistant to proteolytic digestion. As a final measurecells possess serpins that prevent further activation.
A second important group of zymogens are the serineproteases of the blood-clotting cascade (Figure 7.74)that circulate as inactive forms until a stimulusnormally in the form of injury to a blood vessel. Thisrapidly leads to the formation of a clot that preventsfurther bleeding via the aggregation of platelets withinan insoluble network of fibrin. Fibrin is produced fromfibrinogen, a soluble blood protein, through the actionof thrombin, a serine protease. Thrombin is the last ofa long series of enzymes involved in the coagulationprocess that are sequentially activated by proteolysis oftheir zymogens. Sequential enzyme activation leads toa coagulation cascade and the overall process allowsrapid generation of large quantities of active enzymesin response to biological stimuli.
The concept of programmed cell death or apoptosisis vital to normal development of organisms. Forexample, the growing human embryo has fingers thatare joined by web-like segments of skin. The removal
1 122
12216151
1 13 16 122 136 146 149 201 245
136
136
201
201
245
245
1481471514
Figure 7.73 Activation of chymotrypsinogen proceeds through proteolytic processing and the removal of shortpeptide sequences.

COVALENT MODIFICATION 239
ThrombinProthrombin
FibrinFibrinogen
Cross linked fibrin (clot)
Factor XaFactor X
Factor IXaFactor IX
Factor XIII
Factor XIIIa
Factor V
Factor Va
FactorVIIIa
FactorVIII
Factor XIaFactorXI
Figure 7.74 The cascade of enzyme activation duringthe intrinsic pathway of blood clotting involvesprocessing of serine proteases
of these skin folds and the generation of correct patternformation (i.e. fingers) relies on a family of intracellularenzymes called caspases. The term caspase refers tothe action of these proteins as cysteine-dependentaspartate-specific proteases. Their enzymatic propertiesare governed by specificity for substrates containingAsp and the use of a Cys285 sidechain for catalysingpeptide-bond cleavage located within a conserved motifof five residues (QACRG).
The first level of caspase regulation involves theconversion of zymogens to active forms in response
to inflammatory or apoptotic stimuli although secondlevels of regulation involve specific inhibition of activecaspases by natural protein inhibitors. Caspases aresynthesized as inactive precursors and their activity isfully inhibited by covalent modification (Figure 7.75).It is important to achieve complete inhibition of activitysince the cell is easily destroyed by caspase activity.Caspases are synthesized joined to additional domainsknown as death effector domains because their removalinitiates the start of apoptosis where a significant stepis the formation of further active caspases.
Yet another form of enzyme modification is thereversible phosphorylation/dephosphorylation of ser-ine, threonine or tyrosine side chains. The phospho-rylation of serine, threonine or tyrosine is particularlyimportant in the area of cell signalling. Phosphoryla-tion of tyrosine is an important covalent modificationcatalysed by enzymes called protein tyrosine kinases(PTKs). These enzymes transfer the γ phosphate ofATP to specific tyrosine residues on protein substratesaccording to the reaction
Protein + ATP −−−⇀↽−−− Protein-P + ADP
This reaction modulates enzyme activity and leadsto the creation of new binding sites for downstreamsignalling proteins. At least two classes of PTKs arepresent in cells: the transmembrane receptor PTKs andenzymes that are non-receptor PTKs.
Receptor PTKs are transmembrane glycoproteinsactivated by ligand binding to an extracellular sur-face. A conformational change in the receptor resultsin phosphorylation of specific tyrosine residues on theintracellular domains of the receptor (Figure 7.76) – aprocess known as autophosphorylation – and initi-ates activation of many signalling pathways con-trolling cell proliferation, differentiation, migration
α βα b
α b
Figure 7.75 The covalent modification by addition of large protein domains to caspases leads to an inactiveenzyme. Their removal allows the association of the α and β domains in an active heterotetramer

240 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
Figure 7.76 Domain organization of receptor tyrosine kinases. The extracellular domain at the top contains avariety of globular domains arranged with modular architecture. In contrast the cytoplasmic segment is almostexclusively a tyrosine kinase domain (reproduced with permission from Hubbard, S.R. & Till, J.R. Ann. Rev. Biochem.2000, 69, 373–398. Annual Reviews Inc)
Figure 7.77 Domain organization found within non-receptor protein kinases (reproduced with permission fromHubbard, S.R. & Till, J.R. Ann. Rev. Biochem. 2000, 69, 373–398. Annual Reviews Inc)
and metabolism. The transmembrane receptor tyrosinekinase family includes the receptors for insulin andmany growth factors such as epidermal growth fac-tor, fibroblast growth factor and nerve growth factor.Receptors consist of large extracellular regions con-structed from modules of globular domains linkedtogether. A single transmembrane helix links to a cyto-plasmic domain possessing tyrosine kinase activity.
In contrast non-receptor PTKs (Figure 7.77) are alarge family of proteins that are integral components
of the signal cascades stimulated by RTKs as wellas G-protein coupled receptors. These kinases phos-phorylate protein substrates causing a switch betweenactive and inactive states. These tyrosine kinaseslack large extracellular ligand-binding regions togetherwith transmembrane regions and most are locatedin the cytoplasm although some bear N-terminalmodifications such as myristoylation or palmitoyla-tion that lead to membrane association. In addi-tion to kinase activity this large group of proteins

ISOENZYMES OR ISOZYMES 241
possesses domains facilitating protein–protein, pro-tein–lipid or protein–DNA interactions and a commonset of domains are the Src homology 2 (SH2) and Srchomology 3 (SH3) domains.
Phosphotyrosine binding has been studied in SH2domains where the binding site consists of a deeppocket and centres around interaction of the phosphatewith an arginine side chain at the base. The sidechains of phosphoserine and phosphothreonine are bothtoo short to form this interaction thereby accountingfor the specificity of binding between SH2 andtarget sequences.
One of the first forms of phosphorylation to be rec-ognized was that occurring during the activation ofenzymes involved in glycogen metabolism. Glycogenphosphorylase plays a pivotal role in controlling themetabolism of the storage polysaccharide glycogen.Glycogen is primarily a series of α 1→4 glycosidicbonds formed between glucose under conditions wherethe supply of glucose is plentiful. Glycogen phospho-rylase catalyses the first step in the degradation ofglycogen by releasing glucose-1-phosphate from a longchain of glucose residues for use by muscle tissue dur-ing contraction.
Glycogen(n residues)
+Pi −−−⇀↽−−− Glucose-1-phosphate + Glycogen(n−1) residues
As an allosteric enzyme the inactive T state is switchedto the active R state by a change in conforma-tion controlled by phosphorylation of a single ser-ine residue (Ser14) in a polypeptide chain of ∼850residues. Reversible phosphorylation leads to transi-tion between the two forms of the enzyme calledphosphorylase a and phosphorylase b. The two formsdiffer markedly in their allosteric properties withthe ‘a’ form generally uncontrolled by metaboliteswith the exception of glucose whilst the ‘b’ formis activated by AMP and inhibited by glucose-6-phosphate, glucose, ATP and ADP. Conversion is stim-ulated by hormone action, particularly the release ofadrenaline and glucagon, where these signals indi-cate a metabolic demand for glucose that is medi-ated by intracellular production of cAMP by adenylatecyclase. Adenylate cyclase activates cAMP dependentkinases – enzymes causing the phosphorylation of aspecific kinase – that subsequently modifies phospho-rylase b at Ser14. This cascade is also regulated byphosphatases.
The structural basis of phosphorylation of Ser14 hasbeen established from crystallographic studies of thea and b forms. Glycogen phosphorylase is a dimerof two identical chains. In the absence of Ser14phosphorylation the N-terminal residues are poorlyordered but upon modification a region of ∼16 residuesassembles into a distorted 310 helix that interactswith the remaining protein causing an increase indisorder at the C terminal. Phosphorylation ordersthe N-terminal residues and creates major changes inthe position of Ser14 largely from new interactionsformed with Arg43 on an opposing subunit and withArg69 within the same chain. These interactions triggerchanges in the tertiary and quaternary structure thatlead to activation of a catalytic site located ∼4.5 nmfrom the phosphoserine. The subunits move closertogether in the presence of AMP or phosphoserine14 and are accompanied by opening of active siteregions to allow substrate binding and catalysis. Thestructure of glycogen phosphorylase suggested thatinhibition of the enzyme by glucose-6-phosphate andADP occur by excluding AMP from the bindingsite and from preventing conformational movementsof the N-terminal region of the protein containingthe phosphorylated serine residue. It has becomeclear that phosphorylation is a major mechanism forallosteric control of enzyme activity not only inmetabolic systems but also in ‘housekeeping’ proteinssuch as those involved in cell division and thecell cycle.
Isoenzymes or isozymes
The term ‘isoenzyme’ is poorly defined. It commonlyrefers to multiple forms of enzymes arising fromgenetically determined differences in primary struc-ture. In other words isoenzymes are the results ofdifferent gene products. A second form of isoen-zyme involves multimeric proteins where combiningsubunits in different ratios alters the intrinsic cat-alytic properties. Cytochromes P450 are a large groupof proteins that catalyse the oxidation of exogenousand endogenous organic substrates via a commonreaction.
RH + 2H+ + O2 + 2e− −−−⇀↽−−− ROH + H2O

242 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
OC
CH3
COO
OHC
CH3
COO
H
NADH NAD
Lactate Dehydrogenase
Pyruvate Lactate
Figure 7.78 Conversion of pyruvate to lactate bylactate dehydrogenase
Located in the endoplasmic reticulum of cells theseenzymes are responsible for the detoxification of drugsand xenobiotics as well as the transformation ofsubstrates such as steroids and prostaglandins. They aredescribed as isoenzymes but arise from different genescontaining different primary sequences with active sitehomology.
An excellent example of an isoenzyme in the secondgroup is lactate dehydrogenase. In the absence of oxy-gen the final reaction of glycolysis is the conversion ofpyruvate to lactic acid catalysed by the enzyme lactatedehydrogenase (LDH) with the conversion of NADHto NAD (Figure 7.78).
In skeletal muscle, where oxygen deprivation iscommon during vigorous exercise, the reaction is effi-cient and large amounts of lactate are generated. Underaerobic conditions the reaction catalysed by lactatedehydrogenase is not efficient and pyruvate is pref-erentially converted to acetyl-CoA. The subunit com-position of lactate dehydrogenase is not uniform butvaries in different tissues of the body. Five major LDHisoenzymes are found in different vertebrate tissues andeach molecule is composed of four polypeptide chainsor subunits. LDH is a tetramer and it has been shownthat three different primary sequences are detected inhuman tissues. The three types of LDH chains areM (LDH-A) found predominantly in muscle tissues,H (LDH-B) found in heart muscle together with amore limited form called LDH-C present in the testesof mammals. LDH-C is clearly of restricted distribu-tion within the body and is not considered further. TheM and H subunits of LDH are encoded by differentgenes and as a result of differential gene expressionin tissues the tetramer is formed by several differ-ent combinations of subunits. Significantly, isoenzymes
of LDH were originally demonstrated by their dif-ferent electrophoretic mobilities under non-denaturingconditions. Five different forms of LDH were detected;LDH-1, LDH-2, LDH-3, LDH-4, and LDH-5 withLDH-1 showing the lowest mobility. The reasons forthese differences in mobility are clear when one com-pares the primary sequences of the M and H subunits(Figure 7.79).
The H subunit contains a greater number of acidicresidues (Asp,Glu) than the M polypeptide. In heartmuscle the gene for the H subunit is more active thanthe gene for the M subunit leading to an LDH enzymecontaining more H subunit. Thus, LDH isoenzyme 1is the predominant form of the enzyme in cardiacmuscle. The reverse situation occurs in skeletal musclewhere there is more M than H polypeptide producedand LDH-5 is the major form of the enzyme. Moreimportantly, the conversion of pyruvate to lactateincreases with the number of M chains and LDH5 in skeletal muscle contains 4 M subunits and isideally suited to the conversion of pyruvate to lactate.In contrast LDH-1 has 4 H subunits and is lesseffective at converting pyruvate into lactate. Lactatedehydrogenase, like many other enzymes, is also foundin serum where it is the result of normal cell death.Dying or dead cells liberate cellular enzymes intothe bloodstream. The liberation of enzymes into thecirculatory system is accelerated during tissue injuryand the measurement of LDH isoenzymes in serum hasbeen used to determining the site and nature of tissueinjury in humans. For example, when the blood supplyto the heart muscle is severely reduced, as occurs in aheart attack, muscle cells die and liberate the enzymeLDH-1 into the bloodstream. An increase in LDH-1in serum is indicative of a heart attack and may beused diagnostically. In contrast, muscular dystrophy isaccompanied by an increase in the levels of LDH-5derived from dying skeletal muscle cells.
SummaryEnzymes, with the exception of ribozymes, are catalyticproteins that accelerate reactions by factors up to1017 when compared with the corresponding ratein the uncatalysed reaction. All enzymes can be

SUMMARY 243
* 20 * 40 * 60 * 80M subunit : ATLKDQLIYNLLKEEQT-PQNKITVVGVGAVGMACAISILMKDLADELALVDVIEDKLKGEMMDLQHGSLFLRTPKIVSGKDH subunit : ATLKEKLIAPVAEEEATVPNNKITVVGVGQVGMACAISILGKSLADELALVDVLEDKLKGEMMDLQHGSLFLQTPKIVADKD
* 100 * 120 * 140 * 160M subunit : YNVTANSKLVIITAGARQQEGESRLNLVQRNVNIFKFIIPNVVKYSPNCKLLIVSNPVDILTYVAWKISGFPKNRVIGSGCNH subunit : YSVTANSKIVVVTAGVRQQEGESRLNLVQRNVNVFKFIIPQIVKYSPDCIIIVVSNPVDILTYVTWKLSGLPKHRVIGSGCN
* 180 * 200 * 220 * 240M subunit : LDSARFRYLMGERLGVHPLSCHGWVLGEHGDSSVPVWSGMNVAGVSLKTLHPDLGTDKDKEQWKEVHKQVVESAYEVIKLKGH subunit : LDSARFRYLMAEKLGIHPSSCHGWILGEHGDSSVAVWSGVNVAGVSLQELNPEMGTDNDSENWKEVHKMVVESAYEVIKLKG
* 260 * 280 * 300 * 320M subunit : YTSWAIGLSVADLAESIMKNLRRVHPVSTMIKGLYGIKDDVFLSVPCILGQNGISDLVKVTLTSEEEARLKKSADTLWGIQKH subunit : YTNWAIGLSVADLIESMLKNLSRIHPVSTMVKGMYGIENEVFLSLPCILNARGLTSVINQKLKDDEVAQLKKSADTLWDIQK
*M subunit : ELQF--H subunit : DLKDL-
Figure 7.79 The primary sequences of the M and H subunits of lactate dehydrogenase showing sequence identity(blocks) and the distribution of positively charged side chains (red) and negatively charged side chains (blue) ineach sequence. Careful summation of the charged residues reveals that the H subunit has more acidic residues
grouped into one of six functional classes eachrepresenting the generic catalytic reaction. Theseclasses are oxidoreductases, transferases, hydrolases,lyases, isomerase and lyases.
Many enzymes require co-factors to perform effec-tive catalysis. These co-factors can be tightly boundto the enzyme including covalent linkage or moreloosely associated with the enzyme. Co-factors includemetal ions as well as organic components such as pyri-doxal phosphate or nicotinamide adenine dinucleotide(NAD). Many co-factors or co-enzymes are derivedfrom vitamins.
All elementary chemical reactions can be describedby a rate equation that describes the progress (utiliza-tion of initial starting material or formation of product)with time. Reactions are generally described by first orsecond order rate equations.
Enzymes bind substrate forming an enzyme–sub-strate complex which then decomposes to yield product.A plot of the velocity of an enzyme-catalysed reac-tion as a function of substrate concentration exhibits ahyperbolic profile rising steeply at low substrate concen-trations before reaching a plateau above which furtherincreases in substrate have little effect on overall rates.
The kinetics of enzyme activity are usually describedvia the Michaelis–Menten equation that relates theinitial velocity to the substrate concentration. This anal-ysis yields several important parameters such as Vmax
the maximal velocity that occurs when all the enzymeis found as ES complex, Km the substrate concentra-tion at which the reaction velocity is half maximal andkcat/Km, a second order rate constant that represents thecatalytic efficiency of enzymes.
Enzymes catalyse reactions by decreasing the acti-vation free energy (�G‡), the energy associated withthe transition state. Enzymes use a wide range of cat-alytic mechanisms to convert substrate into product.These mechanisms include acid–base catalysis, cova-lent catalysis and metal ion catalysis. Within activesites the arrangement of functional groups allowsnot only the above catalytic mechanisms but furtherenhancements via proximity and orientation effectstogether with preferential binding of the transition statecomplex. The latter effect is responsible for the greatestenhancement of catalytic activity in enzymes.
The catalytic mechanisms of numerous enzymeshave been determined from the application of a combi-nation of structural, chemical modification and kinetic

244 ENZYME KINETICS, STRUCTURE, FUNCTION, AND CATALYSIS
analysis. Enzymes such as lysozyme, the serine pro-tease family including trypsin and chymotrypsin, triosephosphate isomerase and tyrosinyl tRNA synthetaseare now understood at a molecular level and withthis level of understanding comes an appreciation ofthe catalytic mechanisms employed by proteins. Vir-tually all enzymes employ a combination of mecha-nisms to achieve effective (rapid and highly specific)catalysis.
Enzyme inhibition represents a vital mechanismfor controlling catalytic function. In vivo proteininhibitors bind tightly to enzymes causing a lossof activity and one form of regulation. A com-mon form of enzyme inhibition involves the compe-tition between substrate and inhibitor for an activesite. Such inhibition is classically recognized froman increase in Km whilst Vmax remains unaltered.Other forms of inhibition include uncompetitive inhi-bition where inhibitor binds to the ES complexand mixed (non-competitive) inhibition were bind-ing to both E and ES occurs. Different modes ofinhibition are identified from Lineweaver–Burk orEadie–Hofstee plots.
Irreversible inhibition involves the inactivation of anenzyme by an inhibitor through covalent modificationfrequently at the active site. Irreversible inactivationis the basis of many forms of poisoning but is alsoused beneficially in therapeutic interventions with, forexample, the modification of prostaglandin H2 synthaseby aspirin alleviating inflammatory responses.
Allosteric enzymes are widely distributed through-out many different cell types and play important rolesas regulatory units within major metabolic pathways.Allosteric enzymes contain at least two subunits andoften possess many more.
Changes in quaternary structure arise as a resultof ligand (effector) binding causing changes inenzyme activity. The conformational changes fre-quently involve rotation of subunits relative to oneanother and may produce large overall movement. Suchmovements are the basis for the transitions betweenhigh (R state) and low (T state) affinity states of theenzyme with conformational changes leading to largedifferences in substrate binding. The ability to mod-ify enzymes via allosteric effectors allows ‘fine-tuning’of catalytic activity to match fluctuating or dynamicconditions.
Alternative mechanisms of modifying enzyme activ-ity exist within all cells. Most important are covalentmodifications that result in phosphorylation or removeparts of the protein, normally the N-terminal region,that limit functional activity. Covalent modification isused in all cells to secrete proteolytic enzymes as inac-tive precursors with limited proteolysis revealing thefully active protein. Caspases are one group of enzymesusing this mechanism of modification and play a vitalrole in apoptosis. Apoptosis is the programmed destruc-tion of cell and is vital to normal growth and develop-ment. Premature apoptosis is serious and caspases mustbe inactivated via covalent modification to preventedunwanted cell death.
Problems
1. Consult an enzyme database and find three examplesof each major class of enzyme. (i.e. three from eachof the six groupings).
2. Describe the effect of increasing temperature and pHon enzyme-catalysed reactions.
3. How can some enzymes work at pH 2.0 orat 90 ◦C. Illustrate your answer with selectiveexample enzymes.
4. Draw a transition state diagram of energy versusreaction coordinate for uncatalysed and an enzymecatalysed reactions.
5. How is the activity of many dehydrogenases mostconveniently measured. How can this method beexploited to measure activity in other enzymes?
6. Identify nucleophiles used by enzymes in catalysis?
7. Why does uncompetitive inhibition lead to a decreasein Km? What happens to the Vmax?
8. An enzyme is found to have an active site cysteineresidue that plays a critical role in catalysis. Atwhat pH does this cysteine operate efficiently innucleophilic catalysis? Describe how proteins alterthe pK of this side chain from its normal value in

PROBLEMS 245
solution. What would you expect the effect to be onthe catalysed reaction?
9. Construct a plot of initial velocity against substrateconcentration from the following data. Estimatevalues of Km and Vmax. Repeat the calculation of Km
and Vmax to yield improved estimates.
Substrateconcentration
(µM)
Initialvelocity
(µmol s−1)
25.0 80.050.0 133.375.0 171.4
100.0 200.0200.0 266.6300.0 300.0400.0 320.0600.0 342.8800.0 355.5
10. In a second enzyme kinetic experiment the initialvelocity of an enzyme catalysed reaction was fol-lowed in the presence of native substrate and atfive different concentrations of an inhibitory sub-strate. The following data were obtained. Interpretthe results.
Substrateconcentration(µM)
Initialvelocity
(µmol s−1)
Inhibitor50 µM
Inhibitor150 µM
Inhibitor250 µM
Inhibitor350 µM
Inhibitor450 µM
20 90.91 62.50 38.46 27.78 21.74 17.8640 166.67 117.65 74.07 54.05 42.55 35.0960 230.77 166.67 107.14 78.95 62.50 51.7280 285.71 210.53 137.93 102.56 81.63 67.80100 333.33 250.00 166.67 125.00 100.00 83.33125 384.62 294.12 200.00 151.52 121.95 102.04150 428.57 333.33 230.77 176.47 142.86 120.00200 500.00 400.00 285.71 222.22 181.82 153.85250 555.56 454.55 333.33 263.16 217.39 185.19300 600.80 500.83 375.78 300.70 250.62 214.85400 666.67 571.43 444.44 363.64 307.69 266.67500 714.29 625.00 500.00 416.67 357.14 312.50600 750.00 666.67 545.45 461.54 400.00 352.94800 800.00 727.27 615.38 533.33 470.59 421.051000 833.33 769.23 666.67 588.24 526.32 476.19


8Protein synthesis, processing
and turnover
Uncovering the cellular mechanisms resulting in sequen-tial transfer of information from DNA (our genes)to RNA and then to protein represents one of majorachievements of biochemistry in the 20th century.Beginning with the discovery of the structure of DNAin 1953 (Figure 8.1) by James Watson, Francis Crick,Maurice Wilkins and Rosalind Franklin this area of bio-diversity has expanded dramatically in importance. Theinformation required for synthesis of proteins resides inthe genetic material of cells. In most cases this geneticmaterial is double-stranded DNA and the underlyingprinciples of replication, transcription and translationremain the same throughout all living systems.
The information content of DNA resides in theorder of the four nucleotides (adenine, cytosine,guanine and thymine) along the strands making upthe famous double helix. The structure of DNA isone of the most famous images of biochemistry andalthough this chapter will focus on the reactionsoccurring during or after protein synthesis it is worthremembering that replication and transcription involvethe concerted action of many diverse proteins. Thetransfer of information from DNA to protein can bedivided conveniently into three major steps: replication,transcription and translation, with secondary stepsinvolving the processing of RNA and the modificationand degradation of proteins (Figure 8.2).
Cell cycle
Cell division is one part of an integrated series ofreactions called appropriately the cell cycle. The cellcycle involves replication, transcription and translationand is recognized as a universal property of cells.Bacteria such as Escherichia coli replicate and dividewithin 45 minutes whilst eukaryotic cell replicationis slower and certainly more complicated, with ratesof division varying dramatically. Typically, eukaryoticcells pass through distinct phases that combine to makethe cell cycle and lead to mitosis. The first phase isG1, the first gap phase, and occurs after cell divisionwhen a normal diploid chromosome content is present.G1 is followed by a synthetic S phase when DNA isreplicated, and at the end of this synthetic period thecell has twice the normal DNA content and enters asecond gap or G2 phase. The combined G1, S and G2
phase make up the interphase, a period first recognizedby cytogeneticists studying cell division. After the G2
phase cells enter mitosis, with division giving twodaughter cells each with a normal diploid level of DNAand the whole process can begin again (Figure 8.3).
Mitosis is accompanied by the condensation ofchromosomes into dense opaque bodies, the disinte-gration of the nuclear membrane and the formationof a specialized mitotic spindle apparatus containing
Proteins: Structure and Function by David Whitford 2005 John Wiley & Sons, Ltd

248 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
Figure 8.1 The structure of DNA. The bases liehorizontally between the two sugar-phosphate chains.The positions of the major and minor grooves aredefined by the outline of phosphate/oxygen atoms(orange/red) arranged along the sugar-phosphatebackbone. The pitch of the helix is ∼3.4 nm andrepresents the distance taken for each chain tocomplete a turn of 360◦
microtubule elements that directs chromosome move-ments so that daughter cells receive one copy of eachchromosome pair. After mitotic division the cell cycleis complete and normally a new G1 phase starts,although in some cells G1 arrest is observed and a statecalled G0 is recognized.
Proteins with significant roles in the cell cycle werediscovered from cyclical variations in their concentra-tion during cell division and called cyclins (Figure 8.4).The first cyclin molecule was discovered in 1982 fromsea urchin (Arbacia) eggs, but subsequently at leasteight cyclins have been identified in human cells.
G2(Gap 2)
M (Mitosis)
G1(Gap 1)Cell Cycle
S phaseDNA synthesis
Figure 8.3 Cell cycle diagram showing the G1, S, G2
phases in a normal mitotic cell cycle
Figure 8.4 The levels of protein vary according tothe stage of the cell cycle and gave rise to the name ofthis protein. Cyclins are abundant during the stage ofthe cell cycle in which they act and are then degraded
Cyclins have diverse sequences, but functionallyimportant regions called cyclin boxes show high levelsof homology. This homology has facilitated domainidentification in other proteins such as transcrip-tion factors. Cyclin boxes confer binding specificitytowards protein partners. They are found with other
replication DNA RNA RNAtranscription processing translation
Protein
degradation orturnover
post translationalmodification
Figure 8.2 From DNA to protein

CELL CYCLE 249
D box
Consensus sequenceRXXLXXIXD/N
Cyclin box
Helical bundle
450
Cyclin B
1
Figure 8.5 Organization of cyclins showing D box and cyclin box segments
characteristic motifs such as the cyclin destruction boxwith a minimal consensus sequence of Arg-Xaa-Xaa-Leu-Xaa-Xaa-Ile-Xaa-Asn/Asp (Figure 8.5).
The first ‘box’ structure was determined for atruncated construct of cyclin A representing the final200 residues of a 450 residue protein. The structurehas two domains each with a central core of fiveregular α helices that is preserved between cyclinsdespite low levels of sequence identity. Five charged,invariant, residues (Lys266, Glu295, Leu255, Asp240,and Arg21) form a binding site for protein partnerssuch as cyclin-dependent kinases.
Cyclins regulate the activity of cyclin-dependentkinases (Cdks) by promoting phosphorylation of asingle Thr side chain. Cdks were originally identifiedvia genetic analysis of yeast cell cycles and purificationof extracts stimulating the mitotic phase of frog andmarine invertebrate oocytes. However, Cdks are foundin all eukaryotic cell cycles where they control themajor transitions of the cell cycle. Studies of the cellcycle were aided by the identification of mutant strainsof fission yeast (Schizosaccharomyces pombe) and
budding yeast (Saccharomyces cerevisiae) containinggenetic defects (called lesions) within key controllinggenes. In S. cerevisiae Leland Hartwell identified manygenes regulating the cell cycle and coined the term‘cell division cycle’ or cdc gene (Figure 8.6). Onegene, cdc28, controlled the important first step of thecell cycle – the ‘start’ gene. Cells normally go througha number of ‘checkpoints’ to ensure cell divisionproceeded correctly; at each of these points mutantswere identified in cell cycle genes. These studiesallowed a picture of the cell cycle in terms of genes andcells would not advance further in the cycle until the‘block’ was overcome by repairing defects. Cancerouscells turn out to be cells that manage to evade these‘checkpoints’ and divide uncontrollably.
Using the fission yeast, S. pombe, Paul Nurseidentified a gene (cdc2 ) whose product controlledmany reactions in the cell cycle and was identified byhomology to cyclin-dependent kinase 1 (Cdk1) foundin human cells. The human gene could substitute for theyeast gene in Cdk− mutants. The genes cdc2 and cdc28from S. pombe and S. cerevisiae encode homologous
Figure 8.6 The role of mutants and the identification of key genes in the cell cycle of yeast. The genes cdc28,cdc24, cdc31 and cdc7 were subsequently shown to code for cdk2, a guanine nucleotide exchange factor, centrin – aCa-binding protein, and another cyclin-dependent kinase

250 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
cyclin-dependent protein kinases occupying the samepivotal role within the cell cycle.
Cdks are identified in all cells and retain activitywithin a single polypeptide chain (Mr ∼ 35 kDa).This subunit contains a catalytic core with sequencesimilarity to other protein kinases but requires cyclinbinding for activation together with the phosphorylationof a specific threonine residue. Molecular details ofthis activity were uncovered by crystallographic stud-ies of Cdks, cyclins and the complex formed betweenthe two protein partners. Cdks catalyse phosphorylation(and hence activation) of many proteins at differentstages in the cell cycle: including histones, as partof chromosomal DNA unpackaging; lamins, proteinsforming the nuclear envelope that must disintegrateprior to mitosis; oncoproteins that are often transcrip-tion factors; and proteins involved in mitotic spindleformation. Cdks control reactions occurring within thecell by the combined action of a succession of phos-phorylated Cdk–cyclin complexes that elicit activationand deactivation of enzymes. It is desirable that thesesystems represent an ‘all or nothing’ signal in whichthe cell is compelled to proceed in one, irreversible,direction. The reasons for this are fairly obvious inthat once cell division has commenced it would beextremely hazardous to attempt to reverse the proce-dure. Cdks trigger cell cycle events but also enhanceactivity of the next cyclin–Cdk complex with a cascadeof cyclin–Cdk complexes operating during the G1/S/G2
phases in yeast and human cells (Figure 8.7).
The structure of Cdk and its role inthe cell cycle
Understanding the structure of Cdks uncovered themechanism of action of these enzymes with the firststructure obtained in 1993 for Cdk2 in a complexwith ATP (Figure 8.8). A bilobal protein comprisinga small lobe at the N terminal together with a muchlarger C-domain resembled other protein kinases, suchas cAMP-dependent protein kinase. The comparativelysmall size reflected a ‘minimal’ enzyme. Within thissmall structure two regions of Cdk2 stood out as‘different’ from other kinases. A single helical segment,called the PSTAIRE helix from the sequence ofconserved residues Pro-Ser-Thr-Ala-Ile-Arg-Glu-, was
G1 G2S M
G1 S M
H.sapiens
CycDCdk4/6 CycE
CycA
Cib3, 4Cib5, 6
Cln 1,2
Cdk2
Cib1,2Cic28
CycB
S.cerevisiae
Figure 8.7 Major Cdk-cyclin complexes involved incell cycle control in humans and budding yeast. Purplearrows indicate approximate timing of activation andduration of the Cdk–cyclin complexes. Cyclins areshown as coloured rectangles with the cognate Cdkshown by circles. The S and M phases overlap in S.cerevisiae
located in the N-terminal lobe and was unique to Cdks.The second distinctive region contained the site ofphosphorylation (Thr160) in Cdk and was comparableto other regulatory elements seen in protein kinases.This region was called the T or activation loop.
The smaller N-terminal lobe has the PSTAIRE helixand a five stranded β sheet whilst the larger lobe hassix α helices and a small section of two-stranded β
sheet. ATP binding occurs in the cleft between lobeswith the adenine ring located between the β sheet ofthe small lobe and the L7 loop between strands 2 and5. A glycine-rich region interacts with the adenine ring,whilst Lys33, Asp145, and amides in the backbone ofthe glycine-rich loop interact with phosphate groups(Figure 8.9).
The basic fold of Cdk2 contains the PSTAIRE helixand T loop positioned close to the catalytic cleft, but isan inactive form of the enzyme. Activation by a factorof ∼10 000 occurs upon cyclin binding and is generallyassessed via the ability of Cdk2 to phosphorylateprotein substrates such as histone. Further enhancementof activity occurs with phosphorylation of Thr160 inCdk2 and suggested that conformational changes mightmodulate biological activity. The origin and extentof these conformational changes were determined bycomparing the structures of Cdk2–cyclin A complexesin phosphorylated (fully active) and unphosphorylated

THE STRUCTURE OF CDK AND ITS ROLE IN THE CELL CYCLE 251
Figure 8.8 Structure of isolated Cdk2 and its interaction with Mg2+-ATP. The ATP is shown in magenta, the Mg2+ion in light green. The secondary structure elements are shown in blue and green with the PSTAIRE helix shown inyellow and the T loop in red. The L12 helix can be seen adjacent to the PSTAIRE helix in the active site of Cdk2
Asp 145
Asn 132
ATP
Mg2+
Figure 8.9 Binding of Mg2+-ATP to monomeric Cdk2.Magnesium bound in an octahedral environment withsix ligands provided by phosphates of ATP, Asp145,Asn132 and a bound water molecule
(partially active) states together with the isolated(inactive) Cdk2 subunit.
In the Cdk2–cyclin A complex conformationalchanges were absent in cyclin A with structural
perturbations confined to Cdk2. The binding sitebetween cyclin A and Cdk2 involved the cyclin Abox containing Lys266, Glu295, Leu255, Asp240, andArg211 and a binding site located close to the catalyticcleft. In proximity to the cleft hydrogen bonds withGlu8, Lys9, Asp36, Asp38, Glu40 and Glu42 wereimportant to complex formation as well as hydropho-bic interactions that underpinned movement of the‘PSTAIRE’ helix and T loop. Large conformationalchanges upon cyclin binding occur for the PSTAIREhelix and the T loop with the helix rotated by ∼90◦
and the loop region displaced. In the absence of cyclinthe T loop is positioned above the catalytic cleftblocking the active site ATP to polypeptide substrates.Cyclin binding moves the T loop opening the cat-alytic cleft and exposing the phosphorylation site atThr160. Figures 8.10 and 8.11 show the structure ofthe Cdk2–cyclin A–ATP complex.
Threonine phosphorylation in the T loop is per-formed by a second kinase called CAK (Cdk-activatingkinase), and in an activated state the complex phos-phorylates downstream proteins modifying serine orthreonine residues located within target (S/T-P-X-K/R)sequences.

252 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
Figure 8.10 Structure of the Cdk2–cyclin A–ATPcomplex. The structure shows the binding of cyclinA (purple, left) to the Cdk2 protein (green, right). Thecolour scheme of Figure 8.8 is used with the exceptionthat the β-9 strand of Cdk (formerly the L12 helix) isshown in orange
Figure 8.11 Space-filling representation of thecyclinA (purple) and Cdk2 (green) interaction.Phosphorylated Thr160 (red) of Cdk2 in the T looptogether with ATP (magenta) are shown
Cdk–cyclin complex regulationCyclin binding offers one level of regulation of Cdkactivity but other layers of regulatory control existwithin the cell. Phosphorylation of Cdk at Thr160by CAK is a major route of activation but furtherphosphorylation sites found on the surface of Cdklead to enzyme inhibition. In Cdk1 the most importantsites are Thr14 and Tyr15. Further complexity ariseswith the observation of specialized Cdk inhibitorsthat regulate most kinase activity in the form oftwo families of inhibitory proteins: the INK4 familybind to free Cdk preventing association with cyclinswhilst the CIP family act on Cdk–cyclin complexes(Figure 8.12). The INK4 inhibitors are specific for theG1 phase Cdks whereas CIP inhibitors show a broaderpreference. Regulation has become an important issuewith the observation that certain tumour lines showaltered patterns of Cdk regulation as a result ofinhibitor mutation. One inhibitor of CDK4 and CDK6is p16INK4a, and in about one third of all cancercases this tumour suppressor is mutated. Another CDKinhibitor, p27Cip2, is present at low levels in cancersthat show poor clinical prognosis whilst mutation ofCdk4 in melanoma cells leads to a loss of inhibitionby Ink4 proteins.
Cdks are protein kinases transferring the γ-phosphateof ATP onto Ser/Thr side chains in target proteins. Thistype of phosphorylation is a common regulatory event
Cdk
inactive
CAK
Cycin
Cdk-Cyclin
Cdk~P-Cyclin
Cipinhibitors
INK4inhibitors
Partially active
fully active
Figure 8.12 The interaction of cyclin–Cdk complexeswith INK4 and CIP inhibitors

DNA REPLICATION 253
within cells and underpins signal transduction mediatedby cytokines, kinases and growth factors as well as theevents within the cell cycle.
In yeast the cell cycle is controlled by one essentialkinase known as Cdk1 whilst multicellular eukaryoteshave Cdk1 and Cdk2 operating in the M phaseand S phase, respectively, along with two additionalCdks (Cdk4 and Cdk6) regulating G1 entry and exit.Despite the presence of additional Cdks in highereukaryotes the molecular mechanisms controlling cellcycle events show evolutionary conservation operatingsimilarly in yeasts, insects, plants, and vertebrates,including humans. Research aimed at understandingthe cell cycle underpins the development of cancersince the failure to control these processes accuratelyis a major molecular event during carcinogenesis. Itis likely that discoveries in this area will have anenormous impact on molecular medicine. Knowledgeof the structure of Cdks together with the structuresof Cdk–cyclin–inhibitor complexes will be translatedinto the design of drugs that modulate Cdk activity andeliminate unwanted proliferative reactions.
DNA replication
A major function of the cell cycle is to integrateDNA synthesis within cell division. The doublehelix structure of DNA suggested conceptually simplemethods of replication; a conservative model where anew double-stranded helix is synthesized whilst theoriginal duplex is preserved, or a semi-conservativescheme in which the duplex unfolds and each strandforms half of a new double-stranded helix.
The semi-conservative’ model (Figure 8.13) wasdemonstrated conclusively by Matthew Meselson andFranklin Stahl in 1958 by growing bacteria (E. coli ) onmedia containing the heavy isotope of nitrogen (15N)for several generations to show that the DNA was mea-surably heavier than that from cells grown on the nor-mal 14N isotope. Small differences in density betweenthese two forms of DNA were demonstrated exper-imentally by density gradient centrifugation. IsolatedDNA from bacteria grown on 15N for many genera-tions gives a single band corresponding to a density of1.724 g ml−1 whilst DNA from bacteria grown on nor-mal (14N) nitrogenous sources yielded a lighter density
15N DNA (heavy)
14N DNA(light)
Hybrid DNA1 Generation +
++
+
+2 Generations
3 Generations
4 Generations
34
78
14
+ 18
Figure 8.13 Semi-conservative model of DNA repli-cation (reproduced with permission from Voet, D Voet,J.G & Pratt, C.W. Fundamentals of Biochemistry. JohnWiley & Sons Inc, Chichester, 1999)
single band of 1.710 g ml−1. Comparing DNA isolatedfrom bacteria grown on heavy (15N) nitrogenous mediaand then transferred to 14N-containing media for a sin-gle generation led to the observation of a single DNAspecies of intermediate density (1.717 g ml−1). Thiswas only consistent with the semi-conservative model.
The semi-conservative mechanism suggested thateach strand of DNA acted as a template for a new chain.A surprisingly large number of enzymes are involvedin these reactions including:
1. Helicases. These proteins bind double-strandedDNA catalysing strand separation prior to synthesisof new daughter strands.
2. Single-stranded binding proteins. Tetrameric pro-teins bind DNA stabilizing single-stranded structureand enhancing rates of replication.
3. Topoisomerases. This class, sometimes called DNAgyrases, assist unwinding before new DNA synthesis.
4. Polymerases. DNA Polymerase I was the firstenzyme discovered with polymerase activity. It

254 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
is not the primary enzyme involved in bacterialDNA replication, a reaction catalysed by DNApolymerase III. DNA polymerase I has exonucleaseactivities useful in correcting mistakes or repairingdefective DNA. DNA polymerase can be isolatedas a fragment exhibiting the 5′ –3′ polymerase andthe 3′ –5′ exonuclease activity but lacking the 5′ –3′exonuclease of the parent molecule. This fragment,known as the Klenow fragment, was widely usedin molecular biology prior to the discovery ofthermostable polymerases.
5. Primase. DNA synthesis proceeds by the formationof short RNA primers that lead to sections ofDNA known as Okazaki fragments. The initialpriming reaction requires a free 3′ hydroxyl groupto allow continued synthesis from these primers.Specific enzymes known as RNA primases catalysethis process.
6. Ligase. Nicks or breaks in DNA strands occurcontinuously during replication and these gaps arejoined by the action of DNA ligases.
The synthesis of RNA primers in E. coli requiresthe concerted action of helicases (DnaB ) and primases(DnaG) in the primosome complex (see Table 8.1;Mr ∼ 600 000) whilst a second complex, the replisome,contains two DNA polymerase III enzymes engagedin DNA synthesis in a 5′ –3′ direction. Figure 8.14summarizes DNA replication in E. coli.
Table 8.1 Primosome proteins
Protein Organization Subunit mass
PriA Monomer 76PriB Dimer 11.5PriC Monomer 23DnaT Trimer 22DnaB Hexamer 50DnaC Monomer 29DnaG (primase) Monomer 60
The complex lacking DnaG is called a pre-primosome. Derived fromKornberg, A. & Baker, T.A. DNA Replication, 2nd edn. Freeman,New York, 1992.
Transcription
The first step in the flow of information from DNAto protein is transcription. Transcription forms comple-mentary messenger RNA (mRNA) from DNA throughcatalysis by RNA polymerase. RNA polymerase copiesthe DNA coding strand adding the base uracil (U) inplace of thymine (T) into mRNA. Most studies of tran-scription focused on prokaryotes where the absence ofa nucleus allows RNA and protein synthesis to occurrapidly, in quick succession and catalysed by a sin-gle RNA polymerase. In eukaryotes multiple RNApolymerases exist and transcription is more complexinvolving larger numbers of accessory proteins.
Structure of RNA polymerase
Most prokaryotic RNA polymerases have five subunits(α, β, β′, σ and ω) with two copies of the α subunitfound together with single copies of the remainingsubunits (Table 8.2). From reconstitution studies the ω
subunit is not required for holoenzyme assembly orfunction. Similarly the σ subunit is easily dissociatedto leave a ‘core’ enzyme retaining catalytic activity. Adecrease in DNA binding identified a role for the σ
subunit in promoter recognition where it reduces non-specific binding by a factor of ∼104.
Table 8.2 Subunit composition of RNA polymerase ofE. coli; the σ subunit is one member of a group ofalternative subunits that have interchangeable roles
Subunit Mr Stoichiometry Function
α 36 500 2 Chain initiation.Interaction withtranscription factorsand upstreampromoter elements
β 151 000 1 Chain initiation andelongation
β′ 155 000 1 DNA bindingσ 70 000 1 Promoter recognitionω 11 000 1 Unknown
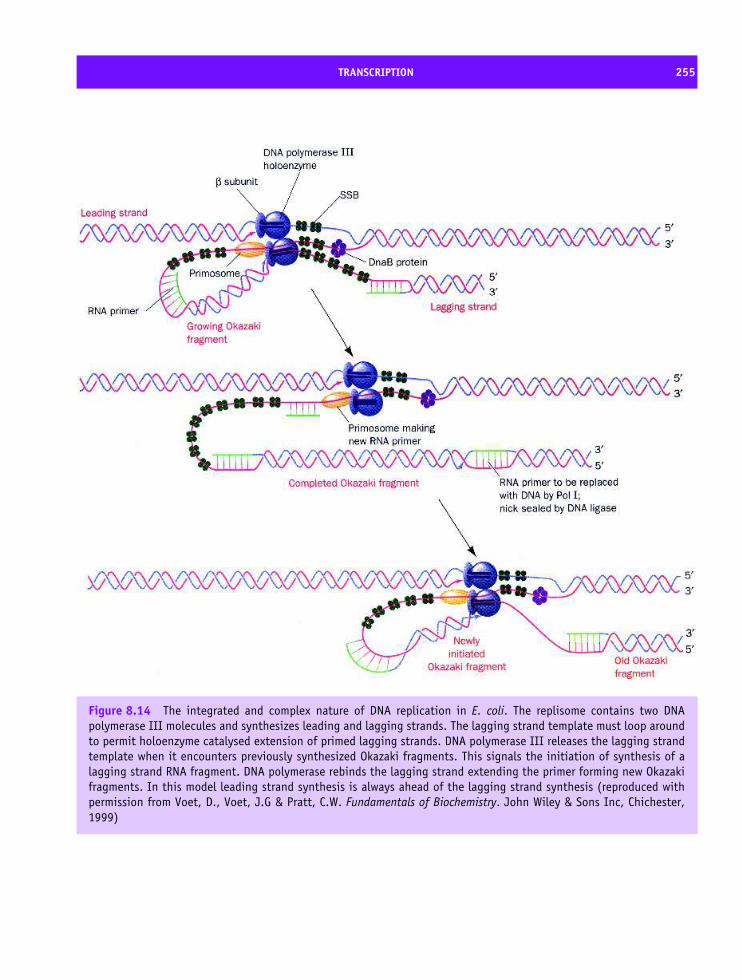
TRANSCRIPTION 255
Figure 8.14 The integrated and complex nature of DNA replication in E. coli. The replisome contains two DNApolymerase III molecules and synthesizes leading and lagging strands. The lagging strand template must loop aroundto permit holoenzyme catalysed extension of primed lagging strands. DNA polymerase III releases the lagging strandtemplate when it encounters previously synthesized Okazaki fragments. This signals the initiation of synthesis of alagging strand RNA fragment. DNA polymerase rebinds the lagging strand extending the primer forming new Okazakifragments. In this model leading strand synthesis is always ahead of the lagging strand synthesis (reproduced withpermission from Voet, D., Voet, J.G & Pratt, C.W. Fundamentals of Biochemistry. John Wiley & Sons Inc, Chichester,1999)

256 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
One of the best-characterized polymerases is frombacteriophage T7 (Figure 8.15) and departs fromthis organization by containing a single polypeptidechain (883 residues) arranged as several domains.It is organized around a cleft that is sufficient toaccommodate the template of double-stranded DNAand has been likened to the open right hand dividedinto palm, thumb and finger regions. The palm, fingersand thumb regions define the substrate (DNA) bindingand catalytic sites.
The thumb domain is a helical extension from thepalm on one side of the binding cleft and stabilizesthe ternary complex formed during transcription bywrapping around template DNA. Mutant T7 RNApolymerases with ‘shortened thumbs’ have less activ-ity as a result of lower template affinity. The palmdomain consisting of residues 412–565 and 785–883
Figure 8.15 The structure of T7 RNA polymerase. Aspace-filling representation with synthetic DNA (shownin orange and green) has a thumb region representedby residues 325–411 (red), a palm region underneaththe DNA and largely obscured by other domains(purple) whilst fingers surround the DNA and are shownin cyan to the right. The N-terminal domain is shownin dark blue (PDB: 1CEZ)
is located at the base of the deep cleft, an inte-gral part of T7 RNA polymerase, and is surroundedby the fingers and the thumb. It contains three β
strands, a structurally conserved motif in all poly-merases with conserved Asp residues (537 and 812)participating directly in catalysis. The finger domainextends from residues 566–784 and contains a speci-ficity loop involved in direct interactions with specificbases located in the major groove of the promoter DNAsequence. This region also has residues that form partof the active site such as Tyr639 and Lys631. Finallythe N-terminal domain forms the front wall of thecatalytic cleft and leads to the enzyme’s character-istic concave appearance. This domain interacts withupstream regions of promoter DNA and the nascentRNA transcript.
Consensus sequences exist for DNA promoterregions (Figure 8.16) and David Pribnow identifieda region rich in the bases A and T approximately−10 bp upstream of the transcriptional start site.A second region approximately 35 bp upstream ofthe transcriptional start site was subsequently iden-tified with the space between these two sites hav-ing an optimal size of 16 or 17 bases in length.Although consensus sequences for the −35 and −10regions have been identified no natural promoter pos-sesses exactly these bases although all show strongsequence conservation.
Figure 8.16 The organization of the promoter regionof genes in E. coli for recognition by RNA polymerase.The consensus sequence TATAAT at a position ∼10 bpupstream of the transcription start site is thePribnow box

TRANSCRIPTION 257
Eukaryotic RNA polymerases
In eukaryotes polymerase binding occurs at the TATAbox and is more complex. Three nuclear RNApolymerases designated as RNA polymerase I, IIand III, together with polymerases located in themitochondria and chloroplasts are known. The nuclearpolymerases are multi-subunit proteins whose activitiesfrequently require the presence of accessory proteinscalled transcription factors (Table 8.3).
In eukaryotes RNA polymerases I and III tran-scribe genes coding for ribosomal (rRNA) and transferRNA (tRNA) precursors. RNA polymerase I producesa large transcript – the 45S pre-rRNA – consisting oftandemly arranged genes that after synthesis in thenucleolus are processed to give 18S, 5.8S and 28SrRNA molecules (Figure 8.17). The 18S rRNA con-tributes to the structure of the small ribosomal subunitwith the 28S and 5.8S rRNAs associating with pro-teins to form the 60S subunit. RNA polymerase III
Table 8.3 The role of eukaryotic RNA polymerases andtheir location
RNA
Polymerase
Location Role
I Nuclear
(nucleolus)
Pre rRNA except 5S
rRNA
II Nuclear Pre mRNA and some
small nuclear
RNAs
III Nuclear Pre tRNA, 5S RNA
and other small
RNAs
Mitochondrial Organelle
(matrix)
Mitochondrial RNA
Chloroplast Organelle
(stroma)
Chloroplast RNA
Figure 8.17 The genes for rRNAs exist as tandemly repeated copies separated by non-transcribed regions. The 45Stranscript is processed to remove the regions shown in tan to give the three rRNAs

258 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
Table 8.4 Basal transcription factors required by eukaryotic RNA polymerase II
Factor Subunits Mass(kDa)
Function
TFIID - TBP 1 38 TATA box recognition and TFIIB recruitment.TAFs 12 15–250 Core promoter recognitionTFIIA 3 12,19,35 Stabilization of TBP bindingTFIIB 1 35 Start site selection.TFIIE 2 34,57 Recruitment and modulation of TFIIH activityTFIIF 2 30,74 Promotor targeting of polymerase and destabilization of non
specific RNA polymerase-DNA interactionsTFIIH 9 35–90 Promoter melting via helicase activity
Adapted from Roeder, R.G. Trends Biochem. Sci. 1996, 21, 327–335. Subunit composition and mass are those of human cells but homologuesare known for rat, Drosophila and yeast. TBP, TATA binding protein; TAFs, TATA binding associated factors.
synthesizes the precursor of the 5S rRNA, the tRNAsas well as a variety of other small nuclear and cytoso-lic RNAs.
RNA polymerase II transcribes DNA into mRNAand is the enzyme responsible for structural gene tran-scription. However, effective catalysis requires addi-tional proteins with at least six basal factors involved inthe formation of a ‘pre-initiation’ complex with RNApolymerase II (Table 8.4). These transcription factorsare called TFIIA, TFIIB, TFIID, TFIIE, TFIIF andTFIIH. The Roman numeral ‘II’ indicates their involve-ment in reactions catalysed by RNA polymerase II.
The assembly of the pre-initiation complex beginswith TFIID (TBP) identifying the TATA consen-sus sequence (T-A-T-A-A/T-A-A/T) and is followedby the coordinated accretion of TFIIB, the non-phosphorylated form of RNA polymerase II, TFIIF,TFIIE, and TFIIH (Figure 8.18). Before elongationcommences RNA polymerase is phosphorylated andremains in this state until termination when a phos-phatase recycles the polymerase back to its initial form.The enzyme can then rebind TATA sequences allowingfurther transcriptional initiation.
Basal levels of transcription are achieved with TBP,TFIIB, TFIIF, TFIIE, TFIIH, RNA polymerase and thecore promoter sequence, and this system has been used
to demonstrate minimal requirements for initiation andcomplex assembly (Figure 8.19). A cycle of efficientre-initiation of transcription is achieved when RNApolymerase II re-enters the pre-initiation complexbefore TFIID dissociates from the core promoter. Non-basal rates of transcription require proximal and distalenhancer regions of the promoter and proteins thatregulate polymerase efficiency (TAFs).
TBP (and TFIID) binding to the TATA box isa slow step but yields a stable protein–DNA com-plex that has been structurally characterized from sev-eral systems. The plant, yeast, and human TBP incomplexes with TATA element DNA share similarstructures (Figure 8.20) suggesting conserved mecha-nisms of molecular recognition during transcription.The three-dimensional structure of the conserved por-tion of TBP is strikingly similar to a saddle; an obser-vation that correlates precisely with function whereprotein ‘sits’ on DNA creating a stable binding plat-form for other transcription factors. DNA bindingoccurs on the concave underside of the saddle with theupper surface (seat of the saddle) binding transcrip-tional components.
DNA binding is mediated by the curved, antiparallelβ sheet whose distinctive topology provides a largeconcave surface for interaction with the minor DNA

TRANSCRIPTION 259
Figure 8.18 The formation of pre-initiation complexes between RNA polymerase II plus TATA sequence DNA
groove of the 8-bp TATA element. The 5′ end of DNAenters the underside of the molecular saddle whereTBP causes an abrupt transition from the normal Bform configuration to a partially unwound form bythe insertion of two Phe residues between the firstT:A base step. The widened minor groove adoptsa conformation comparable to the underside of themolecular saddle allowing direct interactions betweenprotein side chains and the minor groove edges ofthe central 6 bp. A second distortion induced byinsertion of another two Phe residues into a regionbetween the last two base pairs of the TATA elementmediates an equally abrupt return back to the normalB-form DNA.
TFIIB is the next protein to enter the pre-initiationcomplex and its interaction with TBP was sufficientlystrong to allow the structure of TFIIB-TBP-TATAcomplexes to be determined by Ronald Roeder andStephen Burley in 1995 (Figure 8.21). A core regionbased around the C-terminal part of the molecule(cTFIIB) shows homology to cyclin A and binds toan acidic C-terminal ‘stirrup’ of TBP using a basiccleft on its own surface. The protein also interactswith the DNA sugar-phosphate backbone upstreamand downstream of the TATA element. The firstdomain of cTFIIB forms the downstream surfaceof the cTFIIB–TBP–DNA ternary complex and inconjunction with the N-terminal domain of TFIIB this
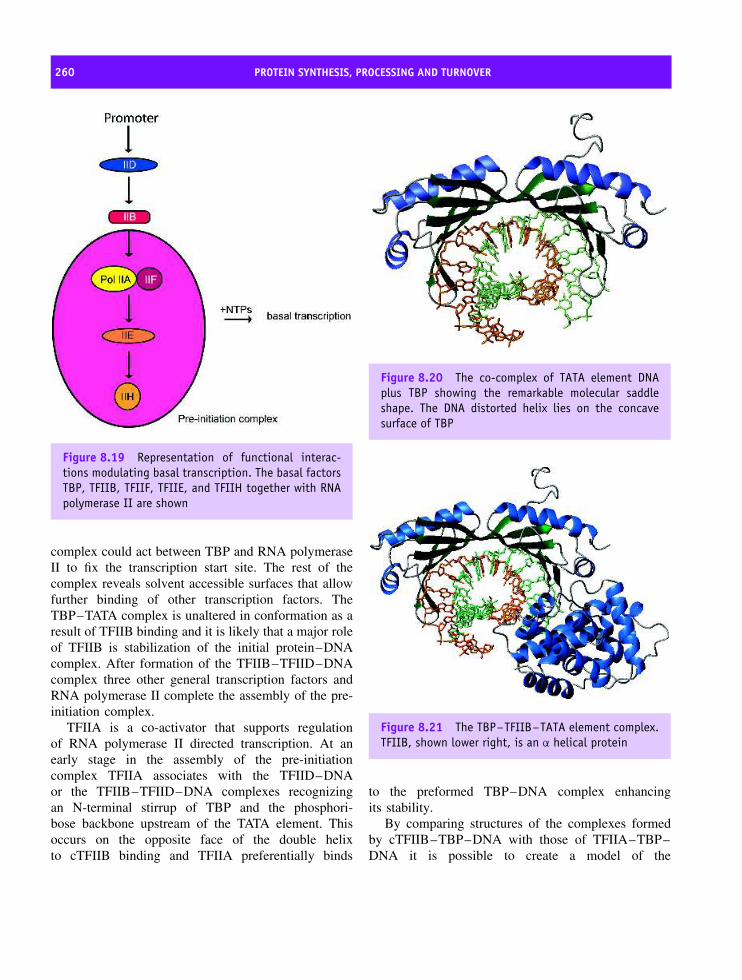
260 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
Figure 8.19 Representation of functional interac-tions modulating basal transcription. The basal factorsTBP, TFIIB, TFIIF, TFIIE, and TFIIH together with RNApolymerase II are shown
complex could act between TBP and RNA polymeraseII to fix the transcription start site. The rest of thecomplex reveals solvent accessible surfaces that allowfurther binding of other transcription factors. TheTBP–TATA complex is unaltered in conformation as aresult of TFIIB binding and it is likely that a major roleof TFIIB is stabilization of the initial protein–DNAcomplex. After formation of the TFIIB–TFIID–DNAcomplex three other general transcription factors andRNA polymerase II complete the assembly of the pre-initiation complex.
TFIIA is a co-activator that supports regulationof RNA polymerase II directed transcription. At anearly stage in the assembly of the pre-initiationcomplex TFIIA associates with the TFIID–DNAor the TFIIB–TFIID–DNA complexes recognizingan N-terminal stirrup of TBP and the phosphori-bose backbone upstream of the TATA element. Thisoccurs on the opposite face of the double helixto cTFIIB binding and TFIIA preferentially binds
Figure 8.20 The co-complex of TATA element DNAplus TBP showing the remarkable molecular saddleshape. The DNA distorted helix lies on the concavesurface of TBP
Figure 8.21 The TBP–TFIIB–TATA element complex.TFIIB, shown lower right, is an α helical protein
to the preformed TBP–DNA complex enhancingits stability.
By comparing structures of the complexes formedby cTFIIB–TBP–DNA with those of TFIIA–TBP–DNA it is possible to create a model of the

EUKARYOTIC TRANSCRIPTION FACTORS: VARIATION ON A ‘BASIC’ THEME 261
TFIIA–TFIIB–TBP–DNA quaternary complex wherethe mechanism of synergistic action between TFIIBand TFIIA is rationalized by the observation that thetwo proteins do not interact directly but instead usetheir basic surfaces to make contact with the phosphori-bose backbone on opposite surfaces of the double helixupstream of the TATA element.
Eukaryotic RNA polymerases interact with a num-ber of transcription factors before binding to the pro-moter and transcription commences. Thus although thefundamental principles of transcription are similar inprokaryotes and eukaryotes the latter process is char-acterized by the presence of multiple RNA polymerasesand additional control processes modulated by a varietyof transcription factors.
Structurally characterizedtranscription factors
The prokaryotic transcription factors cro and λ areexamples of proteins containing helix-turn-helix (HTH)motifs that function as DNA binding proteins. Thebasis for highly specific interactions relies on preciseDNA–protein interactions that involve side chainsfound in the recognition helix of HTH motifs andthe bases and sugar-phosphate regions of DNA. Asubstantial departure in the use of the HTH motif tobind DNA is seen in the met repressor of E. coli.A homodimer binds to the major groove via a pairof symmetrically related β strands, one from eachmonomer, that form an antiparallel sheet composedof just two strands. Other repressor proteins such asthe arc repressor from a Salmonella bacteriophage(P22) were subsequently shown to use this DNArecognition motif.
Eukaryotic transcription factors:variation on a ‘basic’ theme
Eukaryotic cells require a greater range of transcrip-tion factors, and because genes are often selectivelyexpressed in a tissue-specific manner these processesrequire an efficient regulatory mechanism. This isachieved in part through a diverse class of proteins.Included in this large and structurally diverse group of
proteins are Zn finger DNA binding motifs, leucinezippers, helix loop helix (HLH) motifs (reminiscentof the HTH domains of prokaryotes) and proteinscontaining a β scaffold for DNA binding. Eukary-otic transcription factors contain distinct domains eachwith a specific function; an activation domain bindsRNA polymerase or other transcription factors whilsta DNA binding domain recognizes specific bases nearthe transcription start site. There will also be nuclearlocalization domains that direct the protein towardsthe nucleus after synthesis in the cytosol. The follow-ing sections describe briefly the structural propertiesof some of the important DNA binding motifs foundin eukaryotes.
Zn finger DNA binding motifs
The first Zn finger domain characterized was transcrip-tion factor IIIA (TFIIIA) from Xenopus laevis oocytes(Figure 8.22). Complexed with 5S RNA was a pro-tein of Mr 40 000 required for transcription in vitro thatyielded a series of similar sized fragments (Mr ∼ 3000)
containing repetitive primary sequence after limitedproteolysis. Sequence analysis confirmed nine repeatunits of ∼25 residues each containing two cysteine andtwo histidine residues. Each domain contained a singleZn2+ ion.
The proteolytic fragments were folded in the pres-ence of Zn which was ligated in a tetrahedral geom-etry to the side chains of two invariant Cys and twoinvariant His residues. In subsequently characterizeddomains it was found that the Cys2His2 ligation pat-tern varied with sometimes four Cys residues used tobind a single Zn2+ ion (Cys4) or 6 Cys residues used tobind two Zn2+ ions. The Zn-binding domains formedsmall, compact, autonomously folding structures lack-ing extensive hydrophobic cores, and were christenedZn fingers.
The structures of simple Zn finger domains (Figure8.23) reveal two Cys ligands located in a short strandor turn region followed by a regular α helix containingtwo His ligands. From the large number of repeatingZn finger domains and their intrinsic structures itis clear that the ends of the polypeptide chain arewidely separated and that complexes with DNA mightinvolve multiple Zn finger domains wrapping aroundthe double helix. This arrangement was confirmed

262 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
* 20 * 40 * 60 *TFIIIA : MAAKVASTSSEEAEGSLVTEGEMGEKALPVVYKRYICSFADCGAAYNKNWKLQAHLCKHTGEKPFPCKEEGCEKG
80 * 100 * 120 * 140 *TFIIIA : FTSLHHLTRHSLTHTGEKNFTCDSDGCDLRFTTKANMKKHFNRFHNIKICVYVCHFENCGKAFKKHNQLKVHQFS
160 * 180 * 200 * 220TFIIIA : HTQQLPYECPHEGCDKRFSLPSRLKRHEKVHAGYPCKKDDSCSFVGKTWTLYLKHVAECHQDLAVCDVCNRKFRH
* 240 * 260 * 280 * 300TFIIIA : KDYLRDHQKTHEKERTVYLCPRDGCDRSYTTAFNLRSHIQSFHEEQRPFVCEHAGCGKCFAMKKSLERHSVVHDP
* 320 * 340 * 360TFIIIA : EKRKLKEKCPRPKRSLASRLTGYIPPKSKEKNASVSGTEKTDSLVKNKPSGTETNGSLVLDKLTIQ
Figure 8.22 The primary sequence of transcription factor TFIIIA from Xenopus laevi oocytes. The regions highlightedin yellow are units of 25 residues containing invariant Cys and His residues. Zn binding domains have a characteristicmotif CX(2–3)CX(12)HX(3–4)H
Figure 8.23 A Zn finger domain with helix and eitheran antiparallel β strand or a loop region
by crystallographic studies of the mouse transcriptionfactor Zif268 where three repeated Zn finger domainscomplex with DNA by wrapping around the doublehelix with the β strands of each finger positioning theα helix in the major groove.
Zn finger domains are widely distributed throughoutall eukaryotic cells and in the human genome there maybe over 500 genes encoding Zn finger domains. Zif268,shown in Figure 8.24, has three such repeats whilethe homologous Drosophila developmental controlproteins known as Hunchback and Kruppel have fourand five Zn finger domains, respectively. TFIIIA has a
Figure 8.24 The structure of the Zn finger domainsof mouse transcription factor Zif268 in a complex withDNA (PDB:1AIH). Three base pairs form hydrogen bondswith the N terminal region of the helix in one strand ofthe double helix. The periodicity of positively chargedside chains allows interactions with phosphate groups
comparatively large number of domains (nine) althoughincreasingly proteins are being found with much greaternumbers of ‘fingers’. Binuclear Zn clusters exist asCys6 Zn fingers. In GAL4, a yeast transcriptionalactivator of galactose (Figure 8.25), an N terminal

EUKARYOTIC TRANSCRIPTION FACTORS: VARIATION ON A ‘BASIC’ THEME 263
ZnCysCys
Cys
Cys ZnCys
Cys
Figure 8.25 The GAL4 DNA binding domain. Tetrahe-dral geometry formed by six cysteine side chains bindstwo zinc ions
region containing six Cys residues coordinates two Znions. Each Zn cation binds to four Cys side chains in atetrahedral environment with the central two cysteinesligating both Zn ions.
Leucine zippersMany transcription factors contain sequences withleucine residues occurring every seventh position. Thisshould strike a note of familiarity because coiledcoils such as keratin have similar arrangements calledheptad repeats. The heptad repeat is a unit of sevenresidues represented as (a-b-c-d-e-f -g)n in whichresidues a and d are frequently hydrophobic. Thesequences promote the formation of ‘coiled coils’ bythe interaction of residues ‘a’ and ‘d’ in neighbouringhelices. The observation that many DNA bindingproteins contained heptad repeats suggested that theseregions assist in dimerization – a view supported bythe structure of the ‘leucine zipper’ region of theyeast transcription factor GCN4 (Figure 8.26). Thehydrophobic regions do not bind DNA but insteadpromote the association of subunits containing DNAbinding motifs into suitable dimeric structures.
Close inspection of the interaction between helicesreveals that the term zipper is inappropriate. The sidechains do not interdigitate but show a simpler ‘rungsalong a ladder’ arrangement with stability enhanced byhydrophobic interactions between residues at positions‘a’ and ‘d’. Although misleading in terms of organi-zational structure the name ‘leucine zipper’ remainswidely used in the literature. Dimerization of theleucine zipper domains allows the N-terminal regionsof GCN4, rich in basic residues, to lie in the majorgroove of DNA. The combination of basic and zipperregions yields a class of proteins with bZIP domains.
Helix-loop-helix motifsThe HLH motif is similar in both name and function tothe HTH motif seen in prokaryotic repressors such ascro. The HLH motif is frequently found in transcrip-tion factors along with leucine zipper and basic (DNAbinding) motifs (see Figure 8.27). As the name impliesthe structure consists of two α helices often arranged assegments of different lengths – one short and the otherlong – linked by a flexible loop of between 12 and 28residues that allows the helices to fold back and packagainst each other. The effect of folding is that eachhelix lies in parallel plane to the other and is amphi-pathic, presenting a hydrophobic surface of residues onone side with a series of charged residues on the other.
Eukaryotic HTH motifsThe fruit fly Drosophila melanogaster is a popularexperimental tool for studying gene expression and reg-ulation via changes in developmental patterns. Devel-opmental genes control the pattern of structural geneexpression and many Drosophila genes share com-mon sequences known as homeodomains or home-oboxes (Figure 8.28). These sequences occur in otheranimal genomes with an increase in the number ofHox genes from one cluster in nematodes, two clus-ters in Drosophila, with the human genome having39 homeotic genes organized into four clusters. TheDrosophila engrailed gene encodes a polypeptide thatbinds to DNA sequences upstream of the transcrip-tion start site and imposes its pattern of regulation onother genes as a transcription factor. Crystallization ofa 61-residue homeodomain in a complex with DNAshowed a HTH motif comparable to the HTH domainsof prokaryote repressors such as λ.

264 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
* 20 * 40 * 60Yeast_GCN4 : KPNSVVKKSHHVGKDDESRLDHLGVVAYNRKQRSIPLSPIVPESSDPAALKRARNTEAAR : 60
* 80 * 100Yeast_GCN4 : RSRARKLQRMKQLEDKVEELLSKNYHLENEVARLKKLVGER : 101
Figure 8.26 A leucine zipper motif such as that found in GCN4 shows a characteristic sequence known as the heptadrepeat where every seventh side chain is hydrophobic and is frequently a leucine residue. Interaction between twohelices forms a coiled coil and facilitates dimerization of the proteins. The structure of the leucine zipper region ofGCN4 complexed with DNA and a detailed view of the interaction between helices (derived from PDB:1YSA and 2TZA)
Figure 8.27 The structure determined for theMax–DNA complex. A basic domain (blue) at the Nterminal interacts with DNA. The first helix of the HLHmotif (orange) is followed by the loop region (red) andsecond helix (purple). The second helix flows into theleucine zipper (cyan) or dimerization region. This classof transcription factor are often called bHLHZ proteinreflecting the three different types of domains namelybasic, HLH and zipper (PDB: 1AN2)
Homeodomains are variations of HTH motifs con-taining three α helices, where the last two constitute aDNA binding domain. Two distinct regions make con-tact with TAAT sequences in complexes with cognateDNA. An N-terminal arm fits into the minor grooveof the double helix with the side chains of Arg3 andArg5 making contacts near the 5′ end of this ‘core con-sensus’ binding site. The second contact site involvesan α helix that fits into the major groove with the sidechains of Ile47 and Asn51 interacting with the basepairs near the 3′ end of the TAAT site. The ‘recognitionhelix’ is part of a structurally conserved HTH unit, butwhen compared with the structure of the λ repressorthe helices are longer and the relationship between theHTH unit and the DNA is significantly different.
It has been estimated that mammals contain over 200different cell types. These cell lines arise from highlyregulated and specific mechanisms of gene expressioninvolving the concerted action of many transcriptionfactors. The human genome project has identified thou-sands of transcription factors and many carry exoticfamily names such as Fos and Jun, nuclear factor-κB,

THE SPLICEOSOME AND ITS ROLE IN TRANSCRIPTION 265
Figure 8.28 A typical homeobox domain showingthe HTH motif in contact with its cognate DNA.The structure shown is that of a fragment of theubx or ultrabithorax gene product showing the HTHmotif. The recognition helix is shown in yellow lyingin the major groove (PDB: 1B8I). Similar structuresexist in homeodomain proteins such as antennapedia(antp) or engrailed (eng). All play a role in thedevelopmental programme of Drosophila and each ofthese proteins acts an a transcriptional activator,binding to upstream regulatory sites and interactingwith the RNA polymerase II/TFII complex
Pax and Hox. Growing importance is attached to thestudy of transcription factors with the recognition thatmutations occur in many cancer cells. Wilm’s tumour isone of the most common kidney tumours and is preva-lent in children as a result of mutation in the WT-1gene, a gene coding for a transcription factor contain-ing Zn finger motifs. p53 is another transcription factorand is found in a mutated state in over 50 percent ofall human cancers. These examples emphasize the linkbetween transcription factors and human disease, wherestructural characterization has elucidated the basis ofDNA–protein interaction.
The spliceosome and its role intranscriptionEukaryotic DNA contains introns or non-codingregions whose function remains enigmatic despite
widespread occurrence within chromosomal DNA. Ini-tial mRNA transcripts are processed to remove intronsin a series of reactions that occur on the spliceo-some. Spliceosomes are ribonucleoprotein complexeswith sedimentation coefficients between 50 and 60Sand a size and complexity comparable to the ribosome.
Before splicing many eukaryotic RNAs are modifiedat the 5′ and 3′ ends by capping with GTP and addingpolyA tails. GTP is added in a reversed orientationcompared to the rest of the polynucleotide chain and isoften accompanied by methylation at the N7 position ofthe G base. Together with the first two nucleotides theadded G base forms a 5′-cap structure (Figure 8.29).Further modifications remove nucleotides from the 3′end adding ‘tails’ of 50 to 200 adenine nucleotides.The mechanistic reasons for modification are unclearbut involve mRNA stability, mRNA export from thenucleus to the cytoplasm and initiation of translation.
The spliceosome associates with the primary RNAtranscript in the nucleus and consists of four RNA–protein complexes called the U1, U2, U4/U6 and U5or small nuclear ribonucleoproteins (snRNPs, read as‘snurps’). The snRNPs are named after their RNA com-ponents so that U1 snRNP contains U1 small nuclearRNA. The U signifies uridine-rich RNAs, a featurefound in all small nuclear RNA molecules. The U4 andU6 snRNAs are found extensively base paired and aretherefore referred to collectively as the U4/U6 snRNP.The snRNPs pre-assemble in an ordered pathway toform a complex that processes mRNA transcripts.
CH2O
OO
O
O−
PO O P
O−
O
O
O−P
O
O
CH3
CH3
CH2
O
N N
NNH
O
NH2
OH
OHbase1
5′
5′
7-methylguanylate
Figure 8.29 Methylation and G-capping of mRNA wasdiscovered with the purification of mRNA

266 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
Figure 8.30 Splice sites are specific mRNA sequences and assist intron removal
The first requirement for effective mRNA splicing isto distinguish exons from introns. This is achieved byspecific base sequences that signal exon–intron bound-aries to the spliceosomes and define precisely the 5′ and3′ regions to be removed from mRNA. Many intronsbegin with −5′ GU. . . and end with −3′ AG and in ver-tebrates the consensus sequence at the 5′ splice site ofintrons is AGGUAAGU (Figure 8.30). This sequencebase pairs with complementary spliceosomal RNAaligning pre-mRNA and assisting removal of the intron.A second important signal called the branch point isfound in the intron sequence as a block of pyrimidinebases together with a special adenine base approxi-mately 50 bases upstream from the 3′ splice site.
Using this sequence information the spliceosomeperforms complex catalytic reactions to excise theintrons in two major reactions. The first step involvesthe 2′ OH at the branch site and a nucleophilic attackon the phosphate group at the 5′ end of the intronto be removed. For this reaction to occur the mRNAis rearranged to bring two distant sites into closeproximity. This movement breaks the pre mRNA at the5′ end of the intron to leave, at this stage, a branch sitewith three phosphodiester bonds. The reaction createsa free OH group at the 3′ end with exon 1 no longerattached to the intron although it is retained by thespliceosome. The free OH group at the 3′ end of exon1 attacks the phosphodiester bond between exon 2 andthe intron, with the result that both exons are joinedtogether in the correct reading frame (Figure 8.31). Theresulting lasso-like entity is called a lariat structureand is removed from the mRNA (Figure 8.31). Theprocessed mRNA is then exported from the nucleusand into the cytoplasm for translation.
TranslationConversion of genetic information into proteinsequences by ribosomes is called translation and
involves the use of a genetic code residing in theorder of nucleotide bases. In theory, four nucleotidebases could be used in a variety of different codingsystems, but theoretical considerations proposed byGeorge Gamow in the 1950s led to the idea that atriplet of bases is the minimum requirement to codefor 20 different amino acids. With the realization that atriplet of bases was involved two experiments criticallydefined the nature of the genetic code.
The first showed that the genetic code was not‘punctuated’ or ‘overlapping’ but involved a con-tinuous message. Using the bacteriophage T4 Fran-cis Crick and Sydney Brenner showed that inser-tion or deletion of one or two bases resulted inthe formation of nonsense proteins. However, whenthree bases were added or removed a protein wasformed with one residue deleted or added. Thisdemonstrated a code that was an unpunctuated seriesof triplets arising from a fixed starting point. Thesecond experiment identified codons correspondingto individual amino acids through the use of cell-free extracts, capable of protein synthesis, contain-ing ribosomes, tRNA, amino acids and amino acylsynthetases. When synthetic polynucleotide templatessuch as polyU were added a polymer of pheny-lalanine residues was produced. It was deduced thatUUU coded for Phe. Similar experiments involvingpolyC and polyA sequences pointed to the respec-tive codons for proline and lysine. Refinement of thisexperimental approach allowed the synthesis of polyri-bonucleotide chains containing repeating sequencesthat varied according to the frame in which thesequence is read. For example AAGAAGAAGAA-GAAG, i.e. (AAG)n can be read as three differentcodons (Figure 8.32).
In a variation of this last experiment Philip Lederand Marshall Nirenberg showed that synthetic tripletswould bind to ribosomes triggering the binding ofspecific tRNAs. UUU and UUC lead to Phe-tRNAs

TRANSFER RNA (tRNA) 267
Figure 8.31 Splicing of two exons by intron removal. Exons are drawn in different shades of blue, intron in orange
Figure 8.32 Translation of the synthetic polyribonu-cleotide (AAG)n leads to the production of threedifferent amino acids in cell free translation systems.The translation of (AAG)n leads to three possiblesequences depending on the ‘frame’ used to readthe message and can yield polylysine, polyarginineor polyglutamate
binding to ribosomes and this approach decipheredthe remaining codons of the genetic code confirmingdegeneracy in the 64 possible triplets.
The genetic code (Figure 8.33) is usually describedas universal with the same codons used for the sameamino acid in every organism. The ability of bacteria toproduce eukaryotic proteins derived from the transla-tion of eukaryotic mRNAs testifies to this universality.However, a few rare exceptions to this universality aredocumented (Table 8.5) and include alternative use ofcodons by the mitochondria, organelles with their owntranscription and translation systems.
Transfer RNA (tRNA)In the 1950s Francis Crick postulated that adap-tor molecules should contain enzymatically appendedamino acids and recognize mRNA codons. The

268 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
Second base positionU C A G
UUU UCU UAU UGU UUUC
Phe UCC UAC
Tyr UGC
Cys C
UUA UCA UAA Stop UGA Stop AUUG
Leu UCG
Ser
UAG Stop UGG Trp GCUU CCU CAU CGU UCUC CCC CAC
His CGC C
CUA CCA CAA CGA ACUG
Leu
CCG
Pro
CAG Gln
CGG
Arg
GAUU ACU AAU AGU UAUC ACC AAC
Asn AGC
Ser C
AUA
Ile
ACA AAA AGA AAUG Met ACG
Thr
AAG Lys
AGGArg
GGUU GCU GAU GGU UGUC GCC GAC
Asp GGC C
GUA GCA GAA GGA A
Firs
t pos
ition
(5’
end
)
GUG
Val
GCG
Ala
GAG Glu
GGG
Gly
G
Thr
id p
ositi
on (
3’ e
nd)
U
C
A
G
Figure 8.33 The genetic code. The series of triplets found in mRNA coding for the twenty amino acids are shownwith the chain termination signals highlighted in red and the start codon in green
Table 8.5 Rare modifications to the genetic code
Codon Normaluse
Alternativeuse
Organism
AGA, AGG Arg Stop, Ser Some animalmitochondria;someprotozoans
AUA Ile Met MitochondriaCGG Arg Trp Plant
mitochondriaCUX Leu Thr Yeast
mitochondriaAUU Ile Start codon Prokaryotic cellsGUG ValUUG Leu
‘adaptor’ molecule was small soluble RNA now calledtransfer RNA (tRNA). The structure of yeast alanyl-tRNA was reported in 1965 by Robert Holley andcontained 76 nucleotides. Base pairing of nucleotidesled to a cloverleaf pattern and all tRNAs have lengthsbetween 60 and 95 nucleotides showing comparable
structure and function. The cloverleaf structure hasdistinct regions represented by arms, stems and loops(Figures 8.34 and 8.35).
At the 5′ end the short acceptor stem 7bp in lengthforms non-Watson–Crick base pairs with bases at the3′ end of the chain and links via a short stem of 3–4nucleotides to a region known as the D arm because itcontains a modified base, dihydrouridine.1 A 5bp stemfrom the D arm leads to the anticodon arm containing asequence of three bases complementary to the mRNAcodon. A variable loop from 3 to 21 nucleotides inlength leads to a final arm containing a sequence ofbases TψC where ψ represents a modified base calledpseudouridine. This region is the T or TψC arm andrejoins an acceptor stem terminated by the sequence-CCA to which amino acids are added by amino acyltRNA synthetases.
Covalent coupling of amino acids to the tRNAacceptor stem involves activation by reaction with ATPforming an amino adenylate complex, followed bycoupling to tRNA in a reaction catalysed by aminoacyl tRNA synthetases.
Aminoacyl-AMP + tRNA −−−⇀↽−−− aminoacyl-tRNA
+ AMP1Non-Watson–Crick base pairs involve, for example, the pairing ofG with U.

THE COMPOSITION OF PROKARYOTIC AND EUKARYOTIC RIBOSOMES 269
Figure 8.34 The structure of yeast tRNA with thebase sequence drawn as a cloverleaf structurehighlighting the acceptor stem and four arms; theTψC arm, the variable arm, the anticodon arm andthe D arm. Red lines indicate base pairings in thetertiary structure with conserved and semi-conservednucleotides indicated by solid and dashed circlesrespectively. The 3′ end is shown in red, the 5′ end ingreen with the acceptor stem in yellow. The anticodonstem is shown in light green, the D loop is white withthe variable loop shown in orange and the TψC armin cyan
Two classes of synthetases exist: class I enzymes areusually monomeric and attach the carboxyl group oftheir target amino acid to the 2′ OH of A76 in the tRNAmolecule; class II enzymes are dimeric or tetramericand attach amino acids to the 3′ OH of cognatetRNA (Figure 8.36) with the exception of Phe-tRNAsynthetase which uses the 2′ OH. Enzymes within thesame class (Table 8.6) show considerable variations instructure and subunit composition.
Table 8.6 Classification of amino acyl tRNA synthetases
Class 1 Class 2
AminoAcid
Subunitstructure
and numberof residues
AminoAcid
Subunitstructure
and numberof residues
Arg α 577 Ala α4 875Cys α 461 Asn α2 467Gln α 551 Asp α2 590Gln α 471 Gly α2β2 303/689Ile α 939 His α2 424Leu α 860 Lys α2 505Met α2 676 Pro α2 572Trp α2 325 Phe α2β2 327/795Tyr α2 424 Ser α2 430Val α 951 Thr α2 642
The composition of prokaryoticand eukaryotic ribosomesIn E. coli ribosomes may account for 25 percent of thedry mass and are relatively easily isolated from cellsto reveal large structures 25 nm in diameter containingRNA and protein in a 60:40 ratio. The ribosome isalways found as two subunits, one with approximatelytwice the mass of the other.
Prokaryotic ribosomes are described by sedimen-tation coefficients of 70S with two unequal subunitshaving individual S values of 50S (large subunit) and30S (small subunit). Three ribosomal RNA componentsare identified on the basis of sedimentation coeffi-cients – the 5S, 16S and 23S rRNAs. The small subunithas a single 16S rRNA containing ∼1500 nucleotidesand the large subunit contains two rRNA molecules – a23S rRNA of about 2900 nucleotides and a muchsmaller 5S rRNA 120 nucleotides length. When com-bined with over 50 different proteins the prokaryoticribosome has a mass of ∼2.5 MDa.
Eukaryotic ribosomes are larger than their prokary-otic counterparts containing more RNA and protein.They have greater sedimentation coefficients (80S vs70S) with subunits of 60S and 40S. The large subunit

270 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
Figure 8.35 The structure of tRNA showing wireframe and spacefilling models for tRNA (PDB:6TNA). The arms areshown in different colours: acceptor stem (yellow), CCA region including A76 (cerise), variable loop (dark green), Darm (blue), the TψC arm (purple) and the anti-codon arm (red)
OCH2
O OH
AdenineO P O
O
O
tRNA
C
CR
NH3
H
O
2′3′
Figure 8.36 Esterification of an amino acid to the 3′OH group of A76 in tRNA
contains 5S, 5.8S and 28S rRNA composed of 120,156and 4700 nucleotides respectively and about 50 pro-teins. In contrast the small subunit has a single 18SrRNA about 1900 nucleotides in length and 32 pro-teins. Despite increased complexity their architectureis fundamentally the same.
By 1984 the sequences of 52 different polypep-tide chains were identified in E. coli ribosomeswith those from the large subunit designated asL1, L2 . . . etc. and those from the small subunitproteins S1 . . . S21 (Figure 8.37). The number ofproteins found in ribosomes from different species
varies and their homology is much lower than therRNA components. The E. coli large subunit con-tains 31 different polypeptides each occurring once,with the exception of L7 where four copies arefound. Subunit nomenclature refers to migration pat-terns in two-dimensional electrophoresis and as aresult of partial acetylation L7 can have differentmobility and was originally thought to be a dis-tinct subunit (L12). The L7/L12 subunits associatewith the L10 subunit forming a stable complex thatwas also mistakenly ‘identified’ as a separate pro-tein (L8).
The small subunit contains 21 polypeptides, lead-ing to a total of 52 different ribosomal proteinsvarying in size from small fragments with <100residues to polypeptide chains containing in excess of550 residues. Isolated ribosomal proteins were shownto have a high percentage of lysine and arginineresidues, a low aromatic content and a topologybased around antiparallel β sheets where the strandsare connected by regions of α helix. This struc-ture known as the RNA recognition motif (RRM) isseen in L7 and L30 but was also observed in otherRNA binding proteins such as spliceosomal U1-snRNP(Figure 8.38).
Recognition motifs are widely found in RNA bind-ing proteins in prokaryotes and eukaryotes suggesting

THE COMPOSITION OF PROKARYOTIC AND EUKARYOTIC RIBOSOMES 271
Figure 8.37 Assembly of the prokaryotic ribosome. In prokaryotes the 50 and 30S subunits combine to form afunctional 70S ribosome. Association is favoured by increasing concentrations of Mg2+30S + 50S −−−⇀↽−−− 70S. In vivoa substantial proportion of the ribosomes are dissociated and this is important during initiation of protein synthesis
Figure 8.38 The structure of two ribosomal proteins and the U1-snRNP protein showing the homology based aroundthree β strands and two α helices. The structures show the C terminal domain of E. coli ribosomal subunit L7/L12(PDB: 1CTF), the L30 domain of the large ribosomal subunit of T. thermophilus (PDB:1BXY) and the N-terminal domainof the spliceosome RNA binding protein U1A (PDB:1FHT)
evolution from an ancestral RNA binding protein. AllRRMs consist of a domain of 80–90 residues wherethe four-stranded antiparallel β sheet and helices occurin the order β-α-β-β-α-β. RNA binds on the flat sur-face presented by the β sheet a region carrying manyconserved Arg/Lys residues on its edge to facilitateinteractions with the RNA sugar-phosphate backbone.Groups of non-polar, often aromatic, residues exposedin the sheet region interact directly with purine andpyrimidine bases and in strand 1 a conserved sequenceof K/R-G-F/Y-G/A-F-V-x-F/Y is found with alternateresidues exposed (blue). Similarly in strand 3 a con-sensus hexameric sequence L/I-F/Y-V/I-G/K-N/G-L/Mis observed.
Low-resolution studies of the ribosome
Despite differences in RNA and protein compositionthe architecture of all ribosomes is similar with ashape revealed by electron microscopy to containlobes, ridges, protuberances and even the suggestionof channels as ‘anatomical’ features (Figure 8.39).The ribosome contains single copies of each proteinand allowed antibodies to be raised against exposedepitopes. In combination with electron microscopy thedistribution of proteins through the large and smallsubunits was mapped to enhance the picture of theribosome (Figure 8.40).

272 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
Figure 8.39 A three-dimensional model of the E. coli ribosome derived from electron microscopy. (Reproducedwith permission from Voet, D., Voet, J.G & Pratt, C.W. Fundamentals of Biochemistry. John Wiley & Sons Inc,Chichester, 1999)
Figure 8.40 A positional map of surface epitopes forlarge and small E. coli ribosomal subunits. The smallsubunit is shown on the left and dashed lines indicatethe positions for subunits lying of the reverse surface.The symbols 16S 3′ and 16S 5′ mark the two ends of the16S rRNA molecule. In the large subunit P indicates thepeptidyl transferase site; E marks the site of emergencefor the nascent polypeptide from the 50S subunit andM specifies the ribosome’s membrane anchor site. The3′ and 5′ sites are indicated for 5S rRNA. (Reproducedwith permission from Voet, D., Voet, J.G., and Pratt,C.W. Fundamentals of Biochemistry. John Wiley & Sons,Chicheter, 1999)
However, delineation of the catalytic sites of proteinsynthesis required higher levels of resolution – a majorexperimental problem – for an assembly with over50 different proteins, several RNA molecules and amass in excess of 2.5 MDa. Ribosome crystals wereproduced in the 1960s but yielded two-dimensionalarrays of poor diffraction quality. Improved crystalquality stemmed from use of ribosomes derived fromthermophiles (Thermus thermophilus and Haloarculamarismortui ) where protein and nucleic acid stabil-ity was enhanced when compared with mesophilesand from advances in technology that introducedsynchrotron radiation, high efficiency area detectorsand crystal freezing techniques that limited radia-tion damage.
A structural basis for proteinsynthesis
The first structures for the ribosome were published in2000 by Tom Steitz and Peter Moore and representedthe culmination of decades of intensive effort. Thestructure of the large subunit was followed by structuresfor the small (30S) subunits from both E. coli and

AN OUTLINE OF PROTEIN SYNTHESIS 273
O
CO
CHR
NH
C
C
O
NH
R
tRNA
O
CO
CHR
NH2
tRNA
n − 1
n
n
n + 1
Peptidyl-tRNA
P site
Aminoacyl-tRNA
A site
OH
tRNA
O
CO
CHR
NH
C
C
O
NH
R
NH
O
CO
CHR
tRNAn
n+1
Uncharged tRNA
n
n−1
P site
Growing polypeptide chain + tRNA
Figure 8.41 The peptidyl transferase reaction at the A and P sites
T. thermophilus. However, a broad understanding ofthe functional properties of prokaryotic ribosomes arosefrom many years of biochemical studies long beforestructural data was available. These studies establishedthe self-assembly of ribosomes, inactivation of proteinsynthesis by antibiotics, the involvement of accessoryproteins in initiation, elongation and termination, a rolefor GTP in protein synthesis, the nature of tRNA bindingvia codon/anticodon recognition and the presence ofdiscrete binding sites within the ribosome.
An outline of protein synthesisTranslation of mRNA consists of three stages –initiation, elongation and termination – with the mostimportant process being elongation where nascentpolypeptides are extended by the addition of aminoacids. Protein synthesis results in the addition of aminoacid residues to a growing polypeptide chain in adirection that proceeds from the N to C terminals asmRNA is read in a 5′ –3′ direction. Synthesis proceedsat surprisingly high rates for a complex reaction withup to 20 residues per second added to a growing chain.
During elongation the peptidyl transferase reactioncatalyses movement and covalent linkage of an aminoacid onto a growing polypeptide chain in a processinvolving two specific sites on the ribosome. The A
site is the location for amino acyl tRNA binding tothe ribosome and is proximal to the peptidyl (P) site.Displacement of extending polypeptide chains from theribosome by high salt treatment leaves a peptidyl-tRNAspecies as the final ‘residue’. This results from transferof the peptidyl tRNA to the incoming amino acyl tRNAto form a peptidyl tRNA with one additional residuelocated in the A site. The next stage is translocationto create P site peptidyl tRNA, a vacant A site withuncharged tRNA transferred to the E or exit site(Figure 8.41).
Chain initiationMethionine, a relatively uncommon residue, is fre-quently found as the first residue of a polypeptidechain modified by the addition of formic acid. Inmost cases the N -formylmethionine group is removed(Figure 8.42), but occasionally the reaction does pro-ceed efficiently and methionine appears to be thefirst residue.
The initiation site is not solely defined by the AUGor start codon because a mRNA sequence will normallycontain other AUG triplets for internal methionineresidues. Additional structural elements such as theShine–Dalgarno sequence located ∼10 nucleotidesupstream of the start codon are complementary to a

274 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
NC
C
OCH2
CH2
S
CH3
H
C OO
H
tRNAmet
f
Figure 8.42 N-formylmethionine complexed totRNAf
met
pyrimidine-rich sequence found at the 3′ end of 16SrRNA of the 30S subunit, and facilitates ribosomalselection of the correct AUG codon (Figure 8.43).
Protein synthesis requires additional proteins knownas initiation factors (IF) that exhibit dynamic and tran-sient association with the ribosome (Table 8.7). Ini-tiation starts after completion of polypeptide synthe-sis with the ribosome as an inactive 70S complex.IF-3 binding to the 30S subunit promotes complexdissociation and is assisted by IF-1 (Figure 8.44). Ina dissociated state mRNA, IF-2, GTP and fMet-tRNAbind to the small subunit. IF-2 is a G protein and is
obligatory for binding fMet-tRNA to the 30S subunitin a reaction that is unique in not requiring mRNA andinteractions between codon and anticodon. In this statemRNA binds to the small subunit completing primingreactions and the complex is capable of binding the50S subunit. The association of the 50S subunit resultsin conformational change, GTP hydrolysis by IF-2 andthe release of all initiation factors. The result is a ribo-some primed with fMet-tRNA (P site) and a vacant Asite poised to accept the next amino acyl tRNA spec-ified by the second codon in an event that marks thestart of chain elongation.
Chain elongation
Elongation involves the addition of a new residue tothe terminal carboxyl group of an existing polypeptidechain. Elongation is divided into three key events:aminoacyl tRNA binding, the peptidyl transferasereaction, and translocation. Elongation factors assist inmany stages of these reactions (Figure 8.45).
Charged amino acyl tRNA is escorted to the vacantA site by an elongation factor -EF-Tu. EF-Tu isyet another G protein involved in protein synthesisand after depositing amino acyl tRNA at the A sitehydrolysis of GTP releases the EF-Tu–GDP complex
Figure 8.43 Translation initiation sequences aligned with the start codon recognized by E. coli ribosomes. The RNAsequences are aligned at the start (AUG) codon highlighted in blue. The Shine–Dalgarno sequence is highlighted inred and is complementary to a region at the 3′ end of the 16S rRNA. This sequence is shown in green and involvesG-U pairing. (Reproduced with permission from Voet et al., John Wiley & Sons, Ltd, Chichester, 1998)

AN OUTLINE OF PROTEIN SYNTHESIS 275
Table 8.7 Soluble protein factors involved in E. coliprotein synthesis
Factor Mass (kDa) Role
InitiationIF-1 9 Assist in IF-3 bindingIF-2 97 Binds initiator tRNA and
GTPIF-3 22 Dissociates 30S subunit
from inactive ribosomeand aids mRNA binding
ElongationEF-Tu 43 Binds amino acyl tRNA
and GTPEF-Ts 74 Displaces GTP from
EF-TuEF-G 77 Promotes translocation by
binding GTP toribosome. Molecularmimic ofEF-Tu + tRNA.
TerminationRF-1 36 Recognizes UAA and
UAG stop codonsRF-2 38 Recognizes UAA and
UGA stop codonsRF-3 46 G protein (binds GTP) and
enhances RF-1 andRF-2 binding
from the ribosome. At this stage the amino acyl tRNAis checked in a process known as ‘proof-reading’ andif incorrect a new tRNA is reloaded to the A site. Theactivity of EF-Tu is aided by EF-Ts – a protein thatbinds to EF-Tu as a binary complex to facilitate furtherrounds of GTP binding and further cycles of elongation(Figure 8.46).
The structure of Ef-Tu determined by crystallogra-phy in a complex with GDP and a slowly hydrolysing
analogue of GTP known as guanosine-5′-(β, γ-imido)-triphosphate (GDPNP) reveals three domains con-nected by short flexible peptide linkers. The 210residue N-terminal domain with a GTP/GDP bindingsite undergoes structural reorganization when GTP ishydrolysed, changing orientation with respect to theremaining two domains by ∼90◦. The Phe-tRNA–EF-Tu/GDPNP complex is stable and shows two macro-molecules arranged into the shape of a ‘corkscrew’(Figure 8.47). The ‘handle’ consists of EF-Tu and theacceptor region (-CCA) of Phe-tRNA. The remainingpart of the Phe-tRNA molecule forms the ‘screw’ andby comparing individual tRNA and EF-Tu structureswith the complex it is clear that structural perturba-tions are small. In the complex formed with tRNAmost interactions involve the amino acyl region ofthe tRNA – the CCA arm – and side chains of EF-Tu. These interactions also explain the observation ofpoor binding between uncharged tRNA molecules andEF-Tu.
The peptidyl transferase reaction is the secondphase of elongation and involves nucleophilic reactionsbetween uncharged amino groups of A site amino acyl-tRNA and the carbonyl carbon of the final residue ofP site tRNA arranged in close proximity in the 50Ssubunit. Evidence points towards a site and mechanismof protein synthesis performed entirely by RNA.Ribosomes consist predominantly of highly conservedrRNA (Figure 8.48),1 the proteins of large and smallsubunits show little conservation of sequence, almostall of the proteins of the large subunit of the ribosomeof T. aquaticus could be removed without loss ofactivity, and mutations conferring antibiotic resistancewere localized to ribosomal RNA genes.
In the final phase of elongation (translocation),uncharged tRNA is moved to the E site whilst recentlyextended tRNA in the A site is moved to the P site. TheA site becomes vacant and will accept the next chargedtRNA. Ribosomes move along the mRNA chain in the3′ direction so that a new codon occupies the A site.This reaction involves an additional G protein, EF-G, whose structure reveals a remarkable similarity tothe amino acyl-tRNA/EF-Tu/GTP complex despite anabsence of sequence homology and RNA. This is not
1The sequence of rRNA is so highly conserved that it is usedin taxonomic studies to identify new species or to highlightevolutionary lineage.
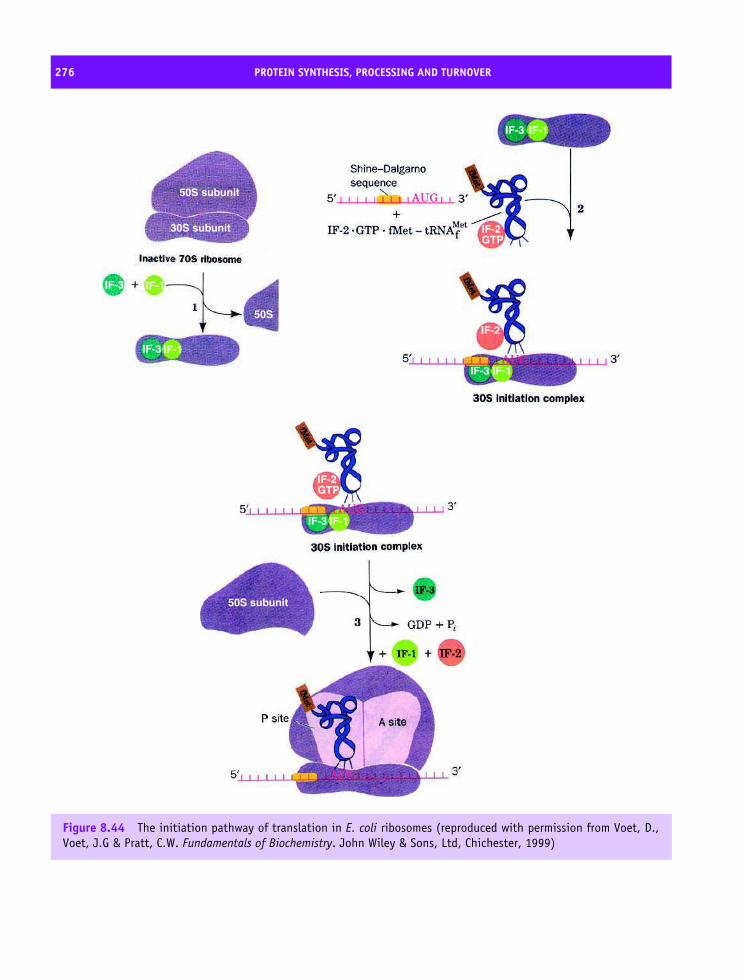
276 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
50S subunit
50S subunit
30S subunit
50S
IF-3
IF-3
IF-3 IF-1
IF-2
IF-2GTP
IF-1
IF-3
IF-2
IF-1IF-3
GTP
IF-1
IF-1
Figure 8.44 The initiation pathway of translation in E. coli ribosomes (reproduced with permission from Voet, D.,Voet, J.G & Pratt, C.W. Fundamentals of Biochemistry. John Wiley & Sons, Ltd, Chichester, 1999)
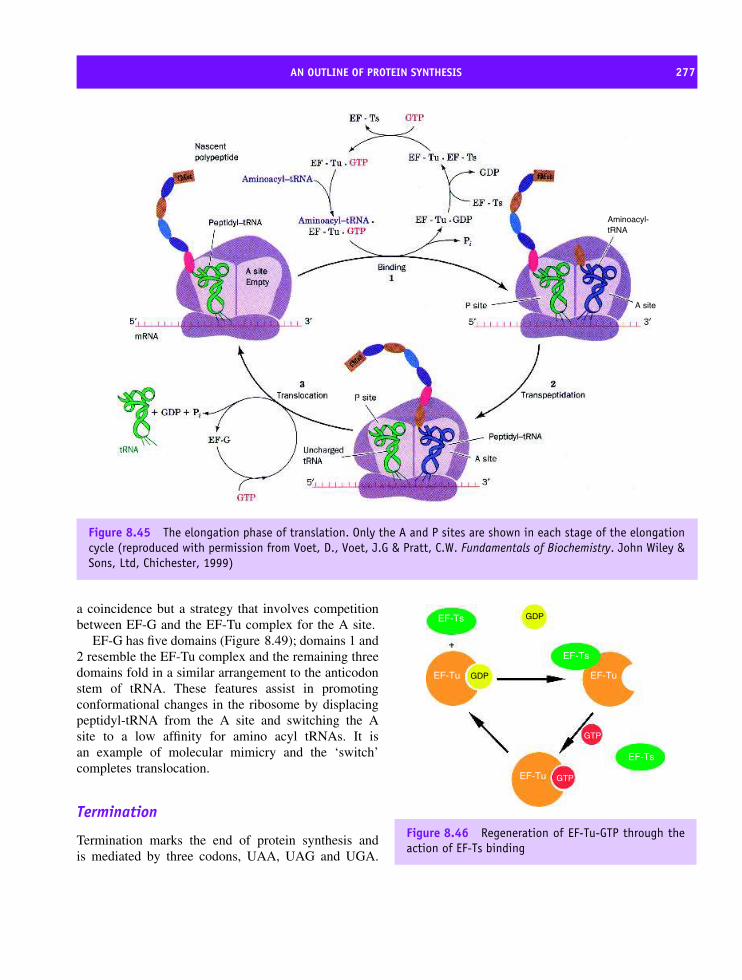
AN OUTLINE OF PROTEIN SYNTHESIS 277
Aminoacyl-tRNA
A site
Figure 8.45 The elongation phase of translation. Only the A and P sites are shown in each stage of the elongationcycle (reproduced with permission from Voet, D., Voet, J.G & Pratt, C.W. Fundamentals of Biochemistry. John Wiley &Sons, Ltd, Chichester, 1999)
a coincidence but a strategy that involves competitionbetween EF-G and the EF-Tu complex for the A site.
EF-G has five domains (Figure 8.49); domains 1 and2 resemble the EF-Tu complex and the remaining threedomains fold in a similar arrangement to the anticodonstem of tRNA. These features assist in promotingconformational changes in the ribosome by displacingpeptidyl-tRNA from the A site and switching the Asite to a low affinity for amino acyl tRNAs. It isan example of molecular mimicry and the ‘switch’completes translocation.
Termination
Termination marks the end of protein synthesis andis mediated by three codons, UAA, UAG and UGA.
EF-Ts
EF-Ts
EF-Tu EF-Tu
EF-Tu
EF-Ts
GDP
GTP
GTP
GDP
Figure 8.46 Regeneration of EF-Tu-GTP through theaction of EF-Ts binding

278 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
Figure 8.47 The structure of EF-Tu in a complex with Phe-tRNA (PDB: 1TTT). Domain 1 is shown in blue, domain 2in red and domain 3 in green. The CCA arm of the Phe-tRNA is bound between the blue and red subunits
Figure 8.48 tRNA environment within the ribosomedefined by cross-linking experiments. The cross linkswere from defined nucleotide positions within a tRNAto ribosomal proteins in the large and small subunits(denoted by prefix L and S). The A site was defined bytriangular symbols, the P site by circles and the E siteby squares. (Reproduced and adapted from Wower, J.Biochimie 1994, 76, 1235–1246)
There are no tRNAs for termination codons but proteinrelease factors RF-1, RF-2 and RF-3 identify the
codons. RF-1 recognizes UAA and UAG whereas RF-2 identifies UAA and UGA. The third protein RF-3is a G protein and in the presence of GTP stimulatesRF-1 and RF-2 binding to the large subunit. With RF-3 and either RF-1 and RF-2 bound to the A site thepeptidyl transferase reaction adds water to the end ofthe growing chain releasing the peptide into the cytosolalong with an uncharged tRNA.
Antibiotics provide insight intoprotein synthesisSome antibiotics block protein synthesis and areused to probe initiation, elongation and termina-tion. Their effectiveness in halting prokaryotic pro-tein synthesis coupled with their relative ineffective-ness in eukaryotic systems has allowed their use inmedicines. Amongst the antibiotics used to interferewith protein synthesis in microbes are tetracycline,erythromycin, puromycin, streptomycin and chloram-phenicol. These reagents block different parts of proteinsynthesis and from analysis of their chemical struc-tures their effects on ribosome function can be ratio-nalized. Chloramphenicol (Figure 8.50), for example,blocks the peptidyl transferase reaction by actingas a competitive inhibitor in which the secondary

STRUCTURAL STUDIES OF THE RIBOSOME 279
Figure 8.49 The five domains of EF-G. Domains 3–5 mimic the conformation of tRNA complexed to EF-Tu(PDB: 2EFG)
amide resembles the normal peptide bond. Similarlypuromycin (Figure 8.50) contains within its structurea portion resembling the 3′ end of the amino acyltRNA. As a result it enters the A site and is trans-ferred to extending peptide chains causing prematurerelease from the ribosome.
Affinity labelling and RNA‘footprinting’Chemical probes such as dimethyl sulfate or carbodi-imides attack accessible bases of rRNA but in thepresence of ligands such as antibiotics, mRNA ortRNA some regions will remain unmodified and leavea ‘footprint’ when analyzed by electrophoresis. In 23SRNA A2451and A2439 were protected by the acylregion of tRNA at the A and P sites whilst the 3′
terminal protected G2252 and G2253. These baseswere universally conserved (or very nearly so) acrossthe phylogenetic spectrum and the results point to func-tional importance. Similarly regions of the 16S rRNAknown as helix 44, the 530 loop and helix 34 areinvolved in the 30S decoding site. Genetic studies fur-ther identified two bases A1492 and A1493 from theiruniversal conservation and requirement for viability. Afew ‘hot spots’ or critical bases effectively modulateprotein synthesis and these experiments allowed crys-tallographers to assess structures on the basis of theknown importance of these bases.
Structural studies of the ribosomeThe structure of the large ribosomal subunit of Haloar-cula marismortui defined sites of protein synthesisand highlighted potential mechanisms for the peptidyl
CH2 C C
H
NH3 O
NH
OCH2OH
H H
OH
HH
N N
NN
N
H
CH3CH3
OMeCH
OH
CH
CH2OH
NH
O2N
C O
CHCl2
Figure 8.50 The structures of chloramphenicol and puromycin

280 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
Domain VI
Domain I
Domain V
Domain IV
Domain III
Domain II
5S rRNA
Figure 8.51 Computer-based secondary structureprediction for 23S and 5S rRNA sequences. (Reproducedwith permission from Ramakrishnan, V. & Moore, P.B.Curr. Opin. Struct. Biol. 2001, 11, 144–154. Elsevier)
transferase reaction with coordinates for RNA and pro-tein representing over 90 percent of the subunit.
The secondary structure of 23S rRNA (Figure 8.51)shows regions of helix formed by base pairing as wellas extended regions but provides little information onhow the RNA folds to give the structure adopted bythe large subunit. The 23S rRNA has six helical richdomains linked via extended loops.
The tertiary structure of 23S rRNA (Figure 8.52)showed domains that are not widely separated but areinterwoven via helical interactions to form a compactmonolithic structure. The 23S rRNA is a highly convo-luted structure dictating the overall shape of the ribo-some as well as defining the peptidyl transferase site.
Large subunit proteins (Figures 8.53 and 8.54) arelocated towards the ribosome’s surface, often exposedto solvent, or on the periphery between the twosubunits. Many proteins have multiple RNA bindingsites and a principal function is directed towardsstabilization of RNA folds. One surprising observationwas that some proteins were not globular domains andfor those located in crevices between rRNA heliceswere often extended structures. Many were basicproteins – an unsurprising observation in view of thelarge number of phosphate groups present within RNA.
Catalytic mechanisms within thelarge subunitUsing substrate analogues of the -CCA region oftRNA the location of the peptidyl-transferase site
was shown to lie at the bottom of a deep cleft atthe interface between large and small subunits. Thepeptidyl transferase reaction involves the bimolecularreaction between two substrates, namely the A siteamino acyl-tRNA and the P site peptidyl-tRNA, and toenhance reactivity the molecules are constrained and inclose proximity.
Transition state analogues based on puromycinpinpointed the location of the CCA arms of the Aand P site tRNA molecules on the large subunit. Inthe presence of CCdA-phosphate-puromycin (the Yarusinhibitor, Figure 8.55) the peptidyl transferase reactionwas inhibited after transfer of peptide chains from P sitebound tRNA to the α amino group of puromycin. Thepeptidyl transferase site was based around nucleotidesbelonging to the central loop of 23S rRNA domain V aregion called the ‘peptidyl transferase loop’ and knownto bind CCdA-phosphate-puromycin. No proteins werelocated close to the puromycin complex and catalysiswas mediated entirely by rRNA.
To facilitate formation of a tetrahedral intermediatean active functional group is required. The nearestsuitable residue was the N3 atom of A2486 (A2451in E. coli ) located ∼3 A from the phosphoramideoxygen of the CCdA-phosphate-puromycin. No otherfunctional group was within 5 A of this reaction siteand more significantly there were no atoms derivedfrom polypeptide chains within 18 A of this centre.This residue had previously been implicated in peptidyltransferase activity from genetic, footprinting andcrosslinking studies.
Under normal conditions the N1 atom of adeninemonophosphate has a pK of ∼3.5 with the N3 centreobserved to be an even weaker base, with a pK twounits lower. The crystal structure (Figure 8.56) sug-gested a higher pK for the N3 atom of A2486 becausethe distance between the N atom and the oxygen is 3 Aand appears as a formal hydrogen bond – an interactionthat would only occur if the N atom is protonated. Thecrystallization performed at pH 5.8 suggested a valuefor the pK of the N3 group above pH 6.0 to accountfor N3 protonation.
Many details of the catalytic process remain tobe clarified but a charge relay network is believedto elevate the pK of the N3 atom of A2486 via anetwork of hydrogen bonds with G2482 and G2102

STRUCTURAL STUDIES OF THE RIBOSOME 281
L1
Interface view
L1
Back view
Figure 8.52 The tertiary structure of the 23S rRNA sequence. The colours utilize those shown in Figure 8.51 for thesecondary structure. (Reproduced with permission from Ramakrishnan, V. & Moore, P.B. Curr. Opin. Struct. Biol. 2001,11, 144–154. Elsevier)
Figure 8.53 The structure of the large subunit of the ribosome of H. marismortui. A space-filling model of the 23Sand 5S rRNA together with the proteins is shown looking down on the active site cleft. The bases are white and thesugar phosphate backbones are orange. The numbered proteins are blue with the backbone traces shown for the L1and L11 proteins. The central protuberance is labelled CP. (Reproduced with permission from Nissen, P. et al. Science2000, 289, 920–930)

282 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
(Figure 8.57). A2486 and G2102 are conserved inthe 23S rRNA sequences analysed from all threekingdoms.1
With this level of structural characterization itwas proposed that peptidyl transferase involved theabstraction of a proton from the α amino group ofthe amino acyl tRNA by the N3 atom of A2486. Inturn the NH2 group of amino acyl tRNA acts as anucleophile attacking the carbonyl group of peptidyl-tRNA. The protonated N3 stabilizes the tetrahedralcarbon intermediate by hydrogen bonding to theoxyanion centre with subsequent proton transfer from
L3
L2
L4
L19
N
C
L15
L39e
L37e
L21e L44e
Figure 8.54 The globular domains and extendedregions of proteins found in the large subunit. Thelong extensions would probably prove destabilizing inisolated proteins. Globular domains are shown in greenwith the extended region in red. (Reproduced withpermission from Ramakrishnan, V. & Moore, P.B. Curr.Opin. Struct. Biol. 2001, 11, 144–154. Elsevier)
1G2482 is conserved at a level of ∼98 percent in all sequences butis replaced by an A in some archae sequences and deleted altogetherin some eubacterial sequences.
the N3 to the peptidyl tRNA 3′ OH occurring asthe newly formed peptide deacylates. The reactionmechanism described is not firmly established andfuture work will undoubtedly seek to experimentallytest the proposed mechanism and role of A2486; apossible mechanism is shown in Figure 8.58.
The structure and function of the30S subunit
Although the A and P sites are located on thelarge subunit the small subunit plays a crucial andobligatory role in protein synthesis. Protein synthesis is
NO
N
N
ON
N
NH2
O
N
O
N
N
O
O
N
H2N
O
O
N
O
P
OO−
Figure 8.55 The transition state analogue CCdA-phosphate-puromycin
NN
NN
NH2
O
OCH2
O P O
O−
O
Figure 8.56 The N1 and N3 of adenine monophos-phate; the N1 is shown in blue and the N3 in red

STRUCTURAL STUDIES OF THE RIBOSOME 283
Figure 8.57 A skeletal representation with dashedhydrogen bonds showing G2482, G2102, and A2486,as well as the buried phosphate that may result ina charge relay through G2482 to the N3 of A2486.(Reproduced with permission from Nissen, P. et al.Science 2000, 289, 920–930)
a multi-step process and starts with IF-3 binding to thesmall subunit. The 30S subunit plays a direct role in‘decoding’ mRNA by facilitating base pairing betweenmRNA codon and the anticodon of relevant tRNAs.
The small subunit from T. thermophilus was crys-tallized in the 1980s but poor crystal diffraction prop-erties limited use for many years until improvementsin resolution occurred with removal of the S1 subunitfrom the ribosome prior to crystallization. Structures,initially at a resolution of 5.5 A were followed byresolutions below ∼3 A and detailed studies of the30S ribosomal subunit were largely the results oftwo groups headed by Ada Yonath and V. Rama-krishnan. The structure defined ordered regions of16S rRNA and confirmed the general shape of thesmall subunit deduced previously using microscopy.All morphological features were derived from RNAand not protein.
N
N
N
N
NH2
N
O
R
O
H
H
tRNA
N
O
R
O
tRNA
peptide chainpeptide chain
N
N
N
N
NH2
H
NHO
R
O tRNAN
O−
R
OtRNA
N
N
N
N
NH2
OtRNA
H
peptide chainNH
O
R
OtRNA
NH
O
R
+
Figure 8.58 A possible mechanism for the peptidyl transferase involving the N3 atom of A2486 (adapted fromNissen, P. et al. Science 2000, 289, 920–930)

284 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
The 16S RNA structure
The 16S rRNA contributes a significant proportion ofthe mass and volume of the 30S subunit and consistsof approximately 50 elements of double stranded helixinterspersed with irregular single stranded loops. The16S RNA fold consists of four domains based oninter-helical packing and interactions with proteins(Figure 8.59). Anatomical features define the 30Sstructure with a head region containing a beak thatpoints away from the large subunit. This structure ison top of a shoulder region whilst at the bottom a spuror projection is observed with a main interface definedby body and platform areas (Figure 8.60).
The 5′ domain of 16S rRNA forms the body region,the central domain and most of the platform of the 30S
Figure 8.59 Secondary structure diagram of 16S RNAshowing the various helical elements. The 5′ domainis shown in red; a central domain in green; a majordomain of the 3′ region in orange–yellow and a minordomain in this region in cyan
Figure 8.60 The tertiary structure of 16S RNA withthe same colour scheme for the domains as inFigure 8.59. The model shows H, head; Be, beak; N,neck; P, platform; Sh, shoulder; Sp, spur; Bo, bodyregions of the 30S subunit from the perspective ofthe 50S subunit. (Reproduced with permission fromRamakrishnan, V. & Moore, P.B. Curr. Opin. Struct. Biol.144–154. 2001, 11, Elsevier)
subunit. In contrast, the 3′ major domain constitutesthe bulk of the head region whilst the 3′ minor domainforms part of the body at the subunit interface. The fourdomains of the 16S rRNA secondary structure radiatefrom a central point in the neck region of the subunitand are closely associated in this functionally importantregion of the 30S subunit.
Proteins of the small subunit
Small differences in composition of 30S subunits werenoticed between T. thermophilus and the previouslydissected E. coli ribosome. Thermus lacks subunit S21but contains an additional short 26 residue peptidefragment. As a result of S1 removal the 30S structurescontain only subunits S2–S20 along with the short 26residue peptide fragment. Many proteins are located atjunctions between helices of the RNA. For example,the S4 subunit binds to a junction formed by fivehelices in the 5′ domain whilst S7 binds tightly toa junction formed from four rRNA helices in the3′ major domain. Both proteins are important in the

STRUCTURAL STUDIES OF THE RIBOSOME 285
S12S11
S10
S9
S13
S6
S14
Figure 8.61 The globular domains and extended regions of some of the proteins found in the 30S subunit. Longextensions are probably destabilizing in isolated proteins. S14 is a Zn-binding protein, the cation is shown by agreen sphere. (Reproduced with permission from Brodersen, D.E. et al. Cold Spring Harbor Symp. 2001, 66, 17–32.CSHL Press)
assembly of the 30S subunit forming parts of thebody and head, respectively. Table 8.8 summarizes thepolypeptide structures of the small subunit.
Almost all of the proteins contain one or more glob-ular domains and common topologies such as the β
barrel are found in subunit S12 and S17. The packingof α-helices against an extended β-sheet is observed inseveral proteins such as S3, S10, S6 and S11. How-ever, a defining characteristic is the relatively longextended regions found in subunits. These extensionsare ordered, occur at the N or C terminal and may con-tain helical regions such as hairpin structures (S2) orC-terminal helices (S13). The extensions include loopsor long β hairpins (S10 and S17) or ‘tails’ to proteinssuch as S4, S9, S11, S12, S13 and S19 (Figure 8.61). Inalmost all cases their role is to stabilize the RNA fold.In the 30S subunit the extensions reach far into cavities
surrounded by RNA and make contact with severalRNA elements. The extensions are well suited to thisrole since they are narrow, allowing close approach todifferent RNA elements, and they have basic patchesto counter the highly charged sugar-phosphate back-bone of RNA. Although protein–RNA interactions areobviously important, some of the protein subunits inter-act with each other via hydrophobic contacts. S3, S10and S14 form a tight cluster held together by hydropho-bic interactions. Other subunits interact via electrostaticand hydrogen bonding interactions and these includeS4, S5 and S8.
Functional activity in the 30S subunit
The major function of the small subunit is decodingand matching the anticodon of tRNA with the mRNA

286 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
Table 8.8 Summary of the structural properties of polypeptide chains in the 30S subunit. (Adapted from Wimberley,B.T. et al. Nature 2000, 407, 327–339. Macmillan).
Protein Residues No. of
domains
Secondary structure
in domains
Protein
interaction
Unusual features
S2 256 2 α2, α1β5α3 None Extended α hairpin
S3 239 2 α2b3, α2β4 S10, S14 N-terminal tail
S4 209 3 Zn finger, α4, α3β4 S5 N-terminal Zn finger
S5 154 2 α1β3, α2β4 S4, S8 Extended β hairpin
S6 101 1 α2β4 S18 C-terminal tail
S7 156 1 α6β2 S9, S11 Extended β hairpin
S8 138 2 β2α3, α1β3 S5, S12, S17
S9 128 1 βα3β4 S7 Long C-terminal tail
S10 105 1 α2β4 S3, S14 Long β hairpin
S11 129 1 α2β5 S18, S7 Long N-terminal tail
S12 135 1 α1β5 S8, S17 Long N-terminal tail and
extended β-hairpin loops
S13 126 1 α3 S19 Long C-terminal tail
S14 61 1 none S3, S10 Zn module mostly extended
S15 89 1 α4 none
S16 88 1 β1α2β4 none C terminal tail
S17 105 1 β5 S12 C-terminal helix + β hairpin
loops
S18 88 1 α4 S6 β strand extends S11 sheet
S19 93 1 α1β3 S13
S20 105 1 α3 none
Partial structural information was available for S4–S8, and S15–S19 and aided their identification in crystals of 30S subunits.
codon. Within the 30S subunit the A, P and E siteswere defined by RNA elements derived from differentdomains (45 different helical domains are found in 16SrRNA designated as H1–H45). The A and P sites aredefined predominantly by RNA with helix 44, helix 34and the 530 loop together with S12 forming part of theA site. In addition, the extended polypeptide chains ofS9 and S13 intrude into the tRNA binding sites andmay form interactions with tRNA. In contrast, the Esite is largely defined by protein.
A combination of molecular genetics, sequenceanalysis and biochemical studies highlighted A1492as an important base to the decoding process. Thestructure of the small subunit confirmed the importanceof A1492 along with its neighbour and conserved
base A1493. The crux of the decoding processis the ability to discriminate between cognate andnear cognate tRNAs and this activity lies in theunique conformation of bases found at the A site.Discrimination against non-cognate tRNA results intwo or three mismatches in base pairing at thecodon–anticodon level with the high energetic costmaking the process unfavourable.
The structure of the small subunit is split intodomains, unlike the 50S subunit, with each possessingindependent mobility and an involvement in conforma-tional changes that influence ribosomal activity. The Esite is predominantly protein and formed by the S7 andS11 subunits, a small interface between subunits formsthe anticodon stem loop binding site whilst an extended

POST-TRANSLATIONAL MODIFICATION OF PROTEINS 287
β hairpin structure in S7 plays a role in dissociation ofthe vacant tRNA molecule from the ribosome.
Strong sequence conservation of rRNA suggests thatRNA topology is likely to be maintained within theeukaryotic ribosomal subunits although there is clearlyan insertion of elements with the formation of 5.8S, 18Sand 28S rRNA. Yeast has a 18S rRNA 256 nucleotideslonger than the 16S rRNA (E. coli ) although thepattern of stem loops and major domains seen in16S rRNA is mirrored by rRNA of yeast and othereukaryotes. However, the 40S and 60S subunits possessincreased numbers of polypeptides relative to their E.coli counterparts, there are functional differences in themechanism of initiation, elongation involves a greatervariety and number of accessory proteins whilst themechanisms of antibiotic inhibition are not shared withprokaryotes.
The remarkable confirmation that the ribosome isa ribozyme defined a new era in structural biology.The structures produced for the 50 and 30S subunitsassimilates and unifies four decades of biochemicaldata on the ribosome and provides a wealth of newinformation about RNA and protein structure, theirrespective interactions and ribosome assembly. In 50years our knowledge has progressed from the initialelucidation of the atomic structure of DNA to a nearcomplete description of replication, transcription andtranslation.
Post-translational modificationof proteinsThe initial translation product may not representthe final, mature, form of the protein with somepolypeptide chains undergoing additional reactionscalled post-translational modification. These reactionsinclude processing to remove sequences normally atthe ends of the molecule, the addition of new groupssuch as phosphate or sugars, and the modification ofexisting groups such as the oxidation of thiol groups. Inalmost all cases these modifications are vital to proteinstructure and function.
Proteolytic processing
The digestive enzymes chymotrypsin, trypsin andpepsin function in the alimentary tracts of animals
to degrade proteins. These enzymes are initiallytranslated as inactive forms called zymogens (chy-motrypsinogen, trypsinogen and pepsinogen) to preventunwanted degradation. Zymogens are converted intoactive enzyme by removal of a short ‘pro’ sequences.Such processes are important in the production of pan-creatic proteases and mechanisms such as the bloodclotting cascade (see Chapter 7).
Peptide hormones as examples of processing
Peptide hormones are frequently synthesized as longerderivatives. Angiotensin, a hormone involved in thecontrol of vasoconstriction and insulin, with a role inthe maintenance of blood glucose concentrations are‘processed’ hormones. The ‘pro’ sequences may alsohave additional sequences located at the N-terminalto facilitate targeting to specific intracellular compart-ments. For example insulin, is synthesized as preproin-sulin (Figure 8.62).
The ‘pre’ sequence transfers insulin across theendoplasmic reticulum (ER) membrane into the lumenwhere cleavage by proteases leaves the ‘pro’ insulinmolecule (Figure 8.63). In this state folding starts withdisulfide bridge formation stabilizing tertiary structure.A fragment known as the C peptide is released(Figure 8.64) and persists in secretory granules seenin pancreatic cells. The insulin remains inactiveas a hormone and in high concentrations in thepancreas. The chain requires further modificationand a second proteolytic reaction cuts the primary
Figure 8.62 Preproinsulin contains 110 residueswithin a single polypeptide. Each circle representsa single amino acid residue

288 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
Figure 8.63 Signal sequence removal yields proin-sulin. The A chain has an intramolecular disulfidebridge as well as two intermolecular disulfide bridgeswith the B chain
sequence at pairs of Lys-Arg and Arg-Arg residuesbetween the A and B regions creating two separatepolypeptides.
A very dramatic example of processing is the pep-tide hormone opiomelanocortin (POMC). The POMCgene is expressed in the anterior and intermediate lobesof the pituitary gland where a 285-residue precursorundergoes differential processing to yield at least eighthormones (Figure 8.65).
Disulfide bond formation
The formation of disulfide bonds between cysteineresidues is a common post-translational modificationresulting in strong covalent bonds. The covalent bondrestricts conformational mobility in a protein andnormally occurs between residues widely separated in
the primary sequence. For example, in ribonucleasefour disulfide bridges form between cysteine residues26–84, 40–95, 58–110 and 55–72.
Disulfide bond formation occurs post-translationallyin the ER of eukaryotes or across the plasma mem-brane of bacteria in proteins destined for secretion. Theformation of disulfide bonds is often a rate-limitingstep during folding, and sometimes the ‘wrong’ disul-fide bond forms where there is a potential for multiplebridges. In all cells enzymes catalyse efficient forma-tion of disulfide bonds and correct ‘wrong’ disulfidebonds by rapid isomerization. These enzymes are col-lectively called disulfide oxidoreductases.
Eukaryotes and prokaryotes catalyse disulfide for-mation with different groups of enzymes. In prokary-otes the Dsb family of enzymes are identified in Gram-negative bacteria and carry names such as DsbA, DsbB,DsbD etc. In E. coli DsbA contains a consensus motifCys-Xaa-Xaa-Cys and shows a similar fold to thiore-doxin despite a low sequence identity (<10 percent). Inthe cytoplasm of E. coli thioredoxin reduces disulfideswhilst in the periplasm DsbA oxidizes thiol groups.Since both thioredoxin and DsbA contain the Cys-Xaa-Xaa-Cys motif the ability to oxidize disulfide bondscentres around the properties of this catalytic centre.In DsbA the first cysteine (Cys30) has a low pK of3.5, compared with a normal value around 8.5, andthe sidechain forms a reduced thiolate anion S− repre-senting a highly oxidizing centre that allows DsbA tocatalyse disulfide formation.
In eukaryotes comparable reactions are performedby protein disulfide isomerase (PDI). Again the activityof PDI depends on the diagnostic motif Cys-Xaa-Xaa-Cys and when the active-site cysteines are present as a
Figure 8.64 The processed (active) form of insulin plus proteolytic fragments

POST-TRANSLATIONAL MODIFICATION OF PROTEINS 289
Figure 8.65 The processing of pro-opiomelanocortin yields additional bioactive peptides. Initial processing ofpro-opiomelanocortin in the anterior and intermediate lobes of the pituitary yields an N-terminal fragment; ACTH,adrenocorticotrophic hormone (39 residues); and β-lipotrophin. Further processing of ACTH in the intermediatelobes yields a melanocyte stimulating hormone – a 13-residue peptide acetylated at the N-terminal and amidatedat the C-terminal residue together with CLIP, a corticotropin-like intermediate lobe peptide of 21 residues. Furtherprocessing of β lipotrophin yields γ-lipotrophin and β endorphin. The peptides are 59 and 26 residues in lengthrespectively with the β endorphin acetylated at the N terminal
disulfide the enzyme transfers disulfide bonds directlyto substrate proteins acting as a dithiol oxidase. Undermore reducing conditions with thiols in the active-site the enzyme reshuffles disulfides (isomerase) intarget proteins.
Hydroxylation
Hydroxylation is another example of a post-transla-tional modification. It is particularly important in thematuration of collagen where hydroxylation of pro-line and lysine residues found in the Gly-Xaa-Xaamotif occurs in procollagen in the ER as part ofthe normal secretory pathway. The reaction main-tains structural rigidity in collagen enabling a role
N CH
H2C CH2
CO
HO H
Figure 8.66 4-Hydroxyproline, a common post-translational modification observed in collagen
as a biological scaffold or framework. Proline ishydroxylated most commonly at the γ or fourth carbonby the enzyme prolyl-4-hydroxylase in reactions requir-ing oxygen, ascorbate (vitamin C) and α-ketoglutarate(Figure 8.66).
The requirement of vitamin C for effective foldingand stability of collagen emphasizes the physiologicalconsequences of scurvy. Scurvy is a disease arisingfrom a lack of ascorbate in the diet, and was frequentlyexperienced by sailors in the 16th and 17th centuriesduring long maritime voyages with the absence offresh fruit or vegetables containing high levels ofvitamin C. Scurvy can also occur under conditions ofmalnourishment and is sometimes seen in the moredisadvantaged regions of the world where famine isprevalent. Hydroxylation of proline residues stabilizescollagen by favouring additional hydrogen bondingand in its absence the microfibrils are weaker. Oneobserved effect is a weakening of connective tissuesurrounding the teeth, a common symptom of scurvy.Proline residues are also hydroxylated at the β or C3position with lower frequency.
In an entirely analogous manner lysine residuesof collagen are hydroxylated in a reaction conferringincreased stability on triple helices by allowing the

290 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
NH CH CO
CH2
CH2
CHOH
CH2
NH3+
Figure 8.67 The hydroxylation of lysine at the C5 orδ position
subsequent attachment of glycosyl groups that formcross links between microfibrils. Lysine residues arehydroxylated at the δ carbon (C5) by the enzyme lysyl-5-hydroxylase (Figure 8.67).
Phosphorylation
The phosphorylation of the side chains of specificserine, threonine or tyrosine residues is a generalphenomenon known to be important to functionalactivity of proteins involved in intracellular signalling.The addition of phosphoryl groups occurs through theaction of specific enzymes (kinases) that utilize ATP asa donor whilst the removal of these groups is controlledby specific phosphatases. A generalized scheme forphosphorylation is
Protein + ATP −−−⇀↽−−− protein-P + ADP
and results in the products phosphoserine, phosphotyro-sine, and phosphothreonine (Figure 8.68). More rarely
CH2 CHOO− P
O−
O
O− P O
O−
O
CH2C
CH3
CH
H
OO− P
O−
O
CH2 CH
Figure 8.68 The phosphorylation of serine, tyrosineand threonine residues
other residues are phosphorylated such as His, Aspand Lys.
Glycosylation
Many proteins found at the cell surface are anchoredto the lipid membrane by a complex series of glycosyl-phosphatidyl inositol (GPI) groups. These groups aretermed GPI anchors and they consist of an arrayof mannose, galactose, galactosamine, ethanolamineand phosphatidyl inositol groups. All eukaryotic cellscontain cell-surface proteins anchored by GPI groupsto the membrane. These proteins have diverse func-tions ranging from cell-surface receptors to adhe-sion molecules but are always located on exteriorsurfaces.
The term GPI was first introduced for the membraneanchor of the variant surface glycoprotein (VSG) ofTrypanosoma brucei, a protozoan parasite that causessleeping sickness in humans (a disease that is fatal ifleft untreated and remains of great relevance to sub-Saharan Africa). GPI anchors are particularly abundantin protozoan parasites. The overall biosynthetic path-way of GPI precursor is now well understood withsignals in the form of consensus sequences occurringwithin a polypeptide chain as sites for covalent attach-ment of GPI anchors. This site is often a region of ∼20residues located at the C-terminal, and specific pro-teases cleave the signal sequence whilst transamidasescatalyse the addition of the GPI anchor to the newlygenerated C-terminal residue. The outline organizationof GPI anchors (Figure 8.69) involves an ethanolaminegroup linked to a complex glycan containing mannose,galactose and galactosamine and an inositol phosphatebefore linking to fatty acid chains embedded in thelipid bilayer.
Alongside GPI anchors the principal post-transla-tional modification involving glycosylation involvesthe addition of complex sugars to asparagine sidechains (N-linked) or threonine/serine side chains (O-linked) in the form of glycosidic bonds. In N-linkedglycosylation an oligosaccharide is linked to the sidechain N of asparagine in the sequence Asn-X-Thr orAsn-X-Ser, where X is any residue except proline.The first sugar to be attached is invariably N -acetyl-glucosamine and strictly occurs co-translationally as

POST-TRANSLATIONAL MODIFICATION OF PROTEINS 291
Protein
Phospho-ethanolamine
Core tetrasaccharide
Phosphatidylinositol
C
O
O
O
Man Man Man
H
H H
HH
H
O
O
O C
O
O
C R1
R2OH
OH
HO HC
H2C
O
O
O OP
GlcNH2
6a1,2 a1,6
a1,6
a1,4
OP−O
NH
CH2
CH2
CH2
Figure 8.69 The attachment of GPI anchors to proteins together with the molecular organization of these complexoligosaccharides. GPI anchors are present in many proteins including enzymes, cell adhesion molecules, receptorsand antigens. They are found in virtually all mammalian cell types and share a core structure of phosphatidylinositolglycosidically linked to non-acetylated glucosamine (GlcN). Glucosamine is usually found acetylated or sulfated formand thus the presence of non-acetylated glucosamine is an indication of a GPI anchor
the polypeptide chain is synthesized. This attachment isthe prelude for further glycosylation which can involvenine mannose groups, three glucose groups and two N -acetyl-glucosamine groups being covalently linked toa single Asn side chain. Further processing removessome of these sugar groups in the ER and in the Golgithrough the action of specific glucosidases and man-nosidases, whilst in some instances fucose and sialicacid groups can be added by glucosyl transferases.These reactions lead to considerable heterogeneity anddiversity in glycosylation reactions occurring withincells. However, all N-linked oligosaccharides have acommon core structure (Figure 8.70).
In O-linked glycosylation the most common modifi-cation involves a disaccharide core of β -galactosyl(1 →3)-α-N -acetylgalactosamine forming a covalent bondwith the side chain O of Thr or Ser. Less frequently,galactose, mannose and xylose form O-linked glycosidic
NH
CH
C
O
CH2CO
N
X
Ser/Thr
NAcGlu
Asn
Figure 8.70 Common core structure for N-linkedoligosaccharides
bonds. Whilst N-linked glycosylation proceeds throughrecognition of Asn-containing motifs, within primarysequences O-linked glycosylation has proved moredifficult to identify from specific Ser/Thr-containingsequences and appears to depend more on tertiary struc-ture and a surface accessible site.
A single protein may contain N- and O-linkedglycosylation at multiple sites and the effect of linking

292 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
large numbers of oligosaccharide units is to increasemolecular mass dramatically whilst also increasingthe surface polarity or hydrophilicity. The function ofadded oligosaccharide groups has proved difficult todelineate and in some instances proteins can functionperfectly well without glycosylated surfaces. Generally,many cell-surface proteins contain glycosylation sitesand this has raised the possibility that oligosaccharidesfunction in molecular recognition events althoughmechanistic roles have yet to be uncovered.
N- and C-terminal modifications
Post-translational modification of the N-terminal ofproteins includes acetylation, myristoylation – theaddition of a short fatty acid chain containing 14 car-bon atoms, and the attachment of farnesyl groups.Many eukaryotic proteins are modified by acetylationof the first residue and the donor atoms are provided byacetyl-CoA and involve N -acetyltransferase enzymes.Acetylation is a common covalent modification at theN-terminal but other small groups are attached to thefree amino group including formyl, acyl and methylgroups. The precise reasons for these modifications areunclear but may be correlated with protein degrada-tion since chains with modified residues are frequentlymore resistant to turnover than unmodified proteins.
In contrast the attachment of myristoyl units hasa clearly defined structural role. As a long aliphaticand hydrophobic chain with 14 carbon units it causesproteins to associate with membranes although theseproteins become soluble when this ‘anchor’ is removed.Myristoylated proteins have properties typical of glob-ular proteins and remain distinct from integral mem-brane proteins with which they are often associated.Important examples of myristoylated proteins includesmall GTPases that function in many intracellular sig-nalling pathways.
Myristoylation is actually a co-translational mod-ification, since the enzyme N -myristoyltransferase isoften bound to ribosomes modifying nascent polypep-tide as they emerge. The substrate is myristoyl-CoA with glycine being the preferential N-terminalamino group. When the residue after glycine con-tains Asn, Gln, Ser, Val or Leu myristoylation isenhanced, whilst Asp, Phe, or Tyr are inhibitory. Thesequence specificity appears efficient with all proteins
containing N-terminal glycine followed by a stimula-tory residue observed to be myristoylated. Palmitoy-lation follows many of the patterns of myristoylationwith the exception that a C16 unit is attached to pro-teins post-translationally in the cytoplasm. The reactionis catalysed by palmitoyl transferase and recognizes asequence motif of Cys-Aliphatic-Aliphatic-Xaa whereXaa can be any amino acid. As progressively more pro-teins are purified it is clear that the range of modifica-tions extents to iodination and bromination of tyrosineside chains, adenylation, methylation, sulfation, amida-tion and side chain decarboxylation.
Rough ER
Golgisystem
Transportvesicles
Protein destined forsecretion
Figure 8.71 The movement of vesicles as part of thenormal route for transfer of proteins from the ER to theGolgi apparatus and on to other destinations. Transferthrough the Golgi occurs with protein processing beforefurther vesicle budding from the trans Golgi region.Vesicles from the trans Golgi are either directed to theplasma membrane, for secretion or to lysosomes.

PROTEIN SORTING OR TARGETING 293
L L L L PL CL A AF
L L L LLP L A AL
A F QL LL CI I Y
V L LF G FL S L I
C L VV GL SI V V
I L VL P L LA AL CL F L
A L LL L L VA LI L A
L F L V A I
L I L L L L
L
S L L FL LIF
Figure 8.72 The N-terminal sequence of secretory preproteins from eukaryotes. The sequences show basic residuesin blue and the hydrophobic cores in brown. The signal peptidase cleavage site is indicated
Protein sorting or targetingThe cell sorts thousands of proteins into specific loca-tions through the use of signal sequences locatednormally at the N-terminal. The hypothesis that signalsequences direct proteins towards cell compartmentswas proposed by Gunter Blobel and David Saba-tini in the early 1970s to explain the journey ofsecreted proteins from cytoplasmic sites of synthesisthrough the ER to the cell’s exterior (Figure 8.71).Signal sequences direct proteins first to the ER mem-brane in a process that is best described as a co-translational event.
A fundamental part of sorting is the interaction ofthe ribosome with a macromolecular complex knownas the signal recognition particle (SRP). The SRP iden-tifies a signal peptide sequence on nascent polypeptidechains emerging from the ribosome and by associationtemporarily ‘halts’ protein synthesis. The whole ensem-ble remains ‘halted’ until the SRP–ribosome complexbinds to further receptors (SRP receptors) in the ERmembrane. Chain elongation resumes and the growingpolypeptide is directed towards the lumen.
Proteins with signal sequences recognized by theSRP have a limited number of destinations thatincludes insertion of membrane proteins directly intothe bilayer, transfer to the ER lumen as a solubleprotein confined to this compartment, transfer via the
ER lumen to the Golgi apparatus, lysosomal targeting,or secretion from the cell. Protein targeting to thenucleus, mitochondrion and chloroplast is based on aSRP-independent pathway.
The SRP-mediated pathway
The presence of additional residues at the N terminusof nascent polypeptide chains not found in the matureform of the protein provided a clue that signalsequences existed (Figure 8.72). Edman sequencingmethods showed that the primary sequences of signalmotifs lacked homology but shared physicochemicalproperties. The overall length of the sequence rangedfrom 10 to 40 residues and was divided into regionswith different properties. A charged region betweentwo and five residues in length occurs immediately afterthe N-terminal methionine residue. The basic region isfollowed by a series of residues that are predominantlyhydrophobic such as Ala, Val, Leu, Ile, and Phe andare succeeded by a block of about five hydrophilic orpolar residues.
Protein synthesis of ∼80–100 residues exposes thesignal peptide through the large subunit channel andresults in SRP binding and the cessation of furtherextension until the ribosomal–mRNA–peptide–SRPcomplex associates with specific receptors in the

294 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
DD
T
T
T
T
TranslocanRibosomenascent chain
SRP SRP receptor
Figure 8.73 Co-translational targeting by SRP. Cycles of GTP binding and hydrolysis occur during the SRP cycle.GTPase activity is associated with SRP54 and the SRP receptor subunits. Activity is modulated by the ribosome andthe translocon. The SRP complex is shown in blue and the receptor in green. SRP D reflects GDP binding, SRP Treflects a GTP-bound state. The signal sequence is shown in yellow for the nascent chain (reproduced with permissionfrom Keenan, R.J. et al. Ann. Rev. Biochem. 2001, 70, 755–75. Annual Reviews Inc.)
ER membrane. This is known as co-translationaltargeting (Figure 8.73) and is conveniently divided intotwo processes; recognition of a signal sequence andassociation with the target membrane.
The structure and organization of the SRP
SRPs are found in all three major kingdoms (archae,eubacteria and eukaryotes) with the mammalian SRPconsisting of a 7S rRNA and six distinct polypep-tides called SRP9, SRP14, SRP19, SRP54, SRP68 andSRP72 and named according to their respective molec-ular masses. With the exception of SRP72 all bind tothe 7S rRNA and SRP54 has a critical role in bind-ing RNA and signal peptide. The diversity of signalsequences plays a significant part in determining themode of binding since introduction of a single chargedresidue destroys interaction with SRP54, whilst a blockof hydrophobic residues remains critical to associa-tion. In SRP54 the M domain is important in molec-ular recognition of signal peptide. The name derivesfrom a high percentage of methionine residues andis conserved in bacterial and mammalian homologuesThe homologous E. coli protein Ffh (Ffh = fifty four
homologue) has ∼16 percent of residues in the Mdomain as methionines – a frequency six times greaterthan its typical occurrence in proteins.
The E coli SRP has a simpler composition – theFfh protein and a 4.5S RNA – and evolutionary con-servation is emphasized by the ability of human SRP54to bind 4.5S RNA. Bacterial SRPs are minimal homo-logues of the eukaryotic complex and are attractive sys-tems for structural studies. The crystal structure derivedfor Ffh from T. aquaticus (Figure 8.74) revealed a C-terminal helical M domain forming a long hydrophobicgroove that is exposed to solvent and is made fromthree helices together with a long flexible region knownas the finger loop linking the first two helices. Thedimensions of the groove are compatible with signalsequence binding in a helical conformation and, as pre-dicted, the surface of the groove is lined entirely withphylogenetically conserved hydrophobic residues.1
Homologous domains from E. coli and mam-malian SRP54 have similar structures (Figure 8.75),
1In T. aquaticus, a thermophile living above 70 ◦C, many of theconserved Met residues found in SRP54/Ffh are replaced by Leu,Val and Phe, possibly reflecting a need for increased flexibility tocounter increased thermal mobility.

PROTEIN SORTING OR TARGETING 295
Figure 8.74 The structure of the full length FfHsubunit from Thermus aquaticus. The M domainencompassing residues 319–418 is shown in yellow,the N domain, residues 1–86, in blue and the largestG domain, residues 87–307, in green (PDB: 2FFH)
signal sequencebinding
RNA binding
h1
h2 h3
h4
h2b
N
C
Figure 8.75 Superposition of E. coli (light blue), T.aquaticus (magenta), and human (green) M domainsshowing similar structures (reproduced with permissionfrom Batey, R.T. et al. J. Mol. Biol. 2001, 307,229–246. Academic Press)
although the finger loop region adopts differentconformations – a variation that reflects intrinsic flex-ibility. Additional N-terminal and GTPase domainsinteract with the SRP receptor.
SRP54 is a GTPase and the catalytic cycle involvesGTP/GDP exchange and GTP hydrolysis although sig-nal sequence binding does not require GTP hydrolysis.Ribosome association stimulates GTP binding by SRP54and is followed by hydrolysis upon interaction with the
membrane receptors. The SRP receptor contains twosubunits designated α and β with the α subunit showinghomology to the GTPase domain of SRP54 and inter-acting with the β subunit, a second GTPase, that showsless similarity to either SRP54 or the α subunit.
The SRP receptor structure and functionMammalian SRP receptors are membrane proteincomplexes but the corresponding bacterial receptors aresimpler containing a single polypeptide known as FtsY(Figure 8.76). FtsY is a GTPase loosely associated withthe bacterial inner membrane that can substitute forthe α subunit of the SRP receptor. FtsY is composedof N and G domains comprising the GTPase catalyticunit and adopting a classic GTPase fold based on fourconserved motifs (I–IV) arranged around a nucleotide-binding site. Motif II is contained within a uniqueinsertion known as the insertion box domain and thisextends the central β sheet of the domain by two strandsand is characteristic for the SRP GTPase subfamily.
The N and G domains of SRP54 (Ffh) interactwith the α subunit of the SRP receptor (FtsY) throughtheir respective, yet homologous, NG domains. Theinteraction occurs when each protein binds GTPand the interaction is weaker in the apo protein.Nucleotide dependent changes in conformation arethe basis for the association between SRP and itscognate receptor and GTP hydrolysis causes SRP todissociate from the receptor complex. SRP lackingan NG domain fails to target ribosome–nascentchain complexes to the membrane and converselythe NG domain of FtsY when fused to unrelatedmembrane proteins promotes association. The primaryrole of the SRP receptor is to ‘shuttle’ the ribosomal-mRNA–nascent chain from a complex with SRP toa new interaction with a membrane-bound complexknown as the translocon. The translocon is a collectionof three integral membrane proteins, Sec61α, Sec61β
and Sec61γ, that form pores within the membraneallowing the passage of nascent polypeptide chainsthrough to the ER lumen.
Signal peptidases have a critical rolein protein targetingAfter translocation across the ER membrane polypep-tides reach the lumen where a peptidase removes the

296 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
N domain N domain
G domainGTPase
FfH NG
Insertion boxdomain
N
NIVIV
I
I
IIII
III
III
CC
Figure 8.76 The structures of E. coli FtsY show homology to Ffh. The apo-form of the NG domain shows theN-terminal N domain (blue) packing tightly against the GTPase fold (green). The conserved insertion box domain(orange) is unique to the SRP family of GTPases. The four conserved GTPase sequence motifs are indicated (I–IV).(Reproduced with permission from Keenan, R.J. et al. Ann. Rev. Biochem. 2001, 70, 755–75. Annual Reviews Inc.)The structure of Ffh is show for comparison in similar colours
‘signal peptide’ by cleavage at specific sites. Sites ofcleavage are determined by local sequence composi-tion after the ‘polar’ portion of the signal peptide. TheER signal peptidase shows a preference for residueswith small side chains at positions −1 and −3 from thecleavage site. Consequently Gly, Ala, Ser, Thr and Cysare common residues whilst aromatic, basic or largeside chains at the −3 position inhibits cleavage. Ala-nine is the most common residue found at the −1 and−3 positions and this has given rise to the Ala-X-Alarule or −1, −3 rule for cleavage site identification.
Post-lumenal targeting is directed byadditional sequence-dependent signals
When proteins reach the lumen other sequence depen-dent signals dictate targeting to additional sites ororganelles. One signal pathway involves the KDELsequence named after the order of residues found atthe C terminal of many soluble ER proteins. In mam-mals proteins with the sequence Lys-Asp-Glu-Leu (i.e.KDEL) are marked for recovery from transport path-ways. One example is PDI and altering this sequence
causes the protein to be permanently secreted from theER. The KDEL sequence binds to specific receptorslocated in the ER membrane and in small secretoryvesicles that bud off from the ER en route for the Golgi.The regulation of this receptor is puzzling but it rep-resents a very efficient mechanism for restraining andrecovering ER proteins.
An interesting variation of this pattern of trans-port occurs in channel proteins such as the K/ATPasecomplex consisting of two different subunits each con-taining a KDEL motif. Individually these subunits areprevented from leaving the ER but their assembly into afunctional channel protein leads to the occlusion of theKDEL motif from the receptor. Thus once assembledthese subunits are exported from the ER and this repre-sents an effective form of ‘quality control’ ensuring thatonly functional complexes enter the transport pathway.
After synthesis, partially processed proteins destinedfor the plasma membrane, lysosomes or secretion areobserved in the Golgi apparatus – a series of flattenedmembrane sacs. Within the Golgi apparatus progressiveprocessing of proteins is observed in the form ofglycosylation and once completed proteins are sent

PROTEIN SORTING OR TARGETING 297
to their final destination through the use of clathrincoated vesicles.
Protein targeting to the mitochondrionand chloroplast
Mitochondria and chloroplasts have small genomes andmost proteins are nuclear coded. This requires thatthe majority of chloroplast and mitochondrial proteinsare transferred to the organelle from cytoplasmic sitesof synthesis. Both organelles have outer and innermembranes and targeting involves traversing severalbilayers. The import of proteins by mitochondriaand chloroplasts is similar to that operating in theER with N-terminal sequences directing the proteinto organelles where specific translocation pathwaysoperate to assist movement across membranes. Themitochondria and chloroplast present unique targetingsystems in view of the number of potential locations.For the mitochondrion this includes locations inthe outer or inner membranes, a location in theinter-membrane space, or a location in the matrix.Unsurprisingly the number of potential locations evenwithin a single organelle places stringent demandson the ‘signal’ directing nuclear coded proteins tospecific sites.
Mitochondrial targeting
In the mitochondrion translocation of precursor pro-teins is an energy-dependent process requiring ATPutilization and complexes in the inner and outer mem-branes. For small proteins (<10 kDa) the outer mem-brane has pores that may allow entry but in mostcases the targeting information for precursor proteinsresides in an N-terminal sequence extension. Mitochon-drial signal sequences vary in length and compositionalthough they have a high content of basic residues andresidues with hydroxyl side chains but lack acidic sidechains. An important property of these signal sequencesis the capacity to form amphipathic helices in solu-tion and this is probably related to the requirement tointeract with membranes, receptors and aqueous envi-ronments. The majority of mitochondrial proteins aresynthesized in the cytosol and chaperones play a rolein maintaining newly synthesized protein in an ‘import
Table 8.9 Proteins identified as components of theyeast outer membrane translocase system
Protein Proposed function
Tom5 Component of GIP, transfer of preproteinfrom Tom20/22 to GIP
Tom6 Assembly of the GIP complexTom7 Dissociation of GIP complexTom20 Preprotein receptor, preference for
presequence-containing preproteinsTom22 Preprotein receptor, cooperation with
Tom20, part of GIP complexTom37 Cooperation with Tom machinery (Tom70)Tom40 Formation of outer membrane translocation
channel (GIP)Tom70 Preprotein receptor, preference for
hydrophobic or membrane preproteinsTom72 None (homologue of Tom70)
Adapted from Voos, W. et al. Biochim. Biophys. Acta 1999, 1422,235–254.
competent’ state that is not necessarily the native foldbut a form that can be translocated across membranes.
Translocation involves specific complexes in themembrane given the abbreviations Tom (translocationouter membrane) and Tim (translocation innermembrane). The first step of translocation involvesprecursor protein binding to a translocase systemconsisting of at least nine integral membrane proteins(see Table 8.9). Tom20, Tom22 and Tom70 are theprincipal proteins involved in import. Tom20 hasa single transmembrane domain at the N-terminuswith a large cytosolic domain recognizing precursorproteins from their targeting signals. Tom20 cooperateswith Tom22 a protein with an exposed N terminaldomain that also binds targeting sequences. This mayindicate autonomous rather than cooperative functionbut import is driven by a combination of hydrophobicand electrostatic interactions between pre-sequenceand receptor. Tom70, the other major receptor has alarge cytosolic domain preferentially interacting withpreproteins carrying ‘internal’ targeting information.Specific recognition by Tom 20, 22, and 70 leads to

298 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
Figure 8.77 Schematic model of the mitochondrialprotein import machinery of S. cerevisiae. (Reproducedwith permission from Voos, W. et al. Biochim. Biophys.Acta 1999, 1422, 235–254. Elsevier)
a second step involving pre-sequence insertion into theTom40 hydrophilic channel.
The inner mitochondrial membrane import path-way (Figure 8.77) completes the sequence of eventsstarted by the Tom assembly. The pathway is com-posed of Tim proteins (Table 8.10) with an innermembrane import channel formed by Tim23, Tim17and Tim44. Tim23 is a membrane protein contain-ing a small exposed acidic domain that may act asa potential binding site for sequences translocatedthrough the outer membrane. Tim23 is hydrophobicand with Tim17 forms the central channel of theinner membrane import pathway. Tim44 is a peripheralprotein that interacts with mitochondrial chaperones
Table 8.10 Proteins identified as components ofthe yeast inner mitochondrial membrane translocasesystem
Protein Possible function
Tim8 Cooperation with Tim9-Tim10Tim9 Complex with Tim10, guides hydrophobic
carrier proteins through inter membranespace.
Tim10 Complex with Tim9, guides hydrophobiccarrier proteins through inter membranespace.
Tim12 Complex with Tim22 and Tim54, innermembrane insertion of carrier proteins.
Tim13 Cooperation with Tim9-Tim10Tim17 Complex with Tim23, translocation of
preproteins through inner membrane intomatrix
Tim22 Inner membrane insertion of carrierproteins
Tim23 Complex with Tim17, translocation ofpreproteins through inner membrane intomatrix
Tim44 Membrane anchor for mitochondrial Hsp70Tim54 Complex with Tim22, inner membrane
insertion of carrier proteinsTim11 Unclear role.
Adapted from Voos, W. et al. Biochim. Biophys. Acta 1999, 1422,235–254.
(Hsp70) to regulate protein folding and prevent aggre-gation. Since proteins are translocated in an extendedconformation the role of chaperones is important tomediate controlled protein folding in the mitochon-drial matrix.
Closely coupled to translocation across the innermembrane is removal of the N-terminal signalsequence by mitochondrial processing proteases (MPP)located in the matrix. An interesting, but unexplained,observation, and one that will be recalled by the carefulreader, is that MPPs constituted the ‘core’ proteins ofcomplex III found in mitochondrial respiratory chains.

PROTEIN SORTING OR TARGETING 299
The import pathway will target proteins into the matrixof mitochondria but to reach other destinations thebasic import pathway is supplemented by additionalsorting reactions.
The Rieske FeS protein is synthesized with anN-terminal matrix targeting sequence that is cleavedby the normal processing peptidase and in vitro thisprotein is detected in the matrix, a foreign locationfor this protein. In vivo additional sorting mechanismstarget the protein back to the inner membrane. Incontrast cytochrome c1 has a dual targeting sequence.A typical matrix targeting sequence is supplementedby a second sorting signal that has a hydrophobiccore and resembles bacterial export signals. Forproteins in the inter-membrane space an incrediblydiverse array of pathways exist. Cytochrome c istargeted to this location without obvious targetingsequences whilst yeast flavocytochrome b2 (lactatedehydrogenase) is located in this compartment by adual targeting sequence.
Targeting of proteins to the chloroplast
The chloroplast (Figure 8.78) uses similar principlesof sorting and targeting to the mitochondrion. Tar-geting sequences contain significant numbers of basicresidues, a high content of serine and threonine residuesand are highly variable in length ranging from 20 to120 residues. In direct analogy to their mitochondrialcounterparts the translocating channel of the outer and
Figure 8.78 A schematic diagram of a chloroplast
inner chloroplast membranes are given the nomencla-ture Toc and Tic.
Although in vitro the Toc and Tic translocationsystems are separable the two complexes coordinateactivities in vivo to direct proteins from the cytosolto the stroma. Two proteins, Toc159 and Toc75, actas receptors and form a conducting channel witha diameter of 0.8–0.9 nm that requires proteins totraverse outer membranes in an extended conformation.The Tic proteins involved in precursor import areTic110, Tic55, Tic40, Tic22 and Tic20, but littleis known about the mechanism of import or theirstructural features.
Stromal proteins such as ribulose bisphosphate car-boxylase, ferredoxin or ferredoxin-NADP reductase donot require additional sorting pathways. They are syn-thesized with a single signal sequence that is cleavedby a stromal processing peptidase. Thylakoid pro-teins such as the light-harvesting chlorophyll com-plexes, reaction centres, and soluble proteins suchas plastocyanin must utilize additional targeting path-ways. Targeting is via a bipartite N-terminal signalsequence where the first portion ensures transfer intothe stromal compartment whilst a second part directsthe peptide to the thylakoid membrane where furtherproteolysis in the lumen removes the second signalsequence.
Nuclear targeting and nucleocytoplasmictransport
The final targeting system involves nuclear import andexport. Nuclear proteins are required for unpacking,replication, synthesis and transcription of DNA as wellas forming the structure of the nuclear envelope. Thereare no ribosomes in the nucleus so all proteins areimported from cytosolic sites of synthesis. This createsobvious problems with the nucleus being surrounded byan extensive double membrane. Proteins are importedthrough a series of large pores that perforate the nuclearmembrane and result from a collection of proteinsassembled into significant macromolecular complexes.
Specific nuclear import pathways were demonstratedwith nucleoplasmin, a large pentameric protein com-plex (Mr ∼ 165 kDa), which accumulates in the solublephase of frog oocyte nuclei. Extraction of the protein

300 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
followed by introduction into the cytosol of oocytes ledto rapid accumulation in the nucleus. The rate of accu-mulation was far greater than expected on the basis ofdiffusion and involved active transport mechanisms andsequence dependent receptors. The sequence specificimport was demonstrated by removing the tail regionsfrom nucleoplasmin pentamers. This region contained a‘signal’ directing nuclear import and in tail-less proteinno accumulation within the nucleus was observed.
Nuclear proteins have import signals defined byshort sequences of 4–10 residues that lack homologybut show a preference for lysine, arginine and pro-line. Nuclear localization signals (NLS) are sensitive tomutation and linking sequences to cytoplasmic proteinsdirects import into the nucleus. Today it is possibleto use computers to ‘hunt’ for NLS within primarysequences and their identification may indicate a poten-tial role for a protein as well as a nuclear localization(Table 8.11). More complex bipartite sequences occurin proteins where short motifs of two to three basicresidues are separated by a linker region of 10–12residues from another basic segment.
Nucleoplasmin-coated gold particles defined theroute of protein entry into the nucleus when electron-dense particles were detected by microscopy aroundpore complexes shortly after injection into the cytosolof oocytes. These observations suggested specific asso-ciation between cytoplasmic-facing proteins of thenuclear pore complex and NLS containing protein.Nuclear transport is bi-directional (Figure 8.79) withRNA transcribed in the nucleus exported to the cyto-plasm as ribonucleoprotein whilst the disassemblyof the nuclear envelope during mitosis requiresthe re-import of all proteins. Many proteins shuttle
Table 8.11 Sequences identified to act as signals forlocalization to the cell nucleus
Protein NLS sequence
SV40 T antigen Pro Lys Lys Lys Arg Lys ValAdenovirus E1a Lys Arg Pro Arg ProNucleoplasmin Lys Arg Pro Ala Ala Thr Lys
Lys Ala Gly Gln Ala LysLys Lys Lys
Figure 8.79 A schematic representation of nuclearimport involving importins, cargo and Ran-GTP(reproduced with permission from Cullen, B.R. Mol.Cell. Biol. 2000, 20, 4181–4187)
continuously between the nucleus and cytoplasm andthe average cell has an enormous level of nucleocy-toplasmic traffic – some estimates suggest that over103 macromolecules are transferred between the twocompartments every second in a growing mammaliancell.
The first proteins discovered with a role in shuttlingproteins from the cytosol to nucleus were identifiedfrom their ability to bind to proteins containing theNLS. The protein was importin-α (Figure 8.80) andhomologues were cloned from other eukaryotes todefine a family of importin-α-like proteins. Importin-αfunctions as a heterodimer with a second protein calledimportin-β. A typical scenario for nuclear transportinvolves cargo recognition via the NLS by importin-α and complex formation with importin-β followedby association and translocation through the nuclearpore complex in a reaction requiring GTP. In the

PROTEIN SORTING OR TARGETING 301
Figure 8.80 The super-helix formed by collections of armadillo motifs in mouse importin-α. A unit of three helicesis shown in green (PDB: 1AIL)
nucleus the complex dissociates in reactions catalysedby monomeric G proteins such as Ran-GTP. Ran-GTP binds specifically to importin-β allowing it to berecycled back to the cytoplasm in events again linkedto GTP hydrolysis. Importin-α and the cargo dissociatewith the protein ‘delivered’ to its correct destination.
The structure of importin-α, determined in a com-plex with and without NLS sequence, reveals twodomains within a polypeptide of ∼60 kDa. A basicN-terminal domain binds importin-β and a much largersecond domain composed of repeating structural unitsknown as armadillo repeats (Figure 8.80). Each unitis composed of three α helices and the combinedeffect of each motif is a translation of ∼0.8–1.0 nmand a rotation of ∼30◦, forming a right-handed super-helical structure.
Importin-β binds to the N-terminal of importin-α but also interacts with the nuclear pore complexand Ran GTPases. The interaction with Ran proteinsis located at its N-terminal whilst interaction withimportin-α is centred around the C-terminal region.Importin-β (Figure 8.81) also contains repeated struc-tural elements – the HEAT motif (the proteins in whichthe motif was first identified are Huntingtin pro-tein–Elongation factor (EF3)–A subunit of proteinphosphatase and the TOR protein). The motif possessesa pattern of hydrophobic and hydrophilic residues andis predicted to form two helical elements of secondarystructure. In a complex with the N-terminal fragment(residues 11 to 54) of the α subunit the HEAT motifsarrange in a convoluted snail-like appearance where the
Figure 8.81 Structure of importin-β plus IBB domainfrom a view down the superhelical axis. A and Bhelices of each HEAT motif are shown in red andyellow respectively. The acidic loop with the DDDDDWmotif in HR8 is shown in blue. (Reproduced withpermission from Cingolani, G. et al. Nature 1999, 399,221–229. Macmillan)

302 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
extended IBB domain is located at the centre of theprotein. Importin-β is composed of 19 HEAT repeatsforming a right-handed super-helix where each HEATmotif, composed of A and B helices connected by ashort loop, arranges into an outer layer of ‘A’ helicesdefining a convex surface and an inner concave layer of‘B’ helices. The HEAT repeats vary in length from 32to 61 residues with loop regions containing as many as19 residues. HEAT repeats 7–19 bind importin-α withthe N-terminal fragment of importin-α bound on theinner surface forming an extended chain from residues11–23 together with an ordered helix from residues24–51. The axis of this helix is approximately coinci-dent to that of importin-β super-helix. The N-terminalregion of the importin-α interacts specifically with along acidic loop linking helices 8A and 8B that con-tains five aspartate residues followed by a conservedtryptophan residue. HEAT repeats 1–6 are implicatedin the interaction with Ran.
The third protein with a critical role in nuclear–cytoplasmic transport is Ran – a GTPase essential fornuclear transport. Ran is a member of the Ras-like family of GTP binding proteins and switchesbetween active GTP bound forms and an inactivestate with GDP (Figure 8.82). Ran is similar to othermonomeric GTPases but it also possesses a long C-terminal extension that is critical for nuclear transport
Figure 8.82 The structure of Ran-GDP
function and is terminated by a sequence of acidicresidues (DEDDDL).
Ran consists of a six-stranded β sheet surroundedby five α helices. GDP and GTP are bound via theconserved NKXD motif with Lys123 and Asp125interacting directly with the base, the ribose portionexposed to the solvent and the phosphates binding viaa P loop motif and a large number of polar interactionswith the sequence GDGGTGKT. The loop regionacts as a switch through nucleotide-exchange inducedconformational changes and dictates interactions withother import proteins such as importin-β.
Ran itself is ‘regulated’ by a cytosolic GTPaseactivating protein termed Ran GAP1 that causesGTP hydrolysis and inactivation. In the nucleus achromatin-bound nucleotide exchange factor calledRCC1 functions to exchange GDP/GTP. The inter-play of factors controlling nucleotide exchange andhydrolysis generates gradients of Ran-GTP that directnuclear–cytoplasmic transport.
Ran binds to the N terminal region of importin-β, but importantly the C-terminal of Ran is able toassociate with other proteins largely through chargedresidues in the tail (DEDDDL). These charges mediateRan-GTP/importin-β complexes interaction with Ranbinding proteins in the nuclear pore complex butalso offer a mechanism of dissociating complexes inthe nucleus. Gradually many of the molecular detailsof protein import via nucleocytoplasmic transportmachinery have become clear with structures forthe above proteins and complexes formed betweenRan and RCC1 as well as the ternary complexof Ran–RanBP1-RanGAP. Ran, importin-α and -βsubunits are exported back to the cytosol for futurerounds of nucleocytoplasmic transport.
Proteins identify themselves to transport machinerywith two signals. In addition to the positively chargedNLS some proteins have nuclear export signals (NES)based around leucine-rich domains.
The nuclear pore assemblyIn higher eukaryotes the nuclear pore complex is amassive macromolecular assembly containing nearly100 different proteins with a combined mass in excessof 120 MDa. In yeast this assembly is simpler withapproximately 30 different polypeptides forming the

PROTEIN TURNOVER 303
CytoplasmicFilaments
CentralChannel
InternalFilaments Transporter
InternalFilaments
NucleoplasmicCoaxial Ring
CytoplasmicCoaxial Ring
CytoplasmicCoaxial Ring
Nuclear Basket
NE
Lamina
Radial armsSpoke
ComplexSpoke
ComplexNE lumen
Lamina
Figure 8.83 A cutaway representation of the nuclear pore complex (reproduced with permission from Allen, T.D.et al. J. Cell Sci. 2000, 113, 1651–1659. Company of Biologists Ltd)
complex. The increased size of the vertebrate poreprobably reflects complexity associated with metazoanevolution, but the basic structural similarities betweenthe pore complexes of yeast and higher eukaryotesindicates a shared mechanism of translocation that canbe discussed within a single framework.
The proteins of the nuclear pore complex arecalled nucleoporins or ‘nups’. As the role of Ran,importins and other import proteins were uncoveredover the last decade emphasis has switched to thestructure and organization of the nuclear pore complex.Electron microscopy reveals the pore complex as asymmetrical assembly containing a pore at the centrethat presents a barrier to most proteins. The nucleoporincomplex (NPC) spans the dual membrane of thenuclear envelope and is the universal gateway formacromolecular traffic between the cytoplasm and thenucleus. The basic framework of the NPC consists ofa central core with a spoke structure (Figure 8.83).From this central ring long fibrils 50–100 nm in lengthextend into the nucleoplasm and the cytoplasm whilstthe whole NPC is anchored within the envelope by thenuclear lamina.
Clarification of nuclear pore organization camefrom studies of the yeast complex where genetic
malleability coupled with genome sequencing revealed30 nucleoporins that are conserved across phyla andthus allowed identification of homologues in metazoaneukaryotes. Once identified nups were cloned andlocalized within the envelope using a combination ofimmunocytochemistry, electron microscopy and cross-linking studies. The yeast nuclear pore complex has beenredefined in terms of nucleoporin distribution. Nup358is equated with the RanBP2 protein described previouslywhilst p62, p58, p54 and p45 are proteins found atthe centre of the nuclear pore complex and gp210 andPOM121 form all or part of the spoke assembly.
Protein turnover
Within the cell sophisticated targeting systems directnewly translated proteins to their correct destinations.However, proteins have finite lifetimes and normal cellfunction requires specialized pathways for degradationand recycling. The half-life of most proteins ismeasured in minutes although some, such as actin orhaemoglobin, are more stable with half-lives in excessof 50 days. At some point all proteins ‘age’ as aresult of limited proteolysis, covalent modification ornon-enzymatic reactions leading to a decline in activity.

304 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
* 20 * 40 * 60 *HUMAN : MQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRGG : 76MOUSE : MQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRGG : 76YEAST : MQIFVKTLTGKTITLEVESSDTIDNVKSKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRGG : 76SOYABEAN : MQIFVKTLTGKTITLEVESSDTIDNVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLADYNIQKESTLHLVLRLRGG : 76GARDEN PEA : MQIFVKTLTGKTITLEVESSDTIDNVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLADYNIQKESTLHLVLRLRGG : 76CHICKEN : MQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRGG : 76FRUIT FLY : MQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRGG : 76FROG : MQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRGG : 76
Figure 8.84 The primary sequence of ubiquitin from plants, animal and microorganisms shows only four changesin sequence at positions 19, 24, 28 and 57
These proteins are degraded into constituent aminoacids and re-cycled for further synthetic reactions. Theproteasome is a large multimeric complex designedspecifically for controlled proteolysis found in allcells. In eukaryotes the ubiquitin system contributesto this pathway by identifying proteins destined fordegradation whilst organelles such as the lysosomeperform ATP-independent protein turnover.
The ubiquitin system and proteindegradation
In eukaryotes ubiquitin (Figure 8.84) occupies a pivotalrole in the pathway of protein degradation. It is a highlyconserved protein found in all eukaryotic organismsbut never in either eubacteria or archae. The levelof homology exhibited by ubiquitin is high with only3 differences in primary sequence between yeast andhuman protein. The lack of evolutionary divergencepoints to functional importance and suggests that mostresidues in ubiquitin have essential roles.
In view of its importance to protein degradationthe structure of ubiquitin (Figure 8.85) is unspectacularcontaining five β strands together with a single α
helix. The final three C-terminal residues are Arg-Gly-Gly and extend away from the globular domaininto bulk solvent. This arrangement is critical tobiological function as the C-terminal glycine formsa peptide bond with the ε-amino group of lysinein target proteins. Covalent modification by addingubiquitin in a process analogous to phosphorylationhas led to the terminology ubiquitination (also calledubiquitinylation). Multiple copies of ubiquitin attachto target proteins acting as ‘signals’ for degradation bythe proteasome.
Figure 8.85 The structure of ubiquitin showing foursignificant β strands (1–7,10–17, 40–45, 64–72) anda single α helix (23–34) whilst shorter strand and helixregions exists from 48–50 and 56–59, respectively.The residues Arg 74, Gly75 and Gly76 are shown alongwith Lys48
Many enzymes catalyse ubiquitin addition toproteins and include E1 enzymes or ubiquitin-activating enzymes, E2 enzymes, known as ubiquitin-conjugating enzymes, and E3 enzymes, which act asligases. Ubiquitin is activated by E1 in a reactionhydrolysing ATP and forming ubiquitin adenylate.The activated ubiquitin binds to a cysteine residue inthe active site of E1 forming a thiol ester. E2, theproximal donor of ubiquitin to target proteins, transfersubiquitin to the acceptor lysine forming a peptidebond, although E3 enzymes also participate by formingcomplexes with target protein and ubiquitin-loaded E2.Figure 8.86 shows the ubiquitin-mediated degradationof cytosolic proteins.

PROTEIN TURNOVER 305
SHE1
HO
C CH2
O
N Ubiquitin
ATP
AMP PPi
SE1 C CH2
O
N Ubiquitin
+
+
H
H
SE1 C CH2
O
N Ubiquitin
E2S
H
E1S
H
SE2 C CH2
O
N Ubiquitin
H
H
Figure 8.86 The first step in the ubiquitin-mediated degradation of cytosolic proteins is ATP dependent activationof E1 to form a thiol ester derivative with the C-terminal glycine of ubiquitin. It is followed by transfer of ubiquitinfrom E1 to the E2 enzyme, the proximal donor of ubiquitin to proteins
Multiple ubiquitination is common and involvespeptide bond formation between the carboxyl groupof Gly76 and the ε-amino group of Lys48 on asecond ubiquitin molecule. Substitution of Lys48with Cys results in a protein that does not supportfurther ubiquitination and is incapable of targetingproteins for proteolysis. Four copies of ubiquitin arerequired to target proteins efficiently to the 26S
proteasome with ‘Ub4’ units exhibiting structuralcharacteristics that enhance recognition. Ubiquitindoes not degrade proteins but tags proteins forfuture degradation. A view of ubiquitin as a simpletag may be an over-simplification since a role forubiquitin enhancing association between proteins andproteasome is implied by the observation that withoutubiquitin proteins interact with the proteasome butquickly dissociate.
Alternative strategies exist for degrading proteinswithin cells. One strategy used by cells is to inactivateproteins by oxidizing susceptible side chains such asarginine, lysine and proline. This event is enough to‘mark’ proteins for degradation by cytosolic proteases.A second method of protein turnover is identifiedwith sequence information and ‘short-lived’ proteinscontain sequences rich in proline, glutamate, serine andthreonine. From the single letter code for these residuesthe sequences have become known as PEST sequences.Comparatively few long-lived proteins contain PEST-rich regions and the introduction of these residues
into stable proteins increases turnover although thestructural basis for increased ability is unclear alongwith the relationship to the ubiquitin pathway. Athird mechanism of controlling degradation lies inthe composition of N-terminal residues. A correlationbetween the half-life of a protein and the identity of theN-terminal residue gave rise to the ‘N-end’ rule. Semi-quantitative predictions of protein lifetime from theidentity of the N-terminal residue suggests that proteinswith Ser possess half-lives of 20 hours or greaterwhilst proteins with Asp have on average half-livesof ∼3 min. The mechanism that couples recognitionof the N-terminal residue and protein turnover isunknown but these correlations are also observed inbacterial systems lacking the ubiquitin pathway ofdegradation.
The proteasomeIntracellular proteolysis occurs via two pathways:a lysosomal pathway and a non-lysosomal, ATP-dependent, pathway. The latter pathway degrades mostcell proteins and involves the proteasome first identifiedas an endopeptidase with multicatalytic activities frombovine pituitary cells. The multicatalytic protease wascalled the proteasome, reflecting its complex structureand proteolytic role.
The proteosome degrades proteins via peptide bondscission, and multiple catalytic functions are seen via

306 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
Figure 8.87 Superior views of α and β heptameric rings showing central cavity
the hydrolysis of many synthetic and natural substrates.The eukaryotic proteasome has activities described byterms such as ‘chymotryptic-like’ activity (preferencefor tyrosine or phenylalanine at the P1 position),‘trypsin-like’ activity (preference for arginine or lysineat the P1 position) and ‘post-glutamyl’ hydrolysingactivity (preference for glutamate or other acidicresidues at the P1 position). The variety of catalyticactivities created a confusing functional picture sinceproteasomes cleave bonds after hydrophobic, basicand acidic residues. In the proteasome the myriadof catalytic activities suggested a unique site. Theproteasome was first identified in eukaryotes butcomparable complexes occur in prokaryotes where thesystem can be deleted without inducing lethality. Asusual, eukaryotic proteasomes have complex structureswhilst those from prokaryotes are simpler.
The hyperthermophile Thermoplasma acidophilumproteasome contains two subunits of 25.8 kDa (α) and22.3 kDa (β) arranged in a 20S proteasome containing28 subunits: 14 α subunits and 14 β subunits arrangedin four stacked rings. The two ends of the cylindereach consist of seven α subunits whilst the twoinner rings had seven β subunits – each ring is ahomoheptamer. The α7β7β7α7 assembly forms a three-chambered cylinder with two antechambers located oneither sides of a central cavity (Figure 8.87). Sequencesimilarity exists between α and β subunits and there
is a common fold based around an antiparallel arrayof β strands surrounded by five helices. Helices 1 and2 are on one side of the β sandwich whilst helices3, 4 and 5 are on the other. In the case of the α
subunits an N-terminal extension of ∼35 residues leadsto further helical structure that fits into a cleft in theβ strand sandwich. In the β subunits this extensionis absent and the cleft remains ‘open’ forming partof the active site. The crystal structure of the 20Scomplex (Figure 8.88) showed a cylindrical assembly(length ∼15 nm, diameter 11.3 nm) with the channelrunning the length of the assembly but widening toform three large internal cavities separated by narrowconstrictions. The cavities between the α subunit andβ subunit rings are the ‘antechambers’ (∼4 × 5 nmdiameter) with the third cavity at the centre of thecomplex containing the active sites. Between theantechamber and the central cavity access is restrictedto approximately 1.3 nm by a loop region containinga highly conserved RPXG motif derived from the α
subunit. In this loop a Tyr residue protrudes furthestinto the channel to restrict access. Further into thechannel side chains derived from the β subunits existat the entrance to the central cavity and restrict thewidth to ∼2.2 nm. Together, these systems form agating system controlling entry of polypeptide chainsto those that thread their way into the central cavity. Ascheme of proteolysis involving protein unfolding and

PROTEIN TURNOVER 307
Figure 8.88 The three-dimensional structure of the 20S proteasome from the archaebacteria T. acidophilum (PDB:1PMA). The structure – a 673 kDa protease complex has a barrel-shaped structure of four stacked rings. An individualα subunit is shown top left, an individual β subunit bottom left. The colour scheme is maintained in the wholecomplex and shows only the distribution of secondary structure for clarity
the formation of extended structure prior to degradationis likely from the organization of the proteasome.
The active site is located on the β subunit wherethe structure reveals a β sandwich with one side pre-dominantly open. This side faces the inside pointingtowards the central cavity. The α subunits possess ahighly conserved N-terminal extension with residues1–12 remaining invisible in the crystal structure pos-sibly due to mobility whilst part of this extension isseen as a helix (residues 20–31) that occupies the cleftregion in the β sandwich. Although the precise func-tion of this extension is unknown its strategic locationcoupled with sequence conservation suggests an impor-tant role in translocation of substrate to the proteasomeinterior. The β subunits lack these N-terminal exten-sions but have pro-sequences of varying length thatare cleaved during assembly of the proteasome. The
most important function of this proteolytic processingis the generation of the active site residues.
The 14 identical catalytically active (β) subunitsshow highest activity for bond cleavage afterhydrophobic residues but extensive mutagenesisinvolving all serines, two histidines, a single cysteineand two conserved aspartate residues in the β subunitfailed to inhibit enzyme activity. The results suggesteddegradation was not associated with four classicalforms of protease action, namely serine, cysteine,aspartyl and metalloproteinases. Further mutagenesisrevealed that deletion of the N-terminal threonine orits replacement by alanine resulted in inactivation.Mechanistic studies showed that N -acetyl-Leu-Leu-norLeu-CHO (Figure 8.89) was a potent inhibitor ofproteolysis, and crystallography of the proteasome-inhibitor adduct located the peptide bound via the

308 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
NH
CH
CH2
CH2
CH
NHCH
CH2
CH
C
CH3CH3
CH2
CH
O
C
CH3CH3
O
NHC2H5
CHO
CH3
CH3
Figure 8.89 The potent inhibitor of proteasomeactivity N-acetyl-Leu-Leu-norLeu-CHO
aldehyde group to the –OH group of the N-terminal threonine.
Further insight into catalytic mechanism came withobservations that the metabolite lactocystin derivedfrom Streptomyces bound to the eukaryotic complexresulting in covalent modification of Thr1.1 The resultsimplied the involvement of N-terminal Thr residuesin catalytic reactions. Initially the proteasome foldwas thought to be a unique topology but graduallymore members of this family of proteins have beenuncovered with a common link being that they are allN-terminal nucleophile hydrolases or NTN hydrolases.
Fourteen genes contribute to the eukaryotic 20Sproteasome (Figure 8.90). Each subunit has a similarstructure but is coded by a separate gene. Crystallogra-phy and cryo-EM studies have shown that the patternsof subunit organization extend to eukaryotic protea-somes and when viewed in the electron microscopearchaeal, bacterial, and eukaryotic 20S proteasomesform similar barrel-shaped particles ∼15 nm in heightand ∼11 nm in diameter consisting of four heptamericrings (Figure 8.91).
The 20S assembly of the proteasome identifiedin eukaryotes, and first structurally characterized inT. acidophilum, is one component of a much largercomplex found in eukaryotes. The 20S proteasomerepresents the core region of a larger assembly that iscapped by a 19S regulatory complex. The attachmentof a regulatory ‘unit’ to the 20S proteasome results
1Surprisingly this inhibitor does not inactivate the Thermoplasmaproteasome.
in the formation of a 2.1 MDa complex called the26S proteasome.
With two 19S regulatory complexes attached tothe central 20S proteasome the 26S proteasomes fromDrosophila, Xenopus, rat liver and spinach cells havesimilar shapes with an approximate length of 45 nmand a diameter of 20 nm. The significance of the 19Sregulatory complex in association with the 20S assem-bly is that it confers ATP- and ubiquitin-dependentproteolysis on the proteasome; it is this pathway thatperforms most protein turnover within eukaryotic cellsdegrading over 90 percent of cellular proteins.
The organization of the 19S complex remains anactive area of research and is relatively poorly under-stood in comparison to the 20S assembly. The complexfrom yeast contains at least 18 different proteins whosesequences are known and reversible interactions occurwith proteins in the cytosol complicating compositionalanalysis. Within the group of 18 different subunits are sixpolypeptides hydrolysing ATP (ATPases) and showinghomology to other members of the AAA superfamily.The AAA superfamily represents ATPases associatedwith various cellular activities and are found in all organ-isms where a defining motif is the presence of one or moremodules of ∼230 residues that includes an ATP-bindingsite and the consensus sequence A/G-x4-G-K-S/T.
A general working model for the action of the pro-teasome envisages that the cap assembly binds ubiquiti-nated proteins, removes the ubiquitin at a later stage forrecycling and unfolds the polypeptide prior to entry intothe 20S core particle. The proteolytic active sites of theproteasome are found in the core particle and the capassembly does not perform proteolysis. However, free20S proteasome does not degrade ubiquitinated proteinconjugates and shows lower activity suggesting that the19S cap assembly is crucial to entry of substrate, activityand ubiquitin dependent hydrolysis.
Lysosomal degradation
Lysosomes contain hydrolytic enzymes including pro-teases, lipases, and nucleases. Any biomolecule trappedin a lysosome is degraded, and these organelles areviewed as non-specific degradative systems whilstthe ubiquitin system is specific, selective and ‘fine-tuned’ to suit cellular demand. Lysosomes are oftendescribed as enzyme sacs and defects in lysosomal

PROTEIN TURNOVER 309
Figure 8.90 The structural organization of the proteasome.
ThermoplasmaProteasome
RhodococcusProteasome
SaccharomycesProteasome
Figure 8.91 Cryo-EM derived maps of the assembly of 20S proteasomes from archaea, eubacteria and eukaryoticcells. The α type subunits are colored in red and the β type subunits in blue. The different shades of these colorsindicate that two or seven distinct α and β-type subunits form the outer and inner rings of the Rhodococcus orSaccharomyces proteasome respectively (reproduced with permission from Vosges, D. et al. Ann. Rev. Biochem. 1998,68, 1015–1068. Annual Reviews Inc)
proteins contribute to over 40 recognizable disorders,collectively termed lysosomal storage diseases.
Individually, the disease states are rare but lead toprogressive and severe impairment resulting from adeficiency in the activity of specific enzymes. The loss
of activity impedes the normal degradative functionof lysosomes and contributes to the accumulation ofunwanted biomolecules. Included in the collection ofidentified lysosomal based diseases are: (i) Hurler syn-drome, a deficiency in α-L-iduronidase required to

310 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
degrade mucopolysaccharides; (ii) Gauchers’ disease, adeficiency in β-glucocerebrosidase needed in the degra-dation of glycolipids; (iii) Fabry disease, caused bya deficiency in α-galactosidase, an enzyme degrad-ing glycolipids; (iv) Pompe disease, a deficiency ofα-glucosidase required to break down glycogen; and(v) Tay–Sachs disease, a lysosomal defect occurringas a result of a mutation in one subunit of the enzymeβ-hexosaminidase A that leads to the accumulation ofthe GM2 ganglioside in neurones.
Apoptosis
Apoptosis is the process of programmed cell death.Whilst superficially this appears to be an unwantedprocess more careful consideration reveals that apopto-sis is vital during development. A developing embryogoes through many changes in structure that can onlybe achieved by programmed cellular destruction. Sim-ilarly, the development of insects involves the reorga-nization of tissues between the larval and adult states.The concept of apoptosis is vital to normal develop-ment and although many details, such as control andinitiation remain to be elucidated, the importance ofa family of intracellular proteases called caspases iswell documented.
Caspases are synthesized as zymogens with theiractivity inhibited by covalent modification, in whichthe active enzyme is synthesized as a precursor proteinjoined to additional death effector domains. Thisconfiguration is vital to achieve complete inhibitionof activity since the cell is easily destroyed by theactivity of caspases. Activation involves a large numberof ‘triggers’ that excise the death effector domains,process the zymogen and form active caspases. Onceactivated caspases catalyse the formation of othercaspases, leading to a proteolytic cascade of cellulardestruction. Several methods of regulating caspaseactivity and hence apoptosis exist within the cell.Zymogen gene transcription is regulated whilst anti-apoptotic proteins such as those of the Bcl-2 familyoccur in cells and block activation of certain pro-caspases.
The first members of the caspase family identifiedwere related to human interleukin-1 converting enzyme(ICE) and the product of the nematode cell-death gene
CED3. To date 11 caspases have been identified inhumans although mammals may possess 13 differentcaspases, Drosophila melanogaster contains seven andnematodes such as Caenorhabditis elegans containthree. Thus, there appears to have been an expansion intheir number with increasing cellular complexity. Theterm caspase refers to the action of these proteins ascysteine-dependent aspartate-specific proteases. Theirenzymatic properties are governed by specificity forsubstrates containing Asp and the use of a Cys285sidechain for catalysing peptide-bond cleavage locatedwithin a conserved motif of five residues (QACRG).Whereas the activity of the proteasome governsthe day to day turnover of proteins the activityof caspases signals the end of the cell with allproteins degraded.
SummaryFifty years have passed since the discovery of thestructure of DNA. This event marking the beginningof molecular biology expanded understanding of allevents in the pathway from DNA to protein. Thisincluded structural and functional descriptions of pro-teins involved in the cell cycle, DNA replication,transcription, translation, post-translational events andprotein turnover. In many instances determining struc-ture has uncovered intricate details of their biologi-cal function.
The cell cycle is characterized by four distinctstages: a mitotic or M phase is followed by a G1 (gap)phase that represents most of the cell cycle, a period ofintense synthetic activity called the S phase, and finallya short G2 phase as the cell prepares for mitosis.
Genetic studies of yeast identified mutants in whichcell division events were inhibited. Control of the cellcycle is mediated by protein kinases known as Cdkswhere protein phosphorylation regulates cellular activ-ity. Activity of Cdks is dependent on cyclin bindingwith optimal activity occurring for Cdk2 in a com-plex with cyclin A and phosphorylation of Thr160.The structure of this complex results in the criticalmovement of the T loop, a flexible region of Cdk2,governing accessibility to the catalytic cleft and theactive site threonine.
Transcription is DNA-directed synthesis of RNAcatalysed by RNA polymerase. Transcription proceeds

SUMMARY 311
from a specific sequence (promoter) in a 5′ –3′ directionuntil a second site known as the transcriptionalterminator is reached. In eukaryotes three nuclear RNApolymerases exist with clearly defined functions. RNApolymerase II is concerned with the synthesis of mRNAencoding structural genes.
Sequences associated with transcriptional elementshave been identified in prokaryotes and eukaryotes. Ineukaryotes the TATA box is located upstream of thetranscriptional start site and governs formation of apre-transcriptional initiation complex. The TATA boxresembles the Pribnow box or −10 region found inprokaryotes.
Specific TATA binding proteins have been identifiedand structural studies reveal that basal transcriptionrequires in addition to RNA polymerase the formationof pre-initiation complexes of TBP, TFIIB, TFIIE,TFIIF and TFIIH.
In eukaryotes transcription is followed by mRNAprocessing that involves addition of 5′ G-caps and 3′polyA tails. Non-coding regions of mRNA known asintrons are removed creating a coherent translation-effective mRNA. Introns are removed by the spliceo-some.
The spliceosome contains snRNA complexed withspecific proteins. Processing initial mRNA transcriptsinvolves cutting at specific pyrimidine rich recognitionsites followed by splicing them together to createmRNA that is exported from the nucleus for translationat the ribosome.
Translation converts mRNA into protein and occursin ribonucleoprotein components known as the ribo-somes. Ribosomes convert the genetic code, a series ofthree non-overlapping bases known as the codon, intoa series of amino acids covalently linked together in apolypeptide chain.
All ribosomes are composed of large and smallsubunits based predominantly on highly conservedrRNA molecules together with over 50 differentproteins. Biochemical studies identified two major sitesknown as the A and P sites. The P site (peptidyl)contains a growing polypeptide chain attached totRNA. The A site (aminoacyl) contains charged tRNAspecies bearing a single amino acid that will be addedto extending chains.
Protein synthesis is divided into initiation, elon-gation and termination. All stages involve accessory
proteins such as IF1, IF2 and IF3 together with elon-gation and release factors such as EF1, EF2, EF3, RF1,RF2 and RF3.
Elongation is the most extensive process in proteinsynthesis and is divided into three steps. Theseprocesses are amino acyl tRNA binding at the A site,peptidyl transferase activity and translocation.
Structures for 50 and 30S subunits revolutionizedunderstanding of ribosome function. The structureof the large subunit confirmed conclusively that thepeptidyl transferase reaction is catalysed entirely byRNA; the ribosome is a ribozyme.
Initial translation products undergo post-translationalmodification that vary dramatically in type from oxi-dation of thiol groups to the addition of new covalentgroups such as GPI anchors, oligosaccharides, myris-tic acid ‘tails’, inorganic groups such as phosphate orsulfate and larger organic skeletons such as heme.
The removal of peptide ‘leader’ sequences in theactivation of zymogens is another important post-translational modification and converts inactive proteininto an active form. Many enzymes such as proteolyticdigestive enzymes, caspases and components of theblood clotting cascade are activated in this typeof pathway.
N-terminal signal sequences share physicochemicalproperties and are recognized by a SRP. The SRPdirects nascent chains to the ER membrane or cellmembrane of prokaryotes. Signal sequences do notexhibit homology but have a basic N-terminal regionfollowed by a hydrophobic core and a polar C-terminalregion proximal to the cleavage site.
SRPs are found in the archae, eubacteria andeukaryotes. Mammalian SRP is the most extensivelycharacterized system consisting of rRNA and sixdistinct polypeptides. The SRP directs polypeptidechains to the ER membrane and the translocon, amembrane bound protein-conducting channel.
Other forms of intracellular protein sorting existwithin eukaryotes. Proteins destined for the mitochon-dria, chloroplast and nucleus all possess signals withintheir polypeptide chains. For the nucleus protein importrequires the presence of a basic stretch of residuesarranged either as a single block or as a bipartite struc-ture anywhere within the primary sequence.
NLS are recognized by specific proteins (importins)that bind the target protein and shuttle the ‘cargo’

312 PROTEIN SYNTHESIS, PROCESSING AND TURNOVER
towards the nuclear pore complex. The formation ofimportin–cargo complexes is controlled by G proteinssuch as Ran.
Proteins are not immortal – they are degraded withturnover rates varying from minutes to weeks. Turnoveris controlled by a complex pathway involving ubiquitinlabelling.
Ubiquitin is a signal for destruction by the protea-some. The proteasome has multiple catalytic activitiesin a core unit based around four heptameric rings.The 20S proteasome from T. acidophilum has anα7β7β7α7 assembly forming a central channel guardedby two antechambers. The central chamber catalyses
proteolysis based around the N terminal threonineresidue (Thr1) where the side chain acts as anucleophile attacking the carbonyl carbon of peptidebonds.
In prokaryotes the proteasome degrades proteins inubiquitin independent pathways. In eukaryotes the 20Sproteasome catalyses ubiquitin-dependent proteolysisonly in the presence of a cap assembly.
Further pathways of protein degradation existin the lysosome where defects are responsible forknown metabolic disorders, such as Tay–Sachs dis-ease, and by caspases that promote programmed celldeath (apoptosis).
Problems
1. Using knowledge about the genetic code describethe products of translation of the following syntheticnucleotide . . . . . .AUAUAUAUAUAUA. . .. in a cellfree system. Explain the results and describe the criticalrole of alternating oligonucleotides in determining thegenetic code. Using the data presented in Table 8.6how would this translation product differ using asystem reconstituted from mitochondria.
2. Arrange the following macromolecules in decreasingorder of molecular mass: tRNA, subunit L23, tetra-cycline, 23S rRNA, the large ribosomal subunit, 5SRNA, spliceosome protein U1A, E. coli DNA poly-merase I, E. coli RNA polymerase. Obtain infor-mation from databases or information given withinChapter 8.
3. Recover the structure of the cyclinA: cdk2 binarycomplex from a protein database. Using any moleculargraphics package highlight the following residuesLys266, Glu295, Leu255, Asp240, and Arg211 ofcyclin A and Glu 8, Lys 9, Asp36, Asp38 Glu40and Glu42 of cdk2. Describe the arrangement ofthese residues.
4. Puromycin binds to the A site and prevents furtherchain elongation. From the structure presented inFigure 8.54 account more fully for this observation.
5. The Yarus inhibitor is described as a transition stateanalogue that mimics the tetrahedral intermediategenerated when the α amino group of the A site boundamino acyl tRNA attacks the carbonyl group of theester linking a peptide to the peptidyl tRNA at the Psite. Draw the normal assumed tetrahedral intermediatemost closely linked to this inhibitor highlighting thetetrahedral carbon centre.
6. An N-terminal photo-affinity label has been usedto map the tunnel of the large ribosomal subunit.Describe how you might perform this experiment andwhat results might you expect to obtain from sucha procedure?
7. Unlike the mitochondria and chloroplast signal se-quence nuclear localization signals are not removed.What might be the reasons underlying this observation?
8. Caspases are proteolytic enzymes with an active sitecysteine. Discuss possible enzyme mechanisms in viewof the known activity of serine proteases.

9Protein expression, purification
and characterization
To study the structure of any protein successfully itis necessary to purify the molecule of interest. This isoften a formidable task especially when some proteinsare present within cells at low concentration, perhaps asfew as 10 molecules per cell. Frequently, this involvespurifying a single protein from a cell paste containingover 10 000 different proteins. The ideal objective isto obtain a single protein retaining most, if not all,of its native (in vivo) properties. This chapter focuseson the methods currently employed to isolate andpurify proteins.
Although functional studies often require smallamounts of protein (often less than 1 ng or 1 pmol),structural techniques require the purification of proteinson a larger scale so that 10 mg of pure protein issometimes needed. Taken together, the requirement forpurity and yield often places conflicting demands on theexperimentalist. However, the successful generation ofhigh-resolution structures is testament to the increasedefficacy of methods of isolation and purificationused today in protein biochemistry. Many of thesemethods evolved from simple, comparatively crude,protocols into sophisticated computer-controlled andenhanced procedures where concurrent advances inbioinformatics and material science have supportedrapid progress in this area.
The isolation and characterizationof proteins
Two broad alternative approaches are available todayfor isolating proteins. We can either isolate the proteinconventionally by obtaining the source cell or tissuedirectly from the host organism or we can usemolecular biology to express the protein of interest,often in a host such as Escherichia coli. Todaymolecular biology represents the most common routewhere DNA encoding the protein of interest is insertedinto vectors facilitating the high level expression ofprotein in E. coli.
Recombinant DNA technologyand protein expression
Prior to the development of recombinant proteinexpression systems the isolation of a protein frommammalian systems required either the death of theorganism or the removal of a small selected piece of tis-sue in which it was suspected that the protein was foundin reasonable concentrations. For proteins such ashaemoglobin removal of small amounts of blood does
Proteins: Structure and Function by David Whitford 2005 John Wiley & Sons, Ltd

314 PROTEIN EXPRESSION, PURIFICATION AND CHARACTERIZATION
not present ethical or practical problems. Given thathaemoglobin is found in a soluble state and at very highnatural concentrations (∼145 g l−1 in humans) it is notsurprising that this protein was amongst the first to bestudied at a molecular level. However, many proteinsare found at much lower concentrations and in tissuesor cells that are not easily obtained except post mortem.
Recombinant systems of protein expression havealleviated most problems of this type and allow thestudy of proteins that were previously inaccessible.Once DNA encoding the protein of interest has beenobtained it is relatively routine to obtain proteins viarecombinant hosts in quantities one could previouslyonly dream about (i.e. mg–g amounts). This does notmean that it is always possible to express proteinsin E. coli. For example, membrane proteins stillpresent numerous technical problems in heterologousexpression, as do proteins rich in disulfide bondsor those bearing post-translational modifications suchas myristoylation and glycosylation. Eukaryotic cellscarry out many of these modifications routinely and thishas led to the development of alternative expressionsystems besides E. coli. Alternative expression hostsinclude simple eukaryotes such as Saccharomycescerevisiae and Pichia pastoris, as well as more complexcell types. The latter group includes cultured insectcells such as Spodoptera frugiperda infected withbaculovirus vectors, Drosophila cell-based expression
systems and mammalian cell lines often derived fromimmortalized carcinomas.
However, the most common approach and the routegenerally taken in any initial investigation is to express‘foreign’ proteins in E. coli. These methods haverevolutionized studies of protein structure and functionby allowing the expression and subsequent isolation ofproteins that were formerly difficult to study perhapsas a result of low intrinsic concentration withincells or tissues. Of importance to the developmentof recombinant DNA technology was the discoveryof restriction enzymes (see Chapter 7) and their usewith DNA ligases to create new (recombinant) DNAmolecules that could be inserted into a carrier moleculeor vector and then introduced into host cells such asE. coli (Figure 9.1). The process of constructing andpropagating new DNA molecules by inserting into hostcells is referred to as ‘cloning’. A clone is an exactreplica of the parent molecule, and in the case of E.coli this results, after several cycles of replication, inall cells containing new and identical DNA molecules.
The generation of new recombinant DNA moleculesinvolves at least five discernable steps (Figure 9.2):
1. Preparation of DNA. Today DNA is usually gener-ated using the polymerase chain reaction (PCR). Thetechnique, described in Chapter 6, involves the actionof a thermostable DNA polymerase with forward and
Figure 9.1 The use of restriction enzymes to generate DNA fragments and the use of DNA ligase to join togethervector and insert together to form a new recombinant (chimeric) circular DNA molecule. The diagram shows thejoining together of a cut vector and a cut fragment described as having sticky ends. This type of end occurswith most restriction enzymes and is contrasted with the smaller group of type II restriction endonucleases thatproduce blunt-ended fragments (reproduced with permission from Voet, D. Voet, J.G and Pratt, C.W. Fundamentals ofBiochemistry. John Wiley & Sons Ltd., Chichester, 1999)

RECOMBINANT DNA TECHNOLOGY AND PROTEIN EXPRESSION 315
Figure 9.2 Generalized scheme for the creation of recombinant DNA molecules
reverse primers in a series of cycles involving denat-uration, annealing and extension. The result is largequantities of amplified and copied DNA. TemplateDNA is preferably derived from cDNA via the actionof reverse transcriptase on processed mRNA. Thisavoids the presence of introns in amplified sequences.In other instances DNA is derived from the productsof restriction enzyme action.
2. Restriction enzyme cleavage. The generation ofDNA fragments of specific length and with ‘tai-lored’ ends was vital to the success of recombi-nant DNA technology. This allowed the ‘insert’ (thename given to the DNA fragment) to be joined into avector containing compatible sites (see Figure 9.1).Today the generation of PCR DNA fragments nor-mally includes sites for restriction enzymes thatfacilitate subsequent cloning reactions and today awide variety of restriction enzymes are commer-cially available.
3. Ligation of DNA fragments. As part of the overallcloning process insert and vector DNA are joined ina process called ligation. Without covalent linkageof pieces of DNA and the formation of closed
circular DNA subsequent transformation reactions(see step 4) occur with very low efficiency.
4. Transformation. The propagation of recombinantDNA molecules requires the introduction of cir-cular DNA (ligated vector + insert) into a suitablehost cell. Replication of the cells generates manycopies of the new recombinant DNA molecules.The whole process is called transformation and rep-resents the uptake of circular DNA by host cellswith the acquisition of new altered properties. Theexact process by which cells take up DNA is unclearbut two common methods used for E. coli involvethe application of a high electric field (electropo-ration) or a mild heat shock. The result is thatmembranes are made ‘leaky’ and take up smallcircular DNA molecules. For E. coli the processof transformation requires special treatment andcells are often described as ‘competent’ meaningthey are in a state to be transformed by plas-mid DNA.
5. Identification of recombinants. After transformationcells such as E. coli can be grown on nutrient richplates and it is here that marker genes found in many

316 PROTEIN EXPRESSION, PURIFICATION AND CHARACTERIZATION
vectors are most useful. Normal E. coli cells usedin the laboratory lack resistance to antibiotics suchas ampicillin, tetracycline or kanamycin. However,transformation with a vector containing one or moreantibiotic resistance genes leads to resistant cells. Asa result when cells are grown on agar plates contain-ing an antibiotic only those cells transformed with aplasmid conferring resistance will grow. This selec-tion procedure allows the screening of cells so thatonly transformed cells are studied further. By trans-forming with known amounts of plasmid DNA it ispossible to calculate transformation efficiencies andit is not unusual for this value to reach 109 –1010
transformants/µg DNA. Further identification of therecombinant DNA might involve isolation of theplasmid DNA and its digestion with restrictionenzymes to verify an expected pattern of fragments.Definitive demonstration of the correct DNA frag-ment would involve nucleotide sequencing.
One of the first vectors to be developed was calledpBR322 and it is the ‘ancestor’ of many vectorsstill used today for cloning. The vector pBR322(Figure 9.3) contains distinctive sequences of DNAthat includes an origin of replication (ori ) gene as
well as a gene (rop) that control DNA replication andthe number of copies of plasmid found within a cell.In addition pBR322 contains two ‘marker’ genes thatcode for resistance to the antibiotics tetracycline andampicillin. These useful markers are denoted by tetR
and ampR.The next step requires the transfer of DNA from the
cloning vector to a vector optimized specifically forprotein expression. A cloning vector such as pBR322will allow several copies of the plasmid to be presentin cells and may facilitate sequence analysis or analysisof restriction sites but they are rarely used for proteinexpression. Other common cloning vectors used widelyin molecular biology and frequently described in sci-entific literature are pUC, pGEM, pBluescript. Eachvector has many variants containing different restric-tion enzyme sites that allow the orientation of the insertin either direction as well as common features suchas one or more antibiotic resistance genes, multiplecloning sites, an origin of replication, and a medium tohigh copy number.
In contrast, expression vectors are designed withadditional features beneficial to protein production. Thevectors retain many of the features of ‘cloning vehicles’
EcoRI (4360)
PstI (3612)
ClaI (25)
HindIII (30)
BamHI (376)
AvaI (1426)
rop
pBR322
4361 bp
ori
tet (R)amp(R)
Figure 9.3 Plasmid vector pBR322. The small circular DNA molecule contains 4361 bp, the rop, ori, ampR and tetR
genes as well as numerous sites for restriction enzymes such as EcoRI, BamHI, AvaI, PstI, ClaI and HindIII

RECOMBINANT DNA TECHNOLOGY AND PROTEIN EXPRESSION 317
such as multicloning sites, antibiotic resistance genesand the rop and ori genes but have in most cases:
1. An inducible promoter. Expressed proteins mayprove toxic to the host organism. To limit this effectthe activity of the promoter can be ‘turned on’by an inducer. Although the mechanism of actionof inducers varies from one system to another themost common system in use involves the promoterderived from the lac operon of E. coli. In manysystems the sequence of this promoter has beenmodified to contain a ‘consensus’ sequence of basesfor optimal activity (see Chapters 3 and 8). The lacoperon (Figure 9.4) is a group of three genes – β-galactosidase, lactose permease and thiogalactosidetransacetylase – whose activities are induced whenlactose is present in the medium. The lac repres-sor is a homotetrameric protein with an N-terminaldomain containing the HTH motif found in manytranscription factors. It specifically binds DNAsequences in the operator region inhibiting tran-scription. A non-metabolized structural analogueof lactose, the natural inducer, is used to induceprotein expression. Isopropylthiogalactose (IPTG)binds the lac repressor removing the inhibitionof transcription (Figure 9.5). This system is thebasis for most inducible expression vectors used ingenetic engineering studies today.
OCH2OH
S
H
H OH
OH
HOH H
CH3
CH3
H
Figure 9.5 The structure of isopropylthiogalactose(IPTG)
2. Restriction enzyme sites. A large number of restric-tion sites are often clustered together in so calledmulti-cloning sites. These sites allow the insert to beplaced ‘in frame’ with the start codon and in a cor-rect relationship with the Shine–Dalgarno sequence.Two restriction enzymes, NcoI and NdeI, cut at thesequence C↓CATGG and CA↓TATG respectively.Careful inspection of this sequence reveals that ATGis part of the start codon and fragments cut withNcoI/NdeI will, if ligated correctly, be in frame withthe start codon. This will yield the correct proteinsequence upon translation.
3. A prokaryotic ribosome binding site containing theShine–Dalgarno sequence. The Shine–Dalgarnosequence has a consensus sequence of AGGAGAlocated 10–15 bp upstream of the ATG codon.
Figure 9.4 The expression of the lac operon. In the absence of inducer (IPTG) the repressor binds to the operatorpreventing transcription of the three genes of the lac operon. IPTG binds to the repressor allowing transcription toproceed. Most expression vectors contain the lacI gene together with the lac promoter as their inducible transcriptionsystem. (Reproduced with permission Voet, D. Voet, J.G and Pratt, C.W. Fundamentals of Biochemistry 1999. JohnWiley & Sons Ltd., Chichester)

318 PROTEIN EXPRESSION, PURIFICATION AND CHARACTERIZATION
Successful expression requires the use of the hostcells synthetic machinery and in particular forma-tion of an initiation complex for translation. The 16SrRNA binds to purine rich sequences (AGGAGA)as a result of sequence complementarity enhanc-ing formation of the initiation complex. The com-position and length of the intervening sequencesbetween the ribosomal binding site and the startcodon are important with structures such as hairpinloops decreasing expression.
4. A transcriptional terminator. In an ideal situationmRNA is transcribed only for the desired proteinand is then halted. To achieve this aim many expres-sion vectors contain transcriptional terminators. Ter-minators are DNA sequences that cause the disas-sembly of RNA polymerase complexes, limit theoverall mRNA length and are G-C rich and palin-dromic. The formation of RNA hairpin structuresdestabilizes DNA–RNA transcripts causing termi-nation. In other cases termination results from theaction of proteins like the Rho-dependent termina-tors in E. coli.
5. Stop codons. In frame stop codons prevent transla-tion of mRNA by ribosomes and their presence aspart of the ‘insert’ or as part of the expression vectoris desirable to ensure the absence of extra residuesin the expressed protein.
6. Selectable markers. One or more selectable markerssuch as the genes for ampicillin or tetracycline resis-tance allows the expression vector to be selected andmaintained.
Extensive discussion of all of these features isbeyond the scope of this book but one set of widelyused expression systems are the pET-based vectors.These vectors are based on the bacteriophage T7 RNApolymerase promoter, a tightly regulated promoter,that allows high levels of protein expression. A largenumber of variants exist on a basic theme. The pETvector is approximately 4600 bp in size, contains eitherthe AmpR and KanR antibiotic resistance genes, aswell as the normal ori and rop genes. The pETvectors (Figure 9.6) have regions containing sitesfor many Type II restriction enzymes and genesare cloned downstream under control of T7 RNApolymerase promoter. It might be thought that host
RNA polymerases would initiate transcription from thispromoter but it turns out that this promoter is highlyselective for T7 RNA polymerase. One consequenceis that background protein expression in the absenceof T7 RNA polymerase is extremely low and this isdesirable when expressed proteins are toxic to hostcells. However, expression of foreign cDNA requiresT7 RNA polymerase – an enzyme not normally foundin E. coli – and to overcome this problem all hostcells contain a chromosomal copy of the gene for T7RNA polymerase. The addition of IPTG to culturesof E. coli (DE3) leads to the production of T7 RNApolymerase that in turn allows transcription of thetarget DNA.
One of the major advantages of bacterial cellsis their ease of culture allowing growth in a ster-ile manner, in large volumes and with short dou-bling times (∼40 minutes under optimal conditions).E. coli cells are easy to transform with vector DNA,especially when compared with eukaryotic cells, andgrowth media are relatively cheap. Large amounts ofexpressed protein can be obtained from E. coli andit is not unusual for the expressed protein to exceed10 percent of the total cell mass. The success ofgenetic engineering has been demonstrated by pro-duction of large amounts of therapeutically importanteukaryotic proteins using E. coli expression systems.Human insulin, growth hormones, blood coagulationfactors IX and X as well as tissue plasminogen acti-vator have all been successfully expressed and usedto treat diseases such as diabetes, haemophilia, strokesand heart attacks.
Purification of proteinsAdvances in recombinant DNA technology allow large-scale protein expression in host cells. Demonstra-tion of protein expression is usually confirmed bySDS–polyacrylamide gel electrophoresis (see below)and is then followed by the question: how is theprotein to be purified? An effective purification strat-egy requires protein recovery in high yield andwith high purity. Achieving these two objectives canbe difficult and is usually resolved in an empiri-cal manner.
The first step in any purification strategy involvesthe fractionation of cells by mechanical disruption

PURIFICATION OF PROTEINS 319
pET-21a(+)(5443bp)
Xho I (158)Not I (166)Eag I (166)Hind III (173) Sal I (179)Sac I (190)EcoR I (192)BamH I(198)
lacI(714-1793)
Ap(3988-4845)
ori (3227)
f1 origin(4977-5432)
Sty I(57)
ScaI (4538)
EcoR V(1514)
Sph I (539)
ApaI (1275)
Sap I (3049)
Nde I (237)
Figure 9.6 The pET series of vectors. Identifiable elements within the vector include the T7 promoter (311–327),T7 transcription start (310), T7 Tag coding sequence (207–239), Multiple cloning sites (BamHI–XhoI) (158–203),His-Tag coding sequence (140–157), T7 terminator region (26–72), lacI coding sequence (714–1793), pBR322origin of replication (3227), and the antibiotic resistance coding sequence (3988–4845)
or the use of chemical agents such as enzymes.Mechanical disruption methods can involve sonica-tion where high frequency sound waves (ultrasoundfrequencies > 20 kHz) disrupt membrane structure.Other methods include mechanical shearing by ruptur-ing membranes by passing cells through narrow orificesunder extremely high pressure or grinding cells withabrasive material such as sand or glass beads to achievesimilar results.
A different method of disruption involves the useof enzymes such as lysozyme to disrupt cell wallsin Gram-negative bacteria like E. coli or chitinase todegrade the complex polysaccharide coats associatedwith yeast. Lysozyme acts by splitting the complex
polysaccharide coat between N -acetylmuramic acidand N -acetylglucosamine units in the peptidoglycanlayer (Figures 9.7 and 9.8). Multiple glycosidic bondcleavage weakens the cell envelope and the E. colimembrane ‘sac’ is easily broken by osmotic shockor mild sonication to release cytoplasmic contentsthat are separated from membranes by differentialcentrifugation.
Purification strategies normally exploit the physicalproperties of the protein as a means to isolation. Ifprimary sequence information is available then predic-tions about overall charge, disulfide content, solubil-ity, mass or hydrophobicity can be obtained. Bioin-formatics may allow recognition of similar proteins

320 PROTEIN EXPRESSION, PURIFICATION AND CHARACTERIZATION
- -
Figure 9.7 A diagram of the organization of the peptidoglycan layer in Gram-positive and Gram-negative bacteria.The peptidoglycan layer is a complex structure consisting of covalently linking carbohydrate and protein componentsand is more extensive in gram positive bacteria. Bacteria are defined as Gram-positive or -negative according towhether heat fixed cells retain a dye (crystal violet) and iodine after destaining with alcohol. Gram-positive bacteriaallow the dye into the cells and heat fixing causes changes in the peptidoglycan layer that prevent the escape of thedye during destaining. In contrast the smaller peptidoglycan layer of Gram-negative bacteria allows the escape ofdye during destaining (reproduced with permission Voet, D. Voet, J.G and Pratt, C.W. Fundamentals of Biochemistry.John Wiley & Sons Ltd., Chichester, 1999)
and therefore the implementation of comparable purifi-cation methods. However, in some circumstances theprotein is ‘new’ and sequence information is absent.To obtain sequence information it is necessary topurify the protein yet to purify the protein it is oftenadvantageous to have sequence information! Exploit-ing physical properties and judicious use of selec-tive methods allows the purification of a proteinfrom cell extracts containing literally thousands ofunwanted proteins to a single protein (homogeneity)in a few steps.
CentrifugationThe principle of centrifugation is to separate parti-cles of different mass. Strictly speaking this is notentirely accurate because centrifugation depends onother factors such as molecular shape, temperatureand solution density. Heavy particles sediment fasterthan light particles – a fact demonstrated by mixingsand and water in a container and waiting for a fewminutes after which time the sand has settled to thebottom of the container. This reflects our everydayexperience that heavier particles are pulled down-wards by the earth’s gravitational force. However,
a close inspection of the solution would reveal thatsmall (microscopically small) grains remain suspendedin solution. This also happens to macromoleculessuch as proteins where, as a result of Brownian dif-fusion, their buoyant density counteracts the effectof gravity.
Fractionation procedures (Figure 9.9) are importantin cell biology and are used extensively to purifycells, subcellular organelles or smaller componentsfor further biochemical analysis. Disruption of E. colicells produces membrane and cytosolic fractions, eachcontaining particles of varying mass. These particlesare separated by centrifugation, a process enhancingrates of sedimentation by increasing the force acting onparticles within solutions. Centrifugation involves therotation of solutions in tubes within specially designedrotors at frequencies ranging from 100 revolutionsper minute up to ∼80 000 r.p.m. More commonlycentrifuges are used at much lower g values and aforce of 20 000 g is sufficient to pellet most membranesfound in cells as well as larger organelles such asmitochondria, chloroplasts, and the ER systems ofanimal and plant cells.
In the preceding section centrifugation has beendescribed as a preparative tool but it also finds an

CENTRIFUGATION 321
Figure 9.8 The arrangement of β(1–4) linked N-acetylglucosamine (NAG) and N-acetylmuramic acid (NAM) togetherwith the linkage to the tetrapeptide bridge of L-Ala, D-isoglutamate, L-Lys and D-Ala peptidoglycan. The tetrapeptidebridges are joined together by five Gly residues that extend from the carboxyl group of one tetrapeptide to theε-amino group of lysine in another. The isoglutamate residue is so called because the side chain or γ carboxyl groupforms a peptide bond with the next amino group. The arrangement of units within the peptidoglycan layers hasbeen studied particularly extensively for Staphylococcus aureus but considerable variation in structure can occur. Thepresence of D-amino acids renders the peptidoglycan layer resistant to most proteases (Reproduced with permissionfrom Voet, D. Voet, J.G. Pratt, C.W. Fundamentals of Biochemistry. John Wiley & Sons Ltd., Chichester, 1999)
application as a precise analytical tool. Centrifugedsolutions are subjected to strong forces that vary alongthe length of the tube or more properly the radius(r) about which rotation occurs. The centrifugal forceacting on any particle of mass m is the productof a particle’s mass multiplied by the centrifugalacceleration (from Newton’s second law of motion).This centrifugal acceleration is itself the product of theradius of rotation and the angular velocity (rω2). Forany particle the net force acting on it will be a balancebetween centrifugal forces causing particles to pelletand buoyant forces acting in the opposite direction.Particles that are less buoyant than the solvent will sinkwhilst those that are lighter than the solvent will float.This results in the following expression describing the
forces acting on a particle during centrifugation:
Net force = centrifugal force − buoyant force (9.1)
= ω2rm − ω2rms (9.2)
= ω2rm − ω2rνρ (9.3)
where ms is the mass of solvent displaced by theparticle, ν is the volume of the particle and ρ isthe density of the solvent. If, for the moment, thesecond term on the right hand side of the equationis neglected the centrifugal force is simply the angularvelocity multiplied by the radius of rotation. Usually,the value of the force applied to particles duringcentrifugation is compared to the earth’s gravitationalforce (g, ∼9.8 m2 s−1) and the dimensionless quantity

322 PROTEIN EXPRESSION, PURIFICATION AND CHARACTERIZATION
Figure 9.9 The fractionation of cells. After initial tissue maceration the solution is filtered to remove large debrisand centrifuged at low speeds to remove unbroken cells (<1000 g). Progressive increases in centrifugal forces pelletsmaller cellular organelles and eventually at the highest speeds large protein complexes may be isolated
called the relative centrifugal force (RCF) is used.
RCF = centrifugal force/force due to gravity (9.4)
= ω2rm/m g (9.5)
= ω2r/g (9.6)
To determine the maximum relative centrifugal forceit is therefore only necessary to know the radius ofthe rotor and the speed of rotation. The radius isfixed for a given rotor and the speed of rotationis a user-defined parameter. Although RCF is adimensionless quantity it is traditionally quoted as anumerical value; for example 5000 g meaning 5000 ×the earth’s gravitational force. Since ω the angularvelocity, in radians per second, is the number ofrevolutions per second multiplied by 2π the RCF ofa 10 cm radius rotor rotating at 8000 r.p.m. is
RCF = (2π × 8000/60)2 × 0.1/9.8 ∼ 7160 g (9.7)
This is actually the g force at the end of the rotortube. More careful consideration reveals that the gforce will vary along the radius of the tube. Inthe above example the g force at a position 5 cmalong the tube is approximately 3580 g and this hasfrequently led to the use of a term gav to reflect averagecentrifugal force.
In analytical methods of centrifugation the secondterm of Equation 9.2, the buoyant force, must beconsidered. In most applications this term is ‘invisible’since rotor speeds are such that membrane or largeorganelles are forced to sediment whilst the lighterparticles remain in solution. An extension of thisrelationship allows the size or molecular mass ofproteins to be determined.
During centrifugation at high speeds the initialacceleration of a particle due to the net force isrelatively short in duration (∼1 ns), after which timethe particle moves at a constant velocity. This constantvelocity arises because the solution exerts a frictionalforce (f ) on the particle that is proportional to thesedimentation velocity (dr/dt). Once the steady stateis reached we can rewrite Equation 9.3 as
f dr/dt = ω2rm − ω2rνρ (9.8)
In Equation 9.8 the volume of a particle is a difficultquantity to measure and is replaced by a term calledthe partial specific volume (ν). This is defined as theincrease in volume when 1 g of solute is dissolved ina large volume of solvent. The quantity mν is definedas the increase in volume caused by the addition ofone molecule of mass m and it is equal to the volume

SOLUBILITY AND ‘SALTING OUT’ AND ‘SALTING IN’ 323
of the particle.1 Introduction of these terms allowsEquation 9.8 to be simplified
f dr/dt = ω2rm − ω2rm νρ (9.9)
f dr/dt = ω2rm (1 − νρ) (9.10)
and rearrangement leads to
S = dr/dt
ω2r= m (1 − νρ)
f(9.11)
where the term dr/dt /ω2r is defined as the sedimen-tation coefficient (S called the Svedburg unit after thepioneer of ultra-centrifugation Theodor Svedburg). TheSvedburg has units of 10−13 s, and as an examplehaemoglobin has a sedimentation coefficient of 4 ×10−13 s or 4 S. Since the mass m can be describedas the molecular weight of the solute divided by Avo-gadro’s constant (M/No) and by assuming the particleto be spherical we can utilize Stokes law to describethe frictional coefficient, f , as
f = 6π η rs (9.12)
where rs is the radius of the particle and η is theviscosity of the solvent (normally water).2 This leadsto the following equation
S = M(1 − νρ)
No 6π η rs(9.13)
that can be rearranged to give
M = SNo (6π η rs)/(1 − νρ) (9.14)
If we do not make any assumptions about shape wecan use the relationship
D = kBT /f (9.15)
1For most proteins ν has a value of 7.4 × 10−4 m3/kg or 0.74 ml/g.2For non-spherical particles correction of this relationship mustconsider anisotropy where one axis may be much longer than theother two. An example would be a cylindrically shaped molecule.Hydrodynamic analysis can in some instances allow the shape ofthe molecule to be deduced or at least distinguished from the simplespherical situation.
where D is the diffusion coefficient, f is the frictionalcoefficient, kB is Boltzmann’s constant and T theabsolute temperature. This leads to
M = SRT/D (1 − νρ) (9.16)
and allows the molecular weight to be calculated ifS, the sedimentation velocity, is measured since all ofthe remaining terms are constants or are derived fromother measurements. From Equation 9.11 S is defined
asdr
dt
1
ω2ror
S dt = 1/ω2dr/r (9.17)
with integration of the above equation leading to therelationship
ln r/r0 = S t ω2 (9.18)
By measuring the time taken for the particle totravel between two points, ro (at time t = 0) and r
(at time t) and knowing ω the angular velocity (inradians/s) it is relatively straightforward to calculateS. Measurement of protein sedimentation rates isperformed by recording changes in refractive index asthe protein migrates through an optical cell. Table 9.1lists the sedimentation coefficients for various proteins.
A more direct way of measuring molecular weightusing ultracentrifugation is to measure not the rateof sedimentation but the equilibrium established aftermany hours of centrifugation at relatively low speeds.At equilibrium a steady concentration gradient willoccur with the flow of proteins towards sedimentationbalanced by reverse flow as a result of diffusion. It canbe demonstrated that for a homogeneous protein thegradient is described by the equation
c(r)/c(r0) = exp M(1 − νρ)ω2 (r2 − r02)/2RT
(9.19)
where c(r) is the concentration at a distance r from theaxis of rotation and c(r0) is the concentration of proteinat the meniscus or interface (r0). From Equation 9.19a plot of ln c(r) against (r2 − r0
2) will yield a straightline whose slope is M(1 − νρ)ω2/2RT, from which M
is subsequently estimated (Figure 9.10).
Solubility and ‘salting out’ and‘salting in’One of the most common and oldest methods of proteinpurification is to exploit the differential solubility of

324 PROTEIN EXPRESSION, PURIFICATION AND CHARACTERIZATION
Table 9.1 Sedimentation coefficients associateddifferent proteins
Protein Molecularmass(kDa)
Sedimentationcoefficient
(S20,w)
Lipase 6.7 1.14Ribonuclease A 12.6 2.00Myoglobin 16.9 2.04Concanavalin B 42.5 3.50Lactate
Dehydrogenase150 7.31
Catalase 222 11.20Fibrinogen 340 7.63Glutamate
Dehydrogenase1015 26.60
Turnip yellow mosaicvirus
3013 48.80
Large ribosomalsubunit
1600 50
Notice the ‘abnormal’ value for fibrinogen and the absence of asimple correlation between mass and S value (after Smith in SoberH.A. (ed.) Handbook of Biochemistry and Molecular Biology, 2ndedn. CRC Press).
proteins at various ionic strengths. In concentratedsolutions the solubility of many proteins decreasesdifferentially leading to precipitation whilst othersremain soluble. This allows protein separation sinceas solubility decreases precipitation occurs with theprecipitant removed from solution by centrifugation.The use of differential solubility to fractionate proteinsis often used as one part in a strategy towards overallpurification.
The solubility of proteins in aqueous solutionsdiffers dramatically. Although membrane proteins areclearly insoluble in aqueous solution many structuralproteins such as collagen are also essentially insolubleunder normal physiological conditions. For smallglobular proteins with masses below 10 kDa intrinsicsolubility is often high and concentrations of 10 mM
are easily achieved. This corresponds to approximately100 g l−1 or about 10 percent w/v. In solution globular
In c
(r)
r 2 – r02
Figure 9.10 A plot of ln c(r) against (r2 − r02) yieldsa straight line that allows direct estimation of M, theprotein molecular weight. The slope of the line is givenby M(1 − νρ)ω2/2RT
proteins are surrounded by a tightly bound layerof water that differs in properties from the ‘bulk’water component. This water, known as the hydrationlayer, is ordered over the surface of proteins andappears to interact preferentially with charged sidechains and polar residues. In contrast, this wateris rarely found close to hydrophobic patches onthe surface of proteins. Altering the properties ofthis water layer effects the solubility of proteins.Similarly protein solubility is frequently related to theisoelectric point or pI . As the pH of the aqueoussolution approaches the pI precipitation of a proteincan occur. As the pH shifts away from the pI
the solubility of a protein generally increases. Thisis most simply correlated with the overall chargeof the protein and its interaction with water. Sincethe pI of proteins can vary from pH 2–10 it isclear that pH ranges for protein solubility will alsovary widely.
The ionic strength (I ) is defined as
I = 1/2 � mizi2 = 1/2 (m+z+2 + m−z−2) (9.20)
where m and z are the concentration and chargeof all ionic species in solution. Thus a solution of0.5 M NaCl has an ionic strength of 0.5 whilst asolution of 0.5 M MgCl2 has an ionic strength of

SOLUBILITY AND ‘SALTING OUT’ AND ‘SALTING IN’ 325
1.5. The concept of ionic strength was introduced byG.N. Lewis to account for the non-ideal behaviour ofelectrolyte solutions that arose from the total numberof ions present along with their charges rather thanthe chemical nature of each ionic species. A physicalinterpretation of properties of electrolytes from thestudies of Peter Debye and Erich Huckel, althoughmathematical and beyond the scope of this book,assumed that electrolytes are completely dissociatedinto ions in solution, that concentrations of ions weredilute below 0.01 M and that on average each ionwas surrounded by ions of opposite charge. This lastconcept was known as an ionic atmosphere and isrelevant to changes in protein solubility as a functionof ionic strength. Trends in protein solubility varyfrom one protein to another and show a strongdependence on the salts used to alter the ionic strength(Figure 9.11).
At low ionic strength a protein is surrounded byexcess counter-ions of the opposite charge knownas the ionic atmosphere. These counter-ions screencharges on the surface of proteins. Addition of extraions increases the shielding of surface charges with theresult that there is a decrease in intermolecular attrac-tion followed by an increase in protein solubility asmore dissolves into solution. This is the ‘salting-in’phenomenon (Figure 9.12). However, the addition of
lonic Strength
Na2SO4
(NH4)2SO4
KCL
NaCl
Log
solu
bilit
y
Figure 9.11 The solubility of haemoglobin indifferent electrolytes illustrates the ‘salting in’ and‘salting out’ effects as a function of ionic strength.Derived from original data by Green, A.A. J. Biol.Chem. 1932, 95, 47
Supersaturated solutions
Saturation curve
Non saturated solutions
Ionic strength (I0.5)
Pro
tein
sol
ubili
ty (
loga
rithm
ic s
cale
)
Figure 9.12 Model profile for solubility as a functionof ionic strength. Saturation occurs when solid andsolution phases are in equilibrium. ‘Salting-out’ is seenon the right-hand side of the diagram where there is areduction in protein solubility as the concentration ofsalt increases whilst ‘salting in’ is apparent on the left-hand side of the diagram where there is an increase inprotein solubility with ionic strength
more ions, reverses this trend. When the ionic concen-tration is very high (>1 M) each ion must be hydratedwith the result that bulk solvent (water) is sequesteredfrom proteins leading to decreases in solubility. If con-tinued decreased solubility leads to protein aggrega-tion and precipitation. Ionic effects on protein solubil-ity were first recognized by Franz Hofmeister around1888, and by analysing the effectiveness of anions andcations in precipitating serum proteins he establishedan order or priority that reflected each ions ‘stabilizing’properties.
Cations: N(CH3)3+ > NH4
+ > K+ > Na+
> Li+ > Mg2+ > Ca2+ > Al3+
> guanidinium
Anions: SO42− > HPO4
2− > CH3COO−
> citrate > tartrate > F− > Cl− > Br−
> I− > NO3− > ClO4
− > SCN−
The priority is known as the Hofmeister series(Table 9.2); the first ions in each series are themost stabilizing with substituted ammonium ions,ammonium itself and potassium being more stabilizing

326 PROTEIN EXPRESSION, PURIFICATION AND CHARACTERIZATION
Table 9.2 Properties of the Hofmeister series ofcations and anions
Stabilizing ions ↔ Destabilizing ionsStrongly hydrated anions ↔ Strongly hydrated
cationsWeakly hydrated cations ↔ Weakly hydrated anionsKosmotropic ↔ ChaotropicIncrease surface tension ↔ No effect on surface
tensionDecrease solubility of
non-polar side-chains↔ Increase solubility of
non-polar side-chainsSalting out ↔ Salting in
than calcium or guanidinium, for example. Similarlyfor anions of the Hofmeister series sulfate, phosphateand acetate are more stabilizing than perchlorate orthiocyanate.
At the heart of this series is the effect of ionson the ordered structure of water. Ordered structurein water is perturbed by ions since they disrupt nat-ural hydrogen-bond networks. The result is that theaddition of ions has a similar effect to increasing tem-perature or pressure. Ions with the greatest disruptiveeffect are known as structure-breakers or chaotropes.In contrast, ions exhibiting strong interactions withwater molecules are called kosmotropes. It is immedi-ately apparent that molecules such as guanidinium thio-cyanate are extremely potent chaotropes whilst ammo-nium sulfate is a stabilizing molecule or kosmotrope.Na+ and Cl− are frequently viewed as the ‘border zone’having neutral effects on proteins.
Ammonium sulfate is commonly used to selectivelyprecipitate proteins since it is very soluble in waterthereby allowing high concentrations (>4 M). Underthese conditions harmful effects on proteins such asirreversible denaturation are absent and NH4
+ andSO4
2− are both at the favourable, non-denaturing,end of the Hofmeister series. The mechanism of‘salting out’ proteins resides in the disruption ofwater structure by added ions that lead to decreasesin the solubility of non-polar molecules. By usingammonium sulfate it is possible to quantitativelyprecipitate one protein from a mixture. The remaining
proteins are left in solution and such methods arewidely used to purify soluble proteins from crudecell extracts.
ChromatographyChromatographic methods form the core of mostpurification protocols and have a number of commonfeatures. First, the protein of interest is dissolved in amobile phase normally an aqueous buffered solution.This solution is often derived from a cell extractand may be used directly in the chromatographicseparation. The mobile phase is passed over a resin(called the immobile phase) that selectively absorbscomponents in a process that depends on the type offunctional group and the physical properties associatedwith a protein. These properties include charge,molecular mass, hydrophobicity and ligand affinity.The choice of the immobile phase depends stronglyon the properties of the protein exploited duringpurification.
The general principle of chromatography involvesa column containing the resin into which bufferis pumped to equilibrate the resin prior to sampleapplication. The sample is applied to the columnand separation relies on interactions between proteinand the functional groups of the resin. Interactionslows the progress of one or more proteins throughthe column whilst other proteins flow unhinderedthrough the column. As a result of their faster progressthrough the column these proteins are separatedfrom the remaining protein (Figure 9.13). In someinstances protein mixtures vary in their interaction withfunctional groups; a strong interaction results in slowprogress through the column, a weak interaction leadsto faster rates of progress whilst no interaction resultsin unimpeded flow through the resin.
Pioneering research into the principles of chro-matography performed by A.J.P. Martin and R.L.M.Synge in the 1940s developed the concept of theoreticalplates in chromatography. In part this theory explainsmigration rates and shapes of eluted zones and viewsthe chromatographic column as a series of continu-ous, discrete, narrow layers known as theoretical plates.Within each plate equilibration of the solute (protein)between the mobile and stationary (immobile) phasesoccurs with the solute and solvent moving through the

CHROMATOGRAPHY 327
5000
4000
3000
2000
1000
0
0 1 2 3 4 5 6 7 8Elution parameter (time or volume)
Inte
nsi
ty (
Ab
sorb
ance
or
Flu
ore
scen
ce)
9 10 11 12 13 14 15
Figure 9.13 An ‘ideal’ separation of a mixture of proteins. The peaks representing different components areseparated with baseline resolution. A less than ideal separation will result in peak overlap
column in a stepwise manner from ‘plate’ to ‘plate’.The resolution of a column will increase as the theo-retical plate number increases. Practically, there are anumber of ways to increase the theoretical plate size,and these include making the column longer or decreas-ing the size of the beads packing the column. The valueof increasing resolution is that two similarly migratingsolutes may be separated with reasonable efficiency.
Today, sophisticated equipment is available toseparate proteins. This equipment, controlled via acomputer, includes pumps that allow flow rates to beautomatically adjusted over a wide range from perhapsas low as 10 µl min−1 to 100 ml min−1. In addition,complex gradients of solutions can be constructedby controlling the output from more than one pump,and under some circumstances these gradients vastlyimprove chromatographic separations. The addition ofthe sample to the top of the column is controlled viaa system of valves whilst material flowing out of thecolumn (eluate) is detected usually via fluorescence- orabsorbance-based detectors (Figure 9.14).
A major advance has been the development ofresins that withstand high pressures (sometimes in
excess of 50 MPa) generated using automated orhigh pressure liquid chromatography (HPLC) systems(Figure 9.15). Higher pressures are advantageous inallowing higher flow rates and decreased separationtimes. Previously, manual chromatographic methodsrelied on the flow of solution through the column underthe influence of gravity and resulted in separationstaking several days. Today it is possible to routinelyperform chromatographic separations with high levelsof resolution in less than 10 min.
Modern chromatographic systems contain many ofthe components shown schematically in Figure 9.14.Two or more pumps drive buffer onto columns at con-trolled rates usually in a range from 0.01–10 ml min−1.The pumps are linked to the column via chemicallyresistant tubing that conveys buffers to the column.Between the pump and the column is a ‘mixer’ thatadjusts the precise volumes dispensed from the pumpsto allow the desired buffer composition. This is par-ticularly important in establishing buffer gradients ofionic strength or pH. Buffer of defined compositionis pumped through a series of motorized valves that

328 PROTEIN EXPRESSION, PURIFICATION AND CHARACTERIZATION
Figure 9.14 Schematic representation of chromatographic systems indicating flow of mobile phase and sampletogether with the flow of information between all devices and a computer workstation
Figure 9.15 A modern automated chromatographysystem embodying the flow scheme of Figure 9.14.(Reproduced courtesy of Amersham Biosciences)
allow addition of sample to the column or its diver-sion to a waste outlet. The column itself is made ofprecision-bored glass, stainless steel or titanium andcontains chromatographic resin.
Elution of material from the column leads to a detec-tion system that usually measures absorbance basedaround the near UV region at 280 nm for proteins.Frequently, detection systems will also measure pHand ionic strength of the material eluting from the col-umn. The detection system is interfaced with a fractioncollector that allows samples of precise volume to becollected and peak selection on the basis of rates ofchange of absorbance with time. In any system all ofthe components are controlled via a central computerand the user inputs desired parameters such as flowrate, fraction volume size, wavelength for detection,run duration, etc.
Ion exchange chromatography
Ion exchange chromatography separates proteins on thebasis of overall charge and depends on the relativenumbers of charged side chains. Simple calculationsof the number of charge groups allow an estimationof the isoelectric point and an assessment of thecharge at pH 7.0. Anionic proteins have a pI < 7.0whilst cationic proteins have isoelectric points >7.0.In ion exchange chromatography anionic proteins bindto cationic groups of the resin in the process of

CHROMATOGRAPHY 329
anion exchange chromatography. Similarly, positivelycharged proteins bind to anionic groups within asupporting matrix in cation exchange chromatography.
The first ion exchangers developed for use withbiological material were based on a supporting matrixof cellulose. Although hydrophilic and non-denaturingcellulose was hindered by its low binding capacityfor proteins due to the small number of functionalgroups attached to the matrix, biodegradation andpoor flow properties. More robust resins were requiredand the next generation were based around cross-linked dextrans, cross-linked agarose, or cross-linkedacrylamide in the form of small, bead-like, particles.These resins offered significant improvements in termsof pH stability, flow rates and binding capacity.The latest resins are based around extremely smallhomogeneous beads of average diameter <10 µmcontaining hydrophilic polymers. One implementationof this technology involves polystyrene cross-linkedwith divinylbenzene producing resins resistant tobiodegradation, usable over wide pH ranges (from1–14) and tolerating high pressures (>10 MPa).
For cation exchange chromatography the commonfunctional groups (Figure 9.16) involve weak acidicgroups such as carboxymethyl or a strong acidicgroup such as sulfopropyl or methyl sulfonate. Sim-ilarly for anion exchange, the functional groups arediethylaminoethyl and the stronger exchange group ofdiethyl-(2-hydroxypropyl)amino ethyl. The binding ofproteins to either anion or cation exchange resins willdepend critically on pH and ionic strength. The pH ofthe solution remains important since it influences theoverall charge on a protein. Fairly obviously, it is not
sensible to perform ion exchange chromatography at apH close to the isoelectric point of a protein. At thispH the protein is uncharged and will not stick to anyion exchange resin.
In ion exchange chromatography samples areapplied to columns at low ionic strengths (normallyI < 0.05 M) to maximize interactions between proteinand matrix. The column is then washed with fur-ther solutions of constant pH and low ionic strengthto remove proteins lacking any affinity for the resin.However, during any purification protocol many pro-teins are present in a sample and exhibit a range ofinteractions with ion exchange resins. This includesproteins that bind very tightly as well as those withvarying degrees of affinity for the resin. These proteinsare separated by slowly increasing the ionic strengthof the solution. Today’s sophisticated chromatographysystems establish gradients where the ionic strength isincreased from low to high levels with the ions com-peting with the protein for binding sites on the resin.As a result of competition proteins are displaced fromthe top of the column and forced to migrate down-wards through the column. Repetition of this processoccurs continuously along the column with the resultthat weakly bound proteins are eluted. Eventually athigh ionic strengths even tightly bound proteins aredisplaced from the resin and the cumulative effect isthe separation of proteins according to charge. Collect-ing proteins at regular intervals with a fraction collectorand recording their absorbance or fluorescence estab-lishes an elution profile and assists in locating theprotein of interest.
O−MatrixO
CH2
C
O
MatrixCH2
CH2
CH2S O
O−
O−
O−CH2
S
O
MatrixO−
MatrixO
CH2
CH2NH+
(CH2CH3)
(CH2CH3)
Figure 9.16 The common functional groups found in ion exchange resins. The terms strong and weak exchangersrefer to the extent of ionization with pH. Strong ion exchangers such as QAE and SP are completely dissociated overa very wide pH range whereas with the weak exchangers (DEAE and CM) the degree of ionization varies and this isreflected in their binding properties

330 PROTEIN EXPRESSION, PURIFICATION AND CHARACTERIZATION
Size exclusion or gel filtration chromatography
Size exclusion separates proteins according to theirmolecular size. The principle of the method relieson separation by the flow of solute and solvent overbeads containing a series of pores. These pores are ofconstant size and are formed by covalent cross-linkingof polymers. High levels of cross-linking lead to asmall pore size. In a mixture of proteins of differentmolecular mass some will be small enough to enterthe pores and as a result their rate of diffusion throughthe column is slowed. Larger proteins will rarely enterthese regions with the result that progress through theresin is effectively unimpeded. Very large proteins willelute in the ‘void volume’ and all proteins that failto show an interaction with the pores will elute inthis void volume irrespective of their molecular size.Migration rates through the column reflect the extentof interaction with the pores; small proteins spend aconsiderable amount of time in the solvent volumeretained within pores and elute more slowly than thoseproteins showing marginal interactions with the beads.
It is clear that this process represents an effective wayof discriminating between proteins of different sizes(Figure 9.17).
In an ideal situation there is a linear relationshipbetween the elution volume and the logarithm of themolecular mass (Figure 9.18). This means that if asize exclusion column is calibrated using proteins ofknown mass then the mass of an unknown protein canbe deduced from its elution position. A reasonablyaccurate estimation of mass can be obtained if theunknown protein is of similar shape to the standardset. This occurs because the process of size exclusionor gel filtration is sensitive to the average volumeoccupied by the protein in solution. This volumeis often represented by a term called the Stokesradius of the protein and this parameter is verysensitive to overall shape. So, for example, elongatedmolecules such as fibrous proteins show anomalousrates of migration. A rigorous description of sizeexclusion chromatography would view the separationas based on differences in hydrodynamic radius. Today,size exclusion chromatography is rarely used as an
Figure 9.17 The principle of gel filtration showing separation of three proteins of different mass at the beginning,middle and end of the process. Resins are produced by cross linking agarose, acrylamide and dextran polymersto form different molecular size ranges from 100–10 000 (peptide–small proteins), 3000–70 000 (small proteins),104 –105 (larger proteins, probably multi-subunit proteins), and 105 –107 (macromolecular assemblies such as largeprotein complexes, viruses, etc

CHROMATOGRAPHY 331
Low molecular weight limit ~Vt
"Standard" proteins of known Mr
Elu
tion
volu
me
(Ve)
High molecular weight limit V0
log Mr
Figure 9.18 Estimation of molecular mass usingsize exclusion chromatography. The volume (or time)necessary to achieve elution from a column is plottedagainst the logarithm of molecular weight to yield astraight line that deviates at extreme (low and high)molecular weight ranges. These extremes correspondto the total volume of the column (Vt) and the voidvolume respectively (Vo)
analytical technique to estimate subunit molecular massbut it is frequently used as a preparative method toeliminate impurities from proteins. One common use isto remove salt from a sample of protein or to exchangethe protein from one buffer to another.
Hydrophobic interaction chromatography
Hydrophobic interaction chromatography (HIC) isbased on interactions between non-polar side chainsand hydrophobic resins. At first glance it is not obvi-ous how HIC is useful for separating and purify-ing proteins. In globular proteins the folded statesare associated with buried hydrophobic residues wellaway from the surrounding aqueous solvent. How-ever, close inspection of some protein structuresreveals that a fraction of hydrophobic side chainsremain accessible to solvent at the surface of foldedmolecules. In some cases these residues produce adistinct hydrophobic patch whose composition andsize varies from one protein to another and leads tolarge differences in surface hydrophobicity. HIC sep-arates proteins according to these differences. As a
technique it exploits interactions between hydropho-bic patches on proteins and hydrophobic groupscovalently attached to an inert matrix. The mostpopular resins are cross-linked agarose polymers con-taining butyl, octyl or phenyl (hydrophobic) func-tional groups.
The hydrophobic interaction depends on solutionionic strength as well as chaotropic anions/cations. Asa result HIC is usually performed under conditionsof high ionic strength in solutions containing 1 M
ammonium sulfate or 2 M NaCl. High ionic strengthincreases interactions between hydrophobic ligand andprotein with the origin of this increased interactionarising from decreased protein solvation as a result ofthe large numbers of ions each requiring a hydrationshell. The net result is that hydrophobic surfacesinteract strongly as ions ‘unmask’ these regionson proteins.
Samples are applied to columns containing resinequilibrated in a solution of high ionic strength andproteins containing hydrophobic patches stick to theresin whilst those lacking hydrophobic interactionsare not retained by the column. Elution is achievedby decreasing favourable interactions by loweringthe salt concentration via a gradient or a sudden‘step’. Proteins with different hydrophobicity areseparated during elution with the most hydrophobicprotein retained longest on the column. However, theprediction of the magnitude of surface hydrophobicityon proteins remains difficult and the technique tendsto be performed in an empirical manner where avariety of resins are tried for a particular purificationprotocol. One advantage of HIC is the relativelymild conditions that preserve folded structure andactivity.
Reverse phase chromatography
A second chromatographic method that exploitshydrophobicity is reverse phase chromatography (RPC)although the harsher conditions employed in thismethod can lead to denaturation. The basis of RPC isthe hydrophobic interaction between proteins/peptidesand a stationary phase containing alkyl or aromatichydrocarbon ligands immobilized on inert supports.The original support polymer for reverse phasemethods was silica to which hydrophobic ligands were

332 PROTEIN EXPRESSION, PURIFICATION AND CHARACTERIZATION
linked. Although still widely used as a base matrix ithas a disadvantage of instability at alkaline pH and thisrestricts its suitability in biological studies. As a result,synthetic organic polymers based on polystyrene beadshave proved popular and show excellent chemicalstability over a wide range of conditions.
The separation mechanism in RPC depends onhydrophobic binding interactions between the solutemolecule (protein or peptide) in the mobile phase andthe immobilized hydrophobic ligand (stationary phase).The initial mobile phase conditions used to promotebinding in RPC are aqueous solutions and lead to ahigh degree of organized water structure surroundingprotein and immobilized ligand. The protein bindsto the hydrophobic ligand leading to a reduction inexposure of hydrophobic surface area to solvent. Theloss of organized water structure is accompanied bya favourable increase in system entropy that drivesthe overall process. Proteins partition between themobile and stationary phases in a process similarto that observed by an organic chemist separatingiodine between water and methanol. The distributionof the protein between each phase reflects the bindingproperties of the medium, the hydrophobicity of theprotein and the overall composition of the mobilephase. Initial experimental conditions are designed tofavour adsorption of the protein from the mobile phaseto the stationary phase. Subsequently, the mobile phasecomposition is modified by decreasing its polarityto favour the reverse process of desorption leadingto the protein partitioning preferentially back intothe mobile phase. Elution of the protein or peptideoccurs through the use of gradients that increase thehydrophobicity of the mobile phase normally throughthe addition of an organic modifier. This is achievedthrough the addition of increasing amounts of non-polarsolvents such as acetonitrile, methanol, or isopropanol.At the beginning a protein sample is applied to acolumn in 5 percent acetonitrile, 95 percent water,and this is slowly changed throughout the elutionstage to yield at the end of the process a solutioncontaining, for example, 80 percent acetonitrile/20percent water.
For both silica and polystyrene beads the hydropho-bic ligands are similar and involve linear hydrocarbonchains (n-alkyl groups) with chain lengths of C2, C4,
OSi
CH2
CH3
CH3
CH3O
Si(CH2)7
CH3
CH3
CH3
OSi
(CH2)17
CH3
CH3
CH3
Figure 9.19 The hydrophobic C2, C8 and C18 ligandswidely used in RPC
C8 and C18 (Figure 9.19). C18 is the most hydropho-bic ligand and is used for small peptide purificationwhilst the C2/C4 ligands are more suitable for pro-teins. Although proteins have been successfully puri-fied using RPC the running conditions often lead todenaturation and reverse phase methods are more valu-able for purifying short peptides arising from chemicalsynthesis or enzymatic digestion of larger proteins (seeChapter 6).
Affinity chromatography
An important characteristic of some proteins is theirability to bind ligands tightly but non-covalently. Thisproperty is exploited as a method of purification withthe general principle involving covalently attachmentof the ligand to a matrix. When mobile phase ispassed through a column containing this group proteinsshowing high affinity for the ligand are bound and theirrate of progress through the column is decreased. Mostproteins will not show affinity for the ligand and flowstraight through the column. This protocol thereforeoffers a particularly selective route of purificationand affinity chromatography has the great advantageof exploiting biochemical properties of proteins, asopposed to a physical property.
Affinity chromatography relies on the interactionbetween protein and immobilized ligand. Bindingmust be sufficiently specific to discriminate betweenproteins but if binding is extremely tight (highaffinity) it can leads to problems in recoveringthe protein from the column. Most methods ofrecovery involve competing for the binding site onthe protein. Consequently, a common method of

POLYACRYLAMIDE GEL ELECTROPHORESIS 333
elution is to add exogenous or free ligand to competewith covalently linked ligand for the binding site ofthe protein. A second method is to change solutionconditions sufficiently to discourage protein–ligandbinding. Under these conditions the protein no longershows high affinity for the ligand and is eluted. Thelast method must of course not use conditions thatpromote denaturation of the protein, and frequently itinvolves only moderate changes in solution pH or ionicstrength. Enzymes are particularly suited to this form ofchromatography since co-factors such as NAD, NADP,and ADP are clear candidate ligands for use in affinitychromatography.
One limitation in the development of affinitychromatography is that methods must exist to linkthe ligand covalently to the matrix. This modificationmust not destroy its affinity for the protein and mustbe reasonably stable i.e not broken under moderatetemperature, pH and solution conditions used duringchromatography. Recently, a great expansion in thenumber and type of affinity based chromatographicseparations has occurred due to the development ofexpression systems producing proteins with histidinetags or fused to other protein such as glutathione-S-transferase or maltose binding protein (Table 9.3).Histidine tags can be located at either the N- orC-terminal regions of a protein and bind to metalchelate columns containing the ligand imidoaceticacid. Similarly glutathione-S-transferase is a proteinthat binds the ligand glutathione which itself can becovalently linked to agarose based resins.
Dialysis and ultrafiltration
The basis of dialysis and ultrafiltration is the pres-ence of a semipermeable membrane that allows theflux of small (low molecular weight) compoundsat the same time preventing the diffusion of largermolecules such as proteins. Both methods are com-monly used in the purification of proteins and in thecase of ultrafiltration particularly in the concentrationof proteins.
Dialysis is commonly used to remove low molecularweight contaminants or to exchange buffer in a solutioncontaining protein. A solution of protein is placedin dialysis tubing that is sealed at both ends and
added to a much larger volume of buffer (Figure 9.20).Low molecular weight contaminants diffuse acrossthe membrane whilst larger proteins remain trapped.Similarly, the buffers inside and outside the dialysistubing will exchange by diffusion until equilibrium isreached between the compartments. This equilibriumis governed by the Donnan effect, which maintainselectrical neutrality on either side of the membrane.For polyvalent ions such as proteins this means thations of the opposite charge will remain in contact withprotein and the solution ionic strength will not fall toexactly that in the bulk solution. For dilute solutions ofprotein coupled with higher exterior ion concentrationsthe Donnan effect becomes negligible but for highconcentrations of protein or low ionic strength buffersit remains a significant effect on the colligativeproperties of ions. Despite this complication, bufferinside the dialysis tubing is gradually replaced bythe solution found in the larger exterior volume. Byreplacing the exterior volume of buffer at regularintervals the process rapidly achieves the removal oflow molecular weight contaminants or the exchangeof solvent.
Ultrafiltration is used to concentrate solutions ofproteins by the application of pressure to a sealedvessel whose only outlet is via a semipermeable mem-brane (Figure 9.20). The molecular weight limit forthe semipermeable membrane can be closely con-trolled. Solvent and low molecular weight moleculespass through the membrane whilst larger moleculesremain inside the vessel. As solvent is forcedacross the membrane proteins inside the ultrafiltra-tion cell are progressively concentrated. Frequently,ultrafiltration is used to concentrate a protein dur-ing the final stages of purification when it may berequired at high concentrations; in crystallization trialsfor example.
Polyacrylamide gel electrophoresisThe most common method of analysing the purityof an isolated protein is to use polyacrylamide gelelectrophoresis (PAGE) in the presence of the detergentsodium dodecyl sulfate (SDS) and a reductant ofdisulfide bridges such as β-mercaptoethanol. Thetechnique has the additional advantage of allowing themonomeric (subunit) molecular mass to be determined

334 PROTEIN EXPRESSION, PURIFICATION AND CHARACTERIZATION
Table 9.3 Ligands used for affinity chromatography together with the natural co-factor/substrate and theproteins/enzymes purified by these methods
Aim of affinity reaction Ligand/natural substrate Example
To bind nucleotide
binding domains
Natural ligands are NADP or NAD. Most
affinity-based methods covalently bind either
ADP or AMP to resins and this is sufficient
to bind the nucleotide binding domain
Dehydrogenases bind NAD or NADH
and are frequently purified via this
route
Separation of proteins
based on the ability of
some side chains to
chelate divalent metal
ions
Iminodiacetic acid covalently linked to gel
binds metal ions such as Ni2+ or Zn2+
leaving a partially unoccupied coordination
sphere. Histidine residues are the usual
target but tryptophan and cysteine residues
can also bind
Any recombinant protein containing a
His-tag. Binding occurs via imidazole
nitrogen and protonation destroys
binding and hence is a route towards
elution
Immobilized lectins such
as concanavalin A
Binds α-D-mannose, α-D-glucose and related
sugars via interaction with free OH groups
Separation of glycoproteins particularly
viral glycoproteins and cell surface
antigens
Glutathione binding
proteins
Glutathione is the natural substrate and can be
covalently linked to column
Fusion proteins containing
glutathione-S -transferase
Affinity of monoclonal
and polyclonal IgG for
protein A, protein G
or a fusion protein,
protein A/G
Protein A is a protein of molecular weight
42 000 derived from S. aureus. It consists of
six major regions five of which bind to IgG.
Other affinity columns exploit the use of
protein G – a protein derived from
streptococci. Both proteins exhibit affinity
for the Fc region of immunoglobulins
Separation of IgG subclasses from serum
or cell culture supernatants
Reversible coupling of
proteins containing a
free thiol group
Thiol group immobilized on column matrix.
Forms mixed disulfide with protein
containing free–SH group. Reversed by
addition of excess thiol or other reductant
Cysteine proteinases a family of enzymes
with reactive thiols at their catalytic
centres are purified by these methods
Affinity for
oligonucleotides
Polyadenylic acid linked via N6 amino group
or polyuridylic acid linked to matrix
Purification of viral reverse transcriptases
and mRNA binding proteins
(a) (b)
Figure 9.20 Dialysis (a) and ultrafiltration (b) assemblies used in protein purification

POLYACRYLAMIDE GEL ELECTROPHORESIS 335
with reasonable accuracy. SDS–PAGE is widely usedto assess firstly if an isolated protein is devoidof contaminating proteins and secondly whether thepurified protein has the expected molecular mass. Bothof these parameters are extremely useful during proteinpurification.
The principle of SDS–PAGE is the separation of pro-teins (or their subunits) according to molecular massby their movement through a polyacrylamide gel ofclosely defined composition under the influence of anelectric field. Looking at the individual componentsof this system allows the principles of electrophore-sis to be illustrated and to emphasize the potential ofthis technique to provide accurate estimations of mass,composition and purity. The gel component is a porousmatrix of cross-linked polyacrylamide. The most com-mon method of formation involves the reaction betweenacrylamide and N , N ′-methylenebisacrylamide (called‘bis’; Figure 9.21) catalysed by two additional com-pounds ammonium persulfate (APS) and N ,N ,N ′,N ′-tetramethylethylenediamine (TEMED). Polyacrylamidegels form when a dissolved mixture of acrylamide andthe bifunctional ‘bis’ cross-linker polymerizes into long,covalently linked, chains. The polymerization of acry-lamide is a free-radical catalysed reaction in which APSacts as an initiator. Initiation involves generating a per-sulfate free-radical that activates the quaternary amineTEMED, which in turns acts as a catalyst for the poly-merization of acrylamide monomers. The concentrationof acrylamide can be varied to alter ‘resolving power’and this contributes to the definition of a ‘pore’ sizethrough which proteins migrate under the influence ofan electric field. A smaller pore size is generated byhigh concentrations of acrylamide with the result thatonly small proteins migrate effectively. This action ofthe gel is therefore similar to size exclusion chromatog-raphy where there is an effective filtration or molecularsieving process by pores.
The mobility of a protein through polyacrylamidegels is determined by a combination of overall charge,molecular shape and molecular weight. Native PAGE(performed in the absence of SDS) yields the massof native proteins and is subject to deviations causedby non-spherical shape and residual charge as wellas interactions between subunits. The technique isless widely used than SDS–PAGE. In the presenceof SDS the parameters of shape and charge become
CHC
O
NH
CH2
CHC
O
NH
H2C CH2
CHC
O
NH2
H2C Acrylamide
Bisacrylamide
Figure 9.21 The structure of acrylamide andbisacrylamide
unimportant and separation is achieved solely on thebasis of protein molecular weight. The underlyingreason for this observation is that SDS binds toalmost all proteins destroying native conformation(Figure 9.22). SDS causes proteins to unfold, formingrod-like protein micelles that migrate through gels.Since almost all proteins form this rod-like unfoldedstructure with an excess of negative charge due tothe bound SDS the effect of shape and charge iseliminated. SDS binds to proteins at a relativelyconstant ratio of 1.4 g detergent/g protein leading in aprotein of molecular weight 10 000 (i.e ∼100 residues)to approximately one SDS molecule for every twoamino acid residues.
A consequence of detergent binding is to coat allproteins with negative charge and to eliminate thecharge found on the native protein. As a result ofSDS binding all proteins become highly negativelycharged, adopt an extended rod-like conformation andmigrate towards the anode under the influence of anelectric field.
A protein mixture is applied to the top of a gel andmigrates through the matrix as a result of the elec-tric field with ‘lighter’ components migrating fasterthan ‘heavier’ components. Over time the component
CH3 O SO3− Na+
Figure 9.22 Sodium dodecyl sulfate, also calledsodium lauryl sulfate, has the structure of a longacyl chain containing a charged sulfate group; it is anextremely potent denaturant of proteins

336 PROTEIN EXPRESSION, PURIFICATION AND CHARACTERIZATION
proteins are separated and the resolving power of thetechniques is sufficiently high that heterogeneous mix-tures of proteins can be separated and distinguishedfrom each other. In practice most gels contain twocomponents – a short ‘focusing’ gel containing a lowpercentage of acrylamide (<5 percent) that assists inthe ordering and entry of polypeptides into a longer‘resolving’ gel (Figure 9.23). The above discussion hasassumed a constant acrylamide concentration through-out the gel but it is now possible to ‘tailor’ gel per-formance by varying the concentration of acrylamide.This creates a gradient gel where acrylamide concentra-tions vary linearly, for example, from 10 to 15 percent.The effect of this gradient is to enhance the resolvingpower of the gel over a narrower molecular weightrange. Although an initial investigation is usually per-formed with gels containing a constant acrylamide con-centration (say 15 percent) increased resolution can beobtained by using gradient gels. The range of gradientusually depends on the mass of the polypeptides underinvestigation.
The migration of proteins through a gel will dependon the voltage/current conditions used as well asthe temperature and it is most common to compare
the mobility of an unknown protein or mixture ofproteins with pure components of known molecularmass. For example, in Figure 9.24 the migrationof proteins in an extract derived from recombinantE. coli is compared to the mobility of standardproteins whose molecular masses are known andrange from 6 kDa to 200 kDa. The fractionation ofthe protein of interest can be followed clearly ateach stage.
A large number of proteins ranging in mass fromless than 10 kDa to greater than 100 kDa are seen inthe starting material. Progressive purification removesmany of these proteins and allows bands of similarmonomeric mass to be identified.
After separation the gel is colourless but profilessuch as those of Figure 9.24 result from gel stain-ing that locates the position of each protein band.A number of different stains exist but the two mostcommon dyes are Coomassie Brilliant Blue R250(see Chapter 3) and silver nitrate. For routine useCoomassie blue staining is preferable and has a detec-tion limit of approximately 100 ng protein. CoomassieBrilliant Blue reacts non-covalently with all proteinsand the success of the staining procedure relies on
Figure 9.23 A conventional polyacrylamide gel. A top ‘stacking’ gel containing a lower concentration of acrylamidefacilitates entry of SDS-coated polypeptides into the ‘resolving’ gel where the bands separate according to molecularmass. Separation is shown by the different migration of bands (shown in red) within the gel

POLYACRYLAMIDE GEL ELECTROPHORESIS 337
1 2 3 4 5 6 7 8
200.0
116.397.4
66.3
55.4
36.5
31.0
21.5
14.4
6.0
Figure 9.24 A ‘real’ gel showing the fractionationof a foreign protein expressed originally as a fusionprotein from E. coli cell lysates. The gel was stainedusing Coomassie blue. From left to right the proteinundergoes progressive purification to eventually yielda pure protein. The cell lysates are shown in lanes 1and 2, lanes 3 and 4 show the non bound materialfrom application to a glutathione affinity columnwhilst lane 5 represents the bound material elutedfrom this column. Lane 6 represents the proteinreleased from the GST fusion by limited proteolysiswhilst lane 7 shows the result of further purificationby anion exchange chromatography. Lane 8 showsmolecular weight markers over a range of 200–6 kDa(reproduced with permission from Chaddock, J.A. et al.Protein Expression and Purification. 2002, 25, 219–228.Academic Press)
‘fixing’ by precipitation the migrated proteins withacetic acid followed by removal of excess dye usingmethanol–water mixtures. Silver nitrate has the advan-tage of increased sensitivity with a detection limitof ∼1 ng of protein. The molecular basis for silverstaining remains less than clear but involves addingsilver nitrate solutions to the gel previously washedunder mildly acidic conditions. Today, purpose-builtmachines have automated the running and staining ofgels (Figure 9.25).
Figure 9.25 An automated electrophoresis system.The left-hand compartment performs electrophoreticseparation of proteins on thin, uniformly made, gelswhilst the second compartment performs the stainingreaction to visualize protein components. The gels arerun horizontally. (Reproduced courtesy of AmershamBiosciences)
Further use of polyacrylamide gels in studyingproteins occurs in the techniques of Western blottingand two-dimensional (2D) electrophoresis. In the firstarea SDS–PAGE is combined with ‘blotting’ tech-niques to allow bands to be identified via further assays.These procedures involve enzyme-linked immunosor-bent assays (ELISA), in which antibody binding islinked to a second reaction involving an enzyme-catalysed colour change. Since the reaction will onlyoccur if a specific antigen is detected this processis highly specific. This combination of techniques iscalled Western blotting (Figure 9.26). Western blot-ting was given its name as a pun on the techniqueSouthern blotting, pioneered by E.M. Southern, whereDNA is transferred from an agarose gel and immobi-lized on nitrocellulose matrices. In Western blotting theprotein is eluted/transferred from SDS–polyacrylamidegels either by capillary action or by electroelution.Electroelution results in the movement of all of theseparated proteins out of the gel and onto a sec-ond supporting media such as polyvinylidene diflu-oride (PVDF) or nitrocellulose. Immobilized on thissecond supporting matrix the proteins undergo furtherreactions that assist with identification (Figure 9.27).A common technique reacts the eluted protein withpreviously raised antibodies. Since antibodies will onlybind to a specific antigen this reaction immediately
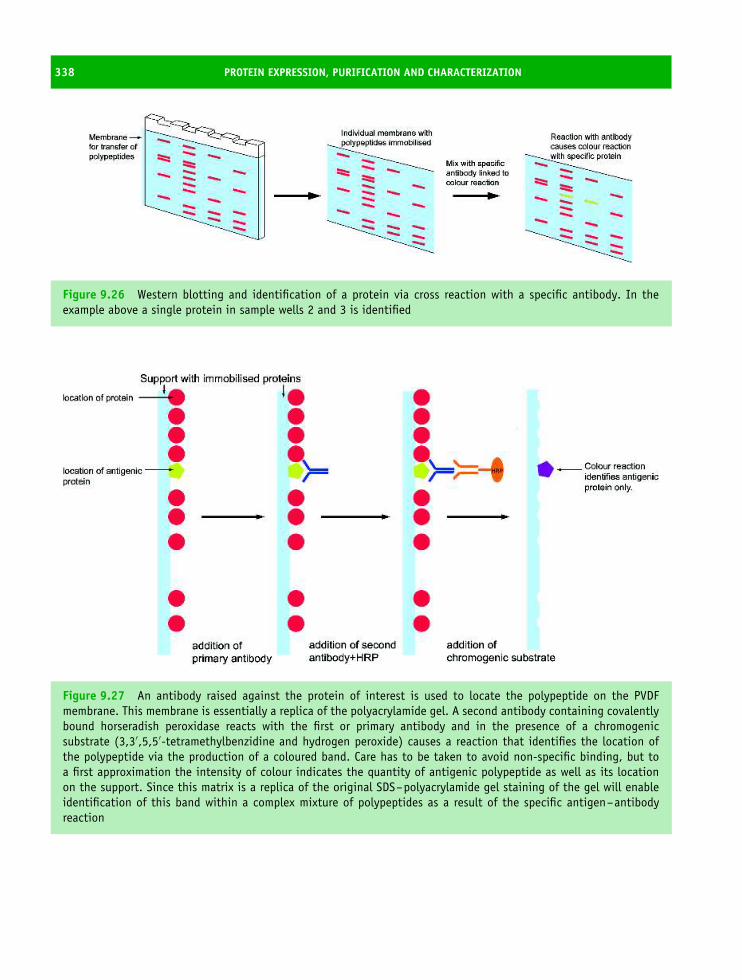
338 PROTEIN EXPRESSION, PURIFICATION AND CHARACTERIZATION
Figure 9.26 Western blotting and identification of a protein via cross reaction with a specific antibody. In theexample above a single protein in sample wells 2 and 3 is identified
Figure 9.27 An antibody raised against the protein of interest is used to locate the polypeptide on the PVDFmembrane. This membrane is essentially a replica of the polyacrylamide gel. A second antibody containing covalentlybound horseradish peroxidase reacts with the first or primary antibody and in the presence of a chromogenicsubstrate (3,3′,5,5′-tetramethylbenzidine and hydrogen peroxide) causes a reaction that identifies the location ofthe polypeptide via the production of a coloured band. Care has to be taken to avoid non-specific binding, but toa first approximation the intensity of colour indicates the quantity of antigenic polypeptide as well as its locationon the support. Since this matrix is a replica of the original SDS–polyacrylamide gel staining of the gel will enableidentification of this band within a complex mixture of polypeptides as a result of the specific antigen–antibodyreaction

POLYACRYLAMIDE GEL ELECTROPHORESIS 339
identifies the protein of interest. However, by itselfthe reaction does not lead to any easily detectablesignal on the supporting matrix. To overcome thisdeficiency the antibody reaction is usually carriedout with a second antibody cross-reacting to the ini-tial antibody and coupled to an enzyme. Two com-mon enzymes linked to antibodies in this fashionare horseradish peroxidase (HRP) and alkaline phos-phatase. These enzymes were chosen because the addi-tion of substrates (benzidine derivatives and hydro-gen peroxide for HRP together with 5-bromo-4-chloro-3-indoyl phosphate or 4-chloro-1-naphthol substrateswith alkaline phosphatase) leads to an insoluble prod-uct and a colour reaction that is detected visually. Theappearance of a colour on the nitrocellulose supportallows the position of the antibody–antigen complexto be identified and to be correlated with the result ofSDS–PAGE.
An alternative procedure to the reaction with anti-bodies is to identify proteins absorbed irreversibly toa nitrocellulose sheet using radioisotope-labelled anti-bodies. Iodine-125 is commonly used to label anti-bodies and the protein is identified from the exposureof photosensitive film. Together these two techniquesallow a protein to be detected on the basis of molec-ular mass (SDS–PAGE) and also native conformation(Western blotting) at very low levels. These techniquesare now the basis for many clinical diagnostic tests.
A second route of analysis involves spreadingprotein separation into a second dimension. Thistechnique is called two-dimensional gel electrophoresisand separates proteins according to their isoelectricpoints in the first dimension followed by a secondseparation based on subunit molecular mass usingSDS–PAGE.
The bioinformatics revolution currently underwayin protein biochemistry is providing ever-increasingamounts of sequence data that requires analysis atthe molecular level. The area of genomics has lednaturally to proteomics – the wish to characterizeproteins expressed by the genome. This involvesanalysis of the structure and function of all proteinswithin a single organism, including not only individualproperties but also their collective properties throughinteractions with physiological partners. Proteomicsrequires a rapid way of identifying many proteinswithin a cell, and in this context 2D electrophoresis
Figure 9.28 Separation of proteins using 2D elec-trophoresis. The proteins separate according to isoelec-tric point in dimension 1 and according to molecularmass in dimension 2. Often the proteins are labelledvia the incorporation of radioisotopes and exposure toa sensitive photographic film allows identification ofeach ‘spot’
is advancing as a powerful method for analysis ofcomplex protein mixtures (Figure 9.28). This techniquesorts proteins according to two independent properties.The first-dimension relies on isoelectric focusing (IEF)and centres around the creation of a pH gradient underthe influence of an electric field. A protein will movethrough this pH gradient until it reaches a point whereits net charge is zero. This is the pI of that proteinand the protein will not migrate further towards eitherelectrode. The separation of protein mixtures by IEF isusually performed with gel ‘strips’ which are layeredonto a second SDS gel to perform the 2D step. Theresult is a 2D array where each ‘spot’ should representa single protein species in the sample. Using thisapproach thousands of different proteins within a cell ortissue can be separated and relevant information such asisoelectric point (pI ), subunit or monomeric mass andthe amount of protein present in cells can be derived.
Although the value of 2D electrophoresis as ananalytical technique was recognized as long ago as

340 PROTEIN EXPRESSION, PURIFICATION AND CHARACTERIZATION
1975 it is only in the last 10 years that technicalimprovements allied to advances in computers haveseen this approach reach the forefront of research. Theobservation of a single spot on 2D gels is sufficient toallow more detailed analysis by eluting or transferringthe ‘spot’ and subjecting it to microsequencing methodsbased on the Edman degradation, amino acid analysisor even mass spectrometry. Computers and softwareare now available that allow digital evaluations ofthe complex 2D profile and electrophoresis resultscan be readily compared between different organisms.One obvious advantage of these techniques is theirability to detect post-translational modifications ofproteins. Such modifications are not readily apparentfrom protein sequence analysis and are not easilypredicted from genome analysis. 2D electrophoresis isalso permitting differential cell expression of proteinsto be investigated, and in some cases the identificationof disease markers from abnormal profiles.
Mass spectrometryMass spectrometry has become a valuable tool instudying covalent structure of proteins. This hasarisen because mass determination methods havebecome more accurate with the introduction of newtechniques such as matrix assisted laser desorptiontime of flight (MALDI-TOF) and electrospray methods.This allows accurate mass determination for primarysequences as well as the detection of post-translationalmodifications, the analysis of protein purity and whereappropriate the detection of single residue mutations ingenetically engineered proteins.
In mass spectrometry the fundamental observedparameter is the mass to charge ratio (m/z) of gasphase ions. Although this technique has its origins instudies originally performed by J.J. Thomson at thebeginning of the 20th century, over the last 20 yearsthe technique has advanced rapidly and specificallyin the area of biological analysis. The technique isimportant to protein characterization, as demonstratedby the award of the Nobel prize for Chemistry in 2002to two pioneers of this technique, John Fenn and KoichiTanaka, for the development of methods permittinganalysis of structure and identification of biologicalmacromolecules and for the development of ‘soft’desorption–ionization methods in mass spectrometry.
All mass spectrometers consist of three basic com-ponents: an ion source, a mass analyser and a detec-tor. Ions are produced from samples generating chargedstates that the mass analyser separates according to theirmass/charge ratio whilst a detector produces quantifiablesignals. Early mass spectrometry studies utilized elec-tron impact or chemical ionization methods to generateions and with the extreme conditions employed this fre-quently led to the fragmentation of large molecules intosmaller ions (Table 9.4). Improvements in biochemicalmass spectrometry arise from the development of ‘soft’ionization methods that generate molecular ions withoutfragmentation (Table 9.4). In particular, two methods ofsoft ionization are widely used in studies of proteinsand these are matrix-assisted laser desorption ionization(MALDI) and electrospray ionization (ESI).
An initial step in all mass spectrometry measure-ments is the introduction of the sample into a vacuum
Table 9.4 The various mass spectrometry methods have different applications
Ionization method Analyte Upper limit formass range (kDa)
Electron impact Volatile ∼1Chemical ionization Volatile ∼1Electrospray ionization Peptides and proteins ∼200Fast atom bombardment Peptides, small proteins ∼10Matrix assisted laser desorption Peptides or proteins ∼500
ionization (MALDI)

MASS SPECTROMETRY 341
OH
O
O
O
HO
CH3
CH3
Figure 9.29 Structure of sinapinic acid used asmatrix in MALDI methods of ionization. Sinapinic acid(3,5-dimethoxy-4-hydroxycinnamic acid) is the matrixof choice for protein of Mr > 10 000, whilst for smallerproteins α-cyano 4-hydroxycinnamic acid is frequentlypreferred as a matrix
and the formation of a gas phase. Since proteins arenot usually volatile it was thought that mass spectrom-etry would not prove useful in this context. Althoughearly studies succeeded in making volatile derivativesby modifying polar groups the biggest advance hasarisen through the use of alternative strategies. One ofthe most important of these is the MALDI-TOF methodwhere the introduction of a matrix assists in the forma-tion of gas phase protein ions (Figure 9.29). A commonmatrix is sinapinic acid (Mr 224) where the protein(analyte) is dissolved in the matrix at a typical molarratio of ∼1:1000. The protein is frequently present at aconcentration of 1–10 µM. This mixture is dried ona probe and introduced into the mass spectrometer.The next stage involves the use of a laser beam toinitiate desorption and ionization and in this area thematrix prevents degradation of the protein by absorbingenergy. The matrix also assists the irradiated sample tovaporize by forming a rapidly expanding matrix plumethat aids transfer of the protein ions into the massanalyser. Most commercially available mass spectrom-eters coupled to MALDI ion sources are ‘time-of-flight’instruments (TOF-MS) in which the processes of ionformation, separation and detection occur under a verystringent high vacuum. The ionization of proteins doesnot cause fragmentation of the protein but generates aseries of positively charged ions the most common ofwhich occurs through the addition of a proton and isoften designated as the (M + H)+ ion. A second ion-ized species with half the previous m/z ratio denoted as(M + 2H)2+ is also commonly formed. If Na+ or K+ions are present in samples then additional molecular
ions can form such as (M + Na)+ and (M + K)+ andwill be observed with characteristic m/z ratios. Thegaseous protein ions formed by laser irradiation arerapidly accelerated to the same (high) kinetic energyby the application of an electrostatic field and thenexpelled into a field-free region (called the flight tube)where they physically separate from each other accord-ing to their m/z ratios. A detector at the end of the flighttube records the arrival time and intensity of signals asgroups of mass-resolved ions exit the flight tube. Smallmolecular ions will exit the flight tube first followedby proteins with progressively greater m/z ratios. Com-monly, the matrix ions will be the first ions to reach thedetector followed by low molecular weight impurities.To estimate the size of each molecular ion samples ofknown size are usually measured as part of an internalcalibration. By estimating the TOF for these standardproteins the TOF and hence the mass for an unknownprotein can be estimated with very high accuracy.
The mass of the intact protein is one of the mostuseful attributes in protein identification procedures.Although SDS–PAGE can yield a reasonably accuratemass for a protein such measurements are prone toerror. Some proteins run with anomalous mobility onthese gels leading to either over or under estimationof mass that can range in error from 5 to 25 percent.As a result, mass spectrometry has been used torefine estimations of molecular weight by excisionof a protein from 1 or 2D gels. MALDI-TOF isthe mass spectrometry technique of choice for rapiddetermination of molecular weights for gel ‘spots’ andcan be performed on samples directly from the gel,from electroblotting membranes or from the proteinextracted into solution. In fast-atom bombardment(FAB) a high-energy beam of neutral atoms, typicallyXe or Ar, strikes a solid sample causing desorptionand ionization. It is used for large biological moleculesthat are difficult to get into the gas phase. FAB causeslittle fragmentation of the protein and usually gives alarge molecular ion peak, making it ideal for molecularweight determination. A third method of ion generationis electrospray ionization (ESI) mass spectrometry andinvolves a solution of protein molecules suspended in avolatile solvent that are sprayed at atmospheric pressurethrough a narrow channel (a capillary of diameter∼0.1 mm) whose outlet is maintained within an electricfield operating at voltages of ∼3–4 kV. This field

342 PROTEIN EXPRESSION, PURIFICATION AND CHARACTERIZATION
causes the liquid to disperse into fine droplets that canpass down a potential and pressure gradient towardsthe mass analyser. Although the exact mechanismof droplet and ion formation remains unclear furtherdesolvation occurs to release molecular ions intothe gas-phase. Like the MALDI-TOF procedure theelectrospray method is a ‘soft’ ionization techniquecapable of ionizing and transferring large proteinsinto the gas-phase for subsequent mass spectrometerdetection. In the previous discussion the mass analyserhas not been described in detail but three principaldesigns exist: the magnetic and/or electrostatic sectormass analyser, the time of flight (TOF) analyser andthe quadrupole analyser. Each analyser has advantagesand disadvantages (Table 9.5).
The principle of using mass spectrometry in proteinsequencing is shown for a 185 kDa protein derivedfrom SDS–PAGE and digested with a specific protease(Figure 9.30). One peptide species is ‘selected’ forcollision (m/z 438) and the derived fragments ofthis peptide are measured to give the product massspectrum. Some of these fragments differ in theirmass by values equivalent to individual amino acids.The sequence is deduced with the exception thatleucine and isoleucine having identical masses arenot distinguished. This sequence information is usedtogether with the known specificity of the protease tocreate a ‘peptide sequence tag’. Such tag sequences canthen be compared with databases of protein sequencesto identify either the protein itself or related proteins.
Mass spectrometry has very rapidly become anessential tool for all well-funded protein biochemistry
Table 9.5 Different methods of detection employedin mass spectrometry
Analyser Features of system
Quadrupole Unit mass resolution, fastscan, low cost
Sector (magnetic and/orelectrostatic)
High resolution, exact mass
Time-of-flight (TOF) Theoretically, no limitationfor m/z maximum, highflux systems
laboratories and the range of applications is increasingsteadily as the proteomics revolution continues.
How to purify a protein?Armed with the knowledge described in precedingsections it should be possible to attempt the isolationof any protein with the expectation that a highlyenriched pure sample will be obtained. In practice,numerous difficulties present themselves and oftenrender it difficult to achieve this objective. Assuminga protein is soluble and found either in the cytoplasmor a subcellular compartment then perhaps the majorproblem likely to be encountered in isolation is thepotentially low concentration of some proteins intissues. The use of molecular biology avoids thisproblem and allows soluble proteins to be over-expressed in high yields in hosts such as E. coli(Table 9.6). If cDNA encoding the protein of interesthas been cloned it is logical to start with an expressionsystem as the source of biological material.
The first step will involve disrupting the host cells(E. coli ) using either lysozyme or mild sonicationfollowed by centrifugation to remove heavier compo-nents such as membranes from the soluble cytoplasmicfraction which is presumed to contain the protein ofinterest. At this stage with the removal of substantialamounts of protein it may be possible to assay for thepresence of the protein especially if it has an enzymaticactivity that can be readily measured.
Alternatively the protein may have a chromophorewith distinctive absorbance or fluorescence spectrathat can be quantified. However, it is worth notingthat at this stage the protein is just one of manyhundreds of soluble proteins found in E. coli. Thesemeasurements remain important in any purificationbecause they define the initial concentration of protein.All subsequent steps are expected to increase theconcentration of protein at the expense of unwantedproteins. Concurrently, most investigators would alsorun SDS–PAGE gels of the initial starting material toobtain a clearer picture of the number and size range ofproteins present in the cell lysate (Figure 9.31). Again,successive steps in the purification would be expectedto remove many, if not all, of these proteins leavingthe protein of interest as a single homogeneous bandon a gel.

HOW TO PURIFY A PROTEIN? 343
Figure 9.30 Mass spectrometric identification of an unknown protein can be obtained from determining thesequence of proteolytic fragments and comparing the sequence with available databases (reproduced and adaptedwith permission from Rappsilber, J. and Mann, M. Trends Biochem. Sci. 2002, 27, 74–78. Elsevier)
Table 9.6 Example of the purification of an over-expressed enzyme from E. coli
Purification of the enzyme NADH-cytochrome b5 reductase
Purification step Activity (units) Protein(mg)
Specific activity(units/mg)
Recovery(% total)
Enrichment(over initial level)
Cell supernatant 26 247 320 82 100 1Ammonium sulfate
precipitation (50 %)19 340 93 208 74 3
Ammonium sulfateprecipitation (90 %)
18 627 72 259 71 3
Affinity 12 762 10 1276 49 16Size exclusion 11 211 8 1400 43 17
The enzyme’s activity is measured at all stages along with the total protein concentration. The total protein falls during purification butthe specific activity increases significantly. Activity involved measurement of the rate of oxidation of NADH (Data adapted from Barber,M. and Quinn, G.B. Prot. Expr. Purif. 1996, 8, 41–47). The enzyme binds NAD and can therefore be purified by affinity chromatographyusing agarose containing covalently bound ADP

344 PROTEIN EXPRESSION, PURIFICATION AND CHARACTERIZATION
94-
67-
43-
30-
20-
Figure 9.31 Progressive purification of a proteinmonitored by SDS–PAGE. Analysis was carried out on12 % acrylamide gels with molecular mass markersshown in the first lane. The lanes contain fromleft to right total extract; polypeptide compositionof supernatant after centrifugation at 14 000 g;polypeptide composition of pellet fraction aftercentrifugation at 14 000 g; material obtained afterelution from an anion-exchange column and finallymaterial obtained after gel filtration. The final proteinis nearly pure, although a few impurities can still beseen (reproduced with permission from Karwaski, M.F.et al. Protein Expression and Purification. 2002, 25,237–240. Academic Press)
It is rare to achieve purification in one step using asingle technique. More frequently the strategy involves‘capture’ of the material from a crude lysate ormixture of proteins followed by one or more stepsdesigned to enrich the protein of interest followed bya final ‘polishing’ stage that removes the last traces ofcontaminants.
It is not unreasonable to ask which method shouldbe used first? In the absence of any preliminaryinformation there are no set rules and the purifica-tion must be attempted in an empirical fashion byassaying for protein presence via enzyme activity, bio-logical function or other biophysical properties at eachstep of the purification procedure. However, it is rarethat purifications are attempted without some ancillary
information obtained from other sources. Such infor-mation should generally be used to guide the procedurein the direction of a possible purification strategy. So,for example, if a sequentially homologous protein hasbeen previously purified using anion exchange chro-matography and size exclusion to homogeneity it isalmost certain to prove successful again.
Membrane proteins present major difficulties inisolation. Removing the protein from the lipid bilayerwill invariably lead to a loss of structure and func-tion whilst attempting many of the isolation proceduresdescribed above with hydrophobic proteins is more dif-ficult as a result of the conflicting solvent requirementsof such proteins. However, it is by no means impossibleto overcome these difficulties and many hydrophobicproteins have been purified to homogeneity by com-bining procedures described in the above sections withthe judicious use of detergents. In many instancesdetergents allow hydrophobic proteins to remain insolution without aggregating and to be amenable tochromatographic procedures that are applicable to sol-uble proteins.
Summary
Purification requires the isolation of a protein froma complex mixture frequently derived from celldisruption. The aim of a purification strategy is theisolation of a single protein retaining most, if notall, biological activity and the absence of contaminat-ing proteins.
Purification methods have been helped enormouslyby the advances in cloning and recombinant DNAtechnology that allow protein over-expression in for-eign host cells. This allows proteins that were diffi-cult to isolate to be studied where previously this hadbeen impossible.
Methods of purification rely on the biophysical prop-erties of proteins with the properties of mass, charge,hydrophobicity, and hydrodynamic radius being fre-quently used as the basis of separation techniques.Chromatographic methods form the most commongroup of preparative techniques used in proteinpurification.
In all cases chromatography involves the use ofa mobile, usually aqueous, phase that interact with

PROBLEMS 345
an inert support (resin) containing functional groupsthat enhance interactions with some proteins. Inion exchange chromatography the supporting matrixcontains negatively or positively charged groups.Similar methods allow protein separation on thebasis of hydrodynamic radius (size exclusion), ligandbinding (affinity), and non-polar interactions (HIC andRPC).
Alongside preparative techniques are analyticalmethods that establish the purity and mass of the prod-uct. SDS–PAGE involves the separation of polypep-tides under the influence of an electric field solelyon the basis of mass. This technique has proved ofwidespread value in ascertaining subunit molecularmass as well as overall protein purity.
An extension of the basic SDS–PAGE technique isWestern blotting. This method allows the identificationof an antigenic polypeptide within a mixture ofsize-separated components by its reaction with aspecific antibody.
2D electrophoresis allows the separation of proteinsaccording to mass and overall charge. Consequently,through the use of these methods for individual
cell types or organisms, it is proving possible toidentify large numbers of different proteins withinproteomes of single-celled organisms or individualcells.
Of all analytical methods mass spectrometry hasexpanded in importance as a result of technicaladvances permitting accurate identification of themass/charge ratio of molecular ions. The most popularmethods are MALDI-TOF and electrospray spectrome-try. Using modern instrumentation the mass of proteinscan be determined in favourable instances to within1 a.m.u.
In combination with 1 and 2D gel methods massspectrometry is proving immensely valuable in charac-terization of proteomes. The expansion of proteomicsin the post-genomic revolution has placed greaterimportance on preparative and analytical techniques.When the methods described here are combined withthe techniques such as NMR spectroscopy, X-ray crys-tallography or cryo-EM it is possible to go from geneidentification to protein structure within a compara-tively short space of time.
Problems
1. Calculate the relative centrifugal force for a rotor spunat 5000 r.p.m at distances of 9, 6 and 3 cm from thecentre of rotation. Calculate the forces if the rotor isnow rotating at 20 000 r.p.m.
2. Subunit A has a mass of 10 000, a pI of 4.7; subunitB has a mass of 12 000, a pI of 10.2, subunit C has amass of 30 000, a pI of 7.5 and binds NAD; subunit Dhas a mass of 30 000, a pI of 4.5 and subunit E has amass of 100 000, with a pI of 5.0. Subunits A, B andE are monomeric, subunits C and D are heptameric. Apreliminary fractionation of a cell lysate reveals thatall proteins are present in approximately equivalentquantities. Outline an efficient possible route towardspurifying each protein. How would you assesseffective homogeneity?
3. Why is it helpful to know amino acid compositionprior to Edman sequencing?
4. A colleague has failed to sequence using Edmandegradation an oligopeptide believed to be part
of a larger polypeptide chain. Provide reasonableexplanations why this might have happened. Suggestalternative strategies that could work. After furtherinvestigation a partial amino acid sequence Cys-Trp-Ala-Trp-Ala-Cys-CONH2 is obtained. Does thishighlight additional problems. Describe the methodsyou might employ to (i) identify this protein and (ii)isolate the complete protein?
5. Discuss the underlying reasons for the use ofammonium sulfate in protein purification.
6. It has been reported that some proteins retain residualstructure in the presence of SDS. What are the impli-cations of this observation for SDS–PAGE. Whatother properties of proteins might cause problemswhen using this technique?
7. In gel filtration the addition of polyethylene glycol tothe running buffer has been observed to make proteinselute at a later stage as if they were of smaller size.Explain this observation. Suggest other co-solventsthat would have a similar effect.

346 PROTEIN EXPRESSION, PURIFICATION AND CHARACTERIZATION
8. How would you confirm that a protein is normallyfound as a multimer?
9. Four mutated forms of protein X have been expressedand purified. Mutant 1 is believed to contain thesubstitution Gly>Trp, Mutant 2 Glu>Asp, Mutant 3Ala>Lys, and Mutant 4 Leu>Ile. How would you
confirm the presence of these substitutions in theproduct protein?
10. Describe the equipment a well-equipped laboratorywould require to isolate and purify to homogeneity aprotein from bacteria.

10Physical methods of determiningthe three-dimensional structure
of proteins
Introduction
Advances in biochemistry in the 20th century includedthe development of methods leading to protein structuredetermination. Atomic level resolution requires thatthe positions of, for example, carbon, nitrogen, andoxygen atoms are known with precision and certainlywith respect to each other. By knowing the positionsof most, if not all, atoms sophisticated ‘pictures’ ofproteins were established as highlighted by many ofthe diagrams shown in previous chapters.
The impact of structural methods on descriptionsof protein function includes understanding the mech-anism of oxygen binding and allosteric activity inhaemoglobin as well as comprehending the catalyticactivity of simple enzymes such as lysozyme. It wouldextend further to understanding large enzymes such ascyclo-oxygenase, type II restriction endonucleases, andamino acyl tRNA synthetases. Structural techniquesare also applied to membrane proteins such as photo-synthetic reaction centres, together with complexes ofrespiratory chains. More recently, the role of macro-molecular systems such as the proteasome or ribosomewere elucidated by the generation of superbly refined
atomic structures. All of these advances stem fromunderstanding the arrangement of atoms within pro-teins and how these topologies are uniquely suited totheir individual biological roles.
At the beginning of 2004 over 22 000 protein struc-tures (22 348) were deposited in the Protein Data-Bank. Over 86 percent of all experimentally derivedstructures (∼19 400) were the result of crystallo-graphic studies, with most of the remaining struc-tures solved using nuclear magnetic resonance (NMR)spectroscopy. Slowly a third technique, cryoelectronmicroscopy (cryo-EM) is gaining ground on the estab-lished techniques and is proving particularly suitablefor asymmetric macromolecular systems. Although thischapter will focus principally on the experimentalbasis behind the application of X-ray crystallogra-phy and NMR spectroscopy to the determination ofprotein structure the increasing impact of electroncrystallography warrants a discussion of this tech-nique. This approach was originally used in deter-mining the structure of bacteriorhodopsin and islikely to expand in use in the forthcoming years asstructural methods are applied to increasingly com-plex structures.
Proteins: Structure and Function by David Whitford 2005 John Wiley & Sons, Ltd

348 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Figure 10.1 Bar showing the distribution of wavelengths associated different regions of the electromagneticspectrum used in the study of protein structure
These methods are the only experimental techniquesyielding ‘structures’ but other biophysical methodsprovide information on specific regions of a protein.Spectrophotometric methods such as circular dichroism(CD) provide details on the helical content of proteinsor the asymmetric environment of aromatic residues.UV-visible absorbance spectrophotometry assists inidentifying metal ions, aromatic groups or co-factorsattached to proteins whilst fluorescence methods indi-cate local environment for tryptophan side chains.
The use of electromagnetic radiationIt is useful to appreciate the different regions ofthe electromagnetic spectrum involved in each of theexperimental methods described in this chapter. Theelectromagnetic spectrum extends over a wide range offrequencies (or wavelengths) and includes radio waves,microwaves, the infrared region, the familiar ultravioletand visible regions of the spectrum, eventually reachingvery short wavelength or high frequency X-rays. Theenergy (E) associated with radiation is defined byPlanck’s law
E = hν where ν = c/λ (10.1)
where c is the velocity of light, and h, Planck’sconstant, has a magnitude of 6.6 × 10−34 Js, and ν
is the frequency of the radiation. The product offrequency times wavelength (λ) yields the velocity – aconstant of ∼3 × 108 m s−1. From the relationshipc = νλ it is apparent that X-rays have very shortwavelengths of approximately 0.15 × 10−9 m or less.
At the opposite end of the frequency spectrum radiowaves have longer wavelengths, often in excess of10 m (Figure 10.1). The frequency scale is significantbecause each domain is exploited today in biochemistryto examine different atomic properties or motionspresent in proteins (Table 10.1).
Radio waves and microwaves cause changes to themagnetic properties of atoms that are detected by sev-eral techniques including NMR and electron spin reso-nance (ESR). In the microwave and infrared regionsof the spectrum irradiation causes bond movementsand in particular motions about bonds such as ‘stretch-ing’, ‘bending’ or rotation. The ultraviolet (UV) andvisible regions of the electromagnetic spectrum are ofhigher energy and probe changes in electronic structurethrough transitions occurring to electrons in the outershells of atoms. Fluorescence and absorbance meth-ods are widely used in protein biochemistry and arebased on these transitions. Finally X-rays are used toprobe changes to the inner electron shells of atoms.These techniques require high energies to ‘knock’ innerelectrons from their shells and this is reflected in thefrequency of such transitions (∼1018 Hz).
All branches of spectroscopy involve either absorp-tion or emission of radiation and are governed by afundamental equation
�E = E2 − E1 = hν (10.2)
where E2 and E1 are the energies of the two quantizedstates involved in the transition. Most branches ofspectroscopy involve the absorption of radiation withthe elevation of the atom or molecule from a groundstate to one or more excited states. All spectral

X-RAY CRYSTALLOGRAPHY 349
Table 10.1 The frequency range and atomic parameters central to physical techniques used to study protein structure
Technique Frequency range (Hz) Measurement
NMR ∼0.6 – 60 × 107 Nucleus’ magnetic fieldESR ∼1–30 × 109 Electron’s magnetic fieldMicrowave ∼0.1–60 × 1010 Molecular rotationInfrared ∼0.6–400 × 1012 Bond vibrations and bendingUltraviolet/visible ∼7.5–300 × 1014 Outer core electron transitionsMossbauer ∼3–300 × 1016 Inner core electron transitionsX-ray ∼1.5–15 × 1018 Inner core electron transitions
lines have a non-zero width usually defined by abandwidth measured at half-maximum amplitude. Iftransitions occur between two discrete and well-definedenergy levels then one would expect a line of infiniteintensity and of zero width. This is never observed, andHeisenberg’s uncertainty principle states that
�E�t ≈ h/2π (10.3)
where �E and �t are the uncertainties associated withthe energy and lifetimes of the transition. Transitions donot occur between two absolutely defined energy levelsbut involve a series of sub-states. Thus if the lifetimeis short (�t is small) it leads to a correspondinglylarge value of �E, where �E defines the width ofthe absorption line (Figure 10.2).
Figure 10.2 Theoretical absorption line of zero widthand a line of finite width (�E)
X-ray crystallography
X-ray crystallography is the pre-eminent techniquein the determination of protein structure progressingfrom the first low-resolution structures of myoglobin tohighly refined structures of macromolecular complexes.X-rays, discovered by Willem Roentgen, were shownto be diffracted by crystals in 1912 by Max vonLaue. Of perhaps greater significance was the researchof Lawrence Bragg, working with his father WilliamBragg, who interpreted the patterns of spots obtainedon photographic plates located close to crystals exposedto X-rays. Bragg realized ‘focusing effects’ ariseif X-rays are reflected by series of atomic planesand he formulated a direct relationship between thecrystal structure and its diffraction pattern that is nowcalled Bragg’s law. All crystallography since Bragghas centred around a basic arrangement of an X-ray source incident on a crystal located close to adetector (Figure 10.3). Historically, detection involvedsensitive photographic films but today’s detectionmethods include charged coupled devices (CCD) andare enhanced by synchrotron radiation, an intensesource of X-rays.
Bragg recognized that sets of parallel lattice planeswould ‘select’ from incident radiation those wave-lengths corresponding to integral multiples of thiswavelength. Peaks of intensity for the scattered X-rays are observed when the angle of incidence is equalto the angle of scattering and the path length differ-ence is equal to an integer number of wavelengths.From diagrams such as Figure 10.4 it is relatively

350 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Figure 10.3 The basic crystallography ‘set-up’ usedin X-ray diffraction. Monochromatic X-rays ofwavelength 1.5418 A, sometimes called Cu Kα X-rays,denote the dislodging of an electron from the K shelland the movement of an electron from the nextelectronic shell (L). After passing through filters toremove Kβ radiation the X-rays strike the crystal
Figure 10.4 X-rays scattered by a crystal lattice
straightforward to establish the path difference (nλ)
using geometric principles as
nλ = 2d sin θ (10.4)
This equality, a quantitative statement of Bragg’slaw, allows information about the crystal structureto be determined since the wavelength of X-rays is
closely controlled. From the Bragg equation diffractionmaxima are observed when the path length differencefor the scattered X-rays is a whole number ofwavelengths. The arrangement of atoms can be re-drawn to make this equality more clear. The pathdifference is equivalent to
d cos θi − d cos θr = nλ (where n = 1, 2, 3, . . .)
(10.5)This formulation is readily extended into three di-mensions and is called the Laue set of equations(Figure 10.5). The Laue equations must be satisfiedfor diffraction to occur but are more cumbersometo deal with and lack the elegant simplicity of theBragg equation. For each dimension Laue equationsare written as
a(cos αi − cos αr) = hλ (where h = 1, 2, 3, . . .) (10.6)
b(cos βi − cos βr) = kλ (where k = 1, 2, 3, . . .) (10.7)
c(cos γi − cos γr) = lλ (where l = 1, 2, 3, . . .) (10.8)
where a, b and c refers to the spacing in each of thethree dimensions (shown by d in Figure 10.5).
Within any crystal the basic repeating pattern isthe unit cell (Figure 10.6) and in some crystals morethan one unit cell is recognized. In these instances thesimplest unit cell is chosen governed by a series ofselection rules. The unit cell can be translated (movedsideways but not rotated) in any direction within thecrystal to yield an identical arrangement (Figure 10.7).
The unit cell, the basic building block of a crystal,is repeated infinitely in three dimensions but ischaracterized by three vectors (a, b, c) that formthe edges of a paralleliped. The unit cell is alsodefined by three angles between these vectors (α,the angle between b and c; β, the angle between a
Figure 10.5 The Laue equations

X-RAY CRYSTALLOGRAPHY 351
Figure 10.6 Possible unit cells in a two-dimensionallattice
Figure 10.7 A unit cell for a simple molecule
and c; γ, the angle between a and b, Figure 10.8).The recognition of different arrangements within aunit cell was recognized by Auguste Bravais duringthe 19th century.1 In two dimensions there are fivedistinct Bravais lattices, whilst in three dimensions thenumber extends to 14, usually classified into crystaltypes. Any crystal will belong to one of seven possibledesigns and the symmetry of these systems introduces
1First described by Frankenheim in 1835 who incorrectly assigned15 different structures as opposed to the correct number of 14,recognized by Bravais which to this day carries only his name.
α
γ
β
a
b
c
z
x
y
Figure 10.8 The angles and vectors defining anyunit cell
constraints on the possible values of the unit cellparameters. The seven crystal systems are triclinic,monoclinic, orthorhombic, tetragonal, rhombohedral,hexagonal and cubic (Table 10.2). The description ofunit cells and lattice types owes much to the origins ofcrystallography within the field of mineralogy.
In biological systems the unit cell may possessinternal symmetry containing more than one proteinmolecule related to others via axes or planes of sym-metry. A series of symmetry operations allows thegeneration of coordinates for atoms in the next unit celland includes operations such as translations in a plane,rotations around an axis, reflection (as in a mirror),and simultaneous rotation and inversion. Collection ofsymmetry operations that define a particular crystallo-graphic arrangement are known as space groups, andwith 230 recognized space groups published by theInternational Union of Crystallography crystals havebeen found in most, but not all, arrangements.
Scattering depends on the properties of the crystallattice and is the result of interactions between theincident X-rays and the electrons of atoms withinthe crystal. As a result metal atoms such as iron orcopper and atoms such as sulfur are very effectiveat scattering X-rays whilst smaller atoms such asthe proton are ineffective. The end result of X-raydiffraction experiment is not a picture of atoms, butrather a map of the distribution of electrons in the

352 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Table 10.2 The parameters (a, b, c and α, β, γ) governing the different crystal lattices together with some of theirsimpler arrangements
Crystal system Representation Crystal system Representation
Number of Bravais lattices. Number of Bravais lattices.
Vector and angle properties. Vector and angle properties
Monoclinic Triclinic
2 1
α = γ = 90◦
β
No restrictionsβ α
γ
Cubic Orthorhombic
3 4
a = b = c; α = β = γ = 90◦α = β = γ = 90◦
Tetragonal Hexagonal
2 1
a = b; α = β = γ = 90◦ a = b; α = β = 90◦, γ = 120◦
Rhombohedral
1
a = b = c; α = β = γ
α
αα
Several permutations are possible within groups and leads to descriptions of primitive unit cells, face-centred containing an additional point in the center ofeach face, body-centred contains an additional point in the centre of the cell and centred, with an additional point in the centre of each end.
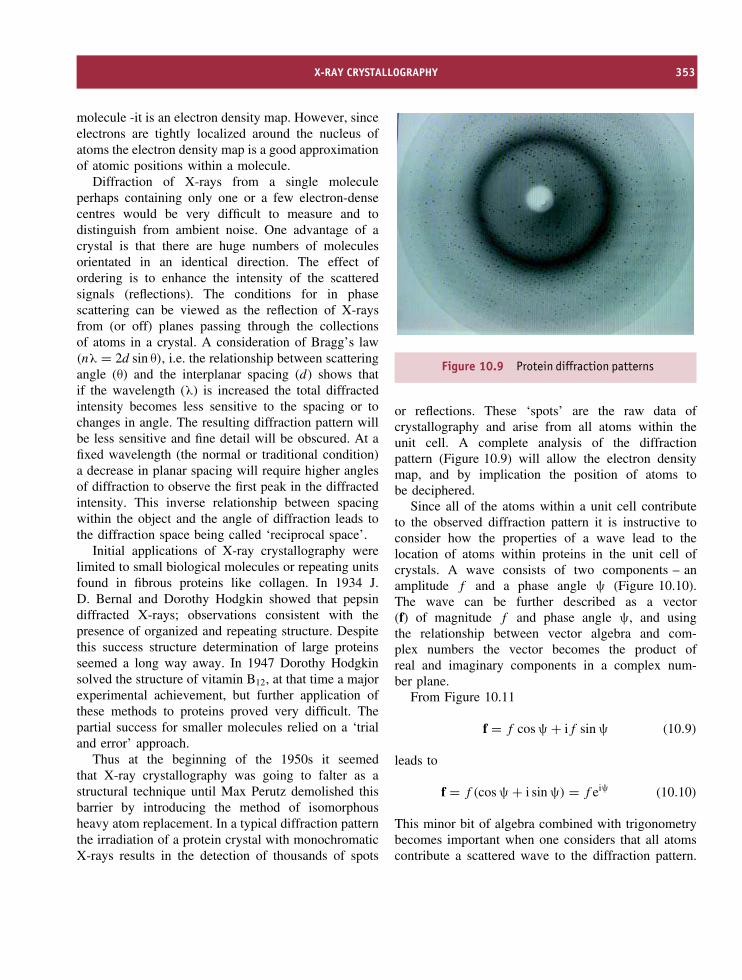
X-RAY CRYSTALLOGRAPHY 353
molecule -it is an electron density map. However, sinceelectrons are tightly localized around the nucleus ofatoms the electron density map is a good approximationof atomic positions within a molecule.
Diffraction of X-rays from a single moleculeperhaps containing only one or a few electron-densecentres would be very difficult to measure and todistinguish from ambient noise. One advantage of acrystal is that there are huge numbers of moleculesorientated in an identical direction. The effect ofordering is to enhance the intensity of the scatteredsignals (reflections). The conditions for in phasescattering can be viewed as the reflection of X-raysfrom (or off) planes passing through the collectionsof atoms in a crystal. A consideration of Bragg’s law(nλ = 2d sin θ), i.e. the relationship between scatteringangle (θ) and the interplanar spacing (d) shows thatif the wavelength (λ) is increased the total diffractedintensity becomes less sensitive to the spacing or tochanges in angle. The resulting diffraction pattern willbe less sensitive and fine detail will be obscured. At afixed wavelength (the normal or traditional condition)a decrease in planar spacing will require higher anglesof diffraction to observe the first peak in the diffractedintensity. This inverse relationship between spacingwithin the object and the angle of diffraction leads tothe diffraction space being called ‘reciprocal space’.
Initial applications of X-ray crystallography werelimited to small biological molecules or repeating unitsfound in fibrous proteins like collagen. In 1934 J.D. Bernal and Dorothy Hodgkin showed that pepsindiffracted X-rays; observations consistent with thepresence of organized and repeating structure. Despitethis success structure determination of large proteinsseemed a long way away. In 1947 Dorothy Hodgkinsolved the structure of vitamin B12, at that time a majorexperimental achievement, but further application ofthese methods to proteins proved very difficult. Thepartial success for smaller molecules relied on a ‘trialand error’ approach.
Thus at the beginning of the 1950s it seemedthat X-ray crystallography was going to falter as astructural technique until Max Perutz demolished thisbarrier by introducing the method of isomorphousheavy atom replacement. In a typical diffraction patternthe irradiation of a protein crystal with monochromaticX-rays results in the detection of thousands of spots
Figure 10.9 Protein diffraction patterns
or reflections. These ‘spots’ are the raw data ofcrystallography and arise from all atoms within theunit cell. A complete analysis of the diffractionpattern (Figure 10.9) will allow the electron densitymap, and by implication the position of atoms tobe deciphered.
Since all of the atoms within a unit cell contributeto the observed diffraction pattern it is instructive toconsider how the properties of a wave lead to thelocation of atoms within proteins in the unit cell ofcrystals. A wave consists of two components – anamplitude f and a phase angle ψ (Figure 10.10).The wave can be further described as a vector(f) of magnitude f and phase angle ψ, and usingthe relationship between vector algebra and com-plex numbers the vector becomes the product ofreal and imaginary components in a complex num-ber plane.
From Figure 10.11
f = f cos ψ + if sin ψ (10.9)
leads to
f = f (cos ψ + i sin ψ) = f eiψ (10.10)
This minor bit of algebra combined with trigonometrybecomes important when one considers that all atomscontribute a scattered wave to the diffraction pattern.

354 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Phase angle Ψ
Wavelength
Am
plitu
de
Figure 10.10 A wave described by its amplitude and phase angle. A cosine and sine wave are simply related by aphase shift of 90◦ or π/2 radians
Figure 10.11 Vector representation of a wave andits expression as a complex number
Diffracted waves of intensity (amplitude, f ) and phase(ψ) are summed together and described by the vectorFhkl known as the structure factor. Equation 10.10becomes
Fhkl =∑
f cos ψ +∑
if sinψ (10.11)
and leads to
Fhkl = Fhkl(cos ϕhkl + i sin ϕhkl) = Fhkleiϕhkl (10.12)
Fhkl is the square root of the intensity of the observed(measured) diffraction spot often called Ihkl, whilst the
ϕhkl term represents the summation of all phase termscontributing to this spot.
The next problem is to relate the structure factorFhkl to the three-dimensional distribution of electronsin the crystal – the distribution of atoms. The structurefactor is the Fourier transform of the electron densityand is a vector defined by intensity Fhkl and phase ϕhkl.The value of the electron density at a real-space latticepoint (x, y, z) denoted by ρ (x, y, z) is equivalent to
ρ(x, y, z) = 1/V
+∞∑hkl=−∞
Fhkle−2πi(hx+ky+lz) (10.13)
where ρ is the value of the electron density at the real-space lattice point (x, y, z) and V is the total volume ofthe unit cell. This is rearranged using Equation 10.12to give
ρ(x, y, z) = 1/V
+∞∑hkl=−∞
Fhkleiϕhkle−2πi(hx+ky+lz)
(10.14)
where ϕhkl is the phase information.To obtain ‘3D pictures’ of molecules the crystal is
rotated while a computer-controlled detector producestwo-dimensional electron density maps for each angle

X-RAY CRYSTALLOGRAPHY 355
of rotation and establishes a third dimension. A rotatingCu target is the source of X-rays and this generatoris normally cooled to avoid excessive heating. X-rays pass via a series of slits (monochromators) tothe crystal mounted in a goniometer. The crystal isheld within a loop by surface tension or attachedby glue to a narrow fibre and can be rotated inany direction. Nowadays crystals are flash cooled to∼77 K (liquid nitrogen temperatures) with benefitsarising from reduced thermal vibrations leading tolower conformational disorder and better signal/noiseratios. Cryocooling also limits radiation damage of thecrystal and it is possible to collect complete data setsfrom a single specimen. Finally, a detector recordsthe diffraction pattern where each spot represents areflection and has parameters of position, intensityand phase. For a typical crystal there may be 40 000reflections to analyse, and although this remains a time-consuming task computers facilitate the process. Ingeneral all of the diffraction apparatus is controlledvia computer interfaces. The data collected is a seriesof frames containing crystal diffraction patterns as itrotates in the X-ray beam. From these frames dataanalysis yields a list of reflections (positions) and theirintensities (amplitudes) but what remains unknown arethe relative phases of the scattered X-rays.
A useful scheme with which to understand theoccurrence of reflections is the Ewald construction. Ifa sphere of radius 1/λ centred around the crystal isdrawn then the origin of the reciprocal lattice lies wherethe transmitted X-ray beam passes straight throughthe crystal and is described as at the edge of theEwald sphere (Figure 10.12). Diffraction spots occuronly when the Laue or Bragg equations are satisfiedand this condition occurs when the reciprocal latticepoint lies exactly on the Ewald sphere. At any oneinstant the likelihood of observing diffraction is smallunless the crystal is rotated to bring more points in thereciprocal lattice to lie on the Ewald sphere by rotationof the crystal (Figure 10.13).
A detection device perpendicular to the incomingbeam records diffraction on an arbitrary scale with themost obvious attributes being position and intensityof the ‘spots’. The intensity is proportional to thesquare of the structure factor magnitude according tothe relationship
Ihkl = k|Fobs|2 (10.15)
Figure 10.12 Scattering of X-rays by atoms withina crystal
where k is a constant that depends on several factorssuch as the X-ray beam energy, the crystal volume,the volume of the unit cell, the angular velocity of thecrystal rotation, etc.
From Equation 10.14 the relationship between inten-sity, phase and the fractional coordinates of each atom(x, y and z) is clearly emphasized and from thisequation it is clear that if we know the structure we cangenerate Fhkl. However, in crystallography the aim isto determine the position of atoms, i.e. x, y and z fromFhkl – the inverse problem. A complete description ofeach reflection – its position, intensity and phase – isrepresented by the structure factor, Fhkl and if this
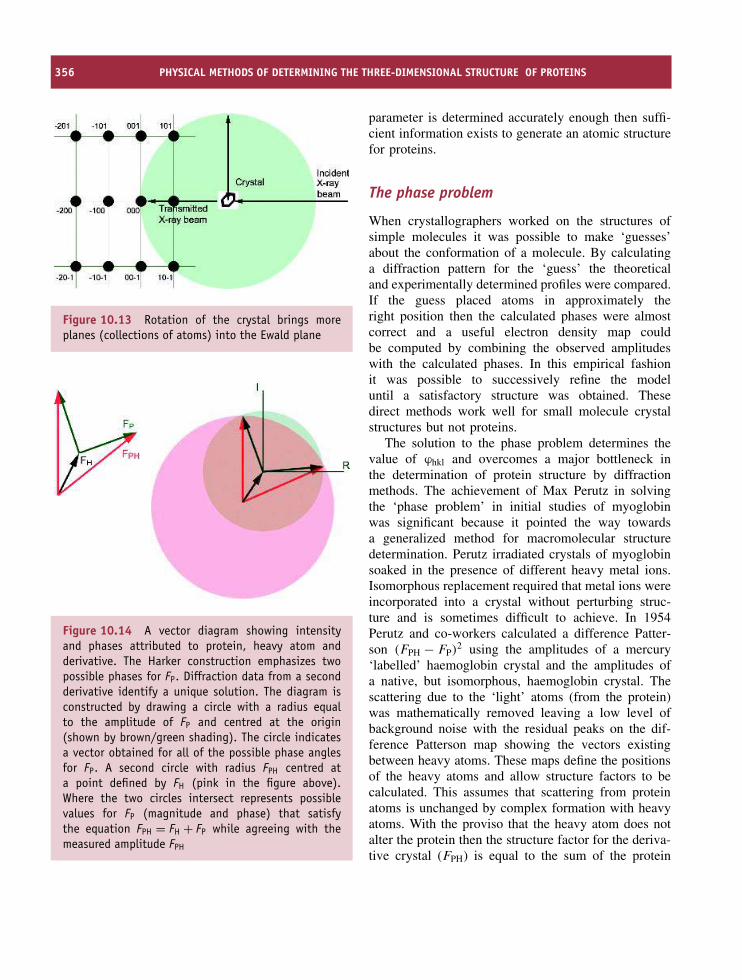
356 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Figure 10.13 Rotation of the crystal brings moreplanes (collections of atoms) into the Ewald plane
Figure 10.14 A vector diagram showing intensityand phases attributed to protein, heavy atom andderivative. The Harker construction emphasizes twopossible phases for FP. Diffraction data from a secondderivative identify a unique solution. The diagram isconstructed by drawing a circle with a radius equalto the amplitude of FP and centred at the origin(shown by brown/green shading). The circle indicatesa vector obtained for all of the possible phase anglesfor FP. A second circle with radius FPH centred ata point defined by FH (pink in the figure above).Where the two circles intersect represents possiblevalues for FP (magnitude and phase) that satisfythe equation FPH = FH + FP while agreeing with themeasured amplitude FPH
parameter is determined accurately enough then suffi-cient information exists to generate an atomic structurefor proteins.
The phase problem
When crystallographers worked on the structures ofsimple molecules it was possible to make ‘guesses’about the conformation of a molecule. By calculatinga diffraction pattern for the ‘guess’ the theoreticaland experimentally determined profiles were compared.If the guess placed atoms in approximately theright position then the calculated phases were almostcorrect and a useful electron density map couldbe computed by combining the observed amplitudeswith the calculated phases. In this empirical fashionit was possible to successively refine the modeluntil a satisfactory structure was obtained. Thesedirect methods work well for small molecule crystalstructures but not proteins.
The solution to the phase problem determines thevalue of ϕhkl and overcomes a major bottleneck inthe determination of protein structure by diffractionmethods. The achievement of Max Perutz in solvingthe ‘phase problem’ in initial studies of myoglobinwas significant because it pointed the way towardsa generalized method for macromolecular structuredetermination. Perutz irradiated crystals of myoglobinsoaked in the presence of different heavy metal ions.Isomorphous replacement required that metal ions wereincorporated into a crystal without perturbing struc-ture and is sometimes difficult to achieve. In 1954Perutz and co-workers calculated a difference Patter-son (FPH − FP)
2 using the amplitudes of a mercury‘labelled’ haemoglobin crystal and the amplitudes ofa native, but isomorphous, haemoglobin crystal. Thescattering due to the ‘light’ atoms (from the protein)was mathematically removed leaving a low level ofbackground noise with the residual peaks on the dif-ference Patterson map showing the vectors existingbetween heavy atoms. These maps define the positionsof the heavy atoms and allow structure factors to becalculated. This assumes that scattering from proteinatoms is unchanged by complex formation with heavyatoms. With the proviso that the heavy atom does notalter the protein then the structure factor for the deriva-tive crystal (FPH) is equal to the sum of the protein

X-RAY CRYSTALLOGRAPHY 357
structure factor (FP) and the heavy atom structure fac-tor (FH), or
FPH = FP + FH (10.16)
The structure factor is a vector and leads to a repre-sentation called the Harker construction (Figure 10.14).Since the length and orientation of one side (FH) isknown along with the magnitude of FPH and FP thereare two possible solutions for the phase of FP.
Heavy metal atom derivatives must give minimalperturbations of protein structure and retain the lat-tice structure of the unmodified crystal. The deriva-tives must also perturb reflection intensities sufficientlyto allow calculation of phases. Protein crystals areusually prepared with heavy metal atoms such asuranium, platinum or mercury introduced at specificpoints within the crystal with thiol groups representingcommon high affinity sites. Amongst the compoundsthat have been used are potassium tetrachloroplati-nate(II), p-chloromercuribenzoate, potassium tetrani-troplatinate(II), uranyl acetate and cis-platinum (II)diamine dichloride.
Modern diffraction systems are highly specializedexploiting advances in computing, material science,semiconductor technology and quantum physics toallow the generation of accurate and precise proteinstructures. All of the originally tedious calculationsare performed computationally whilst all equipment iscontrolled via microprocessors. A further enhancementof X-ray diffraction has been the use of highlyintense or focused beams such as those obtained fromsynchrotron sources. This has allowed new solutionsto the phase problem involving the use of synchrotronradiation at multiple wavelengths. Today one of themethods of choice in X-ray crystallography is calledmultiple anomalous dispersion (MAD). Just as the useof heavy metal derivatives allowed the extrapolationof phase information and permitted the determinationof electron density from observed diffraction data theuse of multiple wavelengths near the absorption edgeof a heavy atom achieves a comparable effect. Thisis commonly performed by replacing methionine withselenomethionine during protein expression. Nowadayscrystallography centres are based around the locationof synchrotron sources and the speed and qualityof structure generation has improved dramatically(Figure 10.15).
Figure 10.15 The arrangement of data collectioncentres about a synchrotron storage ring (reproducedwith permission from Als-Nielsen, J. & McMorrow, D.Elements of Modern X-ray Physics. John Wiley & Sons)
Fitting, refinement and validationof crystal structures
The initial electron density map (Figure 10.16) doesnot resolve individual atoms and in the early stages sev-eral ‘structures’ are compatible with the data. Higherresolution allows the structure to be assigned. Interpre-tation of electron density maps requires knowledge ofthe primary sequence since the arrangement of heavyatoms for the side chains dictates the fitting process.Resolution is not a constant even between similarlysized proteins because crystal lattices have motion fromthermal fluctuations and mobility contributes to thefinal overall resolution. One estimate of mobility withina crystal is the B factor (Debye–Waller factor) thatreflects spreading or blurring of electron density. TheB factor represents the mean square displacement ofatoms and has units of A2.
Structures before refinement are often at a resolu-tion >4.5 A where only α helices are observed andthe identification of side chains is unlikely. At higherresolutions of 2.5–3.5 A the polypeptide backbone is

358 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Figure 10.16 An electron density map
traced via electron density located primarily on car-bonyl groups and helices, strands and aromatic sidechains such as tryptophan are defined. Around 2.0 Aalmost all of the structure will be identified includingthe conformations associated with side chains. Spe-cialized computer programs fit electron density mapsand the process is assisted by assuming standard bondlengths and angles. Refining models in an iterativefashion progressively improves the agreement withexperimental data. A structure is judged by the crys-tallographic R-factor, defined as the average fractionalerror in the sum of the differences between calculatedstructural factors (Fcal) and observed structural factors(Fobs) divided by the sum of the observed structuralfactors.
R =∑
|Fobs − Fcal|/∑
Fobs (10.17)
A value of 0.20 is often represented as an R factor of20 percent and ‘good’ structures have R-factors rangingfrom 15 to 25 percent or approximately 1/10th theresolution of the data. A structure of resolution 1.9 Ais expected to yield an R factor of <0.19. One resultof protein structure determination is the generationof a file that lists x, y, and z coordinates for allheavy atoms whose locations are known. These filesare deposited in protein databanks and the PDB fileslisted throughout this book represent the culminationof this analysis.
Protein crystallization
One of the slowest steps in protein crystallographyis the production of protein crystals. The methodsemployed in crystal production rely on the ordered pre-cipitation of proteins. The first protein (urease) wascrystallized by James Sumner as long ago as 1926,and was followed by the crystallization of pepsinin 1930 by John Northrop. In the next 20 yearsover 40 additional proteins were crystallized, includ-ing lyzosyme, trypsin, chymotrypsin, catalase, papain,ficin, enolase, carbonic anhydrase, carboxypeptidase,hexokinase and ribonuclease, although structure deter-mination had to wait until the advances of Perutz andKendrew (Figure 10.17).
Crystallization requires the ordered formation oflarge (dimensions greater than 0.1 mm along eachaxis), stable crystals with sufficient long-range orderto diffract X-rays. Structures produced by X-raydiffraction are only as good as the crystals from whichthey are derived.
To form a crystal protein molecules assemble intoa periodic lattice from super-saturated solutions. Thisinvolves starting with solution of pure protein at a con-centration between 0.5 and 200 mg ml−1 and addingreagents that reduce protein solubility close to thepoint of precipitation. These reagents perturb pro-tein–solvent interactions so that the equilibrium shiftsin favour of protein–protein association. Further con-centration of the solution results in the formation of

X-RAY CRYSTALLOGRAPHY 359
Figure 10.17 Examples of protein crystals. Top row:azurin from Pseudomonas aeruginosa, flavodoxin fromDesulfovibrio vulgaris, rubredoxin from Clostridiumpasteurianum. Bottom row: azidomethemerythrin fromthe marine worm Siphonosoma funafatti, lampreyhaemoglobin and bacteriochlorophyll a protein fromProsthecochloris aesturii. The beauty of the abovecrystals is their colour arising from the presence of lightabsorbing co-factors such as metal, heme, flavin orchlorophyll (reproduced with permission from Voet, D.,Voet, J.G. & Pratt, C.W. Fundamentals of BiochemistryJohn Wiley & Sons Ltd. Chichester, 1999)
nucleation sites, a process critical to crystal forma-tion and is the first of three basic stages of crys-tallization common to all systems. It is followed byexpansion and then cessation as the crystal reaches alimiting size.
Two experimental methods used to form crystalsfrom protein solutions are vapour diffusion and equilib-rium dialysis. Vapour diffusion is the standard methodused for protein crystallization. It is suitable for usewith small volumes, easy to set-up and to monitorreaction progress. A common format involves ‘hang-ing drops’ containing protein solution plus ‘precipitant’at a concentration insufficient to precipitate the pro-tein (Figure 10.18). The drop is equilibrated againsta larger reservoir of solution containing precipitantand after sealing the chamber equilibration leads tosupersaturating concentrations that induce protein crys-tallization in the drop.
Figure 10.18 The ‘hanging drop’ or vapour diffusionmethod of protein crystallization. As little as 5 µlof concentrated solution (protein + solvent) may besuspended on the coverslip
Figure 10.19 Equilibrium dialysis can be achievedwith many different ‘designs’ although the basicprinciple involves the separation of protein solutionfrom the precipitant by a semipermeable membrane.Diffusion across the membrane promotes orderedcrystallization
A second method is equilibrium dialysis and is usedfor crystallization of proteins at low and high ionicstrengths (Figure 10.19). Small volumes of proteinsolution are placed in a container separated fromprecipitant by a semi-permeable membrane. Slowly theprecipitant causes crystal formation within the wellcontaining the protein solution.
To perform large numbers of crystallization trialsit is common to use robotic systems to automate the

360 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
process and to eliminate labour-intensive manipulationswhilst crystallization is performed in temperature-controlled rooms, free of vibration, leading to crystalsappearing over a period of 4–10 days. When the abovetechniques work successfully X-ray diffraction yieldsunparalleled resolution for protein structures. There islittle doubt that the technique will continue to representthe main method of protein structure determination inthe exploration of new proteomes.
Nuclear magnetic resonancespectroscopy
NMR spectroscopy as a tool for determining proteinstructure arose more recently than X-ray crystallogra-phy. With the origins of X-ray diffraction lying in thediscoveries of Roentgen, Laue and the Braggs at thebeginning of the 20th century it was not until 1945that a description of the NMR phenomenon was givenby Felix Bloch and Edward Purcell. NMR spectra areobserved upon absorption of a photon of energy andthe transition of nuclear spins from ground to excitedstates. The observation of signal associated with thesetransitions and the use of radio frequency irradiationto elicit the response marked the start of NMR spec-troscopy.
This discovery would have remained insignifi-cant except for subsequent observations showing thatnuclear transitions differed in frequency from onenucleus to another but also showed subtle differencesaccording to the nature of the chemical group. So forthe proton this property meant that proteins exhibitedmany signals with, for example, the methyl protons res-onating at different frequencies to amide protons whichin turn are different to the protons attached to the α orβ carbons. In 1957 the first NMR spectrum of a protein(ribonuclease) was recorded but progress as a structuraltechnique remained slow until Richard Ernst describedthe use of transient techniques. Transient signals pro-duced after a pulse of radio frequency radiation are con-verted into a normal spectrum by the mathematical pro-cess of Fourier transformation – this technique wouldpave the way towards important advances, particularlymulti-dimensional NMR spectroscopy, that are now thebasis of all biomolecular structure determination.
Table 10.3 Spin properties and abundance ofimportant nuclei in protein NMR studies
Nuclei Spin Abundance
(% total)
Magnetogyric ratio
(γ) (×107 T−1s−1)
Ratio
(γn/γH)
1H 1/2 99.985 26.7520 1.002H 1 0.015 4.1066 0.1513C 1/2 1.108 6.7283 0.2514N 1 99.630 1.9338 0.0715N 1/2 0.370 2.712 0.1018O 5/2 0.037 3.6279 0.1431P 1/2 100.000 10.841 0.41
NMR phenomena
Underlying the NMR phenomenon is a property ofall atomic nuclei called ‘spin’. Spin describes thenature of a magnetic field surrounding a nucleus and ischaracterized by a spin number, I, which is either zeroor a multiple of 1/2 (e.g. 1/2, 2/2, 3/2, etc.). Nucleiwhose spin number equals zero have no magneticfield and from a NMR standpoint are uninteresting.This occurs when the number of neutrons and thenumber of protons are even. Nuclei with non-zero spinnumbers have magnetic fields that vary considerablyin complexity (Table 10.3). Spin 1/2 nuclei representthe simplest situation and arise when the number ofneutrons plus the number of protons is an odd number.2
The most important spin 1/2 nucleus is the proton witha high natural abundance (∼100 %) and its occurrencein all biomolecules.
For nuclei such as 12C the most common isotope isNMR ‘silent’ and the ‘active’ spin 1/2 nucleus (13C)has a low natural abundance of ∼1.1 percent. Similarly15N has a natural abundance of ∼0.37 percent.However, advances in molecular biological techniquesenable proteins to be expressed in host cells grown onmedia containing labelled substrates. Growing bacteria,yeast or other cell cultures on substrates containing 15Nor 13C (ammonium sulfate and glucose are commonsources) allows uniform enrichment of proteins in
2A third possibility exists when the number of neutrons and thenumber of protons are both odd and this leads to the nucleus havingan integer spin (i.e. I = 1, 2, 3, etc.).

NUCLEAR MAGNETIC RESONANCE SPECTROSCOPY 361
these nuclei. Biomolecular NMR spectroscopy requiresproteins enriched with 13C or 15N or ideally both nuclei.
Although the shape of the magnetic field of anucleus is described by the parameter I (spin number)the magnitude is determined by the magnetogyric ratio(γ). A nucleus with a large γ has a stronger magneticfield than a nucleus with a small γ. The proton (1H)has the largest magnetogyric ratio and coupled with itshigh natural abundance this contributes to its popularityas a common form of NMR spectroscopy.
When samples are placed in magnetic fields nuclearspins become polarized in the direction of the fieldresulting in a net longitudinal magnetization. In thenormal or macroscopic world two bar magnets can bealigned in an infinite number of arrangements but at anatomic level these alignments are governed by quantummechanics and the number of possible orientations is2I + 1. For spin 1/2 nuclei this gives two orientationsthat in the absence of an external magnetic field are ofequal energy. For spin 1/2 nuclei such as 1H, 13C, or15N application of a magnetic field removes degeneracyand the energy levels split into parallel and antiparallelorientations (Figure 10.20).
Spins aligned parallel with external magnetic fieldsare of slightly lower energy than those aligned in anantiparallel orientation. The concept of two energylevels allows one to envisage transitions between lowerand higher energy levels analogous to that seen in otherforms of spectroscopy. The difference in population(nupper/nlower) between each level is governed by
Figure 10.20 An energy level diagram reflecting thealignment of spin 1/2 nuclei in applied magnetic fields
Boltzmann’s distribution
nupper/nlower = e−(�E/kBT ) (10.18)
When �E ∼ kBT , as would occur with two closelyspaced energy levels, the ratio nupper/nlower approaches1. At thermal equilibrium the number of nuclei in thelower energy level slightly exceeds those in the higherenergy level. As a result of this small inequality itis possible to elicit transitions between states by theapplication of short, intense, radio frequency pulses.
Instead of considering a single spin it is moreuseful to consider the magnetic ensemble of spins. Inthe presence of the applied magnetic field (B0) spinpolarization occurs and a vector model of NMR viewsthe net magnetization lying in the direction of the z-axis. Irradiation of the sample by a radiofrequency(rf) field denoted as B1 along the x-axis rotatesthe macroscopic magnetization into the xy plane(Figure 10.21).
Most frequently the pulse length is calculated totip ‘magnetization’ through 90◦ (π/2 radians). Thexy plane lies perpendicular to the magnetic field andcauses transverse magnetization to precess under theinfluence of the applied magnetic field. Precessionin the xy plane at the Larmor frequency of thenuclei under investigation induces a current in thedetector coil that is the observable signal in all NMRexperiments.
Transverse magnetization decays exponentially withtime in the form of a signal called a free inductiondecay (FID) eventually reaching zero. All NMR spectra(such as the one shown in Figure 10.22) are derivedby converting the FID signal of intensity versus timeinto a profile of intensity versus frequency via Fouriertransformation (FT).
Figure 10.21 Application of a radiofrequency pulse(blue arrow) along the y-axis rotates macroscopicmagnetization (red arrow) into the xy plane

362 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Figure 10.22 A simple NMR spectrum for themolecule ethanol
Simple NMR experiments involve repetitive appli-cation of rf pulses with a suitable recovery periodbetween pulses allowing the return of magnetizationto equilibrium. Each FID is acquired, stored on com-puter and added to the previous FID permitting signalaveraging. In this manner high signal to noise ratiospectra are acquired on protein samples in a few min-utes. Unlike most forms of spectroscopy where theincident radiation is slowly scanned through the spec-tral range FT-NMR involves the application of rf pulsesthat excite all nuclear spins simultaneously.
In the construction of magnets for use in NMRspectroscopy the magnetic field is achieved through theuse of superconducting materials operating at liquidhelium temperatures. Magnets are described in termsof their field strength with designations such as 14.1 T(where 1 T or Tesla is equivalent to 104 Gauss and1 G is equivalent to the earth’s magnetic field). TheLarmor frequency is obtained from the relationshipωo = −γB0 where ωo = 2πν0 and ν0 is the Larmorfrequency. A quick calculation of the Larmor frequencyof the proton (where B0, the field strength = 14.1 T; γ,the magnetogyric ratio of 1H = 26.7520 × 107 T s−1)shows that ν0 (1H) occurs at a frequency of ∼600 MHz.As a result spectrometers are often referred to as 400,600, 750, 800 or 900s reflecting the proton Larmorfrequency in MHz at a given magnetic field strength.
Parameters governing NMR signals
The use of NMR spectroscopy as a tool to determineprotein structure is based around several related param-eters that influence the observation of signals. Theseparameters include the chemical shift (δ), spin-spin
coupling constants (J ), the spin lattice or T1 relax-ation time (sometimes denoted as R1 the spin latticerelaxation rate = 1/T1), the spin–spin or T2 relaxationtime (R2 = 1/T2), the peak intensity and the nuclearOverhauser effect (NOE). Since these effects are vitalto all aspects of NMR spectroscopy a brief descriptionis given to highlight their respective importance.
The peak intensity represented formally by theintegrated area reflects the number of nuclei involved inthe signal. The 1H NMR spectrum of ethanol illustratesthis by containing three resonances due to the hydroxylgroup, the methylene group and the methyl group.The integrated areas under each line are in the ratioof 1:2:3. Within proteins methyl resonances exhibitintensities three times greater than the Hα proton.Although the peak height is frequently used as anindicator of intensity many parameters can modify peakheight so the integrated area is always the best indicatorof the number of protons forming each peak.
The chemical shift denotes the position of aresonance along a frequency axis and is uniquelysensitive to the environment in which a nucleusis located. The chemical shift is defined relativeto a standard such as 2,2-dimethyl-2-silapentane-5-sulfonate (DSS) and is quoted in p.p.m. units. Sinceresonant frequencies are directly proportional to thestatic field minor variations between instruments makeit very difficult to compare spectra obtained on differentspectrometers. To avoid this problem all resonances aremeasured relative to a standard defined as having achemical shift of 0 p.p.m. For a resonance the chemicalshift (δ) is measured as
δ = (� − �ref)/ω0 × 106 (10.19)
where � and �ref are the offset frequencies of thesignals of interest and reference respectively. Theresonance attributable to the protons of water occursat ∼4.7 p.p.m. at ∼20 ◦C.
The fundamental equation of NMR, ω = −γBo,suggests that all nuclei subjected to the same magneticfield will resonate at the same frequency. This does notoccur – nuclei experience different fields due to theirlocal magnetic environment. This allows the equationto be recast as
ω = γ(Bo − σBo) (10.20)

NUCLEAR MAGNETIC RESONANCE SPECTROSCOPY 363
where σ represents a screening or shielding constantthat reflects the different magnetic environments foundin molecules. Another way of envisaging this effectis to use an effective field at the nucleus Beff that isreduced by an amount proportional to the degree ofshielding.
Beff = B0(1 − σ) (10.21)
Spin–spin coupling constants or J values aredefined by interactions occurring through bonds. Thesescalar interactions are field independent and occur as aresult of covalent bonds between nuclei linked by threeor less bonds. The scalar coupling constants contributeto the fine structure observed for resonances in 1HNMR spectra of small molecules although they arerarely resolved in studies of proteins. Scalar couplingleads to the splitting of the methylene signal of ethanolinto a quartet as a result of interactions with eachof the three protons of the methyl group. Similarlythe methyl proton resonance is seen as a triplet dueto its interactions with each methylene proton. Asthe molecular weight increases this fine structure isfrequently obscured by line broadening effects.
A correlation exists between the magnitude ofthe spin–spin coupling constant (3J ) and the torsionangles found for vicinal protons in groups of thepolypeptide chain. The Karplus equation derived fromtheoretical studies of 1H–C–C–1H couplings suggestsa relationship of the form
3J = A cos2 θ + B cos θ + C (10.22)
and in proteins the 3JNHα coupling constant and torsionangle φ are related by
3JNHα = 6.51 cos2 θ − 1.76 cos θ + 1.60
(where θ = φ − 60) (10.23)
If the coupling constant 3JNHα is measured withsufficient accuracy the torsion angle can be estimatedand used as a structural restraint. Many coupling con-stants between nuclei are measured using heteronuclearand homonuclear methods (Table 10.4).
The longitudinal relaxation time (T1) reflects therate at which magnetization returns to the longitudinalaxis after a pulse and has the units of seconds.If Mo is the magnetization at thermal equilibriumthen at time t after a pulse the recovery of Mo isexpressed as
Mz = Mo(1 − e−t/T1) (10.24)
where T1 is the longitudinal relaxation time. In solu-tion T1 is correlated with the overall rate of tumblingof a macromolecule but is also modulated by internalmolecular motion arising from conformational flexibil-ity. Both T1 and T2 depend on the correlation time τc
a factor closely linked with molecular mass, and esti-mated assuming a spherical protein of hydrodynamicradius r in a solution of viscosity, η, via the Stokesequation as
τc = 4πηr3/3kBT (10.25)
Macromolecules such as proteins have longer correla-tion times than small peptides. A typical value for a100 residue spherical protein is 3–5 ns.
The transverse or spin–spin relaxation time (T2)
describes the decay rate of transverse magnetizationin the xy plane. T2 is always shorter than T1
and is correlated with dynamic processes occurringwithin a protein. T2 governs resonance linewidthand decreases with increasing molecular mass. For
Table 10.4 One, two and three bond coupling constants
Coupling Magnitude (Hz) Coupling Magnitude (Hz)
1H–13C (1J ) 110–130 13Cα –13CO (1J ) 551H–15N (1J ) 89–95 13Cα –Hα and 13Cβ –Hβ (1J ) 130–140H–C–C–H vicinal (3J ) 2–14 HN–HA (3J ) 2–12H–C–H geminal (3J ) −12–−15 13Cα –15N (1J ) 4515N–13CO 15 13Cα –15N (2J ) 70

364 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Lorentzian lineshapes the linewidth at half maximumamplitude is
�ν1/2 = 1/πT2 (10.26)
with decreases in T2 leading to broader lines. NMRspectroscopy of large proteins is often described as‘limited by the T2 problem’. Many factors influenceT2 and the most important are molecular mass, temper-ature, solvent viscosity, and exchange processes.
Probably the most important measurable parameterin NMR experiments for the determination of proteinstructure is the nuclear Overhauser effect (NOE). Thisis the fractional change in intensity of one resonance asa result of irradiation of another resonance. As a resultof dipolar or ‘through space’ interactions the irradiationof one resonance perturbs intensities of neighbouringresonances. The NOE is expressed as
η = (I − Io)/Io (10.27)
where Io is the intensity without irradiation and I isthe intensity with irradiation. The NOE effect is rapidlyattenuated by distance and declines as the inverse sixthpower of the distance between two nuclei.
η ∝ r−6 (10.28)
The NOE phenomenon, like the relaxation times T1 andT2, varies as a function of the product of the Larmorfrequency and the rotational correlation time. Armedwith an appreciation of these parameters it is possible toextract much information on the structure and dynamicsof regions of the polypeptide chain in proteins.
Practical biomolecular NMR spectroscopy
NMR signals are obtained by placing samples intostrong, yet highly homogeneous, magnetic fields. Thesemagnetic fields arise from the use of superconductingmaterials (niobium–tin and niobium–titanium alloywires) wound around a ‘drum’ maintained at tempera-tures of ∼4 K. High field strengths (11.7–21.5 T) arisefrom the influence of current flowing through the wiresand the constant temperature ensures the field strengthdoes not fluctuate. Samples contained within a quartztube are lowered via a cushion of compressed air intothe centre (bore) of the magnet where a ‘probe’ is main-tained at a consistent temperature (±0.1 ◦C), usually
between 5 and 40 ◦C. Electronics located in the probeallow excitation of nuclei and detection of signals andare linked to computer-controlled devices that permitthe generation of rf pulses of defined timing, phase,amplitude and duration whilst allowing the detectedsignal (FID) to be amplified, filtered and subjectedto further processing (Figure 10.23). This processingincludes storage of the raw data as well as Fouriertransformation.
Experiments may continue for 3–4 days and requireprotein stability for this period. To observe magneti-zation involving exchangeable protons such as amides(NH) in proteins it is necessary to perform experimentsin water. This brings additional problems of intensesignals due to the high concentration of water protons(110 M) that far exceeds the concentration of ‘signals’from the protein (∼10−3 M). Sophisticated methods ofsolvent (water) suppression allied to post-acquisitionprocessing eliminate these signals effectively from pro-tein spectra.
Chemical shifts reflect the magnetic microenviron-ments of groups. The amide (NH) proton of a polypep-tide backbone has a chemical shift between 8.0 and9.0 p.p.m., methyl groups have chemical shifts between0 and 2 p.p.m. whilst the Hα proton has a valuebetween 4.0 and 4.6 p.p.m. 1H chemical shifts werederived from an analysis of short unstructured modelpeptides of the form Gly-Gly-X-Ala, where X was eachof the 20 residues (Table 10.5).
Within folded proteins some chemical shifts deviatefrom their expected values reflecting magnetic envi-ronments that depend on conformation. This is seenin the 1H NMR spectrum of ubiquitin where peaksabove 9.0 and below 0 p.p.m. reflect tertiary struc-ture although many resonances are found close totheir ‘random coil’ conformations. The dispersion overa much wider frequency range offers the possibil-ity of identifying resonances in a protein – a processcalled assignment.
The assignment problem in NMR spectroscopy
The assignment problem for a protein of ∼100 residuesand perhaps 700 protons requires identifying which res-onance belongs to a particular proton. The assignmentproblem remains the crux of determining protein

NUCLEAR MAGNETIC RESONANCE SPECTROSCOPY 365
Figure 10.23 A state-of-the-art NMR spectrometer operating at 900 MHz and a block diagram representing a NMRspectrometer. The major components are the magnet containing the probe together with ‘shim’ or tuning coils tomaintain homogeneity and electronics to ‘lock’ the field at a given frequency. Outside of the magnet are radiofrequency transmitters with pulses designed via a pulse programmer and a frequency synthesizer together with adetection system of numerous amplifiers, filters and analogue–digital converters. All systems are controlled viaa computer (Reproduced courtesy Varian Inc)
structure by NMR spectroscopy and is perhaps anal-ogous to the phase problem of X-ray crystallogra-phy. Without first ‘assigning’ the protein we cannotmove on to derive restraints important in the derivationof molecular structure. The starting point involvesassigning a signal to a specific atom or group of atoms(e.g. HA proton, CH3 protons, etc.). Spectral over-lap in 1D spectra usually prevents complete assign-ment even in small proteins and as shown in the 1HNMR spectra of ubiquitin (Figure 10.24) – a proteinwith 76 residues – many overlapping peaks precludeabsolute identification.
Historically the major advance in this area was tospread conventional 1D spectra into a second dimen-sion that reduces spectral overlap. In the last twodecades these methods have expanded dramatically andalmost all structural investigations of proteins startwith the acquisition of 2D spectra. A normal 1DNMR spectrum involves a pulse followed by mea-surement of the resulting FID. 2D NMR spectroscopyinvolves the application of successive pulses that leadto magnetization transfer between nuclei. The mech-anism of transfer proceeds either by a through bond
(scalar mechanism) or by a through space (dipolar)interaction. A 2D experiment consists of four timeperiods; the preparation, evolution, mixing and acqui-sition periods often preceeded by a relaxation delay.The preparation period consists of a single pulse ora series of pulses and delays and its purpose is tocreate the magnetization terms that will make up theindirect dimension. These terms evolve during theevolution or t1 period. The evolution of magnetiza-tion during the t1 period is followed by a secondpulse that initiates the mixing period and results inmagnetization transfer to other spins, normally viathrough bond or through space interactions; transfercan also occur via chemical exchange. The decay ofthe FID is detected during the acquisition or t2 period.The whole pulse scheme is repeated to allow for sig-nal averaging and other instrumental factors includ-ing relaxation. The experiment is then repeated byincrementing the t1 period to build up a series ofFIDs recorded at different t1 intervals. A 2D exper-iment contains two time variables, t1 and t2, andFourier transformation of this dataset, S(t1, t2) yieldsa 2D contour plot, S(ω1, ω2), where the precession

366 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Table 10.5 The 1H chemical shifts of the amino acids residues in a random coil conformation
Chemical shift (p.p.m.)
Residue HN HA HB others
Ala 8.25 4.35 1.39 –
Asp 8.41 4.76 2.84, 2.75 –
Asn 8.75 4.75 2.83, 2.75 7.59, 6.91 (sc amide)
Arg 8.27 4.38 1.89, 1.79 1.70 (HG), 3.32 (HD), 7.17, 6.62 (sc NH)
Cys 8.31 4.69 3.28, 2.96 –
Gln 8.41 4.37 2.13, 2.01 2.38 (HG), 6.87,7.59 (sc NH2)
Glu 8.37 4.29 2.09, 1.97 2.31,2.28 (HG)
Gly 8.39 3.97 – –
His 8.41 4.63 3.26, 3.20 8.12 (2H), 7.14 (4H)
Ile 8.19 4.23 1.90 1.48, 1.19 (HG=CH2), 0.95 (HG=CH3), 0.89 (HD)
Leu 8.42 4.38 1.65 1.64 (HG), 0.94,0.90 (HD)
Lys 8.41 4.36 1.85, 1.76 1.45 (HG), 1.70 (HD), 3.02 (HE), 7.52 (sc NH3)
Met 8.42 4.52 2.15, 2.01 2.64 (HG), 2.13(HE)
Phe 8.23 4.66 3.22, 2.99 7.30 (2,6H), 7.39 (3,5H), 7.34 (4H)
Pro – 4.44 2.28, 2.02 2.03 (HG), 3.68,3.65 (HD)
Ser 8.38 4.50 3.88, 3.88 –
Thr 8.24 4.35 4.22 1.23
Trp 8.09 4.70 3.32, 3.19 7.24(2H), 7.65(4H), 7.17(5H), 7.24(6H), 7.50(7H),
10.22 (indole NH)
Tyr 8.18 4.60 3.13, 2.92 7.15 (2,6H), 6.86 (3,5H)
Val 8.44 4.18 2.13 0.97,0.94 (HG)
Adapted from NMR of Proteins and Nucleic Acids. Wuthrich, K. (ed.) Wiley Interscience, 1986.
frequencies occurring during the evolution and detec-tion periods determine peak positions (ω1 and ω2) inthe 2D plot.
In general, three homonuclear 1H-NMR experimentsare used for assignment of proteins. The COSY (corre-lated spectroscopy) and TOCSY3 (total correlated spec-troscopy) describe magnetic interactions between scalarcoupled nuclei – resonances linked via ‘through bond’interactions (Figure 10.25). When the COSY exper-iments are performed on protein dissolved in H2O,
3The TOCSY experiment is sometimes called HOHAHA(Homonuclear Hartmann-Hahn) after the discoverers of an origi-nal solid state experiment demonstrating cross-polarization betweennuclei.
cross peaks are observed in 2D spectra reflecting, forexample, connectivity between NH and HA protons.
In 1H NMR through bond interactions (Figure 10.26)as a result of coherence transfer are limited to twoor three bonds and are restricted to intra-residuecorrelations. The four-bond interaction between theHN(i+1),HA(i) is too weak to be observed. Cross peakscan also occur between HA and HB protons via 3Jcouplings and it is soon apparent that characteristicpatterns of connectivity are observed for differentresidues (Figure 10.27).
Performing experiments in D2O (2H2O) removesmost HN signals since protons exchange for deuteronsleading to the loss of the left-hand side of the 2Dspectra. These protons are described as labile.
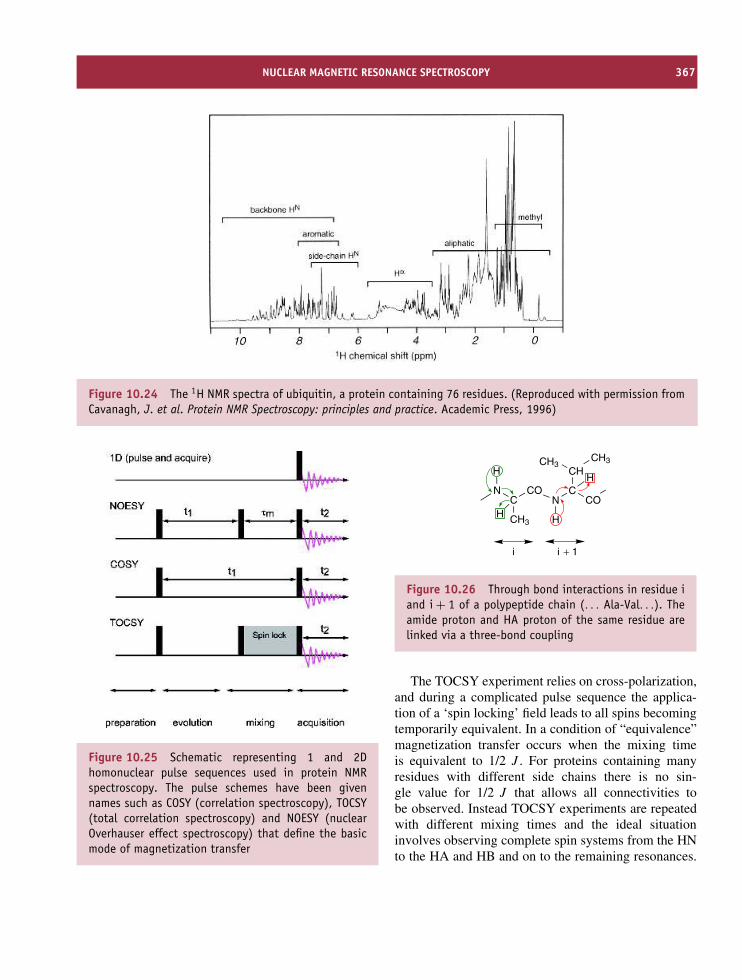
NUCLEAR MAGNETIC RESONANCE SPECTROSCOPY 367
Figure 10.24 The 1H NMR spectra of ubiquitin, a protein containing 76 residues. (Reproduced with permission fromCavanagh, J. et al. Protein NMR Spectroscopy: principles and practice. Academic Press, 1996)
Figure 10.25 Schematic representing 1 and 2Dhomonuclear pulse sequences used in protein NMRspectroscopy. The pulse schemes have been givennames such as COSY (correlation spectroscopy), TOCSY(total correlation spectroscopy) and NOESY (nuclearOverhauser effect spectroscopy) that define the basicmode of magnetization transfer
NC
CO
CHCH3CH3
COC
N
CH3
H
HH
H
i i + 1
Figure 10.26 Through bond interactions in residue iand i + 1 of a polypeptide chain (. . . Ala-Val. . .). Theamide proton and HA proton of the same residue arelinked via a three-bond coupling
The TOCSY experiment relies on cross-polarization,and during a complicated pulse sequence the applica-tion of a ‘spin locking’ field leads to all spins becomingtemporarily equivalent. In a condition of “equivalence”magnetization transfer occurs when the mixing timeis equivalent to 1/2 J . For proteins containing manyresidues with different side chains there is no sin-gle value for 1/2 J that allows all connectivities tobe observed. Instead TOCSY experiments are repeatedwith different mixing times and the ideal situationinvolves observing complete spin systems from the HNto the HA and HB and on to the remaining resonances.

368 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Figure 10.27 Patterns of connectivity seen in 2D COSY and TOCSY spectra of eight of the 20 amino acid residues.*The pattern shown by Cys is similar for all other residues in which the spin system involves a HN–HA and twonon-degenerate HB protons. This is likely to include the aromatic residues Phe, Tyr and Trp as well as Asp, Asn andSer. Similarly, the profile shown by Glu is shared by Gln and Met. The pattern does not take any account of peak finestructure that occurs in COSY type experiments
CCN
HH
C
CH
N
O
H
C
CH3CH3
C
CH3
O
N
H
OH
dNN
dαN
dβN
dNN
dαN
dβN
dNN
dβN
dαN
residue i − 1 residue i residue i + 1
Figure 10.28 The pattern of sequential NOEsexpected within a polypeptide chain for threesuccessive residues
Connectivity patterns are sometimes diagnosticfor individual residues and having identified spinsystem type it is necessary to establish its locationwithin the primary sequence (Figure 10.28). Thisis achieved through NOESY experiments. The 2Dhomonuclear NOESY experiment relies on through
space interactions between nuclei (1H) separated byless than 6 A. By establishing sequential connectivitybetween resonances of a known spin system type itis possible to identify clusters of residues that areunique within the primary sequence. In Figure 10.28valine and alanine exhibit sequential connectivity andif these residues occur sequentially only once in theprimary sequence then these residues are identifiedunambiguously. If the dipeptide Val-Ala occurs inmore than one region of the polypeptide chain thenthe identity of residues i − 1 and i + 2 becomesimportant in establishing conclusive sequence specificassignments.
Some sequential NOEs are indicative of particularunits of secondary structure (Table 10.6). For example,the regular periodicity of helices leads to the closeapproach of HA and HN between residues (i,i + 2),(i,i + 3) and (i,i + 4) that do not occur in antiparallelor parallel β strands. More significantly the observationof dαN (i,i + 4) is strongly indicative of a regular α

NUCLEAR MAGNETIC RESONANCE SPECTROSCOPY 369
Table 10.6 Regular secondary structure gives characteristic NOEs
Interaction α helix 310 helix Antiparallelβ strand
Parallel β
strandType I turn Type II
turn
dαN 2.7 2.7 2.8 2.8 2.8 2.7dαβ 2.2–2.9 2.2–2.9 2.2–2.9 2.2–2.9 2.2–2.9 2.2–2.9dβN 2.2–3.4 2.2–3.4 2.4–3.7 2.6–3.8 2.2–3.5 2.2–3.4dαN(i,i + 1) 3.5 3.4 2.2 2.2 3.4 2.2dNN(i,i + 1) 2.8 2.6 4.3 4.2 2.6 4.5dβN(i,i + 1) 2.5–2.8 2.9–3.0 3.2–4.2 3.7–4.4 2.9–4.1 3.6–4.4dαN(i,i + 2) 4.4 3.8 – – 3.6 3.3dNN(i,i + 2) 4.2 4.1 – – 3.8 4.2dαN(i,i + 3) 3.4 3.3 – – 3.1–4.2 3.8–4.7dαβ (i,i + 3) 2.5–4.4 3.1–5.1 – – – –dαN(i,i + 4) 4.2 – – – – –
Distances involving β protons vary due to the range of distances possible. The first three sets of distances refer to intra-residue connectivity.All distances are in A
helix whilst the presence of only dαN (i,i + 2) and dαN
(i,i + 3) may indicate more tightly packed 310 helices.Similarly, intense NOEs between dβN (i,i + 1) indicatesa β strand. Since both strands and helices tend topersist for several residues the observation of blocksof sequential NOEs with these connectivities allowssecondary structure to be defined.
The NOESY spectrum not only resolves sequen-tially connected residues but also represents the basis ofprotein structure determination using NMR data. Inte-grating the volumes associated with the cross peaksin NOESY spectra quantifies this interaction and fromintra-residue and sequential NOEs allows the volumesto be converted into distances, since regular secondarystructure is associated with relatively fixed separationdistances (see Table 10.6). Some NOE cross peaksarise as a result of long-range interactions (i.e betweenresidues widely separated in the primary sequence)and these cross peaks play a major role in determin-ing the overall fold of a protein since by definitionthey represent separation distances of less than 6 A.Using this approach Kurt Wuthrich pioneered proteinstructure determination for BUSI IIA – a proteinaseinhibitor from bull seminal plasma in 1984 – andit marked a landmark in the progression of NMR
Table 10.7 Some of the first proteins whosestructures were determined by homonuclear NMRspectroscopy
Protein Date Mass
BUSI IIA 1985 6050Lac repressor headpiece 1985 5500BPTI 1987 6500Tendamistat 1986 8000BDS-I 1989 5000Human complement protein C3a 1988 8900Plastocyanin 1991 10 000Thioredoxin 1990 11 700Epidermal growth factor 1987 5800
spectroscopy from analytical tool to structural tech-nique (Figure 10.29).
Between 1984 and 1990 structures for many small(<10 kDa) proteins were determined via 2D NMRtechniques (Table 10.7). To compare the power ofcrystallography and NMR spectroscopy Wuthrich and

370 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Figure 10.29 The structure of BUSI IIA – the firstprotein structure determined using NMR spectroscopy.Backbone atoms are shown in blue, side chains ingreen
Robert Huber independently determined the struc-ture of tendamistat, a protein inhibitor of α-amylasewith 74 residues (Figure 10.30). The results showedemphatically that crystal and solution state structures
were comparable in almost every respect and testifiedto the ability of NMR spectroscopy in deriving3D structures.
However, larger proteins containing more than 100residues presented new and increasingly difficult prob-lems that arose from the greater number of reso-nances. This resulted in spectral overlap with increasedcorrelation times producing broader linewidths. Theanswer was heteronuclear NMR spectroscopy.
Heteronuclear NMR spectroscopy
Resonance assignment is vital to protein structuredetermination. Additional spin 1/2 nuclei allow alter-native assignment strategies and new pulse techniquesexploiting heteronuclear scalar couplings present in13C/15N enriched proteins (Figure 10.31). Experimentsinvolved the transfer of magnetization from 1H to 13Cand/or 15N through large one-bond scalar couplings.The magnitude of these couplings (30–140 Hz) ismuch greater than the 1H–1H 3J couplings (2–10 Hz).
The simplest heteronuclear 2D experiments correlatethe chemical shift of the 15N nucleus with its attachedproton. In 2D 15N–1H heteronuclear spectra cross peaksare spread out according to the nitrogen chemical shift
Figure 10.30 The structures of tendamistat derived by NMR and crystallography are superimposed for thepolypeptide backbone. The NMR structure is shown in red and the crystallographic structure is shown in blue.(PDB files: 2AIT and 1HOE). With the minor exception of the N terminal region the two structures agree very closely.Tendamistat is a protein based largely on β sheet (right)

NUCLEAR MAGNETIC RESONANCE SPECTROSCOPY 371
CNC
O
CC
C
H
N
O
CH3 CH3
H
CH3
N
H H
H~90Hz~140Hz
~55Hz~55Hz
~15Hz
~90Hz
~35Hz
~35Hz
~35Hz
~90Hz
Figure 10.31 Heteronuclear coupling between 13C,1H and 15N within proteins together with themagnitude of these one bond coupling constants (1J)
as well as the proton chemical shift (Figure 10.32).Many residues have characteristic 15N and 13C chem-ical shifts (Tables 10.8 and 10.9). For example, Gly,Ser and Thr residues show 15N chemical shifts below115 p.p.m. along with the side chain amides of Glnand Asn. Observation of a cross peak in heteronuclearspectra in these regions is most probably attributed toone of these residues.
Heteronuclear 2D versions of the NOESY, COSYand TOCSY are often recorded but spectral overlapcan remain a problem especially in NOE spectra whereboth intra- and inter-residue cross peaks are observed.This problem was resolved by further pulse sequencesthat involved three time variables (t3, t2 and t1) andwhere Fourier transformation leads to a cube or 3D
Table 10.8 Nitrogen chemical shifts for the backboneand side chain amides of residues found in proteins
Residue 15N Residue 15N
Ala 125.04 Leu 122.37Arg 121.22 Lys 121.56Asn sc 119.02 Met 120.29Asp 119.07 Phe 120.69Cys 118.84 Pro –Glu 120.23 Ser 115.54Gly 107.47 Thr 111.9Gln sc 120.46 Trp indole 122.08His 118.09 Tyr 120.87Ile 120.35 Val 119.31
Determined from position in a sequence AcGGXGG-NH2 in 8 M urea.Adapted from J. Biomol. NMR 2000, 18, 43–48.
plot. The major advantage of these techniques is that2D spectra are extended into a third dimension usu-ally the 15N or 13C chemical shift. A panoply of newtechniques based on heteronuclear J couplings mas-sively enhanced NMR spectroscopy as a tool for ‘large’protein structure determination. Ambiguities seen pre-viously were eliminated by new pulse sequences thatidentified spin systems with high sensitivity. These
12 11 10 9
H ppm
N ppm
8 7 6
132
130
128
126
124
122
120
118
116
114
112
110
108
106
Figure 10.32 A 2D heteronuclear 15N–1H correlation spectra for a protein

372 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Table 10.9 13C chemical shifts for the backbone and side chain amides of residues found in proteins
Residue CO or C′ Cα Cβ Cγ Cδ Other
Ala 175.8 50.8 17.7Arg 175.0 54.6 28.8 25.7 41.7Asn 173.1 51.5 37.7 175.6 (amide)Asp 174.2 52.7 39.8 178.4 (carboxylate)Cys 175.7 57.9 26.0Gln 174.0 54.1 28.1 32.2 179.0 (amide)Glu 174.8 54.9 28.9 34.6 182.8 (carboxylate)Gly 172.7 43.5His 172.6 53.7 28.0 135.2 (C2), 118.7 (C4), 130.3 (C5)Ile 174.8 59.6 36.9 25.4, 15.7 11.3Leu 175.9 53.6 40.5 25.2 23.1, 21.6Lys 174.7 54.4 27.5 23.1 31.8Met 175.0 53.9 31.0 30.7 15.0 (Cε)Phe 176.0 57.4 37.0 136.2 (C1), 130.3 (C2/C6), 130.3 (C3/C5),
128.6 (C4)Pro 175.2 61.6 30.6 25.5 48.2 trans configuration,
61.3 33.1 23.2 48.8 cis shows small differences with exception of Cβ
Ser 172.6 56.6 62.3Thr 172.7 60.2 68.3 20.0Trp 176.7 56.7 27.4 C3 (108.4), 112.8 (C7), 137.3 (C8), 127.5 (C9)Tyr 176.0 57.4 37.0 128.0 (C1), 130.0 (C2/C6), 117.0 (C3/C5),
156.0 (C4)Val 174.9 60.7 30.8 19.3, 18.5
The chemical shifts were determined in linear pentapeptides of the form GGXGG. Data adapted from Wuthrich, K. NMR of Proteins andNucleic Acids. John Wiley & Sons, 1986.
techniques carry names that highlight the different cor-relation made during experiments. For example a 3DHNCO correlates the NH of residue i with the CO ofthe proceeding residue (i − 1). Triple resonance exper-iments (Table 10.10) greatly increase the number ofassignments, and armed with a larger number of assign-ments comes the possibility of deriving greater num-bers of structural restraints from NOEs and couplingconstant data.
The use of multi-dimensional NMR methods hasseen larger proteins assigned and it is now feasibleto attempt to completely assign proteins of molecular
mass in excess of 30 kDa. As of 2003 the NMRderived structures (coordinates) of over 2500 proteinshave been deposited in the Protein Databank. Of theseproteins the vast majority have masses below 15 000and a search of databases suggests that structures forproteins with more than 150 residues are increasinglysteadily. In 2002 a backbone assignment for a 723-residue protein (malate synthase) was reported andbears testament to the effectiveness of these new multi-dimensional NMR methods. Assignment is, however,only the first step in structure determination and forany protein (large or small) it is necessary to obtain

NUCLEAR MAGNETIC RESONANCE SPECTROSCOPY 373
Table 10.10 A small selection of common triple resonance experiments used for sequential assignment in proteins
Experiment Observed correlation Magnetization transfer J couplings
HNCA 1HNi–15Ni–13Cα
i
N C
H
C
C
H O
N C C
H
C
O
H H H H
H
1JNH, 1JNC, 2JNCα
1HNi–15Ni–13Cα
i−1
HNCO 1HNi–15Ni–13COi−1
N C
H
C
C
H O
N C C
H
C
O
H H H H
H
1JNH, 1JNCO
CBCANH 13Cβi/
13Cαi–15N–1HN
I
N
H
C
C
C
H O
N C C
H
C
O
H H H H
H
1JCH, 1JCαCβ, 1JNCα,2JNCα, 1JNH
13Cβi/
13Cαi–15Ni+1–1HN
I+1
CBCA(CO)NH 13Cβi/
13Cαi–15Ni+1–1HN
I+1
N C
H
C
C
H O
N C C
H
C
O
H H H H
H
1JCH, 1JCαCβ, 1JCαCO,1JNC, 1JNH
a sufficient number of structurally significant NOEs orother conformational restraints to define structure withreasonable precision.
Generating protein structuresfrom NMR-derived constraints
The information derived from NMR spectroscopyabout protein conformation can be divided into twomajor classes. One class of conformational restraints
reflect angular information whilst a second classare distance dependent. It is self evident that ifsufficient angles and distances between atoms aredefined then, in theory at least, it is possible to defineoverall conformation. The majority of conformationalrestraints are obtained via homo- and heteronuclearNOESY experiments and convert the NOE cross peakvolumes derived from the relationship
NOEij ∝ 1/rij6 (10.29)

374 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
into distances that represent the separation between twonuclei i and j. Most schemes categorize NOE crosspeak volumes into three classes of restraints (strong,medium and weak with sometimes a fourth grouptermed very weak). Strong NOEs represent distancesbetween nuclei that are close together ranging from1.9 to 2.9 A; medium NOEs represent distances from2.9–3.5 A and weak NOEs represent distances from3.5–5.0 A. Very weak NOEs are generally assumedto include separation distances in a range 5.0–6.5 A.In effect these distances represent upper and lowerlimits for the separation distances. Until recentlyNOEs were the only experimental restraints used todetermine protein structure since the number of torsionangles measurable with accuracy was often limited.However, heteronuclear NMR methods have permittedincreased numbers of restraints based on torsionangles derived from the measurement of couplingconstants between backbone atoms as well as someside chains.
Most approaches (see Figure 10.33) derive proteinstructures from NMR data using distance geometry andsimulated annealing. In all cases the methods satisfythe conformational data yielding a structure consistentwith experimental data and parameters governing bondlengths and angles. Unfortunately almost all NMRrestraints include a range of possible values. So forexample, an NOE cross peak volume may be consistentwith distances ranging between 3.5 and 5.0 A whilst a3JHNHA coupling constant of <5 Hz is consistent witha torsion angle (φ) between 0 and −120◦. When this isrepeated for every NOE and torsion angle many proteinconformations are consistent with the data and there isno uniquely defined structure.
The approach used to calculate structures from NMRdata involves the generation of an ensemble of ‘lowenergy’ conformations all consistent with the data thatare progressively refined during calculation. Structurecalculations based on experimental data derived fromNMR spectroscopy yield a family of closely relatedstructures all of which are consistent with the inputdata. The three-dimensional structures of interleukin-1β
and thioredoxin derived using NMR spectroscopy areshown in Figure 10.34, and like their crystallographiccounterparts these structures are models of clarityproviding a detailed insight into the mechanism ofprotein function.
Figure 10.33 The approach used to derive proteinstructures from torsion angle and distance restraintsobtained via NMR spectroscopy
Structure calculations yield families of relatedconformations all of low overall energy. Although amean structure calculated from a family of 20–50closely related structures is often shown this structureis of no more relevance than any of the other structuresof low energy generated via structure calculation. Thegeneration of a mean structure allows a root meansquare deviation to be calculated from differences inthe centre of mass of atoms in a standard (mean)structure with that in family of molecules. A smalldifference in position for each atom between the meanand test structures leads to a low rmsd value and

CRYOELECTRON MICROSCOPY 375
Figure 10.34 The solution structures of interleukin-1β and thioredoxin derived using NMR spectroscopy. Thestructures show the 50 lowest energy structures for the polypeptide backbone together with the secondary structuredistribution of the lowest energy structure (bottom row) (PDB: 7I1B and 1TRU). Interleukin-1β contains ∼150residues and has a mass of ∼17 500, whilst thioredoxin contains 105 residues and has a mass of ∼11 500.Reproduced courtesy of FEI Company www.feicompany.com
implies structures are similar.
rmsd =√√√√ 1
N
N∑i=1
(rmean − rtest)2 (10.31)
The precision of NMR structures is ultimatelyrelated to the number of experimental restraintsdefining conformation. If there are no restraintsbetween side chains then structure is poorly defined.Most frequently backbones are defined with highestprecision followed by the side chains. In high qual-ity NMR derived structures as many as 50 restraintsper residue may be obtained leading to a backbonermsd of 0.3–0.5 A. Advances in molecular biology,
instrumentation, heteronuclear pulse sequences andcomputational methods are now permitting structuresto be determined using NMR spectroscopy of equalprecision to those obtained by X-ray diffraction. How-ever, one group of proteins for which NMR and toa lesser extent X-ray diffraction struggle to providedetailed structures are large asymmetric complexes ormacromolecular assemblies.
Cryoelectron microscopy
Visualizing structure has provided enormous impetusto understanding biological processes, and electronmicroscopy has provided many views of cells and

376 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
subcellular organelles through the use of a transmissionelectron microscope (Figure 10.35; TEM).
Pioneering work using electron microscopy to studyviral organization in the tail assembly of bacteriophageT4 by Aaron Klug laid the foundation for determining3D structure from series of 2D images or projectionsof an object’s electron density formed at an imageplane (Figure 10.35). Three-dimensional informationcould be recovered if a number of views of the objectwere recorded at different angles of observation. This isusually done with a tilting stage that elevates specimensto angles of ∼70◦. For structures of high rotationalsymmetry such as helices it is possible to use just asingle or small number of orientations to reconstructa 3D model. The discovery had wider implicationssince it was exploited in the field of medicine as amedical imaging technique called computerized axialtomography (CAT). This work also allowed Hendersonand Unwin to determine the orientation of helices inbacteriorhodopsin and pointed the way forward towards
the use of electron microscopy to study proteins athigher levels of atomic resolution. From continuousdevelopment of instrumentation, improved specimenpreparation, massive advances in data processing andincreased computational power electron microscopyhas become a technique capable of providing structuralinformation. The technique is useful for proteins notamenable to study by X-ray crystallography or NMR.
Early electron microscopy studies involved ‘fixing’biological preparations using cross-linking agents fol-lowed by staining with heavy metal (electron dense)compounds; treatments that could distort structureby introducing artifacts. This problem persisted untilKenneth Taylor and Robert Glaeser reported electrondiffraction data recorded at a temperature of ∼100 Kfrom frozen thin films containing hydrated catalasecrystals. In one swoop the technique advanced allowingbiological ‘units’ to be imaged under representativeconditions. The technique became known as cryo-electron microscopy (cryo-EM). Sample preparation
Figure 10.35 An electron diffraction pattern together with a picture of a modern TEM used for structural analysis.(Reproduced by permission of FEI Company.)

CRYOELECTRON MICROSCOPY 377
is important to cryo-EM and plunging a thin layersupported on a carbon grid into a bath of ethane cooledby liquid nitrogen leads to rapid and efficient cool-ing. Liquid ethane, a very efficient cryogen, produces ametastable form of water called the vitrified state. Thevitrified state is amorphous and lacks the crystallineice-like lattice. Although metastable the vitrified formis maintained indefinitely at low temperatures (∼77 K)and all cryo-EM methods use this approach. Images arerecorded at ∼100 K using ‘low dose’ techniques (0.1electrons A−2) to identify areas of interest and to limitradiation damage. Areas of interest are then capturedat greater magnification with a higher dose of electrons(5–10 A−2). The data has a low signal/noise ratio andrequires extensive averaging and processing in derivingstructural information.
The early successes of cryo-EM centred mainly onviral structure determination and many viral capsidshave been defined. One example involved determina-tion of the hepatitis B capsid protein structure by thegroups of R.A. Crowther and A. Steven (Figure 10.36).At a resolution of 7.6 A this study identified elementsof secondary structure within subunits.
Figure 10.36 The reconstructed 3D structure of thehepatitis B capsid (adapted with permission fromBaumeister, W. & Steven, A.C. Trends Biochem. Sci.2000, 25, 625–631. Elsevier)
Single-particle analysis deals with large proteins orprotein complexes and is growing in its applicationto biological problems largely through the efforts ofJoachim Frank in the study of ribosome structure. Inthis method macromolecular complexes are trapped inrandom orientations within vitreous ice and imagesfrom all possible orientations are combined andaveraged to provide enhanced views.
From a sample preparation point of view singleparticle analysis is straightforward – it requires a well-dispersed, homogeneous complex frozen in amorphousice and aligned over the holes of a carbon grid.Structure determination from differently oriented par-ticles requires the combination of data from manyimages using methods that distinguish ‘good’ from‘bad’ particles, determine the orientation relative toa reference point and evaluate the quality of theresulting data. These methods are intensely computa-tional and beyond the scope of this text. Although themethod has not produced data comparable in resolu-tion to X-ray diffraction or NMR spectroscopy meth-ods the technique has allowed ribosomal subunits tobe determined at resolutions approaching 7.0 A. Sig-nificantly, the results of crystallography and/or NMRcan often be assimilated into the cryo-EM data tobuild models based on more than one structural tech-nique.
The power of electron crystallography has beendemonstrated for bacteriorhodopsin but success hasalso been achieved with a variety of proteins includingtubulin, light harvesting complexes associated with thereaction centres of photosynthetic organisms, aquaporina transport protein of the red blood cell membrane andthe acetylcholine receptor of neurones.
Tubulin is a key protein of the microtubule assemblyregulating intracellular activity particularly cytoskeletalfunction and eukaryotic cell division where it facilitatesthe separation of chromatids as part of mitosis. Inthis role the ability of microtubules to polymerize anddepolymerize is important. The microtubule contains13 protofilaments arranged in parallel to form ahollow structure approximately 24 nm in diameter.The protofilaments are composed of alternating α-and β-tubulin monomers each composed of a singlepolypeptide chain (the α and β subunits) of ∼55 kDa.Within the microfibril each protofilament is staggered
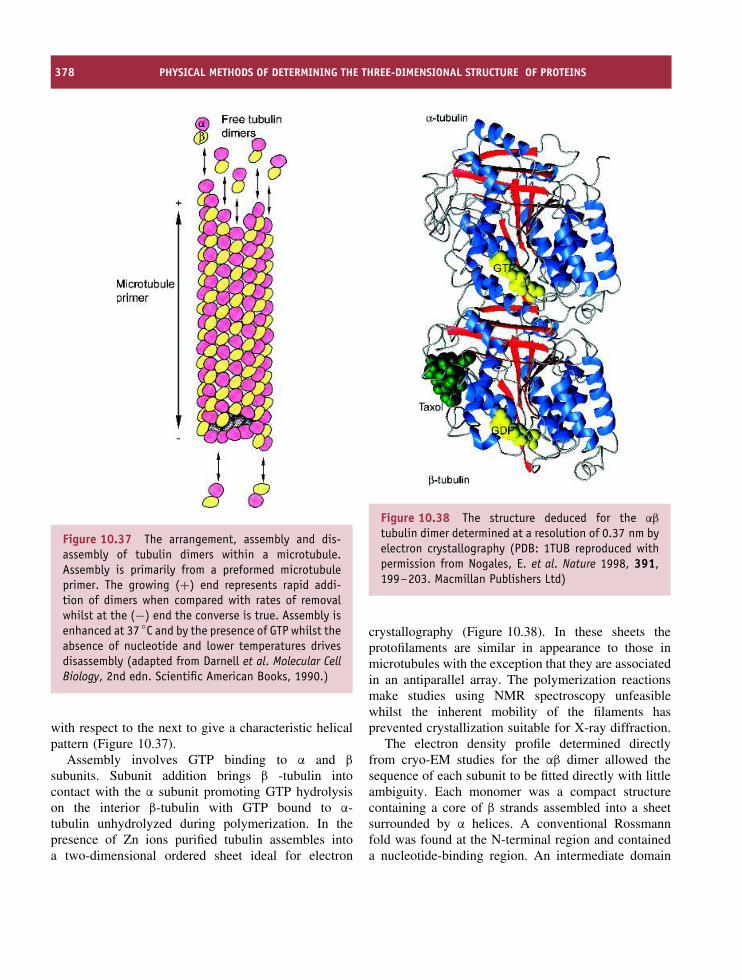
378 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Figure 10.37 The arrangement, assembly and dis-assembly of tubulin dimers within a microtubule.Assembly is primarily from a preformed microtubuleprimer. The growing (+) end represents rapid addi-tion of dimers when compared with rates of removalwhilst at the (−) end the converse is true. Assembly isenhanced at 37 ◦C and by the presence of GTP whilst theabsence of nucleotide and lower temperatures drivesdisassembly (adapted from Darnell et al. Molecular CellBiology, 2nd edn. Scientific American Books, 1990.)
with respect to the next to give a characteristic helicalpattern (Figure 10.37).
Assembly involves GTP binding to α and β
subunits. Subunit addition brings β -tubulin intocontact with the α subunit promoting GTP hydrolysison the interior β-tubulin with GTP bound to α-tubulin unhydrolyzed during polymerization. In thepresence of Zn ions purified tubulin assembles intoa two-dimensional ordered sheet ideal for electron
Figure 10.38 The structure deduced for the αβ
tubulin dimer determined at a resolution of 0.37 nm byelectron crystallography (PDB: 1TUB reproduced withpermission from Nogales, E. et al. Nature 1998, 391,199–203. Macmillan Publishers Ltd)
crystallography (Figure 10.38). In these sheets theprotofilaments are similar in appearance to those inmicrotubules with the exception that they are associatedin an antiparallel array. The polymerization reactionsmake studies using NMR spectroscopy unfeasiblewhilst the inherent mobility of the filaments hasprevented crystallization suitable for X-ray diffraction.
The electron density profile determined directlyfrom cryo-EM studies for the αβ dimer allowed thesequence of each subunit to be fitted directly with littleambiguity. Each monomer was a compact structurecontaining a core of β strands assembled into a sheetsurrounded by α helices. A conventional Rossmannfold was found at the N-terminal region and containeda nucleotide-binding region. An intermediate domain

OPTICAL SPECTROSCOPIC TECHNIQUES 379
contained a four-stranded β sheet and three helicesalong with a Taxol-binding site. A third domain of twoantiparallel helices crossing the N- and intermediateregions represented a binding surface for additionalmotor proteins. A Mg ion was bound near the GTPbinding site whilst a binding site for Zn ions wasidentified at the interface between protofilaments.
Cryo-EM methods are now firmly established asa third method for macromolecular structure determi-nation. The list of successes is continuing to expandwith increasingly detailed pictures of the ribosome, thethermosome, viral capsids, transport proteins, amyloidfibres and receptors obtained using cryo-EM methods.There is little doubt that cryo-EM will increase inimportance as a method of three-dimensional structuredetermination for macromolecular complexes.
Neutron diffraction
Neutron diffraction requires the crystallization ofproteins followed by measurement of the diffraction ofneutrons. Neutron beams were traditionally generatedvia atomic reactors but increasingly sources are derivedvia synchrotron beam lines. Until the introduction ofsynchrotron radiation a major problem was achievingsufficiently high neutron fluxes to enable adequatedata collection times. Although less widely used thetechnique has proved very useful for locating hydrogenatoms within protein crystals something that is difficultto achieve with X-rays.
The usefulness of neutron diffraction to biologi-cal structure determination in the post-genomic erainvolves its ability to probe dynamic properties involv-ing protons. Hydrogen exchange underscores manybiological reactions and neutron diffraction is adept atlocating protons that exchange for deuterium withinenzyme active sites. Such observations help to establishcatalytic mechanisms and the exchange of hydrogen fordeuterium leads to a large change in neutron scatter-ing factors.
Two noteworthy examples where neutron diffractionhas proved of value are the catalytic mechanisms ofserine proteases and lysozyme. In serine proteases suchas trypsin the catalytic mechanism revolves around thecatalytic triad of invariant Ser, His and Asp residues(Figure 10.39). The nucleophilic Ser residue forms two
CH2
N
N
His57
H
H2CC
O
O
Asp102
H
CH2
N
N
His57
CH2O
Ser195
H
H2CC
O
O−
Asp102
CH2O
Ser195
H
Figure 10.39 The exchangeable proton in thecatalytic site of serine proteases was shown to remainattached to His57 and was not transferred to Asp102
tetrahedral intermediates during bond cleavage and isassisted by increased imidazole basicity caused byhydrogen bonding to the aspartate. Hydrogen bondingcould arise from protonation of the imidazole group orprotonation of aspartyl side chains.
Neutron protein crystallography showed that theproton remained attached to the imidazole ring of His57 and not the carboxylate of Asp 102 and clarifiedthe reaction mechanism. Similarly in lysozyme, thedeuterium atom was found to reside on the carboxylside chain of Glu 35, rather than Asp 52, againemphasizing a key aspect of the catalytic mechanism.
Optical spectroscopic techniques
Absorbance
Transitions between different electronic states occurin the ultraviolet, visible and near infrared regionsof the electromagnetic spectrum and are widely usedfor studying protein structure. Absorbance involvestransitions of outer shell electrons between variouselectronic states and is governed by the rules ofquantum mechanics. The absorbance of light excitesan electron from the ground state to a higher excitedstate (Figure 10.40).
Absorbance is governed by several rules; thefrequency of the incident radiation must match the

380 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Figure 10.40 Absorbance and fluorescence resultingfrom excitation and emission between the ground andfirst excited state
quantum of energy necessary for transition from groundto excited states (i.e. hν = �E). When the resonancecondition is satisfied transitions can occur if furthercomplex selection rules are obeyed. Selection rulesare divided into high probability or allowed transitionsand forbidden transitions of much lower probability.Within the latter set of transitions are spin-forbiddenand symmetry-forbidden transitions. Spin forbiddentransitions involve a change in spin multiplicitydefined as (2S + 1) where S is the electron spinnumber (analogous to I , the nuclear spin quantumnumber). Spin multiplicity reflects electron pairing (seeTable 10.11). For a favourable transition there is nochange in multiplicity (�S = 0).
Table 10.11 Spin multiplicity for atoms and molecules
No. of unpairedelectrons.
Electronspin S
(2S + 1) Multiplicity
0 0 1 Singlet1 1/2 2 Doublet2 1 3 Triplet3 3/2 4 Quartet
Symmetry-forbidden transitions reflect redistribu-tion of charge during transitions in a quantity calledthe transition dipole moment. Differences in dipolemoment arise from the different electron distributionsof ground and excited states and when these differ-ences approximate to zero the transition is forbidden.To express this slightly differently absorbance requiresa change in dipole moment.
Absorbance is normally observed by peaks of finiteintensity and definitive linewidth in plots against fre-quency. The intensity is reflected by a Boltzmann dis-tribution that describes the population of each energylevel. In molecules atomic interactions lead to the asso-ciation of electrons with neighbouring nuclei whilstthe nuclei move relative to each other in the formof quantized vibrational or rotational motion. As aresult molecules possess ground and excited states thatsplit into a variety of sub-states, each correspond-ing to different vibrational modes, and then to fur-ther rotational sub-states. Absorbance of light leadsto transitions from the lowest ground state to a vari-ety of energy levels in the first excited state. In theabsence of chemical reactions or energy transfer theelectrons return to the ground state. Absorbance spec-tra of proteins, when recorded at room temperature,consist of several closely related transitions each ofsimilar, but non-identical, frequency leading to distinctbandwidths, reflecting the statistical sum of all individ-ual transitions. Despite broad bands absorbance spectraare frequently characterized by maxima diagnostic ofa particular transition and chromophore (e.g. A280 forproteins). In proteins transitions often involve aromaticside chains (Figure 10.41) but in the near UV/visibleregions (350–750 nm) co-factors such as heme, flavin,or chlorophyll will also give intense absorbance bands.Using molecular orbital theory electrons are definedaccording to the orbitals in which they reside as eitherσ, π or n (non-bonding) with the corresponding anti-bonding orbitals denoted as σ∗, π∗ or n∗. Transitionsbetween σ → σ∗ lie in the far UV region (large energydifference) and are not normally observed by opticalmethods, but transitions between π → π∗ and n → π∗are frequently observed in the UV and visible region ofthe electromagnetic spectrum. Physicists and chemistsplot the spectra of molecules in the form of intensityversus frequency, where the frequency is expressed as
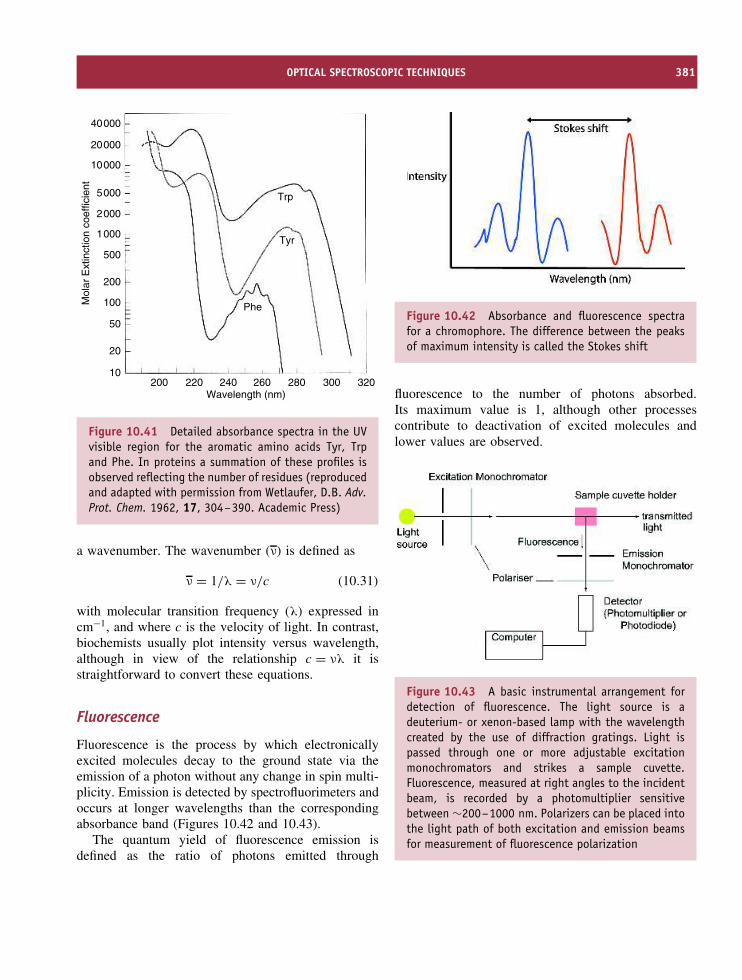
OPTICAL SPECTROSCOPIC TECHNIQUES 381
40000
20000
10000
5000
2000
1000
500
200
100
50
20
10200 220 240 260
Wavelength (nm)
Mol
ar E
xtin
ctio
n co
effic
ient
280 300 320
Phe
Tyr
Trp
Figure 10.41 Detailed absorbance spectra in the UVvisible region for the aromatic amino acids Tyr, Trpand Phe. In proteins a summation of these profiles isobserved reflecting the number of residues (reproducedand adapted with permission from Wetlaufer, D.B. Adv.Prot. Chem. 1962, 17, 304–390. Academic Press)
a wavenumber. The wavenumber (ν) is defined as
ν = 1/λ = ν/c (10.31)
with molecular transition frequency (λ) expressed incm−1, and where c is the velocity of light. In contrast,biochemists usually plot intensity versus wavelength,although in view of the relationship c = νλ it isstraightforward to convert these equations.
Fluorescence
Fluorescence is the process by which electronicallyexcited molecules decay to the ground state via theemission of a photon without any change in spin multi-plicity. Emission is detected by spectrofluorimeters andoccurs at longer wavelengths than the correspondingabsorbance band (Figures 10.42 and 10.43).
The quantum yield of fluorescence emission isdefined as the ratio of photons emitted through
Figure 10.42 Absorbance and fluorescence spectrafor a chromophore. The difference between the peaksof maximum intensity is called the Stokes shift
fluorescence to the number of photons absorbed.Its maximum value is 1, although other processescontribute to deactivation of excited molecules andlower values are observed.
Figure 10.43 A basic instrumental arrangement fordetection of fluorescence. The light source is adeuterium- or xenon-based lamp with the wavelengthcreated by the use of diffraction gratings. Light ispassed through one or more adjustable excitationmonochromators and strikes a sample cuvette.Fluorescence, measured at right angles to the incidentbeam, is recorded by a photomultiplier sensitivebetween ∼200–1000 nm. Polarizers can be placed intothe light path of both excitation and emission beamsfor measurement of fluorescence polarization

382 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
In proteins the principal fluorophore is the indoleside chain of tryptophan with a contribution thatexceeds the other aromatic residues, and dominatesthe fluorescence of proteins between 300 and 400 nm.Folded states of proteins show different fluorescencespectra to unfolded states due to the influence oflocal environment and solvent. In the folded statetryptophan emission shows a blue shift towards 330 nmfrom a ‘free’ or unfolded value of ∼350 nm. Changesin intensity and wavelength reflect the influenceof local environment on emission. Other aromaticsside chains or molecules such as heme may quenchfluorescence whilst collisional quenching by smallmolecules such as oxygen, iodide and acrylamide isan important effect.
Collisional quenching leads to excited state deac-tivation and is a nanosecond process occurring viadiffusion-controlled encounters. Aromatic moleculeson protein surfaces are quenched more effectively thanburied groups with the fluorescence intensity decayingexponentially with time. The intensity at time t isgiven by
I = Ioe−t/τ (10.33)
where Io is the intensity at time t = 0 and τ is themean lifetime of the excited state. The mean lifetimeis therefore the time decay for the fluorescence intensityto fall to 1/e. Measurement of fluorescent lifetimesis possible with sensitive photon-counting fluorimetersand is reflected by time constants ranging from afew picoseconds to hundreds of nanoseconds. Thesemeasurements shed light on molecular motion andfluorophore environments.
Collisional quenching can determine the accessibil-ity of fluorophores in proteins via the Stern–Volmeranalysis. It is modelled as an additional processdeactivating the excited state. Collisional quenchingdepopulates the excited state leading to decreased fluo-rescence lifetimes. The dependence of emission inten-sity, F , on quencher concentration [Q] is given by theStern–Volmer equation
Fo/F = τo/τ = 1 + kqτo[Q] (10.34)
= 1 + KSV[Q] (10.35)
where τ and τo are the lifetime in the presenceand absence of quencher, kq is the bimolecular rate
constant for the dynamic reaction of the quencher withthe fluorophore and the product kqτo is called theStern–Volmer constant or KSV. A low value for KSV
indicates that residues (fluorophores) have low solventexposure and are buried within a protein.
In many proteins tyrosine fluorescence is effectivelyquenched by tryptophan as a result of energy transferoccurring primarily from dipole–dipole (acceptor–donor) interactions where the magnitude is governedby the distance, intervening media and orientation.Semiquantitative models of fluorescence resonanceenergy transfer allow estimation of distances betweendonor and acceptor. Energy transfer involving a dipolar(through space) coupling mechanism of the donorexcited state and the acceptor is known as Forsterenergy transfer and varies as the inverse sixth powerof the intervening distance.
Fluorescence polarization measurement associatedwith emission intensity and fluorescence anisotropyprovide insight into the mobility of fluorophoresin proteins. Fluorescence polarization occurs whenlinearly polarized light is used to excite molecules.Molecules oriented in parallel to the direction ofpropagation are preferentially excited; for fixed orrigid molecules arranged in parallel this will yieldmaximum polarization whilst motion associated withthe fluorophore leads to randomized orientations anddepolarization. Depolarization is also sensitive tothe rotational motion or ‘tumbling’ of molecules.This Brownian motion is described by a rotationalcorrelation time (τc) that to a first approximation isestimated assuming spherical proteins of known radius.The correlation time reflects a relaxation process andif it is shorter than the fluorescence decay timeleads to molecular reorientation between the events ofabsorbance and emission.
The degree of fluorescence polarization, P , isdefined as
P = (I|| − I−)/(I|| + I−) (10.37)
where I|| is the fluorescence intensity measured withpolarization parallel to the absorbed plane-polarizedradiation, and I− is the fluorescence intensity measuredperpendicular to the absorbed radiation. A rigid systemleads to a maximum value of 1.0 whilst a systemexhibiting complete depolarization due to molecular

OPTICAL SPECTROSCOPIC TECHNIQUES 383
tumbling has a value of −1. These values aretheoretical and are rarely, if ever, observed in solution.Typical rotational correlation times for proteins insolution are of the order of 1–100 ns.
Frequently, proteins show partial polarization orare completely unpolarized as a result of tumbling.In addition, internal motion, protein concentration,solvent viscosity, temperature, as well as resonanceenergy transfer can lead to substantial alterations inpolarization. The most common method of analysingfluorescence polarization involves the Perrin equation
1/P − 1/3 = (1/Po − 1/3)(1 + 3τ/τc) (10.38)
where τ is the fluorescence lifetime, τc is a correlationtime reflecting the rotational relaxation rate due tomolecular tumbling and is equivalent to
τc = 4πηr3/kBT (10.39)
where η is the solvent viscosity and r is the hydro-dynamic radius. P0 represents the intrinsic polarizationobserved under conditions promoting least molecularmotion such as high solvent viscosity and low proteinconcentration. For non-spherical proteins the equationshould be re-written as τc = 3ηV/RT where V isthe volume.
Fluorescence polarization depends on the rotationalcorrelation time and the fluorescence lifetime and isdifficult to use if there is no knowledge of the lifetime(τ) or the limiting value of Po. Fluorescence anisotropydecays as the sum of exponentials and is used in timeresolved measurements. Anisotropy is defined as theratio of the difference between the emission intensityparallel to the polarization of the electric vector of theexciting light (I||) and that perpendicular (I−) dividedby the total intensity (IT), where IT = I|| + 2I−
A = (I|| − I−)/(I|| + 2I−) (10.40)
The emission anisotropy (A) is related to the correlationtime of the fluorophore (τc) through the Perrin equation
Ao/A = 1 + τ/τc (10.41)
where Ao, is the limiting anisotropy of the fluorophoreand depends on the angle between the absorption and
emission transition dipoles, and τ is the fluorescencelifetime. Fluorescence anisotropy is used to probetryptophan residues in proteins as well as motion ofcovalently attached fluorophores. Measurement of tryp-tophan anisotropy reveals that proteins often exhibitadditional motion associated with Trp residues and hasbeen attributed to rotation of the indole side chain aboutthe CA–CB bond with models involving precession ofthe side chain within a cone.
Green fluorescent protein
The green fluorescent protein (GFP) is responsible forthe natural green ‘colour’ exhibited by the jellyfish,Aequorea victoria and other coelenterates, whereit gives cells a characteristic glow visible on thesurface of sea at sunset. Details of the structureof GFP (Figure 10.44) have revealed an 11-strandedantiparallel β sheet forming a barrel-like structureof diameter 3.0 nm and length 4.0 nm and that hasbeen likened to a ‘lantern’. GFP is a remarkablystable protein, resistant to proteases and denaturedonly under extreme conditions such as 6 M guanidinehydrochloride at 90 ◦C. The lantern functions to protectthe ‘flame’ which is positioned in a single α helixlocated towards the centre of the β barrel. The ‘flame’responsible for the intrinsic fluorescence of GFP isp-hydroxy-benzylideneimidazolinone and results fromcyclization of the tripeptide Ser65-Tyr66-Gly67 and1,2-dehydrogenation of the Tyr.
GFP absorbs blue light around 395 nm preferen-tially but also exhibits a smaller absorbance peak at475 nm (ε of ∼30 000 and 7000 M−1cm−1 respec-tively). In the coelenterate A. victoria GFP absorbsblue light by fluorescence energy transfer from a sec-ond protein aequorin converting the incident ‘blue’light via energy transfer into green light. Aequoreaare luminescent jellyfish observed to glow around themargins of their umbrellas as a result of light pro-duced from photogenic cells in this region. Expo-sure to physical stress causes the intracellular cal-cium concentrations to increase leading to an excitedstate of aequorin generated through Ca2+ bindingto the pigment coelenterazine. The light producedby aequorin generates GFP fluorescence by energytransfer, although the physiological basis of these reac-tions is unclear (Figure 10.45).

384 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
HN
CH2
C O
N CH
CH2
OC
OH
N
CH
H2C
OH
O
C
Cyclisation
1,2 dehydrogenation
Ser
Tyr
Gly
H
126
23
H
Figure 10.44 The structure of GFP showing 11-stranded β barrel (PDB:1EMA). The cyclic p-hydroxy-benzylideneimidazolinone fluorophore
Figure 10.45 GFP fluorescence. In vitro mixing of aequorin and Ca2+ produces blue light (λmax ∼ 470 nm). Theblue fluorescent form of aequorin represents an excited state that decays to the ground state. In the presence ofGFP energy transfer occurs with a green luminescence observed comparable to that seen in the living animal (right)(reproduced with permission from Yang F., Moss L.G. & Phillips G.N. Nat. Biotechnol. 1996, 14, 1246–1251)

OPTICAL SPECTROSCOPIC TECHNIQUES 385
Figure 10.46 The structure at the active site of GFP associated with the 398 and 475 nm absorbance bands(reproduced with permission from Brejc, K. et al. Proc. Natl Acad. Sci. 1997, 94, 2306–2311)
Absorbance of GFP around 398 nm leads to afluorescence emission peak at 509 nm with a quantumyield between 0.72–0.85, and hence the green colourobserved in vivo and in vitro. The intensity ratiobetween the absorbance peaks is sensitive to factorssuch as pH, temperature, and ionic strength, suggestingthe presence of two different forms of the chromophore.It has been shown that the two forms differ in theirprotonation states with the 398 nm band reflecting aprotonated fluorophore whilst the 475 nm band reflectsdeprotonation of this group (Figure 10.46).
The unique fluorescence of GFP allows theexpressed protein to be fused to other domainscreating a chimeric protein with a fluorescent tagthat can be measured and has been used in N-and C-terminal fusions to follow gene expression,protein–protein interactions, cell sorting pathways, andintracellular signalling.
Circular dichroism
Circularly polarized light travels through opticallyactive media with different velocities due to differentindices of refraction for right (dextro) and left (laevo)components (Figure 10.47). This is called opticalrotation and the variation of optical rotation withwavelength is known as optical rotary dispersion(ORD). ORD is normally measured at a specificwavelength and temperature and is usually denoted
Figure 10.47 The resultant rotation of ε when εR andεL make unequal angles with the x-axis. A levorotatorychiral centre exists causing a rotation of angle α
by a parameter α, the angle of rotation of polarizedlight.
The right and left circularly polarized componentsare also absorbed differentially at some wavelengthsdue to differences in extinction coefficients for the twopolarized components (Figure 10.48). The addition ofthe left and right components yields ε at every pointfrom the relationship ε = εL + εR. When this light is

386 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Figure 10.48 The variation of ε for a circular dichroicsample; the vector traces out an ellipse
passed through an optically active medium the left andright circularly polarized waves are rotated at differentspeeds due to small differences in molar absorptivityfor the left and right components. The differentialabsorption of the left and right circularly polarized lightgives rise to the technique of circular dichroism (CD).
CD spectroscopy measures differences in absorbanceof right and left circularly polarized light. In proteinsthe optically active absorption bands tend to be locatedin the far-UV or near-UV regions of the spectrumand involve π → π∗, n → π∗, and σ → σ∗ transitions.The chromophores responsible for these transitionsare either intrinsically asymmetric or more frequentlyasymmetric as a result of their interactions with theprotein. CD spectroscopy is of considerable impor-tance to the study of proteins as a quantitative toolin the estimation of secondary structure. This arisesbecause regularly repeating helix or sheet produce dif-ferent CD signals. In a protein CD signals reflect thesum of the secondary structure components and it hasproved possible to estimate the proportions of helix,strand or turn in a protein of unknown structure withremarkable precision.
Historically, ellipticity is the unit of CD and isdefined as the tangent of the ratio of the minor to majorelliptical axis. An axial ratio of 1:100 will therefore
result in an ellipticity of 0.57◦. In CD spectroscopythe measured parameter is the rotation of polarizedlight expressed with the units of millidegrees. ModernCD instrumentation is capable of precision down tothousandths of a degree (1 millidegree). There are anumber of ways of expressing the CD signal of asample although frequently many incorrect forms areused in the literature. The observed CD signal (S) isexpressed in millidegrees and is normally convertedto �εm or �εmrw, where �εm is the molar CDextinction coefficient and �εmrw is the mean residueCD extinction coefficient, respectively. In any CDexperiment it is vital to know the protein concentrationextremely accurately to avoid significant error. It canbe shown that
�εm = S/(32980Cl) (10.42)
where C is the concentration in mol dm−3 (or M)and l is the path length in cm. The light path isusually a value between 0.1 and 1.0 cm. The units of�εm are therefore M−1cm−1 and the analogy with themolar extinction coefficient determined via absorbancemeasurement is clear. Alternatively, expressing theellipticity as a mean residue CD extinction coefficientleads to
�εmrw = Smrw/(32980Cl) (10.43)
where the mean residue weight (mrw ) is the molecularweight divided by the number of residues. In thisinstance C is expressed in mg ml−1. CD intensities aresometimes reported as molar ellipticity ([θ]M) or meanresidue ellipticity ([θ]mrw). These terms are calculatedfrom the following equalities
[θ]M = S/(10Cl) (10.44)
where C is again the concentration in mol dm−3 or
[θ]mrw = Smrw/(10Cl) (10.45)
Both [θ]M and [θ]mrw have the units degrees cm2
dmol−1. [θ] and �ε may be inter-converted using therelationship
[θ] = 3298�ε (10.46)

VIBRATIONAL SPECTROSCOPY 387
CD extinction coefficients are the most logical unitsince they are direct analogs of extinction coefficient inabsorbance measurements and lead to values of �εmrw
in an approximate range ±20 whilst the correspondingvalues for �ε range from ±3000. Using the aboverelationships it will be apparent that values of [θ]mrw
values are in the range ±70 000.In the determination of secondary structure content
one approach is to assume that spectra are linear com-binations of each contributing secondary structure type(‘pure’ α helix, ‘pure’ β strand, Figure 10.49) weightedby its relative abundance in the polypeptide confor-mation. Unfortunately the problem with this approachis that there are no standard reference CD spectra for‘pure’ secondary structure. More significantly synthetichomopolypeptides are poor models of secondary struc-ture and most homopolymers do not form helices nor isthere a good example of a ‘model’ β strand. An empir-ical approach involved determining the experimentalCD spectra of proteins for which the structures arealready known. Using knowledge of the structure thecontent of helix, turn and strand is defined accuratelyand by using a database of reference proteins thesemethods prove accurate and reliable when applied tounknown samples. A number of different mathematicalprocedures have been adopted using reference proteinsof varying size and secondary structure content and allof these methods give similar results.
A variation of CD occurs when samples are placedin magnetic fields maintained at low temperatures(<77 K). Under these conditions all molecules exhibitCD spectra and the technique is called magneticcircular dichroism (MCD). In most cases the spec-tra of proteins are too complex to interpret fullybut for metalloproteins the MCD technique providesa powerful tool with which identify ligands andmetal ions.
Vibrational spectroscopyThe vibrational spectra of proteins are extremelycomplex and lie at lower frequencies than electronicspectra within the infrared (IR) region. It is usefulto divide the IR region into three sections, thenear, middle and far IR regions with the mostinformative zone located at wavenumbers between4000 and 600 cm−1 (Table 10.12). Historically, IR
Figure 10.49 CD spectra of proteins with diversesecondary structure content. Top: extensively α helicalproteins; centre: proteins rich in β strands; bottom:protein with (α + β) structure (reproduced with per-mission from Circular Dichroism – the conformationalanalysis of biomolecules, Fasman, G.D (ed) KluwerAcademic 1996.)

388 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Table 10.12 The relationship between wavenumberand wavelength for the near, middle and far IR regions
Region Wavelengthrange (µm)
Wavenumberrange (cm−1)
Near 0.78–2.5 12 800–4000Middle 2.5–50 4000–200Far 50–1000 200–1000
It is worth noting that 750 nm is located at the far red endof the visible spectrum. This is 0.75 µm or a wavenumber of1/0.75 × 10−4 cm−1 or 13 333 cm−1.
spectroscopy uses wavenumbers to denote frequency.In any molecule the atoms are not held in rigid orfixed bonds but move in a manner that is reminiscentof two bodies attached by a spring. Consequently allbonds can either bend or stretch about an equilibriumposition that is defined by their standard bond lengths(Figure 10.50).
Exposure to IR radiation with frequencies between300 and 4000 cm−1 leads to the absorption of energyand transition from the lowest vibrational state to ahigher excited state. In a simple diatomic moleculethere is only one direction of stretching or vibrationand this leads to a single band of IR absorption. Atomsthat are held by weak bonds require less energy toreach excited vibrational modes and this is reflectedin the frequencies associated with IR absorbance.As molecules increase in complexity more modes ofvibrations exist with the appearance of more complexspectra. For a linear molecule with n atoms, there are
3n − 5 vibrational modes whilst if it is non-linear itwill have 3n − 6 modes. Water is a non-linear moleculecontaining three atoms and has three modes of vibrationin IR spectra (Figure 10.51).
There is one more condition that must be met for avibration to be IR active and this requires changes tothe electric dipole moment during vibration (dµ/dr =0). This arises when two oppositely charged atomsmove positions. By treating a diatomic molecule as asimple harmonic oscillator Hooke’s law can be usedto calculate the frequency of radiation necessary tocause a transition. For a simple harmonic oscillator the
Figure 10.51 The IR spectrum of water showingthree fundamental modes of vibration. At higherresolution each peak will show fine structure as aresult of simultaneous transitions between differentrotational levels
Figure 10.50 Bond stretching and bending in a simple molecule (H2O). Ignoring fine structure arising fromsimultaneous transitions the IR spectrum of water has three peaks at 3943, 3832 and 1648 cm−1

RAMAN SPECTROSCOPY 389
frequency of vibration (ν) is given by
ν = 1
2π
√k
µ(10.47)
where k is the force constant determined by bondstrength, and µ is the reduced mass. The term µ, thereduced mass is equivalent to (m1 + m2)/m1m2 andleads to
ν = 1
2π
√k m1m2
m1 + m2(10.48)
From the above equation it can be seen that a strongbond between atoms leads to bands at higher frequency;IR spectra of C≡C bonds absorb between 2300 and2000 cm−1, C=C bonds occur at 1900–1500 cm−1
whilst C–C occur below 1500 cm−1 and the spectralregion becomes crowded and complex. Despite com-plexity it is called the fingerprint region and is diag-nostic for small biomolecules.
Fourier transform IR spectroscopy measures thevibrations associated with functional groups and highlypolar bonds within proteins. In proteins these groupsprovide a biochemical ‘fingerprint’ made up of thevibrational features of all contributing components.Vibrational spectra associated with proteins are com-plex but the common components contributing to theIR spectrum include at least seven amide bands denotedas amide I–amide VII representing different vibra-tional modes associated with the peptide bond. Formost observation on proteins only the first three amidebands denoted as amide I, amide II and amide IIIare of significant interest. Of these the amide I bandis most widely used for secondary structure analy-ses. Amide I is the result of C=O stretching of theamide group coupled to the bending of the N–H bondand the stretching of the C–N bond. These vibrationalmodes give IR bands between 1600–1700 cm−1 andare sensitive to hydrogen bonding and to the secondarystructure environment. Amide I bands centred around1650 to 1658 cm−1 are usually believed to be char-acteristic of groups located within α helices. Unfortu-nately, disordered regions of polypeptides as well asturns can also give amide I bands in this region andthis can complicate assignment. In contrast β strandsgive highly diagnostic bands in the region located from1620–1640 cm−1. Parallel and antiparallel β strands
are sometimes distinguishable with antiparallel regionsshowing a large splitting of the amide I band due tothe interactions between strands.
Water (H2O) is a very strong infrared absorberwith prominent bands centred at wavenumbers of∼3800–3900 cm−1 (H–O stretching band), 2125 cm−1
(water association band) and one at 1640 cm−1 (theH–O–H bending vibration) lying in a major spectralregion of interest (the conformationally sensitive amideI vibrations). Despite the high s/n ratios, accurate fre-quency determination, speed and reproducibility ofmodern instrumentation it is frequently necessary toperform measurements in aqueous solution around1640 cm−1 in D2O where no strong bands are closeto the amide I frequency. Measurements performedon the amide I band in D2O and hence on deuteriumsubstituted proteins are often distinguished as amideI′. One advantage IR spectroscopy shares with CD isthat the technique is readily performed on relativelysmall amounts of material (often ∼100 µl) and repre-sents a good starting point for new structural inves-tigation of a protein. For both methods increasinglysuccessful attempts to extract quantitative informationon protein secondary structure have been made. In IRspectroscopy analysis of the amide I bands has provedsuccessful and has permitted deconvolution of spectrainto components attributable to helical, strand and turnelements of secondary structure. As a result IR like CDprovides a means of estimating the secondary structurepresent in proteins.
Raman spectroscopyRaman spectroscopy is concerned with a change offrequency of light when it is scattered by molecules.The Raman technique owes its name to ChandrasekharaVenkata Raman who discovered in 1928 that light inter-acts with molecules via absorbance, transmission orscattering. Scattering can occur at the same wavelengthwhen it is known as Rayleigh scattering or it can occurat altered frequency when it is the Raman effect (seeFigure 10.52).
Raman’s discovery was that when monochromaticlight is used to irradiate a sample the spectrum ofscattered light shows a pattern of lines of shiftedfrequency known as the Raman spectrum. The shiftsare independent of the excitation wavelength and are

390 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Figure 10.52 Relationship between Raman, Rayleigh,absorbance and fluorescence (adapted from Wang, Y. &van Wart H.E. Methods Enzymol. 1993, 226, 319–373.Academic Press)
characteristic for the molecule under investigation.Collision of a photon with a molecule leads toelastic and inelastic scattering; the former contributesto the Rayleigh line whilst inelastic collisions causequantum transitions of lower and higher frequency andare responsible for the negative and positive shiftsobserved in Raman spectroscopy corresponding tovibrational and rotational transitions within scatteringmolecules. The two types of Raman transitions aresometimes called Stokes lines. These changes arenormally observed directly in the IR region of thespectrum but they can also be observed as frequency
shifts within the more accessible UV or visibleregions. Raman signals from aqueous protein samplesare easily measured with signals from water beingrelatively weak. A much greater problem derives fromfluorescence associated with proteins that swampssmaller Raman signals.
A variation on the basic technique of Ramanspectroscopy is called resonance Raman spectroscopy.The technique is often applied to metalloproteins whereelectronic transitions associated with chromophoreligands to metal centres produce enhancements to thebasic Raman effect. Vibrational modes associated withthe ligand are dramatically enhanced often by factorsof 103 or more and have proved extremely useful inexamining the structural organization of metal centreswithin proteins. Since these centres are often foundat the catalytic core of an enzyme resonance Ramanhas facilitated understanding the chemistry occurringwithin these sites. The techniques of fluorescence,Raman and absorbance are related via excitation of anelectron to an excited state followed by its decay backto the ground state (Figure 10.52).
ESR and ENDOR
Electron spin resonance (ESR) spectroscopy measuresa comparable process to NMR spectroscopy with theexception that changes in electron magnetic momentare measured as opposed to changes in nuclearspin properties. Despite this difference much of thebasic theory of NMR applies to ESR spectroscopy.4
Electrons possess a property called spin that leadsto the generation of a magnetic moment when anexternal magnetic field is applied. Spin is quantizedwith an electron spin quantum number Ms = ±1/2. Inthe presence of an applied magnetic field degeneracyis removed and leads to transitions between thetwo states (Figure 10.53). The resonance condition isexpressed as
�E = hν = gβH (10.49)
where g is a dimensionless constant (often called theLande g factor and is equal to 2.00023) and β is the
4The terms ESR and EPR are used interchangeably and both termsare acceptable and used by authors in the subject literature. EPR =electron paramagnetic resonance.

ESR AND ENDOR 391
Figure 10.53 The resonance condition for ESRspectroscopy. Such a transition would give rise toa single line. For instrumental reasons ESR spectra arepresented as first derivative spectra
Bohr magneton. The electron magnetic moment is 600times greater than that of the proton. ESR experimentsare normally carried out at a field strength of 3400 G(0.34 T) and a frequency of ∼9 GHz (known as theX-band) that lies in the microwave region of theelectromagnetic spectrum. In ESR the frequency isusually fixed and spectra are acquired by sweeping theapplied magnetic field whilst measuring concomitantchanges in resonance absorption.
A fundamental requirement for ESR is the pres-ence of one or more unpaired electrons. In proteins thisoccurs frequently for transition metals such as Fe, Co,Mn, Ni and Cu with unpaired d electrons. RoutinelyESR spectra are recorded at low temperatures, usuallybelow 50 K, through the use of liquid helium cooledprobes. Rapid spin lattice relaxation rates at elevatedtemperatures are slowed at lower temperatures leadingto increased signal intensity and less saturation. Aque-ous samples absorb microwaves strongly and freez-ing results in the formation of ice a less efficientmicrowave absorber.
Iron exists in three different oxidation states inbiological systems. Usually the ferrous (d6) and fer-ric (d5) configurations are encountered but occasionallyenzymes such as peroxidases or cytochromes P450
2.2
0.30 0.32 0.34 0.36 0.38
2.1 2.0
g factor
B (T)
2 [4Fe–4S]1+
[3Fe–4S]1+
[4Fe–4S]1+
[4Fe–4S]3+
[2Fe–2S]1+
1.9 1.8
Figure 10.54 Representative ESR spectra for iron–sulfur clusters found in ferredoxins. From top to bottomthe spectra reveal the properties of the iron centresin proteins from Clostridium pasteurianum, Desulfovibriogigas, Bacillus stearothermophilus, Chromatium vinosumhigh potential protein and Mastogocladus laminosus.All spectra were recorded at X-band frequencies at atemperature between 10 and 20 K
have a ferryl (d4) state. The ferric iron form occursas an unpaired high spin (S = 5/2) state or a partiallypaired low spin (S = 1/2) state that are distinguishedusing ESR spectrometry. In ferredoxins, metallopro-teins containing iron–sulfur clusters, ESR has revealeda variety of different clusters {[Fe-Cys4], [2Fe-2S],[3Fe-3S] and [4Fe-4S] centres}, interacting redox cen-tres, redox state transitions, as well as ligand identity,coordination and geometry (Figure 10.54).

392 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
Table 10.13 International and national facilities for X-ray crystallography and NMR spectroscopy
Facility and role Web address
Synchrotron sourcesDaresbury UK. Crystallography and CD http://www.srs.ac.uk/srs/Cornell USA. Crystallography http://www.chess.cornell.edu/European facility Grenoble France. X-ray and
Neutron diffractionhttp://www.esrf.fr/
NMR facilitiesMadison USA. Ultra high field NMR spectroscopy http://www.nmrfam.wisc.edu/Frankfurt Germany. EU funded large scale facility
for NMR and ESRhttp://www.biozentrum.uni-frankfurt.de/LSF/
Although ESR spectroscopy has been most widelyused to characterize iron-containing proteins it hasalso been used to study: (i) the metal centre ofNi2+ (d8) in enzymes like urease, hydrogenasesand carbon monoxide dehydrogenase; (ii) the widevariety of Cu2+ (d9) centres such as those found insuperoxide dismutase, tyrosinase and haemocyanin;(iii) Mo centres such as that found in enzymes likexanthine oxidase; and (iv) Mn centres found withinphotosynthetic systems involved in oxygen evolution.
Recent developments in ESR spectroscopy utilizeadvantages of pulse methods for acquiring spectra asopposed to sweeping the magnetic field. ESR spec-troscopy has traditionally been a continuous wave(CW) technique but techniques such as ESEEM (elec-tron spin echo envelope modulation) or ENDOR(electron-nuclear double resonance spectroscopy) probeelectron–nuclear interactions in metalloproteins unsuit-able for study using crystallography or NMR. The tech-niques can derive relaxation times and information onmetal–ligand mobility, distance and orientation.
Armed with several of the above spectroscopictools the structure of almost all proteins can begradually determined. The time scale from isolationof a protein to determination of its three-dimensionalstructure has decreased dramatically from severalyears to perhaps a few months. In many casesacquisition of experimental data takes only a fewdays whilst analysis and interpretation of the raw datais often a longer process. Unfortunately the cost of
instrumentation required to perform these studies atthe highest level has become extremely prohibitive.Ultra high field NMR spectrometers, powerful electronmicroscopes and synchrotron beam lines are beyondthe scope of most individual institutions, with a singleinstrument costing millions of pounds. Increasinglythis instrumentation is being organized around corenational, and sometimes internationally coordinatedcentres of excellence where these facilities serve manyresearch groups (Table 10.13).
Summary
By exploiting different regions of the electromagneticspectrum and the interaction of atoms with radiationconsiderable information can be acquired on thestructure and dynamics of proteins.
The plethora of techniques available for studyingproteins means that if one technique is unsuitable thereis almost certainly another method that can be applied.
X-ray crystallography and multi-dimensional NMRspectroscopy yield detailed pictures of protein structureat an atomic level. X-ray crystallography relies onthe diffraction of X-rays by electron dense atomsconstrained within a crystal. Some proteins fail tocrystallize and this represents the major limitation ofthe technique.
The ‘phase problem’ is overcome through the useof isomorphous replacement but a basic requirement

PROBLEMS 393
is that addition of heavy metal atoms does notalter protein structure. The structures derived for theproteasome, the ribosome and viral capsids serve toemphasize the success of the technique.
Membrane proteins or proteins with extensivehydrophobic domains are notoriously difficult to crys-tallize, although several structures now exist within theProtein Databank.
NMR spectroscopy measures nuclear spin reorien-tation in an applied magnetic field most frequently forspin 1/2 nuclei. In proteins this centres around the 1Hbut with isotopic labelling can also include 15N and13C nuclei.
A major hurdle in NMR spectroscopy is the assign-ment problem – identifying which resonances belongto a given residue. Sophisticated multidimensional het-eronuclear NMR methods have been developed to facil-itate this process based around the interaction of nuclei‘through bonds’ and ‘through space’.
Solution structure is defined from the use of acombination of torsion angle and distance restraints.The former are obtained from J coupling experimentswhilst the r−6 dependence of the NOE is the basis ofdistance constraints.
NMR structures are usually presented as a familyor ensemble of closely related protein topologies.Although most structures for proteins determined byNMR spectroscopy have masses below ∼20 kDa thesize limit is increasing steadily with the introductionof new methods.
The vast majority of structures deposited in the Pro-tein Databank (>95 percent) have been determinedusing crystallography or NMR spectroscopy, butslowly a third technique of cryo-EM is gainingprominence.
Cryo-EM methods have the advantage of requiringlittle sample preparation and are particularly suitable tovery large protein complexes. The technique relies onelectron diffraction by particles immersed in a frozen
lattice. Single particle analysis is the current ‘hot’ areaand involves trapping macromolecular complexes inrandom orientations within vitreous ice and combiningthousands of images to provide an enhanced picture ofthe system.
Optical techniques based around electronic transi-tions provide information on the absorbance and flu-orescence of chromophores found in proteins usuallyaromatic residues. Tryptophan exhibits a significantlygreater molar extinction coefficient when compared toeither tyrosine or phenylalanine with the result thatmost of a protein’s absorbance or fluorescence reflectsthe relative abundance of Trp within a polypeptidechain.
Time-resolved measurements allow changes in flu-orescence or absorbance to be followed on very shorttime scales sometimes in the sub-nanosecond range.This allows measurements of protein mobility such asthe motion of aromatic side chains.
CD involves the measurement of differential absorp-tion of right and left circularly polarized light as afunction of wavelength. In proteins the far UV regionfrom 260–180 nm is dominated by the CD signals dueto elements of secondary structure such as helix, turnsand strands.
Proteins composed extensively of helical structurehave very different CD spectra to those containingproportionally more β strands. CD spectra allow thesecondary structure content of unknown proteins to bepredicted with reasonable accuracy.
IR spectroscopy of proteins has been used to mea-sure changes in the vibrational states associated withthe amide bond. Amide bonds in helices, turns andstrands show characteristic transitions in a regionknown as the amide I band between 1600 and1700 cm−1. Experimental spectra are fitted as combina-tions of helices and strands and can be used to estimatesecondary structure content in a comparable manner toCD spectroscopy.
Problems1. Why are fluorescence maxima observed at longer
wavelengths than absorbance maxima?2. Why do peaks in absorbance spectra become narrower
when the temperature is lowered to 77 K?

394 PHYSICAL METHODS OF DETERMINING THE THREE-DIMENSIONAL STRUCTURE OF PROTEINS
3. What is the wavelength range of IR studies conductedat 300–4000 cm−1.
4. Show that an axial ratio of 1:100 leads to anobserved rotation of circularly polarized light of0.57◦.
5. The absorbance of a 10 µM solution of trypto-phan in buffer at 280 nm is 0.06. The buffer alonegives an absorbance of 0.004 at 280 nm. Assum-ing a path length of 1 cm calculate the extinc-tion coefficient of tryptophan. What are the expectedabsorbance values for pathlengths of 1, 2 and20 mm?
6. Using the data provided in Table 10.4 calculate theLarmor frequency of the following nuclei 1H, 13Cand 15N at field strengths of 20, 18.1, 16, 11.7and 8 T.
7. Sketch the high resolution 1D 1H-NMR spectra ofalanine, glycine and threonine.
8. Draw the expected pattern of connectivities in 2Dhomonuclear TOCSY and COSY spectra for theresidues not included in Figure 10.27. (ignore finestructure for cross peaks).
9. Explain how heavy metal derivatives of proteinsmight be detected using non-denaturing electrophoretictechniques. What might be the limitations to thistechnique? Using Harker diagrams show how the useof a second heavy metal derivative leads to a uniquesolution for the intensity and phase.
10. The structure of cytochrome c′ from Chromatiumvinosum has been determined and the data hasbeen deposited in protein databanks. Locate therelevant PDB file and from the primary citationidentify the methods used to determine the structuretogether with the unit cell dimensions, the numberof molecules per unit cell, the resolution achieved.Describe the structure determined and any new featuresdiscovered.

11Protein folding In vivo and In vitro
Introduction
To address the mechanisms of protein folding it isnecessary to understand thermodynamic and kineticparameters governing the formation of the native state.Folding is a reversible reaction defined most simply as
Uk1−−−⇀↽−−−k2
N (11.1)
where the equilibrium lies to the right (Keq > 1 =[N]/[U]) for the native state (N) to be defined asmore stable than the unfolded state (U). Quite clearlythe respective rates of the forward (k1) and reverse(k−1) reactions are related to the value Keq and tounderstand folding requires estimating the magnitudeof these rate constants.
Although many proteins fold in vitro at comparablerates to those observed in vivo the environment andthe presence of ‘helper’ proteins play critical rolesin folding within the cell. The realization that someproteins are assisted to find the correct folded structureby molecular chaperones has expanded the area ofprotein folding.
Protein misfolding is no longer viewed as an exper-imental artefact. It results from gene mutation andunderpins diseases such as Alzheimer’s, collagen baseddiseases such as osteogenesis imperfecta, cystic fibro-sis, familial amyloidotic polyneuropathy – a compara-tively rare disease arising from mutations in the protein
transthyretin – as well as more familiar diseases suchas emphysema.
Studies of protein folding have revolutionizedour ideas of disease transmission. Until recentlyit was thought that nucleic acid uniquely carriedinformation ‘directing’ the transmission of a disease.This occurred either through a mutation in the hostgenome, integration of viral DNA into a genome orby infection directed by a pathogen’s genome. Todaythere is compelling evidence that some diseases arisesolely from a protein and a misfolded one at that!
Factors determining the protein fold
Globular proteins fold into conformations of orderedsecondary and tertiary structure where hydrophobicside chains are buried on the inside of the protein andthe polar/charged side chains are solvent accessible.Interactions governing the formation of secondary andtertiary structure involve the formation of hydrogenbonds, disulfide bridges, charge–pair interactions andnon-polar or hydrophobic effects. The cumulativeeffect of these forces is that in any folded proteinthe magnitude of favourable interactions outweighs thesum of the unfavourable ones.
These interactions are disrupted by extremes oftemperature, immersing proteins in acidic or alkalinesolutions or adding solvents such as alcohol in aprocess known as denaturation that results in a loss
Proteins: Structure and Function by David Whitford 2005 John Wiley & Sons, Ltd
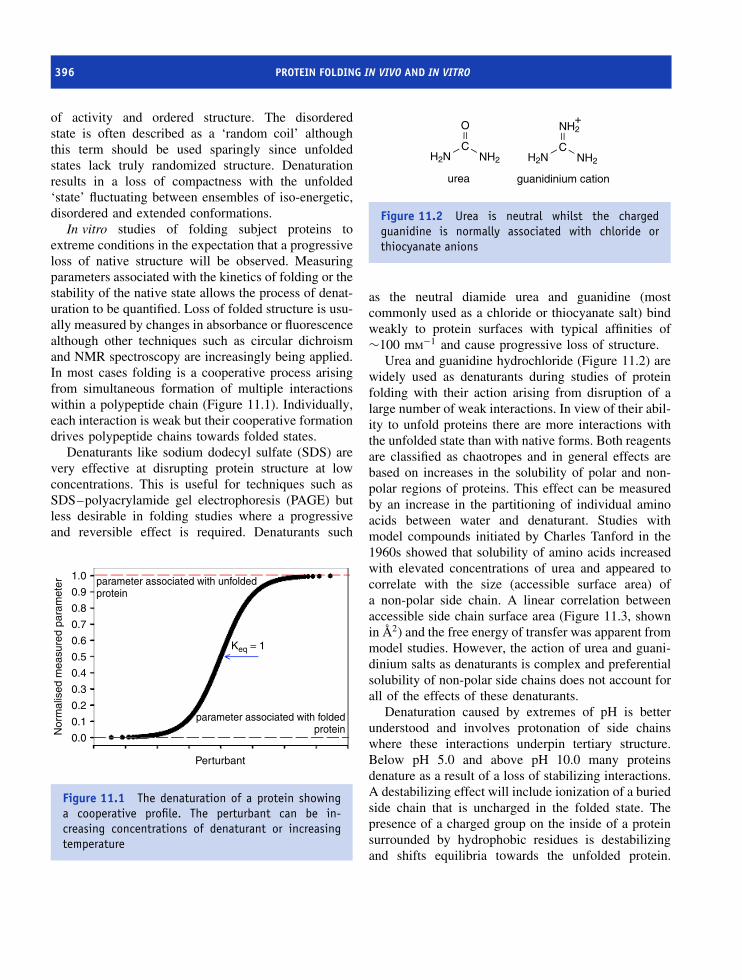
396 PROTEIN FOLDING IN VIVO AND IN VITRO
of activity and ordered structure. The disorderedstate is often described as a ‘random coil’ althoughthis term should be used sparingly since unfoldedstates lack truly randomized structure. Denaturationresults in a loss of compactness with the unfolded‘state’ fluctuating between ensembles of iso-energetic,disordered and extended conformations.
In vitro studies of folding subject proteins toextreme conditions in the expectation that a progressiveloss of native structure will be observed. Measuringparameters associated with the kinetics of folding or thestability of the native state allows the process of denat-uration to be quantified. Loss of folded structure is usu-ally measured by changes in absorbance or fluorescencealthough other techniques such as circular dichroismand NMR spectroscopy are increasingly being applied.In most cases folding is a cooperative process arisingfrom simultaneous formation of multiple interactionswithin a polypeptide chain (Figure 11.1). Individually,each interaction is weak but their cooperative formationdrives polypeptide chains towards folded states.
Denaturants like sodium dodecyl sulfate (SDS) arevery effective at disrupting protein structure at lowconcentrations. This is useful for techniques such asSDS–polyacrylamide gel electrophoresis (PAGE) butless desirable in folding studies where a progressiveand reversible effect is required. Denaturants such
parameter associated with unfoldedprotein
Keq = 1
Perturbant
Nor
mal
ised
mea
sure
d pa
ram
eter
1.0
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0.0
parameter associated with foldedprotein
Figure 11.1 The denaturation of a protein showinga cooperative profile. The perturbant can be in-creasing concentrations of denaturant or increasingtemperature
CNH2
O
H2NC
NH2
NH2
H2N
urea guanidinium cation
+
Figure 11.2 Urea is neutral whilst the chargedguanidine is normally associated with chloride orthiocyanate anions
as the neutral diamide urea and guanidine (mostcommonly used as a chloride or thiocyanate salt) bindweakly to protein surfaces with typical affinities of∼100 mM−1 and cause progressive loss of structure.
Urea and guanidine hydrochloride (Figure 11.2) arewidely used as denaturants during studies of proteinfolding with their action arising from disruption of alarge number of weak interactions. In view of their abil-ity to unfold proteins there are more interactions withthe unfolded state than with native forms. Both reagentsare classified as chaotropes and in general effects arebased on increases in the solubility of polar and non-polar regions of proteins. This effect can be measuredby an increase in the partitioning of individual aminoacids between water and denaturant. Studies withmodel compounds initiated by Charles Tanford in the1960s showed that solubility of amino acids increasedwith elevated concentrations of urea and appeared tocorrelate with the size (accessible surface area) ofa non-polar side chain. A linear correlation betweenaccessible side chain surface area (Figure 11.3, shownin A2) and the free energy of transfer was apparent frommodel studies. However, the action of urea and guani-dinium salts as denaturants is complex and preferentialsolubility of non-polar side chains does not account forall of the effects of these denaturants.
Denaturation caused by extremes of pH is betterunderstood and involves protonation of side chainswhere these interactions underpin tertiary structure.Below pH 5.0 and above pH 10.0 many proteinsdenature as a result of a loss of stabilizing interactions.A destabilizing effect will include ionization of a buriedside chain that is uncharged in the folded state. Thepresence of a charged group on the inside of a proteinsurrounded by hydrophobic residues is destabilizingand shifts equilibria towards the unfolded protein.
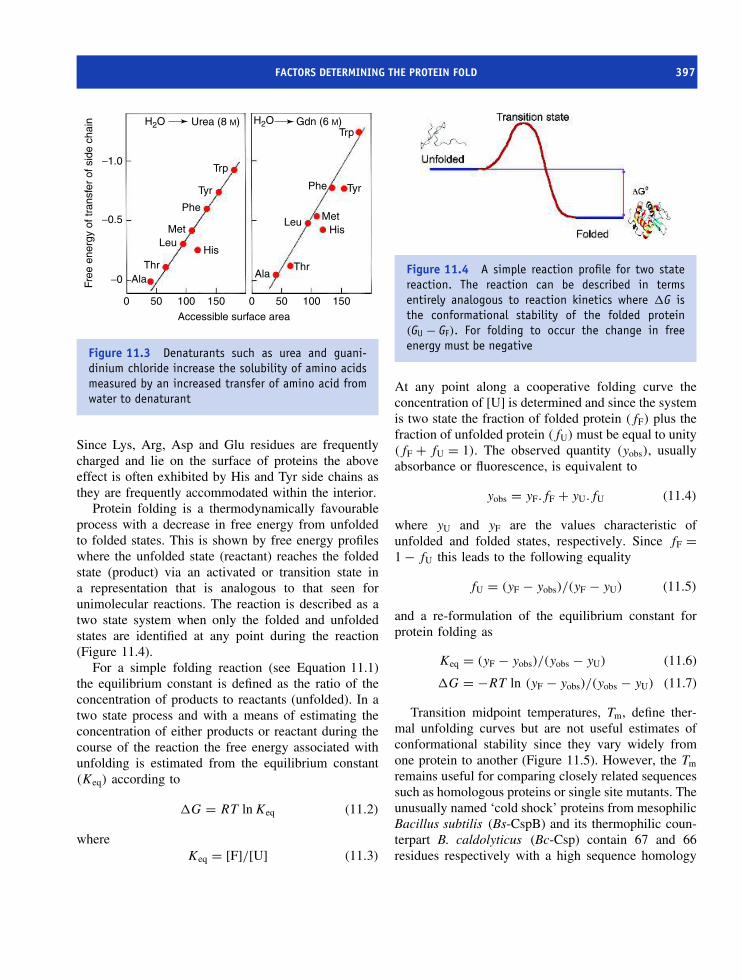
FACTORS DETERMINING THE PROTEIN FOLD 397
H2O Urea (8 M) H2O Gdn (6 M)
−1.0
−0.5
−0
0 50 100 150 0 50 100 150
Ala AlaThrThr
HisLeu
Met
Phe
Tyr
Trp
HisLeu Met
Phe Tyr
Trp
Accessible surface area
Free
ene
rgy
of tr
ansf
er o
f sid
e ch
ain
Figure 11.3 Denaturants such as urea and guani-dinium chloride increase the solubility of amino acidsmeasured by an increased transfer of amino acid fromwater to denaturant
Since Lys, Arg, Asp and Glu residues are frequentlycharged and lie on the surface of proteins the aboveeffect is often exhibited by His and Tyr side chains asthey are frequently accommodated within the interior.
Protein folding is a thermodynamically favourableprocess with a decrease in free energy from unfoldedto folded states. This is shown by free energy profileswhere the unfolded state (reactant) reaches the foldedstate (product) via an activated or transition state ina representation that is analogous to that seen forunimolecular reactions. The reaction is described as atwo state system when only the folded and unfoldedstates are identified at any point during the reaction(Figure 11.4).
For a simple folding reaction (see Equation 11.1)the equilibrium constant is defined as the ratio of theconcentration of products to reactants (unfolded). In atwo state process and with a means of estimating theconcentration of either products or reactant during thecourse of the reaction the free energy associated withunfolding is estimated from the equilibrium constant(Keq) according to
�G = RT ln Keq (11.2)
whereKeq = [F]/[U] (11.3)
Figure 11.4 A simple reaction profile for two statereaction. The reaction can be described in termsentirely analogous to reaction kinetics where �G isthe conformational stability of the folded protein(GU − GF). For folding to occur the change in freeenergy must be negative
At any point along a cooperative folding curve theconcentration of [U] is determined and since the systemis two state the fraction of folded protein (fF) plus thefraction of unfolded protein (fU) must be equal to unity(fF + fU = 1). The observed quantity (yobs), usuallyabsorbance or fluorescence, is equivalent to
yobs = yF.fF + yU.fU (11.4)
where yU and yF are the values characteristic ofunfolded and folded states, respectively. Since fF =1 − fU this leads to the following equality
fU = (yF − yobs)/(yF − yU) (11.5)
and a re-formulation of the equilibrium constant forprotein folding as
Keq = (yF − yobs)/(yobs − yU) (11.6)
�G = −RT ln (yF − yobs)/(yobs − yU) (11.7)
Transition midpoint temperatures, Tm, define ther-mal unfolding curves but are not useful estimates ofconformational stability since they vary widely fromone protein to another (Figure 11.5). However, the Tm
remains useful for comparing closely related sequencessuch as homologous proteins or single site mutants. Theunusually named ‘cold shock’ proteins from mesophilicBacillus subtilis (Bs-CspB) and its thermophilic coun-terpart B. caldolyticus (Bc-Csp) contain 67 and 66residues respectively with a high sequence homology

398 PROTEIN FOLDING IN VIVO AND IN VITRO
1
0.8
0.6
0.4
0.2
0
20 40 60 80 100
temperature (°C)
frac
tion
nativ
e pr
otei
n
1: Bc-Csp2: Bc-Csp L66E3: Bc-Csp R3E4: Bc-Csp R3E/L66E
4 3 2 1
20 40 60 80 100
temperature (°C)
1
0.8
0.6
0.4
0.2
0
5: Bs-CspB6: Bs-CspB E66L7: Bs-CspB E3R8: Bs-CspB E3R/E66L
5 6 8
7
Figure 11.5 Thermal stability curves. Left: Unfolding transitions of wild type Bc-Csp and three destabilizedvariants. Right; Unfolding transitions of wild type Bs-CspB and three stabilized variants. The fractions of nativeprotein obtained after a two-state analysis of the data are shown as a function of temperature with the continuouslines showing the results of the analysis (reproduced with permission from Perl, D. et al. Nat. Struct. Biol. 2000, 7,380–383. Macmillan)
seen by a difference in only 12 residues. The proteinshave similar 3D structures and all of the differentresidues are located on the molecular surface. Theseresidues contribute to a transition midpoint temperatureof nearly 77 ◦C for Bc-Csp, approximately 23◦ higherthan that observed in Bs-CspB. Using site directedmutagenesis the 12 residues were replaced in Bc-Cspby the corresponding residues found in Bs-CspB. Onlytwo out of the 12 residues made significant contribu-tions to thermal stability. These residues were Arg3 andLeu66, with each contributing to enhanced electrostatic(Arg) and hydrophobic (Leu) interactions. A recurringtheme for elevated thermostability is increased num-bers of charged residues and hydrophobic side chainswhilst the number of polar uncharged residues usu-ally decreases.
Analysis of thermal induced unfolding curves cen-tres around substitution of the equation
�GU = �HU − T �SU (11.8)
Since Keq = e−�G/RT this leads to
Keq = e−�HU/RT +�SU/R (11.9)
where �H and �S are the enthalpy and entropyassociated with unfolding. The van’t Hoff analysis ofthe variation in equilibrium constant with temperature
ln Keq = −�H/RT + �S/R (11.10)
is widely used in chemistry to estimate reactionenthalpy and entropy. Over a narrow temperature rangea straight line defines the enthalpy and entropy in plotsof ln Keq versus 1/T . These plots in protein unfoldingtransitions are non-linear, indicating that �H is nottemperature independent. In proteins �H and �S aretemperature dependent with non-negligible variationsover a typical range of 70–100 K.
The heat capacity is defined as the change inenthalpy with temperature
Cp = ∂H/∂T = T ∂S/∂T (11.11)
and during protein unfolding the product and reactantshave different heat capacities leading to significantchanges in enthalpy. Recasting Equation 11.11 leads to
∂H/∂T = Cp(U) − Cp(F ) = �Cp (11.12)
�H(T2) = �H(T1) + �Cp (T2 − T1) (11.13)
where Cp(U) and Cp(F ) are the heat capacities of theunfolded and native states respectively and �Cp is thechange in heat capacity accompanying unfolding. Fromthese equations calculating �G at any temperature T
requires knowledge of �Cp and �H . The enthalpy isbest estimated at a single temperature, and Tm providesthe most convenient estimation point in the thermalunfolding curve since at this point
�G(Tm) = 0 = �Hm − Tm�Sm (11.14)

FACTORS DETERMINING THE PROTEIN FOLD 399
To calculate �G at any temperature T is given by
�G(T ) = �Hm (1 − T /Tm) − �Cp [(Tm − T )
+ T ln (T /Tm)] (11.15)
where Tm is estimated from the unfolding curve; �Hm
is estimated from the gradient of the straight line ofln Keq versus 1/T around Tm – at this point �Hm =Tm �S, since �G = 0, and this leaves �Cp as theonly parameter remaining to be estimated.
Although generally true that proteins with a highTm are more stable than those with lower Tm valuesthis parameter is not a definitive measure of stability.Changes in protein stability reflected by small changesin Tm are approximated through the relationship
��GU ≈ Tm �Sm ≈ �Hm �Tm/Tm (11.16)
If either the entropy or enthalpy associated with unfold-ing is unknown an estimate of ��GU may be obtainedwith reasonable precision from Equation 11.16. Intu-itively it is clear that as the temperature increases aprotein unfolds. The basic equations of state relating toprotein folding reveal that as T increases the term T �S
dominates to such an extent that it becomes greater than�H . At high temperatures the entropy of the unfoldedstate dominates and favours unfolding.
The heat capacity change is most accurately mea-sured by differential scanning calorimetry (DSC). DSC
enables direct measurement of Tm, �H and �Cp fromincreases in heat transfer that occur with unfoldingas a function of temperature. The instrument is basedaround an adiabatic chamber where one cell containsthe sample (protein plus solvent) at a concentrationof ∼1 mg ml−1 whilst another cell acts as a refer-ence and is normally filled with an identical volumeof solvent (Figure 11.6). Both cells are heated withthe temperature difference between the two cells con-stantly measured as a complex feedback loop increasesor decreases the sample cell’s power input via a heaterin an effort to keep the temperature difference close tozero. Since the masses and volumes of the two cellsare matched the power added or subtracted by the sys-tem is a direct measure of the difference between theheat capacity of sample and reference solutions – inother words the heat capacity of the protein. Despitefirst impressions it is surprisingly difficult to achieveperfect matching of sample and reference cells withthe result that experimental scans are observed witha baseline offset. This ‘constant’ is usually subtractedfrom the data to give a corrected profile and accurateestimates of Cp (Figure 11.7).
Although �Cp can be estimated from a singlethermogram it is generally obtained from a seriesof profiles obtained at different pH values. The Tm
decreases as the pH is lowered from small enthalpychanges and a plot of �Hcal against Tm has a slopeof �Cp. From Equation 11.11 integration of the heat
Figure 11.6 In a differential scanning calorimeter the sample cell contains protein plus solvent whilst the referencecell lacks protein. Volumes of ∼0.5 ml are used in each cell

400 PROTEIN FOLDING IN VIVO AND IN VITRO
heat
cap
acity
-cha
nge
(Cp
kJ/m
ol)
20 40 60 80 100
Temperature (K)
Buffer profile
Proteinendotherm
Tm
∆Cp
15
10
5
0
20 40 60 80 100
Temperature (K)
Corrected calorimetricprofile
Figure 11.7 DSC profile for protein unfolding showing pre- and post-denaturational regions and baseline correctionto achieve accurate estimation of �Cp(U−F)
capacity curve yields the enthalpy (�H) and measuresthe total heat absorbed by the protein during the processof unfolding. Calorimetric studies of protein unfoldingshow that the heat capacities associated with unfoldedstates are larger than those of native states, althoughboth are positive values.
In small soluble domains the value of �Cp rangesbetween 7000 and 10 000 J mol−1 K−1. Systematicanalysis of these values in many proteins suggests thata rule of thumb can be constructed for ‘estimating’�Cp – it involves multiplying the number of residuesin a protein by 50 J mol−1 K−1. It is common to com-pare the enthalpy obtained from the van’t Hoff plot(�Hvh) with that determined calorimetrically (�Hcal).A ratio (�Hvh/�Hcal) of 1.0 confirms that the processis two state whilst a value of less than 1 may be usedas evidence of an intermediate and departure from asimple two-state analysis. A value of �Hvh/�Hcal > 1often indicates polymerization at some point in theheating process.
Over a wide temperature range the variation inequilibrium constant is defined by a curve as opposedto a straight line. This curve has two points wherethe equilibrium constant is 1 and the concentrationsof folded and unfolded forms are equal (Figure 11.8).The first point is the thermal denaturation temperature(Tm) whilst the second point reflects a low temperaturedenaturation point. The �G for unfolding is dominatedby the entropy component at high and low temperatures
260 280 300 320 340 360 380
Temperature (K)
20000
0
−20000
−40000
−60000
Free
ene
rgy
(∆G
) (J
mol
−1)
Figure 11.8 The temperature dependence of proteinunfolding. The two curves show the variation in �Gwith temperature in two systems with different Tm, �Hand �Cp
although the cold denaturation point lies below thefreezing point of water and is not routinely accessible.
An alternative is to promote unfolding usingdenaturants such as urea or guanidine hydrochloride(GdnHCl) with the advantage that unfolding curvesare generally simpler to interpret. In direct analogyto Tm a midpoint in the unfolding transition is giventhe symbol Cm and can be determined with reason-able accuracy (±0.05 M). For similar proteins suchas those involving single site-specific mutations thisapproach offers a valid means of comparison but
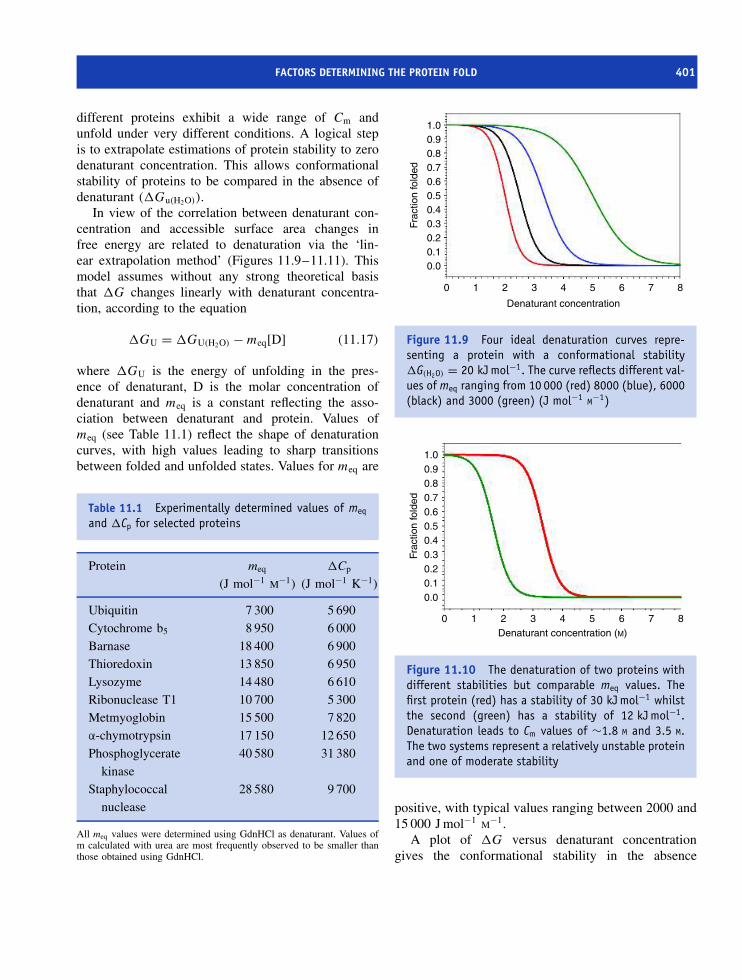
FACTORS DETERMINING THE PROTEIN FOLD 401
different proteins exhibit a wide range of Cm andunfold under very different conditions. A logical stepis to extrapolate estimations of protein stability to zerodenaturant concentration. This allows conformationalstability of proteins to be compared in the absence ofdenaturant (�Gu(H2O)).
In view of the correlation between denaturant con-centration and accessible surface area changes infree energy are related to denaturation via the ‘lin-ear extrapolation method’ (Figures 11.9–11.11). Thismodel assumes without any strong theoretical basisthat �G changes linearly with denaturant concentra-tion, according to the equation
�GU = �GU(H2O) − meq[D] (11.17)
where �GU is the energy of unfolding in the pres-ence of denaturant, D is the molar concentration ofdenaturant and meq is a constant reflecting the asso-ciation between denaturant and protein. Values ofmeq (see Table 11.1) reflect the shape of denaturationcurves, with high values leading to sharp transitionsbetween folded and unfolded states. Values for meq are
Table 11.1 Experimentally determined values of meq
and �Cp for selected proteins
Protein meq
(J mol−1 M−1)�Cp
(J mol−1 K−1)
Ubiquitin 7 300 5 690Cytochrome b5 8 950 6 000Barnase 18 400 6 900Thioredoxin 13 850 6 950Lysozyme 14 480 6 610Ribonuclease T1 10 700 5 300Metmyoglobin 15 500 7 820α-chymotrypsin 17 150 12 650Phosphoglycerate
kinase40 580 31 380
Staphylococcalnuclease
28 580 9 700
All meq values were determined using GdnHCl as denaturant. Values ofm calculated with urea are most frequently observed to be smaller thanthose obtained using GdnHCl.
1.00.90.80.70.60.50.40.30.20.10.0
0 1 2 3 4 5 6 7 8
Denaturant concentration
Frac
tion
fold
ed
Figure 11.9 Four ideal denaturation curves repre-senting a protein with a conformational stability�G(H2O) = 20 kJ mol−1. The curve reflects different val-ues of meq ranging from 10 000 (red) 8000 (blue), 6000(black) and 3000 (green) (J mol−1 M−1)
0 1 2 3 4 5 6 7 8Denaturant concentration (M)
1.00.90.80.70.60.50.40.30.20.10.0
Frac
tion
fold
ed
Figure 11.10 The denaturation of two proteins withdifferent stabilities but comparable meq values. Thefirst protein (red) has a stability of 30 kJ mol−1 whilstthe second (green) has a stability of 12 kJ mol−1.Denaturation leads to Cm values of ∼1.8 M and 3.5 M.The two systems represent a relatively unstable proteinand one of moderate stability
positive, with typical values ranging between 2000 and15 000 J mol−1 M−1.
A plot of �G versus denaturant concentrationgives the conformational stability in the absence

402 PROTEIN FOLDING IN VIVO AND IN VITRO
0 1 2 3 4 5 6 7 8
Denaturant concentration (M)
30
20
10
0
−10
−20
−30
−40
−50
−60
Free
ene
rgy
(kJ
mol
−1)
Figure 11.11 The linear extrapolation methodapplied to the data of Figure 11.9
of denaturant and experimental studies suggest thatusing either urea and guanidine hydrochloride andthe same ‘test’ protein yields a common intercept atzero denaturant and a constant value of �GU(H2O)
(Figure 11.11).A conclusion from many denaturation studies is
that overall stability of a protein (�G, GU − GF) issmall with the folded state being marginally morestable than the unfolded state. This leads to valuesfor protein conformational stability ranging between 10and 75 kJ mol−1, with most proteins exhibiting valuestowards the lower end of this range (Table 11.2).
Table 11.2 The conformational stability of proteins
Protein Chainlength
Conformationallystability
(kJ mol−1)
λ repressor 80 12.5Cytochrome c 104 74.0Cold shock protein CspB 67 12.6SH3 domain of α spectrin 62 12.1CI2 64 29.3U1A spliceosomal protein 102 38.9Ubiquitin 76 15.9CD2 98 33.5
Ribonuclease defines the ‘outline’ of proteinfolding in vitro
Ribonuclease has contributed much to our understandingof protein folding in vitro through the landmark studiesof Christian Anfinsen who posed the question: ‘whatis the origin of the information necessary for folding?’Ribonuclease, with 124 amino acid residues and fourdisulfide bridges located between cysteines 26–84,40–95, 58–110, and 65–72, catalyses the hydrolysisof RNA. Reduction of the disulfide bridges to thiols bymercaptoethanol in the presence of urea results in proteinunfolding and a concomitant loss of activity.
Anfinsen noticed that when ribonuclease was oxi-dized (by standing in air) and the urea removed by dial-ysis that enzyme activity slowly recovered as a resultof protein folding, the reformation of tertiary struc-ture and most importantly the active site. Repeatingthese reactions in the presence of denaturant (oxida-tion of the thiols in 8 M urea) led to the regenerationof less than 1 percent of the total enzyme activity.Urea preventing correct disulfide pairings resulting ina ‘scrambled’ ribonuclease whilst in its absence correctdisulfide bridge formation allowed the folded and ther-modynamically most stable state to be reached. Theseclassic studies showed that all of the information nec-essary for protein folding resides within the primarysequence. The intervening years have demonstrated thegenerality of Anfinsen’s results and although a fewcaveats must be included the maxim ‘sequence definesconformation’ remains relevant today.
One of the first caveats in this picture of proteinfolding is the protease subtilisin E from B. amyloliq-uefaciens. The enzyme is synthesized as a preproen-zyme of ∼380 residues to permit secretion and toavoid premature proteolysis. Folding studies involvingthe removal of the pro sequence showed that its losswas coupled to a lack of subsequent protein refold-ing. In contrast unfolding the full length preproproteinresulted in a folded enzyme. The results were inter-preted along the lines that the pro-sequence partici-pated in folding reactions guiding subtilisin towardsthe native state. In its absence the native state couldnot be reached and emphasized that it is not neces-sarily the ‘final’ protein sequence that encodes fold-ing information. The second caveat arose with theobservation that ‘helper’ proteins assist folding in vivo

FOLDING PROBLEM AND LEVINTHAL’S PARADOX 403
by preventing molecular aggregation. These helperproteins, molecular chaperones, derive their name fromtheir ability to prevent unwanted interactions betweennewly synthesized chains and other proteins.
Factors governing protein stability
The native state is the most stable form of a proteinbut what factors contribute to this stability? From theprevious section it might be thought that disulfidebridges or covalent bonds in general are majordeterminants of conformational stability. However, inthe unfolded protein these bonds remain intact anddo not contribute to conformational stability. To putthis in a slightly different way the covalent bondscontribute equally to the stability of the folded and
−T∆S
− +0
−T∆S
∆H
∆H
∆H
∆G
Sum
FoldedUnfolded
Figure 11.12 Contributions to the free energy offolding of soluble proteins
unfolded proteins and it is necessary when addressingthis concept to focus on non-covalent interactions.
Non-covalent interactions include hydrogen bonds,hydrophobic forces and interactions between chargedgroups that cumulatively contribute to increased sta-bility of the native state (Figure 11.12). The entropiccontribution to folding is generally unfavourable sinceit involves transitions from large numbers of unstruc-tured conformations to a single ordered structure. Thedecrease in entropy works against protein folding andthis conformational entropy is largely responsible forfavouring the unfolded state. To fold proteins mustovercome conformational entropy from entropic andenthalpic contributions that derive from interactionsbetween charged groups, hydrophobic effects, hydro-gen bonding and van der Waals interactions.
A major contribution to thermodynamic stabilityarises from the hydrophobic interaction. Protein foldingresults in the burial of hydrophobic side chains awayfrom water and their interaction with similar sidechains on the inside of the molecule. In thermodynamicterms ordered water molecules specifically arrangedin unfolded state are released from this state andare free to move with the result that the entropyincreases and makes favourable contributions to theoverall free energy change. Favourable enthalpic termsinclude the formation of stabilizing interactions inthe native state via charge interactions and hydrogenbonding. The magnitude of �S and �H varies fromone protein to another but the result appears to belargely compensatory with �G values confined to anarrow range generally between 15 and 40 kJ mol−1.
Folding problem and Levinthal’sparadox
The Ramachandran plot reveals that many dihedralangle combinations are compatible with stable sec-ondary structure with the result that in an averageprotein of 100 residues the number of possible con-formations is enormous. Some idea of the magnitudeof this value is obtained by assuming that only threeconformations exist for each residue in a protein of 100residues and that each conformation can be sampled in10−13 s or 0.1 ps. To sample all conformations would

404 PROTEIN FOLDING IN VIVO AND IN VITRO
Microswitch
Computer
Waste
Valve
drive direction
PneumaticDrive
LightSource
MixingCellMono 1
Loadingsyringes
Mono 2 Detector
Stopsyringe
Figure 11.13 Schematic arrangement of a stopped flow system (lower figure reproduced with permission AppliedPhotophysics Limited)
take 3100 × 10−13 s – an impossibly long period – thatemphasizes that proteins do not sample all possibleconformations.1 This problem commonly known asLevinthal’s paradox has focussed attention on the kinet-ics of protein folding.
Kinetics of protein folding
A consideration of Levinthal’s paradox quickly elimi-nates random models of protein folding but a relevant
1There are far more than three conformations per residue, some pro-teins contain more than 100 residues and 10−13 is an unrealisticallyshort period in which to sample each conformation.
question to ask is what is the time scale of proteinfolding? Experimentally, these studies involve placingunfolded protein into a buffer system that promotesfolding and by mixing rapidly the reaction can befollowed on time scales ranging from 1 ms to ∼10 min.A common method is the stopped flow kinetic tech-nique where reactants combine in a mixing cell ina process followed using changes in fluorescence orabsorbance. In modern instrumentation it is possibleto measure changes in CD signals with time but theseexperiments are technically difficult and less sensitive.In stopped flow experiments (Figure 11.13) pressure-driven syringes force reactants into a mixing cell wherethe reactants mix displacing resident solution within

FOLDING PROBLEM AND LEVINTHAL’S PARADOX 405
a dead time of ∼1 ms. This initiates measurement offolding reactions and kinetic profiles usually followsingle exponential decays that are fitted to obtain theobserved rate constant (kobs) and the maximum ampli-tude (Ai)
kobs = Aie−kt (11.15)
For a two-state unfolding system kinetic analysis isstraightforward since the equilibrium constant Keq issimply the ratio of forward (kf) and reverse (ku)rates defining the folding and unfolding reactions.Combining Equations 11.2 (�GU(H2O) = −RT ln Keq)and 11.14 (�GU = �GU(H2O) − meq[D]) with therelationship
Keq = kfw/kuw (11.16)
allows the dependence of the folding and unfoldingrates on denaturant concentration to be expressed as
ln ku = ln kuw + mu[D] (11.17)
ln kf = ln kfw + mf[D] (11.18)
where D is the molar concentration of denaturant.2
Plotting the logarithm of the rate of folding againstdenaturation concentration yields a linear relationshipthat when combined with the dependence of unfoldingrates on denaturant concentration leads to a characteris-tic profile known as a chevron plot (Figure 11.14). Cm
is defined by the point where ku = kf and the unfold-ing and refolding rates in the absence of denaturantare extrapolated by extending each limb of the plotto zero denaturant. At any point the observed rate iscalculated from
kobs = ku + kf (11.19)
kobs = kuw exp(mu[D]) + kfw exp(mf[D]) (11.20)
wheremeq = (mu − mf)/RT (11.21)
The parameters mu and mf are kinetic m values todistinguish from the equilibrium m value (meq) and areused to estimate the position occupied by the transitionstate along the reaction coordinate from U to F often
2The parameters mku = RT mu and mkf = RT mf and kuw/kfw is therate of unfolding/folding in the absence of denaturant.
−8
−6
−4
−2
0
0 1
Folding region
Cm
Unfolding region
2 3
Denaturant concentration [M]
4 5 6 7
In k
obs
(s−1
) 2
4
6
8
Figure 11.14 A chevron plot describes kinetics ofprotein folding for a simple soluble domain as afunction of denaturant concentration
given the symbol α and equal to mf/(mu + mf). Avalue of 0.5 suggests a transition state midway betweenfolded and unfolded states.
Barnase and CI2 are two proteins that have beenactively studied by Alan Fersht to provide quantitativeinsight into the mechanism of folding. CI2 is a 64-residue inhibitor of chymotrypsin or subtilisin witha structure based around a single α helix fromresidues 12 to 24, and five β strands arranged inparallel and antiparallel sheets (Figure 11.15). The α
helix lies between strands 2 and 3 and CI2 formsa single folding unit that exhibits two state kineticsand a t1/2 of 13 ms at room temperature for themajor phase of protein folding. Smaller contributionsattributed to cis– trans peptidyl proline isomerizationare observed but remain a minor element in the overallfolding pathway that lacks intermediates in kineticor equilibrium-based measurements. CI2 representsthe simplest kinetic pathway for folding and onewidely observed to occur in other small solubledomains.
Barnase is a small, monomeric, soluble, extracel-lular ribonuclease secreted by B. amyloliquefacienscontaining 110 residues and lacking disulfide bonds.The protein exhibits reversible unfolding under awide variety of denaturing conditions. The structure(Figure 11.16) shows a five-stranded β sheet with threehelices (α1 –α3) located between loops and outside of

406 PROTEIN FOLDING IN VIVO AND IN VITRO
Figure 11.15 The structure of CI2 (PDB: 2CI2). Somestrands are poorly defined
L3
L1
L2
L4
β2
β3
β4
α1
α3
α2
β1
Figure 11.16 The structure of barnase showingdistribution of secondary structural elements. L1–L4are loop regions whilst β strands extend from residues50–55, 70–76, 85–91, 94–99 and 106–108
the sheet region. The major helix (α1) is located at theN-terminal and extends from residues 6–18 where itsinteraction with the β sheet forms the major hydropho-bic core. A smaller core is formed from the interactionsbetween loop2, α2 and α3 helices and the first β strand.
As a system for studying folding and stabilitybarnase represents an excellent paradigm. With the α
and β structures in different regions of the molecule,
as opposed to a more mixed arrangement, it ispossible to study either fragments of the proteinor individual mutations confined to one secondarystructural element. To understand protein folding, andin particular the pathway involved, it is necessary todetermine the structure and energetics of the initialunfolded state, all intermediate states, the final foldedstates and importantly the transition states occurringalong this pathway.
One approach that has shed considerable light onthe mechanism of protein folding as well as thenon-covalent interactions involved in these pathwaysintroduced small mutational changes to barnase thatremoved specific interactions. By measuring changesin stability (i.e. �G) alongside kinetic measurementsestimating the activation energy (�G‡) and other prop-erties associated with the transition state it is possi-ble to begin to describe the energetic consequencesof mutation.
The difference in conformational stability betweenthe folded states of the wild type and mutant formsis difficult to measure directly but can be exploitedvia relationships that invoke Hess’ law. The basisof the method is a mutational cycle (Figure 11.17)in which the introduction of specific mutations intobarnase is used to estimate kinetic and thermodynamicparameters. The mutation acts as a sensitive reporterof local events during the pathway of protein folding.The mutational cycle measures the free energy of
N
NM UM
U
∆GN ∆GU
∆GM
∆G
Figure 11.17 Application of Hess’s law to theunfolding free energy of wild type and mutant proteins

FOLDING PROBLEM AND LEVINTHAL’S PARADOX 407
unfolding for wild type (N) and mutant (M) proteinsin the horizontal reactions whilst the vertical reactionsrepresent experimentally indeterminable or ‘virtual’free energy changes. They are the free energy changesthat arise in the folded or unfolded protein as a resultof mutation.
Hess’s Law states that the overall enthalpy changefor a chemical reaction is independent of the route bywhich the reaction takes place as long as the initialand final states are identical (Figure 11.17). It is simplya statement of the law of energy conservation. Usingthe mutational cycle approach it was shown that theenergetic effects of mutation in barnase are evaluatedfrom the relationship
�G + �GU = �GN + �GM (11.22)
Re-arranging leads to
�G − �GM = �GN − �GU (11.23)
where ��GN−U is the change in free energy due tomutation (M) and the experimental quantity one wouldwish to access directly.
Mutations do not have a single effect on one‘folding’ parameter but can alter solvent exposure,hydrophobic contacts, hydrogen bonding – the list isalmost endless. As a result of the large number ofparameters altered by a mutation it is very difficultto ascribe the changes to a single energetic parameter.One way to eliminate this problem is to borrow a trickfrom physical organic chemistry and use the Brønstedequation. The Brønsted equation relates changes inrate constant k and equilibrium constant K to aparameter β as a consequence of altering a non-reactingfunctional group
log k = constant + β log K (11.24)
In physical organic chemistry this involves makingseries of derivatives whilst in the area of protein foldingthis involves the creation of mutants. In each case theeffect on rate and equilibrium constants for foldingare measured. For protein folding this equation wasmodified to
�G‡ = constant + φU�GU (11.25)
in view of the relationship of free energy terms �G‡
and �GU to log k and log K respectively whilst the
parameter φ replaced β from the Brønsted relation.This scheme allows two measurable reactions to becompared since the constant can be cancelled bysubtraction to give
�G‡U − �G‡
N = constant + φU �GU
− constant + φU �GN (11.26)
��G‡ = φU ��GU (11.27)
φU = ��G‡/��GU (11.28)
where ��GU is the difference in conformationalstability between folded and unfolded protein. A valueof φ of 0 (measured from the unfolded state and in thedirection of folding) implies that the structure at thesite of mutation in the transition state is comparableto the structure in the unfolded state. Alternatively aφ value of 1 suggests that the structure in the vicinityof the mutation in the transition state is as folded asthat found in the native structure. Fractional values ofφ imply a mixture of states.
Kinetic analysis of the refolding of barnase was con-sistent with one major intermediate and a linear schemeU ⇀↽ I ⇀↽ N, where the rate-limiting step was conver-sion of I to N. The t1/2 for the observable step ofrefolding was ∼30 ms, although the kinetics of the steppreceding I occur within the dead time of stopped flowstudies. Since �GU and the forward and reverse rateswere measured a partial energy diagram for barnasefolding was constructed where kinetic and thermody-namic analysis exposed the properties of the transitionstate and major, late folding, intermediate in wild typeand mutant forms of the protein (Figure 11.18).
Residues mutated at the C-terminal end of the α1
helix show values of φ of ∼1.0 for the transitionand folded states. This implies that this region of thehelix-ordered structure is formed rapidly. Interestingly,residues such as Thr6 at the beginning of the helixshow intermediate values suggesting disorder is presentbefore formation of native structure. In a similarfashion residues in loop 3 rapidly form structurecomparable to the folded state whilst loops 1,2 and4 remain disordered throughout the folding processuntil the later stages. Residues at the centre of theβ sheet form rapidly but those located at the edgesdo not fold until later. More significantly, residues

408 PROTEIN FOLDING IN VIVO AND IN VITRO
Figure 11.18 Partial free energy diagram describing the folding of wild type and mutant forms of barnase.Although not strictly necessary for simplicity the unfolded forms of the wild type and mutant barnase are shown asiso-energetic. The remaining energy terms describe the differences between the intermediate, transition state andfolded forms of wild type and mutant barnase
1.0
0.8
0.6
0.4
0.2
0
1.0
0.8
0.6
0.4
0.2
0
Unfolded Intermediate Transition Folded Unfolded Intermediate Transition Folded
1.0
0.8
0.6
0.4
0.2
0
Unfolded Intermediate Transition Folded
Residues in centre of β sheet
Figure 11.19 φ-values found for mutation of residues in the loop, β sheet and helix regions of barnase
located in the core region between the helix and sheetshowed φ values dependent on location. Residues atthe centre of the core exhibit φ values close to 1 in theintermediate and subsequent states whilst those locatedat the periphery of this core showed a gradation ofvalues (Figure 11.19). In this fashion an extraordinarydetailed picture of the pathway and energetics ofprotein folding and its sensitivity to mutation wasestablished for barnase.
Models of protein folding
Exceptionally rates of protein folding are observed withtime constants less than 5 ms but most domains withless than 100 residues fold over slower time scalesranging from 10 ms to 1 s. In most small domainsfolding is a cooperative process but for a smaller groupof proteins folding is characterized by the transientpopulation of detectable intermediates. These proteins

MODELS OF PROTEIN FOLDING 409
offer the possibility of characterizing partially foldedstates or sub-domains. Initial studies of protein foldingfocused extensively on helical domains or domainscontaining mixtures of helix and strand (α/β proteins).Gradually, it has been shown that small β sheet proteinsalso fold in a highly cooperative manner whilst larger β
sheet domains especially those with complex topologyfrequently exhibit intermediates during folding. Thus,models of protein folding must account for processesspread over a wide range of time scales as well as thepresence in some cases of intermediates or partiallyfolded structures.
Helix formationThe formation of segments of helical structure occurswith time constants around 1 µs. The reactions proceedwith a slow initial stage as the polypeptide chainstruggles to form hydrogen bonds between donor andacceptor separated by three or four residues in adisordered chain. This is followed by faster rates thatreflect progressively rapid extension of helices from anordered structure sometimes called a nucleation site.Most experiments on helix formation have been derivedusing synthetic homopolypeptides and a major problemis that whilst considerable data has been accumulatedfor helical regions the formation of β structure relieson more than one element of secondary structure forstabilization and there is no satisfactory model system.
Although helices have regular repeating hydrogenbonds coupled with a uniformity of bond lengths andangles this periodicity masks their marginal stability.In an isolated state most helices will unfold and onlysynthetic polyalanine helices are reasonably describedas stable. Initiation of helix formation is a slow andunfavourable process. This arises because five residuesmust be precisely positioned to define the first hydrogenbond between residues 1 and 5. Hydrogen bondsbetween residue i, i + 4 characterize regular α heliceswhilst the intervening three residues must be arrangedto form a turn as well as arranged in a geometryfavouring the formation of future hydrogen bonds.There is a large entropic penalty in forming the firstturn whilst addition of a subsequent residue to the helixis far less severe in terms of energetic cost. At the sametime dipoles associated with the peptide bond in a helixinfluence the energetics since for the first turn theiralignment lies in the same direction (parallel) and is
an unfavourable arrangement. With the completion ofthe first turn subsequent formation of peptide dipolesseparated by 3 to 4 residues leads to a head to tailarrangement that may assist helix propagation.
The molten globule
During studies of protein folding it was recognized thatintermediate states showing many of the properties ofthe native fold could be identified. This state becameknown as the molten globule and is characterizedby a compact structure, containing most elementsof secondary structure as helices, turns and strands,although many long range or tertiary contacts arelacking. The molten globule has a hydrophobic coreconsistent with burial of non-polar side chains awayfrom the solvent.
Studies of α-lactalbumin using circular dichro-ism (CD) under denaturing low pH (<4) conditionsrevealed formation of a stable intermediate state thatwas distinguishable from the native and unfolded statesby possessing far-UV CD spectra resembling the nativefold but an unfolded-like near-UV CD spectrum. Inother words it appeared to possess considerable sec-ondary structure but no tertiary structure. This state wasgiven the name molten globule and similar forms havebeen identified during the folding of other proteins.Proteins showing molten globule-like states includemany familiar soluble proteins such as myoglobin, α-lactalbumin, β-lactoglobulin, BPTI, cytochrome c andazurin. These proteins differ considerably in their sec-ondary structure content as well as tertiary folds. Incontrast, some proteins fold without evidence of moltenglobule intermediates, and lysozyme is one interestingexample, especially since lysozyme and α-lactalbuminare homologous proteins.
Although the molten globule state was identified byits stability and presence at equilibrium under partiallydenaturing conditions similar states have been detectedin kinetic experiments. In particular stopped-flow CDstudies appear to show kinetic intermediates populatedduring folding possessing high levels of secondarystructure yet lacking complete side chain packing andextensive tertiary structure. These studies have led tothe suggestion that molten-globule like intermediatesform as part of the overall process of reaching thenative state and have provided a picture in which the

410 PROTEIN FOLDING IN VIVO AND IN VITRO
formation of elements of secondary structure occursprior to consolidation of the tertiary fold.
Hydrophobic interactions
Hydrophobic interactions are intrinsically weak forcesbetween non-polar side chains that lead to dramaticordering of water. In globular proteins these inter-actions lead to charged/polar side chains residing onthe surface of a protein whilst non-polar side chainsare buried on the inside of the molecule. The flu-orescence probe anthraquinone naphthalene sulfonate(ANS) binds effectively to exposed hydrophobic sur-faces and exhibits a characteristic λmax that changeswhen the probe is placed in a polar environment. Bymeasuring changes in fluorescence of ANS during pro-tein folding in a rapid mixing experiment it is possibleto monitor the formation of hydrophobic cores. Frommany studies of folding involving proteins with dif-ferent topologies it has been recognized that differentevents can occur during the formation of the nativestate. These events include some or all of the follow-ing reactions:
1. Formation of all native contacts in a highlycooperative transition;
2. Folding via a molten globule intermediate;
3. Folding via kinetic intermediates resembling moltenglobules;
4. Condensation of hydrophobic cores prior to forma-tion of secondary and tertiary structure.
A major problem for experimentalists and theoreticianshas been to reconcile all of these different views withina single scheme of protein folding allowing formationof the folded state on time scales between 10 ms and1 s.
A hierarchic model has been proposed where struc-ture is determined by local interactions with confor-mational preferences among short sections of peptidechain guiding the polypeptide towards forming largerunits of secondary structure. An obvious weaknesswith this model is that few short peptides in iso-lation form helices and none will form β strands.Although at first these elements of secondary structure
are only metastable it is envisaged that interactionswith neighbouring units reinforce stability. Early stagesof protein folding are characterized by the interac-tion of secondary structure via a purely diffusionalmodel in which elements collide to allowing favourableinteractions and stable association. These interactionsaccumulate cooperatively to form larger collections ofsecondary structure that can acquire stabilizing tertiarystructure and long-range order at longer time intervals.The detection of intermediates or transition states con-taining secondary but not tertiary structure is perhapssupportive of a hierarchic model whilst the fact thatsecondary structure can be predicated from sequenceis also used as a supportive line of evidence.
Recently, considerable attention has focused on aconcept called contact order. Although the length of aprotein does not show a simple correlation with ratesof protein folding the topology of a protein does seemto influence this process. To reflect the concept oftopology the term contact order (CO) can be definedas the average sequence distance between all pairs ofcontacting residues normalized by the total sequencelength of the protein and is described as
CO = 1
LN
N∑�Si,j (11.29)
where N is the total number of contacts, �Si,j isthe sequence separation in residues between contactingresidues i and j , and L is the total number ofresidues within the protein. Providing the structure ofa protein has been determined at a reasonable levelof resolution the CO can be calculated. A correlationexists between CO and observed rates of proteinfolding. This leads, for example, to proteins that areextensively α helical having low CO and folding morequickly than proteins containing higher proportions ofβ strand. These proteins are characterized by higherCO. The observation that proteins with low CO tendto fold rapidly could suggest that local contacts arethe most effective route towards rapidly reaching thenative state.
Irrespective of the precise details during the earlyevents of protein folding a common objective achievedby all schemes is to restrict the conformational spacesampled by the protein and therefore to avoid theworst excesses of Levinthal’s paradox. Restricting

AMIDE EXCHANGE AND MEASUREMENT OF PROTEIN FOLDING 411
A
B
N
Figure 11.20 The route towards the folded con-formation for protein A and B differ. Real energylandscapes are undoubtedly much more complex inprofiles
the number of conformations leads to remarkablyrapid folding. A new picture of protein foldingrepresented by contoured energy landscapes directsthe unfolded protein towards the final (low energy)conformation by a ‘funnel’ (Figure 11.20). A three-dimensional plot suggests several routes may be usedin reaching the native fold with local energy minimaexisting along pathways. The shape of the funneldirects all conformations towards the native stateyet avoids the need to sample all of the possibleconformations.
A new view of protein folding is one of energy land-scapes, multiple pathways and multiple intermediateswhere folding leads towards the most stable state in aprocess influenced by kinetic events.
Amide exchange and measurementof protein folding
The measurement of amide exchange rates is apowerful way of assessing the protection (resistance
−1
1
1 3 5 7 9 11
pH
3
5
Gua(R)
eNH+3 (k)
aNH+3
NH(bb)
SH(C)
COOH
OH(S,T)
OH(y)
Im(H)CONH2(N,Q)
Indole(w)
7
9
log
k int
r (m
in−1
)Figure 11.21 Rates of amide exchange for differentfunctional groups in proteins. Backbone amide groupsshow minimum exchange at pH 3.5 with time constantsof ∼1 h whilst at pH 10 the rate is increased leadingto exchange on the ms time scale
to exchange) of these groups as a protein folds fromdenatured to native states. Protection occurs becausethe amide is involved a hydrogen bond or is buriedwithin the hydrophobic interior of a protein. In NMRexperiments based around the observation of a protonthe substitution of H by 2H (D) leads to a loss ofa signal. Exchange rates are dominated by pH andproteins show profiles where the rate has a minimumaround pH 3.5 but increases either side of thisminimum due to acid and base catalysis (Figure 11.21).The intrinsic rate of exchange (kex or kintr) reflects asummation of acid- and base-catalysed rates and differsaccording to the type of functional group.
kintr = kOH[OH−] + kH[H+] + kw (11.30)
The rates for each of the functional groups foundin proteins have different pH optima and are stronglyinfluenced by pH, local chemical environment, solvent,side chain identity, neighbouring residues and temper-ature. In proteins NH exchange can be extremely slow

412 PROTEIN FOLDING IN VIVO AND IN VITRO
and may take months or years to reach completion. Aprotection factor is described via a parameter, θp, where
θp = kintrinsic/kobs (11.31)
and a slow rate of exchange is seen by values of θp of106 –107.
A theory of amide exchange in proteins developedby Kai Linderstrom-Lang involves open conformationswhere the protein is exposed to the solvent inequilibrium with a closed structure.
closedkop−−−⇀↽−−−kclo
openkintrinsic
−→ hydrogen exchange
The closed state is often equated with folded moleculesand the open state with denatured proteins. Protectedamide hydrogens are ‘closed’ to exchange but becomeaccessible to exchange through formation of the ‘open’state at rates comparable to those observed for unstruc-tured peptides. Two kinetic regimes are recognizedfor hydrogen exchange in the above scheme. The firstregime is called the EX1 mechanism and occurs whenkintrinsic � kclo. The observed rate is therefore approx-imated by the rate of protein ‘opening’ such thatkobs = kop. In contrast the more common mechanismfound in proteins occurs when kintrinsic � kclo and theobserved rate (kobs) is therefore equivalent to the prod-uct kopkintrinsic. This is called the EX2 mechanism andoccurs when a protein is relatively stable and exchangerates are comparatively slow.
In proteins the most important exchangeable protonis attached to the secondary amide of the polypeptidebackbone. Amide exchange rates in proteins are readilymeasured in protein uniformly enriched in 15N in a‘pulse hydrogen’ exchange experiment. The principleof the experiment involves transferring a protein fromwater-supported buffers to those based on 2H2O (D2O)and leads to a loss of cross-peak intensity in 2D NMRspectra as a result of proton/deuteron (H/D) exchange(Figure 11.22). By observing the rate of change inthe intensity of cross peaks amide exchange ratesare estimated.
Denatured protein in 6 M guanidinium HCl insolutions of 2H2O is allowed to fold in solutionscontaining 2H2O and leads to protein ‘labelled’ withdeuterium. Placing the samples at high pH favoursrapid exchange for accessible amides and is followed
by ‘quenching’ and a lowering of the solution pH.Folding is allowed to continue to completion and themethod samples amides that are rapidly ‘removed’from exchange reactions by forming hydrogen bonds,or more probably from burial on the inside of theprotein. After folding has been completed the patternof NH and ND labels is analysed by 2D NMRspectroscopy (Figure 11.23). Increasing the refoldingtime (tf) allows a greater number of exchangeable sitesto be protected. Using this approach it was possible tosample the protection of over 50 different amide groupsin the enzyme lysozyme. The results showed twogroups of amides existed; approximately 50 percentwere completely protected from exchange within about200 ms whilst the remaining group remained accessibleeven after 1 s. Further analysis of rapidly protectedamides revealed a location within a domain basedaround four α helices whilst the more accessible amideswere contained in the β sheet rich domain.
This picture of amide protection provides a viewof protein folding in which each domain of lysozymeacts as a separate folding unit (Figure 11.24). Studieswith other proteins reinforced this view and domainsare often defined as ‘folding units’. It might be arguedthat the rates of protection simply reflect the units ofsecondary structure but working against this idea isthe observation that 310 helices found in each domainshowed different rates of protection. The differenttechnique used in the study of the kinetics of proteinfolding are listed in Table 11.3.
Kinetic barriers to refolding
Kinetic studies highlight the presence of transientintermediates during folding that represent partiallyfolded forms along the pathway between denaturedand folded states (Figure 11.25). In ‘off’ pathwayintermediates it may be necessary to unfold beforecompleting a folding pathway. When this type ofreaction is performed in vitro the rate of reaction isslow. In vivo a different picture emerges with specificproteins catalysing the elimination of ‘unwanted’ folds.
Additional barriers to efficient protein folding exist.One major kinetic barrier to protein folding involvescis– trans isomerization of the amide bond preceding

KINETIC BARRIERS TO REFOLDING 413
Figure 11.22 The principal of measuring H/D exchange by NMR spectroscopy. Initial cross-peaks are shown inred. Those exhibiting altered intensities are shown in orange and eventually exchange leads to a complete lossof intensity
Table 11.3 Techniques used to study kinetics of protein folding
Method Structural information
Intrinsic fluorescence Primarily environment of Trp and Tyr residues. Can also include environment of
co-factors such as heme or flavin
Absorbance Environment of aromatic groups or other conjugated systems
Near UV CD Asymmetry of aromatic residues within tertiary structure
Far UV CD Formation of secondary structure
H/D exchange NMR Formation of persistent hydrogen bonds or amide groups protected from solvent
H/D exchange MS Detection of folding intermediates and different populations
FTIR Formation of hydrogen bonds
ANS fluorescence Accessibility of hydrophobic residues

414 PROTEIN FOLDING IN VIVO AND IN VITRO
Figure 11.23 The principle of pulsed H/D exchange in a protein during folding
proline residues. Proline has unique properties amongthe 20 naturally occurring building blocks of proteinsstemming from the cyclic side chain and the absence ofan amide proton (NH). The peptide bond immediatelypreceding proline is unlike other peptide bonds in thatthe ratio of cis– trans isomers is ∼4. Unfolding removesmany of the constraints placed on peptide bonds andleads to randomization of prolyl peptide bonds so thatin a protein containing five Pro residues it is expectedthat on average one prolyl peptide bond will exist inthe less favourable cis conformation. The potential forproline cis– trans isomerization to inhibit the rate ofprotein folding arose from studies by Robert Baldwinshowing that kinetically heterogeneous mixtures ofmolecules existed in refolding denatured ribonuclease.An explanation for this observation provided by John
Brandts centred around the cis– trans isomeric state ofprolyl peptide bonds. The refolding of a proportion ofprotein with prolyl peptide bonds in an unfavourableconformation is characterized by slow rates of formationof the native state superimposed on normal rates offolding. Kinetic heterogeneity is observed and arisesbecause molecules with bonds arranged favourablyfold rapidly whilst a fraction with unfavourable cispeptide bonds will fold more slowly. As a resultprotein refolding involving cis– trans isomerization ischaracterized by one or more slow refolding phases.
This view has been supported through the useof model peptides containing proline residues andthe observation of slow refolding rates in proteinsincluding insulin, tryptophan synthase, T4 lysozyme,staphylococcal nuclease, chymotrypsin inhibitor CI2,

IN VIVO PROTEIN FOLDING 415
Figure 11.24 Structure of lysozyme showing the rapidly formed helix rich domain (blue) along with the slowly‘protected’ sheet-rich region (green)
X1 X2
U I1 I2 F
Figure 11.25 On and off pathway intermediatesduring protein folding. I1 and I2 represent on pathwayintermediates such as the molten globule like statewhilst X1 and X2 represent off pathway forms ofthe protein
barnase, yeast iso-1 and iso-2 cytochromes c, pepsino-gen, thioredoxin as well as calcium binding pro-teins such as parvalbumin and calbindin. One directobservation is the refolding of three homologousparvalbumins (Ca binding proteins found in muscle).Two of the parvalbumins contain a single prolineresidue and exhibit refolding reactions with a slowcomponent whilst this phase is absent in a third par-valbumin lacking proline.
Cis– trans proline isomerization is one of the majorcauses of kinetic heterogeneity in protein refolding anddeviations away from a purely two-state system. Thepresence of such effects during refolding are normally
confirmed by site directed mutagenesis, by kineticanalysis using ‘double mixing’ stopped flow techniquesand from analysis of the temperature dependencyassociated with each folding reaction. Those associatedwith cis–trans isomerization of Xaa–Pro peptide bondsare characterized by activation energies ranging 80 to100 kJ/mol.
In vivo protein folding
Proteins fold to form the native state in vitro butdo similar pathways or mechanisms apply in vivo?Translation occurs at rates of ∼20 residues per second,yet many small soluble proteins fold completely within100 ms. The disparity between optimal translation ratesand folding suggests that in vivo limitations exist in thelatter process.
Peptidyl prolyl isomerases (PPI) catalyse one oftwo important but slow reactions associated with fold-ing. The reaction is cis–trans isomerization of prolylpeptide bonds. The first enzyme was isolated frompig kidney in 1984 but during this period other stud-ies looking at the binding of the immunosuppres-sant drug cyclosporin A (a vital immunosuppressantduring transplant surgery preventing the rejection of

416 PROTEIN FOLDING IN VIVO AND IN VITRO
donor organs) discovered its intracellular target andnamed this protein cyclophilin. Subsequent proteinsequencing showed that PPI isolated from pig kidneyand cyclophilin were identical. The precise basis forcyclosporin A binding to PPIs remains to be establishedbut it may be unrelated to the activity of the immuno-suppressant in decreasing T-cell proliferation and activ-ity. Peptidyl prolyl isomerase overcomes kinetic bar-riers presented by incorrectly oriented peptide bondsduring protein folding.
Additional prolyl isomerases exist within cells andare grouped within one of at least three major proteinfamilies. Besides cyclophilins FK506-binding proteinsand parvulins represent different families that are nothomologous but share a common biochemical function.A plethora of PPIs exist in the human genome withat the last count 11 cyclophilins, 18 FK506 bindingproteins and two parvulins encoded.
The second ‘slow’ reaction of protein folding is theformation of disulfide bonds. This reaction is catalysedby the enzyme protein disulfide isomerase (PDI). Sinceboth reactions (cis–trans proline isomerization anddisulfide bond formation) were shown to inhibit rapidprotein folding in vitro the presence of enzymes withincells to specifically catalyse these ‘slow’ reactions iscompelling evidence that similar mechanisms exist forfolding in vitro and in vivo. Protein disulfide isomerasecontains the conserved active site motif of Cys-Xaa-Xaa-Cys found in thioredoxin and shares a similarrole to the Dsb family of proteins found in E. coli.Together with molecular chaperones these systemsallow proteins to fold effectively in the cell.
Chaperones
The fundamental experiments of Anfinsen showed thatdenatured ribonuclease folds in vitro with the pri-mary sequence directing folding to the native state.It was widely assumed that folding of all newlysynthesized proteins in vivo would proceed similarly.Nascent polypeptide chains would not require addi-tional assistance to fold efficiently. With hindsight thisassumption seems foolish, and gradually observationsestablished that specialized proteins within the cytosolassist formation of native states.
One of the first observations was that E. colicontaining a defective operon called groE could notassemble wild type bacteriophage λ. This was despitethe fact that all protein components of mature λ wereencoded by the viral genome. A second observationwas that in the mitochondrial matrix translocatedpolypeptide associated with a protein (Hsp60). Thename reflected its mass and that production increasedapproximately two-fold after heat shock. The name ismisleading because experiments showed that deletionof the Hsp60 gene in yeast was lethal and the proteinwas essential to the cell under all growth conditions.This is expected for a protein playing an important rolein the folding of mitochondrial proteins.
The heat-shock or cell-stress response (i.e. changesin the expression of intracellular proteins) is a commonevent, with increased protein production an essentialsurvival strategy that allows responses to diverse stim-uli, including heat or cold, osmotic imbalance, toxinsand heavy metals as well as pathophysiological signals.The proteins synthesized in response to such envi-ronmental stresses are collectively called heat-shockproteins (or HSPs), stress proteins or, more commonlytoday, molecular chaperones. The response of cellsto stress is a primitive mechanism that appears tobe evolutionarily ancient as well as essential to sur-vival. The ancient nature is demonstrated by significantsequence homology in molecular chaperones from awide range of organisms. Systematic work over the last20 years has identified many proteins with chaperone-like activity. Chaperonins refer to a sub-group of theseproteins that function as components of multimericsystems.
Many newly synthesized proteins reach their foldedstates in vivo spontaneously and without assistance,in processes analogous to the folding of ribonucleasein vitro. However, folding efficiency may be limitedby side-reactions such as aggregation (Figure 11.26)promoted by transiently exposed hydrophobic surfaces.In E. coli aggregation of proteins is readily detectedby SDS–PAGE after heat shock. Cells respond toheat shock and the production of significant amountsof unfolded protein by the synthesis of new systemsdesigned to promote refolding. These systems are themolecular chaperones.

IN VIVO PROTEIN FOLDING 417
Aggregates
U I N
Figure 11.26 Possible routes of aggregation forunfolded and partially-folded protein. Chaperones limitthe extent of aggregation reactions by binding to Uand I forms
Several molecular chaperone systems have beencharacterized in E. coli including the GroES-GroEL,DnaK-DnaJ-GrpE and ClpB molecular chaperone sys-tems. The acronym Hsp (heat-shock protein) is stillused to describe these chaperones and the literatureis full of terms such as Hsp60, Hsp70 and Hsp100for families of homologous proteins. Today theseterms are used interchangeably and the Hsp60 namecould refer to GroEL – one component of the best-characterized chaperone.
Molecular chaperones are distributed in all cellsranging from archaebacteria through to complexeukaryotic cells. Although many chaperone systemshave not been structurally characterized the GroEL-ES, the thermosome of Thermoplasma acidophilumand small HSPs from Methanococcus jannaschii rep-resent exceptions.
The GroEL-ES system
The GroEL-ES chaperone system is essential for E.coli growth under all conditions with mutations in theGroE operon proving lethal. The GroEL-ES complexof E. coli has formed the basis of understandingchaperone function as a result of detailed structuralstudies using crystallography and electron microscopy.In vivo GroES is composed of seven identical subunits(Mr∼10 000) whilst GroEL is composed of 14 largersubunits each with a mass of ∼60 000. This leads toGro-EL being called chaperonin60 (cpn60) and Gro-ESchaperonin10 (cpn10).
A model for the organization of GroEL obtainedfrom negative staining EM studies showed a doublering cylindrical structure with a diameter of ∼14 nmand height ∼16 nm. The seven-fold symmetrical com-plexes were of comparable size to the ribosome with acentral cavity of ∼6 nm that bound polypeptide priorto ATP binding or GroES attachment. The first detailedstructure for GroEL was determined by Paul Siglerand provided beautiful detail of the toroidal architec-ture indicated from electron microscopy. GroEL hada substantial central cavity in a cylindrical structurecomposed of two stacked heptameric rings arrangedwith seven-fold rotational symmetry. The rings arearranged back to back, contacting each other through aregion known as the equatorial interface that is one ofthree distinct domains found in each GroEL subunit.The remaining domains are the apical and intermediatedomains; these zones are simply designated as E, I andA domains (Figure 11.27).
GroEL contains a single polypeptide chain of 550residues starting at the E domain extending to theintermediate region before the central part of thesequence makes up the A domain. The sequence thenreturns to form part of the I domain before terminatingin the E domain. The E domain is a helical rich regionthat provides the ATP/ADP binding site whilst theapical domain shows greater mobility and has a lowerpercentage of regular secondary structure. Schematicallythe GroEL subunit is a tripartite structure (Figure 11.28).
The structure of GroES was determined indepen-dently of GroEL to confirm seven-fold symmetryarranged in a dome-shaped architecture. The domecontained seven subunits each of 110 residues andformed a core β barrel structure with two prominentloop structures (Figure 11.29).
One remarkable feature of the GroEL structure isa massive conformational change caused by bindingco-chaperonin (GroES) and nucleotides. Revealed bycryo-EM studies of the ternary complex formedbetween GroEL/ADP/GroES image reconstructionsshowed a small elongation of the cylinder upon ATPbinding but a very substantial elongation at one endof the GroEL complex upon GroES binding. Thiswas accompanied by increases in diameter at theend cavities with the GroES ring observed as a

418 PROTEIN FOLDING IN VIVO AND IN VITRO
Figure 11.27 The monomeric structure of GroELshowing relative positions of E, I and A domains(PDB: 1GRL)
E
Figure 11.28 The tripartite structure of a GroELsubunit showing a schematic arrangement of apical(A), intermediate (I) and equatorial domains
GroEL GroEL-ATP GroEL-GroES-ATP
Figure 11.30 3D reconstructions of the surfaces ofGroEL, GroEL-ATP and GroEL-GroES-ATP from cryo-EM.The GroES ring is seen as a disc above the GroELand contributes to the bullet-like shape. (Reproducedwith permission from Chen, S. et al. Nature 1994, 371,261–264. Macmillan)
flat, disc-like object above the neighbouring GroELheptamer (Figure 11.30). In the presence of adeninenucleotides GroES binds to the stacked rings ofGroEL forming a ‘bullet-like’ asymmetric structurewith GroES attached to only one of the heptamericrings of GroEL (the cis ring).
The chaperonin-assisted catalysis of protein foldingproceeded through cycles of ATP binding and hydrol-ysis. Nucleotide binding modulates the interactionbetween GroEL and GroES binding. ATP binding toGroEL occurs initially with low affinity (Kd ∼ 4 mM)
Figure 11.29 The structure of GroES within the heptameric ring seen in side and superior views. One subunit withinthe ring is shown in red. The arrangement of secondary structure showing the β barrel core is shown from the sidein the centre for an individual subunit (PDB: 1G31)

IN VIVO PROTEIN FOLDING 419
to one subunit but as a result of positive coopera-tivity binding occurs with substantially higher affinityto the remaining subunits (Kd∼10 µM). The cooper-ative nature of ATP binding establishes GroEL as anallosteric enzyme. In GroEL with a total of 14 sub-units in two heptameric rings it is observed that tightnucleotide binding occurs only to one of the two avail-able rings. ATP binding to the remaining ring is inhib-ited but promotes GroES association and a change inconformation for the cis ring subunits. The change inconformation is central to the switch in binding modesfrom an allosteric T state with low affinity for ATPand high affinity for non-native chains to an R statethat shows the opposite trends.
The molecular basis for some of the conformationalchanges have been understood by comparing thestructures of Gro-EL complexes in the presence ofGro-ES as well as ADP, ATP and AMP-PNP, usingcryo-EM methods. Although the resolution of thesestudies was relatively low by crystallographic standardsthe large size of the complex meant that levels ofresolution were sufficient to identify key catalyticfeatures. Nucleotide binding in the equatorial domainpocket leads to substantial changes in the GroELheptameric ring structures and movement in particularfor the apical domain. The result of domain movementis the existence of several distinct structures.
Crystallographic analysis of the GroEL-GroES-(ADP)7 complex showed a similar bullet shape and bycomparing this structure with the un-liganded GroELstructure the significant conformational perturbationswere shown to arise from ‘en bloc’ movements ofI and A domains with respect to the E domainin the cis ring capped by GroES (Figure 11.31).The overall architecture of GroEL and the GroEL-GroES-(ADP)7 complex emphasized the bullet shape
E
Figure 11.31 Domain movement in GroEL as a resultof GroES and ATP binding
(Figure 11.32) and allowed insight into the mechanismof binding partially folded proteins and preventingprotein aggregation.
The reorganization of the cis ring of GroEL resultsfrom domain re-arrangement involving intermediateand apical domains with the I domain swingingdownwards towards the E domain by approximately25◦. One effect of this movement is to close theoccupied nucleotide (ATP) binding site located onthe top inner surface of the equatorial domain. TheA domain shows greater movements, swinging ∼60◦
upwards relative to the equator, but also twisting aboutits long axis by ∼90◦ to form new interfaces withneighbouring A domains. These movements lead tointeractions between the A domain of GroEL andmobile loops on GroES. In comparison with theA domain the cis equatorial (E) domains do notshow large conformational shifts, with smaller inwardmovement of the cis assembly by ∼4◦. Since theseregions interact with the neighbouring heptameric ringthere is a complementary outwards tilt in the Edomains of the trans ring. These movements haveimportant functional consequences in the overall cycleof catalysis. In contrast to the large conformationalchanges occurring in GroEL, the structure of the GroESring within the complex is similar to that observed inthe standalone structure. A minor exception to this rulewould include mobile and disordered loops that becomestructured within the complex as a result of interactionwith the A domain.
The chaperonin catalytic cycle
With the structure of the ‘inactive’ GroEL and ‘active’GroEL-GroES-ATP complexes known it was possibleto attempt to understand how this macromolecularcomplex assists in protein folding. At the simplestlevel the complex catalyses cyclic binding and releaseof target polypeptides. This process is divided intofour distinct stages or phases (Figure 11.33). PhaseI involves polypeptide binding; phase II the releaseof the polypeptide into the central channel and theinitiation of folding; phase III involves hydrolysis ofATP, reorganization of the cis heptameric ring andthe start of product (GroES, folded peptide and ADP)release; finally, phase IV involves ATP binding toGroEL subunits of the trans ring providing the trigger
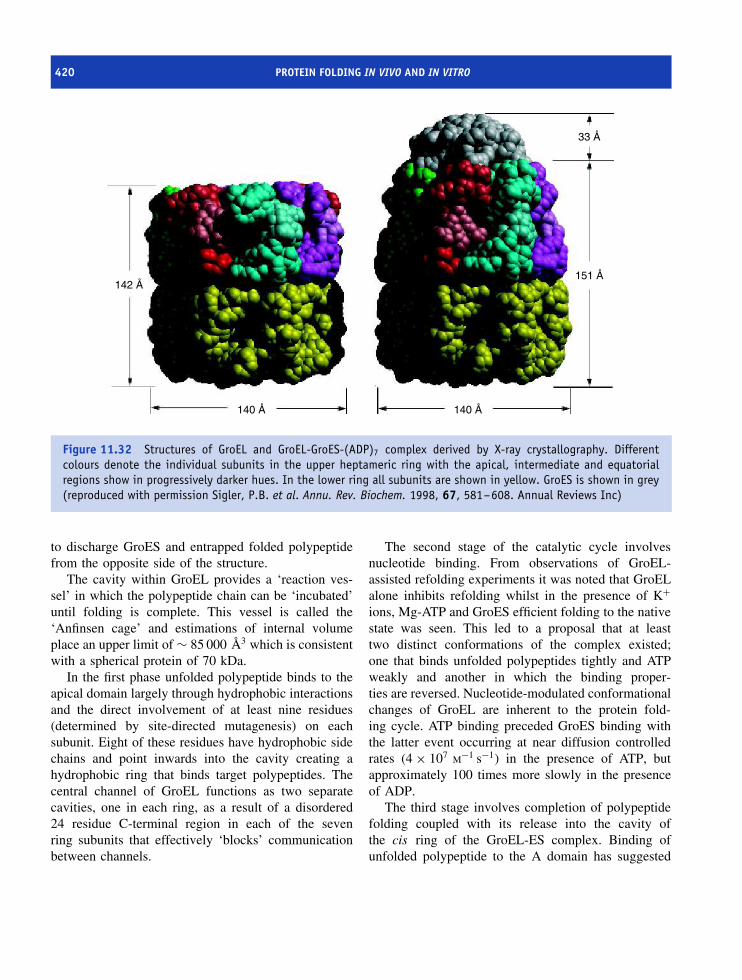
420 PROTEIN FOLDING IN VIVO AND IN VITRO
142 Å
140 Å 140 Å
151 Å
33 Å
Figure 11.32 Structures of GroEL and GroEL-GroES-(ADP)7 complex derived by X-ray crystallography. Differentcolours denote the individual subunits in the upper heptameric ring with the apical, intermediate and equatorialregions show in progressively darker hues. In the lower ring all subunits are shown in yellow. GroES is shown in grey(reproduced with permission Sigler, P.B. et al. Annu. Rev. Biochem. 1998, 67, 581–608. Annual Reviews Inc)
to discharge GroES and entrapped folded polypeptidefrom the opposite side of the structure.
The cavity within GroEL provides a ‘reaction ves-sel’ in which the polypeptide chain can be ‘incubated’until folding is complete. This vessel is called the‘Anfinsen cage’ and estimations of internal volumeplace an upper limit of ∼ 85 000 A3 which is consistentwith a spherical protein of 70 kDa.
In the first phase unfolded polypeptide binds to theapical domain largely through hydrophobic interactionsand the direct involvement of at least nine residues(determined by site-directed mutagenesis) on eachsubunit. Eight of these residues have hydrophobic sidechains and point inwards into the cavity creating ahydrophobic ring that binds target polypeptides. Thecentral channel of GroEL functions as two separatecavities, one in each ring, as a result of a disordered24 residue C-terminal region in each of the sevenring subunits that effectively ‘blocks’ communicationbetween channels.
The second stage of the catalytic cycle involvesnucleotide binding. From observations of GroEL-assisted refolding experiments it was noted that GroELalone inhibits refolding whilst in the presence of K+ions, Mg-ATP and GroES efficient folding to the nativestate was seen. This led to a proposal that at leasttwo distinct conformations of the complex existed;one that binds unfolded polypeptides tightly and ATPweakly and another in which the binding proper-ties are reversed. Nucleotide-modulated conformationalchanges of GroEL are inherent to the protein fold-ing cycle. ATP binding preceded GroES binding withthe latter event occurring at near diffusion controlledrates (4 × 107 M−1 s−1) in the presence of ATP, butapproximately 100 times more slowly in the presenceof ADP.
The third stage involves completion of polypeptidefolding coupled with its release into the cavity ofthe cis ring of the GroEL-ES complex. Binding ofunfolded polypeptide to the A domain has suggested

IN VIVO PROTEIN FOLDING 421
ATP ATP
ATP ATP
ADP ADP
ADP ADP
ATP ATP
E
A A
E
I
A
+ ADP +
I
I
Figure 11.33 The chaperonin catalytic cycle (phases II–IV). ATP and GroES (purple subunits) bind to the GroELring causing change in conformation. ATP is trapped inside complex. In this state non-native polypeptide chains willno longer bind to apical domain but are released into central channel. ATP on cis ring is hydrolysed. Protein folds incentral cavity. ATP binds to trans ring causing conformational change in opposite ring that releases GroES, ADP andfolded peptide
the involvement of residues Leu234, Leu237, Val259,Leu263, and Val264. Mutation of any of these residuesinhibits correct protein folding and in some instancesproves lethal to E. coli. ATP and GroES bindingcause ‘en bloc’ movements of the E, I and Adomains and lead to competition for these sites withinthe complex and the rotation and displacement ofthe heptameric rings in opposite directions. As aresult, unfolded chains are no longer bound andare released into the cavity of the cis ring wherefolding occurs. The non-polar side chains of residuesresponsible for binding the non-native polypeptidethrough hydrophobic interactions in the cavity ofthe un-liganded ring are now buried in the cis ringassembly. This leads to their replacement by polarresidues on the walls of the cavity. This change isvery significant because it now drives folding wherethe released polypeptide chain is able to re-initiatefolding in an enlarged cavity whose lining is nowhydrophilic.
The fourth phase represents the dissociation of theligands (GroES and ADP) from the complex. Thedisassembly of the cis ring complex is triggered byhydrolysis of ATP to ADP with the latter formingweaker interactions at the nucleotide binding site asa result of the loss of a phosphate group. Weakerinteractions breakdown the GroEL–GroES complexwith the susceptibility to disassemble increased fur-ther by binding ATP to the opposite ring. This eventalso initiates the start of another cycle of bind-ing of the non-native polypeptide chain and GroESassociation.
In summary, the GroEL-ES complex representsa remarkable molecular machine designed to ensureprotein folding occurs efficiently and without thepotential for complicating side reactions. As with manyimportant protein complexes it appears that nature hasseized the ‘design’ and used it in other chaperonin-based systems. Whilst GroEL and GroES are respec-tively the prototypical chaperonin and co-chaperonin

422 PROTEIN FOLDING IN VIVO AND IN VITRO
there exist in other cells chaperonins that form similarring-shaped assemblies. The most convincing evidencederives from the structure of the corresponding com-plex from hyperthermophilic bacteria.
It is likely that only a fraction of all cytosolicproteins found in E. coli , or indeed in other cells,are processed via molecular chaperonins. Some pro-teins will be too large to be accommodated withinthe central cavity of chaperonins whilst other nascentpolypeptides, particular small soluble domains, can fold‘safely’ without aggregating and without the interven-tion of other proteins. Estimates of the number ofproteins acting as substrates for GroEL vary but atleast 300 translated polypeptides including essentialenzymes of the cell have been identified as using thissystem. About one-third of these proteins unfold withinthe cell and repeatedly return to GroEL for conforma-tional maintenance. It appears that GroEL substratesconsist preferentially of two or more domains of theα/β class containing mixtures of helices and buried β
sheets with extensive hydrophobic surfaces. Such pro-teins are expected to fold slowly and to be aggregation-prone whilst the hydrophobic binding regions of GroELare well suited to interaction with non-native states ofα/β proteins.
The thermosome of T. acidophilum
The characterization of the GroEL complex as atoroidal structure based on two heptameric ringscapped by a dome like GroES ring defined a majorgroup of chaperonins found widely throughout cells.Systems based on this design are called Type Ichaperonins. Sequence homology studies show thateukaryotic cells have similar protein complexes. Thesecomplexes are located within the mitochondria andchloroplasts with Hsp60, and Cpn60 representing twoprominent systems. This similarity is not surprising inview of the endosymbiont origin of these organellesfrom eubacteria.
In T. acidophilum comparable reactions arecatalysed by thermosomes that show variations inorganization and are example of Type II chaperonins.The structure of the thermosome from T. acidophilumwas the first Type II chaperonin to be described in
detail. It is a hexadecamer composed of two eightmembered rings. Each ring contained an alternatingseries of two polypeptide chains denoted as (αβ)4(αβ)4.Group II chaperonins in archaea and in the eukaryoticcytosol have been shown to use the same mechanismas type I chaperonins, with the binding of substrate toa central cavity and ATP dependent substrate release.The thermosome shares many of the properties of theGroEL rings although the lid structure is derived fromthe secondary structure elements of the apical domainsand does not involve GroES like subunits to cap acentral cavity.
Strands S12 and S13 and most of the N-terminalpart of helix H10 protrude toward the pseudo eight-fold molecular axis at the ends of the particle toblock the entrance to the central cavity and form alid domain. The β strand (S13) from each A domainforms a circularly closed β sheet of eight strandsand leaves a central pore of diameter ∼2.0 nm. Thetriangle of structural elements, strands S12 and S13and helix H10, pack against corresponding elements inthe neighbouring two subunits forming a hydrophobiccore with nine non-polar side chains and a threonineresidue from each subunit (Figure 11.34). The coreis hydrophobic with a third of all lid segmentresidues buried completely and these interactions assistin maintaining the lid structure in a conformationcomparable to that observed in the GroEL-ES complex.
Membrane protein folding
The preceding sections have dealt largely with thefolding of soluble proteins in vitro and the assistanceof macromolecular assemblies in governing foldingin vivo. As with much of biochemistry the details offolding in soluble proteins has largely outpaced under-standing of the same reactions in membrane boundsystems. A key observation made in 1982 by H. GobindKhorana was that bacteriorhodopsin could be dena-tured and refolded in a similar fashion to ribonuclease.This confirmed that membrane proteins possess all ofthe information necessary for folding and formation ofthe native state within their primary sequences. In thecase of bacteriorhodopsin this involved the formation

MEMBRANE PROTEIN FOLDING 423
Figure 11.34 The structural arrangement of thethermosome E, I and A domains with the extendedhelix–strand–strand regions of H10, S12 and S13shown in red (PDB: 1AG6)
of seven transmembrane helices, but similar reactionshave not been demonstrated for all membrane proteins.Most data currently available refers to the β-barrelrich domains of porins and the helix-rich domains ofrhodopsin-like proteins.
Soluble proteins represent the minimum free energyconformation, as defined in Anfinsen’s original experi-ments, and membrane proteins are no exception. How-ever, the mechanism of reaching this folded statein vitro, and especially in vivo, is less obvious. Insoluble proteins thermodynamic studies of the fold-ing process in response to denaturing agents providedinformation on forces stabilizing tertiary structure. Thisapproach is not successful for membrane proteinsas they are very resistant to denaturation. Bacteri-orhodopsin does not unfold completely in SDS, retain-ing helical structure, and denaturation arises primarilyfrom subunit dissociation, loss of interactions between
secondary structure elements and from unfolding ofdomains located either in the aqueous phase orat interfaces.
As a membrane protein bacteriorhodopsin is rep-resentative of proteins with seven transmembranehelices – a very large family – with the added advan-tage of a chromophore that is exquisitely sensitive tochanges in physical properties. In the purple membranebacteriorhodopsin is found as a two-dimensional crys-talline lattice where the protein aggregates as collec-tions of trimers. Two thermal transitions with enthalpiesof denaturation (�Hd) of ∼30 and ∼400 kJ/mol havebeen linked to a dissociation of the lattice and disper-sion into individual trimers at 80 ◦C followed by irre-versible disruption of trimers via the loss of helix–helixinteractions between monomers at 100 ◦C.
One framework for describing membrane proteinfolding is the two-stage model proposed by DonaldEngelman that involves in the first step the indepen-dent formation of hydrophobic helices of ∼25 residuesupon insertion into the membrane. Stage II involvesthe ordering of structure where interhelix interactionsdrive proteins towards the native state. Insertion andordering are believed to be separate and indepen-dent events. Supporting evidence for this model wasobtained using fragments of the polypeptide chain ofbacteriorhodopsin where five helices (A–E) assembledin the absence of the rest of the protein. This confirmedthe ability of fragments to form stable secondary struc-ture in membranes.
These studies emphasize the thermodynamics ofprotein folding, but an equally relevant aspect of theoverall process is the kinetic parameters associatedwith formation of the native state. Folding kinetics ofbacteriorhodopsin measured using CD and absorbancespectrophotometry indicate a rate limiting step isthe formation of transmembrane helices with theformation of one or more helices allowing helix–helixpacking and rapid completion of protein folding.Completion occurs upon retinal binding and a pathwaydefined by a series of intermediates with near nativesecondary structure but lacking the tertiary organizationis observed (Figure 11.35).
Intermediate I1 is formed within a few hundred mswhilst the transition from I1 to I2 is rate limiting andequated with the formation of transmembrane helices.

424 PROTEIN FOLDING IN VIVO AND IN VITRO
BOpsin I1 I2 IR BRhodopsin
R
Figure 11.35 Sequential folding pathway for assembly of bacteriorhodopsin from co-factor (R) and apo-protein(bacterio-opsin)
It occurs slowly on a time scale of 10–100 s. Thesekinetics distinguish the process from those seen insoluble proteins that are frequently complete within1 s. It seems unlikely that these reactions could occurso slowly in vivo, although data is lacking.
Fairly obviously, folding schemes derived fromstudies of helical membrane proteins may not beof great relevance to systems based around the β
barrel, such as porins. Despite different structuresexperimental evidence suggests similarities extend tothe processes of insertion and assembly. Porin OmpAis monomeric and this simplifies protein foldingsince there is no assembly into trimers. OmpA issynthesized with a signal sequence that is cleaved inthe periplasm to leave a mature protein that inserts intomembranes. In vitro OmpA can be completely refoldedin artificial lipid bilayers and because of the moderatehydrophobicity of the individual strands of the β barrelthese proteins are easily extracted from membranes inan unfolded state with urea or guanidinium chloride. Inthis state OmpA has been shown by CD spectroscopyto be unfolded at high denaturant concentrations.Rapid denaturant dilution in the presence of lipidleads to folded, membrane-inserted, conformationswith several ‘events’ identified during a foldingpathway distinguished by time constants of severalminutes in vitro. The formation of individual β strandsis unlikely and has not been described for anyisolated sequence, suggesting that insertion of OmpAinto the membrane may involve partial folding andthe interaction of several strands. Despite a slowseries of reactions the broad similarity to the foldingof bacteriorhodopsin – a protein with very differenttopology – is reassuring and suggests that an outlineof the basic pathway of membrane protein folding hasbeen uncovered. Gradually, the folding of membraneproteins is becoming understood and it is clear thatno new principles are involved in the formation of thenative state (see Table 11.4).
Translocons and in vivo membraneprotein folding
The details of membrane protein folding in vivoinvolve co-translational insertion of nascent proteinsinto the ER membrane at sites termed translocons.Translocons consist of membrane proteins that forma pore into which newly synthesized polypeptidechains enter as part of the normal cell traffickingpathways. Helical membrane proteins are targetedto the ER membrane in eukaryotic cells and tothe plasma membrane in bacterial cells by a signalsequence composed of approximately 20–25 residues.Interaction between the signal sequence and thesignal recognition particle (SRP) arrests translation andpromotes binding to receptors with the direction ofpolypeptide chains into the translocon followed byintegration into the membrane.
Uncovering the organizational structure of thetranslocon relied on yeast genetic studies of proteinsecretion (SEC ) genes. Using electron microscopy itwas possible to identify the cytological event perturbedby lesions and one set of experiments identifiedmutants (SEC61 ) defective in transport into the ERwith unprocessed proteins accumulating in the cytosol.Subsequent cloning of the mammalian version ofSEC61 established a close homology with yeast andbetween prokaryote and eukaryote. The SEC61 geneencoded the main channel-forming subunits withinthe translocon.
The translocon or Sec61 complex consists ofthree integral membrane proteins Sec61α, Sec61β andSec61γ. Sec61α has many transmembrane heliceswhilst Sec61β and γ contain only single helical seg-ments crossing the bilayer. In 1993 elegant reconsti-tution experiments by Tom Rapoport showed that itwas possible to mimic protein translocation in artificialmembranes containing the incorporated Sec61 complexalthough the catalytic efficiency of the reconstituted

MEMBRANE PROTEIN FOLDING 425
Table 11.4 Some of the membrane proteins whose folding has been studied
Protein Structure and function Protein denaturation Formation of folded
protein
Bacteriorhodopsin 7 TM helices and retinal
co-factor; light-driven
protein pump
Apoprotein fully
denatured in
trifluoroacetic acid;
apoprotein partly
denatured in SDS,
leaving ∼55 % of
native helix
Transfer from organic acid
to SDS and then folded
by mixing with lipid
vesicles
LHC-II 3 TM α-helices and 1
short amphipathic helix
at membrane surface.
Photosynthetic
light-harvesting protein
Apoprotein partly
denatured in SDS,
leaving about 30 % of
native helix content
Reconstituted with
thylakoid membrane
extracts by freeze–thaw.
Reconstituted in
micelles containing
pigments, lipid and
detergent
E. coli DGK Unknown structure Apoprotein slightly
denatured by SDS,
leaving ∼85 % of
native helix content
Refolded in DM micelles
E. coli OmpA Membrane domain of 170
amino acids in
8-stranded β barrel.
Exact function
unknown, but likely
channel
Protein completely
denatured in urea
Folding in lipid vesicles by
mixing with
urea-denatured state
E. coli OmpF 16-stranded β barrel.
Trimer forms pore in
outer membrane
Completely denatured in
urea or GdnHCl
Poor folding in lipid
vesicles on mixing the
urea-denatured state.
Folding increased if
detergent used in mixed
micelles
Adapted from Booth, P. et al. Biochem. Soc. Trans. 2001, 29, 408–413. Abbreviations: TM, transmembrane; DM, dodecylmaltoside; GdnHCl, guanidinehydrochloride; LDS, lithium dodecylsulphate; OG, octylglucoside; PG, phosphatidyl-D,L-glycerol; DGDG, digalactosyl diacylglycerol; LHC-II, light harvestingcomplex of higher plants; DGK, diacylglycerol kinase
complex was low. With structural characterizationof the SRP and its receptor greater emphasis hasbeen placed on the organization of the translocon(Figure 11.36). Electrophysiological studies emphasize
the presence of an ion channel in the translocon whilststudies with fluorescent labels attached to residueswithin a nascent polypeptide chain indicate changes inenvironment and conformation. Much work remains to

426 PROTEIN FOLDING IN VIVO AND IN VITRO
Figure 11.36 A model for the organization ofthe translocon. The signal recognition particle(SRP)–ribosome complex binds to receptor (α/β) inthe ER membrane. The translocon consists of theSec61 proteins (α, β, γ) and accessory proteins.Translocation requires GTP hydrolysis and the growingpolypeptide chain is pushed across the membranetowards the lumen
be done in the characterization of the translocon andits role in membrane protein folding.
A key event in membrane protein folding is iden-tification of a signal peptide by the SRP, but mem-brane proteins may contain many blocks of 20–25non-polar residues each reminiscent of such a peptide.This could lead to unwanted transfer into the ER lumenand therefore requires specific recognition mechanisms.The translocon complex must also deal with inte-gral proteins that differ considerably in topology. Oneanswer to this problem lies in the presence of sequenceswithin membrane proteins that control their assem-bly within the ribosome–translocon complex. Thesesequences are known as topogenic signals and includeconventional N-terminal signal peptides, signal-anchorsequences, reverse signal-anchor sequences, and start-transfer/stop-transfer sequences.
Proteins with more than one transmembrane seg-ment are possibly threaded into membranes through theeffect of start- and stop-transfer sequences. In eukary-otes the main features of these transfer sequences aregroupings of hydrophobic residues flanked by residueswith positively charged side chains. The effect of thesesequences is to promote and arrest the transfer ofresidues through the translocon (Figure 11.37). Thisprocess aids the formation of transmembrane helices
but is also vital to the formation of important loopregions that frequently link such domains.
The mechanism by which transmembrane domainsexit the translocon pore to reach the bilayer remainsunclear. Evidence suggests that transmembrane helicesfold within this pore and then exit to the bilayer eitherindividually or in pairs. This scheme would lead to thefinal helix packing reactions occurring in the membranebilayer after translocation.
Protein misfolding and the diseasestateWith a greater understanding of folding in vivo andin vitro has come the realization that diseases arise asa consequence of protein misfolding. Mutation withincoding regions of genes can result in the insertionof a stop codon and the failure to synthesize full-length, folded, protein. Alternatively, mutations changethe identity of one or more residues leading to aprotein with altered folding properties. Ultimately, thisresearch is traced back to studies of sickle cell anaemiaidentifying mutant haemoglobin as the basis fordisease. Similar origins for disease (Table 11.5) occurin protein folding where mutations cause defectivefolding, aberrant assembly and incomplete processing.
Intracellular sorting and the emergenceof defective protein trafficking
A key requirement for cellular function is that proteinsare targeted to the correct compartment. In eukaryoticcells compartmentalization requires specialized sortingpathways and the ER and Golgi apparatus form part ofthe endocytic pathway where a key event is the budding(endocytosis) of membranes as part of the traffickingprocess. Defective protein folding often results inan inability to transfer mutant domains through theendocytic pathway.
One of the best examples described to date occursin cystic fibrosis. Cystic fibrosis is very common inCaucasian populations with an incidence of approxi-mately 1 in 20. In a homozygous state this results ina defective CFTR protein and a disease characterizedby an inability to transport chloride across membraneseffectively and problems achieving correct ion balance.

PROTEIN MISFOLDING AND THE DISEASE STATE 427
Cytoplasm
Translocating Translocating
Translocating
TM2Stop Transfer
TM1Stop transfer
TM2Signal Sequence
Signalsequence
TM1
TM1
TM2
ER lumen
Cytoplasm
ER lumen
Figure 11.37 The function of topogenic sequences in membrane protein insertion. In the first mechanism TM1functions as the signal sequence to initiate translocation of the carboxy-terminal region whilst TM2 functions as astop transfer sequence halting translocation until the next start signal. Alternative mechanisms appear to exist suchas that shown in the second pathway where TM2 initiates translocation of the N-terminal region and TM1 acts asthe stop transfer signal (Reproduced with permission from Dalbey, R.E, Chen, M.Y., Jiang, F. & Samuelson, J.C. Curr.Opin. Cell Biol. 2000, 12, 435–442. Elsevier)
Over 600 different mutations have been identified inthe CFTR protein but the most common mutationinvolves deletion of one residue (Phe508). This doesnot lead to a truncated form of CFTR but insteadresults in a full-length protein missing a single residue.The mutant CFTR protein (�F508) is expressed andthe translated product is easily detected. However,the protein is not processed further and is unable tocomplete post-translational glycosylation and does nottransfer through the Golgi apparatus. Most of the pro-tein becomes ubiquitinated and marked for destructionby the proteasome but a small amount (∼1 percent)reaches the surface where it retains anion selectivityand conductance properties comparable to the wild typeprotein but has a decreased probability of ion channelopening. The result highlights how small ‘defects’ leadto major physiological impairment.
Protein aggregation and amyloidosis
Enormous interest has focussed on the appearanceof large protein aggregates within tissues in diversedisease states. The appearance of protein deposits inclose association with neurofibrillary networks in thecells of brain tissue has long been known to occur inAlzheimer’s disease. Protein aggregation also underliesother diseases and may occur in tissues such as the liverand spleen, besides the brain. The protein aggregatesare linked by formation of elongated fibrils (amyloid)and diseases showing this property are collectivelygrouped together by the term amyloidosis.3
3The term ‘amyloid’ was originally used to describe the resemblanceof protein aggregates associated with disease states to the appearanceof starch chains (amylose).

428 PROTEIN FOLDING IN VIVO AND IN VITRO
Table 11.5 Diseases arising from ‘folding’ defects
Disease Protein or precursor
involved in disease
Cystic fibrosis CFTR
Primary systemic amyloidosis Immunoglobulin
light chain
Medullary carcinoma of the
thyroid
Calcitonin
Osteogenesis imperfecta Collagen (type I
procollagen)
Lung, colon and other forms of
cancer
p53 transcription
factor
Maple syrup disease α-ketoacid
dehydrogenase
complex
Amyotrophic lateral scoliosis Superoxide
dismutase
Creutzfeldt–Jakob disease, vCJD,
scrapie, fatal familial insomnia
Prion protein
Alzheimer’s disease β-amyloid protein
Cataracts Crystallins
Atrial amyloidosis Atrial natriuretic
factor (ANF)
Senile systemic amyloidosis,
Familial amyloidosis
Transthyretin
Tay–Sachs disease β-hexosaminidase
Hereditary emphysema α-antitrypsin
Retinitis pigmentosa Rhodopsin
Hereditary non-neuropathic
systemic amyloidosis
Lysozyme
Transmissible spongiform
encephalopathy (TSEs)
Prion protein
Huntington’s chorea Huntingtin
Familial hypercholesterolaemia LDL receptor
Amyloidosis is apparent in many clinical condi-tions besides Alzheimer’s and includes familiar dis-eases such as Parkinson’s disease, type II diabetestogether with less common conditions such as thespongiform encephalopathies. Some of these diseasesare genetically determined (i.e. inherited) whilst others
are acquired (sporadic) and others may be infectiouslytransmitted. In each amyloid disease a different pro-tein or fragment aggregates, forming a fibril, and insystemic forms of amyloidosis aggregation leads to thedeposition of kg quantities of protein. Ultimately, theprogressive deposition of fibrils will cause death, espe-cially when it occurs in a vital organ.
The association between formation of protein fibrilsand irreversible protein aggregation was deduced froma number of studies but particularly those givingrise to a condition known as familial amyloidoticpolyneuropathy (FAP). The disease was recognizedaccurately in the 1950s in a Portuguese family asan inherited condition. This disease centres aroundmutations in transthyretin, a protein involved inbinding the hormone thyroxine.4 Transthyretin is foundin a binary complex with retinol binding protein(RBP) and acts as a soluble homotetrameric protein(Figures 11.38 and 11.39).
Each subunit contains 127 residues and a series ofeight β strands that adopt a β barrel conformation.Seven of the strands (A–H) are seven to eight residuesin length whilst the D strand is shorter and only threeresidues long (Figure 11.40). The eight strands formtwo sheets; the first is formed between strands DAGHand the second between CBEF. Unsurprisingly, thehigh content of β structure arranged in a barrel liketopology contributes to significant stability.
Analysis of the molecular defects occurring in thetransthyretin gene in individuals with FAP identifiedover 80 different mutations. The clinical symptomsof FAP usually begin from the third to fourthdecades and are characterized by local neurologicimpairment leading to wider autonomic dysfunctionand death within 10 years. A common mutationnoted in Portuguese, Japanese and Swedish kindredsis the transition Val30>Met. The identification andprevalence of this mutation led to an analysis ofthe structural and functional properties of Val30Mettransthyretin especially since the mutant protein wasknown to be amyloidogenic.
Residue 30 lies on the inside of the monomer andthe bulkier Met side chain displaces one set of fourβ strands from the remaining set of four. However,the mutant protein was folded with a conformation
4Transthyretin was formerly called prealbumin as a consequence ofits migration pattern on gels just ahead of albumin.

PROTEIN MISFOLDING AND THE DISEASE STATE 429
Choroidplexus of
brain
Synthesis bychoroid plexus
and liverDegradationin liver and
kidney
Liver
Kidney
Circulation in plasma
KeyRBP
TTR tetramer
HDLs
Figure 11.38 Synthesis, distribution and uptake of transthyretin. Transthyretin tetramer circulates in plasma boundto retinol-binding protein (RBP) providing a transport function for vitamin A and thyroxine with a small proportionbinding high-density lipoproteins (reproduced with permission from Saraiva, M.J.M. Expert Rev. Mol. Med. CambridgeUniversity Press, Cambridge, 2002)
Figure 11.39 The complex of transthyretin andretinol binding protein. RBP is shown in yellow withthe tetramer of transthyretin shown in blue and green.The larger RBP binds to a site formed by two of thesubunits (PDB: 1RLB)
very similar to the wild type. This initially puzzlingresult was interpreted with the recognition that wildtype protein aggregates to form fibrils under someconditions–an event associated with senile systemicamyloidosis, where fibril deposition in the heart leadsto cardiomyopathy at the age of ∼80. The resultsemphasized that amyloidosis is associated with allforms of the protein.
Mutations are viewed as exacerbating diseaseby promoting more active forms of amyloidosis.
Figure 11.40 Transthyretin structure showing strandsA–H and the sandwich formed between DAGH and CBEFin each monomer. Strands H and H′ interact at the inter-face (PDB: 1BMZ)
Structural studies of wild type protein at low pH(∼4.5) show transthyretin dissociation to a monomericbut amyloidogenic intermediate of different tertiarystructure involving rearrangement in the vicinity ofthe C strand–loop–D strand region. Dissociation ofthe tetramer is an unfavourable event under nor-mal physiological conditions but these rates are

430 PROTEIN FOLDING IN VIVO AND IN VITRO
Figure 11.41 Scheme for initial formation of transthyretin-based fibrils
enhanced at low pH and for the Val30Met mutanta lower activation barrier increased the likelihoodof amyloidogenic intermediates. Other mutants suchas Leu55Pro confirmed this trend and led to theconclusion that mutants associated with FAP areless stable than wild type protein and form theamyloidogenic intermediate more quickly.
Individuals with the FAP mutations are kineticallyand thermodynamically predisposed to amyloid fib-ril formation. Mutation increases the steady-state con-centration of the amyloidogenic intermediate with theresult that individuals with these defects are prone toamyloid formation and disease. The biophysical datamirrored the pathological outlook of the disease. Onsetof the disease in the eighth, third and second decadescorrelated with the relative stabilities of wild-type,V30M and L55P forms of the transthyretin with themutations shifting equilibria in favour of amyloido-genic intermediates and decreasing the age of appear-ance of FAP.
Fibril formation by the association of transthyretinresults in comparable aggregates to those observed inother diseases and suggested a similar structure forall amyloid fibrils (Figure 11.41). Alzheimer’s disease
represents aggregates of a short fragment known asthe Aβ peptide. Since the Aβ peptide and transthyretindo not share any sequence homology and are notrelated in any way the formation of similar fibrilstructures was remarkable and significant. The structureof amyloid fibrils examined using X-ray diffractionand cryo-EM methods reveal a β helix structure. Infibrils resulting from six precursor proteins presentin different diseases a common structure existed andyet none of these proteins have sequence homology.5
The morphology and properties of amyloid fibrilswere based on a β scaffold with strands runningtransversely across a helix axis. In the direction ofthe helix axis the strands align to form β sheets.Four discrete chains are wound into the β helix(Figure 11.42).
5The systems studied were the Aβ peptide; the λ immunoglobulinlight chain (monoclonal protein systemic amyloidosis); lysozyme(hereditary lysozyme amyloidosis); calcitonin (medulary carcinomaof the thyroid); insulin (diabetes and insulin related amyloid disease);β2 microglobulin (haemodialysis related amyloidosis), together withsynthetic peptides of transthyretin (FAP) and the prion protein(transmissible spongiform encephalopathies).

PROTEIN MISFOLDING AND THE DISEASE STATE 431
Figure 11.42 Model of a generic amyloid fibrilstructure. The structure of the amyloid fibrils is madeup of four β sheets. Sheets run parallel to the axis of theprotofilament with component β strands perpendicular.The twisting of β strands yields a series of parallelstrands around a common helical axis that coincideswith the axis of the protofilament and leads to a pitchof 11.55 nm containing 24 β strands (reproduced withpermission from Sunde, M. et al., J. Mol. Biol. 1997,273, 729–739. Academic Press)
These structures can be induced in protein notknown to form amyloid fibrils in any documenteddisease phenotype. The SH3 domain (Figure 11.43)of the p85α subunit of phosphatidylinositol-3′-kinasewas induced to form amyloid aggregates. The normalstructure of the 84 residue domain is based arounda β sandwich of five strands but under low pHconditions this domain unfolds in a reaction promotingfibril formation.
Mutating or ‘mistreating’ soluble proteins inducesaggregation and the formation of fibrils. Fibrils derivedfrom unfolded SH3 domains cannot form by simplylinking together several units of the native fold becausethe diameter of the protein is too large to fit properly
into the fibril structure. The SH3 domain must unfoldto adopt a longer and thinner shape.
The assembly of β strands into a helix is a featureof amyloid fibrils and, although unusual, is known tooccur in structural databases, with examples in foldedproteins including alkaline protease, pectate lyases andthe p22 tailspike protein. Collectively, these studiespoint to protein misfolding leading to aggregation andamyloid deposits.
As these studies progressed it became apparent thatan obscure collection of neurodegenerative conditionsresulted in a recognizable event, protein aggregation.These diseases were to become known as the prion-based diseases.
Prions and protein foldingDisease transmission requires the intervention ofgenetic material (DNA or RNA) in order to estab-lish an infection. Most commonly, this event centresabout bacterial or viral infections and the conversionof genetic information into protein with unpleasantconsequences to the host. Gradually, convincing evi-dence has been acquired that a non-nucleic acid basedagent is involved in transmission of a restricted numberof diseases. These ‘agents’ were called ‘proteinaceousinfectious particles’ or prions and were identified, afterexhaustive analysis, as the active components in com-municable and inherited forms of disease.
These ideas met with considerable scepticism andeven hostility from some sections of the scientific andmedical communities but there is good evidence thatprions lie at the heart of neurodegenerative disordersassociated with a pathological collection of diseasesknown as transmissible spongiform encephalopathies(TSEs). The common link between these diseases isdevelopment of gross morphological changes to tissuewithin the brain characterized by the formation ofvacuoles and a sponge-like appearance (Figure 11.44).Disease progression is accompanied by the appearanceof amyloid plaques within the tissue whilst in some statesstructural changes occur to cells leading to a loss ofsynaptic contacts. Three disease states showing theseproperties are scrapie, Creutzfeldt–Jakob disease (CJD)and Kuru.
Elucidation of the role of prions stems from attemptsto understand the biochemical basis of these threeobscure, and at first glance, unrelated diseases. Scrapie
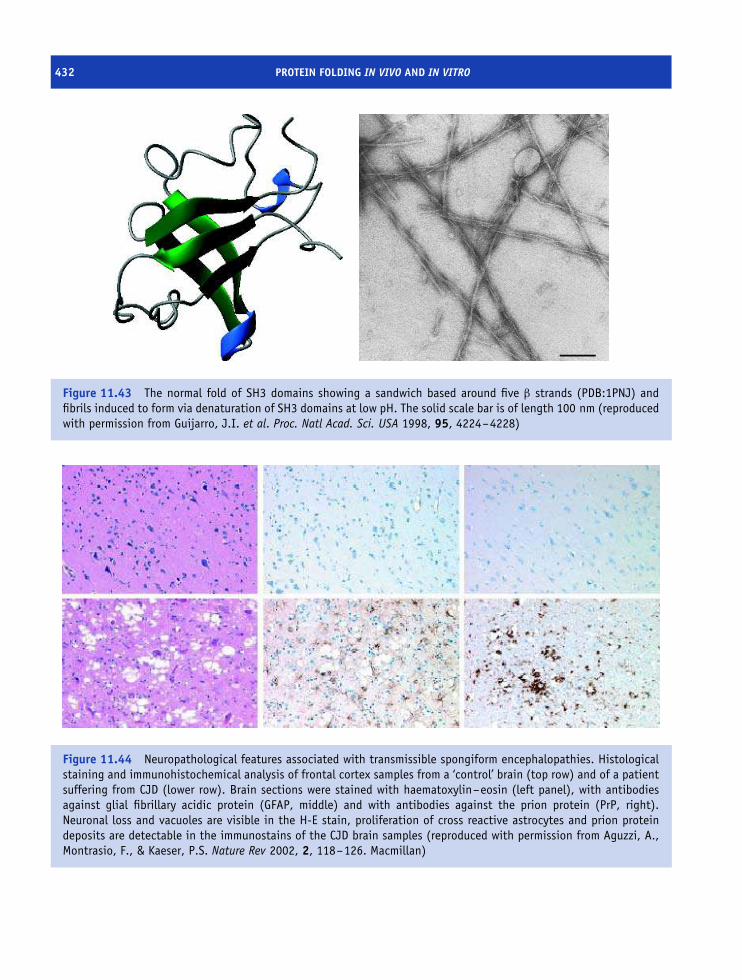
432 PROTEIN FOLDING IN VIVO AND IN VITRO
Figure 11.43 The normal fold of SH3 domains showing a sandwich based around five β strands (PDB:1PNJ) andfibrils induced to form via denaturation of SH3 domains at low pH. The solid scale bar is of length 100 nm (reproducedwith permission from Guijarro, J.I. et al. Proc. Natl Acad. Sci. USA 1998, 95, 4224–4228)
Figure 11.44 Neuropathological features associated with transmissible spongiform encephalopathies. Histologicalstaining and immunohistochemical analysis of frontal cortex samples from a ‘control’ brain (top row) and of a patientsuffering from CJD (lower row). Brain sections were stained with haematoxylin–eosin (left panel), with antibodiesagainst glial fibrillary acidic protein (GFAP, middle) and with antibodies against the prion protein (PrP, right).Neuronal loss and vacuoles are visible in the H-E stain, proliferation of cross reactive astrocytes and prion proteindeposits are detectable in the immunostains of the CJD brain samples (reproduced with permission from Aguzzi, A.,Montrasio, F., & Kaeser, P.S. Nature Rev 2002, 2, 118–126. Macmillan)

PROTEIN MISFOLDING AND THE DISEASE STATE 433
occurs in sheep and is defined by a progressive loss ofmotor coordination that leads to an inability to standunsupported. Animals develop an intense itch that leadsto wool scraping from skin and gives the disease itsname. Scrapie has been recognized since the 17th centuryin the United Kingdom, with similar conditions notedmore recently in animals such as mink, deer and elk.
CJD was first identified in the 1920s by H.GCreutzfeldt and then independently by A. Jakob.Patients exhibit dementia with poor motor coordina-tion, poor perception and reasoning. The incidence ofCJD is low, probably less than 1 case in two million,and typically occurs in individuals above 50 years ofage. It is found throughout the world with little obviousassociation with economic, racial, or social patterns.Most frequently the disease arises sporadically, but in10–15 percent of cases the disease is inherited in anautosomal dominant manner, and very rarely CJD isspread by iatrogenic transmission where contaminationoccurs most probably via donated tissue.
Kuru is an unusual disease confined to tribal peopleof the highlands of Papua, New Guinea where it iscalled the ‘laughing death’. It was comprehensivelydescribed in 1957 as a fatal disease marked by ataxiaand progressive dementia. A link was made betweendisease transmission and ritual cannibalism wheretribes people honoured their dead by eating body partsincluding brain. A decline in cannibalism led to adecline in Kuru although the precise causative agentremained at that time unknown.
The three diseases are linked by the observationthat transmission occurs when diseased brain extractsare injected into healthy animals. Of these diseasesscrapie is experimentally accessible and initial studiesattempted to identify the causative agent. Exhaustiveanalysis of scrapie infected brain tissue purified asingle protein devoid of nuclei acid that was infectiouswhen injected into hamster brains. It was called theprion protein (PrP) and the pioneering work in thisarea was attributable to the work of Stanley Prusinerand his extraordinary quest to define and characterizethese agents.
Prion purification and the demonstration of infectivityraised a number of questions. How was PrP encoded?Was undetected DNA lurking in the ‘pure’ proteinpreparation? What is the origin of PrP? Major insight intothe molecular events underpinning the generation of PrP
Table 11.6 Structural differences between PrPC and PrPSc
Property PrPc (cellular) PrPSc (scrapie)
Polypeptide
chain
1–231 1–231
Protease
resistant
No Stable core residues
90–231
Disulfide
bridges
Yes (179–214) Yes
Solubility Soluble when
expressed -GPI
Very insoluble
except to all but
the strongest
‘solvents’
Aggregation
state
Monomeric Multimeric
Stability 25–40 kJ mol−1 More stable than
PrPc
Structure α helical Increase in
proportion of β
strands
Adapted from Cohen, F. & Prusiner, S.B. Annu. Rev. Biochem. 1998, 67,793–819.
arose with N terminal sequencing of the isolated protein.This sequence allowed the synthesis of degenerateoligonucleotide probes that hybridized to chromosomesfound in a wide range of mammals including mice,hamsters and most importantly humans. The probesdid not hybridize to the isolated PrP eliminating thepossibility of trace amounts of DNA associated with thisfraction. A gene encoding PrP was found on the shortarm of chromosome 20 in humans where it coded for aglycoprotein of mass 33–35 kDa.
Most individuals never develop any form of thedisease yet the prion gene is found in the humangenome. Prusiner suggested one explanation. PrPwas produced in two forms – a normal form andan abnormal one that generated disease. Immediatesupport for this view came with the demonstration thatPrP found in infected brains was resistant to proteolysiswhilst the normal form remained sensitive to proteases.The two forms are often called cellular PrP (normal)and scrapie PrP (infectious) – abbreviated as PrPc and

434 PROTEIN FOLDING IN VIVO AND IN VITRO
PrPSc (Table 11.6). The different proteolytic sensitivityof PrPc and PrPSc suggested that each form of theprotein possessed a different conformation.
Protein expression studies from the Syrian hamster,mice and humans showed monomeric species withlittle tendency to form aggregates whilst the full-lengthinfectious conformer readily formed aggregates. PrPis composed of ∼250 residues with a signal sequenceof 22 residues, and close to the C-terminal residue isa GPI membrane anchor. Additional post-translationalmodifications include two N-linked glycosylation sitesand a single disulfide bridge. In the absence of the GPIanchor the protein is soluble and does not partitionin the membranes; this occurs when the protein isexpressed in prokaryotic systems. As a result of itssolubility it has been possible to subject the protein todetailed structural analysis.
The solution structure of a fragment containingresidues 121–231 for the mouse PrPc protein showeda folded domain with three α helices and two shortβ-strands (Figure 11.45). Structures of the analogoushuman protein (residues 23–231) and longer fragmentsfrom the mouse and Syrian hamster indicated thatthe prion protein consisted of a structured domainfrom residues 121–231 and a less ordered N-terminaldomain. Very little secondary structure was detectedin the N-domain. One reason for the absence ofstructure in the N-terminal region is seen in the primarysequence of the prion protein (Figure 11.46) wherea series of five octapeptide repeats occur with thesequence PHGGGWGQ. The presence of this repeatis an unusual feature of the sequence.
Figure 11.45 The structure of 121–231 fragment ofthe mouse PrPc determined using NMR spectroscopy(PDB: 1AG2)
Prions become infectious particles capable of trans-mitting disease (scrapie) by corrupting the conforma-tion of other protein molecules and initiating a seriesof events that lead to the formation of protein fibrils oramyloid deposits within tissues.
Two additional TSEs, Gerstmann–Straussler–Scheinker (GSS) disease and fatal familial insomonia(FFI), also cause spongiform changes in the brain,neuronal loss, astrocytosis and amyloid plaque for-mation. GSS disease is an inherited neurodegenera-tive condition that offered the possibility of identifyingmutations. Five different TSEs originate from differentmutation sites within the prion gene (Table 11.7).
10 20 30 40 50 60 70 80PrPc : MANLGCWMLVLFVATWSDLGLCKKRPKPGGWNTGGSRYPGQGSPGGNRYPPQGGGGWGQPHGGGWGQPHGGGWGQPHGGG : 80
90 100 110 120 130 140 150 160PrPc : WGQPHGGGWGQGGGTHSQWNKPSKPKTNMKHMAGAAAAGAVVGGLGGYMLGSAMSRPIIHFGSDYEDRYYRENMHRYPNQ : 160
170 180 190 200 210 220 230 240PrPc : VYYRPMDEYSNQNNFVHDCVNITIKQHTVTTTTKGENFTETDVKMMERVVEQMCITQYERESQAYYQRGSSMVLFSSPPV : 240
250PrPc : ILLISFLIFLIVG : 253
Figure 11.46 Primary sequence of the prion protein (PrPc). The N-terminal signal sequence is highlighted by thegreen block whilst the blue block represents the structured domain of 5 helices and three strands. Shown in yellowis the unstructured region and within this sequence are five octapeptide repeats highlighted by red text

SUMMARY 435
Table 11.7 Mutations of the PNRP gene associatedwith inherited forms of transmissible spongiformencephalopathy
Mutation Disease
plus phenotype
Insertion of 24, 48, 96, 120, 144,
168, 192, or 216 base pairs
between codons 51 and 91 of
PNRP gene leads to repeat of
octapeptide motif in PrP
CJD, GSS, or
atypical
dementias
Pro102Leu, Pro105Leu,
Ala117Val, Gly131Val,
His187Arg, Asp202Asn,
Phe198Ser, Gln212Pro,
Glu217Arg
GSS: classical
ataxic form plus
other
phenotypes
Tyr145stop, Thr183Ala Alzheimer-like
dementia
Asp178Asn CJD (where
residue 129 is V
on mutant
allele)
Asn178Asn FFI (where residue
129 is M on
mutant allele)
Val180Ile, Val203Ile, Arg208His,
Val210Ile, Glu211Gln,
Glu200Lys, Met232Arg
CJD
FFI = fatal familial insomnia; CJD = Creutzfeldt–Jakob disease; GSS =Gerstmann–Straussler–Scheinker syndrome.
Most mutations (Figure 11.47) occur in the struc-tured C-domain of PrP and GSS is caused primarilyby the substitution of Pro102>Leu, whilst in CJD theprevalent mutation is at residue 178 and involves thereplacement of Asp by Asn. A slight complication tothis genetic pattern is the existence of polymorphismswithin the PNRP gene where Met or Val is found atresidue 129. By itself this change does not result indisease but when combined with mutation at residue178 the polymorphism contributes to either FFI (Met)or CJD (Val).
The identification of all diseases as the pathologicalconsequence of mutations within the PNRP gene hasunified this area of study but still leaves the awkwardquestion of how does the prion cause a change inconformation and spread the disease? A major problemin this area has been the unambiguous demonstrationof conversion of PrPc into PrPsc.
Strong evidence that conversion of prion pro-tein from normal to abnormal states was responsi-ble for neurodegenerative disease was obtained using‘knockout’ mice where the PNRP gene was deleted.With the normal PNRP gene present the injec-tion of mice with PrPSc resulted in the transmis-sion of the disease; mice showed familiar symp-toms of ataxia and cerebellar lesions within approx-imately 150 days. When knockout mice were sub-jected to the same experiment they did not developthe disease. In other words the disease requiresnative PrP protein encoded by the relevant gene topropagate into the development of amyloid fibrils.Two hypotheses known as the ‘refolding’ and ‘seed-ing’ models attempt to explain prion propagation(Figure 11.48).
Summary
The folding of individual polypeptide chains fromless structured states to highly organized topologiesis vital to biological function. All of the informationdirecting protein folding resides within the primarysequence.
Thermodynamically the folded state is the globalenergy minimum with the free energy decreasing inthe transition from unfolded to native protein. Nativeproteins are marginally more stable than unfolded stateswith estimates of conformational stability ranging from10–70 kJ mol−1.
A number of quantifiable factors have been shownto influence protein stability in globular proteins. Thesefactors include conformational entropy, enthalpic con-tributions from non-covalent interactions and thehydrophobic interaction. When the sum of all favoura-ble interactions outweighs the sum of unfavourableinteractions a protein is stable.
Protein denaturation is the loss of ordered struc-ture and occurs in response to elevated temperature,
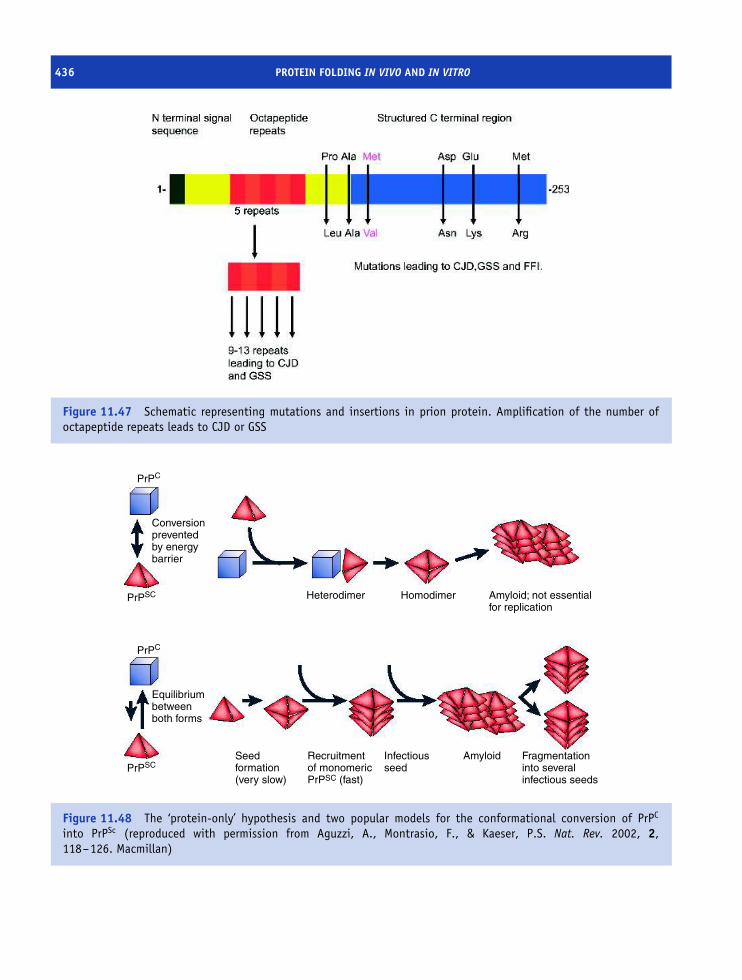
436 PROTEIN FOLDING IN VIVO AND IN VITRO
Figure 11.47 Schematic representing mutations and insertions in prion protein. Amplification of the number ofoctapeptide repeats leads to CJD or GSS
PrPC
Conversionpreventedby energybarrier
Heterodimer
Seedformation(very slow)
Recruitmentof monomericPrPSC (fast)
Infectiousseed
Amyloid Fragmentationinto severalinfectious seeds
Homodimer Amyloid; not essentialfor replication
Equilibriumbetweenboth forms
PrPC
PrPSC
PrPSC
Figure 11.48 The ‘protein-only’ hypothesis and two popular models for the conformational conversion of PrPC
into PrPSc (reproduced with permission from Aguzzi, A., Montrasio, F., & Kaeser, P.S. Nat. Rev. 2002, 2,118–126. Macmillan)

PROBLEMS 437
extremes of pH or the addition of reagents such as ureaor guanidine hydrochloride. Denaturation involves thedisruption of interactions such as hydrophobic interac-tions, salt bridges and hydrogen bonds that normallystabilize tertiary structure.
Experimental measurements of protein stabilityinclude differential scanning calorimetry, absorbanceand fluorescence optical methods, NMR spectroscopyand circular dichroism.
Kinetic methods allow protein folding to be fol-lowed as a function of time. Small soluble proteins(<100 residues) fold within 1 s and the observation ofslower folding rates is normally associated with disul-fide bond formation or cis-trans proline isomerization.To overcome slow in vivo folding specific enzymescatalyse reactions such as peptide prolyl isomerizationand protein disulfide isomerization.
Critical events in the formation of the native fold arethe acquisition of ordered stable secondary structure,the formation of hydrophobic cores, and the exclusionof water from the protein interior.
For proteins such as lysozyme, barnase and chy-motrypsin inhibitor 2 the reaction pathway has beendefined to identify properties of intermediates and tran-sition states. In barnase it has proved possible to quan-tify the folding process in a pathway containing atleast one intermediate between denatured and orderedstates. A hydrophobic core and the formation of theβ sheet are two early events in the folding pathway.In lysozyme each domain folds separately within theobservation of multiple kinetic pathways for each fold-ing unit.
Chaperones are multimeric systems found in allcells that prevent incorrect protein folding based
around a toroidal structure of seven, eight or ninesubunits. The toroidal structure is capped at either endand forms a cavity binding unfolded polypeptide viahydrophobic surfaces with ATP-driven conformationalchanges propelling the peptide towards the native state.
The kinetics of membrane protein folding in vitroare much slower than rates of folding for globulardomains but the principles governing formation of thenative state remain similar.
Incorrect folding leads to a loss of activity that isthe basis of many disease states. Misfolding leads toincorrect protein trafficking within the cell and mutantproteins of CFTR are a good example.
Incorrect protein folding is also a key eventin amyloidosis – the accumulation of long irre-versibly aggregated protein within fibrils. Amyloido-genic diseases include many neurodegenerative dis-orders such as Alzheimer’s, transmissible spongiformencephalopathies and hereditary forms of systemicamyloidosis. Fibrils from a range of amyloid proteinshave a common structure based on collections of β
strands that align into parallel sheets twisted into heli-cal conformations known as β helices.
Neurodegenerative disorders such as CJD, BSEand scrapie are linked via the spongiform appearanceof neuronal tissue and the accumulation of amyloiddeposits. The disease arises from changes in the prionprotein generating conformations with different sec-ondary and tertiary structure. The abnormal form pro-motes amyloidosis by inducing other prion proteinsto change conformation. These events occur sponta-neously at very low frequency leading to sporadicoccurrences of disease but are facilitated by mutationswithin the PNRP gene.
Problems1. Draw the cis and trans conformations associated with
a Xaa–Pro peptide bond.
2. The 310 helix is named because there are three residuesper turn and 10 atoms including hydrogen betweenthe donor and acceptor atoms forming an intra-chainhydrogen bond. Describe the α helix using such
notation. How does the helix compare with the 310
helix? Repeat this analysis for the π helix.
3. Experimental studies of protein folding reveal thefollowing trends of denaturation.Calculate the conformational stability and estimate anyother relevant parameters.

438 PROTEIN FOLDING IN VIVO AND IN VITRO
Absorbance Denaturantconcentration
(M)
Absorbance Denaturantconcentration
(M)
0.985 0 0.375 1.80.985 0.1 0.325 1.90.985 0.2 0.285 2.00.980 0.3 0.220 2.10.975 0.4 0.175 2.20.970 0.5 0.140 2.40.960 0.6 0.120 2.50.945 0.8 0.115 2.60.890 1.0 0.110 2.70.760 1.2 0.105 2.80.625 1.4 0.105 2.90.510 1.6 0.100 3.0
4. Assume that in order to reach the native state a proteinneeds to sample only 10 conformations per residuewith each conformation taking 0.1 ns. Estimate howlong it might take for a 100 residue protein to fold.
Now assume that the each block of 10 residues canfold independently of the remaining residues and byonly sampling three conformations per residue. Howlong does folding take to occur? Comment on thetwo values?
5. Explain how disulfide bonds affect protein stability.What would you expect to be the effect on proteinstability of introducing a disulfide bond using mutage-nesis? What would be the effect of removing a disulfidebond on protein stability?
6. What factors contribute to the observation of het-erogeneous folding kinetics of proteins? How wouldyou attempt to evaluate the importance of thesefactors.
7. ‘You cannot unscramble an egg’. Discuss this state-ment in the light of protein denaturation, refolding,modification and the known presence of chaper-ones.
8. Explain what might be the consequence of deletingthe octapeptide repeats of PrP on prion infection in forexample a host such as the mouse.

12Protein structure and a molecular
approach to medicine
Introduction
The introduction to this book made clear that onemajor reason for studying protein structure and functionis to obtain insight into diseases afflicting moderncivilizations. These diseases range in diversity fromcancer to cholera, from human immunodeficiencyvirus (HIV) to hepatitis, from malaria to myocardialinfarction to name a few. One potential impact ofimproved structural knowledge is the ability to treatmany diseases with highly specific drugs. For bacterialand viral infections immunization has been refined toallow the successful introduction of live, but attenuated,biological samples or components of the active protein.Inoculation elicits an immune reaction that conferslifelong or substantial immunity and has played amajor role in improved health care. In this mannerdiseases such as smallpox have been eradicated fromall parts of the world whilst polio and measles havebeen drastically limited in distribution.
For diseases such as cholera treatments have beendevised that limit routes of infection. Cholera spreadsvia a faecal–oral infection route and the presence ofthe bacterium Vibrio cholerae. By establishing cleanwater supplies infection routes via contaminated wateror food is limited. Infection leads to colonization ofthe intestines and the production of toxin that results
in chronic diarrhea which will, if left untreated, resultin death as a result of fluid loss leading to shock andacidosis. Victims die of dehydration unless water andsalts are replaced.
Cholera represents one example of how diseaseorigin is traced back to the action of one proteinin this case a toxin. By studying the structure andfunction of the cholera toxin we gain insight into themolecular events that underpin disease. The toxin isa hexameric protein of 87 kDa (subunit compositionof AB5) that binds to a ganglioside known as GM1via the B subunits and transfers the A subunit acrossmembranes via receptor mediated endocytosis. Insidethe cell reductive cleavage of a disulfide bond releasesa 195 residue fragment from the A subunit that isresponsible for pathogenesis.
The fragment catalyses the transfer of ADP-ribosefrom NAD+ to the side chain of Arg187 in the Gα
subunit of the heterotrimeric Gs protein; a processknown as ADP-ribosylation (Figure 12.1). GTP bind-ing leads to dissociation into Gα and Gβγ each ofwhich activates further cellular components. G pro-tein activation is usually transitory because Gα is aGTPase that hydrolyses GTP to GDP and promotesG protein re-association. Gα-GTP activates adenylatecyclase with the cAMP acting as a second messen-ger and regulating the activity of cAMP dependent
Proteins: Structure and Function by David Whitford 2005 John Wiley & Sons, Ltd

440 PROTEIN STRUCTURE AND A MOLECULAR APPROACH TO MEDICINE
N+
CO
NH2
O
PO
O
O
P OO
O
OCH2
H
OH
H
OH
H H
AdenosineCH2
CH2
CH2
NH
NH2
+
NH2
Gα subunit
CH2
CH2
CH2
NHNH2
+
NH O
P O
O
O
PO O
O
O CH2
H
OH
H
OH
HH
Adenosine
NH
+
CO
NH2
Gα subunit
+
+NAD+
Nicotinamide
ADP-ribosylation of Gα subunit
Arginine side chain
Figure 12.1 Cholera toxin promotes ADP-ribosylation of the Gα subunit of heterotrimeric G proteins
protein kinase. ADP ribosylation of the Gα subunithas a critical effect; it continues to activate adeny-late cyclase whilst inhibiting the GTPase activity ofGα. Adenylate cyclase remains in a permanently acti-vated state along with Gα and cellular cAMP lev-els increase dramatically. Intestinal cells respond toincreased cAMP levels by activating sodium pumpsand the secretion of Na+. To compensate chloride,bicarbonate and water are also secreted but the neteffect is the loss of enormous quantities of fluid anddehydration.
A molecular approach to the study of cholera eluci-dated the basis of toxicity and showed that dehydrationis counteracted by replenishment of fluids containing
salts. Significantly, other bacterial toxins such as E. coliheat stable enterotoxins and the pertussis toxin from thebacterium Bordetella pertussis act via similar mecha-nisms. Pertussis is responsible for whooping cough inyoung babies and is effectively countered in econom-ically advantaged countries by vaccination of youngbabies around 6 months of age using an inactivatedfragment of the whole toxin.
The fundamental events contributing to two dis-eases have been elucidated through their commonmode of action and effective therapeutic strategies havebeen devised – rehydration and vaccination. How-ever, this approach is not always possible. Increas-ingly the techniques of molecular and cell biology

SICKLE CELL ANAEMIA 441
are providing insights into the underlying causesof disease giving rise to the new field of molec-ular medicine. Molecular medicine became a pos-sibility with the completion of genome sequencingprojects and the identification of many inherited dis-eases arising from mutated genes. Gene screeningis now established as an important diagnostic toolwith DNA extracted from biological samples and‘searched’ for defects. Defective genes can be assmall as a simple base change or may be largerinvolving missing, duplicated or translocated segmentsof genes.
The availability of ‘predictive’ gene tests hasincreased dramatically in the last decade. Currentlygenetic tests are available for the most commondiseases such as cystic fibrosis, neurodegenerativedisorders such amylotrophic lateral sclerosis (LouGehrig’s disease), Tay–Sachs disease, Huntington’sdisease, familial hypercholesterolaemia (a disease caus-ing catastrophically high cholesterol levels and myocar-dial infarction), childhood eye cancer (retinoblastoma),Wilms’ tumour (a kidney cancer), Li–Fraumeni syn-drome, familial adenomatous polyposis (an inheritedpredisposition to form pre-cancerous polyps and associ-ated with colon cancer), and BRCA1 (a gene implicatedin familial breast cancer susceptibility). The list couldbe extended for many more pages. Over 3000 mutatedgenes are known to be involved in inherited disordersor human disease.
Alongside molecular medicine there have beenmajor advances in biotechnology to produce protein-based drugs for the treatment of disease. Todayhuman insulin is given as fast and slow acting formsin the treatment of Type I diabetes arising fromdamage to the β cells of the islets of Langerhansfound in the pancreas. Human insulin is produced byrecombinant DNA methods and avoids many problemsfaced previously where insulin was extracted from thepancreas of pigs or cows.
Molecular medicine will occupy a pivotal role inclinical practice throughout the 21st century. Thischapter deals with a selective group of proteinsimplicated in disease states where there is a growingunderstanding of the biological problem. As such itrepresents a series of case studies, by no meanscomplete, described to provide a flavour of modernapproaches to molecular medicine.
Sickle cell anaemia
The discovery of Ingram in 1956 that sickle cellanaemia arose from the substitution of glutamate byvaline at position 6 of the β subunit of haemoglobinwas the first example of a molecular defect contributingto human disease. It represents a key event, perhapseven the origin, of molecular medicine. Mutationsin haemoglobin were identified because the proteinis obtained from individuals in sufficient quantitiesfor peptide sequencing; this situation rarely extendsto other diseases where samples are normally onlyobtained via invasive biopsies or post-mortem.
Substitution of valine by glutamate alters the sur-face properties of the β subunit leading to aggre-gates of protein in the deoxy form of haemoglobin S
(HbS). The hydrophobic side chain of Val6 projects outinto solution fitting precisely into a small hydropho-bic pocket on the surface of a neighbouring β sub-unit formed by Phe85 and Leu88. The pocket favoursaggregation whilst oxygenation removes this pocketby conformational change. In normal haemoglobinthe charged side chain of glutamate is not accom-modated within a non-polar surface region. Sud-denly the molecular basis of sickle cell anaemiais clear.
Long polymers or fibres of deoxy HbS ∼20 nm indiameter extend the length of the erythrocyte deforminga normal biconcave cell into a sickle shape wheredistortion leads to cell rupture (Figure 12.2). Thegreatest danger of aggregation occurs in the capillarieswhere oxygen is ‘off loaded’ to tissues and theconcentration of deoxy-HbS is higher. Precipitationprevents blood flow through the capillaries causingtissue necrosis and life threatening complications.
Sickle cell anaemia occurs when an individual car-ries two mutated forms of the β subunit gene. A sin-gle copy leads to the sickle cell trait and is gener-ally asymptomatic. Generally, ‘harmful’ mutations arepresent at very low frequency and it is unusual fora mutant gene to be present at high frequency unlessit confers a specific evolutionary advantage. In cer-tain regions of West and Central Africa approximately25 percent of the population exhibit heterozygosity, orthe sickle cell trait. This is unexpected since harm-ful mutations are normally selectively weeded out by

442 PROTEIN STRUCTURE AND A MOLECULAR APPROACH TO MEDICINE
Figure 12.2 Electron micrograph of deoxy-HbS fibresspilling out from a ruptured erythrocyte
evolution. It is clear that the sickle cell trait corre-lates closely with the prevalence of malaria. Malariakills over 1 million individuals each year mostly youngchildren – a staggering and regrettable statistic – andis caused by the mosquito-borne protozoan parasitePlasmodium falciparum. The parasite resides in theerythrocyte during its life cycle and causes adhesionof red blood cells to capillary walls. Individuals withthe sickle cell trait possess red blood cells containingabnormal haemoglobin and these tend to sickle wheninfected by the parasite. The result is that infected cellsflow through the spleen and are rapidly removed fromcirculation. In other words individuals with sickle celltrait deal with the parasite more effectively than normalindividuals – the consequence is that the mutant genepersists in malaria-endemic regions.
The gene encoding the β subunit is located on chro-mosome 11 – a region containing many other globingenes and known as the β globin cluster. Polymorphismsin this region are equated with disease severity andseveral distinct haplotypes are identified in sickle cell
anaemia and given names reflecting their origins withinAfrica, such as Senegal haplotype, the most benign formof sickle cell disease, the Benin haplotype and the Cen-tral African Republic haplotype–one of the severestforms of sickle cell anaemia.
Screening of individuals is routinely performed usingelectrophoresis, where the sickle cell haemoglobinshows altered migration properties when comparedwith the normal protein. Tests are cheap, quick andreliable and a positive result points towards furtherdiagnostic investigation. Tests provide prospectiveparents with realistic information on the inheritanceof the trait. When both parents carry the sickle celltrait there is a 25 percent chance of the offspringhaving sickle cell anemia, a 50 percent chance ofthe sickle cell trait and a 25 percent chance of anormal individual. Despite good screening proceduresthe treatment of sickle cell anaemia is problematic andhighlights a major dilemma of molecular medicine.Diseases can be identified but frequently there is nosimple ‘cure’. Before the introduction of antibioticsindividuals with sickle cell anemia frequently diedin childhood. Modern care involves treatment withpenicillins and significantly reduces infant mortality.
Viruses and their impact on health asseen through structure and function
Viral infections are well known to all of us sincethey are responsible for the common cold as wellas many more serious diseases. The term virus isderived from the Latin word for poison and seemsentirely appropriate in view of the devastating illnessescaused in humans from viral infections. The diseasesattributed to viruses are vast and include influenza(flu), measles, mumps, smallpox, yellow fever, rabies,poliomyelitis, as well as acquired immune deficiencysyndrome (AIDS), haemorrhagic fever and certainforms of cancers.
The existence of sub-microscopic infectious agentswas suspected by the end of the 19th century whenthe Russian botanist Dimitri Ivanovsky showed thatsap from tobacco plants infected with mosaic diseasecontained an agent capable of infecting other tobaccoplants even after passing through filters known toretain bacteria. Plant and animal cells are not the only

HIV AND AIDS 443
living cells subject to viral attack. In 1915 FrederickTwort isolated filterable entities that destroyed bacterialcultures and produced cleared areas on bacterial lawns.The entities were bacteriophages, often abbreviated tophage, that attach to bacterial cell walls introducinggenetic material. The result is rapid replication ofnew viral progeny with cell lysis releasing newinfectious particles.
Crystallization of tobacco mosaic virus (TMV) byW.M. Stanley in 1934 represented one of the firstsystems to be crystallized but also emphasized thehigh inherent order present in viruses. SubsequentlyTMV has been a popular experimental subject. Oneimportant conclusion from studying TMV was thatviruses were constructed using identical subunitspacked efficiently around helical, cubic or icosahedralpatterns of symmetrical organization. To clarify theterminology for virus components a number of termswere introduced. The capsid denotes the protein shellthat encloses the nucleic acid and is based on asubunit structure. The subunit is normally the smallestfunctional unit making up the capsid. The capsidtogether with its enclosed nucleic acid (either DNAor RNA) constitutes the nucleocapsid. In some virusesthe nucleocapsid is coated with a lipid envelopethat can be derived from host cells. All of thesestructures form part of the virion – the completeinfective virus particle.
Viruses lack normal cellular structure and donot perform typical cell-based functions such asrespiration, growth and division. Instead they areviewed as ‘parasites’ that at one stage of a life cycleare free and infectious but once entered into a livingcell are able to sequester the host cell’s machinery forreplication. The conversion of viral genetic materialinto new viruses or the incorporation of the viralDNA into the host genome can occur via a varietyof different routes and depends on the organization ofthe viral genome.
The Baltimore classification (Table 12.1) is basedon nuclei acid type and organization and uses therelationship between viral mRNA and the nucleic acidfound in the virus particle. In this system viral mRNAis designated as the plus (+) strand and a RNA or DNAsequence complementary to this strand is designated asthe minus (−) strand. Production of mRNA requires theuse of either DNA or RNA (−) strands as templates
and viruses can exist as double-stranded DNA, single-stranded + or − DNA, double-stranded RNA, andeither (−) or (+) single strands of RNA. Almostall viruses can be classified in this scheme. RNA-based viruses form DNA using the enzyme reversetranscriptase and represent retroviruses. Retrovirusesare involved in many diseases, transforming cellsinto cancerous states by the conversion of geneticmaterial (RNA) into DNA followed by integrationinto the host cell genome (proviral state) and theproduction of transcripts and translation products viahost cell machinery.
The two most important retroviruses in terms of theireffect on man are the human influenza virus (flu) andHIV. These viruses are responsible for the two greatestpandemics affecting humans over the last 100 years.
Viruses, like bacterial infective agents, act asantigens in the body and elicit antibody formationin infected individuals. As a result vaccines can bedeveloped against viruses most frequently based on acomponent of the virus coat or an inactivated formof the virus. Although diseases such as smallpox havebeen effectively eliminated through the use of vaccinesthis is not always an effective treatment route. Forinfluenza and HIV this is particularly difficult andlies at the heart of the pandemics caused by theseagents. These retroviruses are of considerable relevanceto health care in the modern era and are describedas a result of their importance in two major diseasesaffecting the world today.
HIV and AIDS
HIV has been the subject of intense research since 1983when Luc Montagnier, Robert Gallo and colleaguesidentified it as the causative agent contributing toa complex series of illnesses grouped under theterm ‘acquired immune deficiency syndrome’ (AIDS).The disease was originally recognized in the UnitedStates at the beginning of the 1980s as one affectingprimarily homosexual men and led to a systematicdecrease in the ability of the body to fight infectionarising from a progressive loss of lymphocytes. Thedecline in the immune response led to the nameAIDS. Lymphocyte loss rendered the body defenselessagainst infection, with individuals succumbing to rare
444 PROTEIN STRUCTURE AND A MOLECULAR APPROACH TO MEDICINE
Table 12.1 A scheme of classifying viruses according to their mode of replication and genomic material sometimescalled Baltimore classification
Genome organization Examples Details of replication
ds DNA Adenoviruses
Herpesviruses
Adenoviruses replicate in the nucleus using cellular
proteins. Poxviruses replicate in the cytoplasm making
their own enzymes for replication
Poxviruses
ss (+) sense DNA Parvoviruses Replication occurs in the nucleus, involving the
formation of a (−) sense strand, which serves as a
template for (+) strand RNA and DNA synthesis
ds RNA Reoviruses Viruses have segmented genomes. Each genome segment
is transcribed separately to produce monocistronic
mRNAs
Birnaviruses
ss (+) sense RNA Picornaviruses
Togaviruses
(a) Polycistronic mRNA, e.g. picornaviruses means
naked RNA is infectious. Translation results in the
formation of a polyprotein product, which is cleaved
to make mature proteins
(b) Complex transcription, e.g. togaviruses. Two or more
rounds of translation are necessary to produce the
genomic RNA
ss (−) sense RNA Orthomyxoviruses
Rhabdoviruses
Must have a virion particle RNA directed RNA
polymerase
(a) Segmented, e.g. orthomyxoviruses. First step in
replication is transcription of the (−) sense RNA
genome by the virion RNA-dependent RNA
polymerase to produce monocistronic mRNAs, which
also serve as the template for genome replication
(b) Non-segmented, e.g. rhabdoviruses. Replication
occurs as above and monocistronic mRNAs are
produced
ss (+) sense RNA with DNA
intermediate in life-cycle
Retroviruses Genome is (+) sense but does not act as mRNA. Instead
acts as template for reverse transcription
ds DNA with RNA
intermediate
Hepadnaviruses Viruses also rely on reverse transcription. Unlike
retroviruses this occurs inside the mature virus
particle. On infection the first event to occur is repair
of gapped genome, followed by transcription
ds = double stranded, ss = single stranded.

HIV AND AIDS 445
opportunistic infections or diseases such as pneumoniaand tuberculosis (TB). In some cases rare forms ofskin cancer (Kaposi’s sarcoma) were associated withadvanced HIV infection along with other cancers.
In the immune system a defining event in HIV-mediated disease is a fall in the number of circulatinghelper T cells and this feature is used as a marker forimmunodeficiency. Once the absolute T-cell count fallsbelow a threshold of 200 cells per mm3 in the peripheralblood individuals are vulnerable to AIDS-definingopportunistic infections. The virus preferentially infectshelper T cells, as well as other cells such as macrophagesand dendritic cells, because of the presence of CD4, acell surface protein that acts as a high affinity receptorfor the virus. The interaction between CD4 and virusmediates entry of the virus into the host cell and is acritical step in the overall process of HIV pathogenesis.
Subsequently, the disease was shown to exist inother populations; it was not restricted to homosexualsnor was it confined to the United States (Figure 12.3).HIV infection is responsible for significant mortalityin all sexually active populations and is presentthroughout the world. In many countries the absenceof good health care systems has meant that AIDShas become the fourth leading cause of mortalitythroughout the world and is responsible for majordecreases in life expectancy. Infection with HIValso occurs from intravenous drug use and is also
Figure 12.3 The sites of key events in thedevelopment of the HIV epidemic. This world maphighlights the first known cases of infection withHIV-1 and HIV-2 in West and sub-Saharan Africa
observed as a result of blood transfusions using infected(unscreened) donated samples.
The spectre of progressive HIV infection spread pre-dominantly through sexual intercourse has dominatedhealth care issues in all areas of the world. In 2003it was estimated by the World Health Organizationthat over 40 million individuals were affected by HIVinfection – a sombre and immensely problematic issue.There is little doubt that HIV infection and its sequelaewill be responsible for massive increases in mortal-ity rates, especially in sexually active populations andwill also affect the young as a result of fetal or neona-tal transmission. The pandemic nature of the diseasemeans that dealing with HIV-infected individuals is amajor health care issue in all areas of the world.
The HIV genome
HIV is a member of the lentivirus genus that includesretroviruses with complex genomes. An early objectivein studying HIV was to sequence the genome tounderstand the size, function and properties of thevirally encoded proteins. The HIV genome is small(∼9.4 Kb) and encodes three structural proteins (MA,CA, and NC), two envelope proteins (gp41, gp120),three enzymes (RT, protease and integrase), and sixaccessory proteins (Tat, Rev, Vif, Vpr, Nef & Vpu)(Figures 12.4 and 12.5).
These viruses exhibit cone-shaped capsids coveredwith a lipid envelope derived from the membraneof the host cell. Puncturing the exposed surface areglycoproteins called gp120 – a trimeric complex ofproteins anchored to the virus via interactions with atransmembrane protein called gp41. Both gp120 andgp41 are derived from the env gene product. Insidethis layer is a shell, often called the matrix, composedof the matrix protein (MA). The matrix protein linesthe inner surface of the viral membrane and a conicalcapsid core forms from the major capsid protein (CA).This complex is at the centre of the virus and surroundstwo copies of the unspliced viral genome that arestabilized by interaction with the nucleocapsid protein(NC). One major region of the HIV genome is the polgene (Figure 12.5); its product is a polyprotein of threeessential enzymes. These enzymes are protease, reversetranscriptase and integrase.

446 PROTEIN STRUCTURE AND A MOLECULAR APPROACH TO MEDICINE
Figure 12.4 A picture of the organization of protein and RNA within the intact human immunodeficiency virus
Figure 12.5 Organization of the HIV genome and the proteins derived from the genome
Of the accessory proteins three function within thehost cell by facilitating transcription (Tat), assistingin transport of viral DNA to the cell nucleus (Rev),and acting as a positive factor for viral expression(Vif). These proteins are not found within the virion.The other three proteins are packaged with the virus
and play important roles in regulating viral promoters(Vpr), acting as a negative regulatory factor for viralexpression (Nef) and promoting viral release and CD4degradation (Vpu) (Table 12.2).
The first phase of infection results in virus binding toCD4+ lymphocytes. The helper T cells are the primary

HIV AND AIDS 447
Table 12.2 Functional role of HIV encoded gene products
Protein Gene Role
MA (p17) Gag Matrix proteinCA (p24) Gag Matrix proteinNC (p7) Gag Nucleocapsid proteingp120 Env Extracellular glycoprotein binds to CD4 receptorgp41 Env Transmembrane proteinReverse transcriptase (RT) Pol Conversion of RNA > DNAIntegrase Pol Insertion of DNA into host genome.Protease Pol Processing of translated polyproteinRev Rev Regulate expression of HIV proteinsNef Nef Negative regulatory factorVpr Vpr Viral (HIV) promoter regulatorVpu Vpu Viral release and CD4 degradationVif Vif Viral infectivity factorTat Tat Transcriptional activator
The three viral encoded enzymes play a major role in the HIV replication cycle and the formation of new virions andhave formed the basis of most drug-based treatments of HIV infection. In part this has arisen from the highly resolvedstructural models available for reverse transcriptase, the integrase and protease of HIV.
target and binding leads to a series of intracellularevents that causes their destruction with a half-life ofless than two days. During initial stages of infection theviral genome becomes integrated into the chromosomeof the host cell and is followed by a later phase ofregulated expression of the proviral genome leading tonew virion production. The stages of viral infectionrequire the concerted involvement of viral proteins;this includes, for example, gp120-mediated binding toCD4+ cells, conversion of viral RNA into DNA byreverse transcriptase, integration of viral DNA into thehost chromosome by integrase, nuclear localization ofviral DNA by Vpr, and processing of viral mRNAs byTat, Rev and Nef.
The structure and function of Rev, Nefand Tat proteins
Nef is a single polypeptide chain of 206 residuesoriginally recognized as a ‘negative factor’ where itsabsence caused low viral loads in cells. After viral
infection the protein is expressed in high concentrationsand inoculation of Rhesus monkeys with a Nef-deletion strain in the closely related SIV (simianimmunodeficiency virus) did not lead to disease.Nef has at least two distinct roles during infection;it enhances viral replication, and by an unknownmechanism it reduces the number of CD4 receptorsfound on the surface of infected lymphocytes. Theprotein is unusual in having a myristoylated N-terminalresidue indicating membrane association; removal ofthis modification inhibits activity. The structure ofthe core domain of Nef (residues 71–205) has beendetermined by NMR and crystallographic methods,with both techniques suggesting a fold containing ahelix-turn-helix (HTH) motif with three α helices,a five-stranded antiparallel β sheet, an unusual left-handed poly-L-proline type-II helix and a 310 helix.
The role of Rev is to regulate expression of HIVproteins by controlling the export rate of mRNAs fromthe nucleus to the cytoplasm where translation occurs.The Rev protein contains 116 residues and within thissequence are nuclear localization and export sequences

448 PROTEIN STRUCTURE AND A MOLECULAR APPROACH TO MEDICINE
(NLS and NES) consistent with its role in mRNAexport, but a structure has not been determined.
The final non-structural protein synthesized bythe virus and packaged within the core is Tat–atranscriptional activator that enhances transcription ofintegrated proviral DNA. Transcription starts at theHIV promoter–a region of DNA located within the 5′long terminal repeat region with the promoter bindingRNA polymerase II and other transcription factors.Transcription frequently terminates prematurely dueto abortive elongation and the Tat protein overcomesthis problem by binding to stem–loop sites on thenascent RNA transcript. Description of the Tat proteinis complicated by translation via two exons leadingto a protein containing between 86 and 101 residues;the first exon encodes residues 1–72 and the secondexon translates as residues 73–101. Structural studieshave utilized an 86-residue fragment despite mostHIV strains containing full-length protein, but haveyet to yield a detailed picture for Tat. The proteinis divided into four distinct regions; an N-terminalcysteine-rich domain, a core region, a glutamine-richsegment and a basic zone. The basic region contains thesequence R49KKRRQRRR57 essential for recognitionand binding to RNA.
The role of Vif, Vpr and Vpu
A similar lack of structural information pertains to Vif,Vpr and Vpu. These three proteins range in size from15 to 23 kDa and structures are unavailable, althoughconsiderable functional data has been acquired. Itis known that Vpr, a 96-residue accessory proteinpresent early in the viral life cycle, plays an importantregulatory role during replication. Vpr localizes to thenucleus and is implicated in cell cycle arrest at theG2/M interface. Vpr also enhances transcription fromthe HIV-1 promoter structure and cellular promoters.
Vif stands for viral infectivity factor and is a basicprotein (23 kDa) whose precise function is unknown.Naturally occurring strains of HIV strains bearingmutations in Vif are known to replicate at significantlylower levels when compared with the wild-type virus.In some instances this is associated with low viralloads, high CD4 T-cell counts and slower progressionto advanced forms of the disease. These resultsemphasize the importance of Vif and homologous
proteins have been recognized in other lentivirusesfrom a highly conserved – SLQSLA motif locatedbetween residues 144–149. Vif is required during laterstages of virus formation since virions produced inthe absence of Vif cannot complete proviral DNAintegration into host chromosomes. Unlike Vpr, Vifis primarily found in the cytoplasm as a membrane-associated protein.
Vpu is a small integral membrane protein that reg-ulates virus release from the cell surface and degra-dation of CD4 in the endoplasmic reticulum. Thesetwo distinct biological activities represent independentmechanisms attributed to the presence of distinct struc-tural domains within Vpu. A transmembrane domainis associated with virus release from the cell whereasCD4 degradation involves interaction with the cyto-plasmic domain.
In contrast to the above proteins considerableunderstanding of HIV has come from structural studiesof the remaining components of the virion particularlythe enzymes and envelope proteins. Molecular biologyhas allowed individual proteins, or more frequentlydomains derived from whole proteins, to be cloned,expressed and subjected to detailed structural analysis.These studies assist in providing a picture of theirroles during the viral life cycle and understandingthe interdependence of structure and function in theseproteins represents a key effort in the attempts tocombat the disease. Alongside a large body of data forthe reverse transcriptase and protease there also existsstructural information for gp120, the integrase, domainsof the transmembrane protein gp41 (the TM domain)and the matrix, capsid and nucleocapsid proteins.
Structural proteins of HIV
Structural proteins are derived from the Gag geneproduct by proteolytic cleavage to yield the MA,CA and NC proteins (Figure 12.6). With a combinedmass of ∼55 kDa the Gag polyprotein yields a matrix(MA) protein of 17 kDa, the capsid protein (CA) of24 kDa and the nucleocapsid protein (NC) of ∼7 kDa.The structure of the MA protein consists of five α
helices, two short 310 elements and a three-stranded β
sheet. Although monomeric in solution several lines ofevidence indicate trimeric structures represent ‘buildingblocks’ for the matrix shell within a mature virion.

HIV AND AIDS 449
MA
SU NC CA (N-term)
CA (C-term)
IN (N-term)
IN (C-term)
IN (core)
PR
RT
Nef (core)
TM (ectodomain)
Figure 12.6 The structure of HIV. Drawing ofthe mature HIV virion surrounded by ribbonrepresentations of the structurally characterized viralproteins and fragments. The protein structures havebeen drawn to approximately the same scale whilstthe TM ectodomain shown is that determined for theclosely related SIV (reproduced with permission fromTurner, B.G. & Summers, M.F. J. Mol. Biol. 1999, 285,1–32. Academic Press)
The capsid protein occurs at ∼2000 copies permature virion forming a cone-shaped, dense, structureat the virus centre that encapsulates RNA as wellas enzymes, NC protein and accessory proteins.Two copies of unspliced RNA are packaged ina ribonucleoprotein complex formed with the NCprotein. The CA protein has proved difficult to isolateand no structure is available for the intact protein. Inan N-terminal domain seven helices, two β hairpinsand an exposed partially ordered loop are observedwith the domain forming an arrowhead shape. Thisdomain associates with similar regions as part of thecondensation of the capsid core.
The nucleocapsid or NC protein is involved inrecognition and packaging of the viral genome. Retro-viral NC proteins contain the Cys-X2-Cys-X4-His-X4-Cys array characteristic of Zn finger proteins, and in
HIV the NC protein contains two such elements. Thestructure of this small protein is defined by these twoZn finger elements where reverse turns form a metalbinding centre that leads via a loop to a C terminal 310
helix. In the NC protein the two metal-binding centresmay interact although this is thought to be a flexible,dynamic interaction and both play a role in bindingRNA via conserved hydrophobic pockets.
Reverse transcriptase and viral protease
Two of the three enzymes, reverse transcriptase andprotease, are amenable targets for drug therapy largelyas a result of detailed structural analysis. Reversetranscriptase is responsible for the conversion of RNAto DNA whilst the protease catalyses processing ofthe initial polyprotein (Gag-Pol) product into separatefunctional proteins.
HIV protease The protease was the first HIV-1protein to be structurally characterized and is translatedinitially as a Gag-Pol fusion protein; its role involvesproteolytic cleavage of the polyprotein to release activecomponents. In an autocatalytic reaction analogousto that seen in zymogen processing the proteaseliberates subunits found in the mature virus. TheGag precursor forms the structural proteins whilst Polproduces enzymes – reverse transcriptase, integrase,and protease.
As a protease the HIV enzyme is unique is catalysingbond scission between a tyrosine/phenylalanine anda proline residue with no human enzyme showing asimilar specificity. Despite unique specificity the HIVprotease is structurally similar to proteases of thepepsin or aspartic protease family and this has assistedcharacterization. Historically, pepsin was the secondenzyme to be crystallized (after urease) and structuresare available for pepsins from different sources. Allreveal bilobal structures with two aspartate residueslocated close together in the active site. These residuesactivate a bound water molecule for use in proteolyticcleavage of substrate.
The bilobal structure of pepsin is based around asingle polypeptide chain of ∼330 residues where onelobe contains the first 170 residues and the remainingresidues are located in a second lobe. Each lobe isrelated by an approximate two-fold axis of symmetry

450 PROTEIN STRUCTURE AND A MOLECULAR APPROACH TO MEDICINE
Figure 12.7 The overall topology of pepsin showingstrand-rich secondary structure arranged within twolobes. Secondary structure elements for residues 1–170of the first lobe are shown in orange with the remainingsecondary structure shown for the second lobe in blue.Asp32 and 215 of the active site are shown in red
and at the centre, between each lobe, is the activesite (Figure 12.7). It seems probable that this topologyarose by gene duplication because each lobe contributesa single Asp side chain to the active site foundwithin an Asp-Thr/Ser-Gly- motif preceded by twohydrophobic side chains. In pepsin the catalytic Aspresidues are at positions 32 and 215. Each side chaininteracts via a complex network of hydrogen bonds(Figure 12.8) with at least one molecule of water. The
N
CH
C
O
CH2
C O
O−
N
CH
C
O
CH2
CO
O
H
OHH
Asp32 Asp215
Figure 12.8 The interaction between Asp32 and 215in pepsin involves a bound water molecule and twoprotonation states for the Asp side chain. A watermolecule bridges between two oxygen atoms, one oneach of the carboxyl groups of the side chain
side chains exhibit very different protonation profiles;one aspartate has a low pK value around 1.5 whereasthe other exhibits a higher than expected value of 4.8.These values vary from one enzyme to another but theprinciple of two different pK values for aspartyl sidechains is conserved in all proteins.
Pepsin shows optimal activity around pH 2.0and substrates bind to the active site of pepsinwith the C=O group of the scissile bond situatedbetween the Asp side chains with the oxygen atomin a position comparable to that of the displacedwater molecule. The unionized side chain of Asp215protonates the carbonyl group allowing the displacedwater molecule, hydrogen bonded to the side chainof Asp32, to initiate a nucleophilic attack on thecarbonyl carbon atom by transferring a proton to Asp32 (Figure 12.9). The resulting proton transfer forms atetrahedral intermediate that promptly decomposes withprotonation of the NH group.
HIV protease uses a slightly different trick toachieve the same catalytic function. The protein isnot a single polypeptide chain but is a symmetricalhomodimer with each chain containing 99 residues(Figure 12.10). In itself this organization supports apicture of aspartyl proteases arising from duplication ofan ancestral gene. Crystallography shows the structureof this dimer as two identical chains each subunitcontaining a four-stranded β sheet formed by the N-and C-terminal strands together with a small α helix.The enzyme’s active site is unusual in that it is formedat the interface of the two subunits. However, again theactive site is based around a group of three residuesin each subunit with the sequence Asp25-Thr26-Gly27containing the catalytically relevant acidic side chainthat is responsible for bond cleavage. The residues inthe second subunit are often denoted as Asp25′, Thr26′ and Gly27′.
The cavity region is characterized by two flexibleflaps at the top of each monomer formed by anantiparallel pair of β strands linked by a four residueβ turn (49–52). These flaps are also seen in pepsinand guard the top of the catalytic site with the activesite Asp residues located at the bottom of this cleft.These ‘flap’ regions are flexible, and from studiesof HIV protease in the presence of inhibitors itwas observed that significant conformational changesoccur in this region. Some backbone Cα atoms show

HIV AND AIDS 451
Asp215 Asp215
Asp32
Asp215
Asp32
Asp215
RHO
NHR'
OH
RHO
NHR'
OH
O−
O
CH2
O O
OHH
RO
NHR'
CH2
O
O
Asp32
CH2
CH2
O O−
CH2
O
O
Asp32
CH2
CH2
O O−
O−
O
CH2
O O
RO
OH
NH2R'
1
2
3
4
H
H
H
H
Figure 12.9 The catalytic activity of aspartylproteases such as pepsin. The reaction mechanisminvolves the two aspartyl side chains interactingdirectly with substrate and a bound water molecule.Asp32 accepts a proton from water whilst Asp215donates a proton to the substrate (1), a tetrahedralintermediate formed breakdown via the donation andacceptance of protons (2) with the resulting productyielding a free amino group and a free carboxyl groupwithin the substrate (3). The dissociation of productsallows the pepsin to start the reaction cycle again (4)
displacements of ∼0.7 nm when the bound and freestates of the enzyme are compared around the activesite region.
Catalysis is mediated by twin Asp25 residuescharacterized by pK values that deviate considerablyfrom a normal value around 4.3. One carboxyl groupexhibits an unusually low pK of 3.3 whilst the otherdisplays an unusually high pK of 5.3. The catalyticdiad interacts with the amide bond of the targetsubstrate in a manner analogous to that describe forpepsin. Inhibiting the protease represents one potential
Figure 12.10 The HIV protease homodimer. The twomonomers are shown in orange and blue
method of fighting HIV infection since this willprevent processing of the Gag-Pol polyprotein andthe generation of proteins necessary for new virusformation. Many crystal structures of HIV-1 proteasetogether with inhibitor complexes are known as part ofdrug development aimed at catalytic inhibition. In viewof the preference of HIV protease for Tyr-Pro and Phe-Pro sequences within target substrates inhibitors havebeen designed to mimic the transition state complexand to prevent, by competitive inhibition, the turnoverof the protease with native substrate (Figure 12.11).
The protease inhibitors are known as peptidomimeticdrugs because of their imitation of natural peptide sub-strates. These drugs mimic the enzyme’s transitionstate tetrahedral complex and attempt to reproduce thehydrophobic side chain of phenylalanine, the restrictedmobility of prolyl side chains and a carbonyl centrethat is not easily protonated.
N
O
HN
O
NH
O
CH3
N
R
R'
H
Figure 12.11 Ala-Phe-Pro sequence containing thetarget peptide bond (substrate) for HIV protease. Thebond to be cleaved is shown in red

452 PROTEIN STRUCTURE AND A MOLECULAR APPROACH TO MEDICINE
N
O NH
NHN
OH
NH
O
CH2
O
NH2OH
H
Figure 12.12 The structure of saquinavir
Several drugs were rapidly brought from the labo-ratory through clinical trials and into effective use in1995 and included ritonavir, indinavir, nelfinavir andsaquinavir (Figure 12.12). The presence of additionalfunctional groups such as OH groups close to the targetpeptide bond coupled with aromatic groups results ininhibitors that bind with high affinity (KI < 1 nM) anddecrease viral protease activity by acting as compet-itive inhibitors. Although these drugs are responsiblefor remarkable increases in life expectancy they do noteliminate virus from the body. In general, the proteaseinhibitors are given when treatment with drugs aimedagainst reverse transcriptase fails or may be given incombination with other drug therapies. Unsurprisingly,these drugs have side effects but have proved effectiveat limiting viral proliferation. A major problem withthis approach is resistance due to mutations in HIVprotease that prevent the inhibitor from binding withoptimal efficiency. Mutations within HIV protease leadto a loss of inhibitor binding but protein that retains cat-alytic activity. The mutations arise from the high errorrate associated with activity of reverse transcriptase.
Reverse transcriptase The other major target forantiviral drug therapy is reverse transcriptase. The viralgenome of RNA is transcribed into DNA by the actionof reverse transcriptase followed by the integrationof viral DNA into host cell chromosomes by theintegrase. Reverse transcriptase is derived from theGag-Pol precursor by proteolytic cleavage producingtwo homodimers of mass 66 000 (p66). The p66 subunitis bifunctional with a polymerase activity located inthe larger of two domains and RNase activity locatedin a second smaller domain (called RNaseH). Themature, i.e. active reverse transcriptase is unusual in
that it requires proteolytic removal of one of the RNasedomains from a p66 subunit to form a heterodimer ofp66 and p51 subunits. This heterodimer represents thefunctional enzyme of HIV.
Inhibiting the action of reverse transcriptase shouldslow down new virus production and by implicationdisease progression. To understand how this might beachieved it was necessary to understand the structureand function of the enzyme, as well as its interac-tion with substrates before effective inhibitors couldbe designed. From the structure of p66 (Figure 12.13)and p51 (Figure 12.14) subunits in the presence ofDNA and inhibitors as well as unliganded statesa number of important facts have emerged. Thepolymerase domain is composed of ‘finger’, ‘palm’,‘thumb’ and ‘connecting’ regions and this is rem-iniscent of the organization of other RNA poly-merases. Significantly the p66 subunit shows struc-tural similarity to the Klenow fragment of E. coliDNA polymerase I suggesting that conservative pat-terns of structure and organization exist within thisclass of enzymes.
Studies of both p66 and p51 subunits suggestedthat binding DNA does not perturb complex structuresignificantly, but that p51 and p66 differ in their
Figure 12.13 The organization of reverse transcrip-tase p66 subunit is shown. The finger domain is shownin red, the thumb in dark red, the palm in orangeand the connecting region in gold. The active siteaspartate residues lie at the base of the cleft betweenthumb and fingers

HIV AND AIDS 453
Figure 12.14 The p51 subunit of reverse transcrip-tase. The different conformations are clearly seen withthe relative orientations of sub-domains forming aclosed conformation in p51 and an open state in p66
structures largely as a result of the reorientation ofdomains or sub-domains within each subunit. The‘fingers’ sub-domain is composed of β strands andthree α helices whilst the ‘palm’ sub-domain possessesfive β strands hydrogen bonded to four β strands atthe base of the “thumb” region. The remainder of the‘thumb’ region is predominantly α helical and links to a‘connection’ sub-domain between the polymerase andRNaseH domains. The ‘connection’ domain is basedaround a large extended β sheet with two solitaryhelices. In p66 subunits the sub-domains are arrangedin an open configuration exposing the active sitecontaining three critical Asp side chains at residues110, 185 and 186. In contrast, structural analysisof the p51 subunit reveals that, despite identicalsequences, the polymerase sub-domains are packeddifferently with the fingers covering the palm and byimplication burying the catalytically important activesite residues. Unsurprisingly, functional studies revealthe p51 domain to be catalytically inactive although itinteracts with the RNaseH domain of the p66 subunitand is important to the enzyme’s overall conformation.
Inhibitors of reverse transcriptase fall into twoclasses. The first group based around the naturalnucleotide substrate bind to the active site pocketwhilst the second group, generally described as non-nucleoside inhibitors, function by binding at sitesremote from the catalytic centre. Taken together these
inhibitors represent two very different mechanisms ofaction. The first, and archetypal, reverse transcriptaseinhibitor is 3′-azido-3′ deoxythymidine (AZT) andwas the first HIV antiretroviral to be licensed foruse. It contains an azido group in place of a 3′OH group in the deoxyribose sugar. In a comparablefashion to dideoxynucleotides AZT acts as a chainterminator because it lacks a 3′ OH group. When thedrug enters cells it is phosphorylated at the 5′ endand incorporated into growing DNA chains by theaction of reverse transcriptase on the RNA template(HIV genome). The absence of a 3′ OH ensures thatpolymerization cannot continue. Although it mightbe expected that host DNA polymerases would beinhibited with severe consequences it appears fortuitousthat host cell polymerases do not bind these inhibitorswith high affinity. As a result AZT is effective againstviral replication by inhibiting only reverse transcriptaseactivity. Competitive inhibition has proved extremelyeffective in the primary treatment of HIV with AZT,introduced in 1987 and complemented by additionalnucleoside inhibitors, the accepted treatment option(Figure 12.15).
The non-nucleoside inhibitors (Figure 12.16) bindto a hydrophobic pocket remote from the active site.The effect of binding is to shift three β strandsof the palm sub-domain containing the active siteaspartyl residues towards a conformation seen in thep51 subunit. In other words, these inhibitors favour
OCH2OH
H
H
H
H
H H
Hypoxanthine
OCH2OH
H
N
H
H
H H
Thymine
N+
N
OCH2OH
H
H
H
H
HH
Cytosine
Figure 12.15 Active site (nucleoside inhibitors)of reverse transcriptase. Besides AZT, 2′,3′-dideoxycytidine (ddC) and 2′3′-dideoxyinosine (ddI)are other commonly used nucleoside analogues forfighting HIV infection

454 PROTEIN STRUCTURE AND A MOLECULAR APPROACH TO MEDICINE
NNN
O
NH
Nevirapine
Figure 12.16 Nevirapine, a non-nucleoside inhibitorof HIV reverse transcriptase
formation of inactive states by distorting the catalyticsite as the basis for pharmacological action.
To date these drugs represent the most effectiveroute of controlling HIV infection and are particu-larly important in the continual fight against viralproliferation. The combined use of nucleoside andnon-nucleoside inhibitors in a cocktail known ashighly active antiretroviral therapy (HAART) drasti-cally reduces viral load (the number of copies of virusfound within blood) and is associated with improvedhealth. Unfortunately the spectre of drug resistancemeans that additional drugs or alternative regimes areneeded to effectively combat the virus over a sustainedperiod of time.
The introduction of antiviral drugs has drasticallyimproved the quality of life and life expectancy ofindividuals infected with HIV but the prohibitive costof providing these medicines has restricted treatmentto countries with advanced forms of health care. Thisis particularly regrettable since HIV infection rates anddeaths attributable to advanced HIV disease are mostprevalent in the less economically advantaged areasof the world. Currently, many millions of individualsinfected with HIV are unable to obtain effective lifeextending treatment.
The surface glycoproteins of HIVand alternative strategies to fighting HIV
An alternative approach to fighting HIV infection is tocounter the ability of the virus to enter cells via theinteraction between surface viral glycoproteins and thehost cell receptors. The surface of HIV is coated with
spikes that represent part of the envelope glycoproteingp120. This exposed surface glycoprotein is anchoredto the virus via interactions with the transmembraneprotein gp41. Both proteins are synthesized initiallyas a precursor polyprotein – gp160. The envelopeproteins gp41 and gp120 have been characterizedas part of systematic attempts to understand themechanism of entry into cells.
Viral entry is initiated by gp120 binding to receptorssuch as CD4, a protein containing immunoglobulin-likefolds, expressed on the surface of a subset of T cellsand primary macrophages. The gp120 protein bindsto CD4 with high affinity exhibiting a dissociationconstant (Kd) of ∼4 nM and is a major route of HIVentry into cells although additional membrane proteins(the chemokine receptors CXCR4 and CCR5) can alsooperate in viral entry.
Structural studies of gp120 are aimed at understand-ing the molecular basis of protein–protein interactions.However, gp120 has proved a difficult subject becauseextensive glycosylation has prevented crystallization.Studies have therefore involved partially glycosylatedcore domains containing N- and C-terminal deletionsand truncated loop regions. Immunoglobulin-like vari-able domains exist within gp120 in the form of fiveregions (V1–V5) and are found in all HIV strains.These variable zones are interspersed with five con-served regions. Four of these variable regions arelocated as surface-exposed loops containing disulfidelinks at their base. Despite substantial deletions the coregp120 binds to CD4 and presents a credible version ofthe native interaction.
The gp120 core structure has ‘inner’ and ‘outer’domains linked by a four-stranded β sheet that acts as abridge. The inner domain contains two α helices, a five-strand β sandwich and several loop regions, whilst thelarger outer domain comprises two β barrels of six andseven strands, respectively. The core is often describedas heart shaped with the N- and C-terminals located atthe inner domain (top left-hand corner, Figure 12.17),the V1 and V2 loop regions located at the base of theinner domain and the V3 loop region located at thebase of the outer domain. The V4 and V5 loop regionsproject from the opposite side of the outer domain.
The inner domain is directed towards the virus andfrom mutagenesis studies the interaction site betweengp120 and CD4 is defined broadly at the interface
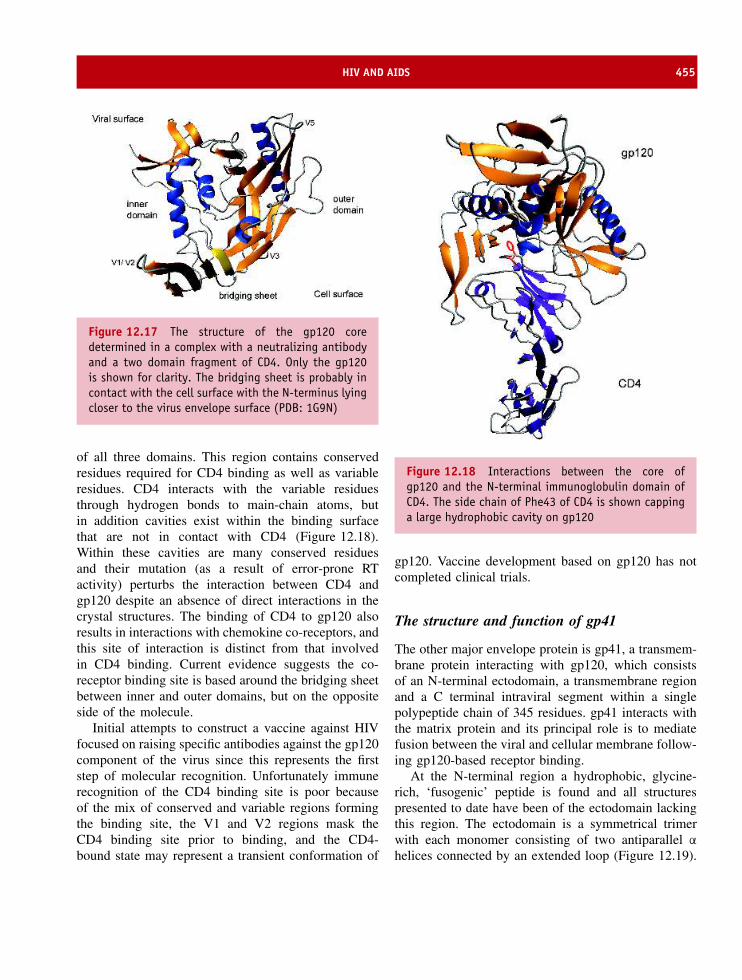
HIV AND AIDS 455
Figure 12.17 The structure of the gp120 coredetermined in a complex with a neutralizing antibodyand a two domain fragment of CD4. Only the gp120is shown for clarity. The bridging sheet is probably incontact with the cell surface with the N-terminus lyingcloser to the virus envelope surface (PDB: 1G9N)
of all three domains. This region contains conservedresidues required for CD4 binding as well as variableresidues. CD4 interacts with the variable residuesthrough hydrogen bonds to main-chain atoms, butin addition cavities exist within the binding surfacethat are not in contact with CD4 (Figure 12.18).Within these cavities are many conserved residuesand their mutation (as a result of error-prone RTactivity) perturbs the interaction between CD4 andgp120 despite an absence of direct interactions in thecrystal structures. The binding of CD4 to gp120 alsoresults in interactions with chemokine co-receptors, andthis site of interaction is distinct from that involvedin CD4 binding. Current evidence suggests the co-receptor binding site is based around the bridging sheetbetween inner and outer domains, but on the oppositeside of the molecule.
Initial attempts to construct a vaccine against HIVfocused on raising specific antibodies against the gp120component of the virus since this represents the firststep of molecular recognition. Unfortunately immunerecognition of the CD4 binding site is poor becauseof the mix of conserved and variable regions formingthe binding site, the V1 and V2 regions mask theCD4 binding site prior to binding, and the CD4-bound state may represent a transient conformation of
Figure 12.18 Interactions between the core ofgp120 and the N-terminal immunoglobulin domain ofCD4. The side chain of Phe43 of CD4 is shown cappinga large hydrophobic cavity on gp120
gp120. Vaccine development based on gp120 has notcompleted clinical trials.
The structure and function of gp41
The other major envelope protein is gp41, a transmem-brane protein interacting with gp120, which consistsof an N-terminal ectodomain, a transmembrane regionand a C terminal intraviral segment within a singlepolypeptide chain of 345 residues. gp41 interacts withthe matrix protein and its principal role is to mediatefusion between the viral and cellular membrane follow-ing gp120-based receptor binding.
At the N-terminal region a hydrophobic, glycine-rich, ‘fusogenic’ peptide is found and all structurespresented to date have been of the ectodomain lackingthis region. The ectodomain is a symmetrical trimerwith each monomer consisting of two antiparallel α
helices connected by an extended loop (Figure 12.19).

456 PROTEIN STRUCTURE AND A MOLECULAR APPROACH TO MEDICINE
Figure 12.19 Part of the ectodomain region of gp41is a coiled coil structure consisting of three pairs ofhelical segments
The combined action of gp120 and gp41 is the firstcontact point between virus and host cell. Unsurpris-ingly, if this stage can be inhibited then infection byHIV will be limited. Extensive proteolysis of a recom-binant ectodomain revealed the existence of stable sub-domains and a trimeric helical assembly composed oftwo discontinuous peptides termed N36 and C34 thatare resistant to denaturation. The N- and C-peptidesoriginate from the N-terminal and C-terminal regions ofthe gp41 ectodomain. The N-terminal derived peptidesform a central trimeric ‘coiled-coil’ with the helical C-peptides surrounding the coiled coil in an antiparallelarrangement. This structural model was confirmed byX-ray crystallography for both HIV-1 and SIV peptidesand is described as a trimer of hairpins (Figure 12.20).
In the trimer the C-peptides bind to the outside of thecoiled-coil core interacting with a conserved hydropho-bic groove formed by two helical N-terminal peptides.Although the structure of the intervening sequence ofgp41 is unclear it would be required to loop aroundfrom the base of the coiled coil to allow the C-peptideto fold back to the same end of the molecule.
The mechanism of viral fusion to cells is slowlybeing unraveled and it may be very similar to fusion
Figure 12.20 The trimer of hairpins exhibited bygp41 core domains (Reproduced by with permissionfrom Eckert, D.E. & Kim, P.S. Annu. Rev. Biochem.2001, 70, 771–810. Annual Reviews Inc)
Figure 12.21 The organization of haemaglutinnin and HIV gp41/gp120 based around interacting helical regionsor coiled coils

THE INFLUENZA VIRUS 457
processes exhibited by the influenza virus during hostcell infection. Some similarities exist between thesequences of gp41 and haemagglutinin (Figure 12.21)and may indicate potentially similar mechanisms existin vivo.
However, this analogy cannot be taken too far,because HIV, unlike influenza virus fuses directlywith cell membranes in a pH-independent manner.The trimer of hairpins is most probably the fusogenicstate and is supported by the observation that gp41-derived peptides or synthetic peptides inhibit HIV-1infection. Peptides derived from the C-terminal regionare much more effective than those derived from N-peptides with inhibition noted at nM concentrations.These peptides act by binding close to the N-peptide region and preventing the hairpin structure.The observed inhibition of HIV by these peptidesoffers another route towards the development ofantiviral drugs. Drugs that target additional steps inthe viral life cycle, such as viral entry, are valuablein providing alternative therapies that may prove lesstoxic and less susceptible to viral resistance thancurrent regimes.
The influenza virus
Influenza (or ’flu) is often considered a harmlessdisease causing symptoms slightly worse than a cold.Common symptoms are pyrexia, myalgia, headacheand pharyngitis and the infection ranges in severityfrom mild, asymptomatic forms to debilitating statesrequiring several days of bed rest. The virus spreadsby contact specifically through small particle aerosolsthat infiltrate the respiratory tract and with a shortincubation period of 18 to 72 hours virus particlesappear in body secretions soon after symptoms startcausing further infection. In the respiratory tract thevirus infects epithelial cells causing their destructionand susceptibility to infections such as pneumonia.
Modern medicines alleviate the worse symptoms offlu but this disease has killed millions in recent historyand continues to present a major health problem.Periodically virulent outbreaks of influenza occur andone of the worst events occurring around 1918 andknown as Spanish flu is conservatively estimatedto have killed 20 million people. This flu pandemicresulted in more fatalities than all of the battles of
vRNA Segments Gene products
PB2
PA
HA HA
NA
NS
M
NPNANP
M1
NS1NEP/NS2
M2
PB1PB2
PAPB1-F2
PB1
Figure 12.22 The influenza A virus. Nucleoprotein (green) associates with each RNA segment to form RNP complexesthat contain PB1, PB2 and PA (red). The viral glycoproteins, haemagglutinin (blue) and neuraminidase (yellow),are exposed as trimers and tetramers. Matrix protein (purple) forms the inner layer of the virion along with thetransmembrane M2 protein (orange). Non-structural proteins are also found (grey). (Reproduced with permissionfrom Ludwig, S. et al Trends Mol. Medicine 9, 46–52, 2003. Elsevier)

458 PROTEIN STRUCTURE AND A MOLECULAR APPROACH TO MEDICINE
Table 12.3 The coding of different viral proteins and their functions by the segmented RNA genome
Segment Size Name Function
1 2341 PB2 Transcriptase; Cap binding2 2341 PB1 Transcriptase; Elongation3 2233 PA Transcriptase; Protease activity?4 1788 HA Haemagglutinin; membrane fusion5 1565 NP Nucleoprotein RNA binding; part of transcriptase complex;
nuclear/cytoplasmic transport of vRNA6 1413 NA Neuraminidase: release of virus7 1027 M1 Matrix protein: major component of virion
M2 Integral membrane protein – ion channel8 890 NS1 Non-structural: nucleus; role in on cellular RNA transport, splicing,
translation. Anti-interferon protein.NS2 NS2 Non-structural: function unknown
World War I combined and emphasizes the seriousnature of influenza outbreaks. Despite drugs and the useof vaccination influenza propagates around the worldat regular intervals. There remains genuine fear offurther major and lethal outbreaks striking again witha severity comparable to the events of 1918. Whenanother pandemic occurs there is little doubt that weare in a much better position to deal with influenzathan previous generations. This position of strengtharises because the influenza virus has been extensivelystudied especially in relation to structural organizationand interaction with cells.
The virus is a member of the orthomyxovirusfamily based around a (-) strand of RNA composedof eight segments in a total genome size of ∼14 kbflanked by short 5′ and 3′ terminal repeat sequences(Figure 12.22). The segments bind to a nucleoprotein(NP) and associate with a polymerase complex ofthree proteins (PA, PB1 and PB2) that constitutes aribonucleoprotein particle (RNP) and is located insidea shell. The shell is formed by the matrix (M1)protein and is itself surrounded by a lipid membranethat is derived from the host cell but contains twoimportant glycoproteins – haemagglutinin (HA) andneuraminidase (NA) – together with a transmembrane-channel protein (M2). Two non-structural components
(NS1 and NS2) are also encoded by the viral genometo make a total of 10 proteins1 (Table 12.3).
Viral classification is based around the M and NPantigens and is used to determine strain type – A,B or C. Most major outbreaks of influenza areassociated with virus types A or B with type Bstrain causing milder forms of the disease. Theexternal antigens haemagglutinin and neuraminidaseshow greater sequence variation.
Viral infection and entry into the cell
The virus binds to sialic-acid-containing cell surfacereceptors via interactions with haemagglutinin enteringthe cell by endocytosis where fusion of the viraland endosomal membranes allows the release of viralRNA. After transfer to the nucleus the viral genome istranscribed and amplified before translation of mRNAin the cytoplasm. Late in the infection cycle the viralgenome is transported out of the nucleus in RNPcomplexes and packaged with the structural proteins.Subsequent release of new virions completes a cycle
1For many years this was thought to be the complete complementof viral proteins but in 2002 further examination of the genomerevealed an alternate reading frame in PB1 that allowed productionof an eleventh protein (F2).

THE INFLUENZA VIRUS 459
Figure 12.23 The lifecycle of the influenza A virus and the pathway of viral entry and exit from host cells.(Reproduced with permission from Ludwig, S. et al Trends Mol. Medicine 9, 46–52, 2003 Elsevier)
that occurs over a time span between 6 to 16 h(Figure 12.23). Of critical importance to viral entryare the characteristic “spikes” of haemagglutinin andtogether with the second major envelope protein,neuraminidase, these proteins have been the targets fordrug treatments to fight “flu”.
Haemagglutinin structure and function
Haemagglutinin was identified from the ability of fluviruses to agglutinate red blood cells via the interactionbetween HA and sialic acid groups present on cellsurface receptors. Haemagglutinin is anchored to themembrane by a short tail but limited proteolysiswith enzymes such as bromelain or trypsin yield asoluble domain (Figure 12.24). The soluble domainwas amenable to crystallographic analysis and in 1981a structure of haemagglutinin determined by JohnSkehel, Donald Wiley and Ian Wilson provided the firstdetailed picture of any viral membrane protein.
Haemagglutinin is a cylindrically shaped homotrimerapproximately 135 A in length. It is synthesized as aprecursor polypeptide (HA0) that associates to forma trimer but is first cleaved by proteases to givetwo disulfide linked subunits -HA1 and HA2. HA1and HA2 are necessary for membrane fusion andvirus infectivity and generally the polypeptide chainsare separated by just one residue, a single arginineresidue, removed from the C terminus of HA1 by car-boxypeptidase activity. It is the HA1/HA2 assemblythat is commonly, but slightly inaccurately, referredto as a monomer and haemagglutinin contains threeHA1 and three HA2 chains. The membrane-anchoringregion is located at the C terminal region of HA2but structural studies were performed on tail lessprotein.
The crystallographic structure shows the free ends,at the cleavage site between HA1 and HA2, areseparated by 20 A and within the structure two distinctregions were readily identified; a globular head regionresponsible for binding sialic acid residues on host cell

460 PROTEIN STRUCTURE AND A MOLECULAR APPROACH TO MEDICINE
HA1 (1-328)
HA
BHA1 1
1 14 328
328 137 175
23 38 137 185 211
HA2 (1-221)
TMS - S
S - S
Figure 12.24 The membrane bound and protease treated form of haemagglutinin showing HA1 and HA2 regions.BHA refers to bromelain (protease) treated haemagglutinin
Figure 12.25 The individual HA1 and HA2 units of haemagglutinin
receptors and a longer extended stem-like region thatcontains a fusogenic sequence (Figure 12.25).
The three monomers assemble into a central α-helical coiled-coil region that forms the stem-like
domain whilst at the top are three globular headscontaining sialic acid-binding sites and responsible forcell adhesion or binding. The stem region consisting ofa triple helix extends for ∼76 A from the membrane

THE INFLUENZA VIRUS 461
surface and is formed predominantly by the HA2polypeptide. The globular region is based mainlyon the HA1 portion of the molecule and eachglobular domain consists of a swiss roll (jelly roll)motif of eight anti-parallel β-strands by residues116–261. At the distal tips receptor (sialic acid)binding pockets were identified. The binding sitesreside in small depressions on the surface and weredetected from crystallographic studies in the presenceof different sialic acid derivatives. Amongst theresidues forming the active sites are Tyr98, Ser136Trp153, His183, Glu190, Leu194, Tyr195, Gly225 andLeu226 with site directed mutagenesis suggesting thatTyr98, His183, and Leu194 are particularly importantin forming critical hydrogen bonds with the substrate(Figure 12.26).
Sialic acid groups bind with one side of the pyra-nose ring facing the base of the site whilst theaxial carboxylate, the acetamido nitrogen, and the 8-and 9-OH groups of the sugar form hydrogen bondswith polar groups of conserved residues. Histidine183 and glutamate 190 form hydrogen bonds withthe 9-OH group, tyrosine 98 forms hydrogen bonds
Globular head regionsof HA1 domain Active site
residues
Figure 12.26 The active site residues of haemagglu-tinin are shown in the spacefilling representations ofthe globular HA1 domain. The view is from above theglobular trimer
with the 8-OH group whilst the 5-acetamido nitro-gen forms a hydrogen bond with the backbone car-bonyl of residue 135 and the methyl group makesvan der Waals contact with the phenyl ring of tryp-tophan 153. The 4-OH group projects out of thesite and does not appear to participate in bindinginteractions.
A key observation in understanding the structureand function of haemagglutinin was the demonstra-tion that the molecule underwent a large conforma-tional change when subjected to low pH. The HAproteolytic fragment does not bind to membranesand is functionally inactive but lowering the pH toa value comparable to that occurring in endosomes(∼pH 5–6) during viral entry caused haemagglutininto become a membrane protein. Electron microscopyshowed protein aggregation into “rosettes” with anelongated and thinner structure. These observationssuggested major conformational changes and perform-ing crystallography on the ‘low pH’ form resolvedthe extent and location of structural perturbations(Figures 12.27 & 12.28).
At low pH the fusion region or peptide waslocated at the end of an elongated coiled-coil chainwith domains near the C-terminal interacting withthose at the N terminus. The low pH form is thethermodynamically stable form of HA2 and the initialstate is best described as a metastable configuration.Exposure to low pH converts this metastable form intothe thermodynamically stable form of HA2.
In total the structures of the ectodomain of haemag-glutinin have been determined for the single-chain pre-cursor (HA0), the metastable neutral-pH conformationand the fusion active, low pH-induced conformation.These structures provide a framework for interpretingresults of experiments on receptor binding, the cycli-cal generation of new and re-emerging epidemics asa result of HA sequence variation as the process ofmembrane fusion during viral entry.
Neuraminidase structure and function
Neuraminidase is vital to viral proliferation. Theenzyme is a glycosyl hydrolase cleaving glycosidiclinkages between N-acetyl neuraminic acid and adja-cent sugar residues by destroying oligosaccharidesunits present on host cell receptors. Viral neuraminidase
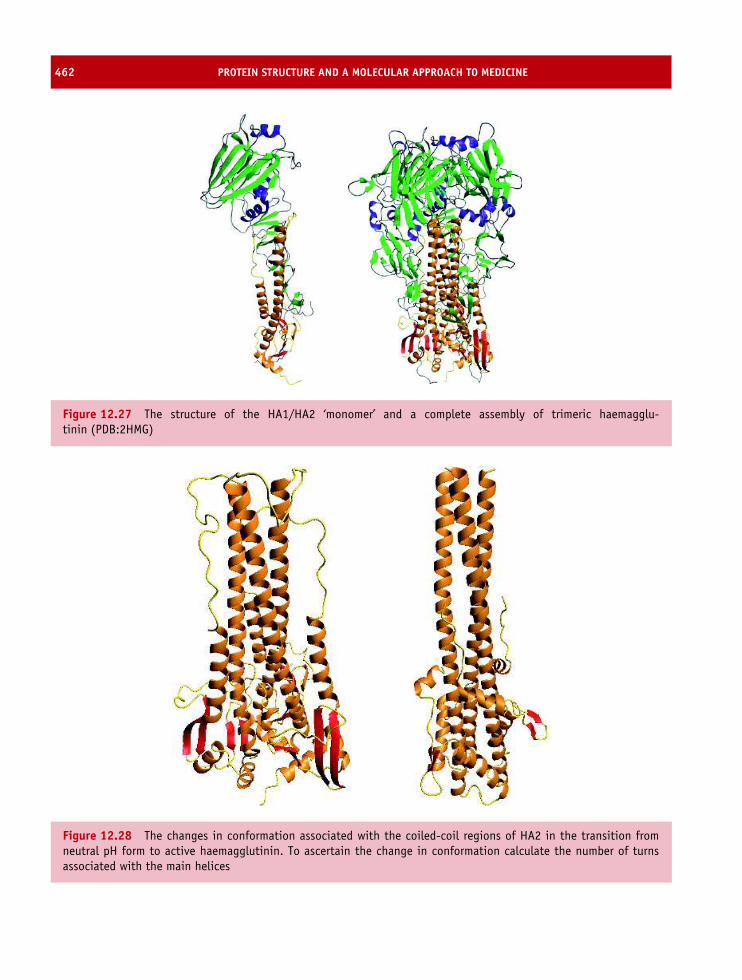
462 PROTEIN STRUCTURE AND A MOLECULAR APPROACH TO MEDICINE
Figure 12.27 The structure of the HA1/HA2 ‘monomer’ and a complete assembly of trimeric haemagglu-tinin (PDB:2HMG)
Figure 12.28 The changes in conformation associated with the coiled-coil regions of HA2 in the transition fromneutral pH form to active haemagglutinin. To ascertain the change in conformation calculate the number of turnsassociated with the main helices

THE INFLUENZA VIRUS 463
Figure 12.29 The box-like arrangement of the tetrameric state of neuraminidase shown alongside a spacefillingmodel of the same view
causes a loss of agglutination and allows subsequentinfection of other host cells.
The amphipathic protein is a single polypeptidechain of ∼470 residues with a hydrophobic mem-brane binding domain of ∼35 residues connected toa larger polar domain of ∼380 residues via a hyper-variable stalk of ∼50 residues. Limited proteolysisremoves the N terminal membrane anchor and facil-itated crystallization. The structure of the large frag-ment of neuraminidase revealed a homo-tetramer withmonomers consisting of six topologically related units(Figure 12.29). Each unit contained a four-strandedsheet arrayed like the petals of a flower or more fre-quently described as one of the blades of a propeller(Figure 12.30).
Neuraminidase is an example of the β pro-peller motif. The four strands connect by reverse turnswith the first strand of one sheet interacting across thetop of the motif to the fourth strand of the adjacentmotif. Disulfide bonds between adjacent strands con-tribute to a distortion of regular sheet conformation andintroduce a distinct twist to each blade.
Figure 12.30 The neuraminidase monomer. Whilstthe structure of the larger soluble fragment has beendetermined the structure of the membrane spanningregion is unknown. The structure shows one monomerwith six topologically related units (blades) composedof four β strands; each blade is shown in a differentcolour. (PDB:2BAT)

464 PROTEIN STRUCTURE AND A MOLECULAR APPROACH TO MEDICINE
OH
OH
O
NH
O
OH
HO
HO
HO
O
Figure 12.31 The structure of sialic acid (N-acetylneuraminic acid, Neu5Ac)
The monomeric units assemble into a box-shapedhead with dimensions of ∼100 A × 100 A × 60 Aattached to a slender stalk region. The tetramericenzyme has circular 4-fold symmetry stabilized inpart by metal ions bound on the symmetry axis witheach subunit interacting via an interface dominated byhydrophobic interactions and hydrogen bonds.
Definition of the active site of neuraminidase provedimportant in designing inhibitors that interfere with
neuraminidase activity and numerous crystal structuresexist for enzyme-inhibitor complexes. Initial studiesfocused on a transition state analogue, 2-deoxy-2, 3-dehydro-N -acetylneuraminic acid (Neu5Ac2en), thatinhibited the enzyme at micromolar concentrationsbut further refinement yielded inhibitors effective atnanomolar levels (Figures 12.31 & 12.32).
One of these compounds 4-guanidino-Neu5Ac2en(Zanamivir) inhibits viral proliferation as a result ofsignificant numbers of hydrogen bonds formed betweendrug and conserved residues in the active site ofneuraminidase (Figure 12.33).
Strategies to combat influenza pandemics
Influenza infection leads to a pronounced antibodyresponse against haemagglutinin and neuraminidasebased on the surface epitopes presented by the infect-ing virus. Ordinarily individuals have protective immu-nity but in the case of flu frequent changes in thesequences of these two antigens compromises the nor-mal immune response. Vaccines against flu viruses are
Arg371
Arg292
Arg152
Arg118
Trp178
His 274
Glu276
Glu119
Sialic acid
Sialic acid
Figure 12.32 The active site of neuraminidase showing substrate bound to active site (PDB:1MWE). A second sialicacid binding site was determined on the surface of neuraminidase

THE INFLUENZA VIRUS 465
Figure 12.33 The molecular structure of Zanamivir. Apotent inhibitor of neuraminidase that is now acceptedas clinical treatment for flu in most countries
recommended by the WHO2 to contain haemagglutinincomponents from two A strains and a single B strainand to be representative of viruses currently circulatingthe world. Unfortunately although vaccines are devel-oped from purified HA the virus alters the sequence ofresidues around the epitopes without impairing proteinfunction. This occurs via point mutations and leads toaltered surface properties that eliminate cross-reactionwith antibodies developed against previous forms of thevirus. Two distinct mechanisms contribute to variationin surface properties of the HA. Point mutations causeantigenic drift whilst a second, potentially more seri-ous, mechanism for variation is antigenic shift wherethe exchange of genes between influenza viruses cre-ates new strains.
Haemagglutinin has at least fifteen different anti-genic sub-types (H1–H15) whilst neuraminidase hasnine antigenic subtypes (N1–N9). Most subtypes oftype A virus infect avian species with sub-types H1,H2 and H3 infecting humans. The HA1 domains showgreater variability than HA2 consistent with their roleas binding sites for receptors and antibodies. Withthe recognition of these sub-types viral outbreaks areclassified according to the virus class (usually A orB), the species from which the virus was isolated, itsgeographical region, the year of isolation and the Hand N sub-group. The 20th century witnessed threeinfluenza pandemics; Spanish influenza (defined asan H1N1 virus) in 1918, Asian influenza (defined as
2WHO = World Health Organization.
an H2N2 virus) in 1957, and Hong Kong influenza(H3N2 virus) in 1968. These global outbreaks causedsevere illness with high mortality rates. DNA sequenc-ing showed that the HA and NA genes of the H1N1viruses emerged from an avian reservoir with subtlechanges in primary sequence leading to altered anti-genic properties. In contrast Asian and Hong Kongoutbreaks were attributed to hybrid viruses arising fromnew combinations of avian and human viral genes.Aquatic birds are believed to be the source of influenzaviruses affecting all other animal species. Recentlyin 1997, a highly pathogenic avian H5N1 influenzavirus was transmitted directly from poultry to humansin Hong Kong. Of the 18 people infected, six diedand this heightened worries about new pandemics inthe future.
Influenza and HIV represent two viruses with anenormous impact on human health. They are not uniquein their mechanism of infection but in both cases theyhave an unfortunate ability to mutate rapidly causingchanges in antigenicity or loss of drug activity. Thisproperty creates pandemics and limits our ability tofight infection through vaccines and drugs. It is veryunlikely that these viruses will remain the only threatto human health. The next decades are likely to seethe emergence of new strains and new viruses, perhapsinvolving those crossing the species boundary, whoseaction will need to be countered through exploitationof knowledge about the structure and function of theirconstituent proteins.
Neurodegenerative disease
In comparison with influenza neurodegenerative dis-eases are less common, less easily transmitted andmost of us will escape neurodegenerative disordersentirely. Despite a low incidence in all populationgroups there is evidence to suggest that these diseasesare of greater importance to human health than pre-viously thought. Many of these diseases have cleargenetic links that enable insight into their molecu-lar origin.
Prion based diseases are linked by the proliferativeformation of insoluble aggregates of protein arisingfrom mutations within the PrP gene. Creutzfeldt-Jakob disease, fatal familial insomnia, kuru andscrapie were linked to the PrP gene and lead to

466 PROTEIN STRUCTURE AND A MOLECULAR APPROACH TO MEDICINE
neurodegenerative conditions. All of these conditionslead to abnormal brain tissue and the presence ofamyloid deposits. Collectively the term amyloidogenicdisease stressed the link between protein misfoldingand the development of neurodegenerative disorders.
BSE and new variant CJD
Scrapie was first reported in 1732 as a diseaseinfecting sheep in England and has been presentcontinuously since this time. It did not appear toeffect bovine livestock. However, in the 1980s anew disease spread through cattle in the UnitedKingdom at an alarming rate producing symptomsnormally associated with TSEs. The disease in cowsled to abnormal gait and posture, poor body condition,nervous behaviour, decreased milk production and aprogressive loss of mass. Diseased animals lost theability to support themselves with death occurring2–25 weeks after the appearance of symptoms. Afeature of the disease was brain tissue of spongyappearance bearing large vacuoles. The disease waschristened bovine spongiform encephalopathy (BSE)
but its alarming rise in frequency yielded anothersobriquet of ‘mad cow disease’ (Figure 12.34). Theappearance of BSE raised a number of importantquestions. What was the origin of the disease, how wasit transmitted and given the role of cattle in the humanfood chain was there a possibility of human infection?
BSE is a chronic degenerative disease and with theobservation of scrapie like symptoms it was logicalto look for a prion-like particle. At the time (mid1980s) the concept of prions was controversial with thepioneering work of Prusiner slow to gain acceptance.The appearance of the disease in cattle was puzzlinggiven the absence of simple routes of transmissionbut epidemiological data suggested BSE arose fromanimal feed containing contaminated protein sources.BSE had a substantial impact on the livestock industryin the United Kingdom with approximately 0.3 % ofthe total UK herd infected and continued to riseuntil 1992.
In 1988 the introduction of a total ban on the useof rendered ruminant based feed to cattle minimizedtransmission and was combined with the large scaleslaughtering of infected cattle. In 1989 these preven-tative actions were supplemented by a ban on human
0
5000
10000
15000
20000
25000
30000
35000
40000
Rep
ort
ed c
ases
1986 1987 1988 1989 1990 1991 1992 1993 1994 1995 1996
YEAR
Figure 12.34 The yearly totals for clinical diagnosis of BSE in the UK herd (data derived from BSE Inquiry Report,Crown copyright 2000)

THE INFLUENZA VIRUS 467
0
10
20
30
40
50
60
70
80
90
1990 1991 1992 1993 1994 1995 1996 1997 1998 1999 2000 2001 2002
vCJD
GSS
FFI
IATROGENIC
SPORADIC
Figure 12.35 The incidence of all CJD cases in the United Kingdom (Data derived from CJD Surveillance unit,Edinburgh http://www.cjd.ed.ac.uk). From 1995 the number of cases of vCJD rise reaching a peak in 2000 beforeshowing small decreases in the subsequent years
consumption of tissues shown to contain high levels ofthe infectious agent (when injected into mice brains)and therefore posing a potential danger to humans.Unfortunately this was not the end of the story becausethe long incubation period of BSE3 ranging from 2 to8 years meant that human consumption of infected cat-tle occurred throughout the 1980’s.
In March 1996 the UK Spongiform EncephalopathyAdvisory Committee announced the identification of 10new cases of Creutzfeldt-Jakob disease (Figure 12.35).CJD is a rare disease and the observation of 10 caseswas startling. Although direct evidence was lacking itseemed possible that exposure by ingestion to BSEagents was responsible for increase numbers of CJDcases. In 1994 initial speculation that BSE might bethe causative agent of new cases of CJD was aired anddespite a lack of definitive evidence the idea gainedcredibility and created widespread unease about the useof beef as a culinary product.
The 10 cases were unusual in terms of their highfrequency and the clinical and pathological phenotypesassociated with the condition. The new forms ofCJD exhibited an onset of symptoms at a youngage with teenagers or young adults most susceptible.
3The time span between an animal becoming infected and thenshowing the first signs of disease.
The disease was associated with neurodegenerationand brain tissue showed lesions characteristic ofspongiform encephalopathies. The new forms of CJDwere called variant CJD (vCJD) and stemmed fromexposure to BSE before the introduction of a specifiedbovine offal ban in 1989. Definitive proof of thelink between BSE and vCJD required comparing bothdiseases at a molecular level to demonstrate that theetiological agents were prion proteins.
Comparing four different types of CJD (genetic, spo-radic, iatrogenic, and “new variant”) as well as BSEusing SDS-PAGE showed that prion proteins exhibitdistinct banding patterns when studied via Westernblotting methods after treatment with proteinase K, aproteolytic enzyme that normally effectively degradesproteins into very small fragments but in the caseof infectious prions produced protease resistant frag-ments. The pattern and intensity of classic forms ofCJD (genetic, sporadic, or iatrogenic) were similar butvariant CJD yielded a unique banding pattern suggest-ing a different genotype. The pattern was comparableto the profile seen in BSE infected organisms. Fur-ther analysis of the DNA sequence of individuals withvCJD showed that all were heterozygous at residue 129(Valine/Methionine) suggesting that this transition con-ferred a genetic predisposition for contracting vCJD

468 PROTEIN STRUCTURE AND A MOLECULAR APPROACH TO MEDICINE
whereas the homozygous (Valine/Valine) state was pos-sibly resistant to infection.
Alzheimer’s disease–a neurodegenerativedisorder
Amyloidosis or protein aggregation is associated withAlzheimer’s disease although like prion-based dis-eases little impact has been made towards effectivetreatment. An understanding of the molecular eventsinvolved in disease pathogenesis offers the possibilityof future therapy.
Alzheimer’s disease first recognized by AloisAlzheimer in the first decade of the 20th century isa slow progressive form of dementia normally seenin the later stages of life and resulting in intellectualimpairment. Today a common diagnostic marker forphysicians is memory impairment accompanied by adecline in language and decision-making ability, judge-ment, attention span, cognitive functions and person-ality. Diagnosis is usually confirmed by post mortemstudies and the microscopic examination of brain tissue
where a diagnostic feature is the presence of neurofib-rillary tangles, aggregates of a single protein withinbrain tissue. The brain is seen to contain plaques andalthough plaques are detected in all brain tissue withage their number is drastically elevated in individualswith Alzheimer’s disease.
Two types of Alzheimer’s disease are recognized.Early onset disease results in symptoms before the ageof 60 and has a genetic basis seen by its presencewithin families and an autosomal dominant form ofinheritance. It accounts for less than 10 % of all formsof Alzheimer’s. In contrast late onset forms are morecommon but the genetic basis is less well defined. Lateonset disease develops in people above 65 with anincidence of 5–10 %.
The amyloid plaques found in Alzheimer’s diseaseare composed of a 4.2 kD peptide called the Aβ protein.The plaques result from the transformation of solubleAβ protein monomers into insoluble aggregates. Thepeptide called Aβ because of its amyloid propertiesand a partial β-stranded structure occurring within a28-residue portion of the sequence is found in at leasttwo forms; a 40 residue peptide and a 42 residue
* 20 * 40 * 60 * 80 * 100APP : MLPGLALLLLAAWTARALEVPTDGNAGLLAEPQIAMFCGRLNMHMNVQNGKWDSDPSGTKTCIDTKEGILQYCQEVYPELQITNVVEANQPVTIQNWCKR
* 120 * 140 * 160 * 180 * 200APP : GRKQCKTHPHFVIPYRCLVGEFVSDALLVPDKCKFLHQERMDVCETHLHWHTVAKETCSEKSTNLHDYGMLLPCGIDKFRGVEFVCCPLAEESDNVDSAD
* 220 * 240 * 260 * 280 * 300APP : AEEDDSDVWWGGADTDYADGSEDKVVEVAEEEEVAEVEEEEADDDEDDEDGDEVEEEAEEPYEEATERTTSIATTTTTTTESVEEVVREVCSEQAETGPC
* 320 * 340 * 360 * 380 * 400APP : RAMISRWYFDVTEGKCAPFFYGGCGGNRNNFDTEEYCMAVCGSAMSQSLLKTTQEPLARDPVKLPTTAASTPDAVDKYLETPGDENEHAHFQKAKERLEA
* 420 * 440 * 460 * 480 * 500APP : KHRERMSQVMREWEEAERQAKNLPKADKKAVIQHFQEKVESLEQEAANERQQLVETHMARVEAMLNDRRRLALENYITALQAVPPRPRHVFNMLKKYVRA
* 520 * 540 * 560 * 580 * 600APP : EQKDRQHTLKHFEHVRMVDPKKAAQIRSQVMTHLRVIYERMNQSLSLLYNVPAVAEEIQDEVDELLQKEQNYSDDVLANMISEPRISYGNDALMPSLTET
* 620 * 640 * 660 * 680 * 700APP : KTTVELLPVNGEFSLDDLQPWHSFGADSVPANTENEVEPVDARPAADRGLTTRPGSGLTNIKTEEISEVKMDAEFRHDSGYEVHHQKLVFFAEDVGSNKG
* 720 * 740 * 760 *APP : AIIGLMVGGVVIATVIVITLVMLKKKQYTSIHHGVVEVDAAVTPEERHLSKMQQNGYENPTYKFFEQMQN
Figure 12.36 The primary sequence of the APP protein and its relationship to the Aβ peptide and other domains.The Aβ peptide extends from residues 672-711/713 (green) and overlaps with the single membrane spanning regionfrom 700–723 (blue lettering and red block). The yellow block indicates a signal sequence (1–17) whilst the cyanblock indicates the location of a domain showing homology to BPTI or Kunitz type inhibitors (291–341). Themagenta blocks highlight poly-glutamate rich domains 230–260, threonine rich regions 265–282, heparin bindingregions 391–424 and 491–522 and collagen binding domain 523–541

THE INFLUENZA VIRUS 469
DAEFGHDSGFEVRHQKLVFFAEDVGSKKGAIIGLMVGGVVI
Ab peptide
Ab1 Ab11 Ab17
ab
b
......VKM ATVIVI.......Ab40 Ab42
g
g
Figure 12.37 Schematic diagram of the Aβ peptide derived from the amyloid precursor protein. The amino acidsequence is shown with single letter codes and the α, β and γ secretase cleavage sites are shown along thepolypeptide chain. The α secretase cleaves at the 17th residue whilst the β secretase cuts at the 1st and more rarely11th residues and the γ secretase cuts at the 40th or 42nd residue
peptide designated as Aβ1−40 and Aβ1−42 respectively.Sequencing and database analysis showed that thepeptides derived from a longer protein expressed inmany tissues – the amyloid precursor protein (APP)-and DNA sequencing indicated a protein of ∼770residues with multiple domains (Figure 12.36).
APP undergoes extensive post-translational modifi-cation as a glycoprotein but despite domain recognitiona clearly defined function for APP is absent although arole in cell adhesion is currently favoured. The APPgene was mapped to chromosome 21, contained 19exons, with alternative splicing mechanisms giving riseto multiple transcripts (APP395, APP563, APP695,APP751, APP770). APP695 is the major isoform inneuronal tissue with APP751 dominating elsewhere.
The Aβ amyloid peptide (residues 672-711/713) isderived from a region of the protein that includes asmall extracellular region together with its membranespanning sequence (700–723). Understanding the ori-gin of the Aβ peptide lies at the heart of discoveringthe molecular pathology of Alzheimer’s disease. Theamyloid peptide located within the full length proteinarises from endoproteolysis of APP (Figure 12.37). Theamyloid peptide was formed by cutting at residues 670and 711/713 at sites associated with enzymes calledβ and γ secretase whilst the enzyme α secretase cutsAPP at residue 687 within the amyloidogenic pep-tide. The properties and identity of these enzymes wereinitially obscure (hence their names) but slowly eachsystem has been shown to occupy an important role inAPP processing.
The demonstration that endoproteolysis of APPwas associated with two different enzymes suggestedthat defining these ‘activities’ in more detail aswell as the associated genes/proteins would assist
understanding the molecular events contributing todisease. β secretase was identified as a new member ofthe aspartic protease family with the unusual propertyof being membrane tethered. The α secretase is amember of the ADAM family of proteins (standingfor a disintegrin and metalloprotease) and finally γ
secretase began to give up its secrets by suggestingsimilarity to a membrane protein called presenillin 1.
Although initially called β secretase the enzymepurified by different groups was given alternativenames of BACE (β-site APP cleaving enzyme) andmemapsin-2 (membrane-associated aspartic protease2) and both are widely quoted in the literature.The enzyme, a single polypeptide chain of 501
Figure 12.38 The organization of secondary struc-ture into two lobes and the active site aspartateresidues in β secretase (PDB:1M4H) shows similaritiesto previous aspartyl proteases

470 PROTEIN STRUCTURE AND A MOLECULAR APPROACH TO MEDICINE
residues synthesized as a preproprotein, contains asingle transmembrane region from residues 458 to478 followed by a short cytoplasmic facing regionof ∼20 residues. The majority of residues are locatedin a large extracellular domain from residues 45–457and this region of the enzyme has been studied toprovide information on kinetics, substrate specificityand structure (Figure 12.38).
As an aspartyl protease the enzyme contains twoconserved aspartate within a bilobal structure andexhibits an optimal pH for catalytic activity at pH4.0 with substrates bound with Km values between1−10 mM and turnover rates ranging from 0.5–3 s−1.The active site of β secretase is more open and lesshydrophobic than corresponding sites in other humanaspartic proteases but by defining the active site ofthis enzyme through substrate specificity and transitionstate analogues offers the hope of designing effectiveinhibitors. Inhibitors of β secretase are importantpharmaceutical drug targets since they would preventamyloid peptide formation.
The genetic basis of Alzheimer’s disease hasassisted understanding of disease pathology. Missensemutations are present in the APP gene in familialAlzheimer’s disease and these mutations are frequentlyfound in close proximity to the α-, β-, or γ secretaseprocessing sites. In a Swedish pedigree a doublemutation Lys → Asn and Met → Leu near the β-secretase cleavage site (671–672) leads to elevatedproduction of the Aβ peptide (both Aβ40 and Aβ42)and therefore presumably accounts for the early onsetof the disease. Defective genes are also responsiblefor early onset disease with mutations to presenilin1 and presenilin 2 observed in families and leadingto increased deposition of the Aβ peptide. Over 70mutations are known most located in presenilin 1.
Despite a lack of a clearly defined function for APPthe Aβ peptide aggregates into larger amyloid fibrils,destroying nerve cells and decrease levels of neuro-transmitters. The correct balance of neurotransmittersis critical to the brain’s activity and the levels of acetyl-choline, serotonin, and noradrenaline are all altered inAlzheimers disease. With both structural defects andchemical imbalance in the brain Alzheimer’s diseaseeffectively disconnects areas of neuronal tissues thatnormally work cohesively together.
An interesting question is why have neurodegenera-tive diseases become more prominent in recent times?Undoubtedly part of the answer lies with longer lifeexpectancy. In 1900 it is estimated that only 1 % ofthe world’s population was above 65 years of age.By 1992 that figure had risen to nearly 7 % and in2050 it is estimated that ∼25 % of the population willbe >65 years of age. The introduction of antibiotics,improved living conditions particularly in relation towater quality and food supply coupled with effectivehealth care has meant that at least in the economicallyadvantaged regions of the world average lifetimes formen and women are well above 80 years of age. Oneconsequence of longer life expectancy is that diseasessuch as neurodegenerative disorders can develop overmany years where previously an individual’s short life-time would preclude their appearance. Susceptibility toneurodegenerative diseases may be an inevitable con-sequence associated with longevity but as moleculardetails concerning these disease states are uncoveredthere is optimism about favourable treatment optionsin the future.
p53 and its role in cancerCancer involves the uncontrolled growth of cells thathave lost their normal regulated cell cycle function.In many instances the origin of diseases lie in themutation of genes controlling cell growth and divisionso that cancer is accurately described as a cell cyclebased disease. Mutations alter the normal function ofthe cell cycle, with three classes of genes identifiedclosely with cancer. Oncogenes ‘push’ the cell cycleforward, promoting or exacerbating the effect of genemutation. In contrast, tumour suppressor genes functionby normally applying the ‘brakes’ to cell growth anddivision. Mutation of tumour suppressor genes causesthe cell to lose their ability to control coordinatedgrowth. Finally, repair genes keep DNA intact bypreserving their unique sequence. During the cell cyclemutagenic events result in DNA modification that inmost circumstances are repaired unless the mutationresides in genes coding for repair enzymes.
One of the most important systems in the area ofcancer and cell cycle control is the protein p53, anddetails of the action of this protein were largely theresult of the research by Bert Vogelstein, David Lane

p53 AND ITS ROLE IN CANCER 471
and Arnold Levine. The p53 gene is located on theshort arm of chromosome 17 where the open readingframe codes for a protein of 393 residues via 11 exons.The protein identified before the gene was originallycharacterized through co-purification with the large Tantigen in SV40 virus-transformed cells and had a massof ∼53 kDa. Since the presence of the protein appearedto correlate with viral transformation of cells p53 wasoriginally labelled as an oncogene. By the late 1980sit was clear that the gene product of cloned p53 wasa mutant form of the protein and that normal (wild-type) p53 was the product of a tumour suppressor gene.Further modification of ideas concerning p53 functionoccurred with the demonstration that the protein causedG1 cell cycle arrest but also activated genes by bindingto specific DNA sequences – it was a transcriptionfactor. The discovery that one transcriptional target ofp53 was an inhibitor (p21) of cyclin dependent kinases(Cdk) provided a direct link to the cell cycle since p21complex formation with cdk2 prevents cell division andrepresents one point of control. Mutant p53 does notbind effectively to DNA with the consequence that p21is not produced and is unavailable to act as the ‘stopsignal’ for cell division.
p53 has been called the ‘Guardian of the Genome’and it is normally present in cells at low concentrationin an inactive state or one of intrinsically low activity.When the DNA of cells is damaged p53 levels risefrom their normal low levels and the protein is switched‘on’ to play a role in cell cycle arrest, transcription andapoptosis. p53 regulates the cell cycle as a transcriptionfactor so that when damage to DNA is detected cellcycle arrest occurs until the DNA can be repaired.Once repaired the normal cell cycle can occur butthis process serves to ensure that damaged DNA isnot replicated during mitosis and is not ‘passed’ onto daughter cells. If repair cannot take place apoptosisoccurs (Figure 12.39).
With an important role within cells the structuralorganization of p53 was of considerable interest tomany different fields ranging from cell biology tomolecular medicine. The protein contains a singlepolypeptide chain that is divided into three discretedomains (Figure 12.40). These domains present amodular structure that has facilitated their individualstudy in the absence of the rest of the protein and areconcerned with transcriptional activation, DNA binding
Figure 12.39 An outline of possible p53 actions withnormal and damaged cells
N terminaldomain
1 100 300 393
Core domain C terminaldomain
Transactivation Proline richdomain
DNA binding domain Tetramerisation Regulatoryand non-specificDNA binding
Figure 12.40 The organization of domains within thetumour suppressor p53
and tetramerization. An N-terminal transactivationdomain extends from residues 1–99 and is followedby the largest domain–a central core region fromresidues 100–300 that binds specific DNA sequences.The third and final region of p53 is a C-terminal domain(residues 301–393) that includes both a tetramerizationdomain (from residues 325–356) and a regulatoryregion (363–393) (Figure 12.41). As a result of the twofunctional activities in this region p53 is sometimesdescribed as containing four as opposed to threecomposite domains.
The presence of discrete domains linked by flexiblelinkers creates a conformationally dynamic moleculeand p53 has proved difficult to crystallize as the

472 PROTEIN STRUCTURE AND A MOLECULAR APPROACH TO MEDICINE
* 20 * 40 * 60 * 80 * 100
p53:
* 120 * 140 * 160 * 180 * 200
p53:
* 220 * 240 * 260 * 280 * 300
p53:
* 320 * 340 * 360 * 380 *
p53: PGSTKRALPNNTSSSPQPKKKPLDGEYFTLQIRGRERFEMFRELNEALELKDAQAGKEPGGRAHSSHLKSKKGQSTSRHKKLMFKTEGPDSDS
MEEPQSDPSVEPPLSQETFSDLWKLLPENNVLSPLPSQAMDDLMLSPDDIEQWFTEDPGPDEAPRMPEAAPPVAPAPAAPTPAAPAPAPSWPLSSSVPSQ
KTYQGSYGFRLGFLHSGTAKSVTCTYSPALNKMFCQLAKTCPVQLWVDSTPPPGTRVRAMAIYKQSQHMTEVVRRCPHHERCSDSDGLAPPQHLIRVEGN
LRVEYLDDRNTFRHSVVVPYEPPEVGSDCTTIHYNYMCNSSCMGGMNRRPILTIITLEDSSGNLLGRNSFEVRVCACPGRDRRTEEENLRKKGEPHHELP
Figure 12.41 The primary sequence of p53 showing the partition of the primary sequence into discrete domains,using the colour codes of Figure 12.40
Figure 12.42 The structure of the isolated coredomain of p53 showing β sandwich structure togetherwith a bound Zn ion (purple)
full length molecule. This almost certainly resultsfrom molecular heterogeneity and structural insightinto p53 followed a modular approach determiningthe structure of isolated domains. One incisive setof results were obtained by Nikolai Pavletich usingproteolytic digestion of the full-length protein toyield a protease-resistant fragment of ∼190 residues(residues 102–292) that corresponded to the centralportion of p53 involved in sequence-specific DNA-binding. DNA binding was inhibited by metal chelatingagents and unsurprisingly within the core domain aZn ion was identified (Figure 12.42). The central coreextending from residues 100–300 contains the DNAbinding residues and also those residues representingmutational hot spots (see below) within the p53 gene.
As a DNA binding protein the core domain binds toboth the major and minor groove of the double helixthrough the presence of charged side chains arrangedprecisely in conserved loops that link elements of β
strands (Figures 12.43 and 12.44). Numerous arginineand lysine side chains form specific interactions withDNA. Most DNA binding proteins are based onthe helix-turn-helix (HTH), HLH or leuzine zippermotifs, but p53 is essentially a β sandwich formed bythe interaction of antiparallel four- and five-strandedelements of β sheet.
Figure 12.43 The structure of p53 core domainbound to DNA showing role of Arg175, Arg273 andGly245 (red wireframe) in these interactions as well asthe position of a bound Zn ion. The Zn ion is bound ina tetrahedral environment formed by the side chains ofthree cysteine residues together with a single histidinearranged as part of two loop regions and a single helix
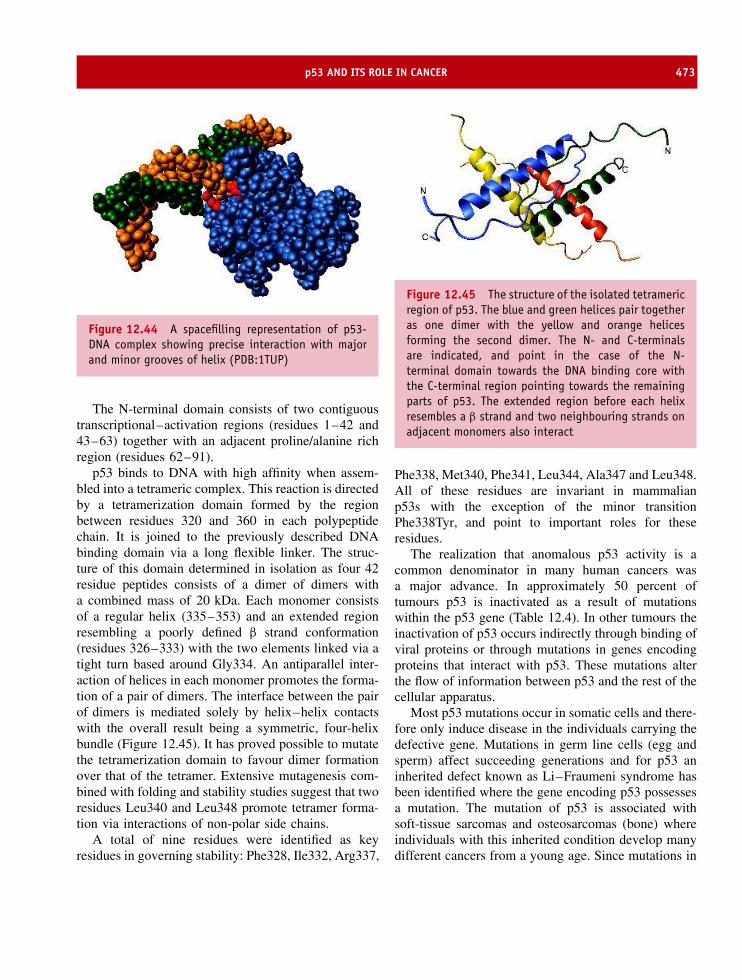
p53 AND ITS ROLE IN CANCER 473
Figure 12.44 A spacefilling representation of p53-DNA complex showing precise interaction with majorand minor grooves of helix (PDB:1TUP)
The N-terminal domain consists of two contiguoustranscriptional–activation regions (residues 1–42 and43–63) together with an adjacent proline/alanine richregion (residues 62–91).
p53 binds to DNA with high affinity when assem-bled into a tetrameric complex. This reaction is directedby a tetramerization domain formed by the regionbetween residues 320 and 360 in each polypeptidechain. It is joined to the previously described DNAbinding domain via a long flexible linker. The struc-ture of this domain determined in isolation as four 42residue peptides consists of a dimer of dimers witha combined mass of 20 kDa. Each monomer consistsof a regular helix (335–353) and an extended regionresembling a poorly defined β strand conformation(residues 326–333) with the two elements linked via atight turn based around Gly334. An antiparallel inter-action of helices in each monomer promotes the forma-tion of a pair of dimers. The interface between the pairof dimers is mediated solely by helix–helix contactswith the overall result being a symmetric, four-helixbundle (Figure 12.45). It has proved possible to mutatethe tetramerization domain to favour dimer formationover that of the tetramer. Extensive mutagenesis com-bined with folding and stability studies suggest that tworesidues Leu340 and Leu348 promote tetramer forma-tion via interactions of non-polar side chains.
A total of nine residues were identified as keyresidues in governing stability: Phe328, Ile332, Arg337,
Figure 12.45 The structure of the isolated tetramericregion of p53. The blue and green helices pair togetheras one dimer with the yellow and orange helicesforming the second dimer. The N- and C-terminalsare indicated, and point in the case of the N-terminal domain towards the DNA binding core withthe C-terminal region pointing towards the remainingparts of p53. The extended region before each helixresembles a β strand and two neighbouring strands onadjacent monomers also interact
Phe338, Met340, Phe341, Leu344, Ala347 and Leu348.All of these residues are invariant in mammalianp53s with the exception of the minor transitionPhe338Tyr, and point to important roles for theseresidues.
The realization that anomalous p53 activity is acommon denominator in many human cancers wasa major advance. In approximately 50 percent oftumours p53 is inactivated as a result of mutationswithin the p53 gene (Table 12.4). In other tumours theinactivation of p53 occurs indirectly through binding ofviral proteins or through mutations in genes encodingproteins that interact with p53. These mutations alterthe flow of information between p53 and the rest of thecellular apparatus.
Most p53 mutations occur in somatic cells and there-fore only induce disease in the individuals carrying thedefective gene. Mutations in germ line cells (egg andsperm) affect succeeding generations and for p53 aninherited defect known as Li–Fraumeni syndrome hasbeen identified where the gene encoding p53 possessesa mutation. The mutation of p53 is associated withsoft-tissue sarcomas and osteosarcomas (bone) whereindividuals with this inherited condition develop manydifferent cancers from a young age. Since mutations in

474 PROTEIN STRUCTURE AND A MOLECULAR APPROACH TO MEDICINE
Table 12.4 p53 malfunctions that may lead to human cancers
Mechanism of p53inactivation
Typical tumours Effect of inactivation
Mutation of residues inDNA binding domain
Colon, breast, lung, bladder,brain, pancreas, stomach,oesophagus and manyothers.
Prevents p53 binding to specific target DNAsequences and activating adjacent genes.
Deletion of C terminaldomain
Occasional tumours at manydifferent sites
Prevents the formation of p53 tetramers.
mdm2 gene multiplication Sarcomas, brain Enhances p53 degradation.Viral infection Cervix, liver, lymphomas Products of viral oncogenes bind to p53
causing inactivation and enhanceddegradation.
Deletion of p14ARF gene. Breast, brain, lung andothers especially whenp53 is not mutated.
Failure to inhibit mdm2 with the result thatp53 degradation is uncontrolled
Mislocalisation of p53 tocytoplasm.
Breast, neuroblastomas p53 functions only in the nucleus.
Adapted from Vogelstein, B. et al. Nature 2000, 408, 307–310.
p53 create genetic instability by the inability to repairdamaged DNA or a failure to perform apoptosis defec-tive DNA accumulates making individuals with theLi–Fraumeni syndrome very susceptible to cancer.
Of the mutations identified within p53 almost all arelocated in DNA binding and tetramerization domainsand a vast array of different mutations have beenidentified. Databases exist where mutations of p53found within tumours have been categorized; over14 000 tumour-associated mutations of p53 have beendescribed as of 2003. Within this vast panoply ofmutations exist ‘hot spots’ along the gene. Threemutational hot spots are residues 175 (Arg), 248 (Arg)and 273 (Arg) of the protein. A distribution of allmutations identified to date suggest approximately 20percent of mutations involve these three residues withresidue 248 being the site of highest mutation (∼7.5percent). Along with a further three residues (245/Gly,249/Arg and 282/Arg) these residues will account for30 percent of all p53 mutations found in tumours.
If humans have an in-built tumour suppressor genesuch as p53 why is cancer so common? Ignoringinherited conditions the answer lies in the influenceof environmental conditions on p53. Mutagens alterthe DNA sequence of p53 leads to base changes thatare eventually reflected in an altered and functionallyless active protein. One potent example of thisenvironmental influence is the effect of cigarettesmoking on p53. There is no doubt today aboutepidemiological data showing the overwhelminglystrong correlation between lung cancer and cigarettesmoking, yet despite emphatic data a molecular basisfor cancer development has taken longer to establish.One poisonous ingredient of cigarette smoke is aclass of organic molecules known as benzopyrenes.Exposure of the p53 gene sequence to these mutagenscaused alterations in base sequence most frequentlythe substitution of G by T. These mutagens covalentlycross-link with DNA at the G residues found at codons175, 248 and 273. Since p53 is a tetramer the mutation

EMPHYSEMA AND α1-ANTITRYPSIN 475
of just one subunit perturbs the activity throughout thecomplex resulting in impaired DNA binding.
Another mechanism for cancer lies in the inactiva-tion of p53 by the human adenovirus and papillomavirus. These viruses bind to p53 inactivating the proteinfunction and preventing a successful repair of dam-aged DNA. At residue 72 of p53 two alleles exist witheither arginine or proline found in the wild type protein.Initial studies suggested these variants were function-ally equivalent but current evidence suggests the Arg72variant is more susceptible to viral mediated proteoly-sis. For the human papillomavirus (HPV18) – a majorfactor in cervical carcinoma – this involves the prefer-ential interaction of Arg72 p53 with the E6 protein ofthe virus in vitro and in vivo. From these studies it wassuggested that cancer of the cervix is approximatelyseven times higher in individuals homozygous for theArg72 allele.
Other cancers involve amplification of mdm2 – aprotein that inactivates p53 by binding to the transcrip-tional domain. High levels of mdm2 cause a perma-nent inactivation of p53 and an increased likelihoodthat mutations will be passed on to daughter cells.In view of its role ‘controlling’ p53 mdm2 is widelybelieved to represent the next avenue for intensive can-cer research.
The amount of information existing on all aspectsof p53 function and mutant protein expression inhuman cancers is vast and reflects a key role inpathogenesis. p53 is just one component of a net-work of events that culminate in tumour formation.Although inactivation of p53 either through director indirect mechanisms is most probably respon-sible for the majority of human cancers it mustbe remembered that genes other than p53 are tar-gets for carcinogens. The identification and subse-quent characterization of these genes/proteins willincrease the molecular approach to the treatmentof cancer.
Emphysema and α1-antitrypsinEmphysema is a genetic, as well as an acquired disease,characterized by over-inflation of structures in thelungs known as alveoli or air sacs. This over-inflationresults from a breakdown of the walls of the alveoliand this results in decreased respiratory function. As
a result breathlessness is a common sign associatedwith emphysema, and together with chronic bronchitis,emphysema comprises a condition known as chronicobstructive pulmonary disease (COPD). FrequentlyCOPD is associated with lung disease arising fromsmoking but in some instances emphysema has beenidentified as a genetically inherited condition.
Emphysema is defined by destruction of airwaysdistal to the terminal bronchiole with destruction ofalveolar septae and the pulmonary capillary regionleading to decreased ability to oxygenate blood. Thedestruction of the alveolar sacs is progressive andbecomes worse with time. The body compensateswith lowered cardiac output and hyperventilation. Thelow cardiac output leads to the body suffering fromtissue hypoxia and pulmonary cachexia. A commonoccurrence is for sufferers to develop muscle wastingand weight loss and they are often identified as ‘pinkpuffers’. In the United States it is estimated that two-thirds of all men and one-quarter of all women haveevidence of emphysema at death with total sufferersapproaching ∼2 million. As part of COPD it isresponsible for the fourth leading cause of death inthe United States and represents a major demand onhealth service care.
As a genetic condition emphysema has beenattributed to a deficiency in a protein called α1-antitrypsin. Unlike the common form of emphysemaseen in individuals who have smoked for many yearsthis form of the disease is often labelled as α1-antitrypsin deficiency form of emphysema. The diseasecan occur at an unusually young age (before 40) afterminimal or no exposure to tobacco smoke and hasbeen shown to arise in families. The latter observationpointed to a genetic link.
The biological role of α1-antitrypsin is as an enzymeinhibitor of serine proteases and despite its nameits main target for inhibition is the enzyme elastase.It is a member of a group of proteins known asserpins or serine protease inhibitors. Collectively theseproteins inhibit enzymes by mimicking the naturalsubstrate and binding to active sites. Serpins have beenidentified in a wide variety of viruses, plants, andanimals with more than 70 different serpins identified.Members of the serpin family have very importantroles in the inflammatory, coagulation, fibrinolytic andcomplement cascades. Consequently malfunctioning

476 PROTEIN STRUCTURE AND A MOLECULAR APPROACH TO MEDICINE
serpins have been shown to lead to several forms ofdisease including cirrhosis, thrombosis, angio-oedema,emphysema as described here and dementia.
The site of action of α1-antitrypsin is the serineprotease elastase. Elastase is commonly found inneutrophils and is structurally homologous to trypsinor chymotrypsin (see Chapter 7). Neutrophils representthe primary defence mechanism against bacteria byphagocytosis of pathogens followed by degradation andin this latter reaction elastase is involved. At sites ofinflammation hydrolytic enzymes of neutrophil originare detected and this includes the enzyme elastase.
α1-antitrypsin is the major serine protease inhibitorpresent in mammalian serum. It is synthesized inthe liver and represents the major protein found in theα1-globulin component of plasma. In the serum theconcentration of α1-antitrypsin is very high (∼2 g l−1)and this reflects its important role. A deficiency incertain individuals of α1-antitrypsin was identified inthe 1960s by detailed analysis of gel electrophoreticprofiles but as with most diseases attention has recentlybeen directed towards identifying the gene and itslocation within the human genome.
The gene is called the PI (protease inhibitor) geneand is located on the long arm of chromosome 14.Genetic screening of individuals presenting with earlyonset emphysema has allowed many mutations to beidentified. The normal allele is the M allele and doesnot result in the disease. Genetic analysis of the Mallele has shown that several different forms designatedas M1, M2, M3, etc. can be distinguished with M1 rep-resenting approximately ∼70 percent of the populationand distinguished by two variants; Val213 and Ala213.These two forms represent nearly all of the M1 pheno-type and occur in a ratio of ∼2:1. Alternative allelesare attributed to an increased incidence of disease inindividuals possessing one or more copies and the twomost common are the S and Z forms. The Z allele ismost frequently associated with codon 342 and resultsin the non-conservative substitution of glutamate bylysine as a result of a single base change from GAGto AAG. This mutation is estimated to be present inthe north European population at ∼1 in 60. Individualshomozygous for the Z allele (often written as PI ZZ)are at greatly increased risk of emphysema and liverdisease. The second form is the S allele and is asso-ciated with the substitution of glutamate by valine at
codon 264. Individuals homozygous for the S allele (PISS) do not display symptoms of the disease but indi-viduals with one copy of the Z allele and one copy ofthe S allele (PI SZ) may develop emphysema. Over 70different mutations have been identified and it appearsthat these mutations can be grouped into three majorcategories. Some mutations disrupt the serpin functionof the protein, whilst rarer mutations are described asnull or silent mutations and a third group appear tocause drastic reduction in the amount of α1-antitrypsinpresent in the serum as opposed to its activity.
The single polypeptide chain of α1-antitrypsin con-tains 394 amino acid residues after removal of a 24-residue signal sequence. The presence of three glyco-sylation sites leads to higher than expected molecularweight (Mr ∼52 000) when serum fractions are anal-ysed by gel electrophoresis but the carbohydrate is notnecessary for catalytic function. Within the polypep-tide chain the sequence Pro357-Met358-Ser359-Ile360represents a ‘substrate’ for target protease. This regionof the polypeptide effectively forms a ‘bait’ for thecognate protease, in this case leucocyte elastase, andthe reaction between serpin and protease leads to com-plete inactivation.
The structure of α1-antitrypsin reveals three sets ofβ sheets designated A–C and nine helical elements.The basis of inhibition revolves around interactionbetween cognate serine protease and serpin by thepresentation of a region of the serpin polypeptidechain that resembles natural substrates of the protease.Binding occurs at an exposed region of the polypeptidechain known as the reactive centre loop a region heldat the apex of the protein and based around the criticalMet358 residue (Figure 12.46). The mobile reactivecentre loop presents a peptide sequence as the substratefor the target protease. Docking of the loop of theserpin with the protease is accompanied by cleavageat the P1 –P′
1 bond and the enzyme is locked into anirreversible and extremely stable complex. In the caseof α1-antitrypsin the P1 –P′
1 bond (i.e. the residueseither side of the bond to be cleaved) lies betweenMet358 and Ser359 in the loop located at the apex ofthe protein.
Upon protease binding large conformational re-arrangement occurs in which the two major structuraltransitions occur. The reactive centre loop is splitbetween Met358 and Ser359 and the Aβ sheet is able to

EMPHYSEMA AND α1-ANTITRYPSIN 477
Figure 12.46 Two conformational states of α1-antitrypsin showing conformational changes uponcleavage. Residues 350–394 are coloured orange todistinguish from the remainder of the molecule wherethe strands are shown in green and the helices in blue(PDB:2 PSI and 7API)
Figure 12.47 Space filling model of interactionbetween serpin and protease. Met358 and Ser359 canbe seen at either end of the serpin (green)
open to accommodate much of the reactive centre loopregion as a fifth β strand. This conformational change isvital for efficient inhibition and leads to Met358 beinglocated at the foot of the molecule (Figure 12.47).In this conformation the α1-antitrypsin and elastase
are irreversibly bound in a stable complex thatprevents further reactivity. Conformational mobility ofthese regions results in sensitivity to mutation andsignificantly the Z allele of α1-antitrypsin is locatedwithin the reactive loop region (effectively as P′
4).In many individuals with α1-antitrypsin deficiency
liver disease is detected as an associated condition. Anexplanation for this association has now been found. Inthe homozygous ZZ condition only about 15 percentof the normal level of α1-antitrypsin is secreted intothe plasma. The normal site of synthesis is the liverand it was observed that the remaining 85 percentaccumulates in the ER of the hepatocyte. Althoughmuch is degraded the remainder aggregates forminginsoluble intracellular inclusions. These aggregates areresponsible for liver injury in approximately 12–15percent of all individuals with this genotype. It isalso observed that about 10 percent of newborn ZZhomozygotes develop liver disease leading to fatalchildhood cirrhosis. Studies based on the structureof α1-antitrypsin have demonstrated the molecularpathology underlying protein accumulation.
The principal Z mutation in antitrypsin results inthe substitution of glutamate by lysine at residue 362and allows a unique molecular interaction betweenthe reactive centre loop of one molecule and the‘gap’ in the A-sheet of another serpin. Effectively thereactive centre loop of one α1-antitrypsin inserts intothe Aβ sheet region of a second molecule in a processknown as loop to sheet polymerization. In mutantα1-antitrypsin this occurs favourably at 37 ◦C and isvery dependent on concentration and temperature. Thepolymerization is blocked by the insertion of a specificpeptide into the A-sheet of α1-antitrypsin.
α1-antitrypsin is an acute phase protein and for mutantprotein it will undergo a significant increase in asso-ciation with temperature as might arise during boutsof inflammation. The result is harmful protein inclu-sions in the hepatocyte that may overwhelm the cell’sdegradative mechanisms, especially in the newborn. Thecontrol of inflammation and pyrexia in ZZ homozy-gote infants is very important to overall managementof the disease. The serpins show considerable homol-ogy and the mechanism of loop–sheet polymerizationis the basis of deficiencies associated also with muta-tions of C1-inhibitor (the activated first component

478 PROTEIN STRUCTURE AND A MOLECULAR APPROACH TO MEDICINE
of the complement system), antithrombin III, and α1-antichymotrypsin implicated in other disease states. Col-lectively, these diseases could be called serpinopathies.
It has been shown that replacement of deficient pro-tein by intravenous infusion of purified α1-antitrypsincan limit disease progression. Unlike many of the previ-ously described conditions this treatment represents aneffective protocol that limits much of the necrosis of lungtissue associated with hereditary forms of emphysema.This form of emphysema is currently treated with proteinisolated from plasma fractions, but with characteriza-tion of the cDNA encoding the protein α1-antitrypsinhas been expressed to allow high level production inthe milk of transgenic sheep. Although this form of α1-antitrypsin has yet to complete clinical trials it will offera better alternative to conventionally purified protein.The underlying genetic condition will always remain butconsiderable improvement results from treatment withpurified α1-antitrypsin. The high concentration of α1-antitrypsin in serum means that on average a patient mayrequire over 200 g of highly purified proteins per yearas the basis of a therapeutic regime.
For individuals with this genetic condition a lifetimeavoidance of smoking is highly recommended since theeffects of smoking as a primary cause of emphysemaare well documented. In this context it has beenshown that one effect of smoking can be to causeoxidation of the Met358 residue within α1-antitrypsin.The oxidation of the methionine side chain to forma sulfoxide results in permanent irreversible damageto α1-antitrypsin and eliminates its biological roleas a serpin. A loss of inhibition allows uncheckedelastase activity and promotes alveolar sac destructionthat lead to emphysema. When combined with agenetic tendency towards emphysema it is clear thatthis situation must be avoided to prevent extremelung damage. Genetically linked forms of emphysemarepresented one of the first molecular defects tobe extensively characterized and the highly detailedstructural and functional studies of serpins such as α1-antitrypsin highlight the diagnostic, preventative andtherapeutic powers of molecular medicine.
SummaryMolecular medicine represents a multidisciplinaryapproach to understanding human disease through
the use of structural analysis, pharmacology, genetechnology and even gene therapy. In 1955 sickle cellanaemia was identified as arising from a single aminoacid change within haemoglobin. Many other diseasesarise from similar genetic mutations.
Unprecedentedprogresshasbeenmade inunderstand-ing the molecular basis of many diseases including new,emerging, ones such as HIV and vCJD alongside familiarconditions such as influenza. Understanding at a molec-ular level offers the prospect of better disease diagnosisand in some cases improved therapeutic intervention.
Viruses represent a persistent human health problem,being responsible for many diseases. Vaccinationusing attenuated viruses or purified proteins effectivelyeradicated infections such as smallpox and polio butthreats from rapidly mutating viruses such as influenzaand HIV continue.
HIV continues to contribute to significant, world-wide, mortality in the 21st century whilst influenzareturns periodically in the form of severe outbreaks orpandemics. In each virus the ability to mutate surfaceantigens rapidly compromises therapeutic drug actionor antibody directed immune response.
The protein p53 has been called the ‘Guardian of thegenome’. Its major role is to prevent the perpetuation ofdamaged DNA by causing cell cycle arrest or apoptosis.The protein is a conformationally flexible moleculeconsisting of an N-terminal transcriptional activationdomain, a central or core DNA binding domain and athird C terminal domain that contains multiple functionsincluding the ability to promote tetramerization.
The core DNA binding domain is based on a β
sheet scaffold that binds to major and minor grooves ofthe double helix through the presence of charged sidechains arranged precisely in conserved loops that linkelements of β strands.
Cancer and p53 are inextricably linked through thedemonstration that many tumours have mutations inp53. Most, but not all, are located in the centralDNA binding domain with mutational hot spotsinvolving residues participating in DNA binding. Thecell requires exquisite control pathways to modulatethe activity of p53 and corruption of these pathwayscan often lead to the development of cancer.
α1-antitrypsin is a serpin that acts to limit theunregulated activity of neutrophil elastase. The proteininhibits elastase by forming a tightly bound complex. It

PROBLEMS 479
is found at high concentrations in the globulin fractionof serum where its abundance helped to identifyindividuals deficient in serum α1-antitrypsin.
Deficiency arises from a gene mutation and is aninherited condition leading to emphysema as a result
of damage caused to the alveolar lining of the lungs.Mutant α1-antitrypsin fails to bind elastase effectively.
Mutated forms of α1-antitrypsin show a tendencyto exhibit loop–sheet polymerization – a process thatcauses the irreversible association of serpins.
Problems1. Why should individuals with sickle cell anemia avoid
intense exercise or exposure to high altitudes?
2. Vaccines have been developed against smallpox,measles and many other viral based diseases. So whatis the problem with developing a vaccine for fluand HIV?
3. The ionization of maleic acid (COOH–CH2–CHOH–COOH) occurs with pK values of 1.9 and 6.2. Discussthe reasons for these values and what is the relevanceto the activity of aspartic proteases.
4. Using the known properties of the active site ofneuraminidase describe why Zanamivir is an efficientinhibitor of enzyme activity especially when comparedwith Neu5Acen. Can you suggest potentially otherbeneficial drug design.
5. Is vCJD a new disease or merely an old one firmlyrecognized and identified?
6. Identify the conformationally sensitive strands toinhibitor binding and active site aspartates in HIVprotease.


Epilogue
It is 50 years since the discovery of the double helicalstructure of DNA, an event that marks the beginningof molecular biology. The structure of DNA representsthe seed that germinated, grew rapidly, flowered andbore fruit. It has led to descriptions of the flow ofinformation from gene to protein. Molecular biology,expanded from a small isolated research area into amajor, all embracing, discipline. As this book is beingpublished close to the 50th anniversary it is timely toreflect that during this period biochemistry has beenmarked by several defining discoveries based on thestructure and function of proteins. The list of significantdiscoveries could provide the bulk of the content ofanother book but a brief and selective description ofthese events might include:
• The elucidation of the structure of myoglobin andhaemoglobin by Perutz and Kendrew using X-raycrystallography. This work paved the way forall future crystallographic studies of proteins andestablished the basis for allostery.
• The structure of the first enzyme, lysozyme, byPhillips. Structural characterization defined an activesite and the geometry of residues that facilitatedbiological catalysis and pointed towards molecularenzymology.
• Protein folding is encoded entirely by the primarysequence. Anfinsen proved that proteins could foldto reach the native state whilst in cells largemacromolecular complexes known as chaperoneswere shown much later to assist folding by formingenvironments known as the Anfinsen cage that limitunfavourable protein interactions.
• Crystallization of the photosynthetic reaction cen-tre by Michel, Diesenhofer and Huber. The heli-cal structure of transmembrane segments was con-firmed and showed that membrane proteins couldbe crystallized and subjected to the same high res-olution methods applied previously to soluble pro-teins.
• The architecture of the ribosome. A daunting exper-imental problem revealed catalysis of the peptidyltransferase was performed by RNA and the ribo-some is a ribozyme. The structure of the ribosomeconfirmed that biological catalysis could proceed inthe absence of protein-based enzymes redefining ourtraditional view of nucleic acid and proteins.
• p53 and the molecular basis of cancer. The demon-stration that over 60 percent of all tumours are asso-ciated with mutations in p53 elucidated a direct linkbetween molecular defects in a protein and subse-quent development of cancer.
• The prion hypothesis. Consequently the prionhypothesis showed that some proteins ‘corrupt’native conformations promoting protein aggregation;a hallmark of many neurodegenerative disorders.
By reading the preceding 12 chapters, where thesediscoveries are described in more detail, I hope thereader will gain an impression of the rapidly expandingand advancing area of protein biochemistry. This areaoffers the potential to revolutionize treatment of humanhealth and to eradicate diseases in all avenues of life.The next 50 years will see the complete description of
Proteins: Structure and Function by David Whitford 2005 John Wiley & Sons, Ltd

482 EPILOGUE
URL Description
www.rcsb.org/pdb The repository for the deposition of biomolecular
structures mainly nucleic acids and proteins
www.hgmp.mrc.ac.uk The human genome mapping project centre in the UK
www.nhgri.nih.gov The human genome mapping research institute of the
NIH
www.expasy.ch The ExPASy (Expert Protein Analysis System)
proteomics server of the Swiss Institute of
Bioinformatics (SIB)
www.ebi.ac.uk European Bioinformatics Institute containing access to
databases and software for studying proteins
www.ncbi.nlm.nih.gov A national resource for molecular biology information in
USA
www.srs.ebi.ac.uk Sequence retrieval system for a wide variety of databases
rebase.neb.com/rebase/rebase.html Restriction enzyme database
bimas.dcrt.nih.gov/sql/BMRBgate.html Biomolecular Nuclear Magnetic Resonance databank
tolweb.org/tree/phylogeny.html Tree of Life web page
www.ncbi.nlm.nih.gov/Omim On line Mendelian Inheritance in Man
www.biochem.ucl.ac.uk/bsm/biocomp/index.html A collection biocomputing resources at University
College London
www.ensembl.org A database annotating sequenced genomes
www.embl-heidelberg.de/predictprotein/predictprotein.html Protein prediction server for secondary structure
www-nbrf.georgetown.edu/pirwww/pirhome3 Protein Information resource centre
archive.uwcm.ac.uk/uwcm/mg/hgmd0.html/ Human gene mutation database
gdbwww.gdb.org/ Another genome database
www.ornl.gov/hgmis US Human Genome Project Information
See home page of this book for latest web-based or on-line resources.
other proteomes and promises the possibility of equallydevastating discoveries to match those of the previous50 years.
The following universal resource locators (URL)represent web addresses of sites that offer very usefulinformation of relevance to the subject of this book.
The author is not responsible for their content and dueto the transitory nature of the web these sites maynot always be accessible, maintained or presenting thelatest information. However, all (as of April 2004) hadpotentially useful information that provides an ideallearning resource.

Glossary
α helix regular unit of secondary structure shown bypolypeptide chains characterised by 3.6 residues per turn ina right-handed helix with a pitch of 0.54 nm.
β sheet collection of β strands assembled into planar sheetlike structure held together by hydrogen bonds.
3d10 helix a form of secondary structure containing threeresidues per turn and hydrogen bonds separated by 10backbone atoms.
Ab initio from the beginning (Latin).
Acid–base catalysis reactions in which the transfer of aproton catalyses the overall process.
Acidic solution a solution whose pH is less than pH 7.0.
Active site part of an enzyme in which the aminoacid residues form a specific three-dimensional structurecontaining the substrate binding site.
ADP adenosine diphosphate.
Affinity chromatography. separation of proteins on thebasis of the specific affinity of one protein for an immobilizedligand covalently attached to an inert matrix.
AIDS disease ultimately resulting from infection with HIV,sometimes called advanced HIV disease.
Allosteric effectors molecules which promote allosterictransitions in a protein.
Allostery with respect to proteins, a phenomenon where theactivity of an enzyme’s active site is influenced by bindingof an effector to a different part of the enzyme.
Alzheimer’s a disease characterized by formation ofprotein deposits with the brain and classified as a neurode-generative disorder.
Amino acid an organic acid containing an amino group,carboxyl group, side chain and hydrogen on a central α
carbon. The building blocks of proteins.
Amphipathic for a molecule, the property of having bothhydrophobic and hydrophilic portions. Usually one end orside of the molecule is hydrophilic and the other end or sideis hydrophobic.
Ampholyte a substance containing both acidic and basicgroups.
Amyloid accumulation of protein into an insoluble aggre-gate often a fibre in tissue such as the brain.
Anabolism process of synthesis from small simple mole-cules to large often polymeric states.
Antigen a substance that can elicit a specific immuneresponse.
Anaerobe organism capable of living in the absenceof oxygen.
Antibody protein component of immune system producedin response to foreign substance and consisting of two heavyand two light chains.
Anticodon sequence of three nucleotide bases found intRNA that recognizes codon via complementary base pairinginteractions.
Antigenic determinant a specific part of an antigen thatelicits antibody production.
Antiparallel running in opposite directions as in DNAstrands or β sheets.
Apoprotein protein lacking co-factor or coenzyme. Apopro-tein often show decreased or negligible activity.
Apoptosis programmed cell death.
Archae one of the two major groupings within theprokaryotes, also called archaebacteria.
Asymmetric centre a centre of chirality. A carbon atomthat has four different substituents attached to it.
Proteins: Structure and Function by David Whitford 2005 John Wiley & Sons, Ltd

484 GLOSSARY
ATP adenosine triphosphate.
ATPase an enzyme hydrolysing ATP to ADP and Pi.
Association constant/Affinity constant given by ‘K’, inthe oxygen binding reaction of myoglobin, K is calculatedas the concentration of myoglobin bound to oxygen dividedby the product of the free oxygen concentration and the freemyoglobin concentration.
B cells lymphatic cells produced by B lymphocytes.
Backbone part of the polypeptide chain consisting ofN–Cα–C portion (distinct from side chain)
Bacteriophage virus that specifically infects a bacterium.
Basic solution a solution whose pH is greater than pH 7.0.
BSE bovine spongiform encephalopathy, a prion-baseddisease first seen in cattle in the UK.
bp base pair, often used to describe length of DNAmolecule.
Buffer a mixture of an acid and its conjugate base at a pHnear to their pK. Buffer solution composed of an acid andconjugate base resist changes in pH.
Cahn–Ingold–Prelog system of unambiguous nomencla-ture of molecules with one or more asymmetric centres via apriority ranking of substituents. Also known as RS system.
Cap a 7-methyl guanosine residue attached to the 5′ end ofeukaryotic mRNA.
Capsid protein coat or covering of nucleic acid in viralparticles.
Catabolism metabolic reactions in which larger moleculesare broken down to smaller units. Proteins are broken downinto amino acids.
cDNA complementary DNA derived from reverse tran-scription of mRNA.
Chaotrope a substance that increases the disorder (chaos).Frequently used to describe agents that cause proteins todenature. An example is urea.
Chaperonins proteins which assist in the assembly ofprotein structure. They include heat-shock proteins andGroEL/ES of E. coli.
Charge–charge interactions interactions between posi-tively and negatively charged side chain groups.
Chiral possessing an asymmetric center due to differentsubstituent groups and exist in two different configurations.
Chloroplasts plant organelles performing photosynthesis.
Clathrate structures hydrophobic molecules that dissolvein aqueous solutions form regular icelike structures calledclathrate structures rather than the hydration shells formedby hydrophilic molecules.
Cloning process of generating an exact copy.
Coding strand analogous to sense strand.
Co-factor a small organic molecule or sometimes a metalion necessary for the catalytic activity of enzymes.
Coiled coil arrangement of polypeptide chains where twohelices are wound around each other.
Configuration arrangement of atoms that cannot be alteredwithout breaking and reforming bonds.
Conformational entropy a protein folding process, whichinvolves going from a multitude of random-coil conforma-tions to a single, folded structure. It involves a decrease inrandomness and thus a decrease in entropy.
Codon sequence of three nucleotide bases found in mRNAthat determines a single amino acid.
Conformation proteins and other molecules occur indifferent spatial arrangements because of rotation aboutsingle bonds that leads to a variety of different, closerelated states.
Conservative amino acid changes Mutations in codingsequences for proteins which convert a codon for one aminoacid to a codon for another amino acid with very similarchemical properties.
Cooperative transition a transition in a multipart structuresuch that the occurrence of the transition in one part ofthe structure makes the transition likelier to happen inother parts.
Covalent bonds the chemical bonds between atoms in anorganic molecule that hold it together are referred to ascovalent bonds.
Cristae invaginations of inner mitochondrial membraneinvolved in oxidative phosphorylation.
Cryo-EM cryoelectron microscopy a technique for thevisualization of macromolecules achieved by rapid freezingof a suspension of the biomolecule without the formationof ice.
Cystine the amino acid cysteine can form disulfide bondsand the resulting structure is sometimes called a cystine,particularly in older textbooks.

GLOSSARY 485
Denaturation loss of tertiary and secondary structureof a protein leading to a less ordered state that isfrequently inactive.
Da Dalton or a unit of atomic mass equivalent to 1/12ththe mass of the 12C atom.
Debye–Huckel radius a quantitative expression of thescreening effect of counterions on spherical macro ions.
Dihedral angle an angle defined by the bonds between foursuccessive atoms. The backbone dihedral angle φ is definedby C′–N–Ca–C′.
Dimer assembly consisting of two subunits.
Dipeptide a molecule containing two amino acids joinedby a single peptide bond.
Dipolar ion term synonymous with zwitterions.
Dipole Moment molecules which have an asymmetricdistribution of charge are dipoles. The magnitude of theasymmetry is called the dipole moment of the molecule.
Disulfide bond a covalent bond between two sulfuratoms formed from the side chains of cysteine residues,for example.
DNA deoxyribose nucleic acid – the genetic material ofalmost all systems.
Domain a compact, locally folded region of tertiarystructure in a protein.
Elution removal of a molecule from chromatographicmatrix.
Edman degradation procedure for systematically sequenc-ing proteins by stepwise removal and identification of Nterminal amino acid residue.
EF elongation factor, one of several proteins involved inprotein synthesis enhancing ribosomal activity.
Enantiomers also called optical isomers or stereoisomers.The term optical isomers arises from the fact that enantiomersof a compound rotate polarized light in opposite directions.
Endergonic reaction process that has a positive overallfree energy process (�G > 0).
Enzymes catalytic proteins.
Electrophile literally electron-lover and characterized byatoms with unfilled electron shells.
Eubacteria one of the two major groupings within theprokaryotes.
Eukaryote a cell containing a nucleus that retains thegenetic material in the form of chromosomes. Often mul-ticellular and with cells showing compartmentation.
Exons a region in the coding sequence of a gene that istranslated into protein (as opposed to introns, which are not).The name comes from the fact that exons are the only partsof an RNA transcript that are seen outside the nucleus ofeukaryotic cells.
Fc fragment a proteolytic fragment of an antibodymolecule
Fibrous proteins a class of proteins distinguished by afilamentous or elongated three dimensional structure suchas collagen.
First order a reaction whose rate is directly proportionalto the concentration (activity) of a single reactant.
FT Fourier transform.
FT-IR Fourier transform infrared spectroscopy.
g estimate of centrifugal force so that 5000 g is 5000 timesthe force of gravity.
G protein a protein that binds guanine nucleotides suchas GTP/GDP.
Gel filtration chromatography also called size exclusionchromatography. Separates biomolecules such as proteinsthrough the use of closely defined pore sizes within an innertmatrix according to the molecular mass.
Globular proteins proteins containing polypeptide chainsfolded into compact structures that are not extended orfilamentous and have little free space on the inside.
Heme prosthetic oxygen binding site of globin (and other)proteins. Is a complex of protoporphyrin IX and Fe (II).Carries oxygen in globin proteins
Henderson–Hasselbach equation describes the dissocia-tion of weak acids and bases according to the equationpH = pKa + log ([A−]/[HA]).
Heterodimer a complex of two polypeptide chains inwhich the two units are non-identical.
HIV human immunodeficiency virus; retrovirus responsi-ble for AIDS.
HLH helix-loop-helix motif found in several eukaryoticDNA binding proteins.
Hormone molecule often but not exclusively protein thatis secreted into blood stream and carried systemically whereit elicits a physiological response in another tissue.

486 GLOSSARY
Hydrolysis cleavage of covalent chemical bond involv-ing water.
Homeobox a DNA binding motif that is widely found ineukaryotic genomes where it encodes a transcription factorwhose activity modulates the development, identity and fateof embryonic cell lines.
Homodimer a complex of two units in which both unitsare identical.
Hsp heat-shock protein – name given to a large group ofmolecular chaperone proteins.
HTH helix-turn-helix motif found in several DNA bindingproteins in which the two helices cross at an angle of ∼120◦.
Huntingtin the mutant protein contributing to Hunting-ton’s disease.
Hydrogen bond an attractive interaction between thehydrogen atom of a donor group, such as OH or =NH, anda pair of nonbonding electrons on an acceptor group, such asO=C. The donor group atom that carries the hydrogen mustbe fairly electronegative for the attraction to be significant.
Hydrophilic refers to the ability of an atom or a moleculeto engage in attractive interactions with water molecules.Substances that are ionic or can engage in hydrogen bondingare hydrophilic. Hydrophilic substances are either soluble inwater or, at least, wettable.
Hydrophobic the molecular property of being unable toengage in attractive interactions with water molecules.Hydrophobic substances are nonionic and nonpolar; they arenonwettable and do not readily dissolve in water.
Hydrophobic effect stabilization of protein structure re-sulting from association of hydrophobic groups with eachother, away from water.
IF initiation factor involved in start of protein synthesis andribosomal assembly and activation.
Ig immunoglobulin and another name for an antibodygroup such as IgG.
Importins generic group of proteins with homology toimportin α and β that function as heterodimer binding NLS-proteins prior to import into nucleus.
In vitro normally means in the laboratory, but literallyin glass.
In vivo in a living organism.
Integral protein a membrane bound protein that can onlybe removed from the lipid bilayer by extreme treatment. Alsocalled intrinsic protein.
Intron(s) a region in the coding sequence of a gene that isnot translated into protein. Introns are common in eukaryoticgenes but are rarely found in prokaryotes. They are excisedfrom the RNA transcript before translation.
Ionic strength an expression of the concentration of allions. In the Debye–Huckel theory I = 1/2 �miz
2i
Isoelectric focusing a technique for separating ampholytesand polyampholytes based on their pI . Also used todetermine pI .
Isoelectric point the pH at which an ampholyte orpolyampholyte has a net charge of zero. Same as pI .
Isoenzymes also called isozymes. Represent different pro-teins from the same species that catalyse identical reactions.
kDa kilodaltons; equivalent to 1000 daltons or approxi-mately 1000 times the mass of a hydrogen atom.
Keratins major fibrous proteins of hair and fingernails.They also compose a major fraction of animal skin.
Kinases enzymes that phosphorylate or transfer phospho-rus groups to substrates including other proteins (e.g. pro-tein kinases)
Km the Michaelis constant. It is the concentration ofsubstrate at which the enzyme-catalysed reaction proceedswith half maximal velocity
Leader sequence a short N-terminal hydrophobic sequencethat causes the protein to be translocated into or through amembrane often called a signal sequence.
Mesophile an organism living at normal temperatures incomparison with a thermophile.
Module a sequence motif of between 40–120 residuesthat occurs in unrelated proteins or as multiple unitswithin proteins.
Molten globule state intermediate structures of a proteinin which the overall tertiary framework of the protein isestablished, but the internal side chains are still free tomove about.
Mutation a change in the sequence of DNA.
NES nuclear export signal – a signal consisting of severalleucine residues found in proteins and indicating export.
NLS nuclear localization signal – a short stretch of basicamino acid residues that targets proteins for import into the

GLOSSARY 487
nucleus. The sequence can have bipartite structure consistingof two short sequences of basic residues separated by #10intervening residues.
NMR spectroscopy acronym for nuclear magnetic reso-nance spectroscopy, a technique useful in the elucidation ofthe three-dimensional structure of soluble proteins.
Non-covalent interactions attractive or repulsive forces,such as hydrogen bonds or charge–charge interactions,which are non-covalent in nature, are called non-covalentinteractions.
NPC nuclear pore complex – a very large assembly ofprotein concerned with regulating flux of macromoleculesbetween nucleus and cytoplasm.
Nucleophile atom or group that contains an unsharedpair(s) of electrons and is attracted to electrophilic (electrondeficient) groups.
Nucleoporins proteins found in the nuclear pore complex.
Oncogene a gene which in a mutated form gives rise toabnormal cell growth or differentiation.
Oncoprotein the product of an oncogene that fails toperform its normal physiological role.
Operator element of DNA at the transcriptional site thatbinds repressor.
Operon a genetic unit found in prokaryotes that istranscribed as a single mRNA molecule and consisting ofseveral genes of related function.
Oxidative phosphorylation process occurring in mito-chondria and bacteria involving oxidation of substrates andthe generation of ATP.
Oxidoreductase catalyse redox reactions.
PAGE polyacrylamide gel electrophoresis – a techniquefor the electrophoretic separation of proteins through poly-acrylamide gels.
PCR polymerase chain reaction – a method involving theuse of thermostable DNA polymerases to amplify in a cyclicseries of primer driven reactions specific DNA sequencesfrom DNA templates.
PDI protein disulfide isomerase – an enzyme catalysingdisulfide bond formation or re-arrangement.
Peptide molecules containing peptide bonds are referred togenerically as peptides usually less than 40 residues in length.
Peptidase an enzyme that hydrolyses peptide bonds.
Peptide bond the bond that links successive amino acidsin a peptide; it consists of an amide bond between thecarboxyl group of one amino acid and the amino group ofthe next.
Peripheral protein also called extrinsic protein and refersto protein weakly associated with membrane.
pH the negative logarithm of the hydrogen ion concentra-tion in an aqueous solution.
Phage shortened version of bacteriophage.
pI the isoelectric point or the pH at which an ampholyteor polyampholyte has a net charge of zero.
Pitch the spacing distance between individual adjacentcoils of a helix.
pK a measure of the tendency of an acid to donate a proton;the negative logarithm of the dissociation constant for anacid. Also called pKa
Polypeptide a polypeptide is a chain of many amino acidslinked by peptide bonds.
Polyprotic acids acids which are capable of donating morethan one proton.
Porphyrins a class of compounds found in chlorophyll, thecytochrome proteins, blood, and some natural pigments. Theyare responsible for the red color of blood and the green colorof plants.
Prebiotic era the time span between the origin of theearth and the first appearance of living organism (∼ 4.6 ×109 –3.6 × 109 years ago)
Preinitiation complex the multiprotein complex of tran-scription factors bound to DNA that facilitates transcriptionby RNA polymerase.
Preproprotein a protein contain a prosequence in additionto the signal sequence.
Preprotein a protein containing a signal sequence that iscleaved to yield the active form.
Pribnow box prokaryotic promoter region located 10 basesupstream of the transcription start site with a consensussequence TATAAT.
Primary structure for a nucleic acid or a protein, thesequence of the bases or amino acids in the polynucleotideor polypeptide.
Protoporphyrin IX a tetrapyrrole ring which chelatesFe(II) and other transition metals.

488 GLOSSARY
Primary structure or sequence the linear order orsequence of amino acids along a polypeptide chain ina protein.
Prions a class of proteins that causes serious diseasewithout the involvement of DNA/RNA.
Procollagen a newly translated form of collagen in whichhydroxylation and addition of sugar residues has occurred,but the triple helix has not formed.
Prokaryote a simple normally unicellular organism thatlacks a nucleus. All bacteria are prokaryotes.
Prosequence region of a protein at the N terminal designedto keep the enzyme inactive. The pro sequence is removedin zymogen processing.
Prosthetic group a co-factor such as a metal ion or smallmolecule such as a heme group. It can be bound covalentlyor non-covalently to a protein and is usually essential forproper protein function.
Protease a generic group of enzymes that hydrolyse peptidebonds cleaving polypeptide chains into smaller fragments(the term proteinase is also used interchangeably). Oftenshow a specificity for a particular amino acid sequence.
Proteasome assembly of proteins based on a core structureof four heptameric rings that functions to degrade proteinsinto small peptide fragments.
Proteins biomolecule composed of one or more polypep-tide chains containing amino acid residues linked togethervia peptide bonds.
Proto-oncogene the normal cellular form of an oncogenewith the potential to be mutated. Mutation of the gene yieldsan oncogene and may lead to cancer.
PrP the protein believed to be responsible for transmittingthe disease of prions. The protein is encoded by the host’sgenome and exists in two forms, only one of which causesthe disease.
Purine planar, heterocyclic aromatic rings with adenineand guanine being two important bases found in cells.
Quantum a packet of energy
Quaternary structure the level of structure that resultsbetween separate, folded polypeptide chains (subunits) toproduce the mature or active protein.
R group one of the 20 side chains found attached to thebackbone of amino acids.
R state the relaxed state describing the activity of anallosteric enzyme or protein.
Ramachandran plot usually shown as a plot of dihedralangle φ against ψ.
Random coil refers to a linear polymer that has nosecondary or tertiary structure but instead is wholly flexiblewith a randomly varying geometry. This is the state of adenatured protein or nucleic acid.
Redox reduction–oxidation reactions.
Renaturation refolding of a denatured protein to assumeits active or native state.
RER rough endoplasmic reticulum – characterized by ribo-somes attached to this membrane involved in cotransla-tional targeting.
Residue a name for a monomeric unit with a polymer suchas an amino acid within a protein.
Reverse turn a short sequence of 3–5 residues that leadsto a polypeptide chain altering direction and characterisedby occurrence of certain amino acid residues with distinctdihedral angles. Also called a β bend.
Ribozyme a enzyme based on RNA capable of catalysinga chemical reaction.
S Svedburg unit of sedimentation with the units of 10−13 s.An example is the 30S ribosome particle. It is an estimate ofhow rapidly a protein or protein complex sediments duringultracentrifugation.
Salting in the effect of moderate amounts of ions, whichincreases the solubility of proteins in solution.
Salting out the effect of an extreme excess of ions whichmakes proteins precipitate from solution.
Scurvy a condition that occurs with vitamin C deficiencyand reflects deficiency in connective tissue and collagencross linking.
Secondary structure the spatial relationship of amino acidresidues in a polypeptide chain that are close together in theprimary sequence.
Serpin serine protease inhibitor.
Sheet a fundamental protein secondary structure (ribbon-like) discovered by Linus Pauling. It contains two amino acidresidues per turn and forms hydrogen bonds with residues onadjacent chains.

GLOSSARY 489
Site-directed mutagenesis technique for altering thesequence of a DNA molecule. If the alteration occurs in aregion coding for protein, the amino acid sequence of theprotein may be altered as a consequence.
Snurps proteins found in spliceosomes with small nuclearRNAs.
SRP signal recognition particle. A ribonucleoprotein com-plex involved in cotranslational targeting of nascent polypep-tide chains to membranes.
Stereoisomers molecules containing a center of asymmetrythat possess same chemical formula but exist with differentconfiguration or arrangement of atoms.
Substrate a reactant in an enzyme catalysed reaction thatbinds to active site and is converted into product.
T cells cells of the immune system derived from the thymusand concerned with fighting pathogens based on two typeskiller: T cells and helper T cells.
TATA box A/T rich region of genes that is involved in thebinding of RNA polymerase to eukaryotic DNA sequences.
Tertiary structure large-scale folding structure in a linearpolymer that is at a higher order than secondary structure.For proteins and RNA molecules, the tertiary structure is thespecific three-dimensional shape into which the entire chainis folded.
Thermophile bacteria capable of living at high tempera-tures sometimes in excess of 90 ◦C.
Thermosome name given to the proteasome in ther-mophiles such as T. acidophilum.
Tic analogous system to Tim found in chloroplast innermembrane
Tim translocation inner membrane – a collection of pro-teins forming a protein import pathway in the inner mito-chondrial membrane.
TMV tobacco mosaic virus.
Toc analogous system to Tom found in chloroplast outermembrane
Tom translocation outer membrane – a collection of pro-teins forming a protein import pathway in the outermitochondrial membrane.
Torsion angle also known as dihedral angle.
Transcription the process of RNA synthesis from a DNAtemplate performed by RNA polymerase and associatedproteins known as transcription factors.
Transition state all reactions proceed through a transitionstate that represents the point of maximum free energy in areaction coordinate linking reactants and products.
Transition state analogue a stable molecule that resemblesclosely the transition state complex formed at the active siteof enzymes during catalysis.
Translation the process of converting the genetic codeas specified by the nucleotide base sequence of mRNAinto a corresponding sequence of amino acids within apolypeptide chain.
Transmembrane a protein or helix that completely spansthe membrane.
Tropocollagen basic unit of collagen fibre. It is a triplehelix of three polypeptide chains, each about 1000 residuesin length.
TSE transmissible spongiform encephalopathy – any agentcausing spongiform appearance in brain.
UV region of the electromagnetic spectrum extending from∼200 to ∼400 nm.
van der Waals interactions weak interactions betweenuncharged molecular groups that help stabilize a pro-tein’s structure.
Variable domain a part of an immunoglobulin that variesin amino acid sequence and tertiary structure from oneantibody to another.
vCJD new variant CJD that arose from BSE and is atransmissible spongiform encephalopathy.
Vmax the maximal velocity in an enzyme-catalysed reaction.
vo the initial velocity associated with an enzyme-catalysedreaction
Zwitterion a molecule containing both positively andnegatively charged groups but has no overall charge. Aminoacids are zwitterionic at ∼pH 7.0.
Zymogen an inactive precursor (proenzyme) of a prote-olytic enzyme.


Appendices
Appendix 1The International System(SI) of units related to proteinstructure
Physical quantity SI unit Symbol
Length Metre mTime Second sTemperature Kelvin KElectric potential Volt VEnergy Joule JMass Kilogram Kg
Appendix 2Prefixes associated with SI units
Prefix Power of 10 (e.g. 10n)
Tera 12Giga 9Mega 6Kilo 3Milli −3Micro −6Nano −9Pico −12Femto −15Atto −18
Frequently when discussing protein structure bondlengths will be expressed in nanometres (nm). Forexample, the average distance between two carbonatoms in an aliphatic side chain is ∼0.14 nm or0.14 × 10−9 m. Occasionally a second (non SI) unit isused and is named after the Swedish physicist, AndersJ Angstrom. It is called the Angstrom (A) and isequivalent to 0.1 nm or 10−10 m. Both units are widelyand interchangeably used in protein biochemistry andin this textbook.
Appendix 3Table of important physicalconstants used in biochemistry
Planck constant h 6.6260755 × 10−34 J sh/2π 1.05457266 × 10−34 J s
Boltzmann constant k 1.380658 × 10−23 J K−1
Elementary charge e 1.60217733 × 10−19 CAvogadro number N 6.0221367 × 1023
particles/molSpeed of light c 2.99792458 × 108 ms−1
1H gyromagnetic ratio 2.67515255 × 108 T s−1
Atomic mass unit amu 1.66057 × 10−27 kgGas constant R 8.31451 J mol−1 K−1
Faraday constant F 96485.3 C mol−1
Proteins: Structure and Function by David Whitford 2005 John Wiley & Sons, Ltd

492 APPENDICES
Appendix 4Derivation of the Henderson–Hasselbalch equation concerning thedissociation of weak acids and bases
The Henderson–Hasselbalch equation reflects thelogarithmic transformation of the expression for thedissociation of a weak acid or base.
HA ↔ A− + H+
K = [H+] [A−]/[HA]
Rearranging this equation leads to
[H+] = K [HA]/[A−]
and by taking negative logarithms this leads to
− log [H+] = − log K − log [HA]/[A−]
Since pH = − log [H+] and we can define pK as− log K this leads to the following equation
pH = pK − log [HA]/[A−]
that is related to the Henderson–Hasselbalch equationby a simple changing of signs
pH = pK + log [A−]/[HA]
Expressed more generally and using the BronstedLowry definition of an acid as a proton donor anda base as a proton acceptor this equation can bere-written as
pH = pK + log [proton acceptor]/[proton donor]
The Henderson–Hasselbalch equation is fundamentalto the application of acid–base equilibria in proteinsor any other biological system. It is used to calculatethe pH formed by mixing known concentrations of
proton acceptor and donor whose pKa’s are known.Alternatively this equation can be used to calculate themolar ratio of donor and acceptor given the pH andpK , or to calculate the pK at a particular pH giventhe concentrations relative or absolute of proton donorand acceptor.
Appendix 5Easily accessible moleculargraphic software
1. Koradi, R., Billeter, M., and Wuthrich, K.(MOLMOL: a superlative program for display andanalysis of macromolecular structures. (obtainablefrom http://www.mol.biol.ethz.ch/groups/Wuthrichgroups/software/) J. Mol. Graphics 1996,14, 51–55.
2. Roger Sayle developed Rasmol although no formalcitation exists. Rasmol is a very suitable introduc-tion to molecular visualization software. An adap-tation of Rasmol for use in web browsers calledChime is available from http://www.mdli.com orhttp://www.mdli.co.uk.
3. Kraulis, P.J. MOLSCRIPT: A Program to produceboth Detailed and Schematic Plots of ProteinStructures. J. Appl. Crystallogr. 1991, 24, 946–950.
4. Molecular graphic software such as VMD pro-duced by the Theoretical Biophysics group, an NIHResource for Macromolecular Modeling and Bioin-formatics, at the Beckman Institute, University ofIllinois at Urbana-Champaign.
5. Guex, N. and Peitsch, M.C. SWISS-MODEL andthe Swiss-PdbViewer: An environment for compar-ative protein modeling Electrophoresis 1997, 18,2714–2723. Official site for software http://www.expasy.ch/spdbv.
A wide range of commercial software has also beenproduced.

APPENDICES 493
Appendix 6 Enzyme nomenclature
Enzyme classes and subclasses
EC 1 OxidoreductasesEC 1.1 Acting on the CH-OH group of donorsEC 1.2 Acting on the aldehyde or oxo group of
donorsEC 1.3 Acting on the CH-CH group of donorsEC 1.4 Acting on the CH-NH2 group of donorsEC 1.5 Acting on the CH-NH group of donorsEC 1.6 Acting on NADH or NADPHEC 1.7 Acting on other nitrogenous compounds as
donorsEC 1.8 Acting on a sulfur group of donorsEC 1.9 Acting on a heme group of donorsEC 1.10 Acting on diphenols and related
substances as donorsEC 1.11 Acting on a peroxide as acceptorEC 1.12 Acting on hydrogen as donorEC 1.13 Acting on single donors with incorporation
of molecular oxygen (oxygenases)EC 1.14 Acting on paired donors, with incorpo-
ration or reduction of molecular oxygenEC 1.15 Acting on superoxide radicals as acceptorEC 1.16 Oxidising metal ionsEC 1.17 Acting on CH2 groupsEC 1.18 Acting on reduced ferredoxin as donorEC 1.19 Acting on reduced flavodoxin as donorEC 1.97 Other oxidoreductases
EC 2 TransferasesEC 2.1 Transferring one-carbon groupsEC 2.2 Transferring aldehyde or ketonic groupsEC 2.3 AcyltransferasesEC 2.4 GlycosyltransferasesEC 2.5 Transferring alkyl or aryl groups, other
than methyl groupsEC 2.6 Transferring nitrogenous groupsEC 2.7 Transferring phosphorus-containing groups
EC 2.8 Transferring sulfur-containing groupsEC 2.9 Transferring selenium-containing groups
EC 3 HydrolasesEC 3.1 Acting on ester bondsEC 3.2 GlycosylasesEC 3.3 Acting on ether bondsEC 3.4 Acting on peptide bonds (peptidases)EC 3.5 Acting on carbon-nitrogen bonds, other
than peptide bondsEC 3.6 Acting on acid anhydridesEC 3.7 Acting on carbon-carbon bondsEC 3.8 Acting on halide bondsEC 3.9 Acting on phosphorus-nitrogen bondsEC 3.10 Acting on sulfur-nitrogen bondsEC 3.11 Acting on carbon-phosphorus bondsEC 3.12 Acting on sulfur-sulfur bonds
EC 4 LyasesEC 4.1 Carbon–carbon lyasesEC 4.2 Carbon–oxygen lyasesEC 4.3 Carbon–nitrogen lyasesEC 4.4 Carbon–sulfur lyasesEC 4.5 Carbon–halide lyasesEC 4.6 Phosphorus–oxygen lyasesEC 4.99 Other lyases
EC 5 IsomerasesEC 5.1 Racemases and epimerasesEC 5.2 cis-trans-isomerasesEC 5.3 Intramolecular isomerasesEC 5.4 Intramolecular transferases (mutases)EC 5.5 Intramolecular lyasesEC 5.99 Other isomerases
EC 6 LigasesEC 6.1 Forming carbon–oxygen bondsEC 6.2 Forming carbon–sulfur bondsEC 6.3 Forming carbon–nitrogen bondsEC 6.4 Forming carbon–carbon bondsEC 6.5 Forming phosphoric ester bonds


Bibliography
General reading
Alberts B., Bray, D., Lewis, J., Raff, M., Roberts, K. &Watson J.D. Molecular Biology of the Cell , 3rd edn.Garland Publishing, New York, 1994.
Barrett, G. C. Chemistry and Biochemistry of Amino Acids.Chapman & Hall, London, 1985.
Branden, C. & Tooze, J. Introduction to Protein Structure.Garland Publishing, New York, 1991.
Cornish-Bowden, A. Fundamentals of Enzyme Kinetics. But-terworths, Oxford, 1979.
Creighton, T. E. Proteins Structures and Molecular Proper-ties, 2nd edn. W.H. Freeman, London, 1993.
Darnell, J., Lodish, H. & Baltimore, D. Molecular CellBiology, 2nd edn. Scientific American Books, New York,1990.
Fersht, A. Enzyme Structure and Mechanism, 2nd edn. W.H.Freeman, New York, 1985.
Frausto da Silva, J. J. R. & Williams, R. J. P. The BiologicalChemistry of the Elements. Oxford University Press,Oxford, 1991.
Gutfreund, H. Enzymes: Physical Principles. Wiley-Interscience, New York, 1972
Lehninger, A., Nelson, D. L. & Cox, M. M. (eds) Principlesof Biochemistry, 3rd edn. Worth Publishers, New York,2000.
Lippard, S. J. & Berg, J. M. Principles of BioinorganicChemistry. University Science Books, 1994.
Voet, D. Voet, J. G. & Pratt, C. W. Fundamentals of Bio-chemistry. John Wiley & Sons, Chichester, 1999.
Watson, J. D. et al. Molecular Biology of the Gene. BenjaminCummings, New York, 1988.
Chapter 1
Ingram, V. A case of sickle-cell anemia. Biochem. Biophys.Acta 1989, 1000, 147–150.
Kendrew, J. The Thread of Life. G. Bell, 1966.Mirsky, A. E. The discovery of DNA. Sci. Am. 1968, 218,
78–88.Olby, R. E. The Path to the Double Helix: the Discovery of
DNA. Dover Publications, 1995.Rutherford, N. J. A Documentary History of Biochemistry
1770–1940. Fairleigh Dickinson University Press, 1992.Schrodinger, E. What is Life? Cambridge University Press,
Cambridge, 1944.
Chapter 2
Barrett, G.C. (ed) Chemistry and Biochemistry of aminoacids. Chapman & Hall, New York, 1985.
Barrett, G.C. & Elmore, D.T. Amino Acids and Peptides.Cambridge University Press, 1998.
Creighton, T.E. Proteins: Structure and molecular properties,2nd edn. Chapters 1–7. W.H. Freeman, New York, 1993.
Creighton, T.E. (ed) Protein Function: A practical approach.IRL Press, Oxford, 1989.
Hirs, C.H.W. & Timasheff, S.N. (eds) Enzyme Structurepart 1. Methods Enzymology , 91. Academic Press, 1983.
Jones, J. Amino Acids and Peptide Synthesis (Oxford Chem-istry Primers). Oxford University Press, 2002.
Lamzin, V.S., Dauter, Z. & Wilson, K.S. How nature dealswith stereoisomers. Curr. Opin. Struct. Biol. 1995, 5,830–836.
Means, G.E. & Feeney, R.E. Chemical Modification of Pro-teins. Holden-Day, 1973.
Chapter 3
Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G.,Bhat, T.N., Weissig, H., Shindyalov, I.N. & Bourne, P.E.The Protein Data Bank. Nucl. Acids Res. 2000, 28,235–242.
Proteins: Structure and Function by David Whitford 2005 John Wiley & Sons, Ltd

496 BIBLIOGRAPHY
Davies, D.R., Padlan, E.A. & Sheriff, S. Antibody-antigencomplexes. Annu. Rev. Biochem. 1990, 59, 439–473.
Doolittle, R.F. Proteins. Sci.Am. 1985, 253, 88–96.Goodsell, D.S. & Olson, A.J. Soluble proteins: Size, shape
and function. Trends Biochem. Sci. 1993, 18, 65–68.Kuby, J. Immunology. W.H. Freeman, London, 1997.Lesk, A.M. Introduction to protein architecture. Oxford
University Press, 2001.Perutz, M.F. Hemoglobin structure and respiratory transport .
Sci. Am. 1978, 239, 92–125.Richardson, J.S. The anatomy and taxonomy of protein
structure. Adv. Prot. Chem. 1981, 34, 167–339.Trabi, M., & Craik, D.J. Circular proteins – no end in sight.
Trends Biochem. Sci. 2002, 27, 132–138.
Chapter 4
Baum, J. & Brodsky, B., Folding of peptide models ofcollagen and misfolding in disease. Curr. Opin. Struct.Biol. 1999, 9, 122–128.
Downing, A. K., Knott, V., Werner, J. M., Cardy, C. M.,Campbell, I. D., & Handford, P. A. Solution structure ofa pair of Ca2+ binding epidermal growth factor-likedomains: implications for the Marfan syndrome and othergenetic disorders. Cell 1996, 85, 597–605.
Glover, J. N., Harrison, S. C. Crystal structure of the het-erodimeric bZIP transcription factor c-Fos c-Jun bound toDNA. Nature 1995, 373, 257.
Handford, P. A. Fibrillin-1, a calcium binding protein ofextracellular matrix. Biochim. Biophys. Acta 2000, 1498,84–90.
Kaplan, D., Adams, W. W., Farmer, B. & Viney, C. SilkPolymers. American Chemical Society, 1994.
Lupas, A. Coiled coils new structures and new functions.Trends Biochem. Sci. 1996, 21 375–382.
Martin, G. R. Timple, R., Muller, P. K. & Kuhn, K. Thegenetically distinct collagens. Trends Biochem. Sci. 1985,10, 285–287.
O’Shea, E. K. Rutkowski, R. & Kim, P. S. Evidence thatthe leucine zipper is a coiled coil. Science 1989, 243,538–542.
Parry, D. A. D. The molecular and fibrillar structure ofcollagen and its relationship to the mechanical propertiesof connective tissue. Biophys. Chem. 1988, 29, 195–209.
Porter, R. M. & Lane E. B. Phenotypes, genotypes and theircontribution to understanding keratin function. TrendsGenet. 2003, 19, 278–285.
Prockop, D. J. & Kivirikko, K. I. Collagens – molecular-biology, diseases, and potentials for therapy. Annu. Rev.Biochem. 1995, 64, 403–434.
Royce, P.M. & Steinmann, B. (eds) Connective Tissue andIts Heritable Disorders: Molecular, Genetic, and MedicalAspects . John Wiley & Sons, 2002.
Van der Rest, M. & Bruckner, P. Collagens: Diversity at themolecular and supramolecular levels. Curr. Opin. Struct.Biol. 1993, 3, 430–436.
Chapter 5
Benz R. (ed) Bacterial and Eukaryotic Porins: Structure,Function, Mechanism . John Wiley & Sons, 2004.
Blankenship, R.E. Molecular Mechanism of Photosynthesis .Blackwell Science, 2001.
Capaldi, R.A & Aggeler, R. Mechanism of the F 1F0-typeATP synthase, a biological rotary motor . Trends Biochem.Sci. 2002, 27, 154–160.
Gennis, R.B. Biomembranes . Springer Verlag, New York,1989.
Hamm, H.E. The many faces of G protein signaling. J. Biol.Chem. 1998, 273, 669–672.
Lehninger, A. Principles of Biochemistry . 3rd edn. (Nel-son, D.L. and Cox, M.M., eds.) Oxidative Phosphorylationand Photophosphorylation, pp. 659–690. Worth Publishers
Nicholls, D.G. & Ferguson, S.J. Bioenergetics 2 , AcademicPress, 1992.
Scheffler, I.E. Mitochondria . John Wiley & Sons Inc, 1999.Vance, D.E. & Vance, J.E. Biochemistry of lipids, lipopro-
teins and membranes . 4th edn. Elsevier, 2002.Torres, J., Stevens, T.J. & Samso, M. Membrane proteins:
the ‘Wild West’ of structural biology . Trends Biochem. Sci.2003, 28, 137–144.
Von Jagow, G., Schaegger, H., & Hunte, C. (eds) Mem-brane Protein Purification and Crystallization: A PracticalGuide. Academic Press, 2003.
Chapter 6
Baxevanis, A.D. & Ouellette, B.F.(eds) Bioinformatics: Apractical guide to the analysis of genes and proteins . JohnWiley & Sons Inc, 2004.
Findlay, J.B.C. & Geisow, M.J. (eds) Protein Sequencing. Apractical approach . IRL press 1989.
Margulis, L & Sagan, C. What is Life. Simon & Schuster,New York, 1995.
Miller, S.J. & Orgel, L.E. The Origins of Life. Prentice-Hall,New Jersey, 1975.
Mount, D.W. Bioinformatics: Sequence and Genome Analy-sis . Cold Spring Harbor Laboratory Press, 2001.
Orengo, C.A., Thornton, J.M & Jones, D.T. Bioinformatics(Advanced Texts Series). BIOS Scientific Publishers, 2002.

BIBLIOGRAPHY 497
Primrose, S.B. Principles of Genome Analysis: A Guide toMapping and Sequencing DNA from Different Organisms .Blackwell Science, 1998.
Sanger, F. Sequences, sequences, sequences . Annu. Rev.Biochem. 1988, 57, 1–28.
Volkenstein, M.V. Physical Approaches to Biological Evolu-tion . Springer Verlag, Berlin, 1994.
Webster, D.M. Protein Structure Prediction: Methods andProtocols . Humana Press, 2000.
Chapter 7
Bairoch, A. The enzyme databank. Nucl. Acids. Res. 1994,22, 3626–3627.
Fersht, A.R. Structure and Mechanism in Protein Science:Guide to Enzyme Catalysis and Protein Folding . W.H.Freeman and Company, New York, 1999.
Gutfreund, H. Kinetics for the Life Sciences: Receptors,Transmitters and Catalysts. Cambridge University Press,Cambridge, 1995.
Kovall, R.A. & Matthews, B.W. Type II restriction endonu-cleases: structural, functional and evolutionary relation-ships. Curr. Opin. Chem. Biol. 1999, 3, 578–583.
Kraut, J. How do enzymes work? Science 1988, 242,533–540.
Martins, L.M. & Earnshaw, W.C. Apoptosis: Alive andkicking in 1997. Trends Cell Biol. 1997, 7, 111–114.
Moore, J.W. & Pearson, R.G. Kinetics and Mechanism . JohnWiley & Sons, Chichester, 1981.
Vaux, D.L. & Strasser, A. The molecular biology of apopto-sis. Proc. Natl. Acad. Sci. USA 1996, 93, 2239–2244.
Wold, F. & Moldave, K. Posttranslational modifications.Methods Enzymol. 106 and 107. Academic Press, 1985.
Chapter 8
Dodson, G. & Wlodawer, A. Catalytic triads and theirrelatives. Trends Biochem. Sci. 1998, 23, 347–352.
Kornberg, A., & Baker. T.A. DNA Replication , 2d ed. W. H.Freeman, San Francisco, 1992.
Murray A.W., & Hunt T, eds. The Cell Cycle: An Introduc-tion . Oxford University Press. 1993.
Norbury C, & Nurse P. Animal Cell Cycles and TheirControl. Annu. Rev. Biochem. 1992, 61, 441–470.
Spirin, A.S. Ribosomes. Kluwer Academic, New York, 1999.Stein G. S, (ed.) The Molecular Basis of Cell Cycle and
Growth Control . John Wiley & Sons Inc, New York, 1999.Stillman, B. (ed.) The Ribosome. Cold Spring Harbor Symp.
66. Cold Spring Harbor Laboratory Press, 2001.
Trends Biochem. Sci. 1997, 22, 371–410. Issue devoted toproteasome and proteolytic processes.
Watson, J.D. The Double Helix: Personal Account of theDiscovery of the Structure of DNA. Penguin 1999.
Zwickl, P. & Wolfgang Baumeister, W. The Proteasome-Ubiquitin Protein Degradation Pathway. Springer Verlag,Berlin, 2002.
Chapter 9
Brown, T.A. Gene Cloning and DNA Analysis: An Introduc-tion. Blackwell, 4th edn, 2001.
Deutscher, M.P., Colowick, S.P. & Simon, M.I. (eds). Guideto protein purification. Methods Enzymol. 182. AcademicPress, London, 1990.
Dunn, B.M. Speicher, D.W., Wingfield, P.T. & John E. Col-igan, J.E. (eds) Short Protocols in Protein Science. JohnWiley & Sons, 2003.
Freifelder, D. Physical Biochemistry: Applications to Bio-chemistry and Molecular Biology. W.H.Freeman,1982.
Hames, B.D. & Rickwood. (eds) Gel Electrophoresis ofProteins, A practical approach, 2nd edn. IRL press, 1990.
Jansen, J-C. & Ryden, L. (eds) Protein Purification: Prin-ciples, High Resolution Methods and Applications. JohnWiley & Sons Inc, 1998.
Meyer, V.R. Practical High-Performance Liquid Chromatog-raphy, 4th edn. John Wiley & Sons, 2004.
Scopes, R. Protein purification: Principles and Practice.Springer-Verlag, Berlin, 1993.
Tanford, C. The hydrophobic effect; formation of micellesand biological membranes, 2nd edn. John Wiley & Sons,Chichester, 1980.
Voet, D., Voet, J.G. & Pratt, C.W. Fundamentals of Biochem-istry, John Wiley & Sons, Ltd, Chichester, 1999.
Chapter 10
Campbell, I.D. & Dwek, R. Biological Spectroscopy . Ben-jamin Cummings, New York, 1984.
Cavanagh, J., Fairbrother, W.J., Palmer III, A.G. & Skel-ton, N.J. Protein NMR spectroscopy: Principles and Prac-tice. Academic Press, 1996.
Chang, R. Chemistry . 8th edn. McGraw-Hill Education,2004.
Drenth, J. Principles of Protein X-ray Crystallography .Springer-Verlag, New York, 1994
Fasman, G.D. (ed) Circular Dichroism and the Conforma-tional Analysis of Biomolecules. Plenum Publishers, 1996.
Ferry, G. Dorothy Hodgkin: A life. Granta Books, 1999.

498 BIBLIOGRAPHY
Harrison, S.C. Whither structural biology? Nat. Struct. Mol.Biol. 2004, 11, 12–15.
Nat. Struct. Biol. 1997, 4, 841–865 and Nat. Struct. Biol.1998, 5, 492–522. (Series of short NMR reviews)
Rhodes, G. Crystallography made crystal clear . AcademicPress 2nd edn, San Diego, 2000.
Wider, G. & Wuthrich, K. NMR spectroscopy of largemolecules and multimolecular assemblies in solution.Curr. Op. Struct. Biol. 1999, 9, 594–601.
Chapter 11
Caughey, B. (ed) Prion Proteins. Adv. Protein Chem. 57.Academic Press, 2001
Creighton, T.E. Protein Folding. W.H.Freeman, New York,1992.
Daggett, V. & Fersht, A.R. Is there a unifying mechanism forprotein folding? Trends Biochem. Sci. 2003, 28, 18–25.
Horwich, A.L. (ed) Protein folding in the cell. Adv. ProteinChem. 59. Academic Press, 2001.
Dobson, C.M. Protein Misfolding, Evolution and DiseaseTrends Biochem. Sci. 1999, 24, 329–332.
Ladbury, J.E & Chowdhry, B.Z. (eds) Biocalorimetry: Appli-cations of Calorimetry in the Biological Sciences . JohnWiley & Sons, Chichester, 1998.
Matthews, B.W. Structural and genetic analysis of proteinstability. Annu. Rev. Biochem. 1993, 62, 139–160.
Matthews, C.R. (eds) Protein Folding Mechanisms. Adv.Protein Chem. 2000, 53.
Pain, R. (ed) Mechanisms of Protein Folding (Frontiers inMolecular Biology Series). Oxford University Press, 2000.
Saibil, H.R. & Ranson, N.A. The chaperonin foldingmachine. Trends Biochem. Sci. 2002, 27, 627–632.
Chapter 12
Crystal, R. G. The α1-antitrypsin gene and its deficiencystates. Trends Genet. 1989, 5, 411–417.
Culotta, E. & Koshland, D. E., Jr. p53 sweeps through cancerresearch. Science 1993, 262, 1958–1959.
Fauci, A. S. HIV and AIDS: 20 years of science. NatureMedicine 2003, 9, 839–843. (and subsequent articles)
Lane, D. & Lain, S. Therapeutic exploitation of the p53pathway. Trends Molec. Med. 2002, 8, S38–S42.
Gouras, G.K. Current theories for the molecular and cellu-lar pathogenesis of Alzheimer’s disease. Expert Rev. Mol.Med. 2001. http://www.expertreviews.org/01003167h.htm
Perutz, M.F. Protein Structure: New Approaches to Diseaseand Therapy . W.H. Freeman, 1992.
Rowland-Jones, S.L. AIDS pathogenesis: what have twodecades of HIV research taught us? Nature Rev. Immunol.2003, 3, 343–348.
Prusiner, S. Prion diseases and the BSE crisis. Science 1997,278, 245–251.
Varmus, H. Retroviruses. Science 1988, 240, 1427–1435.Wiley, D.C. & Skehel, J.J. The Structure and Function of
the Haemagglutinin Membrane Glycoprotein of InfluenzaVirus. Annu. Rev. Biochem. 1987, 56, 365–394.

References
Abrahams, J. P., Leslie, A. G. W., Lutter, R. & Walker,J. E. Structure at 2.8 A resolution of F1-ATPase frombovine heart mitochondria. Nature 1994, 370, 621–628.
Abramson, J., Svensson-Ek, M., Byrne, B. & Iwata, S.Structure of cytochrome c oxidase: a comparison of thebacterial and mitochondrial enzymes Biochim . Biophys.Acta 2001, 1544, 1–9.
Agarraberes, F. A., & Dice, J. F. Protein translocation acrossmembranes. Biochim. Biophys. Acta 2001, 1513, 1–24.
Agarwal, M. L., Taylor, W. R., Chernov, M. V., Cher-nova, O. B. & Stark, G. R. The p53 network. J. Biol.Chem. 1998, 273, 1–4.
Aguzzi, A., Glatzel, M., Montrasio, F., Prinz, M. & Hepp-ner, F. L. Interventional strategies against prion diseases.Nature Rev. Neurosciences 2001, 2, 745–749.
Aguzzi, A., Montrasio, F., & Kaeser, P. S. Prions: Healthscare and biological challenge. Nature Reviews MolecularCell Biology 2002, 2, 118–126.
Albright, R. A., & Matthews, B. W. How Cro and λ
repressor Distinguish Between Operators: The StructuralBasis Underlying a Genetic Switch. Proc. Natl. Acad. SciUSA 1996, 95, 3431–3436.
Allen, T. D., Cronshaw, J. M., Bagley, S. Kiseleva, E. &Goldberg, M. W. The nuclear pore complex: mediator oftranslocation between nucleus and cytoplasm. J. Cell Sci.2000, 113, 1651–1659.
Als-Nielsen, J. & McMorrow, D. Elements of Modern X-rayPhysics. John Wiley & Sons, Chichester, 2001.
Altman, S. & Kirsebom, L. Ribonuclease P. in Geste-land, R. F., Cech, T. R. and Atkins, J. F. (eds), The RNAWorld. Cold Spring Harbor Laboratory Press, Cold SpringHarbor, New York 1999.
Altschul, S. F. Amino acid substitution matrices from aninformation theoretic perspective. J. Mol. Biol. 1991, 219,555–665.
Amit, A. G., Mariuzza, R. A., Phillips, S. E. V. & Pol-jak, R. J. Three dimensional structure of an antigen-antibody complex at 2.8A resolution. Science 1986, 233,747–750.
Andrade, C., A peculiar form of peripheral neuropathy:familial atypical generalised amyloidosis with specialinvolvement of peripheral nerves. Brain 1952, 75,408–427.
Andrews, D. W. & Johnson, A. E. The translocon: more thana hole in the ER membrane? Trends Biochem. Sci. 1996,21, 365–369.
Anfinsen, C. B. Principles that govern the folding of proteinchains. Science 1973, 181, 223–230.
Arakawa, T & Timasheff, S. N. Theory of protein solubility.Methods Enzymol. 1985, 114, 49–77, Academic Press.
Babcock, G. & Wikstrom, M. Oxygen activation and theconservation of energy in cellular respiration. Nature 1992,356, 301–309.
Baldwin, R. L. & Rose. G. D, Is protein folding Hierarchic?I. Local structure and peptide folding. Trends Biochem. Sci.1999 24, 26–33 II. Folding Intermediates and transitionstates. Trends Biochem. Sci. 1999 24, 77–83.
Baltimore. D. Our genome unveiled. Nature 2001, 409,814–816.
Ban, N. Nissen, P., Hansen, J., Moore, P. B. & Steitz,T. A. The complete atomic structure of the largeribosomal subunit at 2.4 A resolution. Science 2000, 289,905–920.
Barber, M & Quinn, G. B. High-Level Expression inEscherichia coli of the Soluble, Catalytic Domain of RatHepatic Cytochrome b5 Reductase. Protein expression andpurification 1996, 8, 41–47.
Bardwell, J. C. A. & Beckwith, J. The bonds that tie:catalyzed disulfide bond formation Cell 1993, 74,769–771.
Batey, R. T., Sagar M. B., & Doudna J. A. Structural andenergetic analysis of RNA recognition by a universally
Proteins: Structure and Function by David Whitford 2005 John Wiley & Sons, Ltd

500 REFERENCES
conserved protein from the signal recognition particle. J.Mol. Biol. 2001, 307, 229–246.
Baum, J. & Brodsky, B. Folding of peptide models ofcollagen and misfolding in disease. Curr. Opin. Struct.Biol. 1999, 9, 122–128.
Baumeister, W. & Steven, A. C. Macromolecular electronmicroscopy in the era of structural genomics. TrendsBiochem. Sci. 2000, 25, 625–631.
Bax, A. Multi-dimensional nuclear magnetic resonancemethods for protein studies. Curr. Opin. Struct. Biol. 1994,4, 738–744.
Bergfors, T. Protein Crystallization Techniques, Strategies,and Tips. A Laboratory Manual 1999.
Berry, E. A., Guergova-Kuras, M., Huang, L. -S. &Antony R. Crofts, A. R. Structure and function ofcytochrome bc complexes Annu. Rev. Biochem. 2000, 69,1005–1075.
Billeter, M., Kline, A. D., Braun, W., Huber, R., & Wuth-rich, K. Comparison of the High-Resolution Structuresof the α-Amylase Inhibitor Tendamistat Determined byNuclear Magnetic Resonance in Solution and by X-rayDiffraction in Single Crystals. J. Mol. Biol. 1989, 206,677–687.
Blobel, G. & Dobberstein, B. Transfer of proteins acrossmembranes. I. Presence of proteolytically processed andunprocessed nascent immunoglobulin light chains onmembrane-bound ribosomes of murine myeloma, J. CellBiol. 1975, 67, 835–851.
Bockaert, J., & Pin, J. P. Molecular tinkering of G protein-coupled receptors: an evolutionary success. EMBO J. 1999,18, 1723–1729.
Bokman S. H., Ward W. W. Renaturation of Aequorea greenfluorescent protein. Biochem. Biophys. Res. Commun.1981, 101, 1372–1380.
Bolton, W. & Perutz. M. F. Three dimensional Fouriersynthesis of horse deoxyhaemo-globin at 2.8 A resolution.Nature 1970, 228, 551–552.
Booth, P., Templer, R. H., Curran, A. R. & Allen,S. J. Can we identify the forces that drive the folding ofintegral membrane proteins? Biochem Soc.Trans. 2001, 29,408–413.
Boriack-Sjodin, P. A., Zeitlin, S., Chen, H. -H., Cren-shaw, L., Gross, S., Dantanarayana, A., Delgado, P.,May, J. A., Dean, T. & Christianson, D. W. Structuralanalysis of inhibitor binding to human carbonic anhydraseII. Protein Science 1998, 7, 2483–2489.
Bowie, J. U., Clarke, N. D., Pabo, C. O. & Sauer,R. T. Deciphering the message in protein sequence: tol-erance to amino acid substitutions. Science 1990, 247,1306–1310.
Boyer, P. D. The binding change mechanism for ATPsynthase – Some probabilities and possibilities. Biochim.Biophys. Acta 1993, 1140, 215–250.
Boyer, P. D., The ATP synthase – a splendid molecularmachine, Annu. Rev. Biochem. 1997, 66, 717–749.
Braig, K., Menz R. I., Montgomery M. G., LeslieA. G., & Walker J. E. Structure of bovine mitochondrialF1-ATPase inhibited by Mg2+ ADP and aluminium fluo-ride. Structure 2000, 8, 567–573.
Braig, K., Otwinowski, Z., Hegde, R., Boisvert, D. C.,Joachimiak, A., Horwich, A. L & Sigler, P. B. The crys-tal structure of the bacterial chaperonin GroEL at 2.8 A.Nature 1994, 371, 578–586.
Brandts, J. F., Halvorson, H. R., & Brennan, M. Considera-tion of the possibility that the slow step in protein denat-uration reactions is due to cis-trans isomerism of prolineresidues. Biochemistry 1975, 14, 4953–4963.
Brandts, U. Bifurcated ubihydroquinone oxidation in thecytochrome bc 1 complex by protongated charge transfer.FEBS Lett. 1996, 387, 1–6.
Brejc, K., Sixma, T. K., Kitts, P. A., Kain, S. R., Tsien,R. Y., Ormo, M., Remington, S. J. Structural basis fordual excitation and photoisomerization of the Aequoreavictoria green fluorescent protein. Proc. Natl. Acad. Sci US A 1997, 94, 2306–2311.
Brenner, S., Jacob, F. & Meselson, M. An unstableintermediate carrying information from genes to ribosomesfor protein synthesis. Nature 1961, 190, 576–581.
Brodersen, D. E., Carter, A. P., Clemons Jr, W. M.,Morgan-Warren, R. J., Murphy IV, F. V. Ogle, J. M.,Tarry, M. J. Wimberley, B. T. & Ramakrishnan, V.Atomic Structures of the 30S Subunit and Its Complexeswith Ligands and Antibiotics. Cold Spring Harbor Symp.2001, 66, 17–32.
Browner, M. F. & Fletterick, R. J. Phosphorylase: a Biolog-ical Transducer. Trends Biochem. Sci. 1992, 17, 66–71.
Burley, S. K. The TATA box binding protein. Curr. Opin.Struct. Biol. 1996, 6, 69–75.
Butler, P. J. G., Klug, A. The assembly of a virus. Sci. Amer.Nov. 1978
Byrne, B. & Iwata, S. Membrane protein complexes. Curr.Opin. Struct. Biol. 2002, 12, 239–243.
Cahn, R. S., Ingold, C. K. & Prelog, V. Specification ofMolecular Chirality. Angew. Chem. 1966, 78, 413–447.
Cammack, R. & Cooper, C. E. Electron paramagneticresonance spectroscopy of iron complexes and iron-containing proteins. Methods Enzymol. 1993, 22, 353–384,Academic Press.
Campbell, I. D. & Dwek, R. Biological Spectroscopy.Benjamin Cummings, New York, 1984.
Capaldi, R. A. Structure and function of cytochrome oxidase.Annu. Rev. Biochem. 1990, 59, 569–96.

REFERENCES 501
Carter, C. W. Cognition, Mechanism, and EvolutionaryRelationships in Aminoacyl-tRNA Synthetases. Ann. Rev.Biochem. 1993, 62, 717–748.
Cavanagh, J., Fairbrother, W. J., Palmer III, A. G. &Skelton, N. J. Protein NMR spectroscopy: Principles andPractice. Academic Press, 1996.
Chaddock, J. A., Herbert, M. H., Ling, R. J., Alexander,F. C. G., Fooks, S. J., Revell, D. F., Quinn, C. P.,Shone, C. C. & Foster, K. A. Expression and purificationof catalytically active, non-toxic endopeptidase derivativesof Clostridium botulinum toxin type A. Protein Expressionand Purification 2002, 25, 219–228.
Chalfie, M., Tu, Y., Euskirchen, G., Ward W. W., PrasherD. C. Green fluorescent protein as a marker for gene
expression. Science 1994, 263, 802–805.Cheetham, G. M., Jeruzalmi, D. & Steitz, T. A. Structural
basis for initiation of transcription from an RNApolymerase- promoter complex Nature 1999, 399, 80–84.
Chen, S., Roseman, A. M., Hunter, A. S., Wood,S. P., Burston, S. G., Ranson, N. A., Clarke, A. R. &Helen R. Saibil, H. R. Location of a folding protein andshape changes in GroEL–GroES complexes imaged bycryo-electron microscopy Nature 1994, 371, 261–264.
Chevet, E., Cameron, P. H., Pelletier, M. F., ThomasD. Y. & Bergeron, J. J. M. The endoplasmic reticulum:integration of protein folding, quality control, signal-ing and degradation. Curr. Opin. Struct. Biol. 2001, 11,120–124.
Cho, Y., Gorina, S., Jeffrey, P. D., Pavletich, N. P. Crystalstructure of a p53 tumor suppressor-DNA complex:understanding tumorigenic mutations. Science 1994, 265,346–355.
Chothia, C & Finkelstein, A. V. The classification andorigins of protein folding patterns. Annu. Rev. Biochem.59, 1007–1039, 1990.
Chou, P. Y. & Fasman, G. D. Empirical Predictions ofProtein Conformation. Annu. Rev.Biochem. 1978, 47,251–276.
Cingolani, G., Petosa, C., Weis, K., & Muller, C. W.Structure of importin-β bound to the IBB domain ofimportin-α. Nature 1999, 399, 221–229.
Cohen, C. & Parry, D. A. D. α helical coiled coils andbundles: how to design an α helical protein. Prot. Struct.Funct. Genet. 1990, 7, 1–15.
Cohen, F. E. & Prusiner, S. B. Pathologic conformations ofprion proteins. Annu. Rev. Biochem. 1998, 67, 793–819.
Collinge, J. Human prion diseases and bovine spongiformencephalopathy (BSE). Hum. Mol. Genet. 1997, 6,1699–1705.
Coux, O., Tanaka, K. & Goldberg, A. L. Structure andFunctions of the 20S and 26S Proteasomes. Annu. Rev.Biochem. 1996, 65, 801–847.
Cowan, S. W., Schirmer, T., Rummel, G., Steiert, M.,Ghosh, R., Pauptit, R. A., Jansonius N. J. & Rosen-busch, J. P. Crystal structures explain functional propertiesof two E. coli porins. Nature 1992, 358, 727–733.
Crick, F. H. C. The packing of α helices: simple coiled coils.Acta Cryst. 1953, 6, 689–697.
Crick, F. H. C., Barnett, L., Brenner, S. & Watts-Tobin,R. J. General nature of the genetic code for proteins.
Nature 1961, 192, 1227–1232.Crystal, R. G. The α1-antitrypsin gene and its deficiency
states. Trends Genet. 1989, 5, 411–417.Cserzo, M., Wallin, E., Simon, I., von Heijne, G. &
Elofsson, A. Prediction of transmembrane α helices inprokaryotic membrane proteins: the Dense AlignmentSurface method. Prot. Eng. 1997, 10, 673–676.
Cullen, B. R, Nuclear RNA export pathways. Mol. Cell. Biol.2000, 20, 4181–4187.
Dalbey, R. E, Chen, M. Y., Jiang, F. & Samuelson, J. C.In vivo Assembly of Transporters and other MembraneProteins. Curr. Opin. Cell Biol. 2000, 12, 435–442.
Dalbey, R. E. & Robinson, C. Protein translocation intoand across the bacterial plasma membrane and the plantthylakoid membrane. Trends Biochem. Sci. 1999, 24,17–24.
Danna, K. & Nathans, D. Specific cleavage of Simianvirus 40 DNA restriction endonuclease of Haemophilusinfluenzae. Proc. Natl. Acad. Sci. USA 1971, 68,2913–2917.
Davie, E. W. Introduction to the blood clotting cascade andthe cloning of blood coagulation factors. J. Prot. Chem.1986, 5, 247–253.
Davies, D. R. The structure and function of the asparticproteinases. Ann. Rev. Biophys. Biophys. Chem. 1990, 19,189–215.
Davies, D. R. & Chacko, S. Antibody structure. Acc. Chem.Res. 1993, 26, 421–427.
Dayhoff, M. The origin and evolution of protein superfami-lies. FASEB J. 1976, 35, 2132–2138.
Dayhoff, M. O., R. M. Schwartz and B. C. Orcutt. 1978.A model of evolutionary change in proteins. In Atlasof Protein Sequence and Structure Vol. 5 suppl. 2 (ed.M. O. Dayhoff), 345–352. National Biomedical ResearchFoundation, Washington DC.
DeBondt, H. L., Rosenblatt, J., Jancarik, J., Jones,H. D., Morgan, D. O. & Kim, S. H. Crystal struc-ture of cyclin-dependent kinase 2. Nature 1993, 363,595–602.
Deisenhofer, J., Epp, O., Miki, K., Huber, R. & Michel, H.Structure of the protein subunits in the photosyntheticreaction centre of R. viridis at 3 A resolution. Nature 1985,318, 618–624.

502 REFERENCES
Deisenhofer, J., Epp, O., Miki, K., Huber, R. & Michel, H.X-ray structure analysis of a membrane protein complex.Electron density map at 3 A resolution and a model ofthe chromophores of the photosynthetic reaction centerfrom Rhodopseudomonas viridis. J. Mol. Biol. 1984, 180,385–398.
Deisenhofer, J., Epp, O., Sinning, I. & Michel, H. Crystallo-graphic refinement at 2.3 A resolution and refined modelof the photosynthetic reaction centre from Rhodopseu-domonas viridis. J. Mol. Biol. 1995, 246, 429–457.
DeRose, V. J. & Hoffman, B. M. Protein structure andmechanism studied by electron nuclear double resonancespectroscopy Methods Enzymol. 1995, 246, 554–589Academic Press.
Dill, K. A. & Chan, H. S. From Levinthal to pathways tofunnels. Nature Struct. Biol. 1997, 4, 10–19.
Dill, K. A. Dominant Forces in Protein Folding. Biochem-istry 1990, 29, 7133–7155.
Dinner, A. R. & Karplus. M. The roles of stability andcontact order in determining protein folding rates. NatureStruct. Biol. 2001, 8, 21–22.
Ditzel, L., Lowe, J. Stock, D., Stetter, K. -O., Huber,H., Huber, R. & Steinbacher, S. Crystal structure of thethermosome, the archaeal chaperonin and homolog ofCCT. Cell 1998, 93, 125–138.
Dobson, C. M. Protein Misfolding, Evolution and DiseaseTrends Biochem. Sci. 1999, 24, 329–332.
Dobson, C. M., Evans, P. A., & Radford, S. E. Understand-ing How Proteins Fold: The Lysozyme Story so Far, TrendsBiochem. Sci. 1994, 19, 31–37.
Dodson, G. & Wlodawer, A. Catalytic triads and theirrelatives. Trends Biochem. Sci. 23, 347–352, 1998.
Dohlman et alBiochemistry 1987, 26, 2660–2666.Doolittle, R. F. The multiplicity of domains in proteins.
Annu. Rev. Biochem. 1995, 64, 287–314.Doudna, J. A. & Batey, R. T. Structural insights into the
signal recognition particle. Annu. Rev. Biochem. 2004, 73,539–557.
Downing, A. K., Knott, V., Werner, J. M., Cardy, C. M.,Campbell, I. D., & Handford, P. A., Solution structureof a pair of Ca2+ binding epidermal growth factor-likedomains: implications for the Marfan syndrome and othergenetic disorders. Cell 1996, 85, 597–605.
Drenth, J. Principles of Protein X-ray Crystallography,Springer-Verlag. New York 1994
Dunbrack, Jr. R. L., & Karplus, M. Conformational analysisof the backbone-dependent rotamer preferences of proteinsidechains. Nature Struct. Biol. 1, 334–340, 1994.
Eckert, D. M. & Kim, P. S. Mechanisms of viral membranefusion and its inhibition. Annu. Rev. Biochem. 2001, 70,777–810.
Edman, P. & Begg, G. A protein sequenator. Eur. J.Biochem. 1967, 1, 80–91.
Eisenberg, D. The discovery of the α-helix and β-sheet, theprincipal structural features of proteins. Proc. Natl. Acad.Sci. USA. 2003. 100, 11207–11210.
Ellenberger, T. E., Brandl, C. J., Struhl, K. & Harrison,S. C. The GCN4 basic region leucine zipper binds DNAas a dimer of uninterrupted alpha helices: crystal structureof the protein-DNA complex. Cell 1992, 71, 1223–1237.
Ellis, R. J. Chaperone substrates inside the cell. TrendsBiochem. Sci. 2000, 25, 210–212.
Elrod-Erickson, M., Benson, T. E., Pabo, C. O.: High-resolution structures of variant Zif268-DNA complexes:implications for understanding zinc finger-DNA recogni-tion. Structure 1998, 6, 451–464.
Englander S. W., Mayne, L., Bai, Y. & Sosnick T. R.Hydrogen exchange: the modern legacy of Linderstrom-Lang. Protein Sci. 1997, 6, 1101–1109.
Englander, S. W. & Kallenbach, N. R. Hydrogen exchangeand structural dynamics of proteins and nucleic acids.Quart. Rev. Biophys. 1984, 16, 521–655.
Evans, J. N. S. Biomolecular NMR spectroscopy.Oxford University Press. 1995.
Evans, P. R. Structural aspects of allostery. Curr. Opin.Struct. Biol. 1991, 1, 773–779.
Farquhar, M. G. Progress in Unraveling Pathways of GolgiTraffic. Annu. Rev. Cell Biol. 1985, 1, 447–488.
Ferguson, M. A. J. & Williams, A. F. Cell surface anchoringof proteins via glycosyl-phosphatidylinositol structures.Annu. Rev. Biochem. 1988, 57, 285–320.
Ferre-D’Amare, A. R., Prendergast, G. C., Ziff, E. B., Bur-ley, S. K. Recognition by Max of its cognate DNA througha dimeric b/HLH/Z domain. Nature 1993, 363, 38–45.
Ferrell, K. Wilkinson, C. R. M., Dubiel, W., & Gordon. C.Regulatory subunit interactions of the 26S proteasome, acomplex problem Trends Biochem. Sci. 2000, 25, 83–88.
Fersht, A. R. Protein folding and stability: the pathway offolding of barnase. FEBS Lett. 1993, 325, 5–16.
Fersht, A. R., Knill-Jones, J. W., Bedouelle, H., & Win-ter G. Reconstruction by site-directed mutagenesis of thetransition state for the activation of tyrosine by the tyrosyl-tRNA synthetase: A mobile loop envelopes the transitionstate in an induced-fit mechanism. Biochemistry 1988, 27,1581–1587.
Fersht, A. R., Matouschek, A. & Serrano, L Folding ofan enzyme: Theory of protein engineering of stabilityand pathway of protein folding. J. Mol. Biol. 1992,224, 771–782, 783–804, 805–818, 819–835, 836–846,847–859.
Findlay, J. B. C. & Geisow, M. J. (eds) Protein Sequencing.A practical approach IRL press 1989.

REFERENCES 503
Fischmann, T. O., Bentley, G. A., Bhat, T. N., Boulot,G., Mariuzza, R. A., Phillips, S. E. V., Tello, D., & Pol-jak, R. J. Crystallographic Refinement of the Three-dimensional Structure of the FabD1.3-Lysozyme Com-plex at 2.5-A Resolution. J. Biol. Chem. 1991, 266,12915–12920.
Frankel, A. D. & Young, J. A. T. HIV-1: Fifteen Proteinsand an RNA. Annu. Rev. Biochem. 1998, 67, 1–25.
Gesteland, R. F., Cech, T. R., & Atkins, J. F. The RNAWorld. Cold Spring Harbor Press, New York 1999.
Gether, U. Uncovering Molecular Mechanisms Involved inActivation of G Protein-Coupled Receptors. EndocrineRev. 2000, 21, 90–113.
Gething, M. J. & Sambrook, J. Protein folding in the cell.Nature 1992, 355, 33–45.
Glover, J. N., & Harrison, S. C., Crystal structure of theheterodimeric bZIP transcription factor c-Fos c-Jun boundto DNA. Nature 1995, 373, 257–260.
Gorlich, D. & Rapoport, T. A, Protein translocation intoproteoliposomes reconstituted from purified componentsof the endoplasmic reticulum membrane. Cell 1993, 75615–630.
Gould, K. L. & Nurse, P. Tyrosine phosphorylation of thefission yeast cdc2 + protein kinase regulates entry intomitosis. Nature 1991, 342, 39–45.
Green, A. A., J. Biol. Chem. 95, 47, 1932.Griffiths, W. J., Jonsson, A. P., Liu, S., Rai, D. K. &
Wang, Y. Electrospray and tandem mass spectrometry inBiochemistry. Biochem. J. 2001, 355, 545–561.
Grigorieff, N., Ceska, T. A., Downing, K. H., Baldwin,J. M. & Henderson, R. Electron-crystallographic refine-ment of the structure of bacteriorhodopsin. J. Mol. Biol.1996, 259, 393–421.
Guidotti, G. Membrane proteins. Annu. Rev. Biochem. 1972,41, 731–752.
Guijarro, J. I. Guijarro, I., Sunde, M., Jones, J. A., Camp-bell, I. D. & Dobson, C. M. Amyloid fibril formation byan SH3 domain. Proc. Natl. Acad. Sci. USA. 1998, 95,4224–4228.
Hames, B. D. & Rickwood, D. (Eds.), Gel Electrophoresis ofproteins. A Practical Approach (2nd Ed). 1990 IRL press.
Handford, P. A. Fibrillin-1, a calcium binding protein ofextracellular matrix. Biochim. Biophys. Acta, 2000, 1498,84–90.
Hansen, J. C., Lebowitz, J. & Demeler, B. Analyticalultracentrifugation of complex macromolecular systems.Biochemistry 1994, 33, 13155–13163.
Hartl F. U. & Hayer-Hartl, M. Molecular chaperones in thecytosol: from nascent chain to folded protein. Science2002, 295, 1852–1888.
Hartl. F. U. Molecular chaperones in cellular protein foldingNature 1996, 381, 571–580.
Heijne, von G. Patterns of amino acids near signal cleavagesites. Eur. J. Biochem. 1983, 133, 17–21.
Henderson, R. & Unwin, P. N. T. Three-dimensional modelof purple membrane obtained by electron microscopy.Nature 1975, 257, 28–32.
Henderson, R., Baldwin, J. M., Ceska, T. A., Zemlin, F.,Beckmann, E. & Downing, K. H. Model for the Structureof Bacteriorhodopsin Based on High-Resolution ElectronCryo-microscopy. J. Mol. Biol. 1990, 231, 899–929.
Henikoff, S. & Henikoff, J. G. Amino acid substitutionmatrices from protein blocks. Proc. Natl. Acad. Sci. USA1992, 89, 10915–10919.
Hensley, P. Defining the structure and stability ofmacromolecular assemblies in solution: the re-emergenceof analytical ultracentrifugation as a practical tool.Structure 1996, 4, 367–373. 1996.
Hershko, A., & Ciechanover, A. The ubiquitin system. Annu.Rev. Biochem. 1998, 67, 425–479.
Hill, A. F., Desbruslais, M., Joiner, K., Sidle, C. L., Gow-land, I., Collinge, J., Doey, L. J. & Lantos, P. The sameprion strain cause nvCJD and BSE. Nature 1997, 389,448–450.
Hochstrasser, M. Ubiquitin-dependent protein degradation.Annu. Rev. Genet. 1996, 30, 405–439.
Hofmeister, F. On the understanding of the effects ofsalts. Arch. Exp. Pathol. Pharmakol. (Leipzig) 1888, 24,247–260.
Holley, R. W., Everett, G. A., Madison, J. T. & Zamir,A. Nucleotide sequences in yeast alanine transfer RNA.
J. Biol. Chem., 1965, 240, 2122–2127.Homans, S. W. A dictionary of concepts in NMR. Oxford
Science Publication. 1992.Hong, L., Turner, R. T., Koelsch, G., Shin, D., Ghosh,
A. K. & Tang, J. Crystal Structure of Memapsin2 (β-Secretase) in Complex with Inhibitor Om00-3Biochemistry 2002, 41, 10963–10967.
Hubbard, S. R. & Till, J. R. Protein tyrosine kinase structureand function Annu. Rev. Biochem. 2000, 69, 373–398.
Huffman, J. L. & Brennan, R. G. Prokaryotic transcriptionregulators: more than just the helix-turn-helix motif. Curr.Opin. Struct. Biol. 2002, 12, 98–106.
Hunkapiller, M. W., Strickler, J. E., & Wilson, K. E. Con-temporary methodology for protein structure determina-tion. Science 1984, 226, 304–311.
Ingram, V. A case of sickle-cell anemia. Biochem. Biophys.Acta 1989, 1000, 147–150.
Iwata, S., et al. Complete structure of the 11-subunit bovinemitochondrial cytochrome bc1 complex. Science 1998,281, 64–71.

504 REFERENCES
Iwata, S., Ostermeier, C., Ludwig, B., & Michel, H. Struc-ture at 2.8 A resolution of cytochrome c oxidase fromParacoccus denitrificans. Nature 1998, 376, 660–669.
Jackson, S. E. & Fersht, A. R. Folding of chymotrypsininhibitor 2, 1: Evidence for a two-state transition.Biochemistry 1991, 30, 10428–10435.
Jeffrey, P. D., Russo, A. A., Polyak, K., Gibbs, E., Hur-witz, J., Massague, J. & Pavletich, N. P.: Mechanism ofCDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 1995, 376, 313–320.
Jencks, W. P. Economies of enzyme catalysis. Cold SpringHarbor Symp. Quant. Biol. 1987, 52, 65–73.
Jimenez, J. L., Tennent, G., Pepys, M & Saibil, H. R.Structural Diversity of ex vivo Amyloid Fibrils Studiedby Cryo-electron Microscopy. J. Mol. Biol. 2001, 311,241–247.
Johnson L. N. Jenkins J. A. Wilson K. S. Stura E. A. &Zanotti G. Proposals for the catalytic mechanism ofglycogen phosphorylase b prompted by crystallographicstudies on glucose 1-phosphate binding. J. Mol. Biol. 1980,140, 565–580.
Johnson, W. C. Jr. Protein secondary structure and circulardichroism. A practical guide. Proteins Struct. Funct. Genet.1990, 7, 205–214.
Jones, D. T., Taylor, W. R. & Thornton. J. M. The rapidgeneration of mutation data matrices from proteinsequences. Computer Applied Biosciences 1992, 8,275–282.
Jordan, P., Fromme, P., Witt, H. T., Klukas, O., Saenger, W.,& Krauss, N. Three-dimensional structure of cyanobacte-rial photosystem I at 2.5 A resolution. Nature 2001, 411,909–917.
Kalies, K. -U. & Hartmann, E. Protein translocation intothe endoplasmic reticulum (ER) Two similar routes withdifferent modes. Eur. J. Biochem. 1998, 254, 1–5.
Karwaski, M. F., Wakarchuk, W. W. & Gilbert, M. High-level expression of recombinant Neisseria CMP-sialic acidsynthetase in Escherichia coli . Protein Expression andPurification 2002, 25, 237–240.
Kauzmann, W. Some factors in the interpretation of proteindenaturation. Adv. Prot. Chem. 1959, 14, 1–63.
Kay, L. E., Clore, G. M., Bax, A & Gronenborn,A. M. Four-dimensional heteronuclear triple-resonanceNMR spectroscopy of interleukin – 1β in solution. Science1990, 249, 411–414.
Kay, L. E., D. Marion, D. & Bax, A. Practical aspectsof three-dimensional heteronuclear NMR of proteins. J.Magn. Reson. 1989, 84, 72–84.
Kay, L. E., Ikura, M., Tschudin, R. & Bax, A. Three-dimensional triple resonance NMR spectroscopy ofisotopically enriched proteins. J. Magn. Reson. 1990, 89,496–514.
Keenan, R. J., Freymann, D. M., Stroud, R. M., & Wal-ter, P. The signal recognition particle. Annu. Rev. Biochem.2001, 70, 755–775.
Keleti, T. Two rules of enzyme kinetics for reversibleMichaelis-Menten mechanisms. FEBS Lett. 1986, 208,109–112.
Kelly, J. W. Alternative conformations of amyloidogenicproteins govern their behavior, Curr. Opin. Struct. Biol.1996 6, 11–17.
Kelly, J. W. Towards an understanding of amyloidosis.Nature Struct. Biol. 2002, 5, 323–324.
Kendrew, J. C., Bodo, G., Dintzis, H. M., Parrish, R. G.,Wyckoff, H., and Phillips, D. C. A Three-DimensionalModel of the Myoglobin Molecule Obtained by X-rayAnalysis. Nature, 1958, 181, 662.
Kim P. S. & Baldwin R. L. Intermediates in the FoldingReactions of Small Proteins Annu Rev. Biochem. 1990, 59,631–660.
King, R. W., Deshaies, R. J., Peters, J. M. & Kirschner,M. W. How proteolysis drives the cell cycle. Science
1996, 274, 1652–1659.Kirby, A. J. The lysozyme mechanism sorted – after
50 years. Nat. Struct. Biol. 2001, 8, 737–739.Knoll, A. H. The early evolution of eukaryotes: A geological
perspective, Science 1992, 256, 622–627.Knowles, J. R & Albery, W. J. Perfection in enzyme
catalysis, the energetics of triose phosphate isomerase. Acc.Chem. Res. 1977, 10, 105–111.
Knowles, J. R. Tinkering with enzymes: what are welearning? Science 1987, 236, 1252–1257.
Kohlstaedt, L. A., Wang, J., Friedman, J. M., Rice, P. A. &Steitz, T. A. Crystal structure at 3.5 A resolution of HIV-1reverse transcriptase complexed with an inhibitor. Science1992, 256, 1783–1790.
Kraut, J. How do enzymes work? Science 1988, 242,533–540.
Kwong, P. D., Wyatt, R., Majeed, S., Robinson, J. Sweet,R. W., Sodroski, J. & Hendrickson, W. A. Structures of
HIV-1 Gp120 Envelope Glycoproteins from Laboratory-Adapted and Primary Isolates. Structure 2000, 8,1329–133X.
Kyte J., Doolittle R. F. A simple method for displaying thehydropathic character of a protein. J. Mol. Biol. 1982, 157,105–132.
Ladbury, J. E & Chowdhry, B. Z. (eds) Biocalorimetry:Applications of Calorimetry in the Biological Sciences.John Wiley & Sons Chichester 1998.
Laemmli, U. K. Cleavage of structural proteins during theassembly of the head of bacteriophage T4. Nature 1970,227, 680–685.

REFERENCES 505
Lander, E. S. Linton, L. M., Birren, B et al. InternationalHuman Genome Sequencing Consortium. Initial sequenc-ing and analysis of the human genome. Nature 2001, 409,860–921.
Lander, E. S. The new genomics: global view of biology.Science 1996, 274, 536–539.
Larrabee, J. A. & Choi, S. Fourier transform infraredspectroscopy. Methods Enzymol. 1993, 226, 289–305,Academic Press.
Lashuel, H. A., Lai, Z. & Kelly, J. W. Characterization ofthe transthyretin acid denaturation pathways by analyticalultracentrifugation: implications for wild-type, V30M, andL55P amyloid fibril formation, Biochemistry 1998, 37,17851–17864.
Leatherbarrow, R. J., Fersht, A. R., & Winter, G. Transition-state stabilization in the mechanism of tyrosyl-tRNAsynthetase revealed by protein engineering. Proc. Natl.Acad. Sci. USA 1985, 82, 7840–7844.
Lee, A. G. A calcium pump made visible. Curr. Opin. Struct.Biol. 2002, 12, 547–554.
Lemmon M. A., Flanagan J. M., Treutlein H. R., Zhang J.,& Engelman D. M. Sequence specificity in the dimeriza-tion of transmembrane alpha-helices Biochemistry 1992,31, 12719–12725.
Lin. L. N. & Brandts, J. F. Isomerization of proline-93during the unfolding and refolding of ribonuclese A.Biochemistry 1983, 22, 559–563.
Lipschutz, R. J. & Fodor, S. P. A. Advanced DNA technolo-gies. Curr. Opin. Struct. Biol. 1994, 4, 376–380.
Lipscomb, W. N. Aspartate Transcarbamylase fromEscherichia Coli : Activity and Regulation Adv. Enzymol.1994, 73, 677–751.
Ludwig, S., Pleschka, S., Planz, O., & Wolff, T. Influenzavirus induced signalling cascades: targets for antiviraltherapy? Trends Mol. Medicine 2003, 9, 46–52.
Luecke, H., Schobert, B., Richter, H. T., Cartailler, P. &Lanyi, J. K. Structure of bacteriorhodopsin at 1.55 Aresolution. J. Mol. Biol. 1999, 291, 899–911.
Luecke, H., Schobert, B., Richter, H. T., Cartailler, P. &Lanyi, J. K. Structural changes in bacteriorhodopsinduring ion transport at 2 A resolution. Science 1999, 286,255–260.
Luong, C., Miller, A., Barnett, J., Chow J., Ramesha C., &Browner M. F. Flexibility of the NSAID binding site inthe structure of human cyclooxygenase-2. Nature Struct.Biol. 1996, 3, 927–933.
Lupas, A. Coiled coils new structures and new functions.Trends Biochem. Sci. 1996, 21, 375–382.
Lynch, D. R. & Synder, S. H. Neuropeptides: multiplemolecular forms, metabolic pathways and receptors. Annu.Rev. Biochem. 1986, 55, 773–799.
Malkin, D., Li, F. P., Strong, L. C., Fraumeni, J. F., Jr.,Nelson, C. E., Kim, D. H., Kassel, J., Gryka,M. A., Bischoff, F. Z., Tainsky, M. A. & Friend, S. H.Germ line p53 mutations in a familial syndrome of breastcancer, sarcomas, and other neoplasms. Science 1990, 250,1233–1238.
Mallucci, G. R., Ratte, S., Asante, E. A., Linehan, J., Gow-land, I., Jefferys, J. G. R., Collinge, J. Post-natal knockoutof prion protein alters hippocampal CA1 properties, butdoes not result in neurodegeneration. EMBO J. 2002, 21,202–210.
Mann, M., Hendrickson, R. C & Pandey, A. Analysis ofproteins and proteomes by mass spectrometry. Annu. Rev.Biochem. 2001, 70, 437–473.
Margulis, L & Sagan, C. What is Life. Simon & Schuster.1995.
Marquart, M., Walter, J., Deisenhofer, J., Bode, W., &Huber, R. The Geometry of the Reactive Site and ofthe Peptide Groups in Trypsin, Trypsinogen and itsComplexes with Inhibitors Acta Crystallogr., Sect. B 1983,39, 480–484.
Martin, G. R., Timple, R., Muller, P. K. & Kuhn, K. Thegenetically distinct collagens. Trends Biochem. Sci. 1985,10, 285–287.
Martoglio, B. & Dobberstein, B. Snapshots of membrane-translocating proteins. Trends Cell Biol. 1996, 6, 142–147.
Masters, C. L.; Simms, G.; Weinman, N. A.;Multhaup,G.; McDonald, B. L.; Beyreuther, K. Amyloid plaque coreprotein in Alzheimer disease and Down syndrome. Proc.Nat. Acad. Sci. USA 1985, 82, 4245–4249.
McPherson, A. Crystallization of Biological Macromolecules.Cold Spring Harbor Laboratory Press, 1999.
McRee, D. E. Practical Protein Crystallography. 2nd edn.Academic Press. 1999.
Meselson, M. & Stahl. F. The replication of DNA inEscherichia coli . Proc. Natl Acad. Sci. USA 1958, 44,671–682.
Michel, H. Three dimensional crystals of a membraneprotein complex. The photosynthetic reaction centre fromRhodopseudomonas viridis. J. Mol. Biol. 1982, 158,567–572.
Miller, S. Cold Spring Harbor Symp. Quant Biol. 1988, 52,17–28.
Miller, S. J. & Orgel, L. E. The Origins of Life, Prentice-Hall, New Jersey, 1975.
Miranker, A., Radford, S. E., Karplus, M., & Dobson, C. M.Demonstration by NMR of Folding Domains in Lysozyme.Nature, 1991, 349, 633–636.
Miranker, A., Robinson, C. V., Radford, S. E., AplinR. T., Dobson C. M. Detection of Transient Protein Fold-ing Populations by Mass Spectrometry. Science 1993, 262,896–900.

506 REFERENCES
Moore, P. B. & Steitz, T. A. The structural basis of largeribosomal subunit function. Annu. Rev. Biochem. 2003, 72,813–850.
Moore, P. B. The ribosome at atomic resolution. Biochem-istry 2001, 40, 3243–3250.
Morgan, D. A. Cyclin dependant kinases: Engines, Clocks,and Microprocessors. Ann Rev. Cell Dev. Biol. 1997, 13,261–291.
Morgan. D. G., Menetret, J. F., Neuhof, A., et al, Structureof the Mammalian Ribosome–Channel Complex at 17 AResolution. J. Mol. Biol. 2002, 324, 871–886.
Morimoto, R., Tissieres, A & Georgopoulos, C, (eds). Thebiology of heat shock proteins and molecular chaperones.Cold Spring Harbour Laboratory Press 1994.
Morise, H., Shimomura, O., Johnson, F. H., & Winant,J. Intermolecular energy transfer in the bioluminecentsystem of Aequorea . Biochemistry 1974, 13, 2656–2662.
Mullan, M., Crawford, F., Axelman, K., Houlden, H., Lil-ius, L., Winblad, B. & Lannfelt, L. A pathogenic mutationfor probable Alzheimer’s disease in the APP gene at theN-terminus of β-amyloid. Nature Genet. 1992, 1, 345–347.
Newman, M., Strzelecka, T., Dorner, L. F., Schildkraut,I. & Aggarwal, A. K. Structure of restriction endonucle-
ase BamHI and its relationship to EcoRI, Nature 1994,368, 660–664.
Nikolov, D. B. & and Burley, S. K. RNA polymerase IItranscription initiation: A structural view. Proc. Natl. AcadSci. 1997, 94, 15–22.
Nissen, P., Hansen, J., Ban, N., Moore, P. B. & Steitz,T. A. The structural basis of ribosome activity in peptide
bond synthesis. Science 2000, 289, 920–930.Noel, J. P. Hamm, H. E. & Sigler, P. B. The 2.2 A
crystal structure of transducin-α complexed with GTPγS.Nature 1993, 366, 654–658.
Nogales, E., Wolf, S. G. & Downing. K. H. Structure of theaβ-tubulin dimer by electron crystallography. Nature 1998,391, 199–203.
Nogales. E. Structural insight into microtubule function.Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 397–420.
Noiva, R., & Lennarz, W. J. Protein Disulfide Isomerase – AMultifunctional Protein Resident in the Lumen of theEndoplasmic Reticulum. J. Biol. Chem. 1992, 267,3553–3556.
Nugent, J. H. A. Oxygenic photosynthesis: electron transferin photosystem I and photosystem II. Eur. J. Biochem.1996, 237, 519–531.
Nurse, P. Genetic control of cell size at cell division in yeast.Nature 1975, 256, 547–551.
O’Farrell, P. H. High resolution two dimensional elec-trophoresis. J. Biol. Chem. 1975, 250, 4007–4021.
O’Shea, E. K. Rutkowski, R. & Kim, P. S., Evidence thatthe leucine zipper is a coiled coil. Science 1989, 243,538–542.
Oliver, J., Jungnickel, B., Gorlich, D., Rapoport, T., &High S. The Sec61 complex is essential for the insertion ofproteins into the membrane of the endoplasmic reticulum.FEBS Lett. 1995, 362, 126–30.
Onuchic, J. N., Wolynes, P. G., Luthey-Schulten, Z. &Socci, N. D. Towards an Outline of the Topography ofa Realistic Protein-Folding Funnel. Proc. Natl. Acad. SciUSA 1995, 92, 3626–3630.
Orengo, C. A., Michie, A. D., Jones, S., Jones, D. T.,Swindells, M. B., & Thornton, J. M. CATH – A Hierar-chic Classification of Protein Domain Structures. Structure1997, 5, 1093–1108.
Orgel, L. E. Molecular replication. Nature 1992, 358,203–209.
Orlova, E. V. & Saibil, H. R. Structure determination ofmacromolecular assemblies by single-particle analysis ofcryo-electron micrographs. Curr. Opin Struct. Biol. 2004,14, 584–590.
Pace, C. N. Conformational stability of proteins. TrendsBiochem. Sci. 1990, 15, 14–17.
Pace, C. N., and Scholtz, J. M. A Helix Propensity ScaleBased on Experimental Studies of Peptides and Proteins.Biophys. J. 1998, 75, 422–427.
Padlan, E. Anatomy of the Antibody Molecule. MolecularImmunology 1994, 31, 169–178.
Parry, D. A. D. The molecular and fibrillar structure ofcollagen and its relationship to the mechanical propertiesof connective tissue. Biophys. Chem. 1988, 29, 195–209.
Passner, J. M., Ryoo, H. D., Shen, L., Mann, R. S. &Aggarwal, A. K. Structure of a DNA-bound Ultrabithorax-Extradenticle homeodomain complex. Nature 1999, 397,714–719.
Pauling, L. & Corey, R. B. Atomic Coordinates andStructure Factors for Two Helical Configurations ofPolypeptide Chains. Proc. Natl. Acad. Sci. USA 1951, 37,235–240.
Perl, D. Welker, C., Schindler, T., Schroder, K., Marahiel,M. A., Jaenicke, R., and Schmid, F. X. Conservation ofrapid two-state folding in mesophilic, thermophilic andhyperthermophilic cold shock proteins. Nature StructuralBiology 2000, 7, 380–383.
Perutz, M. F., Rossmann, M. G., Cullis, A. F., Muir-head, H., Will, G. & North, A. C. T. Structure ofhaemoglobin. A three-dimensional Fourier synthesis at5.5 A resolution, obtained by X-ray analysis. Nature 1960,185, 416–422.
Perutz, M. F., Wilkinson, A. J., Paoli, M., & Dodson, G.The stereochemical mechanism of cooperative effects in

REFERENCES 507
hemoglobin revisted. Annu Rev. Biophys. Biomol. Structure1998, 27, 1–34.
Pfanner, N. & Neupert, W. The mitochondrial protein importapparatus. Annu. Rev. Biochem. 1990, 59, 331–353.
Phillips, M. A. & Fletterick, R. J. Proteases. Cur. Opinion.Struct. Biol. 1992, 2, 713–720.
Picot, D., Loll P. J., & Garavito R. M. The X-ray crystalstructure of the membrane protein prostaglandin H2synthase-l. Nature 1994, 367, 243–249.
Plaxco, K. W., Simons, K. T., & Baker, D. Contact order,transition state placement and the refolding rates of singledomain proteins. J. Mol. Biol. 1998, 277, 985–994.
Poignard, P., Saphire, E. O., Parren, P. W. H. I. & Bur-ton, D. R. GP120: Biologic Aspects of Structural Features.Annu. Rev. Immunol. 2001, 19, 253–74.
Ponder, J. W. & F. M. Richards. Tertiary templates forproteins. Use of packing criteria in the enumeration ofallowed sequences for different structural classes. J. Mol.Biol. 1987, 193, 775–791.
Popot, J.-L. & Engelman, D. M. Helical membrane proteinfolding, stability and evolution Annu. Rev. Biochem. 2000,69, 881–922.
Privalov, P. L. Stability of proteins: Small globular proteins.Adv. Protein Chem. 1979, 33, 167–241.
Privalov, P. L. & Gill, S. J. Stability of protein structureand hydrophobic interaction. Adv. Prot. Chem. 1988, 39,191–234.
Prusiner, S. B., Novel proteinaceous infectious particlescause scrapie. Science 1982, 216, 136–144.
Radford. S. Protein folding: progress made and promiseahead. Trends Biochem. Sci. 2000, 25, 611–618.
Ramachandran, G. N. & Sasiskharan, V. Conformation ofpolypeptides and proteins. Adv. Protein Chem. 1968, 23,283–437.
Ramakrishnan, V. & Moore, P. B. Curr. Opin. Struct. Biol.2001, 11, 144–154, 2001
Rao, S. T. & Rossmann M. G. Comparison of super-secondary structure in protein J. Mol. Biol. 1973, 76,241–256.
Rapoport, T. A., Jungnickel, B. & Kutay, U. Protein trans-port across the eukaryotic endoplasmic reticulum and bac-terial inner membranes, Annu. Rev. Biochem. 1996, 65,271–303.
Rappsilber, J. & Mann, M. What does it mean to identifya protein in proteomics? Trend Biochem. Sci. 2002, 27,74–78.
Rath, V. L., Silvian, L. F., Beijer, B., Sproat, B. S., Steitz,T. A. How glutaminyl-tRNA synthetase selects glutamine.Structure 1997, 6, 439–449.
Rechsteiner, M. & Rogers, S. W. PEST sequences andregulation by proteolysis. Trends Biochem. Sci. 1996, 21,267–271.
Riek, R., Hornemann, S., Wider, G., Billeter, M., Glockshu-ber, R. & Wuthrich, K. NMR structure of the mouseprion protein domain PrP (121–231). Nature 1996, 382,180–184.
Roder, H., Elove, G., & Englander, S. W. Structural char-acterization of folding intermediates in cytochrome c byH-exchange labeling and proton NMR. Nature 1888, 335,700–704.
Roeder, R. G. The role of general initiation factors intranscription by RNA polymerase II. Trends Biochem. Sci.1996, 21, 327–335.
Rose, G. N. Turns in peptides and proteins Adv. ProteinChem. 1985, 37, 1–109.
Rosenberg, J. M. Structure and function of restrictionendonucleases. Curr. Opin. Struct. Biol. 1991, 1, 104–113.
Rould, M. A., Perona, J. J., Soll, D., & Steitz, T. A. Struc-ture of E. coli glutaminyl-tRNA synthetase complexedwith tRNA(Gln) and ATP at 2.8 A resolution. Science1989, 246, 1135–1141.
Rowland-Jones, S. L. AIDS pathogenesis: what have twodecades of HIV research taught us? Nature Rev. Immunol.2003, 3, 343–348.
Rupp. B., http://www-structure.llnl.gov.Russell, P. & Nurse, P. cdc25+ functions as an inducer in the
mitotic control of fission yeast. Cell 1986, 45, 145–153.Russo, A. A., et al. Nat. Struct. Biol. 3, 696–XXX 1996Rutherford, A. W. & Faller, P. The heart of photosynthesis
in glorious 3D. Trends Biochem. Sci. 2001, 26, 341–344.Saibil, H. Molecular chaperones: containers and surfaces for
folding, stabilizing or unfolding proteins. Current Opinionin Struct. Biol. 2000, 10, 251–258.
Sambrook, J. & Russell, D. Molecular Cloning: A laboratorymanual. Cold Spring Harbor Laboratory Press, 2001
Sambrook, J., Fritsch, E. F. & Maniatis, T. MolecularCloning. Cold Spring Harbor Laboratory Press, 1989.
Sanger, F. Sequences, sequences, sequences. Annu. Rev.Biochem. 1988, 57, 1–28.
Saraiva, M. J. M. Hereditary transthyretin amyloidosis:molecular basis and therapeutical strategies. ExpertRev. Mol. Med. 2002. http://www.expertreviews.org/02004647h.htm
Schekman, R. Dissecting the membrane trafficking system.Nature Medicine 2002, 8, 1055–1058.
Schellman, J. A. The thermodynamic stability of proteins.Ann. Rev. Biophys. Biophys. Chem. 1987, 16, 115–137.
Schmid F. X., Mayr L. M., Mucke, M., & Schonbrun-ner, E. R. Prolyl isomerases: role in protein folding. Adv.Prot. Chem. 1993, 44, 25–66.
Schopf, J. W. Microfossils of the early Archaen Apex chert:New evidence of the antiquity of life. Science 1993, 260,640–646.

508 REFERENCES
Schramm, V. L. Enzyme transition states and transition stateanalog design. Annu. Rev. Biochem. 1998, 67, 693–720.
Schulz, G. E. Structure of porin refined at 1.8 A resolution.J. Mol. Biol. 1992, 227, 493–509.
Schuster, T. M. & Toedtt, J. M. New revolutions in theevolution of analytical ultracentrifugation. Curr. Opin.Struct. Biol. 1996, 6, 650–658.
Scopes, R. Protein purification: principles and practice.Springer-Verlag, Berlin 1993.
Selkoe, D. J. Amyloid β-protein and the genetics ofAlzheimer’s disease. J. Biol. Chem. 1996, 271,18295–18298.
Shine, J. & Dalgarno, L. The 3′ terminal sequence of E.coli16S rRNA : Complementary to nonsense triplets andribosome binding sites. Proc. Natl. Acad. Sci USA 1974,71, 1342–1346.
Sidransky, D & Hollstein, M. Clinical implications of thep53 gene. Annu. Rev. Med. 1996, 47, 285–30.
Siebert, F. Infrared spectroscopy applied to biochemicaland biological problems. Methods Enzymol. 1995, 246,501–526 Academic Press.
Sigler, P. B., Xu, Z. Rye, H. S. Burston, S. G. , Fenton,W. A & Horwich, A. L. Structure and function in GroEL-mediated protein folding Annu . Rev. Biochem. 1998, 67,581–608.
Silver, P. A. How proteins enter the nucleus. Cell 1991, 64,489–497.
Silverman, G. A. et al. The Serpins Are an ExpandingSuperfamily of Structurally Similar but FunctionallyDiverse Proteins. J. Biol. Chem. 2001, 276, 33293–33296.
Simons K. T., Ruczinski, I., Kooperberg, C., Fox, B.,Bystroff, C., & Baker, D. Improved Recognition ofNative-like Protein Structures using a Combination ofSequence-dependent and Sequence-independent Featuresof Proteins. Proteins 1999, 34, 82–95.
Singer, S. J. The molecular organization of membranes.Annu. Rev. Biochem. 1974, 805–833.
Singer, S. J. & Nicolson, G. The fluid mosaic model of thestructure of cell membranes. Science 1972, 175, 720–731.
Skehel, J. J., Bayley, P. M., Brown, E. B., Martin,S. R., Waterfield, M. D., White, J. M., Wilson, I. A., andWiley, D. C. Changes in the conformation of influenzavirus haemagglutinin at the pH optimum of virus-mediatedmembrane fusion Proc. Natl. Acad. Sci. USA 1982, 79,968–972.
Skehel, J. J. & Wiley, D. C. Receptor binding and membranefusion in virus entry: the influenza haemagglutinin. Annu.Rev. of Biochem. 2000, 69, 531–569.
Smith, W. L., DeWitt, D. L. & Garavito, R. M. Cyclo-oxygenase: Structural, Cellular, and Molecular BiologyAnnu . Rev. Biochem. 2000, 69, 145–182.
Song, L., Hobaugh, M. R., Shustak, C., Cheley, S., Bay-ley, H., & Gouaux J. E. Structure of staphylococcal α-hemolysin, a heptameric transmembrane pore, Science1996, 274, 1859–1866.
Steinhauer, D. A. & Skehel, J. J. Genetics of influenzaviruses. Annu. Rev. Genet. 2002, 36, 305–332.
Steitz, T. A. & Schulman, R. G. Crystallographic and NMRstudies of the serine proteases. Annu. Rev. Biophys.Bioenerg. 1982, 11, 419–464.
Stock, D., Gibbons, C., Arechaga, I., Leslie, A. G. W. &Walker, J. E. The rotary mechanism of ATP synthase.Curr. Opin. Struc. Biol. 2000, 10, 672–679.
Stock, D., Leslie, A. G. W., & Walker, J. E. MolecularArchitecture of the Rotary Motor in ATP Synthase. Science1999, 286, 1700–1705.
Stoeckenius, W. Bacterial rhodopsins: Evolution of amechanistic model for the ion pumps Prot. Sci., 1999, 8,447–459.
Storey, A., Thomas, M., Kalita, A., Harwood, C., Gar-diol, D., Mantovani, F., Breuer, J., Leigh, I. M., Mat-lashewski, G., & Banks, L. Role of a p53 polymorphismin the development of human papilloma-virus-associatedcancer. Nature 1998, 393, 229–234.
Studier, F. W., Rosenberg, A. H., Dunn, J. J., &Dubendorff, J. W. Use of T7 RNA polymerase to directexpression of cloned genes. Methods in Enzymology 1990,185, 60–89, Academic Press.
Sunde, M., Serpell, L. C., Bartlam, M., Fraser, P. E., Pepys,M. B., & Blake C. C. Common core structure of amyloidfibrils by synchrotron X-ray diffraction. J. Mol. Biol. 1997,273, 729–739.
Tanford, C. The Hydrophobic Effect; formation of micellesand biological membranes. 2nd ed. Wiley 1980.
Tarn W.-Y. & Steitz. J. A. Pre-mRNA splicing: thediscovery of a new spliceosome doubles the challengeTrends Biochem. Sci. 1997, 22, 132–137.
Taylor, K. A. & Glaeser, R. M. Electron diffraction offrozen, hydrated protein crystals Science 1974, 186,1036–1037.
Toyoshima, C., Nakasako, M., Nomura, H. & Ogawa,H. Crystal structure of the calcium pump of sarcoplasmicreticulum at 2.6 A resolution. Nature 2000, 405 647
Trabi, M., & Craik, D. J. Circular proteins – no end in sight.Trends Biochem. Sci. 2002, 27, 132–138.
Tsukihara, T. Aoyama, H., Yamashita, E., Tomizaki, T.,Yamaguchi, H., Shinzawa-Itoh, K., Nakashima, R.,Yaono, R., & Yoshikawa, S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A. Science,1996, 272, 1136–1144.
Tugarinov, V., Hwang, P. M. & Kay, L. E. Nuclear mag-netic resonance spectroscopy of high-molecular weightproteins. Annu. Rev. Biochem. 2004, 73, 107–146.

REFERENCES 509
Turner, B. G. & Summers, M. F. Structural biology of HIV.J. Mol. Biol. 1999, 285, 1–32.
Unger, V. M. Electron cryomicroscopy methods. Curr. Opin.Struct. Biol. 2001, 11, 548–554.
Vane, J. R. Inhibition of prostaglandin synthesis as amechanism of action for the aspirin-like drugs. Nature,1971, 231, 232–235.
Varghese, J. N., Colman, P. M., van Donkelaar, A., Blick,T. J., Sahasrabudhe, A., & McKimm-Breschkin, J. L.
Structural evidence for a second sialic acid binding sitein avian influenza virus neuraminidases. Proc Natl AcadSci USA 1997, 94, 11808–11812.
Varghese, J. N., McKimm-Breschkin, J. L., Caldwell, J. B.,Kortt, A. A. & Colman, P. M. The structure of thecomplex between influenza virus neuraminidase and sialicacid, the viral receptor. Proteins 1992, 14, 327–332.
Varghese, J. N. & Colman, P. M. Three-dimensional struc-ture of the neuraminidase of influenza virus A/Tokyo/3/67at 2.2 A resolution. J. Mol. Biol. 1991, 221, 473–486.
Varghese, J. N., Laver, W. G. & Colman P. M. Structure ofthe influenza virus glycoprotein antigen neuraminidase at2.9 A resolution. Nature 1983, 303, 35–40.
Varshavsky. A., The ubiquitin system. Trends Biochem. Sci.1997, 22, 383–387.
Vermeglio, A & Joliot, P. The photosynthetic apparatusof Rhodobacter sphaeroides Trends Microbiol. 1999, 7,435–440.
Viadiu, H., & Aggarwal, A. K. The role of metals in catalysisby the restriction endonuclease BamHI. Nat. Struct. Biol.1998, 5, 910–6.
Vogelstein, B., Lane, D. & Levine, A. J. Surfing the p53network. Nature 2000, 408, 307–310.
Voges, D., Zwickl, P. & Baumeister, W. The 26S protea-some: a molecular machine designed for controlled pro-teolysis Annu. Rev. Biochem. 1998, 68, 1015–1068.
Voos, W. Martin, H., Krimmer, T. & Pfanner, N., Mecha-nisms of protein translocation into mitochondria Biochim.Biophys. Acta, 1999, 1422, 235–254.
Walter, P. & Johnson, A. E. Signal sequence recogniton andprotein targeting to the endoplasmic reticulum membrane.Annu. Rev. Cell Biol. 1995, 10, 87–119.
Wang, J., Smerdon, S. J., Jager, J., Kohlstaedt, L. A., Rice,P. A., Friedman, J. M., Steitz, T. A. Structural basis ofasymmetry in the human immunodeficiency virus type 1reverse transcriptase heterodimer. Proc Natl Acad Sci USA1994, 91, 7242–7246.
Wang, Y. & van Wart, H. E. Raman and resonance Ramanspectroscopy Methods Enzymol. 1993, 226, 319–373,Academic Press.
Warren, A. J. Eukaryotic transcription factors. Curr. Opin.Struct. Biol. 2002, 12. 107–114.
Watson J. D. & Crick. F. H. C. Molecular structure ofnucleic acids: a structure for deoxyribose nucleic acid.Nature 1953, 171, 737–738.
Weis, W., Brown, J. H., Cusack, S., Paulson, J. C., Ske-hel, J. J., & Wiley, D. C. Structure of the influenza virushaemagglutinin complexed with its receptor, sialic acidNature 1988, 333, 426–431.
Weis. K. Importins and exportins: how to get in and out ofthe nucleus Trends Biochem. Sci. 1998, 23, 185–189.
Weiss M. S., Olson, R., Nariya, H., Yokota, K., Kamio, Y.,& Gouaux, E. Crystal structure of StaphylococcalLukf delineates conformational changes accompanyingformation of a transmembrane channel. Nat. Struct. Biol.1999, 6, 134–140.
Weissmann, C., Enari, M., Klohn, P.-C., Rossi, D. &E. Flechsig. E. Transmission of prions. Proc. Natl. Acad.Sci. USA 2002, 99, 16378–16383.
Weissmann, C., Molecular Genetics of Transmissible Spongi-form Encephalopathies. J . Biol. Chem. 1999, 274, 3–6.
Wetlaufer, D. B. Ultraviolet spectra of proteins and aminoacids. Adv. Prot. Chem. 1962, 17, 303–390.
White, S. H. & Wimley, W. C. Membrane protein foldingand stability: Physical principles. Ann. Rev. Biophys.Biomol. Struct. 1999, 28, 319–6.
Wiley D. C., & Skehel J. J. The structure and function of thehaemagglutinin membrane glycoprotein of influenza virus.Annu. Rev. Biochem. 1987, 56, 365–94.
Wilmot, C. M. & Thornton, J. M. Analysis and predictionof the different types of beta-turn in proteins. J. Mol. Biol.1988, 203, 221–232.
Wimberley, B. T., Brodersen, D., Clemons, W., Morgan-Warren, R., Carter, A., Vonrhein, C., Hartsch, T. &Ramakrishnan, V. Structure of the 3OS RibosomalSubunit. Nature 2000, 407, 327–339.
Wimley, W. C. The versatile β-barrel membrane proteinCurr. Opin. Struct. Biol. 2003, 13, 404–411.
Wlodawer. A. & Erickson, J. W. Structure based inhibi-tors of HIV-1 proteinase. Annu. Rev. Biochem. 1993, 62,543–585.
Woody, R. W. Circular dichroism. Methods Enzymol. 1995,246, 34–71, Academic Press.
Wower, J. Rosen, K. V., Hixson, S. S. & Zimmermann,R. A. Recombinant photoreactive tRNA molecules asprobes for cross-linking studies. Biochimie 1994, 76,1235–1246.
Wuthrich, K. NMR of Proteins and Nucleic Acids. JohnWiley & Sons. 1984.
Xu, Z., Horwich A. L., & Sigler P. B. The crystal structureof the asymmetric GroEL-GroES-(ADP)7 chaperonincomplex. Nature. 1997, 388, 741–750.
Yates. J. R. Mass spectrometry: from genomics to pro-teomics. Trends Genet. 2000, 16, 5–8.

510 REFERENCES
Yoshida, M., Muneyuki, E., & Hisabori, T. ATP synthase–amarvellous rotary engine of the cell. Nat. Rev. Mol. CellBiol. 2001, 2, 669–77.
Yoshikawa, S. Beef heart cytochrome oxidase. Curr. Opin.Struct. Biol. 1997, 7, 574–579.
Zahn, R., Liu, A., Luhrs, T., Riek, R., von Schroetter, C.,Lopez Garcıa, F., Billeter, M., Calzolai, L., Wider, G. &Wuthrich, K. NMR solution structure of the human prionprotein. Proc. Natl. Acad. Sci USA 2000, 97, 145–150.
Zhang, D., Kiyatkin, A., Bolin, J. T. & Low, P. S. Crystal-lographic Structure and Functional Interpretation of theCytoplasmic Domain of Erythrocyte Membrane Band 3.Blood 2000, 96, 2925–2933.
Zouni, A., Horst-Tobias, W., Kern, J., Fromme, P., Krauss, N.,Saenger, W., & Orth, P. Crystal structure of photosys-tem II from Synechococcus elongatus at 3.8 A resolution.Nature 2001, 409, 739–743.

INDEX
Entries are arranged alphabetically with page numbers in italic indicating a presence in a figure whilst boldtype indicates appearance in a table. Greek letters and numbers are sorted as if they were spelt out; β sandwichbecomes beta-sandwich, 5S rRNA appears where fiveS rRNA is normally located in listings. Positional charactersare ignored; so 2-phosphoglycolate appears under phosphoglycolate.
A site, 273Aβ protein, 430, 468–470AAA superfamily, 308Ab intio, 186Abrin, 228Absorbance, 379–381
aromatic amino acids, 381Beer-Lambert’s Law, 32data on aromatic amino acids, 30excitation and emission, 380heme groups, 67spin multiplicity, 380vibrational states, 380wavenumber, 381
Acetazolamide, 205Acetylation, 270, 292Acetylcholinesterase, 191
specificity constant, 201Acid base catalysis, 203–204Acid base properties of amino acids,
14–15Acquired immune deficiency syndrome,
443–457recognition and identification, 443
Acrylamide, 335, 381Activated state (complex), 196Activation energy, 195–196Active site
Gln-tRNA synthetase, 221lysozyme, 210serine proteases, 212triose phosphate isomerase, 216tyrosyl tRNA synthetase, 219
Acyl chains, 107unsaturation, 106
Acyl-enzyme intermediate, 214Adenine, 247
A76, 269–270
A2451, 279A2439, 279A2486, 282
Adenosine monophosphatepK of N1 atom, 280structure of AMP, 282
Adenosine triphosphate, synthesisallosteric activation, 234transition state analogue, 148
Adenylate cyclase, 116–117, 439–440signaling pathway, 120
Adenylation, 2925-adenylyl-imidophosphate, 148Adiabatic chamber, 399Adrenaline, 116Aequorea victoria, 383–384Aerobic, 126Affinity chromatography, 332–334
types of ligands, 334Affinity labeling, 216, 279Aggregation, 416–417
in disease states, 427–428Agonists, 116AIDS, see Acquired immune deficiency
syndromeAlanine
chemical properties, 24D isomer, 35data, 18genetic code specification, 268helix/sheet propensity, 41hydropathy index, 112optical rotation, 34prevalence in secondary structure,
180spatial arrangement of atoms, 16stereoisomers, 34titration curve, 14
Albumin, 1, 33Aldol condensation, 97Alignment methods, 172–173Aliphatic amino acids, 24Alkaline phosphatase, 207, 339Allele, 475, 476Allosteric regulation, 72–74, 231–237Allostery, 189Allysine, 97α helix, 41–45
dihedral angles, 43dimensions, 43dipole moment, 56formation, 409hydrogen bonding, 41–43side chains, 42–43space-filling view, 4structure, 42
α-secretase, 469α1-antitrypsin, 475–478α-haemolysin, 130–131Alveoli, 475Alzheimer’s disease, 422, 427, 430,
468–470diagnostic markers, 468genetic basis, 468–470
Amidation, 292Amide exchange, 411–412Amide, 25–26, 43, 56Amino acid residue, 17Amino acids
acid-base properties, 14–15chemical and physical properties, 23–32detection of, 32–34formation of peptide bonds, 16–17genetic code specification, 268non-standard, 35–36optical rotation, 34
Proteins: Structure and Function by David Whitford 2005 John Wiley & Sons, Ltd

512 INDEX
Amino acids (continued )pK values for ionizable groups, 15quantification, 34reaction with tRNA,single letter codes, 40solubility, 13stereochemical representation, 15stereoisomerism, 34–35three letter codes, 40
Amino acyl AMP, 268Amino acyl tRNA synthetases, 218,
268–269catalysis, 268class I and class II tRNA synthetases, 220classification, 220, 269glutaminyl tRNA synthetase, 220–221transition state analogue, 221tyrosyl tRNA synthetase, 218–220
AMP, see Adenosine monophosphate,Amphiphiles, 122Ampicillin, 315Amplification (PCR), 169–170Amplitude, 353–354AMP-PNP, see 5-adenylyl-imidophosphateAmyloid precursor protein, 469Amyloid, 427
structure of fibril, 431plaques, 468
Amyloidogenesis, 427–431diseases arising from, 428involving transthyretin, 428–430prion based, 431–435
Anacystis sp., 164Analytical centrifugation, 322–323Anfinsen cage, 420Anfinsen, C., 402, 416, 423Angiotensin, 287Angstrom, 18Angstrom, A.J., 478Anisotropy, 383Ankyrin, 111Anoxygenic photosynthesis, 128Antagonists, 116Antechambers, 306Antibiotics, 278–279,
selection of transformants, 315studying ribosomal structure, 279
Antibody, 75–80 see immunoglobulinsAnticodon loop, 268–269Anticodon, 283Antigen, 75
flu virus antigens, 464Antigenic determinants, 77Antigenic drift, 464Antigenic shift, 464Antigenic sub-types, 465Antimycin A, 134Antiport, 154Apoenzyme, 192Apoptosis, 238–239, 310, 471
APP see amyloid precursor protein,468–469
Aquaporin, 377Arabidopsis thaliana, 169Arachidonic acid, 107, 229–230Arber, W., 221Archae, 128, 304, 309, 417, 422Archaebacteria see archaeArginine
charge-charge interactions, 55–56chemical properties, 28–29data, 18genetic code specification, 268helix/sheet propensity, 41hydropathy index, 112optical rotation, 34prevalence in secondary structure, 180trypsin substrate, 227
Armadillo motifs, 301Aromatic amino acids,Aromatic residues, 30Arrhenius equation, 195Arrhenius, S.A., 195Arylation, 29Ascorbate, 289Asparagine chemical properties, 25
data, 18helix/sheet propensity, 41hydropathy index, 112genetic code specification, 268prevalence in secondary structure, 180location in turns, 47–48N linked glycosylation, 98, 290
Aspartatebinding domain in ATCase, 236chemical properties, 25data, 18genetic code specification, 268helix/sheet propensity, 41hydropathy index, 112in HIV protease, 450–452in lysozyme catalysis, 211–212in T7 RNA polymerase, 256prevalence in secondary structure, 180
Aspartate transcarbamoylase, 6, 234–237allosteric behaviour, 234binding of carbamoyl phosphate, 234conformational change, 237feedback inhibition, 235quaternary structure, 235
Aspartic acid see aspartateAspartyl protease, 450–452, 470Aspirin, 228–230AspN, 166Astbury, W.T., 92Astrocytosis, 434Asymmetric centres, 37Ataxia, 433Atherosclerosis, 99ATP synthase, see ATP synthetase
ATP synthetase, 132, 144–152, 173Boyer model, 146chemical modification, 146, 151composition, 145conformational changes in, 146–148cooperativity, 146γ subunit, 150mechanism of ATP synthesis, 146–147nucleotide binding site, 149proton translocation, 149structure of F1 ATPase, 147–151structure of Fo, 150–152yeast enzyme, 156
ATP, see adenosine triphosphateATPase family, 152–156, 308Autolysis, 238, 449AvaI, 2223′-azido-3−deoxythymidine, see AZTAZT, 453Azurin, 359
B cells, 75BACE, see β secretaseBacillus caldolyticus, 397–398Bacillus subtilis, 397–398Bacterial reaction centre, 119–126
crystallization, 123cytochrome subunit, 125electron transfer, 121–122transmembrane helices, 125
Bacteriochlorophyll, 121protein crystallization of, 359
Bacteriophage λ, 64defective groE operon, 416
Bacteriophage T4, 266Bacteriophage T7, 256Bacteriorhodopsin, 114–115
light driven pump, 115protein folding, 422–425structure, 117transmembrane organization, 116
Baldwin, R., 414BamHI, 222
signature sequence, 224Band 3 protein, 111Barnase, 405–408Beer Lamberts Law, quantification of amino
acids, 32Benzamidine, 227Benzene, 55Benzopyrene, 454Bernal, J.D., 353Berzelius, J. J., 1β adrenergic receptor, 117β barrel, 47, 180
GFP, 383gp120 454–455Gro-ES, 417α-haemolysin, 130–131

INDEX 513
ODCase, 205porins, 128–132triose phosphate isomerase, 47
β blocker, 116β endorphin, 289β helix, 61–62, 430–431β lactamase, 181β lipotrophin, 289β meander, 59, 61, 222β propeller motif, 61, 180, 463β sandwich, 59, 180
gp120, 454–455p53, 472
β scaffold, 430β secretase, 469–470β sheet, 45–47β strand, 45–46
dimensions, 43β turn, 47β-N -oxalyl L-α, β-diamino propionic acid,
36B factor, 357Bilayers, 107–109Bimolecular reactions, 194–195, 208,
382Bioinformatics, 50, 184–187Biotin, 1932,3-bisphosphoglycerate, 73, 74Bisacrylamide, 335Biuret, 33Blobel, G., 293Bloch, F., 360BLOCKS database, 175Blood clotting cascade, 237, 239Blood clotting factors, 318, 239BLOSUM, 175Blue green algae, 126Boat conformation, 211Bohr effect, 74Bohr, C., 74Boltzmann distribution, 380Boltzmann’s constant, 196, 323Bombyx mori, 92, 93Bordetella pertussis, 440Bovine pancreatic trypsin inhibitor, 53, 224,
468Bovine serum albumin, 33Bovine spongiform encephalopathy,
466–468Boyer, P.D., 146BPTI, see Bovine pancreatic trypsin
inhibitorBragg, W.H., 349Bragg, W.L. 349Branch point, 266Brandts, J.F., 414Bravais Lattices, 352Brenner, S., 266Briggs, G.E., 199Bromelain, 459, 460
Bromoaspirin, 228Brønsted equation, 407Brownian diffusion, 320, 382Browsers, 185BSA, see bovine serum albuminBSE, see bovine spongiform encephalopathyBuchner, E., 2, 189Buoyant density, 320Burley, S.K., 259Burnet, M., 75Bursa of Fabricius, 75
C peptide, 456Caenorhabditis elegans, collagen genes, 93
genome, 169Cahn Ingold Prelog, 34Calcium binding domains, 101,415Calmodulin, 25Calorimetry, 109
differential scanning, 399–400Canavanine, 36Cancer, 11, 441
cdk-cyclin complex regulation, 252–253cervical, 475involvement of p53, 474Kaposi’s sarcoma, 445
Cannibalism, 433Capsid, 377
TMV, 443HIV, 445, 446p17, 446–447
Carbamoyl phosphate, 234Carbonic anhydrase, 74, 179
catalysis by, 204–207imidazole ligands in active site, 206pH dependent catalysis, 205specificity constant, 201
Carboxypeptidase A, 25, 192, 193Cardiac glycosides, 154Cargo proteins, 300Carotenoids, 121Cartoon representation, 13, 46Cascade, 237, 239CASP, 186Caspases, 238–239, 310Catabolism, 189Catalase, sedimentation coefficient, 324
specificity constant, 201Catalysts, 197Catalytic mechanisms, 202–209
acid-base catalysis, 202–203covalent catalysis, 203electrostatic catalysis, 205metal ion catalysis, 204–205preferential binding of transition state,
207–209proximity and orientation effects,
206–207rate enhancements, 197
Catalytic triad, 213CBCA(CO)NH, 373CBCANH, 373CCD, see charge coupled deviceCCK motif, 81CCM, see common core motifCD4, 446cdc genes, 249Cdks see Cyclin dependant kinases,Celebrex 231Cell cycle, 247–250
DNA replication, 253mitosis, 247
Cell disruption, 319Cell division, 249, 253Cell membrane, 110Centrifugation, 320–323,Chair conformation, 211Chaotropes, 326Chaperones, 416–422
catalytic cycle, 419–422crystal structure, 420domain movements, 418electron microscopy, 417–419GroEL-GroES, 417–422mitochondrial, 297–298organization, 418small heat shock proteins, 417thermosome, 422
Chaperonin, 416Charge coupled devices, 170,
349Charge-charge interactions, 55–56Checkpoints, 249Chemical kinetics, 192–195Chemical modification, 29, 205
in RNase A, 202Chemical shift, 362
table of 13C chemical shifts, 372table of 15N chemical shifts, 371table of 1H chemical shifts, 366
Chemiosmosis, 145Chevron plot, 405χ angle, 43Chiral, 19, 34–35Chitinase, 319Chloroamphenicol, 278, 279Chlorophyll, 121
in photosystem II, 127special pair, 125voyeur, 125
Chloroplastlipid:protein ratios, 109RNA polymerases, 257schematic diagram, 299sorting and targeting, 299stromal proteins, 299Tic system, 299Toc complexes, 299
Cholera toxin, 120, 439–440

514 INDEX
Cholera, 439Chou, P.Y., 182Chou-Fasman algorithm, 183–184Chromatography, 326–333
affinity, 332–333cation exchange, 167,gel filtration principle, 330hydrophobic interaction, 331ideal separation, 327ion exchange, 329–330molecular mass estimation, 331protein hydrolysate, 167reverse phase, 331–332size exclusion, 330–331system and instrumentation, 328
Chromophore, 33, 121Chromosome, 247Chronic obstructive pulmonary disease,
(COPD) 475Chymotrypsin, 46, 287
active site substrate specificity, 212arrangement of catalytic triad, 213hydrolysis of esters, 201inhibitor, 406PDB file, 51proteolytic activation, 237–238structural homology, 177, 179TPCK binding, 213
cI repressor, 64CIP family, 252circe effect, 205Circular dichroism, 385–387
folding of bacteriorhodopsin, 423folding of OmpA, 424for studying protein folding, 404,
409magnetic circular dichroism, 387spectra, 387
cis isomercis ring, 418, 419Cis-trans peptidyl proline isomerase, 59
in protein folding, 415–416parvulins, 416FK506 binding proteins, 416
Citric acid cycle, 190Citrulline, 38CJD see Creutzfeldt Jacob diseaseClathrates, 54Clathrin coated vesicles, 297Cleland’s reagent , see also dithiothreitol,
28Cloning, 314–316Clostripain, 166Cloverleaf structure, 268, 269CMC, see critical micelle concentrationCobalt, 193Codon, 268,Coelenterazine, 383Coenzyme Q, 193, see also ubiquinoneCo-factors, 192–193
Coiled coil, 86–90ATP synthetase, 147, 150gp41 of HIV, 456haemagglutinin, 456non-keratin based motifs, 90parallel and antiparallel coils, 87
Cold shock proteins, 398Collagen, 92–100
abundance, 93biosynthesis, 97–99connective tissue, 93dimensions, 94disease states associated with, 99–100genes, 93hydroxylation, 289processing, 98procollagen, 289related disorders, 100–102structure and function, 94–97thermal denaturation, 96triple helix, 95types, 94
Common core motif, 222Competent cells, 315Competitive enzyme inhibition, 225–226,
453Complementarity determining regions
(CDRs), 77Complex I, see NADH-ubiquinone
oxidoreductaseComplex II, see succinate dehydrogenaseComplex III, see cytochrome bc1 complexComplex IV, see cytochrome oxidaseConformational stability, 396–404
estimations from ideal denaturationcurves, 401
linear extrapolation method, 402table of selected proteins, 402
Connective tissue disorders,Conotoxins, 178Conserved residues, 172Contact order, 410Conus, 178Convergent evolution, 179Coomassie Blue, 33, 336Cooperative binding curve, 69–70, 396Cooperativity, 70,Copper, 7
binuclear cluster, 139co-factor role, 193
Correlation time, 363Coulomb’s Law, 55Coupling constants, 363Covalent catalysis, 204Covalent modification, 237–241Cowpox, 75Cox, G.B., 146COX-1, see also cyclo-oxygenases, 230COX-2, see also cyclo-oxygenases, 230CPK models, 13
Creutzfeldt, H.G., 433Creutzfeldt-Jakob disease, 433Crick, F.H.C., 2, 87, 247, 266, 267Critical micelle concentration, 107–108Cro repressor, 66Crowther, R.A., 377Cryoelectron microscopy, 375–379
amyloid fibril structure, 430–431ATP synthetase, 149hepatitis B capsid, 377instrumentation, 376proteasome, 308, 309, 418ribosome, 271sample preparation, 376–377tubulin structure, 377–379
Crystal lattices, 352Crystal violet, 319Crystallization, 358–360Crystallography, 349–360Cu, 9
centres in cytochrome oxidase, 138Cyanide, 228Cyanobacteria, 126, 164Cyanogen bromide, 26Cyclic AMP, 439–440
in signaling, 116–119Cyclic GMP, 118Cyclic nucleotides, 202Cyclic proteins, 81Cyclin dependant kinases, 249–253
ATP binding, 251conformational changes, 251–252inhibitors, 252interaction with p53, 471regulation, 252structure and function, 250–253T loop activation, 251–252
Cyclins, 248–249binding with cdk, 251–252conformational changes, 251–252cyclin boxes, 249cyclin-cdk complexes in cell cycle, 250regulation, 252tertiary structure, 252threonine phosphorylation, 252
Cyclohexane, 53Cyclo-oxygenases, 228–230
active site, 230tertiary structure, 229isoforms, 230
Cyclophilin, 416Cyclosporin A, 81, 416Cyclotides, 81Cysteine
active site of caspases, 310chemical properties, 26–28data, 19genetic code specification, 268helix/sheet propensity, 41hydropathy index, 112

INDEX 515
ligands in Zn fingers, 261, 263location in turns, 47optical rotation, 34prevalence in secondary structure, 180reaction with Ellman’s reagent, 28
Cystic Fibrosis, 9, 426–427Cytochrome b, 132–137
absorbance maxima, 133ligands, 137redox potential, 133separation distances, 137subunit mass, 137
Cytochrome b5, 58, 60, 173, 181, 401Cytochrome b562, 59Cytochrome bc complex, see cytochrome
bc1 complexCytochrome bc1 complex, 132–137
crystal structure, 136cytochrome c1, 136cytochrome b, 137electron transfer reactions, 135gating, 137inhibitor binding studies, 133–134Q cycle in, 135Reiske protein, 133, 136subunit composition, 137 subunit mass,
137ubiquinone, 134
Cytochrome cprokaryotic, 176sequence homology, 175sorting pathway, 299structural homology, 176
Cytochrome c1, 132–137orientation, 136signal sequence, 299sorting pathway, 299subunit mass, 137
Cytochrome oxidase, 138–144catalytic cycle, 138–139channels, 144CuA centre, 139CuB center, 140enzyme from P.denitrificans, 141isoforms, 141mammalian enzyme, 140–141mitochondrial coded subunits, 139monomeric core, 141oxygen binding site, 143proton pump, 142–143subunit structure, 141
Cytochrome P450, 241Cytosine, 247Cytoskeleton, 111
role of tubulin, 377Cytotoxic T lymphocytes, 79
D amino acids, 34, 36, 321Dansyl chloride, 33, 167
Databases, 184Dayhoff. M.O, 174ddATP, 171ddCTP, 171ddGTP, 171ddI, 453ddTTP, 171Deamidation, 25Death effector domains, 239Debye, P., 325Debye-Huckel Law, 324–325Degradation, see protein degradation�Cp, 55
measurements, 399–400table for selected proteins, 401
Denaturantconcentration dependence, 400–401guanidine hydrochloride, 396solubility of amino acids, 397thermal, 96urea, 396
Denaturation in PCR, 169Denaturation, 396–398
ideal curves, 401linear extrapolation method, 402
Deoxyadenosine, 170Detergents, 122, 425
amphiphiles, 122critical micelle concentration, 107–108SDS binding, 335use of LDAO, 121
Dextrorotatory, 342′3′-dideoxyinosine, see ddIDiabetes, 422, type-I, 441Dialysis, 333, 359Dideoxyadenosine, 170Dideoxynucleotide, 169–170, 171, 453Diesenhofer, J.E., 126Differential scanning calorimetry, 399
profiles, 400Diffusion controlled rate, 195, 201DIFP, see DiisopropylfluorophosphateDihedral angle, 41, 43, 46Dihydrouridine, 268Diisopropylfluorophosphate, 212, 228Dipalmitoylphosphatidylcholine, 107DIPF see di-isopropylphosphofluoridateDipole moment, 41, 56D-isoglutamate, 35Dissociation of weak acids, 478–479Distal histidine, 68Disulfide, bond formation, 288–289
bridges in keratin, 91–92bridges in prions, 433bridges, 53chemical properties, ribonuclease,
neuraminidase, 463oxidoreductase, protein disulfide
isomerase, 288–289thioredoxin, 288
Dithiothreitol, 27–28DNA binding proteins, 64–66, 258–261
arc repressor, 261cro repressor, 66eukaryotic transcription factors, 261–265met repressor, 261molecular saddle, 259–260recognition helix, 65restriction endonucleases, 221–224sequence homology, 66
DNA gyrase, 253DNA ligase, 254
ligation, 315DNA polymerase, 253–254
cloning, 314–315enzyme nomenclature, 191PCR, 170specificity constant, 201
DNA replication, 253–254,semi-conservative model,
DNA sequencing, 168–170profiles from, 171
DNA structure, 248major groove, 222
dNTP, 170Domains
b5-like proteins, 60death effector, 239definition, 58–59EGF-like, 101extracellular domains in receptor tyrosine
kinases, 240folding domains in lysozyme, 415gp120 inner and outer, 454–455Gro-EL, 418immunoglobulin domain, 77non receptor tyrosine kinases, 240nucleotide binding, 62, 148, 155p53, 471tyrosine kinases, 240
Donnan effect, 333Dopamine, 36Double helix, 247–248Double reciprocal plot,DPPC, see dipalmitoylphosphatidylcholineDrosophila melanogaster, 168, 263, 308Drug resistance, 454Drugs, 205, 228, 231, 451, 453, 454Dsb family, 288
E site, 272, 286Eadie Hofstee plot, 199, 225Earth, 127EcoRI, 221–224
cleavage site, 222DNA distortion, 223major groove binding, 223role of divalent ions in catalysis, 223sequence specificity and hydrogen
bonding, 223

516 INDEX
EcoRI (continued )signature sequence, 224tertiary structure, 223
Ectodomain, 455–456Edelman, G., 76Edman degradation, 165, 340Edman, P., 165EF-G, 275, 279EF-Ts, 275,EF-Tu, 274–275
regeneration, 277structure with Phe-tRNA, 278
EGF, see epidermal growth factorEhlers-Danos syndrome, 99–100Eighteen 18S rRNA, 257Elastase
active site substrate specificity, 212emphysema, 476structural homology, 177
Elastin, 101Electromagnetic spectrum, 348Electron density, 108, 356, 358Electron microscope, 376Electron microscopy, see also cryoelectron
microscopy, nuclear pore complex, 303Electron spin resonance, 390–392
ENDOR, 392ESEEM, 392iron sulfur proteins, 391
Electron, 31Electrophile, 205
in covalent catalysis, 203–205Electrophoresis, 333–340
isoelectric focusing, 339SDS-PAGE, 335–338two dimensional electrophoresis, 339
Electroporation, 315Electrospray ionization, 340Electrostatic catalysis, 205–207Elements, 9ELISA, 337–338Ellipticity, 386Ellman’s reagent, 27, 28, 33Elongation factors, see also EF-G, EF-Tu
and EF-Ts, 274–277Eluate, 327Emphysema, 475–478Enantiomers, 34Encounter complex, 195Endergonic process, 195Endocytosis, 98, 458–459, 459Endoplasmic reticulum, 164
collagen processing, 97–99disulfide bond formation, 288–289hydroxylation reactions, 289–290insulin processing, 287–288lumen, 295, 296, 427translocon/SRP interaction, 424–425unwanted transfer, 426
Endoproteolysis, 469
Endosomes, 460Enediol intermediate, 208Energy transfer, 382, 383Engelman, D., 423Engrailed, 263Enteropeptidase, 237Enthalpy, 54Entropy, 54
activation parameters, 397–398Enzymes, 188–245
allosteric regulation, 231–237catalytic efficiency, 201catalytic mechanisms, 202–209covalent modification, 237–241databases, 192inhibition and regulation, 224–228irreversible inhibition, 227–231isoenzymes, 241–242kinetics, 197–202multicatalytic activities, 305–308nomenclature, 189, 478–479preferential binding of transition state,
207–209purification, 343rotary motion, 146steady state approximation, 199–200subsites, 208
Epidermal growth factor, 101–102, 369Epitopes, 77, 272Equilibrium constant, 194Equilibrium dialysis, 359Ernst, R., 360Error rate, 169Erythrocyte, 110–114
band, 3protein, 111glycophorin, 112–114lipid: protein ratio, 109membrane organization, 110–114
Erythromycin, 278Escherichia coli
ATP synthetase, 144–145cell division, 247competent cells, 315expression host, 313genome, 9K12 strain, 168lac operon, 317met repressor, 261OmpA, 425OmpF, 425phosphofructo-kinase, 232porins, 128–132protein expression, 314protein purification, 343replication of DNA, 253restriction endonucleases from, 222ribosomes, 270RNA polymerase, 254SRP54 M domain, 295
ESR see Electron spin resonanceEthanolamine, 106Eukaryotic cells, 164Eukaryotic RNA polymerases, 257–261
basal transcription factors, 258role and location, 257processing, 257
Evolution, 164EX1 mechanism, 412EX2 mechanism, 412Excited state, 380Exergonic reaction, 195Exocytosis, 98Exon, 267Exonuclease activity, 254Exportins, 300Expression vectors, 316–318Extinction coefficient, see Molar
absorptivity coefficientExtrinsic proteins, 110Eye, damage in glaucoma, 205Eyring, H., 195
F1 complex, 146structure, 147–150
Fab fragments, 76–79Fabry disease, 308FAD, see flavin adenine dinucleotideFamilial amyloidotic neuropathy, 428FAP mutations, 430FAP, see familial amyloidotic neuropathyFasman, G.D., 182FASTA, 175Fatal familial insomnia, 428, 434–435, 465Fatty acids, 105–109Fc, 76Feedback mechanisms, 189Fenn, J., 340Ferredoxin, 128, 299
types of clusters, 391Ferredoxin-NADP reductase, 299Ferrimyoglobin, 68Fersht, A.R., 405Fibrillin, 101–102
modular organization, 102Fibrils, transthyretin based, 430Fibrin, 238–239Fibrinogen, 239
sedimentation coefficient, 324Fibroins, 92Fibrous proteins, 85–103
amino acid composition, 8615N, 253,50S subunit, see large subunit, 279–282First order reaction, 192–194Fischer, E., 2, 1895.8S rRNA, 2575S rRNA, 261, 269
secondary structure prediction, 280

INDEX 517
FK506 binding proteins, 416Flavin adenine dinucleotide, 9, 132–133,
162Flavin, 380Flavodoxin, 181, 359Flu, see influenzaFluid mosaic model, 109–110Fluorescamine, 33Fluorescence, 381–385
emission anisotropy, 383Green fluorescent protein, 383–385instrumental setup, 382Perrin equation, 383profile from DNA sequencing, 171Stern-Volmer analysis, 382collisional quenching, 382Stokes shift, 381
Fluoroscein, 167, 170Flurodinitrobenzene, 33, 37Fo, 145,Folding, see protein foldingFolic acid, 193Folin-Ciocalteau’s reagent, 33Folin–Lowry method, 33Formylmethionine, 273–274Forster energy transfer, 3824.5S rRNA, 294Fourier transform, 354, 360, 389Franck, J., 377Franklin, R., 2, 247Free energy, 195–197
contributions to protein folding, 403transition state profile, 196
Free induction decay, 361–362Fructose-6-phosphate, 232–233FtsY, 295Fumarase, specificity constant, 201Fumaroles, 162Funnel, 411Fusogenic peptide, 455, 457
G protein coupled receptor, 115–121cDNA isolation, 115
G protein, 117–119heterotrimeric, 118Gα structure, 119Gβ/Gγ, 118, 120, 439EF-Tu, 274Ran, 302Ran binding proteins, 302
G1 phase, of cell cycle, 247–248G2 phase, of cell cycle, 247–248GABA, see γ amino butyric acidGAL4, transcription factor, 262–263Galactolipids, 107Gallo, R., 443γ crystallin, 59γ secretase, 469γ turn, 46–48
γ amino butyric acid, 36γ lipotrophin, 289Gamow, G., 266Garavito, R., 228Gaucher’s disease, 310GCN4, transcription factor, 263–264Gel electrophoresis, 333–340
enzyme linked immunosorbent assay, 337instrumentation, 337polyacrylamide SDS, 336two dimensional, 339Western Blotting, 337–338
Gel state, 109Gene fusion, 181Gene,
amyloid precursor protein, 469seven transmembrane helices receptors,
118cdc, 249defective, 441engrailed, 263env,gag,pol, 445–446globin cluster, 442p53 location, 471PNRP, 433, 435screening, 441tumour suppressor, 470secretion, 424
Genetic code, 268in organelles, 268translation of synthetic nucleotides, 267start codon, 169,
Genetic engineering, 222Genomes, 168–169
Haemophilus influenzae, 168Helicobacter pylori, 168pathogens, 395RNA based, 444segmented, 444, 457sequencing projects, 168, 185
Genomics, 183Germ line mutations, 473Gerstmann–Straussler–Scheinker, 434–435GFP, see green fluorescent protein.Ghost membranes, 110Glaesser, R., 376Glaucoma, 205Gln-tRNA synthetase, 220
active site pocket with analogue, 221discrimination between substrates, 220transition state analogue, 221
Globin evolution, 182Globin gene cluster, 442Glucokinase, 200Glucose isomerase, specificity constant, 201Glucose-6-phosphate, 241Glutamate, 25
chemical properties, 25data, 19genetic code specification, 268
helix/sheet propensity, 41optical rotation, 34prevalence hydropathy index, 112prevalence in secondary structure,
180Glutamine, 25–26
chemical properties, 25–26data, 19genetic code specification, 268helix/sheet propensity, 41hydropathy index, 112prevalence in secondary structure, 180
Glutaminyl tRNA synthetase, 220–221Glutathione binding proteins, 334Glutathionine-S-transferase, 334,
SDS-PAGE, 337Glycerol backbone, 105Glycerophospholipid, 106Glycine
chemical properties, 24data, 19genetic code specification, 268helix/sheet propensity, 41hydropathy index, 112prevalence in secondary structure, 180in collagen structure, 94–96location in turns, 47poly(Gly), 48
Glycogen phosphorylase, 231, 241Glycogen storage diseases, 309–310Glycogen, 241Glycolysis, 190Glycophorin, 113
domain labeling, 113–114dimerization, 114
Glycophosphatidyl inositol, see GPI anchorsGlycoprotein
amyloid precursor protein, 469prion protein, 433transmembrane receptors, 239variant surface, 290
Glycosidic bonds, 210, 211cleavage by neuraminidase, 462–463
Glycosylation, 98–99, 290–292adrenergic receptor, 115glycophorin, 113N-linked glycosylation, 98prion, 434
GM1, 439GM2, 310Golgi apparatus, 99, 292, 296gp120
domain organization, 455env gene product, 446function, 447interactions with CD4 receptor, 455location within HIV, 446structure, 449surface glycoproteins of HIV, 454–455vaccine development, 455

518 INDEX
gp41, 90ectodomain region, 456–457env gene product, 446function, 447structure, 449fusogenic sequence, 456location within HIV, 446trimer of hairpins, 457
GPCR, see G protein coupled receptorGPI anchors, 291
in prion protein, 433Gradient gel electrophoresis, 336Gram negative, 128, 319Gram positive, 35, 319Gramicidin A, 36Greek key motif, 59, 61Green fluorescent protein, 383–385GroEL-ES, 417–422
catalytic cycle, 419–421domain movements, 418–419hydrophobic interactions, 420–421negative staining EM, 417tripartite structure, 418
GroES structure, 418Ground state, 380GTP
analogue, 275GTP binding proteins, 295hydrolysis of GTP, 274, 294–295, 378use in capping, 265
Guanidine hydrochloride, 396–397, 400,412
Guanidinium thiocyanate, 326Guanidino, 28Guanine, 247
H subunit (Reaction centre), 124H/D exchange, 413HAART, see highly active retroviral therapyHaemoglobin, 69–74
absorbance spectra, 67allosteric regulation of, 72–74α subunit, 70, 172β subunit, 70, 172Bohr effect, 74conformational change, 71–72cooperativity, 72–73deoxy state, 71evolution of globin chains, 181–182haemoglobin S, 441hydrophobic pocket, 441lamprey protein crystals, 359mechanism of oxygenation, 71–72met form, 67R state, 73sequence homology, 172spin state changes, 71structural homology, 172solubility of, 325
Haemophilus influenzae, 168Hair, see keratinsHairpin, 456–457Haldane, J.B.S., 199Half-chair conformation, 211Half-life, 193–194, 305Haloarcula marismortui, 272Halobacterium halobium, 114Halophile, 114Hanging-drop, 359Haplotype, 442Harker construction, 356–357Hartwell, L., 249Heat capacity, 55
changes in protein foldingHEAT motifs, 301Heat shock proteins, 298, 416Heavy chain, 76Heavy metal derivatives, 357Heisenberg, W., 349Helicases, 253, 258Helicobacter pylori, 168Helix, 41–45
α helix, 41–44other conformations, 45π helix, 44PSTAIRE, 250–251reaction center, 115, 125restriction endonucleases, 222–223
Helix loop helix motif, 263, 264Helix propensity, 41Helix turn helix motif, 64–66
eukaryotic, 263–265Helper T cells, 445, 446Haemagglutinin, 459–462
antigenic subtypes, 465comparison of membrane bound and
protease treated forms, 460conformational changes, 462crystal structure, 461HA1 monomer, 461HA2 monomer, 461low pH forms, 462organization, 460sialic acid binding and the active site,
460–462Heme a, 133, 138Heme a3, 133, 138, 140Heme, 9, 66, 67
complex III, 132–133cytochrome oxidase, 138edge to edge distance, 137
Henderson, R., 114, 376Henderson-Hasselbalch equation, 14,
477–478Hepatitis B, 377Heptad repeat, 87, 89, 263Hess’ Law, 406–407Heteronuclear NMR spectroscopy, 370–373Heterotropic effector, 234
Hexokinase, 191, 200Hierarchy, 180Highly active retroviral therapy, 454Hill coefficient, 231Hill equation, 231HindIII, 222Hippocrates, 228His-tags, 333–334Histamine, 36Histidine
acid catalysis in RNaseA, 203chemical modification in ribonuclease,
202chemical properties, 29–30data, 20general base catalysis, 202–203genetic code specification, 268helix/sheet propensity, 41hydropathy index, 112imidazole ligand in globins, 68imidizolate form in catalysis, 215–217optical rotation, 34prevalence in secondary structure, 180pros and tele, 30reaction with TPCK in chymotrypsin,
213tagged proteins, 333–334Zn fingers, 261
Histone, 250HIV protease, 446, 449–452
bond specificity, 449catalytic mechanism, 450inhibitors, 452structure, 451substrates, 451
HIV see human immunodeficiency virusHLH motif, see Helix loop helix motifHNCA, 373HNCO, 373Hodgkin, D., 353Hofmeister series, 325–326Hofmeister, F., 2, 325Holley, R.W., 268Holoenzyme, 260Homedomains, 264Homeobox, 265Homeostasis, 189Homology, 170–175Homopolymer, 40Homoserine lactone, 27Homotropic effector, 234Hooke’s Law, 388Hormone, 116, 228, 287Horseradish peroxidase, 338, 339Host-guest interaction, 40HTH, see Helix turn helix motifHuber, R., 51, 126, 370Huckel, E., 325Human diseases and disorders, 10–11, 428
Alzheimer’s, 468–470

INDEX 519
amyloidogenesis, 427–431cancer, 10, 470–475cervical carcinoma, 475cholera, 439–440collagen based, 99–100Creutzfeldt-Jakob disease, 433cystic fibrosis, 10, 416–427diabetes, 441emphysema, 475–477familial amyloidotic polyneuropathy,
428–430folding diseases, 428glaucoma, 205HIV, 10, 443–457influenza, 457–465kuru, 433misfolding and disease, 426–435neurodegenerative disease, 441, 465–470p53 based, 470–474prion based diseases, 431–435, 466–468sickle cell anemia, 10 441–442skin cancer, 445
Human Genome Sequencing project, 9Human immunodeficiency virus, 11,
443–457epidemic, 445gene products, 447genome organization, 446gp120, 454–455gp41 structure, 455–457helper T cells, 445history, 444–445HIV-2, 446opportunistic infections, 445protease, 63, 449–452reverse transcriptase, 452–454role of Vpu, Vif, Vpr, 448structural proteins, 448–449structure and function of Rev, Nef, Tat,
447–448surface glycoproteins, 454–457virus, 446
Hybridization, 31Hydration layer, 324Hydrogen bonding, 56
examples in proteins, 56helices, 41–44in β sheets, 45–46restriction endonuclease specificity, 223substrate discrimination in tRNA
synthetases, 220Hydrogen exchange, 379Hydrolases, 191, 308Hydrolysis, 202
bromophenol esters, 206–208of acyl enzyme intermediate, 214–215of ATP,152of NAG-NAM polymers, 211
Hydropathy index, 112Hydropathy plot, 111–113
Hydrophobic effect, 53–55energetics, 54folding, 410in chromatography, 331interactions in proteins, 54–55, 403,
410–411Hydrophobic interaction chromatography,
331Hydroxylation, 29, 289–290Hydroxylysine, 97, 289–290Hydroxyproline, 96, 289Hyperbolic binding curve, 69, 232Hyperthermophile, 306Hypervariable regions, 77
Iatrogenic transmission, 433, 467Ibuprofen, 228Ice, 54IF-1, 275IF-2, 274, 275IF-3, 274, 275IgG, 76–80Imidazole, 29, 30
catalytic mechanism of triose phosphateisomerase, 216–217
groups in nucleophilic reactions, 203increased basicity, 214ligands in active site of carbonic
anhydrase, 206Immune response, 74–76Immunity, 76Immunoblotting, 337–339Immunoglobulins, 74–81
CDRs, 77classes, 78–80disulfide bridges, 76heavy and light chains, 74hypervariable regions, 77immunoglobulin fold, 77interaction with lysozyme, 78–79monoclonal antibody, 79structure, 76–78VL and VH domains, 77
Importins, 300–302interaction between α and β 301schematic representation, 300
Indole, 32Influenza, 457–465
avian strains, 465classification, 458entry into cells, 459epitopes, 464genome, 458haemagglutinin, 459–462Hong Kong strain, 465neuraminidase, 462–464segmented RNA genome and coding of
proteins, 458organization, 457
strategies to combat influenza pandemics,464–465
symptoms, 457virus, 457
Infrared spectroscopy, 387–389amide bands, 389fingerprint region, 389regions of interest, 388spectrum of water, 388
Infrared, 348frequency range and measurement,
349Ingram, V., 10, 441Inhibition, of enzyme activity, 224–228Initiation factors, 274–275INK4 family, 252Inositol, 106Insulin, 6,
disulfide bridge formation, 287proteolytic processing, 287expression, 318
Integral proteins, 109–110Integrase, 445Interleukin-1β, structure, 374–375Intermediate filaments, 88–89
classification of, 89Intermembrane space, 298, 299Interphase, 247Intracellular messengers, 116–119Intrinsic pathway, 239Intrinsic proteins, 109–111Introns, 265–267Invariant residues, 39Iodide, 381Ion exchange chromatography, 329–330Ion pumps, 152–156Ion, solvation, 324–325Ionic atmosphere, 325Ionic strength, 324–325IPTG, see IsopropylthiogalactosideIron sulfur protein, 132
ESR spectra, 391Iron, 7, 8, 58
observation by ESR, 391Islets of Langerhans, 441Isoalloxine, 9Isoelectric focusing, 339Isoelectric point, 14Isoenzymes, 205, 241–242Isoleucine
chemical properties, 24data, 20genetic code specification, 268
helix/sheet propensity, 41hydropathy index, 112occasional use as start codon, 268optical rotation, 34prevalence in secondary structure,
180Isomerases, 191

520 INDEX
Isomorphous, 26Isoprenoid units, 134Isopropylthiogalactoside, inducer, 317Isozymes, 205 see also isoenzymesIUPAC, 107,Ivanofsky, D., 442Iwata, S., 132
Jakob, A., 433Jelly roll motif, 61Jencks, W.P., 205Jenner, E., 75Jerne, N., 75
Kanamycin, 316Kaposi’s sarcoma, 445Karplus Equation, 363
kcat /Km see specificity constant, 201KDEL sequence, 296Kendrew, J., 2, 358Keq , (equilibrium constant), 194
in oxygen binding, 69Keratins, 86–92
acidic, 88α-keratin, 86–87basic, 88β-keratin, 88coiled coils, 87–89Distribution of type I and II, 92mutations in, 91structural organization, 89
Kevlar, 92Khorana, H.G., 422Killer T cells, 79Kinase, 172Kinetic analysis, 192–195
of protein folding, 405heterogeneity, 414
Kinetic techniques, 413Kinetics, 192–195Klenow fragment, 254, 452Klug, A. 376Km, Michaelis constant, 200kobs , 195Kohler, G., 75Kosmotropes, 326Kuru, 432, 433, 465
L isomers, 34–35L subunit (reaction centres), 124–125Lac operon, 317Lac repressor, 263, 317Lactate dehydrogenase, 61, 191, 241–242
clinical diagnosis, 242different isoenzymes, 242sedimentation coefficient, 324
Lactocystin, 308Laevorotatory, 34λ-repressor, 59, 65
Lane, D.P., 470Large (50S) ribosomal subunit, 279–282
peptidyl transferase mechanism, 283sedimentation coefficient, 324structure of globular proteins, 282tertiary structure, 281transition state analogue, 280, 28223S rRNA structure, 280
Lariat structure, 267Larmor frequency, 361–362Lathyrism, 36Lauryl, 107LDAO, 121Lecithin, 105Leder, P., 267Lentivirus, 448Leonard-Jones potential, 57Leucine, 24
chemical and physical properties, 24data, 20genetic code specification, 268helix/sheet propensity, 41hydropathy index, 112optical rotation, 34prevalence in secondary structure, 180
Leucine zipper, 87, 263in DNA binding, 264
Leucocyte, 476Levine, A.P., 470Levinthal’s paradox, 403–404, 410Lewis acid, 224Lewis, G.N., 325Li-Fraumeni syndrome, 441, 473–474Ligands, 262
in affinity chromatography, 334in bacterial reaction center, 125in reverse phase chromatography, 332
Ligases, 191, 254Ligation, 315Light (L) chain, 76Light harvesting chlorophyll complexes,
121, 127, 299Light, 118Linderstrom-Lang, K., 412Lineweaver-Burk plot, 199
inhibition, 225–226Linoleic acid, 107Linolenic acid, 107Lipid,
phase transition temperatures, 109protein ratio, 109fluidity, 109–109bilayer, 108
Lipid:protein ratios, 109Lipscomb, W., 234Liquid crystalline state, 110Long terminal repeats (LTRs), 446Longitudinal relaxation time, 363Loop-sheet polymerization, 476–477Lumen, 296, 303
Luminescence, 384Lyases, 191Lymphocytes, 75–76, 443
B cells, 75CD4 bearing, 446
Lysinechemical properties, 28–29cofactor binding in bacteriorhodopsin,
114crosslinks, 96–97data, 20genetic code specification, 268helix/sheet propensity, 41hydropathy index, 112hydroxylation, 97, 289–290in collagen structure, 96–97oxidation of side chain, 97prevalence in secondary structure, 180
Lysogenic phase, 64Lysosome, 296, 308–310Lysozyme, 79
catalysis, 209–212cell disruption, 319folding domains, 415glycosyl intermediate, 212interaction with antibody, 78–79neutron diffraction, 379specificity constant, 201systemic amyloidogenesis, 428tertiary structure, 210
Lysyl hydroxylase, 290Lysyl oxidase, 97Lytic phase, 64
M subunit, 124–125Macrophages, 445, 454Mad cow disease, 466Magnesium, 221, 271
in cytochrome oxidase, 139–140Magnetic moment, 391Magnetic nuclei, 360Magnetogyric ratio, 360Malaria, 442Malate synthase, 372MALDI-TOF, 341–343Malonate, 226–227Maltose binding protein, 334Manganese, 193Marfan’s syndrome, 100–102Martin, A.J.P., 326Mass spectrometry, 340–343Matrix protein, 446, 447
structure, 448in influenza virus, 458
Matrix protein, 446–449, 457Mdm2, 475Measles, 442Membrane proteins, 105–159
bacteriorhodopsin, 423–424

INDEX 521
crystallography, 119–123expression, 314, 344folding, 422–426integral, 110,OmpA, 424peripheral, 110porins, 128–132respiratory complexes, 132–144topology, 110two stage model, 423with globular domains, 111–114
Membranes, 105–110fluid mosaic model, 109–110ghosts, 110molecular organization, 105–108
Memory cells, 75Menaquinone, 125Menten, M., 198
meq , dependence of �G on denaturantconcentration, 400–402, 405
table for selected proteins, 401Mercaptoethanol, 27Meselson, M., 254Meselson-Stahl experiment, 254Messenger RNA capping, 265
methylaton, 265viral, 443plus (+)strand, 443
Metal ion catalysis, 204–205Metal ions, 6, 8,
activity of restriction endonucleases, 221,223
carbonic anhydrase, 205 ribosomeassembly, 271
catalysis, 204–205co-factors, 192, 193
Metalloenzyme, 193, 204Metalloproteinase, 99, 136, 469Metarhodopsin II, 118Metazoa, 133Methanococcus jannaschii, 417Methanol, 55Methionine, 26–28
alternative genetic code, 268chain initiation, 273–274chemical and physical properties,
26–27data, 21genetic code specification, 268helix/sheet propensity, 41hydropathy index, 112interactions with heavy metal derivatives,
26prevalence in secondary structure, 180reaction with cyanogens bromide, 27side chain oxidation, 26SRP54, 294
Methylation, 29, 292by restriction endonucleases, 221lysine sidechains, 28
RNA transcripts, 2657-methylguanylate, 265
Metmyoglobin, 68Micelles, 107Michaelis, L., 198Michaelis-Menten equation, 199Michel, H., 120, 126Microfibril, 88, 89, 101Microwaves, 348
frequency range and measurement, 349use in ESR, 391
Miller, S., 162Milstein, C., 75Mitchell, P., 145Mitochondria, 132–144
complex I, 132–133complex II, 133cristae, 145cytochrome bc1 complexes, 132–137cytochrome oxidase, 138–144inner mitochondrial membrane
translocase system, 298inter-membrane space, 133lipid:protein ratio, 109matrix, 133outer-membrane, 133proteases, 298respiratory complexes, 132–144RNA polymerases, 257targeting, 297–299
Mitosis, 247–248Mixed disulfide, 28Molar absorptivity coefficient, 32,Molar extinction coefficients, 30–31, 385
globins, 67Molecular evolution, 161–165, 189Molecular graphics, 52, 478Molecular mass, 7Molecular medicine, 441Molecular orbitals, 31
theory, 380Molecular saddle, 259–260Molecular weight, see molecular massMolten globule, 409–410Molybdenum, 7, 58, 193
in sulfite oxidase, 181Monochromators, 355, 404Monod, Wyman and Changeaux (MWC)
model, 71Montagnier, L., 443Moore, P., 272Moore’s Law, 185Motifs, 173mRNA see messenger RNAMulder, G. J., 1Multiple anomalous dispersion, 357Mumps, 442Mutagenesis, 307, 406–408Mutational hot spots, 474Myelin sheath, 109
Myoglobin, 67–69absorbance spectrum, 67evolution of globins, 181–182human and sperm whale, 40oxygen binding curve, 69sedimentation coefficient, 324structure, 67superposition of polypeptide chains, 70
Myristoylation, 240, 292of Nef, 447
Myxathiazol, 134,135
N acetyl glucosamine, 209–210, 321N acetyl muramic acid, 209–210, 321N peptide, 456N terminal nucleophile hydrolases, 303N terminal see Amino terminalNa+-K+ ATPase, 153–155
reaction stoichiometry, 153subunit composition, 153transport of ions, 154use of cardiac glycosides, 154
N-acetyl neuraminic acid, 462NAD+, see Nicotinamide adenine
dinucleotideNADH/NADPH, 132NADH-ubiquinone oxidoreductase,
132–133NAG, see N acetyl glucosamineNAM, see N acetyl muramic acidNathans, D., 221N -bromosuccinimide, 32NcoI, 222, 317NdeI, 317Nef, 446–448Nestin, 87Neuraminidase, 61, 462–464
active site, 464crystal structure, 463enzyme activity, 462tetrameric state, 463
Neurodegenerative disease, 441, 465–470Alzheimer’s, 468–470BSE, 466Lou Gehrig’s disease, 441scrapie, 466vCJD, 467
Neurotoxicity, 212Neurotoxins, 212Neutron diffraction, 379Nevirapine, 454Niacin, 193Nicolson, G., 109Nicotinamide adenine dinucleotide, 9, 162,
193, 439–44019S regulatory complex, 308Ninhydrin, 33, 167Nirenberg, M., 267Nitrate reductase, 173, 181

522 INDEX
Nitrocellulose, 337, 339Nitrothiobenzoate, 27Nitrotyrosine, 32N-linked glycosylation, 290NMR structures, BUSI IIa, 370
interleukin 1β, 375tendamistat, 370thioredoxin, 375
NMR, see Nuclear magnetic resonancespectroscopy
N -myristoyltransferase, 292Nobel Prize, 2, 4–5, 126, 340NOE, see also Nuclear Overhauser effect,
364regular secondary structure, 369
Non steroidal anti-inflammatory agents, 230Non Watson-Crick base pairs, 268Non-competitive enzyme inhibition,
226–227Non-competitive inhibition, 226,Non-covalent interactions, 403Non-nucleoside inhibitors, 454Nonsense codons, 268Nostoc sp., 164NSAIDs, see non steroidal
anti-inflammatory agentsNTN hydrolases, see N terminal nucleophile
hydrolasesNuclear envelope, 299,Nuclear localization signals, 300Nuclear magnetic resonance spectroscopy,
360–37513C chemical shifts for amino acid
residues, 37215N 2D connectivity patterns, 3681H chemical shifts for amino acid
residues, 366amide exchange, 411–412assignment problem, 364–370chemical shifts for amino acid residues,
371COSY, 368generating protein structures, 373–375heteronuclear methods, 370–373instrumentation, 365magnetogyric ratio, 360national facilities, 392NOESY, 368nuclear Overhauser effect, 364, 373–374practical biomolecular NMR, 364relaxation, 363–364spectra, 367, 371structure calculations in, 374–375,TOCSY, 367–368triple resonance experiments, 373
Nuclear Overhauser effect, 364Nuclear pore complexes, 302–303Nucleation,
crystal formation, 359helix formation, 409
Nuclei, 360Nucleocapsid, 443Nucleocytoplasmic transport, 299–302
importin α structure, 301importin β structure, 301involvement of Ran binding proteins, 302localization signals, 300pores, 299, 302–303Ran-GDP structure, 302
Nucleophile, 32, 202–205Nucleoplasmin, 299–300Nucleoporins, 302–303Nucleoprotein, 457Nucleoside analogues, 453Nucleoside inhibitors, 453Nucleotides, 169Nucleus, 299Nups, see nucleoporinsNurse, P., 249
O linked glycosylation, 290–291Octapeptide repeat, 434, 436ODAP see β-N -oxalyl L-α, β-diamino
propionic acidODCase, see Orotidine 5-monophosphate
decarboxylaseOkazaki fragments, 254–255Oleic acid, 107Oligomerization domain, 473Oligomycin, 145OmpA, 424OmpF, 129–131Oncogene, 470Oncoprotein, 250Operon, 317Opiomelanocortin, 288
processing of, 289Opsin, 117Optical isomers, 34Optical rotation, 34Optical spectroscopy, 379–390
absorbance, 381–385absorbance spectra, 67, 381circular dichroism, 385–387fluorescence, 381–385fluorescence polarization, 382–383Raman, 389–390
Orbitals, 31, 380Organization, 37Orientation effects, 206–207Orotidine 5-monophosphate decarboxylase,
205Osteogenesis imperfecta, 100, 428Osteoporosis, 99Oubain, 154Oxidoreductases, 191Oxygen, 8,
binding site in cytochrome oxidase, 143binding to myoglobin, 69
binding to hemoglobin, 69oxidation of methionine side chains,
26production from water splitting, 126
Oxygenic photosynthesis, 126–128Ozone, 32
P site, 273p17, 446–449P22 tail spike protein, 61p51, see reverse transcriptasep53, 11, 265, 470–475
crystal structure, 472–473domain structure, 472function, 471Li Fraumeni syndrome, 473–474linked to cancer, 474malfunctions, 474mutation, 474organization, 471primary sequence, 472regulation, 474smoking, 474transcription factor, 265
p66, see reverse transcriptaseP870, 121PALA, 235–236Palindromic sequence, 222Palmitic acid, 107Palmitoyl transferase, 292Palmitoylation, 240, 292PAM matrices, 174Pancreas, 237, 287, 441Pandemics, 443, 445, 457Papain, 76Para substitution, 31Paracoccus denitrificans, 138, 140
cytochrome oxidase, 141, 144Parvalbumin, 415Parvulins, 416Pasteur, L., 75Pauling, L. 14, 41, 67Pavletich, N., 472pBR322, 316p-bromophenylacetate, 206PCR, see polymerase chain reaction,PDI see protein disulfide isomerase,Penicillin, 228Pepsin, 237, 287
catalytic mechanism, 450specificity constant, 201structure, 449–450
Peptide bond formation, 16–17cis/trans isomerization, 23ribosome, properties, 17–23
Peptide fragments, 166Peptidoglycan layer, 320, 321Peptidomimetic, 451Peptidyl proline isomerase, 415–416

INDEX 523
Peptidyl transferase, 273, 275inhibition of, 280mechanism, 282
Periodic table of elements, 8Peripheral proteins, 110Periplasm, 121, 288, 320Perrin equation, 383Pertussis toxin, 440Perutz, M.F., 2, 71, 352, 356, 358PEST sequences, 305pET vector, 319PFK, see phosphofructokinasePGH synthase, 229pH, 14–15Phase angle, 353–354Phase problem, 356–357Phenylalanine
absorbance spectrum, 381chemical properties, 31data, 21DNA distortion, 259genetic code specification, 268helix/sheet propensity, 41hydropathy index, 112mutation in CFTR gene, 427optical rotation, 34π-π interactions, 31secondary structure preference,
180spectroscopic properties, 30
Phenylalanine ammonia lyase, 191Phenylisothiocyanate, 165–166Pheophytin, 121, 124Phe-tRNA, 278φ angle, 41–44Phillips, D., 2102-phosphoglycolate, 209, 233Phosphatase, 258, 290Phosphatidylcholine, 105Phosphatidylethanolamine, 105Phosphatidylglycerol, 105Phosphatidylinositol, 105, 291Phosphatidylserine, 105Phosphodiester bond, 267
cleavage, 224Phosphodiesterase, 118Phosphoenolpyruvate, 233Phosphofructokinase, 231–234
domain organization and movement,232–233
regulation by ATP/ADP, 232–233transition state analogue, 232
Phosphorylase a, 241Phosphorylase b, 241Phosphorylation, 153–154
as post translational event, 290enzyme, 241
Phosphoserine, 25, 290in glycogen phosphorylase, 241
Phosphotyrosine, 25, 241, 290
Photobleaching, 115Photocycle, 115Photon, 121Photosynthetic reaction centres, 119–128
bacterial reaction center, 123–126chromophores, 121cytochrome subunit, 125electron transfer pathway, 122, 125fractionation with detergents, 121H subunit, 123–124kinetics, 122L and M subunits, 124−125organization, 121photosystems I and II, 126–128special pair, 121, 125structure, 123–126
Photosystem I, 126–127Photosystem II, 126π helix, 43–44pI, see isoelectric pointPichia pastoris, expression system,
314Pigment, 115Ping-pong mechanism, 215Pituitary, 288pK, 14–15Planck’s constant, 196, 348Planck’s Law, 348Plaques, 431, 432, 434Plasma membrane, 109, 164, 320Plasmids, 315–316Plasmodium falciparum, 442Plastocyanin, 59, 299, 369Pneumonia, 445, 457Polarization, 382–383Polyacrylamide gel electrophoresis,
335–338polymerization reactions, 335
Polyalanine, 40Polymerase chain reaction, 169–170Polymorphism, 442Polypeptide, 39–50
cleavage by enzymes, 166–167folding kinetics, 408–410hydrolysis, 167permutations, 40
Polyprotein, 289, 445, 448,449Polysaccharide coat, 319Pompe disease, 308Porins, 128–132
biological unit, 130channel selectivity, 128constriction site, 130folding properties, 424hydrophobic residues, 129OmpA, 424OmpF, 129–130PhoE, 130secondary structure, 129trimeric unit, 130
Porter, R., 76Post translational modification, 287–293
covalent modification, 237–241Ppi, see peptidyl proline isomerasePre-steady state, 200Prebiotic synthesis, 161–163
demonstration of, 162yields of biomolecules from, 162
Precipitation, 343Pre-initiation complex, 258–260Preproprotein, 287, 402Preproteins, 287
N terminal sequences, 293Presenilins, 470Pribnow box, 64, 256Primary sequence, 39–40
cytochromes c, 175haemoglobin, 172myoglobins, 40unknown, 167ubiquitin, 304
Primase, 254Primers, 169–170Primosome, 254–255Prion, 431–435
cellular, 433–434glycosylation sites, 434knockout mice, 435mutations of PNRP gene, 435primary sequence, 434scrapie, 433–434structural properties, 433structure, 434
Prokaryotic cellsProline chemical properties, 29
cis-trans isomerization, 412–415collagen structure, 94–96data, 21genetic code specification, 268globin helices, 68helix/sheet propensity, 41hydropathy index, 112hydroxylation, 96, 289location in turns, 47peptidyl isomerases, 415–416poly(Pro), 48prolyl hydroxylase, 289pyrrolidone ring, 94, 289transmembrane helices, 124
Promoter, 256inducible, 317prokaryotic, 64–66
Proof reading, 169, 275Propranolol, 116Pros, 30Prostaglandins, 228–239Protease inhibitors, 451–452, 476Proteasome, 305–308
heptameric ring structures, 306multiple catalytic activities, 305

524 INDEX
Proteasome (continued )mutagenesis, 30719S regulatory complex, 308regulation of activity, 308structural organization, 309subunit structure, 307three dimensional structure, 307
Protein crystallization, 358–360Protein DataBank, 50–51, 105, 180, 347Protein databases, 180–181Protein degradation, lysosomal, 308–310
proteasome, 305–308ubiquitin, 305
Protein disulfide isomerase, 288–289in vivo protein folding, 416sorting pathway, 296
Protein evolution,convergent, 179gene duplication, 181rates of, 178
Protein expression, 313–318Protein folding, 395–438
ab initio, 185–186amide exchange, 411–412barnase, 405–408Brønsted equation, 407databases, 180denaturant dependence, 405diversity, 161–187energy landscapes, 411GroEL-ES mediated, 417–422Hess’ Law, 406–407in vivo, 415–422intermediates, 415kinetics, 404–406membrane proteins, 422–426misfolding and disease, 426–435models, 408–411molten globule, 409–410motifs, 173stopped flow kinetics, 404structure prediction, 186techniques, 413temperature dependence, 400
Protein purification, 313–346affinity chromatography, 332–333centrifugation, 320–323chromatography, 326–333dialysis, 333electrophoresis, 333–340ion-exchange chromatography, 328–329reverse phase methods, 331–332salting-out, 323–326size exclusion chromatography, 330–331ultrafiltration, 333
Protein solubility, 325Protein sorting and targeting, 293–303
chloroplast, 299mitochondrion, 297–299nuclear targeting, 299–302
Protein stabilityconformational stability of proteins,
402contributions to free energy, 403factors governing, 403
Protein synthesis,chain initiation, 273–274elongation, 274–277molecular mimicry in, 277outline of reaction, 273role of soluble factors, 275termination, 277–278
Protein turnover, 303–310half-lives, 303–304
Protein/lipid ratio, 108Proteinase K, 467Proteinase, see proteaseProteins,
cyclic, 81estimation of, 33function, 5–6isolation and characterization, 313size, 6
Proteolysis, 304limited proteolysis, 237–239
Proteolytic processing, 287Proteomics, 184, 339Protofilaments, 88, 89, 431Proton pumping, 114–115, 142–144Protonation, 30Protoporphyrin IX, 9, 66–67Proximal histidine, 68Prusiner, S., 433Pseudomonas aeruginosa, 168ψ angle, 41–44PTC, see phenylisothiocyanatePterin, 9Purcell, E., 360Puromycin, 278, 279, 282Purple membrane, 114Pyridoxal phosphate, 29, 193Pyrimidine biosynthesis, 207, 234Pyrrolidine ring, 16, 21, 29
Q cycle, 135Quality control, 296Quantum mechanics, 379Quantum yield, 30Quaternary structure of proteins, 62–81
hemoglobin, 70aspartate transcarbamoylase, 235
Quench cooling, 355, 377Quenching, 381–382Quinone, 105, 124–125,
R factor, 358R group, 23–32
properties, 18–22R state, 71, 235
R/S isomers, 24, 34, 38Racker, E., 114Radiation, 348Radio waves, 348Ramachandran plot, 48, 186Ramachandran, G.N. 48Ramakrishnan, V., 283Raman spectroscopy, 389–390Raman, C.V., 389Ran binding proteins, 302Ran, 302Random coil, 396Rapoport, T., 424Rate constant, 192
first order, 193pseudo-first order, 195second order, 195
Rayleigh scattering, 389RCF, 322Reaction centres, type I and type II, 128Reaction controlled rate, 195Reaction coordinate, 195–196Reaction rate, 194
enhancement of, 202–209Reactive center loop, 476–477Receptor, 115
acetylcholine, 377adrenergic, 115bacterial, 295chemokine, 454genes, 119hormone binding, 119seven transmembrane, 115tyrosine kinases, 239–240SRP54, 295
Recognition helix, 263Recombinant DNA technology, 313–318Red blood cell, see erythrocyteRedox reactions, 193
and metal ion catalysis, 203Reiske protein, 126, 133–137
targeting sequence, 299Relaxation times, 363–364Release factors, see RF-1, RF-2 RF-3Replication origin, 316, 318Repressor, see also DNA binding proteins,
64–66Residue, 17Resins, 326
polystyrene, 332Resolving gel, 336Resonance, 23, 28Respiratory complexes, 132–144Restriction endonucleases, 221–224
cleavage sites, 315multicloning sites, 317signature sequence, 224table of selected enzymes with cleavage
sites, 222type II in cloning, 314

INDEX 525
Retinal, 114isomerase, 11811-cis, 117–118trans, 117–118
Retinol binding protein, 428,429Retrovirus, 443Rev, 446–448Reverse phase chromatography, 331–332Reverse transcriptase, 452–454
AZT binding, 453p51 crystal structure, 453p66 crystal structure, 452
RF-1, 278RF-2, 278RF-3, 278Rheumatoid arthritis, 99Rhodamine 6G, 170Rhodobacter viridis, 120Rhodopsin, 117–119Riboflavin, 193Ribonuclease, 6, 288
active site chemistry, 202–203protein folding, 402–403sedimentation coefficient, 324structure, 203
Ribonucleoprotein, 265, 458Ribosomal RNA, 257, 261Ribosome, 269–287
A site, 273affinity labeling and footprinting, 279anatomical features, 271assembly, 271complex with SRP, 293composition of prokaryotic, 269–271,composition of eukaryotic, 269–270E site, 272, 28618S rRNA, 257electron microscopy of, 2725.8S rRNA, 2575S rRNA, 269Haloarcula marismortui, 272large and small subunit, 282–287mechanism of protein synthesis, 208–282P site, 273,peptidyl transferase site, 273–275RNA recognition motifs, 270role of antibiotics, 278–279sedimentation coefficient, 32416S rRNA, 269soluble factors interacting with, 275surface epitopes, 272Thermus thermophilus, 272transition state analogue, 28228S rRNA, 25723S rRNA, 269,
Ribosylation, 439Ribozymes, 162, 192, 242, 287Ricin, 228RNA binding protein, 271RNA hydrolysis, 202
RNA polymerase, 64T7 enzyme, 256eukaryotic, 257
RNA recognition motif, 270conserved sequences, 271
RNA transcript, 265RNase A, see ribonucleaseRNase H, 452–453Roeder, R.G., 259Rontgen, W., 349Rossmann fold, 58, 148, 180, 218, 220, 378Rossmann, M. 60Rotamer, 43, libraries, 49–50Rotary motion, 146Rotational states, 380rRNA, see ribosomal RNARS (Cahn Ingold Prelog) system, 34
S (Svedburg), 323S phase, of cell cycle, 247Sabatini, D., 293Saccharomyces cerevisiae,
expression system, 314genome sequencing, 9secretion mutants, 424
Salicylate, 230Salt bridges, in chymotrypsin, 55Salting in, 323–326Salting out, 323–326Sanger, F., 169Saquinavir, 452Sarcoplasmic reticulum, 155
Ca2+ transport, 155–156lipid:protein ratio, 109
Saturation curve, 198, 325Scalar coupling, 363Scattering, 351Schiff base, 29, 203Schizosaccharomyces pombe, 249Scissile bond, 212Scrapie, 433–434, 466Scurvy, 289SDS PAGE, 111, 335–337
identification of vCJD, 467purification using, 344principles of, 335–336use with mass spectrometry, 342
SDS, use in protein folding, 396, 423Sea urchin, 248Sec61, 295, 424–426Second messengers, 119Second order reaction, 195Secondary structure, 37–50
additional, 48α helix, 41β strand, 45dihedral angles, 43other helical conformations, 44prediction, 181–183
Ramachandran plot, 48regular NOEs and distances, 369supersecondary, 58translation distance, 43turns as elements of, 46
Secretory granules, 287Sedimentation coefficient, 323–324
identification of rRNA, 269Sedimentation velocity, 323Selectable markers, 318Selection rules, 380Selenomethionine, 357Semi-conservative model, 253Semiquinone, 134Sequence homology, 170–176
c type cytochromes, 175cro repressor, 66
Sequence identity, 222Sequencing, protein, 165–168
DNA, 168–171Serine
active site serine in cyclo-oxygenases,230
catalytic triad, 25, 177, 179, 212–213chemical properties, 25covalent catalysis by, 214genetic code specification, 268helix/sheet propensity, 41hydropathy index, 112lipid headgroup, 106location in turns, 47nucleophile, 25, 214O linked glycosylation, 290–291phosphorylation, 25proteases, 212–215secondary structure preferences, 180
Serine carboxypeptidase II, 179Serine proteases, 25, 177, 212–215
active site, 212catalytic mechanism, 214–215catalytic triad, 212convergent evolution, 179inactivation by DIPF, 213reaction of chymotrypsin with TPCK,
213studied using neutron diffraction, 379superposition, 177
Serpinopathies, 478Serpins, 224, 2387S rRNA, 294Seven transmembrane helices, 115SH2 domains, 241SH3 domains, 172, 241, 433
formation of amyloid fibrils, 431Shine-Dalgarno sequence, 273–274,
317SI units, 477Sialic acid, 458, 464Sickle cell anemia, 9, 441–442
haplotypes, 442

526 INDEX
Sigler, P., 417Sigmoidal binding curve, 69, 231, 232, 234Signal hypothesis, 293Signal peptidase, 295–296Signal peptide, 288, 293
SRP54 binding, 294Signal recognition particle, 293–295
E.coli homologue, 294pathway, 293receptor structure and function, 295signal peptide, 293Signal sequence peptidase, 293, 295–296SRP54, 294–295structure and organization, 294translocon interaction, 424–426
Signature sequence, 224Silk, 86, 92–93Silver staining, 337Simian immunodeficiency virus, 447, 456Sinapinic acid, 341Singer, S.J., 109Single particle analysis, 377Singlet, 380SIV, see simian immunodeficiency virus16S rRNA, 269
complementarity, 318helix44, 279the 530 loop, 279,helix34, 279secondary structure, 284tertiary structure, 284surface location of ends of molecule, 272
Size exclusion chromatography, 330–331Skehel, J., 459Sleeping sickness, 290SmaI, 222Small subunit (30S), 282–286
anatomical features, 284decoding process, 286functional activity, 285–287IF-3 binding, 283protein composition, 284–28616S rRNA structure, 284
Smallpox, 75, 442Smith, H.O., 221Smoking, effects on p53, 474
emphysema, 478SN2-type reaction, 211snRNPs, 265Sorting, see protein sorting and targetingSouthern Blotting, 337Southern, E.M., 337Soyabean lipoxygenase, 44Spanish flu, 457Special pair, 121Specificity constant, 201Sphingolipids, 107Spin multiplicity, 380Spin state, 71Spin-forbidden transitions, 380
Spin-lattice relaxation time, 363, 391Spin-spin relaxation time, 363–364Splice sites, 266Spliceosome, 265–267
branch point, 266composition, 265consensus sequence, 266lariat structure, 267
SR Ca2+ ATPase, 155–156Src homology domains, 172SRP, see signal recognition particleSRP54, 294–295Stacking gel, 336Stahl, F., 254Stanley, W.M., 443Staphylococcus aureus, 35, 130
hemolysin structure, 130–131Start codon, 268, 273–274Start sequences, 426–427Steady state approximation, 199–200Stearic, 107Steitz, T., 272Stem loop structure, 269Stereochemistry, 15–16, 34–35Stereogenic, 35Stern-Volmer equation, 382Steven, A.C. 377Sticky ends, 222, 314Stigmatellin, 135, 137Stoeckenius, W., 114Stokes Law, 323, 363
radius, 330Stokes shift, 381Stop codons, 268, 318Stop transfer sequences, 426–427Stopped flow analysis, 404–405
double mixing experiments, 415Streptomycin, 278Stroma, 128, 299Structural homology, 175–177Structure factor, 355Subsites, 208, 212Substitution reactions, 32Substrate discrimination, 220Subtilisin, 179, 402Succinate dehydrogenase, 226Succinate, 227Sulfation, 292Sulfite oxidase, 58, 181Sulfolipids, 107Sulfonamide, 205Sulfone, 26Sulfoxide, 26Sumner, J., 358Super secondary structure, 37, 58Superconducting magnet, 364Superfamily, 180Superoxide dismutase, 193,SV40 T antigen, 299Svedburg constant, 323
Svedburg, T., 323Swiss roll motif, 61, 460Symmetry-forbidden transitions, 380Symport, 154Synchotron, 357Synechococcus sp., 164
photosystem structure, 126Synge, R.L.M., 326Syrian hamster, 434
T cells, 75, 443, 445T loop, 251T state, 71T1, 363T2, 363–364T4 DNA ligase, 191T7 RNA polymerase, 256, 313
structure, 256Tanaka, K., 340Tanford, C., 396Taq polymerase, 169Targeting, see protein sorting and targetingTat, 446–448TATA binding protein (TBP), 258–261TATA box, 64, 256
recognition by transcription factors, 258Tautomerization, 202Taxol, 378–379Taylor, K., 376Tay–Sachs disease, 309–310, 428, 441TCA cycle, see citric acid cycletele, 30Template, 169Tertiary structure, 50–62
globular proteins, 52hydrogen bonding, 56hydrophobic effect, 53–55reformation, 402stabilizing interactions, 53–58van der Waals interactions, 56–58
Tetracyline, 315Tetrahedral intermediates, 214Tetramerization domain, 473Tetrapyrrole,TFIIA, 258–261TFIIB, 258–261TFIID, 258–261TFIIE, 258–261TFIIF, 258–261TFIIH, 258–261TFIIIA, 173, 262Thermal denaturation, 97Thermoplasma acidophilum, 306
thermosome, 422Thermosome, 422–423Thermus aquaticus, 169
Ffh subunit, 295M domain, 295
Thermus thermophilus, 272

INDEX 527
Thiamine, see vitamin B1Thiol, 26–28, 288
formation of mixed disulfides, 27ionization, 27
Thiolate, 27Thioredoxin, 59, 369, 375, 401
conserved motif, 41630S subunit, see also small subunit,
282–286Thomson, J.J. 340Threading, 185310 helix, 43–44
in glycogen phosphorlase, 241Threonine
[2S-3R], 35chemical properties, 25data, 22genetic code specification, 268helix/sheet propensity, 41hydropathy index, 112O linked glycosylation, 290–291optical rotation, 34phosphorylation in cdk, 251–252secondary structure preference, 180use as nucleophile in catalysis, 303
Thrombin, 212, 238–239Thylakoid membrane, 128, 299Thymine, 247Thyroxine, 36, 428TIM, see triose phosphate isomeraseTm, see transition temperatureTobacco mosaic virus, 443Topogenic sequences, 426–427Topoisomerases, 253Torsion angle, 41, 48–49Tosyl-L-phenylalanine chloromethylketone,
211, 228Toxin, 228TPCK, see tosyl-L-phenylalanine
chloromethylketoneTrace elements, 7trans ring, 419Transaminase, 193Transcription factors, 258
basal components, 258–261structures, 261–265TBP, 258–261
Transcription, 254–265control in prokaryotes, 64–65cI repressor, 64cro repressor, 66terminators, 318
Transducin, 118–119Transfer RNA (tRNA), 267–270
acceptor stem, 268–269anticodon, 269binding to amino acyl tRNA CCA region,
220, 270D arm and loop, 269discrimination against, 286
structure, 267–269synthetases, 220variable loop, 268–269
Transferases, 191Transformation, 315Transition metals, 7, 26, 193Transition state analogue, 209, 221, 233,
282Transition state, 195–197
bond energies, 207preferential binding to enzymes, 207–209two stage reactions, 197
Transition temperature, 108phase changes for diacyl-phospholipids,
109protein denaturation, 397–398,
Transitions, 380in CD spectra, 386
Translation distance, 94Translation, 266–287
arrest, 424of synthetic polynucleotides, 267
Translocases, 297–299tom, 297tim, 298tic, 299toc, 299
Translocon, 295, 424–426organization, 426
Transmissable spongiform encephalopathies,431, 434
BSE, 466–468incidence in UK, 467inherited forms associated with PNRP
mutations, 435UK advisory committee, 467
Transport, 153Transthyretin, 428–430
mutations and disease progression, 430complex with RBP, 429
Tricarboxylic acid cyle (TCA) see Citricacid cycle
Triglycerides see TriacylglycerolsTriose phosphate isomerase, 179, 215–218
catalytic fine tuning, 218catalytic reaction, 209imidizolate in catalysis, 215–217mechanism of conversion of DHAP,
217pH dependence of catalysis, 216transition state analogues, 208
Triple helices, 94–97cross-linking, 97thermal denaturation, 96structure, 95
Triple resonance experiments, 373Triplet, 380
of genetic code, 268tRNA, see transfer RNATropocollagen, 6, 94
Trypanosoma brucei, 290Trypsin, 287
activation of other proteolytic enzymes,238
active site substrate specificity, 212catalytic mechanism, 212–215inhibitor, 224protein sequencing, 166, 167structural homology, 177
Tryptophanaborbance spectrum, 381anisotropy, 383blue shift, 381chemical properties, 32data, 22fluorescence, 381genetic code specification, 268helix/strand propensity, 41hydropathy index, 112optical rotation, 34secondary structure preference, 180spectroscopic properties, 30
TSEs see transmissible spongiformencephalopathies
Tuberculosis, 445Tubulin, 377–379Tumour suppressor genes, 252, 470, 474Turns, 46–48
β-bends, 48γ-turns, 47
28S rRNA, 25723S rRNA, 269
secondary structure prediction, 280tertiary structure, 281
Two dimensional gel electrophoresis,270
Two dimensional NMR spectroscopy,365–370
H/D exchange measurements, 412–413Twort, F., 443Tyrosine kinases, 239–241Tyrosine
aborbance spectrum, 381chemical properties, 31–32data, 22formation of tyrosyl adenylate, 218genetic code specification, 268helix/strand propensity, 41hydropathy index, 112phosphorylation, 239reaction with nucleophiles, 31secondary structure preference, 180spectroscopic properties, 30
Tyrosyl tRNA synthetase, 218–221active site structure, 219binding affinities for substrates, 218–219characteristic motifs, 219–220mutagenesis of critical residues, 219three dimensional enzyme structure,
218

528 INDEX
U1-snRNA, 265, 270U2-snRNA, 265U4/U6-snRNA, 265U5-snRNA, 265Ubiquinol-cytochrome c oxidoreductase, see
also cytochrome bc1 complex,132–137
Ubiquinone, 133–134, 193Ubiquitin, 304-305
addition to CFTR, 427ATP dependent activation, 305E1 enzymes, 304E2 enzymes, 304E3 enzymes, 304NMR spectrum, 367sequence, 304tertiary structure, 304
Ultracentrifugation, 322–323Ultrafiltration, 333Uncertainty principle, 349Unimolecular reaction, 192, 195, 208Uniport, 154Unit cell, 351Unsaturation, 107Unwin, N., 114, 376Urea, 193, 396, 400Urease, 207, 358Urey, H., 162Uridine monophosphate, 207
Vaccination, 75, 440Valine
chemical properties, 24data, 22genetic code specification, 268helix/strand propensity, 112hydropathy index, 41secondary structure preference, 180
Van der Waals interactions, 53, 56–58in collagen structure, 95
Van der Waals volume, 18–22Vapour diffusion, 359Variable domain, in gp120, 454–455Variable loop, 268–269Variant CJD, 466–468vCJD, see variant CJD
Vectors, 313–319pBR322, 316pET, 319
Vibrational states, 380Vibrio cholerae, 439Vif, 446, 448Viral fusion, 456–457,Viruses, 442–443
Baltimore classification, 444flu virus, 458human papilloma, 475tobacco mosaic, 443
Viscosity, 383Visual cycle, 118Vitamin B6 (pyridoxine), 29, 193Vitamin C and scurvy, 193, 429Vitamin K3, 105, 193Vitamins, 193Vitrified water, 377Vmax, maximal enzyme catalysed velocity,
198–202competitive inhibition, 225–226non-competitive inhibition, 226–227
Vogelstein, B., 470Voyeur chlorophylls, 125Vpr, 446, 448Vpu, 446, 448
Walker, J.E., 147Water elimination of, 16Water splitting reaction, 193Water, metastable form, 377Watson, J.D., 2, 247Wavelength, 348Wavenumber, 381Waves, 353–354Western Blotting, 337, 465WHO, see world health organizationWhooping cough, see pertussis,Wiley, D., 459Wilkins, M.F., 2, 247Wilm’s tumour, 265Wilson, I., 459Wohler, F., 1World Health Organization, 11,
465
World wide web, 185Wuthrich, K., 369
Xenopus laevis, 261, 308X-ray crystallography, 349–359
Bravais lattices and unit cells, 351–352experimental set-up, 350frequency range and measurement, 349isomorphous replacement, 356–357multiple anomalous dispersion, 357national facilities, 392phase problem, 356–357protein crystallization, 358–360R factor, 358refinement of structures, 357–358synchotron radiation, 357wavelengths, 350
X-ray, 348
Yarus inhibitor, 280Yeast genome sequencing, 9
flavocytochrome b2, 299secretion mutants, 424tRNA structure, 269
Yersenia pestis, 168YO2 (fractional saturation with O2), 68–70Yonath, A., 283Yoshikawa, S., 138
Zanamivir, 464, 465Zif268, 262Zinc fingers, 261–263
HIV proteins, 449primary sequence, 262
Zinc ioncarbonic anhydrase, 206carboxypeptidase, 192, 193cytochrome oxidase, 139p53, 472proteases, 136regulation of activity of aspartate
trans-carbamoylase, 235tubulin assembly, 377–379
Zn fingers, 261Zwitterion, 13, 15Zymogens, 237–238, 287, 310




















